Настоящее изобретение относится к новым конъюгатам на основе хитозана, например, наноносителям, содержащим производное биосовместимого полимера хитозана, конъюгированного с фотосенсебилизирующим агентом, и их применению в фотохимической интернализации (ФХИ) и в фотодинамической терапии (ФДТ). Изобретение также относится к применению новых конъюгатов по изобретению при лечении или профилактике заболеваний, в частности, рака, и в целях вакцинации.
Наноматериалы обладают особыми физико-химическими свойствами, которые включают соотношение небольшого размера и большой площади поверхности на единицу массы и высокую активность по сравнению с сыпучими веществами аналогичного состава. Эти уникальные свойства могут улучшить и преодолеть некоторые ограничения, существующие в традиционной медицине. Применение наноматериалов дает возможность изменять такие свойства, как растворимость, коэффициент диффузии, время полужизни в кровотоке, характеристики высвобождения лекарственного средства и иммуногенность. В последние два десятилетия для лечения заболеваний были разработаны ряд агентов на основе наночастиц для терапевтических и диагностических применений.
Использование наноматериалов может обеспечить более эффективные и более удобные способы введения, низкую токсичность, минимальные побочные эффекты, повышенную биодоступность и расширенный жизненный цикл продукта в системе. В качестве систем доставки лекарственных средств наночастицы или наноносители могут обеспечить адресную доставку и контролируемое высвобождение. Кроме того, они могут быть использованы в диагностических целях. Они могут, например, позволить обнаруживать предраковые клетки, вирусные фрагменты и маркеры заболеваний, которые не могут быть обнаружены традиционными широко известными методами.
В настоящее время природные и синтетические полимеры наряду с липосомами являются основными платформами наночастиц, встречающимися в литературе (Peer et al., 2007 г., Natl., 2(12), p751-760). Другие популярные платформы включают дендримеры, масляные наноэмульсии, мезопористые кремнеземные наночастицы и наночастицы оксида железа.
Новые конъюгаты по настоящему изобретению, которые могут являться наноносителями, включают производные биосовместимого полимера хитозана, который является производным хитина. Хитин (поли(β-(1→4)-N-ацетил-D-глюкозамин) является природным полисахаридом и представляет собой материал покровных тканей насекомых и ракообразных. Хитин является вторым наиболее распространенным природным полисахаридом на земле после целлюлозы. Обычно его получают из источников, таких как клетки крабов и креветок. Структурно хитин подобен целлюлозе, но вместо гидроксильной группы в положении C2 полимерного скелета содержит ацетамидную группу.
Хитозан является наиболее важным производным хитина, обычно получаемый путем удаления ацетильных групп щелочными способами. В то время как наиболее часто встречающихся природные полимеры являются нейтральными или кислыми по природе, хитозан представляет собой сильно основный полисахарид. Атом азота в положении С2 обеспечивает возможность модифицировать полимер синтетическими методами, придавая молекуле определенные желаемые свойства, например, повышенную растворимость и улучшенные биологические свойства.
Хитозановый полимер состоит из β-(1→4) связанных D-глюкозаминовых звеньев с различными степенями деацетилирования (СД), где оставшиеся ацетильные группы распределены по блокам или хаотично по всей линейной полимерной цепи. Хитозан растворим в разбавленных кислотах, таких как уксусная кислота, благодаря положительному заряду на амино группе в кислых условиях. Хотя СД может быть очень различной, она почти никогда не бывает 100%. Отличительная номенклатура хитина в сравнении с хитозаном с точки зрения СД не была установлена, но СД для хитозана может варьироваться от 40 до 100%. Молекулярная масса может составлять вплоть до 2000 кДа, но те хитозаны, у которых она менее 50 кДа, иногда рассматриваются как олигохитозаны.
В последние десятилетия к хитину и хитозану было привлечено внимание в связи с возможностью применения их в медицине. С целью повышения растворимости и дальнейшего улучшения их физических, химических и биологических свойств были разработаны и синтезированы различные производные хитозана.
Помимо производного хитозана, конъюгаты по настоящему изобретению также включают фотосенсибилизирующий агент, который конъюгирован с хитозаном. Благодаря этому конъюгаты имеют, в частности, применение в способах с участием фотосенсибилизации.
Фотосенсибилизация представляет собой процесс передачи энергии поглощенного света. После поглощения энергия передается (выбранным) реагентам. Фотосенсибилизаторами являются соединения, которые способны переводить энергию света в химические реакции II порядка. Высокореакционные конечные продукты этих способов приводят к цито- и сосудистой токсичности.
Фотосенсибилизаторы могут оказывать свое действие путем целого ряда механизмов, прямо или косвенно. Так, например, некоторые фотосенсибилизаторы непосредственно становятся токсичными при активации светом, в то время как другие действуют, вырабатывая токсичные образцы, например, окисляющие агенты, такие как синглетный кислород или другие свободные радикалы - производные кислорода, которые являются крайне разрушительными для клеточного материала и биомолекул, таких как липиды, белки и нуклеиновые кислоты.
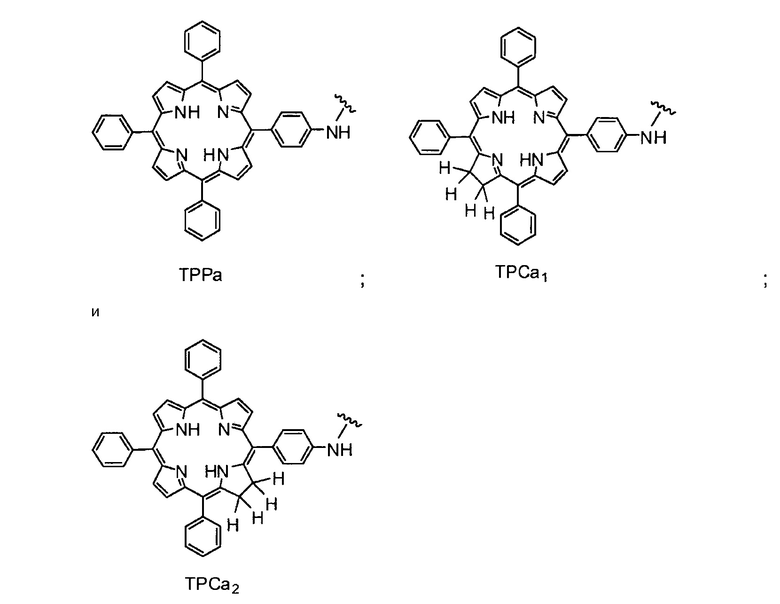
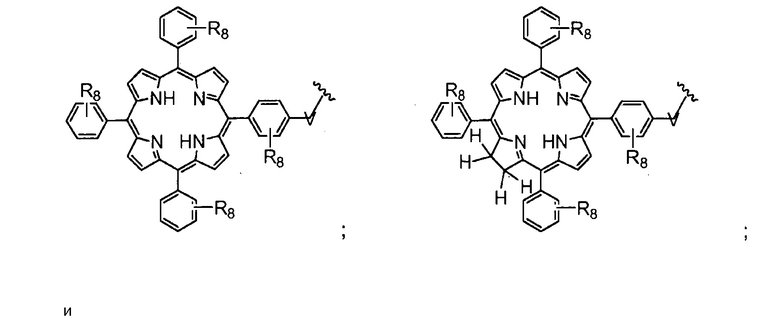
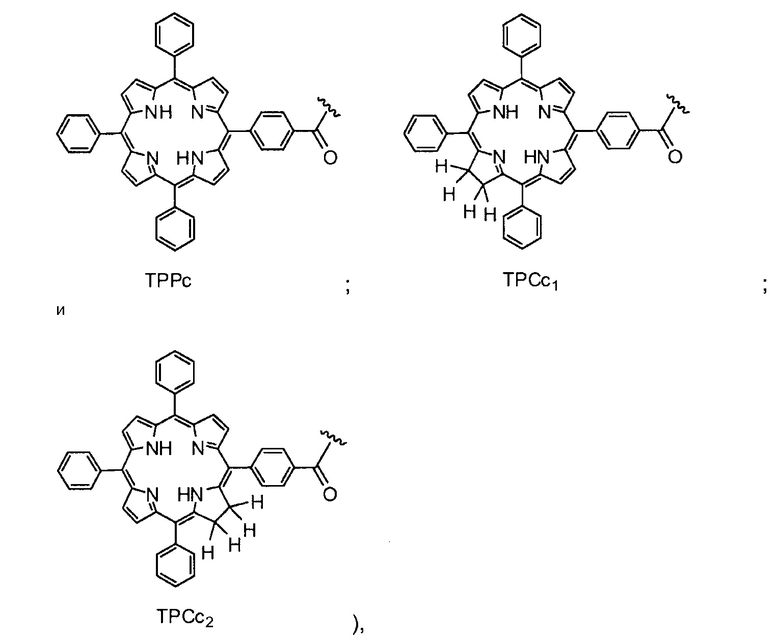
Существует много известных фотосенсибилизирующих агентов, включая порфирины, фталоцианины, пурпурины, хлорины, бензопорфирины, лизомотропные слабые основания, нафталоцианины, катионные красители и тетрациклины и их производные (Berg et al., (1997), J. Photochemistry and Photobiology, 65, 403-409). Другие фотосенсибилизирующие агенты включают тексафирины, феофорбиды, порфицены, бактериохлорины, кетохлорины, производные гематопорфирина и эндогенные фотосенсибилизаторы, индуцируемые 5-аминолевулиновой кислотой. Как обсуждается далее, в молекулах на основе хитозана по настоящему изобретению используются порфирины и хлорины, в частности, тетрафенилпорфирин (TPP) и тетрафенилхлорин (TPC).
Порфирины являются наиболее широко изученными фотосенсибилизирующими агентами. Их молекулярная структура включает в себя четыре пиррольных кольца, связанных вместе посредством метиновых мостиков. Они представляют собой природные соединения, которые зачастую способны образовывать металлокомплексы. Например, в случае кислородного транспорта белка гемоглобина атом железа вводится в порфириновое ядро гема B.
Хлорины представляют собой большие гетероциклические ароматические кольца, которые содержат в ядре три пиррола и один пирролин, соединенные посредством четырех метиновых связей. В отличие от порфиринов хлорин поэтому является в значительной степени ароматическим, но не ароматическим по всей окружности кольца.
Фотосенсибилизирующие агенты используются в методах фотодинамической терапии (ФДТ) и фотохимической интернализации (ФХИ). ФДТ представляет собой двухстадийный процесс, включающий введение фотосенсибилизатора системно или местно с последующим облучением светом с соответствующей длиной волны. Для того, чтобы имело место цитотоксическое действие, должен также присутствовать молекулярный кислород. Когда эти три фактора успешно сочетаются (т.е. фотосенсибилизатор, свет и кислород), протекает фотодинамическая реакция. Фотодинамическая реакция приводит к образованию цитотоксических образцов, которые вызывают гибель клеток и повреждение тканей.
ФДТ используется для лечения, например, рака. Радикалы промежуточных соединений из фотодинамических реакций захватываются кислородом в биологических тканях с образованием обладающих высокой реакционной способностью форм кислорода (АФК), таких как синглетный кислород (1O2). 1O2 представляет собой короткоживущую форму кислорода с высоким цитотоксическим потенциалом. Таким образом может быть достигнуто высоко избирательное цитотоксическое лечение, при котором в значительной степени можно избежать системных побочных эффектов.
ФХИ основан на том же принципе, что и ФДТ, но производит меньше АФК (например, при использовании уменьшенных световых доз), чтобы индуцировать высвобождение удерживаемых лекарственных средств и макромолекул из эндосом в цитозоль без значительной гибели клеток из-за АФК. В ФХИ световое возбуждение приводит к опосредованному АФК селективному повреждению лизосомальных и/или эндосомальных мембран и высвобождению удерживаемых гидрофильных лекарственных средств и макромолекул. Тем самым могут быть высвобождены эндоцитированные молекулы для достижения цели их воздействия до того, как они деградируют в лизосомах.
Было показано, что ФХИ повышает биологическую активность большого разнообразия макромолекул и других молекул, которые не так легко проникают через плазматическую мембрану, в том числе рибосом-инактивирующие белки I типа, иммунотоксины, химиотерапевтические агенты, такие как блеомицин (Blenoxane®) и доксорубицин, плазмиды, кодирующие гены и олигонуклеотиды. Было установлено, что он индуцирует цитотоксичность в более глубоких слоях тканей, чем соответствующий ФДТ. Благодаря сочетанию целевой терапии с активируемой светом цитозольной доставкой, индуцированной фотосенсибилизаторами, предпочтительно накапливающимися в солидных опухолях, ФХИ может быть весьма специфичным, и это также способствует повышению противоопухолевой эффективности.
Одной из основных проблем, связанной с ФДТ при клиническом применении, является фоточувствительность кожи и неблагоприятное биораспространение фотосенсибилизатора. В качестве способа уменьшения побочных эффектов и улучшения фармакокинетики при ФДТ были применены наноносители, такие как дендримеры, липосомы и полимерные мицеллы.
Сохраняется потребность в улучшенных наноносителях и фотосенсибилизирующих агентах для использования их в методах ФХИ и ФДТ. Настоящее изобретение касается решения этой задачи. Авторы настоящего изобретения разработали новые соединения, которые основаны на конъюгате фотосенсибилизатора и хитозана. Новые молекулы имеют неожиданно высокую эффективность в методах ФХИ, как показано в приведенных далее примерах, которые демонстрируют удивительно хорошую эффективность как in vitro, так и in vivo.
Таким образом, в первом аспекте настоящее изобретение относится к соединению, например, наноносителю, содержащему конъюгат фотосенсибилизатора и хитозана, где указанное соединение представляет собой соединение формулы (I):

где
n обозначает целое число, большее чем или равное 3,
R содержится n раз в указанном соединении и
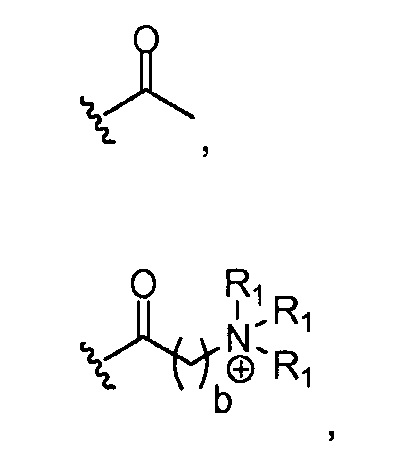
в 0,5%-99,5% из указанных всех Rn групп, каждый R представляет собой группу A, выбранную из:
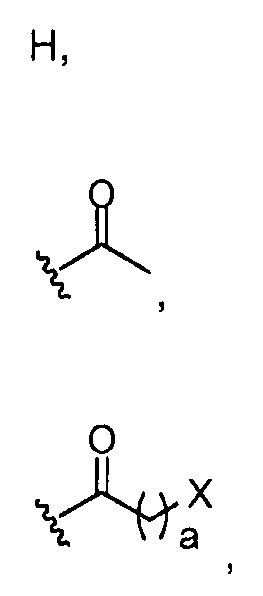
где a обозначает 1, 2, 3, 4 или 5; и X представляет собой Br, Cl или OH;
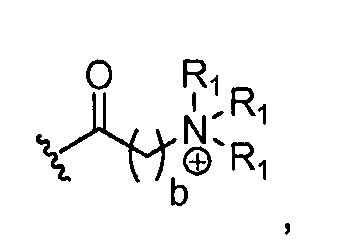
где каждый R1, который может быть одинаковым или разным, выбран из H, CH3 и -(CH2)c-CH3; b обозначает 1, 2, 3, 4 или 5; и c обозначает 0, 1, 2, 3, 4 или 5 (для которого противоионом может быть, например, Cl-);
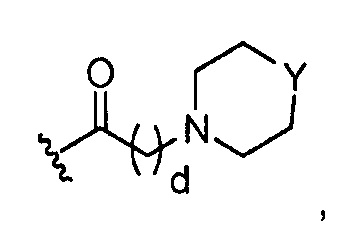
где Y представляет O; S; SO2; -NCH3; или-N(CH2)eCH3; d=1, 2, 3, 4 или 5; и e=1, 2, 3, 4 или 5;
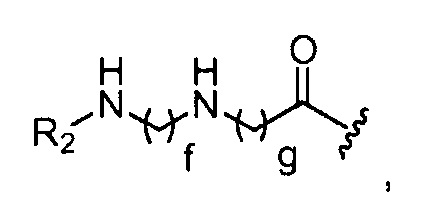
где R2 представляет собой -(CH2)h-CH3 или-CO-(CH2)h-CH3; f обозначает 1, 2, 3, 4 или 5; g обозначает 1, 2, 3, 4 или 5; и h обозначает 0, 1, 2, 3, 4 или 5;
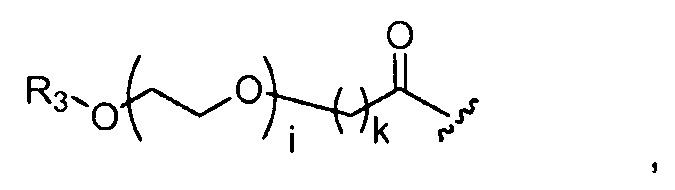
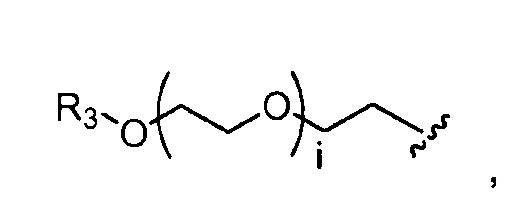
где R3 представляет собой -(CH2)j-CH3, i обозначает целое число от 1 до 200, предпочтительно, от 1 до 50 или от 1 до 10, например, 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10, или, по меньшей мере, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100 или 200; j обозначает 0, 1, 2, 3, 4 или 5; и k обозначает 1, 2, 3, 4 или 5;
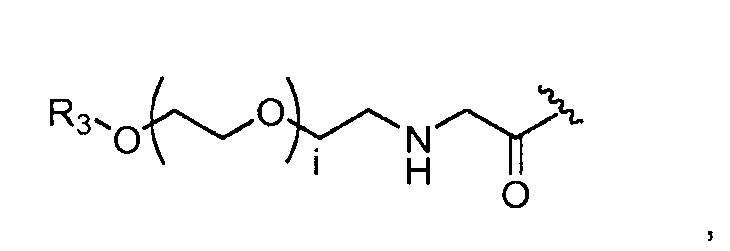
где R3 представляет собой -(CH2)j-CH3, i обозначает целое число, как определено выше; и j обозначает 0, 1, 2, 3, 4 или 5;
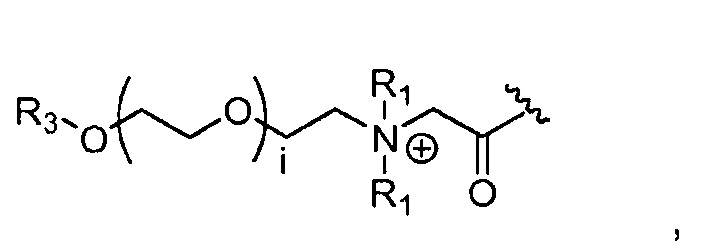
где R3 представляет собой -(CH2)j-CH3, i обозначает целое число, как определено выше; j обозначает 0, 1, 2, 3, 4 или 5; и каждый R1, который может быть одинаковым или разным, выбран из H, CH3 и -(CH2)с-CH3; и c обозначает 0, 1, 2, 3, 4 или 5;
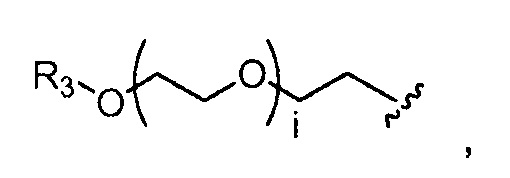
где R3=-(CH2)j-CH3, i обозначает целое число, как определено выше; и j обозначает 0, 1, 2, 3, 4 или 5;
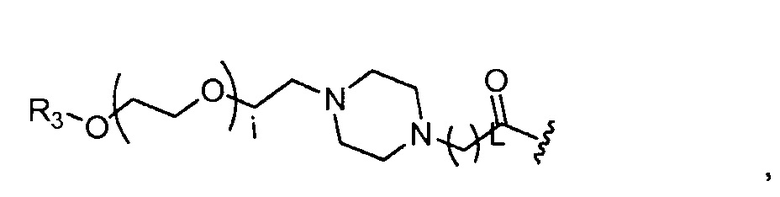
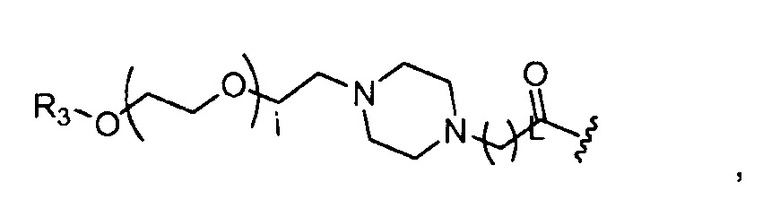
где R3=-(CH2)j-CH3, i обозначает целое число, как определено выше; L обозначает 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10; и j обозначает 0, 1, 2, 3, 4 или 5;
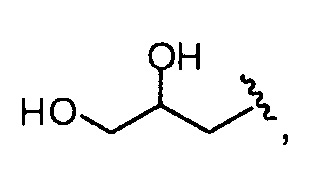
и
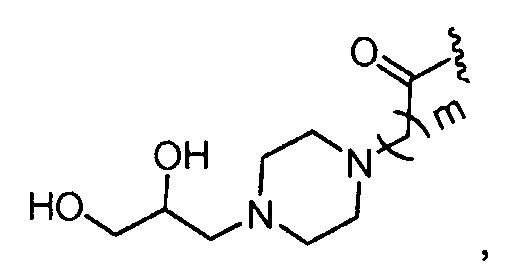
где m обозначает 1, 2, 3, 4 или 5;
где каждая группа R может быть одинаковой или разной; и
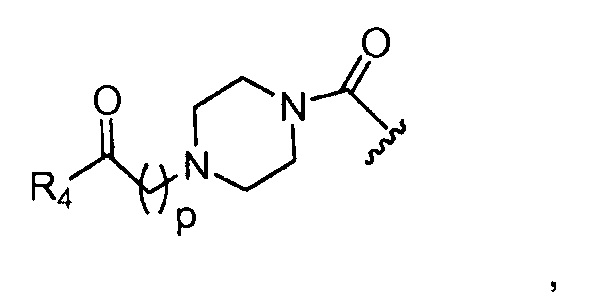
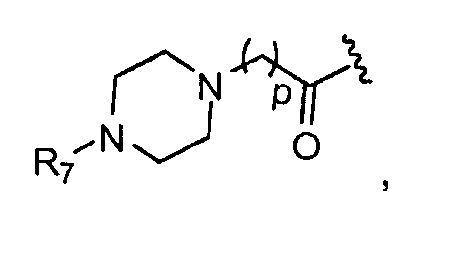
в 0,1%-99,9% из всех указанных Rn групп, каждый R представляет собой группу B, выбранную из:
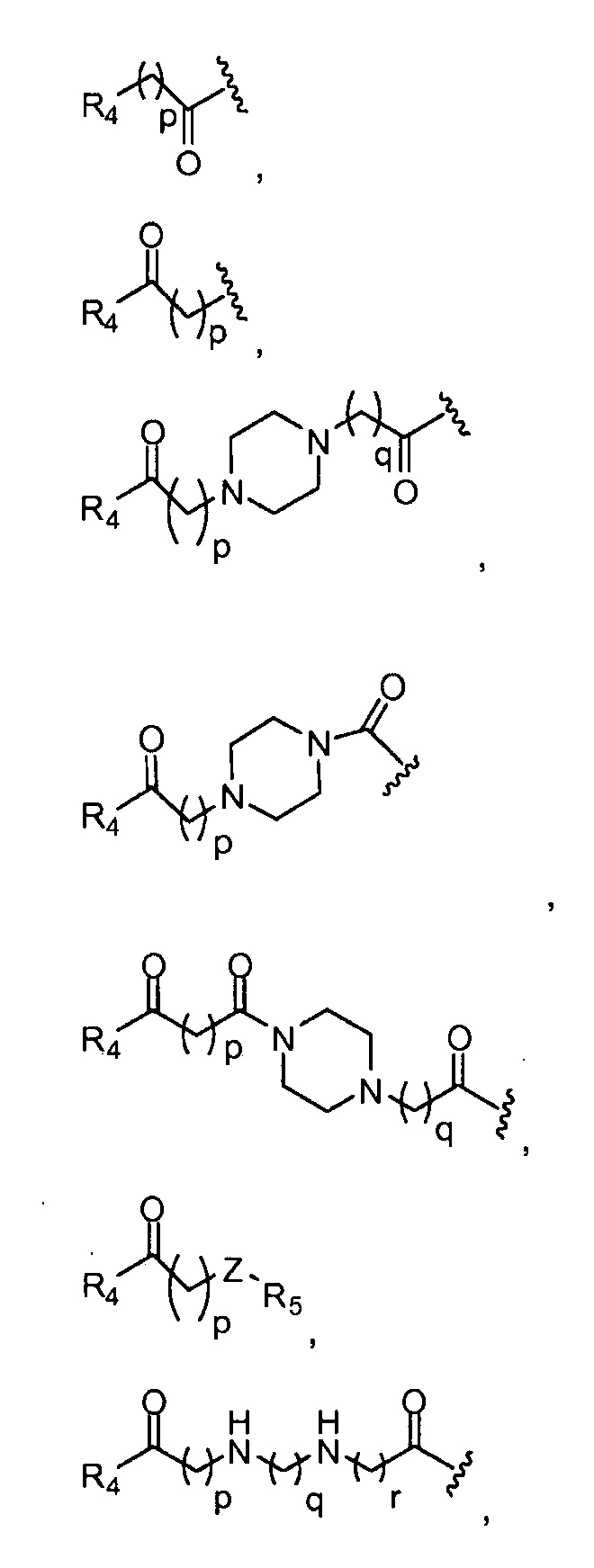
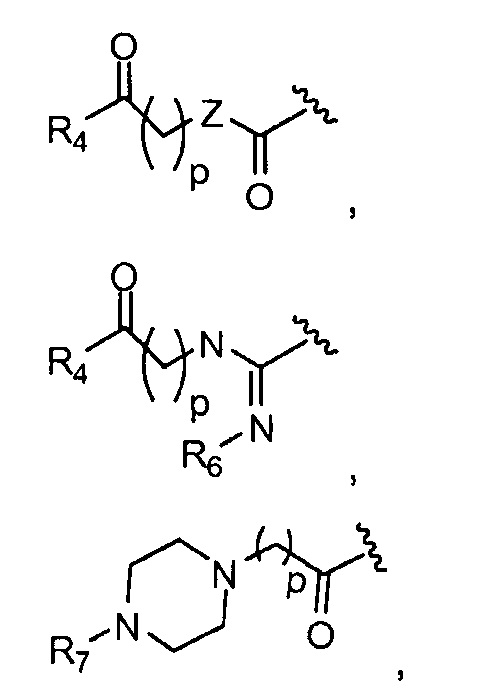
и
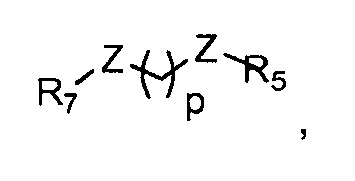
где
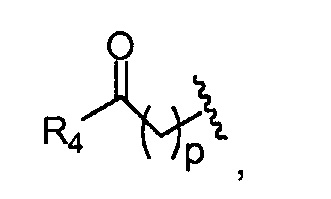
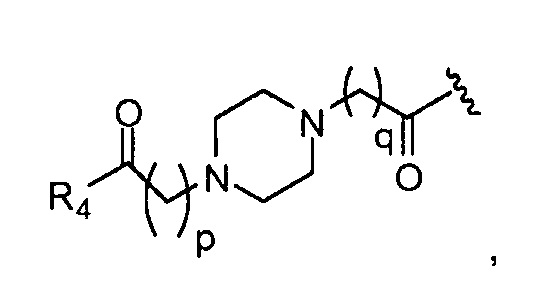
p обозначает 0, 1, 2, 3, 4 или 5; q обозначает 1, 2, 3, 4 или 5; и r обозначает 1, 2, 3, 4 или 5;
R4 представляет собой группу, выбранную из:
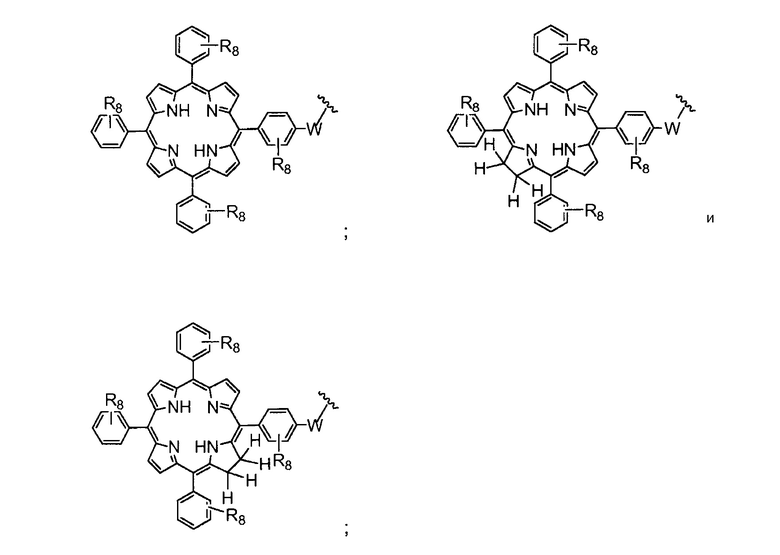
(предпочтительно, R4 выбран из:
W представляет собой группу, выбранную из O, S, NH или N(CH3);
R5 представляет собой группу, выбранную из: -(CH2)s-CO-; -(CH2)s-Z-(CH2)t-CO- и -(CH2)S-Z-(CH2)t-CO-, где s обозначает 0, 1, 2, 3, 4 или 5; t обозначает 0, 1, 2, 3, 4 или 5;
Z представляет собой NH, O, S или SO2
R6 представляет собой группу, выбранную из -CN и CH3;
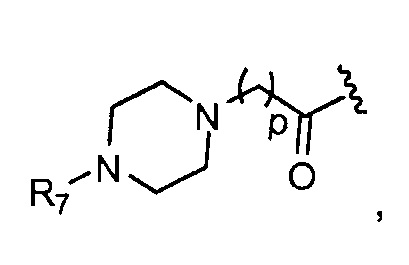
R7 представляет собой группу, выбранную из:
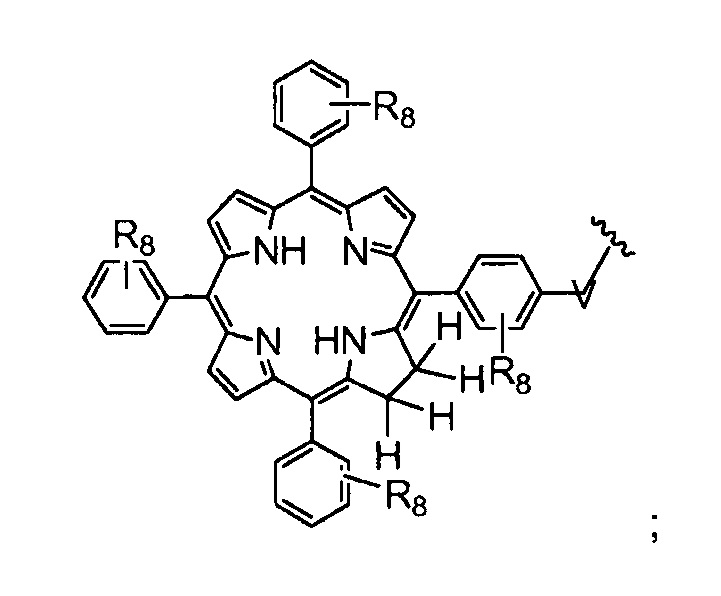
V представляет собой группу, выбранную из CO, SO2, PO, PO2H или CH2;
(предпочтительно, R7 выбран из:
R8 представляет собой группу (заместитель в о-, м- или п- положении), которая может быть одинаковой или разной, выбранную из H, -OH, -OCH3, -CH3, -COCH3, C(CH3)4, -NH2, -NHCH3, -N(CH3)2 и -NCOCH3 (где, предпочтительно, каждый R8 представляет собой H, или, по меньшей мере, один R8 не представляет собой H);
где каждая группа R может быть одинаковой или разной.
Хитозановый полимер содержит, по меньшей мере, 3 звена (n=3). Однако, предпочтительно, n обозначает, по меньшей мере, 10, 20, 50, 100, 500, 1000, например, от 10 до 100 или от 10 до 50.
Как уже упоминалось выше, фотосенсибилизаторы, используемые в конъюгатах, представляют собой производные порфирина и хлорина, в частности, TPPa, TPCa1, TPCa2, TPPc, TPCc1 и TPCc2. Предпочтительно, указанное производное фотосенсибилизатора R4 или R7 представляет собой TPCa1, TPCa2, TPCc1 или TPCc, особенно предпочтительно, TPCa1 или TPCa2.
Производное хитозанового конъюгата может иметь различные степени замещения (СЗ) вышеуказанными группами R. Например, когда они имеются, одна или несколько из групп R, описанные выше, могут включать менее чем 1%, предпочтительно от 0,1 до 1,0%, или более чем 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 96, 97, 98, 99, 99,5 или 99,9% хитозановых замещений. Как отмечалось выше, группа A и группа B из групп R, каждая, содержат 0,1%-99,9% (предпочтительно, от 0,5 до 99,5%) от всех Rn групп. Предпочтительно, группа A содержит, по меньшей мере, 50%, предпочтительно, по меньшей мере, 60, 70, 80, 90 или 95% всех Rn групп. Особенно предпочтительно, группа A содержит от 50 до 95%, например, от 70 и 95% всех Rn групп. Предпочтительно, группа B содержит менее 50%, например, менее 40, 30, 20, 10 или 5% всех Rn групп. Особенно предпочтительно, группа B содержит в интервале от 5 до 30%, например, 10-25% всех Rn групп.
Группа A из групп R (или группа B из групп R) может быть одинаковой или разной. В предпочтительных аспектах, когда имеются, например, разные группы, в группе A из групп R соотношение групп может меняться. Например, одна группа R может быть представлена в диапазоне, например, от 75 до 95% (от всех Rn групп) в качестве основного компонента, в то время как другая группа R может быть представлена в диапазоне, например, от 0,1 до 10% (от всех Rn групп) в качестве второстепенного компонента. Однако, в качестве альтернативы, основной компонент может быть представлен при более низких уровнях (например, от 50 до 90%), и второстепенный компонент может присутствовать при более высоких уровнях (например, от 0,1 до 50%). Предпочтительно, что, когда A из групп R, которые присутствуют, отражает ацетилирование хитозановой молекулы, т.е. R представляет собой
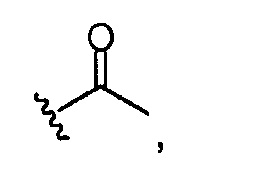
то он представлен в <1% от всех Rn групп. Эта группа R служит отражению степени ацетилирования (DA) исходных хитозановых молекул, используемых для получения соединений по изобретению, и, следовательно, ее доля может изменяться. Предпочтительно, она составляет <60% всех Rn групп, предпочтительно, менее 30% или 20%, например, между 0,1 и 30% от всех Rn групп.
Как будет понятно, суммарный % содержания всех групп A и B из групп R составляет 100%, и выбор в пределах вышеупомянутых диапазонов производится соответственно.
Следует отметить, что, в зависимости от способа синтеза, могут присутствовать некоторые примеси или альтернативные продукты в небольшом количестве, например, в конечном продукте могут оставаться следовые количества других групп R или остаточные защитные группы (например, TBDMS). Однако, такие следовые компоненты или соединения, если они присутствуют, имеются в количестве <1% от общего количества (предпочтительно, <0,1%) и не влияют на функциональные группы. Соединения или композиции, включая такие следовые компоненты или следовые соединения, входят в объем изобретения.
В предпочтительных аспектах, описанных в данном документе, предложен % выбранной R группы как часть всех Rn групп. В таких случаях диапазон % является предпочтительным (например, чтобы отразить изменения при получении). Предпочтительно, диапазон составляет 5%, т.е. если ссылка делается на 90% из R групп, имеющих определенную структуру, это распространяется на соединения с 85 до 95% данного A из группы R, и так далее.
Предпочтительными A из групп R являются:
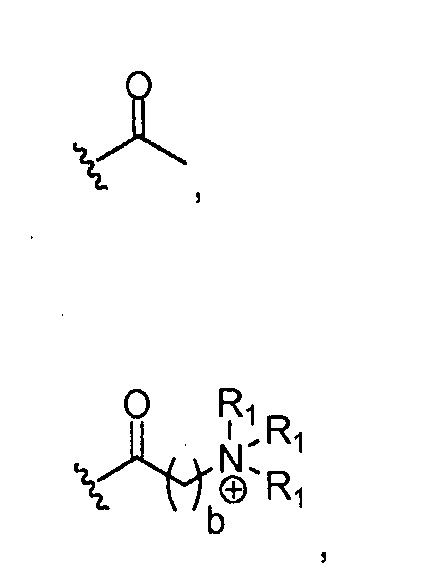
где, предпочтительно, каждый R1 представляет собой CH3, и b обозначает 1;
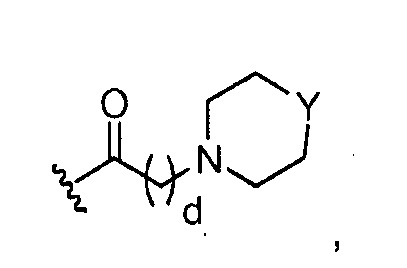
где, предпочтительно, Y представляет собой -NCH3, и d обозначает 1;
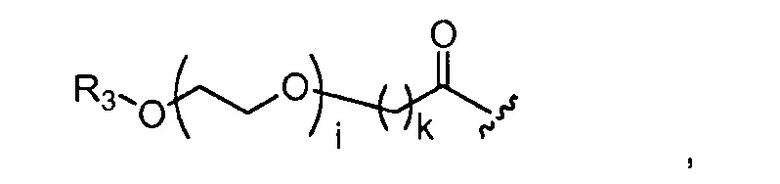
где, предпочтительно, j обозначает 0 или 1; i обозначает 3 или 6, и k обозначает 1;

где, предпочтительно, j обозначает 1, и i обозначает 2;
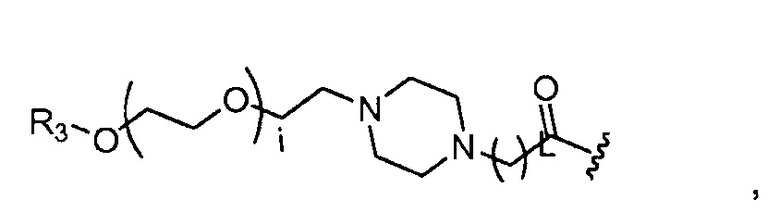
где, предпочтительно, j обозначает 0 или 1, и i обозначает 2, 4 или 5 (например, 2 или 4), и L обозначает 1;
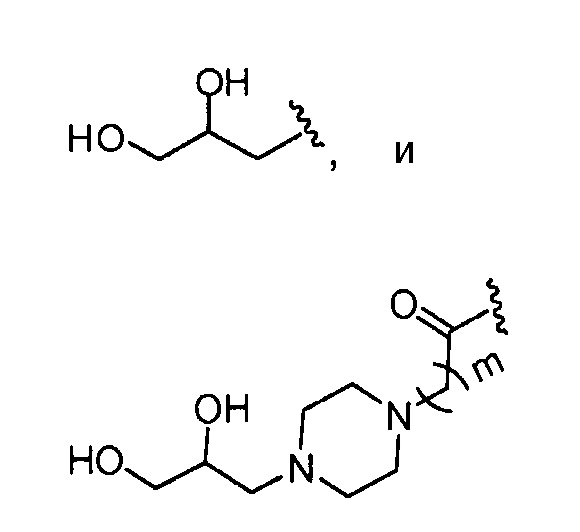
где, предпочтительно, m обозначает 1.
Предпочтительно, вышеуказанные группы R представлены в качестве основных компонентов (то есть более 75% от всех Rn групп (как обсуждалось выше) или второстепенных компонентов (т.е. менее 10% от всех Rn групп (как обсуждалось выше)). Предпочтительно, группа 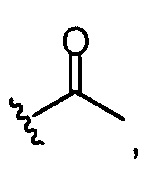 , присутствует только в качестве второстепенного компонента.
, присутствует только в качестве второстепенного компонента.
Предпочтительные B из R групп представляют собой:
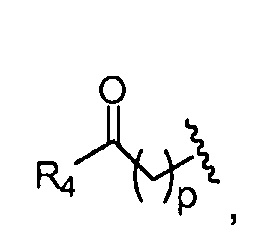
где, предпочтительно, р обозначает 1;
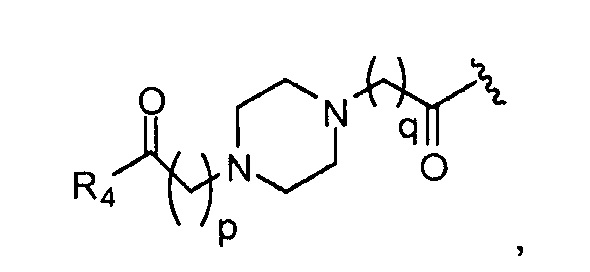
где, предпочтительно, p обозначает 1, и q обозначает 1;
где, предпочтительно, р обозначает 1;
и
где, предпочтительно, р обозначает 1.
Особенно предпочтительные A из R групп представляют собой:
где, предпочтительно, каждый R1 представляет собой CH3, и b обозначает 1;
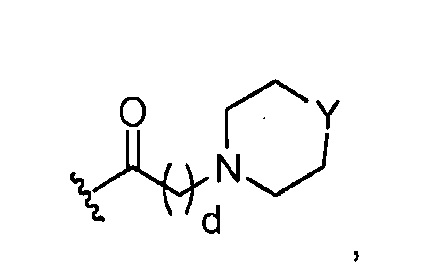
где, предпочтительно, Y представляет собой -NCH3, и d обозначает 1;
где, предпочтительно, j обозначает 1, и i обозначает 2; и
где, предпочтительно, j обозначает 0 или 1, и i обозначает 2, 4 или 5 (например, 2 или 4), и L обозначает 1.
Особенно предпочтительные B из групп R представляют собой:
где, предпочтительно, р обозначает 1;
где, предпочтительно, р обозначает 1, и q обозначает 1; и
где, предпочтительно, р обозначает 1.
Предпочтительные R группы и их относительное преобладание во всех Rn группах приведены в таблице ниже, в которой показаны различные возможные R группы, которые связаны с хитозаном. Преобладание каждого типа R группы указано в скобках и может меняться в пределах 5% в обе стороны от указанного значения. Группа B из R групп имеет присоединенные группы фотосенсибилизатора R4 или R7.
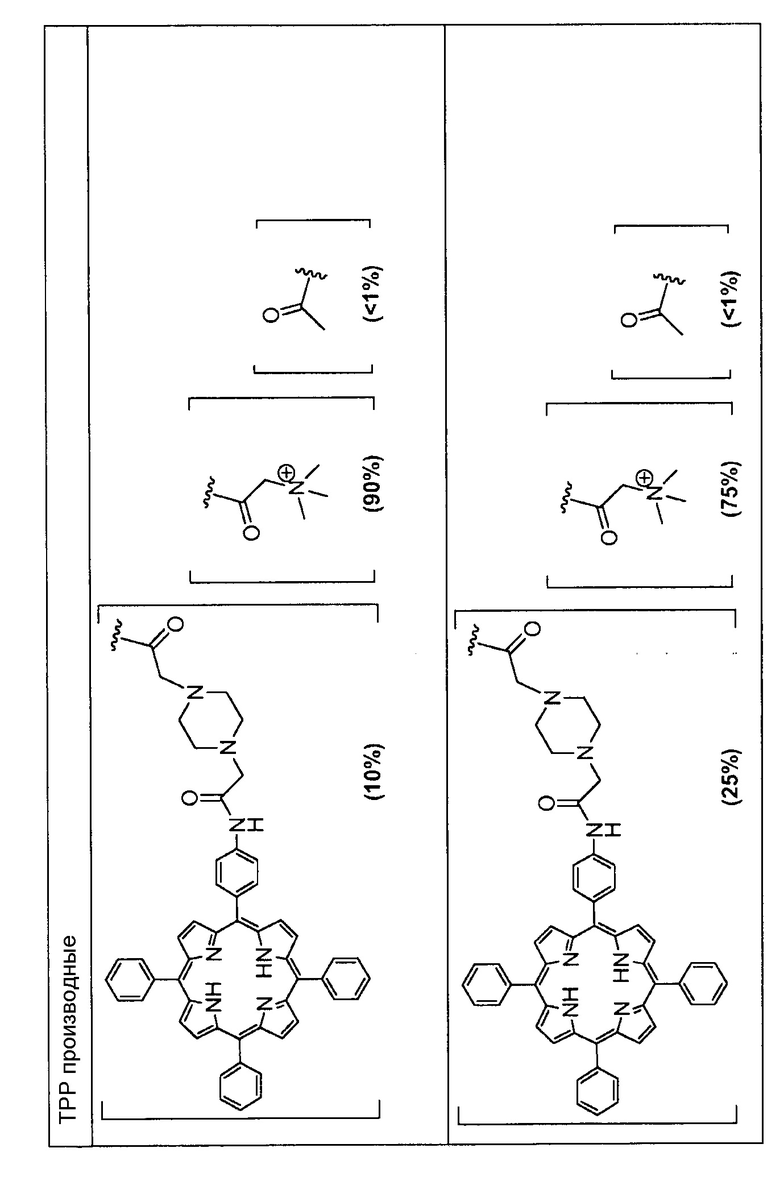
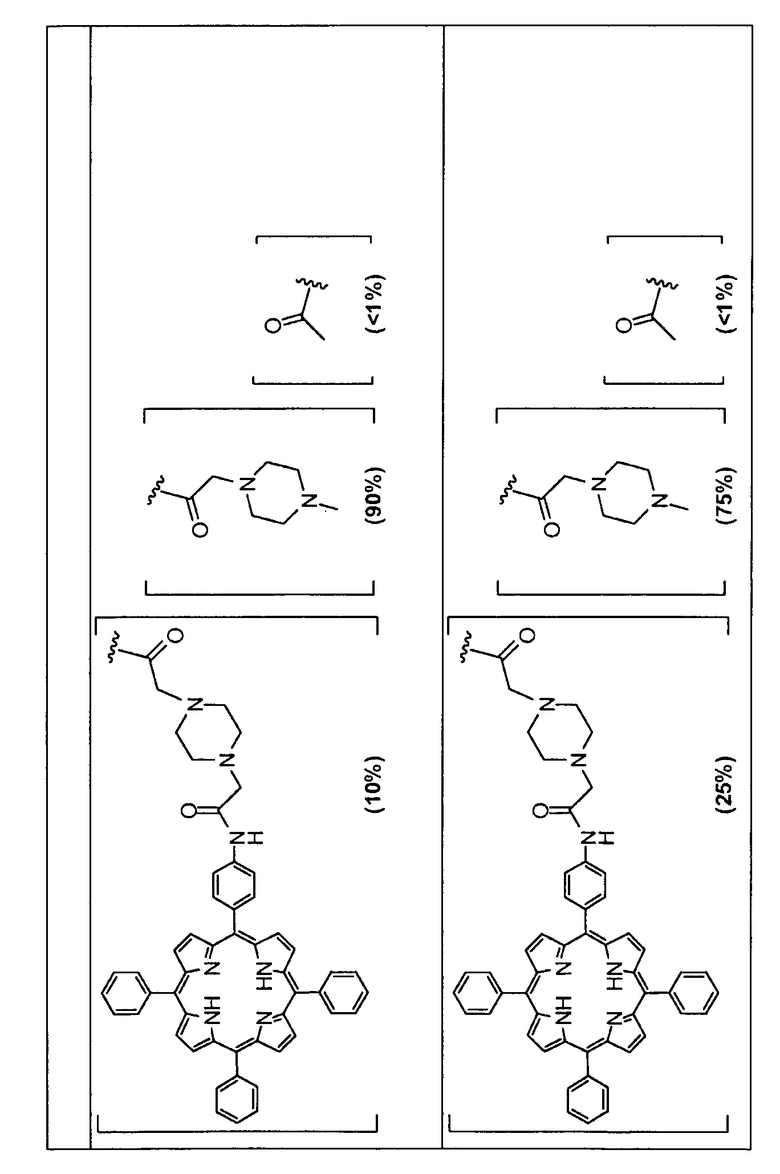
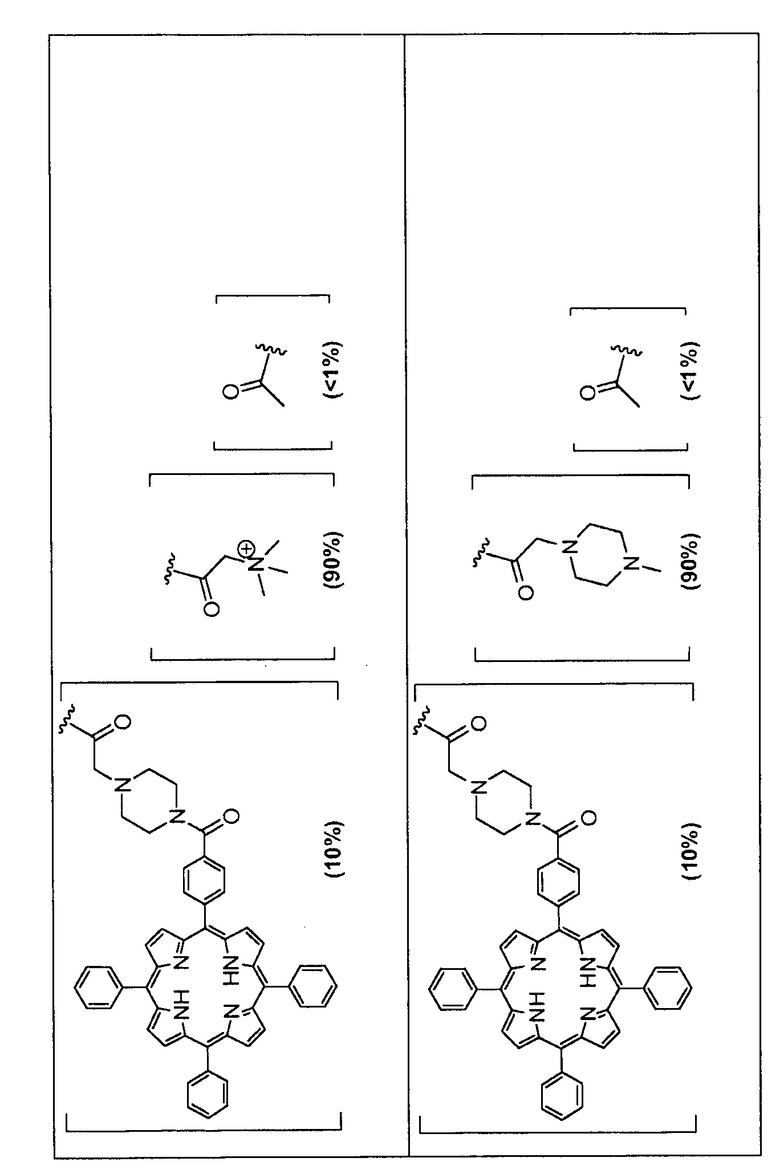
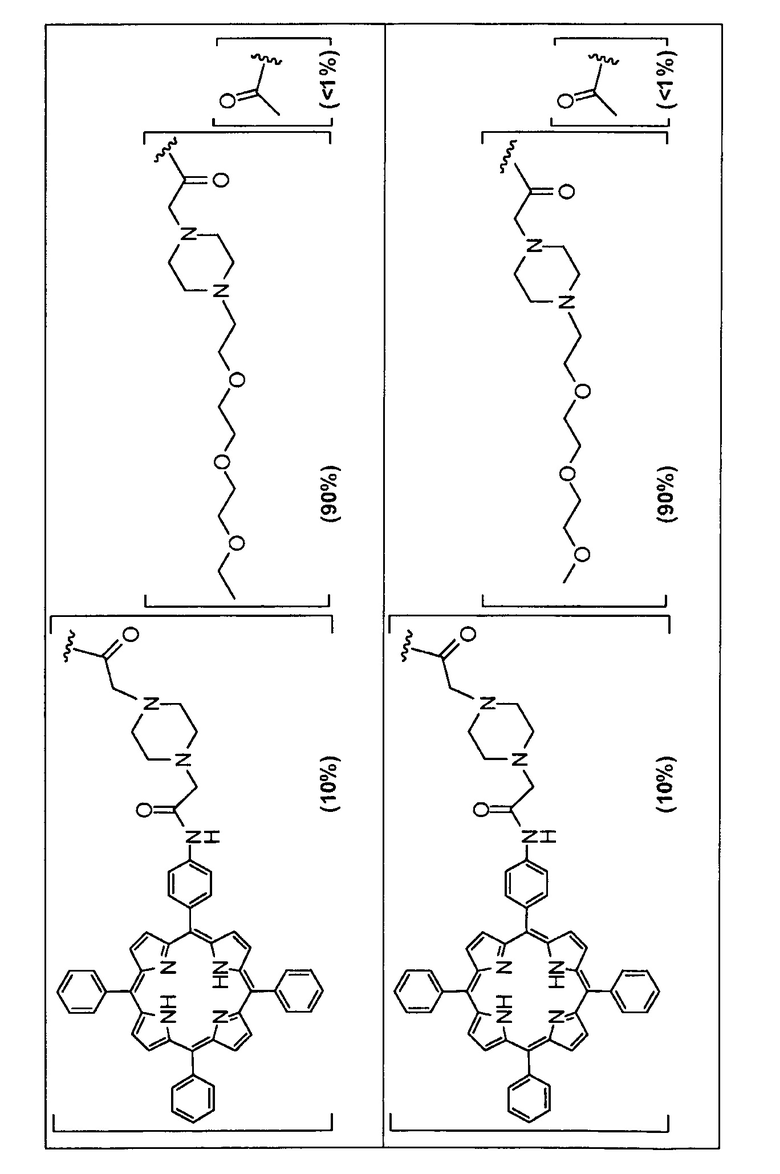
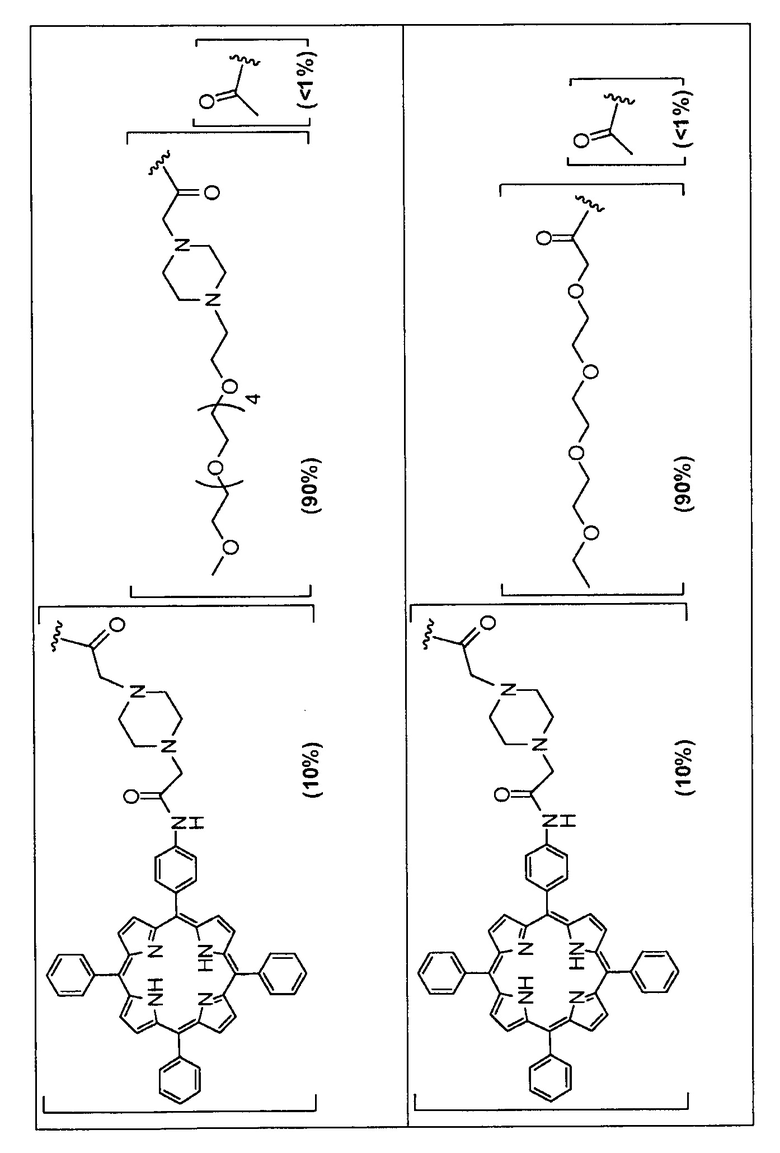
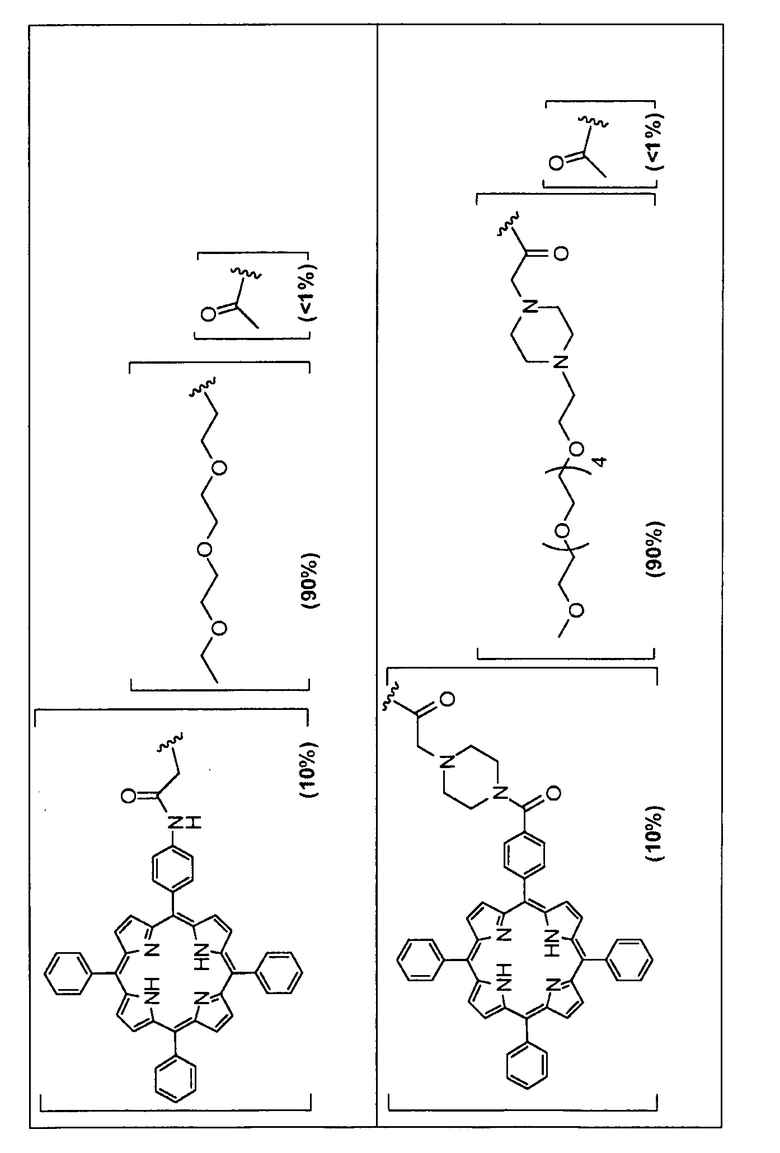
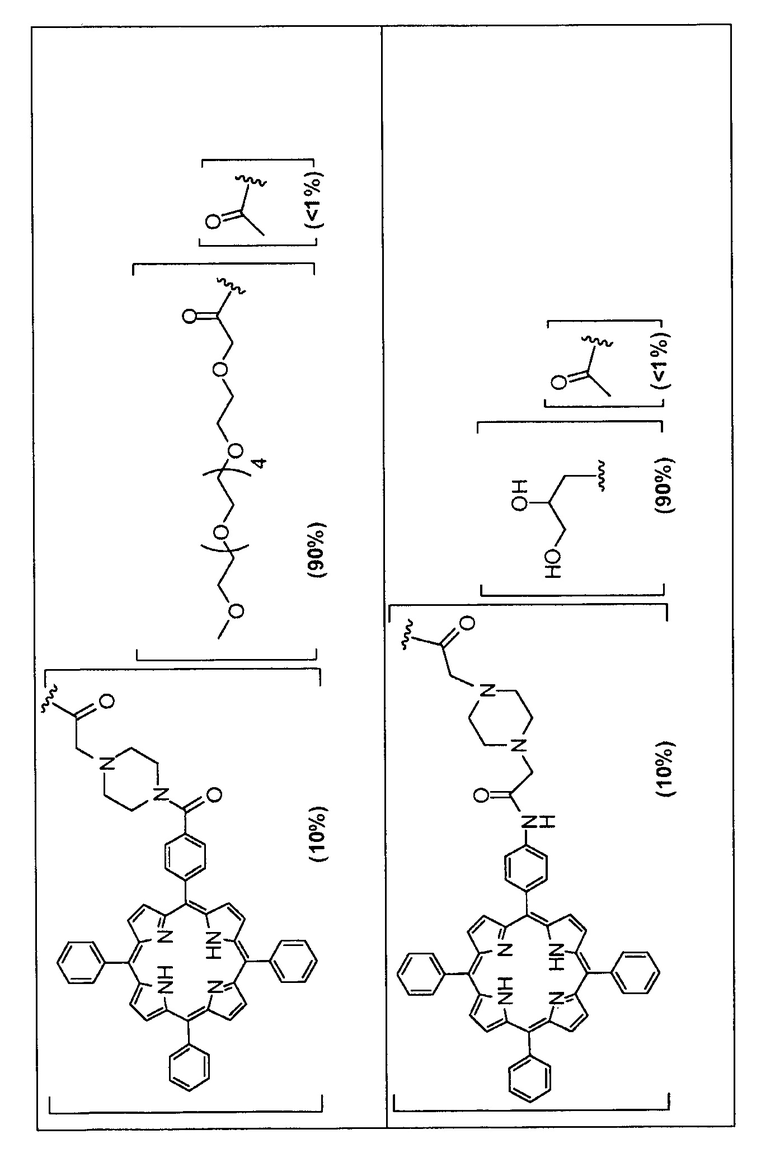
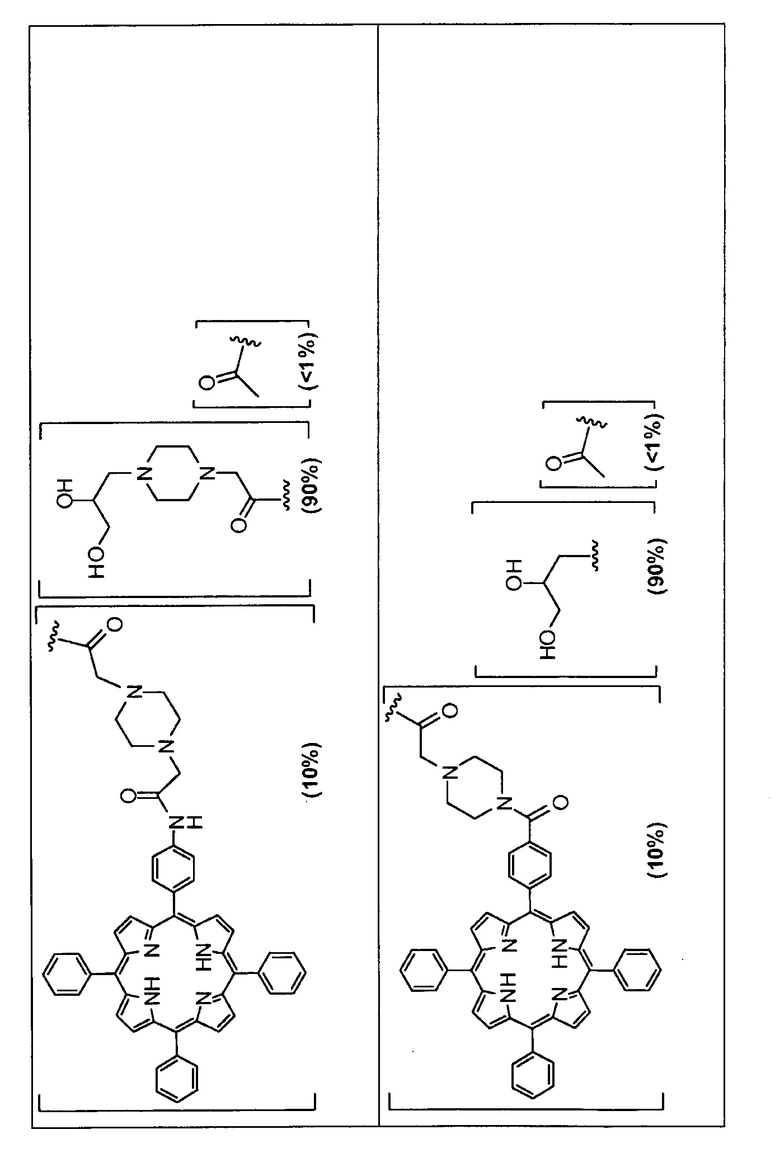
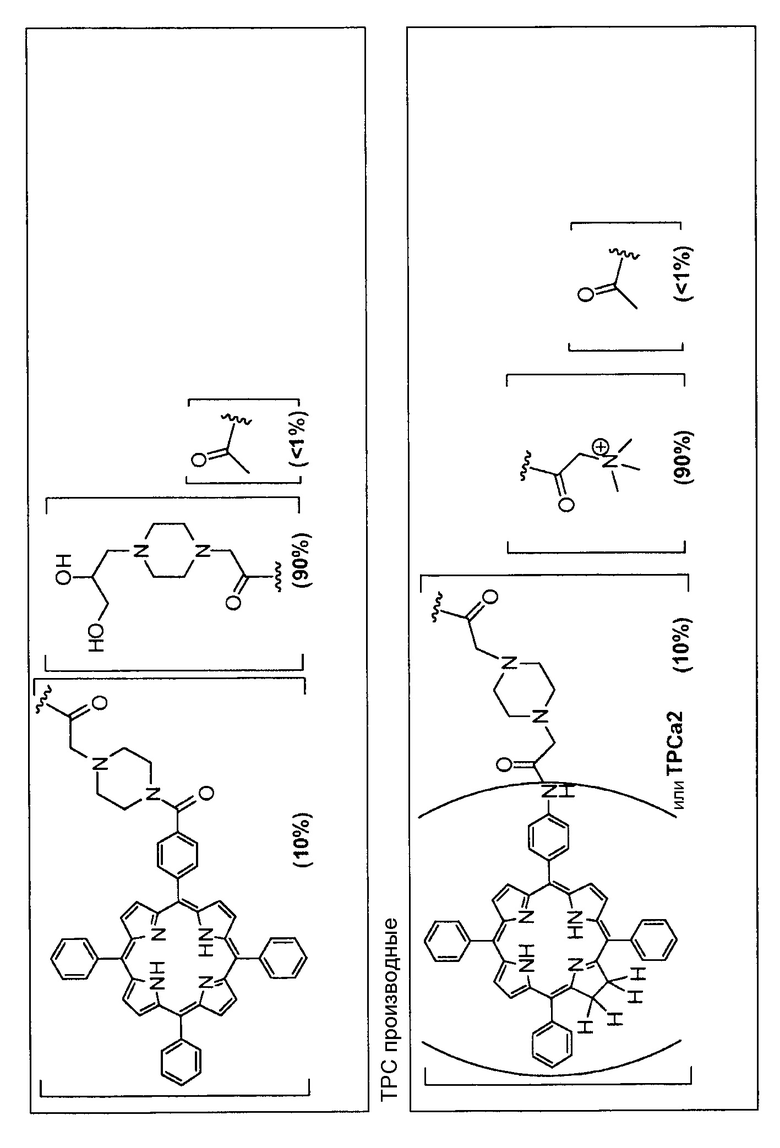
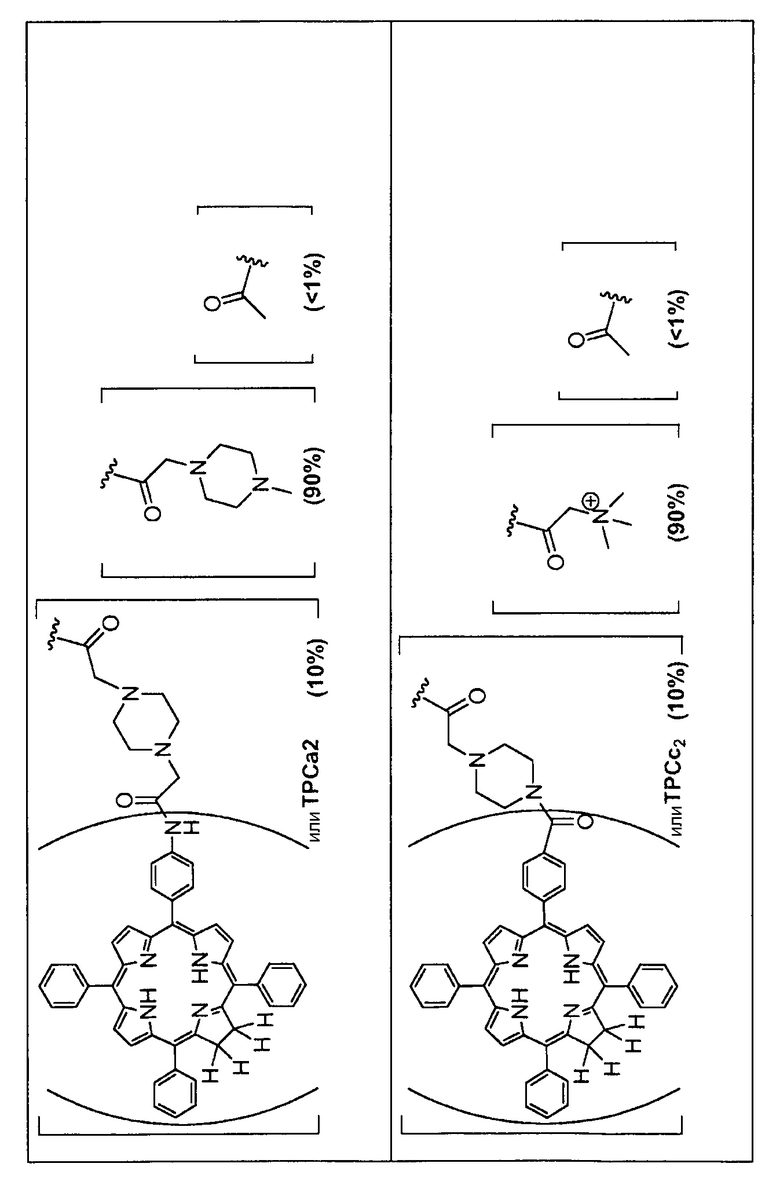
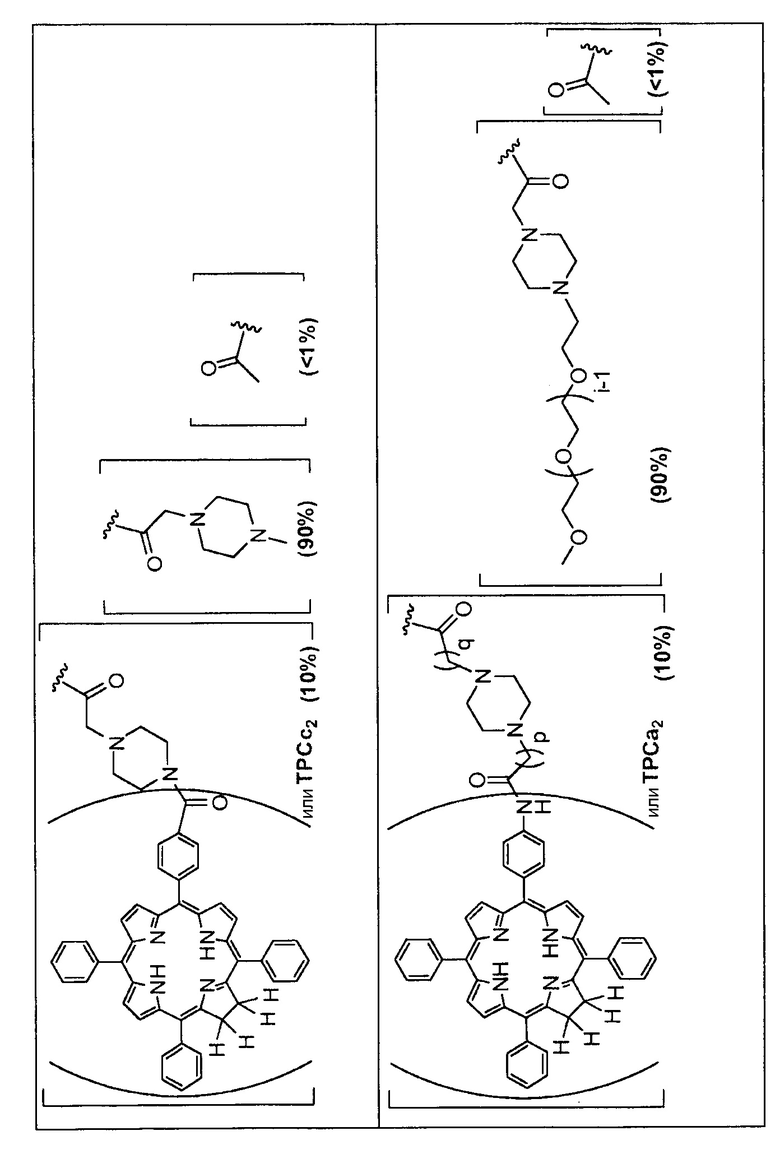
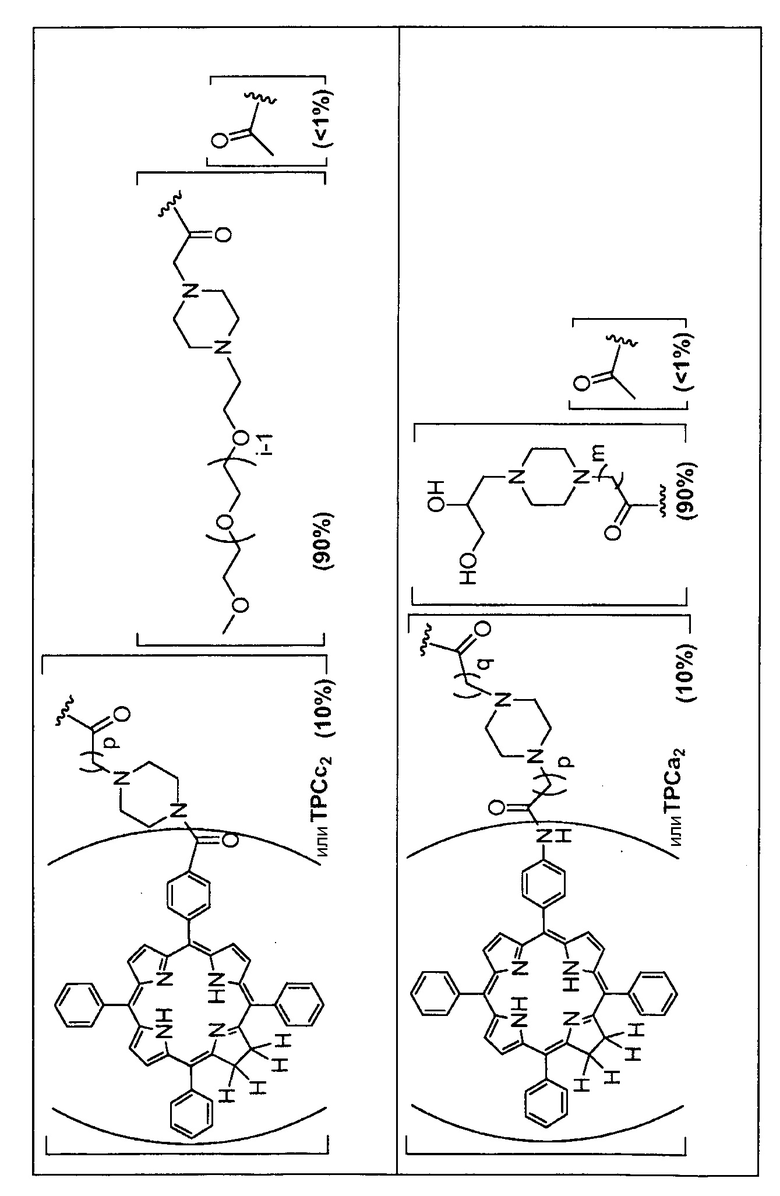
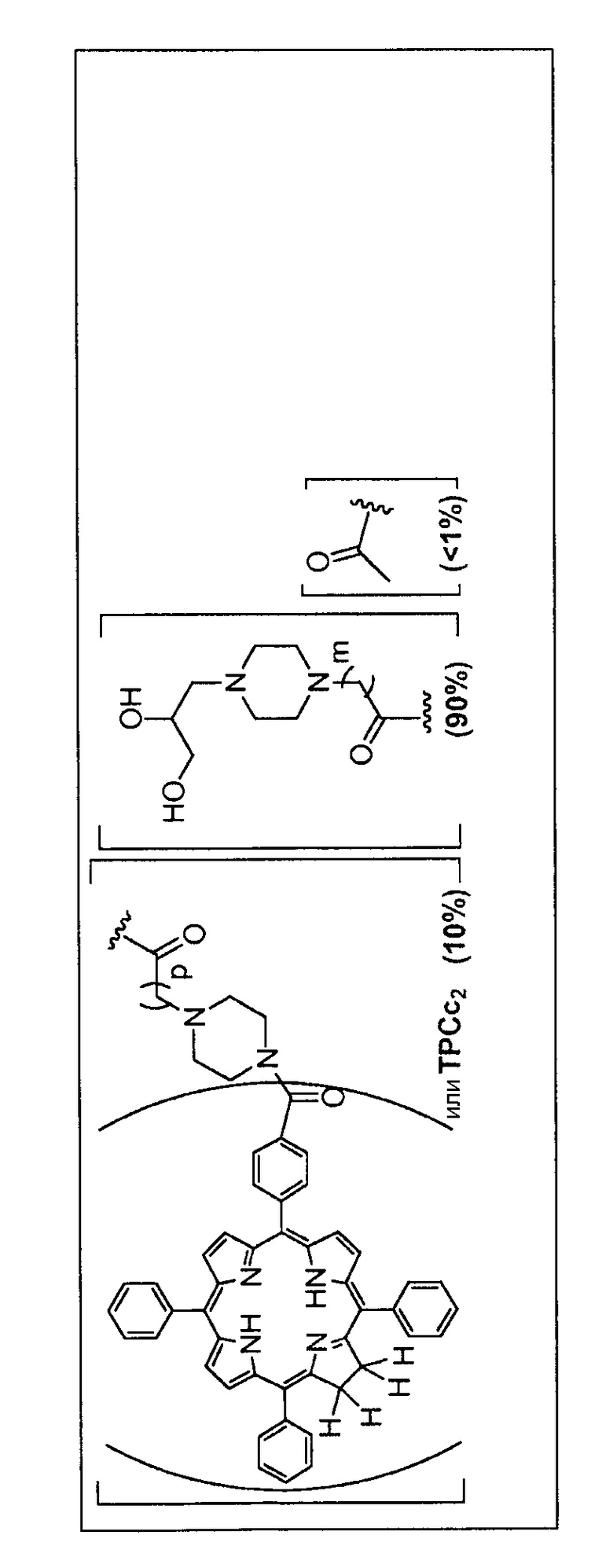
Особенно предпочтительными соединениями по изобретению являются также показанные на фигуре 1, для которых указано преобладание R групп, но оно может варьироваться до 5% в обе стороны от указанного значения. В каждом случае R группа с присоединенным фотосенсибилизатором имеет более низкое указанное преобладание.
Соединения по изобретению могут быть получены как описано в данном документе в примерах. В способах синтеза используются стандартные методы уровня техники, которые хорошо известны специалисту, и которые описаны в примерах ниже. Пегилирование и Тегилирование для обеспечения релевантных R групп могут быть осуществлены в соответствии со стандартными способами данной области техники. Настоящее изобретение также охватывает способы получения соединений по изобретению, например, как описано в примерах и схемах, описанных в данном документе.
Соединения по изобретению имеют низкую токсичность и, следовательно, подходят для широкого круга медицинских показаний.
Соединения по настоящему изобретению являются особенно подходящими для использования в методах ФХИ. Как проиллюстрировано в настоящих примерах, было показано, что соединения по настоящему изобретению обладают удивительно хорошей эффективностью при интернализации молекул в клетку. В примере 3 показано, что эффективность соединения по данному изобретению была значительно лучше, чем эффективность, достигаемая только с сенсибилизатором (в этом случае был использован фотосенсибилизатор TPCS2a, который был специально разработан для использования в ФХИ, и который находится на стадии клинической разработки для лечения рака (Berg et al. 2011, Photochem. Photobiol. Sci., 10, p.1637-1651)). Если сравнивать с TPCS2a, то соединения по изобретению были, по меньшей мере, вплоть до 10 раз более активными, то есть даже при использовании конъюгатов в 10 раз более низкой концентрации достигалось значительно большее усиления трансфекции, чем наблюдаемое с TPCS2a (см. фигуры 19 и 20).
Основной метод фотохимической интернализации (ФХИ) описан в WO 96/07432 и WO 00/54802, которые включены в настоящий документ посредством ссылки. Суммируя, молекула, предназначенная для интернализации, и фотосенсибилизирующий агент, в данном случае фотосенсибилизатор в виде части соединения по настоящему изобретению, приводят в контакт с клеткой. Фотосенсибилизирующий агент и молекула, предназначенная для интернализации, захватываются связанным с клеточной мембраной субкомпартментом внутрь клетки. При облучении клетки светом с соответствующей длиной волны фотосенсибилизирующий агент активируется, что прямо или косвенно создает реакционно активные виды, которые разрушают мембраны внутриклеточных компартментов. Это позволяет поглощенной молекуле высвободиться в цитозоль.
В этих методах фотохимический эффект используется в качестве механизма введения молекул, в ином случае не проникающих (или плохо проникающих) через мембрану в цитозоль клетки, который, таким способом, не приводит к обширному разрушению клеток или гибели клеток, если способ пригодным образом скорректирован, чтобы избежать чрезмерной продукции токсичных частиц, например, путем снижения времени воздействия света или дозы фотосенсибилизатора.
По существу, изобретение также касается способа введения молекулы в цитозоль клетки, включающего контактирование указанной клетки с молекулой, предназначенной для введения, и соединением по изобретению, и облучение клетки светом с длиной волны, эффективной для активации фотосенсибилизирующего агента соединения. После активации внутриклеточные компартменты в указанной клетке, содержащей указанное соединение, высвобождают молекулу, содержащуюся в этих компартментах, в цитозоль. Использование соединения по изобретению для интернализации молекулы, которая желательна для поглощения, также составляет часть изобретения.
ФХИ может осуществлять трансфекцию различных типов молекул в клетку. Например, молекула может быть выбрана из ДНК или антисмысловой ДНК; олиго(дезокси)нуклеотидов; РНК, такой как мРНК, миРНК, двухцепочечная (дс)РНК, одноцепочечная (он)антисмысловая РНК или РНК; ПНК, сахаров; белков; пептидов; мембранонепроницаемых лекарственных средств; других мембранонепроницаемых молекул, либо ковалентно, либо не ковалентно связанных комбинаций вышеупомянутых молекул. Предпочтительно, молекулы, предназначенные для введения, не интернализуются (то есть в цитозоль) значительно без помощи ФХИ, например, их интернализация ограничена, так что меньше, чем 50% (например, менее 30 или 10%) клеток, к которым они применяются, поглощают одну или несколько молекул в цитозоль в течение 4-часового периода.
Молекула, подлежащая интернализации, может быть выбрана с целью достижения различных результатов, например, для изменения, например, уменьшения или увеличения экспрессии мишеневого гена, или чтобы оказать влияние на свойства или жизнеспособность клеток. Чтобы влиять на экспрессию генов, молекула, подлежащая трансфекции, может представлять собой смысловой или антисмысловой олиго- или поли-нуклеотид, например, генную последовательность, например, в плазмиде или антисмысловом олигонуклеотиде или молекуле миРНК. Такие методы могут быть использованы при лечении или профилактике заболеваний и расстройств, таких как рак, а также могут быть использованы в приложениях генной терапии.
Способ по изобретению обеспечивает перенос молекулы, подлежащей интернализации, в цитозоль. Однако, будет понятно, что захват абсолютно каждой молекулы, контактирующей с клеткой, в цитозоль является не достижимым. Однако, достижимым является значительное и улучшенное поглощение относительно фонового уровня, при котором не используется ФХИ или соединение по изобретению.
Предпочтительно способы по изобретению позволяют осуществить интернализацию молекул на достаточном уровне, чтобы их действие проявлялось, например, в продуктах, экспрессированными этими клетками, или путем воздействия на клетки. Соответствующие концентрации молекулы для контактирования с клеткой могут быть скорректированы для достижения этой цели, например, при некоторых применениях может быть желательным, чтобы достичь повышения или снижения экспрессии мишеневого гена (или введенного гена) или гибели клеток после введения цитотоксической молекулы. Уменьшение или гибель клеток может составлять, по меньшей мере, 10%, например, по меньшей мере, уменьшение на 20, 30, 40 50, 60, 70, 80 или 90% (например, при экспрессии одного или нескольких белков, кодируемых мишеневым геном), либо гибель клеток после инкубации с клетками в течение, например, 24, 48, 72 или 96 часов (например, от 24 до 48 часов). Повышение экспрессии может быть оценено относительно существующих уровней, которые могут быть нулевыми, когда используется не-эндогенная молекула, и могут быть достигнуты численно одинаковые уровни с упомянутыми выше по уменьшению. Аналогичным образом, вид соединения (по изобретению) и/или концентрация и время облучения могут быть скорректированы, чтобы добиться уменьшения, описанного выше.
Уровни экспрессированных продуктов могут быть определены, например, путем определения уровня белка в клетке с использованием стандартных методов, известных в данной области техники, таких как вестерн-блоттинг. Уровень снижения белка зависит от времени полужизни белка, т.е. ранее существующий белок будет удален в соответствии с его временем полужизни. Гибель клеток может быть определена любым подходящим средством.
Воздействие введенного генетического материала также может быть определено в терминах уровней экспрессии, например, мРНК, которая имеется в клетке, например, метод может быть осуществлен для достижения повышения или снижения уровней мРНК, по меньшей мере, на 10%, например, по меньшей мере на 20, 30, 40, 50, 60, 70, 80 или 90% повышение или снижение после инкубации с клетками, например, в течение 24, 48, 72 или 96 часов, например, от 24 до 48 часов, относительно уровней мРНК мишени или введенной последовательности в тот же момент времени без добавления генетического материала. Также его можно измерять с использованием стандартных способов, известных в данной области, таких как способы гибридизации или блоттинга и ОТ-ПЦР (RT-PCR).
Термин "клетка" используется в настоящем документе с включением всех эукариотических клеток (включая клетки насекомых и клетки грибов). Представительные "клетки", таким образом, включают все типы относящихся и не относящихся к млекопитающим клеток животных, клеток растений, клеток насекомых, клеток грибов и простейших. Предпочтительными являются, однако, клетки млекопитающих, например, клетки кошек, собак, лошадей, ослов, овец, свиней, коз, коров, мышей, крыс, кроликов, морских свинок, но наиболее предпочтительны, клетки человека.
Как используется в настоящем документе "контактирование" к осуществлению физического контакта клеток и фотосенсибилизирующего агента, содержащего соединение по изобретению и/или молекулу, предназначенную для ведения, в условиях, подходящих для интернализации в клетки, например, предпочтительно, при температуре 37°C в соответствующей питательной среде, например, при температуре от 25 до 39°C.
Термин "облучение" клетки с целью активирования фотосенсибилизирующего агента относится к воздействию светом прямо или не прямо, как описано далее. Таким образом клетки могут быть освещены источником света, например, прямо (например, отдельные клетки in vitro) или не прямо, например, in vivo, когда клетки расположены под поверхностью кожи или находятся в виде слоя клеток, не все из которых прямо освещаются, т.е. без экранирования другими клетками.
Стадия облучения светом для активации фотосенсибилизирующего вещества может осуществляться в соответствии с методами и способами, хорошо известными в данной области техники. Длина волны и интенсивность света выбираются в зависимости от используемого фотосенсибилизирующий агента. Подходящие источники искусственного света хорошо известны в данной области техники, например, используя синюю (450-475 нм) или красную (620-750 нм) длины волн света.
Время, в течение которого клетки подвергаются воздействию света в способах по настоящему изобретению, может меняться. Эффективность интернализации молекулы в цитозоль возрастает с увеличением интенсивности освещения до максимума, после которого увеличивается повреждение клеток и, таким образом, гибель клеток.
Предпочтительная длительность стадии облучения зависит от таких факторов, как мишень, фотосенсибилизатор (в соединении по изобретению), количество фотосенсибилизатора, накопившегося в мишеневых клетке или ткани, и перекрывания между спектром поглощения фотосенсибилизирующего вещества и спектром испускания источника света. Обычно длительность стадии облучения составляет порядка от нескольких секунд до нескольких минут или вплоть до нескольких часов, например, предпочтительно до 60 минут, например, от 0,25 или 1 до 30 минут, например, от 0,5 до 3 минут, или от 1 до 5 минут, или от 1 до 10 минут, например, от 3 до 7 минут, и, предпочтительно, приблизительно 3 минуты, например, от 2,5 до 3,5 минут. Также может быть использовано более короткое время облучения, например, от 1 до 60 секунд, например, 10-50, 20-40 или 25-35 секунд.
Соответствующие дозы света могут быть подобраны специалистом в данной области техники, и они также будут зависеть от фотосенсибилизатора (в соединении по изобретения) и от количества фотосенсибилизатора, накопленного в мишеневых клетке или ткани. Например, доза света, обычно используемая для фотодинамической терапии рака с фотосенсибилизатором Фотофрином и предшественником протопорфирина 5-аминолевулиновой кислотой, находится в диапазоне 50-150 Дж/см2 с диапазоном плотности потока менее 200 МВт/см2 с целью избежать гипертермии. Дозы света обычно являются более низкими, когда используют фотосенсибилизаторы с более высокими коэффициентами экстинкции в красной области видимого спектра. Для методов ФХИ могут быть использованы более низкие дозы, например, доза света в диапазоне 5-25 Дж/см2 с диапазоном плотности потока 75-150 МВт/см2. Кроме того, для лечения нераковых тканей с меньшим количеством накапливающегося фотосенсибилизатора общее требуемое количество света может быть по существу более высоким, чем в случае лечения рака. Более того, если необходимо сохранить жизнеспособность клетки, необходимо избегать образования чрезмерных уровней токсичных частиц, и соответствующие параметры могут быть скорректированы соответствующим образом.
Методы ФХИ по изобретению могут неизбежно приводить к гибели некоторого количества клеток в результате фотохимической обработки, то есть под действием ФДТ посредством образования токсичных частиц при активации фотосенсибилизирующего агента. В зависимости от предлагаемого использования, эта гибель клеток может не быть следствием и, в действительности, может быть целесообразной для некоторых применений (например, при лечении рака).
В одном варианте осуществления изобретение относится к способу достижения гибели клеток, включающему контактирование указанных клеток с соединением по изобретению, и облучение клеток светом с длиной волны, эффективной для активации фотосенсибилизирующего агента соединения к образованию активных форм кислорода, которые вызывают гибель указанных клеток. Когда гибель клеток (ФДТ) будет достигнута, время, интенсивность и длина волны на стадии облучения выбираются соответствующим образом для оптимального достижения гибели клеток-мишеней.
В некоторых вариантах осуществления настоящего изобретения, однако, гибели клеток удается избежать, например, когда желательно ингибирование экспрессии гена в отсутствие клеточной токсичности, или, если гибель клетки вместо этого достигается путем внедренной молекулы. Например, при некоторых применениях является очень удобным достигать ингибирования экспрессии генов или экспрессии в отсутствие основной клеточной токсичности или влиять на жизнеспособность клеток, например, в некоторых подходах генной терапии. Способы по изобретению могут быть модифицированы таким образом, чтобы часть или доля выживших клеток регулировалась посредством подбора световой дозы в зависимости от концентрации фотосенсибилизирующего агента (в соединениях по изобретению). Опять же, такие методы известны в данной области техники.
В способах применения, в которых жизнеспособные клетки являются желательными, практически все клетки, или значительное их большинство (например, по меньшей мере, 50%, более предпочтительно, по меньшей мере, 60, 70, 80 или 90% клеток) не уничтожаются. Жизнеспособность клеток после обработки посредством ФХИ может быть определена стандартными методами, известными в данной области техники, такими как тест MTS.
Независимо от величины гибели клеток, индуцируемой путем активации фотосенсибилизатора, в некоторых применениях важно, чтобы доза света регулировалась таким образом, чтобы некоторые из отдельных клеток, где проявляется эффект ФХИ, не уничтожались посредством фотохимической обработки отдельно (хотя они могут быть впоследствии уничтожены молекулами, введенными в клетки, если эти молекулы оказывают цитотоксическое действие).
Цитотоксические эффекты могут быть достигнуты с помощью, например, введения цитотоксической молекулы (например, цитотоксического пептида, такого как гелонин или блеомицин), или генной терапии, при которой агент, например, ген, антисмысловой олигонуклеотид или молекула миРНК, интернализуется в опухолевую клетку способом по настоящему изобретению.
Соединения и способы по настоящему изобретению могут быть использованы in vitro или in vivo, например, либо для обработки in situ, либо для лечения ex vivo с последующим введением обработанных клеток в организм, для различных целей, включая ингибирование или повышение экспрессии определенных продуктов генов, например, в методах генной терапии.
Таким образом, в дальнейшем аспекте изобретение относится к композиции (например, фармацевтической композиции), содержащей соединение или конъюгат по изобретению, и, необязательно, отдельно, молекулу, предназначенную для интернализации. Когда указанная композиция является фармацевтической композицией, она содержит один или несколько фармацевтически приемлемых разбавителей или вспомогательных веществ.
В следующем аспекте изобретение относится к указанному соединению или композиции для применения в терапии.
В настоящем изобретении предложен набор, включающий соединение или композицию по настоящему изобретению, как описано в данном документе, и молекулу, предназначенную для интернализации. Предпочтительно, указанный набор (или продукт) предназначен для одновременного, раздельного или последовательного применения в медицинском лечении, предпочтительно, в лечении рака или, как описано более подробно далее, в целях вакцинации.
Таким образом, дальнейший аспект касается соединения или композиции, и, необязательно, молекулы, предназначенной для интернализации, как определено в данном документе, для использования при лечении или предотвращении у субъекта заболевания, расстройства или инфекции, в которых, предпочтительно, проявляется анормальный или чрезмерный рост клеток, или в которых происходит анормально повышенная или подавленная экспрессия генов, особенно предпочтительно, когда указанным заболеванием является рак. Способы лечения или профилактики заболевания, расстройства или инфекции у субъекта (что соответствует применениям, описанным в настоящем документе) путем введения соединения или композиции по изобретению и, необязательно, молекулы, предназначенной для интернализации. Предпочтительно, указанное лечение или профилактика достигаются с помощью способа, описанного в настоящем документе.
Указанный способ может быть осуществлен с использованием методов ФХИ, или методы ФХИ или ФДТ могут быть использованы, когда гибель клеток является конечной целью.
Таким образом, метод ФХИ может быть осуществлен, как описано выше, т.е. путем контактирования клеток субъекта с молекулой, которая должна быть введена, и указанного соединения или композиции, и облучением клеток светом с длиной волны, эффективной для активации фотосенсибилизирующего агента соединения. Предпочтительно, молекула, которая должна быть введена, представляет собой цитотоксическую молекулу, предпочтительно, блеомицин.
Метод ФДТ может осуществляться путем контактирования клеток у субъекта с указанным соединением или композицией, и облучением клеток светом с длиной волны, эффективной для активации фотосенсибилизирующего агента соединения с получением активных форм кислорода, которые вызывают гибель клеток.
Альтернативно, настоящее изобретение относится к применению соединения или композиции, описанных в настоящем документе, и, необязательно, молекулы, предназначенную для интернализации в клетку, при получении лекарственного средства для лечения или предупреждения заболевания, расстройства или инфекции. Также предложено применение указанного соединения или композиции при получении лекарственного средства для указанного лечения или профилактики, где указанное лечение или профилактика описаны в настоящем документе. При указанных заболевании, расстройстве или инфекции, предпочтительно, проявляется анормальный или чрезмерный рост клеток или аномальное повышение или подавление экспрессии генов, и/или являлось бы целесообразным уменьшение роста клеток или подавление или повышение экспрессии одного или нескольких генов.
Как указано в настоящем документе, термин аномальное или чрезмерное относится к тому, что считается нормальным в соответствии с возрастом и полом для нормальных индивидуумов или, с другой стороны, для нормальных частей организма некоторых индивидуумов. Аномальный рост может, таким образом, относиться к раковым заболеваниям, доброкачественным опухолям, и чрезмерный рост клеток может касаться таких состояний кожи, как актинический кератоз, бородавки и родинки и т.д. Раковые заболевания, к которым могут быть применены эти методы, включают рак головы и шеи, рак желчных протоков, рак мозга, меланому, кожные метастазы (при различных видах рака), рак легких, мезотелиому, рак поджелудочной железы, рак желудка, рак прямой кишки, рак анального канала, рак полового члена, рак вульвы и рак пищевода. Таким образом, лекарственное средство может быть использовано для лечения рака. Когда используется молекула, предназначенная для интернализации, она может представлять собой противораковое химиотерапевтическое средство.
Аномально повышенная или подавленная экспрессия генов может быть улучшена путем изменения экспрессии одного или более генов-мишеней у указанного субъекта, например, когда молекула, предназначенная для интернализации, представляет собой ген, антисмысловой олигонуклеотид или молекулу миРНК. Предпочтительно, указанное лекарственное средство предназначено для генной терапии, то есть для лечения или предупреждения заболевания, расстройства или инфекции, которые характеризуются аномальной экспрессией генов, или для которых целесообразным является подавление одного или нескольких генов. Указанное изменение включает даун-регуляцию указанной экспрессии.
Когда используется метод ФХИ, соединение (или композиции по изобретению), и молекула, предназначенная для интернализации, могут контактировать с клетками или тканью пациента (или субъекта) одновременно или последовательно, и указанные клетки облучают светом с длиной волны, эффективной для активации фотосенсибилизирующего агента указанного соединения, и облучение проводят до, в процессе или после клеточного захвата указанных соединения и молекулы во внутриклеточный компартмент, содержащий указанный фотосенсибилизирующий агент, предпочтительно, до клеточного захвата указанной перемещаемой молекулы во внутриклеточный компартмент.
Также рассмотрены способы, в которых клетки, вводимые субъекту, подвергаются обработке. Таким образом, в альтернативном аспекте изобретение относится к способу лечения или профилактики заболевания, расстройства или инфекции у пациента, включающий введение соединения (или композиции) по изобретению и, необязательно, молекулы, предназначенной для интернализации, в одну или несколько клеток in vitro, in vivo или ex vivo в соответствии со способами, описанными выше, и, в случае необходимости (т.е., когда трансфекция проводится in vitro или ex vivo), введение указанных клеток указанному пациенту. Поученные таким образом клетки могут быть использованы в терапии или для конкретного использования, как описано выше.
Как указано в настоящем документе, субъектом является животное, предпочтительно, млекопитающее, например, корова, лошадь, овца, свинья, коза, кролик, кошка, собака, особенно предпочтительно, человек.
Как определено в данном документе, термин "лечение" относится к уменьшению, смягчению или устранению одного или нескольких симптомов заболевания, расстройства или инфекции, которые подвергают лечению, относительно симптомов до начала лечения. Термин "профилактика" означает замедление или предотвращение появления симптомов заболевания, расстройства или инфекции.
Композиции по настоящему изобретению могут также включать клетку, содержащую молекулу, которая была интернализована в цитозоль указанной клетки способом по изобретению. Изобретение, кроме того, охватывает такие композиции для применения в терапии, в частности, в терапии раковых заболеваний или в генной терапии.
Таким образом, в еще одном аспекте изобретения предложены клетка или популяция клеток, содержащих молекулу, которая была интернализована в цитозоль указанной клетки, где клетка может быть получена способом по настоящему изобретению.
В еще одном аспекте изобретения предложено применение такой клетки или популяции клеток при получении композиции или лекарственного средства для использования в терапии, как описано выше, предпочтительно, в терапии раковых заболеваний или в генной терапии.
В изобретении, кроме того, предложен способ лечения или профилактики пациента, включающий введение указанному пациенту клеток или композиций по настоящему изобретению, то есть способ, включающий стадии введения молекулы в клетку, как описано выше, и введение полученной таким образом указанной клетки указанному пациенту. Предпочтительно, указанные способы используются для лечения раковых заболеваний или в генной терапии (или при вакцинации, как описано далее).
In vivo может быть использован любой способ, распространенный или стандартный в данной области, например, инъекция, инфузия, местное введение, трансдермальное введение, как на внутреннюю, так и внешнюю поверхности тела и т.д. Для применения in vivo изобретение может быть использовано в отношении любой ткани, которая содержит клетки, в которых помещены содержащее фотосенсибилизирующий агент соединение или молекула, предназначенная для интернализации, включая области расположения физиологических жидкостей организма, а также твердые ткани. Все ткани могут быть подвергнуты лечению при условии, что фотосенсибилизатор захватывается клетками-мишенями, и свет может быть доставлен должным образом.
Таким образом, композиции по изобретению могут быть получены любым удобным образом в соответствии с методиками и способами, известными в фармацевтической области, например, с использованием одного или нескольких фармацевтически приемлемых разбавителей, носителей или инертных наполнителей. Термин «фармацевтически приемлемый» в данном документе относится к ингредиентам, которые совместимы с другими ингредиентами композиций, а также являются физиологически приемлемыми для реципиента. Тип композиции и носителей или инертных наполнителей, дозировки и так далее могут быть выбраны общепринятым образом в соответствии с выбранным и требуемым путем введения, целью лечения и так далее. Дозировки аналогично могут быть определены общепринятым способом, и они могут зависеть от природы молекулы, цели лечения, возраста пациента, способа введения и так далее. В связи с использованием фотосенсибилизирующего агента следует также принимать во внимание их активность/способность разрушать мембраны при облучении.
Следующее применение соединений и композиций по настоящему изобретению заключается в протоколах вакцинации, так как методы ФХИ могут быть использованы, чтобы предоставить или экспрессировать антигены на поверхность клетки. Таким образом, после переноса и высвобождения молекулы, подлежащей интернализации в цитозоль клетки с помощью ФХИ, она может быть перенесена на поверхность, где она может находиться вне клетки, т.е. на поверхности клетки. Этот метод особенно полезен в области вакцинации, когда компоненты вакцины, т.е. антигены или иммуногены, могут быть введены в ячейку для презентации на поверхности для того, чтобы побудить, облегчить или усилить иммунный ответ.
Настоящее изобретение, таким образом, относится к способу экспрессии антигенной молекулы (например, антигена) или ее части на поверхность клетки, предпочтительно, антиген-презентирующей клетки, где указанный метод включает введение молекулы в цитозоль клетки с помощью ФХИ, используя соединения и способы, как описано в настоящем документе, где молекула или ее часть, впоследствии представляется на поверхности указанной клетки.
Выражаясь альтернативно, настоящее изобретение относится к способу экспрессии антигенной молекулы или ее части на поверхность клетки, включающему контактирование указанной клетки с указанной антигенной молекулой и соединением, как определено в настоящем документе, и облучение клеток светом с длиной волны, эффективной для активации фотосенсибилизирующего агента соединения, где антигенная молекула высвобождается в цитозоль клетки, и антигенная молекула или ее часть достаточного размера для стимуляции иммунного ответа представлена на клеточной поверхности.
Как используется в настоящем документе, термин "экспрессия" относится к наличию антигенной молекулы или ее части на поверхности указанной клетки так, что по меньшей мере часть этой молекулы становится открытой и доступной для среды, окружающей клетку. Экспрессия на "поверхность" может быть достигнута, когда молекула, подлежащая экспрессии, находится в контакте с клеточной мембраной и/или с компонентами, которые могут присутствовать или обеспечивать присутствие в этой мембране.
Такая презентация антигена может успешно приводить к стимуляции иммунного ответа, предпочтительно, иммунного ответа, который обеспечивает защиту против последующей атаки структурой, включающей или содержащей указанную антигенную молекулу или ее часть, и, следовательно, изобретение может найти конкретное применение в качестве способа вакцинации.
Более конкретно, в этом аспекте изобретение относится к способу экспрессии антигенной молекулы или ее части на поверхность клетки, где указанный метод включает:
контактирование указанной клетки с указанной антигенной молекулой и с соединением по настоящему изобретению, где указанная молекула и указанное соединение, каждый, поглощаются в ограничивающую компартмент внутриклеточную мембрану указанной клетки; и
облучение указанной клетки светом с длиной волны, эффективной для активации фотосенсибилизирующего агента соединения, так что мембрана указанного внутриклеточного компартмента разрушается, высвобождая указанную молекулу в цитозоль клетки, не убивая клетку,
где указанная высвобожденная антигенная молекула, или ее часть впоследствии презентуются на поверхности указанной клетки.
Как используется в данном документе, термин "разрушенный" компартмент относится к разрушению целостности мембраны этого компартмента либо временно, или постоянно, достаточно, чтобы позволить высвободить содержащуюся в нем антигенную молекулу.
Выражаясь альтернативно, настоящее изобретение относится также к соединению или композиции для использования при экспрессии антигенной молекулы или ее части на поверхность клетки, например, при лечении или профилактике заболевания, расстройства или инфекции у субъекта, предпочтительно, генерируя или стимулируя иммунный ответ, предпочтительно, в методе вакцинации. Указанная композиция, предпочтительно, включает антигенную молекулу и соединение по настоящему изобретению. Предпочтительно, указанная композиция является фармацевтически приемлемой и содержит также фармацевтически приемлемый инертный наполнитель, носитель или разбавитель, как описано выше. Предпочтительно, указанное лечение или профилактика достигаются с использованием способа, описанного в настоящем документе.
В следующем аспекте изобретение относится также к применению антигенной молекулы и/или соединения по настоящему изобретению при получении лекарственного средства для применения в экспрессии указанной антигенной молекулы или ее части на поверхность клетки, например, для лечения или профилактики заболевания, расстройства или инфекции у субъекта, предпочтительно, для генерации или стимулирования иммунного ответа, предпочтительно, в способе вакцинации. Предпочтительно, указанное лечение или профилактика достигаются с помощью метода, описанного в настоящем документе. Предложен также соответствующий способ лечения или профилактики путем введения указанной антигенной молекулы и соединения по настоящему изобретению.
В еще одном аспекте настоящего изобретения предложен продукт, содержащий антигенную молекулу и соединение по настоящему изобретению в качестве комбинированного препарата для одновременного, раздельного или последовательного использования в экспрессии указанной антигенной молекулы или ее части на поверхность клетки, например, для лечения или профилактики заболевания, расстройства или инфекции у субъекта, предпочтительно, для стимуляции иммунного ответа.
В еще одном аспекте настоящего изобретения предложен набор для использования в экспрессии антигенной молекулы или ее части на поверхность клетки, где указанный набор включает
первый контейнер, содержащий указанную антигенную молекулу; и
второй контейнер, содержащий соединение по настоящему изобретению.
Согласно настоящему изобретению антигенная молекула может представлять собой любую молекулу, если такая молекула или ее часть способна к стимулированию иммунного ответа в случае ее попадания соответствующим образом в иммунную систему. Целесообразно поэтому, чтобы антигенная молекула была бы антигеном или компонентом вакцины, таким как структура, содержащая полипептид.
Многие такие антигены или компоненты антигенных вакцин известны в технике и включают все возможные варианты бактериальных и вирусных антигенов, причем либо истинных антигенов, либо антигенных компонентов любых патогенных видов, включая простейших или высшие организмы. Традиционно, антигенные компоненты вакцин включают целые организмы (либо живые, либо убитые или ослабленные), то есть цельноклеточные вакцины, кроме того, широко исследовались субъединичные вакцины, то есть вакцины, построенные на определенных антигенных компонентах организма, например, на белках или пептидах, или даже на углеводах, и результаты этих исследований описаны в литературе. В соответствии с настоящим изобретением любой такой «субъединичный» компонент вакцины может быть использован в качестве антигенной молекулы.
Однако, изобретение в особенности может найти применение в области пептидных вакцин. Таким образом, предпочтительной антигенной молекулой согласно настоящему изобретению является пептид (который, по определению настоящего изобретения, включает пептиды, как короткие, так и длинные, то есть пептиды, олигопептиды или полипептиды, а также белковые молекулы или их фрагменты, то есть пептиды, содержащие 5-500 аминокислотных остатков, например, от 10 до 250, такие как от 15 до 75 или от 8 до 25 аминокислот). Части антигенных молекул, которые презентируются или экспрессируются, предпочтительно, включают такие части, которые генерируются внутри клетки с помощью механизма антигенного процессинга. Эти части, однако, могут генерироваться при помощи других средств, что достигается созданием определенных антигенных конструкций (например, рН-чувствительных участков) или посредством других механизмов клеточного процессинга. Соответственно, такие части характеризуются размером, достаточным для генерирования иммунного ответа, например, в случае пептидов, они имеют длину более 5, например, больше, чем 10 или 20 аминокислот.
В литературе описано огромное количество пептидов, которые могут быть кандидатами на использование в составе вакцин, например, при лечении вирусных заболеваний и инфекций, таких как СПИД/ВИЧ инфекции или грипп, парвовирус собак, вирус лейкоза крупного рогатого скота, гепатит и пр. (см., например, Phanuphak et al., Asian Pac. J. Allergy. Immunol. 1997, 15(1), 41-8; Naruse, Hokkaido Igaku Zasshi 1994, 69(4), 811-20; Casal et al., J. Virol., 1995, 69(11), 7274-7; Belyakov et al., Proc. Natl. Acad. Sci. USA, 1998, 95(4), 1709-14; Naruse et al., Proc. Natl. Sci. USA, 1994 91 (20), 9588-92; Kabeya et al., Vaccine 1996, 14(12), 1118-22; Itoh et al., Proc. Natl. Acad. Sci. USA, 1986, 83(23) 9174-8. Аналогично могут использоваться бактериальные пептиды, если такие пептидные антигены могут быть получены из других организмов или видов.
В дополнение к антигенам, полученным из патогенных организмов, были также предложены пептиды для использования в качестве вакцин против рака или других заболеваний, таких как множественный склероз. Так, например, пептиды мутантного онкогена являются очень многообещающими в качестве антигенов противораковых вакцин, действующих в направлении стимуляции цитотоксических Т-лимфоцитов (Schirrmacher, Journal of Cancer Research and Clinical Oncology, 1995, 121, 443-451; Curtis Cancer Chemotherapy and Biological Response Modifiers, 1997, 17, 316-327). Была также исследована синтетическая пептидная вакцина, применяемая для лечения метастазов меланомы (Rosenberg et al., Nat. Med. 1998, 4(3), 321-7). Пептидная вакцина на основе рецептора Т-клеток для лечения множественного склероза описана Wilson et al., J. Neuroimmunol. 1997, 76(1-2), 15-28. Любой компонент такой пептидной вакцины может использоваться в качестве антигенной молекулы согласно настоящему изобретению, если такие пептиды описаны или предложены в качестве пептидных вакцин в литературе. Следовательно, такой пептид может быть синтетическим, либо он может быть выделен или иным образом получен из организма.
Клетка, которая является объектом данных способов, вариантов использования и т.п. согласно настоящему изобретению, может представлять собой любую клетку, которая способна к экспрессии или презентации на своей поверхности молекулы, которая вводится или переносится в ее цитозоль.
Клетка, кстати, представляет собой иммунную эффекторную клетку, т.е. клетку, участвующую в иммунном ответе. Однако другие клетки также могут презентировать антиген для иммунной системы, и они также входят в объем настоящего изобретения. Клетки в соответствии с настоящим изобретением, таким образом, преимущественно представляют собой антиген-презентирующие клетки. Антиген-презентирующая клетка может участвовать в любом аспекте или "ответвлении" иммунного ответа, включая как гуморальный, так и клеточный иммунитет, например, в стимуляции выработки антител, или в стимуляции цитотоксических или киллерных клеток, которые могут распознавать и разрушать (или иным образом элиминировать) клетки, экспрессирующие на своей поверхности "чужие" антигены. Термин "стимулирование иммунного ответа", таким образом, включает все типы иммунного ответа и механизмы их стимулирования.
Для стимуляции цитотоксичных клеток или клеток, образующих антитела, требуется, чтобы антигены, подлежащие презентации клетками, определенным образом стимулировались антиген-презентирующими клетками, например, презентирование молекулами MHC класса I (например, для активации цитотоксических T-лимфоцитов CD8+ требуется презентирование антигенов MHC-1).
Антиген-презентирующие клетки известны из уровня техники и описаны в литературе и включают, например, лимфоциты (T и B клетки), дендритные клетки, макрофаги и др. Другие включают, например, раковые клетки, например, клетки меланомы.
Для презентации антигена антиген-презентирующими клетками в цитотоксические T-лимфоциты (CTL) антигенной молекуле необходимо войти в цитозоль антиген-презентирующей клетки (Germain, Cell, 1994, 76, 287-299). В настоящем изобретении предложен эффективный способ доставки антигенной молекулы в цитозоль.
Антигенная молекула после высвобождения в цитозоле с помощью фотохимического процесса интернализации может быть процессирована посредством механизма антигенного процессинга клетки и соответствующим образом презентирована на клеточной поверхности, например, посредством MHC класса I. Такой процессинг может включать деградацию антигена, например, деградацию белкового или полипептидного антигена до пептидов, причем указанные пептиды затем образуют комплексы с молекулами MHC для целей презентации. Таким образом, антигенные молекулы, экспрессируемые или презентируемые на поверхности клетки согласно настоящему изобретению, могут представлять собой часть или фрагмент антигенной молекулы, которая поглощается клеткой (эндоцитозированной).
Антигены могут быть поглощены антиген-презентирующими клетками посредством эндоцитоза и далее подвергнуты деградации во внутриклеточных везикулах до пептидов. Указанные пептиды могут связываться с молекулами MHC класса II в эндосомах и далее могут быть перемещены на клеточную поверхность, где комплекс пептид-MHC класса II может быть распознан CD4+ T-хелперными клетками, и индуцировать иммунный ответ. Альтернативно, белки в цитозоле могут быть разложены на составляющие части, например, с помощью протеосом с последующим транспортом в эндоплазматический ретикулум с помощью TAP (переносчик, ассоциированный с презентацией антигена), при этом пептиды могут связываться с молекулами MHC класса I и далее переноситься на клеточную поверхность (Yewdell and Bennink, 1992, Adv. Immunol. 52: 1-123). Если происхождение пептида связано с чужеродным антигеном, то комплекс пептид-MHC класса I будет распознаваться CD8+ цитотоксичными Т-клетками (CTL). CTL будут связываться комплексом пептид-MHC (HLA) класса I и будут, таким образом, активироваться, запускать пролиферацию и образовывать клон CTL. Целевая клетка и другие целевые клетки, включающие тот же комплекс пептид-MHC класса I на клеточной поверхности, могут быть элиминированы клоном CTL. Иммунитет против чужеродного антигена может быть установлен, если в цитозоль вводится достаточное количество антигена (Yewdell and Bennink, 1992, supra; Rock, 1996, Immunology Today, 17: 131-137). Указанный механизм представляет собой основу для развития, в том числе, и противораковых вакцин. Одной из самых больших практических проблем является необходимость введения достаточных количеств антигенов (или частей антигена) в цитозоль. Указанная проблема может быть решена в соответствии с настоящим изобретением.
Эти способы могут быть использованы in vitro или in vivo, как описано выше.
Таким образом, в следующем аспекте изобретение относится к антиген-презентирующей клетке, экспрессирующей антигенную молекулу или его часть на свою поверхность, где клетка может быть получена (или была получена) способом, описанным выше. В других аспектах изобретения предложена популяция или культура таких клеток, особенно жизнеспособной и функционально интактной популяции или культуры таких клеток, а также к применению такой клетки (или популяции или культуры клеток), в терапии, в частности для стимулирования иммунного ответа, и, особенно, для стимуляции CTL.
Также предложено применение такой клетки (или популяции или культуры клеток) при получении лекарственного средства (например, композиция вакцины) для стимулирования иммунного ответа, и, особенно, для стимуляции CTL.
Изобретение описано далее более подробно с помощью следующих неограничивающих примеров, со ссылкой на следующие чертежи, где:
На фигуре 1 представлены особенно предпочтительные соединения по настоящему изобретению (TPC=тетрафенилхлорин).
На фигуре 2 представлена схема 1: путь синтеза для получения соединения 5. Реагенты и условия: (a) пропионовая кислота, кипячение с обратным холодильником, 1 час (20%); (b) NaNO2 (1,8 экв.), ТФУ, комн. темп., 3 мин. 67%); (c) SnCl2·2H2O, конц. HCl, 60°C, 1 час (88%); (d) бромацетилбромид, Et3N, CH2Cl2, комн. темп., 1 час (64%) (e) пиперазин, CH2Cl2, комн. темп., 1 час (94%).
На фигуре 3 представлена схема 2. Синтез N-модифицированного производных хитозана (TPP-CS-ТМА и TPP-CS-MP). Здесь A представляет собой 1ую партию соединений, и B представляет собой 2ую партию соединений. Реагенты и условия: (a) MeSO3H/H2O, 10°C, комн. темп., 1 час, (90%); (b) TBDMSCl, имидазол, ДМСО, комн. темп., 24 часа (96%); (c) Бромацетил бромид, Et3N, CH2Cl2, -20°C, 1 час (92%); (d) соединение 5, т.е. TPP-NH-Pip (0,1 или 0,25 экв.), Et3N, CHCl3 комн. темп., 2 час (92-90%); (e) NMe3 или 1-метил-пиперазин, CHCl3, комн. темп., 24 часа; (f) TBAF, NMP, 55°C, 24 часа, или конц. HCl/MeOH, комн. темп., 24 часа.
На фигуре 4 представлено характерное наложение ИК-спектра с Фурье-преобразованием всех промежуточных соединений и конечного соединения в синтезе TPPp0,1-CS-MP0,9 (18A): (A) TPP-NH-Pip 5; (B) BrA-DiTBDMS-C 9; (C) TPPp0,1-CH2CO-DiTBDMS-C 10A; (D) TPPp0,1-DiTBDMS-CS-MP0,9 14A; (E) TPPp0,1-CS-MP0,9 18A.
На фигуре 5 представлено характерное наложение 1H ЯМР спектра всех промежуточных соединений и конечного соединения в синтезе TPPp0,1-CS-MP0,9 (18A): (A) DiTBDMS-C 8; (B) BrA-DiTBDMS-C 9; (C) TPPp0,1-CH2CO-DiTBDMS-C 10A; (D) TPPp0,1-DiTBDMS-CS-MP0,9 14A; (E) TPPp0,1-CS-MP0,9 18A.
На фигуре 6 показано разделение соединений, растворенных в двухфазной системе смеси вода:CHCl3. (А) TPP(p-NH2)1 3 (B) TPPNH-Pip 5 (C) TPPp0,25-CS-TMA0,75 17A (D)TPPp0,25-CS-MP0,75 19A. Водные фазы указаны на крышке.
На фигуре 7 представлен 13C ЯМР спектр в твердом теле представительных конечных соединений TPPp0,25-CS-TMA0,75 (17B) и TPPp0,25-CS-MP0,75 (19B).
На фигуре 8 представлены (I) 1H ЯМР спектр TPPp0,25-CS-TMA0,75 17B в растворителях: (A) ДМСО-d6: D2O (98:2); (B) ДМСО-d6: D2O (75:25); (C) ДМСО-d6: D2O (50:50); (D) ДМСО-d6: D2O (25:75); (E) ДМСО-d6: D2O (0:100); (II) спектр УФ и видимой области. Наложение спектров поглощения TPPp0,25-CS-TMA0,75 17B при постоянной концентрации (0,3 мг/л) в совместных растворителях: (A) ДМСО: H2O (100:0); (B) ДМСО: H2O (75:25); (C) ДМСО: H2O (50:50); (D) ДМСО: H2O (25:75); (E) ДМСО: H2O (0:100); (III) спектры испускания флуоресценции с наложением TPPp0,25-CS-TMA0,75 17B при постоянной абсорбции (0,85-0,90; данные не показаны) и при возбуждении при 419 нм (входная щель 3-3) в совместных растворителях: (A) ДМСО: H2O (100:0); (B) ДМСО: H2O (75:25); (C) ДМСО: H2O (50:50); (D) ДМСО: H2O (25:75); (E) ДМСО: H2O (0:100).
На фигуре 9 показаны (A) РЭМ-визуализация TPP-CS-ТМА изолированных наночастиц (B) РЭМ-визуализация TPP-CS-ТМА дендритных наноагрегатов (C) график контактных углов производных TPP-CS.
На фигуре 10 представлена схема 3 - схема синтеза соединений 1, 3, 20 и 21. Реакции и условия: ((a) пропионовая кислота, кипячение с обратным холодильником, 1 час, (20%); (b) NaNO2 (1,8 экв.), ТФУ, комн. темп., 3 мин.; (c) SnCl2·2H2O, конц. HCl, 60°C, 1 час, (54%); (d1) п-толуолсульфонилгидразид, K2CO3, пиридин, кипячение с обратным холодильником, 24 часа; (d2) о-хлоранил, CH2Cl2, комн. темп., (80%); (e) хлорацетил хлорид, Et3N, CH2Cl2, комн. темп., 2 час, in situ; (f) Пиперазин, CH2Cl2, комн. темп., 12 час, (61%). Все производные соединения 20 и 21 будут содержать изомер TPCa1 и TPCa2. Однако на схеме и на рисунках структур показана только структура TPCa1.
На фигуре 11 представлена схема 4 - схема синтеза соединений 22-28. Реакции и условия: (a) ацетил хлорид, MeOH, кипячение с обратным холодильником, 24 часа, (87%); (b) BF3·Et2O, CHCl3, комн. темп., п-хлоранил, 48 ч, (14%); (c) 2н KOH (в MeOH), ТГФ:пиридин (10:1), кипячение с обратным холодильником, 24 часа (71%); (d1) п-толуолсульфонилгидразид, K2CO3, пиридин, кипячение с обратным холодильником, 24 часа; (d2) о-хлоранил, CH2Cl2:MeOH (75:25), комн. темп. (70%); (e) EDCl·HCl, HOBT, Et3N, N-Boc-пиперазин 5, ДМФ, комн. темп., 24 часа (54%) (f) ТФУ, CH2Cl2, комн. темп., 1 час (89%). Все производные соединения 26-28 будут содержать изомер TPCc1 и TPCc2. Однако на схемах на изображениях структур показана только структура TPCc1.
На фигуре 12 представлена схема 5 - синтез соединений 7-9. Реагенты и условия: (a) MeSO3H/H2O, 10°C, комн. темп., 1 ч, (90%); (b) TBDMSCl, имидазол, ДМСО, комн. темп., 24 ч, (96%); (c) Бромацетил бромид, Et3N, CH2Cl2, -20°C, 1 ч, (92%).
На фигуре 13 представлена схема 6A и 6B. Реагенты и условия (6а): (а) соединение 21, то есть TPC-NH-Pip (0,1 экв.), Et3N, CHCl3, комн. темп., 2 часа (78%) (b) NMe3 или 1-метил-пиперазин, CHCl3, комн. темп., 24 часа. Реагенты и условия (6b): (а) соединение 28, то есть TPC-CO-Pip (0,1 экв.), Et3N, NMP, 75°C, 12 часов (89%) (b) NMe3 или 1-метилпиперазин, CHCl3, комн. темп., 24 часа.
На фигуре 14 представлен 1H ЯМР спектр TPC-CO-Pip (28) в CDCl3. Показаны два изомера.
На фигуре 15 представлен 1H ЯМР спектр соединения 21 (TPC-NH-Pip) в CDCl3 (Показаны два изомера).
На фигуре 16 представлен 1H ЯМР спектр соединения 29 в CDCl3. Это соединение содержит изомеры TPCa1 и TPCa2.
На фигуре 17 представлен 1H ЯМР спектр соединения 34 в CDCl3. Это соединение содержит изомеры TPCc1 и TPCc2.
На фигуре 18 показаны ЯМР спектр конечных соединений носителя (37, 38, 32 и 33) в d6-ДМСО/D2O.
На фигуре 19 показана трансфекция с pEGFP-N1 в клетки HCT116/LUC. Трансфекцию измеряли через 48 часов после освещения методом проточной цитометрии. Выживаемость клеток измерялась с помощью МТТ-теста. (a) 16А. 0,1 мкг/мл TPP. (b) 16B. 0,1 мкг/мл TPP. (c) 17А. 0,01 мкг/мл TPP. (d) 19А. 0,01 мкг/мл TPP. (e) ТОС2а. 0,1 мкг/мл.
На фигуре 20 показана трансфекция с pEGFP-N1 в клетки HCT116/LUC. Трансфекцию измеряли через 48 часов после освещения методом проточной цитометрии. Выживаемость клеток измерялась с помощью МТТ-теста, (a) соединение 37. 0,05 мкг/РНБ TPC. (b) соединение 38. 0,05 мкг/мл TPC. (c) соединение 32. 0,05 мкг/мл TPC. (d) соединение 33. 0,05 мкг/мл TPC. (e) ТPCS2а. 0,1 мкг/мл.
На фигуре 21 показана трансфекция с pEGFP-N1 в клетки HCT116/LUC. Трансфекцию измеряли через 48 часов после освещения методом проточной цитометрии. Выживаемость клеток измерялась с помощью МТТ-теста. Соединение 54 было использовано в концентрации 0,1 мкг/мл.
На фигуре 22 показан биолюминесцентный имиджинг in vivo после лечения методом ФХИ животных с опухолями хитозан-конъюгатами и блеомицином. Животных обрабатывали, как описано в разделе материалы и методы примера 3. Указана обработка для каждого животного и время имиджинга (дни после введения инъекции фотосенсибилизатора).
На фигуре 23 показан рост опухолей после лечения методом ФХИ животных с опухолями соединением 37, 38 или 33 и блеомицином. Животных обрабатывали, как описано в разделе Материалы и способы в примере 3. ФС: фотосенсибилизатор.
На фигуре 24 представлена схема 7 - синтез реагентов для Тегилирования и Пегилирования. Условия реакции: (a) KOH, п-TsCl, ТГФ/H2O, комн. темп., 12 часов; (b) пиперазин, CH3CN, комн. темп., 12 ч, (41%); (c) окисление по Сверну: (COCl)2, CH2Cl2, ДМСО, Et3N, -78°C.
На фигуре 25 представлена схема 8 - Тегилирование DiTBDMS-хитозана. Реагенты и условия: (a) MeSO3H/H2O, от 10°C до комн. темп., 1 час, (90%); (b) TBDMSCI, имидазол, ДМСО, комн. темп., 24 часа (96%); (c) TEG-OTs 42, Cs2CO3, NMP, KI, 50°C, 24 часа; (d) HCl/MeOH (30% об./об.), комн. темп., 24 часа; (e) бромацетил бромид, Et3N, CH2Cl2, -20°C, 1 час (92%), (f) TEG-Pip 46 Et3N, CH2Cl2, комн. темп., 24 часа; (g) HCl/MeOH (30% об./об.), комн. темп., 24 часа.
На фигуре 26 представлена схема 9 - синтез Тегилированных хитозан-TPP конъюгатов. Реагенты и условия: (a) бромацетил бромид, Et3N, CH2Cl2, -20°C, 1 час (92%), (b) TPP-NH-Pip 5 (0,1 экв.), CH2Cl2, Et3N, комн. темп. (c) TEG-PIP (соединение 46) (2 экв.), CH2Cl2, Et3N, комн. темп., 24 часа (d) 30% (об./об.) HCl в MeOH, комн. темп., 12 часов; (e) соединение 4 (0,25 экв.), NMP, 50°C, Cs2CO3, 24 часа; (f) TEG-моноэтилэфир-тозилат (соединение 42), NMP, 50°C, KI, 24 часа; (g) 30% (об./об.) HCl в MeOH, комн. темп., 12 часов.
На фигуре 27 показан 1H ЯМР (400 МГц, ДМСО-d6:D2O, 96:4) соединения 54.
На фигуре 28 показана продукция ИЛ-2 в первичных мышиных макрофагах, которые инкубировали с соединением 32 (А) и 38 (B) и антигеном пептида овальбумина OVA 257-264 в антиген-специфичных T-клетках с овальбумин-специфичным (OVA 257-264) CD8+ T-клеточным клоном. Продукцию ИЛ-2 из активированных CD8+ T-клеток анализировали с помощью ELISA.
Пример 1. Синтез наноносителей на основе мезо-тетрафенилпорфирин-хитозана
Хорошо растворимые в воде хитозановые наноносители, связанные с фотосенсибилизатором мезо-тетрафенилпорфирином (TPP) были синтезированы 7 стадийным способом исходя из 3,6-ди-O-трет-бутилдиметилсилил-хитозана (DiTBDMS-CS) и 5-(п-аминофенил)-10,15,20-трифенилпорфирина [TPP(п-NH2)1] в качестве исходных продуктов. DiTBDMS-CS хорошо растворим в CH2Cl2, и поэтому высоко липофильный фотосенсибилизатор может быть введен в количественную реакцию с получением 0,1 и 0,25 степени замещения. За этим следовало введение триметиламмонийной и или 1-метилпиперазинильной групп в полимерную цепь для повышения растворимости в воде конечного носителя с удаленными защитными группами. Было показано, что способ хорошо воспроизводится, и что полученное вещество может быть полностью охарактеризовано с помощью ЯМР твердого тела, ИК-спектра с Фурье-преобразованием, и 1H ЯМР. Спектр УФ и видимой области, флуоресценция и ЯМР исследования показали, что носители являлись динамичными структурами, которые в водном растворе образуют подобные наночастицам структуры с ядром из полутвердых π-связанных фотосенсибилизаторов. В липофильных средах существует вероятность того, что указанные структуры могут разворачиваться, и части фотосенсибилизатора, таким образом, могут быть вставлены в мембрану клетки.
Общие материалы и способы
Хитозановый полимер был предоставлен фирмой Genis EHF, Iceland, и был использован для синтеза [хитозановый полимер GO 30626-2 (95%СД, 95 сП)]. Все растворители и реагенты были приобретены коммерчески и использовались без дополнительной очистки. Спектры ЯМР были получены на ЯМР спектрометре DRX 400 МГц Bruker при 298 K, и химические сдвиги регистрировались относительно используемого дейтерированного растворителя для ЯМР [1H ЯМР: CDCl3 (7,26 м.д.), ДМСО-d6 (2,50 м.д.); 13C ЯМР: CDCl3 (77,16 м.д.), ДМСО-d6 (39,52 м.д.)]. Пик ацетона (2,22 м.д.) был использован в качестве внутреннего стандарта для D2O в качестве растворителей. Протоны (орто, мета, пара) на фенильных кольцах порфиринов определялись с точки зрения их положения относительно системы порфиринового кольца, а не в отношении заместителя на фенильном кольца.
13C ЯМР в твердом состоянии соединений 17B и 19B были получены от Department of Chemistry, Durham University. Указанный спектр был получен с помощью спектрометра Varian VNMRS, работая при 100,56 МГц для 13C. Эксперименты кросс-поляризации с вращением под магическим углом проводились с 6 мм (наружный диаметр ротора) зондом. Спектры были записаны при угловой скорости вращения 6,8 кГц, при времени контакта 1 мсек, 1,5 сек задержки рецикла и с смесителем "TOSS" с подавлением зеркального канала. Спектральные сдвиги даны относительно внешнего образца неразбавленного тетраметилсилана, внесенного не напрямую путем установки высокочастотного сигнала от адамантана до 38,5 м.д.
Масс-спектры записывали на приборе Bruker Autoflex III или Bruker micrOTOF-Q11. Измерения ИК-спектра с Фурье-преобразованием проводили на аппарате AVATAR 370 FT-IR (Thermo Nicolet Corporation, Madison, USA). Образцы (2-3 мг) тщательно смешивали с KBr. Образец прессовали в гранулы с помощью компрессора Specac (Specac Inc., Smyrna, США). Точки плавления определяли на приборе Buchi Melting Point B-540. Полимерные образцы диализовали с использованием диализной мембраны Spectra/Por (MWCO: 3500).
Спектры поглощения и стационарной флуоресценции
Спектр УФ и видимой области получали на спектрометре Perkin-Elmer Lambda 25 UV-Vis, оснащенном программой Peltier Temperature Programmer. Флуоресцентные спектры испускания были получены с помощью спектрометра SPEX FluoroMax, используя ячейку со спектральным диапазоном 170-2200 нм (Spectrocell Corporation, Oreland, PA, USA). Спектры поглощения регистрировались при 20°C, и флуоресцентные спектры испускания регистрировались при комнатной температуре с использованием кварцевой кюветы с 10 мм-овой длиной пути. Все спектры флуоресценции регистрировались при постоянной ширине щели, 1 нм для возбуждения и 1 нм для излучения, и спектры флуоресценции усреднялись по трем сканированиям для исследования квантового выхода. Однако, на фигуре 5 (III) спектры флуоресценции были получены при ширине щели 3 нм для возбуждения и 3 нм для излучения.
Квантовые выходы флуоресценции соединений 3, 5, 16А-19B (все возбуждения при λex.=λmax=419 нм) определяли относительно стандартного разбавленного раствора антрацена (ΦF=0,27, λex=365,5 нм) в абсолютном этаноле, используя способ стационарного сравнения с помощью следующего уравнения:
ФX=ФST (GradX/GradST) (ηX2/ηST2)
где подстрочные индексы ST и X обозначают стандартные и тест, соответственно, Φ обозначает квантовый выход флуоресценции, Grad - градиент от участка интегральной интенсивности флуоресценции по сравнению с поглощением, и η - показатель преломления растворителя.
Исследование стабильности:
Для исследования физической стабильности 17А растворяли в H2O (1 мг/мл), сонифицировали в течение 30 минут, центрифугировали (на HERMLE Z-320 при 4000 об. в мин. в течение 10 мин), декантировали и обертывали алюминиевой фольгой. Спектр УФ и видимой области поглощения были измерены при λmax=419 нм в H2O в течение 0-90 дней.
Определение степени замещения (СЗ) с помощью 1H ЯМР.
Для расчета степени замещения TPP-NH-Pip использовали 1H ЯМР целевого соединения. Для расчета СЗ целевых соединений 16А-19B использовали объединение значения пиков TPP (от TPP-NH-Pip части) и значений пиков H-1 (от хитозановой части). Был рассмотрен интеграл TPP (четыре пика из ароматической области), в то время как интегралы группы H-1 были вычислены и использованы в следующих уравнениях:
СЗ=(∫ароматические пики TPP/27)/(∫H-1 пик/1)
В этих условиях полимерный скелет частично перекрывается пиком HDO, но СЗ может быть определен с хорошей точностью от относительного соотношения интегралов пиков H-1 и пиков TPP.
Для расчета СЗ TPC-NH-Pip и TPC-CO-Pip были использованы промежуточные соединения 29 и 34 и объединение значений пиков TPC (от TPC-NH-Pip или TPC-CO-Pip в ароматической области, а также объединение α-пиррольных NH пиков) и объединение пиков TBDMS (от хитозана) в следующем уравнении:
СЗ=(∫ароматические TPC пики+α-пиррольный NH пик/27)/(∫TBDMS пики/30)
Для расчета СЗ Тегилирования для различных производных хитозана использовали соответствующий 1H ЯМР. Было рассмотрено объединение значения пика H-1 и объединение значения CH3 (из конечного этилового пика TEG) на хитозановом скелете. Объединенное значение для этилового конца триплета могло быть равным 3, если степень замещения составляла 100%. Таким образом, уравнение является следующим:
СЗ (%)=(∫этиловый пик (концевой триплет, CH3)/3)/(∫H-1 пик/1)×100%
РЭМ и элипсометрия
Растворы были нанесены в качестве покрытия методом центрифугирования при 1000 об/мин на чистые кремниевые <100> субстраты (15 мм×15 мм) с помощью обычного спиннера в условиях чистого (производственного) помещения класса 100. Кремний имел слой собственного оксида толщиной приблизительно 15 Å. Более того, 10 мкл того же раствора были добавлено с помощью пипетки непосредственно на кремниевые субстраты, и они были высушены на воздухе при комнатной температуре. Изображение покрытых подложек было получено с помощью сканирующего электронного микроскопа Zeiss LEO 1550 при возрастании напряжения 10 КЭВ и рабочем расстоянии 2 мм с помощью встроенного в детектор объектива.
Измерение угла контакта с водой
Углы контакта с водой были определены с помощью оптического измерителя углов контакта KSV CAM 200 (KSV Instruments). 5 мкл деионизированных мелких капель воды помещали в центр кремниевой пластины и определяли углы контакта с водой на основе уравнения Юнга-Лапласа. Измерения проводились при комнатной температуре и во влажной окружающей среде.
Синтез
См. схему 1 на фигуре 2 для синтеза TPP-NH-Pip
Мезо-Тетрафенилпорфирин (1). Следуя способу, описанному в литературе (Adler, J Organic Chem 1967, 32:476).
5-(4-Нитрофенил)-10,15,20-трифенилпорфирин [TPP(п-NO2)1] (2). Следуя способу, описанному в литературе (Luguya R et al. Tetrahedron 2004, 60:2757).
5-(4-Аминофенил)-10,15,20-трифенилпорфирин [TPP(п-NH2)1] (3). Следуя способу, описанному в литературе (Luguya R et al. Tetrahedron 2004, 60:2757).
5-(4α-Бромацетиламидофенил)-10,15,20-трифенилпорфирин (4). Соединение 3 (500 мг, 0,793 ммоль) растворяли в CH2Cl2 (15 мл) и перемешивали в атмосфере N2. Добавляли триэтиламин (0,24 мл, 1,75 ммоль), затем при комнатной температуре по каплям бромацетилбромид (0,097 мл, 1,11 ммоль), и перемешивание продолжали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли CH2Cl2 (45 мл), промывали водой (2×25 мл) и насыщенным солевым раствором (20 мл). Органический слой затем сушили над Na2SO4 и концентрировали в вакууме. Сырой продукт очищали путем колоночной хроматографии на силикагеле, используя CH2Cl2 и гексан в качестве элюента, что давало 385 мг (64%) желаемого продукта 4. ТСХ (гексан/CH2Cl2 3:7): Rf=0,17; ИК-спектр с Фурье-преобразованием, v см-1: 3313 (N-H), 3049, 3019 (арил C-H), 1687, 1594 (CONH), 1556, 1514, 1471, 1439, 1399, 1348, 1177, 1153, 964, 798, 726, 699; 1H ЯМР (CDCl3): δ=8,88-8,91 (м, 8Н, β-пиррольные H), 8,41 (ушир. с, 1H, TPPNHCO), 8,22-8,27 (м, 8H, тетрафенил H0), 7,91 (д, J =8Гц, 2H, CONH-фенил-Hм), 7,75-7,80 (м, 9H, трифенил-Hм,п), 4,15 (с, 2H, COCH2Br), -2,70 (ушир. с, 2H, α-пиррольные NH) м.д.; 13C ЯМР (CDCl3): δ=163,78, 142,26, 139,20, 136,78, 135,25, 134,68, 131,23, 127,87, 126,83, 120,42, 120,38, 119,24, 118,34, 29,72 м.д.; MS (ESI): m/z вычисл. для C46Ч33BrN50 ([M+H]+) 750,1863 найдено 750,1864; спектр УФ и видимой области (ДМСО): λmax: 417, 517, 542, 597, 650 нм.
5-(4α-Пиперазинацетиламидофенил)-10,15,20-трифенилпорфирин [TPP-NH-Pip] (5). Соединение 4 (275 мг, 0,366 ммоль) и избыток пиперазина (158 мг, 1,83 ммоль) смешивали вместе в CH2Cl2 (10 мл) и перемешивали при комнатной температуре в течение 1 час в атмосфере N2. После завершения реакции реакционную смесь разбавляли CH2Cl2 (85 мл) и промывали водой (2×40 мл) и насыщенным солевым раствором (35 мл). Органический слой сушили над Na2SO4 и упаривали в вакууме. Сырой продукт очищали путем колоночной хроматографии на силикагеле с использованием в качестве элюента смеси 1:12 MeOH:CH2Cl2, с получением указанного в заголовке соединения 5 (260 мг, 94%) в виде пурпурного твердого вещества. ТСХ (CH2Cl2/MeOH, 9:1): Rf =0,15 с; ИК-спектр с Фурье-преобразованием (KBr): v 3442 (pip. NH), 3312 (арил N-H), 3052, 3022 (арил C-H), 2903, 2816 (алифатический СН2), 1691, 1596 (CONH), 1557, 1517, 1471, 1439, 1400, 1349, 1309, 1179, 1153, 1071, 1001, 965, 799, 728, 700 см-1; 1H ЯМР(CDCl3): δ=9,51 (ушир. с, 1H, TPP-NHCO), 8,89-8,93 (м, 8H, β-пиррольные H), 8,23-8,26 (м, 8H, тетрафенильные H0), 8,01 (д, J=8,0 Гц, 2H, Pip-NH-фенил-Hм), 7,75-7,81 (м, 9H, трифенил-Hм,п), 3,31 (с, 2H, COCH2-Pip.), 3,11 (т, 4H), 2,77 (ушир. т, 4H), 2,60 (ушир. с, 1H, пиперазиновый NH), -2,71 (ушир. с, α-пиррольные 2H) м.д.; 13C ЯМР (CDCl3): δ=168,75, 142,30, 138,26, 137,44, 135,33, 134,68, 131,26, 127,86, 126,83, 120,32, 119,66, 117,89, 62,87, 54,53, 46,24 м.д.; MS (ESI): m/z вычисл. для C50H42N7O ([M+H]+) 756,3445 найдено 756,3467; спектр УФ и видимой области (ДМСО): λmax: 417, 517, 542, 597, 650 нм.
См. схему 2 на фигуре 3 и схему 5 на фигуре 12
Мезилат хитозана (7). Синтезирован согласно ранее опубликованному авторами способу (Song et al. Carbohydrate Polymers 2010, 81:140).
3,6-O-ди-трет-бутилдиметилсилил хитозан [DiTBDMS-CS] (8). Синтезирован согласно ранее опубликованному авторами способу (Song et al. Carbohydrate Polymers 2010, 81:140).
N-бромацетил-3,6-0-DiTBDMS-CS [BrA-DiTBDMS-CS] (9). DiTBDMS-CS 8 (1 г, 2,60 ммоль) растворяли в сухом CH2Cl2 (15 мл) в круглодонной колбе в атмосфере N2. Затем реакционную смесь охлаждали до -20°C на бане со смесью лед/соль. Добавляли Et3N (1,81 мл, 13 ммоль) с последующим медленным добавлением по каплям бромацетилбромида (0,91 мл, 10 ммоль). Перемешивали в течение 1 часа. Реакционную смесь разбавляли CH2Cl2 и концентрировали в вакууме. Сырой продукт перемешивали в ацетонитриле, фильтровали и промывали свежим ацетонитрилом. Сухой материал разбавляли и экстрагировали CH2Cl2, промывали водой и насыщенным солевым раствором и сушили над Na2SO4, упаривали в вакууме. Получали 1,2 г (92%-ный выход) порошкообразного продукта 9 слегка желтого цвета. ИК-спектр с Фурье-преобразованием (KBr): v 3402 (шир., NH), 2957, 2931, 2886, 2858 (с, C-H TBDMS), 1682 (vs, C=O амид I), 1530 (vs, C=O амид II), 1473, 1391, 1362, 1311, 1259, 1101, 1005, 837, 777 (Si-C), 669 см-1; 1H ЯМР (CDCl3) δ м.д.: 4,40 (ушир. с, H-1), 4,02-3,26 (м, H-2 GlcN, H-3, H-4, H-5, H-6, H-6' и 2H GluNH-C=OCH2Br), 0,90 и 0,88 (ушир. с, (CH3)3C), 0,13 и 0,07 (ушир. с, (CH3)2Si) м.д.
(N-TPP-NH-Pip-ацетил)0,1-(N-бромацетил)0,9-DiTBDMS-CS [TPPp0,1-BrA0,9-DiTBDMS-CS] (10А). Соединение 9 (700 мг, 1,38 ммоль) и соединение NH-pip-TPP 5 (105 мг, 0,138 ммоль) растворяли в CH2Cl2 в атмосфере N2. Добавляли точное эквимолярное количество Et3N (19,3 мкл, 0,130 ммоль) относительно 5, и реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Полное расходование исходного материала подтверждали методом ТСХ. Реакционную смесь разбавляли CH2Cl2, экстрагировали и промывали водой и насыщенным солевым раствором. Органический слой сушили над Na2SO4, упаривали в вакууме с получением 730 мг (92%) продукта 10А. ИК-спектр с Фурье-преобразованием (KBr): v 3324 (шир., NH), 2955, 2929, 2884, 2856 (с, C-H TBDMS), 1678 (vs, C=O амид I), 1600, 1524 (vs, C=O амид II), 1472, 1403, 1361, 1311, 1256, 1098, 1004, 966, 837, 801, 778, 701, 670, 550 см-1; 1H ЯМР (CDCl3) δ м.д.: 9,30(с, TPPNHCO), 8,85 (м, β-пиррольные H), 8,23-8,20 (м, фенил-Ho и Pip-NHTPP-фенил-Hм), 7,97 (д, J=8,0 Гц, RNTPP-фенил-Ho), 7,79-7,73 (м, трифенил–Hм,п), 4,41 (ушир. с, H-1), 4,13-3,50 (м, H-2 GlcN, H-3, H-4, H-5, H-6, H-6' и 2H GluNH-C=O, TPPNHCOCH2pip, CH2CONGlc и (CH2)2 пиперазина), 2,81-2,86 (м, пиперазин-H), 0,92, 0,89 (ушир. с, (CH3)3C), 0,14, 0,07 (ушир. с, (CH3)2Si), -2,77 (ушир. с, α-пиррольный NH) м.д.; спектр УФ и видимой области (ДМСО): λmax: 417, 517, 542, 597, 650 нм.
TPPp0,1-BrA0,9-DiTBDMS-CS (10B): Соединение 10B (1,3 мг, 91%) получали точно так, как описано в предыдущем способе, с использованием промежуточного соединения 5 (180 мг, 0,24 ммоль) и 9 (1,2 г, 2,4 ммоль).
TPPp0,25-BrA0,75-DiTBDMS-CS (11А). Соединение 9 (550 мг, 1,09 ммоль) и соединение NH-pip-TPP 5 (206 мг, 0,273 ммоль) растворяли в CH2Cl2 в атмосфере N2. Добавляли точное эквимолярное количество Et3N (38 мкл, 0,27 ммоль) относительно 5, и реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Полное расходование исходного материала подтверждали методом ТСХ. Реакционную смесь разбавляли CH2Cl2, экстрагировали и промывали водой и насыщенным солевым раствором. Органический слой сушили над Na2SO4 и упаривали в вакууме с получением 670 мг (91%) продукта 11A. ИК-спектр с Фурье-преобразованием (KBr): v 3317 (шир., NH), 2952, 2926, 2883, 2855 (с, C-H TBDMS), 1680 (vs, C=O амид I), 1598, 1520, 1471, 1440, 1402, 1361, 1309, 1254, 1096, 1002, 966, 837, 800, 778, 730, 701, 667, 558 см-1; 1H ЯМР (CDCl3) δ м.д.: 9,30 (с, TPPNHCO), 8,85 (м, β-пиррольные H), 8,22-8,20 (м, фенил-Ho и Pip-NHTPP-фенил-Hм), 7,97 (д, J=8,0 Гц, RNTPP-фенил-Ho), 7,80-7,75 (м, трифенил-Hм,п), 4,40 (ушир. с, H-1), 4,06-3,63 (м, H-2 GlcN, H-3, H-4, H-5, H-6, H-6' и 2H GluNH-C=O, TPPNHCOCH2pip, CH2CONGlc и (CH2)2 пиперазина), 2,81-2,86 (м, пиперазин-H), 0,92, 0,89 (ушир. с, (CH3)3C), 0,14, 0,10, 0,07 и 0,02 (ушир. с, (CH3)2Si), -2,77 (ушир. с, α-пиррольные NH) м.д.; спектр УФ и видимой области (ДМСО): λmax: 417, 517, 542, 597, 650 нм.
TPPp0,25-BrA0,75-DiTBDMS-CS (11B): Соединение 11B (1,35 г, 92%) получали точно так, как описано в предыдущем способе с использованием промежуточного соединения 5 (415 мг, 0,55 ммоль) и 9 (1,1 г, 2,18 ммоль).
Общий способ синтеза соединений 12A, 12B, 13A и 13B (N-TPP-NH-Pip-ацетил)0,1-(N-(N,N,N-триметиламмоний)ацетил)0,9-DiTBDMS-CS [TPPp0,1-DiTBDMS-CS-TMA0,9] (12A). Соединение 10A (350 мг, 0,61 ммоль) растворяли в CH2Cl2 в атмосфере N2. Добавляли избыточное количество раствора триметиламина, и реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Растворитель удаляли в вакууме. Сырой продукт сушили в высоком вакууме с получением сырого продукта 12A (420 мг, 99%) в виде твердого вещества пурпурного цвета. ИК-спектр с Фурье-преобразованием (KBr): v 3426, 3021, 3011, 2962, 2855, 27,7, 2560, 2438, 1749, 1689, 1599, 1563, 1482, 1443, 1401, 1360, 1288, 1254, 1053, 1010, 966, 922, 901, 837, 797, 779, 744, 701, 671, 552 см-1.
TPPp0,1-DiTBDMS-CS-TMA0,9 (12B). Общий способ A осуществляли с использованием 10В (600 мг, 1,04 ммоль) и NMe3 с получением 12B в виде сырого твердого вещества (720 мг, 99%).
TPPp0,25-DiTBDMS-CS-TMA0,75 (13А). Общий способ A осуществляли с использованием 11A (300 мг, 0,44 ммоль) и NMe3 с получением 13А сырого твердого вещества (340 мг, 98%). FTIR (KBr): v 3415, 3021, 3011, 2962, 2854, 1749, 1686, 1598, 1522, 1482, 1442, 1402, 1360, 1311, 1288, 1252, 1053, 1010, 966, 922, 901, 837, 798, 779, 744, 701, 671, 558 см-1.
TPPp0,25-DiTBDMS-CS-TMA0,75 (13B). Общий способ A осуществляли с использованием 11B (600 мг, 0,89 моль) и NMe3 с получением 13B в виде сырого твердого вещества (685 мг, 99%)
Общий способ B получения соединений 14A, 14B, 15A и 15B
(N-TPP-NH-Pip-ацетил)0,1-(N-(N-метилпиперазинил)ацетил)0,9-DiTBDMS-CS [TPPp0,1-DiTBDMS-CS-MP0,9] (14а). Соединение 10A (350 мг, 0,61 ммоль) растворяли в CH2Cl2 в атмосфере N2. Добавляли избыточное количество 1-метилпиперазина, и реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Растворитель удаляли в вакууме. Затем сырой продукт сушили в высоком вакууме с получением соответствующего сырого продукта, 14A (425, 105%). ИК-спектр с Фурье-преобразованием (KBr): v 3378, 2950, 2930, 2884, 2854, 2798, 2694, 2608, 2477, 2223, 1678, 1617, 1519, 1461, 1394, 1371, 1293, 1253, 1168, 1092, 1051, 1003, 966, 939, 920, 837, 801, 779, 701, 671, 612 см-1.
TPPp0,1-DiTBDMS-CS-MP0,9 (14B). Общий способ B осуществляли с использованием 10В (600 мг, 1,04 моль) и 1-метилпиперазина с получением 14B в виде сырого твердого вещества (710 мг, 102%).
TPPp0,25-DiTBDMS-CS-MP0,75 (15A). Общий способ B осуществляли с использованием 11A (250 мг, 0,37 ммоль) и 1-метилпиперазина с получением 15A в виде сырого твердого вещества (290 мг, 104%). ИК-спектр с Фурье-преобразованием (KBr): v 3380, 2950, 2930, 2884, 2854, 2800, 2480, 1677, 1598, 1519, 1461, 1400, 1394, 1371, 1284, 1252, 1168, 1089, 1050, 1003, 966, 939, 920, 837, 801, 779, 732, 701, 671, 591 см-1.
TPPp0,25-DiTBDMS-CS-MP0,75 (15B). Общий способ B осуществляли с использованием 11B (650 мг, 0,96 моль) и 1-метилпиперазина с получением 15B в виде сырого твердого вещества (735 мг, 102%).
Общий способ C удаления защитных групп в TBDMS для соединений 16А, 17A, 18A и 19A (1ая партия соединений).
TPPp0,1-CS-ТМА0,9 (16А). Соединение 12A (350 мг, 0,50 ммоль) растворяли в MeOH (5-10 мл) с последующим добавлением концентрированной HCl (2-3 мл). Реакционную смесь перемешивали в течение 12 часов при комнатной температуре. В реакционную смесь добавляли избыточное количество деионизированной воды и перемешивали в течение 30 минут, затем проводили диализ против 8% NaCl в течение одного дня, и против деионизированной воды в течение 3 дней. В течение этого времени цвет раствора постепенно менялся от темно-зеленого до красного. Раствор красного цвета затем лиофилизовали с получением губчатого вещества коричневого цвета. Соединения опять подвергали удалению защитных групп, ионному обмену, диализу и лиофилизации. Способ повторяли с использованием тех же условий, чтобы удалить следовые количества TBDMS с получением 16А (210 мг, 89%) в виде губчатого вещества коричневого цвета. ИК-спектр с Фурье-преобразованием (KBr): v 3419 (шир., O-H), 3064 (C-H), 1683, 1558, 1506, 1488, 1473, 1403, 1296, 1153, 1112, 1068, 1032, 966, 911, 800, 730, 701, 618 см-1; 1H ЯМР (ДМСО-d6: D2O 96:4) δ м.д.: 8,81 (ушир. м, β-пиррольные H), 8,12-8,16 (ушир. м, фенил-Ho и Pip-NHTPP-фенил-Hм), 8,04 (ушир. д, J=8,0 Гц, RNTPP-фенил-Ho), 7,75-7,84 (м, трифенил-Hм,п), 4,66 (ушир. с, H-1), 4,21 (ушир. с, BrCH2C=O), 3,83-3,54 (м, H-2 GlcNAc, H-3, H-4, H-5, H-6, H-6'), 3,32 (с, +N(CH3)3) м.д.
TPPp0,25-CS-ТМА0,75 (17A). Общий способ C осуществляли с использованием 13А (240 мг, 0,30 ммоль) и конц. HCl/MeOH, с получением 17A в виде твердого вещества пурпурного цвета (120 мг, 71%). ИК-спектр с Фурье-преобразованием (KBr): v 3392, 3061, 2950, 1683, 1559, 1506, 1489, 1473, 1402, 1350, 1296, 1154, 1112, 1068, 1032, 966, 911, 800, 730, 701, 619 см-1; 1H ЯМР (ДМСО-d6: D2O 98:2) δ м.д.: 8,81 (ушир. м, β-пиррольные H), 8,12-8,16 (ушир. м, фенил-Ho и Pip-NHTPP-фенил-Hм), 8,04 (ушир. д, J=8,0 Гц, RNTPP-фенил-Ho), 7,79-7,87 (м, трифенил-Hм,п), 4,50 (ушир. с, H-1), 4,15 (ушир. с, BrCH2C=O), 3,27-3,65 (м, H-2 GlcNAc, H-3, H-4, H-5, H-6, H-6'), 3,24 (с, +N(CH3)3)) м.д.
TPPp0,1-CS-МP0,9 (18A). Общий способ C осуществляли с использованием 14A (350 мг, 0,52 ммоль) и конц. HCl/MeOH, с получением 18A в виде твердого вещества пурпурного цвета (200 мг, 87%). ИК-спектр с Фурье-преобразованием (KBr): v 3419 (шир., O-H), 3057 (C-H), 1683, 1558, 1474, 1403, 1296, 1234, 1154, 1112, 1068, 1032, 966, 930, 911, 799, 730, 701 см-1; 1H ЯМР (ДМСО-d6: D2O 96:4) δ м.д.: 8,83 (ушир. м, β-пиррольные H), 8,13-8,19 (ушир. м, фенил-Ho и Pip-NHTPP-фенил-Hм), 8,08 (ушир. д, J=8,0 Гц, RNTPP-фенил-Ho), 7,79-7,87(м, трифенил-Hм,п), 4,48 (ушир. с, H-1), 3,24-3,78 (м, H-2 GlcNAc, H-3, H-4, H-5, H-6, H-6'), 3,92 (дд, J=12 Гц, Pip-CH2C=O), 2,30-2,67 (м, пиперазиновые (CH2)4), 2,48 (ушир. с, пиперазин, N-CH3) м.д.
TPPp0,25-CS-MP0,75 (19A). Общий способ C осуществляли с использованием 15A (200 мг, 0,26 ммоль) и 1-метилпиперазина с получением 19A в виде твердого вещества пурпурного цвета (80 мг, 58%). ИК-спектр с Фурье-преобразованием (KBr): v 3392, 3056, 2947, 1683, 1558, 1520, 1489, 1472, 1458, 1400, 1349, 1309, 1248, 1154, 1068, 1031, 1001, 966, 911, 800, 729, 701, 658 см-1. 1H ЯМР (ДМСО-d6: D2O 96:4) δ м.д.: 8,83 (ушир. м, β-пиррольные H), 8,14-8,20 (ушир. м, фенил-Ho и Pip-NHTPP-фенил-Hм), 8,08 (ушир. д, J=8,0 Гц, RNTPP-фенил-Ho), 7,78-7,86 (м, трифенил-Hм,п), 4,50 (ушир. с, H-1), 3,24-3,78 (м, H-2 GlcNAc, H-3, H-4, H-5, H-6, H-6'), 3,92 (дд, J=12 Гц, Pip-CH2C=O), 2,28-2,67 (м, пиперазиновые (CH2)4), 2,48 (ушир. с, пиперазин, N-CH3) м.д.
Общий способ D удаления TBDMS защитных групп в соединениях 16B, 17B, 18B и 19B (2ая партия соединений). (Соединение 16B в качестве представительного).
TPPp0,1-CS-ТМА0,9 (16B): Соединение 12B (600 мг, 0,86 ммоль) растворяли в N-метил-2-пирролидоне (NMP) (5-10 мл) с последующим добавлением избыточного количества тетра-н-бутиламмонийфторида (TBAF). Реакционную смесь перемешивали 24 часа при 55°C и охлаждали и подкисляли разбавленной HCl, затем перемешивали в течение 30 минут, после чего проводили диализ против 8% NaCl (масс./об.) в деионизированной воде в течение двух дней и против деионизированной воды в течение 3 дней. В течение этого времени цвет раствора постепенно менялся от серого до красного. Раствор красного цвета затем лиофилизовали с получением губчатого вещества коричневого цвета. Соединения опять подвергали удалению защитных групп, ионному обмену, диализу и лиофилизации. Способ повторяли с использованием тех же условий, чтобы удалить следовые количества TBDMS с получением 16B (350 мг, 87%) в виде губчатого вещества коричневого цвета. ИК (KBr): v 3405, 2943, 2817, 1655, 1528, 1459, 1401, 1375, 1308, 1248, 1153, 1111, 1068, 1031, 966, 832, 799, 729, 702 см-1; 1H ЯМР (ДМСО-d6: D2O 96:4) δ м.д.: 8,82 (ушир. м, β-пиррольные H), 8,12-8,16 (ушир. м, фенил-Ho и Pip-NHTPP-фенил-Hм), 8,04 (ушир. д, J=8,0 Гц, RNTPP-фенил-Ho), 7,75-7,84(м, трифенил-Hм,п),4,47 (ушир. с, H-1), 4,04 (ушир. с, BrCH2C=O), 3,24-3,55 (м, H-2 GlcNAc, H-3, H-4, H-5, H-6, H-6'), 3,19 (ушир. с, +N(CH3)3)) м.д.
TPPp0,25-CS-ТМА0,75 (17B): Общий способ C осуществляли с использованием 13B (650 мг, 0,84 ммоль) и TBAF/NMP с получением 17B в виде твердого вещества пурпурного цвета (400 мг, 87%). ИК (KBr): v 3396, 2942, 2829, 1662, 1526, 1458, 1441, 1401, 1310, 1249, 1068, 1032, 1002, 966, 800, 730, 701 см-1; 1H ЯМР (ДМСО-d6: D2O 98:2) δ м.д.: 8,81 (ушир. м, β-пиррольные H), 8,12-8,16 (ушир. м, фенил-Ho и Pip-NHTPP-фенил-Hм), 8,08 (ушир. д, J=8,0 Гц, RNTPP-фенил-Ho), 7,74-7,86 (м, трифенил-Hм,п), 4,51 (ушир. с, H-1), 4,10 (ушир. с, BrCH2C=O), 3,26-3,55 (м, H-2 GlcNAc, H-3, H-4, H-5, H-6, H-6'), 3,22 (ушир. с, +N(CH3)3)) м.д.; в твердом состоянии 13C ЯМР (100,56 МГц): δ 170,85, 164,68, 128,11, 119,45, 101,53, 75,39, 61,09, 55,47.
TPPp0,1-CS-МP0,9 (19B): Общий способ C осуществляли с использованием 14B (600 мг, 0,90 ммоль) и TBAF/NMP с получением 18B в виде твердого вещества пурпурного цвета (315 мг, 80%). ИК (KBr): v 3396, 3315 (шир., OH, NH), 2941, 1655 (vs, C=O амид I), 1534 (vs, C=O амид II), 1522, 1471, 1440, 1401, 1375, 1310, 1244, 1069, 1030, 1001, 966, 800, 729, 701 см-1; 1H ЯМР (ДМСО-d6: D2O 98:2) δ м.д.: 8,81 (ушир. м, β-пиррольные H), 8,13-8,19 (ушир. м, фенил-Ho и Pip-NHTPP-фенил-Hм), 8,07 (ушир. д, J=8,0 Гц, RNTPP-фенил-Ho), 7,79-7,85 (м, трифенил-Hм,п), 4,48 (ушир. с, H-1), 3,24-3,72 (м, H-2 GlcNAc, H-3, H-4, H-5, H-6, H-6'), 3,92 (дд, J=12 Гц, Pip-CH2C=O), 2,33-2,67 (м, пиперазиновые (CH2)4), 2,48 (ушир. с, пиперазин, N-CH3) м.д.
TPPp0,1-CS-МP0,9 (19B): Общий способ C осуществляли с использованием 15B (700 мг, 0,93 ммоль) и TBAF/NMP с получением 19B в виде твердого вещества пурпурного цвета (415 мг, 85%). FTIR (KBr): v 3396, 3315 (шир., OH, NH), 2941, 1655 (vs, C=O амид I), 1534 (vs, C=O амид II), 1522, 1471, 1440, 1401, 1375, 1310, 1244, 1069, 1030, 1001, 966, 800, 729, 701 см-1; 1H ЯМР (ДМСО-d6: D2O 95:5) δ м.д.: 8,82 (ушир. м, β-пиррольные H), 8,13-8,19 (ушир. м, фенил-Ho и Pip-NHTPP-фенил-Hм), 8,07 (ушир. д, J=8,0 Гц, RNTPP-фенил-Ho), 7,79-7,87 (м, трифенил-Hм,п), 4,49 (ушир. с, H-1), 3,24-3,78 (м, H-2 GlcNAc, H-3, H-4, H-5, H-6, H-6'), 3,92 (дд, J=12 Гц, Pip-CH2C=O), 2,30-2,67 (м, пиперазиновые (CH2)4), 2,40 (ушир. с, пиперазин, N-CH3) м.д.; в твердом состоянии 13C ЯМР (100,56 МГц): δ 171,63, 138,74, 127,97, 119,74, 101,93, 75,54, 61,54, 55,42, 45,34 м.д.
Результаты и их обсуждение
Нуклеофильное промежуточное соединение TPP 5. В данном исследовании, TPP 1 был синтезирован в больших масштабах и использовался в качестве исходного материала. Функциональная группа моно-нитро TPP(п-NO2)1 2 была введена региоизбирательно с помощью 1,8 эквивалентов NaNO2 в ТФУ с последующим восстановлением SnCl2·2H2O и получением моно-аминопорфирина TPP(п-NO2)1 3. Ранее описанный способ был упрощен путем отмены трудоемкой очистки сырого продукта 2 перед восстановлением. Аминопорфирин 3 затем был получен после очистки без потери общего выхода (54%).
Известно, что аминопорфирин TPP(п-NO2)1 3 является слабо нуклеофильным, и попытки связать это соединение с электрофильным BrA-DiTBDMS-CS-9 путем SN2 атаки не давало желаемых результатов в первоначальном исследовании авторов, даже в самых жестких условиях. Таким образом, для того, чтобы преобразовать это производное фотосенсибилизатора в более сильный нуклеофил TPP(п-NO2)1 сначала ацилировали с получением бромацил-TPP 4, затем подвергали нуклеофильному замещению с избытком пиперазина с получением промежуточного нуклеофильного порфирина TPP-NH-Pip 5. Пиперазиновый фрагмент имел положительный заряд в физиологических условиях (в воде pH 7,4). Пиперазиновая группа также являлась подходящей в качестве спейсера из-за низкой токсичности и биотрансформаций, которые включали в себя несколько известных метаболических реакций. Общий путь синтеза для получения TPP-NH-Pip показан на схеме 1 на фигуре 2.
Электрофильное промежуточное соединение хитозана 9. Хитозан был модифицирован, как описано ранее, чтобы получить DiTBDMS-CS 8. После защиты гидроксильных групп растворимость биополимера резко изменялась, и он становится легко растворимым в обычных органических растворителях, таких как CH2Cl2, что облегчало его модификацию с высоко липофильными группами. Таким образом, электрофильный промежуточный BrA-DiTBDMS-CS 9 был получен путем взаимодействия 8 с 2-бромацетилбромидом (схема 2 на фигуре 3). Эта реакция требует точного контроля времени реакции, температуры и соотношения эквивалентов различных реагентов, чтобы избежать побочных реакций, таких как удаление защитных групп и этерификация. Реакция 8 с бромацетилбромидом при температуре 0°C в течение 1,5 час давала продукт, нерастворимый в CH2Cl2, и анализ ИК-спектроскопией с Фурье-преобразованием показывал пик сложного эфира при 1760 см-1 вместе с пиком амида при 1680 см-1 (данные не показаны). Таким образом, необходимо было тщательно оптимизировать условия проведения реакции, чтобы избежать побочных реакций и получить легко растворимый продукт. Оптимизированные условия для этой реакции требуют использования ровно 4 эквивалентов бромацетилбромида и 5 эквивалентов Et3N и поддержания температуры реакции при -20°C в течение 1 час. Полученный промежуточный 9 был полностью растворим в CH2Cl2, и точная его структура могла быть подтверждена анализами 1H ЯМР и ИК-спектроскопии с Фурье-преобразованием. Соединение 9 затем было использовано в качестве ключевого промежуточного электрофильного продукта в синтезе хитозановых носителей.
TPP производные хитозана (16A-19Б). Всего восемь TPP производных хитозановых соединений было синтезировано в двух отдельных партиях из четырех различных соединений. Первая партия (с пометкой "A") была синтезирована в масштабе 80-200 мг, и вторая партия (с маркировкой "B") была синтезирована в немного большем масштабе 300-450 мг для того, чтобы подтвердить воспроизводимость и стабильность способа.
Липофильный ФС был ковалентно присоединен к основному скелету полимера путем взаимодействия BrA-DiTBDMS-CS 9 с нуклеофильным промежуточным соединением TPP-NH-Pip 5. Реакция была количественной, что облегчало хороший контроль степени замещения в полученном продукте. Предварительные исследования показали, что носители с высокой степенью замещения (СЗ) порфиринов были нерастворимы в водном растворе, и поэтому модификация ограничивалась 0,1 и 0,25 СЗ (на мономерное звено глюкозамина). Таким образом, TPP-NH-Pip 5 реагировал контролируемым образом при 0,1 или 0,25 эквивалентов в расчете на мономерные единицы электрофильного хитозанового промежуточного соединения 9 с получением желаемых продуктов TPPp0,1-BrA0,9-DiTBDMS-CS (10A/10B) или TPPp0,25-BrA0,75-DiTBDMS-CS (11A/11B), соответственно. Ход реакции контролировали методом ТСХ, и реакцию останавливали, когда 5 больше не присутствовал, для того, чтобы избежать побочных реакций. 1H ЯМР анализ этих продуктов соответствовал количественной реакции ковалентного присоединения TPP к хитозану.
Катионные группы были затем внедрены в TPP-замещенный хитозановый скелет в целях повышения растворимости в воде носителей и обеспечения афинности к эндоцитозной мембране. Таким образом, соединения 10А/10B и 11A/11B подвергали взаимодействию с избыточным количеством Me3N (ТМА) с получением TPPp0,1-DiTBDMS-CS-ТМА0,9 (12A/12B) и TPPp0,25-DiTBDMS-CS-TMA0,75 (13А, 13B), соответственно, с фиксированным катионным зарядом. Аналогичным образом, соединения 10А/10B и 11A/11B подвергали взаимодействию с избыточным количеством 1-метил-пиперазина (MP) с получением TPPp0,1-DiTBDMS-CS-MP0,9 (14A/14B) и TPPp0,25-DiTBDMS-CS-MP0,75 (15А, 15B) соответственно. Сырые продукты использовали напрямую в конечных стадиях удаления защитных групп.
Наконец, сырые продукты из первой партии 12A-15A были подвергнуты удалению защитных групп с помощью конц. HCl в MeOH при комнатной температуре, 12 час, тогда как сырые продукты со второй партии 12B-15B были подвергнуты удалению защитных групп с помощью TBAF/NMP при температуре 60°C, 24 час, способы давали конечные продукты 16A-19A и 16B-19B, соответственно. В обоих случаях стадию удаления защитных групп повторяли для того, чтобы удалить следовые количества TBDMS (1,5-7%), которые все еще имелись после первой стадии удаления защитных групп. Удаление защитных групп с помощью конц. HCl в MeOH было использовано ранее, но в последнее время мягкое удаление TBAF/NMP защитных групп из хитозановых производных DiTBDMS включают для того, чтобы избежать очень кислых условий, которые могут способствовать разложению скелета полимера. Однако недостатком последнего способа является то, что он требует длительного процесса диализа для удаления растворителя NMP. Производные триметиламмония (Мне3N+) [TPPp0,10-CS-ТМА0,9 (16А, 16B) и TPPp0,25-CS-TMA0,75 (17А, 17B)] растворяются в воде быстрее, чем производные 1-метил-пиперазина [TPPp0,1-CS-MP0,9 (18A, 18B), TPPp0,25-CS-MP0,75 (19A, 19B)]. Это может быть связано с наличием фиксированного ионного заряда в последних соединениях носителей.
Характеристика наноносителей.
Анализ ИК-спектроскопией с Фурье-преобразованием: Сравнение спектров ИК-спектров с Фурье-преобразованием различных промежуточных соединений при синтезе характерного конечного соединения TPPp0,1-CS-MP0,9 (18A) показано на фигуре 4. Характеристические пики для порфиринового промежуточного соединения TPP-NH-Pip 5 (фигура 4A) проиллюстрированы в качестве N-H валентных колебаний в области 3310-3450 см-1 и с ароматическими и алифатическими C-H валентными колебаниями при 3022 и 2816 см-1. Пики при 1691 и 1595 см-1 согласуются с амидными связями и три сильных пика при 966, 799 и 700 см-1 характерны для TPP кольцевой системы (показано стрелками).
Основной характеристический пик для промежуточного соединения BrA-DiTBDMS-CS 9 наблюдается при 2858-2957 см-1, показывая C-H валентные колебания Si-CH3, амидные пики при 1682 и 1530 см-1. Кроме того, пики при 836 и 777 см-1 представляют Si-C валентные колебания и CH3 колебание (эти два последних пика указаны стрелками на фигуре 4B).
Появление характеристических пиков TPP в спектрах промежуточного TPPp0,1-BrA0,9-DiTBDMS-CS 10А (фигура 4C) подтверждает ковалентное присоединение порфирина (TPP-NH-Pip 5) к хитозану (9). Не наблюдалось заметных изменений в спектрах, когда его преобразуют в сырой TPPp0,1-DiTBDMS-CS-MP0,9 14A. Однако окончательное удаление TBDMS защитных групп у продукта с получением TPPp0,1-CS-MP0,9 18A, безусловно, свидетельствует об отсутствии характеристических пиков Si-CH3 и появлении широкого пика O-H, в то время как характеристические пики TPP остаются неизменными (фигура 4E).
Анализ 1H и 13C (твердое состояние) ядерно-магнитно-резонансной спектроскопии (ЯМР):
1H ЯМР анализ: Представительный пример перекрывания 1H ЯМР спектров всех промежуточных продуктов и конечного продукта в синтезе TPP0,1-CS-MP0,9 18A показан на фигуре 5. После защиты с помощью TBDMS продукт (DiTBDMS-CS 8) становится полностью растворимым в CDCl3 и также показывает хорошо разрешенные пики (фигура 5А) каждого протона в хитозановом скелете. Пики при 0,90-0,89 (ушир. с) и 0,13-0,05 (ушир. с) могут быть отнесены к (CH3)3C-Si и (CH3)2Si, соответственно.
При бромацетилировании DiTBDMS-CS с получением BrA-DiTBDMS-CS 9 отмечали наличие сдвига пика H-2 (GluN). Также, все пики скелета хитозана от H-1 до H-6 вместе с пиками COCH2Br проявляются вместе при 4,40-3,26 м.д., в то время как пики TBDMS показывают отсутствие значительных изменений в их положениях (фигура 5B).
1H ЯМР спектры промежуточного соединения TPPp0,1-BrA0,9-DiTBDMS-CS 10А (фигура 5C) подтверждают характеристические пики группы TPP, можно увидеть в ароматической области, а также пик при -2,7 [не показано на фигуре 5C], который обычно связывается с двумя пиррольными внутренними протонами аминов в ядре свободного основания порфирина, могут быть также определены. Кроме того, одним из ярко выраженных пиков, которые могут быть отнесены к половине циклической -CH2 группы пиперазинового фрагмента можно наблюдать при 2,81-2,86 (м) м.д., и оставшаяся циклическая -CH2 группа пиперазина подпадает под широкую область (3,3-4,5 м.д.) хитозанового фрагмента.
В спектре TPPp0,1-DiTBDMS-CS-MP0,9 14A новые пики для N-CH3 1-метилпиперазиновой группы (фигура 5D) не наблюдались. Очистка на этой стадии не проводилась, и спектр, таким образом, показывает превышение 1-метил-пиперазина.
После конечного удаления защитных групп был получен продукт с отличной растворимостью в воде. В 1H ЯМР спектре конечного соединения TPPp0,1-CS-MP0,9 18A все пики хитозанового скелета наряду с пиками 1-метил-пиперазина могут быть четко идентифицированы; однако нет наблюдаемого сигнала ЯМР для TPP группы (фигура 5E). Аналогичные результаты наблюдались для всех подвергнутых удалению защитных групп конечных соединений (17A-19B). В d6-ДМСО пики TPP можно было бы наблюдать, но в случае хитозанового скелета пики были уширены и четко не идентифицировались. Растворимость носителя также была слабой в этом растворителе, и было установлено, что наилучшие результаты могут быть получены со смесью этих двух растворителей (см. обсуждение далее). TPPp0,1-CS-MP0,9 18A имеет темно-красный цвет, что подтверждает наличие ФС. Эксперимент по разделению показал, что высоко липофильные группы ФС TPP-NH2 и TPP-NH-Pip не уходили в водную фазу двухфазной системы H2O/CHCl3 (фигура 6), тогда как конечные (полярные) носители TPP-CS-ТМА и TPP-CS-MP были высоко полярными. Они были полностью растворимыми в водной фазе и не уходили в органическую фазу.
Анализ 13C ЯМР в твердом теле: углеродные пики конечных носителей можно наблюдать в ЯМР в твердом теле (фигура 7). Углероды C2-C6 глюкозамина и углерод COCH2 наблюдались в диапазоне 60-80 м.д. и пик C1 явно был проявлен при -102 м.д. Пики между 115-155 м.д. могут быть отнесены как Cmeso (~119), Cм,п (~128), Cβ (~131), Co (~138), Ci (~140) и Cα (~150) группы TPP, и это соотнесение вполне сопоставимо с 13C ЯМР спектром TPP-NH-Pip 5 в виде раствора. Карбонил (CS-NH-C=O и TPP-NH-C=O) проявляется при 160-169 м.д. В N-(2-(N,N,N-триметиламмоний) ацетил) производном TPPp0,25-CS-TMA0,75 (17B) четвертичные метильные углероды (+N(CH3)3) можно было наблюдать в виде интенсивного пика при 55,5 м.д. В N-метил-пиперазинильном производном TPPp0,25-CS-MP0,75 (19B) метильный углерод (N-CH3) можно наблюдать при 45 м.д. и циклические метиленовые углероды (-CH2-N) объединены с глюкозаминовыми сигналами в области 50-55 м.д. 13C ЯМР в твердом состоянии, следовательно, был в соответствии с носителями с ковалентно связанными ФС.
Степень замещения (СЗ) и эффективность преобразования. В таблице 1, приведенной далее, показана СЗ для конечных соединений носителей. Цель состояла в контроле СЗ с эквивалентным соотношением между соединениями 5 и 9 на стадии реакции "e" (схема 2 на фигуре 3). Окончательная СЗ совпадает с целевым значением на основе эквивалентности в реакции (0,1 или 0,25) c 2% колебанием и 100%-ной эффективностью в реакции. Ранее было исследовано ковалентное связывание различных липофильных групп с хитозаном. Это включает в себя лекарственные средства, такие как доксорубицин и паклитаксел; эндогенные биомолекулы, такие как 5α-желчная кислота, 5β-желчная кислота, деоксихолиновая кислота и холестерин, и фотосенсибилизаторы, как хлорин е6 (Ce6) и протопорфирин IX (PplX). В большинстве случаев СЗ является значительно меньшей, чем сообщалось в данном документе для высоко липофильной TPP. Предыдущие исследователи также отметили, что эффективность конъюгации значительно снижается по мере увеличения степени замещения. Настоящий способ имеет то преимущество, что в нем используется защищенный хитозан, который хорошо растворим в органических растворителях и поэтому может быть эффективно объединен с липофильной группой ФС в мягких условиях. Превосходная воспроизводимость реакции также подтверждалась, поскольку СЗ в двух партиях (A и B) совпадали в пределах того же колебания в 2%.
Степень замещения (СЗ) для TPP модифицированных хитозановых носителей 16А-19B
Анализ самоагрегации носителей с образованием наночастиц и развертывание (анфолдинг) наночастиц носителей
Ароматические порфирины могут образовывать π-π сопряженные системы, которые определяются как агрегаты типа J (красное смещение) или типа H (синее смещение). Периферийные группы заместителей могут способствовать механизмам агрегации. Эту агрегацию можно определить методами ЯМР, спектроскопией УФ и видимой области и флуоресцентной спектроскопией. Таким образом, экстремальные уширение пиков и исчезновение пиков в 1H ЯМР сообщалось для карбоксифенил-порфириновых (TCPP) агрегатов, п-сульфонатофенильных и фенильных мезо-замещенных порфиринов и трех типов катионных порфиринов (TOPyP, TMPyP и APP). Аналогичные наблюдения исчезновения сигнала из-за иммобилизации были сделаны при дисперсионной сополимеризации липофильного н-бутилметакрилата с поли(этиленоксид)макромономером.
В настоящей работе отсутствие пиков ароматической области, в 1H ЯМР D2O конечных соединений 16А-19B предполагало возможность агрегации TPP групп в D2O за счет π-π сопряжения и гидрофобного взаимодействия. ЯМР-исследования представительного соединения TPPp0,1-CS-TMA0,9 16B в смеси ДМСО-d6:D2O показано на фигуре 8. В чистом D2O пики группы TPP отсутствуют, и наблюдаются только очень слабые пики в ароматической области в смеси ДМСО-d6:D2O (25:75), тогда как пики скелета хитозана можно было наблюдать. Когда содержание ДМСО-d6 далее повышается до ДМСО-d6:D2O (50:50), можно было наблюдать широкие пики (фигура 8IC). Относительная интенсивность ароматических пиков возрастала по мере увеличение содержания в смеси ДМСО-d6, и в почти чистом d6-ДМСО (только 2% D2O об./об.) ароматические пики TPP были хорошо видны наряду с H-1 и остальными пиками хитозанового скелета и пиком +NMe3 (фигура 8IA). Это также свидетельствует о том, что носитель полностью подвергался анфолдингу в этих условиях. Эта интерпретация была затем подтверждена исследованиями спектров УФ и видимой области и флуоресценции (фигура 8).
Наноносители, растворенные в воде, показывали широкую полосу Соре для фотосенсибилизатора с π-π сопряжением при максимуме абсорбции 417 нм (фигура 8II). Этот пик постепенно становился острее и немного смещался (к 420 нм в 100% ДМСО), когда концентрации ДМСО повышалась, и структуры подвергались анфолдингу.
В чистой воде флуоресценция почти полностью угасала, что также согласуется с агрегацией групп фотосенсибилизаторов. Но интенсивность флуоресценции резко увеличивается, когда содержание ДМСО повышается до 50%, наблюдается резкое увеличение интенсивности флуоресценции с последующим постепенным увеличением по мере повышения концентрации ДМСО до 100% (фигура 8III).
Была предпринята попытка дальнейшей характеризации частиц наноносителя методом сканирующей (растровой) электронной микроскопии. Капельки раствора наноносителя наносили пипеткой на поверхность силиконовой пластины и давали высохнуть (фигура 9A,B) с помощью РЭМ (растровая электронная микроскопия). В некоторых образцах могут наблюдаться наночастицы с размером дисперсных частиц в 10-200 нм, либо выделенные (фигура 9A), либо кластерные или кристаллизованные в дендритные структуры (фигура 9B), подобные тем, что обычно наблюдается при сушке суспензий наночастиц. Для того, чтобы избежать агрегации частиц, было опробовано нанесение покрытия методом центрифугирования на образцы, но в этом случае наблюдались бесцветная пленка и небольшое количество частиц. Толщина пленки после нанесения покрытия методом центрифугирования была значительно меньше, чем 10 нм, как оценивалось с помощью эллипсометрии. Максимальная толщина слоев, образующихся после высыхания растворов на поверхности, однако, составляла порядка 100 нм. Измерение контактного угла с водой выявило относительно гидрофобную поверхность пленки и то, что гидрофобность пленки зависит от концентрации применяемого образца носителя (фигура 9C). Эти результаты соответствуют динамическим наночастицам, которые подвергаются анфолдингу при покрытии, и тому, что гидрофильный полимерный скелет будет удерживаться на поверхности полярного диоксида кремния, открывая гидрофобные группы фотосенсибилизаторов из ядра частицы. Обычно наблюдается агрегация наночастиц и физическая нестабильность, ведущие к образованию нерастворимых агрегатов. Физическая стабильность 1 мг/мл водного раствора TPPp0,25-CS-MP0,75 (17A) контролировалась на протяжении трех месяцев. Не наблюдалось никакого осадка этого соединения.
Квантовые выходы флуоресценции
Здесь, квантовые выходы флуоресценции TPP(п-NH2)1, TPP-NH-Pip и хитозановых производных TPP (16A-19B) были исследованы в ДМСО (возбуждение при 419 нм). Квантовый выход TPP(п-NH2)1 был меньше, чем у его производного TPP-NH-Pip, а также TPP-CS-ТМА производных и TPP-CS-MP производных. Это свидетельствует о высокой степени гашения возбужденного состояния TPP(п-NH2)1 по сравнению с его производными. ΦF был почти равным для всех соединений носителей (16А-19B) с незначительным колебанием ±0,004 относительно значения, полученного для промежуточного соединения TPP-NH-Pip.
Однако значения ΦF для TPP(п-NH2)1 (0,0002) были ниже по сравнению с некоторыми опубликованными в литературе данными. Bhaumik et al. (J Org Chem 2009, 74:5894) сообщали, что он составляет 0,05 при возбуждении при 418 нм в ДМФ. Bhaumik et al сообщали, что максимум флуоресцентной эмиссии наблюдался при 664 нм, что отличается от обнаруженного авторами данного изобретения в настоящее время показателя при 650 нм. TPP был использован в качестве стандарта, в то время как авторы настоящего изобретения в качестве стандарта использовали антрацен. Низкий ΦF в настоящей работе может быть вызван самоассоциацией фотосенсибилизаторов. В таблице 2, приведенной далее, показан квантовый выход флуоресценции (ΦF) модифицированных TPP хитозановых носителей 16А-19B.
хитозана
λ - длина волны
Вывод
Авторами изобретения было показано, что DiTBDMS хитозан может быть использован для высокоэффективного синтеза хитозана, связанного с мезо-тетрафенилпорфирином на основе наноносителей. Синтез этих носителей был полностью воспроизводимым, и способ позволял точно контролировать степень замещения для достижения высокой липофильности фотосенсибилизатора. Исследования ЯМР, флуоресценция и спектроскопия УФ и видимой области показали соответствие с самоассоциацией групп фотосенсибилизаторов. Носители были полярными и показывали хорошую растворимость в воде и физическую стабильность. В ДМСО фотосенсибилизатор диссоциировался от самоассоциации и, следовательно, становился люминесцентным. В водном растворе носители объединялись в структуры, подобные наночастицам с катионной группой и полимерным скелетом, образуя наружную оболочку вокруг ядра, состоящую из агрегированных (π-π сопряжение) липофильных TPP групп. Катионные группы и полимерный скелет обладали относительно свободной способностью движения в жидкости и, следовательно, могут наблюдаться методом ЯМР. Напротив, ядро TPP является полутвердым с очень ограниченным движением, и поэтому может быть обнаружено только методом ЯМР в твердом теле. Эта структура находится в динамическом равновесии с развернутой и флуоресцентной формой, которая становится доминирующей в ДМСО, что позволяет обнаружить TPP методом ЯМР. Когда носитель находится в контакте с клеткой или эндоцитозной мембраной, структура развертывается (анфолдинг) и липофильные группы вставляются в эндоцитозную мембрану, создавая возможность для возбуждения и фотосенсибилизации, что приводит к эффектам ФДТ и ФХИ.
Пример 2. Синтез наноносителей на основе TPC-хитозана
Общие материалы и способы были такими, как в примере 1, где это уместно.
Синтез
См. схему 3 на фигуре 10 для синтеза TPC-NH-Pip
Мезо-Тетрафенилпорфирин (1). Соединение 1 (TPP) получали способом, описанным в Journal of Organic Chemistry 1967, 32, 476.
5-(4-Аминофенил)-10,15,20-трифенилпорфирин [TPP(p-NH2)1] (3). Соединение 3 было получено в соответствии с литературным способом, описанным в Tetrahedron 2004, 60, 2757.
5-(4-Аминофенил)-10,15,20-трифенилхлорин: TPC(p-NH2)1 (20). Соединение 3 (1,5 г, 2,38 ммоль) растворяли в пиридине в атмосфере N2 и в темноте. Добавляли K2CO3 (2,96 г, 21,5 ммоль) и п-толуолсульфонилгидразид (0,887 г, 4,77 ммоль), и полученную реакционную смесь нагревали при температуре кипения с обратным холодильником. Добавляли дополнительное количество п-толуолсульфонилгидразида (0,887 г, 4,77 ммоль) с интервалом в 2, 4, 6 и 8 часов. Перемешивание продолжали при температуре кипения в течение 24 часов. Реакционную смесь добавляли в смесь 1:1 EtOAc:H2O (2:1, 900 мл) и кипятили с обратным холодильником в течение 1 часа. После охлаждения до комнатной температуры органическую фазу отделяли и промывали 2н HCl (3×200 мл) с последующим промыванием водой (2×100 мл) и насыщенным водным раствором NaHCO3 (2×150 мл). Органическую фазу затем сушили над Na2SO4 и концентрировали в вакууме с получением 1,3 г сырой смеси. Анализ видимого спектра сырого продукта показал, что он является смесью хлорина и бактериохлорина (полосы 651, 738, соответственно). Кроме того, анализ с помощью 1H ЯМР-спектров показал, что нет никаких следов остатков исходного порфиринового продукта.
Сырой материал (1,3 г) (смесь хлорин/бактериохлорин), полученный в вышеописанной стадии реакции, растворяли в CH2Cl2 (100 мл). Затем одной порцией к перемешиваемому растворителю при комнатной температуре добавляли орто-хлоранил (420 мг, 2,7 ммоль), и протекание реакции одновременно контролировали с помощью оптической спектроскопии. Сразу после полного уменьшения пика бактериохлорина (738 нм) реакционную смесь промывали 5% водным раствором гидросульфита натрия (2×125 мл) с последующим промыванием водой (100 мл), 5% NaOH (2×150 мл) и, наконец, водой (150 мл). Органическую фазу затем сушили над Na2SO4 и концентрировали в вакууме с получением исключительно указанного в заголовке соединения хлорина 20 (1,2 г, 80%) в виде твердого вещества коричневого цвета. Как оказалось, соединение 3 было в виде более одного изомера.
ТСХ (гексан/CH2Cl2 3:7): Rf=0,23, 1H ЯМР (CDCl3): δ=7,86-8,66 (м, 14H, β-пиррол-H и фенил-Ho), 7,63-7,73 (м, 9H, трифенил-Hм,п), 7,00 (д, J=8Гц, 2H, NH2-фенил-Hм), 4,14-4,23 (м, 4H, хлорин β-пиррол-CH2), 3,95 (ушир. с, 2H, NH2), -1,38 и -1,46 (ушир. с, 2H, α-пиррол-NH) м.д.; MS (ESI): m/z вычисл. для C44H34N5 ([M+H]+) 632,2809, найдено 632,2792; спектр УФ и видимой области (ДМСО): λmax: 422, 524, 553, 600, 652 нм.
Синтез промежуточного TPC-NH-pip (21). Соединение 20 (600 мг, 0,95 ммоль) растворяли в CH2Cl2 (15 мл) и перемешивали в атмосфере N2. Добавляли Et3N (0,32 мл, 2,27 ммоль), затем по каплям добавляли хлорацетил хлорид (0,092 мл, 1,15 ммоль) при комнатной температуры и продолжали перемешивание при комнатной температуре. Через 2 час in situ добавляли избыточное количество пиперазина (0,328 г, 3,8 ммоль) и перемешивание продолжали в течение ночи. Реакционную смесь разбавляли CH2Cl2 (85 мл), экстрагировали, промывали водой (3×35 мл), насыщенным солевым раствором (35 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт затем очищали путем колоночной хроматографии на силикагеле. Требуемый продукт выделяли смесью MeOH:CH2Cl2 (8:92) в качестве элюента с получением указанного в заголовке промежуточного соединения 21 (440 мг, 61%) в виде твердого вещества коричневого цвета.
ТСХ (CH2Cl2:MeOH, 9:1): Rf=0,15; 1H ЯМР (CDCl3): δ=9,34, 9,39 (с, 1H, TPC-NH), 7,86-8,65 (м, 16H, β-пиррол-H, фенил-Ho и R-NHTPC-фенил-Hо,м), 7,66-7,73 (м, 9H, трифенил-Hм,п), 4,18-4,19 (ушир. с, 4H, хлорин β-пиррол-CH2), 3,30 (с, 2H, ArNHCOCH2-pip), 3,17 (ушир. м, 4Н, пиперазиновое кольцо-CH2), 2,81 (ушир. м, 4Н, пиперазиновое кольцо-CH2), -1,37 (ушир. с, 2H, α-пиррол-NH); 13C ЯМР (CDCl3): δ=168,37, 167,48, 152,61, 143,14, 142,22, 140,86, 139,20, 138,32, 137,19, 136,99, 135,33, 134,64, 133,98, 133,01, 132,37, 132,12, 131,96, 128,17, 127,69, 126,81, 123,56, 123,38, 122,79, 122,08, 119,22, 117,94, 112,41, 111,65, 62,63, 53,50, 45,59, 35,90 м.д.; Спектр УФ и видимой области (ДМСО): λmax: 421, 521, 549, 598, 651 нм.
См. схему 4 на фигуре 11
Синтез трет-бутил пиперазин-1-карбоксилата (22). Пиперазин (6 г, 69,6 ммоль) растворяли в CH2Cl2 (120 мл). Раствор охлаждали до 0°C и добавляли по каплям Boc2O (7,6 г, 34,8 ммоль) в CH2Cl2 (80 мл). Реакционную смесь оставляли нагреваться до комнатной температуры, и перемешивание продолжали в течение 24 часов. Осадок отфильтровывали и промывали CH2Cl2 (2×20 мл), и объединенный фильтрат отделяли и промывали водой (3×40 мл), насыщенным солевым раствором (30 мл) и сушили над Na2SO4 и концентрировали в вакууме, с получением указанного в заголовке соединения 22 (6,5 г, 50%) в виде твердого вещества белого цвета.
Т.пл. 44-46°C (лит. т.пл. 46-47°C); 1H ЯМР (CDCl3): δ=3,32 (т, J=4Гц, 4H), 2,74 (т, J=4Гц, 4H), 1,39 (с, 9H); 13C ЯМР (CDCl3): δ=154,85, 80,00, 79,52, 45,96, 44,45, 28,45 м.д.; MS (ESI): m/z вычисл. для C9H19N202 ([M+H]+) 187,1441 найдено 187,1412.
Синтез п-(метоксикарбонил)бензальдегида (23). 4-Карбоксибензальдегид (4 г, 26,6 ммоль) суспендировали в 60 мл безводного MeOH и перемешивали в атмосфере N2. Реакционную смесь охлаждали до 0°C и добавляли по каплям ацетил хлорид (9,5 мл, 133 ммоль). Реакционную смесь перемешивали в течение 12 ч при комнатной температуре. MeOH удаляли в вакууме, и сырую смесь разбавляли EtOAc (120 мл). Органическую фазу промывали 1н водным раствором NaOH (5×30 мл) и насыщенным солевым раствором (2×25 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой твердый продукт затем перекристаллизовывали с использованием EtOAc и петролейного эфира с получением сложного эфира 23 (3,8 г, 87%) в виде твердого вещества белого цвета.
ТСХ (гексан: CH2Cl2, 3:7): Rf=0,36; Т.пл.: 61-63°C (лит. т.пл. 59-64°C); 1H ЯМР (CDCl3): δ=10,06 (с, 1H, CHO), 8,15 (д, J=8,4Гц, 2H), 7,91 (д, J=8,4Гц, 2H), 3,92 (с, 3H) м.д.; 13C ЯМР (CDCl3): δ =191,66, 166,07, 139,21, 135,13, 130,23, 129,55, 52,62 м.д.
Синтез 5-(4-метоксикарбонилфенил)-10,15,20-трифенилпорфирина TPP(п-CO2Me)1 (24). Следовали способу, описанному в литературе (J. Am. Chem. Soc. 2008, 130, 4236-4237).
Синтез 5-(4-карбоксифенил)-10,15,20-трифенилпорфирина TPP(п-CO2H)1 (25).
Соединение 24 (1,2 г, 1,78 ммоль) растворяли в смеси ТГФ: пиридин (10:1, 100 мл). Добавляли 2н раствор KOH в метаноле (120 мл), и реакционную смесь кипятили с обратным холодильником в течение 24 часов. Затем реакционную смесь охлаждали до комнатной температуры и нейтрализовали насыщенным водным раствором лимонной кислоты. Затем реакционную смесь концентрировали в вакууме до полного удаления MeOH и ТГФ. Сырую смесь разбавляли CH2Cl2 (150 мл) и водой (120 мл), и водную фазу экстрагировали CH2Cl2 (3×50 мл). Объединенную органическую фазу промывали водой (2×40 мл) и насыщенным солевым раствором (35 мл), сушили над Na2SO4 и концентрировали в вакууме. Сырую смесь очищали с помощью колоночной хроматографии на силикагеле, используя MeOH:CH2Cl2 (от 100:0 до 96:4 в качестве элюента) с получением указанной в заголовке кислоты 25 (0,83 г, 71%) в виде твердого вещества пурпурного цвета.
ТСХ (CH2Cl2: MeOH 95:5): Rf=0,54; 1H ЯМР (ДМСО-d6): δ=8,84 (ушир. с, 8H, β-пиррол-H), 8,33-8,39 (м, 4H, R-COTPP-фенил-Hо,м), 8,21-8,23 (м, 6H, трифенил-Ho), 7,81-7,88 (м, 9H, трифенил-Hм,п), -2,92 (с, 2H, NH) м.д.; MS (ESI): m/z вычисл. для C45H3N4O2 ([M+H]+) 659,2442, найдено 659,2420.
Синтез 5-(4-карбоксифенил)-10,15,20-трифенилхлорина TPC(p-CO2H)1 (26).
Соединение 25 (600 мг, 0,9 ммоль) и безводный K2CO3 (1,13 г, 8,2 ммоль) растворяли в пиридине (42 мл) в атмосфере N2 и в темноте. Затем добавляли толуол-4-сульфонилгидразид (340 мг, 1,8 ммоль), и смесь перемешивали при температуре кипения с обратным холодильником. С интервалом 2, 4, 6, 8 и 10 час протекания реакции были добавлены дополнительные количества толуол-4-сульфонилгидразида (340 мг, 1,8 ммоль) в 3 мл пиридина. Перемешивание продолжали при кипячении с обратным холодильником в течение 24 часов. После охлаждения до комнатной температуры добавляли EtOAc (500 мл) и деионизированную H2O (250 мл), и реакционную смесь опять кипятили с обратным холодильником в течение 1 часа. После охлаждения до комнатной температуры органическую фазу отделяли и промывали 2н HCl (2×150 мл) и затем водой (2×150 мл), сушили над Na2SO4 и концентрировали в вакууме с получением 565 мг сырой смеси. Анализ видимого спектра неочищенного продукта показал, что он представляет собой смесь хлорина и бактериохлорина (полосы при 651, 738, соответственно). Кроме того, анализ спектра 1H ЯМР показал, что нет никаких следов остатков исходного порфиринового продукта.
Сырой продукт (смесь хлорин/бактериохлорин, 565 мг), полученный на стадии, представленной выше, полностью растворяли в смеси CH2Cl2:MeOH (75:25). Затем одной порцией добавляли орто-хлоранил (180 мг, 0,7 ммоль) в перемешиваемый органический раствор при комнатной температуре и одновременно отслеживали протекание реакции с помощью спектроскопии УФ и видимой области. Сразу после уменьшения пика абсорбции бактериохлорина (738 нм) органическую фазу промывали 5%-ным водным раствором гидросульфита натрия (2×150 мл) с последующим промыванием водой (100 мл), затем 5%-ным водным NaOH (2×150 мл) и, наконец, водой (120 мл). Если не наблюдалось эмульсии, органическую фазу промывали насыщенным водным раствором лимонной кислоты. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением исключительно указанного в заголовке хлоринового соединения 26 (420 мг, 70%) в виде твердого вещества коричневого цвета. Соединение 9 было в виде более одного изомера.
ТСХ (CH2Cl2: MeOH, 95:5): Rf=0,54, 1H ЯМР (ДМСО-d6): δ=7,91-8,58 (м, 16H, β-пиррол-H, фенил-Ho и R-COTPC-фенил-Ho,м), 7,68-7,77 (м, 9H, трифенил-Hм,п), 4,12-4,13 (м, 4H, хлорин β-пиррол-CH2), -1,53 и -1,60 (2 ушир. с, 2H, α-пиррол-NH); 1H ЯМР (CDCl3): δ=7,87-8,60 (м, 16H, β-пиррол-H, фенил-Ho и R-COTPC-фенил-Hо,м), 7,64-7,74 (м, 9H, трифенил-Hм,п), 4,16-4,18 (м, 4H, хлорин β-пиррол-CH2), -1,39 и -1,49 (2 ушир. с, 2H, α-пиррол-NH) м.д.; MS (ESI) вычисл. для C45H33N4O2 ([M+H]+) 661,2598, найдено 661,2566; Спектр УФ и видимой области (ДМСО): λmax: 420, 520, 547, 599, 651 нм.
Синтез промежуточного трет-бутил-N-[пиперазин-1-карбоксилат]-5-(4-карбоксифенил)-10,15,20-трифенилхлорина (27). Хлориновое соединение 26 (500 мг, 0,76 ммоль) и трет-бутил пиперазин-1-карбоксилат 22 (155 мг, 0,83 ммоль) растворяли в ДМФ (4 мл) в атмосфере N2 в темноте. В реакционную смесь добавляли EDCI-HCl (174 мг, 0,91 ммоль) и HOBT (123 мг, 0,91 ммоль), затем при комнатной температуре добавляли Et3N (0,26 мл, 1,82 ммоль). Реакционную смесь затем при комнатной температуре перемешивали в течение ночи, затем медленно выливали в перемешиваемую воду (100 мл). Твердое вещество отфильтровывали, промывали большим количеством воды и хорошо высушивали. Сырой продукт очищали путем колоночной хроматографии на силикагеле (CH2Cl2:MeOH от 100:0 до 99:1) с получением указанного в заголовке соединения 27 (340 мг, 54%) в виде твердого вещества коричневого цвета.
ТСХ (CH2Cl2:MeOH 99:1): Rf=0,74; 1H ЯМР (CDCl3): δ=7,74-8,59 (м, 16H, β-пиррол-H, фенил-Ho и R-COTPC-фенил-Ho,м), 7,65-7,72 (м, 9H, трифенил-Hм,п), 4,16-4,17 (м, 4H, хлорин β-пиррол-CH2), 3,78-3,86 (ушир. м, 4Н, пиперазиновое кольцо-CH2), 3,63 (ушир. м, 4Н, пиперазиновое кольцо-CH2), 1,53 (с, 9H, boc-(CH3)3), -1,39 и -1,47 (2 ушир. с, 2H, α-пиррольных-NH) м.д.
Синтез промежуточного TPC-CO-pip (28). Соединение 27 (320 мг, 0,39 ммоль) растворяли в CH2Cl2 (8 мл) в атмосфере N2 в темноте. Добавляли CH2Cl2:ТФУ (1:1, 4 мл) и перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли CH2Cl2 (40 мл) и промывали водой (2×15 мл), насыщенным водным раствором NaHCO3 (2×15 мл) и насыщенным солевым раствором (15 мл). Органический слой сушили над Na2SO4 и концентрировали в вакууме. Сырой продукт затем очищали с помощью колоночной хроматографии на силикагеле (CH2Cl2:MeOH от 100:0 до 92:8 в качестве элюента) с получением указанного в заголовке промежуточного соединения 28 (250 мг, 89%) в виде твердого вещества коричневого цвета.
ТСХ (CH2Cl2: MeOH, 9:1): Rf=0,35, 1H ЯМР (CDCl3): δ=7,74-8,59 (м, 16H, β-пиррол-H, фенил-Ho и R-COTPC-фенил-Hо,м), 7,64-7,72 (м, 9H, трифенил-Hм,п), 4,16-4,17 (м, 4H, хлорин β-пиррол-CH2), 3,73-3,90 (ушир. м, 4Н, пиперазиновое кольцо-CH2), 3,04 (ушир. м, 4Н, пиперазиновое кольцо-CH2), -1,40 и -1,47 (2 ушир. с, 2H, α-пиррол-NH) м.д.; MS (ESI) вычисл. для C49H42N6O ([M+2H]+/2) 365,1705, найдено 365,1707; спектр УФ и видимой области (ДМСО): λmax: 420, 520, 546, 599, 651 нм.
Мезилатная соль хитозана (7). Синтезирована согласно ранее опубликованным авторами настоящего изобретения способам (Carbohydrate Polymers 2010, 81:140-144).
N-Бромацетил-3,6-O-DiTBDMS-CS (BrA-DiTBDMS-CS, 9). DiTBDMS-CS 8 (1 г, 2,60 ммоль) растворяли в сухом CH2Cl2 (15 мл) в круглодонной колбе в атмосфере N2. Реакционную смесь охлаждали до -20°C на бане со смесью лед/соль. Добавляли Et3N (1,81 мл, 13 ммоль), затем медленно по каплям добавляли бромацетилбромид (0,91 мл, 10 ммоль). Перемешивание продолжали в течение 1 часа. Реакционную смесь разбавляли CH2Cl2 и концентрировали в вакууме. Сырой продукт перемешивали в ацетонитриле, фильтровали и промывали свежеперегнанным ацетонитрилом. Сухой продукт разбавляли и экстрагировали CH2Cl2, промывали водой и насыщенным солевым раствором, сушили над Na2SO4, концентрировали в вакууме с получением указанного в заголовке соединения 26 (1,2 г, 92%) в виде порошкообразного твердого вещества бледно-желтого цвета.
ИК-спектр с Фурье-преобразованием (KBr): v 3402 (шир., NH), 2957, 2931, 2886, 2858 (с, C-H TBDMS), 1682 (vs, C=O амид I), 1530 (vs, C=O амид II), 1473, 1391, 1362, 1311, 1259, 1101, 1005, 837, 777 (Si-C), 669 см-1; 1H ЯМР (CDCl3) δ м.д.: 4,40 (ушир. с, H-1), 4,02-3,26 (м, H-2 GlcN, H-3, H-4, H-5, H-6, H-6' и 2H GluNH-C=OCH2Br), 0,90 и 0,88 (ушир. с, (CH3)3C-Si), 0,13 и 0,07 (ушир. с, (CH3)2Si) м.д.
См. схему 6А на фигуре 13
Синтез промежуточного соединения 29
(N-TPC-NH-Pip-ацетил)0,1-(N-бромацетил)0,9-DiTBDMS-CS(TPCNPNP0,1-BrA0,9-DiTBDMS-CS, 29). Соединение 9 (800 мг, 1,58 ммоль) и соединение TPC-NH-Pip 21 (120 мг, 0,158 ммоль), растворяли в CH2Cl2 (25 мл) в атмосфере N2 и в темноте. Добавляли точное эквимолярное количество Et3N (22 мкл, 0,158 ммоль) относительно 21, и реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Полное расходование исходного материала подтверждали методом ТСХ. Реакционную смесь разбавляли CH2Cl2 (55 мл) и промывали водой (2×25 мл) и насыщенным солевым раствором (25 мл). Органическую фазу сушили над Na2SO4, концентрировали в вакууме с получением соединения 29 (700 мг, 78%).
1H ЯМР (CDCl3): δ=9,21, 9,25 (с, TPCNHCO), 7,86-8,60 (м, β-пиррол-H, фенил-Ho и R-NHTPC-фенил-Ho,м), 7,65-7,73 (м, трифенил-Hм,п), 3,35-4,50 [ушир. м, хитозан (H-1, H-2 GlcN, H-3, H-4, H-5, H-6, H-6' и 2H GluNH-C=O, CH2CONGlc), TPCNHCOCH2-pip, пиперазиновое кольцо-CH2 и хлорин β-пиррол-CH2], 2,77-2,83 (м, пиперазиновое кольцо-CH2), 0,88-0,89 [ушир. с, (CH3)3C-Si], 0,02-0,13 [(ушир. м, (CH3)2Si], -1,44 (ушир. с, 2H, α-пиррол-NH) м.д.
См. схему 6B на фигуре 13
Синтез промежуточного соединения 34
(N-TPC-CO-Pip-ацетил)0,1-(N-бромацетил)0,9-DiTBDMS-CS (TPCCP0,1-BrA0,9-DiTBDMS-CS, 34). Соединение 9 (800 мг, 1,58 ммоль) растворяли в NMP (25 мл) в атмосфере N2 и в темноте. При комнатной температуре добавляли TPC-CO-Pip 28 (125 мг, 0,173 ммоль) и NaHCO3 (0,29 г, 3,45 ммоль). Реакционную смесь затем нагревали при 75°C и перемешивали в течение ночи, затем охлаждали и выливали в перемешиваемую воду. Твердый продукт отфильтровывали, промывали большим количеством воды и сушили с отсасыванием. Полученный твердый продукт затем растворяли в CH2Cl2, фильтровали и сушили над Na2SO4, и растворитель удаляли в вакууме с получением соединения 34 (810 мг, 89%) в виде твердого вещества коричневого цвета.
1H ЯМР (CDCl3): δ=7,75-8,60 (м, β-пиррол-H, фенил-Ho и R-COTPC-фенил-Hо,м), 7,64-7,71 (м, трифенил-Hм,п), 3,38-4,5 [шир. м, хитозан (H-1, H-2 GlcN, H-3, H-4, H-5, H-6, H-6' и 2H GluNH-C=O, CH2CONGlc), пиперазиновое кольцо-CH2 и хлорин β-пиррол-CH2], 2,76-2,84 (м, пиперазиновое кольцо-CH2), 0,89-0,92 [ушир. с, (CH3)3C-Si], 0,02-0,10 [(ушир. м, (CH3)2Si)], -1,40 и -1,48 (ушир. с, α-пиррольных-NH) м.д.
Общий способ A для соединений 30 и 35
(N-TPC-NH-Pip-ацетил)0,1-(N-(N,N,N-триметиламмоний)ацетил)0,9-DiTBDMS-CS(TPCNP0,1-DiTBDMS-CS-TMA0,9 30). Соединение 29 (350 мг, 0,61 ммоль) растворяли в CH2Cl2 (15 мл) в атмосфере N2 и в темноте. Добавляли избыточное количество раствора триметиламина, и реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Растворитель удаляли в вакууме. Сырой продукт сушили в высоком вакууме с получением сырого продукта 30 (355 мг, 94%) в виде твердого вещества коричневого цвета. Сырое соединение использовали как таковое на следующей стадии.
(N-TPC-CO-Pip-ацетил)0,1-(N-(N,N,N-триметиламмоний)ацетил)0,9-DiTBDMS-CS(TPCCP0,1-DiTBDMS-CS-TMA0,9 35). Общий способ A осуществляли с использованием 34 (350 мг, 0,61 моль) и раствора триметиламина с получением 35 в виде сырого твердого продукта (360 мг, 94%). Сырое соединение использовали как таковое на следующей стадии.
Общий способ B для соединений 31 и 36
(N-TPC-NH-Pip-ацетил)0,1-(N-(N-метилпиперазинил)ацетил)0,9-DiTBDMS-CS(TPCNP0,1-DiTBDMS-CS-MP0,9 31). Соединение 29 (350 мг, 0,61 ммоль) растворяли в CH2Cl2 (15 мл) в атмосфере N2 и в темноте. Добавляли избыточное количество 1-метилпиперазина, и реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Растворитель удаляли в вакууме. Затем сырой продукт сушили в высоком вакууме с получением соответствующего сырого продукта 31 (330, 89%). Сырое соединение использовали как таковое на следующей стадии.
(N-TPC-СО-Pip-ацетил)0,1-(N-(N-метилпиперазинил)ацетил)0,9-DiTBDMS-CS(TPCCP0,1-DiTBDMS-CS-MP0,9 36). Общий способ B осуществляли, используя 34 (250 мг, 0,38 ммоль) и 1-метилпиперазин с получением 36 в виде сырого твердого вещества (265 мг, 93%). Сырое соединение использовали как таковое на следующей стадии.
Синтез конечных продуктов (32, 33, 37 и 38)
Заключительное удаление защитных групп было достигнуто, следуя общей процедуре C:
Соединения (30/31/35/36), растворяли в MeOH в атмосфере N2 и в темноте. Реакционную смесь дегазировали путем продувки N2 в течение 5 минут и затем охлаждали до 0°C, после чего добавляли 4 мл конц. HCl. Реакционной смеси позволяли нагреться до комнатной температуры, и перемешивали в течение 12 часов, затем полностью упаривали в вакууме. Сырой остаток снова растворяли в MeOH и дегазировали в атмосфере N2 и в темноте. Реакционную смесь охлаждали до 0°C, затем добавляли 2 мл конц. HCl. Реакционной смеси позволяли нагреться до комнатной температуры, и перемешивали в течение 12 часов. Реакционную смесь затем разбавляли и подвергали ионному обмену добавлением 5% водного раствора NaCl в течение 1 часа, затем диализовали против 8% водного раствора NaCl в течение 24 часов и затем против деионизированной воды в течение 3 дней. Чистый раствор коричневого цвета впоследствии подвергали сушке вымораживанием в течение ночи с получением конечных продуктов (32/33/37/38, соответственно) в виде пушистого вещества коричневого цвета.
TPCNP0,1-CS-ТМА0,90 (32). Общий способ C проводили с использованием 30 (325 мг, 0,52 ммоль) и конц. HCl/MeOH с получением 32 в виде твердого вещества коричневого цвета (175 мг, 85%). 1H ЯМР (ДМСО-d6:D2O 98:2): δ=7,83-8,62 (м, β-пиррол-H, фенил-Ho и R-NHTPC-фенил-Hо,м), 7,69-7,77 (м, трифенил-Hм,п), 4,52 (ушир. с, H-1), 4,11-4,14 (м, -СН2CONGlc и хлорин β-пиррол-CH2), 3,26-3,67 (ушир. м, частично перекрывающихся пиком HDO, H-2 GlcNAc, H-3, H-4, H-5, H-6, H-6', 2H GluNH-C=O, TPCNHCOCH2-pip, пиперазиновое кольцо-CH2] 3,24 (с, +N(CH3)3)) м.д.; спектр УФ и видимой области (ДМСО): λmax: 421, 520, 549, 599, 651 нм.
TPCNP0,1-CS-MP0,90 (33). Общий способ C проводили с использованием 31 (300 мг, 0,45 ммоль) и конц. HCl/MeOH с получением 33 (165 мг, 84%) в виде твердого вещества коричневого цвета. 1H ЯМР (ДМСО-d6:D2O 98:2): δ=7,83-8,62 (м, β-пиррол-H, фенил-Ho и R-NHTPC-фенил-Hо,м), 7,66-7,75 (м, трифенил-Hм,п), 4,50 (ушир. с, H-1), 4,10-4,14 (м, хлорин β-пиррол-CH2), 2,92-3,55 (м, частично перекрывающихся пиком HDO, H-2 GlcNAc, H-3, H-4, H-5, H-6, H-6', 2H GluNH-C=O, CH2CONGlc, TPCNHCOCH2-pip), 2,33-2,63 (м, частично перекрывающихся с пиком ДМСО-d6, пиперазиновое кольцо-CH2, пиперазин-N-CH3) м.д.; спектр УФ и видимой области (H2O): λmax: 412, 430, 531, 560, 611, 664 нм; спектр УФ и видимой области (ДМСО): λmax: 421, 521, 548, 596, 651 нм.
TPCCP0,1-CS-ТМА0,90 (37).
Общий способ C осуществляли, используя 35 (300 мг, 0,48 ммоль) и конц. HCl/MeOH, с получением 37 в виде твердого вещества коричневого цвета (170 мг, 89%).
ИК-спектр с Фурье-преобразованием (KBr): v 3353, 3061, 2950, 1683, 1580, 1473, 1440, 1376, 1291, 1154, 1112, 1067, 1032, 970, 911, 794, 703 см-1; 1H ЯМР (ДМСО-d6:D2O 98:2): δ=7,89-8,62 (м, β-пиррол-H, фенил-Ho и R-COTPC-фенил-Hо,м), 7,67-7,76 (м, трифенил-Hм,п), 4,50 (ушир. с, H-1), 4,06-4,16 (м, CH2CONGlc и хлорин β-пиррол-CH2), 3,26-3,75 (м, частично перекрывающихся с пиком HDO, H-2 GlcNAc, H-3, H-4, H-5, H-6, H-6', 2H GluNH-C=O, пиперазиновое кольцо-CH2), 3,24 (с, +N(CH3)3)) м.д.; спектр УФ и видимой области (ДМСО): λmax: 420, 520, 547, 599, 651 нм.
TPCCP0,1-CS-МP0,90 (38). Общий способ C осуществляли, используя 36 (240 мг, 0,38 ммоль) и конц. HCl/MeOH, с получением 38 в виде твердого вещества коричневого цвета (85 мг, 52%).
ИК-спектр с Фурье-преобразованием (KBr): v 3349, 2927, 1644, 1580, 1461, 1440, 1374, 1285, 1070, 1043, 985, 945, 794, 719, 703 см-1; 1H ЯМР (ДМСО-d6:D2O 98:2): δ=7,86-8,63 (м, β-пиррол-H, фенил-Ho и R-COTPC-фенил-Hо,м), 7,67-7,76 (м, трифенил-Hм,п), 4,50 (ушир. с, H-1), 4,08-4,14 (м, хлорин β-пиррол-CH2), 2,92-3,55 (м, частично перекрывающихся с пиком HDO, H-2 GlcNAc, H-3, H-4, H-5, H-6, H-6', 2H GluNH-C=O, CH2CONGlc) 2,27-2,63 (м, частично перекрывающихся с пиком ДМСО-d6, пиперазиновое кольцо-CH2, пиперазин-N-CH3) м.д.; спектр УФ и видимой области (ДМСО): λmax: 421, 520, 547, 599, 651 нм.
TPP аналоги соединений 32, 33, 37 и 38
Неожиданные результаты (обратное окисление соединений TPC до соединений TPP с помощью TBAF/NMP) наблюдались при использовании следующего общего способа удаления защитных TBDMS групп в конечных соединениях 32, 33, 37 и 38).
Пример: TPP аналог соединения 32 (TPPNP0,1-CS-ТМА0,9): Продукт 30 (600 мг, 0,86 ммоль) растворяли в N-метил-2-пирролидоне (NMP) (5-10 мл) с последующим добавлением избыточного количества тетра-н-бутиламмонийфторида (TBAF). Реакционную смесь перемешивали 24 часа при 55°C и охлаждали и подкисляли разбавленной HCl, затем перемешивали в течение 30 минут, после чего диализовали против 8% NaCl (масс./об.) в деионизированной воде в течение двух дней и против деионизированной воды в течение 3 дней. В течение этого времени цвет раствора постепенно менялся от серого до красного. Раствор красного цвета затем лиофилизовали с получением губчатого вещества коричневого цвета. Продукт вновь подвергали удалению защитных групп, ионному обмену, диализу и лиофилизации. Однако, неожиданно, из-за обратного окисления соединения были преобразованы обратно в свои TPP аналоги, что подтверждалось данными спектроскопии УФ и видимой области (поскольку характерный пик при 650 уменьшался почти полностью). (Данные не показаны)
Результаты
В таблице 3 далее показаны СЗ для конечных соединений носителей. В таблице 4 приведены квантовые выходы флуоресценции (ΦF) TPC модифицированных хитозановых носителей. На фигуре 14 представлен 1H ЯМР спектр TPC-CO-Pip (28) - обоих изомеров.
На фигурах 15-17 представлен эквивалентный спектр соединений 21, 29 и 34, соответственно.
На фигуре 18 представлен спектр ЯМР конечных соединений носителей 37, 38, 32 и 33 в d6-ДМСО/D2O.
**СЗ, определенная с помощью 1H ЯМР промежуточного соединения 34
хитозана
λ - длина волны
Пример 3 – Исследования in vitro и in vivo
Материалы
Клеточная линия карциномы толстого кишечника человека HCT116/LUC (перманентно трансфицированная с геном, кодирующим люциферазу) была любезно предоставлена доктором Mohammed Amarzguioui, siRNAsense, Oslo, Norway. MTT (3-[4,5-диметилтиазол-2-ил]-2,5-дифенилтетразолия бромид) был получен от компании Sigma-Aldrich (MO, USA; номер по каталогу M 2128), растворен в PBS до концентрации 5 мг/мл, стерилизован фильтрованием и хранился при температуре 4°С.
Плазмида pEGFP-N1 была приобретена на фирме Clontech Laboratories Inc. (CA, USA; номер по каталогу 6085-1), производства ELIM Biopharmaceuticals, Inc. (CA, USA) (лот № 1002) и доставлена в концентрации 2 мг/мл в стерильной воде. Этот стоковый раствор разделяли на аликвоты и хранили при -20°С. Поли-L-лизин HBr (ММ 15000-30000) был получен от фирмы Sigma-Aldrich (MO, USA, номер по каталогу P 7890). Поли-L-лизин HBr растворяли и разбавляли в дистиллированной воде, стерилизовали фильтрованием и хранили при температуре -20°C.
Исследования in vitro
Культивирование клеток
HCT116/LUC культивировали в среде DMEM (Lonza, Veviers, Belgium) с добавкой 10% фетальной сыворотки теленка (PAA Laboratories GmbH, Pasching, Austria), 100 Ед/мл пенициллина и 100 мкг/мл стрептомицина (Sigma-Aldrich, MO, USA) при температуре 37°C и 5% CO2 во влажной среде.
Обработка клеток
Клетки HCT116/LUC (1,5×105 клеток на лунку для измерений трансфекции, 3,75×105 клеток на лунку для МТТ) высевали на 6-луночный (трансфекция) и 24-луночный (МТТ) планшеты (Nunc, Roskilde, Denmark) и инкубировали в течение 24 ч (5% CO2, 37°C). Затем к клеткам добавляли фотосенсибилизатор ТPCS2а или конъюгаты хитозана (16А, 16B, 17А, 19А, 37, 38, 32 или 33), и клетки инкубировали в течение 18 часов (5% CO2, 37°C). Клетки затем промывали три раза клеточной культуральной средой и инкубировали в течение 4 часов (5% CO2, 37°C) в среде, содержащей плазмидный комплекс. Затем клетки промывали один раз. После добавления свежей среды клетки освещали различными дозами света. Спустя 48 часов инкубации с помощью проточной цитометрии анализировали экспрессию EGFP (Enhanced Green Fluorescent Protein - улучшенный зеленый флуоресцентный белок). Уровень выживания клеток измеряли по методу МТТ в параллельных опытах. Клетки подвергались световому облучению прибором LumiSource® (PCI Biotech, Oslo, Norway). LumiSource® подавал свет с помощью панели из четырех световых трубок (4×18 W Osram L 18/67, синий), испуская в основном синий свет с пиковой длиной волны в области 420-435 нм.
Получение комплексов плазмида/поли-L-лизин
Комплексы плазмида/поли-L-лизин с соотношением зарядов 2,2 получали при осторожном смешивании плазмидной ДНК и раствора поли-L-лизина. 2,5 мкл ДНК (стоковый раствор 2 мкг/мл) разводили в 47,5 мкл воды, и 6,92 мкл поли-L-лизина (1 мкг/мкл) разводили в 43,08 мкл воды. После смешивания раствор инкубировали при комнатной температуре в течение 30 мин, затем разбавляли культуральной средой до конечного объема 1 мл и добавляли к клеткам (1 мл на лунку).
Измерение трансфекции
Клетки трипсинизировали 100 мкл трипсина (трипсин-ЭДТА, Sigma-Aldrich, MO, USA), ресуспендировали в 500 мкл клеточной культуральной среды и фильтровали в 5-ти миллиметровую полистирольную круглодонную пробирку с колпачком-ситечком для клеток (BD Falcon) (нейлоновая сетка фильтра в 50 мкм), затем анализировали с помощью проточного цитометра BD LSR (Becton Dickinson, CA, USA). EGFP измеряли при использовании фильтра на 425-475 нм после возбуждения при 488 нм, и пропидий йодид (Calbiochem Corporation, CA, USA) определяли с использованием фильтра на 600-620 нм после возбуждения при 561 нм. Пропидий йодид (1 мкг/мл) был использован для отделения отмерших клеток от жизнеспособных клеток, и осуществляли преобразование импульсов для отделения клеточных дублетов от единичных клеток. 10000 событий были собраны для каждого образца, и данные были проанализированы с использованием программного обеспечения BD FACSDiva (Becton Dickinson, CA, USA).
Определение жизнеспособности клеток
Жизнеспособность клеток определяли с помощью метода, основанного на восстановлении растворимой в воде соли тетразолия (МТТ) до пурпурного нерастворимого продукта формазана с помощью митохондриальных дегидрогеназ, присутствующих в живых метаболически активных клетках. 0,5 мл среды, содержащей 0,125 мг МТТ, добавляли к клеткам, после чего 2 часа инкубировали при температуре 37°C, 5% CO2. Полученные кристаллы формазана растворяли путем добавления 500 мкл ДМСО (Sigma-Aldrich, MO, USA) на лунку. Планшеты считывали с использованием микропланшетного спектрофотометра PowerWave XS2 (BioTek Instruments, VT, США). Жизнеспособность клеток рассчитывали в процентах от контролей (параллели без световой обработки).
Исследования in vivo
Животные. Hsd:бестимусных самок мышей с мутацией по гену nude-Foxn1nu разводили в отделении животных при Norwegian Radium Hospital. Мышей содержали в специальных условиях отсутствия патогенов. Воду и корм они получали вволю. Все процедуры, связанные с мышами, были проведены в соответствии с протоколами, утвержденными комитетом по уходу за животными при Norwegian Radium Hospital в рамках руководящих принципов национального комитета по этике (National Ethical Committee), касающихся благополучия животных.
Масса мышей была 22-25 г (5-8 недель), когда они были включены в эксперимент. Перед трансплантацией клетки HCT116/LUC культивировали при температуре 37°C и 5% CO2 во влажной среде. 1,5×106 клеток вводили подкожно в правое бедро каждой мыши. Размер опухоли измеряли два или три раза в неделю путем определения двух взаимно перпендикулярных диаметров. Объем опухоли рассчитывали с помощью следующей формулы:
V=(W2×L)/2
где W обозначает ширину и L - длину диаметра измеряемой опухоли.
Обработка
Хитозаны разбавляли до 1,25 мг/мл TPC в PBS (соединение 37) и 3% Tween 80 (соединения 38 и 33). Когда опухоли достигли объема 60-100 мм3, в хвостовую вену внутривенно вводили 88-100 мкл (последняя доза 5 м/кг). ТPСS2а разбавляли до 1,25 мг/мл в 3% Твин 80 и, когда опухоли достигли объема 60-100 мм3, в хвостовую вену вводили внутривенно 88-100 мкл (последняя доза 5 м/кг). Через 96 часов после введения фотосенсибилизатор подвергали воздействию света при 652 нм диодным лазером (Ceramoptec GmbH, Bonn, Germany) с интенсивностью 90 МВт/см2 и со световой дозой 15 Дж/см2. Животным, получающим ФХИ+лечение блеомицином, внутрибрюшинно вводили 1500 МЕ блеомицина (европейские единицы) в 100 мкл. Опухоли подвергали воздействию света 30 мин после BLM инъекций при 652 нм диодным лазером (Ceramoptec GmbH, Bonn, Germany) с интенсивностью 90 МВт/см2. Животные были укрыты алюминиевой фольгой, за исключением области над опухолью, где отверстие в фольге был сделано диаметром на 2 мм больше, чем опухоль.
Система визуализации in vivo
Биолюминесценция была измерена с помощью IVIS Lumina 100 серии фирмы Caliper Life Sciences, MA, США. Животных анестезировали (золетил), и внутрибрюшинно вводили 200 мкл D-люциферина (Caliper Life Sciences) (20 мг/мл в PBS). Изображения были сделаны через 10 мин после инъекций D-люциферина. Биолюминесценцию измеряли примерно раз в неделю, начиная от 11 дня после инъекции ФС. Животных забивали, когда опухоль достигла объема >1000 мм3, или когда животное проявляло признаки боли или аномальное поведение.
Данные были проанализированы с помощью программного обеспечения Living Image 4,2 (Caliper Life Sciences).
Биологическое действие конъюгатов TPP-хитозана 16A и 16B была исследовано в экспериментах, где конъюгаты были использованы в качестве фотосенсибилизирующих агентов в фотохимической интернализации для улучшения доставки генов. Экспериментальные подробности описаны в разделе Материалы и способы. Как видно на фигуре 19 ((a) и (b)) конъюгаты 16A и 16B являлись превосходными фотосенсибилизаторами для ФХИ в том, что существенное повышение трансфекции можно было наблюдать уже при низких световых дозах. Эффект был повышенным относительно того, который получался с фотосенсибилизатором ТPСS2а, который был специально разработан для использования в ФХИ, и который находится на стадии клинической разработки для лечения раковых заболеваний (Berg et al. 2011, выше). Таким образом, с ТPСS2а аналогичные уровни трансфекции не были достигнуты даже при применении более высоких световых доз (фигура 19 (e)).
Аналогичные эксперименты были проведены с TPP-хитозанами 17A и 19A (фигура 19(c) и (d)). Можно видеть, что эти конъюгаты являлись даже более мощными, чем 16A и 16B. Таким образом, по сравнению с ТPСS2а они были, по меньшей мере, в 10 раз более активным, поскольку даже при использовании в 10 раз меньших концентраций этих конъюгатов усиление трансфекции было значительно большим, чем то, что наблюдалось с ТPСS2а. Это было весьма неожиданно, поскольку можно было бы предположить, что близость сенсибилизаторных молекул в конъюгатах хитозана должна была бы приводить к фотосенсибилизирующему действию, делая конъюгаты менее эффективными, чем свободные сенсибилизаторные молекулы, которые не были бы объектом таких гасящих эффектов. Таким образом, кажется, что фотосенсибилизатор-хитозановые конъюгаты взаимодействуют с эндоцитотическими мембранами, неизвестно каким образом, что делает их особенно хорошо подходящими для использования в ФХИ и связанных с ним методов.
Как можно видеть на фигуре 20, аналогичные результаты были получены с TPC-хитозановыми конъюгатами (соединения 37, 38, 32 и 33), которые по сравнению с TPP-конъюгатами имеют то преимущество, что они могут быть активированы также при освещении красным светом, позволяя осуществлять лучшее проникновение в ткани при использовании in vivo. Это является важной особенностью при лечении крупных поражений in vivo (например, больших опухолей), но это и недостаток в тех случаях, когда желателен только неглубокий фотохимический эффект, например, при подходах вакцинации, где желаемый эффект должен быть в верхних слоях кожи.
Как видно на фигуре 21, аналогичные результаты были также получены с соединением 54, показывая, что PEG-содержащие конъюгаты тоже эффективны в стимулировании ФХИ эффекта.
TPC-конъюгаты также были изучены в in vivo экспериментах, исследуя, являются ли конъюгаты активными в терапевтических подходах на основе ФДТ и ФХИ. На фигуре 22 представлены фотографии освещенных мышей с опухолями, получавших конъюгаты 38 и 33 либо отдельно, либо вместе с цитотоксическим противораковым препаратом блеомицином (подробнее см. раздел Материалы и способы). Необработанные животные и животные, которым вводили ТPСS2а+блеомицин, были использованы в качестве контроля. Используемые раковые клетки перманентно трансфицировали для экспрессии люциферазы, так что размер опухолей можно было контролировать с помощью визуализации биолюминесценции после инъекции люциферина. Как показано на фигуре 22, необработанные контрольные и обработанные ТPСS2а+блеомицином животные (8 и 7, соответственно) проявляли сильную флуоресценцию через 11 и 15 дней после введения фотосенсибилизатора (через 7 и 11 дней после воздействия света), что указывало на наличие большого количества живых раковых клеток в опухоли. У этих животных опухоли выросли настолько большими, что животных пришлось умертвить из гуманных соображений после 15 дня. В отличие от них у животных, получавших конъюгаты хитозана, была только слабая флуоресценция только у одного из животных (животное 3) в день 11, что показывало, что чистая фотохимическая обработка (только ФХИ, аналогично обработке ФДТ) и ФХИ+блеомицин комбинированное лечение сильно уменьшала количество раковых клеток в опухоли. Можно видеть, что у животных, леченных только ФХИ (животные 3 и 5), флуоресценция увеличивалась в дни от 15 до 20, показывая, что только ФХИ фотохимическая обработка была не достаточной, чтобы убить все раковые клетки. Напротив, животные, получавшие ФХИ+блеомицин (животные 4 и 6), показали по существу отсутствие флуоресценции даже на 20-й день, что свидетельствовало о том, что эта комбинация была значительно более эффективной, чем только ФХИ, демонстрируя, что конъюгаты хитозана вызывали сильный эффект фотохимической интернализации; и гораздо сильнее, чем тот, который вызывал фотосенсибилизатор ТPСS2а, находящийся на стадии клинической разработки для лечения раковых заболеваний.
На фигуре 23 показаны кривые роста опухоли у обработанных животных (включая соединение 37 конъюгата хитозана), которые подтверждают результаты визуализации, представленные на фигуре 22, и показывают, что у животных, получавших соединение 38+блеомицин или 33+блеомицин, опухоли кажутся полностью ликвидированными. Эти результаты еще раз показывают, что конъюгаты хитозана являются гораздо более эффективными агентами для ФХИ, чем ТPСS2а (который в комбинации с блеомицином не оказывал никакого влияния на рост опухоли в эксперименте). Кроме того, можно видеть, что добавление блеомицина для графика лечения значительно улучшало терапевтический эффект для всех трех исследуемых конъюгатов хитозана (по сравнению с только фотохимической обработкой), что указывает на индуцирование сильного фотохимического эффекта интернализации этими конъюгатами. Эта повышенная эффективность является весьма неожиданной, поскольку основной причиной для проектирования фотосенсибилизаторных полимерных конъюгатов является получение лучшей селективности лечения ввиду так называемого ЭПР-эффекта. Следует, однако, ожидать, что после потенциально повышенной селективности будет следовать более низкая эффективность из-за большой молекулярной массы конъюгатов, приводящей к гораздо более медленной диффузии через ткани, чем в случае небольших молекул фотосенсибилизаторов, и, следовательно, к снижению концентрации сенсибилизатора в опухолевых клетках, и к трудности в доставке сенсибилизатора ко всем клеткам в опухоли. Ясно, что это не случай с конъюгатами хитозанов, описанных в данном изобретении.
Пример 4
Общие материалы и методы были такими, как в примере 1, где это уместно.
См. схему 7 на фигуре 24
Синтез монометилового эфира триэтиленгликоля тозилата (40) (также называемого 2-(2-(2-метоксиэтокси)этокси)этил-4-метилбензолсульфонат).
Монометиловый эфир триэтиленгликоля 39 (4,87 мл, 30,43 ммоль) растворяли в ТГФ (25 мл). Добавляли водный раствор (25 мл) гидроксида калия (3,7 г, 65,95 ммоль), и полученную смесь охлаждали до 0°C. Затем в течение 30 минут добавляли по каплям с помощью капельной воронки п-толуолсульфохлорид (6,86 г, 48,77 ммоль), растворенный в ТГФ (50 мл). Реакционную смесь перемешивали в течение более 2 час при 0°C и затем оставляли перемешиваться при комнатной температуре в течение ночи. Реакционную смесь концентрировали в вакууме для удаления ТГФ, затем разбавляли EtOAc (40 мл) и водой (30 мл) и экстрагировали EtOAc (2×75 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением соединения 40 (7,13 г) в виде маслянистого продукта мутно-серого цвета.
ИК-спектр с Фурье-преобразованием: 2878, 1598, 1453, 1356, 1177, 1097 см-1; 1H ЯМР (400 МГц, CDCl3): δ=7,80 (д, J=8Гц, 2H), 7,34 (д, J=8Гц, 2H), 4,16 (т, 2H, CH2OTs), 3,67-3,70 (м, 2H, -CH2-CH2OTs), 3,58-3,62 (м, 6H, TEG OCH2'S), 3,37 (с, 3H, OCH3), 2,44 (с, 3H, Ar-CH3) м.д.; 13C ЯМР (400 МГц, CDCl3): δ=144,91, 133,19, 129,94, 128,12, 72,05, 70,90, 70,71, 69,36, 68,83, 59,17, 21,78 м.д.
Синтез моноэтилового эфира триэтиленгликоля тозилата (42) (также называемого 2-(2-(2-этоксиэтокси)этокси)этил-4-метилбензолсульфонат)
Моноэтиловый эфир триэтиленгликоля 41 (4,9 мл, 28,1 ммоль) растворяли в ТГФ (30 мл). Добавляли водный раствор (25 мл) гидроксида калия (3,7 г, 65,95 ммоль). Полученную смесь охлаждали до 0°C. Затем в течение 30 минут добавляли по каплям с помощью капельной воронки п-толуолсульфохлорид (6,86 г, 48,77 ммоль), растворенный в ТГФ (50 мл). Реакционную смесь перемешивали в течение более 2 час при 0°C и затем оставляли перемешиваться при комнатной температуре в течение ночи. Реакционную смесь концентрировали в вакууме для удаления ТГФ, затем разбавляли EtOAc (40 мл) и водой (30 мл) и экстрагировали EtOAc (2×75 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением соединения 42 (6,83 г) в виде маслянистого продукта мутно-серого цвета.
ИК-спектр с Фурье-преобразованием: 2870, 2975, 1598, 1453, 1358, 1177, 1110 см-1; 1H ЯМР (400 МГц, CDCl3): δ=7,77 (д, J=8Гц, 2H), 7,31 (д, J=8Гц, 2H), 4,13 (т, 2H, CH2OTs), 3,64-3,67 (м, 2H, -CH2CH2OTs), 3,52-3,59 (м, 8H, TEG OCH2'S), 3,49 (кв, J=8Гц, 2H, -OCH2CH3), 2,42 (с, 3H, Ar-CH3), 1,17 (т, J=8Гц, 3H, -OCH2CH3) м.д.
Синтез метокси полиэтиленгликоль тозилата (44)
Монометиловый эфир полиэтиленгликоля 43 (5 г, 14,29 ммоль, средняя ММ: 350 Da) растворяли в ТГФ (50 мл). Добавляли водный раствор (25 мл) гидроксида калия (1,76 г, 31,44 ммоль). Реакционную смесь затем охлаждали до 0°C. Затем в течение 30 минут добавляли по каплям с помощью капельной воронки п-толуолсульфохлорид (3,27 г, 17,14 ммоль), растворенный в ТГФ (50 мл). Реакционную смесь перемешивали в течение более 2 часов при 0°C и затем оставляли перемешиваться в течение ночи при комнатной температуре. Реакционную смесь концентрировали в вакууме для удаления ТГФ, затем разбавляли EtOAc (40 мл) и водой (30 мл) и экстрагировали EtOAc (2×75 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением соединения 44 (5,91 г) в виде маслянистого продукта мутно-серого цвета.
1H ЯМР (400 МГц, CDCl3): δ=7,74 (д, J=8Гц, 2H), 7,29 (д, J =8Гц, 2H), 4,11 (ушир. т, 2H, CH2OTs), 3,49-3,65 (м, 28H, -CH2-CH2OTs и PEG OCH2'S), 3,32 (с, 3H, OCH3), 2,39 (с, 3H, Ar-CH3) м.д.
Синтез триэтиленгликоль моноэтилового эфира пиперазина (46) (также называемый 1-(2-(2-(2-этоксиэтокси)этокси)этил)пиперазин (46) (TEG-Pip)
Пиперазин (10,4 г, ~120 ммоль) растворяли в ацетонитриле (150 мл) в атмосфере азота. Добавляли по каплям соединение 42 (5 г, 15,04 ммоль), растворенное в ацетонитриле (30 мл). Полученную смесь перемешивали при комнатной температуре в течение 12 часов, затем концентрировали в вакууме для удаления ацетонитрила. Сырой продукт очищали с помощью флэш хроматографии на колонке с силикагелем, используя смесь MeOH:CH2Cl2 (8:92) в качестве элюента, с получением чистого соединения 46 (1,52 г, 41%) в виде бесцветной жидкости.
1H ЯМР (400 МГц, CDCl3): δ=3,54-3,64 (м, 10H, TEG OCH2'S), 3,50 (кв, J=8Гц, 2H, -OCH2CH3), 3,43 (с, 1H, NH), 2,88 (т, 4Н, пиперазиновое кольцо-CH2), 2,55 (т, 2H, OCH2-CH2-пиперазин), 2,46 (ушир. м, 4Н, пиперазиновое кольцо-CH2), 1,18 (т, J=8Гц, 3H, -OCH2CH3) м.д.; 13C ЯМР: 70,76, 70,70, 70,47, 69,92, 68,86, 66,73, 58,47, 54,89, 50,55, 45,99, 15,25 м.д.; MS (ESI) вычисл. для C12H27N2O3 ([M+H]+) 247,2016, найдено 247,2014.
Синтез 2-(2-(2-метоксиэтокси)этокси)ацетальдегида (45)
Оксалилхлорид (5 мл, 58,24 ммоль) растворяли в CH2Cl2 (75 мл) в атмосфере азота. Полученную смесь охлаждали до -78°С с помощью бани со смесью сухой лед/ацетон, затем осторожно по каплям добавляли ДМСО (5 мл), разбавленный CH2Cl2 (15 мл). После завершения добавления реакционную смесь перемешивали в течение 10 минут, затем добавляли по каплям монометиловый эфир триэтиленгликоля 39 (5 мл, 31 ммоль), растворенный в CH2Cl2 (30 мл). После завершения добавления реакционную смесь перемешивали в течение 15 минут. Затем в течение 20 минут добавляли по каплям Et3N (20 мл, 143 ммоль), растворенный в CH2Cl2 (20 мл), и перемешивали еще в течение 30 минут при температуре -78°C, затем оставляли нагреваться до комнатной температуры. Белую "молочного" цвета реакционную смесь затем промывали водой (2×45 мл) и насыщенным солевым раствором (40 мл). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали в вакууме. Полученный сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя смесь MeOH/EtOAc (0:100-10:90) в качестве элюента, получая альдегид 45 (2,40 г). 1H ЯМР анализ показывал, что соединение 47 было загрязнено небольшим количеством неотделяемой примеси.
ИК-спектр с Фурье-преобразованием: 3436, 2879, 1734, 1454, 1353, 1108, 1028 см-1; 1H ЯМР (400 МГц, CDCl3) δ м.д.: 9,73 (с, 1H, CHO), 4,16 (с, 2H), 3,54-3,76 (м, 10H), 3,38 (с, 3H).
(Продукт загрязнен исходным материалом. Продукт составляет приблизительно 50%.)
Синтез 2-(2-(2-этоксиэтокси)этокси)ацетальдегида (47)
Тот же способ, что и для описанного выше соединения; за исключением того, что вместо монометилового эфира триэтиленгликоля (39) исходным продуктом являлся моноэтиловый эфир триэтиленгликоля (41). Соединение 47 (выход: 2,34 г, ~42%), загрязненное небольшим количеством неотделяемой примеси.
1H ЯМР (400 МГц, CDCl3) δ м.д.: 9,67 (с, 1H, CHO), 4,10 (с, 2H), 3,52-3,69 (м, 10H), 3,45 (кв, 2H), 1,15 (т, 3H). (Продукт загрязнен исходным материалом. Продукт составляет приблизительно 42%).
Тегилирование хитозана путем прямых N-модификаций 3,6-ди-O-TBDMS-хитозана
См. схему 8 на фигуре 25.
Синтез N-(2-(2-(2-этоксиэтокси)этокси)этиламино хитозана TEG0,41-CS (49a)
DiTBDMS-хитозан 8 (300 мг, 0,77 ммоль) растворяли в NMP (5 мл) и нагревали до 50°C в реакционном сосуде. Добавляли Cs2CO3 (1 г, 3,07 ммоль) и каталитическое количество йодида калия. Затем реакционный флакон герметически закрывали, и реакционную смесь перемешивали в течение 2 час, после этого с помощью шприца добавляли соединение 42 (767 мг, 2,31 ммоль). Реакционную смесь перемешивали в течение 48 часов, затем охлаждали и выливали в ледяную воду, и полученный осадок отфильтровывали и сушили в вакуумном шкафу. Сырой продукт 48а (270 мг) получали в виде желтого порошка и использовали как таковой на следующей стадии удаления защитных групп, как описано далее.
Для удаления защитных групп гидроксила (удаление силильных групп) сырое соединение 48а суспендировали в MeOH (15 мл). При комнатной температуре медленно добавляли концентрированную HCl (2 мл), и полученную смесь перемешивали в течение 12 часов, затем концентрировали в вакууме. Полученный сырой продукт вновь суспендировали в MeOH (15 мл) и добавляли конц. HCl (2 мл) и перемешивали в течение 12 часов, затем разбавляли и подвергали ионному обмену в водном растворе NaCl (5%, 35 мл), после чего диализовали против деионизированной воды в течение 3 дней. После завершения диализа растворимый в воде продукт лиофилизовали с получением 49а (125 мг) в виде липкого вещества белого цвета.
ИК-спектр с Фурье-преобразованием: 3418, 2874, 1712, 1631, 1536, 1378, 1077 см-1; 1H ЯМР (400 МГц, D2O) δ=4,68 и 4,27 (с, 1H, хитозан H-1 и H-1'), 3,14-3,97 (ушир. м, 10H, хитозан от H-2 до H-6, TEG (~41% СЗ) OCH2'S и O-CH2CH3), 2,96-3,14 (ушир. м, 1H, H-2 и H-2'), 1,21 (т, 1,24 Н (~41% СЗ) TEG O-CH2CH3) м.д.
Синтез N-(2-(2-(2-этоксиэтокси)этокси)этиламино хитозана TEG0,27-CS (51b) (27% СЗ)
Соединения 49b было получено с использованием того же способа, как описан выше, за исключением того, что вместо 3 эквивалентов использовали 1,5 эквивалента реагента триэтиленгликоля тозилата (42). Соединение 49b (27 мг) было получено в виде липкого продукта белого цвета.
ИК-спектр с Фурье-преобразованием: 3417, 2876, 1602, 1382, 1259, 1078, 599 см-1; 1H ЯМР (400 МГц, D2O:DCl, 95:5) δ=5,13 и 4,95 (с, 1H, хитозан H-1 и H-1'), 3,24-3,99 (ушир. м, 12H, хитозан от H-2 до H-6, H-2', TEG (27% СЗ) OCH2 и O-CH2CH3), 1,21 (т, 0,89H, TEG O-CH2CH3) м.д.
Синтез N-ацетилбром-3,6-O-diTBDMS-хитозана (9)
DiTBDMS-хитозан 8 (3 г, 7,70 ммоль) (предварительно синтезированный Ingólfur Magnússon) растворяли в CH2Cl2 (30 мл) в атмосфере азота. Полученную смесь охлаждали до -20°C, затем с помощью шприца добавляли триэтиламин (5,30 мл, 38 ммоль). После 10 минут перемешивания с помощью шприца добавляли по каплям бромацетилбромид (2,65 мл, 30,51 ммоль), растворенной в CH2Cl2 (2,5 мл). Реакционную смесь перемешивали в течение 1 часа при -20°C, после чего разбавляли CH2Cl2 (70 мл) и сразу упаривали в вакууме. Затем густой продукт коричневого цвета растирали и промывали ацетонитрилом (3×35 мл), сушили, после чего повторно растворяли в CH2Cl2 (40 мл) и помещали в делительную воронку, где его промывали водой (2×25 мл) и насыщенным солевым раствором (35 мл). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением промежуточного бромацила 9 (2,57 г) в виде твердого вещества коричневого цвета.
ИК-спектр с Фурье-преобразованием (KBr): v 3404 (шир., NH), 2956-2858 (с, C-H TBDMS), 1682 (vs, C=O амид I), 1530 (vs, C=O амид II), 1473, 1391, 1362, 1311, 1259, 1101, 1005, 837, 777 (Si-C), 669 см-1; 1H ЯМР (CDCl3) δ м.д.: 4,40 (ушир. с, 1H, H-1), 3,26-4,02 (м, 8H, H-2 GlcN, H-3, H-4, H-5, H-6, H-6' и GluNH-C=OCH2Br), 0,90 и 0,88 (ушир. с, 18H, (CH3)3C-Si), 0,13 и 0,07 (ушир. с, 12H, CH3-Si) м.д.
Пегилирование хитозана путем нуклеофильного замещения на N-ацетил-бром-3,6-diTBDMS-хитозан
N-(ацетил-пиперазин-TEG)-хитозан (51)
(i) Ацетил бромхитозан 9 (200 мг, 0,397 ммоль) растворяли в CH2Cl2 (25 мл) в атмосфере азота. При комнатной температуре прибавляли по каплям соединение 46 (204 мг, 0,828 ммоль), растворенное в CH2Cl2 (10 мл). Полученную смесь перемешивали в течение 10 минут, затем добавляли триэтиламин (115 мкл, 0,828 ммоль). Перемешивание продолжали при комнатной температуре в течение 24 часов, и полное расходование исходного продукта 46 подтверждали контролем ТСХ в MeOH:CH2Cl2 (1:9). Затем реакционную смесь помещали в делительную воронку, и органическую фазу промывали водой (2×35 мл) и насыщенным солевым раствором (35 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением соединения 50 (214 мг) в виде неочищенной жидкости, которую использовали как таковую на следующей стадии удаления защитных групп.
(ii) Для удаления защитных групп гидроксильной группы (удаление силильных групп) сырое соединение 50 суспендировали в MeOH (15 мл). При комнатной температуре медленно добавляли концентрированную HCl (2 мл), и полученную смесь перемешивали в течение 12 часов, после чего концентрировали в вакууме. Полученный сырой продукт вновь суспендировали в MeOH (15 мл) и добавляли конц. HCl (2 мл) и перемешивали в течение 12 часов, затем разбавляли и подвергали ионному обмену водным раствором NaCl (5%, 35 мл), и затем диализовали против деионизированной воды в течение 3 дней. После завершения диализа растворимый в воде продукт сушили в вакууме с получением 51 (112 мг) в виде желтоватого прозрачного вещества.
1H ЯМР (400 МГц, D2O) δ=4,63 (с, 1H, хитозан H-1), 2,74-3,76 (м, 30H, хитозан от H-2 до H-6, GlcNHCOCH2-Pip-TEG, TEG OCH2'S и O-CH2CH3, пиперазиновое кольцо-CH2'S), 1,21 (т, 3 H, TEG O-CH2CH3) м.д.; СЗ=100%.
Синтез N-ацетил-пиперазин тетрафенилпорфирина (TPP-NH-Pip) (5)
Осуществляют как описано выше в примере 1 и показано на схеме 1.
Синтез TPP-NH-CO-CH2-TBDMS-хитозана (55)
См. схему 9 на фигуре 26.
DiTBDMS-хитозан (100 мг, ммоль) растворяли в NMP (10 мл) при температуре 55°C. Добавляли карбонат цезия (500 мг, 1,54 ммоль), и реакционную смесь перемешивали в течение 15 минут, затем добавляли соединение 4 (72,2 мг, 0,096 ммоль). Добавляли каталитическое количество йодида калия, и перемешивание продолжали в течение 24 часов, затем добавляли каталитическое количество DMAP и перемешивали в течение 24 ч, после чего охлаждали и выливали в воду. Осадок отфильтровывали, сушили в вакуумной печи с получением соединения 55 в виде сырого твердого продукта. Однако наблюдаемый красный цвет водного раствора указывал, что небольшое количество соединения оставалось в воде. Это могло быть из-за NMP; поэтому на данной стадии может быть использован диализ.
1H ЯМР (400 МГц, D2O) δ м.д.: 8,84-8,86 (ушир. м, β-пиррольные H), 8,68 (с, TPPNHCO), 8,21-8,22 (м, тетрафенил-Ho), 7,99 (д, J=8,0 Гц, RNHTPP-фенил-Hм), 7,72-7,79 (м, трифенил-Hм,п), 2,72-4,80 (м, хитозан H2-H6, TPPNHCOCH2), 0,89-0,90 (ушир. c, (CH3)3C-Si), 0,05-0,07 (ушир. с, 12H, CH3-Si). -2,78 (с, α-пиррольные NH); ((СЗ TPP-NHCOCH2=~5%)
Синтез TPPp0,1-BrA0,9-DiTBDMS-CS (52)
Соединение 9 (800 мг, 1,587 ммоль) растворяли в CH2Cl2 (40 мл) в атмосфере азота при комнатной температуре. Добавляли TPP-NH-Pip 5 (120 мг, 0,158 ммоль), затем добавляли триэтиламин (30 мкл, 0,216 ммоль). Реакционную смесь перемешивали 24 часа при комнатной температуре. Полное расходование исходного продукта 5 подтверждали методом ТСХ (MeOH:CH2Cl2, 1:9). Реакционную смесь разбавляли CH2Cl2 (75 мл) и промывали водой (2×35 мл) и насыщенным солевым раствором (25 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением 52 (выход: 716 мг) в виде твердого вещества пурпурного цвета.
1H ЯМР (CDCl3) δ м.д.: 9,31 (с, TPPNHCO), 8,84-8,86 (м, β-пиррольные H), 8,20-8,23 (м, тетрафенил-Ho), 7,97 (д, J=8,0Гц, RNHTPP-фенил-Hм), 7,74-7,79 (м, трифенил-Hм,п), 4,43 (ушир. с, H-1), 3,50-4,14 (м, хитозан H-2 GlcN, H-3, H-4, H-5, H-6, H-6' и H-2 GluNHCO, TPPNHCOCH2-Pip, -CH2CONGlc и пиперазиновое кольцо-CH2'S), 2,81-2,86 (м, пиперазиновое кольцо-CH2'S), 0,91, 0,87 (ушир. с, (CH3)3C-Si), 0,07, 0,14 (ушир. с, CH3-Si), -2,77 (ушир. с, α-пиррольные NH) м.д.; (СЗ TPP-NH-Pip=~10%).
Тегилирование и удаление защитных групп TPP-конъюгированного DiTBDMS-CS
Синтез Тегилированного N-(TPP-NH-CO-CH2)-хитозана (57)
(i) Сырое соединение 55 (350 мг, 0,833 ммоль) растворяли в NMP (10 мл) при 50°C в реакционном сосуде. Добавляли Cs2CO3 (1,09 г, 3,33 ммоль), и реакционную смесь перемешивали в течение 30 минут, затем добавляли соединение 42 (1,108 г, 4,16 ммоль) и каталитическое количество йодида калия. Перемешивание продолжали в течение 12 часов, затем реакционную смесь охлаждали, разбавляли водой (30 мл) и диализовали против деионизированной воды в течение 3 дней, после чего лиофилизовали с получением соединения 56, которое было использовано как таковое на следующей стадии удаления защитных групп.
(ii) Сырое соединение 56 суспендировали в MeOH (15 мл). При комнатной температуре медленно добавляли концентрированную HCl (2 мл), и полученную смесь перемешивали в течение 12 часов, затем упаривали в вакууме. Полученный сырой продукт вновь суспендировали в MeOH (15 мл) и добавляли конц. HCl (2 мл) и перемешивали в течение 12 часов, затем разбавляли и подвергали ионному обмену с водным раствором NaCl (5%, 35 мл) и затем диализовали против деионизированной воды в течение 3 дней. После завершения диализа частично растворимый в воде продукт сушили в вакууме с получением 57 (115 мг) в виде твердого вещества коричневого цвета.
Синтез Тегилированного TPP-NH-Pip-хитозана TPPp0,1-CS-TEG0,9 (54)
(i) Соединение 52 (400 мг, 0,799 ммоль) растворяли в CH2Cl2 (35 мл) в атмосфере азота. Добавляли соединение 46 (394 мг, 1,56 ммоль), затем добавляли Et3N (222 мкл, 1,56 ммоль), и реакционную смесь перемешивали 24 часа при комнатной температуре. Затем под контролем ТСХ добавляли Et3N (222 мкл, 1,56 ммоль) для полного расходования исходного материала 46, и перемешивание продолжали более 12 часов. Затем реакционную смесь концентрировали в вакууме с получением 53 (417 мг) в виде сырого продукта, который использовали как таковой на следующей стадии удаления защитных групп.
(ii) Сырое соединение 53 суспендировали в MeOH (15 мл). При комнатной температуре медленно добавляли концентрированную HCl (2 мл), и полученную смесь перемешивали в течение 12 часов, затем упаривали в вакууме. Полученный сырой продукт вновь суспендировали в MeOH (15 мл) и добавляли конц. HCl (2 мл) и перемешивали в течение 12 часов, затем разбавляли и подвергали ионному обмену с водным раствором NaCl (5%, 35 мл) и затем диализовали против деионизированной воды в течение 3 дней. После завершения диализа растворимый в воде продукт лиофилизовали с получением конечного соединения 54 (252 мг) в виде твердого вещества пурпурно-красно-коричневого цвета.
ИК-спектр с Фурье-преобразованием: 3431, 2869, 1665, 1529, 1442, 1308, 1109, 1070, 1029, 800, 701, 559 см-1; 1H ЯМР (ДМСО-d6:D2O 96:4) δ м.д.: 8,83 (ушир. м, β-пиррольные H), 8,15-8,22 (м, тетрафенил-Ho), 8,11 (д, J=8,0 Гц, RNHTPP-фенил-Hм), 7,80-7,88 (м, трифенил-Hм,п), 4,55 (ушир. с, H-1), 2,54-3,65 (ушир. м, частично перекрывающийся с пиком HDO, хитозан H-2 GlcN, H-3, H-4, H-5, H-6, H-6' и H-2 GluNHCO, TPPNHCOCH2-pip, CH2CONGlc, пиперазиновое кольцо-CH2'S. TEG OCH2'S и TEG OCH2CH3), 1,09 (т, TEG OCH2CH3) м.д.; (СЗ TPP-NH-Pip=~10% и СЗ TEG=~90%)
Структурные данные были подтверждены методами ЯМР, ИК-спектроскопии с Фурье-преобразованием и масс-анализа (данные не показаны). Характерный ЯМР спектр соединения 54 показан на фигуре 27.
Пример 5
Анализ in vitro активности T клеток
Для исследования влияния хитозан-конъюгатов 32 и 38 на MHC класса l-ограниченный по антиген-презентации и активации CD8+ T клеток первичные мышиные макрофаги инкубировали с конъюгатами 32 и 38 и антигеном овальбумина пептида OVA 257-264 в антиген-специфичных T клетках с овальбумин-специфичным (OVA 257-264) CD8+ T клеточным клоном. Продукцию ИЛ-2 из активированных CD8+ T клеток анализировали с помощью ELISA.
Макрофаги, полученные из костного мозга (BMDM), были использованы в качестве антиген-презентирующих клеток (АПК) в антиген-специфичных Т лимфоцитах с овальбумин-специфическими Т лимфоцитными гибридомами. BMDM были получены путем культивирования клеток костного мозга мышей в течение, по меньшей мере, 5 дней в среде, дополненной 20% супернатанта клеточной линии L-292.
30000 АСУ на лунку инкубировали в течение ночи в 96-луночных планшетах с хитозан-конъюгатами 32 и 38 (или без них) при концентрации конъюгатов, дающих концентрацию TPC 0,05 мкг/мл. На следующий день АСУ инкубировали с 2 мкг/мл антигенного пептида (OVA 257-264, фирмы Anaspec) в течение 4 час (все стимуляции трехкратно).
Клетки промывали и подвергали воздействию различных доз синего света (0; 30, 60, 90, 180 сек), затем добавляли 100000 овальбумин-специфических T клеток на лунку и совместно культивированных с АСУ в течение ночи. Был использован клон CD8+ T клеток RF33,70 (MHC l-ограниченный по распознаванию OVA 257-264).
После совместного культивирования в течение ночи CD8+ T клеток и АСУ собирали супернатанты из культур клеток. Супернатанты анализированы на продукцию интерлейкина (ИЛ)-2 из активированных T лимфоцитов с помощью стандартных мышиных ИЛ-2 ELISA (IL-2 Duoset ELISA, системы RnD, анализ дубликатов из каждой лунки T клеточной культуры, 25 мкл неразбавленного супернатанта анализировали в каждой лунке ELISA).
Результаты (фигура 28) показали, что с обоими конъюгатами продукция ИЛ-2 CD8+ антиген-специфическими T-клетками значительно увеличивалась в освещенных клетках (например, удвоение для соединения конъюгата 32, фигура 28А). В освещенных контрольных клетках, инкубированных с конъюгатами без антигена, такой рост не наблюдался (данные не показаны). Это показывает, что наблюдаемый эффект был антиген-зависимым, и не благодаря некоторым неспецифическим эффектам фотохимической обработки, демонстрируя улучшенную презентацию антигенных пептидов на поверхность АСУ в качестве причины наблюдаемого увеличения продукции ИЛ-2.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2010 |
|
RU2581367C2 |
| СОЕДИНЕНИЯ | 2008 |
|
RU2461559C2 |
| НОВЫЕ АМФИФИЛЬНЫЕ ПРОИЗВОДНЫЕ AЛЬФА-C-ФЕНИЛ-N-ТРЕТ-БУТИЛНИТРОНА | 2003 |
|
RU2364602C2 |
| УГЛЕВОДНО-ГЛИКОЛИПИДНЫЕ КОНЪЮГИРОВАННЫЕ ВАКЦИНЫ | 2013 |
|
RU2649009C2 |
| 6-, 7- ИЛИ 8-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ХИНАЗОЛИНОНА И КОМПОЗИЦИИ, ВКЛЮЧАЮЩИЕ ИХ, И СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ | 2008 |
|
RU2476432C2 |
| НУКЛЕИНОВЫЕ КИСЛОТЫ, МОДИФИЦИРОВАННЫЕ АМИНОКИСЛОТАМИ | 1995 |
|
RU2154638C2 |
| СОЕДИНЕНИЯ ИМИДАЗОЛОТИАЗОЛА ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ | 2007 |
|
RU2441011C2 |
| НЕСТЕРОИДНЫЕ МОДУЛЯТОРЫ РЕЦЕПТОРА ПРОГЕСТЕРОНА | 2003 |
|
RU2309155C2 |
| СОЕДИНЕНИЯ НА ОСНОВЕ ПЛАТИНЫ И ЛИПИДОВ И НАНОЧАСТИЦЫ | 2014 |
|
RU2737735C2 |
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ | 2007 |
|
RU2525392C2 |
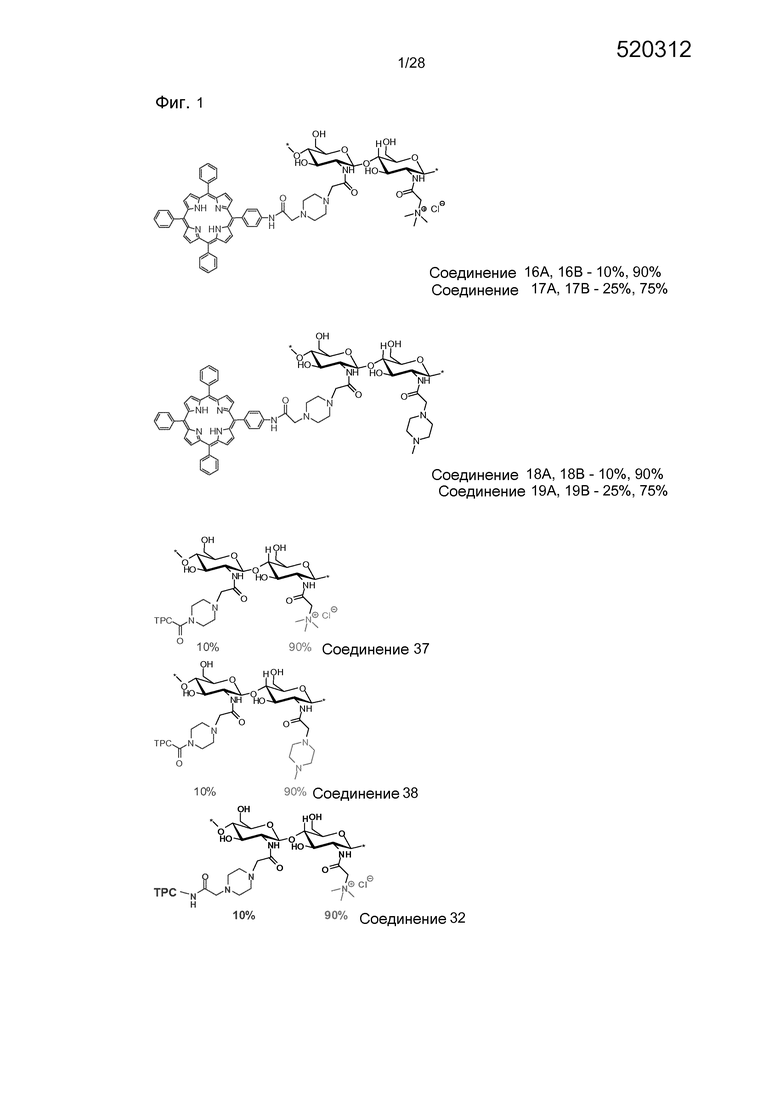
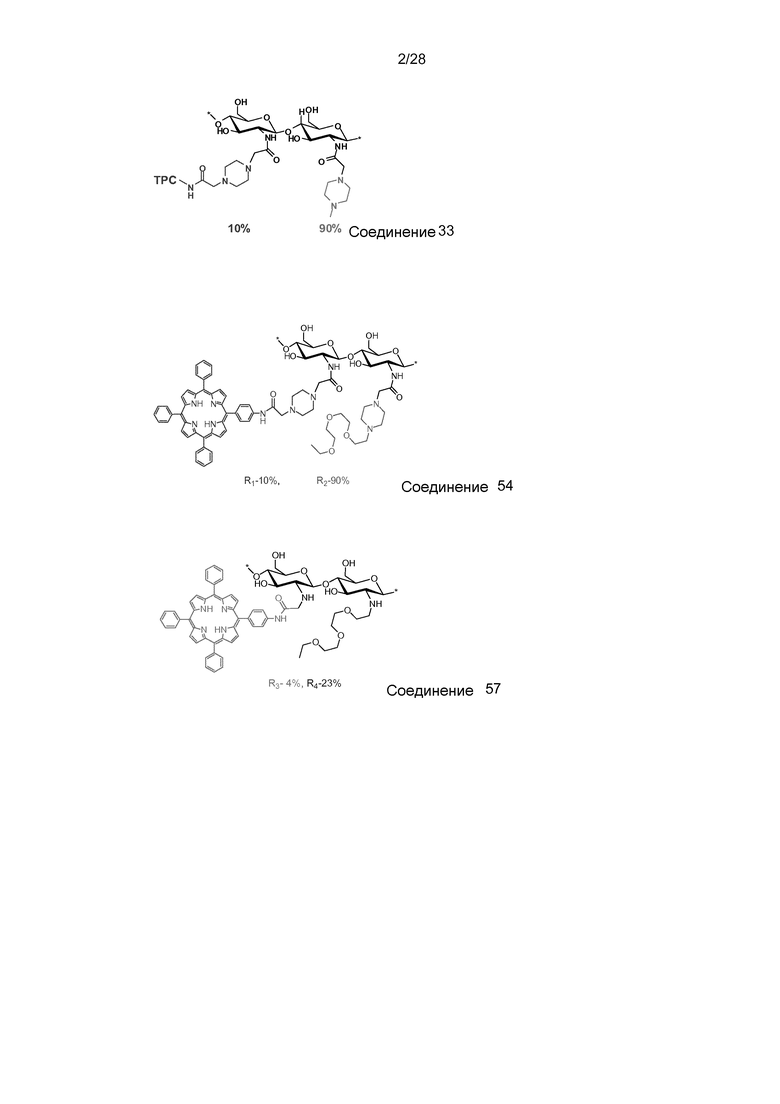
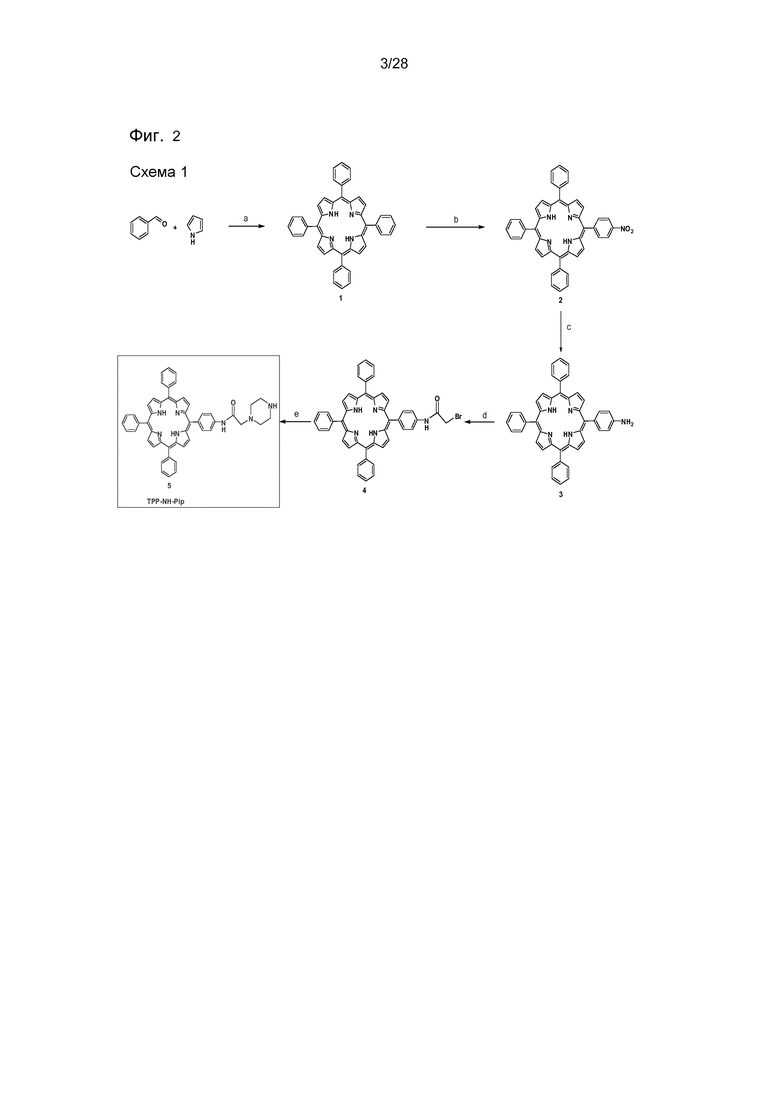
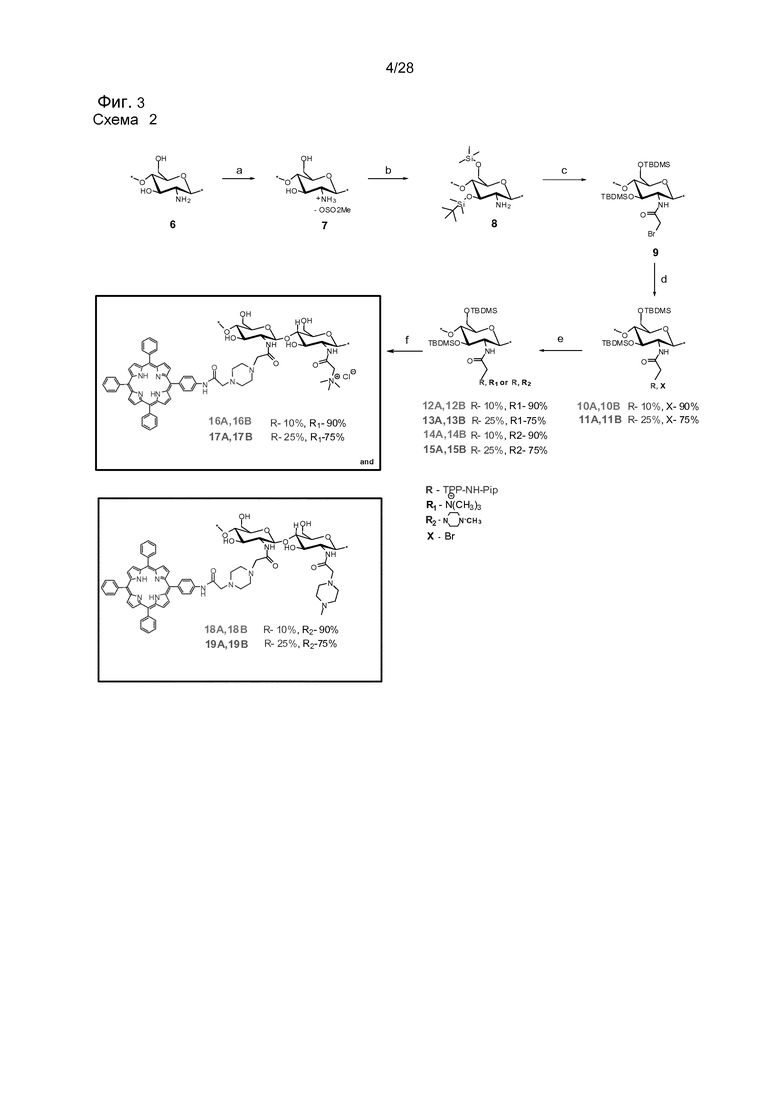
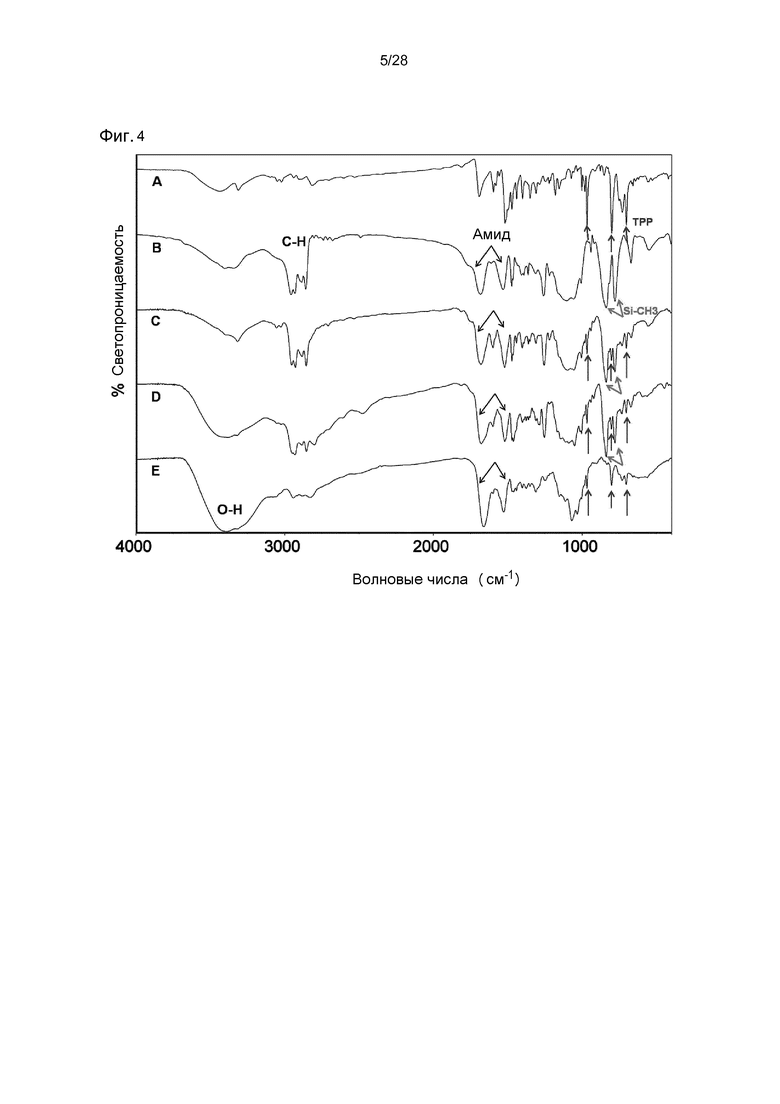
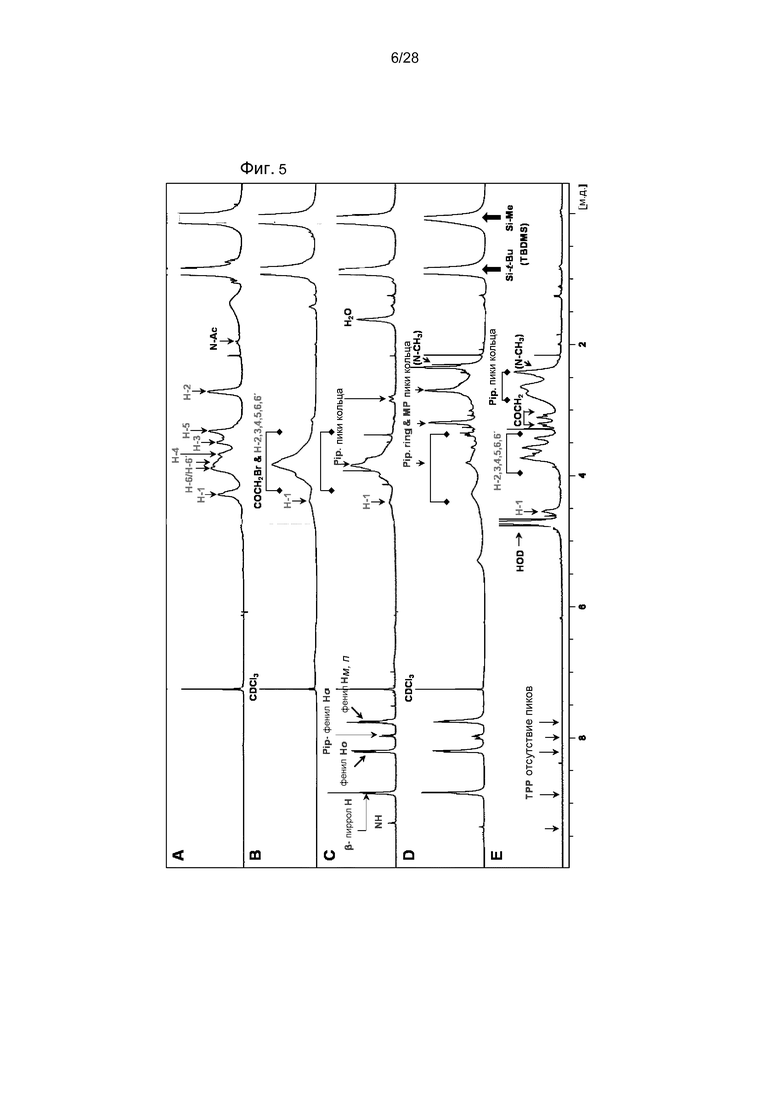

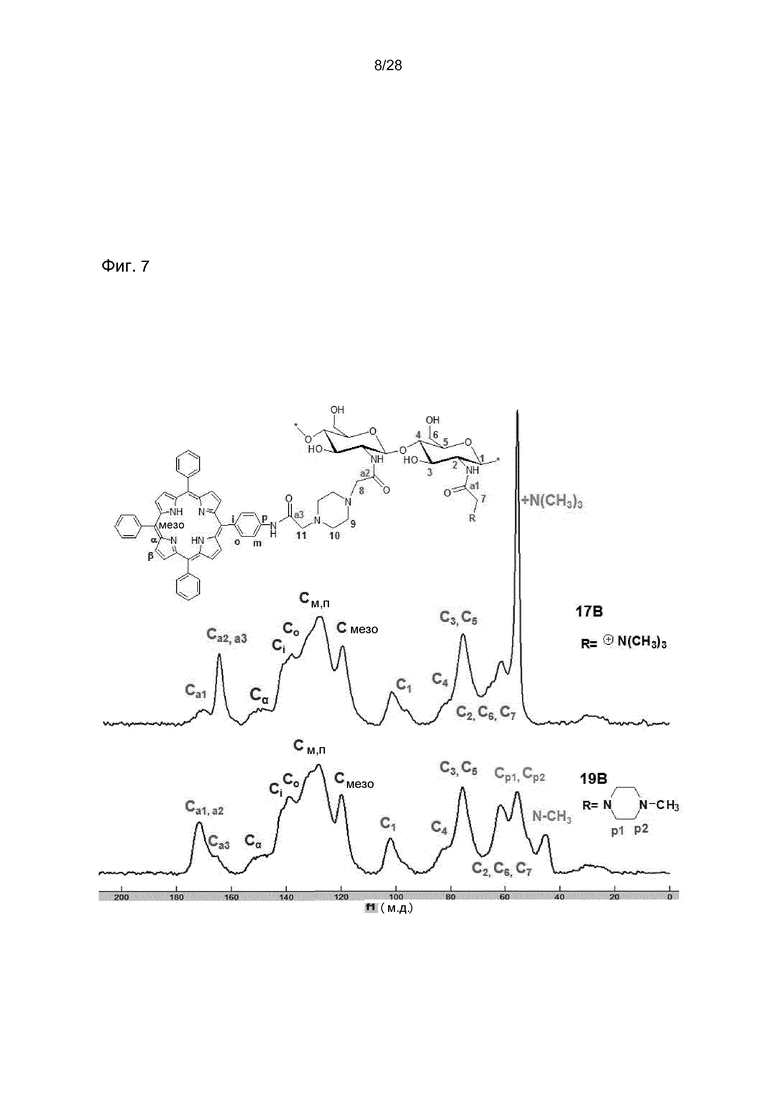
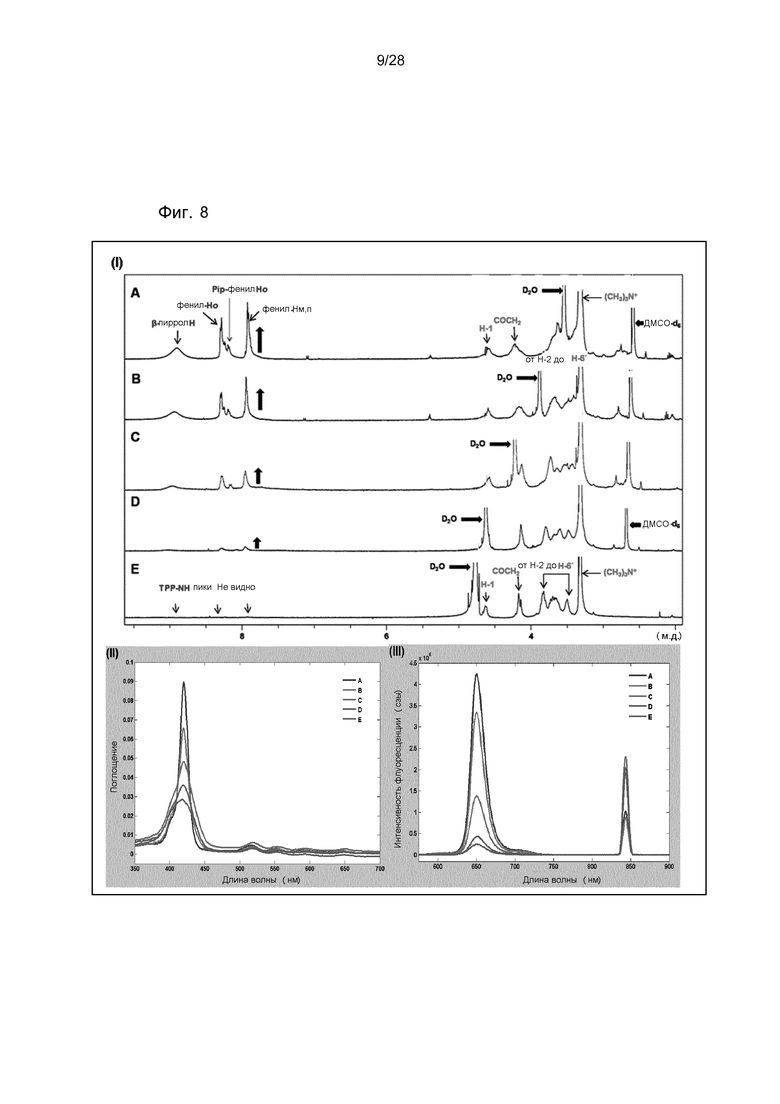
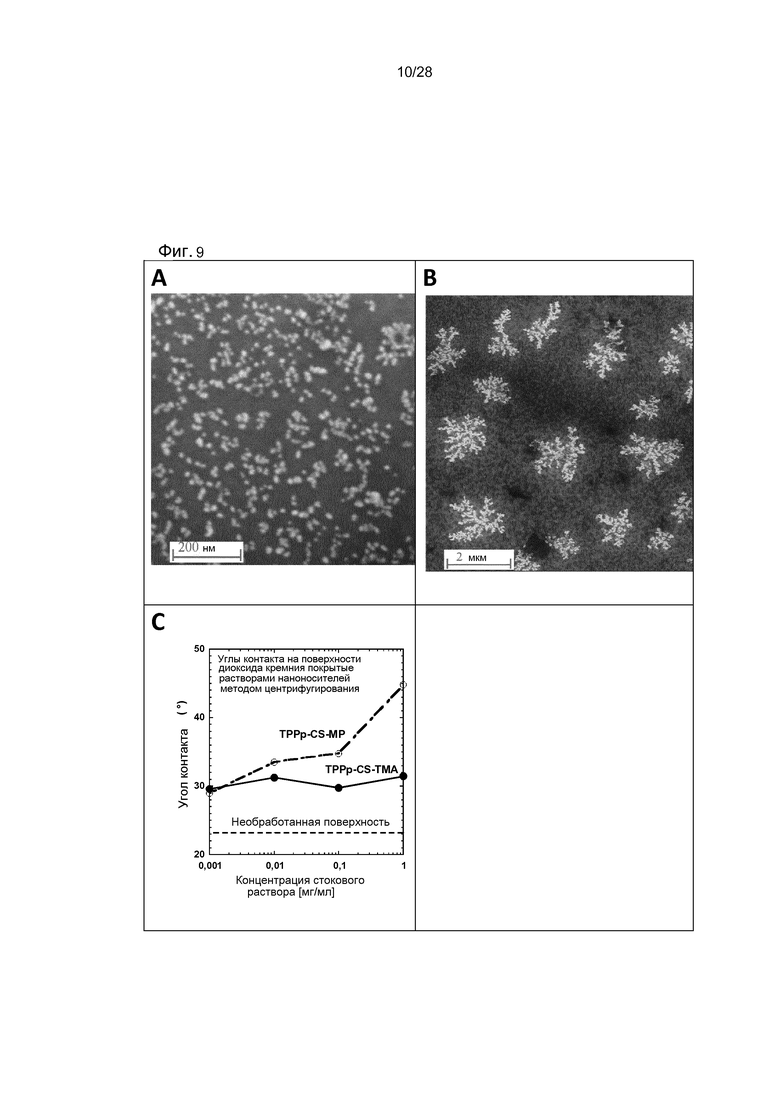
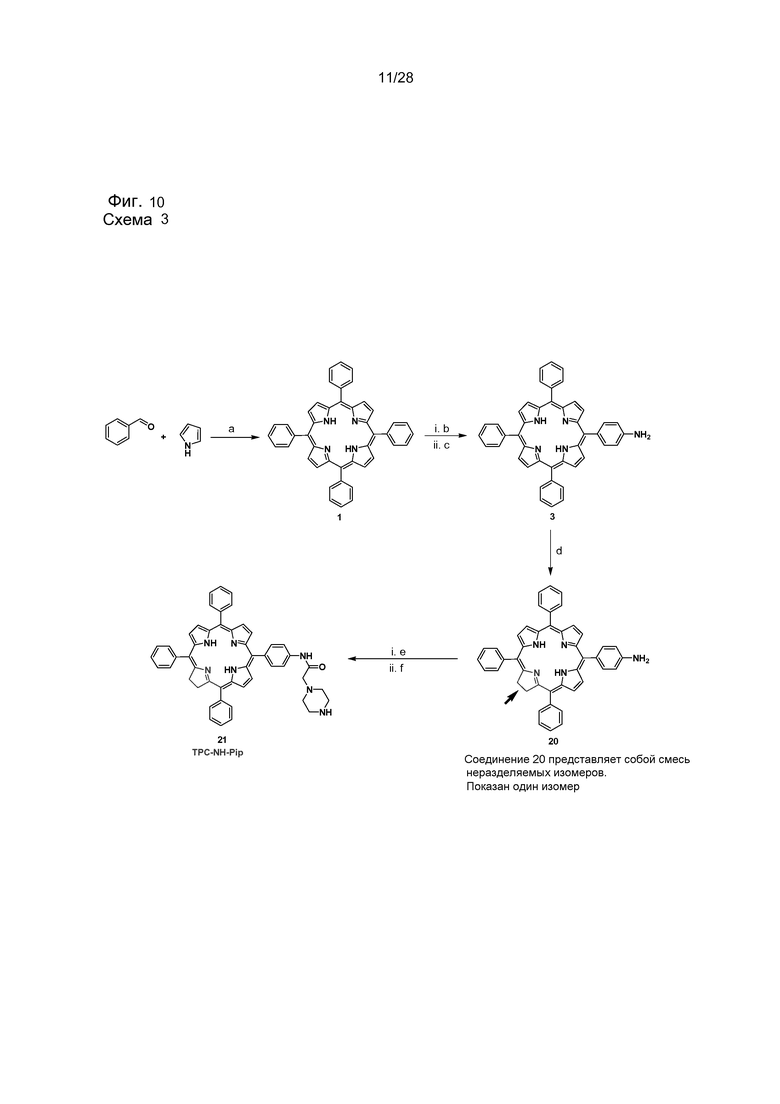
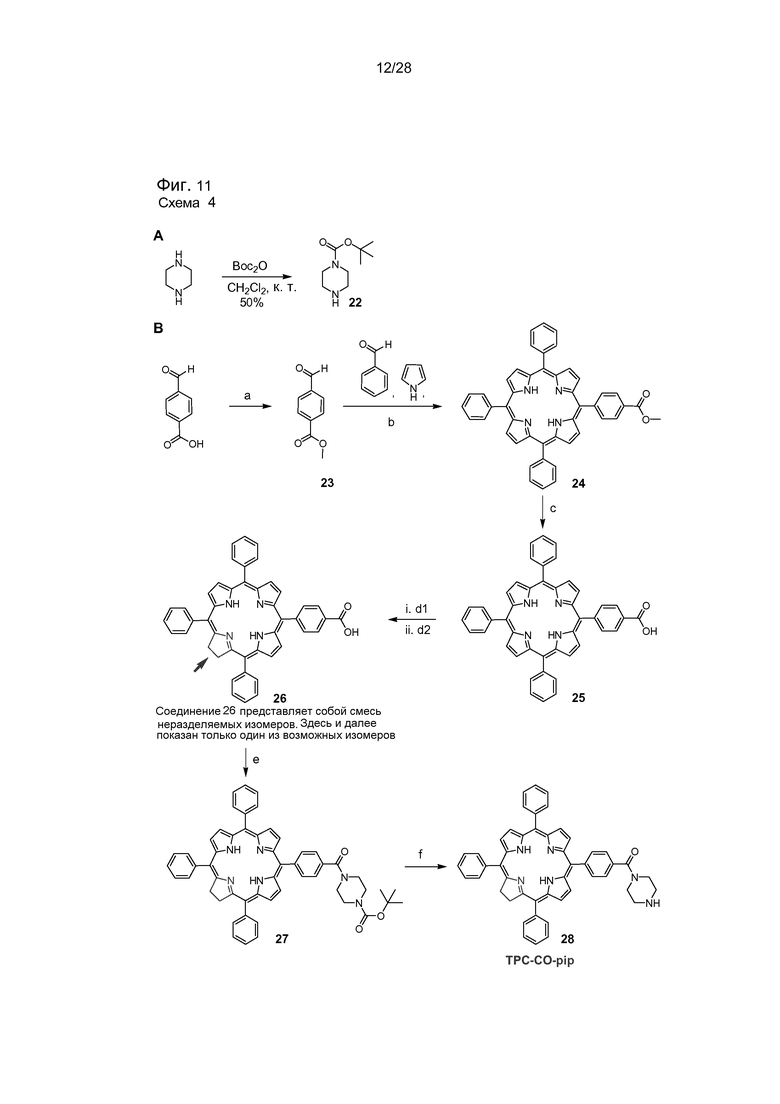
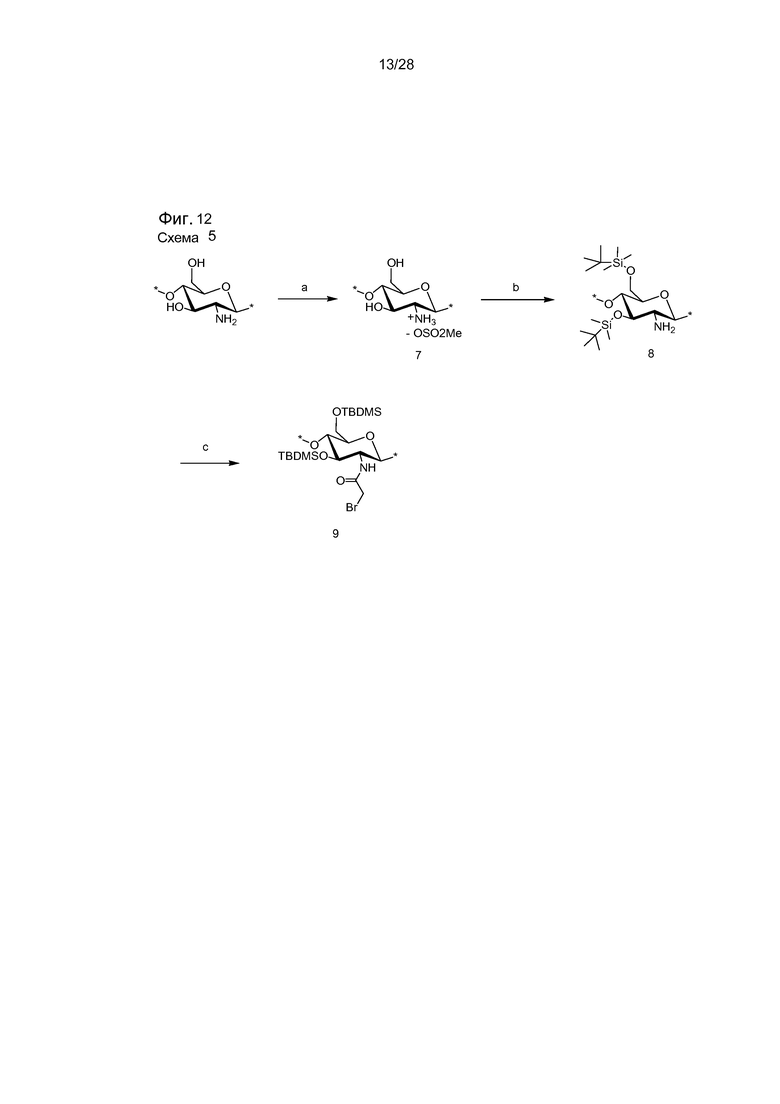
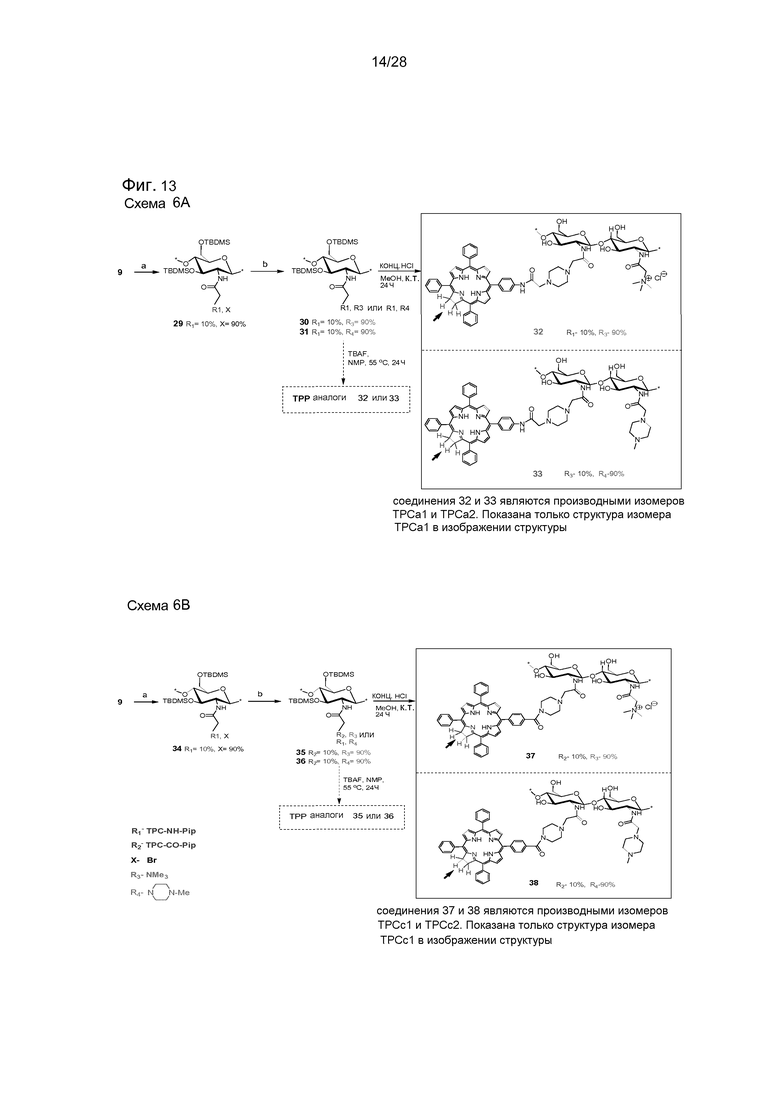
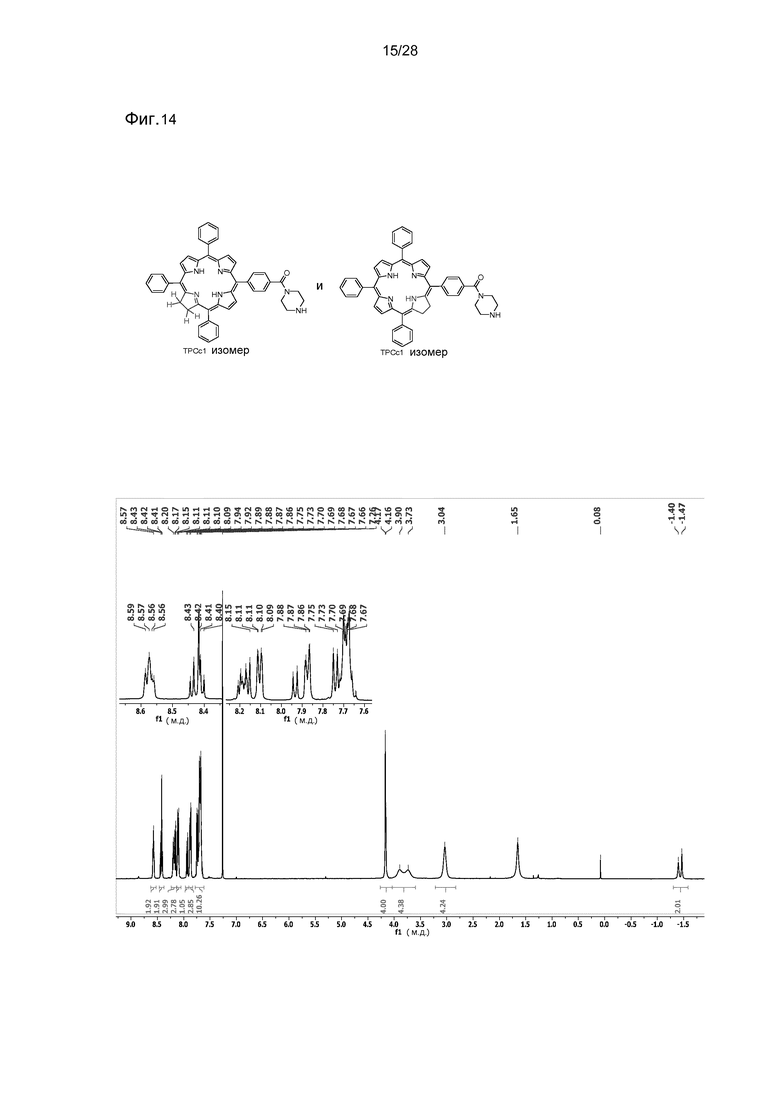
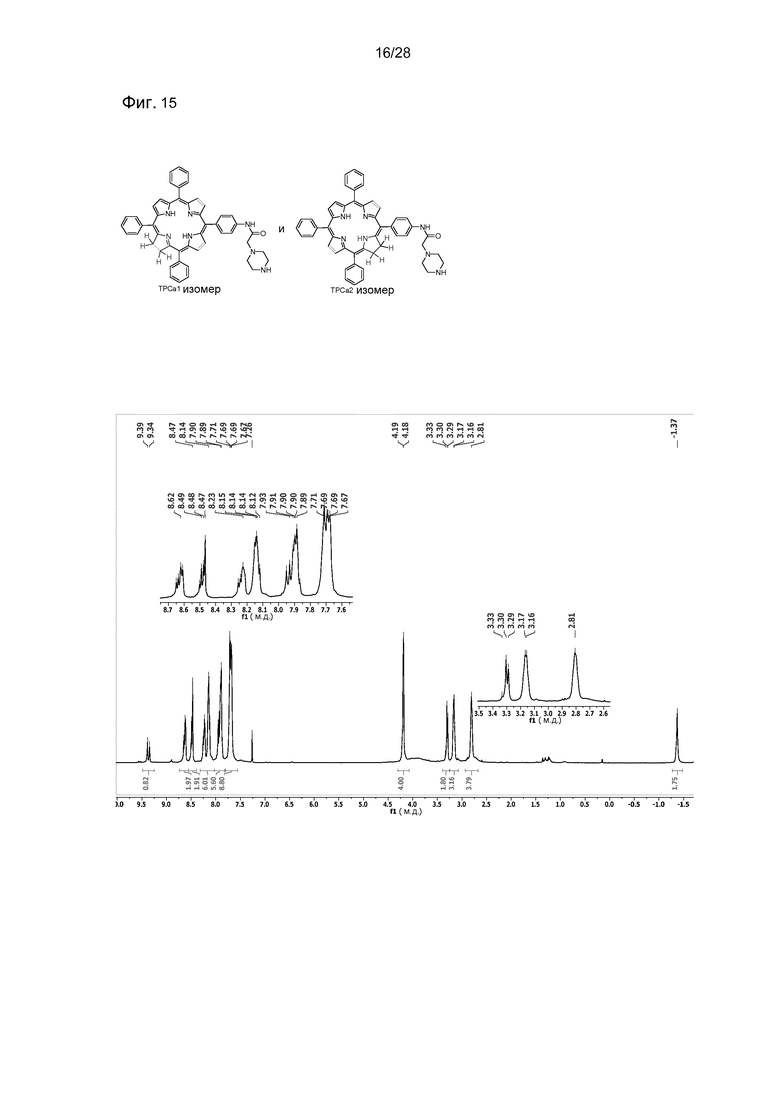
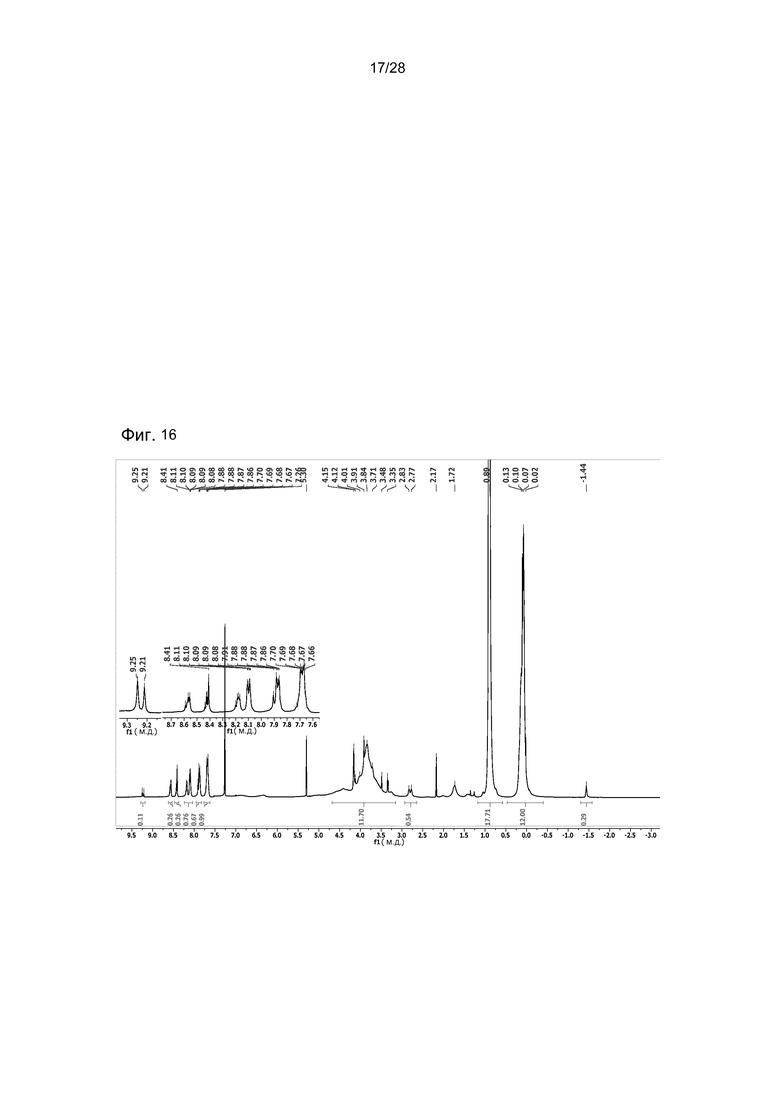
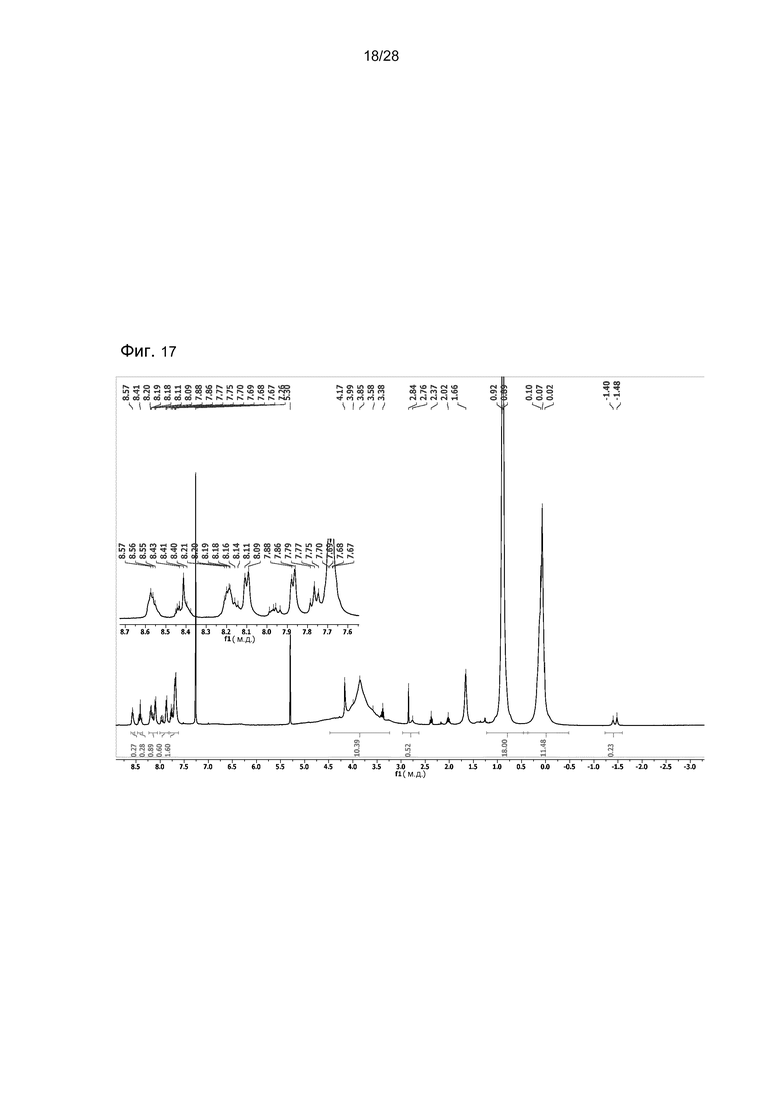
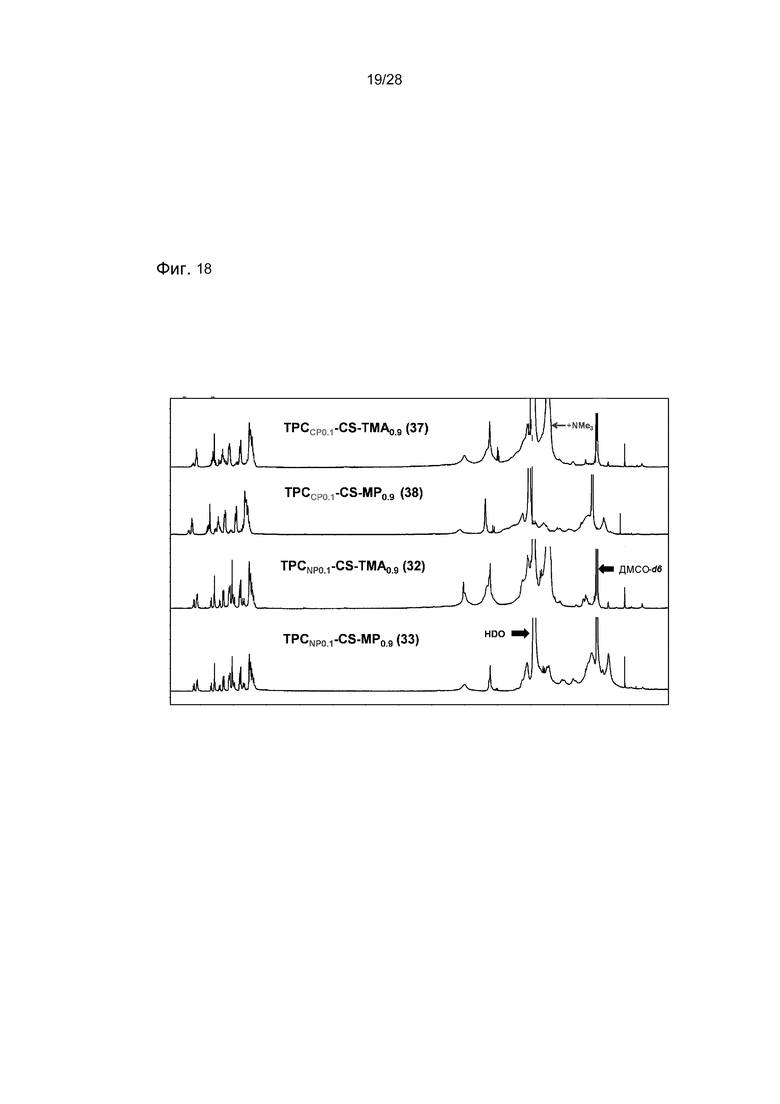
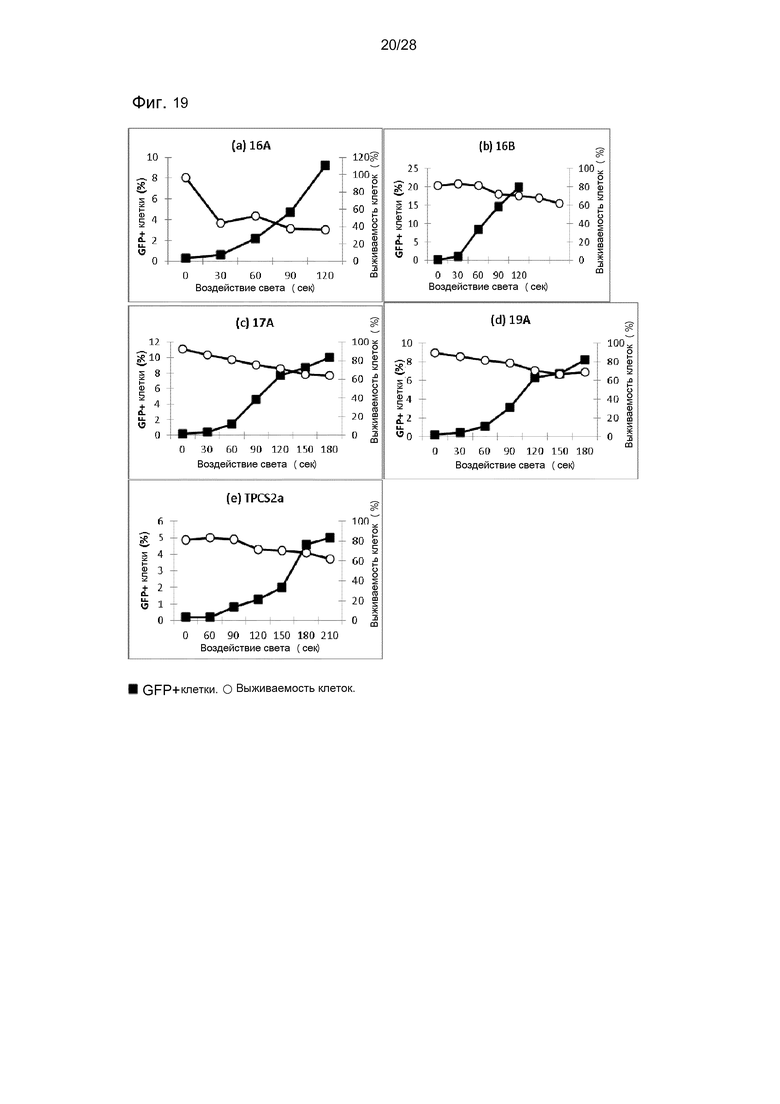
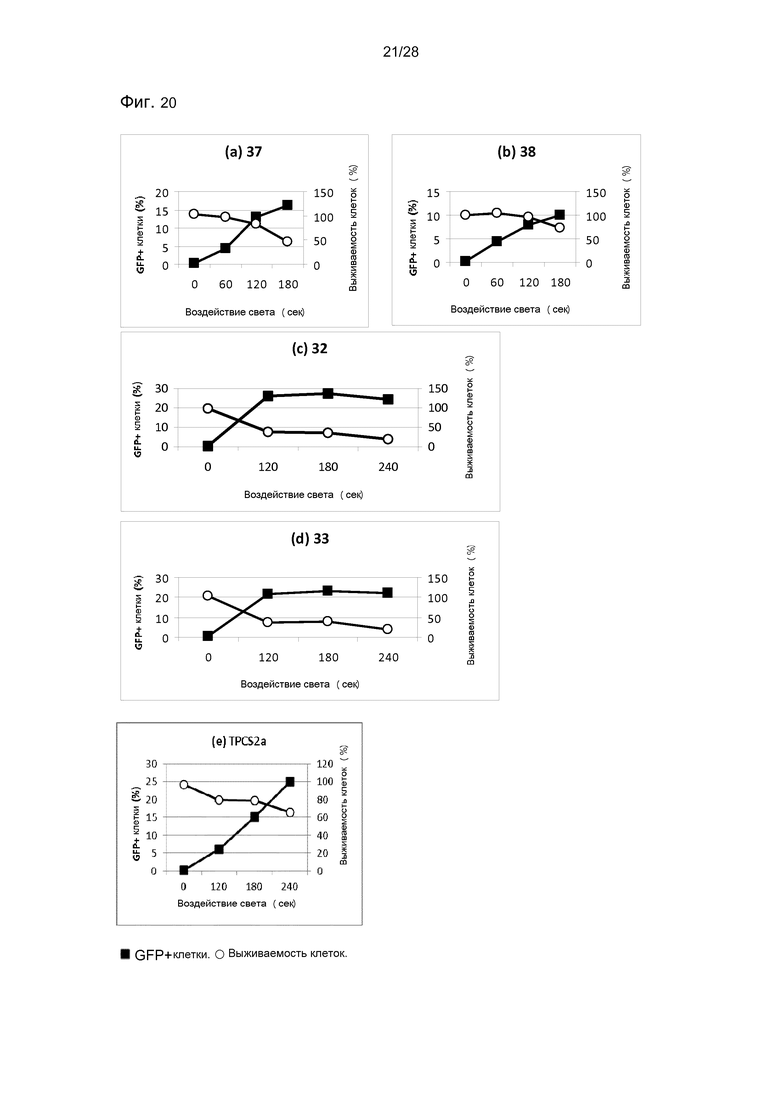
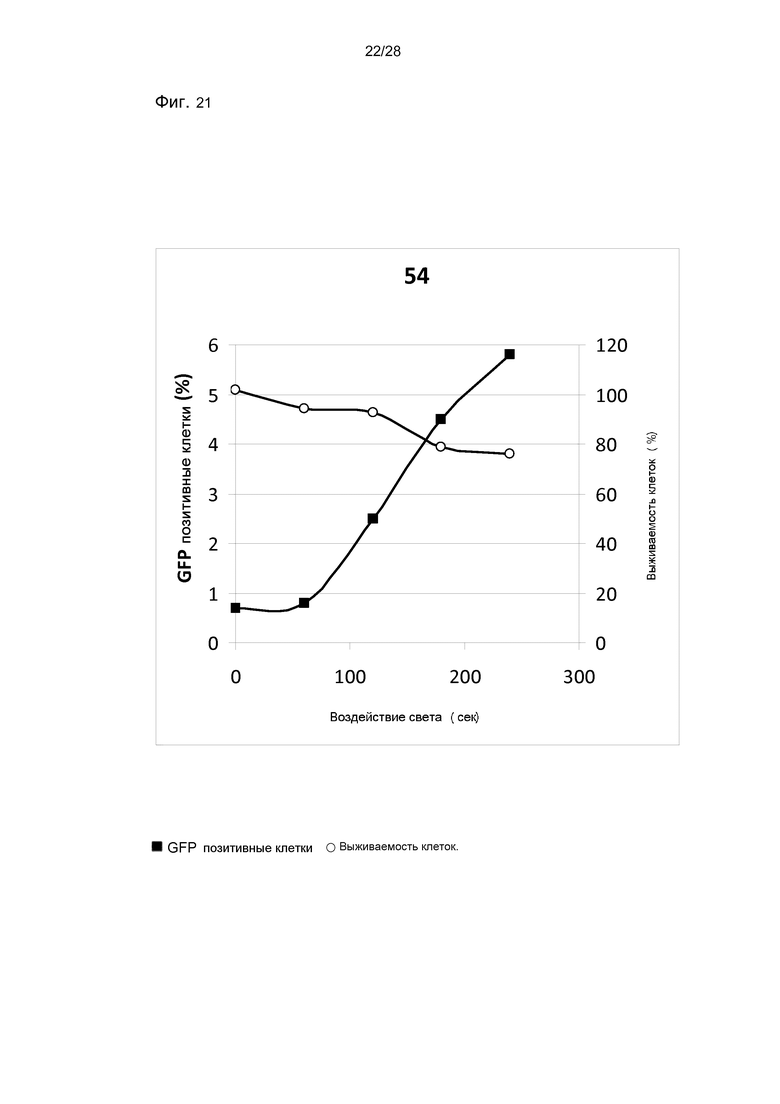

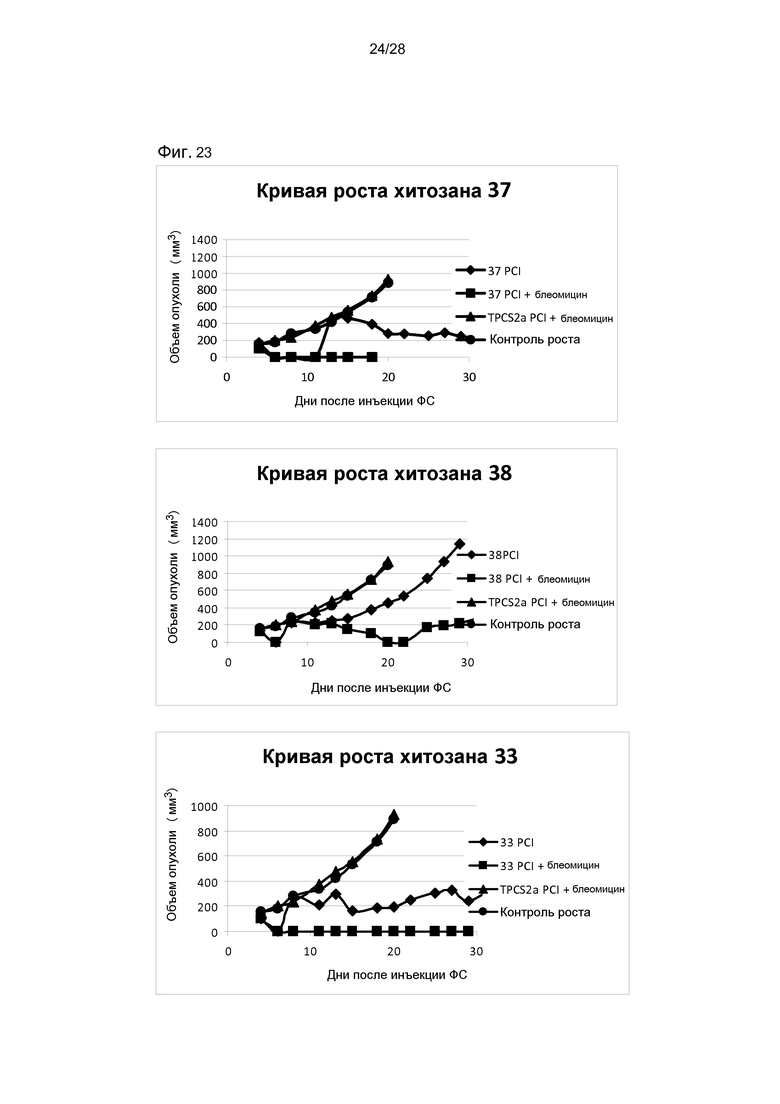
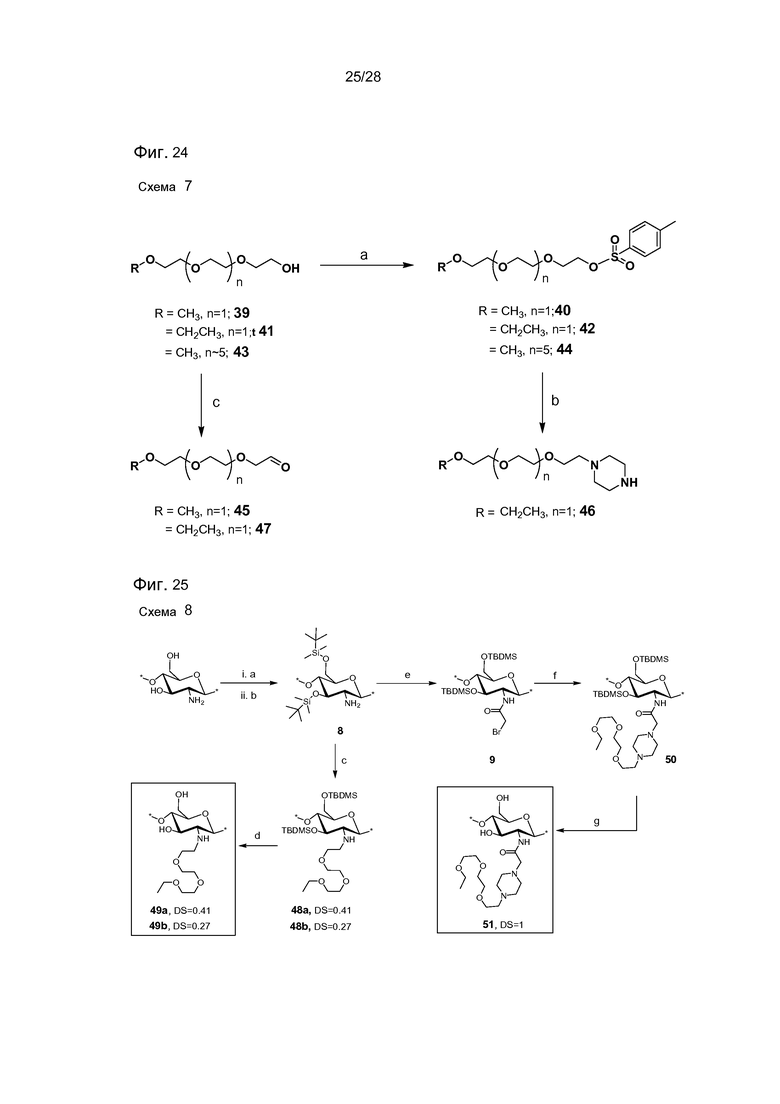
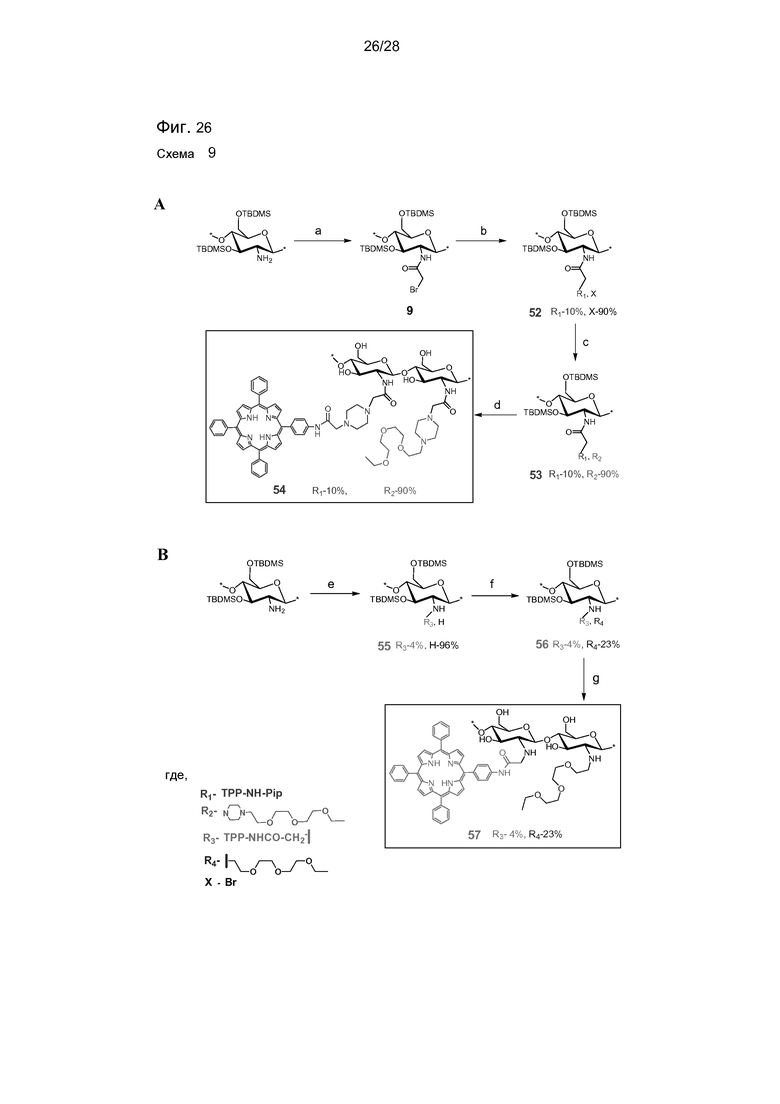
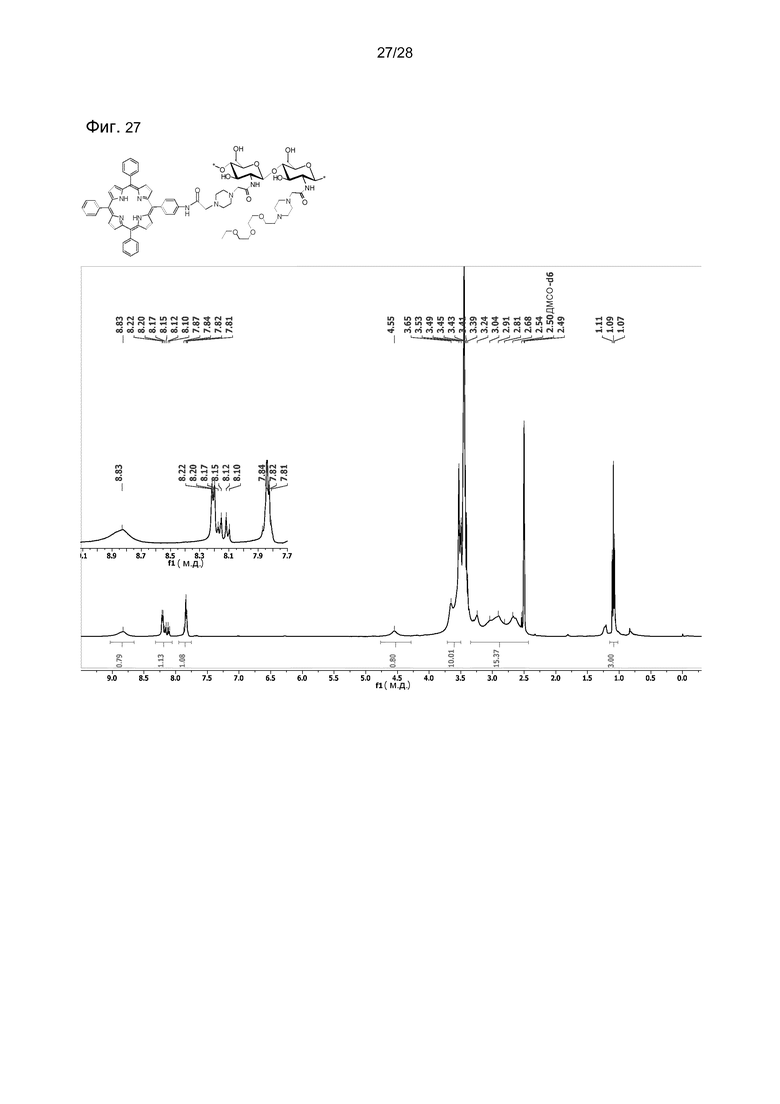
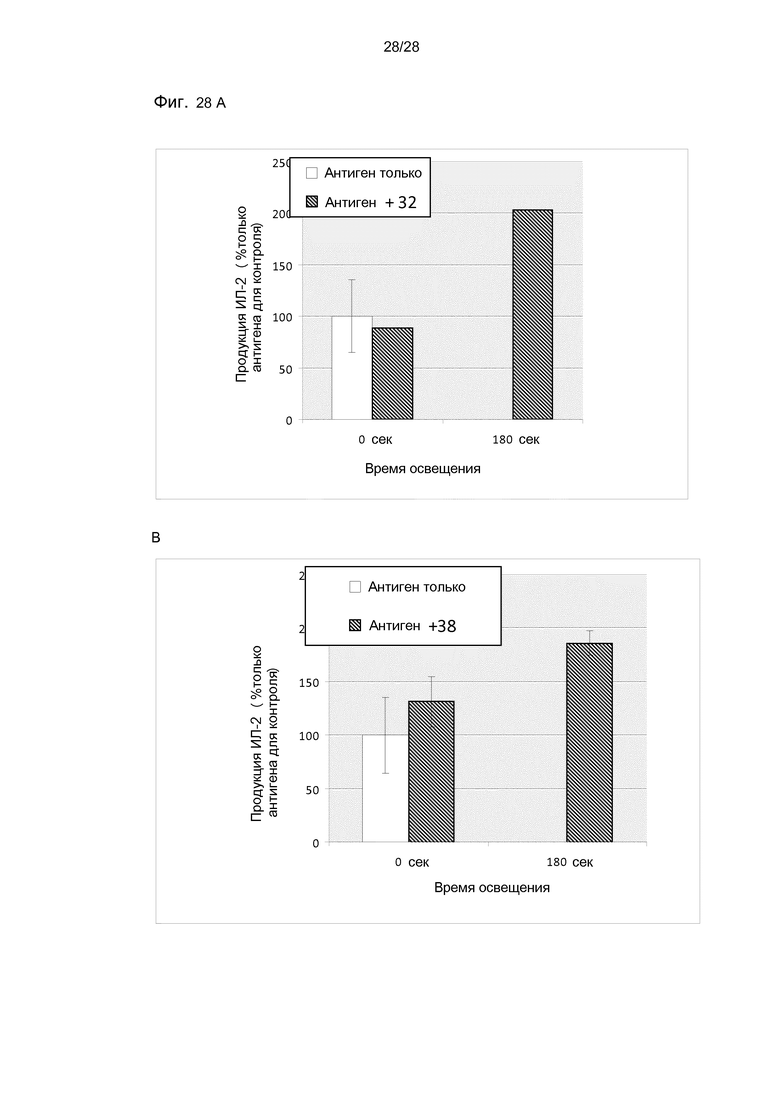
Группа изобретений относится к медицине, конкретно к новым конъюгатам на основе хитозана, например наноносителям, содержащим производное биосовместимого полимера хитозана, конъюгированного с фотосенсибилизирующим агентом, и их применению в фотохимической интернализации (ФХИ) и фотодинамической терапии (ФДТ). Изобретение также относится к применению новых конъюгатов по изобретению при лечении или профилактике заболеваний, в частности раковых заболеваний, и в целях вакцинации. Способ позволяет осуществить интернализацию молекул на достаточном уровне, чтобы их действие проявлялось в продуктах, экспрессированных этими клетками или путем воздействия на клетки. 8 н. и 18 з.п. ф-лы, 28 ил., 5 пр., 4 табл.
1. Соединение, содержащее конъюгат фотосенсибилизатора и хитозана, где указанное соединение представляет собой соединение формулы (I)

где n обозначает целое число, большее или равное 3,
R содержится n раз в указанном соединении и
в 0,5-99,5% из указанных всех Rn групп каждый R представляет собой группу A, выбранную из:
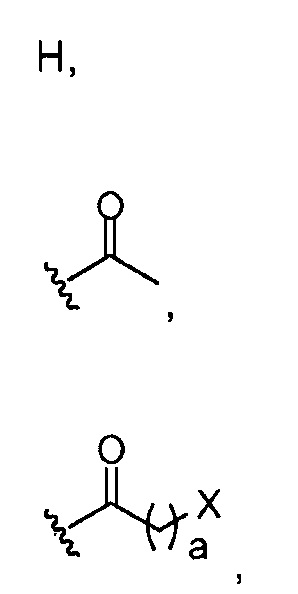
где a обозначает 1, 2, 3, 4 или 5 и X представляет собой Br, Cl или OH;
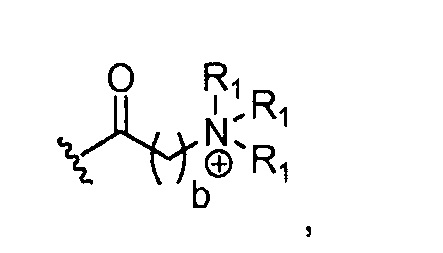
где каждый R1, который может быть одинаковым или разным, выбран из H, CH3 и -(CH2)C-CH3; b обозначает 1, 2, 3, 4 или 5 и c обозначает 0, 1, 2, 3, 4 или 5;
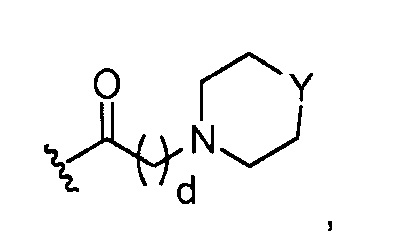
где Y представляет O, S, SO2, -NCH3 или -N(CH2)eCH3; d=1, 2, 3, 4 или 5 и e=1, 2, 3, 4 или 5;
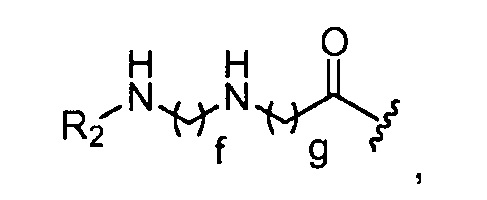
где R2 представляет собой -(CH2)h-CH3 или -CO-(CH2)h-CH3; f обозначает 1, 2, 3, 4 или 5; g обозначает 1, 2, 3, 4 или 5 и h обозначает 0, 1, 2, 3, 4 или 5;
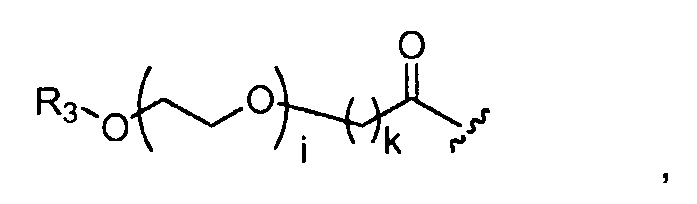
где R3 представляет собой -(CH2)j-CH3, i обозначает целое число от 1 до 200, предпочтительно от 1 до 10; j обозначает 0, 1, 2, 3, 4 или 5 и k обозначает 1, 2, 3, 4 или 5;
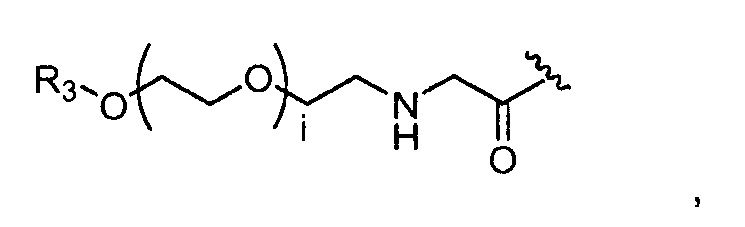
где R3 представляет собой -(CH2)j-CH3, i обозначает целое число от 1 до 200, предпочтительно от 1 до 10 и j обозначает 0, 1, 2, 3, 4 или 5;
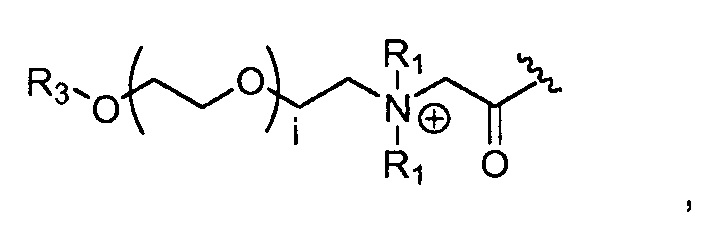
где R3 представляет собой -(CH2)j-CH3, i обозначает целое число от 1 до 200, предпочтительно от 1 до 10; j обозначает 0, 1, 2, 3, 4 или 5; каждый R1, который может быть одинаковым или разным, выбран из H, CH3 и -(CH2)C-CH3 и c обозначает 0, 1, 2, 3, 4 или 5;
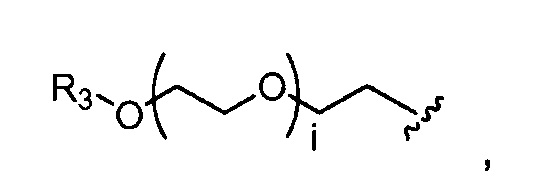
где R3 = -(CH2)j-CH3, где i обозначает целое число от 1 до 200, предпочтительно от 1 до 10, и j обозначает 0, 1, 2, 3, 4 или 5;
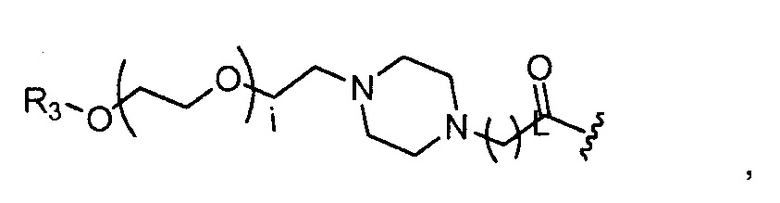
где R3 = -(CH2)j-CH3, i обозначает целое число от 1 до 200, предпочтительно от 1 до 10; L обозначает 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 и j обозначает 0, 1, 2, 3, 4 или 5;
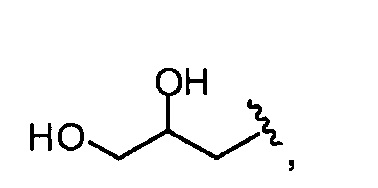
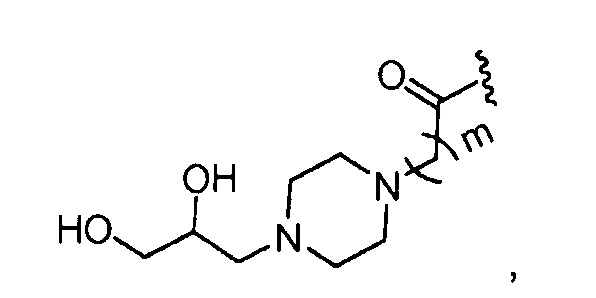
где m обозначает 1, 2, 3, 4 или 5;
где каждая группа R может быть одинаковой или разной и
в 0,5-99,5% из указанных всех групп Rn каждый R представляет собой группу B, выбранную из:
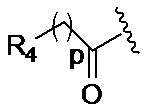 ,
,
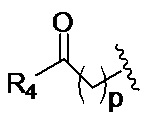 ,
,
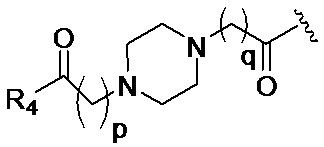 ,
,
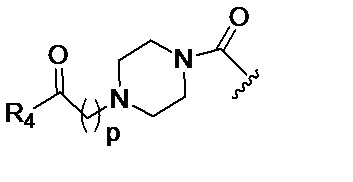 ,
,
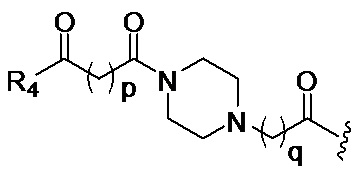 ,
,
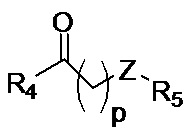 ,
,
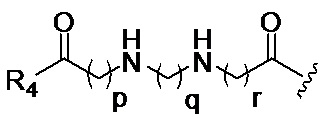 ,
,
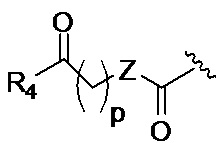 ,
,
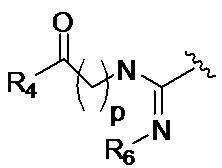 ,
,
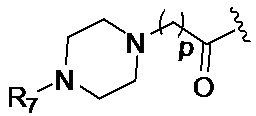 ,
,
и
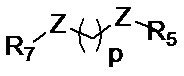 ,
,
где p обозначает 0, 1, 2, 3, 4 или 5; q обозначает 1, 2, 3, 4 или 5 и r обозначает 1, 2, 3, 4 или 5;
R4 представляет собой группу, выбранную из:
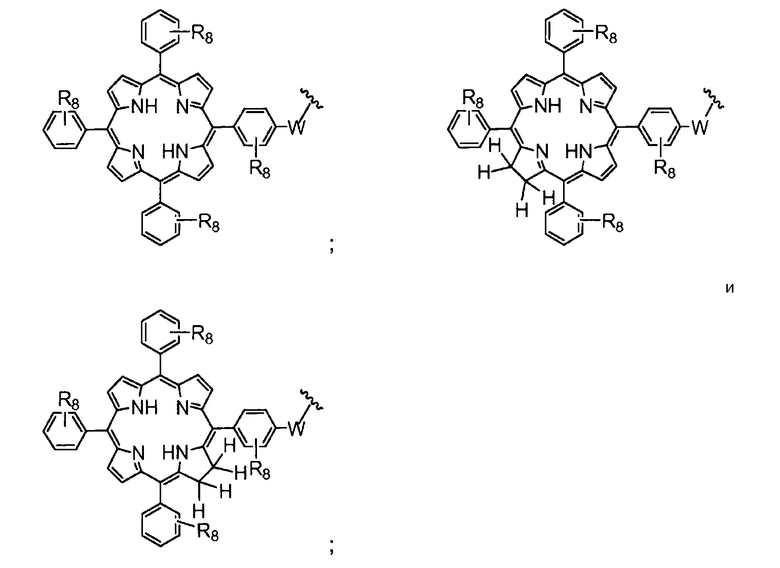
W представляет собой группу, выбранную из O, S, NH или N(CH3);
R5 представляет собой группу, выбранную из: -(CH2)s-CO-; -(CH2)s-Z-(CH2)t-CO- и -(CH2)S-Z-(CH2)t-Z-CO-, где s обозначает 0, 1, 2, 3, 4 или 5; t обозначает 0, 1, 2, 3, 4 или 5;
Z представляет собой NH, O, S или SO2;
R6 представляет собой группу, выбранную из -CN и CH3;
R7 представляет собой группу, выбранную из:
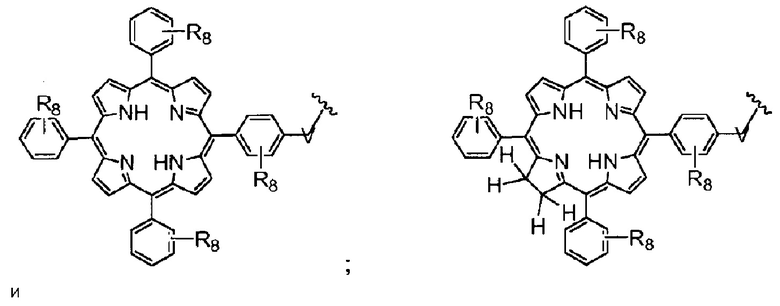
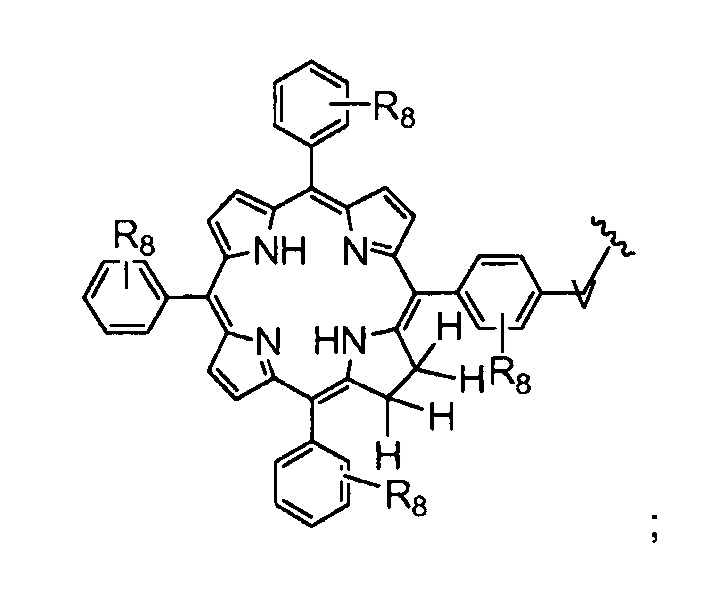
V представляет собой группу, выбранную из CO, SO2, PO, PO2H или CH2, и
R8 представляет собой группу (заместитель в о, м или п положении), которая может быть одинаковой или разной, выбранную из H, -OH, -OCH3, -CH3, -COCH3, C(CH3)4, -NH2, -NHCH3, -N(CH3)2 и -NCOCH3;
где каждая группа R может быть одинаковой или разной.
2. Соединение по п.1, где n обозначает целое число от 10 до 100.
3. Соединение по п.1, где R4 выбран из
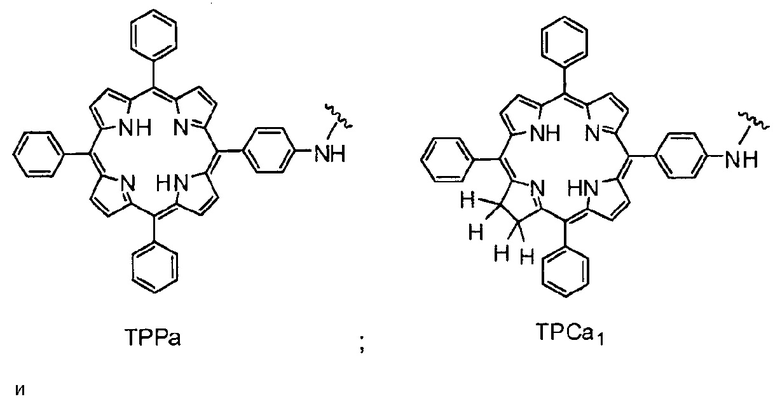
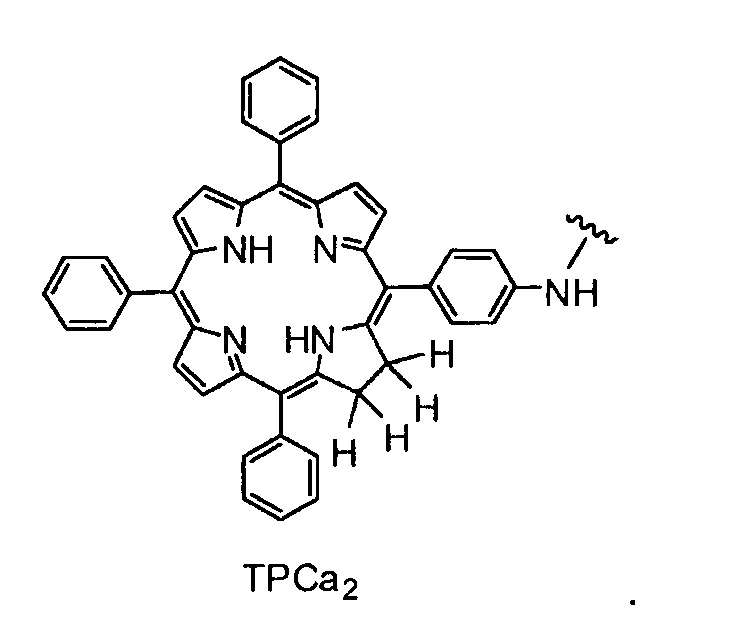
4. Соединение по п.1, где R7 выбран из
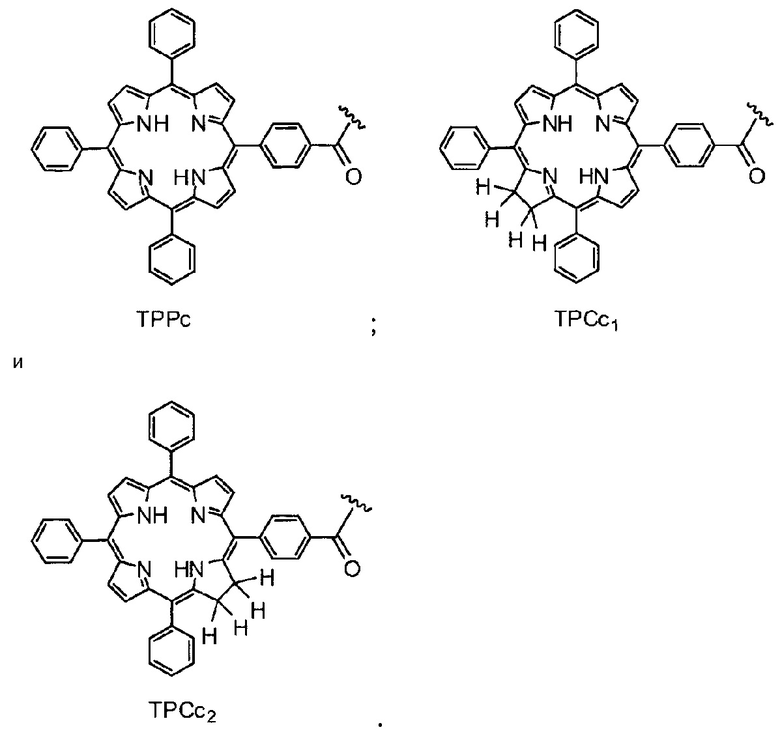
5. Соединение по п.3, где R4 представляет собой TPCa1 или TPCa2.
6. Соединение по любому из пп.1-5, где группа A составляет от 70 до 95% от всех групп Rn и группа B составляет от 5 до 30% от всех групп Rn.
7. Соединение по любому из пп.1-5, где каждая группа A из группы R выбрана из:
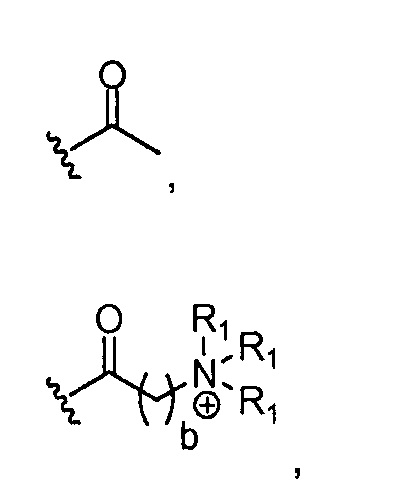
где, предпочтительно, каждый R1 представляет собой -CH3 и b обозначает 1;
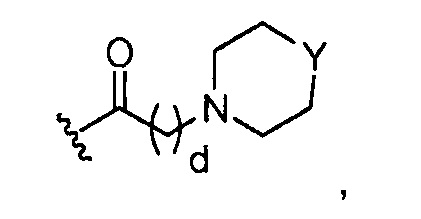
где, предпочтительно, Y представляет собой -NCH3 и d обозначает 1;
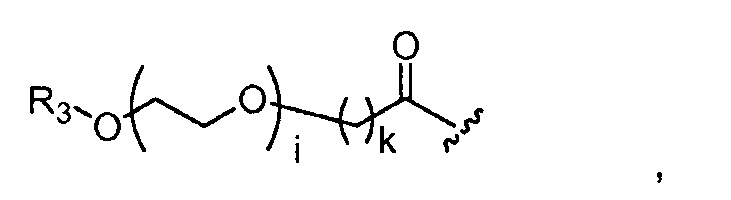
где, предпочтительно, j обозначает 0 или 1; i обозначает 3 или 6 и k обозначает 1;
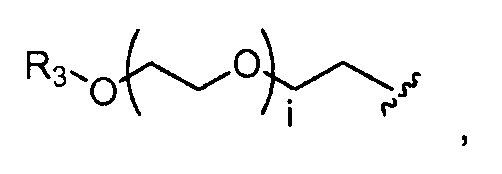
где, предпочтительно, j обозначает 1 и i обозначает 2;
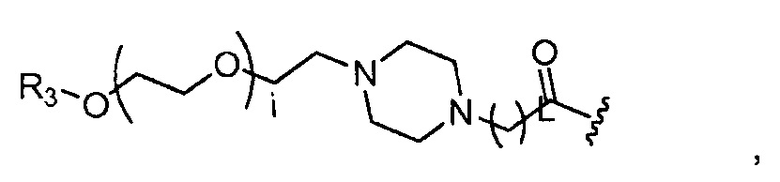
где, предпочтительно, j обозначает 0 или 1, i обозначает 2, 4 или 5 и L обозначает 1;
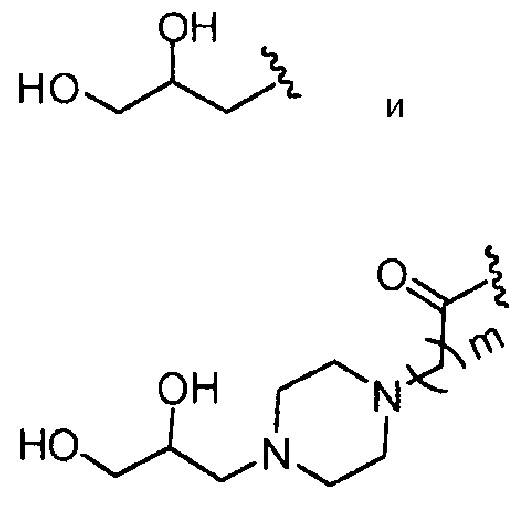 ,
,
где, предпочтительно, m обозначает 1, и каждая из групп R может быть одинаковой или разной.
8. Соединение по любому из пп.1-5, где каждая группа B из группы R выбрана из:
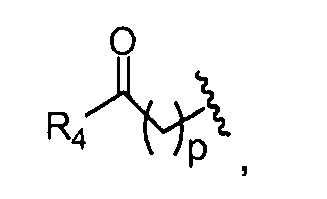
где, предпочтительно, р обозначает 1;
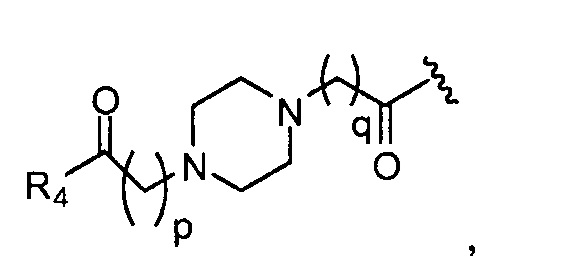
где, предпочтительно, p обозначает 1 и q обозначает 1;
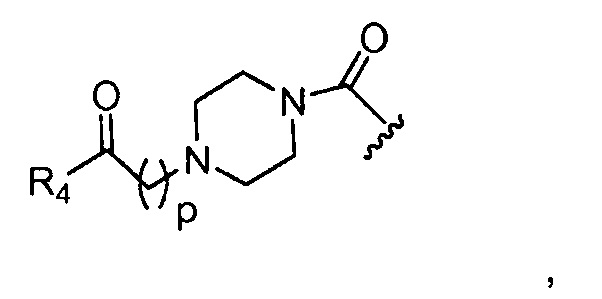
где, предпочтительно, р обозначает 1, и
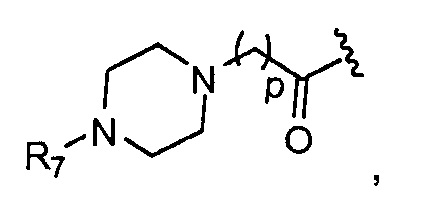
где, предпочтительно, р обозначает 1, и каждая из групп R может быть одинаковой или разной.
9. Соединение по любому из пп.1-5, где указанное соединение выбрано из соединений, показанных на фигуре 1.
10. Способ введения молекулы в цитозоль клетки, включающий контактирование указанной клетки с молекулой, подлежащей введению, и соединением, как определено в любом из пп.1-9, и облучение клетки светом с длиной волны, эффективной для активации фотосенсибилизирующего агента соединения, тем самым высвобождая молекулу в цитозоль, где указанную молекулу, подлежащую введению, выбирают из полинуклеотида, антигенной молекулы и цитотоксической молекулы.
11. Способ достижения гибели клетки, включающий контактирование указанной клетки с соединением, как определено в любом из пп.1-9, и облучение клетки светом с длиной волны, эффективной для активации фотосенсибилизирующего агента соединения для получения активных форм кислорода, которые вызывают гибель указанной клетки.
12. Способ экспрессии антигенной молекулы или ее части на поверхность клетки, включающий контактирование указанной клетки с указанной антигенной молекулой и соединением, как определено в любом из пп.1-9, и облучение клетки светом с длиной волны, эффективной для активации фотосенсибилизирующего агента соединения, где указанная антигенная молекула высвобождается в цитозоль клетки и антигенная молекула или ее часть достаточного размера для стимуляции иммунного ответа представлена на поверхности клетки.
13. Фармацевтическая композиция для стимулирования иммунного ответа, содержащая соединение, как определено в любом из пп.1-9, и один или несколько фармацевтически приемлемых разбавителей, носителей или эксципиентов.
14. Фармацевтическая композиция по п.13, дополнительно содержащая молекулу, подлежащую интернализации, где указанную молекулу, подлежащую интернализации, выбирают из полинуклеотида, антигенной молекулы и цитотоксической молекулы.
15. Соединение или композиция, как определено в любом из пп.1-5, 13 и 14, для применения в терапии, предпочтительно в терапии рака, в генной терапии или для стимуляции иммунного ответа.
16. Соединение или композиция, как определено в любом из пп.1-5, 13 и 14, для применения при лечении или профилактике заболевания, расстройства или инфекции у субъекта, предпочтительно, в котором проявляется аномальный или чрезмерный рост клеток или в котором проявляется аномально повышенная или подавленная экспрессия генов, особенно предпочтительно, когда указанным заболеванием является рак.
17. Соединение или композиция по п.16, где молекулу, подлежащую интернализации, также используют при указанном лечении или профилактике, где указанную молекулу, подлежащую интернализации, выбирают из полинуклеотида, антигенной молекулы и цитотоксической молекулы.
18. Соединение или композиция для применения, как заявлено в п.17, где указанное лечение или профилактика включает контактирование клеток субъекта с указанной молекулой, подлежащей интернализации, и указанного соединения или композиции и облучение клеток светом с длиной волны, эффективной для активации фотосенсибилизирующего агента соединения.
19. Соединение или композиция для применения, как заявлено в п.18, где указанная молекула, подлежащая интернализации, представляет собой цитотоксическую молекулу, предпочтительно блеомицин.
20. Соединение или композиция для применения, как заявлено в п.16 или 17, где указанное лечение или профилактика включает контактирование клеток субъекта с указанным соединением или композицией и облучение клеток светом с длиной волны, эффективной для активации фотосенсибилизирующего агента соединения для получения активных форм кислорода, которые вызывают гибель указанных клеток.
21. Соединение или композиция, как определено в любом из пп.1-5, 13 или 14, для применения при лечении или профилактике заболевания, расстройства или инфекции у субъекта путем генерирования иммунного ответа, предпочтительно, методом вакцинации.
22. Соединение или композиция для применения, как заявлено в п.21, где указанное лечение или профилактика включает контактирование клеток субъекта с указанным соединением или композицией и антигенной молекулой и облучение клеток светом с длиной волны, эффективной для активации фотосенсибилизирующего агента соединения, где указанная антигенная молекула высвобождается в цитозоль клетки и антигенная молекула или ее часть достаточного размера для стимуляции иммунного ответа представлена на поверхности клетки.
23. Клетка или популяция клеток для стимулирования иммунного ответа, получаемые способом, как определено в любом из пп.10-12.
24. Способ лечения или профилактики рака у пациента, включающий введение соединения или композиции, как определено в любом из пп.1-9, 13 и 14, в одну или несколько клеток in vivo способом, как определено в п.10.
25. Способ по п.24, дополнительно включающий введение молекулы, подлежащей интернализации, в указанную одну или несколько клеток, где указанная молекула, подлежащая интернализации, представляет собой цитотоксическую молекулу.
26. Набор, содержащий соединение или композицию, как определено в любом из пп.1-9, 13 и 14, и молекулу, подлежащую интернализации, предпочтительно для одновременного, раздельного или последовательного применения при лечении или профилактике рака, где указанная молекула, подлежащая интернализации, представляет собой цитотоксическую молекулу.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| KR 20120045155 A, 09.05.2012. | |||

