УРОВЕНЬ ТЕХНИКИ
Известно, что нитроксил (HNO) оказывает положительные кардиоваскулярные эффекты в in vitro и in vivo моделях сердечной недостаточности. Однако при физиологических значениях pH нитроксил димеризуется с образованием азотноватистой кислоты, которая затем дегидрируется до оксида азота; в связи с нестабильностью, нитроксил для терапевтического применения должен образовываться in situ из донорских соединений. Различные соединения, способные быть донорами нитроксила, были описаны и предложены для использования в лечении нарушений, которые известно, что реагируют или предположительно реагируют на лечение нитроксилом. Смотреть, например, патенты США №№ 6936639; 7696373; 8030356; 8268890; 8227639; и 8318705 и патентные заявки США (опубликованные через 18 месяцев с даты приоритета) № 2009/0281067; 2009/0298795; 2011/0136827; и 2011/0144067. Хотя все эти соединения способны быть донорами нитроксила, они отличаются по физико-химическим свойствам, и сохраняется потребность в установлении доноров нитроксила, которые обладают физико-химическим свойствами наиболее подходящими для лечения определенных клинических состояний с использованием определенных путей введения.
В патенте США № 8030056 описывается синтез производных соединений типа кислот Пилоти, которые способны быть донорами нитроксила в физиологических условиях и которые можно использовать в лечении сердечной недостаточности и ишемически-реперфузионного повреждения. Донор нитроксила CXL-1020 (N-гидрокси-2-метансульфонилбензол-l-сульфонамид) оценивали в фазе I исследовании безопасности на здоровых добровольцах и в фазе IIa плацебо-контролируемом, двойном слепом, с увеличением дозы исследовании, проведенном в нескольких больницах. Sabbah et at, "Nitroxyl (HNO) a novel approach for the acute treatment of heart failure", Circ Heart Fail., опубликовано он-лайн 9 октября 2013 (Online ISSN: 1941-3297, Print ISSN: 1941-3289). Исследования показали, что у пациентов с систолической сердечной недостаточностью, CXL-1020, при внутривенном введенении в виде водного раствора с pH = 4, снижал давление заполнения как левого, так и правого желудочка и общее сосудистое сопротивление, в то же время увеличивая сердечный индекс и индекс систолического объема кровотока. Таким образом, исследования показали, что CXL-1020 усиливает функцию миокарда у пациентов, страдающих от сердечной недостаточности. Однако обнаружили, что при использовании пороговых доз CXL-1020, необходимых для получения гемодинамических эффектов, соединение вызывает побочные эффекты, включая неприемлемые уровни воспалительного раздражения в месте внутривенного введения и дистальнее его, и авторы сообщают, что в связи с указанными побочными эффектами соединение не является конкурентоспособным кандидатом в виде лекарственного средства для применения у человека. Таким образом, существует потребность в создании новых соединений-доноров нитроксила и композиций, которые можно использовать для лечения сердечной недостаточности, и которые имеют подходящий токсикологический профиль. Создание таких соединений предусматривает понимание фармакокинетического профиля, связанного со способностью соединения быть донором нитроксила, и факторов, влияющих на токсикологический профиль. Непонимание этих факторов препятствует созданию соединений-доноров нитроксила, предназначенных для клинического использования.
Соответственно, существует необходимость в создании композиций, содержащих соединения-доноры нитроксила, для парентерального и/или перорального введения, которые достаточно стабильны и имеют подходящие фармакологические и токсикологические профили.
Кроме того, включение в состав смеси соединений-доноров нитроксила оказалось сложной задачей. Многие из существующих доноров нитроксила нерастворимы в водных растворах и/или недостаточно стабильны. Проблемы, связанные с растворимостью и стабильностью, часто исключают использование таких соединений в виде фармацевтических композиций, предназначенных для парентерального и/или перорального введения.
Цитирование какой-либо ссылки в разделе 1 настоящего приложения не следует истолковывать как допущение, что такая ссылка является прототипом настоящей заявки.
2. СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее описание относится к изобретению соединений-доноров нитроксила, которые высокоэффективны в лечении сердечно-сосудистых заболеваний (например, сердечной недостаточности) и имеют соответствующий токсикологический профиль.
В конкретном варианте осуществления соединение-донор нитроксила по изобретению представляет собой соединение формулы (1):
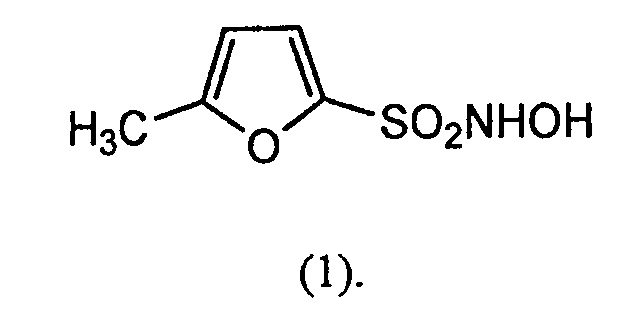
В другом варианте осуществления соединение-донор нитроксила по изобретению представляет собой соединение формулы (2):
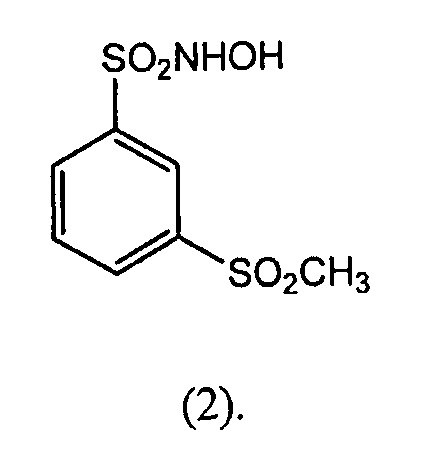
В другом варианте осуществления изобретение относится к соединениям формулы (3):
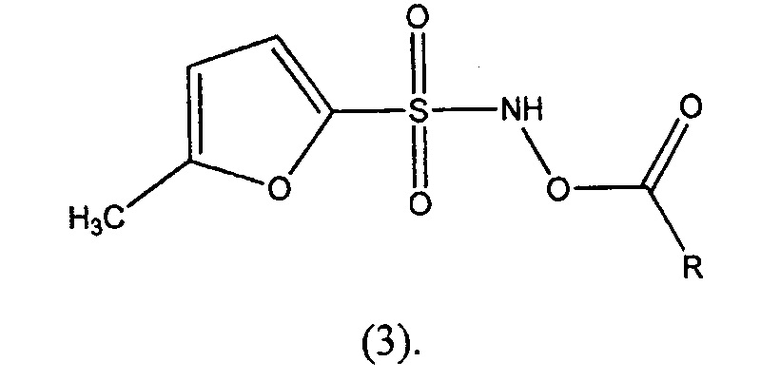
где R представляет собой водород, -(C1-C6)алкил, -(C2-C4)алкенил, фенил, бензил, циклопентил, циклогексил, -(C5-C7)гетероциклоалкил, бензилокси, -O-(C1-C6)алкил, -NH2, -NH-(С1-C4)алкил, или -N((С1-C4)алкил)2, где указанные -(C1-C6)алкил, -(C2-C4)алкенил, фенил, бензил, циклопентил, циклогексил, -(C5-C7)гетероциклоалкил, бензилокси, -O-(C1-C6)алкил,-NH-(С1-C4)алкил, или -N((С1-C4)алкил)2 могут быть незамещены или замещены одним или несколькими заместителями, выбранными из галогена, -(С1-C6)алкила, -(C2-C4)алкенила, -(C2-C3)алкинила, -(5- или 6-членного)гетероарила, -O- (C1-C6)алкила, -S-(C1-C6)алкила, -C(галоген)3, -CH(галоген)2, -CH2(галоген), -CN, -NО2, -NH2, -NH-(C1-C4)алкила, -N(-(-(C1-C4)алкил)2, -C(=О)(C1-C4)алкила, -C(=О)О(C1-C4)алкила, -OC(=О)-(C1-C4)алкила, -OC(=О)NH2, -S(=О)(C1-C4) алкила, или -S(=О)2(C1-C4)алкила. В конкретных вариантах осуществления R представляет собой метил, этил, бензил или фенил. В конкретных вариантах осуществления R представляет собой метил или этил. В конкретных вариантах осуществления R представляет собой метил. В конкретных вариантах осуществления R представляет собой этил. В конкретных вариантах осуществления R представляет собой бензил или фенил. В конкретных вариантах осуществления, R представляет собой бензил. В конкретных вариантах осуществления, R представляет собой фенил.
В другом варианте осуществления изобретение относится к соединениям формулы (4):

где R и его варианты осуществления имеют значения, указанные выше для соединения формулы (3). Соединения по изобретению имеют или предполагается, что имеют, весьма благоприятный терапевтический индекс. В частности, соединения формулы (1) и формулы (2) имеют подходящие гемодинамические профили и токсикологические профили. Токсикологические профиль соединений формулы (1) и формулы (2) существенно улучшается по сравнению с соединением-кандидатом для клинического исследования CXL-1020. Установлено, что благоприятный токсикологический профиль соединений формулы (1) и формулы (2) отчасти связан со временем полураспада соединений и выявлением оптимального диапазона периода полураспада таких доноров нитроксила. Соединение формулы (1) имеет период полураспада приблизительно 68 минут при измерении в аэрированном фосфатно-солевом буферном растворе (PBS) при pH 7,4, и приблизительно 65 минут при измерении в плазме человека при pH 7,4 в присутствии антикоагулянта (например, гепарина или цитрата натрия), каждое из измерений проводили в условиях, указанных в Примере 4. Соединение формулы (2) имеет период полураспада приблизительно 50 минут при измерении в аэрированном фосфатно-солевом буферном растворе (PBS) при pH 7,4, и приблизительно 37 минут при измерении в плазме человека при pH 7,4 в присутствии антикоагулянта (например, гепарина или цитрата натрия), каждое из измерений проводили в условиях, указанных в Примере 4.
Кроме того, соединения формулы (1) и формулы (2) стабильны в водных растворах и хорошо растворимы в воде; таким образом, они подходят как для парентерального, так и для перорального введения. Соединение формулы (1) имеет значение равновесной растворимости в воде выше 100 мг/мл, в то время как соединение формулы (2) имеет значение равновесной растворимости в воде приблизительно 10 мг/мл (например, в условиях, указанных в Примере 5).
Соединения по изобретению можно использовать для лечения различных состояний, которые реагируют на терапию нитроксилом. Например, соединение-донор нитроксила по изобретению можно использовать для лечения или профилактики сердечно-сосудистых заболеваний. В конкретных вариантах осуществления соединение-донор нитроксила по изобретению можно использовать для лечения сердечно-сосудистого заболевания, ишемически-реперфузионного повреждения, легочной гипертензии или другого состояния, реагирующего на лечение нитроксилом. В других вариантах осуществления соединение-донор нитроксила по изобретению можно использовать для лечения сердечной недостаточности. В конкретном варианте осуществления соединение по изобретению можно использовать для лечения декомпенсированной сердечной недостаточности (например, острой декомпенсированной сердечной недостаточности). В определенных вариантах осуществления соединения по изобретению можно использовать для лечения систолической сердечной недостаточности. В конкретных вариантах осуществления соединения по изобретению можно использовать для лечения диастолической сердечной недостаточности.
В одном аспекте соединения по изобретению могут быть введены путем парентерального (например, подкожного, внутримышечного, внутривенного или внутрикожного) введения. Соединения по изобретению не вызывают нежелательных местных побочных эффектов (например, раздражения и/или воспаления) во время или после парентерального введения в дозах, способных обеспечить требуемый уровень эффективности.
В вариантах осуществления, в которых соединение по изобретению вводят парентерально, его, как правило, вводят в виде водного раствора или суспензии. Водный раствор или суспензия могут иметь значение pH приблизительно от 4 до 6,5. В конкретных вариантах осуществления соединение по изобретению может быть включено в состав для парентеральной инъекции со значением pH приблизительно от 4 до 5. В других вариантах осуществления соединение по изобретению может быть включено в состав для парентеральной инъекции со значением pH приблизительно от 5 до 6. В некоторых вариантах осуществления препарат для парентерального введения может содержать вещество, увеличивающее стабильность.
При введении парентерально (например, внутривенно) человеческому индивиду, соединение по изобретению можно вводить со скоростью приблизительно от 5 мкг/кг/мин до 100 мкг/кг/мин. В определенных вариантах осуществления соединение по изобретению можно вводить человеческому индивиду со скоростью приблизительно от 10 мкг/кг/мин до 70 мкг/кг/мин. В определенных вариантах осуществления соединение по изобретению можно вводить человеческому индивиду со скоростью приблизительно от 15 мкг/кг/мин до 50 мкг/кг/мин. В определенных вариантах осуществления соединение по изобретению можно вводить человеческому индивиду со скоростью приблизительно от 20 мкг/кг/мин до 40 мкг/кг/мин.
В другом варианте осуществления соединения по изобретению могут быть включены в состав для перорального введения. Соединения для перорального введения могут быть созданы в виде жидких или твердых лекарственных форм. В конкретных вариантах осуществления, где соединение-донор нитроксила создают в виде пероральной жидкой лекарственной формы, полиэтиленгликоль 300 (PEG300) может служить в качестве типичного вспомогательного вещества.
3. КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг. 1 представлен гемодинамический профиль CXL-1020 и двух соединений по изобретению (соединения формулы (1) и формулы (2)), полученный с использованием модели сердечной недостаточности, вызванной стимуляцией тахикардии (смотреть Пример 6). Каждое соединение вводили внутривенно со скоростью 100 мкг/кг/мин. Гемодинамические параметры получали через 180 мин после введения соответствующего соединения.
На фиг. 2 представлен гемодинамический профиль соединения формулы (1) при использовании в различных дозах в модели сердечной недостаточности, индуцированной тахикардией для бодрствующих животных (смотреть Пример 6).
На фиг. 3 представлен гемодинамический профиль соединения формулы (1) после индукции сердечной недостаточности у собак. Гемодинамику оценивали с использованием модели сердечной недостаточности, вызванной микроэмболизацией у собак (смотреть Пример 7). Данные представлены для конечного момента времени при проведении инфузии (180 мин.) для двух значений скорости инфузии.
На фиг. 4 представлена оценка токсикологического профиля CXL-1020 и двух соединений-доноров нитроксила по изобретению (соединения формулы (1) и формулы (2)) после 24-часовой инфузии в различных дозах с использованием модели введения лекарств в периферические вены собак (смотреть Пример 9). Основные маркеры воспаления, которые измеряли, включали количество лейкоцитов (WBC), фибриноген, и C-реактивный белок (CRP).
На фиг. 5 показаны значения показателей воспаления, полученные в модели с имплантацией собакам центрального катетера на 72 часа, при использовании различных доз CXL-1020 и соединений формул (1) и (2) (смотреть Пример 9).
4. ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Изобретение включает в себя следующее.
(1.) Соединение формулы (1):
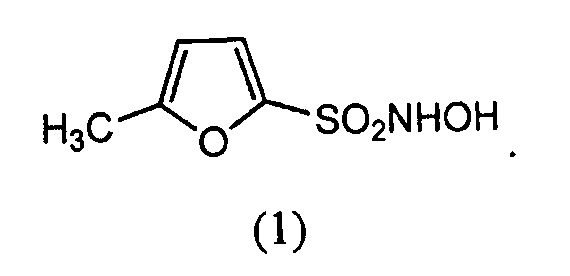
(2.) Соединение формулы (2):

(3.) Фармацевтическая композиция, содержащая соединение по указанному выше п. (1.) или указанному выше п. (2.) и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
(4.) Фармацевтическая композиция по указанному выше п. (3.), где фармацевтическая композиция предназначена для внутривенного введения.
(5.) Фармацевтическая композиция по указанному выше п. (3.) или п. (4.), где фармацевтическая композиция имеет значение pH приблизительно от 4 до 6.
(6.) Фармацевтическая композиция по любому из вышеуказанных пунктов (3.)-(5.), где фармацевтическая композиция имеет значение pH приблизительно от 4 до 5.
(7.) Фармацевтическая композиция по любому из вышеуказанных пунктов (3.)-(6.), где фармацевтическая композиция имеет значение pH приблизительно 4.
(8.) Способ лечения сердечно-сосудистого заболевания, включающий введение эффективного количества соединения по указанному выше п. (1.) или п. (2.) или фармацевтической композиции по любому из указанных выше пп. (3.)-(7.) пациенту, который в этом нуждается.
(9.) Способ по указанному выше п. (8.), в котором сердечно-сосудистое заболевание представляет собой сердечную недостаточность.
(10.) Способ по указанному выше п. (8.) или п. (9.), в котором сердечно-сосудистое заболевание представляет собой острую декомпенсированную сердечную недостаточность.
(11.) Способ по любому из указанных выше пп. (8.)-(10,), в котором соединение или фармацевтическую композицию вводят внутривенно.
(12.) Способ по любому из указанных выше пп. (8.)-(11.), в котором соединение или фармацевтическую композицию вводят в дозе приблизительно от 20 мкг/кг/минуту соединения формулы (1) или (2) до 40 мкг/кг/минуту соединения формулы (1) или (2).
(13.) Способ по любому из указанных выше пп. (8.)-(10,), в котором соединение или фармацевтическую композицию вводят перорально.
(14.) Набор, содержащий соединение по указанному выше п. (1.) или п. (2.) в сухом виде или фармацевтическую композицию по любому из указанных выше пп. (3.)-(7.) в сухом виде; и фармацевтически приемлемый жидкий разбавитель.
(15.) Использование соединения по указанному выше п. (1.) или п. (2.) или использование фармацевтической композиции по любому из указанных выше пп. (3.)-(7.) для изготовления лекарственного средства, применяемого для лечения сердечно-сосудистого заболевания.
(16.) Использование соединения по указанному выше п. (1.) или п. (2.) или использование фармацевтической композиции по любому из указанных выше пп. (3.)-(7.) для изготовления лекарственного средства, применяемого для лечения сердечной недостаточности.
(17.) Использование соединения по указанному выше п. (1.) или п. (2.) или использование фармацевтической композиции по любому из указанных выше пп. (3.)-(7.) для изготовления лекарственного средства, применяемого для лечения острой декомпенсированной сердечной недостаточности.
(18.) Соединение по указанному выше п. (1.) или п. (2.) или фармацевтическая композиция по любому из указанных выше пп. (3.)-(7.), предназначенное для использования в лечении сердечно-сосудистого заболевания.
(19.) Соединение по указанному выше п. (1.) или п. (2.) или фармацевтическая композиция по любому из указанных выше пп. (3.)-(7.), предназначенное для использования в лечении сердечной недостаточности.
(20.) Соединение по указанному выше п. (1.) или п. (2.) или фармацевтическая композиция по любому из указанных выше пп. (3.)-(7.), предназначенное для использования в лечении острой декомпенсированной сердечной недостаточности.
4.1 Определения
Если явно не указано иное, следующие термины, которые используются в описании, имеют значения, указанные ниже.
Термин "фармацевтически приемлемая соль" относится к соли любого терапевтического агента, описанного в настоящем документе, указанная соль может включать любой из множества органических и неорганических противоионов, известных в данной области, и указанная соль является фармацевтически приемлемой. Если терапевтический агент содержит кислотную функциональную группу, различные иллюстративные варианты осуществления противоионов представляют собой натрий, калий, кальций, магний, аммоний, тетраалкил аммония и т.п. Если терапевтический агент содержит щелочную функциональную группу, фармацевтически приемлемая соль может включать в качестве противоиона, в качестве примера, органическую или неорганическую кислоту, такую как гидрохлорид, гидробромид, тартрат, мезилат, ацетат, малеат, оксалат и т.п. Иллюстративные соли включают, но не ограничиваются ими, соли сульфата, цитрата, ацетата, хлорида, бромида, иодида, нитрата, бисульфата, фосфата, кислого фосфата, лактата, салицилата, кислого цитрата, тартрата, олеата, танната, пантотената, битартрата, аскорбата, сукцината, малеата, бесилата, фумарата, глюконата, глюкароната, сахарата, формиата, бензоата, глутамата, метансульфоната, этансульфоната, бензолсульфоната и п-толуолсульфоната. Соответственно, соли могут быть получены из соединения любой из формул, описанных в настоящем документе, имеющего кислую функциональную группу, такую как функциональная группа карбоновой кислоты, и фармацевтически приемлемого неорганического или органического основания. Подходящие основания включают, но не ограничиваются ими, гидроксиды щелочных металлов, таких как натрий, калий и литий; гидроксиды щелочно-земельных металлов, таких как кальций и магний; гидроксиды других металлов, таких как алюминий и цинк; аммиак, органические амины, такие как незамещенные или гидрокси-замещенные моно-, ди-, или триалкиламины; дициклогексиламин; трибутиламин; пиридин; N-метил-N-этиламин; диэтиламин; триэтиламин; моно-, ди- или три-(2-гидрокси-низшие алкиламины), такие как моно-, бис-, или трис-(2-гидроксиэтил)амин, 2-гидрокси-трет-бутиламин или трис-(гидроксиметил) метиламин, N,N-ди-низший алкил-N-(гидрокси-низший алкил)-амины, такие как N,N-диметил-N-(2-гидроксиэтил) амин, или три-(2-гидроксиэтил)амин; N-метил-D-глюкамин; и аминокислоты, такие как аргинин, лизин и т.п. Соль также может быть получена из соединения любой из формул, описанных в настоящем документе, имеющего щелочную функциональную группу, такую как функциональная аминогруппа, и фармацевтически приемлемой неорганической или органической кислоты. Подходящие кислоты включают гидросульфат, лимонную кислоту, уксусную кислоту, соляную кислоту (НСl), бромоводородную кислоту (HBr), иодоводородную кислоту (HI), азотную кислоту, фосфорную кислоту, молочную кислоту, салициловую кислоту, винную кислоту, аскорбиновую кислоту, янтарную кислоту, малеиновую кислоту, бензолсульфоновую кислоту, фумаровую кислоту, глюконовую кислоту, глюкуроновую кислоту, муравьиную кислоту, бензойную кислоту, глутаминовую кислоту, метансульфоновую кислоту, этансульфоновую кислоту, бензолсульфоновую кислоту и п-толуолсульфокислоту.
Термин "фармацевтически приемлемое вспомогательное вещество" относится к любому веществу, не к самому терапевтическому агенту, используемому в качестве носителя, разбавителя, адъюванта, связующего вещества, и/или средства для доставки терапевтического агента пациенту, или добавляемого к фармацевтической композиции для улучшения его обработки или свойств при хранении или для обеспечения или упрощения создания соединения или фармацевтической композиции в стандартной дозированной форме для введения. Фармацевтически приемлемые вспомогательные вещества известны в фармацевтической области и описаны, например, в Gennaro, Ed., Remington: The Science and Practice of Pharmacy, 20 thEd. (Lippincott Williams & Wilkins, Baltimore, MD, 2000) and Handbook of Pharmaceutical excipients, American Pharmaceutical Association, Washington, D.C., (например, 1-е, 2-е и 3-е издания, 1986, 1994 и 2000, соответственно). Как известно специалистам в данной области, фармацевтически приемлемые вспомогательные вещества могут обеспечивать различные функции и могут быть описаны как смачивающие реагенты, буферные реагенты, суспендирующие реагенты, смазывающие вещества, эмульгаторы, разрыхлители, абсорбенты, консерванты, сурфактанты, красители, ароматизаторы, и подсластители. Примеры фармацевтически приемлемых вспомогательных веществ включают, без ограничения: (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал и картофельный крахмал; (3) целлюлоза и ее производные, такие как натрийкарбоксиметилцеллюлоза, этилцеллюлоза, ацетат целлюлозы, гидроксипропилметилцеллюлоза, и гидроксипропилцеллюлоза; (4) порошкообразный трагакант; (5) солод; (6) желатин; (7) тальк; (8) вспомогательные вещества, такие как масло какао и воски для суппозиториев; (9) масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар; (14) буферные реагенты, такие как гидроксид магния и гидроксид алюминия; (15) альгиновую кислоту; (16) апирогенную воду; (17) изотонический физиологический раствор; (18) раствор Рингера; (19) этиловый спирт; (20) pH-забуференные растворы; (21) сложные полиэфиры, поликарбонаты и/или полиангидриды; и (22) другие нетоксичные совместимые вещества, используемые в фармацевтических препаратах.
Термин "единичная дозированная форма" относится к физически дискретной единице, подходящей в качестве однократной дозы для человека или животного. Каждая единичная дозированная форма может содержать заданное количество терапевтического агента, рассчитанное на получение желаемого эффекта.
Если явно не указано иное, термин "пациент" относится к животному, такому как млекопитающее, включая, но без ограничения человека. Таким образом, способы, описанные в настоящем документе, можно использовать в лечении человека и в ветеринарии. В конкретных вариантах осуществления пациент представляет собой млекопитающее. В определенных вариантах осуществления пациент является человеком.
"Эффективное количество" относится к такому количеству терапевтического агента или его фармацевтически приемлемой соли, которое в сочетании с его параметрами эффективности и потенциальной токсичности, а также на основе знаний практикующего специалиста, должно быть эффективным в данной терапевтической форме. Как известно в данной области, эффективное количество может быть введено в одной или нескольких дозах.
"Лечение", "проведение лечения" и т.п. представляет собой подход, направленный на получение благоприятного или желаемого результата, включая клинические результаты. Применительно к описанию, благоприятные или желаемые результаты включают в себя, но не ограничиваются ими, ингибирование и/или подавление наступления и/или развития состояния или уменьшение тяжести такого состояния, например, уменьшение числа и/или тяжести симптомов, связанных с состоянием, улучшение качества жизни пациентов, страдающих от состояния, снижение дозы других лекарственных средств, необходимых для лечения состояния, увеличение эффекта другого лекарственного средства, которое пациент принимает из-за состояния, и/или увеличение срока выживаемости пациентов, имеющих состояние.
"Предотвратить", "предотвращение" и т.п. относится к снижению вероятности развития состояния у пациента, который не имеет состояния, но имеет риск развития состояния. Пациент, "имеющий риск", может иметь или не иметь выявляемое состояние, и у него может проявляться или не проявляться выявляемое состояние до применения способов лечения, описанных в настоящем документе. "Имеющий риск" означает, что пациент имеет один или несколько так называемых факторов риска, которые представляют собой измеряемые параметры, которые коррелируют с развитием состояния и которые известны в данной области. Пациент, имеющий один или несколько указанных факторов риска, имеет более высокую вероятность развития заболевания, чем пациент, не имеющий фактор(ов) риска.
"Положительный инотроп" относится к агенту, который вызывает увеличение сократительной функции миокарда. Типичные положительные инотропы представляют собой агонист бета-адренорецепторов, ингибиторы активности фосфодиэстеразы, и сенсибилизаторы кальция. Агонисты бета-адренорецепторов включают, среди прочего, допамин, добутамин, тербуталин, и изопротеренол. Аналоги и производные таких соединений также предполагаются. Например, в патенте США № 4663351 описано a пролекарство добутамин, которое можно вводить перорально.
Состояние, которое "реагирует на лечение нитроксилом" включает любое состояние, при котором введение соединения, которое служит донором эффективного количества нитроксила в физиологических условиях, лечит и/или предотвращает состояние, в том виде, как эти термины определены в описании. Состояние, симптомы которого подавляются или уменьшаются после введения донора нитроксила, является состоянием, которое реагирует на лечение нитроксилом.
"Легочная гипертензия" или "PH" относится к состоянию, при котором повышается давление в легочной артерии. Согласно принятому в настоящее время гемодинамическому определению PH представляет собой состояние, при котором среднее значение давления в легочной артерии (MPAP) в состоянии покоя большее или равное 25 мм рт.ст.. Badesch et al., J. Amer. Coll. Cardiol. 54(Suppl.):S55-S66 (2009).
"N/A" означает “не оценивали”.
"(C1-C6)алкил" относится к насыщенным линейным или разветвленным углеводородным структурам, имеющим 1, 2, 3, 4, 5 или 6 атомов углерода. Если указывается алкильный остаток, имеющий определенное число атомов углерода, предполагается, что охватываются все геометрические изомеры, имеющие указанное число атомов углерода, таким образом, например, "пропил" включает н-пропил и изо-пропил, и "бутил" включает н-бутил, сек-бутил, изо-бутил и терт-бутил. Примеры (C1-C6)алкильных групп включают метил, этил, н-пропил, изо-пропил, н-бутил, терт-бутил, н-гексил и т.п. "(C1-C4)алкил" относится к насыщенным линейным или разветвленным углеводородным структурам, имеющим 1, 2, 3, или 4 атома углерода. Примеры (C1-C4)алкильных групп включают метил, этил, н-пропил, изо-пропил, н-бутил, терт-бутил.
"(C3-C5)алкил" относится к насыщенным линейным или разветвленным углеводородным структурам, имеющим 3, 4, или 5 атомов углерода. Если указывается алкильный остаток, имеющий определенное число атомов углерода, предполагается, что охватываются все геометрические изомеры, имеющие указанное число атомов углерода, таким образом, например, "пропил" включает н-пропил и изо-пропил и "бутил" включает н-бутил, сек-бутил, изо-бутил и терт-бутил. Примеры (C3-C5)алкильных групп включают н-пропил, изо-пропил, н-бутил, терт-бутил, n-пентил и т.п.
"(C2-C4)алкенил" относится к линейному или разветвленному ненасыщенному углеводородному радикалу, имеющему 2, 3, или 4 атомов углерода и двойную связь в любом положении, например, этенил, 1-пропенил, 2-пропенил (аллил), 1-бутенил, 2-бутенил, 3-бутенил, 1-метилэтенил, 1-метил-1-пропенил, 2-метил-2-пропенил, 2-метил-1-пропенил, 1-метил-2-пропенил и т.п.
"(C2-C3)алкинил" относится к линейному нециклическому углеводороду, имеющему 2 или 3 атома углерода и содержащему по меньшей мере одну двойную углерод-углеродную связь. Примеры (C2-C3)алкенилов включают винил, аллил, и 1-проп-1-енил.
"(C5-C7)гетероциклоалкил" относится к 5-, 6-, или 7-членному, насыщенному или ненасыщенному, содержащему мостиковые связи, моно- или бициклическому гетероциклу, содержащему в кольце 1, 2, 3, или 4 гетероатома, каждый из которых независимо выбран из азота, кислорода и серы. Примеры (C5-C7) гетероциклоалкильных групп включают пиразолил, пирролидинил, пиперидинил, пиперазинил, тетрагидро-оксазинил, тетрагидрофуран, тиолан, дитиолан, пирролин, пирролидин, пиразолин, пиразолидин, имидазолин, имидазолидин, тетразол, пиперидин, пиридазин, пиримидин, пиразин, тетрагидрофуранон, γ-бутиролактон, α-пиран, γ-пиран, диоксолан, тетрагидропиран, диоксан, дигидротиофен, пиперазин, триазин, тетразин, морфолин, тиоморфолин, диазепан, оксазин, тетрагидро-оксазинил, изотиазол, пиразолидин и т.п.
"(5- или 6-членный)гетероарил" относится к моноциклическому ароматическому гетероциклическому кольцу, состоящему из 5 или 6 членов, т.е. к моноциклическому ароматическому кольцу, содержащему по меньшей мере один кольцевой гетероатом, например, 1, 2, 3, или 4 кольцевых гетероатома, каждый из которых независимо выбран из азота, кислорода и серы. Примеры -(5- или 6-членных)гетероарилов включают пиридил, пирролил, фурил, имидазолил, оксазолил, имидазолил, тиазолил, изоксазолил, 1,2,3-оксадиазолил, 1,3,4-оксадиазолил, 1,2,5-оксадиазолил, 1,2,3-триазолил, пиразолил, изотиазолил, пиридазинил, пиримидил, пиразинил, 1,2,3-тиадиазолил, 1,3,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,5-триазинил и тиофенил.
“Галоген” относится к -F, -CI, -Br или -I.
“Сульфо-н-бутиловый эфир β-циклодекстрина" относится к β-циклодекстрину, имеющему по меньшей мере одну группу -OH, который получают путем замещения атома водорода -(CH2)4-S(О)2-OH или -(CH2)4-S(О)2-O-Z+ для получения -О-(CH2)-S(О)2-OH или -О-(CH2)4-S(О)2-O-Z+ группы, соответственно, где Z+ представляет собой катион, такой как натрий, калий, аммоний, тетраметиламмоний, и тому подобное. В одном варианте осуществления каждый Z представляет собой натрий.
4.2 Соединения-доноры нитроксила с улучшенным терапевтическим индексом
В одном аспекте изобретение относится к новым соединениям, подходящим для лечения сердечно-сосудистых заболеваний (например, сердечной недостаточности). В частности, изобретение предлагает соединения-доноры нитроксила, которые обладают сочетанием свойств, которые делают их подходящими для использования в качестве лекарственного средства для человека. В частности, соединения-доноры нитроксила по изобретению иметь соответствующие периоды полураспада, благоприятный терапевтический индекс, являются хорошо растворимыми в воде и обладают достаточной стабильностью в твердом состоянии. В таблице 1 представлены два конкретных соединения N-гидроксисульфонамида, являющихся донорами нитроксила, по изобретению, которые обладают такими желательными свойствами и, следовательно, подходят для терапевтического применения у человека.
Соединения-доноры нитроксила по изобретению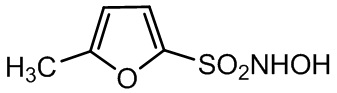
(1)
N-гидрокси-5-метилфуран-2-сульфонамид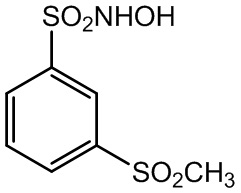
(2)
N-гидрокси-3-метансульфонилбензол-1-сульфонамид
В конкретных вариантах осуществления соединения-доноры нитроксила, приведенные в табл. 1, могут быть использованы в качестве фармацевтически приемлемой соли.
В других вариантах осуществления N-гидрокси-группа соединений, перечисленных в таблице 1, может быть этерифицирована для получения пролекарств соединений.
Например, изобретение предлагает соединения формулы (3):
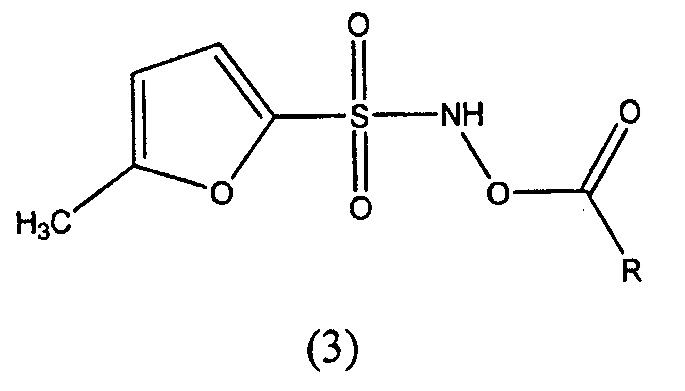
где R представляет собой водород, -(С1-C6)алкил, -(C2-C4)алкенил, фенил, бензил, циклопентил, циклогексил, -(C5-C7) гетероциклоалкил, бензилокси, -О-(C1-C6)алкил, -NH2, -NH-(C1-C4)алкил, или -N((C1-C4)алкил)2, где указанный -(С1-C6)алкил, -(C2-C4) алкенил, фенил, бензил, циклопентил, циклогексил, -(C5-C7)гетероциклоалкил, бензилокси, -О-(С1-C6)алкил, -NH-(C1-C4)алкил, или -N((C1-C4)алкил)2 могут быть не замещенными или замещенными одним или несколькими заместителями, выбранными из галогена, -(С1-C6)алкила, -(C2-C4)алкенила, -(C2-C3)алкинила, -(5- или 6-членного)гетероарила, -O-(C1-C6)алкила, -S-(С1-C6)алкила, -C(галоген)3, -CH(галоген)2, -CH2(галоген), -CN, -NО2, -NH2, -NH-(C1-C4)алкила, -N(-(C1-C4)алкил)2, -C(=О)(С1-C4)алкила, -C(=О)О(С1-C4)алкила, -OC(=О)(С1-C4)алкила, -OC(=О)NH2, -S(=О)(С1-C4)алкила, или -S(=О)2(C1-C4)алкила. В конкретных вариантах осуществления R представляет собой метил, этил, бензил или фенил.
В конкретных вариантах осуществления, где соединение по изобретению представляет собой соединение формулы (3), R представляет собой метил. В других вариантах осуществления, где соединение имеет формулу (3), R представляет собой этил. В определенных вариантах осуществления, где соединение по изобретению представляет собой соединение формулы (3), R представляет собой метил или этил. В других вариантах осуществления, где соединение имеет формулу (3), R представляет собой фенил. В других вариантах осуществления, где соединение имеет формулу (3), R представляет собой бензил. В конкретных вариантах осуществления, где соединение по изобретению представляет собой соединение формулы (3), R представляет собой бензил или фенил. В других вариантах осуществления, где соединение имеет формулу (3), R представляет собой -NH2. В каждом из вышеупомянутых вариантов осуществления в данном параграфе, R является незамещенным в одном варианте осуществления, монозамещенным в другом варианте осуществления, двузамещенным двумя независимо выбранными заместителями в дополнительном варианте осуществления, или трехзамещенным тремя независимо выбранными заместителями в дополнительном варианте осуществления. В различных вариантах осуществления каждого из вышеупомянутых вариантов осуществления в данном параграфе, заместитель представляет собой -галоген, -NH2, -NHCH3, -CF3 и -OCH3, или заместители независимо выбирают из -галогена, -NH2, -NHCH3, -CF3 и -OCH3.
Например, изобретение предлагает соединения формулы (4):
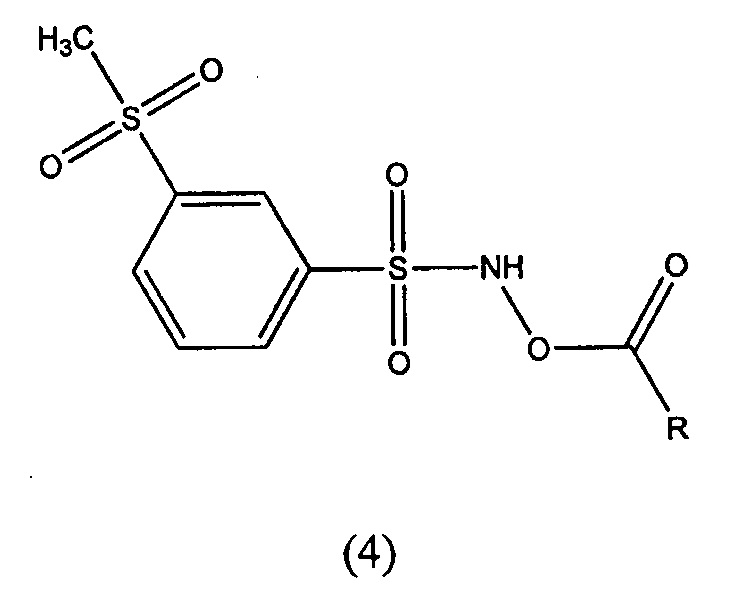
где R и его необязательный заместитель(ли) является такими, как определено выше, в отношении соединения формулы (3).
В конкретных вариантах осуществления, где соединение по изобретению представляет собой соединение (4), R представляет собой метил. В других вариантах осуществления, где соединение имеет формулу (4), R представляет собой этил. В определенных вариантах осуществления, где соединение по изобретению представляет собой соединение (4), R представляет собой метил или этил. В других вариантах осуществления, где соединение имеет формулу (4), R представляет собой фенил. В других вариантах осуществления, где соединение имеет формулу (4), R представляет собой бензил. В конкретных вариантах осуществления, где соединение по изобретению представляет собой соединение (4), R представляет собой бензил или фенил. В других вариантах осуществления, где соединение имеет формулу (4), R представляет собой -NH2. В каждом из вышеупомянутых вариантов осуществления в данном параграфе, R является незамещенным в одном варианте осуществления, монозамещенным в другом варианте осуществления, двузамещенным двумя независимо выбранными заместителями в дополнительном варианте осуществления, или трехзамещенным тремя независимо выбранными заместителями в дополнительном варианте осуществления. В различных вариантах осуществления каждого из вышеупомянутых вариантов осуществления в данном параграфе, заместитель представляет собой -галоген, -NH2, -NHCH3, -CF3 и -OCH3, или заместители независимо выбирают из -галогена, -NH2, -NHCH3, -CF3 и -OCH3.
Неожиданно было обнаружено, что соединения-доноры нитроксила по изобретению обеспечивают уровни эффективности, сходные с CXL-1020, при введении пациентам, но с существенно сниженными побочными эффектами, а именно местными побочными эффектами (например, раздражение и/или воспаление) (смотреть Примеры 8 и 9). Кроме того, соединения-доноры нитроксила по изобретению обеспечивают наступление гемодинамических эффектов в течение 1 часа или меньше, что является желательным с клинической точки зрения.
Вне связи с какой-либо теорией, эксперименты, описанные в разделе “Примеры” настоящего описания, позволяют предположить, что доноры нитроксила, имеющие периоды полураспада по существу короче, чем 15 минут при измерении в PBS или плазме человека (смотреть Пример 4), такие как CXL-1020, создают высокую местную концентрацию нитроксила после введения, и что высокая местная концентрация нитроксила является причиной наблюдаемых нежелательных побочных эффектов. Известно, что нитроксил в высокой концентрации димеризуется, что приводит к образованию азотноватистой кислоты, которая способна производить гидроксильные радикалы. Альтернативно или дополнительно, пероксид, выделяемый лейкоцитами, может реагировать с нитроксилом с образованием гидроксильных радикалов. Гидроксильные радикалы могут быть токсичными для эндотелиальных клеток, что приводит к воспалению и/или непереносимости. В то время как соединения нитроксила с более длительными периодами полураспада теоретически производят гидроксильные радикалы за счет сходных механизмов, и можно ожидать, что образование таких радикалов будет снижено благодаря низким концентрациям нитроксила, соответственно снизится способность нитроксила димеризоваться или реагировать с пероксидом.
Поэтому предполагается, что соединения с очень большими периодами полураспада (например, больше чем 95 минут при измерении в плазме человека в соответствии со способом, описанным в Примере 4) имеют благоприятный токсикологический профиль; однако, поскольку предполагается, что указанные соединения удаляются из кровотока и/или разбавляются до образования нитроксила в существенных количествах, ожидается, что такие соединения имеют низкую эффективность.
Как описано в Примере 4, соединения формул (1) и (2) имеют периоды полураспада больше чем приблизительно 10 минут и меньше чем 95 минут при измерении в аэрированном фосфатно-солевом буферном растворе (PBS) при pH 7,4 и при измерении в плазме человека при pH 7,4 в присутствии антикоагулянта (например, гепарина или цитрата натрия), каждое измерение проводили в условиях, указанных в Примере 4. В частности, соединение формулы (1) имеет период полураспада приблизительно 68 минут при измерении в аэрированном фосфатно-солевом буферном растворе (PBS) при pH 7,4, и приблизительно 65 минут при измерении в плазме человека при pH 7,4 в присутствии антикоагулянта (например, гепарина или цитрата натрия), каждое измерение проводили в условиях, указанных в Примере 4. Соединение формулы (2) имеет период полураспада приблизительно 50 минут при измерении в аэрированном фосфатно-солевом буферном растворе (PBS) при pH 7,4, и приблизительно 37 минут при измерении в плазме человека при pH 7,4 в присутствии антикоагулянта (например, гепарина или цитрата натрия), каждое измерение проводили в условиях, указанных в Примере 4. Кроме того, как описано в примере 5, каждое из соединений формул (1) и (2) хорошо растворимы в воде, и, таким образом, пригодны для парентерального или перорального введения. Соединения можно включать в состав без добавления солюбилизирующего агента. Кроме того, как показано в Примерах 10-12, соединения формулы (1) и формулы (2) имеют отличную стабильность в фармацевтических композициях для парентерального (например, внутривенного) введения.
4.3 Измерение способности отдавать нитроксил
Способность соединений отдавать нитроксил, можно легко проверить с помощью обычных экспериментов. Хотя обычно затруднительно непосредственно измерить отдается ли нитроксил, несколько аналитических подходов одобрены, в качестве подходящих для определения является ли соединение донором нитроксила. Например, исследуемое соединение модно поместить в раствор, например, в фосфотно-солевой буферный раствор (PBS) или в забуференный фосфатом раствор со значением pH приблизительно 7,4, в герметичном контейнере. По истечении времени, достаточного для диссоциации, например, от нескольких минут до нескольких часов, газ, находящийся в свободном пространстве над продуктом, собирают и анализируют с целью определения его состава, например, с помощью газовой хроматографии и/или масс-спектрометрии. Если образуется N2О (что происходит при димеризации HNO), результат теста в отношении способности отдавать нитроксил является положительным, и соединение считают донором нитроксила.
Величина способности отдавать нитроксил может быть выражена в виде процента от теоретического стехиометрического максимального значения для соединения. Соединение, которое отдает "существенное количество нитроксила" обозначает, в различных вариантах осуществления, соединение, которое отдает приблизительно 40% или больше, приблизительно 50% или больше, приблизительно 60% или больше, приблизительно 70% или больше, приблизительно 80% или больше, приблизительно 90% или больше, или приблизительно 95% или больше от теоретического максимального количества нитроксила. В конкретных вариантах осуществления, соединение-донор нитроксила по изобретению отдает приблизительно от 70% до 90% теоретического максимального количества нитроксила. В конкретных вариантах осуществления соединение-донор нитроксила отдает приблизительно от 85% до 95% теоретического максимального количества нитроксила. В конкретных вариантах осуществления соединение-донор нитроксила отдает приблизительно от 90% до 95% теоретического максимального количества нитроксила. Соединения, которые дают меньше, чем приблизительно 40% или меньше чем приблизительно 50% от теоретического максимального количества нитроксила, так же являются донорами нитроксила, и могут быть использованы в описанных способах. Соединение, которое дает меньше, чем приблизительно 50% от теоретического максимального количества нитроксила, может быть использовано в описанных способах, но может предусматривать более высокие уровни доз, по сравнению с соединением, которое дает более высокий уровень нитроксила.
При необходимости, способность отдавать нитроксил также можно оценить путем воздействия исследуемого соединения на метмиоглобин (Mb3+). Смотреть Bazylinski et al., J Amer. Chem. Soc. 107(26):7982-7986 (1985). Нитроксил реагирует с Mb3+ с образованием комплекса Mb2+-NO, который можно обнаружить с помощью изменений в ультрафиолетовом/видимом спектре или с помощью электронного парамагнитного резонанса (EPR). Комплекс Mb2+-NO имеет сигнал EPR, основанный на g-значении приблизительно 2. Оксид азота, с другой стороны, реагирует с Mb3+ с образованием комплекса Mb3+-NO, который имеет незначительный, если таковой имеется, сигнал EPR. Таким образом, если соединение реагирует с Mb3+ с образованием комплекса, обнаруживаемого обычными методами, такими как определение ультрафиолетового/видимого спектра или EPR, то тест является положительным в отношении способности отдавать нитроксил.
Тестирование способности отдавать нитроксил можно осуществить при физиологически значимом уровне pH. Соединения-доноры нитроксила по изобретению способны отдавать нитроксил при физиологическом значении pH (т.е. при pH приблизительно 7,4) и нормальной температуре (т.е. при температуре приблизительно 37°C) (вместе "физиологические условия"). В конкретных вариантах осуществления соединение-донор нитроксила по изобретению может отдавать приблизительно 40% или больше от теоретического максимального (т.е. 100%) количества нитроксила в физиологических условиях. В конкретных вариантах осуществления соединение-донор нитроксила по изобретению может отдавать приблизительно 50% или больше от теоретического максимального количества нитроксила в физиологических условиях. В конкретных вариантах осуществления соединение-донор нитроксила по изобретению может отдавать приблизительно 60% или больше от теоретического максимального количества нитроксила в физиологических условиях. В конкретных вариантах осуществления соединение-донор нитроксила по изобретению может отдавать приблизительно 70% или больше от теоретического максимального количества нитроксила в физиологических условиях. В конкретных вариантах осуществления, соединение-донор нитроксила по изобретению может отдавать приблизительно 80% или больше от теоретического максимального количества нитроксила в физиологических условиях. В конкретных вариантах осуществления соединение-донор нитроксила по изобретению может отдавать приблизительно 90% или больше от теоретического максимального количества нитроксила в физиологических условиях.
Следует понимать, что соединение-донор нитроксила по изобретению также может отдавать ограниченное количество оксида азота, поскольку количество отдаваемого нитроксила превышает количество отдаваемого оксида азота. В определенных вариантах осуществления, соединение-донор нитроксила может отдавать приблизительно 25 моль% или меньше оксида азота в физиологических условиях. В конкретных вариантах осуществления соединение-донор нитроксила может отдавать приблизительно 20 моль% или меньше оксида азота в физиологических условиях. В конкретных вариантах осуществления, соединение-донор нитроксила может отдавать приблизительно 15 моль% или меньше оксида азота в физиологических условиях. В конкретных вариантах осуществления, соединение-донор нитроксила может отдавать приблизительно 10 моль% или меньше оксида азота в физиологических условиях. В конкретных вариантах осуществления, соединение-донор нитроксила может отдавать приблизительно 5 моль% или меньше оксида азота в физиологических условиях. В конкретных вариантах осуществления, соединение-донор нитроксила может отдавать приблизительно 2 моль% или меньше оксида азота в физиологических условиях. В конкретных вариантах осуществления соединение-донор нитроксила может отдавать незначительное количество (например, приблизительно 1 моль% или меньше) оксида азота в физиологических условиях.
4.4 Фармацевтические композиции
Изобретение также охватывает фармацевтические композиции, содержащие соединение-донор нитроксила формул (1), (2), (3) или (4) и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество. Примеры фармацевтически приемлемых вспомогательных веществ включают вещества, описанные выше, такие как носители, сурфактанты, загустители или эмульгаторы, твердые связующие вещества, дисперсионные или суспензионные средства, солюбилизаторы, красители, вкусовые добавки, покрытия, разрыхлители, смазывающие вещества, подсластители, консерванты, изотонические агенты и любые их комбинации. Отбор и использование фармацевтически приемлемых вспомогательных веществ описано, например, в Troy, Ed., Remington: The Science and Practice of Pharmacy, 21 <st>Ed. (Lippincott Williams & Wilkins, Baltimore, MD, 2005). В различных вариантах осуществления по меньшей мере одно фармацевтически приемлемое вспомогательное вещество содержит по меньшей мере один вид циклодекстрина. В конкретном варианте осуществления циклодекстрин представляет собой циклическую структуру, имеющую глюкозные остатки, связанные α(l-4) связями. В другом варианте осуществления циклодекстрин представляет собой β-циклодекстрин, т.е. циклическую структуру, имеющую семь глюкозных остатков, связанных α(l-4) связями. В другом варианте осуществления циклодекстрин химически модифицируют путем дериватизации любой комбинации трех доступных гидроксильных групп на каждом глюкопиранозном остатке.
В некоторых вариантах осуществления, где фармацевтически приемлемое вспомогательное вещество содержит по меньшей мере один вид циклодекстрина, циклодекстрин представляет собой сульфо(C1-C6)алкилэфирное производное β-циклодекстрина. В некоторых из этих вариантов осуществления, циклодекстрин представляет собой сульфо(C1-C6)алкилэфирное производное β-циклодекстрина, имеющее приблизительно от шести до семи групп сульфо(C1-C6)алкильного эфира на молекулу циклодекстрина. В различных вариантах осуществления циклодекстрин представляет собой сульфо(C1-C6)алкилэфирное производное β-циклодекстрина, содержащее в среднем приблизительно от шести до семи сульфо(C1-C6)алкилэфирных групп на молекулу циклодекстрина. В другом таком варианте осуществления, циклодекстрин представляет собой сульфо(C1-C6)алкилэфирное производное β-циклодекстрина, имеющее шесть или семь сульфо(C1-C6)алкилэфирных групп на молекулу циклодекстрина.
В конкретном ряду вариантов осуществления, где фармацевтически приемлемое вспомогательное вещество содержит по меньшей мере один вид циклодекстрина, циклодекстрин представляет собой сульфо(C3-C5)алкилэфирное производное β-циклодекстрина. В одном таком варианте осуществления циклодекстрин представляет собой сульфо(C3-C5)алкилэфирное производное β-циклодекстрина, имеющее приблизительно от шести до семи сульфо(C3-C5)алкилэфирных групп на молекулу циклодекстрина. В различных таких вариантах осуществления, циклодекстрин представляет собой сульфо(C3-C5) алкилэфирное производное β-циклодекстрина, имеющее в среднем приблизительно от шести до семи сульфо(C3-C5)алкилэфирных групп на молекулу циклодекстрина. В другом таком варианте осуществления, циклодекстрин представляет собой сульфо(C3-C5) алкилэфирное производное β-циклодекстрина, имеющий шесть или семь сульфо(C3-C5)алкилэфирных групп на молекулу циклодекстрина.
В конкретных вариантах осуществления, где фармацевтически приемлемое вспомогательное вещество содержит по меньшей мере один вид циклодекстрина, циклодекстрин представляет собой сульфобутилэфирное производное β-циклодекстрина. В некоторых из таких вариантов осуществления, циклодекстрин представляет собой сульфобутилэфирное производное β-циклодекстрина, имеющий приблизительно от шести до семи сульфобутилэфирных групп на молекулу циклодекстрина. В другом таком варианте осуществления, циклодекстрин представляет собой сульфобутилэфирное производное β-циклодекстрина, имеющий в среднем приблизительно от шести до семи сульфобутилэфирных групп на молекулу циклодекстрина. В другом таком варианте осуществления, циклодекстрин представляет собой сульфобутилэфирное производное β-циклодекстрина, имеющее шесть или семь сульфобутилэфирных групп на молекулу циклодекстрина.
В определенных вариантах осуществления, где фармацевтически приемлемое вспомогательное вещество содержит по меньшей мере один вид циклодекстрина, циклодекстрин представляет собой сульфобутилэфирное производное β-циклодекстрина. В одном таком варианте осуществления, циклодекстрин представляет собой сульфобутилэфирное производное β-циклодекстрина, имеющее приблизительно от шести до семи сульфобутилэфирных групп на молекулу циклодекстрина. В другом таком варианте осуществления циклодекстрин представляет собой сульфобутилэфирное производное β-циклодекстрина, имеющее в среднем приблизительно от шести до семи сульфобутилэфирных групп на молекулу циклодекстрина. В другом таком варианте осуществления циклодекстрин представляет собой сульфобутилэфирное производное β-циклодекстрина, имеющее шесть или семь сульфобутилэфирных групп на молекулу циклодекстрина.
В различных конкретных вариантах осуществления, где фармацевтически приемлемое вспомогательное вещество содержит по меньшей мере один вид циклодекстрина, циклодекстрин содержит множество отрицательных зарядов при физиологически совместимых значениях pH, например, при pH приблизительно от 5,0 до 6,8 в некоторых вариантах осуществления, приблизительно от 5,5 до 6,5 в некоторых вариантах осуществления, приблизительно от 5,7 до 6,3 в некоторых вариантах осуществления, приблизительно от 5,8 до 6,2 в некоторых вариантах осуществления, приблизительно от 5,9 до 6,1 в некоторых вариантах осуществления, и приблизительно 6,0 в конкретных вариантах осуществления. В одном таком варианте осуществления, по меньшей мере одно фармацевтически приемлемое вспомогательное вещество содержит циклодекстрин CAPTISOL® (Ligand Pharmaceuticals, La Jolla, CA).
Фармацевтические композиции могут быть созданы для введения в твердой или жидкой форме, в том числе предназначенные для перечисленного ниже: (1) пероральное введение, например, в виде орошений (например, водных или неводных растворов или суспензий), таблеток (например, предназначенных для буккальной, сублингвальной и системной абсорбции), каплет, пилюль, порошков, гранул, паст для нанесения на язык, твердых желатиновых капсул, мягких желатиновых капсул, спреев для рта, пастилок, лепешек, гранул, сиропов, суспензий, эликсиров, жидкостей, эмульсий и микроэмульсий; или (2) парентеральное введение, например, с помощью подкожной, внутримышечной, внутривенной или эпидуральной инъекции, в виде, например, стерильного раствора или суспензии. Фармацевтические композиции могут быть предназначены для немедленного, пролонгированного и контролируемого высвобождения.
В одном конкретном варианте осуществления фармацевтическая композиция создана для внутривенного введения. В другом варианте осуществления фармацевтическая композиция создана для внутривенного введения с помощью непрерывной инфузии.
В другом варианте осуществления фармацевтическая композиция составлена для перорального введения. Соединения для перорального введения могут быть составлены в виде жидких или твердых лекарственных форм. В конкретных вариантах осуществления, где соединения-доноры нитроксила включены в состав в виде пероральных жидких лекарственных форм, полиэтиленгликоль 300 (PEG300) может эффективно использоваться в качестве вспомогательного вещества.
Соединения и фармацевтические композиции, описанные в настоящем документе, могут быть изготовлены в любой подходящей стандартной лекарственной форме, например в виде капсул, саше, таблеток, порошка, гранул, раствора, суспензии в водной жидкости, суспензии в неводной жидкости, жидкие эмульсии масло-в-воде, жидкие эмульсии вода-в-масле, липосомы или болюс.
Таблетки могут быть изготовлены прессованием или формованием, необязательно с одним или несколькими дополнительными ингредиентами. Прессованные таблетки могут быть получены прессованием в соответствующей машине терапевтического агента или агентов в свободно-текучей форме, таких как порошок или гранулы, необязательно смешанного со связующим веществом, смазывающим веществом, инертным разбавителем, консервантом, поверхностно-активным или диспергирующим агентом. Формованные таблетки могут быть изготовлены формованием в соответствующем аппарате смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. Таблетки необязательно могут быть покрыты или на них могут быть нанесены риски, и они могут быть созданы таким образом, чтобы обеспечить медленное или контролируемое высвобождение активного ингредиента. Способы создания таких композиций с замедленным или контролируемым высвобождением фармацевтически активных ингредиентов, таких как описанные в настоящем документе терапевтические агенты и другие соединения, известные в данной области, известны в данной области и описаны в опубликованных патентах США, некоторые из которых включают, но без ограничения патенты США №№ 4369174, 4842866 и ссылки, цитируемые в них. Покрытия могут быть использованы для доставки соединений в кишечник (смотреть, например, патенты США. №№ 6638534, 5217720, 6569457 и ссылки, цитируемые в них). Специалисту в данной области понятно, что наряду с таблетками, другие лекарственные формы могут быть созданы для обеспечения замедленного или контролируемого высвобождения активного ингредиента. Такие лекарственные формы включают, но не ограничиваются ими, капсулы, гранулы и гель-капсулы.
Фармацевтические композиции, подходящие для местного применения включают, без ограничения леденцы, содержащие ингредиенты в ароматизированной основе, такие как сахароза, гуммиарабик и трагакант; и пастилки, содержащие активный ингредиент в ароматизированной основе, либо в инертной основе, такой как желатин и глицерин.
Различные варианты осуществления фармацевтических композиций, подходящих для парентерального введения, включают, без ограничения либо стерильные водные растворы для инъекций, либо неводные стерильные инъекционные растворы, каждый из которых содержит, например, антиоксиданты, буферы, бактериостатические факторы и растворенные вещества, которые придают композиции изотоничность крови предполагаемого получателя; и водные стерильные суспензии и неводные стерильные суспензии, каждая из которых содержит, например, суспендирующие агенты и загустители. Композиции могут быть предоставлены в однодозовых или многодозовых контейнерах, например, в запечатанных ампулах или флаконах, и могут храниться в высушенном сублимацией (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, такого как вода, непосредственно перед применением.
Фармацевтические композиции для парентерального введения могут быть введены в кислом, нейтральном или щелочном растворе. В одном варианте осуществления фармацевтические композиции, содержащие соединение-донор нитроксила по изобретению, могут быть приготовлены в виде кислотного раствора, имеющего значение рН приблизительно от 4 до 5, например, значение pH приблизительно 4, приблизительно 4,5, приблизительно 4,8, или приблизительно 5, в том числе значения между ними. В то время как значение рН приблизительно 4 принято считать оптимальным для создания композиций, отдающих нитроксил, с целью достижения адекватной стабильности соединения, установлено, что создание композиции в кислой среде потенциально может вызвать или усилить венозное раздражение после парентерального введения. Степень раздражения может быть снижена путем включения соединений-доноров нитроксила в менее кислые или даже нейтральные растворы (смотреть фиг. 4). Соответственно, в конкретных вариантах осуществления соединения-доноры нитроксила по изобретению могут быть включены в состав для парентерального применения при pH приблизительно от 5 до 6,2 (например, pH приблизительно 5, приблизительно 5,5, приблизительно 5,8, приблизительно 6, или приблизительно 6,2, в том числе значения там между ними).
4.5 Способы применения соединений и фармацевтических композиций по изобретению
В одном аспекте изобретение относится к способу увеличения in vivo уровней нитроксила, способ включает введение пациенту, который в этом нуждается, эффективного количества соединения или фармацевтической композиции, как описано в настоящем документе. В различных вариантах осуществления пациент имеет, предположительно имеет, или имеет риск наличия или развития состояния, которое реагирует на терапию нитроксилом.
В конкретных вариантах осуществления раскрытие относится к способу лечения, профилактики или замедления наступления и/или развития состояния, способ включает введение пациенту (включая пациента, идентифицированного как нуждающегося в таком лечении, профилактике или замедлении наступления заболевания) эффективного количества соединения или фармацевтической композиции, как описано в настоящем документе. Идентификация пациента, нуждающегося в перечисленном выше, может быть основана на оценке врача, персонала медицинского учреждения, сотрудников аварийно-спасательных служб или других специалистов в области медицины и может быть субъективной (например, мнение) или объективной (например, измеряемой с помощью теста или диагностического метода). Конкретные состояния, охватываемые способами, описанными в настоящем документе, включают без ограничения сердечно-сосудистые заболевания, ишемически-реперфузионное повреждение, и легочную гипертензию (PH).
4.5.1 Сердечно-сосудистые заболевания
В одном варианте осуществления изобретение относится к способу лечения сердечно-сосудистого заболевания, способ включает введение эффективного количества соединения или фармацевтической композиции, как описано в настоящем документе, пациенту, который в этом нуждается.
Примеры сердечно-сосудистых заболеваний и симптомов, которые можно эффективно лечить с помощью соединений и композиций, описанных в настоящем документе, включают сердечно-сосудистые заболевания, которые реагируют на терапию нитроксилом, коронарные обструкции, заболевание коронарной артерии (CAD), стенокардию, сердечный приступ, инфаркт миокарда, высокое кровяное давление, ишемическую кардиомиопатию и образование инфаркта, застой крови в легких, отек легких, фиброз сердца, заболевание клапанов сердца, заболевание перикарда, застойные сосудистые состояния, периферический отек, асциты, болезнь Шагаса, гипертрофию желудочков, заболевание сердечных клапанов, сердечную недостаточность, диастолическую сердечную недостаточность, систолическую сердечную недостаточность, застойную сердечную недостаточность, острую застойную сердечную недостаточность, острую декомпенсированную сердечную недостаточность, и гипертрофию сердца.
4.5.1.1 Сердечная недостаточность
Соединения-доноры нитроксила и композиции по изобретению можно использовать для лечения пациентов, страдающих от сердечной недостаточности. Сердечная недостаточность может быть любого типа или вида, включая любую сердечную недостаточность, описанную в настоящем документе. Неограничивающие примеры сердечной недостаточности включают раннюю стадию сердечной недостаточности, сердечную недостаточность I, II, III и IV стадии, острую сердечную недостаточность, застойную сердечную недостаточность (CHF) и острую застойную сердечную недостаточность. В одном варианте осуществления соединения и композиции по изобретению можно использовать для лечения острой декомпенсированной сердечной недостаточности.
В вариантах осуществления, где соединения-доноры нитроксила и композиции по изобретению используют для лечения пациентов, страдающих от сердечной недостаточности, другой активный агент, который используют для лечения сердечной недостаточности, также может быть введен. В одном таком варианте осуществления донор нитроксила может быть введен в сочетании с положительным инотропом, таким как бета-агонист. Примеры бета-агонистов включают без ограничения допамин, добутамин, изопротеренол, аналоги и производные таких соединений. В другом варианте осуществления донор нитроксила может быть введен в сочетании с антагонистом бета-адренергического рецептора (также обозначаемый здесь как бета-антагонист или бета-блокатор). Примеры бета-антагонистов включают без ограничения пропранолол, метопролол, бисопролол, буциндололом и карведилол.
Как описано в Примерах 6 и 7, различные модели сердечной недостаточности были использованы для оценки гемодинамических профилей нескольких соединений-доноров нитроксила по изобретению. Как показано на фиг. 1-3, которые обсуждаются в примерах 6 и 7, соединения формулы (1) и формулы (2) вызывали, например, существенное усиление сократимости и расслабления миокарда, и умеренное снижение артериального давления без тахикардии. Кроме того, наступление существенных гемодинамических эффектов было быстрым (например, в течение 1 часа) и эффект, близкий к максимальному, достигался в течение 2 часов.
В то время как показатели гемодинамической активности соединений формулы (1) и формулы (2), аналогичны показателям композиций, содержащих донор нитроксила CXL-1020, при внутривенном введении, токсикологический профиль соединений формулы (1) и формулы (2), которые имеют более продолжительный период полураспада, чем CXL-1020, значительно улучшается по сравнению с композициями, содержащими CXL-1020 (смотреть Пример 9 и фиг. 4 и 5). Например, "дозы, не вызывающие наблюдаемых нежелательных эффектов" (NOAEL) соединений формулы (1) и формулы (2) были существенно выше, чем NOAEL для CXL-1020 (смотреть Пример 9 для описания определения NOAEL). В частности, соединение формулы (1) имеет наиболее благоприятный токсикологический профиль среди всех доноров нитроксила типа N-гидроскисульфонамида, тестированных на данный момент, и не проявляет нежелательных явлений, исходя из клинических маркеров воспаления, при внутривенном введении в концентрациях по меньшей мере до 30 мкг/кг/мин (фиг. 4). Для сравнения, CXL-1020 начинает демонстрировать нежелательные побочные эффекты уже в концентрации 0,3 мкг/кг/мин.
4.5.1.2 Ишемически-реперфузионное повреждение
В другом варианте осуществления, раскрытый предмет изобретения относится к способу лечения, профилактики или замедления наступления и/или развития ишемически-реперфузионного повреждения, способ включает введение эффективного количества соединения или фармацевтической композиции, как описано в настоящем документе индивиду, нуждающемуся в этом. В конкретном варианте осуществления способ предназначен для предотвращения ишемически-реперфузионного повреждения.
В конкретном варианте осуществления соединение или фармацевтическую композицию по изобретению вводят до начала ишемии. В конкретном варианте осуществления фармацевтическую композицию по изобретению вводят перед началом процедур, во время которых может произойти ишемия миокарда, например ангиопластика или операция, например, операция аортокоронарного шунтирования. В конкретном варианте осуществления фармацевтическую композицию по изобретению вводят после ишемии, но до реперфузии. В конкретном варианте осуществления фармацевтическую композицию по изобретению вводят после ишемии и реперфузии.
В другом варианте осуществления фармацевтическая композиция по изобретению может быть введена пациенту, который подвержен риску ишемического события. В конкретном варианте осуществления фармацевтическую композицию по изобретению вводят пациенту, подверженному риску ишемического события в будущем, но не имеющему признаков ишемии в данный момент. Определение подверженности пациента риску ишемического события может быть осуществлено любым способом, известным в данной области, например, путем проверки пациента или истории болезни пациента. В конкретном варианте осуществления пациент имел предшествующее ишемическое событие. Таким образом, пациент может быть подвержен риску первого или последующего ишемического события. Примеры пациентов с риском ишемического события включают пациентов с известной гиперхолестеринемией, изменениями ЭКГ, связанными с ишемией (например, заостренные или инвертированные T-волны или увеличение или уменьшение ST-сегмента в соответствующем клиническом контексте), с нарушением ЭКГ, не связанным с активной ишемией, с повышенным CKMB, с клиническими признаками ишемии (например, сдавливающая боль за грудиной или боль в руке, одышка и/или повышенное потоотделение), инфаркт миокарда в анамнезе, повышенный уровень холестерина в сыворотке крови, малоподвижный образ жизни, ангиографические данные, свидетельствующие о частичной обструкции коронарных артерий, эхокардиографические свидетельства повреждения миокарда, или любые другие данные, свидетельствующие о риске ишемического события в будущем. Примеры ишемических событий включают без ограничения инфаркт миокарда (MI) и нейроваскулярную ишемию, такую как a нарушение мозгового кровообращения (CVA).
В другом варианте осуществления предметом лечения является орган, который нужно трансплантировать. В конкретном варианте осуществления фармацевтическая композиция по изобретению может быть введена перед реперфузией органа реципиенту трансплантата. В конкретном варианте осуществления фармацевтическая композиция по изобретению может быть введена перед извлечением органа у донора, например, с помощью перфузионных канюль, используемых в процессе извлечения органа. Если донор является живым донором, например, донором почки, соединения или фармацевтические композиции по изобретению можно вводить донору. В конкретном варианте осуществления соединения или фармацевтические композиции по изобретению вводят путем хранения органа в растворе, содержащем соединение или фармацевтическую композицию. Например, соединение или фармацевтическая композиция по изобретению могут быть включены в раствор для сохранения органа, такой как раствор Висконсинского университета "UW", который представляет собой раствор, содержащий гидроксиэтилкрахмал, по существу свободный от этиленгликоля, этиленхлоргидрина и ацетона (смотреть патент США № 4798824). В конкретном варианте осуществления фармацевтическая композиция по изобретению, которую вводят, является такой, что ишемически-реперфузионное повреждение тканей органа уменьшается после реперфузии у реципиента трансплантированного органа. В конкретном варианте осуществления способ уменьшает некроз тканей (размер инфаркта) в тканях, подверженных риску.
Ишемически-реперфузионное повреждение может повреждать ткани, помимо тканей миокарда, и описываемый объект изобретения охватывает способы лечения или профилактики такого повреждения. В различных вариантах осуществления ишемически-реперфузионное повреждение не является миокардиальным. В конкретных вариантах осуществления способ уменьшает повреждение, вызванное ишемией/реперфузией в ткани головного мозга, печени, кишечника, почек, желудка, или любой другой части тела, кроме миокарда. В другом варианте осуществления пациент имеет риск такого повреждения. Отбор индивида, имеющего риск немиокардиальной ишемии, определение показателей, используемых для оценки риска ишемии миокарда. Однако другие факторы могут указывать на риск ишемии/реперфузии в других тканях. Например, хирургические пациенты часто испытывают ишемию, связанную с операцией. Таким образом, пациентов, которым назначена хирургическая операция, можно рассматривать, как имеющих риск ишемического события. Следующие факторы риска развития инсульта (или подгруппа таких факторов риска) могут продемонстрировать риск развития у пациента ишемии ткани мозга: гипертония, курение, стеноз сонной артерии, отсутствие физической активности, сахарный диабет, гиперлипидемия, транзиторная ишемическая атака, фибрилляция предсердий, коронарно-артериальное заболевание, застойная сердечная недостаточность, перенесенный инфаркт миокарда, дисфункция левого желудочка при наличии пристеночного тромба, и стеноз митрального клапана. Ingall, Postgrad. Med. 107(6):34-50 (2000). Кроме того, осложнения нелеченной инфекционной диареи у пожилых могут включать ишемию миокарда, почек, сосудов головного мозга и кишечника. Slotwiner-Nie et al, Gastroenterol. Clin. N. Amer. 30(3):625-635 (2001). Альтернативно пациенты могут быть отобраны на основе факторов риска развития ишемической болезни кишечника, почек и/или печени. Например, лечение может быть начато у пожилых пациентов с риском гипотензивных эпизодов (например, потеря крови во время операции). Таким образом, пациенты, имеющие указанный признак, будут рассматриваться как имеющие риск ишемического события. В другом варианте осуществления пациент имеет любое одно или несколько состояний, перечисленных в описании, таких как сахарный диабет и гипертония. Другие состояния, которые могут привести к ишемии, такие как церебральная артериовенозная мальформация, могут свидетельствовать о риске развития у пациента ишемического события.
4.5.2 Легочная гипертензия
В другом варианте осуществления соединения или фармацевтическую композицию по изобретению можно использовать для предотвращения или замедления наступления и/или развития легочной гипертензии. В одном таком варианте осуществления соединения или фармацевтическая композиция по изобретению могут быть использованы для предотвращения или замедления наступления и/или развития легочной артериальной гипертензии (PAH).
В другом варианте осуществления раскрытый предмет изобретения относится к способу снижения давления в легочной артерии (MPAP), включающему введение эффективного количества соединения или фармацевтической композиции, описанных в настоящем документе пациенту, который в этом нуждается. В другом варианте осуществления MPAP уменьшается примерно на 50%. В другом варианте осуществления MPAP уменьшается примерно на 25%. В другом варианте осуществления MPAP уменьшается примерно на 20%. В другом варианте осуществления, MPAP уменьшается примерно на 15%. В другом варианте осуществления MPAP уменьшается примерно на 10%. В другом варианте осуществления MPAP уменьшается примерно на 5%. В другом варианте осуществления MPAP уменьшается таким образом, что составляет от приблизительно 12 мм рт.ст. до приблизительно 16 мм рт.ст. В другом варианте осуществления MPAP уменьшается таким образом, что составляет приблизительно 15 мм рт.ст.
4.6 Способы, схемы введения и уровни доз
Соединения и фармацевтические композиции по изобретению могут быть введены с помощью парентерального (например, подкожного, внутримышечного, внутривенного или внутрикожного) введения. В определенных вариантах осуществления соединение или фармацевтическую композицию вводят с помощью внутривенной инфузии. В других вариантах осуществления соединения и фармацевтические композиции по изобретению могут быть введены путем перорального введения. Когда вводят фармацевтическую композицию, содержащую соединение по настоящему изобретению, дозы выражают на основе количества активного фармацевтического ингредиента, т.е. количества соединения(й)-доноров нитроксила по изобретению, представленного в фармацевтической композиции.
При внутривенном введении доза может быть удобно выражена на единицу времени, либо в виде фиксированного количества на единицу времени, либо в виде количества, рассчитанного по весу на единицу времени.
В различных вариантах осуществления соединение или фармацевтическую композицию по изобретению вводят внутривенно в количестве по меньшей мере приблизительно 0,1 мкг/кг/мин, по меньшей мере приблизительно 0,2 мкг/кг/мин, по меньшей мере приблизительно 0,3 мкг/кг/мин, по меньшей мере приблизительно 0,4 мкг/кг/мин, по меньшей мере приблизительно 0,5 мкг/кг/мин, по меньшей мере приблизительно 1 мкг/кг/мин, по меньшей мере приблизительно 2,5 мкг/кг/мин, по меньшей мере приблизительно 5 мкг/кг/мин, по меньшей мере приблизительно 7,5 мкг/кг/мин, по меньшей мере приблизительно 10 мкг/кг/мин, по меньшей мере приблизительно 11 мкг/кг/мин, по меньшей мере приблизительно 12 мкг/кг/мин, по меньшей мере приблизительно 13 мкг/кг/мин, по меньшей мере приблизительно 14 мкг/кг/мин, по меньшей мере приблизительно 15 мкг/кг/мин, по меньшей мере приблизительно 16 мкг/кг/мин, по меньшей мере приблизительно 17 мкг/кг/мин, по меньшей мере приблизительно 18 мкг/кг/мин, по меньшей мере приблизительно 19 мкг/кг/мин, по меньшей мере приблизительно 20 мкг/кг/мин, по меньшей мере приблизительно 21 мкг/кг/мин, по меньшей мере приблизительно 22 мкг/кг/мин, по меньшей мере приблизительно 23 мкг/кг/мин, по меньшей мере приблизительно 24 мкг/кг/мин, по меньшей мере приблизительно 25 мкг/кг/мин, по меньшей мере приблизительно 26 мкг/кг/мин, по меньшей мере приблизительно 27 мкг/кг/мин, по меньшей мере приблизительно 28 мкг/кг/мин, по меньшей мере приблизительно 29 мкг/кг/мин, по меньшей мере приблизительно 30 мкг/кг/мин, по меньшей мере приблизительно 31 мкг/кг/мин, по меньшей мере приблизительно 32 мкг/кг/мин, по меньшей мере приблизительно 33 мкг/кг/мин, по меньшей мере приблизительно 34 мкг/кг/мин, по меньшей мере приблизительно 35 мкг/кг/мин, по меньшей мере приблизительно 36 мкг/кг/мин, по меньшей мере приблизительно 37 мкг/кг/мин, по меньшей мере приблизительно 38 мкг/кг/мин, по меньшей мере приблизительно 39 мкг/кг/мин, или по меньшей мере приблизительно 40 мкг/кг/мин.
В различных вариантах осуществления соединение или фармацевтическую композицию по настоящему изобретению вводят внутривенно в количестве не больше, чем приблизительно 100 мкг/кг/мин, не больше, чем приблизительно 90 мкг/кг/мин, не больше, чем приблизительно 80 мкг/кг/мин, не больше, чем приблизительно 70 мкг/кг/мин, не больше, чем приблизительно 60 мкг/кг/мин, не больше, чем приблизительно 50 мкг/кг/мин, не больше, чем приблизительно 49 мкг/кг/мин, не больше, чем приблизительно 48 мкг/кг/мин, не больше, чем приблизительно 47 мкг/кг/мин, не больше, чем приблизительно 46 мкг/кг/мин, не больше, чем приблизительно 45 мкг/кг/мин, не больше, чем приблизительно 44 мкг/кг/мин, не больше, чем приблизительно 43 мкг/кг/мин, не больше, чем приблизительно 42 мкг/кг/мин, не больше, чем приблизительно 41 мкг/кг/мин, не больше, чем приблизительно 40 мкг/кг/мин, не больше, чем приблизительно 39 мкг/кг/мин, не больше, чем приблизительно 38 мкг/кг/мин, не больше, чем приблизительно 37 мкг/кг/мин, не больше, чем приблизительно 36 мкг/кг/мин, не больше, чем приблизительно 35 мкг/кг/мин, не больше, чем приблизительно 34 мкг/кг/мин, не больше, чем приблизительно 33 мкг/кг/мин, не больше, чем приблизительно 32 мкг/кг/мин, не больше, чем приблизительно 31 мкг/кг/мин, или не больше, чем приблизительно 30 мкг/кг/мин.
В некоторых вариантах осуществления соединение или фармацевтическую композицию по настоящему изобретению вводят внутривенно в количестве, изменяющемся от приблизительно 0,1 мкг/кг/мин до приблизительно 100 мкг/кг/мин, от приблизительно 1 мкг/кг/мин до приблизительно 100 мкг/кг/мин, от приблизительно 2,5 мкг/кг/мин до приблизительно 100 мкг/кг/мин, от приблизительно 5 мкг/кг/мин до приблизительно 100 мкг/кг/мин, от приблизительно 10 мкг/кг/мин до приблизительно 100 мкг/кг/мин, от приблизительно 1,0 мкг/кг/мин до приблизительно 80 мкг/кг/мин, от приблизительно 10,0 мкг/кг/мин до приблизительно 70 мкг/кг/мин, от приблизительно 20 мкг/кг/мин до приблизительно 60 мкг/кг/мин, от приблизительно 15 мкг/кг/мин до приблизительно 50 мкг/кг/мин, от приблизительно 0,01 мкг/кг/мин до приблизительно 1,0 мкг/кг/мин, от приблизительно 0,01 мкг/кг/мин до приблизительно 10 мкг/кг/мин, от приблизительно 0,1 мкг/кг/мин до приблизительно 1,0 мкг/кг/мин, от приблизительно 0,1 мкг/кг/мин до приблизительно 10 мкг/кг/мин, от приблизительно 1,0 мкг/кг/мин до приблизительно 5 мкг/кг/мин, от приблизительно 70 мкг/кг/мин до приблизительно 100 мкг/кг/мин, или от приблизительно 80 мкг/кг/мин до приблизительно 90 мкг/кг/мин. В конкретных вариантах осуществления соединение или фармацевтическую композицию по настоящему изобретению вводят внутривенно в количестве, изменяющимся от приблизительно 10 мкг/кг/мин до приблизительно 50 мкг/кг/мин, от приблизительно 20 мкг/кг/мин до приблизительно 40 мкг/кг/мин, от приблизительно 25 мкг/кг/мин до приблизительно 35 мкг/кг/мин, или от приблизительно 30 мкг/кг/мин до приблизительно 40 мкг/кг/мин. В конкретных вариантах осуществления соединение или фармацевтическую композицию по настоящему изобретению вводят внутривенно в количестве от приблизительно 20 мкг/кг/мин до приблизительно 30 мкг/кг/мин.
В различных вариантах осуществления, включая различные варианты осуществления с пероральным введением, соединения или фармацевтические композиции по изобретению вводят согласно суточной схеме применения, рассчитанной на основе веса, либо в виде однократной суточной дозы (QD), любо в виде нескольких отдельных доз, вводимых, например, два раза в день (BID), три раза в день (TID), или четыре раза в день (QID).
В определенных вариантах осуществления соединение-донор нитроксила или фармацевтическую композицию по изобретению вводят в дозе по меньшей мере приблизительно 0,5 мг/кг/д, по меньшей мере приблизительно 0,75 мг/кг/д, по меньшей мере приблизительно 1,0 мг/кг/д, по меньшей мере приблизительно 1,5 мг/кг/д, по меньшей мере приблизительно 2 мг/кг/д, по меньшей мере приблизительно 2,5 мг/кг/д, по меньшей мере приблизительно 3 мг/кг/д, по меньшей мере приблизительно 4 мг/кг/д, по меньшей мере приблизительно 5 мг/кг/д, по меньшей мере приблизительно 7,5 мг/кг/д, по меньшей мере приблизительно 10 мг/кг/д, по меньшей мере приблизительно 12,5 мг/кг/д, по меньшей мере приблизительно 15 мг/кг/д, по меньшей мере приблизительно 17,5 мг/кг/д, по меньшей мере приблизительно 20 мг/кг/д, по меньшей мере приблизительно 25 мг/кг/д, по меньшей мере приблизительно 30 мг/кг/д, по меньшей мере приблизительно 35 мг/кг/д, по меньшей мере приблизительно 40 мг/кг/д, по меньшей мере приблизительно 45 мг/кг/д, по меньшей мере приблизительно 50 мг/кг/д, по меньшей мере приблизительно 60 мг/кг/д, по меньшей мере приблизительно 70 мг/кг/д, по меньшей мере приблизительно 80 мг/кг/д, по меньшей мере приблизительно 90 мг/кг/д, или по меньшей мере приблизительно 100 мг/кг/д.
В определенных вариантах осуществления, соединение-донор нитроксила или фармацевтическую композицию по изобретению вводят в дозе не больше, чем приблизительно 100 мг/кг/д, не больше, чем приблизительно 100 мг/кг/д, не больше, чем приблизительно 90 мг/кг/д, не больше, чем приблизительно 80 мг/кг/д, не больше, чем приблизительно 80 мг/кг/д, не больше, чем приблизительно 75 мг/кг/д, не больше, чем приблизительно 70 мг/кг/д, не больше, чем приблизительно 60 мг/кг/д, не больше, чем приблизительно 50 мг/кг/д, не больше, чем приблизительно 45 мг/кг/д, не больше, чем приблизительно 40 мг/кг/д, не больше, чем приблизительно 35 мг/кг/д, не больше, чем приблизительно 30 мг/кг/д. В различных вариантах осуществления доза составляет от приблизительно 0,001 мг/кг/д до приблизительно 10,000 мг/кг/д. В определенных вариантах осуществления, доза составляет от приблизительно 0,01 мг/кг/д до приблизительно 1,000 мг/кг/д. В определенных вариантах осуществления доза составляет от приблизительно 0,01 мг/кг/д до приблизительно 100 мг/кг/д. В определенных вариантах осуществления доза составляет от приблизительно 0,01 мг/кг/д до приблизительно 10 мг/кг/д. В определенных вариантах осуществления доза составляет от приблизительно 0,1 мг/кг/д до 1 мг/кг/д. В определенных вариантах осуществления доза составляет меньше, чем приблизительно 1 мг/кг/д.
В определенных вариантах осуществления соединение или фармацевтическую композицию по изобретению вводят в диапазоне доз, в котором нижний предел диапазона составляет любое количество от приблизительно 0,1 мг/кг/день до приблизительно 90 мг/кг/день и верхний предел диапазона составляет любое количество от приблизительно 1 мг/кг/день до приблизительно 100 мг/кг/день (например, от приблизительно 0,5 мг/кг/день до приблизительно 2 мг/кг/день в одних сериях вариантов осуществлений и от приблизительно 5 мг/кг/день до приблизительно 20 мг/кг/день в других сериях вариантов осуществлений).
В конкретных вариантах осуществления соединение или фармацевтическую композицию по изобретению вводят в диапазоне доз от приблизительно 3 до приблизительно 30 мг/кг, вводимых от одного раза в день (QD) до трех раз в день (TID).
В определенных вариантах осуществления соединения или фармацевтические композиции по изобретению вводят в соответствии с постоянной (т.е. не зависящей от веса) схемой применения, либо в виде однократной дневной дозы (QD), либо в виде нескольких отдельных доз, вводимых, например, два раза в день (BID), три раза в день (TID), или четыре раза в день (QID). В различных вариантах осуществления соединение или фармацевтическую композицию по изобретению вводят в дозе по меньшей мере приблизительно 0,01 грамм/день (г/д), по меньшей мере приблизительно 0,05 г/д, по меньшей мере приблизительно 0,1 г/д, по меньшей мере приблизительно 0,5 г/д, по меньшей мере приблизительно 1 г/д, по меньшей мере приблизительно 1,5 г/д, по меньшей мере приблизительно 2.0 г/д, по меньшей мере приблизительно 2,5 г/д, по меньшей мере приблизительно 3,0 г/д, или по меньшей мере приблизительно 3,5 г/д.
В различных вариантах осуществления соединение или фармацевтическую композицию по изобретению вводят в дозе не больше, чем приблизительно 5 г/д, не больше, чем приблизительно 4,5 г/д, не больше, чем приблизительно 4 г/д, не больше, чем приблизительно 3,5 г/д, не больше, чем приблизительно 3 г/д, не больше, чем приблизительно 2,5 г/д, или не больше, чем приблизительно 2 г/д. В определенных вариантах осуществления соединение или фармацевтическую композицию по изобретению вводят в дозе от приблизительно 0,01 грамм в день до приблизительно 4,0 грамм в день. В определенных вариантах осуществления соединение или фармацевтическую композицию по изобретению можно вводить в дозе, в которой нижний предел диапазона составляет любое количество от приблизительно 0,1 мг/день до приблизительно 400 мг/день и верхний предел диапазона составляет любое количество от приблизительно 1 мг/день до приблизительно 4000 мг/день. В определенных вариантах осуществления соединение или фармацевтическую композицию вводят в дозе от приблизительно 5 мг/день до приблизительно 100 мг/день. В различных вариантах осуществления соединение или фармацевтическую композицию вводят в дозе от приблизительно 150 мг/день до приблизительно 500 мг/день. Интервал дозирования при парентеральном или пероральном введении может быть скорректирован в соответствии с потребностями пациента. При более длительных интервалах между введениями, можно использовать препараты с замедленным высвобождением или депо-препараты.
Соединение или фармацевтическую композицию, которые описаны в настоящем документе, можно вводить до, по существу одновременно с, или после введения дополнительного терапевтического агента. Схема применения может включать предшествующее лечение и/или совместное введение с дополнительным терапевтическим агентом. В таком случае, соединение или фармацевтическую композицию и дополнительный терапевтический агент можно вводить одновременно, раздельно или последовательно.
Примеры схем применения включают без ограничения: введение каждого соединения, фармацевтической композиции или терапевтического агента в последовательном порядке; и совместное введение каждого соединения, фармацевтической композиции или терапевтического агента по существу одновременно (например, в виде дозированной лекарственной формы для однократного приема) или в виде нескольких отдельных дозированных лекарственных форм каждого соединения, фармацевтической композиции или терапевтического агента. Специалистам в данной области понятно, что "эффективное количество" или "доза" ("уровень дозы") будет зависеть от различных факторов, таких как конкретный способ введения, режим введения, выбранное соединение и фармацевтическая композиция, а также конкретное состояние и пациент, который получает лечение. Например, соответствующий уровень дозы может варьировать в зависимости от активности, скорости выведения и потенциальной токсичности конкретного соединения или используемой фармацевтической композиции; возраста, массы тела, общего состояния здоровья, пола и диеты пациента, получающего лечение; частоты введения; другого терапевтического агента(ов), который вводят совместно; и типа и тяжести состояния.
4.7 Наборы, содержащие соединения или фармацевтические композиции
Изобретение относится к наборам, содержащим соединение или фармацевтическую композицию, описанным в настоящем документе. В конкретном варианте осуществления набор содержит соединение или фармацевтическую композицию, описанные в настоящем документе, каждый в сухом виде, и фармацевтически приемлемый жидкий разбавитель.
В конкретных вариантах осуществления, либо соединение в сухом виде, либо фармацевтическая композиция в сухом виде содержит приблизительно 2,0% или меньше воды по весу, приблизительно 1,5% или меньше воды по весу, приблизительно 1,0% или меньше воды по весу, приблизительно 0,5% или меньше воды по весу, приблизительно 0,3% или меньше воды по весу, приблизительно 0,2% или меньше воды по весу, приблизительно 0,1% или меньше воды по весу, приблизительно 0,05% или меньше воды по весу, приблизительно 0,03% или меньше воды по весу, или приблизительно 0,01% или меньше воды по весу. Фармацевтически приемлемые жидкие разбавители известны в данной области и включают, но не ограничиваются ими, стерильную воду, солевые растворы, водный раствор декстрозы, глицерин, растворы глицерина и т.п. Другие примеры подходящих жидких разбавителей описаны Nairn, "Solutions, Emulsions, Suspensions and Extracts," pp. 721-752 in Remington: The Science and Practice of Pharmacy, 20th Ed. (Lippincott Williams & Wilkins, Baltimore, MD, 2000). В одном варианте осуществления набор дополнительно содержит инструкции по применению соединения или фармацевтической композиции. Инструкции могут быть в любой подходящей форме, такой как письменная или электронная форма. В другом варианте осуществления инструкции могут представлять собой письменные инструкции. В другом варианте осуществления инструкции содержатся в электронном носителе информации (например, магнитная дискета или оптический диск). В другом варианте осуществления инструкции включают информацию относительно соединения или фармацевтической композиции и способа введения соединения или фармацевтической композиции пациенту. В другом варианте осуществления инструкции относятся к способу применения, описанному в настоящем документе (например, лечение, профилактика и/или замедление наступления и/или развития состояния, выбранного из сердечно-сосудистых заболеваний, ишемически-реперфузионного повреждения, легочной гипертензии и других состояний, реагирующих на лечение нитроксилом).
В другом варианте осуществления набор дополнительно включает подходящую упаковку. Если набор содержит больше чем одно соединение или фармацевтическую композицию, соединения или фармацевтические композиции могут быть аккуратно упакованы в отдельные контейнеры, или соединены в одном контейнере, если позволяет перекрестная реактивность и срок годности.
5. ПРИМЕРЫ
Следующие примеры представлены только в иллюстративных целях и не должны служить для ограничения объема раскрытого предмета изобретения.
5.1 Синтез соединений
Соединения, описанные в настоящем документе, могут быть получены в соответствии со способами, описанными ниже, или по способам, известным в данной области. Исходные вещества для реакций могут быть коммерчески доступными или могут быть получены с помощью известных способов или их очевидных модификаций. Например, некоторые из исходных веществ доступны у коммерческих поставщиков, таких как Sigma-Aldrich (St. Louis, MO). Другие вещества могут быть получены с помощью способов, или их очевидных модификаций, описанных в стандартных справочниках, таких как March's Advanced Organic Chemistry (John Wiley and Sons) и Larock's Comprehensive Organic Transformations (VCH Publishers).
Пример 1. Получение N-гидрокси-5-метилфуран-2-сульфонамида (1)
К раствору гидроксиламина (0,92 мл 50% водного раствора; 13,8 ммоль) в THF (6 мл) и воды (2 мл), охлажденной до 0°C, добавляли 5-метилфуран-2-сульфонилхлорид (1 г, 5,5 ммоль) в виде раствора в THF (6 мл) по каплям для поддержания температуры ниже 10°C. Реакционную смесь перемешивали в течение 5 мин, после указанного времени наблюдали, что TLC (1:1 гексан:этилацетат (H:EA)) практически полностью поглощал сульфонилхлорид. Реакционную смесь разбавляли в два раза 50 мл дихлорметана (DCM) и органическую часть отделяли и промывали водой (10 мл). Органическую часть высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Продукт хроматографировали на силикагеле, элюируя смесью гептана:EtOAc с последующим растиранием с гептаном получая указанное в заголовке соединение в виде желтого твердого вещества (0,59 г, выход 61%). ЖХ-МС tR=0,91 мин; 1H ЯМР (ДМСО, 500 МГц) δ м.д. 9,82 (1H, д, J=3,1 Гц), 9,64 (1H, д, J=3,2 Гц), 7,10 (1H, д, J=3,4 Гц), 6,36 (1H, д, J=3,4 Гц), 2,36 (3H, с).
Пример 2. Получение N-гидрокси-3-метансульфонилбензол-1-сульфонамида (2)
3-метансульфонилбензол-1-сульфонилхлорид
Промежуточный продукт 3-метансульфонилбензол-1-сульфонилхлорид синтезировали по способам, описанным Park et al, J. Med. Chem. 51(21):6902-6915 (2008). При этом метилсульфонилбензол (110 г, 0,7 моль) нагревали в течение 18 часов при 90°C в хлорсульфоновой кислоте (450 мл, 6,7 моль), после чего реакционную смесь охлаждали до температуры приблизительно 21°C, перед тем как медленно вылить на измельченный лед. Полученную суспензию дважды экстрагировали в EtOAc (2 л для каждой экстракции). Органические части объединяли, промывали солевым раствором (50 мл) затем высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением промежуточного продукта сульфонилхлорида в виде беловатого твердого вещества (125 г, выход 75%). 1H ЯМР (400 МГц, CDC13) δ м.д. 8,61 (1Н, т, J=1,7 Гц), 8,35-8,31 (2H, м), 7,90 (1H, т, J=7,9 Гц), 3,15 (3H, с).
N-гидрокси-3-метансульфонильбензол-1-сульфонамид
К раствору водного гидроксиламина (16 мл 50% водного раствора, 245 ммоль) в THF (150 мл) и воды (25 мл), охлажденному до -5°C, медленно добавляли 3-метансульфонилбензол-1-сульфонилхлорид (25 г, 98 ммоль), поддерживая температуру реакции ниже 10°C. Реакционную смесь выдерживали при данной температуре до тех пор, пока не наблюдали по существу полного вхождения в реакцию сульфонилхлорида (приблизительно 5 мин), после чего реакционную смесь разбавляли DCM (250 мл), органическую часть отделяли и отмывали дважды 50 мл воды. Водные экстракты объединяли и дважды повторно отмывали DCM (250 мл для каждой отмывки). Все органические части объединяли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде твердого вещества бежевого цвета. Растирание осуществляли с использованием смеси гептаны:EtOAc (1:1; об:об) с получением указанного в заголовке соединения в виде твердого вещества бежевого цвета (14 г, выход 56%). ЖХ-МС tR = 0,90 мин; масс-спектроскопия высокого разрешения (HRMS): теоретическое значение (C7H9NO5S2) = 249,9844, измеренное значение = 249,9833; 1H ЯМР (500 МГц, ДМСО-d6) δ м.д. 9,85 (2H, кв, J=3,3 Гц), 8,31 (1H, дт, J=1,6 Гц), 8,28 (1H, дт, J=7,8, 1,3 Гц), 8,14-8,19 (1H, м), 7,93 (1H, т, J=7,9 Гц), 3,32 (3H, с).
5.2 Пример 3. Количественное определение образования нитроксила с помощью N2О
Закись азота (N2О) образуется при димеризации и дегидратации HNO, и она является наиболее распространенным маркером образования нитроксила (Fukuto et al, Chem. Res. Toxicol. 18:790-801 (2005)). Однако частично нитроксил может взаимодействовать с кислородом, с образованием продукта, который не превращается в N2O (смотреть Mincione et al, J. Enzyme Inhibition 13:267-284 (1998); и Scozzafava et al, J. Med. Chem. 43:3677-3687 (2000)). Используя газ закись азота или соль Анджели (AS) в качестве эталона, определяли относительные количества N2O, выделившиеся из соединений по изобретению, осуществляя анализ объема газа над продуктом с помощью газовой хроматографии (GC).
Процедура определения относительных количеств N2O, выделившихся из соединений по изобретению описана ниже. GC осуществляли на газовом хроматографе Agilent, оснащенном инжектором для ввода проб с делением потока (деление 10:1), детектором электронного захвата, и капиллярной колонкой с молекулярными ситами HP-MOLSIV 30 м × 0,32 мм × 25 мкм. Гелий использовали в качестве газа-носителя (4 мл/мин) и азот использовали в качестве вспомогательного газа (20 мл/мин). Термостат инжектора и термостат детектора устанавливали на 200°C и 325°C соответственно. Все анализы содержания закиси азота осуществляли при установленной постоянной температуре колоночного термостата 200°C.
Все инъекции газа осуществляли с помощью автоматизированного анализатора паровой фазы. Подача во флакон осуществлялась под давлением 15 фунтов/кв.дюйм. Термостат для образцов, кран-дозатор, и переходную линию анализатора выдерживали при 40°C, 45°C, и 50°C, соответственно. Показатели времени стабилизации термостата, заполнения флакона под давлением, заполнения петли, уравновешивания петли, и инъекции образца составляли 1,00 мин, 0,20 мин, 0,20 мин, 0,05 мин, и 1,00 мин соответственно.
Для всех измерений использовали партию флаконов для парофазного анализа с номинальным объемом 20 мл, при этом объемы предварительно измеряли в целях достижения однородности образцов (фактический объем флаконов изменялся на ≤ 2,0% относительного стандартного отклонения (n=6)). Средний объем флакона в партии определяли для шести случайно выбранных флаконов путем вычисления разности веса между закрытым и запечатанным пустым (т.е. заполненным воздухом) флаконом и закрытым и запечатанным флаконом, заполненным деионизированной водой, используя известное значение плотности деионизированной воды, с последующим определением среднего значения. Холостые пробы получали путем закрывания и запечатывания двух флаконов, при этом осторожно продували каждый флакон в течение 20 секунд струей аргона. Стандарты нитроксила получали путем закрывания и запечатывания четырех флаконов, при этом осторожно продували каждый флакон в течение 1 минуты струей газа из газового баллона, содержащего стандарт нитроксила в концентрации 3000 м.д.
"Стандарты" CXL-1020 (N-гидрокси-2-метансульфонилбензен-1-сульфонамид) готовили в дубликатах, аккуратно взвешивая 10±0,5 мг CXL-1020 и добавляя в каждый 4 мл флакон. С использованием автоматической пипетки 1 мл обработанного аргоном безводного DMF (Sigma-Aldrich) добавляли в каждый 4 мл флакон для получения стокового раствора CXL-1020 для каждого образца и флаконы закрывали и встряхивали и/или обрабатывали ультразвуком, чтобы обеспечить полное растворение, исходя из визуального наблюдения. С использованием автоматической пипетки 20 мл флаконы заполняли 5 мл PBS (обработанными в течение по меньшей мере 30 мин. аргоном перед использованием), продували аргоном в течение по меньшей мере 20 сек и закрывали резиновой пробкой. С использованием 50 мкл шприца, 50 мкл стокового раствора CXL-1020 инъецировали в каждый 20 мл флакон, содержащий PBS.
Подготовку образцов осуществляли, как указано ниже. В дубликатах, 18±1 мг каждого образца аккуратно взвешивали в каждый 4 мл флакон. С использованием автоматической пипетки 1 мл обработанного аргоном безводного DMF добавляли в каждый 4 мл флакон для получения образца стокового раствора для каждого образца, и флаконы закрывали и встряхивали и/или обрабатывали ультразвуком, чтобы обеспечить полное растворение, исходя из визуального наблюдения. С использованием автоматической пипетки, 20 мл флаконы заполняли 5 мл PBS (обработанными в течение по меньшей мере 30 мин. аргоном перед использованием), продували аргоном в течение по меньшей мере 20 сек, и закрывали резиновой пробкой. Флаконы выдерживали в течение по меньшей мере 10 мин. при 37°C в сухом термостате. Затем с использованием 50 мкл шприца 50 мкл образца стокового раствора инъецировали в каждый 20 мл флакон, содержащий PBS. Затем флаконы выдерживали при 37°C в сухом термостате в течение периода времени, такого, что сумма времени, проведенного в сухом термостате, плюс времени, проведенного в термостате автоматизированного анализатора паровой фазы перед инъекцией образца, была равна желаемому времени инкубации.
Автоматическое введение осуществляли в следующей последовательности: 1-й дубликат холостой пробы, 2-й дубликат холостой пробы, 1-й дубликат стандарта N2О, 2-й дубликат стандарта N2О, 1-й дубликат стандарта CXL-1020, 2-й дубликат стандарта CXL-1020, 1-й дубликат образца 1, 2-й дубликат образца 1, 1-й дубликат образца 2, 2-й дубликат образца 2, и т.д., заканчивая 3-м дубликатом стандарта N2О, и 4-м дубликатом стандарта N2О. Электронную таблицу EXCEL используют для ввода данных, установленных таким образом, и рассчитывают для каждого образца относительное значение выхода N2О в процентах для каждого значения времени инкубации. Полученные результаты представлены в таблице 2.
Результаты анализа N2О в паровой фазе
Для соединений формул (3) и (4), определение проводится так, как описано выше, за исключением того, что также готовят образцы, активированные ферментом, как описано ниже: (i) точно взвешивают 50 мг эстеразы свиной печени (PLE, E3019-20KU, не обработанная, Sigma-Aldrich) в 20 мл флакон для парофазного анализа; (ii) с использованием автоматической пипетки добавляют 5 мл обработанного аргоном безводного PBS для получения стокового раствора PLE; (iii) флакон закрывают и встряхивают, чтобы обеспечить полное растворение, исходя из визуального наблюдения; (iv) образцы доноров нитроксила подготавливают, как описано выше за исключением того, что добавляют 4,75 мл PBS вместо 5 мл; и (v) с использованием автоматической пипетки в 20 мл флаконы затем помещают 250 мкл стокового раствора PLE перед добавлением образца. Автоматическое введение осуществляют в следующей последовательности: 1-й дубликат холостой пробы, 2-й дубликат холостой пробы, 1-й дубликат стандарта N2О, 2-й дубликат стандарта N2О, 1-й дубликат стандарта CXL-1020, 2-й дубликат стандарта CXL-1020, образец 1 (без PLE) 1-й дубликат, образец 1 (без PLE) 2-й дубликат, образец 1 (с PLE) 1-й дубликат, образец 1 (с PLE) 2-й дубликат, образец 2 (без PLE) 1-й дубликат, образец 2 (без PLE) 2-й дубликат, образец 2 (с PLE) 1-й дубликат, образец 2 (с PLE) 2-й дубликат и т.д., заканчивая 3-м дупликатом стандарта N2О, и 4-м дупликатом стандарта N2О.
Другой способ определения относительных количеств N2О, выделяемого соединениями по изобретению, описан ниже. GC осуществляют на хроматографе Varian CP-3800, оборудованном ручным инжектором 1041, детектором захвата электронов, и 25 м капиллярной колонкой с молекулярным ситом 5A. Азот марки 5,0 используют как в качестве газа-носителя (8 мл/мин), так и в качестве вспомогательного газа (22 мл/мин). Термостат инжектора и термостат детектора устанавливают на 200°C и 300°C соответственно. Все анализы закиси азота осуществляли при установленной постоянной температуре колоночного термостата 200°C.. Все анализы содержания закиси азота осуществляют при установленной постоянной температуре колоночного термостата 150°C. Все инъекции газа осуществляли с помощью 100 мкл газонепроницаемого шприца с контролем объема образца. Образцы готовят в 15 мл оранжевых флаконах для парофазного анализа, объемы которых предварительно измеряют в целях достижения однородности образцов (фактический объем флакона изменяется от 15,19 до 15,20 мл). Флаконы заполняют 5 мл PBS, содержащего диэтилентриаминпентауксусный ангидрид (DTPA), продувают аргоном и закрывают резиновой пробкой. Флаконы оставляют по меньшей мере на 10 мин при 37°C в сухом термостате. Готовят 10 мМ стоковый раствор AS в 10 мМ растворе гидроксида натрия, и готовят растворы доноров нитроксила либо в ацетонитриле, либо в метаноле и используют непосредственно после получения. Вводят 50 мкл указанных стоковых растворов в отдельные термически уравновешенные флаконы для парофазного анализа с помощью 100 мкл газонепроницаемого шприца с контролем объема образца для получения конечных концентраций субстрата 0,1 мМ. Затем субстраты инкубируют в течение 90 мин или 360 мин. Затем берут образцы паровой фазы (60 мкл) и вводят последовательно пять раз в хроматограф GC с помощью газонепроницаемого шприца с контролем объема образца. Указанную процедуру повторяют для двух или больше флаконов для каждого соединения-донора.
5.3. Пример 4. Стабильность in vitro доноров нитроксила в плазме
Тестировали стабильность в плазме соединения (1), соединения (2) и CXL-1020. Система тестирования включала в себя (i) PBS или плазму крысы, собаки или человека (объединенная плазма, полученная по меньшей мере от 3 доноров мужского пола) с pH 7,4, и (ii) для тестов, осуществляемых в плазме, антикоагулянт (гепарин натрия или цитрат натрия). Каждое тестируемое соединение (5 мкМ) инкубировали в PBS или плазме при 37°C в THERMOMIXER® при встряхивании. Получали по три образца (n=3) для каждой из семи временных точек забора образцов: 0, 10, 30, 60, 90, 180 и 360 мин. Образцы немедленно объединяли с 3 объемами (т.е. с 3-кратным объемом PBS или плазмы) ацетонитрила, содержащего 1% муравьиной кислоты и внутренний стандарт, для остановки реакции. Анализ ЖХ/МС/МС тестируемых соединений осуществляли с помощью устройства AB SCIEX API 3000 без стандартной кривой.
Периоды полураспада (Τ1/2) тестируемых соединений определяли из графиков процента оставшихся значений с использованием коэффициента отклика детектора, выраженного в виде площади пика. Установленные периоды полураспада представлены в таблице 3.
Периоды полураспада (Τ1/2) доноров нитроксила
Для измерения периодов полураспада соединений формулы (3) или формулы (4), стоковый раствор эстеразы печени свиньи (PLE) добавляют к PBS или плазме перед добавлением указанного соединения
5.4. Пример 5. Растворимость доноров нитроксила
Сначала значения растворимости соединений формулы (1) и формулы (2) измеряли путем визуальной оценки в концентрациях 100 мкг/мл и 1000 мкг/мл в буфере со значением pH 4. Буфер получали путем смешивания 660 мл раствора A (10,5023 г лимонной кислоты, растворенной в 1 л воды) и 450 мл раствора B (14,7010 г трехосновного дигидрата цитрата натрия, растворенного в 1 л воды). Значение pH буфера, измеренное с помощью pH-метра, составляло 3,98.
Каждое соединение перемешивали встряхиванием в течение приблизительно 5 мин в буферном растворе с pH 4, приготовленном как указано выше, для двух значений концентрации (100 мкг/мл и 1000 мкг/мл), и определяли растворимость визуально. Полученные результаты представлены в таблице 4.
Растворимость соединений в буфере со значением pH 4 в концентрациях 100 мкг/мл и 1000 мкг/мл
Y = растворимо после перемешивания в течение 5 мин в буфере с pH 4
Кроме того, получали образец соединения формулы (1) в воде для определения приблизительного значения растворимости соединения в отсутствие вспомогательных веществ (например, CAPTISOL®). Достигали значения концентрации приблизительно 300 мг/мл, без учета увеличения объема за счет объема соединения. Определяли, что значение pH образца составляло 2,8, указанный показатель доводили до заданного значения 4,0 с помощью 0,1 N раствора NaOH. После доведения рН, наблюдали осаждение небольшого количества твердого вещества. Прозрачный раствор разбавляли в ацетонитриле и проводили анализ с помощью ВЭЖХ, в результате получали измеренную концентрацию раствора 268 мг/мл. Аналогичный анализ осуществляли для соединения формулы (2). Соединение формулы (2) имеет растворимость приблизительно 10 мг/мл.
5.5 Пример 6. Гемодинамическая эффективность доноров нитроксила у здоровых собак и собак с сердечной недостаточностью (модель сердечной недостаточности, индуцированной тахикардией)
5.5.1 Материалы и методы
Сердечно-сосудистые эффекты доноров нитроксила проверяли с помощью анализа кривой (контура) давление-объем (PV), проведенного на бодрствующих, фиксированных в станке собаках породы бигль. Животные имели свободный доступ к питьевой воде и собачьему корму, представленному на рынке, в стандартных лабораторных условиях. Люминисцентное освещение обеспечивалось с помощью автоматического таймера в течение приблизительно 12 часов в день. В отдельных случаях, ночной цикл периодически прерывали в связи с действиями, предусмотренными исследованиями. Температуру и влажность контролировали и регистрировали ежедневно и поддерживали в максимально возможной степени в интервале от 17,8°С до 29°С и от 30% до 70% соответственно. Собаки проходили акклиматизацию в течение по меньшей мере 1 недели перед операцией. После операции и восстановления животные привыкали к фиксирующему станку в течение 4,5 часов. Животных не кормили в течение ночи перед операцией.
Хирургические процедуры
Анестезия
Постоянный венозный катетер помещали в периферическую вену (например, головную) для введения анестетика. Общий наркоз индуцировали внутривенно (струйное введение) бупренорфином (приблизительно 0,015 мг/кг) с последующим внутривенным струйным введением пропофола (приблизительно 6 мг/кг). Кроме того, после индукции профилактически вводили антибиотик (цефазолин от 20 до 50 мг/кг в/в). Трахеальную трубку вводили и использовали для осуществления механической вентиляции легких 100% О2 с помощью аппарата искусственной вентиляции легких животных с переключением по объему (приблизительно 12 вдохов/минуту с дыхательным объемом приблизительно 12,5 мл/кг) для поддержания значения PaCО2 в пределах физиологического диапазона. Анестезию обеспечивали ингаляцией изофлурана (от 1% до 3%).
Оборудование для определения показателей сердечно-сосудистой системы
После достижения стабильного (хирургического) уровня наркоза осуществляли левостороннюю торакотомию (в строго асептических условиях) и каждому животному вживляли сономикрометрические кристаллы, обеспечивающие измерение размеров/объема левого желудочка (LV).
Кроме того, заполненный жидкостью катетер и полупроводниковый манометр помещали в левый желудочек для контроля давления. Заполненный жидкостью катетер помещали в правый желудочек (RV) и аорту (Ao) для контроля давления/введения тестируемого препарата. Гидравлический обтуратор (In-Vivo Metrics) помещали/закрепляли вокруг нижней полой вены (IVC), для обеспечения контролируемого сужения вены при построении кривых давление-объем LV во время гетерометрической саморегуляции. Катетеры/провода асептически прокладывали и выводили наружу между лопатками. В ходе исследования, заполненные жидкостью катетеры регулярно (по крайней мере, один раз в неделю) промывали блокирующим раствором, чтобы предотвратить образование сгустков и бактериальный рост (2-3 мл раствора тауролидина-цитрата, TCS-04; Access Technologies).
Имплантация кардиостимулятора
После установки оборудования для определения показателей сердечно-сосудистой системы, правую яремную вену осторожно выделяли и канюлировали биполярным водителем ритма/катетером (CAPSUREFIX Novus; Medtronic). Под флюороскопическим контролем направления, указанный водитель ритма продвигали в прямом направлении в правый желудочек и активно прикрепляли (путем внедрения) к эндокарду в апикальной части. Проксимальный конец водителя ритма прикрепляли к устройству для стимуляции сердечного ритма (Kappa 900; Medtronic). Затем водитель ритма помещали/закрепляли в подкожном кармане на шее. Учитывая, что доступ к сердцу осуществляли с помощью торакотомии, биполярный электрод кардиостимулятора помещали в средней части миокарда правого желудочка. Указанный водитель ритма проводили/выводили на поверхность между лопатками, и использовали в сочетании с внешним генератором импульсов/кардиостимулятором. Вживленный эндокардиальный кардиостимулятор использовали в качестве дублера внешнего/эпикардиального кардиостимулятора.
Восстановление после операции
Перед закрытием грудной клетки после торакотомии, помещали плевральную дренажную трубку для оттока жидкости и/или газа, которые скопились в результате хирургического вмешательства. Аспирацию через трубку осуществляли два раза в день, пока количество удаляемой жидкости не составляло меньше чем 35 мл на аспирацию за приблизительно 24-часовой период. Затем плевральную дренажную трубку удаляли. Всем животным профилактически вводили антибиотик (цефазолин от 20 до 50 мг/кг в/в) и обезболивающее (мелоксикам в дозе приблизительно 0,2 мг/кг в/в). При необходимости, также применяли дополнительное обезболивающее средство, которое включало пластырь с фентанилом (от 25 до 50 мкг/час). Все хирургические разрезы закрывали послойно; подлежащие мышцы соединяли рассасывающимся шовным материалом и кожу соединяли скобками. После операции животные восстанавливались в течение по меньшей мере 14 дней.
Цефалексин (от 20 до 50 мг/кг) вводили перорально BID в течение по меньшей мере 7 дней и мелоксикам (0,1 мг/кг) вводили SID перорально или подкожно в течение по меньшей мере 2 дней после операции. В течение фазы восстановления, у животных ежедневно отмечали обычные признаки восстановления, и область раны осматривали в отношении появления признаков возможных инфекций. Если животные испытывали боль, дистресс и/или инфекции, то об этом сообщали лечащему ветеринару и директору исследования. Скобы на кожном разрезе не удаляли в течение по меньшей мере 7 дней после операции.
Индукция сердечной недостаточности
По истечении периода послеоперационного восстановления и/или периода отмывки после приема донора нитроксила, животных подвергали 3-недельному протоколу искусственного ускорения сердечного ритма (210 импульсов/мин), предназначенному для развития дисфункции левого желудочка/ремоделирования, сопоставимого с синдромом сердечной недостаточности. Вкратце, с помощью имплантированного кардиостимулятора/правожелудочкого электрода, желудочек(и) заставляли сокращаться асинхронно и непрерывно с частотой 210 ударов в минуту (уд/мин). Ремоделирование левого желудочка (и индукцию сердечной недостаточности) подтверждали по изменениям эхокардиограммы (например, снижение фракции выброса (EF) от приблизительно 60% до намеченного значения приблизительно 35%, дилатация левого желудочка (LV)) и по нейрогуморальным изменениям (например, увеличение N-терминального фрагмента мозгового натрийуретического пептида (NT-proBNP) свыше 1800 пМ/л от исходного значения приблизительно 300 пМ/л) после приблизительно 3 недель электрокардиостимуляции. Эхокардиограммы и образцы крови получали при отсутствии стимуляции (в течение по меньшей мере 15 мин).
5.5.2 Результаты
Оценки гемодинамической эффективности
Изучали животных (здоровых или с сердечной недостаточностью) во время лечения носителем (контроль) и донором нитроксила (CXL-1020, соединение формулы (1), или соединение формулы (2)). В каждый период лечения осуществляли непрерывный мониторинг бодрствующих животных, закрепленных в станке, в течение двух-трех часов. После стабилизации гемодинамики начинали инфузию носителя. Вскоре после этого, преднапряжение левого желудочка резко снижали путем кратковременных пережатий полой вены (с помощью транзиторного наполнения сосудистого обтуратора) в целях получения ряда кривых/петель давление-объем; осуществляли до трех пережатий, с учетом гемодинамического восстановления между тестами. Продолжали инфузию носителя, и через 30 мин получали другой (исходный) набор гемодинамических данных. После получения гемодинамических данных в исходном состоянии, начинали инфузию соединения-донора нитроксила, которое тестировали, и определяемые гемодинамические/функциональные параметры получали/выполняли для четырех (4) моментов времени, выбранных из следующих моментов: 30, 60, 90, 120, и 180 мин после начала инфузии носителя/тестируемого соединения. В группе плацебо или в группе “время-контрольное лечение” каждому животному проводили инфузию соответствующего плацебо в течение 180 мин. Во всех случаях тестируемое соединение доставляли при постоянной скорости внутривенной инфузии 1 мл/кг/час и сравнивали со скоростью введения дозы в молярном эквиваленте.
Полученные данные относительно давления и объема левого желудочка анализировали с целью определения зависимостей, отражающих сократительную функцию и энергетическое состояние миокарда. Регистрировали систолическое артериальное давление (SAP), диастолическое артериальное давление (DAP), и среднее артериальное давление (MAP). Механические и/или геометрические индексы левого желудочка получали, исходя из сигнала давления (ESP, EDP, dP/dt max/min, времени - константы релаксации - тау [на основании моноэкспоненциального затухания с ненулевой асимптотой]) и объема (конечно-систолический объем (ESV), конечно-диастолический объем (EDV), ударный объем (SV)). Кроме того, следующие измерения получали из данных давление-объем левого желудочка (петлей PV), определяемых во время коротких периодов уменьшения преднагрузки: область давление-объем (PVA) и систолическая работа (SW), зависимость между конечно-систолическим давлением (ESPVR) и конечно-диастолическим давлением (EDPVR) и объемом, и зависимость между конечно-систолическим давлением и ударным объемом (артериальная эластичность (Ea)). Репрезентативные данные, полученные в исследованиях, проведенных на здоровых собаках и собаках с сердечной недостаточностью, представлены в таблице 5 и таблице 6, соответственно. Снижение SVR (системное сосудистое сопротивление) коррелирует с вазодилатацией.
Гемодинамические параметры при использовании доноров нитроксила у здоровых собак (% изменения от исходного значения)
кг/мин)
HR: частота сердечных сокращений. Увеличение HR вследствие рефлекторного ответа на низкое кровяное давление или вследствие первичного действия лекарства на сердце, является неблагоприятным признаком.
ESP: конечно-систолическое давление - сходно с показателем MAP, описанным ниже.
EDP или LVEDP: конечно-диастолическое давление (левого желудочка). Коррелирует со значениями легочного давления.
Снижение показателя указывает на уменьшение застоя крови в легких (основная цель терапии острой сердечной недостаточности)
Тау - показатель расслабления миокарда, или релаксации сердца во время диастолы. Снижение является положительным признаком и указывает на улучшение диастолической функции миокарда.
SW: ударная работа, показатель работы сердца по созданию прямого потока указанной величины.
ESPVR: соотношение между конечно-систолическим давлением и объемом
ERSW: соотношение между ударной работой и конечно-диастолическим объемом - аналогично показателю ESPVR, описанному выше
SV: Ударный объем. Количество крови, изгоняемое из левого желудочка с каждым ударом сердца. Инотроп должен увеличивать данный показатель, при наличии идентичных условий нагрузки.
MAP или MBP: среднее артериальное давление или среднее давление крови. Небольшие снижения показателей являются положительными и свидетельствуют о вазодилатации.
EDV или LVEDV: Конечно-диастолический объем (левого желудочка). Индекс степени наполнения в диастолу. Снижение указывает на уменьшение объемной перегрузки.
Гемодинамические параметры при использовании доноров нитроксила у собак с сердечной недостаточностью (% изменения от исходного значения)
мин)
HR, частота сердечных сокращений; ESP, конечно-систолическое давление; EDV, конечно-диастолический объем; Тау - константа времени релаксации; SW, ударная работа; ESPVR: соотношение между конечно-систолическим давлением и объемом; ERSW: соотношение между ударной работой и конечно-диастолическим объемом.
На фиг. 1 представлен гемодинамический профиль, полученный в течение 180 минут после введения CXL-1020 и двух соединений по изобретению (соединения формулы (1) и формулы (2)) с использованием модели сердечной недостаточности, вызванной тахикардией. Каждое соединение вводили внутривенно со скоростью 100 мкг/кг/мин. На фиг. 2 представлен гемодинамический профиль соединения формулы (1) в различных дозах, полученный с использованием модели сердечной недостаточности, вызванной тахикардией. Результаты показывают, что соединения формулы (1) и (2) имеют гемодинамическую активность, сравнимую с активностью CXL-1020 в моделях здоровых собак и собак с сердечной недостаточностью. В частности, соединения формулы (1) и (2) вызывают значительное улучшение сократимости и расслабления миокарда и небольшое снижение артериального давления.
5.6. Пример 7. Гемодинамическая эффективность доноров нитроксила у собак с сердечной недостаточностью (модель сердечной недостаточности, вызванной микроэмболизацией коронарных артерий у собак)
5.6.1 Материалы и методы
Сердечную недостаточность вызывали у здоровых, соответствующих требованиям, специально выведенных беспородных собак (20-26 кг) с использованием модели последовательной микроэмболизации. Микроэмболизацию коронарных артерий до тех пор, пока LV-фракция выброса (определяемая ангиографически под наркозом) не составляла приблизительно 30% или ниже. Затем в течение двух недель после проведения последней микроэмболизации каждое животное проходило стабилизацию перед началом экспериментов. Начальное исследование подбора дозы (2-100 мкг/кг/мин в течение 40 мин) осуществляли с участием 3 собак для определения терапевтически релевантных доз соединения формулы (1). На основании полученных данных изучали первичную группу из шести животных, получавших 3 или 10 мкг/кг/мин соединения формулы (1) в течение 4-часового периода, с последующим периодом отмывки в течение 1 часа. В определенный день изучали действие только одной дозы, действие другой дозы изучали по меньшей мере через одну неделю, и рандомизировали порядок введения доз. Гемодинамические, вентрикулографические и эхокардиографические измерения получали при осуществлении катетеризации левой и правой части сердца собак, находящихся под наркозом (индукция наркоза: гидроморфон (0,22 мг/кг в/в и диазепам (0,17 мг/кг в/в), поддержание наркоза: 1-2% изофлуран).
LV конечно-систолический объем (ESV) и конечно-диастолический объемы (EDV) рассчитывали на основании вентрикулограмм с использованием метода “площадь-длина”. Пиковую скорость аортального кровотока получали в восходящем отделе аорты с использованием допплеровского картирования кровотока для измерений индекса пиковой мощности (PPI). Фракционное изменение площади LV при укорочении (FAS) измеряли по изображению короткой оси LV на уровне папиллярных мышц, полученному в 2-мерных эхокардиограммах. Измеренные показатели диастолической функции LV включали время замедления скорости потока при прохождении митрального клапана (DT), отношение интеграла начальной митральной скорости потока (Ei) к скорости потока во время сокращения предсердий (Ai) (Ei/Ai) и LV конечно-диастолическое циркулярное напряжение стенки (EDWS).
Измерения потребления миокардом кислорода осуществляли в исходном состоянии и через 4 часа после 10 мкг/кг/мин инфузии. В частности, образцы крови брали одновременно из аоры и коронарного синуса в начале исследования и конце каждого момента времени исследования. Содержание кислорода определяли с помощью гемоксиметра. Скорость кровотока в коронарной артерии измеряли с помощью датчика для допплеровского исследования скорости потока, помещенного в огибающую ветвь левой коронарной артерии проксимально к первой маргинальной ветви, или в переднюю нисходящую ветвь левой коронарной артерии проксимально к первой диагональной ветви. Кровоток оценивали путем вычисления площади поперечного сечения коронарной артерии в месте измерения скорости с помощью коронарной ангиографии. Принимали, что общий коронарный кровоток равнялся двойной величине потока, измеренного в огибающей ветви или в передней нисходящей ветви коронарной артерии. Потребление кислорода левым желудочком (MVО2) рассчитывали в виде произведения общего коронарного кровотока и разности между содержанием кислорода в артериальной и венозной синусовой крови.
5.6.2 Результаты
Оценка гемодинамической эффективности
Животных исследовали в процессе лечения носителем и донором нитроксила. На фиг. 3 представлен гемодинамический профиль соединения формулы (1) после индукции сердечной недостаточности у собак, который получали с использованием модели сердечной недостаточности, вызванной микроэмболизацией. Данные представлены для конечного момента времени при проведении инфузии с двумя скоростями. Результаты показали, что соединение формулы (1) имело гемодинамическую активность, сравнимую с активностью CXL-1020.
5.7 Токсикологические исследования с использованием доноров нитроксила
5.7.1 Пример 8. Испытания CXL-1020 in vivo
При осуществлении испытаний in vivo донора нитроксила, CXL-1020 (N-гидрокси-2-метансульфонилбензол-1-сульфонамида), проводили 14-дневное исследование оценки переносимости у собак, получавших непрерывные инфузии CXL-1020 со скоростью введения до 90 мкг/кг/мин. Первое исследование показало, что животные переносили CXL-1020 при введении доз со скоростью 60 мкг/кг/мин. Однако неожиданно наблюдали клинические патологические изменения, соответствующие процессу воспаления, которые отражались в изменениях клинических патологических маркеров воспаления, при введении доз со скоростью 60 мкг/кг/мин. Для дальнейшего исследования этого нежелательного побочного эффекта, инициировали последующее 14-дневное исследование на собаках. Последующее исследование пришлось прекратить уже после 4 дней в связи с появлением других нежелательных побочных эффектов: неожиданное появление значительного отека и воспаления задних конечностей собак, где были хирургически имплантированы инфузионные катетеры, которые в редких случаях мешали нормальной функции конечности; обесцвечивание кожи в паховой области; снижение активности; отсутствие аппетита; и в группе, получавшей самую высокую дозу, холодная на ощупь кожа.
Для выявления причины воспаления и отека задних конечностей, проводили поисковые исследования с сериями 72-часовых непрерывных инфузий в течение следующих 6 месяцев. Результаты исследований показали, что CXL-1020 при введении в препарате со значением pH 4, содержащем в молярном отношении 1:1 CXL-1020:CAPTISOL®, разведенный в растворе 5% декстрозы в воде, вызывал у собак клинические патологические изменения, соответствующие процессу воспаления при скоростях введения доз, превышавших или равных 0,03 мкг/кг/мин. Сосудистое воспаление наблюдали вокруг места внедрения катетера в бедренную вену (на 15 см выше от кончика катетера), около кончика катетера, и ниже кончика катетера. Первый участок воспаления, место внедрения катетера, обуславливал отек задних конечностей у собак и воспаление, наблюдаемое в досрочно завершенном дополнительном исследовании. Увеличение значения pH инфузата от 4 до 6 снижало воспаление, улучшало воспалительный профиль приблизительно в 3 раза. Однако значительные нежелательные побочные эффекты по-прежнему наблюдали, если дозы CXL-1020 вводили собакам со скоростями, превышавшими или равными 3 мкг/кг/мин.
Чтобы избежать побочных эффектов, связанных с введением катетера, и чтобы определить возникало ли сосудистое воспаление за счет конструкции имплантированного катетера, осуществляли исследование с проведением собакам 24-часовой непрерывной инфузии, с использованием чрескожного катетера, помещенного в периферическую (подкожную латеральную) вену. После 6 часов инфузии наблюдали значительный отек в верхней передней конечности, расположенный ниже кончика катетера. После 24 часов инфузии обнаружили клинические патологические изменения, аналогичные тем изменениям, которые наблюдали в предыдущих исследованиях с использованием имплантированного центрального катетера. Также обнаружили микроскопическую патологию, демонстрирующую тяжелый тромбофлебит около кончика катетера и улучшающуюся с градиентом уменьшения тяжести в нисходящем направлении от кончика катетера. Чтобы определить, может ли возникнуть местный флебит у человека после длительного введения препарата, осуществляли исследование длительного введения препарата здоровым добровольцам. Исследование с длительным введением препарата включало в себя исследование с увеличением дозы, в котором когортам из 10 добровольцев последовательно проводили 24-часовую непрерывную инфузию CXL-1020 со скоростями введения 10, 20, и 30 мкг/кг/мин с проведением оценки безопасности между всеми когортами. Каждая когорта состояла из 2 добровольцев, получавших плацебо, и 8 добровольцев, получавших препарат, с сигнальной парой из 1 добровольца, получавшего препарат, и 1 добровольца, получавшего плацебо, и последующей основной группой из 1 добровольца, получавшего плацебо, и 7 добровольцев, получавших препарат. Инфузию осуществляли через чрезкожный катетер, введенный в вену предплечья. Катетер переставляли на другую руку после 12 часов инфузии. Обнаружили, что введение дозы со скоростью 10 мкг/кг/мин в течение 24 часов переносилось хорошо. Во второй когорте, которой вводили дозу 20 мкг/кг/мин в течение 24 часов, не наблюдали нежелательных эффектов у 2 добровольцев, получавших плацебо, но наблюдали небольшие нарушения (клинические признаки и/или изменения в клинической патологии) у всех 8 индивидов, соответствующие флебиту в месте инфузии. На основании полученных результатов длительное исследование безопасности было прекращено.
Поисковое исследование продолжали, чтобы определить причину нежелательных побочных эффектов при использовании CXL-1020 в более высоких, но клинически желательных дозах. Исследования, проведенные с побочным продуктом CXL-1020, фрагментом, который остается после отдачи нитроксила, дали отрицательный результат, что показало, что побочные эффекты CXL-1020 были обусловлены либо исходным соединением, CXL-1020, либо HNO, полученным из него. Проводили исследования с альтернативными донорами нитроксила, которые структурно не связаны с CXL-1020, но имели сходные периоды полураспада при отдаче нитроксила (периоды полураспада приблизительно 2 мин). В случае указанных доноров самая высокая концентрация нитроксила была около кончика катетера и непосредственно ниже по ходу вены, в которую был вставлен катетер. Во всех случаях наблюдали местные сосудистые побочные эффекты рядом с кончиком катетера. Результаты свидетельствуют, что воспаление вызвано нитроксилом, который быстро освобождался из доноров нитроксила с коротким периодом полужизни.
5.7.2 Пример 9. Соединения по изобретению обладают улучшенным токсикологическим профилем по сравнению с CXL-1020
Исследования проводили на собаках породы бигль обоего пола. Животным обеспечивали свободный доступ к воде и собачьему корму, представленному на рынке, в стандартных лабораторных условиях. Животных не кормили перед забором образцов крови, время которых указано в протоколе исследования. Люминисцентное освещение обеспечивалось с помощью автоматического таймера в течение приблизительно 12 часов в день. В отдельных случаях, ночной цикл периодически прерывали в связи с действиями, предусмотренными исследованиями. Температуру и влажность контролировали и регистрировали ежедневно и поддерживали в максимально возможной степени в интервале от 17,8°С до 29°С и от 30% до 70% соответственно. Собаки проходили акклиматизацию в течение по меньшей мере 1 недели. В течение указанного периода животных взвешивали раз в неделю и наблюдали за общим состоянием здоровья и появлением каких-либо признаков заболевания. Животные привыкали к ношению корсета в течение по меньшей мере трех дней до введения доз. Кроме того, животные также привыкали к ношению “елизаветинского” воротника (e-воротник) во время адаптации к корсету.
Хирургические процедуры и процедура введения доз
Животных катетеризировали за день до введения доз. Чрескожный катетер помещали (с использованием метода асептики и наложения стерильной повязки) в подкожную латеральную вену передней конечности дистальнее локтя. Животные свободно двигались в своих клетках во время непрерывного инфузионного введения дозы. Для облегчения непрерывного инфузионного введения дозы, периферийный катетер подсоединяли к удлинительной инфузионной линии, помещенной под корсет и затем подсоединенной к кабельной инфузионной системе. Для предотвращения доступа/удаления животными периферически размещенного чрескожного катетера, на участок катетеризации накладывали повязку Vet Wrap и надевали на собак e-воротник на время лечения (т.е. на катетеризационный период). Во время подготовительного периода, в венозный катетер непрерывно вводили со скоростью приблизительно 2-4 мл/час 0,9% раствор хлорида натрия для инъекций, USP (физиологический раствор) с целью поддержания проходимости катетера. Перед введением дозы инфузионную систему предварительно заполняли (с помощью медленной болюсной инфузии) соответствующим раствором для введения, чтобы гарантировать, что введение дозы начнется, как только начнет работать инфузионный насос. Инфузионную систему соединяли с резервуаром, содержащим контрольное или тестируемое соединение, и начинали инфузию. Тестируемые композиции вводили непрерывно, с заданной постоянной скоростью инфузии (1 или 2 мл/кг/ч), в течение 24 часов и сравнивали скорости введения молярных эквивалентов доз.
Клинические наблюдения, клиническая патология и микроскопическая патология
Подробный клинический осмотр каждого животного проводили два раза в день, и результаты измерения температуры тела и образцы крови для определения клинической патологии получали от всех животных перед введением дозы и через 6 часов, 12 часов, 24 часов и 72 часа после начала инфузии композиции. В конце исследования, всех животных подвергали эвтаназии на этапе запланированной некропсии и проводили полное исследование секционного материала. Отдельные ткани собирали, фиксировали и хранили для предполагаемого будущего микроскопического исследования. Подкожную латеральную вену конечности, содержащую инфузионный катетер, иссекали в неизменном виде вместе с плечевой веной и исследовали по всей длине. Положение кончика катетера отмечали на нефиксированном образце. После фиксации образец иссекали и обрабатывали для получения препаратов поперечных гистологических срезов, представляющих ткани, в месте расположения кончика катетера, и окружающие ткани, расположенные проксимальнее и дистальнее кончика катетера (т.е. на 1 см дистальнее кончика катетера, в месте расположения кончика катетера, и на 1, 5, 10, 15, и 20 см проксимальнее кончика катетера). По отношению к кончику катетера "проксимальный" означал расположенный ближе к сердцу, и "дистальный" означал расположенный дальше от сердца.
Оценка безопасности
Клинические патологические изменения, соответствующие воспалительному синдрому, наблюдали для одинаковых скоростей введения соединений формулы (1), формулы (2) и CXL-1020. Каждое соединение было включено в состав с CAPTISOL® (7% масс/об) в стерильной воде при pH 4. Наиболее чувствительными маркерами воспаления являлись: (1) число лейкоцитов (WBC, полученное в виде (число лейкоцитов)/мкл, путем умножения значений, представленных в правой части фиг. 4, на 103), (2) концентрация фибриногена (представлена в мг/дл, справа на фиг. 4), и (3) концентрация С-реактивного белка (CRP) (представлена в мг/л справа на фиг. 4). Тяжесть изменений зависела от самого соединения и скорости, с которой вводили соединение (фиг. 4). На фиг. 4, оценку, в диапазоне от 0 (низкая степень тяжести) до 2 (высокая степень тяжести), задавали для каждого из биомаркеров воспаления, которые показаны в правой части фигуры. Совокупную оценку рассчитывали как сумму оценок указанных маркеров. Значения NOAEL, которые определяли на основании указанных клинических патологических маркеров и выражали в виде скорости введения молярного эквивалента дозы (мкг/кг/мин) к CXL-1020, представлены в таблице 7.
Значения уровней, не вызывающих видимых неблагоприятных изменений (NOAEL), для доноров нитроксила
В случае CXL-1020, наблюдали существенное увеличение WBC, фибриногена и CRP, даже при использовании в концентрациях до 0,03 мкг/кг/мин. Соединение формулы (1) и соединение формулы (2), каждое, имеет значение NOAEL, существенно более высокое, чем в случае CXL-1020. Соединение формулы (1) имеет наиболее благоприятный токсикологический профиль, не показывая никаких неблагоприятных эффектов при дозах, не проявляет неблагоприятных эффектов при использовании в дозах по меньшей мере до 20 мкг/кг/мин. Данный показатель более чем в 660 раз выше, по сравнению с аналогичным показателем для CXL-1020.
В совокупности, результаты позволяют предположить, что инфузия CXL-1020 вызывает воспалительный синдром, который является существенно меньше при использовании соединения формулы (1) и соединения формулы (2).
Результаты показывают, что нежелательные сосудистые побочные эффекты, связанные с CXL-1020, наблюдаемые рядом с кончиком катетера, ниже кончика катетера и при определенных обстоятельствах, выше кончика катетера, возникали вследствие локального воспаления, вызванного высвобождением нитроксила. Кроме того, было высказано предположение, что воспаление в данных участках может быть значительно уменьшено при использовании доноров нитроксила с более длительным периодом полураспада. Подтверждение было получено путем оценки действия доноров нитроксила с помощью подробного гистопатологического исследования сосудов в месте введения в бедренную вену (15 см дистальнее кончика катетера), вдоль по ходу катетера до кончика катетера, и ниже кончика катетера, вниз по ходу вены на 20 см. Микроскопические патологические признаки отека, кровоизлияния, сосудистого воспаления и периваскулярного воспаления определяли для конкретных значений скорости введения доноров нитроксила.
На фиг. 5 представлена "карта интенсивности", на которой показана суммарная лабораторная оценка микроскопических патологических признаков, где тяжесть сосудистого воспаления, кровоизлияния, тромбоза сосудов и дегенерации/регенерации оценивали в баллах в срезах сосудов, как описано выше. Признаки (1) отека, (2) сосудистого и периваскулярного воспаления и (3) кровоизлияния оценивали в баллах (каждому признаку присваивали значение, выбранное из 0 = в пределах нормы; 1 = минимальное изменение, 2 = небольшое изменение; 3 = умеренное изменение, 4 = тяжелое изменение) в срезах сосудов, начиная на 1 см дистальнее (выше) кончика катетера, продвигаясь на 20 см проксимальнее (ниже) кончика катетера. Суммарную лабораторную оценку рассчитывали как сумму баллов указанных признаков. На фиг. 5 сводная гистологическая суммарная лабораторная оценка изменяется от 0-2 (низкая степень тяжести) до 11-12 (высокая степень тяжести). Установили, что степень тяжести микроскопических изменений и расстояние от кончика катетера, на котором их выявляли, зависели от самого донора нитроксила и от величины дозы, в которой вводили донор нитроксила. Значения NOAEL, установленные на основании указанных микроскопических патологических маркеров для серий доз доноров нитроксила, выраженных в виде скорости введения молярного эквивалента дозы (мкг/кг/мин) относительно CXL-1020, представлены в таблице 8.
Значения уровней, не вызывающих видимых неблагоприятных изменений (NOAEL), для доноров нитроксила 180 180
180 180
Результаты, представленные в таблице 8, являются дополнительным свидетельством того, что соединения формулы (1) и (2) имеют существенно улучшенный токсикологический профиль по сравнению с CXL-1020. Тяжесть сосудистых побочных эффектов при любой дозе уменьшалась в виде функции расстояния от кончика катетера, и тяжесть сосудистых побочных эффектов уменьшалась со снижением дозы. Результаты подтвердили значительный резерв безопасности соединений формул (1) и (2), который реализуется в широком терапевтическом индексе при использовании у человека, и в пригодности для внутривенного введения в терапевтически эффективных дозах и скоростях введения доз.
5.8 Стабильность растворов для внутривенного введения
5.8.1 Пример 10: Соединение формулы (1) - раствор для введения, хранящийся при 25°C
Стабильность растворов для введения соединения формулы (1), полученных из концентрата CAPTISOL®, разведенного в коммерчески доступных разбавителях для в/в, оценивали при 25°C в течение 48 часов, с проведением анализа для временных точек 0, 8, 12, 16, 24, и 48 часов после разведения. В связи с требуемыми для анализа временными точками, выполняли два исследования с отдельными наборами растворов для введения. Первое исследование (группа A) охватывало все моменты времени кроме временной точки 16 часов. Второе исследование (группа B) включало проведение анализа только в 0 и 16 часов. Концентраты, использованные для приготовления двух наборов растворов для введения, получали из двух отдельных флаконов, взятых из одной партии лиофилизированного лекарственного препарата (24 мг/мл соединения формулы (1)/30% CAPTISOL®).
Получение концентрата
Один флакон лиофилизированного лекарственного препарата (24 мг/мл соединения формулы (1)/30% CAPTISOL®, pH 4) восстанавливали 10 мл воды качества “вода для инъекций” (WFI) для получения каждого концентрата (для получения раствора для введения групп A и B). Измеряли значения pH полученных растворов, и они составляли приблизительно 3,9 в обоих флаконах. Корректирование значений pH не проводили. Концентраты разводили и анализировали с помощью ВЭЖХ (колонка XBridge Phenyl (Waters); детектор УФ при 272 нм; подвижная фаза - ступенчатый градиент водного ацетонитрила, содержащего 0,1% (об/об) муравьиной кислоты), и определяли, что оба раствора содержали 20-21 мг/мл соединения формулы (1), вместо номинального значения 24 мг/мл, по-видимому, в результате увеличения общего объема раствора за счет растворения API и CAPTISOL®.
Приготовление разбавителей
Для оценки выбирали коммерчески доступные растворы ацетата калия и фосфата калия. Ацетат калия приобретали коммерчески, и a раствор фосфата калия степени чистоты USP готовили в соответствии с инструкцией Hospira product для коммерческого продукта. Каждый раствор разводили до 10 мМ в 5% декстрозе (D5W) и 2,5% декстрозе (D2.5W). Коммерчески доступный раствор D5W разводили в 2 раза водой качества WFI для получения раствора D2.5W. Измеряли значение pH для каждого концентрированного и разведенного раствора; результаты представлены в таблице 9.
Результаты измерения pH выбранных разбавителей
Получение растворов для введения доз
Концентрат соединения формулы (1) разводили объемным методом по 5 мл шкале в 10 мМ растворах разбавителей для получения концентраций 8, 1, и 0,1 мг/мл соединения формулы (1), как указано в таблице 10. Каждый образец получали в дубликатах. Содержание декстрозы в 10% растворе CAPTISOL® уменьшали с целью обеспечения в большой степени изотоничности растворов для введения. Каждый раствор хранили при 25°C.
Получение растворов для введения с целью оценки стабильности
Анализ образцов
Образцы анализировали после приготовления и после 8, 12, 16, 24, и 48 часов хранения при 25°C. Отмечали внешний вид каждого образца, измеряли pH, и каждый образец анализировали с помощью ВЭЖХ в отношении концентрации и наличия главного деграданта, соединения формулы (5), представленного ниже, образующегося после высвобождения группы HNO.
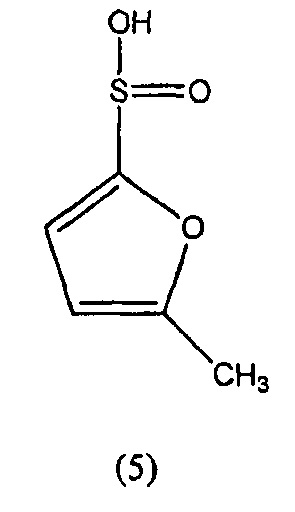
Результаты
Результаты оценки стабильности представлены в таблицах 11, 12 и 13. Наличие пика, соответствующего деграданту (соединение формулы (5)), в образце обозначено "X".
В целом, результаты были сопоставимы для каждого дубликата в паре, и между соответствующими растворами для введения, приготовленными в группах A и B. Наблюдали разницу в извлечении между дубликатами, полученными во временных точках 24 и 48 часов, для образцов, содержащих 0,1 мг/мл соединения формулы (1) в фосфатном растворе.
Полное извлечение (в пределах точности метода ВЭЖХ) и отсутствие детектируемой величины пика соединения формулы (5) сохранялось в течение 48 часов для всех образцов, содержащих 8 мг/мл соединения формулы (1) в разбавителях на основе ацетата и фосфата. Указанные образцы по существу содержали приблизительно 7 мг/мл соединения формулы (1), что соответствует концентрации 20-21 мг/мл соединения формулы (1) в концентрате. В обоих разбавителях, стабильность была выше в образцах, содержащих 8 мг/мл соединения формулы (1), чем в образцах, приготовленных с более низкими концентрациями. Безотносительно к какой-либо теории, более высокую стабильность указанных образцов, по сравнению с образцами, содержащими более низкие концентрации соединения формулы (1), можно объяснить более высокой концентрацией CAPTISOL® (10% в разведенных растворах).
Все образцы оставались прозрачными и бесцветными на протяжении 48 часов хранения. Значение pH всех образцов уменьшалось с течением времени. Известный деградант (соединение формулы (5)) обнаруживали в t0 (непосредственно после получения образца) во всех образцах, приготовленных с концентрацией 0,1 мг/мл соединения формулы (1), и во всех последующих временных точках во всех образцах, приготовленных с концентрацией 0,1 мг/мл и 1 мг/мл соединения формулы (1).
В целом, стабильность уменьшалась со снижением концентрации соединения формулы (1). Безотносительно к какой-либо теории, снижение стабильности вероятно связано со снижением процентного содержания CAPTISOL® в растворах для введения. Начальная степень деградации (в течение 16 часов) была сходной в образцах, приготовленных с содержанием 0,1 мг/мл соединения формулы (1) в разбавителях на основе ацетата и фосфата. Однако образцы, приготовленные с содержанием 1 мг/мл, показали существенно более высокую стабильность в ацетате, чем в фосфате.
Результаты оценки стабильности раствора для введения при 25°C, процент извлечения
(группа В)
Результаты оценки стабильности раствора для введения при 25°C, pH
D5W
Результаты оценки стабильности раствора для введения при 25°C - определение образования соединения формулы (5)
5.8.2 Пример 11. Соединение формулы (1)- хранение раствора для введения при 2°C-8°C с последующим хранением при 25°C
Растворы для введения соединения формулы (1), приготовленные из концентрата CAPTISOL®, разведенного в коммерчески доступных разбавителях для в/в, получали, как описано в Примере 10. Стабильность растворов оценивали при хранении при 2°C-8°C в течение 24 часов с последующим хранением при 25°C в течение 48 часов. Как показано в таблице 14, показатели извлечения соединения формулы (1), в целом, были выше, чем для соответствующих образцов, хранившихся при 25°C, для всех растворов для введения (смотреть таблицу 12 в предыдущем примере), что свидетельствует о повышенной стабильности растворов для введения, полученных и хранившихся при 2°C-8°C перед хранением при 25°C.
Результаты оценки стабильности растворов для введения, хранившихся при 2°C-8°C и 25°C, процент извлечения
25°C
25°C
25°C
5.8.3 Пример 12. Соединение формулы (2) - раствор для введения, хранившийся при 25°C
Оценивали серию растворов для введения соединения формулы (2), предназначенных для в/в введения. Выбранный концентрат соединения формулы (2), приготовленный в концентрации 30 мг/мл в носителе 30% CAPTISOL® с pH 4,0, оценивали в низкой, средней и высокой концентрациях (0,1, 1 и 5 мг/мл, соответственно) после разведения до получения различных растворов для введения. В случае разведения соединения формулы (2) до 0,1 и 1 мг/мл, оценивали три раствора для введения: (1) D5W, (2) D5W, содержащий 5 мМ K-фосфат (pH - 6), и (3) D5W, содержащий 20 мМ K-фосфат (pH = 6). Для поддержания изоосмолярности разведений соединения формулы (2) с концентрацией 5 мг/мл, концентрацию декстрозы в растворах для введения уменьшали до 2,5% (масс/об). Таким образом, оценивали следующие растворы для введения: (1) D2.5W, (2) D2.5W, содержащий 5 мМ K-фосфат (pH = 6), и (3) D2.5W содержащий 20 мМ K-фосфат (pH = 6).
Потенциальные растворы для введения оценивали в отношении внешнего вида, pH, осмолярности, и концентрации и чистоты с помощью ВЭЖХ (колонка XBridge Phenyl (Waters); УФ-детектор абсорбции при 272 нм; подвижная фаза - ступенчатый градиент водного ацетонитрила, содержащего 0,1% (об/об) муравьиной кислоты) после приблизительно 0, 16, 24, и 48 часов хранения при 25°C. Все образцы представляли собой прозрачные бесцветные растворы - с одним исключением, образец 5 мг/мл соединения формулы (2) в D2.5W, содержащем 5 мМ фосфат, который имел прозрачный светло-желтый вид после 48 часов хранения при 25°C. Все растворы были изоосмотическими (290 +/- 50 мОсм/кг) - с одним исключением, образец 1 мг/мл соединения формулы (2) в D5W, содержащем 20 мМ фосфат, который имел осмолярность приблизительно 350 мОсм/кг. Кроме того, с одним исключением, образец 5 мг/мл соединения формулы (2) в растворе D2.5W, содержащем 5 мМ фосфат, все другие растворы для введения обеспечивали соединение формулы (2) в заданных концентрациях 0,1, 1 и 5 мг/мл в течение 48 часов. Кроме того, известный деградант, соединение формулы (6), которое представлено ниже, образующийся после высвобождения активной нитроксильной группы, обнаруживали после 16-часового хранения при 25°C в небольших количествах с помощью ВЭЖХ в растворах для введения, содержащих фосфатный буфер.
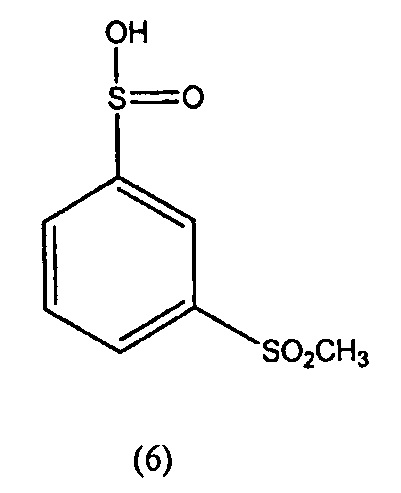
Наблюдаемое количество соединения формулы (6) соответствовало порядку предела обнаружения метода.
Стабильность растворов для введения, содержащих 5 мг/мл соединения формулы (2), дополнительно оценивали в виде функции значения pH и буфера. Концентрированный раствор соединения формулы (2), приготовленный в концентрации 30 мг/мл в носителе 30% CAPTISOL® при pH 4,0, разводили до 5 мг/мл в четырех потенциальных растворах для введения. Оценивали четыре раствора для введения: (1) D2.5W, 5 мМ K-фосфат (pH = 6,0), (2) D2.5W, 5 мМ K-цитрат (pH = 6,0), (3) D2.5W, 5 мМ K-цитрат (pH = 5,0), и (4) D2.5W, 5 мМ K-ацетат (pH = 5,0). Все растворы для введения соединения формулы (2) были изоосмотическими (290 +/- 50 мОсм/кг). Приблизительно через 24 и 48 часов хранения при 25°C оценивали растворы для введения в отношении внешнего вида, pH, и с помощью ВЭЖХ оценивали концентрацию и чистоту. Растворы для введения, не содержащие фосфат, были прозрачными, бесцветными и сохраняли заданную концентрацию 5 мг/мл соединения формулы (2) на протяжении 48 часов; в то время как с учетом требований отбора растворов для введения, раствор для введения, содержащий 5 мг/мл соединения формулы (2) в D2.5W с 5 мМ фосфата (pH 6,0), был прозрачным, светло-желтого цвета, только с 60% извлечения соединения формулы (2) после 48 часов. Кроме того, известный деградант, соединение формулы (6), обнаруживали в небольших количествах с помощью ВЭЖХ во всех образцах за исключением образца 5 мг/мл соединения формулы (2) в растворе D2.5W, содержащем 5 мМ цитрат (pH 5,0).
После 7 дней хранения при 25°C растворы для введения, не содержащие фосфат, оставались прозрачными и бесцветными. Измеряли минимальное увеличение кислотности в течение 7 дней в случае раствора для введения, содержащего 5 мг/мл соединения формулы (2) в D2.5W, 5 мМ цитрата, pH 6,0, тогда как раствор для введения, содержащий D2.5W, 5 мМ цитрата, pH 5,0 имел минимальное увеличение pH в течение первых 24-48 часов. Кроме того, после 14 дней хранения при 25°C образцы раствора для введения, содержащие 5 мМ цитрат, pH 6,0, представляли собой прозрачные, бесцветные растворы, тогда как растворы для введения, содержащие либо 5 мМ цитрат, либо 5 мМ ацетат, pH 5,0, представляли собой прозрачные желтые растворы. Результаты обобщены в таблице 15.
Извлечение соединения формулы (2) из 5 мг/мл растворов для введения
Специалистам в данной области очевидно, что конкретные варианты осуществления описанного объекта изобретения могут быть направлены на один или больше из выше и ниже указанных вариантов осуществления в любой комбинации.
В то время как изобретение описано в некоторых деталях посредством иллюстраций и примеров в целях ясности понимания, специалистам в данной области очевидно, что могут быть внесены различные изменения и могут быть заменены эквиваленты в пределах существа и объема изобретения. Таким образом, описание и примеры не следует истолковывать, как ограничивающие объем изобретения.
Все ссылки, публикации, патенты и патентные заявки, описанные в настоящем документе, включены в данное описание посредством ссылки во всей своей полноте.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ДОНОРЫ НИТРОКСИЛА | 2014 |
|
RU2684916C2 |
| ПРОИЗВОДНЫЕ N-ГИДРОКСИЛСУЛЬФОНАМИДА В КАЧЕСТВЕ НОВЫХ ФИЗИОЛОГИЧЕСКИ ПОЛЕЗНЫХ ДОНОРОВ НИТРОКСИЛА | 2007 |
|
RU2616292C2 |
| ПРОИЗВОДНЫЕ N-ГИДРОКСИЛСУЛЬФОНАМИДА В КАЧЕСТВЕ НОВЫХ ФИЗИОЛОГИЧЕСКИ ПОЛЕЗНЫХ ДОНОРОВ НИТРОКСИЛА | 2007 |
|
RU2448087C2 |
| ПРОИЗВОДНЫЕ N-ГИДРОКСИЛСУЛЬФОНАМИДА КАК НОВЫЕ ФИЗИОЛОГИЧЕСКИ ПРИМЕНИМЫЕ ДОНОРЫ НИТРОКСИЛА | 2008 |
|
RU2485097C2 |
| СОСТАВЫ ВНУТРИВЕННЫХ РАСТВОРОВ ПОЗАКОНАЗОЛА, СТАБИЛИЗИРОВАННЫЕ ПОСРЕДСТВОМ ЗАМЕЩЕННОГО БЕТА-ЦИКЛОДЕКСТРИНА | 2011 |
|
RU2575768C2 |
| АЭРОЗОЛЬНЫЕ ФТОРХИНОЛОНЫ И ИХ ПРИМЕНЕНИЯ | 2006 |
|
RU2428986C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ЭПИЛЕПСИИ ИЛИ ЭПИЛЕПТИЧЕСКОГО СТАТУСА | 2013 |
|
RU2667010C2 |
| НАНОЧАСТИЦЫ, НЕСУЩИЕ ХИМИОТЕРАПЕВТИЧЕСКОЕ ПРОТИВОРАКОВОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2012 |
|
RU2616494C2 |
| КРИСТАЛЛИЧЕСКАЯ (2S,4R)-5-(5'-ХЛОР-2'-ФТОР-[1,1'-БИФЕНИЛ]-4-ИЛ)-2-(ЭТОКСИМЕТИЛ)-4-(3-ГИДРОКСИИЗОКСАЗОЛ-5-КАРБОКСАМИДО)-2-МЕТИЛПЕНТАНОВАЯ КИСЛОТА И ЕЕ ПРИМЕНЕНИЯ | 2017 |
|
RU2756223C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДНЫЕ РАСТВОРЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2803464C2 |
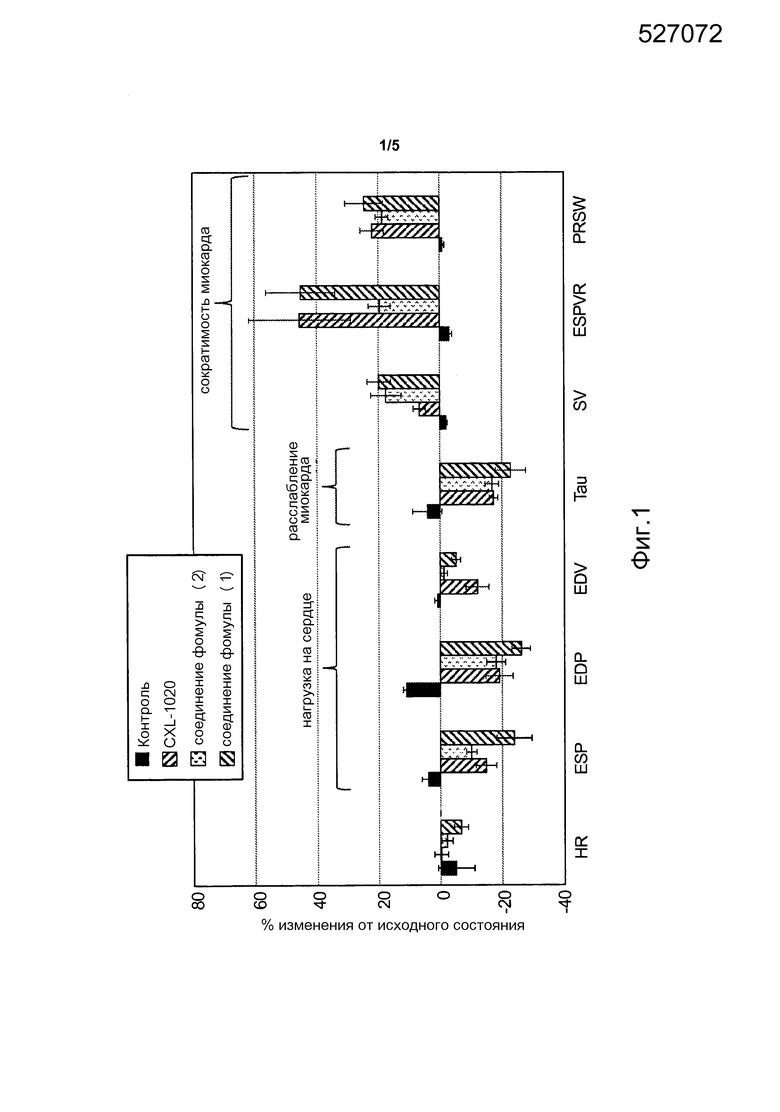
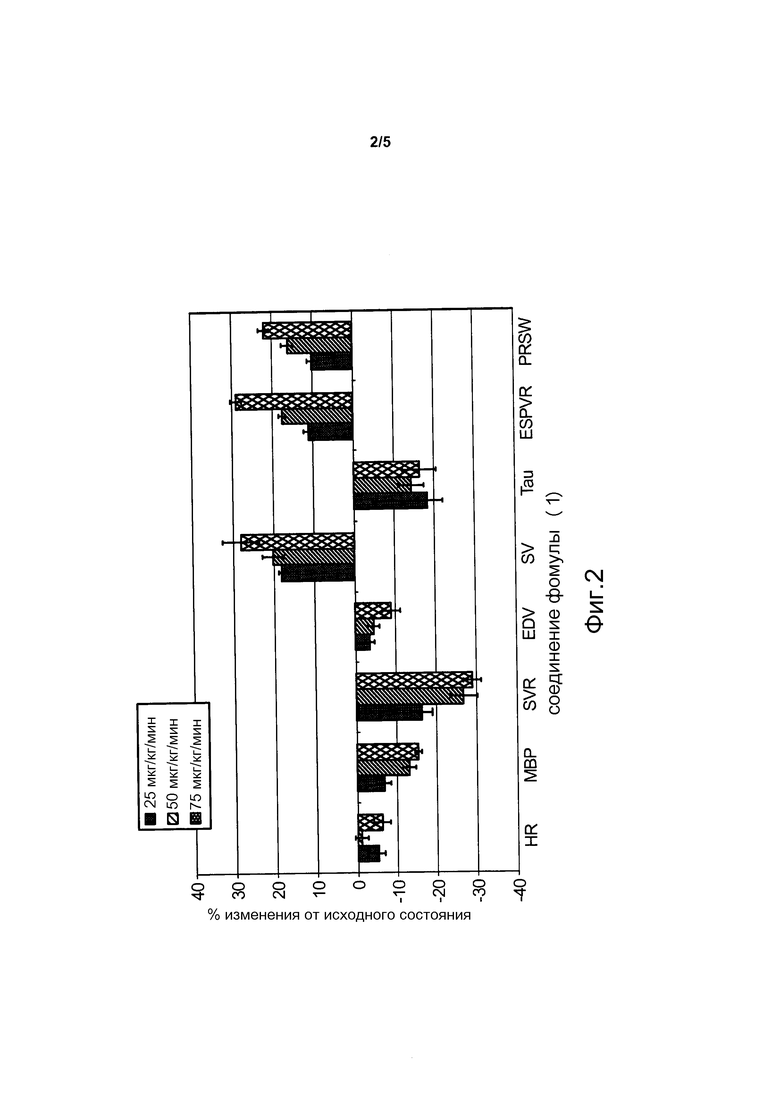
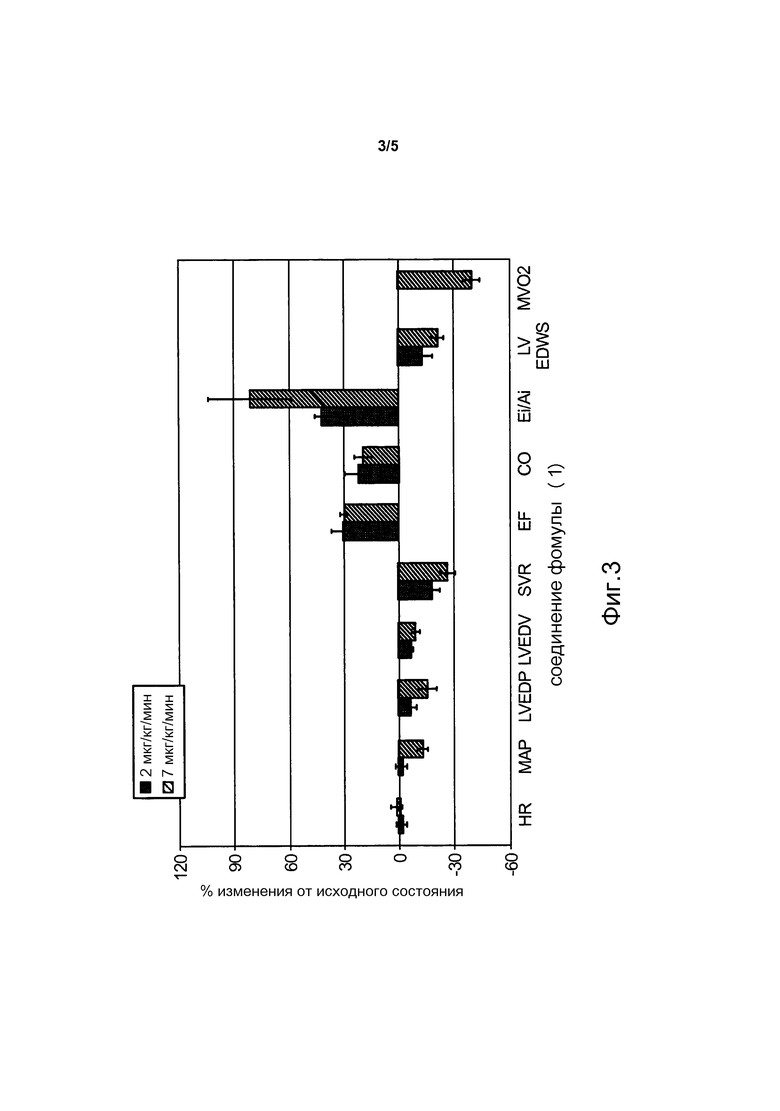

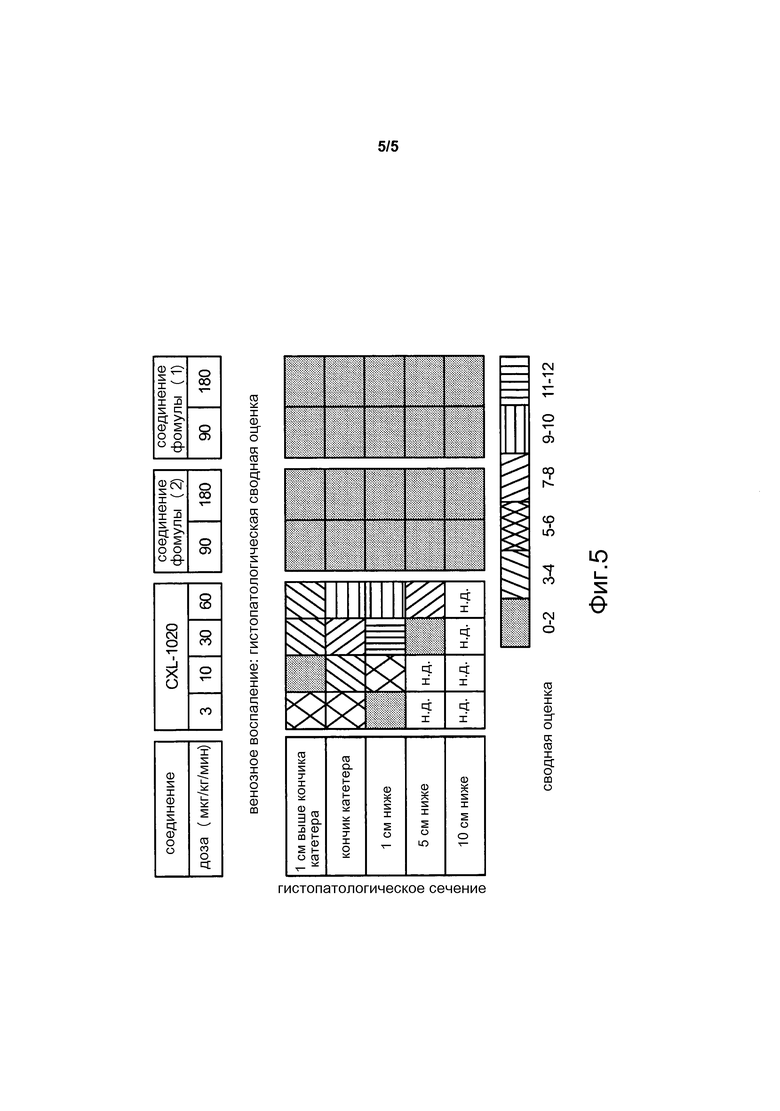
Настоящие изобретение относится к N-замещенным соединениям, производным гидроксиламина формул 1 и 2. Изобретение также относится к фармацевтическим композициям, предназначенным для лечения сердечной недостаточности, и наборам, содержащим такие соединения или фармацевтическую композицию в сухом виде. 6 н. и 12 з.п. ф-лы, 5 ил., 15 табл., 3 пр.

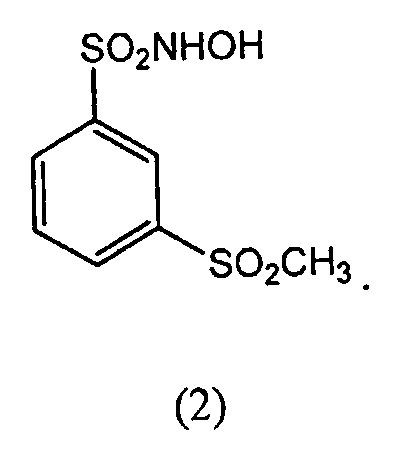
1. Соединение формулы (1):
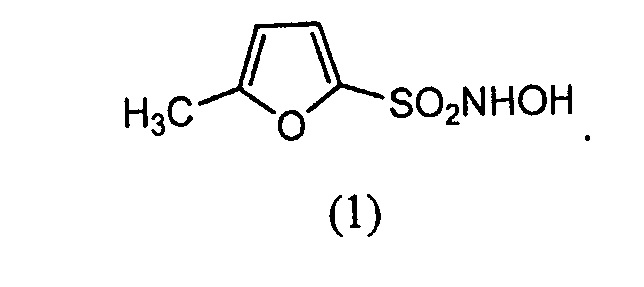
2. Соединение формулы (2):
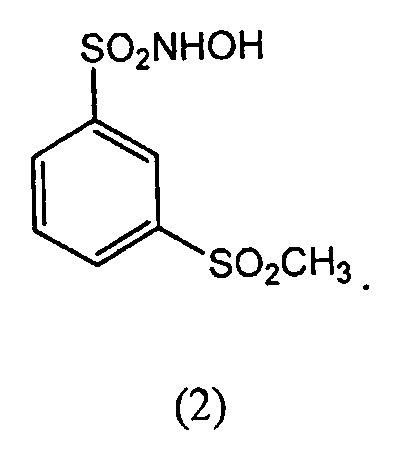
3. Фармацевтическая композиция для лечения сердечно-сосудистого заболевания, реагирующего на терапию нитроксилом, содержащая соединение по п.1 или 2 и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
4. Фармацевтическая композиция для лечения сердечной недостаточности, содержащая соединение по п.1 или 2 и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
5. Фармацевтическая композиция для лечения острой декомпенсированной сердечной недостаточности, содержащая соединение по п.1 или 2 и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
6. Фармацевтическая композиция по любому из пп.3-5, где фармацевтическая композиция является подходящей для внутривенного введения.
7. Фармацевтическая композиция по любому из пп.3-6, где фармацевтическая композиция имеет значение pH от приблизительно 4 до приблизительно 6.
8. Фармацевтическая композиция по любому из пп.3-7, где фармацевтическая композиция имеет значение pH от приблизительно 4 до приблизительно 5.
9. Фармацевтическая композиция по любому из пп.3-8, где фармацевтическая композиция имеет значение pH приблизительно 4.
10. Набор, содержащий
соединение по п.1 или 2 в сухом виде или фармацевтическую композицию по любому из пп.3-9 в сухом виде; и
фармацевтически приемлемый жидкий разбавитель.
11. Соединение по п.1 или 2 для использования в лечении сердечно-сосудистого заболевания, реагирующего на терапию нитроксилом.
12. Соединение по п.1 или 2 для использования в лечении сердечной недостаточности.
13. Соединение по п.1 или 2 для использования в лечении острой декомпенсированной сердечной недостаточности.
14. Соединение по п.1 или 2 для использования как определено в любом из пп.11-13, где соединение вводят внутривенно.
15. Соединение по п.1 или 2 для использования как определено в любом из пп.11-13, где соединение вводят внутривенно в количестве от 10 мкг/кг/мин соединения формулы (1) или (2) до 50 мкг/кг/мин соединения формулы (1) или (2).
16. Соединение по п.1 или 2 для использования как определено в любом из пп.11-13, где соединение вводят внутривенно в количестве не более 30 мкг/кг/мин соединения формулы (1) или (2).
17. Соединение по п.1 или 2 для использования как определено в пп.11-13, где соединение вводят внутривенно в количестве:
(а) по меньшей мере, 2,5 мкг/кг/мин соединения формулы (1) или (2);
(b) по меньшей мере, 5 мкг/кг/мин соединения формулы (1) или (2);
(с) по меньшей мере, 7,5 мкг/кг/мин соединения формулы (1) или (2);
(d) по меньшей мере, 12 мкг/кг/мин соединения формулы (1) или (2); или
(е) по меньшей мере, 15 мкг/кг/мин соединения формулы (1) или (2).
18. Фармацевтическая композиция по любому из пп.3-9, где композицию вводят внутривенно в количестве:
(а) по меньшей мере, 2,5 мкг/кг/мин соединения формулы (1) или (2);
(b) по меньшей мере, 5 мкг/кг/мин соединения формулы (1) или (2);
(с) по меньшей мере, 7,5 мкг/кг/мин соединения формулы (1) или (2);
(d) по меньшей мере, 12 мкг/кг/мин соединения формулы (1) или (2); или
(е) по меньшей мере, 15 мкг/кг/мин соединения формулы (1) или (2).
| WO 2007109175 A1, 27.09.2007 | |||
| WO 2011063339 A1, 26.05.2011 | |||
| US 20110144067 A1, 16.06.2011 | |||
| RU 99101350 A, 20.11.2000 | |||
| ЕА 200300674 A1, 24.06.2004. |

