Настоящее изобретение относится к полиморфам доцетаксела, способам их приготовления и способам их использования для приготовления других полиморфов доцетаксела.
Настоящее изобретение также предусматривает способ получения доцетаксела. Доцетаксел - принятое наименование лекарственного соединения, имеющего химическое название (2R,3S)-N-карбокси-3-фенилизосерин, N-трет-бутиловый сложный эфир, 13-сложный эфир с 5β-20-эпокси-1,2α,4,7β,10β,13α-гексагидрокситакс-11-ен-9-он-4-ацетат-2-бензоатом, и структурную формулу I.

Доцетаксел представляет собой противоопухолевый агент, принадлежащий к семейству таксоидов и доступный на рынке под торговой маркой TAXOTERE® в форме стерильных, апирогенных инъекций во флаконах на одну дозу, содержащих 20 мг (0,5 мл) или 80 мг (2 мл) лекарственного препарата.
Ряд аналогов таксола были описаны F.Gueritte-Voegelein et al., "Relationships between the Structure of Taxol Analogues and Their Antimitotic Activity", Journal of Medicinal Chemistry, Vol.34, pages 992-998, 1991.
Патент США №4814470 раскрывает доцетаксел, его стереоизомерные формы, фармацевтические композиции, содержащие доцетаксел и их использование при лечении острых лейкемий и солидных опухолей.
Патент США №6197980 раскрывает доцетаксел тригидрат и способ его получения.
Способы получения доцетаксела тригидрата были описаны в патенте США №6022985 и публикации патентной заявки США №2006/0217436.
Публикация патентной заявки США №2005/0065138 раскрывает ацетоновый сольват диметоксидоцетаксела и способ его получения.
Публикация международной заявки №WO 2005/061474 раскрывает способ получения безводной, аморфной и тригидратной форм доцетаксела.
Патент США №6838569 раскрывает способ очистки доцетаксела тригидрата, включающий растворение доцетаксела в ацетонитриле с последующим осаждением очищенной водой.
Патент США №6002025 раскрывает способ очистки таксанов с помощью хроматографии на колонке, содержащей фенилалкильную смолу.
Патент США №5476954 раскрывает способ получения доцетаксела и его производных.
Патент США №5532388 раскрывает способ получения таксоида.
Регулятивные органы власти требуют, чтобы предпринимались усилия по идентификации всех полиморфных форм, например кристаллических, аморфных, сольватных и т.д., новых лекарственных веществ, поскольку полиморфные формы могут различаться по своим химическим и физическим свойствам. Эти различия часто приводят к отличиям по биодоступности, стабильности и другим свойствам между производственными партиями изготовленных в соответствии с рецептурами фармацевтических продуктов.
Однако существование и возможное число полиморфных форм данного соединения нельзя предсказать. Дополнительно, не существует "стандартных" процедур, которые могут быть использованы для приготовления полиморфных форм вещества.
Таким образом, существует необходимость в получении новых твердых форм лекарственного вещества и в способах их получения.
В соответствии с настоящим изобретением предлагается удобный способ получения доцетаксела и его полиморфов с желательной чистотой и выходом путем использования улучшенных методов получения, являющихся простыми, экологически безопасными, экономически эффективными, надежными и хорошо пригодными для использования в промышленных масштабах.
Сущность изобретения
Настоящее изобретение относится к полиморфам доцетаксела и способам их получения. Настоящее изобретение также предусматривает способ получения доцетаксела.
В одном аспекте, настоящее изобретение относится к полиморфам доцетаксела и способу их получения.
В варианте исполнения, настоящее изобретение предусматривает кристаллические полиморфы доцетаксела и способ их получения. Эти полиморфы называются далее Формой I, Формой II, Формой III, Формой IV, Формой V, Формой VI, Формой VII, Формой VIII и Формой IX.
Другой вариант исполнения настоящего изобретения относится к способу получения аморфной формы доцетаксела, включающему осаждение аморфного доцетаксела из раствора доцетаксела в тетрагидрофуране (ТГФ) путем объединения с антирастворителем, таким как углеводород, и выделение осажденного аморфного твердого вещества.
В другом аспекте, настоящее изобретение предусматривает способ получения доцетаксела, включающий:
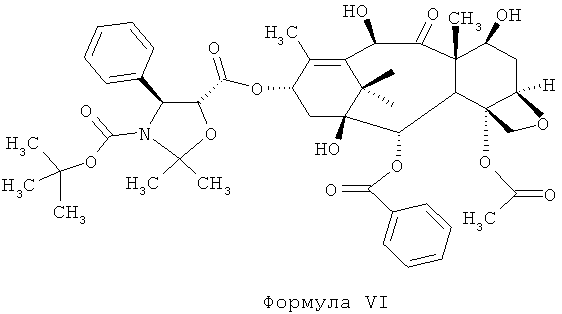
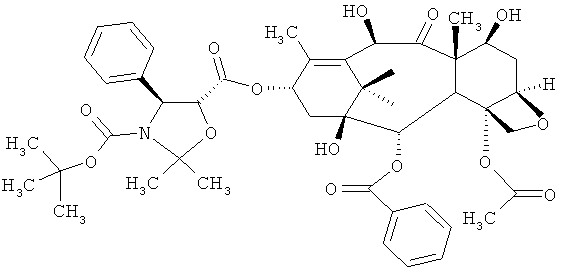
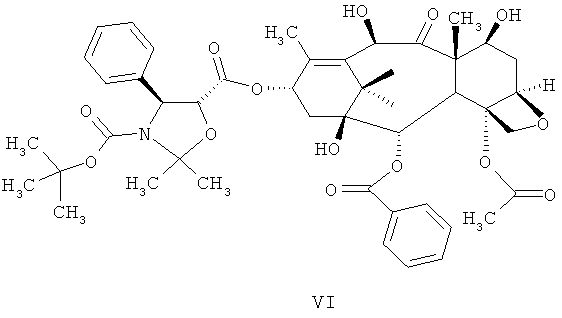
а) проведение реакции соединения 4-ацетокси-2α-бензоилокси-5β,20-эпокси-1-гидрокси-9-оксо-7β,10β-бис(2,2,2-трихлорэтоксикарбонилокси)-такс-11-ен-13-α-ил-(4S,5R)-3-т-(бутоксикарбонил)-2,2-диметил-4-фенил-5-оксазолидин-карбоксилата (DCT-II) формулы V с цинком и уксусной кислотой для получения соединения 4-ацетокси-2α-бензоилокси-5β,20-эпокси-1β,7β,10β-тригидрокси-9-оксо-такс-11-ен-13-α-ил-(4S,5R)-3-т-(бутоксикарбонил)-2,2-диметил-4-фенил-5-оксазолидинкарбоксилата (DCT-III) формулы VI;

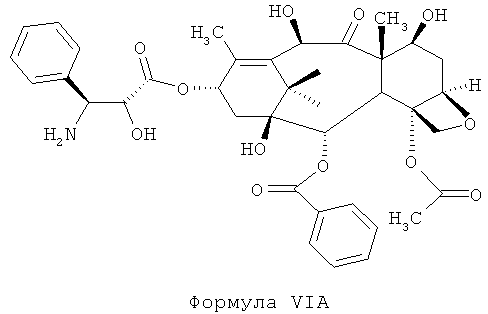
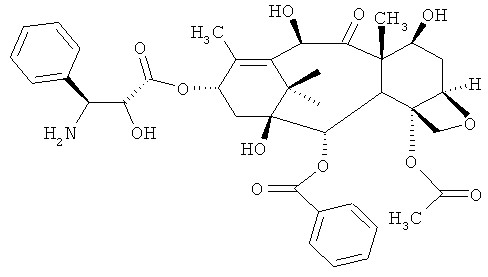
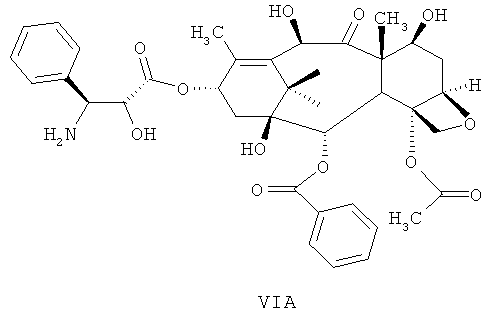
b) проведение реакции соединения DCT-III формулы VI с кислотой для получения соединения 4-ацетокси-2α-бензоилокси-5β,20-эпокси-1β,7β,10β-тригидрокси-9-оксо-такс-11-ен-13-α-ил-(2R,3S)-3-амино-2-гидрокси-3-фенил-пропионата (DCT-IV) формулы VIA; и
с) проведение реакции соединения DCT-IV формулы VIA с ди-т-бутилдикарбоната для получения доцетаксела формулы I.
Далее, аспект настоящего изобретения предусматривает фармацевтическую композицию, включающую одну или больше из кристаллических Формы I, Формы II, Формы III, Формы IV, Формы V, Формы VI, Формы VII, Формы VIII и Формы IX и аморфной формы, доцетаксела вместе с одним или больше фармацевтически приемлемыми эксципиентами.
Полиморфы доцетаксела по настоящему изобретению являются стабильными и хорошо пригодными для фармацевтических композиций, полезных для лечения болезни, включая, без ограничения, неопластические опухоли.
В варианте исполнения, изобретение предусматривает способ получения доцетаксела, включающий проведение реакции соединения, имеющего формулу
с кислотой, для получения соединения, имеющего формулу
В другом варианте исполнения, изобретение предусматривает способ получения кристаллической Формы 1 доцетаксела, включающий объединение раствора доцетаксела в кетоне с антирастворителем.
Следующий вариант исполнения изобретения предусматривает способ получения кристаллической Формы II доцетаксела, включающий объединение раствора доцетаксела в ацетонитриле с водой.
В следующем варианте исполнения, изобретение предусматривает способ получения кристаллической Формы III доцетаксела, включающий суспендирование твердого доцетаксела в изопропиловом спирте.
Следующий вариант исполнения изобретения предусматривает способ получения кристаллической Формы IV доцетаксела, включающий объединение раствора доцетаксела в N,N-диметилформамиде с водой.
В дополнительном варианте исполнения, изобретение предусматривает способ получения кристаллической Формы V доцетаксела, включающий объединение раствора доцетаксела в тетрагидрофуране с толуолом.
Вариант исполнения изобретения предусматривает способ получения кристаллической полиморфной формы доцетаксела, включающий:
а) обеспечение раствора доцетаксела в органическом растворителе;
б) удаление растворителя из раствора а) для образования кристаллов; и
в) выделение твердого кристаллического полиморфа доцетаксела.
Другие варианты исполнения изобретения предусматривают: кристаллическую Форму I доцетаксела; кристаллическую Форму II доцетаксела; кристаллическую Форму III доцетаксела; кристаллическую Форму IV доцетаксела; кристаллическую Форму V доцетаксела; кристаллическую Форму VI доцетаксела; кристаллическую Форму VII доцетаксела; кристаллическую Форму VIII доцетаксела; и кристаллическую Форму IX доцетаксела.
В следующем варианте исполнения, изобретение предусматривает способ получения твердого аморфного доцетаксела, включающий объединение раствора доцетаксела в тетрагидрофуране с антирастворителем.
В следующем варианте исполнения, изобретение предусматривает способ получения твердого аморфного доцетаксела, включающий удаление растворителя из раствора доцетаксела в спирте.
Краткое описание чертежей
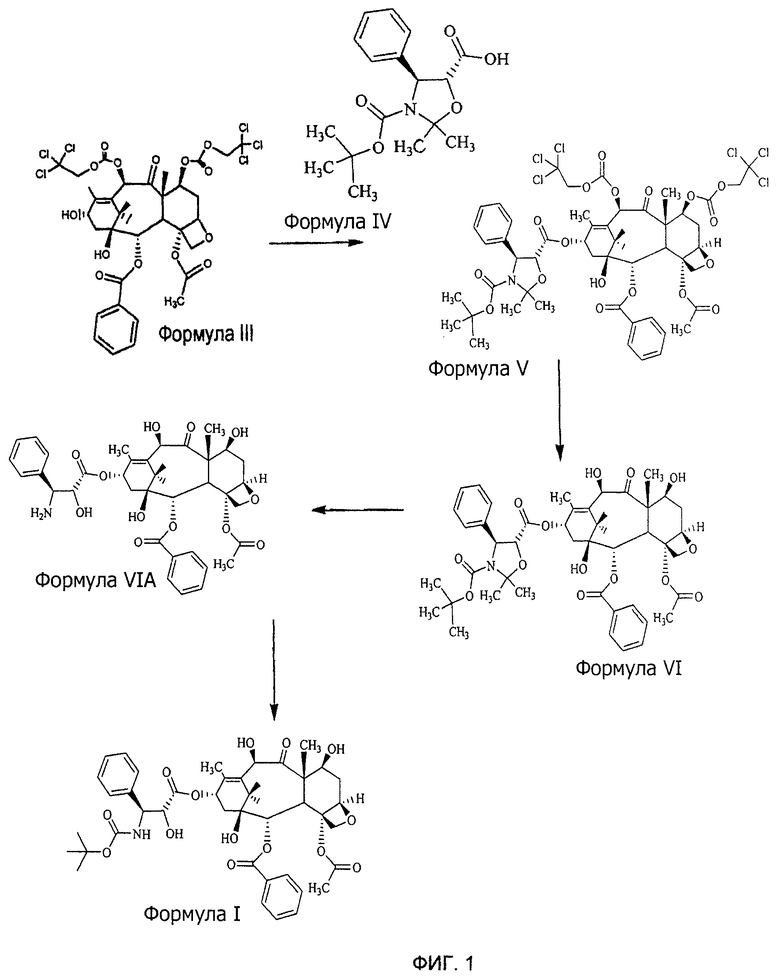
Фиг.1 является схематическим представлением способа получения доцетаксела.
Фиг.2 представляет собой рентгеновскую порошковую дифрактограмму (XRPD) Формы I доцетаксела, приготовленной в соответствии с Примером 18.
Фиг.3 изображает кривую дифференциальной сканирующей калориметрии (ДСК) Формы I доцетаксела, приготовленной в соответствии с Примером 18.
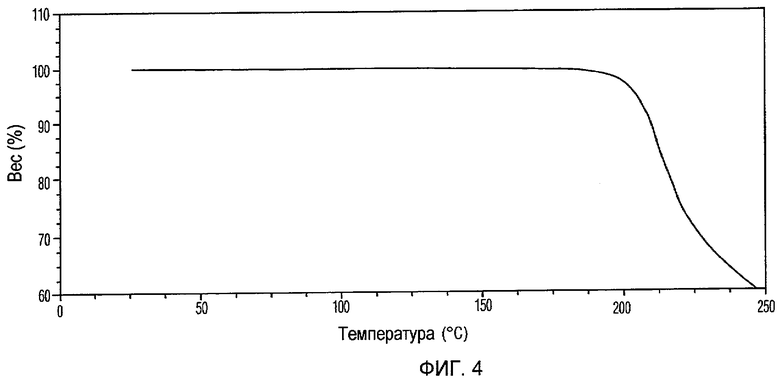
Фиг.4 изображает кривую термогравиметрического анализа (ТГА) Формы I доцетаксела, приготовленной в соответствии с Примером 18.
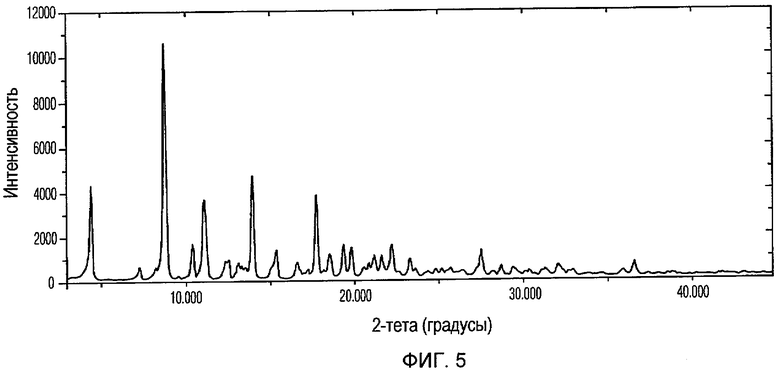
Фиг.5 изображает XRPD рентгенограмму Формы II доцетаксела, приготовленной в соответствии с Примером 2.
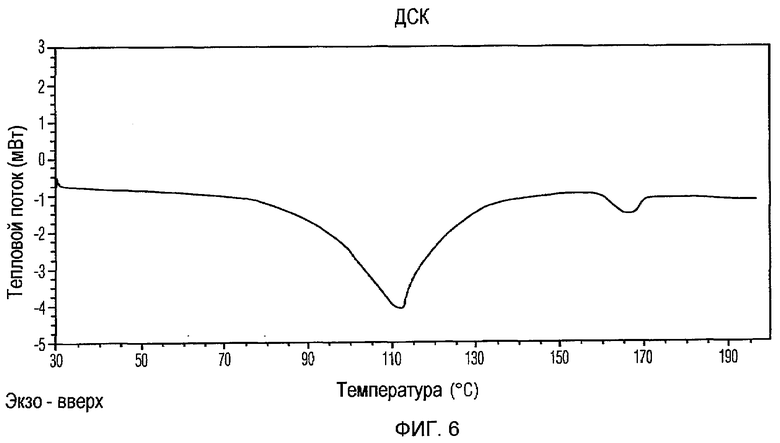
Фиг.6 изображает кривую ДСК Формы II доцетаксела, приготовленной в соответствии с Примером 2.
Фиг.7 изображает кривую ТГА Формы II доцетаксела, приготовленной в соответствии с Примером 2.
Фиг.8 изображает XRPD рентгенограмму Формы III доцетаксела, приготовленной в соответствии с Примером 3.
Фиг.9 изображает кривую ДСК Формы III доцетаксела, приготовленной в соответствии с Примером 3.
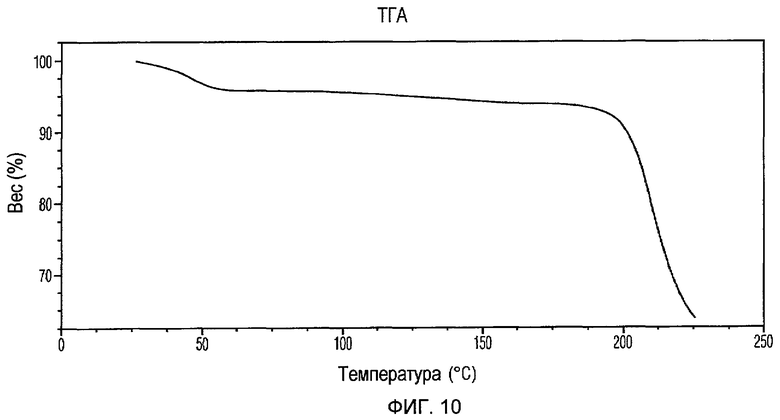
Фиг.10 изображает кривую ТГА Формы III доцетаксела, приготовленной в соответствии с Примером 3.
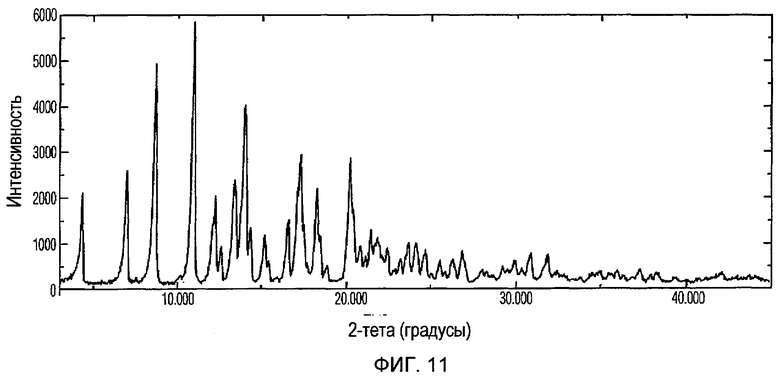
Фиг.11 изображает XRPD рентгенограмму Формы IV доцетаксела, приготовленной в соответствии с Примером 4.
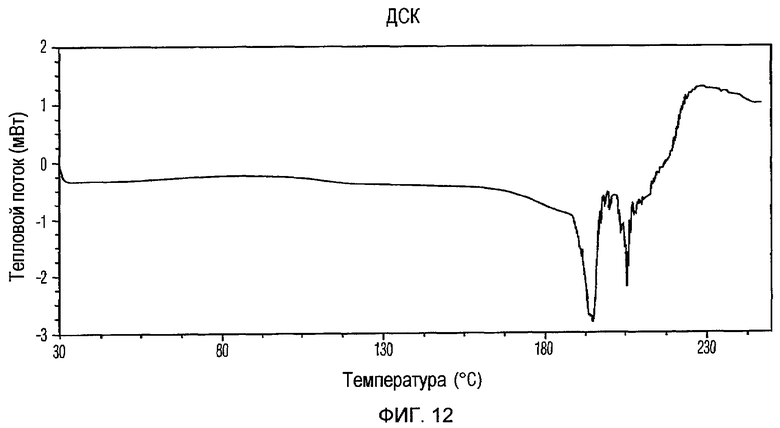
Фиг.12 изображает кривую ДСК Формы IV доцетаксела, приготовленной в соответствии с Примером 4.
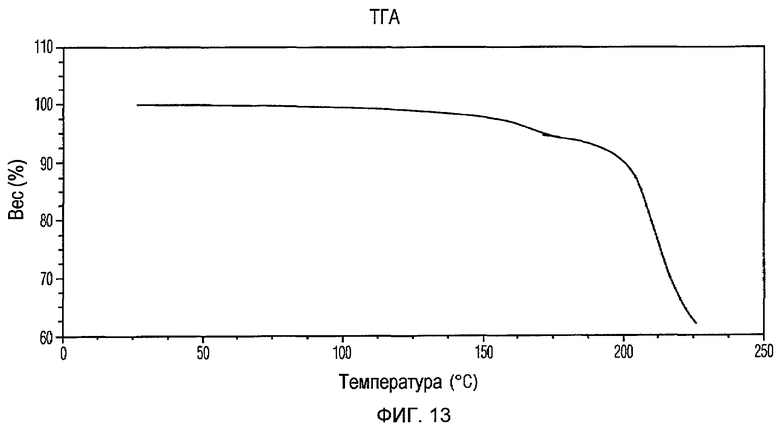
Фиг.13 изображает кривую ТГА Формы IV доцетаксела, приготовленной в соответствии с Примером 4.
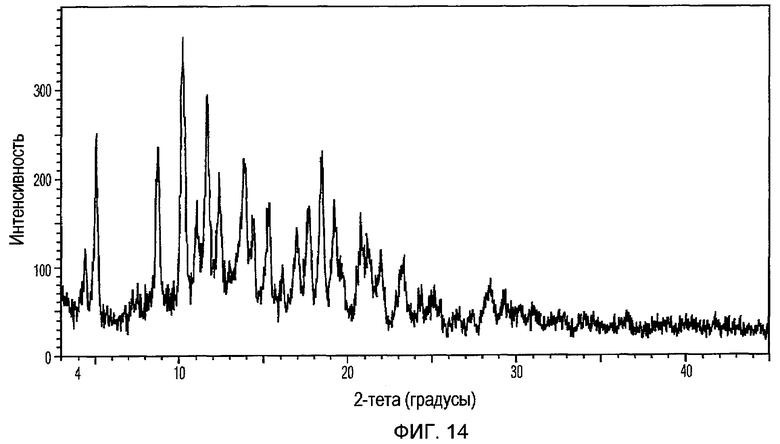
Фиг.14 изображает XRPD рентгенограмму Формы V доцетаксела, приготовленной в соответствии с Примером 5.
Фиг.15 изображает кривую ДСК Формы V доцетаксела, приготовленной в соответствии с Примером 5.
Фиг.16 изображает кривую ТГА Формы V доцетаксела, приготовленной в соответствии с Примером 5.
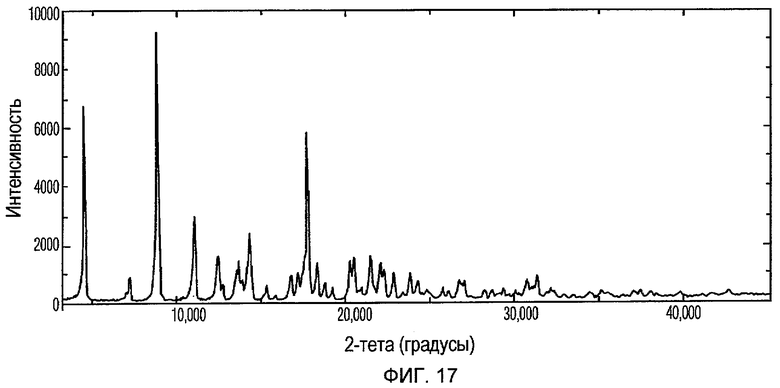
Фиг.17 изображает XRPD рентгенограмму Формы VI доцетаксела, приготовленной в соответствии с Примером 8.
Фиг.18 изображает кривую ДСК Формы VI доцетаксела, приготовленной в соответствии с Примером 8.
Фиг.19 изображает кривую ТГА Формы VI доцетаксела, приготовленной в соответствии с Примером 8.
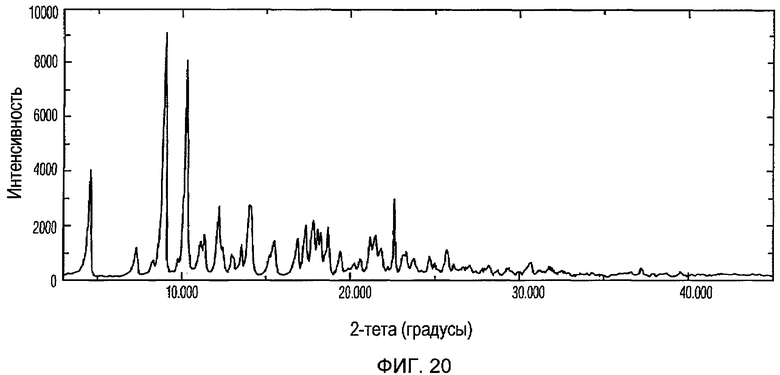
Фиг.20 изображает XRPD рентгенограмму Формы VII доцетаксела, приготовленной в соответствии с Примером 9.
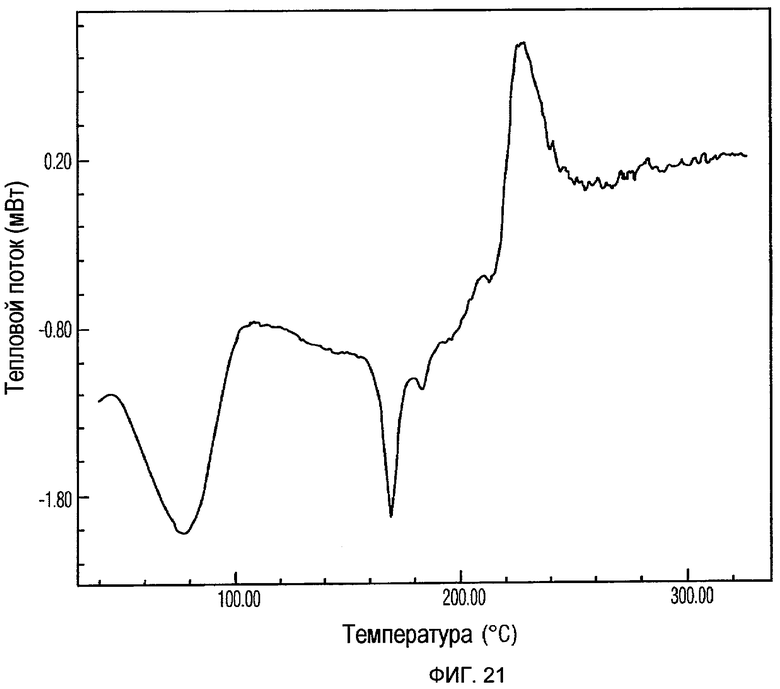
Фиг.21 изображает кривую ДСК Формы VII доцетаксела, приготовленной в соответствии с Примером 9.
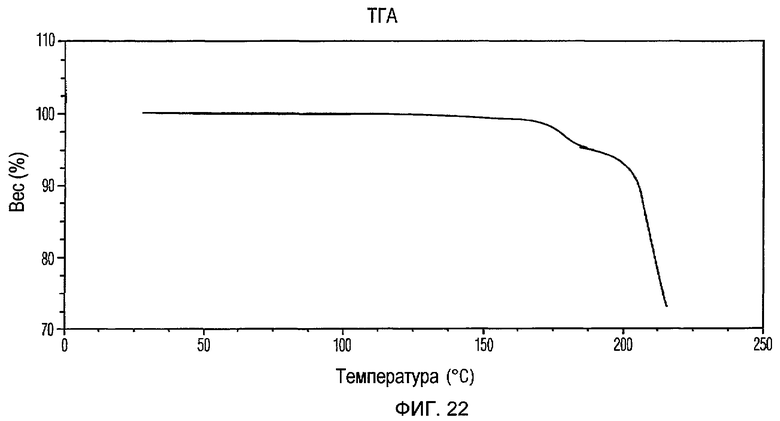
Фиг.22 изображает кривую ТГА Формы VII доцетаксела, приготовленной в соответствии с Примером 9.
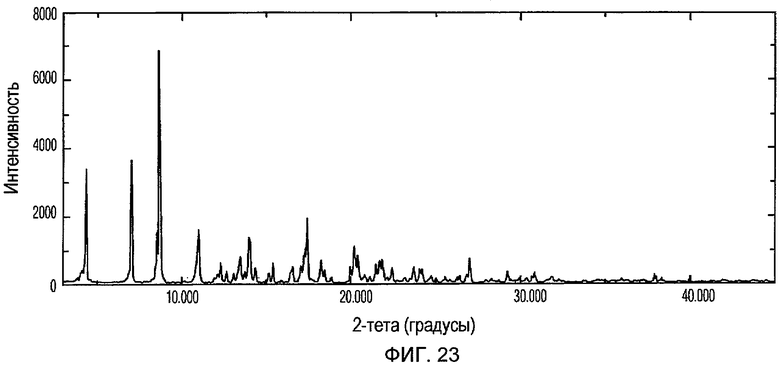
Фиг.23 изображает XRPD рентгенограмму Формы VIII доцетаксела, приготовленной в соответствии с Примером 10.
Фиг.24 изображает кривую ДСК Формы VIII доцетаксела, приготовленной в соответствии с Примером 10.
Фиг.25 изображает кривую ТГА Формы VIII доцетаксела, приготовленной в соответствии с Примером 10.
Фиг.26 изображает XRPD рентгенограмму Формы IX доцетаксела, приготовленной в соответствии с Примером 11.
Фиг.27 изображает кривую ДСК Формы IX доцетаксела, приготовленной в соответствии с Примером 11.
Фиг.28 изображает кривую ТГА Формы IX доцетаксела, приготовленной в соответствии с Примером 11.
Фиг.29 изображает ORTEP рентгенограмму Формы VIII доцетаксела, приготовленной в соответствии с Примером 10.
Фиг.30 изображает ORTEP рентгенограмму Формы IX доцетаксела, приготовленной в соответствии с Примером 11.
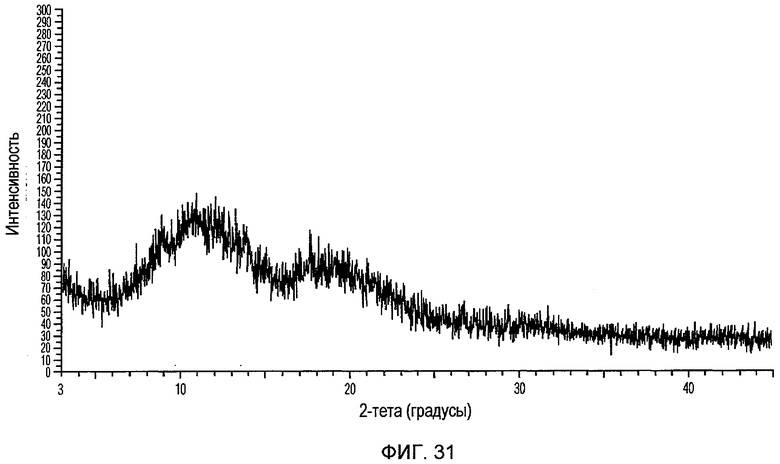
Фиг.31 изображает XRPD рентгенограмму аморфного доцетаксела, приготовленного в соответствии с Примером 6.
Детальное описание изобретения
Настоящее изобретение относится к полиморфным формам доцетаксела и способам их получения. Настоящее изобретение также предусматривает способ получения доцетаксела.
В одном варианте исполнения, настоящее изобретение предусматривает кристаллические полиморфы доцетаксела и способ их получения. Эти полиморфы называются ниже Формой I, Формой II, Формой III, Формой IV, Формой V, Формой VI, Формой VII, Формой VIII и Формой IX.
Кристаллические полиморфы, полученные способом по настоящему изобретению, если не указывается иное, характеризуются их рентгеновскими порошковыми дифрактограммами (XRPD), кривыми дифференциальной сканирующей калориметрии (ДСК) и кривыми термогравиметрического анализа (ТГА).
Все XRPD данные, описанные в настоящем изобретении, были получены с использованием Cu-Кα излучения, имеющего длину волны 1,541 А, и были получены с помощью порошкового рентгеновского дифрактометра Bruker AXS D8 Advance.
Дифференциальный сканирующий калориметрический анализ проводился с использованием инструмента DSC Q1000 фирмы ТА Instruments со скоростью нагрева 5°С/минуту при времени модуляции 60 секунд и температуре модуляции ±1°С. Начальная температура равнялась 0°С, и конечная температура равнялась 200°С.
ТГА анализ проводят с помощью инструмента TGAQ500V64 Build 193 со скоростью нагрева 10°С/минуту до 250°С.
Кристаллическая Форма I доцетаксела по настоящему изобретению характеризуется его XRPD рентгенограммой, которая по существу соответствует Фиг.2. Кристаллическая Форма I доцетаксела по настоящему изобретению характеризуется его XRPD рентгенограммой, имеющей характеристические пики при углах дифракции 2-тета примерно 8,0, 11,3, 12,5, 13,8, 15,4, 16,9, 20,3 и 23,3, ±0,2 градуса.
Кристаллическая Форма I доцетаксела по настоящему изобретению далее характеризуется его кривой ДСК, по существу соответствующей Фиг.3, имеющей эндотермический пик при примерно 165,05°С.
Кристаллическая Форма I доцетаксела по настоящему изобретению далее характеризуется его кривой ТГА, по существу согласно Фиг.4, соответствующей потере веса примерно 0,2% мас./мас.
Настоящее изобретение предусматривает способ получения кристаллической Формы I доцетаксела, включающий объединение раствора доцетаксела в кетоне с антирастворителем.
Раствор доцетаксела получают растворением доцетаксела в пригодном кетоне, таком как ацетон, метилизобутилкетон, метилэтилкетон и.т.д.
Концентрация доцетаксела в растворе не является критичной при условии использования достаточного количества растворителя для обеспечения полного растворения. Количество используемого растворителя обычно поддерживают как можно меньшим во избежание чрезмерных потерь продукта во время кристаллизации и выделения.
Количество растворителя, используемого для приготовления раствора, зависит от природы растворителя и температуры, принятой для приготовления раствора. Концентрация доцетаксела в растворе может обычно составлять от примерно 0,01 до примерно 0,15 г/мл в растворителе.
Пригодные температуры приготовления раствора могут составлять от примерно 20 до 120°С или от примерно 25 до примерно 35°С, в зависимости от используемого растворителя. Любая другая температура также является приемлемой, пока это не создает угрозы для стабильности доцетаксела.
Доцетаксел кристаллизуют из раствора путем объединения с антирастворителем. Пригодные антирастворители включают, без ограничения: эфиры, такие как диэтиловый эфир, диизопропиловый эфир, 1,4-диоксан, диметоксиэтан, метилтретбутиловый эфир и.т.д.; углеводороды, такие как н-пентан, н-гексан, н-гептан, циклогексан, бензол, толуол и.т.д.; низкокипящие смеси углеводородов, такие как петролейный эфир и.т.д.; и их комбинации.
Соотношение между растворителем и антирастворителем в растворе составляет от примерно 1:1 до примерно 1:10 или примерно 1:3 по объему.
Полученную Форму I, необязательно, суспендируют в пригодном растворителе для снижения содержания органических летучих примесей. Пригодные растворители, используемые для приготовления суспензии, включают н-гептан, н-гексан, циклогексан и.т.д. Пригодные температуры могут составлять от 20 до примерно 40°С или от примерно 25 до примерно 35°С. Суспензия может выдерживаться в течение периода времени от примерно 20 минут до примерно 24 часов или больше.
Кристаллическая Форма II доцетаксела по настоящему изобретению характеризуется его XRPD рентгенограммой, которая по существу соответствует Фиг.5. Кристаллическая Форма II доцетаксела по настоящему изобретению характеризуется его XRPD рентгенограммой, имеющей характеристические пики при углах дифракции 2-тета примерно 4,4, 7,2, 8,8, 10,4, 11,1, 14, 17,8 и 19,4, ±0,2 градуса.
Кристаллическая Форма II доцетаксела далее характеризуется его кривой ДСК, которая по существу соответствует Фиг.6, имеющей эндотермические пики при примерно 112 и 166°С.
Кристаллическая Форма II доцетаксела по настоящему изобретению имеет кривую ТГА, по существу совпадающую с Фиг.7, соответствующую потере веса примерно 6% мас./мас. Она имеет водосодержание примерно 7% мас./мас. при определении по метуду К.Фишера (KF).
Настоящее изобретение предусматривает способ получения кристаллической Формы II доцетаксела, включающий объединение раствора доцетаксела в ацетонитриле с водой при пригодной температуре.
Раствор доцетаксела получают путем растворения доцетаксела в ацетонитриле. Температуры приготовления раствора могут находиться в интервале значений от примерно 20 до 120°С или от примерно 40 до примерно 45°С. Концентрация доцетаксела в растворе может обычно составлять от примерно 0,05 до примерно 0,5 г/мл или 0,1 г/мл.
Доцетаксел кристаллизуют из раствора путем объединения с антирастворителем, таким как вода. Соотношение между ацетонитрилом и антирастворителем в растворе составляет от примерно 1:1 до примерно 1:10 или от примерно 1:4 до примерно 1:5 по объему.
Пригодные температуры кристаллизации Формы II доцетаксела составляют от примерно 25 до примерно 70°С или от примерно 40 до примерно 45°С.
Кристаллическая Форма III доцетаксела по настоящему изобретению характеризуется его XRPD рентгенограммой, которая по существу соответствует Фиг.8. Кристаллическая Форма III доцетаксела по настоящему изобретению характеризуется его XRPD рентгенограммой, имеющей характеристические пики при углах дифракции 2-тета примерно 4,3, 7,0, 8,7, 11, 12,3, 13,3, 14, 17,2, 17,3, 18,4 и 20,4, ±0,2 градуса.
Кристаллическая Форма III доцетаксела далее характеризуется его кривой ДСК, которая по существу соответствует Фиг.9, имеющей эндотермические пики при примерно 104 и 162°С.
Кристаллическая Форма III доцетаксела по настоящему изобретению имеет характеристическую кривую ТГА, по существу совпадающую с Фиг.10, соответствующую потере веса примерно 6% мас./мас. Она имеет содержание воды примерно 6% мас./мас. при определении по методу К.Фишера.
Настоящее изобретение предусматривает способ получения кристаллической Формы III доцетаксела, включающий суспендирование доцетаксела в изопропиловом спирте в течение периода времени от примерно 30 минут до примерно 5 часов или примерно 1 часа или дольше.
Количество изопропилового спирта может обычно составлять от примерно 1 л до примерно 5 л или примерно 2 л на 1 кг доцетаксела в растворе.
Пригодная температура суспендирования находится в интервале от примерно 20 до примерно 60°С или от примерно 25 до примерно 35°С.
Кристаллическая Форма IV доцетаксела по настоящему изобретению характеризуется его XRPD рентгенограммой, которая по существу соответствует Фиг.11. Кристаллическая Форма IV доцетаксела по настоящему изобретению характеризуется его XRPD рентгенограммой, имеющей характеристические пики при углах дифракции 2-тета примерно 4,3, 7,0, 8,7, 10,9, 12,2, 13,4, 14, 17,1, 17,2, 18,2, 20,2 и 20,4, ±0,2 градуса.
Кристаллическая Форма IV доцетаксела далее характеризуется его кривой ДСК, которая по существу соответствует Фиг.12, имеющей эндотермические пики при примерно 114 и 195°С.
Кристаллическая Форма IV доцетаксела по настоящему изобретению имеет характеристическую кривую ТГА, по существу совпадающую с Фиг.13, соответствующую потере веса примерно 6% мас./мас. Она имеет водосодержание примерно 1% мас./мас. при определении по методу К.Фишера.
Настоящее изобретение предусматривает способ получения кристаллической Формы IV доцетаксела, включающий объединение раствора доцетаксела в N,N-диметилформамиде (ДМФ) с водой.
Раствор доцетаксела может быть приготовлен растворением доцетаксела в ДМФ. Количество растворенного доцетаксела зависит от объема растворителя и от температуры. Концентрация доцетаксела в растворе может находиться в интервале значений от примерно 0,1 до примерно 1 г/мл или примерно 0,5 г/мл.
Кристаллическую Форму IV доцетаксела осаждают путем объединения воды с раствором доцетаксела в ДМФ.
Соотношение между ДМФ и водой может находиться в интервале значений от примерно 1:1 до примерно 1:15 или до примерно 1:10, по объему.
Пригодные температуры для образования Формы IV доцетаксела составляют примерно 20 до примерно 60°С или от примерно 25 до примерно 35°С.
Кристаллическая Форма V доцетаксела по настоящему изобретению характеризуется его XRPD рентгенограммой, которая по существу соответствует Фиг.14. Кристаллическая Форма V доцетаксела по настоящему изобретению характеризуется его XRPD рентгенограммой, имеющей характеристические пики при углах дифракции 2-тета примерно 4,4, 5,1, 8,8, 10,3, 11,1, 11,7, 12,4, 13,9, 14,4, 15,3, 17,0, 17,7, 18,5, 19,3, 20,8, 21,2 и 22, ±0,2 градуса.
Кристаллическая Форма V доцетаксела далее характеризуется его кривой ДСК, которая по существу соответствует Фиг.15, имеющей эндотермические пики при примерно 96 и 167°С.
Кристаллическая Форма V доцетаксела по настоящему изобретению имеет характеристическую кривую ТГА, по существу совпадающую с Фиг.16, соответствующую потере веса примерно 3% мас./мас. Она имеет водосодержание примерно 4% мас./мас. при определении по методу К.Фишера.
Настоящее изобретение предусматривает способ получения кристаллической Формы V доцетаксела, включающий объединение раствора доцетаксела в тетрагидрофуране с толуолом.
Раствор доцетаксела может быть приготовлен растворением доцетаксела в тетрагидрофуране. Количество растворенного доцетаксела зависит от объема растворителя и от температуры. Концентрация доцетаксела в растворе может находиться в интервале значений от примерно 0,1 до примерно 0,5 г/мл или составлять примерно 0,25 г/мл.
Осаждение кристаллической Формы V доцетаксела может быть осуществлено путем объединения раствора доцетаксела с толуолом.
Соотношение между ТГФ и толуолом может находиться в интервале значений от примерно 1:1 до примерно 1:35 или от примерно 1:20 до примерно 1:30, по объему.
Пригодные температуры для образования кристаллической Формы V доцетаксела составляют от примерно 20 до примерно 60°С или от примерно 25 до примерно 35°С.
Кристаллическая Форма VI доцетаксела по настоящему изобретению характеризуется его XRPD рентгенограммой, которая по существу соответствует Фиг.17. Кристаллическая Форма VI доцетаксела по настоящему изобретению характеризуется его XRPD рентгенограммой, имеющей характеристические пики при углах дифракции 2-тета примерно 4,3, 8,7, 10,8, 12,2, 14,1, 17,4, 17,6, 20,3, 21,3 и 43,7, ±0,2 градуса.
Кристаллическая Форма VI доцетаксела далее характеризуется его кривой ДСК, которая по существу соответствует Фиг.18, имеющей эндотермический пик при примерно 200°С.
Кристаллическая Форма VI доцетаксела по настоящему изобретению имеет характеристическую кривую ТГА, которая по существу совпадает с Фиг.19, соответствующую потере веса примерно 4% мас./мас.
Кристаллическая Форма VII доцетаксела по настоящему изобретению характеризуется его XRPD рентгенограммой, которая по существу соответствует Фиг.20. Кристаллическая Форма VII доцетаксела по настоящему изобретению характеризуется его XRPD рентгенограммой, имеющей характеристические пики при углах дифракции 2-тета примерно 4,6, 9,1, 10,3, 12,2, 14,1, 17,4, 17,8, 18,1, 18,7 и 22,6, ±0,2 градуса.
Кристаллическая Форма VII доцетаксела далее характеризуется его кривой ДСК, которая по существу соответствует Фиг.21, имеющей эндотермический пик при примерно 183°С.
Кристаллическая Форма VII доцетаксела по настоящему изобретению имеет характеристическую кривую ТГА, по существу совпадающую с Фиг.22, соответствующую потере веса примерно 4% мас./мас.
Кристаллическая Форма VIII доцетаксела характеризуется его XRPD рентгенограммой, которая по существу соответствует Фиг.23. Кристаллическая Форма VIII доцетаксела характеризуется его XRPD рентгенограммой, имеющей характеристические пики при углах дифракции 2-тета примерно 4,4, 7,0, 8,7, 11,0, 14,0, 17,5 и 20,3, ±0,2 градуса.
Кристаллическая Форма VIII доцетаксела далее характеризуется его кривой ДСК, которая по существу соответствует Фиг.24, имеющей эндотермический пик при примерно 193°С.
Кристаллическая Форма VIII доцетаксела по настоящему изобретению имеет характеристическую кривую ТГА, которая по существу совпадает с Фиг.25, соответствующую потере веса примерно 1% мас./мас.
Кристаллическая Форма VIII доцетаксела далее характеризуется его данными рентгеновской дифракции на монокристалле ("ORTEP"), по существу соответствующими Фиг.29, и имеет следующие характеристики:
Параметры элементарной ячейки
Информация о пространственной группе
Кристаллическая Форма IX доцетаксела характеризуется его XRPD рентгенограммой, по существу соответствующей Фиг.26. Кристаллическая Форма IX доцетаксела характеризуется его XRPD рентгенограммой, имеющей характеристические пики при углах дифракции 2-тета примерно 4,6, 9,2, 11,3, 12,5, 14,2, 15,4, 17,1, 17,5, 18,4, 18,6, 18,8, 20,6 и 21,0, ±0,2 градуса.
Кристаллическая Форма IX доцетаксела далее характеризуется его кривой ДСК, которая по существу соответствует с Фиг.27, имеющей эндотермический пик при примерно 173°С.
Кристаллическая Форма IX доцетаксела имеет характеристическую кривую ТГА, по существу совпадающую с Фиг.28, имеющую потерю веса, равную примерно 4% мас./мас.
Кристаллическая Форма IX доцетаксела по настоящему изобретению далее характеризуется данными его рентгеновской дифракции на монокристалле (ORTEP), по существу соответствующей Фиг.30, и имеет следующие характеристики.
Параметры элементарной ячейки
Информация о пространственной группе
Настоящее изобретение предлагает способ получения кристаллических Формы VI, Формы VII, Формы VIII и Формы IX доцетаксела, включающий:
а) обеспечение раствора доцетаксела в пригодном органическом растворителе;
б) удаление растворителя из раствора стадии а) для образования кристаллов; и
в) выделение желательного кристаллического полиморфа доцетаксела в твердом состоянии.
Стадия (а) включает обеспечение раствора доцетаксела в органическом растворителе в пригодных условиях.
Раствор доцетаксела может быть получен путем растворения соединения в органическом растворителе. Для приготовления раствора приемлема любая форма доцетаксела, такая как любая кристаллическая или аморфная форма доцетаксела.
Органические растворители, которые могут быть использованы для приготовления раствора доцетаксела, включают, без ограничения: диметилсульфоксид (ДМСО), ацетонитрил, N,N-диметилформамид (ДМФ), н-бутанол и т.д.
В варианте исполнения, кристаллическую Форму VI доцетаксела (ДМСО-сольват) получают при использовании ДМСО в качестве растворителя.
В другом варианте исполнения кристаллическую Форму VII доцетаксела (ацетонитрильный сольват) получают при использовании ацетонитрила в качестве растворителя.
В следующем варианте исполнения, кристаллическую Форму VIII доцетаксела (ДМФ-сольват) получают при использовании ДМФ в качестве растворителя.
В еще одном варианте исполнения кристаллическую Форму IX доцетаксела (н-бутанольный сольват) получают при использовании н-бутанола в качестве растворителя.
Концентрация доцетаксела в растворе не является критичной при условии использования достаточного количества растворителя для обеспечения полного растворения для получения гомогенного раствора. Количество растворителя, используемого для растворение доцетаксела, может находиться в интервале значений от примерно 1- до примерно 25-кратного по отношению к весу взятого доцетаксела.
Температура растворения доцетаксела может находиться в интервале значений от примерно 0°С до примерно 100°С или до температуры кипения с обратным холодильником используемого растворителя.
Полученный раствор, необязательно, может быть профильтрован, например, путем пропускания через фильтровальную бумагу, фильтровальную ткань, стекловолокно или другие мембранные материалы или через слой осветляющего агента, такого как целит, для обеспечения желательного гомогенного раствора.
Стадия (б) включает удаление растворителя из раствора стадии (а) в пригодных условиях для образования кристаллов.
Удаление растворителя может быть пригодно осуществлено с использованием испарения, перегонки при атмосферном давлении или перегонки под вакуумом при перемешивании или без перемешивания раствора.
Выпаривание растворителя может быть проведено при температуре от примерно 0°С до примерно 150°С. Может быть использована любая температура при условии, что концентрированно происходит без увеличения уровня содержания примесей.
Время выпаривания растворителя может находиться в интервале значений от примерно 1 часа до примерно 48 часов или дольше, в присутствии вакуума или без него и в присутствии инертной атмосферы, такой как азот, аргон, гелий и т.д., или без нее.
Стадия (с) включает выделение твердого вещества со стадии (б), которое представляет собой желательную полиморфную форму доцетаксела формулы I.
Кристаллическое состояние соединений может быть однозначно описано несколькими кристаллографическими параметрами: размеры элементарной ячейки, пространственная группа и положения всех атомов соединения по отношению к началу координат его элементарной ячейки. Эти параметры определяются экспериментально рентгеновским анализом монокристалла.
Результаты рентгеновского анализа монокристалла ограничены, как следует из названия методики, одним кристаллом, помещенным в пучок рентгеновских лучей. Кристаллографические данные для большой группы кристаллов дают информацию о рентгеновской порошковой дифракции. Если порошок состоит из чистого кристаллического соединения, то получают простую порошковую рентгенограмму. Для сравнения результатов анализа монокристалла и порошкового рентгеновского анализа могут быть выполнены простые расчеты для преобразования анализа монокристалла и порошковой рентгенограммы. Это преобразование является возможным потому, что эксперимент на монокристалле позволяет легко определить размеры элементарной ячейки, пространственную группу и положения атомов. Эти параметры создают основу для расчета рентгенограммы совершенного порошка. Сравнение этой расчетной порошковой рентгенограммы с порошковой рентгенограммой, полученной экспериментально для большой группы кристаллов, показывает, совпадают ли результаты для двух методик. Эта процедура была выполнена для сольватированных монокристаллов доцетаксела, имеющих Форму VIII и Форму IX.
Размер элементарной ячейки определяется тремя параметрами: длина сторон ячейки, относительные углы между сторонами по отношению друг к другу и объем ячейки. Длины сторон элементарной ячейка обозначаются a, b и с. Относительные углы между сторонами ячейки обозначаются α, β и γ. Объем ячейки обозначается V.
В варианте исполнения, настоящее изобретение предусматривает способ получения аморфной формы доцетаксела, включающий осаждение аморфного доцетаксела из раствора доцетаксела в ТГФ с помощью углеводородного антирастворителя и выделение осажденного аморфного твердого вещества.
Раствор доцетаксела может быть приготовлен путем растворения доцетаксела в ТГФ. Концентрация доцетаксела в растворе не является критичной при условии использования достаточного количества ТГФ для обеспечения полного растворения. Количество используемого ТГФ обычно поддерживают небольшим во избежание чрезмерных потерь продукта во время кристаллизации и выделения.
Количество ТГФ, используемого для приготовление аморфного доцетаксела, часто составляет от примерно 1- до примерно 12-кратного к весу доцетаксела.
Раствор может быть приготовлен при температуре в интервале значений от примерно 0°С до примерно 100°С. В зависимости от количества взятого растворителя данное количество доцетаксела может растворяться при температуре окружающей среды или раствор может потребовать нагревания до повышенной температуры, такой как от примерно 25°С до 100°С.
Аморфный доцетаксел может быть получен путем объединения раствор доцетаксела с антирастворителем.
Пригодные антирастворители, которые могут быть использованы при получении аморфного доцетаксела, включают, без ограничения: линейные или разветвленные или циклические алканы, содержащие от 4 до примерно 10 атомов углерода, такие как н-гексан, н-гептан, циклогексан, циклогептан и.т.д.; ароматические углеводороды, такие как бензол, толуол, ксилол и.т.д.; и их смеси.
Аморфная форма доцетаксела, полученная по настоящему изобретению, может быть выделена любым способом, таким как декантация растворителя или методом фильтрации, или путем испарения растворителя.
Настоящее изобретение также предусматривает другой способ получения аморфной формы доцетаксела, включающий удаление растворителя из раствора доцетаксела в спирте.
Раствор доцетаксела может быть приготовлен растворением доцетаксела в спирте, таком как этанол, метанол, н-бутанол и т.д. или любые их комбинации.
Растворитель может быть удален любыми способами, такими как перегонка, испарение под вакуумом, распылительная сушка, ATFD (сушка в тонком слое с перемешиванием), лиофилизация, мгновенное испарение и.т.д.
Пригодные температуры для образования аморфного доцетаксела составляют от примерно 25 до примерно 70°С или от примерно 35 до примерно 50°С.
Влажный осадок, полученный после удаления растворителя, необязательно, может быть дополнительно высушен. Высушивание может быть пригодно осуществлено в лотковой сушилке, вакуумной печи, сушильном шкафу, сушилке с псевдоожиженным слоем, вращающейся распылительной сушилке, распылительной сушилке и.т.д. Высушивание может быть осуществлено при температурах от примерно 35°С до примерно 70°С. Высушивание может проводиться в течение любых периодов времени, необходимых для получения желательной чистоты, таких как от примерно 1 до 25 часов или дольше.
Исходный материал, который может быть использован для приготовления полиморфных форм по настоящему изобретению, может быть неочищенным или чистым доцетакселем, полученным любым известным специалистам способом. Исходный материал для любого процесса может иметь любую полиморфную форму, такую как кристаллические формы доцетаксела, аморфный доцетаксел или смеси аморфной и кристаллических форм доцетаксела в любых пропорциях, полученных любым способом.
Выделение может быть осуществлено с помощью таких методик, как фильтрование, декантация, центрифугирование и.т.д., или путем фильтрования под инертной атмосферой с использованием таких газов, как, например, азот и.т.д.
В другом аспекте, настоящее изобретение также предусматривает способ получения доцетаксела, включающий:
а) проведение реакции соединения 4-ацетокси-2α-бензоилокси-5β,20-эпокси-1-гидрокси-9-оксо-7β,10β-бис(2,2,2-трихлорэтоксикарбонилокси)такс-11-ен-13-α-ил-(4S,5R)-3-т-(бутоксикарбонил)-2,2-диметил-4-фенил-5-оксазолидин-карбоксилат (DCT-II) формулы V с цинком и уксусной кислотой для получения соединения 4-ацетокси-2α-бензоилокси-5β,20-эпокси-1β,7β,10β-тригидрокси-9-оксо-такс-11-ен-13-α-ил-(4S,5R)-3-т-(бутоксикарбонил)-2,2-диметил-4-фенил-5-оксазолидинкарбоксилата (DCT-III) Формулы VI;

б) проведение реакции соединения DCT-III формулы VI с кислотой для получения соединения 4-ацетокси-2α-бензоилокси-5β,20-эпокси-1β,7β,10β-тригидрокси-9-оксо-такс-11-ен-13-α-ил-(2R,3S)-3-амино-2-гидрокси-3-фенил-пропионата (DCT-IV) формулы VIA; и
в) проведение реакции соединения DCT-IV формулы VIA с ди-т-бутилдикарбонатом для получения доцетаксела формулы I.
Стадия а) включает проведение реакции DCT-II формулы V с цинком и уксусной кислотой для получения DCT-III формулы VI.
Количество цинка, используемого на стадии (а), может находиться в интервале значений от примерно 1 до примерно 10 молярных эквивалентов или примерно 8 молярных эквивалентов на молярный эквивалент DCT-II формулы V. Цинк может быть использован в любой форме, такой как порошок, стружки, гранулы и т.д.
Концентрация уксусной кислоты, используемой на стадии (а), составляет от примерно 95 до примерно 100%. Количество уксусной кислоты может находиться в интервале значений от примерно 1 до примерно 15 л на кг DCT-II формулы V.
После завершения реакции реакционная смесь может быть профильтрована для удаления цинка, и затем твердое вещество выделяют путем объединения раствора с антирастворителем, таким как вода.
Необязательно, полученное твердое вещество может быть растворено в пригодном растворителе и затем твердое вещество повторно осаждают с антирастворителем для получения желательной чистоты.
Растворители, которые могут быть использованы для растворения, включают, без ограничения, этилацетат, изобутилацетат, н-бутилацетат, н-пропилацетат, изопропилацетат и.т.д. Антирастворители, которые могут быть использованы для осаждения, включают, без ограничения, линейные или разветвленные алканы или циклоалканы, состоящие из соединений от С4 до примерно С10, таких как н-пентан, н-гексан, н-гептан, циклогексан и т.д., или ароматические углеводороды, такие как бензол, толуол, ксилол и т.д.
DCT-III формулы VI, полученный описанным выше способом по настоящему изобретению, имеет чистоту не менее примерно 88% или примерно 90%, при определении методом высокоэффективной жидкостной хроматографии (ВЭЖХ).
Стадия (б) включает проведение реакции DCT-III формулы VI с кислотой для получения DCT-IV формулы VIA.
Пригодные кислоты, которые могут быть использованы на стадии (б), включают, без ограничения, муравьиную кислоту, уксусную кислоту, трифторуксусную кислоту и т.д.
Количество кислоты, используемой на стадии (б), могут находиться в интервале значений от примерно 1 до примерно 25 л на 1 кг DCT-III формулы VI.
Пригодные температуры для проведения реакции составляют от примерно 10 до примерно 50°С или от примерно 25 до 30°С.
После завершения реакции реакционную смесь концентрируют и продукт экстрагируют пригодным растворителем, а затем рН устанавливают с помощью основания в пригодном растворителе.
Концентрирование проводят при температуре от примерно 35 до примерно 65°С или от примерно 40 до примерно 45°С для образования остатка.
Добавляют к остатку воду и полученный раствор промывают пригодным органическим растворителем, таким как метилизобутилкетон (MIBK), этилацетат, изобутилацетат, н-бутилацетат, н-пропилацетат, изопропилацетат, дихлорметан, хлороформ и т.д.
Регулируют рН водного слоя с помощью пригодного основания до значения между 7,5 и примерно 10. Пригодные основания, используемые для регулирования рН, включают, без ограничения, неорганические основания, такие как гидроксид натрия, карбонат натрия, бикарбонат натрия, ацетат натрия, гидроксид калия, карбонат калия, бикарбонат калия и т.д.; органические основания, такие как триэтиламин, диизопропиламин и т.д.
Соединение DCT-IV формулы VIA, необязательно, не выделяют из реакционной смеси. Оно может быть использовано непосредственно для превращения в доцетаксел на следующей стадии.
Стадия (в) включает проведение реакции DCT-IV формулы VIA с ди-т-бутилдикарбонатом для получения доцетаксела формулы I.
Количество ди-т-бутилдикарбоната, используемого для получения доцетаксела, составляет от примерно 1 до примерно 4 молярных эквивалентов или примерно 3 молярных эквивалента, на молярный эквивалент DCT-III формулы VI (если DCT-IV не выделяют) или на молярный эквивалент DCT-IV формулы VIA.
Пригодные растворители, которые могут быть использованы для получения доцетаксела, включают, без ограничения, воду, этилацетат, изобутилацетат, н-бутилацетат, н-пропилацетат, изопропилацетат, дихлорметан, хлороформ и т.д. и их смеси.
Стадия с) может быть осуществлена при температурах от примерно 10 до примерно 65°С или от примерно 25 до примерно 35°С.
После завершения реакции органический слой отделяют и концентрируют до пригодного объема. Концентрирование может быть пригодно осуществлено с использованием таких методов, как выпаривание, перегонка при атмосферном давлении, перегонка под вакуумом или сушка в тонком слое при перемешивании ("ATFD"). Концентрирование типично прекращают, когда концентрация доцетаксела достигает от примерно 0,1 г/мл до примерно 0,5 г/мл или примерно 0,3 г/мл.
Твердое вещество может быть выделено из концентрированного реакционного раствора путем объединения с антирастворителем. Пригодные антирастворители включают, без ограничения, линейные или разветвленные алифатические алканы или циклоалканы от C4 до примерно С10, такие как н-гексан, н-гептан, циклогексан и т.д., или ароматические углеводороды, такие как бензол, толуол, ксилол и т.д.
Доцетаксел, полученный способом по настоящему изобретению, типично имеет чистоту не менее примерно 75% или примерно 80%, при определении методом ВЭЖХ.
Полученный доцетаксел по настоящему изобретению может быть очищен путем хроматографии на колонке с гелем диоксида кремния при элюировании с помощью элюента и затем снова очищен путем перекристаллизации из пригодных растворителей.
Гель диоксида кремния, который может быть использован для очистки, может иметь диапазон размера частиц, такой как, например, 230-400 меш, 10С-200 меш, 60-100 меш или 500-750 меш.
Пригодные элюенты включают, без ограничения, этилацетат, изобутилацетат, н-бутилацетат, н-пропилацетат, изопропилацетат, н-гептан, н-гексан, циклогексан и их комбинации.
Твердое вещество может быть выделено из чистых фракций элюента такими методами, как выпаривание, перегонка при атмосферном давлении, перегонка под вакуумом или сушка в тонком слое при перемешивании ("ATFD") и т.д.
Доцетаксел, полученный описанным выше способом хроматографии на колонке по настоящему изобретению, типично имеет чистоту не менее примерно 90% или примерно 94%, при определении методом ВЭЖХ.
Полученный в результате хроматографии на колонке доцетаксел может быть далее очищен путем объединения раствора доцетаксела в кетоне с антирастворителем.
Раствор доцетаксела может быть приготовлен путем растворения доцетаксела в кетонном растворителе. Пригодные кетонные растворители включают, без ограничения, ацетон, метилизобутилкетон, метилэтилкетон и т.д.
Концентрация доцетаксела в растворе не является критичной, но количество используемого растворителя обычно поддерживают минимальным во избежание чрезмерных потерь продукта во время кристаллизации твердого вещества. Концентрация доцетаксела в растворе может обычно составлять от примерно 0,01 до примерно 0,25 г/мл в растворителе.
Раствор может быть приготовлен при температурах в интервале значений от примерно 25°С до 100°С. В зависимости от количества взятого растворителя доцетаксел можно растворять при температуре от 25 до 35°С, или раствор может требовать нагрева до повышенной температуры в интервале от примерно 40°С до 55°С.
Обработка обесцвечивающим углем может быть необязательно проведена или при температуре растворения, или после охлаждения раствора до более низкой температуры.
Твердое вещество может кристаллизоваться из реакционного раствора путем объединения с антирастворителем. Пригодные антирастворители включают эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран (ТГФ), 1,4-диоксан, диметоксиэтан, метилтретбутиловый эфир и т.д.
Пригодные температуры для кристаллизации твердого вещества могут находиться в интервале значений от примерно 20 до примерно 80°С или от примерно 25 до примерно 35°С.
Вышеуказанный процесс перекристаллизации может быть повторен один или больше раз для получения чистоты, типично, более или равной примерно 99% или примерно 99,5% мас. при определении с помощью ВЭЖХ.
Полученный доцетаксел, необязательно, суспендируют в пригодном растворителе для получения желательного уровня содержания остаточных растворителей, часто выражаемого в ppm (млн-1), при определении методом газовой хроматографии (ГХ), Пригодные растворители включают н-гептан, н-гексан, циклогексан и т.д.
Пригодные температуры составляют от 20 до примерно 40°С или от примерно 25 до примерно 35°С.
Суспензия может выдерживаться в течение периода времени от примерно 20 минут до примерно 4 часов или дольше.
Влажное твердое вещество может, необязательно, быть далее высушено. Высушивание может быть осуществлено при температурах от примерно 35°С до примерно 70°С в течение любых периодов времени, необходимых для получения желательной чистоты, таких как от примерно 1 до 25 часов или дольше.
Полиморфы доцетаксела по настоящему изобретению являются стабильными и хорошо пригодными для использования в изготовлении фармацевтических композиций. Фармацевтические композиции в соответствии с настоящим изобретением включают, без ограничения, твердые дозированные формы для перорального приема, такие как таблетки, капсулы, порошки и т.д.; жидкие дозированные формы для перорального приема, такие как растворы, дисперсии, суспензии, эмульсии и т.д.; дозированные формы для парентерального введения (включая внутримышечное, подкожное, внутривенное), такие как инъецируемые дозы в виде растворов или суспензий, или дисперсий, или стерильных порошков для восстановления; системы трансдермальной доставки; системы направленной доставки и т.д.
Композиции твердых дозированных форм для перорального приема включает эксципиенты, включающие, без ограничения, разбавители, дезинтегрирующие агенты, связующие, смазывающие вещества, вкусовые агенты, красящие агенты и т.д. Для жидких дозированных форм для перорального приема композиции включают, без ограничения, фармацевтически приемлемые водные или неводные носители и т.д., вкусовые агенты, консерванты, солюбилизаторы, эмульгаторы и т.д.
Фармацевтические композиции, предназначенные для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостатики и растворенные вещества, делающие композицию изотонической с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители. Композиции могут находиться в разовых или многоразовых контейнерах, например герметизированных ампулах и флаконах, и могут храниться в высушенном вымораживанием (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например воды для инъекций, непосредственно перед использованием. Растворы и суспензии для немедленных инъекций могут быть приготовлены из стерильных порошков, гранул и таблеток.
Фармацевтические композиции могут быть предназначены для местного введения, включая, без ограничения, жидкие мази, кремы, суспензии, лосьоны, порошки, растворы, пасты, гели, спреи, аэрозоли или масла.
Фармацевтические композиции могут быть далее упакованы во флаконы или ампулы, изготовленные из стекла, контейнеры и крышечки из полиэтилена высокой плотности (HDPE), полиэтилена низкой плотности (LDPE) и/или полипропилена, и/или стекла, и блистерные упаковки или полоски (strips), состоящие из алюминия или полипропилена высокой плотности. Эти перечни не должны считаться исчерпывающими, поскольку другие материалы и типы упаковки также являются пригодными.
Способ получения доцетаксела и способы получения полиморфов доцетаксела по настоящему изобретению являются простыми, дают продукт, имеющий высокую точку плавления и более высокую стабильность, и являются экономически эффективными, воспроизводимыми, надежными и промышленно масштабируемыми.
Определенные конкретные аспекты и варианты исполнения настоящего изобретения будут объяснены более подробно со ссылкой на следующие примеры, которые приводятся только для иллюстрации и не должны истолковываться как каким-либо образом ограничивающие объем изобретения.
ПРИМЕР 1: ПОЛУЧЕНИЕ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ I ДОЦЕТАКСЕЛА
Растворяют 126 г доцетаксела в 1,35 л ацетона и осаждают путем добавления 8,1 л петролейного эфира. Смесь перемешивают в течение 90 минут при 27°С и суспензию твердого вещества фильтруют. Твердое вещество высушивают в течение 3 часов при 30°С под вакуумом 600 мм рт.ст., получая 121 г указанного в заголовке соединения. Чистота: 99,6% по методу ВЭЖХ. Водосодержание: 1,98% мас./мас. при определении по методу К.Фишера.
ПРИМЕР 2: ПОЛУЧЕНИЕ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ II ДОЦЕТАКСЕЛА
Растворяют 300 г доцетаксела в 3 л этанола при 27°С и фильтруют. Фильтрат концентрируют до сухого остатка при 50°С под вакуумом 680 мм рт.ст. в течение 1 часа. Полученное твердое вещество высушивают при 35°С под вакуумом 650 мм рт.ст. в течение 4 часов. Твердое вещество суспендируют в 3 л ацетонитрила и нагревают до 40°С. Полученный раствор немедленно загружают в 14,5 л предварительно нагретой воды при 40°С. Смесь перемешивают в течение 4 часов при 45°С и затем суспензию твердого вещества фильтруют. Полученное твердое вещество промывают 2 л воды. Влажное твердое вещество загружают в 8,7 л воды и перемешивают в течение 1 часа при 25°С. Суспензию фильтруют и промывают 2 л воды. Вышеописанную процедуру суспендирования в воде повторяют 3 раза и полученное твердое вещество высушивают при 35°С в течение 2 часов под вакуумом 680 мм рт.ст. Полученное твердое вещество выдерживают при 80±2°С относительной влажности (RH) и температуре 25±2°С в течение примерно 36 часов, получая 273 г указанного в заголовке соединения.
Чистота: 99,67% по методу ВЭЖХ.
Водосодержание: 6,7% мас./мас. по методу К.Фишера.
ПРИМЕР 3: ПОЛУЧЕНИЕ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ III ДОЦЕТАКСЕЛА
Загружают 1 г доцетаксела (Форма II) и 5 мл изопропилового спирта в чистую и сухую круглодонную колбу и перемешивают в течение 1 часа. Полученное суспензию твердого вещества фильтруют и высушивают под вакуумом 680 мм рт.ст. при 50°С в течение 24 часов, получая 0,65 г указанного в заголовке соединения.
Чистота: 99,69% по методу ВЭЖХ.
Водосодержание: 6,3% мас./мас. по методу К.Фишера.
ПРИМЕР 4: ПОЛУЧЕНИЕ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ IV ДОЦЕТАКСЕЛА
Загружают 1 г доцетаксела (Форма II) в чистую и сухую круглодонную колбу. Загружают в колбу 2 мл ДМФ и смесь перемешивают в течение 1 часа. Полученный прозрачный раствор осаждают путем добавления 20 мл воды. Полученную реакционную суспензию фильтруют и высушивают под вакуумом 680 мм рт.ст. в течение 24 часов при 50°С, получая 0,85 г указанного в заголовке соединения.
Чистота: 98,47% по методу ВЭЖХ.
Водосодержание: 1,0% мас./мас. по методу К.Фишера.
ПРИМЕР 5: ПОЛУЧЕНИЕ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ V ДОЦЕТАКСЕЛА
Загружают 70 мл толуола в чистую и сухую круглодонную колбу при 27°С. Раствор доцетаксела (0,6 г Формы II, растворенной в 2,5 мл ТГФ) прибавляют к толуолу на протяжении 10 минут и перемешивают в течение 1 часа при 27°С.
Полученный осадок фильтруют и высушивают под вакуумом 680 мм рт.ст. в течение 30 часов при 50°С, получая 0,514 г указанного в заголовке соединения.
Чистота: 99,6% по методу ВЭЖХ.
Водосодержание: 4,28% мас./мас. по методу К.Фишера.
ПРИМЕР 6: ПОЛУЧЕНИЕ АМОРФНОГО ДОЦЕТАКСЕЛА ПУТЕМ ОСАЖДЕНИЯ
Загружают 70 мл н-гексана в чистую и сухую круглодонную колбу при 27°С. Раствор доцетаксела приготовлен растворением 0,6 г доцетаксела в 2,5 мл ТГФ, прибавляют к н-гексану на протяжении 10 минут и перемешивают в течение 1 часа для осаждения. Полученный осадок фильтруют и высушивают под вакуумом 680 мм рт.ст. при 50°С в течение 30 часов, получая 0,532 г указанного в заголовке соединения.
Чистота: 99,63% по методу ВЭЖХ.
ПРИМЕР 7: ПОЛУЧЕНИЕ АМОРФНОГО ДОЦЕТАКСЕЛА С ПОМОЩЬЮ ПЕРЕГОНКИ
Загружают 1 г доцетаксела и 10 мл этанола в чистую и сухую круглодонную колбу при 27°С. Полученный раствор нагревают до 50°С и прозрачный раствор концентрируют до сухого остатка под вакуумом 650 мм рт.ст. Вышеописанную процедуру повторяют 3 раза и затем подключают вакуум 600 мм рт.ст. на 30 минут для получения сухого твердого вещества. Полученное твердое вещество высушивают под вакуумом 600 мм рт.ст. при 50°С в течение 24 часов, получая 0,82 г указанного в заголовке соединения.
Чистота: 99,45% по методу ВЭЖХ.
ПРИМЕР 8: ПОЛУЧЕНИЕ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ VI ДОЦЕТАКСЕЛА
Загружают 5 мл ДМСО и 1 г доцетаксела в круглодонную колбу с одновременным перемешиванием при 27°С и перемешивают в течение 10 минут. Полученный раствор оставляют стоять без вмешательства при 27°С в течение 7 дней для испарения ДМСО из раствора. Полученные кристаллы фильтруют и высушивают с отсосом в течение 2 дней под вакуумом 650 мм рт.ст. при 30°С, получая указанное в заголовке соединение.
ПРИМЕР 9: ПОЛУЧЕНИЕ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ VII ДОЦЕТАКСЕЛА
Загружают 5 мл ацетонитрила и 1 г доцетаксела в круглодонную колбу при перемешивании при 27°С. Раствор перемешивают в течение 10 минут. Полученный раствор was стоять без вмешательства при 27°С в течение 6 дней для испарения ацетонитрила из раствора. Полученные кристаллы фильтруют под вакуумом 680 мм рт.ст. и высушивают с отсосом в течение 3 дней под вакуумом 600-680 мм рт.ст. при 25-30°С, получая кристаллическую Форму VII.
ПРИМЕР 10: ПОЛУЧЕНИЕ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ VIII ДОЦЕТАКСЕЛА
Загружают 5 мл ДМФ и 1 г доцетаксела в круглодонную колбу при перемешивании при 27°С. Раствор перемешивают в течение 10 минут. Раствор оставляют стоять без вмешательства при 27°С в течение 5 дней для испарения ДМФ из раствора, затем кристаллы фильтруют и высушивают с отсосом в течение 50 часов при 27°С под вакуумом 650 мм рт.ст., получая указанное в заголовке соединение.
Фиг.29 представляет собой ORTEP-изображение продукта сольвата доцетаксела и ДМФ (1:1), полученное при 50% уровне вероятности для неводородных атомов. Водородные атомы опущены для ясности.
ПРИМЕР 11: ПОЛУЧЕНИЕ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ IX ДОЦЕТАКСЕЛА
Загружают 5 мл н-бутанола и 1 г доцетаксела в круглодонную колбу при перемешивании при 27°С и перемешивают в течение 10 минут. Полученный раствор оставляют стоять без вмешательства 27°С в течение 6 дней для испарения н-бутанола из раствора, затем кристаллы фильтруют и высушивают с отсосом в течение 50 часов при 27°С под вакуумом 650 мм рт.ст., получая указанное в заголовке соединение.
Фиг.30 представляет собой ORTEP-изображение продукта сольвата доцетаксела и н-бутанола (1:1), полученное при 50% уровне вероятности для неводородных атомов.
ПРИМЕР 12: ПОЛУЧЕНИЕ 4-АЦЕТОКСИ-2α-БЕНЗОИЛОКСИ-5β,20-ЭПОКСИ-1β,13α-ДИГИДРОКСИ-9-ОКСО-7β,10β-БИС(2,2,2-ТРИХЛОРЭТОКСИ-КАРБОНИЛОКСИ)-11-ТАКСЕНА ФОРМУЛЫ III
Загружают 125 мл пиридина и 25 г 10-DAB III (10-деацетилбаккатин III) в чистую и сухую круглодонную колбу. Перемешивают раствор в течение 15 минут под атмосферой азота и медленно прибавляют раствор Troc-Cl (2,2,2-трихлорэтоксикарбонил хлорид), приготовленный разведением 22,6 мл Troc-Cl в 250 мл дихлорметана, на протяжении 1 часа под атмосферой азота. Перемешивают смесь в течение 5 минут и проверяют степень завершения реакции с помощью тонкослойной хроматографии (ТСХ) для подтверждения расходования исходного материала. Добавляют к реакционной смеси 500 мл деминерализированной воды и перемешивают в течение 5 минут. Отделяют органический слой от смеси и затем трижды промывают органический слой 1750 мл 10% раствор HCl. Органический слой промывают 500 мл насыщенного раствора NaHCO3, а затем 500 мл деминерализированной воды, после чего полученный органический слой концентрируют до объема 97,5 мл при 45°С под вакуумом 650 мм рт.ст. Добавляют к концентрату 250 мл толуола и снова концентрируют до объема 87,5 мл. Концентрат перемешивают в течение 2 часов при 0°С, а затем твердое вещество отделяют методом фильтрации, получая 36 г (выход: 87,8%) указанного в заголовке соединения.
Чистота: 96,76% по методу ВЭЖХ.
ПРИМЕР 13: ПОЛУЧЕНИЕ DCT-II ФОРМУЛЫ V
Помещают 1500 мл дихлорметана и 80,96 г (4S,5R)-3-трет-(бутокси-карбонил)-2,2-диметил-4-фенил-5-оксазолидинкарбоксилата в круглодонную колбу. Добавляют к раствору 150 г 4-ацетокси-2α-бензоилокси-5β,20-эпокси-1β,13α-дигидрокси-9-оксо-7β,10β-бис(2,2,2-трихлорэтоксикарбонилокси)-11-таксена (по Примеру 12) под атмосферой азота. Добавляют к раствору 20,5 г диметиламинопиридина (DMAP) и 103,9 г дициклогексилдикарбамида (DCC) и перемешивают в течение 1 часа. Реакционную смесь фильтруют и промывают 300 мл дихлорметана. Полученный фильтрат промывают 750 мл насыщенного раствора NaHCO3 и снова промывают 1500 мл деминерализированной воды. Полученный органический слой концентрируют до объема 450 мл при 45°С под вакуумом 580 мм рт.ст. Фильтруют органический слой для удаления побочного продукта дициклогексилмочевины и твердое вещество промывают 300 мл дихлорметана. Загружают 3000 мл н-гептана в другую круглодонную колбу при 25°С. Полученный фильтрат медленно прибавляют к н-гептану на протяжении периода 40 минут и затем перемешивают в течение 1 часа. Суспензию фильтруют и твердое вещество промывают 300 мл н-гептана. Твердое вещество высушивают при 50°С в течение 4 часов под вакуумом 680 мм рт.ст., получая 182,5 г (выход: 93,7%) указанного в заголовке соединения.
Чистота: 98,84% по методу ВЭЖХ.
ПРИМЕР 14: ПОЛУЧЕНИЕ DCT-III ФОРМУЛЫ VI
Загружают 1250 мл уксусной кислоты и 125 г DCT-II в чистую и сухую круглодонную колбу при перемешивании. Перемешивают раствор в течение 10 минут при 26°С и затем прибавляют к раствору 1250 мл метанола. Прибавляют к раствору 54,3 г цинковой пыли и затем нагревают до 55°С. Перемешивают смесь в течение 20 минут при 58°С. Готовят целитовый слой с помощью 65 г целита в 250 мл метанола. Реакционную смесь фильтруют через слой целита и слой промывают 250 мл метанола. Фильтрат медленно прибавляют к 8,75 л воды на протяжении периода 5 минут и затем суспензию перемешивают в течение 1 часа при 27°С. Суспензию фильтруют и твердое вещество промывают 250 мл воды. Твердое вещество растворяют в 1250 мл этилацетата и затем слой этилацетата промывают 125 мл воды. Органический слой фильтруют и затем фильтрат концентрируют до объема 500 мл при 50°С. Медленно прибавляют 2,5 л н-гептана на протяжении периода 15 минут при 27°С и затем суспензию перемешивают в течение 1 часа. Суспензию фильтруют и твердое вещество промывают 250 мл н-гептана и высушивают при 50°С в течение 4 часов, получая 76,6 г (выход: 86,7%) указанного в заголовке соединения.
Чистота: 90,54% по методу ВЭЖХ.
ПРИМЕР 15: ПОЛУЧЕНИЕ ДОЦЕТАКСЕЛА ФОРМУЛЫ I
Помещают 500 мл муравьиной кислоты в круглодонную колбу и охлаждают до 22°С. Прибавляют 50 г DCT-III и перемешивают в течение 1,5 часов. Раствор концентрируют при 42°С под вакуумом 580 мм рт.ст. до получения остатка. Прибавляют к остатку 500 мл MIBK и 500 мл воды и затем перемешивают в течение 10 минут. Водный слой отделяют и промывают 500 мл MIBK. Водный слой снова промывают этилацетатом (2×500 мл) и затем рН was доводят до 8,4 путем добавления 54 г NaHCO3. Прибавляют к водному слою 20 мл ди-т-бутилдикарбоната и перемешивают в течение 30 минут. Прибавляют к смеси 500 мл этилацетата и 20 мл ди-т-бутилдикарбоната и смесь перемешивают в течение 30 минут. Органический слой отделяют и концентрируют до объема 100 мл. Твердое вещество осаждают путем добавления 500 мл н-гептана и перемешивания в течение 1 часа. Суспензию фильтруют и твердое вещество высушивают в течение 30 минут при 27°С, получая 34 г указанного в заголовке соединения.
Чистота: 80,43% по методу ВЭЖХ.
ПРИМЕР 16: ОЧИСТКА ДОЦЕТАКСЕЛА С ПОМОЩЬЮ ХРОМАТОГРАФИИ НА КОЛОНКЕ
Колонку набивают 625 г геля диоксида кремния в 2 л 20% этилацетата в н-гептане. 25 г доцетаксела, приготовленного в соответствии с Примером 15, растворяют в 50 мл этилацетата и помещают в колонку. Колонку элюируют смесью этилацетата и н-гептана: 2 л 20% этилацетата и 20 л 50% этилацетата. После элюирования 11 л собирают 8,5 л очищенной фракции. Очищенную фракцию полностью концентрируют при 47°С под вакуумом 680 мм рт.ст., получая 16,2 г указанного в заголовке соединения.
Чистота: 94,47% по методу ВЭЖХ.
ПРИМЕР 17: ОЧИСТКА ДОЦЕТАКСЕЛА
Загружают 1370 мл ацетона и 137 г доцетаксела в чистую и сухую круглодонную колбу. Смесь нагревают до 45°С и затем перемешивают в течение 30 минут. Раствор фильтруют и фильтрат охлаждают до 27°С. Прибавляют к раствору 4110 мл диизопропилового эфира и 100 мг чистого доцетаксела. Суспензию перемешивают в течение 1,5 часов и затем фильтруют. Твердое вещество промывают 275 мл диизопропилового эфира и высушивают при 60°С в течение 4 дней под вакуумом 680 мм рт.ст., получая 92 г указанного в заголовке соединения.
Чистота: 99,38% по методу ВЭЖХ.
ПРИМЕР 18: ПОЛУЧЕНИЕ КРИСТАЛЛИЧЕСКОЙ ФОРМЫ I ДОЦЕТАКСЕЛА
Загружают 140 мл ацетона и 7 г доцетаксела в круглодонную колбу. Раствор перемешивают в течение 10 минут и фильтруют. Фильтрат и 420 мл диизопропилового эфира загружают в круглодонную колбу. Прибавляют в вышеуказанную колбу 50 мг доцетаксела, полученного в соответствии с Примером 1, и затем перемешивают в течение 90 минут. Суспензию фильтруют и твердое вещество промывают 35 мл диизопропилового эфира. Твердое вещество высушивают в течение 2 дней при 27°С под вакуумом 680 мм рт.ст., получая 5,4 г указанного в заголовке соединения.
Загружают в круглодонную колбу 45 мл н-гептана и 4,5 г доцетаксела, полученного выше. Суспензию перемешивают в течение 15 минут и затем фильтруют. Твердое вещество промывают 4,5 мл н-гептана. Вышеописанный процесс повторяют еще два раза и затем твердое вещество высушивают при 50°С в течение 48 часов под вакуумом 680 мм рт.ст., получая 4,25 г указанного в заголовке соединения.
Чистота: 99,82% по методу ВЭЖХ.
Содержание диизопропилового эфира: 96 ppm.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПРИГОТОВЛЕНИЯ ПОЛИМОРФНОЙ ФОРМЫ B ДИЭТАНОЛАМИНОВОЙ СОЛИ ТРЕПРОСТИНИЛА | 2019 |
|
RU2778124C2 |
| ТВЁРДЫЕ ФОРМЫ ИНГИБИТОРА КИНАЗЫ РЕЦЕПТОРА ЭПИДЕРМАЛЬНОГО ФАКТОРА РОСТА | 2013 |
|
RU2673077C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ VI И VII КАЛЬЦИЕВОЙ СОЛИ АТОРВАСТАТИНА | 2002 |
|
RU2304139C2 |
| СОЛИ ИНГИБИТОРА КИНАЗЫ РЕЦЕПТОРА ЭПИДЕРМАЛЬНОГО ФАКТОРА РОСТА | 2013 |
|
RU2711077C2 |
| ПОЛИМОРФЫ И СОЛЬВАТЫ ГИДРОХЛОРИДА 4-[2-[[5-МЕТИЛ-1-(2-НАФТАЛИНИЛ)-1Н-ПИРАЗОЛ-3-ИЛ]ОКСИ]ЭТИЛ]МОРФОЛИНА | 2011 |
|
RU2560150C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ (R)-5-[3-ХЛОР-4-(2, 3-ДИГИДРОКСИПРОПОКСИ)БЕНЗ[Z]ИЛИДЕН]-2-([Z]-ПРОПИЛИМИНО)-3-о-ТОЛИЛТИАЗОЛИДИН-4-ОНА | 2009 |
|
RU2519548C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ { [1-ЦИАНО-5-(4-ХЛОРОФЕНОКСИ)-4-ГИДРОКСИИЗОХИНОЛИН-3-КАРБОНИЛ]-АМИНО} -УКСУСНОЙ КИСЛОТЫ | 2014 |
|
RU2666144C2 |
| СПОСОБ ПОЛУЧЕНИЯ БЕЗИЛАТА АМЛОДИПИНА | 1999 |
|
RU2173685C2 |
| ЦИТРАТНАЯ СОЛЬ 9Е-15-(2-ПИРРОЛИДИН-1-ИЛ-ЭТОКСИ)-7,12,25-ТРИОКСА-19,21,24-ТРИАЗАТЕТРАЦИКЛО[18.3.1.1(2,5).1(14,18)]ГЕКСАКОЗА-1(24),2,4,9,14,16,18(26),20,22-НОНАЕНА | 2010 |
|
RU2543721C2 |
| СПОСОБ ПОЛУЧЕНИЯ И ОЧИСТКИ ГЕМЦИТАБИНА ГИДРОХЛОРИДА | 2006 |
|
RU2345087C2 |
















Изобретение относится к способу получения доцетаксела, включающему стадии:
а) проведение реакции соединения формулы V
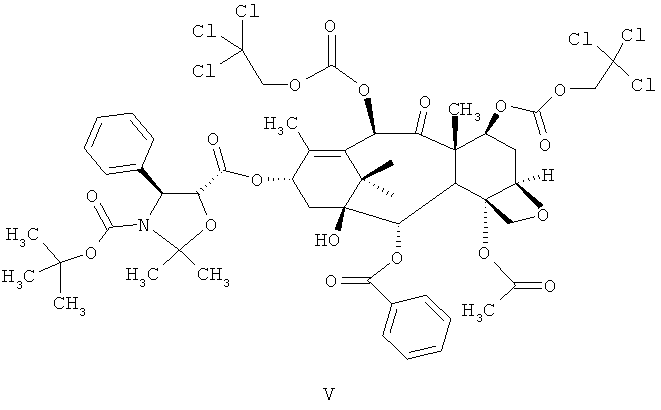
с цинком и уксусной кислотой с получением соединения формулы VI
b) проведение реакции соединения формулы VI с кислотой с получением соединения формулы VIA
и
с) проведение реакции соединения формулы VIA с ди-трет-бутилдикарбанатом с получением доцетаксела. Изобретение также относится к способам получения кристаллических Форм I, II, III, IV, V доцетаксела, к кристаллическим Формам I, II, III, IV, V, VI, VII, VIII, IX доцетаксела, к способам получения твердого аморфного доцетаксела, к способу получения кристаллической Формы I доцетаксела, а также к способу получения кристаллической Формы II доцетаксела. Технический результат - получение доцетаксела и его полиморфов с желательной чистотой и выходом путем использования улучшенных методов получения, являющихся простыми, экологически безопасными, экономически эффективными, надежными и хорошо пригодными для использования в промышленных масштабах. 20 н. и 7 з.п. ф-лы, 31 ил.
1. Способ получения доцетаксела, включающий стадии:
а) проведение реакции соединения формулы V
с цинком и уксусной кислотой с получением соединения формулы VI
b) проведение реакции соединения формулы VI с кислотой с получением соединения формулы VIA
и
с) проведение реакции соединения формулы VIA с ди-трет-бутилдикарбанатом с получением доцетаксела.
2. Способ по п.1, в котором соединение формулы VIA не выделяют перед проведением последующей реакции.
3. Способ получения кристаллической Формы I доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.2, включающий объединение раствора доцетаксела в кетоне с антирастворителем.
4. Способ по п.3, в котором антирастворитель представляет собой простой эфир или углеводород.
5. Способ получения кристаллической Формы II доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.5, включающий объединение раствора доцетаксела в ацетонитриле с водой.
6. Способ получения кристаллической Формы III доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.8, включающий суспендирование твердого доцетаксела в изопропиловом спирте.
7. Способ получения кристаллической Формы IV доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.11, включающий объединение раствора доцетаксела в N,N-диметилформамиде с водой.
8. Способ получения кристаллической Формы V доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.14, включающий объединение раствора доцетаксела в тетрагидрофуране с толуолом.
9. Способ получения кристаллической полиморфной формы доцетаксела, включающий:
a) получение раствора доцетаксела в органическом растворителе;
b) удаление растворителя из раствора а) для образования кристаллов; и
c) выделение твердого кристаллического полиморфа доцетаксела.
10. Способ по п.9, в котором растворитель представляет собой диметилсульфоксид, и получают кристаллическую Форму VI доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.17.
11. Способ по п.9, в котором растворитель представляет собой ацетонитрил, и получают кристаллическую Форму VII доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.20.
12. Способ по п.9, в котором растворитель представляет собой N,N-диметилформамид, и получают кристаллическую Форму VIII доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.23.
13. Способ по п.9, в которой растворитель представляет собой н-бутанол и получают кристаллическую Форму IX доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.26.
14. Кристаллическая Форма I доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.2.
15. Кристаллическая Форма II доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.5.
16. Кристаллическая Форма III доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.8.
17. Кристаллическая Форма IV доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.11.
18. Кристаллическая Форма V доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.14.
19. Кристаллическая Форма VI доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.17.
20. Кристаллическая Форма VII доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.20.
21. Кристаллическая Форма VIII доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.23.
22. Кристаллическая Форма IX доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.26.
23. Способ получения твердого аморфного доцетаксела, который характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.31, включающий объединение раствора доцетаксела в тетрагидрофуране с антирастворителем.
24. Способ по п.23, в которой антирастворитель представляет собой углеводород.
25. Способ получения твердого аморфного доцетаксела, который характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.31, включающий удаление растворителя из раствора доцетаксела в спирте.
26. Способ получения кристаллической Формы I доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.2, включающий:
a) растворение доцетаксела в кетоне, выбранном из ацетона, метилизобутилкетона и метилэтилкетона;
b) объединение полученного раствора с простым эфиром, выбранным из диэтилового эфира, диизопропилового эфира, 1,4-диоксана, диметоксиэтана и метилтретбутилового эфира;
c) выдерживание смеси при температуре 25-35°С; и
d) выделение твердых кристаллов.
27. Способ получения кристаллической Формы II доцетаксела, которая характеризуется рентгеновской порошковой дифрактограммой, как показано на фиг.5, включающий:
a) растворение доцетаксела в ацетонитриле при температуре 40-45°С;
b) объединение полученного раствора с водой;
c) выдерживание смеси при температуре 20-45°С до выпадения осадка; и
d) выделение твердых кристаллов.
| УСТРОЙСТВО для ФИКСАЦИИ КАТУШКИ | 0 |
|
SU212216A1 |
| US 2004116720 A1, 17.06.2004 | |||
| US 2002151579 A1, 17.10.2002 | |||
| WO 2004026230 A2, 01.04.2004 | |||
| KANAZAWA ET AL | |||
| J | |||
| ORG | |||
| CHEM., vol.59, 1994, pages 1238-1240 | |||
| ZASKE ET AL | |||
| MATERIALS SCIENCE FORUM, vol.443-444, 2004, pages 411-414 | |||
| HARPER ET AL | |||
| ACTA CRYSTALLOGRAPHIC SECTION C, no.1, 2001, pages 64-65 | |||
| ПРОИЗВОДНЫЕ 7-(2,2,2-ТРИХЛОР-ТРЕТ.БУТОКСИКАРБОНИЛ)-10-ГИДРОКСИТАКСАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ В СИНТЕЗЕ ТАКСАНОВ | 1996 |
|
RU2162844C2 |

