Настоящая заявка испрашивает приоритет на основании заявки на патент № CN201810804068.3, поданной 20 июля 2018 г.
Область техники
Настоящее изобретение относится к соединению III в качестве ингибитора ЛСД1 и его кристаллической форме, и применению данного соединения и его кристаллической формы для получения лекарственного средства для лечения заболевания, связанного с ЛСД1.
Уровень техники
Эпигенетика регулирует экспрессию генов с помощью разных механизмов, включая ковалентную модификацию гистонов, например метилирование или деметилирование; ковалентную модификацию ДНК, например метилирование или гидроксиметилирование; и реорганизацию ядерного хроматина. Несмотря на то, что данные модификации не изменяют базовую последовательность ДНК, такие эпигенетические изменения могут сохраняться на протяжении жизненного цикла клетки или итерационного процесса клетки при делении клетки [Adrian Bird, Nature, 2007, 396-398]. Таким образом, эпигенетические нарушения могут вызывать и вносить вклад в патологические процессы различных заболеваний (таких как солидные опухоли, гематомы, вирусные инфекции, неврологические расстройства и другие заболевания). В связи с этим эпигенетика стала одним из наиболее активно изучаемых направлений в области развития лекарственных препаратов. Лизин-специфическая деметилаза (ЛСД1, также называемая KDM1A) - первая деметилаза, открытая в 2004 году, - относится к семейству флавинадениндинуклеотид (ФАД)-зависимых аминооксидаз. Структура ЛСД1 состоит из трёх основных доменов: N-концевого домена SWIRM, C-концевого аминооксидазного (AOL) домена и центрального выступающего домена Tower. C-концевой аминооксидазный домен содержит два активных кармана, один из которых служит сайтом связывания с ФДА, а другой является сайтом распознавания и связывания с субстратом. Функции домена SWIRM однозначно не установлены. Он не принимает прямого участия в связывании ФАД или субстратов, однако мутации или удаление данного участка снижают активность ЛСД1. В связи с этим было выдвинуто предположение о том, что данный участок может влиять на активный участок посредством изменения его конформации. Домен tower является тем доменом, по которому ЛСД1 связывается с другими белковыми факторами. ЛСД1 связывается с различными белковыми факторами и оказывает влияние на различные субстраты, тем самым оказывая различные регуляторные эффекты на гистон и экспрессию генов. Например, присоединившись к CoREST, ЛСД1 будет преимущественно действовать на гистон H3K4, удалять отвечающие за активацию маркеры гистона посредством деметилирования и таким образом ингибировать транскрипцию генов; а присоединившись к андрогенному рецепторному белку, рекомбинированная ЛСД1 будет преимущественно действовать на H3K9 и активировать связанную с андрогенным рецептором транскрипцию генов посредством деметилирования. В дополнение к этому, ЛСД1 имеет несколько негистоновых рецепторов, таких как p53, E2F1, DNMT1, MYPT1.
ЛСД1 является ФАД-зависимой аминооксидазой, для которой передача протона считается наиболее вероятным механизмом окисления. Сначала связь N-CH3 в субстрате превращается в иминную связь посредством передачи протона. Данный промежуточный ион имина подвергается гидролизу с образованием деметилированного амина и формальдегида. Во время указанного каталитического цикла ФАД восстанавливается до ФАДН2, которая далее снова окисляется до ФАД молекулой кислорода с образованием при этом молекулы H2O2.
ЛСД1 является незаменимым регулятором в эпигенетике. Она модифицирует гистоны посредством деметилирования и поэтому называется «очищающим» ферментом в организме. ЛСД1 может регулировать экспрессию генов, таким образом регулируя пролиферацию и дифференцировку клеток.
Краткое описание изобретения
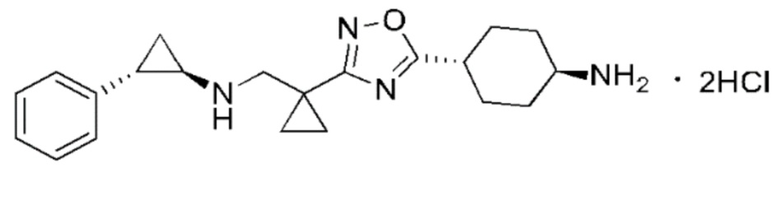
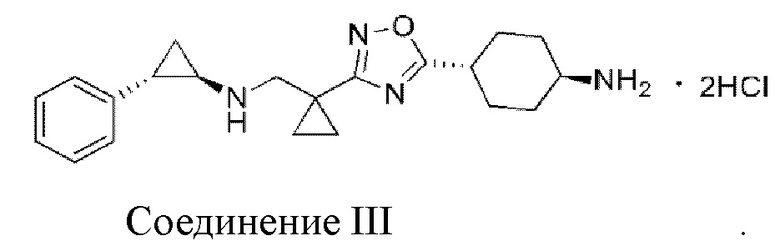
В настоящем изобретении предложено соединение III:
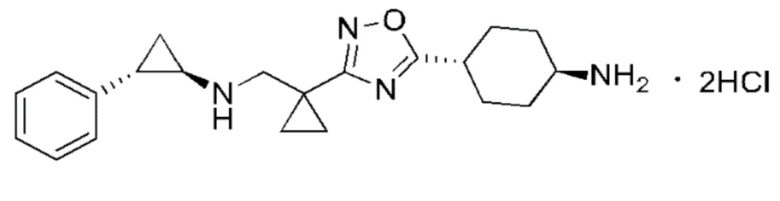
Соединение III
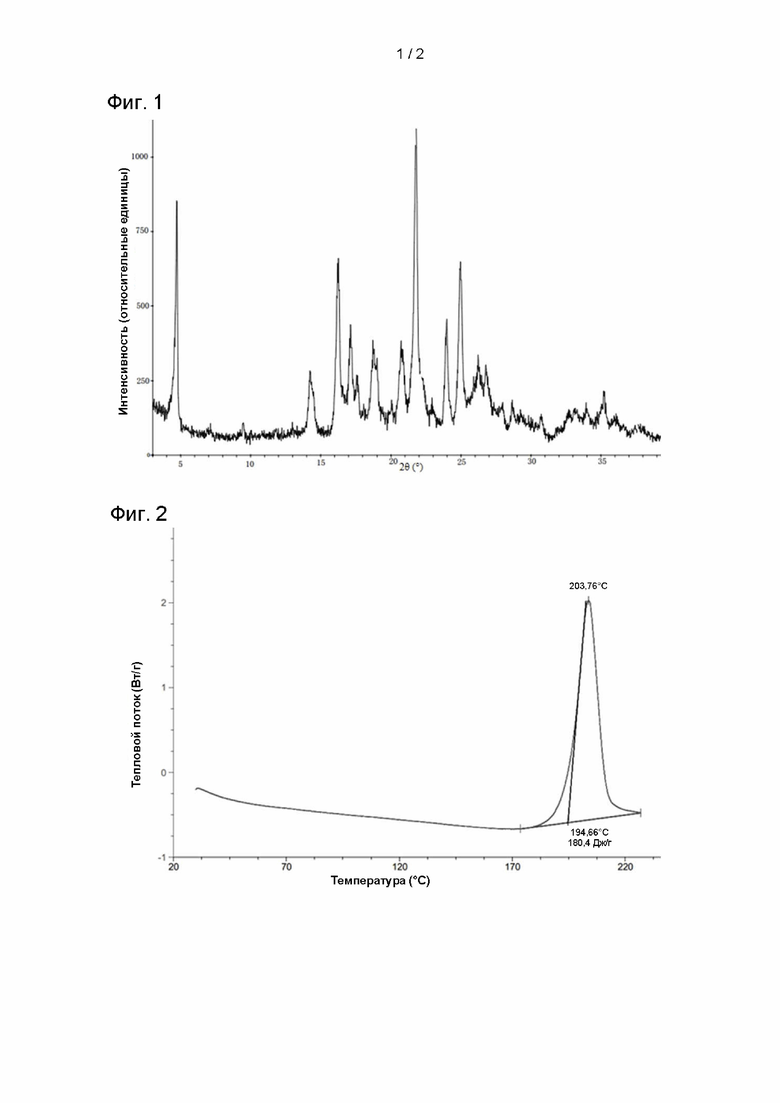
В настоящем изобретении также предложена кристаллическая форма A соединения III с дифрактограммой рентгеновской порошковой дифракции, содержащей характеристические дифракционные пики при значениях угла 2θ: 4,72 ± 0,2°; 14,24 ± 0,2° и 21,78 ± 0,2°.
Согласно некоторым аспектам настоящего изобретения, дифрактограмма рентгеновской порошковой дифракции (XRPD) указанной кристаллической формы A содержит характеристические дифракционные пики при значениях угла 2θ: 4,72 ± 0,2°; 14,24 ± 0,2°; 16,28 ± 0,2°; 17,14 ± 0,2°; 20,72 ± 0,2°; 21,78 ± 0,2°; 23,98 ± 0,2° и 24,96 ± 0,2°.
Согласно некоторым аспектам настоящего изобретения, рентгеновская порошковая дифрактограмма указанной кристаллической формы A содержит характеристические дифракционные пики при значениях угла 2θ: 4,72 ± 0,2°; 14,24 ± 0,2°; 16,28 ± 0,2°; 17,14 ± 0,2°; 17,58 ± 0,2°; 18,70 ± 0,2°; 20,72 ± 0,2°; 21,78 ± 0,2°; 23,98 ± 0,2°; 24,96 ± 0,2° и 26,22 ± 0,2°.
Согласно некоторым аспектам настоящего изобретения, дифрактограмма рентгеновской порошковой дифракции указанной кристаллической формы A содержит характеристические дифракционные пики при значениях угла 2θ: 4,721°; 9,479°; 14,242°; 16,279°; 17,141°; 17,581°; 18,082°; 18,702°; 20,719°; 21,780°; 22,278°; 23,978°; 24,959°; 26,22°; 26,779°; 27,358°; 27,978°; 28,656°; 29,244°; 30,738°; 32,699°; 33,159°; 33,940°; 35,201° и 37,637°.
Согласно некоторым аспектам настоящего изобретения, дифрактограмма XRPD указанной кристаллической формы A по существу соответствует приведенной на Фиг. 1.
Согласно некоторым аспектам настоящего изобретения представлены данные анализа дифрактограммы XRPD указанной кристаллической формы A, которые приведены в Таблице 1:
Таблица 1: Данные анализа XRPD вышеуказанной кристаллической формы A
(°)
костное расстояние (Ангстрем)
сивность
ность (%)
щадь %
рина
Согласно некоторым аспектам настоящего изобретения, кривая, полученная методом дифференциальной сканирующей калориметрии (ДСК) для указанной кристаллической формы А, содержит экзотермический пик при 194,66 ± 3°C.
Согласно некоторым аспектам настоящего изобретения, кривая ДСК, полученная для указанной кристаллической формы А, по существу соответствует приведенной на Фиг. 2.
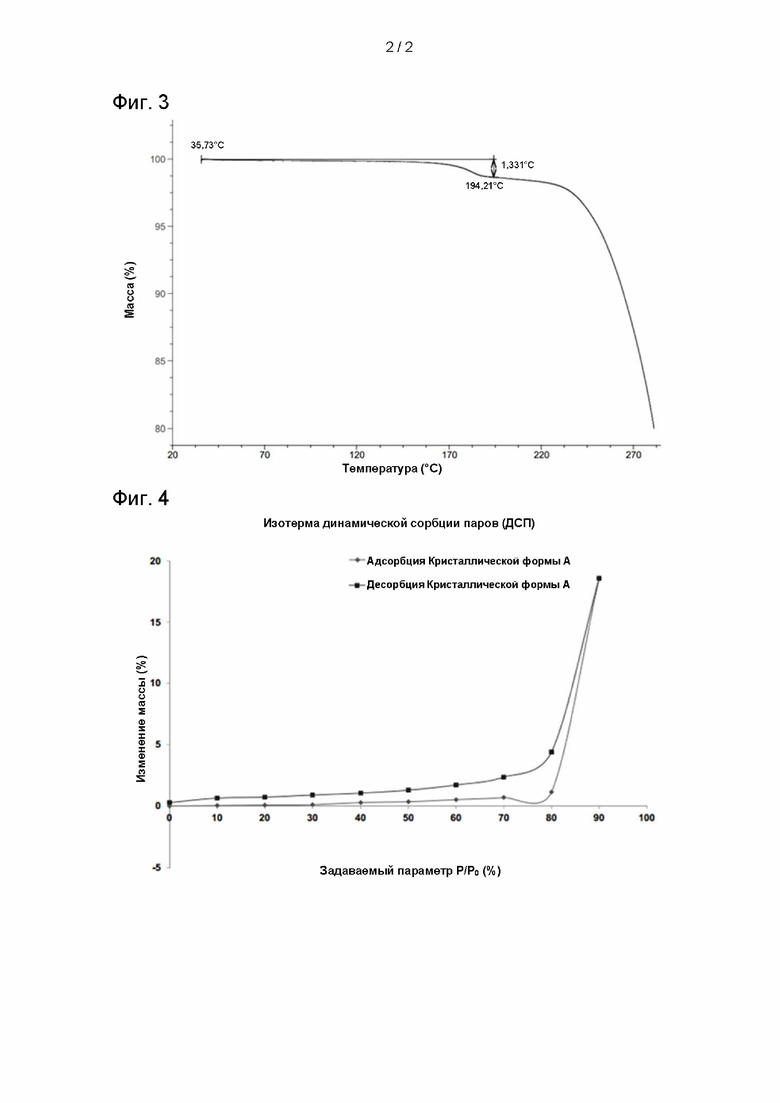
Согласно некоторым аспектам настоящего изобретения, на кривой, полученной методом термогравиметрического анализа (ТГА) для указанной кристаллической формы А, наблюдается потеря массы на 1.331% при 194,21 ± 3°C.
Согласно некоторым аспектам настоящего изобретения кривая ТГА, полученная для указанной кристаллической формы А, по существу соответствует приведенной на Фиг. 3.
В настоящем изобретении также предложено применение указанного соединения III или указанной кристаллической формы A для получения лекарственного средства для лечения заболевания, связанного с ЛСД1.
В настоящем изобретении также предложено применение указанного соединения III или указанной кристаллической формы A для получения лекарственного средства для лечения рака легких, в частности мелкоклеточного рака лёгких.
Технический результат
Соединение III и его кристаллическая форма А согласно настоящему изобретению обладают высокой ингибирующей активностью в отношении ЛСД1 и демонстрируют улучшенное действие in vivo, и по сравнению со свободным основанием и другими солями указанного соединения являются стабильными и обладают высокой растворимостью, а также менее чувствительны к воздействию света и влажности и, таким образом, являются перспективными веществами для получения лекарственного средства.
Определение и описание
Если не указано иное, используемые в данном документе термины и выражения, приведенные ниже, имеют следующие значения. Конкретное выражение или термин не должны считаться неопределенными или неясными, если отсутствует конкретное определение, а должно рассматриваться в своем обычном значении. Если в настоящем изобретении упоминается торговое наименование, оно предназначено для обозначения соответствующего препарата или его действующего вещества.
Промежуточные соединения согласно настоящему изобретению могут быть получены с помощью различных методов синтеза, широко известных специалистам в данной области техники, включая частные варианты реализации, приведенные ниже, а также варианты реализации, согласно которым указанные частные варианты реализации сочетаются с другими методами синтеза, и эквивалентные альтернативные варианты реализации, широко известные специалистам в данной области техники, при этом предпочтительные варианты реализации включают примеры согласно настоящему изобретению, но не ограничивают его каким-либо образом.
Химическая реакция, используемая в частном варианте реализации настоящего изобретения, проводится в подходящем растворителе, и растворитель должен быть подходящим для химического превращения, реагентов и материалов настоящего изобретения. Чтобы получить соединения, предложенные в настоящем изобретении, в некоторых случаях специалистам в данной области необходимо модифицировать или выбрать отдельные стадии синтеза или схем реакций на основании существующих вариантов реализации.
Настоящее изобретение далее будет подробно описано в примерах, и данные примеры не ограничивают настоящее изобретение каким-либо образом.
Все растворители, применяемые в настоящем изобретении, являются коммерчески доступными, и их можно применять без дополнительной очистки.
Все растворители, применяемые в настоящем изобретении, являются коммерчески доступными. В настоящем изобретении используют следующие сокращения: ДХМ означает дихлорметан; ДМФА означает N,N-диметилформамид; ДМСО означает диметилсульфоксид; EtOH означает этанол; MeOH означает метанол; ТФУ означает трифторуксусную кислоту; TsOH означает п-толуолсульфоновую кислоту; тп означает температуру плавления; EtSO3H означает этансульфоновую кислоту; MeSO3H означает метансульфоновую кислоту; АТФ означает аденозин трифосфат; HEPES означает 4-гидроксиэтилпиперазин этансульфоновой кислоты; EGTA означает этиленгликоль-бис-(2-аминоэтиловый эфир)-тетрауксусную кислоту; MgCl2 означает хлорид магния (II); MnCl2 означает хлорид марганца (II); ДТТ означает дитиотреитол; ДЦК означает дициклогексилкарбодиимид; ДМАП означает 4-диметиламинопиридин.
Метод рентгеновской порошковой дифракции (XPDR, с использованием рентгеновского порошкового дифрактометра) согласно настоящему изобретению
Модель прибора: рентгеновский дифрактометр DX-2700BH
Метод исследования: для анализа методом XPDR используется примерно от 10 до 20 мг образца.
Подробные характеристики метода XPDR:
Источник излучения: Cu, k(альфа1) (λ=1,54184Å)
Напряжение рентгеновской трубки: 40 кВ, ток в рентгеновской трубке: 30 мА
Щель расходимости: 1 мм
Первая щель Соллера: 28 мм, вторая щель Соллера: 28 мм
Приёмная щель: 0,3 мм, антирассеивающая щель: 1 мм
Время измерения: 0,5 с
Интервал углов сканирования: 3-40 градусов
Ширина шага: 0,02 градуса
Аналитический метод Дифференциальной Сканирующей Калориметрии (ДСК) согласно настоящему изобретению
Модель прибора: Дифференциальный Сканирующий Калориметр TA Q2000
Метод исследования: взяли навеску образца (около 1 мг) и поместили в алюминиевый тигель для анализа методом ДСК. Образец нагревали от 30°C (комнатная температура) до 300°C (или 350°C) со скоростью 10°C/мин в потоке N2, пропускаемого со скоростью 50 мл/мин.
Метод Термогравиметрического Анализа (ТГА) согласно настоящему изобретению
Модель прибора: термогравиметрический анализатор TA Q5000
Метод исследования: навеску образца (от 2 до 5 мг) и помещали в платиновый тигель для анализа методом ТГА. Образец нагревали от комнатной температуры до 300°C или до потери массы на 20% со скоростью нагрева 10°C/мин в потоке N2, пропускаемого со скоростью 25 мл/мин.
Аналитический метод динамической сорбции паров (ДСП) согласно настоящему изобретению
Модель прибора: анализатор динамической сорбции паров SMS DVS Advantage
Условия исследования: навеску образца (10 ~ 15 мг) и помещали в лодочку для образцов для анализа методом ДСП.
Подробные характеристики метода ДСП:
Температура: 25°C
Равновесие: dm/dt = 0,01%/мин (самое быстрое: 10 мин, самое длительное: 180 мин)
Сушка: сушка при относительной влажности 0% в течение 120 мин
Шаг относительной влажности (%): 10%
Диапазон относительной влажности (%): 0%-90%-0%
Классификация оценки гигроскопичности:
Примечание: ΔW% обозначает увеличение влажности исследуемого образца при 25 ± 1°C и относительной влажности 80 ± 2%.
Описание графических материалов
На Фигуре 1 представлена дифрактограмма XPDR (излучение Cu-Kα) кристаллической формы А соединения III;
На Фигуре 2 представлена кривая ДСК, полученная для кристаллической формы А соединения III;
На Фигуре 3 представлена кривая ТГА, полученная для кристаллической формы А соединения III;
На Фигуре 4 представлена изотерма динамической сорбции паров (ДСП), полученная для кристаллической формы А соединения III.
Подробное описание вариантов реализации
Для лучшего понимания сущности настоящего изобретения далее в описании приведены частные примеры, не ограничивающие настоящее изобретение каким-либо образом.
Пример 1: Получение Соединения I
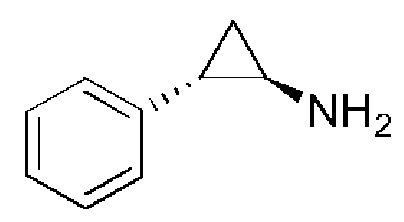
Схема синтеза:
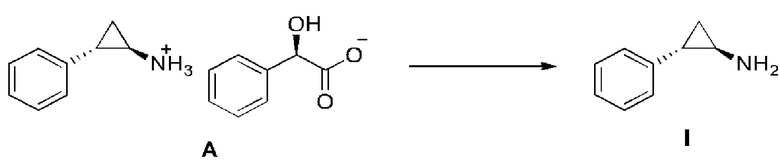
Гидроксид натрия (279 г, 6.99 моль) растворяли в воде (3,00 л), температуру поддерживали около 10°C, и порциями добавляли соединение A (997 г, 3.49 моль). После полного растворения твердого вещества, смесь экстрагировали с помощью этилацетата (2,00 л × 2). Комбинированную органическую фазу промывали водой (1,50 л) и насыщенным раствором соли (1,50 л), постепенно сушили над безводным сульфатом натрия и отфильтровывали. Фильтрат концентрировали при пониженном давлении с получением соединения I. 1H ЯМР (400МГц, CDCl3) δ 7,18-7,14 (м, 2H), 7,07-7,05 (м, 1H), 6,94-6,92 (м, 2H), 2,47-2,44 (м, 1H), 1,78-1,76 (м, 1H), 0,97-0,94 (м, 1H), 0,92-0,89 (м, 1H).
Пример 2: Получение Соединения II
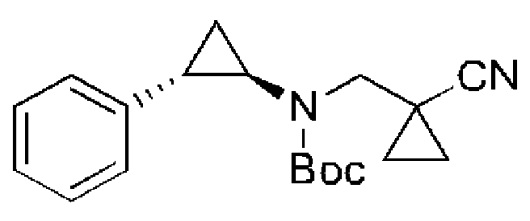
Схема синтеза:
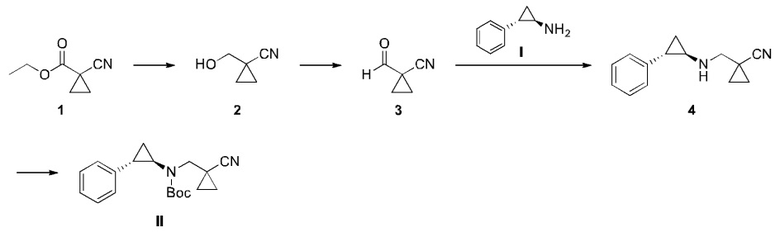
Стадия I
Соединение 1 (260 г, 1,87 моль) растворяли в тетрагидрофуране (2,00 л) и метаноле (200 мл), температуру поддерживали около 20°C, и порциями добавляли боргидрид натрия (70,8 г, 1,87 моль). Реакционную смесь перемешивали при 20°C в течение 18 часов, добавляли по каплям насыщенный раствор боргидрида натрия (2,00 л) при 0°C чтобы охладить реакцию, до тех пор, пока не перестанут образовываться пузыри. Было получено небольшое количество твердого вещества. Реакционную смесь фильтровали. Осадок промывали этилацетатом (1,00 л × 2). К фильтрату добавляли твердый хлорид натрия до перенасыщения и образования слоев. Органическую фазу промыли насыщенным раствором соли (1,00 л). Многокомпонентную водную фазу экстрагировали смесью раствора этилацетата и тетрагидрофурана (этилацетат : тетрагидрофуран = 10:1; 1,00 л × 3). Многокомпонентную органическую фазу промывали насыщенным раствором соли (1,00 л), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, чтобы получить соединение 2. 1H ЯМР (400МГц, CDCl3) δ 3,63 (с., 2H), 2,20 (с., 1H), 1,29 (дд., J1=5,2 Гц, J2=2,0 Гц, 2H), 0,99 (дд., J1=5,2 Гц, J2=2,0 Гц, 2H).
Стадия II
Соединение 2 (101 г, 1,04 моль) растворяли в безводном дихлорметане (1,50 л), температуру поддерживали около 5 - 10°C, и порциями добавили перйодинан Десса-Мартина (486 г, 1,14 моль). Реакционную смесь перемешивали при 25°C в течение 12 часов, затем контролировали при 15°C, и медленно добавляли к насыщенному водному раствору гидрокарбоната натрия (4,00 л) с последующим медленным добавлением насыщенного раствора тиосульфата натрия (4,00 л). Реакционную смесь перемешивали в течение 30 минут и оставляли отстаиваться. Водную фазу экстрагировали с помощью дихлорметана (1,00 л × 3), и сложную органическую фазу последовательно промывали водой (1,00 л × 1) и насыщенным раствором соли (1.00 л × 1), сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 3. 1H ЯМР (400МГц, CDCl3) δ 9,31 (с., 1H), 1,71-1,68 (м, 4H).
Стадия III
Соединение I (97,5 г, 732 ммоль) и соединение 3 (83,5 г, 878 ммоль) растворяли в сухом дихлорметане (1,50 л) и добавляли уксусную кислоту (4,40 г, 73.2 ммоль). Реакционную смесь перемешивали при 26°C в течение 4 часов, добавляли триацетоксиборогидрид натрия (232 г, 1,10 моль) и перемешивали при 25°C в течение 12 часов. После того как медленно добавляли насыщенный раствор гидрокарбоната натрия (3,50 л) до тех пор, пока не перестали образовываться пузыри, реакционную смесь оставляли отстаиваться. Водную фазу экстрагировали с помощью дихлорметана (1,00 л × 1), и сложную органическую фазу концентрировали при низком давлении, чтобы удалить органический растворитель. К оставшейся смеси добавляли воду (800 мл), значение pH которой довели до 3 водным раствором соляной кислоты (1 M), и экстрагировали с помощью трет-бутилметилового эфира (800 м л × 2). Значение рН водной фазы доводили до 8 насыщенным раствором гидрокарбоната натрия, и проводили экстракцию с помощью дихлорметана (1,00 л × 2). Сложную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 4.
1H ЯМР (400МГц, CDCl3) δ 7,29-7,26 (м, 2H), 7,19-7,16 (м, 1H), 7,06-7,04 (м, 2H), 2,83 (с., 2H), 2,51-2,48 (м, 1H), 2,01-1,96 (м, 1H), 1,28-1,24 (м, 2H), 1,18-1,13 (м, 1H), 1,05-1,01 (м, 1H), 0,88-0,79 (м, 2H).
Рассчитанное с помощью метода масс-спектрометрии с ионизацией электрораспылением (MS-ESI) значение [M+H]+ : 213, обнаруженное: 213.
Стадия IV
Соединение 4 (113 г, 534 ммоль) растворяли в тетрагидрофуране (1,20 л) и воде (300 мл), добавляли ди-трет-бутил дикарбонат (128 г, 588 ммоль) и моногидрат гидроксида лития (2,.9 г, 641 ммоль). Реакционную смесь перемешивали при 25°C в течение 12 часов, доводили значение pH до 3 водным раствором соляной кислоты (1 M), и проводили экстракцию этилацетатом (800 мл × 2). Органическую фазу промывали насыщенным раствором соли (1,00 л × 1), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. К остатку добавляли н-гептан (1,00 л), перемешивали в течение 12 часов с получением белого осадка в большом количестве, и отфильтровывали. Отфильтрованный осадок сушили при пониженном давлении с получением соединения II. 1H ЯМР (400МГц, CDCl3) δ 7,23-7,21 (м, 2H), 7,13-7,10 (м, 1H), 7,07-7,05 (м, 2H), 3,42-3,31 (м, 2H), 2,90-2,88 (м, 1H), 2,10-2,05 (м, 1H), 1,37 (s, 9H), 1,28-1,16 (м, 4H), 1,00-0,90 (м, 2H). Рассчитанное с помощью метода MS-ESI значение [M+H]+: 313, измеренное: 313.
Пример 3: Получение Соединения III и его кристаллической формы А
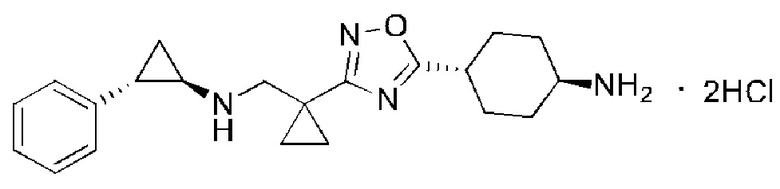
Схема синтеза:
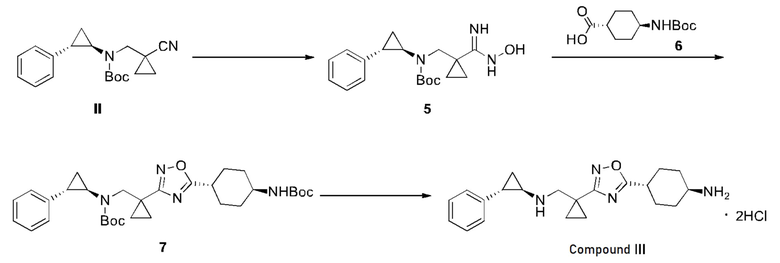
Стадия I
Соединение II (202 г, 647 ммоль) растворяли в абсолютном этаноле (500 мл) при комнатной температуре, добавляли диизопропилэтиламин (209 г, 1,62 моль) и гидроксиламин гидрохлорид (90,0 г, 1,30 моль), нагревали до 80°C и перемешивали в течение 16 часов. Полученный раствор охлаждали до комнатной температуры и концентрировали при низком давлении, чтобы удалить этанол. Остаток растворяли в этилацетате (2,00 л). Органическую фазу промывали водой (500 мл × 3), сушили над безводным сульфатом натрия, фильтровали. Фильтрат концентрировали. Остаток растворяли в этилацетате (200 мл), добавляли н-гептан (2,00L) при перемешивании, и далее перемешивали в течение 12 часов с получением белого осадка. Полученную смесь фильтровали, и отфильтрованный осадок промывали н-гептаном (200 мл) и сушили при 45°C в вакууме в течение 12 часов, чтобы получить соединение 5. 1H ЯМР (400 МГц, ДМСО-д6) δ 8,92 (с., 1H), 7,27-7,23 (м, 2H), 7,16-7,08 (м, 3H), 5,26 (с., 2H), 3,53-3,50 (м, 1H), 3,32-3,28 (м, 1H), 2,78-2,76 (м, 1H), 2,03-2,00 (м, 1H), 1,33 (с., 9H), 1,14-1,11 (м, 2H), 0,71-0,59 (м, 4H). Рассчитанное с помощью метода MS-ESI значение [M+H]+ : 346, измеренное: 346.
Стадия II
Соединение 6 (126 г, 532 ммоль) растворяли в безводном N,N-диметилформамиде N (1,40 л), добавляли карбонилдиимидазол (88,8 г, 557 ммоль) при 30°C в защитной атмосфере азота и перемешивали в течение 3 часов. К реакционной смеси добавляли соединение 5 (175 г, 506 ммоль), нагревали до 110°C и перемешивали в течение 12 часов. Полученный раствор охлаждали до комнатной температуры, медленно охлаждали водой (14 л) при перемешивании с получением белого осадка, и фильтровали. Отфильтрованный осадок промывали водой (3 л × 3) и сушили при 30°C в вакууме, чтобы получить соединение 7. 1H ЯМР (400 МГц, CDCl3) δ 7,28-7,23 (м, 2H), 7,17-7,14 (м, 1H), 7,06-7,05 (м, 2H), 4,43-4,41 (м, 1H), 3,88-3,70 (м, 2H), 3,48-3,47 (м, 1H), 2,76-2,73 (м, 2H), 2,14-2,07 (м, 5H), 1,64-1,61 (м, 2H), 1,46 (с, 9H), 1,41 (с, 9H), 1,24-1,00 (м, 7H). Рассчитанное с помощью метода MS-ESI значение [M+H]+ : 575, измеренное: 575.
Стадия III
Соединение 7 (240 г, 434 ммоль) растворяли в этилацетате (240 мл), добавляли раствор соляной кислоты в этилацетате (4M, 820 мл) при перемешивании при 0°C, перемешивали при температуре от 0°C до 25°C в течение 3 часов, с получением белого осадка и фильтровали. Отфильтрованный осадок промывали этилацетатом (500 м л × 5) и сушили при 40°C в вакууме с получением соединения III. Соединение III смешивали с абсолютным этанолом (1,20 л), нагревали до появления конденсата при перемешивании до растворения всех твердых компонентов и отфильтровывали горячий раствор, чтобы удалить механические примеси. В фильтрате оставалось небольшое количество твердых компонентов. Перегонку проводили до полного растворения всех твердых компонентов, при этом смесь не перемешивали. Фильтрат охлаждали со скоростью 10°C - 20°C в течение 1-2 часов. После охлаждения до 45°C фильтрат оставляли на 12 часов до выпадения большого количества белого осадка. Фильтрат далее снова охлаждали со скоростью 10°C - 20°C в течение 1-2 часов до температуры 25°C, периодически перемешивая, и отфильтровывали. Отфильтрованный осадок промывали изопропанолом (260 м л × 3) и сушили в вакууме при 45°C. Анализ методом рентгеновской порошковой дифракции показал, что была получена кристаллическая форма A соединения III. 1H ЯМР (400МГц, CD3OD) δ 7,32-7,29 (м, 2H), 7,25-7,21 (м, 1H), 7,17-7,14 (м, 2H), 3,70-3,62 (м, 2H), 3,21-3,14 (м, 1H), 3,09-3,05 (м, 1H), 3,01-2,95 (м, 1H), 2,57-2,52 (м, 1H), 2,26-2,22 (м, 2H), 2,18-2,15 (м, 2H), 1,75-1,64 (м, 2H), 1,61-1,54 (м, 3H), 1,44-1,41 (м, 2H), 1,39-1,36 (м, 1H), 1,34-1,32 (м, 2H). Рассчитанное с помощью метода MS-ESI значение [M+H]+ : 353, измеренное: 353.
Пример 4: Анализ содержания хлорид-ионов в кристаллической форме А соединения III
Прибор, применяемый в исследовании: ионный хроматограф ICS5000
Хроматографические параметры:
Подколонка: Dionex IonPac AG11-HC, подколонка 4 x 50 мм
Хроматографическая колонка: Dionex IonPac AS11-HC, подколонка 4 x 250 мм
Температура колонки: 30°C
Режим детектирования: кондуктометрическое детектирование
Скорость потока: 1,0 мл/мин
Подавитель ASRS-4мм 18 мА
Объём вводимой пробы: 25 μл
Время анализа: 20 мин
Подвижная фаза: 7 мМ KOH
Приготовление образца:
Три образца, каждый массой 50 мг, кристаллической формы А соединения III точно взвесили и обозначили как Образец 1, Образец 2 и Образец 3. Их растворили в деионизированной воде и приготовили три раствора с концентрацией 0,2 мг/мл.
Экспериментальные данные:
Таблица 2: Результаты анализа содержания хлорид ионов в кристаллической форме А соединения III
Заключения, сделанные на основе экспериментальных данных:
Измеренное значение содержания хлорид ионов в кристаллической форме А соединения III совпадает с теоретически рассчитанным значением в пределах погрешности менее 0,3%, и данный продукт представляет собой дигидрохлорид.
Пример 5: Анализ гигроскопичности кристаллической формы A соединения III
Оборудование: анализатор динамической сорбции паров SMS DVS Advantage
Методы исследования:
Навеску 10 - 15 мг кристаллической формы A соединения III и помещали в чашку для образцов для анализа методом ДСП.
Результаты исследования:
На Фигуре 4 представлена кривая ДСП, полученная для кристаллической формы A соединения III, δW = 1,14%.
Заключение, сделанное на основе экспериментальных данных:
Кристаллическая форма A соединения III демонстрирует увеличение массы влаги на 1,14% при 25°C и относительной влажности 80%, проявляя низко гигроскопичные свойства.
Пример 6: Анализ стабильности твердой кристаллической формы A соединения III
Согласно "Руководству по исследованию стабильности активного фармацевтического компонента и лекарственных средств" (Китайская фармакопея 2015, Четвертое Издание, Общие принципы 9001) была исследована стабильность кристаллической формы A соединения III в условиях высокой температуры (60°C, на воздухе), высокой влажности (комнатная температура/относительная влажность 92,5%, на воздухе) и на свету (полная освещенность = 1,2 × 106 люкс ⋅ ч/ближний УФ = 200Вт ⋅ ч/м2, на воздухе). Навеску 10 мг кристаллической формы A соединения III и помещали на дно стеклянного сосуда тонким слоем. При анализе образцов, помещенных в условиях высокой температуры (60°C) и высокой влажности (относительная влажность 92,5%), отверстие сосуда запечатывали алюминиевой фольгой, и делали несколько небольших отверстий, с целью обеспечения достаточного контакта образцов с воздухом атмосферы, и помещали образцы в печи с соответствующими постоянными температурой и влажностью. Образцы (на воздухе, не запечатанные алюминиевой фольгой) подвергли воздействию света, и контрольный образец (сосуд с образцом был полностью покрыт алюминиевой фольгой) поместили в световую камеру. В каждый момент времени 2 образца взвешивали, они выступали контрольными образцами. Другую навеску кристаллической формы А соединения III массой 50 мг взяли для исследования методом XPDR. Сосуды с образцами обернули алюминиевой фольгой, и проделали в алюминиевой фольге несколько небольших отверстий. Данные сосуды также помещали в печи с соответствующими постоянными температурой и влажностью. Образец доставали и анализировали (методом XPDR) на 5-ый и 10-ый дни, и результаты анализа сравнивали с исходными результатами, сделанными в самом начале (0 день). Результаты исследования представлены в ниже представленной таблицы 3:
Таблица 3: Результаты анализа стабильности твердой кристаллической формы А соединения III
Заключение: Кристаллическая форма A соединения III стабильна в условиях высокой температуры, высокой влажности и сильной освещенности.
Пример 7: Анализ устойчивости к растворению кристаллической формы А соединения III
Необходимое количество соединения III взвешивали и помещали в различные стеклянные сосуды, и добавили необходимое количество растворителя или смеси растворителей с получением суспензии. В вышеуказанные образцы помещали мешалки и оставляли при комнатной температуре, образцы ставили на магнитную мешалку при постоянной температуре (40°C) и перемешивали в течение 2 дней (в темноте). Для получения требуемой суспензии, количество соединения и растворителя подбираются согласно условиям эксперимента, и даже используемая в исследовании ёмкость может быть заменена во время эксперимента. (Влага из растворенного образца испарилась естественным образом до образования сухого образца). Результаты анализа представлены далее в Таблице 4:
Таблица 4: Результаты анализа устойчивости к растворению кристаллической формы А соединения III
Заключение: Кристаллическая форма A соединения III устойчива к растворению.
Экспериментальный Пример 1: Изучение Ингибирования ЛСД1 для кристаллической формой A соединения III
1.1 Цель эксперимента:
Целью эксперимента являлось определение значений IC50 кристаллической формы A соединения III по отношению к ЛСД1 при 10 значениях концентрации. Эксперимент проводили в двух параллелях с исходной концентрацией 10 мкМ с трехкратным градиентным разбавлением, и такую процедуру повторяли дважды в разные дни.
1.2 Условия исследования:
Состав буферного раствора для ЛСД1: 50 мМ Трис-HCl, pH 7,5, 0,05% (3-[(3-холанидопропил)диметиламмоний]-1-пропансульфонат, 1% ДМСО.
Время реакции: реакция протекала при комнатной температуре в течение 1 часа
Методика проведения реакции:
1.2.1 Добавление фермента к свежеприготовленному буферному раствору
1.2.2 Добавление раствора соединения в ДМСО к смеси фермента с применением Акустической Технологии (Echo 550, LabCyte Inc. Sunnyvale, CA) на уровне нл, и инкубация при комнатной температуре в течение 30 минут
1.2.3 Добавление субстрата к свежеприготовленному буферному раствору
1.2.4 Инкубация при комнатной температуре в течение 1 часа
1.2.5 Подготовка к анализу смеси
1.2.6 Использование Perkin Elmer Envision для считывания данных
1.2.7 Использование программ Excel и GraphPad Prism для анализа данных
1.3 Результаты исследования:
Таблица 5. Ингибирование ЛСД1 кристаллической формой A соединения III
Среднее значение ± стандартное отклонение
Заключение: в настоящем эксперименте оценивали ингибирование ЛСД1 кристаллической формой A соединения III методом коньюгирования с флуоресцентным ферментом. Результаты показывают, что кристаллическая форма А Соединения III обладает значительной ингибирующей активностью в отношении ЛСД1, при этом значение IC50 = 8 нМ.
Экспериментальный Пример 2: Изучение эффективности действия in vivo кристаллической формы А соединения III на мелкоклеточный рак лёгких человека (клетки NCI-H1417 ксенотрансплантата подкожной опухоли) с использованием мышиной модели CB-17 SCID
2.1 Цель эксперимента:
Целью эксперимента является оценка эффективности действия in vivo кристаллической формы А соединения III на мелкоклеточный рак лёгких человека (клетки NCI-H1417 ксенотрансплантата подкожной опухоли) с использованием мышиной модели CB-17 SCID.
2.2 Экспериментальные животные:
Вид: Мышь
Штамм: мыши CB-17 SCID
Возраст в неделях и вес: 6-8 недель, вес: 16-21 грамм
Пол: женский
Поставщик: Shanghai Lingchang Biological Technology Co., Ltd.
2.3 Экспериментальный метод и способ
2.3.1 Культура клеток
Клетки NCI-H1417, вызывающие мелкоклеточный рак легкого человека, выращивали монослойной культурой вне организма при 37°C в среде RPMI-1640 с содержанием фетальной бычьей сыворотки 10%, и CO2 5% для обеспечения роста и переноса культуры. Когда насыщение клетками достигло 80%-90%, клетки собирали с помощью ферментативного гидролиза смесью из трипсина и ЭДТА, считали, затем доводили их содержание до концентрации 10×107 клеток/мл и ресуспендировали в PBS.
2.3.2 Посев опухолевых клеток
0,2 мл (10×106 клеток) клеток NCI-H1417 (в Матригеле, в объёмном соотношении 1:1) вводили подкожно в правую половину спины каждой мыши. Когда средний объём опухоли достиг около 100-150 мм3, мышей разделяли в произвольном порядке на группы и начали введение препарата.
2.3.3 Подготовка исследуемого вещества
Носитель представлял собой 0,5% раствор метилцеллюлозы. Взвешивали 5 г метилцеллюлозы, и растворили в 800 мл сверхчистой воды, перемешали и довели объём до 1000 мл сверхчистой водой. Исследуемое вещество растворяли в носителе, получали однородный раствор определенной концентрации и хранили при 4°C.
2.3.4 Измерение опухоли и индикатор исследования
Индикатор исследования применяли для изучения ингибирования или замедления роста опухоли, или же лечения опухоли. Диаметр опухоли измеряли с помощью штангенциркуля два раза в неделю. Объём опухоли рассчитывали по формуле: V = 0,5a × b2, где a и b обозначают диаметр опухоли по длинной оси и диаметр опухоли по короткой оси, соответственно.
Значение ингибирования роста опухоли TGI (%) используется для оценки противоопухолевого эффекта соединения. TGI (%) показывает скорость ингибирования роста опухоли. TGI (%)=[(1-(Средний объём опухоли по окончании введения препарата в определенной терапевтической группе - средний объём опухоли в начале введения препарата в определенной терапевтической группе))/(Средний объём опухоли по окончании введения препарата в контрольной группе, получающей носитель - средний объём опухоли в начале введения препарата в контрольной группе, получающей носитель)]×100%. Контрольная группа, получающая носитель: носитель (0,5% раствор метилцеллюлозы).
Таблица 6: Оценка противоопухолевого эффекта кристаллической формы A соединения III на мелкоклеточный рак лёгких человека на модели ксенотрансплантата клеток NCI-H1417 подкожной опухоли
(Расчеты сделаны на основе объёма опухоли, измеренного на 35-ый день приёма)
(35ый День)
(35ый День)
Заключение: кристаллическая форма A соединения III согласно настоящему изобретению обладает сильной противоопухолевой активностью в отношении мелкоклеточного рака лёгких человека в ксенотрансплантатной модели опухоли NCI-H1417.
Изобретение относится к соединению III и его кристаллической форме A, характеризующейся дифрактограммой рентгеновской порошковой дифракции (XPDR), содержащей характеристические дифракционные пики при значениях угла 2θ: 4,72 ± 0,2°, 14,24 ± 0,2°, 16,28 ± 0,2°, 17,14 ± 0,2°, 20,72 ± 0,2°, 21,78 ± 0,2°, 23,98 ± 0,2° и 24,96 ± 0,2°. Соединение III по изобретению или его кристаллическую форму A применяют для получения лекарственного средства для лечения заболевания, связанного с ЛСД1. Также соединение III по изобретению или его кристаллическую форму A применяют для получения лекарственного средства для лечения рака легких. Технический результат – соединение III и его кристаллическая форма в качестве ингибитора ЛСД1. 4 н. и 8 з.п. ф-лы, 6 табл., 4 ил., 7 пр.
 Соединение III
Соединение III
1. Соединение III
2. Кристаллическая форма A соединения III, характеризующаяся дифрактограммой рентгеновской порошковой дифракции (XPDR), содержащей характеристические дифракционные пики при значениях угла 2θ: 4,72 ± 0,2°, 14,24 ± 0,2°, 16,28 ± 0,2°, 17,14 ± 0,2°, 20,72 ± 0,2°, 21,78 ± 0,2°, 23,98 ± 0,2° и 24,96 ± 0,2°.
3. Кристаллическая форма A по п. 2, характеризующаяся дифрактограммой XPDR, содержащей характеристические дифракционные пики при значениях угла 2θ: 4,72 ± 0,2°; 14,24 ± 0,2°; 16,28 ± 0,2°; 17,14 ± 0,2°; 17,58 ± 0,2°; 18,70 ± 0,2°; 20,72 ± 0,2°; 21,78 ± 0,2°; 23,98 ± 0,2°; 24,96 ± 0,2° и 26,22 ± 0,2°.
4. Кристаллическая форма A по п. 3, характеризующаяся дифрактограммой XPDR, содержащей характеристические дифракционные пики при значениях угла 2θ: 4,721°; 9,479°; 14,242°; 16,279°; 17,141°; 17,581°; 18,082°; 18,702°; 20,719°; 21,780°; 22,278°; 23,978°; 24,959°; 26,22°; 26,779°; 27,358°; 27,978°; 28,656°; 29,244°; 30,738°; 32,699°; 33,159°; 33,940°; 35,201° и 37,637°.
5. Кристаллическая форма A по п. 4, отличающаяся тем, что указанная дифрактограмма XPDR соответствует приведенной на Фиг. 1.
6. Кристаллическая форма A по любому из пп. 2-5, характеризующаяся кривой дифференциальной сканирующей калориметрии (ДСК), содержащей экзотермический пик при 194,66 ± 3°C.
7. Кристаллическая форма A по п. 6, отличающаяся тем, что указанная кривая ДСК соответствует приведенной на Фиг. 2.
8. Кристаллическая форма A по любому из пп. 2-5, характеризующаяся кривой термогравиметрического анализа (ТГА) с потерей массы на 1.331% при 194,21 ± 3°C.
9. Кристаллическая форма A по п. 8, отличающаяся тем, что указанная кривая ТГА соответствует приведенной на Фиг. 3.
10. Применение соединения III по п. 1 или кристаллической формы A по любому из пп. 2-9 для получения лекарственного средства для лечения заболевания, связанного с ЛСД1.
11. Применение соединения III по п. 1 или кристаллической формы A по любому из пп. 2-9 для получения лекарственного средства для лечения рака легких.
12. Применение по п. 11, отличающееся тем, что указанный рак легких представляет собой мелкоклеточный рак легких.
| CN 103857393 A, 11.06.2014 | |||
| CN 102947265 A, 27.02.2013 | |||
| WO 2017195216 A1, 16.11.2017 | |||
| WO 2017184934 A1, 26.10.2017 | |||
| MINO R.CAIRA, CRYSTALLINE POLYMORPHISM OF ORGANIC COMPOUNDS, TOPICS IN CURRENT CHEMISTRY, 1998, vol.198, p.163-208 | |||
| Richard J.Bastin et al.: "Salt selection and Optimisation Procedures for Pharmaceutical New Chemical Entities", |

