ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к соединению, выбранному из группы, состоящей из соединения формулы (I) и его фармацевтически приемлемых солей, сложных эфиров, пролекарств, гидратов, сольватов и изомеров, и фармацевтической композиции, содержащей их в качестве активного ингредиента для лечения, облегчения или предотвращения заболеваний, вызванных аномальной или неконтролируемой активацией протеинкиназы.
УРОВЕНЬ ТЕХНИКИ
Тирозинкиназа Брутона (BTK) является представителем TEC-семейства нерецепторных тирозинкиназ, состоящих из 650 остатков аминокислот и содержащих плекстрин-гомологичный домен, белковый домен «цинковые пальцы", SH3-домен, SH2-домен и киназный домен. В последнее время наибольший интерес среди указанных доменов в качестве цели для лекарственных средств вызывает киназный домен.
BTK встречается в B-клетках и гемопоэтических клетках, в отличие от некоторых T-клеток, естественных клеток-киллеров, плазматических клеток и т.д. Поскольку BTK стимулируется сигналами мембранного рецептора B-клеток (BCR), которые вызваны различными воспалительными реакциями или раком, BTK играет важную роль в производстве цитокинов, таких как ФНО-α, ИЛ-6 и т.п., а также NF-κВ путем инициирования нисходящих сигналов, таких как фосфолипаза C гамма 2 (PLCγ2).
При лечении воспаления, BTK, как известно, является промежуточным звеном ответа мембранных рецепторов, например, антигенных рецепторов B-клеток, которые обнаруживают вещества, вызывающие воспаление, CD40, TLR-4, БСГ и тому подобные. Кроме того, BTK оказывает сильное влияние на сигнальный механизм воспаления, вызываемое стимуляцией тучных клеток, B-клеток и макрофагов. Таким образом, ингибирование BTK может блокировать сигнализацию IgE, что может замедлить прогрессирование заболеваний, вызванных аномальной активацией BTK. Этот механизм сигнализации представляет собой сложный сигнальный путь секреции иммуновеществ. В этом процессе фосфорилирование и дефосфорилирование белков проходит в многоступенчатом порядке, и, поскольку BTK является одним из высокоуровневых этапов сигнального пути наряду с тирозинкиназой селезенки (SYK), она, таким образом, более эффективна для предотвращения активации факторов, которые вызывают иммунный ответ, чем другие целевые киназы.
Кроме того, известно, что при лечении рака BTK изменяет BCR и поверхностные белки B-клеток, которые генерируют антисамоубийственные сигналы. Таким образом, ингибирование БТК может взывать противоопухолевые эффекты, направленные против видов рака, которые связаны с сигнализацией BCR, таких как лимфома. Действительно, ибрутиниб (PCI-32765), разработанный компанией Pharmacyclics Inc., был недавно одобрен в качестве противоракового средства для лечения хронического лимфоцитарного ЛЛ), и в настоящее время осуществляется фаза III испытаний AVL-292, разработанного компанией Avila Therapeutics для лечения мелкоклеточного лимфоцитарного лейкоза МЛЛ) и ХЛЛ. Было доказано, что эти соединения весьма эффективны против МЛЛ и ХЛЛ, которые представляют собой сравнительно редкие виды рака. Тем не менее, они не смогли добиться удовлетворительных результатов в отношении диффузной крупноклеточной B-клеточной лимфомы (ДКВКЛ), которая является более распространенным видом лимфомы. Таким образом, существует растущая потребность в лекарственном средстве высокого качества.
Механизм действия ингибиторов BTK в качестве противовоспалительных агентов, а также противораковых агентов, был подробно описан в публикации [Nature Chemical Biology 7, (2011), 4].
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В соответствии с этим, задачей настоящего изобретения является обеспечение соединения, выбранного из группы, состоящей из соединения формулы (I), его фармацевтически приемлемых солей, сложных эфиров, пролекарств, гидратов, сольватов и изомеров.
Еще одной задачей настоящего изобретения является обеспечение фармацевтической композиции, содержащей те же соединения в качестве активного ингредиента для лечения, облегчения или предотвращения заболеваний, вызванных аномальной или неконтролируемой активацией протеинкиназы.
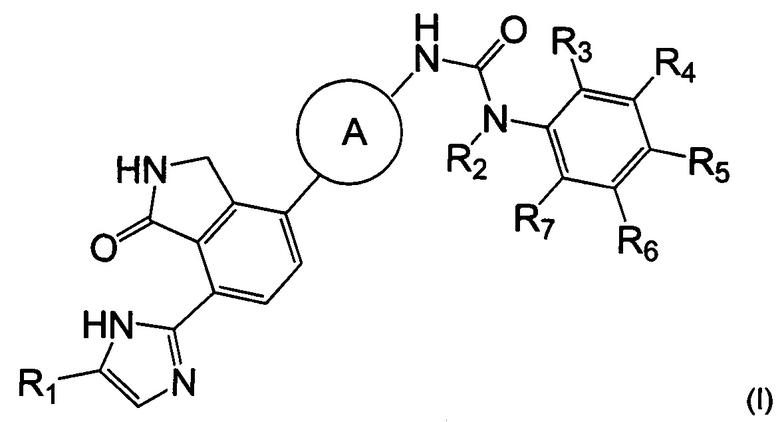
Согласно одному из аспектов настоящего изобретения предложено соединение, выбранное из группы, состоящей из соединения формулы (I) ниже и его фармацевтически приемлемых солей, сложных эфиров, пролекарств, гидратов, сольватов и изомеров:
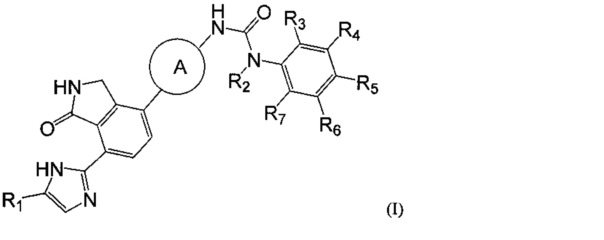
где
A представляет собой 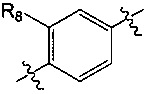 или
или  и R8 представляет собой водород, галоген или C1-алкил,
и R8 представляет собой водород, галоген или C1-алкил,
Каждый из R1 и R2 независимо представляет собой водород или C1-3алкил,
Каждый из R3-R7 независимо представляет собой водород, галоген, циано, нитро или C1-3галогеналкил.
Согласно другому аспекту настоящего изобретения предложена фармацевтическая композиция, содержащая те же соединения в качестве активного ингредиента для лечения, облегчения или предотвращения заболеваний, вызванных аномальной или неконтролируемой активацией протеинкиназы.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Варианты реализации настоящего изобретения подробно объяснены ниже.
Согласно настоящему изобретению предложено соединение, выбранное из группы, состоящей из соединения формулы (I) ниже, и его фармацевтически приемлемых солей, сложных эфиров, пролекарств, гидратов, сольватов и их изомеров:
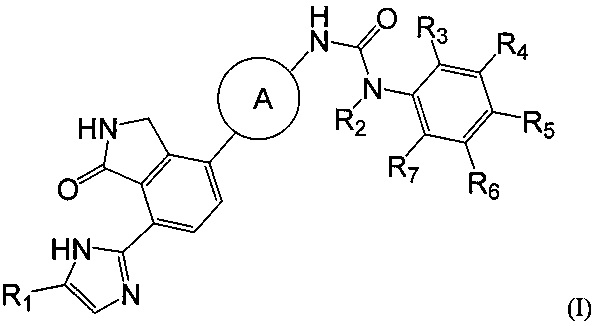
где
A представляет собой 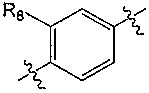 или
или 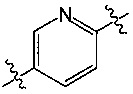 и R8 представляет собой водород, галоген или C1-алкил,
и R8 представляет собой водород, галоген или C1-алкил,
Каждый из R1 и R2 независимо представляет собой водород или C1-алкил,
Каждый из R3-R7 независимо представляет собой водород или электроноакцепторный заместитель, где электроноакцепторный заместитель представляет собой, например, галоген, циано, нитро или C1-3галогеналкил.
Согласно одному конкретному варианту реализации каждый из указанных R3-R7 независимо представляет собой водород, фтор, хлор, бром, йод, циано, нитро, дифторметил или трифторметил.
Согласно другому конкретному варианту реализации,
каждый из указанных R1 и R2 независимо представляет собой водород или метил;
каждый из R3-R7 независимо представляет собой водород, фтор, хлор, циано или трифторметил;
R8 представляет собой водород или фтор.
Используемый в настоящей заявке термин «гало» или «галоген» относится к фтору, хлору, брому или йоду, если не указано иное.
Используемый в настоящей заявке термин «алкил» относится к линейным или разветвленным углеводородным остаткам, если не указано иное.
Согласно настоящему изобретению соединение формулы (I) может образовывать фармацевтически приемлемую соль, являющуюся производным неорганической или органической кислоты, и такая соль может представлять собой фармацевтически приемлемую нетоксичную соль присоединения кислоты, содержащую анион. Например, соль может включать соли присоединения кислоты, образованные неорганическими кислотами, такими как соляная кислота, серная кислота, азотная кислота, фосфорная кислота, бромистоводородная кислота, иодистоводородная кислота и тому подобные; органическими карбоновыми кислотами, такими как винная кислота, муравьиная кислота, лимонная кислота, уксусная кислота, трихлоруксусная кислота, трифторуксусная кислота, глюконовая кислота, бензойная кислота, молочная кислота, фумаровая кислота, малеиновая кислота и тому подобные; и сульфоновыми кислотами, такие как метансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, нафталинсульфоновая кислота и тому подобные. Среди них являются предпочтительными кислотно-аддитивные соли, образованные серной кислотой, метансульфоновой кислотой или галогенводородной кислотой и им подобными.
«Фармацевтически приемлемая соль» соединения формулы (I) может быть получена обычными способами, известными в данной области. В частности, «фармацевтически приемлемая соль» в соответствии с настоящим изобретением может быть получена, например, путем растворения соединения формулы (I) в смешивающемся с водой органическом растворителе, таком как ацетон, метанол, этанол или ацетонитрил и тому подобные; добавления в раствор избыточного количества органической кислоты или водного раствора неорганической кислоты; осаждения или кристаллизации полученной таким образом смеси. Кроме того, она может быть получена путем дальнейшего выпаривания растворителя или избытка кислоты из раствора; и последующей сушки смеси или фильтрованием экстракта с помощью вакуумного фильтра.
Используемый в настоящей заявке термин «сложный эфир» относится к химическому фрагменту, имеющему химическую структуру -(R)n-COOR', где каждый из R и R' независимо выбран из группы, состоящей из алкила, циклоалкила, арила, гетероарила (соединенного с атомом кислорода ароматическим кольцом) и гетероалицикла (соединенного ароматическим кольцом), и n равно 0 или 1, если не указано иное.
Используемый в настоящей заявке термин «про лекарство» относится к соединению-предшественнику, которое будет подвергаться метаболической активации in vivo с образованием исходного лекарственного средства. Пролекарства часто полезны, поскольку в некоторых случаях они могут быть легко введены в сравнении с их исходными лекарственными средствами. Например, некоторые пролекарства являются биодоступными при пероральном приеме, в отличие от исходных лекарственных средств, которые часто демонстрируют плохую биодоступность. Кроме того, пролекарства могут демонстрировать улучшенную растворимость в фармацевтической композиции по сравнению с их исходными лекарственными средствами. Например, соединение формулы (I) может быть введено в форме сложноэфирного пролекарства для повышения эффективности доставки лекарственного средства, поскольку растворимость лекарственного средства может отрицательно повлиять на проницаемость через клеточную мембрану. Затем, когда соединение в форме сложноэфирного пролекарства входит в клетку-мишень, оно может метаболически гидролизоваться до карбоновой кислоты и активного соединения.
Гидраты или сольваты соединения формулы (I) включены в объем настоящего изобретения.
Кроме того, соединение формулы (I) согласно настоящему изобретению может иметь асимметрический атом углерода, и, таким образом, может присутствовать в форме изомера, включая энантиомер, диастереомер или рацемическую смесь, такие полные стереоизомеры и смеси быть включены в объем настоящего изобретения.
Конкретные примеры соединения формулы (I) согласно настоящему изобретению представляют собой:
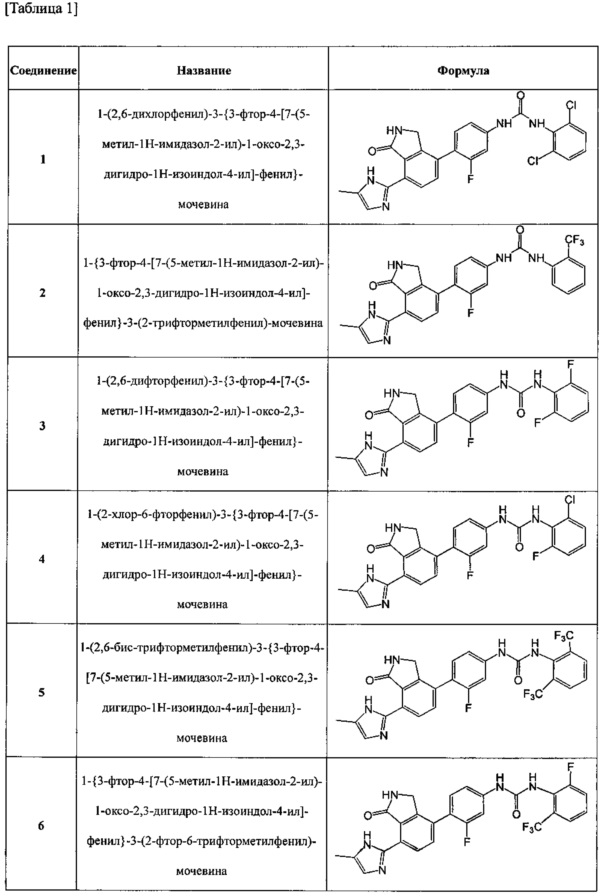
1) 1-(2,6-дихлорфенил)-3-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-мочевину;
2) 1-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-3-(2-трифторметилфенил)-мочевину;
3) 1-(2,6-дифторфенил)-3-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-мочевину;
4) 1-(2-хлор-6-фторфенил)-3-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-мочевину;
5) 1-(2,6-бис-трифторметилфенил)-3-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-мочевину;
6) 1-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-3-(2-фтор-6-трифторметилфенил)-мочевину;
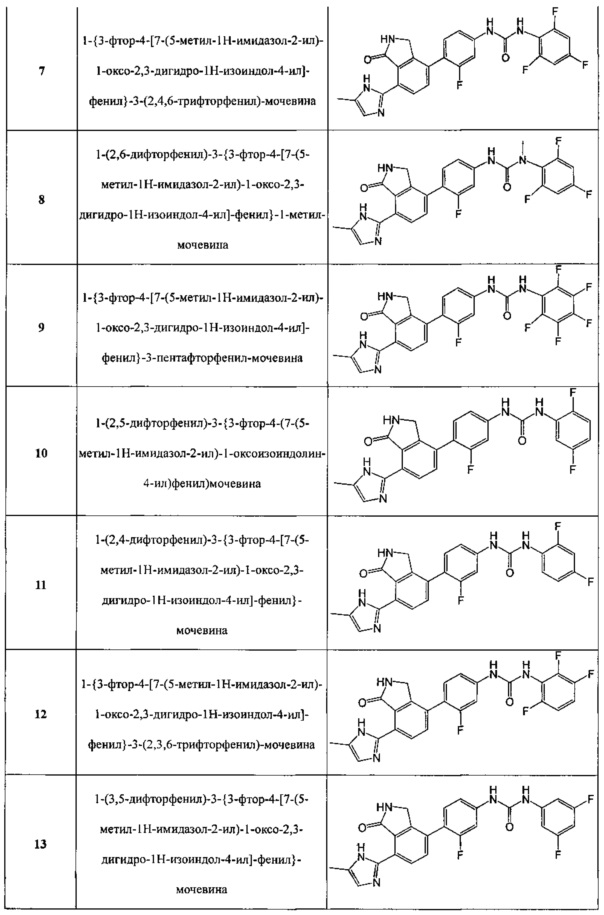
7) 1-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-3-(2,4,6-трифторфенил)-мочевину;
8) 1-(2,6-дифторфенил)-3-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-1-метил-мочевину;
9) 1-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-3-пентафторфенил-мочевину;
10) 1-(2,5-дифторфенил)-3-{3-фтор-4-(7-(5-метил-1H-имидазол-2-ил)-1-оксоизоиндолин-4-ил)фенил)мочевину;
11) 1-(2,4-дифторфенил)-3-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-мочевину;
12) 1-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-3-(2,3,6-трифторфенил)-мочевину;
13) 1-(3,5-дифторфенил)-3-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-мочевину;
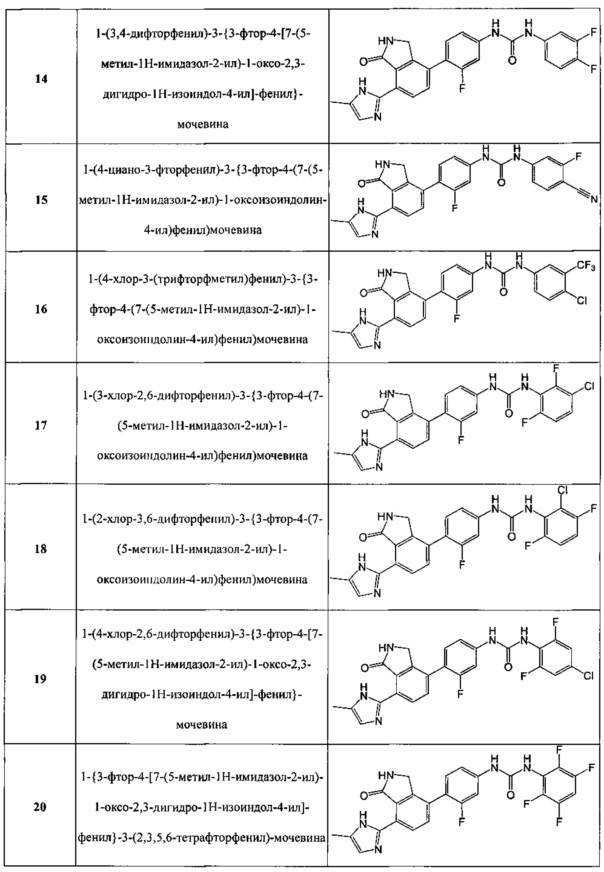
14) 1-(3,4-дифторфенил)-3-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-мочевину;
15) 1-(4-циано-3-фторфенил)-3-{3-фтор-4-(7-(5-метил-1H-имидазол-2-ил)-1-оксоизоиндолин-4-ил)фенил)мочевину;
16) 1-(4-хлор-3-(трифторфметил)фенил)-3-{3-фтор-4-(7-(5-метил-1H-имидазол-2-ил)-1-оксоизоиндолин-4-ил)фенил)мочевину;
17) 1-(3-хлор-2,6-дифторфенил)-3-{3-фтор-4-(7-(5-метил-1H-имидазол-2-ил)-1-оксоизоиндолин-4-ил)фенил)мочевину;
18) 1-(2-хлор-3,6-дифторфенил)-3-{3-фтор-4-(7-(5-метил-1H-имидазол-2-ил)-1-оксоизоиндолин-4-ил)фенил)мочевину;
19) 1-(4-хлор-2,6-дифторфенил)-3-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-мочевину;
20) 1-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-3-(2,3,5,6-тетрафторфенил)-мочевину;
Соединение, выбранное из группы, состоящей из соединения формулы (I), его фармацевтически приемлемых солей, сложных эфиров, пролекарств, гидратов, сольватов и изомеров, может применяться для лечения, облегчения или предотвращения заболеваний, вызванных аномальной или неконтролируемой активацией протеинкиназы, такой как ABL (тирозинкиназа Абельсона), ACK (активированная Cdc42-ассоциированная киназа), AXL, Aurora, BLK (B лимфоидная тирозинкиназа Blk), BMX (сцепленная с X-хромосомой киназа костного мозга), BTK (тирозинкиназа Брутона), CDK (циклинзависимая киназа), CSK (киназа C-Src), DDR (рецептор домена дискоидина), EPHA (эфрин типа A-рецепторная киназа), FER (Fer тирозинкиназа (семейства fps/fes), FES (онкоген саркомы кошек), FGFR (рецептор фактора роста фибробластов), FGR, FLT (fms-подобная тирозинкиназа), FRK (киназа семейства Fyn), FYN, HCK (киназа гемопоэтических клеток), IRR (рецептор семейства инсулиновых рецепторов), ITK (интерлейкин 2-индуцируемая киназа T-клеток), JAK (Янус-киназа), KDR (рецептор, содержащий домен вставки киназы), KIT, LCK (лимфоцит-специфическая протеин тирозинкиназа), LYN, MAPK (митоген-активируемая протеинкиназа), MER (протоонкоген тирозинкиназа с-Mer), MET, MINK (киназа деформированного вида), MNK (MAPK-взаимодействующая киназа), MST (стерильная 20-подобная киназа млекопитающих), MUSK (мышечно-специфическая киназа), PDGFR (рецептора тромбоцитарного фактора роста), PLK (Polo-подобная киназа), RET (перестроенная в ходе трансфекции), RON, SRC (коактиватор стероидного рецептора), SRM (спермидинсинтаза), TIE (тирозинкиназа с иммуноглобулином и повторами EGF), SYK (тирозинкиназа селезенки), TNK1 (нерецепторная тирозинкиназа 1), TRK (тропомиозином-рецепторная киназа), TNIK (киназа взаимодействия TRAF2 и NCK) и тому подобное.
В соответствии с этим, согласно настоящему изобретению предложено применение соединения формулы (I), его фармацевтически приемлемых солей, сложных эфиров, пролекарств, гидратов, сольватов и изомеров для лечения, облегчения или предотвращения заболеваний, вызванных аномальной или неконтролируемой активацией протеинкиназы, и применение соединения для получения лекарственного средства для лечения, облегчения или предотвращения заболеваний.
Кроме того, согласно настоящему изобретению предложен способ лечения, облегчения или предотвращения заболеваний, вызванных аномальной или неконтролируемой активацией протеинкиназы у млекопитающего, включающий введение указанному млекопитающему фармацевтической композиции, содержащей соединение формулы (I), его фармацевтически приемлемых солей, сложных эфиров, пролекарств, гидратов, сольватов и изомеров.
Кроме того, согласно настоящему изобретению предложена фармацевтическая композиция для лечения, облегчения или предотвращения заболеваний, вызванных аномальной или неконтролируемой активацией протеинкиназы у млекопитающего, включающая соединение формулы (I), его фармацевтически приемлемые соли, сложные эфиры, пролекарства, гидраты, сольваты и изомеры.
Указанные заболевания, связанные с активностью киназы, могут включать любое заболевание, вызванное аномальной или неконтролируемой активацией протеинкиназы. Конкретные их примеры могут представлять собой раковое заболевание, воспаление, связанное с ревматоидным артритом и остеоартритом, астму, аллергию, атопический дерматит или псориаз, но не ограничиваются этим.
Примеры указанного раковых заболеваний включают лимфому, лейкоз, рак крови, рак желудка, немелкоклеточный рак легкого, рак печени, рак толстой кишки, рак тонкой кишки, рак поджелудочной железы, рак головного мозга, рак кости, меланому, рак молочной железы, склерозирующий аденоз, рак матки, рак шейки матки, рак яичников, рак головы и шеи, рак пищевода, рак щитовидной железы, рак паращитовидной железы, рак почки, саркому, рак предстательной железы, рак уретры, рак мочевого пузыря, фиброаденому или глиобластому, но не ограничиваются этим.
Фармацевтическая композиция может дополнительно содержать по меньшей мере одну добавку, выбранную из группы, состоящей из антибиотика, алкилирующего агента, антиметаболита, гормонального агента, иммунологического агента, агента интерферонового типа и противоопухолевого агента.
Фармацевтическая композиция согласно настоящему изобретению может быть получена непосредственно, либо дополнительно содержать обычные нетоксичные фармацевтически приемлемые добавки, например, носитель и вспомогательное вещество, для получения в соответствии с любым из традиционных способов, известных в данной области.
Способ лечения, облегчения или предотвращения может включать, например, введение эффективного количества фармацевтической композиции, содержащей соединение согласно настоящему изобретению субъекту, подвергающемуся риску или страдающему от хронической почечной недостаточности, диабета, рака, СПИДа, лучевой терапии, химиотерапии, диализа почек или анемии, вызванной операцией. Согласно одному из вариантов реализации пациент предпочтительно является млекопитающим и, более предпочтительно, человеком.
Эффективное количество фармацевтической композиции согласно настоящему изобретению может быть определено путем проведения обычного теста для выявления наиболее эффективного пути введения и подходящего способа получения. Фармацевтическая композиция согласно настоящему изобретению может быть получена в виде любого состава и системы доставки лекарственных средств любым из традиционных способов, известных в данной области. Фармацевтическая композиция согласно настоящему изобретению может быть получена в виде инъекционных составов, которые могут быть введены путями, включающими интратекальный, внутрижелудочковый, внутривенный, внутрибрюшинный, интраназальный, внутриглазной, внутримышечный, подкожный или внутрикостный. Кроме того, они также могут быть введены перорально или парентерально через прямую кишку, кишечник или слизистую оболочку носовой полости (см Gennaro, A.R., ed. (1995) Remington's Pharmaceutical Sciences). Предпочтительно местное введение композиции вместо энтерального. Например, композиция может быть введена или доставлена с помощью целевой системы доставки лекарственных средств, таких как состав с резервуаром или состав с замедленным высвобождением.
Фармацевтический состав согласно настоящему изобретению может быть получен любым из традиционных способов, известных в данной области, таких как смешивание, растворение, гранулирование, приготовление драже, растирание в порошок, эмульгирование, инкапсулирование, включение или лиофилизация. Как упоминалось выше, композиции согласно настоящему изобретению могут включать один или более физиологически приемлемых носителей, таких как вспомогательные вещества и адъюванты, которые облегчают переработку активных молекул в препараты для фармацевтического применения.
Надлежащий состав зависит от выбранного пути введения. Для инъекции, например, композиция может быть получена в виде водного раствора, предпочтительно в физиологически совместимых буферных растворах, таких как раствор Хенка, раствор Рингера или физиологический солевой буферный раствор. Для трансмукозального или назального введения в композиции применяют проникающие вещества, подходящие для прохождения соответствующего барьера. Такие проникающие вещества широко известны в данной области техники. В предпочтительном варианте реализации настоящего изобретения, соединение согласно изобретению, может быть получено в форме состава для перорального введения. Для перорального введения соединения могут быть легко приготовлены путем объединения активных соединений с фармацевтически приемлемыми носителями, известными в данной области техники. Такие носители позволяют приготовить соединения согласно изобретению в виде таблеток, пилюль, драже, капсул, жидкостей, гелей, сиропов, взвесей, суспензий и т.п., для перорального приема пациентом. Соединения также могут быть получены в виде ректальных композиций, таких как суппозитории или удерживающие клизмы, например, содержащие обычные основы для суппозиториев, такие как масло какао или другие глицериды.
Фармацевтические препараты для перорального применения могут быть получены в виде твердых вспомогательных веществ, необязательно измельченных с получением смеси и обработки смеси гранул после добавления подходящих адъювантов, при желании, с получением таблеток или драже. Подходящие вспомогательные вещества могут представлять собой, в частности, наполнители, такие как сахара, включая лактозу, сахарозу, маннит или сорбит; целлюлозный состав, такой как состав кукурузного крахмала, пшеничного крахмала, рисового крахмала, картофельного крахмала, желатина, трагакантовой камеди, метилцеллюлозы, гидроксипропилметилцеллюлозы, натрийкарбоксиметилцеллюлозы и/или поливинилпирролидона (ПВП). Кроме того, можно применять агенты для улучшения распадаемости, такие как сшитый поливинилпирролидон, агар или альгиновая кислота или ее соль, такая как альгинат натрия. Кроме того, могут быть добавлены смачивающие агенты, такие как додецилсульфат натрия и ему подобные.
Ядра драже снабжают подходящими покрытиями. С этой целью можно применять концентрированные растворы сахара, которые могут необязательно содержать гуммиарабик, тальк, поливинилпирролидон, гель карбопола, полиэтиленгликоль и/или диоксид титана, лаковые растворы и подходящие органические растворители или смеси растворителей. К покрытиям таблеток или драже можно добавлять красители или пигменты для идентификации или описания различных комбинаций доз активных соединений.
Фармацевтические составы для перорального введения могут включать твердые капсулы, изготовленные из желатина, а также мягкие герметичные капсулы, изготовленные из желатина и пластификатора, такого как глицерин или сорбит. Твердые капсулы могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, связующими, такими как крахмалы, и/или смазывающими веществами, такими как тальк или стеарат магния, и, необязательно, стабилизаторами. В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как жирные масла, жидкий парафин или жидкие полиэтиленгликоли. Кроме того, могут быть добавлены стабилизаторы. Все составы для перорального введения должны иметь дозировку, подходящую для такого введения.
Согласно одному из вариантов реализации, соединения согласно настоящему изобретению можно вводить трансдермально, например, через кожный пластырь или местно. Согласно одному из аспектов, составы для трансдермального или местного применения согласно настоящему изобретению могут дополнительно содержать один или несколько усилителей проницаемости или другие эффекторы, в том числе агенты, усиливающие миграцию доставленного соединения. Предпочтительно, составы для трансдермального или местного применения можно применять, например, в ситуациях, в которых желательна доставка к конкретному месту.
Для введения путем ингаляции соединения по настоящему изобретению могут быть удобно доставлены в форме аэрозольного спрея в упаковках под давлением или распылителя с применением подходящего пропеллента, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или любого другого подходящего газа. В случае аэрозоля под давлением, соответствующая доза может быть определена с помощью клапана для доставки отмеренного количества. Для применения в ингаляторе или инсуффляторе могут быть получены капсулы и картриджи, например, из желатина. Они, как правило, содержат порошковую смесь соединения и подходящую порошковую основу, такую как лактоза или крахмал. Композиции для парентерального введения путем инъекции, например, путем болюсного введения или непрерывной инфузии, могут быть представлены в стандартной лекарственной форме, например, в ампулах или в многодозовых контейнерах с добавлением консерванта. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных носителях, и могут содержать вспомогательные агенты, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. Препараты для парентерального введения включают водные растворы или другие композиции в водорастворимой форме.
Суспензии активных соединений также могут быть получены в виде подходящих масляных суспензий для инъекций. Подходящие липофильные растворители или носители могут включают жирные масла, такие как кунжутное масло и синтетические сложные эфиры жирных кислот, такие как этилолеат или триглицериды, или липосомы. Водные суспензии для инъекций могут содержать вещества, которые увеличивают вязкость суспензии, такие как натрий-карбоксиметилцеллюлоза, сорбит или декстран. Суспензия может также необязательно содержать подходящие стабилизаторы или агенты, которые повышают растворимость соединений для обеспечения подготовки высококонцентрированных растворов. В качестве альтернативы активный ингредиент может иметь форму порошка для разведения подходящим носителем, например стерильной апирогенной водой, перед применением.
Как упоминалось выше, композиции согласно настоящему изобретению могут быть также приготовлены в виде состава с резервуарной системой. Такие длительно действующие составы могут быть введены путем имплантации (например, подкожной или внутримышечной) или путем внутримышечной инъекции. Так, например, соединения согласно изобретению могут быть получены с подходящими полимерными или гидрофобными материалами (например, в виде эмульсии в приемлемом масле) или ионообменными смолами, или в виде труднорастворимых производных, например, умеренно растворимой соли.
Для любой композиции, применяемой в существующих способах лечения, терапевтически эффективная доза может быть первоначально оценена с помощью различных методик, известных в данной области. Например, на основе информации, полученной путем анализа культуры клеток, доза может быть сформулирована на животных моделях для достижения диапазона циркулирующих концентраций, который включает IC50. Аналогичным образом могут быть определены диапазоны дозировок, подходящих для человека, например, с использованием данных, полученных путем анализа культур клеток и других исследований на животных.
Терапевтически эффективная доза агента относится к количеству агента, которое приводит к ослаблению симптомов или продлению жизни у пациента. Токсичность и терапевтическая эффективность таких молекул может быть определена с помощью стандартных фармацевтических процедур на клеточных культурах или на подопытных животных, например, путем определения LD50 (дозы, летальной для 50% популяции) и LD50 (дозы, терапевтически эффективной для 50% популяции). Дозовое соотношение между токсическим и терапевтическим эффектами представляет собой терапевтический индекс, который может быть выражен как отношение LD50/ED50. Предпочтительны агенты с высокими терапевтическими индексами.
Дозировки предпочтительно находятся в пределах диапазона циркулирующих концентраций, который включает в себя ED50 с небольшой токсичностью или без токсичности. Дозировки могут варьироваться в пределах этого диапазона в зависимости от применяемой лекарственной формы и способа введения. Точный состав, путь введения, дозировку следует выбирать в соответствии со способами, известными в данной области, с учетом особенностей состояния пациента.
Кроме того, количество введенного агента или композиции будет зависеть от множества факторов, в том числе от возраста, веса, пола, состояния здоровья, степени заболевания пациента, подлежащего лечению, тяжести заболевания, способа введения, и решения лечащего врача.
Далее поясняется примерный способ получения соединения согласно настоящему изобретению.
Различные исходные материалы могут быть получены в соответствии с обычными способами синтеза, известными в данной области техники. Некоторые из исходных материалов коммерчески доступны от изготовителей и поставщиков реагентов, таких как Aldrich, Sigma, TCI, Вако, Канто, Fluorchem, Acros, Alfa, Abocado, Fluka и т.д., но не ограничиваясь ими.
Соединения согласно настоящему изобретению могут быть получены из легко доступных исходных веществ с помощью традиционных способов и процессов, указанных ниже. Для получения соединений согласно изобретению, если не указано иное, можно применять различные способы, если не указаны иные обычные или предпочтительные условия (т.е. температура реакции, время, мольное соотношение реагентов, растворители, давление и т.д.). Оптимальные условия реакции можно варьировать в зависимости от конкретных применяемых реагентов или растворителей. Такие условия, однако, могут быть определены специалистом обычной квалификации в данной области путем обычного процесса оптимизации.
Кроме того, специалистам обычной квалификации в данной области будет понятно, что некоторые функциональные группы могут быть защищены или с них может быть удалена защитная группа с применением различных защитных групп до протекания определенной реакции. Подходящие условия для введения защитной группы и/или снятия защиты с конкретной функциональной группы, а также применение защитных групп известно в данной области.
Например, различные виды защитных групп описаны в T.W. Greene and G.M. Wuts, Protecting Groups in Organic Synthesis, Second edition, Wiley, New York, 1991 и других приведенных выше ссылках.
Согласно одному из вариантов реализации настоящего изобретения, соединение формулы (I) согласно настоящему изобретению может быть получено путем синтеза промежуточного продукта, соединения D, в соответствии со схемой реакции 1, как показано ниже, и последующего подвергания соединения D процедуре, указанной на схемах реакции 2 или 3. Тем не менее, способ синтеза соединения D выше не ограничивается схемой 1.
[Схема реакции 1]
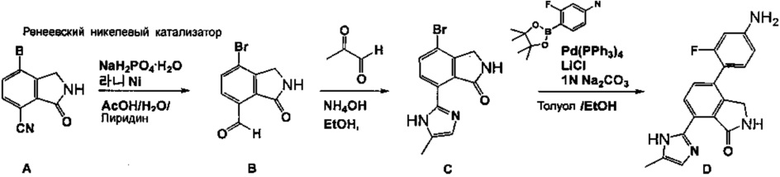
Способ получения исходного материала схемы реакции 1, т.е., соединения A, описан в международной публикации патента WO 2012/014017, и соединение D получали следующими способами.
<1-1> Синтез соединения B
Соединение A (40 г, 168 ммоль) диспергировали в уксусной кислоте (400 мл), добавляли воду (400 мл) и пиридин (800 мл), а затем температуру полученной таким образом смеси снижали до 10°C. К смеси добавляли моногидрат одноосновного фосфата натрия (280 г, 2,01 моль), а затем дополнительно добавляли Ренеевский никелевый катализатор (101 г) в воде (70 мл) с образованием реакционного раствора. Реакционный раствор нагревали до 50°C, давали прореагировать в течение 2 часов. После завершения реакции раствор охлаждали и фильтровали. Раствор промывали этилацетатом (ЭА, 2,5 л), и к фильтрату добавляли воду (800 мл) для извлечения. Полученный таким образом органический слой отделяли и концентрировали при пониженном давлении. К реакционному раствору добавляли охлажденную воду (800 мл), и полученное таким образом твердое вещество фильтровали и сушили с получением соединения B (26,7 г, выход: 66%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 11,10 (s, 1H), 9,15 (s, 1H), 7,95 (d, J=8,1 Гц, 1H), 7,78 (d, J=8,1 Гц, 1H), 4,41 (s, 2H)
ЖХ-МС [М+1]: 241,1
<1-2> Синтез соединения C
Соединение B (26,7 г, 111 ммоль) диспергировали в этаноле (800 мл), и добавляли водные растворы 48% пировиноградного альдегида (67 мл) и 28% аммиака (75 мл). Полученную таким образом реакционный раствор нагревали до 90°C и перемешивали в течение 3 часов. После завершения реакции раствор концентрировали при пониженном давлении для уменьшения объема реакционного раствора до примерно 200 мл, и полученное таким образом твердое вещество отфильтровывали. Твердое вещество промывали этанолом (50 мл) с получением соединения C (17,2 г, выход: 53%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 14,21-14,12 (m, 1H), 9,48 (s, 1H), 8,27 (d, J=8,4 Гц, 1H), 7,83 (d, J=8,4 Гц, 1H), 7,07-6,82 (m, 1H), 4,39 (s, 2H), 2,27-2,18 (m, 3H)
ЖХ-МС [М+1]: 293,1
<1-3> Синтез соединения D
Соединение C (17,2 г, 58,9 ммоль), 3-фтор-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фениламин (19,5 г, 82 ммоль), LiCl (6,9 г, 165 ммоль) и Pd(PPh3)4 (6,8 г, 5,9 ммоль) диспергировали в смешанном растворе толуола (589 мл) и этанола (589 мл), добавляли водный раствор 1 н Na2CO3 (117 мл) и давали прореагировать при 85°C в течение 12 часов. После завершения реакции реакционный раствор полностью концентрировали при пониженном давлении. Добавляли смешанный раствор ацетона (1,2 л) и ацетонитрила (1,2 л) и реакционный раствор перемешивали в течение 2 часов при 80°C, охлаждали, фильтровали и затем промывали ацетонитрилом (0,5 л). Отфильтрованный раствор концентрировали при пониженном давлении для уменьшения объема реакционного раствора до примерно 150 мл и затем фильтровали. Полученное таким образом твердое вещество промывали при пониженном давлении ацетонитрилом (60 мл), н-гексаном (100 мл) и водой (100 мл), соответственно, и сушили с получением соединения D, 4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-она (13,31 г, выход: 70%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 14,44-14,34 (m, 1H), 9,36 (s, 1H), 8,36 (d, J=8,4 Гц, 1H), 7,51 (d, J=7,8 Гц, 1H), 7,16 (t, J=8,7 Гц, 1H), 7,04-6,79 (m, 1H), 6,49-6,42 (m, 2H), 5,65 (s, 2H), 4,36 (s, 2H), 2,28-2,18 (m, 3H)
ЖХ-МС [M+1]: 323,3
Для получения различных соединений, которые могут быть представлены формулой (I) настоящего изобретения, способ синтеза таких соединений с применением промежуточного соединения D подробно описан в схемах 2 и 3 ниже. Тем не менее, это показательный пример получения соединения формулы (I) с применением промежуточного соединения D, и, следовательно, способ получения соединения формулы (I) не ограничивается этим.
[Схема реакции 2]
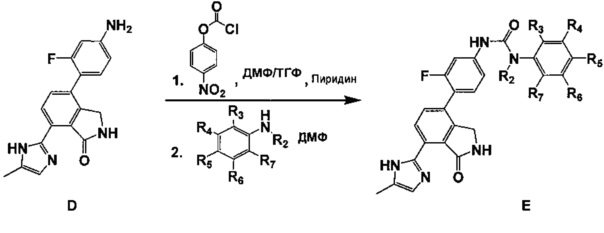
Соединение E, описанное в реакционной схеме 2, получали с помощью следующих способов.
<2-1> Синтез соединения E в соответствии со схемой реакции 2
4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (соединение D, 1 эквивалент), диспергировали в ДМФ/ТГФ (1:4) получением раствора (0,08 м), а затем добавляли к нему пиридин (1,15 эквивалента) и 4-нитрофениловый эфир хлормуравьиной кислоты (1,15 эквивалента) с последующим перемешиванием в течение 4 часов. После завершения реакции (ТСХ) добавляли н-гексан (того же объема, что и ТГФ, реакционный раствор) с последующим перемешиванием в течение 30 минут. Полученное таким образом твердое вещество промывали смешанным растворителем н-гексан : ТГФ = 1:1 (четырехкратный объем ТГФ, реакционный раствор), фильтровали и затем сушили. Высушенное соединение диспергировали в ДМФ с образованием раствора (0,1 м), добавляли замещенный фениламин (от 6 до 15 эквивалентов) и затем перемешивали в течение 20 минут под действием микроволнового излучения (250 Вт, 250psi, 150°C). Реакционный раствор разбавляли этилацетатом, содержащим 5% метанола, а затем промывали насыщенным водным раствором NaHCO3 и водой. Органический слой сушили над безводным сульфатом магния (MgSO4), а затем очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением соединения E.
Соединение E, полученное в соответствии со схемой реакции 2, также можно синтезировать в соответствии со схемой реакции 3 ниже.
[Схема реакции 3]
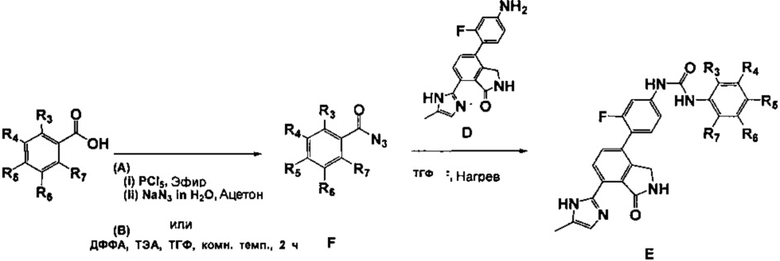
<3-1> (A) Синтез соединения E в соответствии со схемой реакции 3
Замещенную бензойную кислоту (2 эквивалента) диспергировали в диэтиловом эфире с получением смеси (0,08 м), добавляли пентахлорид фосфора (PCl5, 2,2 экв) и затем перемешивали в течение 2 часов. После завершения реакции органический растворитель концентрировали при пониженном давлении при температуре ниже комнатной, а затем реакционный раствор разбавляли (0,08 м) путем добавления ацетона к реагенту. Затем медленно добавляли к реакционному раствору азид натрия (NaN3, 2,4 эквивалента) в воде (1/12 объема ацетона) по каплям при 0°C. После перемешивания в течение 2 часов при комнатной температуре полученный продукт разбавляли этилацетатом и затем водой. Органический слой сушили над безводным сульфатом магния (MgSO4), диспергировали в ТГФ с образованием раствора (0,04 м), добавляли 4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (Соединение D, 1 эквивалент), и перемешивали в течение 4 ч при 90°C. После завершения реакции растворитель концентрировали при пониженном давлении и затем очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением соединения E.
Соединение E, синтезированное в соответствии со схемой реакции 2, также можно синтезировать с применением другого способа в соответствии со схемой реакции 3.
<3-2> (B) Синтез соединения E в соответствии со схемой реакции 3
Замещенную бензойную кислоту (2 эквивалента) диспергировали в ТГФ с образованием раствора (0,05 М) и затем добавляли к нему триэтиламин (4 экв) и дифенилфосфорилазид (ДФФА, 2,3 экв) с последующим перемешиванием в течение 2 часов при комнатной температуре. К реакционному раствору добавляли 4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (Соединение D, 1 эквивалент), а затем перемешивали в течение 4 часов при 90°C. Затем реакционный раствор разбавляли этилацетатом, содержащим 5% метанола, и затем промывали водой и насыщенным водным раствором NaHCO3. Органический слой сушили над безводным MgSO4 и затем концентрировали при пониженном давлении. Полученный таким образом концентрат очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением соединения E.
Далее настоящее изобретение более конкретно описано в следующих примерах, но они приведены только в иллюстративных целях, и настоящее изобретение не ограничивается ими.
Пример 1: Получение 1-(2,6-дихлорфенил)-3-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-мочевины
4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (соединение D, 0,1 г, 0,31 ммоль) диспергировали в диметилформамиде (0,8 мл) и ТГФ (3,4 мл), добавляли пиридин (0,03 мл) и 4-нитрофениловый эфир хлормуравьиной кислоты (0,07 г, 0,36 ммоль) и затем перемешивали в течение 4 часов. После подтверждения завершения реакции с помощью ТСХ добавляли н-гексан (3 мл) и перемешивали в течение 30 минут. Полученное таким образом твердое вещество промывали смешанным растворителем н-гексан : ТГФ = 1:1 (12 мл), фильтровали и затем сушили. Высушенное соединение диспергировали в ДМФ (3 мл), добавляли 2,6-дихлоранилин (0,34 г, 2,08 ммоль) и затем перемешивали в течение 20 минут под действием микроволнового излучения (250 Вт, 250psi, 150°C). Реакционный раствор разбавляли этилацетатом, содержащим 5% метанола, а затем последовательно промывали насыщенным водным раствором NaHCO3 и водой. Органический слой сушили над безводным сульфатом магния, концентрировали, и затем очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,03 г, выход: 19%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 9,54 (s, 1H), 9,36 (s, 1H), 8,58 (s, 1H), 8,43 (d, J=8,1 Гц, 1H), 7,59 (m, 3H), 7,47 (t, J=8,4 Гц, 1H), 7,32 (m, 2H), 7,08 (s, 1H), 4,41 (s, 2H), 2,25 (m, 3H)
ЖХ-МС [М+1]: 511
Пример 2: Получение 1-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-3-(2-трифторметилфенил)-мочевины
4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (соединение D, 0,1 г, 0,31 ммоль) диспергировали в диметилформамиде (0,8 мл) и ТГФ (3,4 мл), добавляли пиридин (0,03 мл) и 4-нитрофениловый эфир хлормуравьиной кислоты (0,07 г, 0,36 ммоль) и затем перемешивали в течение 4 часов. Затем добавляли к смеси н-гексан (3 мл) и перемешивали в течение 30 минут. Полученное таким образом твердое вещество промывали смешанным раствором н-гексан : ТГФ = 1:1 (12 мл), фильтровали и затем сушили. Высушенное соединение диспергировали в ДМФ (2 мл), добавляли 2-трифторметиланилин (0,74 г, 4,65 ммоль) и затем перемешивали в течение 3 минут при комнатной температуре. В реакционный раствор затем последовательно добавляли метанол (6 мл) и насыщенный водный раствор NaHCO3 и перемешивали в течение 30 минут. Полученное таким образом твердое вещество отфильтровывали и промывали водой. После высушивания полученного твердого вещества твердое вещество очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,07 г, выход: 44%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 9,54 (s, 1H), 9,36 (s, 1H), 8,58 (s, 1H), 8,43 (d, J=8,1 Гц, 1H), 7,59 (m, 3H), 7,47 (t, J=8,4 Гц, 1H), 7,32 (m, 2H), 7,08 (s, 1H), 4,41 (s, 2H), 2,25 (m, 3H)
ЖХ-МС [М+1]: 510,0
Пример 3: Получение 1-(2,6-дифторфенил)-3-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-мочевины
2,6-дифтор-бензойную кислоту (0,04 г, 0,248 ммоль) диспергировали в диэтиловом эфире (3 мл), медленно добавляли пентахлорид фосфора (PCl5, 0,057 г, 0,273 ммоль) и затем перемешивали в течение 1 часа. После завершения реакции органический растворитель концентрировали при пониженном давлении при температуре ниже комнатной, а затем реакционный раствор разбавляли путем добавления ацетона (2 мл). Затем медленно добавляли к реакционному раствору азид натрия (NaN3, 0,019 г, 0,298 ммоль), растворенный в воде (0,2 мл) по каплям при 0°C. После перемешивания реакционного раствора в течение 2 ч при комнатной температуре полученный таким образом 2,6-дифтор-бензоилазид разбавляли этилацетатом и затем промывали водой. Органический слой сушили над безводным сульфатом магния, диспергировали в ТГФ (1 мл), добавляли ТГФ (4 мл), содержащий 4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (соединение D, 0,04 г, 0,124 ммоль) и перемешивали в течение 4 ч при 90°C. После завершения реакции растворитель концентрировали при пониженном давлении и затем очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,025 г, выход: 42%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 14,45-14,36 (m, 1H), 9,40-9,36 (m, 2H), 8,42 (d, J=8,1 Гц, 1H), 8,33 (s, 1H), 7,62-7,57 (m, 2H), 7,48 (t, J=8,4 Гц, 1H), 7,37-7,26 (m, 2H), 7,20-6,82 (m, 3H), 4,40 (s, 2H), 2,29-2,19 (m, 3H)
ЖХ-МС [М+1]: 478,4
Пример 4: Получение 1-(2-хлор-6-фторфенил)-3-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-мочевины
2-хлор-6-фтор-бензойную кислоту (0,054 г, 0,31 ммоль) диспергировали в диэтиловом эфире (3 мл), медленно добавляли пентахлорид фосфора (PCl5, 0,074 г, 0,357 ммоль) и затем перемешивали в течение 1 часа. После завершения реакции органический растворитель концентрировали при пониженном давлении при температуре ниже комнатной, а затем реакционный раствор разбавляли путем добавления ацетона (2 мл). Затем медленно добавляли к реакционному раствору азид натрия (NaN3, 0,024 г, 0,372 ммоль), растворенный в воде (0,2 мл) по каплям при 0°C. После перемешивания реакционного раствора в течение 2 ч при комнатной температуре реакционный раствор разбавляли этилацетатом и промывали водой. Органический слой сушили над безводным сульфатом магния, диспергировали в ТГФ (1 мл), добавляли ТГФ (4 мл), содержащий 4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (соединение D, 0,05 г, 0,155 ммоль) и перемешивали в течение 3 ч при 90°C. После завершения реакции растворитель концентрировали при пониженном давлении и затем очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,029 г, выход: 42%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 14,45-14,35 (m, 1H), 9,40-9,35 (m, 2H), 8,42 (d, J=8,1 Гц, 1H), 8,33 (s, 1H), 7,63-7,58 (m, 2H), 7,47 (t, J=8,4 Гц, 1H), 7,41-7,26 (m, 4H), 7,07-6,82 (m, 1H), 4,40 (s, 2H), 2,30-2,20 (m, 3H)
ЖХ-МС [М+1]: 494,4
Пример 5: Получение 1-(2,6-бис-трифторметилфенил)-3-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-мочевины
2,6-бис-трифторметил-бензойную кислоту (0,088 г, 0,31 ммоль) диспергировали в диэтиловом эфире (3 мл), медленно добавляли пентахлорид фосфора (PCl5, 0,068 г, 0,326 ммоль) и затем перемешивали в течение 1 часа. После завершения реакции органический растворитель концентрировали при пониженном давлении при температуре ниже комнатной, а затем реакционный раствор разбавляли путем добавления ацетона (2 мл). Затем медленно добавляли к реакционному раствору азид натрия (NaN3, 0,024 г, 0,372 ммоль), растворенный в воде (0,2 мл) по каплям при 0°C. После перемешивания реакционного раствора в течение 2 ч при комнатной температуре полученный таким образом 2,6-бис-трифторметилбензоилазид разбавляли этилацетатом и затем промывали водой. Органический слой сушили над безводным сульфатом магния, диспергировали в ТГФ (1 мл), добавляли ТГФ (4 мл), содержащий 4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (соединение D, 0,05 г, 0,155 ммоль) и перемешивали в течение 3 ч при 90°C. После завершения реакции растворитель концентрировали при пониженном давлении и затем очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,018 г, выход: 20%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 8,40 (d, J=8,4 Гц, 1H), 8,09-8,06 (m, 2H), 7,76 (t, J=8,1 Гц, 1H), 7,63 (d, J=8,1 Гц, 1H), 7,54 (d, J=12,9 Гц, 1H), 7,38 (t, J=8,4 Гц, 1H), 7,24 (d/d, J=8,4 Гц, 1H), 6,94 (s, 1H), 4,43 (s, 2H), 2,33 (s, 3H)
ЖХ-МС [М+1]: 578,4
Пример 6: Получение 1-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-3-(2-фтор-6-трифторметилфенил)-мочевины
2-фтор-6-трифторметилбензойную кислоту (0,058 г, 0,279 ммоль) диспергировали в диэтиловом эфире (3 мл), медленно добавляли пентахлорид фосфора (PCl5, 0,064 г, 0,307 ммоль) и затем перемешивали в течение 1 часа. После завершения реакции органический растворитель концентрировали при пониженном давлении при температуре ниже комнатной, а затем реакционный раствор разбавляли путем добавления ацетона (2 мл). Затем медленно добавляли к реакционному раствору азид натрия (NaN3, 0,024 г, 0,363 ммоль), растворенный в воде (0,2 мл) по каплям при 0°C. После перемешивания реакционного раствора в течение 2 ч при комнатной температуре реакционный раствор разбавляли этилацетатом и затем промывали водой. Органический слой сушили над безводным сульфатом магния, диспергировали в ТГФ (1 мл) и затем помещали в колбу, содержащую 4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (соединение D, 0,045 г, 0,14 ммоль), растворенный в ТГФ (4 мл) с последующим перемешиванием в течение 4 ч при 90°C. После завершения реакции растворитель концентрировали при пониженном давлении и затем очищали путем колоночной хроматографии на силикагеле (метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,023 г, выход: 32%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 8,41-8,39 (m, 1H), 7,64-7,50 (m, 5H), 7,39 (t, J=8,4 Гц, 1H), 7,25 (m, J=8,4 Гц, 1H), 6,94 (s, 1H), 4,43 (s, 2H), 2,33 (s, 3H)
ЖХ-МС [М+1]: 528,4
Пример 7: Получение 1-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-3-(2,4,6-трифторфенил)-мочевины
2,4,6-трифторбензойную кислоту (0,08 г, 0,45 ммоль) диспергировали в диэтиловом эфире (5,7 мл), медленно добавляли пентахлорид фосфора (PCl5, 0,11 г, 0,52 ммоль) и затем перемешивали в течение 1 часа. После завершения реакции органический растворитель концентрировали при пониженном давлении при температуре ниже комнатной, а затем реакционный раствор разбавляли путем добавления ацетона (3,8 мл). Затем медленно добавляли к реакционному раствору азид натрия (NaN3, 0,035 г, 0,545 ммоль), растворенный в воде (0,28 мл) по каплям при 0°C. После перемешивания в течение 2 ч при комнатной температуре полученный таким образом 2,4,6-трифторбензоилазид разбавляли этилацетатом и затем промывали водой. Органический слой сушили над безводным сульфатом магния, диспергировали в ТГФ (2 мл), добавляли ТГФ (7,5 мл), содержащий 4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (соединение D, 0,073 г, 0,23 ммоль) и перемешивали в течение 3 ч при 90°C. После завершения реакции растворитель концентрировали при пониженном давлении и затем очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,026 г, выход: 23%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 14,46-14,37 (m, 1H), 9,47-9,45 (br m, 1H), 9,37 (s, 1H), 8,45 (d, J=1,8 Гц, 1H), 8,30-8,27 (br m, 1H), 7,63-7,46 (m, 3H), 7,31-7,26 (m, 3H), 7,09-6,84 (m, 1H), 4.42 (s, 2H), 2,31-2,21 (m, 3H)
ЖХ-МС [M+1]: 496,3
Пример 8: Получение 1-(2,6-дифторфенил)-3-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-1-метил-мочевины
4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (соединение D, 0,1 г, 0,31 ммоль) диспергировали в смешанном растворе ДМФ (0,8 мл) и ТГФ (4 мл) и затем добавляли пиридин (0,05 мл) и 4-нитрофениловый эфир хлормуравьиной кислоты (0,07 г, 0,36 ммоль) с последующим перемешиванием в течение 4 часов. После завершения реакции добавляли к полученной смеси н-гексан (3 мл) с последующим перемешиванием в течение 30 минут. Полученное таким образом твердое вещество промывали смешанным растворителем н-гексан : ТГФ = 1:1 (12 мл), фильтровали и затем сушили. Высушенное соединение диспергировали в ДМФ (4 мл), добавляли 2,6-дифторметиланилин (0,294 г, 2,05 ммоль) и затем перемешивали в течение 12 часов при 100°C. Реакционный раствор охлаждали при комнатной температуры, разбавляли этилацетатом, содержащим 5% метанола, и промывали насыщенным водным раствором NaHCO3 и водой. Органический слой и сушили над безводным сульфатом магния, фильтровали и концентрировали. Органический слой очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,018 г, выход: 18%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 14,45-14,36 (m, 1H), 9,36 (s, 1H), 8,91 (s, 1H), 8,42 (d, J=8,1 Гц, 1H), 7,61-7,34 (m, 5H), 7,26-7,21 (m, 2H), 7,07-6,82 (m, 1H), 4,40 (s, 2H), 3,20 (s, 3H), 2,30-2,20 (m, 3H)
ЖХ-МС [М+1]: 492,4
Пример 9: Получение 1-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-3-пентафторфенил-мочевины
Пентафторметил-бензойную кислоту (0,066 г, 0,31 ммоль) диспергировали в диэтиловом эфире (3 мл), медленно добавляли пентахлорид фосфора (PCl5, 0,071 г, 0,341 ммоль) и затем перемешивали в течение 40 минут. После завершения реакции органический растворитель концентрировали при пониженном давлении при температуре ниже комнатной, а затем реакционный раствор разбавляли путем добавления ацетона (3 мл). Затем медленно добавляли к реакционному раствору азид натрия (NaN3, 0,026 г, 0,403 ммоль), растворенный в воде (0,2 мл) по каплям при 0°C. После перемешивания реакционного раствора в течение 1 ч при комнатной температуре реакционный раствор разбавляли этилацетатом и затем промывали водой. Органический слой сушили над безводным сульфатом магния, диспергировали в ТГФ (1 мл) и затем помещали в колбу, содержащую 4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (соединение D, 0,050 г, 0,155 ммоль), растворенный в ТГФ (3 мл) с последующим перемешиванием в течение 3 ч при 90°C. После завершения реакции растворитель концентрировали при пониженном давлении и затем очищали путем колоночной хроматографии на силикагеле (метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,023 г, выход: 28%)
1H-ЯМР спектр (300 МГц, ДМСО-d6): 14,45-14,36 (m, 1H), 9,60 (s, 1H), 9,36 (s, 1H), 8,83 (br s, 1H), 8,43 (d, J=8,1 Гц, 1H), 7,62-7,57 (m, 2H), 7,50 (t, J=8,4Hz, 1H), 7,30 (d, J=8,4 Гц, 1H), 7.08-6,83 (m, 1H), 4,40 (s, 2H), 2,30-2,20 (m, 3H)
ЖХ-МС [M+1]: 532,4
Пример 10: Получение 1-(2,5-дифторфенил)-3-{3-фтор-4-(7-(5-метил-1H-имидазол-2-ил)-1-оксоизоиндолин-4-ил)фенил)мочевины
2,5-дифторбензойную кислоту (0,05 г, 0,32 ммоль) диспергировали в ТГФ (4 мл) и затем добавляли к реакционной смеси триэтиламин (0,088 мл, 0,63 ммоль) и дифенилфосфорилазид (ДФФА, 0,08 мл, 0,36 ммоль) с последующим перемешиванием в течение 2 часов при комнатной температуре. Убедившись, что 2,5-дифторбензоилазид был сформирован, добавляли Соединение D (0,051 г, 0,16 ммоль) с последующим перемешиванием в течение 4 часов при 90°C. После завершения реакции реакционный раствор разбавляли этилацетатом, содержащим 5% метанола, а затем промывали насыщенным водным раствором NaHCO3. Затем органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный таким образом концентрат очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,026 г, выход: 34%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 14,47-14,37 (m, 1H), 9,58 (s, 1H), 9,37 (s, 1H), 8,96 (s, 1H), 8,45 (d, J=8,4 Гц, 1H), 8,06-7,99 (m, 1H), 7,67-7,49 (m, 3H), 7,36-7,09 (m, 2H), 6,89-6,84 (m, 1H), 4,43 (s, 2H), 2,31-2,22 (m, 3H)
ЖХ-МС [М+1]: 478,4
Пример 11: Получение 1-(2,4-дифторфенил)-3-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-мочевины
2,4-дифторбензойную кислоту (0,04 г, 0,248 ммоль) диспергировали в ТГФ (3 мл) и затем добавляли к реакционной смеси триэтиламин (0,069 мл, 0,496 ммоль) и дифенилфосфорилазид (ДФФА, 0,075 мл, 0,273 ммоль) с последующим перемешиванием в течение 2 часов при комнатной температуре. К смеси добавляли 4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (Соединение D, 0,040 г, 0,124 ммоль) с последующим перемешиванием в течение 4 часов при 90°C. После завершения реакции реакционный раствор разбавляли этилацетатом, содержащим 5% метанола, а затем промывали водой и насыщенным водным раствором NaHCO3. Затем органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный таким образом концентрат очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,024 г, выход: 42%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 14,45-14,36 (m, 1H), 9,39-9,35 (m, 2H), 8,64 (s, 1H), 8,43 (d, J=8,1 Гц, 1H), 8,09-8,00 (m, 1H), 7,65-7,59 (m, 2H), 7,49 (t, J=8,4 Гц, 1H), 7,35-7,21 (m, 2H), 7,08-6,83 (m, 2H), 4,41 (s, 2H), 2,30-2,20 (m, 3H)
ЖХ-МС [М+1]: 478,4
Пример 12: Получение 1-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-3-(2,3,6-трифторфенил)-мочевины
2,3,6-трифторбензойную кислоту (0,044 г, 0,248 ммоль) диспергировали в диэтиловом эфире (4 мл), медленно добавляли пентахлорид фосфора (PCl5, 0,057 г, 0,273 ммоль) и затем перемешивали в течение 40 минут. После завершения реакции органический растворитель концентрировали при пониженном давлении при температуре ниже комнатной, а затем реакционный раствор разбавляли путем добавления ацетона (3 мл). Затем медленно добавляли к реакционному раствору азид натрия (NaN3, 0,021 г, 0,322 ммоль) в воде (0,2 мл) по каплям при 0°C. После перемешивания реакционного раствора в течение 1 ч при комнатной температуре реакционный раствор разбавляли этилацетатом и затем промывали водой. Органический слой сушили над безводным сульфатом магния, диспергировали в ТГФ (1 мл) и затем помещали в колбу, содержащую 4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (соединение D, 0,040 г, 0,124 ммоль), растворенный в ТГФ (3 мл) с последующим перемешиванием в течение 6 ч при 90°C. После завершения реакции растворитель концентрировали при пониженном давлении и затем очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,022 г, выход: 36%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 14,45-14,35 (m, 1H), 9,51 (br s, 1H), 9,35 (s, 1H), 8,62 (br s, 1H), 8,43 (d, J=8.1 Hz, 1H), 7,62-7,57 (m, 2H), 7,48 (t, J=8,4 Гц, 1H), 7,44-7,34 (m, 1H), 7,31-7.20 (m, 2H), 7,07-6,83 (m, 1H), 4,40 (s, 2H), 2,30-2,20 (m, 3H)
ЖХ-МС [M+1]: 496,4
Пример 13: Получение 1-(3,5-дифторфенил)-3-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-мочевины
3,5-дифторбензойную кислоту (0,04 г, 0,248 ммоль) диспергировали в ТГФ (4 мл) и затем добавляли к реакционной смеси триэтиламин (0,069 мл, 0,496 ммоль) и дифенилфосфорилазид (ДФФА, 0,075 мл, 0,273 ммоль) с последующим перемешиванием в течение 1 часа при комнатной температуре. К реакционному раствору добавляли 4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (Соединение D, 0,040 г, 0,124 ммоль) с последующим перемешиванием в течение 4 часов при 90°C. Реакционный раствор охлаждали до комнатной температуры, разбавляли этилацетатом и затем промывали водой и насыщенным водным раствором NaHCO3. Органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный таким образом концентрат очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,032 г, выход: 55%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 14,45-14,36 (m, 1H), 9,50 (s, 2H), 9,36 (s, 1H), 8,43 (d, J=8.1 Гц, 1H), 7,64-7,60 (m, 2H), 7,50 (t, J=8.4 Гц, 1H), 7,30-7,20 (m, 3H), 7,08-6,77 (m, 2H), 4,41 (s, 2H), 2,30-2,20 (m, 3H)
ЖХ-МС [М+1]: 478,3
Пример 14: Получение 1-(3,4-дифторфенил)-3-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-мочевины
3,4-дифторбензойную кислоту (0,05 г, 0,32 ммоль) диспергировали в ТГФ (4 мл) и затем добавляли к реакционной смеси триэтиламин (0,088 мл, 0,63 ммоль) и дифенилфосфорилазид (ДФФА, 0,08 мл, 0,36 ммоль) с последующим перемешиванием в течение 2 часов при комнатной температуре. Убедившись, что 3,4-дифторбензоилазид был сформирован, добавляли 4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (Соединение D, 0,051 г, 0,16 ммоль) с последующим перемешиванием в течение 4 часов при 90°C. После завершения реакции реакционный раствор разбавляли этилацетатом, содержащим 5% метанола, а затем промывали насыщенным водным раствором NaHCO3. Затем органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный таким образом концентрат очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,035 г, выход: 46%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 14,40 (br s, 1H), 9,37 (s, 1H), 9,13-9,03 (m, 2H), 8,44 (d, J=8,1 Гц, 1H), 7,71-7,47 (m, 4H), 7,41-7,27 (m, 3H), 7,18-7,16 (m, 1H), 7,00 (s, 1H), 4,43 (s, 2H), 2,26 (s, 3H)
ЖХ-МС [M+1]: 478,4
Пример 15: Получение 1-(4-циано-3-фторфенил)-3-{3-фтор-4-(7-(5-метил-1H-имидазол-2-ил)-1-оксоизоиндолин-4-ил)фенил)мочевины
4-циано-3-фторбензойную кислоту (0,08 г, 0,48 ммоль) диспергировали в ТГФ (6,1 мл) добавляли триэтиламин (0,14 мл, 0,97 ммоль) и дифенилфосфорилазид (ДФФА, 0,12 мл, 0,56 ммоль) и затем перемешивали в течение 2 часов при комнатной температуре. Убедившись, что 4-циано-3-фторбензоилазид был сформирован, добавляли 4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (Соединение D, 0,078 г, 0,24 ммоль) с последующим перемешиванием в течение 4 часов при 90°C. После завершения реакции реакционный раствор разбавляли этилацетатом, содержащим 5% метанола, а затем промывали насыщенным водным раствором NaHCO3. Затем органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный таким образом концентрат очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,012 г, выход: 10%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 14,40 (br s, 1H), 9,89 (s, 1H), 9,64 (s, 1H), 9,37 (s, 1H), 8,44 (d, J=8,7 Гц, 1H), 7,94-7,15 (m, 7H), 7,01-6,99 (m, 1H), 4,43 (s, 2H), 2,26 (s, 3H)
ЖХ-МС [M+1]: 485,4
Пример 16: Получение 1-(4-хлор-3-(трифторфметил)фенил)-3-{3-фтор-4-(7-(5-метил-1H-имидазол-2-ил)-1-оксоизоиндолин-4-ил)фенил)мочевины
4-хлор-3-трифторметилбензойную кислоту (0,08 г, 0,35 ммоль) диспергировали в ТГФ (4,5 мл) и затем добавляли к реакционной смеси триэтиламин (0,1 мл, 0,71 ммоль) и дифенилфосфорилазид (ДФФА, 0,09 мл, 0,41 ммоль) с последующим перемешиванием в течение 2 часов при комнатной температуре. Убедившись, что 4-хлор-3-трифторметилбензоилазид был сформирован, добавляли 4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (Соединение D, 0,057 г, 0,18 ммоль) с по следующим перемешиванием в течение 4 часов при 90°C. После завершения реакции реакционный раствор разбавляли этилацетатом, содержащим 5% метанола, а затем промывали насыщенным водным раствором NaHCO3. Затем органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный таким образом концентрат очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,012 г, выход: 10%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 14,47-14,37 (m, 1H), 9,36-9,32 (m, 2H), 9,25 (s, 1H), 8,45 (d, J=8,1 Гц, 1H), 8,12-8,05 (m, 1H), 7,71-7,61 (m, 4H), 7,54-7,48 (m, 1H), 7,33-7,29 (m, 1H), 7,09-6,85 (m, 1H), 4,42 (s, 2H), 2,32-2,22 (m, 3H)
ЖХ-МС [М+1]: 544,3
Пример 17: Получение 1-(3-хлор-2,6-дифторфенил)-3-{3-фтор-4-(7-(5-метил-1H-имидазол-2-ил)-1-оксоизоиндолин-4-ил)фенил)мочевины
3-хлор-2,6-дифторбензойную кислоту (0,08 г, 0,41 ммоль) диспергировали в диэтиловом эфире (5,2 мл), медленно добавляли пентахлорид фосфора (PCl5, 0,099 г, 0,48 ммоль) и затем перемешивали в течение 1 часа. После завершения реакции органический растворитель концентрировали при пониженном давлении при температуре ниже комнатной, а затем реакционный раствор разбавляли путем добавления ацетона (3,5 мл). Затем медленно добавляли к реакционному раствору азид натрия (NaN3, 0,032 г, 0,50 ммоль), растворенный в воде (0,25 мл) по каплям при 0°C. После перемешивания реакционного раствора в течение 2 ч при комнатной температуре полученный таким образом 3-хлор-2,6-дифторбензоилазид разбавляли этилацетатом и с последующим промыванием водой. Органический слой сушили над безводным сульфатом магния, диспергировали в ТГФ (1,6 мл), добавляли ТГФ (1,6 мл), содержащий 4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (соединение D, 0,067 г, 0,21 ммоль) и перемешивали в течение 3 ч при 90°C. После завершения реакции растворитель концентрировали и затем очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,017 г, выход: 16%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 14,47-14,38 (m, 1H), 9,49-9,39 (m, 2H), 8,53 (s, 1H), 8,44 (d, J=8,1 Гц, 1H), 7,63-7,47 (m, 4H), 7,31-7,24 (m, 2H), 7,09-6,84 (m, 1H), 4,42 (s, 2H), 2,31-2,21 (m, 3H)
ЖХ-МС [М+1]: 512,3
Пример 18: Получение 1-(2-хлор-3,6-дифторфенил)-3-{3-фтор-4-(7-(5-метил-1H-имидазол-2-ил)-1-оксоизоиндолин-4-ил)фенил)мочевины
2-хлор-3,6-дифторбензойную кислоту (0,08 г, 0,41 ммоль) диспергировали в диэтиловом эфире (5,2 мл), медленно добавляли пентахлорид фосфора (PCl5, 0,099 г, 0,48 ммоль) и затем перемешивали в течение 1 часа. После завершения реакции органический растворитель концентрировали при пониженном давлении при температуре ниже комнатной, а затем реакционный раствор разбавляли путем добавления ацетона (3,5 мл). Затем медленно добавляли к реакционному раствору азид натрия (NaN3, 0,032 г, 0,50 ммоль), растворенный в воде (0,25 мл) по каплям при 0°C. После перемешивания реакционного раствора в течение 2 ч при комнатной температуре полученный таким образом 2-хлор-3,6-дифторбензоилазид разбавляли этилацетатом и с последующим промыванием водой. Органический слой сушили над безводным сульфатом магния, диспергировали в ТГФ (1,6 мл), добавляли ТГФ (7 мл), содержащий 4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (соединение D, 0,067 г, 0,21 ммоль) и перемешивали в течение 3 ч при 90°C. После завершения реакции растворитель концентрировали при пониженном давлении и затем очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,038 г, выход: 36%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 14,46-14,37 (m, 1H), 9,51 (s, 1H), 9,37 (s, 1H), 8,58 (s, 1H), 8,44 (d, J=8,1 Гц, 1H), 7,63-7,59 (m, 2H), 7,52-7,29 (m, 4H), 7,09-6,84 (m, 1H), 4,42 (s, 2H), 2,31-2,21 (m, 3H)
ЖХ-МС [М+1]: 512,3
Пример 19: Получение 1-(4-хлор-2,6-дифторфенил)-3-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-мочевины
4-хлор-2,6-дифторбензойную кислоту (0,060 г, 0,31 ммоль) диспергировали в диэтиловом эфире (4 мл), медленно добавляли пентахлорид фосфора (PCl5, 0,071 г, 0,341 ммоль) и затем перемешивали в течение 40 минут. После завершения реакции органический растворитель концентрировали при пониженном давлении при температуре ниже комнатной, а затем реакционный раствор разбавляли путем добавления ацетона (3 мл). Затем медленно добавляли к реакционному раствору азид натрия (NaN3, 0,026 г, 0,403 ммоль), растворенный в воде (0,2 мл) по каплям при 0°C. После перемешивания реакционного раствора в течение 1 ч при комнатной температуре реакционный раствор разбавляли этилацетатом и затем промывали водой. Органический слой сушили над безводным сульфатом магния, диспергировали в ТГФ (1 мл) и затем помещали в колбу, содержащую 4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (соединение D, 0,050 г, 0,155 ммоль), растворенный в ТГФ (3 мл) с последующим перемешиванием в течение 4 ч при 90°C. После завершения реакции растворитель концентрировали при пониженном давлении и затем очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,024 г, выход: 30%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 14,46-14,37 (m, 1H), 9,86 (s, 1H), 9,38 (s, 1H), 8,82 (s, 1H), 8,41 (d, J=8,1 Гц, 1H), 7,63-7,59 (m, 2H), 7,52-7,29 (m, 4H), 6,97 (s, 1H), 4,42 (s, 2H), 2,21 (s, 3H)
ЖХ-МС [М+1]: 512,3
Пример 20: Получение 1-{3-фтор-4-[7-(5-метил-1H-имидазол-2-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-фенил}-3-(2,3,5,6-тетрафторфенил)-мочевины
2,3,5,6-тетрафторбензойную кислоту (0,08 г, 0,41 ммоль) растворяли в диэтиловом эфире (5,2 мл), медленно добавляли пентахлорид фосфора (PCl5, 0,099 г, 0,48 ммоль) и затем перемешивали в течение 1 часа. После завершения реакции органический растворитель концентрировали при пониженном давлении при температуре ниже комнатной, а затем реакционный раствор разбавляли путем добавления ацетона (3,4 мл). Затем медленно добавляли к реакционному раствору азид натрия (NaN3, 0,032 г, 0,50 ммоль), растворенный в воде (0,25 мл) по каплям при 0°C. После перемешивания реакционного раствора в течение 2 ч при комнатной температуре полученный таким образом 2,3,5,6-бис-тетрафторметилбензоилазид разбавляли этилацетатом и затем промывали водой. Органический слой сушили над безводным сульфатом магния, диспергировали в ТГФ (1,6 мл), добавляли ТГФ (7 мл), содержащий 4-(4-амино-2-фторфенил)-7-(5-метил-1H-имидазол-2-ил)изоиндолин-1-он (соединение D, 0,066 г, 0,21 ммоль), и перемешивали в течение 3 ч при 90°C. После завершения реакции растворитель концентрировали при пониженном давлении и затем очищали путем колоночной хроматографии на силикагеле (элюент: метиленхлорид: метанол = 20:1) с получением указанного в заголовке соединения (0,014 г, выход: 13%).
1H-ЯМР спектр (300 МГц, ДМСО-d6): 8,45 (d, J=8,1 Гц, 1H), 7,69-7,61 (m, 2H), 7,48-7,33 (m, 3H), 7,00 (s, 1H), 4,48 (s, 2H), 2,38 (s, 3H)
ЖХ-МС [М+1]: 514,3
Соединения, полученные в примерах с 1 по 20, представлены следующей структурной формулой, как показано в таблице 1 ниже.
Количественное определение биологической активности соединений, полученных в соответствии с примерами, проводили следующим образом.
Оценка биологической активности соединений согласно настоящему изобретению может быть проведена любыми обычными способами, известными в данной области. Соответствующие способы испытаний известны в данной области техники. Следующие исследования представляют собой примеры анализа воздействия соединений согласно изобретению на различные киназы, не ограничиваясь ими. Соединения согласно настоящему изобретению демонстрируют свое действие по меньшей мере в одном из следующих исследований.
Экспериментальный пример 1: Анализ ингибирования активности BTK (метод ИФА)
Для того, чтобы оценить активность соединений по настоящему изобретению, для этого эксперимента в качестве ингибитора BTK применяли коммерчески доступную BTK (Promega). Более конкретно, ферментативную реакцию осуществляли путем смешивания 0,4 нМ фермента BTK, 40 мкМ пептидного субстрата биотин-S1 и 50 мкМ АТФ в реакционном буфере (15 мМ Трис-HCl (pH 7,5), 20 мМ MgCl2, 2 мМ MnC12, 2 мМ ДТТ, 0,1 мг/мл БСА). Смесь обрабатывали исследуемыми соединениями в заданных концентрациях и давали прореагировать в течение 20 минут при 30°C. После завершения реакции воздействие исследуемых соединений измеряли методом ИФА. В качестве контрольного значения использовали значение оптической плотности необработанного образца (контроль 100%). Активности ферментов BTK измеряли после обработки различными концентрациями исследуемых соединений и концентрацию исследуемых соединений, приводившую к 50% ингибированию фермента BTK по сравнению с контрольным образцом определяли как IC50 ингибитора BTK.
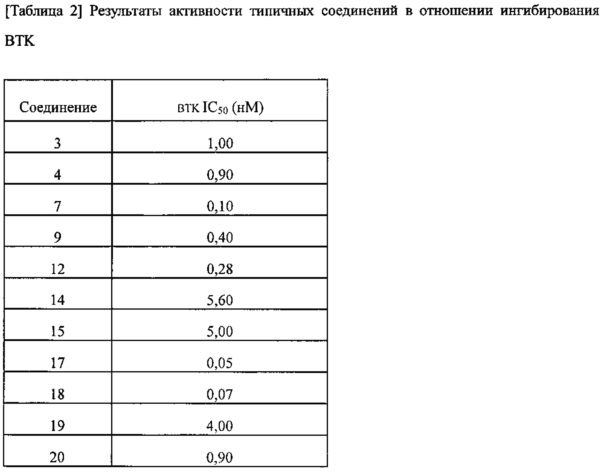
Исследовали активность в отношении ингибирования BTK нескольких случайно выбранных соединений из соединений согласно изобретению. Результаты представлены в Таблице 2 ниже.
Экспериментальный пример 2: Анализ выделения гистамина
По данным Каваками и др. ингибирование активности BTK в тучных клетках уменьшает выработку медиатора (например, гистамина) и липидного медиатора или выделение цитокина. (См. публикацию [J Immunol. 2000 Aug 1; 165 (3): 1210-9. Redundant and opposing functions of two tyrosine kinases, Btk and Lyn, in mast cell activation. Kawakami Y, Kitaura J, Satterthwaite AB, Kato RM, Asai K, Hartman SE, Maeda-Yamamoto M, Lowell CA, Rawlings DJ, Witte ON, Kawakami T]).
Анализы выделения гистамина проводили согласно способу, раскрытому в публикации FEBS Lett. 2002 Sep 11; 527 (1-3):274-8. Silencing of Bruton's tyrosine kinase (Btk) using short interfering RNA duplexes (siRNA). Heinonen JE, Smith CI, Nore BF, и количество гистамина измеряли путем иммуноферментного анализа.
Клеточную линию RBL-2H3, приобретенную у KCLB (Корейский банк клеточных линий), выращивали в минимальной эссенциальной среде Игла, модифицированной по способу Дульбекко с добавлением 10% (об/об) ФБС при 37°C в 5% CO2-инкубаторе в течение 72 часов. Клетки переносили в 96-луночные планшеты с плотностью 10000 клеток на лунку и культивировали при 37°C в 5% CO2-инкубаторе в течение 24 часов.
Клетки обрабатывали 500 нг/мл моноклональными антителами против ДНК-белкового комплекса (Sigma), и концентрациями 0,001, 0,01, 0,1, 1,0 и 10 мкМ исследуемого соединения в 100% (об/об) диметилсульфоксиде (ДМСО). Клетки, которые обрабатывали только 100% (об/об) ДМСО использовали в качестве контрольного образца. Обработанные образцы культивировали при 37°C в 5% CO2-инкубаторе в течение 24 часов. Затем измеряли выделение гистамина в соответствии с инструкциями производителя (комплект ИФА для анализа гистамина, Immunotech). Каждую лунку обрабатывали 50 мкл буфера выделения гистамина и давали прореагировать в течение 30 минут при 37°C. После завершения реакции 100 мкл образца из каждой лунки переносили в новый планшет и затем тщательно смешивали с 25 мкл ацетилирующего буфера и 25 мкл ацетилирующего реагента. 50 мкл полученного таким образом образца ацетилирования переносили в планшет, покрытый антителами, смешанными с 200 мкл конъюгата гистамин-щелочная фосфатаза, и затем давали прореагировать в течение 18 часов при 4°C. После завершения реакции образец удаляли из планшета, а затем три раза добавляли 200 мкл промывающего буфера для промывания. Добавляли 200 мкл субстрата и смесь оставляли реагировать при комнатной температуре в течение 30 минут. Реакцию останавливали добавлением 50 мкл останавливающего раствора и считывали оптическую плотность образцов при 406 нм с использованием Benchmark Plus (Biorad). Уровень выделения гистамина рассчитывали на основе оптической плотности исследуемой группы по отношению к оптической плотности контрольной группы. Значения EC50 (мкМ), при которых исследуемые соединения снижают выделение гистамина на 50%, определяли с помощью графической программы Microsoft Excel.
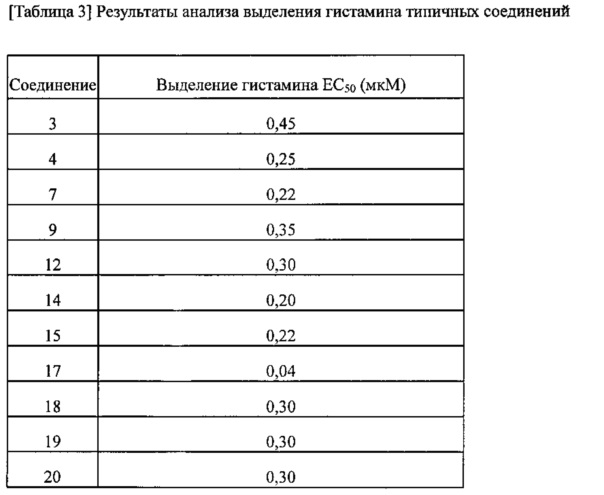
Для оценки эффективности соединений согласно изобретению в качестве противовоспалительного лекарственного средства проводили анализы выделения гистамина для некоторых случайным образом выбранных из них соединений. Значения EC50 соединений приведены в таблице 3. Результаты показывают, что соединения, полученные в примерах согласно настоящему изобретению, имеют превосходную эффективность.
Экспериментальный пример 3: MTS-анализ на основе антипролиферационного анализа
MTS-анализ проводили для оценки анти-пролиферативной активности соединений согласно изобретению путем ингибирования внеклеточно регулируемой киназы (Barltrop, J.A. et al., (1991) 5-(3-carboxymethoxyphenyl)-2-(4,5-dimethylthiazoly)-3-(4-sulfophenyl)tetrazolium, inner salt (MTS) and related analog of 3-(4,5-dimethylthiazolyl)-2,5,-diphenyltetrazolium bromide (MTT) reducing to purple water soluble formazans as cell-viability indicators. Bioorg. Med. Chem. Lett. 1, 611-4; Cory, A.H. et al., (1991) Use of an aqueous soluble tertrazolium/formazan assay for cell growth assays in culture. Cancer Comm. 3, 207-12).
Для исследования в соответствии с процедурой, приведенной ниже, применяли линии клеток лимфомы человека Jeko-1 (ATCC), Mino (ATCC), H9 (корейский банк клеточных линий) и SR (ATCC), и линии клеток лейкоза человека MV4-11 (ATCC), Molm-13 (DSMZ) и Ku812 (ATCC).
Клетки JeKo-1, Мино, H9, SR, MV4-11, Molm-13 и Ku812 переносили в 96-луночные планшеты, содержащие среду RPMI1640 (Gibco, Invitrogen) с добавлением 10% ФБС при плотности 10,000 клеток на лунку и затем инкубировали в течение 24 часов при 37°C и 5% CO2. Лунки обрабатывали концентрациями исследуемых соединений 0,2, 1, 5, 25 и 100 мкМ. Одну из лунок обрабатывали ДМСО в количестве 0,08 масс. %, что является тем же количеством, что и в исследуемых соединениях, и эту лунку использовали в качестве контрольной. Полученные клетки инкубировали в течение 48 часов.
MTS-анализы коммерчески доступны и включают Promega CellTiter 96® Водный нерадиоактивный анализ пролиферации клеток. MTS-анализы проводили для оценки жизнеспособности клеток в присутствии исследуемых соединений. В каждую лунку добавляли 20 мкл смешанного раствора 3-(4,5-диметилтиазол-2-ил)-5-(3-карбоксиметоксифенил)-2-(4-сульфофенил)-2H-тетразолия, внутреннюю соль ("MTS") и феназина метосульфат (ФМС) и затем инкубировали в течение 2 часов при 37°C. Затем считывали оптическую плотность образцов при 490 нм. Уровень антипролиферативной активности рассчитывали на основе оптической плотности исследуемой группы по отношению к оптической плотности контрольной группы. Рассчитывали значения EC50 (мкМ), при которых исследуемые соединения снижают рост раковых клеток на 50%.
Анализ антипролиферативной активности проводили с применением клеток лимфомы Jeko-1, Мино, H9 и SR для оценки эффективности соединений согласно изобретению в качестве противовоспалительного агента а также в качестве противоракового агента. Их значения EC50 представлены в таблице 4 ниже.
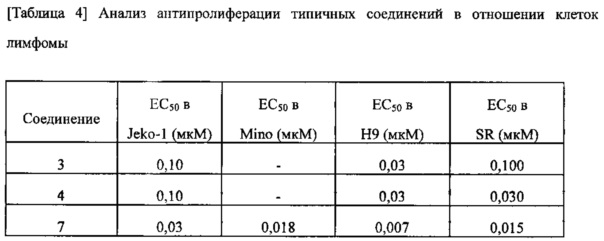
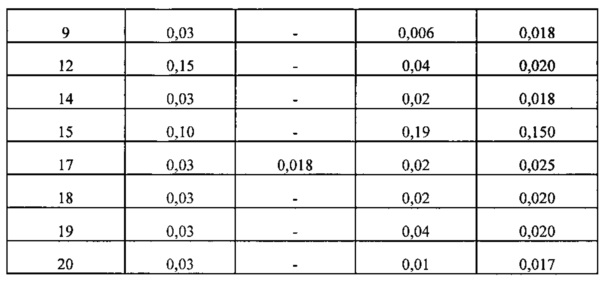
Кроме того, некоторые соединения согласно изобретению, которые показали отличную эффективность в отношении клеток лимфомы дополнительно подвергали анализу антипролиферации в отношении клеток лейкоза для подтверждения их отличной эффективности. Их значения EC50 представлены в таблице 5 ниже.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИЗОИНДОЛИНОНОВЫЕ ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ MDM2-P53, ОБЛАДАЮЩИЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2797295C1 |
| ИЗОИНДОЛИНОНОВЫЕ ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ MDM2-P53, ОБЛАДАЮЩИЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2794333C1 |
| ДИГИДРОНАФТИРИДИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ, ПОДХОДЯЩИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2012 |
|
RU2664055C2 |
| ДИГИДРОНАФТИРИДИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ, ПОДХОДЯЩИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2804468C2 |
| ИЗОИНДОЛИНОВЫЕ СОЕДИНЕНИЯ ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ РАКА | 2009 |
|
RU2527952C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2013 |
|
RU2627269C2 |
| ДИФЕНИЛЬНЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1997 |
|
RU2175319C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ IRAK И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2743360C2 |
| ЦИКЛОАЛКИЛНИТРИЛПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2655380C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, ИХ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2007 |
|
RU2474582C2 |
Изобретение относится к соединению формулы (I), его фармацевтически приемлемым солям, а также к фармацевтическим композициям, содержащим соединение в качестве активного ингредиента; и способу лечения, облегчения или предотвращения заболеваний с применением соединения. Технический результат: получены новые соединения, пригодные для лечения, облегчения или предотвращения заболеваний, вызванных аномальной или неконтролируемой активацией протеинкиназы, такой как BTK. 6 н. и 17 з.п. ф-лы, 5 табл., 20 пр.
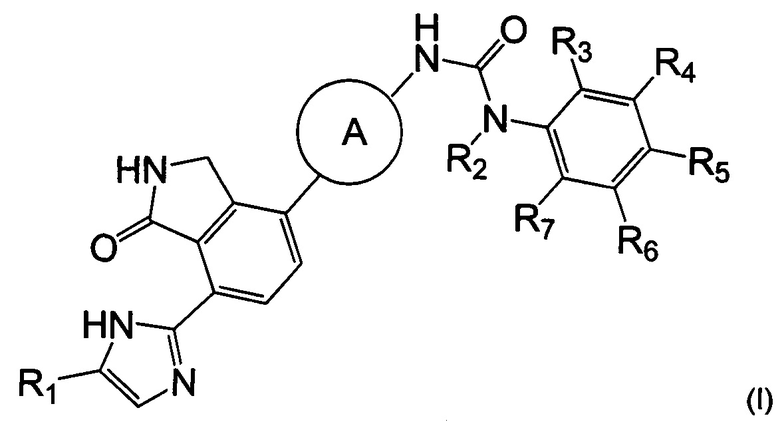
1. Соединение, выбранное из группы, состоящей из соединения формулы (I), или его фармацевтически приемлемая соль:
где
А представляет собой 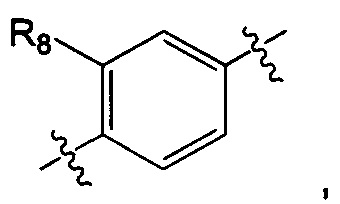 и R8 представляет собой водород или галоген;
и R8 представляет собой водород или галоген;
R1 представляет собой метил; и
R2 представляет собой водород или метил; и
каждый из R3-R7 независимо представляет собой водород, галоген, циано или C1-3галогеналкил, при условии, что по меньшей мере два из R3-R7 независимо друг от друга представляют собой галоген, циано или C1-3галогеналкил.
2. Соединение по п. 1, отличающееся тем, что каждый из R3-R7 независимо представляет собой водород, фтор, хлор, бром, йод, циано, дифторметил или трифторметил.
3. Соединение по п. 1, отличающееся тем, что каждый из R3-R7 независимо представляет собой водород, фтор, хлор, циано или трифторметил; и R8 представляет собой водород или фтор.
4. Соединение по п. 1, отличающееся тем, что соединение формулы (I) выбрано из группы, состоящей из:
1) 1-(2,6-дихлорфенил)-3-{3-фтор-4-[7-(5-метил-1Н-имидазол-2-ил)-1-оксо-2,3-дигидро-1Н-изоиндол-4-ил]-фенил}-мочевины;
3) 1-(2,6-дифторфенил)-3-{3-фтор-4-[7-(5-метил-1Н-имидазол-2-ил)-1-оксо-2,3-дигидро-1Н-изоиндол-4-ил]-фенил}-мочевины;
4) 1-(2-хлор-6-фторфенил)-3-{3-фтор-4-[7-(5-метил-1Н-имидазол-2-ил)-1-оксо-2,3-дигидро-1Н-изоиндол-4-ил]-фенил}-мочевины;
5) 1-(2,6-бис-трифторметилфенил)-3-{3-фтор-4-[7-(5-метил-1Н-имидазол-2-ил)-1-оксо-2,3-дигидро-1Н-изоиндол-4-ил]-фенил}-мочевины;
6) 1-{3-фтор-4-[7-(5-метил-1Н-имидазол-2-ил)-1-оксо-2,3-дигидро-1Н-изоиндол-4-ил]-фенил}-3-(2-фтор-6-трифторметилфенил)-мочевины;
7) 1-{3-фтор-4-[7-(5-метил-1Н-имидазол-2-ил)-1-оксо-2,3-дигидро-1Н-изоиндол-4-ил]-фенил}-3-(2,4,6-трифторфенил)-мочевины;
8) 1-(2,6-дифторфенил)-3-{3-фтор-4-[7-(5-метил-1Н-имидазол-2-ил)-1-оксо-2,3-дигидро-1Н-изоиндол-4-ил]-фенил}-1-метил-мочевины;
9) 1-{3-фтор-4-[7-(5-метил-1Н-имидазол-2-ил)-1-оксо-2,3-дигидро-1Н-изоиндол-4-ил]-фенил}-3-пентафторфенил-мочевины;
10) 1-(2,5-дифторфенил)-3-{3-фтор-4-(7-(5-метил-1Н-имидазол-2-ил)-1-оксоизоиндолин-4-ил)фенил)мочевины;
11) 1-(2,4-дифторфенил)-3-{3-фтор-4-[7-(5-метил-1Н-имидазол-2-ил)-1-оксо-2,3-дигидро-1Н-изоиндол-4-ил]-фенил}-мочевины;
12) 1-{3-фтор-4-[7-(5-метил-1Н-имидазол-2-ил)-1-оксо-2,3-дигидро-1Н-изоиндол-4-ил]-фенил}-3-(2,3,6-трифторфенил)-мочевины;
13) 1-(3,5-дифторфенил)-3-{3-фтор-4-[7-(5-метил-1Н-имидазол-2-ил)-1-оксо-2,3-дигидро-1Н-изоиндол-4-ил]-фенил}-мочевины;
14) 1-(3,4-дифторфенил)-3-{3-фтор-4-[7-(5-метил-1Н-имидазол-2-ил)-1-оксо-2,3-дигидро-1Н-изоиндол-4-ил]-фенил}-мочевины;
15) 1-(4-циано-3-фторфенил)-3-{3-фтор-4-(7-(5-метил-1Н-имидазол-2-ил)-1-оксоизоиндолин-4-ил)фенил)мочевины;
16) 1-(4-хлор-3-(трифторметил)фенил)-3-{3-фтор-4-(7-(5-метил-1Н-имидазол-2-ил)-1-оксоизоиндолин-4-ил)фенил)мочевины;
17) 1-(3-хлор-2,6-дифторфенил)-3-{3-фтор-4-(7-(5-метил-1Н-имидазол-2-ил)-1-оксоизоиндолин-4-ил)фенил)мочевины;
18) 1-(2-хлор-3,6-дифторфенил)-3-{3-фтор-4-(7-(5-метил-1Н-имидазол-2-ил)-1-оксоизоиндолин-4-ил)фенил)мочевины;
19) 1-(4-хлор-2,6-дифторфенил)-3-{3-фтор-4-[7-(5-метил-1Н-имидазол-2-ил)-1-оксо-2,3-дигидро-1Н-изоиндол-4-ил]-фенил}-мочевины;
20) 1-{3-фтор-4-[7-(5-метил-1Н-имидазол-2-ил)-1-оксо-2,3-дигидро-1Н-изоиндол-4-ил]-фенил}-3-(2,3,5,6-тетрафторфенил)-мочевины.
5. Соединение по п. 4, отличающееся тем, что соединение формулы (I) представляет собой 1-{3-фтор-4-[7-(5-метил-1Н-имидазол-2-ил)-1-оксо-2,3-дигидро-1Н-изоиндол-4-ил]-фенил}-3-(2,3,6-трифторфенил)-мочевину или его фармацевтически приемлемую соль.
6. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по п. 1 в качестве активного ингредиента для лечения, облегчения или предотвращения заболеваний, вызванных аномальной или неконтролируемой активацией протеинкиназы, такой как ВТК.
7. Фармацевтическая композиция по п. 6, отличающаяся тем, что протеинкиназа представляет собой ВТК.
8. Фармацевтическая композиция по п. 6, отличающаяся тем, что заболевания, вызванные аномальной или неконтролируемой активацией протеинкиназы, представляют собой раковое заболевание, воспаление, связанное с ревматоидным артритом и остеоартритом, астму, аллергию, атопический дерматит или псориаз.
9. Фармацевтическая композиция по п. 8, отличающаяся тем, что раковое заболевание представляет собой лимфому, лейкоз, рак крови, рак желудка, немелкоклеточный рак легкого, рак печени, рак толстой кишки, рак тонкой кишки, рак поджелудочной железы, рак головного мозга, рак кости, меланому, рак молочной железы, склерозирующий аденоз, рак матки, рак шейки матки, рак яичников, рак головы и шеи, рак пищевода, рак щитовидной железы, рак паращитовидной железы, рак почки, саркому, рак предстательной железы, рак уретры, рак мочевого пузыря, фиброаденому или глиобластому.
10. Фармацевтическая композиция по п. 6, отличающаяся тем, что заболевание, вызванное аномальной или неконтролируемой активацией протеинкиназы, представляет собой раковое заболевание.
11. Фармацевтическая композиция по п. 6, отличающаяся тем, что указанное соединение представляет собой 1-{3-фтор-4-[7-(5-метил-1Н-имидазол-2-ил)-1-оксо-2,3-дигидро-1Н-изоиндол-4-ил]-фенил}-3-(2,3,6-трифторфенил)-мочевину или его фармацевтически приемлемую соль.
12. Способ лечения, облегчения или предотвращения заболеваний, вызванных аномальной или неконтролируемой активацией протеинкиназы, такой как ВТК, у млекопитающего, включающий введение млекопитающему соединения по п. 1.
13. Способ лечения, облегчения или предотвращения ракового заболевания, включающий введение млекопитающему фармацевтической композиции, включающей соединение по п. 1 в качестве активного ингредиента.
14. Способ по п. 13, отличающийся тем, что указанное раковое заболевание представляет собой лимфому, лейкоз, рак крови, рак желудка, немелкоклеточный рак легкого, рак печени, рак толстой кишки, рак тонкой кишки, рак поджелудочной железы, рак головного мозга, рак кости, меланому, рак молочной железы, склерозирующий аденоз, рак матки, рак шейки матки, рак яичников, рак головы и шеи, рак пищевода, рак щитовидной железы, рак паращитовидной железы, рак почки, саркому, рак предстательной железы, рак уретры, рак мочевого пузыря, фиброаденому или глиобластому.
15. Способ по п. 14, отличающийся тем, что указанное раковое заболевание представляет собой лимфому.
16. Способ по п. 14, отличающийся тем, что указанное раковое заболевание представляет собой лейкоз.
17. Способ по пп. 13-16, отличающийся тем, что указанное соединение представляет собой 1-{3-фтор-4-[7-(5-метил-1Н-имидазол-2-ил)-1-оксо-2,3-дигидро-1Н-изоиндол-4-ил]-фенил}-3-(2,3,6-трифторфенил)-мочевину или его фармацевтически приемлемую соль.
18. Применение соединения по п. 1 для получения лекарственного средства для лечения, облегчения или предотвращения заболеваний, вызванных аномальной или неконтролируемой активацией протеинкиназы, такой как ВТК.
19. Применение фармацевтической композиции, содержащей соединение по п. 1 в качестве активного ингредиента, для лечения, облегчения или предотвращения ракового заболевания.
20. Применение по п. 19, отличающееся тем, указанное раковое заболевание представляет собой лимфому, лейкоз, рак крови, рак желудка, немелкоклеточный рак легкого, рак печени, рак толстой кишки, рак тонкой кишки, рак поджелудочной железы, рак головного мозга, рак кости, меланому, рак молочной железы, склерозирующий аденоз, рак матки, рак шейки матки, рак яичников, рак головы и шеи, рак пищевода, рак щитовидной железы, рак паращитовидной железы, рак почки, саркому, рак предстательной железы, рак уретры, рак мочевого пузыря, фиброаденому или глиобластому.
21. Применение по п. 20, отличающееся тем, что указанное раковое заболевание представляет собой лимфому.
22. Применение по п. 20, отличающееся тем, что указанное раковое заболевание представляет собой лейкоз.
23. Применение по пп. 19-22, отличающееся тем, что указанное соединение представляет собой 1-{3-фтор-4-[7-(5-метил-1Н-имидазол-2-ил)-1-оксо-2,3-дигидро-1Н-изоиндол-4-ил]-фенил}-3-(2,3,6-трифторфенил)-мочевину или его фармацевтически приемлемую соль.
| WO2012047017 A2, 12.04.2012 | |||
| WO2004108672 A1, 16.12.2004 | |||
| RU2007131274 A, 27.02.2009. |

