Ссылка на родственные заявки
По настоящей заявке испрашивается приоритет на основании предварительной заявки США №62/909533, поданной 2 октября 2019 года. Полное содержание вышеуказанной заявки включено в настоящий документ посредством ссылки.
Предшествующий уровень техники настоящего изобретения
Активин-рецептороподобная киназа-2 (ALK2) кодируется геном рецептора активина А, типа I (ACVR1). ALK2 представляет собой серин/треониновую киназу в пути морфогенетического белка кости (BMP) (Shore et al., Nature Genetics 2006, 38: 525-27). Ингибиторы ALK2 и мутантные формы ALK2 потенциально могут лечить ряд заболеваний, включая прогрессирующую оссифицирующую фибродисплазию (FOP); гетеротопическую оссификацию (HO), вызванную, например, серьезным хирургическим вмешательством, травмой (например, черепно-мозговой травмой или травмой от взрыва), длительной иммобилизацией или тяжелыми ожогами; диффузную внутреннюю глиому моста (DIPG), редкую форму злокачественной опухоли головного мозга; и анемию, связанную с хроническими воспалительными, инфекционными или неопластическими заболеваниями.
В патенте США №10233186, полное содержание которого включено в настоящий документ посредством ссылки, раскрыты сильнодействующие высокоселективные ингибиторы ALK2 и мутантные формы ALK2. Также в патенте США №10233186 раскрыто Соединение 1 в качестве ключевого промежуточного соединения в синтезе многих раскрытых ингибиторов ALK2. В патенте США №10233186 раскрыты реакции Сузуки для получения промежуточных соединений, таких как Соединение 1, как показано ниже:
Соединение 1
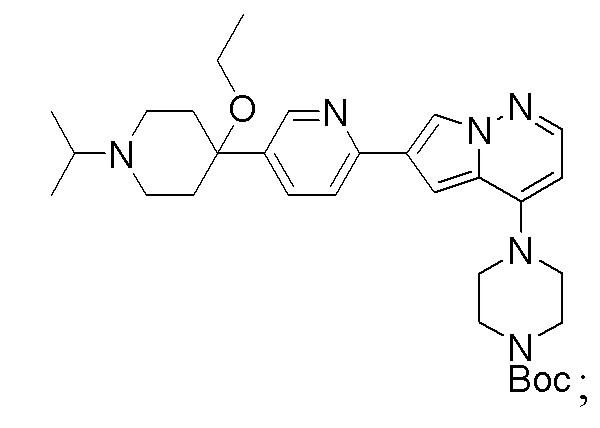
которые включают связывание пирроло-пиридазинового промежуточного соединения с пиперидинил-пиридиновым соединением.
Краткое раскрытие настоящего изобретения
Было обнаружено, что реакция сочетания Сузуки между 6-пирроло-пиридазином и (бис(пинаколато)диборон пиперидинил-пиридином для получения Соединения 1 приводит к сложному профилю чистоты, тогда как образование побочных продуктов в соответствующей реакции сочетания Негиши значительно снижается (Пример 5). Реакция сочетания Негиши имеет дополнительные преимущества, заключающиеся в том, что она требует меньших количеств дорогостоящего исходного вещества 6-пирроло-пиридазина, имеет только одну стадию выделения и использует недорогие реагенты (ZnCl2 и i-пропилмагнийхлорид). Кроме того, на основании реакций малого масштаба, ожидается, что будут получены значительно более высокие выходы, когда для получения Соединения 1 в промышленном масштабе используют реакцию сочетания Негиши, а не реакцию сочетания Сузуки. На основании этих результатов раскрыты новые и улучшенные синтезы важного промежуточного соединения 1.
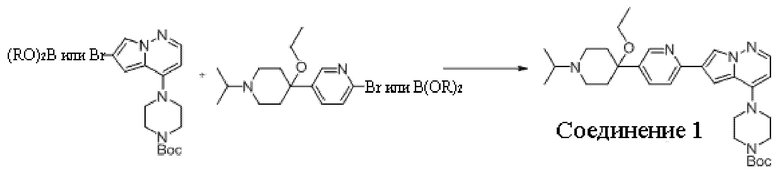
В одном из вариантов осуществления раскрытие относится к способу получения соединения, представленного формулой (I):
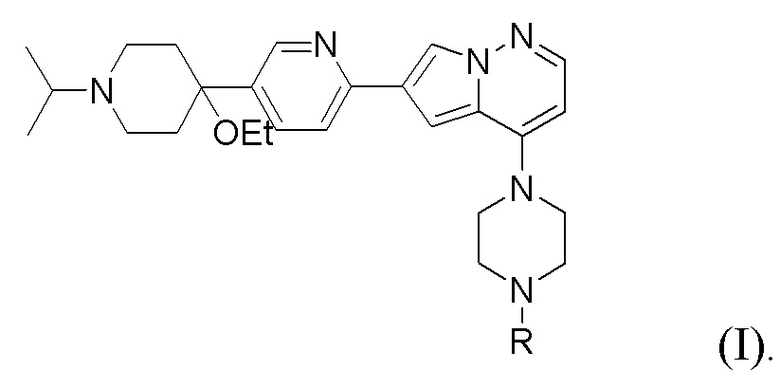
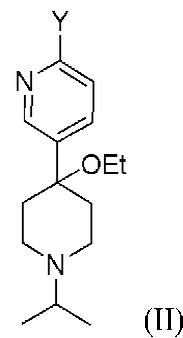
Способ предусматривает реакцию в реакционной смеси первого исходного вещества, представленного формулой (II):
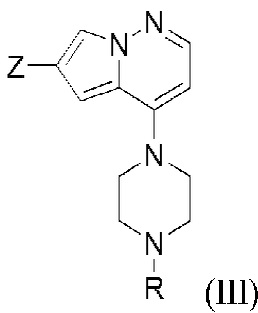
и второго исходного вещества, представленного формулой (III) в условиях Негиши:
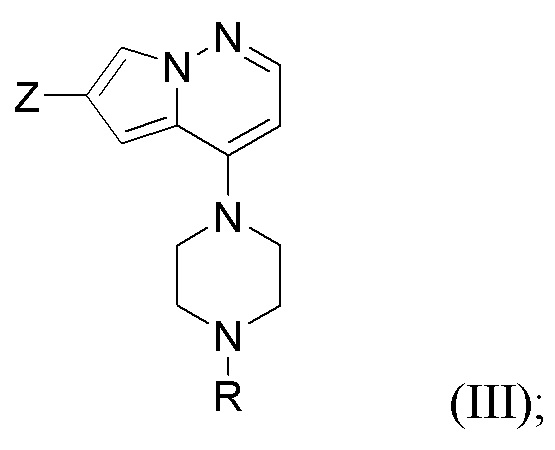
с образованием соединения формулы (I). R представляет собой защитную группу амина; Y представляет собой Cl, Br или I; и Z представляет собой Cl, Br, I или трифлат (предпочтительно, Br). Защитные группы аминов хорошо известны в данной области и раскрыты, например, в T.W. Greene and P. G. M. Wuts “Protective Groups in Organic Synthesis” John Wiley & Sons, Inc., New York 1999. Защитные группы можно включать и удалять с помощью способов, хорошо известных в данной области. Примеры защитных групп аминов включают (9-флуоренилметилкарбамат), Cbz (бензилкарбамат), Boc (трет-бутил карбамат), ацетамид, бензил, тозил (п-толуолсульфонамид). В одном из вариантов осуществления защитная группа амина представляет собой Boc.
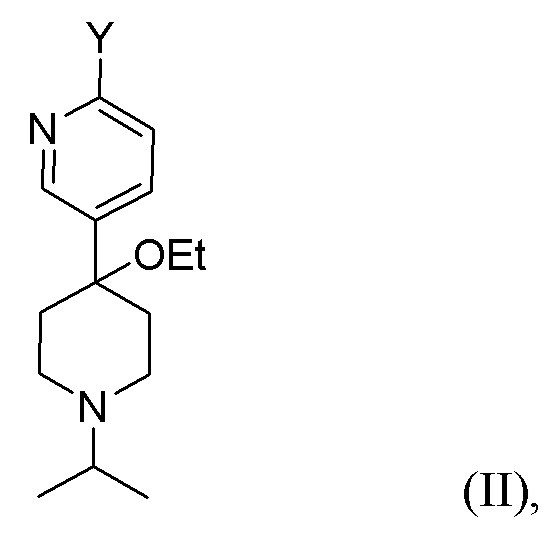
Другой вариант осуществления настоящего раскрытия представляет собой соединение, представленное формулой (II), (II-A) или (II-B):
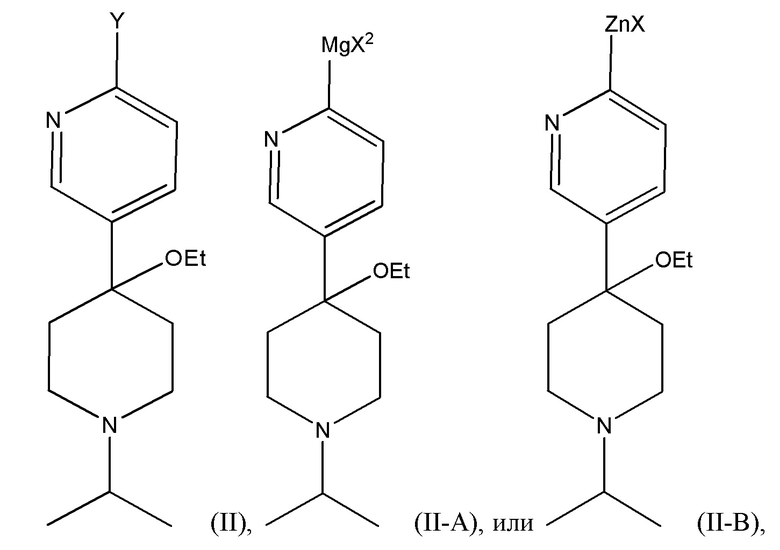
где Y, X и X2 независимо представляют собой Cl, Br или I. В одном из вариантов осуществления Y представляет собой Cl или I. Соединения, представленные формулами (II), (II-A) и (II-B), представляют собой промежуточные соединения в раскрытой реакции Негиши, как описано более подробно далее.
Краткое описание чертежей
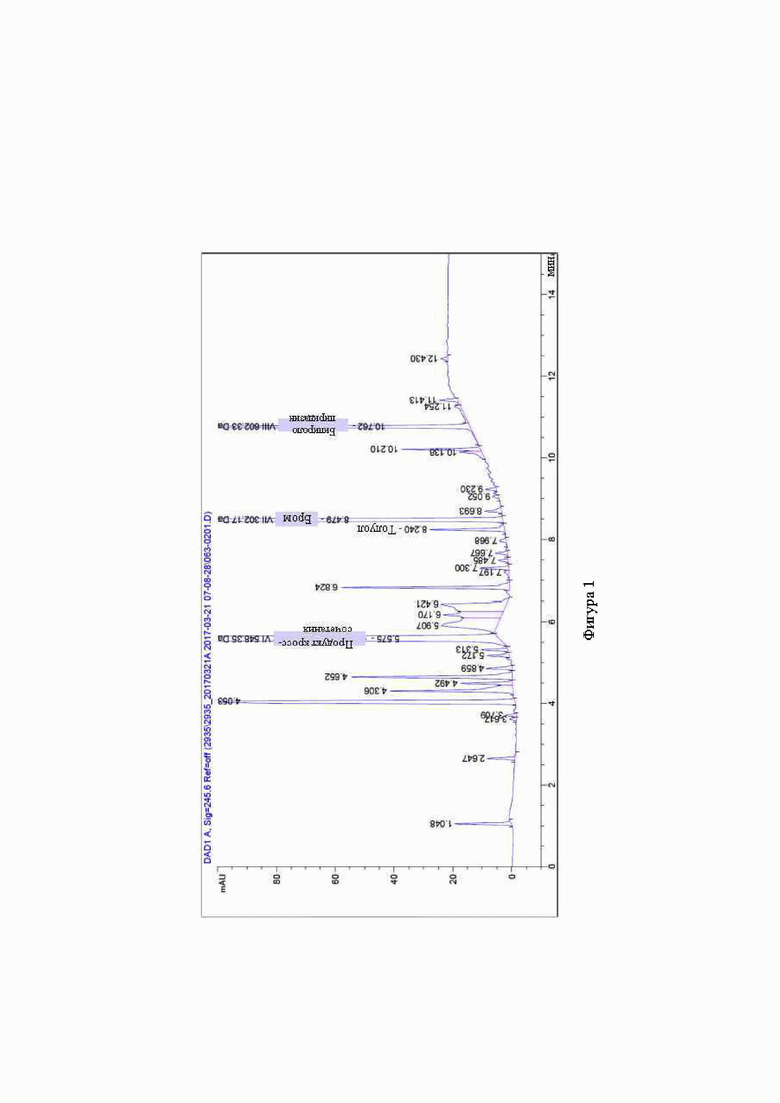
Фигура 1 представляет собой профиль чистоты ионно-парной хроматографии (IPC) для продукта реакции, полученного при получении Соединения 1 с помощью реакции сочетания Сузуки, как раскрыто в патенте США №10233186.
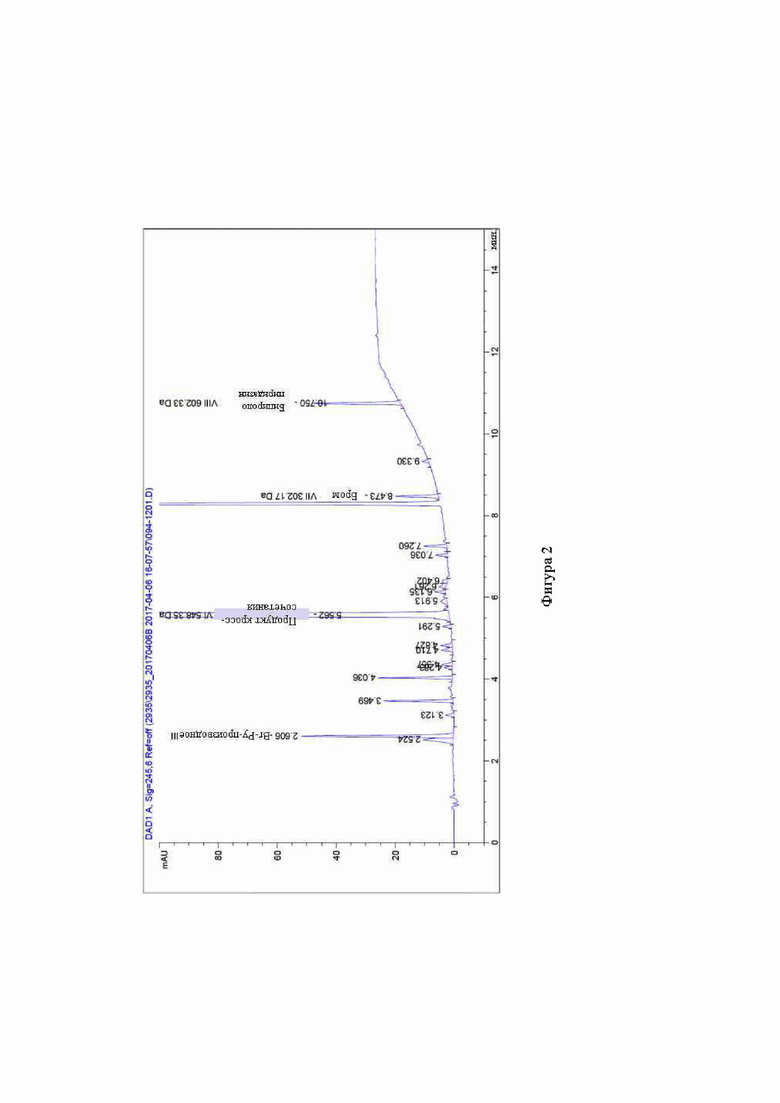
Фигура 2 представляет собой профиль чистоты IPC для продукта реакции, полученного при получении Соединения 1 способом по настоящему изобретению, т.е. реакцией сочетания Негиши.
Подробное раскрытие настоящего изобретения
Настоящее раскрытие относится к улучшенному способу получения Соединения 1 с хорошим выходом и высокой чистотой посредством реакции Негиши (также именуемой в настоящем документе как «реакция сочетания Негиши»).
Реакция Негиши представляет собой реакцию кросс-сочетания, катализируемую переходными металлами. Реакция связывает органические галогениды или трифлаты с цинкорганическими соединениями, тем самым, образуя в процессе углерод-углеродные связи (c-c). Катализатором на основе переходного металла, как правило, является палладий или никель. В случае палладия каталитическим соединением является Pd(0) в форме, например, Pd(X1)2; X1 представляет собой фосфиновый лиганд. Альтернативно, Pd(0) можно получать in situ из разновидностей Pd+2 в форме, например, Pd(X1)2Cl2. Примеры фосфиновых лигандов включают 1,1'-Бис(ди-трет-бутилфосфино)ферроцен (dtbpf), [1,1'-Бис(ди-циклогексилфосфино)ферроцен] (dcypf), 1,1'-Бис(дифенилфосфино)ферроцен (dppf), 2-Ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (t-BuXPhos), [2-(Ди-1-адамантилфосфино)-2',4',6'-триизопропил-3,6-диметоксибифенил][2-(2'-амино-1,1'-бифенил)]палладий(II) метансульфонат (AdBrettPhos), 2-Дициклогексилфосфино-2',6'-диметоксибифенил (SPhos), (2-Дициклогексилфосфино-2',6'-диизопропокси-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладий(II) метансульфонат (RuPhos), [2-Дициклогексилфосфино-2',4',6'-триизопропилбифенил] (XPhos), [(2-Ди-циклогексилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)-2-(2'-амино-1,1'-бифенил)]палладий(II) метансульфонат (BrettPhos), [(2-{Бис[3,5-бис(трифторметил)фенил]фосфин}-3,6-диметокси- 2',4',6'-триизопропил-1,1'-бифенил)-2-(2'-амино-1,1'-бифенил)]палладий(II) метансульфонат (JackiePhos), [(2-Ди-трет-бутилфосфино-3,6-диметокси-2',4',6'-триизопропил-1,1'-бифенил)-2-(2'-амино-1,1'-бифенил)]палладий(II) метансульфонат (t-BuBrettPhos), Мезил(2-(ди-трет-бутилфосфино)-1,1'-бинафтил)[2-(2'-амино-1,1'-бифенил)]палладий (TrixiePhos), (2-бифенил)ди-трет-бутилфосфин, (2-бифенилил)ди-трет-бутилфосфин (JohnPhos), 2'-(Ди-трет-бутилфосфино)-N,N-диметилбифенил-2-амин (t-BuDavePhos), 2-Ди-трет-бутилфосфино-2'-метилбифенил (t-BuMePhos), Хлор(2-дициклогексилфосфино-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладий(II) (CyJohnPhos), 2-Дициклогексилфосфино-2'-(N,N-диметиламино)бифенил (DavePhos), 2-Дициклогексилфосфино-2'-метилбифенил (MePhos), 2-Дициклогексилфосфино-2'-(N,N-диметиламино)бифенил (PhDavePhos), 2-Дициклогексилфосфино-2'-метокси-4',6'-ди-трет-бутилбифенил (VPhos), 2-[(трет-бутил)фенилфосфино]-2',6'-бис(N,N-диметиламино)бифенил (PhCPhos), [(2-Дициклогексилфосфино-2',6'-бис(N,N-диметиламино)-1,1'-бифенил)-2-(2'-амино-1,1'-бифенил)] палладий(II) метансульфонат (CPhos), Метансульфонато[2-диэтилфосфино-2',6'-бис(диметиламино)-1,1-бифенил](2'-амино-1,1'-бифенил-2-ил)палладий(II) (EtCPhos), 2-Ди(трет-бутил)фосфино-2',4',6'-триизопропил-3-метокси-6-метилбифенил (RockPhos), Ди-1-адамантил(4''-бутил-2'',3'',5'',6''-тетрафтор-2',4',6'-триизопропил-2-метокси-мета-терфенил)фосфин (AlPhos) и 2-(трет-бутилфенилфосфино)-2',6'-диметиламино-1,1'-бифенил ((t-Bu)PhCPhos).
Примеры палладиевых катализаторов включают Pd(dppe)2 (Бис[1,2-бис(дифенилфосфино)этан]палладий(0)), Pd(dba)2 (Бис(дибензилиденацетон)палладий(0)), CX-11 (димер 1,3-Бис(2,6-диизопропилфенил)имидазол-2-илиден(1,4-нафтохинон)палладия(0)), CX-12 (димер 1,3-Бис(2,4,6-триметилфенил)имидазол-2-илиден (1,4-нафтохинон)палладия(0)), Pd(t-Bu3P)2 (Бис(три-трет-бутилфосфин)палладий(0)), Pd(PCy3)2 (Бис(трициклогексилфосфин)палладий(0)), Pd(PPh3)4 (Tetrakis(трифенилфосфин)палладий(0)), Pd2(dba)3 (Tris(дибензилиденацетон)дипалладий(0)), Pd(OAc)2 (Палладия (II) ацетат), PdCl2(PPh3)2 (Дихлорбис(трифенилфосфин)палладий(II)), PdCl2(Amphos)2 (Бис(ди-трет-бутил(4-диметиламинофенил)фосфин)дихлорпалладий(II)), Pd(MeCN)2Cl2 (Бис(ацетонитрил)дихлорпалладий(II)), PdCl2(P(o-Tol)3)2 (Дихлорбис(три-o-толилфосфин)палладий(II)), Pd(dppf)Cl2 (1,1'-Бис(дифенилфосфино)ферроцен]дихлорпалладий(II)), Pd(MeCN)4(BF4)2 (Tetrakis(ацетонитрил)палладий(II) тетрафторборат), Pd-PEPPSI-IPent (Дихлор[1,3-бис(2,6-Ди-3-пентилфенил)имидазол-2-илиден](3-хлорпиридил)палладий(II)), Pd-PEPPSI-IPr ([1,3-Бис(2,6-диизопропилфенил)имидазол-2-илиден](3-хлорпиридил)палладий(II) дихлорид) и Pd-PEPPSI-SIPr ((1,3-Бис(2,6-диизопропилфенил)имидазолидон) (3-хлорпиридил) палладий(II) дихлорид).
Альтернативно, палладиевый катализатор выбран из Pd(MeCN)2Cl2, Pd[P(o-Tol)3]2Cl2, PdCl2(Amphos)2 и Pd(dba)2. В другом альтернативном варианте палладиевый катализатор выбран из Pd2(dba)3/P(R1)3, Pd(PPh3)4, Pd(PPh3)2Cl2, Pd(MeCN)2Cl2, Pd[P(o-Tol)3]2Cl2, PdCl2(Amphos)2, Pd(PtBu3)2, Pd(dppf)Cl2, Pd(dba)2, Pd2(dba)3 и Pd(XPhos); каждый R1 представляет собой C1-C6 алкил, C3-C6 циклоалкил, бензил или фенил, и где каждый бензил или фенил необязательно и независимо замещен одной или несколькими группами, выбранными из галогена, C1-C3 алкила и C1-C3 алкокси. В другом альтернативном варианте палладиевый катализатор представляет собой Pd2(dba)3. В еще одном альтернативном варианте палладиевый катализатор представляет собой PdP(tBu)3.
В другом варианте осуществления палладиевый катализатор можно комбинировать с фосфиновым лигандом, например, палладиевый катализатор представляет собой Pd2(dba) в сочетании с P(tBu)3.
В случае никеля каталитическим компонентом является Ni(0); и Ni(0) можно получать in situ из частиц Ni+2, в частности, NiCl2. Примеры никелевых катализаторов включают Ni(acac)2, Ni(cod)2, Ni(PCy3)2Cl2, NiBr2, NiI2, Ni(OAc)2, Ni(OTf)2, Ni(BF4)2, NiCl2(PPh3)2.
Органический галогенид или органический трифлат в реакции Негиши может представлять собой алкенил, арил, аллил, алкинил или пропаргилгалогенид или трифлат; и цинкорганическое соединение представляет собой R-Zn-X, в котором X представляет собой хлорид, бромид или йодид, а R представляет собой группу алкенила, арила, аллила, алкила, бензила, гомоаллила или гомопропаргила. Условия проведения реакции Негиши описаны, например, в Recent Developments in Negishi Cross-Coupling Reactions Diana Haas, Jeffrey M. Hammann, Robert Greiner, Paul Knochel* ACS Catal. 2016, V6(3) p1540-1552; Mild Negishi Cross-Coupling Reactions Catalyzed by Acenaphthoimidazolylidene Palladium Complexes at Low Catalyst Loadings Z. Liu, N. Dong, M. Xu, Z. Sun, T. Tu, J. Org. Chem., 2013, 78, 7436-7444; An Extremely Active Catalyst for the Negishi Cross-Coupling Reaction J. E. Milne, S. L. Buchwald, J. Am. Chem. Soc., 2004, 126, 13028-13032; One-Pot Negishi Cross-Coupling Reactions of In Situ Generated Zinc Reagents with Aryl Chlorides, Bromides, and Triflates S. Sase, M. Jaric, A. Metzger, V. Malakhov, P. Knochel, J. Org. Chem., 2008, 73, 7380-7382; Efficient Negishi Coupling Reactions of Aryl Chlorides Catalyzed by Binuclear and Mononuclear Nickel-N-Heterocyclic Carbene Complexes Z. Xi, Y. Zhou, W. Chen, J. Org. Chem., 2008, 73, 8497-8501; Cross-Coupling of Aryltrimethylammonium Iodides with Arylzinc Reagents Catalyzed by Amido Pincer Nickel Complexes X.-Q. Zhang, Z.-X. Wang, J. Org. Chem., 2012, 77, 3658-3663; Efficient Cross-Coupling of Aryl Chlorides with Arylzinc Reagents Catalyzed by Amido Pincer Complexes of Nickel L. Wang, Z.-X. Wang, Org. Lett., 2007, 9, 4335-4338; Highly Regio- and Stereoselective Synthesis of (Z)-Trisubstituted Alkenes via Propyne Bromoboration and Tandem Pd-Catalyzed Cross-Coupling C. Wang, T. Tobrman, Z. Xu, E.-i. Negishi, Org. Lett., 2009, 11, 4092-4095; Highly Regioselective Synthesis of Trisubstituted Allenes via Lithiation of 1-Aryl-3-alkylpropadiene, Subsequent Transmetalation, and Pd-Catalyzed Negishi Coupling Reaction J. Zhao, Y. Liu, S. Ma, Org. Lett., 2008, 10, 1521-1523; High Temperature Metalation of Functionalized Aromatics and Heteroaromatics using (tmp)2Zn⋅2MgCl2⋅2LiCl and Microwave Irradiation S. Wunderlich, P. Knochel, Org. Lett., 2008, 10, 4705-4707; A Mild Negishi Cross-Coupling of 2-Heterocyclic Organozinc Reagents and Aryl Chlorides M. R. Luzung, J. S. Patel, J. Yin, J. Org. Chem., 2010, 75, 8330-8332; Synthesis of Substituted Cyclopropanecarboxylates via Room Temperature Palladium-Catalyzed α-Arylation of Reformatsky Reagents S. N. Greszler, G. T. Halvorsen, E. A. Voight, Org. Lett., 2017, 19, 2490-2493; and Enantioselective, Palladium-Catalyzed α-Arylation of N-Boc-pyrrolidine K. R. Campos, A. Klapars, J. H. Waldman, P.G. Dormer, C.-Y. Chen, J. Am. Chem. Soc., 2006, 128, 3538-3539.
Арилы цинка можно получать в мягких условиях реакции путем реакции Гриньяра или литийорганического промежуточного соединения с галогенидами цинка, такими как ZnCl2 или ZnBr2. См., например, Recent Developments in Negishi Cross-Coupling Reactions Diana Haas, Jeffrey M. Hammann, Robert Greiner, Paul Knochel* ACS Catal.2016, V6(3) p1540-1552. Giovannini R, Knochel P (1998). "Ni(II)-Catalyzed Cross-Coupling between Polyfunctional Arylzinc Derivatives and Primary Alkyl Iodides". Journal of the American Chemical Society. 120 (43): 11186-11187. doi:10.1021/ja982520o. Jie Jack Li, Chapter 3 - Applications of Palladium Chemistry to the Total Synthesis of Naturally Occurring of Indole Alkaloids in “Alkaloids: Chemical and Biological Perspectives” 14: 437-503 (1999). В некоторых случаях, цинкорганическое соединение можно получать непосредственно путем реакции с ZnCl2 (S.P. Nolan and O. Navarro, 11.01 - C-C Bond Formation by Cross-coupling in “Comprehensive Organometallic Chemistry III” 11: 1-37 (2007).
Термин «в условиях Негиши» означает катализируемую переходными металлами реакцию кросс-сочетания, реакцию, образующую углерод-углеродную связь между органическим галогенидом и цинкорганическим соединением. «В условиях Негиши» также означаетт образование цинкорганического соединения, в частности, путем реакции Гриньяра или литийорганического промежуточного соединения с галогенидом цинка.
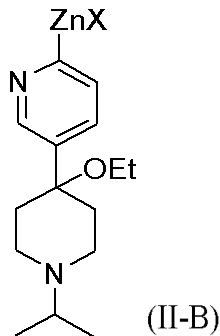
В одном из аспектов, i) первое исходное вещество превращается в цинкорганическое промежуточное соединение, представленное формулой (II-B):
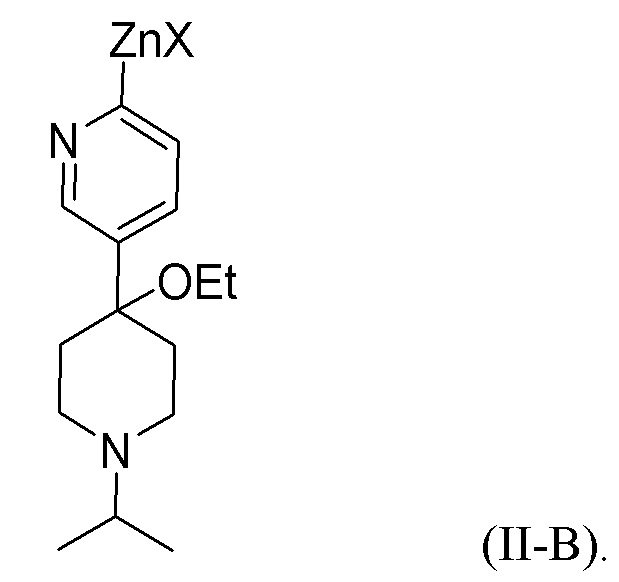
Цинкорганическое промежуточное соединение вступает в реакцию со вторым исходным веществом в присутствии палладиевого катализатора с образованием соединения формулы (I). X представляет собой Cl, Br или I. Альтернативно, X в цинкорганическом промежуточном соединении (формула (II-B) представляет собой Cl; и Y во втором исходном веществе (формула II) представляет собой Br. Подходящие растворители для этой реакции включают эфирные растворители, такие как тетрагидрофуран, метилтетрагидрофуран, анизол и их смесь. Цинкоорганическое промежуточное соединение часто реагирует с исходным вторым веществом без выделения цинкорганического промежуточного соединения.
В одном из аспектов раствор алкоксида или аминового основания в эфирном растворителе объединяют с цинкорганическим промежуточным соединением перед реакцией со вторым исходным веществом. Добавление основания к реакционной смеси имеет то преимущество, что уменьшает образование побочного продукта. Примеры подходящих оснований включают трет-бутоксид калия (KOtBu), морфолин, пиперазин, бензилпиперазин, NH3, NH4Cl и гексаметилдисилазан. Широко используется KOtBu. В одном из аспектов, в пределах от 0,5 до 3,0 эквивалентов основания по отношению к первому исходному веществу. В одном из аспектов, в пределах от 1,5 до 3,0 эквивалентов основания по отношению к первому исходному веществу. Метилтетрагидрофуран широко используется в качестве эфирного растворителя.
В еще одном аспекте реакцию с цинкорганическим промежуточным соединением и вторым исходным веществом проводят в присутствии полярного апротонного растворителя, такого как N-метил-2-пирролидинон, диметилформамид или диметилсульфоксид. Широко используется N-метил-2-пирролидинон. В одном из аспектов, применяют от 0,05 до 1,5 мл полярного апротонного растворителя на грамм исходного вещества.
В другом аспекте реакционную смесь, содержащую продукт реакции цинкорганического промежуточного соединения и второго исходного вещества, экстрагируют основным водным раствором N-ацетил-L-цистеина после образования соединения формулы (I). Концентрация раствора N-ацетил-L-цистеина составляет, в основном, менее 1 г на 5 мл воды. «Экстракция» реакционной смеси относится к промыванию реакционной смеси непосредственно раствором N-ацетил-L-цистеина с образованием водной фазы и органической фазы. Органическую фазу, содержащую Соединение 1, затем отделяют от водной фазы. В качестве альтернативы, «экстракция» относится к остановке реакции в реакционной смеси водным раствором с получением водной фазы и органической фазы. Органическую фазу, содержащую Соединение 1, затем отделяют от водной фазы и затем экстрагируют раствором N-ацетил-L-цистеина. Преимущество экстракции раствором N-ацетил-L-цистеина заключается в снижении остаточного содержания палладия в конечном продукте реакции (Соединение 1).
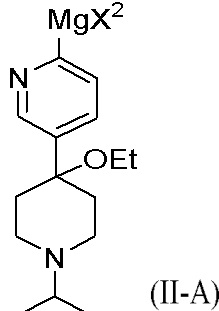
В другом аспекте цинкорганическое промежуточное соединение получают путем реакции первого исходного вещества с реактивом Гриньяра R'MgX2 с образованием металлоорганического промежуточного соединения, представленного формулой (II-A):
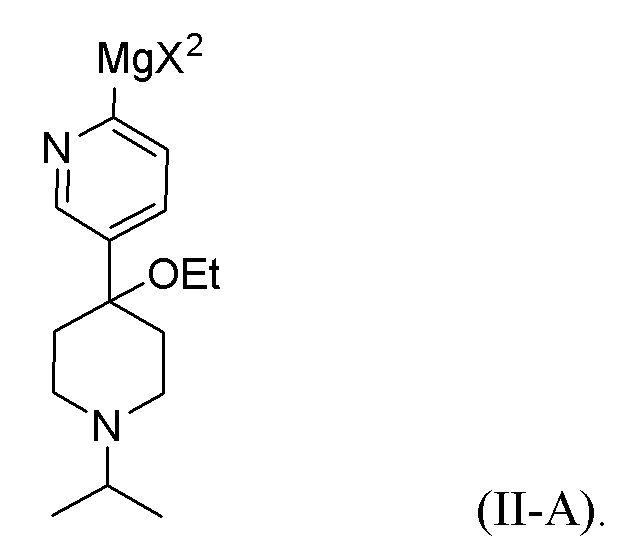
Металлоорганическое промежуточное соединение затем реагирует с ZnX2 с образованием цинкорганического промежуточного соединения. R' представляет собой C1-C6 алкил, C1-C6 алкенил, C1-C6 алкинил, фенил, бензил или моноциклический гетероарил; каждый фенил, бензил или гетероарил необязательно и независимо замещен одной или несколькими группами, выбранными из галогена, C1-C3 алкила и C1-C3 алкоксигруппы; и X2 представляет собой Cl, Br или I. В одном из аспектов реактив Гриньяра представляет собой изопропилхлорид магния (i-PrMgCl). Обычно металлорганическое промежуточное соединение взаимодействует с ZnX2 без выделения металлорганического промежуточного соединения.
Реакцию первого исходного вещества с реактивом Гриньяра можно проводить в эфирных растворителях. Одним из широко используемых эфирных растворителей является тетрагидрофуран. В одном из аспектов, реакцию первого исходного вещества с реактивом Гриньяра проводят в смеси, содержащей анизол и эфирный растворитель. Преимущество использования анизола в реакционной смеси заключается в уменьшении побочных продуктов. В одном из аспектов, реакцию первого исходного вещества с реактивом Гриньяра проводят в смеси, содержащей ароматический растворитель, такой как бензол, толуол, ксилол и их смесь.
Конкретные условия получения Соединения 1 способами настоящего раскрытия представлены в Примерах 1 и 5. Соединение 1 можно легко превратить в ингибиторы ALK-2 путем удаления защитной группы амина и карбамоилирования полученного свободного амина в желаемый ингибитор ALK-2. Подходящие условия для этих двух преобразований раскрыты в патенте США №10233186. Конкретные условия для удаления защитной группы Boc приведены в Примере 2 ниже; и в Примере 3 для карбамоилирования.
Следующие примеры предназначены для иллюстрации и никоим образом не предназначены для ограничения объема раскрытия.
Иллюстративные примеры
Пример 1. Получение трет-бутил 4-(6-(5-(4-Этокси-1-Изопропилпиперидин-4-ил)Пиридин-2-ил)пирроло[1,2-b]пиридазин-4-ил)Пиперазин-1-Карбоксилата
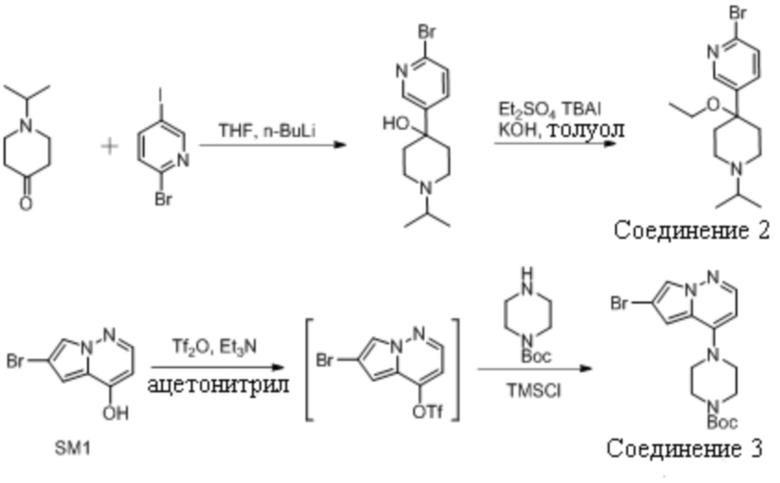
1.1 Получение 2-бромо-5-(4-этокси-1-изопропилпиперидин-4-ил)пиридина (Соединение 2)
Соединение 2 получали из исходных веществ 1-изопропилпиперидин-4-она и 2-бром-5-йодпиридина синтетическим путем, как показано на схеме выше. Оно также было коммерчески доступно у Acceledev.
1.2 Получение трет-бутил 4-(6-бромопирроло[1,2-b]пиридазин-4-ил)пиперазин-1-карбоксилата (Соединение 3)
Смесь 6-бромпирроло[1,2-b]пиридазин-4-ола (5,5 кг, 25,8 моль) и триэтиламина (3,1 кг, 1,2 экв) в ацетонитриле (27 л, 4,9 объемов) перемешивали при температуре -10°C. К этой смеси добавляли трифторметансульфоновый ангидрид (7,1 кг, 0,98 экв) с дополнительным промыванием ацетонитрилом (2 л, 0,36 объемов). Реакцию перемешивали до завершения реакции, после чего добавляли триметилсилилхлорид (0,3 кг, 0,1 экв.). Затем к смеси добавляли триэтиламин (3,7 кг, 1,3 экв) и N-Boc-пиперазин (5,5 кг, 1,2 экв) с дополнительными промываниями ацетонитрилаом (2 л, 0,36 объемов). Реакцию нагревали до 65-75°С до завершения реакции. Реакционную смесь концентрировали при 45-55°С и разбавляли водой перед охлаждением до 15-25°С для кристаллизации продукта. Продукт фильтровали и промывали изопропанолом (2×11 л, 2×2 объема) с получением 7,0 кг (выход 72,4%).
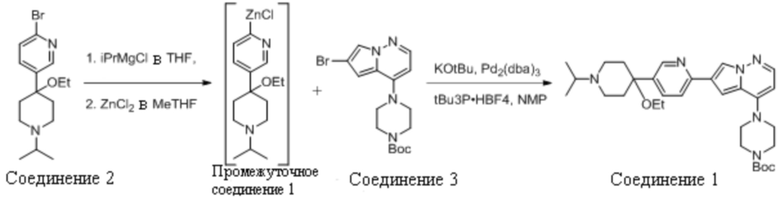
1.3 Получение трет-бутил 4-(6-(5-(4-Этокси-1-Изопропилпиперидин-4-ил)Пиридин-2-ил)пирроло[1,2-b]пиридазин-4-ил)Пиперазин-1-Карбоксилата (Соединение 1)
2-Бром-5-(4-этокси-1-изопропилпиперидин-4-ил)пиридин (Соединение 2, 5,9 кг, приготовленное, как раскрыто в US10233186) растворяли в анизоле (24 л). Полученный раствор нагревали до 90-100°С и частично перегоняли для снижения содержания воды. Оставшуюся реакционную смесь охлаждали до 45-55°C и добавляли изопропилхлорид магния (20% в тетрагидрофуране, 9,6 кг). После того как наблюдали полное превращение, добавляли раствор хлорида цинка (25% в метилтетрагидрофуране, 10,7 кг) при продолжающемся нагревании. В полученный раствор органического цинка (Промежуточное соединение 1) затем загружали трет-бутоксид калия (25% в метилтетрагидрофуране, 17,6 кг), Pd2(dba)3 (18 г) и трет-Bu3P⋅HBF4 (23 г). Дополнительно добавляли N-метил-2-пирролидон (0,6 л), а затем трет-бутил 4-(6-бромпирроло[1,2-b]пиридазин-4-ил)пиперазин-1-карбоксилат (Соединение 3, приготовленное , как раскрыто в US 10233186) в виде раствора в тетрагидрофуране (6,0 кг Соединения 3 в 9 л тетрагидрофурана). Нагревание продолжали до полного превращения в Соединение 1.
Пример 2 Получение 6-(5-(4-этокси-1-изопропилпиперидин-4-ил)пиридин-2-ил)-4-(пиперазин-1-ил)пирроло[1,2-b]пиридазин (Соединение 4)
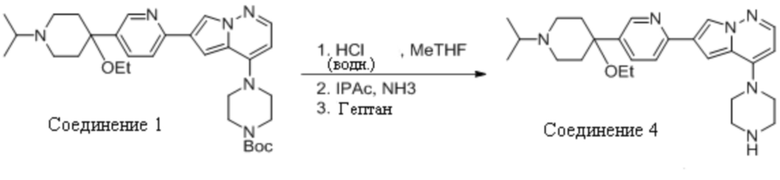
Соединение 1, приготовленное, как описано в примере 1, использовали без очистки или выделения из реакционной смеси. Реакционную смесь разбавляли тетрагидрофураном (8,8 кг) и 33% соляной кислотой (21 кг) в воде (36 кг). Кислую водную смесь, содержащую продукт, отмывали метилтетрагидрофураном (31 кг), а затем изопропилацетатом (2×11 кг). Водный раствор затем разбавляли изопропилацетатом (53 кг) и подщелачивали аммиаком (25%, 33 кг). Органическую фазу отделяли и промывали ацетилцистеином (3×26 кг) и водой (2×18 кг). В органический раствор вносили затравку и добавляли гептан (41,7 кг) для кристаллизации продукта, который затем выделяли фильтрованием с получением приблизительно 3,6 кг Соединения 4 после сушки.
Пример 3 Получение ингибитора ALK-2 из Соединения 4
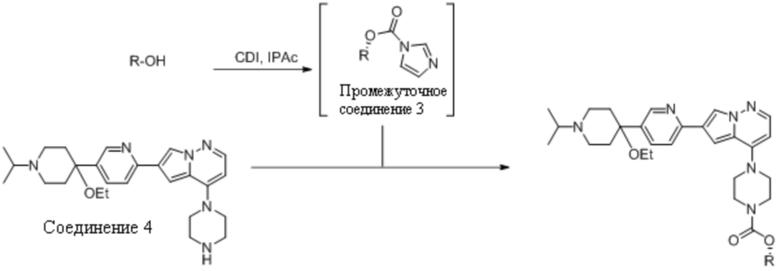
В реактор добавляли 1,1-карбонилдиимидазол (1,51 кг), изопропилацетат (6,4 кг) и спирт R-OH (например, 1,1 эквивалента по отношению к 1,1-карбонилдиимидазолу). Реакционную смесь перемешивали в течение 30 минут при 20-30°C до завершения реакции. Реакцию нагревали до 30-40°С и фильтровали, промывая фильтр изопропилацетатом (7,7 кг). В разбавленную смесь загружали аммиак (25%, 3,2 кг) и Соединение 4 (3,5 кг). Реакцию нагревали до 50-60°С и перегоняли в вакууме. Смесь дополнительно разбавляли изопропилацетатом (6,1 кг) и подтверждали завершение реакции. В это время реакционную смесь разбавляли водой (3,5 кг) и изопропилацетатом (36,9 кг) и перемешивали. Смеси давали осесть, и отделяли органическую фазу. Органический раствор далее промывали водой (5×7 кг). Органическую фазу после окончательного разделения разбавляли изопропилацетатом (7,7 кг) и перегоняли при 50-60°C в вакууме для уменьшения содержания воды. Затем органический растворитель можно удалить, чтобы выделить ингибитор ALK-2.
Пример 4 N-ацетилцистеин значительно более эффективен при удалении палладия из реакции из Примера 1, чем карбонат калия и 2,4,6-тримеркапто1,3,5-триазин (ТМТ)
К 1,15 эквивалентам 2-бром-5-(4-этокси-1-изопропилпиперидин-4-ил)пиридина (Соединение 2) добавляли 6 объемов толуола и смесь нагревали до температуры рубашки охлаждающей жидкости (JT) 145°C1 (1Все эквиваленты и объемы относятся к Соединению 3, в котором один эквивалент составляет 30,4 г. Объем составляет 1 мл/1 грамм эталонного материала, в данном случае Соединения 3). При внутренней температуре (IT) 112-121°С перегоняли 4 объема толуола. При IT 45°C 1,37 эквивалента изопропилхлорида магния (2 М в тетрагидрофуране) добавляли в целом до завершения реакции. В это время 1,20 эквивалента ZnCl2 (2М в тетрагидрофуране) добавляли при 45°C и смесь перемешивали при JT 80°C в течение 14 часов. При IT 45°С добавляли 0,1 объема N-метил-2-пирролдинона, 1 моль% Pd2(dba)3, 4 моль % tBu3P⋅HBF4. Раствор 1,0 эквивалента Соединения 3 (30,4 г) в 2,5 объемах метилтетрагидрофурана (MeTHF) добавляли в течение 30 минут при IT 45°C. Через 1 час добавляли дополнительно 0,15 эквивалента Соединения 3 в 0,4 объема тетрагидрофурана. Смесь перемешивали в течение 2 часов при IT 45°C, переливали в другой реакционный сосуд, промывали 1,6 объемами MeTHF, добавляли в течение 10 минут к раствору 4,1 эквивалента K2CO3 в 25 объемах воды при 24°C. Затем, добавляли 0,43 об. 30%-масс./масс. NaOH, отделяли водную фазу, а органическую фазу разделяли на три части. Каждую порцию отмывали 2×1,6 объема либо (i) карбоната калия, либо (ii) 2,4,6-тримеркапто-1,3,5-триазина (ТМТ), либо (iii) N-ацетилцистеина. Затем добавляли 3,3 объема воды и 0,95 объема 2M HCl и отделяли органическую фазу. Затем добавляли 0,30 объема 30%-масс./масс. NaOH и Norit CGP super (около 0,5 г). Смесь перемешивали при IT 45°C в течение 2 часов, фильтровали и промывали 0,33 объемами MeTHF. Органическую фазу отмывали с 2×1 объема воды. Добавляли Boc2O (0,08 об., 2M) и смесь перемешивали в течение 15 минут при 24°C. MeTHF перегоняли при JT 80-120°C с одновременным добавлением н-гептана (3×1,6 объема). Коричневую взвесь охлаждали до 0°С, перемешивали в течение 15 минут, фильтровали, промывали 2×0,7 объемами н-гептана (повторные промывания взвеси).
Реакционную смесь с остановленной реакцией делили на три порции, и содержание палладия, определенное методом масс-спектрометрии с индуктивно связанной плазмой (IPC-MS), составляло: Эталон (K2CO3): 1400 м.д. Pd; 2 х экстракция с ТМТ: 1000 м.д. Pd; и 2 х экстракция с N-ацетилцистеином: 120 м.д. Pd.
Пример 5. Способ получения Соединения 1 по Негиши дает выход продукта с меньшим количеством примесей, чем соответствующий способ Сузуки
В сосуд, содержащий Соединение 4 (1,0 г) в воде 2,4 мл (2,4 объема) и 1,4-диоксан (12,0 мл, 12 объемов) добавляли 0,68 г (1,0 экв.) 2-бром-5-(4-этокси-1-изопропилпиперидин-4-ил)пиридина (Соединение 2), K2CO3 (0,57 г, 2,0 экв.) и Pd(PPh3)4 (0,12 г , 0,05 экв.). Реакцию нагревали до 80°С до завершения реакции, наблюдаемого по ВЭЖХ (фигура 1).
К 3,27 г 2-бром-5-(4-этокси-1-изопропилпиперидин-4-ил)пиридина (Соединение 2) добавляли 6 объемов толуола и 1,2 экв. iPrMgCl (2М в THF). Смесь нагревали до JT 65°C. После завершения реакции добавляли 1,3 экв ZnCl2 (1,9М в MeTHF) и реакцию проводили при 20-25°С. При 45°С добавляли Соединение 3 с PdP(tBu3)2 (0,1 экв.) с THF (3 объема) и смесь перемешивали до завершения реакции, наблюдаемого по ВЭЖХ (фигура 2).
Из фигур 1 и 2 видно, что получение Соединения 1 по способу настоящего раскрытия проходит значительно чище и производит гораздо меньше примесей, чем соответствующая реакция сочетания Сузуки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ХИМИЧЕСКИЙ СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИРИМИДИНА И ИХ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2015 |
|
RU2703300C2 |
| СПОСОБ ПОЛУЧЕНИЯ 1-[2-(2,4-ДИМЕТИЛФЕНИЛСУЛЬФАНИЛ)ФЕНИЛ]ПИПЕРАЗИНА | 2012 |
|
RU2608307C2 |
| СПОСОБ КРОСС-СОЧЕТАНИЯ ИНДОЛОВ | 2005 |
|
RU2430916C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2,3-ДИЗАМЕЩЕННЫХ ИНДОЛОВ | 2006 |
|
RU2466126C2 |
| СПОСОБЫ КРОСС-СОЧЕТАНИЯ | 2017 |
|
RU2777977C2 |
| СИНТЕЗ ТРЕТ-БУТИЛОВОГО СЛОЖНОГО ЭФИРА (4-ФТОР-3-ПИПЕРИДИН-4-ИЛ-БЕНЗИЛ)-КАРБАМИНОВОЙ КИСЛОТЫ И ЕГО ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ | 2010 |
|
RU2543483C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ОКСАЗОЛИДИНОНОВ И СОДЕРЖАЩИХ ИХ КОМПОЗИЦИЙ | 2009 |
|
RU2556234C2 |
| Способ получения N,N-диметил-4-бифениламина и его производных | 2022 |
|
RU2794095C1 |
| ПОДГОТОВКА И ИСПОЛЬЗОВАНИЕ ИНГИБИТОРА КИНАЗЫ | 2016 |
|
RU2691401C2 |
| СПОСОБ ПОЛУЧЕНИЯ 5-(1-ПИПЕРАЗИНИЛ)БЕНЗОФУРАН-2-КАРБОКСАМИДА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2000 |
|
RU2266292C2 |
Группа изобретений относится к области органической химии и включает способ получения соединения формулы (I) и промежуточные соединения формул (II-A), (II-B). Способ предусматривает взаимодействие соединения формулы (II) с соединением формулы (III) в условиях реакции Негиши с получением соединения формулы (I). В формулах (I), (II), (III) R представляет собой защитную группу, выбранную из 9-флуоренилметилкарбамата, бензилкарбамата, трет-бутоксикарбонила, ацетамида, бензила, и тозила; Y представляет собой Cl, Br или I; Z представляет собой Cl, Br, I или трифлат. В формулах (II-A), (II-B) X и X2 независимо представляют собой Cl, Br или I. Технический результат – усовершенствование способа получения соединения формулы (I) за счет использования меньшего количества дорогого исходного материала, уменьшения количества стадий и обеспечения более высокого выхода и чистоты получаемого продукта. 2 н. и 24 з.п. ф-лы, 2 ил., 5 пр.
1. Способ получения соединения, представленного формулой (I)
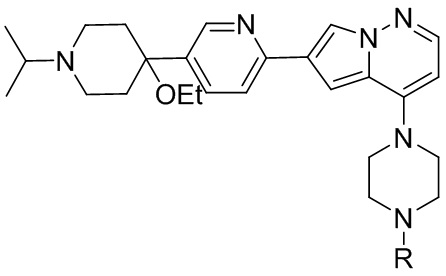 (I),
(I),
предусматривающий реакцию в реакционной смеси первого исходного вещества, представленного формулой (II)
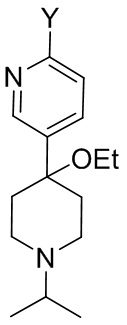 (II),
(II),
и второго исходного вещества, представленного формулой (III), в условиях Негиши
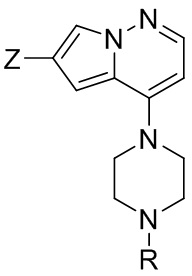 (III),
(III),
с получением соединения формулы (I), где R представляет собой защитную группу амина, выбранную из Fmoc (9-флуоренилметилкарбамата), Cbz (бензилкарбамата), Boc (трет-бутилоксикарбонила), ацетамида, бензила и тозила (п-толуолсульфонамида);
Y представляет собой Cl, Br или I; и
Z представляет собой Cl, Br, I или трифлат.
2. Способ по п. 1, в котором реакция опосредуется палладиевым катализатором.
3. Способ по п. 2, в котором:
i) первое исходное вещество формулы (II) превращают в цинкорганическое промежуточное соединение, представленное формулой (II-B)
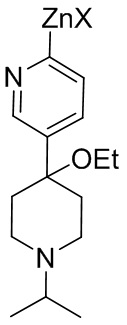 (II-B); и
(II-B); и
ii) проводят реакцию цинкорганического промежуточного соединения формулы (II-B) со вторым исходным веществом формулы (III) в присутствии палладиевого катализатора с получением соединения формулы (I), где X представляет собой Cl, Br или I.
4. Способ по любому из пп. 1-3, в котором X представляет собой Cl, а Y представляет собой Br.
5. Способ по п. 3 или 4, в котором проводят реакцию цинкорганического промежуточного соединения формулы (II-B) со вторым исходным веществом формулы (III) без выделения цинкорганического промежуточного соединения формулы (II-B).
6. Способ по любому из пп. 2-5, в котором палладиевый катализатор выбран из Pd(X1)2 и Pd(X1)2Cl2, причем каждый X1 независимо представляет собой фосфиновый лиганд.
7. Способ по п. 6, в котором фосфиновый лиганд выбран из dtbpf, dcypf, dppf, t-BuXPhos, AdBrettPhos, SPhos, RuPhos, XPhos, BrettPhos, JackiePhos, t-BuBrettPhos, TrixiePhos, JohnPhos, t-BuDavePhos, t-BuMePhos, CyJohnPhos, DavePhos, MePhos, PhDavePhos, VPhos, PhCPhos, CPhos, EtCPhos, RockPhos, AlPhos и (t-Bu)PhCPhos.
8. Способ по п. 6, в котором палладиевый катализатор выбран из
Pd(dppe)2 (бис[1,2-бис(дифенилфосфино)этан]палладий(0)),
Pd(dba)2 (бис(дибензилиденацетон)палладий(0)),
CX-11 (димер 1,3-бис(2,6-диизопропилфенил)имидазол-2-илиден(1,4-нафтохинон)палладия(0)),
CX-12 (димер 1,3-бис(2,4,6-триметилфенил)имидазол-2-илиден (1,4-нафтохинон)палладия(0)),
Pd(t-Bu3P)2 (бис(три-трет-бутилфосфин)палладий(0)), Pd(PCy3)2 (бис(трициклогексилфосфин)палладий(0)),
Pd(PPh3)4 (тетракис(трифенилфосфин)палладий(0)), Pd2(dba)3 (трис(дибензилиденацетон)дипалладий(0)),
Pd(OAc)2 (палладия (II) ацетат),
PdCl2(PPh3)2 (дихлорбис(трифенилфосфин)палладий(II)),
PdCl2(Amphos)2 (бис(ди-трет-бутил(4-диметиламинофенил)фосфин)дихлорпалладий(II)),
Pd(MeCN)2Cl2 (бис(ацетонитрил)дихлорпалладий(II)),
PdCl2(P(o-Tol)3)2 (дихлорбис(три-o-толилфосфин)палладий(II)),
Pd(dppf)Cl2 (1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладий(II)),
Pd(MeCN)4(BF4)2 (тетракис(ацетонитрил)палладия(II) тетрафторборат),
Pd-PEPPSI-IPent (дихлор[1,3-бис(2,6-ди-3-пентилфенил)имидазол-2-илиден](3-хлорпиридил)палладий(II)),
Pd-PEPPSI-IPr (дихлорид [1,3-бис(2,6-диизопропилфенил)имидазол-2-илиден](3-хлорпиридил)палладия(II)) и
Pd-PEPPSI-SIPr (дихлорид (1,3-бис(2,6-диизопропилфенил)имидазолиден) (3-хлорпиридил) палладия(II)).
9. Способ по п. 6, в котором палладиевый катализатор выбран из Pd(MeCN)2Cl2, Pd[P(o-Tol)3]2Cl2, PdCl2(Amphos)2 и Pd(dba)2.
10. Способ по п. 6, в котором палладиевый катализатор выбран из Pd2(dba)3/P(R1)3, Pd(PPh3)4, Pd(PPh3)2Cl2, Pd(MeCN)2Cl2, Pd[P(o-Tol)3]2Cl2, PdCl2(Amphos)2, Pd(PtBu3)2, Pd(dppf)Cl2, Pd(dba)2, Pd2(dba)3 и Pd(XPhos), причем каждый R1 представляет собой C1-C6 алкил, C3-C6 циклоалкил, бензил или фенил, причем каждый бензил или фенил необязательно и независимо замещен одной или несколькими группами, выбранными из галогена, C1-C3 алкила и C1-C3 алкокси.
11. Способ по п. 6, в котором палладиевый катализатор представляет собой Pd2(dba)3.
12. Способ по п. 6, в котором палладиевый катализатор представляет собой PdP(tBu)3.
13. Способ по любому из пп. 2-12, в котором первое исходное вещество формулы (II) превращают в цинкорганическое промежуточное соединение формулы (II-B) за счет реакции первого исходного вещества формулы (II) с реагентом Гриньяра R'MgX2 с получением металлоорганического промежуточного соединения, представленного формулой (II-A)
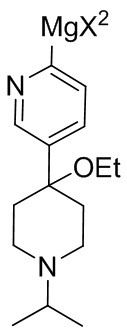 (II-A),
(II-A),
и проводят реакцию металлоорганического промежуточного соединения формулы (II-A) с ZnX2 с получением цинкорганического промежуточного соединения формулы (II-В), причем R’ представляет собой C1-C6 алкил, C1-C6 алкенил, C1-C6 алкинил, фенил, бензил или моноциклический гетероарил, причем каждый фенил, бензил или гетероарил необязательно и независимо замещен одной или несколькими группами, выбранными из галогена, C1-C3 алкила и C1-C3 алкоксигруппы, и X2 представляет собой Cl, Br или I.
14. Способ по п. 13, в котором реагент Гриньяра представляет собой i-PrMgCl.
15. Способ по п. 13, в котором проводят реакцию металлоорганического промежуточного соединения формулы (II-A) с ZnX2 без выделения металлоорганического промежуточного соединения формулы (II-A).
16. Способ по любому из пп. 13-15, в котором проводят реакцию первого исходного вещества формулы (II) с реагентом Гриньяра в смеси, содержащей анизол.
17. Способ по п. 16, в котором проводят реакцию первого исходного вещества формулы (II) с реагентом Гриньяра в смеси анизола и эфирного растворителя.
18. Способ по п. 17, в котором эфирный растворитель представляет собой тетрагидрофуран.
19. Способ по любому из пп. 3-18, в котором KOtBu в эфирном растворителе объединяют с цинкорганическим промежуточным соединением формулы (II-В) перед реакцией со вторым исходным веществом формулы (III).
20. Способ по п. 19, в котором цинкорганическое промежуточное соединение формулы (II-В) объединяют с KOtBu в тетрагидрофуране.
21. Способ по п. 19 или 20, в котором реакцию цинкорганического промежуточного соединения формулы (II-В) и второго исходного вещества формулы (III) проводят в присутствии N-метил-2-пироллидинона.
22. Способ по любому из пп. 1-21, дополнительно предусматривающий экстрагирование реакционной смеси после образования соединения формулы (I) при помощи основного водного раствора N-ацетил-L-цистеина.
23. Способ по любому из пп. 1-22, в котором R представляет собой трет-бутоксикарбонил.
24. Способ по любому из пп. 1-23, в котором Z представляет собой Br.
25. Соединение, представленное формулой (II-A) или (II-B)
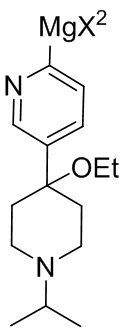 (II-A) или
(II-A) или 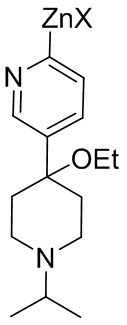 (II-B),
(II-B),
где каждый X и X2 независимо представляет собой Cl, Br или I.
26. Способ по любому из пп. 13-15, в котором проводят реакцию первого исходного вещества формулы (II) с реагентом Гриньяра в смеси, содержащей ароматический растворитель, выбранный из группы, состоящей из бензола, толуола и ксилола.
| US 10233186 B2, 19.03.2019 | |||
| WO 1996040684 A1, 19.12.1996 | |||
| Способ выращивания и подготовки растений для посадочных машин | 1929 |
|
SU20847A1 |

