Перекрестная ссылка на родственную заявку
Настоящая заявка испрашивает приоритет и преимущество U.S.S.N. 61/969546, поданной 24 марта 2014 года, содержание которой включаются в настоящий документ в качестве ссылки во всей своей полноте.
Область техники, к которой относится изобретение
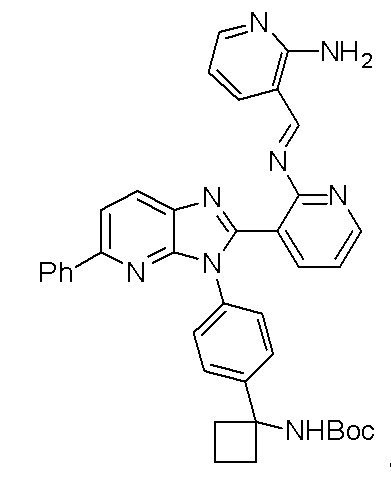
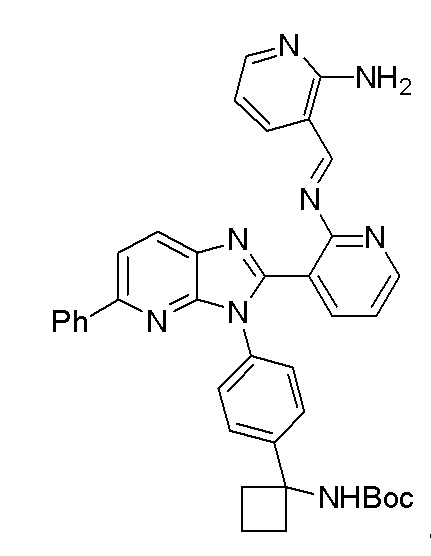
Настоящее изобретение направлено на способы синтеза 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина.
Уровень техники
Рак представляет собой вторую ведущую причину смерти в Соединенных Штатах, превышаемую только сердечными заболеваниями (Cancer Facts and Figures 2004, American Cancer Society, Inc.). Не смотря на последние успехи в диагностике и лечении рака, хирургия и лучевая терапия могут излечивать, если рак обнаруживается рано, но современная лекарственная терапия для метастатического заболевания является по большей части паллиативной и редко дает долговременное излечение.
Семейство AKT регулирует клеточное выживание и метаболизм посредством связывания и регуляции множества нижележащих эффекторов, например, ядерного фактора κB, белков семейства Bcl-2 и двойных генов грызунов 2 (MDM2). Akt1, как известно, играет роль в клеточном цикле. Кроме того, активированный Akt1 может сделать возможной пролиферацию и выживание клеток, которые потенциально сохраняют мутагенное воздействие, и, по этой причине, они могут вносить в клад в осуществление мутаций в других генах. Akt1 могут также участвовать в ангиогенезе и развитии опухоли. Исследования показали, что дефицит Akt1 усиливает патологический ангиогенез и рост опухоли, связанный с аномалиями матрикса в коже и кровеносных сосудах. Поскольку он может блокировать апоптоз, и тем самым способствовать выживанию клеток, Akt1 является главным фактором при многих типах рака.
Соединение 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амин (также известное как соединение 7), как показано, модулирует гены AKT и лечит пролиферационные расстройства, включая рак (US 2011/0172203 A1, в настоящем документе далее упоминается как заявка '203). Сведения о мелкомасштабном синтезе 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина (соединения 7) недавно опубликованы в заявке '203. Синтез заявки '203 является непрактичным при получении больших количеств соединения и имеет несколько недостатков.
Соответственно, имеется необходимость в улучшенном способе синтеза 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина (соединения 7), который является пригодным для коммерческого производства, которое является безопасным и простым.
Сущность изобретения
Настоящее изобретение относится к способу получения 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина:
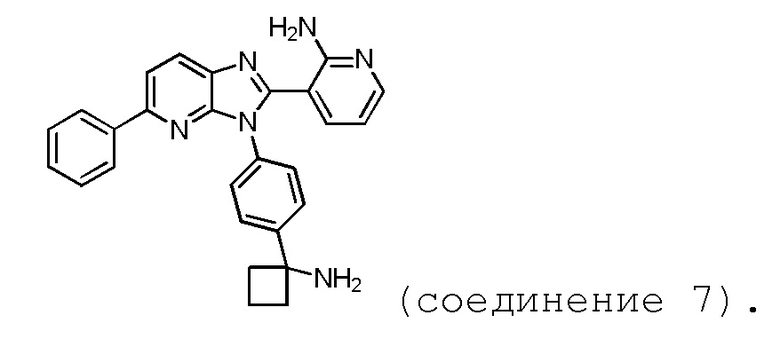
В одном из аспектов, настоящее изобретение относится к способу получения соединения 7, включающему четырехстадийный синтез. В одном из аспектов, настоящее изобретение относится к способу получения соединение 7, включающему трехстадийный синтез.
Подробное описание изобретения
Настоящее изобретение направлено на способ получения 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина (соединения 7). Способ по настоящему изобретению изображен ниже на репрезентативных Схемах.
Схема 1
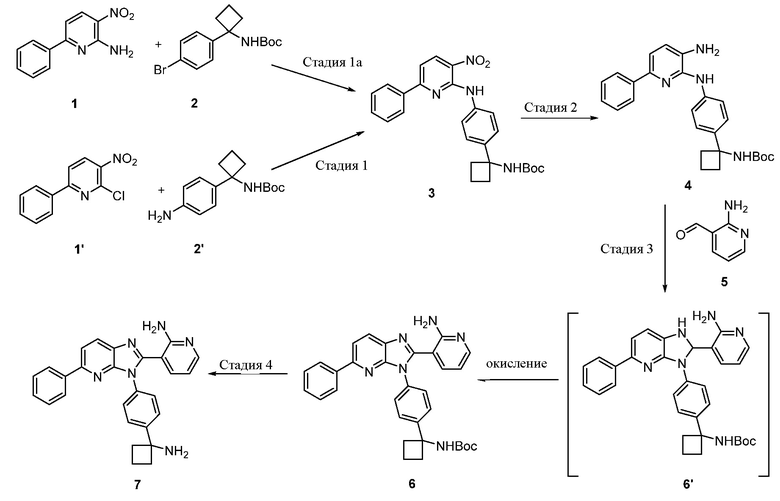
Схема 1'
О способах по настоящему изобретению никогда не сообщалось в данной области.
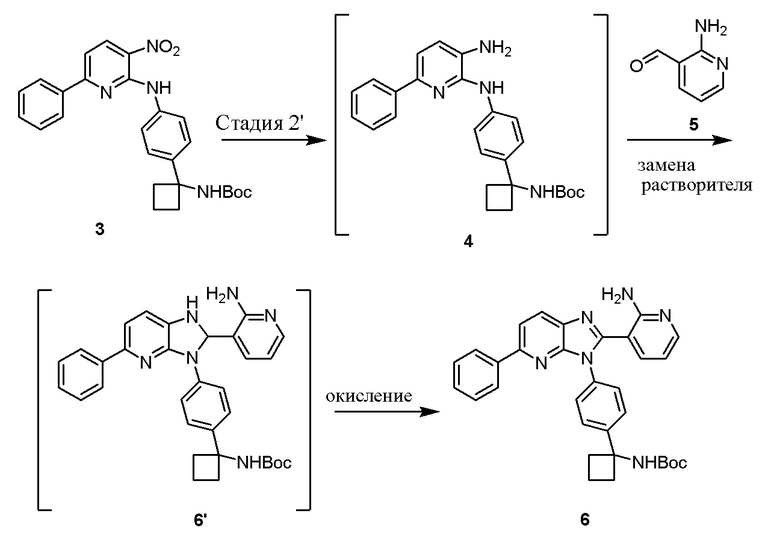
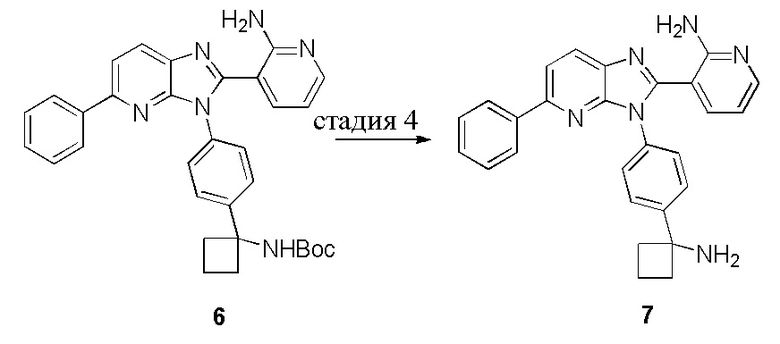
В одном из вариантов осуществления, способ по настоящему изобретению включает в себя четыре стадии (Схема 1). Первая стадия представляет собой реакцию замещения 1' и 2' с получением соединения 3 (Стадия 1) или, альтернативно, реакцию перекрестного связывания 1 и 2 для генерирования соединения 3 (Стадия 1a). Вторая стадия представляет собой восстановление соединения 3 с образованием анилинового соединения 4. Третья стадия представляет собой циклизацию соединений 4 и соединения 5 (2-аминоникотинальдегида) с получением циклизированного промежуточного соединения 6', которое окисляется in situ с образованием соединения 6. Четвертая стадия представляет собой снятие защиты с соединения 6 с получением 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина (соединения 7).
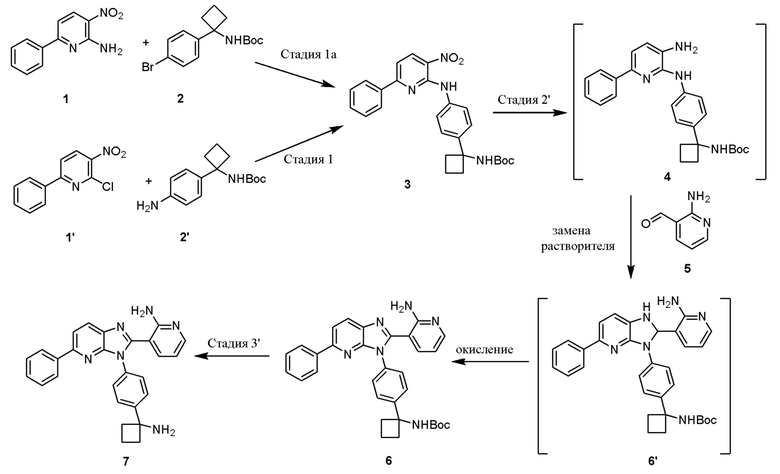
В одном из вариантов осуществления, способ по настоящему изобретению включает в себя три стадии (Схема 1'). В одном из вариантов осуществления, вторая и третья стадии, описанные на Схеме 1, объединяются в оптимальный способ (Стадия 2'). Первая стадия представляет собой реакцию замещения 1' и 2' с получением соединения 3 (Стадия 1) или, альтернативно, реакцию перекрестного связывания 1 и 2 для генерирования соединения 3 (Стадия 1a). Вторая стадия включает восстановление соединения 3 с образованием промежуточного анилинового соединения 4, которое, после замены полярного апротонного растворителя полярным протонным растворителем, взаимодействует с соединением 5 с образованием соединения 6', которое окисляется in situ с получением соединения 6 (Стадия 2'). Третья стадия представляет собой снятие защиты с соединения 6 с получением 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина (соединения 7) (Стадия 3').
В одном из вариантов осуществления, настоящее изобретение относится к способу получения 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина (соединения 7), включающему стадию:
Стадию 3 взаимодействия трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4) с 2-аминоникотинальдегидом (соединением 5) в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил (1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамата (соединения 6).
В одном из вариантов осуществления, способ по настоящему изобретению относится к получению 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина (соединения 7), включающему стадии:
Стадию 2 обработки трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 3) с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4) и
Стадию 3 взаимодействия трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4) с 2-аминоникотинальдегидом (соединением 5) в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил (1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамата (соединения 6).
В одном из вариантов осуществления, способ по настоящему изобретению относится к получению 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина (соединения 7), включающему стадии:
Стадию 1 взаимодействия 2-хлор-3-нитро-6-фенилпиридина (соединения 1') с трет-бутил (1-(4-аминофенил)циклобутил)карбаматом (соединением 2') в присутствии основания в полярном апротонном растворителе с образованием трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 3);
Стадию 2 обработки трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 3) с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4) и
Стадию 3 взаимодействия трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4) с 2-аминоникотинальдегидом (соединением 5) в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил (1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамата (соединения 6).
В одном из вариантов осуществления, способ по настоящему изобретению относится к получению 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина (соединения 7), включающему стадии:
Стадию 1a связывания 3-нитро-6-фенилпиридин-2-амина (соединения 1) с трет-бутил (1-(4-бромфенил)циклобутил)карбаматом (соединением 2) в присутствии палладиевого катализатора и фосфорного лиганда в полярном апротонном растворителе с образованием трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 3);
Стадию 2 обработки трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 3) с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4) и
Стадию 3 взаимодействия трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4) с 2-аминоникотинальдегидом (соединением 5) в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил (1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамата (соединения 6).
В одном из вариантов осуществления, способ по настоящему изобретению относится к получению 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина, включающему стадии:
Стадию 1 взаимодействия 2-хлор-3-нитро-6-фенилпиридина (соединения 1') с трет-бутил (1-(4-аминофенил)циклобутил)-карбаматом (соединением 2') в присутствии основания в полярном апротонном растворителе с образованием трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 3);
Стадию 2 обработки трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 3) с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4);
Стадию 3 взаимодействия трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4) с 2-аминоникотинальдегидом (5) в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил (1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамата (соединения 6) и
Стадию 4 обработки трет-бутил (1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)-карбамата (соединения 6) кислотой в полярном апротонном растворителе с образованием 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина (соединения 7).
В одном из вариантов осуществления, способ по настоящему изобретению относится к получению 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина, включающему стадии:
Стадию 1a связывания 3-нитро-6-фенилпиридин-2-амина (соединения 1) с трет-бутил (1-(4-бромфенил)циклобутил)карбаматом (соединением 2) в присутствии палладиевого катализатора и фосфорного лиганда в полярном апротонном растворителе с образованием трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 3);
Стадию 2 обработки трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 3) с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4);
Стадию 3 взаимодействия трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4) с 2-аминоникотинальдегидом (соединением 5) в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил (1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамата (соединения 6) и
Стадию 4 обработки трет-бутил (1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамата (соединения 6) кислотой в полярном апротонном растворителе с образованием 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина (соединения 7).
В одном из вариантов осуществления, способ по настоящему изобретению относится к получению 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина (соединения 7), включающему стадию:
Стадию 2' обработки трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 3) с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4), замены полярного апротонного растворителя полярным протонным растворителем и взаимодействия трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4) с 2-аминоникотинальдегидом (соединением 5) в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил (1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамата (соединения 6).
В одном из вариантов осуществления, способ по настоящему изобретению относится к получению 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина (соединения 7), включающему стадии:
Стадию 1 взаимодействия 2-хлор-3-нитро-6-фенилпиридина (соединения 1') с трет-бутил (1-(4-аминофенил)циклобутил)-карбаматом (соединением 2') в присутствии основания в полярном апротонном растворителе с образованием трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 3) и
Стадия 2' обработки трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 3) с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4), замены полярного апротонного растворителя полярным протонным растворителем и взаимодействия трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4) с 2-аминоникотинальдегидом (соединением 5) в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил (1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамата (соединения 6).
В одном из вариантов осуществления, способ по настоящему изобретению относится к получению 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина (соединения 7), включающему стадии:
Стадию 1a связывания 3-нитро-6-фенилпиридин-2-амина (соединения 1) с трет-бутил (1-(4-бромфенил)циклобутил)карбаматом (соединением 2) в присутствии палладиевого катализатора и фосфорного лиганда в полярном апротонном растворителе с образованием трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 3) и
Стадию 2' обработки трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 3) с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4), замены полярного апротонного растворителя полярным протонным растворителем и взаимодействия трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4) с 2-аминоникотинальдегидом (соединением 5) в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил (1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамата (соединения 6).
В одном из вариантов осуществления, способ по настоящему изобретению относится к получению 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина, включающему стадии:
Стадию 2' обработки трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 3) с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4), замены полярного апротонного растворителя полярным протонным растворителем и взаимодействия трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4) с 2-аминоникотинальдегидом (соединением 5) в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил (1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамата (соединения 6) и
Стадию 3' обработки трет-бутил (1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)-карбамата (соединения 6) кислотой в полярном апротонном растворителе с образованием 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина (соединения 7).
В одном из вариантов осуществления, способ по настоящему изобретению относится к получению 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина, включающему стадии:
Стадию 1 взаимодействия 2-хлор-3-нитро-6-фенилпиридина (соединения 1') с трет-бутил (1-(4-аминофенил)циклобутил)карбаматом (соединением 2') в присутствии основания в полярном апротонном растворителе с образованием трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)-циклобутил)карбамата (соединения 3);
Стадию 2' обработки трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 3) с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4), замены полярного апротонного растворителя полярным протонным растворителем и взаимодействия трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4) с 2-аминоникотинальдегидом (соединением 5) в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил (1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамата (соединения 6) и
Стадию 3' обработки трет-бутил (1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)-карбамата (соединения 6) кислотой в полярном апротонном растворителе с образованием 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина (соединения 7).
В одном из вариантов осуществления, способ по настоящему изобретению относится к получению 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина, включающему стадии:
Стадию 1a связывания 3-нитро-6-фенилпиридин-2-амина (соединения 1) с трет-бутил (1-(4-бромфенил)циклобутил)карбаматом (соединением 2) в присутствии палладиевого катализатора и фосфорного лиганда в полярном апротонном растворителе с образованием трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 3);
Стадию 2' обработки трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 3) с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4), замены полярного апротонного растворителя полярным протонным растворителем и взаимодействия трет-бутил (1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата (соединения 4) с 2-аминоникотинальдегидом (соединением 5) в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил (1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамата (соединения 6) и
Стадию 3' обработки трет-бутил (1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)-карбамата (соединения 6) кислотой в полярном апротонном растворителе с образованием 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина (соединения 7).
В одном из вариантов осуществления, способ по настоящему изобретению включает Стадию 3. Стадия 3 представляет собой циклизацию соединений 4 и соединения 5 (2-аминоникотинальдегида) с получением промежуточного соединения 6', которое окисляется с образованием соединения 6:
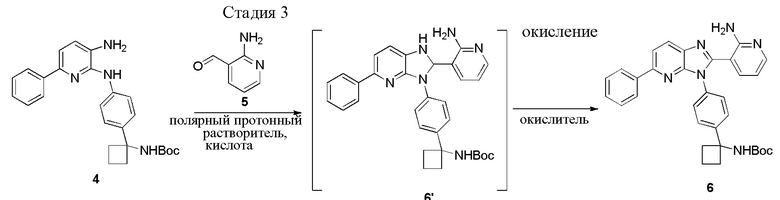
В одном из вариантов осуществления, полярный протонный растворитель представляет собой C1-4 спирт. В другом варианте осуществления, полярный протонный растворитель выбирается из группы, состоящей из метанола, этанола, н-пропанола, изопропанола, н-бутанола, втор-бутанола и трет-бутанола. В другом варианте осуществления, полярный протонный растворитель представляет собой метанол.
В одном из вариантов осуществления, кислота представляет собой органическую кислоту. В другом варианте осуществления, кислота выбирается из группы, состоящей из муравьиной кислоты, уксусной кислоты и пропановой кислоты. В другом варианте осуществления, кислота представляет собой уксусную кислоту. В одном из вариантов осуществления, отношение кислоты к растворителю находится в диапазоне примерно от 1:25 примерно до 25:1, примерно от 1:20 примерно до 20:1, примерно от 1:15 примерно до 15:1, примерно от 1:1 примерно до 15:1, примерно от 3:1 примерно до 12:1 или примерно от 5:1 примерно до 10:1. В другом варианте осуществления, отношение кислоты к растворителю составляет примерно 9:1. В другом варианте осуществления, отношение уксусной кислоты к метанолу составляет примерно 9:1.
В одном из вариантов осуществления, окислитель представляет собой воздух. В другом варианте осуществления окислитель представляет собой соль на основе металла или неметалла или катализатор. В другом варианте осуществления, окислитель выбирается из группы, состоящей из ацетата металла, пербората металла, хлорида металла, катализатора на основе палладия и их гидратов. В другом варианте осуществления, окислитель выбирается из группы, состоящей из пербората щелочного металла и его гидратов. В другом варианте осуществления, окислитель выбирается из группы, состоящей из ацетата меди, пербората натрия, хлорида железа (III), палладия на угле и их гидратов. В другом варианте осуществления, окислитель выбирается из группы, состоящей из Cu(OAc)2⋅H2O, NaBO3⋅4H2O, FeCl3⋅6H2O, и 10% Pd/C. В другом варианте осуществления, окислитель представляет собой NaBO3⋅4H2O.
В одном из вариантов осуществления, температура реакционной смеси составляет примерно от 10°C примерно до 30°C. В другом варианте осуществления, температура составляет примерно от 15°C примерно до 25°C. В другом варианте осуществления, температура составляет примерно 20°C. В другом варианте осуществления, температура реакционной смеси составляет примерно от 10°C примерно до 60°C. В другом варианте осуществления, температура составляет примерно от 30°C примерно до 50°C. В другом варианте осуществления, температура составляет примерно 40°C.
В одном из вариантов осуществления, реакционную смесь перемешивают в течение примерно 40 часов - примерно 50 часов. В другом варианте осуществления, реакционную смесь перемешивают в течение примерно 43 часов - примерно 46 часов. В другом варианте осуществления, реакционную смесь перемешивают в течение примерно 45 часов. В другом варианте осуществления, реакционную смесь перемешивают в течение примерно 10 часов - примерно 18 часов. В другом варианте осуществления, реакционную смесь перемешивают в течение примерно 12 часов - примерно 16 часов. В одном из вариантов осуществления, реакционную смесь перемешивают в течение примерно 12 часов, примерно 13 часов, примерно 14 часов или примерно 15 часов.
В одном из вариантов осуществления, окисление завершается примерно через 40 часов - примерно 50 часов. В другом варианте осуществления, окисление завершается примерно через 43 часа - примерно 46 часов. В другом варианте осуществления, окисление завершается примерно через 45 часов. В другом варианте осуществления, окисление завершается в течение примерно 10 часов - примерно 18 часов. В другом варианте осуществления, окисление завершается примерно через 12 часов - примерно 16 часов. В одном из вариантов осуществления, окисление завершается примерно через 12 часов, примерно через 13 часов, примерно через 14 часов или примерно через 15 часов.
В одном из вариантов осуществления, окисление завершается до того как образуется значительное количество переокисленной примеси N-оксида (M+16) соединения 6. В другом варианте осуществления, количество переокисленной примеси N-оксида (M+16) ниже 10% AUC (площади под кривой), 9% AUC, 8% AUC, 7% AUC, 6% AUC, 5% AUC, 4% AUC, 3% AUC, 2% AUC, 1% AUC, 0,9% AUC, 0,8% AUC, 0,7% AUC, 0,6% AUC, 0,5% AUC, 0,4% AUC, 0,3% AUC, 0,2% AUC, 0,1% AUC, 0,09% AUC, 0,08% AUC, 0,07% AUC, 0,06% AUC, 0,05% AUC, 0,04% AUC, 0,03% AUC, 0,02% AUC или 0,01% AUC, когда окисление завершается. В другом варианте осуществления, количество переокисленной примеси N-оксида (M+16) ниже 3% AUC, 2% AUC, 1% AUC, 0,9% AUC, 0,8% AUC, 0,7% AUC, 0,6% AUC, 0,5% AUC, 0,4% AUC, 0,3% AUC, 0,2% AUC, 0,1% AUC, 0,09% AUC, 0,08% AUC, 0,07% AUC, 0,06% AUC, 0,05% AUC, 0,04% AUC, 0,03% AUC, 0,02% AUC, или 0,01% AUC, когда окисление завершается.
В одном из вариантов осуществления, выделение соединения 6 включает концентрирование реакционной смеси, содержащей соединение 6. В одном из вариантов осуществления, выделение соединения 6 включает добавление основания. В одном из вариантов осуществления, выделение соединения 6 включает добавление основания после концентрирования соединения 6. В одном из вариантов осуществления, основание представляет собой гидроксид (например, NaOH, KOH). В одном из вариантов осуществления, гидроксид представляет собой KOH. В одном из вариантов осуществления, соединение 6 выделяется из 2-метилтетрагидрофурана и изопропилацетата. В одном из вариантов осуществления, выделение соединения 6 включает промывку смеси, содержащей соединение 6, 2-MeTHF. В одном из вариантов осуществления, выделение соединения 6 включает удаление водного слоя после промывки с получением органического слоя. В одном из вариантов осуществления, выделение соединения 6 включает промывку органического слоя насыщенным раствором соли и удаление полученного в результате водного слоя. В одном из вариантов осуществления, стадии промывки насыщенным раствором соли и удаление полученного в результате водного слоя осуществляют один раз, два раза или три раза. В одном из вариантов осуществления, выделение соединения 6 включает добавление IPAc к органическому слою после стадии промывки. В одном из вариантов осуществления, IPAc смешивается с 2-MeTHF. В одном из вариантов осуществления, добавление IPAc к органическому слою приводит к формированию суспензии. В одном из вариантов осуществления, соединение 6 промывают изопропилацетатом, смесью изопропилацетат/гептан и гептаном. В одном из вариантов осуществления, смесь изопропил/гептан находится при отношении 1:1.
В одном из вариантов осуществления, соединение 6 очищают, включая растворение соединения 6 в DCM и элюирование растворенного соединения 6 через силикагель с помощью DCM. В одном из вариантов осуществления, гель промывают EtOAc.
В одном из вариантов осуществления, способ по настоящему изобретению включает Стадию 2. Стадия 2 представляет собой восстановление соединения 3 с образованием анилинового соединения 4:
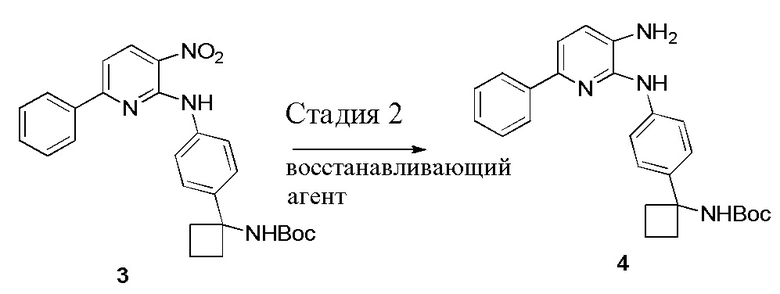
В одном из вариантов осуществления, способ по настоящему изобретению включает Стадии 2 и 3. В одном из вариантов осуществления, восстанавливающий агент на Стадии 2 представляет собой газообразный водород над каталитическим количеством Pd/C. В одном из вариантов осуществления, полярный апротонный растворитель на Стадии 2 представляет собой EtOAc, тетрагидрофуран или 2-метилтетрагидрофуран. В одном из вариантов осуществления, выделение соединения 4 на Стадии 2 включает фильтрование реакционной смеси через Celite®. В одном из вариантов осуществления, выделение дополнительно включает добавление метанола и концентрирование реакционной смеси досуха.
В одном из вариантов осуществления, способ по настоящему изобретению включает Стадию 1. Стадия 1 представляет собой реакцию замещения 1' и 2' с получением соединения 3:
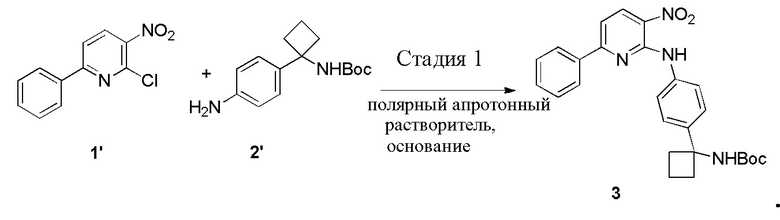
В одном из вариантов осуществления, способ по настоящему изобретению включает Стадии 1, 2 и 3. В одном из вариантов осуществления, полярный апротонный растворитель на Стадии 1 представляет собой диметилацетамид. В одном из вариантов осуществления, основание на Стадии 1 представляет собой Na2CO3. В одном из вариантов осуществления, температура реакционной смеси на Стадии 1 составляет примерно от 90°C примерно до 110°C. В одном из вариантов осуществления, температура составляет примерно от 95°C примерно до 105°C. В одном из вариантов осуществления, температура составляет примерно 100°C. В одном из вариантов осуществления, 1' очищается посредством перемешивания вместе со спиртом с образованием суспензии. В одном из вариантов осуществления, спирт представляет собой метанол.
В одном из вариантов осуществления, способ по настоящему изобретению включает Стадию 1a. Стадия 1a представляет собой реакцию перекрестного связывания 1 и 2 для генерирования соединения 3:
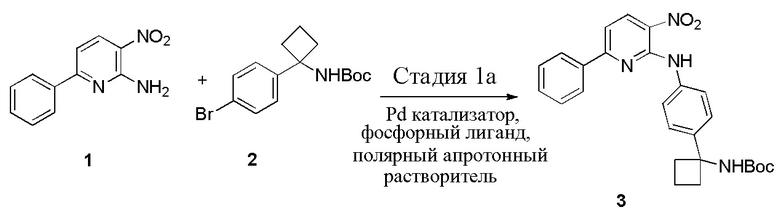
В одном из вариантов осуществления, способ по настоящему изобретению включает Стадии 1a, 2 и 3. В одном из вариантов осуществления, палладиевый катализатор на Стадии 1a представляет собой катализатор Pd(II). В одном из вариантов осуществления, катализатор Pd(II) представляет собой Pd2(dba)3. В одном из вариантов осуществления, фосфорный лиганд на Стадии 1a представляет собой 4,5-бис(дифенилфосфино)-9,9-диметилксантен. В одном из вариантов осуществления, полярный апротонный растворитель на Стадии 1a представляет собой тетрагидрофуран. В одном из вариантов осуществления, температура реакционной смеси на Стадии 1 составляет примерно от 60°C примерно до 80°C. В одном из вариантов осуществления, температура реакционной смеси составляет примерно от 65°C примерно до 75°C. В одном из вариантов осуществления, температура реакционной смеси составляет примерно 70°C.
В одном из вариантов осуществления, способ по настоящему изобретению включает Стадию 4. Стадия 4 представляет собой снятие защиты с соединения 6 с получением соединения 7:
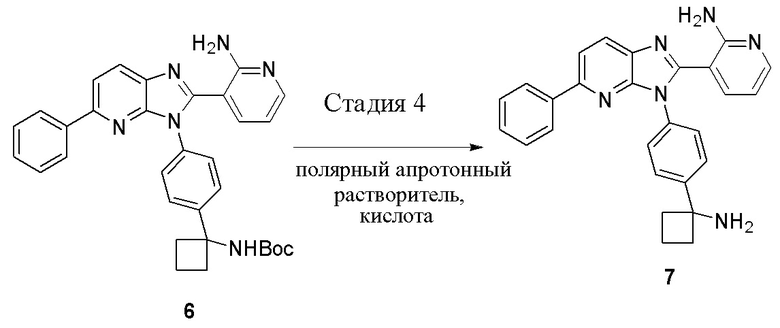
В одном из вариантов осуществления, способ по настоящему изобретению включает Стадии 1, 2, 3 и 4. В одном из вариантов осуществления, способ по настоящему изобретению включает Стадии 1a, 2, 3 и 4. В одном из вариантов осуществления, полярный апротонный растворитель на Стадии 4 представляет собой дихлорметан. В одном из вариантов осуществления, кислота на Стадии 4 представляет собой метансульфоновую кислоту. В одном из вариантов осуществления, отношение кислоты к соединению 6 на Стадии 4 составляет примерно 5:1. В одном из вариантов осуществления, реакция в смеси на Стадии 4 завершается примерно через 1,5 часа - примерно 3 часа. В одном из вариантов осуществления, реакция в смеси завершается примерно через 2 часа - примерно 2,5 часа. В одном из вариантов осуществления, реакция в смеси завершается примерно через 2 часа.
В одном из вариантов осуществления, на Стадии 4 образуется суспензия. В одном из вариантов осуществления, выделение соединения 7 включает добавление воды к суспензии и удаление полученного в результате водного слоя и удерживание слоя DMC. В одном из вариантов осуществления, выделение соединения 7 включает добавление воды к слою DMC и удаление водного слоя. В одном из вариантов осуществления, выделение соединения 7 включает объединение водных слоев и промывку объединенного водного слоя DCM. В одном из вариантов осуществления, выделение соединения 7 включает добавление основания. В одном из вариантов осуществления, основание представляет собой гидроксид (например, NaOH, KOH). В одном из вариантов осуществления, гидроксид представляет собой NaOH. В одном из вариантов осуществления, выделение соединения 7 включает сушку органического слоя после добавления основания с получением твердого соединения 7. В одном из вариантов осуществления, выделение соединения 7 включает концентрирование раствора после добавления основания и добавления IPAc.
В одном из вариантов осуществления, способ по настоящему изобретению включает Стадию 2'. Стадия 2' представляет собой восстановление соединения 3 с образованием промежуточного анилин соединение 4, которое, после замены полярного апротонного растворитель полярным протонным растворителем, взаимодействует с соединением 5 с образованием соединения 6', которое окисляется in situ с получением соединения 6:
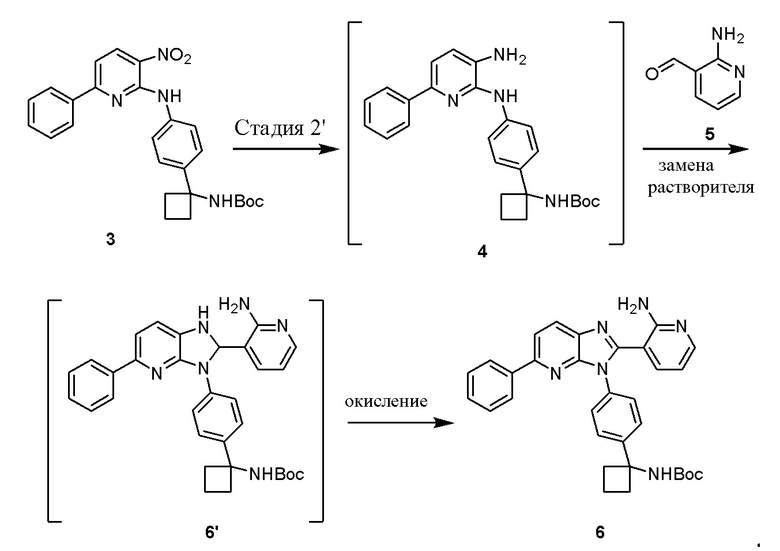
В одном из вариантов осуществления, способ по настоящему изобретению включает Стадии 1 и 2'. В одном из вариантов осуществления, Стадия 2' включает замену полярного апротонного растворителя, такого как THF полярным протонным растворителем, таким как MeOH. В одном из вариантов осуществления, полярный апротонный растворитель, используемый при восстановлении соединения 3 с образованием анилинового соединения 4 на Стадии 2', представляет собой этилацетат, THF или 2-MeTHF. В одном из вариантов осуществления, растворитель представляет собой THF. В одном из вариантов осуществления, восстанавливающий агент на Стадии 2' представляет собой газообразный водород над каталитическим количеством Pd/C. В одном из вариантов осуществления, газообразный водород находится при умеренных давлениях примерно от 20 примерно до 50 фунт/кв. дюйм (1,2-3,1 кг/кв. см). В одном из вариантов осуществления, выделение соединения 4 включает фильтрование реакционной смеси через Celite®. В одном из вариантов осуществления, выделение дополнительно включает добавление метанола и концентрирование реакционной смеси досуха.
В одном из вариантов осуществления, полярный протонный растворитель, используемый в реакции соединения 4 и соединения 5 на Стадии 2', представляет собой C1-4 спирт. В другом варианте осуществления, полярный протонный растворитель выбирается из группы, состоящей из метанола, этанола, н-пропанола, изопропанола, н-бутанола, втор-бутанола и трет-бутанола. В другом варианте осуществления, полярный протонный растворитель представляет собой метанол. В другом варианте осуществления, кислота, используемая в реакции соединения 4 и соединении 5 на Стадии 2', представляет собой органическую кислоту. В другом варианте осуществления, кислота выбирается из группы, состоящей из муравьиной кислоты, уксусной кислоты и пропановой кислоты. В другом варианте осуществления, кислота представляет собой уксусную кислоту. В одном из вариантов осуществления, отношение кислоты к растворителю, используемому на Стадии 2', находится в диапазоне примерно от 1:25 примерно до 25:1, примерно от 1:20 примерно до 20:1, примерно от 1:15 примерно до 15:1, примерно от 1:1 примерно до 15:1, примерно от 3:1 примерно до 12:1 или примерно от 5:1 примерно до 10:1. В другом варианте осуществления, отношение кислоты к растворителю составляет примерно 9:1. В другом варианте осуществления, отношение уксусной кислоты к метанолу составляет примерно 9:1.
В одном из вариантов осуществления, окислитель, используемый на Стадии 2', представляет собой соль на основе металла или неметалла или катализатор. В другом варианте осуществления, окислитель выбирается из группы, состоящей из ацетата металла, пербората металла, хлорида металла, катализатора на основе палладия и их гидратов. В другом варианте осуществления, окислитель выбирается из группы, состоящей из пербората щелочного металла и его гидратов. В другом варианте осуществления, окислитель выбирается из группы, состоящей из ацетата меди, пербората натрия, хлорида железа (III), палладия на угле и их гидратов. В другом варианте осуществления, окислитель выбирается из группы, состоящей из Cu(OAc)2⋅H2O, NaBO3⋅4H2O, FeCl3⋅H2O и 10% Pd/C. В другом варианте осуществления, окислитель представляет собой NaBO3⋅H2O.
В одном из вариантов осуществления, температура реакционной смеси в реакции соединения 4 и соединении 5 на Стадии 2' составляет примерно от 10°C примерно до 30°C. В другом варианте осуществления, температура составляет примерно от 15°C примерно до 25°C. В другом варианте осуществления, температура составляет примерно 20°C. В другом варианте осуществления, температура реакционной смеси составляет примерно от 10°C примерно до 60°C. В другом варианте осуществления, температура составляет примерно от 30°C примерно до 50°C. В другом варианте осуществления, температура составляет примерно 40°C. В одном из вариантов осуществления, реакционную смесь перемешивают в течение примерно от 40 часов примерно до 50 часов. В другом варианте осуществления, реакционную смесь перемешивают в течение примерно от 43 часов примерно до 46 часов. В другом варианте осуществления, реакционную смесь перемешивают в течение примерно 45 часов. В другом варианте осуществления, реакционную смесь перемешивают в течение примерно от 10 часов примерно до 18 часов. В другом варианте осуществления, реакционную смесь перемешивают в течение примерно от 12 часов примерно до 16 часов. В одном из вариантов осуществления, реакционную смесь перемешивают в течение примерно 12 часов, примерно 13 часов, примерно 14 часов или примерно 15 часов.
В одном из вариантов осуществления, окисление завершается примерно через 40 часов - 50 часов. В другом варианте осуществления, окисление завершается примерно через 43 часа - примерно 46 часов. В другом варианте осуществления, окисление завершается примерно через 45 часов. В другом варианте осуществления, окисление завершается в течение примерно 10 часов - примерно 18 часов. В другом варианте осуществления, окисление завершается примерно через 12 часов - примерно 16 часов. В одном из вариантов осуществления, окисление завершается примерно через 12 часов, примерно 13 часов, примерно 14 часов или примерно 15 часов. В одном из вариантов осуществления, окисление завершается до того как образуется значительное количество переокисленной примеси N-оксида (M+16) соединения 6. В другом варианте осуществления, количество переокисленной примеси N-оксида (M+16) ниже 10% AUC, 9% AUC, 8% AUC, 7% AUC, 6% AUC, 5% AUC, 4% AUC, 3% AUC, 2% AUC, 1% AUC, 0,9% AUC, 0,8% AUC, 0,7% AUC, 0,6% AUC, 0,5% AUC, 0,4% AUC, 0,3% AUC, 0,2% AUC, 0,1% AUC, 0,09% AUC, 0,08% AUC, 0,07% AUC, 0,06% AUC, 0,05% AUC, 0,04% AUC, 0,03% AUC, 0,02% AUC или 0,01% AUC, когда окисление завершается. В другом варианте осуществления, количество переокисленной примеси N-оксида (M+16) ниже 3% AUC, 2% AUC, 1% AUC, 0,9% AUC, 0,8% AUC, 0,7% AUC, 0,6% AUC, 0,5% AUC, 0,4% AUC, 0,3% AUC, 0,2% AUC, 0,1% AUC, 0,09% AUC, 0,08% AUC, 0,07% AUC, 0,06% AUC, 0,05% AUC, 0,04% AUC, 0,03% AUC, 0,02% AUC или 0,01% AUC, когда окисление завершается.
В одном из вариантов осуществления, выделение соединения 6 включает концентрирование реакционной смеси, содержащей соединение 6. В одном из вариантов осуществления, выделение соединения 6 включает добавление основания. В одном из вариантов осуществления, выделение соединения 6 включает добавление основания после концентрирования соединения 6. В одном из вариантов осуществления, основание представляет собой гидроксид (например, NaOH, KOH). В одном из вариантов осуществления, гидроксид представляет собой KOH. В одном из вариантов осуществления, соединение 6 выделяется из 2-метилтетрагидрофурана и изопропилацетата. В одном из вариантов осуществления, выделение соединения 6 включает промывку смеси, содержащей соединение 6, с помощью 2-MeTHF. В одном из вариантов осуществления, выделение соединения 6 включает удаление водного слоя после промывки с получением органического слоя. В одном из вариантов осуществления, выделение соединения 6 включает промывку органического слоя насыщенным раствором соли и удаление полученного в результате водного слоя. В одном из вариантов осуществления, стадии промывки насыщенным раствором соли и удаления полученного в результате водного слоя осуществляют один раз, два раза или три раза. В одном из вариантов осуществления, выделение соединения 6 включает добавление IPAc к органическому слою после стадии промывки. В одном из вариантов осуществления, IPAc смешивается с 2-MeTHF. В одном из вариантов осуществления, добавление IPAc к органическому слою приводит к формированию суспензии. В одном из вариантов осуществления, соединение 6 промывают изопропилацетатом, смесью изопропилацетат/гептан и гептаном. В одном из вариантов осуществления, смесь изопропил/гептан находится в отношении 1:1.
В одном из вариантов осуществления, соединение 6 очищают, включая растворение соединения 6 в DCM и элюирование растворенного соединения 6 через силикагель с помощью DCM. В одном из вариантов осуществления, гель промывают EtOAc.
В одном из вариантов осуществления, способ по настоящему изобретению включает Стадию 3'. Стадия 3' представляет собой снятие защиты с соединения 6 с получением соединения 7:
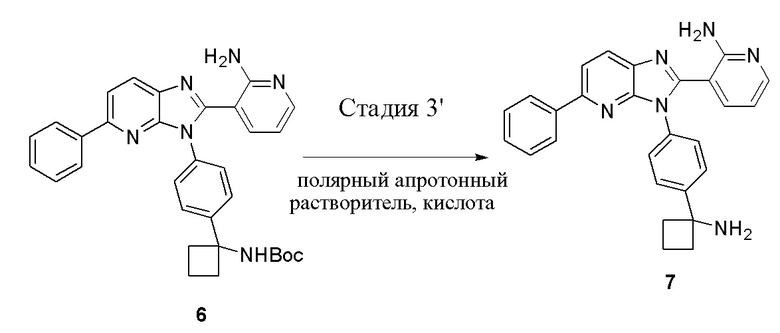
В одном из вариантов осуществления, способ по настоящему изобретению включает Стадии 1, 2' и 3'. В одном из вариантов осуществления, полярный апротонный растворитель на Стадии 3' представляет собой дихлорметан. В одном из вариантов осуществления, кислота на Стадии 3' представляет собой метансульфоновую кислоту. В одном из вариантов осуществления, отношение кислоты к соединению 6 составляет примерно 5:1. В одном из вариантов осуществления, реакционная смесь на Стадии 3' завершается примерно через 1,5 часа - примерно 3 часа. В одном из вариантов осуществления, реакция на Стадии 3' завершается примерно через 2 часа - примерно 2,5 часа. В одном из вариантов осуществления, реакция на Стадии 3' завершается примерно через 2 часа.
В одном из вариантов осуществления, на Стадии 3' образуется суспензия. В одном из вариантов осуществления, выделение соединения 7 включает добавление воды к суспензии и удаление полученного в результате водного слоя и удерживание слоя DMC. В одном из вариантов осуществления, выделение соединения 7 включает добавление воды к слою DMC и удаление водного слоя. В одном из вариантов осуществления, выделение соединения 7 включает объединение водных слоев и промывку объединенного водного слоя DCM. В одном из вариантов осуществления, выделение соединения 7 включает добавление основания. В одном из вариантов осуществления, основание представляет собой гидроксид (например, NaOH, KOH). В одном из вариантов осуществления, гидроксид представляет собой NaOH. В одном из вариантов осуществления, выделение соединения 7 включает сушку органического слоя после добавления основания с получением твердого соединения 7. В одном из вариантов осуществления, выделение соединения 7 включает концентрирование раствора после добавления основания и добавление IPAc.
Недостатки предыдущего способа
Способ по настоящему изобретению представляет собой усовершенствование по сравнению со способом, описанным в предыдущей заявке '203. Способ получения гидрохлорида соединения 7 в патенте '203 изображен на Схеме 2:
Схема 2
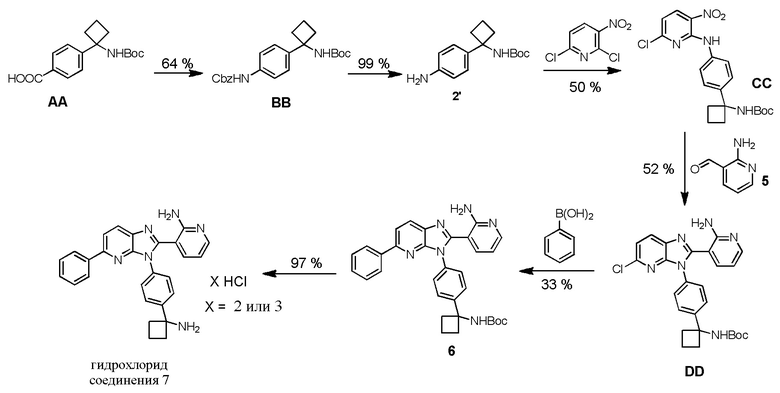
Способ заявки '203, как на Схеме 2, начинается с карбоновой кислоты AA, которая подвергается воздействию перегруппировки Курциуса с использованием дифенилфосфорилазида (DPPA) с последующим поглощением изоцианата с помощью бензилового спирта, который генерирует защищенное Cbz промежуточное соединение BB. Снятие защиты при условиях гидрогенолиза дает анилин 2'. Добавление анилина 2' к 2,6-дихлор-3-нитропиридину происходит с получением сырого CC. После очистки с помощью колоночной хроматографии, CC подвергается воздействию восстанавливающих условий и циклизируется с помощью 2-аминоникотинальдегида (5) с получением циклизированного соединения DD. Связывание Судзуки циклизированного продукта с бензолбориновой кислотой дает соединение 6. После снятия защиты с 6 с помощью HCl в диоксане, желаемая гидрохлоридная соль соединения 7 выделяется в виде некристаллического твердого продукта.
Способ заявки '203 является сложным для масштабирования, дорогостоящим при осуществлении и непригодным для производства в коммерческом масштабе. Недостатки способа заявки '203 являются, по меньшей мере, следующими:
1. он следует линейному способу с общим выходом 5%,
2. использует потенциально взрывоопасный азидный химический механизм,
3. требует дорогостоящей очистки с помощью колоночной хроматографии,
4. использует палладиевый химический механизм для получения предпоследнего промежуточного соединения 6, что приводит к неприемлемому уровню палладиевой примеси в соединении 7,
5. вводит дорогостоящие материалы в начале синтеза, и
6. использует сложный окислительно-восстановительный химический механизм с использованием Na2S2O4.
Способ по настоящему изобретению представляет собой превосходный способ получения 7 и преодолевает перечисленные выше недостатки. Например, способ по настоящему изобретению вводит стадии, которые используют палладий в способе на более ранней стадии, что уменьшает количество палладиевой примеси, если она вообще присутствует, в конечном продукте, соединении 7. Например, реакция для генерирования соединения 1 (Схема 1 или Схема 1') и реакция перекрестного связывания для генерирования 3 (Схема 1 или Схема 1') с использованием палладия в способе по настоящему изобретению вводятся на более ранней стадии. В противоположность этому, способ '203 использует палладиевый химический механизм для получения предпоследнего промежуточного соединения 6, что приводит к возникновению проблем с примесями в конечном продукте, соединении 7.
Способ по настоящему изобретению является также более компактным с уменьшенным количеством стадий и устраняет необходимость в азидном химическом механизме и Na2S2O4 (смотри Схему 2, получение соединения DD). Азиды, как известно, являются опасными и токсичными. Na2S2O4 представляет собой воспламеняемый твердый продукт и может воспламениться в присутствии влажности и воздуха. По этой причине, устранение необходимости в азидном химическом механизме и Na2S2O4 делает способ по настоящему изобретению безопасным и более практичным.
Способ по настоящему изобретению может осуществляться в большом масштабе, в то время как способ заявки '203 является дорогостоящим и сложным при масштабировании. Например, получение соединения 2' с использованием способа '203 включает воздействие на соединение BB перегруппировки Курциуса с использованием DPPA, с последующим поглощением изоцианата избытком бензилового спирта (смотри Схема 2, получения соединения BB). Хотя этот химический механизм является применимым в малом масштабе, его осуществление в большом масштабе является сложным и создает множество проблем. В малом масштабе, защищенное Cbz соединение BB получается только при самом умеренном выходе 62% в два захода с помощью, как преципитации, так и колоночной очистки, что является трудоемким и чрезмерно дорогостоящим для его осуществления в большом масштабе.
Способ по настоящему изобретению использует соединение 3 в качестве синтетического промежуточного соединения (Схема 1 или Схема 1'), что является аналогичным получению соединения CC (Схема 2) в синтезе '203. Получение соединения CC способа '203, как правило, только обеспечивает выход 50%, при этом соединение 3 с использованием способа по настоящему изобретению дает выход 86%.Конкретно, соединение 3 получают в реакции 1' и 2' в DMA в присутствии Na2CO3.
Другой пример недостатка способа '203 включает непосредственное снятие защиты с 6 посредством обработки с помощью безводной HCl в диоксане с получением гидрохлоридной соли 7 в виде некристаллического твердого продукта (Схема 2). Необходим большой избыток HCl (10 экв.) в диоксане. Во время снятия защиты, соль 6 непосредственно преципитирует из раствора, делая реакцию медленной и доставляющей проблемы при мониторинге из-за своей гетерогенной природы. Продукт, как он выделяется, представляет собой смесь бис и трис-солей (HCl), поскольку ионно-хроматографический анализ дает значение, которое находится в пределах между теоретическими значениями для бис и трис солей.
Попытки применения некоторых реагентов и условий способа '203 в настоящем способе также являются неудачными. Например, применение методологии заявки '203 для получения соединения 6, начиная с соединения 3, включает множество осложнений. Конкретно, использование условий для генерирования соединения DD из CC на Схеме 2 для преобразования 3 в 6 по настоящему изобретению имеет множество недостатков (Схема 3).
Схема 3
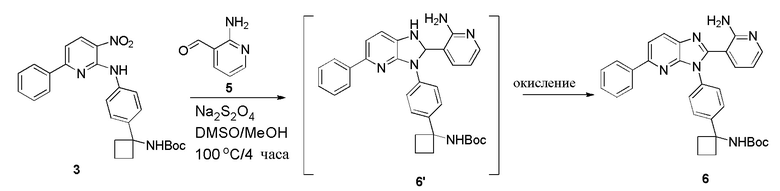
Способ '203 для преобразования CC в DD является сложным и осуществляется как реакция в одной емкости (Схема 2). При применении условий способа '203, нитро остаток 3 (по настоящему изобретению) восстанавливается до анилинового производного, которое затем взаимодействует с альдегидом 5 с образованием предполагаемого иминового промежуточного соединения. Внутримолекулярное добавление дает циклизированное промежуточное соединение 6', которое является неожиданно стабильным и может наблюдаться посредством анализа с помощью ЖХ-МС в ходе реакции. Окисление 6' дает 6. Возникает ряд проблем для реакции по ходу ее осуществления. Наиболее сложной проблемой при этих условиях является значительное количество 6 со снятой защитой. Во время извлечения, определяется, что pH погашенных водных фаз является сильно кислотным (например, pH=3), вероятно, внося вклад в появление большого количества продукта со снятой защитой. Преципитация 6 осложняет манипуляции при извлечении и делает масштабирование менее вероятным. Эти условия, как они используются, не являются благоприятными для будущих возможностей развития. Из-за осложнений при применении условий способа '203 в настоящем способе синтеза, разрабатываются новые условия для преодоления описанного выше осложнения.
Альтернативно, вместо применения методологии '203 для преобразования 3 в 6, которая дает в результате снятие защиты с 6 и осложнение при извлечении, заявляемый способ представляет собой новый подход, который использует двухстадийный способ синтеза 6 из 4 (Схема 4).
Схема 4
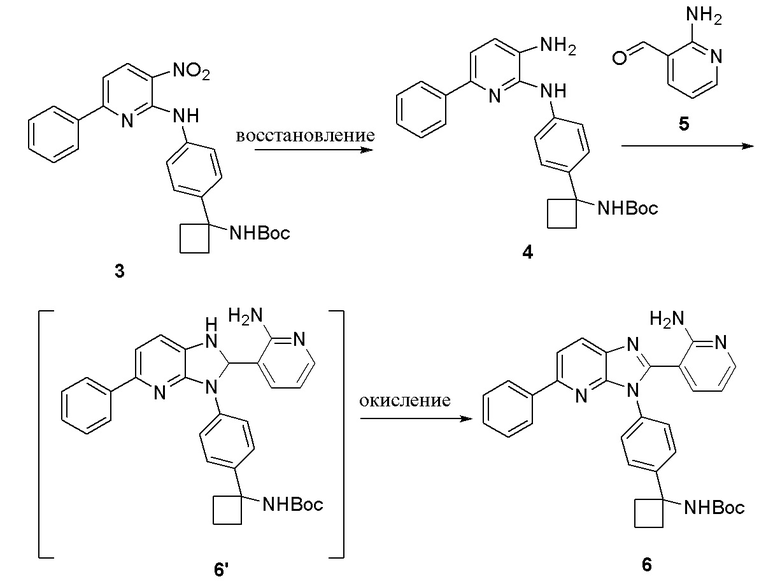
Первая стадия представляет собой дискретное восстановление 3 до анилина 4 с последующим образованием имина, циклизацией, а затем окислением. Гидрогенолиз 3 с помощью Pd/C (например, 10%) дает соединение 4 с высоким выходом (например, с количественным выходом). Затем осуществляют ряд реакций с использованием разнообразных условий реакции для определения возможности циклизации (смотри Таблицу 4) Примера 2. Соединение 4 легко преобразуется в соединение 6; и в большом масштабе, соединение 6 выделяется с выходом примерно 86%.
Способ по настоящему изобретению устраняет недостатки способа '203 с получением способа синтеза, который является безопасным для крупномасштабного получения.
Разработка и оптимизация способа по настоящему изобретению
Разработка и оптимизация способа по настоящему изобретению включает синтез соединений 1, 1', 2, 2', 3, 4, 6 и 7. Стадии обсуждаются в следующем порядке: Стадия 1 (синтез соединения 3), Стадия 2 на Схеме 1 (синтез соединения 4), Стадия 3 на Схеме 1 (синтез соединения 6), Стадия 2' на Схеме 1' (синтез промежуточного соединения 4, а затем соединения 6), Стадия 4 на Схеме 1 или Стадии 3' на Схеме 1' (синтез соединения 7), а затем синтез исходных соединений, 1, 1', 2 и 2'. Наконец, обсуждается очистка 6 и 7 при высоком уровне Pd.
Стадия 1: Синтез соединения 3
В одном из вариантов осуществления, соединение 3 может синтезироваться с использованием реакции замещения и/или реакции перекрестного связывания (Схема 5).
Схема 5
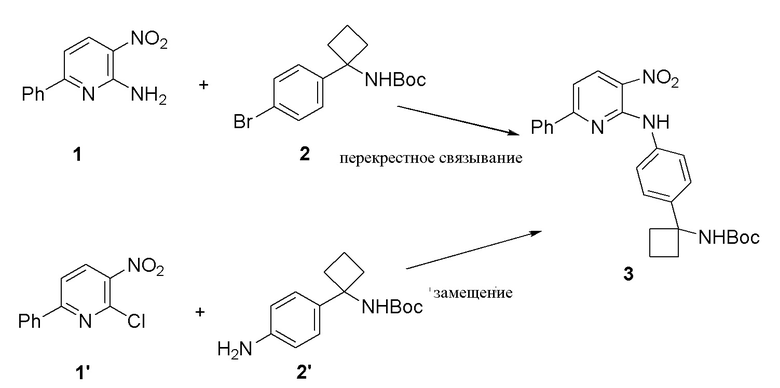
В одном из вариантов осуществления, с использованием реакции замещения, получение соединения 3 осуществляется посредством нагревания соединения 1' и соединения 2' в полярном апротонном растворителе (например, DMA) вместе с основанием (например, Na2CO3, 2 экв.) примерно до 100°C в течение ночи. После завершения реакции, реакционную смесь, как правило, охлаждают до температуры окружающей среды, и добавляют примерно 3% водного раствора NaCl и EtOAc. В одном из вариантов осуществления, слой EtOAc сушат с помощью Na2SO4 и концентрируют до масла. Сырое соединение 3 может повторно растворяться в EtOAc и промываться дополнительной водой для удаления остаточного DMA. В одном из вариантов осуществления, реакция осуществляется в большем масштабе (например, 30 г), и соединение 3, как правило, преципитирует из раствора во время извлечения. Соединение 3 может выделяться с выходом примерно 64%. В одном из вариантов осуществления, экстракционный растворитель представляет собой 2-MeTHF. В одном из вариантов осуществления, может добавляться гептан в качестве антирастворителя для повышения выделяемого выхода без уменьшения чистоты. В одном из вариантов осуществления, соединение 3 кристаллизуется из смеси 2-MeTHF/гептан (например, из раствора 50/50 2-MeTHF/гептан (18 объемов)) с выходом примерно 85%. Таблица 6 в Примере 3 содержит подробное обсуждение анализа растворимости соединения 3 в 2-MeTHF и гептане.
В другом варианте осуществления, осуществляется реакция перекрестного связывания соединения 1 и соединения 2 с образованием соединения 3. В одном из вариантов осуществления, используемое количество катализатора составляет примерно 5% моль и используемое количество фосфорного лиганда составляет примерно 5% моль. В одном из вариантов осуществления, соединение 3, как правило, получают в виде кристаллического твердого продукта с выходом примерно 81% при превосходной чистоте (>99% AUC). В одном из вариантов осуществления, когда количество используемого катализатора меньше примерно, чем 2,5% моль и количество используемого фосфорного лиганда составляет меньше примерно, чем 2,5% моль, реакция может давать максимум 73% (AUC) примерно через 23 часа. В одном из вариантов осуществления, добавление примерно 1% моль Pd2(dba)3 и примерно 2% моль Xantphos дает в результате полное преобразование соединения 3 примерно через 47 часов, что приводит к получению выхода примерно 75% соединения 3 (98,98% AUC) в виде темно-красного кристаллического твердого продукта. Пример 4 приводит подробное обсуждение начальных экспериментов, направленных на реакцию перекрестного связывания.
Как реакция замещения, так и реакция перекрестного связывания дают соединение 3 (все подробности в Примере 1). Однако для реакции перекрестного связывания идентифицируются несколько недостатков. Эти недостатки представляют собой
(1) кинетика реакция в THF является медленной;
(2) часто требуется перезагрузка катализатора и лиганда для завершения реакции;
(3) требуется замена растворителя с THF на EtOAc во время извлечения и выделения;
(4) необходима обработка активированного угля для удаления примесей;
(5) соединение 2 исходных материалов используется в избытке, а кроме того, имеется значительное количество соединения 2, остающееся после завершения реакции.
В одном из вариантов осуществления, реакционный растворитель заменяют с THF на 2-MeTHF, пытаясь решить проблемы, перечисленные выше. Это дает возможность осуществления реакции при более высокой температуре, а также упрощает извлечение, поскольку 2-Me-THF является несмешиваемым с водой, и замены растворителя на EtOAc не требуется. Однако эта модификация не решает всех проблем, перечисленных выше. Пример 5 приводит все подробности оптимизации реакции перекрестного связывания.
В целом, реакция перекрестного связывания является медленной и необходима перезагрузка катализатора и лиганда для завершения реакции; процедура выделения является трудоемкой; удаление примесей, относящихся к 2A, доставляет проблемы (смотри Пример 9) и при получении с использованием подхода с перекрестным связыванием присутствуют повышенные уровни остаточного палладия в соединении 3. Очистка 7 с использованием поглотителя Pd необходима для достижения приемлемых уровней Pd в конечном активном фармацевтическом ингредиенте (смотри Пример 10). По этой причине, в способе по настоящему изобретению, осуществляется реакция замещения 1' и 2' для генерирования соединения 3.
Стадия 2: Синтез соединения 4
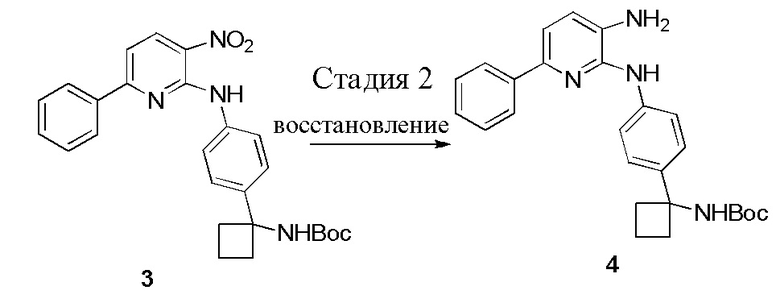
В одном из вариантов осуществления, синтез соединения 4 осуществляется посредством каталитического гидрирования соединения 3 с помощью газообразного водорода при умеренном давлении. Каталитическое гидрирование соединения 3 может осуществляться в полярном апротонном растворителе (например, EtOAc, THF, 2-MeTHF) с помощью Pd/C (например, 10%, 10% масс), как правило, при 40 фунт/кв. дюйм (2,5 кг/кв. см)газообразного водорода. Как правило, примерно через 3 часа, реакция завершается согласно анализу с помощью ВЭЖХ. В одном из вариантов осуществления, соединение 4 может выделяться с количественным выходом в виде пены посредством концентрирования фильтрата досуха после удаления катализатора посредством фильтрования через Celite®. Реакция с использованием газообразного водорода при умеренном давлении, как правило, имеет высокий выход. Пример 6 приводит другие условия реакции, который исследуются.
Стадия 3: Синтез соединения 6
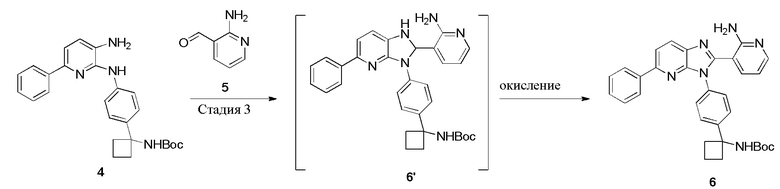
В одном из вариантов осуществления, синтез соединения 6 осуществляется посредством взаимодействия соединения 4 с соединением 5 (2-аминоникотинальдегидом) в присутствии окислителя и кислоты (например, уксусной кислоты) в полярном протонном растворителе (например, метаноле). Исследуют множество реакций оптимизации, чтобы прийти к условиям, используемым в способе по настоящему изобретению. Смотри Пример 7. Например, исследуют растворитель, такой как EtOH, PrOH, толуол и DMSO, но реакции является медленными. Исследуют смеси HOAc/MeOH при различных отношениях и температурах для определения соответствующих условий реакции. В одном из вариантов осуществления, отношение кислоты к растворителю примерно 9:1 (объем/объем) дает соединение 6 с хорошим выходом. В одном из вариантов осуществления, отношение уксусной кислоты к метанолу составляет примерно 9:1 (объем/объем). Если температура реакции повышается примерно до 50°C, можно наблюдать примесь 7:
Примесь 7
В одном из вариантов осуществления, используют 10 объемов AcOH/MeOH (примерно 9:1) при температуре окружающей среды. В другом варианте осуществления, перемешивание соединения 4 (1,0 экв.) и соединения 5 (1,05 экв.) в смеси AcOH/MeOH (10 объемов) в течение ночи при температуре окружающей среды на открытом воздухе дает почти полное преобразование в соединение 6. В одном из вариантов осуществления, соединение 4 и соединение 5 взаимодействуют в присутствии окислителя, выбранного из группы, состоящей из ацетата металла, пербората металла, хлорида металла, катализатора на основе палладия и их гидратов. В другом варианте осуществления, соединение 4 и соединение 5 взаимодействуют в присутствии пербората щелочного металла и его гидратов. В другом варианте осуществления, соединение 4 и соединение 5 взаимодействуют в присутствии окислителя, выбранного из группы, состоящей из ацетата меди, пербората натрия, хлорида железа (III), палладия на угле и их гидратов. В другом варианте осуществления, соединение 4 и соединение 5 взаимодействуют в присутствии окислителя, выбранного из группы, состоящей из Cu(OAc)2⋅H2O, NaBO3⋅4H2O, FeCl3⋅6H2O и 10% Pd/C. В другом варианте осуществления, соединение 4 и соединение 5 взаимодействуют в присутствии NaBO3⋅4H2O.
Выделение соединения 6 является нетривиальным, и оно требует обширных исследований с целью определения соответствующих условий для выделения 6. Смотри Пример 7. В одном из вариантов осуществления, после завершения реакции в смеси, реакционную смесь затем концентрируют (55°C) до тех пор, пока не прекратится дистилляция. В одном из вариантов осуществления, добавляют 2-MeTHF, с последующим добавлением 20% KOH до получения pH >13. В одном из вариантов осуществления, водный слой удаляют, и органический слой промывают 5% насыщенным раствором соли. В одном из вариантов осуществления, водный слой после первой промывки удаляют, и осуществляют вторую промывку 5% насыщенным раствором соли. В одном из вариантов осуществления, водный слой удаляют после второй промывки. В одном из вариантов осуществления, к органическому раствору добавляют IPAc (0,5% масс 2-MeTHF), что приводит к образованию суспензии. В одном из вариантов осуществления, сырое соединение 6 затем фильтруют и промывают IPAc, смесью IPAc/н-гептан (1/1), а затем н-гептаном. В одном из вариантов осуществления, после сушки, соединение 6 на фильтре в течение 2 часов, соединение 6 переносят в вакуумную печь и сушат в течение ночи примерно при 40°C. В одном из вариантов осуществления, соединение 6 выделяют с выходом примерно 86% (учитывая содержание растворителя), 97,3% (AUC) в виде светло-желтого твердого продукта. В одном из вариантов осуществления, 1H ЯМР (CDCl3) показывает, что выделенное соединение 6 содержит 0,8% масс IPAc, 0,7% масс 2-MeTHF и не содержит гептана. В другом варианте осуществления, главная примесь представляет собой N-оксид (M+16), который присутствует при 2,3%.
В одном из вариантов осуществления, очистка соединения 6 осуществляется посредством растворения 6 в DCM и элюирования растворенного 6 через предварительно набитый слой силикагеля (DCM). В одном из вариантов осуществления, колонку затем промывают EtOAc. Две фракции, как правило, собирают и анализируют согласно ВЭЖХ. В одном из вариантов осуществления, не наблюдают примеси N-оксида. В одном из вариантов осуществления, фракции объединяют и частично концентрируют с получением в результате густой суспензии. В одном из вариантов осуществления, добавляют н-гептан, и смесь перемешивают в течение примерно 15 минут. Очищенное соединение 6 фильтруют и промывают гептаном, и сушат в вакуумной печи примерно при 45°C. В одном из вариантов осуществления, соединение 6 [извлечение примерно 89%, примерно 100% (AUC)] получают в виде беловатого твердого продукта примерно через 15 часов сушки. Как правило, 1H ЯМР показывает только микроскопическое количество EtOAc и отсутствие н-гептана.
Стадия 2' на Схеме 1': Синтез промежуточного соединения 4, а затем соединения 6
В одном из вариантов осуществления, синтез промежуточного соединения 4 осуществляют посредством каталитического гидрирования соединения 3 с помощью газообразного водорода при умеренном давлении. Каталитическое гидрирование соединения 3 может осуществляться в полярном апротонном растворителе (например, EtOAc, THF, 2-MeTHF) с Pd/C (например, 10%,10% масс), как правило, при 40 фунт/кв. дюйм (2,5 кг/кв. см) газообразного водорода. Как правило, примерно через 3 часа, реакция завершается согласно анализу с помощью ВЭЖХ. В одном из вариантов осуществления, промежуточное соединение 4 не выделяют до реакции с соединением 5. Реакция с использованием газообразного водорода при умеренном давлении, как правило, имеет высокий выход. В одном из вариантов осуществления, полярный апротонный растворитель заменяется полярным протонным растворителем.
В одном из вариантов осуществления, синтез соединения 6 осуществляют посредством взаимодействия соединения 4 с соединением 5 (2-аминоникотинальдегидом) в присутствии окислителя и кислоты (например, уксусной кислоты) в полярном протонном растворителе (например, в метаноле). Исследуют множество реакций для оптимизации, чтобы прийти к условиям, используемым в способе по настоящему изобретению. Смотри Пример 7. Например, исследуют растворитель, такой как EtOH, PrOH, толуол и DMSO, но реакции являются медленными. Исследуют смеси HOAc/MeOH при различных отношениях и температурах для определения соответствующих условий реакции. В одном из вариантов осуществления, отношение кислоты к растворителю примерно 9:1 (объем/объем) дает соединение 6 с хорошим выходом. В одном из вариантов осуществления, отношение уксусной кислоты к метанолу составляет примерно 9:1 (объем/объем). Если температура реакции повышается примерно до 50°C, наблюдают примесь 7:
Примесь 7
В одном из вариантов осуществления, используют 10 объемов AcOH/MeOH (примерно 9:1) при температуре окружающей среды. В другом варианте осуществления, перемешивание соединения 4 (1,0 экв.) и соединения 5 (1,05 экв.) в смеси AcOH/MeOH (10 объемов) в течение ночи при температуре окружающей среды на открытом воздухе дает почти полное преобразование в соединение 6. В одном из вариантов осуществления, соединение 4 и соединение 5 взаимодействуют в присутствии окислителя, выбранного из группы, состоящей из ацетата металла, пербората металла, хлорида металла, катализатора на основе палладия и их гидратов. В другом варианте осуществления, соединение 4 и соединение 5 взаимодействуют в присутствии пербората щелочного металла и его гидратов. В другом варианте осуществления, соединение 4 и соединение 5 взаимодействуют в присутствии окислителя, выбранного из группы, состоящей из ацетата меди, пербората натрия, хлорида железа (III), палладия на угле и их гидратов. В другом варианте осуществления, соединение 4 и соединение 5 взаимодействуют в присутствии окислителя, выбранного из группы, состоящей из Cu(OAc)2⋅H2O, NaBO3⋅4H2O, FeCl3⋅6H2O и 10% Pd/C. В другом варианте осуществления, соединение 4 и соединение 5 взаимодействуют в присутствии NaBO3⋅4H2O.
Выделение соединения 6 является нетривиальным, и оно требует обширных исследований для определения соответствующих условий для выделения 6. Смотри Пример 7. В одном из вариантов осуществления, после завершения реакции в смеси, реакционную смесь затем концентрируют (55°C) до тех пор, пока не прекратится дистилляция. В одном из вариантов осуществления, добавляют 2-MeTHF с последующим добавлением 20% KOH до получения pH >13. В одном из вариантов осуществления, водный слой удаляют, и органический слой промывают 5% насыщенным раствором соли. В одном из вариантов осуществления, водный слой после первой промывки удаляют и осуществляют вторую промывку 5% насыщенным раствором соли. В одном из вариантов осуществления, водный слой после второй промывки удаляют. В одном из вариантов осуществления, в органический раствор добавляют IPAc (0,5% масс 2-MeTHF), что приводит к образованию суспензии. В одном из вариантов осуществления, сырое соединение 6 затем фильтруют и промывают IPAc, смесью IPAc/н-гептан (1/1), а затем н-гептаном. В одном из вариантов осуществления, после сушки соединения 6 на фильтре в течение 2 часов, соединение 6 переносят в вакуумную печь и сушат в течение ночи примерно при 40°C. В одном из вариантов осуществления, соединение 6 выделяют с выходом примерно 86% (учитывая содержание растворителя), 97,3% (AUC) в виде светло-желтого твердого продукта. В одном из вариантов осуществления, 1H ЯМР (CDCl3) показывает, что выделенное соединение 6 содержит 0,8% масс IPAc, 0,7% масс 2-MeTHF и не содержит гептана. В другом варианте осуществления, главная примесь представляет собой N-оксид (M+16), который присутствует при 2,3%.
В одном из вариантов осуществления, очистка соединения 6 осуществляется посредством растворения 6 в DCM и элюирования растворенного 6 через предварительно набитый слой силикагеля (DCM). В одном из вариантов осуществления, колонку затем промывают EtOAc. Две фракции, как правило, собирают и анализируют согласно ВЭЖХ. В одном из вариантов осуществления, примесь N-оксида не наблюдают. В одном из вариантов осуществления, фракции объединяют и частично концентрируют с получением в результате густой суспензии. В одном из вариантов осуществления, добавляют н-гептан, и смесь перемешивают в течение примерно 15 минут. Очищенное соединение 6 фильтруют и промывают гептаном, и сушат в вакуумной печи примерно при 45°C. В одном из вариантов осуществления, соединение 6 [извлечение примерно 89%, примерно 100% (AUC)] получают в виде беловатого твердого продукта примерно через 15 часов сушки. Как правило, 1H ЯМР показывает только микроскопическое количество EtOAc и отсутствие н-гептана.
Стадия 4 на Схеме 1 или Стадия 3' на Схеме 1': синтез соединения 7
Преобразование соединения 6 в соединение 7 исследуют с использованием различных кислот, таких как TFA, и в различных растворителях, таких как DCE, анизол и IPA. (Смотри Пример 8). Исследование оптимизации в Примере 8 показывает, что дихлорметан (DCM) и метансульфоновая кислота (MSA) являются пригодными для преобразования соединения 6 в соединение 7.
В одном из вариантов осуществления, синтез соединения 7 осуществляется посредством растворения соединения 6 в DCM, и MSA добавляют в течение примерно 15 минут (например, Tmax=29°C). В другом варианте осуществления, отношение MSA к соединению 6 составляет примерно 5:1. В одном из вариантов осуществления, примерно через 2 часа, получается густая суспензия, и добавляют воду, и смесь перемешивают в течение примерно 40 минут. Водный слой удаляют, и добавляют воду для экстрагирования слоя DCM. Водные слои объединяют, а затем промывают DCM. В одном из вариантов осуществления, к водному слою добавляют DCM, и смесь делают основной (например, с помощью 6 н NaOH), устанавливая pH=13. Слои разделяют, и водный слой повторно экстрагируют DCM. Органический слой, как правило, сушат над Na2SO4, а затем концентрируют, получая в результате преципитацию твердых продуктов. В одном из вариантов осуществления, смесь дополнительно концентрируют, и добавляют IPAc. В одном из вариантов осуществления, смесь опять восстанавливают и добавляют IPAc. Добавляют дополнительный IPAc, и суспензию перемешивают в течение ночи. В одном из вариантов осуществления, соединение 7 фильтруют, промывают IPAc и сушат в вакуумной печи (например, >28 рт. ст.) примерно при 45°C в течение примерно 2 дней. Получают соединение 7 (примерно выход 87%, AUC примерно 99,8%) в виде светло-желтого твердого продукта. В одном из вариантов осуществления, 1H ЯМР (CDCl3) показывает, что выделенное 7 содержит IPAc (0,5% масс) и DCM (<0,1% масс).
Синтез исходных соединений 1, 1', 2 и 2'
Приготовление соединений 1, 1', 2 и 2' требует обширного скрининга и оптимизации, чтобы прийти к безопасной и имеющей высокий выход процедуре. Потребности исследований для получения этих исходных соединений приводятся в Примере 9.
Очистка 6 и 7 с высоким уровнем Pd
В одном из вариантов осуществления, соединение 3 получают посредством способа с перекрестным связыванием, и оно имеет высокий уровень остаточного палладия (например, 1888 м.д.). Если эта загрузка соединения 3 проходит через следующие далее стадии до соединения 6, уровень остаточного палладия в соединении 6 в этой загрузке, как правило, является по-прежнему высоким (например, 281 м.д.). По этой причине, для получения соединения 7, имеющего меньше чем 20 м.д. остаточного палладия, инициируются эксперименты по очистке палладия из 6 и свободного основания соединения 7 для идентификации способа удаления остаточного палладия. Смотри Пример 10.
В одном из вариантов осуществления, поглотители являются более эффективными в случае свободного основания соединения 7, по сравнению с соединением 6. В другом варианте осуществления, поглотитель представляет собой QuadraSil MP. В другом варианте осуществления, QuadraSil MP используется в качестве поглотителя для удаления палладия из образца соединения 7.
Определения
Для удобства, здесь собраны определенные термины, используемые в описании, примерах и прилагаемой формуле изобретения.
Способ по настоящему изобретению относится к любому способу как описано в настоящей заявке.
ВЭЖХ представляет собой высокоэффективную жидкостную хроматографию.
ACN или MeCN представляет собой ацетонитрил.
DMA представляет собой диметилацетамид.
MTBE представляет собой простой метил трет-бутиловый эфир.
EtOH представляет собой этанол.
DMSO представляет собой метилсульфоксид.
DPPA представляет собой дифенилфосфорилазид.
ЯМР представляет собой ядерный магнитный резонанс.
MS представляет собой масс-спектрометрию.
RB - это круглодонная.
DI представляет собой деионизованную воду.
DCM представляет собой дихлорметан.
DCE представляет собой 1,2-дихлорэтан.
TFA представляет собой трифторуксусную кислоту.
MSA представляет собой метансульфоновую кислоту.
THF представляет собой тетрагидрофуран.
2-MeTHF представляет собой 2-метилтетрагидрофуран.
EtOAc представляет собой этилацетат.
IPAc представляет собой изопропилацетат.
IPA представляет собой изопропиловый спирт
Xantphos представляет собой 4,5-Бис(дифенилфосфино)-9,9-диметилксантен.
В описании, формы единственного числа также включают множественное число, если только контекст не диктует четко иного. Если не определено иначе, все технические и научные термины, используемые в настоящем документе, имеют такое значение, как обычно понимается специалистами в области, к которой относится настоящее изобретение. В случае конфликта, настоящая заявка является преобладающей.
Все проценты и отношения, используемые в настоящем документе, если не указано иного, являются массовыми.
Все публикации и патентные документы, цитируемые в настоящем документе, включаются в настоящий документ в качестве ссылок как если бы каждая такая публикация или документ был конкретно и индивидуально указан как включаемый в настоящий документ в качестве ссылки. Цитирование публикаций и патентных документов не является признанием того, что любой из них относится к предыдущему уровню техники, или не представляет собой какого-либо признания относительно их содержания или даты. Изобретение описано к настоящему моменту в качестве письменного описания, специалисты в данной области увидят, что настоящее изобретение может осуществляться в различных вариантах осуществления, и что предшествующее описание и примеры ниже предназначены для целей иллюстрации, а не ограничения формулы изобретения, которая следует далее.
Примеры
Пример 1: Получение соединений 1, 1', 2, 2', 3, 4, 6 и 7
Получение 1
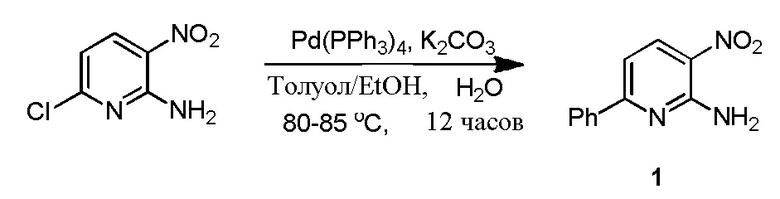
100-л реактор с кожухом, снабженный датчиком температуры, входом для аргона и обратным холодильником, загружают толуолом (30 л, 30 объемов), EtOH (6 л, 6 объемов), 2-амино-3-нитро-6-хлор-пиридином (1,0 кг, 5,76 моль.), фенилбориновой кислотой (772 г, 6,34 моль.), а затем раствором K2CO3 (1,75 кг, 12,67 моль) в DI воде (6,0 л, 6 объемов). Полученную в результате смесь перемешивают при комнатной температуре в течение 10 минут. Реакционную смесь дегазируют с помощью аргона в течение 30 минут перед добавлением в реакционную смесь Pd(PPh3)4 (67,1 г, 1 моль), а затем полученную в результате смесь дегазируют в течение еще 10 минут. Затем реакционную смесь нагревают до 80-85°C. Реакцию считают завершенной согласно ВЭЖХ через 12 часов. Реакционную смесь охлаждают до комнатной температуры и разбавляют водой (10 л, 10 объемов). Органический слой удаляют, и водный слой экстрагируют MTBE (2×10 л, 20 объемов). Объединенные органические слои обрабатывают активированным углем и нагревают до 50°C в течение 1 часа. Горячий раствор фильтруют через слой Celite®, и этот слой промывают горячим (~50°C) MTBE (2 л, 2 объема), и фильтрат сушат над сульфатом натрия. Органический слой концентрируют при пониженном давлении при температуре ниже 50°C с получением темно-коричневого твердого продукта (1,094 кг, 88,9%). Сырое соединение перетирают в гептане (3,5 л, 3,5 объема) в течение 3 часов, отфильтровывают твердые продукты, промывают гептаном (1,5 л, 1,5 объема), и сушат с получением 1 (980,0 г, 79,6%, чистота 89,6%), и соединение характеризуется с помощью 1H ЯМР (CDCl3) и MS.
Получение 1'
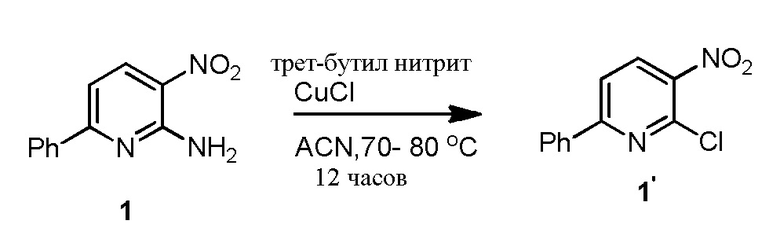
3-л, трехгорлую RB колбу, снабженную мешалкой, входом для аргона, обратным холодильником и термометром загружают ацетонитрилом (1500 мл), Cu(I)Cl (59,7 г, 604,0 ммоль) и трет-бутилнитритом (112,2 мл, 929 ммоль). Смесь нагревают до 40-50°C, а затем добавляют порциями 1 (100,0 г, 467,3 ммоль). Полученную в результате смесь перемешивают при 40-50°C в течение одного часа, и реакцию считают завершенной согласно ВЭЖХ. Реакцию гасят водным раствором хлорида аммония (2,0 л, 20 объемов) и разбавляют MTBE (2,0 л, 20 объемов). Органический слой удаляют, и водный слой экстрагируют MTBE (2 × 1 л, 20 объемов). Объединенные органические слои обрабатывают активированным углем и нагревают до 50°C. Горячий раствор фильтруют через пад из Celite®, и пад Celite® промывают горячим MTBE (1 л, 1 объем), сушат над сульфатом натрия и концентрируют с получением сырого 1' (61,1 г, 60,7%). Сырое соединение перетирают в метаноле (183 мл, 3 объема по отношению к сырой массе) в течение 15 минут. Твердые продукты фильтруют, промывают метанолом (30 мл), и сушат с получением 1' (48,0 г, 43,4%). Его перетирают с гептаном (100 мл, 1 объем) при температуре окружающей среды в течение одного часа, фильтруют и промывают гептаном (25 мл), и сушат с получением 1' в виде светло-желтого твердого продукта (42,02 г, 38,5%, чистота 97,6%). Соединение характеризуется с помощью 1H ЯМР (CDCl3) и MS. Дополнительные навески приготавливают с использованием этой процедуры, и результаты можно увидеть в Таблице 1.
Таблица 1. Получение 1' из 1
(33,0%)
(55-60°C).
(38,5%)
(55-60°C).
(28,2%)
(55-60°C).
Получение 1a из 1
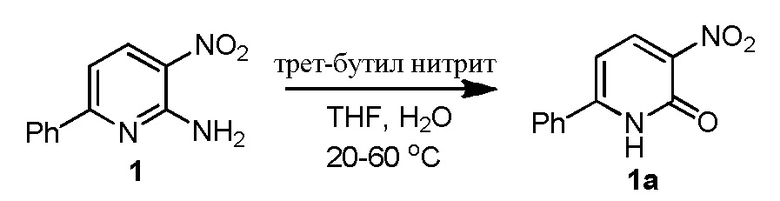
3-л, трехгорлую круглодонную колбу, снабженную мешалкой, входом для аргона, обратным холодильником и термометром, загружают 1 (200,0 г, 929,3 ммоль), THF (1600 мл, 8 объемов) и DI водой (400 мл,2 объема). Полученную в результате смесь перемешивают в течение 10 минут при комнатной температуре, затем добавляют трет-бутилнитрит (110,3 мл, 929,3 ммоль, 1,0 экв.) в течение периода 10 минут. Реакционную смесь нагревают до 55-60°C и перемешивают в течение 14 часов (соединение 1a, как обнаружено, выпадает из раствора в виде твердого продукта в ходе реакции). Через 14 часов, анализ с помощью ВЭЖХ показывает присутствие ~18,7% 1, затем реакционную смесь охлаждают до 40°C и добавляют трет-бутилнитрит (110,3 мл, 929,3 ммоль, 1,0 экв.), затем нагревают до 60°C и перемешивают в течение 20 часов. Через 34 часа ВЭЖХ показывает 5% 1. Затем к реакционной смеси добавляют 0,1 экв. трет-бутилнитрита (11,1 мл, 92,6 ммоль, 0,1 экв.) и перемешивают при 60°C в течение 6 часов. Через 40 часов, ВЭЖХ по-прежнему показывает 5% исходных материалов, затем реакционную смесь охлаждают до комнатной температуры и твердые продукты отфильтровывают, твердые продукты промывают EtOAc (400 мл, 2 объема) и сушат с получением соединения 1a (148,1 г; 73,8%, чистота 95,7%), и характеризуют его с помощью 1H ЯМР (DMSO-d6) и MS.
Получение 1a из 1b и фенилбориновой кислоты
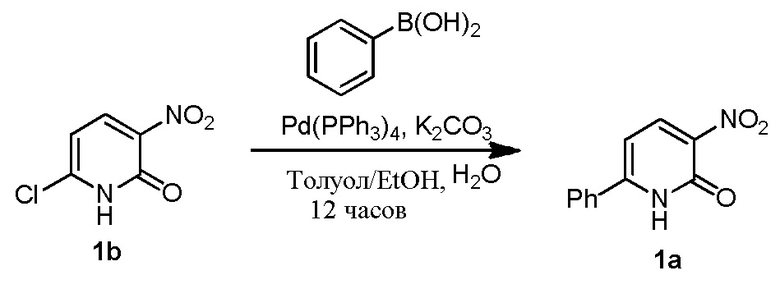
100-л реактор с кожухом, снабженный датчиком температуры, входом для азота и обратным холодильником, загружают толуолом (27,0 л, 30 объемов) и EtOH (5,4 л, 6 объемов), а затем 6-хлор-3-нитропиридин-2(1H)-оном (900,0 г, 5,15 моль) и фенилбориновой кислотой (640,4 г, 5,253 моль). Смесь перемешивают при температуре окружающей среды в течение 15 минут перед добавлением раствора K2CO3 (173,9 г, 11,33 моль) в DI воде (5,4 л, 6 объемов). Реакционную смесь дегазируют с помощью аргона в течение 30 минут при комнатной температуре. Добавляют тетракистрифенилфосфинпалладий (178,2 г, 3% моль), и раствор нагревают до 95-100°C (внутренняя температура составляет 77-79°C) и перемешивают в течение 3 часов. Через 3 часа ВЭЖХ показывает 2,8% исходных материалов и другую отдельную примесь (15,3%, 1,17 RRT). Реакционную смесь поддерживают в течение 3 часов при такой же температуре. Через 6 часов, продвижения реакции нет, и смесь охлаждают до комнатной температуры, дегазируют в течение 30 минут и добавляют еще 5,0 г тетракистрифенилфосфинпалладия, и раствор нагревают до 95-100°C. Реакцию считают завершенной через один час согласно ВЭЖХ. Реакционную смесь охлаждают до комнатной температуры, реакционную смесь разбавляют DI водой (11,7 л, 13 объемов), а затем EtOAc (18,0 л, 20 объемов) и перемешивают в течение 1 часа. Два слоя разделяют, оставляя твердые продукты в водном слое. Водный слой экстрагируют EtOAc (13,5 л, 15 объемов). Объединенные водные слои нейтрализуют с доведением pH до 6,2-6,8 с помощью 3 н HCl, при этом из раствора преципитируют дополнительные твердые продукты, эти твердые продукты отфильтровывают, промывают водой (2 × 2,5 л, 5 объемов) и сушат в вакууме при 45-50°C в течение 48 часов, с получением 1a (761,1 г, выход 68,9%, чистота 78,0%) в виде светло-желтого твердого продукта. Соединение характеризуется с помощью 1H ЯМР (DMSO-d6) и MS.
Объединенные органические (этилацетатные) слои экстрагируют с помощью 3 н NaOH (15 л), при этом образуются твердые продукты. Органический слой отделяют. Затем водный слой подкисляют до pH 5-6 с помощью 3 н HCl, при этом из раствора преципитируют дополнительные твердые продукты, которые отфильтровывают и промывают DI водой (2,0 л), и сушат с получением соединения 1a (140,0 г, чистота 12,7%, 93,8%, 2-ой сбор).
Получение 1' из 1a
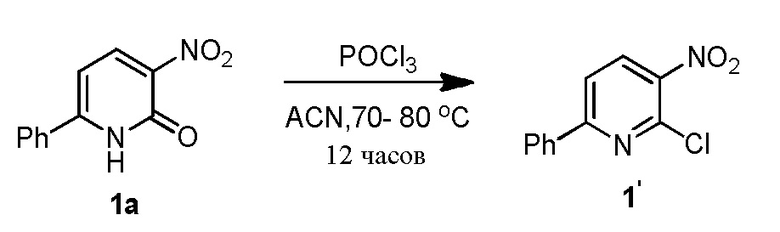
20-л реактор с кожухом, снабженный датчиком температуры, входом для азота и обратным холодильником, загружают с ацетонитрил (6,0 л, 5 объемов), а затем добавляют 1a (1,2 кг, 5,5 моль.), а затем POCl3 (1,2 л, 1 объем) в течение периода 5 минут. Реакционную смесь медленно нагревают до 70-80°C в течение 12-15 часов до тех пор, пока реакция не считается завершенной согласно ВЭЖХ. Реакционную смесь охлаждают до комнатной температуры и гасят водой со льдом (24 л) ниже 10°C и повышают основность до pH: 8-9 с помощью раствора 6 н NaOH (~7,2 л), при температуре ниже 15°C. Преципитировавшие твердые продукты отфильтровывают и промывают DI водой (3,6 л, 3 объема), и сушат с получением 1' в виде темно-коричневого твердого продукта (786 г, 60,8%). Сырой 1' растворяют в EtOAc (12 л, 10 объемов) [Примечание: наблюдают некоторое количество нерастворимых твердых продуктов] и перемешивают в течение 30 минут. Раствор фильтруют через слой Celite® и промывают EtOAc (3 л, 3 объема). Органический раствор обрабатывают активированным углем, отфильтровывают через пад Celite®, и пад Celite® промывают этилацетатом (3 л, 3 объема). Полученный в результате фильтрат концентрируют досуха с получением 1' (688,3 г, 52,9%, чистота 98,07%). Соединение характеризуется с помощью 1H ЯМР (CDCl3) и MS. Сводку характеристик загрузки для получения 1' из 1a можно увидеть в Таблице 2 и 3.
Таблица 2. Получение 1'с использованием 1a и POCl3
(60,4%)
b) Во время реакции не наблюдается примесей при 1,17 RRT (отн. время удерж.).
(48,1%)
(52,9%)
(47,3%)
Суспендирование 1' в метаноле
Соединение 1' [1,78 кг (475,0 г, загрузка 2; 688,0 г, загрузка 3; 617,0 г, загрузка 4)] суспендируют при перемешивании с метанолом (1,8 л, 1 объем) при 20°C. Суспензию перемешивают в течение 30 минут при 20°C перед фильтрованием. Отфильтрованные твердые продукты промывают метанолом с получением 1' (1,71 кг, выход 96%, 99,5% AUC)
Таблица 3. Результаты суспендирования 1'
(ВЭЖХ %AUC)
(475 г, загрузка 2)
(688 г, загрузка 3)
(617 г, загрузка 4)
(Извлечение 96%)
Получение B
Суспензию порошкообразного гидроксида калия (536 г, 9,56 моль, 5,6 экв.) в толуоле (1,54 л) и воде (154 мл) нагревают до 45°C. Затем добавляют тетрабутиламмонийбромид (28 г, 0,85 моль, 0,05 экв.) и 1,3-дибромпропан (379 г, 1,88 моль, 1,1 экв.), с последующим добавлением по каплям раствора A (200 г, 1,7 моль, 1,0 экв.) в толуоле (500 мл) в течение 42 минут. Во время добавления температура повышается до 95°C, а затем смесь нагревают с обратным холодильником, когда добавление A завершается. Полученную в результате розовую суспензию перемешивают при этой температуре в течение 1 часа, за это время реакцию считают завершенной согласно анализу с помощью ВЭЖХ. Затем смесь охлаждают до 20-25°C и фильтруют через пад Celite®. Твердые продукты промывают толуолом (1,0 л), и полученный в результате фильтрат промывают водой (2 × 300 мл), насыщенным раствором соли (150 мл), сушат над MgSO4, фильтруют и концентрируют с получением сырого B (263 г) в виде оранжевого масла. Затем продукт очищают с помощью вакуумной отгонки (температура кипения 105°C/750 миллитор) с получением B [140 г, 52%, 97,7% (AUC)] в виде бесцветной жидкости. Главная присутствующая примесь определяется как B2 (2,3% AUC) (относительно подробностей смотри Пример 9, синтез B).
Получение C
Твердый KNO3 (17,0 г, 0,17 моль, 1,06 экв.) добавляют порциями к H2SO4 (100 мл), поддерживая температуру <15°C. После перемешивания в течение 15 минут, добавляют B (25,0 г, 0,16 моль, 1,0 экв.), поддерживая температуру <15°C. Через час, отбирают образец смеси и анализируют с помощью ВЭЖХ, анализ показывает завершение реакции. Затем смесь выливают на лед и экстрагируют DCM (200 мл). Органический слой промывают 1 M NaOH, насыщенным раствором соли, а затем сушат над MgSO4. После концентрирования, выделяют C [32,1 г, 99%, 95,5% (AUC)] в виде оранжевого/коричневого твердого продукта. 1H ЯМР (CDCl3) говорит, что материал является чуть менее чистым, чем определено с помощью ВЭЖХ.
Получение D
Твердый KNO3 (318,1 г, 3,18 моль, 1,06 экв.) добавляют порциями к H2SO4 (1,9 л), поддерживая температуру <15°C. После перемешивания в течение 15 минут, добавляют соединение B (471,3 г, 3,0 моль, 1,0 экв.) в течение 75 минут, поддерживая температуру <20°C. Через 2 часа, смесь анализируют с помощью ВЭЖХ, анализ показывает завершение реакции (70% C, 30% D). Затем эту реакционную смесь перемешивают при температуре окружающей среды в течение ночи, в этот момент C не остается, согласно анализу с помощью ВЭЖХ. Затем смесь выливают на лед (3 кг) с присутствующим DCM (3 л). Органический слой промывают 1 M NaOH (1,0 л), насыщенным раствором соли (500 мл), а затем сушат над MgSO4. После концентрирования, добавляют гептан (1,5 л) и EtOAc (500 мл), и смесь перемешивают при температуре окружающей среды в течение 4 часов. Затем твердые продукты отфильтровывают и сушат с получением D [365,5 г, 55% в две стадии, ~99% (AUC)] в виде светло-желтого твердого продукта.
Получение D из C
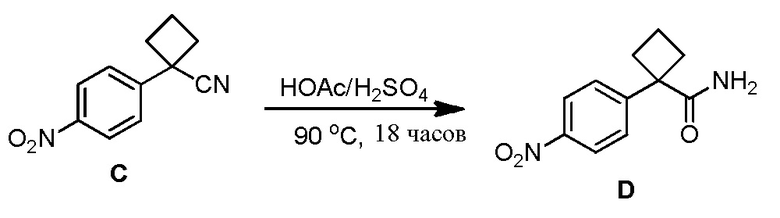
К раствору C (40 г, 0,19 моль, 1,0 экв.) в AcOH (520 мл) медленно добавляют H2SO4 (280 мл), получая в результате значительную экзотермичность (25→65°C). Затем эту смесь нагревают до 90°C в течение ночи, за это время реакцию считают завершенной согласно анализу с помощью ВЭЖХ. Смесь охлаждают до температуры окружающей среды, выливают на лед и экстрагируют DCM. Органический слой промывают насыщенным водным раствором NaHCO3, водой, а затем насыщенным раствором соли. После сушки с помощью MgSO4, раствор частично концентрируют, и добавляют гептан. Дальнейшее концентрирование приводит к преципитации D. Фильтрование и промывка гептаном дает D [30,0 г, 69%, 99% (AUC)] в виде светло-коричневого твердого продукта.
Получение E
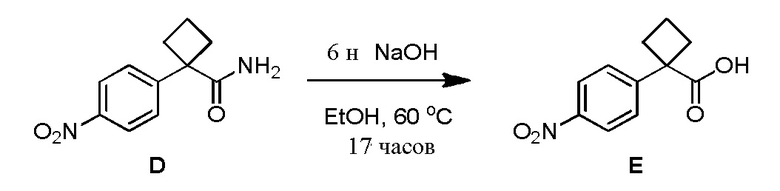
D (10,0 г, 45,4 ммоль, 1,0 экв.) перемешивают в EtOH (50 мл) и 6 M NaOH (60,6 мл, 363,3 ммоль, 8,0 экв.) в течение ночи при 60°C. Через 17 часов, анализ с помощью ВЭЖХ показывает, что реакция завершается. Смесь охлаждают до температуры окружающей среды, разбавляют водой (60 мл) и частично концентрируют для удаления EtOH. После концентрирования, смесь промывают DCM (2 × 100 мл), и затем водный слой подкисляют водным раствором 6 M HCl. Кислый водный слой экстрагируют DCM (3 × 100 мл), и объединенные органические слои промывают насыщенным раствором соли и сушат над MgSO4. После концентрирования, E [10,2 г, 100%, 95% (AUC)] выделяют в виде коричневого твердого продукта.
Получение H
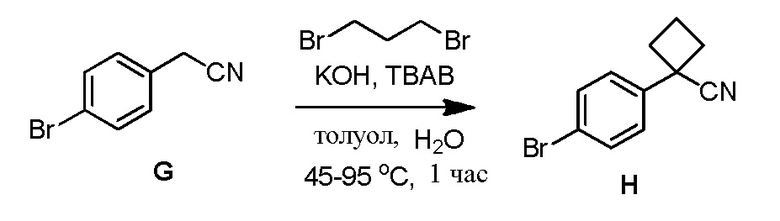
Суспензию порошкообразного гидроксида калия (801 г, 14,3 моль, 5,6 экв.) в толуоле (3,85 л, 7,7 объема) и воде (385 мл, 0,77 объема) нагревают до 50°C. Затем добавляют тетрабутиламмонийбромид (41,1 г, 2,81 моль, 0,05 экв.) и 1,3-дибромпропан (566 г, 2,81 моль, 1,1 экв.). Затем, медленно добавляют раствор G (500 г, 2,6 моль, 1,0 экв.) в толуоле (1,25 л, 2,2 объема) в течение 30 минут, поддерживая при этом температуру 50-85°C. Полученную в результате фиолетовую суспензию нагревают с обратным холодильником (100°C) и перемешивают при этой температуре в течение 1 часа, за это время анализ с помощью ВЭЖХ показывает полное исчезновение G. Смесь охлаждают до 70°C и добавляют гептан (5,2 л). Затем полученную в результате суспензию охлаждают до температуры окружающей среды и фильтруют через пад Celite®. Твердые продукты (значительное количество) промывают толуолом (2,0 л), и полученный в результате фильтрат промывают водой (3 × 500 мл), насыщенным раствором соли (500 мл), сушат над MgSO4, фильтруют и концентрируют. Это дает сырой продукт H [519 г, 86%, 86% (AUC)] в виде красного масла.
Получение I
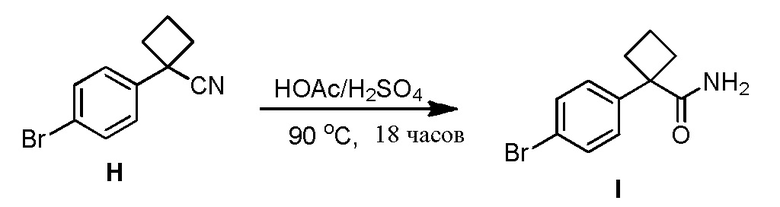
К раствору H (200 г, 0,85 моль, 1,0 экв.) в AcOH (800 мл) медленно добавляют H2SO4 (400 мл), получая в результате значительную экзотермичность (25 → 40°C). Эту смесь нагревают до 90°C в течение ночи, за это время анализ с помощью ВЭЖХ показывает, что реакция завершается. Смесь охлаждают до температуры окружающей среды, а затем медленно добавляют в смесь воды со льдом (3,0 л) и дихлорметана (2,0 л). Двухфазную смесь разбавляют дополнительным дихлорметаном (3,0 л), и кислотный водный слой отделяют. Органический слой промывают водой (2 × 2,5 л), водным раствором 0,5 M NaOH (2 × 2,0 л), а затем насыщенным раствором соли (500 мл). Органический слой сушат над MgSO4, фильтруют и концентрируют с получением сырого I (195 г) в виде коричневого масла. Сырой I очищают с помощью колоночной хроматографии на силикагеле с использованием 80% смеси EtOAc/гептан, с получением I [159 г, 74% от G, >99% (AUC)] в виде белого твердого продукта.
Получение 2
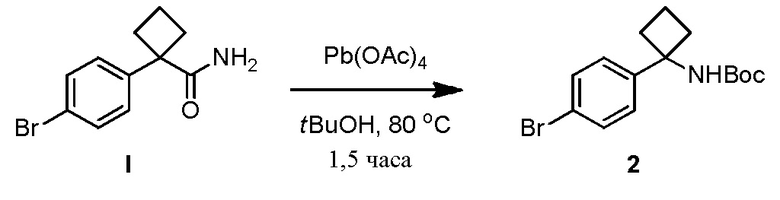
В 3-л трехгорлую колбу добавляют I (250 г, 0,98 моль, 1,0 экв.) и t-BuOH (1250 мл, 5,0 объемов). Суспензию нагревают до 65°C и перемешивают до тех пор, пока все твердые продукты не растворятся (примерно 10 минут). Осторожно добавляют порциями Pb(OAc)4 (40,1 г, 1,13 моль, 1,15 экв.) в течение 35 минут, поддерживая при этом температуру <75°C. Когда добавление Pb(OAc)4 завершается, суспензию перемешивают при 80°C в течение 80 минут, в этот момент реакция завершается согласно анализу с помощью ВЭЖХ. Затем суспензию охлаждают до 25°C и добавляют Na2CO3 (250 г, 1,0 масс экв.), затем MTBE (1,9 л). Суспензию перемешивают в течение 30 минут, а затем твердые продукты удаляют с помощью фильтрования через пад Celite®. Фильтрат промывают водным раствором 10% NaHCO3 (3 × 2,0 л), 10% насыщенным раствором соли (500 мл), сушат над MgSO4, фильтруют и концентрируют с получением сырого 2 [301 г, 94%, 89% (AUC)] в виде бледно-лилового твердого продукта. Сырой 2 очищают посредством повторного суспендирования в 10/90 смеси MTBE/гептан (5,0 объемов) с получением 2 [270 г, 84%, 94% (AUC)] в виде беловатого твердого продукта. Затем 2 [270 г, 0,83 моль, 94% (AUC)] повторно суспендируют 1/1 в смеси ацетонитрил/вода (5,0 объемов) при температуре окружающей среды в течение 22 часов. Твердые продукты фильтруют и сушат с получением соединения 2 [240 г, извлечение 89%, 95,2% (AUC)] в виде белого твердого продукта.
Затем этот материал объединяют с другими навесками из 2 и очищают посредством элюирования через слой диоксида кремния (набивают и элюируют с использованием 1/99 смеси MeOH/DCM). Затем обогащенные фракции концентрируют досуха (525 г 2) и смешивают посредством суспендирования в MTBE (2,0 объема) и гептане (6,0 объемов) при температуре окружающей среды, с получением однородной навески. Это дает 2 [513 г, 97,2% (AUC)] в виде белого твердого продукта.
Получение D
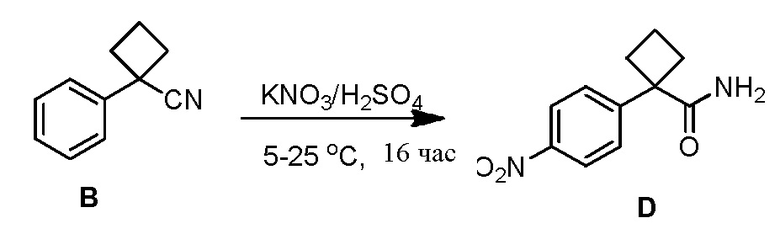
20-л реактор с кожухом, снабженный датчиком температуры, входом для азота, обратным холодильником и капельной воронкой, загружают концентрированной H2SO4 (14 л, 4 объема), и смесь охлаждают до 5-6°C и добавляют порциями KNO3 (3,183 кг, 23,6 моль), поддерживая температуру в пределах между 10 и 15°C. После перемешивания полученной в результате суспензии в течение 15 минут, добавляют B (3,5 кг, 22,27 моль) в течение периода 90 минут, поддерживая внутреннюю температуру в пределах между 10-20°C. Затем реакционную смесь нагревают до температуры окружающей среды и перемешивают в течение 16 часов, пи этом реакция считается завершенной согласно анализу с помощью ВЭЖХ. Затем реакционную смесь выливают в смесь охлажденной воды (~5°C) (35 л, 10 объемов) и DCM (35 л, 10 объемов), поддерживая температуру <15°C. Органический слой отделяют, и водный слой экстрагируют два раза DCM [21 л (6 объемов) и 14 л (4 объема)]. Объединенные органические слои промывают 1 н NaOH (35 л, 10 объемов), насыщенным раствором соли (1,75 л, 0,5 объема), и сушат над безводным Na2SO4. Органический слой концентрируют с получением D в виде беловатого твердого продукта (3,61 кг, выход 72,7%, 83,1% AUC).
Очистка D
Сырой D (3,6 кг) суспендируют в MTBE (7 л,2 объема) и перемешивают при температуре окружающей среды в течение 30 минут. Затем твердые продукты фильтруют, промывают MTBE (700 мл, 0,2 объема), и сушат в вакууме с получением D (2,65 кг, 54,2%, чистота 98,3%) в виде беловатого твердого продукта.
Получение F из D
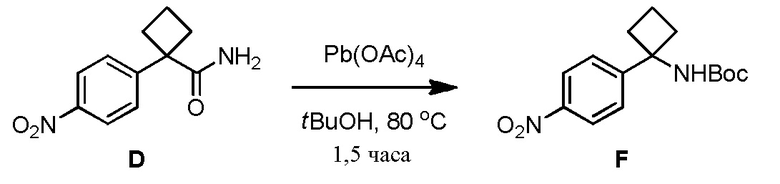
К суспензии D (30 г, 136,2 ммоль, 1,0 экв.) в t-BuOH (230 мл, 6,0 объемов) при 75°C четырьмя порциями добавляют Pb(OAc)4 (69,5 г, 156,7 ммоль, 1,15 экв.) в течение 5 минут. Затем суспензию нагревают до 80°C в течение 90 минут, за это время анализ с помощью ВЭЖХ показывает, что реакция завершается (D не остается). Затем суспензию охлаждают до 25°C и добавляют Na2CO3 (30 г, 1,0 масс эквивалент), а затем MTBE (200 мл). Суспензию перемешивают в течение 30 минут, а затем твердые продукты удаляют с помощью фильтрования через пад Celite®. Фильтрат промывают водным раствором 10% NaHCO3 (3 × 200 мл), насыщенным раствором соли, сушат над MgSO4, фильтруют и концентрируют с получением сырого F [34,0 г, 86%, 92,5% (AUC)] в виде беловатого твердого продукта. Данные 1H ЯМР (CDCl3) соответствуют желаемому продукту. Затем этот материал используют ʺкак естьʺ на следующей стадии без дополнительной очистки.
Получение F из E
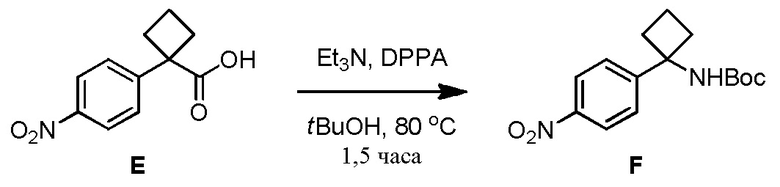
К раствору E (2,0 г, 9,0 ммоль, 1,0 экв.) в t-BuOH (40 мл, 20 объемов) добавляют Et3N (1,10 г, 10,9 ммоль, 1,2 экв.). Этот раствор нагревают до 75°C, в это время добавляют по каплям DPPA (2,71 г, 9,9 ммоль, 1,09 экв.) в течение 5 минут. После перемешивания в течение ночи при 81°C, реакция завершается согласно анализу с помощью ВЭЖХ (E не остается), а затем реакционную смесь охлаждают до температуры окружающей среды. Реакционную смесь концентрируют досуха и анализируют с помощью 1H-ЯМР с использованием внутреннего стандарта (диметилфумарат в CDCl3). На основании этого анализа, общий выход F, как обнаружено, составляет 78%.
Получение F
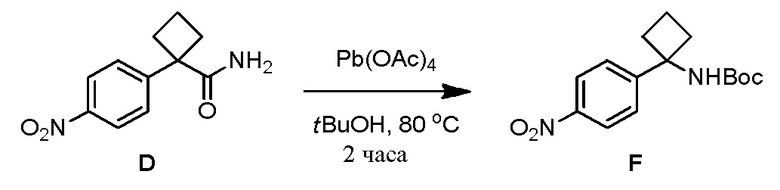
В 20-л чистый и сухой реактор с кожухом, снабженный обратным холодильником, датчиком температуры и входом для азота, загружают D (1,85 кг, 8,174 моль, чистота 98,3%) в t-BuOH (9,25 л, 5 объемов), и полученную в результате смесь нагревают до 50-55°C и перемешивают в течение 45 минут. К этой смеси добавляют четырьмя равными порциями Pb(OAc)4 (4,2 кг, 9,400 моль), и полученную в результате суспензию нагревают до 80°C в течение двух часов. Через два часа реакцию считают завершенной согласно анализу с помощью ВЭЖХ. Реакционную смесь охлаждают до ~25°C и добавляют Na2CO3 (1,85 кг, 17,002 моль), а затем MTBE (10 л, 5,5 объема). Смесь перемешивают в течение 30 минут, а затем твердые продукты удаляют с помощью фильтрования через слой Celite®. Слой Celite® промывают MTBE (5 л, 2,5 объема). Затем фильтрат промывают водным раствором 10% NaHCO3 (20,0 л, 10 объемов), насыщенным раствором соли (5 л, 2,5 объема), сушат над безводным Na2SO4 и концентрируют при пониженном давлении с получением сырого F (1,9 кг, выход 75,8%, 96,1% AUC) в виде беловатого твердого продукта.
Очистка F
Сырой F (1,9 кг) растворяют в EtOH (15,4 л, 8 объемов) при 45°C и перемешивают в течение 15 минут. Медленно добавляют порциями DI воду (11,2 л, 6 объемов) при такой скорости, чтобы поддерживать внутреннюю температуру 45°C. Полученную в результате белую суспензию перемешивают в течение двух часов при температуре окружающей среды. Затем суспензию фильтруют, твердый продукт промывают смесью этанол-вода 4:3 (2 объема) и сушат в вакууме при 40°C, с получением F (1,61 кг, выход 65,6%, 99,4% AUC) в виде беловатого твердого продукта.
Получение 2'
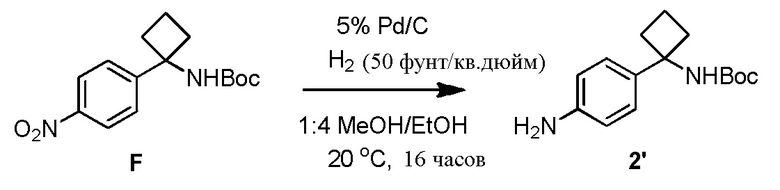
Раствор F (29 г, 99,2 ммоль, 1,0 экв.) в 20/80 смеси MeOH/EtOH (290 мл, 10 объемов) добавляют в стеклянную емкость высокого давления, содержащую 5% Pd/C (1,45 г, нагрузка 5% масс, влажность катализатора 50%). Эту суспензию помещают в атмосферу H2 (45 фунт/кв. дюйм) (2,7 кг/кв. см) и перемешивают при температуре окружающей среды в течение 16 часов. Через 16 часов, реакция завершается согласно анализу с помощью ВЭЖХ, и смесь фильтруют через пад Celite®. Фильтрат концентрируют с получением сырого 2' (34 г) в виде коричневого масла. Затем сырой 2' очищают с помощью колоночной хроматографии (1:1 смесь EtOAc/гептан на силикагеле) с получением 2' [30,4 г, 100%, 97,7% (AUC)] в виде вязкого желтого масла. Единственная примесь в этой навеске соединения 2' представляет собой изопропилкарбаматное производное 2' (2,3% AUC).
Получение 2'
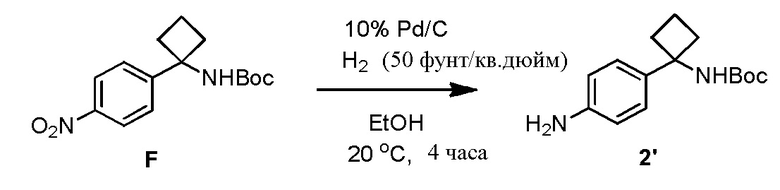
2-л автоклавный реактор из нержавеющей стали, снабженный датчиком температуры, загружают EtOH (5,0 л, 10 объемов), а затем F [500,0 г, 1,71 моль, чистота 99,4%] и 10% Pd/C (25,0 г, 5% масс). Реактор продувают азотом перед заполнением водородом до 45-50 фунт/кв. дюйм (2,8 -3,1 кг/кв. см) и перемешивают при температуре окружающей среды. Через 4 часа реакцию считают завершенной согласно ВЭЖХ. Реакционную смесь фильтруют через пад Celite®, и пад Celite® промывают EtOH (2 л, 4 объема). Фильтрат концентрируют при пониженном давлении с получением 2' (456,5 г, >100%) в виде беловатого полутвердого продукта.
Очистка 2'
Сырой 2' суспендируют в гептане (1 л,2 объема) и перемешивают в течение двух часов при температуре окружающей среды. Суспензию фильтруют, твердые продукты промывают гептаном (250 мл, 0,5 объема), а затем сушат в вакууме при 35-40°C с получением 2' (405,0 г, выход 91,6%, 99,16% AUC) в виде беловатого твердого продукта.
Получение 3 с помощью реакции перекрестного связывания катализируемой Pd
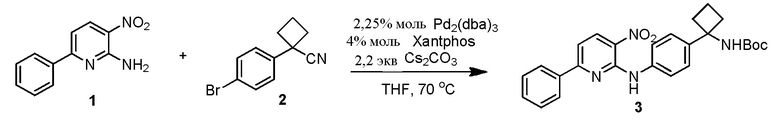
В 1-л реактор с кожухом с притоком N2 добавляют 1 (30,0 г, 139,4 ммоль, 1,0 экв.), 2 (47,75 г, 146,37 ммоль, 1,05 экв.), Cs2CO3 (99,92 г, 306,7 ммоль, 2,2 экв.), а затем THF химического качества (300 мл, 10 объемов, KF=0,024% H2O). Полученную в результате суспензию перемешивают и продувают N2 в течение 15 минут. Затем добавляют Pd2(dba)3 (1,60 г, 1,74 ммоль, 1,25% моль) и Xantphos (2,02 г, 3,49 ммоль, 2,5% моль), и реакционную смесь нагревают при внутренней температуре 60°C при положительном давлении N2. Через 3 часа отбирают образец из реакционной смеси и обнаруживают, что наблюдается только 3% продукта. Добавляют дополнительное количество Pd2(dba)3 (1,60 г, 1,74 ммоль, 1,25% моль) и Xantphos (2,02 г, 3,49 ммоль, 2,5% моль), и реакция переключается с положительного давления N2 на защитный слой N2. Через 23,5 часа имеется 73% продукта, как наблюдается с помощью ВЭЖХ. Добавляют дополнительное количество Pd2(dba)3 (1,40 г, 1,52 ммоль, 1,0% моль), Xantphos (1,80 г, 3,12 ммоль, 2,0% моль) и Cs2CO3 (50,0 г, 153,0 ммоль, 1,1 экв.). Через 30,5 часа ВЭЖХ показывает 96% продукта и 3,1% остающегося 1. Реакционную смесь перемешивают в течение еще 17 часов, и по прохождении общего времени реакции 47 часов остающегося 1 нет. Реакционную смесь охлаждают до 20°C. Добавляют 500 мл EtOAc, а затем 250 мл H2O. После перемешивания двухфазной смеси в течение 15 минут органический слой удаляют. Водный слой экстрагируют 500 мл EtOAc, и объединенные органические слои промывают насыщенным раствором соли (500 мл) и сушат над Na2SO4 и фильтруют. Отфильтрованный раствор выдерживают при 4°C в течение 45 часов, в это время может продолжаться извлечение. К высушенному раствору добавляют 30,0 г активированного угля DARCO (100 меш), и смесь перемешивают при 45°C в течение 1 часа. Смесь фильтруют через пад Celite®, и Celite® промывают EtOAc 2 × 300 мл, а затем концентрируют в вакууме. Полученную в результате красную пену растворяют в 275 мл DCM и через капельную воронку добавляют 1,0 л гептана за период в 10 минут. Полученный в результате красный раствор затравливают 3 (100 мг), а затем перемешивают при 20°C в течение 1 часа. Добавляют гептан (500 мл) в течение периода 15 минут, и полученную в результате суспензию перемешивают при 20°C в течение 18 часов. Концентрация 3 в растворе согласно измерениям составляет 5 мг/мл, а затем суспензию фильтруют. Твердые продукты промывают 200 мл 5% смеси DCM/гептан, а затем 2 × 400 мл гептана. Твердый продукт сушат в вакууме при 20°C в течение 16 часов с получением 38,34 г (60%, 96,5% AUC, SLI 1,1% - ʺM-14ʺ) 3 в виде смеси мелкодисперсных и крупных кристаллических оранжевых твердых продуктов. Во время кристаллизации, ~6 г твердых продуктов оседает в виде покрытия на колбе. Выделение не происходит так, как ожидается на основе пробного эксперимента с 5 г. Твердый продукт на стенках колбы растворяют в DCM, вместе с теми 38 г, которые выделили, и все это объединяют с маточными жидкостями и концентрируют для извлечения всего количества 3. Во время этой неудачной попытки кристаллизации, обнаруживается новый полимроф 3 и получается возможность для второй перекристаллизации из EtOAc.
Перекристаллизация 3 от реакции перекрестного связывания:
Сырой твердый продукт (66,1 г) переносят в 500-мл колбу и добавляют EtOAc 250-мл (4 объема), и смесь нагревают с обратным холодильником в течение 30 минут. Затем раствор охлаждают до 50°C и затравливают 3, и выдерживают при 50°C в течение 15 минут. Затем суспензию охлаждают до 20°C и перемешивают в течение 16 часов. Концентрацию 3 проверяют через 16 часов, и, как обнаружено, она составляет 15 мг/мл. Суспензию фильтруют, фильтрат используют для отмывки колбы, и промывочную жидкость добавляют в воронку с фильтром. Затем твердые продукты промывают 50 мл EtOAc, 50 мл 50% смеси EtOAc/гексан и, наконец, 100 мл гексана. Полученный в результате темно-красный твердый продукт сушат в вакууме при 60°C в течение 3 часов. Это дает 48,4 г 3 (75%, 98,9% AUC, SLI 1,0% - ʺM-14ʺ) в виде темно-красного кристаллического твердого продукта.
Получение 3 с помощью реакции замещения
1' (48,0 г, 1,0 экв.), 2' (59,0 г, 1,1 экв.) и Na2CO3 (43,4 г, 2,0 экв.) загружают в 2-л 3-горлую колбу. Добавляют DMA (310 мл, 6,5 объема), и реакционную смесь нагревают до 100°C. Через 18,5 часа, анализ с помощью ВЭЖХ показывает, что реакция завершается. Реакционную смесь охлаждают до 9°C, и добавляют 2-MeTHF (960 мл, 20 объемов). Добавляют 10% водный раствор NaCl (720 мл, 15 объемов), что приводит в результате к образованию некоторого количества твердых продуктов. Смесь перемешивают в течение одного часа, а затем переносят в разделительную воронку (с промывкой твердых продуктов с помощью 100 мл воды). Слои разделяют, и водный слой (Vaq ~1200 мл) экстрагируют обратно 2-MeTHF (2 × 200 мл). Затем объединенные органические промывают 10% водным раствором NaCl (2 × 250 мл), а затем анализируют с помощью 1H ЯМР на присутствие DMA (0,3% масс). После выдерживания раствора в течение ночи, отбирают аликвоту (6 мл) и промывают (3 мл) водой, что дает в результате хорошее фазовое расслоение (занимает >30 минут). Добавляют воду (650 мл, ½ размера загрузки), и перемешивают в течение 10 минут, а затем переносят в разделительную воронку и позволяют осесть. Через 90 минут, осуществляется частичное расслоение фаз (Vaq=250 мл). Добавляют насыщенный раствор соли (250 мл), что приводит к расслоению фаз. Органический слой (1300 мл, 27 объем, Kf=3,45%) отслаивают и загружают в 3-л RB колбу. Колбу нагревают (при атмосферном давлении) для отгонки некоторой части 2-MeTHF. После того как остается 15 объемов 2-MeTHF (720 мл) (через 30 минут), раствор повторно анализируют на содержание воды (Kf=0,24%). Затем реакционную смесь охлаждают до 50-5°C и окончательно фильтруют через фильтровальную бумагу (присутствует очень малое количество твердых продуктов). Затем раствор повторно загружают в 3-л колбу (после очистки колбы), и раствор отгоняют до 9 объемов (430 мл). Затем раствор нагревают до 70°C и добавляют порциями гептан в течение одного часа. Затем нагрев отключают, и раствору дают возможность для медленного охлаждения до комнатной температуры (через один час температура составляет 48°C). После перемешивания в течение 70 часов, маточную жидкость проверяют согласно анализу с помощью ВЭЖХ в течение 3 (2,7 мг/мл), а затем фильтруют. Твердые продукты промывают 25% раствором 2-MeTHF/гептан (75 мл, суспензия), за этим следует вытеснительная промывка 2 × 240 мл этого же раствора. Осадок промывают еще раз гептаном (240 мл), а затем сушат в вакуумной печи в течение 20 часов при комнатной температуре. 3 (81,1 г, выход 86%, 99,2% AUC) выделяют в виде темно-красного твердого продукта. Анализ 1H ЯМР (CDCl3) показывает отсутствие остаточного растворителя.
Получение 4
Соединение 3 (80,0 г, 1,0 экв.) и 10% Pd/C (4,0 г, 5% масс, 50% смоченного в воде) загружают в 1-л стеклянный реактор. Добавляют THF (400 мл, 5 объемов), и реактор продувают аргоном. Затем реакционную смесь помещают в атмосферу H2 (30 фунт/кв. дюйм) при температуре окружающей среды и перемешивают. Через 2 часа, реакционную смесь нагревают до 30°C и перемешивают. Еще через 4 часа, давление повышают до 40 фунт/кв. дюйм (2,5 кг/кв. см), и перемешивают в течение ночи. Через 16 часов, реакцию считают завершенной согласно анализу с помощью ВЭЖХ (3 не детектируется). Затем реакционную смесь фильтруют через Celite®, и пад промывают THF (3 × 160 мл). Затем органический раствор концентрируют (VF=90 мл), и добавляют MeOH (400 мл, 5 объемов). Смесь концентрируют досуха с получением полутвердого продукта/пены. Добавляют дополнительный MeOH (400 мл, 5 объемов) (растворяются не все твердые продукты), и смесь концентрируют досуха с получением 4 [77,0 г, выход 98% (с учетом растворителя), 98,9% (AUC)] в виде серого твердого продукта. 1H ЯМР (CDCl3) показывает 4,3% масс MeOH и 0,1% масс THF.
Получение 6
Соединение 4 (75,1 г, 1,0 экв.) загружают в 2-л RB колбу, снабженную барботажной трубкой и термопарой. Добавляют AcOH (675 мл, 9 объемов), а затем 5 (22,4 г, 1,05 экв.) и MeOH (75 мл, 1 объем). Затем воздух вводят в реакционную смесь через барботажную трубку. После перемешивания в течение 21 часов при температуре окружающей среды, реакционную смесь анализирует с помощью ВЭЖХ, анализ показывает присутствие 1,7% 4, 79,7% 6 и 18,6% 6'. Реакционную смесь перемешивают в течение еще 24 часов, за это время реакцию считают завершенной (0,3% 4, 1,9% 6'). Затем реакционную смесь концентрируют (55°C) до прекращения отгонки (вычисленное остаточное количество AcOH=86,3 г). Добавляют 2-MeTHF (960 мл, 12,8 объема), а затем 20% KOH (392 г). Требуются дополнительные 210 мл 20% KOH для получения pH >13. Смесь перемешивают в течение 10 минут, а затем она получает возможность для осаждения. Водный слой (650 мл) удаляют, и органические слои промывают 5% насыщенным раствором соли (375 мл, 5 объемов). Водный слой удаляют (390 мл, pH=10) и осуществляют вторую промывку 5% насыщенным раствором соли (375 мл, 5 объемов). Водные слои удаляют (380 мл, pH=7). Третью промывку насыщенным раствором соли не осуществляют, благодаря нейтральному значению pH, получаемому после 2 промывок. Затем осуществляют замену растворителя 2-MeTHF на IPAc (7 объем, 0,5% масс 2-MeTHF), в результате чего образуется суспензия. Образец маточной жидкости, отобранный после перемешивания в течение 65 часов, показывает концентрацию 6 как 6,3 мг/мл. Затем твердые продукты отфильтровывают и промывают IPAc (90 мл, 1,2 объема), смесью IPAc/н-гептан (1/1, 180 мл, 2,4 объема), а затем н-гептаном (90 мл, 1,2 объема). После сушки на фильтре в течение 2 часов, твердые продукты переносят в вакуумную печь и сушат в течение ночи при 40°C, с получением 6 [80,9 г, выход 86% (учитывая содержание растворителя), 97,3% (AUC)] в виде светло-желтого твердого продукта. 1H ЯМР (CDCl3) показывает 0,8% масс IPAc, 0,7% масс 2-MeTHF и отсутствие гептана. Главная примесь представляет собой N-оксид (M+16), который присутствует при 2,3%.
Очистка 6
Соединение 6 (74,2 г) растворяют (присутствуют мелкодисперсные частицы) в DCM (560 мл) и элюируют через предварительно набитый слой силикагеля (DCM) (330 г). Затем колонку промывают EtOAc (3,0 л). Две фракции собирают (2,5 л, 1,0 л), и анализируют их с помощью ВЭЖХ. В обеих фракциях не наблюдают примеси N-оксида. Фракции объединяют и частично концентрируют (VF=620 мл, 8,3 объема), с получением в результате густой суспензии. Добавляют н-гептан (620 мл, 8,3 объема), и смесь перемешивают в течение 15 минут. Образец маточной жидкости показывает концентрацию 6, которая составляет 2,7 мг/мл. Твердые продукты отфильтровывают и промывают гептаном (150 мл, 2 объема), и сушат в вакуумной печи при 45°C. Через 15 часов, получают 6 [извлечение 65,9 г, 89%, ~100% (AUC)] в виде беловатого твердого продукта. 1H ЯМР показывает только микроскопическое количество EtOAc и отсутствие н-гептана.
Получение 7
Соединение 6 (65,4 г) растворяют в DCM (650 мл, 10 объемов) и добавляют MSA (60,0 г, 5,0 экв.) в течение 15 минут (Tmax=29°C). Через 2 часа, получают густую суспензию и отбирают образец реакционной смеси (маточной жидкости), который показывает отсутствие 6 согласно анализу с помощью ВЭЖХ. Добавляют воду (460 мл, 7 объемов), и смесь перемешивают в течение 40 минут. Водный слой удаляют, и добавляют воду (200 мл, 3 объема) для экстрагирования слоя DCM. Водные слои объединяют, а затем промывают DCM (170 мл, 2,5 объема). Добавляют DCM к водному слою (650 мл, 10 объемов), и повышают основность смеси с помощью 6 н NaOH (120 мл) до pH=13. Во время добавления, твердые продукты начинают выпадать из раствора и прилипать к боковым стенкам колбы. В это время, скорость добавления основания значительно увеличивается, заставляя твердые продукты легко растворяться (Tmax= 25°C). Это, вероятно, связано с тем фактом, что 7 преципитирует из раствора до того, как смесь становится достаточно основной для того, чтобы твердые продукты стали растворимыми в DCM. Из-за отсутствия экзотермичности в этот момент, более быстрое добавление основания после появления твердых продуктов выглядит сложным. Слои разделяют, и водный слой повторно экстрагируют DCM (325 мл, 5 объемов). Органический слой сушат над Na2SO4 (Kf=0,17%), а затем концентрируют до 360 мл (5,5 объема), в результате получают преципитацию твердых продуктов. Смесь дополнительно концентрируют (150 мл, 2,3 объема) и добавляют IPAc (900 мл, 13,8 объема). Объем растворителя опять уменьшают (VF=245 мл, 4 объема), и добавляют IPAc (325 мл, 5 объемов). Анализ отношения растворителей показывает, что уровни DCM ниже целевого уровня (1,6% масс). Добавляют дополнительный IPAc (65 мл, 1 объем), и суспензию перемешивают в течение ночи. Количественное определение маточной жидкости показывает, что концентрация 7 ниже целевого уровня (2,0 мг/мл). Твердые продукты фильтруют, промывают IPAc (2 × 130 мл, 2 ×2 объема) и сушат в вакуумной печи (>28 рт. ст.) при 45°C в течение 2 дней. Получают 7 (46,9 г, выход 87%, 99,8% AUC) в виде светло-желтого твердого продукта. 1H ЯМР (CDCl3) показывает присутствие IPAc (0,5% масс) и DCM (<0,1% масс).
Пример 2: Начальная оптимизация для образования соединения 6
Список осуществляемых экспериментов приводится в Таблице 4.
Таблица 4. Оптимизация для образования Соединения 6*
°C**
(объем)
* Все реакции осуществляют в открытых флаконах в масштабе 30-70 мг.
** Репрезентативная температура флакона.
Как можно увидеть из данных в Таблице 4, DMSO представляет собой самый лучший растворитель (смотри пункты 1-3), и использование уксусной кислоты ускоряет реакцию (смотри пункты 4-11). Добавление 3-4 эквивалентов кислоты показывает наилучший профиль реакции. Эта двухстадийная стратегия исключает также гетерогенную природу реакции, наблюдаемую при подходе с использованием одной емкости. Простое окисление в присутствии воздуха необходимо для генерирования 6 после завершения циклизации с получением 6'. Сначала осуществляют две отдельных реакции с использованием условий, определенных в Таблице 4, но, к сожалению, получаются значения выхода более низкие, чем ожидалось, и требуется также очистка на колонке (Таблица 5).
Таблица 5. Синтез 6 с использованием двухстадийной стратегии
°C
(% AUC)
Пример 3: Анализ растворимости 3 в 2-MeTHF и гептане
Осуществляют ряд экспериментов для исследования растворимости 3 в смеси 2-MeTHF и гептана.
Таблица 6. Растворимость 3 в смеси 2-MeTHF/гептан при 25°C
Пример 4: Скрининг и оптимизация реакции с использованием 1 и 2s в реакции перекрестного связывания для генерирования 3
Используют суррогатное соединение (2s) вместо 2 для скрининга условий (с использованием Pd2(dba)3 и Xantphos) и для оптимизации реакции перекрестного связывания. Начальные эксперименты, исследующие эту реакцию, приведены в Таблице 7. Можно увидеть, что сочетание 5% моль Pd2(dba)3 и Xantphos с Cs2CO3 в THF дает в результате в 90% выход выделенного соединения. Выход 90% изначально указывает на этот подход как на жизнеспособную возможность для получения 3.
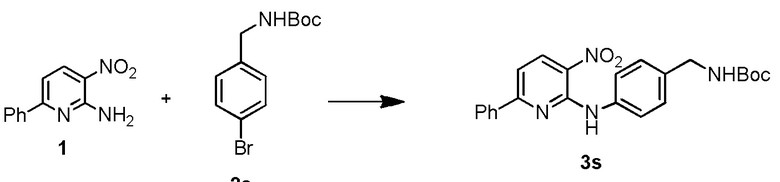
Таблица 7. Начальные результаты для возможности перекрестного связывания 1 и 2s
(77%)
(62%)
Исследуют каталитическую нагрузку реакции перекрестного связывания. Как видно в Таблице 8, количество Pd2(dba)3 может быть уменьшено до 1,25% моль, а Xantphos до 2,5% моль, при этом не наблюдается уменьшения выхода выделенного соединения. Несколько реакций, перечисленных в Таблице 7, также исследуются на устойчивость реакции относительно толерантности к воздуху. Обнаружено, что дегазирование THF посредством барботирования аргона через реакционную смесь перед добавлением катализатора не является необходимым и защитный слой аргона является достаточным для предотвращения окисления катализатора.
Таблица 8. Оптимизация реакции перекрестного связывания 1 и 2s
(AUC)
Пример 5: Эксперимент по реакции перекрестного связывания на 100 г соединения 2 с использованием 2-MeTHF в качестве растворителя
Осуществляют эксперимент по реакции перекрестного связывания на 100 г соединения 2 с использованием 2-MeTHF в качестве растворителя. Ход реакции после добавления 1,25% Pd2(dba)3 и 2,5% Xantphos является медленным. Для доведения реакции до завершения используют вторые загрузки Pd2(dba)3 (1,25%) и Xantphos (2,5%) и Cs2CO3 (1,1 экв.) (в целом 45 часов). Фазовые расслоения во время извлечения являются проблематичными. Мелкодисперсные нерастворимые порошки, всплывающие в растворе, и стенки реактора, покрывающиеся черным остатком, приводят к сложностям во время экстракционного извлечения. Добавление насыщенного раствора соли или подогрев реакционной смеси не решает этой проблемы. Фильтрование загрузки (двухфазной) для удаления всех видов частиц дает удовлетворительное фазовое расслоение, и остальная часть экстракционного извлечения проходит без инцидентов. Конечный продукт выделяют из 2-MeTHF с помощью гептана в качестве антирастворителя (выход: 72%, чистота: 96,98%).
Пример 6: Эксперименты по реакции гидрирования для получения соединения 4
Каталитическое гидрирование 3 изначально осуществляют в EtOAc (15 объемов) с помощью 10% Pd/C (10% масс) при 40 фунт/кв. дюйм (2,5 кг/кв. см) газообразного водорода. Через 3 часа, реакция завершается согласно анализу с помощью ВЭЖХ. Соединение 4 выделяется с количественным выходом в виде пены посредством концентрирования фильтрата досуха после удаления катализатора посредством фильтрования через Celite®.
Хотя реакция имеет высокий выход, количество растворителя необходимого для осуществления реакции ограничивает производительность из-за плохой растворимости 3 в EtOAc. По этой причине, желательно найти альтернативный растворитель для этой реакции с целью предотвращения риска плохого преобразования, в то же время, увеличивая объемную эффективность. Для решения этой проблемы, растворимость 3 оценивается в HOAc и THF, в дополнение к 2-MeTHF. Эти растворители выбираются, поскольку 6 и свободное основание 7 демонстрируют хорошую растворимость по отношению к этим кандидатам и используются на других стадиях способа. Растворимость 3 составляет 24,8 мг/мл в HOAc, 100 мг/мл, в 2-MeTHF и 155 мг/мл, в THF. Эти результаты говорят о том, что может осуществляться гидрирование менее чем в 10 объемах THF. В одном из вариантов осуществления, для реакции гидрирования используют THF. В другом варианте осуществления, для реакции гидрирования используют 2-MeTHF. Когда используют THF, полное растворение 3 наблюдается в 6 объемах растворителя, а 2-MeTHF требует 8 объемов. В одном из вариантов осуществления, когда реакция гидрирования осуществляется при давлении водорода 40 фунт/кв. дюйм (2,5 кг/кв. см) при 40°C, реакция, как правило, завершается за 2-3 часа. Последнее переменное, которое исследуют, представляет собой нагрузку катализатора. Начальная нагрузка 10% масс уменьшается до 5% масс без какого-либо уменьшения времени реакции, выхода или чистоты.
Процедура выделения для 4 разработана в качестве альтернативного подхода для концентрирования досуха. Когда используют либо THF, либо 2-MeTHF, частичное концентрирование с последующей заменой растворителя на 2-PrOH (3-5 объемов) и добавление гептана (10-15 объемов) в качестве антирастворителя дают разумную суспензию, которая при фильтровании дает 4 (выход 95%) в виде светло-серого твердого продукта с высокой чистотой (>99% AUC).
Альтернативно, поскольку соединение 4 является растворимым в HOAc, реакция гидрирования может осуществляться в HOAc (10 объемов) и переноситься непосредственно на стадию циклизации/окисления для непосредственного получения 6. Реакция гидрирования завершается через 4 часа, когда подвергается воздействию давления водорода 40 фунт/кв. дюйм (2,5 кг/кв. см) при температуре окружающей среды. Фильтрование катализатора дает прозрачный раствор 4, который мог бы непосредственно использоваться для преобразования в соединение 6.
Также исследуют синтез соединения 4 с помощью каталитического гидрирования 3 без газообразного водорода. Соединение 3, альдегид 5 (1,05 экв.), NH4COOH (5 экв.) и 10% Pd/C (50% масс) объединяют в спиртовом растворителе (MeOH или EtOH) и нагревают до 65°C. Анализ с помощью ВЭЖХ показывает преобразование в соединение 6, но времена реакции являются очень большими по сравнению со стандартными условиями гидрогенолиза при 40 фунт/кв. дюйм (2,5 кг/кв. см). Реакция гидрирования без газообразного водорода исследуется в AcOH и в смеси AcOH/диоксан с неполным преобразованием в 6. Реакция гидрирования без газообразного водорода также исследуется ступенчатым образом, с исключением альдегида 5. При этих условиях, 3 легко преобразуется в 4 в MeOH меньше чем через 4 часа при температуре окружающей среды. Реакция гидрирования является успешной при использовании 10% раствора MeOH/AcOH. Использование AcOH в качестве растворителя также дает 4, однако реакция дает максимум, который представляет собой растворитель, как продемонстрировано ранее, являющийся успешным при преобразовании 4 в 6. В попытке оптимизации условий реакции, обнаружилось, что реакция может осуществляться с использованием 10% смеси MeOH/AcOH (10 объемов) с NH4COOH (5 экв.) и при нагрузке катализатора 30% масс с получением 4. Затем катализатор отфильтровывают, и альдегид 5 приводит непосредственно к 6. К сожалению, если реакция поддерживается в течение продолжительных периодов времени (>24 часов), генерируются дополнительные примеси. Поскольку осуществление преобразования 3 в 4 при давлении водорода 40 фунт/кв. дюйм (2,5 кг/кв. см) не является проблемой, попытки оптимизации гидрогенолиза без использования газообразного водорода больше не осуществлялись.
Пример 7: Скрининг и оптимизация для преобразования 4 в 6
Осуществляют набор реакций скрининга для исследования преобразования 4 в 6', который мог бы затем окисляться до 6. Оптимизация начинается посредством оценки растворителей EtOH, PrOH, толуола и DMSO, с использованием как 1,1, так и с 3,0 эквивалента альдегида 5. Эти реакции осуществляют на 100 мг 4. Результаты приведены в Таблице 9.
Таблица 9. Скрининг растворителя для преобразования 4 в 6'
(экв.)
Соединение 4 и 5 также нагревают с обратным холодильником в толуоле в атмосфере азота в присутствии фумаровой кислоты и ловушки Дина-Старка для удаления воды. Реакция может быть очень медленной, и профиль чистоты для реакции не является обещающим (Таблица 10).
Таблица 10. Исследование синтеза соединения 6'
(час)
°C
На основании результатов в Таблице 9, трудно предотвратить окисление 6' в 6. Можно преобразовывать 4 в 6 непосредственно при мягких условиях. Реакцию осуществляют в смеси HOAc/MeOH (9/1, 47 объемов). Как можно увидеть из данных в Таблице 11, профиль чистоты улучшается с использованием этой системы растворителей по сравнению с тем, что наблюдается для других систем.
Таблица 11. Непосредственный синтез 6 из 4 в AcOH/MeOH
°C
Дополнительные реакции оптимизации исследуются для уменьшенного объема реакции и небольшого только избытка (1,05-1,1 экв.) соединения 5. При повышенной температуре, реакции завершаются (или почти завершаются) через 4 часа (Таблицы 12-14). Однако генерируется новая примесь, Примесь 7, и она является более значительной в более концентрированных реакционных смесях. При температуре окружающей среды, реакция является медленной, но она дает более благоприятный профиль чистоты, предотвращая образование Примеси 7.
Примесь 7
Для всех собранных данных, объем растворителя, выбранный для дальнейших исследований, составляет 10 объемов AcOH/MeOH (9:1) при температуре окружающей среды. Перемешивание соединения 4 (1,0 экв.) и соединения 5 (1,05 экв.) в смеси AcOH/MeOH (10 объемов) в течение ночи при температуре окружающей среды на открытом воздухе дает почти полное преобразование в соединение 6.
Таблица 12. Преобразование 4 в 6 в смеси AcOH/MeOH (9:1, 20 объемов)
Таблица 13. Преобразование 4 в 6 в смеси AcOH/MeOH (9:1, 10 объемов)
Таблица 14. Преобразование 4 в 6 в смеси AcOH/MeOH (9:1, 5 объемов)
°C
Более конкретно, когда реакционную смесь перемешивают при температуре окружающей среды до тех пор, пока 4 не израсходуется, увеличение температуры реакции до 50°C в течение дополнительных 2 часов способствует преобразованию оставшегося 6' в 6 (Таблица 15). Альтернативно, перемешивание реакционной смеси в течение более длительного периода времени (24 часа) при температуре окружающей среды, в конечном счете, приводит к полному преобразованию в 6. Следовательно, эти данные говорят о том, что после завершения реакции преобразования 4 в 6', общая чистота реакционной смеси не подвергается воздействию нагрева, если он применяется для доведения реакция до завершения, до 6.
Таблица 15. Воздействие температуры реакции (50°C) на более поздние стадии реакции
°C
* В качестве смеси растворителей используется смесь HOAc/MeOH (9:1)
Осуществляют дополнительные исследования для оценки увеличения загрузки 5 (1,2 экв. по сравнению с 1,05) исследуются. Показано, что увеличение эквивалентов 5 оказывает отрицательное воздействие на реакцию. Нет наблюдаемого увеличения скорости реакции, и образуется значительное количество примеси 7.
Осуществляют дополнительные исследования для оценки воздействия изменения отношения растворителей HOAc/MeOH, а также воздействия температуры (50°C по сравнению с 20°C) на реакцию. Как видно в Таблице 16, скорость расходования 4 с получением либо 6', либо 6 является примерно одинаковой через 6 часов при обеих температурах независимо от отношения растворителей, но скорость окисления от 6' до 6 сильнее зависит от температуры, чем от количества уксусной кислоты. Профиль примесей является более благоприятным, когда реакцию осуществляют при 20°C, с <1% неизвестных примесей согласно анализу с помощью ВЭЖХ (AUC).
Таблица 16. Оптимизация условий MeOH/HOAc для преобразования 4 в 6
HOAc*
*Все реакции осуществляются при 10 объемах растворителя с 1,1 эквивалента 5
Фокус разработки этой стадии смещается затем на извлечение. Реакцию для исследования осуществляют со смесью 1:9 MeOH/HOAc (10 объемов) при 50°C с 1,05 эквивалента альдегида 5 в масштабе 5,0 г (4). Конечная точка реакции дает сырую реакционную смесь, которая согласно анализу с помощью ВЭЖХ содержит 1,0% 4, 93% 6, 0,2% 6' и 0,9% 7. После концентрирования для удаления объема уксусной кислоты, остаток растворяют в EtOAc и используют основную водную промывку для удаления остатка HOAc. Затем раствор в EtOAc концентрируют до 5 объемов, и происходит кристаллизация. После перемешивания для суспендирования в течение 4 часов при температуре окружающей среды, твердые продукты выделяют с помощью фильтрования и сушат в вакууме. Этот подход дает 6 в виде беловатого твердого продукта с выходом 67% (98,4% AUC).
Хотя этот начальный опыт является очень обнадеживающим, в дальнейших экспериментах в большем масштабе сталкиваются со сложностями. Одна из проблем заключается в том, что во время нейтрализации уксусной кислоты, часто получаются эмульсии. Исследуют множество систем растворителей и оценивают растворимость 6 для оценки эффективности экстрагирования (Таблица 17).
Таблица 17. Растворимость 6
Когда к реакционной смеси добавляют IPAc и воду, наблюдают соответствующее фазовое расслоение. Однако после того как слой IPAc отделяют и обрабатывают основанием для нейтрализации остаточной уксусной кислоты, образуется эмульсия. Из-за низкой растворимости 6 в EtOAc и IPAc, имеется также проблема с кристаллизацией 6 из раствора до завершения извлечения.
Также исследуется система DCM/вода, и хотя нет проблем с преждевременной кристаллизацией 6, проблемы с эмульсией сохраняются во время нейтрализации HOAc. Делаются попытки нейтрализации реакционной смеси до pH ~5-6 для устранения основного водного слоя. Это процедура устраняет эмульсию, но не является полностью достаточной при нейтрализации AcOH. Добавление дополнительных водных промывок также приводит к проблемам с эмульсией. Предполагается, что основание, используемое для гашения AcOH, также может вносить различия, связанные с растворимостью солей, образующихся в воде. В начальных экспериментах используют водный раствор NaOH. Использование KOH не оказывает какого-либо существенного воздействия, когда в качестве органического растворителя используется DCM.
Третья исследуемая система растворителей представляет собой смесь 2-MeTHF/вода. Сначала, возникает благоприятное фазовое расслоение, хотя требуется большее количество органического растворителя (15 объемов), чем при использовании DCM (10 объемов). Это система страдает от тех же проблем с эмульсией, когда повышают ее основность. Осуществляют также скрининг различных оснований вместе с этой системой, включая KOH и NH4OH. Когда используют NH4OH, возникает значительное количество выделяющихся газов. Когда используют KOH, образование эмульсии улучшается. В результате, KOH выбирается в качестве оптимального основания при извлечении. Проблема с преждевременной кристаллизацией также представляет собой риск для 2-MeTHF. Однако определено, что это составляет риск только если смесь охлаждается во время извлечения. Если смесь нагревают во время гашения (~40-50°C) все твердые продукты остаются в растворе. После завершения нейтрализации HOAc, простая замена растворителя с 2-MeTHF на IPAc дает 6 с хорошим выходом и чистотой [>80%, ~99% (AUC)].
Для уменьшения количества AcOH, который требует нейтрализации, оценивается отгонка части AcOH перед извлечением из водной фазы. Исследование стабильности показывает, что реакционная смесь является стабильной по отношению к концентрированию (от 15 объемом до 4 объемов), а также к продолжительным временам выдерживания (>1 дня). На основе этой информации, осуществляют ряд экспериментов, в которых частично удаляют AcOH (до 4 объемов) перед нейтрализацией. Сводку данных экспериментов можно найти в Таблице 18, ниже. Определено, что отгонка AcOH значительно уменьшает объем извлечения, и это используется в будущих экспериментах.
Таблица 18. Оптимизация растворителя и основания для выделения 6
AcOH
(объем)
(AUC)
Хотя процедура выделения 6 является установленной, примесь с массой M+16 периодически детектируется посредством анализа с помощью ЖХ-МС в некоторых загрузках 6, синтезируемых и выделяемых с использованием этой процедуры (<4% AUC). По-прежнему непонятно, почему и где образуется эта примесь. После образования этой примеси ее сложно удалять посредством перекристаллизации. Перенос примеси на стадию снятия защиты и ее удаление во время выделения 7 также не является успешным. Обнаружено, что использование фильтрования 6 на силикагеле с элюированием EtOAc успешно удаляет эту примесь.
Пример 8: Оптимизация для синтеза 7
Начальные попытки синтеза 7 из 6 включают добавление TFA (10 экв.) к раствору 6 в DCM (10 объемов). Общее преобразование завершается при температуре окружающей среды через 15 часов. Повышение температуры до 40°C понижает время преобразования до 4 часов. Другие растворители, которые оцениваются, представляют собой DCE, анизол и IPA. В последних двух случаях, смесь соединения 6 и 7 преципитирует из раствора (вероятно в виде соли TFA). DCE дает полное преобразование через один час при 80°C. Хотя преобразование в 7 является относительно простым, извлечение является проблематичным, поскольку дает в результате липкие твердые продукты, преципитирующие из раствора, с использованием множества условий.
После того, как устанавливаются условия гашения реакции, осуществляют несколько экспериментов для выделения 7. Экстрагирование водным раствором HCl дает в результате преципитацию твердых продуктов в водном слое. Исследуют другие эксперименты с различными кислотами для устранения преципитации, включая лимонную кислоту и метансульфоновую кислоту (MSA). Обнаружено, что MSA не приводит к преципитации твердых продуктов во время извлечения. Это дает возможность для промывки водного слоя DCM для удаления примесей, включая остаточный 6.
Выделение 7 из водного слоя MSA завершается посредством добавления DCM с последующим повышение основности с помощью водного раствора NaOH для экстрагирования свободного основания 7 в слой DCM. Эта процедура не преципитирует твердые продукты во время процесса экстрагирования, сводя к минимуму встречающиеся ранее проблемы. Разрабатывается выделение из раствора в DCM посредством концентрирования объема DCM, чтобы вызвать кристаллизацию с последующим добавлением гептана в качестве антирастворителя. Последующие эксперименты показывают, что замена растворителя с DCM на 2-PrOH дает хорошо фильтрующуюся суспензию. Возможные кандидаты в растворители, которые могли бы использоваться для эффективного экстрагирования 7, ограничиваются DCM из-за плохой растворимости 7 в разнообразных распространенных растворителях (Таблица 19).
Таблица 19. Растворимость 7 при выборе растворителей
На основе успеха при использовании MSA во время извлечения и выделения 7, делается попытка осуществления снятия защиты с 6 с использованием MSA (5 экв.) вместо TFA. Реакция, опосредуемая MSA, является более простой (один час), чем для TFA, при температуре окружающей среды. Извлечение осуществляют, как описано выше, с помощью процесса экстрагирования DCM.
Пример 9: Исследование синтеза соединений 1, 1', 2 и 2'
9a). Исследование синтеза соединений 1 и 1'
Один из подходов (Схема 6) основывается на реакции Судзуки между коммерчески доступным 6-хлор-2-амино-3-нитропиридином и фенилбориновой кислотой с получением 1. К счастью, 1 не только представляет собой желаемый исходный материал для синтеза 3, но также представляет собой и промежуточное соединение для синтеза 1'.
Схема 6
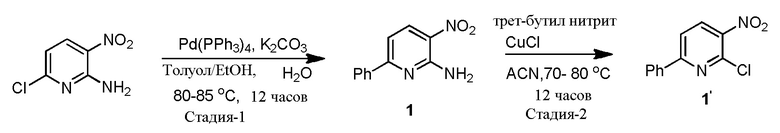
Реакция Судзуки доставляет две главные проблемы для масштабирования реакции и доведения до максимума производительности. Процедура требует больших объемов растворителя (85 объемов), а также использует примерно 2 процента молярных катализатора Pd(PPh3)4.
Для уменьшения количества растворителя и понижения каталитической нагрузки Pd(PPh3)4, осуществляют небольшой ряд оптимизаций. Обнаружено, что количество растворителя может быть уменьшено до 36 объемов, в целом, и каталитическая нагрузка Pd(PPh3)4 может быть понижена до 1% моль без уменьшения выхода или чистоты 1. Результаты приведены в Таблице 20 и репрезентативную процедуру для оптимизированных условий можно найти в разделе Примеры.
Таблица 20. Исследование объема реакционной смеси и каталитической нагрузки для синтез 1
(г)
(выход)
(AUC)
Очищают с помощью колоночной хроматографии
Очищают с помощью колоночной хроматографии
Очищают посредством перетирания в гептане
Очищают посредством перетирания в гептане
Очищают посредством перетирания в гептане
Соединение 1' можно синтезировать с помощью процедуры реакции Зандмейера (Стадия 2 Схемы 6). Хотя можно было бы синтезировать 1', эта реакция имеет низкий выход (выход после выделение 30-40%). Два главных побочных продукта идентифицируются как продукты дегалогенирования и гидролиза 1. Осуществляют краткий скрининг альтернативных условий, и его результаты приведены в Таблице 21. Как показано в Таблице 21, безводный ацетонитрил (MeCN) не влияет на выход при выделении, так же, как и использование альтернативного источника хлорида (TMSCl), свежего CuCl или диоксана вместо MeCN.
Таблица 21. Условия, используемые для исследования и улучшения преобразования 1 в 1' (химический механизм Зандмейера)
(г)
(% AUC)
40°C - 50°C
40°C-50°C
60°C
55°C - 60°C
комн. темп.-60°C
(5 час)
25-60°C (6 час)
используют BF3⋅Et2O
CuCl, 55°C - 60°C
CuCl, 55°C - 60°C
55°C - 60°C
Сложности, встречающиеся при улучшении выхода реакции Зандмейера, приводят в результате к поиску альтернативных условий для получения 1'. Наблюдают, что одна из главных примесей в реакции Зандмейера представляет собой гидрокси производное 1a. Когда 1a обрабатывают POCl3, получают 1' с хорошим выходом (80%). Сначала разрабатывают способ преобразования 1 в 1a перед осуществлением реакции POCl3 для получения 1' (Схема 7). При осуществлении реакции Зандмейера в водном растворе THF, 1a получают при умеренном выходе и чистоте (Таблица 22). Сначала реакцию POCl3 осуществляют без растворителя (5 объемов), но обнаружено, что использование ацетонитрила в качестве растворителя также хорошо работает, и загрузка POCl3 может быть уменьшена до 1 объема (Таблица 23).
Схема 7
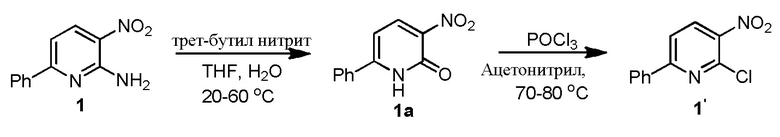
Таблица 22. Получение 1a с использованием химического механизма Зандмейера с помощью 1 в водном растворе THF
(г)
(выход)
(66,1%)
(73,8%)
Таблица 23. Сравнение POCl3 без растворителя со смесью MeCN/POCl3 для преобразования 1a в 1'
(г)
(Выход)
(97,2%)
70-80°C, 8 часов
(81,6%)
70-80°C, 12 часов
Способность преобразования 1a в 1' с использованием POCl3 не только увеличивает выход с выделением 1', но тот факт, что 1b является доступным коммерчески, дает возможность для получения 1a без потребности в диазониевом химическом механизме. Использование реакции Судзуки с фенилбориновой кислотой и 1b дает 1a за одну стадию (Схема 8). Начальные попытки, которые делаются, чтобы приспособить реакцию Судзуки на 6-хлор-3-нитропиридин-2(1H)-оне с фенилбориновой кислотой и Pd(PPh3)4 для получения 1a, приведены в Таблице 24. Как правило, реакция Судзуки является успешной, но во всех случаях наблюдают неидентифицируемую примесь. Эта примесь доставляет проблемы при удалении, но позднее обнаруживается, что эту примесь можно удалить во время выделения 1' после реакции POCl3.
Схема 8
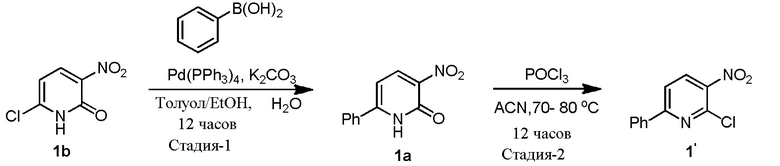
Таблица 24. Оптимизация и осуществление реакции Судзуки 1b и фенилбориновой кислоты
(г)
(выход)
(% AUC)
b) Фенилбориновая кислота, 1,1 экв.
c) Присутствуют 9,8% примеси (при 1,17 RRT)
d) Используют 60 объемов EtOAc для промывки
e) Реакция завершается через 6 часов
93%
(2-ой сбор)
b) Фенилбориновая кислота (1,02 экв.)
c) Через 6 часов, добавляют дополнительно 5,0 г
Pd катализатора.
d) Реакция завершается через 7 часов
e) Используют 35 объемов EtOAc
f) 16,3% примеси при 1,17 RRT
86,3%
(2-ой сбор)
b) Фенилбориновая кислота (1,02 экв.)
c) Реакция завершается через 7 часов
d) 19,1% примеси при 1,17 RRT
83,8%
(2-ой сбор)
b) Фенилбориновая кислота (1,02 экв.)
c) Реакция завершается через 5 часов
d) 16,1% примеси при 1,17 RRT
9b). Разработка соединений 2 и 2'
Схемы 9 и 10 иллюстрируют стадии синтеза для генерирования соединений 2 и 2'.
Схема 9
Схема 10
9bi). Синтез B
Синтез B оценивается следующим образом. Смесь порошкообразного KOH (5,6 экв.), воды (0,77 объема), толуола (7,7 объема) и каталитического количества тетрабутиламмонийбромида (0,05 экв.) нагревают до 45°C. Затем добавляют как одну порцию 1,3-дибромпропан (1,10 экв.), затем медленно добавляют раствор фенилацетонитрила A (1,0 экв.) в толуоле (5,0 объемов). Этот раствор добавляют в течение 45 минут, поддерживая при этом температуру реакции при 55-85°C. Во время добавления соединения A, преципитирует значительное количество белого твердого продукта. Затем смесь нагревают с обратным холодильником (98-102°C) в течение одного часа и анализируют с помощью ВЭЖХ. Через один час, соединение A расходуется и реакцию считают завершенной. На этой стадии, реакционную смесь охлаждают до 70°C, а затем разбавляют н-гептаном (10,4 объема) для преципитации дополнительных неорганических солей. После охлаждения смеси до 20-30°C, твердые продукты удаляют с помощью фильтрования и фильтрат промывают водой и насыщенным раствором соли. После сушки над MgSO4, фильтрат концентрируют с получением сырого B в виде желтого масла (как правило, извлечение >90% сырой массы). Затем это сырое масло очищают с помощью вакуумной отгонки (750 миллитор, температура кипения=105-110°C) с получением B (как правило, с выходом 50-60%). Отогнанный B, как правило, содержит единственную примесь B2 при уровнях, находящихся в пределах 2-4% (ВЭЖХ, AUC). Приготавливают четыре загрузки соединения B в масштабах, находящихся в пределах от 50 г до 500 г, не встречая каких-либо сложностей при масштабировании.
Примесь B2
В качестве альтернативного способа получения C, исследуется указанный выше подход с использованием 4-нитрофенилацетонитрила. Во многих экспериментах, реакционные смеси превращаются в черную смолу после добавления 4-нитрофенилацетонитрила к KOH в толуоле и воде.
9bii). Синтез C и D
Преобразование B в C получают посредством медленного добавления раствора B к смеси KNO3 в H2SO4, поддерживая при этом температуру ниже 15°C. В малом масштабе (25 г), реакция завершается меньше чем за один час и гасится посредством выливания раствора на лед. После экстракционного извлечения, этот подход дает C [выход 99%, 95% (AUC)] в виде свободно сыпучего желто-коричневого твердого продукта. Однако при масштабировании (500 г) сложно остановить реакцию при получении C. Вместо этого, C в дальнейшем самопроизвольно гидролизуется до D. Через два часа, в реакционной смеси не остается детектируемого B, однако с помощью ВЭЖХ наблюдают 70% C и 30% D (AUC). После перемешивания реакционной смеси в течение ночи, преобразование C в D завершается. Смесь выливают на лед и экстрагируют DCM. После концентрирования раствора в DCM, остаток растворяют в горячем EtOAc (500 мл) и гептане (1,5 л) и медленно охлаждают до температуры окружающей среды, чтобы вызвать кристаллизацию. Это дает чистый D [выход 55% для двух стадий из B, 99% (ВЭЖХ, AUC)] в виде светло-желтого твердого продукта. Вероятно, при дальнейшей оптимизации выход этой кристаллизации может быть улучшен, поскольку анализ маточной жидкости показывает по-прежнему присутствие относительно чистого D (92% AUC). Однако осуществляемые попытки выделить второй сбор из этой маточной жидкости являются безуспешными, приводя к выделению масла и к отсутствию кристаллизации.
9biii). Синтез D из C
Поскольку B может легко преобразовывать в D в одной емкости, преобразование C в D исследуется только кратко. Имеется два возможных способа преобразования H в I. Один способ использует 30% водный раствор H2O2 и K2CO3 в DMSO. Во избежание возможных проблем для пероксида в большем масштабе, эти условия не исследуются. Вместо этого, используют условия с использованием HOAc (или TFA) в H2SO4. Поскольку эти условия, как обнаружено, хорошо работают для H, видимо, является разумным ожидать подобного же успеха при применении для преобразования C в D. При нагреве C до 90°C в присутствии HOAc (13,0 объемов) и H2SO4 (7,0 объемов) в течение 19 часов, преобразование в D завершается согласно ВЭЖХ. Затем смесь выливают на лед, и после экстракционного извлечения, D очищают посредством преципитации из DCM и гептана. Это дает D [выход 69%, 99% (AUC) согласно ВЭЖХ)] в виде желто-коричневого твердого продукта. Условия с TFA также исследуются. Хотя реакция может осуществляться при комнатной температуре вместо 90°C, реакция не доходит до завершения через 19 часов. После извлечения, выход с выделением соединения является хорошим (82%), но общая чистота ниже (92% AUC согласно ВЭЖХ).
9biv). Синтез E
Исследуют синтез E с генерированием производного карбоновой кислоты D. Предполагается, что условия перегруппировки Курциуса с использованием E и DPPA в трет-бутаноле могут представлять собой пригодную для использования альтернативу применению Pb(OAc)4 для синтеза F. Обнаружено, что D может легко преобразовываться в E в присутствии EtOH и 6 M NaOH при 60°C. После извлечения в водный слой, E выделяют количественно в виде желто-коричневого твердого продукта (99% AUC). Этот материал можно использовать непосредственно без дополнительной очистки.
9bv). Синтез F с помощью Pb(OAc)
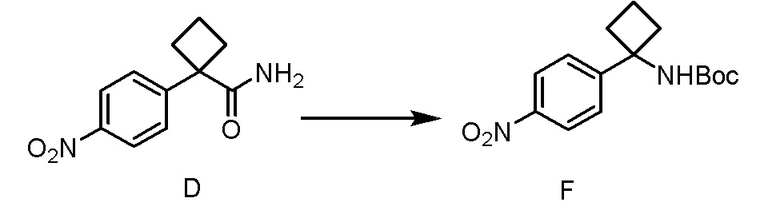
Синтез F осуществляется посредством адаптирования процедуры для получения 2 из I. Способ получения 2 включает добавление порциями Pb(OAc)4 к раствору I в трет-бутаноле (5,0 объемов) при 75°C. Однако D не является настолько же растворимым в трет-бутаноле (5,0-6,0 объемов) при 75°C, насколько I растворим в трет-бутаноле. Когда преобразование D в F осуществляется в малом масштабе (30 г), добавление Pb(OAc)4 является обычным, и реакцию считают завершенной через час при 75°C (более низкая растворимость D не изменяет выход реакции). После извлечения и выделения, получают F с выходом 85%, однако чистота является плохой (92,5%), с двумя значительными примесями. Одна из примесей идентифицируется как изопропилкарбаматное производное F2 (5%), которое, как предполагается, представляет собой результат присутствия микроскопического количества изопропанола в трет-бутаноле. Другая примесь (2,5%) не идентифицируется. Затем этот материал преобразуется непосредственно в 2', и полученный в результате продукт очищают с помощью хроматографии на силикагеле для удаления двух примесей, присутствующих в F.
Соединение F2, примесь изопропилкарбамат
Затем преобразование D в F масштабируется до 200 г и осложнений не встречается. Реакция 200 г дает соединение F [178 г, 67%, 92,0% (AUC)] в виде белого твердого продукта после очистки посредством повторного суспендирования в MTBE (1,5 объема) и гептане (3,0 объема). Единственная самая большая примесь представляет собой изопропилкарбаматное производное F2 [6,5% (AUC)]. Затем фильтрат концентрируют досуха с получением дополнительного F в виде коричневого твердого продукта [48 г, 18%, 72% (AUC)].
В попытке сведения к минимуму образования примеси F2, используют трет-бутанол качества для ВЭЖХ (чистота 99,8%) вместо трет-бутанола химического качества. D (50 г) преобразуется в F [38 г, 57%, 96% (AUC)] и, как обнаружено, содержит только 3,1% примеси F2. Затем оценивается другая реакция при более низкой температуре (45°C по сравнению с 75°C) в попытке подавления образования примеси; однако при 45°C не наблюдается преобразования D в F.
Делаются попытки параллельной разработки способа удаления примеси изопропилкарбамата F2. Перекристаллизация из MeOH или EtOH и воды дает умеренное улучшение чистоты (содержание примеси понижается от 2% до 1%). Пытаются также воздействовать на материал посредством второй перекристаллизации, но эта стратегия не удаляет остаточный F2 (остается ~0,6%). Затем идентифицируется кратковременное решение для удаления этой проблемной примеси посредством исследования различий в химической активности Boc- и изопропилкарбаматных групп. При обработке с помощью HCl, Boc группа F легко отщепляется и образуется водорастворимая соль HCl. Изопропилкарбаматное производное F2 не взаимодействует с HCl и, таким образом, затем отмывается во время извлечения в водном слое, и соединение свободное основание F может извлекаться посредством экстракционного извлечения при pH=11. В масштабе 5 г, соединение свободное основание F выделяется при количественном выходе, оно не содержит соединения примеси F2.
Затем исследуют повторную защиту свободного основания F с использованием стандартных условий (DCM, Boc2O, и Et3N), и, как обнаружено, она является очень проблематичной. В ходе реакции, вместе с желаемым F (80%), в заметных количествах образуется нежелательное симметричное мочевинное производное (20%, фигура 3). Затем осуществляют другую реакцию посредством медленного добавления соединения свободного основания F с избытком Boc2O и Et3N в DCM; однако это неожиданно дает еще больше побочного продукта симметричной мочевины (45%). В конечном счете, идентифицируются условия, которые полностью подавляют образование симметричной мочевины. При двухфазных условиях, водный раствор 1 M NaOH, THF и Boc2O (1,5 экв.) дает желаемый продукт F с хорошим выходом и с высокой чистотой [81%, >99% (AUC)].
Соединения свободного основания F, F-OMe и симметричной мочевины
9bvi). Синтез F с помощью смеси NaOH/бром
Также исследуют синтез F с использованием условий перегруппировки Гофмана. D растворяют в MeOH, обрабатывают 25% масс NaOMe в MeOH (4,3 экв.), а затем охлаждают до 5°C. Добавление по каплям Br2 (1,0 экв.) к реакционной смеси вызывает умеренную экзотермичность. Затем реакционную смесь нагревают до температуры окружающей среды и перемешивают в течение одного часа. За это время реакция завершается согласно анализу с помощью ВЭЖХ, а затем ее гасят посредством медленного добавления насыщенного водного раствора NH4Cl (40 объемов). Во время гашения, продукт (F-OMe) кристаллизуется из реакционной смеси в виде больших хлопьев белого твердого продукта. Эти твердые продукты выделяют и сушат с получением соединения метилкарбаматного производного F-OMe [81%, >99% (AUC)] в виде белого твердого продукта.
Также исследуют второй набор условий, где йодозобензол генерируется in-situ под действием Oxone® на йодобензол либо в воде и ацетонитриле (генерирует соединение свободное основание F), либо в MeOH (генерирует соединение F-OMe). Хотя они являются до некоторой степени эффективными, эти две реакции требуют продолжительных времен реакции (>41 часа) для достижения умеренного преобразования. В воде и ацетонитриле происходит 31% преобразование (ВЭЖХ) в соединение свободного основания F через 41 час. В MeOH происходит 73% преобразование (ВЭЖХ) в соединение F-OMe через 46 часов. Эти две реакции считаются слишком медленными для использования в настоящее время, и дополнительной работы с этой системой реагентов не проводят.
Хотя имеется превосходный способ получения соединения F-OMe, способ преобразования F-OMe обратно в соединение F ранее не был установлен. Исследуется множество условий (Таблица 25), однако только один набор условий (HBr в HOAc) является эффективным. Главный недостаток использования HBr в HOAc представляет собой образование множества побочных продуктов во время снятия защиты. После этих экспериментов, метилкарбаматный способ был оставлен для более перспективных способов.
Таблица 25. Снятие защиты с F-метилкарбамата (F-OMe)
9bvii). Синтез F с помощью DPPA
Оценивают синтез F из E и дифенилфосфорилазида (DPPA) в трет-бутаноле. Использование DPPA потенциально устраняло бы два главных недостатка, связанных с использованием Pb(OAc)4. Первый заключается в том, что с Pb(OAc)4 трудно манипулировать и загружать его порциями в реакцию. Это липкий твердый продукт постепенно ожижается и становится черным при экспонировании для воздуха и/или влажности. Вторая причина для устранения Pb(OAc)4 представляет собой большое количество отходов PbCO3, которое генерируется и которое затем нужно утилизировать. Использование DPPA устранило бы обе эти проблемы, поскольку он представляет собой жидкость, которой легко манипулировать и которая дает дифенилфосфат в качестве побочного продукта. Начальные попытки преобразования E в F осуществлялись посредством медленного добавления DPPA (1,1 экв.) к раствору E (1,0 экв.) и Et3N (1,1 экв.) в трет-бутаноле (20,0 объемов) при 75°C. Через 16 часов при 75°C, реакция завершается согласно анализу с помощью ВЭЖХ. Однако, при попытке очистки F с помощью колоночной хроматографии (5-20% EtOAc в гептане на силикагеле), выделяется только незначительное количество F [выход 26%, 99% (AUC)]. Низкий выход приписывают возможной кристаллизации F во время колоночной хроматографии. Это объяснение является разумным, поскольку позднее определяется, что F имеет низкую растворимость в EtOAc и н-гептане.
По этой причине, реакцию повторяют, и при ее завершении, смесь гасят водным раствором 1 M NaOH и перемешивают при температуре окружающей среды в течение 3 часов. После извлечения, продукт выделяют посредством кристаллизации из EtOH (8,0 объемов) и воды (6,0 объемов). Это способ дает F [55%, 88,3% (AUC)] в виде желто-коричневого твердого продукта, содержащего только единственную примесь [изопропилкарбаматное производное F2, 11,7% (AUC)]. Интересно, что побочные продукты DPPA полностью удаляются посредством кристаллизации. Затем осуществляют конечный эксперимент для количественного определения F, присутствующего в сырой реакционной смеси. После извлечения в воду, количественный ЯМР (CDCl3 с использованием диметилфумарата) показывает интенсивность 78% F, присутствующего в сырой смеси. Изопропилкарбаматная примесь F2 также присутствует [6,8% (AUC)]. Хотя эта реакция работает хорошо, образование доставляющего проблемы изопропилкарбаматного побочного продукта F2 устранить нельзя.
Затем осуществляют несколько дополнительных экспериментов с использованием различных нуклеофилов для захвата промежуточного изоцианата, генерируемого с помощью DPPA. Главная цель этих экспериментов заключается в предотвращении образования изопропилкарбаматной примеси F2 посредством исключения использования коммерческого трет-бутанола. Каждую реакцию (Таблица 26) осуществляют посредством воздействия на E (5 г, 1,0 экв.) DPPA (1,1 экв.) в толуоле или THF (20 объемов) в присутствии Et3N (1,1 экв.).
Таблица 26. Эксперименты по гашению изоцианата для получения F с помощью DPPA
* Реакционную смесь нагревают до 75°C в течение 14 часов после гашения HCl
На основе успеха эксперимента по гашению водным раствором 6 M HCl (Таблица 26), эту реакцию повторяют в большем масштабе. E (11,9 г) преобразуют в изоцианат с использованием DPPA в толуоле с Et3N и гасят водным раствором 6 M HCl. В это раз, однако, преобразование изоцианатного промежуточного соединения в соль F-HCl периодически отслеживают согласно анализу с помощью ВЭЖХ вместо перемешивания в течение ночи при повышенных температурах. После нагрева в присутствии водного раствора 6 M HCl при 75°C в течение 2,5 часа, анализ с помощью ВЭЖХ показывает отсутствие остающегося изоцианатного промежуточного соединения, и реакцию считают завершенной. Затем возникает проблема во время извлечения в водном слое и образуется жесткая эмульсия. Ретроспективно, эта эмульсия может появляться в результате отсутствия перемешивания реакционной смеси, погашенной 6 M HCl, в течение ночи при 75°C (как делалось при эксперименте малого масштаба). Возможно, что при перемешивании только в течение 2,5 часа, побочный продукт DPPA (дифенилфосфат) не гидролизуется полностью до степени реакционной смеси, которую нагревают в течение ночи при 75°C. Решение отслеживать расходование изоцианатного промежуточного соединения согласно анализу с помощью ВЭЖХ может приводить к необратимому частичному гидролизу побочного продукта DPPA. В будущем реакция должна отслеживаться также и относительно расходования побочных продуктов DPPA. Кроме того, проблемы с эмульсией приводят к понижению чистоты свободного основания F, которое затем преобразуется в F, по сравнению с нормальной. Затем, это дает в результате низкий выход и низкую чистоту выделенного F из этого способа [80%, 79% (AUC)]. Этот способ было бы разумным повторно исследовать для проверки этих гипотезы
9bviii) Синтез 2'
Преобразование F в 2' сначала осуществляют посредством воздействия на F (5 г) в MeOH (10 объемов) 5% палладия на угле (50% масс смоченного водой катализатора) при давлении водорода 45 фунт/кв. дюйм (2,7 кг/кв. см). После перемешивания в течение 17 часов, реакция завершается согласно анализу с помощью ВЭЖХ, и смесь фильтруют через пад Celite®. Затем фильтрат концентрируют с получением 2' примерно при количественном выходе (4,6 г) в виде желтого масла. Затем реакцию масштабируют до 29 г F. Делаются попытки осуществления реакции в EtOH вместо MeOH из-за проблем с воспламеняемостью в большем масштабе; однако растворимость F в EtOH является плохой. В качестве компромисса, исследуют смешанную систему растворителей из 20% MeOH в EtOH (10 объемов). После перемешивания в течение 16 часов, реакцию завершают. Фильтрат концентрируют до желтого масла (34 г), а затем объединяют с экспериментом на 5 г для хроматографической очистки (40/60 смесь EtOAc/гептан, силикагель). 2' [30,4 г, выход 99%, 97,7% (AUC)] получают в виде желтого масла. Наконец, осуществляют реакцию в масштабе 170 г с использованием MeOH (10 объемов), и реакцию осуществляют подобно первым двум экспериментам. После фильтрования через Celite®, фильтрат концентрируют до желтого масла, которое отверждается при стоянии в течение ночи при температуре окружающей среды. Это дает 2' [154 г, 100%, 98,3% (AUC)], содержащий только единственную примесь [изопропилкарбаматное производное 2' (1,7%)].
9bix). Синтез H
Процедура включает медленное добавление раствора G (1 экв.) в толуоле (3 объема) к двухфазной смеси KOH (5,6 экв.), воды (0,77 объема), толуола (7,2 объема), 1,3-дибромпропана (1,1 экв.) и тетрабутиламмонийбромида (0,1 экв.) при 50-85°C. Во время добавления, реакционная смесь становится очень вязкой, и присутствует значительное количество белого твердого продукта (предположительно, KBr). После завершения реакции, смесь охлаждают до комнатной температуры и разбавляют гептаном (10,4 объема) для преципитации дополнительного твердого продукта. Затем загрузку фильтруют, и фильтрат промывают водой (3 объем, дважды), сушат над MgSO4, фильтруют и концентрируют. Это дает сырой H [выход >90%, типичная чистота 82-91% (AUC)] в виде фиолетового масла. Затем этот материал используют обычным образом без дополнительной очистки на следующей стадии. Главные присутствующие примеси, вероятно, представляют собой олигомерные побочные продукты и соответствующий олефин из нециклизованного промежуточного соединения, которое подвергается воздействию устранения HBr (H2).
Примесь H2
Первая введенная модификация представляет собой замену порошкообразного KOH 50% водным раствором NaOH. Это устраняет экзотермическое растворение KOH и является более простым при работе в большем масштабе. К сожалению, 50% NaOH является неэффективным и достигается только 16% преобразование в H при образовании многочисленных новых примесей. Вторая исследуемая модификация заключается в увеличении разбавления реакционной смеси в попытке сделать не такой густой вязкую суспензию. Когда объемы толуола и воды удваиваются, наблюдается только небольшое изменение вязкости суспензии. Неожиданный результат удвоения количества воды также вызывает понижение температуры дефлегмации до 95°C (обычно она составляет 100-105°C) и преобразование G в H не происходит. Для достижения 100°C, воду отгоняют при атмосферном давлении (ловушка Дина-Старка) до тех пор, пока температура дефлегмации не достигнет 100°C. При 100°C, реакция доходит до завершения через час и дает H со средним выходом и чистотой. На основе этого результата, процедурные условия используются для масштабирования. Результаты для трех загрузок больших масштабов приведены в Таблице 27
Таблица 27. Крупномасштабное преобразование G в H
9bx). Синтез I
Условия, используемые для получения I, являются идентичными условиям, используемым для получения D. Хотя условия с HOAc и H2SO4 работают хорошо, эти условия доставляют проблемы с безопасностью при нагреве смеси до 90°C в большем масштабе. В попытке устранить нагрев до 90°C, оценивают дополнительные условия с использованием TFA и H2SO4 при температуре окружающей среды. Обработка H (масштаб 10 г) TFA (4 объема) и H2SO4 (1 объем) при температуре окружающей среды дает в результате 95% преобразование (согласно ВЭЖХ) в I через 26 часов. Затем реакционную смесь выливают в воду со льдом и экстрагируют DCM. Органический слой промывают насыщенным NaHCO3, сушат, и концентрируют с получением сырого I, содержащего значительное количество остаточного TFA. Поскольку извлечение в водном слое не удаляют эффективно TFA; эти условия дальше не используются.
Приготавливают четыре загрузки промежуточного масштаба с использованием условий с HOAc и H2SO4. Следуя общей процедуре, H нагревают до 90°C в присутствии HOAc (4 объема) и H2SO4 (2 объема) до тех пор, пока не расходуется H (<1% AUC с помощью ВЭЖХ). Затем реакционную смесь охлаждают до температуры окружающей среды и медленно гасят посредством выливания в воду со льдом. После экстракционного извлечения в DCM, сырой I очищают с помощью хроматографии на силикагеле. Результаты приведены в Таблице 28.
Таблица 28. Крупномасштабное получение I
В одном из случаев (2-ой пункт Таблицы 28), сырой I отверждается при стоянии при температуре окружающей среды. Затем этот материал перекристаллизуется из EtOAc. Эту стратегию перекристаллизации также пытаются использовать для сырого I? выделенного в виде масла, но безуспешно. I также частично кристаллизуется при нагрузке на колонку с силикагелем со смесью EtOAc/гептан. Для устранения этой проблемы, является преимущественным предварительное поглощение сырого I на силикагеле с использованием DCM, а затем концентрирование суспензии с силикагелем досуха перед загрузкой в колонку. В качестве альтернативы хроматографии с использованием EtOAc и гептана, оценивается также колонка со слоем MTBE (3-ий пункт Таблицы 28). К сожалению, колонка со слоем MTBE оценивается только один раз и дает низкое извлечение (51%). Вероятно, из-за того, что I кристаллизуется на силикагеле, а затем не растворяется повторно с легкостью.
9bxi). Синтез 2
Синтез 2 осуществляют с использованием Pb(OAc)4. Как правило, реакция завершается через 90 минут при 80-85°C. Когда преобразование I в 2 завершается, реакционную суспензию охлаждают до температуры окружающей среды и обрабатывают твердым Na2CO3 (1 масс эквивалент), а затем MTBE (7,5 объема). После перемешивания в течение 30 минут, твердые продукты (PbCO3) удаляют с помощью фильтрования, и фильтрат промывают водным раствором NaHCO3. После извлечения в водном слое, сушки и концентрирования, сырой 2 очищают посредством повторного суспендирования в 10/90 смеси MTBE/гептан (5 объемов) при температуре окружающей среды. Этот способ, как правило, дает 2 [выход 64-86%, 94-97% (AUC)] в виде беловатого твердого продукта. Единственная значительная примесь, присутствующая на этой стадии, представляет собой изопропилкарбаматное производное 2 (2A).
Примесь 2A
Этот нежелательный побочный продукт подобен химическому механизму для 2' и, как предполагается, представляет собой результат реакции изоцианатного промежуточного соединения с микроскопическим количеством изопропанола, присутствующего в коммерческом трет-бутаноле. Примесь 2A, как правило, присутствует в 3-4% (AUC) в 2 после повторного суспендирования в 10/90 смеси MTBE/гептан. Таблица 29 приводит результаты получения соединения 2 в больших масштабах.
Таблица 29. Крупномасштабное получение 2
Во время конечного эксперимента (Таблица 29, эксперимент 4) появляется новая главная примесь (RRT=0,75). Причина появления этой новой примеси является неясной, поскольку одни и те же навески трет-бутанола и Pb(OAc)4 используются в каждом эксперименте. Исследование предыдущих реакций подтверждает, что эта примесь, как правило, присутствует, но не при уровнях выше 3-5%. К счастью, эту новую примесь (RRT=0,75) можно удалить с помощью колоночной хроматографии (1/99 смесь MeOH/DCM на силикагеле) с получением 2, содержащего 2A (2,8% AUC) в качестве единственной примеси.
В качестве альтернативы условиям с Pb(OAc)4, исследуют как условия перегруппировки Гофмана, так и условия с йодозобензолом in-situ. Используя стандартные условия перегруппировки Гофмана, I (1 экв.) суспендируют в водном растворе NaOH и обрабатывают по каплям бромом (1 экв.). После добавления брома, реакционную смесь нагревают до 60°C, и начальная жидкая суспензия преобразуется в шар из маслянистых твердых продуктов, который сложно перемешивать. Через 2 часа, реакционную смесь анализируют с помощью ВЭЖХ (после гашения образца с помощью HCl), и анализ показывает сложную смесь с множеством пиков. Имеется также значительное количество (25%) присутствующего непрореагировавшего I, и эту реакцию не используют. Оценивают также второй набор условий с использованием йодозобензола, генерируемого in-situ под действием Oxone® на йодобензол. Кинетика этой реакции, как обнаружено, является очень медленной, и образуется множество соединений согласно ВЭЖХ. В результате, эти условия далее не используются.
9bxii). Очистка 2
Затем исследуют несколько стратегий очистки 2 для удаления изопропилкарбаматной примеси (2A). Первая группа экспериментов предназначается для повторного суспендирования сырого 2 [чистота 95%, содержит 4% 2A (AUC)] в смесях ацетонитрила и воды при температуре окружающей среды (Таблица 30). На основе результатов в Таблице 13, смесь из 25-50% воды в ацетонитриле дает наилучший баланс между извлечением и чистотой.
Таблица 30. Результаты повторного суспендирования соединения 2
Следующий эксперимент предназначен для оценки перекристаллизации 2 из ацетонитрила и воды (Таблица 30). В этом исследовании, используют 2 (чистота 97,3%, 2,7% примеси 2A). При этих перекристаллизациях, нет значительного улучшения чистоты по сравнению с результатами повторного суспендирования в Таблице 31.
Таблица 31. Результаты перекристаллизации соединения 2
Затем оценивают вторую систему растворителей для повторного суспендирования с использованием 2-PrOH и гептана. Хотя наблюдается умеренное улучшение чистоты (Таблица 31), извлечение ниже, чем в соответствующих экспериментах с водой и ацетонитрилом в Таблицах 30 и 31.
Таблица 32. Дополнительные попытки повторного суспендирования соединения 2
Поскольку многие примеси (за исключением примеси 2A) являются более полярными, чем соединение 2, колонка со слоем силикагеля может быть использована в качестве способа предварительной очистки. Для осуществления этой колонки со слоем, сырой 2 растворяют в 1/99 смеси MeOH/DCM, а затем нагружают на колонку с силикагелем, набитой с помощью такой же системы растворителей. Затем соединение 2 быстро элюируется (RF=0,9-1,0), оставляя позади более полярные примеси. Затем обогащенные фракции концентрируют досуха и смешивают посредством повторного суспендирования в MTBE (2 объема) и гептане (6 объемов) при температуре окружающей среды с получением однородной навески. С использованием этого способа можно получить 500 г навеску 2. Это дает 2 [97,2% (AUC)], содержащий соединение 2A (2,8% AUC) в качестве единственной присутствующей примеси.
9c). Перенос способа для 2' на производство
На основе разработки способа получения 2' осуществляют получение в целом 2 кг 2'. Имеются некоторые проблемы относительно масштабирования получения B, связанные с извлечением и отгонкой для получения чистого B. Осуществляют переключение с толуола на DMSO, и хотя реакционная смесь становится более однородной, по-прежнему наблюдается образование B2 и необходима колоночная хроматография. Эту процедуру используют для получения 900 г B, который используется в дальнейшем для получения материала для освоения и получения ~500 г API для снабжения исследований токсичности GLP. Осваивание остальных стадий проходит хорошо, и, неожиданно, нет проблем с изопропилкарбаматной примесью (F2), которая затрудняет разработку и которая наблюдается при 500-г синтезе 2. Возможно, крупномасштабный производитель получает свой трет-бутанол из другого источника, который не содержит никакого 2-PrOH, который мог бы взаимодействовать с образованием F2. 900 г синтезированного B дают 477 г 2' с чистотой 99,5% согласно ВЭЖХ.
Идентифицируют коммерческий источник B, а затем купленный там B используют для получения 1,61 кг 2'. Экспериментaльные процедуры для крупномасштабного синтеза 2' можно найти в разделе Примеры.
Пример 10: Эксперименты по очистки 6 и 7 с высоким уровнем Pd
Малое количество 7 свободного основания получают из аликвоты навески 6, генерируемого из 3, полученного с помощью реакции перекрестного связывания. Уровень палладия понижается до 206 м.д. от 281 м.д. после выделения свободного основания.
Оценивают пять недорогих, коммерчески доступных поглотителей и активированный уголь. Для осуществления процесса скрининга используют, по меньшей мере, 4-кратное количество выбранных поглотителей, по сравнению с вычисленным, для увеличения вероятности успеха за короткий период времени. Для сравнения, также исследуют два ускоренных эксперимента с повышенной нагрузкой (20-кратной и 40-кратной).
Таблица 33. Результат обработки 6 поглотителями
(мг)
В типичном эксперименте, поглотитель (>28 мг, превышает потребность по калькуляции больше чем в 4 раза) добавляют к раствору 6 (или 7, 500 мг) в DCM (5 мл). Смесь перемешивают при 35°C в течение 2,5 часов, охлаждают до температуры окружающей среды, фильтруют через 0,45-мкм диск в заранее взвешенный флакон. Затем фильтрат концентрируют, регистрируют извлечение и анализируют уровень палладия (Таблицы 33 и 34).
Таблица 34. Результат обработки 7 (свободного основания) поглотителями
(мг)
Эти данные говорят о том, что поглотители являются более эффективными в случае свободного основания 7 по сравнению с 6. Как и ожидалось, чем большее количество поглотителя используется, тем ниже извлечение субстрата. Наилучший поглотитель представляет собой QuadraSil MP, который является также самым недорогим поглотителем для обработки свободного основания 7.
Пример 11: Скрининг альтернативных окислителей для получения 6
В исходном способе, в качестве окислителя для получения 6 используют воздух. Хотя воздух в качестве окислителя необходим для ароматизации промежуточного соединения 6' до 6, наблюдается также медленное переокисление конечного продукта 6 до N-оксида (M+16) из-за медленной стадии ароматизации. В процессе достижения завершения реакции, как отмечено, образуется N-оксид (M+16), и его содержание в реакционной смеси увеличивается. Эта конкретная примесь не быть удалена ни на этой стадии, ни дальше, и она составляет главную проблему в этом способе из-за благоприятных для способа процедур кристаллизации/перекристаллизации. Для получения большего контроля на стадии окисления, на этой стадии, рассматриваются альтернативы окислению на воздухе. Целью является селективная ароматизация циклизированного промежуточного соединения 6' до 6, но не переокисление 6 до N-оксида (M+16). Используют различные окислители на основе металлов и неметаллов для катализа/ускорения окисления 6' до 6, включая ацетат меди (Cu(OAc)2⋅H2O), перборат натрия (NaBO3⋅4H2O), хлорид железа (III) (FeCl3⋅6H2O), палладий на угле (10% Pd/C). Реакции осуществляют в 4-драмовых флаконах с закрытыми крышками при комнатной температуре. Для перемешивания реакционной смеси используют бруски магнитной мешалки. Не используют ни внешнего воздуха, ни атмосферы азота. Масштаб реакции выбирают при 100 мг по отношению к 4. Результаты из этого исследования подробно изложены в Таблице 1.
Таблица 35. Получение 6 с использованием внешних окислителей
(% AUC)
(% AUC)
Условия реакции: 1 экв. 4, 1,05 экв. 5, 10 объемов раствора AcOH/MeOH (отношение 9:1), перемешивание при комнатной температуре с различными окислителями (1 экв.).
Реакции, как правило, завершаются через 12-15 часов (по сравнению с 35-40 час при условиях окисления на воздухе). Продукт 6 (чистота 97-98% AUC) преципитирует из реакционной смеси при добавлении воды (10 объемов). В выделенном продукте наблюдают N-оксид (M+16), 0,1-0,5% AUC. Для дальнейшей оптимизации способа, теперь объединяют Стадию 2 и Стадию 3 способа (Стадия 2'). После завершения преобразования 3 в 4, в полученном растворе 4 в THF, растворитель заменяют на MeOH для аккомодации к оптимизированным условиям для преобразования 4 в 6 с перборатом натрия в качестве окислителя. Настоящий способ подробно описывается ниже:
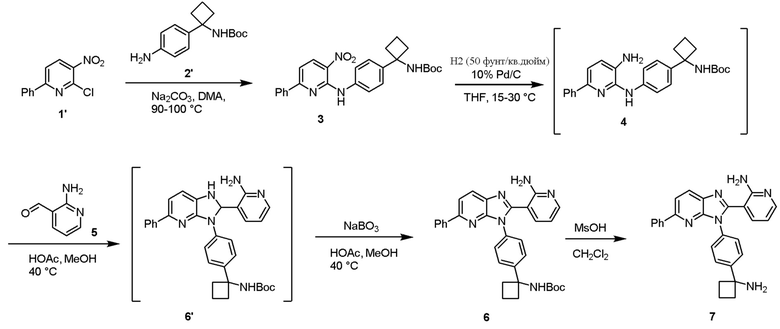
Стадия 1, Синтез 3:
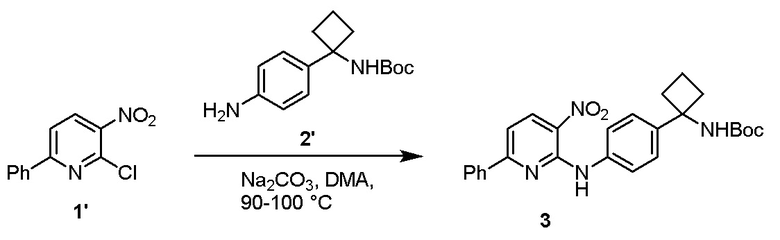
22-л реакционную колбу устанавливают в нагревательный кожух и продувают азотом перед загрузкой 1' (1,20 кг), 2' (1,48 кг), карбонатом натрия (1,09 кг) и диметилацетамидом (7,3 кг). Реакционную смесь нагревают приблизительно до 91°C, и дают возможность для ее перемешивания при этой температуре в атмосфере азота. Через 20 часов отбирают образец смеси для анализа с помощью ВЭЖХ, при этом результат показывает приблизительно 2% AUC (по отношению к продукту) остающегося 1'. Анализ образца через 24 часа показывает приблизительно 1,5% AUC остающегося 1' по отношению к продукту. Нагрев выключают приблизительно после 26 часов нагрева, и реакционной смесь дают возможность для охлаждения в течение ночи (анализ с помощью ВЭЖХ: 1,1% AUC 1'). После охлаждения, реакционную смесь переносят в 100-л реактор. Реакционную колбу промывают 20,6 кг 2-MeTHF в 100-л реакторе, и загрузку промывают 5% водным раствором хлорида натрия (22,1 кг). Слои разделяют, и водный слой экстрагируют обратно с помощью 14,7 кг 2-MeTHF. После разделения слоев имеется значительное количество соли/карбоната натрия, остающегося в реакторе. Водный слой загружают в реактор и нагревают до 30°C. Загружают дополнительно 5,0 кг воды для растворения большей части соли (мутный раствор), и водный слой экстрагируют 15,0 кг 2-MeTHF. При хранении в течение выходных из первого и второго органических слоев кристаллизуется значительное количество продукта. Органические слои загружают в реактор, и оставшийся твердый продукт растворяют в 5,0 кг 2-MeTHF и объединяют с органическим слоем в реакторе. Объединенные органические слои промывают два раза 5% водным раствором хлорида натрия (каждая промывка по 12,0 кг). Анализ органического слоя с помощью 1H-ЯМР показывает 0,1 процента молярного остающегося диметилацетамида. После отгонки органического слоя до 18 л, анализ образца показывает, что уровень влажности составляет 0,15%. Загрузку разбавляют 2-MeTHF (25,5 кг) и охлаждают до 28°C перед окончательным фильтрованием через 0,22-микронный фильтр. 100-л реактор промывают окончательно отфильтрованным 2-MeTHF перед повторной загрузкой отфильтрованной загрузки. Затем загрузку отгоняют в вакууме до 10,8 л и нагревают до 72°C. Гептан (7,4 кг) загружают в течение 75 минут, поддерживая температуру в пределах между 66 и 72°C. После перемешивания при 66°C в течение 16 минут загрузку охлаждают до 25°C в течение 2 часов, 45 мин. Загрузку перемешивают при этой температуре в течение 15,5 часа до отбора образца. Образец фильтруют, и фильтрат анализируют с помощью ВЭЖХ, которая показывает 4 мг/мл продукта в фильтрате. Загрузку фильтруют, промывают дважды 1:3 (объем/объем) смесью 2-MeTHF в гептане (каждая промывка 4,4 кг) и один раз промывают 4,1 кг гептана. Продукт сушат на фильтре в атмосфере азота в течение 1 часа, 18 мин и переносят на сушильные поддоны (влажная масса 2,31 кг). После сушки в течение ночи при 25-30°C масса остается постоянной, и продукт упаковывают с получением 2,17 кг 3 (выход 92%, 99,8% AUC).
Стадия 2', Синтез 6:
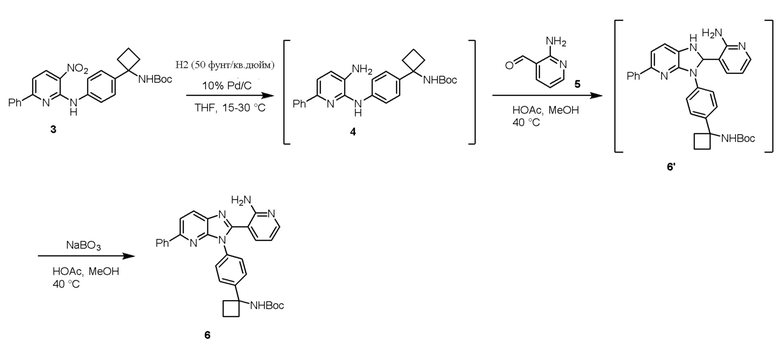 После осуществления проверки давления и создания инертной атмосферы азота, в 10-галлонный реактор загружают 1,89 кг 3, а затем 95 г 10% Pd/C (содержание влажного 50%). Затем реактор продувают три раза азотом перед загрузкой 10,3 кг тетрагидрофурана (THF). Реактор герметизируют и откачивают до -20 дюймов (51 см) рт. ст. перед повышением давления до 30 фунт/кв. дюйм (1,9 кг/кв. см) с помощью водорода. Начальная температура реакции составляет 15°C, и загрузка нагревается самопроизвольно до 30°C в ходе реакции. По прохождении времени реакции равного 3,75 часа отбирают образец загрузки для анализа с помощью ВЭЖХ (во время анализа гидрирование при 30 фунт/кв. дюйм (1,9 кг/кв. см) продолжают). Анализ в это время показывает 0,43% AUC остающихся исходных материалов (3), и через 5 часов общего времени реакции реактор разгерметизируют и продувают азотом. Анализ после фильтрования катализатора показывает 0,23% AUC остающегося исходного материала (3). Реактор промывают 8,0 кг THF, и это промывка также используется для промывки фильтра в загрузку. Загрузку загружают в 100-л реактор с кожухом и используют 1,6 кг THF для промывки вещества в 100-л реакторе с кожухом. Загрузку (~34 л) отгоняют в вакууме при 25°C до 9 л перед загрузкой 6,4 кг метанола. Вакуумную отгонку продолжают до 8 л, загружают 6,6 кг метанола и продолжают отгонку до 8 л. Анализ с помощью 1H-ЯМР показывает, что остается 4,5% моль THF по отношению к метанолу, поэтому осуществляют два дополнительных загрузки (6,6 кг и 6,4 кг) с метанолом, после чего % моль THF по отношению к метанолу составляет 0,1 процента молярного. В загрузку загружают 527 г 5 и 632 г натрия пербората тетрагидрата. Загрузку нагревают до 40°C и перемешивают в течение 2 часов перед отбором образцов для первого IPC. Анализ с помощью ВЭЖХ показывает, что 5,6% AUC 4 остаются непрореагировавшими, перемешивание продолжают при 40°C в течение ночи. Анализ образцов, отобранных через 19 часов и 22 часов, показывает отсутствие изменения в пике (1,6% AUC 6') со временем удерживания сходным с промежуточным соединением 6'. Реакцию гасят водой (29,8 кг) и перемешивают при 35-40°C в течение 1 часа. Загрузку охлаждают в течение 2 часов до 24°C и перемешивают в течение ночи (17 часов) при 15-25°C. Продукт фильтруют, промывают два раза водой (каждая промывка по 13,6 кг), а затем гептаном (9,3 кг). Сушка сырого промежуточного соединения до постоянной массы при 45°C требует 47 часов. Анализ промежуточного соединения показывает, что чистота составляет 97,2% AUC. Этот материал (1,84 кг) загружают в 100-л реактор с кожухом, вместе с изопропилацетатом (IPAc, 12,8 кг) в атмосфере азота. Смесь нагревают до 70°C (необходимо 1 час 12 мин), перемешивают в течение 1 час, а затем охлаждают в течение 10 часов до 20°C. Суспензию перемешивают при 20°C в течение 54 часов перед фильтрованием. Продукт промывают 1,6 кг IPAc, затем 50% (объем/объем) смесью IPAc/гептан (1,8 л). Продукт сушат при 40-45°C в течение 18 часов с получением 1,33 кг 6 (выход 61%, 99,3% AUC)
После осуществления проверки давления и создания инертной атмосферы азота, в 10-галлонный реактор загружают 1,89 кг 3, а затем 95 г 10% Pd/C (содержание влажного 50%). Затем реактор продувают три раза азотом перед загрузкой 10,3 кг тетрагидрофурана (THF). Реактор герметизируют и откачивают до -20 дюймов (51 см) рт. ст. перед повышением давления до 30 фунт/кв. дюйм (1,9 кг/кв. см) с помощью водорода. Начальная температура реакции составляет 15°C, и загрузка нагревается самопроизвольно до 30°C в ходе реакции. По прохождении времени реакции равного 3,75 часа отбирают образец загрузки для анализа с помощью ВЭЖХ (во время анализа гидрирование при 30 фунт/кв. дюйм (1,9 кг/кв. см) продолжают). Анализ в это время показывает 0,43% AUC остающихся исходных материалов (3), и через 5 часов общего времени реакции реактор разгерметизируют и продувают азотом. Анализ после фильтрования катализатора показывает 0,23% AUC остающегося исходного материала (3). Реактор промывают 8,0 кг THF, и это промывка также используется для промывки фильтра в загрузку. Загрузку загружают в 100-л реактор с кожухом и используют 1,6 кг THF для промывки вещества в 100-л реакторе с кожухом. Загрузку (~34 л) отгоняют в вакууме при 25°C до 9 л перед загрузкой 6,4 кг метанола. Вакуумную отгонку продолжают до 8 л, загружают 6,6 кг метанола и продолжают отгонку до 8 л. Анализ с помощью 1H-ЯМР показывает, что остается 4,5% моль THF по отношению к метанолу, поэтому осуществляют два дополнительных загрузки (6,6 кг и 6,4 кг) с метанолом, после чего % моль THF по отношению к метанолу составляет 0,1 процента молярного. В загрузку загружают 527 г 5 и 632 г натрия пербората тетрагидрата. Загрузку нагревают до 40°C и перемешивают в течение 2 часов перед отбором образцов для первого IPC. Анализ с помощью ВЭЖХ показывает, что 5,6% AUC 4 остаются непрореагировавшими, перемешивание продолжают при 40°C в течение ночи. Анализ образцов, отобранных через 19 часов и 22 часов, показывает отсутствие изменения в пике (1,6% AUC 6') со временем удерживания сходным с промежуточным соединением 6'. Реакцию гасят водой (29,8 кг) и перемешивают при 35-40°C в течение 1 часа. Загрузку охлаждают в течение 2 часов до 24°C и перемешивают в течение ночи (17 часов) при 15-25°C. Продукт фильтруют, промывают два раза водой (каждая промывка по 13,6 кг), а затем гептаном (9,3 кг). Сушка сырого промежуточного соединения до постоянной массы при 45°C требует 47 часов. Анализ промежуточного соединения показывает, что чистота составляет 97,2% AUC. Этот материал (1,84 кг) загружают в 100-л реактор с кожухом, вместе с изопропилацетатом (IPAc, 12,8 кг) в атмосфере азота. Смесь нагревают до 70°C (необходимо 1 час 12 мин), перемешивают в течение 1 час, а затем охлаждают в течение 10 часов до 20°C. Суспензию перемешивают при 20°C в течение 54 часов перед фильтрованием. Продукт промывают 1,6 кг IPAc, затем 50% (объем/объем) смесью IPAc/гептан (1,8 л). Продукт сушат при 40-45°C в течение 18 часов с получением 1,33 кг 6 (выход 61%, 99,3% AUC)
Стадия 3', Синтез 7:
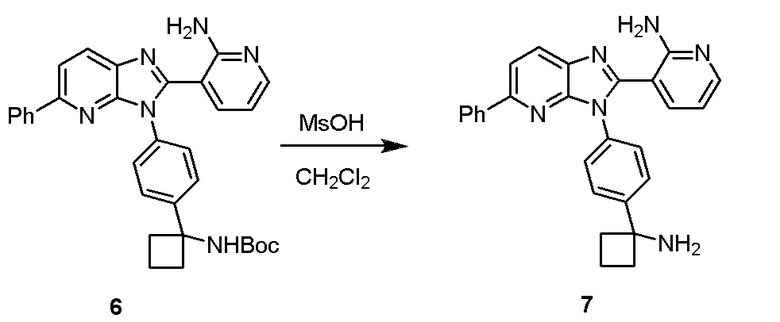
Промежуточное соединение с промежуточной стадии (6, 1,33 кг) загружают в 100-л реактор с кожухом в атмосфере азота с последующим добавлением дихлорметана (18,6 кг). К этому раствору при 20°C, добавляют метансульфоновую кислоту (1,27 кг) в течение 34 минут, в результате чего происходит увеличение температуры до 24°C. Смесь перемешивают при 20-23°C и отслеживают ее с помощью ВЭЖХ. Анализ образца через 4,5 часа показывает 0,3% AUC исходного материала. Воду (1,4 кг) загружают в реакционную смесь, которую перемешивают при 20°C в течение ночи. Загружают дополнительный дихлорметан (9,1 кг) из-за преципитации продукта перед загрузкой 6 н гидроксида натрия (3,0 кг) для доведения pH до 13. После перемешивания в течение 15 минут смесь оседает, и нижний органический слой откачивают. Водный слой экстрагируют дихлорметаном (15,0 кг). Объединенные органические слои промывают водой (8,0 кг). Анализ органического слоя по Карлу Фишеру показывает, что содержание воды составляет влажность 0,2%, так что дополнительной сушки с помощью сульфата натрия не требуется. Quadrasil MP (191 г) загружают в органический слой в 100-л реактор с кожухом, который нагревают до 30°C, и перемешивают при этой температуре в течение 15,5 часа. Поглотитель фильтруют, промывают дважды дихлорметаном (2 × 1,9 кг) и возвращают в очищенный 100-л реактор. Загрузку отгоняют в вакууме приблизительно до 4 л перед загрузкой изопропилацетата (8,6 кг), и продолжают вакуумную отгонку приблизительно до 5 л. После доведения объема до желаемого уровня (~10 л) с помощью изопропилацетата, отбирают образец смеси для 1H-ЯМР. Уровень дихлорметана, как определено с помощью 1H-ЯМР, составляет 2,3 процента молярных. Загружают изопропилацетат (4,7 кг), и вакуумную отгонку продолжают до конечного объема 9 л. Анализ с помощью 1H-ЯМР показывает 0,5 процента молярного остающегося дихлорметана (согласно спецификации, он <1%). Загружают изопропилацетат (1,6 кг), и смесь перемешивают при 20-25°C в течение 16 часов. Затем смесь фильтруют, и твердый продукт промывают на фильтре два раза изопропилацетатом (2,3 кг и 2,5 кг). Твердый продукт сушат при 42°C в течение 1 дня с получением 805 г 7 (выход 70%, 99,5% AUC).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ИМИДАЗО-ПИРИДИНА, ОБЛАДАЮЩИЕ СРОДСТВОМ К РЕЦЕПТОРУ МЕЛАНОКОРТИНА | 2004 |
|
RU2358974C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ НЕКОТОРЫХ 2-(ПИРИДИН-3-ИЛ)ТИАЗОЛОВ | 2013 |
|
RU2647851C2 |
| СПОСОБ ПРИГОТОВЛЕНИЯ 1-(4-МЕТАНСУЛЬФОНИЛ-2-ТРИФТОРМЕТИЛБЕНЗИЛ)-2-МЕТИЛ-1H-ПИРРОЛО[2,3-B]ПИРИДИН-3-ИЛ-УКСУСНОЙ КИСЛОТЫ | 2018 |
|
RU2756507C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ОПРЕДЕЛЕННЫХ 2-(ПИРИДИН-3-ИЛ)ТИАЗОЛОВ | 2013 |
|
RU2647853C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ 2-АМИНОТИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2008 |
|
RU2456285C2 |
| ПОЛИМОРФНАЯ ФОРМА СОЕДИНЕНИЯ 2-АМИНОТИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2012 |
|
RU2491286C1 |
| ИНГИБИТОРЫ ТРАНСГЛУТАМИНАЗЫ 2 (TG2) | 2019 |
|
RU2781370C2 |
| СПОСОБ ПОЛУЧЕНИЯ НЕКОТОРЫХ 2-(ПИРИДИН-3-ИЛ)ТИАЗОЛОВ | 2013 |
|
RU2649000C2 |
| КОМПОЗИЦИИ И СПОСОБЫ, КОТОРЫЕ МОЖНО ИСПОЛЬЗОВАТЬ ДЛЯ ЛЕЧЕНИЯ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2015 |
|
RU2711500C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРА АКТИВИН-РЕЦЕПТОРОПОДОБНОЙ КИНАЗЫ | 2020 |
|
RU2826600C1 |
Изобретение описывает способ получения 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина, включающий стадию 3 взаимодействия трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата с 2-аминоникотинальдегидом в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил(1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)-циклобутил)карбамата. Также описывается вариант способа получения 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина. Технический результат указанных способов заключается в том, что данные способы являются простыми и обеспечивают безопасность ведения процесса. 2 н. и 28 з.п. ф-лы, 35 табл., 11 пр.
1. Способ получения 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина, включающий:
стадию 3 взаимодействия трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата с 2-аминоникотинальдегидом в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил(1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)-циклобутил)карбамата.
2. Способ по п.1, включающий:
стадию 2 обработки трет-бутил(1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)-циклобутил)карбамата и
стадию 3 взаимодействия трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата с 2-аминоникотинальдегидом в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил(1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)-циклобутил)карбамата.
3. Способ по п.2, включающий:
стадию 1 взаимодействия 2-хлор-3-нитро-6-фенилпиридина с трет-бутил(1-(4-аминофенил)циклобутил)карбаматом в присутствии основания в полярном апротонном растворителе с образованием трет-бутил(1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)-циклобутил)карбамата;
стадию 2 обработки трет-бутил(1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)-циклобутил)карбамата и
стадию 3 взаимодействия трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата с 2-аминоникотинальдегидом в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил(1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)-циклобутил)карбамата.
4. Способ по п.2, включающий:
стадию 1a связывания 3-нитро-6-фенилпиридин-2-амина с трет-бутил(1-(4-бромфенил)циклобутил)карбаматом в присутствии палладиевого катализатора и фосфорного лиганда в полярном апротонном растворителе с образованием трет-бутил(1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата;
стадию 2 обработки трет-бутил(1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)-циклобутил)карбамата и
стадию 3 взаимодействия трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата с 2-аминоникотинальдегидом в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил(1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)-циклобутил)карбамата.
5. Способ по п.3, включающий:
стадию 1 взаимодействия 2-хлор-3-нитро-6-фенилпиридина с трет-бутил(1-(4-аминофенил)циклобутил)карбаматом в присутствии основания в полярном апротонном растворителе с образованием трет-бутил(1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)-циклобутил)карбамата;
стадию 2 обработки трет-бутил (1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)-циклобутил)карбамата;
стадию 3 взаимодействия трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата с 2-аминоникотинальдегидом в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил(1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)-циклобутил)карбамата и
стадию 4 обработки трет-бутил(1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамата кислотой в полярном апротонном растворителе с образованием 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина.
6. Способ по п.4, включающий:
стадию 1a связывания 3-нитро-6-фенилпиридин-2-амина с трет-бутил(1-(4-бромфенил)циклобутил)карбаматом в присутствии палладиевого катализатора и фосфорного лиганда в полярном апротонном растворителе с образованием трет-бутил(1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата;
стадию 2 обработки трет-бутил(1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)-циклобутил)карбамата;
стадию 3 взаимодействия трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата с 2-аминоникотинальдегидом в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил(1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)-циклобутил)карбамата и
стадию 4 обработки трет-бутил(1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамата кислотой в полярном апротонном растворителе с образованием 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина.
7. Способ получения 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина, включающий:
стадию 2' обработки трет-бутил(1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата, замены полярного апротонного растворителя полярным протонным растворителем и взаимодействия трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)-циклобутил)карбамата с 2-аминоникотинальдегидом в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил(1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамата.
8. Способ по п.7, включающий:
стадию 2' обработки трет-бутил(1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата, замены полярного апротонного растворителя полярным протонным растворителем и взаимодействия трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)-циклобутил)карбамата с 2-аминоникотинальдегидом в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил(1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамата и
стадию 3' обработки трет-бутил(1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)-карбамата кислотой в полярном апротонном растворителе с образованием 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина.
9. Способ по п.8, включающий:
стадию 1 взаимодействия 2-хлор-3-нитро-6-фенилпиридина с трет-бутил(1-(4-аминофенил)циклобутил)карбаматом в присутствии основания в полярном апротонном растворителе с образованием трет-бутил(1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)-карбамата;
стадию 2' обработки трет-бутил(1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата, замены полярного апротонного растворителя полярным протонным растворителем и взаимодействия трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)-циклобутил)карбамата с 2-аминоникотинальдегидом в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил(1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамата и
стадию 3' обработки трет-бутил(1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)-карбамата кислотой в полярном апротонном растворителе с образованием 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина.
10. Способ по п.8, включающий:
стадию 1a связывания 3-нитро-6-фенилпиридин-2-амин с трет-бутил(1-(4-бромфенил)циклобутил)карбаматом в присутствии палладиевого катализатора и фосфорного лиганда в полярном апротонном растворителе с образованием трет-бутил(1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата;
стадию 2' обработки трет-бутил(1-(4-((3-нитро-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата с помощью восстанавливающего агента в полярном апротонном растворителе с образованием трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)циклобутил)карбамата, замены полярного апротонного растворителя полярным протонным растворителем и взаимодействия трет-бутил(1-(4-((3-амино-6-фенилпиридин-2-ил)амино)фенил)-циклобутил)карбамата с 2-аминоникотинальдегидом в присутствии окислителя и кислоты в полярном протонном растворителе с образованием трет-бутил(1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамата и
стадию 3' обработки трет-бутил(1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)-карбамата кислотой в полярном апротонном растворителе с образованием 3-(3-(4-(1-аминоциклобутил)фенил)-5-фенил-3H-имидазо[4,5-b]пиридин-2-ил)пиридин-2-амина.
11. Способ по п.1 или 7, в котором полярный протонный растворитель представляет собой метанол.
12. Способ по п.1 или 7, в котором кислота представляет собой уксусную кислоту.
13. Способ по п.1 или 7, в котором отношение кислоты к растворителю составляет примерно 9:1.
14. Способ по п.1 или 7, в котором окислитель представляет собой воздух.
15. Способ по п.1 или 7, в котором окислитель представляет собой соль на основе металла или не металла или катализатор.
16. Способ по п.15, в котором окислитель выбирается из группы, состоящей из ацетата металла, пербората металла, хлорида металла, катализатора на основе палладия и их гидратов.
17. Способ по п.16, в котором окислитель выбирается из группы, состоящей из Cu(OAc)2⋅H2O, NaBO3⋅4H2O, FeCl3⋅6H2O и 10% Pd/C и их гидратов.
18. Способ по п.17, в котором окислитель представляет собой NaBO3⋅H2O.
19. Способ по п.2, в котором на стадии 2 полярный апротонный растворитель представляет собой тетрагидрофуран или 2-метилтетрагидрофуран.
20. Способ по п.8, в котором на стадии 2' полярный апротонный растворитель представляет собой тетрагидрофуран или 2-метилтетрагидрофуран.
21. Способ по п.3 или 9, в котором на стадии 1 полярный апротонный растворитель представляет собой диметилацетамид.
22. Способ по п.3 или 9, в котором на стадии 1 основание представляет собой Na2CO3.
23. Способ по п.4 или 10, в котором на стадии 1a фосфорный лиганд представляет собой 4,5-бис(дифенилфосфино)-9,9-диметилксантен.
24. Способ по п.4 или 10, в котором на стадии 1a полярный апротонный растворитель представляет собой тетрагидрофуран.
25. Способ по п.5, в котором на стадии 4 полярный апротонный растворитель представляет собой дихлорметан.
26. Способ по п.5, в котором на стадии 4 кислота представляет собой метансульфоновую кислоту.
27. Способ по п.5, в котором на стадии 4 отношение кислоты к трет-бутил(1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамату составляет примерно 5:1.
28. Способ по п.9, в котором на стадии 3' полярный апротонный растворитель представляет собой дихлорметан.
29. Способ по п.9, в котором на стадии 3' кислота представляет собой метансульфоновую кислоту.
30. Способ по п.9, в котором на стадии 3' отношение кислоты к трет-бутил(1-(4-(2-(2-аминопиридин-3-ил)-5-фенил-3H-имидазо[4,5-b]пиридин-3-ил)фенил)циклобутил)карбамату составляет примерно 5:1.
| US 20110172203 A1, 14.07.2011 | |||
| WO 2007111904 A3, 04.10.2007 | |||
| US 20020151549 A1, 17.10.2002 | |||
| US 20060035921 A1, 16.02.2006 | |||
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ НА ИХ ОСНОВЕ (ВАРИАНТЫ), СПОСОБ ИНГИБИРОВАНИЯ СЕКРЕЦИИ ЖЕЛУДОЧНОЙ КИСЛОТЫ, СПОСОБ ЛЕЧЕНИЯ ЖЕЛУДОЧНО-КИШЕЧНЫХ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И СПОСОБ ЛЕЧЕНИЯ СОСТОЯНИЙ, В КОТОРЫХ ВОВЛЕЧЕНО ИНФИЦИРОВАНИЕ H.PYLORI | 1999 |
|
RU2238271C2 |

