РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке испрашивается приоритет предварительной заявки США № 61/784681, поданной 14 марта 2013, полное содержание которой включено в настоящее изобретение с помощью ссылки во всей своей полноте.
ОБЛАСТЬ ТЕХНИКИ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
В общем, варианты осуществления, описанные в настоящем изобретении, относятся к местному введению фармацевтического соединения или его соли для лечения глазных заболеваний или состояний. Описанные варианты осуществления включают офтальмологические составы фармацевтического соединения или его соли, где состав представляет собой раствор или суспензию. Раствор содержит агент, улучшающий растворимость, и является пригодным для доставки к заднему сегменту глаза субъекта.
УРОВЕНЬ ТЕХНИКИ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Лечение заболеваний или расстройств заднего сегмента глаза путем нанесения местно активного агента является неэффективным из-за неэффективной доставки активного агента в целевое место. Значительное большинство местных лекарственных средств проникает через роговицу. Однако роговица не является одинаково проницаемой для всех применяемых местно активных агентов, поскольку основная структура роговицы определяет относительное проникновение активного агента. В действительности, наибольший барьер для проникновения активного агента создает эпителий роговицы, который содержит много клеточных мембран и, следовательно, более поддается проникновению активных агентов, которые являются липофильными. Напротив, поскольку строма роговицы состоит в основном из воды, активные агенты проходят более быстро через данный самый толстый компонент роговицы, если они являются гидрофильными. Эндотелий представляет собой монослой, который также является липофильным. Активные агенты, которые являются липофильными или амфифильными, то есть они могут вести себя или как заряженные или как незаряженные молекулы, проникают через роговицу наилучшим способом. Аналогично роговице, через эпителий конъюнктивы и кровеносные сосуды в или под эпителием конъюнктивы может проникать тот же тип липофильных или двухфазных агентов. Однако, из-за свойств липофильных мембран в конъюнктиве и ее собственной сосудистой системы, большинство активных агентов обычно не проникают через конъюнктиву и в глаз. Агенты с ограниченным проникновением в сосудистые ткани в конъюнктивальной и субконъюнктивальной областях попадают в общее кровообращение.
Если активный агент проникает через роговицу в переднюю камеру глаза, все еще существуют барьеры для успешной доставки лекарственного средства в ткани заднего сегмента глаза, такие как сетчатка и сосудистая оболочка. Данные барьеры состоят, по меньшей мере, отчасти, из пассивных барьеров, таких как гидродинамика водянистой влаги, хрусталик и ресничный поясок и большой объем стекловидного тела, а также активные барьеры, такие как клеточный транспорт или насосы, расположенные в цилиарном эпителии или ионные градиенты, существующие в глазу.
Помимо проблем с местной доставкой лекарственных средств в задний сегмент глаза, существует несколько преимуществ данного способа доставки по сравнению с системной доставкой и интравитреальной или субконъюнктивальной доставкой. Интравитреальное и субконъюнктивальное введение обычно основано на применении иглы, соединенной со шприцом, для проникновения или через стенку глаза или через конъюнктивальную ткань, доставляя водные фармакологические агенты или водные суспензии агентов (например, стероидов) для неотложного лечения. Однако следует также отметить, что увеличивающееся количество способов может продолжительно доставлять груз посредством транспортных средств, таких как полимеры, органические ячейки или наночастицы, доставляя активный терапевтический агент в течение длительного или продленного периода времени. Местная доставка обеспечивает прямое нанесение на целевой орган - глаз, с относительной простотой нанесения для большинства пациентов, и благодаря направленному нанесению, необходимость меньших доз активного агента, связанных с началом действия, часто приводя к пониженному или несущественному системному воздействию. Недостатки местной доставки включают: загрязнение капель для местного применения, потенциальное требование наличия консервантов, потенциальная токсичность лекарственного средства или консерванта для глазной поверхности, ограничение проникновения большинства местных активных агентов через конъюнктиву, роговицу и переднюю камеру глаза, и риск, хотя и значительно меньший по сравнению с системной доставкой, системного поглощения лекарственных средств, что может воздействовать на другие органы, такие как сердце и легкие. Широко известные осложнения интравитреального введения включают инфекцию, отслоение сетчатки, кровоизлияние и рубцевание. Осложнения субконъюнктивального введения также включают инфекцию, рубцевание, кровоизлияние и случайное проникновение через глазное яблоко.
Из-за ограниченной проходимости многих капель для местного применения через роговичные и конъюнктивальные барьеры, одного из основных недостатков капель для местного применения, может быть необходима большая концентрация активных агентов в местной состава для того, чтобы достигать значимых терапевтических концентраций лекарственного средства в глазу. В зависимости от активного агента, концентрация состава для наружного применения может быть высокотоксичной для переднего сегмента глаза, включая роговицу и хрусталик. Следовательно, лечение заболеваний или расстройств заднего сегмента глаза будет составами, которые обеспечивают низкую биодоступность активного агента в переднем сегменте глаза, будет оказывать благоприятное действие, при этом обеспечивая доступность эффективной концентрации активного агента к заднему сегменту глаза.
Настоящие варианты осуществления изобретения обеспечивают новые состава, которые преодолевают данные проблемы, обнаруживаемые при глазной доставке существующих местных терапевтических агентов. Настоящее изобретение обеспечивает комбинированные эффекты пониженного воздействия лекарственного средства на роговицу и передний сегмент глаза, при этом увеличивая биодоступность к заднем сегменту глаза. Снижая воздействие на роговицу и увеличивая биодоступность к заднему сегменту, состав настоящего изобретения улучшает офтальмологическую переносимость и улучшает терапевтический индекс активного агента.
СУЩНОСТЬ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к фармацевтическим составам в виде раствора и/или суспензии, которые снижают воздействие активного агента в переднем сегменте глаза, например, роговице, при этом увеличивая биодоступность агента в заднем сегменте глаза, например, в центральной сосудистой оболочке глаза и/или центральной зоне сетчатки.
Варианты осуществления обеспечивают две различные формы состава, содержащего активный агент, раствор и суспензию, с улучшенными характеристиками по сравнению с другой композицией в виде геля. Варианты осуществления обеспечивают то, что активный агент в виде раствора и/или суспензии, является лучшим по сравнению с тем же самым активным ингредиентом, сформулированным в виде геля, при доставке активного ингредиента в задний сегмент глаза, при этом снижая воздействие в переднем сегменте глаза. Повышенные концентрации активного агента в переднем сегменте глаза ограничивают глазную переносимость капель для местного применения, содержащих активный агент. Следовательно, пониженная биодоступность активного агента на роговичной или конъюнктивальной поверхности, при этом поддерживая подходящие концентрации, необходимые для связывания соответствующих рецепторов в тканях-мишенях и обеспечения терапевтического эффекта в заднем сегменте глаза, таком как сосудистая оболочка глаза и сетчатка, является очень желательной и представляет собой конечный результат, достигаемый конкретными вариантами осуществления, описанными в настоящем изобретении.
Настоящее изобретение относится к композициям и способам, пригодным для лечения патологических состояний, которые возникают или обостряются глазным ангиогенезом и пропотеванием жидкости через сосуды, например, при диабетической ретинопатии (включая фоновую диабетическую ретинопатию, пролиферативную диабетическую ретинопатию и диабетический отек макулы); возрастной дегенерации макулы (ВМД) (включая неоваскулярную (влажную/экссудативную) ВМД, сухую ВМД и географическую атрофию); патологической неоваскуляризации сосудистой оболочки глаза (ХНВ) любого механизма (т.е. миопии высокой степени, травме, серповидноклеточной анемии; гистоплазмозе глаз, ангиоидных полосах сетчатки, разрыве сосудистого тракта глаза при травме, друзах оптического нерва и некоторых ретинальных дистрофиях); патологической ретинальной неоваскуляризации любого механизма (т.е. серповидно-клеточной ретинопатии, болезни Илза, глазном ишемическом синдроме, каротидно-кавернозном соустье, семейной экссудативной форме витреоретинопатии, синдроме повышенной вязкости крови, идиопатическом окклюзионном артериолите, дробьевидной ретинохороидопатии, ретинальном васкулите, саркоидозе или токсоплазмозе); увеите; окклюзию вены сетчатки (центральной или ветки); глазной травме; эдеме, вызванной хирургической операцией; неоваскуляризации, вызванной хирургической операцией; кистозном макулярном отеке; ишемии глаз; ретинопатии недоношенных; синдроме Коутса; серповидно-клеточной ретинопатии и/или неоваскулярной глаукоме. Состав настоящего изобретения содержит, по меньшей мере, один антиангиогенный агент, противовоспалительный агент или агент, ослабляющий проницаемость сосудов, для применения в лечении ангиогенных глазных расстройств.
Согласно вариантам осуществления настоящего изобретения, активный агент представляет собой ингибитор киназы. Примеры некоторых ингибиторов киназ, которые можно применять, получая полезные терапевтические результаты, включают ингибиторы рецепторных тирозинкиназ, например, без ограничения, VEGFR, FGFR, Tie-2 и эфриновые рецепторы киназ.
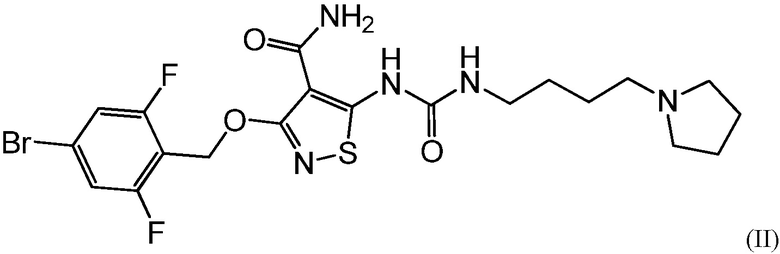
Варианты осуществления настоящего изобретения обеспечивают офтальмический состав для лечения глазной неоваскуляризации активным агентом формулы I:
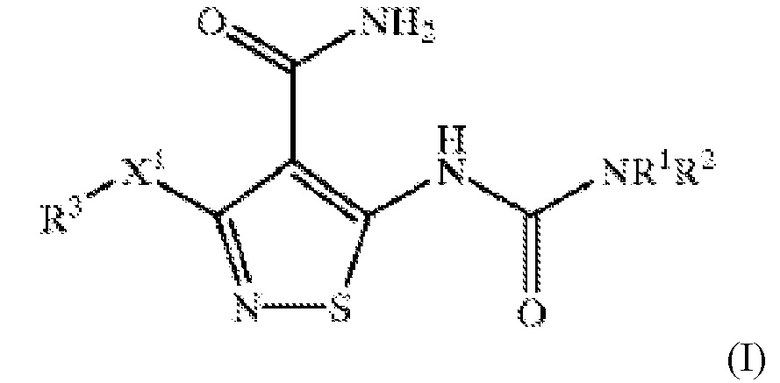
или его фармацевтически приемлемой солью; и фармацевтически приемлемые вспомогательные вещества; активный агент или фармацевтически приемлемая соль присутствует в количестве от приблизительно 0,02% до приблизительно 0,6% масс./об. так, чтобы композиция образовывала раствор или суспензию, и где активный агент определяют как: X1 представляет собой O или S; R1 представляет собой H, C1-C10 алкил, C2-C10 алкенил, C2-C10 алкинил, -C(O)(C1-C10 алкил), -(CH2)t(C6-C10 арил), -(CH2)t(4-10-членный гетероцикл), -C(O)(CH2)t(C6-C10 арил) или -C(O)(CH2)t (5-10-членный гетероцикл), где t представляет собой целое от 0 до 5; алкильная группа необязательно содержит 1 или 2 гетерофрагмента, выбранные из O, S и -N(R6)- при условии, что два O атома, два S атома или O и S атом не соединены непосредственно друг с другом; арильная и гетероциклическая R1 группа необязательно конденсирована с C6-C10 арильной группой, C5-C8 насыщенной циклической группой или 5-10-членной гетероциклической группой; 1 или 2 атома углерода в указанных выше гетероциклических фрагментах необязательно замещены оксо (═O) группой или анионом кислорода; -(CH2)t- фрагменты указанных выше R1 групп необязательно содержат углерод-углерод двойную или тройную связь, где t представляет собой целое от 2 до 5; и указанные выше R1 группы, за исключением H, необязательно замещены 1-3 R4 группами; R2 представляет собой H; R3 представляет собой -(CH2)t(C6 -C10 арил), где t представляет собой целое от 0 до 5; необязательно конденсированный с C6 -C10 арильной группой, C5 -C8 насыщенной циклической группой или 5-10-членной гетероциклической группой; -(CH2)t- фрагменты необязательно содержат углерод-углерод двойную или тройную связь, где t представляет собой целое от 2 до 5, и необязательно замещены 1-5 R4 группами; каждая R4 независимо выбрана из C1-C10 алкила, C2-C10 алкенила, C2-C10 алкинила, галогена, циано, нитро, трифторметила, трифторметокси, азидо, -OR5, -C(O)R5, -C(O)OR5, -NR6C(O)OR5, -OC(O)R5, -NR6 SO2R5, -SO2NR5 R6, -NR6C(O)R5, -C(O)NR5R6, -NR5R6, -S(O)jR7, где j представляет собой целое в диапазоне от 0 до 2, -SO3H, -NR5(CR6R7)tOR6, -(CH2)t(C6-C10 арила), -SO2(CH2)t(C6-C10 арила), -S(CH2)t(C6-C10арила), -O(CH2)t(C6-C10 арила), -(CH2)t(5-10-членного гетероцикла) и -(CR6 R7)mOR6 , где m представляет собой целое от 1 до 5, и t представляет собой целое от 0 до 5; алкильная группа необязательно содержит 1 или 2 гетеро фрагмента, выбранные из O, S и -N(R6)-, при условии, что два O атома, два S атома, или O и S атом не соединены непосредственно друг с другом; арильная и гетероциклическая R4 группы необязательно конденсированы с C6-C10 арильной группой, C5-C8 насыщенной циклической группой или 5-10-членной гетероциклической группой; 1 или 2 атома углерода в указанных выше гетероциклических фрагментах необязательно замещены оксо (═O) группой или анионом кислорода; и алкильный, арильный и гетероциклический фрагменты указанных выше R4 групп необязательно замещены 1-3 заместителями, независимо выбранными из галогена, циано, нитро, трифторметила, трифторметокси, азидо, -NR6SO2R5 , -SO2NR5R6 , -C(O)R5, -C(O)OR5 , -OC(O)R5, -NR6C(O)R5, -C(O)NR5R6, -NR5R6, -(CR6R7)mOR6, где m представляет собой целое от 1 до 5, -OR5 и заместителями, перечисленными в определении R5; и каждый R5, R6, и R7 независимо представляет собой H или C1-C6 алкил.
В другом варианте осуществления настоящего изобретения, R3 в формуле I представляет собой -(CH2)t(C6 -C10 арил), где t представляет собой целое от 1 до 3, и R3 группа необязательно замещена 1-4 R4 группами.
В другом варианте осуществления, R3 в формуле I настоящего изобретения представляет собой бензил, необязательно замещенный 1-4 заместителями, независимо выбранными из галогена и C1-C4 алкила. R3 в формуле I настоящего изобретения представляет собой бензил, замещенный 1-4 заместителями, независимо выбранные из метила, фтора, хлора и брома.
В некоторых вариантах осуществления, R1 в формуле I настоящего изобретения представляет собой -(CH2)t (5-10-членный гетероцикл), где t представляет собой целое от 0 до 5, необязательно замещенный 1 или 2 заместителями, независимо выбранные из C1-C4 алкила, гидрокси и гидроксиметила.
Настоящее изобретение относится к гетероциклическому фрагменту R1 группы в формуле I, выбранному из морфолино, пирролидинила, имидазолила, пиперазинила, пиперидинила и 2,5-диазабицикло[2,2,1]гепт-2-ила, t переменная R1 группы находится в диапазоне от 2 до 5, и R1 группа необязательно замещена одной или более гидрокси группами.
Например, гетероциклический фрагмент R1 группы в формуле I настоящего изобретения представляет собой пирролидин.
В следующих вариантах осуществления настоящего изобретения, активный агент представляет собой:
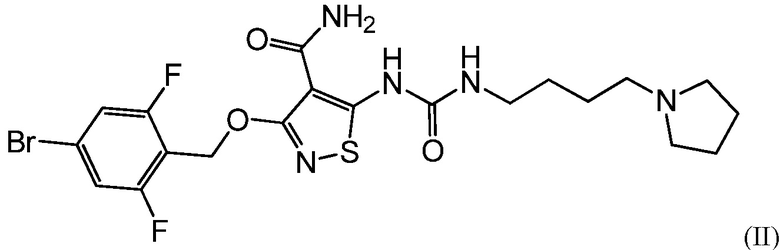 .
.
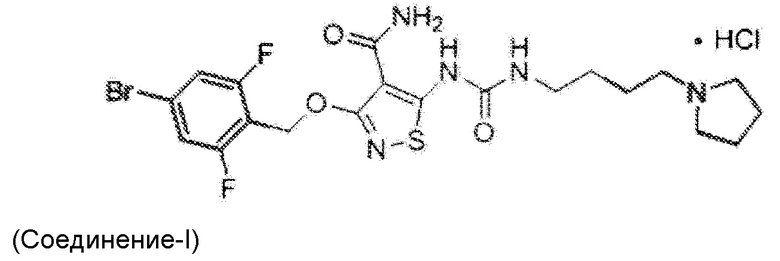
В некоторых вариантах осуществления настоящего изобретения, активный агент представляет собой гидрохлоридную соль соединения формулы II, а именно соединение-I:
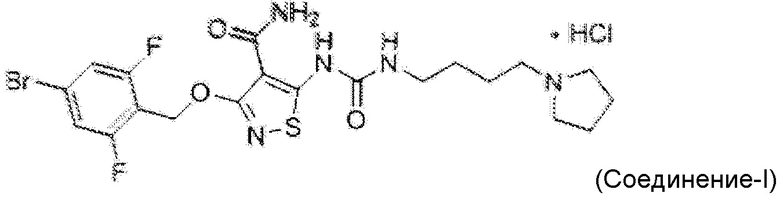 .
.
Варианты осуществления настоящего изобретения относятся к составам, содержащим от приблизительно 0,005% до приблизительно 5,0% масс./об. активного агента формул (I), (II) или его фармацевтически приемлемой соли, например, соединения-I. В некоторых вариантах осуществления концентрация соединения-I или его свободного основания (формула II) в композициях составляет приблизительно 0,005% - приблизительно 0,01%, приблизительно 0,01% - приблизительно 0,05%, приблизительно 0,05% - приблизительно 0,1%, приблизительно 0,1% - приблизительно 0,2%, приблизительно 0,2% - приблизительно 0,3%, приблизительно 0,3% - приблизительно 0,4%, приблизительно 0,4% - приблизительно 0,5%, приблизительно 0,5% - приблизительно 0,6%, приблизительно 0,6% - приблизительно 0,7%, приблизительно 0,7% - приблизительно 0,8%, приблизительно 0,8% - приблизительно 0,9%, приблизительно 0,9% - приблизительно 1,0%, приблизительно 1,1 - приблизительно 2,0%, приблизительно 2,1 - приблизительно 3,0%, приблизительно 3,1 - приблизительно 4,0%, или приблизительно 4,1- приблизительно 5,0% масс./об. для местного введения. В некоторых вариантах осуществления состава содержат приблизительно 0,005%, приблизительно 0,05%, приблизительно 0,1%, приблизительно 0,2%, приблизительно 0,3%, приблизительно 0,4%, приблизительно 0,5%, приблизительно 0,6%, приблизительно 0,7%, приблизительно 0,8%, приблизительно 0,9%, приблизительно 1,0%, приблизительно 2,0%, приблизительно 3,0%, приблизительно 4,0%, или приблизительно 5,0% масс./об. соединения-I или его свободного основания (формула II).
Настоящее изобретение относится к раствору активного агента (например, соединения-I), который содержит один или более агентов, улучшающих растворимость.
Состав, содержащий приблизительно 0,005%, приблизительно 0,05%, приблизительно 0,1%, приблизительно 0,2%, приблизительно 0,3%, приблизительно 0,4%, приблизительно 0,5%, приблизительно 0,6%, приблизительно 0,7%, приблизительно 0,8%, приблизительно 0,9%, приблизительно 1,0%, приблизительно 2,0%, приблизительно 3,0%, приблизительно 4,0% или приблизительно 5,0% масс./об. соединения формулы (I), (II) или его фармацевтически приемлемой соли, например, соединения-I, содержит агент, улучшающий растворимость. Агент, улучшающий растворимость, в составе состава может представлять собой циклодекстрин, например, 2-гидроксипропил-β-циклодекстрин, метил-β-циклодекстрин, произвольно метилированный-β-циклодекстрин, этилированный-β-циклодекстрин, триацетильный-β-циклодекстрин, перацетилированный-β-циклодекстрин, карбоксиметил-β-циклодекстрин, гидроксиэтил-β-циклодекстрин, 2-гидрокси-3-(триметиламмонио)пропил-β-циклодекстрин, глюкозил-β-циклодекстрин, мальтозил-β-циклодекстрин, сульфобутиловый эфир-β-циклодекстрин, разветвленный-β-циклодекстрин, гидроксипропил-β-циклодекстрин, произвольно метилированный-β-циклодекстрин, триметил-β-циклодекстрин или их комбинации.
В одном варианте осуществления, агент, улучшающий растворимость, в составе представляет собой 2-гидроксипропил-β-циклодекстрин или β-циклодекстринсульфобутиловый эфир. Состав содержит один или более из хлорида бензалкония (BAK), хлорида натрия и регулирующего pH агента.
В дополнительных вариантах осуществления, состав, содержащий приблизительно 0,005%, приблизительно 0,05%, приблизительно 0,1%, приблизительно 0,2%, приблизительно 0,3%, приблизительно 0,4%, приблизительно 0,5%, приблизительно 0,6%, приблизительно 0,7%, приблизительно 0,8%, приблизительно 0,9%, приблизительно 1,0%, приблизительно 2,0%, приблизительно 3,0%, приблизительно 4,0%, или приблизительно 5,0% масс./об. активного агента или его фармацевтически приемлемой соли, содержит буфер, например, трометамин. В одном варианте осуществления состав, содержащий приблизительно 0,005%, приблизительно 0,05%, приблизительно 0,1%, приблизительно 0,2%, приблизительно 0,3%, приблизительно 0,4%, приблизительно 0,5%, приблизительно 0,6%, приблизительно 0,7%, приблизительно 0,8%, приблизительно 0,9%, приблизительно 1,0%, приблизительно 2,0%, приблизительно 3,0%, приблизительно 4,0%, или приблизительно 5,0% масс./об. активного агента или его фармацевтически приемлемой соли, содержит приблизительно 0,3% - приблизительно 1,0% масс./об. трометамин и необязательно дополнительно содержит приблизительно 0,005% масс./об. хлорида бензалкония (BAK).
Настоящее изобретение относится к составу, имеющему величину pH от приблизительно 4,5 до приблизительно 7,5 при температуре ниже или равной приблизительно 40°C. В некоторых вариантах осуществления, величина pH состава составляет от приблизительно pH 5,0 до приблизительно 7,0. В одном варианте осуществления величина pH состава равна приблизительно 6,0 при температуре ниже или равной приблизительно 40°C.
Настоящие варианты осуществления относятся к применению состава соединения-I или его свободного основания (формула II) в получении лекарственного препарата для получения доступа к заднему сегменту глаза и/или для лечения и/или облегчения заболевания заднего сегмента глаза, васкулопатического или воспалительного заболевания глаза. Они включают, например, диабетическую ретинопатию (включая фоновую диабетическую ретинопатию, пролиферативную диабетическую ретинопатию и диабетический отек макулы); возрастную дегенерацию макулы (ВМД) (включая неоваскулярную (влажную/экссудативную) ВМД, сухую ВМД и географическую атрофию); патологическую хороидальную неоваскуляризацию (ХНВ) любого механизма (т.е. миопию высокой степени, травму, серповидноклеточную анемию; гистоплазмоз глаз, ангиоидные полосы сетчатки, разрыв сосудистого тракта глаза при травме, друзы оптического нерва и некоторые ретинальные дистрофии); патологическую ретинальную неоваскуляризацию любого механизма (т.е. серповидно-клеточную ретинопатию, болезнь Илза, глазной ишемический синдром, каротидно-кавернозное соустье, семейную экссудативную форму витреоретинопатию, синдром повышенной вязкости крови, идиопатический окклюзионный артериолит; дробьевидную ретинохороидопатию, ретинальный васкулит, саркоидоз и токсоплазмоз); увеит; окклюзию вены сетчатки (центральную или ветки); травму глаз; эдему, вызванную хирургической операцией; неоваскуляризацию, вызванную хирургической операцией; кистозный макулярный отек; ишемию глаз; ретинопатию недоношенных; синдром Коутса; серповидно-клеточную ретинопатию и/или неоваскулярную глаукома.
В некоторых вариантах осуществления, продолжительность воздействия соединения-I составляет от 1 до 90 дней. В некоторых вариантах осуществления, режим дозирования включает несколько курсов местного глазного введения состава, содержащего соединение-I, субъекту в течение от 1 до 90 дней. Например, режим дозирования включает введение состава один раз в день, дважды в день, три раза в день или четыре раза в день в течение от 1 до 90 дней. Например, режим дозирования включает введение состава один, два, три или четыре раза через день (т.е. в 1, 3, 5, 7 день и т.д.) в течение вплоть до 90 дней. Например, режим дозирования включает введение один раз в 1 день, один или два раза в 2-90 день. Например, режим дозирования включает введение один, два, три или четыре раза в 1 день, с последующим введением один раз в день в течение 2-90 дней. Например, режим дозирования включает введение один, два, три, четыре раза в 1 день, с последующим введением один, два, три или четыре раза через день (т.е. в 1, 3, 5, 7 день и т.д.) в течение вплоть до 90 дней. Например, один режим дозирования включает введение один раз в день или дважды в день в течение 1, 2, 3, 4 или 5 последовательных дней. Для режима дозирования два или три раза в день, субъекты получают местную глазную дозу состава, содержащего соединение-I, в 1-й и 4-й дни с интервалом приблизительно 4, 6 или 8 часов. В другом варианте осуществления, субъекты получают местные глазные дозы состава, содержащего соединение-I, приблизительно с интервалом 4, 6 или 8 часов в течение четырех последовательных дней. В некоторых вариантах осуществления субъекты получают одну или две местные глазные дозы состава, содержащего соединения-I, в день в течение 5 последовательных дней. В еще других вариантах осуществления, субъекты получают одну или две местные глазные дозы состава, содержащего соединения-I, в течение 5-90 последовательных дней. В некоторых вариантах осуществления, субъекты получают одну или две местные глазные дозы состава, содержащего соединения-I, в течение, по меньшей мере, 25 последовательных дней. В одном варианте осуществления, субъекты получают одну или две местные глазные дозы в течение, по меньшей мере, 90 последовательных дней или более.
Например, состав, содержащий приблизительно 2 мг/мл соединения-I, вводят дважды в день в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления состав, содержащий приблизительно 3 мг/мл соединения-I, вводят дважды в день в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления состав, содержащий приблизительно 3 мг/мл соединения-I, вводят ежедневно в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления состав, содержащий приблизительно 4 мг/мл соединения-I, вводят дважды в день в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления состав, содержащий приблизительно 4 мг/мл соединения-I, вводят в глаз или оба глаза субъекта ежедневно в течение от 1 до 90 дней. В некоторых вариантах осуществления состав, содержащий приблизительно 5 мг/мл соединения-I, вводят в глаз или оба глаза субъекта дважды в день в течение от 1 до 90 дней.
В некоторых вариантах осуществления состав, содержащий приблизительно 5 мг/мл соединения-I, вводят в глаз или оба глаза субъекта ежедневно в течение от 1 до 90 дней. В некоторых вариантах осуществления состав, содержащий приблизительно 6 мг/мл соединения-I, вводят в глаз или оба глаза субъекта дважды в день в течение от 1 до 90 дней. В некоторых вариантах осуществления состав, содержащий приблизительно 6 мг/мл соединения-I, вводят в глаз или оба глаза субъекта ежедневно в течение от 1 до 90 дней. Режим дозирования в течение от 1 до 90 дней может представлять собой любой из режимов, включающих последовательные или чередующиеся дни введения, описанные в параграфе выше.
В некоторых вариантах осуществления, состав, содержащий соединение формулы (II) или соединения-I, вводят в глаз или оба глаза субъекта. Например, приблизительно 0,2% - приблизительно 0,6% (масс./об.) соединения формулы (II) или приблизительно 0,1%-0,7% (масс./об.) состава, содержащего соединение I, настоящего изобретения вводят один раз в день (QD) или два раза в день (BID) в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления соединение формулы (II) или соединение-I образует комплекс с комплексообразующим агентом, например, циклодекстрином (например, гидроксипропил-β-циклодекстрином (HP-β-CD, KLEPTOSE® HPB) (%)) в соотношении приблизительно 1:8, где приблизительно 2%-13% (масс./об.) циклодекстрина (например, Kleptose® HPB (%)) добавляют к составу. Состав дополнительно содержит приблизительно 0,1% - приблизительно 0,2% буфера, например, 10 мМ фосфатного буфера. Требуемая осмолярность состава составляет приблизительно 200 - приблизительно 300 мОсм, достигаемую добавлением количества, достаточного для достижения осмолярности соли, например, хлорида натрия. pH состава составляет приблизительно 6,0.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Материалы, соединения, состав, изделия и способы, описанные в настоящем изобретении, будут более ясны со ссылкой на следующее подробное описание конкретных аспектов описанного объекта изобретения и примеры, включенные в настоящее изобретение. Перед тем как настоящие материалы, соединения, состав, изделия, устройства и способы будут описаны, ясно, что аспекты, описанные ниже, не ограничивают конкретные способы получения или конкретные реагенты, которые могут изменяться. Также ясно, что терминология, применяемая в настоящем изобретении, служит только для целей описания конкретные аспектов и не предполагается ограничивающей.
Кроме того, во всем описании, ссылаются на различные публикации. Описание данных публикаций включено во всей полноте в настоящее изобретение с помощью ссылки в настоящую заявку для того, чтобы более полно описать состояние настоящего уровня техники, к которому относится описанный объект. Описанные ссылки также отдельно и конкретно включены с помощью ссылки для материала, содержащегося в них, который обсуждается в предложении, на котором основана ссылка.
Настоящее изобретение относится к композициям или составам, которые содержат активный агент для применения в лечении глазных расстройств, вызванные пролиферацией эндотелиальных клеток, повышенной проницаемостью, воспалением или ангиогенезом сосудов. Состав настоящего изобретения являются пригодными в предотвращении или ингибировании неоваскуляризации и пропотевания жидкости через сосуды, связанного с глазным расстройством. В некоторых случаях, состав настоящего изобретения вызывают регресс неоваскуляризацию. Вкратце, в контексте настоящего изобретения, активные агенты следует понимать как любую молекулу, или природную или полученную, которая действует, ингибируя рост сосудов, снижает проницаемость сосудов и/или снижает воспаление. В частности, настоящее изобретение относится к составам, содержащим активный агент, в терапевтически эффективном количестве.
Общие определения
В данном описании и в формуле изобретения, которая следует далее, ссылаются на ряд терминов, которые определяют так, что они имеют следующие значения: все проценты, соотношения и пропорции в настоящем изобретении приведены по весу, если не указано иначе. Все температуры приведены в градусах Цельсия (°C), если не указано иначе.
Под “фармацевтически приемлемым” подразумевают материал, который не является биологически или иначе нежелательным, т.е. материал можно вводить индивиду вместе с соответствующим активным соединением, не вызывая клинически нежелательных биологических эффектов или не взаимодействуя пагубным способом с любым из других компонентов фармацевтической состава или состава, в котором он содержится.
Весовой процент компонента, если специально не указано противоположное, основан на суммарном весе состава, в котором содержится компонент.
“Эффективное количество”, как применяют в настоящем изобретении, обозначает “количество одного или более описанных соединений, эффективное при дозах и в течение периодов времени, необходимых для достижения требуемого или терапевтического результата. ”Эффективное количество” может изменяться в зависимости от факторов, известных в данной области техники, таких как болезненное состояние, возраст, пол и вес человека или животного, которого лечат. Хотя конкретные режимы дозирования можно описать в примерах настоящего изобретения, специалисту в данной области техники ясно, что режим дозирования можно изменять, обеспечивая оптимальный терапевтический ответ. Например, несколько раздельных доз можно вводить ежедневно, или дозу можно пропорционально снижать, как показано требованиями терапевтической ситуации. Кроме того, состав настоящего изобретения можно вводить с такой частотой, которая требуется для получения терапевтического количества.
“Вспомогательное вещество” применяют в настоящем изобретении для включения любого другого соединения, которое может содержаться в или которое можно смешивать с одним или более из описанных ингибиторов, которое не является терапевтически или биологически активным соединением. Как есть, вспомогательное вещество должно быть фармацевтически или биологически приемлемым или подходящим (например, вспомогательное вещество должно быть, в общем, нетоксичным для субъекта). “Вспомогательное вещество” включает отдельное данное соединение, и также предполагается, что оно включает несколько вспомогательных веществ. Для целей настоящего изобретения термин “вспомогательное вещество” и “носитель” применяют взаимозаменяемо во всем описании настоящего изобретения, и указанные термины определяют в настоящем изобретении как “ингредиенты, которые применяют на практике формулирования безопасной и эффективной фармацевтического состава”.
Как применяют в настоящем изобретении, под “субъектом” подразумевают индивида. Таким образом, “субъект” может включать домашних животных (например, кошек, собак и т.д.), сельскохозяйственных животных (например, крупный рогатый скот, лошадей, свиней, овец, коз и т.д.), лабораторных животных (например, мышь, кролика, крысу, морскую свинку и т.д.) и птиц. “Субъект” может также включать примата или человека.
Под “снижать” или другими формами слова, такими как “снижение”, подразумевают ослабление осложнения или снижение характеристики (например, пропотевания жидкости через сосуды или опухания ткани). Ясно, что это обычно относится к некоторой стандартной или ожидаемой величине, другими словами, оно является относительным, но не всегда необходимо ссылаться на стандартную или относительную величину.
Термин “лечить” или другие формы слова, такие как “подвергаемый лечению” или “лечение”, применяют в настоящем изобретении для обозначения того, что введение соединения настоящего изобретения облегчает заболевание или расстройство у хозяина и/или снижает, ингибирует или устраняет конкретную характеристику или осложнение, связанные с расстройством (например, пропотевание жидкости через сосуды).
Поскольку способы настоящего изобретения относятся к предотвращению расстройств, ясно, что термин “предотвращать” не требует, чтобы болезненное состояние полностью устранялось. Скорее, как применяют в настоящем изобретении, термин предотвращение относится к способности специалиста в данной области техники определить популяцию, которая является восприимчивой к расстройству так, чтобы введение соединений настоящего изобретения можно было осуществлять перед возникновением заболевания. Термин доза не подразумевает, что болезненное состояние полностью избегается.
Термин “облегчение” или другие формы слова, такие как “облегчать”, применяют в настоящем изобретении для обозначения того, что введение терапевтического агента настоящего изобретения ослабляет один или более симптомов заболевания или расстройства у хозяина и/или ослабляет, ингибирует или устраняет конкретный синдром, связанный с заболеванием или состоянием перед и/или после введения терапевтического агента.
Описанные соединения влияют на пропотевание жидкости через сосуды или патологическую неоваскуляризацию ингибированием рецепторной тирозинкиназы.
Во всем описании и формуле изобретения настоящего изобретения слово “включает” и другие формы слова, такие как “включая”, обозначают включение, без ограничения, и не предполагается, что они исключают, например, другие добавки или компоненты.
“Необязательный” или “необязательно” обозначает то, что описанное впоследствии событие или обстоятельство может происходить или не происходить, и что описание включает случаи, где событие или обстоятельства возникает и случае, где оно не возникает.
Термин “приблизительно” относится к любому минимальному изменению концентрации или количества терапевтического агента, которое не изменяет эффективность агента при получении состава и лечении заболевания или расстройства. Например, без ограничения, концентрация терапевтического агента будет эффективной, если концентрация изменяется между 0,005%-5,0% (±0,0005%). Термин “приблизительно” относительно диапазона концентраций терапевтических/активных агентов настоящего изобретения также включает любое изменение стандартного количества или диапазона, который будет представлять собой эффективное количество или диапазон.
Диапазоны можно выразить в настоящем изобретении в виде от “приблизительно” одной конкретной величины и/или до “приблизительно” другой конкретной величины. Когда выражают данный диапазон, другой аспект включает от одной конкретной величины и/или до другой конкретной величины. Аналогично, когда величины выражают как приблизительные величины, применяя антецедент “приблизительно”, ясно, что конкретная величина образует другой аспект. Кроме того, ясно, что конечные величины каждого из диапазонов являются важными и относительно другой конечной величины и независимо от другой величины. Также ясно, что имеется ряд величин, описанных в настоящем изобретении, и что каждая величина также описана в настоящем изобретении в виде “приблизительно” данная конкретная величина в добавление к самой величине. Например, если описывают величину “10”, то также описывают “приблизительно 10”. Также ясно, что когда описывают величину, то описывают также “меньше чем или равно” величине, “больше чем или равно величине”, и возможные диапазоны между величинами, как ясно специалисту в данной области техники. Например, если описывают величину “10”, то также описывают “меньше чем или равно 10”, а также “больше чем или равно 10”. Также ясно, что во всей заявке данные приведены в ряде различных форматов и что данные представляют собой конечные величины и исходные величины и диапазоны для любой комбинации значений данных. Например, если описывают конкретное значение “10” и конкретное значение “15”, ясно, что больше чем, больше чем или равно, меньше чем, меньше чем или равно, и равно 10 и 15 считают описанными, а также между 10 и 15. Также ясно, что также описана каждая величина между двумя конкретными величинами. Например, если описаны 10 и 15, то также описаны 11, 12, 13 и 14.
Термин “галоген”, как применяют в настоящем изобретении, если не указано иначе, включает фтор, хлор, бром или йод. Предпочтительные галогеновые группы представляют собой фтор, хлор и бром.
Термин “алкил”, как применяют в настоящем изобретении, если не указано иначе, включает и разветвленные и нормальные насыщенные алифатические углеводородные группы, содержащие конкретное количество атомов углерода. Например, предполагается, что C1-6 алкил включает C1, C2, C3, C4, C5 и C6 алкильные группы. Примеры алкила включают, но не ограничиваются, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, н-пентил, втор-пентил и н-гексил. В конкретных вариантах осуществления, нормальный или разветвленный алкил содержит шесть или меньше атомов углерода в его остове (например, C1-C6 для нормальной цепи, C3-C6 для разветвленной цепи), и в другом варианте осуществления нормальный или разветвленный алкил содержит четыре или меньше атомов углерода. Аналогично, циклоалкилы содержат от трех до восьми атомов углерода в их кольцевой структуре, и в других вариантах осуществления, циклоалкилы содержат пять или шесть атомов углерода в своей кольцевой структуре. Алкил может быть замещен заменой атома водорода одним или более атомами углерода углеводородного остова. Данные заместители могут включать, например, алкил, алкенил, алкинил, галоген, гидроксил, алкилкарбонилокси, арилкарбонилокси, алкоксикарбонилокси, арилоксикарбонилокси, карбоксилат, алкилкарбонил, арилкарбонил, алкоксикарбонил, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, алкилтиокарбонил, алкоксил, фосфат, фосфонато, фосфонито, циано, амино (включая алкиламино, диалкиламино, ариламино, диариламино и алкилариламино), ациламино (включая алкилкарбониламино, арилкарбониламино, карбамоил и уреидо), амидино, имино, сульфгидрил, алкилтио, арилтио, тиокарбоксилат, сульфаты, алкилсульфинил, сульфонато, сульфамоил, сульфонамидо, нитро, трифторметил, циано, азидо, гетероциклил, алкиларил, или ароматический или гетероароматический фрагмент. Циклоалкилы могут быть дополнительно замещены, например, заместителями, описанными выше. “Алкиларильная” или “аралкильная” группа представляет собой алкил, замещенный арилом (например, фенилметил (бензил)).
Термин “алкенил”, как применяют в настоящем изобретении, если не указано иначе, включает ненасыщенный алифатические группы, аналогичные по длине и возможному замещению алкилам, описанным выше, но которые содержат, по меньшей мере, одну двойную связь. Например, термин “алкенил” включает нормальные алкенильные группы (например, этенил, пропенил, бутенил, пентенил, гексенил, гептенил, октенил, ноненил, деценил), разветвленные алкенильные группы, циклоалкенильные (например, алициклические) группы (например, циклопропенил, циклопентенил, циклогексенил, циклогептенил, циклооктенил), циклоалкенильные группы, замещенные алкилом или алкенилом, и алкенильные группы, замещенные циклоалкилом или циклоалкенилом. В конкретных вариантах осуществления, нормальная или разветвленная алкенильная группа содержит шесть или меньше атомов углерода в своем остове (например, C2-C6 для нормальной цепи, C3-C6 для разветвленной цепи). Аналогично, циклоалкенильные группы могут содержать от трех до восьми атомов углерода в своей кольцевой структуре, и в некоторых вариантах осуществления, циклоалкенильные группы содержат пять или шесть атомов углерода в кольцевой структуре. Термин “C2-C6” включает алкенильные группы, содержащие от двух до шести атомов углерода. Термин “C3-C6” включает алкенильные группы, содержащие от трех до шести атомов углерода. Алкенил можно заместить заменой атома водорода одним или более атомами углерода углеводородного остова. Данные заместители могут включать, например, алкильные группы, алкинильные группы, галогены, гидроксил, алкилкарбонилокси, арилкарбонилокси, алкоксикарбонилокси, арилоксикарбонилокси, карбоксилат, алкилкарбонил, арилкарбонил, алкоксикарбонил, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, алкилтиокарбонил, алкоксил, фосфат, фосфонато, фосфонито, циано, амино (включая алкиламино, диалкиламино, ариламино, диариламино и алкилариламино), ациламино (включая алкилкарбониламино, арилкарбониламино, карбамоил и уреидо), амидино, имино, сульфгидрил, алкилтио, арилтио, тиокарбоксилат, сульфаты, алкилсульфинил, сульфонато, сульфамоил, сульфонамидо, нитро, трифторметил, циано, азидо, гетероциклил, алкиларил, или ароматическую или гетероароматическую группу.
Термин “алкинил”, как применяют в настоящем изобретении, если не указано иначе, включает ненасыщенные алифатические группы, аналогичные по длине и возможному замещению алкилам, описанным выше, но которые содержат, по меньшей мере, одну тройную связь. Например, “алкинил” включает нормальные алкинильные группы (например, этинил, пропинил, бутинил, пентинил, гексинил, гептинил, октинил, нонинил, децинил), разветвленные алкинильные группы и алкинильные группы, замещенные циклоалкилом или циклоалкенилом. В конкретных вариантах осуществления, нормальная или разветвленная алкинильная группа содержит шесть или меньше атомов углерода в своем остове (например, C2-C6 для нормальной цепи, C3-C6 для разветвленной цепи). Термин “C2-C6” включает алкинильные группы, содержащие от двух до шести атомов углерода. Термин “C3-C6” включает алкинильные группы, содержащие от трех до шести атомов углерода. Алкинил можно заместить заменой атома водорода одним или более атомами углерода углеводородного остова. Данные заместители могут включать, например, алкильные группы, алкинильные группы, галогены, гидроксил, алкилкарбонилокси, арилкарбонилокси, алкоксикарбонилокси, арилоксикарбонилокси, карбоксилат, алкилкарбонил, арилкарбонил, алкоксикарбонил, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, алкилтиокарбонил, алкоксил, фосфат, фосфонато, фосфонито, циано, амино (включая алкиламино, диалкиламино, ариламино, диариламино и алкилариламино), ациламино (включая алкилкарбониламино, арилкарбониламино, карбамоил и уреидо), амидино, имино, сульфгидрил, алкилтио, арилтио, тиокарбоксилат, сульфаты, алкилсульфинил, сульфонато, сульфамоил, сульфонамидо, нитро, трифторметил, циано, азидо, гетероциклил, алкиларил, или ароматическую или гетероароматическую группу.
Термин “алкокси”, как применяют в настоящем изобретении, если не указано иначе, включает O-алкильные группы, в которых “алкил” представляет собой, как определено выше.
Термин “арил”, как применяют в настоящем изобретении, если не указано иначе, включает 5- и 6-членные “несопряженные” или однокольцевые ароматические группы, которые могут содержать от нуля до четырех гетероатомов, а также “сопряженные” или многокольцевые системы, по меньшей мере, с одним ароматическим кольцом. Примеры арильных групп включают бензол, фенил, пиррол, фуран, тиофен, тиазол, изотиазол, имидазол, триазол, тетразол, пиразол, оксазол, изооксазола, пиридин, пиразин, пиридазин, пиримидин и подобные. Кроме того, термин “арил” включает многокольцевые арильные группы, например, трициклические, бициклические, например, нафталин, бензоксазол, бензодиоксазол, бензотиазол, бензимидазол, бензотиофен, метилендиоксифенил, хинолин, изохинолин, нафтиридин, индол, бензофуран, пурин, бензофуран, деазапурин или индолизин. Арильные группы, содержащие гетероатомы в кольцевой структуре, можно также называть “арильными гетероциклами”, “гетероциклами”, “гетероарилами” или “гетероароматическими кольцами”. Ароматическое кольцо можно замещать по любому одному или более из кольцевых положений заместителями, как описано выше, такими как, например, галоген, гидроксил, алкокси, алкилкарбонилокси, арилкарбонилокси, алкоксикарбонилокси, арилоксикарбонилокси, карбоксилат, алкилкарбонил, алкиламинокарбонил, аралкиламинокарбонил, алкениламинокарбонил, алкилкарбонил, арилкарбонил, аралкилкарбонил, алкенилкарбонил, алкоксикарбонил, аминокарбонил, алкилтиокарбонил, фосфат, фосфонато, фосфонито, циано, амино (включая алкиламино, диалкиламино, ариламино, диариламино и алкилариламино), ациламино (включая алкилкарбониламино, арилкарбониламино, карбамоил и уреидо), амидино, имино, сульфгидрил, алкилтио, арилтио, тиокарбоксилат, сульфаты, алкилсульфинил, сульфонато, сульфамоил, сульфонамидо, нитро, трифторметил, циано, азидо, гетероциклил, алкиларил или ароматическая или гетероароматическая группа. Арильные группы могут также быть конденсированы или соединены мостиком с алициклическими или гетероциклическими кольцами, которые не являются ароматическим и так, чтобы образовывать многокольцевую систему (например, тетралин, метилендиоксифенил).
Термин “4-10-членный гетероцикл”, как применяют в настоящем изобретении, если не указано иначе, включает ароматические и неароматические гетероциклические группы, содержащие один или более гетероатомов, причем каждый выбран из O, S и N, где каждая из гетероциклических групп содержит 4-10 атомов в своей кольцевой системе. Неароматические гетероциклические группы включают группы, содержащие только 4 атома в своей кольцевой системе, но ароматические гетероциклические группы должны содержать, по меньшей мере, 5 атомов в своей кольцевой системе. Примером 4-членной гетероциклической группы является азетидинил (полученный из азетидина). Примером 5-членной гетероциклической группы является тиазолил, и примером 10-членной гетероциклической группы является хинолинил. Примерами неароматических гетероциклических групп являются пирролидинил, тетрагидрофуранил, тетрагидротиенил, тетрагидропиранил, тетрагидротиопиранил, пиперидино, морфолино, тиоморфолино, токсанил, пиперазинил, азетидинил, оксетанил, тиетанил, гомопиперидинил, оксепанил, тиепанил, оксазепинил, диазепинил, тиазепинила, 1,2,3,6-тетрагидропиридинил, 2-пирролинил, 3-пирролинил, индолинил, 2Н-пиранил, 4Н-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинил, имидазолинил, имидазолидинил, 3-азабицикло[3,1,0]гексанил, 3-азабицикло[4,1,0]гептанил, 3Н-индолил и хинолизинил. Примерами ароматических гетероциклических групп являются пиридинил, имидазолил, пиримидинил, пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил, индолизинил, фталазинил, пиридазинил, триазинил, изоиндолил, птеридинил, пуринил, оксадиазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил и фуропиридинил. Указанные выше группы, как получено из соединений, перечисленных выше, могут быть C-присоединенными или N-присоединенными, где это возможно. Например, группа, полученная из пиррола, может представлять собой пиррол-1-ил (N-присоединенная) или пиррол-3-ил (C-присоединенная). “4-10-членная гетероциклическая” группа может быть замещенной.
Фраза “фармацевтически приемлемая соль (соли)”, как применяют в настоящем изобретении, если не указано иначе, включает соли кислых или основных групп, которые могут присутствовать в соединениях формулы (I) или (II). Соединения формулы (I) или (II), которые являются основными по свойствам, способны образовывать широкий диапазон солей с различными неорганическими и органическими кислотами. Кислоты, которые можно применять, получая фармацевтически приемлемые соли присоединения кислоты данных основных соединений формулы (I) или (II), представляют собой кислоты, которые образуют нетоксичные соли присоединения кислоты, т.е. соли, содержащие фармакологически приемлемые анионы, такие как гидрохлорид, гидробромид, гидроиодид, нитратную, сульфатную, бисульфатную, фосфатную, кислую фосфатную, изоникотинатную, ацетатную, лактатную, салицилатную, цитратную, кислую цитратную, тартратную, пантотенатную, битартратную, аскорбатную, сукцинатную, малеатную, гентизинатную, фумаратную, глюконатную, глюкаронатную, сахаратную, формиатную, бензоатную, глутаматную, метансульфонатную, этансульфонатную, бензолсульфонатную, п-толуолсульфонатную и памоатную [т.е. 1,1'-метилен-бис(2-гидрокси-3-нафтоатную)] соли.
Соединения формулы (I), которые являются кислыми по свойствам, способны образовывать основные соли с различными фармакологически приемлемыми катионами. Примеры данных солей включают соли щелочных или щелочноземельных металлов и в частности, соли натрия и калия. В некоторых вариантах осуществления соль представляет собой соль присоединения кислоты, например, HCl соль.
Определенные соединения формулы (I) могут содержать асимметрические центры и, следовательно, существовать в виде различных энантиомерных форм. Настоящее изобретение относится к применению всех оптических изомеров и стереоизомеров соединений формулы (I) и их смесей. Соединения формулы (I) могут также существовать в виде E/Z геометрических изомеров или таутомеров. Настоящее изобретение относится к применению всех данных геометрических изомеров и таутомеров и их смесей.
Настоящее изобретение также включает изотопно-меченные соединения и их фармацевтически приемлемые соли, которые являются идентичными соединениям и их фармацевтически приемлемым солям, перечисленным в формуле (I), но в которых один или более атомов замещен атомом, имеющим атомную массу или атомное число, отличное от атомной массы или массового числа, обычно обнаруживаемых в природе. Примеры изотопов, которые можно вводить в соединения настоящего изобретения, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2H, 3H, 13C, 14C, 15N, 18O, 17O, 35S, 18F и 36Cl, соответственно. Соединения настоящего изобретения, их конъюгаты и фармацевтически приемлемые соли указанных соединений или указанных конъюгатов, которые содержат приведенные выше изотопы и/или другие изотопы других атомов включены в объем настоящего изобретения. Определенные изотопно-меченные соединения настоящего изобретения, например, соединения, в которые включен радиоактивный изотоп, такой как 3H и 14C, являются пригодными в анализах на распределение лекарственного средства и/или субстрата в тканях. Тритиевый, т.е. 3H и углерод-14, т.е. 14C изотопы являются особенно предпочтительными из-за простоты получения и детектируемости. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2H, может давать определенные терапевтические преимущества, являющиеся результатом большей метаболической стабильности, например, повышенного времени полуразложения in vivo или пониженных требуемых доз и, следовательно, может быть предпочтительным в некоторых обстоятельствах. Изотопно-меченные соединения формул (I) настоящего изобретения и эфиры или их липидные конъюгаты можно обычно получить проведением способов, описанных на схемах и/или в примерах и способах получения ниже, заменой изотопно немеченного реагента на легко доступный изотопно-меченный реагент.
Настоящее изобретение также включает фармацевтические составы, содержащие производные соединений формулы (I) или его фармацевтически приемлемых солей. Соединения формулы (I) или их фармацевтически приемлемые соли, содержащие свободные амино или амидо группы, можно превратить в сопряженные производные, в которых аминокислотный остаток, или полипептидная цепь двух или более (например, двух, трех или четырех) аминокислотных остатков ковалентно соединена через амидную или эфирную связь со свободной амино группой соединений формулы (I) или их фармацевтически приемлемых солей. Аминокислотные остатки включают, но не ограничиваются, 20 природных аминокислот, обычно обозначаемых трехбуквенными символами и также включают 4-гидроксипролин, гидроксилизин, демозин, изодемозин, 3-метилгистидин, норвалин, бета-аланин, гамма-аминомасляную кислоту, цитруллин, гомоцистеин, гомосерин, орнитин и метионинсульфон.
Также включены дополнительные типы производных. Амидные и эфирные группы могут вводить группы, включая, но не ограничиваясь, эфирные, аминовые и карбоксидные функциональные группы. Свободные гидрокси группы можно превращать в производные, применяя группы, включая, но не ограничиваясь, гемисукцинаты, фосфатные эфиры, диметиламиноацетаты и фосфорилоксиметилоксикарбонилы, как указано в D. Fleisher, et al., Advanced Drug Delivery Reviews (1996) 19, 115. Также включены карбаматные конъюгаты гидрокси и амино групп, также как карбонатные конъюгаты и сульфатные эфиры гидрокси групп. Также включено получение производных гидрокси групп, таких как (ацилокси)метильные и (ацилокси)этильные эфиры, где ацильная группа может представлять собой алкильный эфир, необязательно замещенный группами, включающими, но не ограничивающимися, эфирные, аминовые и карбоксильные функциональные группы, или, где ацильная группа представляет собой аминокислотный эфир, как описано выше. Производные данного типа описаны в R.P. Robinson et al., J. Medicinal Chemistry (1996) 39, 10.
Термин “киназа” относится к любому ферменту, который катализирует присоединение фосфатных групп к белковому остатку; например, серин и треонинкиназы катализируют присоединение фосфатных групп к сериновому и треониновому остаткам.
Термины “VEGFR киназа,” “VEGFR,” относятся к любому из рецепторов 2 сосудистого эндотелиального фактора роста.
Термины “VEGF передача сигнала” и “VEGF каскад” относятся и к предшествующим и к последующим компонентам VEGF сигнального каскада.
Термин “фармацевтически приемлемый” относится к тому факту, что носитель, разбавитель или вспомогательное вещество должно быть совместимым с другими ингредиентами состава и не вредным для ее реципиента.
Термины “введение соединения” относятся к действию, обеспечивающему нуждающегося в лечении субъекта соединением настоящего изобретения или фармацевтической композицией.
Термин “васкулостаз” относится к поддержанию гомеостатического функционирования сосудов, приводящему к нормальному физиологическому функционированию.
Термин “васкулостатический агент” относится к агентам, которые претендуют на борьбу с заболеваниями, в которых васкулостаз нарушается предотвращением потери или восстановления или поддержания васкулостаза.
В настоящем описании “композиция” и “состав” применяют взаимозаменяемо, и они относятся к общепринято понимаемой, как известно в данной области техники, композиции или составу.
Настоящее изобретение относится к офтальмическому составу. В некоторых вариантах осуществления, офтальмический состав настоящего изобретения представляет собой состав в форме геля или полугелевый состав, или оба.
“Гель” согласно настоящему изобретению представляет собой полутвердую лекарственную форму настоящего изобретения, содержащую суспендированные частицы. Полутвердая форма не является жидкотекучей; она не течет или принимает форму содержащего ее контейнера при комнатной температуре. Полутвердая форма не течет при низком сдвигающем напряжении и обычно проявляет пластическое течение. Коллоидная дисперсия представляет собой систему, в которой частицы коллоидного размера (т.е. обычно от 1 нм до 1 мкм) равномерно распределены по всей жидкости.
В некоторых вариантах осуществления, “гель” представляет собой полутвердую систему, содержащую суспензии или небольших неорганических частиц или органических молекул, наполняющих жидкость. “Гели” классифицируют или как однофазные или как двухфазные системы. “Гели” также состоят из мезофазы, или состояния материи, промежуточного между жидкостью и твердым состоянием, которое представляет собой частично упорядоченную структуру, которая представляет собой состояние для активных агентов в “гелевых каплях” настоящих вариантов осуществления. Двухфазный гель состоит из сети небольших дискретных частиц. В двухфазной системе, гелевую массу иногда называют суспензией (например, бентонитовая суспензия), если размер частиц суспендированного материала является большим. И гели и суспензии являются тиксотропными, образующими полутвердые формы при стоянии и становящиеся жидкостью при встряхивании. Полутвердые составы нужно встряхивать перед введением, обеспечивая гомогенность, и они должны быть маркированы соответствующим способом (см. суспензии). Однофазные гели состоят из органических макромолекул, равномерно распределенных по жидкости таким способом, что не существует видимых границ между макромолекулами и жидкостью. Однофазные гели также могут состоять из низкомолекулярных (LMW) молекул, где компонент, ответственный за образование геля, представляет собой фактический активный ингредиент. Эти так называемые “LMW гидрогели” являются отличными от традиционных агентов, образующих гели, воды, таких как высокомолекулярные синтетические полимеры, полисахариды и белки. Высокомолекулярные гелеобразующие агенты являются высокоупорядоченными и однонаправленными из-за водородных связей, тогда как силы, регулирующие LMW гидрогели, представляют собой в основном неориентированные ван-дер-ваальсовские взаимодействия (гидрофобные). На практике LMW гидрогели наблюдают в виде высокоанизотропных (обычно фибриллярные) структур, которые размещаются в жидкости, образуя физически разветвленную или сложную сеть. Таким образом, гели могут быть неупорядоченными или незначительно упорядоченными, показывая некоторое двойное преломление, жидкокристаллические свойства. Гели вводят местно или, после встряхивания, в виде гидрогеля в виде глазных капель.
Полутвердый “гель” согласно настоящему изобретению представляет собой полутвердое вещество в соответствии с определениями фармакопеи США и приводимой в ней литературе. Динамическая вязкость полутвердого состава увеличивается с концентрацией. Клиническая дозируемая концентрация настоящего состава находится в диапазоне от низкой концентрации ≤1 мг/мл (0,1%) до высокой концентрации ≤6 мг/мл (0,6%). Дозы с низкой концентрацией являются наименее вязкими и подпадают в категорию “раствор”, тогда как большие концентрации является более вязкими и удовлетворяют определению геля.
“Студни” согласно настоящему изобретению представляет собой класс гелей, которые представляют собой полутвердые системы, которые состоят из суспензий, образованных или небольшими органическими частицами или большими органическими молекулами, наполняющими жидкость, в которых структурная сцепляющая матрица содержит большую долю жидкости, обычно воды.
“Раствор” согласно настоящему изобретению представляет собой прозрачную, гомогенную жидкую лекарственную форму, которая содержит одно или более химических веществ, растворенных в растворителе или смеси взаимосмешиваемых растворителей. Раствор представляет собой жидкий препарат, который содержит одно или более растворенных химических веществ в подходящем растворителе или смеси взаимосмешиваемых растворителей. Поскольку молекулы лекарственного вещества в растворе распределены равномерно, применение растворов в виде лекарственных форм, в общем, относится к обеспечению равномерного дозирования после введения и высокой точности при разбавлении или другом способе смешивания раствора.
“Жидкость” согласно настоящему изобретению представляет собой лекарственную форму, состоящую из чистого химического соединения в его жидком состоянии. Жидкость является текучей; она течет и принимает форму емкости при комнатной температуре. Жидкости обладают ньютоновским или псевдопластическим поведением.
“Суспензия” согласно настоящему изобретению представляет собой жидкую лекарственную форму, которая содержит твердые частицы, диспергированные в жидкой среде.
Соединения настоящего изобретения формулируют в виде терапевтических композиций в виде нейтральных или солевых форм. Фармацевтически приемлемые нетоксичные соли включают соли присоединения основания (образованные свободными карбоксильными или другими анионными группами), которые получены из неорганических оснований, таких как, например, гидроксиды натрия, калия, аммония, кальция или железа (III), и таких органических оснований, как изопропиламин, триметиламин, 2-этиламиноэтанол, гистидин, прокаин и подобные. Данные соли получают в виде солей присоединения кислоты с любыми свободными катионными группами и обычно получают с неорганическими кислотами, такими как, например, хлористоводородная, серная кислота, фосфорная кислота, или органическими кислотами, такими как уксусная, лимонная, п-толуолсульфоновая, метансульфоновая кислота, щавелевая кислота, винная кислота, миндальная кислота и подобными. Соли настоящего изобретения включают соли аминов, образованные протонированием амино групп неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, фосфорная кислота и подобными. Соли настоящего изобретения также включают соли аминов, образованные протонированием аминогруппы подходящими органическими кислотами, такими как п-толуолсульфокислота, уксусная кислота и подобные. Дополнительные вспомогательные вещества, которые предполагают для применения на практике настоящего изобретения, представляют собой вспомогательные вещества, доступные специалистам в данной области техники, например, вспомогательные вещества, имеющиеся в Американской Фармакопее том XXII и добавлении к Американской Фармакопее том XVII, U.S. Pharmacopoeia Convention, Inc., Rockville, Md. (1989), соответствующее содержание которых включено в настоящее изобретение с помощью ссылки. Кроме того, полиморфы соединений настоящего изобретения включены в настоящее изобретение.
Варианты осуществления настоящего изобретения обеспечивают офтальмический композицию или состав для лечения глазной неоваскуляризации активным агентом формулы I:
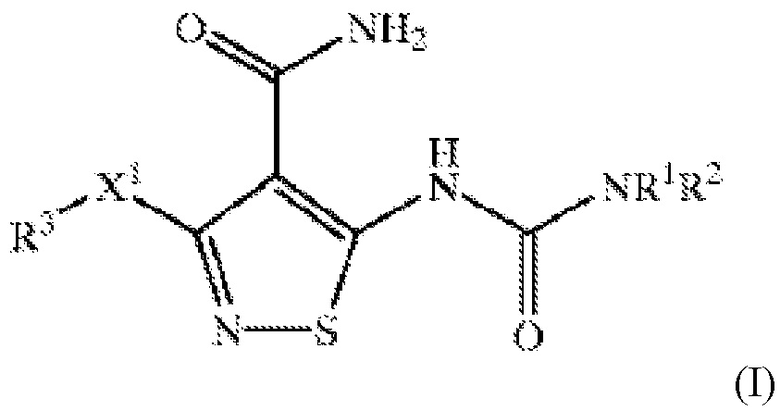
или его фармацевтически приемлемой солью; и фармацевтически приемлемыми вспомогательными веществами; активный агент или фармацевтически приемлемая соль присутствует в количестве от приблизительно 0,02% до приблизительно 1,0% масс./об., и где активный агент определяют как: X1 представляет собой O или S; R1 представляет собой H, C1-C10 алкил, C2-C10 алкенил, C2-C10 алкинил, -C(O)(C1-C10 алкил), -(CH2)t(C6-C10 арил), -(CH2)t(4-10-членный гетероцикл), -C(O)(CH2)t(C6 -C10 арил) или -C(O)(CH2)t (5-10-членный гетероцикл), где t представляет собой целое от 0 до 5; алкильная группа необязательно содержит 1 или 2 гетерофрагмента, выбранные из O, S и -N(R6)-, при условии, что два O атома, два S атома, или O и S атом не соединены непосредственно друг с другом; арильная и гетероциклическая R1 группы необязательно конденсированы с C6 -C10 арильной группой, C5-C8 насыщенной циклической группой или 5-10-членной гетероциклической группой; 1 или 2 атома углерода в указанных выше гетероциклических фрагментах необязательно замещены оксо (=O) группой или анионом кислорода; -(CH2)t- группы указанных выше R1 групп необязательно содержат углерод-углерод двойную или тройную связь, где t представляет собой целое от 2 до 5; и указанные выше R1 группы, за исключением H, необязательно замещены 1-3 R4 группами; R2 представляет собой H; R3 представляет собой -(CH2)t(C6-C10 арил), где t представляет собой целое от 0 до 5; необязательно конденсированный с C6-C10 арильной группой, C5-C8 насыщенной циклической группой или 5-10-членной гетероциклической группой; -(CH2)t-группы необязательно содержат углерод-углерод двойную или тройную связь, где t представляет собой целое от 2 до 5, и необязательно замещены 1-5 R4 группами; каждый R4 независимо выбран из C1-C10 алкила, C2-C10 алкенила, C2-C10 алкинила, галогена, циано, нитро, трифторметила, трифторметокси, азидо, -OR5 , -C(O)R5, -C(O)OR5, -NR6C(O)OR5, -OC(O)R5, -NR6 SO2R5, -SO2NR5 R6, -NR6C(O)R5, -C(O)NR5R6, -NR5R6, -S(O)jR7, где j представляет собой целое в диапазоне от 0 ДО 2, -SO3H, -NR5(CR6R7)tOR6, -(CH2)t(C6-C10 арила), -SO2(CH2)t(C6-C10 арила), -S(CH2)t(C6-C10 арила), -O(CH2)t(C6-C10 арила), -(CH2)t(5-10-членного гетероцикла) и -(CR6 R7)mOR6, где m представляет собой целое от 1 до 5, и t представляет собой целое от 0 до 5; алкильная группа необязательно содержит 1 или 2 гетерофрагмента, выбранные из O, S и -N(R6)-, при условии, что два O атома, два S атома, или O и S атом не соединены непосредственно друг с другом; арильная и гетероциклическая R4 группы необязательно конденсированы с C6 -C10 арильной группой, C5-C8 насыщенной циклической группой или 5-10-членной гетероциклической группой; 1 или 2 атома углерода в указанных выше гетероциклических фрагментах необязательно замещены оксо (=O) группой или анионом кислорода; и алкильные, арильные и гетероциклические фрагменты указанных выше R4 групп необязательно замещены 1-3 заместителями, независимо выбранными из галогена, циано, нитро, трифторметила, трифторметокси, азидо, -NR6SO2R5, -SO2NR5R6, -C(O)R5, -C(O)OR5, -OC(O)R5, -NR6C(O)R5, -C(O)NR5R6, -NR5R6, -(CR6R7)mOR6, где m представляет собой целое от 1 до 5, -OR5 и заместителей, перечисленных в определении R5 ; и каждый R5, R6 и R7 независимо представляют собой H или C1 -C6 алкил.
В следующем варианте настоящего изобретения, R3 в формуле I настоящего изобретения представляет собой -(CH2)t(C6-C10 арил), где t представляет собой целое от 1 до 3, и R3 группа необязательно замещена 1-4 R4 группами.
В другом варианте осуществления, R3 в формуле I настоящего изобретения представляет собой бензил, необязательно замещенный 1-4 заместителями, независимо выбранными из галогена и C1-C4 алкила. Например, R3 в формуле I настоящего изобретения представляет собой бензил, замещенный 1-4 заместителями, независимо выбранными из метила, фтора, хлора и брома.
R1 в формуле I настоящего изобретения представляет собой -(CH2)t (5-10-членный гетероцикл), где t представляет собой целое от 0 до 5, необязательно замещенный 1 или 2 заместителями, независимо выбранными из C1-C4 алкила, гидрокси и гидроксиметила.
Гетероциклический фрагмент R1 группы в формуле I настоящего изобретения выбран из морфолино, пирролидинила, имидазолила, пиперазинила, пиперидинила и 2,5-диазабицикло[2,2,1]гепт-2-ила, переменная t R1 группы находится в диапазоне от 2 до 5, и R1 группа необязательно замещена одной или более гидрокси группами.
Например, гетероциклический фрагмент R1 группы в формуле I настоящего изобретения представляет собой пирролидин.
В следующих вариантах осуществления настоящего изобретения, активный агент представляет собой:
 .
.
Соединение настоящего изобретения представляет собой гидрохлорид 3-[(4-бром-2,6-дифторфенил)метокси]-5-[[[[4-(1-пирролидинил)бутил]амино]карбонил]амино]-4-изотиазолкарбоксамида, молекулярной формулы: C20H24BrF2N5O3S·HCl, молекулярный вес: 568,86 г/моль, и при условии, что молекула не содержит асимметрического центра и не является хиральной. Соединение настоящего изобретения представлено соединением-I:
 .
.
Соединение-I настоящего изобретения представляет собой ингибитор активности тирозинкиназы VEGFR-2, который блокирует VEGF-стимулируемое автофосфорилирование данного рецептора, а также пролиферацию эндотелиальных клеток. Оно является селективным (>500x) относительно концентрации, требуемой для ингибирования тирозинкиназ, рецептора эпидермального фактора роста (EGFR) и инсулинового рецептора (IR). Соединение-I описано в патенте США No 6235764.
Общие свойства
Соединение-I настоящего изобретения обладает характеристиками, как показано в таблице 1. Варианты осуществления относятся к трем композициям соединения-I или его свободного основания - соединения формулы II.
коды: соединение-I
Этанол: 0,7
Ацетонитрил: 0,04
Тетрагидрофуран: 0,02
Гексан: <0,01
0,1 N NaOH: 0,05
pH 9,0 (0,05 М Na2HPO4): 0,7
pH 7,5 (0,05 М NaH2PO4): 1,0
0,1 N HCl: 0,04
Деионизированная вода: 0,5-1,2b
Состав композиций соединения-I перечислен в таблице 1B. Материалы состава перечислены в таблице 1C.
1,0-2,0% глицерин
с или без 0,005% хлорида бензалкония, NF (BAK)
pH ~6,0-7,0
1,0-2,0% глицерин, USP
с или без 0,005% хлорида бензалкония, NF (BAK)
pH ~6,0-7,0
0,1%-0,9% хлорида натрия
pH ~6,0-7,0
или
1%-20% сульфобутилэфир-β-циклодекстрина (SBE-β-CD, CAPTISOL®)
с или без 0,122% трометамина (Tris)
0,1-0,2% фосфата натрия, двухосновный, безводный
0%-0,6% хлорид натрия
pH ~6,0-7,0
или
1%-20% HP-β-CD (KLEPTOSE® HPB или
KLEPTOSE® HP)
0,1- 0,2% фосфата натрия, двухосновный, безводный
0,50%-0,6% хлорида натрия
pH ~6,0-7,0
Офтальмические растворы
Настоящее изобретение относится к составам, содержащим соединение-I и/или его свободное основание (соединение формулы II), полученное в виде раствора с вязкостью, аналогичной воде. Раствор содержит фармацевтически приемлемые агенты/вспомогательные вещества, например, без ограничения, циклодекстрин. Таким образом, полученный раствор представляет собой прозрачный и бесцветный раствор, подходящий для местного введения в глаз.
Растворы настоящего изобретения снижают воздействие на передний сегмент глаза активного агента; посредством этого они обеспечивают повышенную концентрацию активного агента в растворе и повышенную частоту доставки, таким образом, способствуя поддержанию высокой концентрации активного агента в заднем сегменте глаза.
Растворы настоящего изобретения содержат от приблизительно 0,005% до приблизительно 5,0% масс./об. активного агента формулы I или его фармацевтически приемлемой соли, например, соединения-I. В некоторых вариантах осуществления, концентрация соединения-I или его свободного основания (формула II) в растворах составляет приблизительно 0,005% - приблизительно 0,01%, приблизительно 0,01% - приблизительно 0,05%, приблизительно 0,05% - приблизительно 0,1%, приблизительно 0,1% - приблизительно 0,2%, приблизительно 0,2% - приблизительно 0,3%, приблизительно 0,3% - приблизительно 0,4%, приблизительно 0,4% - приблизительно 0,5%, приблизительно 0,5% - приблизительно 0,6%, приблизительно 0,6% - приблизительно 0,7%, приблизительно 0,7% - приблизительно 0,8%, приблизительно 0,8% - приблизительно 0,9%, приблизительно 0,9% - приблизительно 1,0%, приблизительно 1,0 - приблизительно 2,0%, приблизительно 2,0 - приблизительно 3,0%, приблизительно 3,0 - приблизительно 4,0%, или приблизительно 4,0 - приблизительно 5,0% масс./об. для местного введения. В некоторых вариантах осуществления, растворы содержат приблизительно 0,005%, приблизительно 0,05%, приблизительно 0,1%, приблизительно 0,2%, приблизительно 0,3%, приблизительно 0,4%, приблизительно 0,5%, приблизительно 0,6%, приблизительно 0,7%, приблизительно 0,8%, приблизительно 0,9%, приблизительно 1,0%, приблизительно 2,0%, приблизительно 3,0%, приблизительно 4,0%, или приблизительно 5,0% масс./об. соединения-I или его свободного основания (формула II).
В некоторых вариантах осуществления композиция содержит циклодекстрин для улучшения растворимости соединения-I. Циклодекстрин, олигосахарид, образованный шестью - семью мономерами декстрозы, соединенными через одну или четыре связи, повышает растворимость активных агентов, которые имеют плохую или низкую растворимость в воде или водных растворах (например, в PBS буфере). Циклодекстрины образуют гидрофильные комплексы с гидрофобными активными агентами.
Один или более циклодекстринов применяют в растворе настоящего изобретения. Неограничивающие примеры циклодекстринов для применения в составе настоящего изобретения представляют собой, например: 2-гидроксипропил-β-циклодекстрин, метил-β-циклодекстрин, случайным образом метилированный-β-циклодекстрин, этилированный-β-циклодекстрин, триацетил-β-циклодекстрин, перацетилированный-β-циклодекстрин, карбоксиметил-β-циклодекстрин, гидроксиэтил-β-циклодекстрин, 2-гидрокси-3-(триметиламмонио)пропил-β-циклодекстрин, глюкозил-β-циклодекстрин, мальтозил-β-циклодекстрин, сульфобутилэфир-β-циклодекстрин, разветвленный-β-циклодекстрин, гидроксипропил-β-циклодекстрин, случайным образом метилированный-γ-циклодекстрин, триметил-γ-циклодекстрин, или их комбинации.
В некоторых вариантах осуществления, раствор соединения формулы II или соединения-I, содержащий циклодекстрин, представляет собой прозрачный и бесцветный раствор и имеет вязкость, аналогичную воде. Настоящее изобретение относится к раствору, содержащему соединение-I и один или более циклодекстринов для местного применения и является применяемым местно на глазе.
Офтальмический раствор, содержащий циклодекстрин, настоящего изобретения содержит фармацевтические вспомогательные вещества, выбранные при или ниже концентраций, оптимальных для офтальмического раствора. Вспомогательные вещества настоящего изобретения представляют собой, например, хлорид бензалкония (BAK) и NaCl. В некоторых вариантах осуществления, офтальмический раствор содержит приблизительно 0,001 - приблизительно 0,005% масс./об. хлорида бензалкония (BAK). Количество BAK изменяется в зависимости от требований настоящего изобретения.
Офтальмический раствор содержит, например, без ограничения, приблизительно 0,005%-5,0% соединения-I или его свободного основания, приблизительно 2 - приблизительно 25% циклодекстрина, например, без ограничения, гидроксипропил-β-циклодекстрина (HPβCD) или метилциклодекстрина (KLEPTOSE® HPB) и/или сульфобутиловый эфир-β-циклодекстрина (CAPTISOL®), приблизительно 0,1 - приблизительно 0,7% соли, например, без ограничения, NaCl, и/или приблизительно 0,005% противомикробного агента, например, без ограничения, хлорида бензалкония (BAK). Композиция содержит соотношение соединения-I или его свободного основания к циклодекстрину 1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8, 1:9, 1:10 или 1:10 - 1:20. В некоторых вариантах осуществления, офтальмический раствор, содержащий циклодекстрин, дополнительно содержит трометамин (также известный как Tris, трис(гидроксиметил)аминометан или Tris буфер). В некоторых вариантах осуществления офтальмический раствор содержит приблизительно 1% Tris.
Офтальмические растворы настоящих вариантов осуществления содержат, например, без ограничения: приблизительно 0,3% - приблизительно 5,0% соединения-I (приблизительно 3 мг/мл - приблизительно 50,0 мг/мл), приблизительно 0,05% моногидрата моноосновного фосфата натрия, приблизительно 2% глицерина; приблизительно 0,4% соединения-I, приблизительно 7% HPβCD, приблизительно 0,7% NaCl, приблизительно 0,005% BAK; приблизительно 0,4% соединения-I, приблизительно 4% HPβCD, приблизительно 0,7% NaCl, приблизительно 0,005% BAK; приблизительно 0,4% соединения-I, приблизительно 7% HPβCD, приблизительно 1% трометамина, приблизительно 0,4% NaCl, приблизительно 0,005% BAK; и приблизительно 0,6% соединения-I, приблизительно 7% HPβCD, приблизительно 0,7% NaCl, приблизительно 0,005% BAK. Для соединения-I концентрации от приблизительно 0,005% до приблизительно 5,0%, циклодекстрин присутствуют при соответствующем молярном соотношении.
Дополнительные офтальмические растворы содержат, например, без ограничения: приблизительно 0,4% соединения формулы II (свободное основание), приблизительно 7,15% HPβCD, приблизительно 0,7% NaCl; приблизительно 0,1% соединения формулы II (свободное основание), приблизительно 1,79% HPβCD, приблизительно 0,85% NaCl; приблизительно 0,2% соединения формулы II (свободное основание), приблизительно 3,57% HPβCD, приблизительно 0,8% NaCl; приблизительно 0,6% соединения формулы II (свободное основание), приблизительно 10,72% HPβCD, приблизительно 0,6% NaCl; приблизительно 0,4% соединения формулы II (свободное основание), приблизительно 8,41% HPβCD, приблизительно 0,65% NaCl; приблизительно 0,4% соединения-I, приблизительно 10,51 HPβCD, приблизительно 0,65% NaCl; приблизительно 0,4% соединения формулы II (свободное основание), приблизительно 10,51% HPβCD, приблизительно 0,15% NaCl, приблизительно 1,0% трометамина (Tris); и/или приблизительно 0,1% соединения формулы II (свободное основание), приблизительно 2,63% HPβCD, приблизительно 0,8% NaCl; приблизительно 0,6% соединения-I (в виде свободного основания), приблизительно 15,77% HPβCD, приблизительно 0,37% NaCl. Для формулы II концентрации от приблизительно 0,005% до приблизительно 5,0% циклодекстрина присутствует при соответствующем молярном соотношении.
В некоторых вариантах осуществления, офтальмические растворы соединения-I содержат приблизительно 1,0% - приблизительно 25% циклодекстрина. Например, без ограничения, состав соединения-I содержат приблизительно 2,0% - приблизительно 3,0% HPβCD, приблизительно 3,0% - приблизительно 5,0% HPβCD, приблизительно 5,0% - приблизительно 10% HPβCD или приблизительно 10% - приблизительно 25% HPβCD.
В дополнительных вариантах осуществления, офтальмические растворы соединения-I или его свободного основания формулируют в виде, например, без ограничения: приблизительно 8,41% KLEPTOSE® HPB и приблизительно 0,142% фосфата; приблизительно 8,9% KLEPTOSE® HPB и приблизительно 0,142% фосфата; приблизительно 4,88% CAPTISOL® и приблизительно 0,142 фосфата; и/или приблизительно 4,88% CAPTISOL® и приблизительно 0,122% фосфата.
В некоторых вариантах осуществления офтальмические растворы, содержащие циклодекстрины, являются прозрачными и бесцветными и чрезвычайно вязкими, умеренно вязкими, или имеют вязкость, аналогичную воде.
В некоторых вариантах осуществления, офтальмический раствор настоящего изобретения имеет величину pH приблизительно 4,5 до приблизительно 7,5 при ниже или приблизительно 40°C.
Например, офтальмический раствор настоящего изобретения имеет величину pH приблизительно 6,0 при температуре ниже или равной приблизительно 40°C.
В некоторых вариантах осуществления офтальмический раствор настоящего изобретения имеет величину pH от приблизительно 5,0 до приблизительно 7,0 при ниже или приблизительно 40°C.
Офтальмические растворы настоящего изобретения содержат различные вводимые обычно добавки, такие как буферные агенты (например, фосфатные буферы, боратные буферы, цитратные буферы, тартратные буферы, ацетатные буферы, аминокислоты, ацетат натрия, цитрат натрия и подобные), агенты, регулирующие тоничность (например, сахариды, такие как сорбитол, глюкоза и маннитол, многоатомные спирты, такие как глицерин, концентрированный глицерин, PEG и пропиленгликоль, соли, такие как хлорид натрия и т.д.), консерванты или антисептические агенты (например, хлорид бензалкония, п-оксибензоаты, такие как метил п-оксибензоат или этил п-оксибензоат, бензиловый спирт, фенэтиловый спирт, сорбиновую кислоту или ее соль, тимеросал, хлорбутанол и т.д.), агенты, способствующие растворению, или стабилизирующие агенты (например, водорастворимые полимеры, такие как поливинилпирролидон, поверхностно-активные вещества, такие как тилоксапол, полисорбаты и т.д.), pH модификаторы (например, хлористоводоную кислоту, уксусную кислоту, фосфорную кислоту, гидроксид натрия, гидроксид калия, гидроксид аммония и подобные), загустители (например, HEC, гидроксипропилцеллюлозу, метилцеллюлозу, HPMC, карбоксиметилцеллюлозу и их соли), хелатообразующие агенты (например, эдетат натрия, цитрат натрия, конденсированный фосфат натрия и т.д.).
Офтальмические растворы, содержащие циклодекстрин, настоящего изобретения, дополнительно содержат дополнительные вспомогательные вещества, например, без ограничения, приблизительно 0,5% - приблизительно 3% поверхностно-активного соединения и эмульгатора, например, без ограничения, полисорбата 80 или его эквивалентных вспомогательных веществ; приблизительно 0,05 - приблизительно 0,4% неионного жидкого полимера алкиларилполиэфирспиртового типа, например, без ограничения тилоксапола; и/или приблизительно 0,05% - приблизительно 0,6% гидрофильного неионного поверхностно-активного соединения, например, без ограничения, полоксамера, такого как полоксамер 407.
Концентрация в различных тканях глаза, доставленная в виде офтальмического раствора
Офтальмический раствор, содержащий циклодекстрин, улучшает биодоступность активных агентов настоящего изобретения в заднем сегменте глаза. Будучи несвязанными теорией, в одном варианте осуществления, композиция, содержащая циклодекстрин, образует прозрачный и бесцветный раствор, который снижает воздействие на роговицу активного агента, например, воздействие соединения-I, приблизительно в 5-15 раз по сравнению с воздействием на роговицу эквимолярной состава в виде гелевых капель.
Будучи несвязанными теорией, в одном варианте осуществления, офтальмический раствор, содержащий циклодекстрин, увеличивает терапевтический индекс соединения-I при местном глазном введении. После введения, гидрофильный комплекс циклодекстрин-соединение-I является фармакологически инертным для роговицы. Будучи несвязанными теорией, в некоторых вариантах осуществления, комплекс циклодекстрин-соединение-I улучшает переносимость роговицы соединения-I. Будучи несвязанными теорией, в некоторых вариантах осуществления, самопроизвольная диссоциация циклодекстрина от соединения-I в периферической сосудистой системе увеличивает биодоступность в целевой ткани, например, в сосудистой оболочке глаза или сетчатке.
В отличие от других композиций соединения-I, которые в некоторых вариантах осуществления способствуют токсичности в роговице, офтальмический раствор на основе циклодекстрина, содержащий аналогичную концентрацию соединения-I, снижает воздействие на роговицу и, посредством этого, увеличивает терапевтический индекс и соответствующую пользу для пациентов. В одном варианте осуществления, применение раствора на основе циклодекстрина соединения-I относится к приблизительно 10x снижению воздействия на роговицу по сравнению с эквимолярными концентрациями гелевых капель. В некоторых вариантах осуществления, раствор на основе циклодекстрина соединения-I снижает воздействие на роговицу соединения-I на 5×, 20×, 30×, 40× или 50×. В одном варианте осуществления, 1-90 дней или 3-9 месяцев местного офтальмического дозирования приблизительно 0,005% - приблизительно 5,0% соединения-I в виде раствора на основе циклодекстрина не проявляет любого неблагоприятного или токсического воздействия на роговицу, сосудистую оболочку глаза и/или сетчатку. В еще другом варианте осуществления, 1-90 дней местного офтальмического дозирования приблизительно 0,6% - приблизительно 5,0% соединения-I в виде раствора на основе циклодекстрина не проявляет любого неблагоприятного или токсического воздействия на роговицу, сосудистую оболочку глаза и/или сетчатку.
Снижение воздействия на роговицу коррелирует с увеличением биодоступности и терапевтическим индексом активного агента в заднем сегменте, например, в сетчатке или сосудистой оболочке глаза. Например, не наблюдают токсического воздействия, приписываемого активному агенту или подходящему носителю в роговице или других частях глаза, когда приблизительно 0,1% - приблизительно 5,0% состава соединения-I, содержащей циклодекстрин, вводят местно в глаз в течение, по меньшей мере, 30 дней или более чем 60 дней.
В одном варианте осуществления, когда композицию, содержащую приблизительно 0,4% (приблизительно 4 мг/мл) соединения-I или его свободного основания, и циклодекстрин, вводят местно в глаз, концентрация в центральной сосудистой оболочке глаза составляет приблизительно 0,2 мкМ - приблизительно 0,9 мкМ, концентрация в центральной зоне сетчатки активного агента составляет приблизительно 0,02 мкМ - приблизительно 0,4 мкМ, концентрация в водянистой влаге активного агента составляет приблизительно 0,003 мкМ - приблизительно 0,009 мкМ, и концентрация в роговице активного агента составляет 6 мкМ - 30 мкМ. Применяемый в составе циклодекстрин представляет собой, например, без ограничения, KLEPTOSE® HPB или CAPTISOL®.
В некоторых вариантах осуществления, раствор на основе циклодекстрина соединения-I или его свободного основания улучшает биодоступность активного агента в центральной сосудистой оболочке глаза и центральной зоне сетчатки при снижении концентрации в роговице. В некоторых вариантах осуществления, местная доставка соединения-I или его свободного основания, сформулированных в присутствии циклодекстрина, снижает концентрацию в роговице приблизительно в 5 - приблизительно 15 раз по сравнению с концентрацией в роговице эквимолярных гелевых капель.
Будучи несвязанными теорией, в некоторых вариантах осуществления, комбинированные эффекты снижения воздействия лекарственного средства в роговице так, чтобы избежать плохой глазной переносимости при улучшении биодоступности в заднем сегменте, увеличивают терапевтический индекс и соответствующие полезные эффекты для пациентов.
В некоторых вариантах осуществления, продолжительность воздействия соединения-I составляет 1-90 дней. В некоторых вариантах осуществления, режим дозирования включает несколько курсов местного глазного введения состава, содержащего соединение-I, субъекту в течение от 1 до 90 дней. Например, режим дозирования включает введение состава один раз в день, дважды в день, три раза в день или четыре раза в день в течение от 1 до 90 дней. Например, режим дозирования включает введение состава один, два, три или четыре раза через день (т.е. в 1, 3, 5, 7 день и т.д.) в течение вплоть до 90 дней. Например, режим дозирования включает введение один раз в 1 день, один или два раза во 2-90 день. Например, режим дозирования включает введение один, два, три или четыре раза в 1 день, с последующим введением один раз в день в течение 2-90 дней. Например, режим дозирования включает введение один, два, три, четыре раза в 1 день, с последующим введением один, два, три или четыре раза через день (т.е. в 1, 3, 5, 7 день и т.д.) в течение вплоть до 90 дней. Например, один режим дозирования включает один раз в день или дважды в день в течение 1, 2, 3, 4, или 5 последовательных дней. Что касается режима дозирования два или три раза в день, субъекты получают местную глазную дозу состава соединения-I в 1 и 4 день с интервалом приблизительно 4, 6, или 8 часов. В другом варианте осуществления, субъекты получают местные глазные дозы состава соединения-I приблизительно с интервалом 4, 6, или 8 часов в течение четырех последовательных дней. В некоторых вариантах осуществления, субъекты получают одну или две дозы местной глазной дозы состава соединения-I в день в течение 5 последовательных дней. В еще других вариантах осуществления, субъекты получают одну или две дозы местной глазной дозы состава соединения-I в течение 5-90 последовательных дней. В некоторых вариантах осуществления, субъекты получают одну или две дозы местной глазной дозы состава соединения-I в течение, по меньшей мере, 25 последовательных дней. В одном варианте осуществления, субъекты получают одну или две местные глазные дозы в течение, по меньшей мере, 90 последовательных дней или более.
Например, состав, содержащий приблизительно 2 мг/мл соединения-I дважды в день, вводят в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления состав, содержащий приблизительно 3 мг/мл соединения-I дважды в день, вводят в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления состав, содержащий приблизительно 3 мг/мл соединения-I ежедневно, вводят в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления состав, содержащий приблизительно 4 мг/мл соединения-I дважды в день, вводят в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления состав, содержащий приблизительно 4 мг/мл соединения-I ежедневно, вводят в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления состав, содержащий приблизительно 5 мг/мл соединения-I дважды в день, вводят в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления состав, содержащий приблизительно 5 мг/мл соединения-I ежедневно, вводят в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления состав, содержащий приблизительно 6 мг/мл соединения-I дважды в день, вводят в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления состав, содержащий приблизительно 6 мг/мл соединения-I ежедневно, вводят в глаз или оба глаза субъекта в течение от 1 до 90 дней. Режим дозирования в течение от 1 до 90 дней может представлять собой любой из режимов, включающий последовательные или чередующиеся дни, описанных в параграфе выше.
Настоящее изобретение относится к растворам на основе циклодекстрина, содержащим гидроксипропил-бета-циклодекстрин (HP-β-CD, KLEPTOSE® HPB) или CAPTISOL®, которые хорошо переносятся при введении местно в течение 30-90 дней или в течение 4-6 месяцы. В некоторых вариантах осуществления, введение один или дважды в день приблизительно 0,005% - приблизительно 5,0% масс./об. соединения-I или его свободного основания в растворе, содержащем приблизительно 1,0% - приблизительно 25% HP-β-CD или CAPTISOL®, хорошо переносится субъектом.
В некоторых вариантах осуществления, состав, содержащий соединение формулы (II) или соединения-I, вводят в глаз или оба глаза субъекта. Например, приблизительно 0,2% - приблизительно 0,6% (масс./об.) соединения формулы (II) или приблизительно 0,1%-0,7% (масс./об.) состава, содержащего соединение I настоящего изобретения, вводят один раз в день (QD) или два раза в день (дважды в день) в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления, соединение формулы (II) или соединение-I образует комплекс с комплексообразующим агентом, например, циклодекстрином (например, Kleptose® HPB (%)) в соотношении приблизительно 1:8, в котором приблизительно 2%-13% (масс./об.) циклодекстрина (например, Kleptose® HPB (%)) добавляют к состава. Состав дополнительно содержит приблизительно 0,1% - приблизительно 0,2% буфера, например, 10 мМ фосфатного буфера. Требуемая осмолярность состава составляет приблизительно 200 - приблизительно 300 мОсм, достигаемая добавлением количества, достаточного для получения осмолярности, соли, например, хлорида натрия. pH состава составляет приблизительно 6,0 при ниже или приблизительно 40°C. Режим дозирования в течение от 1 до 90 дней может представлять собой любой из режимов, включающих последовательные или чередующиеся дни, описанных в параграфе выше.
Офтальмические суспензии
Настоящие варианты осуществления относятся к суспензиям соединения-I, содержащим соединение и фармацевтически приемлемые вспомогательные вещества. Например, суспензии соединения-I содержат, без ограничения, буферные агенты, кислоты и основания, например, без ограничения, HCl и NaOH. В одном варианте осуществления, суспензии соединения-I или его свободного основания содержат буферный агент, например, без ограничения, трометамин (Tris). Суспензия на основе трометамина соединения формулы II или соединения-I является пригодной для местного введения в глаз.
Суспензии настоящего изобретения содержат от приблизительно 0,005% до приблизительно 5,0% масс./об. активного агента формул (I), (II) или его фармацевтически приемлемой соли, например, соединения-I. Концентрация соединения-I или его свободного основания (формула II) в суспензиях составляет приблизительно 0,005% - приблизительно 0,01%, приблизительно 0,01% - приблизительно 0,05%, приблизительно 0,05% - приблизительно 0,1%, приблизительно 0,1% - приблизительно 0,2%, приблизительно 0,2% - приблизительно 0,3%, приблизительно 0,3% - приблизительно 0,4%, приблизительно 0,4% - приблизительно 0,5%, приблизительно 0,5% - приблизительно 0,6%, приблизительно 0,6% - приблизительно 0,7%, приблизительно 0,7% - приблизительно 0,8%, приблизительно 0,8% - приблизительно 0,9%, приблизительно 0,9% - приблизительно 1,0%, приблизительно 1,0 - приблизительно 2,0%, приблизительно 2,0 - приблизительно 3,0%, приблизительно 3,0 - приблизительно 4,0% или приблизительно 4,0 - приблизительно 5,0% масс./об. для местного введения. В некоторых вариантах осуществления, суспензии содержат приблизительно 0,005%, приблизительно 0,05%, приблизительно 0,1%, приблизительно 0,2%, приблизительно 0,3%, приблизительно 0,4%, приблизительно 0,5%, приблизительно 0,6%, приблизительно 0,7%, приблизительно 0,8%, приблизительно 0,9%, приблизительно 1,0%, приблизительно 2,0%, приблизительно 3,0%, приблизительно 4,0% или приблизительно 5,0% масс./об. соединения-I или его свободного основания (формула II).
Офтальмические суспензии могут содержать различные традиционно включаемые добавки, такие как буферные агенты (например, фосфатные буферы, боратные буферы, цитратные буферы, тартратные буферы, ацетатные буферы, аминокислоты, ацетат натрия, цитрат натрия и подобные), агенты, регулирующие тоничность (например, сахариды, такие как сорбитол, глюкоза и маннитол, многоатомные спирты, такие как глицерин, концентрированный глицерин, PEG и пропиленгликоль, соли, такие как хлорид натрия и т.д.), консерванты или антисептические агенты (например, хлорид бензалкония, п-оксибензоаты, такие как метил п-оксибензоат или этил п-оксибензоат, бензиловый спирт, фенэтиловый спирт, сорбиновую кислоту или ее соль, тимеросал, хлорбутанол и т.д.), агенты, способствующие растворению, или стабилизирующие агенты (например, водорастворимые полимеры, такие как поливинилпирролидон, поверхностно-активные вещества, такие как тилоксапол, полисорбаты и т.д.), pH модификаторы (например, хлористоводоную кислоту, уксусную кислоту, фосфорную кислоту, гидроксид натрия, гидроксид калия, гидроксид аммония и подобные), загустители (например, HEC, гидроксипропилцеллюлозу, метилцеллюлозу, HPMC, карбоксиметилцеллюлозу и их соли), хелатообразующие агенты (например, эдетат натрия, цитрат натрия, конденсированный фосфат натрия и т.д.).
Офтальмическая суспензия настоящего изобретения содержит фармацевтические вспомогательные вещества, выбранные при или ниже концентраций, оптимальных для офтальмического раствора. Вспомогательные вещества настоящего изобретения включают, например, без ограничения, моногидрат фосфата натрия, глицерин и хлорид бензалкония (BAK).
В некоторых вариантах осуществления, приблизительно 0,005%, приблизительно 0,05%, приблизительно 0,1%, приблизительно 0,2%, приблизительно 0,3%, приблизительно 0,4%, приблизительно 0,5%, приблизительно 0,6%, приблизительно 0,7%, приблизительно 0,8%, приблизительно 0,9%, приблизительно 1,0%, приблизительно 2,0%, приблизительно 3,0%, приблизительно 4,0% или приблизительно 5,0% масс./об. формулы (I), (II) или его фармацевтически приемлемой соли, например, соединения-I, представляют собой водный раствор трометамина. Суспензия на основе трометамина соединения-I или его свободного основания содержит дополнительные буферы и вспомогательные вещества, например, без ограничения, фосфатный буфер. Суспензии, полученные в приблизительно 0,2% - приблизительно 1,0% Tris, могут дополнительно содержать один или более поверхностно-активных соединений и эмульгаторов, например, без ограничения, полисорбат 80 или его эквивалентные вспомогательные вещества; один или более неионных жидких полимеров алкиларилполиэфирспиртового типа, например, без ограничения тилоксапол; и/или один или более гидрофильных неионных поверхностно-активных веществ, например, без ограничения, полоксамер, такой как полоксамер 407.
Настоящее изобретение относится к суспензиям агентов настоящего изобретения, сформулированным в присутствии вспомогательных веществ, таких как, без ограничения, повидон, полисорбат 80 (PS80), полиэтиленгликоль (PEG) 400, тилоксапол, полоксамер, глицерин и BAK в Tris буфере.
В одном варианте осуществления суспензия соединения-I или его свободного основания содержит приблизительно 0,1-0,5% фосфатного буфера. В некоторых вариантах осуществления, pH суспензии на основе трометамина составляет pH 4-7, например, pH 6,0. В некоторых вариантах осуществления, суспензии, полученные в Tris, дополнительно содержат приблизительно 0,5% - приблизительно 2% полисорбата 80; приблизительно 0,05 - приблизительно 0,2% тилоксапола; и/или приблизительно 0,05% - приблизительно 0,4% полоксамера 407.
В некоторых данных вариантах осуществления, суспензии настоящего изобретения дополнительно содержат приблизительно 0,01 - приблизительно 1%, или приблизительно 1 - приблизительно 2,0% масс./об. глицерина. В конкретном варианте осуществления, суспензии содержат приблизительно 2% масс./об. глицерина.
В некоторых вариантах осуществления, суспензии настоящего изобретения дополнительно содержат приблизительно 0,001- приблизительно 0,005% масс./об. хлорида бензалкония (BAK). Количество BAK может изменяться в зависимости от любых наблюдаемых неблагоприятных эффектов. BAK может быть повреждающим для клеток на поверхности глаза, и, следовательно, количество в составе можно изменять, получая оптимальное количество для глазного проникновения соединения-I, но не в ущерб целостности клеточного слоя глаза и без повышенной токсичности.
В некоторых вариантах осуществления, суспензия необязательно содержит буферы. Буферы при применении, например, могут представлять собой основный монофосфат натрия, фосфорную кислоту и Tris буфер. Концентрация соединение-I в суспензии составляет приблизительно 0,005% - приблизительно 5,0% масс./об.. Суспензия, полученная без дополнительного буфера, дополнительно содержит приблизительно 0,005% BAK и приблизительно 2% глицерина и с pH 6,0. В другом варианте осуществления суспензия, полученная без дополнительного буфера, содержит приблизительно 1% полисорбата 80, приблизительно 0,1% тилоксапола, приблизительно 0,2% полоксамера 407, приблизительно 0,005% BAK, приблизительно 2,0% глицерина и с pH 6,0.
В суспензиях, полученных в фосфорной кислоте/Tris, суспензия содержит приблизительно 0,14% фосфорной кислоты, приблизительно 0,2% Tris основания, приблизительно 1,0% полисорбата 80, приблизительно 0,005% BAK, приблизительно 2,0% глицерина и с pH 6,0. В одном варианте осуществления, суспензия дополнительно содержит приблизительно 0,2% тилоксапола. pH суспензии изменяется в диапазоне приблизительно pH 6,0-7,2.
Суспензии, полученные только в трометамине (Tris), содержат приблизительно 1% полисорбата 80, приблизительно 0,1% тилоксапола, приблизительно 0,2% полоксамера 407, приблизительно 0,6% Tris, приблизительно 0,005% BAK, и приблизительно 2,0% глицерина с pH 6,0. В другом варианте осуществления, суспензия, полученная в Tris, содержит приблизительно 1% Tris, приблизительно 0,45% NaCl, приблизительно 0,025% EDTA, приблизительно 0,2% HPMC, приблизительно 0,1% полисорбата 80, приблизительно 0,005% BAK с pH 6,0. В данных суспензиях 1 N HCl и/или 1N NaOH применяют для титрования до подходящего pH.
Суспензия настоящего изобретения содержит приблизительно 0,005%, приблизительно 0,05%, приблизительно 0,1%, приблизительно 0,2%, приблизительно 0,3%, приблизительно 0,4%, приблизительно 0,5%, приблизительно 0,6%, приблизительно 0,7%, приблизительно 0,8%, приблизительно 0,9%, приблизительно 1,0%, приблизительно 2,0%, приблизительно 3,0%, приблизительно 4,0% или приблизительно 5,0% активного агента формул (I), (II) или его фармацевтически приемлемой соли, например, соединения-I, и приблизительно 0,01% - приблизительно 0,05%, приблизительно 0,05 - приблизительно 0,09%, или приблизительно 0,09 - приблизительно 0,2% масс./об. моногидрата моноосновного фосфата натрия и/или приблизительно 0,3% - приблизительно 1,0% Tris. В конкретном варианте осуществления суспензия содержит приблизительно 0,14% или приблизительно 0,2% масс./об. Tris-буфера. В дополнительных вариантах осуществления, суспензии получают в приблизительно 0,6% Tris или приблизительно 1,0% Tris. Другие эквивалентные буферные системы, хорошо известные в данной области техники, также применяют в суспензиях настоящего изобретения. В одном варианте осуществления, соединение формулы II или соединение-I формулируют при приблизительно 0,4% активного агента, приблизительно 5% кремофора RH40, приблизительно 2,0% глицерина и приблизительно 0,005% BAK.
В некоторых вариантах осуществления, суспензия настоящего изобретения имеет величину pH приблизительно 4,0-7,5 при ниже или приблизительно 40°C.
Например, суспензия настоящего изобретения имеет величину pH приблизительно 6,0 при ниже или приблизительно 40°C.
В некоторых вариантах осуществления, суспензия настоящего изобретения имеет величину pH приблизительно 5,0 до приблизительно 7,0 при ниже или приблизительно 40°C.
Концентрация в различных глазных тканях, доставляемая в виде офтальмической суспензии
В некоторых вариантах осуществления суспензия соединения-I или его свободного основания обеспечивает аналогичную концентрацию активного агента в центральной сосудистой оболочке глаза и центральной зоне сетчатки по сравнению с концентрацией активного агента, доставляемой в виде гелевых капель (обсуждаемых ниже).
В некоторых вариантах осуществления суспензия на основе Tris соединения-I или его свободного основания улучшает биодоступность активного агента в центральной сосудистой оболочке глаза и центральной зоне сетчатки, при снижении концентрации в роговице. В некоторых вариантах осуществления, местная доставка соединения-I или его свободного основания, сформулированных в Tris-основании, снижает концентрацию в роговице приблизительно в 5-10×, 10-20×, 20-30×, 30-40× или приблизительно в 50-100× по сравнению с концентрацией в роговице эквимолярного соединения-I или его свободного основания, доставляемых в виде гелевых капель.
Комбинированные эффекты снижения воздействие на роговицу лекарственного средства так, чтобы избежать плохой глазной переносимости, при этом поддерживая или улучшая биодоступность в заднем сегменте так, чтобы улучшить ингибирование рецепторной тирозинкиназы (RTK), например, VEGF, значительно улучшает терапевтический индекс и соответствующие полезные эффекты для пациентов.
Введение один или дважды в день приблизительно 0,005% - приблизительно 5,0% масс./об. суспензии соединения-I настоящего изобретения в течение 30-90 дней или 4-6 месяцы хорошо переносится глазом.
Гелевые капли
В некоторых вариантах осуществления офтальмическую композицию или состав настоящего изобретения формулируют в виде гелевых капель. Состав в виде гелевых капель содержит не более чем приблизительно 0,05% моногидрата моноосновного фосфата натрия, обеспечивая требуемую буферную емкость и свободнотекучие, поддающееся фильтрованию состава при приблизительно 0,005% - приблизительно 2,0% соединения-I без необходимости добавлять поверхностно-активное вещество.
Композиция в виде гелевых капель настоящего изобретения содержит от приблизительно 0,005% до приблизительно 2,0% масс./об. активного агента формул (I), (II) или его фармацевтически приемлемой соли, например, соединения-I. Концентрация соединения-I или его свободного основания (формула II) в гелевых каплях может составлять приблизительно 0,005% - приблизительно 0,01%, приблизительно 0,01% - приблизительно 0,05%, приблизительно 0,05% - приблизительно 0,1%, приблизительно 0,1% - приблизительно 0,2%, приблизительно 0,2% - приблизительно 0,3%, приблизительно 0,3% - приблизительно 0,4%, приблизительно 0,4% - приблизительно 0,5%, приблизительно 0,5% - приблизительно 0,6%, приблизительно 0,6% - приблизительно 0,7%, приблизительно 0,7% - приблизительно 0,8%, приблизительно 0,8% - приблизительно 0,9%, приблизительно 0,9% - приблизительно 1,0% или приблизительно 1,0% - приблизительно 2,0% масс./об. для местного введения. В некоторых вариантах осуществления, гелевые капли содержат приблизительно 0,005%, приблизительно 0,05%, приблизительно 0,1%, приблизительно 0,2%, приблизительно 0,3%, приблизительно 0,4%, приблизительно 0,5%, приблизительно 0,6%, приблизительно 0,7%, приблизительно 0,8%, приблизительно 0,9%, приблизительно 1,0%, или приблизительно 2% масс./об. соединения-I или его свободного основания (формула II).
В некоторых вариантах осуществления, офтальмический состав в виде гелевых капель настоящего изобретения содержит глицерин в качестве агента, регулирующего тоничность. Некоторые варианты осуществления настоящего изобретения относятся к офтальмическому составу, содержащей маннитол. Содержание глицерина или маннитола в количестве, предотвращающем любые изменения растворимости соединения-I, и при концентрации приблизительно 2,0 - приблизительно 2,5%, глицерина, обеспечивает осмолярность приблизительно 225 - приблизительно 300 мОсм/кг в зависимости от концентрации фосфата. В дополнительных вариантах осуществления, глицерин составляет приблизительно 2%, и фосфат составляет приблизительно 0,05% офтальмического состава в виде гелевых капель. Концентрации глицерина и фосфата настоящего изобретения представляют собой такое количество, что тоничность офтальмического состава составляет приблизительно 240 мОсм/кг.
Офтальмический состав в виде гелевых капель настоящего изобретения содержит хлорид бензалкония (BAK). В некоторых вариантах осуществления, содержание BAK составляет приблизительно 0,005%, достаточное для сохранения офтальмического состава от загрязнения микроорганизмами. В некоторых вариантах осуществления настоящего изобретения, BAK не требуется для применения офтальмического состава в виде стерильного продукта одноразового применения.
Офтальмический состав в виде гелевых капель соединения-I содержит: приблизительно 0,005% - приблизительно 2,0%, соединения-I или его свободного основания, приблизительно 0,05% фосфата натрия, приблизительно 2% глицерина в качестве агента, регулирующего тоничность, приблизительно 0,005% BAK в качестве консерванта, воду (очищенную, т.е. дистиллированную или деионизированную) в качестве среды и гидроксид натрия, доводящий pH до 6,0. В одном варианте осуществления, не добавляют другие вспомогательные вещества.
Гелевые капли настоящего изобретения содержат приблизительно 0,005% - приблизительно 2,0% активного агента формулы (I) (II) или его фармацевтически приемлемой соли, например, соединения-I и приблизительно 0,01% - приблизительно 0,05%, приблизительно 0,05 - приблизительно 0,09%, или приблизительно 0,09 - приблизительно 0,2% масс./об. моногидрата моноосновного фосфата натрия. В конкретном варианте осуществления гелевые капли содержит приблизительно 0,05%, приблизительно 0,2% или приблизительно 0,2% масс./об. буфера на основе моногидрата моноосновного фосфата натрия. Другие эквивалентные буферные системы, хорошо известные в данной области техники, также применяют в гелевых каплях настоящего изобретения. В одном варианте осуществления, соединение-I или его свободное основание формулируют в виде приблизительно 0,4% - приблизительно 2,0% активного агента, приблизительно 5% кремофора RH40, приблизительно 2,0% глицерина и приблизительно 0,005% BAK.
В одном варианте осуществления, гелевые капли соединения-I содержат приблизительно 0,3% - приблизительно 2,0% (3-20 мг/мл) соединения-I, приблизительно 0,05% - приблизительно 0,2% фосфата натрия и приблизительно 2% глицерина. pH состава составляет pH 5,0-7,0.
Настоящее изобретение относится к гелевым каплям агентов настоящего изобретения, сформулированным в присутствии вспомогательных веществ, таких как, без ограничения, повидон, полисорбат 80 (PS80), полиэтиленгликоль (PEG) 400, тилоксапол, полоксамер, глицерин и BAK, в фосфатном буфере.
Глазные капли
Описанной в настоящем изобретении является состав, содержащий описанные соединения в виде глазных капель, формы доставки лекарственного средства, которая является фармацевтически приемлемой для пациентов, удобной, безопасной, с началом действия через несколько минут. Стандартные глазные капли, применяемые в терапии согласно федеральной регуляторной практики США, являются стерильными, имеют pH приблизительно 6,0-7,4, и, если предполагается применять их более одного раза, содержат консервант, но имеют ограниченный срок годности после вскрытия, обычно один месяц. Если глазные капли упакованы в стерильный дозатор только для однократного применения, то консервант можно не включать.
Один способ формулирования состава в виде глазных капель включает наиболее чистую форму описанного соединения (например, больше чем 99% чистота) и смешивание соединения с буферами и регуляторами тоничности, для получения физиологического pH и осмолярности. Примеры буферных агентов для поддержания или регулирования pH включают, но не ограничиваются, ацетатные буферы, цитратные буферы, фосфатные буферы и боратные буферы. Примеры регуляторов тоничности представляют собой хлорид натрия, маннитол и глицерин. В некоторых вариантах осуществления также добавляют другие фармацевтически приемлемые ингредиенты.
Затем, сформулированный раствор разделяют на аликвоты или в множество дискретных, стерильных картриджей для одноразового применения, каждый из которых является пригодным для единичного дозирования, или в один картридж для единичного дозирования. Данный один картридж для одноразового применения представляет собой, например, конический или цилиндрический дозатор удельного объема, причем боковые стенки контейнера сдавливают в радиальном направлении к продольной оси, обеспечивая распыление содержимого контейнера на одном конце контейнера.
Настоящее изобретение относится к офтальмическим растворам/суспензиям в качестве капель для глаз, упакованным в виде формы с множеством доз или формы с однократной дозой, например, в виде пластиковой бутылки с капельницей. В составах для введения множества доз, консерванты требуются для предотвращения загрязнения микроорганизмами после вскрытия контейнера. Подходящие консерванты включают, но не ограничиваются: хлорид бензалкония, тимеросал, хлорбутанол, метилпарабен, пропилпарабен, фенилэтиловый спирт, эдетат динатрия, сорбиновую кислоту, поликватермиум-1 или другие агенты, известные специалистам в данной области техники, и все из которых предполагаются для применения в настоящем изобретении. Данные консерванты обычно применяют при концентрации от 0,001 до приблизительно 1,0% вес/объем.
Не желая быть связанными теорией, состав настоящего изобретения в виде глазных капель обеспечивает импульсное введение лекарственного средства. Способ, которым соединение-I получает доступ к заднему сегменту, не является направленной диффузией через роговицу с последующей диффузией через водянистую влагу, стекловидное тело, сетчатку и, наконец, сосудистую оболочку глаза. Скорее, соединение-I достигает значительной биодоступности в задней части глаза после местной инсталляции, применяя окружной путь, а не через глазное яблоко.
При определенных клинических заболеваниях, растворы/суспензии в виде глазных капель можно формулировать с другими фармацевтическими агентами для того, чтобы уменьшить раздражение другим ингредиентом и способствовать достижению клинической реакции. Данные агенты включают, но не ограничиваются, сосудосуживающее средство, такое как фенилэфрин, оксиметазолин, нафтазолин или тетрагидрозолин; стабилизатор тучных клеток, такой как олопатадин; антигистамин, такой как азеластин; антибиотик, такой как тетрациклин; стероидный противовоспалительный агент, такой как бетаметазон; нестероидный противовоспалительный агент, такой как диклофенак; иммуномодулятор, такой как имиквимод или интерферонов; и противовирусные средства, такие как валацикловир, цидофовир и трифлуридин. Дозы, применяемые для описанных выше целей, изменяются, но представляют собой эффективное количество для подавления дискомфорта, зуда, раздражения или боли в глазу. При применении композиций местно, “фармацевтически эффективное количество” соединения может обычно находиться в диапазоне концентраций от 0,05 мг/мл до приблизительно 10 мг/мл, в 1-4 каплях, вводимых в виде одной дозы 1-4 раза в день. Самый распространенный способ офтальмической доставки лекарственного средства представляет собой введение капель в роговицу (т.е. “глазные капли”).
Ключевым требованием является то, что композиция должна быть стерильной и быть получена в стерильном окружении. Идеальное описанное соединение для применения в офтальмических растворах/суспензиях должно быть растворимым и/или смешиваемым с водной средой при нормальных глазных pH и тоничности. Более того, описанные соединения должны быть стабильными, нетоксичными, долгодействующими и достаточно эффективными для противодействия разбавлению концентрации лекарственного средства при моргании и слезоотделении.
Лекарственные формы
Состав настоящего изобретения может быть пригоден для офтальмического применения. В одном варианте осуществления состав представляет собой раствор. Раствор настоящего изобретения может представлять собой прозрачный, бесцветный, стерильный, изотонический, забуференный водный свободнотекучий жидкий препарат. Лекарственный продукт имеет pH приблизительно 6,0, и его можно хранить при +5°C. Лекарственный продукт можно обеспечивать в виде контейнерной закрытой системы, состоящей из полупрозрачной офтальмической бутыли - дозатора с наконечником-дозатором и крышкой.
В некоторых вариантах осуществления, клиническая концентрация офтальмического раствора или суспензии соединения-I является равной или меньшей чем приблизительно 0,1 мг/мл, равной или меньшей чем приблизительно 0,2 мг/мл, приблизительно 0,2 - приблизительно 1,0 мг/мл, приблизительно 0,3 - приблизительно 1,0 мг/мл, приблизительно 0,4 - приблизительно 1,0 мг/мл, приблизительно 0,5 - приблизительно 1,0 мг/мл, приблизительно 0,6 - приблизительно 1,0 мг/мл, приблизительно 0,7 - приблизительно 1,0 мг/мл, приблизительно 0,8 - приблизительно 1,0 мг/мл, приблизительно 0,9 - приблизительно 1,0 мг/мл, приблизительно 1,0 - приблизительно 2,0 мг/мл, приблизительно 2,0 - приблизительно 3,0 мг/мл, приблизительно 3,0 - приблизительно 4,0 мг/мл, приблизительно 4,0 - приблизительно 5,0 мг/мл, приблизительно 5,0 - приблизительно 6,0 мг/мл, приблизительно 5,0 - приблизительно 10,0 мг/мл, приблизительно 10 - приблизительно 20 мг/мл, приблизительно 20 - приблизительно 30 мг/мл, приблизительно 30 - приблизительно 40 мг/мл, или приблизительно 40 - приблизительно 50 мг/мл.
В одном варианте осуществления настоящего изобретения концентрация соединения составляет приблизительно 0,005% - приблизительно 5,0% (приблизительно 0,5 - приблизительно 50 мг/мл). Требуемую фармакологическую активность (или концентрацию) состава настоящего изобретения против патологической неоваскуляризации сосудистой оболочки глаза и сетчатки получают после глазного введения композиций, содержащих приблизительно 0,005%, приблизительно 0,05%, приблизительно 0,1%, приблизительно 0,2%, приблизительно 0,3%, приблизительно 0,4%, приблизительно 0,5%, приблизительно 0,6%, приблизительно 0,7%, приблизительно 0,8%, приблизительно 0,9%, приблизительно 1,0%, приблизительно 2,0%, приблизительно 3,0%, приблизительно 4,0%, или приблизительно 5,0% масс./об. соединения-I. Настоящее изобретение обеспечивает то, что после местного глазного введения оптимальной дозы (например, приблизительно 0,005% - приблизительно 5,0%) фармакологически активная концентрация достигается и поддерживается в центральной хориоидальной ткани-мишени. В одном варианте осуществления активный агент (формулы II или соединение-I) формулируют при концентрации приблизительно 0,005 - приблизительно 5,0% масс./об. и дозируют один или два раза в день в глаз в течение более чем 60 последовательных дней. Наблюдаемые концентрации в плазме после местного введения являются существенно меньшими, чем концентрации, которые, как ожидают, обеспечивают системную токсичность.
В некоторых вариантах осуществления, соединение-I проявляет сильное ингибирование активности тирозинкиназы для нескольких рецепторов проангиогенных факторов роста, с IC50, меньше чем приблизительно 100 нМ (см. таблицу 3). Соединение-I также блокирует высокоаффинные VEGF рецепторы, например, VEGFR-1/Flt-1, но с меньшей эффективностью (с IC50 приблизительно 122 нМ (69,41 нг/мл)).
Хотя VEGFR ингибирование, по-видимому, является важным для снижения проницаемости сосудов и предотвращения последующего неоваскулярного роста, одновременное ингибирование VEGF сигнального пути с ингибированием других сигнальных путей факторов роста (например, PDGF и ангиопоэтины/Tie2) может быть связано с уникальными терапевтическими эффектами. Терапевтические эффекты более широкого ингибирования сигнальных путей могут способствовать регрессии вновь образованных патологических сосудов в заднем сегменте глаза.
В некоторых вариантах осуществления, приблизительно 300 нМ (приблизительно 170,67 нг/мл) соединения-I ингибирует VEGFR-2 киназную функцию (см. таблицу 4). Также наблюдается существенная блокировка аналогичного набора рецепторов проангиогенных факторов роста, включая FGFR1-3, Tie-2 и EphB-4. Неожиданный результат заключается в том, что концентрация соединения-I приблизительно 300 нМ ингибирует VEGFR-2 киназную функцию, которая попадает в типичный диапазон, обнаруживаемый в центральной сосудистой оболочке глаза и сетчатке после пяти дней местной офтальмической доставки.
Обзор лекарственного вещества и лекарственного продукта
Лекарственный продукт: офтальмические составы соединения-I для клинических исследований получали при дозируемых концентрациях 0,05%-1,0% (в виде соединения-I). В некоторых вариантах осуществления доза соединения-I в составе составляет 0,05%, 0,1%, 0,2%, 0,3%, 0,4%, 0,6%, 0,8% или 1,0%. Офтальмические составы соединения-I (растворы или суспензии) предполагаются для ежедневного одноразового местного введения в глаз в клинических условиях. В добавление к активному ингредиенту, в некоторых вариантах осуществления лекарственный продукт содержит приблизительно 0,005% BAK в качестве консерванта, очищенную воду в качестве среды и отрегулирован по pH гидроксидом натрия до pH 6,0.
Гелевые капли на основе фосфата натрия
Офтальмические полезные эффекты соединения-I в составе на основе фосфата натрия (перечисленные в таблице 6) являются результатом свойств самообразования геля API в буферах, таких как фосфат натрия. Спонтанное образование самообразующегося тиксотропного геля соединения-I из прозрачного раствора осуществляют повышением концентрации активного агента в фосфате натрия. Когда концентрация активного агента в фосфатном буфере достигает состояния сверхнасыщения, в геле наблюдают нерастворимые частицы соединения-I.
Современное состояние данной области техники предсказывает, что нанесение геля с повышенной вязкостью на поверхность глаза будет увеличивать время удерживания в роговице. Повышенное время удерживания в роговице, в свою очередь, способствует поглощению лекарственного средства в глазе. Как результат, внутриглазные концентрации лекарственного средства вязких гелей будут увеличены по сравнению с невязкими композициями, такими как водные растворы. Один способ увеличения вязкости заключается в применении различных вспомогательных веществ, увеличивающих вязкость, например, карбоксиметилцеллюлозы, которая по существу обеспечивает повышенное внутриглазное поглощение различных лекарственных веществ после глазного введения. Настоящее изобретение относится к тиксотропному гелю соединения-I, полученному в отсутствии вспомогательных веществ, увеличивающих вязкость. Например, когда соединение-I растворяют в простом буфере, таком как фосфат натрия, образуется тиксотропный гель. Тиксотропный гель, который образуется без любых вспомогательных веществ, повышающих вязкость, формулируют в виде гелевых капель.
Настоящее изобретение относится к зависящей от дозы и частоты приема доставке соединения-I в ткани заднего сегмента глаза.
Составы в виде гелевых капель настоящего изобретения (перечисленные в таблице 6) отличаются друг от друга несколькими аспектами, такими как активная концентрация, концентрация фосфата натрия, присутствие или отсутствие агента, регулирующего тоничность (глицерина) или консерванта (хлорида бензалкония/BAK), поверхностно-активных веществ, увеличивающих растворимость (полисорбат 80, тилоксапол, и/или полоксамер) и pH.
Суспензия на основе трометамина
Настоящее изобретение относится к суспензии соединения-I в виде состава на основе трометамина. В некоторых вариантах осуществления, суспензия соединения-I в виде состава на основе трометамина содержит равное или большее чем 95% активного лекарственного вещества в нерастворимой форме. Данная характеристика является отличимой от растворимого или полурастворимого состояния соединения-I в гелевых каплях (гелевые капли (гель), которое не представляет собой полностью растворенное состояние по мере увеличения концентрации активного агента), или в составе на основе циклодекстрина. Составы на основе трометамина соединения-I показывают повышенную мутность при повышении концентрации активного агента. Введение местных капель суспензии соединения-I в глаз, которые представляют собой комбинацию растворимых и нерастворимых компонентов активного агента, является полезным с точки зрения и безопасности/переносимости и эффективности.
Настоящее изобретение обеспечивает соединение-I в суспензии на основе трометамина, доставляемое при концентрациях в ткани-мишени в 10-1000× клеточной IC50 для различных проангиогенных RTK. См., например, таблицу 7.
Безопасность и переносимость роговицы введенного местно соединения-I представляет собой прямое следствие количества растворенного (в противоположность нерастворенному) активного агента, нанесенного на поверхность роговицы и полученной в результате концентрации в ткани роговицы. В некоторых вариантах осуществления, субъекты, которые получают местное глазное введение суспензии на основе трометамина, способны переносить вплоть до больших концентраций активного агента в составе по сравнению с эквимолярнными составами в виде гелевых капель на основе фосфата натрия.
мг/мл
[сосудистая оболочка глаза] нМ
[сетчатка]
нМ
[сетчатка]
нМ
[AH] нМ
нМ
нМ
мг/мл
[сетчатка] нМ
нМ
Настоящее изобретение обеспечивает биодоступность соединения-I в заднем сегменте глаза после введения суспензии на основе трометамина. Биодоступность соединения-I в заднем сегменте глаза является прямо пропорциональной суммарному количеству введенного лекарственного средства (нерастворимое плюс растворимое, см. таблицу 7). Хотя нерастворимые частицы лекарственного средства не являются легко доступными в тканях переднего сегмента глаза; собственные и уникальные физико-химические свойства соединения-I обеспечивают вхождение нерастворимого и растворимого компонентов в ткани заднего сегмента глаза, такие как сосудистая оболочка глаза и сетчатка. Таким образом, даже большие концентрации лекарственного средства, чем концентрации, получаемые с композициями в виде гелевых капель, содержащими эквимолярные количества активного агента, получают с суспензией на основе трометамина. Таким образом, суспензия на основе трометамина обеспечивает: a) улучшенную переносимость роговицы и b) повышенную биодоступность в заднем сегменте глаза, в частности в сосудистой оболочке глаза, первичной целевой ткани для лечения неоваскулярной (влажной) ВМД.
Раствор на основе циклодекстрина
Циклодекстрины, которые представляют собой циклические олигосахариды, образованные шестью-восемью остатками декстрозы (α-, β- и γ-CD), содержанные одной-четырьмя связями, являются хорошо известными из-за их способности действовать в качестве агента, улучшающего растворимость, для относительно нерастворимых лекарственных средств. См. Stella & He, Cyclodextrins, Toxicol. Pathol., 36: 30-42 (2008).
В некоторых вариантах осуществления, 2-гидроксипропил-β-циклодекстрин (HP-β-CD, также известный как KLEPTOSE® HPB) при равном или большем чем 1:6 молярном отношении или сульфобутилэфир-β-циклодекстрин (SBE-β-CD, также известный как CAPTISOL®) при равном или большем чем 1:2 отношении в предложенных клинических композициях, офтальмических растворах соединения-I или его свободного основания, обеспечивает растворимость, которая соответствует концентрациям клинических доз 0,1-1,0% соединения-I.
В некоторых вариантах осуществления, растворы на основе циклодекстрина соединения-I или его свободного основания не только обладают улучшенной растворимостью активного агента в однородном растворе, но, после местного глазного введения, также обладают новыми и ранее не наблюдаемыми характеристиками значительно улучшенного терапевтического индекса активного агента в заднем сегменте глаза. Растворы соединения-I настоящего изобретения снижают воздействие на передний сегмент глаза, посредством этого увеличивая концентрацию активного агента в растворе и повышая частоту его доставки для того, чтобы поддерживать высокие концентрации в заднем сегменте глаза. Обе данные полезные характеристики связаны с известным свойством циклодекстрина образовывать гидрофильные комплексы с гидрофобными лекарственными средствами. См. Stella & He, Cyclodextrins, Toxicol. Pathol., 36: 30-42 (2008). При формулировании с соединением-I или его свободным основанием, циклодекстрин образует прозрачный, бесцветный раствор, который обладает вязкостью, подобной воде. После местного глазного введения, комплекс соединение-I/циклодекстрин имеет фармакологически неактивный и метаболически инертный вид. Комплекс соединение-I/циклодекстрин обеспечивает переносимость роговицы до того момента, как циклодекстрин самопроизвольно диссоциирует от активного агента, таким образом, обеспечивая высокую концентрацию соединения-I в его предполагаемом месте действия в заднем сегменте глаза, например, сосудистой оболочке глаза и сетчатке.
В некоторых вариантах осуществления, растворы на основе циклодекстрина соединения-I снижают воздействие на роговицу соединения-I по сравнению с составами в виде гелевых капель при аналогичных концентрациях лекарственного средства. Применение растворов на основе циклодекстрина соединения-I обеспечивает приблизительно 10x снижение концентраций в роговице по сравнению с дозированием эквимолярных составов гелевых капель. В некоторых вариантах осуществления, после 20-30 дней местного офтальмического дозирования приблизительно 0,2-2,0%, например, приблизительно 0,6%, соединение-I в виде раствора на основе циклодекстрина, неблагоприятные результаты не приписывают испытуемому изделию или среде. Настоящее изобретение обеспечивает более высокие концентрации соединения-I в целевых тканях заднего сегмента глаза, таких как в центральной сосудистой оболочке глаза и центральной зоне сетчатки, когда применяют местно раствор на основе циклодекстрина соединения-I. В некоторых вариантах осуществления, комбинированные эффекты снижения воздействия на роговицу лекарственного средства так, чтобы избежать низкой глазной переносимости, при этом увеличивая биодоступность в заднем сегменте так, чтобы усиливать RTK ингибирование, значительно улучшает терапевтический индекс и соответствующий полезный эффект (эффекты) для подвергаемых лечению субъектов.
Настоящее изобретение обеспечивает расширение терапевтического окна и для составов на основе суспензии (см. пример 3) и для составов на основе циклодекстрина, благодаря значительно сниженному воздействию (приблизительно 10-100× или 1-2 log снижению). Сниженное воздействие улучшает безопасность/переносимость в роговице, что обеспечивает большие концентрации или частоту дозирования соединения-I, вводимого местно. Большая концентрация позволяет соединению-I достигать больших концентраций в задних целевых тканях глаза, что увеличивает терапевтическую эффективность соединения-I.
В некоторых вариантах осуществления, местное глазное дозирование офтальмических гелевых капель связано с высоких воздействием на ткани роговицы (≥100 мкМ) и соответствующими нежелательными результатами наблюдений в переднем сегменте, такими как дискомфорт, воспаление роговицы и конъюнктивы, разрушение эпителия роговицы и/или истончение и дегенерация. Напротив, повторяемое местное глазное дозирование офтальмического раствора соединения-I обеспечивает воздействие на роговицу, которое является примерно в 5-10 раз меньшим, чем эквимолярная доза офтальмических гелевых капель, и часто не сопровождается нежелательными клиническими или гистопатологическими результатами. Местное глазное дозирование офтальмического раствора соединения-I также обеспечивает равное или большее целевое терапевтическое воздействие в центральной сосудистой оболочке глаза по сравнению с эквимолярной дозой офтальмических гелевых капель. В общем, комбинация сниженного воздействия на роговицу и соответствующей повышенной глазной переносимости, при этом одновременно поддерживая или способствуя доставки лекарственного средства в целевые ткани заднего сегмента глаза, вместе с повышенной физико-химической стабильностью, обеспечивает большую пользу субъектам по сравнению с офтальмическим составом в виде гелевых капель.
1- - 5-дневные PK результаты с местным глазным введением соединения-I в виде растворов на основе циклодекстрина
Настоящее изобретение обеспечивает фармакокинетические параметры для глаз различных составов и режимов дозирования соединения-I после местного глазного введения дозы. Три дозируемые концентрации в девяти (9) различных местных офтальмологических составах соединения-I применяют для дозирования или один раз в день (q.d.) или дважды в день (дважды в день) в течение 1, 2, 3, 4 или 5 последовательных дней. Каждый из субъектов получает приблизительно 30 мкл билатеральной местной глазной дозы одной из трех (3) композиций соединения-I или состава со средой, применяя пипетку прямого вытеснения.
Состав каждого состава соединения-I описан в таблице 8A. Все дозы вводили в пределах ±1 часа от запланированного времени введения. В 1 день группы 1, 2, 4-6, 8, 10, 11, 13, 15 и 17 получают одну дозу (q.d.) в течение или одного (1) или четырех (4) дней. В 1-4 дни группы 3, 7, 9, 12, 14 и 16 получают дозу дважды в день с интервалом приблизительно 8 часов в 7:00 AM и 3:00 PM в течение четырех (4) дней. Некоторые субъекты получают дозу дважды в день состава только со средой в течение пяти (5) последовательных дней.
В некоторых вариантах осуществления, отбор глазной пробы проводят через приблизительно 0,5, приблизительно 1, приблизительно 2, приблизительно 3, приблизительно 4, приблизительно 5, приблизительно 6, приблизительно 7, приблизительно 8, приблизительно 9, приблизительно 10, приблизительно 11, приблизительно 12, приблизительно 13, приблизительно 14, приблизительно 15, приблизительно 16, приблизительно 17, приблизительно 18, приблизительно 19, приблизительно 20, приблизительно 21, приблизительно 22, приблизительно 23 или приблизительно 24 часов после введения дозы относительно введения дозы в 1 день. Образцы водянистой влаги, роговицы, центральной и периферической сетчатки и центральной и периферической сосудистой оболочки глаза собирали для отслеживания эффектов обработки. Образцы водянистой влаги, роговицы, центральной зоны сетчатки и центральной сосудистой оболочки анализировали.
В таблице 8A-C перечислены 1- - 5-днвные PK результаты с местным глазным введением соединения-I в растворах на основе циклодекстрина.
В таблице 8B перечислены средние концентрации соединения-I в водянистой влаге, сетчатке, сосудистой оболочке глаза и роговице (LLOQ: нижний предел количественного анализа; LLOQ представляет собой наименьшую концентрацию аналита, которую можно количественно измерить с подходящими точностью и достоверностью).
Центральная зона сетчатки LLOQ = 0,0181 мкМ.
Периферическая сетчатка LLOQ = 0,00873 мкМ (группы 1-8); LLOQ = 0,00898 мкМ (группы 12-16).
Центральная сосудистая оболочка глаза LLOQ = 0,175 мкМ.
Периферическая сосудистая оболочка глаза LLOQ = 0,0349 мкМ (группы 1-8); LLOQ = 0,0359 мкМ (группы 12-16).
LLOQ роговицы = 0,0181 мкМ (группы 1-5); LLOQ = 0,0453 мкМ (группы 6-8, 10-13, 15, 16A17); LLOQ = 0,0873 мкМ (группы 4, 9, 16B).
N/A = не предусмотрено; образцы не анализировали в соответствии с протоколом исследования.
* Среднее на основе n=1.
В таблице 8C приведена сводка средних концентраций в глазных тканях соединения-I в водянистой влаге, центральной и периферической сетчатке, центральной и периферической сосудистой оболочке глаза и роговице для групп 1-10. Любые величины <LLOQ исключали из статистических расчетов. Когда все величины являются <LLOQ для указанного момента времени, <LLOQ приводят в виде среднего.
Центральная зона сетчатки LLOQ = 0,0181 мкМ.
Периферическая сетчатка LLOQ = 0,00873 мкМ (группы 1-8); LLOQ = 0,00898 мкМ (группы 12-16).
Центральная сосудистая оболочка глаза LLOQ = 0,175 мкМ.
Периферическая сосудистая оболочка глаза LLOQ = 0,0349 мкМ (группы 1-8); LLOQ = 0,0359 мкМ (группы 12-16).
LLOQ роговицы = 0,0181 мкМ (группы 1-5); LLOQ = 0,0453 мкМ (группы 6-8, 10-13, 15, 16A17); LLOQ = 0,0873 мкМ (группы 4, 9, 16B).
N/A = не предусмотрено; образцы не анализировали в соответствии с протоколом исследования.
* Среднее на основе n=1.
5-дневные PK результаты с местным глазным введением соединения-I в виде растворов на основе циклодекстрина
Настоящее изобретение относится к фармакокинетическим параметрам для глаз различных режимов дозирования местных офтальмических растворов соединения-I, содержащих гидроксипропил-β-циклодекстрин (“HDβCD”), после глазного введения дозы. Различные местные офтальмические растворы соединения-I вводят или один раз в день (q.d.) или дважды в день (дважды в день) в течение или 4 или 5 последовательных дней. Каждый из субъектов получает 30 мкл билатеральной местной глазной дозы одной из четырех дозируемых концентраций соединения-I.
Все дозы вводили с интервалом ±1 час от запланированного времени дозирования, за исключением некоторых субъектов, которым вводили дозу в 1 день. Отбор образцов тканей глаза после введения соединения-I проводят через один час после первой ежедневной дозы на 5 день для субъектов, за исключением некоторых субъектов, для которых отбор тканей глаза проводят через 24 часов после первой ежедневной дозы на 4 день.
Собирали образцы водянистой влаги, роговицы, центральной и периферической сетчатки и центральной и периферической сосудистой оболочки глаза. Образцы сетчатки, центральной зоны сетчатки и центральной сосудистой оболочки глаза анализировали; образцы водянистой влаги, периферической сетчатки и периферической сосудистой оболочки глаза не анализировали.
В таблице 9 (A-B) приведены 5-дневные PK результаты с местным глазным введением соединения-I в растворах на основе циклодекстрина.
0,7% хлорид натрия
pH 6,5
0,85% хлорид натрия
pH 6,5
0,8% хлорид натрия
pH 6,5
0,6% хлорид натрия
pH 6,5
0,65% хлорид натрия
pH 6,5
0,65% хлорид натрия
pH 6,5
0,15% хлорид натрия
pH 6,5
0,8% хлорид натрия
pH 6,5
0,37% хлорид натрия
pH 6,5
В таблице 9B приведена сводка средних концентраций соединения-I в тканях глаза в центральной зоне сетчатки, центральной сосудистой оболочке глаза и роговице. Все величины <LLOQ исключали из статистических расчетов. Когда все величины были <LLOQ для указанного момента времени, <LLOQ приводили в виде среднего.
Центральная сосудистая оболочка глаза LLOQ = 0,174 мкМ.
LLOQ роговицы = 0,0174 мкМ.
* Среднее на основе n=1.
Концентрации соединения-I (в мкМ) в различных жидкостях и тканях глаза
В некоторых вариантах осуществления, концентрацию активного агента в различных тканях и жидкостях глаза измеряют после местного глазного введения раствора приблизительно 0,4% (приблизительно 4 мг/мл) соединения-I и циклодекстрина. Среднюю концентрацию соединения-I измеряют в центральной сосудистой оболочке глаза, центральной зоне сетчатки, водянистой влаге и роговице. Соединение-I находится в растворе (0,4% или 4 мг/мл) с 8,41% KLEPTOSE® и 0,142% фосфатного буфера; 8,9% KLEPTOSE® HPB и 0,142% фосфат; 4,88% CAPTISOL® и 0,142% фосфата; или 4,88% CAPTISOL® и 0,122% фосфата. См. таблицу 10A-B.
В некоторых вариантах осуществления, после местного глазного введения раствора приблизительно 0,4% (приблизительно 4 мг/мл) соединения-I и циклодекстрина, концентрация соединения-I в центральной сосудистой оболочке глаза составляет от приблизительно 0,2 мкМ до приблизительно 0,8 мкМ. Концентрация соединения-I в центральной зоне сетчатки составляет от приблизительно 0,05 мкМ до приблизительно 0,15 мкМ. В некоторых вариантах осуществления, после местного глазного введения раствора приблизительно 0,4% (приблизительно 4 мг/мл) соединения-I и циклодекстрина, концентрация соединения-I в водянистой влаге составляет приблизительно 0,003 мкМ - приблизительно 0,008 мкМ, и концентрация в роговице соединения-I составляет приблизительно 6,0 мкМ - приблизительно 40 мкМ. KLEPTOSE® HPB или CAPTISOL® применяют в растворе соединения-I, вводимого местно в глаз.
В некоторых вариантах осуществления, средние концентрации соединения-I в тканях глаза после местного дозирования дважды в день офтальмических композиций 0,3% соединения-I в виде гелевых капель с и без хлорида бензилалкония являются наибольшими в роговице с приблизительно 200 мкМ - приблизительно 350 мкМ в роговице, приблизительно 2,0 мкМ - приблизительно 5,0 в периферической сосудистой оболочке глаза, приблизительно 0,2 мкМ - приблизительно 0,7 мкМ в центральной сосудистой оболочка глаза, приблизительно 0,05 мкМ - приблизительно 0,5 мкМ в периферической сетчатке и приблизительно 0,01 мкМ - приблизительно 0,05 мкМ в водянистой влаге.
В некоторых вариантах осуществления, суспензия на основе Tris состава соединения-I является хорошо переносимой, без любых изменений в сетчатке и только с несколькими единичными случаями легкого конъюнктивита. В некоторых вариантах осуществления, средние концентрации соединения-I в тканях глаза, оцененные через 1 час ±15 минут после первой ежедневной местной глазной дозы в 30 день при местном дозировании дважды в день 0,3% суспензии соединение-I на основе Tris с и без хлорида бензилалкония, являются наибольшими в роговице, например, приблизительно 2,00 мкМ - приблизительно 4,0 мкМ. Концентрация в периферической сосудистой оболочке глаза при той же дозе составляет приблизительно 0,7 мкМ - приблизительно 1,5 мкМ; концентрация в центральной сосудистой оболочке глаза составляет приблизительно 0,3 мкМ - приблизительно 0,4 мкМ; концентрация в периферической сетчатке составляет приблизительно 0,08 мкМ - приблизительно 0,09 мкМ; концентрация в центральной зоне сетчатки составляет приблизительно 0,04 мкМ - приблизительно 0,07 мкМ; и концентрация в водянистой влаге составляет приблизительно 0,001 мкМ - приблизительно 0,002 мкМ.
Настоящее изобретение относится к растворам на основе циклодекстрина (например, растворам, содержащим гидроксипропил-бета-циклодекстрин (HP-β-CD, KLEPTOSE® HPB)) соединения-I, которые были хорошо переносимыми при введении местно в течение вплоть до 30 дней, дважды в день при приблизительно 0,1% соединения-I (в растворе с приблизительно 2,0% - приблизительно 2,5% HP-β-CD), дважды в день при приблизительно 0,2% соединения-I (в растворе с приблизительно 4,0% - приблизительно 4,5% HP-β-CD), один раз или дважды в день при приблизительно 0,4% соединения-I (в растворе с приблизительно 8,0% - приблизительно 8,5% HP-β-CD), и один раз или дважды в день при приблизительно 0,6% соединения-I (в растворе с вплоть до приблизительно 14% HP-β-CD) субъектам. Более того, в дополнительных вариантах осуществления, растворы на основе циклодекстрина приблизительно 0,4% масс./об. соединения-I в Kleptose® HPB, Kleptose® HP или Captisol® были хорошо переносимыми при дозировании дважды в день в течение вплоть до 24 дней.
Настоящее изобретение обеспечивают дозолимитирующую токсичность для роговицы, наблюдаемую с офтальмическими композициями соединения-I в виде гелевых капель. В некоторых вариантах осуществления, офтальмические гелевые капли обеспечивают от приблизительно в пять раз до приблизительно в пятнадцать раз большие концентрации в роговице соединения-I по сравнению с раствором на основе циклодекстрина, и от приблизительно в пятнадцать раз до приблизительно в сто раз большие концентрации в роговице соединения-I по сравнению с суспензиями на основе Tris. Суспензии на основе Tris и растворы на основе циклодекстрина соединения I настоящего изобретения хорошо переносятся без проявления явной офтальмической токсичности. В некоторых вариантах осуществления, введение один раз или дважды в день в течение, по меньшей мере, 30 дней приблизительно 0,005% - приблизительно 5,0% масс./об. раствора на основе циклодекстрина или суспензии на основе Tris соединения-I хорошо переносится субъектами. Настоящее изобретение обеспечивает наибольшие концентрации соединения-I в центральной сосудистой оболочке глаза, применяя растворы на основе циклодекстрина по сравнению с эквимолярными дозами гелей и/или композиций на основе Tris.
Сосудистая оболочка глаза LLOQ = 0,184 мкМ.
Сетчатка LLOQ = 0,0229 мкМ.
AH LLOQ = 0,000918 мкМ.
Роговица LLOQ = 0,0918 мкМ.
** Гидроксипропил-β-циклодекстрин (HPβCD) от Roquette.
*** CAPTISOL® представляет собой полианионное β-циклодекстриновое производное с сульфонатом натрия, отделенным от липофильной полости бутилэфирной спейсерной группой, или сульфобутиловым эфиром (SBE).
Таблица 11 показывает концентрации в роговице и центральной сосудистой оболочке глаза композиций соединения-I.
масс./об.%
29 дней
30 дней
(Captisol®)
24 дня
(Kleptose® HP)
24 дня
(Kleptose® HPB)
24 дня
Протокол фазы I для исследования с увеличением дозы на пациентах с неоваскулярной ВМД
Настоящее изобретение относится к исследованию фазы I, включающему двенадцатинедельное многоцентровое исследование с повышением дозы и без контроля плацебо, оценивающему безопасность, переносимость и фармакокинетические свойства после местного глазного введения соединения-I пациентам с неоваскулярной возрастной дегенерацией макулы (ВМД). В сумме, вплоть до 60 пациентов обрабатывали один или два раза ежедневно местным глазным дозированием офтальмического раствора соединения-I в течение трех месяцев, где планировали три варианта монотерапии с повышением дозы и одну вспомогательную терапию, применяя единичное интравитреальное введение Lucentis® плюс максимально переносимая при монотерапии доза (15 пациентов на один вариант обработки). Пациентов, которые удовлетворяют предусмотренному критерию зрительного восприятия и критерию поражения ХНВ, подтвержденным центром по независимой интерпретации данных, обеспечивали одновременно непрерывным местным глазным дозированием и стандартным лечением.
Настоящее изобретение обеспечивает 3 концентрации доз в диапазоне 0,1%-1,0% (масс./об.) (в виде соединения-I) офтальмического раствора для клинических исследований. Концентрации составляют приблизительно 0,1%, приблизительно 0,3%, приблизительно 0,6%, и приблизительно 1,0% (масс./об.) соединения-I HCl.
Получение составов
Неограничивающие примеры составов настоящего изобретения показаны в таблице 12.
1:4, 1:8, 1:10, 1:12
1:6, 1:8, 1:10
1:6, 1:10
1:2, 1:3, 1:4, 1:6, 1:8, 1:10
HPβCD
1:8
0,1, 0,4%
5,5, 6,5
Фосфат, Tris
b KLEPTOSE® HPB.
c CAPTISOL®.
Дозы обработки
Состав настоящего изобретения является эффективным в лечении или предотвращении (т.е. регрессии) неоваскуляризации (НВ) сосудистой оболочки глаза и сетчатки глаза субъекта, являющегося млекопитающим. Соединение-I настоящего изобретения при указанной дозе ингибирует рецепторную тирозинкиназу. В некоторых вариантах осуществления, композиция соединения-I при указанной дозе ингибирует рецепторную тирозинкиназу, включая, VEGFR, FGFR, Tie2 и EphB-4. Ингибирование нескольких RTK композицией настоящего изобретения при указанной дозе является одновременным и обладает синергетическим эффектом, и является эффективным в лечении или регрессии НВ в заднем сегменте глаза.
В следующем варианте, состав настоящего изобретения является эффективным в лечении НВ при введении один, два, три и четыре раза в день местной офтальмической доставкой приблизительно 0,005% - приблизительно 5,0% (приблизительно 0,05 - приблизительно 50 мг/мл) соединения-I. Состав соединения-I или его свободного основания (формула II) для лечения или регрессии НВ, представляет собой раствор, содержащий циклодекстрин, или суспензию, содержащую Tris. Раствор или суспензия при доставке субъекту, подвергаемому воздействию атмосферного кислорода, вызывая ретинопатию, вызванную кислородом (OIR) или НВ, например, способно эффективно уменьшать среднюю площадь преретинальной НВ на сетчатке. Предотвращение или лечение НВ композицией и/или суспензией соединения-I осуществляют ингибированием нескольких рецепторных тирозинкиназ (RTK), включая VEGFR-2.
Любое из описанных заболеваний или состояний, описанных в настоящем изобретении, можно лечить или предотвращать получением концентрации в целевой ткани приблизительно 200 нМ - приблизительно 2 мкМ описанных соединений или их фармацевтически приемлемых солей, составов и/или суспензии. Один вариант осуществления настоящего изобретения относится к способу лечения патологического ангиогенеза в заднем сегменте глаза, получением концентрации в целевой ткани приблизительно 200 нМ - приблизительно 2 мкМ описанных соединений или их фармацевтически приемлемых солей и/или состава. Другая версия данного варианта осуществления обеспечивает получение концентрации в целевой ткани приблизительно 300 нМ - приблизительно 2 мкМ одного или более из описанных соединений или их фармацевтически приемлемых солей и/или состава.
В одном варианте осуществления настоящего изобретения приблизительно 0,2 - приблизительно 1,0% (приблизительно 2 - приблизительно 10 мг/мл) соединения-I, сформулированного в виде раствора или суспензии, после введения могут эффективно ингибировать функционирование VEGFR-2 киназы и обеспечивают существенную блокировку ряда рецепторов проангиогенных факторов роста, включая FGFR 1-3, Tie-2 и EphB-4. Концентрация 2-10 мг/мл соединения-I в состава обеспечивает фармакологически эффективные концентрации лекарственного средства в центральной сосудистой оболочке глаза и сетчатке после 1-5 дней местного офтальмического введения.
В некоторых вариантах осуществления, продолжительность воздействия соединения-I составляет 1 - 90 дней. В некоторых вариантах осуществления, режим дозирования включает несколько курсов местного глазного введения состава, содержащей соединение-I, субъекту в течение от 1 до 90 дней. Например, режим дозирования включает введение состава один раз в день, дважды в день, три раза в день или четыре раза в день в течение от 1 до 90 дней. Например, режим дозирования включает введение состава один, два, три или четыре раза через день (т.е. в 1, 3, 5, 7 день и т.д.) в течение вплоть до 90 дней. Например, режим дозирования включает введение один раз в 1 день, один или два раза во 2 - 90 дни. Например, режим дозирования включает введение один, два, три или четыре раза в 1 день, с последующим введением один раз в день в течение 2-90 дней. Например, режим дозирования включает введение один, два, три, четыре раза в 1 день, с последующим введением один, два, три или четыре раза через день (т.е. в 1, 3, 5, 7 день и т.д.) в течение вплоть до 90 дней. Например, один режим дозирования включает введение один раз в день или дважды в день в течение 1, 2, 3, 4 или 5 последовательных дней. Для режима дозирования два или три раза в день ежедневно, субъекты получают местную глазную дозу состава соединения-I в 1 и 4 день с приблизительно 4, 6, или 8 часовым интервалом. В другом варианте осуществления, субъекты получают местные глазные дозы состава соединения-I приблизительно через 4, 6 или 8 часовые интервалы в течение четырех последовательных дней. В некоторых вариантах осуществления субъекты получают одну или две дозы местной глазной дозы состава соединения-I в день в течение 5 последовательных дней. В еще других вариантах осуществления субъекты получают одну или две дозы местной глазной дозы состава соединения-I в течение 5-90 последовательных дней. В некоторых вариантах осуществления субъекты получают одну или две дозы местной глазной дозы состава соединения-I в течение, по меньшей мере, 25 последовательных дней. В одном варианте осуществления, субъекты получают одну или две местные глазные дозы в течение, по меньшей мере, 90 последовательных дней или более.
В некоторых вариантах осуществления, настоящее изобретение относится к составам соединения-I или его свободного основания, вводимому местно в передний сегмент глаза субъекта для лечения ВМД, патологической ХНВ и/или патологической НВ. Например, композицию вводят в глаз субъекта 1, 2, 3 или 4 раза ежедневно. В конкретных вариантах осуществления, композицию вводят в глаз субъекта 2 или 3 раза ежедневно. Например, композицию вводят в глаз или оба глаза субъекта. Например, приблизительно 1 мг/мл состава, содержащей активный агент настоящего изобретения, вводят два раза в день (дважды в день) в глаз или оба глаза субъекта. В некоторых вариантах осуществления приблизительно 2 мг/мл дважды в день, приблизительно 3 мг/мл один раз в день (QD) или дважды в день, приблизительно 4 мг/мл ежедневно или дважды в день, приблизительно 5 мг/мл ежедневно или дважды в день или приблизительно 6 мг/мл ежедневно или дважды в день вводят в глаз или оба глаза субъекта.
Например, композицию, содержащую приблизительно 2 мг/мл соединения-I дважды в день, вводят в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления композицию, содержащую приблизительно 3 мг/мл соединения-I дважды в день, вводят в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления композицию, содержащую приблизительно 3 мг/мл соединения-I ежедневно, вводят в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления композицию, содержащую приблизительно 4 мг/мл соединения-I дважды в день, вводят в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления композицию, содержащую приблизительно 4 мг/мл соединения-I ежедневно, вводят в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления композицию, содержащую приблизительно 5 мг/мл соединения-I дважды в день, вводят в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления композицию, содержащую приблизительно 5 мг/мл соединения-I ежедневно, вводят в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления композицию, содержащую приблизительно 6 мг/мл соединения-I дважды в день, вводят в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления композицию, содержащую приблизительно 6 мг/мл соединения-I ежедневно, вводят в глаз или оба глаза субъекта в течение от 1 до 90 дней. Режим дозирования в течение от 1 до 90 дней может представлять собой любой из режимов, включающий последовательные или чередующиеся дни введения, описанные в параграфе выше.
Настоящее изобретение относится к композициям, как показано в таблице 13, для введения в глаз или оба глаза субъекта.
день
В некоторых вариантах осуществления композицию формулы (II) или соединения-I вводят в глаз или оба глаза субъекта. Например, приблизительно 0,2% - приблизительно 0,6% (масс./об.) соединения формулы (II) или приблизительно 0,1%-0,7% (масс./об.) состава, содержащей соединение I настоящего изобретения, вводят один раз в день (QD) или два раза в день (дважды в день) в глаз или оба глаза субъекта в течение от 1 до 90 дней. В некоторых вариантах осуществления, соединение формула (II) или соединение-I образует комплекс с комплексообразующим агентом, например, циклодекстрином (например, Kleptose® HPB (%)), в соотношении приблизительно 1:8, где приблизительно 2%-13% (масс./об.) циклодекстрина (например, Kleptose® HPB (%)) добавляют к состава. Композиция дополнительно содержит приблизительно 0,1% - приблизительно 0,2% буфера, например, 10 мМ фосфатного буфера. Требуемая осмолярность состава составляет приблизительно 200 - приблизительно 300 мОсм, получаемая добавлением количества, достаточного для достижения осмолярности, соли, например, хлорида натрия. pH состава составляет приблизительно 6,0 при ниже или приблизительно 40°C. Режим дозирования в течение от 1 до 90 дней может представлять собой любой из режимов, включающий последовательные или чередующиеся дни введения, описанных в параграфе выше.
Способы настоящего изобретения комбинируют со стандартным лечением, включающим, но не ограничивающимся, лазерную терапию и терапию инъецируемыми противонеоваскулярными агентами.
Показания и способы лечения
Описанными являются способы лечения заболеваний или состояний глаза. Описанные способы относятся к лечению, предотвращению или контролированию глазной неоваскуляризации (NV) или лечению заболевания или состояния, которое связано с возникновением НВ, введением субъекту одного или более из описанных соединений и их композиций.
Один аспект описанного способа относится к лечению или предотвращению НВ введением субъекту эффективного количества одного или более из описанных соединений или их фармацевтически приемлемых солей и/или композиций. Один вариант осуществления данного аспекта относится к способу лечения НВ введением субъекту состава: a) эффективного количества одного или более из описанных соединений или их фармацевтически приемлемых солей и/или композиций, и необязательно b) одного или более носителей или совместимых вспомогательных веществ.
Описанные способы относятся к предотвращению или контролированию патологической глазной неоваскуляризации (NV) или лечению заболевания или состояния, которое связано с возникновением НВ, введением субъекту одного или более из описанных соединений, и их композиций.
Настоящие варианты осуществления относятся к применению состава соединения-I или его свободного основания (формула II) в получении лекарственного препарата для лечения субъекта с васкулопатическими или воспалительными заболеваниями заднего сегмента глаза. Они включают, например, диабетическую ретинопатию (включая фоновую диабетическую ретинопатию, пролиферативную диабетическую ретинопатию и диабетический отек макулы); возрастную дегенерация макулы (ВМД) (включая неоваскулярную (влажную/экссудативную) ВМД, сухую ВМД и географическую атрофию); патологическую хороидальную неоваскуляризацию (ХНВ) любого механизма (т.е. миопию высокой степени, травму, серповидноклеточную анемию; гистоплазмоз глаз, ангиоидные полосы сетчатки, разрыв сосудистого тракта глаза при травме, друзы оптического нерва, и некоторые типы ретинальной дистрофии); патологическую неоваскуляризацию сетчатки любого механизма (т.е. серповидно-клеточную ретинопатию, болезнь Илза, глазной ишемический синдром, каротидно-кавернозное соустье, семейную экссудативную форму витреоретинопатии, синдром повышенной вязкости крови, идиопатический окклюзионный артериолит; дробьевидную ретинохороидопатию, ретинальный васкулит, саркоидоз или токсоплазмоз); увеит; окклюзию вены сетчатки (центральной или ветвей); травму глаза; эдему, вызванную хирургической операцией; неоваскуляризацию, вызванную хирургической операцией; кистозный макулярный отек; ишемию глаз; ретинопатию недоношенных; синдром Коутса; серповидно-клеточную ретинопатию и/или неоваскулярную глаукому.
В одном аспект настоящего изобретения композицию применяют в лечении возрастной дегенерации макулы (ВМД) (включая неоваскулярную (влажную/экссудативную) ВМД, сухую ВМД и географическую атрофию). Растворы или суспензии применяют в лечении неоваскулярной (экссудативной или влажной) ВМД. В другом варианте осуществления растворы или суспензии применяют для лечения сухой ВМД. В еще другом варианте осуществления растворы или суспензии применяют для лечения географической атрофии.
Композиция настоящего изобретения предотвращает, задерживает или лечит возникновение патологической хороидальной неоваскуляризации (ХНВ) любого механизма (т.е. миопию высокой степени, травму, серповидноклеточную анемию; гистоплазмоз глаз, ангиоидные полосы сетчатки, разрыв сосудистого тракта глаза при травме, друзы оптического нерва и некоторые типы ретинальной дистрофии) у субъектов.
Композиция настоящего изобретения задерживают возникновение, препятствуют развитию или лечат области с патологической хороидальной неоваскуляризацией (ХНВ) ниже нейросенсорной сетчатки. Композиция настоящего изобретения является эффективной в лечении ХНВ.
Один аспект данного способа относится к лечению или предотвращению глазной неоваскуляризации введением субъекту эффективного количества одного или более из описанных соединений или их фармацевтически приемлемых солей. Один вариант осуществления данного аспекта относится к способу лечения эдемы глаз и неоваскуляризации введением субъекту состава: a) эффективного количества одного или более из описанных соединений или их фармацевтически приемлемых солей и/или композиций, и необязательно b) одного или более носителей или совместимых вспомогательных веществ.
Описанные способы также относятся к предотвращению или контролированию эдемы глаза или лечению заболевания или состояния, которое связанно с возникновением эдемы глаза введением субъекту одного или более из описанных соединений.
Один аспект данного способа относится к лечению или предотвращению эдемы глаза введением субъекту эффективного количества одного или более из описанных соединений или их фармацевтически приемлемых солей. Один вариант осуществления данного аспекта относится к способу лечения эдемы глаза введением субъекту состава: a) эффективного количества одного или более из описанных соединений или их фармацевтически приемлемых солей и/или композиций, и необязательно b) одного или более носителей или совместимых вспомогательных веществ.
Другой описанный способ относится к предотвращению или контролированию эдемы сетчатки или неоваскуляризации сетчатки или лечению заболевания или состояния, которое связано с возникновением эдемы сетчатки или неоваскуляризации сетчатки, введением субъекту одного или более из описанных соединений. Один аспект данного способа относится к лечению или предотвращению эдема сетчатки или неоваскуляризации сетчатки введением субъекту эффективного количества одного или более из описанных соединений или их фармацевтически приемлемых солей. Один вариант осуществления данного аспекта относится к способу лечения эдемы сетчатки или неоваскуляризации сетчатки введением субъекту состава: a) эффективного количества одного или более из описанных соединений или их фармацевтически приемлемых солей и/или композиций, и необязательно b) одного или более носителей или совместимых вспомогательных веществ.
Другой вариант осуществления данного аспекта относится к способу замедления или предотвращения развития непролиферативной ретинопатии до пролиферативной ретинопатии введением субъекту состава a) эффективного количества одного или более из описанных соединений или их фармацевтически приемлемых солей и/или композиций, и необязательно b) одного или более носителей или совместимых вспомогательных веществ.
Один аспект описанных способов относится к заболеваниям, которые являются прямым или косвенным результатом диабета, в том числе, диабетический отек макулы и диабетическая ретинопатия. Сосудистая система глаза диабетика становится нестабильной с течением времени, приводя в результате к заболеваниям, таким как непролиферативная ретинопатия, макулярный отек и пролиферативная ретинопатия. По мере того как жидкость просачивается в центр макулы, часть глаза, за счет которой осуществляется острое прямое зрение, протекает накопление жидкости, и ассоциированный белок начинает откладываться на или под макулой. Это приводит в результате к разбуханию, которое вызывает постепенное нарушение центрального видения субъекта. Данное заболевание называют “макулярным отеком”. Другое заболевание, которое может возникать, представляет собой непролиферативную ретинопатию, при которой можно наблюдать изменения сосудов, такие как микроаневризмы, снаружи макулярной области глаза. При пролиферативной ДР, патологические новые кровеносные сосуды вырастают и распространяются из сетчатка в стекловидное тело, где данные атипичные сосуды могут изменять морфологию сетчатки в макуле, и/или вызывать кровоизлияние в стекловидное тело и закрывать зрительную линию.
Следующий описанный способ относится к лечению, предотвращению или контролированию диабетической ретинопатии или лечению заболевания или состояния, которое связано с возникновением диабетической ретинопатии, введением субъекту одного или более из описанных соединений.
Один аспект описанного способа относится к лечению или предотвращению диабетической ретинопатии введением субъекту эффективного количества одного или более из описанных соединений или их фармацевтически приемлемых солей. Один вариант осуществления данного аспекта относится к способу лечения диабетической ретинопатии введением субъекту состава a) эффективного количества одного или более из описанных соединений или их фармацевтически приемлемых солей, и/или композиций, и необязательно b) одного или более носителей или совместимых вспомогательных веществ.
Диабетическая пролиферативная ретинопатия характеризуется неоваскуляризацией. Новые кровеносные сосуды являются хрупкими и подвержены кровотечению. Результатом является рубцевание сетчатки, а также окклюзия или полная блокировка светового пути через глаз из-за атипичного образования новых кровеносных сосудов. Обычно, субъекты, имеющие диабетический отек макулы, страдают от непролиферативной стадии диабетической ретинопатии; однако, нередки случаи, когда макулярный отек у субъектов начинает проявляться только на пролиферативной стадии.
Еще следующий описанный способ относится к предотвращению или контролированию диабетического отека макулы или лечению заболевания или состояния, которое связано с возникновением диабетического отека макулы, введением субъекту одного или более из описанных соединений.
Один аспект данного способа относится к лечению или предотвращению диабетического отека макулы введением субъекту эффективного количества одного или более из описанных соединений или их фармацевтически приемлемых солей или композиций. Один вариант осуществления данного аспекта относится к способу лечения диабетического отека макулы введением субъекту состава a) эффективного количества одного или более из описанных соединений или их фармацевтически приемлемых солей, и/или композиций, и b) одного или более носителей или совместимых вспомогательных веществ.
Наборы
Также описанными являются наборы описанных соединений и композиций для доставки лекарственного средства человеку, млекопитающему или в клетку. Наборы могут содержать одну или более упакованных одноразовых доз состава, содержащей одно или более из соединений, которые будут доставлять человеку, млекопитающему или в клетку. Ампулы с единичными дозами или многодозовые контейнеры, в которые соединения, которые будут доставлять, упаковывают перед применением, могут включать герметично закрытый контейнер, содержащий количество активного агента или его фармацевтически приемлемой соли или состава, подходящее для его фармацевтически эффективной дозы или множества эффективных доз. Соединения можно упаковывать в виде стерильной состава, и герметично закрытый контейнер разрабатывают так, чтобы он сохранял стерильность состава до применения.
Набор настоящего изобретения содержит одноразовую бутыль-дозатор глазных капель для доставки офтальмической состава. В альтернативном варианте осуществления, набор настоящего изобретения содержит многоразовую бутыль-дозатор глазных капель. Многоразовая бутыль-дозатор дозатор содержит подходящее количество антиинфекционного агента и/или консерванта, например, без ограничения, 0,005% BAK. Офтальмический дозатор настоящего изобретения содержит насадку и колпачок. Контейнер настоящего изобретения содержит полупрозрачную LDPE офтальмическую бутыль-дозатор с LDPE капельным дозатором и HDPE колпачком. Контейнер может быть другого типа и может быть получен при необходимости и/или как применяют в данной области техники.
Следующие примеры являются иллюстративными, но не ограничивающими способы и состава настоящего изобретения. Другие подходящие модификации и варианты ряда условий и параметров, обычно встречающиеся в терапии и которые являются очевидными специалисту в данной области техники, включены в сущность и объем вариантов осуществления.
ПРИМЕРЫ
Соединение-I представляет собой эффективный и селективный низкомолекулярный ингибитор VEGFR-2, вместе с другими проангиогенными RTK, такими как FGF рецепторы (FGFR-1-3), Tie-2 и эфриновый рецептор B4 (EPHB-4). Было показано, что соединение-I ингибирует фосфорилирование специфических RTK, пролиферацию эндотелиальных клеток и патологический ангиогенез после системного введения в моделях роговицы мыши и пластинки роста крысы, а также рост человеческих опухолевых ксенотрансплантатов у бестимусных мышей. Что касается потенциальных офтальмических показаний, примеры настоящего изобретения, описанные ниже, показывали, что местная офтальмическая доставка соединения-I обеспечивала значительное ингибирование патологической неоваскуляризации сетчатки и хороидальной неоваскуляризации в клинически значимых моделях грызунов. Сводка данных представлена ниже.
Следующие исследования проводили для измерения эффекта описанных соединений на пропотевание жидкости из сосудов и неоваскуляризацию ткани сетчатки.
ПРИМЕР 1
Первичные фармакодинамические параметры
In Vitro фармакология эффективности соединения-I. Соединение-I эффективно ингибирует активность тирозинкиназы рецептора 2 сосудистого эндотелиального фактора роста (VEGF-2), а также отобранный подкласс других проангиогенных RTK, в различных in vitro анализах. Конкретно, соединение-I блокирует VEGF-стимулируемое фосфорилирование VEGFR-2 в цельных клетках вместе с пролиферацией выращенных эндотелиальных клеток. Соединение-I ингибировало рекомбинантную активность тирозинкиназы VEGFR-2 и FGFR-2 с 50% ингибирующей концентрацией (IC50) 10,55 нМ (6 нг/мл) и 8,79 нМ (5 нг/мл), соответственно; и ингибировало аутофосфорилирование VEGFR-2 в интактных клетках с IC50=5,27 нМ (3 нг/мл). Данное ингибирование было селективным по сравнению со многими другими тирозинкиназами, например, IC50 VEGFR-2 была приблизительно в 500x и 1000x меньшей, чем IC50 для тирозинкиназ рецептора эпидермального фактора роста (EGFR) и инсулинового рецептора (IR), соответственно (см. таблицу 2).
При применении кривой титрования с 10 точками в диапазоне 257 - 5000 нМ (146 - 2845 нг/мл), соединение-I показывало эффективное ингибирование активности тирозинкиназ для нескольких рецепторов проангиогенных факторов роста, о чем свидетельствует IC50<100 нМ (56,89 нг/мл) (см. таблицу 3). IC50 для данной отобранной группы киназ были следующими: рекомбинантная KDR (человеческая изоформа VEGFR-2) = 1,27 нМ (0,72 нг/мл), Tie-2 = 10,10 нМ (5,75 нг/мл), и FGFR 1-3 = 8,50 нМ (4,84 нг/мл), 3,08 нМ (1,75 нг/мл) и 33,9 нМ (19,29 нг/мл), соответственно. Соединение также блокирует другой высокоаффинный VEGF рецептор, VEGFR-1/Flt-1, но с меньшей эффективностью: IC50=122 нМ (69,41 нг/мл).
Хотя ингибирование VEGFR, по-видимому, является существенным для снижения проницаемости сосудов и предотвращения последующего роста неоваскуляризации, одновременное ингибирование передачи сигнала VEGF с ингибированием других сигнальных путей с участием факторов роста (например, PDGF и ангиопоэтины/Tie2) может быть связано с уникальными терапевтическими эффектами. Терапевтические эффекты более широкого ингибирования сигнальных путей могут способствовать регрессии вновь образовавшихся патологических сосудов в заднем сегменте глаза.
300 нМ (170,67 нг/мл) соединения-I полностью ингибировало VEGFR-2 киназную функцию (см. таблицу 4) и обеспечивало значительную блокировку аналогичного набора рецепторов проангиогенных факторов роста, включая FGFR-1-3, Tie-2 и EphB-4. Неожиданным результатом было то, что концентрация 300 нМ была способна полностью ингибировать VEGFR-2 киназную функцию. Данная концентрация попадает в стандартный диапазон, обнаруживаемый в центральной сосудистой оболочке глаза и сетчатке после пяти дней местного офтальмического введения кроликам и собакам.
Обзор лекарственного вещества и лекарственного продукта
Лекарственное вещество: активный фармацевтический ингредиент (API), гидрохлорид формулы II (соединение-I, CP-547632-01), представляет собой небольшую молекулу одного полиморфа. API вещество систематически получают с чистотой, превышающей 99,7%. Любая примесь в лекарственном веществе ≥ 0,15% надлежаще квалифицируют в токсикологических исследованиях, и современная спецификация для новых неизвестных отдельных примесей установлена на не более чем 0,2%. Конечное лекарственное вещество и лекарственный продукт анализируют, применяя стандартные способы.
Лекарственный продукт: офтальмические состава соединения-I для клинических исследований получали при концентрациях доз 0,05%-1,0% (в виде соединения-I). Концентрации, применяемые для GLP серий, составляют 0% (плацебо), 0,05%, 0,1%, 0,2%, 0,3%, 0,4%, 0,6%, 0,8% и 1,0% соединения-I. Офтальмические состава соединения-I (растворы или суспензии) применяют для ежедневного одноразового местного введения в глаз в клиническом исследовании. В добавление к активному ингредиенту, лекарственный продукт содержит 0,005% BAK в качестве консерванта, очищенную воду в качестве среды, и pH доводят гидроксидом натрия до pH 6,0.
ПРИМЕР 2
Гелевые капли на основе фосфата натрия
Офтальмические полезные эффекты соединения-I в состава на основе фосфата натрия (перечисленные в таблице 6) являются результатом свойств самопроизвольно образовывать гели API в буферах, таких как фосфат натрия. Спонтанное образование самообразующегося тиксотропного геля соединения-I из прозрачного раствора осуществляют повышением концентрации API в фосфате натрия. Данный гель первоначально выглядит прозрачным, и затем показывает повышенную густоту/вязкость при больших концентрациях API, а также становится все более непрозрачным, т.е. мутным. Когда концентрация API в фосфатном буфере достигает пересыщенного состояния, в геле также наблюдают нерастворимые частицы соединения-I.
Нанесение геля с повышенной вязкостью на поверхность глаза увеличивает время удерживания в роговице. Увеличенное время удерживания в роговице, в свою очередь, облегчает поглощение глазом лекарственного средства. Как результат, внутриглазные концентрации лекарственного средства вязких гелей увеличиваются по сравнению с невязкими композициями, такими как водные растворы. Один способ увеличить вязкость заключается в применении различных вспомогательных веществ, увеличивающих вязкость, например, карбоксиметилцеллюлозы, которая по существу достигает повышенного внутриглазного поглощения различных лекарственных веществ после местного глазного введения. Однако в данном исследовании тиксотропный гель соединения-I неожиданно образуется в отсутствии любых вспомогательных веществ, увеличивающих вязкость. Например, когда соединение-I растворяли в простом буфере, таком как фосфат натрия, образовывался тиксотропный гель. Тиксотропный гель, который образовывался без любых вспомогательных веществ, увеличивающих вязкость, формулировали в виде гелевых капель в данном примере.
Гелевые капли соединения-I наносили на глаза голландских кроликов. Гелевые капли соединения-I вводили голландским кроликам в течение 4 или 5 последовательных дней при дозировании три раза в день. Концентрации соединения-I в целевых тканях измеряли через 1 час после последней введенной дозы. Доставка соединения-I в ткани заднего сегмента глаза зависела от дозы и частоты введения дозы.
Состава в виде гелевых капель (перечисленные в таблице 6) отличаются по нескольким аспектам, таким как концентрация API, концентрация фосфата натрия, присутствие или отсутствие агентов, регулирующих тоничность, (глицерина) или консервантов (хлорид бензалкония/BAK), поверхностно-активных веществ, увеличивающих растворимость (полисорбат 80, тилоксапол, и/или полоксамер), и pH.
ПРИМЕР 3
Суспензия на основе трометамина
Соединение-I (от приблизительно 1 мг/мл до приблизительно 10 мг/мл) в состава на основе трометамина образовывало суспензию. Суспензия соединения-I в состава на основе трометамина содержала >95% активного лекарственного вещества в нерастворимой форме. Данная характеристика является отличимой от растворимого или полурастворимого состояния соединения-I в гелевых каплях (гелевые капли (гель) не является полностью растворимым состоянием, по мере увеличения концентрации активного агента) или в состава на основе циклодекстрина. Состава на основе трометамина соединения-I показывали повышенную мутность при повышении концентрации активного агента. Ожидали, что введение местно капли суспензии соединения-I в глаз (которая представляет собой комбинацию компонентов растворимого и нерастворимого активного агента) будет обеспечивать уникальные преимущества относительно и безопасности/переносимости и эффективности.
Суспензию на основе трометамина соединения-I вводили голландским кроликам в течение 4 или 5 последовательных дней при дозировании три раза в день. Концентрации соединения-I в тканях глаза и плазме измеряли через 1 час после последней введенной дозы. Соединение-I в суспензии на основе трометамина доставляло концентрации в целевые ткани 10-1000x их клеточной IC50 для различных проангиогенных RTK. См. таблицу 7.
Безопасность и переносимость для роговицы местного соединения-I представляли собой прямое следствие количества растворенного (в противоположность нерастворенному) активного агента, нанесенного на поверхность роговицы, и полученной в результате концентрации в ткани роговицы. Животные, которые получали местное глазное введение суспензии на основе трометамина, были способны переносить большую концентрацию активного агента в состава по сравнению с эквимолярными композициями гелевых капель на основе фосфата натрия. Результаты, полученные на голландских кроликах и собаках породы бигль, показывают, что офтальмические побочные эффекты, такие как дискомфорт и воспаление и в некоторых случаях, истончение роговицы, более регулярно наблюдали, когда концентрация соединения-I в роговице превышала 100 мкМ.
Введение суспензии на основе трометамина обладало неожиданным эффектом на глазную биодоступность соединения-I в заднем сегменте. Наблюдали, что глазная биодоступность соединения-I в заднем сегменте является непосредственно пропорциональной суммарному количеству введенного лекарственного средства (нерастворимого плюс растворимого, см. таблицу 7). Хотя нерастворимые частицы лекарственного средства являются труднодоступными для тканей переднего сегмента; характерные и уникальные физико-химические свойства соединения-I позволяли и нерастворимым и растворимым компонентам проникать в ткани заднего сегмента, такие как сосудистая оболочка глаза и сетчатка. Следовательно, даже большие концентрации лекарственного средства, чем концентрации, получаемые с композициями в виде гелевых капель, содержащими эквивалентные количества активного агента, получали с суспензией на основе трометамина. Таким образом, суспензия на основе трометамина обеспечивала: a) улучшенную переносимость роговицей и b) повышенную биодоступность в заднем сегменте, в частности в сосудистой оболочке глаза, основной целевой ткани для лечения неоваскулярной (влажной) ВМД.
ПРИМЕР 4
Раствор на основе циклодекстрина
Циклодекстрины, которые представляют собой циклические олигосахариды, образованные шестью - восемью остатками декстрозы (α-, β- и γ-CD), соединенными одной - четырьмя связями, являются хорошо известными за их способность действовать в качестве агента, улучшающего растворимость, для относительно нерастворимых лекарственных средств. См. Stella & He, Cyclodextrins, Toxicol. Pathol., 36: 30-42 (2008).
Клинический состав офтальмического раствора соединения-I или его свободного основания в 2-гидроксипропил-β-циклодекстрине (HP-β-CD, KLEPTOSE® HPB) при молярном отношении равном или большем чем 1:6 или сульфобутилэфир-β-циклодекстрине (SBE-β-CD, CAPTISOL®) при соотношении равном или большем чем 1:2 обеспечивал растворимость при клинических концентрациях доз 0,1-1,0% соединения-I.
Растворы на основе циклодекстрина соединения-I или его свободного основания не только увеличивают растворимость активного агента в однородном растворе, но, после местного глазного введения, также обладают новой и ранее ненаблюдаемой характеристикой значительно улучшенного терапевтического индекса активного агента для заднего сегмента глаза. Раствор снижает воздействие на передний сегмент глаза, посредством этого обеспечивая повышенную концентрацию активного ингредиента в растворе и улучшенную частоту доставки, которая поддерживала высокие концентрации в заднем сегменте. Обе данные полезные характеристики связаны с известным свойством циклодекстрина образовывать гидрофильные комплексы с гидрофобными лекарственными средствами. См. Stella & He, Cyclodextrins, Toxicol. Pathol., 36: 30-42 (2008). При формулировании с соединением-I или его свободным основанием, циклодекстрин образовывал прозрачный бесцветный раствор и обладал вязкостью, подобной воде. После местного глазного введения, комплекс соединение-I/циклодекстрин на вид был фармакологически неактивным и метаболически инертным. Комплекс соединение-I/циклодекстрин обеспечивал переносимость роговицы до того, как циклодекстрин самопроизвольно диссоциировал от активного агента, таким образом, обеспечивая высокую концентрацию соединения-I в месте его предполагаемого действия в заднем сегменте глаза, например, сосудистой оболочке глаза и сетчатке.
В процессе исследований с местным глазным дозированием, длящихся 1-30 дней, на голландских кроликах, растворы на основе циклодекстрина соединения-I показывали существенно пониженное воздействие на роговицу по сравнению с композициями в виде гелевых капель (см. пример 2) при одинаковых концентрациях лекарственного средства. Применение растворов на основе циклодекстрина соединения-I обеспечивало приблизительно 10x снижение концентраций в роговице по сравнению с дозированием эквимолярных композиций гелевых капель. Соответственно, после 30 дней местного офтальмического дозирования 0,6% соединения-I в виде раствора на основе циклодекстрина, нежелательные результаты не приписывали испытуемому изделию или среде. Раствор на основе циклодекстрина соединения-I также обеспечивал равные или значительно большие концентрации лекарственного средства в тканях заднего сегмента, таких как в центральной сосудистой оболочке глаза и центральной зоне сетчатки. Комбинированные эффекты снижения воздействия на роговицу лекарственного средства так, чтобы избежать плохой глазной переносимости, при этом увеличивая биодоступность в заднем сегменте так, чтобы усиливать ингибирование RTK, могут значительно улучшать терапевтический индекс и соответствующий полезный эффект (эффекты) на пациентов.
И для композиций в виде суспензий (см. пример 3) и для композиций на основе циклодекстрина, терапевтическое окно расширяется из-за значительно пониженного воздействия (10-100× или 1-2 log снижение). Пониженное воздействие улучшает безопасность/переносимость роговицы, что обеспечивает большие концентрации или частоту дозирования соединения-I, которое вводят местно. Большие концентрации позволяют достигнуть больших концентраций лекарственного средства в целевой ткани заднего сегмента глаза, что увеличивает терапевтическую эффективность соединения-I.
Данное исследование показало, что на кроликах и собаках местное глазное дозирование офтальмических гелевых капель связано с большим воздействием на ткань роговицы (≥100 мкМ) и соответствующими нежелательными проявлениями в переднем сегменте, такими как дискомфорт, воспаление роговицы и конъюнктивы, разрушение эпителия роговицы и/или истончение и дегенерация. Напротив, повторяемое местное глазное дозирование офтальмического раствора соединения-I обеспечивало воздействие на роговицу, которое приблизительно в 5-10 раз меньше, чем эквимолярная доза офтальмических гелевых капель, и не сопровождается нежелательными клиническими или гистопатологическими нарушениями. Кроме того, местное глазное дозирование офтальмического раствора соединения-I обеспечивало равное или большее целевое терапевтическое воздействие в центральной сосудистой оболочке глаза по сравнению с эквимолярной дозой офтальмических гелевых капель. В общем, комбинация сниженного воздействия на роговицу и соответствующей повышенной глазной переносимости при одновременном сохранении или стимулировании доставки лекарственного средства в целевые ткани заднего сегмента, вместе с повышенной физико-химической стабильностью, будет приносить большую пользу пациентам по сравнению с офтальмической композицией в виде гелевых капель.
ПРИМЕР 5
1- - 5-дневные PK результаты с местным глазным введением соединения-I в виде растворов на основе циклодекстрина
После местного глазного введения дозы различных композиций и режимом дозирования соединения-I на голландских кроликах, исследовали фармакокинетические параметры для глаз. Девять (9) различных местных офтальмологических композиций, содержащих три дозы соединения-I, вводили или один раз в день (q.d.) или дважды в день (дважды в день) в течение 1, 4 или 5 последовательных дней. План исследования (см. таблицы 10A-C) включал семьдесят два (72) кролика, каждому из которых вводили 30 мкл билатеральной местной глазной дозы одной из трех (3) композиций соединения-I или плацебо, применяя пипетку прямого вытеснения.
Состав каждой состава соединения-I описан в таблице 8A. Все дозы вводили в пределах ±1 часа от планового времени дозирования. В 1 день группам 1, 2, 4-6, 8, 10, 11, 13, 15 и 17 вводили одну дозу (q.d.) в течение или одного (1) или четырех (4) дней. В 1-4 дни группам 3, 7, 9, 12, 14 и 16 вводили дозу дважды в день приблизительно через 8 часов в 7:00 AM и 3:00 PM в течение четырех (4) дней. Животным в группах 18 и 19 вводили дозу плацебо дважды в день в течение пяти (5) последовательных дней.
Отбор офтальмических образцов осуществляли для группы 1 через 0,5, 1, 2, 4, 8 или 24 часа после дозирования относительно дозы в 1 день. Отбор офтальмических образцов осуществляли для групп 2 и 6 через 1, 8, и 24 часов после дозирования относительно утренней дозы в 5 день. Отбор офтальмических образцов для групп 3, 7, 8, 11 и 13 осуществляли через 1 и 24 часа после дозирования относительно утренней дозы в 5 день. Отбор офтальмических образцов для групп 4, 9, 10, 12, 14, 15, 16 и 17 осуществляли через 1 час после дозирования относительно утренней дозы на 5 день. Отбор офтальмических образцов для группы 5 осуществляли через 0,5, 1, 2, 4, 8 и 24 часа после дозирования относительно дозы в 1 день. За животными в группах 18 и 19 осуществляли только клиническое наблюдение в течение пяти (5) дней.
Собирали образцы водянистой влаги, роговицы, центральной и периферической сетчатки и центральной и периферической сосудистой оболочки глаза. Затем, анализировали образцы водянистой влаги, роговицы, центральной зоны сетчатки и центральной сосудистой оболочки глаза. Образцы периферической сетчатки и периферической сосудистой оболочки глаза анализировали только для групп 1-8, 12, 14 и 16 согласно протоколу исследования.
Анализировали образцы водянистой влаги, роговицы, центральной и периферической сетчатки и центральной и периферической сосудистая оболочка глаза кроликов. Калибровочные кривые получали в контрольной матрице, определяя концентрацию соединения-I в различных тканях.
ПРИМЕР 6
5-дневные PK результаты с местным глазным введением соединения-I в виде растворов на основе циклодекстрина
После глазного введения доз различных режимов дозирования местных офтальмических растворов соединения-I, содержащих гидроксипропил-β-циклодекстрин (“HDβCD”), голландским кроликам, рассчитывали фармакокинетические параметры для глаз. Девять (9) различных местных офтальмических растворов, содержащих четыре различные дозы соединения-I, вводили или один раз в день (q.d.) или дважды в день (дважды в день) в течение или 4 или 5 последовательных дней. План исследования включал сорок (40) кроликов, каждому из которых вводили 30 мкл билатеральной местной глазной дозы одной из четырех (4) дозируемых концентраций соединения-I, применяя пипетку прямого вытеснения.
Все дозы вводили в пределах ±1 час от планового времени введения, за исключением 1 дня для групп 1, 4 и 10. Первую дозу в 1 день вводили в 12:00 PM, и вторую дозу (для групп 1 и 10) вводили приблизительно через 4 часа. Это объяснялось запоздалым поступлением композиций. Все другие дозирования для данных группы осуществляли, как запланировано. В 1 день группам 4-8 и 11-12 вводили одну дозу (q.d.) в течение четырех (4) дней. В 1-4 дни группам 1-3 и 9-10 вводили дозу дважды в день приблизительно через 8 часов в течение четырех (4) дней.
Отбор офтальмических образцов осуществляли через один час после первой ежедневной дозы в 5 день для всех групп, за исключением групп 5b и 6b, где отбор офтальмических образцов осуществляли через 24 часа после первой ежедневной дозы в 4 день.
Образцы крови для сбора плазмы получали непосредственно перед запланированной эвтаназии всех животных. Собирали образцы водянистой влаги, роговицы, центральной и периферической сетчатки и центральной и периферической сосудистой оболочки глаза. Образцы роговицы, центральной зоны сетчатки и центральной сосудистой оболочки глаза групп 1-12 анализировали. Образцы водянистой влаги, периферической сетчатки и периферической сосудистой оболочки глаза не анализировали.
ПРИМЕР 7
Концентрации соединения-I (в мкМ) в различных глазных жидкостях и тканях
Офтальмический раствор соединения-I, содержащий циклодекстрин, получали и испытывали в различных группах животных. После местного глазного введения раствора 0,4% (4 мг/мл) соединения-I и циклодекстрина, концентрацию активного агента измеряли в различных тканях и жидкостях глаза. Среднюю концентрацию соединения-I измеряли в центральной сосудистой оболочке глаза, центральной зоне сетчатки, водянистой влаге и роговице. Соединение-I было в растворе (0,4% или 4 мг/мл) с 8,41% KLEPTOSE® и 0,142% фосфатного буфера; 8,9% KLEPTOSE® HPB и 0,142% фосфата; 4,88% CAPTISOL® и 0,142% фосфата; или 4,88% CAPTISOL® и 0,122% фосфата. См. таблицу 10A-B.
Концентрация соединения-I в центральной сосудистой оболочке глаза составляла 0,259 мкМ - 0,769 мкМ. См. таблицу 10A. Концентрация соединения-I в центральной зоне сетчатки составляла 0,0531 мкМ - 0,124 мкМ. См. таблицу 10A. Концентрация соединения-I в водянистой влаге составляла 0,00313 мкМ - 0,00656 мкМ. См. таблицу 10A. Концентрация соединения-I в роговице составляла 6,49 мкМ - 30 мкМ. См. таблицу 10A. Циклодекстрины, применяемые для получения растворов, представляли собой KLEPTOSE® HPB или CAPTISOL®. См. таблицу 10B.
ПРИМЕР 8
Токсикологические исследования на глазах
Ограничивающая дозу офтальмическая токсичность характеризовалась роговичными и конъюнктивальными нарушениями у голландских кроликов и собак породы бигль. Данные для глаз из исследовании токсичности многократных доз офтальмических гелевых капель соединения-I были основаны на клинических офтальмических и гистопатологических оценках и ограничивались конъюнктивальной гиперемией, хемозом, конгестией и выделениями, помутнением роговицы и эпителиальной эрозией, и кератоконъюнктивитом. Не наблюдали нежелательных изменений, включающих более глубокие структуры глаза (радужку, хрусталик, ресничное тело, сетчатку, сосудистую оболочку глаза, склеру) или оптический нерв. Функционирование сетчатки было нормальным для всех групп, обрабатываемых испытываемым изделием и средой в процессе полноформатной электроретинограммы, осуществляемой на кроликах.
Цели для поисковых токсикологических исследований на глазах заключались в определении: a) местной офтальмической состава, которая была хорошо переносимой, и b) местной офтальмической состава, которая могла обеспечивать требуемые терапевтические концентрации соединения-I в центральной сосудистой оболочке глаза.
Средние концентрации соединения-I в тканях глаза после местного введения дозы дважды в день офтальмических композиций 0,3% соединения-I в виде гелевых капель с и без хлорида бензилалкония были наибольшими в роговице (236-260мкМ) >> в периферической сосудистой оболочке глаза (2,79-4,10 мкМ), центральной сосудистой оболочке глаза (0,340-0,496мкМ), периферической сетчатке (0,150-0,309 мкМ) и водянистой влаге (0,0197-0,0395 мкМ) у голландских кроликов.
Состава в виде суспензии на основе Tris были хорошо переносимыми, где клинические офтальмические исследования показали существенное отсутствие нарушений, относящихся к роговице, только с несколькими единичными случаями слабого конъюнктивита у голландских кроликов. Более того, глаза животных, которым вводили суспензию на основе Tris 0,3% масс./об. соединения-I дважды в день в течение 30 дней, считали нормальными в процессе микроскопии. Средние концентрации соединения-I в тканях глаза, оцененные через 1 час ±15 минут после первой ежедневной местной глазной дозы в 30 день для местного дозирования дважды в день суспензий на основе Tris 0,3% соединения-I с и без хлорида бензилалкония, были наибольшими в роговице (2,69-3,10мкМ) >> периферической сосудистой оболочке глаза (0,781-1,21 мкМ), центральной сосудистой оболочке глаза (0,303-0,319 мкМ), периферической сетчатке (0,0819-0,0868 мкМ), центральной зоне сетчатки (0,0495-0,0592 мкМ) и водянистой влаге (0,00127-0,00145 мкМ).
Растворы на основе циклодекстрина, применяя гидроксипропил-бета-циклодекстрин (HP-β-CD, KLEPTOSE® HPB), были хорошо переносимыми при введении местно в течение 30 дней, дважды в день при 0,1% соединения-I (2,1% HP-β-CD), дважды в день при 0,2% соединения-I (4,21% HP-β-CD), один раз или дважды в день при 0,4% соединения-I (8,41% HP-β-CD), и один раз или дважды в день при 0,6% соединения-I (вплоть до 12,62% HP-β-CD) голландским кроликам. Более того, в аналогичном исследовании с многократным дозированием растворы на основе циклодекстрина 0,4% масс./об. соединения-I в Kleptose® HPB, Kleptose® HP или Captisol® были хорошо переносимыми при дозировании дважды в день в течение 24 дней. Не было обнаружено явной офтальмической токсичности, связанной с соединением-I или средой, в процессе клинических офтальмические или микроскопических наблюдений в обоих исследованиях.
В 24-дневном исследовании, концентрации тканей глаз, обработанных растворами на основе циклодекстрина соединения-I, оцениваемые через 1 час ±15 минут после первой ежедневной местной глазной дозы в 24 день, были, в порядке убывания, наибольшими в роговице (6,49-30 мкМ) >> центральной сосудистой оболочке глаза (0,212-0,769 мкМ) > центральной сетчатке (0,0531-0,124) > водянистой влаге (0,002-0,007).
Таким образом, дозолимитирующую токсичность в роговице наблюдали для офтальмических композиций в виде гелевых капель соединения-I. Офтальмические гелевые капли обеспечивали в пять - пятнадцать раз большие концентрации в роговице соединения-I по сравнению с раствором на основе циклодекстрина, и в пятьдесят-сто раз большие концентрации в роговице соединения-I по сравнению с суспензиями на основе Tris. Суспензии на основе Tris и растворы на основе циклодекстрина соединения-I были хорошо переносимыми с отсутствием явной офтальмической токсичности. Величины доз, которые были хорошо переносимыми для растворов на основе циклодекстрина или суспензий на основе Tris соединения-I при введении один раз или дважды в день, находились в диапазоне от приблизительно 0,005% до приблизительно 5,0% масс./об. в течение, по меньшей мере, 30 дней. Растворы на основе циклодекстрина также давали наибольшие концентрации соединения-I в центральной сосудистой оболочке глаза при применении эквимолярных доз трех композиций, и соответствовали или превышали требуемые терапевтические концентрации.
ПРИМЕР 9
Протокол I фазы для исследования с увеличением дозы на пациентах с неоваскулярной ВМД
Исследование I фазы представляет собой двенадцатинедельное многоцентровое исследование без контроля плацебо и с повышением дозы, разработанное для оценки безопасности, переносимости и фармакокинетических параметров после местного глазного введения соединения-I пациентам с неоваскулярной возрастной дегенерацией макулы (ВМД). В сумме вплоть до 60 пациентов обрабатывали один - два раза в день ежедневно местным глазным дозированием офтальмического раствора соединения-I в течение трех месяцев, где планировали три монотерапии с повышением дозы и одну вспомогательную терапию, применяя единичное интравитреальное введение Lucentis® плюс максимально переносимая доза для монотерапии (15 пациентов на каждую терапию). Пациентов, которые удовлетворяют предусмотренному критерию зрительного восприятия и критерию поражения ХНВ, подтвержденным центром по независимой интерпретации данных, обеспечивали одновременно непрерывным местным глазным дозированием и стандартным лечением.
Офтальмический раствор соединения-I для клинических исследований получали, по меньшей мере, в виде 3 дозируемых концентраций в диапазоне 0,1%-1,0% (в виде соединения-I). Концентрации для GLP партий представляли собой 0% (плацебо), 0,1%, 0,3%, 0,6% и 1,0% соединения-I HCl. Вплоть до 2- - 3-кратные возрастающие дозы (единичный шаг приблизительно ½ log) вводят последующим группам людей.
ПРИМЕР 10
Местную композицию в виде “негелевого” “невязкого” гомогенного офтальмического раствора, который является и физически и химически стабильным в пределах концентраций лекарственного средства 0,1-1,0% (1-10 мг/мл), получали измерением концентрации соединения-I, концентрации циклодекстрина, являющегося комплексообразующим агентом, pH и тоничности, растворимости и стабильности соединения-I. Подходящая буферная система предотвращает влияние сдвига pH на стабильность при концентрациях меньше чем 1 мг/мл. И фосфат и трометамол (Tris) оценивали в качестве буферных агентов. Хлорид натрия применяли для регулирования тоничности.
Качественные характеристики продукта показаны в таблице 14.
Получение состава
Составы, перечисленные в таблице 12 и 13, получали, применяя приведенный общий способ.
Состав доводили до требуемого объема водой для инъекции и перемешивали в течение 30 минут при 500 об/мин. Конечный pH проверяли и регулировали или NaOH или HCl до требуемого диапазона. Приблизительно 5 мл аликвоты непосредственно фильтровали в полупрозрачные 5 мл LDPE бутыли при непрерывном перемешивании при постоянной скорости с помощью системы труб Pumpsil D компании Ватсон-Марлоу, оснащенной Flexicon наполнителем и соединенной с 0,2 микронным PVDF капсульным фильтром. Образцы хранили при 2-8°C до завершения получения всех образцов. Затем, все образцы предоставляли аналитику для хранения и испытаний.
ВКЛЮЧЕНИЕ С ПОМОЩЬЮ ССЫЛКИ
Полное содержание каждого из патентных документов и научных статей, на которые ссылаются в настоящем изобретении, включено с помощью ссылки для всех целей. В настоящем изобретении основной документ определяют с достаточной подробностью, и материалы, которые имеют отношение к описанию, истолковывают на основе контекста ссылки. Цитирование публикаций и патентных документов не предполагается как допущение того, что любой из них относится к предшествующему уровню техники, и не отражает также любое допущение относительно их содержания или даты. Хотя настоящее изобретение описано здесь посредством письменного описания, специалисту в данной области техники ясно, что настоящее изобретение можно осуществлять на практике в виде ряда вариантов осуществления, и указанное выше описание и примеры приведены для иллюстративных целей и не ограничивают формулу изобретения, которая следует далее.
ЭКВИВАЛЕНТЫ
Настоящее изобретение можно воплощать в виде других конкретных форм, не выходя за пределы его сущности или существенных характеристик. Следовательно, указанные выше варианты осуществления следует рассматривать во всех аспектах иллюстративными, а не ограничивающими описанное настоящее изобретение. Таким образом, объем настоящего изобретения определяется прилагаемой формулой изобретения, а не указанным выше описанием, и все изменения, которые попадают в пределы значения и диапазона эквивалентности формулы изобретения предполагаются включенными в настоящее изобретение.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОФТАЛЬМОЛОГИЧЕСКИЕ СОСТАВЫ ДЛЯ ДОСТАВКИ ЛЕКАРСТВЕННЫХ СРЕДСТВ К ЗАДНЕМУ СЕГМЕНТУ ГЛАЗА | 2014 |
|
RU2768489C2 |
| ОФТАЛЬМОЛОГИЧЕСКИЕ СОСТАВЫ ДЛЯ ДОСТАВКИ ЛЕКАРСТВ И ЗАЩИТЫ ПЕРЕДНЕГО ОТДЕЛА ГЛАЗА | 2015 |
|
RU2768652C1 |
| ОФТАЛЬМОЛОГИЧЕСКИЕ СОСТАВЫ ДЛЯ ДОСТАВКИ ЛЕКАРСТВ И ЗАЩИТЫ ПЕРЕДНЕГО ОТДЕЛА ГЛАЗА | 2015 |
|
RU2704810C2 |
| КОМПОЗИЦИЯ ГЛАЗНЫХ КАПЕЛЬ | 2017 |
|
RU2753511C2 |
| КОМПОЗИЦИЯ ГЛАЗНЫХ КАПЕЛЬ | 2012 |
|
RU2623066C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ВВЕДЕНИЯ СРЕДСТВ, СВЯЗЫВАЮЩИХ ТУБУЛИН, ДЛЯ ЛЕЧЕНИЯ ГЛАЗНЫХ ЗАБОЛЕВАНИЙ | 2004 |
|
RU2359693C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ВВЕДЕНИЯ СРЕДСТВ, СВЯЗЫВАЮЩИХ ТУБУЛИН, ДЛЯ ЛЕЧЕНИЯ ГЛАЗНЫХ ЗАБОЛЕВАНИЙ | 2002 |
|
RU2354398C2 |
| ОФТАЛЬМИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ АСКОМИЦИН | 2002 |
|
RU2313344C2 |
| УСКОРЕННОЕ ЗАЖИВЛЕНИЕ ПОВРЕЖДЕНИЙ ГЛАЗ С ПОМОЩЬЮ АНГИОТЕНЗИНОВЫХ ПЕПТИДОВ | 2014 |
|
RU2674148C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБЫ ПРИМЕНЕНИЯ ПРОИЗВОДНЫХ 4-ПРЕГЕНЕН-11β-17-21-ТРИОЛ-3,20-ДИОНА | 2012 |
|
RU2683775C2 |
Группа изобретений относится к химико-фармацевтической промышленности и представляет собой глазной состав в виде суспензии для наружного применения для лечения заболевания заднего сегмента глаза, причем состав содержит активный агент формулы II или его фармацевтически приемлемую соль и одно или более фармацевтически приемлемое вспомогательное вещество, где активный агент или фармацевтически приемлемая соль присутствует в количестве от 0,1% до 5,0% масс./об. и по меньшей мере 95% активного агента представлено в нерастворимой форме, а также применение состава в получении лекарственного препарата, пригодного для обеспечения доступа к заднему сегменту глаза и/или для лечения и/или облегчения заболевания заднего сегмента глаза. Группа изобретений позволяет усовершенствовать способ доставки активного агента в задний сегмент глаза без явного его накопления в переднем сегменте, что достигается благодаря наличию более 95% активного агента в нерастворимой форме. 2 н. и 35 з.п. ф-лы, 20 табл., 10 пр.
1. Глазной состав в виде суспензии для наружного применения для лечения заболевания заднего сегмента глаза, причем состав содержит:
активный агент формулы II
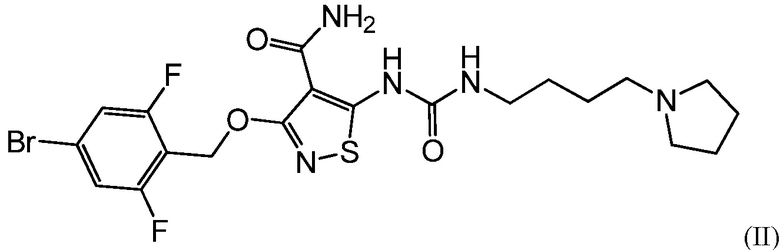 ,
,
или его фармацевтически приемлемую соль и
одно или более фармацевтически приемлемое вспомогательное вещество;
где активный агент или фармацевтически приемлемая соль присутствует в количестве от 0,1% до 5,0% масс./об. и по меньшей мере 95% активного агента представлено в нерастворимой форме.
2. Состав по п.1, содержащий один или несколько фармацевтически приемлемых эксципиентов, выбранных из гидроксиэтилцеллюлозы (HEC) и гидроксипропилметилцеллюлозы (HPMC).
3. Состав по п.1, содержащий один или несколько фармацевтически приемлемых эксципиентов, выбранных из НРМС и карбоксиметилцеллюлозы и их солей.
4. Состав по любому из пп.1-3, содержащий неионный жидкий полимер алкиларилполиэфирспиртового типа.
5. Состав по п.4, где неионным жидким полимером алкиларилполиэфирспиртового типа является тилоксапол.
6. Состав по любому из пп.1-5, содержащий гидрофильное неионное поверхностно-активное вещество.
7. Состав по п.6, где гидрофильное неионогенное поверхностно-активное вещество представляет собой полоксамер.
8. Состав по п.7, где полоксамер представляет собой полоксамер 407.
9. Состав по п.1, содержащий один или несколько фармацевтически приемлемых эксципиентов, выбранных из НРМС и карбоксиметилцеллюлозы и их солей, неионный жидкий полимер типа алкиларилового простого полиэфира и гидрофильное неионогенное поверхностно-активное вещество.
10. Состав по п.1, содержащий один или несколько фармацевтически приемлемых эксципиентов, выбранных из НРМС и карбоксиметилцеллюлозы и их солей, тилоксапола и полоксамера.
11. Состав по любому из пп.1-10, где фармацевтически приемлемая соль представляет собой гидрохлоридную соль, представленную формулой
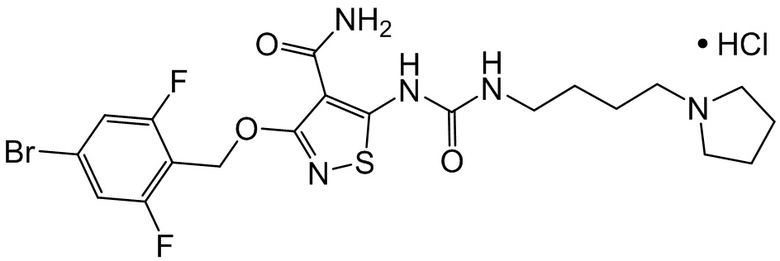 (Соединение-I).
(Соединение-I).
12. Состав по любому из пп.1-11, содержащий один или более из хлорида бензалкония (BAK), хлорида натрия и регулирующего pH агента.
13. Состав по любому из пп.1-12, содержащий от 0,2% до 1% масс./об. трометамина.
14. Состав по п.13, содержащий 0,3-1% масс./об. трометамина.
15. Состав по п.14, содержащий 0,6% масс./об. трометамина.
16. Состав по любому пп.1-15, содержащий 0,005% масс./об. хлорида бензалкония (BAK).
17. Состав по любому из пп.1-16, содержащий от 1% до 2% глицерина.
18. Состав по п.17, содержащий 2% глицерина.
19. Состав по п.1, содержащий от 0,2% до 1% масс./об. трометамина, от 1% до 2% глицерина и HEC или HPMC.
20. Состав по п.1, содержащий от 0,2% до 1% масс./об. трометамина, от 1% до 2% глицерина и НРМС и карбоксиметилцеллюлозу и их соли.
21. Состав по любому из пп.1-20, содержащий цитратный буфер.
22. Состав по любому из пп.1-21, содержащий от 0,1% до 2,0% масс./об. активного агента или его фармацевтически приемлемой соли.
23. Состав по любому из пп.1-21, содержащий от 0,1% до 1,0% масс./об. активного агента или его фармацевтически приемлемой соли.
24. Состав по любому из пп.1-21, содержащий 0,1%, 0,2%, 0,3%, 0,4%, 0,5%, 0,6%, 0,7%, 0,8%, 0,9%, 1,0%, 2,0%, 3,0% 4,0% или 5,0% масс./об. активного агента или его фармацевтически приемлемой соли.
25. Состав по любому из пп.1-24, имеющий величину pH от 4,5 до 7,5 при температуре ниже или равной 40°C.
26. Состав по любому из пп.1-24, имеющий величину pH 6,0 при температуре ниже или равной 40°C.
27. Состав по любому из пп.1-24, имеющий величину pH от 5,0 до 7,0 при температуре ниже или равной 40°C.
28. Состав по любому из пп.1-27, где состав пригоден для обеспечения доступа к заднему сегменту глаза и/или для лечения и/или облегчения глазной неоваскуляризации.
29. Применение состава по любому из пп.1-28 в получении лекарственного препарата, пригодного для обеспечения доступа к заднему сегменту глаза и/или для лечения и/или облегчения заболевания заднего сегмента глаза, выбранного из группы, состоящей из: диабетической ретинопатии, возрастной дегенерации макулы (ВМД), патологической хороидальной неоваскуляризации (ХНВ), патологической неоваскуляризации сетчатки, увеита, окклюзии вены сетчатки, травмы глаз, эдемы, вызванной хирургической операцией, неоваскуляризации, вызванной хирургической операцией, кистозного макулярного отека, ишемии глаз, ретинопатии недоношенных, синдрома Коутса, серповидно-клеточной ретинопатии и неоваскулярной глаукомы.
30. Применение по п.29, где диабетическая ретинопатия представляет собой фоновую диабетическую ретинопатию, пролиферативную диабетическую ретинопатию или диабетический отек макулы.
31. Применение по п.29, где ВМД представляет собой неоваскулярную ВМД, сухую ВМД или географическую атрофию.
32. Применение по п.31, где неоваскулярная ВМД представляет собой влажную или экссудативную ВМД.
33. Применение по п.29, где ХНВ относится к миопии высокой степени, травме, серповидноклеточной анемии, гистоплазмозу глаз, ангиоидным полосам сетчатки, разрыву сосудистого тракта глаза при травме, друзам оптического нерва или некоторым видам ретинальной дистрофии.
34. Применение по п.29, где патологическая неоваскуляризация сетчатки относится к серповидно-клеточной ретинопатии, болезни Илза, глазному ишемическому синдрому, каротидно-кавернозному соустью (фистула), семейной экссудативной форме витреоретинопатии, синдрому повышенной вязкости крови, идиопатическому окклюзионному артериолиту, дробьевидной ретинохороидопатии, ретинальному васкулиту, саркоидозу или токсоплазмозу.
35. Применение по п.29, где окклюзия вены сетчатки представляет собой центральную окклюзию или окклюзию ветки.
36. Применение по п.29, включающее введение состава 1, 2, 3 или 4 раза ежедневно в течение последующих дней или через день.
37. Применение по п.29, включающее введение состава 2 или 3 раза ежедневно в течение последующих дней или через день.
| US 2005074497 A1, 07.04.2005 | |||
| WO 2004011461 A1, 05.02.2005 | |||
| US 2007020336 A1, 25.01.2007 | |||
| Beebe JS, Jani JP, Knauth E, Pharmacological characterization of CP-547,632, a novel vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for cancer therapy// CANCER RESEARCH 63,November 1, 2003 - 7301-7309 | |||
| RU 2008125986 A, 20.01.2010. |

