Данная заявка заявляет приоритет предварительной заявки США № 60/853135, зарегистрированной 19 октября 2006 г., которая введена посредством ссылки во всей своей полноте.
1. Область техники, к которой относится изобретение
Предложено несколько гетероарильных соединений, композиции, включающие эффективное количество одного или более таких соединений, и способы лечения или предупреждения рака, воспалительных патологических состояний, иммунологических патологических состояний, метаболических патологических состояний и патологических состояний, которые лечатся или предупреждаются путем ингибирования пути метаболизма киназы, включающие введение нуждающемуся в этом пациенту эффективного количества гетероарильного соединения.
2. Уровень техники
Связь между аномальным фосфорилированием белка и причиной или следствием заболеваний известна в течение свыше 20 лет. В соответствии с этим протеинкиназы становятся очень важной группой мишеней лекарственных средств. См. Cohen, Nature, 1:309-315 (2002). Различные ингибиторы протеинкиназы применяют клинически при лечении широкого разнообразия заболеваний, таких как рак и хронические воспалительные заболевания, включающие диабет и инсульт. См. Cohen, Eur. J. Biochem., 268:5001-5010 (2001).
Протеинкиназы представляют собой большое и разнообразное семейство ферментов, которые катализируют фосфорилирование белка и играют критическую роль в клеточной передаче сигнала. Протеинкиназы могут оказывать положительные или отрицательные регуляторные воздействия, в зависимости от целевого белка. Протеинкиназы вовлекаются в специфические пути передачи сигнала, которые регулируют клеточные функции, такие как, но не ограничиваются перечисленными, метаболизм, последовательность клеточного цикла, клеточная адгезия, сосудистая функция, апоптоз и ангиогенез. Нарушения клеточной передачи сигнала связано со многими заболеваниями, наиболее характерные из которых включают рак и диабет. Регулирование сигнальной трансдукции цитокинами и ассоциирование сигнальных молекул с протоонкогенами и генами подавления опухоли хорошо документировано. Подобным же образом продемонстрирована связь между диабетом и аналогичными патологическими состояниями и нерегулируемыми уровнями протеинкиназ. См. например, Sridhar et al., Pharmaceutical Research 17(11):1345-1353 (2000). Вирусные инфекции и относящиеся к ним патологические состояния также связаны с регулированием протеинкиназ. Park et al., Cell 101 (7):777-787 (2000).
Протеинкиназы можно разделить на широкие группы, основанные на идентичности аминокислот(ы), на которые они нацелены (серин/треонин, тирозин, лизин и гистидин). Например, тирозинкиназы включают в себя рецепторные тирозинкиназы (RTK), такие как факторы роста, и нерецепторные тирозинкиназы, такие как семейство src киназы. Имеются также протеинкиназы двойной специфичности, которые нацелены как на тирозин, так и на серин/треонин, такие как циклин-зависимые киназы (CDK) и митоген-активируемые протеинкиназы (МАРК).
Киназы IκB (IKK) представляют собой ключевые регуляторные молекулы передачи сигнала, координирующие активацию NF-κB. IKK-1 и IKK-2 представляют собой структурно уникальные киназы, содержащие N-концевой домен киназы с двойной петлей активации серина, домен лейцина типа «застежка-молния» и С-концевой домен спираль-петля-спираль и сериновый кластер. Показано, что многие иммунные и воспалительные медиаторы, включая TNFα, липополисахарид (LPS), IL-1, анти-CD28, CD40L, Fasl, вирусную инфекцию и окислительный стресс, приводят к активации NF-κB. Хотя рецепторные комплексы, которые осуществляют трансдукцию этих разнообразных стимулов, по-видимому, очень различные в их белковых компонентах, понятно, что каждый из этих случаев стимулирования приводит к активации IKK и NF-κB.
Данные позволяют предполагать, что небольшие молекулы ингибиторов IKK-2 обладают противовоспалительными свойствами. Catley et al. Mol. Pharmacol. 70:697-705 (2006). IKK-2 активируется в ответной реакции на несколько воспалительных стимулов и путей передачи сигнала, многие из которых играют важную роль в респираторном заболевании, включая IL-1β, LPS, TNFα, CD3/CD28 (антигенная презентация), CD40L, вирусную инфекцию и окислительный стресс. Убиквитарная экспрессия NF-κB вместе с его ответной реакцией на несколько стимулов означает, что почти все клеточные типы, присутствующие в легких, представляют собой потенциальную мишень для анти-NF-κB/IKK-2 терапии. Типы включают альвеолярный эпителий, тучные клетки, фибробласты, сосудистый эндотелий и инфильтратные лейкоциты; нейтрофилы, макрофаги, лимфоциты, эозинофилы и базофилы. Полагают, что ингибиторы IKK-2 демонстрируют широкую противовоспалительную активность путем ингибирования экспрессии генов, таких как циклооксигеназа-2 и 12-липоксигеназа (синтез медиаторов воспаления), транспортер пептида ТАР-1 (антигенный процессинг), инвариантные цепи МНС класса I H-2K и класса II (антигенная презентация), Е-селектин и молекула сосудистой клеточной адгезии (рекрутмент лейкоцита), интерлейкины-1, 2, 6, 8 (цитокины), RANTES, эотаксин, GM-CSF (хемокины) и супероксид дисмутаза и NADPH оксидоредуктаза хинона (реакционноспособные кислородные частицы).
mTOR (мишень рапамицина млекопитающего), которая также называется FRAP, RAFTI или SEPT, представляет собой 2549-аминокислотную Ser/Thr протеинкиназу, которая, как показано, представляет собой один из наиболее критических белков в пути PI3K/Akt, который регулирует клеточный рост и пролиферацию. Georgakis and Younes, 2006, Expert Rev. Anticancer Ther. 6(1):131-140. Вследствие того, что PI3K и Akt вовлекаются в регулирование нескольких клеточных функций, возможно, имеются токсичности, связанные с ингибированием этих киназ, что делает ингибирование mTOR более перспективным подходом. Id. В настоящее время три ингибитора mTOR находятся в клинических испытаниях для лечения рака. Имеются CCI-779 (рак почки, рак молочной железы, лимфома клеток головного мозга, мультиформа глиобластомы и метастатическая меланома), RAD001 (рефрактерные твердые опухоли, запущенные гематологические опухоли, GIST и запущенный немелкоклеточный рак легких, и АР23573 (твердые опухоли, гематологическое новообразование и саркома). Id. Преклинический успех этих соединений демонстрирует применимость ингибиторов mTOR при лечении рака и необходимость дополнительных соединений с активностью ингибитора mTOR.
Вследствие того, что протеинкиназы регулируют почти каждый клеточный процесс, включая метаболизм, клеточную пролиферацию и выживание клеток, они представляют собой привлекательные мишени для терапевтического воздействия при различных болезненных состояний. Например, подавление (контроль) клеточного цикла и ангиогенез, в котором протеинкиназы играют ведущую роль, представляют собой клеточные процессы, связанные с несколькими болезненными состояниями, такими как, но не ограничивающимися перечисленными, рак, воспалительные заболевания, аномальный ангиогенез и аналогичные им заболевания, атеросклероз, макулярная дегенерация, диабет, ожирение и боль.
Протеинкиназы становятся привлекательными мишенями для лечения раковых заболеваний. Fabbro et al., Pharmacology & Therapeutics 93:79-98 (2002). Предполагают, что вовлечение протеинкиназ в развитие новообразований у человека может происходить путем: (1) геномных перегруппировок (например, BCR-ABL при хроническом миелоидном лейкозе), (2) мутаций, приводящих к активности конститутивно активной киназы, такой как острый миелоидный лейкоз и желудочно-кишечные опухоли, (3) отсутствия регулирования активности киназы путем активации онкогенов или потери функций подавления опухоли, таких как при раковых заболеваниях с онкогенами RAS, (4) отсутствия регулирования активности киназы путем сверхэкспрессии, как в случае EGFR, и (5) эктопической экспрессии факторов роста, которые могут вносить вклад в развитие и поддержку неопластического фенотипа. Fabbro et al., Pharmacology & Therapeutics 93:79-98 (2002).
Разъяснение сложности путей метаболизма протеинкиназы и сложности взаимоотношения и взаимодействия среди и между различными протеинкиназами и путями метаболизма киназы подчеркивает важность развития фармацевтических агентов, способных действовать как модуляторы протеинкиназы, регуляторы или ингибиторы, которые оказывают полезное действие на несколько киназ или несколько путей метаболизма киназ. В соответствии с этим сохраняется необходимость в новых модуляторах киназы.
Цитирование или идентификация любой ссылки в разделе 2 данной заявки не должно быть истолковано как допущение, что ссылка представляет собой известный посредством ссылки уровень техники для данной заявки.
3. Сущность изобретения
Предложены соединения, имеющие следующую формулу (I)

и их фармацевтически приемлемые соли, полиморфы, клатраты, сольваты, гидраты, стереоизомеры и пролекарства, где R1, R3, R4, L, X, Y, A и В представляют собой, как раскрыто в описании.
Соединения формулы (I) или их фармацевтически приемлемые соли, клатраты, сольваты, гидраты, стереоизомеры или пролекарства (причем каждое обозначено как «гетероарильные соединения») применимы для лечения или предупреждения рака, воспалительных патологических состояний, иммунологических патологических состояний, метаболических патологических состояний и состояний, подвергаемых лечению или предупреждаемых ингибированием пути метаболизма киназы. В одном варианте осуществления путь метаболизма киназы представляет собой путь метаболизма IKK-2, mTOR, PI3K, SYK или TYK2. В другом варианте осуществления путь метаболизма киназы представляет собой путь метаболизма PI3Kα, PI3Kβ, PI3Kδ, Aurora, Abl, KDR, MLK1, CaMKIV, GSK3α, GSK3β, ATM, ATX или DNA-PK.
Далее предложены композиции, включающие эффективное количество гетероарильного соединения, и композиции, включающие эффективное количество гетероарильного соединения и фармацевтически приемлемый носитель или наполнитель. Композиции применяют для лечения или предупреждения рака, воспалительных патологических состояний, иммунологических патологических состояний, метаболических патологических состояний и патологических состояний, которые лечатся или предупреждаются ингибированием пути метаболизма киназы, в одном варианте осуществления пути метаболизма IKK-2, mTOR, PI3K, SYK или TYK2.
Далее предложены способы для лечения или предупреждения рака, воспалительных патологических состояний, иммунологических патологических состояний, метаболических патологических состояний и патологических состояний, подвергаемых лечению или предупреждаемых ингибированием пути метаболизма киназы, в одном варианте осуществления пути метаболизма IKK-2, mTOR, PI3K, SYK или TYK2, включающие в себя введение пациенту, нуждающемуся в лечении или предупреждении, эффективного количества гетероарильного соединения.
Данные варианты осуществления могут быть понятны более полно путем ссылки на подробное описание и примеры, которые предназначены для иллюстрации неограничивающих вариантов осуществления.
4. ПОДРОБНОЕ ОПИСАНИЕ
4.1 ОПРЕДЕЛЕНИЯ
«С1-8алкильная» группа представляет собой нециклический углеводород с цепью нормального или разветвленного строения, имеющий от 1 до 8 углеродных атомов. Репрезентативные -(С1-8)алкилы включают -метил, -этил, -н-пропил, -н-бутил, -н-пентил, -н-гексил, -н-гептил и -н-октил, в то время как насыщенные алкилы разветвленного строения включают -изопропил, -втор-бутил, -изобутил, -трет-бутил, -изопентил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и тому подобное. -(С1-8Алкильная) группа может быть замещенной или незамещенной. Например, С1-8алкильная группа может быть замещена фенилом с образованием бензильной группы.
«С2-8алкенильная» группа представляет собой нециклический углеводород с цепью нормального или разветвленного строения, имеющий от 2 до 8 углеродных атомов и включающий, по меньшей мере, одну углерод-углеродную двойную связь. Репрезентативные (С2-С8)алкенилы с цепью нормального или разветвленного строения включают -винил, -аллил, -1-бутенил, -2-бутенил, -изобутенил, -1-пентенил, -2-пентенил, -3-метил-1-бутенил, -2-метил-2-бутенил, -2,3-диметил-2-бутенил, -1-гексенил, -2-гексенил, -3-гексенил, -1-гептенил, -2-гептенил, -3-гептенил, -1-октенил, -2-октенил, -3-октенил и тому подобное. Двойная связь алкенильной группы может быть неконъюгированной или конъюгированной с другой ненасыщенной группой. Алкенильная группа может быть незамещенной или замещенной.
«С2-8алкинильная» группа представляет собой нециклический углеводород с цепью нормального или разветвленного строения, имеющий от 2 до 8 углеродных атомов и включающий по меньшей мере одну углерод-углеродную тройную связь. Репрезентативные -(С2-С8)алкинилы с цепью нормального и разветвленного строения включают -ацетиленил, -пропинил, -1-бутинил, -2-бутинил, -1-пентинил, -2-пентинил, -3-метил-1-бутинил, -4-пентинил, -1-гексинил, -2-гексинил, -5-гексинил, -1-гептинил, -2-гептинил, -6-гептинил, -1-октинил, -2-октинил, -7-октинил и тому подобное. Алкинильная группа может быть незамещенной или замещенной.
Термины «галоген» и «галогено» означают фтор, хлор, бром и йод.
«Арильная» группа представляет собой ненасыщенную ароматическую карбоциклическую группу из 6-14 углеродных атомов, имеющую одно кольцо (например, фенил) или несколько конденсированных колец (например, нафтил или антрил). Конкретные арилы включают фенил, бифенил, нафтил и тому подобное. Арильная группа может быть замещенной или незамещенной.
«Гетероарильная» группа представляет собой арильную кольцевую систему, имеющую от одного до четырех гетероатомов (например, О, S или N) в качестве атомов кольца в гетероароматической кольцевой системе, где остальные атомы представляют собой атомы улерода. Подходящие гетероатомы включают кислород, серу и азот. В некоторых вариантах осуществления гетероциклическая кольцевая система представляет собой моноциклическую или бициклическую. Неограничивающие примеры включают ароматические группы, выбранные из следующих групп:

где Q представляет собой СН2, СН=СН, О, S или NH. Следующие репрезентативные примеры гетероарильных групп включают, но не ограничиваются перечисленными, бензофуранил, бензотиенил, индолил, бензопиразолил, кумаринил, фуранил, изотиазолил, имидазолил, изоксазолил, тиазолил, триазолил, тетразолил, тиофенил, пиримидинил, изохинолинил, хинолинил, пиридинил, пирролил, пиразолил, 1Н-индолил, 1Н-индазолил, бензо[d]тиазолил и пиразинил. Следующие репрезентативные примеры гетероарильных групп включают группы предложенных в описании соединений. Гетероарилы могут быть связаны у любого кольцевого атома (например, у любого углеродного атома или гетероатома гетероарильного кольца). Гетероарильная группа может быть замещенной или незамещенной. В одном варианте осуществления гетероарильная группа представляет собой С3-10гетероарильную группу.
«Циклоалкильная» группа представляет собой насыщенное или ненасыщенное неароматическое карбоциклическое кольцо. Репрезентативные циклоалкильные группы включают, но не ограничиваются перечисленными, циклопропил, циклобутил, циклопентил, циклопентадиенил, циклогексил, циклогексенил, 1,3-циклогексадиенил, 1,4-циклогексадиенил, циклогептил, 1,3-циклогептадиенил, 1,3,5-циклогептатриенил, циклооктил и циклооктадиенил. Циклоалкильная группа может быть замещенной или незамещенной. В одном варианте осуществления циклоалкильная группа представляет собой С3-8циклоалкильную группу.
«Гетероциклоалкильная» группа представляет собой неароматический циклоалкил, в котором от одного до четырех кольцевых углеродных атомов независимо заменены гетероатомом из группы, состоящей из О, S и N. Репрезентативные примеры гетероциклоалкильной группы включают, но не ограничиваются перечисленными, морфолинил, пирролил, пирролидинил, тиенил, фуранил, тиазолил, имидазолил, пиразолил, триазолил, пиперазинил, изотиазолил, изоксазолил, (1,4)-диоксан, (1,3)-диоксолан, 4,5-дигидро-1Н-имидазолил и тетразолил. Гетероциклоалкилы также могут быть связаны у любого кольцевого атома (например, у любого углеродного атома или гетероатома гетероарильного кольца). Гетероциклоалкильная группа может быть замещенной или незамещенной. В одном варианте осуществления гетероциклоалкил представляет собой 3-7-членный гетероциклоалкил.
Когда описанные группы называют «замещенные или незамещенные», в тех случаях, когда замещенные, группы могут быть замещены одним (или более) любым заместителем. Примеры заместителей представляют собой такие, которые находятся в описанных иллюстративных соединениях и вариантах осуществления, а также галоген (например, хлор, йод, бром или фтор), С1-8алкил; С2-8алкенил; С2-8алкинил; С1-8алкоксил; гидроксил; амино; нитро; тиол; простой тиоэфир; имин; циано; амидо; фосфонато; фосфин; карбоксил; карбамоил; карбамат; ацеталь; мочевину; тиокарбонил; сульфонил; сульфонамид; кетон; альдегид; сложный эфир; ацетил; ацетокси; окси (=О); галогеналкил (например, трифторметил); замещенный аминоацил и аминоалкил; карбоциклический циклоалкил, который может быть моноциклическим или конденсированным или неконденсированным полициклическим (например, циклопропилом, циклобутилом, циклопентилом или циклогексилом), или гетероциклоалкилом, который может быть моноциклическим или конденсированным или неконденсированным полициклическим (например, пирролидинилом, пиперидинилом, пиперазинилом, морфолинилом, фуранилом или тиазинилом); карбоциклическим или гетероциклическим, моноциклическим или конденсированным или неконденсированным полициклическим арилом (например, фенилом, нафтилом, пирролилом, индолилом, фуранилом, тиенилом, имидазолилом, оксазолилом, изоксазолилом, тиазолилом, триазолилом, тетразолилом, пиразолилом, пиридинилом, хинолинилом, изохинолинилом, акридинилом, пиразинилом, пиридазинилом, пиримидинилом, бензимидазолилом, бензотиенилом или бензофуранилом); амино (первичной, вторичной или третичной); -О-низший алкил; -О-арил; арил; арил-низший алкил; СО2СН3; CONH2; OCH2CONH2; NH2; N(C1-4алкил)2; NHC(O)C1-4алкил; SO2NH2; SO2C1-4алкил, OCHF2; CF3; OCF3; и такие части также могут быть необязательно замещены конденсированной кольцевой структурой или мостиком, например, -ОСН2О- или -О-низший алкилен-О-. Такие заместители необязательно могут быть дополнительно замещены заместителем, выбранным из таких групп.
Применяемый термин «фармацевтически приемлемая соль(и)» относится к соли, полученной из фармацевтически приемлемой нетоксичной кислоты или основания, включающего неорганическую кислоту и основание и органическую кислоту и основание. Подходящие фармацевтически приемлемые аддитивные соли гетероарильных соединений с основанием включают, но не ограничиваются перечисленными, соли металлов, такие как алюминиевые, кальциевые, литиевые, магниевые, калиевые, натриевые и цинковые соли, или органические соли, полученные из лизина, N,N'-дибензилэтилендиамина, хлорпрокаина, холина, диэтаноламина, этилендиамина, меглумина (N-метилглюкамина) и прокаина. Подходящие нетоксичные кислоты включают, но не ограничиваются перечисленными, неорганические и органические кислоты, такие как уксусная, альгиновая, антраниловая, бензолсульфоновая, бензойная, камфорсульфоновая, лимонная, этенсульфоновая, муравьиная, фумаровая, фуровая, галактуроновая, глюконовая, глюкуроновая, глутаминовая, гликолевая, бромистоводородная, хлористоводородная, изетионовая, молочная, малеиновая, яблочная, манделовая, метансульфоновая, слизевая, азотная, памовая, пантотеновая, фенилуксусная, фосфорная, пропионовая, салициловая, стеариновая, янтарная, сульфаниловая, серная, винная кислота и п-толуолсульфоновая кислота. Специфические нетоксичные кислоты включают хлористоводородную, бромистоводородную, фосфорную, серную и метансульфоновую кислоту. Таким образом, примеры специфических солей включают хлористоводородную и мезилатную соли. Другие хорошо известны в области техники, см., например, Remington's Pharmaceutical Sciences, 18th eds., Mack Publishing, Easton PA (1990) или Remingtonc: The Science and Practice of Pharmacy, 19th eds. Mack Publishing, Easton PA (1995).
Применяемый термин «полиморф(ы)» и аналогичные термины относятся к солевым формам гетероарильных соединений, обладающим различными физическими свойствами, как результат расположения молекул в кристаллической решетке. Различия в физических свойствах, проявляемых твердыми формами, воздействуют на фармацевтические параметры, такие как стабильность при хранении, сжимаемость и плотность (важные при изготовлении фармацевтической лекарственной формы и изготовлении продукта) и скорости растворения (важного фактора в определении биологической доступности). Различия в стабильности могут являться следствием изменений в химической реакционной способности (например, различного окисления, такого, что лекарственная форма обесцвечивается более быстро, при сравнении одной твердой формы, чем при сравнении другой твердой формы) или механических изменений (например, разрушения таблеток при хранении, как следствие превращения кинетически благоприятного полиморфа в термодинамически более стабильную твердую форму), или обоих изменений (например, таблетки одной твердой формы более чувствительны к разрушению при высокой влажности). Как результат различий растворимости/растворения, в предельном случае, несколько превращений твердой формы могут приводить к потере эффективности или, при другом предельном случае, к токсичности. Помимо этого физические свойства кристалла могут быть важными при обработке, например, одна твердая форма может быть более подходящей для образования сольватов или может быть трудной для фильтрования и отмывания примесей (т.е. форма частицы и распределение по размерам могут отличаться между частицами, одна твердая форма по отношению к другой).
Применяемый термин «клатрат», и если не указано иное, означает гетероарильное соединение или его соль в форме кристаллической решетки, которая содержит зоны (например, каналы), которые имеют внутри захваченную молекулу гостя (например, растворителя или воды), или кристаллической решетки, в которой гетероарильное соединение представляет собой молекулу гостя.
Применяемый термин «гидрат», и если не указано иное, означает гетероарильное соединение или его соль, которое дополнительно включает стехиометрическое или нестехиометрическое количество воды, связанной нековалентными межмолекулярными силами.
Применяемый термин «сольват», и если не указано иное, означает гетероарильное соединение или его соль, которое дополнительно включает стехиометрическое или нестехиометрическое количество растворителя, связанного нековалентными межмолекулярными силами.
Применяемый термин «пролекарство», и если не указано иное, означает производное гетероарильного соединения, которое гидролизуется, окисляется или иным образом подвергается реакции в биологических условиях (in vitro или in vivo) с получением активного соединения, конкретно, гетероарильного соединения. Примеры пролекарств включают, но не ограничиваются перечисленными, производные и метаболиты гетероарильного соединения, которое включает биогидролизуемые части, такие как биогидролизуемые амиды, биогидролизуемые сложные эфиры, биогидролизуемые карбаматы, биогидролизуемые карбонаты, биогидролизуемые уреиды и биогидролизуемые фосфатные аналоги. В некоторых вариантах осуществления пролекарства соединений с карбоксильными функциональными группами представляют собой сложные низшие алкильные эфиры карбоновой кислоты. Сложные эфиры карбоксилатов представляют собой подходящим способом образованные этерификацией любой из частей карбоновой кислоты, присутствующей в молекуле. Пролекарства обычно можно изготавливать с применением хорошо известных способов, таких как способы, описанные Burger's Medicianal Chemistry and Drug Discovery 6th ed.(Donald J. Abraham ed., 2001, Wiley) и Design and Application of Prodrags (H. Bundgaard ed., 1985, Harwood Academic Publisher Gmfh).
Применяемый термин «стереоизомер» или «стереоизомерно чистый», и если не указано иное, означает один стереоизомер гетероарильного соединения, который по существу свободен от других стереоизомеров такого соединения. Например, стереоизомерно чистое соединение, имеющее один хиральный центр, будет по существу свободно от противоположного энантиомера соединения. Стереоизомерно чистое соединение, имеющее два хиральных центра, будет по существу свободно от других диастереомеров соединения. Типичное стереомерно чистое соединение включает более чем приблизительно 80% массы одного стереоизомера соединения и менее чем приблизительно 20% массы других стереоизомеров соединения; более чем приблизительно 90% массы одного стереоизомера соединения и менее чем приблизительно 10% массы других стереоизомеров соединения; более чем приблизительно 95% массы одного стереоизомера соединения и менее чем приблизительно 5% массы других стереоизомеров соединения или более чем приблизительно 97% массы одного стереоизомера соединения и менее чем приблизительно 3% массы других стереоизомеров соединения. Гетероарильные соединения могут иметь хиральные центры и могут находиться в виде рацематов, индивидуальных энантиомеров или диастереомеров и их смесей. Все такие изомерные формы включены в предлагаемые в описании варианты осуществления, включая их смеси.
Различные гетероарильные соединения содержат один или более хиральных центров и могут существовать в виде рацемических смесей энантиомеров, смесей диастереомеров или энантиомерно или оптически чистых соединений. Применение стереоизомерно чистых форм таких гетероарильных соединений, а также применение смесей таких форм включены посредством предложенных вариантов осуществления. Например, смеси, включающие равные или неравные количества энантиомеров конкретного гетероарильного соединения, можно применять в предложенных способах и композициях. Такие изомеры можно синтезировать асимметрическим синтезом или разделять с применением стандартных методов, таких как хиральные колонки или хиральные разделяющие агенты. См., например, Jacques, J., et al., Enantiomers, Racemates and Resolutions (Wiley-Interscience, New York, 1981); Wilen, S.H., et al., Tetrahedron 33:2725 (1977); Eliel, E.L., Stereochemistry of Carbon Compounds (McGraw-Hill, NY, 1962); and Wilen, S.H., Tables of Resolving Agents and Optical Resolutions p.268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dam, IN, 1972).
Следует также отметить, что гетероарильные соединения могут включать Е- и Z-изомеры или их смеси и цис- и транс-изомеры или их смеси. В некоторых вариантах осуществления гетероарильные соединения выделяют в виде либо Е-, либо Z-изомера. В других вариантах осуществления гетероарильные соединения представляют собой смеси Е и Z-изомеров.
Термин «эффективное количество» в связи с гетероарильным соединением может означать количество, способное лечить или предупреждать описанное заболевание, такое как рак, воспалительные патологические состояния, иммунологические патологические состояния, метаболические патологические состояния или патологические состояния, которые лечатся или предупреждаются путем ингибирования пути метаболизма киназы, в одном варианте осуществления пути метаболизма IKK-2, mTOR или PI3K.
Термин «пациент» включает животное, но не ограничивается перечисленными, такое как корова, обезьяна, лошадь, овца, свинья, цыпленок, индюк, куропатка, кошка, собака, мышь, крыса, кролик или морская свинка, в одном варианте осуществления млекопитающее, в другом варианте осуществления человек.
4.2 Гетероарильные соединения
Предложены гетероарильные соединения, имеющие следующую формулу (I)

и их фармацевтически приемлемые соли, полиморфы, клатраты, сольваты, гидраты, стереоизомеры, энантиомеры и пролекарства, где:
R1 представляет собой замещенный или незамещенный С1-8алкил, замещенный или незамещенный арил, замещенный или незамещенный гетероарил, замещенный или незамещенный циклоалкил или замещенный или незамещенный гетероциклоалкил;
-Х-А-В-Y-, взятые вместе, образуют -N(R2)CH2C(O)NH-, -N(R2)C(O)CH2NH-, -N(R2)C(O)NH-, -N(R2)C=N- или -C(R2)=CHNH-;
L представляет собой прямую связь, NH или О;
R2 представляет собой замещенный или незамещенный С1-8алкил, замещенный или незамещенный арил, замещенный или незамещенный гетероарил, замещенный или незамещенный циклоалкил или замещенный или незамещенный гетероциклоалкил; и
R3 и R4 независимо представляют собой Н или С1-8алкил.
В одном варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где -Х-А-В-Y-, взятые вместе, образуют -N(R2)CH2C(O)NH-.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где -Х-А-В-Y-, взятые вместе, образуют -N(R2)C(O)CH2NH-.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где -Х-А-В-Y-, взятые вместе, образуют -N(R2)C(O)NH-.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где -Х-А-В-Y-, взятые вместе, образуют N(R2)C=N-.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где -Х-А-В-Y-, взятые вместе, образуют -C(R2)=CHNH-.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где L представляет собой прямую связь.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где R1 представляет собой замещенный арил, такой как замещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где R1 представляет собой замещенный или незамещенный гетероарил, такой как замещенный или незамещенный пиридин, замещенный или незамещенный индол или замещенный или незамещенный хинолин.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где R1 представляет собой замещенный или незамещенный циклоалкил, такой как замещенный или незамещенный циклопентил.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где -Х-А-В-Y-, взятые вместе, образуют -N(R2)C(O)NH-, и R1 представляет собой арил, такой как фенил.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где -Х-А-В-Y-, взятые вместе, образуют -N(R2)C(O)NH-, и R1 представляет собой замещенный или незамещенный гетероарил, такой как замещенный или незамещенный пиридин, замещенный или незамещенный индол или замещенный или незамещенный хинолин.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где -Х-А-В-Y-, взятые вместе, образуют -N(R2)C(O)NH-, и R1 представляет собой замещенный или незамещенный циклоалкил, такой как замещенный или незамещенный циклопентил.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где R2 представляет собой замещенный С1-8алкил, такой как -СН2С6Н5.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где R2 представляет собой С1-8алкил, такой как незамещенный метил.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где R2 представляет собой замещенный или незамещенный арил, такой как замещенный или незамещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где R2 представляет собой замещенный арил, такой как галоген, галогеналкил или алкоксизамещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где R2 представляет собой замещенный или незамещенный циклоалкил, такой как замещенный или незамещенный циклогексил или замещенный или незамещенный циклогептил.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где R2 представляет собой замещенный гетероциклоалкил, такой как замещенный пиперидин.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где R3 и R4 представляют собой Н.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где -Х-А-В-Y-, взятые вместе, образуют -N(R2)C(O)NH-, и R2 представляет собой незамещенный арил, такой как незамещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где -Х-А-В-Y-, взятые вместе, образуют -N(R2)C(O)NH-, R1 представляет собой замещенный или незамещенный гетероарил, такой как замещенный или незамещенный пиридин, и R2 представляет собой замещенный или незамещенный арил, такой как замещенный или незамещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где -Х-А-В-Y-, взятые вместе, образуют -N(R2)C(O)NH-, R1 представляет собой замещенный или незамещенный гетероарил, такой как замещенный или незамещенный пиридин, R2 представляет собой замещенный или незамещенный арил, такой как замещенный или незамещенный фенил, и R3 и R4 представляют собой Н.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где -Х-А-В-Y-, взятые вместе, образуют -N(R2)C(O)NH-, L представляет собой прямую связь, R1 представляет собой замещенный или незамещенный гетероарил, такой как замещенный или незамещенный пиридин, R2 представляет собой замещенный или незамещенный арил, такой как замещенный или незамещенный фенил, и R3 и R4 представляют собой Н.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где -Х-А-В-Y-, взятые вместе, образуют -N(R2)C(O)NH-, R1 представляет собой замещенный или незамещенный арил, такой как замещенный или незамещенный фенил, и R2 представляет собой замещенный или незамещенный арил, такой как замещенный или незамещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где -Х-А-В-Y-, взятые вместе, образуют -N(R2)C(O)NH-, R1 представляет собой замещенный или незамещенный арил, такой как замещенный или незамещенный фенил, R2 представляет собой замещенный или незамещенный арил, такой как замещенный или незамещенный фенил, и R3 и R4 представляют собой Н.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где -Х-А-В-Y-, взятые вместе, образуют -N(R2)C(O)NH-, L представляет собой прямую связь, R1 представляет собой замещенный или незамещенный арил, такой как замещенный или незамещенный фенил, R2 представляет собой замещенный или незамещенный арил, такой как замещенный или незамещенный фенил, и R3 и R4 представляют собой Н.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где -Х-А-В-Y-, взятые вместе, образуют -N(R2)C(O)NH-, R1 представляет собой замещенный или незамещенный гетероарил, L представляет собой прямую связь и R2 представляет собой замещенный или незамещенный С1-8алкил или замещенный или незамещенный циклоалкил.
В другом варианте осуществления гетероарильные соединения формулы (I) представляют собой такие, где -Х-А-В-Y-, взятые вместе, образуют -N(R2)C(O)NH-, R1 представляет собой замещенный или незамещенный арил, L представляет собой прямую связь и R2 представляет собой замещенный или незамещенный С1-8алкил или замещенный или незамещенный циклоалкил.
В другом варианте осуществления гетероарильные соединения формулы (I) не включают 8,9-дигидро-8-оксо-9-фенил-2-(3-пиридинил)-7Н-пурин-6-карбоксамид, 8,9-дигидро-8-оксо-9-фенил-2-(3-пиридинил)-7Н-пурин-6-карбоксамид, 8,9-дигидро-8-оксо-9-фенил-2-(3-пиридинил)-7Н-пурин-6-карбоксамид, 2-(4-цианофенил)-8-оксо-9-фенил-8,9-дигидро-7Н-пурин-6-карбоксамид, 2-(4-нитрофенил)-8-оксо-9-фенил-8,9-дигидро-7Н-пурин-6-карбоксамид, 9-бензил-2-(4-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид, 2-метил-8-оксо-9-фенил-8,9-дигидро-7Н-пурин-6-карбоксамид, 9-бензил-9Н-пурин-2,6-дикарбоксамид, 9-[2,3-бис[(бензоилокси)метил]циклобутил]-2-метил-9Н-пурин-6-карбоксамид, 9-бензил-2-метил-9Н-пурин-6-карбоксамид, 9-(2-гидроксиэтил)-2-метил-9Н-пурин-6-карбоксамид, 9-(2-гидроксиэтил)-2-(трифторметил)-9Н-пурин-6-карбоксамид, 9-(2-гидроксиэтил)-2-(проп-1-енил)-9Н-пурин-6-карбоксамид, 9-(2-гидроксиэтил)-2-фенил-9Н-пурин-6-карбоксамид, 9-(3-гидроксипропил)-2-метил-9Н-пурин-6-карбоксамид, 9-(3-гидроксипропил)-2-(трифторметил)-9Н-пурин-6-карбоксамид, 2-метил-9-фенилметил-9Н-пурин-6-карбоксамид или 2-метил-9-β-D-рибофуранозил-9Н-пурин-6-карбоксамид.
В другом варианте осуществления гетероарильные соединения формулы (I) не включают соединения, где R2 представляет собой замещенный фуранозид.
В другом варианте осуществления гетероарильные соединения формулы (I) не включают соединения, где R2 представляет собой замещенный или незамещенный фуранозид.
В другом варианте осуществления гетероарильные соединения формулы (I) не включают (2'R)-2'-дезокси-2'-фтор-2'-С-метилнуклеозиды.
В следующем варианте осуществления предложены гетероарильные соединения, имеющие формулу (II):

и их фармацевтически приемлемые соли, полиморфы, клатраты, сольваты, гидраты, стереоизомеры, энантиомеры и пролекарства, где:
R1 представляет собой замещенный или незамещенный С1-8алкил, замещенный или незамещенный арил, замещенный или незамещенный гетероарил, замещенный или незамещенный циклоалкил или замещенный или незамещенный гетероциклоалкил;
R2 представляет собой замещенный или незамещенный С1-8алкил, замещенный или незамещенный арил, замещенный или незамещенный гетероарил, замещенный или незамещенный циклоалкил или замещенный или незамещенный гетероциклоалкил, и
R3 и R4 независимо представляют собой Н или С1-8алкил.
В одном варианте осуществления гетероарильные соединения формулы (II) представляют собой такие, где R1 представляет собой замещенный арил, замещенный или незамещенный гетероарил, такой как замещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (II) представляют собой такие, где R1 представляет собой замещенный или незамещенный гетероарил, такой как замещенный или незамещенный пиридин, замещенный или незамещенный индол или замещенный или незамещенный хинолин.
В другом варианте осуществления гетероарильные соединения формулы (II) представляют собой такие, где R1 представляет собой замещенный или незамещенный циклоалкил, такой как замещенный или незамещенный циклопентил.
В другом варианте осуществления гетероарильные соединения формулы (II) представляют собой такие, где R2 представляет собой замещенный С1-8алкил, такой как -СН2С6Н5.
В другом варианте осуществления гетероарильные соединения формулы (II) представляют собой такие, где R2 представляет собой незамещенный С1-8алкил, такой как незамещенный метил.
В другом варианте осуществления гетероарильные соединения формулы (II) представляют собой такие, где R2 представляет собой замещенный или незамещенный арил, такой как замещенный или незамещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (II) представляют собой такие, где R2 представляет собой замещенный арил, такой как галоген, галогеналкил или алкоксизамещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (II) представляют собой такие, где R2 представляет собой замещенный или незамещенный циклоалкил, такой как замещенный или незамещенный циклогексил или замещенный или незамещенный циклогептил.
В другом варианте осуществления гетероарильные соединения формулы (II) представляют собой такие, где R2 представляет собой замещенный гетероциклоалкил, такой как замещенный пиперидин.
В другом варианте осуществления гетероарильные соединения формулы (II) представляют собой такие, где R3 и R4 представляют собой Н.
В другом варианте осуществления гетероарильные соединения формулы (II) не включают 8,9-дигидро-8-оксо-9-фенил-2-(3-пиридинил)-7Н-пурин-6-карбоксамид, 8,9-дигидро-8-оксо-9-фенил-2-(3-пиридинил)-7Н-пурин-6-карбоксамид, 8,9-дигидро-8-оксо-9-фенил-2-(3-пиридинил)-7Н-пурин-6-карбоксамид, 2-(4-цианофенил)-8-оксо-9-фенил-8,9-дигидро-7Н-пурин-6-карбоксамид, 2-(4-нитрофенил)-8-оксо-9-фенил-8,9-дигидро-7Н-пурин-6-карбоксамид, 9-бензил-2-(4-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид, 9-фенилметил-9Н-пурин-2,6-дикарбоксамид или 2-метил-8-оксо-9-фенил-8,9-дигидро-7Н-пурин-6-карбоксамид.
В другом варианте осуществления гетероарильные соединения формулы (II) не включают соединения, где R2 представляет собой замещенный фуранозид.
В другом варианте осуществления гетероарильные соединения формулы (II) не включают соединения, где R2 представляет собой замещенный или незамещенный фуранозид.
В другом варианте осуществления гетероарильные соединения формулы (II) не включают (2'R)-2'-дезокси-2'-фтор-2'-C-метилнуклеозиды.
В следующем варианте осуществления предложены гетероарильные соединения, имеющие следующую формулу (III):

и их фармацевтически приемлемые соли, полиморфы, клатраты, сольваты, гидраты, стереоизомеры, энантиомеры и пролекарства, где:
 представляет собой -С(R2)=CH-NH- или -N(R2)-CH=N-;
представляет собой -С(R2)=CH-NH- или -N(R2)-CH=N-;
R1 представляет собой замещенный или незамещенный С1-8алкил, замещенный или незамещенный арил, замещенный или незамещенный гетероарил, замещенный или незамещенный циклоалкил или замещенный или незамещенный гетероциклоалкил;
R2 представляет собой замещенный или незамещенный С1-8алкил, замещенный или незамещенный арил, замещенный или незамещенный гетероарил, замещенный или незамещенный циклоалкил или замещенный или незамещенный гетероциклоалкил; и
R3 и R4 независимо представляют собой Н или С1-8алкил.
В одном варианте осуществления гетероарильные соединения формулы (III) представляют собой такие, где R1 представляет собой замещенный арил, такой как замещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (III) представляют собой такие, где R1 представляет собой замещенный или незамещенный гетероарил, такой как замещенный или незамещенный пиридин, замещенный или незамещенный индол или замещенный или незамещенный хинолин.
В другом варианте осуществления гетероарильные соединения формулы (III) представляют собой такие, где R1 представляет собой замещенный или незамещенный циклоалкил, такой как замещенный или незамещенный циклопентил.
В другом варианте осуществления гетероарильные соединения формулы (III) представляют собой такие, где R2 представляет собой замещенный С1-8алкил, такой как -СН2С6Н5.
В другом варианте осуществления гетероарильные соединения формулы (III) представляют собой такие, где R2 представляет собой незамещенный С1-8алкил, такой как незамещенный метил.
В другом варианте осуществления гетероарильные соединения формулы (III) представляют собой такие, где R2 представляет собой замещенный или незамещенный арил, такой как замещенный или незамещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (III) представляют собой такие, где R2 представляет собой замещенный арил, такой как галоген, галогеналкил или алкоксизамещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (III) представляют собой такие, где R2 представляет собой замещенный или незамещенный циклоалкил, такой как замещенный или незамещенный циклогексил или замещенный или незамещенный циклогептил.
В другом варианте осуществления гетероарильные соединения формулы (III) представляют собой такие, где R2 представляет собой замещенный гетероциклоалкил, такой как замещенный пиперидин.
В другом варианте осуществления гетероарильные соединения формулы (III) представляют собой такие, где R3 и R4 представляют собой Н.
В другом варианте осуществления гетероарильные соединения формулы (III) представляют собой такие, где группа представляет собой -С(R2)=CH-NH-, и R2 представляет собой замещенный арил, такой как замещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (III) представляют собой такие, где группа представляет собой -N(R2)-CH=N- и R2 представляет собой замещенный арил, такой как замещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (III) представляют собой такие, где R1 представляет собой замещенный арил, такой как фенил, и R2 представляет собой замещенный арил, такой как замещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (III) не включают 9-бензил-9Н-пурин-2,6-дикарбоксамид, 9-[2,3-бис[(бензоилокси)метил]циклобутил]-2-метил-9Н-пурин-6-карбоксамид, 9-бензил-2-метил-9Н-пурин-6-карбоксамид, 9-(2-гидроксиэтил)-2-метил-9Н-пурин-6-карбоксамид, 9-(2-гидроксиэтил)-2-(трифторметил)-9Н-пурин-6-карбоксамид, 9-(2-гидроксиэтил)-2-(проп-1-енил)-9Н-пурин-6-карбоксамид, 9-(2-гидроксиэтил)-2-фенил-9Н-пурин-6-карбоксамид, 9-(3-гидроксипропил)-2-метил-9Н-пурин-6-карбоксамид, 9-(3-гидроксипропил)-2-(трифторметил)-9Н-пурин-6-карбоксамид, 9-фенилметил-9Н-пурин-2,6-дикарбоксамид, 2-метил-9-фенилметил-9Н-пурин-6-карбоксамид или 2-метил-9-β-D-рибофуранозил-9Н-пурин-6-карбоксамид.
В другом варианте осуществления гетероарильные соединения формулы (III) не включают соединения, где R2 представляет собой замещенный циклобутил, где группа представляет собой -N(R2)-CH=N-.
В другом варианте осуществления гетероарильные соединения формулы (III) не включают соединения, где R2 представляет собой замещенный фуранозид, где группа представляет собой -N(R2)-CH=N-.
В другом варианте осуществления гетероарильные соединения формулы (III) не включают соединения, где R2 представляет собой замещенный пиримидин, где группа представляет собой -С(R2)=CH-NH-.
В другом варианте осуществления гетероарильные соединения формулы (III) не включают соединения, где R2 представляет собой замещенный оксетан, где группа представляет собой -N(R2)-CH=N-.
В другом варианте осуществления гетероарильные соединения формулы (III) не включают соединения, где R2 представляет собой замещенный циклопентил или гетероциклопентил, где группа представляет собой -N(R2)-CH=N-.
В следующем варианте осуществления предложены гетероарильные соединения, имеющие следующую формулу (IV):

и их фармацевтически приемлемые соли, полиморфы, клатраты, сольваты, гидраты, стереоизомеры, энантиомеры и пролекарства,
где
R1 представляет собой замещенный или незамещенный С1-8алкил, замещенный или незамещенный арил, замещенный или незамещенный гетероарил, замещенный или незамещенный циклоалкил или замещенный или незамещенный гетероциклоалкил;
R2 представляет собой замещенный или незамещенный С1-8алкил, замещенный или незамещенный арил, замещенный или незамещенный гетероарил, замещенный или незамещенный циклоалкил или замещенный или незамещенный гетероциклоалкил, и
R3 и R4 независимо представляют собой Н или С1-8алкил.
В одном варианте осуществления гетероарильные соединения формулы (IV) представляют собой такие, где R1 представляет собой арил, такой как замещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (IV) представляют собой такие, где R1 представляет собой замещенный или незамещенный гетероарил, такой как замещенный или незамещенный пиридин, замещенный или незамещенный индол или замещенный или незамещенный хинолин.
В другом варианте осуществления гетероарильные соединения формулы (IV) представляют собой такие, где R1 представляет собой замещенный или незамещенный циклоалкил, такой как замещенный или незамещенный циклопентил.
В другом варианте осуществления гетероарильные соединения формулы (IV) представляют собой такие, где R2 представляет собой замещенный С1-8алкил, такой как -СН2С6Н5.
В другом варианте осуществления гетероарильные соединения формулы (IV) представляют собой такие, где R2 представляет собой незамещенный С1-8алкил, такой как незамещенный метил.
В другом варианте осуществления гетероарильные соединения формулы (IV) представляют собой такие, где R2 представляет собой замещенный или незамещенный арил, такой как замещенный или незамещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (IV) представляют собой такие, где R2 представляет собой замещенный арил, такой как галоген, галогеналкил или алкоксизамещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (IV) представляют собой такие, где R2 представляет собой замещенный или незамещенный циклоалкил, такой как замещенный или незамещенный циклогексил или замещенный или незамещенный циклогептил.
В другом варианте осуществления гетероарильные соединения формулы (IV) представляют собой такие, где R2 представляет собой замещенный гетероциклоалкил, такой как замещенный пиперидин.
В другом варианте осуществления гетероарильные соединения формулы (IV) представляют собой такие, где R3 и R4 представляют собой Н.
В следующем варианте осуществления предложены гетероарильные соединения, имеющие формулу (V):
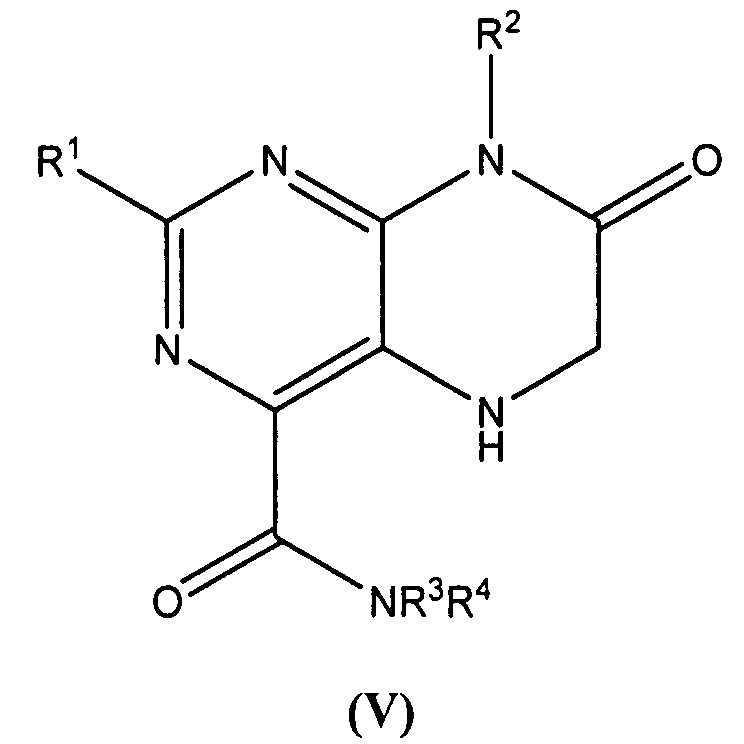
и их фармацевтически приемлемые соли, полиморфы, клатраты, сольваты, гидраты, стереоизомеры, энантиомеры и пролекарства,
где
R1 представляет собой замещенный или незамещенный С1-8алкил, замещенный или незамещенный арил, замещенный или незамещенный гетероарил, замещенный или незамещенный циклоалкил или замещенный или незамещенный гетероциклоалкил;
R2 представляет собой замещенный или незамещенный С1-8алкил, замещенный или незамещенный арил, замещенный или незамещенный гетероарил, замещенный или незамещенный циклоалкил или замещенный или незамещенный гетероциклоалкил; и
R3 и R4 независимо представляют собой Н или С1-8алкил.
В одном варианте осуществления гетероарильные соединения формулы (V) представляют собой такие, где R1 представляет собой замещенный арил, такой как замещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (V) представляют собой такие, где R1 представляет собой замещенный или незамещенный гетероарил, такой как замещенный или незамещенный пиридин, замещенный или незамещенный индол или замещенный или незамещенный хинолин.
В другом варианте осуществления гетероарильные соединения формулы (V) представляют собой такие, где R1 представляет собой замещенный или незамещенный циклоалкил, такой как замещенный или незамещенный циклопентил.
В другом варианте осуществления гетероарильные соединения формулы (V) представляют собой такие, где R2 представляет собой замещенный С1-8алкил, такой как -СН2С6Н5.
В другом варианте осуществления гетероарильные соединения формулы (V) представляют собой такие, где R2 представляет собой незамещенный С1-8алкил, такой как незамещенный метил.
В другом варианте осуществления гетероарильные соединения формулы (V) представляют собой такие, где R2 представляет собой замещенный или незамещенный арил, такой как замещенный или незамещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (V) представляют собой такие, где R2 представляет собой замещенный арил, такой как галоген, галогеналкил или алкоксизамещенный фенил.
В другом варианте осуществления гетероарильные соединения формулы (V) представляют собой такие, где R2 представляет собой замещенный или незамещенный циклоалкил, такой как замещенный или незамещенный циклогексил или замещенный или незамещенный циклогептил.
В другом варианте осуществления гетероарильные соединения формулы (V) представляют собой такие, где R2 представляет собой замещенный гетероциклоалкил, такой как замещенный пиперидин.
В другом варианте осуществления гетероарильные соединения формулы (V) представляют собой такие, где R3 и R4 представляют собой H.
Репрезентативные гетероарильные соединения представлены в таблице 1 ниже по тексту.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
4.3 Способы получения гетероальных соединений
Гетероарильные соединения может получать любой специалист в области техники с применением стандартного органического синтеза и коммерчески доступных веществ. С помощью примеров и без ограничения гетероарильное соединение можно получать, как описано на схемах 1-8, представленных ниже по тексту, в также в примерах, изложенных в разделе 5.1. Следует отметить, что любой специалист в области техники может модифицировать методики, представленные на иллюстративных схемах и в примерах, чтобы прийти к требуемому веществу.
Схема 1:

Схема 2:

Схема 3:

Схема 4:

Схема 5:

Схема 6:

Схема 7:

Схема 8:

Фармацевтически приемлемые соли гетероарильных соединений можно получать стандартными и известными методами, такими как взаимодействие гетероарильного соединения с подходящей кислотой, как указано выше в описании. Такие соли обычно получают с высокими выходами при умеренных температурах и часто получают просто выделением соединения из подходящего кислотного промывочного раствора на конечной стадии синтеза. Солеобразующую кислоту можно растворить в соответствующем органическом растворителе или водном органическом растворителе, таком как спирт, кетон или сложный эфир. С другой стороны, если гетероарильное соединение требуется в форме свободного основания, его извлекают на конечной стадии из щелочного промывочного раствора, согласно известным методам. Например, обычный метод получения хлористоводородной соли представляет собой растворение свободного основания в подходящем растворителе и тщательное высушивание раствора, например, над молекулярными ситами, перед пропусканием через него газообразного хлористого водорода.
4.4 Способы применения
Описанные гетероарильные соединения применяют в качестве фармацевтических препаратов для лечения или предупреждения заболевания у животных или людей. Затем, описанные гетероарильные соединения являются активными против киназ (например, протеинкиназ), включая киназы, связанные с раком, воспалительными патологическими состояниями, иммунологическими патологическими состояниями, нейродегенеративными патологическими заболеваниями, сердечно-сосудистыми заболеваниями и метаболическими патологическими состояниями. Не ссылаясь на теорию, существует мнение, что гетероарильные соединения являются эффективными для лечения и предупреждения рака, воспалительных патологических состояний, иммунологических патологических состояний, нейродегенеративных заболеваний, сердечно-сосудистых заболеваний и метаболических патологических состояний вследствие их способности модулировать (например, ингибировать) киназы, которые вовлечены в этиологию таких патологических состояний. Соответственно, предложено много применений гетероарильных соединений, включающих лечение или предупреждение таких заболеваний, изложенных ниже по тексту. Предложенные способы включают введение эффективного количества одного или нескольких гетероарильных соединений нуждающемуся в этом пациенту.
Репрезентативные иммунологические патологические состояния, для лечения или предупреждения которых применимы гетероарильные соединения, включают, но не ограничиваются перечисленными, ревматоидный артрит, ревматоидный спондилит, остеоартрит, рассеянный склероз, волчанку, воспалительное кишечное заболевание, язвенный колит, болезнь Крона, тяжелую псевдопаралитическую миастению, болезнь Грейвса и диабет (например, диабет I типа).
Репрезентативные воспалительные патологические состояния, для лечения или предупреждения которых применимы гетероарильные соединения, включают, но не ограничиваются перечисленными, псориаз, астму и аллергический ринит, бронхит, хроническое обструктивное легочное заболевание, муковисцидоз, воспалительное кишечное заболевание, синдром раздраженного кишечника, болезнь Крона, колит слизистой оболочки, язвенный колит, диабет (например, диабет I типа и диабет II типа) и ожирение.
Репрезентативные сердечно-сосудистые заболевания, для лечения или предупреждения которых применимы гетероарильные соединения, включают, но не ограничиваются перечисленными, рестеноз, инсульт, инфаркт миокарда или ишемическое повреждение сердца, легких, кишечника, почки, печени, поджелудочной железы, селезенки или головного мозга.
Репрезентативные метаболические патологические состояния, для лечения или предупреждения которых применимы гетероарильные соединения, включают, но не ограничиваются перечисленными, ожирение и диабет (например, диабет II типа).
Репрезентативные нейродегенеративные заболевания, для лечения или предупреждения которых применимы гетероарильные соединения, включают, но не ограничиваются перечисленными, болезнь Гентингтона, болезнь Альцгеймера и ВИЧ-ассоциированный энцефалит.
В конкретном варианте осуществления предложены способы лечения или предупреждения устойчивости к инсулину. В некоторых вариантах осуществления предложены способы лечения или предупреждения устойчивости к инсулину, которая приводит к диабету (например, диабету II типа).
В другом варианте осуществления предложены способы лечения или предупреждения синдрома Х или метаболического синдрома.
В другом варианте осуществления предложены способы лечения или предупреждения диабета.
В другом варианте осуществления предложены способы лечения или предупреждения диабета II типа, диабета I типа, диабета I типа с замедленным проявлением, несахарного диабета (например, несахарного нейрогенного диабета, несахарного нефрогенного диабета, несахарного диабета, сопровождающегося жаждой, или несахарного гестагенного диабета), сахарного диабета, сахарного диабета у беременных, синдрома поликистоза яичников, диабета у взрослых, ювенильного диабета, инсулин-зависимого диабета, неинсулин-зависимого диабета, диабета, связанного с нарушением питания, диабета, сопровождающегося кетозом и пронацией, преддиабета (например, ухудшения метаболизма глюкозы), диабета, связанного с муковисцидозом, гемохроматозом и кетозо-резистентного диабета.
В другом варианте осуществления предложены способы лечения или предупреждения фибротических заболеваний и расстройств. В конкретном варианте осуществления описаны способы лечения или предупреждения идиопатического легочного фиброза, миелофиброза, фиброза печени, стеатофиброза и стеатогепатита.
Репрезентативные раковые заболевания, для лечения или предупреждения которых применимы гетероарильные соединения, включают, но не ограничиваются перечисленными, раковые заболевания головы, шеи, глаза, рта, горла, пищевода, бронхов, гортани, глотки, грудной клетки, кости, легких, толстой кишки, прямой кишки, желудка, простаты, мочевого пузыря, матки, шейки матки, молочной железы, яичников, яичка или других репродуктивных органов, кожи, щитовидной железы, крови, лимфатических узлов, почки, печени, поджелудочной железы и головного мозга или центральной нервной системы. Гетероарильные соединения также применимы для лечения или предупреждения твердых опухолей и гематогенных опухолей.
Конкретные раковые заболевания, которые входят в объем описанных способов, включают заболевания, связанные с киназами IKK-2, mTOR, PI3K, SYK или TYK2 и их мутантами или изоформами. Другие раковые заболевания, которые входят в объем описанных способов, включают заболевания, связанные с путями метаболизма следующих киназ: PI3Kα, PI3Kβ, PI3Kδ, Aurora, Abl, KDR, MLK1, CaMKIV, GSK3α, GSK3β, ATM, ATX или ДНК-РК киназы и их мутанты или изоформы.
Более конкретно, раковые заболевания и аналогичные расстройства, которые можно лечить или предупреждать описанными способами и композициями, включают, но не ограничиваются перечисленными, следующие: лейкозы, такие как, но не ограничивающиеся перечисленными, острый лейкоз, острый лимфоцитарный лейкоз, острый миелоидный лейкоз, такой как миелобластный, промиелоцитарный, миеломоноцитарный, моноцитарный, эритролейкоз, лейкоз и синдром миелодисплазии (или ее симптом, такой как анемия, тромбоцитопения, нейтропения, бицитопения или панцитопения), рефрактерная анемия (RA), RA с кольцевыми сидеробластами (RARS), RA с избыточными бластными клетками (RAEB), RAEB в трансформации (RAEB-T), предлейкоз и хронический миеломоноцитарный лейкоз (CMML), хронический лейкоз, такой как, не ограниченный перечисленным, хронический миелоцитарный (гранулоцитарный) лейкоз, хронический лимфоцитарный лейкоз, лейкозный ретиколоэндотелиоз; истинная полицитемия, лимфома, такая как, но не органиченная перечисленным, болезнь Ходжкина, неходжкинская (болезнь) лимфома; множественная миелома, такая как, но не ограниченная перечисленным, тлеющая (вялотекущая) множественная миелома, несекреторная миелома, остеосклеротическая миелома, лейкоз клеток плазмы, солитарная плазмацитома и экстрамедулярная плазмацитома; макроглобулинемия Вальденстрема; моноклональная гаммопатия неопределенного значения (смысла); доброкачественная моноклональная гаммопатия; болезнь тяжелой цепи; саркома костной и соединительной ткани, такая как, но не ограниченная перечисленными, костная саркома, остеосаркома, хондросаркома, саркома Юинга, злокачественная гигантоклеточная опухоль, фибросаркома кости, хордома, периостальная саркома, саркома мягкой ткани, ангиосаркома (гемангиосаркома), фибросаркома, саркома Капоши, лейомиосаркома, липосаркома, лимфангиосаркома, метастические раковые заболевания, невринома, рабдомиосаркома, синовиальная саркома; опухоли головного мозга, такие как, но не ограниченные перечисленными, глиома, астроцитома, стволовая глиома головного мозга, эпендимома, олигодендроглиома, неглиальная опухоль, акустическая невринома, краниофарингиома, медуллобластома, менингиома, пинеоцитома, пинеобластома, первичная лимфома головного мозга; рак молочной железы, включающий, но не ограничивающийся перечисленным, аденокарциному, лобулярную (малоклеточную) карциному, внутрипротоковую карциному, медуллярный рак молочной железы, слизеобразующий рак молочной железы, тубулярный рак молочной железы, папиллярный рак молочной железы, первичные раковые заболевания, болезнь Педжета и воспалительный рак молочной железы; рак надпочечника, такой как, не ограничивающийся перечисленным, феохромоцитома и адренокортикальная карцинома; рак щитовидной железы, такой как, но не ограничивающийся перечисленными, папиллярный или фолликулярный рак щитовидной железы, медуллярный рак щитовидной железы и анапластический рак щитовидной железы; рак поджелудочной железы, такой как, но не ограничивающийся перечисленным, инсулинома, гастринома, глюкагонома, випома, соматостатин-секретирующая опухоль и карциноид или опухоль островковых клеток; раковые заболевания гипофиза, такие как, но не ограничивающиеся перечисленными, болезнь Кушинга, пролактин-секретирующая опухоль, акромегалия и несахарный диабет; раковые заболевания глаза, такие как, но не ограничивающиеся перечисленными, глазная меланома, такая как меланома радужной оболочки, хороидальная меланома и меланома ресничного тела, и ретинобластома; вагинальные раковые заболевания, такие как карцинома сквамозных клеток, аденокарцинома и меланома; рак влагалища, такой как карцинома сквамозных клеток, меланома, аденокарцинома, карцинома базальных клеток, саркома и болезнь Педжета; цервикальные раковые заболевания, такие как, но не ограничивающиеся перечисленными, карцинома сквамозных клеток и аденокарцинома; раковые заболевания матки, такие как, но не ограничивающиеся перечисленными, эндометриальная карцинома и карцинома матки; раковые заболевания яичников, такие как, но не ограничивающиеся перечисленными, эпителиальная карцинома яичников, пограничные опухоли, гоноцитома и стромальная опухоль; эзофагеальные раковые заболевания, такие как, но не ограничивающиеся перечисленными, сквамозный рак, аденокарцинома, аденокистозная карцинома, мукоэпидермоидная карцинома, аденосквамозная карцинома, саркома, меланома, плазмацитома, веррукозная карцинома и овсяно-клеточная (малоклеточная) карцинома; раковые заболевания желудка, такие как, но не ограничивающиеся перечисленными, аденокарцинома, грибовидная (полипоидная), язвенная, поверхностная, диффузная, злокачественная лимфома, липосаркома, фибросаркома и карциносаркома; раковые заболевания толстой кишки; ректальные раковые заболевания; раковые заболевания печени, такие как, но не ограничивающиеся перечисленными, гепатоцеллюлярная карцинома и гепатобластома, раковые заболевания желчного пузыря, такие как аденокарцинома; холангиокарцинома, такая как, но не ограничивающаяся перечисленными, папиллярная, нодулярная и диффузная; раковые заболевания легких, такие как, немелкоклеточный рак легких, карцинома сквамозных клеток (эпидермоидная карцинома), аденокарцинома, крупноклеточная карцинома и мелкоклеточный рак легких; тестикулярные раковые заболевания, такие как, но не ограничивающиеся перечисленными, опухоль зародыша, семинома, анапластическая, классическая (типичная), относящаяся к сперматоцитам, несеминома, эмбриональная карцинома, тератома карцинома, хориокарцинома (опухоль желточного пузыря); раковые заболевания простаты, такие как, но не ограничивающиеся перечисленными, аденокарцинома, лейомиосаркома и рабдомиосаркома; пенитенциарные раковые заболевания; ротовые раковые заболевания, такие как, но не ограничивающиеся перечисленными, карцинома сквамозных клеток; базальные раковые заболевания; раковые заболевания слюнных желез, такие как, но не ограничивающиеся перечисленными, аденокарцинома, мукоэпидермоидная карцинома и аденоцистная карцинома; раковые заболевания глотки, такие как, но не ограничивающиеся перечисленными, рак сквамозных клеток и веррукоз; раковые заболевания кожи, такие как, но не ограничивающиеся перечисленными, карцинома базальных клеток, карцинома сквамозных клеток и меланома, поверхностная меланома, нодулярная меланома, злокачественная меланома лентиго, относящаяся к периферии лентикулярная меланома; раковые заболевания почки, такие как, но не ограничивающиеся перечисленными, рак клеток почечного эпителия, аденокарцинома, гипернефрома, фибросаркома, переходно-клеточный рак (почечной лоханки и/или матки); опухоль Вильмса; раковые заболевания мочевого пузыря, такие как, но не ограничивающиеся перечисленными, переходно-клеточная карцинома, рак сквамозных клеток, аденокарцинома, карциносаркома. Помимо этого раковые заболевания включают в себя миксосаркому, остеогенную саркому, эндотелиосаркому, лимфангио-эндотелиосаркому, мезотелиому, синовиому, гемангиобластому, эпителиальную карциному, цистаденокарциному, бронхогенную карциному, карциному потовых желез, карциному сальных желез, папиллярную карциному и папиллярную аденокарциному (обзор таких расстройств см. Fishman et al., 1985, Medicine, 2d Ed., J.B. Lippincott Co., Philadelphia and Murphy et al., 1997, Informed Decisions: The Complete Book of Cancer Diagnosis, Treatment and Recovery, Viking Penguin, Penguin Books U.S.A., Inc., United States of America).
В соответствии с этим, предложенные способы и композиции также применимы для лечения или предупреждения множества раковых заболеваний или других аномальных пролиферативных заболеваний, включающих (но не ограничивающихся перечисленными) такие, как карцинома, включающая заболевание мочевого пузыря, молочной железы, толстой кишки, почки, печени, легкого, яичника, поджелудочной железы, желудка, шейки матки, щитовидной железы и кожи, включая карциному сквамозных клеток; гемопоэтические опухоли лимфоидной линии, включая лейкоз, острый лимфоцитарный лейкоз, острый лимфобластный лейкоз, В-клеточную лимфому, Т-клеточную лимфому, лимфому Беркетта, кроветворные опухоли миелоидной линии, включая острый и хронический миелогенный лейкоз и промиелоцитарный лейкоз; опухоли мезенхимального происхождения, включающие фибросаркому и рабдомиосаркому; другие опухоли, включающие меланому, семиному, тетратокарциному, нейробластому и глиому; опухоли центральной и периферической нервной системы, включая астроцитому, многообразную глиобластому, нейробластому, глиому и шванному; твердые и кроветворные опухоли; опухоли мезенхимального происхождения, включая фибросаркому, рабдомиосаркому и остеосаркому; и другие опухоли, включающие меланому, xenoderma pegmentosum, кератоакантому, семиному, фолликулярный рак щитовидной железы и тератокарциному. Ожидают также, что раковые заболевания, вызванные аберрацией в апоптозе, также можно лечить описанными способами и композициями. Такие раковые заболевания могут включать в себя, но не ограничиваются перечисленными, фолликулярную лимфому, карциному с р53 мутациями, гормон-зависимые опухоли молочной железы, простаты и яичников, и предраковые патологические изменения, такие как семейный аденоматозный полипоз и синдромы миелодисплазии. В конкретных вариантах осуществления подвергают лечению или предупреждают злокачественные или диспролиферативные изменения (такие как метаплазия и дисплазия) или гиперпролиферативные расстройства в яичниках, мочевом пузыре, молочной железе, толстой кишке, легком, коже, поджелудочной железе, почке или матке. В других специфических вариантах осуществления подвергают лечению или предупреждают саркому, меланому или лейкоз.
В конкретном варианте осуществления предложенные способы и композиции также применимы для лечения, предупреждения или управления различными типами лимфомы (например, гетерогенной группой неоплазм, возникающих в ретикулоэндотелиальной и лимфатической системах), такими как неходжкинская лимфома (NHL) (например, злокачественная моноклональная пролиферация лимфоидных клеток в участках иммунной системы, включающих в себя лимфатические узлы, костный мозг, селезенку, печень и желудочно-кишечный тракт). NHL, для лечения или предупреждения которых применимы гетероарильные соединения, включают, но не ограничиваются перечисленными, лимфому клеток коры головного мозга, MCL, лимфоцитарную лимфому промежуточной дифференциации, промежуточную лимфоцитарную лимфому, ILL, диффузную недостаточно дифференцированную лимфоцитарную лимфому, PDL, центроцитарную лимфому, диффузную лимфому мало расщепляемых клеток, DSCCL, фолликулярную лимфому и любой тип лимфомы клеток коры головного мозга, которые можно наблюдать под микроскопом (узелковую, диффузную, бластную лимфому и лимфому зоны коры головного мозга).
В другом варианте осуществления предложенные способы и композиции применяют также для введения пациентам, нуждающимся в трансплантации костного мозга для лечения злокачественного заболевания (например, пациентов, страдающих острым лимфоцитарным лейкозом, острым миелогенным лейкозом, хроническим миелогенным лейкозом, хроническим лимфоцитарным лейкозом, синдромом миелодисплазии («предлейкозом»), синдромом моносомии 7, неходжкинской лимфомой, нейробластомой, опухолями головного мозга, множественной миеломой, опухолью тестикулярных зародышевых клеток, раком молочной железы, раком легкого, раком яичников, меланомой, глиомой, саркомой или другими твердыми опухолями), пациентам, которым требуется трансплантация костного мозга для лечения незлокачественного заболевания (например, пациентам, страдающим гематологическими расстройствами, врожденным иммунодефицитом, заболеваниями мукополисахаридозом, липидозом, остеопорозом, гистоцитозом клеток Лангерханса, синдромом Леш-Нихана или гликогенозом), пациентам, которых подвергают химиотерапии или лучевой терапии, тем, которых подготавливают для воздействия химиотерапии или лучевой терапии, и тем, которых ранее подвергали химиотерапии или лучевой терапии.
В другом варианте осуществления предложены способы для лечения миелопролиферативных нарушений или синдромов миелодисплазии, включающие введение нуждающемуся в этом пациенту эффективного количества гетероарильного соединения или его композиции. В некоторых вариантах осуществления миелопролиферативное нарушение представляет собой истинную полицитемию, первичную тромбоцитемию, хронический миелогенный лейкоз, острый или хронический гранулоцитарный лейкоз, острый или хронический миеломоноцитарный лейкоз, миелофибро-эритролейкоз или ангиогенную миелоидную метаплазию.
В другом варианте осуществления предложены способы лечения рака и опухолей, резистентных к другим ингибиторам киназы, таким как иматиниб мезилат (STI-571 или GleevecTM), лечение, включающее введение нуждающемуся в этом пациенту эффективного количества гетероарильного соединения или его композиции. В конкретном варианте осуществления предложены способы лечения лейкоза, включающие, но не ограничивающиеся перечисленными, желудочно-кишечную стромальную опухоль (GIST), острый лимфоцитарный лейкоз или хронический миелоцитарный лейкоз, резистентный к иматиниб мезилату (STI-571 или GleevecTM), лечение, включающее в себя введение нуждающемуся в этом пациенту эффективного количества гетероарильного соединения или его композиции.
В конкретном варианте осуществления предложены способы лечения или предупреждения заболевания или нарушения, связанного с ингибированием IKK-2, mTOR, PI3K, SYK или TYK2. Конкретные заболевания, которые подвергают лечению или предупреждают ингибированием IKK2, mTOR, PI3K, SYK или TYK2, включают, но не ограничиваются перечисленными, ревматоидный артрит, ревматоидный спондилит, остеоаритрит, подагру, астму, бронхит, аллергический ринит, хроническое обструктивное легочное заболевание, муковисцидоз, воспалительное кишечное заболевание, синдром раздраженного кишечника, колит слизистой оболочки, язвенный колит, болезнь Крона, болезнь Гентингтона, гастрит, эзофагит, гепатит, панкреатит, нефрит, рассеянный склероз, красная волчанка, диабет II типа, ожирение, атеросклероз, рестеноз после пластической операции на сосудах, гипертрофию левого желудочка, инфаркт миокарда, инсульт, ишемическое повреждение сердца, легкого, кишки, почки, печени, поджелудочной железы, селезенки и головного мозга, острое или хроническое отторжение трансплантированного органа, сохранение органа для трансплантации, повреждение органа или потерю конечности (например, включающее, но не ограничивающееся перечисленными, такие, которые являются следствием ишемического реперфузного повреждения, травмы, значительного повреждения тела, автомобильного несчастного случая, размозжения или неудачной трансплантации), заболевания «трансплантат против хозяина», эндотоксиновый шок, множественное расстройство органов, псориаз, ожог вследствие воздействия огня, химических средств или излучения, экзему, дерматит, кожную трансплантацию, ишемию, ишемические патологические состояния, связанные с операцией или травматического повреждения (например, дорожно-транспортным травматизмом, огнестрельной раной или разрушением конечности), эпилепсию, болезнь Альцгеймера, болезнь Паркинсона, иммунологическую ответную реакцию на бактериальную или вирусную инфекцию, кахексию, ангиогенное и пролиферативное заболевание, твердую опухоль и раковые заболевания ряда тканей, таких как толстая кишка, прямая кишка, простата, печень, легкое, бронхи, поджелудочная железа, головной мозг, голова, шея, желудок, кожа, почка, шейка матки, кровь, гортань, пищевод, рот, горло, мочевой пузырь, яичник или матка.
В специфическом варианте осуществления предложены способы лечения или предупреждения лейкоза (например, злокачественных неоплазм кроветворных тканей), включающего, но не ограничивающегося перечисленным, хронический лимфоцитарный лейкоз, хронический миелоцитарный лейкоз, острый лимфобластный лейкоз, острый миелогенный лейкоз и острый миелобластный лейкоз. Лейкоз может быть рецидивирующим, рефрактерным или резистентным к общепринятой терапии. Термин «рецидивирующий» относится к ситуации, когда пациенты, которые имели ремиссию лейкоза после терапии, имеют возврат лейкозных клеток в костном мозге и снижение нормальных кровяных клеток. Термин «рефрактерный или резистентный» относится к обстоятельству, когда пациенты даже после интенсивного лечения имеют оставшиеся лейкозные клетки в их костном мозге.
Различные типы раковых заболеваний описаны в предварительной заявке США № 60/380842, зарегистрированной 17 мая 2002 года, которая приведена во всей полноте посредством ссылки (см., например, раздел 2.2 Типы раковых заболеваний). Специфические раковые заболевания включают, но не ограничиваются перечисленными, лейкозы, такие как хронический лимфоцитарный лейкоз, хронический миелоцитарный лейкоз, острый лимфобластный лейкоз, острый миелогенный лейкоз и острый миелобластный лейкоз; прогрессирующее злокачественное заболевание, амилоидоз, нейробластому, менингиому, гемангиоперицитому, множественные метастазы головного мозга, многообразную глиобластому, глиобластому, стволовую глиому головного мозга, злокачественную опухоль головного мозга с плохим прогнозом, злокачественную глиому, рецидивирующую злокачественную глиому, анапластическую астроцитому, анапластическую олигодендроглиому, нейроэндокринную опухоль, аденокарциному прямой кишки, колоректальный рак Дукс С & D, нерезектабельную колоректальную карциному, метастатическую печеночно-клеточную карциному, саркому Капоши, каротипический острый миелобластный лейкоз, лимфому Ходжкина, неходжкинскую лимфому, кожную Т-клеточную лимфому, кожную В-клеточную лимфому, диффузную лимфому больших В-клеток, фолликулярную лимфому низкой степени, злокачественную меланому, злокачественную мезотелиому, синдром злокачественной плевральной распространенной мезотелиомы, перитонеальную карциному, папиллярную серозную карциному, гинекологическую саркому, саркому мягких тканей, склеродермию, кожный васкулит, гистиоцитоз клеток Лангерханса, лейомиосаркому, прогрессирующую костеобразующую фибродисплазию, гормональный рефрактерный рак простаты, резецируемую с высоким риском саркому мягких тканей, нерезектабельную печеночно-клеточную карциному, макроглобулинемию Вальденстрома, тлеющую миелому, безболезненную миелому, рак фаллопиевой трубы, андроген-независимый рак простаты, андроген-зависимый неметастатический рак простаты IV стадии, гормон-нечувствительный рак простаты, нечувствительный к химиотерапии рак простаты, папиллярную карциному щитовидной железы, фолликулярную карциному щитовидной железы, медуллярную карциному щитовидной железы и лейомиому. В одном варианте осуществления рак представляет собой первичный или метастатический. В другом варианте осуществления рак представляет собой рецидивирующий, рефрактерный или резистентный к химиотерапии или облучению; в особенности, рефрактерный к талидомиду.
Далее предложены способы лечения пациентов, которых ранее подвергали лечению рака, но являющихся невосприимчивыми к стандартным методам терапии, а также и пациентов, которых ранее не подвергали лечению. Также предложены способы лечения пациентов независимо от возраста пациента, хотя некоторые раковые заболевания представляют собой более распространенные в конкретных возрастных группах. Далее предложены способы лечения пациентов, которых подвергали хирургическому вмешательству с попыткой получить результат лечения рака, а также и таких, которых не подвергали операции. Вследствие того, что пациенты, болеющие раком, имеют разнородные клинические проявления и различные клинические результаты, лечение, предоставляемое пациенту, может различаться в зависимости от его прогноза. Специалист-клиницист будет способен легко определить без чрезмерного экспериментирования специфические дополнительные агенты, типы хирургического вмешательства и типы не основанной на лекарственном средстве терапии, которые можно эффективно применять для лечения отдельного пациента с раковым заболеванием.
В другом варианте осуществления предложены способы лечения или предупреждения заболевания или нарушения (например, рака или опухоли), связанного с ингибированием PI3Kα, PI3Kβ, PI3Kδ, Aurora, Abl, KDR, MLK1, CaMKIV, GSK3α, GSK3β, ATM, ATX или ДНК-РК.
В конкретном варианте осуществления предложены способы лечения или предупреждения заболевания или нарушения, связанного с ингибированием mTOR, включающего, но не ограничивающегося перечисленными, синдромы опухоли, появляющиеся в результате непосредственно или не непосредственно из генетических дефектов в PTEN (делеции фосфатазной и тензиновой гомологии на хромосоме 10), TSC1 (туберозного склероза 1), TSC2 (туберозного склероза 2), NF1 (нейрофибромина 1), АМРК (АМР-зависимой протеинкиназы STK11, серин/треонинкиназы 11) и LKB1. Не основываясь на теории, полагают, что генетические дефекты, связанные с такими белками, приводят к гиперактивности пути метаболизма mTOR. Конкретные заболевания, которые подвергают лечению или предупреждают посредством ингибирования пути метаболизма mTOR, включают, но не ограничиваются перечисленными, болезнь Коудена, синдром Коудена, Коуден-подобный синдром, синдром Баннаяна-Зонана, синдром Баннаян-Рилей-Рувалсаба, болезнь Лермитте-Дуклос, эндометриальную карциному, карциному простаты и злокачественную меланому, комплекс туберозного склероза, лимфангиолейомиоматоз, нейрофиброматоз 1, семейную гипертрофическую кардиомиопатию, синдром Пеутц Ождегерс, карциному почечных клеток и поликистозное почечное заболевание.
В конкретном варианте осуществления предложены способы лечения или предупреждения заболевания или нарушения, связанного с модулированием, например ингибированием, киназы, включающей, но не ограничивающейся перечисленными, такие как тирозин-протеинкиназу (ZAP-70), протеин-тирозинкиназу 2 бета (PYK2), киназу 1 фокальной адгезии (FAK), киназу В лимфоцита (BLK), киназу кроветворных клеток (НСК), вирусную саркому v-yes-1 Ямагучи, аналогичную гомологу онкогена (LYN), Т-клеточноспецифичную протеин-тирозинкиназу (LCK), протоонкогенную тирозин-протеинкиназу (YES), протоонкогенную тирозин-протеинкиназу (SRC), протоонкогенную тирозин-протеинкиназу (FYN), прот-онкогенную тирозин-протеинкиназу (FGR), протоонкогенную тирозин-протеинкиназу (FER), протоонкогенную тирозин-протеинкиназу (FES), С-SRC киназу, протеин-тирозинкиназу (CYL), тирозин-протеинкиназу (CSK), мегакариоцит-ассоциированную тирозин-протеинкиназу (СТК), рецептор тирозин-протеинкиназы (ЕРН), эфрин типа-А рецептор 1, эфрин типа-А рецептор 4 (ЕРНА4), эфрин типа-В рецептор 3 (ЕРНВ3), эфрин типа-А рецептор 8 (ЕРНА8), рецептор нейротрофической тирозинкиназы, типа 1 (NTRK1), протеин-тирозинкиназу (РТК2), syk-аналогичную тирозинкиназу (SRK), протеин-тирозинкиназу (СТК), tyro3 протеин-тирозинкиназу (TYRO3), тирозинкиназу брутон агаммаглобулинемии (ВТК), тирозинкиназу лейкоцитов (LTK), протеин-тирозинкиназу (SYK), протеин-тирозинкиназу (STY), tek-тирозинкиназу (ТЕК), elk-аналогичную тирозинкиназу (ERK), тирозинкиназу с иммуноглобулином и доменами, гомологичными фактору egf (TIE), протеин-тирозинкиназу (TKF), рецептор, типа 3 нейротрофической тирозинкиназы, (NTRK3), протеинкиназу-3 смешанной последовательности (MLK3), протеинкиназу митоген-активированную 4 (PRKM4), протеинкиназу митоген-активированную 1 (PRKM1), протеин-тирозинкиназу (РТК7), протеин-тирозинкиназу (ЕЕК), гомолог мини-головного мозга (дрозофилы) (MNBH), киназу костного мозга, х-связанную (ВМХ), eph-подобную тирозинкиназу 1 (ЕТК1), макрофаг стимулирующий 1 рецептор (MSTIR), btk-ассоциированный белок, 135 kd, лимфоцит-специфическую протеин-тирозинкиназу (LCK), рецептор-2 фактора роста фибробласта (FGFR2), протеин-тирозинкиназу-3 (TYK3), протеин-тирозинкиназу (ТХК), tec протеин-тирозинкиназу (ТЕС), протеин-тирозинкиназу-2 (TYK2), лиганд 1 eph-аналогичной рецепторной тирозинкиназы (EPLG1), Т-клеточную тирозинкиназу (ЕМТ), eph тирозинкиназу 1 (ЕРНТ1), zona pellucida рецепторную тирозинкиназу, 95 кд (ZRK), протеинкиназу, митоген-активированную, киназу 1 (PRKMK1), eph тирозинкиназу 3 (ЕРНТ3), специфичный для задержки роста ген-6 (GAS6), рецептор домена вставки киназы (KDR), axl рецепторную тирозинкиназу (AXL), рецептор-1 фактора роста фибробласта (FGFR1), гомолог 2 онкогена v-erb-b2 птичьего эритробластного вируса лейкоза (ERBB2), fms-подобную тирозинкиназу-3 (FLT3), нейроэпителиальную тирозинкиназу (NEP), аналогичную рецептору-3 нейротрофическую тирозинкиназу (NTRKR3), лиганд 5 epf-аналогичной рецепторной тирозинкиназы (EPLG5), нейротрофическую тирозинкиназу, рецептор, типа 2 (NTRK2), рецептор-подобную тирозинкиназу (RYK), тирозинкиназу, специфичную для b-лимфоцита (BLK), epf тирозинкиназу 2 (ЕРНТ2), лиганд 2 epf-аналогичную рецепторную тирозинкиназу (EPLG2), болезни VIII накопления гликогена, лиганд 7 epf-аналогичную рецепторную тирозинкиназу (EPLG7), janus киназу 1 (JAK1), fms-аналогичную тирозинкиназу-1 (FLT1), протеинкиназу, camp-зависимую, регуляторную, типа I альфа (PRKAR1A), wee-1 тирозинкиназу (WEE1), epf-подобную тирозинкиназу 2 (ЕТК2), рецепторную тирозинкиназу musk, инсулиновый рецептор (INSR), janus киназу 3 (JAK3), fms-аналогичной тирозинкиназы-3 лиганд протеинкиназы с, бета 1 (PRKCB1), тирозинкиназы-типа клеточный поверхностный рецептор (HER3), janus киназу 2 (JAK2), lim домен киназу 1 (LIMK1), фосфатазу 1 двойной специфичности (DUSP1), киназу кроветворной клетки (НСК), белок активации тирозин 3-монооксигеназы/триптофан 5-монооксигеназы, eta полипептид (YWHAH), ret протоонкоген (RET), белок активации тирозин 3-монооксигеназы/триптофан 5-монооксигеназы, зета полипептид (YWHAZ), белок активации тирозин 3-монооксигеназы/триптофан 5-монооксигеназы, бета полипептид (YWHAB), трансмембранную киназу гепатомы (НТК), map киназу киназы 6, фосфатидилинозит 3-киназы, каталитический, альфа полипептид (PIK3CA), ингибитор 3 циклин-зависимой киназы (CDKN3), диацилглицерин киназу, дельта, 130 кд, протеин-тирозинфосфатазу, нерецепторный тип, 13 (PTPN13), онкогенный гомолог вируса abelson мышиного лейкоза (ABL1), диацилглицерин киназу, альфа (DAGK1), киназу 2 фокальной адгезии, рецептор 1 эпителиального дискоидного домена (EDDR1), киназу анапластической лимфомы (ALK), фосфатидилинозит 3-киназы, каталитический гамма полипептид (PIK3CG), регуляторную субъединицу фосфатидилинозит 3-киназы (PIK3R1), eph гомологичную киназу-1 (ЕНК1), гомолог онкогена кошачьего вируса v-kit hardy-zuckerman 4 (KIT), рецептор-3 фактора роста фибробластов (FGFR3), сосудистый эндотелиальный фактор с роста (VEGFC), рецептор эпидермального фактора роста (EGFR), онкоген (TRK), белок-7 рецептор-связанного фактора роста (GRB7), ras p21 активатор белка (RASA2), met протоонкоген (МЕТ), src-подобный адаптер (SLA), сосудистый эндотелиальный фактор роста (VEGF), рецептор сосудистого эндотелиального фактора роста (VEGFR), рецептор фактора роста нерва (NGFR), рецептор тромбоцитарного фактора роста (PDGFR), бета рецептор тромбоцитарного фактора роста (PDGFRB), тирозин-(Y)-фосфорилированием регулируемую киназу 2 двойной специфичности (DYRK2), регулируемую тирозин-(Y)-фосфорилированием киназу 3 двойной специфичности (DYRK3), регулируемую тирозин-(Y)-фосфорилированием киназу 4 двойной специфичности (DYRK4), регулируемую тирозин-(Y)-фосфорилированием киназу 1А двойной специфичности (DYRK1A), регулируемую тирозин-(Y)-фосфорилированием киназу 1В двойной специфичности (DYRK1B), CDC-подобную киназу 1 (CLK1), протеин-тирозинкиназу STY, CDC-подобную киназу 4 (CLK4), CDC-подобную киназу 2 (CLK2) или CDC-подобную киназу 3 (CLK3).
В другом варианте осуществления предложены способы лечения или предупреждения заболевания или нарушения, связанного с модулированием, например ингибированием, серин/треонин киназы или аналогичных молекул, включающих, но не ограничивающихся перечисленными, Akt/протеинкиназу В, протеинкиназу А (РКА), СК2, циклин-зависимую киназу 7 (CDK7), rac серин/треонин протеинкиназу, серин-треонин протеинкиназу n (PKN), серин/треонин протеинкиназу 2 (STK2), zipper протеинкиназу (ZPK), протеин-тирозинкиназу (STY), тирозинкиназу bruton агаммаглобулинемии (ВТК), mkn28 киназу, протеинкиназу, х-связанную (PRKX), elk-аналогичную тирозинкиназу (ERK), рибосомной протеин-s6-киназы, 90 кд, полипептид 3 (RPS6KA3), ассоциированную с гибелью протеинкиназу 1 болезни VIII накопления гликогена (DAPK1), pctaire протеинкиназу 1 (РСТК1), протеинкиназу, интерферон-индуцибельной РНК с двойной спиралью (PRKR), типа II-подобную киназу 1 рецептора активина (ACVRLK1), протеинкиназу, camp-зависимую, каталитическую, альфа (PRKACA), протеинкиназу, y-связанную (PRKY), G белок-связанную рецепторную киназу 2 (GPRK21), протеинкиназу с, тэта формы (PRKCQ), lim домена киназу 1 (LIMK1), фосфоглицерат киназу 1 (PGK1), lim домена киназу 2 (LIMK2), c-jun киназы, активина а рецептор, тип II-подобную киназу 2 (ACVRLK2), janus киназу 1 (JAK1), elk1 мотив киназу (ЕМК1), киназу, ассоциированную с мужской половой клеткой (МАК), альфа-примированную субъединицу казеинкиназы 2 (CSNK2A2), бета полипептид казеинкиназы 2 (CSNK2B), альфа 1 полипептид казеинкиназы 2 (CSNK2A1), ret прото-онкоген (RET), убиквитарную киназу кроветворного предшественника киназы 1, с сохраненной (формой) спираль-петля-спираль (CHUK), казеинкиназу 1, дельта (CSNK1D), казеинкиназу 1, эпсилон (CSNK1E), гомолог 1 онкогена мышиного вируса тимомы v-akt (АКТ1), опухолевый белок р53 (ТР53), регуляторную (ингибиторную) субъединицу 2 протеинфосфатазы 1 (PPP1R2), онкоген pim-1 (PIM1), бета рецептор трансформирующего фактора роста, тип II (TGFBR2), бета рецептор трансформирующего фактора роста типа I (TGFBR1), онкогенный гомолог b1 вируса v-raf мышиной саркомы (BRAF), костный морфогенетический рецептор типа II (BMPR2), гомолог 1 онкогена вируса v-raf мышиной саркомы 3611 (ARAF1), гомолог 2 онкогена вируса v-raf мышиной саркомы 3611 (ARAF2), протеинкиназа С (РКС), гомолог онкогена вируса v-kit hardy-zuckerman 4 кошачьей саркомы (KIT) или с-KIT рецептор (KITR).
В другом варианте осуществления предложены способы лечения или предупреждения заболевания или нарушения, связанного с модулированием, например ингибированием, МАР киназы, включающей, но не ограничивающиеся перечисленными, такие как митоген-активированная протеинкиназа 3 (МАРК3), p44erk 1, p44mapk, митоген-активируемая протеинкиназа 3 (МАР киназа 3; р44), ERK1, PRKM3, P44ERK1, P44MAPK, митоген-активируемая протеинкиназа 1 (МАРК1), митоген-активируемая протеинкиназа киназа 1 (МЕК1), МАР2К1 протеин-тирозинкиназа ERK2, митоген-активируемая протеинкиназа 2, внеклеточным сигналом-регулируемая киназа 2, протеин-тирозинкиназа 2 ERK2, митоген-активируемая протеинкиназа 2, внеклеточным сигналом-регулируемая киназа 2, ERK, p38, p40, p41, ERK2, ERT1, MAPK2, PRKM1, PRKM2, P42MAPK, р41 mapk, митоген-активируемая протеинкиназа 7 (МАРК7), ВМК1 киназа, внеклеточным сигналом-регулируемая киназа 5, ВМК1, ERK4, ERK5, PRKM7, nemo-подобная киназа (NLK), вероятный ортолог мышиной nemo-подобной киназы, митоген-активируемая протеинкиназа 8 (МАРК8), протеинкиназа JNK1, JNK1 бета протеинкиназа, JNK1 альфа протеинкиназа, с-Jun N-концевая киназа 1, стресс-активируемая протеинкиназа JNK1, JNK, JNK1, PRKM8, SAPK1, JNK1A2, JNK21B1/2, митоген-активируемая протеинкиназа 10 (МАРК10), с-Jun киназа 3, JNK3 альфа протеинкиназа, с-Jun N-концевая киназа 3, стресс-активируемая протеинкиназа JNK3, стресс-активируемая протеинкиназа бета, митоген-активируемая протеинкиназа 9 (МАРК9), МАР киназа 9, с-Jun киназа 2, с-Jun N-концевая киназа 2, стресс-активируемая протеинкиназа JNK2, JNK2, JNK2A, JNK2B, PRKM9, JNK-55, JNK2BETA, p54aSAPK, JNK2ALPHA, митоген-активируемая протеинкиназа 14 (МАРК14), р38 МАР киназа, МАР киназа Mxi2, Csaids связывающий белок, МАХ-взаимодействующий белок 2, стресс-активируемая протеинкиназа 2А, р38 митоген-активируемая протеинкиназа, цитокин супрессивный противовоспалительный белок, связывающий лекарственное средство, RK, p38, EXIP, Mxi2, CSBP1, CSBP2, CSPB1, PRKM14, PRKM15, SAPK2A, p38ALPHA, митоген-активируемая протеинкиназа 11 (МАРК11), стресс-активируемая протеинкиназа-2, стресс-активируемая протеинкиназа-2b, митоген-активируемая протеинкиназа р38-2, митоген-активируемая протеинкиназа р38бета, Р38В, SAPK2, p38-2, PRKM11, SAPK2B, p38Бета, Р38БЕТА2, митоген-активируемая протеинкиназа 13 (МАРК13), стресс-активируемая протеинкиназа 4, митоген-активируемая протеинкиназа р38 дельта, SAPK4, PRKM13, р38дельта, митоген-активируемая протеинкиназа 12 (МАРК12), р38гамма, стресс-активируемая протеинкиназа 3, митоген-активируемая протеинкиназа 3, ERK3, ERK6, SAPK3, PRKM12, SAPK-3, P38ГАММА, митоген-активируемая протеинкиназа 6 (МАРК6), изоформа р97 МАР киназы, митоген-активируемая 5 протеинкиназа, митоген-активируемая 6 протеинкиназа, внеклеточным сигналом-регулируемая киназа 3, внеклеточным сигналом-регулируемая киназа р97, ERK3, PRKM6, p97MARK, митоген-активируемая протеинкиназа 4 (МАРК4), Erk3-аналогичная протеинкиназа, митоген-активируемая 4 протеинкиназа (МАР киназа 4; р63), PRKM4, p63MAPK, ERK3-аналогичная или ERK3-внеклеточным сигналом-регулируемая киназа 8 (ERK7).
Гетероарильное соединение можно объединять с другими фармакологически активными соединениями («вторыми активными агентами») в описанных способах и композициях. Полагают, что некоторые комбинации могут действовать синергически при лечении конкретных типов заболеваний или нарушений, и патологических состояний и симптомов, связанных с такими заболеваниями или нарушениями. Гетероарильное соединение может также действовать для облегчения неблагоприятных действий, связанных с некоторыми вторыми активными агентами и vice versa.
В описанных способах и композициях можно применять один или несколько вторых активных ингредиентов или агентов. Вторые активные агенты могут представлять собой большие молекулы (например, белки) или небольшие молекулы (например, синтетические неорганические, органометаллические или органические молекулы).
Примеры больших молекул активных агентов включают, но не ограничиваются перечисленными, кроветворные факторы роста, цитокины и моноклональные и поликлональные антитела. Специфические примеры активных агентов представляют собой анти-CD40 моноклональные антитела (такие как, например, SGN-40); ингибиторы гистон деацетилазы (такие как, например, SAHA и LAQ824); ингибиторы белка-90 теплового шока (такие как, например, 17-AAG); ингибиторы рецепторной киназы инсулиноподобного фактора-1 роста; ингибиторы рецепторной киназы сосудистого эндотелиального фактора роста (такие как, например, РТК787); ингибиторы рецепторов инсулинового фактора роста; ингибиторы ацилтрансферазы лизофосфатидовой кислоты; ингибиторы IkB киназы; р38МАРК ингибиторы; EGFR ингибиторы (такие как, например, гефитиниб и ерлотиниб HCl); HER-2 антитела (такие как, например, трастузумаб (Herceptin®) и пертузумаб (OmnitargTM)); VEGFR антитела (такие как, например, бевацизумаб (AvastinTM)); VEGFR ингибиторы (такие как, например, flk-1 специфичные ингибиторы киназы, SU5416 и ptk787/zk222584); PI3K ингибиторы (такие как, например, вортманнин); С-Met ингибиторы (такие как, например, РНА-665752); моноклональные антитела (такие как, например, ритуксимаб (Rituxan®), тозитумомаб (Bexxar®), едреколомаб (Panorex®) и G250); и анти-TNF-α антитела. Примеры небольших молекул активных агентов включают, но не ограничиваются перечисленными, небольшие молекулы противораковых агентов и антибиотиков (например, кларитромицин).
Специфические вторые активные соединения, которые можно объединять с гетероарильным соединением, различаются в зависимости от специфического показания при лечении, предупреждении или руководстве лечением.
Например, для лечения, предупреждения или руководства при лечении рака вторые активные агенты включают, но не ограничиваются перечисленными, семаксаниб, циклоспорин, этанерцепт, доксициклин, бортезомиб, активицин, акларубицин, акодазол гидрохлорид, акронин, адозелезин, алдезлейкин, алтретамин, амбомицин, аметантрон ацетат, амсакрин, анастрозол, антрамицин, аспарагиназа, асперлин, азацитидин, азетепа, азотомицин, батимастат, бензодепа, бикалутамид, бисантрен гидрохлорид, биснафид димезилат, бизелесин, блеомицин сульфат, бреквинар натрий, бропиримин, бусульфан, кактиномицин, калустерон, карасемид, карбетимер, карбоплатин, кармустин, карубицин гидрохлорид, карзелезин, цедефингол, целекоксиб, хлорамбуцил, циролемицин, цисплатин, кладрибин, криснатол мезилат, циклофосфамид, цитарабин, дакарбазин, дактиномицин, даунорубицин гидрохлорид, децитабин, дексормаплатин, дезагуанин, дезагуанин мезилат, диазиквон, доцетаксел, доксорубицин, доксорубицин гидрохлорид, дролоксифен, дролоксифен цитрат, дромостанолон пропионат, дуазомицин, эдатрексат, эфлорнитин гидрохлорид, элсамитруцин, энлоплатин, энпромат, эпипропидин, эпирубицин гидрохлорид, эрбулозол, эзорубицин гидрохлорид, эстрамустин, эстрамустин фосфат натрий, этанидазол, этопосид, этопосид фосфат, этоприн, фадрозол гидрохлорид, фазарабин, фенретинид, флоксуридин, флударабин фосфат, фторурацил, фторцитабин, фосквидон, фостриецин натрий, гемцитабин, гемцитабин гидрохлорид, гидроксимочевина, идарубицин гидрохлорид, ифосфамид, илмофосин, ипроплатин, иринотекан, иринотекан гидрохлорид, ланреотид ацетат, летрозол, лейпролид ацетат, лиарозол гидрохлорид, лометрексол натрий, ломустин, лозоксантрон гидрохлорид, масопрокол, майтанзин, меклоретамин гидрохлорид, мегестрол ацетат, меленгестрол ацетат, мелфалан, меногарил, меркаптопурин, метотрексат, метотрексат натрий, метоприн, метуредепа, митиндомид, митокарцин, митокромин, митогиллин, митомалцин, митомицин, митоспер, митотан, митоксантрон гидрохлорид, микофенольная кислота, нокодазол, ногаламицин, ормаплатин, оксисуран, паклитаксель, пегаспаргас, пелиомицин, пентамустин, пепломицин сульфат, перфосфамид, пипоброман, пипосульфан, пироксантрон гидрохлорид, пликамицин, пломестан, порфимер натрий, порфиромицин, преднимустин, прокарбазин гидрохлорид, пуромицин, пуромицин гидрохлорид, пиразофурин, рибоприн, сафингол, сафингол гидрохлорид, семустин, симтразен, спарфосат натрий, спарсомицин, спирогерманий гидрохлорид, спиромустин, спироплатин, стрептонигрин, стрептозоцин, сулофенур, талисомицин, текогалан натрий, таксотер, тегафур, телоксантрон гидрохлорид, темопорфин, тенипосид, тероксирон, тестолактон, тиамиприн, тиогуанин, тиотепа, тиазофурин, тирапазамин, торемифен цитрат, трестолон ацетат, трицирибин фосфат, триметрексат, тримексетрат глюкуронат, трипторелин, тубулозол гидрохлорид, урацил мустард, уредепа, вапреотид, вертепорфин, винбластин сульфат, винкристин сульфат, виндесин, виндесин сульфат, винепидин сульфат, винглицинат сульфат, винлейросин сульфат, винорелбин тартрат, винросидин сульфат, винзолидин сульфат, ворозол, зениплатин, зиностатин и зорубицин гидрохлорид.
Другие вторые агенты включают, но не ограничиваются перечисленными: 20-эпи-1,25-дигидроксивитамин Д3, 5-этинилурацил, абиратерон, акларубицин, ацилфульвен, адеципенол, адозелесин, алдезлейкин, ALL-TK антагонисты, алтретамин, амбамустин, амидокс, амифостин, аминолевулиновая кислота, амрубицин, амсакрин, анагрелид, анастрозол, андрографолид; ингибиторы ангиогенеза, антагонист D, антагонист G, антареликс, анти-дорсалазиновый морфогенный белок-1, антиандроген карциномы простаты, антиэстроген, антинеопластон, антисмысловые олигонуклеотиды, афидиколин глицинат, модуляторы апоптоза гена, регуляторы апоптоза, апуриновая кислота, ara-CDP-DL-PTBA, аргинин деаминаза, асулакрин, атаместан, атримустин, аксинастатин 1, аксинастатин 2, аксинастатин 3, азасетрон, азатоксин, азатирозин, производные баккатина III, баланол, батимастат, BCR/ABL антагонисты, бензохлорины, бензоилстауроспорин, производные бета-лактама, бета-алетин, бетакламицин В, бетулиновая кислота, bFGF ингибитор, бикалутамид, бисантрен, бисазиридинилспермин, биснафид, бистратен А, бизелесин, брефлат, бропиримин, будотитан, бутионин сульфоксимин, кальципотриол, калфостин С, производные камптотецина, капецитабин, карбоксамидаминотриазол, карбоксиамидотриазол, CaRest M3, CARN 700, хрящевой ингибитор, карзелесин, ингибиторы казеинкиназы (ICOS), кастаноспермин, секропин В, цетрореликс, хлорины, хлорхиноксалин сульфонамид, цикапрост, цис-порфирин, кладрибин; клатромицин, аналоги кломифена, клотримазол, коллисмицин А, коллисмицин В, комбретастатин А4, аналог комбретастатина, конагенин, крамбесцидин 816, криснатол, криптофицин 8, производные криптофицина А, курацин А, циклопентантрахиноны, циклоплатам, ципемицин, цитарабин окфосфат, цитолитический фактор, цитостатин, дакликсимаб, децитабин, дегидродидемнин В, деслорелин, дексаметазон, дексифосфамид, дексразоксан, дексверапамил, диазиквон, дидемнин В, дидокс, диэтилнорспермин, дигидро-5-азацитидин, дигидротаксол 9, диоксамицин, дифенилспиромустин, доцетаксель, докозанол, долазетрон, доксифлуридин, доксорубицин, дролоксифен, дронабинол, дуокармицин SA, эбселен, экомустин, эделфосин, эдреколомаб, эфлорнитин, элемен, эмитефур, эпирубицин, эпристерид, аналог эстрамустина, агонисты эстрогена, антагонисты эстрогена, этанидазол, этопосид фосфат, эксеместан, фадрозол, фазарабин, фенретинид, филграстим, финастерид, флавопиридол, флезеластин, флуастерон, флударабин, фтордауноруницин гидрохлорид, форфенимекс, форместан, фостриецин, фотемустин, гадолиний тексафирин, галлий нитрат, галоцитабин, ганиреликс, ингибиторы желатиназы, гемцитабин, ингибиторы глутатиона, гепсульфам, герегулин, гексаметиленбисацетамид, гиперицин, ибандроновая кислота, идарубицин, идоксифен, идрамантон, илмофосин, иломастат, иматиниб (Gleevec®), имихимод, иммуностимулирующие пептиды, ингибитор рецептора инсулиноподобного фактора-1 роста, агонисты интерферона, интерфероны, интерлейкины, иобенгуан, йододоксирубицин, ипомеанол 4, ироплакт, ирсогладин, изобенгазол, изогомохаликондрин В, итасетрон, жасплакинолид, кахалалид F, ламелларин-N триацетат, ланреотид, лейнамицин, ленограстим, лентинан сульфат, лептолстатин, летрозол, фактор ингибирования лейкоза, интерферон альфа лейкоцита, лейпролид+эстроген+прогестерон, лейпрорелин, левамисол, лиарозол, аналог линейного полиамина, пептид липофильного дисахарида, липофильные платиновые соединения, лиссоклинамид 7, лобаплатин, ломбрицин, лометрексол, лонидамин, лозоксантрон, локсорибин, луртотекан, лютеций тексафирин, лизофиллин, литические пептиды, майтансин, манностатин А, маримастат, масопрокол, маспин, ингибиторы матрилизина, ингибиторы матричной металлопротеиназы, меногарил, мербарон, метерелин, метиониназа, метоклопрамид, MIF ингибитор, мифепристон, милтефосин, миримостим, митогуазон, митолактол, аналоги митомицина, митонафид, митотоксиновый фактор роста фибробластов-сапонин, митоксантрон, мофаротен, молграмостим, эрбитукс, хорионический гонадотропин человека, монофосфорильный липид А+миобактериальная клеточная стенка sk, мопидамол, горчичный противораковый агент, микапероксид В, экстракт микобактериальной клеточной стенки, мириапорон, N-ацетилдиналин, N-замещенные бензамиды, нафарелин, нагрестип, налоксон+пентазоцин, напавин, нафтерпин, нартограстим, недаплатин, неморубицин, неридроновая кислота, нилутамид, нисамицин, модуляторы оксида азота, нитроксидный антиоксидант, нитруллин, облимерсен (Genasense®), О6-бензилгуанин, октреотид, окиценон, олигонуклеотиды, онапристон, ондансетрон, орацин, пероральный цитокиновый индуктор, ормаплатин, осатерон, оксалиплатин, оксауномицин, паклитаксель, аналоги паклитакселя, производные паклитакселя, палауамин, пальмитоилризоксин, памидроновая кислота, панакситриол, паномифен, парабактин, пазеллиптин, пегаспаргаза, пелдесин, пентозан полисульфат натрий, пентостатин, пентрозол, перфлуброн, перфосфамид, перилловый спирт, феназиномицин, фенилацетат, ингибиторы фосфатазы, пицибанил, пилокарпин гидрохлорид, пирарубицин, пиритрексим, плацетин А, плацетин В, ингибитор активатора плазминогена, платиновый комплекс, платиновые соединения, комплекс платина-триамин, порфимер натрий, порфиромицин, преднизон, пропилбисакридон, простагландин J2, ингибиторы протеасомы, иммунный модулятор на основе белка А, ингибитор протеинкиназы С, ингибиторы протеинкиназы С, микроалгал, ингибиторы протеин-тирозинфосфатазы, ингибиторы пурин нуклеозид фосфорилазы, пурпурины, пиразолакридин, конъюгат пиридоксилированный гемоглобин полиоксиэтилен, антагонисты raf, ралтитрексед, рамосетрон, ингибиторы ras фарнезил-протеинтрансферазы, ингибиторы ras, ингибитор ras-GAP, деметилированный ретеллиптин, рений Re 186 этидронат, ризоксин, рибозимы, RII ретинамид, рохитукин, ромуртид, рохинимекс, рубигинон В1, рубоксил, сафингол, саинтопин, SarCNU, саркофитол А, сарграмостим, Sdi 1 миметики, семустин, сенесценсный ингибитор 1 (старения), смысловые олигонуклеотиды, ингибиторы сигнальной трансдукции, сизофиран, собузоксан, натрий борокаптат, натрий фенилацетат, солверол, соматомедин-связывающий белок, сонермин, спарфосивая кислота, спикамицин D, спиромустин, спленопентин, спонгистатин 1, скваламин, стипиамид, стромелизиновые ингибиторы, сульфинозин, антагонист сверхактивного вазоактивного кишечного пептида, сурадиста, сурамин, свайнсонин, таллимустин, тамоксифен метиодид, тауромустин, тазаротен, текогалан натрий, тегафур, теллурапирилий, ингибиторы теломеразы, темопорфин, тенипозид, тетрахлордекаоксид, тетразомин, талибластин, тиокоралин, тромбопоэтин, миметик тромбопоэтина, тималфазин, агонист рецептора тимопоэтина, тимотринан, гормон, стимулирующий щитовидную железу, этилолово этиопурпурин, тирапазамин, титаноцен бихлорид, топсентин, торемифен, ингибиторы трансляции, третиноин, триацетилуридин, трицирибин, триметрексат, трипторелин, трописетрон, туростерид, ингибиторы тирозинкиназы, тирфостины, UBC ингибиторы, убенимекс, фактор ингибирования роста мочеполового синуса, антагонисты рецепторов урокиназы, вапреотид, вариолин В, веларесол, верамин, вердины, вертепорфин, винорелбин, винксалтин, витаксин, ворозол, занотерон, зениплатин, зиласкорб и зиностатин стималамер.
Специфические вторые активные агенты включают, но не ограничиваются перечисленными, 2-метоксиэстрадиол, теломестатин, индукторы апоптоза в клетках множественной миеломы (такие как, например, TRAIL), бортезомиб, статины, семаксаниб, циклоспорин, этанерцепт, доксициклин, бортезомиб, облимерсен (Genasense®), ремикад, доцетаксел, целекоксиб, мелфалан, дексаметазон (Decadron®), стероиды, гемцитабин, цисплатинум, темозоломид, этопозид, циклофосфамид, темодар, карбоплатин, прокарбазин, глиадел, тамоксифен, топотекан, метотрексат, Arisa®, таксол, таксотер, фторурацил, лейковорин, иринотекан, кселода, СРТ-11, интерферон альфа, пегилированный интерферон альфа (например, PEG INTRON-A), капецитабин, цисплатин, тиотепа, флударабин, карбоплатин, липосомный даунорубицин, цитарабин, доксетаксол, паклитаксел, винбластин, IL-2, GM-CSF, дакарбазин, винорелбин, золедроновая кислота, пальмитронат, биаксин, бусульфан, преднизон, бисфосфонат, триоксид мышьяка, винкристин, доксорубицин (Doxil®), паклитаксел, ганцикловир, адриамицин, эстрамустин натрий фосфат (Emcyt®), сулиндак и этопозид.
Аналогично примеры специфических вторых агентов в соответствии с показаниям для лечения, предупреждения или управляемого лечения можно найти в следующих ссылках, каждая из которых введена во всей своей полноте: патент США №№ 6281230 и 5635517, заявка США №№ 10/411649, 10/483213, 10/411656, 10/693794, 10/699154 и 10/981189 и предварительная заявка США №№ 60/554923, 60/565172, 60/626975, 60/630599, 60/631870 и 60/533862.
Примеры дополнительных вторых активных агентов включают, но не ограничиваются перечисленными, общепринятые терапевтические средства, применяемые для лечения или предупреждения боли, такие как антидепрессанты, противосудорожные агенты, антигипертензивные агенты, анксиолитики, блокаторы кальциевых каналов, миорелаксанты, не наркотические анальгетики, опиоидные анальгетики, противовоспалительные агенты, ингибиторы сох-2, иммуномодулирующие агенты, альфа-адренергические рецепторные агонисты или антагонисты, иммуносупрессивные агенты, кортикостероиды, гипербарический кислород, кетамин, другие обезболивающие агенты, антагонисты NMDA и другие терапевтические средства, найденные, например, в Physician's Desk Reference 2003. Специфические примеры включают, но не ограничиваются перечисленными, ацетат салициловой кислоты (Aspirin®), целекоксиб (Celebrex®), Enbrel®, кетамин, габапентин (Neurontin®), фенитоин (Dilantin®), карбамазепин (Tegretol®), окскарбазепин (Trileptal®), вальпроевая кислота (Depaken®), морфин сульфат, гидроморфон, преднизон, гризеофульвин, пентоний, алендронат, дифенгидрамид, гуанетидин, кеторолак (Acular®), тирокальцитонин, диметилсульфоксид (ДМСО), клонидин (Catapress®), бретилий, кетансерин, резерпин, дроперидол, атропин, фентоламин, бупивакаин, лидокаин, ацетаминофен, нортриптилин (Pamelor®), амитриптилин (Elavil®), имипрамин (Tofranil®), доксепин (Sinequan®), кломипрамин (Anafranil®), флуоксетин (Prozac®), сертралин (Zoloft®), нефазодон (Serzone®), венлафаксин (Effexor®), тразодон (Desyrel®), бупропион (Wellbutrin®), мексилетин, нифедипин, пропранолол, трамадол, ламотригин, зиконотид, кетамин, декстрометорфан, бензодиазепины, баклофен, тизанидин и феноксибензамин.
Примеры дополнительных вторых активных агентов включают, но не ограничиваются перечисленными, стероид, светосенсибилизатор, интегрин, антиоксидант, интерферон, производное ксантина, гормон роста, нейротрофический фактор, регулятор образования новых сосудов, анти-VEGF антитела, простагландин, антибиотик, фитоэстроген, противовоспалительное соединение или соединение против ангиогенеза или их сочетание. Специфические примеры включают, но не ограничиваются перечисленными, вертепорфин, пурлитин, ангиостатический стероид, rhuFab, интерферон-2у, пентоксифиллин, олово этиопурпурин, мотексафин лютеций, 9-фтор-11,21-дигидрокси-16, 17-1-метилэтилиденбис(окси)прегна-1,4-диен-3,20-дион, латанопрост (см. патент США № 6225348), тетрациклин и его производные, рифамицин и его производные, макролиды, метронидазол (патент США №№ 6218369 и 6015803), генистеин, генистин, 6'-O-Mal-генистин, 6'-O-Ac-генистин, даидзеин, даидзин, 6'-O-Mal-даидзин, 6'-O-Ac-даидзин, глицитеин, глицитин, 6'-O-Mal-глицитин, биочанин А, формононетин (патент США № 6001368), триамцинолон ацетомид, дексаметазон (патент США № 5770589), талидомид, глутатион (патент США № 5632984), фактор роста фибробластов основного характера (bFGF), трансформирующий фактор b роста (TGF-b), нейротрофический фактор головного мозга (BDNF), фактор типа 2 активатора плазминогена (PAI-2), EYE101 (Eyetech Pharmaceuticals), LY333531 (Eli Lilly), Miravant и RETISERT implant (Bausch & Lomb). Все цитированные выше по тексту ссылки введены во всей своей полноте посредством ссылки.
Примеры дополнительных вторых активных агентов включают, но не ограничиваются перечисленными, кератолитические средства, ретиноиды, α-гидроксикислоты, антибиотики, коллаген, ботулиновый токсин, интерферон и иммуномодулирующие агенты. Специфические примеры включают, но не ограничиваются перечисленными, 5-фторурацил, мазопрокол, трихлоруксусную кислоту, салициловую кислоту, молочную кислоту, лактат аммония, мочевину, третиноин, изотретиноин, антибиотики, коллаген, ботулиновый токсин, интерферон, кортикостероид, трансретиноевую кислоту и коллагены, такие как плацентарный коллаген человека, плацентарный коллаген животного, дермалоген, аллодерм, фасция, циметра, аутологен, зидерм, зипласт, резопласт и изолаген.
Примеры дополнительных вторых активных агентов включает, но не ограничивается перечисленными, антикоагулянты, диуретики, сердечные гликозиды, блокаторы кальциевых каналов, вазодилататоры, аналоги простациклина, антагонисты эндотелина, ингибиторы фосфодиэстеразы (например, ингибиторы PDE V), ингибиторы эндопептидазы, агенты, снижающие липиды, ингибиторы тромбоксана и другие терапевтические средства, известные для снижения легочного артериального давления. Специфические примеры включают, но не ограничиваются перечисленными, варфарин (Coumadin®), диуретик, сердечный гликозид, дигоксин-кислород, дилтиазем, нифедипин, вазодилататор, такой как простациклин (например, простагландин I2 (PGI2), эпопростенол (EPO, Floran®), трепростинил (Remodulin®), оксид азота (NO), бозентан (Tracleer®), амлодипин, эпопростенол (Floran®), трепростинил (Remodulin®), простациклин, тадалафил (Cialis®), симвастатин (Zocor®), омапатрилат (Vanlev®), ирбесартан (Avapro®), правастатин (Pravachol®), дигоксин, L-аргинин, илопрост, бетапрост и силденафил (Viagra®).
Примеры дополнительных вторых активных агентов включают, но не ограничиваются перечисленными, антрациклин, платинум, алкилирующий агент, облимерсен (Genasense®), цисплатинум, циклофосфамид, темодар, карбоплатин, прокарбазин, глиадел, тамоксифен, топотекан, метотрексат, таксотер, иринотекан, капецитабин, цисплатин, тиотепа, флударабин, карбоплатин, липосомный даунорубицин, цитарабин, доксетаксол, паклитаксел, винбластин, IL-2, GM-CSF, дакарбазин, винорелбин, золедроновая кислота, пальмитронат, биаксин, бусульфан, преднизон, бисфосфонат, триоксид мышьяка, винкристин, доксорубицин (Doxil®), паклитаксел, ганцикловир, адриамицин, блеомицин, гиалуронидазу, митомицин С, мепакрин, тиотепа, тетрациклин и гемцитабин.
Примеры дополнительных вторых активных агентов включают, но не ограничиваются перечисленными, хлорохин, хинин, хинидин, пириметамин, сульфадиазин, доксициклин, клиндамицин, мефлохин, галофантрин, примахин, гидроксихлорохин, прогуанил, атоваквон, азитромицин, сурамин, пентамидин, меларсопрол, нифуртимокс, бензнидазол, амфотерицин В, пентавалентные соединения сурьмы (например, стибоглюкуронат натрий) интерферон гамма, итраконазол, комбинация мертвых промастиготов и BCG, лейковорин, кортикостероиды, сульфонамид, спирамицин, IgG (серологический), триметоприм и сульфаметоксазол.
Примеры дополнительных вторых активных агентов включают, но не ограничиваются перечисленными, антибиотики (терапевтические или профилактические), такие как, но не ограниченные перечисленными, ампициллин, кларитромицин, тетрациклин, пенициллин, цефалоспорины, стрептомицин, канамицин и эритромицин; антивирусные средства, такие как, но не ограниченные перечисленными, амантадин, римантадин, ацикловир и рибавирин; иммуноглобулин; плазму; иммунологические усиливающие лекарственные средства, такие как, но не ограниченные перечисленными, левамисол и изоприносин; биологические средства, такие как, но не ограниченные перечисленными, гаммаглобулин, фактор переноса, интерлейкины и интерфероны; гормоны, такие как, но не ограниченные перечисленными, тимусный; и другие иммунологические агенты, такие как, но не ограниченные перечисленными, В-клеточные стимуляторы (например, BAFF/BlyS), цитокины (например, IL-2, IL-4 и IL-5), факторы роста (например, TGF-y), антитела (например, анти-CD-40 и IgM), олигонуклеотиды, содержащие неметилированные мотивы CpG, и вакцины (например, вирусные и опухолевые пептидные вакцины).
Примеры дополнительных вторых активных агентов включают, но не ограничиваются перечисленными, допаминовый агонист или антагонист, такой как, но не ограничивающийся перечисленными, леводопа, L-DOPA, кокаин, α-метилтирозин, резерпин, тетрабеназин, бензотропин, паргилин, фенодолпам мезилат, каберголин, прамипексол дигидрохлорид, ропинорол, амантадин гидрохлорид, селегилин гидрохлорид, карбидопа, перголид мезилат, синемет CR и симметрел; ингибитор МАО, такой как, но не ограниченный перечисленными, ипрониазид, клоргилин, фенелзин и изокарбоксазид; ингибитор СОМТ, такой как, но не ограниченный перечисленными, толкапон и энтакапон; ингибитор холинестеразы, такой как, но не ограниченный перечисленными, физостигмин салицилат, физостигмин сульфат, физостигмин бромид, меостигмин бромид, неостигмин метилсульфат, амбеноним хлорид, эдрофоний хлорид, такрин, пралидоксим хлорид, обидоксим хлорид, тримедоксим бромид, диацетилмоноксим, эндрофоний, пиридостигмин и демекариум; и противовоспалительный агент, такой как, но не ограниченный перечисленными, напроксен натрий, диклофенак натрий, диклофенак калий, целекоксиб, сулиндак, оксапрозин, дифлунизал, этодолак, мелоксикам, ибупрофен, кетопрофен, набуметон, рефекоксиб, метотрексат, лефлуномид, сулфазалазин, соли золота, Rho-D иммуноглобулин, микофенилат мофетил, циклоспорин, азатиоприн, такролимус, базиликсимаб, даклизумаб, салициловую кислоту, ацетилсалициловую кислоту, метилсалицилат, дифлунизал, салсалат, олсалазин, сульфазалазин, ацетаминофен, индометацин, сулиндак, мефенамовую кислоту, меклофенамат натрий, толметин, кеторолак, диклофенак, флурбинпрофен, оксапрозин, пироксикам, мелоксикам, ампироксикам, дроксикам, пивоксикам, теноксикам, фенилбутазон, оксифенбутазон, антипирин, аминопирин, апазон, зилейтон, ауротиоглюкоза, тиомалат натрий золото, ауранофин, метотрексат, колхицин, аллопуринол, пробенецид, сулфинпиразон и бензбромарон или бетаметазон, и другие глюкокортикоиды; и противорвотный агент, такой как, но не ограниченный перечисленными, метоклопромид, домперидон, прохлорперазин, прометазин, хлорпромазин, триметобензамид, ондансетрон, гранисетрон, гидроксизин, ацетиллейцин моноэтаноламин, ализаприд, азасетрон, бензхинамид, биетанаутин, бромоприд, буклизин, клебоприд, циклизин, дименгидринат, дифенидол, доласетрон, меклизин, металлатал, метопимазин, набилон, оксиперидил, пипамазин, скополамин, сулпирид, тетрагидроканнабинол, тиэтилперазин, тиопроперазин, трописетрон и их смесь.
Примеры дополнительных вторых активных агентов включают, но не ограничиваются перечисленными, иммуномодуляторные агенты, иммуносупрессивные агенты, антигипертензивные средства, противосудорожные средства, фибринолитические агенты, антитромбоцитарные агенты, антипсихотические агенты, антидепрессанты, бензодиазепины, буспирон, амантадин и другие известные или общепринятые агенты, применяемые для пациентов с CNS травмой/повреждением и аналогичными синдромами. Специфические примеры включают, но не ограничиваются перечисленными, стероиды (например, глюкокортикоиды, такие как, но не ограниченные перечисленными, метилпреднизолон, дексаметазон и бетаметазон), противовоспалительный агент, включающий, но не ограниченный перечисленными, напроксен натрий, диклофенак натрий, диклофенак калий, целекоксиб, сулиндак, оксапрозин, дифлунизал, этодолак, мелоксикам, ибупрофен, кетопрофен, набуметон, рефекоксиб, метотрексат, лефлуномид, сульфазалазин, соли золота, Rho-D иммуноглобулин, микофенилат мофетил, циклоспорин, азатиоприн, такролимус, басиликсимаб, даклизумаб, салициловую кислоту, ацетилсалициловую кислоту, метилсалицилат, дифлунисал, салсалат, олсалазин, сулфазалазин, ацетаминофен, индометацин, сулиндак, мефенамовую кислоту, меклофенамат натрий, толметин, кеторолак, диклофенак, флурбинпрофен, оксапрозин, пироксикам, мелоксикам, ампироксикам, дроксикам, пивоксикам, теноксикам, фенилбутазон, оксифенбутазон, антипирин, аминопирин, апазон, зилейтон, ауротиоглюкоза, тиомалат натрий золото, ауранофин, метотрексат, колхицин, аллопуринол, пробенецид, сулфинпиразон и бензбромарон, аналог сАМР, включающий, но не ограниченный перечисленным, db-cAMP, агент, включающий метилфенидатное лекарственное средство, которое включает в себя 1-трео-метилфенидат, d-трео-метилфенидат, dl-трео-метилфенидат, l-эритрометилфенидат, d-эритрометилфенидат, dl-эритрометилфенидат и их смесь, и диуретический агент, такой как, но не ограниченный перечисленными, маннит, фуросемид, глицерин и мочевину.
Примеры дополнительных вторых активных агентов включает, но не ограничиваются перечисленными, трициклический антидепрессантный агент, селективный ингибитор повторного поглощения серотонина, противоэпилептический агент (габапентин, прегабалин, карбамазепин, окскарбазепин, левитирацетам, топирамат), антиаритмический агент, агент, блокирующий натриевый канал, селективный ингибитор медиатора воспаления, опиоидный агент, второе иммуномодуляторное соединение, комбинированный агент и другие известные общепринятые агенты, применяемые в терапии сна. Специфические примеры включают, но не ограничиваются перечисленными, нейронтин, оксиконтин, морфин, топирамат, амитриптилин, нортриптилин, карбамазепин, леводопа, L-DOPA, кокаин, α-метилтирозин, резерпин, тетрабеназин, бензотропин, паргилин, фенодолпам мезилат, каберголин, прамипексол дигидрохлорид, ропинорол, амантадин гидрохлорид, селегилин гидрохлорид, карбидопа, перголид мезилат, синемет CR, симметрел, ипрониазид, клоргилин, фенелзин, изокарбоксазид, толкапон, энтакапон, физостигмин салицилат, физостигмин сульфат, физостигмин бромид, меостигмин бромид, неостигмин метилсульфат, амбеноним хлорид, эдрофоний хлорид, такрин, пралидоксим хлорид, обидоксим хлорид, тримедоксим бромид, диацетил моноксим, эндрофоний, пиридостигмин, демекариум, напроксен натрий, диклофенак натрий, диклофенак калий, целекоксиб, сулиндак, оксапрозин, дифлунизал, этодолак, мелоксикам, ибупрофен, кетопрофен, набуметон, рефекоксиб, метотрексат, лефлуномид, сульфазалазин, соли золота, Rho-D иммуноглобулин, микофенилат мофетил, циклоспорин, азатиоприн, такролимус, базиликсимаб, даклизумаб, салициловую кислоту, ацетилсалициловую кислоту, метилсалицилат, дифлунизал, салсалат, олсалазин, сульфазалазин, ацетаминофен, индометацин, сулиндак, мефенамовую кислоту, меклофенамат натрий, толметин, кеторолак, диклофенак, флурбинпрофен, оксапрозин, пироксикам, мелоксикам, ампироксикам, дроксикам, пивоксикам, теноксикам, фенилбутазон, оксифенбутазон, антипирин, аминопирин, апазон, зилейтон, ауротиоглюкозу, тиомалат натрий золото, ауранофин, метотрексат, колхицин, аллопуринол, пробенецид, сулфинпиразон, бензбромарон, бетаметазон и другие глюкокортикоиды, метоклопромид, домперидон, прохлорперазин, прометазин, хлорпромазин, триметобензамид, ондансетрон, гранисетрон, гидроксизин, ацетиллейцин моноэтаноламин, ализаприд, азасетрон, бензхинамид, биетанаутин, бромоприд, буклизин, клебоприд, циклизин, дименгидринат, дифенидол, доласетрон, меклизин, металлатал, метопимазин, набилон, оксиперндил, пипамазин, скополамин, сулпирид, тетрагидроканнабинол, тиэтилперазин, тиопроперазин, тропизетрон и их смесь.
Примеры дополнительных вторых активных агентов включают, но не ограничиваются перечисленными, интерлейкины, такие как IL-2 (включая рекомбинантный IL-II (“rIL2”) и канарипокс IL-2), IL-10, IL-12 и IL-18; интерфероны, такие как интерферон альфа-2а, интерферон альфа-2b, интерферон альфа-n1, интерферон альфа-n3, интерферон бета-Ia и интерферон гамма-Ib; и G-CSF; гидроксимочевину, бутираты или производные бутирата; оксид азота; HEMOXINTM (NIPRISANTM, см. патент США № 5800819); антагонисты Gardos-канала, такие как клотримазол и производные триарилметана; дефероксамин; протеин С; и переливания крови или кровезаменители, такого как HemospanTM или HemospanTM PS (Sangart).
Введение гетероарильного соединения и второго активного агента пациенту может происходить одновременно или последовательно одинаковыми или различными путями введения. Пригодность конкретного пути введения, используемого для конкретного активного агента, будет зависеть от самого активного агента (например, можно ли будет его вводить перорально без разложения перед введением в кровоток) и заболевания, подвергаемого лечению. Предпочтительным путем введения гетероарильных соединений является пероральный путь. Предпочтительные пути введения для вторых активных агентов или ингредиентов изобретения известны среднему специалисту в области техники. См. Physicians' Desk Reference, 1755-1760 (56th ed., 2002).
В одном варианте осуществления второй активный агент вводят внутривенно или подкожно и однократно или дважды в день в количестве от приблизительно 1 до приблизительно 1000 мг, от приблизительно 5 до приблизительно 500 мг, от приблизительно 10 до приблизительно 350 мг или от приблизительно 50 до приблизительно 200 мг. Специфическое количество второго активного агента будет зависеть от специфического применяемого агента, типа заболевания, подвергаемого лечению, или управляемого лечения, тяжести и стадии заболевания и количества гетероарильного соединения и любых необязательных дополнительных активных агентов, одновременно вводимых пациенту.
Далее предложены способы уменьшения, лечения и/или предупреждения неблагоприятных или нежелательных действий, связанных с общепринятой терапией, включающие, но не ограничивающиеся перечисленными, хирургическое вмешательство, химиотерапию, лучевую терапию, гормональную терапию, биологическую терапию и иммунотерапию. Гетероарильные соединения и другие активные ингредиенты можно вводить пациенту до, во время или после возникновения неблагоприятного действия, связанного с общепринятой терапией.
4.5 Фармацевтические композиции и пути введения
Гетероарильные соединения можно вводить пациенту перорально или парентерально в общепринятой форме препаратов, таких как капсулы, микрокапсулы, таблетки, гранулы, порошок, пастилки, пилюли, суппозитории, инъекции, суспензии и сиропы. Подходящие препараты можно получать обычно применяемыми способами с использованием общепринятых органических или неорганических добавок, таких как эксципиент (например, сахароза, крахмал, маннит, сорбит, лактоза, глюкоза, целлюлоза, тальк, фосфат кальция или карбонат кальция), связующее средство (например, целлюлоза, метилцеллюлоза, гидроксиметилцеллюлоза, полипропилпирролидон, поливинилпирролидон, желатин, аравийская камедь, полиэтиленгликоль, сахароза или крахмал), дезинтегрирующее средство (например, крахмал, карбоксиметилцеллюлоза, гидроксипропилкрахмал, низкозамещенная гидроксипропилцеллюлоза, бикарбонат натрия, фосфат кальция или цитрат кальция), смазывающее средство (например, стеарат магния, неплотная безводная кремниевая кислота, тальк или лаурилсульфат натрия), корригирующий агент (например, лимонная кислота, ментол, глицин или апельсиновый порошок), консервант (например, бензоат натрия, бисульфит натрия, метилпарабен или пропилпарабен), стабилизатор (например, лимонная кислота, цитрат натрия или уксусная кислота), суспендирующий агент (например, метилцеллюлоза, поливинилпирролидон или стеарат алюминия), диспергирующий агент (например, гидроксипропилметилцеллюлоза), разбавитель (например, вода) и восковая основа (например, масло какао, белый петролатумный парафин или полиэтиленгликоль). Эффективное количество гетероарильного соединения в фармацевтической композиции может быть на уровне, который будет выполнять требуемое действие, например, приблизительно 0,005 мг/кг массы тела пациента до приблизительно 10 мг/кг массы тела пациента в единице дозы как для перорального, так и парентерального введения.
Доза гетероарильного соединения для введения пациенту довольно широко различается и может быть предметом для решения заботящегося о здоровье врача-практика. Обычно гетероарильные соединения можно вводить в организм пациента от одного до четырех раз в день в дозе от приблизительно 0,005 мг/кг массы тела пациента до приблизительно 10 мг/кг массы тела пациента, но вышеуказанную дозу можно должным образом изменять в зависимости от возраста, массы тела и болезненного состояния пациента и типа введения. В одном варианте осуществления доза составляет от приблизительно 0,01 мг/кг массы тела пациента до приблизительно от 5 мг/кг массы тела пациента, от приблизительно от 0,05 мг/кг массы тела пациента до приблизительно от 1 мг/кг массы тела пациента, от приблизительно от 0,1 мг/кг массы тела пациента до приблизительно от 0,75 мг/кг массы тела пациента или от приблизительно от 0,25 мг/кг массы тела пациента до приблизительно от 0,5 мг/кг массы тела пациента. В одном варианте осуществления дают одну дозу в день. В любом определенном случае количество вводимого гетероарильного соединения будет зависеть от таких факторов, как растворимость активного компонента, применяемого препарата и пути введения.
В другом варианте осуществления предложены способы лечения или предупреждения заболевания или нарушения, включающие введение от приблизительно 0,375 мг/день до приблизительно 750 мг/день, от приблизительно 0,75 мг/день до приблизительно 375 мг/день, от приблизительно 3,75 мг/день до приблизительно 75 мг/день, от приблизительно 7,5 мг/день до приблизительно 55 мг/день или от приблизительно 18 мг/день до приблизительно 37 мг/день гетероарильного соединения нуждающемуся в этом пациенту.
В другом варианте осуществления предложены способы лечения или предупреждения заболевания или нарушения, включающие введение от приблизительно 1 мг/день до приблизительно 1200 мг/день, от приблизительно 10 мг/день до приблизительно 1200 мг/день, от приблизительно 100 мг/день до приблизительно 1200 мг/день, от приблизительно 400 мг/день до приблизительно 1200 мг/день, от приблизительно 600 мг/день до приблизительно 1200 мг/день, от приблизительно 400 мг/день до приблизительно 800 мг/день или от приблизительно 600 мг/день до приблизительно 800 мг/день гетероарильного соединения нуждающемуся в этом пациенту. В конкретном варианте осуществления описанные способы включают введение 400 мг/день, 600 мг/день или 800 мг/день гетероарильного соединения нуждающемуся в этом пациенту.
В другом варианте осуществления, предложенном в описании, имеются единицы дозы препаратов, которые включают от приблизительно 1 мг до приблизительно 200 мг, от приблизительно 35 мг до приблизительно 1400 мг, от приблизительно 125 мг до приблизительно 1000 мг, от приблизительно 250 мг до приблизительно 1000 мг или от приблизительно 500 мг до приблизительно 1000 мг гетероарильного соединения.
В конкретном варианте осуществления, предложенном в описании, имеется единица дозы препарата, включающая приблизительно 100 мг или 400 мг гетероарильного соединения.
В другом варианте осуществления, предложенном в описании, имеются единицы дозы препаратов, которые включают в себя 1 мг, 5 мг, 10 мг, 15 мг, 20 мг, 30 мг, 35 мг, 50 мг, 70 мг, 100 мг, 125 мг, 140 мг, 175 мг, 200 мг, 250 мг, 280 мг, 350 мг, 500 мг, 560 мг, 700 мг, 750 мг, 1000 мг или 1400 мг гетероарильного соединения.
Гетероарильное соединение можно вводить однократно, дважды, три, четыре или более раз в день. В конкретном варианте осуществления вводили дозы 600 мг или менее как однократную суточную дозу, и дозы более чем 600 мг вводили дважды в день в количестве, равном половине полной суточной дозы.
Гетероарильное соединение можно вводить перорально по причинам удобства. В одном варианте осуществления, когда вводили перорально, гетероарильное соединение вводили с пищей или водой. В другом варианте осуществления гетероарильное соединение диспергировали в воде или соке (например, яблочном соке или апельсиновом соке) и вводили перорально в виде суспензии.
Гетероарильное соединение можно также вводить внутрикожно, внутримышечно, внутрибрюшинно, чрескожно, внутривенно, подкожно, внутрь носа, эпидурально, подъязычно, интрацеребрально, интравагинально, чрескожно, ректально, через слизистую оболочку, путем ингаляции или местно в уши, нос, глаза или кожу. Способ введения прекращают по усмотрению заботящегося о здоровье врача-практика, и он может зависеть отчасти от очага болезненного состояния.
В одном варианте осуществления предложены капсулы, содержащие гетероарильное соединение без дополнительного носителя, эксципиента или наполнителя.
В другом варианте осуществления предложены композиции, включающие эффективное количество гетероарильного соединения и фармацевтически пригодный носитель или наполнитель, где фармацевтически приемлемый носитель или наполнитель может включать в себя эксципиент, разбавитель или их смесь. В одном варианте осуществления композиция представляет собой фармацевтическую композицию.
Композиции могут быть в форме таблеток, жевательных таблеток, капсул, растворов, парентеральных растворов, пастилок, суппозиториев и суспензий, и тому подобного. Композиции могут быть изготовлены так, что содержат суточную дозу или удобную фракцию суточной дозы, в единице дозы, которая может быть цельной таблеткой или капсулой или удобным объемом жидкости. В одном варианте осуществления растворы приготавливают из водорастворимых солей, таких как хлористоводородная соль. Обычно все композиции приготавливают согласно методам, известным в фармацевтической химии. Капсулы можно изготавливать смешиванием гетероарильного соединения с подходящим носителем или разбавителем и наполнением надлежащего количества смеси в капсулы. Обычные носители и разбавители включают, но не ограничиваются перечисленными, инертные порошкообразные вещества, такие как крахмал многих различных сортов, порошкообразная целлюлоза, в особенности кристаллическая и микрокристаллическая целлюлоза, сахара, такие как фруктоза, маннит и сахароза, крупчатная мука и подобные съедобные порошки.
Таблетки можно изгатавливать прямым прессованием, влажным гранулированием или сухим гранулированием. В такие препараты обычно вводят разбавители, связующие средства, смазывающие средства и дезинтегрирующие средства, а также соединение. Типичные разбавители включают, например, различные типы крахмала, лактозу, маннит, каолин, фосфат или сульфат кальция, неорганические соли, такие как хлористый натрий и порошкообразный сахар. Порошкообразные производные целлюлозы также применимы. В одном варианте осуществления фармацевтическая композиция не содержит лактозы. Типичные связующие средства для таблетки представляют собой такие вещества, как крахмал, желатин и сахара, такие как лактоза, фруктоза, глюкоза и тому подобное. Пригодны также природные и синтетические смолы, включающие аравийскую камедь, альгинаты, метилцеллюлозу, поливинилпирролидин и тому подобное. Полиэтиленгликоль, этилцеллюлоза и воски также могут служить в качестве связующих средств.
Смазывающее средство может быть необходимо в препарате в форме таблетки для предохранения от склеивания в матрице. Смазывающее средство можно выбрать из таких скользящих твердых веществ, как тальк, стеарат магния и кальция, стеариновая кислота и гидрированные растительные масла. Дезинтегрирующие средства для таблетки представляют собой вещества, которые набухают при увлажнении, для разрушения таблетки и высвобождения соединения. Они включают крахмалы, глины, целлюлозы, альгинаты и смолы. Более конкретно, можно применять, например, кукурузный и картофельный крахмалы, метилцеллюлозу, агар, бентонит, древесную целлюлозу, порошкообразную природную губку, катионообменные смолы, альгиновую кислоту, гуаровую камедь, цитрусовую пульпу и карбоксиметилцеллюлозу, а также лаурилсульфат натрия. Таблетки могут быть покрыты сахаром в качестве корригента и изолирующим средством, или пленкообразующими защищающими агентами для модификации растворяющих свойств таблетки. Композиции также можно изготавливать как жевательные таблетки, например, с применением в препарате таких веществ, как маннит.
Когда требуется для введения гетероарильного соединения в виде суппозитория, можно применять типичные основы. Масло какао представляет собой традиционную основу суппозитория, которое можно модифицировать добавлением восков для незначительного повышения точки плавления. Несмешивающиеся с водой основы суппозитория, включающие, конкретно, полэтиленгликоль различной молекулярной массы, представляют собой широко применимые.
Действие гетероарильного соединения можно замедлить или продлить соответствующим препаратом. Например, можно изготовить медленно растворимую гранулу гетероарильного соединения и вводить в таблетку или капсулу, или в качестве имплантируемого устройства с медленным высвобождением. Метод также включает изготовление гранул с различными скоростями растворения и наполнение капсулы смесью гранул. Таблетки или капсулы можно покрывать пленкой, которая устойчива к растворению в течение заранее заданного периода времени. Даже парентеральные препараты можно изготовить с длительным растворением или суспендированием гетероарильного соединения в масляном или эмульгированном наполнителе, чтобы обеспечить возможность медленно диспергироваться в сыворотке.
5. ПРИМЕРЫ
Следующие примеры представлены путем иллюстрации без ограничения.
5.1 Синтетические примеры
Общий метод А. В круглодонной колбе растворяли в ацетонитриле 2,3-диаминомалеонитрил и перемешивали при комнатной температуре. Добавляли требуемый изоцианат и реакционную смесь перемешивали при комнатной температуре в течение ночи. Полученный продукт собирали фильтрованием, промывали небольшим количеством ацетонитрила с последующим простым диэтиловым эфиром. Отфильтрованное вещество сушили в высоком вакууме при 60°С в течение ночи с получением требуемой мочевины.
Общий метод В. Исходное вещество мочевину растворяли в метаноле и перемешивали при комнатной температуре до однородного состояния. Последовательно добавляли требуемый альдегид и триэтиламин. Реакционный раствор перемешивали при комнатной температуре в течение ночи. Образовавшуюся неоднородную смесь фильтровали, промывали ацетонитрилом и простым диэтиловым эфиром с получением требуемого вещества.
Общий метод С. В круглодонной колбе растворяли в тетрагидрофуране этил-2,6-дихлор-5-нитропиримидин-4-карбоксилат и перемешивали при комнатной температуре. К реакционной смеси добавляли требуемый амин при -78°С в инертной атмосфере. Затем в течение нескольких минут добавляли по каплям диизопропилэтиламин. Реакционную смесь перемешивали в течение ночи. Образовавшееся вещество концентрировали при пониженном давлении и очищали с применением подходящих хроматографических методов.
Обший метод D. Замещенный 5-нитропиримидин и дигидрат хлорида олова(II) растворяли в смеси этанола и ДМФА. Реакционной смеси предоставляли возможность перемешиваться в течение от 24 до 48 ч. Образовавшуюся неоднородную смесь концентрировали при пониженном давлении и растирали с этилацетатом с получением в результате вещества.
Общий метод D2. Замещенный 5-нитропиримидин, уксусную кислоту и железо объединяли и нагревали до 65°С. Проводили мониторинг реакционной смеси путем тонкослойной хроматографии. Получившуюся в результате неоднородную смесь концентрировали при пониженном давлении и распределяли между этилацетатом и водным раствором карбоната калия. Органические вещества сушили над сульфатом магния, фильтровали и удаляли (растворитель) при пониженном давлении с получением названного соединения.
Общий метод Е. Замещенный 5-аминопиримидин, бороновую кислоту, фосфат калия, дициклогексил-(2',6'-диметоксибифенил-2-ил)фосфин и ацетат палладия(II) добавляли к тетрагидрофурану и воде в микроволновой колбе. Реакционную смесь нагревали при 120°С в течение 30 мин в микроволновом реакторе Biotage Emrys Optimizer. Реакционную смесь фильтровали и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с применением подходящих хроматографических методов. Фракции, содержащие вещество, нейтрализовали карбонатом калия (насыщенный водный раствор), экстрагировали этилацетатом и сушили над сульфатом магния с получением названного соединения.
Общий метод F. Диамин растворяли в метиленхлориде и добавляли 1,1'-карбонилдиимидазол. Реакционную смесь нагревали термическим способом для кипячения с обратным холодильником в течение от 2 до 48 часов или с применением микроволнового реактора Biotage Emrys Optimizer при 120°С в течение 30 мин. Растворитель удаляли при пониженном давлении и неочищенное вещество очищали подходящими хроматографическими методами с получением названного соединения.
Общий метод G. Раствор требуемого карбоксилата добавляли к безводному метанолу и охлаждали до -78°С. Затем раствор насыщали газообразным аммиаком. Реакционный сосуд герметизировали при -78°С и постепенно предоставляли возможность нагреваться до комнатной температуры. Через 24 часа реакционную смесь охлаждали до -78°С и открывали для контакта с атмосферой. Летучие продукты выпаривали и полученное в результате вещество суспендировали в метаноле и фильтровали. Осадок сушили в высоком вакууме с получением названного соединения, которое далее очищали с применением подходящих хроматографических методов.
5.1.1 ПРИМЕР 1
СИНТЕЗ 9-БЕНЗИЛ-8-ОКСО-2-(ПИРИДИН-3-ИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 1-((Z)-2-Амино-1,2-дициановинил)-3-бензилмочевина. Бензилизоцианат (1,3 г, 9,7 ммоль) и 2,3-диаминомалеонитрил (1,0 г, 9,3 ммоль) подвергали реакции в ацетонитриле, согласно общему методу А. Вещество растирали с ацетонитрилом/простым диэтиловым эфиром. Образовавшееся в результате твердое вещество отфильтровали и сушили с получением названного соединения в виде оранжевого твердого вещества (0,83 г, 4,4 ммоль, выход 37%); МС (ESI) m/z 242,1 [M+1]+.
В. 9-Бензил-8-оксо-2-(пиридин-3-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид. 1-((Z)-2-Амино-1,2-дициановинил)-3-бензилмочевину (0,1 г, 0,4 ммоль) и 3-пиридинкарбоксальдегид (0,1 г, 0,9 ммоль) подвергали реакции согласно общему методу В. Продукт реакции очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (5-60% ацетонитрил + 0,1% TFA в Н2О + 0,1 TFA, в течение 39 мин). Фракции, содержащие требуемое вещество, объединяли и концентрировали при пониженном давлении перед пропусканием через ионообменную колонку Strata-XC с водой, метанолом и 5% гидроксидом аммония в метаноле. Полученное в результате твердое вещество сушили в высоком вакууме при 60°С с получением названного соединения в виде белого твердого вещества (0,082 г, 0,24 ммоль, выход 59%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,76 (с, 1Н), 9,70 (д, J=1,6, 1H), 8,86 (дт, J=8,2, 2,0, 1H), 8,67 (дд, J=4,7, 1,6, 1Н), 8,58 (с, 1Н), 7,96 (с, 1Н), 7,53 (дд, J=7,8, 5,1, 1Н), 7,44 (д, J=7,0, 2Н), 7,35 (т, J=7,4, 2H), 7,26-7,31 (м, 1Н), 5,12 (с, 2Н); МС (ESI) m/z 347,1 [M+1]+; т.пл. 334-335°С.
5.1.2 ПРИМЕР 2
СИНТЕЗ 2-(4-ГИДРОКСИФЕНИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевина. В круглодонной колбе растворяли в ацетонитриле (60 мл) 2,3-диаминомалеонитрил (3,41 г, 31,62 ммоль) и перемешивали при комнатной температуре. Добавляли 2-метоксифенилизоцианат (5,0 г, 33,5 ммоль) и раствор перемешивали при комнатной температуре в течение 16 часов. Полученный в результате мочевинный продукт собирали фильтрованием, промывали небольшими порциями ацетонитрила, с последующим простым диэтиловым эфиром. Отфильтрованное вещество сушили в высоком вакууме при 60°С в течение ночи с получением названного соединения (4,10 г, 51%). МС (ESI) m/z 258,0 [M+1]+.
В. 2-(4-Гидроксифенил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (0,200 г, 0,778 ммоль) растворяли в метаноле (15 мл) и перемешивали при комнатной температуре до однородности. Затем последовательно добавляли триэтиламин (0,15 мл) и 4-гидроксибензальдегид (0,208 г, 1,71 ммоль). Раствору предоставляли возможность перемешиваться при окружающей температуре в течение 16 ч. Образовавшуюся в результате неоднородную смесь фильтровали, промывали добавляемым ацетонитрилом с последующим простым диэтиловым эфиром с получением названного соединения (0,091 г, 31%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,61 (с, 1Н), 9,79 (с, 1Н), 8,42 (с, 1Н), 8,18 (д, J=8,7, 2H), 7,94 (c, 1Н), 7,53 (т, J=7,99, 1H), 7,47 (д, J=7,59, 1H), 7,28 (д, J=7,9, 1H), 7,15 (т, J=7,59, 1H), 6,78 (д, J=8,79, 2H); МС (ESI) m/z 378,1 [M+1]+; т.пл. 362-363°С.
5.1.3 ПРИМЕР 3
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-9-О-ТОЛИЛ-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевина. Диаминомалеонитрил (600 мг, 5,55 ммоль) и о-толилизоцианат (0,729 мл, 5,88 ммоль) подвергали реакции в ацетонитриле согласно общему методу А с получением названного соединения (604,8 мг, 42%). МС (ESI) m/z 242,2 [M+1]+.
В. 2-(3-Гидроксифенил-8-оксо-9-о-толил-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (0,2 г, 0,83 ммоль), 3-гидроксибензальдегид (0,221 г, 1,81 ммоль) и триэтиламин (0,1 мл) подвергали реакции согласно общему методу В. Образовавшийся осадок растворяли в ДМФА и добавляли воду, чтобы вызвать осаждение. Этот осадок отфильтровывали и сушили в вакууме с получением названного соединения 95% чистоты (0,188 г, 63%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,79 (с, 1Н), 9,47 (с, 1Н), 8,40 (с, 1Н), 7,98 (с, 1Н), 7,89 (д, J=8,00, 1Н), 7,67 (с, 1Н), 7,49-7,43 (перекрывающиеся м, 4Н), 7,22 (т, J=8,00, 1H), 6,81 (д, J=6,05, 1H), 2,16 (c, 3H). МС (ESI) m/z 362,1 [M+1]+; т.пл. 366-367°С.
5.1.4 ПРИМЕР 4
СИНТЕЗ 2-(1Н-ИНДОЛ-4-ИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(1Н-Индол-4-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,2 г, 0,78 ммоль), индол-4-карбоксальдегид (0,246 г, 1,7 ммоль) и триэтиламин (0,1 мл) подвергали реакции согласно общему методу В. Образовавшийся в результате осадок растворяли в ДМФА и добавляли воду, чтобы вызвать осаждение. Этот осадок отфильтровывали и сушили в вакууме с получением названного соединения 98,8% чистоты (0,122 г, 39%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,68 (с, 1Н), 11,19 (с, 1Н), 8,23 (м, 2Н), 8,02 (с, 1Н), 7,57 (м, 2Н), 7,48 (д, J=8,20, 1H), 7,31 (м, 2Н), 7,17 (м, 2Н), 7,01 (с, 1Н), 3,76 (с, 3Н); МС (ESI) m/z 401,3 [M+1]+, т.пл. 312-313°С.
5.1.5 ПРИМЕР 5
СИНТЕЗ 2-(1Н-ИНДОЛ-6-ИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(1Н-Индол-6-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,2 г, 0,78 ммоль), индол-6-карбоксальдегид (0,246 г, 1,7 ммоль) и триэтиламин (0,1 мл) подвергали взаимодействию согласно общему методу В. Образовавшийся в результате осадок растворяли в ДМФА и добавляли воду, чтобы вызвать осаждение. Этот осадок отфильтровывали и сушили в вакууме с получением названного соединения 97,5% чистоты (0,116 г, 37%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,62 (c, 1H), 11,18 (с, 1Н), 8,41 (с, 1Н), 8,32 (с, 1Н), 8,17 (дд, J=9,76, 1,37, 1H), 7,97 (с, 1Н), 7,55 (м, 3Н), 7,41 (т, J=2,6, 1Н), 7,31 (д, J=8,2, 1Н), 7,18 (т, J=7,7, 1Н), 6,43 (с, 1Н), 3,76 (с, 3Н); МС (ESI) m/z 401,3 [M+1]+, т.пл. 203-204°С.
5.1.6 ПРИМЕР 6
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛ)-9-(4-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-(4-метоксифенил)мочевина. В круглодонной колбе растворяли в ацетонитриле (25 мл) 2,3-диаминомалеонитрил (2,0 г, 18,50 ммоль) и перемешивали при комнатной температуре. Добавляли 4-метоксифенилизоцианат (2,92 г, 19,61 ммоль) и раствор перемешивали при комнатной температуре в течение 16 часов. Образовавшийся в результате мочевинный продукт собирали фильтрованием, промывали небольшими порциями ацетонитрила с последующим простым диэтиловым эфиром. Отфильтрованное вещество сушили в высоком вакууме при 60°С в течение ночи с получением названного соединения (4,39 г, 92%). МС (ESI) m/z 258,3 [M+1]+.
В. 2-(3-Гидроксифенил)-9-(4-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (0,250 г, 0,972 ммоль) растворяли в метаноле (15 мл) и перемешивали при комнатной температуре до однородного состояния. Затем последовательно добавляли триэтиламин (0,2 мл) и 3-гидроксибензальдегид (0,261 г, 2,13 ммоль). Раствору предоставляли возможность перемешиваться в течение 16 часов при окружающей температуре. Полученную в результате неоднородную смесь фильтровали, промывали добавляемым ацетонитрилом с последующим простым диэтиловым эфиром с получением неочищенного кристаллизующегося продукта. Полученное в результате твердое вещество растирали с диметилформамидом/водой наряду с ультразвуковой обработкой. Это твердое вещество снова растирали с метанолом/водой наряду с ультразвуковой обработкой с получением названного соединения (0,087 г, 23%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,69 (c, 1Н), 9,47 (с, 1Н), 8,36 (с, 1Н), 7,75 (с, 1Н), 7,58 (д, J=7,99, 1H), 7,25 (т, J=7,99, 1H), 7,15 (т, J=7,99, 2H), 6,83 (д, J=7,99, 1H); МС (ESI) m/z 378,5 [M+1]+, т.пл. 386-388°С.
5.1.7 ПРИМЕР 7
СИНТЕЗ 2-(2-ГИДРОКСИПИРИДИН-4-ИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(2-Гидроксипиридин-4-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,2 г, 0,78 ммоль), 2-гидроксиизоникотинальдегид (0,209 г, 1,7 ммоль) и триэтиламин (0,1 мл) подвергали взаимодействию согласно общему методу В. Образовавшийся в результате осадок растворяли в ДМФА и добавляли воду, чтобы вызвать осаждение. Этот осадок отфильтровывали и сушили в вакууме с получением названного соединения 97,6% чистоты (0,2 г, 68%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,91 (с, 1Н), 11,64 (с, 1Н), 8,57 (с, 1Н), 7,98 (с, 1Н), 7,55 (т, J=7,9, 1Н), 7,49 (д, J=7,8, 1Н), 7,40 (д, J=6,4, 1Н), 7,29 (м, 2Н), 7,14 (м, 2Н), 3,74 (с, 3Н); МС (ESI) m/z 379,4 [M+1]+; т.пл. 360-362°С.
5.1.8 ПРИМЕР 8
СИНТЕЗ 9-(2-ХЛОРФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-хлорфенил)мочевина. 2-Хлорфенилизоцианат (1,0 г, 9,2 ммоль) и 2,3-диаминомалеонитрил (1,5 г, 9,7 ммоль) подвергали взаимодействию в ацетонитриле согласно общему методу А. Образовавшийся в результате твердый продукт отфильтровывали и сушили в высоком вакууме (1,1 г, 4,2 ммоль, выход 45%); МС (ESI) m/z 262,1 [M+1]+.
В. 9-(2-Хлорфенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-5-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-хлорфенил)мочевину (0,25 г, 1,0 ммоль) и 3-гидроксибензальдегид (0,13 г, 1,1 ммоль) подвергали взаимодействию согласно общему методу В. Продукт очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (5-60% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 39 мин). Фракции, содержащие требуемое вещество, объединяли и концентрировали при пониженном давлении до пропускания через ионообменную колонку Strata-XC с водой, метанолом и 5% гидроксида аммония в метаноле. Полученное в результате твердое вещество сушили в высоком вакууме при 60°С с получением названного соединения в виде белого твердого вещества (0,030 г, 0,079 ммоль, выход 8%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,90 (с, 1Н), 9,48 (с, 1Н), 8,43 (с, 1Н), 8,02 (с, 1Н), 7,89 (д, J=7,8, 1H), 7,79 (дд, J=7,4, 2,0, 1H), 7,74 (дд, J=7,0, 2,3, 1H), 7,59-7,68 (м, 3Н), 7,23 (т, J=8,0, 1H), 6,82 (дд, J=8,0, 2,5, 1Н); МС (ESI) m/z 382,0 [M+1]+, т.пл. 360-364°С.
5.1.9 ПРИМЕР 9
СИНТЕЗ 9-(2-ФТОРФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-фторфенил)мочевина. Диаминомалеонат (1,0 г, 9,25 ммоль) и 2-(фторфенилизоцианат (1,10 мл, 9,71 ммоль) подвергали взаимодействию в ацетонитриле (20 мл) согласно общему методу А. Вещество сушили в вакуумном сушильном шкафу в течение ночи с получением названного соединения в виде твердого вещества (0,34 г, 19%). МС (ESI) m/z 234,2 [M+1]+.
В. 9-(2-Фторфенил)-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид. Раствор (Z)-1-(2-амино-1,2-дициановинил)-3-(2-фторфенил)мочевины (0,25 г, 1,24 ммоль), 3-гидроксибензальдегида (0,33 г, 2,72 ммоль) и триэтиламина (0,2 мл) в метаноле (15 мл) подвергали взаимодействию согласно общему методу В. Полученный в результате продукт помещали в ДМФА (3 мл) и растирали с деионизированной водой. Полученный в результате осадок отфильтровали, промывали деионизированной водой и сушили в высоком вакууме при 60°С с получением названного соединения (0,18 г, 39%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,89 (с, 1Н), 9,48 (с, 1Н), 8,41 (с, 1Н), 8,00 (с, 1Н), 7,91 (д, J=7,8, 1Н), 7,69-7,74 (м, 2Н), 7,61-7,68 (м, 1Н), 7,55 (тд, J=9,3, 1,0, 1Н), 7,46 (тд, J=7,6, 1,2, 1H), 7,24 (т, J=7,8, 1H), 6,85 (д, J=1,6, 1H), 6,83 (дд, J=2,7, 0,8, 1H). МС (ESI) m/z 366,3 [M+1]+.
5.1.10 ПРИМЕР 10
СИНТЕЗ 9-(2,6-ДИФТОРФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2,6-дифторфенил)мочевина. Диаминомалеонитрил (600 мг, 5,55 ммоль) и 2,6-дифторфенилизоцианат (0,912 мл, 5,88 ммоль) подвергали взаимодействию в ацетонитриле согласно общему методу А с получением названного соединения (550 мг, 38%); МС (ESI) m/z 264,2 [M+1]+.
В. 9-(2,6-Дифторфенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2,6-дифторфенил)мочевину (0,2 г, 0,76 ммоль), 3-гидроксибензальдегид (0,202 г, 1,66 ммоль) и триэтиламин (0,1 мл) подвергали взаимодействию согласно общему методу В. Полученный в результате осадок растворяли в ДМФА и добавляли воду, чтобы вызвать осаждение. Этот осадок отфильтровывали и сушили в вакууме с получением названного соединения 99,7% чистоты (0,135 г, 46%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,09 (с, 1Н), 9,49 (с, 1Н), 8,43 (с, 1Н), 8,04 (с, 1Н), 7,91 (д, J=7,81, 1H), 7,76 (м, 1Н), 7,71 (с, 1Н), 7,49 (т, J=8,0, 2H), 7,25 (т, J=8,0, 1H), 6,84 (д, J=8,2, 1H); МС (ESI) m/z 384,4 [M+1]+, т.пл. 355-358°С.
5.1.11 ПРИМЕР 11
СИНТЕЗ 9-ЦИКЛОГЕПТИЛ-8-ОКСО-2-(3-ПИРИДИЛ)-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-циклогептилмочевина. 2,3-Диаминомалеонитрил (1,0 г, 9,25 ммоль) и циклогептилизоцианат (1,29 мл, 9,71 ммоль) подвергали взаимодействию в ацетонитриле (20 мл) при 50°С согласно общему методу А. Вещество сушили в высоком вакууме при 60°С в течение ночи с получением названного соединения в виде твердого вещества (0,80 г, 35%). МС (ESI) m/z 248,4 [M+1]+.
В. 9-Циклогептил-8-оксо-2-(3-пиридил)-7-гидропурин-6-карбоксамид. Раствор (Z)-1-(2-амино-1,2-дициановинил)-3-циклогептилмочевины (0,25 г, 1,01 ммоль), 3-гидроксибензальдегида (0,21 г, 2,22 ммоль) и триэтиламина (0,2 мл) в метаноле (15 мл) подвергали взаимодействию согласно общему методу В. Полученный в результате продукт помещали в ДМФА (3 мл) и растирали с деионизированной водой. Этот осадок отфильтровывали, промывали деионизированной Н2О и сушили в высоком вакууме при 60°С с получением названного соединения (0,02 г, 7%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,56 (с, 1Н), 9,70 (д, J=1,2, 1Н), 8,86 (дт, J=8,1, 1,8, 1Н), 8,68 (д, J=3,5, 1H), 8,54 (с, 1Н), 7,91 (с, 1Н), 7,55 (дд, J=7,8, 4,7, 1H), 4,43-4,51 (м, 1Н), 2,33-2,45 (м, 2Н), 1,86-1,93 (м, 3Н), 1,81-1,85 (м, 1Н), 1,61-1,72 (м, 4Н), 1,49-1,60 (м, 2Н). МС (ESI) m/z 353,5 [M+1]+.
5.1.12 ПРИМЕР 12
СИНТЕЗ 9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-2-(ХИНОЛИН-5-ИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 9-(2-Метоксифенил)-8-оксо-2-(хинолин-5-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,250 г, 0,972 ммоль) растворяли в метаноле (15 мл) и перемешивали при комнатной температуре до однородного состояния. Затем добавляли триэтиламин (0,2 мл) и хинолин-5-карбоксальдегид (0,305 г, 1,945 ммоль) и раствору предоставляли возможность перемешиваться в течение 16 часов. Полученную в результате неоднородную смесь фильтровали, промывали добавляемым ацетонитрилом с последующим простым диэтиловым эфиром с получением неочищенного кристаллизующегося продукта. Твердый продукт растирали с диметилформамидом/водой наряду с ультразвуковой обработкой. Это твердое вещество снова растирали с метанолом/водой наряду с ультразвуковой обработкой с получением названного соединения (0,184 г, 46%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,88 (c, 1Н), 9,00 (д, J=8,79, 1H), 8,90 (м, 1Н), 8,21 (м, 2Н), 8,10 (д, J=8,39, 1H), 8,00 (с, 1Н), 7,83 (т, J=7,59, 1H), 7,51 (м, 3Н), 7,28 (д, J=7,59, 1H), 7,12 (т, J=7,60, 1H); МС (ESI) m/z 413,0 [M+1]+, т.пл. 335-337°С.
5.1.13 ПРИМЕР 13
СИНТЕЗ 2-ЦИКЛОПЕНТИЛ-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-Циклопентил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. К раствору, содержащему (Z)-1-(2-амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,250 г, 0,972 ммоль) в метаноле (15 мл) и триэтиламин (0,2 мл), добавляли циклопентанкарбоксальдегид (0,190 г, 1,94 ммоль). Раствору предоставляли возможность перемешиваться в течение 16 часов при окружающей температуре. Образовавшуюся в результате неоднородную смесь фильтровали, промывали добавляемым ацетонитрилом с последующим простым диэтиловым эфиром с получением неочищенного кристаллизующегося продукта. Твердое вещество растирали с диметилформамидом/водой наряду с ультразвуковой обработкой. Затем твердое вещество промывали добавляемой водой с последующей небольшой частью метанола с получением названного соединения после сушки (0,146 г, 42%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,52 (с, 1Н), 8,01 (с, 1Н), 7,89 (с, 1Н), 7,51 (т, J=7,19, 1H), 7,41 (д, J=7,99, 1H), 7,23 (д, J=8,39, 1H), 7,10 (т, J=7,19, 1H), 3,72 (с, 3Н), 3,14 (м, 1Н), 1,90 (м, 2Н), 1,80 (м, 2Н), 1,69 (м, 2Н), 1,56 (м, 2Н); МС (ESI) m/z 354,0 [M+1]+, т.пл. 244-246°С.
5.1.14 ПРИМЕР 14
СИНТЕЗ 9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-2-(3-ТРИФТОРМЕТИЛ)ФЕНИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 9-(2-Метоксифенил)-8-оксо-2-(3-(трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,40 г, 1,6 ммоль) и 3-(трифторметил)бензальдегид (0,28 г, 1,6 ммоль) подвергали взаимодействию согласно общему методу В. Образовавшуюся в результате неоднородную смесь фильтровали и затем растирали с диметилформамидом/водой наряду с ультразвуковой обработкой. Полученное в результате твердое вещество промывали простым диэтиловым эфиром и затем сушили в высоком вакууме при 60°С с получением названного соединения в виде белого твердого вещества (0,047 г, 0,11 ммоль, выход 7%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,90 (с, 1Н), 9,48 (с, 1Н), 8,43 (с, 1Н), 8,02 (с, 1Н), 7,89 (д, J=7,8, 1Н), 7,79 (дд, J=7,4, 2,0, 1H), 7,74 (дд, J=7,0, 2,3, 1H), 7,59-7,68 (м, 3Н), 7,23 (т, J=8,0, 1H), 6,82 (дд, J=8,0, 2,5, 1Н); МС (ESI) m/z 430,0 [M+1]+, т.пл. 271-275°С.
5.1.15 ПРИМЕР 15
СИНТЕЗ 2-(6-МЕТОКСИ(3-ПИРИДИЛ))-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. 2-(6-Метокси(3-пиридил))-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид. Раствор (Z)-1-(2-амино-1,2-дициановинил)-3-(2-метоксифенил)мочевины (см. пример 2.А) (0,25 г, 0,97 ммоль), 6-метокси-3-пиридинкарбоксальдегида (0,30 г, 2,14 ммоль) и триэтиламина (0,2 мл) в метаноле (10 мл) подвергали взаимодействию согласно общему методу В. Образовавшийся в результате продукт помещали в ДМФА (3 мл) и растирали с деионизированной водой. Этот осадок отфильтровывали, перемешивали в метаноле при кипячении с обратным холодильником в течение 15 минут и фильтровали горячим. Этот осадок впоследствии сушили в вакуумном сушильном шкафу с получением названного соединения (0,22 г, 58%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,73 (с, 1Н), 9,15 (д, J=2,3, 1Н), 8,53-8,57 (м, 2Н), 7,96 (с, 1Н), 7,53-7,58 (м, 1Н), 7,49 (дд, J=7,6, 1,8, 1Н), 7,29 (дд, J=8,4, 1,0, 1Н), 7,15 (тд, J=7,6, 1,2, 1Н), 6,87 (д, J=9,0, 1H), 3,90 (с, 3Н), 3,75 (с, 3Н); МС (ESI) m/z 393,2 [M+1]+.
5.1.16 ПРИМЕР 16
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-9-(4-(ТРИФТОРМЕТИЛ)ФЕНИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 1-((Z)-2-Амино-1,2-дициановинил)-3-трифторметилфенил)мочевина. 4-(Трифторметил)фенилизоцианат (1,0 г, 9,2 ммоль) и 2,3-диаминомалеонитрил (1,5 г, 9,7 ммоль) подвергали взаимодействию согласно общему методу А. Полученное в результате желто-зеленое соединение отфильтровывали и сушили в высоком вакууме (1,1 г, 4,2 ммоль, выход 45%). МС (ESI) m/z 262,1 [M+1]+.
В. 2-(3-Гидроксифенил)-8-оксо-9-(4-(трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид. 1-((Z)-2-Амино-1,2-дициановинил)-3-трифторметилфенил)мочевину (0,30 г, 1,0 ммоль) и 3-гидроксибензальдегид (0,13 г, 1,1 ммоль) подвергали взаимодействию согласно общему методу В. Неочищенный остаток растирали с диметилформамидом/водой наряду с ультразвуковой обработкой. Полученный в результате продукт сушили в высоком вакууме при 60°С с получением названного соединения в виде желтого твердого вещества (146 мг, 0,352 ммоль, выход 35%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 1,90 (с, 1Н), 9,51 (с, 1Н), 8,44 (с, 1Н), 8,00-8,07 (м, 5Н), 7,98 (д, J=8,2, 1Н), 7,80-7,82 (м, 1Н), 7,27 (т, J=8,0, 1Н), 6,86 (дд, J=7,6, 2,9, 1Н). МС (ESI) m/z 382,0 [M+1]+; т.пл. 360-364°С.
5.1.17 ПРИМЕР 17
СИНТЕЗ 9-БЕНЗИЛ-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 9-Бензил-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. 1-((Z)-2-Амино-1,2-дициановинил)-3-бензилмочевину (см. пример 1.А) (0,50 г, 2,1 ммоль) и 3-гидроксибензальдегид (0,27 г, 2,2 ммоль) подвергали взаимодействию согласно общему методу В. Полученную в результате неоднородную смесь фильтровали и затем промывали добавляемым ацетонитрилом и простым диэтиловым эфиром с получением неочищенного кристаллизующегося продукта. Твердый продукт растирали с диметилформамидом/водой наряду с ультразвуковой обработкой. Продукт фильтровали и сушили в высоком вакууме при 60°С с получением названного соединения в виде белого твердого вещества (0,082 г, 0,24 ммоль, выход 59%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,66 (с, 1Н), 9,53 (с, 1Н), 8,35 (с, 1Н), 8,02 (д, J=7,8, 1H), 7,95 (с, 1Н), 7,90-7,92 (м, 1Н), 7,43 (с, 1Н), 7,41 (с, 1Н), 7,35 (т, J=7,4, 2H), 7,28 (тд, J=7,5, 2,9, 2H), 6,87 (дд, J=7,4, 2,0, 1H), 5,09 (с, 2Н); МС (ESI) m/z 362,1 [M+1]+; т.пл. 362-366°С.
5.1.18 ПРИМЕР 18
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-9-[2-(ТРИФТОРМЕТОКСИ)ФЕНИЛ]-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-(трифторметокси)фенил)мочевина. Диаминомалеонат (0,5 г, 4,63 ммоль) и 2-(трифторметил)фенилизоцианат (0,73 мл, 4,86 ммоль) подвергали взаимодействию в ацетонитриле (15 мл) согласно общему методу А. Вещество сушили в высоком вакууме при 60°С в течение ночи с получением названного соединения в виде твердого вещества (0,94 г, 57%). МС (ESI) m/z 312,2 [M+1]+.
В. 2-(3-Гидроксифенил)-8-оксо-9-[2-(трифторметокси)фенил]-7-гидропурин-6-карбоксамид. Раствор (Z)-1-(2-амино-1,2-дициановинил)-3-(2-(трифторметокси)фенил)мочевину (0,25 г, 0,80 ммоль), 3-гидроксибензальдегид (0,22 г, 1,78 ммоль) и триэтиламин (0,2 мл) в метаноле (15 мл) подвергали взаимодействию согласно общему методу В. Полученный в результате продукт помещали в ДМФА (3 мл) и растирали с деионизированной водой. Образовавшийся в результате осадок отфильтровывали и сушили в вакуумном сушильном шкафу с получением названного соединения (0,22 г, 64%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,92 (с, 1Н), 9,49 (с, 1Н), 8,44 (с, 1Н), 8,03 (с, 1Н), 7,90 (д, J=7,8, 1H), 7,80 (дд, J=7,8, 1,6, 1H), 7,64-7,76 (м, 4Н), 7,23 (т, J=8,0, 1H), 6,83 (ддд, J=6,6, 1,6, 1,2, 1H); МС (ESI) m/z 432,4 [M+1]+.
5.1.19 ПРИМЕР 19
СИНТЕЗ 9-(2,4-ДИХЛОРФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2,4-дихлорфенил)мочевина. В круглодонной колбе растворили в ацетонитриле (20 мл) 2,3-диаминомалеонитрил (1,0 г, 9,26 ммоль) и перемешивали при комнатной температуре. Добавляли 2,4-дихлорфенилизоцианат (1,82 г, 9,72 ммоль) и раствор перемешивали при комнатной температуре в течение 16 часов. Полученное в результате соединение мочевины собирали фильтрованием, промывали небольшими порциями ацетонитрила с последующим простым диэтиловым эфиром. Отфильтрованное вещество сушили в высоком вакууме при 60°С в течение ночи с получением названного соединения (2,15 г, 79%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 8,41 (д, J=4,8, 2Н), 8,14 (д, 8,7, 1Н), 7,63 (д, J=2,1, 1Н), 7,39 (дд, 1Н), 7,32 (ушир.с, 2Н).
В. 9-(2,4-Дихлорфенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. К раствору, содержащему (Z)-1-(2-Амино-1,2-дициановинил)-3-(2,4-дихлорфенил)мочевину (0,500 г, 1,69 ммоль) в метаноле (25 мл) и триэтиламин (0,33 мл), добавляли 3-гидроксибензальдегид (0,413 г, 3,38 ммоль). Раствору предоставляли возможность перемешиваться в течение 16 часов. Полученную в результате неоднородную смесь фильтровали, промывали добавляемым ацетонитрилом с последующим простым диэтиловым эфиром с получением неочищенного кристаллизующегося продукта. Твердое вещество растирали с диметилформамидом/водой наряду с ультразвуковой обработкой с получением названного соединения после сушки (0,207, 29%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,94 (с, 1Н), 9,47 (с, 1Н), 8,43 (с, 1Н), 8,01 (м, 2Н), 8,10 (д, J=7,99, 1H), 7,79 (д, J=8,79, 1H), 7,72 (дд, 1Н), 7,68 (м, 1Н), 7,23 (д, J=7,99, 1Н), 6,82 (дд, 1Н); МС (ESI) m/z 418,0 [M+1]+, т.пл. 375-377°С.
5.1.20 ПРИМЕР 20
СИНТЕЗ 9-(2-МЕТОКСИФЕНИЛ)-2-(3-НИТРОФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 9-(2-Метоксифенил)-2-(3-нитрофенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,2 г, 0,78 ммоль), 3-нитробензальдегид (0,256 г, 1,7 ммоль) и триэтиламин (0,1 ммоль) подвергали взаимодействию согласно общему методу В. Полученный в результате осадок растворяли в ДМФА и добавляли воду, чтобы вызвать осаждение. Этот осадок отфильтровывали и сушили в вакууме с получением названного соединения 99,7% чистоты (0,118 г, 37%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,91 (с, 1Н), 9,02 (с, 1Н), 8,88 (д, J=7,8, 1Н), 8,69 (с, 1Н), 8,29 (д, J=8,0, 1H), 8,04 (с, 1Н), 7,75 (т, J=8,0, 1Н), 7,58 (т, J=7,9, 1Н), 7,52 (д, J=7,8, 1Н), 7,32 (д, J=8,2, 1Н), 7,17 (т, J=7,6, 1Н), 3,76 (с, 3Н); МС (ESI) m/z 407,3 [M+1]+; т.пл. 295-296°С.
5.1.21 ПРИМЕР 21
СИНТЕЗ 9-(3-ФТОРФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 1-((Z)-2-Амино-1,2-дициановинил)-3-(фторфенил)мочевина. 3-Фторфенилизоцианат (1,0 г, 9,3 ммоль) и 2,3-диаминомалеонитрил (1,3 г, 9,7 ммоль) подвергали взаимодействию согласно общему методу А. Полученное в результате коричневое твердое вещество отфильтровывали и сушили в высоком вакууме (2,0 г, 8,2 ммоль, выход 89%). МС (ESI) m/z 246,1 [M+1]+.
В. 9-(3-Фторфенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. 1-((Z)-2-Амино-1,2-дициановинил)-3-(фторфенил)мочевину (0,50 г, 2,0 ммоль) и 3-гидроксибензальдегид (0,26 г, 2,1 ммоль) подвергали взаимодействию согласно общему методу В. Полученную в результате неоднородную смесь фильтровали и растирали с диметилформамидом/водой наряду с ультразвуковой обработкой. Твердое вещество растирали второй раз с применением диметилформамида/простого диэтилового эфира. Полученное в результате твердое вещество сушили в высоком вакууме при 95°С в течение ночи с получением названного соединения в виде белого твердого вещества (0,27 г, 0,74 ммоль, выход 37%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,83 (с, 1Н), 9,50 (с, 1Н), 8,40 (с, 1Н), 7,98 (с, 1Н), 7,95 (ддд, J=8,0, 1,2, 1,0 1H), 7,78 (дд, J=2,3, 1,6, 1H), 7,62-7,70 (м, 3Н), 7,33-7,38 (м, 1Н), 7,26 (т, J=8,0, 1Н), 6,85 (ддд, J=8,0, 2,5, 0,8, 1H); МС (ESI) m/z 366,0 [M+1]+; т.пл. >375°С.
5.1.22 ПРИМЕР 22
СИНТЕЗ 9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-2-(2-(ТРИФТОРМЕТИЛ)ФЕНИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 9-(2-Метоксифенил)-8-оксо-2-(2-(трифторметил)фенил-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,50 г, 1,9 ммоль) и 2-(трифторметил)бензальдегид (0,36 г, 2,0 ммоль) подвергали взаимодействию согласно общему методу В. Продукт очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (5-60% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA в течение 39 мин). Фракции, содержащие требуемое вещество, объединяли и концентрировали при пониженном давлении перед пропусканием через ионообменную колонку Strata-XC с водой, метанолом и 5% гидроксидом аммония в метаноле. Полученное в результате твердое вещество сушили в высоком вакууме при 95°С с получением названного соединения в виде белого твердого вещества (0,079 г, 0,18 ммоль, выход 9%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,89 (с, 1Н), 8,02 (с, 1Н), 7,95 (с, 1Н), 7,72-7,83 (м, 3Н), 7,65 (т, J=7,2, 1H), 7,50 (дд, J=7,0, 1,2, 1H), 7,44 (дд, J=7,8, 1,6, 1Н), 7,24 (дд, J=8,4, 1,0, 1H), 7,09 (тд, J=7,6, 1,2, 1H), 3,73 (с, 3Н). МС (ESI) m/z 430,0 [M+1]+; т.пл. 237-240°С.
5.1.23 ПРИМЕР 23
СИНТЕЗ 2-(5-ФТОР-(3-ПИРИДИЛ))-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. 2-(5-Фтор-(3-пиридил))-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид. Раствор (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевины (см. пример 2.А) (0,25 г, 1,00 ммоль), 3-фтор-5-формилпиридин (0,27 г, 2,14 ммоль) и триэтиламин (0,2 моль) в метаноле (10 мл) подвергали взаимодействию согласно общему методу В. Полученный в результате продукт помещали в ДМФА (3 мл) и растирали с деионизированной водой. Полученный в результате осадок отфильтровывали, перемешивали с метанолом при кипячении с обратным холодильником в течение 15 мин и фильтровали горячим. Продукт помещали в ДМСО и нагревали до превращения в однородный раствор и затем предоставляли возможность стоять в течение ночи. Кристаллы отфильтровывали и промывали охлажденным на льду ДМСО с получением названного соединения (0,01 г, 2%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,92 (с, 1Н), 9,29 (т, J=1,6, 1H), 8,74 (с, 1Н), 8,68 (дд, J=2,9, 1,8, 1H), 8,64-8,67 (м, 1Н), 8,02 (с, 1Н), 7,57 (ддд, J=8,7, 7,3, 1,6, 1H), 7,51 (дд, J=7,8, 1,6, 1Н), 7,31 (дд, J=8,4, 1,0, 1H), 7,16 (тд, J=7,6, 1,2, 1H), 3,76 (с, 3Н); МС (ESI) m/z 381,1 [M+1]+.
5.1.24 ПРИМЕР 24
СИНТЕЗ 9-(2-МЕТОКСИФЕНИЛ)-8-0КСО-2-[1-БЕНЗИЛ(4-ПИПЕРИДИЛ)]-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. 9-(2-Метоксифенил)-8-оксо-2-[1-бензил(4-пиперидил)]-7-гидропурин-6-карбоксамид. Раствор (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевины (см. пример 2.А) (0,5 г, 1,95 ммоль), N-бензилпиперидин-4-карбоксальдегида (0,85 мл, 4,28 ммоль) и триэтиламина (0,5 мл в метаноле (20 мл) подвергали взаимодействию согласно общему методу В. Полученный в результате продукт помещали в ДМФА (3 мл) и растирали с деионизированной водой. Этот осадок отфильтровывали, перемешивали с метанолом при кипячении с обратным холодильником в течение 15 мин и фильтровали горячим с получением названного соединения (0,08 г, 10%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,90 (с, 1Н), 7,49-7,55 (м, 1Н), 7,41 (дд, J=7,8, 1,6, 1H), 7,21-7,32 (м, 6Н), 7,11 (т, J=7,2, 1H), 3,72 (с, 3Н), 3,44 (с, 2Н), 2,81-2,87 (м, 2Н), 2,61-2,70 (м, 1Н), 1,94-1,99 (м, 1Н), 1,80 (д, J=2,0, 2H), 1,78 (с, 2Н); МС (ESI) m/z 459,6 [M+1]+.
5.1.25 ПРИМЕР 25
СИНТЕЗ БЕНЗИЛ-4-(6-КАРБАМОИЛ-8-ОКСО-2-(ПИРИДИН-3-ИЛ)-7Н-ПУРИН-9(8Н)-ИЛ)ПИПЕРИДИН-1-КАРБОКСИЛАТ
А. (Z)-Бензил-4-(3-(2-амино-1,2-дициановинил)уреидо)пиперидин-1-карбоксилат. 2,3-Диаминомалеонитрил (0,28 г, 2,60 ммоль) и бензил-4-изоцианатотетрагидропиридинкарбоксилат (0,7 г, 2,69 ммоль) подвергали взаимодействию в ацетонитриле (15 мл) согласно общему методу А. Вещество сушили в высоком вакууме при 60°С в течение ночи с получением названного соединения в виде твердого вещества (0,36 г, 37%). МС (ESI) m/z 369,3 [M+1]+.
В. Бензил-4-(6-карбамоил-8-оксо-2-(пиридин-3-ил)-7Н-пурин-9(8Н)-ил)пиперидин-1-карбоксилат. Раствор (Z)-бензил-4-(3-(2-амино-1,2-дициановинил)уреидо)пиперидин-1-карбоксилата (0,35 г, 0,95 ммоль), 3-пиридинкарбоксальдегида (0,2 мл, 2,09 ммоль) и триэтиламина (0,3 мл) в метаноле (15 мл) подвергали взаимодействию согласно общему методу В. Полученный в результате продукт помещали в метанол, нагревали в течение 10 минут, предоставляли возможность охладиться до комнатной температуры, фильтровали и сушили в высоком вакууме с получением названного соединения (0,20 г, 44%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,68 (с, 1Н), 9,69 (д, J=1,6, 1H), 8,83 (дт, J=7,8, 2,0, 1H), 8,69 (дд, J=4,9, 1,8, 1Н), 8,57 (с, 1Н), 7,95 (с, 1Н), 7,52 (дд, J=8,2, 5,1, 1H), 7,30-7,42 (м, 5Н), 4,58 (тт, J=12,1, 4,1, 1Н), 4,19 (д, J=12,9, 2H), 3,04 (с, 2Н), 2,45 (с, 2Н), 1,83 (д, J=10,9, 2H). МС (ESI) m/z 474,4 [M+1]+.
5.1.26 ПРИМЕР 26
СИНТЕЗ 9-ЦИКЛОГЕКСИЛ-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-циклогексилмочевина. 2,3-Диаминомалеонитрил (1,00 г, 9,25 ммоль) и циклогексилизоцианат (1,33 мл, 9,71 ммоль) подвергали взаимодействию в ацетонитриле (20 мл) согласно общему методу А. Для взаимодействия потребовалось нагревание при 50°С в течение ночи для превращения приблизительно 70% (мониторинг ЖХ-МС) исходного вещества в требуемый продукт. При обработке следовали общему методу А. Вещество сушили в вакуумном сушильном шкафу в течение ночи с получением названного соединения в виде твердого вещества (1,17 г, 55%). МС (ESI) m/z 234,4 [M+1]+.
В. 9-Циклогексил-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Раствор (Z)-1-(2-амино-1,2-дициановинил)-3-циклогексилмочевины (0,25 г, 1,08 ммоль), 3-гидроксибензальдегида (0,28 мл, 2,36 ммоль) и триэтиламина (0,3 мл) в метаноле (10 мл) подвергали взаимодействию согласно общему методу В. Полученный в результате продукт помещали в смесь этилацетат:метанол (3:1), нагревали в течение 10 минут, фильтровали и сушили в высоком вакууме с получением названного соединения (0,08 г, 21%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,50 (с, 1Н), 9,56 (с, 1Н), 8,33 (с, 1Н), 8,01 (д, J=7,8, 1H), 7,92 (с, 1Н), 7,89-7,91 (м, 1Н), 7,29 (т, J=7,8, 1Н), 6,86-6,90 (м, 1Н), 4,26 (тт, J=12,3, 3,8, 1Н), 2,32-2,43 (м, 2Н), 1,89 (д, J=12,9, 2Н), 1,80 (д, J=10,5, 2Н), 1,73 (д, J=12,1, 1Н), 1,35-1,45 (м, 2Н); МС (ESI) m/z 354,4 [M+1]+.
5.1.27 ПРИМЕР 27
СИНТЕЗ 9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-2-(3-(ТРИФТОРМЕТОКСИ)ФЕНИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 9-(2-Метоксифенил)-8-оксо-2-(3-(трифторметилфенил)-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,50 г, 1,9 ммоль) и 3-(трифторметокси)бензальдегид (0,52 г, 2,0 ммоль) подвергали взаимодействию согласно общему методу В. Полученную в результате неоднородную смесь фильтровали и растирали с метанолом/простым диэтиловым эфиром наряду с ультразвуковой обработкой. Коричневое твердое вещество, извлеченное фильтрованием, растирали второй раз с применением метанола/простого диэтилового эфира с получением желтого порошка, который впоследствии сушили в высоком вакууме при 60°С с получением названного соединения (0,19 г, 0,43 ммоль, выход 22%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,83 (с, 1Н), 8,66 (с, 1Н), 8,40 (с, 1Н), 8,34 (д, J=8,2, 1H), 8,01 (с, 1Н), 7,53-7,60 (м, 2Н), 7,50 (дд, J=7,6, 1,4, 1H), 7,44 (д, J=8,2, 1H), 7,30 (д, J=8,2, 1H), 7,16 (т, J=7,4, 1H), 3,75 (с, 3Н); МС (ESI) m/z 446,1 [M+1]+; т.пл. 269-272°С.
5.1.28 ПРИМЕР 28
СИНТЕЗ 9-ФЕНИЛ-2-(3-ПИРИДИЛ)ПУРИН-6-КАРБОКСАМИДА
А. Метил-2,6-дигидрокси-5-нитропиримидин-4-карбоксилат. Данное соединение можно получить, как описано в публикации J. Med. Chem., 42(11), 1951-1964, 1999, которая введена в качестве ссылки во всей своей полноте.
В. Метил-2,6-дихлор-5-нитропиримидин-4-карбоксилат. Данное соединение можно получить, как описано в публикации J. Med. Chem., 42(11), 1951-1964, 1999, которая введена в качестве ссылки во всей своей полноте.
С. Метил-2-хлор-5-нитро-6-(фениламино)пиримидин-4-карбоксилат. Раствор метил-2,6-дихлор-5-нитропиримидин-4-карбоксилата (0,72 г, 2,87 ммоль) в безводном ТГФ (14 мл) охлаждали до -78°С в атмосфере азота. Затем добавляли по каплям раствор анилина (0,288 мл, 3,16 ммоль) и диизопропилэтиламина (1,50 мл, 8,61 ммоль) в безводном ТГФ (10 мл) при перемешивании в течение 10 минут. Реакционную смесь перемешивали при -78°С в течение 1 часа и затем предоставляли возможность нагреваться до комнатной температуры. Через 90 минут растворитель выпаривали и полученный в результате остаток очищали с применением хроматографии на силикагелевой колонке с нормальной фазой (0-10% этилацетата в гексане). Фракции, содержащие продукт, объединяли и растворитель выпаривали с получением названного соединения (0,545 г, 1,76 ммоль, выход 61%). МС (ESI) m/z 309,4 [M+1]+.
D. Метил-5-амино-2-хлор-6-(фениламино)пиримидин-4-карбоксилат. К раствору метил-2-хлор-5-нитро-6-(фениламино)пиримидин-4-карбоксилата (0,259 г, 0,839 ммоль) в ДМФА (3,0 мл) и этаноле (13 мл) добавляли дигидрат хлорида олова(II) (0,568 г, 2,52 ммоль). Смесь перемешивали при комнатной температуре в течение 70 минут, фильтровали и выпаривали летучие вещества. Полученный в результате остаток очищали с применением хроматографии на силикагелевой колонке с нормальной фазой (1-10% метанола в дихлорметане). Фракции, содержащие продукт, объединяли и растворитель выпаривали с получением названного соединения (0,190 г, 0,683 ммоль, выход 82%). МС (ESI) m/z 279,3 [M+1]+.
Е. Метил-5-амино-6-(фениламино)-2-(3-пиридил)пиримидин-4-карбоксилат. К раствору метил-5-амино-2-хлор-6-(фениламино)пиримидин-4-карбоксилата (0,189 г, 0,68 ммоль) в безводном ДМФА (3,5 мл) добавляли 3-(трибутилстаннил)пиридин (1,252 г, 3,4 ммоль) и дихлорбис(трифенилфосфин)палладий(II) (0,239 г, 0,34 ммоль). Через раствор продували азот, затем нагревали в микроволновом реакторе Emrys Optimizer в течение 30 минут при 120°С. Летучие вещества выпаривали и остаток очищали с применением хроматографии на силикагелевой колонке с нормальной фазой (0-10% метанола в дихлорметане). Фракции, содержащие продукт, объединяли и растворитель выпаривали. Вещество повторно очищали с применением препаративной ВЭЖХ с обращенной фазой (30-80% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 минут). Фракции, содержащие требуемое вещество, объединяли и концентрировали при пониженном давлении перед пропусканием через ионообменную колонку Strata-XC с водой, метанолом и 5% гидроксидом аммония в метаноле. Полученное в результате твердое вещество сушили в высоком вакууме при 60°С с получением названного соединения (0,125 г, 0,389 ммоль, выход 57%). МС (ESI) m/z 322,4 [M+1]+.
F. 5-Амино-6-(фениламино)-2-(3-пиридил)пиримидин-4-карбоксамид. Раствор метил-5-амино-6-(фениламино)-2-(3-пиридил)пиримидин-4-карбоксилата (0,123 г, 0,383 ммоль) в безводном метаноле (15 мл) охлаждали до -78°С. Затем раствор насыщали газообразным аммиаком. Реакционный сосуд герметизировали при -78°С и предоставляли возможность нагреваться до комнатной температуре. Через 18 часов реакционную смесь охлаждали до -78°С и открывали для контакта с атмосферой. Летучие вещества выпаривали и полученные в результате твердые вещества сушили в вакууме с получением названного соединения (0,117 г, 0,383 ммоль, выход 100%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,50 (д, J=1,6, 1Н), 8,93 (с, 1Н), 8,67 (дт, J=8,0, 2,0, 1Н), 8,58 (дд, J=4,8, 1,6, 1Н), 8,43 (с, 1Н), 7,85 (д, J=8,4, 2H), 7,63 (с, 1Н), 7,42-7,47 (м, 3Н), 7,23 (с, 2Н), 7,11 (т, J=7,6, 1H). МС (ESI) m/z 307,3 [M+1]+; т.пл. 285-288°С.
G. 9-Фенил-2-(3-пиридил)пурин-6-карбоксамид. Суспензию 5-амино-6-(фениламино)-2-(3-пиридил)пиримидин-4-карбоксамида (0,061 г, 0,199 ммоль) в триэтилортоформиате (6 мл) перемешивали при 130°С в течение 3 часов. Затем реакционную смесь охлаждали до комнатной температуры и разбавляли простым этиловым эфиром. Полученные в результате твердые вещества собирали фильтрованием, промывали простым этиловым эфиром и сушили в вакууме при 60°С с получением названного соединения (0,056 г, 0,177 ммоль, выход 89%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,70 (д, J=1,6, 1H), 9,20 (с, 1Н), 8,84 (дт, J=8,0, 2,0, 1Н), 8,72 (дд, J=4,8, 1,6, 1Н), 8,61 (с, 1Н), 8,12 (с, 1Н), 8,05 (д, J=8,4, 2Н), 7,71 (т, J=6,4, 2Н), 7,54-7,61 (м, 2Н); МС (ESI) m/z 317,4 [M+1]+; т.пл. 298-299°С.
5.1.29 ПРИМЕР 29
СИНТЕЗ 6-ОКСО-8-ФЕНИЛ-2-(3-ПИРИДИЛ)-5,7,8-ТРИГИДРОПТЕРИДИН-4-КАРБОКСАМИДА
А. Этил-2-{[2-хлор-6-(этоксикарбонил)-5-нитропиримидин-4-ил]фениламино}ацетат. Раствор этил-2,6-дихлор-5-нитропиримидин-4-карбоксилата (1,5 г, 5,67 ммоль) в безводном ТГФ (20 мл) охлаждали до -78°С в азоте. Затем добавляли по каплям раствор этил-2-(фениламино)ацетата (1,11 г, 6,22 ммоль) и диизопропилэтиламина (3,0 мл, 17,01 ммоль) в безводном ТГФ (10 мл) при перемешивании в течение 10 мин. Реакционную смесь перемешивали при -78°С в течение 6 часов с последующим добавлением водного раствора бикарбоната натрия (насыщенный, 10 мл). Смесь экстрагировали этилацетатом (3×10 мл). Органическую фазу сушили над сульфатом натрия и концентрировали с получением остатка, который очищали хроматографией на силикагелевой колонке с нормальной фазой (0-10% этилацетата в гексанах). Фракции, содержащие продукт, объединяли и растворитель выпаривали с получением названного соединения (1,1 г, 2,89 ммоль, выход 41%). МС (ESI) m/z 409,5 [M+1]+.
В. Этил-2-хлор-6-оксо-8-фенил-5,7,8-тригидроптеридин-4-карбоксилат. К раствору этил-2-{[2-хлор-6-(этоксикарбонил)-5-нитропиримидин-4-ил]фениламино}ацетата (1,7 г, 4,16 ммоль) в ледяной уксусной кислоте (20,0 мл) добавляли порошкообразное железо (1,2 г, 20,8 ммоль). Серую суспензию нагревали до 60°С в течение 12 часов. В течение следующих 24 часов добавляли дополнительно порошок железа (суммарно 3,4 г). Уксусную кислоту удаляли при пониженном давлении и остаток суспендировали в метаноле, фильтровали через небольшой слой целита и концентрировали при пониженном давлении с получением остатка. Полученный в результате остаток очищали хроматографией на силикагелевой колонке с нормальной фазой (20-40% этилацетата в гексанах). Фракции, содержащие продукт, объединяли и растворитель выпаривали с получением названного соединения (0,595 г, 1,78 ммоль, выход 43%); МС (ESI) m/z 333,2 [M+1]+.
С. Этил-6-оксо-8-фенил-2-(3-пиридил)-5,7,8-тригидроптеридин-4-карбоксилат. К раствору 2-хлор-6-оксо-8-фенил-5,7,8-тригидроптеридин-4-карбоксилата (0,150 г, 0,45 ммоль) в безводном ДМФА (3,0 мл) добавляли 3-(трибутилстаннил)пиридин (0,828 г, 2,25 ммоль) и дихлорбис(трифенилфосфин)палладий(II) (0,158 г, 0,225 ммоль). Раствор продували азотом, затем нагревали в микроволновом реакторе Emrys Optimizer в течение 30 минут при 120°С. Летучие вещества выпаривали и полученный в результате остаток очищали хроматографией на силикагелевой колонке с нормальной фазой (1-10% метанола в дихлорметане). Фракции, содержащие продукт, объединяли, и растворитель выпаривали. Вещество повторно очищали с применением препаративной ВЭЖХ с обращенной фазой (10-70% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Фракции, содержащие требуемое вещество, объединяли и концентрировали при пониженном давлении с получением остатка. Остаток растворяли в метиленхлориде (100 мл), который промывали водным раствором карбоната калия (насыщенный, 10 мл) и сушили над сульфатом натрия. Органическую фазу концентрировали и полученное в результате твердое вещество сушили в высоком вакууме при 60°С с получением названного соединения (0,082 г, 0,12 ммоль, выход 48%). МС (ESI) m/z 376,4 [M+1]+.
D. 6-Оксо-8-фенил-2-(3-пиридил)-5,7,8-тригидроптеридин-4-карбоксамид. Раствор этил-6-оксо-8-фенил-2-(3-пиридил)-5,7,8-тригидроптеридин-4-карбоксилата (0,038 г, 0,101 ммоль) в безводном метаноле (15 мл) охлаждали до -78°С. Затем раствор насыщали газообразным аммиаком. Реакционный сосуд герметизировали при -78°С и предоставляли возможность нагреваться до комнатной температуры. Через 18 часов реакционную смесь охлаждали до -78°С и открывали для контакта с атмосферой. Летучие вещества выпаривали и полученные в результате твердые вещества сушили в вакууме с получением названного соединения (0,027 г, 0,078 ммоль, выход 76%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,4 (с, 1Н), 9,33 (д, J=1,2, 1H), 8,83 (с, 1H), 8,59 (дд, J=4,9, 1,6, 1H), 8,52 (дт, J=8,0, 3,9, 1H), 8,23 (с, 1H), 7,59-7,49 (м, 4H), 7,45-7,33 (м, 2H), 4,67 (с, 2H); MC (ESI) m/z 347,4 [M+1]+; т. пл. 294-296°C.
5.1.30 ПРИМЕР 30
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛ)-9-(2-МЕТОКСИФЕНИЛ)-9Н-ПУРИН-6-КАРБОКСАМИДА
A. Этил-2-хлор-6-(2-метоксифениламино)-5-ниагропиримидин-4-карбоксилат. В 250 мл круглодонную колбу помещали этил-2,6-дихлор-5-нитропиримидин-4-карбоксилат (2 г, 7,52 ммоль) в тетрагидрофуране (50 мл) и смесь охлаждали до -78°С. Добавляли по каплям раствор 2-метоксианилина (0,763 мл, 6,77 ммоль) и диизопропилэтиламина (1,313 мл, 7,52 ммоль) в 4 мл тетрагидрофурана и реакционной смеси предоставляли возможность нагреваться до комнатной температуры в течение ночи. Растворитель удаляли при пониженном давлении и неочищенное вещество очищали колоночной хроматографией (SiO2, 90% н-гексанов в этилацетате) с получением названного соединения в виде твердого оранжевого вещества (2,42 г, 6,86 ммоль, выход 91%). МС (ESI) m/z 353,3 [М+1]+.
B. Этил-5-амино-2-хлор-6-(2-метоксифениламино)пиримидин-4-карбоксилат. Этил-2-хлор-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (622 мг, 1,763 ммоль) суспендировали в смеси этанола (13 мл) и ДМФА (3 мл) и добавляли дигидрат хлорида олова(II) (1,19 г, 5,29 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, фильтровали и летучие соединения выпаривали. Полученный в результате остаток очищали хроматографией на силикагелевой колонке с нормальной фазой (50% н-гексанов в этилацетате). Фракции, содержащие продукт, объединяли и растворитель выпаривали с получением названного соединения (0,465 г, 1,441 ммоль, выход 82%) в виде желтого твердого вещества. МС (ESI) m/z 323,3 [M+1]+.
С. Этил-5-амино-2-(3-гидроксифенил)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. В микроволновую колбу помещали этил-5-амино-2-хлор-6-(2-метоксифениламино)пиримидин-4-карбоксилат (404 мг, 1,252 ммоль), 3-гидроксифенилбороновую кислоту (259 мг, 1,878 ммоль), фосфат калия (531 мг, 2,504 ммоль), дициклогексил(2',6'-диметоксибифенил-2-ил)фосфин (77 мг, 0,188 ммоль) и ацетат палладия(II) (42,2 мг, 0,188 ммоль) в тетрагидрофуране (20 мл) и воду (2 мл) и реакционную смесь нагревали при 120°С в течение 20 мин в микроволновом реакторе. Реакционную смесь фильтровали и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией (SiO2, 80-60% н-гексанов в этилацетате) и полупрепаративной ВЭЖХ (20-100% ацетонитрил + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Фракции, содержащие продукт, нейтрализовали карбонатом калия (насыщенный водный раствор), экстрагировали этилацетатом и сушили над сульфатом магния с получением названного соединения (57,8 мг, 0,152 ммоль, выход 12%). МС (ESI) m/z 381,4 [M+1]+.
D. 5-Амино-2-(3-гидроксифенил)-6-(2-метоксифениламино)пиримидин-4-карбоксамид. Раствор этил-5-амино-2-(3-гидроксифенил)-6-(2-метоксифениламино)пиримидин-4-карбоксилата (57,8 мг, 0,152 ммоль) в безводном метаноле (5 мл) охлаждали до -78°С. Затем раствор насыщали газообразным аммиаком. Реакционный сосуд герметизировали при -78°С и предоставляли возможность нагреваться до комнатной температуры. Через 18 ч реакционную смесь охлаждали до -78°С и открывали для контакта с атмосферой. Летучие вещества выпаривали и полученное в результате вещество очищали колоночной хроматографией (SiO2, от 2 до 5% метанола в метиленхлориде) с получением названного соединения 98,3% чистоты (53 мг, 0,151 ммоль, выход 99%). 1Н ЯМР (400 МГц, CD3OD) δ 8,41 (м, 1Н), 7,79 (дт, J=8,00, 1,20, 1Н), 7,73 (м, 1Н), 7,21 (т, J=8,00, 1Н), 7,06 (м, 3Н), 6,79 (ддд, J=8,00, 2,54, 0,98, 1H), 3,94 (с, 3Н); МС (ESI) m/z 352,2 [M+1]+; т.пл. 230-231°С.
Е. 2-(3-Гидроксифенил)-9-(2-метоксифенил)-9Н-пурин-6-карбоксамид. 5-Амино-2-(3-гидроксифенил)-6-(2-метоксифениламино)пиримидин-4-карбоксамид (30 мг, 0,085 ммоль) суспендировали в триэтилортоформиате (3 мл) и перемешивали при 130°С в течение 2 ч. Затем реакционную смесь охлаждали до комнатной температуры и разбавляли простым этиловым эфиром. Полученные в результате твердые вещества собирали фильтрованием, промывали простым этиловым эфиром и сушили в вакууме при 60°С с получением названного соединения 98,3% чистоты (20 мг, 0,054 ммоль, выход 64%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,59 (с, 1Н), 8,85 (с, 1Н), 8,48 (с, 1Н), 8,12 (с, 1Н), 7,90 (д, J=7,81, 1H), 7,84 (с, 1Н), 7,69 (д, J=7,81, 1H), 7,61 (т, J=8,20, 1H), 7,39 (д, J=8,20, 1H), 7,31-7,223 (перекрывание м, 2Н), 6,87 (д, J=7,61, 1Н), 3,81 (с, 3Н). МС (ESI) m/z 362,0 [M+1]+, т.пл. 257-259°С.
5.1.31 ПРИМЕР 31
СИНТЕЗ 9-ЦИКЛОПЕНТИЛ-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Этил-2-хлор-6-(циклопентиламино)-5-нитропиримидин-4-карбоксилат. В 250-мл круглодонную колбу помещали этил-2,6-дихлор-5-нитропиримидин-4-карбоксилат (1,5 г, 5,64 ммоль) в тетрагидрофуране (30 мл) и смесь охлаждали до -78°С. Добавляли по каплям раствор циклопентиламина (0,501 мл, 5,07 ммоль) и диизопропилэтиламина (0,985 мл, 5,64 ммоль) в 4 мл тетрагидрофурана и реакционной смеси предоставляли возможность нагреваться до комнатной температуры в течение ночи. Растворитель удаляли при пониженном давлении и неочищенное вещество очищали колоночной хроматографией (0-2% этилацетата в гексанах) с получением названного соединения (1,26 г, 4,00 ммоль, выход 71%). МС (ESI) m/z 315,2 [M+1]+.
В. Этил-5-амино-2-хлор-6-(циклопентиламино)пиримидин-4-карбоксилат. Этил-2-хлор-6-(циклопентиламино)-5-нитропиримидин-4-карбоксилат (283 мг, 0,899 ммоль) суспендировали в смеси этанола (8 мл) и ДМФА (1,85 мл) и добавляли дигидрат хлорида олова(II) (609 мг, 2,70 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, фильтровали и летучие вещества выпаривали. Полученный в результате остаток очищали хроматографией на силикагелевой колонке с нормальной фазой (50% этилацетата в гексанах). Фракции, содержащие продукт, объединяли и растворитель выпаривали с получением названного соединения (212,7 мг, 0,747 ммоль, выход 83%) в виде белого твердого вещества. МС (ESI) m/z 285,2 [M+1]+.
С. Этил-5-амино-6-(циклопентиламино)-2-(3-гидроксифенил)пиримидин-4-карбоксилат. В микроволновую колбу помещали этил-5-амино-2-хлор-6-(циклопентиламино)пиримидин-4-карбоксилат (212 мг, 0,745 ммоль), 3-гидроксифенилбороновую кислоту (154 мг, 1,12 ммоль), фосфат калия (316 мг, 1,49 ммоль, дициклогексил(2',6'-диметоксибифенил-2-ил)фосфин (45,8 мг, 0,112 ммоль) и ацетат палладия(II) (25,1 мг, 0,112 ммоль) в тетрагидрофуране (3 мл) и воду (0,3 мл) и реакционную смесь нагревали при 120°С в течение 30 мин в микроволновом реакторе. Реакционную смесь фильтровали и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией (25-43% этилацетата в гексанах и второй раз хроматографировали с применением 0-2% метанола в метиленхлориде) с получением названного соединения (76,3 мг, 0,223 ммоль, выход 30%). МС (ESI) m/z 343,2 [M+1]+.
D. Этил-9-циклопентил-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат. Этил-5-амино-6-(циклопентиламино)-2-(3-гидроксифенил)пиримидин-4-карбоксилат (76,3 мг, 0,223 ммоль) растворяли в метиленхлориде (5 мл) и добавляли 1,1'-карбонилдиимидазол (361,6 мг, 2,23 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 1 ч и затем перемешивали при комнатной температуре в течение 5 дней. Растворитель удаляли при пониженном давлении и неочищенное вещество очищали колоночной хроматографией (50% этилацетата в гексанах) с получением названного соединения (49,7 мг, 0,135 ммоль, выход 60%). МС (ESI) m/z 369,4 [M+1]+.
Е. 9-Циклопентил-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Раствор этил-9-циклопентил-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилата (49,7 мг, 0,135 ммоль) в безводном метаноле (5 мл) охлаждали до -78°С. Затем раствор насыщали газообразным аммиаком. Реакционный сосуд герметизировали при -78°С и предоставляли возможность нагреваться до комнатной температуры. Через 18 ч реакционную смесь охлаждали до -78°С и открывали для контакта с атмосферой. Летучие вещества выпаривали и полученное в результате вещество повторно суспендировали в метаноле и фильтровали. Осадок сушили в высоком вакууме с получением названного соединения 100% чистоты (33 мг, 0,151 ммоль, выход 72%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,48 (с, 1Н), 9,52 (с, 1Н), 8,31 (с, 1Н), 7,99 (д, J=7,81, 1Н), 7,89 (д, J=12,88, 1Н), 7,28 (т, J=8,00, 1Н), 6,87 (дд, J=8,00, 1,66, 1Н), 4,83 (м, 1Н), 2,22 (м, 2Н), 2,01 (м, 4Н), 1,69 (м, 2Н); МС (ESI) m/z 340,0 [M+1]+, т.пл. 360-361°С.
5.1.32 ПРИМЕР 32
СИНТЕЗ 9-ТРЕТ-БУТИЛ-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 6-трет-Бутиламино-2-хлор-5-нитропиримидин-4-карбоксилат. трет-Бутиламин (0,26 г, 3,6 ммоль), 2,6-дихлор-5-нитропиримидин-4-карбоксилат (1,0 г, 3,8 ммоль) и диизопропилэтиламин (1,5 г, 11 ммоль) подвергали взаимодействию согласно общему методу С и очищали с применением хроматографии на силикагеле Biotage (0-35% этилацетата в гексанах) с получением названного соединения (0,73 г, 2,4 ммоль, выход 91%). МС (ESI) m/z 303,5 [M+1]+, 304,5 [M+2]+.
В. 5-Амино-6-трет-бутиламино-2-хлорпиримидин-4-карбоксилат. 6-трет-Бутиламино-2-хлор-5-нитропиримидин-4-карбоксилат (0,73 г, 2,4 ммоль) суспендировали в этаноле (40 мл) и ДМФА (4 мл) и подвергали взаимодействию с дигидратом хлорида олова(II) (1,6 г, 7,5 ммоль) согласно общему методу D. Неочищенный остаток (0,46 г, 1,69 ммоль, выход 70%) применяли на следующей стадии без дополнительной очистки. МС (ESI) m/z 273,4 [M+1]+.
С. 5-Амино-6-трет-бутиламино-2-(3-гидроксифенил)-пиримидин-4-карбоксилат. В микроволновую колбу помещали 5-амино-6-трет-бутиламино-2-хлорпиримидин-4-карбоксилат (0,46 г, 1,69 ммоль), 3-гидроксифенилбороновую кислоту (0,35 г, 2,53 ммоль), фосфат калия (0,72 г, 3,4 ммоль), дициклогексил(2',6'-диметоксибифенил-2-ил)фосфин (0,10 г, 0,25 ммоль) и ацетат палладия(II) (0,06 г, 0,25 ммоль) в тетрагидрофуране (17 мл) и воду (1,7 мл). Смесь подвергали взаимодействию согласно общему методу Е. Неочищенный остаток очищали с применением хроматографии на силикагеле Biotage (0-35% этилацетата в гексанах) с получением названного соединения (0,26 г, 0,79 ммоль, выход 36%). МС (ESI) m/z 331,4 [M+1]+.
D. 9-трет-Бутил-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксалат. 5-Амино-6-трет-бутиламино-2-(3-гидроксифенил)-пиримидин-4-карбоксилат (0,26 г, 0,79 ммоль) и 1,1'-карбонилдиимидазол (0,64 г, 4,0 ммоль) подвергали взаимодействию согласно общему методу F. Неочищенный остаток очищали с применением хроматографии на силикагеле Biotage (0-50% этилацетата в гексанах) с получением названного соединения (0,13 г, 0,36 ммоль, выход 46%). МС (ESI) m/z 357,4 [M+1]+.
Е. 9-трет-Бутил-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. 9-трет-Бутил-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат (0,13 г, 0,36 ммоль) и газообразный аммиак подвергали взаимодействию согласно общему методу G. Неочищенный остаток очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (10-70% ацетонитрила + 0,1% TFA в Н2О + 0,1%TFA, в течение 39 мин). Фракции, содержащие требуемое вещество, объединяли и концентрировали при пониженном давлении перед пропусканием через ионообменную колонку Strata-XC с водой, метанолом и 5% гидроксидом аммония в метаноле. Полученный в результате остаток сушили в высоком вакууме при 60°С с получением названного соединения в виде белого твердого вещества 100% чистоты (0,013 г, 0,040 ммоль, выход 11%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,33 (с, 1Н), 9,53 (с, 1Н), 8,31 (с, 1Н), 7,97 (д, 1Н), 7,89 (с, 1Н), 7,82-7,87 (м, 1Н), 7,28 (т, J=8,0, 1Н), 6,82-6,90 (м, 1Н), 1,82 (с, 9Н). МС (ESI) m/z 328,1 [M+1]+.
5.1.33 ПРИМЕР 33
СИНТЕЗ [2-(3-ГИДРОКСИФЕНИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-(7-ГИДРОПУРИН-6-ИЛ)]-N-МЕТИЛКАРБОКСАМИДА
А. Метил-2-(3-гидроксифенил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксилат. 2-(3-Гидроксифенил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид (0,49 г, 1,27 ммоль) растворяли в смеси этанола (20 мл) и ДМСО (10 мл). К такой реакционной смеси добавляли 1 н. NaOH (10 мл) и нагревали в течение 2 дней при 90°С. Реакционную смесь охлаждали до комнатной температуры, летучие вещества удаляли и остаток растворяли в смеси этанола и Et2O и устанавливали рН~4 с помощью 3 н. HCl. Органические вещества удаляли вакуумным фильтрованием и твердое вещество собирали. Продукт очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (30-70% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 39 мин). Фракции, содержащие требуемое вещество, собирали и концентрировали при пониженном давлении перед пропусканием через ионообменную колонку Strata-XC с водой, метанолом и 5% гидроксидом аммония в метаноле. Полученное в результате твердое вещество сушили в высоком вакууме при 60°С с получением 2-(3-гидроксифенил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоновой кислоты в виде белого твердого вещества (0,005 г, 0,01 ммоль, выход 1%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,68 (с, 1Н), 7,61 (д, J=7,6, 1H), 7,53 (т, J=8,0, 1H), 7,46 (д, J=6,8, 1H), 7,28 (с, J=8,4, 1Н), 7,20-7,12 (м, 2Н), 6,75 (д, J=6,4, 1H), 3,74 (с, 3Н); МС (ESI) m/z 379,4 [M+1]+, т.пл. 229-230. 2-(3-Гидроксифенил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоновую кислоту (0,6 г, 1,6 ммоль) растворяли в МеОН (20 мл) и добавляли 30% раствор Н2О2 (0,75 мл) и предоставляли возможность перемешиваться при комнатной температуре в течение ночи. После расходования исходного вещества летучие продукты реакции удаляли и оставшуюся смесь нейтрализовали насыщенным NaHCO3. Образовавшийся осадок отфильтровывали и промывали деионизированной водой. Продукт очищали хроматографией на силикагеле с применением градиента метанола в дихлорметане (0-7% МеОН). Чистые фракции собирали и конденсировали с получением названного соединения (0,275 г, 43%). МС (ESI) m/z 379,4 [M+1]+.
В. [2-(3-Гидроксифенил)-9-(2-метоксифенил)-8-оксо-(7-гидропурин-6-ил)]-N-метилкарбоксамид. Метил-2-(3-гидроксифенил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксилат (0,15 г, 0,38 ммоль) и цианид калия (0,025 г, 0,38 ммоль) растворяли в метаноле (10 мл). Добавляли метиламин (1,0 мл) и реакционную смесь герметизировали и нагревали при 60°С в течение ночи. Реакционную смесь охлаждали до комнатной температуры, летучие вещества удаляли и продукт очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (30-70% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 39 мин). Фракции, содержащие требуемое вещество, собирали и концентрировали при пониженном давлении перед пропусканием через ионообменную колонку Strata-XC с водой, метанолом и 5% гидроксидом аммония в метаноле. Полученное в результате твердое вещество сушили в высоком вакууме при 60°С с получением названного соединения в виде белого твердого вещества (0,005 г, 0,01 ммоль, выход 1%). 1Н ЯМР (400 МГц, CD3OD-d4) δ 11,74 (с, 1Н), 9,49 (с, 1Н), 8,96 (д, J=5,2, 1Н), 7,89 (д, J=7,6, 1Н), 7,69 (м, 1Н), 7,55 (дт, J=8,4, 2,0, 1Н), 7,49 (дд, J=8,0, 2,0, 1H), 7,30 (д, J=7,6, 1H), 7,23 (т, J=7,6, 1Н), 7,15 (дт, J=7,6, 1,3, 1H), 6,82 (дд, J=8,0, 2,4, 1H), 3,75 (с, 3Н), 3,92 (д, J=4,8, 3H). МС (ESI) m/z 392,3 [M+1]+, т.пл. 339°С.
5.1.34 ПРИМЕР 34
СИНТЕЗ 9-(2-ИЗОПРОПИЛФЕНИЛ)-2-(4-(5-МЕТИЛ-4Н-1,2,4-ТРИАЗОЛ-3-ИЛ)ФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 4-(Диэтоксиметил)бензонитрил. 4-Формилбензонитрил (1,00 г, 7,63 ммоль) и триэтилортоформиат (2,54 мл, 15,3 ммоль) добавляли к суспензии перхлорной кислоты на силикагеле (полученной перемешиванием 1 мл перхлорной кислоты (60%) с 12 г силикагеля в 50 мл простого эфира в течение 30 мин и затем удалением простого эфира при пониженном давлении) (0,100 г) в этаноле (7,6 мл). Смесь перемешивали при комнатной температуре в течение 30 мин и затем растворители удаляли при пониженном давлении с получением прозрачного, бледно-желтого масла (1,57 г, 100%). Поскольку продукт быстро гидролизуется обратно в исходное вещество, его быстро переносили на следующую стадию без дополнительной очистки. МС (ESI) m/z 206,1 [M+1]+.
В. 3-(4-(Диэтоксиметил)фенил-5-метил-4Н-1,2,4-триазол. 4-(Диэтоксиметил)бензонитрил (500 мг, 2,44 ммоль), ацетогидразид (361 мг, 4,87 ммоль) и карбонат калия (673 мг, 4,87 ммоль) добавляли к 1-бутанолу (2,4 мл) в толстостенном боросиликатном стеклянном сосуде (20 мл). Затем раствор нагревали в микроволновом реакторе Biotag Emrys Optimizer при 150°С в течение 7 ч. Реакционную смесь фильтровали и затем концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией (Biotage) (0-10% метанола в дихлорметане) с получением требуемого продукта (135 мг, 21%) в виде бледно-желтого твердого вещества. МС (ESI) m/z 262,0 [M+1]+.
С. 4-(5-Метил-4Н-1,2,4-триазол-3-ил)бензальдегид. 3-(4-(Диэтоксиметил)фенил)-5- (130 мг, 0,497 ммоль) добавляли к суспензии перхлорной кислоты на силикагеле (100 мг) в смеси 1:1 метанол:вода (1 мл). Реакционную смесь перемешивали в течение 1 ч при комнатной температуре. После фильтрования силикагеля из фильтрата осаждали белое твердое вещество. Твердое вещество собирали и определяли как требуемый продукт (80 мг, 86%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,88, (ушир.с, 1Н), 10,04 (с, 1Н), 8,19 (д, J=8,20, 2Н), 7,98 (д, J=8,59, 2H), 2,44 (с, 3Н).
D. 9-(2-Изопропилфенил)-2-(4-(5-метил-4Н-1,2,4-триазол-3-ил)фенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-изопропилфенил)мочевину (216 мг, 0,801 ммоль), 4-(5-метил-4Н-1,2,4-триазол-3-ил)бензальдегид (75 мг, 0,401 ммоль) и триэтиламин (0,112 мл, 0,801 ммоль) перемешивали в метаноле (7 мл) в течение 24 ч. Полученную в результате неоднородную смесь фильтровали. Осадок собирали и перекристаллизовывали растворением в теплом ДМФА (1 мл) и добавлением по каплям воды. Твердое вещество собирали и сушили в вакууме при 60°С в течение 48 ч с получением не совсем белого твердого вещества (30 мг, 16%). 1Н ЯМР (400 МГц, CD3OD) δ 8,55-8,39 (м, 2Н), 8,08-7,91 (м, 2Н), 7,64-7,54 (м, 2Н), 2,78 (септ., J=6,64, 1Н), 2,49 (с, 2Н), 2,40 (ушир.с, 1Н), 1,22 (д, J=6,64, 3Н), 1,17 (д, J=6,64, 3Н); МС (ESI) m/z 455,1 [M+1]+.
5.1.35 ПРИМЕР 35
СИНТЕЗ [2-(3-ГИДРОКСИФЕНИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-(7-ГИДРОПУРИН-6-ИЛ]-N,N-ДИМЕТИЛКАРБОКСАМИДА
А. [2-(3-Гидроксифенил)-9-(2-метоксифенил)-8-оксо-(7-гидропурин-6-ил)]-N,N-диметилкарбоксамид. Метил-2-(3-гидроксифенил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксилат (0,13 г, 0,32 ммоль) (см. пример 33.А) и цианид калия (0,025 г, 0,38 ммоль) растворяли в метаноле (10 мл). Добавляли диметиламин (1,0 мл) и реакционную смесь герметизировали и нагревали при 60°С в течение ночи. Реакционную смесь охлаждали до комнатной температуры, летучие соединения удаляли и продукт очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (5-60% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 39 мин). Фракции, содержащие требуемое вещество, объединяли и концентрировали при пониженном давлении перед пропусканием через ионообменную колонку Strata-XC с водой, метанолом и 5% гидроксидом аммония в метаноле. Полученное в результате твердое вещество сушили в высоком вакууме при 60°С с получением названного соединения в виде белого твердого вещества (0,005 г, 0,01 ммоль, выход 1%). 1Н ЯМР (300 МГц, CD3OD-d4) δ 11,84 (с, 1Н), 9,51 (с, 1Н), 7,60-7,48 (м, 4Н), 7,30 (д, J=8,1, 1H), 7,25-7,13 (м, 2Н), 6,80 (д, J=7,8, 1H), 3,77 (с, 3Н), 3,19 (с, 3Н), 3,10 (с, 3Н); МС (ESI) m/z 406,0 [M+1]+; т.пл. 290-292°С.
5.1.36 ПРИМЕР 36
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛАМИНО)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Этил-2-(3-гидроксифениламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат. К раствору этил-2-хлор-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилата (см. пример 30.А) (200 мг, 0,567 ммоль) в ДМФА (3 мл) добавляли 3-аминофенол (74,3 мг, 0,680 ммоль) и N,N-диизопропилэтиламин (0,149 мл, 0,851 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и полученный в результате остаток очищали хроматографией на силикагелевой колонке с нормальной фазой (25-43% этилацетата в гексанах). Фракции, содержащие продукт, объединяли и растворитель выпаривали с получением названного соединения (227 мг, 0,534 ммоль, выход 94%) в виде оранжевого твердого вещества. МС (ESI) m/z 426,2 [M+1]+.
В. Этил-5-амино-2-(3-гидроксифениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. Этил-2-(3-гидроксифениламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (227 мг, 0,534 ммоль) растворяли в этаноле (15 мл) и добавляли 10% палладия на угле (56,8 мг, 0,053 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода в течение ночи. Затем реакционную смесь фильтровали через целит, растворитель удаляли при пониженном давлении и очищали колоночной хроматографией (2% метанола в метиленхлориде). Была необходима вторая хроматографическая очистка (50% этилацетата в гексанах). Фракции, содержащие продукт, объединяли и растворитель выпаривали с получением названного соединения (186,8 мг, 0,472 ммоль, выход 89%). МС (ESI) m/z 396,2 [M+1]+.
С. 2-(3-Гидроксифениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Этил-5-амино-2-(3-гидроксифениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (110,4 мг, 0,279 ммоль) растворяли в метиленхлориде (5 мл) и добавляли 1,1'-карбонилдиимидазол (453 мг, 2,79 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 1 ч и затем перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и неочищенное вещество пропускали через слой (пробку) силикагеля с применением смеси 5% метанол/этилацетат в качестве элюента с получением смеси этил-2-(3-гидроксифениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилата и этил-2-(3-(1Н-имидазол-1-карбонилокси)фениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилата. Такую смесь растворяли в безводном метаноле (5 мл) и охлаждали до -78°С. Затем раствор насыщали газообразным аммиаком. Реакционный сосуд герметизировали при -78°С и предоставляли возможность нагреваться до комнатной температуры. Через 18 ч реакционную смесь охлаждали до -78°С и открывали для контакта с атмосферой. Летучие вещества выпаривали и полученное в результате вещество очищали колоночной хроматографией (5% метанола в метиленхлориде) с получением названного соединения 99,4% чистоты (38,1 мг, 0,097 ммоль, выход 35%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,25 (с, 1Н), 9,32 (с, 1Н), 9,27 (с, 1Н), 8,05 (с, 1Н), 7,54-7,48 (м, 2Н), 7,44 (д, J=7,42, 1Н), 7,35 (с, 1Н), 7,23 (д, J=8,20, 1Н), 7,10 (т, J=8,20, 1H), 6,97 (м, 2Н), 6,28 (м, 1Н), 3,75 (с, 3Н). МС (ESI) m/z 393,1 [M+1]+, т.пл. 190-191°С.
5.1.37 ПРИМЕР 37
СИНТЕЗ 2-(4-ГИДРОКСИФЕНИЛАМИНО)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Этил-2-(4-гидроксифениламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат. К раствору этил-2-хлор-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилата (см. пример 30.А) (200 мг, 0,567 ммоль) в ДМФА (3 мл) добавляли 4-аминофенол (74,3 мг, 0,680 ммоль) и N,N-диизопропилэтиламин (0,149 мл, 0,851 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и полученный в результате остаток очищали хроматографией на силикагелевой колонке с нормальной фазой (50% этилацетата в гексанах). Фракции, содержащие продукт, объединяли и растворитель выпаривали с получением названного соединения (220 мг, 0,517 ммоль, выход 91%) в виде оранжевого твердого вещества. МС (ESI) m/z 426,2 [M+1]+.
В. Этил-5-амино-2-(4-гидроксифениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. Этил-2-(4-гидроксифениламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (220 мг, 0,517 ммоль) растворяли в этаноле (10 мл) и добавляли 10% палладия на угле (55 мг, 0,052 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода в течение ночи. Затем реакционную смесь фильтровали через целит, растворитель удаляли при пониженном давлении и очищали колоночной хроматографией (60-100% этилацетата в гексанах). Фракции, содержащие продукт, объединяли и растворитель выпаривали с получением названного соединения (201,3 мг, 0,509 ммоль, выход 98%). МС (ESI) m/z 396,2 [M+1]+.
С. 2-(4-Гидроксифениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Этил-5-амино-2-(4-гидроксифениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (156,8 мг, 0,397 ммоль) растворяли в метиленхлориде (5 мл) и добавляли 1,1'-карбонилдиимидазол (643 мг, 3,97 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 1 ч и затем перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и неочищенное вещество пропускали через слой (пробку) силикагеля с применением смеси 6:4 этилацетат/н-гексаны в качестве элюента с получением смеси этил-2-(4-гидроксифениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилата и этил-2-(4-(1Н-имидазол-1-карбонилокси)фениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилата. Такую смесь растворяли в безводном метаноле (5 мл) и охлаждали до -78°С. Затем раствор насыщали газообразным аммиаком. Реакционный сосуд герметизировали при -78°С и предоставляли возможность нагреваться до комнатной температуры. Через 18 ч реакционную смесь охлаждали до -78°С и открывали для контакта с атмосферой. Летучие соединения выпаривали и полученное в результате вещество очищали колоночной хроматографией (5% метанола в метиленхлориде и повторной хроматографией с применением 100% этилацетата) с получением названного соединения 98,8% чистоты (12,6 мг, 0,097 ммоль, выход 8%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,17 (с, 1Н), 9,09 (с, 1Н), 8,94 (с, 1Н), 7,93 (с, 1Н), 7,50 (м, 2Н), 7,42 (м, 3Н), 7,23 (д, J=8,20, 1H), 7,10 (т, 8,20, 1Н), 6,64 (д, J=8,79, 2H), 3,75 (с, 3Н). МС (ESI) m/z 393,1 [M+1]+; т.пл. 223-224°С.
5.1.38 ПРИМЕР 38
СИНТЕЗ N-МЕТИЛ-8-ОКСО-9-ФЕНИЛ-2-(ПИРИДИН-3-ИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 8-Оксо-9-фенил-2-(пиридин-3-ил)-8,9-дигидро-7Н-пурин-6-карбоновая кислота. 8-Оксо-9-фенил-2-(пиридин-3-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид (0,2 г, 0,55 ммоль) растворяли в смеси ДМСО (3 мл) и водного раствора 6 н. хлористоводородной кислоты (1,2 мл). Смесь нагревали до 90°С в течение 24 ч и затем выливали на смесь лед/вода. Устанавливали рН 5, полученный в результате осадок отфильтровывали и сушили с получением названного соединения в виде твердого вещества (0,108 г, 0,323 ммоль, выход 54%), которое непосредственно применяли на следующей стадии. МС (ESI) m/z 334,1 [M+1]+.
В. N-Метил-8-оксо-9-фенил-2-(пиридин-2-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид. К раствору 8-оксо-9-фенил-2-(пиридин-3-ил)-8,9-дигидро-7Н-пурин-6-карбоновой кислоты (0,150 г, 0,45 ммоль) в ДМСО (2,0 мл) добавляли диизопропилэтиламин (0,17 г, 1,35 ммоль), метиламин (0,280 г, 4,5 мл 2,0 М раствор в тетрагидрофуране, 9 ммоль) и затем бензотриазол-1-илокси-трис(диметиламино)фосфонийгексафторфосфат (0,298 г, 0,68 ммоль). Смесь подвергали ультразвуковой обработке в течение 5 мин для растворения всех компонентов смеси. После перемешивания в течение 10 мин исходное вещество израсходовалось (мониторинг ЖХ-МС). Растворитель удаляли и продукт очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (10-40% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Фракции, содержащие требуемое вещество, нейтрализовали водным раствором карбоната натрия и затем концентрировали до меньшего объема. Полученный в результате осадок отфильтровывали и промывали водой. Полученное в результате твердое вещество сушили в высоком вакууме при 60°С с получением названного соединения в виде твердого вещества (0,070 г, 0,20 ммоль, выход 44%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,0 (с, 1Н), 9,73 (с, 1Н), 9,29-9,20 (ушир.с, 1Н), 8,81-8,76 (м, 1Н), 8,76-8,72 (м, 1Н), 7,86-7,57 (м, 6Н), 3,01 (д, J=4,9, 3Н); МС (ESI) m/z 347,2 [M+1]+; т.пл. >330°С.
5.1.39 ПРИМЕР 39
9-(ТРАНС-4-ГИДРОКСИЦИКЛОГЕКСИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 1-((Z)-2-Амино-1,2-дициановинил)-3-(транс-4-гидроксициклогексил)мочевина. цис-4-Гидроксициклогексанкарбоновую кислоту (2,0 г, 13,87 ммоль), триэтиламин (1,40 г, 13,87 ммоль) и дифенилфосфорилазид (3,81 г, 13,87 ммоль) объединяли в толуоле и перемешивали при комнатной температуре. Мониторинг реакции проводили путем тонкослойной хроматографии (50% этилацетата в гексанах, окрашивание KMnO4). Через 30 мин раствор конденсировали при пониженном давлении и масло разбавляли ацетонитрилом (30 мл) с последующим добавлением диаминомалеонитрила (1,57 г, 14,56 ммоль). Раствор нагревали до 65°С в течение 16 ч. Раствор конденсировали при пониженном давлении и распределяли между водой и этилацетатом (3х), органические слои объединяли, сушили сульфатом магния и растворитель удаляли с получением неочищенного вещества. Неочищенное масло очищали с применением хроматографии на силикагеле Biotage (70-100% этилацетата в гексанах с последующими 10% метанола в этилацетате) с получением названного соединения (1,90 г, 37%). МС (ESI) m/z 250,1 [M+1]+.
В. 9-(транс-4-Гидроксициклогексил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. 1-((Z)-2-Амино-1,2-дициановинил)-3-(транс-4-гидроксициклогексил)мочевину (1,0 г, 4,01 ммоль), 3-гидроксибензальдегид (1,02 г, 8,02 ммоль) и триэтиламин (1,2 мл) в метаноле (35 мл) подвергали взаимодействию согласно общему методу В и очищали с применением препаративной ВЭЖХ с обращенной фазой (5-55% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 39 мин) с получением названного соединения (0,047 г, 3,2%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,47 (с, 1Н), 9,48 (с, 1Н), 8,31 (с, 1Н), 8,01 (д, J=8,0, 1H), 7,91 (м, 1Н), 7,27 (т, J=8,0, 1Н), 6,87 (д, J=8,0, 1H), 4,47 (с, 1Н), 4,26 (м, 1Н), 3,92 (с, 1Н), 2,80 (кв, J=12,4, 2H), 1,84 (д, J=13,2, 2H), 1,57 (т, J=13,6, 2H), 1,49 (д, J=10,4, 2H); МС (ESI) m/z 370,1 [M+1]+; т.пл. 366-368°С.
5.1.40 ПРИМЕР 40
9-(ТРАНС-4-ГИДРОКСИЦИКЛОГЕКСИЛ)-8-ОКСО-2-(ПИРИДИН-3-ИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 9-(транс-4-Гидроксициклогексил)-8-оксо-2-(пиридин-3-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид. 1-((Z)-2-Амино-1,2-дициановинил)-3-(транс-4-гидроксициклогексил)мочевину (см. пример 3.А) (0,900 г, 3,61 ммоль) и 3-пиридилкарбоксальдегид (0,774 г, 7,22 ммоль) и триэтиламин (1,2 ммоль) подвергали взаимодействию согласно общему методу В. Неочищенное вещество очищали с применением препаративной ВЭЖХ с обращенной фазой (10-40% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин) с получением названного соединения (0,037 г, 2,8%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 10,80 (c, 1Н), 9,72 (д, J=1,8, 1Н), 8,89 (дд, J=8,1, 1Н), 8,67 (д, J=5,1, 1H), 8,57 (с, 1Н), 7,91 (с, 1Н), 7,52 (м, 1Н), 4,52 (дд, J=2,1, 1Н), 4,27 (м, 1Н), 3,92 (с, 1Н), 2,84 (кв, J=9,6, 2Н), 1,84 (д, J=13,8, 2Н), 1,54 (м, 5Н); МС (ESI) m/z 355,4 [M+1]+; т.пл. 331-333°С.
5.1.41 ПРИМЕР 41
СИНТЕЗ 9-(ТРАНС-4-ГИДРОКСИЦИКЛОГЕКСИЛ)2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 1-((Z)-2-Амино-1,2-дициановинил)-3-(транс-4-гидроксициклогексил)мочевина. транс-4-Гидроксициклогексанкарбоновую кислоту (3,0 г, 20,80 ммоль), триэтиламин (2,10 г, 20,80 ммоль) и дифенилфосфорилазид (5,72 г, 20,80 ммоль) объединяли в толуоле и перемешивали при комнатной температуре. Мониторинг реакции проводили путем тонкослойной хроматографии (50% этилацетата в гексанах, окрашивали KMnO4). Через 30 мин раствор конденсировали при пониженном давлении и масло разбавляли ацетонитрилом (30 мл) с последующим добавлением диаминомалеонитрила (2,24 г, 21,84 ммоль). Раствор нагревали до 65°С в течение 16 ч. Полученную в результате неоднородную смесь растирали с водой. Полученный в результате осадок отфильтровывали и сушили в вакууме с получением названного соединения (1,60 г, 31%). МС (ESI) m/z 250,1 [M+1]+.
В. 9-(транс-4-Гидроксициклогексил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. 1-((Z)-2-Амино-1,2-дициановинил)-3-(транс-4-гидроксициклогексил)мочевину (0,800 г, 3,21 ммоль), 3-гидроксибензальдегид (0,612 г, 4,81 ммоль) и триэтиламин (1,2 мл) в метаноле (35 мл) подвергали взаимодействию согласно общему методу В и очищали с применением препаративной ВЭЖХ с обращенной фазой (5-55% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA в течение 30 мин) с получением названного соединения (0,082 г, 6,9%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,51 (с, 1Н), 9,56 (с, 1Н), 8,33 (с, 1Н), 8,00 (д, J=8,1, 1Н), 7,89 (д, J=7,2, 2Н), 7,29 (т, J=7,5, 1Н), 6,87 (д, J=7,5, 1Н), 4,72 (д, J=4,2, 1Н), 4,23 (м, 1Н), 3,60 (м, 1Н), 1,97 (д, J=10,4, 2Н), 1,77 (д, J=10,4, 2Н), 1,35 (м, 2Н); МС (ESI) m/z 370,1 [M+1]+; т.пл. 373-375°С.
5.1.42 ПРИМЕР 42
СИНТЕЗ 9-(ТРАНС-4-ГИДРОКСИЦИКЛОГЕКСИЛ)-8-ОКСО-2-(ПИРИДИН-3-ИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 9-(транс-4-Гидроксициклогексил)-8-оксо-2-(пиридин-3-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид. 1-((Z)-2-Амино-1,2-дициановинил)-3-(транс-4-гидроксициклогексил)мочевину (см. пример 41.А) (0,800 г, 3,21 ммоль), 3-пиридилкарбоксальдегид (0,516 г, 4,81 ммоль) и триэтиламин (1,2 мл) подвергали взаимодействию согласно общему методу В. Неочищенное вещество очищали с применением препаративной ВЭЖХ с обращенной фазой (5-40% ацетонитрила + 0,1% TFA в Н2О + 0,1 TFA, в течение 30 мин) с получением названного соединения (0,060 г, 5,3%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,61 (с, 1Н), 9,70 (с, 1Н), 8,85 (д, 1Н), 8,85 (д, J=7,8, 1Н), 8,68 (д, J=4,8, 1Н), 8,56 (с, 1Н), 7,93 (с, 1Н), 7,54 (дд, J=6,0, J=8,1, 1Н), 4,71 (д, J=4,2, 1H), 4,27 (м, 1Н), 3,59 (м, 1Н), 2,0 (д, J=10,5, 2Н), 1,78 (д, J=11,7, 2H), 1,35 (кв, J=11,7, 2H); МС (ESI) m/z 355,4 [M+1]+; т.пл. 343-345°С.
5.1.43 ПРИМЕР 43
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛАМИНО)-9-(2-МЕТОКСИФЕНИЛ)-9Н-ПУРИН-6-КАРБОКСАМИДА
А. 5-Амино-2-(3-гидроксифениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксамид. В герметизируемой трубке этил-5-амино-2-(3-гидроксифениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (см. пример 36.В) (76,4 мг, 0,193 ммоль) растворяли в безводном метаноле (5 мл) и охлаждали до -78°С. Затем раствор насыщали газообразным аммиаком. Реакционный сосуд герметизировали при -78°С и предоставляли возможность нагреваться до комнатной температуры. Через 18 ч реакционную смесь охлаждали до -78°С и открывали для контакта с атмосферой. Летучие вещества выпаривали и оставшееся вещество очищали колоночной хроматографией (100% этилацетата) с получением названного соединения (61,5 мг, 0,168 ммоль, выход 87%). МС (ESI) m/z 367,2 [M+1]+.
В. 2-(3-Гидроксифениламино)-9-(2-метоксифенил)-9Н-пурин-6-карбоксамид. 5-Амино-2-(3-гидроксифениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксамид (61,5 мг, 0,168 ммоль) суспендировали в триэтилортоформиате (5 мл) и перемешивали при 130°С в течение 1 ч. Затем реакционную смесь охлаждали до комнатной температуры и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали колоночной хроматографией (5% метанола в метиленхлориде) и второй раз хроматографировали (0-10% метанола в этилацетате) с получением названного соединения 99,6% чистоты (48,2 мг, 0,128 ммоль, выход 76%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,74 (с, 1Н), 9,18 (с, 1Н), 8,49 (с, 1Н), 8,29 (с, 1Н), 8,09 (с, 1Н), 7,63 (дд, J=7,81, 1,56, 1Н), 7,56 (т, J=8,60, 1Н), 7,33 (д, J=8,59, 1H), 7,26 (д, J=8,59, 1Н), 7,20-7,16 (м, 2Н), 6,97 (т, J=8,10, 1H), 6,33 (д, J=9,76, 1H), 3,81 (с, 3Н); МС (ESI) m/z 377,1 [M+1]+; т.пл. 155-157°С.
5.1.44 ПРИМЕР 44
СИНТЕЗ 9-ИЗОПРОПИЛ-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 6-Изопропиламино-2-хлор-5-нитропиримидин-4-карбоксилат. Изопропиламин (0,34 г, 5,7 ммоль) и 2,6-дихлор-5-нитропиримидин-4-карбоксилат (1,6 г, 6,0 ммоль) подвергали взаимодействию согласно общему методу С и очищали с применением хроматографии на силикагеле Biotage (0-35% этилацетата в гексанах) с получением названного соединения (1,8 г, 4,0 ммоль, выход 68%). МС (ESI) m/z 289,7 [M+1]+, 290,5 [M+2]+.
В. 5-Амино-6-изопропиламино-2-хлорпиримидин-4-карбоксилат. 6-Изопропиламино-2-хлор-5-нитропиримидин-4-карбоксилат (1,2 г, 4,2 ммоль) суспендировали в этаноле (80 мл) и ДМФА (16 мл) и подвергали взаимодействию с дигидратом хлорида олова(II) (2,8 г, 12,3 ммоль) согласно общему медоду D. Неочищенный остаток (0,46 г, 1,69 ммоль, выход 70%) брали на следующую стадию без дополнительной очистки. МС (ESI) m/z 259,1 [M+1]+, 260,1 [M+2]+.
С. 5-Амино-6-изопропиламино-2-(3-гидроксифенил)-пиримидин-4-карбоксилат. В микроволновую колбу помещали 5-амино-6-изопропиламино-2-хлорпиримидин-4-карбоксилат (1,2 г, 4,6 ммоль), 3-гидроксифенилбороновую кислоту (0,96 г, 7,0 ммоль), фосфат калия (3,0 г, 14,0 ммоль), дициклогексил(2',6'-диметоксибифенил-2-ил)фосфин (0,28 г, 0,68 ммоль) и ацетат палладия(II) (0,16 г, 0,71 ммоль) в тетрагидрофуране (25 мл) и воду (4 мл). Смесь подвергали взаимодействию согласно общему методу Е. Неочищенный остаток очищали с применением хроматографии на силикагеле Biotage (0-35% этилацетата в гексанах) с получением названного соединения (0,43 г, 1,4 ммоль, выход 29%). МС (ESI) m/z 317,1 [M+1]+.
D. 9-Изопропил-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат. 5-Амино-6-изопропиламино-2-(3-гидроксифенил)-пиримидин-4-карбоксилат (0,43 г, 1,4 ммоль) и 1,1'-карбонилдиимидазол (1,1 г, 6,8 ммоль) подвергали взаимодействию согласно общему методу F. Неочищенный остаток растирали с метиленхлоридом и холодными гексанами с получением названного соединения в виде белого твердого вещества (0,29 г, 0,85 ммоль, выход 63%). МС (ESI) m/z 343,4 [M+1]+.
Е. 9-Изопропил-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. 9-Изопропил-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат (0,29 г, 0,85 ммоль) и газообразный аммиак подвергали взаимодействию согласно общему методу G. Неочищенный остаток растирали с кипящим метанолом и диэтиловым эфиром. Полученное в результате белое твердое вещество сушили в высоком вакууме при 60°С с получением названного соединения в виде белого твердого вещества 95,8% чистоты (0,029 г, 0,093 ммоль, выход 11%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,48 (с, 1Н), 9,52 (с, 1Н), 8,33 (с, 1Н), 8,01 (д, J=6,6, 1H), 7,91 (с, 1Н), 7,18-7,35 (м, 1Н), 6,87 (д, J=8,6, 1H), 4,55-4,83 (м, 1Н), 1,57 (д, J=6,2, 6Н); МС (ESI) m/z 314,2 [M+1]+; т.пл. 338-342°С.
5.1.45 ПРИМЕР 45
СИНТЕЗ МЕТИЛ-4-(6-КАРБАМОИЛ-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-2-ИЛ)БЕНЗОАТА
А. Метил-4-(6-карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-ил)бензоат. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,65 г, 2,53 ммоль), 4-ацетоксибензальдегид (0,44 г, 2,65 ммоль) и триэтиламин (3,0 мл) подвергали взаимодействию согласно общему методу В. Неочищенный остаток очищали с применением хроматографии на силикагеле Biotage (0-100% этилацетата в гексанах) с получением названного соединения 98,9% чистоты (0,129 г, 0,069 ммоль, выход 51%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,87 (с, 1Н), 8,58 (с, 1Н), 8,51-8,53 (м, J=1,2, 1H), 8,49-8,51 (м, J=3,5, 1H), 8,02 (дд, J=8,4, 4,1, 1H), 7,50-7,60 (м, 1Н), 7,31 (дд, J=7,4, 3,5, 1Н), 7,15-7,20 (м, 1Н), 3,88 (с, 1Н), 3,76 (с, 1Н); МС (ESI) m/z 420,1 [M+1]+; т.пл. 286-290°С.
5.1.46 ПРИМЕР 46
СИНТЕЗ 2-(2-ХЛОР-3-ГИДРОКСИФЕНИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. 2-(2-Хлор-3-гидроксифенил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид. N-(1-Амино-2,2-дициановинил)[(2-метоксифенил)амино]карбоксамид (0,25 г, 1,0 ммоль), 2-хлор-3-гидроксибензальдегид (0,33 г, 2,1 ммоль) и триэтиламин (0,25 мл) подвергали взаимодействию согласно общему методу В. Продукт очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-70% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 39 мин). Фракции, содержащие требуемое вещество, объединяли и концентрировали при пониженном давлении перед пропусканием через ионообменную колонку Strata-XC с водой, метанолом и 5% гидроксидом аммония в метаноле. Полученное в результате твердое вещество сушили в высоком вакууме при 60°С с получением названного соединения в виде белого твердого вещества (0,082 г, 0,19 ммол, выход 20%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,82 (с, 1Н), 10,22 (с, 1Н), 8,00 (д, J=16, 2H), 7,51 (дт, J=7,8, 0,9 1H), 7,45 (дд, J=7,8, 1,2, 1H), 7,25-7,00 (м, 5Н), 3,73 (с, 3Н); МС (ESI) m/z 412,2 [M+1]+; т.пл. 331-334°С.
5.1.47 ПРИМЕР 47
СИНТЕЗ 2-(3-ЦИАНОФЕНИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(3-Цианофенил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,2 г, 0,78 ммоль), 3-формилбензонитрил (0,222 г, 1,7 ммоль) и триэтиламин (0,1 мл) подвергали взаимодействию согласно общему методу В. Полученный в результате осадок растворяли в ДМФА и крошили при добавлении воды. Этот осадок отфильтровывали и сушили в вакууме с получением названного соединения 96,3% чистоты (0,174 г, 0,451 ммоль, выход 58%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,87 (с, 1Н), 8,99 (с, 1Н), 8,76 (с, 1Н), 8,52 (д, J=7,97, 1H), 8,03 (с, 1Н), 7,90 (д, J=7,69, 1Н), 7,67-7,49 (м, 3Н), 7,30 (д, J=7,69, 1H), 7,16 (т, J=7,55, 1Н), 3,75 (с, 3Н); МС (ESI) m/z 387,3 [M+1]+; т.пл. 316-318°С.
5.1.48 Пример 48
СИНТЕЗ 2-(2-ГИДРОКСИФЕНИЛАМИНО)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Этил-2-(2-гидроксифениламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат. К раствору этил-2-хлор-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилата (см. пример 30.А) (300 мг, 0,851 ммоль) в ДМФА (5 мл) добавляли 2-аминофенол (111 мг, 1,021 ммоль) и N,N-диизопропилэтиламин (0,223 мл, 1,276 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и полученный в результате остаток очищали хроматографией на силикагелевой колонке с нормальной фазой (50% этилацетат в гексанах). Фракции, содержащие продукт, объединяли и растворитель выпаривали с получением названного соединения (362 мг, 0,851 ммоль, выход 100%). МС (ESI) m/z 426,2 [M+1]+.
В. Этил-5-амино-2-(2-гидроксифениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. Этил-2-(2-гидроксифениламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (362 мг, 0,851 ммоль) растворяли в этаноле (20 мл) и добавляли 10% палладия на угле (91 мг, 0,085 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение ночи. Затем реакционную смесь фильтровали через целит, растворитель удаляли при пониженном давлении и очищали колоночной хроматографией (50% этилацетата в гексанах). Фракции, содержащие продукт, объединяли и растворитель выпаривали с получением названного соединения (272,6 мг, 0,689 ммоль, выход 81%). МС (ESI) m/z 396,2 [M+1]+.
С. Этил-5-амино-2-(2-(трет-бутилдиметилсилилокси)фениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. Этил-5-амино-2-(2-гидроксифениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (184 мг, 0,465 ммоль) растворяли в метиленхлориде (5 мл) и добавляли трет-бутилдиметилсилилхлорид (77 мг, 0,512 ммоль) и имидазол (38,0 мг, 0,558 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Растворитель удаляли при пониженном давлении и неочищенное вещество очищали колоночной хроматографией (20% этилацетата в гексанах). Фракции, содержащие продукт, объединяли и растворитель выпаривали с получением названного соединения (232,6 мг, 0,456 ммоль, выход 98%) в виде желтого твердого вещества. МС (ESI) m/z 510,5 [M+1]+.
D. Этил-2-(2-(трет-бутилдиметилсилилокси)фениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат. Этил-5-амино-2-(2-(трет-бутилдиметилсилилокси)фениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (232,6 мг, 0,456 ммоль) растворяли в метиленхлориде (5 мл) и добавляли 1,1'-карбонилдиимидазол (740 мг, 4,56 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 1 ч и затем перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и неочищенное вещество очищали колоночной хроматографией (50% этилацетата в гексанах). Фракции, содержащие продукт, объединяли и растворитель выпаривали с получением названного соединения (235,2 мг, 0,439 ммоль, выход 96%) в виде желтого твердого вещества. МС (ESI) m/z 536,4 [M+1]+.
Е. 2-(2-Гидроксифениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Этил-2-(2-(трет-бутилдиметилсилилокси)фениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат (235,2 мг, 0,439 ммоль) растворяли в безводном метаноле (8 мл) и охлаждали до -78°С. Затем раствор насыщали газообразным аммиаком. Реакционный сосуд герметизировали при -78°С и предоставляли возможность нагреваться до комнатной температуры. Через 18 ч реакционную смесь охлаждали до -78°С и открывали для контакта с атмосферой. Летучие вещества выпаривали и полученное в результате вещество ресуспендировали в метиленхлориде. Осадок отфильтровывали и промывали метиленхлоридом и 5% метанолом в метиленхлориде и сушили в высоком вакууме с получением названного соединения 97,4% чистоты (125 мг, 0,319 ммоль, выход 73%) 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,28 (с, 1Н), 9,81 (с, 1Н), 7,94-7,91 (м, 2Н), 7,81 (с, 2Н), 7,53 (т, J=7,00, 1H), 7,45 (дд, J=7,69, 1,65, 1H), 7,26 (д, J=7,42, 1H), 7,12 (т, J=7,42, 1H), 6,82-6,66 (м, 3Н), 3,76 (с, 3Н); МС (ESI) m/z 393,2 [M+1]+; т.пл. 314-315°С.
5.1.49 ПРИМЕР 49
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛ)-9-(4-МЕТОКСИ-2-МЕТИЛФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-(4-метокси-2-метилфенил)мочевина. 4-Метокси-2-метилфенилизоцианат (1,51 г, 9,25 ммоль) и диаминомалеонитрил (1,0 г, 9,25 ммоль) подвергали взаимодействию согласно общему методу А. Полученную в результате неоднородную смесь фильтровали и промывали ацетонитрилом с последующим простым диэтиловым эфиром с получением названного соединения (0,435 г, 17%). МС (ESI) m/z 272,4 [M+1]+.
В. 2-(3-Гидроксифенил)-9-(4-метокси-2-метилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(4-метокси-2-метилфенил)мочевину (0,435 г, 1,60 ммоль), 3-гидроксибензальдегид (0,390 г, 3,2 ммоль) и триэтиламин (0,6 мл) подвергали взаимодействию согласно общему методу В. Осадок растирали с диметилформамидом и водой с получением названного соединения (0,310 г, 49%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,73 (с, 1Н), 9,47 (с, 1Н), 8,38 (с, 1Н), 7,97 (с, 1Н), 7,90 (д, J=8,4, 1H), 7,68 (с, 1Н), 7,36 (д, J=8,4, 1H), 7,27 (т, J=8,4, 1H), 7,03 (д, J=3,0, 1H), 6,95 (дд, J=2,7, J=9,0, 1H), 6,81 (дд, J=8,1, 1H), 3,84 (с, 3Н), 2,1 (с, 3Н); МС (ESI) m/z 392,4 [M+1]+; т.пл. 355-357°С.
5.1.50 ПРИМЕР 50
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-9-(2-(ТРИФТОРМЕТИЛ)ФЕНИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-(трифторметил)фенил)мочевина. α,α,α-Трифтор-о-толилизоцианат (1,73 г, 9,24 ммоль) и диаминомалеонитрил (2,5 г, 23,12 ммоль) подвергали взаимодействию согласно общему методу А. Полученную в результате неоднородную смесь фильтровали и промывали ацетонитрилом с последующим простым диэтиловым эфиром с получением названного соединения (0,410 г, 15%). МС (ESI) m/z 296,4 [M+1]+.
В. 2-(3-Гидроксифенил)-8-оксо-9-(2-(трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-(трифторметил)фенил)мочевину (0,403 г, 1,36 ммоль), 3-гидроксибензальдегид (0,333 г, 2,73 ммоль) и триэтиламин (0,6 мл) подвергали взаимодействию согласно общему методу В. Осадок растирали с диметилформамидом и водой с получением названного соединения (0,206 г, 36%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,89 (с, 1Н), 9,45 (с, 1Н), 8,42 (с, 1Н), 8,0 (м, 3Н), 7,84 (м, 3Н), 7,62 (с, 1Н), 7,21 (т, J=8,1, 1H), 6,81 (дд, J=7,2, 1,5, 1Н); МС (ESI) m/z 416,4 [M+1]+; т.пл. 363-365°С.
5.1.51 ПРИМЕР 51
СИНТЕЗ 2-(4-ЦИАНОФЕНИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(4-Цианофенил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,30 г, 1,2 ммоль), 4-формилбензонитрил (0,16 г, 1,22 ммоль) и триэтиламин (1,5 мл) подвергали взаимодействию согласно общему методу В. Неочищенный остаток растирали с ДМФА и водой с получением светло-коричневого твердого вещества. Это твердое вещество сушили в вакууме при 60°С с получением названного соединения 98,4% чистоты (0,051 г, 0,13 ммоль, выход 11%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,91 (с, 1Н), 8,64 (с, 1Н), 8,56 (д, J=8,2, 1Н), 8,03 (с, 1Н), 7,92 (д, J=8,2, 1Н), 7,48-7,61 (м, 1Н), 7,30 (д, J=8,5, 1Н), 7,16 (т, J=7,6, 1Н), 3,75 (с, 1Н); МС (ESI) m/z 387,1 [M+1]+; т.пл. 335-338°С.
5.1.52 ПРИМЕР 52
СИНТЕЗ СЛОЖНОГО МЕТИЛОВОГО ЭФИРА 4-[6-КАРБАМОИЛ-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-2-ИЛ]БЕНЗОЙНОЙ КИСЛОТЫ
А. 4-[6-Карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-ил]бензойная кислота. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,30 г, 1,2 ммоль), 4-формилбензойную кислоту (0,44 г, 2,65 ммоль) и триэтиламин (3,0 мл) подвергали взаимодействию согласно общему методу В. Неочищенный остаток очищали с применением хроматографии на силикагеле Biotage (0-100% этилацетата в гексанах) с получением названного соединения 99,4% чистоты (0,129 г, 0,069 ммоль, выход 51%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,85 (с, 1Н), 8,56 (с, 1Н), 8,48 (д, J=8,2, 1Н), 8,03 (с, 1Н), 7,99 (д, J=8,5, 1H), 7,49-7,61 (м, 1Н), 7,31 (д, J=8,5, 1Н), 7,17 (тд, J=7,7, 1,1, 1H), 3,76 (с, 1Н); МС (ESI) m/z 420,1 [M+1]+; т.пл. 328-332°С.
5.1.53 ПРИМЕР 53
СИНТЕЗ МЕТИЛ-3-(6-КАРБАМОИЛ-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-2-ИЛ)БЕНЗОАТ
А. Метил-3-(6-карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-ил)бензоат. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,4 г, 1,56 ммоль), метил-3-формилбензоат (0,556 г, 3,39 ммоль) и триэтиламин (0,2 мл) подвергали взаимодействию согласно общему методу В. Полученный в результате осадок растворяли в ДМФА и крошили при добавлении воды. Этот осадок отфильтровывали и сушили в вакууме с получением названного соединения 98,7% чистоты (0,426 г, 1,02 ммоль, выход 65%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,83 (с, 1Н), 8,77-8,74 (м, 2Н), 8,53 (с, 1Н), 8,03-8,00 (м, 2Н), 7,64-7,51 (м, 3Н), 7,32 (д, J=7,69, 1H), 7,17 (т, J=7,69, 1H), 3,86 (с, 3Н), 3,77 (с, 3Н); МС (ESI) m/z 420,1 [M+1]+; т.пл. 265-266°С.
5.1.54 ПРИМЕР 54
СИНТЕЗ 2-(3-АМИНОФЕНИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(3-Аминофенил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. 9-(2-Метоксифенил)-2-(3-нитрофенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид (см. пример 20.А) (68 мг, 0,167 ммоль) растворяли в этаноле (10 мл) и добавляли катализатор 10% палладия на угле (17,81 мг, 0,017 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода в течение ночи. Неочищенную реакционную смесь фильтровали через целит и очищали колоночной хроматографией (SiO2, 2% метанола в дихлорметане) с получением названного соединения 98,8% чистоты (15 мг, 24%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,70 (с, 1Н), 8,27 (с, 1Н), 8,02 (с, 1Н), 7,55 (м, 2Н), 7,49 (м, 2Н), 7,29 (д, J=8,2, 1H), 7,15 (т, J=7,4, 1H), 7,07 (т, J=7,7, 1Н), 6,62 (д, J=7,8, 1Н). 5,10 (с, 2Н), 3,75 (с, 3Н); МС (ESI) m/z 377,1 [M+1]+; т.пл. 284-285°С.
5.1.55 ПРИМЕР 55
СИНТЕЗ 3-(6-КАРБАМОИЛ-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-2-ИЛ)БЕНЗОЙНОЙ КИСЛОТЫ
А. 3-(6-Карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-ил)бензойная кислота. Метил-3-(6-карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-ил)бензоат (50 мг, 0,119 ммоль) растворяли в ДМФА (2 мл) и добавляли 1 мл 5М раствора гидроксида натрия в воде. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворители удаляли при пониженном давлении и остаток нейтрализовали 1 н. HCl. Осадок отфильтровывали, промывали водой и сушили в высоком вакууме с получением названного соединения 97,9% чистоты (48,3 мг, 0,119 ммоль, выход 100%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,81 (с, 1Н), 8,76-8,74 (м, 2Н), 8,51 (с, 1Н), 8,01-7,98 (м, 2Н), 7,61-7,50 (м, 3Н), 7,31 (д, J=8,24, 1Н), 7,17 (т, J=7,69, 1H), 3,76 (с, 3Н); МС (ESI) m/z 406,5 [M+1]+; т.пл. 348-350°С.
5.1.56 ПРИМЕР 56
2-(3-ГИДРОКСИФЕНИЛ)-9-(2-ИЗОПРОПИЛФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИД
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-изопропилфенил)мочевина. 2-Изопропилфенилизоцианат (1,49 г, 9,25 ммоль) и диаминомалеонитрил (1,0 г, 9,25 ммоль) подвергали взаимодействию согласно общему методу А. Полученную в результате неоднородную смесь фильтровали и промывали ацетонитрилом с последующим простым диэтиловым эфиром с получением названного соединения (0,338 г, 14%). МС (ESI) m/z 270,4 [M+1]+.
В. 2-(3-Гидроксифенил)-9-(2-изопропилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-изопропилфенил)мочевину (0,338 г, 1,25 ммоль), 3-гидроксибензальдегид (0,306 г, 2,51 ммоль) и триэтиламин (0,6 мл) подвергали взаимодействию согласно общему методу В. Полученную в результате неоднородную смесь фильтровали и промывали метанолом с последующим простым диэтиловым эфиром с получением названного соединения (0,137 г, 28%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,80 (с, 1Н), 9,48 (с, 1Н), 8,40 (с, 1Н), 7,99 (с, 1Н), 7,88 (д, J=8,1, 1H), 7,66 (с, 1Н), 7,57 (м, 2Н), 7,41 (д, J=4,2, 2H), 7,21 (т, J=7,8, 1H), 6,80 (д, J=7,8, 1H), 2,73 (м, 1Н), 1,11 (т, J=7,2, 6H); МС (ESI) m/z 390,4 [M+1]+; т.пл. 322-324°С.
5.1.57 ПРИМЕР 57
СИНТЕЗ 2-(1Н-ИНДАЗОЛ-6-ИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. 2-(1Н-Индазол-6-ил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,23 г, 0,89 ммоль) и 1Н-индазол-6-карбальдегид (0,29 г, 1,95 ммоль) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение 18 ч. Полученную в результате неоднородную смесь фильтровали и очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% MeCN + 0,1% TFA в Н2О + 0,1% TFA). Летучие вещества удаляли при пониженном давлении, ресуспендированное твердое вещество обрабатывали гидроксидом аммония с помощью ультразвука и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (42 мг, 0,11 ммоль, 12%) в виде не совсем белого твердого вещества. 1Н ЯМР (300 МГц, ДМСО-d6) δ 8,64 (с, 1Н), 8,52 (с, 1Н), 8,36 (дд, J=8,4, 1,5, 1Н), 8,16 (с, 1Н), 8,09 (с, 1Н), 7,86 (д, J=8,4, 1Н), 7,63 (м, 2Н), 7,40 (дд, J=7,2, 1,0, 1H), 7,26 (ддд, J=8,7, 7,5, 1,3, 1H), 3,84 (с, 3Н); МС (ESI) m/z 402,1 [M+1]+; т.пл. 309-310°С.
5.1.58 ПРИМЕР 58
СИНТЕЗ 2-(4-КАРБАМОИЛФЕНИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИД
А. 2-(4-Карбамоилфенил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Метил-4-(6-карбамоил-9-(2-метоксифенил)-8-оксо-8,9-гидро-7Н-пурин-2-ил)бензоат (см. пример 45.А) (0,15 г, 0,36 ммоль) растворили в 30 мл метанола. Температуру раствора доводили до -78°С и в реакционный сосуд пропускали газообразный NH3 в течение 15 мин. Реакционный сосуд герметизировали и перемешивали в течение 6 ч. Через 6 ч добавляли KCN (0,05 г, 0,77 ммоль) и реакционную смесь нагревали при 50°С в течение 24 часов. Неочищеный остаток очищали с применением хроматографии на силикагеле Biotage (0-20% метанола в DCM) и промывали кипящим метанолом (100 мл) с получением требуемого соединения (0,020 г, 0,049 ммоль, выход 14%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,56 (с, 1Н), 8,58 (с, 1Н), 8,51-8,53 (м, J=1,2, 1H), 8,49-8,51 (м, 1Н), 8,02 (дд, J=8,4, 4,1, 1H), 7,50-7,59 (м, 1Н), 7,31 (дд, J=7,4, 3,5, 1H), 7,15-7,22 (м, 1Н), 3,81 (с, 1Н). МС (ESI) m/z 405,1 [M+1]+; т.пл. >350°С.
5.1.59 ПРИМЕР 59
СИНТЕЗ 9-(2-ЭТИЛФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-этилфенил)мочевина. 2-Изопропилфенилизоцианат (1,36 г, 9,25 ммоль) и диаминомалеонитрил (1,0 г, 9,25 ммоль) подвергали взаимодействию согласно общему методу А. Полученную в результате неоднородную смесь фильтровали и промывали ацетонитрилом с последующим простым диэтиловым эфиром с получением названного соединения (1,05 г, 45%). МС (ESI) m/z 256,4 [M+1]+.
В. 9-(2-Этилфенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-этилфенил)мочевину (1,05 г, 4,11 ммоль), 3-гидроксибензальдегид (1,0 г, 8,22 ммоль) и триэтиламин (1,2 мл) подвергали взаимодействию согласно общему методу В. Полученную в результате неоднородную смесь фильтровали и промывали метанолом с последующим простым диэтиловым эфиром с получением неочищенного соединения. Твердое вещество очищали с применением препаративной ВЭЖХ (0,202 г, 13%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,80 (с, 1Н), 9,47 (с, 1Н), 8,40 (с, 1Н), 7,99 (с, 1Н), 7,88 (д, J=8,1, 1H), 7,65 (с, 1Н), 7,51 (м, 2Н), 7,43 (д, J=4,2, 2Н), 7,22 (т, J=7,8, 1Н), 6,80 (д, J=7,8, 1Н), 2,46 (кв, J=7,5, 1H), 1,05 (т, J=7,8, 6Н). МС (ESI) m/z 390,4 [M+1]+; т.пл. 355-357°С.
5.1.60 ПРИМЕР 60
СИНТЕЗ 9-(2,5-ДИХЛОРФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. N-((1Z)-2-Амино-1,2-дициановинил)[(2,5-дихлорфенил)амино]карбоксамид. 2,5-Дихлорбензолизоцианат (1,83 г, 9,71 ммоль) и 2,3-диаминомалеонитрил (1,0 г, 9,3 ммоль) подвергали взаимодействию в ацетонитриле согласно общему методу А. Вещество отфильтровывали и суспендировали в ацетонитриле/простом диэтиловом эфире. Полученное в результате твердое вещество отфильтровывали и сушили с получением названного соединения в виде желтовато-коричневого твердого вещества (2,38 г, 8,07 ммоль, выход 87%); МС (ESI) m/z 296,1 [M+1]+.
В. 9-(2,5-Дихлорфенил)-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид. N-((1Z)-2-Амино-1,2-дициановинил)[(2,5-дихлорфенил)амино]карбоксамид (0,44 г, 1,49 ммоль), 3-гидроксибензальдегид (0,409 г, 3,36 ммоль) и триэтиламин (0,291 мл, 2,08 ммоль) в МеОН (15 мл) подвергали взаимодействию согласно общему методу В. Полученную в результате неоднородную смесь фильтровали, промывали добавляемым ацетонитрилом с последующим МеОН с получением названного соединения (0,365 г, 59%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,90 (с, 1Н), 9,49 (с, 1Н), 8,42 (с, 1Н), 8,06 (с, 1Н), 7,90 (д, J=2,3, 1Н), 7,81-7,88 (м, 1Н), 7,65-7,75 (м, 1Н), 7,23 (т, J=7,6, 1Н), 6,82 (дд, J=7,5, 2,2, 1Н). МС (ESI) m/z 416,1 [M+1]+; т.пл. 358-360°С.
5.1.61 ПРИМЕР 61
СИНТЕЗ 2-(3-КАРБАМОИЛФЕНИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(3-Карбамоилфенил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Метил-3-(6-карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-ил)бензоат (96 мг, 0,229 ммоль) и цианид калия (7,45 мг, 0,114 ммоль) растворяли в сухом метаноле и охлаждали до -78°С. Раствор насыщали газообразным аммиаком и реакционную смесь закрывали и перемешивали при 50°С в течение 48 ч. Анализ ЖХ-МС показал, что реакция не завершена, добавляли дополнительно цианид калия (7,45 мг, 0,114 ммоль) и раствор охлаждали до -78°С и снова насыщали газообразным аммиаком. Реакционную смесь перемешивали при 60°С в течение трех дней. После остывания образовавшийся осадок отфильтровывали и очищали колоночной хроматографией (10% метанола в метиленхлориде). Фракции, содержащие продукт, объединяли с получением названного соединения 97,2% чистоты (44 мг, 0,109 ммоль, выход 47%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,83 (с, 1Н), 8,80 (с, 1Н), 8,60 (с, 1Н), 8,41 (д, J=7,97, 1H), 8,12-8,08 (м, 2Н), 7,93 (д, J=7,42, 1H), 7,58-7,49 (м, 4Н), 7,30 (д, J=7,69, 1H), 7,16 (т, J=7,69, 1H), 3,75 (с, 3Н); МС (ESI) m/z 405,1 [M+1]+; т.пл. 338-339°С.
5.1.62 ПРИМЕР 62
СИНТЕЗ 9-(2,6-ДИХЛОРФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. N-((1Z)-2-Амино-1,2-дициановинил)[(2,6-дихлорфенил)амино]карбоксамид. 2,6-Дихлорбензолизоцианат (1,83 г, 9,71 ммоль) и 2,3-диаминомалеонитрил (1,0 г, 9,3 ммоль) подвергали взаимодействию в ацетонитриле согласно общему методу А. Вещество отфильтровывали и суспендировали в смеси ацетонитрил/простой диэтиловый эфир. Полученное в результате твердое вещество отфильтровывали и сушили с получением названного соединения в виде светло-коричневого твердого вещества (1,74 г, 5,87 ммоль, выход 64%); МС (ESI) m/z 296,1 [M+1]+.
В. 9-(2,6-Дихлорфенил)-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид. N-((1Z)-2-Амино-1,2-дициановинил)[(2,6-дихлорфенил)амино]карбоксамид (0,3 г, 1,02 ммоль), 3-гидроксибензальдегид (0,278 г, 2,28 ммоль) и триэтиламин (0,199 мл, 1,43 ммоль) в МеОН (15 мл) подвергали взаимодействию согласно общему методу В. Продукт очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (30-70% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 39 мин). Фракции, содержащие требуемое вещество, собирали и концентрировали при пониженном давлении до минимального количества воды. Добавляли гидроксид аммония (2 мл) и полученную в результате взвесь обрабатывали ультразвуком и экстрагировали EtOAc. Органический слой сушили и концентрировали с получением названного соединения в виде белого твердого вещества (0,07 г, 0,17 ммоль, выход 17%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 12,2 (c, 1Н), 9,50 (с, 1Н), 8,47 (с, 1Н), 8,07 (с, 1Н), 7,89 (д, J=8,0, 1H), 7,78-7,86 (м, 1Н), 7,63-7,76 (м, 1Н), 7,24 (т, J=7,8, 1H), 6,84 (дд, J=7,6, 2,1, 1H); МС (ESI) m/z 416,1 [M+1]+; т.пл. 290-292°С.
5.1.63 ПРИМЕР 63
СИНТЕЗ 2-(2-ГИДРОКСИФЕНИЛ)-9-(2-МЕТОКСИФЕНИЛ)ПУРИН-6-КАРБОКСАМИДА
А. Этил-5-амино-2-(2-гидроксифенил)-6-[(2-метоксифенил)амино]пиримидин-4-карбоксилат. В микроволновую колбу помещали этил-5-амино-2-хлор-6-(2-метоксифениламино)пиримидин-4-карбоксилат (400 мг, 1,24 ммоль), 3-гидроксифенилбороновую кислоту (260 мг, 1,86 ммоль), фосфат калия (531 мг, 2,504 ммоль), дициклогексил(2',6'-диметоксибифенил-2-ил)фосфин (77 мг, 0,188 ммоль) и ацетат палладия(II) (42,2 мг, 0,188 ммоль) в тетрагидрофуране (20 мл) и воду (2 мл) и реакционную смесь нагревали при 120°С в течение 60 мин в микроволновом реакторе. Реакционную смесь фильтровали и неочищенный продукт адсорбировали на силикагеле. Флэш-хроматографией (40% EtOAc в гексанах) выделяли названное соединение (235 мг, 0,62 ммоль, 50%) в виде светло-коричневого твердого вещества. МС (ESI) m/z 381,3 [M+1]+.
В. 5-Амино-2-(2-гидроксифенил)-6-[(2-метоксифенил)амино]пиримидин-4-карбоксамид. Раствор этил-5-амино-2-(2-гидроксифенил)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (235 мг, 0,61 ммоль) в безводном метаноле (10 мл) охлаждали до -78°С. Затем раствор насыщали газообразным аммиаком в течение 15 мин. Реакционный сосуд герметизировали при -78°С и предоставляли возможность нагреваться до комнатной температуры. Через 18 ч реакционную смесь охлаждали до -78°С и открывали для контакта с атмосферой. Летучие вещества удаляли при пониженном давлении и неочищенный продукт применяли на следующей стадии без очистки.
С. 2-(2-Гидроксифенил)-9-(2-метоксифенил)пурин-6-карбоксамид. 5-Амино-2-(2-гидроксифенил)-6-(2-метоксифениламино)пиримидин-4-карбоксамид (153 мг, 0,61 ммоль) суспендировали в триэтилортоформиате (15 мл) и перемешивали при 130°С в течение 1 ч. Затем реакционную смесь охлаждали до комнатной температуры и разбавляли простым диэтиловым эфиром. Полученные в результате твердые вещества собирали фильтрованием, промывали простым этиловым эфиром и сушили в вакууме при 60°С с получением названного соединения (120 мг, 0,33 ммоль, выход 54%) в виде светло-коричневого твердого вещества. 1Н ЯМР (300 МГц, ДМСО-d6) δ 9,06 (с, 1Н), 8,57 (с, 1Н), 8,54 (с, 1Н), 8,29 (дд, J=7,8, 1,5, 1H), 7,73 (дд, J=7,5, 1,5, 1H), 7,63 (м, 1Н), 7,39 (м, 2Н), 7,26 (ддд, J=8,7, 7,8, 1,2, 1H), 6,95 (м, 2Н), 3,87 (с, 3Н); МС (ESI) m/z 362,1 [M+1]+; т.пл. 278-280°С.
5.1.64 ПРИМЕР 64
СИНТЕЗ 2-(1Н-ИНДАЗОЛ-5-ИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. 2-(1Н-Индазол-5-ил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,37 г, 3,51 ммоль) и 1Н-индазол-5-карбальдегид (0,47 г, 3,21 ммоль) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение 18 ч. Полученную в результате неоднородную смесь фильтровали и очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA). Летучие вещества удаляли при пониженном давлении, суспендированное твердое вещество обрабатывали гидроксидом аммония с помощью ультразвука и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (89 мг, 0,22 ммоль, 15%) в виде не совсем белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,14 (c, 1Н), 11,66 (с, 1Н), 8,80 (с, 1Н), 8,52 (с, 1Н), 8,40 (дд, J=9,2, 1,6, 1H), 8,15 (с, 1Н), 7,98 (с, 1Н), 7,55 (м, 3Н), 7,31 (д, J=7,6, 1H), 7,17 (ддд, J=8,8, 7,6, 1,2, 1H), 3,76 (с, 3Н); МС (ESI) m/z 402,1 [M+1]+; т.пл. 360°С.
5.1.65 ПРИМЕР 65
СИНТЕЗ 9-(2,3-ДИХЛОРФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. N-((Z)-2-Амино-1,2-дициановинил)[(2,3-дихлорфенил)амино]карбоксамид. 2,3-Дихлорбензолизоцианат (0,91 г, 4,85 ммоль) и 2,3-диаминомалеонитрил (0,5 г, 4,62 ммоль) подвергали взаимодействию в ацетонитриле согласно общему методу А. Вещество растирали в смеси ацетонитрил/простой диэтиловый эфир. Полученное в результате твердое вещество отфильтровывали и сушили с получением названного соединения в виде светло-коричневого твердого вещества (0,68 г, 2,58 ммоль, выход 28%); МС (ESI) m/z 297,1 [M+1]+.
В. 9-(2,3-Дихлорфенил)-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид. N-((1Z)-2-Амино-1,2-дициановинил)[(2,3-дихлорфенил)амино]карбоксамид (1,04 г, 3,51 ммоль) и 3-гидроксибензальдегид (0,94 г, 7,72 ммоль) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение 18 ч. Полученную в результате неоднородную смесь фильтровали и 200 мг неочищенного продукта очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA). Летучие вещества удаляли при пониженном давлении, суспендировали твердое вещество, обрабатывали гидроксидом аммония с помощью ультразвука и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (104 мг, 0,25 ммоль) в виде не совсем белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,95 (c, 1Н), 9,46 (с, 1Н), 8,42 (с, 1Н), 8,01 (с, 1Н), 7,92 (дд, J=8,0, 1,2, 1H), 7,88 (дд, J=7,6, 1,0, 1H), 7,77 (д, J=8,0, 1H), 7,65 (м, 2Н), 7,23 (т, J=7,6, 1Н), 6,83 (дд, J=8,0, 2,4, 1H); МС (ESI) m/z 415,9 [M+1]+; т.пл. 352-353°С.
5.1.66 ПРИМЕР 66
СИНТЕЗ 2-[4-(ГИДРОКСИМЕТИЛ)ФЕНИЛ]-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. 2-[4-(Гидроксиметил)фенил]-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид. Метил-4-(6-карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-ил)бензоат (см. пример 45.А) (300 мг, 0,72 ммоль) растворяли в безводном тетрагидрофуране (20 мл). При -78°С добавляли раствор литийалюминийгидрида (2,0 М, 0,72 мл) и предоставляли возможность реакционной смеси нагреваться до комн. темп. в течение 4 ч. Реакционную смесь гасили метанолом и летучие продукты удаляли при пониженном давлении. Неочищенное вещество очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA). Летучие продукты удаляли при пониженном давлении, суспендированное твердое вещество обрабатывали гидроксидом аммония с помощью ультразвука и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (43 мг, 0,11 ммоль, 15%) в виде не совсем белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,71 (с, 1Н), 8,48 (с, 1Н), 8,31 (д, J=8,0, 1H), 7,98 (с, 1Н), 7,55 (ддд, J=8,4, 8,0, 1,6, 1H), 7,50 (дд, J=8,0, 2,0, 1H), 7,36 (д, J=8,4, 1H), 7,29 (дд, J=8,4, 0,8, 1H), 7,17 (ддд, J=8,8, 7,6, 1,2, 1H), 5,25 (т, J=5,6, 1H), 4,53 (д, J=5,6, 2H), 3,74 (с, 3Н); МС (ESI) m/z 392,1 [M+1]+; т.пл. 294-295°С.
5.1.67 ПРИМЕР 67
СИНТЕЗ 2-[3-(ГИДРОКСИМЕТИЛ)ФЕНИЛ]-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. Метил-3-[6-карбамоил-9-(2-метоксифенил)-8-оксо-7-гидропурин-2-ил]бензоат. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,3 г, 1,17 ммоль), метил-3-формилбензоат (0,431 г, 2,63 ммоль) и триэтиламин (0,229 мл, 1,64 ммоль) в МеОН (15 мл) подвергали взаимодействию согласно общему методу В. Полученную в результате неоднородную смесь фильтровали, промывали добавляемым ацетонитрилом с получением названного соединения (0,400 г, 0,95 ммоль, выход 82%); МС (ESI) m/z 420,4 [M+1]+.
В. 2-[3-(Гидроксиметил)фенил]-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид. Метил-3-[6-карбамоил-9-(2-метоксифенил)-8-оксо-7-гидропурин-2-ил]бензоат (0,128 г, 0,3 ммоль) растворяли в тетрагидрофуране и охлаждали до -78°С. Добавляли литийалюминийгидрид (2М в тетрагидрофуране, 0,301 мл, 0,601 ммоль) и реакционной смеси предоставляли возможность нагреваться до комнатной температуры. Через 10 часов реакционную смесь гасили МеОН, соли отфильтровывали и реакционную смесь концентрировали. Продукт очищали с применением препаративной ВЭЖХ с обращенной фазой (30-70% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 40 мин). Фракции, содержащие требуемое вещество, объединяли и концентрировали при пониженном давлении до минимального количества воды. Добавляли EtOAc и органический слой промывали 5 раз насыщ. NaHCO3. Органический слой сушили и концентрировали с получением названного соединения в виде не совсем белого твердого вещества (0,03 г, 0,077 ммоль, выход 26%) 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,75 (с, 1Н), 8,46 (с, 1Н), 8,30 (с, 1Н), 8,19-8,26 (м, 1Н), 8,02 (с, 1Н), 7,53-7,59 (м, 1Н), 7,50 (дд, J=7,8, 1,6, 1H), 7,35-7,42 (м, 1Н), 7,30 (д, J=7,4, 1H), 7,16 (т, J=7,5, 1H), 5,20 (т, J=5,9, 1H), 4,53 (д, J=5,9, 1H), 3,75 (с, 1Н); МС (ESI) m/z 392,3 [M+1]+; т.пл. 280-282°С.
5.1.68 ПРИМЕР 68
СИНТЕЗ 9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-2-(ПИРИДИН-4-ИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 9-(2-Метоксифенил)-8-оксо-2-(пиридин-4-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,25 г, 0,97 ммоль), изоникотинальдегид (0,156 мл, 1,65 ммоль) и триэтиламин (0,271 мл, 1,94 ммоль) подвергали взаимодействию согласно общему методу В. Полученное в результате вещество сушили в вакууме с получением продукта в виде светло-коричневого твердого вещества (0,23 г, 0,64 ммоль, выход 65%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,93 (с, 1Н), 8,66 (AA'XX', JAX=6,05, 2H), 8,62 (ушир.с, 1Н), 8,27 (AA'XX', JAX=6,05, 2H), 8,04 (ушир.с, 1Н), 7,56 (тд, J=7,91, 1,76, 1H), 7,50 (дд, J=7,71, 1,67, 1H), 7,30 (д, J=7,61, 1H), 7,16 (тд, J=7,71, 1,10, 1H), 3,75 (с, 3Н); МС (ESI) m/z 363,1 [M+1]+; т.пл. 318-320°С.
5.1.69 ПРИМЕР 69
СИНТЕЗ 2-(4-ФТОР-3-ГИДРОКСИФЕНИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. 4-Фтор-3-гидроксибензальдегид. 4-Фтор-3-метоксибензальдегид (0,590 г, 3,83 ммоль) растворяли в CH2Cl2 и охлаждали до -78°С. Медленно добавляли трибромид бора (1М в CH2Cl2, 9,58 мл, 9,58 ммоль) и предоставляли возможность реакционной смеси нагрваться до комнатной температуры в течение ночи. К полученной в результате взвеси добавляли смесь лед/вода и перемешивали в течение 30 мин. Слои разделяли и органический слой промывали NaHCO3 (насыщ.) и 2 н. NaOH. Водный слой подкисляли концентрированной HCl и экстрагировали CH2Cl2. Органический слой сушили и концентрировали с получением названного соединения (0,220 г, 1,57 ммоль, выход 41%).
В. 2-(4-Фтор-3-гидроксифенил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,207 г, 0,698 ммоль), 4-фтор-3-гидроксибензальдегид (0,220 г, 1,57 ммоль) и триэтиламин (0,136 мл, 0,977 ммоль) в МеОН (15 мл) подвергали взаимодействию согласно общему методу В. Продукт очищали с применением препаративной ВЭЖХ с обращенной фазой (30-70% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 40 мин). Фракции, содержащие требуемое вещество, объединяли и концентрировали при пониженном давлении до минимального количества воды. Добавляли гидроксид аммония (2 мл) и полученную в результате взвесь обрабатывали ультразвуком и фильтровали. Твердое вещество сушили и концентрировали с получением названного соединения в виде не совсем белого твердого вещества (0,106 г, 0,267 ммоль, выход 38%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,73 (с, 1Н), 9,93 (с, 1Н), 8,42 (с, 1Н), 7,90-8,04 (м, 1Н), 7,85 (дд, J=9,0, 2,3, 1H), 7,52-7,62 (м, 1Н), 7,49 (дд, J=7,6, 1,8, 1H), 7,30 (д, J=7,4, 1H), 7,09-7,23 (м, 1Н), 3,74 (с, 1Н); МС (ESI) m/z 396,4 [M+1]+; т.пл. 344-346°С.
5.1.70 ПРИМЕР 70
СИНТЕЗ 2-(2-ФТОР-3-ГИДРОКСИФЕНИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. 2-Фтор-3-гидроксибензальдегид. 2-Фтор-3-метоксибензальдегид (1,0 г, 6,49 ммоль) растворяли в CH2Cl2 и охлаждали до -78°С. Медленно добавляли трибромид бора (1М в CH2Cl2, 16,22 мл, 16,22 ммоль) и предоставляли возможность реакционной смеси нагреваться до комнатной температуры в течение ночи. К полученной в результате взвеси добавляли смесь лед/вода и перемешивали в течение 30 мин. Слои разделяли и органический слой промывали NaHCO3 (насыщ.) и 2 н. NaOH. Водный слой подкисляли концентрированной HCl и экстрагировали CH2Cl2. Органический слой сушили и концентрировали с получением названного соединения (0,190 г, 1,36 ммоль, выход 21%).
В. 2-(2-Фтор-3-гидроксифенил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,179 г, 0,603 ммоль), 2-фтор-3-гидроксибензальдегид (0,190 г, 1,36 ммоль) и триэтиламин (0,117 мл, 0,844 ммоль) в МеОН (15 мл) подвергали взаимодействию согласно общему методу В. Полученную в результате неоднородную смесь фильтровали и промывали добавляемым ацетонитрилом. Далее твердое вещество растворяли в ДМФА и вызывали образование осадка добавлением воды. Полученное в результате твердое вещество отфильтровывали и сушили с получением названного соединения в виде не совсем белого твердого вещества (0,157 г, 0,396 ммоль, выход 66%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,86 (с, 1Н), 8,12 (с, 1Н), 8,01 (с, 1Н), 7,44-7,61 (м, 1Н), 7,32-7,41 (м, 1Н), 7,27 (д, J=7,6, 1H), 7,14 (т, J=7,4, 1H), 6,99-7,05 (м, 1Н), 3,74 (с, 1Н); МС (ESI) m/z 396,4 [M+1]+; т.пл. >300°С.
5.1.71 ПРИМЕР 71
СИНТЕЗ 2-[4-(1-ГИДРОКСИИЗОПРОПИЛ)ФЕНИЛ]-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИД
А. 2-[4-(1-Гидроксиизопропил)фенил]-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид. Метил-4-(6-карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-ил)бензоат (см. пример 45.А) (500 мг, 1,19 ммоль) растворяли в безводном тетрагидрофуране (20 мл). С помощью шприца добавляли метилмагнийбромид (1,4 М, 2,2 мл, 2,97 ммоль) и перемешивали при комнатной температуре 3 дня. Реакционную смесь гасили метанолом и летучие вещества удаляли при пониженном давлении. Неочищенное вещество очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрил + 0,1% TFA в Н2О + 0,1% TFA). Летучие вещества удаляли при пониженном давлении, суспендированное твердое вещество обрабатывали гидроксидом аммония, ультразвуком и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (150 мг, 0,36 ммоль, 30%) в виде не совсем белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,51 (с, 1Н), 8,25 (с, 1Н), 8,23 (с, 1Н), 7,95 (с, 1Н), 7,54 (м, 1Н), 7,51 (с, 1Н), 7,48 (м, 2Н), 7,29 (дд, J=8,4, 1,2 1H), 7,15 (ддд, J=8,8, 7,6, 1,2, 1H), 5,06 (c, 1H), 3,74 (c, 3H), 1,42 (c, 6H); МС (ESI) m/z 420,4 [M+1]+; т.пл. 293-294°С.
5.1.72 ПРИМЕР 72
СИНТЕЗ 2-[3-(1-ГИДРОКСИИЗОПРОПИЛ)ФЕНИЛ]-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. Метил-3-[6-карбамоил-9-(2-метоксифенил)-8-оксо-7-гидропурин-2-ил]бензоат. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,3 г, 1,17 ммоль), метил-3-формилбензоат (0,431 г, 2,63 ммоль) и триэтиламин (0,229 мл, 1,64 ммоль) в МеОН (15 мл) подвергали взаимодействию согласно общему методу В. Полученную в результате неоднородную смесь фильтровали, промывали добавляемым ацетонитрилом с получением названного соединения (0,400 г, 0,95 ммоль, выход 82%). МС (ESI) m/z 420,4 [M+1]+.
В. 2-[3-(1-Гидроксиизопропил)фенил]-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид. Метил-3-[6-карбамоил-9-(2-метоксифенил)-8-оксо-7-гидропурин-2-ил]бензоат (0,200 г, 0,477 ммоль) растворяли в тетрагидрофуране и добавляли метилмагнийбромид (1,4 М в тетрагидрофуране, 2,73 мл, 3,82 ммоль), и реакционную смесь нагревали при кипячении с обратным холодильником в течение 4 дней. Реакционную смесь гасили МеОН, соли фильтровали и реакционную смесь концентрировали. Продукт очищали с применением препаративной ВЭЖХ с обращенной фазой (20-70% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 40 мин). Фракции, содержащие требуемое вещество, объединяли и концентрировали при пониженном давлении до минимального количества воды. Добавляли EtOAc и органический слой промывали 5 раз насыщ. NaHCO3. Органический слой сушили и концентрировали с получением названного соединения в виде не совсем белого твердого вещества (0,040 г, 0,095 ммоль, выход 20%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,74 (с, 1Н), 8,42 (с, 1Н), 8,18 (д, J=7,8, 1H), 8,04 (с, 1Н), 7,43-7,67 (м, 1Н), 7,23-7,43 (м, 1Н), 7,16 (т, J=7,6, 1H), 5,05 (c, 1H), 3,76 (c, 1H), 1,40 (c, 6H); МС (ESI) m/z 420,4 [M+1]+; т.пл. 149-151°С.
5.1.73 ПРИМЕР 73
СИНТЕЗ 9-(2-МЕТОКСИФЕНИЛ)-2-(2-НИТРОФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. 9-(2-Метоксифенил)-2-(2-нитрофенил)-8-оксо-7-гидропурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,3 г, 1,17 ммоль), 2-нитробензальдегид (0,397 г, 2,63 ммоль) и триэтиламин (0,228 мл, 1,64 ммоль) в 20 мл МеОН подвергали взаимодействию согласно общему методу В. Полученную в результате неоднородную смесь фильтровали и промывали добавляемым ацетонитрилом. Продукт растворяли в CH2Cl2/МеОН и осадок отфильтровывали с получением названного соединения в виде не совсем белого твердого вещества (0,090 г, 0,22 ммоль, выход 19%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,96 (с, 1Н), 8,01-8,18 (м, 1Н), 7,93 (д, J=2,0, 1H), 7,85 (д, J=7,8, 1H), 7,75 (т, J=7,8, 1H), 7,67 (т, J=6,8, 1H), 7,53 (т, J=7,6, 1H), 7,41 (д, J=7,4, 1H), 7,25 (д, J=7,4, 1H), 7,10 (т, J=7,6, 1H), 3,76 (с, 1Н); МС (ESI) m/z 407,4 [M+1]+; т.пл. 319-321°С.
5.1.74 ПРИМЕР 74
СИНТЕЗ 9-(2-МЕТОКСИФЕНИЛ)-2-(4-НИТРОФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. 9-(2-Метоксифенил)-2-(4-нитрофенил)-8-оксо-7-гидропурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,3 г, 1,17 ммоль), 4-нитробензальдегид (0,397 г, 2,63 ммоль) и триэтиламин (0,228 мл, 1,64 ммоль) в 20 мл МеОН подвергали взаимодействию согласно общему методу В. Полученную в результате неоднородную смесь фильтровали и промывали добавляемым ацетонитрилом. Продукт растворяли в CH2Cl2/МеОН и осадок отфильтровывали с получением названного соединения в виде не совсем белого твердого вещества (0,060 г, 0,15 ммоль, выход 13%).1Н ЯМР (400 МГц, ДМСО-d6) δ 11,94 (с, 1Н), 8,63 (м, 1Н), 8,28 (д, J=9,0, 1H), 8,06 (c, 1H), 7,58 (т, J=7,4, 1H), 7,51 (дд, J=7,8, 1,6, 1H), 7,31 (д, J=7,4, 1H), 7,17 (т, J=7,6, 1H), 3,76 (c, 1H); МС (ESI) m/z 407,4 [M+1]+; т.пл. 348-350°С.
5.1.75 ПРИМЕР 75
СИНТЕЗ 2-(5-МЕТОКСИ-4-МЕТИЛ(3-ПИРИДИЛ))-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. 9-(2-Метоксифенил)-2-(2-нитрофенил)-8-оксо-7-гидропурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,124 г, 0,482 ммоль), 5-метокси-4-метилпиридин-3-карбальдегид (0,164 г, 1,09 ммоль) и триэтиламин (0,094 мл, 0,675 ммоль) в 20 мл МеОН подвергали взаимодействию согласно общему методу В. Полученную в результате неоднородную смесь фильтровали и промывали добавляемым ацетонитрилом с получением названного соединения в виде не совсем белого твердого вещества (0,115 г, 0,283 ммоль, выход 59%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,85 (c, 1H), 8,52 (с, 1Н), 8,31 (с, 1Н), 8,21 (с, 1Н), 7,97 (с, 1Н), 7,49-7,58 (м, 1Н), 7,47 (дд, J=7,8, 1,6, 1H), 7,26 (дд, J=8,4, 1,0, 1H), 7,11 (тд, J=7,6, 1,2, 1H), 3,91 (c, 1H), 3,75 (c, 1H), 2,25 (c, 1H); МС (ESI) m/z 407,0 [M+1]+; т.пл. 278-280°С.
5.1.76 ПРИМЕР 76
СИНТЕЗ 9-(2,4-ДИФТОРФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. N-((1Z)-2-Амино-1,2-дициановинил)[(2,4-дифторфенил)амино]карбоксамид. 2,4-Дифторбензолизоцианат (1,15 мл, 9,71 ммоль) и 2,3-диаминомалеонитрил (1,0 г, 9,25 ммоль) подвергали взаимодействию в ацетонитриле согласно общему методу А. Вещество растирали в смеси ацетонитрил/простой диэтиловый эфир. Полученное в результате твердое вещество отфильтровывали и сушили с получением названного соединения в виде светло-коричневого твердого вещества (0,68 г, 2,58 ммоль, выход 28%). МС (ESI) m/z 264,2 [M+1]+.
В. 9-(2,4-Дифторфенил)-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид. N-((1Z)-2-Амино-1,2-дициановинил)[(2,4-дифторфенил)амино]карбоксамид (500 мг, 1,9 ммоль) и 3-гидроксибензальдегид (510 мг, 4,18 ммоль) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение 18 ч. Полученную в результате неоднородную смесь фильтровали и 100 мг неочищенного продукта очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA). Летучие вещества удаляли при пониженном давлении, суспендированное твердое вещество обрабатывали гидроксидом аммония с помощью ультразвука и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (45 мг, 0,13 ммоль) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,91 (с, 1Н), 9,48 (с, 1Н), 8,42 (с, 1Н), 8,01 (с, 1Н), 7,91 (д, J=8,0, 1H), 7,80 (дд, J=8,4, 6,0, 1H), 7,16 (м, 1Н), 7,65 (м, 1Н), 7,38 (м, 1Н), 7,23 (т, J=8,0, 1H), 6,83 (дд, J=7,6, 1,2, 1H); МС (ESI) m/z 384,1 [M+1]+; т.пл. 364°С.
5.1.77 ПРИМЕР 77
СИНТЕЗ 9-(2-МЕТОКСИФЕНИЛ)-2-{3-[(МЕТИЛСУЛЬФОНИЛ)АМИНО]ФЕНИЛ}-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. Метил-3-[(метилсульфонил)амино]бензоат. Метил-3-аминобензоат растворяли в безводном тетрагидрофуране (50 мл) с последующим добавлением триэтиламина (2,8 мл, 19,8 ммоль) и метансульфонилхлорида (0,85 мл, 10,9 ммоль). Реакционную смесь перемешивали в течение ночи. К реакционной смеси добавляли воду (100 мл) и этилацетат (100 мл) и слои разделяли. Водный слой экстрагировали этилацетатом (2×50 мл), сушили сульфатом натрия и адсорбировали на силикагеле. С помощью флэш-хроматографии на силикагеле (40% EtOAc в гексанах) получали названное соединение (1,3 г, 5,6 ммоль, 57%) в виде белого твердого вещества. МС (ESI) m/z 230,1 [M+1]+.
В. [3-(Гидроксиметил)фенил](метилсульфонил)амин. Метил-3-[(метилсульфонил)амино]бензоат (1,2 г, 5,23 ммоль) растворяли в безводном тетрагидрофуране (40 мл) и охлаждали до -78°С. С помощью шприца добавляли раствор литийалюминийгидрида (2,0 М, 5,23 мл, 10,46 ммоль) и предоставляли возможность медленно нагреваться до комнатной температуры. Реакционную смесь гасили метанолом и неочищенный продукт адсорбировали на силикагеле. С помощью флэш-хроматографии (80% EtOAc в гексанах) получали названное соединение (0,85 г, 4,22 ммоль, 81%) в виде белого твердого вещества. МС (ESI) m/z 202,21 [M+1]+.
С. 3-[(Метилсульфонил)амино]бензальдегид. [3-(Гидроксиметил)фенил](метилсульфонил)амин (0,42 г, 2,08 ммоль) растворяли в дихлорметане (20 мл) с последующим добавлением пиридинийхлорхромата (0,67 г, 3,12 ммоль). Реакционную смесь перемешивали при комн. темп. в течение 1 ч. Фильтровали неочищенную реакционную смесь через слой силикагеля и промывали 60% EtOAc/гексан (250 мл), удаляли летучие вещества при пониженном давлении с получением названного соединения (0,40 г, 2,01 ммоль, 97%) в виде белого твердого вещества. МС (ESI) m/z 200,2 [M+1]+.
D. 9-(2-Метоксифенил)-2-{3-[(метилсульфонил)амино]фенил}-8-оксо-7-гидропурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (154 мг, 0,60 ммоль) и 3-[(метилсульфонил)амино]бензальдегид (260 мг, 1,31 ммоль) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение 16 ч. Полученную в результате неоднородную смесь фильтровали и очищали в применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1%TFA в Н2О + 0,1% TFA). Летучие вещества удаляли при пониженном давлении, суспендированное твердое вещество обрабатывали гидроксидом аммония с помощью ультразвука и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (151 мг, 0,33 ммоль, 55%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,80 (с, 1Н), 9,78 (с, 1Н), 8,32 (с, 1Н), 8,20 (д, J=8,0, 1H), 8,06 (м, 2Н), 7,55 (м, 1Н), 7,50 (дд, J=8,0, 4,0, 1H), 7,41 (м, 1Н), 7,31 (м, 2Н), 7,15 (м, 1Н), 3,73 (с, 3Н), 2,96 (с, 3Н); МС (ESI) m/z 455,1 [M+1]+; т.пл. 306°С.
5.1.78 ПРИМЕР 78
СИНТЕЗ 9-(4-ХЛОР-2-ФТОРФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. N-((Z)-2-Амино-1,2-дициановинил)[(4-хлор-2-фторфенил)амино]карбоксамид. 4-Хлор-2-фторбензолизоцианат (0,834 г, 4,86 ммоль) и 2,3-диаминомалеонитрил (0,500 г, 4,63 ммоль) подвергали взаимодействию в ацетонитриле согласно общему методу А. Вещество фильтровали и промывали ацетонитрилом. Полученное в результате твердое вещество сушили с получением названного соединения в виде светло-коричневого твердого вещества.
В. 9-(4-Хлор-2-фторфенил)-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид. N-((Z)-2-Амино-1,2-дициановинил)[(4-хлор-2-фторфенил)амино]карбоксамид (1,29 г, 4,63 ммоль), 3-гидроксибензальдегид (1,27 г, 10,42 ммоль) и триэтиламин (0,903 мл, 6,48 ммоль) в 10 мл МеОН подвергали взаимодействию согласно общему методу В. Полученную в результате неоднородную смесь фильтровали и промывали добавляемым ацетонитрилом. Продукт растворяли в ДМФА и вызывали осаждение водой. Осадок отфильтровывали с получением названного соединения в виде не совсем белого твердого вещества (0,400 г, 1,0 ммоль, выход 22% посредством 2 стадий). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,94 (с, 1Н), 9,48 (с, 1Н), 8,43 (с, 1Н), 8,02 (с, 1Н), 7,92 (дт, J=7,8, 1,2, 1H), 7,85 (дд, J=10,0, 2,1, 1H), 7,77 (т, J=8,4, 1H), 7,71-7,74 (м, 1Н), 7,56-7,62 (м, 1Н), 7,24 (т, J=7,8, 1H), 6,84 (дд, J=8,2, 1,6, 1H); МС (ESI) m/z 400,1 [M+1]+; т.пл. >350°С.
5.1.79 ПРИМЕР 79
СИНТЕЗ 9-(2-ХЛОРФЕНИЛ)-8-ОКСО-2-(3-ПИРИДИЛ)-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. N-((1Z)-2-Амино-1,2-дициановинил)[(2-хлорфенил)амино]карбоксамид. 2-Хлорбензолизоцианат (0,744 г, 4,85 ммоль) и 2,3-диаминомалеонитрил (0,500 г, 4,63 ммоль) подвергали взаимодействию в ацетонитриле согласно общему методу А. Вещество отфильтровывали и промывали ацетонитрилом. Полученное в результате твердое вещество сушили с получением названного соединения в виде светло-коричневого твердого вещества.
В. 9-(2-Хлорфенил)-8-оксо-2-(3-пиридил)-7-гидропурин-6-карбоксамид. N-((1Z)-2-Амино-1,2-дициановинил)[(5-хлор-2-фторфенил)амино]карбоксамид (1,21 г, 4,63 ммоль), пиридин-3-карбальдегид (1,16 г, 10,42 ммоль) и триэтиламин (0,903 мл, 6,48 ммоль) в 10 мл МеОН подвергали взаимодействию согласно общему методу В. Полученную в результате неоднородную смесь фильтровали и промывали добавляемым ацетонитрилом. Продукт очищали с применением препаративной ВЭЖХ с обращенной фазой (10-40% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Фракции, содержащие требуемое вещество, объединяли и концентрировали при пониженном давлении до минимального количества воды, добавляли EtOAc и органический слой промывали 5 раз NaHCO3 (насыщ.). Органический слой сушили и концентрировали с получением названного соединения в виде не совсем белого твердого вещества (0,018 г, 0,05 ммоль, выход 11% посредством 2 стадий). 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,02 (с, 1Н), 9,54 (с, 1Н), 8,58-8,70 (м, 1Н), 8,04 (с, 1Н), 7,77-7,82 (м, 1Н), 7,74 (дд, J=7,4, 2,0, 1H), 7,55-7,68 (м, 1Н), 7,47 (дд, J=8,4, 4,9, 1H), 5,76 (с, 1Н); МС (ESI) m/z 367,4 [M+1]+; т.пл. 296-298°С.
5.1.80 ПРИМЕР 80
СИНТЕЗ 8-ОКСО-2-(3-ПИРИДИЛ)-9-[2-(ТРИФТОРМЕТИЛ)ФЕНИЛ]-7-ГИДРОПУРИН-6-КАРБОКСАМИДА 3
А. N-((1Z)-2-Амино-1,2-дициановинил)[(2-трифторметилфенил)амино]карбоксамид. 2-Трифторметилбензолизоцианат (0,909 г, 4,85 ммоль) и 2,3-диаминомалеонитрил (0,500 г, 4,63 ммоль) подвергали взаимодействию в ацетонитриле согласно общему методу А. Вещество фильтровали и промывали ацетонитрилом. Полученное в результате твердое вещество сушили с получением названного соединения в виде светло-коричневого твердого вещества.
В. 8-Оксо-2-(3-пиридил)-9-[2-(трифторметил)фенил]-7-гидропурин-6-карбоксамид. N-((1Z)-2-Амино-1,2-дициановинил)[(2-трифторметилфенил)амино]карбоксамид (1,37 г, 4,63 ммоль), пиридин-3-карбальдегид (1,16 г, 10,42 ммоль) и триэтиламин (0,903 мл, 6,48 ммоль) в 10 мл МеОН подвергали взаимодействию согласно общему методу В. Полученную в результате смесь концентрировали. Продукт очищали с применением препаративной ВЭЖХ с обращенной фазой (20-60% ацетонитрил + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Фракции, содержащие требуемое вещество, объединяли и концентрировали при пониженном давлении до минимального количества воды. Добавляли EtOAc и органический слой промывали 5 раз NaHCO3 (насыщ.). Органический слой сушили и концентрировали с получением названного соединения в виде не совсем белого твердого вещества (0,072 г, 0,18 ммоль, выход 39% посредством 2 стадий). 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,01 (с, 1Н), 9,51 (д, J=2,0, 1H), 8,65 (c, 1H), 8,62 (дд, J=4,9, 1,8, 1H), 8,58 (дт, J=8,2, 2,0, 1H), 8,01-8,08 (м, 1Н), 7,97 (т, J=7,6, 1H), 7,81-7,89 (м, 1Н), 7,46 (дд, J=8,2, 4,7, 1H); МС (ESI) m/z 400,9 [M+1]+, т.пл. 300-302°С.
5.1.81 ПРИМЕР 81
СИНТЕЗ 9-(3-ХЛОР-2-ФТОРФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. N-((1Z)-2-Амино-1,2-дициановинил)[(3-хлор-2-фторфенил)амино]карбоксамид. 3-Хлор-2-фторбензолизоцианат (0,834 г, 4,86 ммоль) и 2,3-диаминомалеонитрил (0,500 г, 4,63 ммоль) подвергали взаимодействию в ацетонитриле согласно общему методу А. Вещество фильтровали и промывали ацетонитрилом. Полученное в результате твердое вещество сушили с получением названного соединения в виде светло-коричневого твердого вещества.
В. 9-(3-Хлор-2-фторфенил)-2-(3-гидроксифенил)-8-окси-7-гидропурин-6-карбоксамид. N-((1Z)-2-Амино-1,2-дициановинил)[(3-хлор-2-фторфенил)амино]карбоксамид (1,29 г, 4,63 ммоль), 3-гидроксибензальдегид (1,27 г, 10,42 ммоль) и триэтиламин (0,903 мл, 6,48 ммоль) в 10 мл МеОН подвергали взаимодействию согласно общему методу В. Полученную в результате неоднородную смесь фильтровали и промывали добавляемым ацетонитрилом. Продукт очищали с применением препаративной ВЭЖХ с обращенной фазой (30-70% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA посредством 2 стадий). Фракции, содержащие требуемое вещество, объединяли и концентрировали при пониженном давлении до минимального количества воды. Добавляли гидроксид аммония (2 мл) и полученную в результате взвесь обрабатывали ультразвуком и фильтровали. Твердое вещество сушили и концентрировали с получением названного соединения в виде не совсем белого твердого вещества (0,033 г, 0,08 ммоль, выход 18%, посредством 2 стадий). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,97 (с, 1Н), 9,49 (с, 1Н), 8,43 (с, 1Н), 8,02 (с, 1Н), 7,91 (д, J=7,8, 1H), 7,85 (т, J=7,6, 1H), 7,68-7,77 (м, 1Н), 7,51 (т, J=8,2, 1H), 7,24 (т, J=8,0, 1H), 6,84 (дд, J=8,0, 1,8, 1Н), 5,76 (с, 1Н); МС (ESI) m/z 400,1 [M+1]+; т.пл. >350°С.
5.1.82 ПРИМЕР 82
СИНТЕЗ 9-(2-ФТОР-3-ТРИФТОРМЕТИЛФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. N-((1Z)-2-Амино-1,2-дициановинил)[(2-фтор-3-трифторметилфенил)амино]карбоксамид. 2-Фтор-3-трифторметилбензолизоцианат (0,991 г, 4,86 ммоль) и 2,3-диаминомалеонитрил (0,500 г, 4,63 ммоль) подвергали взаимодействию в ацетонитриле согласно общему методу А. Вещество фильтровали и промывали ацетонитрилом. Полученное в результате твердое вещество сушили с получением названного соединения в виде светло-коричневого твердого вещества.
В. 9-(2-Фтор-3-трифторметилфенил)-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид. N-((1Z)-2-Амино-1,2-дициановинил)[(2-фтор-3-трифторметилфенил)амино]карбоксамид (1,45 г, 4,63 ммоль), 3-гидроксибензальдегид (1,27 г, 10,42 ммоль) и триэтиламин (0,903 мл, 6,48 ммоль) в 10 мл МеОН подвергали взаимодействию согласно общему методу В. Полученную в результате неоднородную смесь фильтровали и промывали добавляемым ацетонитрилом. Продукт очищали с применением препаративной ВЭЖХ с обращенной фазой (20-60% ацетонитрил + 0,1% TFA в Н2О + 0,1% TFA, в течение 35 мин). Фракции, содержащие требуемое вещество, объединяли и концентрировали при пониженном давлении до минимального количества воды. Добавляли гидроксид аммония и полученную в результате взвесь обрабатывали ультразвуком и фильтровали. Твердое вещество сушили и концентрировали с получением названного соединения в виде не совсем белого твердого вещества (0,053 г, 0,12 ммоль, выход 27% посредством 2 стадий). 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,00 (с, 1Н), 9,48 (с, 1Н), 8,44 (с, 1Н), 8,10 (т, J=7,2, 1H), 7,97-8,06 (м, 1Н), 7,90 (д, J=8,2, 1H), 7,60-7,77 (м, 1Н), 7,24 (т, J=7,8, 1H), 6,84 (д, J=7,8, 1H), 5,76 (с, 1Н); МС (ESI) m/z 434,3 [M+1]+; т.пл. >350°С.
5.1.83 ПРИМЕР 83
СИНТЕЗ 9-(2,3,4-ТРИФТОРФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. N-((1Z)-2-Амино-1,2-дициановинил)[(2,3,4-трифторфенил)амино]карбоксамид. 2,3,4-Трифторбензолизоцианат (0,836 г, 4,86 ммоль) и 2,3-диаминомалеонитрил (0,500 г, 4,63 ммоль) подвергали взаимодействию в ацетонитриле согласно общему методу А. Вещество фильтровали и промывали ацетонитрилом. Полученное в результате твердое вещество сушили с получением названного соединения в виде светло-коричневого твердого вещества.
В. 9-(2,3,4-Трифторфенил)-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид. N-((1Z)-2-Амино-1,2-дициановинил)[(2,3,4-трифторфенил)амино]карбоксамид (1,3 г, 4,63 ммоль), 3-гидроксибензальдегид (1,27 г, 10,42 ммоль) и триэтиламин (0,903 мл, 6,48 ммоль) в 10 мл МеОН подвергали взаимодействию согласно общему методу В. Полученную в результате неоднородную смесь фильтровали и промывали добавляемым ацетонитрилом с получением названного соединения в виде не совсем белого твердого вещества (0,030 г, 0,075 ммоль, выход 16%, посредством 2 стадий). 1ЯМР (400 МГц, ДМСО-d6) 11,98, (с, 1Н), 9,49 (с, 1Н), 8,44 (с, 1Н), 8,02 (с, 1Н), 7,92 (д, J=8,2, 1H), 7,70-7,79 (м, 1Н), 7,59-7,70 (м, 1Н), 7,25 (т, J=7,8, 1H), 1Н), 6,85 (дд, J=7,6, 2,1, 1H); МС (ESI) m/z 402,0 [M+1]+; т.пл. >350°С.
5.1.84 ПРИМЕР 84
СИНТЕЗ 2-(1Н-БЕНЗО[D]ИМИДАЗОЛ-6-ИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (1Н-Бензо[d]имидазол-6-ил)метанол. Бензимидазол-6-карбоновую кислоту (2,0 г, 12,3 ммоль) суспендировали в безводном тетрагидрофуране (50 мл) и охлаждали до -78°С. Добавляли через шприц раствор литийалюминийгидрида (2,0 М, 12,3 мл, 26,6 ммоль) и предоставляли возможность медленно нагреваться до комнатной температуры при перемешивании в течение ночи. Реакционную смесь гасили метанолом и неочищенный продукт адсорбировали на силикагеле. С помощью флэш-хроматографии (20% МеОН в EtOAc) получали названное соединение (1,42 г, 9,6 ммоль, выход 78%) в виде желтой пены. МС (ESI) m/z 149,1 [M+1]+.
В. 1Н-Бензо[d]имидазол-6-карбальдегид. (1Н-Бензо[d]имидазол-6-ил)метанол (1,0 г, 6,75 ммоль) растворяли в безводном диметилсульфоксиде (30 мл), добавляли через шприц сульфотриоксид-пиридиновый комплекс (3,21 г, 20,2 ммоль) и диизопропилэтиламин (3,5 мл, 20,2 ммоль). Реакционную смесь нагревали до 55°С в течение 3 дней. Реакционную смесь выливали в воду (100 мл), экстрагировали EtOAc (3×100 мл), сушили объединенные органические слои сульфатом натрия и адсорбировали на силикагеле. С помощью флэш-хроматографии (10% МеОН в EtOAc) получали названное соединение (0,11 г, 4,22 ммоль, 11%) в виде белого твердого вещества. МС (ESI) m/z 147,1 [M+1]+.
С. 2-(1Н-Бензо[d]имидазол-6-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (88 мг, 0,34 ммоль) и 1Н-бензо[d]имидазол-6-карбальдегид (110 мг, 0,75 ммоль) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение 16 ч. Полученную в результате неоднородную смесь фильтровали и очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA). Летучие вещества удаляли при пониженном давлении, суспендированное твердое вещество обрабатывали гидроксидом аммония с помощью ультразвука и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (120 мг, 0,30 ммоль, 88%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,69 (с, 1Н), 8,59 (с, 1Н), 8,55 (с, 1Н), 8,43 (с, 1Н), 8,37 (д, J=8,4, 1H), 7,98 (с, 1Н), 7,62 (д, J=8,8, 1H), 7,57 (ддд, J=9,2, 7,6, 1,6, 1H), 7,52 (дд, J=7,6, 1,6, 1H), 7,31 (дд, J=7,2, 1,2, 1H), 7,18 (ддд, J=8,8, 7,6, 1,2, 1H), 3,76 (c, 3H); МС (ESI) m/z 402,1 [M+1]+; т.пл. 306°С.
5.1.85 ПРИМЕР 85
СИНТЕЗ 2-[3-(АЦЕТИЛАМИНО)ФЕНИЛ]-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. 2-[3-(Ацетиламино)фенил]-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид. 2-(3-Аминофенил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид (100 мг, 0,27 ммоль) растворяли в безводном пиридине (4 мл) и охлаждали до 0°С. Добавляли через шприц уксусный ангидрид (0,10 мл, 0,29 ммоль) и предоставляли возможность реакционной смеси нагреваться до комнатной температуры, и перемешивали в течение ночи. При пониженном давлении удаляли растворитель. Остаток очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA). Летучие вещества удаляли при пониженном давлении, суспендированное твердое вещество обрабатывали гидроксидом аммония с помощью ультразвука и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (20 мг, 0,048 ммоль, 18%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,12 (c, 1H), 9,23 (с, 1Н), 8,21 (с, 1Н), 7,94 (д, J=8,0, 1H), 7,87 (д, J=8,0, 1H), 7,62 (с, 1Н), 7,52 (ддд, J=9,6, 8,8, 1,6, 1H), 7,34 (м, 3Н), 7,17 (ддд, J=8,8, 7,6, 1,2, 1H), 3,80 (с, 3Н), 2,11 (с, 3Н); МС (ESI) m/z 419,1 [M+1]+; т.пл. 265°С.
5.1.86 ПРИМЕР 86
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛ)-8-(2-МЕТОКСИФЕНИЛ)-6-ОКСО-5,6,7,8-ТЕТРАГИДРОПТЕРИДИН-4-КАРБОКСАМИДА
А. Этил-2-(2-метоксифениламино)ацетат. о-Анизидин (5,0 г, 40,60 ммоль) и карбонат калия (16,80 г, 121,80 ммоль) объединяли в диметилформамиде (120 мл) и предоставляли возможность перемешиваться при комнатной температуре. К раствору сразу добавляли этилбромацетат (6,78 г, 40,60 ммоль) в диметилформамиде (10 мл) и смесь нагревали до 55°С. Реакционную смесь подвергали мониторингу исчезновения исходных веществ методом тонкослойной хроматографии. Раствор фильтровали через целит и промывали этилацетатом. Фильтрат конденсировали при пониженном давлении и полученное в результате неочищенное масло очищали с применением хроматографии на силикагеле Biotage (0-40% этилацетата в гексанах) с получением названного соединения (6,3 г, 74%). МС (ESI) m/z 210,1 [M+1]+.
В. Этил-2-хлор-6-((2-этокси-2-оксоэтил)(2-метоксифенил)амино)-5-нитро-5,6-дигидропиримидин-4-карбоксилат. Этил-2,6-дихлор-5-нитропиримидин-4-карбоксилат (3,0 г, 11,27 ммоль), этил-2-(2-метоксифениламино)ацетат (2,35 г, 11,27 ммоль) и диизопропилэтиламин (4,36 г, 33,81 ммоль) подвергали взаимодействию согласно общему методу С и распределяли между водой и этилацетатом (3х) с получением названного соединения без очистки (2,05 г, 41%). МС (ESI) m/z 439,2 [M+1]+, 441,4 [M+2]+.
С. Этил-2-хлор-8-(2-метоксифенил)-6-оксо-4а,5,6,7,8,8а-гексагидроптеридин-4-карбоксилат. Этил-2-хлор-6-((2-этокси-2-оксоэтил)(2-метоксифенил)амино)-5-нитро-5,6-дигидропиримидин-4-карбоксилат (2,07 г, 4,72 ммоль), порошок железа (5,27 г, 94,4 ммоль) и уксусную кислоту объединяли и нагревали до 60°С. Реакционную смесь подвергали мониторингу расхода исходного вещества и образования продукта путем тонкослойной хроматографии. Через один час раствор конденсировали при пониженном давлении, разбавляли метанолом и фильтровали через целит. Фильтрат конденсировали при пониженном давлении и полученное в результате масло очищали хроматографией на силикагеле Biotage (5-75% этилацетата в гексанах) с получением названного соединения (1,17 г, 69%). 1Н ЯМР (300 МГц, ДМСО-d6), δ 10,28 (ушир.с, 1Н), 7,39 (м, 2Н), 7,20 (д, J=8, 1H), 7,06 (т, J=7,5, 1H), 4,1 (ушир.с, 1Н), 4,55 (ушир.с, 1Н), 4,37 (кв, J=7,2, 2H), 3,79 (с, 3Н), 1,33 (т, J=6,9, 3H); МС (ESI) m/z 363,4 [M+1]+.
D. Этил-2-(3-гидроксифенил)-8-(2-метоксифенил)-6-оксо-5,6,7,8-тетрагидроптеридин-4-карбоксилат. Этил-2-хлор-8-(2-метоксифенил)-6-оксо-4а,5,6,7,8,8а-гексагидроптеридин-4-карбоксилат (0,500 г, 1,38 ммоль), аддукт дихлор[1,1'-бис(дифенилфосфино)ферроцен]палладия(II) с дихлорметаном (0,112 г, 0,138 ммоль), фосфат калия (0,877 г, 4,19 ммоль) и тетрагидрофуран (15 мл) объединяли и нагревали вместе в микроволновом реакторе Biotage Emrys Optimizer при 120°С в течение 45 мин. Раствор конденсировали при пониженном давлении и распределяли между водным карбонатом калия и этилацетатом (3х), фильтровали и растворитель удаляли с получением неочищенного названного соединения. Неочищенный продукт очищали с применением препаративной ВЭЖХ с обращенной фазой (30-70% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Фракции, содержащие очищенный продукт, пропускали через экстракционную колонку с твердой фазой Phenomenex Strata-X-C для удаления TFA. Продукт выделяли из колонки с применением 2М аммиака в метаноле. Раствор концентрировали при пониженном давлении и сушили в вакууме с получением названного соединения (0,250 г, 43%). МС (ESI) m/z 421,2 [M+1]+.
Е. 2-(3-Гидроксифенил)-8-(2-метоксифенил)-6-оксо-5,6,7,8-тетрагидроптеридин-4-карбоксамид. Этил-2-(3-гидроксифенил)-8-(2-метоксифенил)-6-оксо-5,6,7,8-тетрагидроптеридин-4-карбоксилат (0,250 г, 0,59 ммоль) и метанол (10 мл) подвергали взаимодействию согласно общему методу G и очищали с применением препаративной ВЭЖХ с обращенной фазой (30-70% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Фракции, которые содержали продукт, очищенный методом ВЭЖХ, объединяли и конденсировали при пониженном давлении. Взвесь разбавляли концентрированным гидроксидом аммония (1 мл) для нейтрализации трифторуксусной кислоты. Полученный в результате осадок отфильтровывали, промывали водой и сушили в вакуумном сушильном шкафу с получением названного соединения (0,026 г, 11%). 1H ЯМР (400 МГц, ДМСО-d6) δ 11,24 (c, 1H), 9,39 (c, 1H), 8,57 (c, 1H), 8,18 (c, 1H), 7,61 (д, J=7,99, 1H), 7,43 (м, 3H), 7,22 (д, J=7,59, 1H), 7,09 (м, 2H), 6,79 (д, J=7,59, 1H), 4,5 (ушир.с, 2H), 3,76 (с, 3H); MC (ESI) m/z 392 A [M+l]+; т.пл. 336-338°C.
5.1.87 ПРИМЕР 87
СИНТЕЗ 9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-2-ПИРАЗОЛ-4-ИЛ-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. 9-(2-Метоксифенил)-8-оксо-2-пиразол-4-ил-7-гидропурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (540 мг, 2,10 ммоль) и пиразол-4-карбальдегид (400 мг, 4,16 ммоль) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение 21 ч. Полученную в результате неоднородную смесь фильтровали и очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA). Летучие вещества удаляли при пониженном давлении, суспендированное твердое вещество обрабатывали гидроксидом аммония с применением ультразвука и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (420 мг, 1,19 ммоль, 57%) в виде белого твердого вещества. 1ЯМР (400 МГц, ДМСО-d6) δ 13,04 (с, 1Н), 11,58 (с, 1Н), 8,43 (с, 1Н), 8,34 (с, 1Н), 8,03 (д, J=1,6, 1H), 7,94 (c, 1H), 7,53 (ддд, J=8,4, 8,0, 2,0, 1H), 7,45 (дд, J=7,6, 1,6, 1H), 7,27 (дд, J=8,8, 1,2, 1H), 7,12 (ддд, J=8,8, 7,6, 1,2, 1H), 3,78 (c, 3H); МС (ESI) m/z 352,0 [M+1]+; т.пл. 306°С.
5.1.88 ПРИМЕР 88
СИНТЕЗ 9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-2-ПИРАЗОЛ-3-ИЛ-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. 9-(2-Метоксифенил)-8-оксо-2-пиразол-3-ил-7-гидропурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (540 ммоль, 2,10 ммоль) и пиразол-3-карбальдегид (400 мг, 4,16 ммоль) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение 21 ч. Полученную в результате неоднородную смесь фильтровали и очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA). Летучие вещества удаляли при пониженном давлении, суспендированное твердое вещество обрабатывали гидроксидом аммония с применением ультразвука и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (385 мг, 1,09 ммоль, 52%) в виде белого твердого вещества. 1 ЯМР (400 МГц, ДМСО-d6) δ 13,65 (с, 1Н), 11,85 (с, 1Н), 8,77 (с, 1Н), 8,00 (с, 1Н), 7,45 (м, 3Н), 7,28 (дд, J=8,4, 1,0, 1Н), 7,15 (ддд, J=8,8, 8,0, 1,0, 1H), 3,74 (с, 3Н); МС (ESI) m/z 352,0 [M+1]+; т.пл. 306°С.
5.1.89 ПРИМЕР 89
СИНТЕЗ 9-(4-АМИНОЦИКЛОГЕКСИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. Этил-6-({4-[(трет-бутокси)карбониламино]циклогексил}амино)-2-хлор-5-нитропиримидин-4-карбоксилат. Этил-2,6-дихлор-5-нитропиримидин-4-карбоксилат-2 (1,5 г, 5,64 ммоль), N-(4-аминоциклогексил)(трет-бутокси)карбоксамид (1,09 г, 5,08 ммоль) и диизопропилэтиламин (0,982 мл, 5,64 ммоль) подвергали взаимодействию в тетрагидрофуране (40 мл) согласно общему методу С. Реакционную смесь концентрировали и полученное в результате масло непосредственно применяли без дополнительной очистки. МС (ESI) m/z 444,4 [M+1]+.
В. Этил-5-амино-6-({4-[(трет-бутокси)карбониламино]циклогексил}амино)-2-хлорпиримидин-4-карбоксилат. Этил-6-({4-[(трет-бутокси)карбониламино]циклогексил}амино)-2-хлор-5-нитропиримидин-4-карбоксилат (2,51 г, 5,64 ммоль) и дигидрат хлорида олова(II) (3,82 г, 16,92 ммоль) в этаноле (35 мл) и ДМФА (10 мл) подвергали взаимодействию согласно общему методу D. Полученную в результате неоднородную смесь фильтровали и концентрировали. Продукт очищали хроматографией на силикагеле Biotage (0-60% этилацетата в гексанах) с получением названного соединения (1,08 г, 2,60 ммоль, выход 46% посредством 2 стадий). МС (ESI) m/z 414,4 [M+1]+.
С. Этил-5-амино-6-({4-[(трет-бутокси)карбониламино]циклогексил}амино)-2-(3-гидроксифенил)пиримидин-4-карбоксилат. Этил-5-амино-6-({4-[(трет-бутокси)карбониламино]циклогексил}амино)-2-хлорпиримидин-4-карбоксилат (0,480 г, 1,16 ммоль), 3-гидроксифенилбороновую кислоту (0,239 г, 1,73 ммоль), фосфат калия (0,499 г, 2,32 ммоль), ацетат палладия(II) (0,039 г, 0,174 ммоль) и дициклогексил(2',6'-диметоксибифенил-2-ил)фосфин (0,071 г, 0,174 ммоль) растворяли в тетрагидрофуране (12 мл) и воде (1,2 мл) и подвергали взаимодействию согласно общему методу Е. Полученную в результате реакционную смесь концентрировали. Продукт очищали хроматографией на силикагеле Biotage (0-70% этилацетата в гексанах) с получением названного соединения в виде не совсем белого твердого вещества (0,100 г, 0,212 ммоль, выход 18%). МС (ESI) m/z 472,5 [M+1]+.
D. Этил-5-амино-6-({4-[(трет-бутокси)карбониламино]циклогексил}амино)-2-(3-гидроксифенил)пиримидин-4-карбоксилат. Этил-5-амино-6-({4-[(трет-бутокси)карбониламино]циклогексил}амино)-2-(3-гидроксифенил)пиримидин-4-карбоксилат (0,100 г, 0,212 ммоль) и 1,1'-карбонилдиимидазол (0,344 г, 2,12 ммоль) растворяли в тетрагидрофуране (8 мл) и подвергали взаимодействию согласно общему методу F. Полученную в результате реакционную смесь концентрировали. Продукт очищали хроматографией на силикагеле Biotage (0-60% этилацетата в гексанах) с получением названного соединения в виде не совсем белого твердого вещества (0,100 г, 0,202 ммоль, выход 95%). МС (ESI) m/z 498,5 [M+1]+.
Е. 9-{4-[(трет-Бутокси)карбониламино]циклогексил}-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид. Этил-5-амино-6-({4-[(трет-бутокси)карбониламино]циклогексил}амино)-2-(3-гидроксифенил)пиримидин-4-карбоксилат (0,100 г, 0,201 ммоль) растворяли в МеОН (20 мл) и подвергали взаимодействию согласно общему методу G. Полученную в результате реакционную смесь концентрировали и непосредственно применяли на следующей стадии без дополнительной очистки или характеристики. МС (ESI) m/z 469,4 [M+1]+.
F. 9-(4-Аминоциклогексил)-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид. 9-{4-[(трет-Бутокси)карбониламино]циклогексил}-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид (0,100 г, 0,212 ммоль) растворяли в CH2Cl2 (10 мл) и добавляли TFA (1 мл). Реакционную смесь перемешивали в течение 4 ч и концентрировали. Остаток растворяли в CH2CN и воде и полученный в результате осадок отфильтровывали. Продукт пропускали через ионообменную колонку Strata-XC с водой, метанолом и 5% гидроксидом аммония в метаноле. Продукт элюировали 15-20% гидроксидом аммония в воде, и фракции концентрировали с получением названного соединения в виде белого порошка (0,032 г, 0,087 ммоль, выход 41% посредством 2 стадий). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,54 (с, 1Н), 7,94 (д, J=7,8, 1H), 7,87 (дд, J=2,3, 1,6, 1Н), 7,78 (с, 1Н), 7,27 (т, J=7,8, 1H), 6,83 (дд, J=7,6, 2,1, 1H), 4,14-4,32 (м, 1Н), 2,81-2,89 (м, 1Н), 2,52-2,56 (м, 1Н), 2,41-2,47 (м, 1Н), 1,97 (д, J=12,1, 1H), 1,75 (д, J=10,9, 1H), 1,22-1,38 (м, 1Н); МС (ESI) m/z 369,5 [M+1]+; т.пл. 318-320°С.
5.1.90 ПРИМЕР 90
СИНТЕЗ 2-[3-(ДИФТОРМЕТИЛ)ФЕНИЛ]-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. 3-(Дифторметил)бензальдегид. 3-(Дифторметил)-1-бромбензол (1,0 г, 4,83 ммоль) растворяли в безводном тетрагидрофуране (20 мл) и охлаждали до -78°С. Добавляли н-бутиллитий (1,6М, 3,2 мл, 5,07 ммоль) и реакционную смесь перемешивали в течение 30 мин. Добавляли диметилформамид (1 мл) и раствору предоставляли возможность нагреваться до комнатной температуры. Реакционную смесь гасили насыщенным раствором бикарбоната натрия, экстрагировали простым диэтиловым эфиром (2×75 мл) и сушили сульфатом натрия. Очисткой флэш-хроматографией (20% EtOAc в гекс.) получали желтое масло (560 мг, 3,58 ммоль, 74%). МС (ESI) m/z 157,1 [M+1]+.
В. 2-[3-(Дифторметил)фенил]-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (560 мг, 2,18 ммоль), 3-(дифторметил)бензальдегид (750 мг, 4,80 ммоль), триэтиламин (0,14 мл, 3,27 ммоль) и метанол (30 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали и очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA). Летучие вещества удаляли при пониженном давлении, суспендированное твердое вещество обрабатывали гидроксидом аммония с применением ультразвука и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (45 мг, 0,11 ммоль, 5%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,82 (с, 1Н), 8,28 (дд, J=7,2, 1,0, 1H), 8,21 (с, 1Н), 8,06 (с, 1Н), 7,70 (м, 1Н), 7,53 (ддд, J=8,4, 7,6, 1,6, 1H), 7,47 (дд, J=8,0, 2,0, 1Н), 7,42 (дд, J=10,8, 8,8, 1H), 7,27 (дд, J=8,4, 1,2, 1H), 7,12 (ддд, J=8,8, 7,6, 1H), 7,07 (т, J=55,0, 1H), 3,78 (с, 3Н); МС (ESI) m/z 412,0 [M+1]+; т.пл. 270°С.
5.1.91 ПРИМЕР 91
СИНТЕЗ 2-[5-(ДИФТОРМЕТИЛ)-2-ФТОРФЕНИЛ]-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. 5-(Дифторметил)-2-фторбензальдегид. 1-(Дифторметил)-4-фторбензол (1,0 г, 8,84 ммоль) растворяли в безводном тетрагидрофуране (20 мл) и охлаждали до -78°С. Добавляли через шприц н-бутиллитий (1,6 М, 4,5 мл, 7,18 ммоль) и реакционную смесь перемешивали в течение 30 минут. Добавляли диметилформамид (1 мл) и раствору предоставляли возможность нагреваться до комнатной температуры. Реакционную смесь гасили насыщенным раствором бикарбоната натрия, экстрагировали простым диэтиловым эфиром (2×75 мл) и сушили сульфатом натрия. Очисткой флэш-хроматографией (10% EtOAc в гекс.) получали продукт в виде желтого масла (540 мг, 3,08 ммоль, 45%). МС (ESI) m/z 175,0 [M+1]+.
В. 2-[5-(Дифторметил)-2-фторфенил]-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (510 мг, 2,00 ммоль) и 5-(дифторметил)-2-фторбензальдегид (680 мг, 4,38 ммоль) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение 16 ч. Полученную в результате неоднородную смесь фильтровали и очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA). Летучие вещества удаляли при пониженном давлении, суспендированное твердое вещество обрабатывали гидроксидом аммония с применением ультразвука и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (80 мг, 0,19 ммоль, 10%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,85 (с, 1Н), 8,28 (дд, J=7,2, 1,0, 1H), 8,23 (с, 1Н), 8,06 (с, 1Н), 7,70 (м, 1Н), 7,53 (ддд, J=8,4, 7,6, 1,6, 1Н), 7,47 (дд, J=8,0, 2,0, 1H), 7,42 (дд, J=10,8, 8,8, 1H), 7,27 (дд, J=8,4, 1,2, 1Н), 7,12 (ддд, J=8,8, 7,6, 1,2, 1H), 7,07 (т, J=55,0, 1Н), 3,78 (с, 3Н); МС (ESI) m/z 430,0 [M+1]+; т.пл. 225°С.
5.1.92 ПРИМЕР 92
СИНТЕЗ 2-(1Н-БЕНЗО[D]ИМИДАЗОЛ-4-ИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИД
А. 1Н-Бензо[d]имидазол-4-карбоновая кислота. 2,3-Диаминобензойную кислоту (1,0 г, 6,57 ммоль) суспендировали в триэтилортоформиате (20 мл) и нагревали до 130°С в течение ночи. Затем добавляли простой диэтиловый эфир (100 мл) и полученный в результате осадок отфильтровывали с получением белого твердого вещества (1,04 г, 6,41 ммоль, 98%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,22 (с, 1Н), 7,91 (дд, J=8,0, 1,2, 1H), 7,82 (дд, J=8,0, 1,2, 1H), 7,29 (дд, J=8,0, 7,6, 1H); МС (ESI) m/z 163,0 [M+1]+.
В. (1Н-Бензо[d]имидазол-4-ил)метанол. 1Н-Бензо[d]имидазол-4-карбоновую кислоту (1,04 г, 6,41 ммоль) суспендировали в безводном тетрагидрофуране (80 мл) и охлаждали до -78°С. Добавляли через шприц раствор литийалюминийгидрида в тетрагидрофуране (2,0 М, 6,4 мл). Реакционной смеси предоставляли возможность нагреваться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь гасили метанолом и адсорбировали на силикагеле. Флэш-хроматографией (20% МеОН в EtOAc) получали белое твердое вещество (550 мг, 3,72 ммоль, 58%). МС (ESI) m/z 149,0 [M+1]+.
С. 1Н-Бензо[d]имидазол-4-карбальдегид. (1Н-Бензо[d]имидазол-4-ил)метанол (550 мг, 3,72 ммоль) растворяли в диметилсульфоксиде (30 мл). Добавляли диизопропилэтиламин (1,9 мл, 11,1 ммоль) и сульфотриоксидный комплекс пиридина (1,77 г, 11,1 ммоль) и раствору предоставляли возможность перемешиваться в течение ночи при комнатной температуре. Реакционную смесь выливали в воду (50 мл) и экстрагировали этилацетатом (3×150 мл), сушили сульфатом натрия и концентрировали при пониженном давлении с получением белого твердого вещества (100 мг, 18%). МС (ESI) m/z 147,0 [M+1]+.
D. 2-(1Н-Бензо[d]имидазол-4-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (80 мг, 0,31 ммоль), 1Н-бензо[d]имидазол-4-карбальдегид (80 мг, 0,54 ммоль), триэтиламин (0,06 мл, 0,41 ммоль) и метанол (3 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали и промывали простым диэтиловым эфиром. Твердое вещество сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (30 мг, 0,075 ммоль, 24%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,91 (с, 1Н), 11,75 (с, 1Н), 8,57 (с, 1Н), 8,32 (с, 1Н), 8,13 (с, 1Н), 8,09 (д, J=7,6, 1H), 7,77 (д, J=7,6, 1H), 7,56 (ддд, J=10,4, 9,8, 2,0, 1H), 7,54 (м, 1Н), 7,32 (дд, J=2,0, 7,6, 1H), 7,26 (т, J=7,6, 1H), 7,18 (ддд, J=8,8, 7,6, 1,2, 1H), 3,76 (с, 3Н); МС (ESI) m/z 402,1 [M+1]+; т.пл. 312°С.
5.1.93 ПРИМЕР 93
СИНТЕЗ 2-(6-ГИДРОКСИПИРИДИН-3-ИЛ)-8-ОКСО-9-(2-(ТРИФТОРМЕТИЛ)ФЕНИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(6-Гидроксипиридин-3-ил)-8-оксо-9-(2-(трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-(трифторметилфенил)мочевину (см. пример 50.А) (0,15 г, 0,51 ммоль), 6-гидроксиникотинальдегид (0,13 г, 1,0 ммоль) и триэтиламин (0,10 мл, 0,72 ммоль) в метаноле (7,0 мл) подвергали взаимодействию согласно общему методу В. Полученное в результате вещество осаждали из смеси ДМФА/вода и сушили помещением в вакуумируемое устройство с получением продукта в виде не совсем белого твердого вещества (0,105 г, 0,25 ммоль, выход 49%). 1ЯМР (400 МГц, ДМСО-d6) δ 11,83 (перекрывающийся ушир.с, 2Н), 8,66 (м, 1Н), 8,30 (2, 2Н), 8,02-7,92 (перекрывающийся м, 3Н), 7,85-7,78 (перекрывающийся м, 2Н), 6,39 (д, J=9,9, 2H), МС (ESI) m/z 417,0 [M+1]+; т.пл. 348-352°С (разл.).
5.1. ПРИМЕР 94
СИНТЕЗ 2-(1Н-БЕНЗО[D]ИМИДАЗОЛ-6-ИЛ)-9-(2-ФТОРФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(1Н-бензо[d]имидазол-6-ил)-9-(2-фторфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. N-((1Z)-2-Амино-1,2-дициановинил)[(2-фторфенил)амино]карбоксамид (см. пример 9.А) (67 мг, 0,27 ммоль), 1Н-бензо[d]имидазол-6-карбальдегид (см. пример 84.В) (80 мг, 0,54 ммоль), триэтиламин (0,06 мл, 0,41 ммоль) и метанол (3 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали и очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA). Летучие вещества удаляли при пониженном давлении, суспендированное твердое вещество обрабатывали гидроксидом аммония с применением ультразвука и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу с получением названного соединения (75 мг, 0,20 ммоль, 74%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,53 (с, 1Н), 11,84 (с, 1Н), 8,37 (м, 1Н), 8,27 (с, 1Н), 8,00 (д, J=9,6, 1H), 7,74 (ддд, J=9,2, 8,0, 1,6, 1H), 7,65 (м, 1Н), 7,57 (ддд, J=10,0, 8,4, 1,6, 1H), 7,48 (ддд, J=9,2, 8,0, 1,6, 1H); МС (ESI) m/z 390,1 [M+1]+; т.пл. 278°С.
5.1.95 ПРИМЕР 95
СИНТЕЗ 2-БЕНЗИМИДАЗОЛ-6-ИЛ-8-ОКСО-9-[2-(ТРИФТОРМЕТИЛ)ФЕНИЛ]-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. 2-Бензимидазол-6-ил-8-оксо-9-[2-(трифторметил)фенил]-7-гидропурин-6-карбоксамид. N-((1Z)-2-Амино-1,2-дициановинил){[2-(трифторметил)фенил]амино}карбоксамид (150 мг, 0,51 ммоль), бензимидазол-6-карбальдегид (150 мг, 0,1.02 ммоль), триэтиламин (0,11 мл, 0,77 ммоль) и метанол (8 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали и очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA). Летучие вещества удаляли при пониженном давлении, суспендированное твердое вещество обрабатывали гидроксидом аммония с применением ультразвука и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (31 мг, 0,071 ммоль, 14%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,53 (с, 1Н), 12,46 (с, 1Н), 8,71 (с, 1Н), 8,61 (с, 1Н), 8,54 (с, 1Н), 8,38 (с, 1Н), 8,32 (дд, J=1,6, 8,4, 1H), 8,25 (д, J=3,2, 1H), 7,99 (м, 1Н), 7,86 (м, 1Н), 7,65 (д, J=8,4, 1H), 7,51 (д, J=8,4, 1H); МС (ESI) m/z 440,1 [M+1]+; т.пл. 258°С.
5.1.96 ПРИМЕР 96
СИНТЕЗ 2-(5-ХЛОРПИРИДИН-3-ИЛ)-8-ОКСО-9-(2-(ТРИФТОРМЕТИЛ)ФЕНИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(5-Хлорпиридин-3-ил)-8-оксо-9-(2-трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-(трифторметил)фенил)мочевину (см. пример 50.А) (0,15 г, 0,51 ммоль), 5-хлорникотинальдегид (0,14 г, 0,99 ммоль) и триэтиламин (0,10 мл, 0,72 ммоль) объединяли в метаноле (7,0 мл) и перемешивали при комнатной температуре в течение ночи. Избыток растворителя удаляли при пониженном давлении и полученный в результате остаток очищали препаративной ВЭЖХ с обращенной фазой (30-80% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Чистые фракции нейтрализовали гидроксидом аммония, и растворитель удаляли при пониженном давлении. Полученное в результате вещество помещали в этилацетат, промывали последовательно карбонатом калия, водой и насыщенным раствором соли. Раствор сушили над сульфатом натрия, фильтровали, и растворитель удаляли при пониженном давлении с получением продукта в виде не совсем белого твердого вещества (0,035 г, 0,08 ммоль, выход 16%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,07 (ушир.с, 1Н), 9,23 (с, 1Н), 8,95 (с, 1Н), 8,59 (м, 2Н), 7,93 (м, 2Н), 7,78 (м, 2Н), 7,63 (ушир.с, 1Н); МС (ESI) m/z 435,0 [M+1]+; т.пл. 230-232°С.
5.1.97 ПРИМЕР 97
СИНТЕЗ ТРАНС-4-(6-КАРБАМОИЛ-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-2-ИЛАМИНО)ЦИКЛОГЕКСИЛКАРБАМАТА
А. Этил-2-(транс-4-гидроксициклогексиламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат. Этил-2-хлор-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (см. пример 30.А) (0,300 г, 0,852 ммоль), транс-4-аминоциклогексанол (0,117 г, 1,022 ммоль) и диизопропилэтиламин подвергали взаимодействию согласно общему методу С, за исключением того, что при комнатной температуре и в диметилформамиде (5 мл). Неочищенную реакционную смесь конденсировали и очищали с применением хроматографии (Biotage) (0-100% этилацетата в гексанах) с получением названного соединения (0,303 г, 82%). МС (ESI) m/z 432,5 [M+1]+.
В. Этил-5-амино-2-(транс-4-гидроксициклогексиламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. Этил-2-(транс-4-гидроксициклогексиламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (0,300 г, 0,852 ммоль) растворяли в этаноле (20 мл) и добавляли в колбу 10% палладия на угле (0,073 г) и продували свежими порциями газообразного водорода и предоставляли возможность перемешиваться при комнатной температуре. Через 16 ч реакционную смесь фильтровали через целит и фильтрат конденсировали при пониженном давлении. Неочищенное масло очищали с применением хроматографии (Biotage) (5% метанола в этилацетате) с получением названного соединения (0,240 г, 70%). МС (ESI) m/z 402,4 [M+1]+.
С. Этил-2-(транс-4-(1Н-имидазол-1-карбонилокси)циклогексиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат. Этил-5-амино-2-(транс-4-гидроксициклогексиламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (0,240 г, 0,598 ммоль) и 1,1'-1,1'-карбонилдиимидазол (0,968 г, 5,98 ммоль) в дихлорметане (20 мл) подвергали взаимодействию согласно общему методу F и очищали с применением хроматографии (Biotage) (40-100% этилацетата в гексанах) с получением смеси названного продукта и отщепившегося продукта со свободным гидроксилом. МС (ESI) m/z 522,5 [M+1]+.
D. транс-4-(6-Карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-иламино)циклогексилкарбамат. Этил-2-(транс-4-(1Н-имидазол-1-карбонилокси)циклогексиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат (0,250 г, 0,479 ммоль) и газообразный аммиак подвергали взаимодействию в метаноле (10 мл) согласно общему методу G и очищали с применением препаративной ВЭЖХ с обращенной фазой (10-80% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин) с получением названного соединения (0,135 г, 25%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,899 (с, 1Н), 7,984 (с, 1Н), 7,784 (с, 1Н), 7,47 (т, J=7,19, 1H), 7,36 (д, J=7,99, 1H), 7,19 (д, J=7,59, 1H), 7,06 (т, J=6,39, 1H), 6,78 (д, J=6,79, 1H), 4,37 (с, 1Н), 1,92 (с, 4Н), 1,42 (м, 2Н), 1,22 (м, 2Н); МС (ESI) m/z 442,4 [M+1]+; т.пл. 187-189°С.
5.1.98 ПРИМЕР 98
СИНТЕЗ (R)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-2-(ПИРРОЛИДИН-3-ИЛАМИНО)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (R)-Этил-2-(1-(трет-бутоксикарбонил)пирролидин-3-иламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат. Этил-2-хлор-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (см. пример 30.А) (0,300 г, 0,852 ммоль), (R)1-boc-3-аминопирролидин (0,190 г, 1,022 ммоль) и диизопропилэтиламин (0,164 г, 1,27 ммоль) подвергали взаимодействию согласно общему методу С, за исключением того, что при комнатной температуре и в диметилформамиде (5 мл). Неочищенную реакционную смесь конденсировали и очищали с применением хроматографии (Biotage) (0-100% этилацетата в гексанах) с получением названного соединения (0,403 г, 94%). МС (ESI) m/z 503 [M+1]+.
В. (R)-Этил-5-амино-2-(1-трет-бутоксикарбонил)пирролидин-3-иламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. (R)-Этил-2-(1-(трет-бутоксикарбонил)пирролидин-3-иламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (0,403 г, 0,802 ммоль) растворяли в этаноле (20 мл) и в колбу добавляли 10% палладия на угле (0,080 г) и продували свежими порциями газообразного водорода, и предоставляли возможность перемешиваться при комнатной температуре. Через 16 часов реакционную смесь фильтровали через целит и фильтрат конденсировали при пониженном давлении. Неочищенное масло очищали с применением хроматографии (Biotage) (0-100% этилацетата в гексанах) с получением названного соединения (0,340 г, 89%). МС (ESI) m/z 473,5 [M+1]+.
С.(R)-Этил-2-(1-(трет-бутоксикарбонил)пирролидин-3-иламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат. (R)-Этил-5-амино-2-(1-трет-бутоксикарбонил)пирролидин-3-иламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (0,34 г, 0,720 ммоль) и карбонилдиимидазол (1,16 г, 7,20 ммоль) в дихлорметане (20 мл) подвергали взаимодействию согласно общему методу F и очищали с применением хроматографии (Biotage) (0-100% этилацетата в гексанах) с получением названного соединения (0,333 г, 93%) в виде смеси. МС (ESI) m/z 499,5 [M+1]+.
D. (R)-Этил-9-(2-метоксифенил)-8-оксо-2-(пирролидин-3-иламино)-8,9-дигидро-7Н-пурин-6-карбоксилат. (R)-Этил-2-(1-(трет-бутоксикарбонил)пирролидин-3-иламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат (0,333 г, 0,668 ммоль) растворяли в дихлорметане (3 мл) и трифторуксусной кислоте (1 мл). Раствор перемешивали в течение двух часов и конденсировали при пониженном давлении с получением неочищенного названного соединения (0,300 г, 100%). МС (ESI) m/z 399,3 [M+1]+.
Е. (R)-9-(2-Метоксифенил)-8-оксо-2-(пирролидин-3-иламино)-8,9-дигидро-7Н-пурин-6-карбоксамид. (R)-Этил-9-(2-метоксифенил)-8-оксо-2-(пирролидин-3-иламино)-8,9-дигидро-7Н-пурин-6-карбоксилат (0,300 г) и метанол, насыщенный газообразным аммиаком, (10 мл) подвергали взаимодействию согласно общему методу G и очищали препаративной ВЭЖХ с обращенной фазой (5-60% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин) с получением названного соединения (0,076 г, 30%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,91 (с, 1Н), 7,781 (с, 1Н), 7,466 (м, 1Н), 7,34 (д, J=7,59, 1H), 7,19 (д, J=7,99, 1H), 7,06 (т, J=7,19, 1H), 6,96 (д, J=6,39, 1H), 4,24 (с, 1Н), 3,72 (с, 3Н), 2,94 (м, 2Н), 2,72 (м, 1Н), 2,64 (м, 1Н), 1,95 (м, 1Н), 1,58 (м, 1Н); МС (ESI) m/z 370,2 [M+1]+; т.пл. >400°С.
5.1.99 ПРИМЕР 99
СИНТЕЗ (S)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-2-(ПИРРОЛИДИН-3-ИЛАМИНО)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (S)-Этил-2-(1-(трет-бутоксикарбонил)пирролидин-3-иламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат. Этил-2-хлор-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (см. пример 30.А) (0,300 г, 0,852 ммоль), (S)-1-boc-3-аминопирролидин (0,190 г, 1,022 ммоль) и диизопропилэтиламин (0,164 г, 1,27 ммоль) подвергали взаимодействию согласно общему методу С, за исключением того, что при комнатной температуре и в диметилформамиде (5 мл). Неочищенную реакционную смесь конденсировали и очищали с применением хроматографии (Biotage) (0-100% этилацетата в гексанах) с получением названного соединения (0,424 г, 99%). МС (ESI) m/z 503 [M+1]+.
В. (S)-Этил-5-амино-2-(1-(трет-бутоксикарбонил)пирролидин-3-иламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. (S)-Этил-2-(1-(трет-бутоксикарбонил)пирролидин-3-иламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (0,424 г, 0,844 ммоль) растворяли в этаноле (20 мл) и в колбу добавляли 10% палладия на угле (0,085 г) и продували свежими порциями газообразного водорода, и предоставляли возможность перемешиваться при комнатной температуре. Через 16 ч реакционную смесь фильтровали через целит и фильтрат конденсировали при пониженном давлении. Неочищенное масло очищали с применением хроматографии (Biotage) (0-100% этилацетата в гексанах) с получением названного соединения (0,357 г, 94%). МС (ESI) m/z 473,5 [M+1]+.
С. (S)-Этил-2-(1-(трет-бутоксикарбонил)пирролидин-3-иламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат. (S)-Этил-5-амино-2-(1-(трет-бутоксикарбонил)пирролидин-3-иламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (0,357 г, 0,756 ммоль) и карбонилдиимидазол (1,22 г, 7,56 ммоль) в дихлорметане (20 мл) подвергали взаимодействию согласно общему методу F и очищали с применением хроматографии (Biotage) (0-100% этилацетата в гексанах) с получением названного продукта (0,369 г, 98%) в виде смеси. МС (ESI) m/z 499,5 [M+1]+.
D. (S)-Этил-9-(2-метоксифенил)-8-оксо-2-(пирролидин-3-иламино)-8,9-дигидро-7Н-пурин-6-карбоксилат. (S)-Этил-2-(1-(трет-бутоксикарбонил)пирролидин-3-иламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат (0,369 г, 0,668 ммоль) растворяли в дихлорметане (3 мл) и трифторуксусной кислоте (1 мл). Раствор перемешивали в течение двух часов и конденсировали при пониженном давлении с получением неочищенного названного соединения (0,300 г, 100%). МС (ESI) m/z 399,3 [M+1]+.
Е. (S)-9-(2-Метоксифенил)-8-оксо-2-(пирролидин-3-иламино)-8,9-дигидро-7Н-пурин-6-карбоксамид. (S)-Этил-9-(2-метоксифенил)-8-оксо-2-(пирролидин-3-иламино)-8,9-дигидро-7Н-пурин-6-карбоксилат (0,300 г) и насыщенный газообразным аммиаком метанол (10 мл) подвергали взаимодействию согласно общему методу G и очищали с применением препаративной ВЭЖХ с обращенной фазой (5-60% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин) с получением названного соединения (0,105 г, 31%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,91 (с, 1Н), 7,781 (с, 1Н), 7,466 (м, 1Н), 7,34 (д, J=7,59, 1H), 7,19 (д, J=7,99, 1H), 7,06 (т, J=7,19, 1H), 6,96 (д, J=6,39, 1H), 4,24 (с, 1Н), 3,72 (с, 3Н), 2,94 (м, 2Н), 2,72 (м, 1Н), 2,64 (м, 1Н), 1,95 (м, 1Н), 1,58 (м, 1Н); МС (ESI) m/z 370,2 [M+1]+; т.пл. >400°С.
5.1.100 ПРИМЕР 100
СИНТЕЗ (ЦИС)-4-(6-КАРБАМОИЛ-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-2-ИЛАМИНО)ЦИКЛОГЕКСИЛКАРБАМАТА
А. Этил-2-(цис-4-гидроксициклогексиламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат. Этил-2-хлор-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (см. пример 30.А) (0,300 г, 0,852 ммоль), цис-4-аминоциклогексанол (0,155 г, 1,022 ммоль) и диизопропилэтиламин (0,274 г, 2,13 ммоль) подвергали взаимодействию согласно общему методу С, за исключением того, что при комнатной температуре и в диметилформамиде (5 мл). Неочищенную реакционную смесь конденсировали и очищали с применением хроматографии (Biotage) (0-100% этилацетата в гексанах) с получением названного соединения (0,342 г, 93%). МС (ESI) m/z 432,0 [M+1]+.
В. Этил-5-амино-2-(цис-4-гидроксициклогексиламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. Этил-2-(цис-4-гидроксициклогексиламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (0,340 г, 0,788 ммоль) растворяли в этаноле (20 мл) и добавляли в колбу 10% палладия на угле (0,070 г) и продували свежими порциями газообразного водорода, и предоставляли возможность перемешиваться при комнатной температуре. Через 16 ч реакционную смесь фильтровали через целит, и фильтрат конденсировали при пониженном давлении. Неочищенное масло очищали с применением хроматографии (Biotage) (5% метанола в этилацетате) с получением названного соединения (0,272 г, 70%). МС (ESI) m/z 402,4 [M+1]+.
С. Этил-2-(цис-4-(1Н-имидазол-1-карбонилокси)циклогексиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат. Этил-5-амино-2-(цис-4-гидроксициклогексиламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (0,272 г, 0,678 ммоль) и 1,1'-1,1'-карбонилдиимидазол (1,09 г, 6,78 ммоль) в дихлорметане (20 мл) подвергали взаимодействию согласно общему методу F и очищали с применением хроматографии (Biotage) (60-100% этилацетата в гексанах) с получением названного соединения и не содержащего гидроксильного производного (0,250 г, 71%). МС (ESI) m/z 522,4 [M+1]+.
D. (цис)-4-(6-Карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-иламино)циклогексилкарбамат. Этил-2-(цис-4-(1Н-имидазол-1-карбонилокси)циклогексиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат (0,250 г, 0,479 ммоль) и газообразный аммиак подвергали взаимодействию в метаноле (10 мл) согласно общему методу G и очищали с применением препаративной ВЭЖХ с обращенной фазой (10-80% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин) с получением названного соединения (0,105 г, 24%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,888 (с, 1Н), 7,777 (с, 1Н), 7,47 (т, J=7,19, 1H), 7,36 (д, J=7,99, 1H), 7,19 (д, J=7,59, 1H), 7,07 (т, J=6,39, 1H), 6,86 (с, J=6,79, 1H), 6,33 (с, 1Н), 4,47 (с, 1Н), 3,85 (с, 1Н), 3,70 (с, 3Н), 1,60 (м, 7Н); МС (ESI) m/z 442,4 [M+1]+; т.пл. 157-160°С.
5.1.101 ПРИМЕР 101
СИНТЕЗ 2-(ТРАНС-4-ГИДРОКСИЦИКЛОГЕКСИЛАМИНО)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИД
А. Этил-2-(транс-4-гидроксициклогексиламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат. Этил-2-хлор-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (см. пример 30.А) (0,300 г, 0,852 ммоль), транс-4-аминоциклогексанол (0,117 г, 1,022 ммоль) и диизопропилэтиламин подвергали взаимодействию согласно общему методу С, за исключением того, что при комнатной температуре и в диметилформамиде (5 мл). Неочищенную реакционную смесь конденсировали и очищали с применением хроматографии (Biotage) (0-100% этилацетата в гексанах) с получением названного соединения (0,303 г, 82%). МС (ESI) m/z 432,5 [M+1]+.
В. Этил-5-амино-2-(транс-4-гидроксициклогексиламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. Этил-2-(транс-4-гидроксициклогексиламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (0,300 г, 0,852 ммоль) растворяли в этаноле (20 мл) и добавляли в колбу 10% палладия на угле (0,073 г), продували свежими порциями газообразного водорода и предоставляли возможность перемешиваться при комнатной температуре. Через 16 часов реакционную смесь фильтровали через целит, и фильтрат конденсировали при пониженном давлении. Неочищенное масло очищали с применением хроматографии (Biotage) (5% метанола в этилацетате) с получением названного соединения (0,240 г, 70%). МС (ESI) m/z 402,4 [M+1]+.
С. Этил-2-(транс-4-гидроксициклогексиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат и сложный имидазольный эфир. Этил-5-амино-2-(транс-4-гидроксициклогексиламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (0,240 г, 0,598 ммоль) и 1,1'-1,1'-карбонилдиимидазол (0,968 г, 5,98 ммоль) в дихлорметане (20 мл) подвергали взаимодействию согласно общему методу F. Получали смесь названного соединения и сложного имидазольного эфира и применяли без дополнительной очистки. МС (ESI) m/z 399 [M+1]+ (названное соединение), 522 [M+1]+ (имидазолмочевина).
D. 2-(транс-4-Гидроксициклогексиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Этил-2-(транс-4-гидроксициклогексиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат и сложный имидазольный эфир и газообразный аммиак подвергали взаимодействию в метаноле согласно общему методу G и очищали с применением препаративной ВЭЖХ с обращенной фазой (10-80% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин) с получением названного соединения (0,050 г, 3% посредством 2 стадий). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,822 (с, 1Н), 7,75 (с, 1Н), 7,46 (т, J=8,69, 1H), 7,36 (д, J=7,99, 1H), 7,19 (д, J=8,39, 1H), 7,06 (т, J=7,99, 1H), 6,98 (с, 1Н), 4,64 (с, 1Н), 3,72 (с, 3Н), 1,78 (т, J=15,99, 4H), 1,19 (м, 4Н); МС (ESI) m/z 399,1 [M+1]+; т.пл. 165-167°С.
5.1.102 ПРИМЕР 102
СИНТЕЗ 2-(4-ХЛОРПИРИДИН-3-ИЛ)-8-ОКСО-9-(2-(ТРИФТОРМЕТИЛ)ФЕНИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(4-Хлорпиридин-3-ил)-8-оксо-9-(2-(трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-(трифторметил)фенил)мочевину (см. пример 50.А) (0,15 г, 0,51 ммоль), 4-хлорникотинальдегид (0,14 г, 0,99 ммоль) и триэтиламин (0,10 мл, 0,72 ммоль) объединяли в этаноле (7,0 мл) и перемешивали при комнатной температуре в течение ночи. Избыток растворителя удаляли при пониженном давлении и полученный в результате остаток очищали хроматографией на силикагеле (Biotage) (0-20% метанола в дихлорметане). Чистые фракции объединяли, и растворитель удаляли при пониженном давлении. Полученное в результате вещество сушили в вакуумируемом устройстве с получением продукта в виде не совсем белого твердого вещества (0,075 г, 0,17 ммоль, выход 34%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,11 (с, 1Н), 8,93 (с, 1Н), 8,56 (д, J=5,3, 1H), 8,25 (ушир.с, 1Н), 8,06 (ушир.с, 1Н), 8,01 (д, J=7,8, 1H), 7,93 (м, 1Н), 7,82 (м, 2Н), 7,61 (д, J=5,73, 1H); МС (ESI) m/z 435,0 [M+1]+; т.пл. 244-248°С (разл.).
5.1.103 ПРИМЕР 103
СИНТЕЗ 2-(ЦИС-4-ГИДРОКСИЦИКЛОГЕКСИЛАМИНО)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Этил-2-(цис-4-гидроксициклогексиламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат. Этил-2-хлор-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (см. пример 30.А) (0,300 г, 0,852 ммоль), цис-4-аминоциклогексанол (0,155 г, 1,022 ммоль) и диизопропилэтиламин (0,274 г, 2,13 ммоль) подвергали взаимодействию согласно общему методу С, за исключением того, что при комнатной температуре и в диметилформамиде (5 мл). Неочищенную реакционную смесь конденсировали и очищали с применением хроматографии (Biotage) (0-100% этилацетата в гексанах) с получением названного соединения (0,342 г, 93%). МС (ESI) m/z 432,0 [M+1]+.
В. Этил-5-амино-2-(цис-4-гидроксициклогексиламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. Этил-2-(цис-4-гидроксициклогексиламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (0,340 г, 0,788 ммоль) растворяли в этаноле (20 мл) и добавляли в колбу 10% палладия на угле (0,070 г), продували свежими порциями газообразного водорода и предоставляли возможность перемешиваться при комнатной температуре. Через 16 ч реакционную смесь фильтровали через целит, и фильтрат конденсировали при пониженном давлении. Неочищенное масло очищали с применением хроматографии (Biotage) (5% метанола в этилацетате) с получением названного соединения (0,272 г, 70%). МС (ESI) m/z 402,4 [M+1]+.
С. Этил-2-(цис-4-гидроксициклогексиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат. Этил-5-амино-2-(цис-4-гидроксициклогексиламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (0,272 г, 0,678 ммоль) и 1,1'-1,1'-карбонилдиимидазол (1,09 г, 6,78 ммоль) в дихлорметане (20 мл) подвергали взаимодействию согласно общему методу F. Получали смесь названного соединения и сложного имидазольного эфира и применяли без дополнительной очистки. МС (ESI) m/z 399 [M+1]+ (названное соединение), 522 [M+1]+ (имидазолмочевина).
D. 2-(цис-4-Гидроксициклогексиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Этил-2-(цис-4-гидроксициклогексиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат и сложный имидазольный эфир и газообразный аммиак в метаноле подвергали взаимодействию согласно общему методу G и очищали с применением препаративной ВЭЖХ с обращенной фазой (10-80% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин) с получением названного соединения (0,010 г, 5% посредством 2 стадий). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,75 (с, 1Н), 7,4 (т, J=8,69, 1H), 7,36 (дд, J=7,99, 1H), 7,19 (д, J=8,39, 1H), 7,06 (т, J=7,99, 1H), 6,73 (с, 1Н), 4,23 (д, J=3,19, 1H), 3,68 (с, 1Н), 1,53 (м, 9Н); МС (ESI) m/z 399,1 [M+1]+; т.пл. 295-297°С.
5.1.104 ПРИМЕР 104
СИНТЕЗ 2-(4-((1Н-ИМИДАЗОЛ-1-ИЛ)МЕТИЛ)ФЕНИЛАМИНО)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Этил-2-(4-(гидроксиметил)фениламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат. Этил-2-хлор-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (см. пример 30.А) (0,250 г, 0,710 ммоль), 4-аминобензиловый спирт (0,104 г, 0,852 ммоль) и диизопропилэтиламин (0,137 г, 1,065 ммоль) подвергали взаимодействию согласно общему методу С, за исключением того, что при комнатной температуре и в диметилформамиде (5 мл). Неочищенную реакционную смесь конденсировали и очищали с применением хроматографии (Biotage) (60-100% этилацетата в гексанах) с получением названного соединения (0,374 г, >100%). МС (ESI) m/z 440,0 [M+1]+.
В. Этил-5-амино-2-(4-(гидроксиметил)фениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. Этил-2-(4-(гидроксиметил)фениламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (0,374 г, 0,788 ммоль) растворяли в этаноле (20 мл), добавляли в колбу 10% палладия на угле (0,075 г), обрабатывали газообразным водородом и предоставляли возможность перемешиваться при комнатной температуре. Через 16 ч реакционную смесь фильтровали через целит, и фильтрат конденсировали при пониженном давлении. Неочищенное масло очищали с применением хроматографии (Biotage) (0-100% этилацетата в гексанах) с получением названного соединения (0,180 г, 51%). МС (ESI) m/z 410,5 [M+1]+.
С. Этил-2-(4-((1Н-имидазол-1-ил)метил)фениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат. Этил-5-амино-2-(4-(гидроксиметил)фениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (0,180 г, 0,440 ммоль) и 1,1'-1,1'-карбонилдиимидазол (0,713 г, 4,44 ммоль) в дихлорметане (20 мл) подвергали взаимодействию согласно общему методу F и очищали с применением хроматографии на силикагеле (Biotage) (10% метанола в этилацетате) с получением названного соединения (0,700 г, >100%. МС (ESI) m/z 486,5 [M+1]+.
D. 2-(4-((1Н-Имидазол-1-ил)метил)фениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Этил-2-(4-((1Н-имидазол-1-ил)метил)фениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат (0,700 г) и газообразный аммиак подвергали взаимодействию в метаноле согласно общему методу G и очищали с применением препаративной ВЭЖХ с обращенной фазой (5-60% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин) с получением названного соединения (0,048 г, 24%) 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,24 (с, 1Н), 9,44 (с, 1Н), 7,93 (с, 1Н), 7,70 (с, 1Н), 7,646 (д, J=8,79, 2Н), 7,59 (с, 1Н), 7,50 (т, J=7,19, 1H), 7,43 (д, J=7,99, 1H), 7,23 (д, J=8,39, 1H), 7,15 (д, J=7,19, 2H), 7,09 (т, J=7,59, 2H), 6,87 (с, 1Н), 5,07 (с, 2Н), 3,74 (с, 3Н); МС (ESI) m/z 457,3 [M+1]+; т.пл. 165-170°С.
5.1.105 ПРИМЕР 105
СИНТЕЗ 2-(4-ГИДРОКСИПИРИДИН-3-ИЛ)-8-ОКСО-9-(2-(ТРИФТОРМЕТИЛ)ФЕНИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(4-Гидроксипиридин-3-ил)-8-оксо-9-(2-(трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-(трифторметил)фенил)мочевину (см. пример 50.А) (0,15 г, 0,51 ммоль), 4-гидроксиникотинальдегид (0,13 г, 1,06 ммоль) и триэтиламин (0,10 мл, 0,72 ммоль) объединяли в этаноле (7,0 мл) и перемешивали при комнатной температуре в течение ночи. Избыток растворителя удаляли при пониженном давлении и полученный в результате остаток очищали хроматографией на силикагеле (Biotage) (0-40% метанола в дихлорметане). Чистые фракции объединяли, и растворитель удаляли при пониженном давлении. Полученное в результате вещество сушили в вакуумируемом устройстве с получением продукта в виде не совсем белого твердого вещества (0,07 г, 0,17 ммоль, выход 33%). МС (ESI) m/z 417,0 [M+1]+.
5.1.106 ПРИМЕР 106
СИНТЕЗ (R)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-2-(ПИРРОЛИДИН-2-ИЛМЕТИЛАМИНО)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (R)-Этил-2-((1-трет-бутоксикарбонил)пирролидин-2-ил)метиламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат. Этил-2-хлор-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (см. пример 30.А) (0,300 г, 0,852 ммоль), (R)-1-boc-(аминометил)пирролидин (0,207 г, 1,02 ммоль) и диизопропилэтиламин (0,164 г, 1,27 ммоль) подвергали взаимодействию согласно общему методу С, за исключением того, что при комнатной температуре и в диметилформамиде (95 мл). Неочищенную реакционную смесь конденсировали и очищали с применением хроматографии (Biotage) (0-100% этилацетат в гексанах) с получением названного соединения (0,430 г, 97%). МС (ESI) m/z 517,5 [M+1]+.
В. (R)-Этил-5-амино-2-((1-(трет-бутоксикарбонил)пирролидин-2-ил)метиламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. (R)-Этил-2-((1-(трет-бутоксикарбонил)пирролидин-2-ил)метиламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (0,430 г, 0,802 ммоль) растворяли в этаноле (20 мл) и в колбу добавляли 10% палладия на угле (0,086 г), продували свежими порциями газообразного водорода и предоставляли возможность перемешиваться при комнатной температуре. Через 16 ч реакционную смесь фильтровали через целит, и фильтрат конденсировали при пониженном давлении. Неочищенное масло очищали с применением хроматографии (Biotage) (0-100% этилацетата в гексанах) с получением названного соединения (0,360 г, 89%). МС (ESI) m/z 487,2 [M+1]+.
С. (R)-Этил-2-((1-(трет-бутоксикарбонил)пирролидин-2-ил)метиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат. (R)-Этил-5-амино-2-((1-(трет-бутоксикарбонил)пирролидин-2-ил)метиламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (0,360 г, 0,740 ммоль) и карбонилдиимидазол (1,19 г, 7,40 ммоль) в дихлорметане (20 мл) подвергали взаимодействию согласно общему методу F и очищали с применением хроматографии (Biotage) (0-100% этилацетата в гексанах) с получением названного соединения (0,381 г, 100%). МС (ESI) m/z 513,0 [M+1]+.
D. (R)-Этил-9-(2-метоксифенил)-8-оксо-2-(пирролидин-2-илметиламино)-8,9-дигидро-7Н-пурин-6-карбоксилат. (R)-Этил-2-((1-(трет-бутоксикарбонил)пирролидин-2-ил)метиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат (0,381 г, 0,744 ммоль) растворяли в дихлорметане (3 мл) и добавляли трифторуксусноую кислоту (1 мл). Раствор перемешивали в течение двух часов и конденсировали при пониженном давлении с получением неочищенного названного соединения (0,420 г, >100%). МС (ESI) m/z 413,1 [M+1]+.
Е. (R)-9-(2-Метоксифенил)-8-оксо-2-(пирролидин-2-илметиламино)-8,9-дигидро-7Н-пурин-6-карбоксамид. (R)-Этил-9-(2-метоксифенил)-8-оксо-2-(пирролидин-2-илметиламино)-8,9-дигидро-7Н-пурин-6-карбоксилат (0,420 г) и газообразный аммиак в метаноле подвергали взаимодействию согласно общему методу G и очищали с применением препаративной ВЭЖХ с обращенной фазой (5-60% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение30 мин) с получением названного соединения (0,120 г, 30%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,73 (с, 1Н), 7,45 (т, J=7,99, 3H), 7,31 (д, J=6,79, 1H), 7,19 (д, J=8,39, 1H), 7,05 (т, J=7,19, 1H), 6,76 (с, 1Н), 3,71 (с, 3Н), 3,23 (с, 2Н), 2,88 (с, 1Н), 1,70 (с, 3Н); МС (ESI) m/z 384,4 [M+1]+; т.пл. 155-157°С.
5.1.107 ПРИМЕР 107
СИНТЕЗ (S)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-2-(ПИРРОЛИДИН-2-ИЛМЕТИЛАМИНО)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (S)-Этил-2-((1-(трет-бутоксикарбонил)пирролидин-2-ил)метиламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксалат. Этил-2-хлор-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (см. пример 30.А) (0,300 г, 0,852 ммоль), (S)-1-boc-(аминометил)пирролидин (0,206 г, 1,02 ммоль) и диизопропилэтиламин (0,164 г, 1,27 ммоль) подвергали взаимодействию согласно общему методу С, за исключением того, что при комнатной температуре и в диметилформамиде (5 мл). Неочищенную реакционную смесь конденсировали и очищали с применением хроматографии (Biotage) (0-100% этилацетата в гексанах) с получением названного соединения (0,416 г, 95%). МС (ESI) m/z 517,3 [M+1]+.
В. (S)-Этил-5-амино-2-((1-(трет-бутоксикарбонил)пирролидин-2-ил)метиламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. (S)-Этил-2-((1-(трет-бутоксикарбонил)пирролидин-2-ил)метиламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксалат (0,416 г, 0,802 ммоль) растворяли в этаноле (20 мл), добавляли в колбу 10% палладия на угле (0,083 г), пропускали свежие порции газообразного водорода и предоставляли возможность перемешиваться при комнатной температуре. Через 16 ч реакционную смесь фильтровали через целит, и фильтрат конденсировали при пониженном давлении. Неочищенное масло очищали с применением хроматографии (Biotage) (0-100% этилацетата в гексанах) с получением названного соединения (0,340 г, 84%). МС (ESI) m/z 487,6 [M+1]+.
С. (S)-Этил-2-((1-(трет-бутоксикарбонил)пирролидин-2-ил)метиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат. (S)-Этил-5-амино-2-((1-(трет-бутоксикарбонил)пирролидин-2-ил)метиламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (0,340 г, 0,720 ммоль) и карбонилдиимидазол (1,13 г, 7,40 ммоль) в дихлорметане (20 мл) подвергали взаимодействию согласно общему методу F и очищали с применением хроматографии (Biotage) (0-100% этилацетата в гексанах) с получением названного соединения (0,352 г, 98%). МС (ESI) m/z 513,5 [M+1]+.
D. (S)-Этил-9-(2-метоксифенил)-8-оксо-2-(пирролидин-2-илметиламино)-8,9-дигидро-7Н-пурин-6-карбоксилат. (S)-Этил-2-((1-(трет-бутоксикарбонил)пирролидин-2-ил)метиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат (0,352 г, 0,687 ммоль) растворяли в дихлорметане (3 мл) и добавляли трифторуксусную кислоту (1 мл). Раствор перемешивали в течение двух часов и конденсировали при пониженном давлении с получением неочищенного названного соединения (0,400 г, >100%). МС (ESI) m/z 413,1 [M+1]+.
Е. (S)-9-(2-Метоксифенил)-8-оксо-2-(пирролидин-2-илметиламино)-8,9-дигидро-7Н-пурин-6-карбоксамид. (S)-Этил-9-(2-метоксифенил)-8-оксо-2-(пирролидин-2-илметиламино)-8,9-дигидро-7Н-пурин-6-карбоксилат (0,400 г) и газообразный аммиак в метаноле (10 мл) подвергали взаимодействию согласно общему методу G и очищали с применением препаративной ВЭЖХ с обращенной фазой (5-60% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин) с получением названного соединения (0,140 г, 38%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,72 (с, 1Н), 7,45 (т, J=7,99, 3H), 7,30 (д, J=6,79, 1H), 7,18 (д, J=8,39, 1H), 7,05 (т, J=7,19, 1H), 6,76 (с, 1Н), 3,71 (с, 3Н), 3,22 (с, 2Н), 2,88 (с, 1Н), 1,70 (м, 3Н), МС (ESI) m/z 384,4 [M+1]+; т.пл. 160-165°С.
5.1.108 ПРИМЕР 108
СИНТЕЗ 2-(4-(1Н-1,2,4-ТРИАЗОЛ-3-ИЛ)ФЕНИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. (4-(1Н-1,2,4-Триазол-3-ил)фенил)метанол. 4-(1Н-1,2,4-Триазол-3-ил)бензойную кислоту (1,79 г, 9,46 ммоль) суспендировали в безводном тетрагидрофуране (50 мл) и охлаждали до -78°С. Добавляли раствор литийалюминийгидрида (2,0 М, 23,0 мл, 46,0 ммоль) и реакционной смеси предоставляли возможность медленно нагреваться до комнатной температуры при перемешивании в течение ночи. Реакционную смесь гасили метанолом, и неочищенный продукт адсорбировали на силикагеле. С помощью флэш-хроматографии (10% МеОН в EtOAc) выделяли названное соединение (1,60 г, 9,14 ммоль, 97%) в виде белого твердого вещества. МС (ESI) m/z 176,1 [M+1]+.
В. 4-(1Н-1,2,4-Триазол-3-ил)бензальдегид. (4-(1Н-1,2,4-Триазол-3-ил)фенил)метанол (92 мг, 0,53 ммоль) растворяли в безводном диметилсульфоксиде (1,5 мл) и метиленхлориде (5 мл). К раствору добавляли пиридинийхлорхромат (0,23 г, 1,06 ммоль) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали через силикагель и промывали этилацетатом. Органические вещества выливали в воду (100 мл), экстрагировали EtOAc (3×100 мл), объединенные органические слои сушили сульфатом натрия и концентрировали при пониженном давлении с получением названного соединения (70 мг, 0,27 ммоль, 51%) в виде белого твердого вещества. МС (ESI) m/z 174,1 [M+1]+.
С. 2-(4-(1Н-1,2,4-Триазол-3-ил)фенил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (70 мг, 0,27 ммоль), 4-(1Н-1,2,4-триазол-3-ил)бензальдегид (110 мг, 0,53 ммоль), триэтиламин (0,1 мл, 0,72 ммоль) и метанол (4 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали, промывали простым диэтиловым эфиром и сушили при пониженном давлении с получением названного соединения (68 мг, 0,16 ммоль, 59%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,78 (с, 1Н), 8,55 (с, 1Н), 8,47 (м, 2Н), 8,07 (д, J=8,4, 2H), 8,01 (с, 1Н), 7,57 (ддд, J=9,2, 7,6, 2,0, 1H), 7,51 (дд, J=7,6, 1,6, 1H), 7,31 (дд, J=8,4, 1,2, 1H), 7,17 (ддд, J=8,8, 7,6, 1,2, 1H), 3,76 (с, 3Н); МС (ESI) m/z 429,1 [M+1]+; т.пл. 358°С.
5.1.109 ПРИМЕР 109
СИНТЕЗ 2-(2-ГИДРОКСИЭТИЛАМИНО)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Этил-2-(2-гидроксиэтиламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат. Этил-2-хлор-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (см. пример 30.А) (0,250 г, 0,710 ммоль), этаноламин (0,052 г, 0,852 ммоль) и диизопропилэтиламин (0,137 г, 1,06 ммоль) подвергали взаимодействию согласно общему методу С, за исключением того, что при комнатной температуре и в диметилформамиде (5 мл). Неочищенную реакционную смесь конденсировали и очищали с применением хроматографии (Biotage) (0-100% этилацетата в гексанах) с получением названного соединения (0,250 г, 95%). МС (ESI) m/z 378,5 [M+1]+.
В. Этил-5-амино-2-(2-гидроксиэтиламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. Этил-2-(2-гидроксиэтиламино)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (0,250 г, 0,663 ммоль) растворяли в этаноле (20 мл) и в колбу добавляли 10% палладия на угле (0,050 г), пропускали свежие порции газообразного водорода и предоставляли возможность перемешиваться при комнатной температуре. Через 16 ч реакционную смесь фильтровали через целит, и фильтрат конденсировали при пониженном давлении. Неочищенное масло очищали с применением хроматографии (Biotage) (5% метанола в этилацетате) с получением названного соединения (0,200 г, 87%). МС (ESI) m/z 348,1 [M+1]+.
С. Этил-2-(2-гидроксиэтиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат. Этил-5-амино-2-(2-гидроксиэтиламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (0,200 г, 0,576 ммоль) и карбонилдиимидазол (0,939 г, 5,76 ммоль) в дихлорметане (20 мл) подвергали взаимодействию согласно общему методу F и очищали с применением хроматографии (Biotage) (0-100% этилацетата в гексанах) с получением названного соединения и имидазолкарбамата в виде смеси (0,240 г вместе, 89%). МС (ESI) m/z 374,1 [M+1]+ (названное соединение) и 468,1 [M+1]+ (имидазолкарбамат).
D. 2-(2-Гидроксиэтиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Этил-2-(2-гидроксиэтиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат (0,400 г) и газообразный аммиак в метаноле (10 мл) подвергали взаимодействию согласно общему методу G и очищали с применением препаративной ВЭЖХ с обращенной фазой (5-60% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин) с получением названного соединения (0,015 г, 8%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,49 (с, 1Н), 7,86 (с, 1Н), 7,81 (с, 1Н), 7,48 (т, J=6,79, 1H), 7,36 (д, J=7,99, 1H), 7,20 (д, J=8,39, 1H), 7,07 (т, J=7,59, 1H), 6,78 (с, 1Н), 4,50 (т, J=5,59, 1H), 3,72 (с, 3Н), 3,44 (кв, J=6,39, 2H), 3,31 (м, 2Н); МС (ESI) m/z 345,2 [M+1]+; т.пл. 157-160°С.
5.1.110 ПРИМЕР 110
СИНТЕЗ 9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-2-(2-(ТРИФТОРМЕТИЛ)-1Н-БЕНЗО[D]ИМИДАЗОЛ-6-ИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (2-(Трифторметил)-1Н-бензо[d]имидазол-6-ил)метанол. 2-(Трифторметил)-1Н-бензо[d]имидазол-6-карбоновую кислоту (1,38 г, 6,00 ммоль) суспендировали в безводном тетрагидрофуране (100 мл) и охлаждали до -78°С. Добавляли раствор литийалюминийгидрида (2,0 М, 15 мл, 30,0 ммоль) и полученному в результате раствору предоставляли возможность медленно нагреваться до комнатной температуры при перемешивании в течение ночи. Реакционную смесь гасили метанолом, и неочищенный продукт адсорбировали на силикагеле. С помощью флэш-хроматографии (10% МеОН в EtOAc) получали названное соединение (1,20 г, 5,55 ммоль, 93%) в виде белого твердого вещества. МС (ESI) m/z 217,1 [M+1]+.
В. 2-(Трифторметил)-1Н-бензо[d]имидазол-6-карбальдегид. (2-(Трифторметил)-1Н-бензо[d]имидазол-6-ил)метанол (1,08 мг, 4,99 ммол) растворяли в безводном диметилсульфоксиде (5,0 мл) и метиленхлориде (30,0 мл). К раствору добавляли пиридинийхлорхромат (4,31 г, 20,0 ммоль) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали через силикагель и промывали этилацетатом. Органические вещества выливали в воду (100 мл), экстрагировали EtOAc (3×100 мл), объединенные органические слои сушили сульфатом натрия и концентрировали при пониженном давлении с получением названного соединения (545 мг, 2,54 ммоль, 51%) в виде белого твердого вещества. МС (ESI) m/z 215,1 [M+1]+.
С. 9-(2-Метоксифенил)-8-оксо-2-(2-(трифторметил)-1Н-бензо[d]имидазол-6-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (240 мг, 0,93 ммоль), 2-(трифторметил)-1Н-бензо[d]имидазол-6-карбальдегид (400 мг, 1,86 ммоль), триэтиламин (0,2 мл, 1,40 ммоль) и метанол (6,0 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали и очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Летучие вещества удаляли при пониженном давлении, суспендированное твердое вещество обрабатывали гидроксидом аммония с помощью ультразвука и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (23 мг, 0,049 ммоль, 5%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,75 (с, 1Н), 8,59 (с, 1Н), 8,50 (с, 1Н), 8,00 (с, 1Н), 7,57 (ддд, J=8,0, 7,6, 1,6, 1H), 7,53 (дд, J=8,0, 1,6, 1H), 7,32 (д, J=7,2, 1H), 7,18 (м, 1Н), 3,76 (с, 3Н); МС (ESI) m/z 470,1 [M+1]+; т.пл. 220-222°С.
5.1.111 ПРИМЕР 111
СИНТЕЗ 2-(3-(1Н-1,2,4-ТРИАЗОЛ-3-ИЛ)ФЕНИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. (3-(1Н-1,2,4-Триазол-3-ил)фенил)метанол. 3-(1Н-1,2,4-Триазол-3-ил)бензойную кислоту (2,01 г, 10,62 ммоль) суспендировали в безводном тетрагидрофуране (100 мл) и охлаждали до -78°С. Добавляли раствор литийалюминийгидрида (2,0 М, 26,0 мл, 52,0 ммоль) и полученному в результате раствору предоставляли возможность медленно нагреваться до комнатной температуры при перемешивании в течение ночи. Реакционную смесь гасили метанолом, и неочищенный продукт адсорбировали на силикагеле. С помощью флэш-хроматографии (10% МеОН в EtOAc) выделяли названное соединение (1,0 г, 5,71 ммоль, 54%) в виде белого твердого вещества. МС (ESI) m/z 176,1 [M+1]+.
В. 3-(1Н-1,2,4-Триазол-3-ил)бензальдегид. (3-(1Н-1,2,4-Триазол-3-ил)фенил)метанол (1,0 г, 5,71 ммоль) растворяли в безводном диметилсульфоксиде (4,0 мл) и метиленхлориде (60,0 мл). К раствору добавляли пиридинийхлорхромат (4,92 г, 22,8 ммоль) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали через силикагель и промывали этилацетатом. Выливали органические вещества в воду (100 мл), экстрагировали EtOAc (2×100 мл), сушили объединенные органические слои сульфатом натрия и концентрировали при пониженном давлении с получением названного соединения (660 мг, 3,79 ммоль, 66%) в виде белого твердого вещества. МС (ESI) m/z 174,1 [M+1]+.
С. 2-(3-(1Н-1,2,4-Триазол-3-ил)фенил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (245 мг, 0,95 ммоль), 3-(1Н-1,2,4-триазол-3-ил)бензальдегид (330 мг, 1,90 ммоль), триэтиламин (0,2 мл, 1,43 ммоль) и метанол (8,0 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали и очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Летучие вещества удаляли при пониженном давлении, суспендированное твердое вещество обрабатывали гидроксидом аммония с применением ультразвука и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (72 мг, 0,15 ммоль, 16%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,79 (с, 1Н), 8,82 (с, 1Н), 8,62 (с, 1Н), 8,56 (д, J=8,0, 1H), 8,45 (с, 1Н), 8,07 (м, 2Н), 7,56 (м, 3Н), 7,32 (дд, J=8,4, 0,8, 1H), 7,17 (м, 1Н), 3,77 (с, 3Н); МС (ESI) m/z 429,1 [M+1]+; т.пл. 242-243°С.
5.1.112 ПРИМЕР 112
СИНТЕЗ 9-(БИФЕНИЛ-2-ИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-(бифенил-2-ил)мочевина. В круглодонной колбе в ацетонитриле (35 мл) растворяли 2,3-диаминомалеонитрил (1,0 г, 9,25 ммоль) и перемешивали при комнатной температуре. Добавляли по каплям в течение 10 минут 2-бифенилизоцианат (1,80 г, 9,25 ммоль) в ацетонитриле (5 мл) и реакционную смесь перемешивали при комнатной температуре. Через 16 часов раствор конденсировали при пониженном давлении и полученное в результате твердое вещество очищали с применением хроматографии на силикагеле Biotage (0-100% этилацетата в гексанах) с получением названного соединения (1,48 г, 53%). МС (ESI) m/z 304,3 [M+1]+.
В. 9-(Бифенил-2-ил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(бифенил-2-ил)мочевину (0,500 г, 1,65 ммоль), 3-гидроксибензальдегид (0,403 г, 3,30 ммоль) и триэтиламин (0,6 мл) подвергали взаимодействию согласно общему методу В и растирали со смесью диметилформамид/вода с получением названного соединения (0,160 г, 23%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,63 (с, 1Н), 9,49 (с, 1Н), 8,34 (с, 1Н), 7,95 (с, 1Н), 7,84 (д, J=7,59, 1H), 7,67 (м, 1Н), 7,63 (м, 4Н), 7,20 (м, 5Н), 7,17 (м, 1Н), 6,80 (д, J=7,99, 1H); МС (ESI) m/z 424,2 [M+1]+; т.пл. 293-296°С.
5.1.113 ПРИМЕР 113
СИНТЕЗ 2-(4-(1Н-1,2,4-ТРИАЗОЛ-3-ИЛ)ФЕНИЛ)-9-(2-ФТОРФЕНИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. 2-(4-(1Н-1,2,4-Триазол-3-ил)фенил)-9-(2-фторфенил)-8-оксо-7-гидропурин-6-карбоксамид. N-((1Z)-2-Амино-1,2-дициановинил)[(2-фторфенил)амино]карбоксамид (см. пример 9.А) (290 мг, 1,15 ммоль), 4-(1Н-1,2,4-триазол-3-ил)бензальдегид (см. 108.В) (400 мг, 2,54 ммоль), триэтиламин (0,24 мл, 1,73 ммоль) и метанол (8,0 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали и очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Летучие вещества удаляли при пониженном давлении, суспендированное твердое вещество обрабатывали гидроксидом аммония с применением ультразвука и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (28 мг, 0,067 ммоль, 6%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,95 (c, 1Н), 8,65 (с, 1Н), 8,57 (с, 1Н), 8,48 (д, J=8,4 1H), 8,08 (д, J=8,0, 1H), 8,05 (с, 1Н), 7,73 (ддд, J=9,2, 7,6, 1,6, 1H), 7,64 (м, 2Н), 7,56 (ддд, J=10,0, 8,4, 1,2, 1H), 7,47 (ддд, J=8,8, 7,6, 1,2, 1H); МС (ESI) m/z 417,1 [M+1]+; т.пл. 358°С.
5.1.114 ПРИМЕР 114
СИНТЕЗ 2-(4-(1Н-1,2,4-ТРИАЗОЛ-3-ИЛ)ФЕНИЛ)-9-(2-ИЗОПРОПИЛФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(4-(1Н-1,2,4-Триазол-3-ил)фенил)-9-(2-изопропилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-изопропилфенил)мочевину (см. пример 56.А) (310 мг, 1,15 ммоль), 4-(1Н-1,2,4-триазол-3-ил)бензальдегид (см. 108.В) (440 мг, 2,54 ммоль), триэтиламин (0,24 мл, 1,73 ммоль) и метанол (8,0 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали, промывали простым диэтиловым эфиром и сушили при пониженном давлении с получением названного соединения (225 мг, 0,51 ммоль, 44%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,85 (с, 1Н), 8,64 (с, 1Н), 8,55 (с, 1Н), 8,45 (д, J=7,6, 1H), 8,06 (д, J=8,0, 1H), 8,03 (с, 1Н), 7,58 (м, 2Н), 7,42 (м, 2Н), 2,75 (гепт, J=6,8, 1H), 1,14 (д, J=6,8, 3H), 1,12 (д, J=6,8, 1H); МС (ESI) m/z 441,1 [M+1]+; т.пл. 368°С.
5.1.115 ПРИМЕР 115
СИНТЕЗ 9-(2-МЕТОКСИФЕНИЛ)-2-(2-МЕТИЛ-1Н-БЕНЗО[D]ИМИДАЗОЛ-6-ИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А.(2-Метил-1Н-бензо[d]имидазол-6-ил)метанол. 2-Метил-1Н-бензо[d]имидазол-6-карбоновую кислоту (2,0 г, 11,35 ммоль) суспендировали в безводном тетрагидрофуране (100 мл) и охлаждали до -78°С. Добавляли раствор литийалюминийгидрида (2,0 М, 22,7 мл, 45,4 ммоль) и предоставляли возможность медленно нагреваться до комнатной температуры при перемешивании в течение ночи. Реакционную смесь гасили метанолом, и неочищенный продукт адсорбировали на силикагеле. С помощью флэш-хроматографии (20% МеОН в EtOAc) получали названное соединение (1,11 г, 6,85 ммоль, 60%) в виде белого твердого вещества. МС (ESI) m/z 163,1 [M+1]+.
В. 2-Метил-1Н-бензо[d]имидазол-6-карбальдегид. (2-Метил-1Н-бензо[d]имидазол-6-ил)метанол (1,11 г, 6,85 ммоль) растворяли в безводном диметилсульфоксиде (6,0 мл) и метиленхлориде (60,0 мл). К раствору добавляли пиридинийхлорхромат (5,90 г, 27,4 ммоль) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали через силикагель и промывали этилацетатом. Органические вещества выливали в воду (100 мл), экстрагировали EtOAc (2×100 мл), сушили объединенные органические слои сульфатом натрия и концентрировали при пониженном давлении с получением названного соединения (742 мг, 3,79 ммоль, 67%) в виде белого твердого вещества. МС (ESI) m/z 161,1 [M+1]+.
С. 9-(2-Метоксифенил)-2-(2-метил-1Н-бензо[d]имидазол-6-ил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (290 мг, 1,13 ммоль), 2-метил-1Н-бензо[d]имидазол-6-карбальдегид (400 мг, 2,49 ммоль), триэтиламин (0,24 мл, 1,70 ммоль) и метанол (8,0 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали, промывали простым диэтиловым эфиром и сушили при пониженном давлении с получением названного соединения (265 мг, 0,64 ммоль, 57%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,24 (с, 1Н), 11,64 (с, 1Н), 8,52 (м, 1Н), 8,32 (с, 1Н), 8,22 (м, 1Н), 7,97 (с, 1Н), 7,57 (ддд, J=9,2, 8,8, 1,6, 1H), 7,51 (дд, J=7,6, 1,6, 1H), 7,31 (дд, J=8,4, 0,8, 1H), 7,17 (ддд, J=8,8, 8,0, 1,6, 1H), 3,75 (с, 1Н), 2,48 (с, 3Н). МС (ESI) m/z 416,1 [M+1]+; т.пл. 270°С.
5.1.116 ПРИМЕР 116
СИНТЕЗ 2-(3-(ГИДРОКСИМЕТИЛ)ФЕНИЛАМИНО)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Этил-5-нитро-2-(3-(гидроксиметил)фениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. Этил-2-хлор-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (см. пример 30.А) (0,300 г, 0,852 ммоль), 3-аминобензиловый спирт (0,125 г, 1,2 ммоль) и диизопропилэтиламин (0,219 г) подвергали взаимодействию согласно общему методу С, за исключением того, что при комнатной температуре и в диметилформамиде (5 мл). Неочищенную реакционную смесь конденсировали и очищали с применением хроматографии (Biotage) (60-100% EtOAc в гексанах) с получением названного соединения (0,350 г, 93%). МС (ESI) m/z 440,5 [M+1]+.
В. Этил-5-амино-2-(3-(гидроксиметил)фениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. Этил-5-нитро-2-(3-(гидроксиметил)фениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (0,35 г, 0,797 ммоль) растворяли в этаноле (20 мл) и в колбу добавляли 10% палладия на угле (0,087 г), пропускали свежие порции газообразного водорода и предоставляли возможность перемешиваться при комнатной температуре. Через 16 ч реакционную смесь фильтровали через целит, и фильтрат конденсировали при пониженном давлении. Неочищенное масло очищали с применением хроматографии (Biotage) (50-100% этилацетат в гексанах) с получением названного соединения (0,277 г, 85%). МС (ESI) m/z 410,5 [M+1]+.
С. Этил-5-амино-2-(3-((трет-бутилдиметилсилилокси)метил)фениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. Этил-5-амино-2-(3-(гидроксиметил)фениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (0,277 г, 0,677 ммоль), трет-бутилдиметилсилилхлорид (0,132 г, 0,880 ммоль), имидазол (0,047 г, 0,693 ммоль) объединяли в метиленхлориде (10 мл) и перемешивали в течение 16 ч при комнатной температуре. Раствор конденсировали при пониженном давлении и очищали с применением хроматографии (Biotage) (0-100% EtOAc в гексанах) с получением названного соединения (0,242 г, 68%). МС (ESI) m/z 524,7 [M+1]+.
D. Этил-2-(3-((трет-бутилдиметилсилилокси)метил)фениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат. Этил-5-амино-2-(3-((трет-бутилдиметилсилилокси)метил)фениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (0,242 г, 0,462 ммоль) и 1,1'-1,1'-карбонилдиимидазол (0,525 г, 3,23 ммоль) в дихлорметане (15 мл) подвергали взаимодействию согласно общему методу F и очищали с применением хроматографии (Biotage) (10-90% EtOAc в гексанах) с получением названного соединения (0,180 г, 71%). МС (ESI) m/z 550,5 [M+1]+.
Е. 2-(3-((трет-Бутилдиметилсилилокси)метил)фениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Этил-2-(3-((трет-бутилдиметилсилилокси)метил)фениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат (0,180 г, 3,97 ммоль) растворяли в метаноле (10 мл), насыщали газообразным аммиаком и подвергали взаимодействию согласно общему методу G. Через 16 ч раствор конденсировали при пониженном давлении с получением названного соединения (0,153 г, 91%). МС (ESI) m/z 521,6 [M+1]+.
F. 2-(3-(Гидроксиметил)фениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. 2-(3-((трет-Бутилдиметилсилилокси)метил)фениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид (0,153 г, 0,294 ммоль) растворяли в 4 н. HCl в диоксане (5 мл) и перемешивали при комнатной температуре. Через 30 мин раствор конденсировали при пониженном давлении с получением HCl соли. Соль разбавляли метанолом и пропускали через ионообменную колонку Strata-XC с получением названного соединения в виде свободного основания (0,083 г, 69%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,27 (с, 1Н), 9,43 (с, 1Н), 8,00 (с, 1Н), 7,78 (с, 1Н), 7,54 (ушир.с, 1Н), 7,50 (м, 1Н), 7,45 (д, J=6,39, 1H), 7,42 (д, J=8,79, 1Н), 7,24 (д, J=8,39, 1H), 7,15 (т, J=7,59, 1H), 7,11 (т, J=7,99, 1H), 6,82 (д, J=7,19, 1H), 5,13 (т, J=5,59, 1H), 4,41 (д, J=5,59, 2H), 3,75 (с, 3Н). МС (ESI) m/z 407,4 [M+1]+; т.пл. 237-239°С.
5.1.117 ПРИМЕР 117
СИНТЕЗ 2-(2-(ГИДРОКСИМЕТИЛ)ФЕНИЛАМИНО)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Этил-5-нитро-2-(2-(гидроксиметил)фениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. Этил-2-хлор-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (см. пример 30.А) (0,300 г, 0,852 ммоль), 2-аминобензиловый спирт (0,125 г, 1,2 ммоль) и диизопропилэтиламин (0,296 г) подвергали взаимодействию согласно общему методу С, за исключением того, что при комнатной температуре и в диметилформамиде (5 мл). Неочищенную реакционную смесь конденсировали и очищали с применением хроматографии (Biotage) (60-100% EtOAc в гексанах) с получением названного соединения (0,323 г, 86%). МС (ESI) m/z 440,5 [M+1]+.
В. Этил-5-амино-2-(2-(гидроксиметил)фениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. Этил-5-нитро-2-(2-(гидроксиметил)фениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (0,325 г, 0,740 ммоль) растворяли в этаноле (20 мл) и в колбу добавляли 10% палладия на угле (0,081 г), пропускали свежие порции газообразного водорода и предоставляли возможность перемешиваться при комнатной температуре. Через 16 ч реакционную смесь фильтровали через целит, и фильтрат конденсировали при пониженном давлении. Неочищенное масло очищали с применением хроматографии (Biotage) (50-100% этилацетата в гексанах) с получением названного соединения (0,270 г, 89%). МС (ESI) m/z 410,5 [M+1]+.
С. Этил-5-амино-2-(2-((трет-бутилдиметилсилилокси)метил)фениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. Этил-5-амино-2-(2-(гидроксиметил)фениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (0,227 г, 0,555 ммоль), трет-бутилдиметилсилилхлорид (0,104 г, 0,693 ммоль), имидазол (0,047 г, 0,683 ммоль) объединяли в метиленхлориде (10 мл) и перемешивали в течение 16 ч при комнатной температуре. Раствор конденсировали при пониженном давлении и очищали с применением хроматографии (Biotage) (0-100% EtOAc в гексанах) с получением названного соединения (0,200 г, 69%). МС (ESI) m/z 524,7 [M+1]+.
D. Этил-2-(2-((трет-бутилдиметилсилилокси)метил)фениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат. Этил-5-амино-2-(3-((трет-бутилдиметилсилилокси)метил)фениламино)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (0,200 г, 0,382 ммоль) и 1,1'-1,1'-карбонилдиимидазол (0,433 г, 2,67 ммоль) в дихлорметане (20 мл) подвергали взаимодействию согласно общему методу F и очищали с применением хроматографии (Biotage) (10-90% EtOAc в гексанах) с получением названного соединения (0,190 г, 91%). МС (ESI) m/z 550,5 [M+1]+.
Е. 2-(2-((трет-Бутилдиметилсилилокси)метил)фениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Этил-2-(2-((трет-бутилдиметилсилилокси)метил)фениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат (0,190 г, 3,97 ммоль) растворяли в метаноле (15 мл) и насыщали газообразным аммиаком согласно общему методу G. Через 16 ч раствор конденсировали при пониженном давлении с получением названного соединения (0,150 г, 90%). МС (ESI) m/z 521,6 [M+1]+.
F. 2-(2-(Гидроксиметил)фениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. 2-(2-((трет-Бутилдиметилсилилокси)метил)фениламино)-9-(2-метоксифенил)-8-
оксо-8,9-дигидро-7Н-пурин-6-карбоксамид (0,150 г, 0,294 ммоль) растворяли в растворе 4 н. HCl в диоксане (5 мл) и перемешивали при комнатной температуре. Через 30 мин раствор конденсировали при пониженном давлении с получением HCl соли. Соль разбавляли метанолом и пропускали через ионообменный шприц Strata XC (колонку) с получением названного соединения в виде свободного основания (0,049 г, 41%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,26 (с, 1Н), 8,63 (с, 1Н), 7,89 (с, 1Н), 7,83 (д, J=7,99, 1H), 7,57 (с, 1Н), 7,50 (т, 8,39, 1Н), 7,44 (дд, J=7,59, 1,59, 1H), 7,27 (д, J=7,19, 1Н), 7,24 (д, J=8,39, 1H), 7,19 (т, J=7,59, 1H), 7,11 (т, J=7,59, 1H), 7,97 (т, J=7,99, 1H); МС (ESI) m/z 407,4 [M+1]+; т.пл. 159-162°С.
5.1.118 ПРИМЕР 118
СИНТЕЗ 9-(2-ТРЕТ-БУТИЛФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-трет-бутилфенил)мочевина. 2,3-Диаминомалеонитрил (3,0 г, 27,75 ммоль) и 2-трет-бутилфенилизоцианат (1,61 г, 9,25 ммоль) подвергали взаимодействию согласно общему методу А. Полученный в результате осадок отфильтровывали и промывали ацетонитрилом, и сушили с получением названного соединения (0,391 г, 15%). МС (ESI) m/z 284,3 [M+1]+.
В. 9-(2-трет-Бутилфенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-трет-бутилфенил)мочевину (0,391 г, 1,38 ммоль), 3-гидроксибензальдегид (0,337 г, 2,76 ммоль) и триэтиламин (0,6 мл) подвергали взаимодействию согласно общему методу В и очищали с применением препаративной ВЭЖХ с обращенной фазой (10-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин) с получением названного соединения (0,097 г, 17%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,40 (с, 1Н), 8,00 (с, 1Н), 7,85 (д, J=7,99, 1H), 7,718 (дд, J=8,39, 1,19, 1H), 7,62 (т, J=1,99, 1H), 7,53 (т, J=7,99, 1H), 7,40 (т, J=8,79, 1H), 7,30 (дд, J=7,59, 1,59, 1H), 7,21 (т, J=7,99, 1H), 6,80 (дд, J=7,99, 1,59, 1H), 3,37 (с, 9Н); МС (ESI) m/z 404,1 [M+1]+; т.пл. 294-297°С.
5.1.119 ПРИМЕР 119
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-9-(2-ФЕНОКСИФЕНИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-феноксифенил)мочевина. 2,3-Диаминомалеонитрил (0,3 г, 27,75 ммоль) и 2-феноксифенилизоцианат (1,95 г, 9,25 ммоль) в ацетонитриле (40 мл) подвергали взаимодействию согласно общему методу А. Осадок отфильтровывали и промывали ацетонитрилом, и сушили с получением названного соединения (0,573 г, 19%). МС (ESI) m/z 320,1 [M+1]+.
В. 2-(3-Гидроксифенил)-8-оксо-9-(2-феноксифенил)-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-феноксифенил)мочевину (0,573 г, 1,79 ммоль), 3-гидроксибензальдегид (0,438 г, 3,59 ммоль) и триэтиламин (0,6 мл) подвергали взаимодействию согласно общему методу В с получением названного соединения (0,436 г, 55%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,36 (с, 1Н), 7,96 (с, 1Н), 7,91 (д, J=7,99, 1H), 7,74 (т, J=1,99, 1H), 7,66 (дд, J=7,99, 1,59, 1H), 7,57 (тд, J=7,59, 1,59, 1H), 7,37 (тд, J=7,99, 1,19, 1H), 7,23 (т, 3Н), 7,11 (дд, J=8,39, 1,19, 1H), 7,02 (т, J=7,19, 1H), 6,97 (д, J=7,59, 2H), 6,82 (дд, J=7,99, 1,99, 1H), МС (ESI) m/z 440,1 [M+1]+; т.пл. 329-331°С.
5.1.120 ПРИМЕР 120
СИНТЕЗ 2-(1Н-БЕНЗО[D]ИМИДАЗОЛ-6-ИЛ)-9-(2-ИЗОПРОПИЛФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(1Н-Бензо[d]имидазол-6-ил)-9-(2-изопропилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-изопропилфенил)мочевину (см. пример 56.А) (370 мг, 1,37 ммоль), 1Н-бензо[d]имидазол-6-карбальдегид (см. пример 84.В) (400 мг, 2,73 ммоль), триэтиламин (0,30 мл, 2,10 ммоль) и метанол (6,0 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали, промывали простым диэтиловым эфиром и сушили при пониженном давлении с получением названного соединения (185 мг, 0,45 ммоль, 33%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,48 (с, 1Н), 11,71 (с, 1Н), 8,72 (с, 1Н), 8,52 (м, 1Н), 8,41 (с, 1Н), 8,24 (м, 1Н), 7,95 (д, J=8,0, 1H), 7,59 (м, 3Н), 7,42 (м, 2Н), 2,75 (гепт, J=5,7, 1H), 1,14 (д, J=5,7, 3H), 1,12 (д, J=5,7, 3H). МС (ESI) m/z 414,1 [M+1]+; т.пл. 275°С (разл.).
5.1.121 ПРИМЕР 121
СИНТЕЗ 2-(1Н-ИНДАЗОЛ-4-ИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 4-Бром-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол. 4-Бром-1Н-индазол (1,0 г, 5,07 ммоль) растворяли в безводном тетрагидрофуране (50 мл). К этому раствору добавляли дигидропиран (0,93 мл, 10,14 ммоль) и толуолсульфоновую кислоту (144 мг, 0,76 ммоль). Реакционную смесь нагревали при кипячении с обратным холодильником в течение ночи. Добавляли насыщенный раствор бикарбоната, и водный слой экстрагировали этилацетатом (3×50 мл). Органические фракции собирали, сушили над сульфатом натрия и адсорбировали на силикагеле. С помощью флэш-хроматографии (10% EtOAc в гекс.) выделяли белое твердое вещество (1,3 г, 4,62 ммоль, 91%). МС (ESI) m/z 282,1 [M+1]+.
В. 1-(Тетрагидро-2Н-пиран-2-ил)-1Н-индазол-4-карбальдегид. 4-Бром-1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол (816 мг, 2,90 ммоль) растворяли в безводном тетрагидрофуране (10 мл) и охлаждали до -78°С. Добавляли н-бутиллитий (1,6 М, 4,0 мл, 6,38 ммоль) и реакционную смесь перемешивали в течение 30 мин. Добавляли диметилформамид (0,53 мл) и раствору предоставляли возможность нагреваться до комнатной температуры. Реакционную смесь гасили насыщенным раствором бикарбоната натрия, экстрагировали простым диэтиловым эфиром (2×75 мл) и сушили над сульфатом натрия. Продукт применяли на следующей стадии без дополнительной очистки или характеристики. МС (ESI) m/z 231,2 [M+1]+.
С. 2-(1Н-Индазол-4-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (340 мг, 1,31 ммоль), 1-(тетрагидро-2Н-пиран-2-ил)-1Н-индазол-4-карбальдегид (2,90 ммоль), триэтиламин (0,33 мл, 2,36 ммоль) и метанол (8 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Осадок отфильтровывали и растворяли в растворе 4,0 М HCl/диоксан (0,2 мл) и безводном диоксане (3 мл). Реакционную смесь нагревали до 55°С в течение ночи. Неочищенное вещество выливали в насыщенный раствор бикарбоната натрия и экстрагировали этилацетатом (3×75 мл), сушили сульфатом натрия и концентрировали при пониженном давлении с получением названного соединения (45 мг, 0,11 ммоль) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,16 (с, 1Н), 11,80 (с, 1Н), 8,42 (м, 2Н), 8,40 (с, 1Н), 8,35 (с, 1Н), 7,59 (м, 3Н), 7,35 (дд, J=8,4, 7,2, 1H), 7,35 (дд, J=8,4, 0,8, 1H), 7,20 (ддд, J=8,8, 7,6, 1,2, 1H), 3,75 (с, 3Н); МС (ESI) m/z 402,1 [M+1]+; т.пл. 280°С.
5.1.122 ПРИМЕР 122
СИНТЕЗ 2-(2-ГИДРОКСИПИРИДИН-3-ИЛ)-8-ОКСО-9-(2-(ТРИФТОРМЕТИЛ)ФЕНИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(2-Гидроксипиридин-3-ил)-8-оксо-9-(2-(трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-трифторметил)фенил)мочевину (см. пример 50.А) (0,140 г, 0,474 ммоль), 2-оксо-1,2-дигидропиридин-3-карбоксальдегид (0,130 г, 0,948 ммоль) и триэтиламин (0,1 мл) подвергали взаимодействию согласно общему методу В и очищали с применением хроматографии (Biotage) (5-20% МеОН в дихлорметане) с получением названного соединения (0,078 г, 39%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,127 (с, 1Н), 9,10 (с, 1Н), 8,73 (дд, J=7,59, 1,59, 1H), 7,98 (м, 4Н), 7,79 (м, 3Н), 7,68 (д, J=7,59, 1H), 6,77 (т, J=6,39, 1H); МС (ESI) m/z 417,0 [M+1]+; т.пл. 331-335°С.
5.1.123 ПРИМЕР 123
СИНТЕЗ 2-(1Н-ИМИДАЗО[4,5-В]ПИРИДИН-6-ИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (1Н-Имидазо[4,5-b]пиридин-6-ил)метанол. Имидазо[4,5-b]пиридин-6-карбоновую кислоту (2,02 г, 12,38 ммоль) суспендировали в безводном тетрагидрофуране (100 мл) и охлаждали до -78°С. Добавляли раствор литийалюминийгидрида (2,0 М, 24,7 мл, 49,5 ммоль) и реакционной смеси предоставляли возможность нагреваться до комнатной температуры при перемешивании в течение ночи. Реакционную смесь гасили метанолом, и неочищенный продукт адсорбировали на силикагеле. С помощью флэш-хроматографии (30% МеОН в EtOAc) выделяли названное соединение (0,29 г, 1,95 ммоль, 15%) в виде белого твердого вещества. МС (ESI) m/z 150,1 [M+1]+.
В. 1Н-Имидазо[4,5-b]пиридин-6-карбальдегид. (1Н-Имидазо[4,5-b]пиридин-6-ил)метанол (290 мг, 1,95 ммоль) растворяли в безводном диметилсульфоксиде (4,0 мл) и метиленхлориде (30 мл). К раствору добавляли пиридинийхлорхромат (1,68 г, 7,80 ммоль) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали через силикагель и промывали этилацетатом. Органические вещества выливали в воду (100 мл), экстрагировали EtOAc (3×100 мл), объединенные органические слои сушили сульфатом натрия и концентрировали при пониженном давлении с получением названного соединения (133 мг, 0,89 ммоль, 46%) в виде белого твердого вещества. МС (ESI) m/z 148,1 [M+1]+.
С. 2-(1Н-Имидазо[4,5-b]пиридин-6-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (162 мг, 0,63 ммоль), 1Н-имидазо[4,5-b]пиридин-6-карбальдегид (133 мг, 0,89 ммоль), триэтиламин (0,14 мл, 0,94 ммоль) и метанол (5 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали, промывали простым диэтиловым эфиром и сушили при пониженном давлении с получением названного соединения (7 мг, 0,017 ммоль, 3%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,77 (с, 1Н), 9,43 (с, 1Н), 8,90 (с, 1Н), 8,69 (с, 1Н), 8,57 (с, 1Н), 7,98 (с, 1Н), 7,57 (ддд, J=9,2, 8,0, 1,6, 1H), 7,53 (дд, J=8,0, 1,6, 1H), 7,31 (дд, J=7,6, 1,2, 1H), 7,17 (ддд, J=8,8, 7,6, 1,2, 1H), 3,76 (с, 3Н); МС (ESI) m/z 403,0 [M+1]+; т.пл. 358°С (разл.).
5.1.124 ПРИМЕР 124
СИНТЕЗ 2-(4-(1Н-ИМИДАЗОЛ-1-ИЛ)ФЕНИЛ)-9-(2-ИЗОПРОПИЛФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(4-(1Н-Имидазол-1-ил)фенил)-9-(2-изопропилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-изопропилфенил)мочевину (см. пример 56.А) (360 мг, 1,32 ммоль), 4-(1Н-имидазол-1-ил)бензальдегид (500 мг, 2,90 ммоль), триэтиламин (0,28 мл, 1,98 ммоль) и метанол (10,0 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали и очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Летучие вещества удаляли при пониженном давлении, суспендированное твердое вещество обрабатывали гидроксидом аммония с применением ультразвука и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (150 мг, 0,34 ммоль, 26%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CD3OD) δ 10,77 (с, 1Н), 10,64 (с, 1Н), 10,62 (с, 1Н), 10,16 (с, 1Н), 9,96 (м, 1Н), 9,87 (м, 2Н), 9,74 (м, 2Н), 9,56 (м, 2Н), 9,28 (с, 1Н), 4,90 (гепт, J=6,8, 1H), 3,29 (д, J=6,8, 3H), 3,27 (д, J=6,8, 3H); МС (ESI) m/z 440,1 [M+1]+; т.пл. 232°С.
5.1.125 ПРИМЕР 125
СИНТЕЗ 9-(2-ЦИКЛОГЕКСИЛФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 1-Циклогексил-2-изоцианатобензол. К раствору 2-циклогексилбензойной кислоты (0,97 г, 4,75 ммоль) в CH2Cl2 добавляли оксалилхлорид (0,62 мл, 7,13 ммоль) при комн. темп. Затем смесь кипятили с обратным холодильником в течение 15 мин, охлаждали до комнатной температуры, концентрировали и затем растворяли в диоксане (10 мл). К этому раствору добавляли азид натрия (0,34 г, 5,23 ммоль) в смеси диоксан/вода (10 мл, 1:1 об./об.) Смесь перемешивали в течение 5 мин, разбавляли EtOAc (50 мл) и промывали водой (50 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенный остаток очищали с применением хроматографии на силикагеле (90% гексанов в EtOAc) с получением промежуточного соединения ацилазида с количественным выходом (1,13 г). Промежуточное соединение ацилазид растворяли в толуоле (50 мл) и перемешивали при 100°С в течение 1 ч. Смесь концентрировали с получением названного соединения с количественным выходом (0,96 г). 1Н ЯМР (400 МГц, ДМСО) δ 7,23 (м, 2Н), 7,15 (м, 2Н), 2,81 (т, J=7,2, 1H), 1,80 (м, 5Н), 1,40 (м, 5Н).
В. 9-(2-Циклогексилфенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. 1-Циклогексил-2-изоцианатобензол (0,96 г, 4,75 ммоль) и диаминомалеонитрил (0,51 г, 4,75 ммоль) объединяли вместе в ацетонитриле (30 мл). Смесь перемешивали 3 дня, после чего промежуточное соединение мочевины обнаружили методом ЖХМС. Раствор концентрировали. К неочищенной мочевине добавляли 3-гидроксибензальдегид (1,45 г, 11,88 ммоль) в МеОН (20 мл) и триэтиламине (1 мл). Смесь перемешивали в течение 11 ч. Осадок отфильтровывали с получением названного соединения (0,559 г, 27% посредством двух стадий). 1Н ЯМР (400 МГц, ДМСО) δ 11,78 (с, 1Н), 9,48 (с, 1Н), 8,41 (с, 1Н), 8,00 (с, 1Н), 7,87 (д, J=8,0, 1H), 7,65 (т, J=2,0, 1H), 7,54 (м, 1Н), 7,39 (дд, J=4,4, 1,2, 2H), 7,22 (т, J=7,6, 1H), 6,81 (дд, J=8,0, 1,6, 1H), 2,32 (т, J=12,0, 1H), 1,71 (м, 3Н), 1,56 (м, 2Н), 1,44 (кв, J=9,2, 2H), 1,17 (кв, J=10,0, 2H), 0,95 (м, 1Н); МС (ESI) m/z 430,1 [M+1]+; т.пл. >270°С.
5.1.126 ПРИМЕР 126
СИНТЕЗ 2-(4-(1Н-ИМИДАЗОЛ-2-ИЛ)ФЕНИЛ)-9-(2-ИЗОПРОПИЛФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (4-(1Н-Имидазол-2-ил)фенил)метанол. 4-(1Н-Имидазол-2-ил)бензойную кислоту (2,05 г, 10,9 ммоль) суспендировали в безводном тетрагидрофуране (60 мл) и охлаждали до -78°С. Добавляли раствор литийалюминийгидрида (2,0 М, 21,8 мл, 43,6 ммоль) и реакционной смеси предоставляли возможность медленно нагреваться до комнатной температуры при перемешивании в течение ночи. Реакционную смесь гасили метанолом, и неочищенный продукт адсорбировали на силикаге. С помощью флэш-хроматографии (20% МеОН в EtOAc) получали названное соединение (1,6 г, 9,19 ммоль, 84%) в виде белого твердого вещества. МС (ESI) m/z 175,1 [M+1]+.
В. 4-(1Н-Имидазол-2-ил)бензальдегид. (4-(1Н-Имидазол-2-ил)фенил)метанол (1,6 г, 9,19 ммоль) растворяли в безводном диметилсульфоксиде (5,0 мл) и метиленхлориде (50 мл). К раствору добавляли пиридинийхлорхромат (4,31 г, 20,0 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали через силикагель и промывали этилацетатом. Органические вещества выливали в воду (150 мл), экстрагировали EtOAc (4×100 мл), сушили объединенные органические слои сульфатом натрия и концентрировали при пониженном давлении с получением названного соединения (1,10 г, 6,39 ммоль, 70%) в виде белого твердого вещества. МС (ESI) m/z 173,1 [M+1]+.
С. 2-(4-(1Н-Имидазол-2-ил)фенил)-9-(2-изопропилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-изопропилфенил)мочевину (см. пример 56.А) (363 мг, 1,35 ммоль), 4-(1Н-имидазол-2-ил)бензальдегид (420 мг, 2,44 ммоль), триэтиламин (0,28 мл, 2,03 ммоль) и метанол (10 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали и очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Летучие вещества удаляли при пониженном давлении, суспендированное твердое вещество обрабатывали гидроксидом аммония с применением ультразвука и фильтровали. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (19 мг, 0,043 ммоль, 3,2%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,94 (с, 1Н), 8,63 (с, 1Н), 8,57 (д, J=8,4, 1H), 8,04 (м, 2Н), 7,76 (с, 1Н), 7,59 (м, 2Н), 7,42 (м, 2Н), 2,75 (гепт, J=7,2, 1H), 1,13 (д, J=6,8, 3H), 1,11 (д, J=6,8, 3H); МС (ESI) m/z 440,1 [M+1]+; т.пл. 340°С (разл.).
5.1.127 ПРИМЕР 127
СИНТЕЗ 2-(1Н-БЕНЗО[D]ИМИДАЗОЛ-1-ИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Метил-2-(1Н-бензо[d]имидазол-1-ил)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат. К раствору 1Н-бензо[b]имидазола (0,354 г, 3,0 ммоль) в тетрагидрофуране (20 мл) добавляли гидрид натрия (0,120 г, 60% в минеральном масле, 30 ммоль). Реакционную смесь перемешивали при 60°С в течение 15 мин. Добавляли метил-2-хлор-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (см. пример 30.А) (1,014 г, 3 ммоль) и полученный в результате раствор перемешивали в течение 4 ч. Реакционную смесь экстрагировали EtOAc (3×25 мл) и промывали насыщенным раствором соли (3×25 мл). Органические слои объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенное вещество подвергали хроматографии на силикагеле (25%-40% EtOAc в гексанах). Концентрированием требуемых фракций получали названное соединение в виде желтого твердого вещества (1,0 г). МС (ESI) m/z 421,4 [M+1]+.
В. Метил-5-амино-2-(1Н-бензо[d]имидазол-1-ил)-6-(2-метоксифениламино)пиримидин-4-карбоксилат. К раствору метил-2-(1Н-бензо[d]имидазол-1-ил)-6-(2-метоксифениламино)-5-нитропиримидин-4-карбоксилат (0,500 г, 1,19 ммоль) в метаноле (20 мл) добавляли палладий на угле (0,025 г, 10%). Реакционную смесь обрабатывали газообразным водородом при 25°С в течение 16 ч. Реакционную смесь фильтровали через целит. Концентрированием фильтрата получали названное соединение (0,300 г, 60%). МС (ESI) m/z 417,5 [M+1]+.
С. Метил-2-(1Н-бензо[d]имидазол-1-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат. Раствор метил-5-амино-2-(1Н-бензо[d]имидазол-1-ил)-6-(2-метоксифениламино)пиримидин-4-карбоксилат (0,300 г, 0,769 ммоль) 1,1'-1,1'-карбонилдиимидазол (0,311 г, 1,922 ммоль) и дихлорметан (10 мл) подвергали взаимодействию, как описано в общем методе F. Реакционную смесь фильтровали с получением названного соединения в виде белого твердого вещества (0,200 г, 62%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 8,78 (с, 1Н), 8,44-8,41 (м, 1Н), 7,76-7,73 (м, 1Н), 7,64-7,55 (м, 2Н), 7,38-7,29 (м, 3Н), 6,22-7,16 (м, 1Н), 4,02 (с, 3Н), 3,79 (с, 3Н); МС (ESI) m/z 417,5 [M+1]+.
D. 2-(1Н-Бензо[d]имидазол-1-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Раствор метил-2-(1Н-бензо[d]имидазол-1-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат (0,200 г, 0,48 ммоль) в метаноле (10 мл) насыщали аммиаком и подвергали взаимодействию, как описано в общем методе G. Реакционную смесь выливали на смесь лед-холодная вода. Сбором полученного в результате твердого вещества фильтрованием получали названное соединение в виде белого твердого вещества, 97,5% чистоты (188 мг, 94%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,91 (с, 1Н), 9,54 (с, 1Н), 8,67 (с, 1Н), 8,08-8,04 (м, 2Н), 7,74-7,72 (м, 1Н), 7,64-7,56 (м, 2Н), 7,37-7,35 (м, 1Н), 7,30-7,25 (м, 1Н), 7,22-7,19 (м, 2Н), 3,78 (с, 3Н); МС (ESI) m/z 401,9 [M+1]+; т.пл. 318-319°С.
5.1.128 ПРИМЕР 128
СИНТЕЗ 2-(1Н-ИМИДАЗО[4,5-B]ПИРИДИН-6-ИЛ)-9-(2-ИЗОПРОПИЛФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(1Н-Имидазо[4,5-b]пиридин-6-ил)-9-(2-изопропилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-изопропилфенил)мочевину (см. пример 56.А) (455 мг, 1,69 ммоль), 1Н-имидазо[4,5-b]пиридин-6-карбальдегид (см. пример 123.В) (300 мг, 2,03 ммоль), триэтиламин (0,35 мл, 2,54 ммоль) и метанол (10 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали и очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1 TFA, в течение 30 мин). Летучие вещества удаляли при пониженном давлении, обрабатывали насыщенным раствором NaHCO3 и экстрагировали этилацетатом (4×75 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученное в результате твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (19 мг, 0,045 ммоль, 2,7%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6 δ 13,23 (с, 1Н), 12,68 (с, 1Н), 11,83 (м, 1Н), 9,30 (м, 1Н), 8,70 (м, 1Н), 8,48 (м, 1Н), 7,98 (м, 1Н), 7,61 (м, 2Н), 7,43 (м, 2Н), 2,77 (гепт, J=7,6, 1H), 1,14 (д, J=7,6, 3Н), 1,12 (д, J=7,6, 3H); МС (ESI) m/z 415,0 [M+1]+; т.пл. 320°С.
5.1.129 ПРИМЕР 129
СИНТЕЗ 9-(2-ИЗОПРОПИЛФЕНИЛ)-8-ОКСО-2-(1Н-ПИРРОЛО[2,3-B]ПИРИДИН-5-ИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (1Н-Пирроло[2,3-b]пиридин-5-ил)метанол. 1Н-Пирроло[2,3-b]пиридин-5-карбоновую кислоту (1,0 г, 5,67 ммоль) суспендировали в безводном тетрагидрофуране (75 мл) и охлаждали до -78°С. Добавляли раствор литийалюминийгидрида (2,0 М, 10,6 мл, 17,0 ммоль) и реакционной смеси предоставляли возможность медленно нагреваться до комнатной температуры при перемешивании в течение ночи. Реакционную смесь гасили метанолом, и летучие вещества удаляли при пониженном давлении. Неочищенное вещество применяли на следующей стадии без дополнительной очистки или характеристики. МС (ESI) m/z 175,1 [M+1]+.
В. 1Н-Пирроло[2,3-b]пиридин-5-карбальдегид. (1Н-Пирроло[2,3-b]пиридин-5-ил)метанол (неочищенный продукт из предыдущей реакции) растворяли в безводном метиленхлориде (50 мл). К раствору добавляли пиридинийхлорхромат (3,70 г, 17,0 ммоль) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали через силикагель и промывали этилацетатом. Органические вещества выливали в воду (150 мл), экстрагировали EtOAc (4×100 мл), объединенные органические слои сушили сульфатом натрия и концентрировали при пониженном давлении с получением названного соединения (0,72 г, 4,93 ммоль, 87% посредством 2 стадий) в виде белого твердого вещества. МС (ESI) m/z 147,1 [M+1]+.
С. 9-(2-Изопропилфенил)-8-оксо-2-(1Н-пирроло[2,3-b]пиридин-5-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-изопропилфенил)мочевину (см. пример 56.А) (762 мг, 2,83 ммоль), 1Н-пирроло[2,3-b]пиридин-5-карбальдегид (0,72 мг, 4,93 ммоль), триэтиламин (0,60 мл, 4,24 ммоль) и метанол (10 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали и очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Летучие вещества удаляли при пониженном давлении, обрабатывали насыщенным раствором NaHCO3 и экстрагировали этилацетатом (3×75 мл). Объединенные органические слои сушили сульфатом натрия и концентрировали при пониженном давлении. Полученное в результате твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (358 мг, 0,87 ммоль, 31%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,62 (с, 1Н), 9,15 (м, 1Н), 9,05 (д, J=2,0, 1H), 8,61 (д, J=1,6, 1H), 7,56 (м, 1Н), 7,51 (дд, J=8,0, 1,6, 1H), 7,45 (м, 1Н), 7,42 (м, 1Н), 7,33 (ддд, J=9,2, 7,6, 1,6, 1H), 7,19 (м, 1Н), 6,47 (дд, J=3,2, 1,2, 1H), 2,78 (гепт, J=7,2, 1H), 1,11 (д, J=6,4, 3H), 1,10 (д, J=6,4, 3H); МС (ESI) m/z 414,1 [M+1]+; т.пл. 380°С (разл.).
5.1.130 ПРИМЕР 130
СИНТЕЗ 2-(1Н-ИМИДАЗО[4,5-B]ПИРИДИН-6-ИЛ)-8-ОКСО-9-(2-(ТРИФТОРМЕТИЛ)ФЕНИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Этил-1-(тетрагидро-2Н-пиран-2-ил)-1Н-имидазо[4,5-b]пиридин-6-карбоксилат. Этил-1Н-имидазо[4,5-b]пиридин-6-карбоксилат (2,0 г, 10,4 ммоль), дигидропиран (1,9 мл, 10,9 ммоль) и толуолсульфоновую кислоту (400 мг, 2,10 ммоль) растворяли в безводном тетрагидрофуране (100 мл) и кипятили с обратным холодильником в течение ночи. Неочищенную реакционную смесь адсорбировали на силикагеле и очищали с применением флэш-хроматографии (60% EtOAc в гекс) с получением белого твердого вещества (2,75 г, 10,0 ммоль). МС (ESI) m/z 276,1 [M+1]+.
В. (1-(Тетрагидро-2Н-пиран-2-ил)-1Н-имидазо[4,5-b]пиридин-6-ил)метанол. Этил-1-(тетрагидро-2Н-пиран-2-ил)-1Н-имидазо[4,5-b]пиридин-6-карбоксилат (2,75 г, 10,0 ммоль) суспендировали в безводном тетрагидрофуране (75 мл) и охлаждали до -78°С. Добавляли раствор литийалюминийгидрида (2,0 М, 18,8 мл, 30,0 ммоль) и предоставляли возможность медленно нагреваться до комнатной температуры при перемешивании в течение ночи. Реакционную смесь гасили метанолом, и летучие вещества удаляли при пониженном давлении. Неочищенный продукт применяли на следующей стадии без дополнительной очистки или характеристики. МС (ESI) m/z 234,1 [M+1]+.
С. 1-(Тетрагидро-2Н-пиран-2-ил)-1Н-имидазо[4,5-b]пиридин-6-карбальдегид. (1-(Тетрагидро-2Н-пиран-2-ил)-1Н-имидазо[4,5-b]пиридин-6-ил)метанол (неочищенный продукт из предыдущей реакции) растворяли в безводном метиленхлориде (50 мл). Добавляли пиридинийхлорхромат (6,46 г, 30,0 ммоль) и раствор перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали через силикагель и промывали этилацетатом. Фильтрат выливали в воду (150 мл) и экстрагировали EtOAc (4×250 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали при пониженном давлении с получением названного соединения (0,69 г, 2,98 ммоль, 30% посредством 2 стадий) в виде белого твердого вещества. МС (ESI) m/z 232,1 [M+1]+.
D. 2-(1Н-Имидазо[4,5-b]пиридин-6-ил)-8-оксо-9-(2-(трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-(трифторметил)фенил)мочевину (см. пример 50.А) (580 мг, 1,99 ммоль), 1-(тетрагидро-2Н-пиран-2-ил)-1Н-имидазо[4,5-b]пиридин-6-карбальдегид (690 мг, 2,98 ммоль), триэтиламин (0,50 мл, 3,0 ммоль) и метанол (10 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи и собирали осадок фильтрованием. 8-Оксо-2-(1-(тетрагидро-2Н-пиран-3-ил)-1Н-имидазо[4,5-b]пиридин-6-ил)-9-(2-(трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид (120 мг, 0,23 ммоль) растворяли в диоксане (2,мл), добавляли раствор 4,0 М HCl/диоксан (4,0 мл) и воду (0,3 мл) и полученный в результате раствор перемешивали при комн. темп. в течение ночи. Реакционную смесь нейтрализовали насыщенным раствором NaHCO3, экстрагировали EtOAc (3×50 мл) и сушили над сульфатом натрия. Неочищенный продукт очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрил + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Летучие вещества удаляли при пониженном давлении, обрабатывали насыщенным раствором NaHCO3 и экстрагировали этилацетатом (3×75 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученное в результате твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (40 мг, 0,091 ммоль, 40%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,95 (с, 1Н), 9,41 (с, 1Н), 8,86 (с, 1Н), 8,72 (с, 1Н), 8,73 (с, 1Н), 8,59 (с, 1Н), 8,05 (м, 2Н), 7,98 (м, 1Н), 7,86 (м, 2Н); МС (ESI) m/z 441,0 [M+1]+; т.пл. 320°С.
5.1.131 ПРИМЕР 131
СИНТЕЗ 8-ОКСО-9-ФЕНИЛ-2-(ПИРИДИН-2-ИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-фенилмочевина. Фенилизоцианат (0,700 г, 5,88 ммоль) и 2,3-диаминомалеонитрил (0,600 г, 5,55 ммоль) подвергали взаимодействию в ацетонитриле согласно общему методу А. Вещество растирали в смеси ацетонитрила и простого диэтилового эфира. Полученное в результате твердое вещество отфильтровали и сушили с получением названного соединения в виде оранжевого твердого вещества (0,95 г, 4,2 ммоль, выход 76%), которое применяли непосредственно на следующей стадии.
В. 8-Оксо-9-фенил-2-(пиридин-2-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-фенилмочевину (0,400 г, 1,76 ммоль) и 2-пиридинкарбоксальдегид (0,414 г, 3,87 ммоль) подвергали взаимодействию согласно общему методу В. Продукт очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (10-60% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Фракции, содержащие требуемое вещество, нейтрализовали водным раствором карбоната натрия и затем концентрировали до меньшего объема. Полученный в результате осадок отфильтровывали и промывали водой. Полученное в результате твердое вещество сушили в высоком вакууме при 60°С с получением названного соединения в виде белого твердого вещества (0,129 г, 0,39 ммоль, выход 22%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,87 (с, 1Н), 8,65 (д, J=4,0, 1H), 8,56 (ушир.с, 2Н), 7,98 (ушир.с, 1Н), 7,94-7,88 (м, 1Н), 7,71 (с, 1Н), 7,69 (с, 1Н), 7,59 (т, J=7,6, 2H), 7,52-7,41 (м, 2Н); МС (ESI) m/z 333,2 [M+1]+; т.пл. 347-349°С.
5.1.132 ПРИМЕР 132
СИНТЕЗ 9-(2-МЕТОКСИФЕНИЛ)-2-(2-(МЕТИЛТИО)-1Н-БЕНЗО[D]ИМИДАЗОЛ-5-ИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (2-(Метилтио)-1Н-бензо[d]имидазол-5-ил)метанол. Этил-2-(метилтио)-1Н-бензо[d]имидазол-5-карбоксилат (4,72 г, 20,0 ммоль) суспендировали в безводном тетрагидрофуране (100 мл) и охлаждали до -78°С. Добавляли раствор литийалюминийгидрида (2,0 М, 37,5 мл, 60,0 ммоль) и предоставляли возможность медленно нагреваться до комнатной температуры при перемешивании в течение ночи. Реакционную смесь гасили метанолом, и неочищенное вещество адсорбировали на силикагеле. С помощью флэш-хроматографии (20% МеОН в EtOAc) выделяли названное соединение (4,36 г, 22,5 ммоль, 95%) в виде белого твердого вещества. МС (ESI) m/z 195,1 [M+1]+.
В. 2-Метилтиобензимидазол-5-карбальдегид. (2-(Метилтио)-1Н-бензо[d]имидазол-5-ил)метанол (4,36 г, 22,5 ммоль) растворяли в безводном метиленхлориде (150 мл). Добавляли пиридинийхлорхромат (9,68 г, 44,9 ммоль) и раствор перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали через силикагель и промывали этилацетатом. Добавляли воду и раствор экстрагировали EtOAc (4×250 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали при пониженном давлении с получением названного соединения (2,10 г, 10,9 ммоль, 48%) в виде белого твердого вещества. МС (ESI) m/z 193,1 [M+1]+.
С. 9-(2-Метоксифенил)-2-(2-(метилтио)-1Н-бензо[d]имидазол-5-ил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (1,87 г, 7,29 ммоль), 2-метилтиобензимидазол-5-карбальдегид (2,10 г, 10,93 ммоль), триэтиламин (1,52 мл, 10,9 ммоль) и метанол (40 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали и 120 мг неочищенного продукта очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Летучие вещества удаляли при пониженном давлении, обрабатывали насыщенным раствором NaHCO3 и экстрагировали этилацетатом (3×50 мл). Органические слои объединяли, сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученное в результате твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (70 мг, 0,16 ммоль, 58%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,65 (с, 1Н), 8,52 (с, 1Н), 8,25 (м, 1Н), 7,96 (с, 1Н), 7,56 (м, 1Н), 7,51 (дд, J=10,0, 2,0, 1H), 7,44 (м, 1Н), 7,31 (д, J=11,2, 1H), 7,17 (м, 1Н), 3,75 (с, 3Н), 2,69 (с, 3Н); МС (ESI) m/z 448,0 [M+1]+; т.пл. 244-245°С.
5.1.133 ПРИМЕР 133
СИНТЕЗ 2-(1Н-ИНДОЛ-5-ИЛ)-9-(2-ИЗОПРОПИЛФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(1Н-Индол-5-ил)-9-(2-изопропилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-изопропилфенил)мочевину (см. пример 56.А) (400 мг, 1,48 ммоль), 1Н-индол-5-карбальдегид (431 мг, 2,97 ммоль), триэтиламин (0,50 мл, 3,70 ммоль) и метанол (10 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали и очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Летучие вещества удаляли при пониженном давлении, обрабатывали насыщенным раствором NaHCO3 и экстрагировали этилацетатом (3×50 мл). Органические вещества сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученное в результате твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (75 мг, 0,18 ммоль, 12%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,68 (с, 1Н), 11,20 (с, 1Н), 8,60 (с, 1Н), 8,48 (м, 1Н), 8,13 (дд, J=10,0, 2,0, 1H), 7,98 (м, 1Н), 7,59 (м, 2Н), 7,41 (д, J=3,6, 1H), 7,35 (м, 3Н), 6,48 (м, 1Н), 2,75 (гепт, J=7,2, 1H), 1,13 (д, J=6,4, 3H), 1,11 (д, J=6,4, 3H). МС (ESI) m/z 413,1 [M+1]+; т.пл. 310°С (разл.).
5.1.134 ПРИМЕР 134
СИНТЕЗ 9-(ЦИКЛОГЕКСИЛМЕТИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (Изоцианатометил)циклогексан. Циклогексилуксусную кислоту (1,5 г, 10,54 ммоль) и N-метилморфолин (1,06, 10,54 ммоль) растворяли в тетрагидрофуране (30 мл) и охлаждали до -10°С. Добавляли по каплям этилхлорформиат (1,25 г, 11,60 ммоль) и перемешивали при -10°С. Затем добавляли азид натрия (1,02 г, 15,85 ммоль) и предоставляли возможность перемешиваться в течение дополнительных 30 мин. Затем раствор разбавляли дихлорметаном, распределяли вместе с водой, сушили над сульфатом магния, фильтровали, и растворитель удаляли при пониженном давлении. Полученное в результате масло очищали с применением хроматографии (Biotage) (0-20% EtOAc в гексанах) с получением названного соединения (0,450 г, 26%).
В. (Z)-1-(2-Амино-1,2-дициановинил)-3-(циклогексилметил)мочевина. 2,3-Диаминомалеонитрил (0,119 г, 1,10 ммоль) и (изоцианатометил)циклогексан (0,140 г, 1,00 ммоль) в тетрагидрофуране (2 мл) подвергали взаимодействию согласно общему методу А. К раствору добавляли дихлорметан и гексаны и полученный в результате осадок отфильтровывали и сушили с получением названного соединения (0,200 г, 47%). МС (ESI) m/z 248,2 [M+1]+.
С. 9-(Циклогексилметил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(циклогексилметил)мочевину (0,200 г, 0,809 ммоль), 3-гидроксибензальдегид (0,197 г, 1,62 ммоль) и триэтиламин (0,4 мл) подвергали взаимодействию согласно общему методу В и очищали с применением препаративной ВЭЖХ с обращенной фазой (5-60% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин) с получением названного соединения (0,011 г, 3,7%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,53 (с, 1Н), 9,53 (с, 1Н), 8,33 (с, 1Н), 8,00 (д, J=7,99, 1H), 7,93 (с, 1Н), 7,89 (с, 1Н), 7,28 (т, J=7,59, 1H), 6,87 (д, J=7,99, 1H), 3,74 (д, J=7,19, 2H), 1,90 (ушир.с, 1Н), 1,64 (м, 5Н), 1,16 (м, 3Н), 1,02 (м, 2Н); МС (ESI) m/z 368,2 [M+1]+; т.пл. 368-370°С.
5.1.135 ПРИМЕР 135
СИНТЕЗ 9-(2,3-ДИГИДРО-1Н-ИНДЕН-1-ИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Метил-2-хлор-6-(2,3-дигидро-1Н-инден-1-иламино)-5-нитропиримидин-4-карбоксилат. Метил-2,6-дихлор-5-нитропиримидин (1,3 г, 5,23 ммоль), диизопропилэтиламин (1,68 г, 13,07 ммоль) и 1-аминоиндан (0,734 г, 5,49 ммоль) подвергали взаимодействию согласно общему методу С и очищали с применением хроматографии (Biotage) (0-60% EtOAc в гексанах) с получением названного соединения (0,595 г, 33%). МС (ESI) m/z 349,3 [M+1]+.
В. Метил-5-амино-2-хлор-6-(2,3-дигидро-1Н-инден-1-иламино)пиримидин-4-карбоксилат. 2-Хлор-6-(2,3-дигидро-1Н-инден-1-иламино)-5-нитропиримидин-4-карбоксилат (0,595 г, 1,71 ммоль), железо (тв) (0,477 г, 8,55 ммоль) и уксусную кислоту (20 мл) подвергали взаимодействию согласно общему методу D2 с получением названного соединения (0,205 г, 38%). МС (ESI) m/z 319,3 [M+1]+.
С. Метил-5-амино-6-(2,3-дигидро-1Н-инден-1-иламино)-2-(3-(триизопропилсилилокси)фенил)пиримидин-4-карбоксилат. Метил-5-амино-2-хлор-6-(2,3-дигидро-1Н-инден-1-иламино)пиримидин-4-карбоксилат (0,205 г, 0,604 ммоль), триизопропил(3-(триметилстаннил)фенокси)силан (0,400 г, 0,966 ммоль) и бисдихлор(трифенилфосфин)палладий(0) (0,135 г, 0,193 ммоль) объединяли в диметилформамиде (4 мл) и нагревали до 100°С. Реакционную смесь подвергали мониторингу путем тонкослойной хроматографии. Как только исходные вещества израсходовались, раствор конденсировали при пониженном давлении и очищали с применением хроматографии (Biotage) (0-40% EtOAc в гексанах) с получением названного соединения (0,192 г, 56%). МС (ESI) m/z 533,2 [M+1]+.
D. Метил-9-(2,3-дигидро-1Н-инден-1-ил)-8-оксо-2-(3-(триизопропилсилилокси)фенил)-8,9-дигидро-7Н-пурин-6-карбоксилат. Метил-5-амино-6-(2,3-дигидро-1Н-инден-1-иламино)-2-(3-(триизопропилсилилокси)фенил)пиримидин-4-карбоксилат (0,192 г, 0,360 ммоль) и 1,1'-1,1'-карбонилдиимидазол (0,467 г, 2,88 ммоль) в дихлорметане (15 мл) подвергали взаимодействию согласно общему методу F и очищали с применением хроматографии (Biotage) (0-45% EtOAc в гексанах) с получением названного соединения (0,171 г, 85%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,85 (с, 1Н), 7,76 (д, J=7,99, 1H), 7,64 (с, 1Н), 7,36 (д, J=7,59, 1H), 7,32 (т, J=7,99, 1H), 7,24 (м, 1Н), 7,12 (с, 1Н), 7,11 (с, 1Н), 6,92 (дд, J=7,99, 2,39, 1H), 6,05 (т, J=7,19, 1H), 3,94 (с, 3Н), 3,33 (с, 9Н), 3,06 (м, 1Н), 2,67 (м, 1Н), 1,23 (м, 3Н), 1,08 (м, 18Н).
Е. 9-(2,3-Дигидро-1Н-инден-1-ил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Метил-9-(2,3-дигидро-1Н-инден-1-ил)-8-оксо-2-(3-(триизопропилсилилокси)фенил)-8,9-дигидро-7Н-пурин-6-карбоксилат (0,180 г, 3,97 ммоль) и газообразный аммиак подвергали взаимодействию в метаноле (10 мл) согласно общему методу G. Через 16 ч раствор конденсировали при пониженном давлении, полученный в результате неочищенный продукт разбавляли тетрагидрофураном (10 мл) и добавляли 1,0 М тетрабутиламмонийфторид в тетрагидрофуране (0,76 мл). Через 2 ч полученный в результате осадок отфильтровывали и промывали тетрагидрофураном, за которым следовали гексаны. Очисткой с применением препаративной ВЭЖХ с обращенной фазой (10-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин) получали названное соединение (0,052 г, 3%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,60 (с, 1Н), 9,45 (с, 1Н), 8,31 (с, 1Н), 7,92 (с, 1Н), 7,98 (д, J=7,99, 1H), 7,71 (с, 1Н), 7,38 (д, J=7,59, 1H), 7,23 (м, 2Н), 7,10 (с, 2Н), 6,83 (дд, J=7,99, 1,99, 1H), 6,06 (т, J=6,39, 1H), 3,40 (м, 1Н), 3,05 (м, 1Н), 2,61 (м, 2Н); МС (ESI) m/z 388,0 [M+1]+; т.пл. 341-343°С.
5.1.136 ПРИМЕР 136
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛ)-9-ИЗОБУТИЛ-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Метил-2-хлор-6-(изобутиламино)-5-нитропиримидин-4-карбоксилат. Метил-2,6-дихлор-5-нитропиримидин (1,3 г, 5,23 ммоль), диизопропилэтиламин (2,3 г, 17,85 ммоль) и изобутиламин (0,457 г, 6,24 ммоль) подвергали взаимодействию согласно общему методу С и очищали с применением хроматографии (Biotage) (0-40% EtOAc в гексанах) с получением названного соединения (1,10 г, 64%). МС (ESI) m/z 289,2 [M+1]+.
В. Метил-5-амино-2-хлор-6-(изобутиламино)пиримидин-4-карбоксилат. Метил-2-хлор-6-(изобутиламино)-5-нитропиримидин-4-карбоксилат (1,10 г, 3,80 ммоль), железо (тверд.) (1,06 г, 19,0 ммоль) и уксусную кислоту (25 мл) подвергали взаимодействию согласно общему методу D2 с получением названного соединения (0,726 г, 74%). МС (ESI) m/z 259,1 [M+1]+, 260,1 [M+2]+.
С. Метил-5-амино-6-(2,3-дигидро-1Н-инден-1-иламино)-2-(3-(триизопропилсилилокси)фенил)пиримидин-4-карбоксилат. Метил-5-амино-2-хлор-6-(изобутиламино)пиримидин-4-карбоксилат (0,285 г, 1,11 ммоль), триизопропил(3-(триметилстаннил)фенокси)силан (0,691 г, 1,67 ммоль) и бисдихлор(трифенилфосфин)палладий(0) (0,311 г, 0,444 ммоль) объединяли в диметилформамиде (4 мл) и нагревали до 100°С. Реакционную смесь подвергали мониторингу путем тонкослойной хроматографии. Как только исходные вещества израсходовались, раствор конденсировали при пониженном давлении и очищали с применением хроматографии (Biotage) (0-40% EtOAc в гексанах) с получением названного соединения (0,300 г, 58%). МС (ESI) m/z 473,6 [M+1]+.
D. Метил-9-изобутил-8-оксо-2-(3-(триизопропилсилилокси)фенил)-8,9-дигидро-7Н-пурин-6-карбоксилат. Метил-5-амино-6-(2,3-дигидро-1Н-инден-1-иламино)-2-(3-(триизопропилсилилокси)фенил)пиримидин-4-карбоксилат (0,300 г, 0,635 ммоль) и 1,1'-1,1'-карбонилдиимидазол (0,411 г, 2,54 ммоль) в дихлорметане (25 мл) подвергали взаимодействию согласно общему методу F. Раствор конденсировали при пониженном давлении и распределяли между водой и этилацетатом (3х). Органические фракции объединяли и сушили над сульфатом магния, фильтровали и растворитель удаляли при пониженном давлении. Неочищенное твердое соединение разбавляли метанолом (15 мл) и обрабатывали ультразвуком. Полученный в результате осадок отфильтровывали с получением названного соединения (0,185 г, 85%). МС (ESI) m/z 268,2 [M+1]+.
Е. 2-(3-Гидроксифенил)-9-изобутил-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Метил-9-изобутил-8-оксо-2-(3-(триизопропилсилилокси)фенил)-8,9-дигидро-7Н-пурин-6-карбоксилат (0,185 г, 3,97 ммоль) растворяли в метаноле (10 мл) и подвергали взаимодействию с газообразным аммиаком согласно общему методу G. Через 16 ч раствор конденсировали при пониженном давлении и полученный в результате неочищенный продукт разбавляли тетрагидрофураном (10 мл), и добавляли 1,0 М тетрабутиламмонийфторид в тетрагидрофуране (0,94 мл). Через два часа полученный в результате осадок отфильтровывали и промывали тетрагидрофураном, за которым следовали гексаны. Осадок растирали с метанолом и дихлорметаном с получением названного соединения (0,061 г, 50%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,54 (с, 1Н), 9,52 (с, 1Н), 8,33 (с, 1Н), 8,0 (д, J=7,59, 1H), 7,93 (с, 1Н), 7,89 (с, 1Н), 7,28 (т, J=7,59, 1H), 6,86 (дд, J=7,99, 2,39, 1H), 3,72 (д, J=7,59, 2H), 2,26 (м, 1Н), 0,923 (д, J=6,39, 6H). МС (ESI) m/z 328,1 [M+1]+; т.пл. 374-376°С.
5.1.137 ПРИМЕР 137
СИНТЕЗ 9-(ТРАНС-4-ГИДРОКСИЦИКЛОГЕКСИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Метил-2-хлор-6-(транс-4-метоксициклогексиламино)-5-нитропиримидин-4-карбоксилат. Метил-2,6-дихлор-5-нитропиримидин (0,934 г, 3,71 ммоль), диизопропилэтиламин (1,43 г, 11,13 ммоль) и транс-4-метоксициклогексанамин (0,613 г, 3,71 ммоль) подвергали взаимодействию согласно общему методу С и очищали с применением хроматографии (Biotage) (0-60% EtOAc в гексанах) с получением названного соединения (0,880 г, 69%). МС (ESI) m/z 345,3 [M+1]+, 346,3 [M+2]+.
В. Метил-5-амино-2-хлор-6-(транс-4-метоксициклогексиламино)пиримидин-4-карбоксилат. Метил-2-хлор-6-(транс-4-метоксициклогексиламино)-5-нитропиримидин-4-карбоксилат (0,880 г, 2,55 ммоль), железо (тв) (0,712 г, 12,75 ммоль) и уксусную кислоту (20 мл) подвергали взаимодействию согласно общему методу D2 с получением названного соединения (0,719 г, 90%). МС (ESI) m/z 315,3 [M+1]+, 316,3 [M+2]+.
С. Метил-5-амино-6-(транс-4-метоксициклогексиламино)-2-(3-(триизопропилсилилокси)фенил)пиримидин-4-карбоксилат. Метил-5-амино-2-хлор-6-(транс-4-метоксициклогексиламино)пиримидин-4-карбоксилат (0,300 г, 0,955 ммоль), триизопропил(3-(триметилстаннил)фенокси)силан (0,591 г, 1,43 ммоль) и бисдихлор(трифенилфосфин)палладий(0) (0,200 г, 0,286 ммоль) объединяли в диметилформамиде (6 мл) и нагревали до 100°С. Реакционную смесь подвергали мониторингу путем тонкослойной хроматографии. Как только исходные вещества израсходовались, раствор конденсировали при пониженном давлении и очищали с применением хроматографии (Biotage) (0-60% EtOAc в гексанах) с получением названного соединения (0,261 г, 56%). МС (ESI) m/z 529,6 [M+1]+.
D. Метил-9-((транс-4-метоксициклогексил)-8-оксо-2-(3-(триизопропилсилилокси)фенил)-8,9-дигидро-7Н-пурин-6-карбоксилат. Метил-5-амино-6-(транс-4-метоксициклогексиламино)-2-(3-(триизопропилсилилокси)фенил)пиримидин-4-карбоксилат (0,261 г, 0,494 ммоль) и 1,1'-1,1'-карбонилдиимидазол (0,640 г, 3,95 ммоль) в дихлорметане (20 мл) подвергали взаимодействию согласно общему методу F и очищали с применением хроматографии (Biotage) (0-65% EtOAc в гексанах) с получением названного соединения (0,206 г, 88%). МС (ESI) m/z 555,0 [M+1]+.
Е. 9-(транс-4-Метоксициклогексил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Метил-9-(транс-4-метоксициклогексил)-8-оксо-2-(3-(триизопропилсилилокси)фенил)-8,9-дигидро-7Н-пурин-6-карбоксилат (0,205 г, 370 ммоль) растворяли в метаноле (15 мл), подвергали взаимодействию с газообразным аммиаком согласно общему методу G. Через 16 ч раствор конденсировали при пониженном давлении и полученный в результате продукт разбавляли тетрагидрофураном (15 мл) и добавляли 1,0 М тетрабутиламмонийфторид в тетрагидрофуране (0,92 мл). Через 2 часа полученный в результате осадок отфильтровывали и промывали тетрагидрофураном, за которым следовали гексаны. Продукт очищали с применением препаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин) с получением названного соединения (0,093 г, 66%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,51 (с, 1Н), 9,55 (с, 1Н), 8,33 (с, 1Н), 8,01 (д, J=7,59, 1H), 7,91 (с, 1Н), 7,89 (с, 1Н), 7,29 (т, J=7,99, 1H), 6,7 (дд, J=7,59, 1,59, 1H), 4,27 (м, 1Н), 3,34 (с, 3Н), 3,31 (с, 1Н), 2,18 (д, J=10,79, 1H), 1,82 (д, J=11,19, 2H), 1,31 (кв, J=12,39, 2H); МС (ESI) m/z 384,4 [M+1]+, т.пл. 355-357°С.
5.1.138 ПРИМЕР 138
СИНТЕЗ 9-(ЦИС-4-ГИДРОКСИЦИКЛОГЕКСИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Метил-2-хлор-6-(цис-4-метоксициклогексиламино)-5-нитропиримидин-4-карбоксилат. Метил-2,6-дихлор-5-нитропиримидин (1,14 г, 4,54 ммоль), диизопропилэтиламин (1,75 г, 13,62 ммоль) и цис-4-метоксициклогексанамин (0,750 г, 4,54 ммоль) подвергали взаимодействию согласно общему методу С и очищали с применением хроматографии (Biotage) (0-60% EtOAc в гексанах) с получением названного соединения (0,712 г, 46%). МС (ESI) m/z 345,3 [M+1]+, 346,3 [M+2]+.
В. Метил-5-амино-2-хлор-6-(цис-4-метоксициклогексиламино)пиримидин-4-карбоксилат. Метил-2-хлор-6-(цис-4-метоксициклогексиламино)-5-нитропиримидин-4-карбоксилат (0,712 г, 2,06 ммоль), железо (тв) (0,577 г, 10,34 ммоль) и уксусную кислоту (25 мл) подвергали взаимодействию согласно общему методу D2 с получением названного соединения (0,567 г, 87%). МС (ESI) m/z 315,3 [M+1]+, 316,3 [M+2]+.
С. Метил-5-амино-6-(цис-4-метоксициклогексиламино)-2-(3-(триизопропилсилилокси)фенил)пиримидин-4-карбоксилат. Метил-5-амино-2-хлор-6-(цис-4-метоксициклогексиламино)пиримидин-4-карбоксилат (0,300 г, 0,955 ммоль), триизопропил(3-(триметилстаннил)фенокси)силан (0,591 г, 1,43 ммоль) и бисдихлор(трифенилфосфин)палладий(0) (0,200 г, 0,286 ммоль) объединяли в диметилформамиде (6 мл) и нагревали до 100°С. Реакционную смесь подвергали мониторингу путем тонкослойной хроматографии. Как только исходные вещества израсходовались, раствор конденсировали при пониженном давлении и очищали с применением хроматографии (Biotage) (0-60% EtOAc в гексанах) с получением названного соединения (0,223 г, 44%). МС (ESI) m/z 529,6 [M+1]+.
D. Метил-9-(цис-4-метоксициклогексил)-8-оксо-2-(3-(триизопропилсилилокси)фенил)-8,9-дигидро-7Н-пурин-6-карбоксилат. Метил-5-амино-6-(цис-4-метоксициклогексиламино)-2-(3-(триизопропилсилилокси)фенил)пиримидин-4-карбоксилат (0,223 г, 0,422 ммоль) и 1,1'-1,1'-карбонилдиимидазол (0,547 г, 3,38 ммоль) в дихлорметане (20 мл) подвергали взаимодействию согласно общему методу F и очищали с применением хроматографии (Biotage) (0-65% EtOAc в гексанах) с получением названного соединения (0,210 г, 90%). МС (ESI) m/z 555,5 [M+1]+.
Е. 9-(цис-4-Метоксициклогексил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Метил-9-(цис-4-метоксициклогексил)-8-оксо-2-(3-(триизопропилсилилокси)фенил)-8,9-дигидро-7Н-пурин-6-карбоксилат (0,210 г, 377 ммоль) растворяли в метаноле (15 мл), подвергали взаимодействию с газообразным аммиаком согласно общему методу G. Через 16 ч раствор конденсировали при пониженном давлении и полученный в результате неочищенный продукт разбавляли тетрагидрофураном (15 мл) и добавляли 1,0 М тетрабутиламмонийфторид в тетрагидрофуране (0,92 мл). Через 2 ч полученный в результате осадок отфильтровывали и промывали тетрагидрофураном, за которым следовали гексаны. Продукт очищали с применением препаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин) с получением названного соединения (0,093 г, 64%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,48 (с, 1Н), 9,47 (с, 1Н), 8,32 (с, 1Н), 8,03 (д, J=7,59, 1H), 7,92 (с, 2Н), 7,28 (т, J=7,59, 1H), 6,86 (дд, J=7,99, 2,39, 1H), 4,30 (м, 1Н), 3,49 (с, 1Н), 3,34 (с, 3Н), 2,71 (м, 2Н), 2,06 (с, 1Н), 2,03 (с, 1Н), 1,53 (м, 4Н). МС (ESI) m/z 384,4 [M+1]+, т.пл. 345-347°С.
5.1.139 ПРИМЕР 139
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-9-(5,6,7,8-ТЕТРАГИДРОНАФТАЛИН-1-ИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-(5,6,7,8-тетрагидронафталин-1-ил)мочевина. 5-Изоцианато-1,2,3,4-тетрагидронафталин (0,34 мл, 2,16 ммоль) и 2,3-диаминомалеонитрил (0,234 г, 2,16 ммоль) перемешивали вместе в ацетонитриле (10 мл). Названное соединение осаждали из раствора и собирали фильтрованием. МС (ESI) m/z 321,4 [M+1]+.
В. 2-(3-Гидроксифенил)-8-оксо-9-(5,6,7,8-тетрагидронафталин-1-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(5,6,7,8-тетрагидронафталин-1-ил)мочевину (0,59 г, 2,16 ммоль) смешивали вместе с 3-гидроксибензальдегидом (0,55 г, 5,4 ммоль) в смеси метанол/триэтиламин (9:1 об./об., 15 мл). Через 24 ч смесь концентрировали и затем подвергали препаративной ВЭЖХ (30-80% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Требуемые фракции объединяли и концентрировали с получением названного соединения. Полученное в результате вещество сушили в вакууме в течение ночи с получением названного соединения в виде не совсем белого твердого вещества, 97,5% чистоты (15 мг, 2%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,77 (с, 1Н), 9,50 (с, 1Н), 8,39 (с, 1Н), 7,98 (с, 1Н), 7,89 (д, J=4,8, 1H), 7,67 (т, J=2,0, 2H), 7,26 (м, 4Н), 6,81 (дд, J=8,0, 2,0, 1H), 2,86 (т, J=5,6, 2H), 2,45 (м, 2Н), 1,75 (м, 2Н), 1,67 (м, 2Н); МС (ESI) m/z 402,3 [M+1]+; т.пл. >250°С.
5.1.140 ПРИМЕР 140
СИНТЕЗ 2-(4-(1Н-1,2,4-ТРИАЗОЛ-3-ИЛ)ФЕНИЛ)-9-ЦИКЛОГЕКСИЛ-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(4-(1Н-1,2,4-Триазол-3-ил)фенил)-9-циклогексил-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-циклогексилмочевину (см. пример 26.А) (200 мг, 0,86 ммоль), 4-(1Н-1,2,4-триазол-3-ил)бензальдегид (см. 108.В) (300 мг, 1,72 ммоль), триэтиламин (0,18 мл, 1,29 ммоль) и метанол (10 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали и очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Летучие вещества удаляли при пониженном давлении, обрабатывали насыщенным раствором NaHCO3 и экстрагировали этилацетатом (3×50 мл). Объединенные органические фракции сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученное в результате твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (75 мг, 0,18 ммоль, 12%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,56 (с, 1Н), 8,66 (м, 2Н), 8,49 (м, 1Н), 8,14 (д, J=8,0, 1H), 7,95 (м, 1Н), 4,30 (м, 1Н), 2,67 (м, 1Н), 2,37 (м, 2Н), 2,32 (м, 1Н), 1,88 (м, 2Н), 1,78 (м, 2Н), 1,42 (м, 2Н); МС (ESI) m/z 404,1 [M+1]+; т.пл. 375°С (разл.).
5.1.141 ПРИМЕР 141
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛ)-9-(1Н-ИНДОЛ-4-ИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 1-Бензолсульфонил-1Н-индол-4-иламин. 4-Нитроиндол (1,07 г, 6,6 ммоль) растворяли в ацетонитриле (12 мл) и добавляли диизопропилэтиламин (1,2 мл, 7,92 ммоль). Раствор нагревали до 80°С и затем добавляли бензолсульфонилхлорид (0,93 мл, 7,26 ммоль). Смесь перемешивали при 80°С в течение 1,5 ч. Реакционную смесь охлаждали, разбавляли водой (20 мл) и фильтровали. Неочищенный 1-бензолсульфонил-4-нитро-1Н-индол промывали водой (10 мл) и затем метанолом (5 мл). Затем неочищенный 1-бензолсульфонил-4-нитро-1Н-индол помещали в метанол (50 мл). К этой суспензии добавляли 10% Pd/C (0,5 г) и формиат аммония (1,0 г, 15,6 ммоль). Смесь перемешивали при кипячении с обратным холодильником в течение 1,5 ч, после чего, как показано методом ЖХМС, реакция завершалась. (МС (ESI) m/z 273,1 [M+1]+). Реакционную смесь концентрировали и неочищенный остаток подвергали хроматографии на силикагеле (90% гексанов в EtOAc). Концентрированием требуемых фракций получали названное соединение (1,42 г, 5,22 ммоль, 79%).
В. Метил-5-амино-2-хлор-6-(1-(фенилсульфонил)-1Н-индол-4-иламино)пиримидин-4-карбоксилат. К раствору метил-2,4-дихлор-5-нитропиримидин-6-карбоксилата (2,0 г, 7,9 ммоль) в тетрагидрофуране (20 мл) и диизопропиламине (2,3 мл, 13,05 ммоль) добавляли амин (1,42 г, 5,22 ммоль) в тетрагидрофуране (5 мл) при -78°С. После нагревания до комн. темп. наблюдали образование только требуемого аддукта (МС (ESI) m/z 487,9 [M+1]+). Реакционную смесь разбавляли EtOAc (100 мл) и промывали 5% HCl (водный 100 мл). Слой EtOAc отделяли, сушили (Na2SO4), фильтровали и концентрировали. Неочищенный сложный метиловый эфир 6-(1-бензолсульфонил-1Н-индол-4-иламино)-2-хлор-5-нитропиримидин-4-карбоновой кислоты подвергали хроматографии на силикагеле (9:1 гекс. в EtOAc) с получением 1,32 г (2,71 ммоль, 52%), после концентрирования требуемых фракций. Затем метиловый эфир 6-(1-бензолсульфонил-1Н-индол-4-иламино)-2-хлор-5-нитропиримидин-4-карбоновой кислоты (1,32 г, 2,71 ммоль) растворяли в АсОН (15 мл). К этому раствору добавляли Fe (твердое, 1,4 г). Смесь перемешивали при 60°С в течение 1 ч, после чего анализ ЖХМС показал образование требуемого продукта восстановления (МС (ESI) m/z 458,1 [M+1]+). Реакционную смесь фильтровали и затем концентрировали. Неочищенный остаток подвергали хроматографии на силикагеле (4:1 гекс. в EtOAc). Концентрированием требуемых фракций получали названное соединение (0,85 г, 1,86 ммоль, 69%).
С. Метил-5-амино-2-(3-гидроксифенил)-6-(1-(фенилсульфонил)-1Н-индол-4-иламино)пиримидин-4-карбоксилат. Метил-5-амино-2-хлор-6-(1-(фенилсульфонил)-1Н-индол-4-иламино)пиримидин-4-карбоксилат (0,31 г, 0,68 ммоль), 3-гидроксифенилбороновую кислоту (0,14 г, 1,0 ммоль), ацетат палладия(II) (30 мг, 0,14 ммоль), дициклогексил(2',6'-диметоксибифенил-2-ил)фосфин (60 мг, 0,14 ммоль) и фосфат калия (0,36 г, 2,1 ммоль) вместе подвергали взаимодействию согласно общему методу Е. Реакционную смесь разбавляли EtOAc (40 мл) и промывали 5% HCl (водный, 40 мл). Органический слой отделяли, сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенный остаток подвергали хроматографии на силикагеле (50% гексанов в EtOAc). Концентрированием требуемых фракций получали названное соединение (0,1 г, 0,19 ммоль, 28%). МС (ESI) m/z 516,3 [M+1]+.
D. 5-Амино-2-(3-гидроксифенил)-6-(1-фенилсульфонил)-1Н-индол-4-иламино)пиримидин-4-карбоксамид. Метил-5-амино-2-(3-гидроксифенил)-6-(1-(фенилсульфонил)-1Н-индол-4-иламино)пиримидин-4-карбоксилат (0,1 г, 0,19 ммоль) перемешивали в 7н NH3/MeOH (20 мл) при 55°С в течение 24 ч. Анализ ЖХМС показал образование только требуемого продукта. Смесь концентрировали с получением названного соединения, 50 мг, 0,1 ммоль, 50%. МС (ESI) m/z 501,5 [M+1]+.
Е. 2-(3-Гидроксифенил)-9-(1Н-индол-4-ил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. К раствору 5-амино-2-(3-гидроксифенил)-6-(1-фенилсульфонил)-1Н-индол-4-иламино)пиримидин-4-карбоксамид (50 мг, 0,1 ммоль) в CH2Cl2 (4 мл) и EtOAc (1 мл) добавляли 1,1'-1,1'-карбонилдиимидазол (0,1 г, 0,6 ммоль). Смесь перемешивали при 55°С в течение 1,5 ч, после чего анализ ЖХМС показал образование продукта требуемой массы, МС (ESI) m/z 527,1 [M+1]+. Затем реакционную смесь концентрировали и полученный в результате осадок помещали в метанол (3 мл). Добавляли метоксид натрия (25% в МеОН, 0,5 мл). Смесь перемешивали при 70°С в течение 1 ч, после чего анализ ЖХМС показал завершение реакции. Для нейтрализации смеси добавляли АсОН. Затем смесь подвергали полупрепаративной ВЭЖХ (10-70% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Чистые фракции концентрировали с получением названного соединения 98,3% чистоты (15 мг, 39% посредством 2 стадий). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,75 (с, 1Н), 11,41 (с, 1Н), 9,41 (с, 1Н), 8,34 (с, 1Н), 7,98 (с, 1Н), 7,84 (дт, J=8,0, 1,2, 1H), 7,61 (т, J=2,4, 1H), 7,58 (д, J=8,0, 1H), 7,41 (т, J=2,8, 1H), 7,28 (т, J=8,0, 1H), 7,18 (м, 2Н), 6,78 (дкв, J=7,6, 1,2, 1H), 6,23 (м, 1Н); МС (ESI) m/z 387,3 [M+1]+; т.пл. >260°С.
5.1.142 ПРИМЕР 142
СИНТЕЗ 9-(2-ФТОР-3-МЕТОКСИФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-Фтор-1-изоцианато-3-метоксибензол. 2-Фтор-3-метоксибензойную кислоту (0,61 г, 3,59 ммоль) растворяли в CH2Cl2 (10 мл) и ДМФА (0,5 мл). Медленно добавляли оксалилхлорид (0,41 мл, 4,7 ммоль). Смесь перемешивали при комнатной температуре в течение 15 мин. Затем смесь концентрировали и преобразовывали в диоксане (10 мл). К хлорангидриду кислоты добавляли при 0°С азид натрия (0,26 г, 4,0 ммоль) в смеси вода/диоксан (1:1 об./об., 10 мл). Смеси предоставляли возможность нагреваться до комн. темп. в течение 1 ч. Затем реакционную смесь разбавляли EtOAc (50 мл) и водой (50 мл). Смесь встряхивали и разделяли. Органический слой сушили, фильтровали и концентрировали. Неочищенный ацилазид фильтровали через слой силикагеля (90% гекс в EtOAc). Фильтрат концентрировали с получением промежуточного продукта 2-фтор-3-метоксибензоилазида. 2-Фтор-3-метоксибензоилазид растворяли в толуоле (50 мл) и перемешивали при 100°С в течение 1 ч. Смесь концентрировали с получением названного соединения с количественным выходом.
В. 9-(2-Фтор-3-метоксифенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. 2-Фтор-1-изоцианато-3-метоксибензол (0,6 г, 3,59 ммоль) подвергали взаимодействию с 2,3-диаминомалеонитрилом (0,39 г, 3,59 ммоль) согласно общему методу А. Промежуточное соединение мочевины осаждали из раствора и фильтровали. Промежуточное соединение мочевины подвергали взаимодействию с 3-гидроксибензальдегидом согласно общему методу В. Названное соединение осаждали из раствора и фильтровали с получением 0,184 г (13% посредством 2 стадий, 98,9% чистоты). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,89 (с, 1Н), 9,50 (с, 1Н), 8,38 (с, 1Н), 8,42 (с, 1Н), 8,01 (с, 1Н), 7,90 (д, J=7,6, 1H), 7,70 (т, J=1,8, 1H), 7,40 (д, J=4,8, 1H), 7,38 (д, J=5,2, 1H), 7,25 (м, 2Н), 6,84 (дд, J=8,0, 1,6, 1Н), 3,95 (с, 3Н); МС (ESI) m/z 396,4; [M+1]+ т.пл. >260°С.
5.1.143 ПРИМЕР 143
СИНТЕЗ 9-(2-ФТОР-5-МЕТОКСИФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-фтор-5-метоксифенил)мочевина. 2-Фтор-5-метоксибензойную кислоту (0,62 г, 3,62 ммоль) растворяли в CH2Cl2 (15 мл) и ДМФА (0,5 мл). Добавляли по каплям оксалилхлорид (0,38 мл, 4,32 ммоль). Смесь перемешивали в течение следующих 15 мин. Реакционную смесь концентрировали и помещали в диоксан (10 мл). К хлорангидриду кислоты добавляли при 0°С азид натрия (0,26 г, 3,96 ммоль) в смеси вода/диоксан (1:1 об./об., 10 мл). Смесь перемешивали в течение 10 мин. Реакционную смесь распределяли между EtOAc и водой. Слои встряхивали и разделяли. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенный ацилазид фильтровали через слой силикагеля (90% гексанов в EtOAc). Фильтрат концентрировали. Очищенный ацилазид растворяли в толуоле и перемешивали при 100°С в течение 1 ч. Толуольный раствор концентрировали с получением 1-фтор-2-изоцианато-4-метоксибензола с количественым выходом. 1-Фтор-2-изоцианато-4-метоксибензол (0,6 г, 3,62 ммоль) подвергали взаимодействию с 2,3-диаминомалеонитрилом (0,39 г, 3,62 ммоль) согласно общему методу А с получением названного соединения. МС (ESI) m/z 276,4 [M+1]+.
В. 9-(2-Фтор-5-метоксифенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-фтор-5-метоксифенил)мочевину подвергали взаимодействию с 3-гидроксибензальдегидом согласно общему методу В с получением названного соединения 98,6% чистоты (94 мг, 0,24 ммоль, 7% посредством 2 стадий). 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,50 (с, 1Н), 8,46 (с, 1Н), 7,99 (с, 1Н), 7,90 (д, J=7,6, 1H), 7,72 (с, 1Н), 7,46 (т, J=9,2, 1H), 7,29 (м, 1Н), 7,24 (т, J=8,0, 1H), 7,18 (м, 1Н), 6,83 (д, J=8,0, 1H), 3,80 (с, 3Н); МС (ESI) m/z 396,4 [M+1]+; т.пл. >260°С.
5.1.144 ПРИМЕР 144
СИНТЕЗ 9-ЦИКЛОГЕКСИЛ-2-(1Н-ИМИДАЗО[4,5-B]ПИРИДИН-6-ИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 9-Циклогексил-2-(1Н-имидазо[4,5-b]пиридин-6-ил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-циклогексилмочевину (см. пример 26.А) (215 мг, 0,93 ммоль), 1-(тетрагидро-2Н-пиран-2-ил)-1Н-имидазо[4,5-b]пиридин-6-карбальдегид (см. пример 130.С) (322 мг, 1,39 ммол) и триэтиламин (0,20 мл, 1,39 ммол) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи и полученный в результате осадок собирали фильтрованием. Раствор 5-(1-(2Н-3,4,5,6-тетрагидропиран-2-ил)имидазо[4,5-b]пиридин-6-ил)-3-циклогексил-2-оксо-4-имидазолино[4,5-b]пиридин-7-карбоксамид (316 мг, 0,68 ммоль) в диоксане (2 мл) обрабатывали раствором 4,0 М HCl/диоксан (4,0 мл) и водой (0,3 мл) и перемешивали при комнатной темп. в течение ночи. Реакционную смесь нейтрализатовали насыщенным раствором NaHCO3, экстрагировали EtOAc (3×50 мл) и сушили над сульфатом натрия. Неочищенный продукт очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Летучие вещества удаляли при пониженном давлении, обрабатывали насыщенным раствором NaHCO3 и экстрагировали этилацетатом (3×75 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученное в результате твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (40 мг, 0,091 ммоль, 40%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,49 (с, 1Н), 8,97 (с, 1Н), 8,83 (с, 1Н), 8,49 (с, 1Н), 7,74 (м, 1Н), 4,26 (м, 1Н), 2,67 (м, 1Н), 2,37 (м, 2Н), 2,32 (м, 1Н), 1,88 (м, 2Н), 1,72 (м, 2Н), 1,41 (м, 2Н). МС (ESI) m/z 378,0 [M+1]+; т.пл. 320°С (разл.).
5.1.145 ПРИМЕР 145
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-9-(ТЕТРАГИДРО-2Н-ПИРАН-4-ИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Метил-2-хлор-5-нитро-6-(тетрагидро-2Н-пиран-4-иламино)пиримидин-4-карбоксилат. Метил-2,6-дихлор-5-нитропиримидин (2,49 г, 9,89 ммоль), диизопропилэтиламин (3,18 г, 24,72 ммоль) и тетрагидро-2Н-пиран-4-амин (1,00 г, 9,89 ммоль) подвергали взаимодействию согласно общему методу С и очищали путем хроматографии (Biotage) (0-50% EtOAc в гексанах) с получением названного соединения (2,01 г, 33%). МС (ESI) m/z 317,2 [M+1]+.
В. Метил-5-амино-2-хлор-6-(тетрагидро-2Н-пиран-4-иламино)пиримидин-4-карбоксилат. Метил-2-хлор-5-нитро-6-(тетрагидро-2Н-пиран-4-иламино)пиримидин-4-карбоксилат (2,01 г, 6,36 ммоль), железо (тверд.) (2,48 г, 44,52 ммоль) и уксусную кислоту (35 мл) подвергали взаимодействию согласно общему методу D2 и распределяли между бикарбонатом натрия и этилацетатом (3х) с получением названного соединения (1,60 г, 88%). МС (ESI) m/z 287,3 [M+1]+.
С. Метил-5-амино-2-(3-(трет-бутилдифенилсилилокси)фенил)-6-(тетрагидро-2Н-пиран-4-иламино)пиримидин-4-карбоксилат. Метил-5-амино-2-хлор-6-(тетрагидро-2Н-пиран-4-иламино)пиримидин-4-карбоксилат (0,400 г, 1,39 ммоль), триизопропил-(3-(триметилстаннил)фенокси)силан (0,810 г, 1,95 ммоль) и бисдихлор(трифенилфосфин)палладий(0) (0,292 г, 0,410 ммоль) объединяли в диметилформамиде (6 мл) и нагревали до 100°С. Реакционную смесь подвергали мониторингу путем тонкослойной хроматографии. Как только исходные вещества израсходовались, раствор конденсировали при пониженном давлении и очищали путем хроматографии (Biotage) (0-50% EtOAc в гексанах) с получением названного соединения (0,361 г, 51%). МС (ESI) m/z 501,5 [M+1]+.
D. Метил-2-(3-(трет-бутилдифенилсилилокси)фенил)-8-оксо-9-(тетрагидро-2Н-пиран-4-ил)-8,9-дигидро-7Н-пурин-6-карбоксилат. Метил-5-амино-2-(3-(трет-бутилдифенилсилилокси)фенил)-6-(тетрагидро-2Н-пиран-4-иламино)пиримидин-4-карбоксилат (0,361 г, 0,722 ммоль) и 1,1'-1,1'-карбонилдиимидазол (0,702 г, 4,33 ммоль) в дихлорметане (15 мл) подвергали взаимодействию согласно общему методу F и очищали путем хроматографии (Biotage) (0-75% EtOAc в гексанах) с получением названного соединения (0,270 г, 71%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,80 (с, 1Н), 7,96 (с, 2Н), 7,41 (т, J=8,39, 1H), 7,00 (дд, J=7,99, 2,39, 1H), 4,51 (м, 1Н), 4,01 (дд, J=11,19, 3,59, 2H), 3,94 (с, 3Н), 3,48 (т, J=11,99, 2Н), 2,65 (м, 2Н), 1,75 (д, 9,59, 2Н), 1,33 (м, 3Н), 1,10 (д, J=3,59, 18H).
Е. Метил-2-(3-гидроксифенил)-8-оксо-9-(тетрагидро-2Н-пиран-4-ил)-8,9-дигидро-7Н-пурин-6-карбоксилат. Метил-2-(3-(трет-бутилдифенилсилилокси)фенил)-8-оксо-9-(тетрагидро-2Н-пиран-4-ил)-8,9-дигидро-7Н-пурин-6-карбоксилат (0,270 г, 0,513 ммоль) растворяли в тетрагидрофуране (10 мл) и к раствору добавляли тетрабутиламмонийфторид на силикагеле (0,410 г, 0,615 ммоль). Раствор перемешивали при окружающей температуре в течение двух часов. Методом ЖХМС подтвердили образование продукта и отсутствие исходных веществ. Раствор фильтровали и конденсировали с получением неочищенного названного соединения (0,110 г, 58%), которое применяли на следующей стадии без дополнительной очистки или характеристики.
F. 2-(3-Гидроксифенил)-8-оксо-9-(тетрагидро-2Н-пиран-4-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид. Метил-2-(3-гидроксифенил)-8-оксо-9-(тетрагидро-2Н-пиран-4-ил)-8,9-дигидро-7Н-пурин-6-карбоксилат (0,103 г, 0,277 ммоль) и газообразный аммиак подвергали взаимодействию в метаноле согласно общему методу G. Через 16 часов методом ЖХМС подтвердили образование продукта, и раствор конденсировали при пониженном давлении с получением названного соединения (0,080 г, 81%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,54 (с, 1Н), 9,57 (с, 1Н), 8,35 (с, 1Н), 7,99 (д, J=8,4, 2H), 7,92 (с, 1Н), 7,88 (с, 1Н), 7,29 (т, J=7,8, 2H), 6,87 (д, J=7,8, 1H), 4,51 (м, 1Н), 4,02 (д, J=7,8, 2H), 3,4 (т, J=12,00, 2H), 1,72 (д, J=9,9, 2Н), МС (ESI) m/z 356,5 [M+1]+; т.пл. 361-363°С.
5.1.146 ПРИМЕР 146
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-9-((ТЕТРАГИДРО-2Н-ПИРАН-4-ИЛ)МЕТИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Метил-2-хлор-5-нитро-6-((тетрагидро-2Н-пиран-4-ил)метиламино)пиримидин-4-карбоксилат. Метил-2,6-дихлор-5-нитропиримидин (2,18 г, 8,68 ммоль), диизопропилэтиламин (2,8 г, 21,7 ммоль) и (тетрагидро-2Н-пиран-4-ил)метанамин (1,00 г, 8,68 ммоль) подвергали взаимодействию согласно общему методу С и очищали путем хроматографии (Biotage) (0-50% EtOAc в гексанах) с получением названного соединения (2,14 г, 75%). МС (ESI) m/z 331,3 [M+1]+.
В. Метил-5-амино-2-хлор-6-((тетрагидро-2Н-пиран-4-ил)метиламино)пиримидин-4-карбоксилат. Метил-2-хлор-5-нитро-6-((тетрагидро-2Н-пиран-4-ил)метиламино)пиримидин-4-карбоксилат (2,14 г, 6,45 ммоль), железо (тверд.) (2,50 г, 45,39 ммоль) и уксусную кислоту (35 мл) подвергали взаимодействию согласно общему методу D2 и распределяли между бикарбонатом натрия и этилацетатом (3х) с получением названного соединения (1,69 г, 87%), которое применяли на следующей стадии без дополнительной очистки или характеристики. МС (ESI) m/z 301,4 [M+1]+.
С. Метил-5-амино-2-(3-(трет-бутилдифенилсилилокси)фенил)-6-((тетрагидро-2Н-пиран-4-ил)метиламино)пиримидин-4-карбоксилат. Метил-5-амино-2-хлор-6-((тетрагидро-2Н-пиран-4-ил)метиламино)пиримидин-4-карбоксилат (0,400 г, 1,33 ммоль), триизопропил(3-(триметилстаннил)фенокси)силан (0,772 г, 1,86 ммоль) и бисдихлор(трифенилфосфин)палладий(0) (0,280 г, 0,400 ммоль) объединяли в диметилформамиде (6 мл) и нагревали до 100°С. Реакционную смесь подвергали мониторингу путем тонкослойной хроматографии. Как только исходные вещества израсходовались, раствор конденсировали при пониженном давлении и очищали путем хроматографии (Biotage) (0-50% EtOAc в гексанах) с получением названного соединения (0,276 г, 40%). МС (ESI) m/z 515,6 [M+1]+.
D. Метил-2-(3-(трет-бутилдифенилсилилокси)фенил)-8-оксо-9-((тетрагидро-2Н-пиран-4-ил)метил)-8,8-дигидро-7Н-пурин-6-карбоксилат. Метил-5-амино-2-(3-(трет-бутилдифенилсилилокси)фенил)-6-((тетрагидро-2Н-пиран-4-ил)метиламино)пиримидин-4-карбоксилат (0,276 г, 0,536 ммоль) и 1,1'-1,1'-карбонилдиимидазол (0,522 г, 3,22 ммоль) в дихлорметане (15 мл) подвергали взаимодействию согласно общему методу F и очищали путем хроматографии (Biotage) (0-75% EtOAc в гексанах) с получением названного соединения (0,202 г, 70%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,79 (с, 1Н), 7,98 (с, 1Н), 7,96 (с, 1Н), 7,40 (т, J=8,39, 1H), 7,00 (д, J=7,99, 1H), 3,94 (с, 3Н), 3,8 (м, 4Н), 3,24 (т, J=10,79, 3Н), 2,15 (м, 1Н), 1,57 (д, J=12,39, 2H), 1,31 (м, 5Н), 1,11 (д, J=7,59, 18Н).
Е. Метил-2-(3-гидроксифенил)-8-оксо-9-((тетрагидро-2Н-пиран-4-ил)метил)-8,9-дигидро-7Н-пурин-6-карбоксилат. Метил-2-(3-(трет-бутилдифенилсилилокси)фенил)-8-оксо-9-((тетрагидро-2Н-пиран-4-ил)метил)-8,9-дигидро-7Н-пурин-6-карбоксилат (0,202 г, 0,367 ммоль) растворяли в тетрагидрофуране (10 мл) и к раствору добавляли тетрабутиламмонийфторид на силикагеле (0,294 г, 0,404 ммол). Раствор перемешивали при окружающей температуре в течение 2 ч. Методом ЖХМС подтвердили образование продукта и отсутствие исходных веществ. Раствор фильтровали и конденсировали с получением неочищенного названного соединения (0,110 г, 58%).
F. 2-(3-Гидроксифенил)-8-оксо-9-((тетрагидро-2Н-пиран-4-ил)метил)-8,9-дигидро-7Н-пурин-6-карбоксамид. Метил-2-(3-гидроксифенил)-8-оксо-9-((тетрагидро-2Н-пиран-4-ил)метил)-8,9-дигидро-7Н-пурин-6-карбоксилат (0,083 г, 0,215 ммол) и газообразный аммиак подвергали взаимодействию в метаноле согласно общему методу G. Через 16 ч методом ЖХМС подтвердили образование продукта, и раствор конденсировали при пониженном давлении с получением названного соединения (0,063 г, 81%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,53 (с, 1Н), 9,51 (с, 1Н), 8,31 (с, 1Н), 8,02 (д, J=7,8, 1H), 7,90 (с, 2Н), 7,28 (т, J=8,10, 1H), 6,84 (д, J=8,10, 1H), 3,81 (м, 4Н), 3,31 (с, 1Н), 3,25 (м, 3Н), 2,15 (м, 1Н), 1,55 (д, J=10,8, 2H), 1,33 (м, 2Н); МС (ESI) m/z 356,5 [M+1]+; т.пл. 363-365°С.
5.1.147 ПРИМЕР 147
СИНТЕЗ 9-(2-ЦИКЛОПЕНТИЛФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. N-(2-(Циклопент-2-енил)фенил)ацетамид. 2-Йоданилин (3,07 г, 14 ммол), уксусный ангидрид (1,46 мл, 15,4 ммоль) и триэтиламин (5,9 мл) объединяли в хлороформе (20 мл). Смесь перемешивали в течение ночи при комнатной температуре. Смесь концентрировали и очищали на силикагелевой колонке (15% этилацетата в гексанах) с получением ацилированного анилина (2,91 г, 11,1 ммоль, 80%), который применяли на следующей стадии реакции. N-Ацетил-2-йоданилин (2,91 г, 11,1 ммоль) объединяли с циклопентеном (4,9 мл, 55 ммоль), ацетатом палладия(II) (0,5 г), тетрабутиламмонийхлоридом (3,06 г), трифенилфосфином (0,58 г) и ацетатом калия (3,25 г) в ДМФА (25 мл). Смесь продували азотом и затем перемешивали при 100°С в течение 2 ч. Смесь разбавляли этилацетатом и водой и слои встряхивали. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенный остаток очищали с применением хроматографии на силикагеле (20% этилацетата в гексанах). Концентрированием требуемых фракций получали названное соединение (1,45 г, 65%). 1Н ЯМР (300 МГц, CDCl3) δ 7,83 (д, J=6,6, 1H), 7,41 (ушир.с, 1Н), 7,07-7,26 (м, 3Н), 6,07 (м, 1Н), 5,85 (м, 1Н), 4,00 (м, 1Н), 2,38-2,54 (м, 4Н), 2,15 (с, 3Н); МС (ESI) m/z 202,3 [M+1]+.
В. 2-Циклопентиланилин. N-(2-(Циклопент-2-енил)фенил)ацетамид (1,45 г, 7,2 ммоль), бикарбонат аммония (1,36 г, 21 ммоль) и палладий на угле (0,4 г) объединяли в метаноле (20 мл). Смесь перемешивали при кипячении с обратным холодильником в течение 2 ч. Смесь фильтровали и затем концентрировали. Неочищенный остаток растворяли в этаноле (20 мл) и гидроксиде калия (водн. 4,5 М, 20 мл). Смесь перемешивали при 100°С в течение 24 ч. Смесь концентрировали и разбавляли водой (100 мл). Смесь промывали этилацетатом (2×100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенный остаток очищали хроматографией на силикагеле (25% этилацетата в гексанах). Концентрированием требуемых фракций получали названное соединение (0,3 г, 1,86 ммоль, 25%). МС (ESI) m/z 204,4 [M+1]+.
С. 2-Циклопентилбензойная кислота. 2-Циклопентиланилин (0,64 г, 3,98 ммоль) растворяли в конц. хлористоводородной кислоте (водн). К этому раствору добавляли нитрит натрия (0,27 г, 4,37 ммоль) в воде (3 мл) при 0°С. Смесь перемешивали 15 мин с последующим добавлением йодида калия (4,62 г, 27,8 ммоль) в воде (10 мл). Смеси предоставляли возможность нагреваться до комн. темп. Затем реакционную смесь разбавляли этилацетатом (100 мл) и метабисульфитом натрия (10% водн., 50 мл). Слои встряхивали и разделяли. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали с получением 2-циклопентилйодбензола (0,62 г, 2,3 ммоль, 57%). Арилйодид (0,62 г, 2,3 ммоль) растворяли в тетрагидрофуране. При -78°С добавляли н-бутиллитий (2,2 мл, 1,6 М в гекс., 3,45 ммоль). После перемешивания в течение 10 мин к раствору добавляли раскрошенный сухой лед (1 г). Смесь нагревали до комнатной температуры. Реакционную смесь разбавляли 5% хлористоводородной кислотой (водн., 50 мл) и этилацетатом (50 мл). Слои встряхивали и разделяли. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали с получением названного соединения (0,19 г, 43%). 1Н ЯМР (300 МГц, CDCl3) δ 9,6-10,2 (ушир.с, 1Н), 7,90 (д, J=7,8, 1H), 7,46 (м, 2Н), 7,24 (м, 1Н), 2,12 (м, 2Н), 1,71 (м, 7Н).
D. 1-Циклопентил-2-изоцианатобензол. 2-Циклопентилбензойную кислоту (0,19 г, 1 ммоль) растворяли в метиленхлориде (10 мл) и ДМФА (0,5 мл). Добавляли по каплям оксалилхлорид (0,11 мл, 1,3 ммоль). Через 15 мин смесь концентрировали и полученный в результате остаток разбавляли диоксаном (5 мл). При 0°С добавляли азид натрия (72 мг, 1,1 ммоль) в смеси вода/диоксан (5 мл, 1:1 об./об.). Смесь перемешивали в течение 15 мин с последующим разбавлением этилацетатом (50 мл) и водой (50 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенный ацилазид пропускали через слой силикагеля (10% этилацетата в гексанах). Элюент концентрировали и затем растворяли в толуоле (20 мл). Раствор перемешивали в течение 1 ч при 100°С. Концентрированием реакционной смеси получали названное соединение (0,19 г, колич.).
Е. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-циклопентилфенил)мочевина. Названное соединение получали согласно общему методу А с применением 1-циклопентил-2-изоцианатобензола (0,19 г, 1 ммоль) и 2,3-диаминомалеонитрила (0,11 г, 1 ммоль). Смесь концентрировали с получением названного соединения (0,29 г, колич). МС (ESI) m/z 296,3 [M+1]+.
F. 9-(2-Циклопентилфенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Названное соединение получали согласно общему методу В с применением 3-гидроксибензальдегида (0,31 г, 2,5 ммоль) и (Z)-1-(2-амино-1,2-дициановинил)-3-(2-циклопентилфенил)мочевины (0,29 г, 1 ммоль). Неочищенную реакционную смесь концентрировали. Неочищенный остаток очищали полупрепаративной ВЭЖХ (10-70% ацетонитрила + 0,1 TFA в Н2О + 0,1% TFA, в течение 30 мин). Чистые фракции концентрировали с получением названнго соединения (20 мг, 5%) в виде белого твердого вещества, 100% чистоты. 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,57 (с, 1Н), 8,49 (с, 1Н), 8,07 (с, 1Н), 7,96 (д, J=7,2, 1H), 7,48 (м, 1Н), 7,30 (т, J=7,8, 2H), 6,89 (дд, J=7,8, 2,4, 1H), 2,91 (кв, J=7,5, 1H), 1,44-2,00 (м, 7Н), МС (ESI) m/z 416,1 [M+1]+, т.пл. >260°С.
5.1.148 ПРИМЕР 148
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-9-(ПИПЕРИДИН-4-ИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Этил-6-(1-(трет-бутоксикарбонил)пиперидин-4-иламино)-2-хлор-5-нитропиримидин-4-карбоксилат. Этил-2,6-дихлор-5-нитропиримидин (3,0 г, 11,28 ммоль), диизопропилэтиламин (1,46 г, 11,28 ммоль) и трет-бутил-4-аминопиперидин-1-карбоксилат (2,03 г, 10,15 ммоль) подвергали взаимодействию согласно общему методу С, фильтровали и удаляли растворитель при пониженном давлении с получением названного соединения (5,25 г, 75%). МС (ESI) m/z 430,1 [M+1]+.
В. Этил-5-амино-6-(1-(трет-бутоксикарбонил)пиперидин-4-иламино)-2-хлорпиримидин-4-карбоксилат. Этил-6-(1-(трет-бутоксикарбонил)пиперидин-4-иламино)-2-хлор-5-нитропиримидин-4-карбоксилат (1,5 г, 3,49 ммоль) объединяли с дигидратом хлорида олова(II) (2,36 г, 10,47 ммоль) и этанолом (50 мл). Через 16 ч раствор фильтровали, фильтрат конденсировали и очищали путем хроматографии (Biotage) (0-60% EtOAc в гексанах) (0,788 г, 56%). МС (ESI) m/z 400,1 [M+1]+.
С. Этил-5-амино-6-(1-(трет-бутоксикарбонил)пиперидин-4-иламино)-2-(3-гидроксифенил)пиримидин-4-карбоксилат. Этил-5-амино-6-(1-(трет-бутоксикарбонил)пиперидин-4-иламино)-2-хлорпиримидин-4-карбоксилат (0,400 г, 1,00 ммоль), 3-гидроксифенилбороновую кислоту (0,207 г, 1,5 ммоль), ацетат палладия(II) (0,034 г, 0,15 ммоль), фосфат калия (0,430 г, 2,0 ммоль) и дициклогексил(2,6-диметоксифенил)фосфин (0,062 г, 0,15 ммоль) объединяли в тетрагидрофуране (6 мл) и воде (0,6 мл) и подвергали взаимодействию в микроволновом устройстве при 120°С в течение 30 мин. Реакционную смесь подвергали мониторингу расхода исходных веществ путем тонкослойной хроматографии. Раствор фильтровали и концентрировали, и остаток очищали путем хроматографии (Biotage) (0-60% EtOAc в гексанах) с получением названного соединения (0,120 г, 40%). МС (ESI) m/z 458,5 [M+1]+.
D. Этил-9-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат. Этил-5-амино-6-(1-(трет-бутоксикарбонил)пиперидин-4-иламино)-2-(3-гидроксифенил)пиримидин-4-карбоксилат (0,120 г, 0,260 ммоль) и 1,1'-1,1'-карбонилдиимидазол (0,425 г, 2,62 ммоль) в дихлорметане (10 мл) подвергали взаимодействию согласно общему методу F и очищали путем хроматографии (Biotage) (0-70% EtOAc в гексанах) с получением названного соединения (0,050 г, 19%). МС (ESI) m/z 484,3 [M+1]+.
Е. 2-(3-Гидроксифенил)-8-оксо-9-(пиперидин-4-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид. Этил-9-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат (0,050 г, 0,103 ммоль) подвергали взаимодействию согласно общему методу G. Через 16 ч методом ЖХМС подтвердили образование продукта, и раствор конденсировали при пониженном давлении с получением защищенного продукта. Твердое вещество помещали в HCl (4 н. в диоксанах, 4 мл) и перемешивали при комнатной температуре. Через 2 ч методом ЖХМС подтвердили образование продукта. Раствор конденсировали при пониженном давлении и разбавляли метанолом, обрабатывали ультразвуком и фильтровали с получением названного соединения в виде соли HCl (0,011 г, 28%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,49 (с, 2Н), 8,34 (с, 1Н), 8,07 (д, J=7,59, 2Н), 8,04 (с, 1Н), 7,95 (с, 1Н), 7,28 (т, J=7,99, 2H), 6,89 (дд, J=7,99, 1,59, 2H), 4,63 (м, 3Н), 3,45 (м, 4Н), 3,16 (м, 4Н), 2,75 (м, 3Н), 2,32 (с, 1Н), 1,99 (д, J=13,19, 3H). МС (ESI) m/z 355,2 [M+1]+.
5.1.149 ПРИМЕР 149
СИНТЕЗ 9-(2-ФТОР-4-МЕТОКСИФЕНИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-фтор-4-метоксифенил)мочевина. В 100-мл круглодонную колбу добавляли 2-фтор-4-метоксианилин (0,79 г, 5,60 ммоль) и трихлорметилкарбохлоридат (0,68 мл, 5,60 ммоль) в толуоле (6 мл) с получением пурпурной суспензии. Затем смесь перемешивали при 110°С в течение 3 ч. После нагревания суспензия превратилась в гомогенный раствор. Через 3 ч не осталось исходного вещества (ТСХ, 3:1 гекс./EtOAc). Реакционную смесь концентрировали с получением 2-фтор-1-изоцианато-4-метоксибензола (0,936 г, 5,6 ммоль, колич.) в виде зеленого масла. Затем промежуточное соединение 2-фтор-1-изоцианато-4-метоксибензол (0,936 г, 5,6 ммоль) растворяли в ацетонитриле (20 мл) и подвергали взаимодействию с 2,3-диаминомалеонитрилом (0,605 г, 5,60 ммоль) согласно общему методу А. Продукт фильтровали с получением названного соединения (1,36 г, 88%). МС (ESI) m/z 276,3 [M+1]+.
В. 9-(2-Фтор-4-метоксифенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-фтор-4-метоксифенил)мочевину (1,36 г, 4,94 ммоль) и 3-гидроксибензальдегид (1,509 г, 12,35 ммол) объединяли вместе в метаноле (20 мл) и триэтиламине (4 мл, 4,94 ммоль) согласно общему методу В. Продукт фильтровали, промывали метанолом и сушили (1,08 г, 55%, 97,6% чистоты). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,83 (с, 1Н), 9,49 (с, 1Н), 8,42 (с, 1Н), 8,00 (с, 1Н), 7,91 (т, 8,0, 1Н), 7,71 (т, J=2,0, 1H), 7,59 (т, J=8,8, 1H), 7,24 (т, J=8,0, 1H), 7,17 (дд, J=12, 2,4, 1H), 7,10 (дд, J=9,2, 2,4, 1H), 6,83 (дд, J=8,0, 1,6, 1H), 3,88 (с, 3Н); МС (ESI) m/z 396,3 [M+1]+, т.пл. >260°С.
5.1.150 ПРИМЕР 150
СИНТЕЗ 2-(1Н-БЕНЗО[D]ИМИДАЗОЛ-6-ИЛ)-9-ЦИКЛОГЕКСИЛ-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(1Н-Бензо[d]имидазол-6-ил)-9-циклогексил-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-циклогексилмочевину (см. пример 26.А) (520 мг, 2,23 ммоль), 1Н-бензо[d]имидазол-6-карбальдегид (см. пример 84.В) (652 мг, 4,46 ммоль), триэтиламин (0,34 мл, 3,34 ммоль) и метанол (20 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при окружающей температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали и очищали путем полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Летучие вещества удаляли при пониженном давлении, обрабатывали насыщенным раствором NaHCO3 и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (103 мг, 0,27 ммоль, 12,2%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,47 (с, 1Н), 8,79 (с, 1Н), 8,52 (м, 1Н), 8,50 (м, 1Н), 8,39 (с, 1Н), 7,92 (с, 1Н), 7,69 (д, J=8,0, 1H), 4,30 (м, 1Н), 2,41 (м, 2Н), 1,85 (м, 5Н), 1,36 (м, 4Н); МС (ESI) m/z 378,1 [M+1]+; т.пл. 280°С (разл.).
5.1.151 ПРИМЕР 151
СИНТЕЗ 2-БЕНЗИМИДАЗОЛ-6-ИЛ-9-(ТРАНС-4-МЕТОКСИЦИКЛОГЕКСИЛ)-8-ОКСО-7-ГИДРОПУРИН-6-КАРБОКСАМИДА
А. транс-4-Метоксициклогексанизоцианат. Суспензию транс-4-метоксициклогексиламина (243 мг, 1,46 ммоль) в толуоле (5 мл) обрабатывали дифосгеном (0,18 мл, 1,46 ммоль) и полученный в результате раствор нагревали до 100°С в течение 3 ч. Растворитель удаляли при пониженном давлении и полученное в результате вещество сушили в высоком вакууме в течение ночи.
В. N-((1Z)-2-Амино-1,2-дициановинил)[транс-(4-метоксициклогексил)амино]карбоксамид. Раствор транс-4-метоксициклогексанизоцианата (200 мг, 1,29 ммоль) и (1Z)-1,2-диаминоэтен-1,2-дикарбонитрила (139 мг, 1,29 ммоль) в безводном тетрагидрофуране (13 мл) перемешивали при комнатной температуре в течение ночи. Летучие вещества удаляли при пониженном давлении и очищали путем полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Фракции, содержащие продукт, объединяли и концентрировали. Полученное в результате вещество обрабатывали насыщенным раствором NaHCO3 и экстрагировали EtOAc (4×25 мл). Органические вещества сушили сульфатом натрия и концентрировали с получением названного соединения (200 мг, 0,76 ммоль, 59%) в виде желтого твердого вещества.
С. 2-Бензимидазол-6-ил-9-(транс-4-метоксициклогексил)-8-оксо-7-гидропурин-6-карбоксамид. N-((1Z)-2-Амино-1,2-дициановинил)[(4-метоксициклогексил)амино]карбоксамид (200 мг, 0,76 ммоль), бензимидазол-6-карбальдегид (167 мг, 1,14 ммоль), триэтиламин (0,16 мл, 1,14 ммоль) и метанол (8 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали и очищали путем полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Летучие продукты удаляли при пониженном давлении, обрабатывали насыщенным раствором NaHCO3 и экстрагировали этилацетатом (3×50 мл). Объединенные органические фракции сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученное в результате твердое вещество промывали простым диэтиловым эфиром и сушили в вакумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (0,018 мг, 0,044 ммоль, 5,8%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,42 (с, 1Н), 8,74 (с, 1Н), 8,65 (м, 1Н), 8,54 (м, 1Н), 8,39 (с, 1Н), 7,92 (с, 1Н), 7,69 (д, J=8,0, 1H), 4,30 (м, 1Н), 3,80 (м, 1Н), 3,39 (с, 3Н), 2,41 (м, 2Н), 1,85 (м, 4Н), 1,36 (м, 4Н); МС (ESI) m/z 408,1 [M+1]+; т.пл. 298°С (разл.).
5.1.152 ПРИМЕР 152
СИНТЕЗ 2-(4-(АМИНОМЕТИЛ)ФЕНИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. трет-Бутил-4-(6-карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-ил)бензилкарбамат. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (см. пример 2.А) (0,2 г, 0,777 ммоль), трет-бутил-4-формилбензилкарбамат (0,366 г, 1,555 ммоль) и триэтиламин (0,163 мл, 1,166 ммоль) объединяли в метаноле (15 мл) и перемешивали при комнатной температуре в течение ночи. Избыток растворителя удаляли при пониженном давлении и полученное в результате твердое вещество очищали хроматографией на силикагеле (Biotage) (0-10% метанола в дихлорметане). Чистые фракции объединяли, и растворитель удаляли при пониженном давлении. Полученное в результате вещество сушили в вакуумном устройстве с получением продукта (0,145 г, 0,296 ммоль, выход 38,0%) в виде желтого твердого вещества. МС (ESI) m/z 491,5 [M+1]+.
В. 2-(4-(Аминометил)фенил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. Раствор трет-бутил-4-(6-карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-ил)бензилкарбамата (0,14 г, 0,285 ммоль) и HCl (3,0 мл, 3,00 ммоль, 1 М в простом диэтиловом эфире) в дихлорметане (10 мл) перемешивали при комнатной температуре 2,5 ч. Растворитель удаляли при пониженном давлении, и вещество помещали в смесь метанол/дихлорметан и ДМСО. Раствор фильтровали через ионообменную колонку Strata-XC. Продукт выделяли гидроксидом аммония (5% в метаноле), и растворитель удаляли при пониженном давлении с получением названного соединения (0,045 г, 115 ммоль, выход 40%) после сушки в вакуумном устройстве. 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,62 (ушир.с, 1Н), 8,28 (AA'XX', JAX=8,20, 2H), 7,90 (с, 1Н), 7,53 (тд, J=7,13, 1,56, 1H), 7,46 (дд, J=7,71, 1,46, 1H), 7,39 (AA'XX', JAX=8,20, 2Н), 7,28 (м, 1Н), 7,14 (тд, J=7,61, 1,17, 1H), 3,79 (с, 2Н), 3,74 (с, 3Н); МС (ESI) m/z 391,3 [M+1]+.
5.1.153 ПРИМЕР 153
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛ)-9-(ЦИС-4-(МЕТОКСИМЕТИЛ)ЦИКЛОГЕКСИЛ)-8-ОКСО-8,8-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Трет-Бутил-цис-4-(гидроксиметил)циклогексилкарбамат. трет-Бутил-цис-4-аминоциклогексанкарбоновую кислоту (2,0 г, 8,23 ммоль) разбавляли тетрагидрофураном (20 мл) и охлаждали до -10°С. Добавляли N-метилморфолин (0,31 г, 8,23 ммоль) и изобутилхлорформиат (1,12 г, 8,23 ммоль) и перемешивали в течение десяти минут. Затем добавляли одной порцией борогидрид натрия (0,938 г, 24,69 ммоль). Через десять минут добавляли по каплям метанол (5 мл) и затем реакционную смесь перемешивали при 0°С дополнительно тридцать минут. Затем раствор разбавляли дихлорметаном и распределяли с помощью 5% раствора гидроксида натрия (3х), сушили над сульфатом магния, фильтровали и растворитель удаляли при пониженном давлении с получением названного соединения без очистки (2,2 г, количественный выход).
В. трет-Бутил-цис-4-(метоксиметил)циклогексилкарбамат. Гидрид натрия (0,216 г, 8,98 ммоль) суспендировали в тетрагидрофуране (30 мл) и перемешивали при 0°С. Затем добавляли цис-4-(гидроксиметил)циклогексилкарбамат (1,0 г, 5,99 ммоль) и простой 15-краун-5-эфир (1,385 г, 6,29 ммоль), реакционную смесь перемешивали в течение 30 мин. Затем добавляли метилйодид (0,393 мл, 8,98 ммоль) и раствор перемешивали при комнатной температуре в течение 16 ч. Дополнительно добавляли гидрид натрия (1,0 экв.) и продолжали перемешивание. После израсходования исходного вещества (мониторинг путем тонкослойной хроматографии) раствор конденсировали при пониженном давлении и распределяли между этилацетатом и водой (3х). Органические вещества объединяли, сушили над сульфатом магния, фильтровали и растворитель удаляли при пониженном давлении. Полученное в результате масло очищали путем хроматографии (Biotage) (0-50% EtOAc в гексанах) с получением названного соединения (0,498 г, 45%). МС (ESI) m/z 188,3 [M+1]+.
С. цис-4-(Метоксиметил)циклогексанамин гидрохлорид. трет-Бутил-цис-4-(метоксиметил)циклогексилкарбамат (1,0 г, 4,11 ммоль) растворяли в 1,4-диоксане (5 мл), добавляли 4,0н HCl в диоксане (2 мл) и раствор перемешивали с получением названного соединения (0,573 г, 78%). МС (ESI) m/z 144,3 [M+1]+.
D. цис-1-Изоцианат-4-(метоксиметил)циклогексан. Цис-4-(метоксиметил)циклогексанамин гидрохлорид (0,423 г, 2,36 ммоль) разбавляли толуолом (8 мл) и к раствору добавляли трихлорметилкарбохлоридат (0,207 г, 1,05 ммоль) в толуоле (5 мл). Смесь перемешивали при 100°С в течение трех часов. Раствор конденсировали с получением масла и применяли без очистки (количественный выход).
Е. 1-((Z)-2-Амино-1,2-дициановинил)-3-(цис-4-(метоксиметил)циклогексил)мочевина. цис-1-Изоцианат-4-(метоксиметил)циклогексан (0,398 г, 2,35 ммоль) растворяли в тетрагидрофуране (10 мл), с последующим добавлением диаминомалеонитрила (0,508 г, 4,70 ммоль). Раствору предоставляли возможность перемешиваться при комнатной температуре в течение 16 ч. Методом ЖХМС подтвердили образование продукта. Раствор очищали путем препаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин) с получением названного соединения (0,405 г, 62%). МС (ESI) m/z 278,5 [M+1]+.
F. 2-(3-Гидроксифенил)-9-(цис-4-(метоксиметил)циклогексил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. 1-((Z)-2-Амино-1,2-дициановинил)-3-(цис-4-(метоксиметил)циклогексил)мочевину (0,405 г, 1,46 ммоль), 3-гидроксибензальдегид (0,268 г, 2,19 ммоль) и триэтиламин (0,611 мл, 4,38 ммоль) объединяли в метаноле (10 мл). Раствор перемешивали при окружающей температуре в течение 16 ч. Полученный в результате осадок отфильтровывали и промывали ацетонитрилом и сушили при пониженном давлении с получением названного соединения (0,089 г, 15%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,52 (с, 1Н), 9,50 (с, 1Н), 8,32 (с, 1Н), 7,99 (д, J=7,99, 1H), 7,90 (м, 2Н), 7,27 (т, J=7,59, 1H), 6,89 (дд, J=6,79, 1,59), 4,26 (м, 1Н), 3,63 (д, J=7,59, 2H), 3,79 (с, 3H), 1,98 (c, 1H), 1,85 (м, 2Н), 1,59 (м, 4Н); МС (ESI) m/z 398,1 [M+1]+, т.пл. 324-326°С.
5.1.154 ПРИМЕР 154
СИНТЕЗ 9-(ТРАНС-4-АМИНОЦИКЛОГЕКСИЛ)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. Этил-6-(транс-4-(трет-бутоксикарбониламино)циклогексиламино)-2-хлор-5-нитропиримидин-4-карбоксилат. Этил-2,6-дихлор-5-нитропиримидин (1,5 г, 5,64 ммоль), диизопропилэтиламин (0,729 г, 5,64 ммоль) и трет-бутил-транс-4-аминоциклогексилкарбамат (1,09 г, 5,07 ммоль) подвергали взаимодействию согласно общему методу С с получением названного соединения (3,08 г, количественный выход). МС (ESI) m/z 444 [M+1]+.
В. Этил-5-амино-6-(транс-4-(трет-бутоксикарбониламино)циклогексиламино)-2-хлорпиримидин-4-карбоксилат. Этил-6-(транс-4-(трет-бутоксикарбониламино)циклогексиламино)-2-хлор-5-нитропиримидин-4-карбоксилат (2,5 г, 5,63 ммоль) растворяли в этаноле (25 мл) и диметилформамиде (6 мл). Добавляли дигидрат хлорида олова (2,52 г, 11,16 ммоль) и раствор перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь фильтровали и конденсировали. Остаток очищали путем хроматографии (Biotage) (0-55% EtOAc в гексанах) (1,83 г, 78%). МС (ESI) m/z 414 [M+1]+.
С. Этил-5-амино-6-(транс-4-(трет-бутоксикарбониламино)циклогексиламино)-2-(3-гидроксифенил)пиримидин-4-карбоксилат. Этил-5-амино-6-(транс-4-(трет-бутоксикарбониламино)циклогексиламино)-2-хлорпиримидин-4-карбоксилат (0,500 г, 1,21 ммоль), 3-гидроксифенилбороновую кислоту (0,249 г, 1,81 ммоль), дициклогексил(2',6'-диметоксибифенил-2-ил)фосфин (0,075 г, 0,182 ммоль), ацетат палладия (0,041 г, 0,182 ммоль) и фосфат калия (0,52 г, 2,42 ммоль) объединяли в тетрагидрофуране (7 мл) и воде (0,6 мл) и нагревали до 120°С в микроволновом реакторе Biotage Emrys Optimizer в течение тридцати минут. Реакционный раствор фильтровали и концентрировали. Остаток очищали путем хроматографии (Biotage) (0-60% этилацетата в гексанах) с получением названного соединения (0,150 г, 26%). МС (ESI) m/z 472 [M+1]+.
D. Этил-9-(транс-4-(трет-бутоксикарбониламино)циклогексил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат. Этил-5-амино-6-(транс-4-(трет-бутоксикарбониламино)циклогексиламино)-2-(3-гидроксифенил)пиримидин-4-карбоксилат (0,150 г, 0,317 ммоль) и 1,1'-1,1'-карбонилдиимидазол (0,514 г, 3,17 ммоль) в дихлорметане (10 мл) подвергали взаимодействию согласно общему методу F и очищали путем хроматографии (Biotage) (0-60% EtOAc в гексанах) с получением названного соединения (0,070 г, 44%). МС (ESI) m/z 498 [M+1]+.
F. трет-Бутил-транс-4-(6-карбамоил-2-(3-гидроксифенил)-8-оксо-7Н-пурин-9(8Н)-ил)циклогексилкарбамат. Этил-9-(транс-4-(трет-бутоксикарбониламино)циклогексил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксилат (0,070 г, 0,140 ммоль) и аммиак в метаноле подвергали взаимодействию согласно общему методу G. Через 16 ч методом ЖХМС подтвердили образование продукта, и раствор конденсировали при пониженном давлении с получением названного соединения (0,040 г, 60%). МС (ESI) m/z 469 [M+1]+.
G. 9-(транс-4-Аминоциклогексил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. трет-Бутил-транс-4-(6-карбамоил-2-(3-гидроксифенил)-8-оксо-7Н-пурин-9(8Н)-ил)циклогексилкарбамат (0,090 г, 0,212 ммоль) разбавляли дихлорметаном (10 мл) с последующей трифторуксусной кислотой (1 мл). Раствор концентрировали и очищали путем препаративной ВЭЖХ с обращенной фазой (10-40% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 35 мин) с получением названного соединения (0,030 г, 38%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,4 (ушир.с, 1Н), 8,00 (д, J=7,99, 1H), 7,90 (c, 1Н), 7,87 (с, 1Н), 7,26 (т, J=7,59, 1H), 6,86 (д, J=7,59, 1H), 4,24 (м, 1Н), 3,2 (с, 1Н), 2,75 (м, 2Н), 1,73 (м, 4Н), 1,53 (с, 1Н), 1,52 (с, 1Н); МС (ESI) m/z 398,1 [M+1]+; т.пл. 299-301°С.
5.1.155 ПРИМЕР 155
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛ)-9-(2-ИЗОБУТИЛФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 1-(2-Метилпроп-1-енил)-2-нитробензол. Раствор изопропилтрифенилфосфонийбромида (1,405 г, 4,6 ммоль) в тетрагидрофуране (20 мл) охлаждали на бане со льдом. Добавляли по каплям н-бутиллитий (3,2 мл, 5,1 ммоль в виде 1,6 М в гекс.). Смесь перемешивали 5 мин и затем добавляли 2-нитробензальдегид в тетрагидрофуране (10 мл). Реакционную смесь подвергали мониторингу расходования исходного вещества. Через 1 ч реакционную смесь гасили водой (100 мл) и экстрагировали EtOAc (100 мл). Органический слой промывали насыщенным раствором соли (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Полученный в результате осадок очищали путем (хроматографии) Biotage (95% гексанов в EtOAc; колонка 40+S). Концентрированием требуемых фракций получали требуемый продукт (0,48 г, 59%). 1Н ЯМР (300 МГц, CDCl3) δ 7,93 (дд, J=6,0, 0,9, 1H), 7,54 (тд, J=5,4, 0,9, 1H), 7,26-7,38 (м, 2Н), 6,49 (с, 1Н), 1,93 (д, J=0,9, 3H), 1,70 (д, J=0,9, 3H).
В. 2-Изобутиланилин. К раствору 1-(2-метилпроп-1-енил)-2-нитробензола (0,48 г, 2,71 ммоль) в этаноле (15 мл) добавляли палладий на угле (10%, 0,3 г). Реакционную смесь перемешивали в атмосфере водорода в течение 1-2 ч. Анализ ЖХМС через 2 ч показал (М+1=150,6), что реакция завершилась. Смесь фильтровали через слой целита, промывали этанолом и концентрировали. Остаток очищали (хроматографией) Biotage (90% гексанов в EtOAc; колонка 40+S). Концентрированием требуемых фракций получали продукт (0,34 г, 85%). МС (ESI) m/z 150,6 [M+1]+.
С. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-изобутилфенил)мочевина. В 50-мл круглодонной колбе объединяли 2-изобутиланилин (0,34 г, 2,300 ммоль) и трихлорметилкарбохлоридат (0,28 мл, 2,32 ммоль) в толуоле (3 мл). Смесь кипятили с обратным холодильником при перемешивании в течение 1 ч. Затем раствор концентрировали. Остаток помещали в ацетонитрил (10 мл). Добавляли 2,3-диаминомалеонитрил (0,249 г, 2,300 ммоль). Смесь перемешивали при комнатной температуре в течение 24 ч. Реакционную смесь фильтровали с получением названного соединения (0,6 г, 2,1 ммоль, 90%). МС (ESI) m/z 284,5 [M+1]+.
D. 2-(3-Гидроксифенил)-9-(2-изобутилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-изобутилфенил)мочевину (0,595 г, 2,100 ммоль) и 3-гидроксибензальдегид (0,641 г, 5,25 ммоль) объединяли в метаноле (10 мл) и триэтиламине (1 мл) согласно общему методу В. Реакционную смесь концентрировали и полученный в результате осадок очищали (хроматографией) Biotage (50% гексанов в EtOAc; колонка 40+S). Концентрированием требуемых фракций получали названное соединение (0,22 г, 25%, 97,9% чистоты). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,79 (с, 1Н), 9,50 (с, 1Н), 8,43 (с, 1Н), 8,01 (с, 1Н), 7,91 (д, J=8,0, 1H), 7,71 (т, J=2,0, 1H), 7,59 (т, J=8,8, 1H), 7,24 (т, J=8,0, 1H), 7,17 (дд, J=12, 2,4, 1H), 7,10 (дд, J=9,2, 2,4, 1H), 6,83 (дд, J=8,0, 1,6, 1H), 2,37 (д, J=7,2, 2H), 1,68 (септ., J=2,8, 1H), 0,72 (д, J=6,8, 3H), 0,67 (д, J=6,4, 3H), МС (ESI) m/z 404,3 [M+1]+; т.пл. >260°С.
5.1.156 СИНТЕЗ 156
СИНТЕЗ (R)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-9-(ТЕТРАГИДРОФУРАН-3-ИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (R)-3-Изоцианатотетрагидрофурантолуолсульфонатная соль. Соль (R)-тетрагидрофуран-3-аминотолуолсульфоновой кислоты (0,75 г, 2,91 ммоль) разбавляли толуолом (15 мл). К раствору добавляли трихлорметилкарбохлоридат (0,577 г, 2,91 ммоль) и смесь нагревали до 100°С. Через 3 ч раствор конденсировали при пониженном давлении и применяли без очистки (выход количественный).
В. (R,Z)-1-(2-Амино-1,2-дициановинил)-3-(тетрагидрофуран-3-ил)мочевина. (R)-3-Изоцианатотетрагидрофурантолуолсульфонатную соль (0,327 г, 2,89 ммоль) и диаминомалеонитрил (0,625 г, 5,78 ммоль) подвергали взаимодействию согласно общему методу А в тетрагидрофуране (10 мл) и очищали путем препаративной ВЭЖХ с обращенной фазой (0-10% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 35 мин) с получением названного соединения (0,404, 63%). МС (ESI) m/z 222,2 [M+1]+.
С. (R)-2-(3-Гидроксифенил)-8-оксо-9-(тетрагидрофуран-3-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид. (R,Z)-1-(2-Амино-1,2-дициановинил)-3-(тетрагидрофуран-3-ил)мочевину (0,404 г, 2,26 ммоль), 3-гидроксибензальдегид (0,554 г, 4,53 ммоль) и триэтиламин (0,63 мл, 4,53 ммоль) в метаноле (15 мл) подвергали взаимодействию согласно общему методу В с получением названного соединения (0,094 г, 12%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,75 (с, 1Н), 9,53 (с, 1Н), 8,34 (с, 1Н), 7,99 (д, J=9,19, 1H), 7,93 (с, 1Н), 7,87 (с, 1Н), 7,28 (т, J=7,99, 1H), 6,88 (д, J=7,99, 1H), 5,07 (м, 1Н), 4,25 (кв, J=7,99, 1H), 3,99 (м, 3Н), 2,30 (м, 1Н); МС (ESI) m/z 342,1 [M+1]+; т.пл. 353-355°С.
5.1.157 ПРИМЕР 157
СИНТЕЗ (S)-2-(3-ГИДРОКСИФЕНИЛ)-8-ОКСО-9-(ТЕТРАГИДРОФУРАН-3-ИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. (S)-3-Изоцианатотетрагидрофурантолуолсульфонатная соль. Соль (S)-Тетрагидрофуран-3-аминотолуолсульфоновой кислоты (0,75 г, 2,91 ммоль) разбавляли толуолом (15 мл). К раствору добавляли трихлорметилкарбохлоридат (0,577 г, 2,91 ммоль) и смесь нагревали до 100°С. Через 3 ч раствор конденсировали при пониженном давлении и применяли без очистки (выход количественный).
В. (S,Z)-1-(2-Амино-1,2-дициановинил)-3-(тетрагидрофуран-3-ил)мочевина. (S)-3-Изоцианатотетрагидрофурантолуолсульфонатную соль (0,825 г, 7,29 ммоль) и диаминомалеонитрил (1,57 г, 14,59 ммоль) подвергали взаимодействию согласно общему методу А в тетрагидрофуране (10 мл) и очищали путем препаративной ВЭЖХ с обращенной фазой (0-10% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 35 мин) с получением названного соединения (0,636, 39%). МС (ESI) m/z 222,2 [M+1]+.
С. (S)-2-(3-Гидроксифенил)-8-оксо-9-(тетрагидрофуран-3-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид. (S,Z)-1-(2-Амино-1,2-дициановинил)-3-(тетрагидрофуран-3-ил)мочевину (0,636 г, 3,57 ммоль), 3-гидроксибензальдегид (0,872 г, 7,14 ммоль) и триэтиламин (1,0 мл, 4,53 ммоль) в метаноле (15 мл) подвергали взаимодействию согласно общему методу В с получением названного соединения (0,120 г, 10%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,75 (с, 1Н), 9,53 (с, 1Н), 8,34 (с, 1Н), 7,99 (д, J=9,19, 1H), 7,93 (с, 1Н), 7,87 (с, 1Н), 7,28 (т, J=7,99, 1H), 6,88 (д, J=7,99, 1H), 5,07 (м, 1Н), 4,25 (кв, J=7,99, 1H), 3,99 (м, 3Н), 2,30 (м, 1Н); МС (ESI) m/z 342,1 [M+1]+; т.пл. 354-356°С.
5.1.158 ПРИМЕР 158
СИНТЕЗ 2-(4-(1Н-1,2,3-ТРИАЗОЛ-5-ИЛ)ФЕНИЛ)-9-(2-ИЗОПРОПИЛФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. ((4-(Диэтоксиметил)фенил)этинил)триметилсилан. 20-мл пробирки для микроволновой реакции наполняли ДМФА (10 мл). Растворитель дегазировали в течение 5 мин перед добавлением 1-бром-4-(диэтоксиметил)бензола (3,89 г, 15,0 ммоль), триметилсилилацетилена (6,31 мл, 45,0 ммоль), хлорида бис(трифенилфосфин)палладия(II) (0,316 г, 0,450 ммоль), йодида меди(I) (0,171 г, 0,900 ммоль) и 1,1,3,3-тетраметилгуанидина (5,65 мл, 45,0 ммоль). Полученную смесь дегазировали в течение последующих 2 мин и затем разделили на две 20-мл пробирки для микроволновой реакции, по объему. Реакционную смесь нагревали при 150°С в течение 20 мин в микроволновом реакторе Biotage Emrys Optimizer. Две реакционные смеси объединяли и затем фильтровали для удаления каких-либо твердых веществ. Объем жидкости уменьшали наполовину при пониженном давлении. К оставшейся жидкости добавляли воду (20 мл) и полученный в результате раствор экстрагировали метиленхлоридом (3×30 мл). Органические слои объединяли, сушили над сульфатом магния и фильтровали через целит. Растворитель удаляли при пониженном давлении. Темно-коричневое масло применяли на следующей стадии без дополнительной очистки. 1Н ЯМР (300 МГц, CDCl3) δ 7,46 (д, J=8,4, 2H), 7,40 (д, J=8,4, 2H), 5,50 (с, 1Н), 3,60-3,50 (м, 4Н), 1,25-1,20 (м, 6Н), 0,25 (с, 9Н).
В. 1-(Диэтоксиметил)-4-этинилбензол. В 50-мл круглодонную колбу добавляли ((4-(диэтоксиметил)фенил)этинил)триметилсилан (4,15 г, 15,0 моль) и тетрабутиламмонийфторид на силикагеле (4,31 г, 16,5 ммоль) в тетрагидрофуране (15 мл) с получением коричневой суспензии. Смесь перемешивали при комнатной температуре в течение 5 ч и затем фильтровали для удаления твердых веществ. К реакционной смеси добавляли воду (20 мл), затем экстрагировали метиленхлоридом (3×40 мл). Объединенные экстракты сушили над сульфатом магния, и растворитель удаляли при пониженном давлении. Темно-коричневый остаток очищали с применением колоночной хроматографии Biotage (градиент 0-10% EtOAc в гексанах), получая прозрачное желтое масло (1,92 г, 63% посредством двух стадий). 1Н ЯМР (300 МГц, CDCl3) δ 7,49 (д, J=8,4, 2H), 7,44 (д, J=8,4, 2H), 5,50 (с, 1Н), 3,61-3,51 (м, 4Н), 3,08 (с, 1Н), 1,24 (т, J=6,9, 6H).
С. 4-(4-(Диэтоксиметил)фенил)-1Н-1,2,3-триазол. 35-мл Герметизируемую пробирку наполняли диметилформамидом (7,27 мл) и метанолом (0,808 мл). Добавляли 1-(диэтоксиметил)-4-этинилбензол (0,825 г, 4,04 ммоль), триметилсилилазид (0,804 мл, 6,06 ммоль) и йодид меди(I) (0,038 г, 0,202 ммоль) и реакционную смесь перемешивали при 100°С в течение 24 ч. Осадки удаляли, и фильтрат концентрировали при пониженном давлении. Масло растворяли в этилацетате (20 мл) и промывали водой (20 мл). Водный слой экстрагировали этилацетатом (2×20 мл). Органические слои объединяли и сушили над сульфатом магния. Растворитель выпаривали при пониженном давлении, получая светло-коричневое масло. Очисткой с применением колоночной хроматографии Biotage (градиент 0-20% МеОН в CH2Cl2) получали белое твердое вещество (0,600 г, 60%).
D. 4-(1Н-1,2,3-Триазол-4-ил)бензальдегид. 4-(4-(Диэтоксиметил)фенил)-1Н-1,2,3-триазол (0,600 г, 2,43 ммоль) перемешивали в растворе 4 М HCl в диоксане (15 мл, 4,85 ммоль) в течение 4 ч. Бледно-желтое твердое вещество, которое осаждалось, собирали и промывали гексанами. Твердое вещество добавляли к воде (10 мл) и добавляли 1 М NaOH до тех пор, пока рН не будет между 7 и 8. Раствор экстрагировали этилацетатом (3×20 мл) и сушили над сульфатом магния. Растворители удаляли при пониженном давлении с получением прозрачного бледно-желтого масла (0,250 г, 60%). Продукт был более 95% чистоты и применялся на следующей стадии без дополнительной очистки. 1Н ЯМР (300 МГц, CDCl3) δ 10,06 (с, 1Н), 8,08 (с, 1Н), 8,03 (д, J=6,6, 2H), 7,98 (д, J=6,6, 2H).
Е. 2-(4-(1Н-1,2,3-Триазол-5-ил)фенил)-9-(2-изопропилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-изопропилфенил)мочевину (0,311 мг, 1,16 ммоль) растворяли в метаноле (7 мл) и перемешивали при комнатной температуре до гомогенного состояния. Последовательно добавляли 4-(1Н-1,2,3-триазол-5-ил)бензальдегид (0,100 г, 0,577 ммоль) и триэтиламин (0,121 мл, 0,866 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 24 ч. Полученную в результате неоднородную смесь фильтровали. Осадок собирали и перекристаллизовывали растворением в теплом ДМФА (1 мл) и добавлением по каплям воды. Твердое вещество собирали и сушили в вакуумном сушильном шкафу в течение 48 ч с получением бледно-желтого твердого вещества (0,080 г, 32% посредством двух стадий). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,84 (с, 1Н), 8,56 (с, 1Н), 8,44 (д, J=6,3, 2H), 8,31 (с, 1Н), 8,02 (с, 1Н), 7,92 (д, J=6,3, 2H), 7,62-7,55 (м, 2Н), 7,42-7,39 (м, 2Н), 2,75 (септет, J=7,2, 1H), 1,13 (д, J=7,2, 3H), 1,12 (д, J=7,2, 3H); МС (ESI) m/z 441,1 [M+1]+.
5.1.159 ПРИМЕР 159
СИНТЕЗ 2-(3-(АМИНОМЕТИЛ)ФЕНИЛ)-9-(2-МЕТОКСИФЕНИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. трет-Бутил-3-(6-карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-ил)бензилкарбамат. (Z)-1-(2-Амино-1,2-дициановинил)-3-(2-метоксифенил)мочевину (0,200 г, 0,777 ммоль) растворяли в метаноле (5 мл) и перемешивали при комнатной температуре до гомогенного состояния. Последовательно добавляли трет-бутил-3-формилбензилкарбамат (0,183 г, 0,777 ммоль) и триэтиламин (0,108 мл, 0,777 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 16 ч. Полученную в результате неоднородную смесь фильтровали и промывали холодным метанолом с получением белого твердого вещества (0,247 г, выход 65%). МС (ESI) m/z 491,6 [M+1]+.
В. 2-(3-(Аминометил)фенил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. трет-Бутил-3-(6-карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-ил)бензилкарбамат (0,100 г, 0,204 ммоль) перемешивали в растворе 4 М HCl в диоксане (4,0 мл, 16 ммоль) в течение 5 ч. Полученную в результате неоднородную смесь фильтровали и промывали метиленхлоридом. Полученное в результате твердое вещество добавляли к воде (1 мл) и нейтрализовали насыщенным раствором бикарбоната натрия. Осадок собирали и перекристаллизовывали растворением в теплом ДМФА (1 мл) и добавлением по каплям воды. Твердое вещество сушили в вакуумном сушильном шкафу в течение 48 ч с получением белого твердого вещества (0,050 г, 63%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,62 (с, 1Н), 8,36 (с, 1Н), 8,14 (дт, J=7,3, 1,6, 1H), 7,95 (с, 1Н), 7,56-7,52 (м, 1Н), 7,46 (дд, J=7,8, 2,0, 1H), 7,41-7,34 (м, 2Н), 7,28 (дд, J=8,6, 1,2, 1H), 7,14 (тд, J=7,5, 1,4, 1H), 3,83 (с, 2Н), 3,74 (с, 3Н); МС (ESI) m/z 391,0 [M+1]+.
5.1.160 ПРИМЕР 160
СИНТЕЗ 2-(4-(1Н-1,2,4-ТРИАЗОЛ-3-ИЛ)ФЕНИЛ)-9-(ЦИС-4-МЕТОКСИЦИКЛОГЕКСИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. цис-(1-Изоцианато-4-метоксициклогексан. Суспензию цис-4-метоксициклогексанамина (1,89 г, 11,41 ммоль) в толуоле (50 мл) и дифосгена (2,25 г, 11,41 ммоль) нагревали до 100°С в течение 5 ч. Растворитель удаляли при пониженном давлении и продукт сушили в высоком вакууме в течение ночи. Это вещество применяли без дополнительной очистки или характеристики.
В. 1-((Z)-2-Амино-1,2-дициановинил)-3-(цис-4-метоксициклогексил)мочевина. Раствору цис-1-изоцианато-4-метоксициклогексана (1,77 г, 11,41 ммоль) и (1Z)-1,2-диаминоэтен-1,2-дикарбонитрила (1,23 г, 11,41 ммоль) в безводном тетрагидрофуране (50 мл) предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Летучие вещества удаляли при пониженном давлении и полученное в результате твердое вещество очищали путем полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Фракции, содержащие продукт, объединяли и растворитель удаляли. Добавляли насыщенный раствор бикарбоната натрия и продукт экстрагировали EtOAc (4×25 мл). Объединенные органические вещества сушили над сульфатом натрия и концентрировали с получением названного соединения (1,12 г, 4,25 ммоль, 37%) в виде желтого твердого вещества.
С. 2-(4-(1Н-1,2,4-Триазол-3-ил)фенил)-9-(цис-4-метоксициклогексил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. 1-((Z)-2-Амино-1,2-дициановинил)-3-(цис-4-метоксициклогексил)мочевину (375 мг, 1,42 ммоль), 4-(1Н-1,2,4-триазол-3-ил)бензальдегид (см. 108.В) (450 мг, 2,60 ммоль), триэтиламин (0,32 мл, 2,28 ммоль) и метанол (8 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали и очищали путем полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Летучие вещества удаляли при пониженном давлении, обрабатывали насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом (3×50 мл), сушили органические вещества сульфатом натрия и концентрировали при пониженном давлении. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (0,037 мг, 0,085 ммоль, 6,0%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,54 (с, 1Н), 8,68 (м, 2Н), 8,14 (д, J=8,0, 2H), 7,94 (м, 2Н), 4,34 (м, 1Н), 3,51 (м, 1Н), 3,38 (с, 3Н), 2,74 (м, 2Н), 2,06 (м, 4Н), 1,55 (м, 4Н); МС (ESI) m/z 435,1 [M+1]+; т.пл. 350°С (разл.).
5.1.161 ПРИМЕР 161
СИНТЕЗ 2-(1Н-БЕНЗО[D]ИМИДАЗОЛ-6-ИЛ)-9-(ЦИС-4-МЕТОКСИЦИКЛОГЕКСИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(1Н-Бензо[d]имидазол-6-ил)-9-(цис-4-метоксициклогексил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. 1-((Z)-2-Амино-1,2-циановинил)-3-(цис-4-метоксициклогексил)мочевину (см. пример 160.В) (375 мг, 1,42 ммоль), 1Н-бензо[d]имидазол-6-карбальдегид (см. пример 84.В) (416 мг, 2,85 ммоль), триэтиламин (0,29 мл, 2,14 ммоль) и метанол (20 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи. Полученную в результате неоднородную смесь фильтровали и очищали путем полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Летучие вещества удаляли при пониженном давлении, обрабатывали насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом (3×50 мл). Объединенные органические вещества сушили над сульфатом натрия и концентрировали при пониженном давлении. Твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (0,039 мг, 0,095 ммоль, 6,7%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,56 (с, 1Н), 11,43 (с, 1Н), 8,94 (с, 1Н), 8,68 (с, 1Н), 8,51 (м, 2Н), 8,29 (м, 2Н), 7,89 (м, 2Н), 7,73 (д, J=8,0, 1H), 7,58 (д, J=8,0, 1H), 4,33 (м, 1Н), 3,52 (м, 1Н), 3,42 (с, 3Н), 2,79 (м, 2Н), 2,07 (м, 4Н), 1,56 (м, 4Н); МС (ESI) m/z 408,1 [M+1]+; т.пл. 345°С (разл.).
5.1.162 ПРИМЕР 162
СИНТЕЗ 2-(1Н-ИМИДАЗО[4,5-B]ПИРИДИН-6-ИЛ)-9-(ЦИС-4-МЕТОКСИЦИКЛОГЕКСИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(1Н-Имидазо[4,5-b]пиридин-6-ил)-9-(цис-4-метоксициклогексил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. 1-((Z)-2-Амино-1,2-дициановинил)-3-(цис-4-метоксициклогексил)мочевину (см. пример 160.В) (375 мг, 1,42 ммоль), 1-(тетрагидро-2Н-пиран-2-ил)-1Н-имидазо[4,5-b]пиридин-6-карбальдегид (см. пример 130.С) (644 мг, 2,80 ммоль), триэтиламин (0,4 мл, 2,80 ммоль) и метанол (20 мл) подвергали взаимодействию согласно общему методу В. Раствору предоставляли возможность перемешиваться при комнатной температуре в течение ночи и полученный в результате осадок собирали фильтрованием с получением 9-(цис-4-метоксициклогексил)-8-оксо-2-(1-(тетрагидро-2Н-пиран-2-ил)-1Н-имидазо[4,5-b]пиридин-6-ил)-8,9-дигидро-7Н-пурин-6-карбоксамида. Этот ТНР-защищенный промежуточный продукт (235 мг, 0,48 ммоль) растворяли в диоксане (2 мл) и добавляли 4,0 М раствор HCl/диоксан (4,0 мл) и воду (0,3 мл). Полученный в результате раствор перемешивали при комнатной температуре в течение ночи. Реакционную смесь нейтрализовали насыщенным раствором бикарбоната натрия, экстрагировали EtOAc (3×50 мл) и объединенные органические вещества сушили над сульфатом натрия. Неочищенный продукт очищали путем полупрепаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Летучие вещества удаляли при пониженном давлении, обрабатывали насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом (3×75 мл). Органические слои объединяли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученное в результате твердое вещество промывали простым диэтиловым эфиром и сушили в вакуумном сушильном шкафу при 60°С в течение ночи с получением названного соединения (15 мг, 0,036 ммоль, 7,5%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, MeOD-d3) δ 9,56 (c, 1H), 8,50 (м, 1Н), 7,80 (м, 1Н), 4,31 (м, 1Н), 3,51 (м, 1Н), 3,39 (с, 3Н), 2,77 (м, 2Н), 2,07 (м, 4Н), 1,53 (м, 4Н). МС (ESI) m/z 409,0 [M+1]+; т.пл. 360°С (разл.).
5.1.163 ПРИМЕР 163
СИНТЕЗ 2-(2-ХЛОРПИРИДИН-3-ИЛ)-8-ОКСО-9-ФЕНИЛ-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(2-Хлорпиридин-3-ил)-8-оксо-9-фенил-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-фенилмочевину (см. пример 131.А) (0,153 г, 0,67 ммоль) и 2-хлорникотинальдегид (0,191 г, 1,35 ммоль) подвергали взаимодействию согласно общему методу В. Продукт очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (0-50% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Фракции, содержащие требуемое вещество, нейтрализовали водным раствором карбоната натрия и затем концентрировали до меньшего объема. Полученный в результате осадок отфильтровывали и промывали водой. Полученное в результате твердое вещество сушили в высоком вакууме при 60°С с получением названного соединения в виде белого твердого вещества (0,070 г, 0,19 ммоль, выход 28%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 8,94 (дд, J=4,4, 2,0, 1H), 8,22-8,10 (м, 1Н), 7,77 (д, J=7,6, 3H), 7,55-7,46 (м, 3Н), 7,38-7,28 (м, 1Н); МС (ESI) m/z 367,2 [M+1]+; т.пл. 283-286°С.
5.1.164 ПРИМЕР 164
СИНТЕЗ 2-(2-МЕТОКСИПИРИДИН-3-ИЛ)-8-ОКСО-9-ФЕНИЛ-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(2-Метоксипиридин-3-ил)-8-оксо-9-фенил-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-фенилмочевину (см. пример 131.А) (0,400 г, 1,61 ммоль) и 2-метоксиникотинальдегид (0,44 г, 3,24 ммоль) подвергали взаимодействию согласно общему методу В. Продукт перекристаллизовывали из кипящего этанола, фильтровали и сушили с получением белого твердого вещества (0,290 г, 0,8 ммоль, выход 50%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,93 (с, 1Н), 8,37-8,31 (м, 2Н), 8,31-8,25 (ушир.с, 1Н), 8,09-8,03 (ушир.с, Н), 7,87-7,81 (м, 2Н), 7,70-7,62 (м, 2Н), 7,57-7,49 (м, 1Н), 7,19 (дд, J=7,4, 4,9, 1H), 3,99 (с, 3Н); МС (ESI) m/z 363,4 [M+1]+; т.пл. 279-281°С.
5.1.165 ПРИМЕР 165
СИНТЕЗ N,N-ДИМЕТИЛ-8-ОКСО-9-ФЕНИЛ-2-(ПИРИДИН-3-ИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. N,N-Диметил-8-оксо-9-фенил-2-(пиридин-3-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид. К раствору 8-оксо-9-фенил-2-(пиридин-3-ил)-8,9-дигидро-7Н-пурин-6-карбоновой кислоты (см. пример 38.А) (0,290 г, 0,87 ммоль) в ДМСО (2 мл) добавляли диизопропилэтиламин (1,6 г, 12,4 ммоль), диметиламин гидрохлорид (0,633 г, 7,81 ммоль) и бензотриазол-1-илокси-трис(диметиламино)фосфонийгексафторфосфат (0,580 г, 1,30 ммоль). На смесь воздействовали ультразвуком в течение 5 мин для растворения всех компонентов смеси. После перемешивания в течение 10 мин исходное вещество израсходовалось (мониторинг ЖХМС). Растворитель удаляли и продукт очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (10-50% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Фракции, содержащие требуемое вещество, объединяли и концентрировали при пониженном давлении перед пропусканием через ионообменную колонку Strata-XC с водой, метанолом и 5% гидроксидом аммония в метаноле. Полученное в результате твердое вещество сушили в высоком вакууме при 60°С с получением названного соединения в виде белого твердого вещества (0,037 г, 0,10 ммоль, выход 12%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 12,05 (с, 1Н), 9,35 (д, J=2,1, 1H), 8,65 (дд, J=4,8, 1,8, 1H), 8,49 (дт, J=8,1, 2,1, 1H), 7,75 (д, J=7,8, 2H), 7,62 (т, J=7,5, 2H), 7,55-7,45 (м, 2Н), 3,17 (с, 3Н), 3,11 (с, 3Н); МС (ESI) m/z 361,2 [M+1]+; т.пл. 250-252°С.
5.1.166 ПРИМЕР 166
СИНТЕЗ 9-МЕТИЛ-8-ОКСО-2-(ПИРИДИН-3-ИЛ)-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 9-Метил-8-оксо-2-(пиридин-3-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-метилмочевину (0,730 г, 4,42 ммоль) и 3-пиридинкарбальдегид (1,04 г, 9,73 ммоль) подвергали взаимодействию согласно общему методу В. Продукт очищали с применением полупрепаративной ВЭЖХ с обращенной фазой (10-40% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Фракции, содержащие требуемое вещество, нейтрализовали водным раствором карбоната натрия и затем концентрировали до меньшего объема. Полученный в результате осадок отфильтровывали и промывали водой. Полученное в результате твердое вещество сушили в высоком вакууме при 60°С с получением названного соединения в виде твердого вещества (0,073 г, 0,268 ммоль, выход 6%). 1Н ЯМР (300 МГц, ДМСО-d6) δ 11,6 (с, 1Н), 9,73 (д, J=1,9, 1H), 8,89 (дт, J=10,1, 2,0, 1H), 8,68 (дд, J=4,8, 1,7, 1H), 8,58 (ушир.с, 1Н), 7,96 (с, 1Н), 7,53 (дд, J=7,8, 4,9, 1H), 3,41 (с, 3Н); МС (ESI) m/z 271,5 [M+1]+; т.пл. >350°С.
5.1.167 ПРИМЕР 167
СИНТЕЗ 2-(3-ЦИАНОФЕНИЛ)-8-ОКСО-9-ФЕНИЛ-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. 2-(3-Цианофенил)-8-оксо-9-фенил-8,9-дигидро-7Н-пурин-6-карбоксамид. (Z)-1-(2-Амино-1,2-дициановинил)-3-фенилмочевину (см. пример 131.А) (0,25 г, 1,10 ммоль) и 3-цианокарбоксальдегид (0,314 г, 2,4 ммоль) подвергали взаимодействию согласно общему методу В. Продукт очищали в применением полупрепаративной ВЭЖХ с обращенной фазой (30-80% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин). Фракции, содержащие требуемое вещество, нейтрализовали водным раствором карбоната натрия и затем концентрировали до меньшего объема. Полученный в результате осадок отфильтровывали и промывали водой. Полученное в результате твердое вещество сушили в высоком вакууме при 60°С с получением названного соединения в виде белого твердого вещества (0,078 г, 0,22 ммоль, выход 20%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,92 (с, 1Н), 8,88 (ушир.с, 2Н), 8,61 (д, J=8,0, 1H), 7,98-7,86 (м, 2Н), 7,75 (д, J=7,6, 2H), 7,67 (т, J=8,0, 1H), 7,60 (т, J=8,0, 2H), 7,49-7,42 (м, 1Н); МС (ESI) m/z 358,0 [M+1]+; т.пл. 328-330°С.
5.1.168 ПРИМЕР 168
СИНТЕЗ 6-ОКСО-8-ФЕНИЛ-2-(ПИРИДИН-4-ИЛ)-5,6,7,8-ТЕТРАГИДРОПТЕРИДИН-4-КАРБОКСАМИДА
А. Этил-2-хлор-6-((2-этокси-2-оксоэтил)(фенил)амино)-5-нитропиримидин-4-карбоксилат. Раствор этил-2,6-дихлор-5-нитропиримидин-4-карбоксилата (1,5 г, 5,67 ммоль) в безводном тетрагидрофуране (20 мл) охлаждали до -78°С в атмосфере азота. Затем добавляли по каплям раствор этил-2-(фениламино)ацетата (1,1 г, 6,22 ммоль) и диизопропилэтиламина (3,0 мл, 17,01 ммоль) в безводном тетрагидрофуране (10 мл) при перемешивании в течение 10 мин. Реакционную смесь перемешивали при -78°С в течение 6 ч, с последующим добавлением водного раствора бикарбоната натрия (насыщенный раствор, 10 мл). Смесь экстрагировали этилацетатом (3×10 мл). Органическую фазу сушили над сульфатом натрия и концентрировали до получения остатка, который очищали хроматографией на силикагелевой колонке с нормальной фазой (0-10% этилацетата в гексанах). Фракции, содержащие продукт, объединяли, и растворитель выпаривали с получением названного соединения (1,1 г, 2,89 ммоль, выход 41%). МС (ESI) m/z 409,5 [M+1]+.
В. Этил-2-хлор-6-оксо-8-фенил-5,6,7,8-тетрагидроптеридин-4-карбоксилат. К раствору этил-2-хлор-6-((2-этокси-2-оксоэтил)(фенил)амино)-5-нитропиримидин-4-карбоксилата (1,7 г, 4,16 ммоль) в ледяной уксусной кислоте (20,0 мл) добавляли порошок железа (1,2 г, 20,8 ммоль). Серую суспензию нагревали до 60°С в течение 12 ч. В течение следующих 24 ч добавляли дополнительно порошок железа (суммарно 3,4 г). Уксусную кислоту удаляли при пониженном давлении и остаток суспендировали в метаноле, фильтровали через невысокий слой целита и концентрировали при пониженном давлении до получения остатка. Полученный в результате остаток очищали хроматографией на силикагелевой колонке с нормальной фазой (20-40% этилацетата в гексанах). Фракции, содержащие продукт, объединяли и растворитель выпаривали с получением названного соединения (0,595 г, 1,78 ммоль, выход 43%). МС (ESI) m/z 333,2 [M+1]+.
С. Этил-6-оксо-8-фенил-2-(пиридин-4-ил)-5,6,7,8-тетрагидроптеридин-4-карбоксилат. К раствору этил-2-хлор-6-оксо-8-фенил-5,6,7,8-тетрагидроптеридин-4-карбоксилата (0,160 г, 0,48 ммоль) в безводном ДМФА (6,7 мл) добавляли 4-(трибутилстаннил)пиридин (0,884 г, 2,40 ммоль) и дихлорбис(трифенилфосфин)палладий(II) (0,170 г, 0,24 ммоль). Через раствор продували азот при нагревании в микроволновом реакторе Biotage Emrys Optimizer в течение 30 мин при 120°С. Летучие вещества выпаривали и полученный в результате остаток очищали хроматографией на силикагелевой колонке с нормальной фазой (1-10% метанола в дихлорметане). Фракции, содержащие продукт, объединяли и растворитель выпаривали. Вещество повторно очищали препаративной ВЭЖХ с обращенной фазой (10-70% ацетонитрила + 0,1 TFA в Н2О + 0,1% TFA в течение 30 мин). Фракции, содержащие требуемое вещество, объединяли и концентрировали при пониженном давлении с получением остатка. Остаток, растворенный в метиленхлориде (100 мл), промывали водным раствором карбоната калия (насыщенный раствор, 10 мл) и сушили над сульфатом натрия. Органическую фазу концентрировали и полученное в результате твердое вещество сушили в высоком вакууме при 60°С с получением названного соединения (0,090 г, 0,24 ммоль, выход 50%). МС (ESI) m/z 376,4 [M+1]+.
D. 6-Оксо-8-фенил-2-(пиридин-4-ил)-5,6,7,8-тетрагидроптеридин-4-карбоксамид. Раствор этил-6-оксо-8-фенил-2-(пиридин-4-ил)-5,6,7,8-тетрагидроптеридин-4-карбоксилата (0,037 г, 0,098 ммоль) в безводном метаноле (15 мл) охлаждали до -78°С. Затем раствор насыщали газообразным аммиаком. Реакционный сосуд герметизировали при -78°С и предоставляли возможность нагреваться до комнатной температуры. Через 18 ч реакционную смесь охлаждали до -78°С и сосуд соединяли с атмосферой. Летучие вещества выпаривали и полученные в результате твердые вещества сушили в вакууме с получением названного соединения (0,029 г, 0,083 ммоль, выход 85%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,4 (с, 1Н), 8,83 (с, 1Н), 8,62 (д, J=4,5, 2H), 8,25 (с, 1Н), 8,11-8,09 (м, 2Н), 7,55-7,50 (м, 4Н), 7,39-7,35 (м, 1Н), 4,67 (с, 2Н). МС (ESI) m/z 347,2 [M+1]+; т.пл. 298-304°С.
5.1.169 ПРИМЕР 169
СИНТЕЗ 2-(3-ГИДРОКСИФЕНИЛ)-9-((1R,4R)-4-(МЕТОКСИМЕТИЛ)ЦИКЛОГЕКСИЛ)-8-ОКСО-8,9-ДИГИДРО-7Н-ПУРИН-6-КАРБОКСАМИДА
А. трет-Бутил-(1R,4R)-4-(гидроксиметил)циклогексилкарбамат. трет-Бутил-транс-4-аминоциклогексанкарбоновую кислоту (1,5 г, 6,17 ммоль) разбавляли тетрагидрофураном (20 мл) и охлаждали до -10°С. Добавляли N-метилморфолин (0,624 г, 6,17 ммоль) и изобутилхлорформиат (1,12 г, 8,23 ммоль) и реакционную смесь перемешивали в течение десяти мин. Затем добавляли одной порцией борогидрид натрия (0,842 г, 6,17 ммоль). Через две мин добавляли по каплям метанол (5 мл) и реакционную смесь перемешивали при 0°С в течение 30 мин. Затем раствор разбавляли дихлорметаном, распределяли с 5% раствором гидроксида натрия (3х), сушили над сульфатом магния, фильтровали и растворитель удаляли при пониженном давлении с получением названного соединения без очистки (1,85 г, количественный выход).
В. трет-Бутил-(1R,4R)-4-(метоксиметил)циклогексилкарбамат. Гидрид натрия (0,216 г, 8,98 ммоль) суспендировали в тетрагидрофуране (30 мл) и перемешивали при 0°С. Затем добавляли (1R,4R)-4-(гидроксиметил)циклогексилкарбамат (1,0 г, 5,99 ммоль) и простой 15-краун-5-эфир (1,385 г, 6,29 ммоль), перемешивали в течение 30 мин. Затем добавляли метилйодид (0,393 мл, 8,98 ммоль) и раствор перемешивали при окружающей температуре в течение 16 ч. Реакция не завершилась. Добавляли дополнительно гидрид натрия (1,0 экв.) и продолжали перемешивание. Как только исходное вещество израсходовалось (мониторинг методом ТСХ), раствор конденсировали при пониженном давлении и распределяли между этилацетатом и водой (3х). Органические вещества объединяли, сушили сульфатом магния, фильтровали и растворитель удаляли при пониженном давлении. Полученное в результате масло очищали путем хроматографии (Biotage) (0-50% этилацетата в гексанах) с получением названного соединения (0,280 г, 26%). МС (ESI) m/z 188,3 [M+1]+.
С. (1R,4R)-(4-Метоксиметил)циклогексанамин. трет-Бутил(1R,4R)-4-(метоксиметил)циклогексилкарбамат (0,28 г, 1,15 ммоль) растворяли в 1,4-диоксане (3 мл). Добавляли 4,0 н. хлористоводородную кислоту в диоксане (2 мл) и раствор перемешивали при окружающей температуре в течение 16 ч. Раствор конденсировали при пониженном давлении с получением хлористоводородной соли названного соединения (0,207 г, количественный выход). МС (ESI) m/z 144,3 [M+1]+.
D. (1R,4R)-1-Изоцианато-4-(метоксиметил)циклогексан. (1R,4R)-4-(метоксиметил)циклогексанамин гидрохлорид (0,207 г, 1,15 ммоль) разбавляли толуолом (15 мл) и к раствору добавляли трихлорметилкарбохлоридат (0,228 г, 1,15 ммоль) в толуоле (5 мл). Смесь перемешивали при 105°С в течение 3 ч. Раствор конденсировали с получением масла и применяли без очистки (количественный выход).
Е. 1-((Z)-2-Амино-1,2-дициановинил)-3-((1R,4R)-4-(метоксиметил)циклогексил)мочевина. (1R,4R)-1-Изоцианато-4-(метоксиметил)циклогексан (0,195 г, 1,15 ммоль) растворяли в тетрагидрофуране (10 мл) с последующим добавлением диаминомалеонитрила (0,125 г, 1,15 ммоль). Раствору предоставляли возможность перемешиваться при окружающей температуре в течение 16 ч. Раствор очищали препаративной ВЭЖХ с обращенной фазой (20-100% ацетонитрила + 0,1% TFA в Н2О + 0,1% TFA, в течение 30 мин) с получением названного соединения (0,186 г, 62%). МС (ESI) m/z 278,5 [M+1]+.
F. 2-(3-Гидроксифенил)-9-((1R,4R)-4-(метоксиметил)циклогексил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид. 1-((Z)-2-Амино-1,2-дициановинил)-3-((1R,4R)-4-(метоксиметил)циклогексил)мочевину (0,180 г, 0,649 ммоль), 3-гидроксибензальдегид (0,159 г, 1,29 ммоль) и триэтиламин (0,2 мл, 1,43 ммоль) объединяли в метаноле (10 мл). Раствор перемешивали при окружающей температуре в течение 16 ч. Продукт в виде осадка отфильтровывали, промывали ацетонитрилом и сушили при пониженном давлении с получением названного соединения (0,042 г, 16%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,50 (с, 1Н), 9,55 (с, 1Н), 8,32 (с, 1Н), 8,01 (д, J=7,99, 1H), 7,90 (м, 2Н), 7,28 (т, J=7,59, 1H), 6,87 (дд, J=6,79, 1,59, 1H), 4,24 (м, 1Н), 3,27 (с, 3Н), 3,23 (д, J=6,39, 2H), 2,42 (м, 2Н), 1,90 (д, J=11,59, 2H), 1,81 (д, J=11,59, 2H), 1,70 (с, 1Н), 1,35 (кв, J=14,79, 2H); МС (ESI) m/z 398,1 [M+1]+; т.пл. 354-356°С.
5.2 БИОЛОГИЧЕСКИЕ ПРИМЕРЫ
5.2.1 Испытание MG63 pS6 MesoScale
Нижеследующее представляет собой пример испытания, которое можно применять для определения противораковой активности тестируемого соединения.
В данном испытании применяли клетки остеосаркомы человека MG63 (АТСС: CRL-1427) (пассаж 7-15). Клетки сохраняли с применением DMEM (высокое содержание глюкозы с L-глутамином), 10% FBS и Pen/Strep. Применяли следующие буферы: полный лизисный буфер Tris (для 10 мл применяли: 100 мкл ингибитора I фосфатазы (100Х исходного раствора), 100 мкл ингибитора II фосфатазы (100Х исходного раствора), 1 таблетку Complete Mini (не содержащего ЭДТА), 40 мкл PMSF, все тщательно смешивали в течение 5 минут при комнатной температуре); 1Х трис буфером (для 250 мл применяли: 25 мл 10Х трис буфером, 225 мл деионизированной воды, сохраняли при комнатной температуре); MSD блокирующий раствор-А (для 20 мл применяли: 20 мл 1Х трис буфером и 600 мг MSD блокатора А, сохраняли на льду); буфер для разведения антитела (для 3 мл применяли: 1 мл блокирующего раствора-А, 1,82 мл 1Х трис буфером, 150 мкл 2% MSD блокатора D-М, 30 мкл 10% MSD блокатора D-R, сохраняли на льду).
В первый день испытания днем клетки помещали в 96-луночные плоскодонные планшеты для клеточной культуры 5000 клеток/на лунку в 100 мкл объема. На 2 день утром тестируемые соединения разводили до требуемой концентрации и добавляли к клеткам. Клетки обрабатывали соединением в течение 16-24 часов при 37°С, 0,5% СО2.
Планшеты блокировали приблизительно 5 минут перед завершением обработки соединением путем добавления 150 мкл MSD блокирующего раствора-А в планшет и инкубирования при энергичном встряхивании при комнатной температуре в течение 1 часа.
Клетки собирали и получали лизаты удалением среды многоканальной пипеткой, промыванием 1Х охлажденной на льду PBS (не содержащей Ca, не содержащей Mg), добавлением 50 мкл/на лунку полного лизисного буфера трис и инкубировали со встряхиванием при 4°С в течение 1 часа.
Образцы лизата добавляли к MSD многолуночному планшету пипетированием клеточных лизатов вверх-вниз приблизительно 4-5 раз, переносили 25 мкл/на лунку к MSD многолуночному планшету (RA для отрицательного контроля и RB для положительного контроля) (лизисный буфер добавляли только к фоновым лункам) и инкубировали с энергичным встряхиванием при комнатной температуре в течение 2 часов.
Добавляли обнаруживаемые антитела разведением анти-pS6 антитела (меченые SULFO-TAG, чувствительные к свету) к 3 мл охлажденного буфера для разведения антитела до конечной концентрации 10 нМ, добавляли 25 мкл/на лунку 10 нМ обнаруживаемого антитела к MSD планшету, инкубировали с энергичным встряхиванием при комнатной температуре в темноте в течение 1 часа и промывали планшет 4 раза 1Х трис буфером.
Планшет считывали добавлением 150 мкл/на лунку 1Х трис буфером (с поверхностно-активным веществом) и применением, например, устройства для считывания планшетов MSD SECTOR и соответствующей программы для анализа данных.
5.2.2 Испытание mTOR HTR-FRET
Нижеследующее представляет собой пример испытания, которое можно применять для определения mTOR ингибирующей активности тестируемого соединения. Реагенты приготавливали следующим образом: «Simple TOR buffer» (применяемый для разведения TOR фракции с высоким содержанием глицерина): 10 мМ трис pH 7,4, 100 мМ NaCl, 0,1% Tween-20, 1 мМ DTT (из 1 М исходного раствора, замороженного при -20°С непосредственно перед применением). Для удобства большое количество «Simple TOR buffer» мас./об. DTT можно сохранять при 4°С. Буфер можно приводить к комнатной температуре и добавлять DTT непосредственно перед разведением TOR фракции.
Раствор 5XKB/5XMn/5XATP (применяемый для разбавления субстрата GST-p70S6kin 81 a.a. непосредственно перед применением) (40 мл, представляющее собой количество для скрининга):
Раствор фермента: разведенная TOR фракция 1:14 в “Simple TOR Buffer”. Для данной части это представляет собой 640 мкг/мл TOR фракции, разведенной 14Х с получением 45,7 мкг/мл TOR в буфере (т.е. 7,85 мл TOR объединенной фракции + 102,1 мл Simple TOR буфера = 110 мл 14Х разведенной TOR фракции). Каждая часть фермента должна быть QC'd перед испытанием.
Раствор субстрата: раствор можно приготовить непосредственно перед испытанием, если это предпочтительно. Разводят 5,3 мг/мл исходного фрагмента GST-p70S6 киназы до 3,5 мкг/мл (97 нМ) рабочего исходного раствора в растворе 5XKB/5XMn/5XATP (т.е. 26,41 мкл (5,3 мг/мл) GST-p70S6 + 40 мл 5XKB/5XMn/5XATP = 40 мл 3,5 мкг/мл (97 нМ)).
Буфер для испытания (для разведения антител, применяемых в реагенте для детектирования антитела):
Реагент для детектирования антитела (данный реагент должен быть приготовлен непосредственно перед добавлением к испытуемым планшетам):
С применением программы PlateTrak (Screen) или Matrix Pipettor (SAR) добавляли 19,5 мкл разведенной TOR фракции к испытуемому планшету во все лунки: теста, ссылочного или положительного контроля. Добавляли 19,5 мкл “Simple TOR buffer” ко всем лункам отрицательного контроля. Если обрабатывали многократные планшеты такими же соединениями, можно было увеличить объем фермента до кратных количеств, 19,5 мкл, в высоком 384-луночном полипропиленовом планшете.
С применением ЕР3, 0,5 мкл теста, ссылочного или контроля, в ДМСО добавляли к каждой лунке при перемешивании. Планшеты инкубировали в течение 30 минут при комнатной температуре.
С применением PlateTrak программы (Screen) или Matrix Pipettor (SAR) раствор 5 мкл субстрата 5XKB/5XMn/5XATP/5X добавляли к каждой лунке испытательного планшета для начальной реакции. Растворы смешивали в лунках и инкубировали в течение 2 часов при комнатной температуре.
С применением PlateTrak программы (Screen) или Matrix Pipettor добавляли 5 мкл 60 мМ ЭДТА для остановки реакции. Растворы смешивали в лунке и предоставляли возможность выстаивания в течение 15-20 минут перед следующей стадией.
С применением PlateTrak программы (Screen) или Matrix Pipettor добавляли 10 мкл реагента для детектирования антитела. Растворы смешивали в лунке и инкубировали в течение 5 часов до O/N для предоставления возможности антителам образовывать комплексы с фосфорилированным субстратом.
Планшеты считывали на AnalystHT с применением протокола Multi-Method protocol.
5.2.3 Протокол испытания IKK2EE 33 P
Нижеследующее представляет собой пример испытания, которое можно применять для определения IKK2EE ингибиторной активности тестируемого соединения.
Приготавливали реакционный буфер (рН 7,8), содержащий HEPES (20 мМ), MgCl2 (10 мМ), ЭДТА (0,1 мМ), DTT (1 мМ) и Triton X-100 (0,004%). Применяли IKK2EE (0,75 мкг/мл). В качестве субстрата применяли GST-IKBα (50 мкг/мл). Применяли аденозин-5'-трифосфат (1,5 мкМ с 33Р-АТР 5 нКи/мкл). Реакции останавливали трихлоруксусной кислотой (6,25%).
К испытуемому планшету добавляли подходящие количества тестируемых соединений или контролей в 100% ДМСО. Раствор пептидного субстрата (PSS) приготавливали добавлением пептидного исходного раствора к подходящему объему буфера (концентрация пептида в PSS будет составлять приблизительно 100 мкг/мл). Раствор фермент-пептид (EPS) приготавливали добавлением исходного раствора фермента к подходящему объему PSS (концентрация фермента в EPS будет составлять приблизительно 1 мкг/мл). Раствор АТР приготавливали добавлением исходного АТР к подходящему объему буфера (концентрация АТР будет составлять 15 мкМ).
К испытуемым лункам 96-луночных полипропиленовых планшетов добавляли 85 мкл EPS (или PSS для лунок положительного контроля). Раствор АТР затем укомплектовывали добавлением 33Р-АТР (до конечной концентрации 50 мкКи/мл).
Реакцию инициировали добавлением 10 мкл раствора АТР к каждой лунке. Планшеты встряхивали приблизительно 12 секунд и предоставляли возможность инкубирования при комнатной температуре в течение 1 часа. Для остановки реакции добавляли 100 мкл раствора трихлоруксусной кислоты (12,5%) и планшеты инкубировали при комнатной температуре в течение, по меньшей мере, 10 минут. Испытательные планшеты собирали на Millipore Multiscreen harvest Plate FC c применением подходящего сборщика и планшеты сбора промывали раствором 1Х PBS в течение приблизительно 10 секунд (непрерывный поток). Планшетам сбора предоставляли возможность тщательного высыхания и донные части планшетов герметизировали. К каждой лунке добавляли 20-50 мкл Microscint20, верхние части герметизировали и планшеты считывали с применением подходящего сцинтилляционного счетчика.
5.2.4 Протокол испытания IKK2EE NEMO HTRE
Нижеследующее представляет собой пример испытания, которое можно применять для определения IKK2EE ингибиторной активности тестируемого соединения.
Приготавливали реакционный буфер (рН 7,6), содержащий HEPES (20 мМ), MgCl2 (10 мМ), ЭДТА (0,05 мМ), DTT (мМ) и Tritin X-100) (0,004%). Применяли IKK2EE-NEMO (0,7 мкг/мл). В качестве субстрата применяли GST-IKBα (0,5 мкг/мл). Применяли аденозин-5'-трифосфат (1,5 мкМ). Применяемые реагенты для детектирования представляли собой мышиный анти-Р-IKBα (30 нг/мг), Eu антимышиный (300 нг/мл) и Cy5 анти-GST (11,5 мкг/мл). Реакции останавливали ЭДТА (20 мМ).
Раствор фермента (ES) приготавливали добавлением исходного раствора фермента к подходящему объему буфера (концентрация фермента в ES будет составлять приблизительно 1,7 нг/мл). Детектирующую смесь приготавливали добавлением в буфер АТР (конечн. конц. 3,75 мкМ), мышиный анти-P-IKBα (конечн. конц. 75 нг/мл), GST-IKBα (конечн. конц. 1,26 мкг/мл), антимышиный Eu в буфере (конечн. конц. 750 нг/мл).
Добавляли 5 мкл соединения (растворенного в 10% ДМСО) к испытуемым лункам (или 10% ДМСО к контрольным лункам) реакционных планшетов для нижнего связывания. Добавляли 10 мкл ES к испытуемым лункам (или буфер к контрольным лункам) и инициировли реакцию добавлением 10 мкл DM ко всем лункам. Лунки инкубировали при комнатной температуре в течение 45 минут. Смесь для остановки реакции приготавливали добавлением ЭДТА (конечн. конц. 70 мМ) и Су5 анти-GST (конечн. конц. 40 мкг/мл) к подходящему объему буфера. Добавляли смесь для остановки реакции и планшеты встряхивали в течение 20 секунд и инкубировали при комнатной температуре в течение, по меньшей мере, 3 часов (предпочтительно в темноте). Планшеты считывали на Analyst HT с применением Multi-Method HTRF.
5.2.5 Протокол испытания Tyk2 HTRF (с опцией АТР сдвига)
Нижеследующее представляет собой пример испытания, которое можно применять для определения Tyk2 ингибиторной активности тестируемого соединения.
25 мкл/на лунку ДМСО добавляли к колонкам 2 и 14 (за исключением того, что 28,5 мкл добавляли к лунке Р14 384-луночного полипропиленового планшета Greiner). 20 мкл/на лунку ДМСО добавляли ко всем остальным лункам.
5 мМ растворов соединения добавляли путем добавления 5 мкл 30 мМ соединения к 25 мкл ДМСО в колонки 2 и 14 планшета. 1,5 мМ ссылочного контроля приготавливали добавлением 1,5 мкл 30 мМ JAK3 ингибитор VI к 28,5 мкл ДМСО в лунку Р14.
Затем осуществляли серийное разведение следующими стадиями: (i) соединения в колонке 2 смешивали пипетированием 20 мкл вверх-вниз 6Х; (ii) 10 мкл/на лунку соединений в ДМСО переносили из одной колонки в следующую колонку для колонок 2-11; (iii) содержимое лунок смешивали пипетированием 20 мкл вверх-вниз 6Х; (iv) верхние концы промывали 3×25 мкл ДМСО, 2×25 мкл снова ДМСО; (v) стадии i-iv повторяли для колонок 14-23.
Приготавливали следующие буферы:
Буфер для испытания: 50 мМ HEPES pH 7,6; 1 мМ DTT; 10 мМ MgCl2; 0,01% Triton Х100; 0,01% BSA и 0,1 мМ ЭДТА.
Киназа в буфере для испытания: 450 нг/мл TYK2 KD (Carna Biosciences 08-147 Lot 06CBS-3022D).
Смесь для субстрата/детектирования (1Х АТР) в буфере для испытания: 188 нМ DyLight 647-стрептавидин (Pierce 21824); 5 мкМ Biotin-EQEDEPEGDYFEWLE (Lyn субстратный пептид); 750 нг/мл Eu-анти-фосфо-тирозин (Perkin Elmer AD0069); 62,5 мкМ АТР; 80 нМ субстратного пептида (American Peptide Company 332722).
Смесь для субстрата/детектирования (20Х АТР) в буфере для испытания: 188 нМ DyLight 647-стрептавидин; 5 мкМ Biotin-EQEDEPEGDYFEWLE; 750 нг/мл Eu-анти-фосфо-тирозин; 1250 мкг АТР; 80 нМ субстратного пептида.
14,5 мкл/на лунку ферментной смеси или буфера для разведения (фоновые контроли) добавляли к 384-луночным черным планшетам Costar.
Добавление соединения и смешивание осуществляли следующими стадиями: (i) 0,5 мкл/на лунку ДМСО/соединений в ДМСО переносили из 384-луночного полипропиленового планшета Greiner в планшет, содержащий 14,5 мкл/на лунку ферментной смеси и буфера для разведения; (ii) смешивали пипетированием 10 мкл вверх-вниз 4Х; (iii) верхние концы промывали 4×10 мкл в ДМСО, 2×20 мкл еще раз ДМСО; (iv) стадии i-iii повторяли до тех пор, пока все планшеты не укомплектовывали.
10 мкл/на лунку смеси для субстрата/детектирования добавляли и инкубировали при комнатной температуре 2 часа (на шейкере в течение первых 2+ минут).
Добавляли 10 мкл/на лунку 50 мМ ЭДТА/0,01% Triton X100 и инкубировали >15 минут при комнатной температуре (на шейкере в течение первых 2+ минут).
Планшеты считывали при 665 нм и 620 нм испускания на устройстве Analyst Gt протокол HTRF-SP-A (импульсы=665/620 х 10000).
5.2.6 Протокол испытания Syk HTRF
5 мкл/на лунку ДМСО добавляли к колонке 2 лунки А-О и 29,5 мкл к лунке Р2 384-луночного полипропиленового планшета Greiner. 20 мкл/на лунку ДМСО добавляли к колонкам 1 и 3-12.
25 мМ растворов соединения приготавливали добавлением 25 мкл 30 мМ соединения к колонке 2 и 0,5 мкл 30 мМ ссылочного контроля к лунке Р2.
Затем осуществляли серийное разведение следующими стадиями: (i) соединения в колонке 2 смешивали пипетированием 20 мкл вверх-вниз 6Х; (ii) 10 мкл/на лунку соединений в ДМСО переносили из одной колонки в следующую колонку для колонок 2-11; (iii) содержимое лунок смешивали пипетированием 20 мкл вверх-вниз 6Х; (iv) верхние концы промывали 3×25 мкл ДМСО, 2×25 мкл снова ДМСО (фигура 1).
Приготавливали следующие буферы:
Буфер для разведения: 50 мМ HEPES pH 7,6; 1 мМ DTT; 10 мМ MgCl2; 0,01% Triton Х100; 0,01% BSA; 0,1 мМ ЭДТА.
Ферментная смесь в буфере для разведения: 8,621 нг/мл Syk (Carna Biosciences 08-176).
Стартовая смесь в буфере для разведения: 87,5 мкМ АТР; 80 нМ субстратного пептида (American Peptide Company 332722) (фигура 2).
14,5 мкл/на лунку ферментной смеси или буфера для разведения (фоновые контроли) добавляли к 384-луночным черным планшетам Costar.
Добавление соединения и смеси осуществляли следующими стадиями: (i) 0,5 мкл/на лунку ДМСО/соединения в ДМСО переносили из 384-луночного планшета Greiner к левой половине испытуемого планшета, содержащей 14,5 мкл/на лунку ферментной смеси и буфер для разведения; (ii) смешивали пипетированием 10 мкл вверх-вниз 4Х; (iii) верхние концы промывали 4×10 мкл в ДМСО, 2×20 мкл еще раз в ДМСО: (iv) стадии i-iii повторяли с переносом в правую половину испытуемого планшета; (v) стадии i-iv повторяли с каждым соединением/испытуемым планшетом до тех пор, пока не укомплектовывали все планшеты (фигура 3).
Добавляли 10 мкл/на лунку стартовой смеси и инкубировали при комнатной температуре на шейкере в течение 2 минут (общее время реакции 1 час).
Приготавливали следующие буферы:
Останавливающий раствор в буфере для разведения: 120 мМ ЭДТА.
Смесь антител в буфере для разведения: 4,86 мкг/мл DyLight 647 стрептавидин (Pierce 21824); 1 мкг/мл Lance Eu-антифосфотирозин (PerkinElmer AD0069).
Добавляли 5 мкл/на лунку/в буфере для разведения и инкубировали при комнатной температуре на шейкере в течение 2 минут.
Добавляли 10 мл/на лунку смеси антител и инкубировали при комнатной температуре на шейкере в течение 2 минут (общее время 4 часа, до ночи).
Планшеты считывали при 665 нм и 620 нм испускания на Analyst GT протокол HTRF-SP-A или EnVision протокол Steve's TR-FRET.
5.2.7 Протокол функционального испытания Syk (экспрессии CD69 в анти-IgM стимулированных первичных В-клетках
Клетки: Первичные В-клетки очищали из светлого слоя кровяных сгустков клеточных препаратов, полученных от здоровых человеческих доноров в San Diego Blood Bank (SDBB). Клетки сохраняли в RPIM/10% FBS.
Реагенты: AffiniPure F(ab') фрагмент козьего против человеческого IgM (Jackson, кат. 109-006-129, 1,3 мг/мл); РЕ меченный против человеческого СВ69 (BD Pharmingen, кат. 555531, 2 мл); 7AAD (BD Pharmingen, кат. 559925, 2 мл); RosetteSep B-клеткой обогащенный реагент (Stem Cell Technologies, кат. 15064, 10 мл); Ficoll-Paque Plus (Amersham, кат. 17-440-02; FBS окрашивающий буфер (BD Pharmingen).
Протокол: (i) Клеточный препарат светлого слоя пронумерован заранее от SDBB (обычно пронумерованы два в случае трудности, неожиданно появившейся с одним их них); (ii) В-клетки очищали с применением метода RosetteStep отрицательного отбора следующим образом:
а. 2,0 мл Реагента RosetteStep добавляли к 40 мл светлого слоя кровяного сгустка. Каждый светлый слой кровяного сгустка обычно составлял 80-100 мл. Смесь осторожно смешивали и предоставляли возможность оставаться при комнатной температуре в течение 20 минут (может происходить некоторое отстаивание).
b. В колбе с тканевой культурой 40 мл светлого слоя кровяного сгустка смешивали с равным объемом стерильного фильтрованного 2% FBS в PBS (кальций/магний отсутствуют).
с. 35 мл Названного разведенного светлого слоя кровяного сгустка добавляли к каждой из пяти 50-мл пропиленовых конических пробирок. 14 мл Ficoll Paque медленно добавляли под светлый слой кровяного сгустка и на дно каждой пробирки (осторожно, чтобы не смешать со светлым слоем кровяного сгустка).
d. Пробирки центрифугировали со скоростью 2200 об/мин в течение 20 мин в настольной центрифуге Solvall с торможением.
е. После вращения клетки должны быть видимыми на поверхности раздела сыворотка/Ficoll. Сыворотку осторожно отсасывали в точке вблизи поверхности раздела. С применением пипетки Пастера и управляющего устройства pipetteman клеточный слой осторожно удаляли из поверхности раздела с удалением немного Ficoll, насколько возможно.
f. Извлеченные клетки разбавляли (приблиз. 10 раз) в 100 мл 2% FBS в PBS, центрифугировали со скоростью 1200 об/мин в течение 5 минут и клеточную гранулу ресуспендировали в 5-10 мл RPMI среде для выращивания, в зависимости от извлечения ожидаемых клеток.
Клетки считали и клеточную плотность устанавливали до 1 млн/мл в среде выращивания RPMI. Предварительно обработанный соединением планшет, с 96 лунками круглодонной формы, приготавливали с достаточным объемом клеток для покрытия требуемого количества лунок, рассчитанных 50 мкл клеток/на лунку в обрабатываемом планшете. В отдельном 96-луночном планшете соединения разбавляли 1:50 в среде для выращивания RPMI. 22 мкл разбавленного соединения добавляли к 200 мкл клеток в обработанном соединением планшете. Смесь помещали в инкубатор для тканевой культуры на 30-60 минут.
Приготавливали раствор 20 мкг/мл анти-IgM в среде для выращивания RPMI. Добавляли 50 мкл раствора анти-IgM на лунку в новый 96-луночный планшет круглодонной формы (планшет клеточной стимуляции). Включали только контроли использованной культуральной среды. 50 мкл предварительно обработанных соединением клеток добавляли к анти-IgM содержащему планшету с применением многоканального пипеттора. Смесь помещали обратно в инкубатор тканевой культуры на 12-14 часов.
Планшет центрифугировали со скоростью 1200 об/мин в течение 5 минут. Среду выгружали и планшет осторожно переворачивали для сливания, и подвергали блоттингу сухим. Приготавливали достаточное количество раствора антител для покрытия планшета, задавая 100 мкл окрашивающего буфера, содержащего 5 мкл CD69 антител/на лунку. Добавляли 100 мкл раствора антител на лунку, планшет осторожно постукивали для смешивания, планшет покрывали алюминиевой фольгой и помещали в ящик при комнатной температуре на 30 минут.
Планшет центрифугировали, разгружали и подвергали блоттингу. Планшет промывали однократно 250 мкл окрашивающего буфера, центрифугировали, разгружали и подвергали блоттингу. Конечную клеточную гранулу ресуспендировали в 100 мкл окрашивающего буфера и считывали на цитометре.
5.2.8 Syk функциональное испытание S.O.P. (IgE-зависимая секреция бета-гексозаминидазы из LAD2 линии тучных клеток человека)
Обзор: LAD2 клетки помещали в планшет 96-луночного формата, сенсибилизировали посредством FcepsilonR c NP-IgE и дегранулировали сшивкой с NP16-BSA. Супернатанты собирали и измеряли компоненты секреторных гранул, включающих бета-гексозаминидазу различными колориметрическими анализами.
Клетки: LAD2 клетки получали Metcalf lab у NIH. Для подробного описания дериватизации, характеристики и выращивания/хранения таких клеток имеется ссылка на оригинальную публикацию (Kirshrnbaum, et al., Leukemia Research 27:677-682, 2003). Клетки выращивали совсем медленно, с удваиванием каждые 10-14 дней, и поэтому необходимо питать путем полудеплеции каждую неделю и нечасто разделять. Среда для выращивания: StemPro-34 плюс сывороточный конглютинин (Invitrogen) c 100 нг/мл рекомбинантного человеческого SCF (BioSource). Клетки можно выдерживать в культуре приблизительно 15 пассажей перед морфологическими и функциональными изменениями.
Реагенты: химерный человеческий нитрофенил-IgE (Serotec, MCA333S, 20 мкг/мл исходного раствора); NP16-BSA (Bioseach Technologies, N5050-10 мг, 10 мг/мл исходного раствора); PNAG субстрат (п-нитрофенил-N-ацетил-β-D-глюкозаминид; Sigma N-9376) 0,004 М = 1,37 мг/мл; приготавливали 1,37 мг/мл в цитрат/фосфатном буфере, 150 мкл/в образце (будет требоваться 30-60 мин при 37°С с обычным интенсивным перемешиванием)); цитрат/фосфатный буфер (0,04 М безводной лимонной кислоты (FW 192 г/моль); 2 мл 1 М лимонной кислоты (Hampton Research); 0,02 M Na2HPO4; 2 мл 0,5 М Na2HPO4 (SIGMA), применяли 5н NaOH до рН 4,6 (приблизит. 1 мл) на 50 мл раствора); модифицированный Tyrod'e буфер (Tyrode's Buffer Powder (SIGMA, T2145) одна пробирка на 1 л дистиллированной воды; предоставляли возможность порошку растворяться и затем добавляли следующие компоненты: 1 М HEPES буфера рН 7,8 до 20 мМ конечн. конц. (1:50), 0,5 М Na2HPO4 до 0,5 мМ конечн. конц. (1:1000), 0,04% BSA (400 мг/л), рН должен быть 7,4); глициновый останавливающий раствор (0,32 М глицина, 2,4 г/100 мл; 0,2 М карбоната натрия (FW 106 г/моль), 2,5 г/100 мл).
Протокол: LAD2 осторожно удаляли из культуральной колбы, собирали и центрифугировали при 1200 об//мин в течение 5 минут. Использованную культуральную среду удаляли и сохраняли. Клетки ресуспендировали при 0,8-1 миллион/мл в сохраненной культуральной среде. 100 мкл 0,5 мкг/мл NP-IgE засевали в сохраненной культуральной среде в круглодонный 96-луночный планшет. Примечание: раствор IgE нуждается в осветлении для удаления агрегатов центрифугированием в течение 10 мин при >10000 об/мин при 4°С. 100 мкл клеток добавляли к планшету и помещали обратно в инкубатор для тканевой культуры на 12-14 часов для сенсибилизации клеток и нагрузки FcepsilonR рецепторов. Холодному модифицированному буферу Tyrode's предоставляли возможность нагреваться до комнатной температуры в течение ночи.
Следующим утром планшет центрифугировали при 1200 об/мин в течение 5 минут. Среды удаляли многоканальным пипеттором. Клеточную гранулу ресуспендировали в 100 мкл модифицированного буфера Tyrode's с осторожным растиранием (5 ударов). Клеткам предоставляли возможность находиться в покое в течение 3,5 часов в инкубаторе для тканевой культуры. Примечание: В течение этого времени необходимо нагреть цитрат/фосфатный буфер до 37°С и затем ресуспендировать субстрат PNAG до 1,3 мг/мл при периодическом интенсивном перемешивании. Серии соединений разбавляли 1:50 в модифицированном буфере Tyrode's и затем добавляли 11 мкл соединения без дополнительного перемешивания к каждой лунке (получая 0,2% ДМСО конечную концентрацию). Соединение предварительно инкубировали в течение 30-60 минут в инкубаторе для тканевой культуры.
Добавляли 12 мкл 1,0 мкг/мл NP16-BSA, разбавленного в модифицированном Tyrode's. Суммарный объем теперь составлял 123 мкл. Можно добавлять иономицин при 100 нМ конечн. конц. вместо NP-BSA в качестве Syk-независимого контроля, для стимуляции. Инкубировали в инкубаторе для тканевой культуры в течение 90 минут.
Планшет центрифугировали при 1200 об/мин в течение 5 минут, 75 мкл супернатанта (SN) переносили в пустой 96-луночный планшет для сохранения. Оставшийся SN удаляли из планшета с клетками и отбрасывали. Добавляли к клеточной грануле 125 мкл 0,1% triton X-100 в модифицированном буфере Tyrode's, пипетировали вверх-вниз для лизиса клеток и смесь инкубировали на льду в течение 15 мин.
Добавляли 30 мкл супернатанта из сохраненного планшета или 5 мкл лизата клеточной гранулы плюс 25 мкл раствора 0,1% Triton к новым 96-луночным плоскодонным планшетам в идентичном расположении для конечного считывания планшета. Ко всем лункам добавляли 150 мкл субстрата PNAG. Планшет инкубировали при 37°С в бактериальном инкубаторе в течение 1 часа.
К каждой лунке добавляли 50 мкл останавливающего раствора. Лунки с наибольшей активностью будут ярко желтыми. Планшет считывали немедленно при 405 нм.
Вычисляли % высвобождения на лунку (после вычитания фона из всех лунок) = 100 × (SN/(SN + 6 × клеточный лизат)). Чистый % высвобождения = 100 × (SN стим. - SN PBS)/(SN стим. + клеточный лизат стим. - SN PBS).
Критерий количественного контроля испытания: 3 главных параметра осуществления испытания: 1) величина процента высвобждения должна быть между 10-20% в IgE- и ДМСО-обработанных лунках (40% высвобождение с 100 нМ иономицина); 2) величина IC50 с Syk средством соединений должна быть в интервале 50-200 нМ; 3) Z' для испытания должен быть >0,55.
5.2.9 Протокол испытания с Syk биомаркером (измерение фосфоBLNK с помощью PhosFlow в анти-IgM стимулированных Ramos)
Клетки: Ramos В-леточную лимфому (клон RA1, CRL1596) из АТСС быстро выращивали и для поддерживания подвергали необходимому разделению 1:20 каждые 3-4 дня. Клетки выращивали в RPMI/10% FBS.
Реагенты: AffiniPure F(ab') фрагмент козьего против человеческого IgM (Jackon, кат. 109-006-129, 1,3 мг/мл); РЕ мышиный анти-фосфоBLNK (pY84, BD Pharmingen, кат. 558442); CytoFix реагент (BD Pharmingen, кат. 554655); Perm/Wash буфер I (BD Pharmingen, кат. 557885, 10X раствор); окрашивающий буфер BSA (BD Pharmingen, кат. 554657).
Протокол: Ramos клетки разделяли 1:1 с помощью свежеприготовленных сред для выращивания за день до эксперимента. В день эксперимента клетки центрифугировали при 1200 об/мин в течение 5 минут. Все использованные культуральные среды сохраняли. Клетки ресуспендировали при 1 млн/мл в используемых культуральных средах. Предварительно обработанный соединением планшет приготавливали в 96-луночном круглодонной формы планшете с достаточным объемом клеток для покрытия требуемого количества лунок, рассчитывая 50 мкл клеток/на лунку в обработанном планшете, например, добавляли на 4 лунки 200 мкл клеток. В отдельном 96-луночном планшете соединения разбавляли 1:50 в используемых культуральных средах. 22 мкл разбавленного соединения добавляли к 200 мкл клеток в предварительно обработанный соединением планшет. Засеянный планшет помещали обратно в инкубатор для тканевой культуры на 30-60 минут. Реагент CytoFix предварительно нагревали на водяной бане при 37°С перед стимулированием клеток.
Приготавливали 40 мкг/мл анти-IgM раствора в используемых культуральных средах. Добавляли 50 мкл анти-IgM раствора на лунку в новый 96-луночный круглодонный планшет (планшет клеточной стимуляции). Контроли включали в себя только расходуемые культуральные среды. С применением многоканального пипеттора 50 мкл предварительно обработанных соединением клеток быстро добавляли к планшету, содержащему анти-IgM, и планшет помещали обратно в инкубатор для культуры ткани на 10 минут.
Равные объемы (100 мкл) предварительно нагретого реагента CytoFix добавляли ко всем лункам планшета для стимулирования клеток. Планшет помещали обратно в инкубатор для культуры ткани на 10 минут, центрифугировали при 1200 об/мин в течение 5 минут и среды осторожно сливали, а планшет подвергали блоттингу сухим.
Добавляли ко всем лункам 100 мкл Perm/Wash буфер I. Планшет оставляли при комнатной температуре в течение 10 минут, центрифугировали при 1200 об/мин в течение 5 минут и среды осторожно сливали, а планшет подвергали блоттингу сухим. Клетки промывали три раза 200 мкл окрашивающего буфера BSA. Планшет центрифугировали, переворачивали и подвергали блоттингу.
Приготавливали достаточное количество раствора антител для покрытия планшета, рассчитывая 100 мкл содержащих окрашивающий буфер 5 мкл pBLNK антител/на лунку. Добавляли 100 мкл раствора антител на лунку, планшет осторожно постукивали для смешивания и покрывали алюмиевой фольгой, и помещали в ящик при комнатной температуре на 30 минут.
Планшет центрифугировали, переворачивали и подвергали блоттингу. Планшет промывали один раз 200 мкл окрашивающего буфера. Планшет центрифугировали, переворачивали и подвергали блоттингу. Конечную клеточную гранулу ресуспендировали в 100 мкл окрашивающего буфера и считывали на цитометре.
Обнаружено, что соединения в таблице 1 имеют следующие значения скрининговых испытаний mTOR и IKK-2.
В представленной выше таблице применяли следующую систему: ***** равно 0,1-5 мкМ, **** равно 5,1-10 мкМ, *** равно 10,1-20 мкМ, ** равно 20,1-30 мкМ, * равно >30 мкМ. “ND” означает, что соединение не тестировали против такого конкретного фермента.
Описанные варианты осуществления не ограничиваются в объеме специфическими вариантами осуществления, описанными в примерах, которые предназначены в качестве иллюстрации немногих аспектов описанных вариантов осуществления, и любые варианты осуществления, которые представляют собой функционально эквивалентные, включены в объем посредством настоящего описания. Фактически различные модификации описанных вариантов осуществления, помимо тех, которые продемонстрированы и описаны, будут становиться очевидными специалисту в области техники, и находятся в пределах объема приведенной формулы изобретения.
Процитирован ряд ссылок, описания которых введены посредством ссылки во всей своей полноте.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ПИРРОЛЫ, АКТИВНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2013 |
|
RU2666538C2 |
| 7-ЗАМЕЩЕННЫЕ СУЛЬФОНИМИДОИЛПУРИНОНЫ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИРУСНОЙ ИНФЕКЦИИ | 2017 |
|
RU2838538C1 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2840815C2 |
| 7-ЗАМЕЩЕННЫЕ СУЛЬФОНИМИДОИЛПУРИНОНЫ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИРУСНОЙ ИНФЕКЦИИ | 2017 |
|
RU2751349C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2528046C2 |
| НОВЫЕ АЛКИЛИРУЮЩИЕ СРЕДСТВА | 2013 |
|
RU2632206C2 |
| ТРИЦИКЛИЧЕСКИЕ ПИРРОЛО ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2012 |
|
RU2591191C2 |
| НОВЫЕ ИНДОЛЬНЫЕ И ПИРРОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2693404C2 |
| СПОСОБ МОДУЛЯЦИИ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2525115C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2556984C2 |



Изобретение относится к новым гетероарильным соединениям общей формулы (I) и его фармацевтически приемлемым солям, обладающим свойствами ингибитора протеинкиназы, такой как mTOR, IKK-2, Tyk2, Syk-киназ. Соединения могут найти применение для лечения или предупреждения рака, воспалительных патологических состояний, иммунологических патологических состояний, метаболических патологических состояний. К таким заболеваниям относятся, в частности, рак почки и диабет. В формуле (I)

R1 представляет собой замещенный или незамещенный С1-8алкил, замещенный или незамещенный арил, выбранный из фенила, замещенный или незамещенный 5-6-членный гетероарил с 1-3 атомами азота в цикле, выбранный из пиридина, пиразола, индола, индазола, триазола, бензимидазола, 2-(1H-имидазо-[4,5-b]пиридина, замещенный или незамещенный 5-7-членный циклоалкил или замещенный или незамещенный гетероциклоалкил, выбранный из пирролидинила; -Х-А-В-Y-, взятые вместе, образуют -N(R2)CH2C(O)NH-, -N(R2)C(O)CH2NH-, -N(R2)C(O)NH-, -N(R2)ON- или -C(R2)=CHNH-; L представляет собой прямую связь, NH или О; R2 представляет собой замещенный или незамещенный С1-8алкил, замещенный или незамещенный арил, выбранный из фенила, тетрагидронафталина, незамещенный 5-7-членный моно- или 8-членный бициклоалкил; и R3 и R4 представляют собой независимо Н или С1-8-алкил. Заместители в замещенных группах выбираются из одного или более галогена, С1-8алкила, гидроксила, амино, нитро, тиола, простого С1-4 алкилового тиоэфира, циано, карбоксила, С1-4 алкилового сложного эфира, галогеналкила, С6-циклоалкила или гетероарила, выбранного из пиридила, триазола, О-низшего алкила, арила, выбранного из фенила, фенил-низшего алкила, CO2CH3, CONH2, OCHF2, CF3 или OCF3 групп, где CONH2 группа может быть замещена циклогексилом. 5 н. и 19 з.п. ф-лы, 3 ил., 2 табл., 169 пр.
1. Соединение, имеющее следующую формулу:

и его фармацевтически приемлемые соли, где
R1 представляет собой замещенный или незамещенный С1-8алкил, замещенный или незамещенный арил, выбранный из фенила, замещенный или незамещенный 5-6-членный гетероарил с 1-3 атомами азота в цикле, выбранный из пиридина, пиразола, индола, индазола, триазола, бензимидазола, 2-(1Н-имидазо-[4,5-b]пиридина), замещенный или незамещенный 5-7-членный циклоалкил или замещенный или незамещенный гетероциклоалкил, выбранный из пирролидинила;
-X-A-B-Y-, взятые вместе, образуют -N(R2)CH2C(O)NH-, -N(R2)C(O)CH2NH-,
-N(R2)C(O)NH-, -N(R2)C=N- или -C(R2)=CHNH-;
L представляет собой прямую связь, NH или О;
R2 представляет собой замещенный или незамещенный С1-8алкил, замещенный или незамещенный арил, выбранный из фенила, тетрагидронафталина, незамещенный 5-7-членный моно- или 8-членный бициклоалкил; и
R3 и R4 представляют собой независимо Н или С1-8алкил;
где группы, которые являются замещенными, замещены одним или более галогеном, С1-8алкилом, гидроксилом, амино, нитро, тиолом, простым С1-4алкиловым тиоэфиром, циано, карбоксилом, С6алкиловым сложным эфиром, галогеналкилом, С6циклоалкилом или гетероарилом, выбранным из пиридила, триазола, О-низшим алкилом, арилом, выбранным из фенила, фенил-низшим алкилом, CO2CH3, CONH2, OCHF2, CF3 или OCF3 группами, где CONH2 группа может быть замещена циклогексилом;
при условии, что R2 не является замещенным или незамещенным фуранозидом и соединение не является
8,9-дигидро-8-оксо-9-фенил-2-(3-пиридинил)-7Н-пурин-6-карбоксамидом,
8,9-дигидро-8-оксо-9-фенил-2-(3-пиридинил)-7Н-пурин-6-карбоксамидом,
8,9-дигидро-8-оксо-9-фенил-2-(3-пиридинил)-7Н-пурин-6-карбоксамидом,
2-(4-цианофенил)-8-оксо-9-фенил-8,9-дигидро-7Н-пурин-6-карбоксамидом,
2-(4-нитрофенил)-8-оксо-9-фенил-8,9-дигидро-7Н-пурин-6-карбоксамидом,
9-бензил-2-(4-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамидом,
2-метил-8-оксо-9-фенил-8,9-дигидро-7Н-пурин-6-карбоксамидом,
9-бензил-9Н-пурин-2,6-дикарбоксамидом,
9-[2,3-бис[(бензоилокси)метил]циклобутил]-2-метил-9Н-пурин-6-карбоксамидом,
9-бензил-2-метил-9Н-пурин-6-карбоксамидом,
9-(2-гидроксиэтил)-2-метил-9Н-пурин-6-карбоксамидом,
9-(2-гидроксиэтил)-2-(трифторметил)-9Н-пурин-6-карбоксамидом,
9-(2-гидроксиэтил)-2-(проп-1-енил)-9Н-пурин-6-карбоксамидом,
9-(2-гидроксиэтил)-2-фенил-9Н-пурин-6-карбоксамидом,
9-(3-гидроксипропил)-2-метил-9Н-пурин-6-карбоксамидом,
9-(3-гидроксипропил)-2-(трифторметил)-9Н-пурин-6-карбоксамидом,
2-метил-9-фенилметил-9Н-пурин-6-карбоксамидом или
2-метил-9-β-D-рибофуранозил-9Н-пурин-6-карбоксамидом.
2. Соединение по п.1, где -X-A-B-Y-, взятые вместе, образуют -N(R2)C(O)NH-.
3. Соединение по п.1, где R1 представляет собой замещенный или незамещенный 5-6-членный гетероарил с 1-3 атомами азота в цикле, выбранный из пиридина, пиразола, индола, индазола, триазола, бензимидазола, 2-(1Н-имидазо-[4,5-b]пиридина).
4. Соединение по п.3, где R1 представляет собой замещенный или незамещенный пиридин.
5. Соединение по п.1, где R1 представляет собой замещенный или незамещенный фенил.
6. Соединение по п.1, где R1 представляет собой замещенный или незамещенный 5-7-членный циклоалкил.
7. Соединение по п.6, где R1 представляет собой замещенный или незамещенный циклопентил.
8. Соединение по п.1, где R2 представляет собой замещенный или незамещенный арил, выбранный из фенила, тетрагидронафталина.
9. Соединение по п.8, где R2 представляет собой замещенный или незамещенный фенил.
10. Соединение по п.1, где R2 представляет собой замещенный или незамещенный C1-8алкил.
11. Соединение по п.1, где R2 представляет собой незамещенный 5-7-членный моно- или 8-членный бициклоалкил.
12. Соединение по п.1, где -X-A-B-Y-, взятые вместе, образуют -N(R2)C(O)NH-, R1 представляет собой замещенный или незамещенный 5-6-членный гетероарил с 1-3 атомами азота в цикле, выбранный из пиридина, пиразола, индола, индазола, триазола, бензимидазола, 2-(1Н-имидазо-[4,5-b]пиридина), L представляет собой прямую связь, и R2 представляет собой замещенный или незамещенный арил, выбранный из фенила, тетрагидронафталина.
13. Соединение по п.1, где -X-A-B-Y-, взятые вместе, образуют -N(R2)C(O)NH-, R1 представляет собой замещенный или незамещенный арил, выбранный из фенила, L представляет собой прямую связь, и R2 представляет собой замещенный или незамещенный арил, выбранный из фенила, тетрагидронафталина.
14. Соединение по п.1, где -X-A-B-Y-, взятые вместе, образуют -N(R2)C(O)NH-, R1 представляет собой замещенный или незамещенный 5-6-членный гетероарил с 1-3 атомами азота в цикле, выбранный из пиридина, пиразола, индола, индазола, триазола, бензимидазола, 2-(1Н-имидазо-[4,5-b]пиридина), L представляет собой прямую связь, и R2 представляет собой замещенный или незамещенный С1-8алкил или незамещенный 5-7-членный моно- или 8-членный бициклоалкил.
15. Соединение по п.1, где -X-A-B-Y-, взятые вместе, образуют -N(R2)C(O)NH-, R1 представляет собой замещенный или незамещенный арил, выбранный из фенила, L представляет собой прямую связь, и R2 представляет собой замещенный или незамещенный С1-8алкил или незамещенный 5-7-членный моно- или 8-членный бициклоалкил.
16. Соединение или его фармацевтически приемлемая соль, где соединение представляет собой
9-бензил-8-оксо-2-(пиридин-3-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
N-метил-8-оксо-9-фенил-2-(пиридин-3-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
8-оксо-9-фенил-2-(пиридин-2-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(2-хлорпиридин-3-ил)-8-оксо-9-фенил-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(2-метоксипиридин-3-ил)-8-оксо-9-фенил-8,9-дигидро-7Н-пурин-6-карбоксамид;
N,N-диметил-8-оксо-9-фенил-2-(пиридин-3-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-метил-8-оксо-2-(пиридин-3-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(4-гидроксифенил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-гидроксифенил)-8-оксо-9-о-толил-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(1Н-индол-4-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(1Н-индол-6-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-гидроксифенил)-9-(4-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(2-гидроксипиридин-4-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2-хлорфенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2-фторфенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2,6-дифторфенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-циклогептил-8-оксо-2-(пиридин-3-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2-метоксифенил)-8-оксо-2-(хинолин-5-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-циклопентил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2-метоксифенил)-8-оксо-2-(3-(трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2-метоксифенил)-2-(6-метоксипиридин-3-ил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-гидроксифенил)-8-оксо-9-(4-(трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-бензил-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-гидроксифенил)-8-оксо-9-(2-(трифторметокси)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2,4-дихлорфенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2-метоксифенил)-2-(3-нитрофенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-цианофенил)-8-оксо-9-фенил-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(3-фторфенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2-метоксифенил)-8-оксо-2-(2-(трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(5-фторпиридин-3-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(1-бензилпиперидин-4-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
бензил-4-(6-карбамоил-8-оксо-2-(пиридин-3-ил)-7Н-пурин-9(8Н)-ил)пиперидин-1-карбоксилат;
9-циклогексил-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2-метоксифенил)-8-оксо-2-(3-(трифторметокси)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-фенил-2-(пиридин-3-ил)-9Н-пурин-6-карбоксамид;
6-оксо-8-фенил-2-(пиридин-3-ил)-5,6,7,8-тетрагидроптеридин-4-карбоксамид;
6-оксо-8-фенил-2-(пиридин-4-ил)-5,6,7,8-тетрагидроптеридин-4-карбоксамид;
2-(3-аминофенил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-гидроксифенил)-9-(2-метоксифенил)-9Н-пурин-6-карбоксамид;
9-циклопентил-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-трет-бутил-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
[2-(3-гидроксифенил)-9-(2-метоксифенил)-8-оксо-(7-гидропурин-6-ил)]-N-метилкарбоксамид;
[2-(3-гидроксифенил)-9-(2-метоксифенил)-8-оксо-(7-гидропурин-6-ил)]-N,N-диметилкарбоксамид;
2-(3-гидроксифениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(4-гидроксифениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(транс-4-гидроксициклогексил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(транс-4-гидроксициклогексил)-8-оксо-2-(пиридин-3-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-гидроксифениламино)-9-(2-метоксифенил)-9Н-пурин-6-карбоксамид;
9-изопропил-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
метил-4-(6-карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-ил)бензоат;
2-(2-хлор-3-гидроксифенил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
2-(3-цианофенил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(2-гидроксифениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-гидроксифенил)-9-(4-метокси-2-метилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-гидроксифенил)-8-оксо-9-(2-(трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(4-цианофенил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
4-[6-карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-ил]бензойная кислота;
метил-3-(6-карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-ил)бензоат;
3-(6-карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-ил)бензойная кислота;
2-(3-гидроксифенил)-9-(2-изопропилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(1Н-индазол-6-ил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
2-(4-карбамоилфенил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2-этилфенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2,5-дихлорфенил)-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
2-(3-карбамоилфенил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2,6-дихлорфенил)-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
2-(2-гидроксифенил)-9-(2-метоксифенил)пурин-6-карбоксамид;
2-(1Н-индазол-5-ил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
9-(2,3-дихлорфенил)-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
2-[4-(гидроксиметил)фенил]-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
2-[3-(гидроксиметил)фенил]-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
9-(2-метоксифенил)-8-оксо-2-(пиридин-4-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(4-фтор-3-гидроксифенил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
2-(2-фтор-3-гидроксифенил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
2-[4-(1-гидроксиизопропил)фенил]-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
2-[3-(1-гидроксиизопропил)фенил]-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
9-(2-метоксифенил)-2-(2-нитрофенил)-8-оксо-7-гидропурин-6-карбоксамид;
9-(2-метоксифенил)-2-(4-нитрофенил)-8-оксо-7-гидропурин-6-карбоксамид;
9-(2-метоксифенил)-2-(2-нитрофенил)-8-оксо-7-гидропурин-6-карбоксамид;
9-(2,4-дифторфенил)-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
9-(2-метоксифенил)-2-{3-[(метилсульфонил)амино]фенил}-8-оксо-7-гидропурин-6-карбоксамид;
9-(4-хлор-2-фторфенил)-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
9-(2-хлорфенил)-8-оксо-2-(3-пиридил)-7-гидропурин-6-карбоксамид;
8-оксо-2-(3-пиридил)-9-[2-(трифторметил)фенил]-7-гидропурин-6-карбоксамид;
9-(3-хлор-2-фторфенил)-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
9-(2-фтор-3-трифторметилфенил)-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
9-(2,3,4-трифторфенил)-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
2-(1Н-бензо[d]имидазол-6-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-[3-(ацетиламино)фенил]-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
2-(3-гидроксифенил)-8-(2-метоксифенил)-6-оксо-5,6,7,8-тетрагидроптеридин-4-карбоксамид;
9-(2-метоксифенил)-8-оксо-2-пиразол-4-ил-7-гидропурин-6-карбоксамид;
9-(2-метоксифенил)-8-оксо-2-пиразол-3-ил-7-гидропурин-6-карбоксамид;
9-(4-аминоциклогексил)-2-(3-гидроксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
2-[3-(дифторметил)фенил]-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
2-[5-(дифторметил)-2-фторфенил]-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
2-(1Н-бензо[d]имидазол-4-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(6-гидроксипиридин-3-ил)-8-оксо-9-(2-(трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(1Н-бензо[d]имидазол-6-ил)-9-(2-фторфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-бензимидазол-6-ил-8-оксо-9-[2-(трифторметил)фенил]-7-гидропурин-6-карбоксамид;
2-(5-хлорпиридин-3-ил)-8-оксо-9-(2-(трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
транс-4-(6-карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-иламино)циклогексилкарбамат;
(R)-9-(2-метоксифенил)-8-оксо-2-(пирролидин-3-иламино)-8,9-дигидро-7Н-пурин-6-карбоксамид;
(S)-9-(2-метоксифенил)-8-оксо-2-(пирролидин-3-иламино)-8,9-дигидро-7Н-пурин-6-карбоксамид;
(цис)-4-(6-карбамоил-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-2-иламино)циклогексилкарбамат;
2-(транс-4-гидроксициклогексиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(4-хлорпиридин-3-ил)-8-оксо-9-(2-(трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(цис-4-гидроксициклогексиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(4-((1Н-имидазол-1-ил)метил)фениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(4-гидроксипиридин-3-ил)-8-оксо-9-(2-(трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
(R)-9-(2-метоксифенил)-8-оксо-2-(пирролидин-2-илметиламино)-8,9-дигидро-7Н-пурин-6-карбоксамид;
(S)-9-(2-метоксифенил)-8-оксо-2-(пирролидин-2-илметиламино)-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(4-(1Н-1,2,4-триазол-3-ил)фенил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
2-(2-гидроксиэтиламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2-метоксифенил)-8-оксо-2-(2-(трифторметил)-1Н-бензо[d]имидазол-6-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-(1Н-1,2,4-триазол-3-ил)фенил)-9-(2-метоксифенил)-8-оксо-7-гидропурин-6-карбоксамид;
9-(бифенил-2-ил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(4-(1Н-1,2,4-триазол-3-ил)фенил)-9-(2-фторфенил)-8-оксо-7-гидропурин-6-карбоксамид;
2-(4-(1Н-1,2,4-триазол-3-ил)фенил)-9-(2-изопропилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2-метоксифенил)-2-(2-метил-1Н-бензо[d]имидазол-6-ил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-(гидроксиметил)фениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(2-(гидроксиметил)фениламино)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2-трет-бутилфенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-гидроксифенил)-8-оксо-9-(2-феноксифенил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(1Н-бензо[d]имидазол-6-ил)-9-(2-изопропилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(1Н-индазол-4-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(2-гидроксипиридин-3-ил)-8-оксо-9-(2-(трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(1Н-имидазо[4,5-b]пиридин-6-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(4-(1Н-имидазол-1-ил)фенил)-9-(2-изопропилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2-циклогексилфенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(4-(1Н-имидазол-2-ил)фенил)-9-(2-изопропилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(1Н-бензо[d]имидазол-1-ил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(1Н-имидазо[4,5-b]пиридин-6-ил)-9-(2-изопропилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2-изопропилфенил)-8-оксо-2-(1Н-пирроло[2,3-b]пиридин-5-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(1Н-имидазо[4,5-b]пиридин-6-ил)-8-оксо-9-(2-(трифторметил)фенил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2-метоксифенил)-2-(2-(метилтио)-1Н-бензо[d]имидазол-5-ил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(1Н-индол-5-ил)-9-(2-изопропилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(циклогексилметил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2,3-дигидро-1Н-инден-1-ил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-гидроксифенил)-9-изобутил-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(транс-4-метоксициклогексил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(цис-4-метоксициклогексил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-гидроксифенил)-8-оксо-9-(5,6,7,8-тетрагидронафталин-1-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(4-(1Н-1,2,4-триазол-3-ил)фенил)-9-циклогексил-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-гидроксифенил)-9-(1Н-индол-4-ил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2-фтор-3-метоксифенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2-фтор-5-метоксифенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-циклогексил-2-(1Н-имидазо[4,5-b]пиридин-6-ил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-гидроксифенил)-8-оксо-9-(тетрагидро-2Н-пиран-4-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-гидроксифенил)-8-оксо-9-((тетрагидро-2Н-пиран-4-ил)метил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2-циклопентилфенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-гидроксифенил)-8-оксо-9-(пиперидин-4-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(2-фтор-4-метоксифенил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(1Н-бензо[d]имидазол-6-ил)-9-циклогексил-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-бензимидазол-6-ил-9-(транс-4-метоксициклогексил)-8-оксо-7-гидропурин-6-карбоксамид;
2-(4-(аминометил)фенил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-гидроксифенил)-9-(цис-4-(метоксиметил)циклогексил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
9-(транс-4-аминоциклогексил)-2-(3-гидроксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-гидроксифенил)-9-(2-изобутилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
(R)-2-(3-гидроксифенил)-8-оксо-9-(тетрагидрофуран-3-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
(S)-2-(3-гидроксифенил)-8-оксо-9-(тетрагидрофуран-3-ил)-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-(аминометил)фенил)-9-(2-метоксифенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(4-(1Н-1,2,3-триазол-5-ил)фенил)-9-(2-изопропилфенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(4-(1Н-1,2,4-триазол-3-ил)фенил)-9-(цис-4-метоксициклогексил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(1Н-бензо[d]имидазол-6-ил)-9-(цис-4-метоксициклогексил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(1Н-имидазо[4,5-b]пиридин-6-ил)-9-(цис-4-метоксициклогексил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид;
2-(3-гидроксифенил)-9-((1R,4R)-4-(метоксиметил)циклогексил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид или
9-(2-изопропилфенил)-2-(4-(5-метил-4Н-1,2,4-триазол-3-ил)фенил)-8-оксо-8,9-дигидро-7Н-пурин-6-карбоксамид.
17. Фармацевтическая композиция, обладающая свойствами ингибитора протеинкиназы, содержащая эффективное количество соединения, имеющего формулу:
или его фармацевтически приемлемой соли, где R1, -X-A-B-Y-, L, R2, R3 и R4 имеют значения, указанные в п.1, или соединения по п.16 и фармацевтически приемлемый носитель, эксципиент или разбавитель.
18. Фармацевтическая композиция по п.17, пригодная для перорального, парентерального, через слизистую оболочку, чрескожного введения или местного нанесения.
19. Способ лечения или предупреждения рака, воспалительного патологического состояния, иммунологического патологического состояния или метаболического патологического состояния, опосредованного активностью протеинкиназ, таких как mTOR, IKK-2, Tyk2, Syk-киназ, включающий введение нуждающемуся в этом пациенту эффективного количества соединения, имеющего формулу:
или его фармацевтически приемлемой соли, где R1, -X-A-B-Y-, L, R2, R3 и R4 имеют значения, указанные в п.1, или соединения по п.16, или фармацевтической композиции, включающей указанные соединения и фармацевтический носитель, эксципиент или разбавитель, при условии, что R2 не является замещенным или незамещенным фуранозидом.
20. Способ по п.19, где раковое заболевание представляет собой рак почки.
21. Способ по п.19, где воспалительное патологическое состояние представляет собой диабет.
22. Способ по п.19, где иммунологическое патологическое состояние представляет собой диабет.
23. Способ по п.19, где метаболическое патологическое состояние представляет собой диабет.
24. Способ ингибирования киназы в клеточной экспрессии указанной киназы, включающий контактирование указанной клетки с эффективным количеством соединения, имеющего формулу:

или его фармацевтически приемлемой соли, где R1, -X-A-B-Y-, L, R2, R3 и R4 имеют значения, указанные в п.1, или соединения по п.16, и фармацевтической композиции, включающей указанные соединения и фармацевтический носитель, эксципиент или разбавитель.
| MAGDI Е.А | |||
| et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| BRIAN I.BOOTH et al | |||
| “The Reactions of Diaminomaleonitrile with Isocyanates and Either Aldehides or Ketones Revisited" Journal of Organic | |||

