Область техники, к которой относится изобретение
Настоящее изобретение относится к ряду новых соединений, которые проявляют сильное ингибирующее действие против тирозинкиназы Брутона, и следовательно, могут обеспечить потенциальную тактику лечения карциномы человека, включая лимфому B-клеток и аутоиммунные заболевания, такие как ревматоидный артрит, системная красная волчанка, и множественный склероз.
Уровень техники
Тирозинкиназа Брутона (Btk) представляет собой нерецепторную цитоплазматическую тирозинкиназу, принадлежащую к Tec-семейству киназ, члены которого также включают Tec, Itk, Txk и Bmx. Большинство киназ преимущественно экспрессируются в гематопоэтических клетках и играют важную роль при ретрансляции сигнальной трансдукции от рецепторов клеточных поверхностей до непосредственного развития клеток, дифференцировки и других функций (Berg JJ и др. Annual Review of Immunology, 2005; 23:549-600). Btk является важным фактором развития, дифференцировки, созревания и передачи сигнала B-клеток (Mohamed AJ и др. Immunological Reviews, 2009; 228:58-73). Мутации с потерей функции Btk вызывают X-связанную гипогаммаглобулинемию (XLA) у людей и X-связанный иммунодефицит у мышей (Thomas JD и др. Science 1993; 261:355-358). Пациенты с XLA имеют нормальную пре-B-клеточную популяцию в костном мозге, но эти клетки является недостаточно зрелыми, чтобы участвовать в кровообращении. Поэтому у этих пациентов практически отсутствуют циркулирующие B-клетки, и они не способны вырабатывать антитела.
BTK играет главнейшую роль в пролиферации и активации B-клеток при посредничестве B-клеточного рецептора (BCR). При активации BCR Btk перемещается в плазматическую мембрану, где она фосфорилируется и впоследствии инициирует каскад событий передачи сигнала, в том числе активацию фосфолипазы Cγ2 (PLCγ2), и в конечном счёте приводит к восстановлению подвижности кальция и регуляции на уровне транскрипции, включая клеточный ядерный фактор каппа-B (NF κB) (Mohamed AJ и др. Immunological Reviews 2009; 228:58-73). В связи с обязательной ролью в сигнальном пути BCR, предполагается, что киназная активность Btk является весьма важной при развитии и поддержании множества злокачественный развитий B-клеток, в том числе хронической лимфоцитарной лейкемии (CLL) и ряда субтипов неходжкинской лимфомы (NHL), мантийноклеточной лимфомы (MCL), и диффузной лимфомы больших B-клеток (DLBCL) (Ponader S. и др. Blood 2012, 119:1182-1189; Honigberg LA и др. Proceedings of the National Academy of Sciences, 2010, 107:13075-13080). Кроме того, клинически была продемонстрирована роль B-клеток при патогенезе ревматоидного артрита, системной красной волчанки, множественного склероза, и других иммунных расстройствах (Edwards JC и др. The New England Journal of Medicine, 2004, 350:2572-2581; Favas C и др. Nature Review Rheumatology, 2009, 5:711-716; Hauset SL и др. The New England Journal of Medicine, 2008, 358:676-688). Поэтому, позиционирование Btk с помощью небольших молекулярных ингибиторов может обеспечить терапевтический эффект при лечении злокачественного развития B-клеток и аутоиммунных заболеваний.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
В одном замысле, соединения имеют формулу (I), или их фармацевтически приемлемые соли, сольваты, гидраты, метаболиты, или пролекарства:
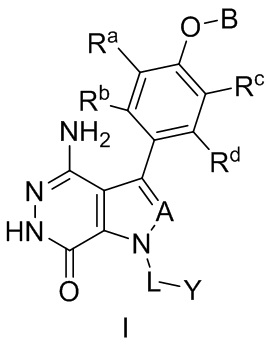
где:
A выбирают из группы, состоящей из CR1 и N; и где R1 выбирают из группы, состоящей из водорода, галогена, и незамещенного или замещенного алкила;
Ra, Rb, Rc и Rd независимо выбирают из группы, состоящей из водорода, галогена, гидроксила, нитро, циано, незамещенного или замещенного алкила, и незамещенного или замещенного алкоксила;
B выбирают из группы, состоящей из водорода, незамещенного или замещенного алкила, незамещенного или замещенного циклоалкила, незамещенного или замещенного гетероциклоалкила, незамещенного или замещенного арила, и незамещенного или замещенного гетероарила;
L представляет собой незамещенный или замещенный алкил, или отсутствует; и
Y выбирают из группы, состоящей из незамещенного или замещенного циклоалкила, незамещенного или замещенного гетероциклоалкила, незамещенного или замещенного арила, и незамещенного или замещенного гетероарила.
Краткое описание чертежей
На фигуре 1 продемонстрировано влияние Btk ингибиторов на рост опухоли для модели ксенотрансплантата TMD-8.
На фигуре 2 продемонстрировано влияние Btk ингибиторов на рост опухоли для модели ксенотрансплантата TMD-8 (окончательная масса опухоли).
Осуществление изобретения
Настоящее изобретение, главным образом, относится к соединениям, которые регулируют активность протеиновой тирозинкиназы, к способам синтеза и применения указанных соединений в терапевтических методах.
Определения
Любые термины в настоящем изобретении, за исключением специально определённых, будут принимать обычные значения, которые понятны специалисту в этой области техники.
Использованные в этом документе формы артиклей единственного числа включают и формы множественного числа, если из контекста, очевидно, не следует другое.
Если не обусловлено иное, все арильные, циклоалкильные, гетероарильные, и гетероциклические группы настоящего изобретения могут быть замещенными, как описано в каждом из соответствующих определений групп. Например, арильный компонент арилалкильной группы, такой как бензил, может быть замещен, как описано в определении термина “арил.”
Термин “алкокси” который используется в этом документе, относится к C1-C10, предпочтительно C1-C6, алкильной группе, присоединенной к функциональной группе исходной молекулы через атом кислорода. Типичные примеры алкокси группы включают (но не ограничиваются приведенным) метокси (CH3O-), этокси (CH3CH2O-), и трет-бутокси ((CH3)3CO-).
Термин “алкил” который используется в этом документе, относится к группе, произведенной из насыщенного углеводорода с линейной или разветвлённой цепью путем удаления водорода от одного из насыщенных атомов углерода. Предпочтительно алкильная группа содержит от одного до десяти атомов углерода, более предпочтительно от одного до шести атомов углерода. Типичные примеры алкильных групп включают (но не ограничиваются приведенным) метил, этил, изопропил, и трет-бутил.
Термин “арил” который используется в этом документе, относится к группе, произведенной из C6-C12, предпочтительно C6-C10, ароматического карбоцикла путем удаления атома водорода из ароматического кольца. Арильная группа может быть моноциклической, бициклической или полициклической. Предпочтительные примеры арильных групп включают фенил и нафтил.
Термин “циано” который используется в этом документе, относится к -CN.
Термин “циклоалкил” который используется в этом документе, относится к группе, произведенной из моноциклического насыщенного карбоцикла, предпочтительно имеющего от трех до восьми, более предпочтительно от трех до шести, атомов углерода, путем удаления атома водорода от насыщенного карбоцикла. Типичные примеры циклоалкильных групп включают (но не ограничиваются приведенным) циклопропил, циклопентил и циклогексил. Когда циклоалкильная группа содержит одну или несколько двойных связей в кольце, однако не ароматического, образуется “циклоалкенильная” группа.
Термины “галоид” и “галоген” которые используются в этом документе, относятся к F, Cl, Br, или I.
Термин “галоидалкокси” который используется в этом документе, относится к C1-C6, предпочтительно C1-C4, галоидалкильной группе, присоединенной к функциональной группе исходной молекулы через атом кислорода.
Термин “галоидалкил” который используется в этом документе, относится к
C1-C10, предпочтительно C1-C6, более предпочтительно C1-C4, алкильной группе, замещенной, по меньшей мере, одним атомом галогена. Галоидалкильная группа может быть алкильной группой, в которой все атомы водорода замещены галогеном. Характерные примеры галоидалкила включают (но не ограничиваются приведенным) трифторметил (CF3-), 1-хлорэтил (ClCH2CH2-), и 2,2,2-трифторэтил (CF3CH2-).
Термин “гетероарил” который используется в этом документе, относится к 5- - 10-членной, моноциклической или бициклической ароматической группе, содержащей один или несколько, предпочтительно от одного до трех, гетероатомов, независимо выбранных из азота, кислорода и серы в ароматическом кольце (кольцах). Как хорошо известно специалистам в этой области техники, гетероарильные кольца имеют менее выраженный ароматический характер, чем их полностью углеродные аналоги. Таким образом, для целей настоящего изобретения необходимо, чтобы гетероарильная группа обладала только в некоторой степени ароматической природой. Иллюстративные примеры гетероарильных групп включают (но не ограничиваются приведенным) пиридил, пиридазинил, пиримидил, пиразил, триазинил, пирролил, пиразолил, имидазолил, пиримидинил, фурил, тиенил, изоксазолил, тиазолил, изоксазолил, oксазолил, индолил, хинолинил, изохинолинил, бензизоксазолил, бензoтиазолил, и бензoтиенил.
Термин “гетероциклическая” который используется в этом документе, относится к 3- - 10-членной моноциклической или бициклической неароматической группе, содержащей один или несколько, предпочтительно от одного до трех, гетероатомов, независимо выбранных из азота, кислорода, и серы в неароматическом кольце (кольцах). Гетероциклические группы настоящего изобретения могут быть присоединены к функциональной группе исходной молекулы через атом углерода или aтом азота в группе. Гетероциклическая группа может быть насыщенной или ненасыщенной, например, содержащей одну или несколько двойных связей в кольце. Примеры гетероциклических групп включают (но не ограничиваются приведенным) морфолинил, оксазолидинил, пиперазинил, пиперидинил, пирролидинил, тетрагидрофурил, тиоморфолинил и индолинил, или тому подобные.
Термины “гидрокси” или “гидроксил” которые используются в этом документе, относятся к -OH.
Термин “нитро” который используется в этом документе, относится к -NO2.
Термин “оксо” который используется в этом документе, относится к “=O”.
Когда указано, что любая группа, например, алкил, алкенил, “циклоалкил”, “арил”, “гетероциклическая” или “гетероарильная”, является “необязательно замещенной” за исключением специально определённых случаев, это означает, что эта группа является замещенной (или незамещенной) одним - пятью, предпочтительно одним - тремя заместителями, которые независимо выбраны из галогена, алкила, алкокси, галоидалкила, галоидалкокси, гидрокси, оксо, ацила, циано, нитро, и амино-группы, или тому подобной, при условии, что такое замещение не будет нарушать общепринятые принципы связывания, известные специалистам в этой области техники. При использовании выражения “необязательно замещенные” до перечня групп это означает, что каждая из перечисленных групп может быть необязательно замещенной.
Соединения настоящего изобретения могут существовать как фармацевтически приемлемые соли или сольваты. Термин “фармацевтически приемлемая соль”, который используется в этом документе, означает любую нетоксичную соль, которая при введении реципиенту, способна образовать соединения или пролекарственные соединения настоящего изобретения. Соли могут быть получены в ходе конечного выделения и очистки соединений или отдельно, путем взаимодействие соответствующего атома азота с подходящей кислотой. Кислоты, которые обычно используют с целью получения фармацевтически приемлемых солей, включают неорганические кислоты, такие как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, фосфорная кислота, сероводородная кислота, а также органические кислоты, такие как пара-толуолсульфоновая кислота, салициловая кислота, винная кислота, кислая винная кислота, аскорбиновая кислота, малеиновая кислота, бензолсульфоновая кислота, фумаровая кислота, глюконовая кислота, глюкуроновая кислота, муравьиная кислота, глутаминовая кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, молочная кислота, щавелевая кислота, пара-бромфенилсульфоновая кислота, угольная кислота, янтарная кислота, лимонная кислота, бензойная кислота, уксусная кислота, и родственные неорганические и органические кислоты.
Основные аддитивные соли могут быть получены в ходе окончательного выделения и очистки соединений, путем взаимодействия карбоксильной группы с подходящим основанием, таким как гидроксид, карбонат, или бикарбонат катиона металла, или с аммиаком, или органическим первичным, вторичным или третичным амином. Катионы фармацевтически приемлемых солей включают (но не ограничиваются приведенным) литий, натрий, калий, кальций, магний, и алюминий, а также нетоксичные катионы четветичного аммония, такие как аммоний, тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, триэтиламин, диэтиламин, этиламин, трибутиламин, пиридин, N,N-диметиланилин, N-метилпиперидин, и N-метилморфолин.
Термин “сольват”, который используется в этом документе, означает физический ассоциат соединения настоящего изобретения с одной или несколькими, предпочтительно от одной до трех, молекулами растворителя, или органического, или неорганического. Этот физический ассоциат включает водородную связь. В определенных случаях сольват может быть выделен, например, когда одна или несколько, предпочтительно от одной до трех, молекул растворителя включаются в кристаллическую решётку кристаллического твёрдого вещества. Характерные сольваты включают (но не ограничиваются приведенным) гидраты, этанолаты, метанолаты и изопропанолаты. Способы сольватации обычно известны из уровня техники.
Термин “терапевтически эффективное количество”, который используется в этом документе, относится к общему количеству каждого активного компонента, которое достаточно для демонстрации показательной полезности для пациента, например, продолжительное уменьшение вирусной нагрузки. Применительно к индивидуальному активному компоненту, введенному отдельно, термин относится только к этому компоненту. Применительно к комбинации, термин относится к объединённому количеству активных компонентов, которые приводят к терапевтическому эффекту, независимо от введения в комбинации, последовательно или одновременно.
Термин “фармацевтически приемлемый”, который используется в этом документе, относится к тем соединениям, материалам, композициям и/или формам дозировки, которые находятся в объеме действующей медицинской оценки, подходящих для применения в контакте с тканями пациентов без чрезмерной токсичности, раздражения, аллергической реакции, или других проблем или соответствующих осложнений с обоснованным отношением полезность/риск, и являются эффективными для использования по назначению.
Термин “пациент” включает людей, а также других млекопитающих.
Термин “лечение” относится к: (i) предупреждению заболевания, расстройства или состояния, проявляющимся у пациента, который может быть предрасположен к заболеванию, расстройству, и/или состоянию, но которое еще не диагностировано у него; (ii) подавление заболевания, расстройства, или состояния, то есть прекращение их развития; и (iii) облегчение заболевания, расстройства или состояния, то есть вызывание регрессии заболевания, расстройства и/или состояния.
В одном варианте осуществления, соединения имеют формулу (I), или ее фармацевтически приемлемые соли, сольваты, гидраты, метаболиты, или пролекарства:
где:
A выбирают из группы, состоящей из CR1 и N; и в котором R1 выбирают из группы, состоящей из водорода, галогена, и незамещенного или замещенного алкила;
Ra, Rb, Rc и Rd независимо выбирают из группы, состоящей из водорода, галогена, гидроксила, нитро, циано, незамещенного или замещенного алкила, и незамещенного или замещенного алкоксила;
B выбирают из группы, состоящей из водорода, незамещенного или замещенного алкила, незамещенного или замещенного циклоалкила, незамещенного или замещенного гетероциклоалкила, незамещенного или замещенного арила, и незамещенного или замещенного гетероарила;
L представляет собой незамещенный или замещенный алкил, или L отсутствует; и
Y выбирают из группы, состоящей из незамещенного или замещенного циклоалкила, незамещенного или замещенного гетероциклоалкила, незамещенного или замещенного арила, и незамещенного или замещенного гетероарила.
В некоторых вариантах осуществления, A выбирают из группы, состоящей из CH, CF, CCl и N.
В некоторых вариантах осуществления, по меньшей мере, один из Ra, Rb, Rc и Rd выбирают из группы, состоящей из водорода, F, Cl, и метоксила. В некоторых вариантах осуществления, Ra, Rb, Rc и Rd представляют собой водород. В некоторых вариантах осуществления, Ra означает F, Cl, или метоксил. В некоторых вариантах осуществления, Rd представляет собой F, Cl, или метоксил.
В некоторых вариантах осуществления, B является незамещенным или замещенным C1-C6 алкилом. В некоторых вариантах осуществления, B является незамещенным или замещенным арилом. В некоторых вариантах осуществления, B представляет собой незамещенный или замещенный фенил. В некоторых вариантах осуществления, B означает фенил. В некоторых вариантах осуществления, B представляет собой фенил, замещенный, по меньшей мере, одним фрагментом, который выбирают из группы, состоящей из галогена, циано, нитро, гидроксила, незамещенного или замещенного алкила, незамещенной или замещенной алкокси-, -NR1R2, -C(O)R3, -C(O)OR4, -C(O)NHR5 и -S(O)2R6 групп. Радикалы R1 и R2 независимо выбирают из группы, состоящей из водорода, незамещенного или замещенного алкила, -C(O)R7, -C(O)OR8,
-C(O)NHR9, -S(O)2R10; и в котором R3, R4, R5, R6, R7, R8, R9, и R10 независимо выбирают из группы, состоящей из незамещенного или замещенного алкила, незамещенного или замещенного циклоалкила, незамещенного или замещенного гетероциклоалкила, незамещенного или замещенного арила, и незамещенного или замещенного гетероарила.
В некоторых вариантах осуществления, B означает фенил, замещенный, по меньшей мере, одним фрагментом, который выбирают из группы, состоящей из F, Cl, и метокси-группы. В некоторых вариантах осуществления, B означает фенил, замещенный двумя атомами F. В некоторых вариантах осуществления, B представляет собой фенил, замещенный двумя атомами Cl. В некоторых вариантах осуществления, B представляет собой фенил, замещенный одним Cl и одной метокси-группой.
В некоторых вариантах осуществления, L отсутствует. В некоторых вариантах осуществления, L является метиленовой группой.
В некоторых вариантах осуществления, Y выбирают из группы, состоящей из незамещенного или замещенного пиперидинила, незамещенного или замещенного фенила, незамещенного или замещенного бицикло[3.2.1]октанила, незамещенного или замещенного азетидинила, и незамещенного или замещенного пирролидинила. В некоторых вариантах осуществления, Y замещен, по меньшей мере, одним фрагментом, который выбирают из группы, состоящей из галогена, -CN, -C(O)R11, -NHC(O)R12,
-S(O)2R13 и -NHS(O)2R14; и в котором R11, R12, R13, и R14 независимо выбирают из группы, состоящей из незамещенного или замещенного алкила, незамещенного или замещенного циклоалкила, незамещенного или замещенного алкенила, незамещенного или замещенного циклоалкенила, и незамещенного или замещенного алкинила.
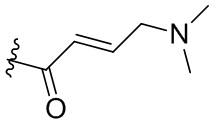
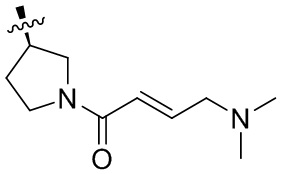
В некоторых вариантах осуществления, Y замещен, по меньшей мере, одним фрагментом, который выбирают из группы, состоящей из F, CN,-C(O)CH=CH2,
-C(O)CH=CHCH2N(CH3)2, -NHC(O)CH=CH2, -NHC(O)CH=CHCH2N(CH3)2,
-C(O)CH=CHCH2N(CH3)(COOC(CH3)3; -C(O)CH=CHCH2NH(CH3), -C(O)CH2CH3,
-C(O)CH2CH2CH3, 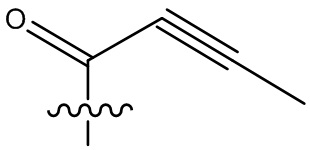 ,
,  ,
,  , -C(O)CH2CN,
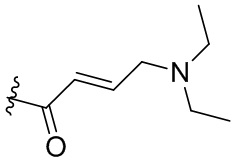
, -C(O)CH2CN, 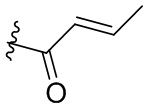 ,
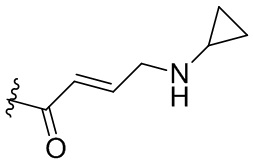
,  ,
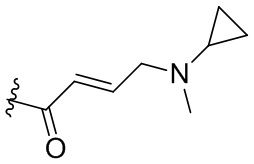
,  ,
,  ,
, 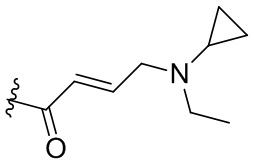 ,
, 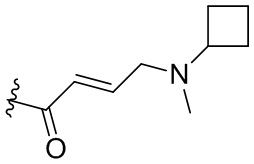 ,
, 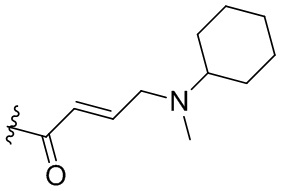 ,
, 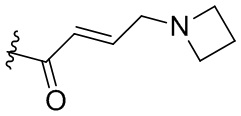 ,
, 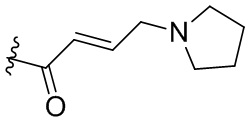 ,
, 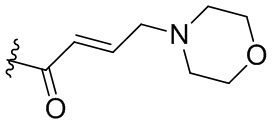 ,
, 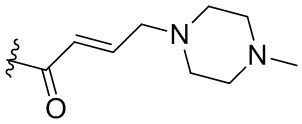 ,
, 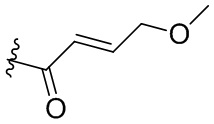 ,
, 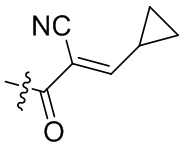 ,
, 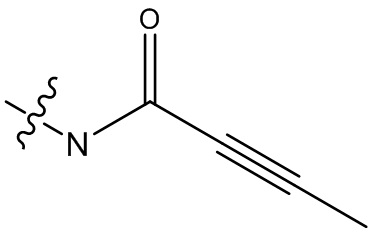 , и
, и 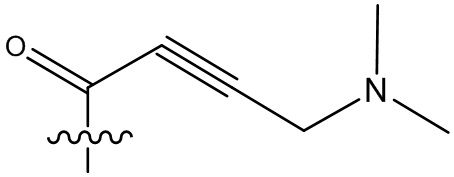 .
.
В некоторых вариантах осуществления соединение имеет S-форму, а также R-форму. В некоторых вариантах осуществления соединение находится больше в R-форме, чем в S-форме. В некоторых вариантах осуществления соединение находится больше в S-форме, чем в R-форме.
В некоторых вариантах осуществления соединение имеет структуру формулы (II):
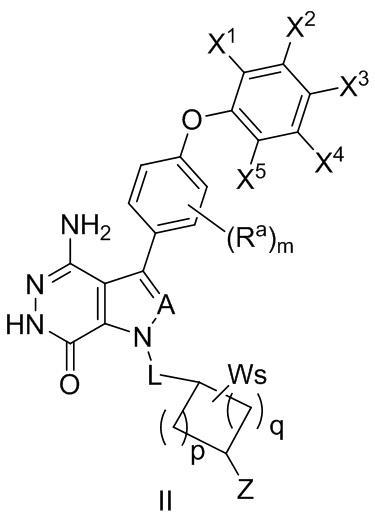 ,
,
где Ra, A, и L определены так же, как в формуле (I);
X1, X2, X3, X4, и X5 независимо выбирают из группы, состоящей из водорода, галогена, циано, нитро, гидроксила, незамещенного или замещенного алкила, незамещенной или замещенной алкокси-, -NR1R2, -C(O)R3, -C(O)OR4, -C(O)NHR5, и
-S(O)2R6 группы;
где R1 и R2 независимо выбирают из группы, состоящей из водорода, незамещенного или замещенного алкила, -C(O)R7, -C(O)OR8, -C(O)NHR9, -S(O)2R10; и в котором R3, R4, R5, R6, R7, R8, R9, и R10 независимо выбирают из группы, состоящей из незамещенного или замещенного алкила, незамещенного или замещенного циклоалкила, незамещенного или замещенного гетероциклоалкила, незамещенного или замещенного арила, и незамещенного или замещенного гетероарила;
W выбирают из группы, состоящей из галогена, гидроксила, незамещенного или замещенного алкила, и незамещенного или замещенного алкоксила; в котором два фрагмента W могут комбинироваться с атомом или атомами, к которому они присоединены, с образованием незамещенного или замещенного C3-12 циклоалкила, незамещенного или замещенного 3- - 12-членного гетероцикла, незамещенного или замещенного C6-12 арила, или незамещенного, или замещенного 5- - 12-членного гетероарила;
m = 0, 1, 2, или 3;
p = 1, 2, или 3;
q = 0, 1, или 2;
s = 0, 1, 2, или 3; и
Z выбирают из группы, состоящей из -NHC(O)R12, и -NHS(O)2R14; и где R12, и R14 независимо выбирают из группы, состоящей из незамещенного или замещенного алкила, незамещенного или замещенного циклоалкила, незамещенного или замещенного алкенила, незамещенного или замещенного циклоалкенила, и незамещенного или замещенного алкинила.
В некоторых вариантах осуществления A представляет собой N, CH, CF или CCl. В некоторых вариантах осуществления L отсутствует или означает группу -CH2-. В некоторых вариантах осуществления p и q независимо равны 1 или 2. В некоторых вариантах осуществления s равно 1 или 2.
В некоторых вариантах осуществления Z выбирают из группы, состоящей из
-NHC(O)CH=CH2, -NHC(O)CH=CHCH2N(CH3)2 и .
В некоторых вариантах осуществления соединение имеет структуру формулы (III):
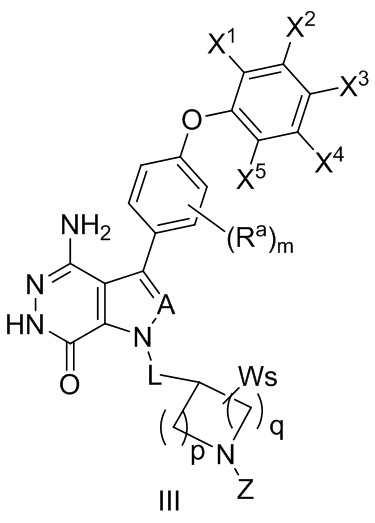 ,
,
где значения Ra, A, L, X1, X2, X3, X4, X5, W, p, q, s и m определены выше.
Z выбирают из -CN, -C(O)R11 и -S(O)2R13; и где R11 и R13 независимо выбирают из группы, состоящей из незамещенного или замещенного алкила, незамещенного или замещенного циклоалкила, незамещенного или замещенного алкенила, незамещенного или замещенного циклоалкенила, и незамещенного или замещенного алкинила.
В некоторых вариантах осуществления, Z выбирают из группы, состоящей из CN,-C(O)CH=CH2, -C(O)CH=CHCH2N(CH3)2, -C(O)CH=CHCH2N(CH3)(COOC(CH3)3;
-C(O)CH=CHCH2NH(CH3), -C(O)CH2CH3, -C(O)CH2CH2CH3, , , , -C(O)CH2CN, , , , , , , , , , , , , , и .
В некоторых вариантах осуществления соединение имеет формулу (IV):
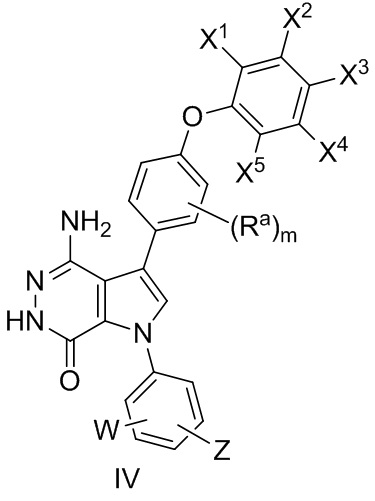
где значения Ra, X1, X2, X3, X4, X5, W, Z, и m определены выше.
В некоторых вариантах осуществления Z выбирают из группы, состоящей из
-NHC(O)R12 и -NHS(O)2R14.
В некоторых вариантах осуществления соединение имеет формулу (V):
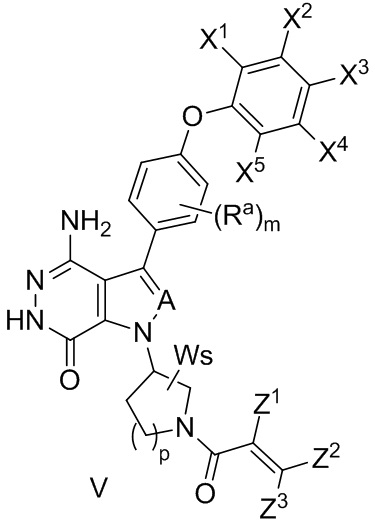 ,
,
где значения Ra, A, X1, X2, X3, X4, X5, W, s, и m определены выше;
Z1 выбирают из группы, состоящей из водорода, галогена, циано, и незамещенного или замещенного алкила; и
Z2 и Z3 независимо выбирают из группы, состоящей из водорода, незамещенного или замещенного алкила, незамещенного или замещенного циклоалкила, незамещенного или замещенного гетероциклоалкила, -CH2OR15, и -CH2NR16R17;
R15 и R16 независимо выбирают из группы, состоящей из водорода, незамещенного или замещенного алкила, незамещенного или замещенного циклоалкила, и незамещенного или замещенного гетероциклоалкила; R17 выбирают из группы, состоящей из незамещенного или замещенного алкила, незамещенного или замещенного циклоалкила, незамещенного или замещенного арила, и незамещенного или замещенного гетероарила,
-C(O)R18, -C(O)OR19, и -S(O)2R20; в котором R18, R19 и R20 независимо выбирают из группы, состоящей из незамещенного или замещенного алкила, и незамещенного или замещенного циклоалкила;
R16 и R17 комбинируются с атомом N, к которому они присоединены, с образованием незамещенного или замещенного 3- - 12-членного гетероцикла, или незамещенного или замещенного 5- - 12-членного гетероарила; и
Z1 и Z2 могут объединяться вместе, с образованием связи, или они комбинируются с атомами, к которым они присоединены, с образованием незамещенного или замещенного C5-12 циклоалкенила, незамещенного или замещенного 5- - 12-членного гетероцикла, незамещенного или замещенного C6-12 арила, или незамещенного или замещенного 5- - 12-членного гетероарила.
В некоторых вариантах осуществления A выбирают из группы, состоящей из CH, CF и CCl.
В некоторых вариантах осуществления Z1, Z2 и Z3 означают H. В некоторых вариантах осуществления Z1 и Z2 объединяются вместе с образованием связи. В некоторых вариантах осуществления Z1 и Z3 означают водород, Z2 представляет собой
-CH2NR16R17.
В некоторых вариантах осуществления 3, или меньше трех из X1, X2, X3, X4 и X5, представляют собой галоген. В некоторых вариантах осуществления X1 означает F. В некоторых вариантах осуществления X2, X3, и X4 означают водород. В некоторых вариантах осуществления X5 выбирают из группы, состоящей из H, F и Cl.
Хотя все приведенные выше структурные формулы изображены для удобства как определенные изомеры, настоящее изобретение может включать все изомеры, такие как, таутомеры, ротамеры, геометрические изомеры, диастереомеры, рацематы, и энантиомеры.
Таутомеры представляют собой структурные изомеры органических соединений, которые легко взаимопревращаются в результате химической реакции, названной таутомеризация. Эта реакция обычно приводить к формальной миграции атома водорода или протона, которая сопровождается переносом простой связи и соседней двойной связи. Примерами обычных таутомерных являются: кетон - енол, лактам - лактим. Примером равновесия лактам - лактим между A и B показан ниже.
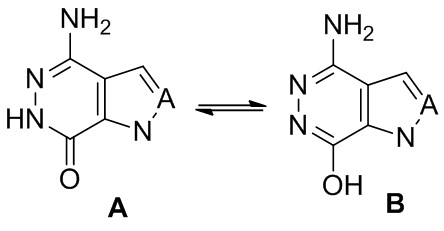
Все соединения настоящего изобретения могут быть изображены или в форме A или форме B. Все таутомерные формы входят в объем настоящего изобретения. Наименование соединений не исключает любые таутомеры.
Фармацевтические композиции или рецептуры настоящего изобретения включают такие, которые пригодны для орального, назального, местного (в том числе трансбуккального и подъязычного), ректального, вагинального и/или парэнтерального введения. Независимо от выбранного способа назначения, активный компонент (компоненты) включается в фармацевтически приемлемые формы дозировки методами, которые известны специалистам в этой области техники.
Количество активного компонента (компонентов), которые могут быть объединены с материалом носителя, с целью получения единичной формы дозировки, может изменяться в зависимости от реципиента, подлежащего лечению, конкретного способа назначения и всех других факторов, описанных выше. Количество активного компонента (компонентов), которое может объединяться с материалом носителя для получения единичной формы дозировки, обычно будет таким количеством активного компонента (компонентов), которое является наименьшей дозой, эффективной для получения терапевтического действия.
Способы получения фармацевтических рецептур или композиций включают стадию введения активного компонента (компонентов) в ассоциацию с носителем и, необязательно, с одним или несколькими дополнительными ингредиентами. Обычно рецептуры получают путем однородного смешивания активного компонента (компонентов) в жидких носителях, или тонко диспергированных твёрдых носителях, или обоими методами, и затем, в случае необходимости, продукт формуют.
Характерные, не ограничивающие примеры рецептур изобретения, пригодных для орального введения, могут находиться в форме капсул, облаток, пилюль, таблеток, лепешек (с использованием вкусовой основы, обычно сахарозы и акации или трагаканта), порошков, гранул, или в виде раствора или суспензии в водной или неводной жидкости, или в виде жидкой эмульсии типа «масло в воде» или «вода в масле», или как эликсир или сироп, или как пастилки (с использованием инертной основы, такой как желатин и глицерин, или сахароза и акация) и/или в виде раствора для полоскания полости рта и тому подобного, где каждый содержит заданное количество активного компонента (компонентов).
В твёрдых формах дозировки изобретения для орального введения (капсулы, таблетки, пилюли, драже, порошки, гранулы и тому подобное), пролекарство (пролекарства), активный компонент (компоненты) (в виде микронных частиц) смешивают с одним или несколькими фармацевтически приемлемыми носителями, известными специалистам в этой области техники. Примеры пригодных водных и неводных носителей, которые могут быть использованы в фармацевтических композициях изобретения, включают воду, этанол, полиолы (такие как глицерин, пропиленгликоль, полиэтиленгликоль, и тому подобное), и их подходящие смеси. Соответствующая текучесть может поддерживаться, например, с использованием покрывающих материалов, таких как лецитин, путем поддержания необходимого размера частиц, и использования поверхностно-активных веществ.
Кроме того, эти композиции могут содержать вспомогательные средства, такие как смачивающие вещества, эмульгаторы и диспергирующие вещества. Также может быть желательным включение в композиции изотонических реагентов, таких как сахара, хлорид натрия и тому подобное. Кроме того, длительная абсорбция впрыскиваемой фармацевтической формы может быть осуществлена путем включения веществ, которые замедляют абсорбцию, таких как моностеарат алюминия и желатин.
В некоторых случаях, с целью продления действия активного компонента (компонентов), желательно замедлить абсорбцию лекарственного препарата из подкожной или внутримышечной инъекции. Это может быть осуществлено с использованием жидкой суспензии кристаллического или аморфного материала, обладающего низкой растворимостью в воде. Тогда скорость абсорбции активного компонента (компонентов) зависит от скорости его растворения, которая, в свою очередь, может зависеть от размера кристаллов и кристаллической формы.
Рецептуры могут быть представлены в герметичных кассетах единичной дозы или множественной дозы, например, в ампулах и бутылочках, и может храниться в лиофилизированном состоянии, требующем только добавления стерильного жидкого носителя, например воды для инъекции, непосредственно перед применением. Непредусмотренные инъекции растворов и суспензий могут быть приготовлены из стерильных порошков, гранул и таблеток описанного выше типа.
Настоящее изобретение включает способ модулирования активности протеиновой тирозинкиназы, который включает в себя контактирование клетки с эффективным количеством любого соединения формул (I)-(V) или их фармацевтически приемлемых солей.
Настоящее изобретение включает способ лечения состояния или заболевания, передаваемого с помощью белковой тирозинкиназы, который включает введение субъекту терапевтически эффективного количества любого соединения формул (I)-(V), или их фармацевтически приемлемых солей. В некоторых вариантах осуществления состояние или заболевание представляет собой рак или аутоиммунные заболевания. В некоторых вариантах осуществления, рак представляет собой злокачественное развитие В-клеток. В некоторых вариантах осуществления, рак означает хроническую лимфоцитарную лейкемию (CLL), мантийноклеточную лимфому (MCL), диффузную лимфому больших B-клеток (DLBCL), множественную миелому (MM), макрофолликулярную лимфому (FL), лимфому пограничной зоны и макроглобулинемию Вальденстрёма (WM). В некоторых вариантах осуществления, аутоиммунным заболеванием является ревматоидный артрит. В некоторых вариантах осуществления, аутоиммунным заболеванием является системная красная волчанка.
Способы синтеза
Сокращения
Ниже приведены сокращения, которые могут быть использованы при описании схем и в примерах:
Cy означает циклогексан
DAST - вместо трифторида диэтиламиносеры;
DCM - вместо дихлорметана;
DIEA или DIPEA - вместо диизопропилэтиламина;
DMAP - вместо N,N-диметиламинопиридина;
DME - вместо диметилового эфира этиленгликоля;
DMF - вместо N,N-диметилформамида;
ДМСО - вместо диметилсульфоксида;
DPPA - вместо дифеноксифосфорилазида;
EDCI или EDC - вместо гидрохлорида 1-(3-диэтиламинопропил)-3-этилкарбодиимида;
ESI - вместо ионизации электрораспылением;
Et - вместо этил;
EtOAc - вместо этилацетат;
г - вместо грамма;
ч - вместо часа;
HATU - вместо O-(7-Азабензoтриазол-1-ил)-N,N,N’,N’-тетраметилуроний гексафторфосфат;
HBTU - вместо гексафторфосфата O-бензoтриазол-N,N,N’,N’-тетраметилурония;
HPLC - вместо жидкостной хроматографии высокого разрешения;
mCPBA - вместо 3-Хлорпербензойной кислоты;
Me - вместо метил;
MeOH - вместо метанол;
мг - вместо миллиграмм;
мин - вместо минут;
МС - вместо масс-спектрометрии;
NBS - вместо N-Бромсукцинимида;
NCS - вместо N-Хлорсукцинимида;
ЯМР - вместо ядерный магнитный резонанс;
Pd(dppf)Cl2 - вместо [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладий(II);
Pd2(dba)3 - вместо трис(дибензилиденацетон)дипалладий(0);
PG - вместо защищающей группы;
Ph - вместо фенила;
PPh3 - вместо трифенилфосфина;
rt - вместо комнатной температуры;
TEA - вместо триэтиламина;
TFA - вместо трифторуксусной кислоты;
THF - вместо тетрагидрофурана;
TLC - вместо тонкослойная хроматография; и
tBOC или Boc - вместо трет-бутилоксикарбонила.
Соединения и способы настоящего изобретения можно лучше понять в связи со следующими схемами синтеза, которые иллюстрируют способы, с помощью которых можно получить соединения изобретения. Другие реакционные схемы могут быть легко разработаны специалистами в этой области техники.
Схема 1
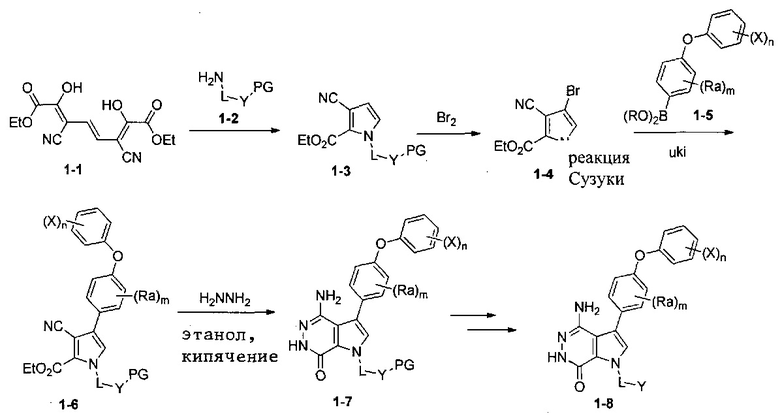
Триен 1-1 и амин 1-2 перемешивают в органических растворителях (например, этилацетат) при повышенной температуре, чтобы получить пиррол 1-3. Бромирование бромом или другими подходящими реагентами дает бромид 1-4, который взаимодействует с легкодоступной бороновой кислотой или эфиром бороновой кислоты 1-5 в условиях реакции Сузуки, чтобы получить 1-6. При кипячении эфира 1-6 и гидразина в этаноле получают ключевое промежуточное соединение 1-7. После удаления защитной группы и ввода группы Z получают 1-8.
Схема 2
ацетат меди, пиридин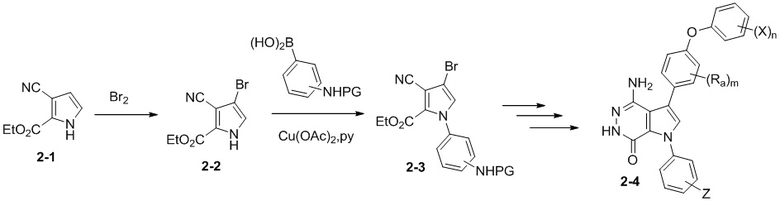
В качестве альтернативы, получен пиррол 2-1 без заместителя при атоме N, который при последующем бромировании дает бромид 2-2. Ароматическое кольцо вводится по реакциям C–N перекрёстного сочетания, которые катализирует медь. Образовавшееся промежуточное соединение 2-3 превращается в конечное соединение
2-4 путем аналогичных последовательностей, показанным в схеме 1.
Cхема 3
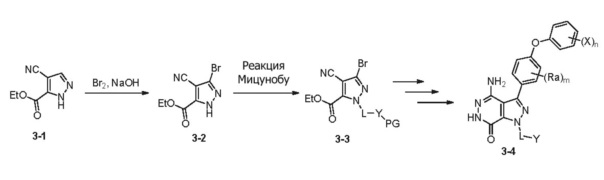
Пиразол 3-1 взаимодействует с Br2 в присутствии щелочи и дает бромид 3-2. Группа Y вводится по реакциям Mitsunobu. Соединение 3-3 превращается в 3-4, следуя аналогичным последовательностям, показанным в схеме 1.
ПРИМЕРЫ
Соединения и способы настоящего изобретения могут быть лучше поняты в связи со следующими примерами, которые предназначены в только качестве иллюстрации, а не для ограничения объема изобретения. Различные изменения и модификации раскрытых вариантов осуществления будут очевидны для специалистов в этой области техники и такие изменения и модификации, включая без ограничения химические структуры, заместители, производные, рецептуры и/или способы изобретения, могут быть выполнены без отклонения от сущности изобретения и объема приложенной Формулы изобретения.
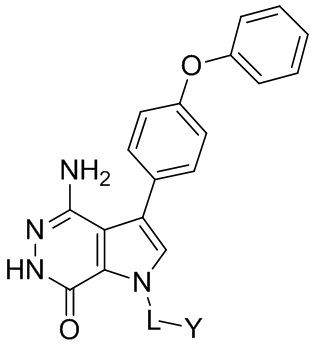
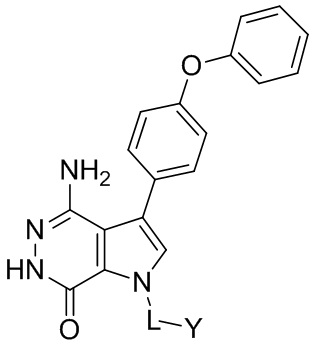
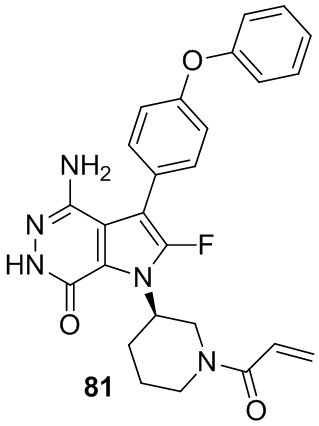
Пример 1. (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-3-(4-феноксифенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
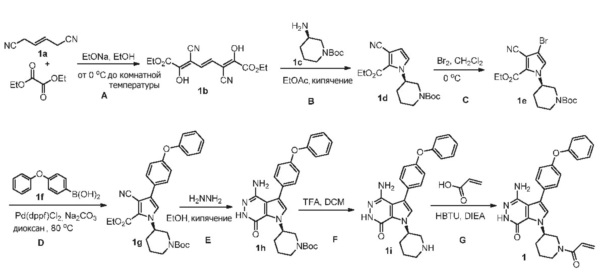
Стадия 1A
К раствору этилата натрия (160 мл, 21%-ный раствор в этаноле, 0.49 ммоль) в этаноле (EtOH, 110 мл) в ледяной ванне добавляют диэтилоксалат (64 мл, 0.47 моль). Смесь перемешивают в течение 30 мин. Добавляют раствор 1a (16 г, 0.15 ммоль) в EtOH (30 мл). Полученную смесь перемешивают в течение ночи при комнатной температуре. После охлаждения в ледяной ванне суспензию фильтруют. Твёрдое вещество промывают небольшим количеством EtOH и затем растворяют в воде (380 мл). Раствор подкисляют HCl до pH ~4. Образовавшееся большое количество твёрдого вещества фильтруют, промывают водой и сушат, получая 1b (11.9 г) в виде желтого твёрдого вещества.
Стадия 1B
К раствору 1b (2.3 г, 7.5 ммоль) в этилацетате (EtOAc, 120 мл) при 60°C по каплям добавляют раствор 1c (2.3 г, 11.4 ммоль) в EtOAc (32 мл). Смесь кипятят в течение 4 ч. После охлаждения до комнатной температуры, растворители удаляются. Остаток очищается методом хроматографии на силикагеле, чтобы получить светло-желтое масло 1d (1.09 г).
Стадия1C
К раствору 1d (1.09 г) в дихлорметане (DCM, 200 мл) медленно добавляют раствор Br2 (6.15 г) в DCM (7 мл) в течение 30 мин. Смесь перемешивают в течение 30 мин, и затем реакцию прерывают, добавляя раствор 10% Na2S2O3 и насыщенный раствор NaHCO3. Две фазы разделяют; водную фазу экстрагируют дихлорметаном. Объединённые органические экстракты обрабатывают избытком ангидрида бутилоксикарбоновой кислоты (Boc2O), сушат над Na2SO4, фильтруют и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить 1e (0.8 г) и 1d (0.3 г).
Стадия 1D
Смесь 1e (0.8 г), 1f (1.2 г), Na2CO3 (2 M, 5 мл), и [1,1′-бис(дифенилфосфино)-ферроцен]дихлорпалладий(II) (Pd(dppf)Cl2, 0.3 г) в 1.4-диоксане (50 мл) перемешивают в атмосфере N2 при 80°C в течение 20 ч. После охлаждения до комнатной температуры, растворители удаляются. Остаток очищается методом хроматографии на силикагеле, чтобы получить бесцветное масло 1g (0.8 г). МС (ионизация электрораспылением - ESI): m/z=516 [M+H]+.
Стадия 1E
Смесь 1g (0.8 г) и N2H4 (8 мл) в EtOH (80 мл) кипятят в течение 28 ч. После охлаждения до комнатной температуры растворители удаляются. Остаток очищается методом хроматографии на силикагеле, чтобы получить бесцветное масло 1h (0.327 г). МС (ESI): m/z=502 [M+H]+.
Стадия 1F
К раствору 1 h (0.327 г) в DCM (15 мл) добавляют TFA (1.5 мл). Смесь перемешивают при комнатной температуре в течение 30 мин и концентрируют, чтобы получить соединение 1i, которое используется непосредственно на следующей стадии.
Стадия 1G
К раствору 1i (10.6 мг, 0.026 ммоль) в DCM (2 мл) добавляют триэтиламин (0.1 мл), акриловую кислоту (5 мг, 0.067 ммоль) и HBTU (19 мг, 0.05 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 0.5 ч и очищают методом препаративной ЖХВР на обращенной фазе, чтобы получить соединение 1, указанное в заголовке (3.5 мг) в виде белого твёрдого вещества. МС (ESI): m/z = 456 [M+H]+.
В примерах 2 - 12 (Таблица 1) соединения получают из 1b и соответствующих аминов (промышленно доступных) в условиях, аналогичных описанным для стадий 1B~1G Примера 1.
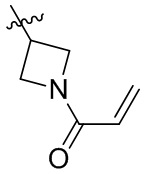
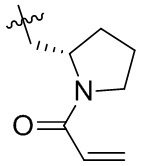
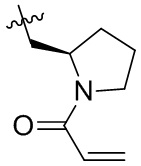
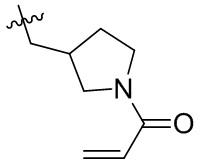
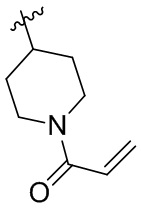
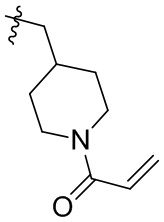
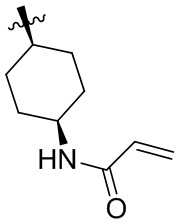
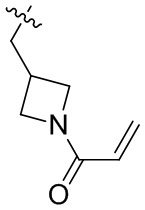
Таблица 1. Соединения формулы:
m/z [M+H]+
Пример 13. 1-(1-акрилоил-4-фторпиперидин-3-ил)-4-амино-3-(4-феноксифенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
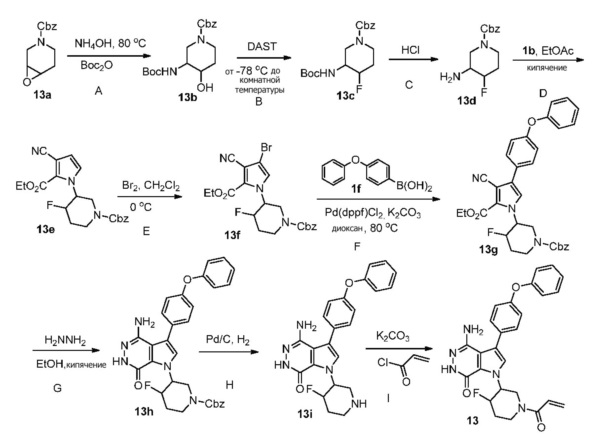
Стадия 13A
Смесь 13a (2.2 г) и NH4OH (14 мл) в EtOH (33 мл) перемешивают в герметизированной трубке при 80°C в течение 18 ч. Растворители удаляются; остаток растворяют в ТГФ (30 мл) и EtOH (30 мл) и добавляют Boc2O (2.46 г). Смесь перемешивают при комнатной температуре в течение 20 ч. Сырой продукт очищают методом хроматографии на силикагеле, чтобы получить белое твёрдое вещество 13b (1.02 г) и нежелательный региональный изомер (1.7 г).
Стадия 13B
Трифторид диэтиламиносеры (DAST, 0.28 мл, 2.14 ммоль) по каплям добавляют к раствору 13b (0.68 г, 1.94 ммоль) в DCM (20 мл) при -78°C. Смеси дают нагреться до комнатной температуры в течение ночи. Реакцию прерывают, добавляя насыщенный раствор NaHCO3, и экстрагируют дихлорметаном. Органические экстракты сушат над Na2SO4, фильтруют и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить бесцветное масло 13c (0.31 г).
Стадия 13C
Смесь 13c (0.31 г) и HCl (4 M в диоксане) перемешивают в течение 2 ч. Растворители удаляются, остаток суспендируют в EtOAc, добавляют 2M раствор K2CO3, чтобы установить значение pH выше 9. Водный слой экстрагируют 3 раза этилацетатом. Объединённые органические экстракты сушат над Na2SO4, фильтруют и концентрируют, чтобы получить светло-желтое масло 13d (0.18 г).
Стадия 13D
К раствору 13d (140 мг) в этилацетате (12 мл) при 60°C добавляют по каплям раствор 1b (140 мг) в этилацетате (3 мл). Смесь кипятят в течение 18 ч. После охлаждения до комнатной температуры растворители удаляются. Остаток очищается методом хроматографии на силикагеле, чтобы получить желтое масло 13e (40 мг).
Стадия 13E
К раствору 13e (40 мг) и AcOH (40 мкл) в DCM (3 мл) при 0°C медленно добавляют Br2 (40 мг). Смеси дают нагреться до комнатной температуры и перемешивают в течение 5 ч, и затем реакцию прерывают, добавляя 10%-ный раствор Na2S2O3 и насыщенный раствор NaHCO3. Две фазы разделяют; водную фазу экстрагируют этилацетатом. Объединённые органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить 13f (18 мг).
Стадия 13F
Смесь 13f (22 мг), 1f (25 мг), K2CO3 (2 M, 0.1 мл), и 13 мг [1,1′-бис(дифенил-фосфино)ферроцен]дихлорпалладия (II) [Pd(dppf)Cl2] в 1.4-диоксане (1 мл) перемешивают в атмосфере N2 при 80°C в течение 20 ч. После охлаждения до комнатной температуры растворители удаляются. Остаток очищается методом хроматографии на силикагеле, чтобы получить бесцветное масло 13g (28 мг). МС (ESI): m/z=568 [M+H]+.
Стадия 13G
Смесь 13g (28 мг) и N2H4 (0.2 мл) в этаноле (2 мл) кипятят в течение двух суток. После охлаждения до комнатной температуры растворители удаляются. Остаток очищается методом хроматографии на силикагеле, чтобы получить 13h (15мг). МС (ESI): m/z=554 [M+H]+.
Стадия 13H
Смесь 13h (15 мг) и Pd/C (10 масс.%, 9 мг) метаноле (1 мл) перемешивают в атмосфере H2 (из баллона) в течение 3 ч. Реакционную смесь фильтруют через слой целита, промывают смесью этилацетат/метанол и концентрируют, чтобы получить белое твёрдое вещество 13i (10 мг).
Стадия 13I
К смеси 13i (5.3 мг) и K2CO3 (2M, 30 мкл) в ТГФ (0.8 мл) при 0°C добавляют раствор акрилоилхлорида (1.4 мг) в ТГФ. Полученную смесь перемешивают при 0°C в течение 0.5 ч, и очищают методом препаративной ЖХВР на обращенной фазе, чтобы получить указанное в заголовке соединение 13 (3 мг) в виде беловатого твёрдого вещества. МС (ESI): m/z = 474 [M+H] +.
Пример 14.(R)-1-(1-акрилоил-5,5-дифторпиперидин-3-ил)-4-амино-3-(4-фенокси-фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
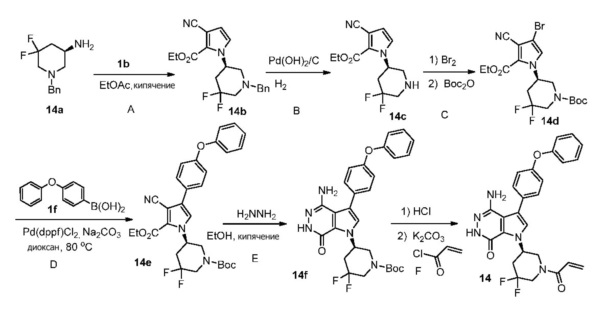
Стадия 14A
К раствору 1b (271 мг) в этилацетате (12 мл) при 60°C по каплям добавляют раствор 14a (223 мг, соединение получено, следуя методикам, описанным в Organic Letters, 2011, том 13, с. 4442-4445 Anne Cochi и др.) в этилацетате (2 мл). Смесь кипятят в течение 18 ч. После охлаждения до комнатной температуры растворители удаляются. Остаток очищается методом хроматографии на силикагеле, чтобы получить светло-желтое масло 14b (76 мг).
Стадия 14B
Смесь 14b (65 мг) и Pd(OH)2/C (10 масс.%, 50 мг) в Метаноле/ТГФ (3/1 мл) перемешивают в атмосфере H2 (из баллона) в течение 20 ч. Реакционную смесь фильтруют через слой целита, промывают смесью этилацетат/метанол и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить белое твёрдое вещество 14c (38 мг).
Стадия 14C
К раствору 14c (38 мг) в DCM (3 мл) при 0°C медленно добавляют Br2 (15 мкл). Смесь перемешивают при комнатной температуре 18 ч. Добавляют избыток TEA и Boc2O. Полученную смесь перемешивают при комнатной температуре в течение 24 ч. Добавляют воду, и смесь экстрагируют этилацетатом. Органические экстракты промывают соленой водой, сушат над Na2SO4, фильтруют и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить 14d (20 мг).
Стадия 14D
Смесь 14d (20 мг), 1f (19 мг), K2CO3 (2M, 0.1 мл), и Pd(dppf)Cl2 (7 мг) в
1.4-диоксане (1.5 мл) перемешивают в атмосфере N2 при 80°C в течение 3 ч. После охлаждения до комнатной температуры, растворители удаляются. Остаток очищается методом хроматографии на силикагеле, чтобы получить бесцветное масло 14e (30 мг). МС (ESI): m/z=552 [M+H]+.
Стадия 14E
Смесь 14e (30 мг) и N2H4 (0.25 мл) в этаноле (2.5 мл) кипятят в течение 20 ч. После охлаждения до комнатной температуры, растворители удаляются. Остаток очищается методом хроматографии на силикагеле, чтобы получить белое твёрдое вещество 14f (15 мг). МС (ESI): m/z=538 [M+H]+.
Стадия 14F
Смесь 14f (15 мг) и раствор HCl (1 мл, 4M в диоксане) перемешивают при комнатной температуре 2 ч и концентрируют, чтобы получить соединение 14g (22 мг), которое используется непосредственно на следующей стадии без дополнительной очистки. К смеси 14g (13 мг) и раствора K2CO3 (2 M, 40 мкл) в ТГФ (1 мл) при 0°C добавляют раствор акрилоилхлорида (4 мг) в ТГФ. Полученную смесь перемешивают при 0°C в течение 20 мин и очищают методом препаративной ЖХВР на обращенной фазе, чтобы получить указанное в заголовке соединение 14 (3.3 мг) в виде белого твёрдого вещества. МС (ESI): m/z = 492 [M+H] +.
Промежуточное соединение 1. трет-Бутил(3-аминобицикло[3.2.1]октан-8-ил)карбамат
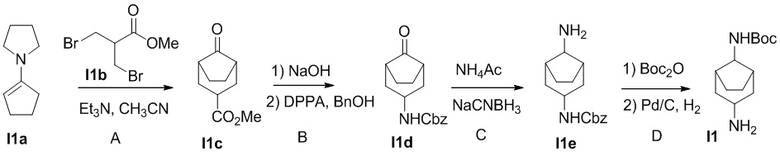
Стадия I1A
Метиловый эфир 3-бром-2-(бромметил)пропановой кислоты I1b (2.61 г) по каплям добавляют к раствору 1-(циклопент-1-ен-1-ил)пирролидина I1a (1.44 г) и триэтиламина (1.46 мл) в ацетонитриле (10 мл). Смесь кипятят в течение 20 ч. Добавляют раствор 5% AcOH в воде (1мл). Смесь кипятят 1.5 ч. После охлаждения до комнатной температуры добавляют этилцетат (15 мл). Суспензию фильтруют. Полученный фильтрат обрабатывают водой и экстрагируют этилацетатом. Объединённые органические экстракты промывают соленой водой, сушат над Na2SO4, фильтруют и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить I1c (1.0 г).
Стадия I1B
Смесь I1c (1.0 г), 2M NaOH (10 мл) и метанола (5 мл) перемешивают при комнатной температуре в течение 1.5 ч. Смесь подкисляют и экстрагируют этилацетатом. Объединённые органические слои промывают соленой водой, сушат над Na2SO4, фильтруют и концентрируют. Полученное масло растворяют в толуоле (14 мл), с последующим добавлением дифеноксифосфорилазида (DPPA 1.7 г) и триэтиламина (0.84 мл). Смесь перемешивают при комнатной температуре 1.5 ч и нагревают до 110°C в течение 2 ч. После добавления бензилового спирта (BnOH 5.7 мл), смесь перемешивают при 110°C в течение 2 суток. Смесь разбавляют этилацетатом и промывают соленой водой, сушат над Na2SO4, фильтруют и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить I1d (3.0 г).
Стадия I1C
К раствору I1d (0.5 г), NH4OAc (0.7 г) в метаноле (5 мл) добавляют NaCNBH3 (0.23 г). Смесь перемешивают при комнатной температуре 2 ч. Реакцию прерывают, добавляя насыщенный раствор NaHCO3, экстрагируют дихлорметаном. Органические экстракты сушат над Na2SO4, фильтруют и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить I1e (45мг).
Стадия I1D
Смесь I1e (0.24 г) и Boc2O (1.2 г) в DCM (10 мл) перемешивают при комнатной температуре 20 ч. После выпаривания растворителей при пониженном давлении, остаток очищается методом хроматографии на силикагеле, чтобы получить 0.14 г бесцветного масла. Это масло растворяют в метаноле (15 мл) и добавляют Pd/C. Смесь перемешивают в атмосфере H2 (из баллона) в течение 20 ч, фильтруют через слой целита и концентрируют, чтобы получить I1 (66 мг). МС (ESI): m/z = 241 [M+H] +.
Пример 15. N-(3-(4-амино-7-оксо-3-(4-феноксифенил)-6,7-дигидро-1H-пирроло[2,3 -d]пиридазин-1-ил)бицикло[3.2.1]октан-8-ил)акриламид
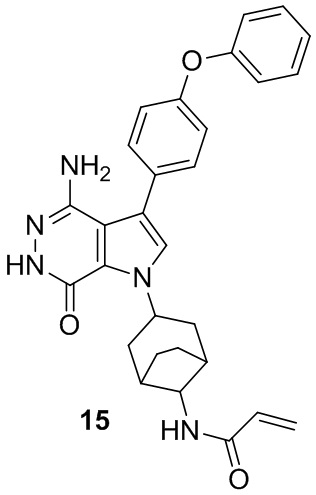
Указанное в заголовке соединение 15 (светло-желтое твёрдое вещество, 4.8 мг) получают из 1b и I1 в условиях, аналогичных описанным для стадий 1B~1G из Примера 1. МС (ESI): m/z = 496 [M+H] +.
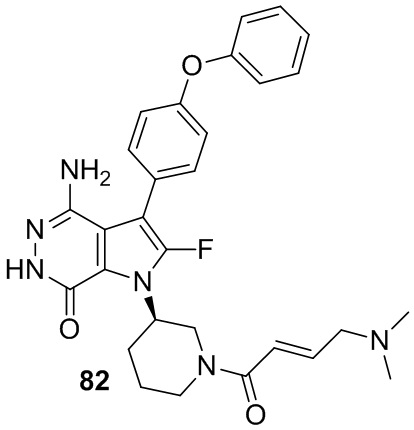
Пример 16. 4-амино-1-[(3R)-1-[(E)-4-[изопропил(метил)амино]бут-2-еноил]-3-пиперидил]-3-(4-феноксифенил)-6H-пирроло[2,3-d]пиридазин-7-он
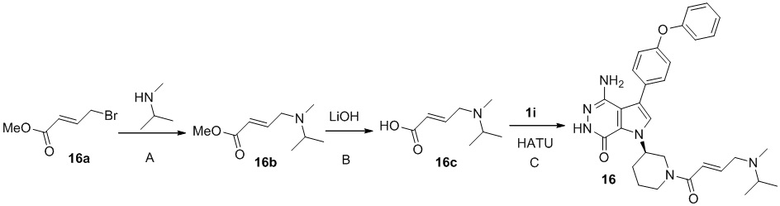
Стадия 16A
Смесь 16a (225 мг) и N-метилпропан-2-амина (190 мг) в ТГФ (3 мл), перемешивают при комнатной температуре в течение 2 суток. Добавляют насыщенный раствор NaCl, смесь экстрагируют 2 раза этилацетатом. Органические экстракты промывают соленой водой, сушат над Na2SO4, фильтруют, и концентрируют, чтобы получить 16b (200 мг).
Стадия 16B
К раствору 16b (195 мг) в ТГФ (5 мл) добавляют раствор LiOH (1M, 2.5 мл). Смесь перемешивают при комнатной температуре 2 ч. Добавляют 2M раствор HCl до pH <5 и концентрируют, чтобы получить 16c (500 мг).
Стадия 16C
К раствору 1i (11 мг) в ДМФ (1 мл) добавляют DIEA (0.05 мл), кислоту 16c (15 мг) и HATU (20 мг). Полученную смесь перемешивают при комнатной температуре в течение 1.5 ч. Удаляют растворители, и остаток очищают методом препаративной ЖХВР на обращенной фазе, чтобы получить указанное в заголовке соединение 16 (4 мг). МС (ESI): m/z = 541 [M+H]+.
Пример 17. трет-Бутил N-[(E)-4-[(3R)-3-[4-амино-7-оксо-3-(4-феноксифенил)-6H-пирроло[2,3-d]пиридазин-1-ил]-1-пиперидил]-4-оксо-бут-2-енил]-N-метилкарбамат
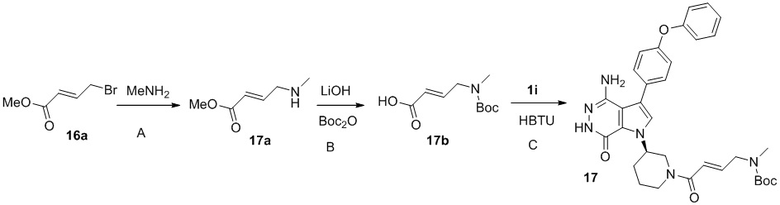
Стадия 17A
К смеси 16a (0.43 г) в ТГФ при -60°C по каплям добавляют 2M раствор метиламина в ТГФ (3 мл). Смесь перемешивают при -60°C в течение 2 ч и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить 17a (0.146 г).
Стадия 17B
К раствору 17a (145 мг) в ТГФ (5 мл) добавляют 2 M раствор NaOH (2 мл) и метанол (0.5 мл). Смесь перемешивают при комнатной температуре 35 мин. Добавляют 1M раствор HCl до pH <5 и концентрируют. Сырой продукт растворяют в смеси DCM/метанол и обрабатывают Boc2O (0.5 г). Смесь перемешивают при комнатной температуре 2 ч и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить 17b (0.11 г).
Стадия 17C
К раствору 1i (10 мг) в DCM (2 мл) добавляют триэтиламин (0.1 мл), кислоту 17b (7 мг) и HBTU (18 мг). Полученную смесь перемешивают при комнатной температуре в течение 0.5 ч и очищают методом хроматографии на силикагеле, чтобы получить указанное в заголовке соединение 17 (10 мг) в виде кремового твёрдого вещество. МС (ESI): m/z = 599 [M+H]+.
Пример 18. 4-амино-1-[(3R)-1-[(E)-4-(метиламино)бут-2-еноил]-3-пиперидил]-3-(4-феноксифенил)-6H-пирроло[2,3-d]пиридазин-7-он
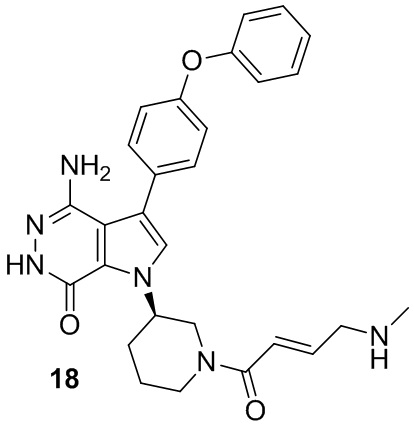
К раствору соединения 17 (6 мг) в DCM (1.6 мл) добавляют TFA (0.2 мл). Смесь перемешивают при комнатной температуре в течение 30 мин и концентрируют. Остаток очищают методом препаративной ЖХВР на обращенной фазе, чтобы получить указанное в заголовке соединение 18 (2.4 мг) в виде белого твёрдого вещества. МС (ESI): m/z = 499 [M+H]+.
Пример 19. 4-амино-3-(4-феноксифенил)-1-(1-пропионилпиперидин-3-ил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
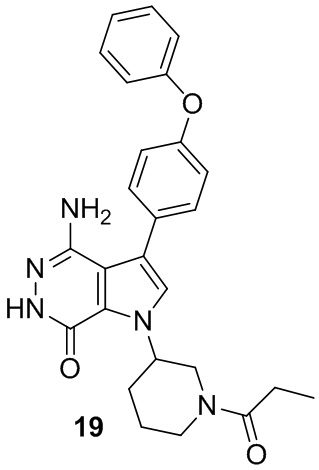
Смесь соединения 2 (5 мг) и Pd/C (10 масс.%, 5 мг) в метаноле (5 мл) перемешивают атмосфере H2 (из баллона) в течение 3 ч. Реакционную смесь фильтруют через слой целита, промывают смесью этилацетат/метанол и концентрируют, чтобы получить указанное в заголовке соединение 19 (2.7 мг) в виде белого твёрдого вещества. МС (ESI): m/z = 458 [M+H]+.
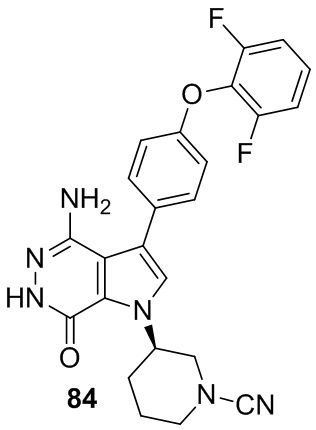
Пример 20. (R)-3-(4-амино-7-оксо-3-(4-феноксифенил)-6,7-дигидро-1H-пирроло[2,3-d]пиридазин-1-ил)пиперидин-1-карбонитрил
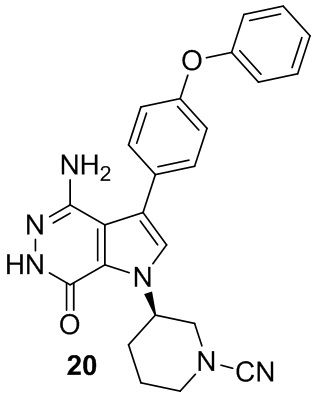
В смесь 1i (3.5 мг) и раствора K2CO3 (2M, 15 мкл) в ацетоне (3 мл) добавляют BrCN (1 мг). Смесь перемешивают при комнатной температуре 2 ч и концентрируют. Остаток очищают методом препаративной ЖХВР на обращенной фазе, чтобы получить указанное в заголовке соединение 20 (1.6 мг) в виде белого твёрдого вещества. МС (ESI): m/z = 427 [M+H]+.
Пример 21. 4-амино-1-(1-(бут-2-иноил)пиперидин-3-ил)-3-(4-феноксифенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
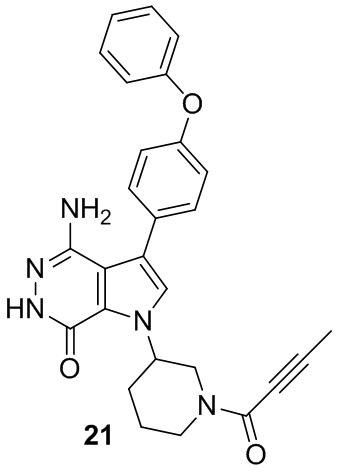
Указанное в заголовке соединение получают из рацемической формы 1i и бут-2-иновой кислоты в условиях, аналогичных описанным для стадии 1G примера 1. МС (ESI): m/z = 468 [M+H]+.
Соединения в примерах 22 - 37 (Таблица 2) были получены из 1i и соответствующих кислот (промышленно доступны или легко получаются) в условиях, аналогичных описанным в примерах 16, 17, 18.
Таблица 2. Соединения формулы:
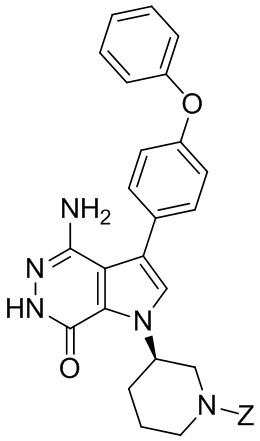
m/z [M+H]
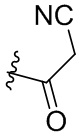
Соединения в примерах 38-49 (Таблица 3) были получены из (E)-4-(диметиламино)бут-2-еновой кислоты и соответствующих аминов (предшествующие соединения предыдущих примеров) в условиях, аналогичных описанным для стадии 16C Примера 16.
Таблица 3. Соединения формулы:
m/z [M+H]+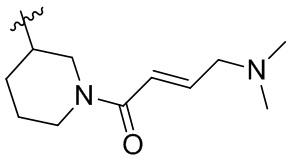
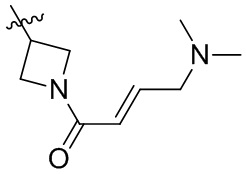
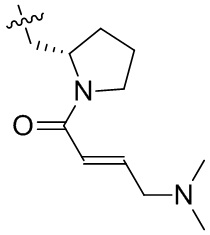
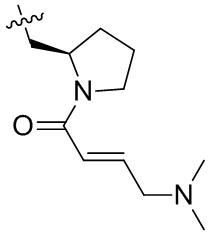
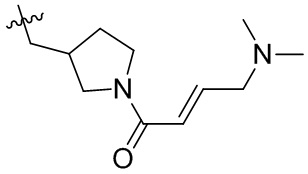
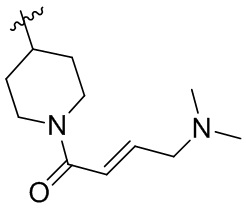
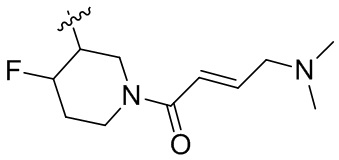
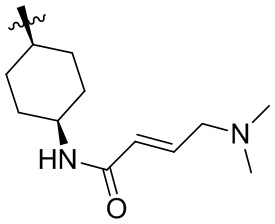
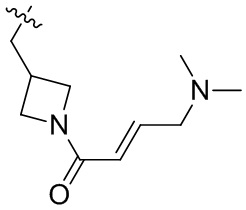
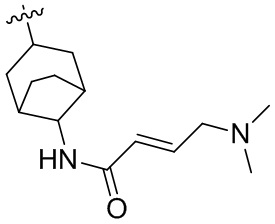
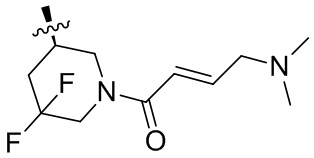
Пример 50. (E)-2-[(3R)-3-[4-амино-7-оксо-3-(4-феноксифенил)-6H-пирроло[2,3-d]пиридазин-1-ил]пиперидин-1-карбонил]-3-циклопропил-проп-2-енонитрил
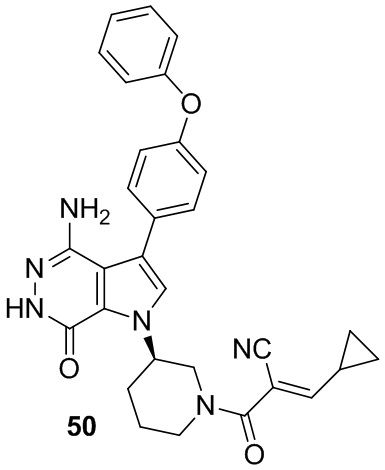
К смеси соединения 24 (9 мг) и пиперидина (2 мг) в метаноле (1 мл) добавляют циклопропанкарбальдегид (2.1 мг). Смесь перемешивают при комнатной температуре 20 ч и концентрируют. Остаток очищают методом препаративной ЖХВР на обращенной фазе, чтобы получить указанное в заголовке соединение 50 (2.7 мг) в виде белого твёрдого вещества. МС (ESI): m/z = 521 [M+H]+.
Пример 51. 1-(1-акрилоилпиперидин-3-ил)-4-амино-3-(4-метоксифенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
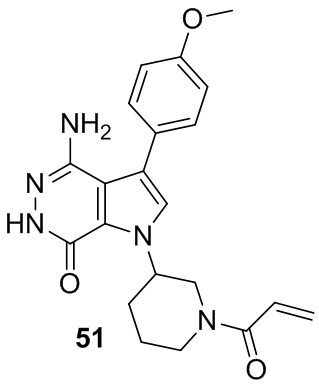
Указанное в заголовке соединение получают из рацемической формы 1e и (4-метоксифенил)бороновой кислоты в условиях, аналогичных описанным для стадий 1D~1G из Примера 1. МС (ESI): m/z = 394 [M+H]+.
Пример 52. 4-амино-1-[1-[(E)-4-(диметиламино)бут-2-еноил]-3-пиперидил]-3-(4-метоксифенил)-6H-пирроло[2,3-d]пиридазин-7-он
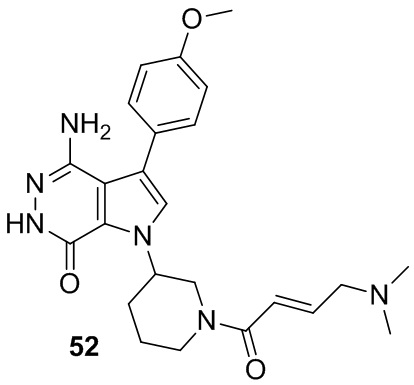
Указанное в заголовке соединение получают из (E)-4-(диметиламино)бут-2-еновой кислоты и соответствующего амина (предшественник из примера 51) в условиях, аналогичных описанным для стадии 16C из Примера 16. МС (ESI): m/z = 451 [M+H]+.
Пример 53. 1-(1-акрилоилпиперидин-3-ил)-4-амино-3-(3-хлор-4-метоксифенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
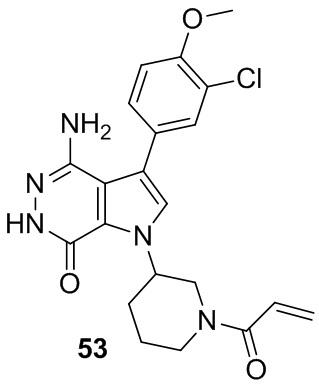
Указанное в заголовке соединение получают из рацемической формы 1e и (3-хлор-4-метоксифенил)бороновой кислоты в условиях, аналогичных описанным для стадий 1D~1G из Примера 1. МС (ESI): m/z = 428 [M+H]+.
Пример 54. 4-амино-3-(3-хлор-4-метокси-фенил)-1-[1-[(E)-4-(диметиламино)бут-2-еноил]-3-пиперидил]-6H-пирроло[2,3-d]пиридазин-7-он
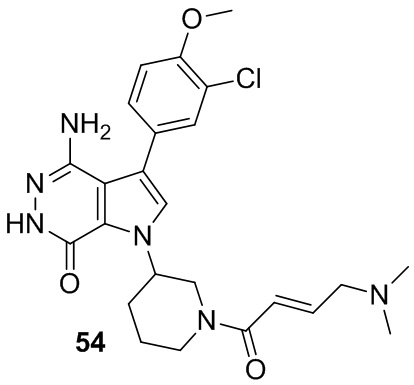
Указанное в заголовке соединение получают из (E)-4-(диметиламино)бут-2-еновой кислоты и соответствующего амина (предшественник из примера 53) в условиях, аналогичных описанным для стадии 16C из Примера 16. МС (ESI): m/z = 485 [M+H]+.
Пример 55. 1-(1-акрилоилпиперидин-3-ил)-4-амино-3-(4-(2-фторфенокси)фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
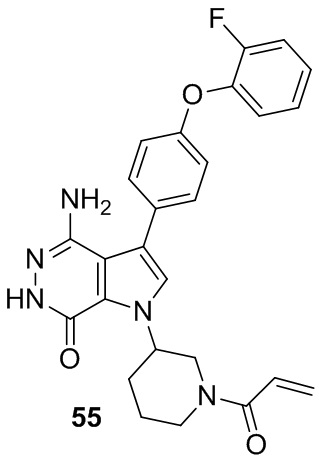
Указанное в заголовке соединение получают из рацемической формы 1e и ((4-(2-фторфенокси)фенил)бороновой кислоты в условиях, аналогичных описанным для стадий 1D~1G из Примера 1. МС (ESI): m/z = 474 [M+H]+.
Пример 56. (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-3-(4-(2-фторфенокси)-фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
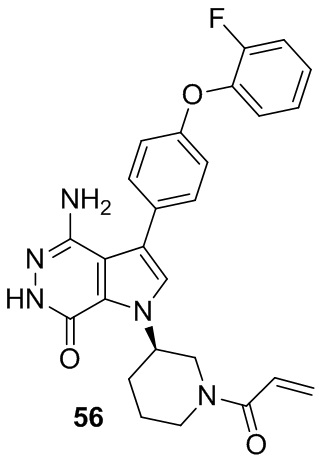
Указанное в заголовке соединение получают из 1e и ((4-(2-фторфенокси)фенил)бороновой кислоты в условиях, аналогичных описанным для стадий 1D~1G из Примера 1. МС (ESI): m/z = 474 [M+H]+.
Пример 57. 4-амино-1-[(3R)-1-[(E)-4-(диметиламино)бут-2-еноил]-3-пиперидил]-3-[4-(2-фторфенокси)фенил]-6H-пирроло[2,3-d]пиридазин-7-он
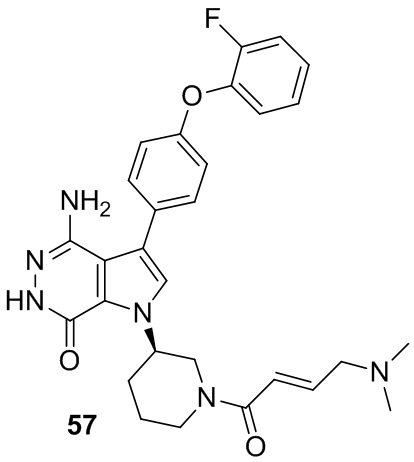
Указанное в заголовке соединение получают из (E)-4-(диметиламино)бут-2-еновой кислоты и соответствующего амина (предшественник из примера 56) в условиях, аналогичных описанным для стадии 16C из Примера 16. МС (ESI): m/z = 531 [M+H]+.
Промежуточное соединение 2
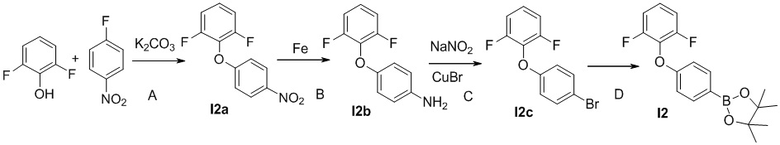
Стадия I2A
Смесь 2,6-дифторфенола (3.0 г, 21.3 ммоль), 1-фтор-4-нитробензола (3.04 г, 23.4 ммоль) и K2CO3 (4.4 г, 32 ммоль) в CH3CN (50 мл) кипятят 16 ч. После охлаждения до комнатной температуры растворители удаляются. Добавляют воду, смесь экстрагируют этилацетатом три раза. Органические экстракты промывают водой, соленой водой, сушат над MgSO4, фильтруют, и концентрируют, чтобы получить масло I2а (4.9 г).
Стадия I2B
Смесь 1,3-дифтор-2-(4-нитрофенокси)бензола I2а (4.9 г, 19.5 ммоль), насыщенного раствора NH4Cl (5 мл) и порошка железа (5.5 г, 97.5 ммоль) в метаноле (40 мл) кипятят 3 ч. Смесь фильтруют. К фильтрату добавляют воду и экстрагируют этилацетатом три раза. Органические экстракты промывают водой, соленой водой, сушат над MgSO4, фильтруют, и концентрируют, чтобы получить светло-желтое масло I2b (4.1 г). МС (ESI): m/z=222.1 [M+H]+.
Стадия I2C
К смеси 4-(2,6-дифторфенокси)анилина I2b (4.1 г, 18.5 ммоль) в 2M растворе H2SO4 (50 мл) при 0°C добавляют раствор NaNO2 (6.4 г, 92.7 ммоль) в воде (20 мл). Смесь перемешивают при 0°C в течение 40 мин и добавляют CuBr (5.3 г, 37 ммоль). Полученную смесь кипятят 16 ч, охлаждают до комнатной температуры и экстрагируют этилацетатом три раза. Органические экстракты промывают водой, соленой водой, сушат над MgSO4, фильтруют, и концентрируют, чтобы получить бесцветное масло I2c (1.6 г).
Стадия I2D
Смесь 2-(4-бромфенокси)-1,3-дифторбензола I2c (1.6 г, 3.6 ммоль), бис(пинаколято)дибора (1.71 г, 6.7 ммоль), ацетата калия (830 мг, 8.4 ммоль) и Pd(PPh3)2Cl2(126 мг, 0.18 ммоль) в 1,4-диоксане (40 мл) перемешивают в атмосфере N2 при 80°C в течение 16 ч. После охлаждения до комнатной температуры растворители удаляются. Остаток очищается методом хроматографии на силикагеле, чтобы получить бесцветное масло I2 (1.6 г).
Промежуточное соединение 3. 2-(4-(4-хлорфенокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан
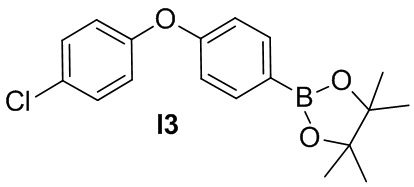
Смесь 1-хлор-4-(4-йодфенокси)бензола (330 мг), бис(пинаколято)дибора (508 мг), KOAc (300 мг) и Pd(PPh3)2Cl2(82 мг) в 1,4-диоксане (10 мл) перемешивают в герметизированной трубке в атмосфере N2 при 100°C в течение 16 ч. После охлаждения до комнатной температуры, твёрдые вещества отфильтровывают и фильтрат концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить желтое масло I3 (150 мг).
Промежуточное соединение 4. 2-(4-(4-фторфенокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан
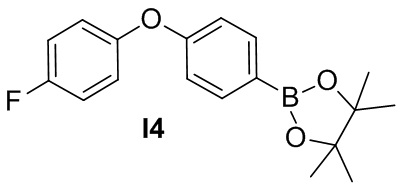
Указанное в заголовке соединение получают из 1-фтор-4-(4-йодфенокси)бензола в условиях, аналогичных описанным для промежуточного соединения 3.
Промежуточное соединение 5. 2-(4-(3-фторфенокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан
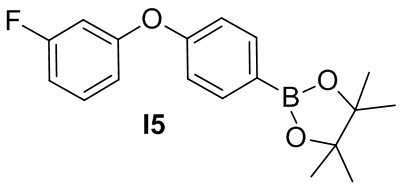
Смесь (3-фторфенил)бороновой кислоты (0.71 г), 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенола (1.02 г), ацетата меди (1.0 г), триэтиламина (1.3мл), и молекулярного сита 4A (3 г) в DCM (20 мл) перемешивают при комнатной температуре 24 ч. Реакционную смесь фильтруют через слой целита, промывают дихлорметаном и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить бесцветное масло I5 (60 мг).
Промежуточное соединение 6. 2-(4-(3-хлорфенокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан
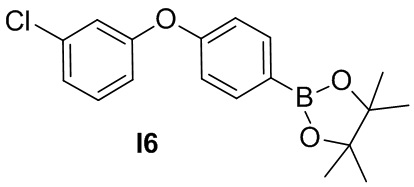
Указанное в заголовке соединение получают из (3-хлорфенил)бороновой кислоты в условиях, аналогичных описанным для промежуточного соединения 5.
Промежуточное соединение 7. 2-(4-(3-хлорфенокси)-3-метоксифенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан
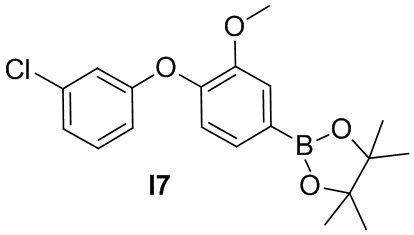
Указанное в заголовке соединение получают из 3-хлорфенола и 1-фтор-2-метокси-4-нитробензола в условиях, аналогичных описанным для промежуточного соединения 2.
Промежуточное соединение 8. 2-(4-(3,4-дихлорфенокси)-3-метоксифенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан
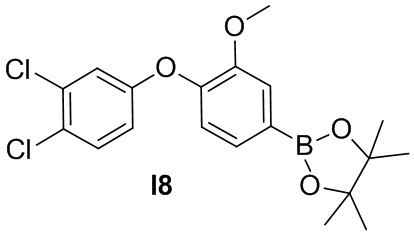
Указанное в заголовке соединение получают из 3,4-дихлорфенола и 1-фтор-2-метокси-4-нитробензола в условиях, аналогичных описанным для промежуточного соединения 2.
Промежуточное соединение 9. 2-(4-(2-хлор-6-фторфенокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан
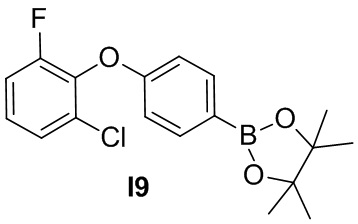
Указанное в заголовке соединение получают из 2-хлор-6-фторфенола и 1-фтор-4-нитробензола в условиях, аналогичных описанным для промежуточного соединения 2.
Промежуточное соединение 10. 2-(4-(2-хлорфенокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан
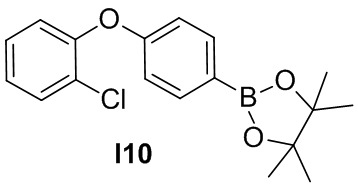
Указанное в заголовке соединение получают из 2-хлорфенола и 1-фтор-4-нитробензола в условиях, аналогичных описанным для промежуточного соединения 2.
Промежуточное соединение 11. 2-(2-фтор-4-феноксифенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан
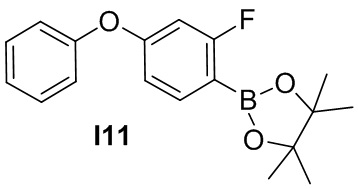
Указанное в заголовке соединение получают из фенола и 2,4-дифтор-1-нитробензола в условиях, аналогичных описанным для промежуточного соединения 2.
Промежуточное соединение 12. (3-фтор-4-феноксифенил)бороновая кислота
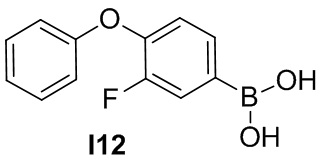
К раствору 4-бром-2-фтор-1-феноксибензола (0.5 г, 3.6 ммоль) в ТГФ (20 мл) при -78°C по каплям добавляют раствор н-бутиллития (2.5 M в гексане, 2.16 мл, 5.4 ммоль). Через 30 мин, по каплям добавляют триизопропилборат (1.15 мл, 5.4 ммоль). Смесь перемешивают при -78°C в течение 1 ч. Реакцию прерывают, добавляя воду, экстрагируют этилацетатом три раза. Объединённые органические экстракты промывают водой, соленой водой, сушат над MgSO4, фильтруют, и концентрируют, чтобы получить светло-желтое масло I12 (0.18 г).
Пример 58. (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-3-(4-(2,6-дифторфенокси)-фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
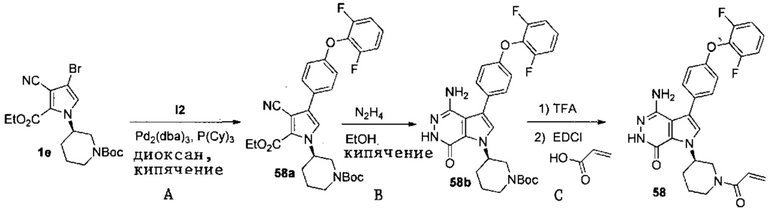
Стадия 58A
Смесь 1e (2.8 г, 6.6 моль), I2 (2.2 г, 6.6 моль) и K3PO4·3H2O (2.6 г, 9.9 моль) в смеси 1,4-диоксан/вода (10 мл/1 мл) дегазируют азотом. Затем добавляют Pd2(dba)3 (300мг, 0.33 ммоль) и трициклогексилфосфин [P(Cy)3 ,185 мг, 0.66 ммоль]. Полученную смесь кипятят 16 ч в атмосфере N2. После охлаждения до комнатной температуры твёрдое вещество отфильтровывают, и фильтрат концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить белое твёрдое вещество 58a (1.2 г). МС (ESI): m/z=552 [M+H]+.
Стадия 58B
Смесь 58a (1.2 г) и N2H4·H2O (1 мл) в этаноле (5 мл) кипятят в течение 16 ч. После охлаждения до комнатной температуры растворители удаляются. Остаток очищается методом хроматографии на силикагеле, чтобы получить белое твёрдое вещество 58b (0.66 г). МС (ESI): m/z=538 [M+H]+.
Стадия 58C
К раствору 58b (880 мг, 1.63 моль) в DCM (5 мл) добавляют трифторуксусную кислоту (1 мл). Смесь перемешивают при комнатной температуре 3 ч и концентрируют, чтобы получить масло 58c (940 мг). К раствору 58c (940 мг) в DCM (5 мл) добавляют акриловую кислоту (210 мг, 2.5 моль), гидрохлорид 1-(3-диэтиламинопропил)-3-этилкарбодиимида (627 мг, 3.3 ммоль) и TEA (340 мг, 3.3 ммоль). Полученную смесь перемешивают при комнатной температуре 18 ч и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить указанное в заголовке соединение 58 (450 мг) в виде белого твёрдого веществ. МС (ESI): m/z = 492 [M+H] +.
Пример 59. 4-амино-3-[4-(2,6-дифторфенокси)фенил]-1-[(3R)-1-[(E)-4-(диметиламино)бут-2-еноил]-3-пиперидил]-6H-пирроло[2,3-d]пиридазин-7-он
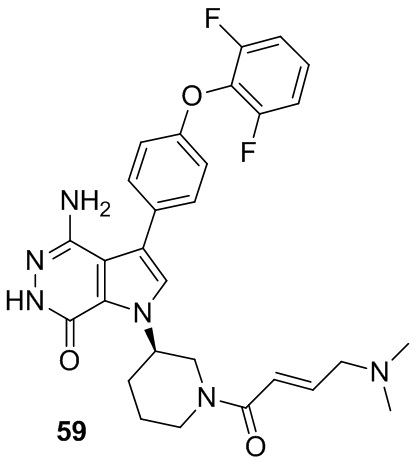
Указанное в заголовке соединение получают из (E)-4-(диметиламино)бут-2-еновой кислоты и амина 58c в условиях, аналогичных описанным для стадии 16C из Примера 16. МС (ESI): m/z = 549 [M+H]+.
Пример 60. 1-(1-акрилоилпиперидин-3-ил)-4-амино-3-(4-(4-фторфенокси)фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
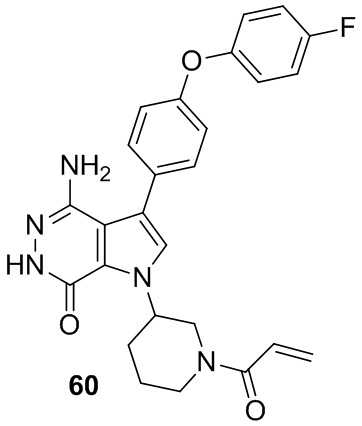
Указанное в заголовке соединение получают из рацемической формы 1e и промежуточного соединения 4 в условиях, аналогичных описанным для стадий 1D~1G из Примера 1. МС (ESI): m/z = 474 [M+H]+.
Пример 61. 4-амино-1-[1-[(E)-4-(диметиламино)бут-2-еноил]-3-пиперидил]-3-[4-(4-фторфенокси)фенил]-6H-пирроло[2,3-d]пиридазин-7-он
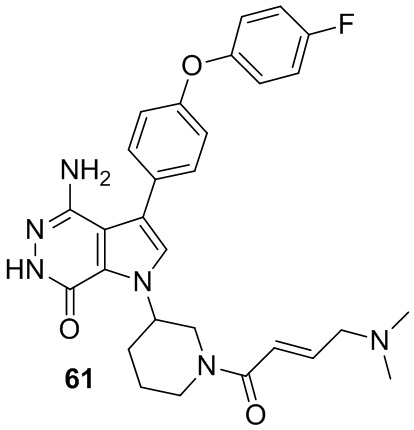
Указанное в заголовке соединение получают из (E)-4-(диметиламино)бут-2-еновой кислоты и соответствующего амина (предшественник из примера 60) в условиях, аналогичных описанным для стадии 16C из Примера 16. МС (ESI): m/z = 531 [M+H]+.
Пример 62. 1-(1-акрилоилпиперидин-3-ил)-4-амино-3-(4-(3-фторфенокси)фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
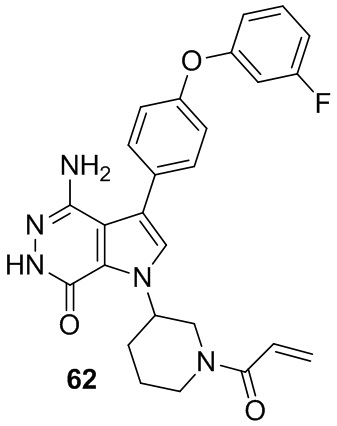
Указанное в заголовке соединение получают из рацемической формы 1e и промежуточного соединения 5 в условиях, аналогичных описанным для стадий 1D~1G из Примера 1. МС (ESI): m/z = 474 [M+H]+.
Пример 63. 4-амино-1-[1-[(E)-4-(диметиламино)бут-2-еноил]-3-пиперидил]-3-[4-(3-фторфенокси)фенил]-6H-пирроло[2,3-d]пиридазин-7-он
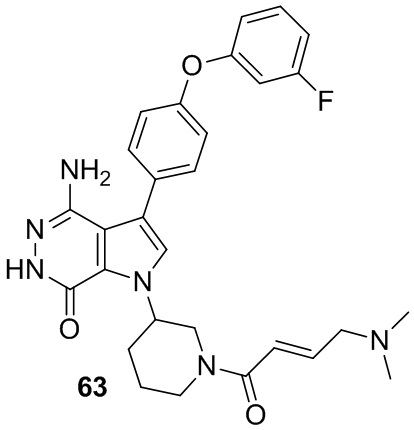
Указанное в заголовке соединение получают из (E)-4-(диметиламино)бут-2-еновой кислоты и соответствующего амина (предшественник из примера 62) в условиях, аналогичных описанным для стадии 16C из Примера 16. МС (ESI): m/z = 531 [M+H]+.
Пример 64. 1-(1-акрилоилпиперидин-3-ил)-4-амино-3-(4-(4-хлорфенокси)фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
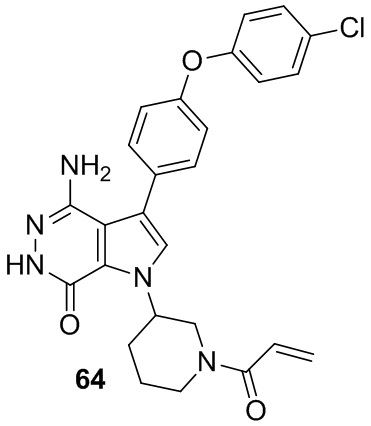
Указанное в заголовке соединение получают из рацемической формы 1e и промежуточного соединения 3 в условиях, аналогичных описанным для стадий 1D~1G из Примера 1. МС (ESI): m/z =490 [M+H]+.
Пример 65. 4-амино-3-[4-(4-хлорфенокси)фенил]-1-[1-[(E)-4-(диметиламино)бут-2-еноил]-3-пиперидил]-6H-пирроло[2,3-d]пиридазин-7-он
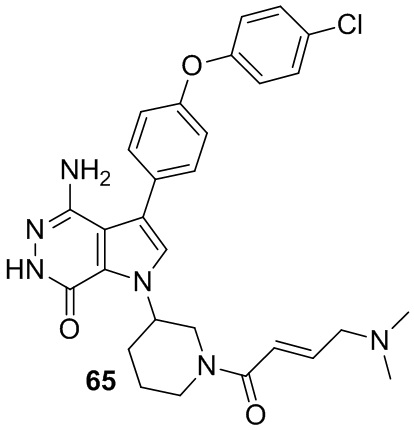
Указанное в заголовке соединение получают из (E)-4-(диметиламино)бут-2-еновой кислоты и соответствующего амина (предшественник из примера 64) в условиях, аналогичных описанным для стадии 16C из Примера 16. МС (ESI): m/z = 547 [M+H]+.
Пример 66. 1-(1-акрилоилпиперидин-3-ил)-4-амино-3-(4-(3-хлорфенокси)фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
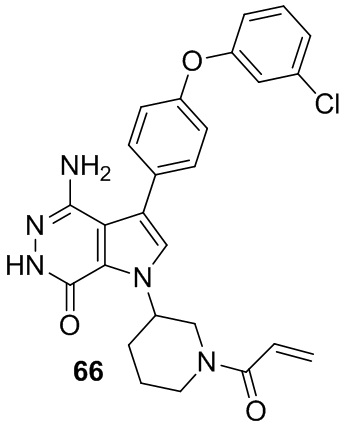
Указанное в заголовке соединение получают из рацемической формы 1e и промежуточного соединения 6 в условиях, аналогичных описанным для стадий 1D~1G из Примера 1. МС (ESI): m/z = 490 [M+H]+.
Пример 67. 4-амино-3-[4-(3-хлорфенокси)фенил]-1-[1-[(E)-4-(диметиламино)бут-2-еноил]-3-пиперидил]-6H-пирроло[2,3-d]пиридазин-7-он
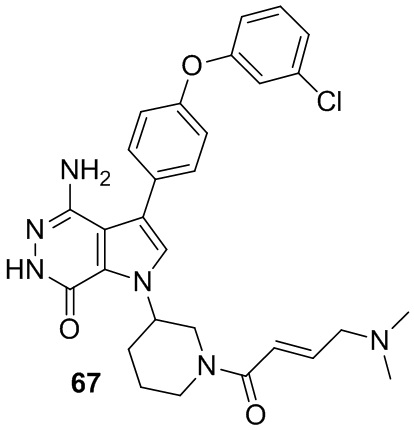
Указанное в заголовке соединение получают из (E)-4-(диметиламино)бут-2-еновой кислоты и соответствующего амина (предшественник из примера 66) в условиях, аналогичных описанным для стадии 16C из Примера 16. МС (ESI): m/z = 547 [M+H]+.
Пример 68. 1-(1-акрилоилпиперидин-3-ил)-4-амино-3-(4-(3-хлорфенокси)-3-метоксифенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
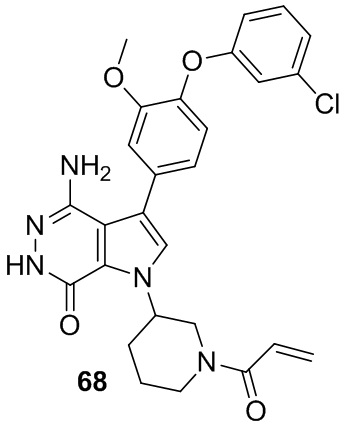
Указанное в заголовке соединение получают из рацемической формы 1e и промежуточного соединения 7 в условиях, аналогичных описанным для стадий 1D~1G из Примера 1. МС (ESI): m/z = 520 [M+H]+.
Пример 69. 4-амино-3-[4-(3-хлорфенокси)-3-метокси-фенил]-1-[1-[(E)-4-(диметиламино)бут-2-еноил]-3-пиперидил]-6H-пирроло[2,3-d]пиридазин-7-он
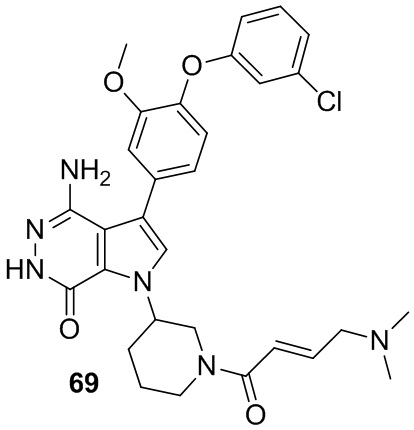
Указанное в заголовке соединение получают из (E)-4-(диметиламино)бут-2-еновой кислоты и соответствующего амина (предшественник из примера 68) в условиях, аналогичных описанным для стадии 16C из Примера 16. МС (ESI): m/z = 577 [M+H]+.
Пример 70. (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-3-(4-(2-хлорфенокси)-фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
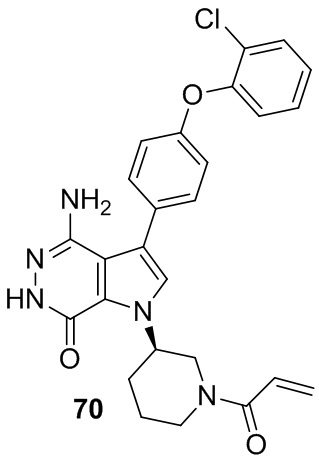
Указанное в заголовке соединение получают из 1e и промежуточного соединения 10 в условиях, аналогичных описанным в примере 58. МС (ESI): m/z = 490 [M+H]+.
Пример 71. 4-амино-3-[4-(2-хлорфенокси)фенил]-1-[(3R)-1-[(E)-4-(диметиламино)бут-2-еноил]-3-пиперидил]-6H-пирроло[2,3-d]пиридазин-7-он
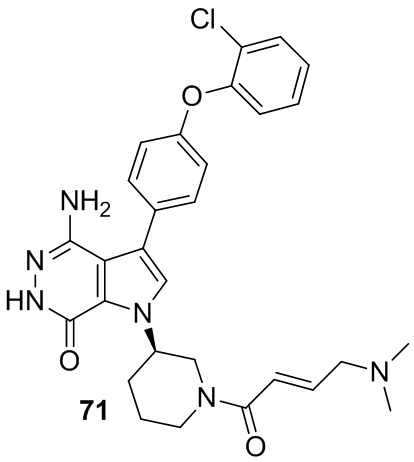
Указанное в заголовке соединение получают из (E)-4-(диметиламино)бут-2-еновой кислоты и соответствующего амина (предшественник из примера 70) в условиях, аналогичных описанным для стадии 16C из Примера 16. МС (ESI): m/z = 547 [M+H]+.
Пример 72. (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-3-(4-(3,4-дихлорфенокси)-3-метоксифенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
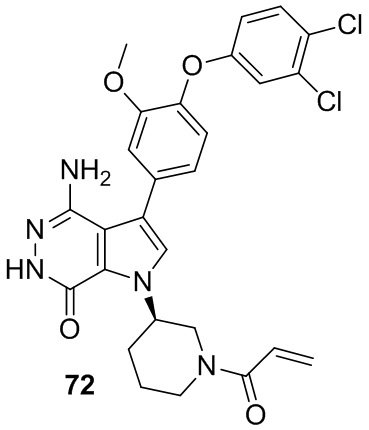
Указанное в заголовке соединение получают из 1e и промежуточного соединения 8 в условиях, аналогичных описанным в примере 58. МС (ESI): m/z = 554 [M+H]+.
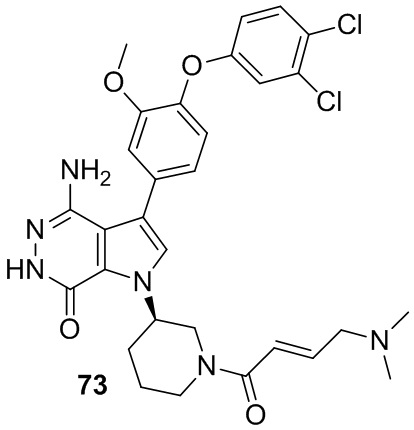
Пример 73. 4-амино-3-[4-(3,4-дихлорфенокси)-3-метокси-фенил]-1-[(3R)-1-[(E)-4-(диметиламино)бут-2-еноил]-3-пиперидил]-6H-пирроло[2,3-d]пиридазин-7-он
Указанное в заголовке соединение получают из (E)-4-(диметиламино)бут-2-еновой кислоты и соответствующего амина (предшественник из примера 72) в условиях, аналогичных описанным для стадии 16C из Примера 16. МС (ESI): m/z = 611 [M+H]+.
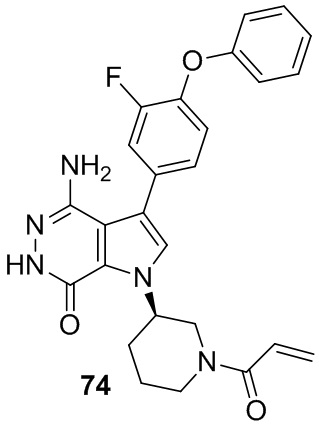
Пример 74. (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-3-(3-фтор-4-феноксифенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
Указанное в заголовке соединение получают из 1e и промежуточного соединения 12 в условиях, аналогичных описанным в примере 58. МС (ESI): m/z = 474 [M+H]+.
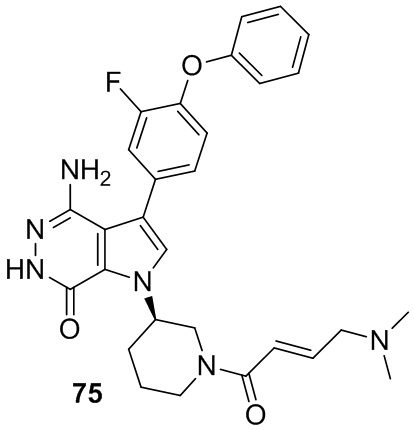
Пример 75. 4-амино-1-[(3R)-1-[(E)-4-(диметиламино)бут-2-еноил]-3-пиперидил]-3-(3-фтор-4-фенокси-фенил)-6H-пирроло[2,3-d]пиридазин-7-он
Указанное в заголовке соединение получают из (E)-4-(диметиламино) бут-2-еновой кислоты и соответствующего амина (предшественник из примера 74) в условиях, аналогичных описанным для стадии 16C из Примера 16. МС (ESI): m/z = 531 [M+H]+.
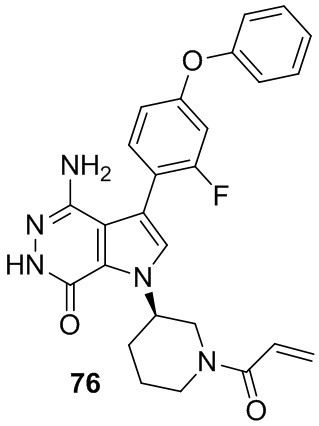
Пример 76. (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-3-(2-фтор-4-феноксифенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
Указанное в заголовке соединение получают из 1e и промежуточного соединения 11 в условиях, аналогичных описанным в примере 58. МС (ESI): m/z = 474 [M+H]+.
Пример 77. 4-амино-1-[(3R)-1-[(E)-4-(диметиламино)бут-2-еноил]-3-пиперидил]-3-(2-фтор-4-феноксифенил)-6H-пирроло[2,3-d]пиридазин-7-он
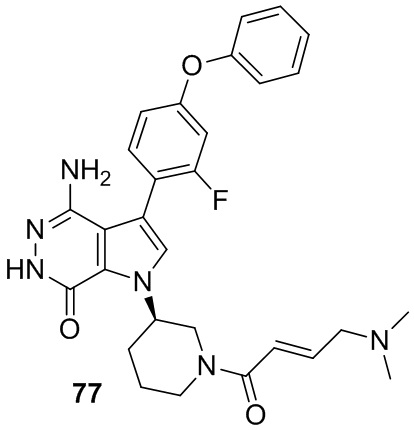
Указанное в заголовке соединение получают из (E)-4-(диметиламино)бут-2-еновой кислоты и соответствующего амина (предшественник из примера 76) в условиях, аналогичных описанным для стадии 16C из Примера 16. МС (ESI): m/z = 531 [M+H]+.
Пример 78. (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-3-(4-(2-хлор-6-фторфенокси)фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
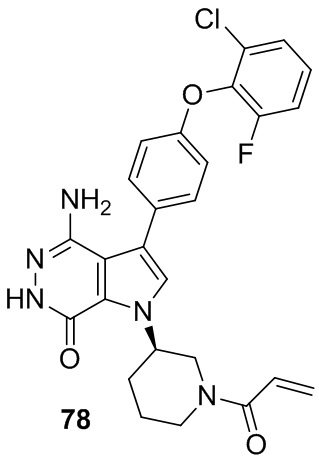
Указанное в заголовке соединение получают из 1e и промежуточного соединения 9 в условиях, аналогичных описанным в примере 58. МС (ESI): m/z = 508 [M+H]+.
Пример 79. 4-амино-3-[4-(2-хлор-6-фтор-фенокси)фенил]-1-[(3R)-1-[(E)-4-(диметиламино)бут-2-еноил]-3-пиперидил]-6H-пирроло[2,3-d]пиридазин-7-он
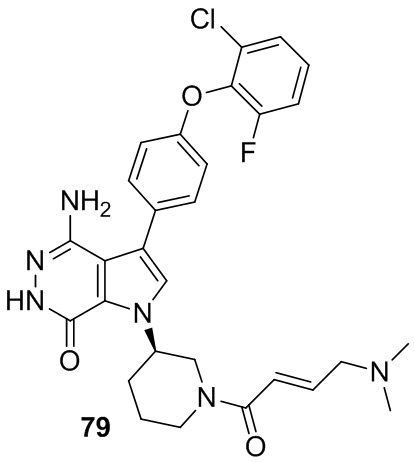
Указанное в заголовке соединение получают из (E)-4-(диметиламино)бут-2-еновой кислоты и соответствующего амина (предшественник из примера 78) в условиях, аналогичных описанным для стадии 16C из Примера 16. МС (ESI): m/z = 565 [M+H]+.
Пример 80. (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-2-хлор-3-(4-феноксифенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
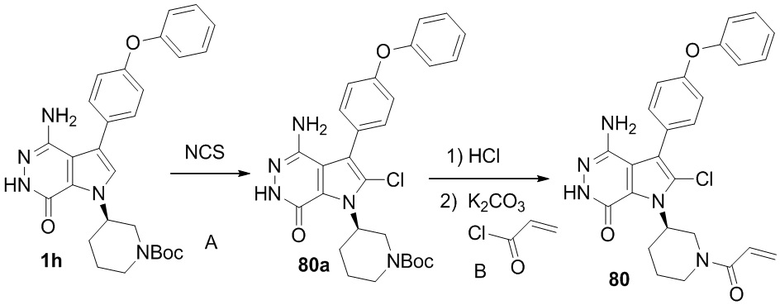
Стадия 80A
Смесь 1h (23 мг) и NCS (15 мг) в DCM (1.5 мл) перемешивают при комнатной температуре в течение 20 ч и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить бежевое твёрдое вещество 80a (13 мг). МС (ESI): m/z = 536 [M+H] +.
Стадия 80B
Смесь 80a (13 мг) и раствора HCl (0.2 мл, 4M в диоксане) в DCM (1 мл) перемешивают при комнатной температуре в течение 0.5 ч и концентрируют. Сырой продукт растворяют в ТГФ и охлаждают в ледяной ванне. К смеси добавляют 2 M раствор K2CO3 (30 мкл) и раствор акрилоилхлорида (5 мг) в ТГФ. Полученную смесь перемешивают при 0oC в течение 30 мин и очищают методом препаративной ЖХВР на обращенной фазе, чтобы получить указанное в заголовке соединение 80 (2.1 мг) в виде кремового твёрдого вещества. МС (ESI): m/z = 490 [M+H]+.
Пример 81. (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-2-фтор-3-(4-феноксифенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
Смесь 1g (120 мг, 0.23 ммоль) и 98.9 мг (0.28 ммоль) Селектфтора (дитетрафторборат 1-фтор-4-хлорметил-1,4-диазониабицикло[2.2.2]октан) в CH3CN (10 мл) кипятят 10 ч. После охлаждения до комнатной температуры растворители удаляются. Добавляют воду, и смесь экстрагируют этилацетатом три раза. Объединённые органические экстракты промывают водой, соленой водой, сушат над MgSO4, фильтруют, и концентрируют, чтобы получить масло 81a (71 мг). МС (ESI): m/z = 534 [M+H] +.
Указанное в заголовке соединение получают из 81a и в условиях, аналогичных описанным для стадий 1E~1G из Примера 1. МС (ESI): m/z = 474 [M+H] +.
Пример 82. 4-амино-1-[(3R)-1-[(E)-4-(диметиламино)бут-2-еноил]-3-пиперидил]-2-фтор-3-(4-феноксифенил)-6H-пирроло[2,3-d]пиридазин-7-он
Указанное в заголовке соединение получают из (E)-4-(диметиламино)бут-2-еновой кислоты и соответствующего амина (предшественник из примера 81) в условиях, аналогичных описанным для стадии 16C из Примера 16. МС (ESI): m/z = 531 [M+H]+.
Пример 83. (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-2-хлор-3-(4-(2-фторфенокси)фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
Указанное в заголовке соединение получают в условиях, аналогичных описанным в примере 80. МС (ESI): m/z=508 [M+H]+.
Пример 84. (R)-3-(4-амино-3-(4-(2,6-дифторфенокси)фенил)-7-оксо-6,7-дигидро-1H-пирроло[2,3-d]пиридазин-1-ил)пиперидин-1-карбонитрил
Указанное в заголовке соединение получают в условиях, аналогичных описанным в примере 20. МС (ESI): m/z=463 [M+H]+.
Пример 85. (R)-3-(4-амино-3-(3-фтор-4-феноксифенил)-7-оксо-6,7-дигидро-1H-пирроло[2,3-d]пиридазин-1-ил)пиперидин-1-карбонитрил
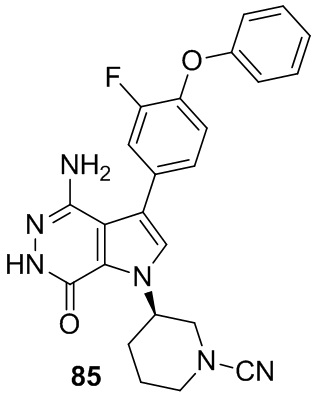
Указанное в заголовке соединение получают в условиях, аналогичных описанным в примере 20. МС (ESI): m/z=445 [M+H]+.
Пример 86. (R)-3-(4-амино-2-хлор-3-(4-(2-фторфенокси)фенил)-7-оксо-6,7-дигидро-1H-пирроло[2,3-d]пиридазин-1-ил)пиперидин-1-карбонитрил
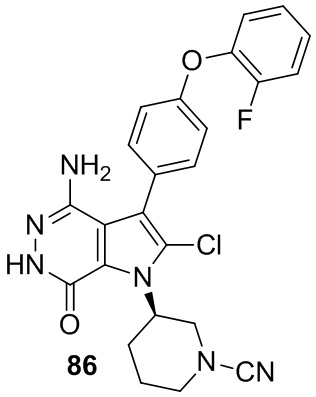
Указанное в заголовке соединение получают в условиях, аналогичных описанным в примерах 20, 80. МС (ESI): m/z=479 [M+H]+.
Пример 87. (R)-3-(4-амино-3-(4-(2-фторфенокси)фенил)-7-оксо-6,7-дигидро-1H-пирроло[2,3-d]пиридазин-1-ил)пиперидин-1-карбонитрил
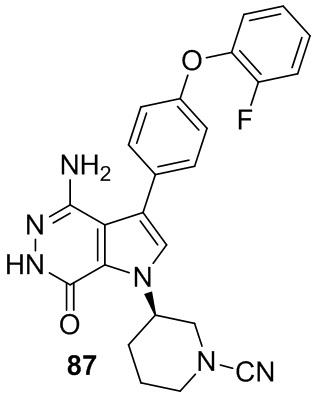
Указанное в заголовке соединение получают в условиях, аналогичных описанным в примере 20. МС (ESI): m/z=445 [M+H]+.
Пример 88. N-(3-(4-амино-7-оксо-3-(4-феноксифенил)-6,7-дигидро-1H-пирроло[2,3-d]пиридазин-1-ил)фенил)акриламид
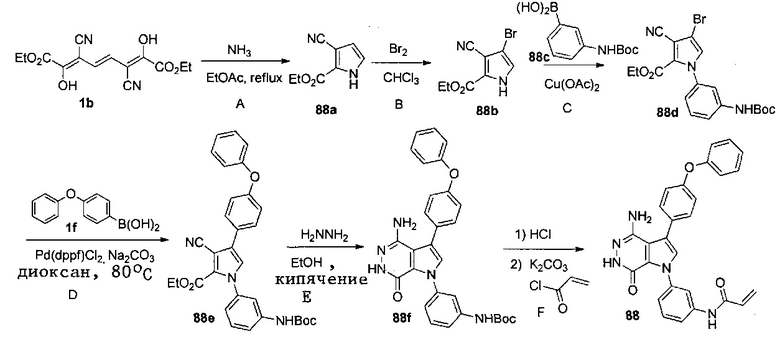
Стадия 88A
К раствору 1b (1.62 г) в этилацетате (30 мл) при 60oC добавляют по каплям 0.5 M раствор NH3 в диоксане (22 мл). Смесь кипятят в течение 18 ч. После охлаждения до комнатной температуры растворители удаляются. Остаток очищается методом хроматографии на силикагеле, чтобы получить светло-желтое твёрдое веществ 88a (0.37 г).
Стадия 88B
К раствору 88a (320 мг) в CHCl3 (17 мл) при -20oC добавляют по каплям раствор Br2 (350 мг) в CHCl3 (3 мл). Смесь перемешивают при температуре ниже 10°C в течение 5 ч, и затем реакцию прерывают, добавляя раствор 10% Na2S2O3 и насыщенный раствор NaHCO3. Две фазы разделяют; водную фазу экстрагируют дихлорметаном три раза. Объединённые органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить 88b (400 мг).
Стадия 88C
К смеси 88b (200 мг, 0.82 ммоль), 88c (390 мг, 1.64 ммоль) в DCM (10 мл) при 0°C добавляют Cu(OAc)2 (224 мг, 1.23 ммоль) и пиридин (185 мкл). Смесь перемешивают при комнатной температуре 20 ч. Добавляют воду, экстрагируют два раза этилацетатом. Органические экстракты промывают соленой водой, сушат над Na2SO4, фильтруют, и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить 88d (360 мг).
Стадия 88D
Смесь 88d (133 мг, 0.306 ммоль), 1f (131 мг, 0.612 ммоль), K2CO3 (2M, 0.5 мл), и Pd(dppf)Cl2 (49 мг, 0.06 ммоль) в 1.4-диоксане (8 мл) перемешивают в атмосфере N2 при 80°C в течение 18 ч. Добавляют воду, и экстрагируют два раза этилацетатом. Органические экстракты промывают соленой водой, сушат над Na2SO4, фильтруют и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить бесцветное масло 88e (135 мг). МС (ESI): m/z=524 [M+H]+.
Стадия 88E
Смесь 88e (130 мг) и N2H4 (1.2 мл) в этаноле (10 мл) кипятят 24 ч. После охлаждения до комнатной температуры растворители удаляются. Остаток очищается методом хроматографии на силикагеле, чтобы получить белое твёрдое вещество 88f
(38 мг). МС (ESI): m/z=510 [M+H]+.
Стадия 88F
Смесь 88f (18 мг) и 4 M раствор HCl (1 мл, в диоксане) перемешивают при комнатной температуре 1 ч и концентрируют. Сырой продукт растворяют в ТГФ и охлаждают в ледяной ванне. К смеси добавляют 2 M раствор K2CO3 (40 мкл) и раствор акрилоилхлорида (4 мг) в ТГФ. Полученную смесь перемешивают при 0°C в течение 20 мин и очищают методом препаративной ЖХВР на обращенной фазе, чтобы получить указанное в заголовке соединение 88 (7.2 мг) в виде белого твёрдого вещества. МС (ESI): m/z = 464 [M+H] +.
Пример 89. N-(3-(4-амино-3-(3-хлор-4-метоксифенил)-7-оксо-6,7-дигидро-1H-пирроло[2,3-d]пиридазин-1-ил)фенил)акриламид
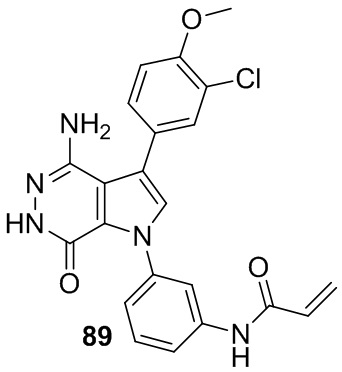
Указанное в заголовке соединение получают из 88d и (3-хлор-4-метоксифенил)бороновой кислоты в условиях, аналогичных описанным для стадий 88D~88F из Примера 88. МС (ESI): m/z = 436 [M+H]+.
Пример 90. N-(4-(4-амино-7-оксо-3-(4-феноксифенил)-6,7-дигидро-1H-пирроло[2,3-d]пиридазин-1-ил)фенил)акриламид
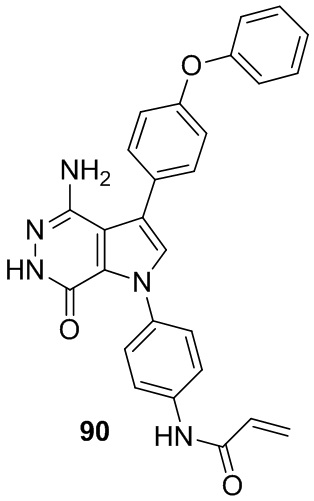
Указанное в заголовке соединение получают из 88b и (4-((трет-бутоксикарбонил)амино)фенил)бороновой кислоты в условиях, аналогичных описанным для стадий 88C~88F из Примера 88. МС (ESI): m/z = 464 [M+H]+.
Пример 91. N-(3-(4-амино-3-(4-(2-фторфенокси)фенил)-7-оксо-6,7-дигидро-1H-пирроло[2,3-d]пиридазин-1-ил)фенил)акриламид
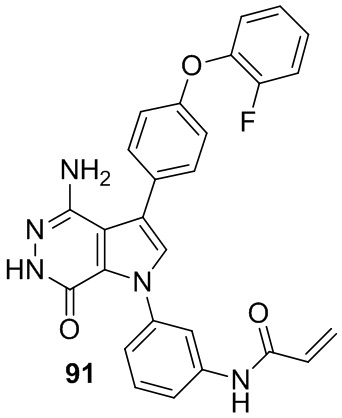
Указанное в заголовке соединение получают из 88d и (4-(2-фторфенокси)фенил)бороновой кислоты в условиях, аналогичных описанным для стадий 88D~88F из Примера 88. МС (ESI): m/z = 482 [M+H]+.
Пример 92. (E)-N-[3-[4-амино-3-[4-(2-фторфенокси)фенил]-7-оксо-6H-пирроло[2,3-d]пиридазин-1-ил]фенил]-4-(диметиламино)бут-2-енамид
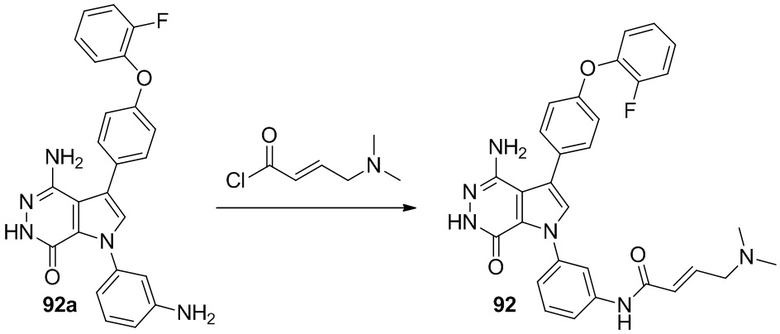
К раствору 92a (10мг) в DCM при 0°C добавляют недавно приготовленный (E)-4-(диметиламино)бут-2-еноилхлорид. Полученную смесь перемешивают при 0°C в течение 0.5 ч и очищают методом препаративной ЖХВР на обращенной фазе, чтобы получить указанное в заголовке соединение 92 (1 мг). МС (ESI): m/z = 539 [M+H] +.
Пример 93. (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
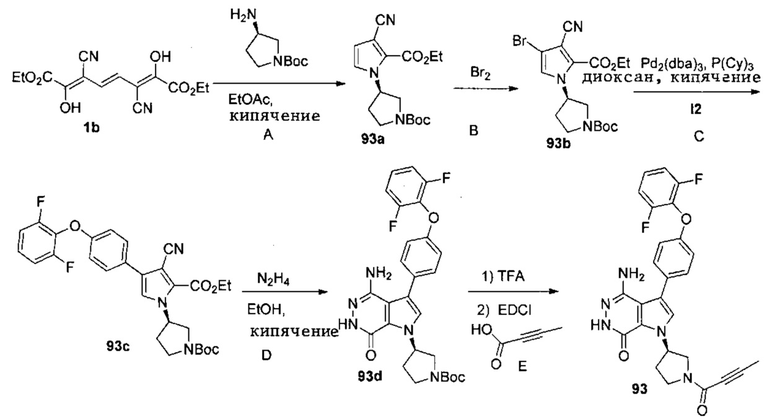
Стадия 93A
К раствору 1b (1.5 г) в этилацетате (84 мл) при 60°C добавляют по каплям раствор (R)-трет-бутилового эфира 3-аминопирролидин-1-карбоновой кислоты (1.41 г) в этилацетате (21 мл). Смесь кипятят 4 ч. После охлаждения до комнатной температуры растворители удаляются. Остаток очищается методом хроматографии на силикагеле, чтобы получить 93a (0.686 г).
Стадия 93B
К раствору 93a (0.686 г) в DCM (120 мл) при 0°C медленно добавляют раствор Br2 (3.7 г) в DCM (5 мл). Смесь перемешивают 1.5 ч, и затем реакцию прерывают, добавляя 10%-ный раствор Na2S2O3 и насыщенный раствор NaHCO3. Две фазы разделяют; водную фазу экстрагируют дихлорметаном. Объединённые органические экстракты обрабатывают избытком Boc2O, сушат над Na2SO4, фильтруют и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить 93b (0.342 г).
Стадия 93C
Смесь 93b (198 мг, 0.48 ммоль), I2 (160 мг, 0.48 ммоль) и K3PO4·3H2O (188 мг, 0.72 ммоль) в смеси 1.4-диоксан/вода (10 мл/1 мл) дегазируют азотом. Затем добавляют Pd2(dba)3 (22 мг, 0.024 ммоль) и трициклогексилфосфин (14 мг, 0.048 ммоль). Полученную смесь кипятят в атмосфере N2 в течение 16 ч. После охлаждения до комнатной температуры твёрдое вещество отфильтровывают, фильтрат концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить белое твёрдое вещество 93c (59 мг). МС (ESI): m/z=538 [M+H]+.
Стадия 93D
Смесь 93c (72 мг, 0.13 ммоль) и N2H4·H2O (1 мл) в этаноле (5 мл) кипятят 16 ч. После охлаждения до комнатной температуры растворители удаляются. Остаток очищается методом хроматографии на силикагеле, чтобы получить белое твёрдое вещество 93d (24 мг). МС (ESI): m/z=524 [M+H]+.
Стадия 93E
К раствору 93d (40 мг, 0.08 ммоль) в DCM (5 мл) добавляют TFA (1 мл). Смесь перемешивают при комнатной температуре 3 ч и концентрируют, чтобы получить масло 93e (49 мг). К раствору 93e (49 мг) в DCM (5 мл) добавляют бут-2-иновую кислоту (13 мг, 0.16 ммоль), EDCI (31 мг, 0.16 ммоль) и TEA (17 мг, 0.16 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 18 ч и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить указанное в заголовке соединение 93 (20 мг) в виде белого твёрдого вещества. МС (ESI): m/z = 490 [M+H] +.
Пример 94. (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2-хлор-6-фторфенокси)фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
Указанное в заголовке соединение получают из 93b и промежуточного соединения 9 в условиях, аналогичных описанным для стадий 93C~93E из Примера 93. МС (ESI): m/z = 506 [M+H] +.
Пример 95. (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2-фторфенокси)фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
Указанное в заголовке соединение получают из 93b и ((4-(2-фторфенокси)фенил)бороновой кислоты в условиях, аналогичных описанным для стадий 93C~93E из Примера 93. МС (ESI): m/z = 472 [M+H] +.
Пример 96. (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2-хлорфенокси)фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
Указанное в заголовке соединение получают из 93b и промежуточного соединения 10 в условиях, аналогичных описанным для стадий 93C~93E из Примера 93. МС (ESI): m/z = 488 [M+H] +.
Пример 97. (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-феноксифенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
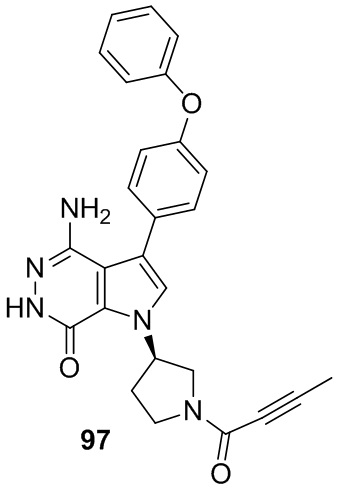
Указанное в заголовке соединение получают из 93b и (4-феноксифенил)бороновой кислоты в условиях, аналогичных описанным для стадий 93C~93E из Примера 93. МС (ESI): m/z = 454 [M+H] +.
Пример 98. 4-амино-1-((3S,4S)-1-(бут-2-иноил)-4-метоксипирролидин-3-ил)-3-(4-(2-фторфенокси)фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он и энантиомер
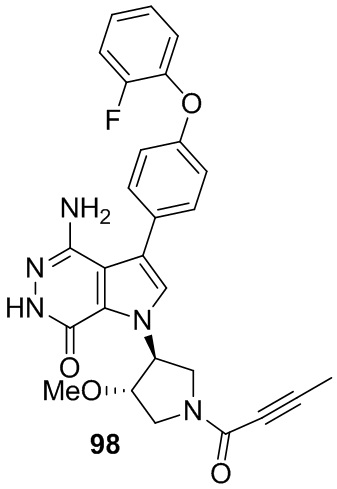
Указанное в заголовке соединение получают из транс-трет-бутилового эфира
3-амино-4-метоксипирролидин-1-карбоновой кислоты в условиях, аналогичных описанным в примере 1. МС (ESI): m/z=502 [M+H]+.
Пример 99. (R)-4-амино-1-(1-(4-(диметиламино)бут-2-иноил)пирролидин-3-ил)-3-(4-(2-фторфенокси)фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
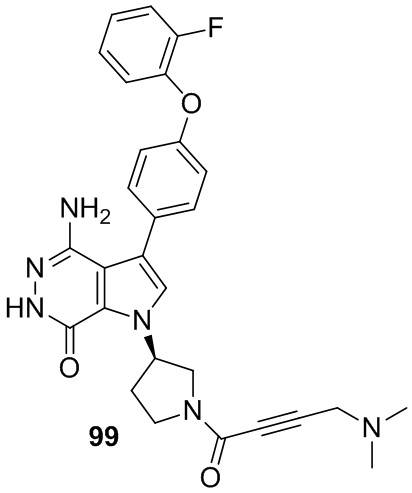
Указанное в заголовке соединение получают по аналогичным методикам, описанным в примере 93. МС (ESI): m/z=515 [M+H]+.
Пример 100. N-(3-(4-амино-3-(4-(2-фторфенокси)фенил)-7-оксо-6,7-дигидро-1H-пирроло[2,3-d]пиридазин-1-ил)циклопентил)бут-2-инамид
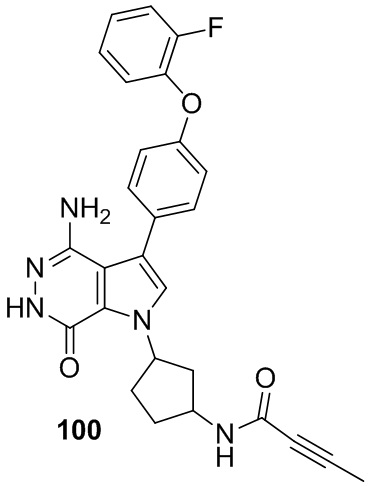
Указанное в заголовке соединение получают по аналогичным методикам, описанным в примере 93. МС (ESI): m/z=486 [M+H]+.
Пример 101. (S)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2-фторфенокси)фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
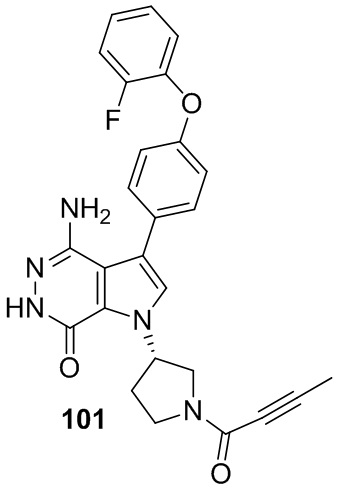
Указанное в заголовке соединение получают по аналогичным методикам, описанным в примере 93. МС (ESI): m/z=472 [M+H]+.
Пример 102. N-((1s,4s)-4-(4-амино-3-(4-(2-фторфенокси)фенил)-7-оксо-6,7-дигидро-1H-пирроло[2,3-d]пиридазин-1-ил)циклогексил)бут-2-инамид
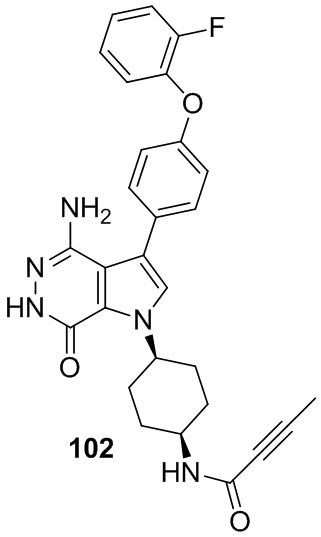
Указанное в заголовке соединение получают по аналогичным методикам, описанным в примере 1. МС (ESI): m/z=500 [M+H]+.
Пример 103. N-((1r,4r)-4-(4-амино-3-(4-(2-фторфенокси)фенил)-7-оксо-6,7-дигидро-1H-пирроло[2,3-d]пиридазин-1-ил)циклогексил)бут-2-инамид
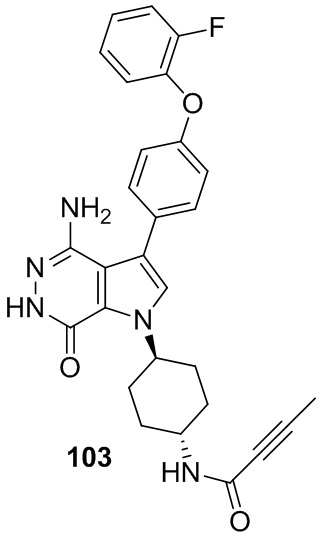
Указанное в заголовке соединение получают по аналогичным методикам, описанным в примере 1. МС (ESI): m/z=500 [M+H]+.
Пример 104. N-((1s,4s)-4-(4-амино-3-(4-(2-фторфенокси)фенил)-7-оксо-6,7-дигидро-1H-пирроло[2,3-d]пиридазин-1-ил)циклогексил)акриламид
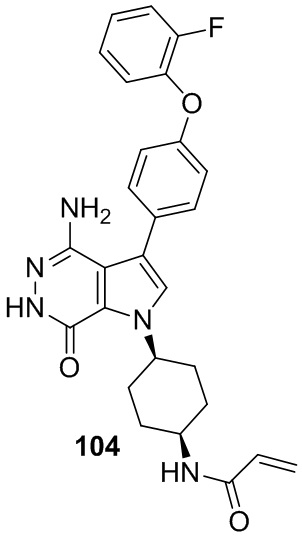
Указанное в заголовке соединение получают по аналогичным методикам, описанным в примере 1. МС (ESI): m/z=488 [M+H]+.
Пример 105. N-((1r,4r)-4-(4-амино-3-(4-(2-фторфенокси)фенил)-7-оксо-6,7-дигидро-1H-пирроло[2,3-d]пиридазин-1-ил)циклогексил)акриламид
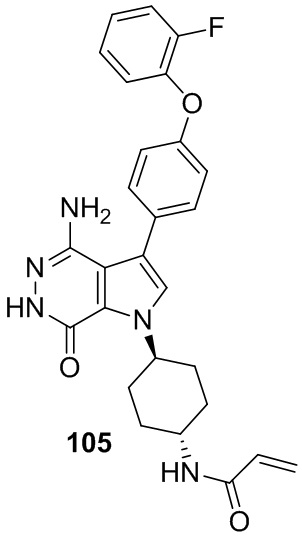
Указанное в заголовке соединение получают по аналогичным методикам, описанным в примере 1. МС (ESI): m/z=488 [M+H]+.
Пример 106. (R)-1-(1-акрилоилпирролидин-3-ил)-4-амино-3-(4-(2-фторфенокси)фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
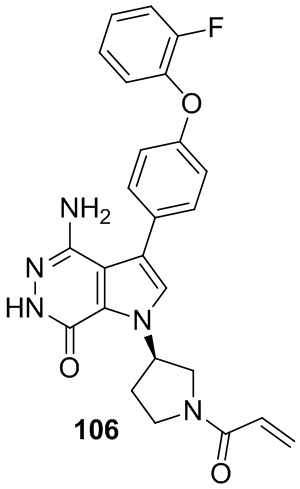
Указанное в заголовке соединение получают по аналогичным методикам, описанным в примере 1. МС (ESI): m/z=460 [M+H]+.
Пример 107. (R)-3-(4-амино-7-оксо-3-(4-феноксифенил)-6,7-дигидро-1H-пирроло[2,3-d]пиридазин-1-ил)пирролидин-1-карбонитрил
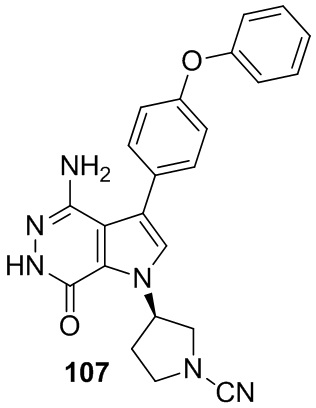
Указанное в заголовке соединение получают в условиях, аналогичных описанным в примере 20. МС (ESI): m/z=413 [M+H]+.
Пример 108. 3-(4-амино-3-(4-(2-фторфенокси)фенил)-7-оксо-6,7-дигидро-1H-пирроло[2,3-d]пиридазин-1-ил)пиперидин-1-карбонитрил
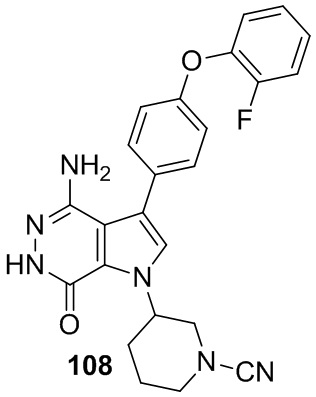
Указанное в заголовке соединение получают в условиях, аналогичных описанным в примере 20. МС (ESI): m/z=445 [M+H]+.
Пример 109. (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-3-(4-(2-фторфенокси)фенил)-1H-пиразол[3,4-d]пиридазин-7(6H)-он
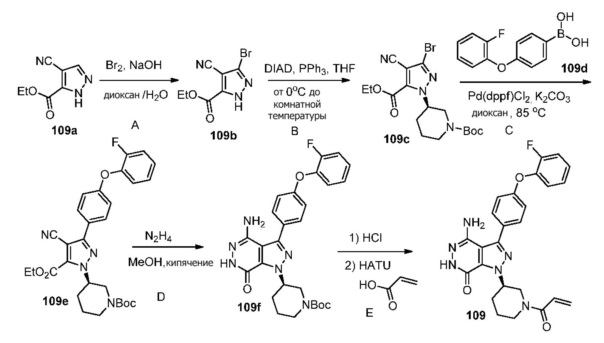
Стадия 109A
К смеси 109a (200 мг) и NaOH (2 M, 1.2 мл) в диоксане (4 мл) при 0°C добавляют раствор Br2 (380 мг) в диоксане (2 мл). Смесь перемешивают при комнатной температуре в течение 1 ч и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить 109b (340 мг).
Стадия 109B
К раствору 109b (50 мг), (S)-трет-бутилового эфира 3-гидроксипиперидин-1-карбоновой кислоты (80 мг) и трифенилфосфина (100 мг) в ТГФ (5 мл) при 0°C добавляют DIAD (80 мг). Смесь перемешивают при комнатной температуре 18 ч и концентрируют. Остаток очищается методом хроматографии на силикагеле, чтобы получить 109c (76 мг).
Стадия 109C
Смесь 109c (70 мг), 109d (114 мг), K2CO3 (113 мг), и Pd(dppf)Cl2 (66 мг) в смеси диоксан/вода (5 мл/0.5 мл) перемешивают в атмосфере N2 при 85°C в течение 3 ч. После охлаждения до комнатной температуры растворители удаляются. Остаток очищается методом хроматографии на силикагеле, чтобы получить 109e (100 мг). МС (ESI): m/z=535 [M+H]+.
Стадия 109D
Смесь 109e (100 мг) и N2H4 (2.5 мл) в метаноле (5 мл) кипятят 3 ч. После охлаждения до комнатной температуры растворители удаляются. Остаток очищается методом хроматографии на силикагеле, чтобы получить 109f (70 мг). МС (ESI): m/z=521 [M+H]+.
Стадия 109E
Смесь 109f (70 мг) и 4M раствора HCl (4 мл, в диоксане) перемешивают при комнатной температуре в течение 0.5 ч и концентрируют, чтобы получить соединение 109g (100 мг), которое используется непосредственно на следующей стадии.
К раствору 109g (9 мг) в ДМФ (1 мл) добавляют DIEA (13 мг), кислоту (6 мг) и HATU (18 мг). Полученную смесь перемешивают при комнатной температуре в течение 0.5 ч и очищают методом препаративной ЖХВР на обращенной фазе, чтобы получить указанное в заголовке соединение 109 (3.1 мг). МС (ESI): m/z = 475 [M+H].
Пример 110. (R)-1-(1-акрилоилпиперидин-3-ил)-4-амино-3-(4-феноксифенил)-1H-пиразол[3,4-d]пиридазин-7(6H)-он
Указанное в заголовке соединение получают из 109c и (4-феноксифенил)бороновой кислоты в условиях, аналогичных описанным для стадий 109C~109E из Примера 109. МС (ESI): m/z = 457 [M+H]+.
Пример 111. (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-феноксифенил)-1H-пиразол[3,4-d]пиридазин-7(6H)-он
Указанное в заголовке соединение получают из 109b и соответствующих реагентов в условиях, аналогичных описанным для стадий 109B~109E из Примера 109. МС (ESI): m/z = 455 [M+H]+.
Пример 112. (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2-фторфенокси)фенил)-1H-пиразол[3,4-d]пиридазин-7(6H)-он
Указанное в заголовке соединение получают из 109b и соответствующих реагентов в условиях, аналогичных описанным для стадий 109B~109E из Примера 109. МС (ESI): m/z = 473 [M+H]+.
Пример 113. (R)-4-амино-1-(1-(бут-2-иноил)пирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1H-пиразол[3,4-d]пиридазин-7(6H)-он
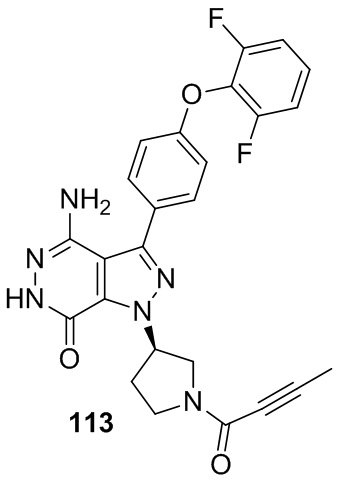
Указанное в заголовке соединение получают из 109b и соответствующих реагентов в условиях, аналогичных описанным для стадий 109B~109E из Примера 109. МС (ESI): m/z = 491 [M+H]+.
Пример 114. (S)-1-(1-акрилоилпиперидин-3-ил)-4-амино-3-(4-(2,6-дифторфенокси)фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
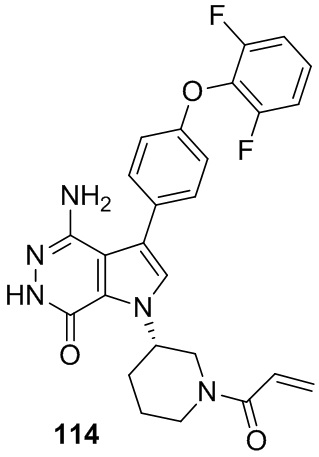
Указанное в заголовке соединение получают в условиях, аналогичных описанным в примере 58. МС (ESI): m/z = 492 [M+H]+.
Пример 115. (R)-4-амино-3-(4-(2,6-дифторфенокси)фенил)-1-(1-пропионилпиперидин-3-ил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
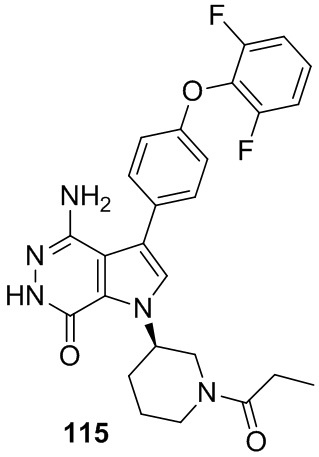
Указанное в заголовке соединение получают из соединения 58 в условиях, аналогичных описанным в примере 19. МС (ESI): m/z = 494 [M+H]+.
Пример 116. 4-амино-3-[4-(2,6-дифторфенокси)фенил]-1-[(3R)-1-[(E)-бут-2-еноил]-3-пиперидил]-6H-пирроло[2,3-d]пиридазин-7-он
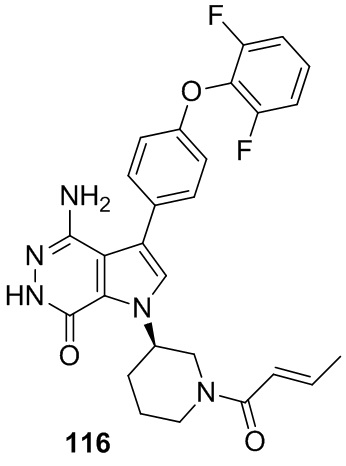
Указанное в заголовке соединение получают из (E)-бут-2-еновой кислоты и амина 58c в условиях, аналогичных описанным для стадии 58C из Примера 58. МС (ESI): m/z = 506 [M+H]+.
Пример 117. (R)-4-амино-1-(1-(бут-2-иноил)пиперидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
Указанное в заголовке соединение получают из бут-2-иновой кислоты и амина 58c в условиях, аналогичных описанным для стадии 58C из Примера 58. МС (ESI):
m/z = 504 [M+H]+.
Пример 118. (R)-4-амино-1-(1-бутирилпирролидин-3-ил)-3-(4-(2,6-дифторфенокси)фенил)-1H-пирроло[2,3-d]пиридазин-7(6H)-он
Указанное в заголовке соединение получают из соединения 93 в условиях, аналогичных описанным в примере 19. МС (ESI): m/z = 494 [M+H]+.
Оценка активности Btk
Методики анализов биохимического и на клеточном уровне:
Анализ Btk киназы – Анализ Btk киназы осуществляется с использованием набора реактивов ADP-Glo Btk киназы, поставляемого фирмой Promega (Madison, WI). Анализ осуществляется согласно протоколам, прилагаемым с набором реактивов. Вкратце проводится ферментативная реакция в реакционном буферном растворе киназы, содержащем Btk (2 нг/мкл), ATP (1.2 µM), полипептид GT (0.3 µM), Дитиотреитол (40 нM), MnCl2 (1.4 мM), и буферный раствор 1×киназ (включен в набор) в присутствии или в отсутствие контрольных изделий при различных концентрациях в планшете с 384-лунками, при комнатной температуре (22 ± 1°C), 60 минут. Окончательный объем реакционной смеси для каждой реакции составляет 10 мкл. Затем в реакционную смесь добавляют 4 мкл реагента ADP-Glo (включен в набор), и планшет дополнительно выдерживают в течение 40 мин, чтобы завершить реакцию и исчерпать оставшийся аденозинтрифосфат (ATФ). Окончательно добавляют в реакционную смесь 10 мкл реагента, обнаруживающего киназу, чтобы одновременно превратить ADP в ATФ и обеспечить измерение вновь синтезированного ATФ с помощью люминометра для прочтения планшетов (мультимаркерный ридер Victor X5 2030, фирмы PerkinElmer). Величины ингибирующей (на 50%) концентрации, IC50, рассчитывали, используя соответствующие программы в пакете GraphPad Prism, из зависимости процента ингибирования от логарифма концентрации, по сравнению с контрольным носителем (ДМСО). Величины IC50 для соединений примеров показаны в Таблице 4.
Анализ клеточной пролиферации. Клетки TMD-8 и SU-DHL-1 выдерживают при 37°C в увлажненной атмосфере с 5% CO2 в рекомендованной среде и концентрации сыворотки. Для анализа клеточной пролиферации клетки высевают при плотности от 5000 до 10000 клеток на лунку в 96-луночные планшеты и культивируют в течение ночи при 37°C в рекомендованной среде с добавкой 5-10% телячьей сыворотки крови. На следующий день в клеточную культуру добавляют контрольные изделия при различной концентрации или контрольный носитель (0.5% ДМСО). Через 5 суток после обработки оценивают рост клеток с помощью CellTiter-Glo® Luminestceaent Cell Viability Assay (фирма Promega). Величины IC50 рассчитывали, используя пакет GraphPad Prism, из зависимости процента ингибирования от логарифма концентрации, по сравнению с контрольным носителем. Величины IC50 для соединений примеров показаны в Таблице 4.
Таблица 4. Результаты биологических испытаний
A ≤0.01 мкM; 0.01 мкM < B ≤1 мкM; 1 мкM < C < 100 мкM
Фармакокинетические испытания. Испытуемые изделия вводили крысам Sprague-Dawley или собакам Бигль внутривенно (В/В) и перорально (П/О). Образцы плазмы были приготовлены из проб крови, взятых в различные моменты времени. Концентрацию плазмы для испытуемых изделий определяли специальными аналитическими методами ЖХ-MС/МС. Фармакокинетические параметры рассчитывали с помощью пакета WinNonlin®. Результаты фармакокинетических (ФК) испытаний для соединений примеров показаны ниже в Таблице 5 (крысы) и Таблице 6 (собаки).
Таблица 5. ФК параметры соединений выбранных примеров на крысах
ППК*- площадь под кривой.
Таблица 6. ФК параметры соединений выбранных примеров на собаках
Исследование эффективности in vivo. Противоопухолевую активность in vivo оценивали на модели ксенотрансплантата TMD-8. Вкратце TMD-8 клетки имплантировали NOD-SCID голым мышам, и давали им вырасти до заданного размера (приблизительно 100-200 мм3) до лечения. Испытуемые изделия вводили перорально при различных уровнях дозы, ежедневно (QD) или дважды в день (BID) в течение 14 последовательных дней. В ходе эксперимента измеряли массы опухоли и тела, причем объем опухоли оценивали по формуле [длина/2] x [ширина2]. Установившаяся опухоль у каждого животного была индивидуально нормализована к ее размеру в начале эксперимента, и данные рассчитывали как изменение объема опухоли относительно объема на день 0 эксперимента, с использованием формулы относительного объема опухоли (ООО), ООО = TVx/TV0, где TVx означает объем опухоли в любой день и TV0 – это объем опухоли в начале дозирования изделия. Отмечено значительное подавление роста опухоли для соединений примера 58 (степень ингибирования роста опухоли 98%) и 93 (степень ингибирования роста опухоли 80%).
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ПИРРОЛО[2,3-D]ПИРИДАЗИН-4-ОНЫ И ПИРАЗОЛО[3,4-D]ПИРИДАЗИН-4-ОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2017 |
|
RU2749038C2 |
| ИНГИБИТОРЫ БРОМДОМЕНА | 2012 |
|
RU2671571C1 |
| ИНГИБИТОРЫ БРОМДОМЕНА | 2012 |
|
RU2647592C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНОНА И ПИРИДАЗИНОНА | 2012 |
|
RU2632915C2 |
| НОВЫЕ АЛКИЛИРУЮЩИЕ СРЕДСТВА | 2013 |
|
RU2632206C2 |
| ПРОИЗВОДНЫЕ БОРОНОВОЙ КИСЛОТЫ | 2015 |
|
RU2717558C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛОПИРИДАЗИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ФОСФОДИЭСТЕРАЗЫ IV, И/ИЛИ ПРОДУЦИРОВАНИЯ TNF | 2003 |
|
RU2328499C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ И ОЧИСТКИ ГЕТЕРОАРИЛЬНЫХ СОЕДИНЕНИЙ | 2010 |
|
RU2557242C2 |
| КОНДЕНСИРОВАННОЕ ХИНОЛИНОВОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2005 |
|
RU2384571C2 |
| СОЕДИНЕНИЯ ПУРИНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2013 |
|
RU2655388C2 |
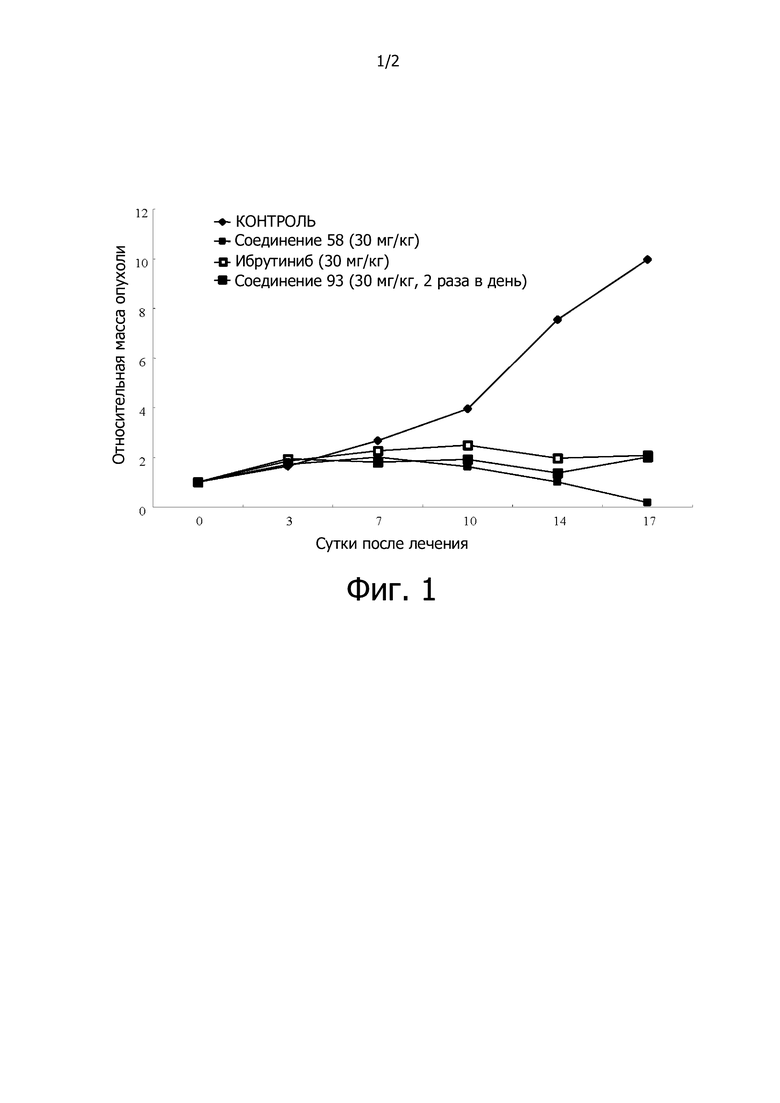
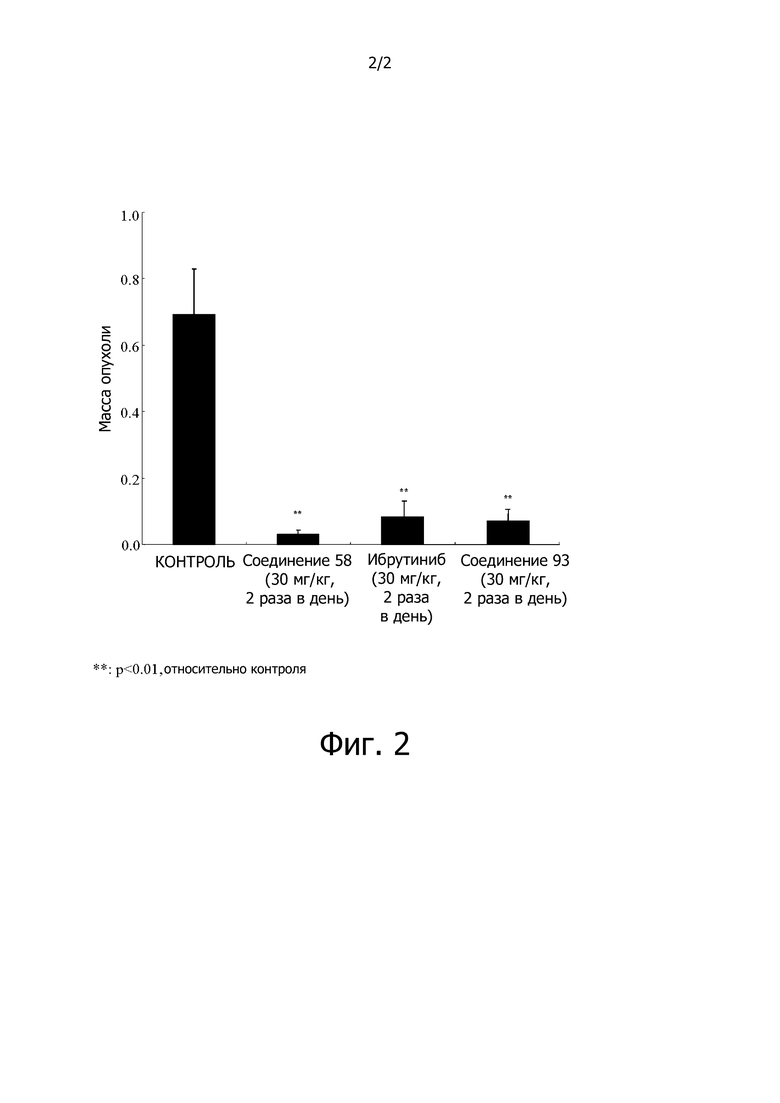
Настоящее изобретение относится к соединению общей формулы (I) или его фармацевтически приемлемой соли
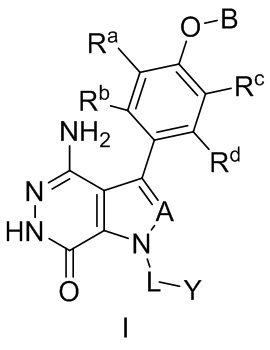 , где A, Ra, Rb, Rc, Rd, B, L, Y имеют значения, указанные в п.1 формулы изобретения. Также раскрыты фармацевтическая композиция, обладающая ингибирующей активностью в отношении протеиновой тирозинкиназы, содержащая указанное выше соединение, способ модулирования активности протеиновой тирозинкиназы и способ лечения состояния или заболевания, передаваемого белковой тирозинкиназой, с использованием указанного выше соединения. Технический результат – получение новых соединений, которые проявляют сильное ингибирующее действие против тирозинкиназы Брутона, и, следовательно, могут обеспечить потенциальную тактику лечения карциномы человека, включая лимфому B-клеток, и аутоиммунные заболевания, такие как ревматоидный артрит, системная красная волчанка и множественный склероз. 5 н. и 34 з.п. ф-лы, 2 ил., 6 табл., 118 пр.
, где A, Ra, Rb, Rc, Rd, B, L, Y имеют значения, указанные в п.1 формулы изобретения. Также раскрыты фармацевтическая композиция, обладающая ингибирующей активностью в отношении протеиновой тирозинкиназы, содержащая указанное выше соединение, способ модулирования активности протеиновой тирозинкиназы и способ лечения состояния или заболевания, передаваемого белковой тирозинкиназой, с использованием указанного выше соединения. Технический результат – получение новых соединений, которые проявляют сильное ингибирующее действие против тирозинкиназы Брутона, и, следовательно, могут обеспечить потенциальную тактику лечения карциномы человека, включая лимфому B-клеток, и аутоиммунные заболевания, такие как ревматоидный артрит, системная красная волчанка и множественный склероз. 5 н. и 34 з.п. ф-лы, 2 ил., 6 табл., 118 пр.
1. Соединение формулы (I) или его фармацевтически приемлемая соль
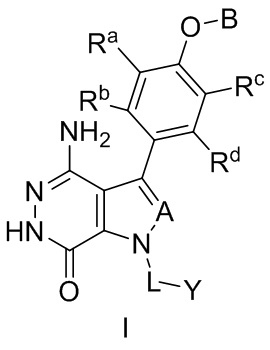
в котором A выбирают из группы, состоящей из CR0 и N; и в котором R0 выбирают из группы, состоящей из водорода и галогена;
Ra, Rb, Rc и Rd независимо выбирают из группы, состоящей из водорода, галогена, C1-C6 алкоксила;
B выбирают из группы, состоящей из C1-C6 алкила, незамещенного арила и арила, замещенного по меньшей мере, одним заместителем, который выбирают из группы, состоящей из галогена, и C1-C6 алкила;
L представляет собой C1-C6 алкилен или L отсутствует; и
Y выбирают из группы, состоящей из C3-C8 циклоалкила, гетероциклоалкила и арила, где Y или не замещен, или замещен, по меньшей мере, одним заместителем, который выбирают из группы, состоящей из галогена, -CN, -C(O)R11, и -NHC(O)R12; и где R11 и R12 независимо выбирают из группы, состоящей из C1-C6 алкила, C3-C8 циклоалкила, C2-C6 алкенила, C3-C8 циклоалкенила, и C2-C6 алкинила, и где гетероциклоалкил означает 3- - 12-членный цикл, содержащий один или несколько гетероатомов, выбранных из азота, и
где арил означает 6- - 12-членный цикл.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, в котором A выбирают из группы, состоящей из CH, CF, CCl и N.
3. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, в котором, по меньшей мере, один из Ra, Rb, Rc и Rd выбирают из группы, состоящей из водорода, F, Cl и метоксила.
4. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, в котором Ra, Rb, Rc и Rd означают водород.
5. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, в котором B представляет собой незамещенный C1-C6 алкил.
6. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, в котором B представляет собой незамещенный или замещенный арил.
7. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, в котором B представляет собой незамещенный или замещенный фенил.
8. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, в котором B означает фенил, замещенный, по меньшей мере, одним фрагментом, который выбирают из группы, состоящей из F и Cl.
9. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, в котором B является фенилом, замещенным двумя атомами F.
10. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, в котором B является фенилом, замещенным двумя атомами Cl.
11. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, в котором B является фенилом, замещенным одним атомом Cl и одним F.
12. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, в котором L отсутствует.
13. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, в котором L представляет собой метилен.
14. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, в котором Y выбирают из группы, состоящей из незамещенного или замещенного пиперидинила, незамещенного или замещенного фенила, незамещенного или замещенного бицикло[3.2.1]октанила, незамещенного или замещенного азетидинила, и незамещенного или замещенного пирролидинила.
15. Соединение по п. 14 или его фармацевтически приемлемая соль, в котором группа Y замещена, по меньшей мере, одним фрагментом, выбранным из группы, состоящей из F, CN,-C(O)CH=CH2,
-C(O)CH=CHCH2N(CH3)2, -NHC(O)CH=CH2, -C(O)CH=CHCH2N(CH3)(COOC(CH3)3;
-C(O)CH=CHCH2NH(CH3), -C(O)CH2CH3, -C(O)CH2CH2CH3,  ,
,
 ,
,  , -C(O)CH2CN, ,
, -C(O)CH2CN, ,  ,
,  ,
,  ,
, 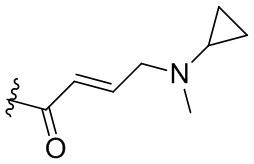 ,
, 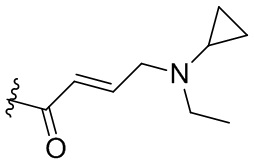 ,
, 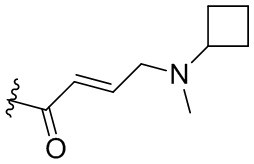 ,
, 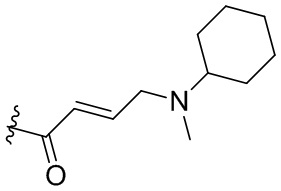 ,
,  ,
,  ,
,  ,
,  ,
, 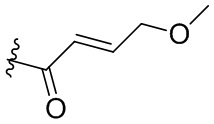 ,
, 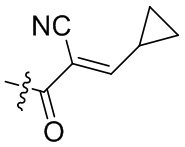 ,
, 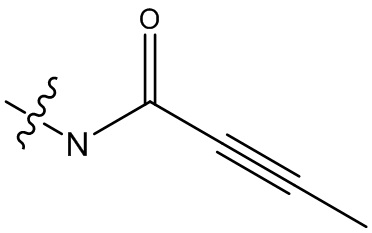 и
и 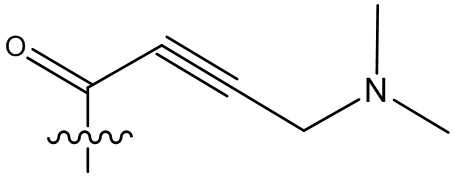 .
.
16. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, которое имеет содержание R-формы, больше чем S-формы.
17. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, которое имеет содержание S-формы, больше чем R-формы.
18. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, где соединение имеет формулу (II):
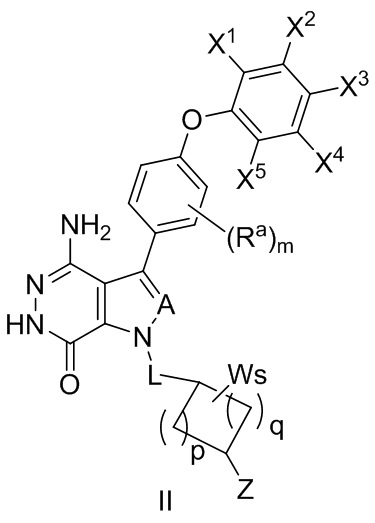
в которой X1, X2, X3, X4, и X5 независимо выбирают из группы, состоящей из водорода, галогена, C1-C6 алкила;
W выбирают из группы, состоящей из галогена и C1-C6 алкила; в котором два заместителя W могут комбинироваться с атомом или атомами, к которым они присоединены, с образованием C3-12 циклоалкила;
m = 0, 1, 2, или 3;
p = 1, 2, или 3;
q = 0, 1, или 2;
s = 0, 1, 2, или 3; и
Z выбирают из группы, состоящей из -NHC(O)R12; и в котором R12 независимо выбирают из группы, состоящей из C1-C6 алкила, C2-C6 алкенила и C1-C6 алкинила.
19. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, в котором соединение имеет формулу (III):
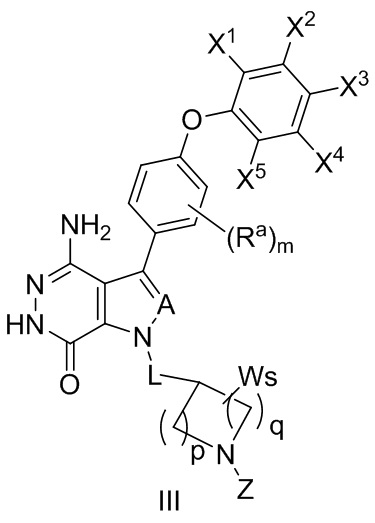
в которой X1, X2, X3, X4, и X5 независимо выбирают из группы, состоящей из водорода, галогена, C1-C6 алкила;
W выбирают из группы, состоящей из галогена, C1-C6 алкила, и C1-C6 алкоксила; в котором два заместителя W могут комбинироваться с атомом или атомами, к которым они присоединены, с образованием 3- - 12-членного гетероцикла, содержащего один или несколько гетероатомов, выбранных из азота;
m = 0, 1, 2, или 3;
p = 1, 2, или 3;
q = 0, 1, или 2;
s = 0, 1, 2, или 3; и
Z выбирают из группы, состоящей из CN и -C(O)R11; и в котором R11 независимо выбирают из группы, состоящей из C1-C6 алкила, C3-C8 циклоалкила, C2-C6 алкенила, и C2-C6 алкинила.
20. Соединение по п. 19 или его фармацевтически приемлемая соль, в котором Z выбирают из группы, состоящей из CN,-C(O)CH=CH2, -C(O)CH=CHCH2N(CH3)2, -C(O)CH=CHCH2N(CH3)(COOC(CH3)3;
-C(O)CH=CHCH2NH(CH3), -C(O)CH2CH3, -C(O)CH2CH2CH3, ,
, , -C(O)CH2CN, , , , , , , , , , , , , , и .
21. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, где соединение имеет формулу (IV):
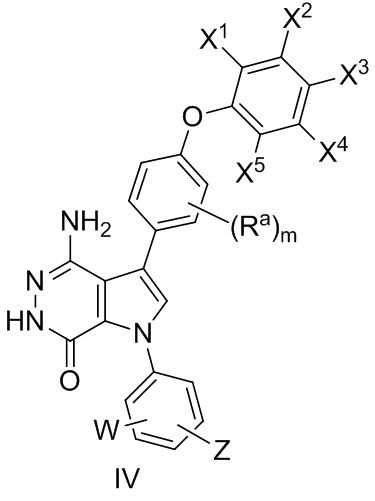
в которой X1, X2, X3, X4, и X5 независимо выбирают из группы, состоящей из водорода, галогена, C1-C6 алкила;
W представляет собой водород;
m = 0, 1, 2, или 3; и
Z выбирают из группы, состоящей из -NHC(O)R12; и в котором R12 независимо выбирают из группы, состоящей из C2-C6 алкенила.
22. Соединение по п. 1 или 2 или его фармацевтически приемлемая соль, где соединение имеет формулу (V):
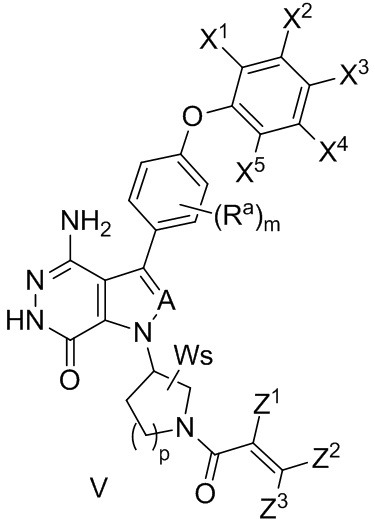
в котором X1, X2, X3, X4, и X5 независимо выбирают из группы, состоящей из водорода, галогена, C1-C6 алкила;
W выбирают из группы, состоящей из галогена и C1-C6 алкоксила;
m = 0, 1, 2, или 3;
p = 1 или 2;
s = 0, 1, 2, или 3;
Z1 выбирают из группы, состоящей из водорода, галогена и циано; и
Z2 и Z3 независимо выбирают из группы, состоящей из водорода, -CH2OR15, -CH2NR16R17;
в котором R15 и R16 независимо выбирают из группы, состоящей из водорода, C1-C6 алкила и C3-C8 циклоалкила; R17 выбирают из группы, состоящей из C1-C6 алкила, C3-C8 циклоалкила, -C(O)OR19; где R19 независимо выбирают из группы, состоящей из C1-C6 алкила;
в котором R16 и R17 объединяются с атомом N, к которому они присоединены, с образованием 3- - 12-членного гетероцикла, содержащего один или несколько гетероатомов, независимо выбранных из азота и кислорода; и
в котором Z1 и Z2 могут объединяются вместе с образованием связи.
23. Соединение по п. 22 или его фармацевтически приемлемая соль, в котором A означает N.
24. Соединение по п. 22 или его фармацевтически приемлемая соль, в котором A выбирают из группы, состоящей из CH, CF и CCl.
25. Соединение по п. 22 или его фармацевтически приемлемая соль, в котором Z1, Z2 и Z3 представляют собой Н.
26. Соединение по п. 22 или его фармацевтически приемлемая соль, в котором Z1 и Z3 представляют собой H, Z2 означает -CH2NR16R17.
27. Соединение по п. 22 или его фармацевтически приемлемая соль, в котором Z1 и Z2 объединяются вместе с образованием связи.
28. Соединение по п. 22 или его фармацевтически приемлемая соль, в котором 3 или меньше, чем три из X1, X2, X3, X4, и X5 представляют собой галоген.
29. Соединение по п. 22 или его фармацевтически приемлемая соль, в котором X1 означает F.
30. Соединение по п. 22 или его фармацевтически приемлемая соль, в котором X2, X3, и X4 означают водород.
31. Соединение по п. 22 или его фармацевтически приемлемая соль, в котором X5 выбирают из группы, состоящей из H, F и Cl.
32. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении протеиновой тирозинкиназы, содержащая эффективное количество соединения по п. 1 или 2, и фармацевтически приемлемый носитель.
33. Способ модулирования активности протеиновой тирозинкиназы, включающий контактирование клетки с эффективным количеством соединения по п. 1 или 2 или его фармацевтически приемлемой соли.
34. Способ лечения состояния или заболевания, передаваемого белковой тирозинкиназой, включающий введение субъекту терапевтически эффективного количества соединения по п. 1 или 2 или его фармацевтически приемлемой соли.
35. Способ по п. 34, в котором состояние или заболевание представляет собой рак или аутоиммунные заболевания.
36. Способ по п. 35, в котором рак представляет собой злокачественные развития В-клеток, которые выбраны из группы, состоящей из хронической лимфоцитарной лейкемии (CLL), мантийноклеточной лимфомы (MCL), диффузной лимфомы больших B-клеток (DLBCL), множественной миеломы (MM), макрофолликулярной лимфомы (FL), лимфомы пограничной зоны, и макроглобулинемии Вальденстрёма (WM).
37. Соединение или его фармацевтически приемлемая соль, в котором соединение выбирают из группы, состоящей из:
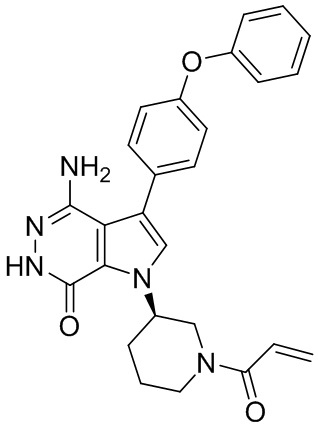 ;
; 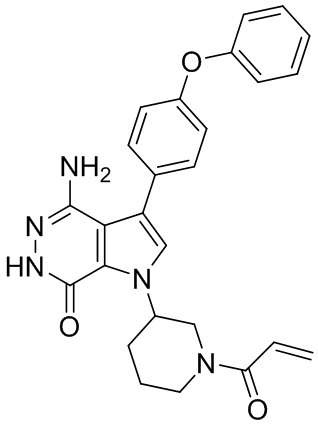 ;
; 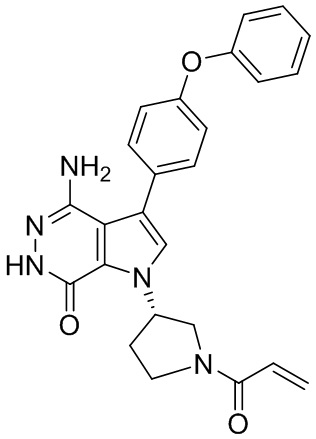 ;
;
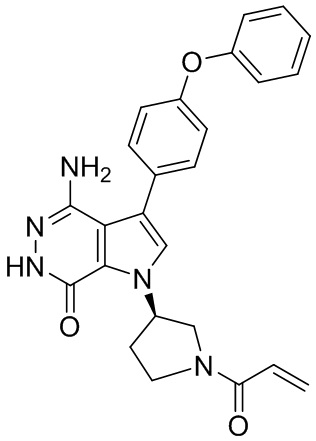 ;
; 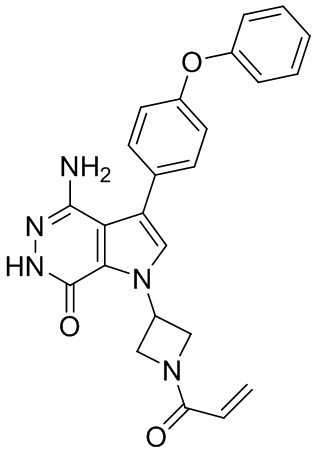 ;
; 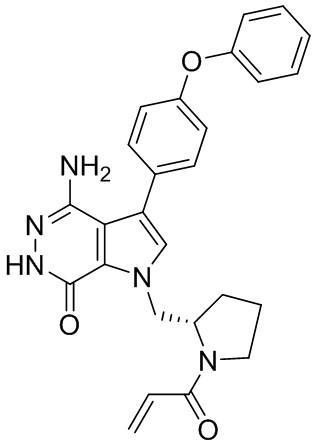 ;
;
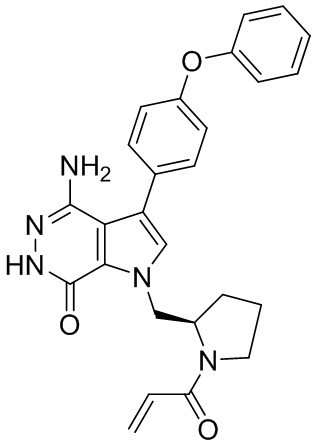 ;
;  ;
; 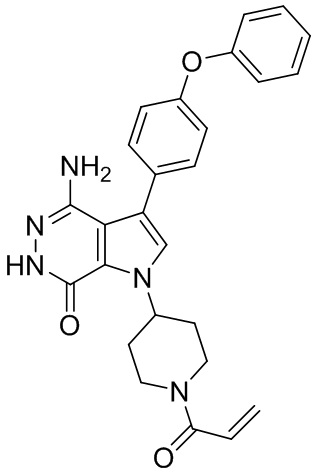 ;
;
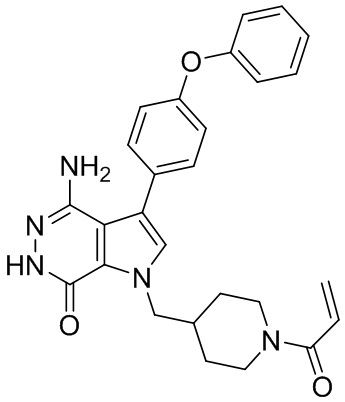 ;
; 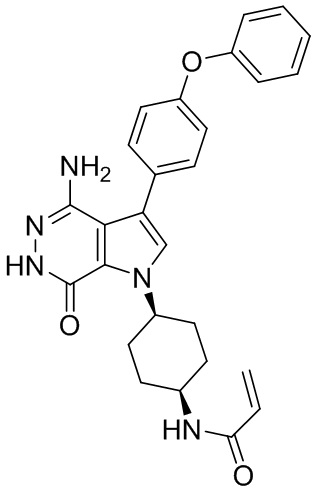 ;
; 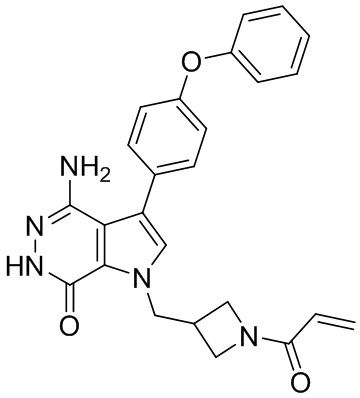 ;
;
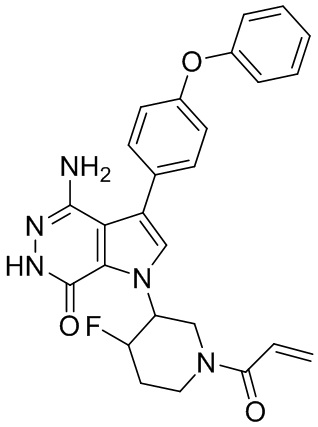 ;
; 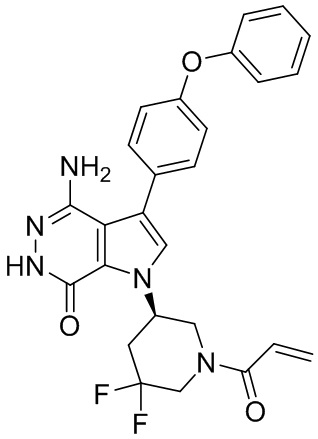 ;
; 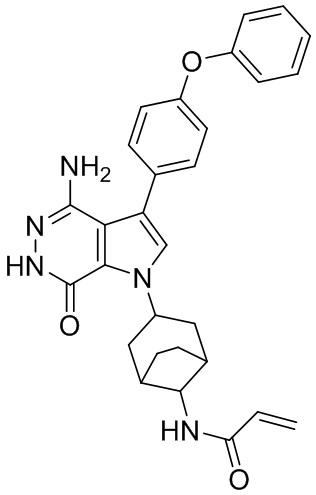 ;
;
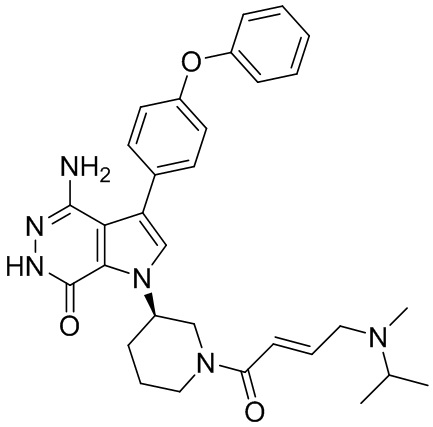 ;
; 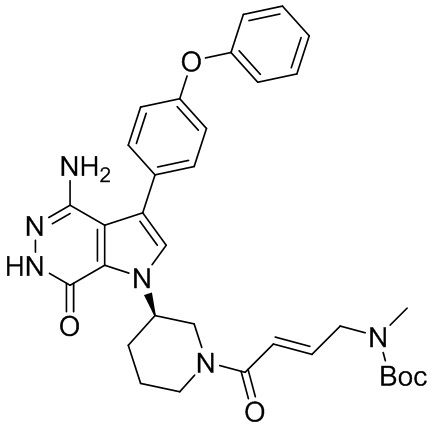 ;
; 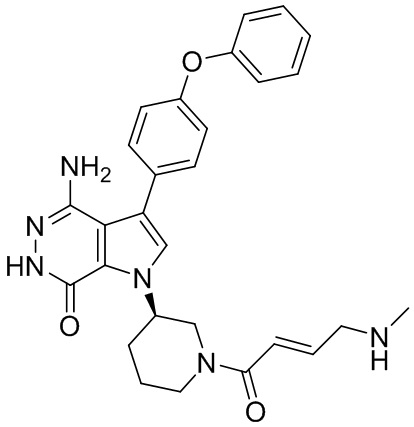 ;
; 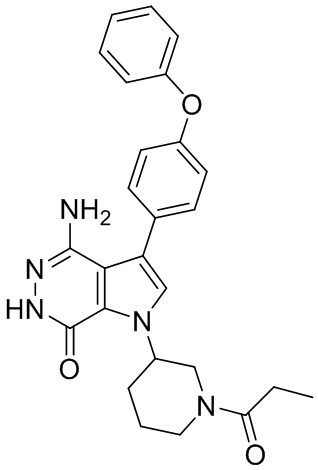 ;
; 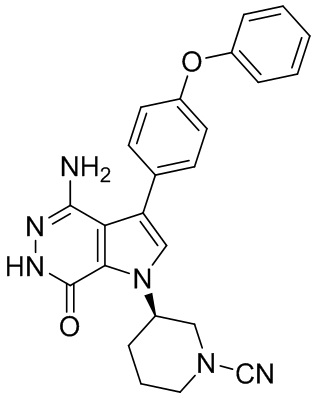 ;
;
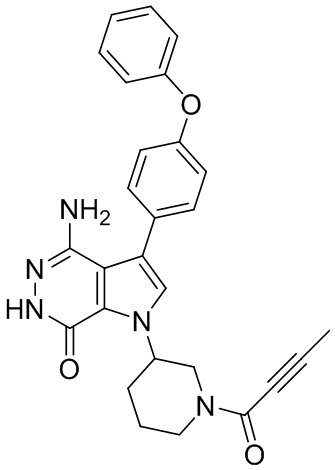 ;
; 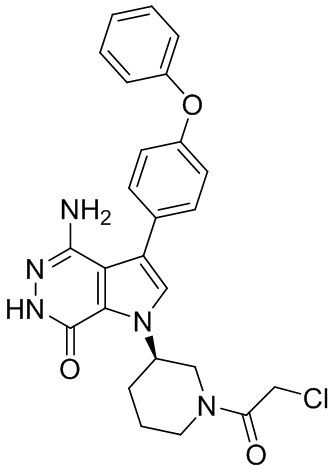 ;
; 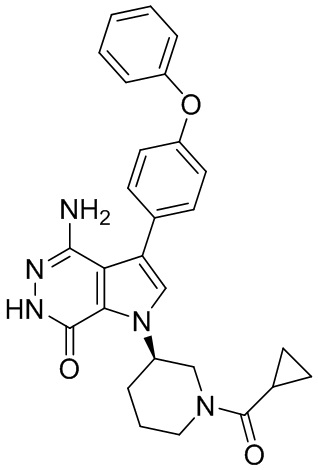 ;
;
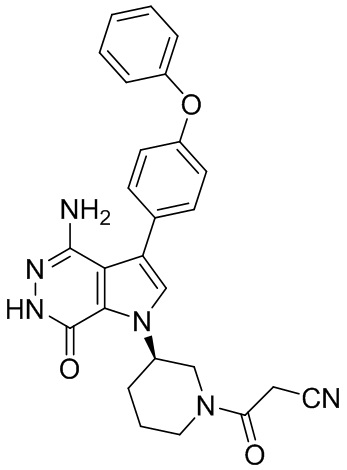 ;
; 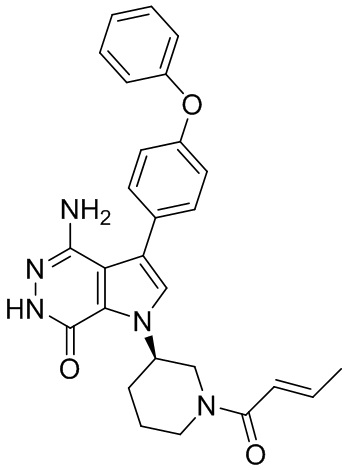 ;
; 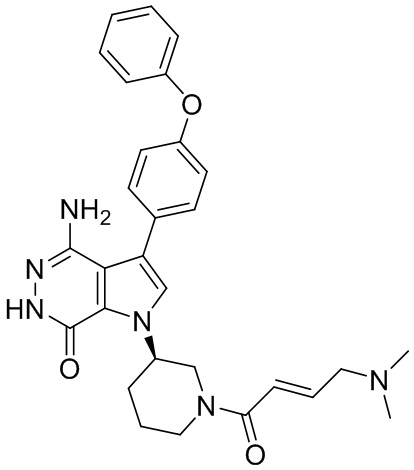 ;
;
 ;
;  ;
; 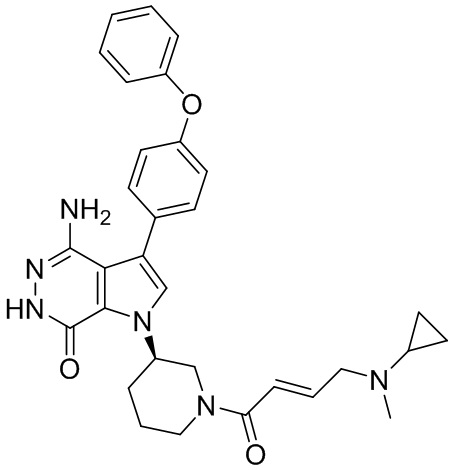 ;
; 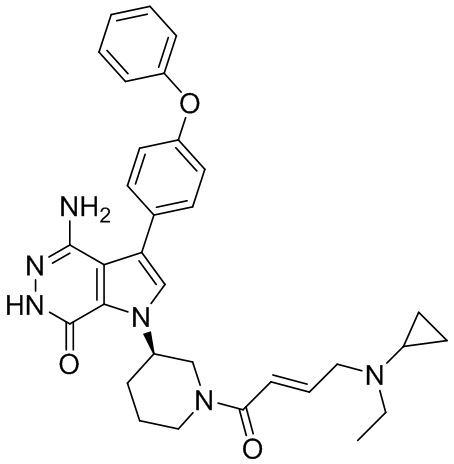 ;
; 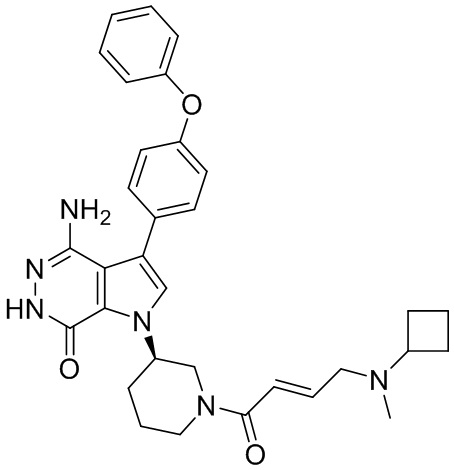 ;
; 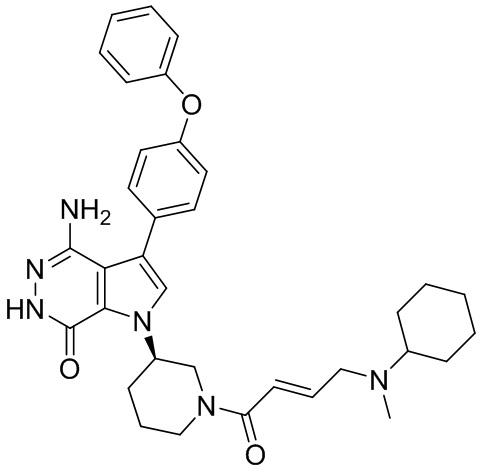 ;
;
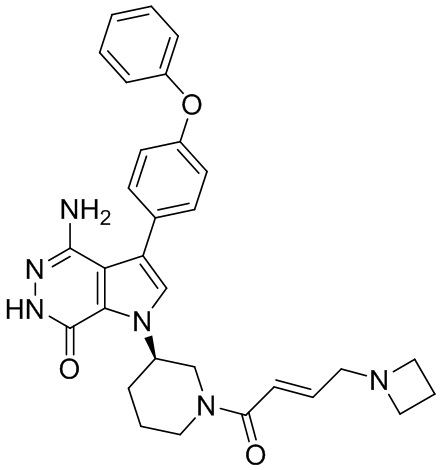 ;
; 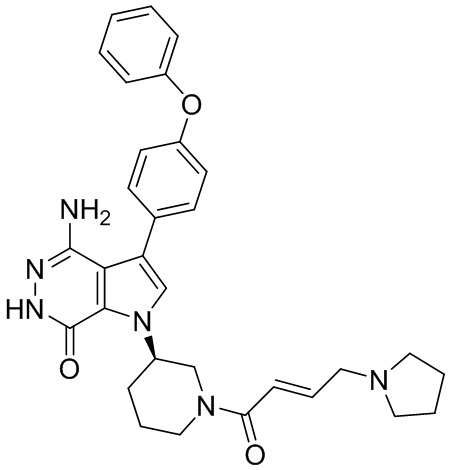 ;
;
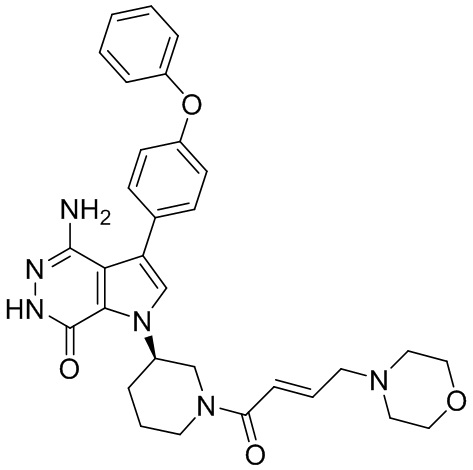 ;
; 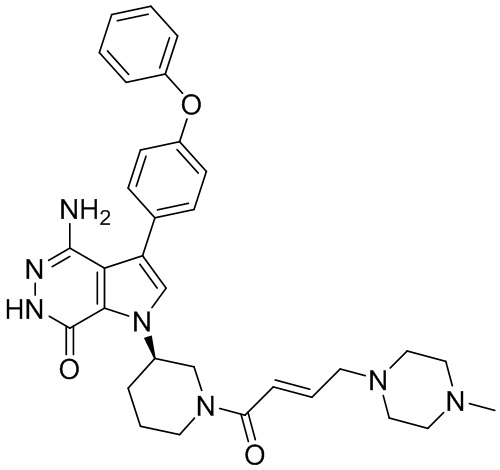 ;
;
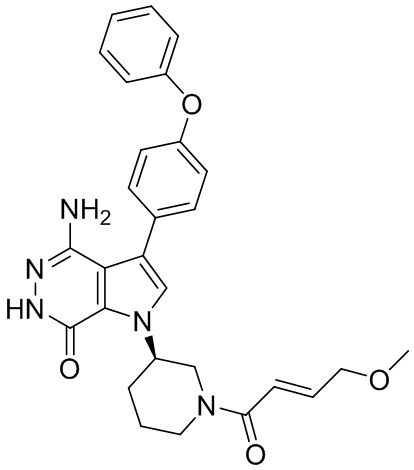 ;
; 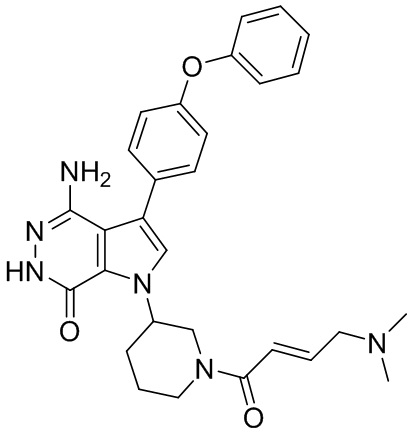 ;
;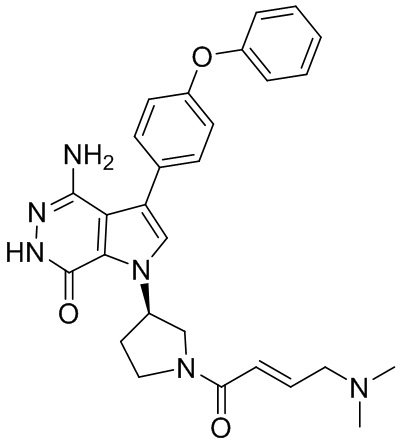 ;
;  ;
; 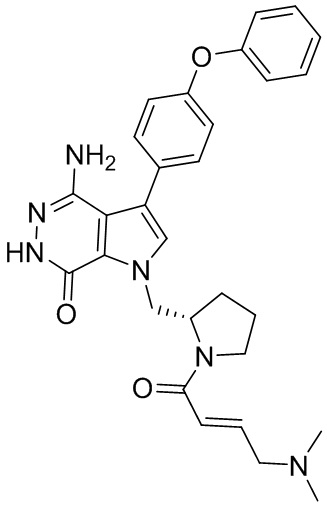 ;
;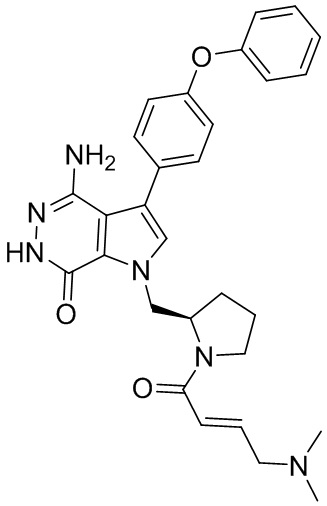 ;
; 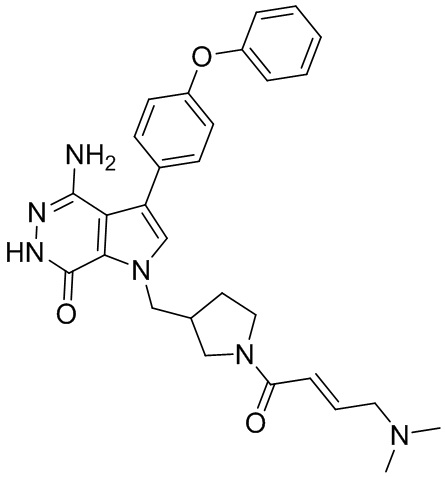 ;
; 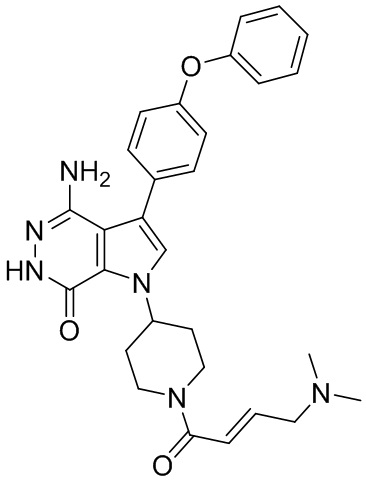 ;
;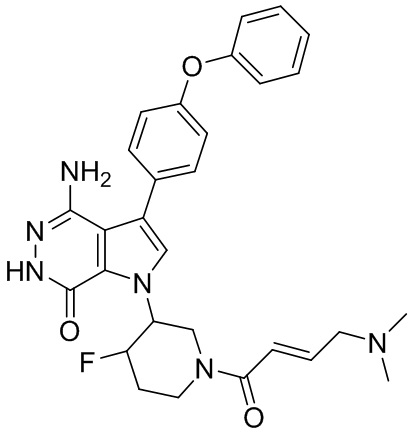 ;
; 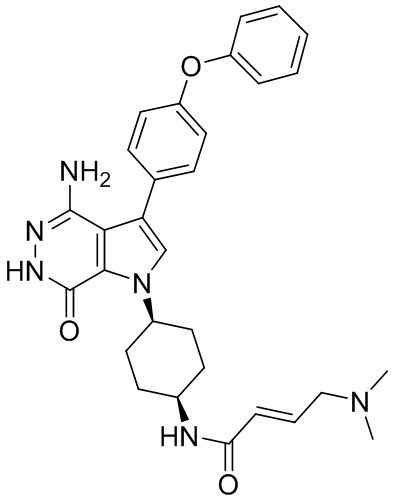 ;
;  ;
; 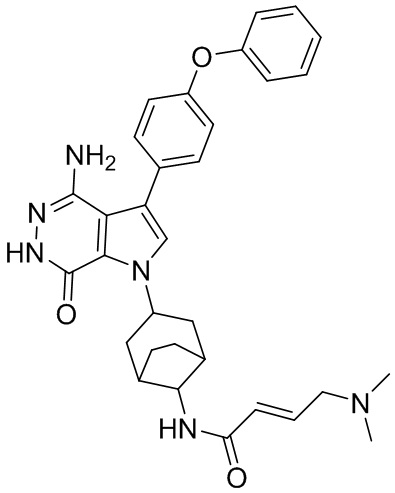 ;
; 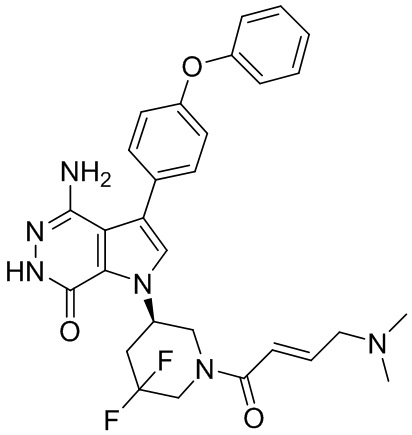 ;
; 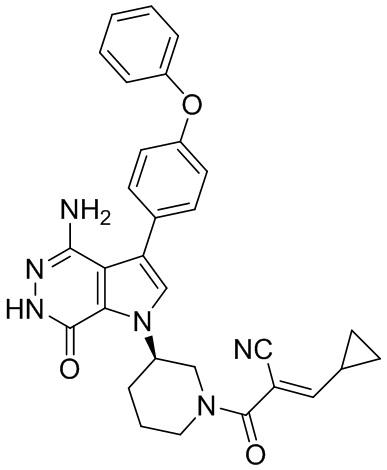 ;
; 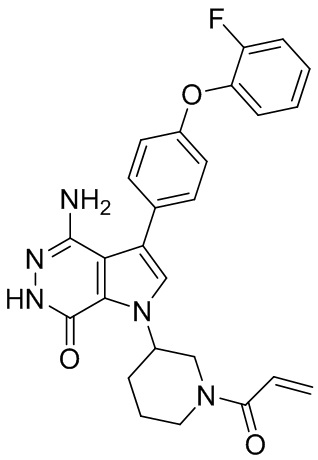 ;
; 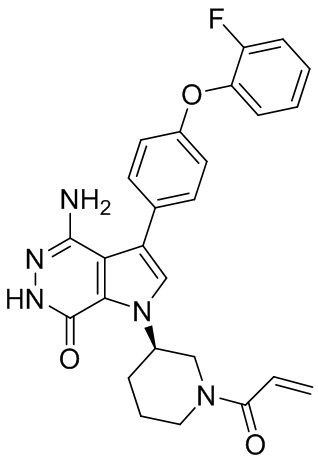 ;
;  ;
;  ;
; ;
;  ;
;
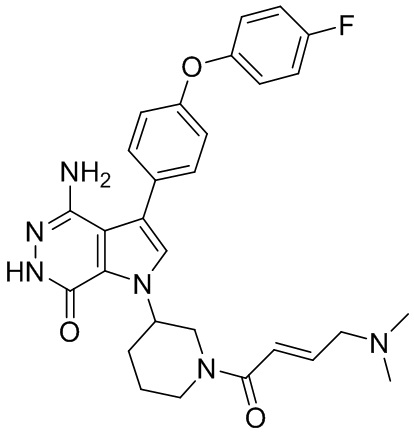 ;
;  ;
;  ;
;  ;
;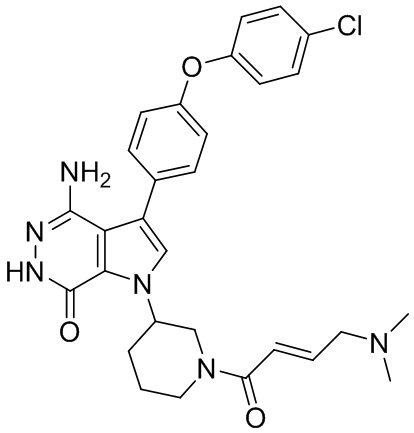 ;
;  ;
;  ;
;  ;
; 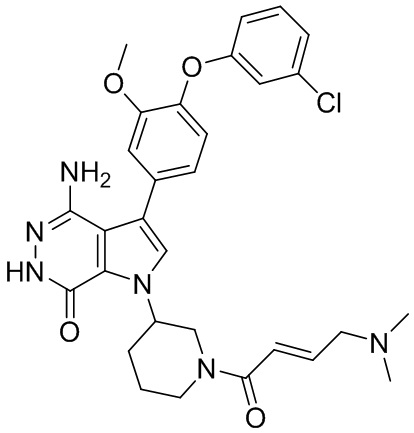 ;
; 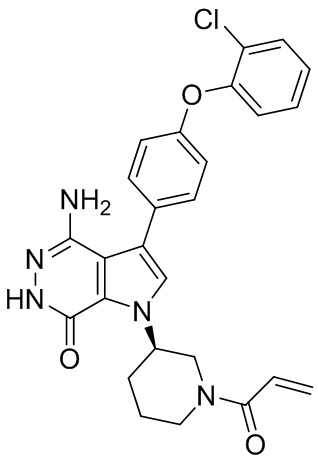 ;
; 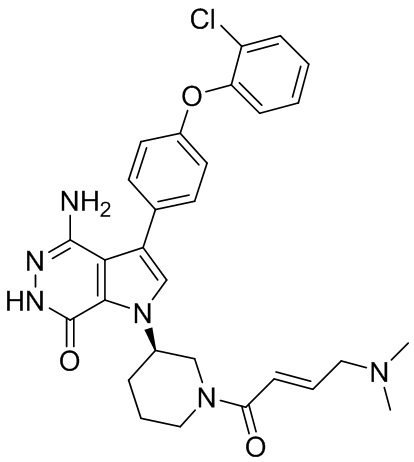 ;
;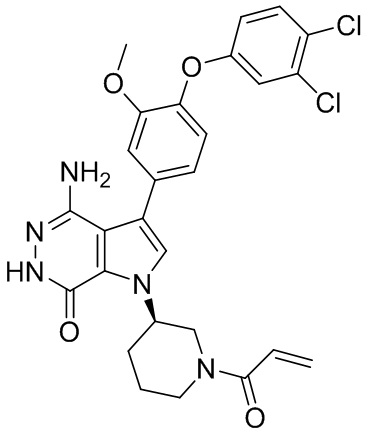 ;
;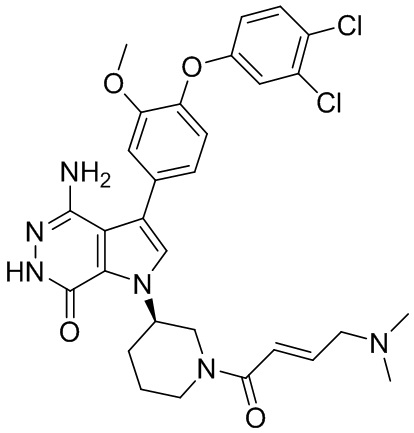 ;
; 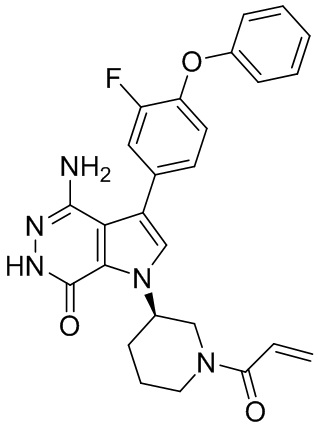 ;
; 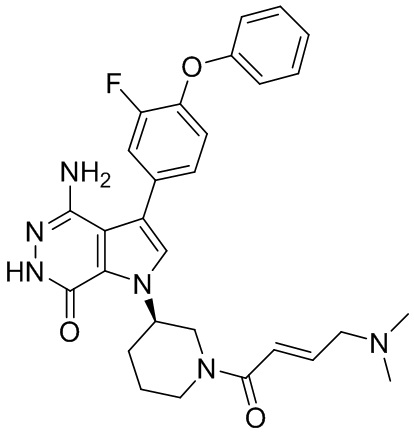 ;
; 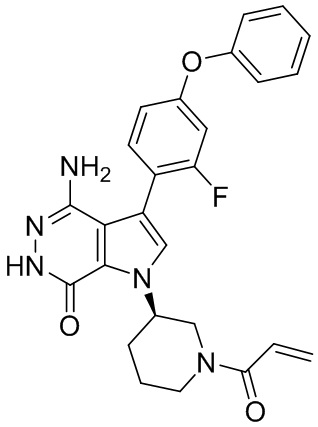 ;
; 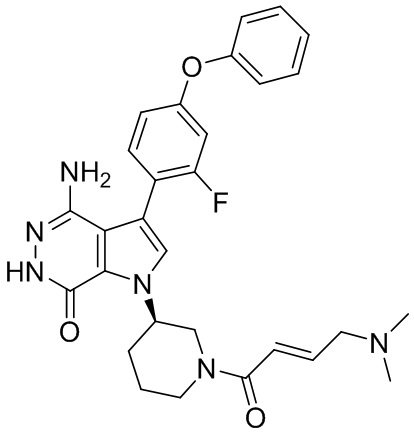 ;
; 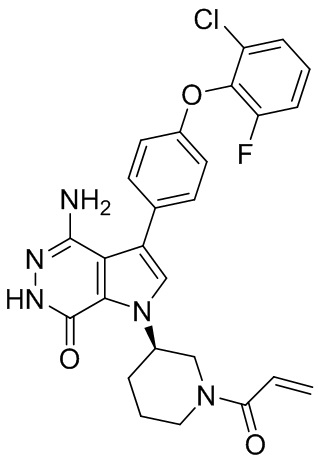 ;
; 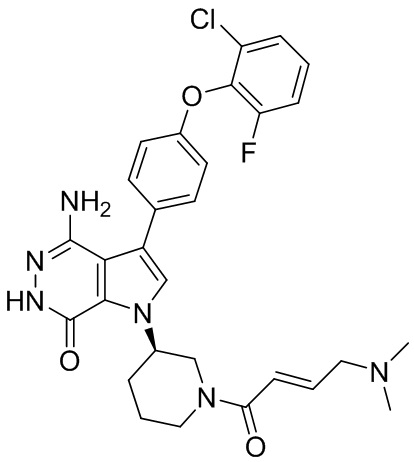 ;
; 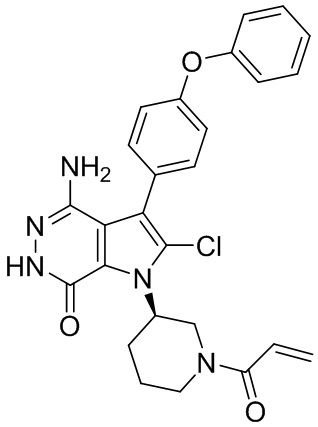 ;
; 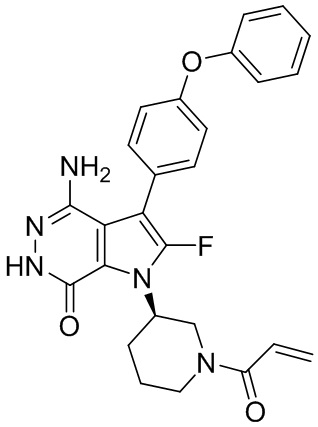 ;
; 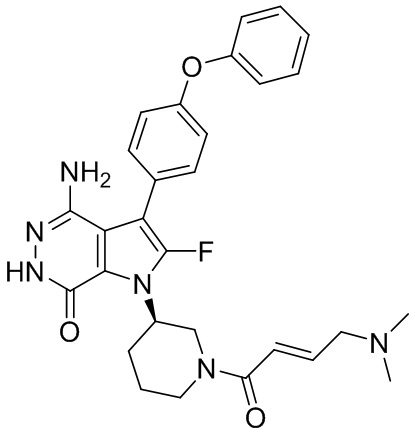 ;
; 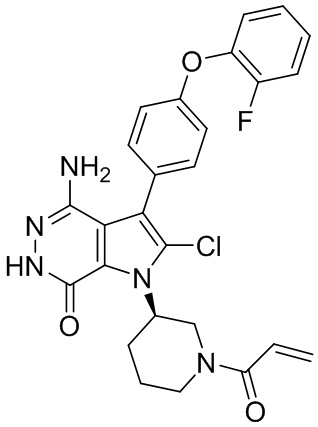 ;
; 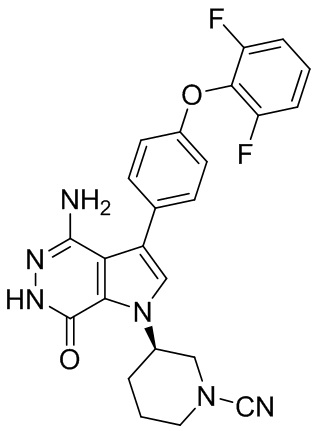 ;
; 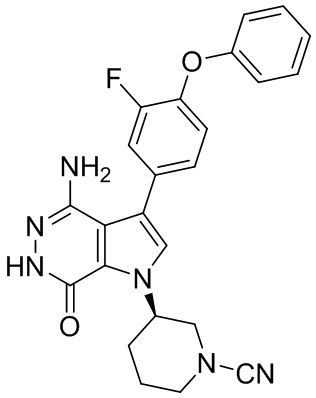 ;
; 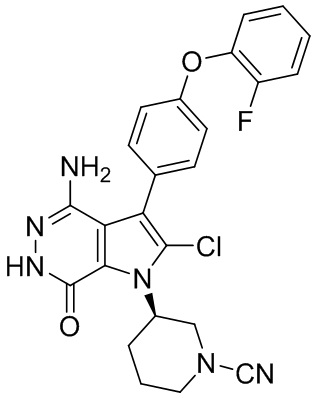 ;
; 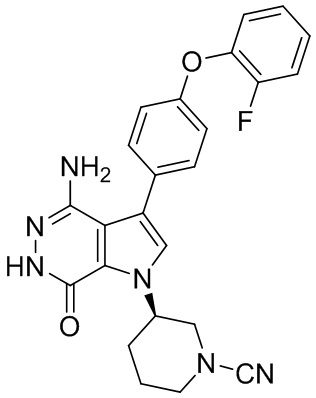 ;
; 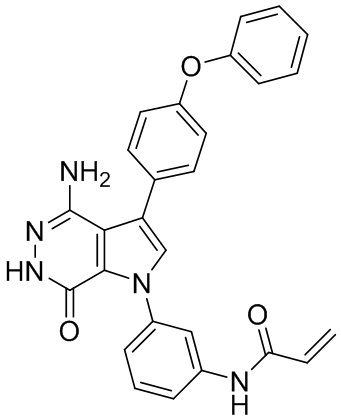 ;
; 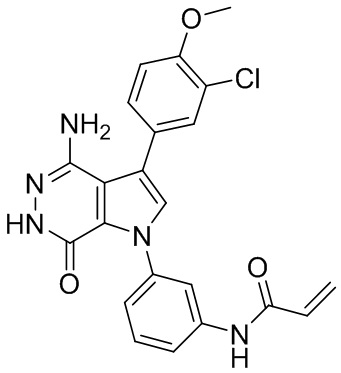 ;
; 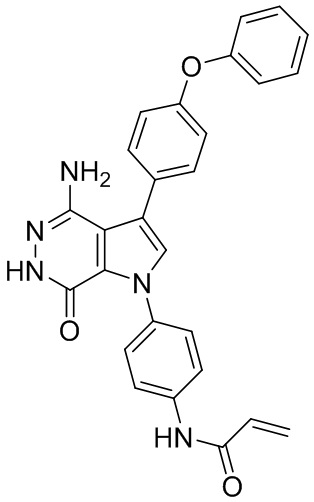 ;
; 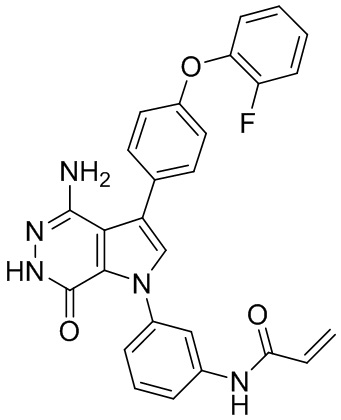 ;
; 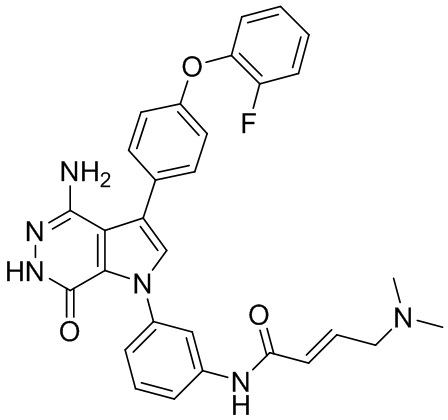 ;
; 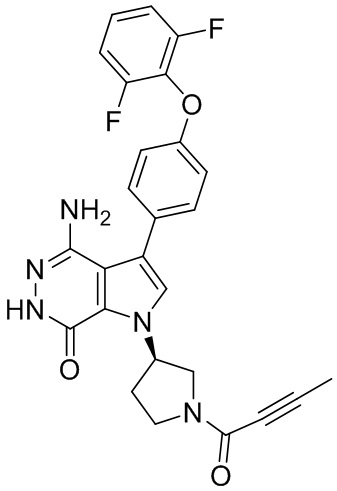 ;
; 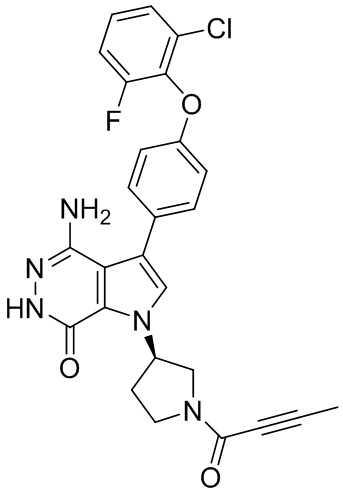 ;
; 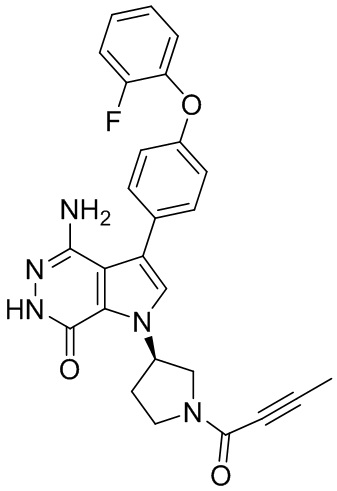 ;
; 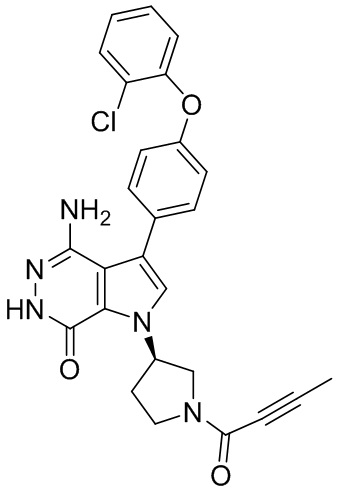 ;
; 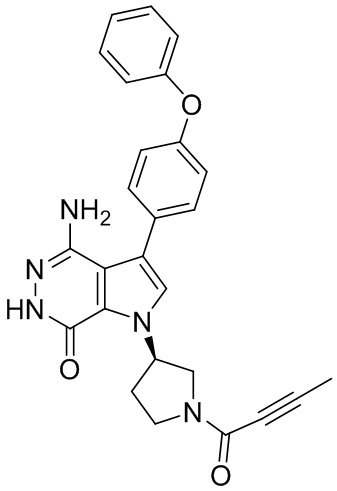 ;
; 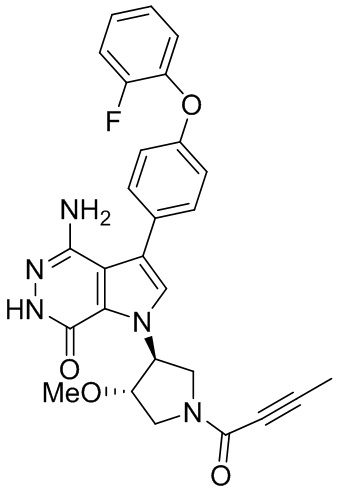 ;
; 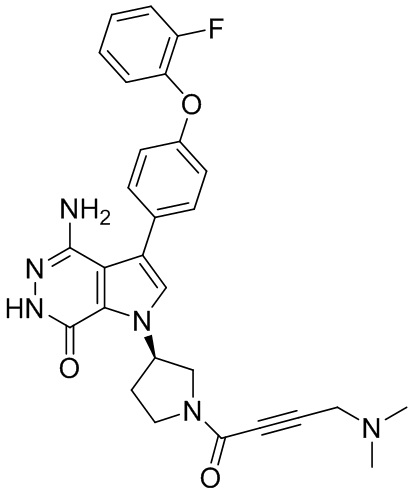 ;
; 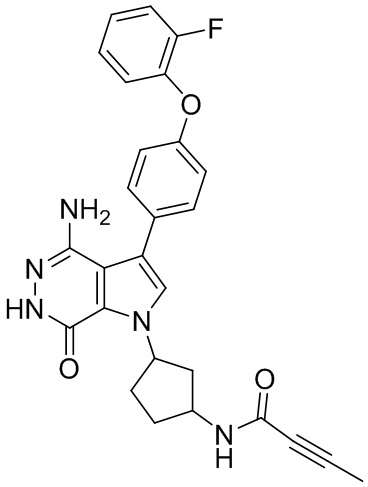 ;
;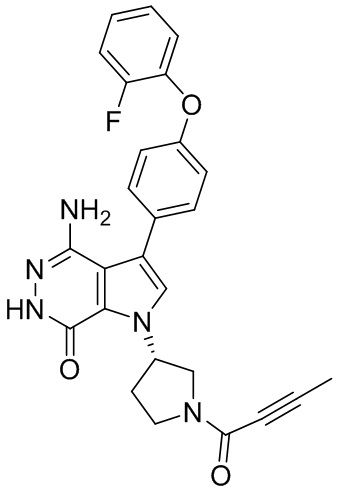 ;
; 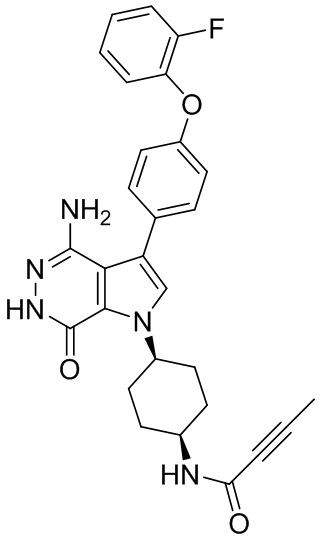 ;
; 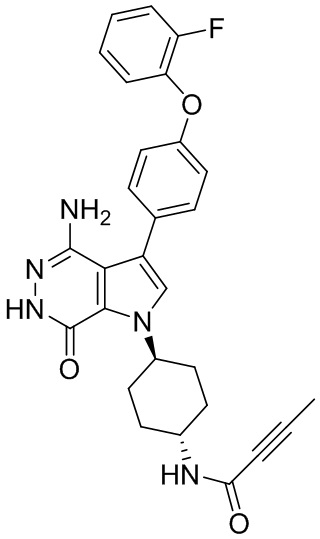 ;
; 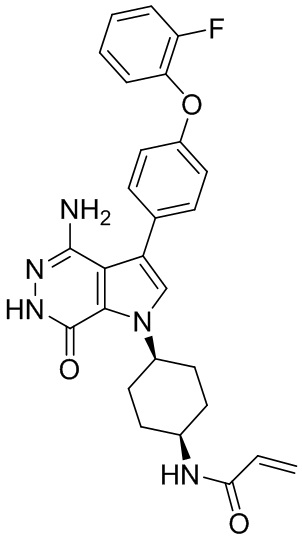 ;
; 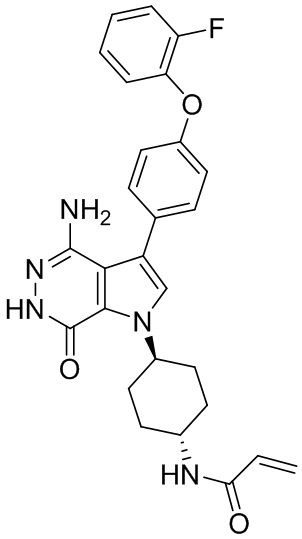 ;
; 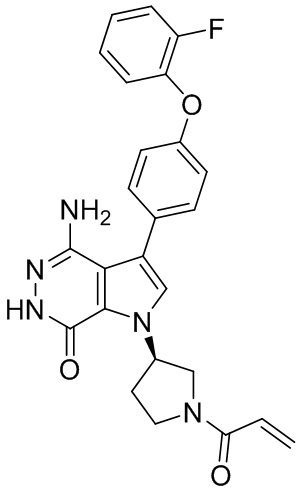 ;
; 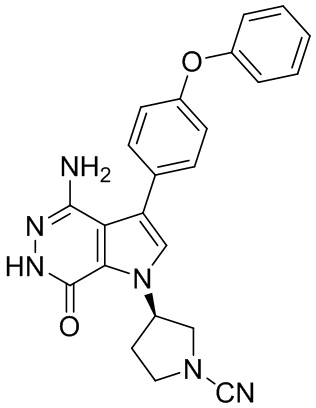 ;
; 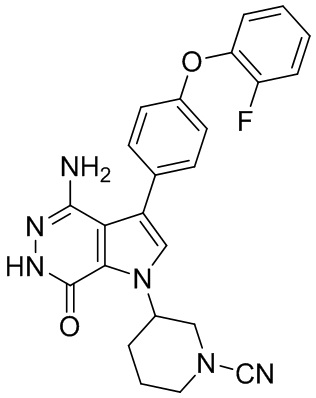 ;
; 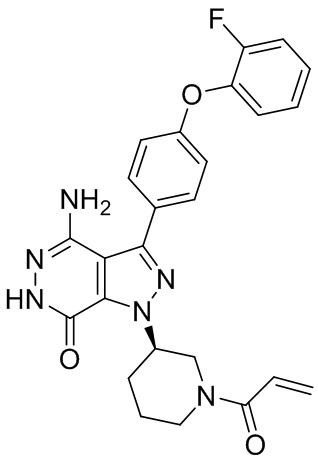 ;
; 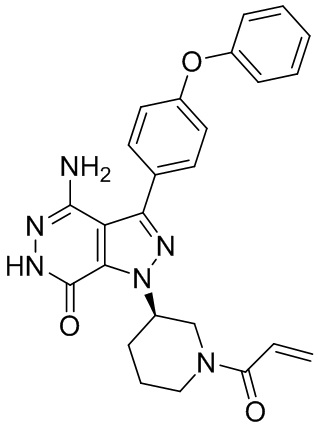 ;
; 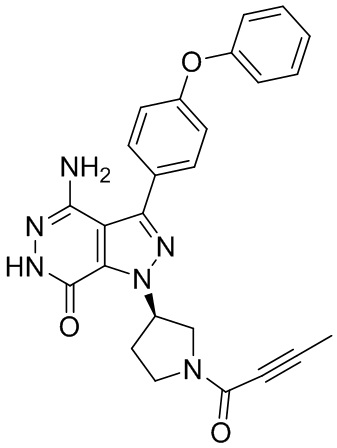 ;
; 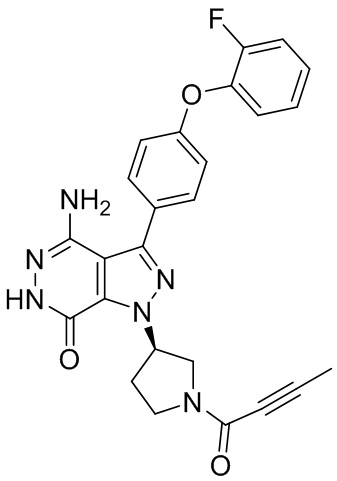 ;
;
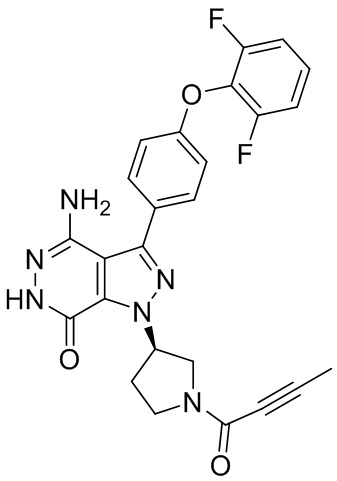 ;
; 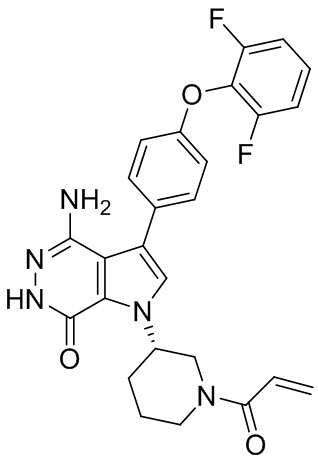 ;
; 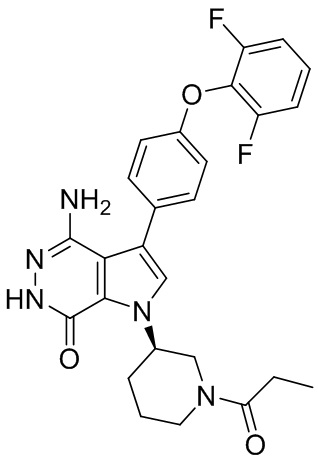 ;
; 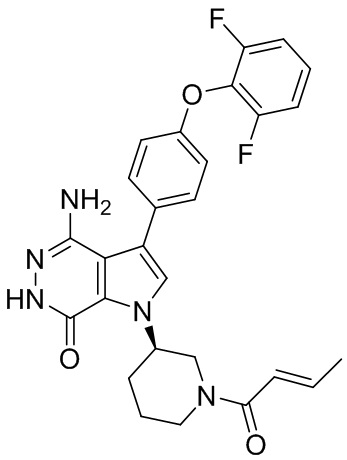 ;
;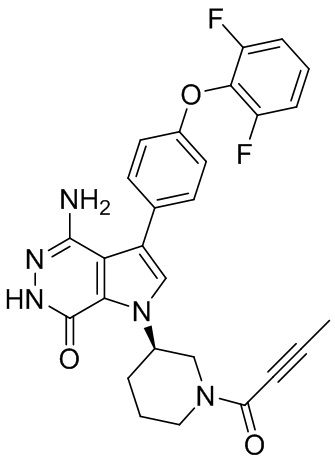 ; и
; и  .
.
38. Соединение по п. 37 или его фармацевтически приемлемая соль, в котором соединение представляет собой
.
39. Соединение по п. 37 или его фармацевтически приемлемая соль, в котором соединение представляет собой
.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| US 8673925 B1, 18.03.2014 | |||
| Колосоуборка | 1923 |
|
SU2009A1 |

