Изобретение относится к медицинским токсикологическим исследованиям, в частности к санитарной токсикологии, и может быть использовано для количественного определения бенз(а)пирена в крови, для оценки риска здоровью человека и разработки мероприятий по обеспечению химической безопасности.
Бенз(а)пирен является одним из наиболее сильных канцерогенов среди полиароматических углеводородов (далее ПАУ). Концентрация бенз(а)пирена в воздухе на уровне 3-6 нг/м3 при длительном воздействии может привести к увеличению частоты рака легкого у населения. Поэтому в России и многих других странах законодательно введены максимально допустимые пределы для бенз(а)пирена, что, в свою очередь, обусловливает необходимость разработки и применения соответствующих методов контроля.
Для определения бенз(а)пирена и других ПАУ в различных средах используют методы газовой и жидкостной хроматографии, масс-спектроскопии и т.д. Наиболее высокую достоверность идентификации и чувствительность имеют методы, основанные на использовании высокоэффективной жидкостной хроматографии с обращенной фазой. Определение бенз(а)пирена в различных объектах окружающей среды сильно осложняется тем, что он присутствует обычно в очень малых концентрациях. При определении возможны потери вещества на различных этапах анализа, в частности в результате улетучивания его в процессе отгонки растворителей.
Анализ литературы показал, что известные химико-аналитические методы касаются в основном определения метаболитов бенз(а)пирена в биологических средах при его экспозиции различными путями. Это связано, вероятнее всего, с тем, что бенз(а)пирен является «латентным» химическим веществом. При попадании в организм он метаболизирует с образованием более токсичных соединений, которые вызывают канцерогенный и другие опасные для организма эффекты.
Известны различные способы определения бенз(а)пирена: в пищевых продуктах, в биологических объектах (жире, мышцах и тканях).
В соответствии с СанПиН 2.3.2.560-96 допустимый уровень содержания бенз(а)пирена в пищевых продуктах составляет 0,001 мг/кг. Загрязнение пищевых продуктов бенз(а)пиреном происходит из воздуха, воды и в результате переработки пищевых продуктов. Применение открытого огня или жаровен, для которых используют древесный уголь, могут вызывать загрязнения ПАУ не только воздуха, но и пищи, которая готовится таким способом.
Например, известен способ определения бенз(а)пирена в пищевых продуктах животного происхождения (копченых мясных и рыбных продуктах) [1]. Пробоподготовка включает следующие процедуры: гомогенизированную пробу экстрагируют хлористым метиленом, экстракт фильтруют через бумажный фильтр и слой безводного сульфата натрия. Фильтрат упаривают на роторном испарителе, перерастворяют в 100 см3 гексана, переносят в делительную воронку емкостью на 1000 см3 и двумя порциями по 50 см3 смеси диметилформамида и воды переэкстрагируют. Отделяют нижний слой, а из верхнего гексанового экстрагируют ПАУ повторно. Гексановый слой отбрасывают, а объединенный диметилформамидный экстракт разбавляют 100 см3 и экстрагируют ПАУ гексаном трижды по 40 см3. Гексановые экстракты объединяют и промывают водой двумя порциями по 50 см3. Фильтруют через бумажный фильтр и слой безводного сульфата натрия помещают в грушевидную колбу на 200 см3. Обезвоженный экстракт упаривают до объема 1-1,5 см3 а остаток растворителя удаляют током азота или воздуха. Высушенный экстракт растворяют в 200 мм3 ацетонитрила и анализируют методом тонкослойной хроматографии. Способ позволяет определить 0,5-1 мкг бенз(а)пирена в 1 кг массы биоматериала.
Также известен метод определения бенз(а)пирена в копченостях методом высокоэффективной жидкостной хроматографии (ВЭЖХ) с флуориметрической и УФ-детекцией [2]. Навеску массой 5 г измельчают, перетирают с 0,2 г безводного сульфата натрия и помещают в коническую колбу. Туда же добавляют 50 мл изопропанола и проводят экстракцию в течение 15 минут. Экстракт фильтруют в круглодонную колбу, добавляют 6 мл воды и 0,2 г гидроксида калия. Проводят гидролиз экстракта в течение 30 минут в колбе с обратным холодильником. Гидролизат фильтруют в стакан, добавляют 60 мл воды и экстрагируют 3×10 мл хлороформа (нижний слой). Органические вытяжки объединяют, осушают над хлоридом кальция и упаривают на роторном испарителе до объема 3-5 мл. Упаренную вытяжку переносят на стеклянную колонку диаметром 1-2 см, заполненную на 5 см силикагелем с зернением 60-140 мкм, и элюируют полиароматические углеводороды 30 мл смеси «гексан-хлороформ» 80:20. Элюат упаривают на роторном испарителе до объема 3-5 мл, переносят в пробирку на 5 мл и отгоняют растворитель током воздуха. Сухой остаток перерастворяют в 500 мкл ацетонитрила (смеси «ацетонитрил-вода» 95:5) и анализируют на колонке с Диасорбом C16 с использованием УФ- и флуориметрической детекции.
Также известны: Способ определения ПАУ в биологических объектах [3] и Способ количественного определения углеводородов в биологических объектах [4]. Однако их недостатком является то, что определение производится только суммы ПАУ, без разделения на составляющие, что делает известные способы неселективными.
Наиболее близким к предлагаемому изобретению является способ определения бенз(а)пирена в биологическом объекте - мышечной ткани, методом ВЭЖХ с флуориметрическим детектором [5]. Для определения бенз(а)пирена 3 г мускула гомогенизировали жидким азотом и извлекали из матрицы 50 см3 гексана при действии ультразвуком в течение 30 минут. Гомогенаты фильтровали через безводный сульфат натрия и концентрировали приблизительно до 1 см3 при 45°C в ротационном испарителе. Сконцентрированный раствор очищали, используя активизированный патрон Florisil. Бенз(а)пирен элюировали с патрона 18 см3 раствора гексана и дихлорметана в соотношении 3:1, элюат высушивали при 45°C, затем растворяли в 1 см3 ацетонитрила и обрабатывали ультразвуком. Затем 20 мм3 фильтрованного раствора анализировали, используя жидкостную хроматографию с детектором флюоресценции. Недостатком указанного известного способа является низкая чувствительность определения (нижний предел определения 1 мкг/кг), поэтому способ применяется в токсикологических исследованиях.
Технический результат, достигаемый предлагаемым изобретением, заключается в обеспечении высокой чувствительности способа при одновременном обеспечении селективности и его доступности для серийных анализов.
Указанный технический результат достигается предлагаемым способом количественного определения бенз(а)пирена в крови методом жидкостной хроматографии, включающим отбор пробы крови, ее твердофазную экстракцию и исследование экстракта путем жидкостной хроматографии, при этом новым является то, что после отбора пробы крови проводят ее консервацию добавлением гепарина, подвергают пробу крови центрифугированию со скоростью 2000 об/мин в течение 5 мин с отделением при этом плазмы и форменных элементов, далее осуществляют раздельную пробоподготовку указанных плазмы и форменных элементов крови, при этом для пробоподготовки плазмы осуществляют твердофазную экстракцию плазмы на сорбенте Oasis HLB путем последовательного пропускания через указанный сорбент ацетонитрила, дистиллированной воды, плазмы, дистиллированной воды, 50%-ного водного раствора ацетонитрила, затем картридж с сорбентом высушивают под вакуумом и далее пропускают через него экстрагент - 100%-ный метиленхлорид, экстракт высушивают в потоке воздуха и сухой остаток перерастворяют в 100%-ном ацетонитриле, получая экстракт плазмы, а пробоподготовку форменных элементов пробы крови осуществляют дисперсионной твердофазной экстракцией путем добавления к ним 100%-ного ацетонитрила, перемешивания, добавления набора солей по методу QuECHeRS для экстракции, перемешивания, дальнейшего центрифугирования, отбора верхней фазы и добавления к ней набора солей по методу QuECHeRS для очистки, перемешивания, повторного центрифугирования этой очищенной верхней части и вновь отбора верхней фазы - экстракта форменных элементов пробы крови для анализа, затем полученные экстракты плазмы и форменных элементов пробы крови анализируют по отдельности методом жидкостной хроматографии, используя в качестве подвижной фазы смесь ацетонитрила и воды при их изменяющемся соотношении от 60:40 об.% до 90:10 об.% соответственно, а также 100%-ный ацетонитрил в градиентном режиме, который осуществляют при хроматографии путем подачи вначале в течение 1 мин подвижной фазы, состоящей из смеси ацетонитрила и воды в соотношении 60:40 об.%, затем путем увеличения в течение 3 мин в подвижной фазе ацетонитрила до 68 об.%, дальнейшего увеличения его концентрации в течение 1,5 мин до 70 об.%, затем увеличения концентрации ацетонитрила в течение 1,5 мин до 90 об.%, а также увеличения ацетонитрила до 100% в течение следующих 4,5 мин. и пропускания такой подвижной фазы в течение еще 1,5 мин с последующим резким снижением объемного количества ацетонитрила до 60 об.% и пропусканием такой подвижной фазы через колонку в течение 4 мин до уравновешивания колонки, а количество бенз(а)пирена в крови устанавливают по градуировочному графику, причем результаты анализов, полученные при хроматографировании экстрактов плазмы и форменных элементов, суммируют.
Скорость потока подвижной фазы составляет 1,5 мл/мин.
Жидкостную хроматографию проводят на жидкостном хроматографе «Agilent 1200» с флуориметрическим детектором при длине волны возбуждения 265 нм и длине волны эмиссии 412 нм при температуре колонки 27°C.
Указанный технический результат достигается за счет следующего.
Благодаря тому, что отобранную пробу крови подвергают консервации путем добавления гепарина, обеспечивается стабилизация цельной крови для дальнейшего разделения на плазму и форменные элементы.
Благодаря тому, что отобранную пробу крови подвергают центрифугированию со скоростью 2000 об/мин в течение 5 мин, происходит разделение пробы на фракции, что обеспечивает раздельный анализ плазмы и форменных элементов при определении бенз(а)пирена.
Экспериментальным путем было установлено, что высокая чувствительность и точность определения бенз(а)пирена в крови достигается лишь при строго определенной последовательности пробоподготовки плазмы и форменных элементов, полученных из пробы.
Пропускание через указанный сорбент Oasis HLB ацетонитрила и дистиллированной воды обеспечивает очистку сорбента от возможного присутствия посторонних примесей и увлажнение сорбента перед пропусканием плазмы (в целом - это подготовка сорбента к анализу). А промывка сорбента после пропускания плазмы дистиллированной водой и 50%-ным водным раствором ацетонитрила необходима для цели удаления с сорбента слабоудерживаемых компонентов биоматериала и повышения селективности извлечения. Использование водного раствора ацетонитрила именно такой концентрации обеспечивает хорошее удаление ненужных компонентов, и в то же время не производится экстракция целевого компонента.
Сушка картриджа с сорбентом под вакуумом необходима для удаления остатков воды, которые мешают дальнейшему проведению анализа (вода переходит в экстракт метиленхлорида, при выпаривании которого капля воды неизвестного объема не испаряется, а остается в пробирке, в которой перерастворяют высушенный экстракт. Таким образом остается неизвестным общий объем анализируемой жидкости, что влияет на точность расчета концентрации бенз(а)пирена в плазме крови.
Сушка экстракта после пропускания через сорбент экстрагента 100%-ного метиленхлорида необходима для перевода аналита в совместимый с подвижной фазой растворитель (ацетонитрил). А перерастворение сухого остатка в 100%-ном ацетонитриле обеспечивает полный переход аналита (бенз(а)пирена) в анализируемый раствор.
Предложенная последовательность пробоподготовки форменных элементов пробы крови также обусловлена указанным выше химизмом.
Проведение последующего анализа экстракта плазмы и форменных элементов методом жидкостной хроматографии с особыми режимами, а именно: используя в качестве подвижной фазы смесь ацетонитрила и воды при их изменяющемся соотношении от 60:40 об.% до 90:10 об.% соответственно, а также 100%-ный ацетонитрил в градиентном режиме, который осуществляют при хроматографии путем подачи вначале в течение 1 мин подвижной фазы, состоящей из смеси ацетонитрила и воды в соотношении 60:40 об.%, затем путем увеличения в течение 3 мин в подвижной фазе ацетонитрила до 68 об.%, дальнейшего увеличения его концентрации в течение 1,5 мин до 70 об.%, затем увеличения концентрации ацетонитрила в течение 1,5 мин до 90 об.%, а также увеличения ацетонитрила до 100% в течение следующих 4,5 мин и пропускания такой подвижной фазы в течение еще 1,5 мин с последующим резким снижением объемного количества ацетонитрила до 60 об.% и пропусканием такой подвижной фазы через колонку в течение 4 мин до уравновешивания колонки, соответствует оптимизации элюирования, а значит, обеспечивает высокую чувствительность предлагаемого способа. Следует пояснить, что изменение объемного соотношения ацетонитрила и воды в подвижной фазе осуществляется за счет работы градиентного насоса Agilent 1200, смешивающего от 2 до 4 компонентов подвижной фазы.
Исследование экстрактов на жидкостном хроматографе «Agilent 1200» с флуориметрическим детектором при длине волны возбуждения 265 нм и длине волны эмиссии 412 нм при температуре колонки 27°C обусловлено достижением максимального сигнала флуориметрического детектора, что влияет на чувствительность и точность определения.
Скорость потока подвижной фазы 1,5 мл/мин является оптимальной при проведении исследований.
Количество бенз(а)пирена в крови устанавливают по градуировочному графику, причем результаты анализов, полученные при хроматографировании экстрактов плазмы и форменных элементов, суммируют. Это позволяет повысить точность определения, т.к. пробоподготовку плазмы и форменных элементов следует производить по-разному.
Для осуществления предлагаемого способа проводят следующие операции в нижеуказанной последовательности:
- производят отбор пробы крови в количестве 2 см3;
- переносят указанную пробу в центрифужную пробирку и центрифугируют со скоростью 2000 об/мин в течение 5 мин;
- верхнюю часть - плазму, отделяют от нижней части - форменных элементов;
- проводят по отдельности их экстракцию.
Алгоритм экстракций
Твердофазную экстракцию плазмы проводят по следующей схеме: кондиционирование картриджа Oasis HLB 1 сс (30 мг) 1 см3 ацетонитрила, 1 см3 дистиллированной воды, далее загрузка 0,5 см3 плазмы на картридж Oasis HLB 1 сс (30 мг), промывка картриджа с нанесенной пробой 1 см3 дистиллированной воды, 0,2 см3 50%-ного водного раствора ацетонитрила, высушивание картриджа в токе воздуха посредством вакуумного насоса, элюирование 1 см3 100%-ным метиленхлоридом. Затем метиленхлорид высушивают в токе воздуха, сухой остаток перерастворяют в 0,5 см3 100%-ного ацетонитрила. Полученный экстракт плазмы в дальнейшем анализируют в количестве 20 мм3 на жидкостном хроматографе.
Дисперсионная твердофазная экстракция форменных элементов: к 0,5 см3 форменных элементов прибавляют 2 см3 100-%-ного ацетонитрила, интенсивно встряхивая 1 мин, при этом белки крови сворачиваются, добавляют 250 мг набора солей по методу QuECHeRS для экстракции (состав: магния сульфат, натрия хлорид, натрия цитрат, динатриевая соль лимонной кислоты), встряхивают интенсивно в течение 1 минуты, центрифугируют 10 минут со скоростью 2000 об/мин, при этом образуется 3 слоя, 1,5 см3 верхнего слоя переносят в другую пробирку, в которой содержится 120 мг набора солей по методу QuECHeRS для очистки (состав: магния сульфат, сорбент С18), встряхивают 1 мин, вновь центрифугируют 10 минут со скоростью 2000 об/мин, отбирают верхний слой экстракта форменных элементов и анализируют 20 мм3 на жидкостном хроматографе.
Условия проведения анализа жидкостной хроматографии для обоих экстрактов соответствуют следующим характеристикам:
длина волны возбуждения
длина волны эмиссии
Количество бенз(а)пирена в крови устанавливают по градуировочному графику, причем результаты анализов, полученные при хроматографировании экстрактов плазмы и форменных элементов, суммируют.
Для построения градуировочного графика в мерную колбу вместимостью 100 см3 вносят 1 см3 стандартного образца (ГСО) бенз(а)пирена в ацетонитриле с концентрацией 100 мкг/см3, доводят содержимое колбы до метки ацетонитрилом и перешивают. Концентрация бенз(а)пирена в исходном растворе (раствор №1) составляет 1 мкг/см3. Затем 2,5 см3 раствора №1 вносят в мерную колбу вместимостью 100 см3 и доводят ацетонитрилом до метки. Концентрация бенз(а)пирена в разбавленном растворе (раствор №2) составляет 25 нг/см3. Используют свежеприготовленный раствор.
Градуировочную характеристику устанавливают методом абсолютной градуировки. Она выражает зависимость площади пика на хроматограмме (мм2) от массовой концентрации бенз(а)пирена в крови (мг/дм3) и строится по 5 сериям рабочих стандартных растворов. Каждую серию, состоящую из 3 стандартных растворов, готовят в мерных пробирках вместимостью 5 см3. Для этого в каждую пробирку вносят растворы бенз(а)пирена для градуировки №1 и 2 в соответствии с таблицей 1, доводят содержимое пробирки до метки 5 см3 цельной кровью и перемешивают.
Отработка оптимальных жидкостно-хроматографических параметров для определения бенз(а)пирена в крови предлагаемым способом осуществлялась с использованием жидкостного хроматографа «Agilent 1200», оснащенного термостатом колонок, градиентным насосом и флуориметрическим детектором.
Полнота разделения бенз(а)пирена в присутствии компонентов матрицы достигнута на колонке Zorbax длиной 50 мм и внутренним диаметром 4,6 мм с сорбентом Eclipse РАН (C18) зернением 3,5 мкм при температуре колонки 27°C, при использовании режимов указанного выше градиентного изменения состава подвижной фазы. Максимальный сигнал флуориметрического детектора получен при длине волны возбуждения 265 нм и длине волны эмиссии 412 нм. В случае, если не был применен градиентный режим элюирования чувствительность способа резко изменялась.
В процессе исследований по выбору оптимальных условий проведения твердофазной экстракции (ТФЭ) бенз(а)пирена из плазмы крови, включающей кондиционирование сорбента Oasis HLB растворителем - 100%-ным ацетонитрилом и дистиллированной водой, промывку картриджа от мешающих компонентов матрицы дистиллированной водой и 50%-ным водным раствором ацетонитрила после нанесения анализируемой пробы плазмы на указанный сорбент и последующую десорбцию бенз(а)пирена 100%-ным метиленхлоридом, были апробированы 2 варианта пробоподготовки: без предварительной деглюкуронизации и с деглюкуронизацией путем добавления в плазму раствора β-глюкуронидазы в ацетатном буфере (2500 ME), перемешивания и термостатирования на водяной бане при температуре 37°C в течение 1 часа. Данные, полученные в ходе испытаний, приведены в таблице 3.
метиленхлоридом
Данные, приведенные в таблице 3, показывают, что максимальная степень экстракции бенз(а)пирена из плазмы крови была достигнута при использовании режима №1, который применяли в дальнейших исследованиях.
Экспериментальным путем также были установлены, наряду с необходимостью применения в качестве экстрагента 100%-ного метиленхлорида, и другие характеристики и режимы предлагаемого способа, обеспечивающие эффективное и селективное извлечение бенз(а)пирена из пробы крови и повышение чувствительности и точности определения, а именно: центрифугирование пробы крови при 2000 об/мин, использование при твердофазной экстракции 1 см3 100%-ного ацетонитрила и 1 см3 дистиллированной воды для конденционирования сорбента, промывание картриджа 1 см3 дистиллированной воды и 0,2 см3 50%-ного водного раствора ацетонитрила после пропускания фракции плазмы крови перед экстракцией 1,0 см3 100%-ного метиленхлорида.
При изучении выбора оптимальных условий извлечения бенз(а)пирена из форменных элементов методом дисперсионной твердофазной экстракции, включающей добавление к клеткам крови 100%-ного ацетонитрила и набора солей по методу QuECHeRS «для экстракции», а также отделение и очистку экстракта набором солей по методу QuECHeRS «для очистки», было установлено, что степень экстракции составляет 64%.
Пример конкретного выполнения предлагаемого способа
Анализируют пробы крови (обследуемая группа населения). Каждую пробу крови анализируют дважды. Свежеотобранную пробу крови объемом 2 см3 центрифугируют со скоростью 2000 об/мин в течение 5 мин. Разделяют на фракции плазмы и форменных элементов. Отбирают 0,5 см3 плазмы и проводят твердофазную экстракцию, последовательно пропуская под вакуумом через картридж с сорбентом Oasis HLB 3 сс 1 см3 100%-ного ацетонитрила, 1 см3 дистиллированной воды, 0,5 см3 плазмы, 1 см3 дистиллированной воды, 0,2 см3 50%-ного водного раствора ацетонитрила. Затем картридж с сорбентом высушивают под вакуумом, переносят в накопительный сосуд (бюкс) и пропускают через сорбент 1,0 см3 100%-ного хлористого метилена. Скорость экстракции (скорость потока) не более см3/мин. Аликвотную часть полученного экстракта отбирают и хроматографируют на жидкостном хроматографе Agilent серии 1200 с флуориметрическим детектором на колонке Zorbax длиной 50 мм и внутренним диаметром 4,6 мм с сорбентом Eclipse РАН C18 при температуре колонки 27°C, при использовании в качестве подвижной фазы смеси ацетонитрила и воды со скоростью потока 1,5 см3/мин и оптимизации элюирования в градиентном режиме (1 мин подача подвижной фазы: смеси 60 об.% ацетонитрила и 40 об.% воды, увеличение ацетонитрила с 60 об.% до 68 об.% в течение 3 мин, увеличение ацетонитрила с 68 об.% до 70 об.% в течение 0,5 мин, увеличение ацетонитрила с 70 об.% до 90 об.% в течение 1,5 мин, увеличение ацетонитрила с 90 об.% до 100 об.% в течение 4,5 мин, подача 100%-ного ацетонитрила в течение 1,5 мин, затем резкое снижение ацетонитрила до 60 об.% и подача 60 об.% ацетонитрила в течение 4 мин до уравновешивания колонки). При этом длина волны возбуждения флуориметрического детектора составляла 265 нм и длина волны эмиссии - 412 нм.
По градуировочному графику определяют содержание бенз(а)пирена в плазме (пробы 1-5). Полученные результаты приведены в таблице 4.
Отбирают 0,5 см3 форменных элементов и проводят дисперсионную твердофазную экстракцию: к 0,5 см3 форменных элементов прибавляют 2 см3 100%-ного ацетонитрила, интенсивно встряхивают в течение 1 мин, добавляют 250 мг набора солей по методу QuECHeRS «для экстракции», встряхивают интенсивно в течение 1 минуты, центрифугируют 10 минут со скоростью 2000 об/мин, при этом образуется 3 слоя, 1,5 см3 верхнего слоя переносят в другую пробирку, в которой содержится 120 мг набор солей по методу QuECHeRS «для очистки», встряхивают 1 мин, вновь центрифугируют 10 минут со скоростью 2000 об/мин, отбирают верхний слой и анализируют 20 мм3 на жидкостном хроматографе Agilent серии 1200 аналогично плазме.
По градуировочному графику определяют содержание бенз(а)пирена в форменных элементах (пробы 1-5). Полученные результаты приведены в таблице 4.
образца
параллельных
определений,
мг/дм3
мг/дм3
параллельных
определений,
мг/дм3
мг/дм3
мг/дм3
%
Чувствительность определения бенз(а)пирена в анализируемом объеме крови (10 мм3) составила 0,2 пг (или 0,00001 мг/дм3). Погрешность определения не превышает 23,6%.
Методика выполнения измерений обеспечивает получение результатов измерений с погрешностью, не превышающей значений, приведенных в таблицах 5 и 6.
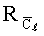
Применение предлагаемого способа позволяет повысить чувствительность определения бенз(а)пирена, как минимум, в 10 раз, например, по сравнению с прототипом.
Заявляемый способ прост и может быть рекомендован для серийных анализов.
Источники информации
1. Патент РФ №2153167.
2. Сычев С.Н. Методы совершенствования хроматографических систем и механизмы удерживания в ВЭЖХ. Орел, Орловский ГТУ, 2000 г., с.211.
3. Патент РФ №2187106.
4. Патент РФ №1337764.
5. Kang Н., Jeong S., Cho М., Cho J. Changes of biomarkers with oral exposure to benzo(a)pyrene, phenanthrene and pyrene in rats // J. Vet. Sci. 2007. - 8(4). - p.361-368.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ БЕНЗ(А)ПИРЕНА В МОЧЕ МЕТОДОМ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2011 |
|
RU2466406C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ КОНЦЕНТРАЦИИ АКРОЛЕИНА В АТМОСФЕРНОМ ВОЗДУХЕ МЕТОДОМ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2014 |
|
RU2556294C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ПЕНТАХЛОРФЕНОЛА В КРОВИ МЕТОДОМ ГАЗОХРОМАТОГРАФИЧЕСКОГО АНАЛИЗА | 2014 |
|
RU2546527C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ 2,4-ДИХЛОРФЕНОЛА В КРОВИ МЕТОДОМ ГАЗОХРОМАТОГРАФИЧЕСКОГО АНАЛИЗА | 2013 |
|
RU2521277C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ ФУМАРОВОЙ И МАЛЕИНОВОЙ КИСЛОТ В ПЛАЗМЕ КРОВИ МЕТОДОМ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2018 |
|
RU2677341C1 |
| Способ определения массовых концентраций фенола и пирокатехина в крови методом высокоэффективной жидкостной хроматографии | 2022 |
|
RU2786509C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ДИМЕТИЛТЕРЕФТАЛАТА В МОЧЕ МЕТОДОМ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2010 |
|
RU2425380C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ АКРИЛОНИТРИЛА В КРОВИ МЕТОДОМ ГАЗОХРОМАТОГРАФИЧЕСКОГО АНАЛИЗА | 2011 |
|
RU2452961C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ N-НИТРОЗОДИМЕТИЛАМИНА И N-НИТРОЗОДИЭТИЛАМИНА В МОЧЕ МЕТОДОМ ГАЗОХРОМАТОГРАФИЧЕСКОГО АНАЛИЗА | 2013 |
|
RU2521711C1 |
| Способ количественного определения N-нитрозаминов в детских кашах | 2015 |
|
RU2613303C1 |
Изобретение относится к медицинским токсикологическим исследованиям, в частности к санитарной токсикологии, и может быть использовано для количественного определения бенз(а)пирена в крови, для оценки риска здоровью человека и разработки мероприятий по обеспечению химической безопасности. Способ осуществляется следующим образом. Каждую пробу крови анализируют дважды. Свежеотобранную пробу крови центрифугируют со скоростью 2000 об/мин в течение 5 мин. Разделяют на фракции плазмы и форменных элементов. Проводят твердофазную экстракцию плазмы путем последовательного пропускания под вакуумом через картридж с сорбентом Oasis HLB 3 сс 100%-ного ацетонитрила, дистиллированной воды, плазмы, дистиллированной воды, 50%-ного водного раствора ацетонитрила. Затем картридж с сорбентом высушивают под вакуумом и пропускают через сорбент 100%-ный метиленхлорид. Аликвотную часть полученного экстракта хроматографируют. Для получения экстракта форменных элементов проводят дисперсионную твердофазную экстракцию: добавляют к ним 100%-ный ацетонитрил, интенсивно встряхивают. Добавляют набор солей QuECHeRS для экстракции, встряхивают интенсивно, центрифугируют 10 минут со скоростью 2000 об/мин, при этом образуется 3 слоя, верхний слой переносят в другую пробирку, в которой содержится набор солей QuECHeRS для очистки, центрифугируют со скоростью 2000 об/мин, отбирают верхний слой. Экстракт плазмы и форменных элементов анализируют на жидкостном хроматографе Agilent серии 1200 с флуориметрическим детектором на колонке Zorbax длиной 50 мм и внутренним диаметром 4,6 мм с сорбентом Eclipse РАН C18 при температуре колонки 27°C, при использовании в качестве подвижной фазы смеси ацетонитрила и воды со скоростью потока 1,5 см3/мин и оптимизации элюирования в градиентном режиме (1 мин подача подвижной фазы 60 об.% ацетонитрила и 40 об.% воды, увеличение ацетонитрила с 60 об.% до 68 об.% в течение 3 мин, увеличение ацетонитрила с 68 об.% до 70 об.% в течение 0,5 мин, увеличение ацетонитрила с 70 об.% до 90 об.% в течение 1,5 мин, увеличение ацетонитрила с 90 об.% до 100 об.% в течение 4,5 мин, подача 100%-ного ацетонитрила в течение 1,5 мин, затем снижение ацетонитрила до 60 об.% и подача 60%-ного ацетонитрила в течение 4 мин до уравновешивания колонки). При этом длина волны возбуждения флуориметрического детектора составляла 265 нм и длина волны эмиссии 412 нм. По градуировочному графику отдельно определяют содержание бенз(а)пирена в плазме и форменных элементах, а результаты суммируют. Изобретение обеспечивает высокую чувствительность способа при одновременном обеспечении селективности и его доступности для серийных анализов. 2 з.п. ф-лы, 6 табл., 1 пр.
1. Способ количественного определения бенз(а)пирена в крови методом жидкостной хроматографии, включающий отбор пробы крови, ее твердофазную экстракцию и исследование экстракта путем жидкостной хроматографии, отличающийся тем, что после отбора пробы крови проводят ее консервацию добавлением гепарина, подвергают пробу крови центрифугированию со скоростью 2000 об/мин в течение 5 мин с отделением при этом плазмы и форменных элементов, далее осуществляют раздельную пробоподготовку указанных плазмы и форменных элементов крови, при этом для пробоподготовки плазмы осуществляют твердофазную экстракцию плазмы на сорбенте Oasis HLB путем последовательного пропускания через указанный сорбент ацетонитрила, дистиллированной воды, плазмы, дистиллированной воды, 50%-ного водного раствора ацетонитрила, затем картридж с сорбентом высушивают под вакуумом и далее пропускают через него экстрагент - 100%-ный метиленхлорид, экстракт высушивают в потоке воздуха и сухой остаток перерастворяют в 100%-ном ацетонитриле, получая экстракт плазмы, а пробоподготовку форменных элементов пробы крови осуществляют дисперсионной твердофазной экстракцией путем добавления к ним 100%-ного ацетонитрила, перемешивания, добавления набора солей по методу QuECHeRS для экстракции, перемешивания, дальнейшего центрифугирования, отбора верхней фазы и добавления к ней набора солей по методу QuECHeRS для очистки, перемешивания, повторного центрифугирования этой очищенной верхней части и вновь отбора верхней фазы - экстракта форменных элементов пробы крови для анализа, затем полученные экстракты плазмы и форменных элементов пробы крови анализируют по отдельности методом жидкостной хроматографии, используя в качестве подвижной фазы смесь ацетонитрила и воды при их изменяющемся соотношении от 60:40 об.% до 90:10 об.% соответственно, а также 100%-ный ацетонитрил в градиентном режиме, который осуществляют при хроматографии путем подачи вначале в течение 1 мин подвижной фазы, состоящей из смеси ацетонитрила и воды в соотношении 60:40 об.%, затем путем увеличения в течение 3 мин в подвижной фазе ацетонитрила до 68 об.%, дальнейшего увеличения его концентрации в течение 1,5 мин до 70 об.%, затем увеличения концентрации ацетонитрила в течение 1,5 мин до 90 об.%, а также увеличения ацетонитрила до 100% в течение следующих 4,5 мин и пропускания такой подвижной фазы в течение еще 1,5 мин с последующим резким снижением объемного количества ацетонитрила до 60 об.% и пропусканием такой подвижной фазы через колонку в течение 4 мин до уравновешивания колонки, а количество бенз(а)пирена в крови устанавливают по градуировочному графику, причем результаты анализов, полученные при хроматографировании экстрактов плазмы и форменных элементов, суммируют.
2. Способ по п.1, отличающийся тем, что скорость потока подвижной фазы составляет 1,5 мл/мин.
3. Способ по п.1, отличающийся тем, что жидкостную хроматографию проводят на жидкостном хроматографе «Agilent 1200» с флуориметрическим детектором при длине волны возбуждения 265 нм и длине волны эмиссии 412 нм при температуре колонки 27°C.
| Kang Н., Jeong S., Cho М., Cho J | |||
| Changes of biomarkers with oral exposure to benzo(a)pyrene, phenanthrene and pyrene in rats // J | |||
| Vet | |||
| Sci | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Способ получения продуктов уплотнения фенолов с альдегидами | 1920 |
|
SU361A1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ БЕНЗ(А)ПИРЕНА В МОЧЕ МЕТОДОМ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ | 2011 |
|
RU2466406C1 |
| Способ количественного определения углеводородов в биологических объектах | 1984 |
|
SU1337764A1 |
| ГОСТ Р | |||

