Объектом изобретения являются новые соединения, являющиеся производными α-C-фенил-N-трет-бутилнитрона, способ их получения и их применение для получения лекарственных средств, предназначенных для профилактики или лечения заболеваний, связанных с оксидативным стрессом.
Патологии, связанные с оксидативным стрессом и с образованием соединений со свободными радикалами кислорода, описаны в работах Croos C.E., Arch. Intern. Med. (1987) 107, 526-545 и Anderson K.M., Ells G., Bonomi P., Harris J.E., Medical Hypotheses (1999) 52, 53-57.
Они многочисленны: более 70 патологий этого типа указано в этом списке, который включает, в частности, иммунные и воспалительные заболевания, синдром ишемии-реперфузии, атеросклероз, болезни Альцгеймера и Паркинсона, повреждения, вызванные ионизирующим и УФ-излучением, определенные формы химического онкогенеза, а также старение клеток.
Реакционно-способные формы кислорода и азота (ROS и RNS) образуются в организме естественным путем, и их регулирование обеспечивается определенными специфическими ферментами, такими как растворимая супероксиддисмутаза (СОД). Улавливание этих чрезвычайно реакционно-способных радикальных соединений жизненно необходимо, так как они вызывают необратимые повреждения в клетках. Если нормальное производство этих радикальных соединений легко регулируется клеткой, перепроизводство свободных радикалов, связанное с внешним оксидативным стрессом (воспалительный шок, синдром ишемии-реперфузии и т.д.) или генетическим недостатком (в частности, аномалией митохондрий), приводит к быстрой деградации клеток. Из-за этого становится невозможной переработка этого значительного количества радикалов организмом человека или животного.
Существует множество механизмов защиты от оксидативного стресса клетки, способных осуществляться на разных уровнях окислительного каскада. Они инициируются обычно перепроизводством пероксидных радикалов, связанным с частичным восстановлением молекулярного кислорода в митохондрии (типичный синдром при ишемии-реперфузии). Этот пероксидный радикал может дисмутировать в перекись водорода. Эти два соединения посредством реакции Фентона в присутствии двухвалентного железа могут дать гидроксильные радикалы, которые имеют особенность очень быстро и неспецифическим образом реагировать с любым составляющим клетки, таким как липиды, ДНК или белки, вызывая в них необратимые повреждения, как это описано в работах Stadtman H. R., Berlett B.S. J. Biol.Chem. (1991) 266, 17201-17211; Floyd R. A. Carcinogenesis (1990) 11, 1447-1450; Gille J.J., Van Berkel C.G., Joenge H. Carcinogenesis (1994) 15, 2695-2699; Halliwell B. Mutat. Res. (1999) 443, 37-52.
Эти радикальные соединения, активируя определенные суицидные гены (гены Bel или p53) посредством фактора NF-kB, также являются следствием клеточного апоптоза, которое описано в работе Siebenlist U., Franzoso G., Brown K. Annu. Rev. Cell. Biol. (1994) 10, 405-455.
Задачей СОД является превращение пероксидных радикалов в перекись водорода, которая затем расщепляется каталазами или глутатион-зависимыми пероксидазами.
Существуют другие уровни клеточной защиты от оксидантов, в частности, на уровне мембраны, которые позволяют ограничить окисление ненасыщенных мембранных фосфолипидов. α-Токоферол и β-каротин являются главными примерами липидных антиоксидантов.
Наиболее многообещающая стратегия в поиске терапии, направленной на предупреждение или лечение заболеваний, связанных с оксидативным стрессом, состоит в том, чтобы как можно раньше вмешаться в этот окислительный каскад, чтобы очень быстро предотвратить повреждения, связанные с очень высокой реакционно-способностью радикальных соединений.
Для этого пытались улавливать эти чрезвычайно реакционно-способные свободные радикалы с помощью молекул, называемых spin trap или спиновыми ловушками, из которых самыми эффективными оказались нитроны.
Терапевтический эффект нитронов в снижении и предотвращении повреждений, вызванных свободными радикалами в биологических системах, был доказан в 1990 в работе Oliver C., Starke-Read P., Stadman E., Liu G., Carney J., Floyd R. Proc. Natl. Acad. Sci. USA (1990) 87, 5144-5147.
Эти авторы смогли показать уменьшение повреждений, вызванных церебральной ишемией, после инъекций α-C-фенил-N-трет-бутилнитрона (PBN) мышам-песчанкам. Церебральные ишемии сопровождаются сильным увеличением продукции свободных радикалов, которые захватываются PBN, чтобы образовать аддукторы спина, намного более стабильные и, следовательно, намного менее активные и токсичные спиновые аддукты. PBN является спиновой ловушкой, что делает его объектом большого числа биологических исследований.
Можно сослаться, например, на работу Hensley K., Carney J. M., Stewart C.A., Tabatabaie T., Pye Q.N., Floyd R. A. Int. Rev. Neurobiol. (1997) 40, 229-317.
Он обладает большой специфичностью церебрального действия, возможно, по причине его сильной гидрофобности, что позволяет ему преодолевать гематоэнцефалический барьер, как это показано в работе Cheng H.Y., Liu T., Feuerstein G., Barone F.C. Free Radic. Biol. Med. (1993) 14, 243-250.
Самыми известными и самыми эффективными из нитронов являются α-C-фенил-N-трет-бутилнитрон (PBN), 5,5-диметилпирролидин-N-оксид (DMPO) и молекулы, открытые позднее: N-бензилиден-1-диэтоксифосфорил-1-метилэтиламин-N-оксид (PBNP) и 5-диэтилфосфоно-5-метилпирролин-N-оксид (DEPMPO).
Можно назвать также дисульфонатное производное PBN, NXY-059
(динатрий-4-[(трет-бутилимино)-метилбензол-1,3-дисульфонат-N-оксид), обладающее более высокой нейрозащитной активностью, чем у PBN, с которым проводятся фармакологические и клинические исследования:
Kuroda S., Tsuchidate R., Smith M.L., Maples K.R., Siesjo B.K. J. Cereb. Blood Flow Metab. (1999) 19, 778-787;
Lees K.R., Sharma A.K., Barer D., Ford G.A., Kostulas V., Cheng Y.F., Odergren T. Stroke (2001) 32, 675-680.
Однако ни одна из названных молекул не обладает эффективностью in vivo или ex vivo, достаточной в малых дозах, даже если их цитотоксическая концентрация повышена: Almli L.M., Hamrick S.E.G., Koshy A.A., Tauber M.G., Ferriero D.M. Dev. Brain Res. (2001) 132, 121-129; Nakao N., Grasbon-Frodl E.M., Widner H., Brundin P. Neuroscience (1996) 73, 185-200. Этот недостаток эффективности связан, возможно, с плохой биологической эффективностью лекарства и проблемой проникновения в клетку.
Таким образом, продолжает существовать потребность в молекуле типа spin trap, или спиновой ловушки, способной захватывать свободные радикалы, которая также способна доставляться организмом человека или животного до ее цели на внутриклеточном уровне.
В частности, в молекуле, способной проходить через клеточную мембрану и, что является еще более трудной и важной задачей, митохондриальную мембрану, чтобы войти в компартмент, где продуцируются пероксидные радикалы.
С этой целью в работах Ouari O., Polidori A., Pucci B., Tordo P., Chalier F. J. Org. Chem. (1999) 64, 3554-3556 и Geromel V., Kadhom N., Cebalos-Picot I., Ouari O., Polidori A., Munnich A., Rötig A., Rustin P. Hum. Моl. Genet. (2001) 10, 1221-1228, было предложено амфифильное перфторуглеродное производное PBN, а именно TA1PBN.
 TA1PBN
TA1PBN
Эту молекулу проверяли на клеточных линиях фибробластов, страдающих от сильного дефицита активности комплекса V дыхательной цепи (АТФазы), и она дала обнадеживающие результаты.
Однако синтез TA1PBN представляет трудности, которые делают его производство на промышленном уровне трудно представимым.
Таким образом, заявитель определяет целью разработку и производство новых молекул, обладающих активностью spin trap, или спиновой ловушки, имеющих повышенную биологическую эффективность по сравнению с молекулами предшествующего уровня техники и которые получаются просто, что позволяет планировать производство на промышленном уровне.
Объектом изобретения являются новые молекулы, отличающиеся тем, что они отвечают приведенной ниже формуле (I):
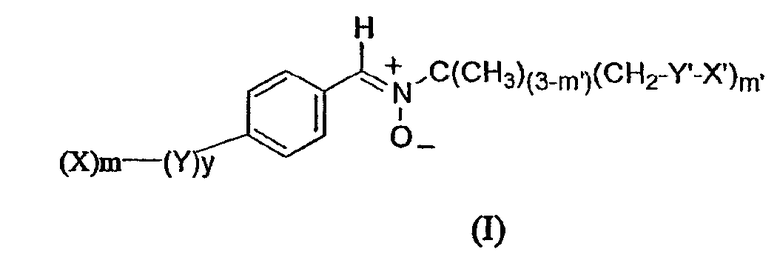
в которой:
X означает гидрофильную группу, выбранную из моно- или полисахарида, а также аминопроизводных моно- или полисахаридов, полиэтиленоксидную цепь, пептидную цепь, ионную полярную группу, выбранную из четвертичного аммония, аминоксида, карнитиновой группы;
m означает целое число, равное 1, 2 или 3;
Y означает связующее звено, предназначенное для соединения ароматического кольца и гидрофильных заместителей X;
Y выбран из функциональных групп сложного эфира, амида, мочевины, уретана, простого эфира, тиоэфира, амина, углеводородных цепей C1-C6, в случае необходимости прерванных одной или несколькими группами сложного эфира, амида, мочевины, уретана и одним или несколькими мостиками простого эфира, амина или тиоэфира;
y означает целое число, равное 0 или 1;
Y' означает группу, выбранную из функциональной группы сложного эфира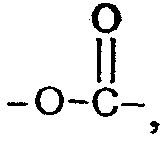 амидной группы
амидной группы 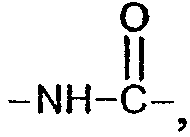 карбамидной группы
карбамидной группы 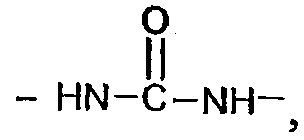 уретановой группы
уретановой группы 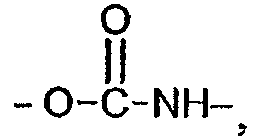 эфирного мостика -O-, тиоэфирного мостика -S-;
эфирного мостика -O-, тиоэфирного мостика -S-;
m' является целым числом, выбранным из 1 и 2;
X' означает атом водорода или алкильную цепь C4-C14, при необходимости замещенную одним или несколькими атомами фтора.
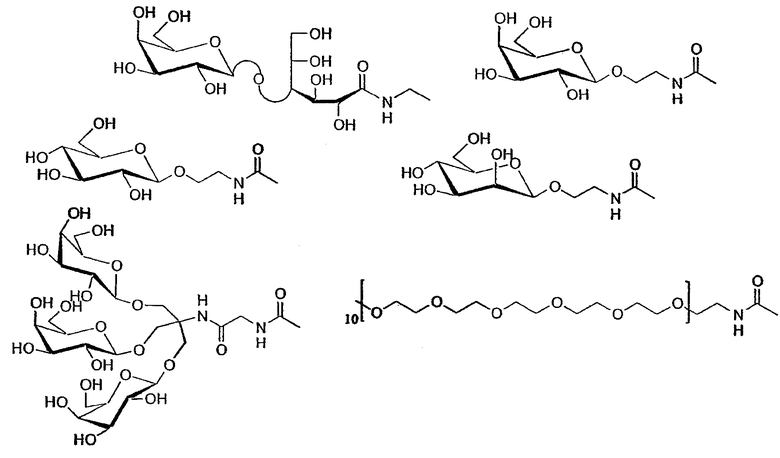
Из моносахаридов, которые могут использоваться в настоящем изобретении, можно назвать глюкозу, лактозу, фруктозу, маннозу, галактозу, рибозу, мальтозу. Из аминопроизводных сахаров можно назвать, в частности, глюкозамин. Из полисахаридов, которые могут использоваться в настоящем изобретении, можно назвать цепи, состоящие из нескольких моносахаридных звеньев, как, например, сахароза и лактобионамид.
Если гидрофильная часть X молекулы формулы (I) является цепью полиэтиленоксида, она содержит предпочтительно от 30 до 100 звеньев этиленоксида, более предпочтительно от 50 до 60 звеньев.
Предпочтительно пептидная цепь составлена из натуральных аминокислот, таких как аланин, аргинин, аспарагин, аспарагиновая кислота, цистеин, глутамин, глутаминовая кислота, глицин, гистидин, изолейцин, лейцин, лизин, метионин, фенилаланин, пролин, серин, треонин, триптофан, тирозин, валин.
Ионные или неионные гидрофильные группы, которые могут использоваться в настоящем изобретении, показаны ниже на схеме 1.
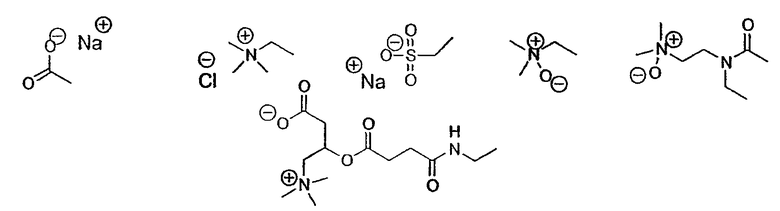
Полярные ионные головки
Полярные неионные головки
Схема 1: Общая структура полярных головок
В зависимости от моно- или полифункциональности связующего звена Y, оно может быть одно- или двукратно замещено группой X.
Группа X' может, например, быть выбрана из следующих радикалов:
- углеводородные радикалы: н-бутил, трет-бутил, изобутил, н-пентил, изопентил, н-гексил, н-гептил, н-октил, н-нонил, н-децил, н-ундецил, н-додецил, н-тридецил, н-тетрадецил и т.д.;
- фторированные углеводородные радикалы: можно назвать радикалы, отвечающие формуле -(CH2)t-(CF2)rF, в которой r и t означают два целых числа, причем 14 ≥ r+t ≥ 4, такие, например, как:
-(CF2)4F; -(CF2)5F; -(CF2)6F; -(CF2)7F; -(CF2)8F; -(CF2)9F; -(CF2)10F; -(CF2)11F; -(CF2)12F; -(CF2)13F; -(CF2)14F; -CH2-(CF2)3F; -CH2-(CF2)4F; -CH2-(CF2)5F; -CH2-(CF2)6F; -CH2-(CF2)7F; -CH2-(CF2)8F; -CH2-(CF2)9F; -CH2-(CF2)10F; -CH2-(CF2)11F; -CH2-(CF2)12F; -CH2-(CF2)13F; -(CH2)2-(CF2)2F; -(CH2)2-(CF2)3F; -(CH2)2-(CF2)4F; -(CH2)2-(CF2)5F; -(CH2)2-(CF2)6F; -(CH2)2-(CF2)7F; -(CH2)2-(CF2)8F; -(CH2)2-(CF2)9F; -(CH2)2-(CF2)10F; -(CH2)2-(CF2)11F; -(CH2)2-(CF2)12F; -(CH2)3-(CF2)1F;… -(CH2)13-(CF2)F.
Предпочтительно удовлетворяется по меньшей мере одно из приведенных ниже условий:
X означает лактобионамидную, карнитиновую группу или цепь полиэтиленоксида;
m означает 1;
m' означает 1 или 2;
X' выбран из октила, децила, додецила, CF3(CF2)rCH2CH2-
8 ≥ r ≥ 6
Соединения по изобретению обладают тем преимуществом по сравнению с соединениями предшествующего уровня техники, что имеют улучшенную биологическую эффективность. Это улучшение биологической эффективности по крайней мере частично связано с амфифильным характером молекул по изобретению.
Объектом изобретения является также способ получения соединений, соответствующих формуле (I), причем этот способ отличается тем, что приводят в реакцию альдегид, отвечающий формуле (II), с гидроксиламином, отвечающим формуле (III), согласно схеме 2 ниже:
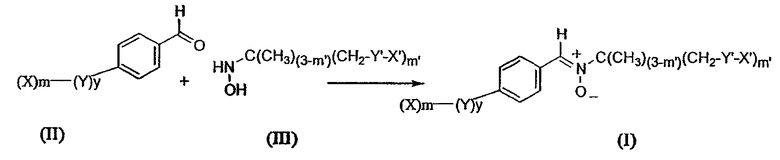
Схема 2
в которой X, y, Y, m, X', m' и Y' имеют то же значение, что и определенные выше.
Соединения формулы (III) получают способом, описываемым в схеме 3 ниже:
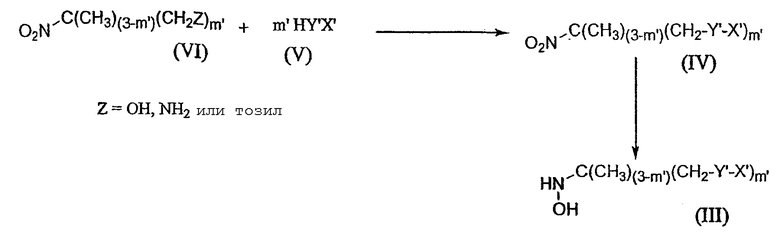
Схема 3
В зависимости от природы липофильной группы схема 3 осуществляется в условиях, которые будут изложены ниже.
a- Гидрофобная одноцепочечная углеводородная или перфторуглеродная часть (фигура 1):
На фигуре 1 показано получение соединений формулы (III), в которой:
m' = 1;
X' = (CH2)2-R с R = C6F13, C8F17, CH3(CH2)n
4 < n <14;
Y' = 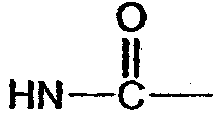 (соединение 5),
(соединение 5), 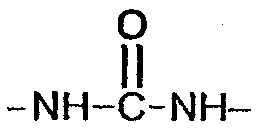 (соединение 6),
(соединение 6), 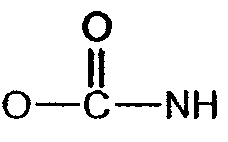 (соединение 2),
(соединение 2), 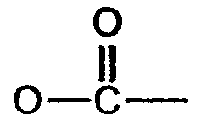 (соединение 1), -S- (соединение 7).
(соединение 1), -S- (соединение 7).
Гидрофобная одноцепочечная часть синтезирована из 2-метил-2-нитропропанола. Спиртовая группа синтона позволяет привить углеводородные и перфторуглеродные цепи непосредственно с помощью связывания сложного эфира реакцией спирта и кислоты в присутствии связующего агента, дициклогексилкарбодиимида и диметиламинопиридина (1).
Спирт может также реагировать с алкилизоцианатом с получением связывания по типу уретана (2).
Спиртовая группа может быть превращена в аминогруппу реакцией с тозилом с последующим замещением нитридом натрия. Реакцией Штаудингера алкилазид преобразуют в амин в присутствии трифенилфосфина и соды.
Этот амин может реагировать с жирной кислотой с получением связывания по амидному типу (5) или с алкилизоцианатом с образованием мочевины (6).
Наконец, тозилат может быть замещен в основной среде тиолом с образованием тиоэфирной связи (7).
Затем нитрогруппу разных гидрофобных синтонов (1-7) восстанавливают в гидроксиламин с помощью 4 эквивалентов реактива Кагана (SmI2) в смеси ТГФ/MeOH или в уксусной кислоте.
Эта реакция описана в работах Girard P., Namy J.L., Kagan H.B. J. Am. Chem. Soc. (1980) 102, 2693-2698 и Namy J.L., Girard P, Kagan H.B. Nouv. J. Chem. (1977) 1, 5.
Эта очень быстрая (3 мин) реакция проводится с переменным выходом от 50 до 100% в зависимости от природы восстанавливаемого нитроалкила.
b- Двухцепочечная гидрофобная углеводородная или перфторуглеродная часть (фигура 2):
На фигуре 2 показано получение соединения формулы (III), в которой:
m'= 2;
X' = (CH2)2-R с R= C6F13, C8F17, CH3(CH2)n
4<n<14
Y' = 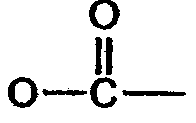 (соединение 8),
(соединение 8), 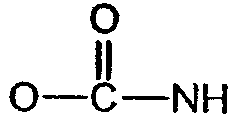 (соединение 9), -S- (соединение 12),
(соединение 9), -S- (соединение 12), 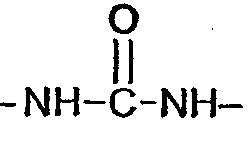 (соединение 14),
(соединение 14),  (соединение 13).
(соединение 13).
Гидрофобные двухцепочечные части синтезированы из 2-нитро-2-метил-1,3-пропандиола. Жирные цепи фиксируются на спиртовых функциональных группах уретановыми (9) или сложноэфирными связями (8). Спиртовые функциональные группы преобразуют в тозилат реакцией с хлоридом тозила. Дитозилат может быть замещен алкилмеркаптаном, чтобы получить тиоэфир (12). Этот дитозилат может быть преобразован в диамин замещением тозилата нитридом натрия и реакцией с трифенилфосфином и основным гидролизом. Этот диамин может реагировать с изоцианатом с получением карбамидной связи (14) или с кислотой с получением амидной связи (13).
Функциональную нитрогруппу "двухантенных" синтонов затем восстанавливают реактивом Кагана с выходом, который может варьировать от 60 до 80% в зависимости от рассматриваемой молекулы.
c- Гидрофильная неионная часть (фигура 3):
На фигуре 3 показано получение соединения формулы (II), в которой:
X означает полярную неионную группу:
Y означает -NH-CH2- (соединение 20), 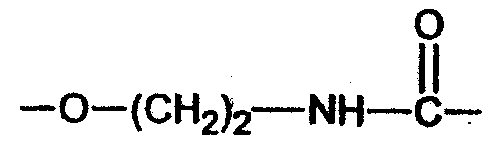 (соединения 21, 22, 23),
(соединения 21, 22, 23),
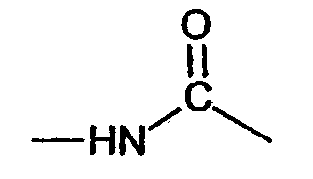 (соединение 24),
(соединение 24),
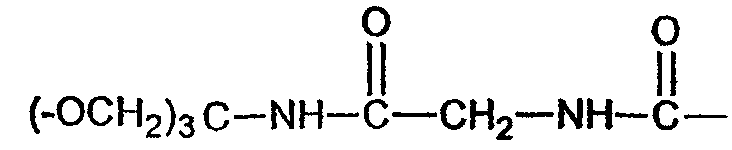 (соединение 25)
(соединение 25)
y=1;
m = 1 (соединения с 20 по 24);
m = 3 (соединение 25).
Гидрофильные неионные головки состоят из сахаров (лактобионамид, галактоза, глюкоза, манноза и т.п.), многоатомных спиртов, гликозилированных или нет (таких, например, как Tris и т.д.), или полиэтиленгликоля. Производные лактобионамида 20 синтезированы из 4-цианобензальдегида и лактобионлактона. После защиты альдегидной функциональной группы ацеталированием (15), а затем восстановлением нитрогруппы полученный амин конденсируют с лактобионлактоном. Ацетилирование спиртовых функциональных групп и удаление защиты у альдегидной функциональной группы избытком ацетальдегида в кислой среде дает полярный синтон 20.
Другие полярные головки синтезированы из 4-карбоксибензальдегида. Глюкозиловые (21), маннозиловые (22), галактозиловые (23) производные получены конденсацией Boc этаноламина с соответствующими ацетобромоглюкозидами (17, 18, 19) в условиях реакции Хельфериха. После удаления защиты у функциональной аминогруппы и конденсации с кислотной группой в присутствии пептидного связующего агента получают 3 полярные гликозилированные головки 21-23.
Пегилированное производное 24 получают в результате конденсации полиэтиленгликоля, функционализованного амином, с кислотной группой 4-карбоксибензальдегида, защищенного ацеталем. Удаление защиты у ацеталя позволяет получить это производное. Наконец, можно получить тригалактозиловое производное 25 конденсацией амина, уже описанного в литературе: Pucci B., Zarif L., Lacombe J-M., Riess J-G., Pavia A.A., Chem. Phys. Lipids (1995) 77, 225-251, с кислотной группой 4-карбоксибензальдегида.
d- Гидрофильная ионная часть (фигура 4):
На фигуре 4 показано получение соединения формулы (II), в которой:
X означает полярную ионную группу,
Y означает -CH2- (соединения 26, 27), 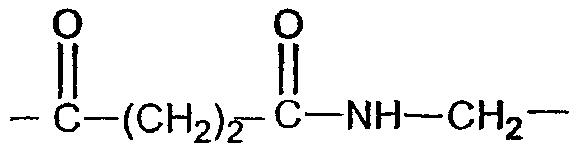 (соединение 28),
(соединение 28),  (соединение 29);
(соединение 29);
y = 1;
m = 1.
Полярные ионные головки состоят из групп четвертичного аммония, оксида амина или карнитина. Аммонийная группа синтезирована из нитрила после восстановления с помощью AlLiH4 и предварительной защиты альдегидной функциональной группы этиленгликоля. Полученный амин перметилируют йодистым метилом в присутствии трибутиламина в ДМФ по способу, описанному в работе Sommer H.Z., Lipp H.I, Jackson L.L. J. Org. Chem. (1971) 36, 824-828.
Кристаллизованный продукт гидролизуют в водном растворе уксусной кислоты, чтобы извлечь производное 26.
Оксид амина 27 получен из того же амина после образования третичного амина в присутствии 2 эквивалентов йодистого метила. Азот окисляют перекисью водорода, 10 объемов в метаноле. После удаления защиты у ацеталя получают соединение 27.
Оксид амина 29 синтезируют по методу McQuade и др. из 4-карбоксибензальдегида, защищенного на его альдегидной функциональной группе ацеталевой группой, и N-этил-N',N'-диметилэтилендиамина. Этот метод был описан в работе McQuade D.T., Quinn M.A., YU S.M., Polans A.S., Krebs M.P., Gellman S.H. Angew. Chem. Int. Ed. (2000) 39, 758-761.
Связывание проводят в присутствии пептидного связующего агента DCC. После окисления функциональной аминогруппы перекисью водорода, 10 объемов, и удаления защиты у ацеталя выделяют соединение 29.
Наконец, производное 28 карнитина получено конденсацией амина 15 с янтарным ангидридом, а затем связыванием с кислотной группой на спиртовой группе карнитина в ДМФ в присутствии DCC. После удаления защиты у функциональной кетальной группы получают продукт 28.
e- Получение одно- (фигура 5) и двухцепочечных (фигура 6) амфифильных нитронов:
Различные амфифильные нитроны получены соединением альдегидной функциональной группы разных полярных синтонов с гидроксиламиногруппой гидрофобных частей. В зависимости от более или менее полярной природы гидрофильных головок, ионные (очень полярные), обозначены I, или неионные гликозилированные (аполярные, так как ацетилированные), обозначены NI, используют полярный протонный (этанол) или апротонный (ТГФ) растворитель. Однако реакция в полярных протонных растворителях более быстрая (2 дня вместо 10 в ТГФ).
Прямоугольник с указанием CH показывает на этих фигурах углеводородную цепь X', в случае необходимости фторированную.
ТГФ применяется с полярными гликозилированными головками, так как он является растворителем, который не вызывает реакцию деацетилирования спиртовых функциональных групп. Все амфифильные гликозилированные нитроны были очищены с помощью ВЭЖХ в обращенной фазе (колонка C18, элюент метанол-вода). Ионные соединения были выделены кристаллизацией. Амфифильные пегилированные нитроны очищены c помощью гель-проникающей хроматографии (сефадекс LH20).
Кроме того, объектом изобретения является использование соединения по формуле (I), такого как определенное выше, в качестве агента против свободных радикалов.
Действительно, было показано, что соединениям согласно настоящему изобретению была придана способность захватывать свободные радикалы, эквивалентная соединениям предшествующего уровня техники.
Это свойство позволяет предусмотреть использование молекул изобретения в различных областях.
- В области терапии продукты по изобретению могут использоваться для профилактики и/или лечения патологий, связанных с оксидативным стрессом и образованием реакционно-способных форм кислорода.
Таким образом, объектом изобретения являются фармацевтические композиции, содержащие соединение согласно изобретению в фармацевтически приемлемом носителе. Его объектом является применение соединения согласно изобретению для получения лекарственного средства, предназначенного для профилактики и/или лечения эффектов, вызванных свободными радикалами.
Объектом изобретения является также применение соединения по изобретению для получения фармацевтической композиции, предназначенной для профилактики и/или лечения патологий, связанных с оксидативным стрессом и с образованием реакционно-способных форм кислорода, в частности иммунных и воспалительных заболеваний, синдрома ишемии-реперфузии, атеросклероза, болезни Альцгеймера, болезни Паркинсона, болезни Гентингтона, поражений, вызванных ионизацирующим или УФ-излучением, раковых заболеваний, старения клеток.
Продукты по изобретению могут быть введены любым путем, известным специалисту, в частности путем внутривенных или внутримышечных инъекций, пероральным или кожным введением. Они могут использоваться индивидуально или в сочетании с другими активными веществами. Их дозировку и суточную дозу устанавливают в зависимости от активности, измеренной для конкретной молекулы, и в зависимости от массы тела пациента.
- В области косметики соединения по изобретению могут использоваться для профилактики и/или лечения эффектов старения, а также эффектов, вызванных солнечным излучением.
Таким образом, объектом изобретения является также косметическая композиция, содержащая соединение по изобретению в косметически приемлемом носителе.
Указанная композиция может быть предназначена для нанесения на кожу или на запретные покровы (ногти, волосы).
Она может быть представлена в виде водного или масляного раствора, эмульсии "вода в масле" или "масло в воде", тройной эмульсии, мази.
Соединения по изобретению могут быть введены в любую косметическую композицию, для которой желательна активность против радикалов: крем для кожи, продукт для защиты от солнца, средство для снятия макияжа, маска для кожи или волос, шампунь, средство для макияжа, такое как губная помада, румяна, основа для краски, лак для ногтей и т.д.
- В области органического синтеза соединения по изобретению могут применяться как ловушки свободных радикалов в радикальных реакциях.
Благодаря своей растворимости в различных средах соединения по изобретению легко использовать и они могут быть использованы в самых разных условиях.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
I- Биологическая оценка:
Соединение A 1 использовали для проведения экспериментов по захвату свободных радикалов. Несколько соединений согласно изобретению были протестированы in vitro на их биологическую антиоксидантную и антирадикальную активность.
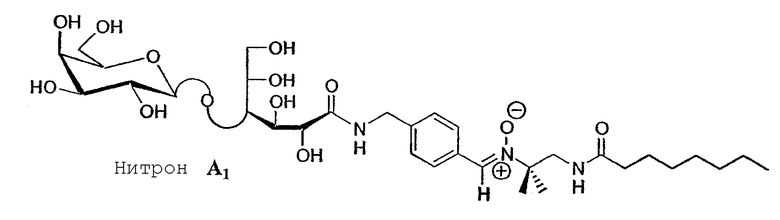
1- Измерение способности улавливать радикальные соединения
Эксперименты по улавливанию свободных радикалов с центром на углероде (радикалы CH3 и CО2) и на кислороде (радикал OH), проведенные на соединении A 1, показали, что функционализация PBN не влияет на способность этих молекул улавливать радикальные соединения. В случае свободных радикалов с центром на углероде можно было наблюдать ЭПР-сигналы, характерные для этих радикальных соединений, как это показано на фигуре 7.
Зато при образовании в системе гидроксильных радикалов регистрировались сигналы ЭПР, характерные для захвата радикалов с центром на углероде. Это вызвано захватом нитроном углеродсодержащих радикалов, полученных на полярных головках реакцией радикалов OH с водородами сахаров.
2- Измерение in vitro антиоксидантной и антирадикальной биологической активности
a- Оценка антиапоптозной активности на нейронах коры головного мозга крыс путем анализа энзиматической активности каспазы III.
Эти предварительные тесты были проведены на амфифильном гликозилированном углеводородном производном нитрона (нитрон A 2). Его активность против апоптоза сравнивали с двумя имеющимися в продаже нитронами, PBN и DMPO.
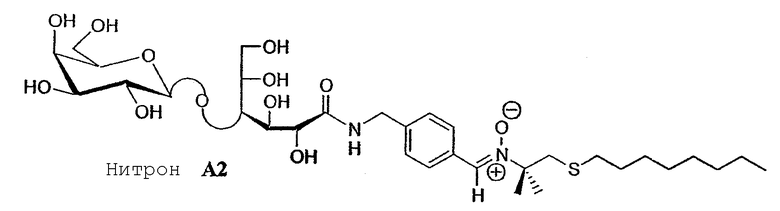
Нейронные клетки крыс подвергали интоксикации в течение 20 мин перекисью водорода в концентрации 100 мкМ на 8-дневной культуре. Эта добавка перекиси водорода вызывает явление апоптоза, как это было описано в работе Whittemore E.R., Loo D.T., Cotman C.W. Neuroreport (1994) 5, 1485-1488 (проверено положительным контролем апоптоза добавлением стауроспорина), оцениваемого колориметрическим анализом при 405 нм специфического фермента его метаболизма - каспазы III, по сравнению с максимальной интоксикацией контроля (описано ранее в работе Nicholson D.W., Ali A., Thombury N.A., Vaillancourt J.P., Ding C.H., Gallant M., Griffin P.R., Labelle M., Lazebnik Y.A., Munday N.A., Raju S.M., Smulson M.E., Yannin T., Yu V.I., Miller D.K. Nature (1995) 376, 37-43).
Различные тестируемые молекулы инкубировали в течение 20 часов при различных нетоксичных концентрациях (10, 100, 200 мкM) перед интоксикацией перекисью водорода. После промывки и сушки в сушильном шкафу клетки лизировали перед колориметрическим анализом. Амфифильный нитрон A 2 обладает значительной цитотоксичностью, начиная с 400 мкМ.
Полученные результаты (показанные на фигуре 8) ясно продемонстрировали очень отчетливое снижение активности каспазы III после интоксикации перекисью водорода концентрацией 100 мкМ в присутствии амфифильного нитрона A 1. Эта активность проявляется гораздо слабее, чем нормальная активность каспазы III на нейронных клетках, не подвергавшихся интоксикации.
Результаты ясно указывают на повышенный уровень защиты по сравнению с измеренным для имеющихся в продаже нитронов PBN и DMPO.
b- Оценка нейропротективной эффективности на совместных культурах нерв-мышца
Протективная оценка этих амфифильных нитронов была измерена на совместных культурах нервы-мышцы после интоксикации перекисью водорода в течение 30 мин.
Мышечные клетки человека, взятые из здоровых поперечно-полосатых мышц, выделяли миграцией сателлитных клеток в подходящую культуральную среду. Эти клетки соединялись в не способные к сокращению миофибры в культуральной среде. Эксплантаты спинного мозга крысиных эмбрионов наносились на мышечные клетки.
Через три недели все мышечные волокна, близкие к эксплантатам, не сокращались и обладали зрелыми нейромышечными связями. После созревания эти культуры отбирали и снимали на видеокамеру, соединенную с микроскопом. Молекулы типа
A (A 1 , A 2 , A 3 , A 4 ) и B (B 1 ) инкубировали в течение 20ч00 при концентрациях 100 и 200 мкМ. Затем клетки подвергали интоксикации в течение 30 мин H2О2 концентрацией 800 мМ, а затем промывали. Через 24ч00 и 48ч00 после того, как возник этот оксидативный стресс, клетки обследовали и снимали на камеру.
Через 20ч00 инкубации оказалось, что ионное соединение B 1 карбоксилатного типа является цитотоксичным и вызывает быструю деградацию мышечных клеток. Другие соединения нетоксичны, но вызывают задержку или ослабление мышечного сокращения, какой бы ни была используемая концентрация (100 и 200 мкМ). Зато для перфторированных соединений A 3 и A 4 при концентрации 100 мкМ и углеводородных соединений A 1 и A 2 при 100 и 200 мкМ наблюдается полное или частичное восстановление сокращений через 48ч00 после интоксикации перекисью водорода (таблица 1). Другие соединения защищают клетки от деградации, но не позволяют восстановление сокращений.
Затем было осуществлено количественное охарактеризование состояния апоптоза после лизиса клеток и центрифугирования при добавлении некоторого количества фрагментов ДНК с помощью устройства "cell death detection ELISA" в супернатанте. После ферментативного проявления были измерены оптические плотности при 405 нм с помощью устройства для считывания планшетов (фигура 9).
Результаты ясно показывают, что все исследованные молекулы, за исключением ионного производного B 1, защищают клетки от апоптоза, вызванного добавлением перекиси водорода. При концентрации 200 мМ карбоксилатное производное B 1 имеет очень слабый ложный сигнал апоптоза, который вызван до клеточного лизиса выделением фрагментов ДНК в культурную среду после смертью клеток в культуре, обработанной этим нитроном.

Измерение сократительной активности клеточных культур нерв-мышца через 48ч00 после интоксикации H2О2 в концентрации 800 мкM
Критерий обнаружения сократительной активности мышечных волокон
c. Оценка антиоксидантной активности на клеточных линиях фибробластов, страдающих сильным дефицитом комплекса V дыхательной цепи: определение клеточной жизнестойкости по тесту MTT
Опыты проводились на клеточных линиях фибробластов, отличающихся мутацией гена NARP, кодирующего белок (субъединица 6) комплекса V митохондриальной цепи. Эти клетки характеризуются аномальной сверхпродукцией фермента супероксиддисмутазы, что позволяет предположить, что генетический дефицит вызывает рост производства пероксидного радикала. Эта сверхпродукция пероксидных радикалов вызывает ускоренный процесс апоптоза клеток (Geromel V., Kadhom N., Cebalos-Picot I., Ouari O., Polidori A., Munnich A., Rötig A., Rustin P. Hum. Mol. Genet. (2001) 10, 1221-1228).
Культуры фибробластов были приготовлены из биопсии кожи двух индивидуумов (контроль) и одного пациента - носителя мутации NARP. Клетки культивировали в среде RPMi 1640 (выпускаемой компанией Life technologies SARL, Cergy Pontoise, Франция), в которую добавили глутамакс (446 мг/л): 10% эмбриональной сыворотки теленка не диализованной, 100 мкг/мл стрептомицина, 100 IU/мл пенициллина, 200 мкМ уридина, 2,5 мМ пирувата натрия. Для тестов на цитотоксичность клетки были засеяны до плотности 3000 клеток на лунку в микрочашке Петри при 37°C и
при 5% CО2. Чтобы вызвать оксидативный стресс, клетки после 24 часов подвергают гипогликемии путем замены глюкозы галактозой в концентрации 10 мМ (селективная среда, обозначенная "ms" на фигурах с 10a по 10f). Через 24 часа клетки были подвергнуты действию в течение 48 часов и 72 часов возрастающих концентраций различных тестируемых соединений в селективной среде, предназначенной для дыхательных клеток (среда RPMi 1640 без глюкозы). В конце сравнения все исследования были проделаны на клетках, собранных после удвоения популяции.
Антиоксидантная активность амфифильных нитронов оценивалась по измерению их способности защищать клетки от апоптоза по тесту MTT.
Тест MTT является колориметрическим методом, позволяющим определить число жизнеспособных клеток в опытах на пролиферативность и цитотоксичность. Лунки оставляли для инкубации с 20 мкл раствора MTT (5 мг/мл в PBS) в течение 1 часа при 37°C. Затем было добавлено 200 мкл изопропанола для экстрагирования формазана MTT, и был измерен коэффициент поглощения каждой лунки при 540 нм с помощью аппарата с автоматическим считывающим устройством.
Результаты, полученные при колориметрическом анализе MTT, показаны на фигурах с 10a по 10f. На этих фигурах соединение A 5 является нитроном типа A, в котором R=OCONH(CH2)5CH3, а соединение H отвечает нижеследующей формуле:
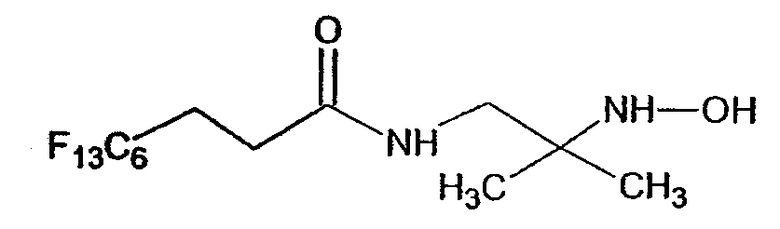
Соединение H
Тест показывает способность TA1PBN в концентрации 50 мкМ защищать клетки NARP от клеточной смерти через апоптоз. Эти результаты хорошо подтверждают, таким образом, анализ, проведенный ранее на TA1PBN (Geromel V., Kadhom N., Cebalos-Picot I., Ouari O., Polidori A., Munnich A., Rötig A., Rustin P. Hum. Mol. Genet. (2001) 10, 1221-1228). Перфторуглеродное соединение B 1 оказывается также эффективным, начиная с концентрации 100 мкМ. Следует отметить, что перфторуглеродное соединение A 4 и углеводородные соединения A 1 , A 2 и A 5 не эффективны на этой клеточной модели. Это отсутствие эффективности можно отнести за счет малой гидрофобности жирных углеводородных цепей. Перфторированное соединение A 4 имеет длину цепи меньше, чем у соединения A 3. Наконец, следует отметить, что TA1PBN имеет более длинную перфторированную цепь C8F17, чем у соединения A 3 (цепь C6F13). Это может объяснить различие в эффективности при обработке клеток NARP этими двумя амфифильными нитронами. Тесты осуществляются на производных, аналогичных A 3, имеющих фторированную
цепь C8F17. В заключении укажем, что степень гидрофобности этих нитронов, по-видимому, играет решающую роль для их биологической активности. Она, возможно, должна быть обусловлена способностью переносить нитрон через цитопластическую мембрану и, возможно, в митохондриальную полость.
II- Примеры синтеза
1. Синтез амфифильного одноцепочечного углеводородного гликозилированного нитрона
a. Синтез 4-метилбензилсульфонат-2-метил-2-нитропропила E3

9,6 г хлорида тозила (0,050 моль - 1,2 экв.) растворяют в 30 мл пиридина. Затем по каплям добавляют 5 г 2-метил-2-нитропропанола (0,042 моль - 1 экв.), растворенного в 30 мл дихлорметана. Во время добавления среду поддерживают при 0°C, а затем при комнатной температуре в течение 48 часов. Реакционную смесь впрыскивают в 150 мл ледяной воды при интенсивном перемешивании. Водную фазу экстрагируют 3 раза, используя по 50 мл дихлорметана. Органические фазы соединяют, промывают 3 раза 3н. HCl порциями по 75 мл, затем 2 раза рассолом порциями по 75 мл рассола, сушат над Na2SО4, наконец, упаривают при пониженном давлении. После перекристаллизации из смеси этилацетат/циклогексан получают соединение E3 в виде легкого белого порошка (9,55 г - 0,035 моль - 83%). Rf: 0,35 (циклогексан/этилацетат 8:2). Т. плавл. = 74-75,5°C.
1H-ЯМР (250 МГц, CDCl3): δ 7,76 (2H, д, J=8,5 Гц, H аром.), 7,37 (2H, д, J=8,5 Гц, H аром.), 4,27 (2H, с, CH2-O), 2,46 (3H, с, CH3 тозила), 1,56 (6H, с, CH3 трет-бутила)
13С-ЯМР (62,86 МГц, CDCl3): 8 146,1 (CIV аром.), 132,6 (CIV аром.), 130,7 и 128,6 (CH аром.), 86,3 (CIV), 73,2 (CH2-O), 23,5 (CH3 трет-бутила), 22,3 (CH3 тозила)
ИК (KBr, см-1): ν(CH аром.) = 3059 и 3005, ν(NO2) = 1543
b. Синтез 1-октансульфанил-2-метил-2-нитропропила E7a

В атмосфере аргона диспергируют 4,1 г трет-бутилата натрия (0,039 моль - 2 экв.) в 30 мл безводного ДМФ. После 20 минут перемешивания по каплям добавляют с помощью бромовой колбы 6,4 мл октантиола (0,039 моль - 2 экв.), растворенного в 10 мл ДМФ. Среда постепенно приобретает белый молочный вид, и через 10 минут медленно добавляют 5 г E3 (0,0183 моль - 1 экв.), растворенного в 20 мл ДМФ. Реакционную среду доводят до 50°C в потоке аргона в течение 4 часов. Смесь впрыскивают в 400 мл ледяного рассола, затем экстрагируют 5 раз, используя по 50 мл циклогексана. Органическую фазу промывают 2 раза рассолом порциями по 100 мл, сушат над Na2SО4 и упаривают при пониженном давлении. После очистки флэш-хроматографией на силикагеле (элюент: циклогексан/дихлорметан от 9:1 до 8:2) получают соединение E7a (4,4 г - 0,0178 моль - 97 %) в виде масла. Rf: 0,65 (циклогексан/этилацетат 8:2).
1Н-ЯМР (250 МГц, CDCl3): δ 3,04 (2H, с, CIV-CH2-S), 2,52 (2H, т, J=7,25 Гц, CH 2-СН2-S), 1,64 (6H, с, CH3 трет-бутила), 1,54 (2H, к.т., J=7,25 Гц, CH 2-S), 1,40-1,20 (10H, м, CH2 цепи), 0,87 (3H, т, J=7 Гц, CH3 цепи).
13C-ЯМР (62,86 МГц, CDCl3): δ 87,4 (CIV), 41,5 (CIV -СH2); 33,2 (CH2-S), 30,8, 28,8, 28,1 и 27,7 (CH2 цепи), 24,4 (CH3 трет-бутила), 21,6 (CH2 цепи), 13,1 (CH3 цепи).
ИК (KBr, см-1): ν(NO2) = 1543
c. Синтез N-(1,1-диметил-2-октилсульфанилэтил)-гидроксиламина E7b
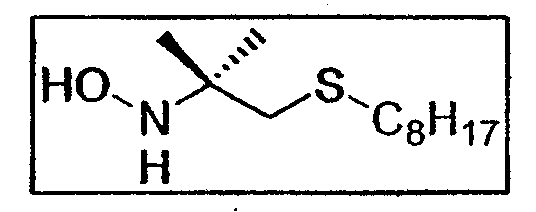
0,247 г нитросоединения E7a (1 ммоль - 0,25 экв.) растворяют в 6 мл смеси ТГФ/MeOH (2:1), предварительно продутой аргоном. Этот раствор за один раз добавляют в реактив Кагана (4 экв.) в инертной атмосфере. За реакцией, почти мгновенной, следует появление зеленовато-серого окрашивания (исчезновение соединения SmI2 с появлением SmI3). После 15 минут перемешивания в реакционную среду добавляют 20 мл 10%-ного раствора Na2S2O3. ТГФ выпаривают при пониженном давлении. Водную фазу разбавляют в 2 раза, затем 3 раза экстрагируют, используя по 30 мл этилацетата. Органическую фазу 2 раза промывают дистиллированной водой порциями по 30 мл, затем упаривают при пониженном давлении. Очистка хроматографией на силикагеле (элюент: циклогексан/этилацетат от 8:2 до 7:3) приводит к гидроксиламину E7b (164 мг - 0,7 ммоль - 70%) в виде полупрозрачного масла.
Также выделяют 50 мг исходного соединения E7a , что позволяет определить степень конверсии в 88%.
1H-ЯМР (250 МГц, ДМСО): δ 7,09 (1H, с, NH), 5,3 (1H, шир.с, OH), 2,63 (2H, с,
CIV-CH2-S), 2,55 (2H, т, J=7,2 Гц, CH2-S), 1,64 (6H, с, CH3 трет-бутила), 1,54 (2H, квинтет, J=6,7 Гц и J=7,2 Гц, CH 2-CH2-S), 1,40-1,20 (10H, м, CH2 цепи), 0,87 (3H, т, J=7 Гц, CH3 цепи)
13C-ЯМР (62,86 МГц, CDCl3): δ 57,2 (CIV), 40,8 (CIV-CH2), 33,2 (CH2-S), 31,2, 29,4, 28,6 и 28,5 (CH2 цепи), 23,7 (CH3 трет-бутила), 22,0 (CH2 цепи), 13,9 (CH3 цепи)
ИК (KBr, см-1): ν(NH) = 3246
d. Синтез углеводородного нитрона A2
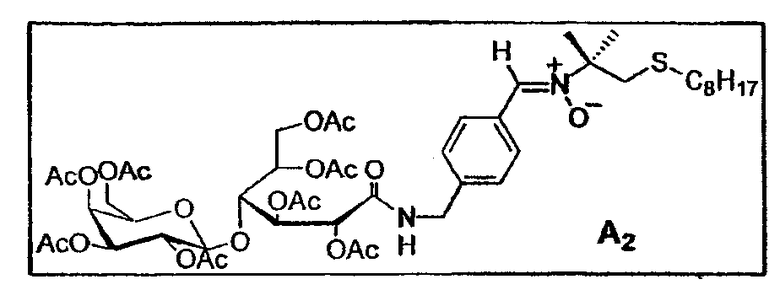
0,5 г гликозилированного альдегида (Ouari O., Chalier F., Pucci B., Tordo P., J. Chem. Soc., Perkin Trans 2 (1998), 2299) (0,615 ммоль - 1 экв.) растворяют в атмосфере аргона в 10 мл безводного и дегазированного ТГФ. Добавляют 0,1 г гидроксиламина E7b (0,430 ммоль - 0,7 экв.), растворенного в 2 мл ТГФ, а также на кончике шпателя молекулярные сита 4 Е. Реакционную смесь доводят до 60°C в атмосфере аргона в отсутствие света. Оба дня добавляли 50 мг гидроксиламина (0,215 ммоль - 0,35 экв.) и на кончике шпателя молекулярные сита 4 Е. Продвижение реакции оценивается по ККМ, и после добавления через 8 дней 1,75 экв. гидроксиламина реакционную смесь фильтруют на целите. После выпаривания испарителя при пониженном давлении неочищенный реакционный продукт очищают флэш-хроматографией на силикагеле (элюент: этилацетат/циклогексан 7:3). Дополнительная очистка проводится на гель-проникающей смоле LH-20 (элюент: метанол/дихлорметан 1:1) и приводит к чистому нитрону A 2 (313 мг - 0,304 ммоль - 50%) и 116 мг фракции, содержащей исходный альдегид (в отношении приблизительно 1/3, определено по 1H-ЯМР). Т. плавл. = 70°C (разложение). [α]D = + 17,8 (c, 1, CH2Cl2).
1H-ЯМР (250 МГц, CDCl3): δ 8,24 (2H, д, J=8,1 Гц), 7,49 (1H, с, CH=N(O)), 7,25 (2H, д, J=8,1 Гц), 6,57 (1H, м, NH), 5,67 (1H, д, J=6,6 Гц, H-2), 5,60 (1H, двойной дуплет, J=3,8 Гц и J=5,8 Гц, H-3), 5,37 (1H, д, J=3 Гц, H-4'), 5,18 (1H, двойной дуплет, J=2,5 Гц и J=10,3 Гц, H-2'), 5,08 (1H, м, H-5), 4,98 (1H, двойной дуплет, J=3,4 Гц и J=10,4 Гц, H-3'), 4,65 (1H, д, J=7,9 Гц, H-1'), 4,62-4,45 (2H, м, 1H-6a и H-7a), 4,42-4,30 (2H, м, H-4 и H-7b), 4,23-3,98 (3H, м, H-6b, H-6'a и H-6'b), 3,90 (1H, т, J=6,5 Гц), 3,00 (2H, с, CIV-CH2-S), 2,41 (2H, т, J=7,25 Гц, CH2-S), 2,13, 2,05, 2,02, 2,01, 1,98, 1,95 (24H, 6с,CH3-CO), 1,61 (6H, с, CH3 трет-бутила), 1,43 (2H, м, CH 2-CH2-S), 1,3-1,1 (10H, м, CH2 цепи), 0,82 (3H, т, J=6,6 Гц, CH3 цепи).
13C-ЯМР (62,86 МГц, CDCl3): δ 170,4, 170,4, 170,0, 170,0, 169,9, 169,7, 169,7, 169,5, 169,2 (CH3-CO), 167,1 (CO-NH), 139,8 (CIV аром.), 131,0 (CH=N(O)), 130,1 (CIV аром.), 129,2, 127,5 (CH аром.), 101,7 (CH-1'), 77,3 (CH-4), 73,4 (CIV), 71,7 (CH-2), 70,9 (CH-5' и CH-3'), 69,7 (CH-5), 69,1 (CH-3), 68,9 (CH-2), 66,8 (CH-4'), 61,6, 60,9 (CH2-OAc), 42,9 (CH2-NH), 42,4 (CIV-CH2-S), 33,3 (CH2-S), 31,7, 29,9, 29,0, 29,0, 28,6 (CH2 цепи), 25,7 (CH3 трет-бутил), 22,5 (CH2 цепи), 20,8, 20,7, 20,6, 20,6, 20,6, 20,5, 20,5, 20,4 (CH3-CO), 14,0 (CH3 цепи).
МС FAB+ (1027,1 г/моль): [M+H]+ = 1027 (5%), [M+Na]+ = 1049 (10%),[C12H25S]+ = 201 (70%).
После деацетилирования сахаров по методу Zemplen получают незащищенный продукт:
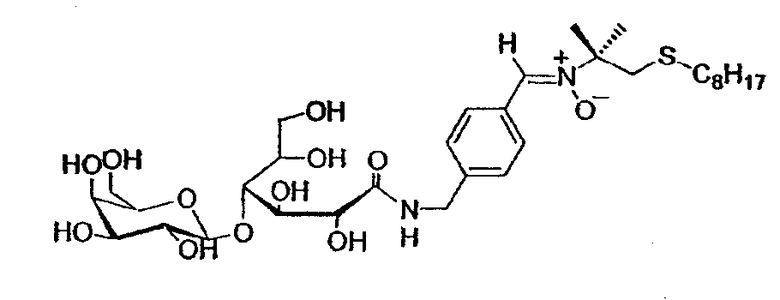
Т. плавл.=115°C (разложение)
Rf: 0,52 (этилацетат/метанол/вода 7:2:1)
[α]D = +17,2 (0,25c, 1, CH3OH)
1H-ЯМР (250 МГц, CD3OD): δ 8,28 (2H, д, J=8,25 Гц), 7,82 (1H, с, CH=N(O)), 7,42 (2H, д, J=8,25 Гц), 4,65-4,35 (4H, м, CH2-NH, H-1', H-2), 4,25 (1H, м, H-3), 4,00-3,35 (10H, м, H-4, H-5, CH2-OH, H-4', H-5', H-3' и H-2'), 3,01 (2H, с, CH2-S), 2,43 (2H, т, J=7,3 Гц, CH2-S), 1,61 (6H, синглет, CH3 трет-бутила), 1,44 (2H, м, CH2), 1,3-1,1 (10H, м, CH2), 0,87 (3H, т, J=6,9 Гц)
13C-ЯМР (62,86 МГц, CD3OD): δ 175,3 (CO-NH), 143,4 (CIV аром.), 136,0 (CH=N(O)), 131,1 (CH аром.), 130,6 (CIV аром.), 128,3 (CH аром.), 105,8 (CH-1'), 83,3 (CH-4), 77,2 (CH-5'), 74,8 (CIV), 74,6 (CH-3' или CH-2'), 74,1 (CH-2), 73,2 (CH-5), 72,8 (CH-3' или CH-2'), 72,5 (CH-3), 70,4 (CH-4'), 63,8, 62,7 (CH2-OH), 43,5, 43,0 (CH2-NH и CH2-S), 34,2 (CH2-S), 32,9, 31,0, 30,3, 30,2, 29,7 (CH2), 26,0 (CH3 трет-бутила), 23,7 (CH2), 14,4 (CH3)
УФ (MeOH, нм): λmax = 299
МС FAB+ (690,8 г/моль): следы [M+H]+, [M+Na]+ = 713 (2,5%), [M+K]+ = 729 (1,5%), [C12H25S]+ = 201 (65%)
МС FAB- (690,8 г/моль): [M-H]- = 689 (очень слабый)
ВЭЖХ (Microsorb C18 - 21,4 мм / 250 мм): вр. удерж. = 11,4 мин
Градиент от 70 MeOH - 30 H2O до 85 MeOH - 15 H2О от t= 0 до t= 5 мин
Изократическое элюирование 85 MeOH -15 H2О, начиная с t =5 мин
Расход 0,6 мл /мин
2. Синтез фторуглеродного нитрона A 4
a. Синтез 1-азидо-2-метил-2-нитропропана
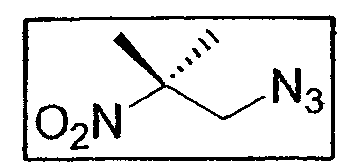
6 г соединения E3 (0,0218 моль - 1 экв.) и 2,3 г нитрида натрия (0,0353 моль - 1,6 экв.) приводят в реакцию в 20 мл ДМФ при активации ультразвуком (большой зонд - импульс 1 сек/покой 2 сек - амплитуда 90%), охлаждая среду ледяной баней. После 3 часов обработки звуком и полного исчезновения исходного продукта реакционную смесь помещают в 50 мл дихлорметана, затем подвергают сильному перемешиванию на 300 мл ледяного рассола. Водную фазу экстрагируют 2 раза, используя по 50 мл дихлорметана, затем собирают органическую фазу, промывают 2 раза рассолом порциями по 75 мл, сушат над Na2SО4 и упаривают при пониженном давлении. После удаления при 50°C в вакууме лопастного насоса остаточных следов ДМФ получают конечное соединение (2,66 г - 0,0185 моль - 85%) в виде желтого прозрачного масла, очень жидкого.
1H-ЯМР (250 МГц, CDCl3): δ 3,74 (2H, с, CH2-N3), 1,60 (6H, с, CH3 трет-бутила)
13C-ЯМР (62,86 МГц, CDCl3): δ 86,7 (CIV), 58,3 (CH2-N3), 23,9 (CH3 трет-бутила)
ИК (KBr, см-1): ν(N3) = 2111, ν(NO2)= 1546
b. Синтез 2-метил-2-нитропропиламина E4
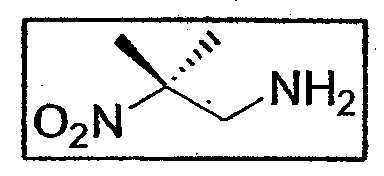
4,08 г азида (0,0283 моль - 1 экв.) растворяют в потоке азота в 10 мл безводного и дегазированного ТГФ. К азиду по каплям добавляют 11,3 г трифенилфосфина (0,0431 моль -1,50 экв.), растворенного в 30 мл ТГФ. Происходит интенсивное выделение газов. После 2 часов перемешивания при комнатной температуре в атмосфере азота добавляют 20 мл 2н. водного раствора соды и среду оставляют на 24 часа. ТГФ выпаривают при пониженном давлении, водную фазу подкисляют до pH 1 добавлением 20 мл 3н. HCl, затем экстрагируют 2 раза, используя по 30 мл этилацетата. Затем водную фазу подщелачивают добавлением твердой соды до pH 10, затем 3 раза экстрагируют, используя по 30 мл дихлорметана. Органическую фазу 2 раза промывают водой порциями по 30 мл, сушат над Na2SO4 и упаривают при пониженном давлении. Получают амин E4 (2,90 г - 0,0245 моль - 87%) в виде желтого масла.
1H-ЯМР (250 МГц, CDCl3): δ 3,07 (2H, с, CH2-NH2), 1,57 (6H, с, CH3 трет-бутила)
13C-ЯМР (62,86 МГц, CDCl3): δ 89,4 (CIV), 51,1 (CH2-NH2), 23,6 (CH3 трет-бутила)
Учитывая его нестабильность, для охарактеризования и сохранения авторы синтезировали соответствующий гидрохлорид аммония.
Амин вводят в 60 мл простого эфира, через который пропускают в виде пузырьков газообразный HCl в течение 10 минут. Среду выдерживают 2 часа при -20 °C, затем фильтруют при пониженном давлении. После удаления следов растворителя лопастным насосом получают гидрохлорид аммония E4 (3,75 г - 0,0243 моль - количественный выход) в виде белого порошка.
1H-ЯМР (250 МГц, D2O): δ 3,62 (2H, с, CH 2-NH3 +Cl-), 1,72 (6H, с, CH3 трет-бутила)
13C-ЯМР (62,86 МГц, D2O): δ 87,0 (CIV), 46,9 (CH2-NH3 +Cl-), 24,8 (CH3 трет-бутила)
ИК (KBr, см-1): ν(NO2)= 1541
c. Синтез 4,4,5,5,6,6,7,7,8,8,9,9,9-тридекафлуорононаноил- (2-метил-2-нитропропил)амида E5a
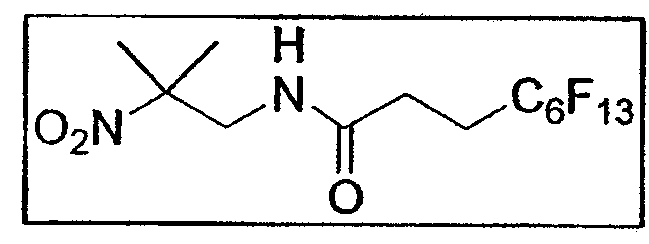
В атмосфере азота 2,47 г растворяют DCC (0,0120 моль - 1,2 экв.) и HOBt на кончике шпателя в 10 мл безводного дихлорметана. 1,41 г амина E4 (0,012 моль - 1,2 экв.), растворенного в 10 мл дихлорметана, добавляют в среду. Раствор дегазируют в течение нескольких минут, затем однократно добавляют 3,86 г фтористого водорода (0,0098 моль - 1 экв.), растворенного в 30 мл этилацетата. Через 36 часов перемешивания реакционную смесь фильтруют, органическую фазу промывают соответственно 2 раза 1н. HCl порциями по 50 мл, 2 раза рассолом порциями по 50 мл, затем сушат над Na2SО4 и упаривают при пониженном давлении. Очистка хроматографией на силикагеле (элюент: циклогексан/этилацетат от 8:2 до 7:3) приводит к соединению E5a (4,4 г - 8,94 ммоль - 91%) в виде белого порошка. Т. плавл. = 87,3-88,8°C. Rf: 0,37 (циклогексан/этилацетат 8:2).
1H-ЯМР (250 МГц, CDCl3): δ 6,11 (1H, масс., NH), 3,76 (2H, д, CIV-CH 2-NH, J=6,75 Гц), 2,55-2,42 (4H, мас., CH2-CH2-Rf), 1,58 (6H, мас., CH3 трет-бутила)
13C-ЯМР (62,86 МГц, CDCl3): δ 170,5 (CO), 88,7 (CIV), 46,1 (CH2-NH), 24,1 (CH3 трет-бутила)
19F-ЯМР (235 МГц, CDCl3): δ -81,1 (CF 3, синглет), -114,8 (CF2-CH2, синглет), -122,1, -123,1 и -123,8 (CF 2, синглет), -126,4 (CF 2-CF3)
ИК (KBr, см-1): ν(NH) = 3280, ν(С-О) = 1664, ν(NО2) = 1547, ν(CF2)= 1246
d. 4,4,5,5,6,6,7,7,8,8,9,9,9-тридекафлуорононаноил-(2-гидроксиамино-2-метилпропил)амид E5b
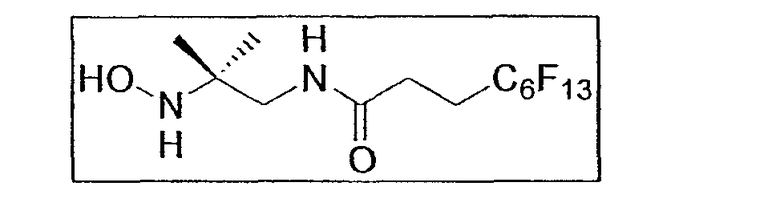
Экспериментальная процедура аналогична использованной для первого гидроксиламина E7b . 1,71 г нитросоединения E7a (3,5 ммоль - 0,25 экв.), растворенного в 21 мл смеси ТГФ/MeOH, добавляют к 140 мл реактива Кагана.
После обработки и очистки хроматографией на силикагеле (элюент: этилацетат/метанол от 10:0 до 9,5:0,5) получают гидроксиламин E7b (0,82 г - 1,7 ммоль - 49%) в виде белого порошка.
Также выделяют 0,56 г исходного соединения E7a , что позволяет определить степень конверсии в 72%. Т. плавл. = 110,5-112,3°C. Rf: 0,58 (этилацетат/метанол 95:5).
1H-ЯМР (250 МГц, ДМСО): δ 7,79 (1H, т, J=6,0 Гц, NH), 6,89 (1H, с, NH), 5,23 (1H, шир.с, OH), 3,05 (2H, д, J=6,0 Гц, CH 2-NH), 2,48 (4H, м, CH2-CH2-Rf), 0,86 (6H, с, CH3 трет-бутила)
13C-ЯМР (62,86 МГц, ДМСО): δ 169,7 (CO), 56,9 (CIV), 44,7 (CIV-CH2); 22,4
(2 CH3 трет-бутила)
19F-ЯМР (235 МГц, ДМСО): δ -80,0 (CF3, синглет), -113,4 (CF2-CH2, синглет), -121,5, -122,5 и -123,0 (CF2, синглет), -125,6 (CF 2-CF3, синглет)
e. Синтез фторуглеродного нитрона A4
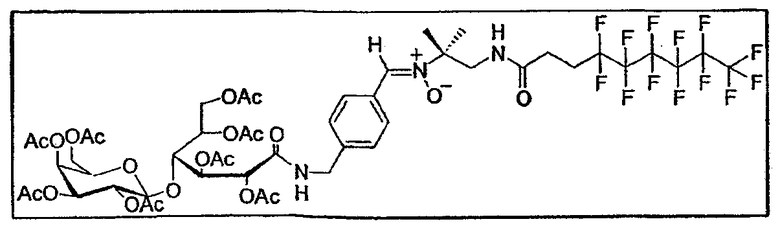
Экспериментальная процедура идентична использованной для первого нитрона.
0,61 г альдегида (0,75 ммоль - 1 экв.) приводят в реакцию с гидроксиламином E7b (0,26 г - 0,7 экв.) в 15 мл ТГФ. Через 10 дней перемешивания и дополнительного добавления 0,35 г гидроксиламина (0,732 ммоль - 0,98 экв.) реакцию останавливают.
Очистку проводят флэш-хроматографией на силикагеле (элюент: этилацетат/метанол от 10:0 до 95:5) и гель-проникающей хроматографией на смоле LH-20 (элюент: метанол/дихлорметан 1:1). Получают чистый нитрон A 4 (0,564 г - 0,443 ммоль - 60%) в виде белой пены. Выделяют также исходный альдегид (115 мг - 0,142 ммоль), что позволяет определить степень конверсии в 73%. Т. плавл. = 95°C (разложение).
1H-ЯМР (250 МГц, CDCl3): δ 8,21 (2H, д, J=8,1 Гц), 7,49 (1H, с, CH=N(O)), 7,31 (2H, д, J=8,1 Гц), 6,95 (1H, т, J=6 Гц, NH), 6,76 (1H, т, J=6 Гц, NH), 5,45-3,80 (15H), 3,69 (2H, д, J = 6,0 Гц, CIV-CH2-NH), 2,70-2,35 (4H, м, CH2-CH2-Rf), 2,17, 2,16, 2,08, 2,07, 2,06, 2,05, 2,04, 1,98 (24H, 8 с, CH3-CO), 1,60 (6H, с, CH3 трет-бутила)
13C-ЯМР (62,86 МГц, CDCl3): δ 170,6 (CO-NH + CH3-CO), 170,4, 170,2, 170,1, 170,0, 169,8, 169,7, 169,3 (CH3-CO), 167,3 (CO-NH), 140,6 (CIV аром.), 131,5 (CH=N(O)), 129,8
(CIV аром.), 129,4, 127,7 (CH аром.), 101,9 (CH-1'), 77,5 (CH-4), 73,4 (CIV), 71,7 (CH-2), 71,0 (CH-5' и CH-3'), 70,0 (CH-5), 69,3 (CH-3), 69,1 (CH-2), 66,9 (CH-4'), 61,8, 60,9 (CH2-OAc), 47,3, 43,1 (CH2-NH), 27,0 (), 24,9 (CH3 трет-бутила), 20,9, 20,8, 20,7, 20,7, 20,6, 20,5 (CH3-CO)
19F-ЯМР (235 МГц, CDCl3): δ -81,1 (CF3, с), -115,0 (CF2-CH2, с), -122,3, -123,2, -123,9 (CF2, с), -126,5 (CF 2-CF3, с)
МС FAB+ (1272,0 г/моль): [M+H] = 1273 (1,5%), [M+Na] = 1295 (3,5%)
После деацетилирования сахаров по методу Zemplen получают незащищенный продукт:
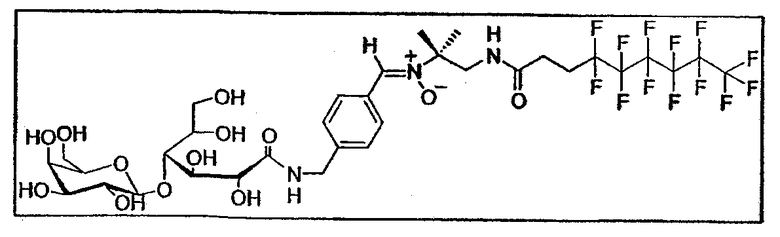
Т. плавл.=150°C (разложение)
[α]D = +14,4 (0,25 c, 1, CH3OH)
УФ (MeOH, нм): λmax = 299
Rf: 0,47(этилацетат/метанол/вода 7:2:1)
1H-ЯМР (250 МГц, CD3OD): δ 8,33 (2H, д, J=8,4 Гц), 7,86 (1H, с, CH=N(O)), 7,47 (2H, д, J=8,5 Гц), 4,65-4,45 (4H, м, CH2-NH, H-1', H-2), 4,3 (1H, м, H-3), 4,05-3,87 (2H, м, H-4 и H-5), 3,87-3,66 (7H, м, CH2-OH, H-4' и CH2-NH), 3,66-3,45 (3H, м, H-5', H-3' и H-2'), 2,55-2,40 (4H, м, CH2-CH2-Rf), 1,59 (6H, синглет, CH3 трет-бутила)
13C-ЯМР (62,86 МГц, CD3OD): δ 174,0, 171,9 (CO-NH), 142,1 (CIV аром.), 134,6 (CH=N(O)), 129,7 (CH аром.), 129,2 (CIV аром.), 126,9 (CH аром.), 104,4 (CH-1'), 82,0 (CH-4), 75,8 (CH-5'), 73,5 (CIV), 73,4 (CH-3' или CH-2'), 72,7 (CH-2), 71,8 (CH-5), 71,4 (CH-3' или CH-2'), 71,2 (CH-3), 69,0 (CH-4'), 62,4 (CH2-6) , 61,3 (CH2-6'), 46,3 (CIV-CH2-NH), 42,1 (CIV аром.-CH2-NH), 26,0 (CH2-CH2-Rf), 23,3 (CH3 трет-бутила)
19F-ЯМР (235 МГц, CD3OD): δ -82,1 (CF3, с), -115,3 (CF2-CH2, с), -122,6, -123,6, -124,3 (CF2, с), -127,0 (CF 2-CF3, с)
МС FAB+ (935,7 г/моль): [C13H13F13NO]+ = 446 (1%), [C9H4F13O]+ = 375 (8%)
МС FAB- (35,7 г/моль): [M-H] = 934 (очень слабый)
Препаративная колонка (Microsorb C18 - 21,4 мм / 250 мм): вр. удерж. = 9.800
Градиент от 70 MeOH - 30 H2O до 80 MeOH - 20 H2O от t = 0 до t = 5 мин
Градиент от 80 MeOH - 20 H2O до 82 MeOH - 18 H2O от t = 5 до t = 8 мин
Изократическое элюирование 82 MeOH -18 H2O, начиная с t = 8 мин
Расход 0,8 мл/мин
3. Синтез ионного углеводородного нитрона B 1
a. Синтез йодистого [4-(1,3-диоксолан)-2-ил-бензил]триметиламмония
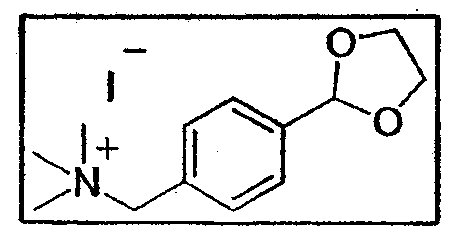
1,25 г амина E15 (7 ммоль - 1 экв.) растворяют в 4 мл ДМФ в запаянной трубке. Затем за один раз добавляют 2,58 г трибутиламина (14 ммоль - 2 экв.) при перемешивании. Среду охлаждают до 0°C и в нее медленно добавляют 5,2 г йодистого метила (35 ммоль - 5 экв.). Запаянную трубку закрывают и продолжают перемешивать при комнатной температуре в течение 20 часов. Неочищенный продукт реакции вводят в AcOEt и полученный осадок фильтруют, вводят в простой эфир, а затем снова фильтруют. Получают аммоний (1,7 г - 4,9 ммоль - 70%) в виде белого порошка.
1H-ЯМР (250 МГц, ДМСО-d6): δ 7,59 (4H, с, H аром.), 5,80 (H, с, H ацеталя), 4,61 (2H, с, CH2-NH), 4,2-3,9 (4H, AA'BB', CH2-O), 3,06 (9H, с, CH3-N)
13C-ЯМР (62,86 МГц, ДМСО-d6): δ 140,6 (CIV аром.), 133,3 (CH аром.) 129,6 (CIV аром.), 127,5 (CH аром.), 102,7 (CH ацеталь), 67,6 (CH2-N), 65,4 (CH2-O), 52,2(CH3-N)
Процентный анализ: (C13H20NO2I, 0,83 H2O): рассчитано C 41,69, H 4,60, N 4,42, найдено C 41,69, H 4,58, N 4,31.
b. Синтез йодистого (4-формилбензил)-триметиламмония E26
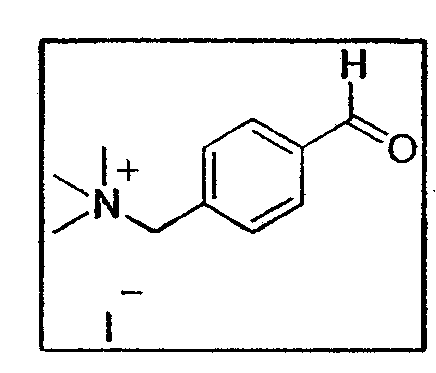
0,38 г диоксалана аммония (1,08 ммоль - 1 экв.) растворяют в 10 мл смеси уксусная кислота/вода 1:1. Через 12 часов перемешивания реакционную смесь выпаривают в вакууме и следы растворителя удаляют лопастным насосом. Получают соединение E26 (0,34 г - 1,08 ммоль - количественный выход) в виде темно-коричневого порошка.
1H-ЯМР (250 МГц, ДМСО-d6): δ 10,12 (1 H, с, CHO), 8,06 (2H, д, J=8 Гц, H аром.), 7,80 (2H, д, J=8 Гц, H аром.), 4,69 (2H, с, CH2-NH), 3,09 (9H, с, CH3-N)
13C-ЯМР (62,86 МГц, ДМСО-d6): δ 193,4 (CHO), 137,6, 134,8 (CIV аром.), 134,1, 130,2 (CH аром.), 62,2 (CH2N), 52,6 (CH3N)
c. Синтез ионного фторуглеродного нитрона B2
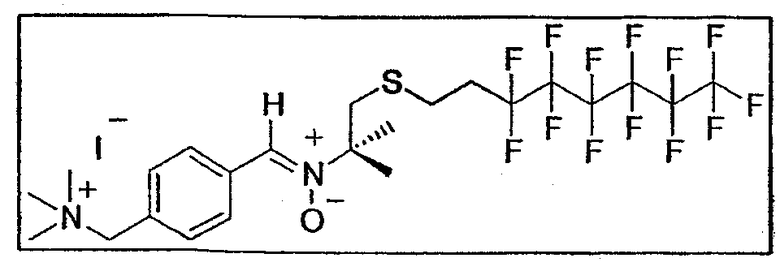
0,25 г соединения E26 (0,82 ммоль - 1 экв.) и 0,48 г гидроксиламина E7b (1,02 ммоль - 1,25 экв.) растворяют в 5 мл пиридина, продутого аргоном. Реакционную среду доводят до 80°C в темноте в атмосфере аргона в течение 42 часов. Затем реакционную смесь выпаривают при пониженном давлении и удаляют следы пиридина лопастным насосом. Получают нитрон в виде белого порошка (0,35 г - 0,47 ммоль - 57%) после двух последовательных кристаллизаций из смеси MeOH/простой эфир. Т. плавл. = 171-173
1Н-ЯМР (250 МГц, CD3OD): δ 8,54 (2H, д, J=8,4 Гц, H аром.), 8,03 (1H, с, CH=N(O)), 7,71 (2H, д, J=8,4 Гц, H аром.), 4,65 (2H, с, CH2-N), 3,18 (11H, с, CH3-N + CIV-CH2-S), 2,7 (2H, м, CH2-S), 2,45 (2H, м, CH2-CH 2-Rf), 1,71 (6H, с, CH3 трет-бутила)
13C-ЯМР (62,86 МГц, CD3OD): δ 133,3 (CIV аром.), 132,8 (CH=N(O)), 132,7 (CH аром.), 129,8 (CIV аром.), 129,7 (CH аром.), 73,9 (CIV), 68,5 (CH2-N), 51,9, 51,9, 51,8 (CH3-N), 41,3 (CH2-S), 31,9 (CH2-СH 2-Rf), 24,6 (CH3 трет-бутила), 23,0 (СH2-CH2-Rf)
19F-ЯМР (235 МГц, CD3OD): δ -82,3 (CF3, синглет), -115,2 (CF2-CH2, синглет), -112,9, -123,9, -124,3 (CF2, синглет), -127,3 (CF2-CF3, синглет)
УФ (MeOH, нм): λmax = 304 нм
МС ВР FAB+ (754,4 г/моль): m/z теоретическое: 755,0838 для C23H29F13IN2OS
([M+H]+)
m/z найдено: 755,0851
МС FAB+ (754,4 г/моль): [2M+H]+ = 1510, [2M-I]+ = 1381 (5%), [M+H]+ = 755 (2,5%), [M-I]+ = 627 (100%), [C12H12F13S]+ = 435 (100%)
4. Синтез двухцепочечного углеводородного нитрона C 1
a. Синтез 3-гептадецилкарбамоилокси-2-метил-2-нитропропилового эфира гептадецилкарбаминовой кислоты E9a

6,31 г стеариновой кислоты (0,022 моль - 3 экв.) диспергируют в 50 мл безводного толуола в атмосфере аргона. Добавляют 2,47 г триэтиламина (0,024 моль - 3,3 экв.) и 6,71 г дифенилфосфорилазида (0,024 моль - 3,3 экв.) и среду доводят до 60°C. Через 2 часа перемешивания переводят в суспензию 1 г 2-нитро-2-метил-1,3-пропандиола (0,0074 моль - 1 экв.) и DABCO на кончике шпателя и перемешивание продолжают в течение 12 часов. Неочищенный продукт реакции разбавляют 100 мл этилацетата, промывают 3 раза 1н. HCl порциями по 50 мл, 3 раза насыщенной NaHCО3 порциями по 50 мл и, наконец, моют 2 раза рассолом порциями по 50 мл. Органическую фазу сушат над Na2SО4 и выпаривают при пониженном давлении. После 3 последовательных кристаллизаций получают соединение E9a (2,02 г - 2,89 ммоль - 40%) в виде белого порошка. Т. плавл.: 75-76,2°C.
Rf: 0,42 (циклогексан/этилацетат 8:2)
1H-ЯМР (250 МГц, CDCl3): δ 4,77 (2H, м, NH), 4,45 (2H, система AB,CH2-O), 3,15 (2H, квинтет, J=9,8 Гц, CH 2-NH), 1,59 (3H, с, CH3 трет-бутила), 1,47 (2H, м, CH 2-CH2-NH), 1,24 (55H, м, CH2 цепи), 0,87 (3H, т, CH3 цепи)
13C-ЯМР (62,86 МГц, CDCl3): δ 155,1 (CO), 88,1 (CIV), 65,1 (CH2-O), 41,3 (CH2-NH), 31,9, 29,8, 29,7, 29,6, 29,5, 29,4, 29,3 (CH2 цепи), 26,7 (СH3 трет-бутила), 22,7, 18,5 (CH2 цепи), 14,1 (CH3 цепи)
ИК (KBr, см-1): ν(NH) = 3392, ν(CO) = 1720 и 1703, ν(NO2) = 1549
b. Синтез гептадецилкарбамоил-3-гептадецилкарбамоилокси-2-гидроксиламино-2-метилпропилового сложного эфира E9b
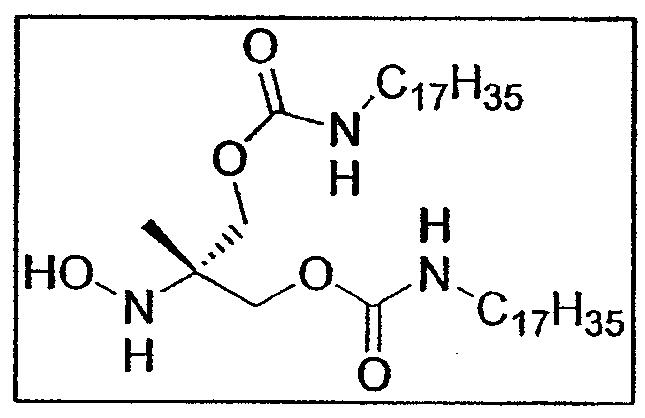
Экспериментальная процедура идентична использованной для синтеза соединений E5b и E7b .
1,39 г нитросоединения E9a (2,0 ммоль - 0,25 экв.), растворенного в 20 мл смеси ТГФ/MeOH, добавляют к 80 мл реактива Кагана.
После обработки и очистки с помощью хроматографии на силикагеле (элюент: дихлорметан/этилацетат от 10:0 до 5:5) получают гидроксиламин E9b (0,8 г - 1,17 ммоль - 60%) в виде белого порошка.
Также выделено 0,24 г исходного соединения E9a , что позволяет определить степень конверсии 71%. Т. плавл. = 80-81,6°C.
Rf: 0,51 (циклогексан/этилацетат 5:5)
1H-ЯМР (250 МГц, CDCl3): δ 4,83 (2H, т, J=NH), 4,45 (4H, система AB,CH2-О), 3,17 (4H, квинтет, J = 9,8 Гц, CH 2-NH), 1,49 (2H, м, CH 2-CH2-NH), 1,25 (55H, м, CH2 цепи), 1,07 (3H, с, CH3 трет-бутила), 0,88 (3H, т, CH3 цепи)
13C-ЯМР (62,86 МГц, CDCl3): δ 156,7 (CO), 88,1 (CIV), 64,7 (CH2-O), 41,2 (CH2-NH), 31,9, 29,8, 29,7, 29,6, 29,5, 29,4, 29,3 (CH2 цепи), 26,8 (CH3 трет-бутила), 22,7, 16,8 (CH2 цепи), 14,1 (CH3 цепи)
c. Синтез "двухантенного" углеводородного нитрона C 1

0,5 г альдегида E20 (0,616 ммоль - 1 экв.) растворяют в присутствии молекулярных сит 4 Е и 0,3 г гидроксиламина E9b (0,44 ммоль - 0,71 экв.) в 6 мл безводного и дегазированного ТГФ. Добавляют 1,2 мл ледяной уксусной кислоты и среду нагревают до 50°C в атмосфере аргона, не допуская света.
Через 48 и 96 часов добавляют 0,2 г гидроксиламина (0,29 ммоль - 0,47 экв.) и реакционную смесь фильтруют на целите по окончании 5 дней реакции.
Очистка флэш-хроматографией на силикагеле (элюент: этилацетат/дихлорметан от 7:3 до 8:2) и гель-проникающей хроматографией на смоле сефадекс LH-20 (элюент: этанол/дихлорметан 1:1) позволяет получить нитрон типа C (0,58 г - 0,392 ммоль - 63%), почти без следов альдегида, в виде белого порошка. Также извлечено 80 мг чистого альдегида, что позволяет оценить степень конверсии в 76 %. [α]D = + 16,9° (c, 1, CHCl3) при 20°C.
Rf: 0,37 (этилацетат/дихлорметан 8:2)
1H-ЯМР (250 МГц, CDCl3): δ 8,28 (2H, д, J = 8,1 Гц, H аром.), 7,45 (1H, с, CH=N(O)), 7,31 (2H, д, J=8,5 Гц, H аром.), 6,65 (1H, т, J=5,8 Гц, NH амид), 5,75-5,55 (2H, м, H-2 и H-3), 5,35 (1H, д, J=3 Гц), 5,25-4,80 (5H, м, H-2', H-5, H-3' и NH уретан), 4,70-4,25 (9H, м, H-1', H-6a и H-7a, H-4, H-7b и CH2-O-CO-NH), 4,20-3,80 (4H, м, H-6b, H-6'a, H-6'b и H-5'), 3,14 (4H, двойной дуплет, J = 6,7 Гц, CH2-NH-CO-O), 2,16, 2,15, 2,09, 2,05, 2,04, 1,98, 1,92 (24H, 8 с, CH3-CO), 1,60 (3H, с, CH3 трет-бутила), 1,55-1,10 (60H, м, CH2 цепи), 0,87 (6H, т, J = 6,4 Гц, CH3 цепи)
13C-ЯМР (250 МГц, CDCl3): δ 170,6, 170,3, 170,2, 170,0, 169,9, 169,8, 169,4, (CH3-СO), 167,3 (CO-NH), 155,7 (O-CO-NH), 140,3 (CIV аром.), 132,4 (CH=N(O)), 130,0 (CIV или CH аром.), 129,6 (CIV или CH аром.), 127,8 (CH аром.), 101,9 (CH-1'), 77,4 (CH-4), 75,1 (CIV), 71,7 (CH-2), 71,1 (CH-5' и CH-3'), 69,9 (CH-5), 69,3 (CH-3), 69,1 (CH-2), 66,9 (CH-4'), 65,5 (CH2-O-CO-NH), 61,8 и 61,0 (CH2-OAc), 43,2 (CH2-NH), 41,3 (CH2-NH-CO-O), 32,0, 29,9, 29,8, 29,7, 29,7, 29,6, 29,4, 29,3 (CH2 цепи), 26,8 (CH3 трет-бутила), 22,8 (CH2 цепи), 20,9, 20,9, 20,8, 20,7, 20,7, 20,6 (CH3-CO), 14,2 (CH3 конец цепи)
МС FAB+ (1477,8 г/моль): [M+H] = 1478 (16%), [M+Na] = 1500 (6%).
После деацетилирования сахаров по методу Zemplen получают незащищенный продукт:
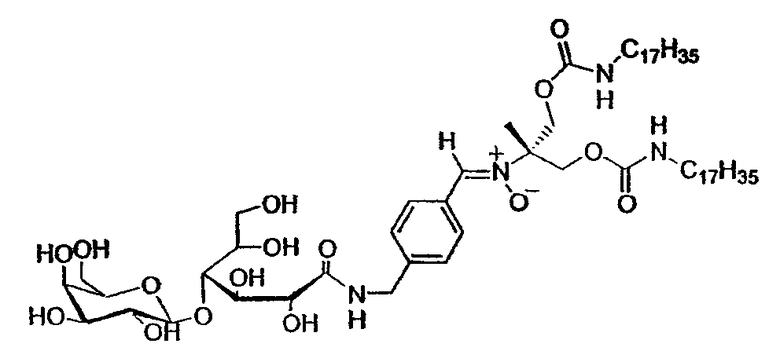
Очистку нитрона C 1 проводят флэш-хроматографией на силикагеле (элюент: хлороформ/метанол/вода 8:2:0,1), а затем гель-проникающей хроматографией на сефадексе LH-20 (элюент: дихлорметан/метанол 7:3).
[α]D = +7,6° (0,25 c, 1, CHCl3)
Rf: 0,28 (хлороформ/метанол/вода 8:2:0,1)
Т. плавл. = 190°C (разложение)
1H-ЯМР (250 МГц, ДМСО-d6): δ 8,28 (2H, д, J=8,2 Гц, H аром.), 8,07 (1H, т, J=6,3 Гц, NH амид), 7,72 (1H, с, CH=N(O)), 7,35 (2H, д, J = 8,3 Гц, H аром.), 7,08 (2H, м, NH уретан), 4,60-4,00 (9H, м, CH2-NH, CH2-O-CO-NH, H-1', H-2 и H-3), 3,78 (2H, м, H-4 и H-5), 3,70-3,40 (8H, м, CH2-OH, H-2', H-3', H-4' и H-5'), 2,94 (4H, м, CH2-NH-CO-O), 1,54 (3H, с, CH3 трет-бутила), 1,45-1,10 (60H, м, CH2), 0,87 (6H, т, J = 6,6 Гц, CH3)
13C-ЯМР (62,86 МГц, ДМСО-d6): δ 173,0 (CO-NH), 156,1 (O-CO-NH), 142,3 (CIV аром.), 132,1 (CH=N(O)), 130,0 (CIV аром.), 129,1 (CH аром.), 127,2 (CH аром.), 105,1 (CH-1'), 83,4 (CH-4), 76,2 (CH-5'), 74,9 (CIV), 73,7 (CH-3' или CH-2'), 72,6 (CH-2), 71,9 (CH-5), 71,6 (CH-3' или CH-2'), 71,1 (CH-3), 68,7 (CH-4'), 65,1 (CH2-O-CO-NH), 62,8, 61,1 (CH2-6 и CH2-6'), 42,2 (CH2-NH), 40,7 (CH2-NH-CO-O), 31,8, 29,8, 29,6, 29,2 (CH2 цепи), 26,7 (CH3 трет-бутила), 22,6 (CH2 цепи), 14,4 (CH3 цепи)
МС FAB+ (1140,77 г/моль): [M + Na] = 1164, [M + H] = 1142
III- Измерение гидрофобности молекул изобретения:
Одной из целей изобретения является изменение ГЛБ ловушек свободных радикалов, чтобы способствовать трансмембранному проходу и транспорту in vivo.
В этой точки зрения важным было определить коэффициент распределения P этих соединений.
Действительно, Ханш и его сотрудники (Hansch, C.; Steward, A.R.; Andersen, S.M.; Benfley, D.J. Med. Chem, 11, 1 (1968)) смогли установить, при изучении эффективности действия разных снотворных в зависимости от их гидрофобности, следующую зависимость:
log 1/C = -k(log P)2 + k'(log P) + k"
C: молярная концентрация, производящая стандартный биологический отклик,
k, k' и k": константы, определенные методом наименьших квадратов.
Таким образом, чем соединение более гидрофобно, тем выше 0 будет значение log P и тем выше будут взаимодействия с липидной фазой.
Авторы определили коэффициент распределения этих нитронов методом высокоэффективной жидкостной хроматографии в обращенных фазах (Lambert, W. J. J. Chromtogr. 656, 469 (1993); Dorsey, J. G.; Kahaledi, M. G. J. Chromtogr. 656, 485 (1993)).
Thomas также использовал этот хроматографический подход для определения гидрофобности спиновых ловушек циклических производных PBN (Fevig, T.L.; Bowen, S.M.; Janowick, D.A.; Jones, B.K.; Munson, H.R.; Ohlweiler, D.F.; Thomas, C.E. J. Med. Chem. 39, 4988 (1996)). Оценка коэффициента разделения октанол/вода методом ВЭЖХ в обращенных фазах (Kow) в высшей степени зависит от времени удерживания соединений и, следовательно, коэффициента емкости k'. Он может быть выражен следующей зависимостью:
log Kow = a log k'+ b,
в которой a и b являются эмпирическими константами, которые характеризуют систему растворителя.
Экспериментально k' определяют по следующей формуле для четырех разных смесей метанол/вода
k' = (t R-t 0)/t 0,
в которой t R означает время удержания образца, а t0 время элюирования подвижной фазы.
Затем нужно линейной регрессией экстраполировать значение k' для фазы, состоящей на 100% из воды, чтобы получить значение k w.
Авторы действовали этим путем для соединений производных лактобионовой кислоты типа A. Также для сравнения и оценки их модели авторами была определена гидрофобность PBN и TA1PBN.
В таблице 3 приведены значения средних времен удержания, полученных минимум из трех значений, определенных минимум в течение 2 разных дней.
Линейная регрессия значений k', полученных в зависимости от использованных подвижных фаз, позволяет получить уравнение прямой типа:
y = ax + b,
в которой y означает log k', a означает log k'w,
x означает долю метанола в элюенте.
Определение значений log k' методом ВЭЖХ
(C в мг/мл)
(0,64 мг/мл)
(0,26 мг/мл)
(0,52 мг/мл)
(0,52 мг/мл)
(0,48 мг/мл)
(0,57 мг/мл)
(0,52 мг/мл)
Для 4 других производных действовали тем же образом, результаты представлены вкратце в следующей таблице (таблица 4).
Определение значений log k'w методом ВЭЖХ
Для ясности значения log k'w различных нитронов приведены на гистограмме (фигура 11).
Полученные результаты позволяют сделать несколько примечаний и выводов:
1- Для соединений с углеводородными цепями полученные значения согласуются с ожиданиями авторов. Чем больше атомов углерода содержит цепь, тем выше уровень гидрофобности. С другой стороны, нельзя пренебречь влиянием на значение log k' природы цепных связей, причем амидная или уретановая связь по своей природе более полярна, чем тиоэфирная связь. Таким образом, в терминах гидрофобности получают следующий возрастающий порядок:
A 5 < A 1 < A 2
2- Для фторированных соединений соединение A 3 имеет большее сродство к липидным средам, чем соединение A 4, что, по-видимому, согласуется с соответствующими значениями их ККМ. Однако соединения A 3 и A 4 имеют близкие значение log k'w, хотя их значения ККМ отличаются более чем вдвое. Это происходит из-за природы фторированной цепи, которая обнаруживает особые поверхностно-активные свойства. Следовательно, следует хорошо разделять понятие поверхностной активности и понятие гидрофобности. Таким образом, в терминах гидрофобности получают следующий возрастающий порядок:
A 5 < A 1 < A 2 < A 4 < A 3
3- Значение log k'w молекулы PBN существенно более слабое, чем у множества синтетических соединений. Согласно теории Ханша, отсюда можно сделать вывод о лучшей трансмембранной проницаемости соединений и, таким образом, лучшей активности в захвате свободных радикалов.
4- Значение log k'w молекулы TA1PBN почти идентично значению для соединения A 3.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПОВЕРХНОСТНО-АКТИВНЫЕ ВЕЩЕСТВА И ИХ ПРИМЕНЕНИЕ | 2004 |
|
RU2395493C2 |
| ПРОИЗВОДНЫЕ 2-АМИНО-2-ФЕНИЛАЛКАНОЛА, ИХ ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2009 |
|
RU2486174C2 |
| ПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ КИСЛОТНЫЕ ГРУППЫ | 2017 |
|
RU2782066C2 |
| ПРОИЗВОДНЫЕ 2-((4-(4-(БЕНЗИЛОКСИ)ПИРИМИДИН-2-ИЛ)ПИПЕРАЗИН-1-ИЛ)МЕТИЛ)-1Н-БЕНЗО[d]ИМИДАЗОЛА В КАЧЕСТВЕ НИЗКОМОЛЕКУЛЯРНЫХ АГОНИСТОВ РЕЦЕПТОРА ГЛЮКАГОНОПОДОБНОГО ПЕПТИДА-1 | 2024 |
|
RU2838323C1 |
| НОВЫЕ ГЕМ-ДИФТОРИРОВАННЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2369612C2 |
| СПОСОБ СИНТЕЗА ЦИКЛОГЕКСЕНОНОВ И ИХ ПРИМЕНЕНИЕ В ПАРФЮМЕРИИ | 2013 |
|
RU2663619C2 |
| ФОСФОИНДОЛЫ КАК ИНГИБИТОРЫ ВИЧ | 2005 |
|
RU2393163C2 |
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ | 2007 |
|
RU2525392C2 |
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ | 2007 |
|
RU2466729C2 |
| ФТОРСОДЕРЖАЩИЕ ЛИГАНДЫ ДЛЯ НАЦЕЛИВАНИЯ ПЕРИФЕРИЧЕСКИХ БЕНЗОДИАЗЕПИНОВЫХ РЕЦЕПТОРОВ | 2007 |
|
RU2468014C2 |
Настоящее изобретение относится к производным α-C-фенил-N-трет-бутилнитрона общей формулы (I), где Х означает лактобионамидную группу, группу четвертичного аммония или группу  ; m означает 1, 2 или 3; у означает 0 или 1; Y обозначает группу -CH2-; m' означает 1; X' означает алкильную цепь C4-C14, при необходимости замещенную одним или несколькими атомами фтора; Y' означает группу, выбранную из функциональной группы амида (-NHC(O)-), уретановой группы
; m означает 1, 2 или 3; у означает 0 или 1; Y обозначает группу -CH2-; m' означает 1; X' означает алкильную цепь C4-C14, при необходимости замещенную одним или несколькими атомами фтора; Y' означает группу, выбранную из функциональной группы амида (-NHC(O)-), уретановой группы
(-OC(O)NH-) или тиоэфирного мостика (-S-). Изобретение относится также к применению заявленных соединений для получения лекарственного средства для лечения патологий, связанных с оксидативным стрессом и с образованием соединений со свободными радикалами кислорода. Кроме того, изобретение относится и к применению соединений формулы (I) для получения косметической композиции для предупреждения или лечения эффектов старения. 5 н. и 4 з.п. ф-лы, 4 табл., 11 ил.
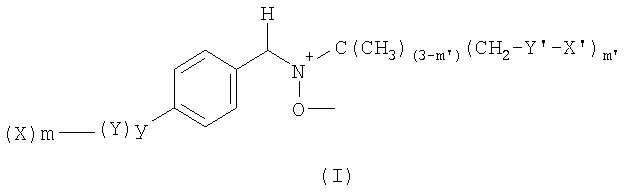
1. Производное α-C-фенил-N-трет-бутилнитрона, отличающееся тем, что оно отвечает формуле (I)
в которой Х означает лактобионамидную группу или группу четвертичного аммония или группу
m означает целое число, равное 1, 2 или 3;
y означает целое число, равное 0 или 1;
Y обозначает группу -CH2-;
m' означает 1;
X' означает алкильную цепь C4-C14, при необходимости замещенную одним или несколькими атомами фтора;
Y' означает группу, выбранную из функциональной группы амида (-NHC(O)-), уретановой группы (-OC(O)NH-) или тиоэфирного мостика (-S-).
2. Соединение по п.1, отличающееся тем, что Х означает лактобионамидную группу.
3. Соединение по п.1, отличающееся тем, что Х означает группу, выбранную из
4. Соединение по п.1, отличающееся тем, что удовлетворяется по меньшей мере одно из нижеследующих условий:
Х означает лактобионамид;
m означает 1;
m' означает 1;
X' выбран из гексила, гептила, октила, CF3(CF2)rCH2CH2-,
r=5.
5. Применение соединения, отвечающего формуле (I), по любому из пп.1-4 для получения лекарственного средства для лечения патологий, связанных с оксидативным стрессом и с образованием соединений со свободными радикалами кислорода.
6. Применение соединения, отвечающего формуле (I), по любому из пп.1-4 для получения лекарственного средства, предназначенного для профилактики и/или лечения эффектов, вызванных свободными радикалами.
7. Применение соединения по любому из пп.1-4 для получения лекарственного средства, предназначенного для профилактики или лечения патологий, связанных с оксидативным стрессом и с образованием соединений со свободными радикалами кислорода.
8. Применение по п.7 для профилактики или лечения патологии, выбранной из иммунных и воспалительных заболеваний, синдрома ишемии-реперфузии, атеросклероза, болезни Альцгеймера, болезни Паркинсона, повреждений, вызванных ионизирующим и УФ-излучением, болезни Гентингтона, онкологических заболеваний, старения клеток.
9. Применение соединения, отвечающего формуле (I), по любому из пп.1-4 для получения косметической композиции для предупреждения или лечения эффектов старения.
| Ouari О | |||
| et al // J | |||
| Chem | |||
| Soc., Perkin Trans | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| ИСПОЛЬЗОВАНИЕ АКЦЕПТОРА СПИНОВ В КОСМЕТИЧЕСКОЙ ИЛИ ДЕРМАТОЛОГИЧЕСКОЙ КОМПОЗИЦИИ И КОСМЕТИЧЕСКАЯ ИЛИ ДЕРМАТОЛОГИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 1994 |
|
RU2139038C1 |
| Химическая энциклопедия | |||
| Научное изд-во «Большая Российская энциклопедия» | |||
| - М., т | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| US 6107315 А, 22.08.2000. | |||

