ПРИТЯЗАНИЕ НА ПРИОРИТЕТ
Данная заявка претендует на приоритет заявки Соединенных Штатов, серийный номер 61/097,716, поданной 17 сентября 2008 г., заявки Соединенных Штатов, серийный номер 61/141,686, поданной 31 декабря 2008 г., и заявки Соединенных Штатов, серийный номер 61/161,387, поданной 18 марта 2009 г., полное раскрытие которых включено в данное описание путем ссылки.
ОБЛАСТЬ ТЕХНИКИ
Изобретение касается, главным образом, фармацевтических композиций, позволяющих улучшить способы доставки, например, пероральной доставки, и способов применения таких композиций.
УРОВЕНЬ ТЕХНИКИ
Технологические приемы, способствующие эффективному проникновению целевого вещества через биологический барьер, представляют значительный интерес в области биотехнологии и медицины. Например, такие технические приемы могут быть применены для транспортировки различных веществ через биологический барьер, регулируемый плотными соединениями (т.е. слизистый эпителий, в том числе кишечный эпителий и эпителий дыхательных путей, и сосудистый эндотелий, в том числе гематоэнцефалический барьер, носовую перегородку, роговицу и другие оболочки глаза, а также мембраны мочеполовой системы). В частности, существует большой интерес к пероральной доставке терапевтических средств во избежание применения более инвазивных средств введения и, следовательно, улучшения удобства для пациентов и соблюдения ими режима терапии.
Использовали разнообразные носители для доставки лекарственных средств, в том числе липосомы, липидные или полимерные наночастицы и микроэмульсии. Они улучшают биодоступность определенных препаратов при пероральном введении, в основном благодаря защитному эффекту, которым они обладают. Однако, для большинства соответствующих препаратов, биодоступность остается очень низкой и не достигает минимальных терапевтических целей.
Следовательно, существует необходимость в эффективных, специфических, неинвазивных средствах с низким уровнем риска, нацеленных на различные биологические барьеры для неинвазивной доставки различных терапевтических средств, таких как пептиды и полипептиды, макромолекулы препаратов и других терапевтических средств, которые включают молекулы небольшого размера, с низкой биодоступностью.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Авторы данного изобретения обнаружили, что абсорбция определенных терапевтических средств у субъекта может быть улучшена при введении в композиции, описанной в данном описании. Например, терапевтическое средство, которое вводят в композицию в соответствии с одним или несколькими вариантами, демонстрирует улучшенную биодоступность (БД) по сравнению с тем же самым терапевтическим средством, которое вводят аналогичным путем, но в композиции, практически не содержащей соли жирной кислоты со средней длиной цепи, описанной в данном описании, или содержащей меньшее количество соли жирной кислоты со средней длиной цепи, описанной в данном описании. Такое улучшение относительной БД может быть выше как минимум приблизительно в 1,5, 2, 3, 5, 10, 50 или 100 раз В некоторых аспектах композиция, описанная в данном описании, улучшает абсорбцию терапевтического средства в желудочно-кишечном тракте (ЖКТ), которое, как правило, характеризуется низкой или нулевой биодоступностью и/или абсорбцией при пероральном введении. Данные терапевтические средства могут иметь низкую или нулевую биодоступность, например, в водном растворе и в других лекарственных формах для перорального введения, известных в данной области. По крайней мере, в одном из аспектов композиция, описанная в данном описании, улучшает биодоступность путем повышения проницаемости перегородки/барьера ЖКТ относительно молекул препарата. Например, композиция, описанная в данном описании, может способствовать абсорбции через проникающую перегородку/барьер ЖКТ главным образом путем вскрытия плотных соединений между эпителиальными клетками ЖКТ, хотя она может также действовать через трансцеллюлярную абсорбцию.
Авторы данного изобретения разработали способ создания фармацевтической композиции (нерасфасованной лекарственной формы препарата), который включает приготовление водорастворимой композиции, содержащей терапевтически эффективное количество как минимум одного терапевтического средства и соли жирной кислоты средней длинны (и другие ингредиенты - см. ниже), высушивание (например, путем лиофилизации) водорастворимой композиции для получения твердого порошка, а также суспендирование лиофилизированного материала (твердого порошка) в гидрофобной (жирной) среде, предпочтительно в касторовом масле или в глицерилтрикаприлате (включая другие ингредиенты, например, ПВП и поверхностно-активные вещества и модификаторы вязкости - см. ниже), для получения суспензии, содержащей в твердой форме терапевтическое средство и соль жирной кислоты средней длинны, тем самым создавая нерасфасованную лекарственную форму препарата, которая должна содержать не менее 10% по массе соли жирной кислоты средней длинны. Твердая форма может содержать частицы (например, состоит в основном из частиц, или состоит из частиц). Частицы могут быть получены путем лиофилизации или грануляции. Нерасфасованная лекарственная форма препарата может быть впоследствии инкапсулирована в капсулы, которые будут покрыты оболочкой, чувствительной к уровню рН, и могут использоваться для перорального применения. Стандартный процесс производства заявленной лекарственной формы показан на фиг. 1, где инсулин представлен в качестве примера активного фармацевтического ингредиента (АФИ), и солью жирной кислоты со средней длиной цепи является октаноат натрия (Na-C8), который также называют каприловокислым натрием.
Настоящее изобретение демонстрирует доставку препарата в кишечник, что является моделью для пероральной доставки, а оттуда в кровь, с высокой биодоступностью.
Таким образом, в одном из аспектов изобретение характеризует композицию. Композиция включает терапевтическое средство и соль жирной кислоты средней длины, объединенные в существенной мере в гидрофобной среде, предпочтительно в касторовом масле, в которой терапевтическое средство и соль жирной кислоты со средней длиной цепи в твердой форме, например, в той же твердой форме, такой как частица, полученная путем высушивания из водной среды, например, лиофилизацией из водной среды, и в которой соль жирной кислоты средней длины составляет 10% по массе или более, предпочтительно 12-15%, например, около 12%, около 13%, около 14% или около 15%, или около 16%, или около 17%, и где композиция включает другие ингредиенты (как описано в данном описании), но практически не содержит "мембрано-разжижающего средства". "Мембрано-разжижающими средствами" являются различные линейные, разветвленные, ароматические и циклические спирты средней длинны, в частности, гераниол и октанол.
Представленные композиции по изобретению не являются эмульсиями. Почти все представленные композиции являются масляными суспензиями, и количество воды в композициях является весьма незначительным; несколько представленных композиций, которые не являются суспензиями, включают большое количество (около 78%) октановой кислоты и являются растворами.
В композициях по изобретению, терапевтическое средство и соль жирной кислоты средней длины находятся в непосредственном контакте с главным образом гидрофобной средой. Например, порошок, включающий терапевтическое средство и соль жирной кислоты со средней длиной цепи, покрывают оболочкой, погружают или суспендируют, в основном, в гидрофобной среде.
В ходе процесса производства водную среду, которая включает терапевтическое средство и соль жирной кислоты средней длины, а также другие ингредиенты, высушивают (например, путем лиофилизации) для получения гидрофильной фракции, которая представляет собой порошок (например, твердую форму, включающую множество частиц), и частица данного порошка включает все ингредиенты, т.е. терапевтическое средство и соль жирной кислоты со средней длиной цепи находятся одновременно в одной частице. Твердая форма может быть, например, гранулированной частицей или лиофилизированной частицей.
В некоторых вариантах терапевтическое средство выбрано из группы, состоящей из пептидов, полисахаридов, полинуклеотидов, и молекул небольшого размера. Терапевтическое средство может быть белком. Например, терапевтическое средство может быть инсулином. В других вариантах терапевтическое средство является полинуклеотидом, например, соединением ДНК или РНК. В некоторых вариантах терапевтическое средство является молекулой небольшого размера, трудно растворимым лекарственным препаратом или в высокой степени кристаллическим лекарственным препаратом. Терапевтическое средство может быть гормоном роста. По крайней мере, в одном из вариантов, терапевтическое средство является терипаратидом. В некоторых вариантах терапевтическое средство может быть лейпролином или алендронатом или октреотидом.
В некоторых вариантах композиция содержит множество солей жирной кислоты со средней длиной цепи и их производных. Например, твердая частица может дополнительно содержать множество солей жирных кислот со средней длиной цепи и их производных.
В некоторых вариантах соль жирной кислоты со средней длиной цепи выбрана из группы, состоящей из гексаноата натрия, гептаноата натрия, октаноата натрия, нонаноата натрия, деканоата натрия, ундеканоата натрия, додеканоата натрия, тридеканоата натрия и тетрадеканоата натрия или их комбинации. В соответствии с одним или несколькими вариантами композиция практически не содержит додеканоат натрия, тридеканоат натрия и тетрадеканоат натрия. В некоторых вариантах жирной кислотой средней длины является октаноат натрия, и октаноат натрия присутствует в концентрации свыше 10%, например, около 11%, до приблизительно 50% масс.
В некоторых вариантах в существенной мере гидрофобная среда включает триглицерид. Например, триглицерид может быть выбран из группы, состоящей из глицерилтрибутирата, глицерилмоноолеата, глицерилмонокаприлата и глицерилтрикаприлата.
В некоторых вариантах в существенной мере гидрофобная среда включает минеральное масло, касторовое масло, оливковое масло, кукурузное масло, кокосовое масло, арахисовое масло, соевое масло, хлопковое масло, кунжутное масло или масло канолы, или их комбинации.
В некоторых вариантах водорастворимая композиция включает соль жирной кислоты средней длины, и гидрофобная среда включает соответствующую жирную кислоту со средней длиной цепи; в некоторых конкретных вариантах солью жирной кислоты со средней длиной цепи является соль октановой кислоты, такая как октаноат натрия, и жирной кислотой со средней длиной цепи является октановая кислота.
В некоторых вариантах водорастворимая композиция включает соль жирной кислоты средней длины, и гидрофобная среда включает соответствующий моноглицерид средней длины или соответствующий триглицерид средней длины или их комбинацию; в некоторых конкретных вариантах солью жирной кислоты средней длины натрия является октаноат и моноглицеридом является глицерилмонокаприлат и триглицеридом является глицерилтрикаприлат.
В некоторых вариантах композиция дополнительно включает один или несколько наполнителей. Наполнители могут быть солью, например, MgCl2, или соединением, содержащим амин, или маннитом. В некоторых вариантах наполнитель находится в такой же твердой форме, что и терапевтическое средство.
В некоторых вариантах наполнитель является стабилизатором. Авторы изобретения неожиданно обнаружили, что несмотря на то, что поливинилпирролидон (ПВП), в частности ПВП-12, известен в данной области как стабилизатор, в композициях по изобретению он служит для усиления эффекта энхансера проницаемости при абсорбции терапевтического средства.
В некоторых вариантах композиция дополнительно включает одно или несколько поверхностно-активных соединений. Например, поверхностно-активное соединение может быть выбрано из группы, состоящей из сорбитан монопальмитата (Спан 40®), полиоксиэтиленсорбитан моноолеата (Твин 80), лецитина и глицерилмоноолеата (ГМО). В одном или нескольких вариантах поверхностно-активное соединение составляет примерно от 0,1% до 6% от веса композиции. В предпочтительных вариантах композиция является лекарственной формой для перорального применения. Например, композицией можно наполнить твердую или мягкую желатиновую капсулу. В некоторых вариантах композиция может быть в форме суппозиториев. В соответствии с одним или несколькими вариантами композиция может быть в виде тающей клизмы (enema fleet).
В некоторых вариантах биодоступность лекарственного средства при введении субъекту составляет как минимум 1,5-2% по сравнению с парентеральным (подкожным или внутривенным) введением. В некоторых вариантах композиция при введении субъекту обеспечивает более 2%, более 3%, более 5%, более 10% или более 20%, или более 30% абсорбции терапевтического средства через биологический барьер. Полученные уровни абсорбции являются терапевтическими уровнями, необходимыми для исследуемых показаний к применению.
В одном аспекте изобретение описывает способ лечения нарушений у субъекта. Способ включает введение субъекту любой одной из композиций, описанных в данном описании.
В некоторых вариантах композицию вводят перорально. В других вариантах композицию вводят ректально, сублингвально или трансбуккально.
В некоторых вариантах расстройством может быть анемия. В соответствии с одним или несколькими вариантами заболеванием является остеопороз. Расстройство может представлять собой женское бесплодие. В других вариантах расстройством является отставание в росте или дефицит гормона роста. По крайней мере, в одном из вариантов расстройством является связанное с ВИЧ снижение массы тела или истощение, акромегалия или диабет.
В некоторых вариантах терапевтическим средством является октреотид, и расстройством является акромегалия, аномальная перистальтика ЖКТ, гастропарез, диарея или портальная гипертензия.
В некоторых вариантах способ может включать инкапсуляцию суспензии в форму капсулы. Способ может дополнительно включать покрытие капсулы оболочкой.
В некоторых вариантах способ может включать предоставление инструкций для введения капсулы субъекту. Инструкции могут быть связаны с введением капсулы субъекту при любом из симптомов, описанных в данном описании.
В одном из аспектов изобретение характеризует капсулы, предоставленные с инструкциями для введения капсулы субъекту при любом из симптомов, описанных в данном описании.
Другие аспекты, варианты и преимущества данных аспектов и вариантов, представленных в качестве примеров, подробно обсуждаются ниже. Кроме того, следует понимать, что вышеизложенная информация и последующее подробное описание являются всего лишь примерами, иллюстрирующими различные аспекты и варианты, и предназначены для обеспечения обзора или основы для понимания природы и характера заявленных аспектов и вариантов. Прилагаемые рисунки включены с целью обеспечения иллюстрации и глубокого понимания различных аспектов и вариантов, они включены и составляют часть данного описания. Фигуры, вместе с остальной частью спецификации служат для объяснения принципов и операций описанных и заявленных аспектов и вариантов.
Во всем тексте данной заявки различные публикации, в том числе и патенты США, приведены путем ссылки на автора и год, и патенты и заявки - на номер. Раскрытие данных публикаций, патентов и патентных заявок во всей их полноте, таким образом, включена путем ссылки на данную заявку с целью более полно описать состояние области, к которой относится данное изобретение.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Различные аспекты, по крайней мере, одного из вариантов рассматриваются ниже со ссылкой на прилагаемые фигуры. На фигурах, которые не предназначены для начертания в масштабе, каждый идентичный или практически идентичный компонент, который иллюстрирует различные фигуры, представлен в виде символа. Для большей ясности не все компоненты могут быть помечены на каждом рисунке. Фигуры приводятся с целью иллюстрации и объяснения результатов и не предназначены для определения пределов изобретения. На фигурах:
фиг. 1 представлен процесс производства композиции лекарственной формы инсулина в соответствии с одним или несколькими вариантами, как указано в сопроводительных примерах;
фиг. 2-5 представляют данные, упоминаемые в сопроводительных Примерах 3-6;
фиг. 6 представляет данные, упоминаемые в сопроводительном Примере 8;
фиг. 7 представляет значения проницаемости для маркера молекулярной массы, упоминаемые в сопроводительном Примере 33,
фиг. 8 представляет значения периода действия проницаемости, упоминаемые в сопроводительном Примере 34; и
фиг. 9 и 10 представлены данные, относящиеся к введению октреотида обезьянам, упоминаемые в сопроводительном Примере 35.
ПОДРОБНОЕ ОПИСАНИЕ
Композиции, описанные в данном описании, могут быть введены субъекту для обеспечения улучшения биодоступности терапевтического средства.
Фармацевтические композиции
Фармацевтические композиции, описанные в данном описании, содержат терапевтическое средство и соль жирной кислоты со средней длиной цепи, которые находятся в непосредственном контакте или в связи с, в существенной мере, гидрофобной средой. Например, терапевтическое средство и жирная кислота со средней длиной цепи или ее производные могут покрывать оболочкой, суспендировать, распылять или погружать в существенной мере гидрофобную среду, для образования суспензии. Композиции по изобретению не являются эмульсиями. Практически все композиции являются масляными суспензиями, и количество воды в композициях является незначительным; несколько из представленных композиций, которые не являются суспензиями, содержат большое количество (около 78%) октановой кислоты и являются растворами, что определяют визуально. Суспензия может быть жидкой суспензией, содержащей твердые вещества, или полужидкой суспензией, содержащей твердые вещества (мазь).
Многие из композиций, описанные в данном описании, содержат суспензию, которая содержит смесь гидрофобной среды и твердой формы, где твердая форма содержит терапевтически эффективное количество терапевтического средства и как минимум одну соль жирной кислоты со средней длиной цепи, и где соль жирной кислоты со средней длиной цепи присутствует в композиции в количестве 10% или более по массе. Твердая форма может содержать частицы (например, состоять в основном из частиц, или состоять из частиц). Частица может быть получена путем лиофилизации или грануляции. В некоторых вариантах, предпочтительно после измельчения, 90% (об./об.) частиц имеют размер менее 130 микрон, и 50% (об./об.) частиц имеют размер менее 45 мкм.
Соединением карго является терапевтическое средство (например, инсулин) или испытуемое соединение (например, высокомолекулярный декстран), которые изготовлены, как описано в данном описании, в виде композиций по изобретению.
Авторы данного изобретения уделяли особое внимание тому, чтобы включать во многие композиции по изобретению только те наполнители, которые по всеобщему признанию являются безопасными, основываясь на имеющиеся данные безопасности относительно применения у человека, а также данные безопасности и нормативные рекомендации относительно применения у животных (например, наполнители, имеющие статус безвредных - GRAS). Некоторые композиции по изобретению могут содержать и другие типы наполнителей (например, не имеющих статуса GRAS). В некоторых вариантах композиции по изобретению содержат такое количество наполнителей, которое находится в пределах максимальной суточной дозы, как указано в соответствующих имеющихся данных для каждого конкретного наполнителя.
Соль жирной кислоты со средней длиной цепи может в целом способствовать или повышать проницаемость и/или абсорбцию терапевтического средства. В некоторых вариантах соли жирных кислот со средней длиной цепи содержат производные солей жирных кислот со средней длиной цепи. Терапевтическое средство и соль жирной кислоты со средней длиной цепи находится в твердой форме, например, в форме твердой частицы, такой как лиофилизированная частица, гранулированная частица, гранула или микросфера. В предпочтительных вариантах терапевтическое средство и соль жирной кислоты со средней длиной цепи находятся в одной и той же твердой форме, например, в одной и той же частице. В других вариантах терапевтическое средство и соль жирной кислоты со средней длиной цепи могут находиться в отдельных твердых формах, например, каждая из них в отдельной частице. Композиции, описанные в данном описании, в существенной мере не содержат каких-либо "мембрано-разжижающих средств" таких как линейные, разветвленные, ароматические и циклические спирты со средней длиной цепи, в частности, гераниол и октанол. Например, композиции предпочтительно не содержат мембрано-разжижающих средств, но некоторые варианты могут включать, например, менее 1% или менее 0,5%, или менее 0,1% по массе мембрано-разжижающих средств.
В отличие от эмульсий, где вода является основным элементом формулы, композиции, описанные в данном описании, находятся в твердой форме, такой как частица, содержащая терапевтическое средство, которое затем связывается с гидрофобной (жирной) средой. Количество воды в композициях составляет, как правило, менее 3% по массе, как правило, менее 2% или приблизительно 1% или менее по массе.
Композиции, описанные в данном описании, являются суспензиями, которые содержат смесь гидрофобной среды и твердой формы, где твердая форма содержит терапевтически эффективное количество терапевтического средства и, по крайней мере, одну соль жирной кислоты со средней длиной цепи. Твердая форма может быть частицей (например, состоять в основном из частиц, или состоять из частиц). Частица может быть получена путем лиофилизации или грануляции. Соль жирной кислоты со средней длиной цепи обычно присутствует в композициях, описанных в данном описании, в количестве от 10% или более по массе. В некоторых вариантах соль жирной кислоты со средней длиной цепи присутствует в композиции в количестве 10%-50%, предпочтительно 11%-18%, или приблизительно 11%-17%, или 12%-16%, или 12%-15%, или 13%-16%, или 13%-15%, или 14%-16%, или 14%-15%, или 15%-16%, или более предпочтительно 15% или 16% по массе, и жирная кислота со средней длиной цепи имеет цепь длиной от 6 до приблизительно 14 атомов углерода, предпочтительно 8, 9 или 10 атомов углерода.
В некоторых вариантах в композициях, описанных выше, твердая форма, которая содержит терапевтическое средство, также содержит стабилизатор (например, стабилизатор структуры белка). Стабилизаторы структуры белка являются соединениями, которые стабилизируют структуру белка в водных или безводных условиях, или которые могут снижать или предотвращать агрегацию терапевтического средства, например, во время процесса сушки, такого как лиофилизация, или другой стадии обработки. Стабилизаторы структуры могут быть полианионными молекулами, такими как фитиновая кислота, поливалентными ионами, такими как Са, Zn или Mg, сахарами, такими как дисахарид (например, трегалоза, мальтоза), или олиго- или полисахаридом, таким как декстрин или декстран, или сахарным спиртом, таким как маннит, или аминокислотой, такой как глицин, или поликатионными молекулами, такими как спермин, или поверхностно-активными веществами, такими как полиоксиэтиленсорбитан моноолеат (Твин 80) или плюрониловая кислота. Незаряженные полимеры, такие как маннит, метилцеллюлоза и поливиниловый спирт, также являются подходящими стабилизаторами.
Несмотря на то что поливинилпирролидон (ПВП) известен в данной области как стабилизатор, авторы изобретения неожиданно установили, что в композициях по изобретению, описанных в данном описании, ПВП, особенно ПВП-12, предназначен для усиления эффекта энхансера проницаемости на синергетической основе; кроме того, повышение уровня ПВП-12 до 10% приводило к повышению уровня абсорбции терапевтического средства в крови в связи с улучшенной активностью лекарственной формы. Авторы изобретения продемонстрировали, что декстран обладает аналогичным (но менее выраженным) эффектом, как и ПВП. Другие матриксы, образованные полимерами, обладают таким же эффектом.
В некоторых вариантах, например, когда терапевтическое средство является молекулой небольшого размера, можно добавить наполнитель, например, маннит или глицин.
В некоторых вариантах композиций, описанных в данном описании, терапевтическое средство представляет собой белок, полипептид, пептид, гликозаминогликан, молекулу небольшого размера, полисахарид или полинуклеотид, в том числе, такие как октреотид, гормон роста, паратиреоидный гормон, аминокислоты паратиреоидный гормона 1-34 [РТН (1-34), имеющий название терипаратид], низкомолекулярный гепарин или фондапаринукс и др. Низкомолекулярные гепарины определяют как соли гепарина со средней молекулярной массой менее 8000 Да и для которых не менее 60% всех цепей имеют молекулярную массу менее 8000 Да.
В конкретном варианте композиций, описанных в данном описании, солью жирной кислоты является октаноат натрия, и гидрофобной средой является касторовое масло; в другом конкретном варианте композиция дополнительно содержит глицерилмоноолеат и сорбитан монопальмитат или глицерилмонокаприлат и глицерилтрикаприлат, и полиоксиэтиленсорбитан моноолеат; в другом конкретном варианте композиция дополнительно содержит глицерилтрибутират, лецитин, этилизовалерат и как минимум один стабилизатор. В конкретных вариантах терапевтическим средством является октреотид, гормон роста, паратиреоидный гормон, терипаратид, интерферон-альфа (IFN-α), низкомолекулярный гепарин, фондапаринукс, siPHK, соматостатин и их аналоги (агонисты), включая пептидомиметики, экзенатид, ванкомицин или гентамицин.
Терапевтические средства
Фармацевтические композиции, описанные в данном описании, могут быть применены с различными терапевтическими средствами (также называемыми активными фармацевтическими ингредиентами = АФИ). В некоторых вариантах фармацевтическая композиция содержит множество терапевтических средств (эффекторов). Терапевтические средства могут находиться в той же самой твердой форме (например, в той же самой частице), или любое из терапевтических средств может находиться в отдельной твердой форме (например, каждое из них в различных частицах). В некоторых вариантах терапевтическое средство находится в форме частицы, например, гранулированной или твердой частицы. Частица связана или находится в непосредственном контакте с, в существенной мере, гидрофобной средой, например, гидрофобной средой, описанной в данном описании.
Терапевтические средства, которые могут быть применены в композициях, описанных в данном описании, содержат любую молекулу или соединение, выступающего в качестве, например, биологического, лечебного, фармацевтического или диагностического средства, включая средство визуализации. Терапевтические средства включают лекарственные препараты и другие средства, включая, не ограничиваясь ими, средства, перечисленные в Фармакопеи США и других известных фармакопеях. Терапевтические средства включены в композиции по изобретению без каких-либо химических модификаций. Терапевтические средства включают белки, полипептиды, пептиды, полинуклеотиды, полисахариды и молекулы небольшого размера.
Термин "молекула небольшого размера" относится к органическому соединению с низкой молекулярной массой, которое можно синтезировать или получить из природных источников, и обычно имеющему молекулярную массу менее 2000 Да, или менее 1000 Да, или даже менее 600 Да, например, менее или около 550 Да, или менее или приблизительно 500 Да, или менее или около 400 Да, или от приблизительно 400 Да до приблизительно 2000 Да, или приблизительно от 400 Да до приблизительно 1700 Да. Примерами молекул небольшого размера являются эрготамин (молекулярная масса = 582 Да), фондапаринукс (молекулярная масса = 1727 Да), лейпролид (молекулярная масса = 1209 Да), ванкомицин (молекулярная масса = 1449 Да), гентамицин (молекулярная масса = 478 Да) и доксорубицин (молекулярная масса = 544).
Термин "полинуклеотид" относится к любой молекуле, состоящей из нуклеотидов ДНК, нуклеотидов РНК или комбинации обоих типов, которая содержит два или более оснований гуанидина, цитозина, тимидина, аденина, урацила или инозина и др. Полинуклеотид может содержать природные нуклеотиды, химически модифицированные нуклеотиды и синтетические нуклеотиды, или их химические аналоги, и может быть одноцепочечным или двухцепочечным. Этот термин включает "олигонуклеотиды" и включает "нуклеиновые кислоты".
Под термином "малая интерферирующая РНК" (siPHK) понимают молекулу РНК (рибонуклеотид), которая снижает или останавливает (предотвращает) экспрессию гена/мРНК его эндогенного или клеточного контрагента. Термин включает "РНК-интерференцию" (PHKi) и" двухцепочечную РНК" (dsPHK).
Под термином "полипептид" понимают молекулу, состоящую из ковалентно связанных аминокислот; термин также включает пептиды, полипептиды, белки и пептидомиметики. Пептидомиметик является соединением, содержащим непептидные структурные элементы, которые способны подражать биологической активности(ям) природных материнских пептидов. Некоторые из классических пептидных характеристик, такие как ферментативное расщепление пептидных связей, как правило, отсутствуют в пептидомиметике.
Термин "аминокислота" относится к молекуле, которая состоит из любой одной из 20 встречающихся в природе аминокислот, аминокислот, которые были химически модифицированы, или синтетических аминокислот.
Под термином "полисахарид" понимают линейный или разветвленный полимер, состоящий из ковалентно связанных моносахаридов; глюкоза является наиболее распространенным моносахаридом и имеет обычно как минимум восемь моносахаридных единиц в полисахариде и, как правило, намного больше. Полисахариды имеют общую формулу Сх(Н2О)y, где х является, как правило, большим числом между 200 и 2500. Учитывая то, что повторяющиеся единицы в основной цепи полимера часто являются моносахаридами с шестью атомами углерода, общая формула может быть представлена как (C6H10O5)n, где 40≤n≤3000, т.е. обычно между 40 и 3000 моносахаридных единиц в полисахариде.
Тликозаминогликан" является полисахаридом, который содержит аминосодержащие сахара.
Примеры анионных терапевтических средств включают полинуклеотиды различного происхождения, и в том числе полученные от человека, вируса, животного, эукариота или прокариота, растений или синтетического происхождения, и т.д., включая системы для терапевтической доставки генов. Полинуклеотид интереса может быть различных размеров, от, например, простого остаточного нуклеотида до фрагмента гена или всего гена. Это может быть вирусный ген или плазмида. Примеры полинуклеотидов, которые служат терапевтическими средствами, включают специфические последовательности ДНК (например, кодирующие гены), специфические последовательности РНК (например, РНК-аптамеры, антисмысловые РНК, малые интерферирующие РНК (siPHK) или специфические ингибиторы РНК (PHKi)) поли CPG или поли I:С синтетические полимеры полинуклеотидов.
Альтернативно, терапевтическое средство может быть белком, например, ферментом, гормоном, инкретином, протеогликаном, рибозимом, цитокином, пептидом, аполипопротеином, фактором роста, биоактивной молекулой, антигеном или антителом, или их фрагментом(ами) и т.д. Пептид может быть пептидом небольшого размера, например, длинной от 2 до 40 аминокислот, примеры включают антагонистов рецептора фибриногена (пептиды, содержащие RGD, которые являются тетрапептидами со средней молекулярной массой около 600). Примерами пептидов являются соматостатин и его аналоги, например, октреотид и ланреотид (Соматулин), которые являются циклическими октапептидами, и пазиреотид (SOM-230), который является циклическим гексапептидом (Weckbecker et al, 2002, Endocrinology 143 (10) 4123-4130; Schmid, 2007, Molecular and Cellular Endocrinology 286, 69-74). Другие примеры пептидов включают глатирамера ацетат (Copaxone®), который является тетрапептидом, терлипрессин, состоящий из 12 аминокислот, который является пептидным аналогом (агонистом) лизина вазопрессина (ADH), и экзенатид, пептид, состоящий из 39 аминокислот, который является инкретин-миметическим агентом, и другие аналоги глюкагон-подобного пептида-1 (GLP-1). Byetta® является торговым названием экзенатида (Eli Lilly and Company/Amylin Pharmaceuticals, Inc). Другие пептиды включают даларгин, который является гексапептидом, и киоторфин, который является дипептидом. Пептиды включают пептиды, высвобождающие гормон, которые являются пептидами, состоящими из приблизительно 12 аминокислот или менее; см., например, пептиды, раскрытые в патентах США №№4411890 (Momany) и 4839344 (Bowers et al.).
Примеры других пептидов, которые могут быть применены в практике данного изобретения, раскрыты в патенте США №4589881 (30 или более остатков аминокислот) у Pierschbacher et al.; патенте США №4544500 (20-30 остатков) у Bittle et al.; и ЕР 0204480 (>34 остатков) у Dimarchi et al. и терипаратид. В некоторых вариантах терапевтическое средство может включать полисахарид, такой как гликозаминогликан. Примеры гликозаминогликанов включают гепарин, производные гепарина, гепарансульфат, хондроитина сульфат, дерматансульфат и гиалуроновую кислоту. Примеры производных гепарина включают, не ограничиваясь ими, низкомолекулярные гепарины, такие как эноксапарин, дальтепарин и тинзапарин. Терапевтическим средством с гепаринподобным эффектом является фондапаринукс.
Другие примеры терапевтических средств включают, не ограничиваясь ими, гормоны, такие как инсулин, эритропоэтин (ЕРО), глюкагон-подобный пептид 1 (GLP-1), меланоцитостимулирующий гормон (альфа-MSH), паратиреоидный гормон (РТН), терипаратид, гормон роста (GH), лейпролид, лейпролида ацетат, фактор VIII, рилизинг фактор гормона роста (GHRH), пептид YY аминокислот 3-36 (PYY(3_36)), кальцитонин, соматотропин, соматостатин, соматомедин, интерлейкины, такие как интерлейкин-2 (IL-2), альфа-1-антитрипсин, колониестимулирующий фактор гранулоцитов/моноцитов (GM-CSF), колониестимулирующий фактор гранулоцитов (G-CSF), Т20, тестостерон, интерфероны, такие как интерферон-альфа (IFN-α) IFN-β и IFN-γ, лютеинизирующий гормон (LH), фолликулостимулирующий гормон (FSH), хорионический гонадотропин человека (hCG), энкефалин, даларгин, киоторфин, основной фактор роста фибробластов (bFGF), гирудин, хирулог, рилизинг фактор лютеинизирующего гормона (LHRH), аналог рилизинг фактора гонадотропина (GnRH), натрийуретический пептид из мозга (BNP), активатор плазминогена ткани (TPA), окситоцин, и их аналоги и комбинации.
Другие примеры терапевтических средств включают, не ограничиваясь ими, анальгетики, средства против мигрени, антикоагулянты, противорвотные средства, сердечно-сосудистые, антигипертензивные и сосудорасширяющие средства, седативные средства, наркотические антагонисты, хелатные средства, антидиуретические средства и противоопухолевые средства.
Средства против мигрени включают, не ограничиваясь ими, наратриптан, напроксен, альмотриптан, буталбитал, фроватриптан, суматриптан, ризатриптан, ацетаминофен, изометептен, буторфанол, дихлоралфеназон, алкалоиды спорыньи, такие как дигидроэрготамин и эрготамин, нестероидные противовоспалительные средства (НСПВС), такие как кетопрофен и кеторолак, элетриптан, буторфанол, топирамат, золмитриптан, кофеин, аспирин и кодеин, а также их аналоги и комбинации.
Антикоагулянты включают, не ограничиваясь ими, гепарин, гирудин, низкомолекулярные гепарины и их аналоги, а также фондапаринукс. Противорвотные средства включают, не ограничиваясь ими, скополамин, ондансетрон, домперидон, метоклопрамид и их аналоги. Сердечно-сосудистые, антигипертензивные и сосудорасширяющие средства включают, не ограничиваясь ими, дилтиазем, клонидин, нифедипин, верапамил, изосорбид-5-мононитрат, органические нитраты, нитроглицерин и их аналоги. Седативные средства включают, не ограничиваясь ими, бензодиазепины, фенотиазины и их аналоги. Наркотические антагонисты включают, не ограничиваясь ими, налтрексон, налоксон и их аналоги. Хелатирующие средства включают, не ограничиваясь ими, дефероксамин и его аналоги. Антидиуретические средства включают, не ограничиваясь ими, десмопрессин, вазопрессин и их аналоги (агонисты), такие как терлипрессин; торговым названием терлипрессина является Глипрессин®. Противоопухолевые средства включают, не ограничиваясь ими, 5-фторурацил, блеомицин, винкристин, прокарбазин, темезоламид, 6-тиогуанин, гидроксимочевину, цитарабин, циклофосфамид, доксорубицин, алкалоид барвинка, эпирубицин, этопозид, ифосфамид, карбоплатин и другие противоопухолевые препараты на основе платины (такие, как карбоплатин [Аплатин®], тетраплатин, оксалиплатин, ароплатин и трансплатин), винбластин, винорельбин, хлорамбуцил, бусульфан, хлорметин, митомицин, дакарбазин, тиотепа, даунорубицин, идарубицин, митоксантрон, эсперамицин А1, дактиномицин, пликамицин, кармустин, ломустин (CCNU), тауромустин, стрептозоцин, мелфалан, дактиномицин, прокарбазин, дексаметазон, преднизолон, 2-хлородеоксиаденозин, цитарабин, доцетаксел, флударабин, гемцитабин, герцептин, гидроксимочевину, иринотекан, метотрексат, ритуксин, семустин, томудекс и топотекан, таксол и таксоло-подобные соединения, а также их аналоги и комбинации.
Дополнительные примеры терапевтических средств включают, не ограничиваясь ими, факторы свертывания и нейротрофические факторы, анти-ФНО антитела и фрагменты рецепторов ФНО.
Терапевтические средства также включают фармацевтически активные средства, выбранные из группы, состоящей из витамина В12, бисфосфоната (например, памидроната динатрия, алендроната, этидроната, тилудроната, ризендроната, золедроновой кислоты, клодроната натрия или ибандроновой кислоты), таксола, каспофунгина или аминогликозидных антибиотиков. Дополнительные терапевтические средства включают токсин или противопатогенное средство, такое, как антибиотик (например, ванкомицин), противовирусное, противогрибковое или противопаразитное средство. Терапевтическое средство само может быть непосредственно активным или может быть активировано композицией in situ определенным веществом или условиями окружающей среды.
В некоторых вариантах композиция может содержать множество терапевтических средств (комбинация препаратов). Например, композиция может содержать фактор VIII и фактор Виллебрандта (vWF), GLP-1 и PYY, IFN-α и нуклеотидные аналоги (т.е. рибавирин), и алендронат или инсулин, а также GLP-1.
В некоторых вариантах композиция может содержать молекулу небольшого размера и пептид или белок. Примеры комбинаций включают сочетание IFN-α и нуклеотидных аналогов (т.е. рибавирин) для лечения гепатита С, терипаратида и алендроната для лечения костных нарушений, сочетание лекарств GH плюс для ВИЧ-терапии (т.е. HAART) для одновременного лечения вирусной инфекции и липодистрофии, сопровождающей ВИЧ или побочных эффектов истощения при СПИДе. Комбинации из двух молекул небольшого размера могут быть применены, когда одна из них обычно имеет недостаточную абсорбцию или биодоступность, даже если вторая обычно имеет эффективную абсорбцию или биодоступность, как некоторые антибиотики (например, сочетание ванкомицина и аминогликозидов, таких как гентамицин). Примеры комбинаций для лечения и профилактики метаболических расстройств, таких как диабет и ожирение, также включают сочетание инсулина и метформина, инсулина и розиглитазона, GLP-1 (или экзенатида) и метформина, и GLP-1 (или экзенатида) и розиглитазона.
Симптомы и состояния, которые могут подвергаться лечению с применением фондапаринукса, сформулированного, как описано в данном описании, включают тромбоз глубоких вен, протезирование бедра или коленного сустава, а также прикованных к постели пациентов.
В некоторых вариантах композиций, описанных в данном описании, композиция включает сочетание белка или пептида с молекулами небольшого размера, любое из которых обладает или не обладает хорошей абсорбцией или биодоступностью. Например, композиция может содержать как минимум одно терапевтическое средство, которое может быть в целом охарактеризовано как обладающее низким уровнем абсорбции или биодоступности. Композиция также может быть применена для введения терапевтических средств, которые абсорбируются в желудке и/или кишечнике, но вызывают раздражение желудка и/или кишечника и, следовательно, тяжело переносятся. В такой ситуации преимущество для субъекта может состоять в том, что биодоступность терапевтического средства будет увеличена или, что большее количество терапевтического средства абсорбируется непосредственно в кровоток; если вводят меньшее количество терапевтического средства, очевидно, что, будет снижена вероятность раздражения желудка и/или кишечника. Таким образом, композиции по изобретению предусматривают содержание двух или более терапевтических средств.
В целом, композиция может содержать от приблизительно 0,01% до приблизительно 50% по массе терапевтического средства, например, приблизительно 0,01, 0,02 0,05, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45 или 50% по массе. Максимальное количество, содержащееся в композиции, обычно находится в диапазоне приблизительно 6%-33% по массе терапевтического средства. В некоторых вариантах композиций, описанных в данном описании, твердая форма, содержащая терапевтическое средство, также содержит стабилизатор (например, стабилизатор структуры белка). Стабилизаторы структуры белка являются соединениями, которые стабилизируют структуру белка в водных или неводных условиях, или могут снижать или предотвращать агрегацию терапевтического средства, например, во время процесса сушки, такой как лиофилизация, или во время другой стадии обработки. Стабилизаторы структуры могут быть полианионными молекулами, такими как фитиновая кислота, поливалентными ионами, такими как Са, Zn или Mg, сахаридами, такими как дисахарид (например, трегалоза, мальтоза), или олиго или полисахаридами, такими как декстрин или декстран, или сахарным спиртом, таким как маннитол, или аминокислотой, такой как глицин, или поликатионными молекулами, такими как спермин, или поверхностно-активными веществами, такими как Твин 80 или Спан 40, или плюрониевая кислота. Незаряженные полимеры, такие как метилцеллюлоза и поливиниловый спирт, также являются подходящими стабилизаторами.
Соль жирной кислоты со средней длиной цепи
Композиции, описанные в данном описании, включают соль жирной кислоты со средней длиной цепи или ее производное в твердой форме. Например, соль жирной кислоты со средней длиной цепи в форме частицы, такой как твердая частица. В некоторых вариантах частица может быть охарактеризована как гранулированная частица. Как минимум в некоторых вариантах твердая форма, как правило, может быть результатом процесса сушки распылением или выпаривания. В предпочтительных вариантах, соль жирной кислоты со средней длиной цепи находится в той же частице, что и терапевтическое средство. Например, терапевтическое средство и соль жирной кислоты со средней длиной цепи могут быть получены вместе путем предварительного приготовления раствора, такого как водный раствор, содержащий как терапевтическое средство, так и соль жирной кислоты со средней длиной цепи, и совместной лиофилизации раствора для обеспечения твердой формы или частицы, которая содержит как терапевтическое средство, так и соль жирной кислоты со средней длиной цепи (и другие ингредиенты). Как описано выше, полученные твердые частицы связаны с гидрофобной средой. Например, твердые частицы могут быть суспендированы или погружены в гидрофобную среду.
В различных вариантах композиций, описанных в данном описании, соль жирной кислоты со средней длиной цепи может быть в той же самой частице или в другой частице, чем АФИ. Было установлено, что биодоступность соединения карго ниже, если жирная кислота со средней длиной цепи находится в другой частице, чем терапевтическое средство, т.е. наблюдается повышенная биодоступность при условии, что соль жирной кислоты со средней длиной цепи и соединение карго были высушены после растворения вместе в гидрофильной фракции. Считается, что если соль жирной кислоты со средней длиной цепи и соединение карго сушат после растворения вместе в гидрофильной фракции, то они находятся в одной частице в порошке конечного продукта.
Соли жирной кислоты со средней длиной цепи включают соли, имеющие длину углеродной цепи от 6 до 14 атомов углерода. Примерами солей жирных кислот являются гексаноат натрия, гептаноат натрия, октаноат натрия (также называемый каприлатом натрия), нонаноат натрия, деканоат натрия, ундеканоат натрия, додеканоат натрия, тридеканоат натрия и тетрадеканоат натрия. В некоторых вариантах соль жирной кислоты со средней длиной цепи содержит катион, выбранный из группы, состоящей из калия, лития, аммония и других одновалентных катионов, например, соль жирной кислоты со средней длиной цепи выбрана из октаноата лития или октаноата калия или октаноата аргинина или других одновалентных солей жирных кислот со средней длиной цепи. Авторы изобретения установили, что повышение количества соли жирной кислоты со средней длиной цепи повышает биодоступность полученной в результате композиции. В частности, повышение количества соли жирной кислоты со средней длиной цепи, в частности, октаноата натрия выше 10%, в диапазоне от 12% до 15%, повышает биодоступность терапевтических средств в фармацевтических композициях, описанных в данном описании.
В целом, количество соли жирной кислоты со средней длиной цепи в композициях, описанных в данном описании, может быть от 10% до 50% по массе нерасфасованной фармацевтической композиции. Например, соль жирной кислоты со средней длиной цепи может присутствовать в количестве приблизительно 10%-50%, предпочтительно приблизительно 11%-40%, наиболее предпочтительно приблизительно 11%-28% по массе, например, приблизительно 12%-13%, 13%-14%, 14%-15%, 15%-16%, 16%-17%, 17%-18%, 18%-19%, 19%-20%, 20%-21%, 21%-22%, 22%-23%, 23%-24%, 24%-25%, 25%-26%, 26%-27% или 27%-28% по массе нерасфасованной фармацевтической композиции. В других вариантах соль жирной кислоты со средней длиной цепи может присутствовать в количестве, как минимум приблизительно 11%, как минимум приблизительно 12%, как минимум приблизительно 13%, как минимум приблизительно 14%, как минимум приблизительно 15%, как минимум приблизительно 16%, по меньшей мере, приблизительно 17%, как минимум приблизительно 18%, как минимум приблизительно 19%, как минимум приблизительно 20%, как минимум приблизительно 21%, как минимум приблизительно 22%, как минимум приблизительно 23%, как минимум приблизительно 24%, как минимум приблизительно 25%, как минимум приблизительно 26%, как минимум приблизительно 27% или как минимум приблизительно 28% по массе нерасфасованной фармацевтической композиции. В конкретных вариантах соль жирной кислоты со средней длиной цепи (натрия, калия, лития или соль аммония или их смесь) составляет приблизительно 12%-21% по массе нерасфасованной фармацевтической композиции, предпочтительно 11%-18% или приблизительно 11%-17%, или 12%-16%, или 12%-15%, или 13%-16%, или 13%-15%, или 14%-16%, или 14%-15%, или 15%-16%, или более предпочтительно 15% или 16%. В конкретных вариантах соль жирной кислоты со средней длиной цепи (с длиной углеродной цепи от 6 до 14 атомов углерода, в частности 8, 9 или 10 атомов углерода) составляет приблизительно 12%-21% по массе нерасфасованной фармацевтической композиции, предпочтительно 11%-18%, приблизительно 11%-17%, или 12%-16%, или 12%-15%, или 13%-16%, или 13%-15%, или 14%-16%, или 14%-15%, или 15%-16% или более предпочтительно 15% или 16%. В конкретных вариантах соль жирной кислоты со средней длиной цепи (например, соли октановой кислоты, соли субериновой кислоты, соли гераневой кислоты) составляет приблизительно 12%-21% по массе нерасфасованной фармацевтической композиции, предпочтительно 11%-18%, приблизительно 11%-17%, или 12%-16%, или 12%-15%, или 13%-16%, или 13%-15%, или 14%-16%, или 14%-15%, или 15%-16%), или более предпочтительно 15% или 16%. В некоторых вариантах соль жирной кислоты со средней длиной цепи присутствует в твердом порошке в количестве от 50% до 90%, предпочтительно в количестве от 70% до 80%.
Один из вариантов изобретения включает композицию, содержащую суспензию, которая состоит в основном из примеси гидрофобной среды и твердой формы, где твердая форма содержит терапевтически эффективное количество терапевтического средства и как минимум одну соль жирной кислоты со средней длиной цепи, и где солью жирной кислоты со средней длиной цепи не является соль натрия. Солью может быть соль другого катиона, например, лития, калия или аммония, предпочтительно это соль аммония.
Полимер формирования матрикса
В некоторых вариантах композиция по изобретению содержит суспензию, которая содержит смесь гидрофобной среды и твердую форму, где твердая форма содержит терапевтически эффективное количество терапевтического средства, как минимум одну соль жирной кислоты со средней длиной цепи и полимер формирования матрикса, и где полимер формирования матрикса присутствует в композиции в количестве 3% или более по массе. В некоторых вариантах композиция содержит суспензию, которая состоит в основном из примеси гидрофобной среды и твердой формы, где твердая форма содержит терапевтически эффективное количество терапевтического средства, как минимум одну соль жирной кислоты со средней длиной цепи и полимер формирования матрикса, и где полимер формирования матрикса присутствует в композиции в количестве 3% или более по массе. В конкретных вариантах полимером формирования матрикса является декстран или поливинилпирролидон (ПВП). В конкретных вариантах поливинилпирролидон присутствует в композиции в количестве от приблизительно 2% до приблизительно 20% по массе, предпочтительно в количестве от приблизительно 3% до приблизительно 18% по массе, более предпочтительно в количестве от приблизительно 5% до приблизительно 15% по массе, наиболее предпочтительно в количестве приблизительно 10% по массе. В некоторых конкретных вариантах поливинилпирролидоном является ПВП-12 и/или имеет молекулярную массу приблизительно 3000. Другие полимеры формирования матрикса имеют такой же эффект в композициях по изобретению; такие полимеры формирования матрикса содержат ионные полисахариды (например, альгиновую кислоту и альгинаты) или нейтральные полисахариды (например, декстран и НРМС), полиакриловую кислоту и полиметакриловую кислоту и высокомолекулярные органические спирты (например, поливиниловый спирт).
Ингибиторы протеазы
Общепринято в данной области относительно доставки белков, полипептидов и пептидов, что ингибиторы протеазы обычно добавляют в композицию для предотвращения распада АФИ. Однако нет необходимости добавлять ингибиторы протеазы в композиции по данному изобретению. Композиции по изобретению придают устойчивость терапевтическому средству относительно распада протеаз в течение периода активности структуры, т.е. композиции по изобретению являются подавляющей средой для активности фермента. Кроме того, авторы изобретения проводили эксперимент, в котором ингибитор протеазы апротинин добавляли в композицию, и это не оказывало положительного влияния на активность. Проводили аналогичный эксперимент, где ингибитор протеазы ε-аминокапроновая кислота добавляли в композицию, и это тоже не оказывало положительного влияния на активность. Таким образом, в некоторых вариантах, фармацевтическая композиция, описанная в данном описании, в существенной мере не содержит ингибитор протеазы.
Гидрофильная фракция
В вариантах изобретения описанные выше соединения, в том числе терапевтическое средство и соль жирной кислоты со средней длиной цепи, растворяли в водной среде, а затем сушили для получения порошка. Процесс сушки можно проводить, например, путем лиофилизации или грануляции. Полученный порошок называют "гидрофильной фракцией". В гидрофильной фракции вода, как правило, присутствует в количестве менее 6%.
Лиофилизацию можно проводить, как показано в примерах данного описания и способами, известными в данной области, например, как описано в Lyophilization: Introduction and Basic Principles, Thomas Jennings, published by Interpharm/CRC Press Ltd (1999, 2002). Лиофилизат можно оптимально измельчать (например, до 150 микрон) или растирать в ступке. В промышленном производстве лиофилизат предпочтительно измельчают перед смешиванием гидрофильной фракции и гидрофобной среды с целью достижения воспроизводимости партий.
Гранулирование можно проводить, как показано в примерах данного описания, и способами, известными в данной области, например, как описано в Granulation, Salman et al, eds, Elsevier (2006) и в Handbook of Pharmaceutical Granulation Technology, 2nd edition, Dilip M. Parikh, ed., (2005).
Различные связывающие средства можно использовать в процессе грануляции, такие как целлюлоза (в том числе микрокристаллическая целлюлоза), лактоза (например, моногидрат лактозы), декстроза, крахмал и маннитол, и другие связывающие средства, описанные в предыдущих двух ссылках.
Гидрофобная среда
Жиры: Как описано выше, в композициях по изобретению, описанных в данном описании, терапевтическое средство и соль жирной кислоты со средней длиной цепи находятся в непосредственном контакте или связаны с гидрофобной средой. Например, один или оба компонента могут быть покрыты оболочкой, суспендированы, погружены или иным образом связаны с гидрофобной средой. Подходящие гидрофобные среды могут содержать, например, алифатические, циклические или ароматические молекулы. Примеры подходящей алифатической гидрофобной среды включают, не ограничиваясь ими, минеральное масло, моноглицериды жирных кислот, диглицериды, триглицериды, простые и сложные эфиры, а также их комбинации. Примерами подходящей жирной кислоты являются октановая кислота, декановая кислота и додекановая кислота, а также жирные кислоты С7 и С9 и двухосновные кислоты, такие как себациновая кислота и субериновая кислота и их производные. Примеры триглицеридов включают, не ограничиваясь ими, триглицериды с длинной цепью, триглицериды со средней длиной цепи и триглицериды с короткой цепью. Например, триглицеридом с длинной цепью может быть касторовое масло, или кокосовое масло, или оливковое масло, триглицеридом с короткой цепью может быть глицерилтрибутират, а триглицеридом со средней длиной цепи может быть глицерилтрикаприлат. Моноглицериды считаются поверхностно-активными веществами и описаны ниже. Примеры сложных эфиров включают этилизовалерат и бутилацетат. Примеры подходящей циклической гидрофобной среды включают, не ограничиваясь ими, терпеноиды, холестерин, производные холестерина (например, холестерина сульфат) и холестериновые эфиры жирных кислот. Неограничивающий пример ароматической гидрофобной среды включает бензилбензоат.
В некоторых вариантах композиций, описанных в данном описании, желательно, чтобы гидрофобная среда содержала множество гидрофобных молекул. В некоторых вариантах композиций, описанных в данном описании, гидрофобная среда также содержит одно или несколько поверхностно-активных веществ (см. ниже).
В некоторых вариантах композиций, описанных в данном описании, гидрофобная среда также содержит один или несколько адгезивных полимеров, таких как метилцеллюлоза, этилцеллюлоза, гидроксипропилметилцеллюлоза (ГПМЦ) или поли(акрилат) производное Карбопола®934Р (Carbopol®934P, С934Р). Такие адгезивные полимеры могут способствовать монолитности композиции и/или способствовать связыванию с поверхностью слизистой оболочки.
Поверхностно-активные вещества (ПАВ)
Композиции по данному изобретению, описанные в данном описании, могут также содержать поверхностно-активные вещества. Например, поверхностно-активное вещество может быть компонентом гидрофобной среды, как описано выше, и/или поверхностно-активное вещество может быть одним из компонентов твердой формы, как описано выше, например, твердой формой или частицей, которая содержит терапевтическое средство.
Подходящие поверхностно-активные вещества включают ионные и неионные поверхностно-активные вещества. Примерами ионных поверхностно-активных веществ является лецитин (фосфатидилхолин), соли желчных кислот и детергенты. Примеры неионных поверхностно-активных веществ включают моноглицериды, кремофор, простой эфир полиэтиленгликоля и жирного спирта, эфир сорбита и жирной кислоты, эфир полиоксиэтилен сорбитана и жирной кислоты, Солютол HS 15, полоксамер или их комбинацию. Примерами моноглицеридов являются глицерилмонокаприлат (также называемый глицерилмонооктаноатом), глицерилмонодеканоат, глицерилмонолаурат, глицерилмономиристат, глицерилмоностеарат, глицерилмонопальмитат и глицерилмоноолеат. Примеры эфиров сорбита и жирной кислоты включают сорбитан монолаурат, сорбитан моноолеат и сорбитан монопальмитат (Спан 40) или их комбинацию. Примеры эфиров полиоксиэтиленсорбитана и жирной кислоты включают полиоксиэтиленсорбитан моноолеат (Твин 80), моностеаратполиоксиэтиленсорбитан, полиоксиэтиленсорбитан монопальмитат или их комбинацию. Коммерческие препараты моноглицеридов, которые были использованы, также содержат различные количества диглицеридов и триглицеридов.
Композиции, описанные в данном описании, содержащие поверхностно-активное вещество, обычно содержат менее 12% по массе общего количества поверхностно-активного вещества (например, менее 10%, менее 8%, менее 6%, менее 4%, менее 2% или менее 1%). В отдельных вариантах изобретения суммарное количество всех поверхностно-активных веществ составляет приблизительно 6%.
Способы создания лекарственного препарата и созданные препараты
Также включены в изобретение способы получения лекарственных препаратов, описанных в данном описании. Таким образом, одним из вариантов изобретения является способ получения фармацевтического препарата, который включает подготовку водорастворимого препарата, содержащего терапевтически эффективное количество, по меньшей мере, одного терапевтического средства и соль жирной кислоты со средней длиной цепи (как описано выше), сушку водорастворимого препарата для получения твердого порошка, а также суспендирование твердого порошка в гидрофобной среде, для получения суспензии, содержащей в твердой форме терапевтическое средство и соль жирной кислоты со средней длиной цепи, и получение фармацевтического препарата, в котором фармацевтический препарат содержит 10% или более по массе соли жирной кислоты со средней длиной цепи.
Одним из вариантов является процесс получения лекарственного препарата, который включает обеспечение твердого порошка терапевтически эффективным количеством, по меньшей мере, одного терапевтического средства, и твердый порошок, содержащий соль жирной кислоты со средней длиной цепи, и суспендирование твердых порошков в гидрофобной среде для получения суспензии, содержащей в твердой форме терапевтическое средство и соль жирной кислоты со средней длиной цепи, тем самым создавая лекарственный препарат, где лекарственный препарат содержит 10% или более по массе соли жирной кислоты со средней длиной цепи.
В одном из вариантов процессов и препаратов, описанных в данном описании, водорастворимый препарат является водным раствором. В некоторых вариантах сушка водорастворимого препарата достигается путем лиофилизации или грануляции. В процессе грануляции связывающее вещество может быть добавлено к водорастворимому препарату перед сушкой. В некоторых вариантах на стадии сушки удаляется достаточное количество воды, чтобы содержание воды в лекарственном препарате было ниже, чем 6% по массе, около 5% по массе, около 4% по массе, около 3%, около 2%, или около 1% по массе. В некоторых вариантах процессов и препаратов, описанных в данном описании, на стадии сушки удаляется количество воды, так что содержание воды в твердом порошке ниже, чем 6%, или 5%, или 4%, или 3%, или, предпочтительно, ниже, чем 2% по массе. Содержание воды является, как правило, незначительным и вода может быть адсорбирована из твердой фазы в процессе лиофилизации, т.е. вода может сохраняться из-за межмолекулярных связей. В некоторых вариантах водорастворимая композиция дополнительно содержит стабилизатор, например, метилцеллюлозу. В предпочтительных вариантах процессов и препаратов, описанных в данном описании, гидрофобная среда является касторовым маслом, глицерилтрикаприлат, глицерилтрибутират, или их комбинация, и она может дополнительно содержать октановую кислоту; в некоторых вариантах гидрофобная среда включает алифатическое, олефиновое, циклическое или ароматическое соединение, минеральное масло, парафин, жирную кислоту, такую как октановая кислота, моноглицерид, диглицерид, триглицерид, эфир или сложный эфир, или их комбинацию. В некоторых вариантах процессов и препаратов, описанных в данном описании, триглицеридом является триглицерид с длинной цепью, триглицерид со средней длиной цепи, предпочтительно глицерилтрикаприлат, или триглицерид с короткой цепью, предпочтительно глицерилтрибутират, и триглицеридом с длинной цепью является касторовое масло или кокосовое масло или их комбинация. В некоторых вариантах процессов и препаратов, описанных в данном описании, гидрофобная среда содержит касторовое масло, глицерилтрикаприлат, глицерилтрибутират или их комбинацию или смесь, и может дополнительно содержать октановую кислоту. В некоторых вариантах процессов и препаратов, описанных в данном описании, гидрофобная среда содержит глицерилтрикаприлат или низкомолекулярный сложный эфир, например, этилизовалерат или бутилацетат. В некоторых вариантах процессов и препаратов, описанных в данном описании, основным компонентом по массе гидрофобной среды является касторовое масло, и она может дополнительно содержать глицерилтрикаприлат. В некоторых вариантах процессов и препаратов, описанных в данном описании, основным компонентом по массе гидрофобной среды является глицерилтрикаприлат, и она может дополнительно содержать касторовое масло.
Основной препарат предоставили как вариант, в котором гидрофобная среда состоит, в основном, из касторового масла, глицерилмоноолеата и глицерилтрибутирата; в следующем варианте основного препарата гидрофильная фракция состоит, в основном, из терапевтического средства, ПВП-12 и октаноата натрия.
Специфический препарат привели как вариант, в котором гидрофобная среда состоит в основном из глицерилтрикаприлата, касторового масла, глицерилмонокаприлата, и Твин 80, и гидрофильная фракция состоит в основном из терапевтического средства (например, октреотида), ПВП-12 и октаноата натрия. Другой специфический препарат привели как вариант, в котором гидрофобная среда включает глицерилтрикаприлат, касторовое масло, глицерилмонокаприлат, и Твин 80, и гидрофильная фракция включает терапевтическое средство (например, октреотид), ПВП-12 и октаноат натрия. В некоторых вариантах гидрофобная среда состоит в основном из глицерилтрикаприлата, и в некоторых вариантах дополнительно содержит касторовое масло и/или глицерилмонокаприлат.
В некоторых вариантах препарат включает суспензию, которая состоит в основном из смеси гидрофобной среды и твердой формы, в которой твердая форма включает терапевтически эффективное количество терапевтического средства и не менее одной соли жирной кислоты со средней длиной цепи, и, в котором соль жирной кислоты со средней длиной цепи присутствует в препарате в количестве 10% или более по массе. В некоторых вариантах гидрофобная среда состоит в основном из касторового масла, глицерилмоноолеата и глицерилтрибутирата; или гидрофобная среда состоит в основном из глицерилтрибутирата и глицерилмонокаприлата; или гидрофобная среда состоит в основном из касторового масла, глицерилтрикаприлата и глицерилмонокаприлата. В некоторых вариантах гидрофобная среда состоит из триглицеридов и моноглицерид, и в некоторых частных вариантах моноглицерид имеет те же радикалы жирной кислоты, что и триглицерид. В некоторых из этих вариантов триглицеридом является глицерилтрикаприлат и моноглицеридом является глицерилмонокаприлат. В некоторых вариантах соль жирной кислоты со средней длиной цепи в водорастворимом препарате имеет такой же радикал жирной кислоты, что и моноглицерид со средней длиной цепи или триглицерид со средней длиной цепи, или их комбинация. В некоторых из этих вариантов солью жирной кислоты со средней длиной цепи является каприлат натрия (октаноат натрия) и моноглицеридом является глицерилмонокаприлат и триглицеридом является глицерилтрикаприлат.
Многие препараты, описанные в данном описании, содержат суспензию, которая включает смесь гидрофобной среды и твердой формы, в которой твердая форма включает терапевтически эффективное количество терапевтического средства и не менее одной соли жирной кислоты со средней длиной цепи, и в которой соль жирной кислоты со средней длиной цепи присутствует в препарате в количестве 10% или более по массе. Твердая форма может быть частицей (например, состоит в основном из частиц, или состоит из частиц). Частица могут получать путем лиофилизации или грануляции.
В конкретном варианте состав включает в основном суспензию, которая содержит смесь гидрофобной среды и твердой формы, в которой твердая форма включает терапевтически эффективное количество терапевтического средства и около 10-20%, предпочтительно 15%, соли жирной кислоты со средней длиной цепи, предпочтительно октаноата натрия, и около 5-10%, предпочтительно 10%, ПВП-12; и где гидрофобная среда содержит около 20-80%, предпочтительно 30-70%, триглицерида, предпочтительно глицерилтрикаприлата, или глицерилтрибутирата, или касторового масла, или их смеси, примерно 3-10% поверхностно-активных веществ, преимущественно около 6%, предпочтительно глицерилмонокаприлата, и Твин 80, и около 1% воды; в конкретных вариантах терапевтическое средство присутствует в количестве менее чем 33%, или менее чем 25%, или менее чем 10%, или менее чем 1%, или менее чем 0,1%. Твердая форма может быть частицей (например, состоит в основном из частиц, или состоит из частиц). Частица может быть получена путем лиофилизации или грануляции. В конкретном варианте твердая форма может быть частицей и может быть получена путем лиофилизации или грануляции.
В еще одном варианте препарат включает в основном суспензию, которая содержит смесь гидрофобной среды и твердой формы, в которой твердая форма включает терапевтически эффективное количество терапевтического средства и около 10-20%, предпочтительно 15%, соли жирной кислоты со средней длиной цепи, предпочтительно октаноата натрия, и около 5-10%, предпочтительно 10%, ПВП-12; и, в котором гидрофобная среда содержит около 20-80%, предпочтительно 30-70%, триглицерида со средней или короткой длиной цепи, предпочтительно глицерилтрикаприлата или глицерилтрибутирата, около 0-50%, предпочтительно 0-30%, касторового масла, около 3-10% поверхностно-активных веществ, предпочтительно около 6%, предпочтительно глицерилмонокаприлата и Твин 80, и около 1% воды; в частных вариантах терапевтическое средство присутствует в количестве менее чем 33%, или менее чем 25%, или менее чем 10%, или менее чем 1%, или менее чем 0,1%.
В конкретном варианте состав включает в основном суспензию, которая содержит смесь гидрофобной среды и твердой формы, в которой твердая форма включает терапевтически эффективное количество терапевтического средства и около 15% октаноата натрия и около 10% ПВП-12; и в которой гидрофобная среда содержит около 41% глицерилтрикаприлата, около 27% касторового масла, около 4% глицерилмонокаприлата, около 2% Твин-80, около 1% воды, и 1% или менее терапевтического средства, когда терапевтическим средством является октреотид, составляющий около 0,058%.
В другом частном варианте состав включает в основном суспензию, которая содержит смесь гидрофобной среды и твердой формы, в которой твердая форма включает терапевтически эффективное количество терапевтического средства и около 15% октаноата натрия, и около 10% ПВП-12; и в которой гидрофобная среда содержит около 68% глицерилтрикаприлата, около 4% глицерилмонокаприлата, около 2% Твин-80, около 15% октаноата натрия, около 10% ПВП-12, около 1% воды, и менее 1% терапевтического средства, когда терапевтическое средство является октреотидом, составляющим примерно 0,058%.
Одним из вариантов является препарат, включающий суспензию, который содержит смесь гидрофобной среды и твердой формы, в которой твердая форма включает терапевтически эффективное количество октреотида и не менее одной соли жирной кислоты со средней длиной цепи; а в следующем варианте соль жирной кислоты со средней длиной цепи присутствует в препарате в количестве 10% или более по массе, предпочтительно 15% по массе, а в следующем варианте твердая форма дополнительно содержит формирующий матрицу полимер. В следующем варианте формирующим матрицу полимером является декстран или поливинилпирролидон (ПВП). В конкретном варианте формирующим матрицу полимером является поливинилпирролидон, и поливинилпирролидон присутствует в препарате в количестве приблизительно от 2% до 20% по массе, предпочтительно около 10% по массе. В конкретном варианте поливинилпирролидон является ПВП-12 и/или поливинилпирролидон имеет молекулярную массу около 3000. В конкретных вариантах гидрофобная среда состоит в основном из глицерилтрикаприлата и твердой формы, дополнительно содержит ПВП-12 и октаноат натрия. В более конкретных вариантах гидрофобная среда дополнительно содержит касторовое масло или глицерилмонокаприлат, или их комбинацию, и поверхностно-активное вещество. В дальнейших конкретных вариантах гидрофобная среда содержит глицерилтрикаприлат, глицерилмонокаприлат, и полиоксиэтиленсорбитан моноолеат (Твин 80). В следующем варианте твердая форма состоит в основном из октреотида, ПВП-12 и октаноата натрия. В конкретном варианте препарат содержит около 41% глицерилтрикаприлата, около 27% касторового масла, около 4% глицерилмонокаприлата, около 2% Твин-80, около 15% октаноата натрия, около 10% ПВП-12, около 1% воды и около 0,058% октреотида. В другом конкретном варианте препарат содержит около 68% глицерилтрикаприлата, около 4% глицерилмонокаприлата, около 2% Твин-80, около 15% октаноата натрия, около 10% ПВП-12, около 1% воды, и около 0,058% октреотида.
Во всех вышеупомянутых препаратах, перечисленные проценты показывают массовое соотношение, и твердая форма может быть частицей (например, состоять в основном из частиц или состоять из частиц). Частица может быть получена путем лиофилизации или грануляции.
При нормальных условиях хранения, терапевтическое средство в препарате изобретения остается стабильным в течение длительного периода времени. Химическое и физическое состояние препарата устойчивое. При введении в кишечник, терапевтическое средство становится защищенным от повреждений средой ЖКТ, так как препараты созданы на масляной основе, и поэтому в кишечнике образовывается отдельная местная окружающая среда, в которой терапевтическое средство содержится в капельках масла, что дает стабильность in vivo.
В некоторых вариантах разрабатывается производство препарата, который состоит в основном из терапевтического средства, соли жирной кислоты со средней длиной цепи, и гидрофобной среды. В вариантах изобретения твердый порошок (твердая форма) состоит в основном из терапевтического средства и соли жирной кислоты со средней длиной цепи. Дальнейшими вариантами изобретения являются лекарственные препараты, продуцированные в процессе, описанном в данном описании. В некоторых лекарственных препаратах терапевтическое средство представляет собой белок, полипептид, пептид, гликозаминогликан, полисахарид, низкомолекулярное соединение или полинуклеотид, и, в частных вариантах терапевтическим средством является инсулин, гормон роста, паратиреоидный гормон, терипаратид, интерферон-альфа (ИФН-α), низкомолекулярный гепарин, лейпролид, фондапаринукс, октреотид, экзенатид, терлипрессин, ванкомицин или гентамицин. Особые варианты изобретения включают лекарственную форму для пероральной доставки, включающую лекарственный препарат, в частности лекарственную форму для пероральной доставки, которая покрыта кишечнорастворимой оболочкой. Дальнейшие варианты изобретения включают капсулу, содержащую препарат изобретения, и в различных вариантах капсулой является жесткая или мягкая желатиновая капсула, и в целом капсула покрыта кишечнорастворимой оболочкой. Другие варианты изобретения включают ректальную лекарственную форму, включающую лекарственный препарат, в частности, свечи, или буккальную лекарственную форму. Набор, включающий инструкции и лекарственную форму, также предусмотрен.
Терапевтическое средство или соль жирной кислоты со средней длиной цепи, или любое сочетание терапевтического средства и других компонентов, таких как белковые стабилизаторы, могут быть приготовлены путем растворения смеси (например, образуя водный раствор или смесь), которые могут быть лиофилизированы совместно, а затем суспендированы в гидрофобной среде. Другие компоненты препарата также могут быть дополнительно лиофилизированы или добавлены во время растворения твердых веществ.
В некоторых вариантах терапевтическое средство растворяли в смеси, например, включая один или несколько дополнительных компонентов, таких как соль жирной кислоты со средней длиной цепи, стабилизатор и/или поверхностно-активное вещество, и растворитель удаляют, чтобы обеспечить получение твердого порошка (твердая форма), который суспендирован в гидрофобной среде. В некоторых вариантах терапевтическое средство и/или соль жирной кислоты со средней длиной цепи может быть сформировано в гранулированной частице, которая затем связывается с гидрофобной средой (например, суспендированы в гидрофобной среде или покрыты гидрофобной средой). В целом, препараты, описанные в данном описании, практически не содержат "мембрано-разжижающих средств", таких как спирты со средней длиной цепи.
"Мембрано-разжижающие средства" определены как спирты со средней длиной цепи, которые имеют длину углеродной цепи от 4 до 15 атомов углерода (например, включая от 5 до 15, от 5 до 12, 6, 7, 8, 9, 10 или 11 атомов углерода). Например, мембрано-разжижающее средство может быть линейным (например, насыщенным или ненасыщенным), разветвленным (например, насыщенным или ненасыщенным), циклическим (например, насыщенным или ненасыщенным), или ароматическим спиртом. Примеры применимых линейных спиртов включают, не ограничиваясь ими, бутанол, пентанол, гексанол, гептанол, октанол, нонанол, деканол, ундеканол, додеканол, тридеканол, тетрадеканол и пентадеканол. Примеры разветвленных спиртов включают, не ограничиваясь ими, гераниол, фарнезол, родинол, цитронеллол. Примеры ароматических спиртов включают, не ограничиваясь ими, ментол, терпинеол, миртенол, периллил и этанол. Примеры применимых ароматических спиртов включают, не ограничиваясь ими, бензиновый спирт, 4-гидроксикоричную кислоту, тимол, стирол гликоль, и фенольные соединения. Примеры фенольных соединений включают, не ограничиваясь ими, фенол, м-крезол, и м-хлоркрезол.
При желании, лекарственный препарат может также содержать небольшие количества нетоксичных вспомогательных веществ, таких как рН буферные агенты, и других веществ, такие как, например, ацетат натрия и триэтаноламинолеат.
По крайней мере, в одном из вариантов, терапевтическое средство, такое как белок, может быть химически модифицировано, чтобы повысить его период полураспада в кровотоке. Например, терапевтическое средство может пройти такой процесс, как пегилирование.
В некоторых вариантах способ получения лекарственного препарата включает подготовку водорастворимой препарата, содержащего терапевтически эффективное количество не менее одного терапевтического средства и соли жирной кислоты со средней длиной цепи, сушку водорастворимого препарата для получения твердого порошка, и растворение твердого порошка в растворе, состоящем в основном из октановой кислоты, тем самым создавая лекарственный препарат, который представляет собой раствор. В некоторых вариантах твердой формой может быть частица (например, состоит в основном из частиц, или состоит из частиц). В некоторых вариантах частица может быть получена путем лиофилизации или грануляции. В некоторых вариантах этого процесса октановая кислота присутствует в препарате в количестве приблизительно от 60% до 90%, или в количестве приблизительно от 70 до 85%, предпочтительно около 78%. В некоторых вариантах этого процесса солью жирной кислоты является октаноат натрия; в других вариантах этого процесса соль жирной кислоты со средней длиной цепи присутствует в препарате в количестве от 11% до 40% по массе, или в количестве примерно от 11% до 28% по массе, или в количестве около 15% по массе. В некоторых вариантах этого процесса препарат дополнительно содержит формирующий матрицу полимер и в частных вариантах этого процесса формирующим матрицу полимером является декстран или поливинилпирролидон (ПВП); в других вариантах этого процесса поливинилпирролидон присутствует в препарате в количестве приблизительно от 2% до 20% по массе, или в количестве приблизительно от 5% до 15% по массе, предпочтительно в количестве примерно 10% от массе. В некоторых вариантах этого процесса поливинилпирролидон ПВП-12 и/или имеет молекулярную массу около 3000. Препарат может также включать поверхностно-активные вещества, описанные выше. Фармацевтическая продукция этих процессов является дальнейшими вариантами изобретения, например, препарат, содержащий октановую кислоту в количестве от 60% до 90%, или в количестве от 70 до приблизительно 85%, предпочтительно около 78%; соль жирной кислоты, предпочтительно октаноат натрия, присутствующая в препарате в количестве от 11% до 40% по массе, или в количестве примерно от 11% до 28% по массе, или в количестве около 15% по массе; формирующий матрицу полимер, например, поливинилпирролидон, предпочтительно ПВП-12, присутствует в препарате в количестве от 2% до 20% по массе, или предпочтительно в количестве от 5% до 15% по массе, предпочтительно в количестве примерно 10% по массе; и поверхностно-активные вещества, как описано выше. Могут также присутствовать небольшие количества других гидрофобных составляющих, как описано выше.
Капсулы
Предпочтительными лекарственными препаратами являются лекарственные формы пероральной доставки или суппозитории. Типичные лекарственные формы включают желатиновые или растительные капсулы, крахмалподобные гидроксилпролилметилцеллюлозные ("ГПМЦ") капсулы, покрытые кишечнорастворимой оболочкой, содержащие нерасфасованную лекарственную форму. Капсулы, которые могут быть применены для инкапсуляции препарата по данному изобретению, известны в данной области и описаны, например, в Pharmaceutical Capsules edited by Podczech and Jones, Pharmaceutical Press (2004) и в Hard gelatin capsules today - and tomorrow, 2nd edition, Steggeman ed published by Capsugel Library (2002).
Дополнительные препараты
Препараты изобретения могут быть составлены с применением дополнительных способов, известных в данной области, например, как описано в следующих публикациях: Pharmaceutical Dosage Forms Vols 1-3 ed. Lieberman, Lachman and Schwartz, published by Marcel Dekker Inc, New York (1989); Water-insoluble Drug Formulation 2nd edition, Liu, editor, published by CRC Press, Taylor and Francis Group (2008); Therapeutic Peptides and Proteins: Formulation, Processing and Delivery Systems, 2nd edition by Ajay K. Banga (author) published by CRC Press, Taylor and Francis Group (2006); Protein Formulation and Delivery, 2nd edition, McNally and Hasted eds, published by Informa Healthcare USA Inc (2008); and Advanced Drug Formulation to Optimize Therapeutic Outcomes, Williams et al eds, published by Informa Healthcare USA (2008).
Препараты изобретения могут быть составлены с применением технологии микрочастиц, например, как описано в Microparticulate Oral Drug Delivery, Gerbre-Selassie ed., published by Marcel Dekker Inc (1994) и в Dey et al, Multiparticulate Drug Delivery Systems for Controlled Release, Tropical Journal of Pharmaceutical Research, September 2008; 7 (3): 1067-1075.
Способы лечения
Препараты, описанные в данном описании, проявляют эффективную энтеральную доставку неизмененных биологически активных веществ (например, терапевтического средства) и, таким образом, имеют множество применений. Например, препараты, описанные в данном описании, могут быть применены для лечения сахарного диабета.
В частности, инсулин для лечения и профилактики субъектов (пациентов), страдающих диабетом типа II (профилактика сахарного диабета), а также для лечения пациентов, страдающих от дисгликемии, предиабета, метаболического синдрома, и других заболеваний, может вводиться в соответствии с одним или несколькими вариантами изобретения. Метаболический синдром представляет собой сочетание медицинских расстройств, которые увеличивают риск развития сердечно-сосудистых заболеваний и диабета. Метаболический синдром представляет собой совокупность различных симптомов: (1) гипергликемия натощак (инсулинорезистентность, диабет II типа, и т.д.); (2) уменьшение уровня холестерол-ЛПВП; (3) повышение уровня триглицеридов; (4) высокое кровяное давление; (5) центральное ожирение; и (6) провоспалительное состояние.
Одним из вариантов изобретения является способ лечения или профилактики субъекта, страдающего от вышеперечисленных состояний, когда количеством инсулина, достаточного для лечения состояния, является небольшая доза инсулина, сформированная в препарате изобретения. Небольшая доза инсулина обеспечена менее чем 300 или менее чем 200 Ед на капсулу, например, 40-200 Ед на капсулу.
Терлипрессин (или другие аналоги вазопрессина) для лечения субъектов (пациентов), страдающих от печеночно-почечного синдрома (ППС), включая ППС I и II, кровотечений расширенных варикозных вен пищевода, портальной гипертензии и других состояний, может вводиться в соответствии с одним или несколькими вариантами изобретению. Такие препараты терлипрессина также могут быть применены для первичной и вторичной профилактики варикозного кровотечения. Препарат изобретения включает суспензию, которая включает смесь гидрофобной среды и твердой формы, где твердая форма включает терапевтически эффективное количество терлипрессина (или другие аналоги вазопрессина) и не менее одной соли жирной кислоты со средней длиной цепи.
Экзенатид для улучшения гликемического контроля у пациентов, страдающих диабетом II типа и для лечения других состояний, таких как ожирение, и для применения при коррекции массы тела, может вводиться в соответствии с одним или несколькими вариантами изобретению.
Интерферон-альфа для лечения субъектов, страдающих хроническим гепатитом С и хроническим гепатитом В, и для лечения других состояний, включая рак, может вводиться в соответствии с одним или несколькими вариантами изобретению.
Копаксон для лечения субъектов, страдающих рассеянным склерозом и для лечения других состояний, включая воспалительные заболевания, может вводиться в соответствии с одним или несколькими вариантами изобретению.
Десмопрессин для лечения субъектов, страдающих от первичного ночного энуреза, центрального несахарного диабета (НД) или от нарушений свертываемости крови (болезнь Виллебрандта и гемофилия) может вводиться в соответствии с одним или несколькими вариантами изобретению. Пероральная форма десмопрессина в препаратах, известных в данной области, обладает крайне низкой биодоступностью при пероральном введении.
Октреотид был впервые синтезирован в 1979 г., и является октапептидом, который фармакологически имитирует естественный соматостатин, хотя это более мощный ингибитор гормона роста, глюкагона и инсулина, чем естественный гормон. Октреотид или другие аналоги соматостатина могут вводиться в соответствии с одним или несколькими вариантами изобретения для применения в лечении или профилактике заболевания или нарушения у субъекта, страдающего от заболеваний, таких как акромегалия, аномальная моторика ЖКТ, приступы гиперемии, связанные с карциноидным синдромом, портальная гипертензия, эндокринная опухоль (например, карциноидная опухоль, ВИПома), гастропарез, диарея, панкреатическое подтекание или поджелудочная псевдокиста. Диарея может возникнуть в результате лучевой терапии или может произойти, например, у субъекта с опухолью поджелудочной железы, клетки которой вырабатывают вазоактивный кишечный полипептид (ВИПома). Кроме того, пациенты, которые перенесли операцию на поджелудочной железе, могут страдать от внешней секреции поджелудочной железы, и они уязвимы к развитию панкреатического подтекания или псевдокисты, которые могут быть подвергнуты лечению октреотидными препаратами по изобретению. Некоторые предпочтительные варианты нацелены на способ лечения субъекта, имеющего заболевание, такое как акромегалия, ненормальная моторика ЖКТ, приступы гиперемии, связанные с карциноидным синдромом, портальная гипертензия, эндокринная опухоль (например, карциноидная опухоль, ВИПома), гастропарез, диарея, панкреатическое подтекание или поджелудочная псевдо-киста, который включает введение субъекту препарата изобретения, где терапевтическим средством является октреотид в количестве достаточном для лечения заболевания. Препараты с октреотидом по изобретению могут также быть применены для первичной и вторичной профилактики варикозного кровотечения, которое может вызвать портальная гипертензия; варикоз может быть желудка или пищевода. Другими областями применения препарата с октреотидом по изобретению является лечение гиповолемического шока (например, геморрагического) или сосудорасширяющего (например, септического) происхождения, печеночно-почечного синдрома (ППС), сердечно-легочной реанимации и анестезии, искусственной артериальной гипотензии. Другие аналоги соматостатина могут быть применены в способах и препаратах, в которых используется октреотид.
Ванкомицин (молекулярный вес 1449 Да) является гликопептидным антибиотиком, который используется для профилактики и лечения инфекций, вызванных грамположительными бактериями. Первоначальным показанием к применению ванкомицина было лечение метициллин-устойчивого золотистого стафилококка (МУЗС). Ванкомицин не стал терапией первой линии для золотистого стафилококка, одна из причин в том, что ванкомицин следует вводить внутривенно. Прежние в данной области препараты ванкомицина необходимо вводить внутривенно при системной терапии, так как ванкомицин не должен пересекать слизистую оболочку кишечника. Это большие гидрофильные молекулы, которые плохо проходят через желудочно-кишечную слизистую оболочку. Единственным указанием для пероральной терапии ванкомицином является лечение псевдомембранозного колита, где он должен использоваться перорально для доступа к очагу инфекции в толстом кишечнике. Ванкомицин для применения в лечении или профилактике инфекции у субъекта может быть введен перорально этому субъекту в соответствии с одним или несколькими вариантами изобретения. Некоторые предпочтительные варианты изобретения направлены на способ лечения или профилактики инфекции у субъекта, который включает введение субъекту препарата изобретения, где терапевтическим средством является ванкомицин, в количестве достаточном для лечения и профилактики инфекции.
Гентамицин (молекулярный вес=478) является аминогликозидным антибиотиком, который используется для лечения многих видов бактериальных инфекций, особенно вызванных грамотрицательными бактериями. Когда гентамицин вводили в пероральной форме, в существующих препаратах данной области, он не обладал системной активностью. Это по причине того, что он не абсорбируется в какой-нибудь заметной степени из тонкого кишечника.
Кроме того, препараты по изобретению также могут быть применены для лечения состояний, связанных с атеросклерозом, образованием тромбов, и эмболией, такими как инфаркт миокарда и инсульт. В частности, препараты могут быть применены для доставки гепарина, низкомолекулярного гепарина, или фондапаринукса через слизистую оболочку эпителия.
Препараты по данному изобретению могут также быть применены для лечения гематологических заболеваний и дефицитных состояниях, таких как анемия и гипоксия, которые восприимчивы к введению гематологических факторов роста. Препараты изобретения могут быть применены для доставки витамина В 12 субъекту с высокой биодоступностью, при которой слизистый эпителий субъекта нуждается в достаточном внутреннем факторе. ГКСФ может также вводиться в соответствии с различными вариантами. Кроме того, препараты по данному изобретению могут быть применены для лечения остеопороза, например, путем энтерального введения ПТГ, терипаратида или кальцитонина один, два раза или более в день.
Гормон роста человека (ГР) для лечения дефицита гормона роста, в частности у детей, может быть введен в соответствии с одним или несколькими вариантами. В некоторых предпочтительных вариантах препарат, описанный в данном описании, включающий гормон роста, может быть введен субъекту для лечения и профилактики метаболических и липидных заболеваний, связанных, например, с ожирением, абдоминальным ожирением, гиперлипидемией или гиперхолестеринемией. Например, препарат изобретения, включающий гормон роста, может быть введен перорально субъекту, таким образом лечить ожирение (например, абдоминальное ожирение). В некоторых предпочтительных вариантах препарат, описанный в данном описании, который содержит гормон роста, вводят субъекту для лечения и профилактики липодистрофии, связанной с ВИЧ (истощение при СПИДе), или для лечения синдрома Прадера-Вилли, при нарушении роста, из-за недостаточной секреции гормона роста (например, связанных с дисгенезией гонад или синдромом Тернера), при нарушении роста у детей препубертатного возраста с хронической почечной недостаточностью, и в качестве заместительной терапии у взрослых с выраженным дефицитом гормона роста. Препараты изобретения, включающие гормон роста, могут быть введены перорально субъекту для содействия заживлению ран и ослабления катаболических реакций при тяжелых ожогах, сепсисе, множественной травме, тяжелых операциях, остром панкреатите и кишечном свище. Многие другие условия, кроме дефицита ГР, приводят к снижению роста, но благоприятное воздействие на рост (увеличение роста) часто хуже, чем при лечении дефицита ГР. Другими примерами низкорослости, которые можно лечить препаратами по изобретению, включающими гормон роста, являются задержка внутриутробного развития и тяжелая идиопатическая низкорослость. Другое потенциальное применение препаратов изобретения, включающих гормон роста, включает лечение для изменения в обратном направлении или предотвращения последствия старения у пожилых людей, для содействия при наращивании мышц, и для лечения фибромиалгии.
Некоторые предпочтительные варианты направлены на способ лечения заболеваний, таких как ожирение, расстройство жирового обмена, связанного с ВИЧ, нарушение обмена веществ, или дефицит роста у субъекта, который включает введение субъекту препарата изобретения, в котором терапевтическим средством (эффектором) является гормон роста, в количестве достаточном для лечения заболевания.
Некоторые предпочтительные варианты направлены на способ лечения костных заболеваний у субъекта, который включает введение субъекту препарата изобретения, в котором терапевтическим средством является терипаратид или паратиреоидный гормон, в количестве достаточном для лечения костных заболеваний.
Некоторые предпочтительные варианты направлены на способ лечения или профилактики коагулирующих заболеваний крови у субъекта, который включает введение субъекту препарата изобретения, в котором терапевтическим средством является гепарин, производная гепарина, или фондапаринукс, в количестве достаточном для лечения и профилактики коагулирующих заболеваний крови.
Лейпролид (агонист ГнРГ), препарат который разработан в варианте изобретения, может использоваться для лечения женского бесплодия (например, доза раз или два раза в день), рака простаты и болезни Альцгеймера.
Один из вариантов изобретения относится к способу лечения субъекта, страдающего от болезни или заболевания, который включает введение субъекту препарата изобретения в количестве, достаточном для лечения этого заболевания. Другой вариант изобретения относится к препарату изобретения для применения в лечении заболевания или нарушения у субъекта. Другой вариант изобретения относится к применению терапевтического средства в производстве лекарственного препарата с помощью процесса изобретения для лечения заболевания.
Схема приема лекарственного средства выбирается в соответствии с целым рядом факторов, включая тип, вид, возраст, вес, пол и состояние здоровья пациента, тяжесть состояния, подлежащего лечению; пути введения, почечную и печеночную функции пациента; и конкретное соединение или соль после применения. Опытный врач или ветеринарный врач может легко определить и назначить эффективное количество препарата, необходимое для предотвращения, противодействия или приостановки прогресса заболевания. Пероральные дозы препарата изобретения, при применении для указанных эффектов, могут быть предоставлены в форме капсул, содержащих 0,001, 0,0025, 0,005, 0,01, 0,025, 0,05, 0,1, 0,25, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 50,0 или 100, 200, 300, 400, 500, 600, 700, 800 или 1000 мг терапевтического средства.
Препараты по настоящему изобретению могут быть введены в однократной суточной дозе, или общая суточная доза может быть введена в дробных дозах два, три, четыре, пять или шесть раз в день. В некоторых вариантах препарат вводят в суточной дозе от 0,01 до 5000 мг/день, например, введение один раз в день (например, утром или перед сном) или два раза или более в день (например, утром и перед сном).
Типичным продуктом изобретения является препарат для перорального применения на основе АФИ, в виде покрытых кишечнорастворимой оболочкой капсул: каждая капсула содержит АФИ ко-лиофилизированный с ПВП-12 и октаноатом натрия, и суспендированый в гидрофобной (липофильной) среде, содержащей: глицерилтрикаприлат, глицерилмонокаприлат и Твин-80; в другом типичном продукте изобретения дополнительно присутствует касторовое масло. Препараты, описанные в данном описании, могут быть введены субъекту, т.е. человеку или животному, для лечения субъекта фармакологически или терапевтически эффективным количеством терапевтического средства, описанным в данном описании. Животным может быть млекопитающее, например, мышь, крыса, свинья, лошадь, корова или овца. Используемый в данном описании термин "фармакологически или терапевтически эффективное количество" означает, что количество лекарства или фармацевтического средства (терапевтического средства), которое вызовет биологическую или медицинскую реакцию ткани, системы, животного или человека, является таким, как требует исследователь или клиницист.
Препараты изобретения позволяют включать лекарственное средство в разработку без химической модификации терапевтического средства. В дополнение, как было показано выше, много различных терапевтических средств успешно составлены в препаратах изобретения, включая полипептиды, нуклеотиды, малые молекулы и даже белки среднего размера. Кроме того, препараты изобретения предусматривают высокую пластичность при введении терапевтического средства. Предельные параметры концентрации зависят от терапевтического средства. На сегодняшний день, границы допустимой концентрации не найдены, однако концентрация до 1,5%, масс (полипептиды) и 6%, масс (малые молекулы) достигнута и предусмотрена более высокая концентрация до 33%. В заключение, препараты изобретения защищают соединения карго от инактивации в окружающей среде ЖКТ, связанной, например, с протеолитической деградацией и окислением.
Функции и преимущества этих и других вариантов будут более полно осознаны из следующих примеров. Эти примеры приводятся с целью наглядности и не предназначены для определения пределов применения систем и способов, рассмотренных в данном описании.
ПРИМЕРЫ
Пример 1: Препараты
А. Композиция инсулинового препарата
Табл. 1А представляет пример препарата в соответствии с одним или несколькими вариантами. Более конкретно, эта композиция является инсулиновым препаратом. Инсулин был получен от Diosynth Biotechnology; октаноат натрия и NaOH от Merck, MgCl2, МС400, Спан 40, лецитин и касторовое масло от Spectrum; ПВП-12 от BASF; этилизовалерат от Merck/Sigma; глицерилтрибутират от Acros/Penta и глицерилмоноолеат от Abitec Corp.

Б. Препарат для лейпролида: Табл. 1Б представляет пример композиции для АФИ (активного фармацевтического ингредиента) в соответствии с одним или несколькими вариантами. Более конкретно, эта композиция является препаратом лейпролида.

В. Препараты со сниженным количеством гидрофобной среды (50% гидрофобной среды)
Табл. 1 В представляет пример композиции для АФИ в соответствии с одним или несколькими вариантами. Более конкретно, эта композиция является препаратом для декстрана (FD4). FD4 представляет собой декстран, меченый ФИТЦ с MB 4,4 кДа (Sigma, FD4), и это декстран использовали во всех примерах, если не указано иное. Данный состав содержит кокосовое масло (Sigma) вместо GTB.


Вышеприведенные составы применяются для широкого спектра терапевтических средств и имеют хорошую биодоступность соединений карго на моделях животных, описанных ниже. Отметим, что общее количество терапевтического средства может изменяться по мере необходимости в любых препаратах и может быть незначительно изменено в препаратах, например, NaOH не всегда используется; кокосовое масло может быть использовано вместо глицерилтрибутирата; MgCl2 не всегда используется (например, с чГР он не используется); все ингредиенты могут быть заменены, как описано выше в спецификации.
Пример 2: Схематическое представление производства инсулинового препарата
Фиг. 1 иллюстрирует способ получения композиции в соответствии с одним или несколькими вариантами. Например, этот способ может осуществлять изготовление композиций, представленных выше в примере 1.
Пример 3: Сочетание твердых частиц, содержащих октаноат натрия, и гидрофобной среды имеет решающее значение для проникающей активности
Фиг. 2 представляет данные, относящиеся к уровню инсулина в сыворотке после ректального введения крысам. Крысам давали наркоз и вводили 100 мкл нерасфасованной лекарственной формы препарата, содержащего дозу инсулина 328 мкг на крысу (9 ME на крысу). Образцы крови были собраны на 0, 3, 6, 10, 15, 25, 30, 40, 60 и 90 мин после введения и сыворотка подготовили для определения человеческого инсулина с помощью комплекта для иммуноанализа без перекрестной реактивности между инсулином крысы и человека.
Данные представлены как среднее значение ± стандартное отклонение, n=5. Левая часть на фиг. 2 относится к введению человеческого инсулина с октаноатом натрия (Na-C8) или твердой гидрофильной фракцией, суспендированной в воде (твердые частицы в воде). Правая часть Фиг. 2 относится к введению полностью инсулинового препарата (твердые частицы в гидрофобной среде). В табл. 2 ниже представлены суммарные AUC значения, рассчитанные из кривой зависимости концентрации от времени.

Данные: среднее значение ± стандартное отклонение
Средняя величина (состоящая из значения AUC) к инсулину после ректального введения инсулина-п/кБ является приблизительно в 50 раз выше, чем величина после введения без гидрофобной среды. Минимальное воздействие обнаружили у крыс при ведении инсулина с октаноатом натрия отдельно или при введении части твердых частиц гидрофильной фракции (как указано в примере 1) суспендированной в воде. Эти данные свидетельствуют о синергизме между твердым октаноатом натрия и гидрофобной средой.
Пример 4: Кишечная абсорбция инсулина после введения инсулина в ЖКТ крыс
Фиг. 3 представляет данные, относящиеся к уровню инсулина в сыворотке и уровню глюкозы в крови после ректального введения раствора инсулина и инсулинового препарата крысам. Крысам давали наркоз и вводили 100 мкл тестируемого препарата (инсулин в составе или инсулин в ФБР), содержащего дозу инсулина 328 мкг на крысу (9 ME на крысу). Образцы крови были собраны на 0, 3, 6, 10, 15, 25, 30, 40, 60 и 90 мин после введения. Уровень глюкозы сразу определяли с помощью глюкометра и сыворотка подготовили для определения человеческого инсулина с помощью набора для иммуноанализа без перекрестной реактивности между инсулином крысы и человека.
Уровни глюкозы представлены как процентное соотношение от основного уровня, измеренного до введения (время 0). Данные Фиг. 3 представлены как среднее значение ± стандартное отклонение, n=5.
Представлены уровни инсулина (левая часть Фиг. 3) и глюкозы (правая часть Фиг. 3) после ректального введения человеческого инсулина, растворенного в ФБР (раствор инсулина) или включенного в препарат. Уровень инсулина резко возрос в сыворотке крыс после ректального введения инсулина в препарате. Максимальные уровни измеряли в течение 6 мин после введения и постепенное снижение обнаружено до достижения исходного уровня, через 90 мин после введения. Такое резкое и значительное повышение уровня инсулина сопровождалось значительным снижением уровня глюкозы, достигшего в среднем 20% от исходного уровня уже на 30 мин после введения. В отличие от этого, ректальное введение инсулина в ФБР вызвало только очень незначительное снижение уровня глюкозы, которое совпадает с наблюдаемым после лечения контрольным ФБР отдельно.
Пример 5: Абсорбция инсулина после ректального введения инсулина в составе крысам
Фиг. 4 представляет данные, касающиеся изменения уровня глюкозы в крови и концентрации инсулина в сыворотке крови в результате п/к (подкожного) введения раствора инсулина (20 мкг на крысу) и ректального введения инсулина в составе (328 мкг на крысу). Образцы крови были собраны на 0, 3, 6, 10, 15, 25, 30, 40, 60 и 90 мин после ректального введения и на 0, 15, 30, 45, 60, 90 мин, 2, 3, и 4 часах после п/к введения. Глюкозу немедленно определяли с помощью глюкометра, и инсулин - с помощью набора для иммуноанализа. Уровни глюкозы представлены как процентное соотношение от основного уровня, измеренного до введения (время 0). Данные на фиг. 4 представлены как среднее значение ± стандартное отклонение, n=5.
Уровни абсорбции инсулина в толстой кишке крыс после введения инсулина в составе сравнивают с уровнями абсорбции инсулина после п/к введения. Концентрацию инсулина рассчитывали из площади кривой зависимости концентрации сыворотки от времени (AUC) и активности в пересчете на относительную биодоступность (оБД) по следующей формуле:
оБД = (ректальная AUC(о-∞) / п/к AUC(о-∞)) * (п/к доза / ректальную дозу)
Проникновение инсулин в кровоток происходит в узком окне времени, обычно в течение примерно 10 мин вводится ректальный инсулин в препарате. Повышение уровня инсулина в сыворотке крови сопровождается падением уровня глюкозы в крови.
В целях получения информации о биодоступности инсулина при присутствии разработанного инсулина в толстой кишке, AUC(о-∞) определяли при ректальном и п/к введении, и объем оБД человеческого инсулина составил 29,4+3,4% с коэффициентом отклонения (КО)=11,4%. Ректальное введение различных инсулин-содержащих препаратов проводили на сотнях животных. Анализ развивали в дальнейшем и квалифицировали как биоанализ для поддержки платформы разработки и испытания выпускаемых серий с линейным участком при 10-200 мкг на крысу, с воспроизводимостью результатов 39%, и внутрилабораторной погрешностью 33%.
Инсулиновый препарат, описанный в данном описании, был опробован в пяти различных исследованиях с использованием в общей сложности 25 крыс. оБД была 34,1±12,6% с КО 28,9%.
Пример 6: Абсорбция инсулина после интраеюнального введения инсулина в препарате крысам
Целевым сайтом абсорбции пероральной платформы изобретения является, как правило, тонкий кишечник. Для проверки активности инсулинового состава в кишечнике крыс рассматривали два основных условия: 1. Капсулы, покрытые кишечнорастворимой оболочкой для крыс, отсутствуют, и поэтому необходимо обойти желудок, вводить непосредственно в тощую кишку. 2. Инсулин интенсивно метаболизируется в печени; у людей 50-80% эндогенного инсулина, секретируемого поджелудочными β-клетками, изолируется в печени, и поэтому не может быть обнаружено в системном кровотоке. Введение инсулина с помощью кишечного путей (посредством инсулинового препарата) имитирует эндогенный способ применения инсулина, так как кишечный кровоток сливается в воротной вене, которая непосредственно ведет к печени. Поэтому для определения абсорбции инсулина, образцы крови должны быть взяты из воротной вены (кровообращение в системе воротной вены, до печени), а также из яремной вены (кровообращение большого круга, после печени).
Специализированная модель на крысах, в которой три различные канюли хирургическим путем имплантировали в анестезированных крыс, была разработана: 1. Тонкокишечная канюля - минуя желудок, позволяет введение инсулинового препарата, 2. Канюля воротной вены - забор крови до печени, определение инсулина, который пересекает перегородку ЖКТ в крови, и 3. Канюля яремной вены - определение общего уровня инсулина. Используя эту модель, определяли биодоступность инсулинового препарата (оБД).
Фиг. 5 представляет данные из репрезентативного исследования, касающиеся уровня инсулина в кровообращении в системе воротной вены и в кровообращении большого круга после интраеюнального введения контрольного инсулина и инсулинового препарата крысам. Крыс (8 крыс в каждой группе) анестезировали, и их тонкая кишка подверглась абдоминальной хирургии. Тонкую кишку, содержащая кишечную петлю, помещали на марлю и оставляли влажной и совершенно неповрежденной на протяжении всего исследования. Временную канюлю вставляли в тонкую кишку, и вводили инсулиновый препарат. Кровь собрали с обеих портальной и яремной вены в тех же временных точках, примерно 4 временные точки на крысу, среднее значение ± стандартное отклонение для каждого момента времени использовали для создания зависимости концентрации в плазме от кривой времени. AUC определяли и рассчитывали оБД.
Уровень инсулина в обоих портальном и системном кровотоке резко возрос после интраеюнального введения инсулинового препарата. Это, в отличие от минимальной абсорбции инсулина, обнаружили, когда ввели контрольный инсулин. Временной интервал абсорбции был коротким и уровень инсулина достиг максимума через 6 мин. Эти параметры аналогичные тем, которые наблюдались после ректального введения инсулинового препарата (см. выше). Более высокие уровни инсулина обнаружили в портальном кровотоке по сравнению с системным кровотоком, с оБД 10,1% по сравнению с 5,6% соответственно.
Пример 7: Дополнительные композиции, содержащие различные соединения карго
Таблица 3А подробно описывает компоненты серии декстрановых препаратов, которые подготовили, как описано в следующих примерах. Соль натрия каприновой кислоты приобрели у Fluka/Sigma, оливковое масло у Fluka, октановую кислоту у Sigma и нефтепродукты у Acros.


Табл. 3Б подробно описывает компоненты серии препаратов терипаратидацетата и лейпролида, которые подготовили, как описано в следующих примерах. Терипаратид приобрели у Novetide, и лейпролид приобрели у Bambio.

Табл. 3В подробно описывает компонентов препаратов чГР, которые подготовили, как описано в следующих примерах. чГР приобрели от PLR, Израиль (GHP-24).
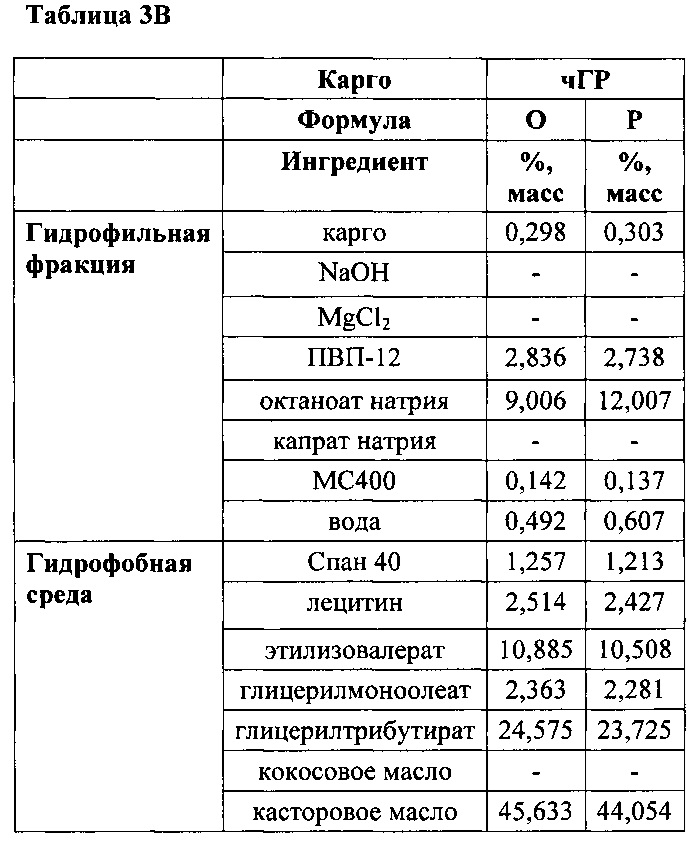
Производственный процесс для всех этих вышеописанных формул происходил в основном, как описано на фиг. 1 и в Примере 11.
Пример 8: Влияние дозы октаноата натрия, включенного в препарат, на активность состава
Влияние увеличения количества октаноата натрия (Na-C8) в препарате на активность состава протестировали с помощью препаратов, содержащих декстран (в среднем MB=4,4 кДа, меченый ФИТЦ) в качестве соединения карго, при различных дозах Na-C8, а именно препараты из табл. 3А (которые содержат 12% октаноата натрия по массе) и аналогичные декстрановые препараты, содержащие различные дозы Na-C8: 9%, 6% и 3% соответственно.
Проверяли активность этих препаратов в тонкой кишке неанестезированных крыс, создали модель крысы, в которой две разные канюли хирургическим путем имплантировали самцам крыс Спрэг-Доули
1 - Тонкокишечная канюля, минуя желудок, позволяет проводить прямое введение препарата в тонкую кишку.
2 - Канюля яремной вены позволяет определить системные уровни веденного декстрана в результате введения в тонкую кишку. Крысы были здоровыми за 4 дня до исследования и их лишали пищи за 18 час до начала исследования.
На фиг. 6 представлены данные из исследования, где определена биодоступность декстрана (4,4 кДа), меченого ФИТЦ, у неанестезированных крыс в результате введения в тонкую кишку препаратов, содержащих различные количества Na-C8 или декстрана меченого ФИТЦ, солюбилизированного с Na-С8 в физиологическом растворе (контроль).
Биодоступность различных препаратов декстрана и контроля оценивали по введению различных препаратов непосредственно в тонкую кишку неанестезированных крыс и по измерениям уровней декстрана в плазме на 3, 6, 10, 25, 60 и 90 мин после введения. Уровни декстрана плазмы после введения декстрана в препарате или в физиологическом растворе сравнивали с уровнями декстрана плазмы после внутривенного введения. Значения системного контакта AUC (0-90) определили для тонкокишечного и внутривенного введения, и абсолютную биодоступность (аБД) рассчитали по следующей формуле:
аБД = (тонкокишечная AUC(0-90)) / (п/к AUC(0-90)) * (п/к доза / тонкокишечная дозу)
Данные представлены как среднее значение±стандартное отклонение (n>5; 5 крыс в группе).
Результаты показывают, что увеличение количества Na-C8, включенного в препарат, улучшает биодоступность декстрана в способе доза-реакция, достигнув почти 30%) аБД при 12%, масс дозе. Введенный декстран с Na-C8 в аналогичных дозах и суспендированный в солевом растворе (т.е. не в препарате), продемонстрировал более низкую биодоступность (-6% аБД). Дальнейшие результаты доза-реакция приведены, как показано в Примере 26.
Пример 9: Влияние соотношения гидрофильной фракции/гидрофобной среде на активность препарата
Влияние на активность препарата изменения соотношения (масс) между гидрофильной фракцией и гидрофобной средой проводили с помощью препаратов, содержащих декстран (в среднем MB=4,4 кДа, меченый ФИТЦ) в качестве соединения карго (препараты А и В в табл. 3А). Модель неанестезированных крыс in vivo, описанная в Примере 8, использовали для того, чтобы сравнить активность описанных препаратов.
В табл. 4 представлены данные биодоступности в результате интраеюнального введения препаратов, содержащих различное соотношение гидрофильной фракции к гидрофобной среде.

Препараты А и В вводили непосредственно в тонкую кишку неанестезированных крыс и уровни декстрана плазмы измеряли через 3, 6, 10, 25, 60 и 90 мин после введения состава. Уровни абсорбции декстрана из тонкой кишки крыс после введения декстрана в препарате сравнивали с уровнями абсорбции декстрана после внутривенного введения. Экспозиционные значения AUC (0-90) определяли для тонкокишечного и внутривенного введения, и абсолютную биодоступность (аБД) рассчитывали по следующей формуле:
аБД = (тонкокишечная AUC(0-90)) / (п/к AUC(0-90)) * (п/к доза / тонкокишечная дозу)
Данные представлены как среднее значение±стандартное отклонение (n>5; 5 крыс в группе).
Результаты показывают, что изменение соотношения между гидрофильной фракцией и гидрофобной средой в этих препаратах с низким % массы терапевтического средства не оказало существенного влияния на биодоступность карго, который предоставляется при наполнении в разработке дополнительных препаратов.
Пример 10: активность препаратов, содержащих различные соединения карго
С целью проверки возможности платформы препарата, активные препараты, содержащие три различных соединения карго (АФИ), испытывали на трех различных моделях животных: еюнальное введение неанестезированным крысам, ректальное введение анестезированным крысам и еюнальное введение неанестезированным свиньям. В табл. 5 приведены результаты представленных экспериментов по проверке биодоступности препаратов, содержащих различные АФИ, в трех различных моделях животных, описанных выше.
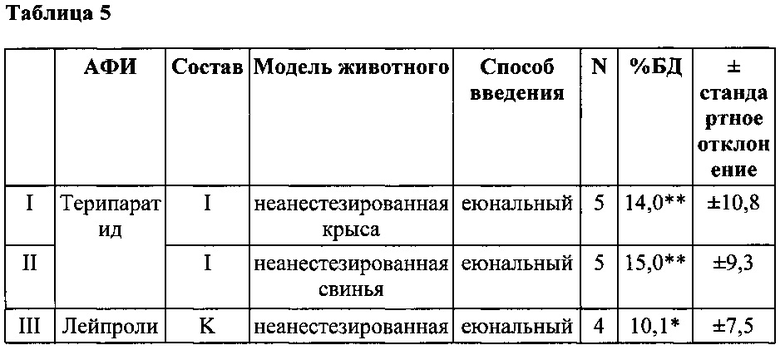

А. Абсорбция лейпролида после еюнального введения лейпролида в препарате крысам
Табл. 5-III представляет данные репрезентативных исследований, связанных с % аБД лейпролида в результате в/в (внутривенного) введения раствора лейпролида (75 мкг/кг) и еюнального введения лейпролида в препарате (450 мкг/кг; препарат К, табл. 3Б), неанестезированным крысам, как описано выше в Примере 8.
Образцы крови взяли из яремной вены на 3, 6, 10, 15, 25, 40, 60 и 90 мин после еюнального введения и на 3, 10, 25, 40, 90 мин, 2, 3,3 и 5 час после в/в введения, подготовили плазму и определили уровни лейпролида в каждом образце. Уровень лейпролида в системном кровотоке резко возрос после еюнального введения лейпролида в препарате. Уровень лейпролида крови достиг максимума на 3 мин после введения. Среднюю аБД, полученную после еюнального введения лейпролида в препарате, рассчитывали, как описано в вышеприведенных Примерах, и средняя аБД составила 10,1%. В контрольном эксперименте, еюнальное введение лейпролида в ФБР продемонстрировало незначительное проникновение в кровоток.
Аналогичный препарат лейпролида, содержащий 12% октаноата натрия, как описано в табл. 1Б, подготовили и испытывали на вышеуказанных моделях и препарат показал следующую биодоступность:
оБД (по сравнению с п/к) = 21,1% ± 12,0 (КО=57%).
Б. Абсорбция терипаратида после ЕЮНАЛЬНОГО введения терипаратида в препарате крысам
Табл. 5-1 представляет данные из репрезентативных исследований, связанных со сравнительными параметрами терапаратида плазмы концентрация-время в результате п/к введения раствора терипаратида (85 мкг на состав) и еюнального введения терипаратида (терипаратид) в препарате (550 мкг/кг; препарат I, табл. 3Б), неанестезированным крысам, как описано выше в Примере 8. Образцы крови взяли из яремной вены на 3, 6, 10, 25, 60 и 90 мин после еюнального введения и на 3, 10, 30, 60, 90 мин, 2 и 3 часах после п/к введения, подготовили плазму и определили уровень терипаратида в каждом образце. Уровень терипаратида в системном кровотоке резко возрос после еюнального введения терипаратида в препарате. Уровень терипаратида достиг максимума на 3-ей мин после введения. Рассчитали среднюю оБД, полученную после еюнального введения терипаратида в препарате, как описано в приведенных выше Примерах, и средняя аБД составила 14,0%. В контрольном эксперименте еюнальное введение терипаратида в физиологическом растворе не показало проникновения в кровоток.
В. Абсорбция терипаратида после еюнального введения терипаратида в препарате свиньям
Табл. 5-II представляет данные репрезентативных исследований, связанных со сравнительными параметрами терипаратида «концентрация в плазме-время» в результате п/к введения раствора терипаратида (10,65 мкг/кг) и еюнального введения терипаратида в препарате (100 мкг/кг; препарат I, таблица 3Б), неанестезированным свиньям. Создали модель свиньи, в которой две разные канюли хирургическим путем имплантировали домашней свинье:
1 - Тонкокишечная канюля, минуя желудок, позволяет проводить прямое введение препарата в тонкую кишку.
2 - Канюля яремной вены позволяет определить систематические уровни веденного карго в результате еюнального введения.
Свиньи были здоровыми за 7 дней до эксперимента и их лишали пищи за 18-20 час до начала эксперимента.
Образцы крови отбирали из яремной вены на 0, 3, 6, 10, 15, 25, 40, 60, 90 мин, 2, 2,5 и 3 часах после еюнального введения и на 0, 3, 6, 10, 15, 20, 30, 45, 60, 90 мин, 2, 2,5, 3 и 4 часах после п/к введения, подготовили плазму и определяли уровни терипаратида в каждом образце. Уровень терипаратида в системном кровотоке резко возрос после еюнального введения терипаратида в препарате. Уровень терипаратида достиг максимума на 10 мин после введения.
Среднюю оБД, полученную после еюнального введения терипаратида в препарате, рассчитывали, как описано в вышеприведенных Примерах, и она составила 15,0%.
Аналогичный эксперимент со свиньей проводили с применением декстрана (FD4, препарат А в табл. 3А) и было установлено, что средняя биодоступность декстрана составила 20% у свиней по сравнению с в/в введением.
Г. Абсорбция чГР после ректального введения чГР в препарате крысам
Табл. 5-IV представляет данные из репрезентативных исследований, связанных со сравнительными параметрами чГР плазмы концентрация-время в результате п/к введения раствора чГР (81 мкг/кг) и ректального введения чГР в препарате (800 мкг/кг; препарат Р, таблица 3В), анестезированным крысам.
Самцов крыс Спрэг-Доули лишали пищи за 18 час до начала эксперимента. Крыс анестезировали раствором кетамина/ксилазина. Препарат (100 мкл на крысу) вводили ректально с применением катетера 14G-venflon. Образцы крови отбирали из яремной вены на 3, 6, 10, 15, 40, 60 и 90 мин после ректального введения и на 15, 30, 45, 60, 90 мин, 2, 3, и 4 час после п/к введения, подготовили плазму и уровень чГР определяли в каждом образце. Уровень чГР в системном кровотоке резко возрос после ректального введения чГР в препарате. Уровень чГР достиг максимума на 15 мин. Среднюю оБД, полученную после ректального введения чГР в препарате, рассчитывали, как описано в вышеприведенных Примерах, и она составила 17,9%. В отдельном эксперименте чГР вводили в тонкую кишку, и аБД была ниже. В контрольном эксперименте ректальное введение чГР в ФБР не показало проникновения в кровоток.
Таким образом, результаты, представленные в табл. 5, показывают, что существенное воздействие получали для всех соединений карго, испытанных на всех тестируемых моделях животных. Полученные результаты показывают, что препараты, описанные в данном описании, позволяют производить доставку широкого спектра различных макромолекул через кишечный эпителий у различных моделей животных.
Пример 11: Подробное описание производственного процесса препарата терипаратида
Получение гидрофильной фракции: К 200 мл воды следующие ингредиенты медленно добавляли по одному (с 2-3 минутным перемешиванием после каждого ингредиента): 172 мг терипаратида, 200 мг MgCl2, 4,0 г ПВП-12, 17,52 г октаноата натрия и 10,0 г 2% водного раствора МС-400, приготовленного следующим образом: 1 г порошка МС-400 добавляют в 50 мл воды при температуре 60±2°C при перемешивании. После 5 мин перемешивания, стакан перемещают на лед для получения прозрачного раствора.
После добавления раствора МС-400, раствор перемешивают в течение еще 5 мин, а затем лиофилизируют в течение приблизительно 24 час. Эта процедура вырабатывает около 22 г гидрофильной фракции.
Получение гидрофобной среды: 2 г Спан 40, 4 г лецитина и 3,8 г ГМО (глицеролмоноолеата) растворяют в 17,3 г этилизовалерата при перемешивании. К этому раствору добавляют 39,1 г GTB (глицерилтрибутирата) и 72,6 г касторового масла. Эта процедура вырабатывает около 136-138 г гидрофобной среды.
Выпуск нерасфасованного лекарственного продукта: смешивание гидрофильной фракции с гидрофобной средой выполняли при температуре 20±2°C.
15,7 г гидрофильной фракции медленно добавляли в 84,3 г гидрофобной среды при перемешивании со скоростью 600±50 об/мин. После добавления всей гидрофильной фракции, скорость перемешивания была увеличена до 2000±200 об/мин в течение 2-10 мин с последующими 4-8 циклами перемешивания в течение 15 мин со скоростью 600±50 об/мин и 2 мин перемешивания со скоростью 2000±200 об/мин. Далее применялась дегазация с помощью вакуума, как указано ниже: 5 мин при давлении 600 мбар, 5 мин при давлении 500 мбар и 30-120 мин при давлении 400 мбар. Полученную суспензию разливали во флаконы из темного стекла объемом 100 мл и хранили при температуре 2-8°C. Эта формула терипаратида, обозначенная «I», описана в табл. 3В.
Все другие препараты, описанные здесь, производились с помощью этого метода, путем изменения самих ингредиентов и их количества в соответствии с деталями, приведенными в соответствующих таблицах (см. например, Пример 29). Диаграмма этого метода (с инсулином в качестве карго) представлена на рис 1.
Пример 12: влияние масла, введенного в препарат, на активность препарата
Было протестировано влияние типа масла, введенного в технологию приготовления лекарственного средства (в гидрофильной среде) на активность лекарственного средства. Лекарственные средства, содержащие декстран (средняя молекулярная масса = 4,4 кДа, ФИТЦ маркированная), как карго и различные типы масел в гидрофобной среде (препараты Е, F и G в табл. 3А), были протестированы на крысах. Для тестирования активности этих препаратов в тонкой кишке неанестезированных крыс, применялась модель, в которой самцам крыс Спрэг-Доули хирургическим путем имплантировали два различных катетера:
1 - катетер тонкой кишки для шунтирования желудка и обеспечения прямого введения препаратов в тонкую кишку.
2 - катетер яремной вены для систематического определения уровня введенного декстрана после введения лекарственного средства в тонкую кишку.
Крыс оставляли для восстановления на протяжении 4 дней перед исследованием и лишали еды на протяжении 18 час перед началом исследования.
Табл. 6 представляет данные исследования у неанестезированных крыс, после введения внутрь тонкой кишки препаратов, содержащих различные масла в гидрофобной среде.


Препараты, содержащие различные масла, вводились непосредственно в тонкую кишку неанестезированных крыс и уровни декстрана в плазме измерялись через 3, 6, 10, 25, 60 и 90 мин после введения препарата. Уровни абсорбции декстрана из тонкого кишечника крыс после введения декстрана в препарате сравнивали с уровнями абсорбированного декстрана после внутривенного введения. Экспозиционные числа и показатели AUC (0-90) определялись для введения в тонкую кишку и внутривенного введения, абсолютная биодоступность определялась в соответствии со следующим уравнением:
Абсолютная биодоступность = (AUC(0-90) для введения в тонкую кишку) / (AUC (0-90) внутривенное введение) * (внутривенная доза/доза, введенная в тонкую кишку). Данные представлены как средний показатель ± стандартное отклонение (n>5 крыс в группе).
Подобная биодоступность достигалась, при включении декстрана в препараты, содержащие касторовое или кокосовое масло. Хорошая биодоступность также достигалась в тонком кишечнике крысы, при применении терипаратида в качестве карго, применяя препараты I и J; которые содержат касторовое масло и GTB, касторовое масло и кокосовое масло, соответственно.
Результаты показали, что препараты, содержащие различные виды масел в гидрофобной среде являются активными, обеспечивающими проницаемость карго (декстрана, терипаратида), переносимого препаратом. Эти данные демонстрируют, что все протестированные масла обеспечивали биодоступность карго. Касторовое масло и кокосовое масло возможно являются лучшими из других протестированных масел.
Пример 13: изготовление препарата с применением грануляции вместо лиофилизации.
Производство гидрофильной фракции: в пластиковые пакеты были добавлены следующие ингредиенты: 1,00 г ПВП-30, 6,70 г октаноата натрия и 13,00 г моногидрата лактозы, в качестве связующего вещества. После 5 мин перемешивания, весь порошок переносили в ступку и растирали. Водный раствор декстрана FD4 был приготовлен следующим образом: 0,42 г декстрана растворяли в 1,2 г воды для инъекций. Затем весь раствор декстрана медленно добавляли в порошок при перемешивании с помощью медленного x растирания пестиком в ступке; перемешивание занимало около 45 мин. Смесь далее перемещали в планшет для лиофилизации и сушили на протяжении 20 час при температуре 50°C. Этим способом получают около 20 г хорошо гранулированной гидрофильной фракции.
Производство гидрофобной среды: 2 г Спан 40 (сорбитан монопальмитат), 4 г лецитина и 3,8 г ГМО растворили в 17,3 г изовалерианового спирта при перемешивании. К этому раствору добавляли 39,1 г ГТБ и 72,6 г касторового масла. Этим способом производится около 136-138 г гидрофобной среды.
Производство нерасфасованного лекарственного препарата: Смешивание гидрофильной фракции и гидрофобной среды проводилось при температуре 20±2°C.
19,00 г (29,58% финального беклометазона дипропионат БДП) гидрофильной фракции медленно добавляли в 45,23 г (70,42%) финального БДП) гидрофобной среды, при перемешивании со скоростью 600±50 об/мин. После добавления всей гидрофобной фракции, скорость перемешивания увеличили до 2000±200 об/мин в течение 2-10 мин с последующими 4-8 циклами 15-минутного перемешивания со скоростью 60±50 об/мин и 2 мин перемешивания со скоростью 2000±200 об/мин.
Далее применялась дегазация с помощью вакуума, как указано ниже: 5 мин при давлении 600 мбар, 5 мин при давлении 500 мбар и 30-120 мин при давлении 400 мбар. Полученную суспензию разливали во флаконы из темного стекла объемом 100 мл и хранили при температуре 2-8°C.
Исследование на крысах: Вышеупомянутая суспензия вводилась ректально крысам, как описано в примерах выше и результаты были следующими: 35% биодоступность, 12,9% допустимое отклонение. Другие партии суспензии, приготовленные гранулированием, как описано выше, готовились и вводились в тонкую кишку крыс, как описано в примерах выше и результаты были следующими: 21,8% биодоступность, 4,0% допустимое отклонение. Ряд препаратов, приготовленных подобным образом, с применением грануляции и введения определенных лекарственных веществ, и изменением количества октаноата натрия.
Пример 14: отбор капсул
Эксперименты in vitro осуществлялись, используя раздельно 3 типа растворов: гидрофильную среду, как описано в примере выше, единственный изовалериановый спирт, и изовалериановый спирт, содержащий 5% каждого из следующих ПАВ: лецитина, Спан 40 и глицерилмоноолеата. Каждую из 3-х типов распечатанных капсул, желатиновую, крахмальную и из ГПМЦ (гидроксипропилметилцеллюлозы), наполняли этим раствором. Наполненные капсулы хранились далее in vitro 29 дней при температуре 22±2°C и относительной влажности 30-50%). Желатиновые капсулы и капсулы из ГПМЦ продемонстрировали наилучшие результаты, а именно отсутствие деформации капсулы.
Подобные эксперименты проводились с использованием тех же 3-х растворов, желатиновых и ГПМЦ капсул. Капсулы наполняли раствором, запаивали (связывали) и хранили на протяжении 8 дней при температуре 22±2°C, относительной влажности 30-50%. Оба типа капсул продемонстрировали стабильность протестированных растворов, например, не было просачиваний и деформации капсул.
Пример 15: влияние различных катионов в соли жирной кислоты с цепью средней длины
Препараты готовились с декстраном (FD4) подобно препарату А табл. 3А за исключением того, что 12% октаноат натрия (0,722 М) замещали октаноатом лития, октаноатом калия или октаноатом аргинина равной молярности (последний, как модель для соли аммония). Эти препараты представлены в табл. 7А ниже.

Каждый из этих препаратов тестировали на модели тонкого кишечника крысы, описанной в примере 8. Были получены результаты и подсчитана биодоступность. Результаты представлены ниже в табл. 7В.

Препарат А, который использовали в вышеприведенном эксперименте, был из другой партии, чем тот, который использовали в примере 8, и поэтому результаты биодоступности, представленные для препарата А, несущественно отличаются от приведенных в табл. 4.
Вышеприведенные результаты показывают, что при замещении 12% октаноата натрия в препарате октаноатом лития или октаноатом калия эквивалентной молярности, препарат все еще обладал биодоступностью, но меньшего уровня. Препарат с октаноатом аргинина проявлял активность, подобную препарату с 12% октаноатом натрия.
Пример 16: влияние добавления спиртов с цепью средней длины (гераниола и октанола) к гидрофобной среде
Препараты, содержащие гераниол (BASF) и октанол (Spectrum/MP) были приготовлены, как описано выше, с использованием ингредиентов, представленных ниже в табл. 8. Додеканоат натрия приобретен у Spectrum/Acros.
Препарат Q - низкий % соли жирной кислоты с цепью средней длины: препарат декстрана (FD4) был приготовлен, в существенной мере как описано в примере 11, с общим содержанием 2,9% соли жирной кислоты с цепью средней длины (октаноат натрия 1,042% + додеканоат натрия 1,869%), и также с содержанием гераниола и октанола в гидрофобной среде, как показано ниже в табл. 8.
Препарат R - более 10% соли жирной кислоты с цепью средней длины: препарат декстрана приготовлен, в существенной мере как описано для препарата А, за исключением того, что гераниол и октанол добавляли в гидрофобную среду, как показано в табл. 8.


Препарат Q (низкий % соли жирных кислот средней длины цепи ЖКСДЦ) был протестирован на внутри-тонкокишечной модели крысы, описанной выше и была рассчитана биодоступность: абсолютная биодоступность = 4,4%, допустимое отклонение = 3,8 (n=12). Препарат R (больше 10% соли ЖКСДЦ) была протестирована на внутри-тонкокишечной модели крысы, описанной выше, была рассчитана биодоступность: абсолютная биодоступность = 22,7%, допустимое отклонение = 1,6 (n=6).
Биодоступность этих препаратов не отличалась значительно от подобных препаратов, описанных в примере выше, которые не содержали гераниол.
Пример 17: препараты для гентамицина и для РНК
Препараты для гентамицина и для РНК, были приготовлены в существенной мере как описано в Примере 11, с ингредиентами нерасфасованного лекарственного продукта, как описано в табл. 9. Гентамицин приобрели у Applichem и РНК была представлена натриевой солью полиинозиновой-полицитидиловой кислоты (Sigma).

Препарат гентамицина был протестирован на модели тонкого кишечника крысы и на ректальной модели крысы, как описано выше (например, Пример 4 и 5). Гентамицин анализировали с применением иммуноанализа (ELISA). Результаты представлены ниже в табл. 9В; % биодоступности посчитан и сравнен с в/в введением. Препаратами была продемонстрирована способность обеспечивать биодоступность гентамицина.

Подобным образом, РНК препарат из табл. 9А тестируется на модели тонкого кишечника крысы и на ректальной модели крысы, как описано выше. РНК анализируется и предполагается, что препарат обеспечивает биодоступность РНК.
Пример 18: влияние на активность препарата поверхностно-активных веществ в гидрофильной среде
Влияние на активность препарата извлекаемых поверхностно-активных веществ из гидрофильной среды тестировали с использованием препаратов, содержащих декстран (средняя молекулярная масса = 4,4 кДа, FITC маркированная) как карго (препараты А и Н в табл. 3А).
Табл. 10 представляет данные исследования на неанестезированных крысах после введения внутрь тонкого кишечника препаратов с или без поверхностно-активных веществ (например Спан 40, лецитина, глицерина моноолеата) в гидрофобной среде.

Препараты с или без ПАВ в гидрофобной среде вводили непосредственно в тонкую кишку неанестезированных крыс и измеряли уровень декстрана в плазме через 3, 6, 10, 25, 60 и 90 мин после введения препарата. Уровень абсорбции декстрана в тонкой кишке крыс после введения декстрана в препарате сравнивали с уровнем абсорбированного декстрана после внутривенного введения.
Определяли экспозиционное число, показатели AUC (0-90), для введения в тонкий кишечник и внутривенного введения и определяли абсолютную биодоступность в соответствии со следующим уравнением:
абсолютная биодоступность = (AUC(0-90) для введения в тонкий кишечник) / (внутривенное AUC (0-90)) * (внутривенная доза/доза введения в тонкий кишечник)
Данные представлены как средняя абсолютная биодоступность ± допустимое отклонение
Меньшая биодоступность достигалась при включении декстрана в препарат, не содержащий поверхностно-активные вещества в гидрофобной среде (препарат Н), в сравнении с препаратом, содержащим поверхностно-активные вещества в гидрофобной среде (препарат А). Результаты демонстрируют, что извлечение из гидрофобной среды поверхностно-активных веществ неблагоприятно влияет на активность препарата.
Пример 19: влияние на активность препарата извлечения жирных кислот с цепью средней длины из гидрофильной фракции
Влияние на активность препарата извлечения жирных кислот с цепью средней длины (ЖКСДЦ) из гидрофильной фракции тестировали с использованием препаратов, содержащих декстран (средняя молекулярная масса = 4,4 кДа, FITC маркированные) как карго.
Табл. 11 представляет данные исследования на неанестезированных крысах после введения препаратов в тонкий кишечник с или без октаноата натрия в гидрофильной фракции (препараты А и D в табл. 3А, соответственно).

Препараты, описанные выше, вводились непосредственно в тонкую кишку неанестезированных крыс и через 3, 6, 10, 25, 60 и 90 мин после введения препарата измерялся уровень декстрана в плазме. Уровень абсорбции декстрана в тонкой кишке крыс после введения декстрана в смеси сравнивали с уровнем абсорбированного декстрана после внутривенного введения.
Определялось экспозиционное число, показатели AUC (0-90), для введения в тонкий кишечник и для внутривенного введения и абсолютную биодоступность определяли в соответствии со следующим уравнением:
абсолютная биодоступность = (AUC(0-90) для введения в тонкий кишечник) / (внутривенное AUC (0-90)) * (внутривенная доза/доза введения в тонкий кишечник)
Данные представлены как средняя абсолютная биодоступность ± допустимое отклонение.
Незначительное проникновение декстрана достигалось, при включении декстрана в препарат с недостатком жирных кислот с цепью средней длины в гидрофильной фракции ((препарат D, % абсолютной биодоступности = 0,6 ± 1,0) в сравнении с препаратом, содержащим 12% октаноата натрия в весовом соотношении в гидрофильной фракции (препарат А, % абсолютной биодоступности = 28,0 ± 6,8).
Результаты демонстрируют, что препарат без жирных кислот цепью средней длины в гидрофильной фракции не является активным.
Подобный эксперимент проводили с использованием октреотида как карго в улучшенном препарате (см. ниже). rBA составила 0,11% (коэффициент вариации = 158%)
Пример 20: влияние на активность препарата упрощения состава
Влияние на активность препарата упрощения состава было протестировано с использованием препарата, содержащего декстран (средняя молекулярная масса = 4,4 кДа, FITC маркированный) или октреотид (Novetide) как карго. Основной препарат, описан в примере выше (например препараты, обозначенные А, I и Р) упрощали тем, что не добавляли MgCl2, и МС 400 к гидрофильной среде и не добавляли Спан 40, лецитин и изовалериановый спирт к гидрофобной среде. Это приводит к увеличению количества глицерина моноолеата (поверхностно-активного вещества) и глицерилтрибутирата, добавленного к гидрофобной среде. Такие препараты представлены ниже в табл. 12А. Эти упрощенные составы не проявляют видимого осаждения, хотя частицы видимы при помощи микроскопа, то есть они являются стабильными суспензиями.

Процесс производства вышеупомянутых упрощенных препаратов в существенной мере такой, как описан на фиг. 1 и в Примере 11 для основных препаратов. Основной препарат октреотида представлен ниже в табл. 12В.


Табл. 13 представляет данные исследования на неанестезированных крысах после введения внутрь тонкого кишечника двух различных препаратов декстрана - препарата А из табл. 3А и упрощенного препарата, представленного в табл. 12А

Представленные выше результаты показывают, что сходные значения AUC были достигнуты, при включении декстрана в препарат, содержащий основной препарат (препарат А) в сравнении с упрощенным препаратом.
Табл. 14 ниже представляет данные исследования на неанестезированных крысах после введения в тонкий кишечник двух различных препаратов октреотида - основной препарат показан в табл. 12В, упрощенный препарат показан в табл. 12А. Были определены уровни абсорбции октреотида в тонкой кишке крыс после введения октреотида с основным препаратом и упрощенным препаратом.
Определяли значения системного контакта, показатели AUC (0-25).

Представленные в табл. 14 результаты показывают, что значения AUC были незначительно меньше, чем при включении октреотида в упрощенный препарат, в сравнении с полным препаратом.
Пример 21: влияние на активность препарата замещения касторового масла октановой кислотой
Влияние на активность препарата замены касторового масла (и глицерилтрибутирата и этилизовалериата) октановой кислотой (Aldritch) было протестировано с использованием препарата, содержащего декстран, как карго. Это было сделано для поддержания фрагмента С8 в препарате, то есть предполагалось, что наличие С8 кислоты в гидрофобной среде в дополнение к С8 соли в гидрофильной фракции может быть благоприятным.
Влияние добавления рицинолеиновой кислоты (Spectrum) было также протестировано с помощью изготовления препарата декстрана, содержащего октановую кислоту/рицинолеиновую кислоту. Рицинолеиновая кислота была избрана, поскольку основной компонент триглицерида в касторовом масле образуется из рицинолеиновой кислоты. Три препарата декстрана были приготовлены, как показано ниже в табл. 15А. Основной препарат декстрана был приготовлен преимущественно, как описано в примере выше. Декстран октанового препарата был приготовлен преимущественно, как описано в примере выше, но касторовое масло, глицерилтрибутират и этилизовалериат были замещены октановой кислотой. Этот препарат визуально представлял собой раствор, но анализы истинной растворимости не проводились. По-видимому, октановая кислота в высокой концентрации (около 78% препарата) растворяет твердую гидрофильную фракцию, с ПВП и октаноатом натрия, растворяющегося в октановой кислоте при высокой концентрации. Декстран препарата рицинолеиновой/октановой кислоты был приготовлен преимущественно, как описано в примере выше, но касторовое масло, глицерилтрибутират и этилизовалериат замещали смесью октановой и рицинолеиновой кислот. Препарат представлял собой суспензию, как большинство препаратов изобретения.

Препараты, описанные выше в табл. 15А вводились непосредственно в тонкую кишку неанестезированных крыс, и уровни декстрана в плазме измерялись после введения препарата. Экспозиционные числа, показатели AUC, определялись для различных препаратов. Эти результаты представлены ниже в табл. 15В.

Результаты, показанные в табл. 15В выше, демонстрируют, что абсорбция декстрана значительно увеличена (больше чем в два раза) в препарате, содержащем октановую кислоту. К тому же, вид графика был изменен, показывая более медленную, но более продолжительную секрецию. Это может быть преимуществом, поскольку обеспечивает более длительное действие АФИ в организме. Показатели рицинолеинового/октанового декстрана продемонстрировали меньшую активность, чем препарат октановой кислоты, но все же были улучшены по сравнению с основным препаратом.
Так как октановая кислота и препараты рицинолеиновой кислоты/октановой кислоты показали высокую активность, подобные препараты были приготовлены с экзенатидом, как карго. Три препарата экзенатида были произведены, как показано ниже в табл. 16А. Базовый препарат экзенатида был приготовлен преимущественно, как описано в примерах выше. Препарат экзенатида/октановой кислоты были приготовлены преимущественно, как описано выше, но с замещением в них касторового масла, глицерилтрибутирата и этилизовалериата октановой кислотой. Этот препарат, содержащий более 78% октановой кислоты, визуально представлял собой раствор, как подобный препарат декстрана, описанный выше. Препараты экзенатида рицинолеиновой/октановой кислоты были приготовлены преимущественно, как описано выше, но с замещением в них касторового масла, глицерилтрибутират и этилизовалериата октановой и рицинолеиновой кислотами.
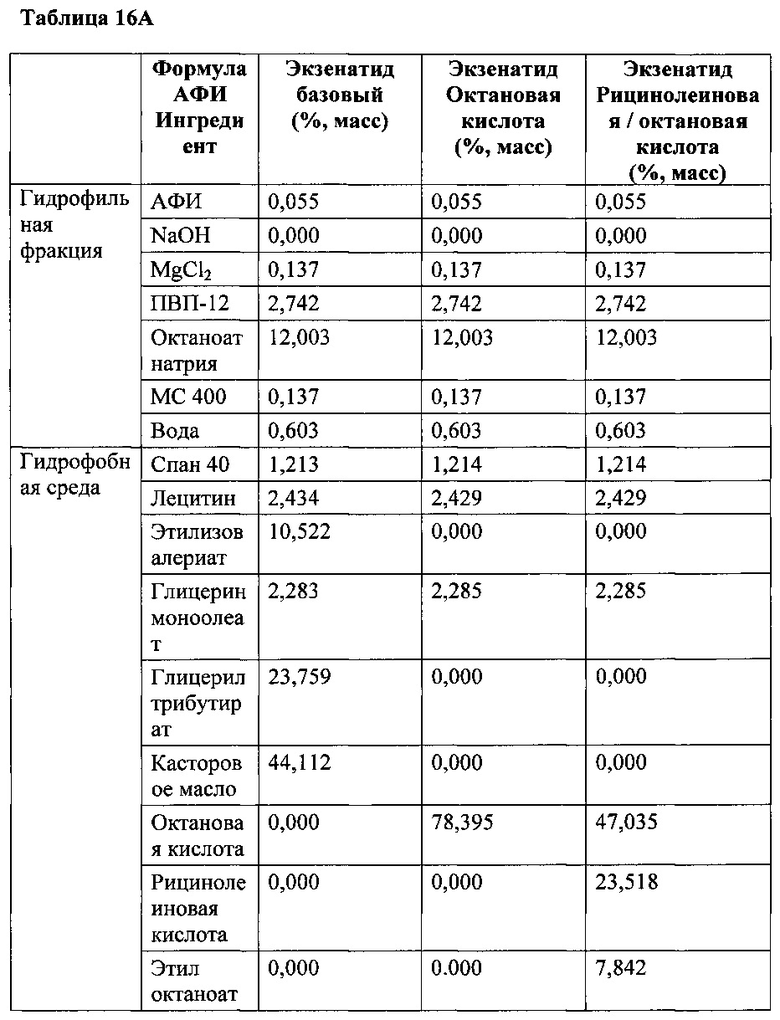
Препараты, описанные в табл. 16А выше, вводились непосредственно в тонкую кишку неанестезированным крысам, и после введения препарата измерялись уровни экзенатида в плазме. Определяли системный контакт, показатели AUC, для различных препаратов. Эти результаты приведены ниже в табл. 16В.

Результаты, приведенные выше в табл. 16В, демонстрируют, что препарат экзенатида, содержащий октановую кислоту, проявлял биодоступность, но абсорбция экзенатида была снижена по сравнению с базовым препаратом. Вид графика был изменен, показывая более медленное, но длительное высвобождение, как в случае с препаратом декстрана октановой кислоты, описанным выше; этот пролонгированный фармакокинетический профиль может быть благоприятным. Заметим, что в случае с препаратом октановой кислоты, AUC 0-180 мин использовали для подсчетов биодоступности из-за пролонгированного фармакокинетического профиля. Препарат экзенатид рицинолеиновой/октановой кислоты обладает меньшей биодоступностью, чем препарат октановой кислоты.
Пример 22: дозозависимый ответ для октановой кислоты
А. Октреотидные препараты: влияние на активность препарата изменения количества октановой кислоты было протестировано с использованием препаратов, содержащих октреотид, как карго. Четыре препарата октреотида были приготовлены с применением 0%, 5%, 10% или 15% октановой кислоты, как показано ниже в табл.. 17. Препараты являются базовыми препаратами октреотида, приготовленные преимущественно, как описано выше, в которых количество октановой кислоты варьируется, как описано, и количество других ингредиентов в гидрофобной среде (этилизовалериат и глицерилтрибутират) было соответственно уменьшено (В этих препаратах гидрофильную фракцию упростили исключением MgCl2 и МС 400)

В. Препарат экзенатида: влияние на активность препарата изменения количества октановой кислоты было протестировано с использованием препарата, содержащего экзенатид, как карго. 5 препаратов экзенатида были приготовлены с применением 0%, 10%, 15%, 20% или 35% октановой кислоты, как показано ниже в табл. 18. Препараты, являющиеся базовой формой экзенатида, приготовлены в существенной мере, как описано выше, где количество октановой кислоты варьируется, как описано, и количество других ингредиентов в гидрофобной среде (этилизовалериат и глицерилтрибутират) было соответственно уменьшено.

Препараты, описанные выше в табл. 17 и 18, вводились непосредственно в тонкую кишку неанестезированным крысам, и уровни плазмы октреотида и экзенатида измерялись после введения состава. Экспозиционные числа, показатели AUC, были определены для различных препаратов. Эти результаты показаны ниже в табл. 19.

Результаты, приведенные выше в табл. 19, демонстрируют, что препарат октреотида показывает увеличение активности, в сравнении с основным препаратом, при увеличении количества октановой кислоты до 15% (максимальное протестированное количество). Кроме того, результаты, приведенные в табл. 19, демонстрируют, что препарат экзенатида показывает увеличение активности в сравнении с основной формулой, при увеличении количества октановой кислоты до 15%, при более высоких уровнях октановой кислоты активность уменьшалась..
Пример 23: влияние различных солей жирных кислот с цепью средней длины
А. Натриевая соль себациновой кислоты (динатриевая соль октандикарбоновой кислоты): было протестировано влияние на активность препарата замещения октаноата натрия себацинатом натрия (динатриевая С10 соль) в препарате декстрана. Себацинат натрия был приготовлен in situ из себациновой кислоты (Aldrich) и гидроокиси натрия. Произведенный препарат описан в табл. 20 ниже. Препарат готовили преимущественно, как описано выше, но 12% октаноат натрия был замещен себацинатом натрия, с такой же молярной концентрацией октаноата натрия, т.е. было использовано эквимолярное количество себацината натрия (а именно, 0,72 М).

Препарат, описанный выше в табл. 20, вводился непосредственно в тонкую кишку не анестезированных крыс, после введения препарата измерялись уровни декстрана в плазме. Экспозиционное число, показатели AUC, определялись для препарата и сравнивались с подобными препаратами, приготовленными с октаноатом натрия. Эти результаты приведены ниже в табл. 21.

Результаты, показанные в табл. 21, демонстрируют, что препарат декстрана, содержащий себацинат натрия, проявлял активность, но абсорбция декстрана в сравнении с препаратом, содержащим эквимолярное количество октаноата натрия, была снижена.
В. Мононатрия суберат или динатрия суберат: октреотид-содержащие препараты были приготовлены с замещением 12% октаноата натрия эквимолярным количеством (0,72 М) мононатрия суберата или динатрия суберата, которые являются солями С8. Эти натриевые соли готовили in situ из субериновой кислоты (Tokyo Chemical Industry Со.) и гидроксида натрия.

Препараты, описанные выше в табл. 22, вводятся непосредственно в тонкую кишку неанестезированным крысам, и уровни октреотида в плазме измеряются после введения препарата. Экспозиционное число, показатели AUC, определяются для препаратов, и сравниваются с подобным препаратом, приготовленным с октаноатом натрия.
С. Соль гераниевой кислоты
Два октреотид-содержащих препарата были приготовлены, как описано выше, в них 12% октаноат натрия замещали 18% натриевой солью гераниевой кислоты (0,95 М) и 14,6% (0,77 М) натриевой солью гераниевой кислоты, которая является 3,7-диметил-2,6-октадиеновой кислотой (получено от SAFC). Произведенные препараты описаны в табл. 22В ниже.

Препараты, описанные выше в табл. 22В, вводились непосредственно в тонкую кишку неанестезированных крыс, и уровни октреотида в плазме измерялись после введения препарата. Экспозиционные числа, показатели AUC, определялись для препаратов и сравнивались с подобными препаратами, приготовленными с октаноатом натрия. Результаты представлены в табл. 22С ниже и демонстрируют, что препарат с 18% гераната натрия обладал такой же активностью, как и препарат с 12% октаноата натрия, и препарат с 14,6% гераната натрия обладал повышенной активностью.

Пример 24: влияние поливинилпирролидона на активность препарата
Влияние на активность препарата замены ПВП-12 маннитолом (Sigma) был протестирован с применением препаратов, содержащих экзенатид, как карго. Было понятно, что ПВП-12 является стабилизатором и мог бы быть замещен в препарате другим стабилизатором, таким как маннитол. Препарат, который представлен в табл. 23 ниже, был приготовлен. Этот препарат является основным препаратом экзенатида, приготовленным преимущественно как описано выше, но в нем ПВП-12 замещен маннитолом.
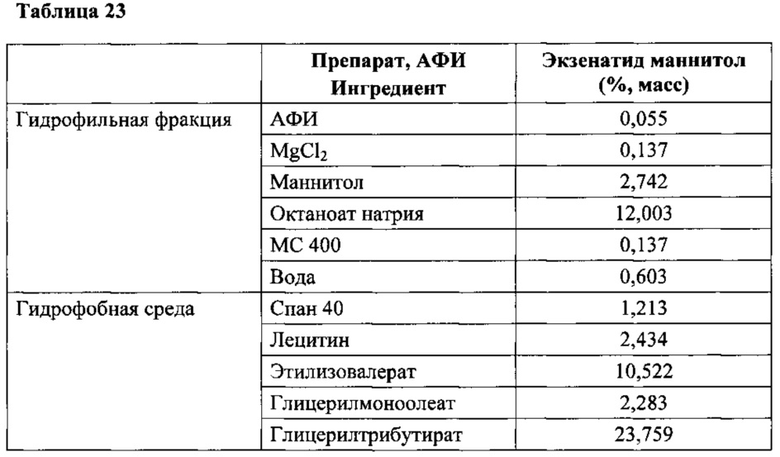

Препарат, описанный выше в табл.23, вводили непосредственно в тонкую кишку неанестезированных крыс, и уровни октреотида в плазме измерялись после введения препарата. Экспозиционные числа, показатели AUC, определялись для препаратов и сравнивались с основным препаратом. Эти результаты представлены ниже в табл. 24

Результаты, показанные выше в табл. 24, демонстрируют удивительный и неожиданный результат, что активность препарата экзенатида без ПВП-12 значительно уменьшилась по сравнению с основным препаратом. Было решено, поэтому, в дальнейшем исследовать влияние ПВП на биодоступность.
Влияние на активность препарата изменения молекулярного веса ПВП было протестировано с применением препаратов, содержащих экзенатид, как карго. 3 препарата экзенатида были приготовлены с использованием или ПВП-12, ПВП-17 или ПВП-25 (все получены от BASF). Все препараты: ПВП-12, ПВП-17 и ПВП-25 являются полимерами поливинилпирролидона; средние молекулярные массы составляют приблизительно 2500-3000, 10000 и 30000 соответственно. Препараты являются основными препаратами экзенатида, приготовленными преимущественно, как описано выше, где ПВП варьируется, как описано и в котором гидрофильную фракцию упростили пренебрежением MgCl2 и МС 400.

Три препарата, описанные выше в табл. 25, вводились непосредственно в тонкую кишку не анестезированным крысам, и уровни экзенатида в плазме измерялись после введения препарата. Для препаратов определялись экспозиционные числа, показатели AUC. Результаты представлены ниже в табл. 26.

Результаты, приведенные выше в табл. 26, демонстрируют, что препараты экзенатида, содержащие ПВП-12 проявляют значительно большую активность, чем препараты экзенатида, содержащие ПВП-17 и ПВП-25. Таким образом, только влияние ПВП-12 было исследовано дополнительно и было решено провести изучения дозозависимой ответной реакции, с использованием ПВП-12. Влияние увеличения количества ПВП-12 в препарате на активность препарата было протестировано с использованием препаратов, содержащих октреотид, как смесь карго и различные дозы ПВП-12, как показано в табл. 27 ниже. Протестированные ПВП-12 дозы составляли 2,75% (стандартные дозы, которые использовали в препаратах выше) и 5,0%, 7,5% и 10,0% ПВП-12; гидрофильную фракцию упростили пренебрежением MgCl2 и МС 400. Препарат, содержащий 10% ПВП, был полужидкий, т.е. представлял собой видимую полужидкую суспензию.

Препараты, описанные выше в табл. 27, вводились непосредственно в тонкую кишку неанестезированным крысам, и уровни октреотида в плазме измерялись после введения препарата. Для четырех различных препаратов определяли экспозиционные числа, показатели AUC. Результаты представлены ниже в табл. 28А.


Результаты, показанные выше в табл. 28А, демонстрируют, что абсорбция октреотида разительно увеличивается, когда в препарате увеличивается количество ПВП. Препарат, содержащий 10% ПВП-12, абсорбировал октреотид в 1,7 раза больше чем препарат, содержащий 2,75% ПВП-12. Улучшенный препарат октреотида, в котором было 10% ПВП-12, но не было октаноата натрия, практически не демонстрировал активности. rBA была 0,11% (CV=158%) n=5.
По-видимому, соль жирных кислот с цепью средней длины действует, как проницаемый усилитель (через облегчение или усиление проницаемости и/или абсорбции терапевтического агента), и что ПВП служит для увеличения влияния проницаемого усилителя в синергетической манере, так как один ПВП фактически не имеет эффекта, см. также пример 31.
Дальнейший эксперимент был проведен для исследования возможности замещения 10% ПВП-12 декстраном с сохранением активности препарата. Декстран производился компанией Fluka; средняя молекулярная масса составляла приблизительно 6000. Препараты были приготовлены преимущественно, как описано выше, ПВП и декстран в них варьировались, как описано, гидрофильную фракцию упростили исключением MgCl2. и МС 400 и количество октаноата натрия было увеличено до 15%; см. также пример 16.

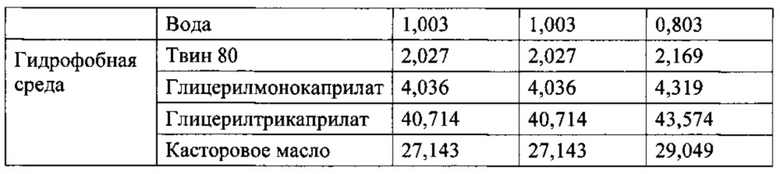
Три препарата, описанные выше в табл. 28В, вводились непосредственно в тонкую кишку не анестезированным крысам, и уровни октреотида в плазме измерялись после введения препарата. Для препаратов определяли экспозиционные числа, показатели AUC. Результаты представлены ниже в табл. 28С.

Результаты, представленные в табл. 28С демонстрируют, что абсорбция октреотида понижалась, когда ПВП в препарате замещали декстраном, но активность все еще оставалась значительной. Препарат, содержащий 10% декстрана, абсорбировал около 75% препарата, содержащего 10% ПВП, и препарат, содержащий 5% декстрана, абсорбировал около 73% октреотида препарата, содержащего 10% ПВП.
Пример 25: сравнительное изучение С8, С9, и С10 солей жирных кислот цепей средней длинны, а именно октаноата натрия, нонаноата натрия и деканоата натрия
Влияние на активность препарата замещения октаноата натрия другими солями жирных кислот натрия с цепью средней длины тестировались с использованием препаратов, содержащих октреотид, как карго.
Были приготовлены 3 препарата октреотида, как показано ниже в табл. 29. Все основные препараты готовили преимущественно, как описано выше, где гидрофильную фракцию упростили исключением MgCl2 и МС 400 и в котором соль жирной кислоты с цепью средней длины является эквимолярным количеством октаноата натрия, нонаноата натрия или деканоата натрия.
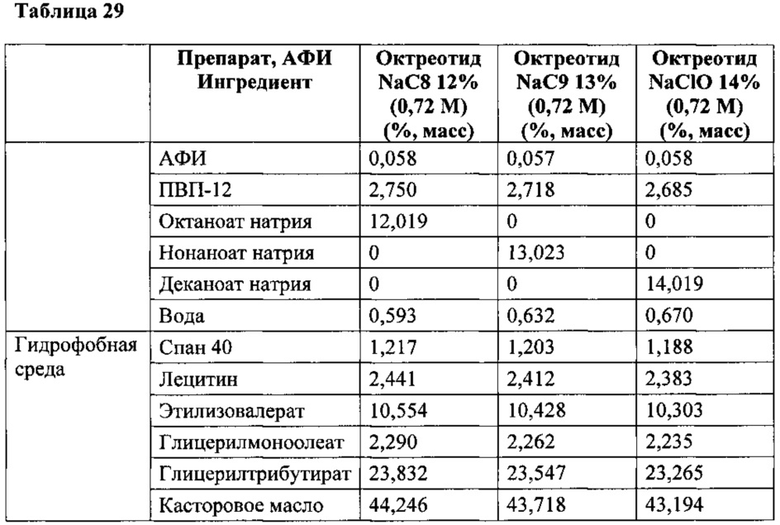
Препараты, описанные выше в табл. 29, вводились непосредственно в тонкую кишку неанестезированным крысам, и уровни октреотида в плазме измерялись после введения препарата. Для препаратов определялись экспозиционные числа, показатели AUC. Результаты представлены ниже в табл. 30.


Результаты, представленные выше в табл. 30 демонстрируют, что при замещении октаноата натрия в препарате нонаноатом натрия или деканоатом натрия сохранялась одинаковая активность. На основании статистических анализов, нет никакой разницы в активности между всеми тремя препаратами.
Пример 26: дозозависимый эффект октаноата натрия
Дозозависимый эффект октаноата натрия при 12%, 15% и 18% был протестирован с помощью приготовления препаратов, показанных в табл. 31. Все основные препараты приготовили преимущественно, как описано выше, в которых гидрофильная фракция была упрощена исключением MgCl2 и МС 400 и карго был октреотид. В дополнение, была откорректирована вязкость препарата, то есть одинаковая или подобная вязкость поддерживалась для всех трех препаратов; что было достигнуто изменением количества касторового масла и глицерилтрибутирата.

Препараты, описанные выше в табл. 31, вводились непосредственно в тонкую кишку не анестезированным крысам, и уровни октреотида в плазме измерялись после введения препарата. Для препаратов определяли экспозиционные числа, показатели AUC. Результаты представлены ниже в табл. 32.

Результаты, представленные выше в табл. 32, демонстрируют, что при увеличении количества октаноата натрия с 12% до 15%, происходит и увеличение в активности, но дальнейшее увеличение количества октаноата натрия до 18% не ведет дальше к повышению активности, более той, которая достигнута при 15%. Таким образом, количество октаноата натрия около 15% является предпочтительным.
Пример 27: исследование влияния изменения гидрофильного/липофильного баланса поверхностно-активных веществ в препарате
Табл. 33 ниже описывает различные препараты октреотида. Первая колонка, препарат(а) - основной препарат, приготовленный преимущественно, как описано выше, в котором гидрофильную фракцию упростили исключением MgCl2 и МС 400, и карго представлен октреотидом. Поверхностно-активные вещества - Спан 40, лецитин и глицерин моноолеат, и при расчете HLB приблизительно 5-6. В других препаратах (препараты b, с и d) HLB изменялся, как индикаторный (до 3,5; 6,7 и 14) замещением Спан 40 и лецитина разными количествами Твин 80 и варьированием количества глицерилмоноолеата.

Препараты, описанные выше в табл. 33, вводились непосредственно в тонкую кишку неанестезированным крысам, и уровни октреотида в плазме измерялись после введения препарата. Для препаратов определялись экспозиционные числа, показатели AUC. Результаты представлены ниже в табл. 34.

Результаты, приведенные в табл. 34, демонстрируют, что все 3 новых препарата, заменяющие Спан 40 и лецитин на Твин 80 [b, с и d] обладали лучшей активностью чем основной препарат [а], даже если бы HLB в [b] был ниже, и [с] был незначительно выше, и в [d] был намного выше, чем HLB поверхностно-активного вещества в (а). В дополнение, активности всех новых препаратов [(b, с, and d] были статистически очень схожи. Таким образом, только HLB из всех поверхностно-активных веществ, по-видимому, не влияет на активность, но возможно, что свойства поверхностно-активного вещества играют важную роль. В частности, замещение Спан 40 и лецитина на Твин 80 благоприятно для активности этих препаратах октреотида.
Пример 28: Препараты октреотида с различным соотношением глицерилтрибутирата к касторовому маслу
Основываясь на накопленных результатах, описанных выше, которые включают результаты ПВП-12 дозозависимого эффекта и результаты для поверхностно-активного вещества в числе прочих, серию препаратов октреотида готовили с применением 10% ПВП-12 и 15% октаноата натрия и варьированием отношения глицерилтрибутирата к касторовому маслу. В дополнение, глицерилмоноолеат и глицерилтрибутират замещали (если использовали) глицерилмонокаприлатом и глицерилтрибутиратом (оба приобретены у Abitec). Это необходимо для сохранения С8 мотива в составе. Таким образом, гидрофильная фракция содержит соль С8 кислоты (октаноат) и гидрофобная среда содержит моноглицериды и триглицериды, включающие одну и ту же С8 кислоту. Авторы изобретения полагают, что использование С8 соединений в гидрофильной фракции и гидрофобной среде может способствовать биодоступности. Количества Твин 80 и глицерилмонокаприлата также варьировались в препарате. Препараты готовили, как представлено в табл. 35А ниже. Препараты I, II, V и VI были полужидкими (несомненно, суспензии) и препараты III и IV были обычными жидкими суспензиями.


Препараты, описанные выше в табл. 35А, вводили непосредственно в тонкую кишку не анестезированным крысам, и уровни октреотида в плазме измеряли после введения препарата. Для составов определяли экспозиционные значения AUC. Результаты представлены ниже в табл. 35 В.


Результаты, представленные выше в табл. 35В, демонстрируют, что препараты I и IV обладают наибольшей активностью. Так как касторовое масло отсутствует в составе IV, показано, что касторовое масло не является существенным для активности.
Вероятно, высокое соотношение глицерилтрикаприлат: касторовое масло, например, 6:4 является предпочтительным с точки зрения активности. В дополнение, так как состав V (который имеет низкую активность) имеет то же соотношение глицерилтрикаприлат/касторовое масло, что и состав I, из этого следует, что дополнительно глицерилмонокаприлат (или другой моноглицерид) предпочтителен с точки зрения активности. В дополнение, готовили состав, подобный препарату I табл. 36, но октаноат натрия исключили. Этот состав фактически не проявлял активности, оБД=0,1%. Нерасфасованный лекарственный продукт препарата IV (улучшенный, без касторового масла) измельчали с помощью 150 микронного сита, и, далее, размер частицы определяли с применением технологии лазерной дифракции Малверна. Предварительные результаты указывают, что 90% (в объемном отношении) частиц имели размер менее 130 микрон, и 50% (в объемном отношении) частиц были менее 45 микрон. Предварительные эксперименты с использованием подобных составов к препарату I, но с варьированием возрастающих количеств октреотида, все показали подобную биологическую активность, т.е. это была приблизительно независимая линейная экспозиция от нагрузки АФИ. Предварительный эксперимент с использованием подобного препарата к препарату IV при высшей нагрузке октреотида - 1,5% (в весовом отношении) - также давал подобную биологическую активность.
Аналогичный улучшенный состав для вышеуказанного препарата I готовили с использованием FD4 как карго, вместо октреотида, и сопоставляли с основным составом. Эти препараты представлены в табл. 36А ниже.

Препараты, описанные выше в табл. 36А, вводили непосредственно в тонкую кишку не анестезированным крысам, и уровни FD4 в плазме измеряли после введения препарата. Для составов определяли экспозиционные значения AUC. Результаты представлены ниже в табл. 36 В.

Результаты, представленные выше в табл. 36В, демонстрируют, что улучшенный состав обладает значительно большей активностью, чем основной состав.
Пример 29: Подробный процесс продуцирования для селекции (улучшения) препарата октреотида
Препарат октреотида в Примере 28 (табл. 6, первая колонка) готовили преимущественно, как описано в Примерах выше. Ниже следует подробное описание процесса получения для этого препарата.
Получение гидрофильной фракции: К 150 мл воды медленно добавляли и перемешивали следующие ингредиенты: 24,05 г октаноата натрия, 16,04 г ПВП-12 и 92,4 г 10 мг/мл водного раствора октреотида. Полученный раствор лиофилизировали.
Получение гидрофильной среды:
3,25 г Твин 80, 6,47 г глицерилмонокаприлата, 65,25 г глицерилтрикаприлата и 43,50 г касторового масла смешивали вместе.
Получение нерасфасованного лекарственного продукта:
26,08 г гидрофильной фракции медленно добавляли к 73,92 г гидрофобной среды при 20±2°C при перемешивании. После добавления всей гидрофильной фракции, скорость перемешивания увеличили. Потом применяли дегазацию вакуумом и полученную суспензию хранили при 2-8°C. Для создания возможности растворения больших количеств октреотида, разработали следующий способ:
1. Количество воды гидрофильной фракции препарата было таким же, как рассчитанный объем конечного нерасфасованного лекарственного продукта.
2. ПВП-12 растворяли в половине количества вышеуказанной воды.
3. Октаноат натрия растворяли во второй половине количества воды.
4. Октреотид растворяли в ПВП-12 растворе (из параграфа 2).
5. Раствор октаноата натрия добавляли в раствор октреотида и ПВП-12.
На этой стадии выпадал небольшой осадок, но он растворялся после перемешивания.
Пример 30: Эксперименты на свиньях с применением капсул
Для теста на активность составов по изобретению, когда вводились капсулы, была установлена модель животного, позволявшая вводить капсулы свиньям (домашним свиньям). Для того, чтобы обойти желудок и разрешить непосредственное введение капсул в тонкую кишку свиньи, хорошо доказанную модель на собаках ("Nipple Valve model"; Wilsson-Rahmberg & О. Jonsson, Laboratory Animals (1997), 31, 231-240) адаптировали к промышленной свинье.
Готовили два препарата октреотида, приведенные ниже в табл. 37. Препарат октреотида (х) готовился непосредственно, как описано выше для основного препарата, в котором гидрофильная фракция была упрощена исключением MgCl2 и МС400. Препарат октреотида (y) готовили непосредственно, как описано выше для улучшенного препарата октреотида. Препараты помещали в желатиновые капсулы (от Capsugel), основной препарат (x) 0,42 мл на капсулу и улучшенный препарат (y) 0,44 мл на капсулу, что приводит к 5 мг нетто содержания октреотида в обоих типах наполненных капсул.
Капсулы не покрывали кишечнорастворимой оболочкой, т.е. они не были покрыты.

Препараты, описанные в табл. 37 выше, вводили непосредственно в тонкую кишку не анестезированным свиньям через обходной желудочный анастомоз, описанный выше, и уровни октреотида в плазме измеряли после введения препарата. Для составов определяли значения системного контакта AUC. Процент БА рассчитывали по сравнению с системным контактом октреотида после подкожного введения. Результаты представлены ниже в табл. 8.

Результаты, представленные в табл. 36В выше, показывают, что в модели свиньи имеется биологическая активность для инкапсулированных составов, как для базовых, так и для улучшенных составов. Биологическая активность улучшенного препарата октреотида была приблизительно в 3 раза выше уровня биологической активности основного препарата.
Результаты, полученные для биодоступности недооценены, потому что время отбора проб не достаточно для того, что бы уровень октреотида вернулся к базовой линии (0 нг/мл). Это было связано с более длинным временем ожидания экспозиции в свиней в сравнении с тем, что предварительно измеряли у крыс. Форму графика изменяли по сравнению с результатами, полученными от крыс, показывающими более длительное время для достижения максимального уровня пика и продолжительного времени, в котором октреотид постоянно присутствует в крови. Это может быть полезным, так как это позволяет октреотиду длительно действовать в организме. Таким образом, фактическая биодоступность у свиней должна быть выше, чем полученные числа.
Основываясь на результатах, полученных от крыс, уровень биодоступности октреотида в свиньях, введенного в водном растворе, экстраполировали до значения около 0,1%. Этот уровень биодоступности ниже уровня чувствительности биоанализов, которые использовали для свиней.
Пример 31: результаты дозозависимого эффекта для ПВП в улучшенном составе
В дополнение к ПВП результатам в Примере 24, исследовали влияние на активность увеличения количества ПВП-12 в улучшенном составе. Улучшенные препараты, приготовленные преимущественно как описано выше, содержали октреотид, как карго, и различные дозы ПВП-12, как представлено в табл. 39 ниже. Тестировали ПВП-12 с дозами 7,5%, 10% и 15% ПВП-12. Препараты, содержащие 10% и 15% ПВП, были полужидкими, то есть они были, несомненно, полужидкими суспензиями, и препарат, содержащий 7,5% ПВП, был вязкой суспензией.

Препараты, описанные выше в табл. 39, вводили непосредственно в тонкую кишку не анестезированным крысам и уровни октреотида в плазме измеряли после введения препарата. Для трех составов определяли экспозиционные значения AUC. Эти результаты представлены ниже в табл. 40.

Результаты, представленные в табл. 40, демонстрируют, что абсорбция октреотида была наибольшей, когда ПВП в составе был 10% и увеличение количества до 15% приводит к значительному уменьшению активности. Это подтверждает выбор 15% ПВП в улучшенной формуле.
Пример 32: Активность АФИ в составе по сравнению с АФИ, введенном вместе с составом
Три различных основных составов трех различных соединений карго готовили (декстран, гентамицин и экзенатид), преимущественно как описано выше (в которых основной состав является основной неупрощенной гидрофильной фракцией). Каждый из трех препаратов вводили непосредственно в тонкую кишку не анестезированных крыс, уровень карго в плазме измеряли после введения препарата. Экспозиционные значения AUC определяли для составов. В дополнение, подобный состав готовили с нехарактерным соединением карго (имитатор препарата). Отдельно, имитатор препарата вводили вместе с декстраном, гентамицином и экзенатидом в водном растворе и определяли экспозиционные значения AUC. Совместное введения проводили с помощью введения карго в водном растворе немедленно после введения имитатора препарата через катетер в тонкой кишке (обходной желудочный анастомоз). Для каждого соединения, экспозицию после введения составленного карго сравнивали с экспозицией после введения не введенного в препарат карго (сопутствующего).
Результаты сравнения представлены ниже в табл. 41. Результаты показывают, что наивысшая активность (биодоступность) достигается, когда карго во всех трех случаях составлен в сравнении с не введенным в препарат (сопутствующим) карго, и что экзенатид показал значительное увеличение активности благодаря препарату.
Заметим, что декстран и гентамицин - соединения, нечувствительные к расщеплению протеазой, тогда как экзенатид, будучи пептидом, является предметом деградации кишечными ферментами. Значительная разница в активности между составленной лекарственной формой экзенатида в сравнении с не введенным в препарат может быть связана с защитным действием препарата от деградации.

Пример 33: Оценка кишечной гиперпроницаемости
А. Размер ограничения: Технология и препараты, описанные выше, предназначены для усиления проницаемости кишечника, позволяющие производить специфическую доставку протеинов, пептидов и других непроницаемых веществ через этот барьер другим способом.
В известной мере неспецифическое проникновение содержимого кишечника может быть результатом побочного действия этого усиления специфической проницаемости. Размер молекул, которые могли бы не специфически проникнуть через кишечник, оценивали с применением маркерных молекул различного размера. Чтобы оценить предел размера молекул увеличенной проницаемости ЖКТ, выбрали пять различных ФИТС-меченых декстранов с различным молекулярным весом, как молекулярных маркеров для теста увеличенной проницаемости кишечника; средняя молекулярная масса 5 видов декстрана составляла 4,4, 10, 20, 40 и 70 кДа, с эквивалентным радиусом 14, 23, 33, 45 и 60 А соответственно. Эти различные по размеру маркеры вводили непосредственно в тонкую кишку не анестезированных крыс, через имплантированный в кишечнике катетер, и показали практическое отсутствие основной проницаемости кишечника, при тестировании по одному. Каждый из этих маркеров вводили непосредственно в тонкую кишку не анестезированных крыс вместе с 300 мл основного препарата, и уровень проницаемости оценивали тестированием уровня декстрана в крови.
Уровни декстрана в плазме измеряли перед введением дозы и на 3, 6, 10, 25, 60, 90 мин после введения препарата. Определяли экспозиционные значения AUC(0-90), результаты показаны на фиг. 7. Данные представлены как среднее значение±допустимое отклонение, n≥4. Результаты показывают, что протестированный наименьший молекулярный маркер (декстран со средним молекулярным весом 4,4 кДа), проникает через кишечник, когда его вводят вместе с составом, который увеличивает молекулярный размер, степень проникновения уменьшается: молекулярный маркер 10 кДа проникает с меньшей степенью и маркер 20 кДа в еще меньшей степени. Молекулярный маркер в 40 кДа дает минимальную проницаемость тогда, как молекулярный маркер в 70 кДа не показывает проницаемости вообще (основная проницаемость). Эти результаты указывают, что 40-70 кДа является предельным размером для усиления неспецифической проницаемости препаратами по изобретению. Таким образом, введения большого объема препарата (300 мл) в кишечник крыс привели к результату усиления проницаемости через кишечный барьер, и эта усиленная проницаемость, ограниченная размером молекулы, имеет предельный размер 40-70 кДа и минимальную проницаемость при 40 кДа.
Опубликованные значения размера опасных молекул (молекулярный вес и радиус), которые могут потенциально присутствовать в кишечнике, показаны ниже в табл. 42.


Табл. 42 демонстрирует, что размер потенциально опасных молекул, присутствующих в кишечнике, больше предельного размера усиленного проникновения тестируемых составов, как показано выше. Таким образом, эти результаты предлагают, что тестируемые препараты не будут облегчать проникновение опасных молекул через кишечный барьер и эти препараты могут, следовательно, рассматривать как безопасные. Другие препараты по изобретению дают подобные результаты.
В. Повторное введение препарата: Чтобы исследовать влияет ли повторное введение препарата на проницаемость кишечника, улучшенный препарат октреотида (12% октаноат натрия с касторовым маслом) вводили крысам последовательно на протяжении 14 дней с применением модели in vivo, описанной выше (крысы с двумя имплантированными в тонкую кишку катетерами). На 1, 7 и 14 день введения, маркер проницаемости декстрана (ФИТС-декстран с молекулярным весом в 4,4 кДа; FD4) вводили через 60 мин после введения препарата. Это делали для оценки проницаемости кишечника по способности проникать FD4 из кишечника в кровь. Не найдено значительной разницы в воздействии FD4 для следующих 14 дней повторяющегося введения препарата. Эти результаты предполагают, что увеличения проницаемости кишечника в результате периода повторяющегося введения нет, и усиленная проницаемость кишечника сохраняет обратимый процесс на протяжении этого периода.
Результаты предполагают, что препараты не приводят к повреждению ткани кишечника, но действуют с помощью специфического открывания кишечного барьера, не показывая эффект усиления дополнительной проницаемости.
Пример 34: Оценка гиперпроницаемости кишечника: динамика и обратимость
В дополнение к изученному в вышеприведенных Примерах, исследование было спланировано, чтобы определить динамику увеличения проницаемости кишечника препаратами по изобретению и обратимость этого процесса, используя декстран как маркер проницаемости.
С целью определения интервала времени для возрастания интенсивности проницаемости, разработали модель на крысах in vivo, в тонкую кишку которых были имплантированы один или два катетера. ФТИС-меченый декстран (средняя молекулярная масса 4,4 кДа, FD4), который фактически не имеет базального кишечного проникновения, служил как молекулярный маркер в тесте на кишечную проницаемость. Был запланирован эксперимент, в котором декстрановый маркер вводили вместе с составом (через катетер, имплантированный в тонкую кишку), или с различными временными интервалами от введения препарата (через вторичный отдельный катетер, имплантированный в тонкую кишку). Кишечную проницаемость рассчитывали тестированием FD4 проницаемости в крови. Крысам вводили основной состав вместе с декстрановым маркером, или основной состав и далее декстрановый маркер с различными интервалами времени (10, 30 и 60 мин). Образцы крови анализировали на концентрацию декстрана перед введением и на 3, 6, 10, 25, 60, и 90 мин после введения декстрана. Результаты представлены на фиг. 8. Данные представлены как среднее значение ± допустимое отклонение, n≥5.
Фиг. 8 демонстрирует, что декстрановый маркер проникает в кишку в наивысшей степени, когда введение проводили вместе с составом. Интервал в 10 мин между введением препарата и введением декстранового маркера имеет результатом значительное уменьшение количества проникновения маркера, и увеличение интервала, затем имеет результатом экспоненциальное уменьшение проникновения маркера.
Эти результаты показывают, что при некоторой степени повышения неспецифической проницаемости препарата, это ограничивается коротким периодом времени после введения препарата. Кишечная проницаемость резко уменьшается со временем, и за 60 мин после введения препарата не наблюдалось проникновения препарата. Таким образом, введение препарата в кишечник крысы имело результатом повышенную проницаемость кишечного барьера в очень короткий период. Другие препараты по изобретению давали подобные результаты.
Пример 35: Пероральное введение октреотида обезьянам
С целью тестирования фармакокинетики октреотида после перорального введения обезьянам препарата октреотида, пятью яванским макакам назначали пероральное введение капсул, содержащих улучшенный состав октреотида в касторовом масле (подобный препарату I из табл. 35, но с более высоким содержанием октреотида). Капсулы, которые использовались, были желатиновыми капсулами, покрытыми 6,7% Acryl-EZE® кишечнорастворимой оболочкой размера 1; эта оболочка предотвращает разрушение капсулы в желудке и позволяет открыться капсуле в тонкой кишке животных. Использованная доза октреотида была 5 мг на капсулу.
Обезьян не кормили ночь перед введением капсул. После перорального введения, образцы крови отбирали через 9,75 часа, выделяли из плазмы и анализировали на содержание октреотида способом жидкостной хроматографии с тандемной масс-спектрометрией (LC/MS/MS способ), см. фиг. 9. Подобные эксперименты проводили с улучшенным составом без касторового масла/GTC (аналогичный препарату IV из табл. 35, но с более значительным содержанием АФИ) и получили аналогичные результаты. Подобные эксперименты также проводили с несколькими различными кишечнорастворимыми оболочками и получали аналогичные результаты. С целью сравнения фармакокинетики октреотида после введения улучшенного препарата октреотида, с фармакокинетикой инъекционно введенного октреотида, раствор ацетата октреотида (0,1 мг на обезьяну) вводили подкожно двум обезьянам от вышеупомянутой группы, которые выступают в качестве эталона. Образцы крови отбирали на протяжении 4 час, отделялись от плазмы и анализировались на содержание октреотида LC/MS/MS способом. Фармакокинетику октреотида сравнивали после перорального применения и подкожной инъекции раствора октреотида (см. Фиг. 9 и 10). Результаты перорального введения препарата показывают абсорбцию на протяжении нескольких час. Форму графика изменяли в сравнении с подкожным введением, показывая более медленное, но длительное высвобождение октреотида в кровь. Это может быть выгодно, так как это позволяет проникать октреотиду в кровь более длительный период, потенциально удлиняя интервал активности.
Утвержденной дозой для инъекции ацетата октреотида у человека является 0,1 мг на пациента. Вышеприведенные результаты в обезьянах предполагают, что улучшенный состав, содержащий около 10 мг октреотида на дозу, будет производить терапевтическое воздействие на человека.
Пример 36: Данные стабильности
Основной и улучшенный состав октреотида по изобретению сохраняли при 4°C и при 25°C и периодически тестировали на содержание октреотида. Оба препарата были признаны стабильными.
Пример 37: Препараты, содержащие ванкомицин, интерферон альфа и терлипресин
А. Ванкомицин: Табл. 43 ниже описывает улучшенный состав ванкомицина, содержащий 10% ПВП и 15% октаноата натрия в гидрофильной фракции, и содержащий глицерилтрикаприлат, как основной компонент гидрофобной среды. Ванкомицин приобрели у Gold Biotechnology.

В предыдущих экспериментах, препараты, описанные ниже в табл. 43, вводили непосредственно в тонкую кишку неанестезированных крыс, и измеряли уровень ванкомицина в плазме после введения препарата. Экспозиционные значения AUC, определяли для составов. Результаты показали, что аБД составляет около 15% (сравнительно с IV, n=6). Когда ванкомицин в солевом растворе вводили в тонкую кишку не анестезированных крыс, биодоступность не обнаружена.
Интерферон-альфа: Табл. 44 ниже описывает улучшенный препарат интерферона-альфа, содержащий 10% ПВП и 15% октаноата натрия в гидрофильной фракции и содержащего глицерилтрикаприлат как основной компонент гидрофобной среды. Интерферон-альфа поставлялся в буфере (от Intas Biopharmaceuticals) и ингредиенты буфера интерферона-альфа в составе отмечены звездочкой (*).
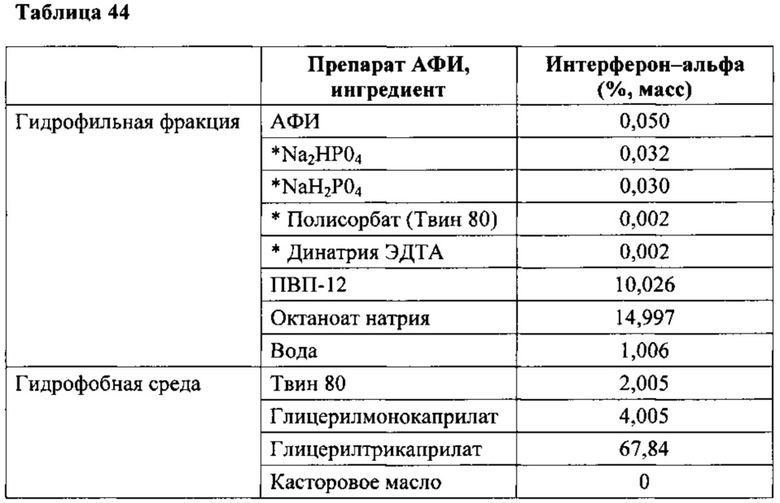
Препараты, описанные выше в табл. 44, вводили непосредственно в тонкую кишку неанестезированных крыс. Измеряли уровни интерферона-альфа в плазме после введения препарата.
С. Терлипресин: Табл. 45 выше описывает терлипресин основного препарата и терлипресин улучшенного препарата, содержащего 10% ПВП и 15% октаноата натрия в гидрофильной фракции, и содержащего глицерилтрикаприлат как основной состав в гидрофобной среде.
Терлипресин приобрели от Bambio. Основной состав готовили преимущественно, как описано выше, и улучшенный состав готовили преимущественно так же, как описывали выше.

Препараты, описанные выше в табл. 45, вводили непосредственно в тонкую кишку не анестезированных крыс. Измеряли уровни терлипресина в плазме после введения состава.
Пример 38: Ингибирование гормона роста октреотидом in vivo
Одним из лучше всего описанных эффектов октреотида является ингибирование высвобождения гормона роста. С целью тестирования эффективности ингибирования гормона роста составом октреотида по изобретению использовали модель крыс, где уровень эндогенного крысиного гормона роста (кГР) контролировали после введения препарата октреотида в тонкую кишку модели не анестезированных крыс (описанной выше).
Введение основного препарата октреотида (содержащего 12% октаноата натрия) в тонкую кишку крыс показало снижение уровня rGH до 87,4% в сравнении с введением солевого контроля. Результаты показали, что октреотид составов, описанных в данном описании, обеспечивает доставку октреотида в его активной форме из кишечной полости в кровоток.
Пример 39: Токсикологические исследования
28-Дневное токсикологическое исследование введения контрольного препарата (только наполнители, не карго) проводили на крысах Wistar. Животным в тест группе ежедневно вводили ректально максимально допустимую дозу препарата (100 мкл на животное в день) на протяжении 28 последующих дней. Тест группу сравнивали с двумя контрольными группами: не подвергнутая воздействию группа (необработанная) и группа, которой вводили солевой раствор, (n=15/группу).
Общие клинические исследования делали дважды в день, и детальные клинические исследования проводили еженедельно. Вес тела и потребления пищи измеряли еженедельно. Клиническую патологию и макропатологию изучали через 1 день после последней обработки. Гистологические исследования проводили на прямой кишке, толстой кишке, печени и почках, и токсические эффекты не обнаружены. Это было гистопатологическое исследование без локальных ЖКТ или общих результатов, клинических результатов, не связанных с препаратами, без изменений гематологических и химических параметров крови, не связанных с макроскопическими результатами при аутопсии и смертности.
В заключении, этот эксперимент показал, что не наблюдалось токсичности во время ежедневного ректального дозирования препарата у крыс на протяжении 28 последующих дней.
Имея, таким образом, описанные несколько аспектов, по меньшей мере, одного из вариантов, понятно, что различные изменения, модификации и улучшения будут легко осуществлены специалистами в данной области. Соответственно такие изменения, модификации и улучшения являются частью этого разглашения и находятся в сфере изобретения. Соответственно, вышеизложенное описание и фигуры представлены только в качестве примера, и сфера изобретения может быть определена от соответствующей конструкции прилагаемых формул изобретения и их эквивалентов.










Изобретение относится к медицине и касается покрытой кишечнорастворимой оболочкой лекарственной формы для перорального применения, содержащей композицию, включающую суспензию, которая содержит смесь гидрофобной среды и твердой формы. Твердая форма содержит терапевтически эффективное количество полипептида, по меньшей мере одну соль жирной кислоты со средней длиной цепи, имеющей длину цепи от 6 до 14 атомов углерода и поливинилпирролидона (ПВП), где поливинилпирролидон присутствует в композиции в количестве от 3% по массе или более и где по меньшей мере одна соль жирной кислоты со средней длиной цепи присутствует в композиции в количестве по меньшей мере 12% по массе или более. Изобретение обеспечивает повышение биологической активности препарата полипептида. 28 з.п. ф-лы, 10 ил., 45 табл., 39 пр.
1. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения, содержащая композицию, включающую суспензию, которая содержит смесь гидрофобной среды и твердой формы, где твердая форма содержит терапевтически эффективное количество полипептида, по меньшей мере одну соль жирной кислоты со средней длиной цепи, имеющей длину цепи от 6 до 14 атомов углерода и поливинилпирролидона (ПВП), где поливинилпирролидон присутствует в композиции в количестве от 3% по массе или более и где по меньшей мере одна соль жирной кислоты со средней длиной цепи присутствует в композиции в количестве по меньшей мере 12% по массе или более.
2. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.1, где соль жирной кислоты со средней длиной цепи присутствует в композиции в количестве от 12% до 40% по массе.
3. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.2, где соль жирной кислоты со средней длиной цепи присутствует в композиции в количестве от 12% до 18% по массе.
4. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.1, где поливинилпирролидон присутствует в композиции в количестве от около 3% до около 20 % по массе.
5. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.4, где поливинилпирролидон присутствует в композиции в количестве от около 5% до около 15 % по массе.
6. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.1, где гидрофобная среда включает глицерилтрикаприлат и твердая форма состоит из полипептида, поливинилпирролидона с молекулярной массой около 3000 и октаноата натрия.
7. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.1, где гидрофобная среда дополнительно включает касторовое масло, или глицерилмонокаприлат, или их комбинацию и поверхностно–активное вещество.
8. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.1, где гидрофобная среда состоит из глицерилтрикаприлата, глицерилмонокаприлата и полиоксиэтиленсорбитана.
9. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.1, где твердая форма состоит по существу из полипептида, поливинилпирролидона с молекулярной массой около 3000 и октаноата натрия.
10. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.1, где композиция включает около 41% грицерилтрикаприлата, около 27% касторового масла, около 4% глицерилмонокаприлата, около 2% полиоксиэтилен сорбитан моноолеата, около 15% октаноата натрия, около 10% поливинилпирролидона с молекулярным весом около 3000 и терапевтически эффективное количество полипептида.
11. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.1, где композиция включает около 68% грицерилтрикаприлата, около 4% глицерилмонокаприлата, около 2% полиоксиэтилен сорбитан моноолеата, около 15% октаноата натрия, около 10% поливинилпирролидона с молекулярным весом около 3000 и терапевтически эффективное количество полипептида.
12. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.1, где композиция включает терапевтически эффективное количество полипептида, около 12-21% октаноата натрия, около 5-10% поливинилпирролидона с молекулярным весом около 3000, около 20-80% грицерилтрикаприлата, около 0-50% касторового масла и около 3-10% поверхностно-активного вещества.
13. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.1, где композиция включает терапевтически эффективное количество полипептида, около 12-21% октаноата натрия, около 5-10% поливинилпирролидона с молекулярным весом около 3000, около 20-80% грицерилтрикаприлата и около 3-10% поверхностно-активного вещества.
14. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.13, где полипептид присутствует в количестве менее 33%.
15. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.14, где композиция включает около 15% октаноата натрия, около 10% поливинилпирролидона с молекулярным весом около 3000, около 30-70% грицерилтрикаприлата и около 6% поверхностно-активного вещества.
16. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.15, где поверхностно-активное вещество представляет собой глицерилмонокаприлат или полиоксиэтиленсорбитан моноолеат.
17. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.1, где твердая фаза включает частицу или множество частиц.
18. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.16, где твердая форма дополнительно включает стабилизатор.
19. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.1, где твердая форма дополнительно включает стабилизатор.
20. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.13, где полипептид присутствует в количестве менее 25%.
21. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.13, где полипептид присутствует в количестве менее 10%.
22. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.13, где полипептид присутствует в количестве менее 1%.
23. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.13, где полипептид присутствует в количестве менее 0,1%.
24. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.1, где поливинилпирролидон имеет молекулярный вес около 3000.
25. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.1, где соль жирной кислоты со средней длиной цепи представляет собой гексаноат натрия, гептаноат натрия, октаноат натрия, нонаноат натрия, деканоат натрия, ундеканоат натрия, додеканоат натрия, тридеканоат натрия, или тетрадеканоат натрия, или соответствующую соль калия, или лития, или аммония, или их комбинацию.
26. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.25, где соль жирной кислоты со средней длиной цепи представляет собой октаноат натрия.
27. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.1, где гидрофобная среда включает минеральное масло, парафин, моноглицерид жирной кислоты, диглицерид, триглицерид, сложный эфир или простой эфир, или их комбинацию.
28. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.1, где гидрофобная среда включает глицерилтрикаприлат.
29. Покрытая кишечнорастворимой оболочкой лекарственная форма для перорального применения по п.1, где композиция дополнительно включает поверхностно-активное вещество.
| US 20070219131 A1, 20.09.2007 | |||
| US 6200602 B1, 13.04.2001 | |||
| US 4234437 A, 18.11.1980 | |||
| ALIAUTDIN RN., et al., [Drug delivery to the brain with nanoparticles] | |||
| [Article in Russian] Eksp Klin Farmakol | |||
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |

