Уровень техники
Антагонисты 5-НТ3-рецепторов представляют группу лекарственных средств, которые функционируют в качестве рецепторных антагонистов 5-НТ3-рецептора, подтипа серотонинового рецептора, находящегося в окончаниях блуждающего нерва и некоторых областях головного мозга. За примечательным исключением алосетрона и цилансетрона, которые используются в лечении синдрома раздраженного кишечника, все антагонисты 5-НТ3-рецепторов представляют собой противорвотные лекарственные средства, используемые в предупреждении и лечении тошноты и рвоты. Они являются особенно эффективными в контроле тошноты и рвоты, вызванных химиотерапией при раке, и рассматриваются в качестве золотого стандарта для данной цели. Ондансетрон является антагонистом 5-НТ3-рецептора, который используется самостоятельно или в комбинации с другими лекарственными средствами для предупреждения тошноты и рвоты, и используется для предупреждения тошноты и рвоты, вызванной лечением рака лекарственными средствами (химиотерапией) и лучевой терапией. Он также применяется для предупреждения и лечения тошноты и рвоты после хирургической операции.
Сущность изобретения
В данной заявке раскрываются твердые лекарственные формы с замедленным высвобождением. Конкретнее, в данной заявке раскрываются твердые лекарственные формы, обладающие противорвотным действием, с замедленным высвобождением для предупреждения тошноты и рвоты. Согласно аспектам, описанным здесь, раскрывается таблетка ондансетрона с замедленным высвобождением, которая включает ядро, содержащее агент для замедленного высвобождения, содержащий ондансетрон или его фармацевтически приемлемую соль, и электролит; агент первой изолирующей оболочки; слой препарата с немедленным высвобождением, окружающий агент первой изолирующей оболочки, содержащий ондансетрон или его фармацевтически приемлемую соль; и агент второй изолирующей оболочки, где слой с немедленным высвобождением выполнен таким образом, чтобы обеспечить высвобождение примерно 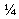 от общей дозы ондансетрона в течение 1 ч после перорального введения, и где выполнен таким образом, чтобы обеспечить высвобождение остальной дозы ондансетрона в течение периода времени до 24 ч посредством высвобождения нулевого порядка. В варианте осуществления ядро содержит примерно 18 мг ондансетрона в виде свободного основания. В варианте осуществления ядро содержит примерно 20 мг ондансетрона в виде свободного основания. В варианте осуществления ядро содержит примерно 28 мг ондансетрона в виде свободного основания. В варианте осуществления электролит представляет дигидроцитрат натрия безводный, находящийся в концентрации в пределах примерно от 50 масс. % до примерно 100 масс. % по отношению к массе агента для замедленного высвобождения. В варианте осуществления агент для замедленного высвобождения представляет собой гидрофильный набухающий матрикс.В варианте осуществления гидрофильный набухающий матрикс ядра представляет METHOCEL™ K4M Premium DC, гипромеллоза первой изолирующей оболочки и второй изолирующей оболочки представляет METHOCEL™ Е5 Premium LV, и гипромеллоза в слое препарата с немедленным высвобождением представляет METHOCEL™ Е5 Premium LV. В варианте осуществления гидрофильный набухающий матрикс ядра представляет METHOCEL™ K4M Premium CR, гипромеллоза первой изолирующей оболочки и второй изолирующей оболочки представляет METHOCEL™ Е5 Premium LV, и гипромеллоза в слое препарата с немедленным высвобождением представляет METHOCEL™ Е5 Premium LV. В варианте осуществления слой препарата с немедленным высвобождением содержит примерно 6 мг ондансетрона.
от общей дозы ондансетрона в течение 1 ч после перорального введения, и где выполнен таким образом, чтобы обеспечить высвобождение остальной дозы ондансетрона в течение периода времени до 24 ч посредством высвобождения нулевого порядка. В варианте осуществления ядро содержит примерно 18 мг ондансетрона в виде свободного основания. В варианте осуществления ядро содержит примерно 20 мг ондансетрона в виде свободного основания. В варианте осуществления ядро содержит примерно 28 мг ондансетрона в виде свободного основания. В варианте осуществления электролит представляет дигидроцитрат натрия безводный, находящийся в концентрации в пределах примерно от 50 масс. % до примерно 100 масс. % по отношению к массе агента для замедленного высвобождения. В варианте осуществления агент для замедленного высвобождения представляет собой гидрофильный набухающий матрикс.В варианте осуществления гидрофильный набухающий матрикс ядра представляет METHOCEL™ K4M Premium DC, гипромеллоза первой изолирующей оболочки и второй изолирующей оболочки представляет METHOCEL™ Е5 Premium LV, и гипромеллоза в слое препарата с немедленным высвобождением представляет METHOCEL™ Е5 Premium LV. В варианте осуществления гидрофильный набухающий матрикс ядра представляет METHOCEL™ K4M Premium CR, гипромеллоза первой изолирующей оболочки и второй изолирующей оболочки представляет METHOCEL™ Е5 Premium LV, и гипромеллоза в слое препарата с немедленным высвобождением представляет METHOCEL™ Е5 Premium LV. В варианте осуществления слой препарата с немедленным высвобождением содержит примерно 6 мг ондансетрона.
Согласно аспектам, описанным здесь, раскрывается таблетка ондансетрона с замедленным высвобождением, которая включает ядро, содержащее гидрофильный набухающий матрикс, содержащий ондансетрон или его фармацевтически приемлемую соль, и гидроцитрат натрия безводный; первую изолирующую оболочку, содержащую гипромеллозу и plasACRYL™; слой препарата с немедленным высвобождением, окружающий первую изолирующую оболочку, содержащий ондансетрон или его фармацевтически приемлемую соль, гипромеллозу и plasACRYL™; и вторую изолирующую оболочку, содержащую гипромеллозу и plasACRYL™, где слой с немедленным высвобождением выполнен таким образом, чтобы обеспечить высвобождение примерно % от общей дозы ондансетрона примерно в течение 1 ч после перорального введения, и где ядро выполнен таким образом, чтобы обеспечить высвобождение остальной дозы ондансетрона в течение периода времени до 24 ч посредством высвобождения нулевого порядка. В варианте осуществления ядро содержит примерно 18 мг ондансетрона в виде свободного основания. В варианте осуществления ядро содержит примерно 20 мг ондансетрона в виде свободного основания. В варианте осуществления ядро содержит примерно 28 мг ондансетрона в виде свободного основания. В варианте осуществления дигидроцитрат натрия безводный находится в концентрации в пределах примерно от 50 масс. % до примерно 100 масс. % по отношению к массе гидрофильного набухающего матрикса. В варианте осуществления гидрофильный набухающий матрикс ядра представляет METHOCEL™ K4M Premium DC, гипромеллоза первой изолирующей оболочки и второй изолирующей оболочки представляет METHOCEL™ Е5 Premium LV, и гипромеллоза в слое препарата с немедленным высвобождением представляет METHOCEL™ Е5 Premium LV. В варианте осуществления гидрофильный набухающий матрикс ядра представляет METHOCEL™ K4M Premium CR, гипромеллоза первой изолирующей оболочки и второй изолирующей оболочки представляет METHOCEL™ Е5 Premium LV, и гипромеллоза в слое препарата с немедленным высвобождением представляет METHOCEL™ Е5 Premium LV. В варианте осуществления слой препарата с немедленным высвобождением содержит примерно 6 мг ондансетрона.
Согласно аспектам, описанным здесь, раскрывается твердая лекарственная форма с замедленным высвобождением, которая включает внутреннюю часть, где внутренняя часть содержит первую дозу, по меньшей мере, одного антагониста серотонина; первую оболочку, где первая оболочка непосредственно инкапсулирует внутреннюю часть твердой лекарственной формы; оболочку слоя препарата, где оболочка слоя препарата непосредственно инкапсулирует первую оболочку, где оболочка слоя препарата содержит вторую дозу, по меньшей мере, одного антагониста серотонина, где оболочка слоя препарата составляет, по меньшей мере, 4 масс. % по отношению к массе твердой лекарственной формы, где вторая доза равна, по меньшей мере, 15 масс. % от общей дозы, по меньшей мере, одного антагониста серотонина в твердой лекарственной форме, и где первая доза равна общей дозе минус вторая доза, и вторую оболочку, где вторая оболочка непосредственно инкапсулирует оболочку слоя препарата, где внутренняя часть имеет растворимость в воде X, где первая оболочка, оболочка слоя препарата и вторая оболочка обладают растворимостью в воде, по меньшей мере, Y, и где X ниже Y. В варианте осуществления, по меньшей мере, один антагонист рецептора серотонина типа 3 является ондансетроном гидрохлоридом. В варианте осуществления вторая доза равна, по меньшей мере, 20 масс. % от общей дозы, по меньшей мере, одного антагониста рецептора серотонина типа 3 в твердой лекарственной форме. В варианте осуществления, по меньшей мере, один антагонист рецептора серотонина типа 3 является ондансетроном гидрохлоридом. В варианте осуществления вторая доза равна, по меньшей мере, 25 масс. % от общей дозы, по меньшей мере, одного антагониста рецептора серотонина типа 3 в твердой лекарственной форме. В варианте осуществления первая оболочка и вторая оболочка содержит гидрофильное вещество. В варианте осуществления слой препарата дополнительно содержит гидрофильное вещество. В варианте осуществления гидрофильное вещество представляет гипромеллозу. В варианте осуществления первая оболочка и вторая оболочка каждая составляет, по меньшей мере, 1,5 масс. % по отношению к массе твердой лекарственной формы. В варианте осуществления отношение гипромеллозы, по меньшей мере, к одному антагонисту рецептора серотонина типа 3 в слое препарата составляет 4:6. В варианте осуществления общее количество гипромеллозы в первой оболочке, слое препарата и второй оболочке равно ниже 4 масс. % по отношению к массе твердой лекарственной формы. В варианте осуществления ядро дополнительно содержит цитрат натрия в количестве ниже 15 масс. % по отношению к массе ядра. В варианте осуществления X существенно ниже Y, в результате вторая доза по существу высвобождается из твердой лекарственной формы в течение менее 12 ч после воздействия на твердую лекарственную форму водной среды, и первая доза по существу высвобождается из твердой лекарственной формы с профилем высвобождения нулевого порядка в течение 12-24 ч после воздействия на твердую лекарственную форму водной среды. В варианте осуществления водная среда имеет pH от 1,5 до pH 7,5. В варианте осуществления твердая лекарственная форма спрессована в таблетку. В варианте осуществления твердая лекарственная форма сформирована в виде капсулы. В варианте осуществления ядро дополнительно содержит глицин в количестве ниже 20 масс. % по отношению к массе ядра.
Согласно аспектам, описанным здесь, раскрывается таблетка ондансетрона с замедленным высвобождением, полученная прессованием ядра таблетки с замедленным высвобождением и затем покрытием ядра таблетки первой изолирующей оболочкой, затем слоем препарата и наконец, второй изолирующей оболочкой, где ядро таблетки содержит гидрофильный набухающий матрикс, содержащий ондансетрон гидрохлорид и дигидроцитрат натрия безводный, где первая изолирующая оболочка содержит гипромеллозу и plasACRYL™, где оболочка с препаратом содержит ондансетрон гидрохлорид, гипромеллозу и plasACRYL™ и где вторая изолирующая оболочка содержит гипромеллозу и plasACRYL™ Т20.
Согласно аспектам, описанным здесь, раскрывается твердая лекарственная форма для перорального введения, которая включает ядро, содержащее матрикс из неионогенного полимера, первое количество первого противорвотного препарата или его фармацевтически приемлемой соли, диспергированное внутри матрикса, и соль, диспергированную внутри матрикса; первую изолирующую оболочку, окружающую ядро, где первая изолирующая оболочка состоит из матрикса из неионогенного полимера; и слой препарата с немедленным высвобождением, окружающий первую изолирующую оболочку, где слой препарата с немедленным высвобождением содержит неионогенный полимер, и второе количество второго противорвотного препарата или его фармацевтически приемлемой соли, диспергированное в нем, где слой препарата выполнен таким образом, чтобы обеспечить высвобождение второго количества противорвотного препарата в течение, по меньшей мере, 1 ч, где твердая лекарственная форма выполнен таким образом, чтобы обеспечить высвобождение первого количества первого противорвотного препарата и второго количества второго противорвотного препарата в течение периода времени минимум 16 ч.
Согласно аспектам, описанным здесь, раскрывается твердая лекарственная форма для перорального введения, которая включает ядро, содержащее гипромеллозу, 18 мг ондансетрона или эквивалентное количество соли ондансетрона, и цитрат натрия безводный; первую изолирующую оболочку, окружающую ядро и содержащую гипромеллозу; и слой препарата с немедленным высвобождением, окружающий первую изолирующую оболочку и содержащий гипромеллозу и 6 мг ондансетрона или эквивалентное количество соли ондансетрона, слой препарата с немедленным высвобождением, приспособленный для высвобождения ондансетрона в течение периода времени, по меньшей мере, 1 ч, где общее количество ондансетрона в лекарственной форме высвобождается в течение 24 ч.
Согласно аспектам, описанным здесь, раскрывается твердая лекарственная форма для перорального введения, которая включает ядро, содержащее матрикс из неионогенного полимера, первое количество ондансетрона или эквивалентное количество соли ондансетрона, диспергированное внутри матрикса, и соль, диспергированную внутри матрикса; первую изолирующую оболочку, окружающую ядро, где первая изолирующая оболочка состоит из матрикса из неионогенного полимера, и слой препарата с немедленным высвобождением, окружающий первую изолирующую оболочку, где слой препарата с немедленным высвобождением содержит неионогенный полимер, и второе количество ондансетрона или эквивалентное количество соли ондансетрона, диспергированное в нем, где твердая лекарственная форма для перорального введения обеспечивает профиль растворения ондансетрона in vitro при определении на аппарате для оценки растворения с 2 лопастями при 37°С в водном растворе, содержащем дистиллированную воду, при 50 об/мин, показывающий следующее: а) примерно от 20% до 50% от общего количества ондансетрона высвобождается через 2,5 ч при определении в аппарате; b) примерно от 50% до 70% от общего количества ондансетрона высвобождается через 5 ч при определении в аппарате; с) не менее примерно 90% от общего количества ондансетрона высвобождается через 15 ч при определении в аппарате.
Согласно аспектам, описанным здесь, раскрывается упакованный фармацевтический препарат, который включает множество твердых лекарственных форм для перорального введения по настоящему изобретению в герметично закрытом контейнере и инструкции по введению лекарственных форм перорально для осуществления предупреждения тошноты и рвоты.
Согласно аспектам, описанным здесь, раскрывается фармацевтический препарат, который включает множество твердых лекарственных форм для перорального введения по настоящему изобретению, каждая в отдельном герметично закрытом контейнере и инструкции по введению лекарственных форм перорально для осуществления предупреждения тошноты и рвоты.
Согласно аспектам, описанным здесь, раскрывается способ контроля тошноты и рвоты, включающий введение твердой лекарственной формы по настоящему изобретению пациенту, где тошнота и рвота контролируются после того, как количество ондансетрона высвободилось из твердой лекарственной формы, достигло системного кровотока у пациента, и всосалось у пациента.
Согласно аспектам, описанным здесь, раскрывается способ снижения побочных эффектов химиотерапии, включающий введение твердой лекарственной формы по настоящему изобретению пациенту, где побочные эффекты, включая тошноту и рвоту, снижаются после того, как количество ондансетрона высвободилось из твердой лекарственной формы, всосалось у пациента и достигло системного кровотока у пациента.
Согласно аспектам, описанным здесь, раскрывается способ снижения побочных эффектов в результате болезни движения, включающий введение твердой лекарственной формы по настоящему изобретению пациенту, где побочные эффекты, включая тошноту и рвоту, снижаются после того, как количество ондансетрона высвободилось из твердой лекарственной формы, всосалось у пациента и достигло системного кровотока у пациента.
Согласно аспектам, описанным здесь, раскрывается способ снижения побочных эффектов анестетиков, включающий введение твердой лекарственной формы по настоящему изобретению пациенту, где побочные эффекты, включая тошноту и рвоту, снижаются после того, как количество ондансетрона высвободилось из твердой лекарственной формы, всосалось у пациента и достигло системного кровотока у пациента.
Краткое описание фигур
Раскрытые варианты осуществления настоящего изобретения будут дополнительно пояснены при обращении к прилагаемым фигурам. Представленные фигуры не обязательно выполнены в масштабе, и вместо этого, в общем, приведены для лучшей иллюстрации принципов раскрытых вариантов осуществления.
На фигуре 1 показаны профили растворения ондансетрона согласно двум вариантам осуществления твердых лекарственных форм с замедленным высвобождением по настоящему изобретению при определении с использованием аппарата для растворения по Фармакопее США 2 (лопастного) при 50 об/мин, при температуре 37±0,5°С с дистиллированной водой в качестве среды для растворения.
На фигуре 2 показан профиль растворения ондансетрона согласно варианту осуществления твердой лекарственной формы с замедленным высвобождением по настоящему изобретению при определении с использованием аппарата для растворения по Фармакопее США 2 (лопастного при 50 об/мин, при температуре 37±0,5°С с 0,1Н раствором HCl и фосфатным буфером с pH 6,8 в качестве среды для растворения.
На фигуре 3 показан профиль растворения ондансетрона согласно варианту осуществления твердой лекарственной формы с замедленным высвобождением по настоящему изобретению при определении с использованием аппарата для растворения по Фармакопее США 2 (лопастного при 50 об/мин, при температуре 37±0,5°С с 0,1Н раствором HCl и фосфатным буфером с pH 6,8 в качестве среды для растворения.
На фигуре 4 показаны профили растворения ондансетрона согласно варианту осуществления твердой лекарственной формы с замедленным высвобождением по настоящему изобретению при определении с использованием аппарата для растворения по Фармакопее США 2 (лопастного при 50 об/мин, при температуре 37±0,5°С с физиологически соответствующими средами с pH в пределах от 1,2 до 7,2, примерные уровни pH, обнаруживаемые в желудочно-кишечном тракте.
На фигуре 5 показаны средние измеренные значения концентрации ондансетрона в плазме крови в зависимости от времени, полученные при введении различных вариантов осуществления твердых лекарственных форм с замедленным высвобождением по настоящему изобретению и референс-препарата.
На фигуре 6 показаны ln-преобразованные средние значения концентрации ондансетрона в плазме крови в зависимости от времени, полученные при введении различных вариантов осуществления твердых лекарственных форм с замедленным высвобождением по настоящему изобретению и референс-препарата.
На фигуре 7 показаны средние значения концентрации в плазме крови в линейном диапазоне измерения в зависимости от времени для тест-препарата на сутки 1, полученные при введении варианта осуществления твердой лекарственной формы с замедленным высвобождением по настоящему изобретению и референс-препарата.
На фигуре 8 показаны средние значения концентрации в плазме крови в линейном диапазоне измерения в зависимости от времени для тест-препарата на сутки 2, полученные при введении варианта осуществления твердой лекарственной формы с замедленным высвобождением по настоящему изобретению и референс-препарата.
На фигуре 9 показаны ln-преобразованные средние значения концентрации в плазме крови в зависимости от времени для тест-препарата на сутки 1, полученные при введении варианта осуществления твердой лекарственной формы с замедленным высвобождением по настоящему изобретению и референс-препарата.
На фигуре 10 показаны ln-преобразованные средние значения концентрации в плазме крови в зависимости от времени для тест-препарата на сутки 2, полученные при введении варианта осуществления твердой лекарственной формы с замедленным высвобождением по настоящему изобретению и референс-препарата.
На фигуре 11 показан общий профиль средних значений концентрации в плазме крови в линейном диапазоне измерений в зависимости от времени для тест-препарата и референс-препарата, полученные при введении варианта осуществления твердого лекарственной формы с замедленным высвобождением по настоящему изобретению и референс-препарата.
На фигуре 12 показан ln-преобразованный общий профиль средних значений концентрации в плазме крови в зависимости от времени для тест-препарата и референс-препарата, полученный при введении варианта осуществления твердого лекарственной формы с замедленным высвобождением по настоящему изобретению и референс-препарата.
Несмотря на то, что вышеуказанные фигуры приведены для раскрытых вариантов осуществления, предусматриваются другие варианты осуществления, как это отмечается в обсуждении. В данном раскрытии представлены иллюстративные варианты осуществления путем представления и без ограничения. Специалисты в данной области могут разработать многочисленные другие модификации и варианты осуществления, не отступая от объема и сущности принципов раскрытых вариантов осуществления настоящего изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В том смысле, в котором здесь используются следующие термины, они имеют определения, которые приведены ниже.
Термин «гидрофобность» относится к шкале характеристик растворимости, объединяющей гидрофобность и гидрофильность аминокислот. Конкретнее, данный термин относится к скользящей шкале, аналогичной шкале значений pH, которая приписывает относительные значения, представляющие относительный баланс гидрофобных и гидрофильных компонентов аминокислоты. Типичная шкала представлена в публикации Pliska et al., J. Chromatog., 216, 79, 1981 под названием «Relative Hydrophobic Character of Amino Acid Side Chains», где глицин имеет значение О, представляя относительно равный баланс гидрофобных и гидрофильных компонентов, и он может быть отнесен к относительно «нейтральному», «сбалансированному», «незначительно гидрофильному» или «слабо гидрофобному, изолейцин, который имеет положительное значение 1,83 и является высоко гидрофобным, и с противоположного конца шкалы, аспарагиновая кислота, которая имеет отрицательное значение -2,15 и может быть охарактеризована, как высоко гидрофильная аминокислота. Такая шкала и индекс гидрофобности, описанные здесь, хорошо известны и понятны специалистам в данной области.
Термин «монолитные» относится к таблеткам, для которых не требуются многочисленные слои, особые формы, осмотические компартменты и/или специализированные покрытия, как правило, без сочленений или швов, и которые можно приготовить таблетированием на современном высокоскоростном оборудовании для изготовления таблеток.
В том смысле, в котором здесь используется термин «бимодальный», он относится к бимодальным профилям высвобождения препарата (быстрое высвобождение/медленное высвобождение).
Термины «антагонист серотонина» или «антагонист 5-НТ3-рецептора» относятся к группе лекарственных средств, пригодных для предупреждения и ослабления тошноты и рвоты, вызванных химиотерапией и анестезией. Полагается, что антагонисты серотонина функционируют посредством блокирования эффектов химического серотонина, который продуцируется в головном мозге и желудке. Антагонисты 5-НТ3-рецептора, эффективные в лечении вызванной химиотерапией рвоты, включают, не ограничиваясь этим, доласетрон, гранисетрон, ондансетрон, палоносетрон, трописетрон.
Обеспечиваются твердые лекарственные формы с замедленным высвобождением. Конкретнее, настоящее изобретение относится к бимодальным твердым лекарственным формам с замедленным высвобождением для предупреждения тошноты и рвоты, вызванной химиотерапией. В варианте осуществления твердая лекарственная форма с замедленным высвобождением включает внутреннюю часть, где внутренняя часть содержит первую дозу ондансетрона; первую оболочку, где первая оболочка непосредственно инкапсулирует внутреннюю часть твердой лекарственной формы; оболочку слоя препарата, где оболочка слоя препарата непосредственно инкапсулирует первую оболочку, где оболочка слоя препарата содержит вторую дозу ондансетрона, где оболочка слоя препарата составляет, по меньшей мере, 4 масс. % по отношению к массе твердой лекарственной формы, где вторая доза равна, по меньшей мере, 15 масс. % по отношению от общей дозы ондансетрона в твердой лекарственной форме, и где первая доза равна общей дозе минус вторая доза; и вторую оболочку, где вторая оболочка непосредственно инкапсулирует оболочку слоя препарата, где внутренняя часть имеет растворимость в воде X, где первая оболочка, оболочка слоя препарата и вторая оболочка обладают растворимостью в воде Y, и где X ниже Y. В варианте осуществления твердая лекарственная форма с замедленным высвобождением способна обеспечивать «взрывное» высвобождение примерно 25% ондансетрона с последующим высвобождением нулевого порядка остального ондансетрона в течение периода времени 16-20 ч. В варианте осуществления твердая лекарственная форма с замедленным высвобождением способна обеспечивать «взрывное» высвобождение примерно 25% ондансетрона с последующим высвобождением нулевого порядка остального ондансетрона в течение периода времени 20-30 ч.
В варианте осуществления твердая лекарственная форма по настоящему изобретению включает лекарственные формы для перорального введения, такие как таблетки, капсулы, таблетки в форме капсул, гранулы. В варианте осуществления твердая лекарственная форма по настоящему изобретению представляет лекарственную форму для ректального введения, такую как суппозиторий.
Ондансетрон
Ондансетрон является эффективным противорвотным лекарственным средством, которое существенно повышает качество жизни у пациентов, проходящих химиотерапию. Обычная доза, вводимая пациентам, составляет 8 мг, 16 мг, 24 мг или 32 мг, которые вводятся один раз в день или в разделенных дозах. Ондансетрон проявляет центральное и/или периферическое действие, преимущественно блокируя серотониновые 5-НТ3-рецепторы. Ондансетрон гидрохлорид (HCl) представляет дигидрат, рацемическую смесь ондансетрона. Ондансетрон имеет эмпирическую форму C18H19N3O⋅2H2O с молекулярной массой 365,9. Ондансетрон HCl дигидрат представляет порошок от белого до не совсем белого цвета, который растворяется в воде и физиологическом растворе.
Внутренняя часть («ядро») твердых лекарственных форм варианта осуществления настоящего изобретения
По мере того, как таблетка проходит по пищеварительному тракту человека, она подвергается воздействию сред со значениями pH в пределах от 1,5 до примерно 7,4. Слюна в ротовой полости имеет нейтральное значение pH, желудок имеет pH в диапазоне примерно 1,5-4,0, и pH кишечника находится в диапазоне pH 5,0-7,5. Для приближения высвобождения препарата к высвобождению нулевого порядка, растворение препарата не должно зависеть от pH окружающей среды. Внутренняя часть («ядро») лекарственной формы по настоящему изобретению может приближаться к высвобождению препарата нулевого порядка.
Внутренняя часть - электролитная основа
В варианте осуществления внутренняя часть («ядро») состоит из гидрофильного набухающего матрикса, в котором находится фармацевтически активный ингредиент («API») и один или более электролитов. «Электролитное ядро» представляет композицию с медленным высвобождением («SR»). Один или более электролитов, в комбинации с API или другой солью, при взаимодействии в водной среде вызывает реакцию затвердевания матрикса. Скорость диффузии, направленной наружу, контролируется воздействием водной среды на внутреннюю часть. В свою очередь, это вызывает реакцию затвердевания, которая протекает в зависимости от времени от внешних границ к внутренним границам внутренней части; в свою очередь, продукт реакции затвердевания ограничивает диффузию API, направленную наружу, по мере того, как проникновение водной среды внутрь вызывает постепенное затвердевание от внешних границ внутренней части в направлении к внутреннему ядру.
Во внутренней части используется феномен коллоидной химии «высаливание» для сдерживания кинетики набухания и эрозии матрикса неионогенного полимера, содержащего API и один или более электролитов. Присутствие таких электролитов в форме ионизируемых солей позволяет образоваться неразъемным диффузионным каналам; агенты для каналообразования на предшествующем уровне техники не были ионизируемыми, следовательно, диффузионные каналы были непредсказуемыми, приводя к профилям слабого высвобождения и отсутствию контроля. Также электролиты вносят свой вклад в сжатие микросреды в таблетке, pH которой опосредуется значением рКа электролита, что приводит к повышению или снижению растворимости самого API. По мере гидратации матрикса электролит и полимер конкурируют за воду в процессе гидратации с API, приводя к обеспечению программируемой скорости высвобождения. Таким образом, внутренняя часть способна к pH-независимому высвобождению API нулевого порядка в течение 24 ч, независимо от растворимости самого API.
Посредством ионного взаимодействия/комплексообразования/мо-лекулярной ассоциации и/или самоассоциации между лекарственным средством и электролитом или комбинаций электролит/лекарственное средство, гомогенно диспергированных в набухающем полимере, таком как гидроксипропилметилцеллюлоза (НРМС), модифицируется динамика скорости набухания матрикса и эрозии набухающего полимера в соответствии с изменениями pH внешней среды в пределах 1,5-7,0. Такие взаимодействия приводят к контролируемому затвердеванию матрикса. Такое затвердевание ответственно за контроль эрозии/растворения полимера и скорости высвобождения препарата. Согласно дизайну растворитель проникает по периферии таблетки и быстрое первоначальное взаимодействие между лекарственным средством и электролитом, включенным в полимерный матрикс, вызывает немедленное затвердевание внешней границы таблетки, скорость затвердевания постоянно снижается по направлению к центру ядра матрикса в зависимости от времени в течение длительного периода времени (например, 24 ч).
Дифференциальная скорость затвердевания матрикса является ведущим принципом для внутренней части, которая зависит от и регулируется скоростью вхождения жидкости в ядро внутренней части. С одновременным зависимым от времени снижением целостности гелевого слоя снижается скорость диффузии препарата. Данное явление компенсирует увеличение длины пути диффузии и снижение площади поверхности «убывающего» ядра, что возникает в результате набухающих свойств полимера. Следовательно, достигается более контролируемое, предпочтительно нулевого порядка высвобождение препарата. Процесс высвобождения препарата можно приспособить до 24 ч. Контроль изменений в прочности ядра и синхронизация эластичности/набухания передней части и описанные «убывающие» фазовые границы, а также эрозия границ растворения передней части (т.е. эрозия периферии таблетки) приводит к контролируемому высвобождению препарата, предпочтительно включая кинетику нулевого порядка. Необязательно затвердевание полимерного матрикса также легко достигается посредством взаимодействия двойных солей. Эта бинарная комбинация солей также равномерно диспергируется в полимерном матриксе, что посредством ионного взаимодействия/комплексообразования/молекулярной ассоциации и/или самоассоциации повышает относительную прочность и твердость матрикса, приводя к контролируемому высвобождению препарата с механизмом, аналогичным описанному выше.
Одним гидрофильным материалом, пригодным для внутренней части, является HPMC K4M. Это неионогенный набухающий гидрофильный полимер производства «The Dow Chemical Сотрапу» под торговым названием «Methocel». HPMC K4M также имеет сокращенное название HPMC K4MP, в котором «Р» относится к простому эфиру целлюлозы высокого качества, предназначенному для использования в композициях с контролируемым высвобождением. Цифра «4» в обозначении предполагает, что полимер имеет номинальную вязкость 4000 (2% в воде). Процент метоксильных и гидроксипропильных групп составляет 19-24 и 7-12 соответственно. В ее физической форме HPMC K4M представляет сыпучий порошок не совсем белого цвета с ограниченным диапазоном размера частиц, составляющим 90% < 100 меш. Имеются другие типы НРМС, такие как K100LVP, K15MP, K100MP, Е4МР и Е10МР CR с номинальной вязкостью соответственно 100, 1500, 100000, 4000 и 10000.
Поскольку внутренняя часть состоит из нековалентно связанного матрикса, то процесс производства является в основном двухстадийным процессом, состоящим из сухого смешивания и прямого прессования.
В варианте осуществления соль диспергирована в матриксе в концентрации в пределах примерно от 50% до 100 масс. % по отношению к массе полимерного матрикса. В варианте осуществления соль выбрана из одного-двух членов группы, состоящей из хлорида натрия, бикарбоната натрия, бикарбоната калия, цитрата натрия, бисульфата натрия, сульфита натрия, сульфата магния, хлорида кальция, хлорида калия и карбоната натрия.
Полагается, что в результате взаимодействия препарата и соли образуется комплекс в окружающем набухающем матриксе в виде слоев, поскольку это происходит в зависимости от времени по мере того, как среда растворителя для высвобождения препарата проникает внутрь таблетки. Аналогично, поскольку катализатором для инициации высвобождения препарата является вхождение жидкости, то скорость высвобождения препарата также регулируется прогрессирующим затвердеванием внутри комплекса соли.
Бинарную солевую систему (например, хлорид кальция и карбонат натрия) также можно использовать в том случае, когда реакция затвердевания зависит от взаимодействия солей. Хлорид кальция может быть включен для образования комплекса с карбонатом натрия. При такой комбинации продуктами реакции являются нерастворимый карбонат кальция и образующий растворимый канал хлорид натрия. Поскольку карбонат кальция сам по себе включается в полимерный матрикс, то он инициирует затвердевание и медленно растворяется с вхождением жидкости и последующим образованием диффузионных каналов по мере диффундирования препарата. Аналогичным путем другие бинарные солевые комбинации проявляют зависимое от времени поведение «затвердевание/размягчение».
Количество используемой соли можно определить с учетом растворимости препарата, свойств полимера и требуемой степени затвердевания матрикса. В случае дилтиазема гидрохлорида в матриксе НРМС, 100 мг бикарбоната натрия обеспечивает подходящее затвердевание матрикса для контролируемого высвобождения нулевого порядка, в то время, как в случае такого же количества препарата в другом полимере, таком как полиэтиленоксид, 50 мг бикарбоната натрия оказалось идеальным для достижения контролируемого высвобождения нулевого порядка.
Фармацевтически активный ингредиент можно легко выбрать из группы, состоящей из апрепитанта (Emend), дексаметазона, доласетрона (Anzemet), дронабинола (Marinol), дроперидола (Insapsine), гранисетрона (Kytril), галоперидола (Haldol), метилпреднизолона (Medrol), метоклопрамида (Reglan), набилона (Cesamet), ондансетрона (Zofran), палоносетрона (Aloxi), прохлорперазина (Procomp) и их фармацевтически приемлемых солей, или их комбинаций.
В варианте осуществления внутренняя часть твердой лекарственной формы по настоящему изобретению представляет матрикс гидрофильного набухающего полимера, содержащий внутри матрикса диспергированное фармацевтически эффективное количество, по меньшей мере, одного антагониста серотонина, степень солюбилизации которого по существу не зависит от pH в пределах значений pH от 1,5 до 7,5, и неорганическую соль, где неорганическая соль находится в концентрации в пределах от 50 масс. % до 100 масс. % по отношению к массе полимерного матрикса. В варианте осуществления неорганическая соль представляет цитрат натрия. В варианте осуществления гидрофильный набухающий полимер матрикса представляет гидроксипропилметилцеллюлозу или полиэтиленоксид.
Внутреннюю часть, описанную выше, можно приготовить способом, раскрытым в патенте США №6090411, который включен здесь в виде ссылки для принципов, раскрытых здесь.
Внутренняя часть - аминокислотная основа
В варианте осуществления внутренняя часть («ядро») состоит из гидрофильного экстрагранулярного полимера, в котором диспергировано множество гранул API, гранулированных, по меньшей мере, с одной аминокислотой, и внутригранулярного полимера. «Аминокислотное ядро» или «АА ядро» представляет композицию с медленным высвобождением («SR»). Гранулы диспергированы в гидрофильном экстрагранулярном полимере с образованием монолитного матрикса. Экстрагранулярный полимер гидратируется быстрее по сравнению с внутригранулярным полимером. Быстрая гидратация экстрагранулярного полимера способствует приближению к линейному профилю высвобождения препарата и способствует практически 100% растворению, несмотря на то, что увеличение продолжительности высвобождения и снижение «взрывного» высвобождения часто характерно для лекарственных форм с замедленным высвобождением. Несмотря на то, что скорость линейного высвобождения можно приспособить для соответствия потребностям в каждом применении путем выбора полимеров для обеспечения различных скоростей растворения, как это понятно специалистам в данной области, время высвобождения в диапазоне от 12 до 24 ч является наиболее предпочтительным.
Внутригранулярный полимер объединяют с API, и, по меньшей мере, с одной аминокислотой с образованием гранул. Внутригранулярный полимер может представлять одно или более из следующего: поливинилацетат, галактоманновый полисахарид, такой как гидроксипропиловый гуар, гуаровую камедь, камедь плодов рожкового дерева, пектин, аравийскую камедь, трагантовую камедь, камедь карайи, простые эфиры целлюлозы, такие как гидроксиропилметилцеллюлозу (НРМС), а также другие камеди и простые эфиры целлюлозы, которые могут выбрать специалисты в данной области по свойствам, согласующимся с принципами данного изобретения. В варианте осуществления внутригранулярный полимер представляет галактоманнановый полисахарид, такой как гуаровая камедь (с вязкостью в пределах 75-6000 сП для 1% раствора при 25°С в воде и размером частиц 100-300 мкм).
Внутригранулярный полимер во внутренней части находится в количестве от 4 масс. % до 45 масс. % по отношению к общей массе лекарственной формы. Конкретный тип внутригранулярного полимера и используемое количество внутригранулярного полимера выбирают в зависимости от обеспечения желаемой скорости высвобождения препарата, вязкости полимера, желаемой нагрузки лекарственным средством и растворимости препарата. Внутригранулярный полимер гидратируется медленнее, чем экстрагранулярный полимер. Относительное различие в скоростях гидратации между двумя полимерами приводит к тому, что внутригранулярный полимер является менее вязким и экстрагранулярный полимер более вязким. Со временем различие в вязкости вносит свой вклад в непрерывную эрозию и дезинтеграцию твердой лекарственной формы.
Аминокислоты пригодны в данном варианте осуществления по двум основным причинам. Первое, аминокислоты являются фактором, определяющим вязкость полимеров. Как уже указывалось выше, со временем разница в вязкости экстрагранулярного и внутригранулярного полимеров вносит свой вклад в непрерывную эрозию и дезинтеграцию ядра, обеспечивая примерно 100% высвобождение препарата. Другим важным аспектом применения аминокислоты в грануле является то, что гидрофобность аминокислоты может использоваться для модуляции растворимости и высвобождения препарата.
Таким образом, аминокислоту выбирают по индексу гидрофобности в зависимости от растворимости активного соединения. Когда соединение является, по меньшей мере, умеренно растворимым в воде, т.е., например, умеренно растворимым, растворимым или обладает более высокой степенью растворимости, как определено Фармакопей США, то используется аминокислота, которая имеет относительно равный баланс гидрофильных и гидрофобных компонентов, т.е. нейтральная или сбалансированная, или близкая к нейтральным свойствам, или относительно более высоко гидрофильная аминокислота.
Например, растворение или высвобождение растворимых или умеренно растворимых ионизированных лекарственных средств, таких как верапамил HCl, можно контролировать включением одной или более аминокислот в гранулы. Не желая связываться с какой-либо конкретной теорией высвобождения и растворения препарата, полагается, что природа процесса гранулирования является такой, что по мере того, как компоненты композиция приходят в тесный молекулярный контакт, гранулирование снижает доступную площадь поверхности частиц, таким образом, приводя к снижению первоначальной скорости гидратации. В гранулированных композициях имеется достаточный период времени для взаимодействия карбоксильных (СООН--) групп и аминогрупп (NH2/NH3+) аминокислот с гидроксильными группами полимера, таким образом опосредуя набухание, вязкость и гелевые свойства полимера и тем самым контроль набухания, опосредованного диффузией препарата. Одновременно карбоксильные группы аминокислот также могут взаимодействовать с подходящими полярными заместителями в молекуле препарата, такими как вторичные или третичные амины. Кроме того, гидрофильные свойства и ионная природа аминокислот приводит к их интенсивной гидратации в водном растворе. Следовательно, аминокислоты способствуют эрозии, но конкурируют с полимером и препаратом за поглощение воды, необходимой для гидратации и растворения.
Однако когда активное соединение является менее чем умеренно растворимым, включая активные соединения, которые слаборастворимы или вовсе не растворимы, то применяется комбинация, по меньшей мере, двух аминокислот, одна из которых является высоко гидрофобной, а другая из которых является относительно более гидрофильной, чем гидрофобный компонент, т.е. примерно нейтральная или от сбалансированной до высокогидрофильной аминокислоты.
Аминокислотный компонент гранул может включать любые фармацевтически приемлемые α-аминокислоты или β-аминокислоты, соли α- или β-аминокислот, или любые их комбинации. Примерами подходящих α-аминокислот являются глицин, аланин, валин, лейцин, изолейцин, фенилаланин, пролин, аспарагиновая кислота, глутаминовая кислота, лизин, аргинин, гистидин, серии, треонин, цистеин, аспарагин и глутамин. Примером β-аминокислоты является β-аланин.
Тип аминокислоты, используемой в данном варианте осуществления внутренней части, может быть охарактеризован как гидрофильные, гидрофобные, соли гидрофильных, гидрофобных аминокислот, или любые их комбинации. Подходящие гидрофобные аминокислоты для применения включают, не ограничиваясь этим, изолейцин, фенилаланин, лейцин и валин. Кроме того, в грануле могут использоваться гидрофильные аминокислоты, такие как глицин, аспартат и глутамат. В конечном итоге, в настоящем изобретении можно использовать любую аминокислоту и любую аминокислоту в комбинации с другой аминокислотой для повышения растворимости лекарственного средства. Подробный перечень аминокислот, которые можно использовать в настоящем изобретении, и индекс гидрофобности для каждой аминокислоты смотри в публикации Albert L. Lehninger et al., Principles of Biochemistry 113 (2nd ed. Worth Publishers, 1993).
Тип и количество аминокислоты можно выбрать в зависимости от желаемой нагрузки препаратом, желаемой скорости высвобождения препарата и растворимости препарата. Как правило, аминокислота в лекарственной форме находится в количестве от 4 масс. % до 45 масс. % по отношению к общей массе лекарственной формы. Однако количество аминокислоты предпочтительно составляет от 11 масс. % до 29 масс. % по отношению к общей массе лекарственной формы.
Гранулы можно необязательно смешать с покровным материалом, например, стеаратом магния или другими гидрофобными производными стеариновой кислоты. Количество используемого покровного материала варьируется в пределах от 1 масс. % до 3 масс. % по отношению к общей массе лекарственной формы. Обычно стеарат магния используется для облегчения обработки, например, в качестве средства, повышающего сыпучесть, но в настоящем изобретении стеарат магния приносит дополнительную пользу, заключающуюся в задержке растворения, за счет гидрофобной природы покровного материала. Следовательно, стеарат магния может использоваться для дополнительного доведения растворимости лекарственной формы и дополнительной задержки высвобождения препарата из гранул.
Для повышения механических свойств и/или оказания влияния на скорость высвобождения препарата гранулы также могут содержать небольшие количества инертных фармацевтических наполнителей и связующих/гранулирующих агентов, как это принято в данной области. Примеры инертных фармацевтических наполнителей включают: лактозу, сахарозу, мальтозу, мальтодекстрины, декстрины, крахмал, микрокристаллическую целлюлозу, фруктозу, сорбит, ди- и трикальцийфосфат. Примеры гранулирующих агентов/связующих агентов включают крахмал, метилцеллюлозу, гидроксипропил- или гидроксипропилметилцеллюлозу, натриевую соль карбоксиметилцеллюлозы или поливинилпирролидон, аравийскую камедь, трагакант и сахарозу. Специалистам в данной области должно быть понятно, что могут использоваться другие подходящие наполнители. В зависимости от физических и/или химических свойств препарата можно использовать процедуру влажного гранулирования (с использованием водной или органической гранулирующей жидкости) или процедуру сухого гранулирования (например, брикетирование или вальцевание).
После гранулирования фармацевтически активного соединения внутригранулярного полимера, аминокислот и необязательно наполнителей и гидрофобных покровных материалов, затем гранулы смешивают с и диспергируют в экстрагранулярном полимере.
Экстрагранулярный полимер может представлять одно или более из следующего: полиэтиленоксид, галактоманнановый полисахарид, такой как гидроксипропиловый гуар, гуаровую камедь, камедь плодов рожкового дерева, пектин, аравийскую камедь, трагантовую камедь, камедь карайи, простые эфиры целлюлозы, такие как гидроксиропилметилцеллюлоза (НРМС), а также другие камеди и простые эфиры целлюлозы, выбранные специалистами в данной области по свойствам, согласующимися с принципами данного изобретения. В варианте осуществления внутригранулярный полимер представляет галактоманнановый полисахарид, такой как гуаровая камедь (с вязкостью в пределах 75-6000 сП для 1% раствора при 25°С в воде и размером частиц 100-300 мкм). Как уже указывалось выше, экстрагранулярный полимер должен гидратироваться быстро и достигать высокой степени вязкости в течение более короткого периода времени по сравнению с внутригранулярным полимером.
Различие в скорости гидратации экстрагранулярного полимера и внутригранулярного полимера обеспечивается тремя основными путями: (1) выбором полимеров на основе различий в размере частиц; (2) выбором полимеров на основе различий в молекулярной массе и химического состава и (3) выбором полимеров на основе комбинации по п.п. (1) и (2). Несмотря на то, что раскрытие в основном фокусируется на полимерах, выбранных на основе различий в размере частиц, является возможным достичь результатов данного изобретения посредством использования внутригранулярного полимера с молекулярной массой и/или химическим составом иными, чем у экстрагранулярного полимера. Например, полиэтиленоксид можно использовать в качестве внутригранулярного полимера и гуаровую камедь в качестве экстрагранулярного полимера.
Размер частиц является еще одной характеристикой промышленно доступной гуаровой камеди, поскольку более крупные частицы обеспечивают быстрое диспергирование, в то время как более мелкие частицы являются идеальными для быстрой гидратации. Следовательно, для достижения желаемого результата по настоящему изобретению в варианте осуществления более мелкие частицы используются для экстрагранулярного полимера и менее мелкие частицы используются в качестве частиц внутригранулярного полимера. В брошюре HERCULES Incorporated под заголовком «Supercol® Guar Gum, 1997» описаны типичные свойства гуаровой камеди различных марок и с различными размерами частиц. Другие быстро гидратирующие экстрагранулярные полимеры, которые могут использоваться, включают: полиэтиленоксид (ПЭО), простые эфиры целлюлозы и полисахариды, такие как гидроксипропиловый гуар, пектин, аравийскую камедь и трагакант, камедь карайи, смеси вышеуказанных полимеров и любые другие полимеры, выбранные специалистами в данной области по свойствам, согласующимся с принципами данного изобретения. Количества и типы экстрагранулярного полимера выбирают в зависимости от желаемой нагрузки лекарственным средством, скорости высвобождения препарата и растворимости препарата. Было установлены, что пределы примерно 4-47 масс. % (к общей массе таблетки) экстрагранулярного полимера являются подходящими, но особенно предпочтительными являются пределы 15-47 масс. %.
Во внутреннюю часть можно включать терапевтическое количество API, например, примерно до 75 масс. % по отношению к общей массе лекарственной формы. При такой нагрузке препаратом внутренняя часть приближается к линейному профилю высвобождения с минимальным «взрывным» эффектом или его отсутствием. Однако, если желательно специалистам в данной области, то экстрагранулярный полимер может содержать дополнительные количества фармацевтически активного соединения для достижения более быстрого высвобождения препарата или индуцированного «взрывного» эффекта, а также содержать аминокислоты для опосредования растворения фармацевтически активного соединения, как описано выше.
Таблетированную лекарственную форму с замедленным высвобождением для перорального введения можно покрыть полимерами, пластификаторами, замутнителями и красителями, как это принято в данной области.
В варианте осуществления внутренняя часть твердой лекарственной формы по настоящему изобретению представляет: (1) множество гранул, содержащих (а) по меньшей мере, один антагонист серотонина; (b) по меньшей мере, одну аминокислоту и (с) внутригранулярный полимер; где внутригранулярный полимер составляет от 4 масс. % до 45 масс. % по отношению к общей массе лекарственной формы, (2) гидрофильный экстрагранулярный полимер, в котором диспергированы гранулы, где экстрагранулярный полимер составляет от 4 масс. % до 47 масс. % по отношению к общей массе лекарственной формы и гидратируется быстрее, чем внутригранулярный полимер, где аминокислота выбрана по свойствам гидрофобности в зависимости от характеристик растворимости, по меньшей мере, одного антагониста серотонина и составляет от 11 масс. % до 29 масс. % по отношению к общей массе лекарственной формы. В варианте осуществления, когда, по меньшей мере, один антагонист серотонина, по меньшей мере, умеренно растворим в воде, то аминокислота имеет относительно равный баланс гидрофобных и гидрофильных компонентов или является относительно более гидрофильной. В варианте осуществления, когда, по меньшей мере, один антагонист серотонина менее чем умеренно растворим в воде, то аминокислота представляет комбинацию, по меньшей мере, двух аминокислот, где одна аминокислота является умеренно или высоко гидрофобной, а другая является относительно более гидрофильной. В варианте осуществления внутригранулярный полимер включает, по меньшей мере, одно из следующего: поливинилацетат, галактоманнановый полисахарид, выбранный из группы, состоящей из гидроксипропилового гуара, гуаровой камеди, камеди плодов рожкового дерева, пектина, аравийской камеди, трагантовой камеди, камеди карайи или простых эфиров целлюлозы. В варианте осуществления аминокислота выбрана из группы, состоящей из: а) α-аминокислот; b) β-аминокислот и с) комбинации α- и β-аминокислот. В варианте осуществления α-аминокислота представляет, по меньшей мере, один член, выбранный из группы, состоящей из глицина, аланина, валина, лейцина, изолейцина, фенилаланина, пролина, аспарагиновой кислоты, глутаминовой кислоты, лизина, аргинина, гистидина, серина, треонина, цистеина, аспарагина и глутамина. В варианте осуществления комбинация α- и β-аминокислот содержит β-аланин и, по меньшей мере, одну α-аминокислоту, выбранную из группы, состоящей из глицина, аланина, валина, лейцина, изолейцина, фенилаланина, пролина, аспарагиновой кислоты, глутаминовой кислоты, лизина, аргинина, гистидина, серина, треонина, цистеина, аспарагина и глутамина. В варианте осуществления аминокислота выбрана из группы, состоящей из: а) сбалансированной аминокислоты, обладающей относительно равным балансом гидрофобных и гидрофильных компонентов, или относительно более гидрофильной аминокислоты или b) комбинации (i) сбалансированной аминокислоты или относительно более гидрофильной аминокислоты и (ii) гидрофобной аминокислоты. В варианте осуществления сбалансированная аминокислота включает глицин. В варианте осуществления внутренняя часть содержит глицин и гидрофобную аминокислоту, выбранную из изолейцина, валина и фенилаланина. В варианте осуществления множество гранул перемешивают с гидрофобным покровным материалом. В варианте осуществления гидрофобным покровным материалом является стеарат магния. В варианте осуществления гидрофобный покровный материал составляет от 1 масс. % до 3 масс. % по отношению к общей массе лекарственной формы.
Внутреннюю часть, описанную выше, можно приготовить способом, описанным в патенте США №6517868, который включен здесь в виде ссылки для принципов, раскрытых здесь.
Первая и вторая оболочка
Первая оболочка и вторая оболочка бимодальной твердой лекарственной формы с замедленным высвобождением по настоящему изобретению являются нефункциональными оболочками, которые действуют в качестве технологических добавок. Первая оболочка и вторая оболочка по существу не оказывают влияния на высвобождение API из лекарственной формы. В варианте осуществления первая и вторая оболочка содержит гидрофильное вещество. В варианте осуществления гипромеллоза представляет Methocel Е5. В варианте осуществления первая и вторая оболочка дополнительно содержит добавку для покрытия plasACRYL™, водную эмульсию глицерилмоностеарата и триэтилцитрата (разработанную Emerson Resources, Inc. of Norristown, PA, США). В варианте осуществления plasACRYL™, используемая в первой и второй оболочках, имеет марку Т20. В варианте осуществления plasACRYL™ Т20 представляет 20% водную суспензию, содержащую антиадгезив, пластификатор и стабилизатор. Гипромеллоза представляет независимый от pH неионогенный полимер, полученный частичным замещением О-метилированной и О-(2-гидроксипропилированной) группами. Марки гипромеллозы могут варьироваться по степени замещения, что влияет на вязкость полимера. НРМС K4M Premium имеет вязкость 3550 мпуаз, в то время как НРМС Е5 Premium LV является полимером с низкой вязкостью, равной 5 мпуаз. Гипромеллоза растворима в холодной воде и образует коллоидную вязкую жидкость.
Верхняя оболочка со слоем препарата
Верхняя оболочка со слоем препарата твердой лекарственной формы с замедленным высвобождением по настоящему изобретению представляет слой препарата с немедленным высвобождением («IR»). В варианте осуществления верхняя оболочка со слоем препарата по существу предназначена для обеспечения «взрывного» высвобождения примерно 25% API, что при пероральном заглатывании твердой лекарственной формы будет приводить к тому, что примерно 25% API высвобождается в желудке. В варианте осуществления верхняя оболочка со слоем препарата или слой препарата с немедленным высвобождением содержит ондансетрон гидрохлорид, гипромеллозу и plasACRYL™. В варианте осуществления гипромеллоза, используемая в слое IR, представляет Metrocel Е5.
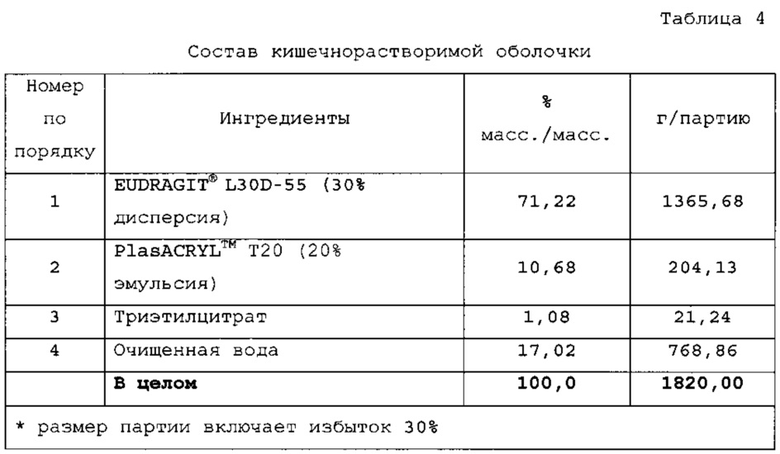
Дополнительные слои - кишечнорастворимая оболочка В варианте осуществления твердая лекарственная форма с замедленным высвобождением по настоящему изобретению дополнительно включает кишечнорастворимую оболочку. В варианте осуществления слой кишечнорастворимой оболочки располагается между первой оболочкой и верхней оболочкой слоя препарата. В варианте осуществления слой кишечнорастворимой оболочки представляет EUDRAGIT® L30D-55. В варианте осуществления слой кишечнорастворимой оболочки представляет EUDRAGIT® FS 30 D. В варианте осуществления слой кишечнорастворимой оболочки представляет SURETERIC®.
Нижеприведенные примеры представлены для того, чтобы предоставить специалисту полное раскрытие и описание того, как осуществить и применить описанное изобретение, и не предназначены для ограничения объема того, что заявители рассматривают как свое изобретение, и не предназначены представлять то, что нижеприведенные эксперименты представляют все или единственные проведенные эксперименты. Предприняты усилия для гарантии точности числовых параметров (например, количеств, температур и т.д.), но следует учитывать некоторые ошибки и отклонения. Если не указано иначе, части представляют собой весовые части, температура дается в °С, а давление является атмосферным или близким к атмосферному.
ПРИМЕРЫ
Пример 1 - Производство внутренних ядер с 18 мг ондансетрона
Аминокислотную композицию («АА ядро») получали с использованием влажного гранулирования с малым усилием сдвига.
Микрокристаллическую целлюлозу Avxcel® РН-102, ондансетрон HCl, глицин и НРМС К15М смешивали в V-образном смесителе емкостью 1 фут3 в течение 10 мин, влажную смесь выгружали и дробили с использованием мельницы Comil, снабженной ситом 20 меш. Затем предварительную смесь гранулировали в Hobart D3 00 при добавлении воды к смеси при перемешивании. После добавления воды материал перемешивали еще в течение 2 мин. Материал соответствующим образом гранулировали, но не покрывали влажным, следовательно, дополнительное количество воды не добавляли. Влажную массу пропускали через сито 8 меш, затем высушивали в печи. Высушенную гранулированную смесь измельчали с использованием мельницы Comil с ситом 18 меш, перемешивали с экстрагранулярной НРМС K100LV и смазывающим агентом. Прессование конечной смеси проводили на таблеточном прессе 36-station Kikusui с использованием модифицированного овального инструмента для таблетирования 0,32''×0,58''.

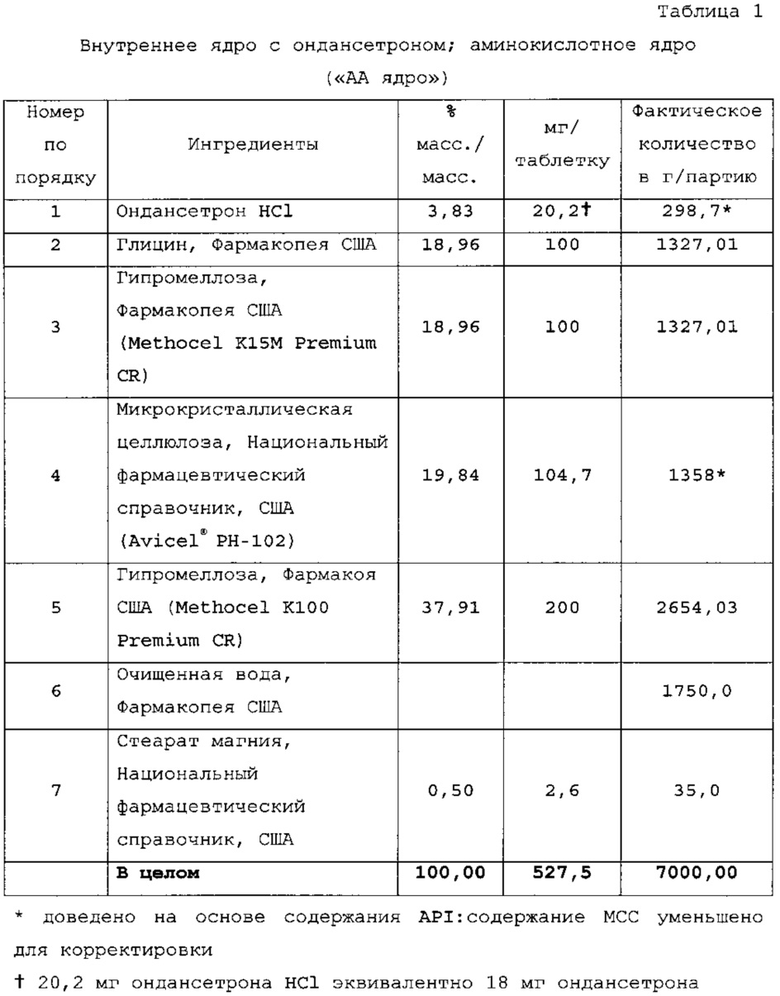
Электролитную композицию («электролитное ядро») получали смешиванием и прессованием. Все вещества просеивали по отдельности через сито 30 меш, вносили в V-образный смеситель и перемешивали в течение 15 мин, затем добавляли смазывающий агент. Прессование конечной смеси проводили на таблеточном прессе 36-station Kikusui с использованием модифицированного овального инструмента для таблетирования 0,28''×0,50''.
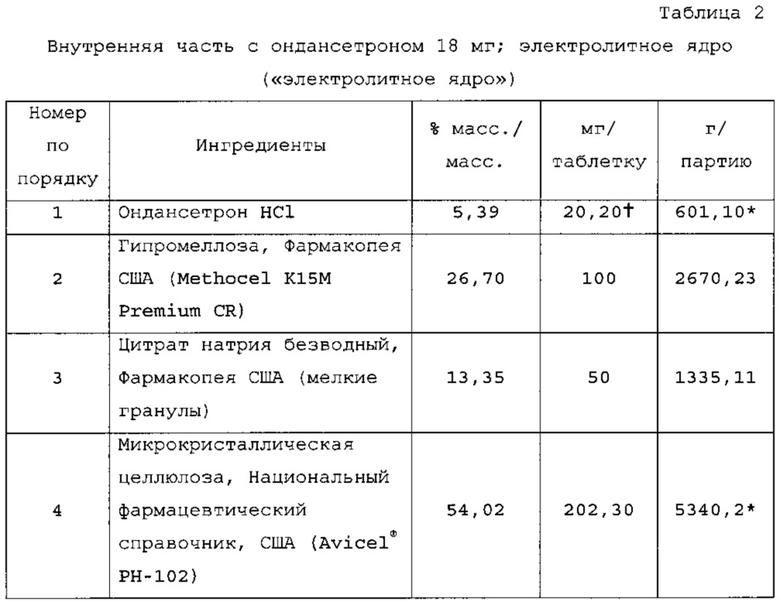
Пример 2 - Первая и вторая изолирующие оболочки; необязательная кишечнорастворимая оболочка
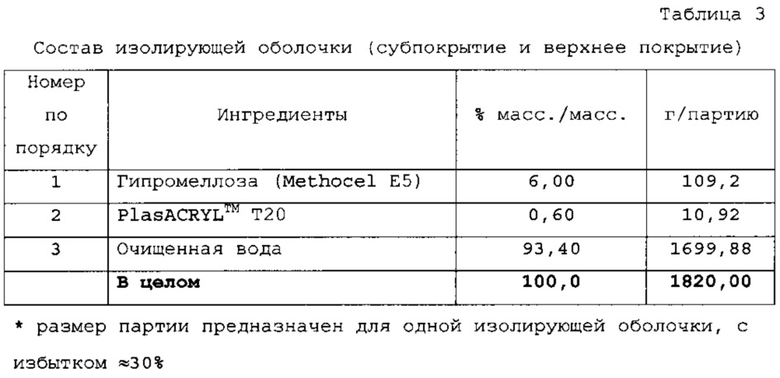
Раствор изолирующей оболочки готовили растворением Methocel Е5 в воде, затем добавлением PlasACRYL™. Суспензию кишечнорастворимой оболочки готовили смешением воды, триэтилцитрата и PlasACRYL™. Добавляли дисперсию EUDRAGIT®; суспензию перемешивали в течение 30 мин, затем пропускали через сито 60 меш. Суспензию активного соединения готовили вначале растворением Methocel Е5 в воде и отдельно диспергированием ондансетрона в воде и гомогенизированием. Затем раствор Methocel добавляли в суспензию препарата и добавляли PlasACRYL™.
Пример 3 - Верхняя оболочка со слоем препарата
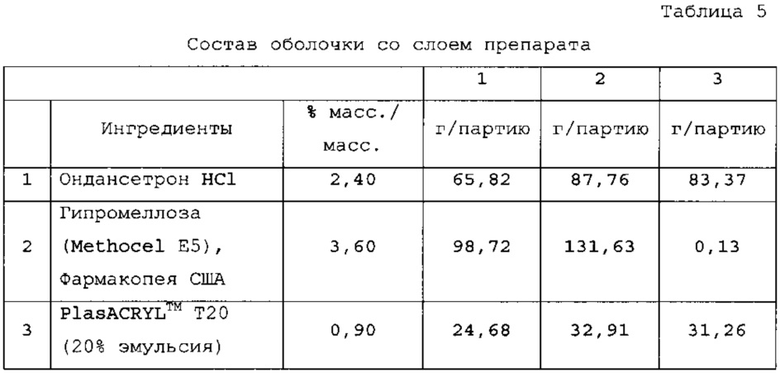

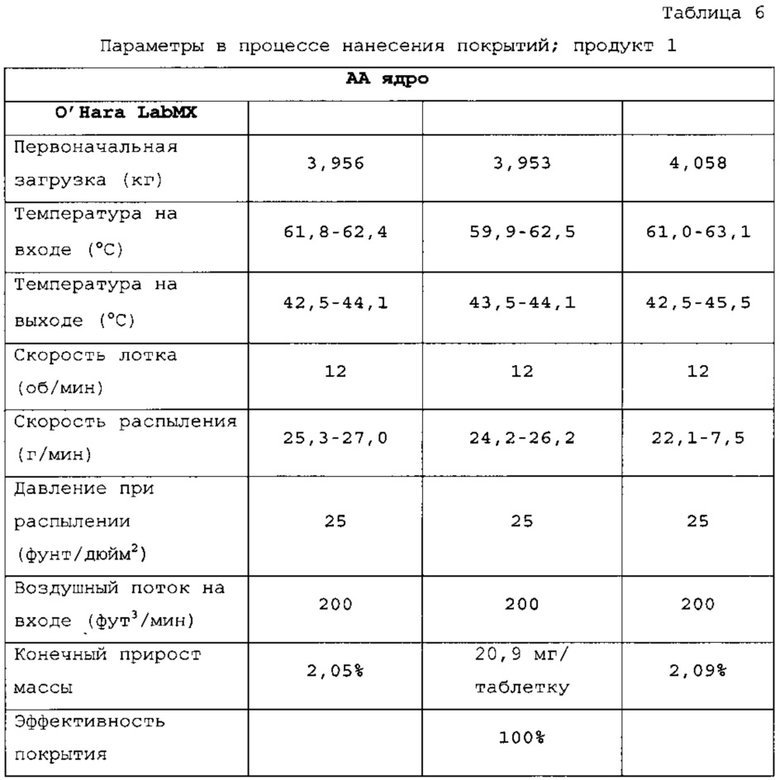
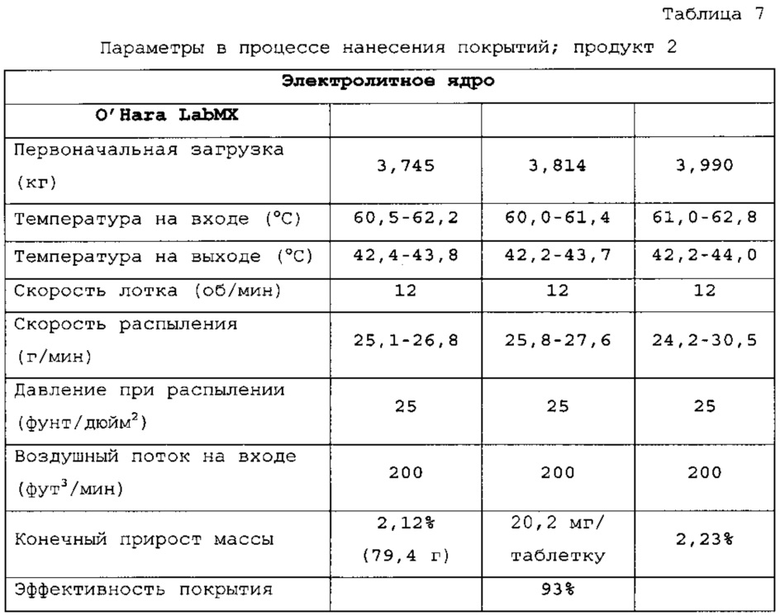
Таблетки покрывали необходимыми оболочками, как представлено в таблицах 6-8. Прибавление массы прослеживали по определению массы 50 таблеток каждые 10 мин. По причине пригодности оборудования первые две партии покрывали с использованием оборудования для нанесения покрытий на таблетки R&D (О'Нага LabMX). Третью партию получали с использованием оборудования cGMP, которое используется для производств СТМ.
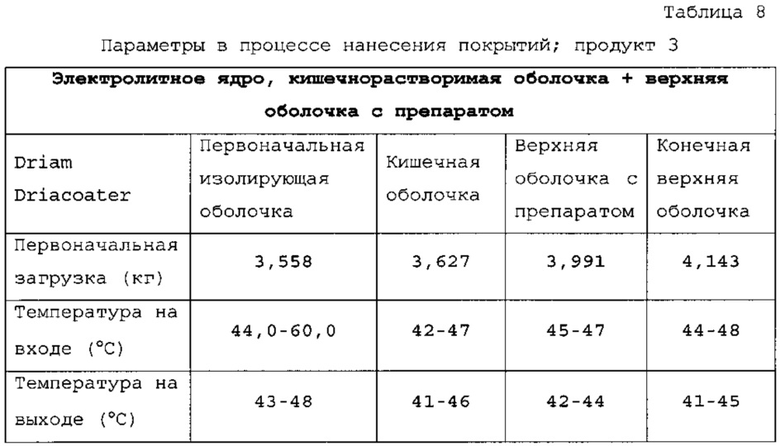
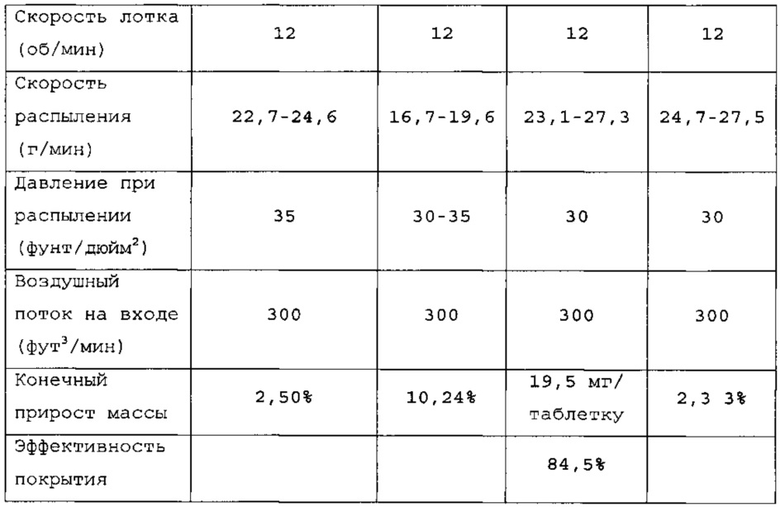

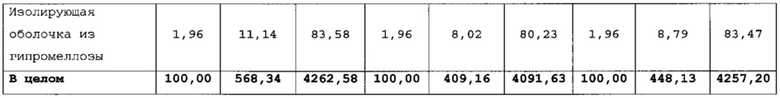
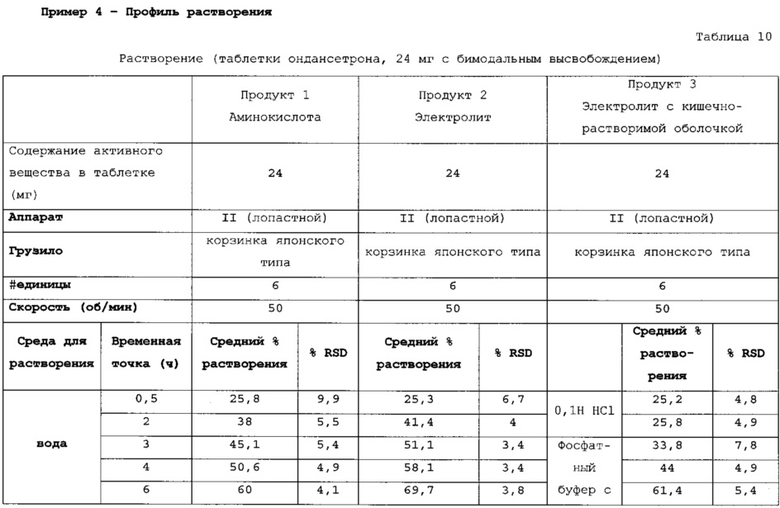

Данные таблицы 10 в сочетании с фигурой 1 и фигурой 2 показывают профиль растворения продуктов 1, 2 и 3. Для продукта 1 характерно первоначальное «взрывное» высвобождение 25% препарата с последующим замедленным высвобождением в течение 24 ч. Для продукта 2 характерно первоначальное «взрывное» высвобождение 25% препарата с последующим замедленным высвобождением в течение 24 ч. Для продукта 3 характерно первоначальное «взрывное» высвобождение 25% препарата с последующим латентным периодом высвобождения в кислоте.
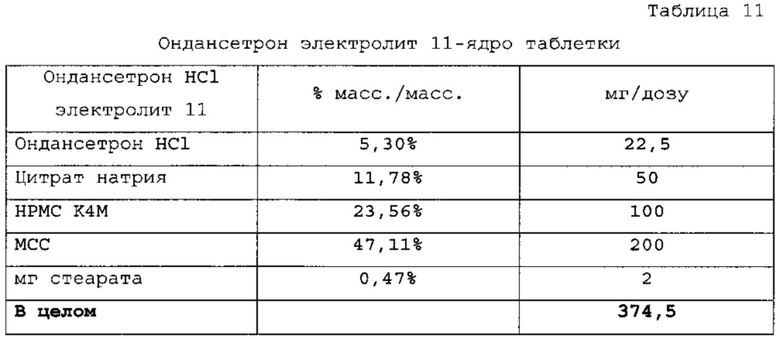
Пример 5 - Получение внутреннего электролитного ядра с ондансетроном
Ядра таблеток с ондансетроном HCl получали смешиванием сухих компонентов и прессованием. Подробное описание состава ингредиентов представлено в таблицах 11 и 12. Профиль растворения (с наличием кишечнорастворимой оболочки и оболочки с немедленным высвобождением препарата 6 мг) для данного состава приведен на фигуре 3.

Пример 6 - Профиль растворения
Оценку растворения in vitro проводили с физиологически соответствующими средами с pH в пределах от 1,2 до 7,2, которые приближаются к значениям pH в пищеварительном тракте. За счет различий в растворимости API ондансетрона HCl при различных значениях pH максимальное поглощение использовали для расчета растворения-высвобождения, а не калибровочную кривую, полученную с раствором API в воде. Результаты тестирования растворения для сред: pH 1,2; 6,8; 7,2 и дистиллированной водой приведены на фигуре 4.
Пример 7 - Тестирование твердых лекарственных форм in vivo
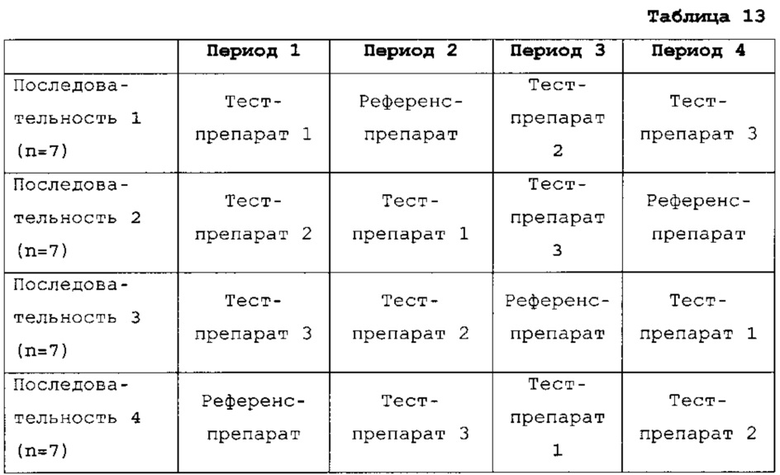
Проводили одноцентровое, рандомизированное, слепое, четырехпериодное, с четырьмя последовательными приемами, перекрестное исследование на здоровых мужчинах и женщинах. Натощак вводили следующие испытуемые продукты:
тест-препарат 1: 1× таблетка ондансетрона с бимодальным высвобождением 24 мг (аминокислотное ядро)
тест-препарат 2: 1× таблетка ондансетрона с бимодальным высвобождением 24 мг (электролитное ядро)
тест-препарат 3: 1× таблетка ондансетрона с бимодальным высвобождением 24 мг (электролитное ядро с кишечнорастворимой оболочкой)
референс-препарат: 3×Zofran® таблетки 8 мг (1хтаблетка 8 мг, три раза в день, с 8-часовым интервалом: утром с последующим 10-часовым ночным периодом голодания, днем и вечером).
Продукты вводили 28 здоровым мужчинам и женщинам по схеме, приведенной в таблице 13.
Выбор доз в исследовании
Дозу выбирали для достижения биодоступности, одинаковой с промышленно доступным препаратом с немедленным высвобождением (Zofran® таблетки 8 мг), который вводился три раза в день.
Выбор и время введения доз для каждого субъекта
Субъекты голодали ночью, по меньшей мере, в течение 10 ч до введения препаратов утром. Тест-препараты 1-3
Одну дозу назначенного тест-препарата вводили перорально примерно с 240 мл воды комнатной температуры, начиная с 07:30, одному субъекту в мин.
Референс-препарат
Назначенный референс-препарат вводили перорально (три раза в день с 8-часовым интервалом) с 240 мл воды комнатной температуры, начиная с 07:30, одному субъекту в мин. Последующее введение препарата проводили днем и вечером соответственно в 15:30 и 23:30.
Голодание продолжали в течение, по меньшей мере, 4 ч после приема препаратов утром, после чего был обычный обед. Обед должен быть заканчиваться не позднее, чем 5 ч после введения утром. Все блюда предоставляли в определенное время, но не позднее 9 ч после приема препарата утром. Ужин предоставляли через 11 ч после утреннего приема препаратов и не позднее, чем 13 ч после утреннего приема препаратов. Кроме того, легкий перекус должен быть съедаться не позднее 13 ч после утреннего приема препаратов. Воду давали без ограничения в период за 1 ч до приема дозы и начиная через 1 ч после каждого приема препаратов.
Оценка эффективности и безопасности и схема
Оценка фармакокинетики
Пробы крови для фармакокинетических исследований отбирали перед и до 32 ч (серийный отбор проб) после каждого приема препаратов утром. Непосредственным анализом в данном исследовании было определение концентрации ондансетрона. Концентрации определяли анализом плазмы крови, полученной из проб цельной крови, отобранных в ходе настоящего исследования.
Общий объем крови, отобранный у каждого субъекта (639 мл у мужчин и 653 мл у женщин), рассматривался в качестве незначительного и не оказывающего влияния на фармакокинетические профили лекарственных средств и оценку биоэквивалентности. Кроме того, полагали, что такой объем оказывает незначительное влияние в отношении безопасности для субъекта.
Определение концентрации препаратов
Тест-препараты 1-3 (21 проба крови):
Первую пробу крови для каждого периода, т.е. контрольную пробу плазмы крови, отбирали до введения препарата, в то время как другие отбирали через 0,25; 0,5; 1; 1,5; 2; 2,5; 3; 3,5; 4; 5; 6; 7; 8; 9; 10; 12; 16; 20; 24 и 32 ч после введения препарата в одну пробирку емкостью 6 мл (вакуумные пробирки с ЭДТА К2).
Референс-препарат (33 пробы крови):
Первую пробу крови для каждого периода, т.е. контрольную пробу плазмы крови, отбирали до введения препарата, в то время как другие отбирали через 0,25; 0,5; 1; 1,5; 2; 2,5; 3; 3,5; 4; 5; 6; 7; 8; 9; 10; 12; 16; 20; 24 и 32 ч после введения препарата утром в одну пробирку емкостью 6 мл (вакуумные пробирки с ЭДТА К2). Пробы на 8 ч и 16 ч отбирали в течение 5 мин до введения препарата (введение днем и вечером).
Ондансетрон - тест-препарат 1 в сравнении с референс-препаратом
В сравнительное исследование тест-препарата 1 и референс-препарата было включено двадцать шесть (26) субъектов. Обобщенные данные по фармакокинетическим параметрам и стандартам для сравнительной биодоступности представлены в таблицах 14 и 15. Средние значения измеренной концентрации в плазме крови в зависимости от времени, полученные после введения тест-препарата 1 и референс-препарата, представлены на фигуре 5, в то время как ln-преобразованные значения средней концентрации в зависимости от времени представлены на фигуре 6.
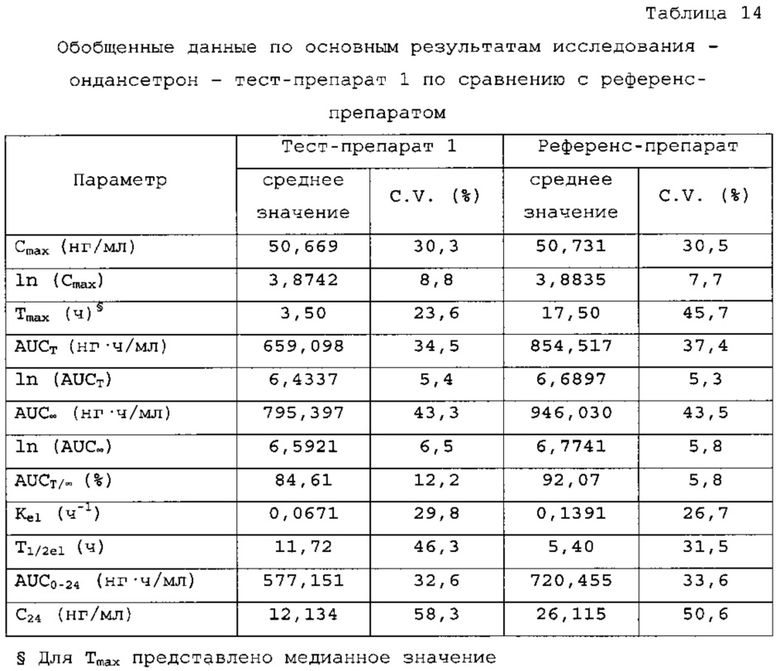
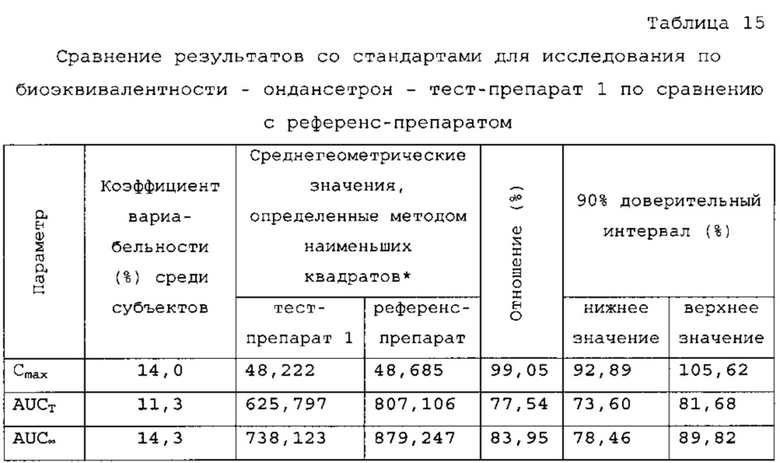
Количество субъектов, включенных в статистический анализ данных параметров, составляло n=24 для тест-препарата 1 и n=26 для референс-препарата. Среднее значение Cmax равнялось 50,669 нг/мл и 50,731 нг/мл соответственно для композиций тест-препарата 1 и референс-препарата. Отношение среднегеометрических значений Cmax, определенных методом наименьших квадратов, тест-препарата 1 к референс-препарату равнялось 99,05% (90% доверительный интервал: от 92,89 до 105,62%). Данные результаты показывают, что отношение и соответствующий 90% доверительный интервал для относительных среднегеометрических значений Cmax, определенных методом наименьших квадратов, тест-препарата 1 к референс-препарату укладывались в заранее установленный интервал биоэквивалентности от 80,00 до 125,00%. Медианное значение Tmax равнялось 3,50 и 17,50 ч соответственно для композиций тест-препарата 1 и референс-препарата. Среднее значение AUCT составляло 659,098 и 854,517 нг⋅ч/мл соответственно для композиций тест-препарата 1 и референс-препарата. Отношение среднегеометрических значений AUCT, определенных методом наименьших квадратов, тест-препарата 1 к референс-препарату равнялось 77,54% (90% доверительный интервал: от 73,60 до 81,68%). Данные результаты показывают, что отношение и соответствующий 90% доверительный интервал для относительных среднегеометрических значений AUCT, определенных методом наименьших квадратов, тест-препарата 1 к референс-препарату не укладывались в заранее установленный интервал биоэквивалентности от 80,00 до 125,00%. Среднее значение Kel равнялось 0, 0671 ч-1 для композиции тест-препарата 1 и 0,1391 ч-1 для композиции референс-препарата. Среднее значение Ti/2el равнялось 11,72 и 5,40 ч соответственно для композиций тест-препарата 1 и референс-препарата. Среднее значение AUG∞ равнялось 795,397 нг⋅ч/мл и 946,030 нг⋅ч/мл соответственно для композиций тест-препарата 1 и референс-препарата. Отношение среднегеометрических значений AUG∞, определенных методом наименьших квадратов, тест-препарата 1 к референс-препарату равнялось 83,95% (90% доверительный интервал: от 78,46 до 89,82%). Данные результаты показывают, что отношение и соответствующий 90% доверительный интервал для относительных среднегеометрических значений AUCT, определенных методом наименьших квадратов, тест-препарата 1 к референс-препарату не укладывались в заранее установленный интервал биоэквивалентности от 80,00 до 125,00%. Среднее значение отношения индивидуальных значений AUCT к AUG∞ (AUCT/∞ равнялось 84,61% и 92, 07% соответственно для тест-препарата 1 и референс-препарата.
Ондансетрон - тест-препарат 2 в сравнении с референс-препаратом
В сравнительное исследование тест-препарата 2 и референс-препарата было включено двадцать шесть (26) субъектов. Обобщенные данные по фармакокинетическим параметрам и стандартам для сравнительной биодоступности представлены в таблицах 16 и 17. Средние значения измеренной концентрации в плазме крови в зависимости от времени, полученные после введения тест-препарата 2 и референс-препарата, представлены на фигуре 5, в то время как ln-преобразованные значения средней концентрации в зависимости от времени представлены на фигуре 6.
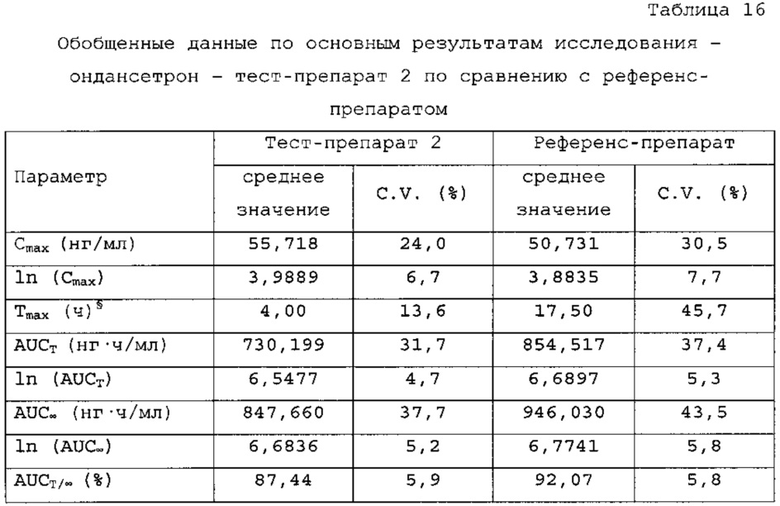
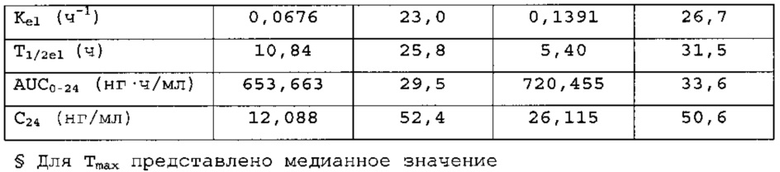
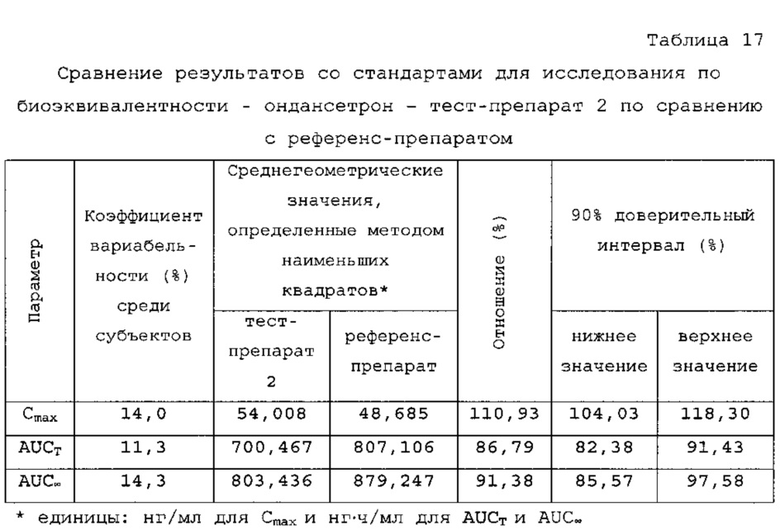
Среднее значение Cmax равнялось 55,718 нг/мл и 50, 731 нг/мл соответственно для композиций тест-препарата 2 и референс-препарата. Отношение среднегеометрических значений Cmax, определенных методом наименьших квадратов, тест-препарата 2 к референс-препарату равнялось 110,93% (90% доверительный интервал: от 104,03 до 118,30%). Данные результаты показывают, что отношение и соответствующий 90% доверительный интервал для относительных среднегеометрических значений Cmax, определенных методом наименьших квадратов, тест-препарата 2 к референс-препарату укладывались в заранее установленный интервал биоэквивалентности от 80,00 до 125,00%. Медианное значение Tmax равнялось 4,00 и 17,50 ч соответственно для тест-препарата 2 и референс-препарата. Среднее значение AUCT равнялось 730,199 и 854,517 нг⋅ч/мл соответственно для композиций тест-препарата 2 и референс-препарата. Отношение среднегеометрических значений AUCT, определенных методом наименьших квадратов, тест-препарата 2 к референс-препарату равнялось 86,79% (90% доверительный интервал: от 82,38 до 91,43%). Данные результаты показывают, что отношение и соответствующий 90% доверительный интервал для относительных среднегеометрических значений AUCT, определенных методом наименьших квадратов, тест-препарата 2 к референс-препарату укладывались в заранее установленный интервал биоэквивалентности от 80, 00 до 125,00%. Среднее значение Kel равнялось 0, 0676 ч-1 для композиции тест-препарата 2 и 0,1391 ч-1 для композиции референс-препарата. Среднее значение T1/2el равнялось 10,84 и 5,40 ч соответственно для композиций тест-препарата 2 и референс-препарата. Среднее значение AUG∞ равнялось 847, 660 нг⋅ч/мл и 946,030 нг⋅ч/мл соответственно для композиций тест-препарата 2 и референс-препарата. Отношение среднегеометрических значений AUG., определенных методом наименьших квадратов, тест-препарата 2 к референс-препарату равнялось 91,38% (90% доверительный интервал: от 85,57 до 97,58%). Данные результаты показывают, что отношение и соответствующий 90% доверительный интервал для относительных среднегеометрических значений AUCT, определенных методом наименьших квадратов, тест-препарата 2 к референс-препарату укладывались в заранее установленный интервал биоэквивалентности от 80,00 до 125,00%. Среднее значение отношения индивидуальных значений AUCT к AUG∞ (AUCT/∞ равнялось 87, 44% и 92, 07% соответственно для композиций тест-препарата 2 и референс-препарата.
Ондансетрон - тест-препарат 3 в сравнении с референс-препаратом
В сравнительное исследование было включено двадцать пять (25) субъектов для тест-препарата 3 и двадцать шесть (26) субъектов для референс-препарата. Обобщенные данные по фармакокинетическим параметрам и стандартам для сравнительной биодоступности представлены в таблицах 18 и 19. Средние значения измеренной концентрации в плазме крови в зависимости от времени, полученные после введения тест-препарата 3 и референс-препарата, представлены на фигуре 5, в то время как ln-преобразованные значения средней концентрации в зависимости от времени представлены на фигуре б
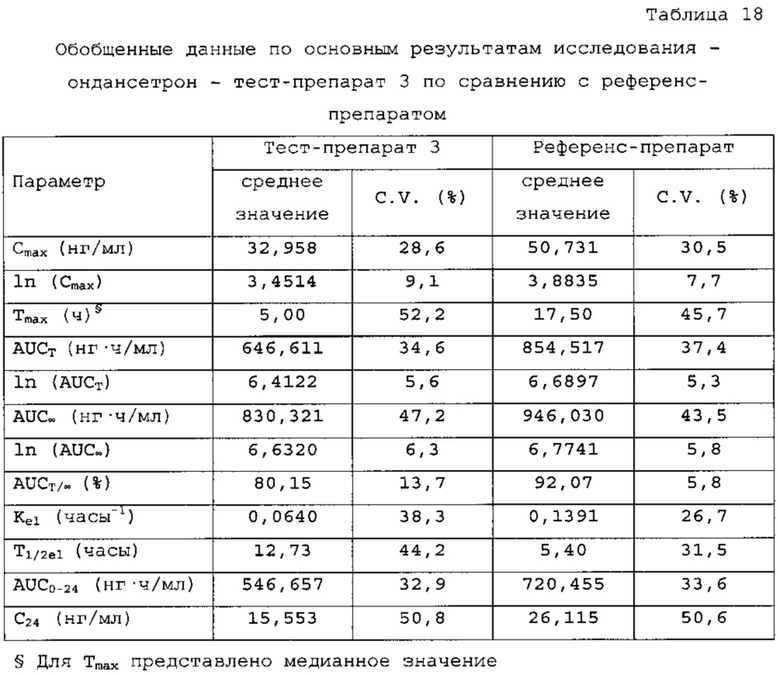
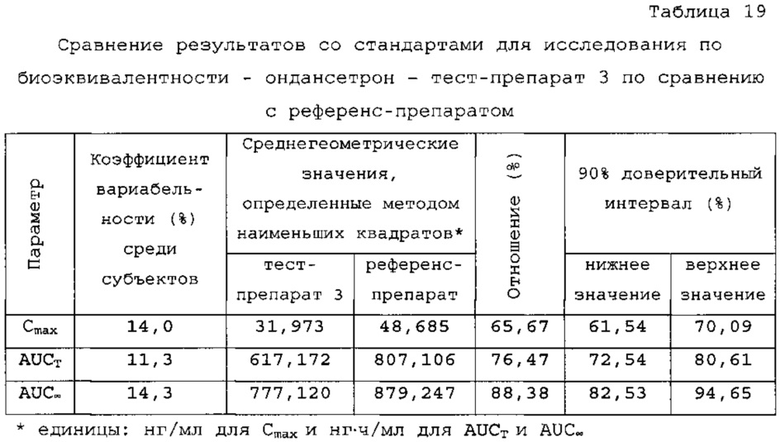
Количество субъектов, включенных в статистический анализ данных параметров, составляло n=23 для тест-препарата 3 и n=26 для референс-препарата. Среднее значение Cmax равнялось 32,958 нг/мл и 50,731 нг/мл соответственно для композиций тест-препарата 3 и референс-препарата. Отношение среднегеометрических значений Cmax, определенных методом наименьших квадратов, тест-препарата 3 к референс-препарату равнялось 65,67% (90% доверительный интервал: от 61,54 до 70,09%). Данные результаты показывают, что отношение и соответствующий 90% доверительный интервал для относительных среднегеометрических значений Cmax, определенных методом наименьших квадратов, тест-препарата 3 к референс-препарату не укладывались в заранее установленный интервал биоэквивалентности от 80,00 до 125,00%. Медианное значение Tmax равнялось 5,00 и 17,50 ч соответственно для композиций тест-препарата 3 и референс-препарата. Среднее значение AUCT равнялось 646,611 и 854,517 нг⋅ч/мл соответственно для композиций тест-препарата 3 и референс-препарата. Отношение среднегеометрических значений AUCT, определенных методом наименьших квадратов, тест-препарата 3 к референс-препарату равнялось 76,47% (90% доверительный интервал: от 72,54 до 80,61%). Данные результаты показывают, что отношение и соответствующий 90% доверительный интервал для относительных среднегеометрических значений AUCT, определенных методом наименьших квадратов, тест-препарата 3 к референс-препарату не укладывались в заранее установленный интервал биоэквивалентности от 80, 00 до 125,00%. Среднее значение Kel равнялось 0,0640 ч-1 для композиции тест-препарата 3 и 0,1391 ч-1 для композиции референс-препарата. Среднее значение T1/2el равнялось 12,73 и 5,40 ч соответственно для композиций тест-препарата 3 и референс-препарата. Среднее значение AUC∞ равнялось 830, 321 нг⋅ч/мл и 946,030 нг⋅ч/мл соответственно для композиций тест-препарата 3 и референс-препарата. Отношение среднегеометрических значений AUG∞, определенных методом наименьших квадратов, тест-препарата 3 к референс-препарату равнялось 88,38% (90% доверительный интервал: от 82,53 до 94,65%). Данные результаты показывают, что отношение и соответствующий 90% доверительный интервал для относительных среднегеометрических значений AUC∞, определенных методом наименьших квадратов, тест-препарата 3 к референс-препарату укладывались в заранее установленный интервал биоэквивалентности от 80,00 до 125,00%. Среднее значение отношения индивидуальных значений AUCT к AUC∞ (AUCT/∞) равнялось 80,15% и 92,07% соответственно для тест-препарата 1 и референс-препарата.
Пример 8 - трехстороннее перекрестное сравнительное исследование биодоступности твердых лекарственных форм
Трехстороннее перекрестное сравнительное исследование биодоступности с введением в течение пяти суток твердых лекарственных форм по настоящему изобретению один раз в день по сравнению с введением в течение двух дней два раза в день таблеток ондансетрона 8 мг с немедленным высвобождением по сравнению с одной дозой таблеток ондансетрона 24 мг с немедленным высвобождением на здоровых добровольцах мужчинах и женщинах натощак.
Цели:
Основной целью данного исследования было сравнение относительной биодоступности, максимальной пиковой и минимальной концентраций двух схем применения промышленно доступных таблеток ондансетрона 8 мг с немедленным высвобождением (схема с Zofran® 8 мг два раза в день при введении в течение 2 дней и схема с Zofran® 24 мг один раз в день при введении трех таблеток Zofran® 8 мг вместе), разрешенных Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов (США), и тест-препарата, таблетки ондансетрона 24 мг с замедленным высвобождением по настоящему изобретению (вводили один раз в день).
Вторичными целями исследования были:
1. Оценить кумуляцию ондансетрона в плазме крови после введения суточных доз тест-препарата пять дней подряд натощак;
2. Оценить безопасность и переносимость композиции с замедленным высвобождением у здоровых добровольцев.
Методология:
Одноцентровое, рандомизированное, открытое, трехпериодное, с тремя последовательными приемами, перекрестное исследование. Количество субъектов (запланированное и анализированное):
Запланировано для включения в исследование: 18
Включено: 18
Исключено: 0
Анализировано: 18
Рассматривалось в фармакокинетическом и статистическом анализе:18
Рассматривалось в анализе безопасности: 18
Диагноз и основные критерии для включения в исследование:
В исследование были включены мужчины и женщины добровольцы, некурящие или бросившие курить, в возрасте, по меньшей мере, 18 лет с индексом массы тела выше или равным 18,50 и ниже 30,00 кг/м2. У субъектов было хорошее состояние здоровья, что было подтверждено анамнезом, полным физическим осмотром (включая витальные показатели), снятием электрокардиограммы в 12 отведениях (ЭКГ) и результатами стандартных лабораторных анализов (общая биохимия, гематология, анализ мочи), включая наличие отрицательного анализа на вирус иммунодефицита человека (ВИЧ), вирус гепатита В и гепатита С, а также отрицательных результатов скрининга на алкоголь, котинин и наркотики, и отрицательных результатов теста на человеческий хорионический гонадотропин (ХГ), качественного теста на беременность (у женщин).
Тест-препарат, доза и способ введения:
Название: ондансетрон
Лекарственная форма/путь введения: таблетка с бимодальным высвобождением по настоящему изобретению (электролитное ядро CDT)/пероральное введение («тест-препарат»)
Схема лечения 1: одна доза 24 мг (1×24 мг) один раз в день в течение 5 дней подряд.
Референс-препарат, доза и способ введения
Название: Zofran®
Лекарственная форма/путь введения: таблетка/пероральное введение
Схема лечения 2: одна доза 8 мг (1×8 мг) дважды в день с 8 часовым интервалом на сутки 1 и с 12-часовым интервалом на сутки 2
Схема лечения 3: одна доза 24 мг (3×8 мг)
Схемы лечения:
Схема лечения 1: тест-препарат один раз в день в течение 5 дней подряд
Схема лечения 2: референс-препарат два раза в день с 8 часовым интервалом на сутки 1 и с 12-часовым интервалом на сутки 2
Схема лечения 3: референс-препарат одна доза 24 мг в виде трех таблеток, принятых одновременно. Периоды лечения:
Период 1: с 2013/08/08 по 2013/08/12 (схема лечения 1)
Период 1: с 2013/08/08 по 2013/08/12 (схема лечения 2)
Период 1: с 2013/08/08 (схема лечения 3)
Период 2: с 2013/08/17 по 2013/08/21 (схема лечения 1)
Период 2: с 2013/08/17 по 2013/08/18 (схема лечения 2)
Период 2: с 2013/08/17 (схема лечения 3)
Период 3: с 2013/08/26 по 2013/08/30 (схема лечения 1)
Период 3: с 2013/08/26 по 2013/08/27 (схема лечения 2)
Период 3: с 2013/08/26 (схема лечения 3)
Продолжительность лечения:
Схема лечения 1: одну дозу ондансетрона 24 мг (1хтаблетка 24 мг с бимодальным высвобождением (электролитное ядро CDT)) («тест-препарат») вводили перорально один раз в день утром после 10-часового ночного периода голодания 5 дней подряд.
Схема лечения 2: одну дозу Zofran® 8 мг (1×таблетка 8 мг) вводили перорально дважды в день два дня подряд с 8 часовым интервалом на сутки 1 и с 12-часовым интервалом на сутки 2 (первая доза утром каждый день после 10-часового ночного голодания и вторая доза днем (сутки 1) или вечером (сутки 2)) (в целом 4 введения препарата).
Схема лечения 3: одну дозу Zofran® 24 мг (3×таблетки 8 мг) вводили перорально после 10-часового ночного голодания.
Период «отмывки» между первыми введениями препаратов в каждом периоде исследования составлял 9 календарных дней.
Точки отбора проб крови:
Во время исследования в целом было отобрано 98 проб крови следующим образом:
Схема лечения 1: на сутки 1 и 2 введения было отобрано 13 проб крови каждый день. Первую пробу крови отбирали до введения препарата (в течение 5 мин), в то время как другие отбирали через 1, 2, 3, 4, 5, 6, 8, 10, 12, 14, 16 и 20 ч после введения препарата;
на сутки 3 и 4 введения было отобрано 8 проб крови каждый день, первую пробу крови отбирали до введения препарата (в течение 5 мин), в то время как другие отбирали через 2, 4, 6, 10, 14 и 18 ч после введения препарата;
на сутки 5 введения было отобрано 10 проб крови каждый день, первую пробу крови отбирали до введения препарата (в течение 5 мин), в то время как другие отбирали через 2, 4, 6, 8, 10, 14, 18, 24 и 48 ч после введения препарата.
В целом было отобрано 52 пробы у каждого субъекта во время данной схемы лечения.
Схема лечения 2: на сутки 1 введения было отобрано 15 проб крови каждый день. Первую пробу крови отбирали до введения препарата утром (в течение 5 мин), в то время как другие отбирали через 1, 2, 3, 4, 5, 6, 8, 9, 10, 11, 12, 14, 16 и 20 ч после введения препарата утром. Пробу крови на 8 ч отбирали в течение 5 мин до введения препарата днем;
на сутки 2 введения было отобрано 17 проб крови каждый день. Первую пробу крови отбирали до введения препарата утром (в течение 5 мин), в то время как другие отбирали через 1, 2, 3, 4, 5, 6, 8, 12, 13, 14, 15, 16, 18, 20, 24 и 48 ч после введения препарата утром. Пробу крови на 12 ч отбирали в течение 5 мин до введения препарата вечером.
В целом было отобрано 32 пробы у каждого субъекта во время данной схемы лечения.
Схема лечения 3: на сутки 1 введения было отобрано 14 проб крови каждый день. Первую пробу крови отбирали до введения препарата (в течение 5 мин), в то время как другие отбирали через 1, 2, 3, 4, 5, 6, 8, 10, 12, 16, 20, 24 и 48 ч после введения препарата.
Критерии оценки:
Аналитический метод:
Анализируемое соединение: ондансетрон в плазме крови человека.
Метод: ВЭЖХ с МС/МС детектированием.
Пределы определения: от 0,500 нг/мл до 3000,000 нг/мл.
Безопасность:
Безопасность оценивали по выявлению побочных эффектов, проведению стандартных лабораторных анализов, витальным признакам, ЭКГ и физическому осмотру.
Математическая модель и статистические методы определения фармакокинетических параметров
Основные параметры всасывания и распределения определяли с использованием некомпартментного метода с допущением наличия log-линейной терминальной фазы. Трапецеидальное правило для установления площади под кривой, установления терминальной фазы основывались на максимизации коэффициента определения. Представляющими интерес фармакокинетическими параметрами были Cmax для каждого дня введения, AUC0-24 для каждого дня введения, Cmin для каждого дня введения и С24 для каждого дня введения. Другими рассчитанными параметрами были Tmax для каждого дня введения, AUCT, AUC∞, AUCT/∞, Kel и T1/2el.
Статистический анализ всех фармакокинетических параметров был основан на параметрической модели случайного дисперсионного анализа ANOVA. Двусторонний 90% доверительный интервал отношения среднегеометрических значений полученных методом наименьших квадратов, получали из ln-преобразованных фармакокинетических параметров.
Во время введения тест-препарата Cmax и AUC0-24 на сутки 2-5 сравнивали со значениями Cmax и AUC0-24 на сутки 1 для оценки возможной кумуляции при повторных введениях.
Кумуляцию композиции тест-препарата оценивали с использованием ln-преобразованных значений Cmax и AUC0-24. Модель дисперсионного анализа (ANOVA) подгоняли с сутками в качестве постоянного эффекта и субъекта в качестве случайного эффекта.
Модель ANOVA для сравнения схем лечения:
- постоянные факторы: последовательность, период, схема лечения
- случайный фактор: субъект (иерархический в последовательности).
Модель ANOVA для оценки кумуляции:
- постоянные факторы: сутки
- случайный фактор: субъект.
Стандарты для сравнительного исследования биоэквивалентности:
Концентрации ондансетрона во времени после введения тест-препарата сравнивали с данными после введения референс-препарата. Одну дозу 24 мг ондансетрона с немедленным высвобождением рассматривали в качестве эффективной для предупреждения тошноты и рвоты, вызванных химиотерапией рака с сильно выраженным рвотным эффектом, и введение в дозе 8 мг дважды в день рассматривали в качестве эффективного для химиотерапии с умеренно выраженным рвотным эффектом. Следовательно, если обнаруживалось, что концентрация ондансетрона после введения тест-препарата была аналогичной или выше, чем после введения одной или обеих схем введения референс-препарата на большую часть временных точек в первые 24 ч исследуемого периода, то можно заключить, что тест-препарат обладал, по меньшей мере, такой же эффективностью с применяемыми схемами для химиотерапии рака с умеренно выраженным рвотным эффектом.
Безопасность:
Описательная статистика.
Обобщение результатов
Результаты по оценке безопасности:
У девяти (9) из 18 субъектов (50,0%), включенных в исследование, в целом отмечали 28 побочных явлений. Все 28 побочных явлений, о которых сообщалось во время исследования, были слабыми по выраженности. Ниже в таблице представлено количество побочных явлений, вызванных лечением, классифицированных по выраженности и причинной связи.
Шесть (6) субъектов (33,3%) сообщали о 12 побочных явлениях (2 различных класса по поражению систем органов и 7 различных предпочтительных терминов) после схемы лечения 1, 5 субъектов (27,8%) сообщали о 7 побочных явлениях (3 различных класса по поражению систем органов и 5 различных предпочтительных терминов) после схемы лечения 2, и 6 субъектов (33,3%) сообщали о 9 побочных явлениях (4 различных класса по поражению систем органов и 7 различных предпочтительных терминов) после схемы лечения 3. Количество субъектов, которые испытывали, по меньшей мере, одно побочное явление во время исследования, было аналогичным для всех 3 схем лечения.
Побочные явления, которые испытывали два или более субъектов с любой схемой лечения (схема лечения 1, схема лечения 2, схема лечения 3), были следующие: аномальные фекалии (2, 0, 0), запор (2, 0, 1), гематомы в месте прокола сосуда при взятии крови (2, 3, 2), боль в месте прокола сосуда при взятии крови (0, 1, 1), головная боль (0, 1, 1) и бессонница (0, 1, 1). Кроме того, связанные с введением препаратов побочные явления, которые испытывали два или более субъектов с любой схемой лечения (схема лечения 1, схема лечения 2, схема лечения 3), были запор (2, 0, 1), головная боль (0, 1, 1) и бессонница (0, 1, 1).
Во время данного исследования не сообщалось о серьезных побочных явлениях или смертях. Кроме того, отсутствовали существенные изменения в результатах лабораторных анализов, витальных признаках, ЭКГ или физическом осмотре.
Отсутствовали серьезные побочные явления, для купирования которых потребовалось бы использование препаратов, после первого введения.
Ни один из субъектов не был исключен из исследования по причинам безопасности.
Результаты по фармакокинетике:
Сравнение схем лечения:
Основные фармакокинетические параметры (Cmin, Cmax, С24 и AUC0-24) в каждой схеме лечения определяли для каждого дня введения. Проводили сравнение между первыми 2 днями введения тест-препарата с 2 днями введения препарата Zofran 8 мг дважды в день и также проводили сравнение между первым днем введения тест-препарата с введением препарата Zofran 24 мг. Обобщенные результаты данных сравнений представлены на фигурах 7-13 и в таблицах 21-24.

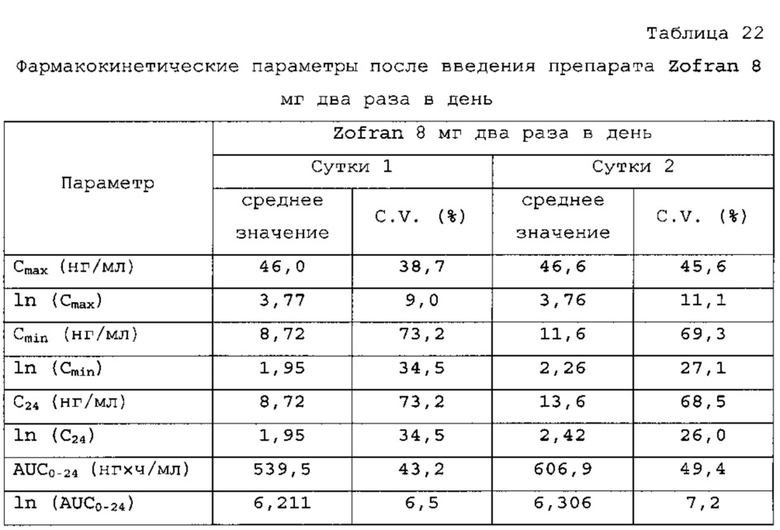
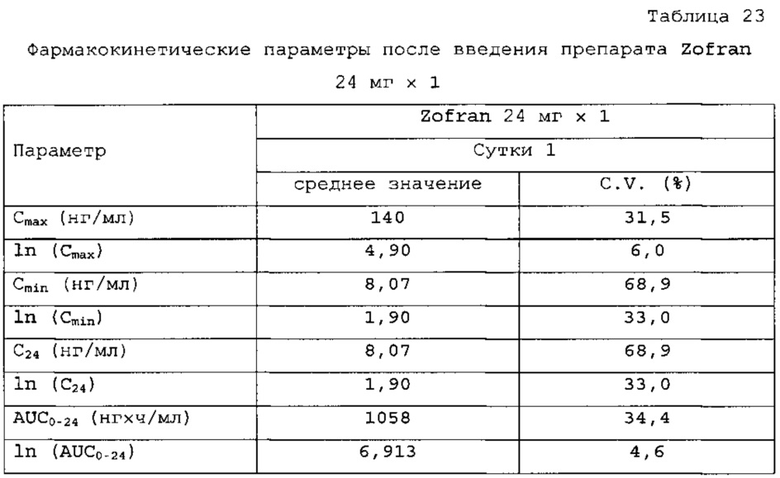

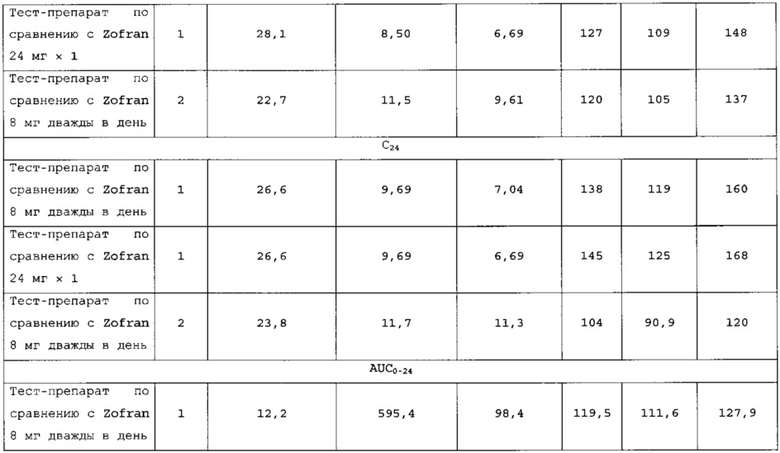

Сравнение концентраций:
Концентрации ондансетрона на выбранные временные точки после введения тест-препарата сравнивали с концентрациями после введения препарата Zofran 8 мг дважды в день и Zofran 24 мг × 1. Измеренные концентрации, достигаемые после введения тест-препарата на 10, 12, 14 и 16 ч после введения для суток 1 и на 20 ч после введения для суток 2, сравнивали с соответствующими измеренными концентрациями ондансетрона, достигаемыми при введении других схем лечения. Обобщенные результаты данных сравнений представлены в последующих таблицах.
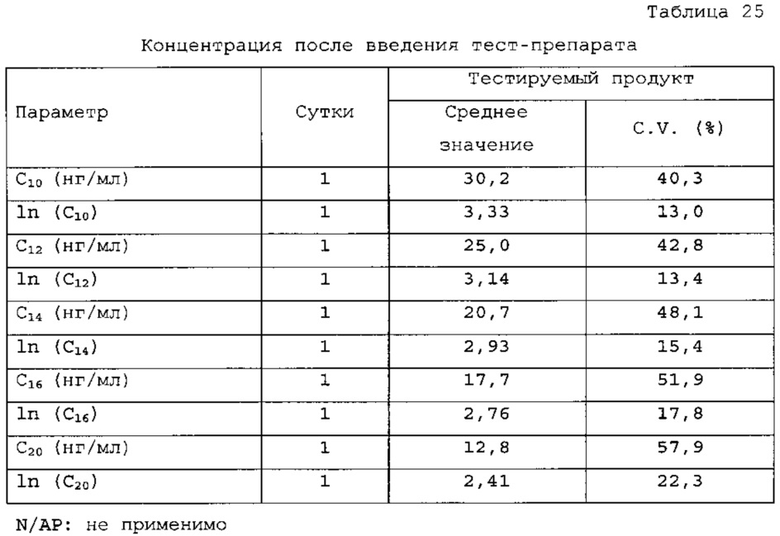
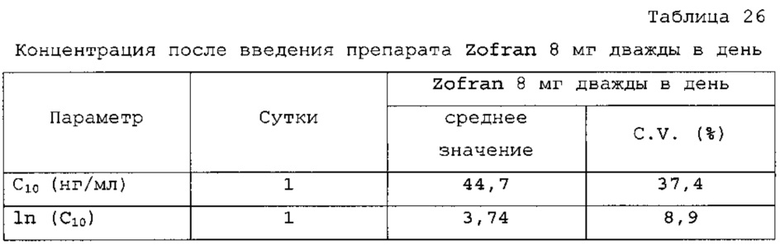
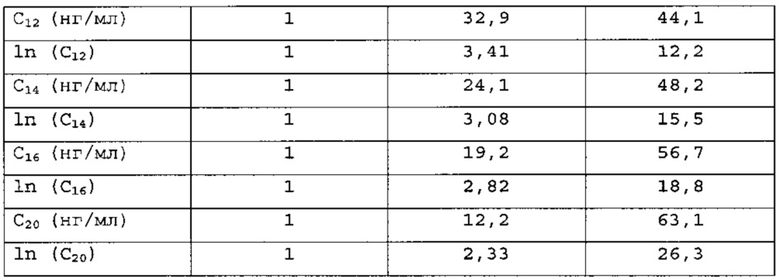
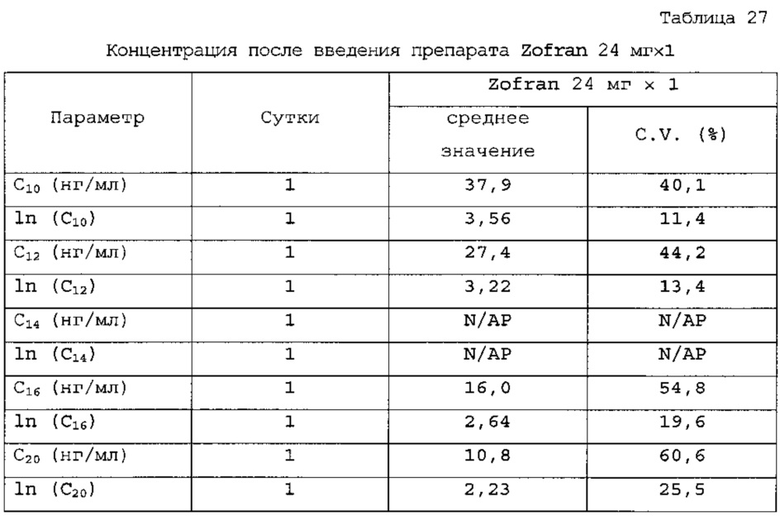
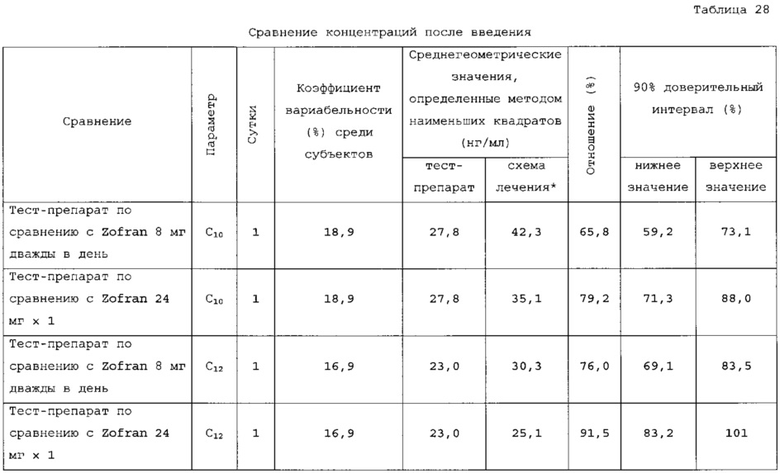
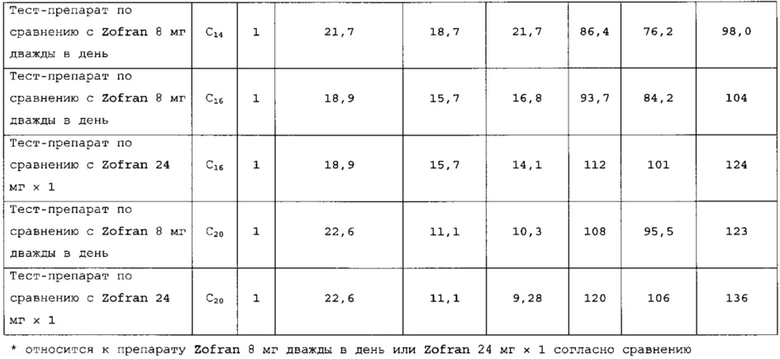
Оценка кумуляции:
Для оценки кумуляции ондансетрона после многократного введения тест-препарата определяли Cmax и AUC0-24 для введения 5 суток подряд и сравнивали с введением в однократной дозе тест-препарата на сутки 1. Обобщенные результаты представлены в последующих таблицах.
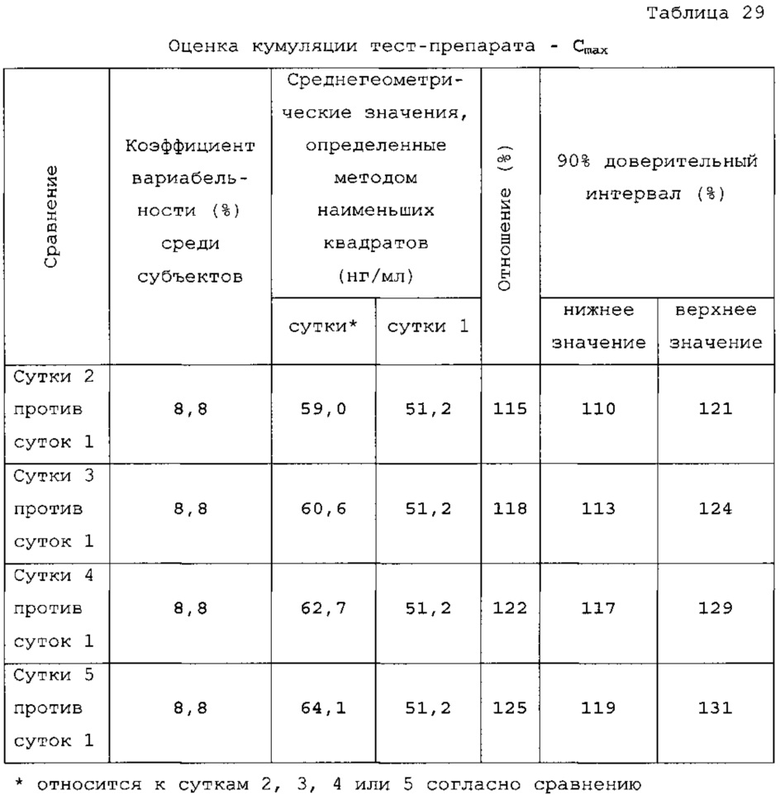
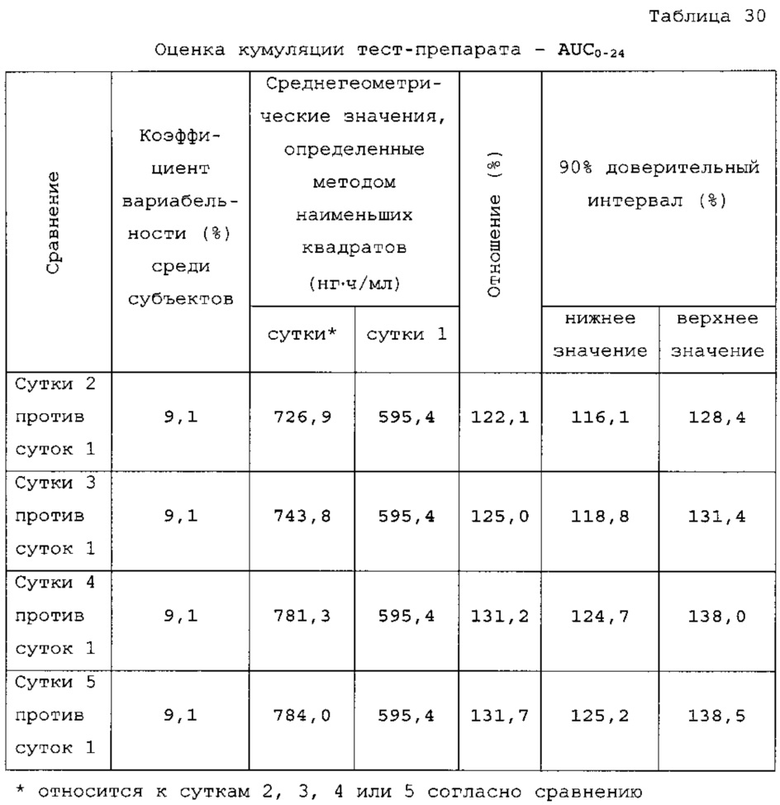
Обсуждение фармакокинетических данных:
Сравнение схем лечения:
- Тест-препарат по сравнению с препаратом Zofran 8 мг два раза в день:
Результаты, представленные здесь, показывают, что значения Cmin и Cmax в течение 24 ч, а также AUC0-24 были выше в течение первых двух суток введения тест-препарата по сравнению с обоими сутками введения препарата Zofran 8 мг дважды в день.
Было установлено, что значение С24 было выше при введении тест-препарата на первые сутки схемы лечения, и было установлено, что оно сравнимо между тест-препаратом и препаратом Zofran 8 мг дважды в день на вторые сутки схемы лечения.
- Тест-препарат по сравнению с препаратом Zofran 24 мг × 1: Значения Cmin и С24 также были выше для первых суток схемы лечения тест-препаратом по сравнению с введением препарата Zofran 24 мг × 1.
Однако значения Cmax и AUC0-24, достигаемые с введением тест-препарата, были примерно на 60% (отношение 38%) и 40% (отношение 59%) ниже, чем значения Cmax и AUC0-24, достигаемые при введении препарата Zofran 24 мг × 1.
Сравнение концентраций:
Тест-препарат по сравнению с препаратом Zofran 8 мг дважды в день:
Измеренные концентрации, начиная с 3 ч по 8 ч после введения первоначальной дозы, были выше после введения тест-препарата. На 10 и 12 ч было установлено, что концентрации были ниже после введения тест-препарата на первые сутки лечения; последующие концентрации были аналогичными между двумя группами на первые сутки.
За счет более позднего введения второй дозы на сутки 2 форма кривой концентрации для препарата Zofran 8 мг дважды в день несколько отличалась от таковой на первые сутки, но в целом результаты были аналогичными.
- Тест-препарат по сравнению с препаратом Zofran 24 мг × 1: Было установлено, что измеренные концентрации до 10 ч были ниже после введения тест-препарата. Было установлено, что измеренные концентрации на 12 и 16 ч были сравнимы между двумя схемами лечения и были выше для тест-препарата на 20 и 24 ч.
Оценка кумуляции
Оценка кумуляции, проведенная по параметру Cmax, показала первое 15% повышение концентрации в период между сутками 1 и сутками 2, и также показала постоянное повышение отношения на основе обратного преобразования разницы средних значений наименьших площадей в период с суток 3 по сутки 5 (118-125% от значения Cmax, которое имело место на сутки 1), указывая на наличие кумуляции ондансетрона после многократных введений тест-препарата. Аналогичное повышение наблюдали для параметра AUC0-24 на сутки 3 и 4 (125-131% от значения AUC0-24, которое имело место на сутки 1) после 22% повышения в период между сутками 1 и сутками 2.
Отношение на основе обратного преобразования разницы средних значений наименьших площадей AUC0-24 было аналогично для суток 4 (131%) и суток (132%), указывая, что стационарное состояние достигалось в период между сутками 4 и 5 при повторных ежедневных введениях тест-препарата.
Заключение:
Сравнительная биоэквивалентность:
Представленные здесь результаты показывают, что биодоступность тест-препарата не была ниже по сравнению с биодоступностью препарата Zofran 8 мг, который использовали по официально разрешенной схеме для предупреждения тошноты и рвоты, вызванной химиотерапией с умеренно выраженным рвотным эффектом.
Ключевые точки в данном сравнительном исследовании:
среднегеометрическое значение AUC0-24 тест-препарата было на 19% выше, чем для препарата Zofran 8 мг дважды в день (90% доверительный интервал 12-28%) на сутки 1 введения, на 33% выше (90% доверительный интервал 24-43%) на сутки 2;
среднегеометрическое значение Cmax тест-препарата было на 18% выше, чем для препарата Zofran 8 мг дважды в день (90% доверительный интервал 9-28%) на сутки 1 введения, на 38% выше (90% доверительный интервал 30-47%) на сутки 2;
оба значения С24 и Cmin тест-препарата были выше, чем эти параметры для препарата Zofran 8 мг дважды в день и Zofran 24 мг × 1;
уровни ондансетрона были аналогичны или выше после введения тест-препарата, чем после введения препарата Zofran 8 мг на все временные точки, за исключением 10 и 12 ч после первоначального введения на сутки 1 и 14-20 ч на сутки 2;
на 10 и 12 ч после введения на сутки 1 уровни ондансетрона после введения тест-препарата были на 107% и 72% выше, чем самый минимальный уровень ондансетрона через 8 ч после введения первоначальной дозы препарата Zofran 8 мг;
на 14-20 ч после первоначального введения на сутки 2 уровни ондансетрона после введения тест-препарата были на 47-159% выше, чем самый минимальный уровень ондансетрона через 12 ч после введения первоначальной дозы препарата Zofran 8 мг;
кроме того, начиная с 12 ч, уровни ондансетрона после введения тест-препарата были аналогичны или выше чем уровни после введения препарата Zofran 24 мг × 1, где эта схема является официально разрешенной для химиотерапии с высоко выраженным рвотным эффектом.
Уровень ондансетрона в плазме крови после введения тест-препарата был аналогичен или выше, чем уровень в плазме крови после введения препарата Zofran 8 мг дважды в день на большинство временных точек, и концентрации на другие временные точки были существенно выше, чем самые минимальные уровни на 8 или 12 ч (соответственно сутки 1 и 2) после введения первоначальной дозы для референтной схемы препарата Zofran 8 мг дважды в день. Следовательно, обоснованно заключить, что эффективность тест-препарата является, по меньшей мере, такой же высокой, что и у препарата Zofran 8 мг дважды в день.
Оценка кумуляции:
Введение один раз день тест-препарата в течение 5 дней подряд натощак подтвердило наличие кумуляции ондансетрона в плазме крови. Максимальные концентрации в плазме крови поднимались с 15% до 25% в период с суток 2 по сутки 5. После первого 15% повышения отмечали примерно 3% прирост максимальной концентрации на каждый следующий дня введения. Значение AUC0-24 повышалось с 22% до 31% в период с суток 2 до суток 4 и затем стабилизировалось на 32% на сутки 5, указывая на возникновение стационарного состояния.
Безопасность и переносимость тест-препарата:
Результаты, представленные здесь, показывают, что введение тест-препарата один раз в день в течение 5 дней подряд было безопасным и хорошо переносилось субъектами, включенными в данное исследование. Кроме того, количество субъектов, испытавших, по меньшей мере, одно побочное явление было сравнимым между всеми группами лечения, и все 28 побочных явления, о которых сообщалось во время исследования, были слабыми по выраженности, свидетельствуя о том, что безопасность и переносимость композиции с замедленным высвобождением, тест-препарата, были аналогичны профилю безопасности других схем лечения.
Несмотря на наличие определенной кумуляции препарата при повторных введениях, отсутствовало доказательство того, что это приводило к проблемам с безопасностью. В частности, незначительное удлинение интервала QTc было более выраженным после введения однократной дозы 24 мг препарата Zofran с немедленным высвобождением, чем после введения 5 суточных доз тест-препарата.
Согласно аспектам, описанным здесь, раскрывается твердая лекарственная форма для перорального введения, которая включает ядро, содержащее матрикс из неионогенного полимера, первое количество первого противорвотного препарата или его фармацевтически приемлемой соли, диспергированное внутри матрикса, и соль, диспергированную внутри матрикса/ первую изолирующую оболочку, окружающую ядро, где первая изолирующая оболочка состоит из матрикса из неионогенного полимера; и слой препарата с немедленным высвобождением, окружающий первую изолирующую оболочку, где слой препарата с немедленным высвобождением содержит неионогенный полимер, и второе количество второго противорвотного препарата или его фармацевтически приемлемой соли, диспергированное в нем, где слой препарата выполнен таким образом, чтобы обеспечить высвобождение второго количества противорвотного препарата в течение периода времени, по меньшей мере, 1 ч, где твердая лекарственная форма для перорального введения выполнен таким образом, чтобы обеспечить высвобождение первого количества первого противорвотного препарата и второго количества второго противорвотного препарата в течение периода времени минимум 16 ч. В варианте осуществления твердая лекарственная форма для перорального введения включает кишечнорастворимую оболочку, окружающую первую изолирующую оболочку. В варианте осуществления твердая лекарственная форма для перорального введения дополнительно включает вторую изолирующую оболочку, окружающую слой препарата с немедленным высвобождением, где вторая изолирующая оболочка состоит из неионогенного полимера. В варианте осуществления первая изолирующая оболочка содержит добавку для покрытия, такую как plasACRYL™. В варианте осуществления соль в ядре диспергирована в матриксе в концентрации в пределах от 50 масс. % до 100 масс. % по отношению к массе матрикса. В варианте осуществления при воздействии на твердую лекарственную форму водной среды соль вызывает затвердевание границ по периферии матрикса, где границы последовательно распространяются внутрь к его центру по мере проникновения водной среды в матрикс, где затвердевание границ ограничивает скорость, с которой противорвотный препарат в матриксе высвобождается из таблетки. В варианте осуществления твердая лекарственная форма для перорального введения выполнена таким образом, чтобы обеспечить высвобождение первого количества противорвотного препарата и второго количества противорвотного препарата в течение периода времени минимум 20 ч. В варианте осуществления твердая лекарственная форма для перорального введения выполнен таким образом, чтобы обеспечить высвобождение первого количества противорвотного препарата и второго количества противорвотного препарата в течение периода времени минимум 24 ч. В варианте осуществления первый противорвотный препарат и второй противорвотный препарат представляют один и тот же препарат. В варианте осуществления первый противорвотный препарат и второй противорвотный препарат каждый представляет ондансетрон или эквивалентное количество соли ондансетрона.
Согласно аспектам, описанным здесь, раскрывается твердая лекарственная форма для перорального введения, которая включает ядро, содержащее гипромеллозу, 18 мг ондансетрона или эквивалентное количество соли ондансетрона и цитрат натрия безводный; первую изолирующую оболочку, окружающую ядро и содержащую гипромеллозу; и слой препарата с немедленным высвобождением, окружающий первую изолирующую оболочку и содержащий гипромеллозу, и 6 мг ондансетрона или эквивалентное количество соли ондансетрона, где слой препарата с немедленным высвобождением выполнен таким образом, чтобы обеспечить высвобождение ондансетрона в течение периода времени, по меньшей мере, 1 ч, где общее количество ондансетрона в лекарственной форме высвобождается в течение 24 ч. В варианте осуществления твердая лекарственная форма для перорального введения дополнительно включает кишечнорастворимую оболочку, окружающую первую изолирующую оболочку. В варианте осуществления твердая лекарственная форма для перорального введения дополнительно включает вторую изолирующую оболочку, окружающую слой препарата с немедленным высвобождением, где вторая изолирующая оболочка состоит из неионогенного полимера. В варианте осуществления первая изолирующая оболочка также содержит добавку для покрытия, такую как plasACRYL™. В варианте осуществления цитрат натрия безводный в ядре диспергирован в гипромеллозе в концентрации в пределах от 50 масс. % до 100 масс. % по отношению к массе гипромеллозы. В варианте осуществления при воздействии на твердую лекарственную форму водной среды цитрат натрия безводный вызывает затвердевание границ по периферии гипромеллозы, где границы последовательно распространяются внутрь к ее центру по мере проникновения водной среды в гипромеллозу, где затвердевание границ ограничивает скорость, с которой ондансетрон в гипромеллозе высвобождается из таблетки. В варианте осуществления, когда твердая лекарственная форма для перорального введения вводится пациенту натощак, то достигается Cmax, по меньшей мере, 50 нг/мл. В варианте осуществления, когда твердая лекарственная форма для перорального введения вводится пациенту натощак, то достигается AUC, по меньшей мере, 600 нг⋅ч/мл.
Согласно аспектам, описанным здесь, раскрывается твердая лекарственная форма для перорального введения, которая включает ядро, содержащее матрикс из неионогенного полимера, первое количество ондансетрона или эквивалентное количество соли ондансетрона, диспергированное внутри матрикса, и соль, диспергированную внутри матрикса; первую изолирующую оболочку, окружающую ядро, где первая изолирующая оболочка состоит из матрикса из неионогенного полимера; и слой препарата с немедленным высвобождением, окружающий первую изолирующую оболочку, где слой препарата с немедленным высвобождением содержит неионогенный полимер, и второе количество ондансетрона или эквивалентное количество соли ондансетрона, диспергированное в нем, где твердая лекарственная форма для перорального введения обеспечивает профиль растворения ондансетрона in vitro при определении на аппарате для оценки растворения с 2 лопастями при 37°С в водном растворе, содержащем дистиллированную воду, при 50 об/мин, показывающий следующее: а) примерно от 20% до 50% от общего количества ондансетрона высвобождается через 2,5 ч при определении в аппарате; b) примерно от 50% до 70% от общего количества ондансетрона высвобождается через 5 ч при определении в аппарате; с) не менее примерно 90% от общего количества ондансетрона высвобождается через 15 ч при определении в аппарате. В варианте осуществления, когда твердая лекарственная форма для перорального введения вводится пациенту натощак в дозе 24 мг ондансетрона, то достигается Cmax, по меньшей мере, 50 нг/мл. В варианте осуществления, когда твердая лекарственная форма для перорального введения вводится пациенту натощак в дозе 24 мг ондансетрона, то достигается AUC, по меньшей мере, 600 нг⋅ч/мл.
Согласно аспектам, описанным здесь, раскрывается упакованный фармацевтический препарат, который включает множество любых твердых лекарственных форм для перорального введения по настоящему изобретению в герметично закрытом контейнере и инструкции по введению лекарственных форм перорально для осуществления предупреждения тошноты и рвоты.
Согласно аспектам, описанным здесь, раскрывается фармацевтический препарат, который включает множество любых твердых лекарственных форм для перорального введения по настоящему изобретению, каждая в отдельном герметично закрытом контейнере и инструкции по введению лекарственных форм перорально для осуществления предупреждения тошноты и рвоты.
Согласно аспектам, описанным здесь, раскрывается разовая лекарственная форма для перорального введения пациенту, где разовая лекарственная форма выполнен таким образом, чтобы обеспечить предупреждение тошноты и рвоты у пациента, и где разовая лекарственная форма включает комбинацию компонента с немедленным высвобождением ондансетрона, содержащего разовую лекарственную форму ондансетрона или его фармацевтически приемлемую соль в пределах от 4 мг до 8 мг; и компонент с контролируемым высвобождением ондансетрона или его фармацевтически приемлемой соли в пределах от 16 мг до 28 мг, где компонент с контролируемым высвобождением ондансетрона содержит матрикс из неионогенного полимера, ондансетрон внутри матрикса и соль, диспергированную внутри матрикса, и где разовая лекарственная форма показывает максимальную концентрацию в плазме крови (Cmax) примерно на 2-5 ч (Tmax) после введения и показывает сравнимые значения Cmax с композицией ондансетрона с неконтролируемым высвобождением при введении три раза в день, без снижения общей биодоступности препарата, определяемой площадью под кривой концентрация-время (AUC), тем самым обеспечивая уменьшение зависимых от концентрации побочных эффектов без снижения эффективности.
Упакованный фармацевтический препарат, который включает множество разовых лекарственных форм по настоящему изобретению, может находиться в герметично закрытом контейнере и включает инструкции по введению лекарственных форм перорально для осуществления предупреждения тошноты и рвоты.
Упакованный фармацевтический препарат, который включает множество разовых лекарственных форм по настоящему изобретению, может находиться в отдельном герметично закрытом контейнере и включает инструкции по введению лекарственных форм перорально для осуществления предупреждения тошноты и рвоты.
Способ предупреждения тошноты и рвоты, включающий стадию введения терапевтически эффективного количества твердой лекарственной формы для перорального введения по настоящему изобретению, пациенту.
Согласно аспектам, описанным здесь, раскрывается композиция для введения один раз в день, которая включает: (а) ядро, содержащее матрикс из неионогенного полимера, первое количество ондансетрона или эквивалентное количество соли ондансетрона, диспергированное внутри матрикса, и соль, диспергированную внутри матрикса; (b) первую изолирующую оболочку, окружающую ядро, где первая изолирующая оболочка состоит из матрикса из неионогенного полимера; и (с) слой препарата с немедленным высвобождением, окружающий кишечнорастворимую оболочку, где слой препарата с немедленным высвобождением содержит неионогенный полимер, и второе количество ондансетрона или эквивалентное количество соли ондансетрона, диспергированное в нем, где слой препарата с немедленным высвобождением выполнен таким образом, чтобы обеспечить высвобождение второго количества ондансетрона в течение периода времени, по меньшей мере, 1 ч, где слой с немедленным высвобождением препарата высвобождает второе количество ондансетрона в верхнем отделе желудочно-кишечного тракта пациента, где ядро высвобождает первое количество ондансетрона в нижнем отделе желудочно-кишечного тракта пациента, где композиция представляет таблетку или капсулу, которая содержит от 24 до 40 мг ондансетрона или эквивалентное количество соли ондансетрона, и обеспечивает профиль в плазме крови in vivo, выбранный из: (а) среднего значения Cmax, по меньшей мере, 50,0 нг/мл; (b) среднего значения AUC0-24, по меньшей мере, 550,0 нг⋅ч/мл и (с) среднего значения Tmax в пределах примерно от 2,0 ч до 5,0 ч. В варианте осуществления композиция для введения один раз день, при введении один раз в день человеку натощак, биоэквивалентна введению человеку натощак разовой лекарственной формы для введения три раза в день, содержащей 8 мг ондансетрона. В варианте осуществления биоэквивалентность устанавливают по тому, что 90% доверительный интервал укладывается в пределы от 0,80 до 1,25 для обоих параметров Cmax и AUC0-24 при введении человеку. В варианте осуществления свойства растворимости и растворения независимы от pH. В варианте осуществления ядро имеет независимый от pH профиль растворения-высвобождения в диапазоне значений pH 1,2-6,8. В варианте осуществления каждый из ядра и слоя с немедленным высвобождением препарата имеет независимый от pH профиль растворения-высвобождения в диапазоне pH 1,2-6,8. В варианте осуществления ядро и слой с немедленным высвобождением препарата окружены изолирующей оболочкой из неионогенного полимера, которая повышает гидрофильность композиции, и в результате профиль растворения композиции не зависит от pH.
Все патенты, заявки на патент и опубликованные источники, цитированные здесь, включены здесь в полном объеме в виде ссылки. Очевидно, понятно, что несколько из раскрытых выше и других признаков и функций, или их альтернативы при желании можно объединить во многие другие различные системы или применения. Специалисты в данной области затем могут осуществить различные непредвиденные в настоящее время или непредполагаемые альтернативы, модификации, вариации или усовершенствования.
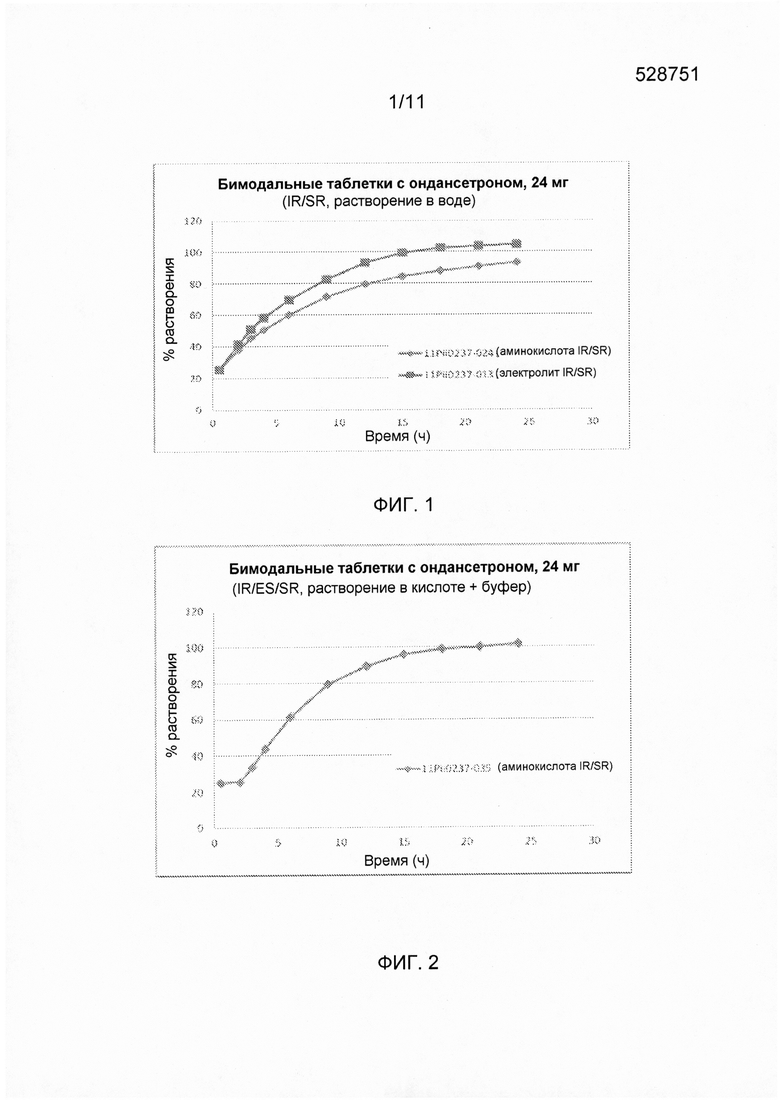
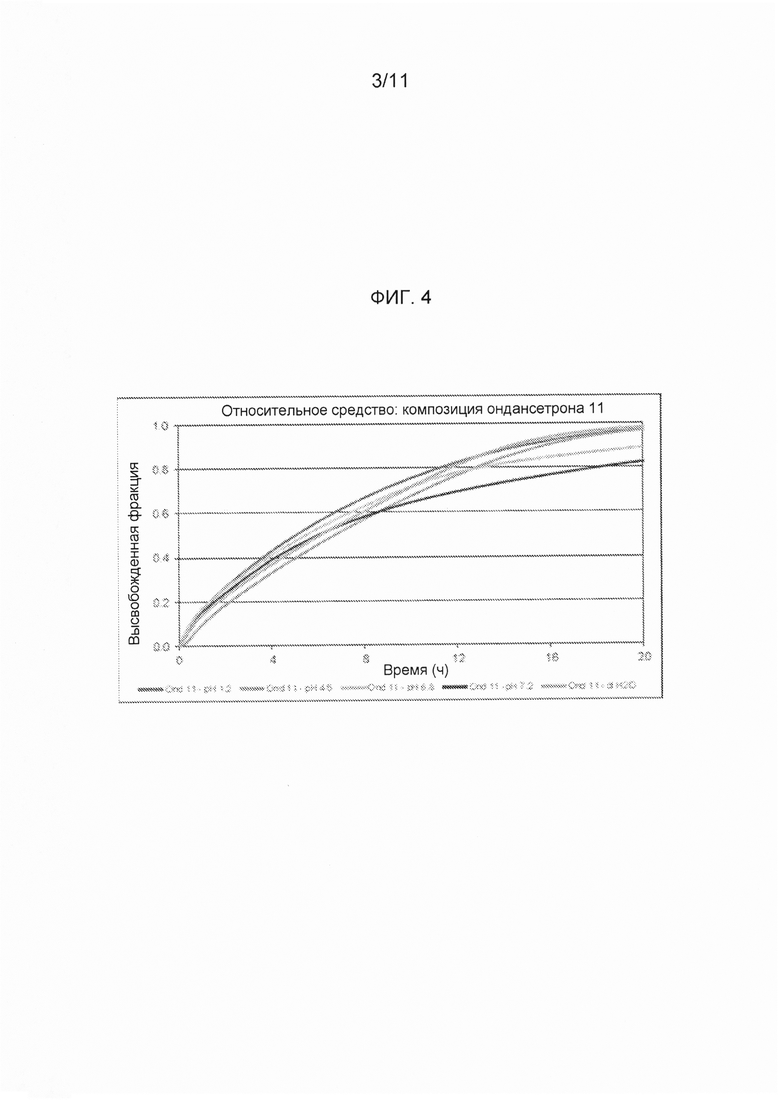
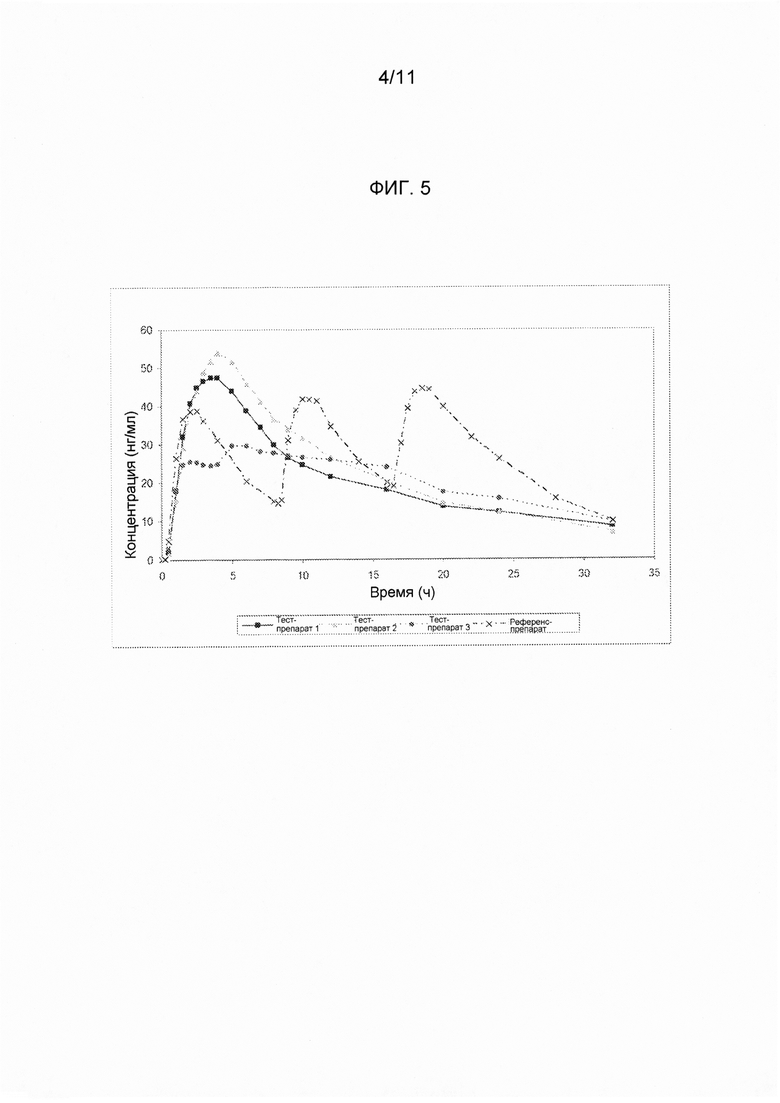
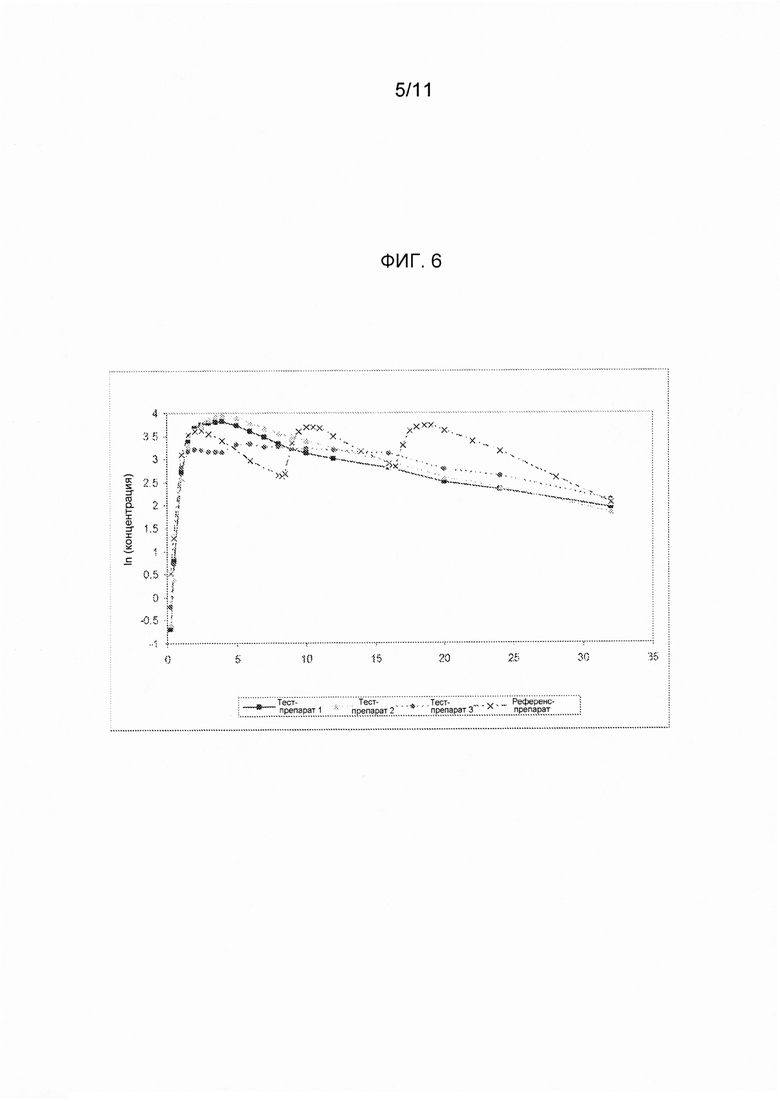
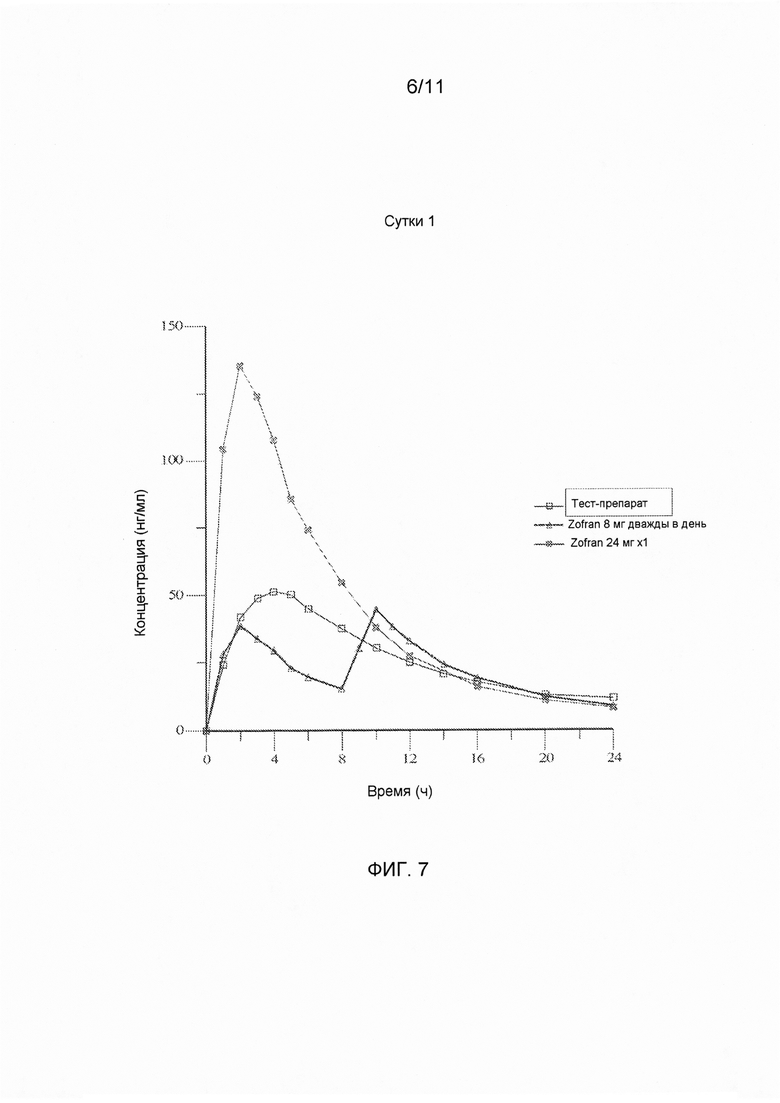
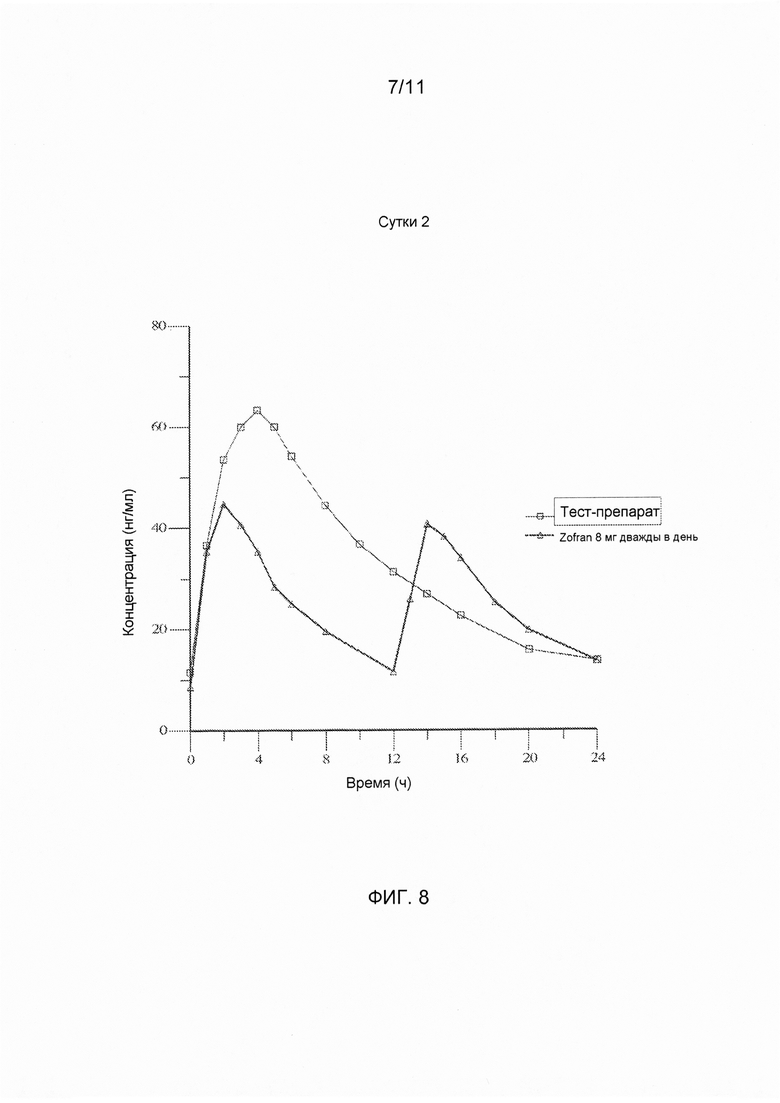
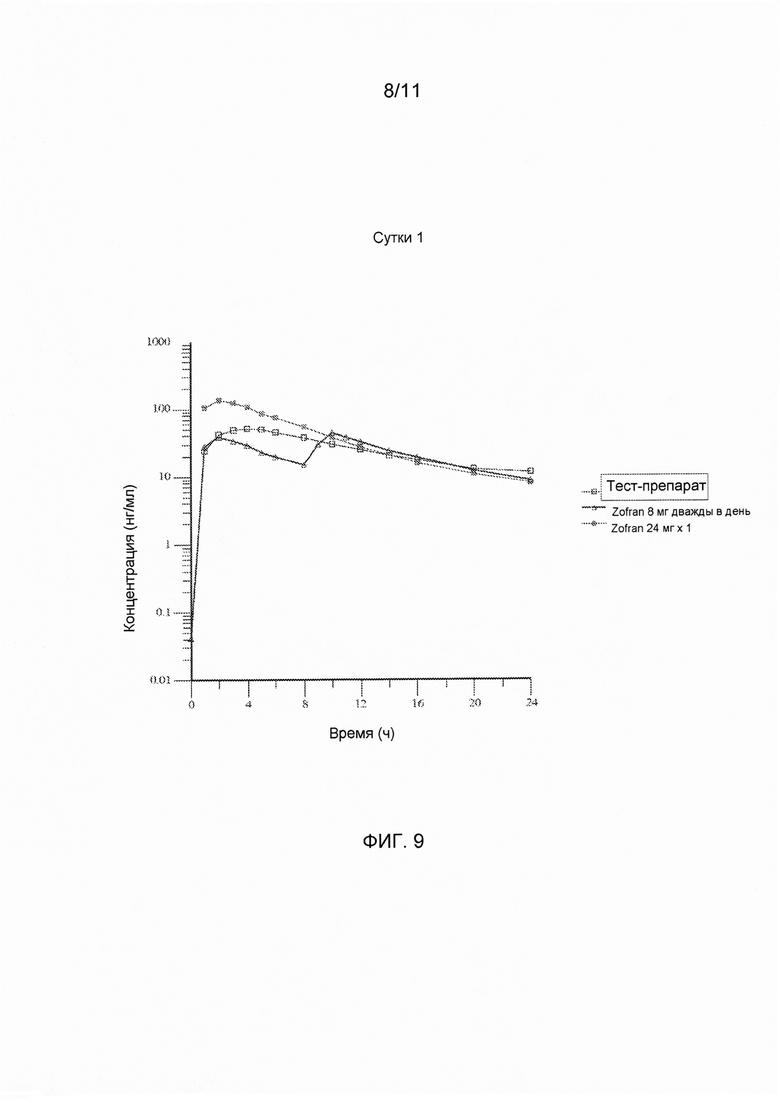
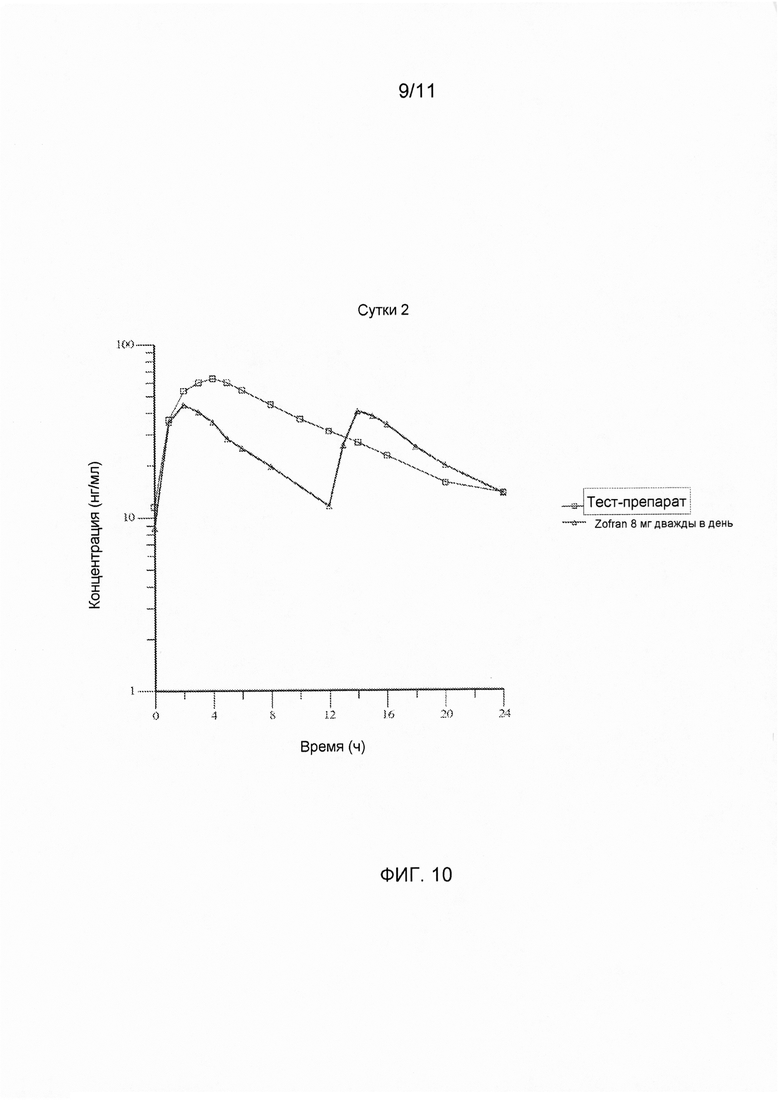
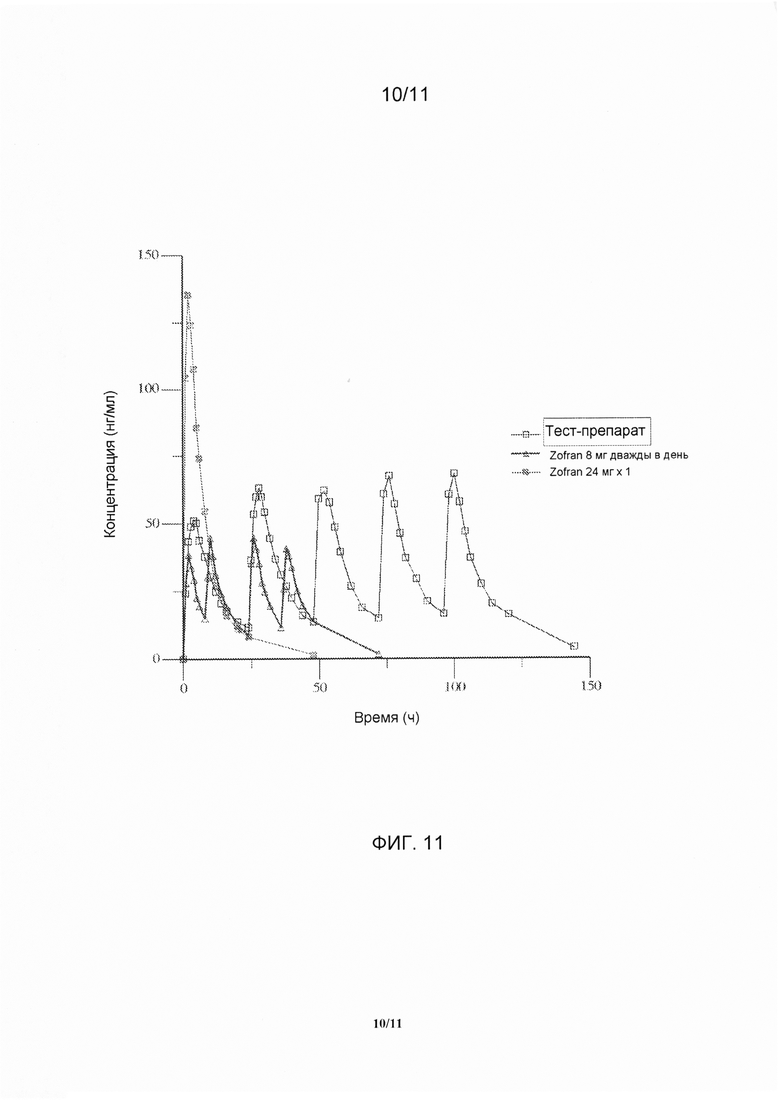

Изобретение относится к медицине, в частности к таблетке ондансетрона для лечения тошноты и рвоты, а также к упакованному фармацевтическому препарату и способу лечения тошноты и рвоты. Таблетка ондансетрона включает ядро, содержащее гипромеллозу, первое количество первого противорвотного препарата или его фармацевтически приемлемой соли, диспергированное внутри гипромеллозы, и безводный цитрат натрия, диспергированный внутри гипромеллозы; первую нефункциональную изолирующую оболочку, окружающую ядро, где первая изолирующая оболочка состоит из гипромеллозы; и слой препарата с немедленным высвобождением, окружающий первую изолирующую оболочку, где слой препарата с немедленным высвобождением содержит гипромеллозу, и второе количество второго противорвотного препарата или его фармацевтически приемлемой соли, диспергированное в нем, где таблетка контролирует тошноту и рвоту в течение по меньшей мере 24 ч. Осуществление изобретения позволяет получить твердую фармацевтическую композицию, обеспечивающую устойчивое контролируемое высвобождение препарата. 12 н. и 17 з.п. ф-лы, 12 ил., 30 табл., 8 пр.
1. Таблетка ондансетрона для лечения тошноты и рвоты, включающая:
ядро, содержащее гипромеллозу, первое количество ондансетрона или его фармацевтически приемлемой соли, диспергированное внутри гипромеллозы, и безводный цитрат натрия, диспергированный внутри гипромеллозы, где безводный цитрат натрия присутствует в количестве, достаточном для образования затвердевающей границы вокруг периферии гипромеллозы при воздействии водной среды, чтобы ограничить скорость, с которой ондансетрон или эквивалентное количество соли ондансетрона, диспергированное внутри гипромеллозы, высвобождалось из ядра, и где ондансетрон или эквивалентное количество соли ондансетрона высвобождаются из ядра с постоянной скоростью;
первую нефункциональную изолирующую оболочку, окружающую ядро, где первая нефункциональная изолирующая оболочка состоит из гипромеллозы, где первая нефункциональная изолирующая оболочка не оказывает влияния на высвобождение ондансетрона из таблетки; и
слой препарата с немедленным высвобождением, окружающий первую изолирующую оболочку, где слой препарата с немедленным высвобождением содержит гипромеллозу, и второе количество ондансетрона или его фармацевтически приемлемой соли, диспергированное в нем,
где таблетка контролирует тошноту и рвоту в течение по меньшей мере 24 часов.
2. Таблетка по п. 1, дополнительно включающая кишечнорастворимую оболочку, окружающую первую изолирующую оболочку.
3. Таблетка по п. 1, дополнительно включающая вторую изолирующую оболочку, окружающую слой препарата с немедленным высвобождением, где вторая изолирующая оболочка состоит из гипромеллозы.
4. Таблетка по п. 1, где первая изолирующая оболочка содержит добавку для покрытия.
5. Таблетка по п. 1, где безводный цитрат натрия в ядре диспергирован в гипромеллозе в пределах от 50 масс.% до 100 масс.% по отношению к массе гипромеллозы.
6. Таблетка по п. 1, где таблетка выполнена таким образом, чтобы обеспечить высвобождение первого количества ондансетрона и второго количества ондансетрона в течение периода времени минимум 24 ч.
7. Упакованный фармацевтический препарат для лечения тошноты и рвоты, включающий множество таблеток по п. 1 в герметично закрытом контейнере и инструкции по введению таблетки для осуществления лечения тошноты и рвоты.
8. Упакованный фармацевтический препарат для лечения тошноты и рвоты, включающий множество таблеток по п. 1, каждая в отдельном герметично закрытом контейнере, и инструкции по введению таблетки для осуществления лечения тошноты и рвоты.
9. Способ лечения тошноты и рвоты, включающий стадию введения терапевтически эффективного количества таблетки по п. 1 пациенту.
10. Таблетка ондансетрона для лечения тошноты и рвоты, включающая:
ядро, содержащее гипромеллозу, 18 мг ондансетрона или эквивалентное количество соли ондансетрона и цитрат натрия безводный, где безводный цитрат натрия присутствует в количестве, достаточном для образования затвердевающей границы вокруг периферии гипромеллозы при воздействии водной среды, чтобы ограничить скорость, с которой ондансетрон или эквивалентное количество соли ондансетрона, диспергированное внутри гипромеллозы, высвобождалось из ядра, и где ондансетрон или эквивалентное количество соли ондансетрона высвобождаются из ядра с постоянной скоростью;
первую нефункциональную изолирующую оболочку, окружающую ядро и содержащую гипромеллозу, где первая нефункциональная изолирующая оболочка не оказывает влияния на высвобождение ондансетрона из таблетки; и
слой препарата с немедленным высвобождением, окружающий первую изолирующую оболочку и содержащий гипромеллозу, и 6 мг ондансетрона или эквивалентное количество соли ондансетрона,
где таблетка контролирует тошноту и рвоту в течение по меньшей мере 24 часов.
11. Таблетка по п. 10, дополнительно содержащая кишечнорастворимую оболочку, окружающую первую изолирующую оболочку.
12. Таблетка по п. 10, дополнительно содержащая вторую изолирующую оболочку, окружающую слой препарата с немедленным высвобождением, где вторая изолирующая оболочка состоит из гипромеллозы.
13. Таблетка по п. 10, где первая изолирующая оболочка также содержит добавку для покрытия.
14. Таблетка по п. 10, где цитрат натрия безводный в ядре диспергирован в гипромеллозе в концентрации в пределах от 50 масс.% до 100 масс.% по отношению к массе гипромеллозы.
15. Таблетка по п. 10, где когда таблетка вводится пациенту натощак, то достигается Cmax по меньшей мере 40 нг/мл.
16. Таблетка по п. 10, где когда таблетка вводится пациенту натощак, то достигается AUC по меньшей мере 450 нг⋅ч/мл.
17. Таблетка по п. 10, где концентрация ондансетрона в крови примерно через 24 ч после перорального введения ондансетрона (Cmin) составляет по меньшей мере 6 нг/мл.
18. Таблетка по п. 10, где отношение максимальной концентрации ондансетрона (Cmax) к концентрации ондансетрона примерно через 24 ч после приема внутрь ондансетрона (Cmin) составляет от 3 до 7.
19. Упакованный фармацевтический препарат для лечения тошноты и рвоты, включающий множество таблеток по п. 10 в герметично закрытом контейнере и инструкции по введению таблеток для осуществления лечения тошноты и рвоты.
20. Упакованный фармацевтический препарат для лечения тошноты и рвоты, включающий множество таблеток по п. 10, каждая в отдельном герметично закрытом контейнере и инструкции по введению таблеток для осуществления лечения тошноты и рвоты.
21. Способ лечения тошноты и рвоты, включающий стадию введения терапевтически эффективного количества таблетки по п. 10 пациенту.
22. Таблетка ондансетрона для лечения тошноты и рвоты, включающая: ядро, содержащее гипромеллозу, первое количество ондансетрона или эквивалентное количество соли ондансетрона, диспергированное внутри гипромеллозы, и безводный цитрат натрия, диспергированный внутри гипромеллозы, где безводный цитрат натрия присутствует в количестве, достаточном для образования затвердевающей границы вокруг периферии гипромеллозы при воздействии водной среды, чтобы ограничить скорость, с которой ондансетрон или эквивалентное количество соли ондансетрона, диспергированное внутри гипромеллозы, высвобождалось из ядра, и где ондансетрон или эквивалентное количество соли ондансетрона высвобождаются из ядра с постоянной скоростью; первую нефункциональную изолирующую оболочку, окружающую ядро, где первая изолирующая оболочка состоит из гипромеллозы, и где первая нефункциональная изолирующая оболочка не оказывает влияния на высвобождение ондансетрона из таблетки; и слой препарата с немедленным высвобождением, окружающий первую изолирующую оболочку, где слой препарата с немедленным высвобождением содержит гипромеллозу, и второе количество ондансетрона или эквивалентное количество соли ондансетрона, диспергированное в нем, где таблетка обеспечивает профиль растворения ондансетрона in vitro при определении на аппарате для оценки растворения с 2 лопастями при 37°С в водном растворе, содержащем дистиллированную воду, при 50 об/мин, показывающий следующее: а) примерно от 20% до 50% от общего количества ондансетрона высвобождается через 2,5 ч при определении в аппарате; b) примерно от 50% до 70% от общего количества ондансетрона высвобождается через 5 ч при определении в аппарате; с) не менее примерно 90% от общего количества ондансетрона высвобождается через 15 ч при определении в аппарате, и
где таблетка контролирует тошноту и рвоту в течение по меньшей мере 24 часов.
23. Таблетка по п. 22, где когда таблетка вводится пациенту натощак в дозе 24 мг ондансетрона, то достигается Cmax по меньшей мере 40 нг/мл.
24. Таблетка по п. 22, где когда таблетка вводится пациенту натощак в дозе 24 мг ондансетрона, то достигается AUC по меньшей мере 450 нг⋅ч/мл.
25. Таблетка по п. 22, где концентрация ондансетрона в крови примерно через 24 ч после перорального введения ондансетрона (Cmin) составляет по меньшей мере 6 нг/мл.
26. Таблетка по п. 22, где отношение максимальной концентрации ондансетрона (Cmax) к концентрации ондансетрона примерно через 24 ч после перорального введения ондансетрона (Cmin) составляет от 3 до 7.
27. Упакованный фармацевтический препарат для лечения тошноты и рвоты, включающий множество таблеток по п. 22 в герметично закрытом контейнере и инструкции по введению таблеток для осуществления лечения тошноты и рвоты.
28. Упакованный фармацевтический препарат для лечения тошноты и рвоты, включающий множество таблеток для перорального введения по п. 22, каждая в отдельном герметично закрытом контейнере, и инструкции по введению таблеток для осуществления лечения тошноты и рвоты.
29. Способ лечения тошноты и рвоты, включающий стадию введения терапевтически эффективного количества таблетки по п. 22 пациенту.
| US 2012128730 A1, 24.05.2012 | |||
| US 2008200508 A1, 21.08.2008 | |||
| WO 03053413 A2, 03.07.2003 | |||
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ ИНФЕКЦИОННЫХ И НЕИНФЕКЦИОННЫХ ДИАРЕЙ | 2009 |
|
RU2427389C2 |

