ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет и преимущество предварительной заявки США 61/655086, поданной 4 июня 2012. Полное содержание этой предварительной заявки включено в настоящую заявку посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Изобретение, раскрытое в данном документе, относится к области способов получения определенных 2-(пиридин-3-ил)тиазолов в качестве промежуточных соединений для синтеза пестицидных тиазоламидов.
УРОВЕНЬ ТЕХНИКИ
Контроль численности сельскохозяйственных вредителей важен для современного сельского хозяйства, хранения продовольствия и гигиены. Существует более десяти тысяч видов сельскохозяйственных вредителей, которые вызывают потери в сельском хозяйстве. Общемировые потери в сельском хозяйстве ежегодно составляют миллиарды долларов США. Известно также, что сельскохозяйственные вредители, такие как термиты, наносят ущерб всем видам частных и государственных структур, приводящий к ежегодным потерям в миллиарды долларов США. Сельскохозяйственные вредители также поедают и загрязняют хранящееся продовольствие, приводя к ежегодным потерям в миллиарды долларов США, а также к утрате необходимого для населения продовольствия.
У некоторых сельскохозяйственных вредителей существует или вырабатывается устойчивость к используемым в настоящее время пестицидам. Сотни видов сельскохозяйственных вредителей устойчивы к одному или более пестицидам. Следовательно, существует постоянная потребность в новых пестицидах и способах получения таких пестицидов.
В WO 2010/129497 (раскрытие которой полностью включено в настоящее описание) описаны некоторые пестициды. Однако способы получения таких пестицидов могут быть дорогостоящими и неэффективными. Соответственно, существует потребность в способах эффективного получения таких пестицидов.
ОПРЕДЕЛЕНИЯ
Приведенные в определениях примеры в целом не исчерпывающие и не могут истолковываться как ограничивающие раскрытое в данном документе изобретение. Понятно, что заместитель должен соответствовать требованиям правил образования химических связей и стерического ограничения на совместимость в отношении конкретной молекулы, к которой он присоединяется.
"Алкенил" означает ациклический, ненасыщенный (по меньшей мере одна углерод-углеродная двойная связь), разветвленный или неразветвленный заместитель, состоящий из углерода и водорода, например винил, аллил, бутенил, пентенил, гексенил, гептенил, октенил, ноненил и деценил.
"Алкенилокси" означает алкенил, дополнительно включающий простую углерод-кислородную связь, например аллилокси, бутенилокси, пентенилокси, гексенилокси, гептенилокси, октенилокси, ноненилокси и деценилокси.
"Алкокси" означает алкил, дополнительно включающий углерод-кислородную простую связь, например метокси, этокси, пропокси, изопропокси, 1-бутокси, 2-бутокси, изобутокси, трет-бутокси, пентокси, 2-метилбутокси, 1,1-диметилпропокси, гексокси, гептокси, октокси, нонокси и декокси.
"Алкил" означает ациклический, насыщенный, разветвленный или неразветвленный заместитель, состоящий из углерода и водорода, например метил, этил, пропил, изопропил, 1-бутил, 2-бутил, изобутил, трет-бутил, пентил, 2-метилбутил, 1,1-диметилпропил, гексил, гептил, октил, нонил и децил.
"Алкинил" означает ациклический, ненасыщенный (по меньшей мере одна углерод-углеродная тройная связь и любые двойные связи), разветвленный или неразветвленный заместитель, состоящий из углерода и водорода, например этинил, пропаргил, бутинил, пентинил, гексинил, гептинил, октинил, нонинил и децинил.
"Алкинилокси" означает алкинил, дополнительно включающий углерод-кислородную простую связь, например пентинилокси, гексинилокси, гептинилокси, октинилокси, нонинилокси и децинилокси.
"Арил" означает циклический, ароматический заместитель, состоящий из водорода и углерода, например фенил, нафтил и бифенил.
"Циклоалкенил" означает моноциклический или полициклический, ненасыщенный (по меньшей мере одна углерод-углеродная двойная связь) заместитель, состоящий из углерода и водорода, например циклобутенил, циклопентенил, циклогексенил, циклогептенил, циклооктенил, циклодеценил, норборненил, бицикло[2.2.2]октенил, тетрагидронафтил, гексагидронафтил и октагидронафтил.
"Циклоалкенилокси" означает циклоалкенил, дополнительно включающий углерод-кислородную простую связь, например циклобутенилокси, циклопентенилокси, циклогексенилокси, циклогептенилокси, циклооктенилокси, циклодеценилокси, норборненилокси и бицикло[2.2.2]октенилокси.
"Циклоалкил" означает моноциклический или полициклический, насыщенный заместитель, состоящий из углерода и водорода, например циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклодецил, норборнил, бицикло[2.2.2]октил и декагидронафтил.
"Циклоалкокси" означает циклоалкил, дополнительно включающий углерод-кислородную простую связь, например циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси, циклогептилокси, циклооктилокси, циклодецилокси, норборнилокси и бицикло[2.2.2]октилокси.
"Циклогалогеналкил" означает моноциклический или полициклический, насыщенный заместитель, состоящий из углерода, галогена и водорода, например 1-хлорциклопропил, 1-хлорциклобутил и 1-дихлорциклопентил.
"Галоген" означает фтор, хлор, бром и йод.
"Галогеналкил" означает алкил, дополнительно включающий от одного до максимально возможного числа одинаковых или различных галогенов, например фторметил, дифторметил, трифторметил, 1-фторэтил, 2-фторэтил, 2,2,2-трифторэтил, хлорметил, трихлорметил и 1,1,2,2-тетрафторэтил.
"Гетероциклил" означает циклический заместитель, который может быть полностью насыщенным, частично ненасыщенным или полностью ненасыщенным, где циклическая структура содержит по меньшей мере один углерод и по меньшей мере один гетероатом, где указанный гетероатом представляет собой азот, серу или кислород, например бензофуранил, бензоизотиазолил, бензоизоксазолил, бензоксазолил, бензотиенил, бензотиазолил, циннолинил, фуранил, индазолил, индолил, имидазолил, изоиндолил, изохинолинил, изотиазолил, изоксазолил, 1,3,4-оксадиазолил, оксазолинил, оксазолил, фталазинил, пиразинил, пиразолинил, пиразолил, пиридазинил, пиридил, пиримидинил, пирролил, хиназолинил, хинолинил, хиноксалинил, 1,2,3,4-тетразолил, тиазолинил, тиазолил, тиенил, 1,2,3-триазинил, 1,2,4-триазинил, 1,3,5-триазинил, 1,2,3-триазолил и 1,2,4-триазолил.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
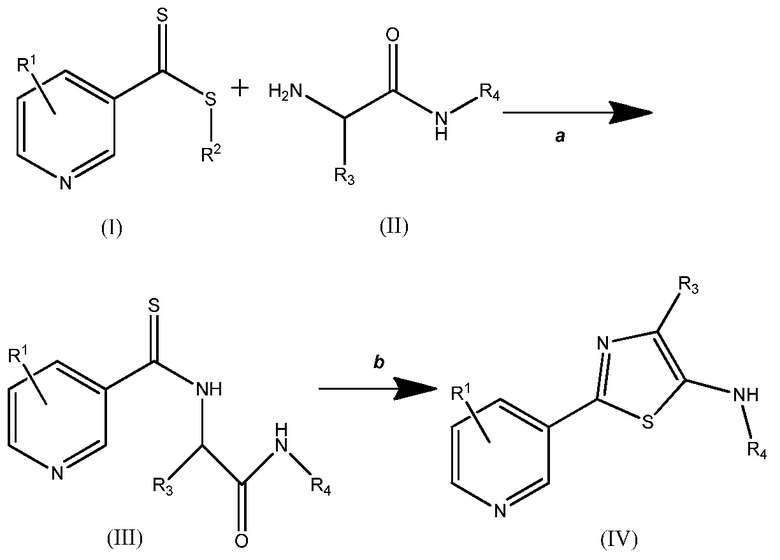
Вариант осуществления настоящего изобретения показан на схеме один
Схема один
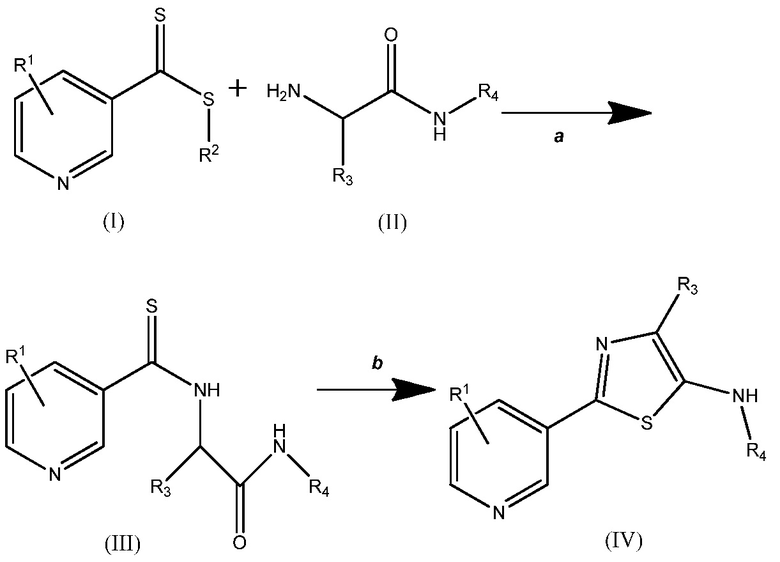
где
(A) каждый R1 независимо выбирают из H, F, Cl, Br, I, CN, NO2 и замещенного или незамещенного (C1-C6)алкила, где каждый замещенный R1 содержит один или более заместителей, независимо выбираемых из F, Cl, Br, I, CN, NO2, (C1-C6)алкила и (C1-C6)галогеналкила;
(B) R2 выбирают из замещенного или незамещенного (C1-C6)алкила, замещенного или незамещенного (C2-C6)алкенила, замещенного или незамещенного (C1-C6)алкокси, замещенного или незамещенного (C2-C6)алкенилокси, замещенного или незамещенного (C3-C10)циклоалкила, замещенного или незамещенного (C3-C10)циклоалкенила, замещенного или незамещенного (C6-C20)арила, замещенного или незамещенного (C1-C6)алкил)(C6-C20)арила и замещенного или незамещенного (C1-C20)гетероциклила, где каждый замещенный R2 содержит один или более заместителей, независимо выбираемых из F, Cl, Br, I, CN, NO2, (C1-C6)алкила, (C2-C6)алкенила, (C1-C6)галогеналкила, (C2-C6)галогеналкенила, (C1-C6)галогеналкилокси, (C2-C6)галогеналкенилокси, (C3-C10)циклоалкила, (C3-C10)циклоалкенила, (C3-C10)галогенциклоалкила, (C3-C10)галогенциклоалкенила, (C6-C20)арила и (C1-C20)гетероциклила;
(C) R3 выбирают из H, замещенного или незамещенного (C1-C6)алкила, замещенного или незамещенного (C3-C10)циклоалкила, замещенного или незамещенного (C1-C6)алкил(C3-C10)циклоалкила, замещенного или незамещенного (C6-C20)арила и замещенного или незамещенного (C1-C6)алкил(C6-C20)арила, где каждый замещенный R3 содержит один или более заместителей, независимо выбираемых из F, Cl, Br и I; и
(D) R4 выбирают из H, замещенного или незамещенного (C1-C6)алкила, замещенного или незамещенного (C3-C10)циклоалкила, замещенного или незамещенного (C1-C6)алкил(C3-C10)циклоалкила, замещенного или незамещенного (C6-C20)арила, замещенного или незамещенного (C1-C6)алкил(C6-C20)арила, замещенного или незамещенного (C1-C6)алкил(C2-C6)алкенила и замещенного или незамещенного (C1-C6)алкил(C2-C6)алкинила, где каждый указанный R4, который является замещенным, содержит один или более заместителей, выбираемых из F, Cl, Br I, CN, NO2, (C1-C6)алкила, (C1-C6)галогеналкила, (C1-C6)алкилокси, (C1-C6)галогеналкилокси, (C3-C10)циклоалкила, (C3-C10)галогенциклоалкила, (C6-C20)арила и (C1-C20)гетероциклила.
В другом варианте осуществления настоящего изобретения каждый R1 независимо выбирают из H, F и Cl.
В другом варианте осуществления настоящего изобретения R1 означает H.
В другом варианте осуществления настоящего изобретения R3 выбирают из H, (C1-C6)алкила, (C1-C6)галогеналкила и (C6-C20)арила.
В другом варианте осуществления настоящего изобретения R3 выбирают из H, CF3, CH2F, CHF2, CH3, CH2CH3, CH(CH3)2 и фенила.
В другом варианте осуществления настоящего изобретения R3 выбирают из H и CH3.
В другом варианте осуществления настоящего изобретения R4 означает (C1-C6)алкил(C3-C10)циклогалогеналкил.
В другом варианте осуществления настоящего изобретения R4 выбирают из H, (C1-C6)алкила, (C1-C6)алкил(C6-C20)арила, (C1-C6)галогеналкила, (C1-C6)алкил(C3-C10)циклоалкила, (C3-C10)циклоалкил-O-(C1-C6)алкила и (C3-C10)циклогалогеналкила.
В другом варианте осуществления настоящего изобретения R4 выбирают из H, CH3, CH2CH3, CH(CH3)2, CH2CH(CH3)2, циклопропила, (C6-C20)арила, CH2-фенила, CH2-фенил-OCH3, CH2OCH2-фенила, CH2CH2CH3, CH2CH2F, CH2CH2OCH3, CH2-циклопропила и циклопропил-O-CH2CH3.
В другом варианте осуществления настоящего изобретения R4 выбирают из H, CH3, CH2CH3, CH(CH3)2, CH2CH(CH3)2, CH2CH2CH3, циклопропила, CH2-циклопропила и CH2CH=CH2, CH2C≡CH.
В другом варианте осуществления настоящего изобретения молекулы, имеющие структуру, соответствующую соединению (III), раскрыты в качестве промежуточных соединений, пригодных для синтеза пестицидных тиазоламидов.
Как правило, S-R2 означает уходящую группу, где R2 представляет собой часть уходящей группы, которая не оказывает существенного и отрицательного влияния на требуемую реакцию. Желательно, чтобы R2 означал группу, которая фактически влияет на летучесть побочного тиопродукта реакции.
На стадии a соединения (I) и (II) подвергают взаимодействию, получая соединение (III). Реакция может быть проведена при комнатной температуре и при давлении окружающей среды, но, при желании, можно применять более высокие или низкие температуры и давления. Реакцию проводят в полярном протонном растворителе. Примеры таких растворителей включают, но не ограничиваются ими, муравьиную кислоту, н-бутанол, изопропанол, н-пропанол, этанол, метанол, уксусную кислоту и воду или их смесь. Как правило, предпочтительным является метанол.
На стадии b соединение (III) циклизуют, используя дегидратирующий агент. Примеры таких дегидратирующих агентов включают, но не ограничиваются ими, POCl3, H2SO4, SOCl2, P2O5, полифосфорную кислоту, п-толуолсульфоновую кислоту и трифторуксусный ангидрид или их смесь. Реакция может быть проведена при комнатной температуре и при давлении окружающей среды, но, при желании, можно применять более высокие или низкие температуры и давления. Обычно предпочтительно использовать температуру выше, чем комнатная температура, предпочтительно, до температуры кипения раствора включительно, например можно использовать температуру от приблизительно 60°C до приблизительно 120°C. Реакцию проводят в полярном протонном растворителе. Как правило, предпочтительным является ацетонитрил.
Одно из преимуществ стадий a и b над уровнем техники состоит в том, что соединения (III) и (IV) обычно получают в виде по существу чистых твердых веществ, которые не нуждаются в дополнительных процедурах очистки. Другое преимущество таких способов состоит в том, что в соединении (IV) - когда R3 означает H, то он может быть галогенированным. Следовательно, на данный момент R3 дополнительно теперь включает F, Cl, Br и I (см. схему два).
Схема два
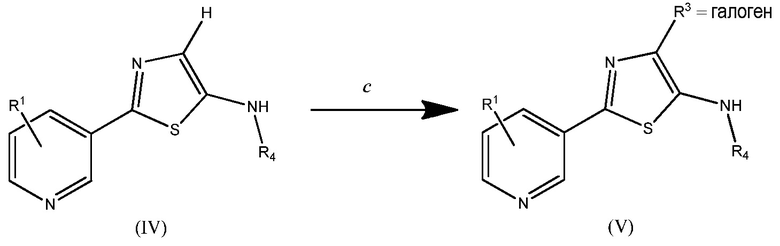
На стадии c может быть использован любой галогенирующий агент, например 1-хлорпирролидин-2,5-дион, N-бромсукцинимид и бис(тетрафторборат) 1-хлорметил-4-фтор-1,4-диазонийбицикло[2.2.2]октана. Могут быть использованы полярные растворители, такие как дихлорметан, тетрагидрофуран, этилацетат, ацетон, диметилформамид, ацетонитрил и диметилсульфоксид. Обычно предпочтительным является дихлорметан. Реакция может быть проведена при комнатных температуре и давлении, но, по желанию, могут быть использованы более высокие или более низкие температуры и давления. Обычно предпочтительными являются температуры от приблизительно 0°C до температуры окружающей среды.
В другом варианте осуществления настоящего изобретения R3 предпочтительно означает Cl.
Соединение (IV) или соединение (V) могут быть дополнительно подвергнуты взаимодействию с образованием определенных пестицидов, описанных в WO 2010/129497 (полное содержание которого включено в описание посредством ссылки).
ПРИМЕРЫ
Примеры приведены в целях иллюстрации и не могут рассматриваться как ограничивающие раскрытое в данном документе изобретение исключительно описанными в этих примерах вариантами осуществления изобретения.
Исходные материалы, реагенты и растворители, которые были получены из коммерческих источников, использовали без дополнительной очистки. Безводные растворители приобретали как Sure/Seal™ от Aldrich и использовали в состоянии поставки. Температуры плавления получали на приборе для определения температуры плавления в капилляре Thomas Hoover Unimelt или с помощью автоматической системы определения температуры плавления OptiMelt от Stanford Research Systems и без учета поправок. Молекулам присвоены общеизвестные названия, указанные в соответствии с программами для присвоения названий по номенклатуре в ISIS Draw, ChemDraw или ACD Name Pro. Если такие программы не способны дать название молекуле, то молекулу называют, используя общепринятые правила наименования. Все данные ЯМР приведены в м.д. (δ) и зарегистрированы при 300, 400 или 600 МГц, если не указано иное.
Пример 1: получение N -(4-хлор-2-(пиридин-3-ил)тиазол-5-ил)- N -этил-2-метил-3-(метилтио)пропанамид:

Стадия 1: получение N -этил-2-(пиридин-3-карботиоамидо)ацетамида
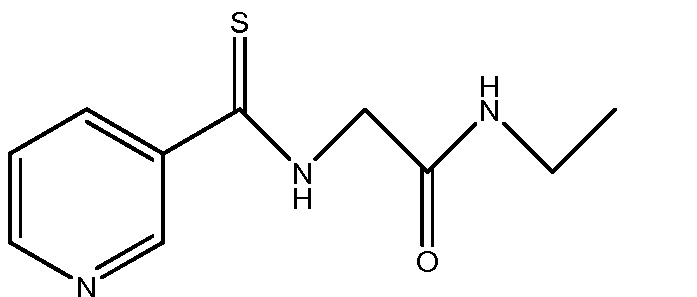
В сухую круглодонную колбу на 3 л, снабженную механической мешалкой, входным отверстием для азота, трехступенчатым последовательным скруббером меркаптана (водный раствор гипохлорита натрия, 30% гидроксид натрия и насыщенный гидроксид калия), термометром и капельной воронкой, загружали гидрохлорид 2-амино-N-этилацетамида (SPECS, каталожный № AS-787, 68,8 г, 500 ммоль) и метанол (500 мл). Реакционную смесь охлаждали до 5°C и добавляли по каплям триэтиламин (50,6 г, 500 ммоль) в метаноле (50 мл) (примечание: слабая экзотермическая реакция, до 10°C). К этой смеси добавляли по каплям метилпиридин-3-карбодитиолат (85,0 г, 500 ммоль) в метаноле (100 мл) и образовавшуюся смесь перемешивали при 5-10°C в течение 2 ч. Реакционной смеси давали нагреться до 25°C и перемешивали в атмосфере азота в течение 2 ч. Реакционную смесь охлаждали до 5°C и добавляли воду (1 л) до тех пор, пока из раствора не осаждалось твердое вещество. Твердое вещество собирали вакуумным фильтрованием, промывали водой (3 л), гексанами (500 мл) и сушили на воздухе в течение 16 ч, получая N-этил-2-(пиридин-3-карботиоамидо)ацетамид в виде рассыпчатого твердого вещества желтого цвета (без малейшего запаха меркаптана), которое сушили в вакууме при 40°C в течение 6 ч. Это давало твердое вещество желтого цвета (77,7 г, выход 70%): т.пл. 143-145°C; 1H ЯМР (400 МГц, CDC13) δ 9,02 (дд, J=2,4, 0,7 Гц, 1H), 8,86 (с, 1H), 8,70 (дд, J=4,8, 1,6 Гц, 1H), 8,15 (ддд, J=8,0, 2,4, 1,7 Гц, 1H), 7,35 (ддд, J=8,0, 4,8, 0,8 Гц, 1H), 6,05 (с, 1H), 4,43 (д, J=4,5 Гц, 2H), 3,38 (дд, J=13,0, 6,4 Гц, 2H), 1,20 (т, J=7,3 Гц, 3H); 13C-ЯМР (101 МГц, CDC13) δ 195,66, 166,90, 152,00, 147,20, 136,32, 134,96, 123,24, 49,45, 34,92, 14,75; Анал. вычислено для C10H13N3OS: C, 53,79; H, 5,87; N, 18,82; S, 14,36. Найдено: C, 53,77: H, 5,79; N, 18,87; S, 14,52.
Стадия 2: получение N -этил-2-(пиридин-3-ил)тиазол-5-амина:
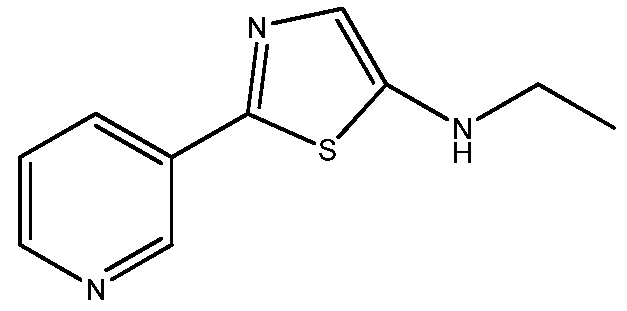
В сухую круглодонную колбу на 1 л, снабженную механической мешалкой, капельной воронкой и обратным холодильником, загружали N-этил-2-(пиридин-3-карботиоамидо)ацетамид (50,0 г, 224 ммоль) и ацетонитрил (400 мл). К этой смеси добавляли по каплям оксихлорид фосфора (103 г, 672 ммоль) и реакционную смесь перемешивали при температуре окружающей среды в течение 20 мин. Реакционную смесь нагревали до 55°C и проводили мониторинг протекания реакции с помощью ВЭЖХ (колонка YMC AQ, от смеси 5% ацетонитрила ("ACN") в 95% воды-0,05% трифторуксусной кислоты ("ТФУК") до смеси 95% ACN в 5% воды с 0,05% ТФУК за 20 мин при 1,0 мл/мин). Через 2 ч реакция практически завершалась. Реакционную смесь охлаждали до 25°C и растворитель удаляли на роторном испарителе, получая густой сироп желтого цвета. Густой сироп желтого цвета осторожно выливали в насыщенный водный раствор бикарбоната натрия (1,5 л) при быстром перемешивании. Величину pH полученного желтого раствора доводили до слабо щелочного (pH 8) с помощью твердого бикарбоната натрия, и из раствора осаждалось твердое вещество желтого цвета. К смеси добавляли дополнительное количество охлажденной воды (1 л) и перемешивали еще в течение 20 мин. Осадок собирали вакуумным фильтрованием и споласкивали водой (1 л) и гексанами (500 мл). Собранное твердое вещество сушили в вакууме при 40°C в течение 16 ч, получая N-этил-2-(пиридин-3-ил)тиазол-5-амин в виде твердого вещества желтого цвета (36,7 г, 80%): т.пл. 97-98°C; 1H ЯМР (400 МГц, CDC13) δ 8,98 (дд, J=2,3, 0,8 Гц, 1H), 8,53 (дд, J=4,8, 1,6 Гц, 1H), 8,07 (ддд, J=8,0, 2,3, 1,6 Гц, 1H), 7,31 (ддд, J=8,0, 4,8, 0,8 Гц, 2H), 6,98 (с, 1H), 3,96 (с, 1H), 3,24 (кв, J=5,8 Гц, 2H), 1,31 (т, J=7,2 Гц, 3H); 13C ЯМР (101 МГц, CDCl3) δ 152,00, 149,21, 149,21, 146,61, 132,17, 130,44, 123,62, 121,84, 43,09, 14,80. Анал. вычислено для C10H11N3S: C, 58,51; H, 5,40; N, 20,47. Найдено: C, 58,34: H, 5,40; N, 20,38; ESIMS m/z 205 ([M+H]+).
Стадия 3: получение гидрохлорида 4-хлор- N -этил-2-(пиридин-3-ил)тиазол-5-амина:
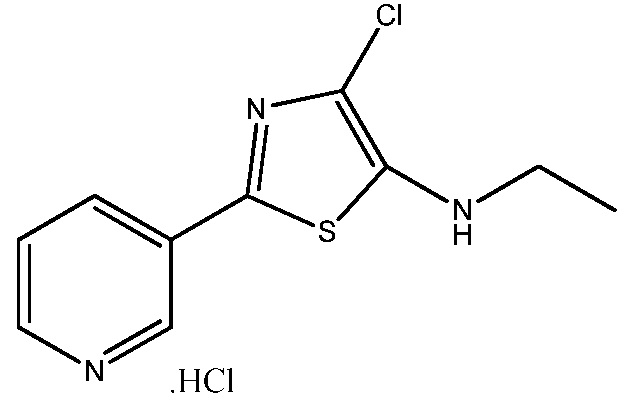
В сухую круглодонную колбу на 500 мл, снабженную магнитной мешалкой, термометром и входным отверстием для азота, загружали N-этил-2-(пиридин-3-ил)тиазол-5-амин (5,1 г, 25 ммоль), простой диэтиловый эфир (200 мл) и диоксан (5 мл). Полученную суспензию (не все твердое вещество растворяется) охлаждали до 5°C и добавляли порциями N-хлорсукцинамид (3,65 г, 27,3 ммоль). После добавления всего хлорирующего агента из раствора осаждалось твердое вещество коричневого цвета. Реакционную смесь перемешивали при 5°C в течение 60 мин, затем анализировали методом ВЭЖХ (колонка YMC AQ, от смеси 5% ACN в 95% воды-0,05% ТФУК до смеси 95% ACN в 5% воды с 0,05% ТФУК за 20 мин при 1,0 мл/мин). Анализ методом ВЭЖХ показывал отсутствие исходного материала, и один основной продукт соответствовал требуемому хлориду. Коричневую суспензию фильтровали через слой Celite® и слой Celite® споласкивали простым диэтиловым эфиром (~20 мл). Фильтрат охлаждали до 5 °C и подкисляли при перемешивании добавлением 6,5 мл 4M HCl в диоксане. Сразу же образовывалось твердое вещество желтого цвета. Твердое вещество собирали вакуумным фильтрованием, споласкивали простым диэтиловым эфиром и сушили в вакууме при 40°C в течение 2 ч. Это давало гидрохлорид 4-хлор-N-этил-2-(пиридин-3-ил)тиазол-5-амина в виде твердого вещества желтого цвета (6,3 г, 92%): т.пл. 180-182°C; 1H ЯМР (400 МГц, ДМСО-d6) δ 9,07 (д, J=2,0 Гц, 1H), 8,70 (дд, J=5,4, 1,3 Гц, 1H), 8,59-8,42 (м, 1H), 7,86 (дд, J=8,2, 5,3 Гц, 1H), 5,27 (с, 5H), 3,20 (кв, J=7,2 Гц, 2H), 1,23 (т, J=7,1 Гц, 3H); 13C ЯМР (101 МГц, ДМСО-d6) δ 148,10, 140,19, 138,58, 137,91, 137,01, 132,06, 127,30, 115,89, 43,43, 13,87; Анал. вычислено для C1011Cl2N3S: C, 43,49; H, 4,01; Cl, 25,67; N, 15,21; S, 11,61. Найдено: C, 43,42; H, 4,01; Cl, 25,55; N, 14,99; S, 11,46.
Стадия 4: получение N -(4-хлор-2-(пиридин-3-ил)тиазол-5-ил)- N -этил-2-метил-3-(метилтио)пропанамида:
В трехгорлую колбу на 1 л, снабженную температурным датчиком J-KEM типа-T, верхнеприводной мешалкой, обратным холодильником и входным отверстием для азота, добавляли гидрохлорид 4-хлор-N-этил-2-(пиридин-3-ил)тиазол-5-амина (75,8 г, 274 ммоль) и дихлорметан (500 мл). К полученной суспензии зеленого цвета добавляли порциями, за 1 мин, пиридин (55 г, 695 ммоль, 2,5 экв.) (примечание: дымление с экзотермической реакцией 20°C-26°C). Реакционная смесь превращалась в темный, раствор зелено-черного цвета. К этому раствору добавляли N,N-диметилпиридин-4-амин (DMAP, 16,5 г, 135 ммоль, 0,5 экв.) (примечание: не было никаких изменений в реакции или температуре) с последующим добавлением 2-метил-3-метилтиопропаноилхлорида (44,3 г, 290 ммоль, 1,06 экв.), который добавляли порциями за 1 мин. Реакция была экзотермической от 17°C до 29°C во время добавления хлорангидрида кислоты. Реакционную смесь нагревали до 35ºC в течение 19 ч и затем охлаждали до 25°C за 4 ч. Анализ методом ВЭЖХ (колонка YMC AQ, от смеси 5% ACN в 95% воды-0,05% трифторуксусной кислоты ("ТФУК") до смеси 95% ACN в 5% воды с 0,05% ТФУК за 20 мин при 1,0 мл/мин) показывал, что реакция завершилась на 95%. Реакционную смесь черного цвета переносили в делительную воронку на 2 л и добавляли дихлорметан (200 мл) и воду (300 мл). Фазы разделяли, водный слой (коричневый) экстрагировали дихлорметаном (100 мл) и дихлорметановые экстракты объединяли. Объединенный дихлорметановый экстракт промывали насыщенным раствором соли (300 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали (40°C, 40 мм Hg, 1 час). Это давало 99,4 г густого масла черного цвета. Густое масло черного цвета растворяли в дихлорметане (100 мл) и подсоединяли вакуум к верхней части картриджа, нагруженного 240 г твердого вещества, содержащего 230 г силикагеля 60. Нагруженный твердым веществом картридж подсоединяли к ISCO companion XL и продукт очищали на заполненной диоксидом кремния колонке Redisep на 1,5 кг, используя подвижную фазу гексан:этилацетат (градиент: 20% этилацетат 5 мин, 20%-90% этилацетат на протяжении 70 мин) при скорости потока 400 мл/мин. Требуемое соединение, элюированное из колонки в диапазоне 30-50 мин, собирали в склянки на 500 мл (фракции 2-16). Фракции 2-9 объединяли (примечание: фракции 8-9, которые были мутными, фильтровали через бумагу) и упаривали на роторном испарителе (40°C, 40 мм Hg, 2 ч). Это давало 56,8 г темно-желтого масла, которое имело 98% чистоту по ВЭЖХ при 254 нм. Фракции 10-15 фильтровали, объединяли, и упаривали на роторном испарителе (40°C, 40 мм Hg, 2 ч), получая золотистое масло (27,51 г). Пробы анализировали методом ВЭЖХ (колонка YMC AQ, от смеси 5% ACN в 95% воды-0,05% ТФУК до смеси 95% ACN в 5% воды с 0,05% ТФУК за 20 мин при 1,0 мл/мин) при 254 нм, что показывало 88% чистоту, содержание 10% исходного вещества тиазоламина и содержание 2% неизвестной высокоподвижной примеси. Золотистое масло (27,5 г, 88% чистота) растворяли в простом эфире (50 мл) и через 1 мин осаждалось твердое вещество желтого цвета. Смесь перемешивали в течение 15 мин при 25°C, затем добавляли гексан (50 мл) и смесь перемешивали еще 15 мин при 25°C. Твердое вещество собирали вакуумным фильтрованием и твердое вещество желтого цвета промывали смесью простой эфир/гексан (1:1, 25 мл). Это давало 19,23 г N-(4-хлор-2-(пиридин-3-ил)тиазол-5-ил)-N-этил-2-метил-3-(метилтио)пропанамида в виде твердого вещества желтого цвета. Анализ методом ВЭЖХ показывал чистоту 97% при 254 нм. Пробу в 56,8 г (золотистое масло, 98% чистота) растворяли в диэтиловом эфире (100 мл), и через 1 мин осаждалось твердое вещество светло-коричневого цвета. Смесь перемешивали в течение 15 мин при 25°C и добавляли гексан (100 мл). Смесь перемешивали еще 15 мин. Твердое вещество собирали вакуумным фильтрованием и промывали смесью простой эфир/гексан (1:1, 2×50 мл). Это давало 49,67 г твердого вещества светло-желтого цвета. Анализ методом ВЭЖХ (колонка YMC AQ, от смеси 5% ACN в 95% воды-0,05% ТФУК до смеси 95% ACN в 5% воды с 0,05% ТФУК за 20 мин при 1,0 мл/мин) при 254 нм показывал чистоту >99%. Маточные жидкости от двукратной перекристаллизации объединяли и упаривали на роторном испарителе (40°C, 40 мм Hg, 1 час). Это давало 11,27 г темно-желтого масла. Масло повторно растворяли в простом эфире (40 мл) и перемешивали в течение 30 мин, за указанное время образовывался темно-желтый осадок. Добавляли гексан (50 мл) и смесь перемешивали в течение 15 мин. Твердое темное вещество собирали вакуумным фильтрованием и промывали смесью простой эфир/гексан (1:1, 2×20 мл). Это давало 5,0 г твердого вещества коричневого цвета, для которого была установлена чистота >99% методом ВЭЖХ при 254 нм. Перекристаллизованные образцы все объединяли и смешивали вручную, получая N-(4-хлор-2-(пиридин-3-ил)тиазол-5-ил)-N-этил-2-метил-3-(метилтио)пропанамид в виде твердого вещества желтого цвета (75 г, 85%): т.пл. 80-81°C; 1H ЯМР (400 МГц, CDC13) δ 9,12 (д, J=1,9 Гц, 1H), 8,72 (дд, J=4,8, 1,4 Гц, 1H), 8,22 (ддд, J=8,0, 2,2, 1,8 Гц, 1H), 7,43 (ддд, J=8,0, 4,8, 0,6 Гц, 1H), 4,03-3,80 (м, 1H), 3,80-3,59 (м, 1H), 2,97-2,68 (м, 2H), 2,60-2,39 (м, 1H), 2,03 (с, 3H), 1,30-1,16 (м, 6H); 13C ЯМР (101 МГц, ДМСО-d6) δ 175,66, 162,63, 151,89, 147,14, 138,19, 133,49 133,23, 128,58, 123,90, 44,81, 38,94, 37,93, 18,16, 16,83, 12,90; Анал. вычислено для C15H18ClN3OS2: C, 50,62; H, 5,10; N, 11,81; S, 18,02. Найдено: C, 50,49; H, 5,21; N, 11,77; S, 17,99.
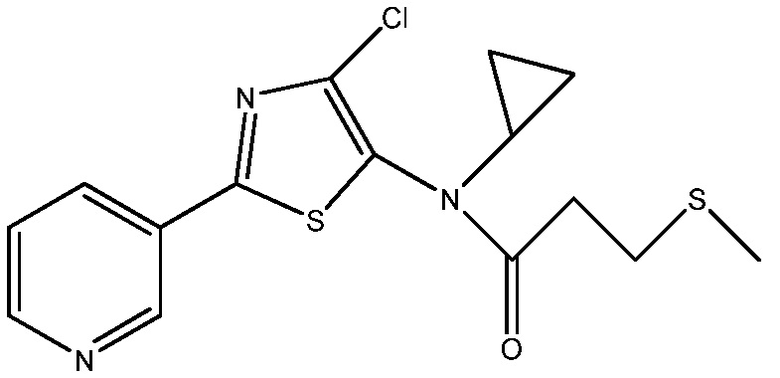
Пример 2: получение N -(4-хлор-2-(пиридин-3-ил)тиазол-5-ил)- N -циклопропил-3-(метилтио)пропанамида
Стадия 1: получение гидрохлорида 2-амино- N -циклопропилацетамида:
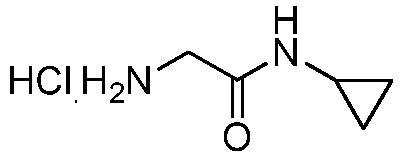
К раствору 3H-[1,2,3]триазоло[4,5-b]пиридин-3-ола (7,77 г, 57,1 ммоль), 2-(трет-бутоксикарбониламино)уксусной кислоты (10 г, 57,1 ммоль), циклопропанамина (3,91 г, 68,5 ммоль) и DMAP (8,37 г, 68,5 ммоль) в ДМФА (28 мл) добавляли гидрохлоридную соль N1-((этилимино)метилен)-N3,N3-диметилпропан-1,3-диамина и смесь перемешивали при комнатной температуре в течение 16 ч. Смесь разбавляли этилацетатом, промывали 0,1 н. водным HCl, водным NaHCO3 и насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали в вакууме, получая трет-бутил-2-(циклопропиламино)2-оксоэтилкарбамат (8,90 г, 41,5 ммоль, 72,8%) в виде светло-желтого масла: 1H ЯМР (400 МГц, CDC13) δ 6,18 (с, 1H), 3,74 (д, J=5,9 Гц, 2H), 2,71 (м, 1H), 1,45 (с, 9H); 0,84-0,7 (м, 2H), 0,56-0,43 (м, 2H); EIMS m/z 214 ([M]+). К раствору трет-бутил-2-(циклопропиламино)2-оксоэтилкарбамата (8,5 г, 39,7 ммоль) в диоксане (20 мл) добавляли HCl (100 ммоль, 25 мл 4 М в диоксане) и смесь перемешивали при 10°C в течение 3 ч. Смесь разбавляли гексанами и фильтровали в вакууме, получая HCl-соль 2-амино-N-циклопропилацетамида в виде твердого вещества белого цвета (5,2 г, 83%): т.пл. 139-142°C; 1H ЯМР (400 МГц, ДМСО-d6) δ 8,66 (уш.с, 1H), 8,19 (уш.с, 3H), 3,46 (с, 2H), 2,73-2,60 (м, 1H), 0,71-0,60 (м, 2H), 0,48-0,36 (м, 2H).
Стадия 2: получение N -циклопропил-2-(пиридин-3-карботиоамидо)ацетамида:
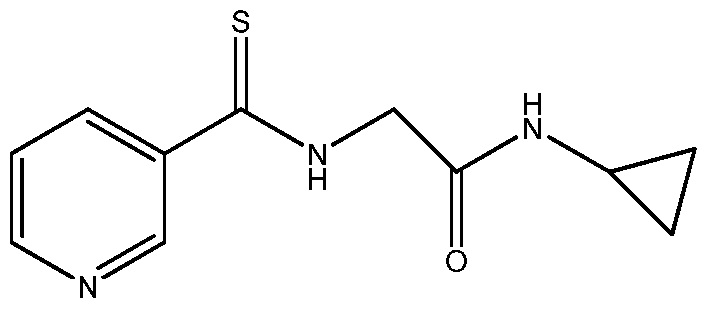
К раствору метилпиридин-3-карбодитиоата (2,97 г, 17,52 ммоль) в метаноле (10 мл) добавляли раствор 2-амино-N-циклопропилацетамида (2 г, 17,52 ммоль) (HCl-соль) и триэтиламина (3,55 г, 35,0 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч и затем разбавляли этилацетатом и промывали насыщенным водным NaHCO3, насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали в вакууме, получая N-циклопропил-2-(пиридин-3-карботиоамидо)ацетамид в виде твердого вещества желтого цвета (3,60 г, 83%): т.пл. 152-153ºC; 1H ЯМР (400 МГц, ДМСО-d6) δ 10,60 (с, 1H), 8,91 (ддд, J=7,0, 2,3, 0,7 Гц, 1H), 8,76-8,56 (м, 1H), 8,10 (м, 2H), 7,57-7,32 (м, 1H), 4,26 (м, 2H), 2,73-2,57 (м, 1H), 0,77-0,61 (м, 2H), 0,50-0,29 (м, 2H); ESIMS (m/z) 234 ([M-H]-).
Стадия 3: получение N -циклопропил-2-(пиридин-3-ил)тиазол-5-амина:
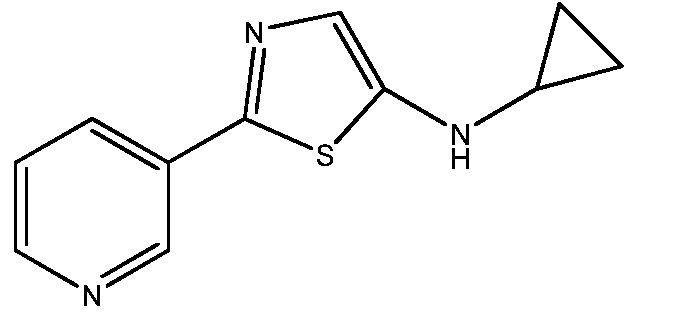
N-циклопропил-2-(пиридин-3-карботиоамидо)ацетамид (1,00 г, 4,25 ммоль) растворяли в ацетонитриле (5 мл) в сухой колбе и добавляли по каплям фосфорилокситрихлорид (3,26 г, 21,25 ммоль). Смесь нагревали до 100°C и перемешивали в течение 1 ч. Смесь охлаждали до комнатной температуры и твердое вещество желтого цвета фильтровали в вакууме. Это твердое вещество промывали ацетонитрилом и сушили в вакууме, получая 0,36 г HCl-соли N-циклопропил-2-(пиридин-3-ил)тиазол-5-амина (ЖХМС и 1H-ЯМР показывали 100% чистоту). Фильтрат разбавляли этилацетатом и осторожно подщелачивали насыщенным водным NaHCO3. Органическую фазу отделяли и промывали насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме, получая N-циклопропил-2-(пиридин-3-ил)тиазол-5-амин в виде коричневого масла (0,45 г, 3,47 ммоль, 82%): 1H ЯМР (400 МГц, CDC13) δ 9,10 (д, J=2,1 Гц, 1H), 8,68 (дд, J=5,5, 1,2 Гц, 1H), 8,63-8,58 (м, 1H), 7,89 (дд, J=8,1, 5,4 Гц, 1H), 7,07 (д, J=7,8, 1H), 2,57 (дт, J=10,0, 3,3 Гц, 1H), 0,85-0,68 (м, 2H), 0,57-0,45 (м, 2H); ESIMS (m/z) 216 ([M-H]-).
Стадия 4: получение 4-хлор- N -циклопропил-2-(пиридин-3-ил)тиазол-5-амина:
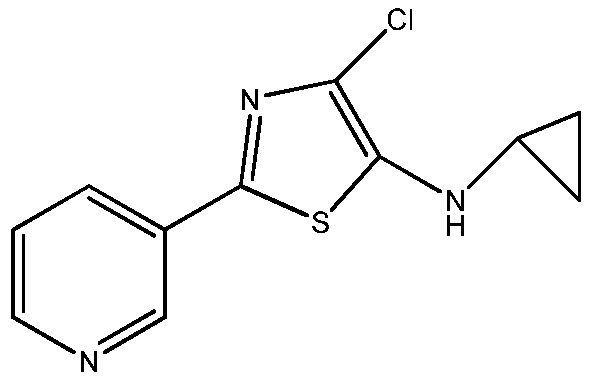
К раствору N-циклопропил-2-(пиридин-3-ил)тиазол-5-амина (1,00 г, 4,60 ммоль) в ацетонитриле (2 мл) добавляли 1-хлорпирролидин-2,5-дион (645 мг, 4,83 ммоль) и смесь перемешивали при 0°C в течение 1 ч. Смесь фильтровали и фильтрат обрабатывали избытком HCl (4М в диоксане), получая 4-хлор-N-циклопропил-2-(пиридин-3-ил)тиазол-5-амин в виде твердого вещества коричневого цвета: т.пл. 56-60°C.
Стадия 5: получение N -(4-хлор-2-(пиридин-3-ил)тиазол-5-ил)- N -циклопропил-3-(метилтио)пропанамида:
К раствору HCl-соли 4-хлор-N-циклопропил-2-(пиридин-3-ил)тиазол-5-амина (288 мг, 1 ммоль) и DMAP (305 мг, 2,500 ммоль) в CH2ClCH2Cl (1 мл) добавляли 3-(метилтио)пропаноилхлорид (166 мг, 1,200 ммоль) и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь разбавляли этилацетатом, смешанным с водным NaHCO3 (10 мл). Органическую фазу отделяли, споласкивали насыщенным раствором соли (2×), сушили над MgSO4 и концентрировали в вакууме, получая желтую смолу. Эту смолу очищали методом хроматографии на колонке с обращенной фазой (C-18, CH3CN/H2O), получая N-(4-хлор-2-(пиридин-3-ил)тиазол-5-ил)-N-циклопропил-3-(метилтио)пропанамид (82 мг, 22%) в виде коричневой смолы: 1H ЯМР (400 МГц, CDC13) δ 9,10 (с, 0,6H), 9,02 (с, 0,4H), 8,71 (с, 6H), 8,61 (д, J=3,4 Гц, 0,4H), 8,21 (д, J=7,6 Гц, 1H), 8,19-8,10 (м, 1H), 7,41 (д, J=5,6, 0,6H), 7,35 (дд, J=8,3, 4,5 Гц, 0,4H), 3,16 (уш.с, 1H), 2,91 (с, 3H), 2,88-2,72 (м, 2H), 2,11 (м, 2H), 0,85 (м, 4H); ESIMS (m/z) 354,56 ([M+H]+).
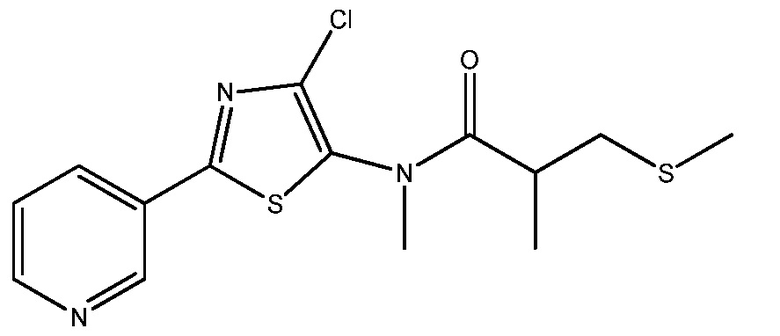
Пример 3: получение N -(4-хлор-2-(пиридин-3-ил)тиазол-5-ил)- N ,2-диметил-3-(метилтио)пропанамида:
Стадия 1: получение N -метил-2-(пиридин-3-карботиоамидо)ацетамида:
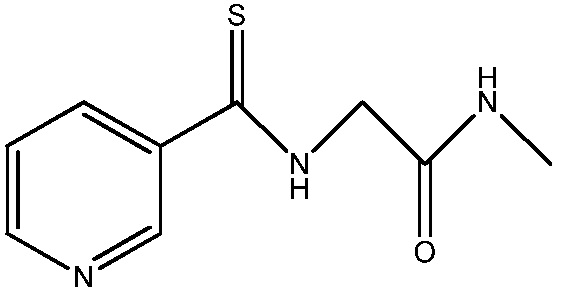
Трехгорлую круглодонную колбу на 5 л снабжают отверстием для входа азота через капельную воронку, механической мешалкой и обратным холодильником. Трубка от верхней части холодильника проведена, достигая дна, через буферную колбу на 1 литр и затем в барботажную трубку в другой трехгорлой колбе с перемешиванием на 5 л с 2,5-литровым слоем на дне 12% водного раствора гипохлорита натрия. Выходное отверстие колбы с водным раствором гипохлорита натрия соединено через насадку-крокодил приблизительно с 250 мл 12% водного раствора гипохлорита натрия. Реактор загружали 2-амино-N-метилацетамидом (160 г, 1,81 моль) и ацетонитрилом (3 л), получая мутный раствор. Капельную воронку загружали метилпиридин-3-карбодитиоатом (307 г, 1,81 моль) и ацетонитрилом (200 мл). Добавление дитиоата занимало примерно 20 мин, и реакционную смесь продували сильной струей азота. При добавлении отмечали слабый экзотермический эффект (около 3°C). После завершения добавления капельную воронку споласкивали 550 мл ацетонитрила, доводя общий объем ацетонитрила до 3750 мл. После перемешивания в течение примерно 10 мин из красного раствора выпадал осадок, дающий твердое вещество, подобное прессованному творогу, которое не перемешивалось. Капельную воронку заменяли прямой трубкой в 1/4 дюйма для барботирования в реактор азота. Реактор медленно нагревали приблизительно до 45-50°C до образования красноватого раствора, которому затем давали медленно охладиться опять до комнатной температуры и кристаллизоваться до тонкодисперсного желтоватого, игольчатого твердого вещества, которое легко перемешивалось. Игольчатые кристаллы собирали вакуумным фильтрованием и промывали 100 мл ацетонитрила. Твердое вещество сушили в вакууме при 40°C в течение 16 ч, получая N-метил-2-(пиридин-3-карботиоамидо)ацетамид (268,6 г, 71%) в виде твердого вещества светло-желтого цвета: т.пл. 135-137°C; 1H ЯМР (400 МГц, ДМСО-d6) δ 10,65 (с, 1H), 8,95 (дд, J=2,4, 0,7 Гц, 1H), 8,67 (дд, J=4,8, 1,6 Гц, 1H), 8,14 (ддд, J=8,0, 2,3, 1,7 Гц, 1H), 7,95 (д, J=4,3 Гц, 1H), 7,48 (ддд, J=7,9, 4,8, 0,7 Гц, 1H), 4,34 (с, 2H), 2,62 (д, J=4,6 Гц, 3H); 13C ЯМР (101 МГц, ДМСО-d6) δ 194,98, 165,77, 150,37, 146,40, 135,25, 134,04, 121,51, 47,66, 24,41; Анал. вычислено для C9H11N3OS: C, 51,65; H, 5,30; N, 20,08; S, 15,32. Найдено: C, 51,47; H, 5,30; N, 20,01; S, 15,53.
Стадия 2: получение N -метил-2-(пиридин-3-ил)тиазол-5-амина:
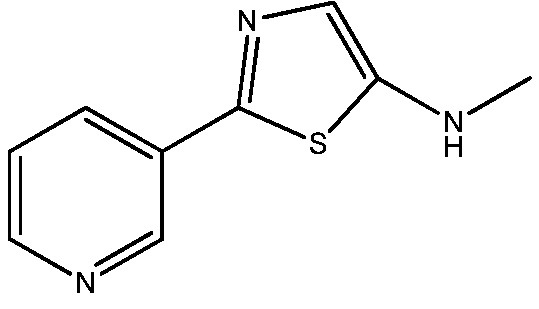
В сухую круглодонную колбу на 2 л, снабженную механической мешалкой, капельной воронкой и обратным холодильником, загружали N-метил-2-(пиридин-3-карботиоамидо)ацетамид (100 г, 478 ммоль) и ацетонитрил (1 л). К этой смеси добавляли порциями за 10 мин оксихлорид фосфора (256 г, 1672 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 10 мин, в течение указанного времени наблюдали слабый экзотермический эффект от 22°C до 34°C (примечание: некоторое количество твердого вещества оставалось нерастворенным в реакционной смеси, и смесь становилась густой, но все еще перемешивалась сравнительно хорошо). Реакционную смесь нагревали до 85°C (осторожное нагревание до температуры кипения с обратным холодильником). Через 3 ч все твердое вещество растворялось, образуя раствор темно-янтарного цвета. Анализ аликвоты методом ТСХ (смесь 70% этилацетат:30% гексаны) спустя 4 ч указывал на то, что реакция практически завершилась. Реакционной смеси давали охладиться до 25°C и растворитель удаляли на роторном испарителе. Остаток растворяли в воде и обрабатывали твердым бикарбонатом натрия до слабощелочного (pH ~8) при непрерывном перемешивании (примечание: никаких усилий не прилагали для регулирования температуры, и колба была немного теплая на ощупь). Спустя несколько минут начинал образовываться осадок. Смесь продолжали перемешивать при 25°C в течение 16 ч. Твердое вещество коричневого цвета собирали вакуумным фильтрованием и промывали водой. Это давало влажный осадок на фильтре твердого вещества желтовато-коричневого цвета (91 г), которое затем сушили в вакууме при 40°C до постоянной массы. Это давало N-метил-2-(пиридин-3-ил)тиазол-5-амин в виде окрашенного в песочный цвет твердого вещества (68,5 г, 75%): т.пл. 140-141°C; 1H ЯМР (400 МГц, CDC13) δ 8,98 (дд, J=2,3, 0,7 Гц, 1H), 8,53 (дд, J=4,8, 1,6 Гц, 1H), 8,07 (ддд, J=8,0, 2,2, 1,7 Гц, 1H), 7,40-7,21 (м, 1H), 6,96 (с, 1H), 4,18 (с, 1H), 2,96 (с, 3H); 13C ЯМР (101 МГц, хлороформ) δ 153,23, 149,15, 146,54, 132,23, 130,47, 123,65, 121,20, 34,48; Анал. вычислено для C9H9N3S: C, 56,52; H, 4,74; N, 21,97; S, 16,77. Найдено: C, 56,31; H, 4,74; N, 21,81; S, 16,96.
Стадия 3: получение 4-хлор- N -метил-2-(пиридин-3-ил)тиазол-5-амина:
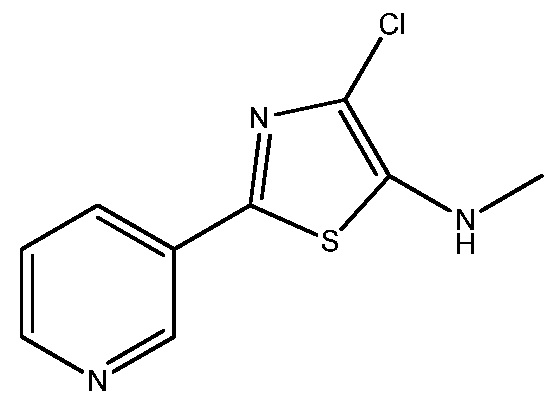
В сухую круглодонную колбу на 100 мл, снабженную магнитной мешалкой, термометром и входным отверстием для азота, загружали N-метил-2-(пиридин-3-ил)тиазол-5-амин (0,528 г, 2,76 ммоль) и дихлорметан (50 мл). Полученный раствор охлаждали до 5°C с последующим добавлением порциями твердого N-хлорсукцинамида. После добавления всего хлорирующего агента образовывался раствор темно-коричневого цвета. Раствор перемешивали при 5°C в течение 20 мин, затем анализировали аликвоту методом ВЭЖХ (колонка YMC AQ, от смеси 5% ACN в 95% воды-0,05% ТФУК до смеси 95% ACN в 5% воды с 0,05% ТФУК за 20 мин при 1,0 мл/мин). Анализ методом ВЭЖХ показывал отсутствие исходного вещества и наличие одного основного продукта. Реакционную смесь выливали в делительную воронку, содержащую дихлорметан (50 мл), и промывали водой (2×10 мл) и затем насыщенным водным раствором хлорида натрия (10 мл). Органическую фазу сушили над безводным сульфатом магния, фильтровали и упаривали на роторном испарителе, получая порошкообразное твердое вещество коричневого цвета (0,51 г). Твердое вещество очищали на ISCO Combiflash Rf (силикагелевый картридж на 80 г, подвижная фаза A=гексан, B=этилацетат, градиент от 0% B до 100% B за 20 мин). Фракции собирали в пробирки на 25 мл. Пробирки, содержащие требуемое вещество, объединяли и упаривали на роторном испарителе, получая 4-хлор-N-метил-2-(пиридин-3-ил)тиазол-5-амин в виде твердого вещества канареечно-желтого цвета (0,32 г, 51%); 1H ЯМР (400 МГц, CDC13) δ 8,97 (дд, J=2,3, 0,7 Гц, 1H), 8,54 (дд, J=4,8, 1,6 Гц, 1H), 8,07 (ддд, J=8,0, 2,3, 1,6 Гц, 1H), 7,45-7,14 (м, 1H), 4,07 (дд, J=40,5, 38,0 Гц, 1H), 3,03 (д, J=5,3 Гц, 3H); 13C ЯМР (101 МГц, CDC13) δ 149,55, 146,03, 145,60, 145,28, 131,73, 129,71, 123,64, 117,37, 35,75; Анал. вычислено для C9H8C1N3S: C, 49,89; H, 3,57; N, 18,62; S, 14,21. Найдено: C, 48,03; H, 3,64; N, 18,42; S, 14,23.
Стадия 4: получение N -(4-хлор-2-(пиридин-3-ил)тиазол-5-ил)- N ,2-диметил-3-(метилтио)пропанамида:
В сухую круглодонную колбу на 500 мл, снабженную магнитной мешалкой, термометром и входным отверстием для азота, добавляли 4-хлор-N-метил-2-(пиридин-3-ил)тиазол-5-амин (22 г, 97 ммоль) и дихлорметан (250 мл). Суспензию перемешивали при комнатной температуре, в то время как добавляли пиридин (8,48 г, 107 ммоль) и DMAP (1,20 г, 9,75 ммоль). К этой суспензии добавляли 2-метил-3-(метилтио)пропаноилхлорид (17,8 г, 117 ммоль) за 5 мин. Во время добавления все твердые вещества переходили в раствор и реакция сопровождалась экзотермическим эффектом от 20°C до 30°C. Реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч. Смесь контролировали с помощью ВЭЖХ (колонка YMC AQ, от смеси 5% ACN в 95% воды-0,05% ТФУК до смеси 95% ACN в 5% воды с 0,05% ТФУК за 20 мин при 1,0 мл/мин), которая показывала полное превращение всего исходного вещества. Реакционную смесь разбавляли дихлорметаном и затем добавляли воду. Смесь выливали в делительную воронку с дихлорметаном и водой и слои разделяли. Органическую фазу промывали насыщенным раствором соли, сушили над безводным сульфатом магния, фильтровали и упаривали на роторном испарителе, получая 33,6 г темного масла. Масло очищали на ISCO Combiflash Rf (силикагелевый картридж на 330 г, подвижная фаза A=гексан, B=этилацетат, градиент от 0% B до 100% B за 20 мин). Фракции собирали в пробирки на 25 мл. Пробирки, содержащие требуемый продукт, объединяли и растворитель удаляли на роторном испарителе. Это давало 22,8 г густой желтой жидкости с 68,4% практическим выходом. Весь образец кристаллизовали и добавляли гексан (200 мл) с получением суспензии. Суспензию фильтровали в вакууме и твердому веществу давали высохнуть на воздухе. Это давало N-(4-хлор-2-(пиридин-3-ил)тиазол-5-ил)-N,2-диметил-3-(метилтио)пропанамид в виде твердого вещества не совсем белого цвета; т.пл. 75-80°C; 1H ЯМР (400 МГц, CDC13) δ 9,12 (д, J=1,4 Гц, 1H), 8,73 (д, J=3,8 Гц, 1H), 8,34-8,09 (м, 1H), 7,43 (дд, J=7,9, 4,9 Гц, 1H), 3,30 (с, 3H), 3,06-2,70 (м, 2H), 2,49 (д, J=7,4 Гц, 1H), 2,04 (с, 3H), 1,21 (д, J=6,4 Гц, 3H); 13C ЯМР (101 МГц, ДМСО-d6) δ 175,22, 162,37, 151,91, 146,53, 136,46, 134,64, 133,35, 127,98, 124,27, 37,47, 36,71, 36,47, 17,56, 15,44; Анал. вычислено для C14H16C1N3OS2: C, 49,18; H, 4,72; N, 12,29; S, 18,76. Найдено: C, 49,04; H, 4,68; N, 12,29; S, 18,68.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ПОЛУЧЕНИЯ НЕКОТОРЫХ 2-(ПИРИДИН-3-ИЛ)ТИАЗОЛОВ | 2013 |
|
RU2647851C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ | 2010 |
|
RU2550352C2 |
| ЗАМЕЩЕННЫЕ ХРОМАНЫ | 2015 |
|
RU2718060C2 |
| СПОСОБ ПОЛУЧЕНИЯ НЕКОТОРЫХ 2-(ПИРИДИН-3-ИЛ)ТИАЗОЛОВ | 2013 |
|
RU2649000C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2013 |
|
RU2623233C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОПИРИДИНИЛ-АМИНОПИРИДИНОВЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ РАКА | 2010 |
|
RU2619463C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И СПОСОБЫ, ОТНОСЯЩИЕСЯ К НИМ | 2012 |
|
RU2605537C2 |
| СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ ПРЕДУПРЕЖДЕНИЯ МЕТАСТАЗОВ РАКОВЫХ КЛЕТОК | 2010 |
|
RU2519123C2 |
| БЕНЗАМИДНЫЕ СОЕДИНЕНИЯ | 2019 |
|
RU2801647C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-5-ФТОР-3-ГАЛОГЕН-6-(ЗАМЕЩЕННЫХ)ПИКОЛИНАТОВ | 2012 |
|
RU2545074C1 |
Изобретение относится к способу получения 2-(пиридин-3-ил)тиазолов. Способ включает (i) взаимодействие соединения (I) с соединением (II) на стадии получения соединения (III), где указанную реакцию проводят в полярном протонном растворителе при давлении окружающей среды, с последующей (ii) циклизацией соединения (III) с использованием дегидратирующего агента, с получением соединения (IV). Дегидратирующий агент выбирают из группы, состоящей из POCl3, H2SO4, SOCl2, P2O5, полифосфорной кислоты, п-толуолсульфоновой кислоты, трифторуксусного ангидрида или их смеси, и указанную циклизацию осуществляют при давлении окружающей среды и температуре от 60°C до 120°C, при этом (A) R1 представляет собой H; (B) R2 представляет собой (C1-C6)алкил; (C) R3 представляет собой H или (C1-C6)алкил; и (D) R4 представляет собой H, (C1-C6)алкил или циклопропил. Способ дополнительно включает галогенирование указанного R3 в соединении (IV) до F, Cl, Br или I в полярном растворителе при температуре от 0°C до температуры окружающей среды. Технический результат - способ получения 2-(пиридин-3-ил)тиазолов, предназначенных в качестве промежуточных соединений для синтеза пестицидных тиазоламидов. 6 з.п. ф-лы, 3 пр.
1. Способ, включающий
Схема один
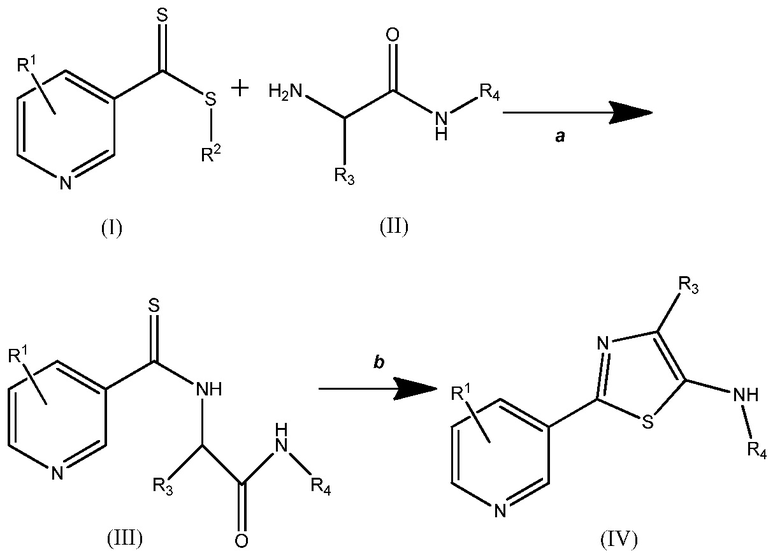 , где
, где
(i) взаимодействие соединения (I) с соединением (II) на стадии получения соединения (III), где указанную реакцию проводят в полярном протонном растворителе при давлении окружающей среды; с последующей
(ii) циклизацией соединения (III) с использованием дегидратирующего агента с получением соединения (IV), где дегидратирующий агент выбирают из группы, состоящей из POCl3, H2SO4, SOCl2, P2O5, полифосфорной кислоты, п-толуолсульфоновой кислоты, трифторуксусного ангидрида или их смеси, и где указанную циклизацию осуществляют при давлении окружающей среды и температуре от 60°C до 120°C;
где (A) R1 представляет собой H;
(B) R2 представляет собой (C1-C6)алкил;
(C) R3 представляет собой H или (C1-C6)алкил; и
(D) R4 представляет собой H, (C1-C6)алкил или циклопропил.
2. Способ по п.1, в котором указанный полярный протонный растворитель представляет собой муравьиную кислоту, н-бутанол, изопропанол, н-пропанол, этанол, метанол, уксусную кислоту и воду или их смесь.
3. Способ по п.1, в котором указанный полярный протонный растворитель представляет собой метанол.
4. Способ по п.1, дополнительно включающий галогенирование указанного R3 в соединении (IV) до F, Cl, Br или I в полярном растворителе при температуре от 0°C до температуры окружающей среды.
5. Способ по п.4, в котором указанное галогенирование осуществляют в полярном растворителе, выбираемом из дихлорметана, тетрагидрофурана, этилацетата, ацетона, диметилформамида, ацетонитрила и диметилсульфоксида.
6. Способ по п.5, в котором указанный полярный растворитель представляет собой дихлорметан.
7. Способ по любому из пп.4, 5 и 6, в котором R3 означает Cl.
| G.C.BARRETT: "Reactions of N-thiobenzoyl-[alpha]-amino-acids and related N-substituted thiobenzamides with trifluoroacetic anhydride", TETRAHEDRON, 1978, vol.34, no.5, p.611-616 | |||
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| US 5219824 A, 15.06.1993 | |||
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| ПРОИЗВОДНЫЕ ПИРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ИНСЕКТИЦИДНАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2083562C1 |

