Область техники к которой относится изобретение
Изобретение относится к нестандартным фармацевтическим композициям, обеспечивающим введение путем пероральной доставки белков/пептидов или их конъюгатов и/или комплексов катион-конъюгат инсулина, демонстрирующих требуемые фармакокинетические профили и активность на эффективных моделях диабета на собаках и у человека. Предпочтительный препарат содержит 0,01÷20 мас.% инсулина, конъюгатов соединений инсулина и/или катион-конъюгатов инсулина, 10÷60 мас.% одного или более компонентов жирных кислот, выбранных из насыщенных или ненасыщенных С4-С12 жирных кислот и/или солей данных жирных кислот, и дополнительно содержит оптимальные количества других фармацевтически приемлемых полимерных наполнителей, которые обеспечивают повышенную растворимость, скорость растворения и эффективную биодоступность слабо растворимых в воде композиций и совместимых профилей высвобождения in-vivo при масштабируемости в процессе изготовления. Другой аспект изобретения характеризует способ получения вышеуказанных препаратов.
Уровень техники
Принятые способы подкожного введения инсулина все в большей степени исследуют в плане замены пероральными механизмами доставки лекарственных препаратов, которые не изменяли бы его физиологической клинической активности. Проблемы, с которыми сталкиваются в данной области техники при создании эффективной системы пероральной доставки лекарственного препарата для биологических макромолекул, в основном приписывают его чувствительности к ферментативному разложению и низкой проницаемости через эпителий. Кроме того, структура и конформация инсулина легко изменяются при приготовлении лекарственного средства и условиях этого процесса, приводя к потере биологической активности. Некоторые подходы к преодолению данных ограничений включают использование аналогов инсулина, введение таких пептидов, как амилин, глюкагоноподобный пептид, С-пептиды, ингаляционные формы, интраназальные формы, которые удовлетворительным образом не направлены индивидуально на ограничения биодоступности.
В области техники существует потребность в фармацевтически приемлемых комплексах, включая дериватизированные конъюгаты инсулина, обладающие повышенной биодоступностью или другими усовершенствованными фармацевтическими свойствами относительно имеющихся конъюгатов. Более того, требуется, чтобы данные усовершенствованные конъюгаты доставлялись в форме стабильного и нестандартного препарата, который легко максимально увеличивает преимущества пероральной доставки белков. В настоящем изобретении применяют комбинированный подход использования усовершенствованного конъюгата инсулина с повышенной биодоступностью и усовершенствованной лекарственной формы в отношении ограничений доставки инсулина.
Примеры соединений инсулина включают человеческий инсулин, лизпро-инсулин, дез30-инсулин, нативный проинсулин, искусственные проинсулины и т.п. Катионный компонент может, например, представлять собой катион двухвалентного металла, выбранный из группы, состоящей из Zn++, Mn++, Ca++, Fe++, Ni++, Сu++, Со++ и Мg++. Комплексы катион-конъюгат соединения инсулина также включают модифицированную группу, связанную (например, ковалентно или ионно) с соединением инсулина с образованием конъюгата соединения инсулина. Кроме того, модифицирующую группу выбирают так, чтобы она делала конъюгат соединения инсулина настолько же или более растворимым, чем соответствующее неконъюгированное соединение инсулина, и чтобы растворимость в воде конъюгата соединения инсулина снижалась при добавлении цинка. Модифицирующую группу выбирают так, чтобы она делала конъюгат соединения инсулина настолько же или более растворимым, чем соответствующее неконъюгированное соединение инсулина; растворимость в воде конъюгата соединения инсулина снижают добавлением цинка, и растворимость в воде комплекса превышает растворимость в воде соединения инсулина.
Примеры подходящих модифицирующих групп и конъюгатов инсулина, используемых для получения композиций, можно найти в патентах США № 7060675, 6303569, 6214330, 6113906, 5985263, 5900402, 5681811, 5637749, 5612640, 5567422, 5405877, 5359030, полные описания которых включены в данном контексте в виде ссылки. Дополнительные примеры данных комплексов катион-конъюгат соединения инсулина приведены в патентных заявках США US 2003/083232, US 2006/0019873 и US 2006/0019874.
В противоположность существующему предшествующему уровню техники соединения, соответствующие настоящему изобретению, проявляют пониженный митогенный потенциал, который почти в три раза ниже, чем у Insugen®.
Инсулин связывается и активирует свой родственный инсулиновый рецептор (ИР) с субнаномолярной аффинностью. Инсулин связывается также с структурно близким рецептором инсулиноподобного фактора роста (рецептор ИФР-1), но с приблизительно в 1000 раз более низкой аффинностью, чем инсулиновый рецептор. Вследствие этого при физиологических концентрациях инсулина рецептор ИФР-1 не играет никакой роли в опосредовании эффектов инсулина. Однако при высоких концентрациях инсулина (у больных диабетом, получающих инсулин) он дает митогенные эффекты посредством рецептора ИФР-1, который передает сигналы роста более эффективно, чем инсулиновый рецептор (см. статью Lammers R, et al., EMBO 1989).
Один из ранее идентифицированных аналогов инсулина с модификацией по остатку аспарагиновой кислоты В10 направлен на получение эффекта инсулина быстрого действия, однако модификация, в свою очередь, привела к существенному повышению митогенности данного аналога [см. статью Drejer, К., The bioactivity of insulin analogues from in vitro receptor binding to in vivo glucose uptake. (Биоактивность аналогов инсулина от связывания рецептора in vitro до поглощения глюкозы in vivo) Diabetes Metab Rev, 1992. 8(3): стр. 259-85]. Исследование проведено с целью изучения связывания ряда аналогов инсулина с рецептором инсулиноподобного фактора роста-1 и инсулиновым рецептором. Константы связывания измеряют и соотносят с метаболической и митогенной активностью исследуемых аналогов инсулина [см. статью Kurtzhals, P., et al., Correlations of receptor binding and metabolic and mitogenic potencies of insulin analogs designed for clinical use. (Корреляции связывания рецептора и метаболической и митогенной активности аналогов инсулина, созданных для применения в клинике), Diabetes, 2000. 49(6): стр. 999-1005.] Согласно проведенным исследованиям, по сравнению с нормальным инсулином инсулин лизпро, инсулин аспарт и инсулин гларгин имели минимальное изменение в связывании инсулинового рецептора, тогда как инсулин детемир был существенно менее активным. Каждый из данных аналогов имел близкие или повышенные скорости диссоциации с рецептором ИФР-1, которые коррелировали с его митогенным поведением.
Митогенная активность белков/пептидов и их конъюгатов, комплексов катион-конъюгат пептида, комплексов катион-конъюгат инсулина представляет собой важную проблему, приписываемую риску повышенной митогенности и росту эпителиальных клеток молочной железы человека. Исследования показали, что связывание ИФР-1 инсулина аспарт аналогично связыванию нативного человеческого инсулина. Инсулин лизпро и инсулин гларгин имеют 1,5÷6,5-кратное повышение аффинности связывания с рецептором ИФР-1, соответственно, позволяя предположить, что инсулин гларгин обладает существенно более сильным митогенным ответом.
Длительные эффекты митогенных свойств аналогов инсулина, его конъюгатов, комплексов катион-конъюгат пептида, комплексов катион-конъюгат инсулина продолжают оставаться важным фактором, который следует принимать во внимание. Пункты, по которым рассматривают документ CPMP/SWP/372/01 касательно неклинической оценки канцерогенного потенциала аналогов инсулина, утверждают: "Нативный человеческий инсулин в дополнение к метаболическим действиям имеет слабый митогенный эффект. Данный эффект становится важным в плане безопасности аналогов инсулина, поскольку структурные модификации молекулы инсулина могли бы повысить его митогенную активность, возможно, приводящую к стимуляции роста ранее существующих новообразований". "Хотя предполагают повышенную активацию рецептора инсулиноподобного фактора роста 1 и/или аберрантную передачу сигнала через инсулиновый рецептор, механизм(ы), ответственный за митогенную активность аналогов инсулина, остается невыясненным".
Поскольку накапливаются доказательства того, что ИФР-1 стимулирует рост рака толстой кишки, молочной железы, простаты и легкого, имеется необходимость создания белков/пептидов и их конъюгатов, комплексов катион-конъюгат пептида, комплексов катион-конъюгат инсулина с минимальным митогенным риском, который оценивают в анализе пролиферации клеток и который можно назвать безопасным в течение длительного лечения.
Настоящее изобретение относится к белкам/пептидам, их конъюгатам и/или комплексам катион-конъюгат полипептида, проявляющим в три раза более низкие митогенные свойства по сравнению с Insugen®. Следующие аспекты настоящего изобретения направлены на тот факт, что наполнители в лекарственном продукте статистически не воздействуют на характеристики митогенной активности лекарственной субстанции.
Другой аспект изобретения относится к нестандартным фармацевтическим композициям, обеспечивающим введение путем пероральной доставки белков/пептидов или их конъюгатов и/или комплексов катион-конъюгат инсулина, демонстрирующих требуемые фармакокинетические профили и активность на эффективных моделях диабета на собаках и у человека.
WO 00/50012 раскрывает твердые пероральные лекарственные формы, содержащие лекарственный препарат и усиливающий агент, причем усиливающий агент представляет собой соль жирной кислоты с цепью средней длины с углеродной цепью из приблизительно 6-20 атомов углерода. US 2006/0018874 покрывает твердые фармацевтические композиции, приготовленные для перорального введения путем проматывания, имеющие 0,1÷75 мас.% компонента жирной кислоты, где компонент жирной кислоты включает насыщенные или ненасыщенные жирные кислоты и/или соли и лечебный фактор.
Несмотря на вышеизложенное, еще существует потребность в изготовлении "жизнеспособных" пероральных препаратов инсулина, которые могут преодолевать проблемы, связанные с потерей биологической активности в процессе изготовления, и в то же самое время проявлять повышенную устойчивость к ферментному разложению in vivo после приема. Настоящее изобретение направлено на оба требования. Вследствие этого изобретение направлено на проблемы, с которыми сталкиваются в области техники при создании высокоэффективного механизма пероральной доставки лекарственных препаратов типа конъюгатов инсулина.
Изобретение демонстрирует различные преимущества в плане дозирования и удобного способа введения. Изобретение создает еще одно преимущество относительно композиций, соответствующих предшествующему уровню техники, поскольку авторы предполагают, что заявленный рационально разработанный пероральный препарат IN-105 с измеренными компонентами других наполнителей и способ изготовления таблеток для перорального применения легко масштабируются. Кроме того, за счет данного рационально разработанного перорального препарата и способа его получения фактор масштабируемости не влияет на действие лекарственного препарата in vivo или его профиль высвобождения in vivo.
Имеющиеся в настоящее время пероральные препараты инсулина проявляют низкие уровни стабильности, исключая возможности перорального введения данных лекарственных средств. Один из наиболее важных аспектов настоящего изобретения характеризуется тем фактом, что настоящий пероральный препарат инсулина стабилен в интервале температур без вредного воздействия на различные параметры стабильности таблеток, такие как твердость, время распада, накопление высокомолекулярных примесей и скорость растворения. Изготовленные таким образом таблетки показывают высокую стабильность даже в ускоренных условиях стабильности при относительной влажности (ОВ) 75% при 40°С (далее 40°С/75%). Свойственные молекуле характеристики стабильности и способы изготовления обусловливают стабильную природу препарата.
Другой аспект изобретения относится к усовершенствованным фармацевтическим композициям комплексов катион-конъюгат инсулина, полученных с помощью масштабируемого способа распылительной сушки, причем указанный способ включает стадии получения водной суспензии комплекса катион-конъюгат инсулина и по меньшей мере одного компонента жирной кислоты необязательно с одним или более фармацевтически приемлемых наполнителей.
В типичном способе получения тонких частиц с использованием способа распылительной сушки материал, такой как ингредиент, который предназначен для образования массы частиц, растворяют в подходящем растворителе с образованием раствора. Альтернативно материал, предназначенный для распылительной сушки, можно суспендировать в нерастворителе с образованием суспензии или эмульсии. На данной стадии необязательно добавляют другие компоненты, такие как лекарственные препараты, фармацевтически приемлемые наполнители или порообразующие агенты. Затем раствор разбрызгивают с образованием тонкого капельного аэрозоля. Капли немедленно проходят в сушильную камеру, в которой они контактируют с сушильным газом. Раствор упаривают из капель в сушильный газ для затвердевания капель с образованием, таким образом, частиц. Затем данные частицы отделяют от сушильного газа и собирают.
При масштабировании данного способа распылительной сушки, например, с масштаба лабораторной или пилотной установки до масштаба промышленной установки, можно столкнуться с рядом проблем. Если скорость сушки и объем сушки не оптимизированы адекватным образом, можно встретиться с нежелательными проблемами, такими как неправильная сушка частиц растворителя, пониженный выход продукта, чистота и т.п. С другой стороны, повышение скорости сушки, которое неадекватно масштабировано, может дать в результате неподходящую морфологию и/или распределение по размеру некоторых частиц продукта, таких как частицы, имеющие критически определенные технические характеристики. Более существенно, что оно может также изменить процесс осаждения материала, образующего твердое вещество, по мере упаривания растворителя, изменяя тем самым структуру (например, пористость) частицы так, что она выходит за границы стандартов технических условий, делая частицу неспособной включать и доставлять надлежащим образом диагностический или лечебный фактор.
В связи с этим в области техники существует необходимость в усовершенствованном способе распылительной сушки, который приводит в результате к получению однородных твердых аморфных суспензий с высокой частотой при улучшенных характеристиках сыпучести, повышенной однородности содержания и повышенной эффективности сбора.
Одним из объектов настоящего изобретения является получение высушенных распылительной сушкой композиций комплексов катион-конъюгат инсулина, включающее способ сушки, который предусматривает улучшенную сушку частиц без вредного воздействия на чистоту, выход и стабильность продукта. Настоящий способ дает однородные высушенные распылительной сушкой твердые частицы направленного лечебного фактора, которые далее готовят с другими необходимыми наполнителями с получением пероральных фармацевтических композиций комплексов катион-конъюгат инсулина.
Другие объекты и преимущества настоящего изобретения будут более очевидны для обычного специалиста в области техники в свете последующего описания и прилагаемой формулы изобретения.
Раскрытие изобретения
Главным объектом настоящего изобретения является разработка твердой фармацевтической композиции для перорального применения в виде комплекса катион-конъюгат инсулина, включающей насыщенные или ненасыщенные С4-С12 жирные кислоты, сложные эфиры жирных кислот или их соли и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, включающей связующие компоненты, разрыхлители, разбавители, смазывающие вещества, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы.
Другим объектом настоящего изобретения является разработка твердой фармацевтической композиции для перорального применения, причем пероральная лекарственная форма находится в форме таблетки, капсулы, частиц, порошка или саше либо сухих суспензий.
Еще одним объектом настоящего изобретения является разработка способа изготовления твердой фармацевтической композиции для перорального применения IN-105.
Еще одним объектом настоящего изобретения является разработка композиции таблетки IN-105.
Еще одним объектом настоящего изобретения является разработка дозы твердой фармацевтической композиции для перорального применения, предназначенной для достижения максимального контроля концентрации глюкозы в крови после приема пищи у больных диабетом в течение 5-60 минут после приема.
Еще одним объектом настоящего изобретения является разработка стабильной фармацевтической композиции для перорального применения IN-105, отличающейся тем, что указанная композиция остается стабильной.
Еще одним объектом настоящего изобретения является разработка способа получения аморфных высушенных распылительной сушкой частиц.
Еще одним объектом настоящего изобретения является разработка фармацевтической композиции IN-105, причем она проявляет пониженную в три раза митогенность по сравнению со своим нативным аналогом.
Еще одним объектом настоящего изобретения является разработка фармацевтической композиции, содержащей комплекс катион-конъюгат пептида, в котором наполнители не влияют на митогенную активность конъюгата.
Настоящее изобретение предусматривает твердую фармацевтическую композицию для перорального применения IN-105, содержащую приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы; фармацевтическую композицию для перорального применения, причем пероральная лекарственная форма находится в виде таблетки, капсулы, частицы, порошка или саше либо сухих суспензий; способ изготовления твердой фармацевтической композиции для перорального применения IN-105, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы, включающий стадии а) перемалывания подходящих насыщенных или ненасыщенных С4-С12 жирных кислот и/или солей данных жирных кислот, b) грануляции результирующей жирной кислоты, полученной на стадии (а), с использованием органического растворителя, с) сушки на воздухе гранул, полученных на стадии (b), d) протирания высушенных гранул через сито для получения гранул с требуемым размером частиц, е) перемешивания гранул жирных кислот с комплексом катион-конъюгат инсулина с другими наполнителями, f) прессования перемешанной смеси для формирования таблеток; способ изготовления твердой фармацевтической композиции для перорального применения IN-105, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы, включающий стадии а) перемалывания подходящих насыщенных или ненасыщенных С4-С12 жирных кислот и/или солей данных жирных кислот и связующего компонента, b) суспендирования комплекса катион-конъюгат инсулина в органическом растворителе при использовании связующего компонента с целью формирования мокрой массы, с) грануляции компонентов, полученных на стадии (b) с использованием связующего компонента, d) перетирания высушенных гранул, полученных на стадии (с), е) смешивания гранул с другими наполнителями, f) прессования перемешанной смеси для формирования таблеток; причем способ, в котором используемый органический растворитель выбран из группы, включающей изопропанол, ацетон, метиловый спирт, метилизобутилкетон, хлороформ, 1-пропанол, 2-пропанол, ацетонитрил, 1-бутанол, 2-бутанол, этиловый спирт, циклогексан, диоксан, этилацетат, диметилформамид, дихлорэтан, гексан, изооктан, метиленхлорид, трет-бутиловый спирт, толуол, четыреххлористый углерод или их комбинации; таблетку массой 5-500 мг твердой фармацевтической композиции для перорального применения IN-105, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы; таблетку массой 50 мг твердой фармацевтической композиции для перорального применения IN-105, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы; таблетку массой 100 мг твердой фармацевтической композиции для перорального применения IN-105, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы; таблетку массой 150 мг твердой фармацевтической композиции для перорального применения IN-105, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10-60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы; таблетка массой 200 мг твердой фармацевтической композиции для перорального применения IN-105, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы; таблетку массой 250 мг твердой фармацевтической композиции для перорального применения IN-105, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы; дозу принимаемой перорально твердой фармацевтической композиции, предназначенную для достижения максимального контроля концентрации глюкозы в крови после приема пищи у больных диабетом в течение 5-60 минут после введения; стабильную принимаемую перорально твердую фармацевтическую композицию IN-105, содержащую приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающую связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы, отличающуюся тем, что указанная композиция остается стабильной после воздействия условий, выбранных из группы, включающей (а) интервал температур приблизительно 2÷40°С, (b) относительную влажность 25±2°С/60±5%, относительную влажность 30±2°С/65±5%, относительную влажность 40±2°С/75±5% в период по меньшей мере 6 месяцев; способ получения аморфных высушенных распылительной сушкой частиц, содержащих приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы, причем указанный способ включает стадии а) получения раствора или суспензии, включающих катион-конъюгат соединения инсулина и компонент жирной кислоты в растворителе, b) разбрызгивания раствора в камере в условиях, которые позволяют удалить существенное количество растворителя, с) представления полученных распылительной сушкой частиц катион-конъюгата соединения инсулина; инсулинотропную фармацевтическую композицию IN-105, содержащую приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы, отличающуюся тем, что она проявляет пониженную в три раза митогенность по сравнению со своим нативным аналогом; и фармацевтическую композицию, содержащую приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающую связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы, причем указанная композиция, отличается тем, что наполнители не влияют на митогенную активность конъюгата.
Настоящее изобретение относится к твердой фармацевтической композиции для перорального применения IN-105, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы.
В другом варианте осуществления настоящего изобретения компонент жирной кислоты представляет каприновую кислоту и/или лауриновую кислоту или их соли.
В еще одном варианте осуществления настоящего изобретения жирная кислота представляет собой капрат натрия.
В еще одном варианте осуществления настоящего изобретения связующий компонент выбран из группы, включающей поливинилпирролидон, карбоксиметилцеллюлозу, метилцеллюлозу, крахмал, желатин, сахара, природные и синтетические камеди или их комбинации.
В еще одном варианте осуществления настоящего изобретения связующий компонент представляет собой поливинилпирролидон.
В еще одном варианте осуществления настоящего изобретения разбавители выбраны из группы, включающей соли кальция, целлюлозу или производные целлюлозы, палатинозу, органические кислоты, сахар и сахароспирты, соли пектиновой кислоты или их комбинации.
В еще одном варианте осуществления настоящего изобретения разбавитель представляет собой маннит.
В еще одном варианте осуществления настоящего изобретения разрыхлитель выбран из группы, включающей поперечно сшитый поливинилпирролидон, карбоксиметилцеллюлозу, метилцеллюлозу, катионобменные смолы, альгиновую кислоту, гуаровую камедь или их комбинации.
В еще одном варианте осуществления настоящего изобретения смазывающее вещество выбрано из группы, включающей стеарат магния, стеарат натрия, бензоат натрия, ацетат натрия, фумаровую кислоту, полиэтиленгликоли, аланин и глицин.
В еще одном варианте осуществления настоящего изобретения смазывающее вещество представляет собой стеарат магния.
В еще одном варианте осуществления настоящего изобретения агент, усиливающий проницаемость, выбран из группы, включающей лаурилсульфат натрия, лаурат натрия, пальмитоилкарнитин, фосфатидилхолин, циклодекстрин и его производные, карнитин и их производные, мукоадгезивные полимеры, ZOT-токсин (токсин zonula occludens - "плотных контактов"), желчные кислоты, жирные кислоты или их комбинации.
В еще одном варианте осуществления настоящего изобретения, агент, усиливающий проницаемость, представляет собой лаурилсульфат натрия.
В еще одном варианте осуществления настоящего изобретения агент, усиливающий проницаемость, представляет собой β-циклодекстрин.
В еще одном варианте осуществления настоящего изобретения пластификатор выбран из группы, включающей полиэтиленгликоль, пропиленгликоль, ацетилцитрат, триацетин, ацетилированный моноглицерид, рапсовое масло, оливковое масло, кунжутное масло, ацетилтриэтилцитрат, глицеринсорбитол, диэтилоксалат, диэтилмалат, диэтилфумарат, дибутилсукцинат, дибутилфталат, диоктилфталат, дибутилсебацинат, триэтилцитрат, трибутилцитрат, глицеролтрибутират, глицерилтриацетат или их смеси.
В еще одном варианте осуществления настоящего изобретения пластификатор представляет собой полиэтиленгликоль.
Настоящее изобретение относится также к фармацевтической композиции для перорального применения, причем пероральная лекарственная форма находится в форме таблетки, капсулы, частиц, порошка или саше либо сухих суспензий.
Настоящее изобретение относится также к способу изготовления твердой фармацевтической композиции в виде комплекса катион-конъюгат инсулина для перорального применения, содержащей насыщенные или ненасыщенные С4-С12 жирные кислоты или их соли и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, содержащей связующие компоненты, разрыхлители, разбавители, смазывающие вещества, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы.
Настоящее изобретение относится также к способу изготовления твердой фармацевтической композиции для перорального применения, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы, включающему стадии
а) перемалывания подходящих насыщенных или ненасыщенных С4-С12 жирных кислот и/или солей данной жирной кислоты;
b) грануляции результирующей жирной кислоты, полученной на стадии (а), с использованием органического растворителя;
с) сушки на воздухе гранул, полученных на стадии (B);
d) протирания высушенных гранул через сито для получения гранул с требуемым размером частиц;
е) перемешивания гранул жирных кислот с комплексом катион-конъюгат инсулина с другими наполнителями;
f) прессования перемешанной смеси для формирования таблеток.
Настоящее изобретение относится также к способу изготовления твердой фармацевтической композиции для перорального применения, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы, включающему стадии
a) перемалывания подходящих насыщенных или ненасыщенных С4-С12 жирных кислот и/или солей данных жирных кислот и связующего компонента;
b) суспендирования комплекса катион-конъюгат инсулина в органическом растворителе при использовании связующего компонента с целью формирования мокрой массы;
c) грануляции компонентов, полученных на стадии (B) с использованием связующего компонента;
d) перетирания высушенных гранул, полученных на стадии (с);
e) смешивания гранул с другими наполнителями;
f) прессования перемешанной смеси для формирования таблеток.
В еще одном варианте осуществления настоящего изобретения органический растворитель выбран из группы, включающей изопропанол, ацетон, метиловый спирт, метилизобутилкетон, хлороформ, 1-пропанол, 2-пропанол, ацетонитрил, 1-бутанол, 2-бутанол, этиловый спирт, циклогексан, диоксан, этилацетат, диметилформамид, дихлорэтан, гексан, изооктан, метиленхлорид, трет-бутиловый спирт, толуол, четыреххлористый углерод или их комбинации.
Настоящее изобретение относится также к таблетке массой 5-500 мг твердой фармацевтической композиции для перорального применения IN-105, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы.
Настоящее изобретение относится также к таблетке массой 50 мг твердой фармацевтической композиции для перорального применения IN-105, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10 - 60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы.
Настоящее изобретение относится также к таблетке массой 100 мг твердой фармацевтической композиции для перорального применения IN-105, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы.
Настоящее изобретение относится также к таблетке массой 150 мг твердой фармацевтической композиции для перорального применения IN-105, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы.
Настоящее изобретение относится также к таблетке массой 200 мг твердой фармацевтической композиции для перорального применения IN-105, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы.
Настоящее изобретение относится также к таблетке массой 250 мг твердой фармацевтической композиции для перорального применения IN-105, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы.
Настоящее изобретение относится также к дозе твердой фармацевтической композиции для перорального применения, предназначенной для достижения максимального контроля концентрации глюкозы в крови после приема пищи у больных диабетом в течение 5-60 минут после введения.
В другом варианте осуществления настоящего изобретения твердая фармацевтическая композиция для перорального применения дает пониженный по меньшей мере на 5% уровень глюкозы в сыворотке у больных людей в течение 120 минут после перорального приема.
Настоящее изобретение относится также к стабильной твердой фармацевтической композиции для перорального применения IN-105, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы, отличающейся тем, что указанная композиция остается стабильной при воздействии условий, выбранных из группы, включающей
(a) интервал температур приблизительно 2-40°С,
(b) относительную влажность 25±2°С/60±5%, относительную влажность 30±2°С/65±5%, относительную влажность 40±2°С/75±5% в период по меньшей мере 6 месяцев.
В другом варианте осуществления настоящего изобретения количество примесей не возрастает больше чем на 5% по сравнению с уровнями примесей во время изготовления.
В еще одном варианте осуществления настоящего изобретения количество примесей не возрастает больше чем на 10% по сравнению с уровнями примесей во время изготовления.
В еще одном варианте осуществления настоящего изобретения результат анализа указанного соединения в композиции не снижается больше чем на 10%.
В еще одном варианте осуществления настоящего изобретения профиль растворения составляет по меньшей мере 75% в любой указанный интервал времени.
В еще одном варианте осуществления настоящего изобретения интервал времени составляет период более 2 лет.
В еще одном варианте осуществления настоящего изобретения разница между распределением твердости составляет не больше чем 1 кг/см2 по сравнению с распределением твердости во время изготовления.
В еще одном варианте осуществления настоящего изобретения по меньшей мере 95±2% композиции остается неразложившейся после воздействия условий, выбранных из группы, содержащей
(a) интервал температур приблизительно 2÷8°С или 25÷40°С,
(b) относительную влажность 25±2°С/60±5%, относительную влажность 30±2°С/65±5%, относительную влажность 40±2°С/75±5% в период по меньшей мере 6 месяцев.
В еще одном варианте осуществления настоящего изобретения по меньшей мере 90±2% композиции остается неразложившейся после воздействия условий, выбранных из группы, содержащей
(a) интервал температур приблизительно 2÷8°С или 25÷40°С,
(b) относительную влажность 25±2°С/60±5%, относительную влажность 30±2°С/65±5%, относительную влажность 40±2°С/75±5% в период по меньшей мере 6 месяцев.
Способ получения аморфных высушенных распылительной сушкой частиц, содержащих приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающих связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы, причем указанный способ включает стадии
а) получения раствора или суспензии, содержащих катион-конъюгат соединения инсулина и компонент жирной кислоты в растворителе;
и) распыления раствора в камере в условиях, которые дают возможность удаления существенного количества растворителя;
с) получения высушенных распылительной сушкой частиц катион-конъюгата соединения инсулина.
В еще одном варианте осуществления настоящего изобретения компонент жирной кислоты выбран из группы, включающей С4-С12 жирную кислоту и/или соли данной жирной кислоты.
В еще одном варианте осуществления настоящего изобретения компонент жирной кислоты представляет собой капрат натрия.
В еще одном варианте осуществления настоящего изобретения растворитель представляет собой воду.
В еще одном варианте осуществления настоящего изобретения раствор или суспензия, содержащие IN-105 и компонент жирной кислоты в растворителе, дополнительно включают разбавитель.
В еще одном варианте осуществления настоящего изобретения разбавитель представляет собой маннит.
В еще одном варианте осуществления настоящего изобретения размер высушенных распылительной сушкой частиц составляет приблизительно 1-100 мкм.
В еще одном варианте осуществления настоящего изобретения раствор или суспензия, содержащие IN-105, высушены распылительной сушкой при температуре, лежащей в интервале 80÷150°С.
В еще одном варианте осуществления настоящего изобретения давление при распылении, используемое для распылительной сушки, лежит в интервале от 49 кПа (0,5 кг/см2) до 147,1 кПа (1,5 кг/см2).
В еще одном варианте осуществления настоящего изобретения чистота высушенной распылительной сушкой композиции IN-105 составляет по меньшей мере 95%.
В еще одном варианте осуществления настоящего изобретения чистота высушенной распылительной сушкой композиции IN-105 составляет по меньшей мере 98%.
В еще одном варианте осуществления настоящего изобретения чистота высушенной распылительной сушкой композиции IN-105 составляет по меньшей мере 99%.
Настоящее изобретение относится к высушенной распылительной сушкой композиции IN-105.
Настоящее изобретение относится также к аморфной форме IN-105. Настоящее изобретение относится также к инсулинотропной фармацевтической композиции IN-105, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы, отличающейся тем, что она проявляет пониженную в три раза митогенность по сравнению со своим нативным аналогом.
В еще одном варианте осуществления настоящего изобретения инсулинотропная фармацевтическая композиция IN 105 снижает пролиферацию клеток in vitro и/или in vivo до по меньшей мере 20±5% от активности своего нативного аналога.
В еще одном варианте осуществления настоящего изобретения инсулинотропная фармацевтическая композиция IN105 снижает пролиферацию клеток in vitro и/или in vivo до минимум 2±0,5% от активности своего нативного аналога.
В еще одном варианте осуществления настоящего изобретения композиция проявляет метаболическую эффективность, близкую эффективности своего нативного аналога.
Настоящее изобретение относится также к фармацевтической композиции, содержащей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере три фармацевтически приемлемых наполнителя, выбранных из группы, состоящей из по меньшей мере 10 мас.% разрыхлителя, 10 мас.% разбавителя, 0,5 мас.% смазывающего вещества, необязательно включающей связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы, причем указанная композиция отличается тем, что наполнители не влияют на митогенную активность конъюгата.
Главной целью изобретения является получение препарата с немедленным высвобождением, включающего белки/пептиды, их конъюгаты и/или комплексы катион-конъюгат полипептида, демонстрирующего требуемые фармакокинетические профили и активности на эффективных моделях диабета у человека при пероральной доставке. В современном состоянии области техники признают нестабильность немодифицированного инсулина в желудочно-кишечном тракте и предпринимают попытки преодолеть данные проблемы путем создания рационально разработанного препарата для немедленного высвобождения.
Комплексы катион-конъюгат пептида, комплексы катион-конъюгат инсулина, соответствующие настоящему изобретению, характеризуются тем, что демонстрируют пониженные митогенные активности. Конъюгаты, описанные в настоящем изобретении, перспективны вследствие того факта, что они несут относительно меньше или не несут риска индукции митогенных реакций даже после длительного применения, и, следовательно, их можно безопасно использовать в долговременном лечении и ведении диабета. Терапевтические соединения, соответствующие настоящему изобретению, характеризуются тем, что демонстрируют уменьшенную в три раза митогенную активность по сравнению с Insugen®.
Согласно одному из существенных аспектов настоящего изобретения наполнители в лекарственном продукте не влияют на митогенные характеристики лекарственной субстанции, и, следовательно, препарат или композиция сохраняют относительно меньшую митогенную активность по сравнению с имеющимся в настоящее время в продаже лекарственными препаратами инсулина.
Другим объектом настоящего изобретения является получение препарата с немедленным высвобождением, включающего белки/пептиды, их конъюгаты и/или комплексы катион-конъюгат полипептида, проявляющие стабильность в области температур в интервалах времени, продолжающихся до 12 месяцев. Разработанный в настоящее время препарат стабилен при комнатной температуре и, кроме того, проявляет стабильность при повышенных температурах.
Согласно одному аспекту изобретения фармацевтическую композицию, изготовленную таким образом, оценивают в различных тест-условиях температур и относительно интервала времени, продолжающегося до 12 месяцев.
Предпочтительный вариант осуществления настоящего изобретения относится к препарату, доставляемому пероральным путем, содержащему 0,01÷20 мас.% инсулина, конъюгатов соединения инсулина и/или катион-конъюгатов инсулина, 10÷60 мас.% одного или более компонентов жирных кислот, выбранных из насыщенных или ненасыщенных С4-С12 жирных кислот и/или солей данных жирных кислот, 10÷60 мас.% разбавителя, 1÷20% разрыхлителя, 0,01÷5% связующего компонента и 0,01÷5% адсорбента, включая, но без ограничения перечисленным, другие подходящие фармацевтически приемлемые носители.
Соответственно, один аспект изобретения относится к препарату, содержащему по меньшей мере одно биологически активное соединение, выбранное из дериватизированных конъюгатов инсулина, аналогов инсулина, комплексов инсулина, включая, но без ограничения перечисленным, другие лечебные факторы с инсулиноподобной активностью.
В еще одном аспекте изобретение предусматривает способ изготовления вышеупомянутых препаратов для пероральной доставки в оптимальных рабочих условиях с подходящими наполнителями, которые повышают устойчивость терапевтически активного фактора к разложению.
В еще одном предпочтительном аспекте способ изготовления таблетки для пероральной доставки, содержащей по меньшей мере одно биологически активное соединение, выбранное из белков/пептидов, их конъюгатов и/или комплексов катион-конъюгат полипептида, обладающих устойчивостью к ферментному разложению, включает стадии:
1. Перемалывания подходящей насыщенной или ненасыщенной С4-С12 жирных кислот и/или солей данной жирной кислоты.
2. Грануляции результирующей жирной кислоты, полученной на стадии (1), с использованием органического растворителя.
3. Сушки на воздухе гранул, полученных на стадии (2).
4. Протирания высушенных гранул через сито для получения гранул с требуемым размером частиц приблизительно 250 мкм.
5. Перемешивания гранул жирных кислот, катион-конъюгат инсулинового соединения с другими наполнителями.
6. Прессования перемешанной смеси для формирования таблеток.
В соответствии с вышеуказанными объектами и другими изобретение направлено отчасти на пероральную твердую лекарственную форму, включающую дозу модифицированного инсулина, которая достигает максимального контроля концентрации глюкозы в крови после приема пищи у больных диабетом в течение 20-30 минут после введения.
Компетентные специалисты легко поймут, что уровни дозы могут варьировать как функция специфического соединения, тяжести симптомов и чувствительности пациента к побочным эффектам. Компетентный специалист в области техники легко определяет предпочтительные дозы для заданного соединения рядом способов. Предпочтительным способом является измерение физиологической активности заданного соединения. Унифицированная лекарственная форма может представлять собой жидкость или твердое вещество, такое как таблетка, капсула или частицы, включая порошок или саше.
Данное изобретение также относится к усовершенствованному способу распылительной сушки, приводящему в результате к образованию высушенных распылительной сушкой частиц размером 1-100 мк, содержащих лекарственный препарат с по меньшей мере одним из фармацевтически приемлемых наполнителей.
Способ включает (а) получение раствора, который содержит лекарственный препарат и, необязательно, разбавитель в концентрации от 0,001 до 20 мас.%, необязательно 10÷60 мас.% одного или более компонентов жирных кислот, выбранных из насыщенных или ненасыщенных С4-С12 жирных кислот и/или солей данных жирных кислот, (B) распылительную сушку полученной в результате суспензии с образованием высушенного распылительной сушкой порошка.
Следующий аспект изобретения относится к комбинированию другого фармацевтического наполнителя с высушенным распылительной сушкой порошком, который прессуют в форме таблетки.
Способы, соответствующие изобретению, могут предусматривать, например, возможность генерировать однородные твердые частицы с лечебным фактором, задавая условия, подходящие для получения повышенной эффективности упаривания при заданной подводимой теплоте.
Способ получения данного порошка частиц в изобретении может включать, например, получение водной суспензии или раствора биоактивного материала, образующих смесь раствора или суспензии, разбрызгивание ультрамелких капель путем сброса давления смеси и сушку капель с получением порошка частиц путем замены газов разбрызгивания сушильными газами, например разбрызгивания в сушильной камере аппарата для распылительной сушки.
Другие объекты и преимущества настоящего изобретения будут более очевидны обычным специалистам в области техники в свете последующего описания и прилагаемой формулы изобретения.
Изобретение далее предусматривает способы лечения диабета, нарушенной переносимости глюкозы, ранней стадии диабета и поздней стадии диабета у животных, предпочтительно у человека, и способ достижения гомеостаза глюкозы, предусматривающий введение одной или больше унифицированных доз лекарственных форм; препарат с немедленным высвобождением, содержащий белки/пептиды, их конъюгаты и/или комплексы катион-конъюгат полипептида с подходящими наполнителями, которые усиливают устойчивость терапевтически активного фактора к разложению.
Определения
В описании и составлении формулы настоящего изобретения будет использована следующая терминология в соответствии с определениями, приведенными в данном контексте.
Термин "катион-конъюгат соединения инсулина" включает компонент любого соединения инсулина. Соединение инсулина может, например, представлять собой соединение инсулина млекопитающего, такого как человеческий инсулин, или производные либо аналоги соединения инсулина.
Термин лечебный фактор специально относится к молекуле IN-105. IN-105 представляет собой молекулу инсулина, конъюгированную по ∈-аминокислоте лизину в положении В29 В-цепи инсулина с амфифильным олигомером структурной формулы CH3O-(С4H2O)3-CH2-CH2-COOH. Молекула может быть моноконъюгирована по А1, В1 и В29, диконъюгирована по различным комбинациям А1, В1 и В29 или триконъюгирована по различным комбинациям А1, В1 и В29.
Термин "терапевтически эффективное количество" относится к количеству инсулина, включенному в лекарственные формы, соответствующие изобретению, которое достаточно для эффективного достижения клинически адекватного контроля концентраций глюкозы в крови у больных диабетом либо в состоянии голодания, либо в состоянии после приема пищи во время интервала дозирования.
Как используют в данном контексте, "разбавители"/"заполнители"/ "наполнители" представляют собой инертные субстанции, добавляемые для увеличения массы препарата, чтобы сделать таблетку удобного для прессования размера. Обычно используемые разбавители среди тех, которые предполагают использовать в настоящем изобретении, включают сахароспирты, органические кислоты, галеновы соединения, палатинозу, целлюлозу и производные целлюлозы, фосфат кальция, сульфат кальция, лактозу, каолин, манит, хлорид натрия, сухой крахмал, порошковый сахар, оксид кремния и т.п.
Как используют в данном контексте, "связующие компоненты" представляют собой компоненты, используемые для придания связующих свойств порошковому материалу. Связующие компоненты или "грануляторы", как их иногда называют, придают связующие свойства составу таблеток, который обеспечивает то, что таблетка остается интактной после прессования, а также улучшают свойства свободной сыпучести препарата гранул требуемой твердости и размера. Материалы, обычно используемые в качестве связующих компонентов, включают карбоксиметилцеллюлозу, метилцеллюлозу, поливинилпирролидон, крахмал; желатин; сахара, такие как сахароза, глюкоза, декстроза, мелассы и лактоза; природные и синтетические камеди, такие как акация, альгинат натрия, экстракт ирландского мха, камедь панвар, камедь гатти, растительный клей из шелухи, вигум, микрокристаллическую целлюлозу, микрокристаллическую декстрозу, амилозу и арабогалактан лиственницы и т.п. Поливинилпирролидон используют в контексте настоящего изобретения.
Как используют в данном контексте, "дезинтегрирующие компоненты" или "разрыхлители" представляют собой субстанции, которые способствуют распаду или способствуют дезинтеграции таблеток после приема. Материалы, служащие разрыхлителями, по химическим признакам разделены на крахмалы, глины, целлюлозы, альгины или камеди. Другие дезинтегрирующие компоненты включают вигум HV, метилцеллюлозу, агар, бентонит, целлюлозу и продукты древесины, природную губку, катионообменные смолы, альгиновую кислоту, гуаровую камедь, мякоть цитрусовых, поперечно-сшитый поливинилпирролидон, карбоксиметилцеллюлозу и т.п.
"Смазывающее вещество" может быть выбрано из группы, включающей стеарат магния, стеарат натрия, бензоат натрия, ацетат натрия, фумаровую кислоту, полиэтиленгликоли (ПЭГ) с молекулярной массой выше 4000, аланин и глицин. Предпочтительным смазывающим веществом в контексте настоящего изобретения являются стеарат магния или стеарат натрия.
Термин "стеарат магния" означает соединение магния со смесью твердых органических кислот, полученных из жиров и в основном состоящих из различных соотношений стеарата магния и пальмитата магния. Его используют как необходимый фармацевтический элемент (смазывающее вещество) при изготовлении прессованных таблеток.
Термин "агенты, усиливающие проницаемость", означает любое соединение, которое повышает проницаемость мембраны и облегчает транспорт лекарственного препарата через биологическую мембрану, повышая таким образом биодоступность доставляемого лечебного фактора. Подходящие агенты, усиливающие проницаемость мембраны, включают поверхностно-активные вещества, такие как лаурилсульфат натрия, лаурат натрия, пальмитоилкарнитин, Лаурет-9, фосфатидилхолин, циклодекстрин и его производные, желчные кислоты, такие как гликохолат натрия, дезоксихолат натрия, таурохолат натрия и фузидат натрия, хелатирующие агенты, включая ЭДТА (этилендиаминтетрауксусную кислоту), лимонную кислоту и салицилаты, и жирные кислоты (например, олеиновую кислоту, лауриновую кислоту, ацилкарнитины, моно- и диглицериды), L-карнитин и производные, мукоадгезивные полимеры, ZOT-токсин или их комбинации.
"Пластификаторы" выбраны из группы, состоящей из ацетил цитрата, триацетина, ацетилированного моноглицерида, рапсового масла, оливкового масла, кунжутного масла, ацетилтриэтилцитрата, глицеринсорбита, диэтилоксалата, диэтилмалата, диэтилфумарата, дибутилсукцината, дибутилфталата, диоктилфталата, дибутилсебацината, триэтилцитрата, трибутилцитрата, глицеролтрибутирата, глицерилтриацетата, полиэтиленгликоля, пропиленгликоля и их смесей. Наиболее предпочтительно, когда пластификатор представляет собой полиэтиленгликоль. Пластификатор может составлять до 40 мас.% пленкообразующего полимера. Репрезентативным пластификатором, используемым в контексте настоящего изобретения, является ПЭГ.
Термин "полиалкиленгликоль" или "ПАГ" относится к замещенным или незамещенным, неразветвленным или разветвленным полимерам полиалкиленгликоля, таким как полиэтиленгликоль (ПЭГ), полипропиленгликоль (ППГ) и полибутиленгликоль (ПБГ), и их комбинации (например, неразветвленные или разветвленные полимеры, включающие комбинации двух или более различных мономеров ПАГ, таких как два или более различных мономеров ПАГ, выбранных из мономеров ПЭГ, ППГ и ПБГ), и включает моноалкилэфир полиалкиленгликоля. Термин мономер ПАГ означает одно звено ПАГ, например, термин "мономер ПЭГ" относится к одному звену полиэтиленгликоля, например -(СН2СН2О)-, "мономер ППГ" означает одно звено полипропиленгликоля, например -(CH2CH2CH2O)-, и "мономер ПБГ" означает одно звено полипропиленгликоля, например -(CH2CH2CH2CH2O)-. ПАГ и/или мономеры ПАГ включают также замещенные ПАГ или мономеры ПАГ, например, ПАГ, включающие алкильные боковые цепи, такие как метиловая, этиловая или пропиловая боковые цепи, или карбониловые боковые цепи, а также ПАГ, включающие один или более разветвленных мономеров ПАГ, такие как изо-ППГ или изо-ПБГ.
"Солюбилизаторы" могут представлять собой любую субстанцию, которая повышает водорастворимость лекарственного препарата.
Как используют в данном контексте, термин "фармацевтически приемлемый носитель" включает один или несколько агентов, выбранных из группы, состоящей из углеводов и модифицированных углеводов и их производных, полиэтилена и/или полипропиленгликоля и его производных, неорганических наполнителей или смазывающих веществ, жирных кислот и их сложных эфиров и солей, консервантов и покрывающих компонентов.
Как указывают в описании, "эффективное количество" означает количество любого агента, который является нетоксичным, но достаточным для получения требуемого местного или системного эффекта и действия при приемлемом соотношении благоприятный эффект/риск, сопровождающим любое лечение. Например, эффективное количество смазывающего вещества представляет собой количество, достаточное для того, чтобы оно действовало в плане смазывания композиции для целей таблетирования без получения каких-либо вредных эффектов.
В контексте настоящего изобретения термин "органические растворители" относятся к любому растворителю неводной природы, включая жидкие полимеры и их смеси. Органические растворители, подходящие для настоящего изобретения, включают ацетон, метиловый спирт, метилизобутилкетон, хлороформ, 1-пропанол, изопропанол, 2-пропанол, ацетонитрил, 1-бутанол, 2-бутанол, этиловый спирт, циклогексан, диоксан, этилацетат, диметилформамид, дихлорэтан, гексан, изооктан, метиленхлорид, трет-бутиловый спирт, толуол, четыреххлористый углерод или их комбинации.
"Анализ" может представлять собой лабораторный тест, предназначенный для обнаружения и измерения количества специфической субстанции.
"Примеси" можно определить как компоненты, не являющиеся частью нативного препарата, которые приводят к ухудшению качества или состояния композиции, в основном приписываемому одному или комбинации любой из следующих характеристик:
а) заражение или загрязнение;
b) отсутствие стабильности или однородности; примесь;
с) что-либо, которое делает что-либо другое загрязненным; второстепенный компонент или добавка.
"Тест на растворение" проводят для определения соответствия с требованиями к растворению, как указано в индивидуальном описании для лекарственной формы в виде таблетки или капсулы. Растворение таблетки является стандартизованным способом измерения скорости высвобождения лекарственного препарата из лекарственной формы. Основную функцию теста на растворение можно кратко выразить с помощью таких характерных свойств, как а) оптимизация терапевтической эффективности при создании продукта и оценка стабильности, b) рутинная оценка качества продукта для обеспечения однородности партий продукта, с) оценка "биоэквивалентности", другими словами, получение одной и той же биологической активности для отдельных партий продукта, полученных от одного или разных производителей.
Растворение можно осуществить, растворяя таблетку в оптимальном объеме среды при приблизительно 37,0±0,5°С и при частоте вращения приблизительно 50 об/мин. Растворение измеряют в разные интервалы времени, начиная с 15 минут. Различные среды, которые можно использовать для проведения тестов на растворение, включают воду, буфер и кислую среду с рН, лежащим в интервале 1-9, более предпочтительно в интервале 2-7.
"Твердость", как правило, измеряют как силу, необходимую для того, чтобы разрушить таблетку в тесте поперечного сжатия. Тест состоит из помещения таблетки между двумя пятами до тех пор, пока таблетка не разрушится. Регистрируют прочность при раздавливании, которая приводит к разрушению таблетки. "Твердость" иногда обозначают как прочность таблетки при раздавливании. Ряд инструментов, используемых при измерении твердости таблетки, включает тестер Стоукса (Monsanto), тестер твердости Кобба, тестер Пфайзера, тестер Эрвека, тестер Хеберлейна (или Шлеунигера), ключевой тестер и тестер Ван дер Кампа. Единицей измерения твердости является кг/см2.
"Распад" определяют как то состояние, при котором любой остаток элемента за исключением фрагментов нерастворимого покрытия или оболочки капсулы, остающихся на экране тест-устройства, представляет собой мягкую массу, не имеющую явного твердого ядра.
Термин "распылительная сушка" используют традиционно, и он в широком смысле относится к способам, включающим разделение жидких смесей на маленькие капли (разбрызгивание) и быстрое удаление растворителя из смеси в аппарате для распылительной сушки, где существует высокая движущая сила, направленная на упаривание растворителя из капель. Способы распылительной сушки и оборудование для распылительной сушки описаны в общем виде в справочнике Perry's Chemical Engineers' Handbook (Справочник для инженеров-химиков Перри), стр. 20-54-20-57 (6 изд., 1984). Более детально способы и оборудование для распылительной сушки описаны в обзоре Marshall, "Atomization and Spray-Drying" (Разбрызгивание и распылительная сушка) 50 Chem. Eng. Prog. Monogram., серия 2 (1954) и монографии Masters, Spray Drying Handbook (Руководство по распылительной сушке) (4 изд., 1985).
Как используют в данном контексте, термин "сушка" относится к каплям или частицам, образованным в результате удаления растворителя из капли или частицы.
Как используют в данном контексте, термин "частица" включает микро-, субмикро- и макрочастицы. Как правило, диаметр или самое длинное измерение частиц составляет приблизительно от 100 нм до 5 мм. Частицы могут представлять собой сферы, капсулы, иметь неправильные формы, представлять собой кристаллы, порошки, агломераты или агрегаты.
Изобретение предусматривает использование покрывающих компонентов, которые могут включать как нефункциональные (инста-покрытие, kolli-покрытие IR), так и энтеросолюбильные покрывающие компоненты, такие как полимеры на основе целлюлозы, пленкообразующие агенты или полиметилакрилат и другие покрывающие компоненты, известные обычному специалисту в области техники.
Настоящее изобретение относится в широком композиционном аспекте к препаратам, включающим ковалентно конъюгированные комплексы лечебных факторов, в которых лечебный фактор ковалентно связан с одной или более молекул полимера, включающего в качестве существенной части указанного полимера гидрофобную группу, например группу полиалкиленгликоля, и липофильную группу, например группу жирной кислоты. В одном предпочтительном аспекте лечебный фактор может быть ковалентно конъюгирован посредством ковалентного связывания с одной или более молекулами линейного полимера полиалкиленгликоля, в который в качестве существенной части инкорпорирована липофильная группа, например группа жирной кислоты.
Модифицированные конъюгаты инсулина, как используют в настоящем изобретении, создают путем присоединения низкомолекулярных полимеров или олигомеров, которые являются амфифильными по природе, включающими полиэтиленгликолевую часть, которая является гидрофильной, тогда как алкильная цепь является липофильной. Присоединение модифицирует растворимость лекарственной молекулы и стабилизирует белок или пептид в отношении ферментного разложения в желудочно-кишечном тракте. Конъюгированный лекарственный препарат более эффективно всасывается через стенку желудочно-кишечного тракта, чем лекарственный препарат в его нативном состоянии. При прохождении в кровяной поток связь между гидрофильной и гидрофобной цепью гидролизуется, оставляя высокоактивное соединение инсулин-ПЭГ циркулировать в крови, таким образом благоприятным образом изменяя фармакокинетику лекарственного препарата.
Настоящее изобретение конкретным образом относится к молекуле IN-105. IN-105 представляет собой молекулу инсулина, конъюгированную по ∈-аминокислоте лизину в положении В29 В-цепи инсулина с амфифильным олигомером структурной формулы CH3O-(C4H2O)3-CH2-CH2-COOH.
Что касается одного из наиболее важных аспектов настоящего изобретения, то комплексы катион-конъюгат пептида, комплексы катион-конъюгат инсулина, соответствующие настоящему изобретению, характеризуются тем, что демонстрируют пониженные митогенные активности. Конъюгаты, описанные в настоящем изобретении, имеют преимущество вследствие того факта, что имеют относительно более низкий риск или у них отсутствует риск вызывать митогенные реакции даже после длительного применения и, следовательно, они могут быть безопасно использованы для длительного лечения и ведения диабета. Лекарственные соединения, соответствующие настоящему изобретению, как считают, демонстрируют пониженную в три раза митогенную активность по сравнению с Insugen®.
Согласно одному важному аспекту настоящего изобретения наполнители в лекарственном продукте не влияют на митогенные характеристики лекарственной субстанции, и, следовательно, препарат или композиция сохраняют относительно более низкую митогенную активность по сравнению с имеющимися в настоящее время в продаже лекарственными препаратами инсулина.
Термин "нативные аналоги" относится к немодифицированному пептиду/белку, проявляющему основную биоактивность in vitro или in vivo, близкую активности модифицированного аналога, т.е. указанная молекула представляет собой пептид/полипептид до того, как он стал объектом какого-либо вида модификации.
Согласно стандартной терминологии термин "митогенный" определяет те субстанции, которые стимулируют деление клеток, которые в ином случае (т.е. без воздействия данной субстанции) делились бы очень ограниченно.
Изобретательское преимущество субстанции, представленной в настоящем изобретении, проявляется в том факте, что, согласно определенному опыту и экспериментам, эксперты считают, что инсулин сам по себе как субстанция является митогенным [см. статьи DeMeyts P; The structural basis of Insulin and Insulin-like growth factor-1 receptor binding and negative cooperativity and its relevance to Mitogenic versus metabolic signaling. (Структурная основа связывания инсулина и рецептора инсулиноподобного фактора роста-1 и отрицательная кооперативность и ее отношение к передаче митогенного сигнала относительно метаболического), Diabetologia 37: S135-S148, 1994, Ish-Shalom D, Christoffersen CT, Vorwerk P, Sacerdoti-Sierra N, Shymko RM, Naor D and De Meyts P; Mitogenic properties of Insulin and Insulin analogues mediated by insulin receptor (Митогенные свойства инсулина и аналогов инсулина, опосредованные инсулиновым рецептором), Diabetologia 40: S25-S31], и белок/пептиды, их конъюгаты и/или комплексы катион-конъюгат полипептида, соответствующие настоящему изобретению, проявляют пониженную в три раза митогенную активность, определяемую по индукции in-vitro измеряемой пролиферации клеток Balb 3T3-A31.
Согласно одному аспекту настоящего изобретения считают, что иллюстративные соединения, соответствующие данному изобретению, демонстрируют "пониженную митогенность" по сравнению с Insugen®, если они индуцируют определяемо более низкие уровни пролиферации клеток, как измеряют по поглощению клетками красителя аламар синий. Компетентные специалисты в области техники будут иметь в виду, что можно использовать другие подходящие способы и что изобретение ни в коей мере не ограничено специфическим анализом пролиферации.
Согласно одному аспекту настоящего изобретения считают, что иллюстративные соединения, соответствующие данному изобретению, являются в существенной степени "немитогенными" по сравнению с Insugen®, если они индуцируют неопределяемо более низкие уровни пролиферации клеток, как измеряют по поглощению клетками красителя аламар синий. Компетентные специалисты в области техники будут иметь в виду, что можно использовать другие подходящие способы и что изобретение ни в коей мере не ограничено специфическим анализом пролиферации.
Колориметрические биоанализы in vitro используют для количественной оценки митогенной активности [см. статью Okajima Т., Nakamura К., Zhang Н., Ling N., Tanabe Т., Yasuda Т. и Rosenfeld R. G. Sensitive Colorimetric Bioassays for Insulin-like Growth Factor (IGF) Stimulation of Cell Proliferation and Glucose Consumption: Use in Studies of IGF Analogs. (Чувствительные колориметрические биоанализы стимуляции инсулиноподобным фактором роста пролиферации клеток и потребления глюкозы, использование в исследованиях аналогов ИФР-1), Endocrinology, 1992, т.120: 2201-2210].
Биологические тесты часто анализируют с помощью метода параллельных линий. Логарифм доз откладывают на горизонтальной оси, тогда как соответствующие ответы представляют на вертикальной оси. Индивидуальные ответы на каждый вариант лечения обозначают синими треугольниками для стандартного препарата и зелеными квадратами для препарата образца.
С помощью модели с использованием параллельных линий тестируют статистическую достоверность следующего предположения:
1. Зависимость доза-ответ является линейной для стандартного препарата и препарата образца.
2. Кривая зависимости доза-ответ имеет значительный наклон.
3. Кривые зависимости доза-ответ стандартного препарата и препарата образца параллельны.
Способ параллельных линий имеет ряд преимуществ по сравнению с традиционным анализом в одной точке. На основании проверки вышеупомянутого предположения
1) линейную корреляцию доза-ответ не только предполагают, но также доказывают,
2) получают независимую от дозы относительную активность.
Воспроизводимость митогенных активностей анализируют при использовании подбора логистической кривой по 4 параметрам на линейной части кривой, состоящей из по меньшей мере четырех следующих друг за другом точек. Результаты показывают статистически пониженные в 3 раза митогенные активности IN105 по сравнению с Insugen®.
Что касается одного существенного аспекта изобретения, то митогенная активность молекулы IN-105 в порошковой форме и диализованном растворе одинаковы. IN-105 в три раза менее митогенный, по сравнению с Insugen®.
Что касается еще одного существенного аспекта изобретения, то митогенная активность молекулы HIM2 в тридцать раз меньше по сравнению с Insugen®.
Без исключения иллюстративные производные, например IN-105, соответствующие данному изобретению, снижают пролиферацию клеток in vitro до по меньшей мере 20%, или по меньшей мере 25%, или по меньшей мере 30%, по меньшей мере 35%, по меньшей мере 40% от уровня их нативных аналогов. Более предпочтительно до уровня по меньшей мере 30%.
Без исключения иллюстративные производные, например HIM-2, соответствующие данному изобретению, снижают пролиферацию клеток in vitro до по меньшей мере 2%, или по меньшей мере 2,5%, или по меньшей мере 3%, по меньшей мере 3,5% от уровня их нативных аналогов. Более предпочтительно до уровня по меньшей мере 3,8%, наиболее предпочтительно до по меньшей мере 4%.
Согласно еще одному аспекту настоящего изобретения оценку связывания инсулина со своим родственным рецептором проводят для сравнения относительных аффинностей связывания лекарственной субстанции формы Insugen®, IN-105 и HIM-2. Наиболее существенный аспект изобретения относится к тому факту, что метаболическая активность иллюстративных соединений, соответствующих настоящему изобретению, остается не поврежденной и неизмененной, несмотря на значительное снижение митогенной активности по сравнению с их нативными аналогами.
Эффект инсулина на клетки как метаболическое действие зависит от способности инсулина связываться с инсулиновым рецептором, и метаболический анализ, приведенный в качестве примера в настоящем изобретении, помогает определить эффект инсулина на поглощение глюкозы дифференцированными адипоцитами, представляя, таким образом, данные для оценки и сравнения метаболической эффективности иллюстративных лекарственных субстанций, соответствующих настоящему изобретению.
Экстракция лекарственной субстанции из лекарственного продукта
Insugen® и иллюстративные соединения, соответствующие настоящему изобретению, например IN 105 и HIM2, используют для получения не содержащей Zn и не содержащей наполнитель лекарственной субстанции. Флаконы осветляют с помощью ледяной уксусной кислоты (рН около 3,4). Осветленный раствор нагружают на С8-обращенно-фазовую колонку с силикагелем и разделяют, используя градиент элюции с 250 мМ уксусной кислоты и 100% этанолом. Фракции собирают во время элюции и объединяют на основе чистоты, превышающей или равной 99%. Данную группу, полученную при элюции, диализуют в течение 15 часов, используя мембрану с отсечением молекулярной массы 1 кД против 10 мМ Трис, рН 8,0. Наконец, диализованный не содержащий Zn и не содержащий наполнитель инсулин, полученный при рН 8,0, анализируют с помощью аналитической ОФ-ВЭЖХ (высокоэффективной жидкостной хроматографии с обращенной фазой).
Клетки HepG2 получают из АТСС (Американской коллекции типовых культур). Среду Дульбекко в модификации Игла (DMEM), инактивированную нагреванием фетальную сыворотку коров (ФСК), 100Х раствор пенициллина-стрептомицина и 100Х раствор солей HEPES приобретают в фирме Invitrogen. Бычий сывороточный альбумин, гидроксид натрия, Тритон-Х 100 и бикарбонат натрия получают в фирме Sigma Aldrich.
Рекомбинантный человеческий инсулин с радиоактивной меткой приобретают в фирме Immunotech (Beckman Coulter) со специфической радиоактивностью 2200 Ки/мМ (No. По каталогу А36474).
Способы:
Клетки HepG2 поддерживают в забуференной 10 мМ HEPES среде DMEM с добавлением 10% ФСК и 1Х раствора пенициллина-стрептомицина в условиях увлажнения при 37°С в атмосфере 5% СO2. Для анализа клетки HepG2 трипсинизируют и высевают с густотой 400000 клеток на лунку 24-луночного планшета. Через 3 дня инкубирования клетки используют для проведения анализов связывания радиолиганда.
Перед проведением анализа среды удаляют и клетки дважды промывают буфером для связывания (DMEM, 2,2 мг/мл бикарбоната натрия, 1 мг/мл бычьего сывороточного альбумина и 50 мМ HEPES) для удаления каких-либо следов факторов роста, присутствующих в среде. Эксперименты по конкурентному связыванию проводят в повторностях с использованием фиксированного количества радиолиганда (0,325 нМ) и варьирующих концентраций немеченой лекарственной субстанции инсулина (от 10-13 М до 10-5 М). Конечный объем реакции доводят до 1 мл. Затем планшеты инкубируют при 15°С в течение ночи на качалке, установленной на 60 об/мин. На следующий день все среды из лунок отбрасывают и каждую лунку промывают дважды ледяным буфером для связывания.
В каждую лунку вносят 1 мл солюбилизационного реагента (0,5 М гидроксида натрия, 0,5% Тритон-Х 100). Солюбилизированные клеточные пеллеты переносят в пробирку для радиоиммуноанализа и читают связанную радиоактивность в γ-счетчике (Stratec BioMedical Systems, Germany). Прибор калиброван и, как показано, имеет эффективность 80%.
Расчеты аффинности связывания: для нормализации числа импульсов в минуту (значений СРМ) при используемых различных концентрациях инсулина процент связывания рассчитывают, используя следующее уравнение:
% связывания=(СРМобразца-СРМбез вещества)/(СРМ контроля-СРМбез вещества)×100.
Когда СРМконтроля представляет собой среднее СРМ лунок, которые содержат клетки с радиоактивным инсулином без добавления какого-либо немеченого инсулина, СРМбез вещества представляет собой среднее СРМ лунок, которые не содержат инсулин с радиоактивной меткой, а также немеченый инсулин, и СРМобразца представляет собой среднее СРМ лунок, которые содержат инсулин с радиоактивной меткой, а также варьирующие концентрации инсулина без метки.
Анализ кривых конкурентного связывания
В экспериментах по конкурентному связыванию измеряют связывание одной концентрации меченого лиганда в присутствии различных концентраций немеченого лиганда. Эксперименты по конкурентному связыванию используют для определения количества и аффинности рецепторов при использовании одного и того же соединения в качестве меченого и немеченого лиганда.
Эксперимент выполняют с использованием одной концентрации радиолиганда. Инкубирование следует проводить до достижения равновесия, причем, как правило, варьирующие концентрации немеченого соединения охватывают приблизительно шесть порядков величины.
Верхняя часть кривой представляет собой плато при значении, равном значению связывания лиганда в отсутствие конкурирующего несвязанного лекарственного вещества. Оно представляет собой общее связывание. Нижняя часть кривой представляет собой плато, равное значению неспецифического связывания. Разница между значениями верхнего и нижнего плато представляет собой значение специфического связывания.
Ось Y можно выразить в СРМ (число импульсов в минуту) или превратить в более удобные единицы, например фмоль связанного вещества/миллиграмм белка или число связанных центров/клетку. Возможна нормализация данных от 100% (в отсутствие конкурирующего агента) до 0% (неспецифическое связывание при максимальных концентрациях конкурирующего агента).
Концентрацию немеченого лекарственного вещества, которая дает в результате равные расстояния значений связывания радиолиганда между верхним и нижним плато, называют IС50 (концентрация, ингибирующая 50%), называемую также ЕС50 (эффективная концентрация 50%). IС50 представляет собой концентрацию немеченого лекарственного вещества, которая блокирует половину специфического связывания.
Если меченый и немеченый лиганд конкурирует за один центр связывания, крутизну кривой конкурентного связывания определяют по закону действующих масс.
Нелинейную регрессию используют для построения кривой конкурентного связывания с целью определения Lоg(IС50). Как правило, используют пакет программ Graph pad Prism Software и с помощью уравнения модели одноцентровой конкуренции определяют Log(IC50).
Для определения наиболее соответствующего значения IС50 (концентрации немеченого лекарственного вещества, которая блокирует 50% специфического связывания радиолиганда) проблема нелинейной регрессии должна давать возможность определять 100% (общее) и 0% (неспецифическое) плато. При использовании данных, собранных для широкого круга концентраций немеченого лекарственного вещества, кривая будет ясно определять нижнее и верхнее плато, и программа не должна сталкиваться с затруднениями при подборе всех трех значений (оба плато и IC50).
Рационально созданная композиция биодоступна при немедленном высвобождении. В конъюгированном контактным образом лечебном факторе вышеописанного типа полимерный компонент может быть подходящим образом сконструирован, модифицирован или соответствующим образом функционализирован, чтобы придать способность к контактному конъюгированию избирательным образом.
В одном аспекте изобретение предусматривает композиции жирных кислот с одной или более насыщенными или ненасыщенными С4, С5, С6, С7, С8, С9 или С10 жирными кислотами и/или солями данных жирных кислот. Предпочтительными жирными кислотами являются каприловая, каприновая, миристиновая и лауриновая. Предпочтительными солями жирных кислот являются натриевые соли каприловой, каприновой, миристиновой и лауриновой кислоты.
Модифицированные группы могут включать другие гидрофильные полимеры. Примеры включают полиоксиэтилированные полиолы, такие как полиоксиэтилированный глицерин, полиоксиэтилированный сорбит и полиоксиэтилированную глюкозу; поливиниловый спирт; декстран; полимеры на углеводной основе и т.п. Полимеры могут представлять собой гомополимеры или случайные или блоксополимеры и тройные сополимеры на основе мономеров вышеуказанных полимеров, неразветвленную цепь или разветвленную.
Общее количество катион-конъюгата соединения инсулина, необходимое для использования, могут определить компетентные специалисты в области техники. Количество лечебного фактора представляет собой количество, эффективное для осуществления цели конкретного активного агента. Количество в композиции представляет собой терапевтически эффективную дозу, т.е. фармакологически или биологически эффективное количество. Однако количество может быть меньше, чем фармакологически или биологически эффективное количество, когда композицию используют в унифицированной лекарственной форме, такой как капсула, таблетка или жидкость, поскольку унифицированная лекарственная форма может содержать множество композиций доставляющего агента/биологически или химически активного компонента или может содержать разделенное фармакологически или биологически эффективное количество. Общие эффективные количества затем могут быть введены в кумулятивных формах, содержащих в целом фармакологически или биологически либо химически активные количества биологически или фармакологически активного агента.
В ряде предпочтительных вариантов осуществления фармацевтическая композиция, содержащаяся в одной или более лекарственных форм, включает от приблизительно 5 мг до приблизительно 800 мг доставляющего агента, предпочтительно от приблизительно 10 мг до приблизительно 600 мг, более предпочтительно от приблизительно 10 мг до приблизительно 400 мг, даже более предпочтительно от приблизительно 25 мг до приблизительно 200 мг, наиболее предпочтительно приблизительно 75 мг, 100 мг или 150 мг. Более предпочтительно, когда композиция дает пик концентрации инсулина в плазме в течение от приблизительно 15 минут до приблизительно 60 минут после перорального введения и более предпочтительно - в течение приблизительно 10-20 минут после перорального введения больным диабетом после приема пищи.
Для целей настоящего изобретения предполагают, что лекарственные формы, соответствующие настоящему изобретению, содержащие терапевтически эффективные количества инсулина, могут включать одну или более унифицированных доз (например, таблетки, капсулы, порошки, полутвердые формы, спреи для полости рта, подъязычные таблетки (например, гелевые капсулы или пленки) для достижения терапевтического эффекта. Кроме того, для целей настоящего изобретения предполагают, что предпочтительным вариантом осуществления лекарственной формы является пероральная лекарственная форма.
В ряде случаев комплексированный конъюгат соединения инсулина будет демонстрировать замедленный или иным образом измененный профиль рК относительно приемлемого с научной точки зрения контроля, такого как соответствующий некомплексированный конъюгат соединения инсулина. Профиль рК можно оценить с использованием стандарта в экспериментах in vivo, например, на мышах, крысах, собаках или человеке. Описанные в данном контексте анализы, предназначенные для оценки свойств комплексов катион-конъюгат соединения инсулина, представляют собой аспект изобретения.
В предпочтительном аспекте способ изготовления перорально доставляемой таблетки, содержащей по меньшей мере одно биологически активное соединение, выбранное из белков/пептидов, их конъюгатов и/или комплексов катион-конъюгат полипептида, обладающих устойчивостью к ферментному разложению, включает стадии:
1. Перемалывания подходящей насыщенной или ненасыщенной С4-С12 жирной кислоты и/или солей данной жирной кислоты.
2. Грануляции результирующей жирной кислоты, полученной на стадии (1), с использованием органического растворителя.
3. Сушки на воздухе гранул, полученных на стадии (2).
4. Протирания высушенных гранул через сито для получения гранул с требуемым размером частиц.
5. Перемешивания гранул жирных кислот с катион-конъюгатом инсулинового соединения, разрыхлителем, связующим компонентом и другими наполнителями.
6. Прессования, полировки и упаковки таблеток.
В еще одном предпочтительном аспекте способ изготовления перорально доставляемой таблетки, содержащей по меньшей мере одно биологически активное соединение, выбранное из белков/пептидов, их конъюгатов и/или комплексов катион-конъюгат полипептида, обладающих устойчивостью к ферментному разложению, включает стадии:
1. Перемалывания подходящей насыщенной или ненасыщенной С4-С12 жирной кислоты и/или солей данной жирной кислоты, таких как капрат натрия, поливинилпирролидон-К-30 (далее ПВП).
2. Суспендирования IN-105 в органическом растворителе с использованием ПВП-К-30 в качестве связующего компонента с образованием мокрой массы.
3. Грануляции результирующих компонентов с использованием ПВП-К-30 в качестве связующего компонента.
4. Протирания высушенных гранул капрата натрия через 45# (355 мкм).
5. Перемешивания гранул капрата натрия с другими наполнителями.
6. Прессования, полировки и упаковки таблеток.
Согласно наиболее существенным аспектам изобретения термин "стабильный", применительно к композиции, соответствующей настоящему изобретению, предназначен для целей изобретения для обозначения того, что препарат остается в неразрушенном виде или не разлагается до неприемлемой степени даже после воздействия на препарат ряда условий хранения, выбранных из группы, включающей (i) температуру приблизительно 2÷8°С, 25°С, 30°С и 40°С, (ii) относительную влажность (ОВ) 25°С/60%, относительную влажность 30°С/65%, относительную влажность 40°С/75% в течение по меньшей мере одного года, или любую комбинацию приведенных примеров условий. В контексте природы настоящего изобретения выражения остающийся в неразрушенном виде или не разлагающийся до неприемлемой степени будут означать, что компоненты дозы хорошо сохраняются в используемых рамках стабильности в течение периодов тестирования.
Согласно одному аспекту изобретения тестирование стабильности проводят в течение 1-12 месяцев, предпочтительно 1 месяца, предпочтительно 2 месяцев, предпочтительно 3 месяцев, предпочтительно 4 месяцев, предпочтительно 5 месяцев, предпочтительно 6 месяцев, предпочтительно 7 месяцев, предпочтительно 9 месяцев, предпочтительно 11 месяцев и наиболее предпочтительно 12 месяцев. Изобретение предполагает, что характеристики стабильности лекарственной формы остаются совместимыми в течение периода, доходящего до приблизительно 2 лет.
Характеристики стабильности фармацевтической композиции, соответствующей настоящему изобретению, оценивают в различных тестируемых условиях температуры, таких как условия пониженной температуры в интервале 2÷8°С, условия комнатной температуры (температуры окружающей среды) 25÷30°С и условия немного повышенной температуры 40°С. Как может быть очевидно компетентному специалисту в области техники, температурные условия и стабильности, установленные в указанных температурных условиях, могут не быть строго ограниченными температурными условиями, протестированными в данном контексте. Изобретение предполагает, что расширение характеристик стабильности, проявляемое в тест-условиях, будет установлено в рамках широкого круга пониженных и повышенных температур. Композиция и ее характеристики стабильности не подвергаются большим изменениям при температурах, лежащих в интервале 2÷40°С.
Характеристики стабильности фармацевтической композиции, соответствующей настоящему изобретению, оценивают в различных тестируемых условиях относительной влажности, таких как относительная влажность 25°С/60%, относительная влажность 30°С/65%, относительная влажность 40°С/75% или комбинация условий, иллюстрируемых в данном контексте. Как может быть очевидно компетентному специалисту в области техники, условия относительной влажности и стабильности, установленные в указанных температурных условиях, могут не быть строго ограниченными условиями относительной влажности, протестированными в данном контексте. Изобретение предполагает, что расширение характеристик стабильности, проявляемое в тест-условиях, будет установлено в рамках широкого круга температур относительной влажности, не раскрытых подробно в данном контексте. Композиция и ее характеристики стабильности не подвергаются большим изменениям при значениях относительной влажности, лежащих в интервале 25°С/60%÷40°С/75%.
Согласно одному аспекту изобретения композиция, соответствующая настоящему изобретению, характеризуется тем, что по меньшей мере 95±2% композиции остается неразложившейся при воздействии условий, выбранных из группы, содержащей
(a) интервал температур приблизительно 2÷8°С или 25÷40°С,
(b) относительную влажность 25°С/60%, относительную влажность 30°С/65%, относительную влажность 40°С/75% в период по меньшей мере 6 месяцев.
Согласно одному аспекту изобретения композиция, соответствующая настоящему изобретению, отличается тем, что по меньшей мере 95%±2% композиции остается неразложившейся при воздействии условий, выбранных из группы, содержащей
(a) интервал температур приблизительно 2÷8°С или 25÷40°С,
(b) относительную влажность 25°С/60%, относительную влажность 30°С/65%, относительную влажность 40°С/75% в период по меньшей мере 6 месяцев.
Согласно еще одному аспекту данного изобретения композицию таблеток тестируют в отношении различных характеристик стабильности, включающих, но без ограничения перечисленным, твердость, время распада, профиль растворения и хроматографическую чистоту.
Таким образом, согласно различным вариантам осуществления изобретение демонстрирует различные преимущества в плане дозирования и удобного способа применения. Изобретение содержит еще одно преимущество относительно композиций, соответствующих предшествующему уровню техники, поскольку авторы предполагают, что заявленный рационально разработанный пероральный препарат IN-105 с измеренными компонентами других наполнителей и способ изготовления таблеток для перорального применения легко масштабируются, не воздействуя на профиль высвобождения in vitro или in vivo. Кроме того, приписываемый данному рационально разработанному пероральному препарату и способу его получения фактор масштабируемости не влияет на действие лекарственного препарата in vivo или его профиль высвобождения in vivo.
Один из важных аспектов изобретения относится к распылительной сушке композиции для получения однородной аморфной смеси комплекса катион-конъюгат инсулина.
Ряд параметров можно оптимизировать с целью модификации среднего размера капель. На размер капель можно воздействовать, например, путем регуляции близкого к суперкритическому давления жидкости или давления газа высокого давления, регуляции давления суспензии или раствора, регуляции скорости потока суспензии или раствора, выбора внутреннего диаметра канала сопла, регуляции температуры сушильного газа, регуляции давления внутри емкости для образования частиц, изменения концентраций составляющих суспензии или раствора и т.п. Например, суспензию или раствор можно подавать в камеру для смешивания со скоростью от приблизительно 0,5 мл/мин до приблизительно 40 мл/мин для разбрызгивания из отверстия сопла с внутренним диаметром 100 мкм, причем более низкие скорости образуют более мелкие капли и более высокие скорости образуют более крупные капли. В способах образования капель предпочтительным является массовый средний диаметр, лежащий в интервале от приблизительно 1 мкм до приблизительно 200 мкм.
В настоящем изобретении температура, при которой эффективно проводят распылительную сушку, в норме находится в интервале от приблизительно 80°С до приблизительно 300°С, предпочтительно от приблизительно 100°С до приблизительно 180°С. Контроль температуры может быть особенно важным для поддержания стабильности частиц продукта, который образуется или содержит чувствительные к высокой температуре субстанции.
Фармацевтические композиции, полученные с использованием ряда вариантов осуществления изобретения, можно ввести любому животному, которое может испытывать благоприятные эффекты соединений, соответствующих изобретению. Главным среди данных животных является человек, хотя не предусматривают, что изобретение ограничено таким образом.
Способ получения частиц порошкового препарата в изобретении может включать, например, получение водной суспензии или раствора биоактивного материала и полиола необязательно с одним или более другими совместимыми фармацевтическими наполнителями, образование смеси раствора или суспензии с газом, находящимся под давлением, разбрызгивание ультратонких капель путем снижения давления в смеси и сушку капель с образованием частиц порошка посредством замены газов для разбрызгивания сушильными газами, например разбрызгиванием в сушильной камере устройства для распылительной сушки.
Термин "растворитель" относится к жидкости, в которой материал, образующий массу высушенных распылением частиц, растворяют, суспендируют или эмульгируют для доставки в распылитель устройства для распылительной сушки и которую упаривают в сушильном газе, независимо от того, растворяет ли или не растворяет жидкость материал. Выбор растворителя зависит от сыпучего материала и формы материала, подаваемого в разбрызгиватель, например, может ли материал растворяться, суспендироваться или эмульгироваться в растворителе. Наиболее предпочтительным растворителем в контексте настоящего изобретения является вода.
При использовании способа распылительной сушки неочищенный материал в форме раствора, содержащего по меньшей мере один из активных ингредиентов, выбранных из группы, состоящей из катион-конъюгат соединения инсулина, в комбинации с разбавителями, сушат распылительной сушкой с целью получения частиц.
Размер частиц раствора лекарственного препарата является функцией разбрызгивателя, используемого для разбрызгивания раствора полимера, давления разбрызгивателя, скорости потока, используемых ингредиентов, их концентрации, типа растворителя, вязкости, температуры разбрызгивания (температуры, как на входе, так и на выходе) и молекулярной массы лечебного фактора. Как правило, чем выше молекулярная масса, тем больше размер частиц, допуская, что концентрация одинакова.
Один из аспектов настоящего изобретения относится к сравнительному исследованию на фиксированных собаках двух прототипных составов пероральных композиций катион-конъюгатов соединения инсулина, полученных способом прямого прессования и способом распылительной сушки.
Проводят следующие измерения: скорость инфузии глюкозы, концентрация инсулина в плазме и уровни глюкозы в плазме (для получения возможности оценки высвобождения эндогенного соединения инсулина). Скорость инфузии глюкозы, необходимая для поддержания эугликемии, дает коэффициент действия соединения инсулина. Скорости инфузии глюкозы и уровни инсулина в плазме оценивают и сравнивают.
Прототипные препараты, полученные способом распылительной сушки, показывают согласующиеся уровни всасывания лекарственного препарата и результирующей скорости инфузии глюкозы без потери стабильности или биологической активности.
Прототипный препарат I (Препарат 862) имеет следующую композицию
Краткое описание чертежей:
Фиг.1: Профиль среднего уровня инсулина в плазме - таблетки препарата IN-105 (FDT-3).
Фиг.2: Нормализованный профиль глюкозы при введении FDT-3.
Фиг.3: Профиль среднего уровня инсулина в плазме - таблетки препарата IN-105 (FDT-19).
Фиг.4: Нормализованный профиль глюкозы при введении FDT-19.
Фиг.5: Профиль среднего уровня инсулина в плазме - таблетки препарата IN-105 (FDT-20).
Фиг.6: Профиль глюкозы после введения FDT-20.
Фиг.7: Профиль инсулина в плазме.
Фиг.8: Профиль скорости инфузии глюкозы.
Фиг.9: Профиль глюкозы в плазме.
Фиг.10: Параллелизм и линейность биоанализа при различных концентрациях Insugen® и IN 105, определенных с помощью пакета программ PLA. Данные представляют собой среднее ± стандартная ошибка (стандартная ошибка) по значениям, полученным в трех повторах эксперимента.
Фиг.11: Митогенная активность Insugen по сравнению с IN-105 и HIM-2 по данным анализа PLA при использовании 4 точек в линейном интервале.
Фиг.12: Параллелизм и линейность биоанализа при различных концентрациях Insugen® относительно IN 105 и Insugen® относительно HIM-2(B), определенных с помощью пакета программ PLA. Данные представляют собой среднее ± стандартная ошибка по значениям, полученным в трех повторах эксперимента.
Фиг.13: Митогенная активность диализованного IN-105 по сравнению с порошком IN-105 для анализа PLA при использовании 4 точек в линейном интервале.
Фиг.14: Сравнение метаболической активности Insugen®, IN 105 и HIM-2.
Фиг.15: Параллелизм и линейность биоанализа при различных концентрациях Insugen® относительно IN 105 и Insugen относительно HIM-2(B), определенных с помощью пакета программ PLA. Данные представляют собой среднее ± стандартная ошибка по значениям, полученным в трех повторах эксперимента.
Фиг.16: Кривые конкуренции в одной области (A) Insugen®, (В) IN 105 и (С) HIM-2 соответственно. Кривые определяют с помощью пакета программ Graph Pad версия 4. Данные представляют собой среднее по значениям, полученным в трех повторах эксперимента.
Осуществление изобретения
Настоящее изобретение далее определяют в следующих примерах. Следует иметь в виду, что данные примеры, хотя показывают предпочтительные варианты осуществления изобретения, приведены только в качестве иллюстрации. Из вышеприведенного обсуждения и данных примеров компетентный специалист в области техники может определить основные характеристики данного изобретения и, не выходя из его сущности и объема, может сделать различные изменения и модификации изобретения, чтобы адаптировать его к различным вариантам применения и условиям.
Изобретение будет более понятным из следующих примеров. Однако компетентные специалисты в области техники поймут, что данные примеры являются иллюстрацией изобретения, которое определяют в формуле изобретения, которая следует за ними. Следующие Примеры представляют предпочтительные варианты осуществления настоящего изобретения.
Пример 1
Получают и тестируют лекарственные препараты в соответствии с настоящим изобретением. Соответственно, требуемое количество измельченного капрата натрия точно отвешивают в планетарный смеситель и гранулируют с 700 мл изопропилового спирта. Рассчитывают количество изопропилового спирта, добавляемого для превращения порошковой смеси в гранулированную форму. Мокрую массу счищают каждые 5 минут, так что смесь не прилипает к стенке планетарного миксера. Мокрую массу пропускают через сито 18# в мокром грануляторе и сушат на воздухе в течение ночи в вытяжном шкафу.
Содержание влаги в гранулах: Масса образца=0,662 г; содержание влаги, %=2,27.
Взвешивают соответствующее количество IN-105, гранул капрата натрия, Коллидон CL и Перлитол и пропускают через сито 60# и смешивают в двухконусном блендере в течение 20 минут при скорости 12 об/мин. После однородного перемешивания в течение 20 минут смесь смазывают с помощью Аэросила и стеарата магния в течение 3 минут при скорости 88-90 об/мин.
Таблетки, полученные согласно примеру 1, тестируют на подвергнутых голоданию в течение 26 часов шести здоровых самцах собак породы бигль. Каждую таблетку вводят с 20 мл воды. Собирают образцы крови для измерения уровней глюкозы в крови и инсулина в плазме. Как представлено на Фиг. 1 и Фиг. 2, таблетка дает в результате быстрое повышение уровней инсулина в плазме, показывая значение Сmах приблизительно 100 мЕд/мл при Тmах 20 минут после времени введения, приводя в результате к соответствующему падению концентрации глюкозы в плазме приблизительно на 35%.
Полученные таким образом таблетки подвергают исследованиям стабильности согласно руководству ICH (Международная конференция по гармонизации). Длительные исследования проводят при 2÷8°С и 25°С, тогда как ускоренное исследование стабильности осуществляют при ОВ 30°С/65% и OВ 40°С/75%.
Пример 2
Измельченный капрат натрия точно взвешивают и вносят в планетарный смеситель и перемешивают в течение 2 минут. Взвешивают требуемые количества IN-105 и добавляют изопропанол с 0,35 мас.% поливинилпирролидона с образованием суспензии и хорошо перемешивают в течение 30 минут, используя магнитную мешалку. В процессе грануляции добавляют соответствующие количества изопропанола.
Гранулы перемешивают в течение 15 минут в планетарном смесителе и пропускают через сито 14# и сушат в канальной печи в течение 2 часов при 30°С.
Скорость планетарного смесителя=4 в течение 15 мин. Объем добавляемого ИПС (изопропилового спирта)=200 мл.
0,7 г (0,35%) ПВП+7,33 г IN-105 суспендируют в 80 мл ИПС. Оставшиеся 6,3 г ПВП суспендируют в 40 мл ИПС. Добавляют оставшееся количество ИПС для образования твердой мокрой массы.
Способ смешивания
Точно взвешенное количество гранул IN-105+Капрат натрия+ПВП К-30, Перлитола и Коллидона CL пропускают через сито 45# и смешивают в октагональном блендере. После однородного перемешивания смесь смазывают с помощью Аэросила и стеарата магния, хорошо перемешивают и прессуют в таблетки.
Таблетки, полученные согласно примеру 2, тестируют на подвергнутых голоданию в течение 26 часов шести здоровых самцах собак породы бигль. Каждую таблетку вводят с 20 мл воды. Собирают образцы крови для измерения уровней глюкозы в крови и инсулина в плазме. Как представлено на Фиг. 3 и Фиг. 4, таблетки дают в результате быстрое повышение уровней инсулина в плазме, показывая значение Сmах приблизительно 75 мЕд/мл при Тmах 20 минут после времени введения. Это приводит в результате к соответствующему падению концентрации глюкозы в плазме приблизительно на 35% относительно исходного уровня.
Полученные таким образом таблетки подвергают исследованиям стабильности согласно руководству IСН. Длительные исследования проводят при 2÷8°С и 25°С, тогда как ускоренное исследование стабильности осуществляют при ОВ 30°С/65% и ОВ 40°С/75%.
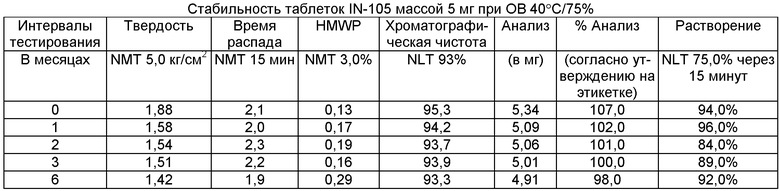
Пример 3
Точно взвешенное количество IN-105, капрата натрия + комплекса β-циклодекстрина, бикарбоната натрия и Коллидона СL пропускают через сито 45# и смешивают в полиэтиленовой емкости. После однородного перемешивания смесь смазывают Аэросилом и стеаратом магния, хорошо перемешивают и прессуют в таблетки.
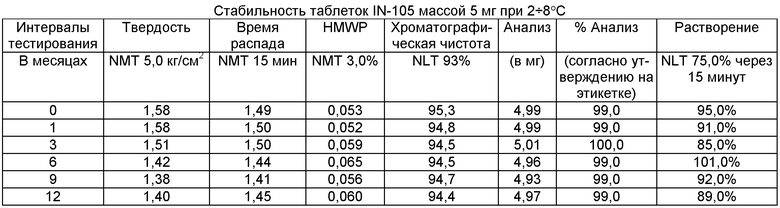
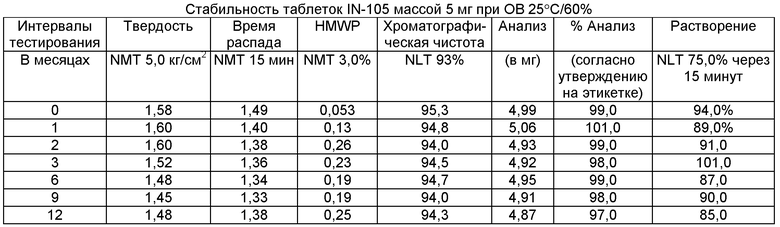
Пример 4
Точно взвешенные количества IN-105, лаурилсульфата натрия, Коллидона CL и Перлитола пропускают через сито 45# и перемешивают в октагональном блендере. После однородного перемешивания смесь смазывают Аэросилом и стеаратом магния, хорошо перемешивают и прессуют в таблетки.
Таблетки, полученные, как описано в примере 4, тестируют на подвергнутых голоданию в течение 26 часов шести здоровых самцах собак породы бигль. Каждую таблетку вводят с 20 мл воды. Собирают образцы крови для измерения уровней глюкозы в крови и инсулина в плазме. Полученные результаты представляют на Фиг. 5 и Фиг. 6.4. Полученные таким образом таблетки подвергают исследованиям стабильности согласно руководству ICH. Длительные исследования проводят при 2÷8°С и 25°С, тогда как ускоренное исследование стабильности осуществляют при OВ 30°С/65% и OВ 40°С/75%.
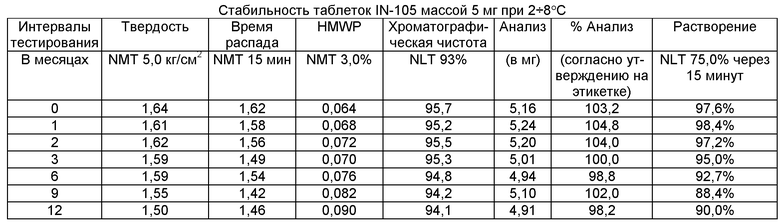
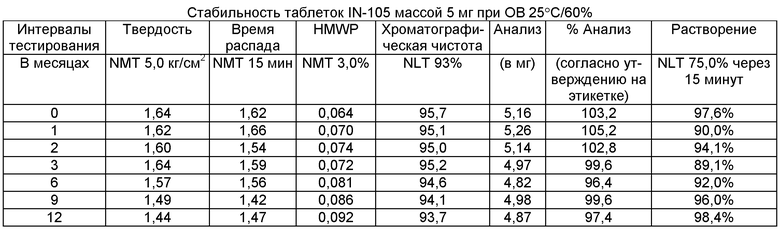
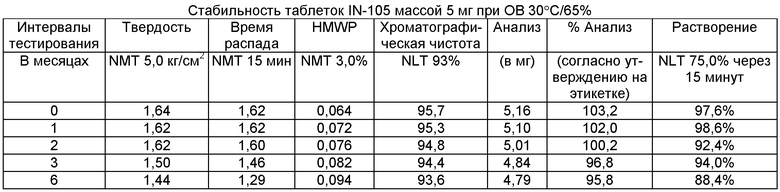
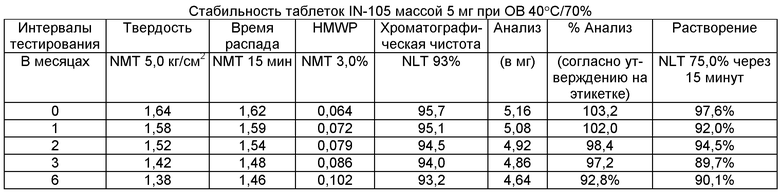
Пример 5
Получение гранул капрата натрия, бикарбоната натрия и Перлитола с ПВП К-30 (2 мас.%) в качестве связующего компонента и ПЭГ 6000 (1 мас.%) в качестве пластификатора и IN-105, добавляемых в процессе грануляции.
Точно взвешенное количество измельченного капрата натрия, бикарбоната натрия и Перлитола вносят в планетарный смеситель, сухое перемешивание проводят в течение 5 мин. Одновременно ПВП К 30 растворяют в ИПС и суспендируют IN-105. ПЭГ 6000 растворяют в воде (5% об./об.). Раствор хорошо перемешивают, используя магнитную мешалку. Капрат натрия, бикарбонат натрия и Перлитол гранулируют, используя ПВПК и раствор ПЭГ в качестве грануляционного компонента. Процесс осуществляют в течение 20 минут. Образованную мокрую массу пропускают через сито 18# в мокром грануляторе и сушат в ламинарном потоке воздуха.
Объем добавляемого ИПС - 130 мл.
100 мл=IN-105+ПВП k. 6 мл=ПЭГ в воде.
Скорость планетарного смесителя=2-4 в течение 20 мин.
Скорость мокрого гранулятора - 200 об/мин.
Содержание влаги в гранулах.
Масса образца - 0,525 г.
% содержания влаги - 1,99%.
Точно взвешенное количество гранул IN-105+Капрат натрия+Бикарбонат натрия+Перлитол+ПВП K30 и ПЭГ пропускают через сито 35# и смешивают с Коллидоном СL - в двухконусном смесителе. После однородного перемешивания смесь смазывают Аэросилом и стеаратом магния, хорошо перемешивают и прессуют в таблетки.
Полученные таким образом таблетки подвергают исследованиям стабильности согласно руководству ICH. Длительные исследования проводят при 2÷8°С и 25°С, тогда как ускоренное исследование стабильности осуществляют при 0В 30°С/65% и ОВ 40°С/75%.
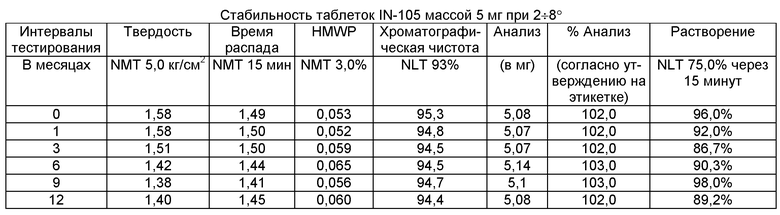
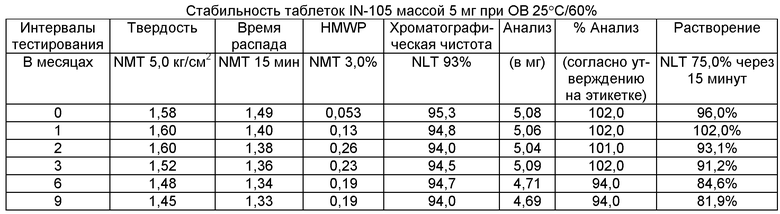
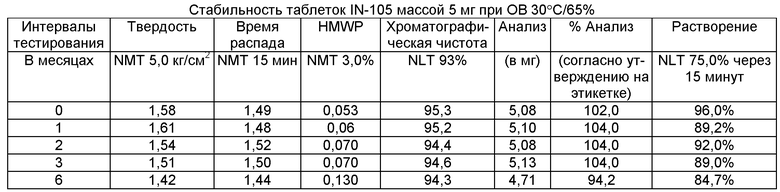
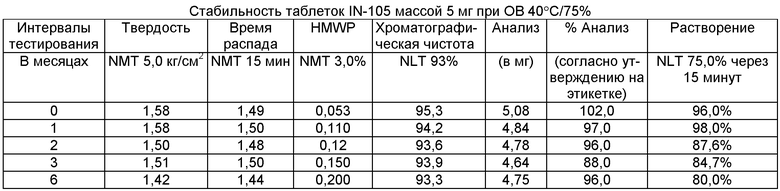
Имеются предпочтительные препараты с пропорциональной дозой, дающие требуемые профили высвобождения лекарственного препарата. Представлены данные препараты, которые обеспечивают приблизительно согласующееся высвобождение IN-105.
Пример 6
1,663 г IN-105 и 50 г капрата натрия растворяют в воде путем подведения рН до 8,28 с помощью 10% гидроксида натрия. Раствор используют для распылительной сушки. Раствор сушат распылительной сушкой при 80°С проточным образом. Используемое давление распыления составляет 147,1 кПа (1,5 кг/см2), и скорость потока, подаваемого на распылительную сушку, составляет 2 мл/мин. Чистота образца, высушенного распылительной сушкой, составляет около 98% и из в целом ожидаемых 52 г выделяют всего 16 г гранул и 1,5 г тонкого порошка.
Данный высушенный распылительной сушкой порошок смешивают с другими наполнителями препарата, такими как маннит, натриевая соль карбоксиметилкрахмала, коллоидный оксид кремния и стеарат магния и прессуют в форме таблеток. Затем данные таблетки тестируют на модели фиксированных собак, чтобы посмотреть фармакокинетический и фармакодинамический ответ.
Пример 7
Исходную суспензию IN 105, имеющую концентрацию 35 г/л, используют для получения образцов, высушенных распылительной сушкой. Используемые наполнители представляют собой маннит и капрат натрия. К смеси 1050 мг капрата натрия и 1015 мг маннита добавляют 20 мл раствора, содержащего 1 мл исходной суспензии, и тщательно перемешивают. Раствор сушат распылительной сушкой при 80°С проточным образом. Используемое давление распыления составляет 147,1 кПа (1,5 кг/см2), и скорость потока, подаваемого на распылительную сушку, составляет 2 мл/мин. Получают выход для образца приблизительно 60%, и чистота полученного распылительной сушкой образца составляет около 98%.
Пример 8
Исходную суспензию IN 105, имеющую концентрацию 35 г/л, используют для получения образцов, высушенных распылительной сушкой. Используемые наполнители представляют собой маннит и капрат натрия. К смеси 1050 мг капрата натрия и 945 мг маннита добавляют 20 мл раствора, содержащего 3 мл исходной суспензии, и тщательно перемешивают. Раствор сушат распылительной сушкой при 80°С проточным образом. Используемое давление распыления составляет 117,7 кПа (1,2 кг/см2), и скорость потока, подаваемого на распылительную сушку, составляет 2 мл/мин. Получают выход для образца приблизительно 60% и позднее определяют, что чистота составляет 98,6%.
Пример 9
Исходную суспензию IN 105, имеющую концентрацию 35 г/л, используют для получения образцов, высушенных распылительной сушкой. Используемые наполнители представляют собой маннит и капрат натрия. К смеси 1050 мг капрата натрия и 1015 мг маннита добавляют 20 мл раствора, содержащего 1 мл исходной суспензии, и тщательно перемешивают. Раствор сушат распылительной сушкой при 120°С проточным образом. Используемое давление распыления составляет 147,1 кПа (1,5 кг/см2), и скорость потока, подаваемого на распылительную сушку, составляет 2 мл/мин. Получают выход для образца приблизительно 60% и позднее определяют, что чистота составляет 99,5%.
Пример 10
Исходную суспензию IN 105, имеющую концентрацию 35 г/л, используют для получения образцов, высушенных распылительной сушкой. Используемые наполнители представляют собой маннит и капрат натрия. К смеси 1050 мг капрата натрия и 945 мг маннита добавляют 20 мл раствора, содержащего 3 мл исходной суспензии, и тщательно перемешивают. Раствор сушат распылительной сушкой при 120°С проточным образом. Используемое давление распыления составляет 117,7 кПа (1,2 кг/см2), и скорость потока, подаваемого на распылительную сушку, составляет 2 мл/мин. Получают выход для образца приблизительно 60% и позднее определяют, что чистота составляет 98,3%.
Пример 11
Исходную суспензию IN 105, имеющую концентрацию 11,5 г/л, используют для получения образцов, высушенных распылительной сушкой. Используемые наполнители представляют собой маннит и капрат натрия. К смеси 3000 мг капрата натрия и 2885 мг маннита добавляют 50 мл раствора, содержащего 10 мл исходной суспензии, и тщательно перемешивают. Раствор сушат распылительной сушкой при 150°С проточным образом. Используемое давление распыления составляет 84,3 кПа (0,86 кг/см2), и скорость потока, подаваемого на распылительную сушку, составляет 4 мл/мин. Определяют, что чистота образца составляет 94,71%.
Два прототипных препарата [Препарат - 862, таблетка, полученная способом прямого прессования] и [Препарат - 872 - таблетка, полученная способом распылительной сушки], которые подвергают скринингу с использованием исследования на фиксированных собаках, показывают, что препарат -872, полученный процессом распылительной сушки, проявляет относительно согласующиеся уровни всасывания лекарственного препарата и результирующие скорости инфузии глюкозы без потери стабильности или биологической активности, делая процесс получения очень экономичным, коммерчески жизнеспособным и масштабируемым способом изготовления композиций катион-конъюгат соединения инсулина.
Пример 12
Диализованную в течение ночи лекарственную субстанцию, приготовленную как Insugen® и IN-105, в 10 мМ Трис, рН 8, получают в концентрациях, определяемых с помощью количественной оценки ВЭЖХ (высокоэффективной жидкостной хроматографии).
Таблица 11: Концентрации диализованного инсулина, используемые для изучения с помощью митогенного анализа
Клетки 3Т3-А31 получают из АТСС (мышиный фибробласт, АТСС CCL-163). Среду Дульбекко в модификации Игла (DMEM), инактивированную нагреванием ФСК, и краситель аламар синий приобретают в фирме Invitrogen. ИФР1 и 1М раствор HEPES получают в фирме Sigma Aldrich. Клетки 3Т3-А31 поддерживают в забуференной 10 мМ HEPES среде DMEM с добавлением 10% ФСК в условиях увлажнения при 37°С в атмосфере 5% СО2. Для анализа клетки 3Т3-А31 трипсинизируют 0,25% трипсин-ЭДТА. Количество клеток подсчитывают с помощью гемоцитометра, используя окрашивание трипаном синим. 10000 клеток/лунку высевают в забуференную 10 мМ HEPES среду DMEM с добавлением 0,5% ФСК в 96-луночные планшеты. Различные концентрации соответственного инсулина добавляют в лунки в трех повторностях. Через 20 часов культивирования с различными концентрациями факторов роста добавляют краситель аламар синий (10 об.%) и планшеты далее инкубируют в течение дополнительных 4 часов в термостате на 37°С. Флуоресценцию (длина волны возбуждения = 530 нм; длина волны эмиссии = 590 нм) измеряют, используя ридер для 96-луночных планшетов.
Пример 13
Диализованную в течение ночи лекарственную субстанцию, приготовленную как Insugen® и IN 105, в 10 мМ Трис, рН 8, получают в концентрациях, определяемых с помощью количественной оценки ВЭЖХ, представленной в таблице 13.
Клетки 3T3-L1 получают из АТСС. Среду Дульбекко в модификации Игла (DMEM), инактивированную нагреванием ФСК, раствор пенициллина-стрептомицина (10Х) и 1М раствор солей HEPES приобретают в фирме Invitrogen. Дексаметазон, изобутилметилксантин, 4-аминоантипирин, N-этил-N-сульфопропил м-толуидин и реагент глюкозооксидаза/пероксидаза получают в фирме Sigma Aldrich.
Клетки 3T3-L1 поддерживают в забуференной 10 мМ HEPES среде DMEM с добавлением 10% ФСК, 100 ед./100 мкг пенициллина/стрептомицина (поддерживающая среда) в условиях увлажнения при 37°С в атмосфере 5% СO2. Для проведения метаболического анализа клетки 3T3-L1 должны дифференцироваться до адипоцитов. Клетки 3T3-L1 трипсинизируют 0,25% трипсин-ЭДТА. Количество клеток подсчитывают с помощью гемоцитометра, используя окрашивание трипаном синим. 25000 клеток/лунку высевают в поддерживающую среду в 96-луночные планшеты. Клеткам дают возможность достигнуть конфлюэнтности в течение следующих 2 дней. В день 3 среду для клеток заменяют средой для дифференцировки (0,5 мМ дексаметазон и 0,25 мкМ изобутилметилксантин в поддерживающей среде в течение дополнительных четырех дней. В день 7 среду для дифференцировки снова заменяют поддерживающей средой в течение трех дней. В день 9 клетки промывают 1Х ФСБ (фосфатно-солевой буфер) и различные концентрации соответственного инсулина получают в пробирках для центрифугирования объемом 15 мл в буфере для анализа среды с низким содержанием глюкозы с добавлением 0,5% ФСК, 2 мМ L-глутамина и 1Х Pen-Strep. Разведения добавляют в лунки в трех повторностях. Через 22 часа культивирования с различными концентрациями инсулина 96-луночные планшеты вынимают из термостата для определения глюкозы. Количество глюкозы оценивают с помощью реагента GOPOD, который превращает глюкозу в среде в глюконовую кислоту и пероксид водорода. Образованный пероксид водорода реагирует с субстратом (4-аминоантипирином и N-этил N-сульфопропил м-толуидином), что дает продукт, окрашенный в фиолетовый цвет, который можно читать при длине волны 550 нм.
Расчеты метаболической активности:
Для представления данных % глюкозы, остающейся в среде, измеряют, принимая величину "без инсулина" за 100%.
Результаты данного анализа показывают, что метаболические активности как Insugen®, так и IN 105 и HIM-2 близки и активности находятся в приемлемых рамках (0,8-1,2) анализа PLA, показывая тем самым, что Insugen® и IN-105 метаболически равно активны, как представлено на фиг. 14. На фиг. 14 показывают, что метаболическая активность Insugen® относительно IN 105 и Insugen®) относительно HIM-2 одинакова. Обнаружено, что соотношение метаболической активности Insugen® и IN 105 составляет 1,166, и обнаружено, что соотношение Insugen® и HIM-2 составляет 1,149. Данные, представляющие полученные в эксперименте определения в трех повторностях, как Среднее ± стандартная ошибка, на фиг. 15 демонстрируют параллелизм и линейность биоанализа при различных концентрациях Insugen (R) и IN-105, определяемые с помощью пакета программ PLA. Данные представляют среднее ± стандартная ошибка по значениям, полученным в трех повторностях эксперимента.
Пример 14
Различные флаконы инсулина осветляют, используя ледяную уксусную кислоту (рН~3,4). Затем осветленный раствор нагружают на С8 обращенно-фазовую колонку с силикагелем, используя градиент элюции с 250 мМ уксусной кислотой и 100% этанолом. Фракции собирают во время элюции и объединяют на основе чистоты, превышающей или равной 99%. Данную группу, полученную при элюции, диализуют в течение 15 часов.
Используют мембрану с отсечением молекулярной массы 1 кД против 10 мМ Трис, рН 8,0. Наконец, диализованный не содержащий Zn и не содержащий наполнитель инсулин, полученный при рН 8,0, анализируют с помощью аналитической ОФ-ВЭЖХ. Диализованная в течение ночи не содержащая цинк лекарственная субстанция в виде Insugen®, IN-105 и HIM-2 в 10 мМ Трис, рН 8 с концентрациями, определенными с помощью количественной ВЭЖХ, представлена в Таблице 15.
Клетки HepG2 получают из АТСС (Номер по каталогу АТСС НВ-8065). Среду Дульбекко в модификации Игла, инактивированную нагреванием ФСК, 100Х раствор пенициллина-стрептомицина и 100Х раствор солей HEPES приобретают в фирме Invitrogen. Бычий сывороточный альбумин, гидроксид натрия, Тритон-Х 100 и бикарбонат натрия получают в фирме Sigma Aldrich.
Рекомбинантный человеческий инсулин с радиоактивной меткой приобретают в фирме Perkin Elmer со специфической радиоактивностью 2200 Ки/ммоль (No. по каталогу 2200 NEX420).
Клетки HepG2 поддерживают в забуференной 10 мМ HEPES среде DMEM с добавлением 10% ФСК и 1Х раствора пенициллина-стрептомицина в условиях увлажнения при 37°С в атмосфере 5% СО2. Для анализа клетки HepG2 трипсинизируют и высевают с густотой 400000 клеток на лунку 24-луночного планшета. Через 3 дня инкубирования клетки используют для проведения анализов связывания радиолиганда.
Перед проведением анализа среды удаляют и клетки дважды промывают буфером для связывания (DMEM, 2,2 мг/мл бикарбоната натрия, 1 мг/мл бычьего сывороточного альбумина и 50 мМ HEPES) для удаления каких-либо следов факторов роста, присутствующих в среде. Эксперименты по конкурентному связыванию проводят в повторностях с использованием фиксированного количества радиолиганда (0,325 нМ) и варьирующих концентраций немеченой лекарственной субстанции инсулина (от 10-12 М до 10-7 М). Конечный объем реакции доводят до 1 мл. Затем планшеты инкубируют при 15°С в течение ночи на качалке, установленной на 100 об/мин. На следующий день все среды из лунок отбрасывают и каждую лунку промывают дважды ледяным буфером для связывания.
В каждую лунку вносят 1 мл солюбилизационного реагента (0,5 М гидроксида натрия, 0,5% Тритон-Х 100). Солюбилизированные клетки переносят в пробирку для радиоиммуноанализа и читают связанную радиоактивность в γ-счетчике (Stratec BioMedical Systems, Germany). Прибор калиброван и, как показано, имеет эффективность 80%.
Расчеты аффинности связывания: для нормализации числа импульсов в минуту (значений СРМ) при используемых различных концентрациях инсулина процент связывания рассчитывают, используя следующее уравнение:
% связывания=(СРМобразца-СРМбез вещества)/(СРМконтроля-СРМбез вещества)×100.
Когда СРМконтроля представляет собой среднее СРМ лунок, которые содержат клетки с радиоактивным инсулином без добавления какого-либо немеченого инсулина, СРМбез вещества представляет собой среднее СРМ лунок, которые не содержат инсулин с радиоактивной меткой, а также немеченый инсулин, и СРМобразца представляет собой среднее СРМ лунок, которые содержат инсулин с радиоактивной меткой, а также варьирующие концентрации инсулина без метки.
Представленное выше описание примеров дано только для легкости понимания. Не следует понимать их как необязательные ограничения, поскольку модификации будут очевидны для компетентных специалистов в области техники, которые признают, что изобретение может быть практически реализовано с модификациями и вариантами, входящими в сущность прилагаемой формулы изобретения.
Предшествующее описание предпочтительного варианта осуществления и лучший способ, соответствующий изобретению, известный заявителю на время подачи заявки, представлены и предназначены для целей иллюстрации и описания. Не предусматривают, что они являются исчерпывающими или ограничивают изобретение точной раскрытой формой, и возможны многие модификации и варианты в свете вышеприведенной идеи. Вариант осуществления выбран и описан для наилучшего объяснения принципов изобретения и его практического применения и для того, чтобы дать возможность другим компетентным специалистам в области техники наилучшим образом использовать изобретение в различных вариантах осуществления и с различными модификациями, которые подходят для конкретного предусматриваемого применения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТВЕРДЫЕ ДИСПЕРСИИ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ВКЛЮЧАЮЩИЕ ЗАМЕЩЕННЫЙ ИНДАН, И СПОСОБЫ ИХ ПРИГОТОВЛЕНИЯ И ПРИМЕНЕНИЯ | 2019 |
|
RU2772693C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕЕ ВВЕДЕНИЕ | 2013 |
|
RU2802442C2 |
| КОМПОЗИЦИИ ПЕПТИДОВ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2009 |
|
RU2517135C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕЕ ВВЕДЕНИЯ | 2013 |
|
RU2692779C2 |
| ПРОТИВОТУБЕРКУЛЕЗНАЯ СТАБИЛЬНАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ В ФОРМЕ ТАБЛЕТКИ С ПОКРЫТИЕМ, СОДЕРЖАЩЕЙ ГРАНУЛИРОВАННЫЙ ИЗОНИАЗИД И ГРАНУЛИРОВАННЫЙ РИФАПЕНТИН, И СПОСОБ ЕЕ ПРИГОТОВЛЕНИЯ | 2014 |
|
RU2682178C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ПИРАЗОЛА | 2020 |
|
RU2829944C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2018 |
|
RU2767872C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2000 |
|
RU2330666C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ С МАСКИРОВАННЫМ ВКУСОМ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2012 |
|
RU2583935C2 |
| ПРЕПАРАТ АКТИВАТОРА ГЛЮКОКИНАЗЫ ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2017 |
|
RU2728824C1 |
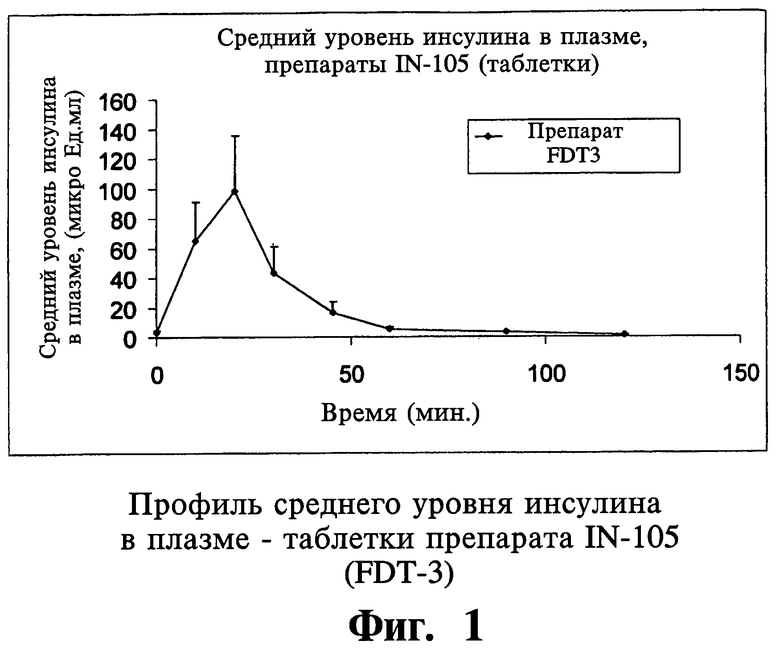
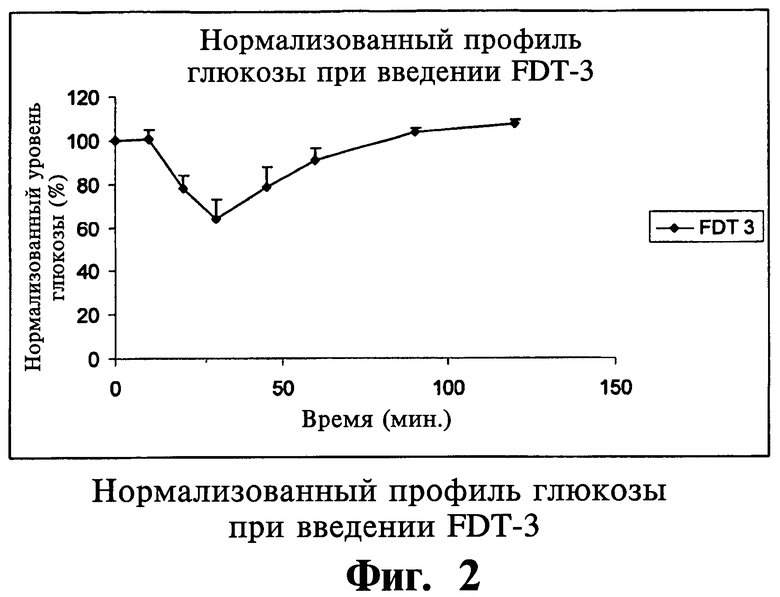
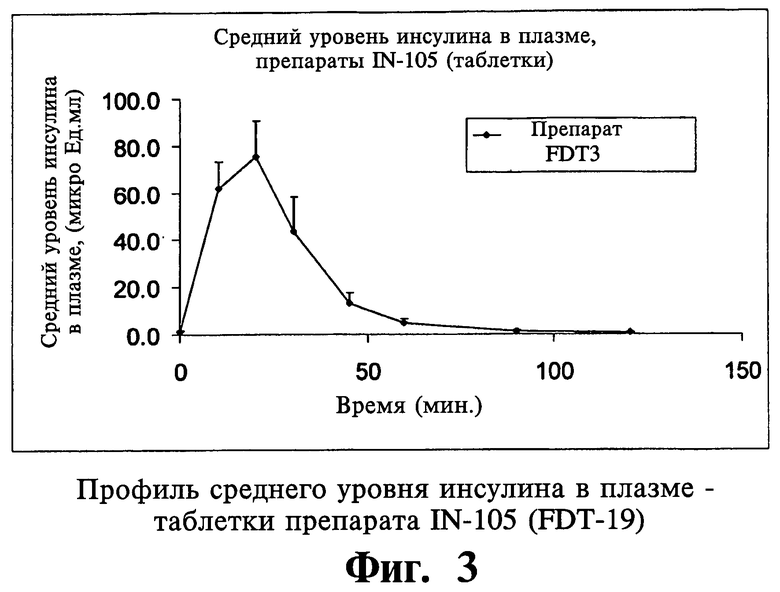
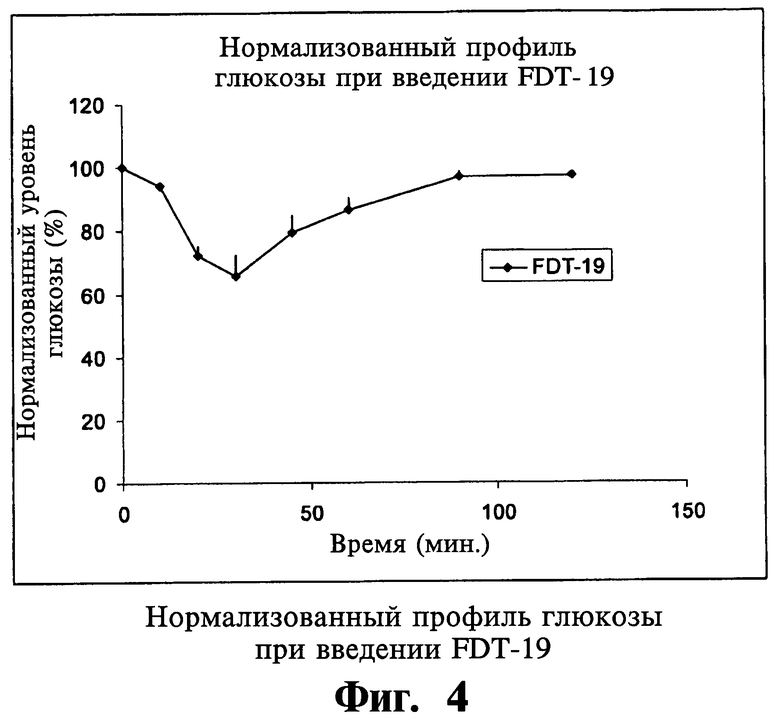
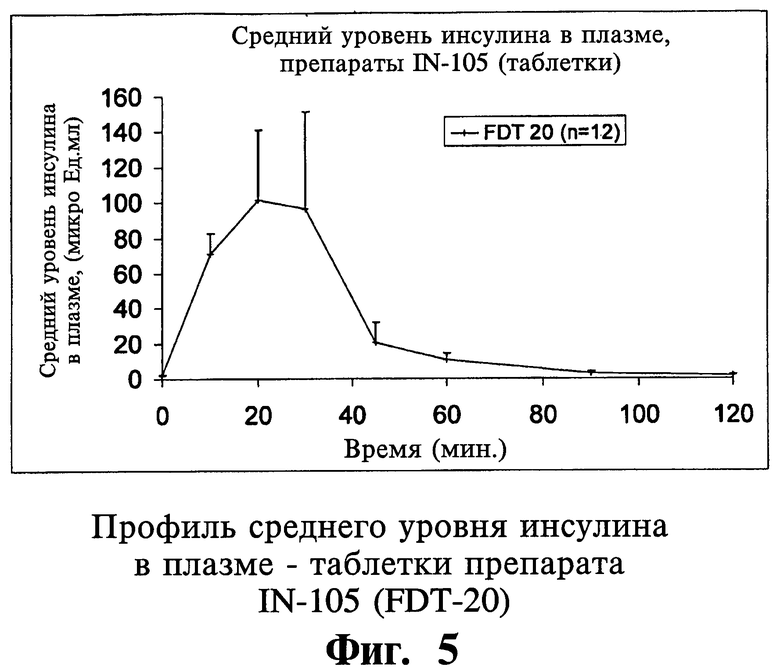
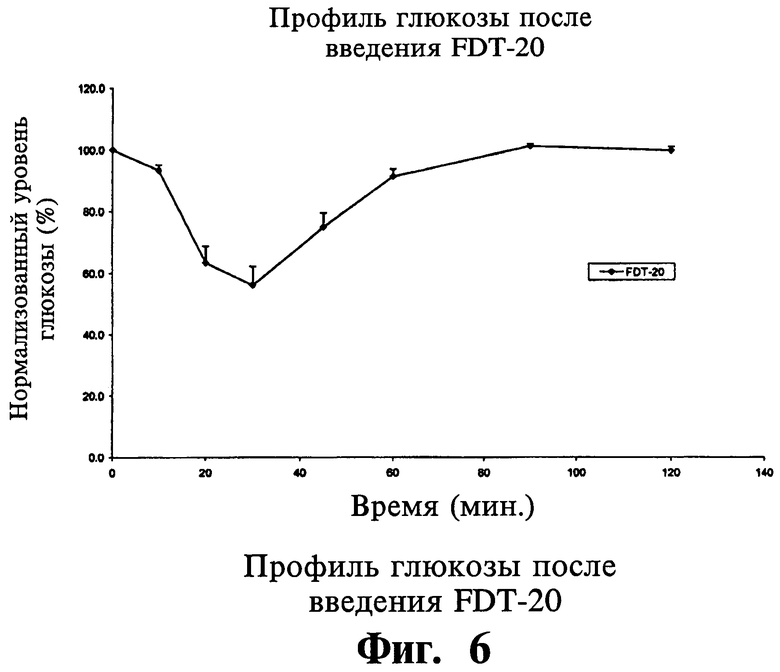
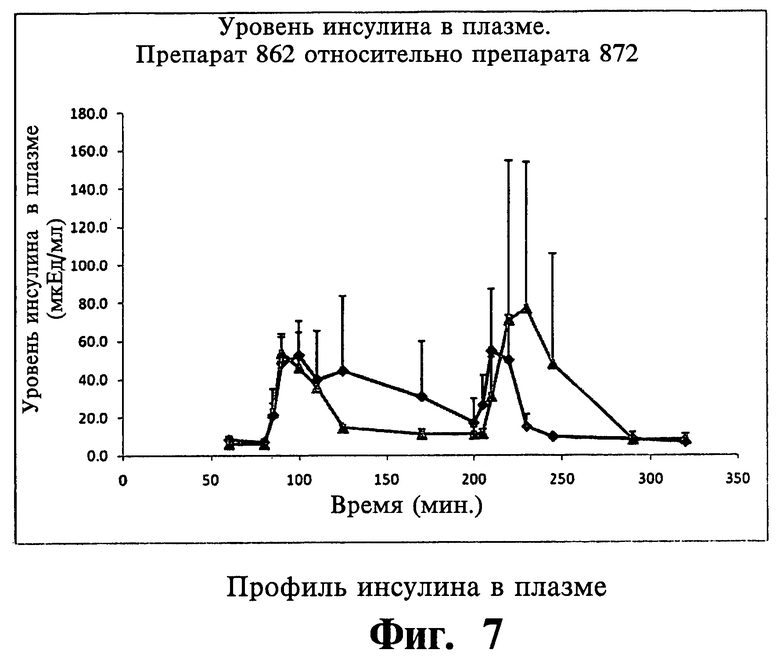
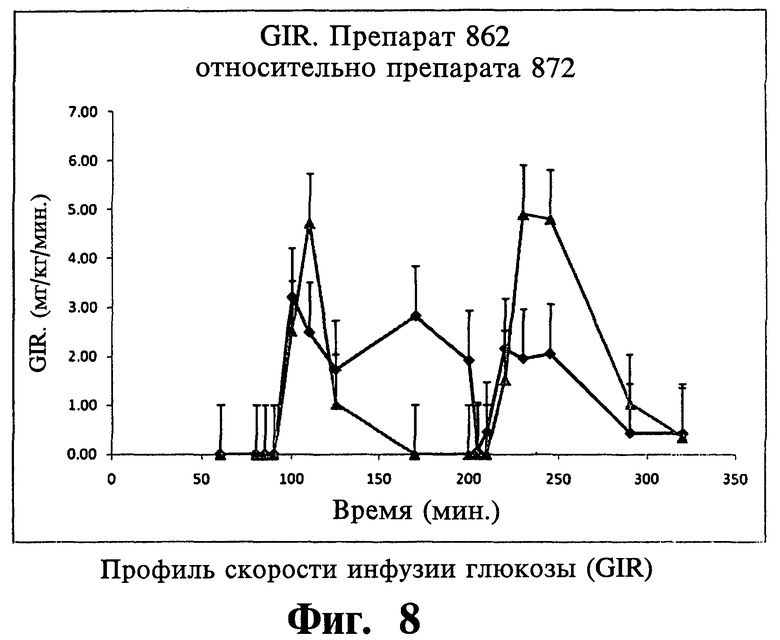
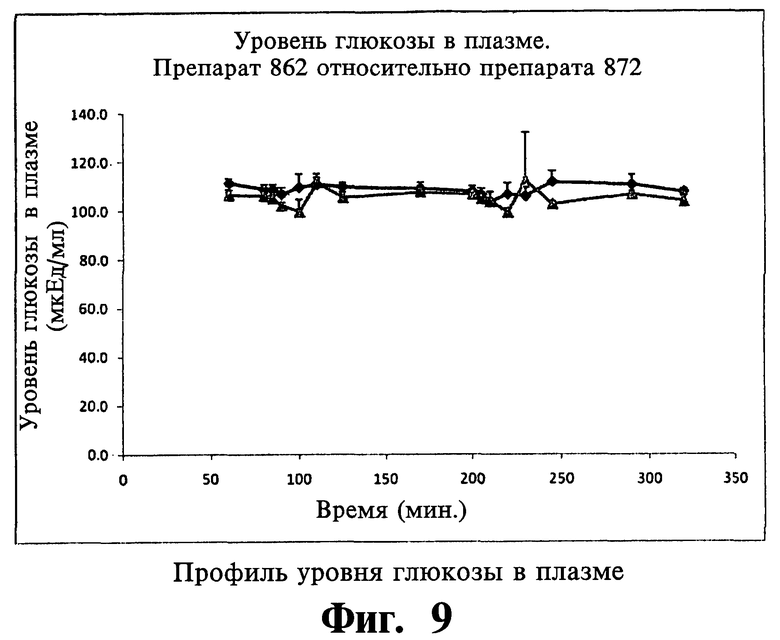
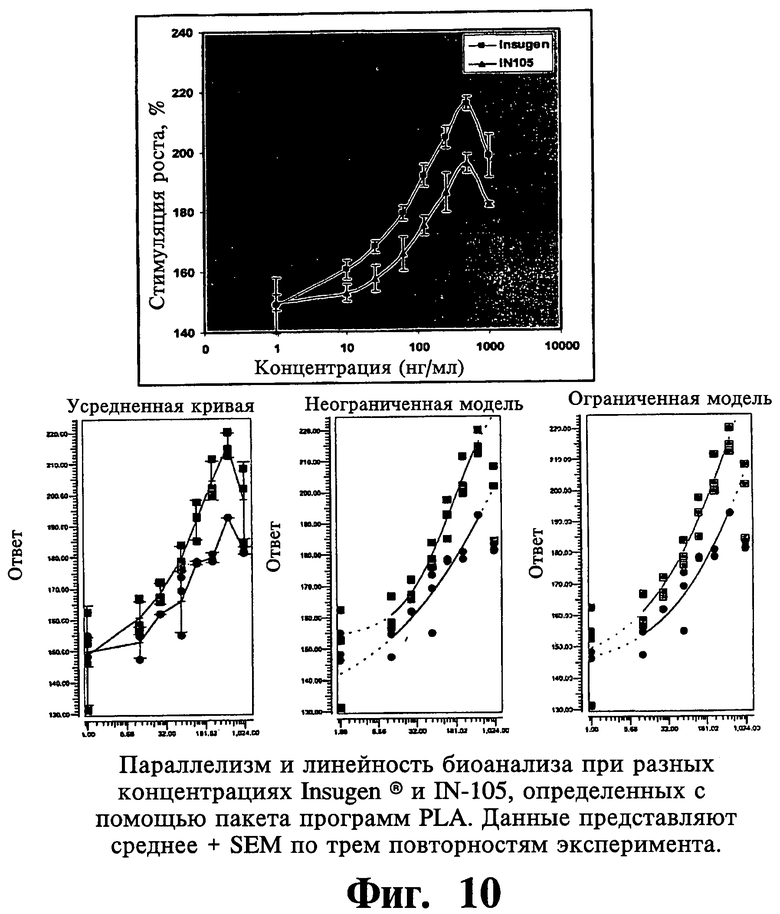
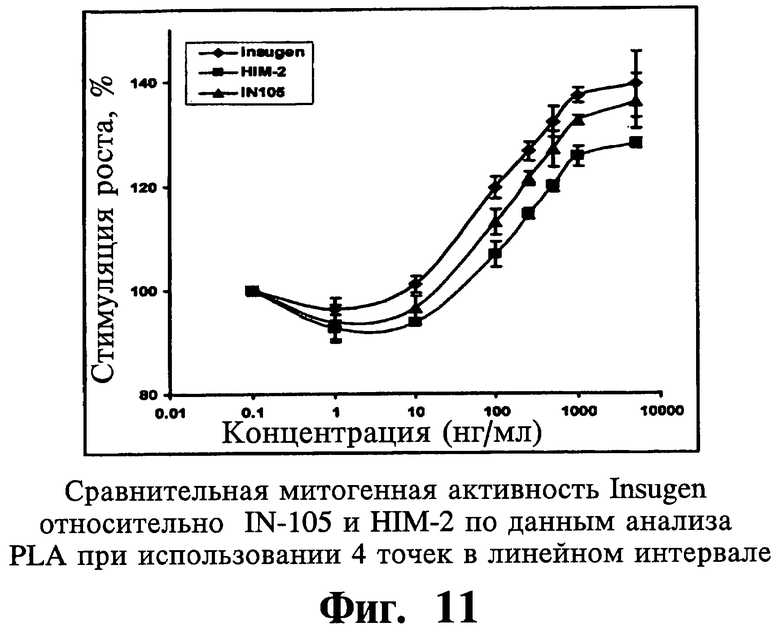
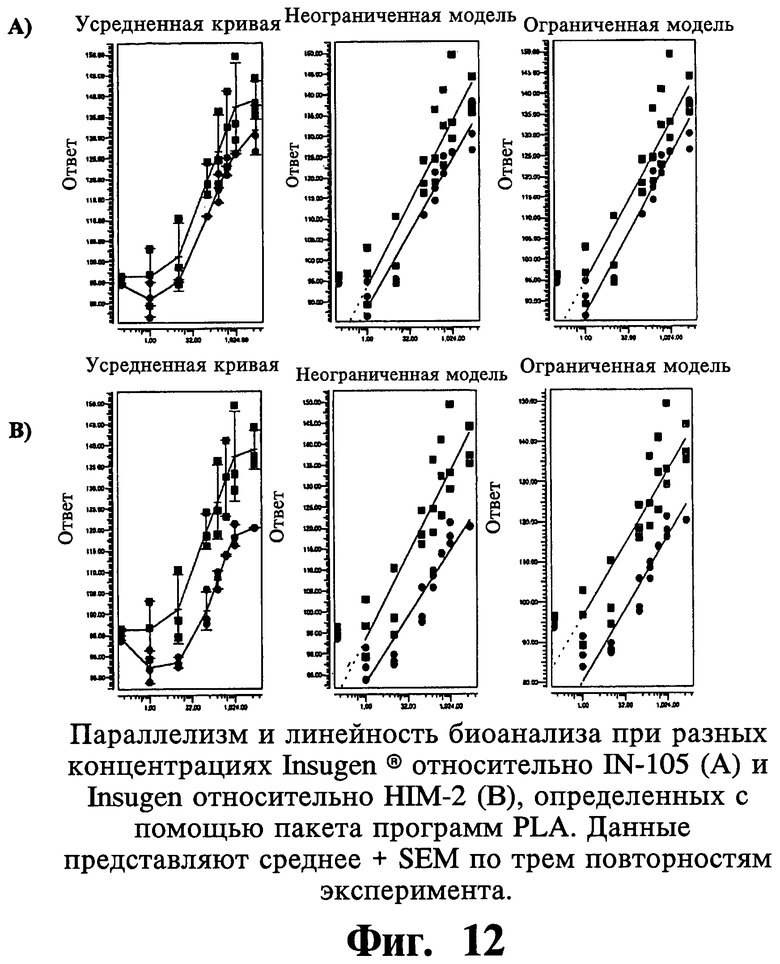
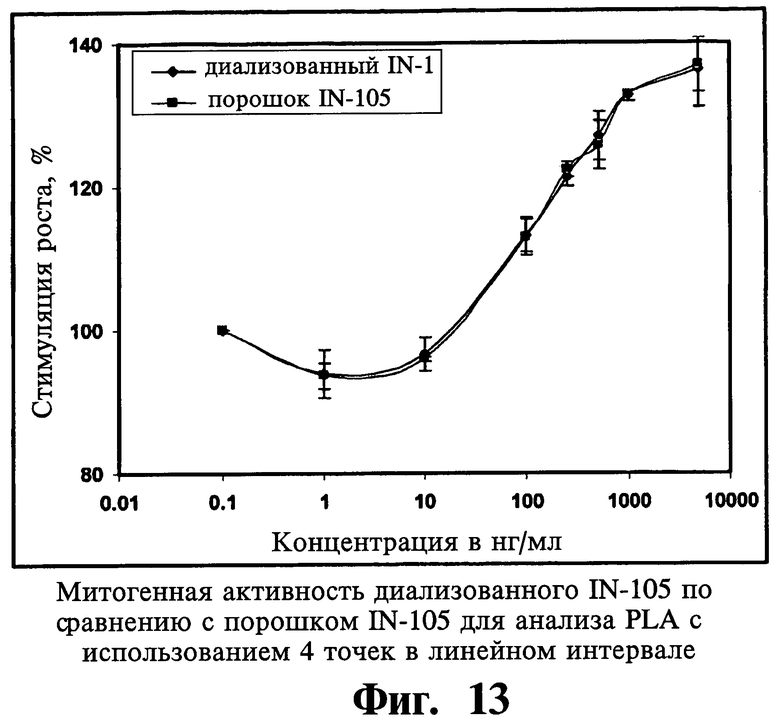
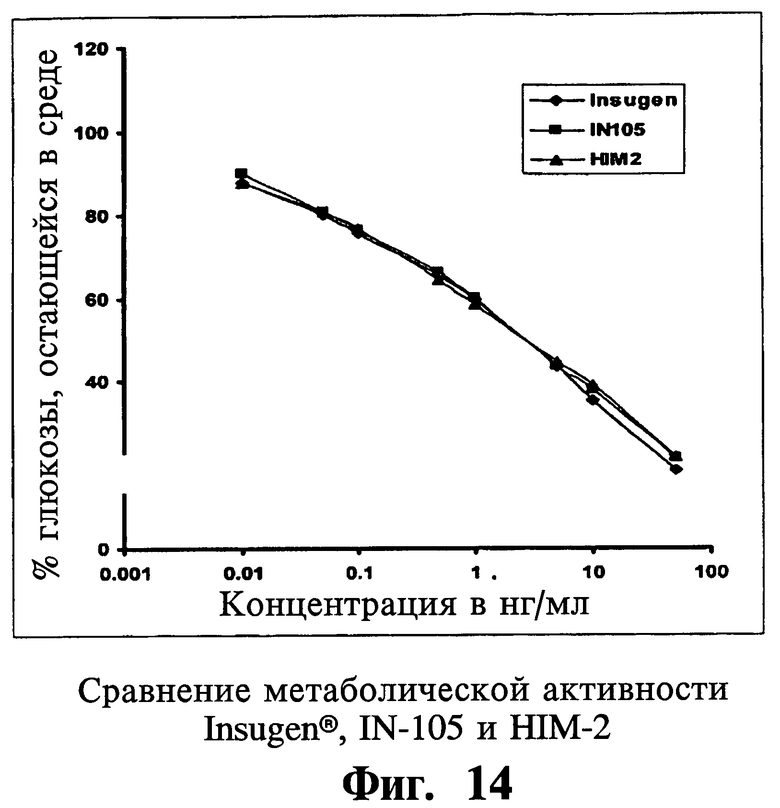
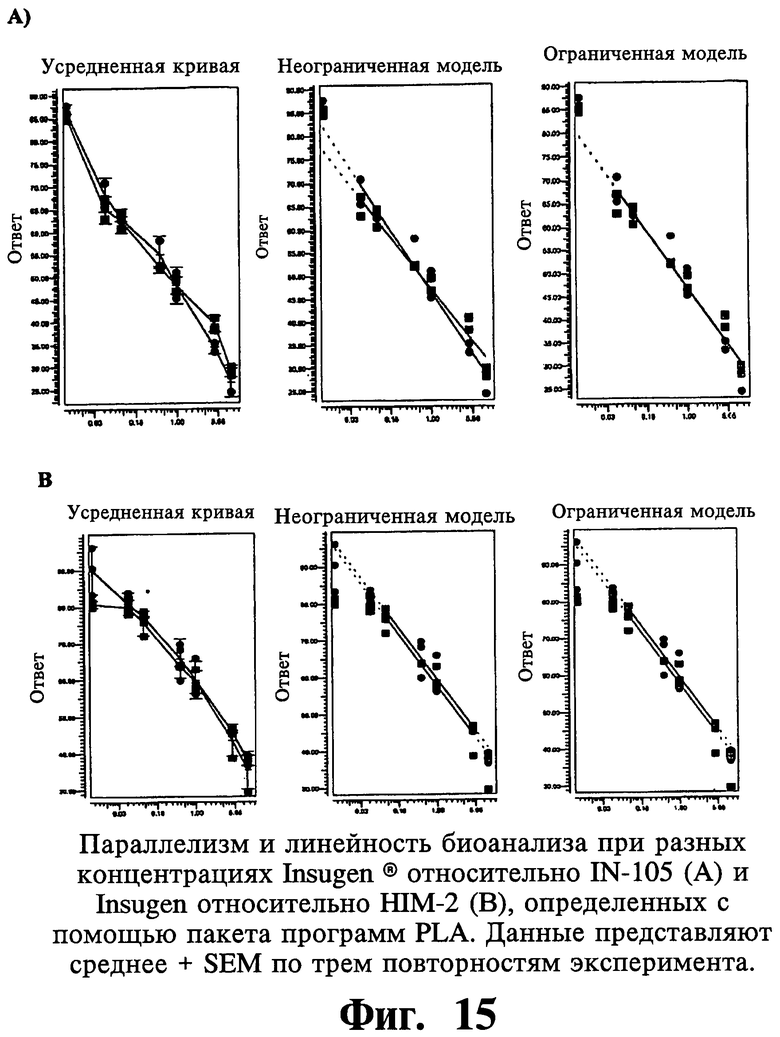
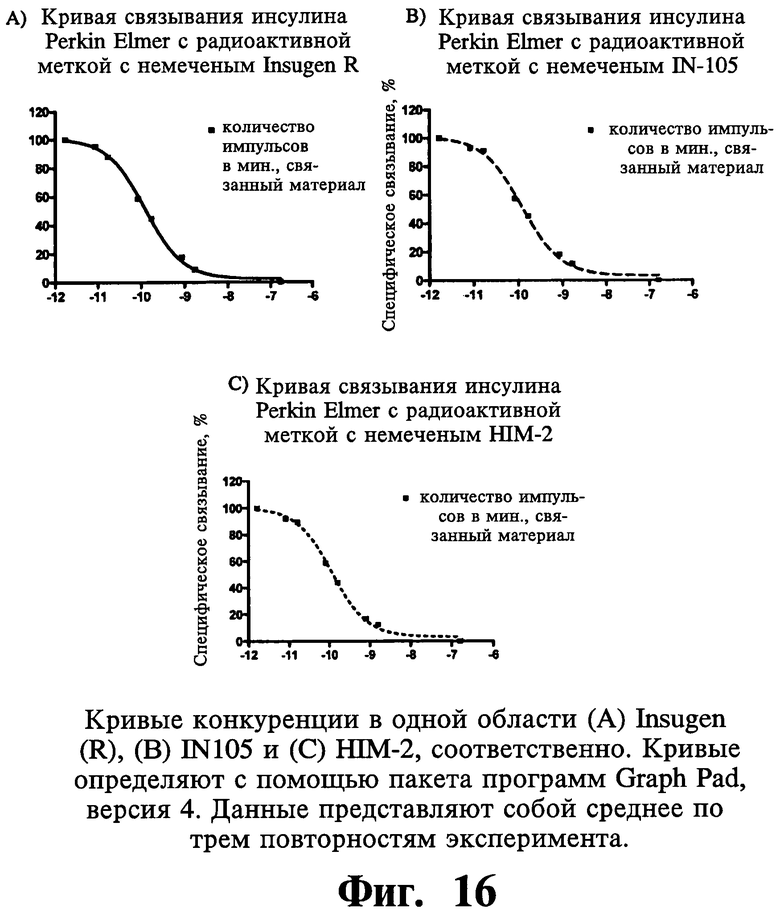
Изобретение относится к области медицины. Предложена твердая фармацевтическая композиция для перорального применения, включающая приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных C4-C12 жирных кислот, их сложных эфиров или солей и по меньшей мере один фармацевтически приемлемый наполнитель, выбранный из группы, включающей по меньшей мере 10 мас.% разрыхлителя, 10 мас.% маннита и 0,5 мас.% смазывающего вещества. Предложены способ контроля концентрации глюкозы в крови с помощью указанной композиции, способы ее получения и таблетки массой 5÷500 мг. Предложены твердая фармацевтическая композиция, стабильная при интервале температур 2÷40°С и разных показателях относительной влажности в течение по меньшей мере 6 месяцев, и инсулинотропная фармацевтическая композиция с пониженной в три раза митогенностью по сравнению со своим нативным аналогом, включающие указанную композицию. Предложен также способ получения аморфных высушенных распылительной сушкой частиц. Изобретение обеспечивает изготовление фармацевтической композиции для перорального применения, включающей конъюгат инсулина с повышенной биодоступностью и пониженной митогенностью. 13 н. и 52 з.п. ф-лы, 16 ил., 16 табл., 14 пр.
1. Твердая фармацевтическая композиция для перорального применения, включающая приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных C4-C12 жирных кислот, их сложных эфиров или солей и по меньшей мере один фармацевтически приемлемый наполнитель, выбранный из группы, включающей по меньшей мере 10 мас.% разрыхлителя, 10 мас.% маннита и 0,5 мас.% смазывающего вещества.
2. Композиция по п.1, которая необязательно включает связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, или солюбилизаторы.
3. Композиция по п.1, в которой жирная кислота представляет собой каприновую кислоту и/или лауриновую кислоту или их соли.
4. Композиция по п.1, в которой соль жирной кислоты представляет собой капрат натрия.
5. Композиция по п.1, в которой соль жирной кислоты представляет собой лаурилсульфат натрия.
6. Композиция по п.2, в которой связующий компонент выбран из группы, включающей поливинилпирролидон, карбоксиметилцеллюлозу, метилцеллюлозу, крахмал, желатин, сахара, природные или синтетические камеди или их комбинации.
7. Композиция по п.6, в которой связующий компонент представляет собой поливинилпирролидон.
8. Композиция по п.1, в которой маннит представляет собой разбавитель.
9. Композиция по п.1, в которой разрыхлитель выбран из группы, включающей поперечно-сшитый поливинилпирролидон, карбоксиметилцеллюлозу, метилцеллюлозу, катионообменные смолы, альгиновую кислоту, гуаровую камедь, натриевую соль карбоксиметилкрахмала, натрий кроскармелозу и их комбинации.
10. Композиция по п.1, в которой смазывающее вещество выбрано из группы, включающей стеарат магния, стеарат натрия, бензоат натрия, ацетат натрия, фумаровую кислоту, коллоидный оксид кремния, полиэтиленгликоль, аланин и глицин.
11. Композиция по п.10, в которой смазывающее вещество представляет собой стеарат магния.
12. Композиция по п.10, в которой смазывающее вещество представляет собой коллоидный оксид кремния.
13. Композиция по п.2, в которой агент, усиливающий проницаемость, выбран из группы, включающей лаурилсульфат натрия, лаурат натрия, пальмитоилкарнитин, фосфатидилхолин, циклодекстрин и его производные, карнитин и его производные, мукоадгезивные полимеры, ZOT-токсин, соли желчных кислот, жирные кислоты и их комбинации.
14. Композиция по п.13, в которой агент, усиливающий проницаемость, представляет собой лаурилсульфат натрия.
15. Композиция по п.13, в которой агент, усиливающий проницаемость, представляет собой β-циклодекстрин.
16. Композиция по п.2, в которой пластификатор представляет собой полиэтиленгликоль.
17. Композиция по п.1, которая представлена в форме таблетки, капсулы, частиц, порошка или саше, либо сухой суспензии.
18. Композиция по п.1, в которой растворимость составляет по меньшей мере 75% в любой момент времени в течение 12 месяцев.
19. Композиция по любому из пп.1-18, которая снижает уровень глюкозы в сыворотке больных людей по меньшей мере на 5% в течение 120 мин после перорального введения.
20. Способ получения твердой фармацевтической композиции для перорального применения, включающей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных С4-C12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере один фармацевтически приемлемый наполнитель, выбранный из группы, включающей по меньшей мере 10 мас.% разрыхлителя, 10 мас.% маннита и 0,5 мас.% смазывающего вещества, включающий стадии перемалывания подходящих насыщенных или ненасыщенных C4-C12 жирных кислот и/или солей данных жирных кислот, грануляции результирующей жирной кислоты, полученной на стадии перемалывания, с использованием органического растворителя, сушки на воздухе гранул, полученных на стадии грануляции, протирания высушенных гранул через сито для получения гранул с требуемым размером частиц, перемешивания гранул жирных кислот с комплексом конъюгата IN-105 с другими наполнителями и прессования перемешанной смеси для формирования таблеток.
21. Способ по п.20, в котором композиция необязательно включает связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы.
22. Способ по п.20, в котором органический растворитель представляет собой изопропанол.
23. Способ получения твердой фармацевтической композиции для перорального применения, включающей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных C4-С12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере один фармацевтически приемлемый наполнитель, выбранный из группы, включающей по меньшей мере 10 мас.% разрыхлителя, 10 мас.% маннита и 0,5 мас.% смазывающего вещества, включающий стадии перемалывания подходящих насыщенных или ненасыщенных C4-C12 жирных кислот и/или солей данных жирных кислот и связующего компонента, суспендирования комплекса конъюгата IN-105 в органическом растворителе с использованием связующего компонента с целью формирования мокрой массы, грануляции компонентов, полученных на стадии суспендирования с использованием связующего компонента, перетирания высушенных гранул, полученных на стадии грануляции, смешивания гранул с другими наполнителями и прессования перемешанной смеси для формирования таблеток.
24. Способ по п.23, в котором композиция необязательно включает связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы.
25. Способ по п.23, в котором используемый органический растворитель выбран из группы, включающей изопропанол, ацетон, метиловый спирт, метил изобутилкетон, хлороформ, 1-пропанол, 2-пропанол, ацетонитрил, 1-бутанол, 2-бутанол, этиловый спирт, циклогексан, диоксан, этилацетат, диметилформамид, дихлорэтан, гексан, изооктан, метиленхлорид, трет-бутиловый спирт, толуол, четыреххлористый углерод или их комбинации.
26. Таблетка массой 5÷500 мг из твердой фармацевтической композиции для перорального применения, включающей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных C4-C12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере один фармацевтически приемлемый наполнитель, выбранный из группы, включающей по меньшей мере 10 мас.% разрыхлителя, 10 мас.% маннита и 0,5 мас.% смазывающего вещества.
27. Таблетка по п.26, в которой композиция необязательно включает связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы.
28. Таблетка из твердой фармацевтической композиции для перорального применения массой 50 мг, содержащая приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных C4-C12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере один фармацевтически приемлемый наполнитель, выбранный из группы, включающей по меньшей мере 10 мас.% разрыхлителя, 10 мас.% маннита и 0,5 мас.% смазывающего вещества.
29. Таблетка по п.28, в которой композиция необязательно включает связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы.
30. Таблетка массой 100 мг из твердой фармацевтической композиции для перорального применения, включающей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных C4-C12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере один фармацевтически приемлемый наполнитель, выбранный из группы, включающей по меньшей мере 10 мас.% разрыхлителя, 10 мас.% маннита и 0,5 мас.% смазывающего вещества.
31. Таблетка по п.30, в которой композиция необязательно включает связующие компоненты, пластификаторы, агенты, усиливающие проницаемость и солюбилизаторы.
32. Таблетка массой 150 мг из твердой фармацевтической композиции для перорального применения, включающей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных C4-C12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере один фармацевтически приемлемый наполнитель, выбранный из группы, включающей по меньшей мере 10 мас.% разрыхлителя, 10 мас.% маннита и 0,5 мас.% смазывающего вещества.
33. Таблетка по п.32, в которой композиция необязательно включает связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы.
34. Таблетка массой 200 мг из твердой фармацевтической композиции для перорального применения, включающей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных C4-C12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере один фармацевтически приемлемый наполнитель, выбранный из группы, включающей по меньшей мере 10 мас.% разрыхлителя, 10 мас.% маннита и 0,5 мас.% смазывающего вещества.
35. Таблетка по п.34, в которой композиция необязательно включает связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы.
36. Таблетка массой 250 мг из твердой фармацевтической композиции IN-105 для перорального применения, включающей приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных C4-C12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере один фармацевтически приемлемый наполнитель, выбранный из группы, включающей по меньшей мере 10 мас.% разрыхлителя, 10 мас.% маннита и 0,5 мас.% смазывающего вещества.
37. Таблетка по п.36, в которой композиция необязательно включает связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы.
38. Способ максимального контроля концентрации глюкозы в крови после приема пищи у больных диабетом, включающий оральное введение композиции по п.1 и контроль указанных уровней в течение 5÷60 мин после введения.
39. Стабильная твердая фармацевтическая композиция для перорального применения, характеризующаяся сохранением стабильности при воздействии условий, выбранных из группы, включающей интервал температур приблизительно 2÷40°С и относительную влажность (60±5)% при (25±2)°С, относительную влажность (65±5)% при (30+2)°С, относительную влажность (75±5)% при (40±2)°С в течение по меньшей мере 6 месяцев, и включающая приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных C4-C12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере один фармацевтически приемлемый наполнитель, выбранный из группы, включающей по меньшей мере 10 мас.% разрыхлителя, 10 мас.% маннита и 0,5 мас.% смазывающего вещества.
40. Композиция по п.39, которая необязательно включает связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы.
41. Композиция по п.39, в которой количество примесей возрастает по большей мере на 5%, по сравнению с количеством примесей во время изготовления.
42. Композиция по п.39, в которой количество примесей возрастает по большей мере на 10%, по сравнению с количеством примесей во время изготовления.
43. Композиция по п.39, в которой количество IN-105 снижается по большей мере на 10%.
44. Композиция по п.39, в которой разница между распределением твердости составляет не более 1 кг/см2 по сравнению с распределением твердости во время изготовления.
45. Композиция по п.39, в которой по меньшей мере (95±2)% композиции остается неразложившейся после воздействия условий, выбранных из группы, включающей интервал температур приблизительно 2÷8°С или 25÷40°С и относительную влажность (60±5)% при (25±2)°С, относительную влажность (65±5)% при (30±2)°С, относительную влажность (75±5)% при (40±2)°С в период по меньшей мере 6 месяцев.
46. Композиция по п.39, в которой по меньшей мере (90±2)% композиции остается неразложившейся после воздействия условий, выбранных из группы, включающей интервал температур приблизительно 2÷8°С или 25÷40°С и относительную влажность (60±5)% при (25±2)°С, относительную влажность (65±5)% при (30±2)°С, относительную влажность (75±5)% при (40±2)°С в период по меньшей мере 6 месяцев.
47. Способ получения аморфных высушенных распылительной сушкой частиц, характеризующихся приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных C4-C12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере одним фармацевтически приемлемым наполнителем, выбранным из группы, включающей по меньшей мере 10 мас.% разрыхлителя, 10 мас.% маннита и 0,5 мас.% смазывающего вещества, включающий стадии получения раствора или суспензии, содержащих конъюгат IN-105 и компонент жирной кислоты в растворителе, распыления раствора в камере в условиях, которые позволяют удалить существенную часть растворителя, и получения высушенных распылительной сушкой частиц конъюгата IN-105.
48. Способ по п.47, в котором композиция необязательно включает связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы.
49. Способ по п.47, в котором компонент жирной кислоты выбран из группы, включающей C4-C12 жирную кислоту и/или соли данной жирной кислоты.
50. Способ по п.49, в котором компонент жирной кислоты представляет собой капрат натрия.
51. Способ по п.47, в котором растворитель представляет собой воду.
52. Способ по п.47, в котором раствор или суспензия, содержащие IN-105 и компонент жирной кислоты в растворителе, дополнительно содержат разбавитель.
53. Способ по п.52, в котором разбавитель представляет собой маннит.
54. Способ по п.47, в котором размер полученных распылительной сушкой частиц составляет приблизительно 1÷100 мкм.
55. Способ по п.47, в котором раствор или суспензию, включающие IN-105, сушат распылительной сушкой при температуре в интервале 80÷150°С.
56. Способ по п.47, в котором давление при распылении, используемое для распылительной сушки, лежит в интервале от 84,3 кПа до 147,1 кПа.
57. Способ по любому из пп.47-56, в котором чистота полученной распылительной сушкой композиции IN-105 составляет по меньшей мере 95%.
58. Способ по любому из пп.47-56, в котором чистота полученной распылительной сушкой композиции IN-105 инсулина составляет по меньшей мере 98%.
59. Способ по любому из пп.47-56, в котором чистота полученной распылительной сушкой композиции IN-105 составляет по меньшей мере 99%.
60. Инсулинотропная фармацевтическая композиция, характеризующаяся пониженной в три раза митогенностью по сравнению со своим нативным аналогом и включающая приблизительно 0,01÷20 мас.% IN-105, приблизительно 10÷60 мас.% насыщенных или ненасыщенных C4-C12 жирных кислот, сложных эфиров жирных кислот или их солей и по меньшей мере один фармацевтически приемлемый наполнитель, выбранный из группы, включающей по меньшей мере 10 мас.% разрыхлителя, 10 мас.% маннита и 0,5 мас.% смазывающего вещества.
61. Композиция по п.60, которая необязательно включает связующие компоненты, пластификаторы, агенты, усиливающие проницаемость, и солюбилизаторы.
62. Композиция по п.60, которая снижает in vitro и/или in vivo уровень пролиферации клеток на по меньшей мере (20±5)% от уровня своего нативного аналога.
63. Композиция по п.60, которая снижает in vitro и/или in vivo уровень пролиферации клеток на по меньшей мере (2±0,5)% от уровня своего нативного аналога.
64. Композиция по любому из пп.60-63, которая проявляет метаболическую эффективность, близкую метаболической эффективности своего нативного аналога.
65. Композиция по п.60, в которой наполнители не влияют на митогенную активность конъюгата.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| ЧУЕШОВ В.И | |||
| и др | |||
| Промышленная технология лекарств, т.2 | |||
| - X.: МТК-Книга, Издательство НФАУ, 2002, 716 с | |||
| ТВЕРДОЕ ИНСУЛИНСОДЕРЖАЩЕЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1997 |
|
RU2117488C1 |
| MASSON M | |||
| et al | |||
| Cyclodextrins as permeation enhancers: some theoretical evaluations and in vitro testing | |||
| Journal of Controlled Release, 1999 May 1, 59(1): 107-118 [Найдено 06.07.2011] | |||

