Перекрестная ссылка на родственную заявку
По этой заявке испрашивается приоритет предварительной заявки на патент США 60/762766, поданной 27 января 2006 года, содержание которой включено в настоящее описание ссылкой.
Область техники
Настоящее изобретение относится к разработке лекарственных форм с модифицированным высвобождением, содержащих одно или более объединений гранул с пульсирующим высвобождением слабоосновного азот-(N)-содержащего терапевтического средства, имеющего pKa в диапазоне от приблизительно 5 до 14 и растворимость не больше, чем 200 мкг/мл при pH 6,8, и одну или более фармацевтически приемлемых органических кислот. Лекарственная форма показывает сравнимые профили высвобождения как активного вещества, так и органической кислоты после заданной задержки времени при исследовании растворения методикой растворения согласно Фармакопее США (USP), используя двухстадийную среду растворения (сначала 2 часа в 0,1 н. HСl с последующим исследованием в буфере при pH 6,8). В соответствии с другим аспектом сообщается о оральных системах доставки лекарственного средства достижения ФК профиля (фармакокинетики, то есть зависимости концентрации в плазме от времени), подходящего для одноразового или двухразового суточного режима дозирования для пациентов, нуждающихся в лечении.
Уровень техники
Большинство терапевтических средств наиболее эффективны, когда они доставляются при постоянных скоростях в участки поглощения или около участков поглощения. Поглощение терапевтических средств, сделанных доступными, таким образом, как правило, приводит к желаемым концентрациям в плазме, приводящим к максимальной эффективности и минимальным токсичным побочным эффектам. Много усилий посвящалось развитию сложных систем доставки лекарственного средства, таких как осмотические устройства для орального применения. Однако есть примеры, в которых поддержание постоянного уровня лекарственного средства в крови нежелательно. Например, основная цель хронотерапии для сердечно-сосудистых заболеваний состоит в том, чтобы доставить лекарственное средство в более высоких концентрациях в момент самой большой потребности, например, в ранние утренние часы, и в меньших концентрациях, когда потребность - меньше, например, поздним вечером и в ранние ночные часы. В дополнение к разработанной должным образом системе доставки лекарственного средства, время приема не менее важно. Может быть рассчитан своеобразный необходимый фармакокинетический профиль, используя компьютерное моделирование и методы моделирования, основанные на знании фармакокинетических параметров, растворимости, поглощения по желудочно-кишечному тракту и периода полувыведения.
В то время как введенная орально фармацевтическая лекарственная форма проходит через человеческий пищеварительный тракт, лекарственное средство должно высвободиться из лекарственной формы и стать доступным в растворенной форме на или около активного центра для того, чтобы произошло поглощение из желудочно-кишечного (ЖК) тракта. Скорость, при которой лекарственное средство входит в раствор и высвобождается из лекарственной формы, важна для кинетики поглощения лекарственного средства. Лекарственная форма и, следовательно, активный ингредиент подвергается варьированию в pH во время прохождения, то есть pH изменяется от приблизительно 1,2 (pH желудка во время лечебного голодания, но может измениться между 1,2 и 4,0 после потребления пищи) до приблизительно 7,4 (pH желчи: 7,0-7,4 и pH кишечника: от 5 до 7). Кроме того, время прохождения лекарственной формы в отдельных участках пищеварительного тракта может значительно изменяться в зависимости от ее размера и преобладающих местных условий. Другие факторы, которые влияют на поглощение лекарственного средства, включают физико-химические свойства самого лекарственного вещества, такие как pKa, растворимость, энергия кристаллизации и определенная площадь поверхности. Местные условия, которые играют наиболее важную роль, включают свойства люминального содержания (pH, поверхностное натяжение, объем, активация и буферность) и изменения после приема пищи. Следовательно, часто трудно достигнуть высвобождения лекарственного средства при постоянных скоростях.
Основные и кислотные лекарственные средства имеют профили растворимости, зависимые от pH, изменяющиеся более чем на 2 порядка величины в физиологическом диапазоне pH. Наиболее трудными кандидатами для работы являются слабоосновные фармацевтически активные вещества, которые практически нерастворимы при pH>6 и нуждаются в том, чтобы большие дозы были терапевтически эффективными. После поступления в кишечную область часть лекарственного средства, высвобожденного из лекарственной формы, может осаждаться при pH неблагоприятных окружающих условий, если скорость поглощения будет не быстрее, чем скорость высвобождения лекарственного средства. Как вариант, лекарственное средство может остаться в состоянии пересыщенного раствора, благодаря присутствию солей желчной кислоты и лецитина в кишечнике. Пересыщение, значительно большее порядка величины, высшего, чем водная растворимость, было очевидным в предшествующем уровне техники. В случае осаждения существует доказательство перерастворения для поглощения на более медленной стадии.
Функциональные полимерные мембраны, включающие подходящие комбинации синтетических полимеров, таких как водорастворимые (например, повидон), водонерастворимые (например, этилцеллюлоза, нерастворимая при физиологических значениях pH), растворимые в желудке (например, эудрагит EPO) или растворимые в кишечнике (например, гастрорезистентный фталат гидроксипропилметилцеллюлозы) полимеры, включающие активное вещество и один или более солюбилизатов для достижения высвобождения лекарственного средства при постоянных скоростях, применялись к ядрам таблеток или драже с ограниченным успехом. Была описана разработка фармацевтических композиций активных веществ, водорастворимых при кислом или щелочном значениях pH, используя фармацевтически приемлемые буферные кислоты, соли буферных кислот и их смеси, чтобы обеспечить высвобождение лекарственного средства в основном при постоянных скоростях. Были использованы органические кислоты, чтобы улучшить биодоступность, уменьшить внутрисубъектную изменчивость и минимизировать влияние пищи на слабоосновные фармацевтически активные вещества. Также описаны в литературе лекарственные формы, состоящие из множества частиц, включающие слабоосновные лекарственные средства для обеспечения профилей с пролонгированным высвобождением. Эти лекарственные формы в основном получаются гранулированием или нанесением лекарственного средства с одной или более органическими кислотами и покрытием комбинацией водонерастворимых и водорастворимых или кишечнорастворимых полимеров.
Хотя высвобождение лекарственного средства в этих формах могло быть незначительно пролонгировано, они имели два недостатка, а именно, невозможность соответствовать требуемому профилю плазмы для достижения одноразового суточного режима дозирования и неполное завершение образования формы соли in situ, таким образом, создающее новую химическую субстанцию. Даже когда внутренние ядра, содержащие органическую кислоту, были покрыты полимерной мембраной с замедленным высвобождением, система доставки не смогла продлить высвобождение кислоты для пролонгированного растворения и конечного поглощения активных веществ, чтобы обеспечить требуемые уровни в плазме в течение 24 часов после орального приема пищи. Кроме того, многие слабоосновные лекарственные средства, как известно, образуют соли в присутствии органических кислот, особенно при совместном растворении в растворителях для получения многослойного лекарственного средства или во время гранулирования. Даже в лекарственных формах, в которых органическая кислота и слои лекарственного средства отделяются мембраной замедленного высвобождения (SR), многослойное образование лекарственного средства содержит органическую кислоту. Следовательно, активное вещество в конечной лекарственной форме находится в частично или полностью нейтрализованной форме соли. Это неприемлемое состояние из соображений регулирования. Контролирующие органы могут рассматривать эти активные вещества как новые лекарственные объекты. Таким образом, есть неудовлетворенная потребность в развитии систем доставки лекарственного средства, включающих слабоосновные лекарственные средства с pKa в диапазоне от приблизительно 5 до 14 и требующих больших доз и органических кислот в неизменной форме для высвобождения активных веществ с тем, чтобы поддерживать расчетные концентрации в плазме Cmax и Cmin для того, чтобы быть пригодными для одноразового суточного режима дозирования. После углубленных исследований с удивлением было обнаружено, что эта неудовлетворенная потребность может быть реализована предотвращением контактирования органической кислоты и слабоосновного активного средства друг с другом с образованием соли во время обработки и/или во время хранения лекарственной формы перед введением в in vitro среду растворения или перед оральным введением. Это могло быть достигнуто использованием мембраны, контролирующей скорость SR между кислотным слоем на инертных ядрах и слоем лекарственного средства, используемым на кислотосодержащих ядрах, чтобы изолировать эти два компонента, а также SR с замедленным высвобождением и/или TPR (покрытие с задержкой времени) мембраны на IR с быстрым высвобождением, чтобы синхронизировать высвобождение кислоты с высвобождением лекарственного средства.
Сущность изобретения
Настоящее изобретение обеспечивает фармацевтические композиции и способы для создания систем пульсирующей доставки, которая включает предотвращение того, чтобы слабоосновное азот-(N)-содержащее терапевтическое средство, имеющее pKa в диапазоне от приблизительно 5 до 14 (в основном растворимого при кислых значениях pH, но практически нерастворимого при нейтральном и щелочном значениях pH) и элиминационный период полувыведения от приблизительно 2 часов или дольше, и фармацевтически приемлемая органическая кислота не контактировали друг с другом с образованием соединения с присоединением кислот. Кроме того, лекарственные формы, описанные в настоящем изобретении, обеспечивают расчетные профили высвобождения лекарственного средства солюбилизированием лекарственного средства до его высвобождения в неблагоприятные окружающие условия кишечника, в котором лекарственное средство практически нерастворимо, таким образом, увеличивая вероятность достижения приемлемой концентрации в плазме до 12-24 часов, пост-дозирование для того, чтобы быть пригодным для двухразового или одноразового суточного режима дозирования.
Другой вариант осуществления изобретения касается фармацевтических композиций, состоящих из множества частиц, включающих одну или более покрытых гранул, содержащих одно или более слабых оснований, азот-(N)-содержащие терапевтические средства, имеющие pKa в диапазоне от приблизительно 5 до 14, растворимость не больше, чем приблизительно 200 мкг/мл при pH 6,8 и отношение оптимальной самой высокой дозы к растворимости при pH 6,8, по меньшей мере, приблизительно 100. Например, если режим дозирования для лекарственной формы с быстрым высвобождением (IR) лекарственного средства с растворимостью 0,05 мг/мл при pH 6,8 составляет 5 мг два раза в день, то оптимальная самая высокая доза составляет 10 мг один раз в день, и отношение оптимальной самой высокой дозы (мг) к растворимости (мг/мл) при pH 6,8 было бы 200. Композиция, состоящая из множества частиц, приготовленная в соответствии с одним аспектом настоящего изобретения, будет включать внутренние ядра, содержащие органическую кислоту и покрытые барьерной мембраной (например, SR (замедленное высвобождение)), на которой слабоосновное терапевтическое средство с pKa в диапазоне от приблизительно 5 до 14 слоится и далее покрывается мембраной SR и/или мембраной с задержкой времени, такой, что как органическая кислота, так и слабоосновное терапевтическое средство показывают сравнимые профили высвобождения лекарственного средства.
Композиции, состоящие из множества частичек, приготовленные в соответствии с одним аспектом настоящего изобретения, включают одно или более покрытые объединения гранул, проявляющие подобные сложные профили высвобождения как органической кислоты, так и слабоосновного азот-(N)-содержащего терапевтического средства при исследовании для растворения с использованием устройства 1 Фармакопеи США (барабан, 100 оборотов в минуту) или устройства 2 (мешалки с лопастью, 50 оборотов в минуту) и двухстадийной методики растворения (проверенной в 700 мл 0,1 н. HCl (соляной кислоты) в течение первых 2 часов и затем в 900 мл раствора при pH 6,8, полученного добавлением 200 мл pH-модификатора). Другой вариант осуществления изобретения касается фармацевтической композиции, составленной из множества частиц, включающей одно или более покрытых объединений гранул, показывающих профиль кислотного высвобождения, который является особенно более медленным по сравнению с высвобождением слабоосновного активного средства, чтобы избежать того, чтобы нерастворенное активное средство осталось после на внутренней части покрытых гранул.
Фармацевтическая композиция, состоящая из множества частиц в соответствии с одним аспектом изобретения, включает объединения гранул, покрытые слабоосновным фармацевтическим средством с pKa в диапазоне от приблизительно 5 до 14, включающие:
a) внутреннее ядро частицы, содержащее органическую кислоту (кристалл органической кислоты, шарик или гранула и т.п.);
b) барьер или мембрану замедленного высвобождения на внутреннем ядре частицы, содержащем органическую кислоту, включающем водонерастворимый полимер или комбинацию водонерастворимого полимера с порообразующим водорастворимым или кишечнорастворимым полимером;
с) слабоосновное лекарственное средство, нанесенное слоем на защитное покрытие внутреннего ядра частицы, содержащего кислоту, и необязательно с защитным покрытием изолирующего слоя для образования гранул быстрого высвобождения (IR);
d) при обеспечении гранул с SR, SR покрывающую мембрану на IR гранулах, включающих водонерастворимый полимер или комбинацию водонерастворимого полимера с водорастворимым полимером, образующим SR гранулу; и/или
e) при обеспечении гранул контролируемого по времени, пульсирующего высвобождения (TPR), покрывающую мембрану с задержкой времени на грануле с поверхностью SR или непосредственно на грануле IR, включающей комбинацию водонерастворимых и кишечнорастворимых полимеров с образованием TPR гранулы.
Композиции в соответствии с определенными аспектами изобретения в основном проявляют желаемые или расчетные профили высвобождения как активного вещества, так и органической кислоты после предопределенной задержки времени, по меньшей мере, 2 часов при исследовании высвобождения лекарственного средства и/или органической кислоты с использованием двухстадийной методики растворения, описанной выше.
Фармацевтическая композиция слабоосновного азот-(N)-содержащего терапевтического средства, имеющего pKa в диапазоне от приблизительно 5 до 14, растворимость не больше, чем приблизительно 200 мкг/мл при pH 6,8 и отношение оптимальной самой высокой дозы к растворимости при pH 6,8 не меньше, чем приблизительно 100, может быть приготовлена заполнением соответствующими гранулами твердой желатиновой капсулы или прессованием в обычную таблетку или в ОРТ (орально распадающуюся таблетку) форму в соответствии с некоторыми вариантами осуществления настоящего изобретения.
Фармацевтическая композиция слабоосновного терапевтического средства в форме ОРТ, приготовленной в соответствии с другим вариантом осуществления настоящего изобретения, распадается при контакте со слюной в полости рта в пределах приблизительно 60 секунд с образованием однородной, легкой для проглатывания суспензии (без привкуса песка и мела). Фармацевтическая композиция слабоосновного фармацевтического активного вещества в форме ОРТ, которая может включать одно или более покрытых объединений гранул со средним размером частицы не больше, чем приблизительно 400 мкм, такие как микрокапсулы с исправленным вкусом, включающие внутренние ядра, содержащие лекарственное средство (кристаллы, гранулы, драже, шарики и т.п.), гранулы с SR и регулируемым по времени, объединения гранул с пульсирующим высвобождением (TPR, TПВ), включающие SR покрытые ядра, содержащие кислоту. Исправление вкуса может быть достигнуто любым способом, известным из предшествующего уровня техники. ОРТ может также включать быстро диспергирующие микрогранулы со средним размером частицы не больше, чем приблизительно 400 мкм, или в некоторых вариантах осуществления не больше, чем приблизительно 300 мкм, включающие дезинтегрирующее вещество (например, кросповидон, кросс-связанный поливинилпирролидон) и сахарный спирт (например, маннит), сахарид (например, лактоза) или их комбинация, причем каждый имеет средний размер частицы не больше, чем приблизительно 30 мкм, и, произвольно, фармацевтически приемлемые наполнители, в основном используемые в ОРТ-образованиях, а именно, ароматические добавки, подсластители, окрашивающие средства и дополнительные дезинтегрирующие вещества.
ПРТ в соответствии с одним вариантом осуществления обладает следующими свойствами:
1) распадается при контакте со слюной в полости рта в течение приблизительно 60 секунд с образованием однородной, легкой для проглатывания суспензии, включающей покрытые частицы (SR и/или TPR гранулы) с замаскированным (направленным) вкусом и/или
2) частицы с замаскированным вкусом, если имеются, обеспечивают быстрым, существенно полным высвобождением дозы после попадания в желудок (например, в основном больше, чем приблизительно 75% в течение приблизительно 60 минут);
3) покрытые частицы (SR и/или TPR гранулы) обеспечивают пролонгированное высвобождение активного вещества для непрерывного поглощения по ЖК тракту.
ПРТ в соответствии с одним вариантом осуществления, включающим микрочастицы с замаскированным вкусом, демонстрирующие эффективную маскировку вкуса высвобождением не больше, чем 10% в течение приблизительно 3 минут (самое длительное время нахождения, ожидаемое для ОРТ в ротовой полости) при исследовании растворения в моделируемой жидкости слюны (pH ~6,8) при высвобождении не меньше, чем приблизительно 50% дозы в течение приблизительно 30 минут при исследовании растворения в 0,1н HCl.
В соответствии с некоторыми вариантами осуществления быстро рассеивающиеся микрогранулы и покрытые гранулы (IR, SR и/или TPR гранулы с замаскированным вкусом) одного или более слабоосновных активных веществ могут быть представлены в массовом отношении от приблизительно 6:1 до 1:1, а конкретнее от приблизительно 4:1 до 2:1, чтобы достичь однородных вкусовых ощущений. В соответствии с некоторыми другими вариантами осуществления покрытые гранулы (с замаскированным вкусом IR, SR и/или TPR гранулы) одного или более слабоосновных активных веществ могут быть покрыты спрессованным покрытием (например, покрытие с псевдоожиженным слоем с пластифицированной водной дисперсией этилцеллюлозы), чтобы минимизировать мембранное разрушение во время прессования с быстро рассеивающимися микрогранулами.
Фармацевтическая композиция слабоосновного фармацевтически активного вещества в форме обычной таблетки в соответствии с другим вариантом осуществления настоящего изобретения может включать одно или более объединений гранул, таких как IR гранулы (кристаллы, гранулы, драже, шарики и т.п.), и гранулы SR и/или TPR гранулы, включающие SR покрытые ядра, содержащие кислоту. Фармацевтическая композиция слабоосновного фармацевтического активного вещества в форме обычной таблетки распадается на составные гранулы (частицы с замаскированным вкусом, SR покрытые гранулы и/или TPR гранулы) при оральном приеме приблизительно за 10 минут. Обычная таблетка может также включать фармацевтически приемлемые наполнители, в основном используемые в распадающихся формах таблетки, таких как спрессованные разбавители, наполнители, окрашивающие средства и, необязательно, смазывающее вещество.
Обычная таблетка, приготовленная в соответствии с одним вариантом осуществления, показывает следующие свойства:
1) распадается при оральном приеме приблизительно за 10 минут на IR частицы и/или покрытые частицы (SR и/или TPR гранулы);
2) IR частицы, если имеются, обеспечивают быстрое, существенно полное высвобождение (например, большее чем приблизительно 95%) дозы в пределах приблизительно 60 минут, особенно большое в пределах приблизительно 30 минут после попадания в желудок;
3) SR и/или TPR гранулы обеспечивают пролонгированное высвобождение активного вещества для непрерывного поглощения по желудочно-кишечному тракту.
Другой вариант осуществления изобретения относится фармацевтической композиции, состоящей из множества частиц, включающих одно или более покрытых объединений гранул, включающих одно или более слабоосновных терапевтических средств, имеющих период полувыведения приблизительно 2 часа или дольше, в которых активное вещество наносится на SR покрытые ядра, содержащие кислоту. Пульсирующая система доставки, разработанная в соответствии с этим аспектом настоящего изобретения, может включать IR гранулу, SR гранулу и объединения гранул с регулируемым по времени, пульсирующим высвобождением (TPR). SR покрытые внутренние ядра, содержащие органическую кислоту, в основном приготавливаются нанесением слоя органической кислоты (например, фумаровой кислоты) на инертные частицы (например, сахарные сферы) из полимерного связующего раствора и покрываются водонерастворимым полимером (например, этилцеллюлозой с вязкостью приблизительно 10 cps), одним или в комбинации с водорастворимым полимером (например, поливинилпирролидоном, повидоном K-25 или полиэтиленгликолем, ПЭГ-400) или кишечнорастворимым полимером (например, фталатом гидропропилметилцеллюлозы, HPMCP или HP-55). IR объединения гранул, включающие SR покрытые внутренние ядра, содержащие кислоту, приготавливаются нанесением слоя лекарственного средства на SR покрытые ядра, содержащие кислоту, из полимерного связующего раствора и обеспечивающие защитный изолирующий слой Opadry Clear или Pharmacoat™ 603. SR и TPR объединения гранул приготавливаются покрытием IR гранул водонерастворимым полимером (например, этилцеллюлозой), одним или в комбинации с водорастворимым полимером (например, PVP K-25 или ПЭГ-400) или кишечнорастворимым полимером (например, фталатом гидропропилметилцеллюлозы, HPMCP или HP-55). IR объединения гранул, включающие SR покрытые внутренние ядра, содержащие кислоту, приготавливаются нанесением слоя лекарственного средства на SR покрытые ядра, содержащие кислоту, из полимерного связующего раствора и обеспечивающие защитный изолирующий слой Opadry Clear. SR и TPR объединения гранул приготавливаются покрытием IR гранул с водонерастворимым полимером (например, этилцеллюлозой), одним или в комбинации с водорастворимым полимером (например, PVP K-25 или ПЭГ-400). В соответствии с одним аспектом изобретения каждое SR или TPR объединение гранул высвобождает как лекарственное средство, так и кислоту при сопоставимых скоростях, в качестве профилей быстрого высвобождения или замедленного высвобождения после предопределенного времени задержки (например, задержка времени до 10 часов) при оральном введении. IR гранулы, если включены в лекарственную форму (капсула или обычная таблетка или орально распадающаяся таблетка), могут включать лекарственное средство, наслоенное непосредственно на инертные ядра и покрытые защитным изолирующим слоем или мембраной исправления вкуса, которая является частью полной дозы, обеспечивает быстрое поглощение (болюсная доза) при оральном введении.
Также обеспечивается способ производства фармацевтической композиции, состоящей из множества частиц, в которой система доставки, разработанная в соответствии с некоторыми вариантами осуществления настоящего изобретения, включает одно или более активных слабоосновных фармацевтических ингредиентов в достаточных количествах, которые нужно орально ввести пациенту при предписанном двухразовом или одноразовом суточном режиме дозирования для обеспечения терапевтической эффективности.
Способ приготовления фармацевтической композиции, состоящей из множества частиц, в соответствии с определенными вариантами осуществления включает наслоение фармацевтически приемлемой органической кислоты, такой как фумаровая кислота, из полимерного связующего раствора на инертные частицы, отобранные из группы, состоящей из сахарных сфер и сфер целлюлозы. Псевдоожиженный слой или смазывание форм могут использоваться для нанесения органической кислоты и полимерного связующего раствора. В соответствии с другими вариантами осуществления частицами ядра могут быть кристаллы с желаемым распределением размера частицы, микрогранулы, драже или гранулы, содержащие одну или более органическую кислоту(ы). В соответствии с некоторыми вариантами осуществления микрогранулы, экструдированные-сферонизированные драже или спрессованные микротаблетки, включающие одну или более органических кислот, полимерное связывающее вещество, которое придает эластичные характеристики высушенным микрогранулам, гидрофильные наполнители/разбавители, и произвольные ароматические добавки, подсластители и/или дезинтегрирующие вещества. Эти частицы, содержащие органическую кислоту, являются защитной полимерной мембраной с покрытием SR (замедленного высвобождения), включающей водонерастворимый полимер (например, этилцеллюлозу со средней вязкостью 10 cps), один или в комбинации с водорастворимым полимером (например, поливинилпирролидон или полиэтиленгликоль) или кишечнорастворимый полимер (например, фталат гидроксипропилметилцеллюлозы (HPMCP или HP-55)). Водонерастворимые и водорастворимые или кишечнорастворимые полимеры могут присутствовать при массовом соотношении от приблизительно 95:5 до приблизительно 50:50, а конкретнее от приблизительно 90:10 до 60:40, а мембранная толщина может изменяться от приблизительно 3% до 50%, а конкретнее от приблизительно 5% до 30% по массе в соответствии с определенными вариантами осуществления.
В соответствии с определенными вариантами осуществления одно или более слабоосновное лекарственное средство (лекарственные средства) наносится на частицы защитного покрытия, содержащие кислоту, из полимерного связующего раствора, а также наносится на гранулы, защитный изолирующий слой с гидрофильным полимером (например, Pharmacoat™ 603 или Opadry® Clear) для получения IR гранул. Загрузка органической кислоты или лекарственного средства зависит от физико-химических, а также фармакологических свойств слабоосновных активных веществ, выбранных для разработки, а лекарственные средства и органическая кислота могут присутствовать при массовом соотношении от приблизительно 5:1 до 1:10, или конкретнее от приблизительно 3:1 до 1:3 в зависимости от того, используются ли кристаллы органической кислоты или ядра, содержащие органическую кислоту в соответствии с некоторыми вариантами осуществления.
В соответствии с некоторыми вариантами осуществления настоящего изобретения IR гранулы, включающие внутренние ядра с защитным покрытием, содержащие кислоту, покрытые полимерной мембраной с покрытием SR, включающей водонерастворимый полимер (например, этилцеллюлозу со средней вязкостью 10 cps), один или в комбинации с водорастворимым полимером (например, поливинилпирролидон или полиэтиленгликоль). Водонерастворимые и водорастворимые полимеры могут присутствовать при массовом соотношении от приблизительно 95:5 до приблизительно 50:50, а конкретнее от приблизительно 90:10 до 60:40, а мембранная толщина может изменяться от приблизительно 3% до 50%, а конкретнее от приблизительно 5% до 30% по массе в соответствии с определенными вариантами осуществления.
В соответствии с другими вариантами осуществления настоящего изобретения гранулы SR, включающие гранулы, покрытые слоем лекарственного средства, покрываются мембраной со временем задержки, включающей комбинацию водонерастворимого полимера (например, этилцеллюлозы со средней вязкостью 10 cps) и кишечнорастворимого полимера (например, фталат гидроксипропилметилцеллюлозы (HPMCP или HP-55)) для получения TPR гранул. В соответствии с некоторыми другими вариантами осуществления, водонерастворимые и кишечнорастворимые полимеры могут присутствовать при массовом соотношении от приблизительно 9:1 до приблизительно 1:4, а конкретнее от приблизительно 3:1 до 1:1, а мембранная толщина может изменяться от приблизительно 5% до 60%, а конкретнее от приблизительно 15% до 50% по массе в соответствии с определенными вариантами осуществления.
Функциональные полимерные системы, применяемые из водных или основанных на растворителе композиций, в основном содержат пластификаторы при подходящих концентрациях. Конечная лекарственная форма может быть капсулой с модифицированным высвобождением (МВ), стандартной (обычной) таблеткой или орально распадающейся таблеткой (ПРТ), включающей покрытые сферические объединения гранул, содержащие активное вещество в чистом виде или комбинацию двух или более покрытых объединений гранул для обеспечения расчетных концентраций в плазме, подходящих для одноразового суточного режима дозирования. Например, одноразовая суточная лекарственная форма активного вещества с периодом полувыведения приблизительно 7 часов может содержать смесь IR объединения гранул, которое обеспечивает быстрое высвобождение, в течение секунды, TPR объединение гранул с более коротким временем задержки (приблизительно 3-4 часа), которое обеспечивает запаздывающее быстрое высвобождение и третье TPR объединение гранул с более длительным временем задержки (приблизительно 7-8 часов), которое обеспечивает в основном запаздывающий профиль замедленного высвобождения свыше приблизительно 8-12 часов, чтобы поддерживать приемлемые концентрации в плазме в течение 12-24 часов, таким образом, увеличивая безопасность, терапевтическую эффективность и соблюдение больным режима и схемы лечения при сокращении стоимости лечения. Как вариант, конечная лекарственная форма может включать IR объединения гранул и, второе, TPR объединения гранул со временем задержки приблизительно 7-8 часов с последующим профилем замедленного высвобождения свыше 10-12 часов. Достигаемая задержка времени зависит от композиции и толщины барьерного покрытия, а также композиции и толщины покрытия с задержкой времени. Определенные факторы, которые могут влиять на достижение оптимальной двухразовой или одноразовой суточных лекарственных форм, включают, но не ограничиваются, pKa терапевтического средства (и его растворимостью выше pH 6,0), элиминационный период полувыведения и повышения растворимости в водном растворе органической кислоты, отобранной из группы, состоящей из аспарагиновой кислоты, лимонной кислоты, фумаровой кислоты, малеиновой кислоты, щавелевой кислоты, янтарной кислоты, винной кислоты и т.п.
В соответствии с некоторыми вариантами осуществления настоящего изобретения также обеспечивается способ производства композиции, состоящей из множества частиц, включающей слабое, азот-(N)-содержащее терапевтическое средство, имеющее pKa в диапазоне от приблизительно 5 до 14 и растворимость не больше, чем 200 мкг/мл при pH 6,8. Метод может включать стадии:
а) приготовление частиц ядра (кристаллы с распределением размера частиц 20-500 мкм, конкретнее 100-300 мкм, гранулы или драже) с одной или более фармацевтически приемлемыми органическими кислотами;
b) покрытие этих внутренних слоев, содержащих кислоту, водонерастворимым полимером или комбинацией водонерастворимого полимера с водорастворимым или кишечнорастворимым полимером для того, чтобы запрограммировать высвобождение кислоты для увеличения массы от приблизительно 3% до 50%;
с) вышеупомянутое наслоение слабоосновного азот-(N)-содержащего терапевтического средства из полимерного связующего раствора и применение защитного изолирующего слоя на гранулы, покрытые слоем лекарственного средства для получения IR гранул;
d) использование защитного покрытия (с замедленным высвобождением) водонерастворимого полимера или комбинации водонерастворимого и водорастворимого полимера для увеличения массы от приблизительно 3% до 30% для получения SR гранул;
e) использование покрытия с задержкой времени (запаздыванием во времени) из комбинации водонерастворимых и кишечнорастворимых полимеров при массовом соотношении от приблизительно 10:1 до 1:4 для увеличения массы от приблизительно 10% до 60% по массе покрытой гранулы для получения TPR гранул; и
f) заполнение в твердые желатиновые капсулы или прессование в обычные таблетки/орально распадающиеся таблетки (ПРТ) после смешивания с фармацевтически приемлемыми наполнителями и одним или более объединениями гранул (например, комбинация из IR гранул, SR гранул и/или TPR гранул при желаемом соотношении).
Композиция, включающая одно или более объединений гранул (например, комбинация IR и TPR объединений гранул) может проявлять следующие свойства:
а) композиция распадается при контакте со слюной в полости рта, образуя однородную, легкую для проглатывания суспензию (если она в форме ОРТ) или распадается в течение приблизительно 10 минут при оральном приеме (если она в форме капсулы или обычной таблетки);
b) IR гранулы, с исправленным вкусом или нет, быстро высвобождают дозу после приема в желудок (например, в основном больше, чем приблизительно 50%, конкретнее больше, чем приблизительно 75%, в течение приблизительно 60 минут);
с) SR или TPR гранулы высвобождают лекарственное средство в течение от приблизительно 4 до 20 часов в синхронизации с высвобождением органической кислоты после предопределенного запаздывания (например, приблизительно до 10 часов) после орального приема;
d) сложный профиль высвобождения лекарственного средства композиции подобен расчетному высвобождению лекарственного средства in vitro/in vivo профилю концентрации в плазме для того, чтобы быть подходящим для двухразового или одноразового суточного режима дозирования.
Эти и другие варианты осуществления, преимущества и особенности настоящего изобретения стали очевидными при детальных описаниях, а примеры представлены в последующих разделах.
Краткое описание чертежей
Фиг.1 иллюстрирует профили рН-растворимости для (а) гидрохлорида ондансетрона, (b) карведилола, (с) дипиридамола, и (d) клоназепама;
Фиг.2 иллюстрирует поперечный срез SR покрытого ядра, содержащего органическую кислоту, в соответствии с одним аспектом изобретения;
Фиг.3 иллюстрирует поперечный срез TPR гранулы, включающей SR покрытое ядро, содержащее органическую кислоту, в соответствии с определенным аспектом изобретения;
Фиг.4 иллюстрирует высвобождение фумаровой кислоты из SR-покрытых кислотных кристаллов, покрытых при различных соотношениях ЕС-10/ПЭГ примера 1;
Фиг.5 иллюстрирует профили высвобождения дипиридамола из TPR гранул примера 2Е;
Фиг.6 иллюстрирует профили высвобождения TPR гранул карведилола примера 5 по отношению со сравнительным примером 7В;
Фиг.7 иллюстрирует профили зависимости высвобождения гидрохлорида ондансетрона из TPR гранул от стабильности (пример 6);
Фиг.8 иллюстрирует профили высвобождения гидрохлорида ондансетрона из TPR гранул, покрытых при различных соотношениях ЕС-10/НР-55/ТЕС при 50% по массе примера 8; и
Фиг.9 иллюстрирует профили зависимости концентрации в плазме от времени исследуемых образований гидрохлорида ондансетрона (ежедневно - QD) и 8 мг зофрана (2 раза в сутки - BID) примера 9.
Подробное описание изобретения
Все цитируемые документы, в соответствующих местах, включаются в настоящее описание ссылкой; цитирование любого документа не должно быть рассмотрено как признание того, что оно предшествует настоящему изобретению.
Термин "слабоосновное фармацевтически активное вещество", использованный в настоящем изобретении, а также и в определенных примерах настоящего изобретения, включает основание, фармацевтически приемлемые соли, полиморфы, их стереоизомеры и смеси. Этот термин, который полностью определен в последующем разделе, относится к азот-N)-содержащему терапевтическому средству, имеющему pKa в диапазоне от приблизительно 5 до 14, а конкретнее средству, имеющему растворимость не больше, чем 200 мкг/мл при pH 6,8.
Термин "быстрое высвобождение", использованный в настоящем изобретении, относится к высвобождению, большему или равному, чем приблизительно 50%, особенно при исправленном вкусе для объединения в орально распадающуюся лекарственную форму таблетки, предпочтительно большему, чем приблизительно 75%, более предпочтительно большему, чем приблизительно 90%, и в соответствии с некоторыми вариантами осуществления большему, чем приблизительно 95% активного вещества в течение приблизительно 2 часов, а конкретнее в течение приблизительно одного часа после приема лекарственной формы. Термин может также относиться к высвобождению активного вещества из лекарственной формы с регулируемым по времени пульсирующим высвобождением, отличающимся быстрым импульсом высвобождения после рассчитанного времени задержки. Термин "время задержки" относится к периоду времени, в котором меньше, чем приблизительно 10%, а конкретнее в основном ни одна из доз (лекарственного средства) не высвобождается, а время задержки, по меньшей мере, от приблизительно 2 до 10 часов достигается покрытием в основном комбинацией водонерастворимых и кишечнорастворимых полимеров (например, этилцеллюлозой и фталатом гидроксипропилметилцеллюлозы).
Если не указано иначе, все проценты и соотношения рассчитаны по массе на основе общей композиции.
Водная или фармацевтически приемлемая среда растворителя может использоваться для приготовления частиц ядра, содержащих органическую кислоту, для нанесения лекарственного средства, а именно, гранул, содержащих кислоту, нанесением кислоты на инертные ядра (например, сахарные сферы) или IR гранул нанесением лекарственного средства на ядра, содержащие кислоту, или непосредственно на сахарные сферы из соответствующего раствора полимерного связующего в аппаратуре с псевдоожиженным слоем. Также, водная дисперсия функциональных полимеров, которые являются доступными в качестве дисперсии или системы растворителей, может использоваться для растворения функциональных полимеров для покрытия гранул, содержащих кислоту, IR гранул или SR гранул.
Многие активные фармацевтические ингредиенты (АФИ) являются слабоосновными в том смысле, что эти активные вещества без ограничений умеренно растворимы при кислых pH, но практически малорастворимы при нейтральных и щелочных значениях pH. Их pKa величины находятся в диапазоне от приблизительно 5 до 14. Данные растворимости в зависимости от pH для в основном слабоосновных активных веществ представлены на фиг.1. Например, растворимость дипиридамола в 0,1 н. HCl (соляная кислота) составляет приблизительно 1 мг/мл, в то время как при pH 6,8 растворимость составляет только 30 мкг/мл. Хотя растворимость карведилола подобным образом зависит от pH и изменяется, это не очевидно из фиг.1, поскольку он быстро подвергается in situ образованию соли с буферным веществом, таким как лимонная, уксусная и соляная кислоты, и, следовательно, наблюдаемая растворимость представляет собой растворимость соли, образованной in situ.
В таблице 1 приводится повышение растворимости слабоосновных активных веществ в органических кислотных буферах. Могут быть определены три различные группы. Активные вещества группы А, как представлено гидрохлоридом ондансетрона, показывают резкое увеличение в растворимости слабоосновного активного вещества в буфере со следовым количеством фумаровой кислоты. Например, растворимость ондансетрона приблизительно 26 мг/мл в буфере, содержащем только 0,05 мг/мл фумаровой кислоты, остается неизменной после увеличения концентрации фумаровой кислоты в буфере до 5 мг/мл. В группе B, представленной дипиридамолом, карведилолом и ламотригином, растворимость слабоосновного лекарственного средства увеличивается с увеличением концентрации кислоты. В группе C, представленной клоназепамом, органическая кислота очень ограничила воздействие, то есть повышение растворимости становится в основном меньшим, чем 3-кратное. Например, растворимость клоназепама составляет приблизительно 11,6 и 6,9 мкг/мл в буферных растворах при pH 2,3 и 6,8, содержащих более высокую и более низкую концентрацию фумаровой кислоты соответственно.
Определенные варианты осуществления изобретения будут далее описаны детально по отношению к сопровождающим фиг. 2 и 3. На фиг.2, SR-покрытое ядро 10, включающее SR-покрытие 12, используемое на ядре, содержащем органическую кислоту, включающем слой фармацевтически приемлемой органической кислоты в связывающем веществе 14 на инертном ядре 16 частицы. Инертное ядро 16 частицы, слой 14, покрывающий органическую кислоту, и SR-слой 12, контролирующий скорость растворения, образуют SR-покрытое ядро 10, содержащий органическую кислоту. Фиг.3 иллюстрирует TPR гранулу. TPR гранула 20 включает покрытие 22 с задержкой времени, применяемой на первичном SR-слое 24, защитный изолирующий слой 26 и слой 28 слабоосновного лекарственного средства, используемого на SR-покрытом ядре 10, содержащем кислоту. Слабоосновное лекарственное средство в основном используется из полимерного связующего раствора. Покрытие SR поддерживает высвобождение лекарственного средства, в то время как покрытие с задержкой времени обеспечивает время задержки (период времени, показывающий меньше, чем приблизительно 10%, а конкретнее в основном, ни одной высвобожденной дозы). Таким образом, покрытие 22 с задержкой времени, внешнее SR покрытие на IR гранулах 24, и внутреннее SR покрытие 12 на ядре, содержащем кислоту, вместе контролируют свойства высвобождения как лекарственного средства, так и кислоты из TPR гранул.
в органических кислотах
Новизна/полезность препаративных форм, разработанных в соответствии с некоторыми вариантами осуществления настоящего изобретения, раскрыта с использованием гидрохлорида ондансетрона, ламотригина, дипиридамола и карведилола в качестве примеров слабоосновных азот-(N)-содержащих терапевтических средств, имеющих pKa в диапазоне от приблизительно 5 до 14 и растворимости не больше, чем приблизительно 200 мкг/мл при pH 6,8. Гидрохлорид ондансетрона, отобранное блокирующее средство серотонинового 5-HT3 рецептора, является средством от тошноты и противорвотным средством. Он хорошо растворим при кислых значениях pH, в то время как практически нерастворим при pH 6,8. Дипиридамол, антитромбоцитарное средство, является химическим производным дипиперидина. Он растворим в разбавленных кислотах и практически нерастворим в воде, нейтральных и щелочных буферных растворах.
Карведилол, бета-блокатор с дополнительными сосудорасширяющими, антипролиферативными свойствами, предназначен для лечения высокого давления (ВД), болезни коронарной артерии и ишемической болезни сердца. Современное коммерческое образование карведилола быстро высвобождается и вводится два раза в сутки. Непосредственная лекарственная форма быстро и экстенсивно поглощается при оральном введении, с предельным элиминационным периодом полувыведения между 7 и 10 часами. Одноразовая суточная дозировка образования карведилола коммерчески желательна и упростила бы режим дозирования и увеличила соблюдение больным режима и схемы лечения. Карведилол существует в виде рацемата и содержит α-гидроксилвторичный амин, с pKa 7,8. Он проявляет предсказуемую водную растворимость, то есть выше pH 9, растворимость будет <1 мкг/мл и его растворимость увеличивается с уменьшением pH и достигает стабильного значения рН, близкого к pH 5; его растворимость составляет приблизительно 23 мкг/мл при pH 7 и приблизительно 100 мкг/мл при pH 5. При более низких значениях pH (pH от 1 до 4), растворимость ограничена растворимостью протонированной формы карведилола или формой его соли, образованной in situ. Форма соли соляной кислоты менее растворима, чем протонированная форма сама по себе. Карведилол поглощается из ЖК тракта межклеточным транспортом. In vivo поглощение уменьшилось в пределах кишечника в следующем порядке: тонкая кишка > подвздошная кишка > толстая кишка. Самое высокое поглощение было достигнуто в тонкой кишке при нейтральном pH. Так как растворение лекарственного средства является скорость-ограничивающим фактором для поглощения карведилола в дистальной части ЖК тракта теоретически из-за уменьшения в растворимости, то одноразовая суточная лекарственная форма в соответствии с одним вариантом осуществления включила бы, по меньшей мере, два объединения гранул - одно IR объединение гранул и другое TPR объединение гранул, включающее SR ядра, покрытые органической кислотой. Илоперидон представляет собой антипсихотическое средство и ламотригин, противосудорожное лекарственное средство, предназначено для лечения эпилепсии.
В соответствии с некоторыми вариантами осуществления настоящего изобретения используется растворимость, увеличивающая качество буферных растворов органической кислоты, и в то же самое время, in situ образование кислотных дополнительных соединений предотвращается наличием мембраны SR покрытия между внутренним слоем органической кислоты и слоем слабоосновного лекарственного средства. Мембрана SR покрытия, нанесенная таким образом, точно контролирует высвобождение органической кислоты таким образом, чтобы никакое лекарственное средство, не осталось в лекарственной форме, чтобы обеспечить отсутствие солюбилизирующей кислоты в TPR грануле. В одном варианте осуществления ядро активного вещества лекарственной формы настоящего изобретения может включать инертную частицу, покрытую органической кислотой, SR покрытие, наслоенное лекарственное средство (IR гранулы), далее барьер, или SR покрытый, и/или покрытый с задержкой времени. Количество органической кислоты и загрузки лекарственного средства в ядре будет зависеть от лекарственного средства, дозы, ее растворимости, зависимой от рH, повышения растворимости и элиминационного периода полувыведения. Специалисты в этой области техники будут способны выбрать соответствующее количество лекарственного средства/кислоты для покрытия ядра для достижения желаемого BID (два раза в сутки) или QD (один раз в сутки) режима дозирования. В одном варианте осуществления инертная частица может быть сахарной сферой, сферой целлюлозы, сферой диоксида кремния или тому подобное. Как вариант, кристаллы органической кислоты с желаемым распределением размера частицы могут функционировать как ядра, особенно для лекарственных средств Группы C, и в этом случае эти кристаллы покрываются мембраной для программирования кислотного высвобождения, которое, в соответствии с некоторыми вариантами осуществления, синхронизируется с высвобождением лекарственного средства, чтобы гарантировать полное высвобождение лекарственного средства до того, как закончится кислота.
В соответствии с одним аспектом настоящего изобретения ядро лекарственной формы может включать кристалл органической кислоты (например, фумаровой кислоты) с желаемым средним размером частицы или инертной частицей, такой как сахарная сфера, слоеная с органической кислотой из полимерного связующего раствора. Кристаллы органической кислоты или ядра, содержащие кислоту, покрываются водонерастворимым полимером, одним или в комбинации с водорастворимым или кишечнорастворимым полимером, а композиция и толщина SR мембраны оптимизируется таким образом, что высвобождение кислоты происходит медленнее или синхронизируется с растворением/высвобождением лекарственного средства из гранулы, таким образом, гарантируя то, что высвобождение кислоты не завершится до окончания высвобождения лекарственного средства. В некоторых аспектах изобретения ядра, содержащие кислоту, могут быть в форме микрогранул или драже, которые могут быть приготовлены ротогранулированием, гранулированием с высоким сдвигом и экструзией-сферонизацией или прессованием (как микротаблетки приблизительно 1-1,5 мм в диаметре) органической кислоты, полимерного связывающего вещества и произвольных наполнителей/разбавителей.
Слабоосновное активное средство, такое как карведилол, наслаивается на SR покрытые гранулы, содержащие фумаровую кислоту, из полимерного связующего раствора (например, провидона) и защитного изолирующего слоя, включающего гидрофильный полимер, такой как Opadry® Сlear или Pharmacoat 603 (гидропропилметилцеллюлоза 2910; 3 cps) для образования IR гранул. В одном варианте осуществления, IR гранулы, содержащие лекарственное средство, могут быть покрыты дважды - внутренний барьер, покрывающий мембрану с водонерастворимым полимером (например, этилцеллюлозой), одним или в комбинации с водорастворимым полимером, и мембрана с покрытием задержки времени водонерастворимого полимера в комбинации с кишечнорастворимым полимером для получения TPR гранул с задержкой времени (высвобождение с запаздывающим началом) от приблизительно 1 до 10 часов при оральном введении. Водонерастворимый полимер и кишечнорастворимый полимер могут присутствовать при массовом соотношении от приблизительно 9:1 до приблизительно 1:4, предпочтительно при массовом соотношении от приблизительно 2:1 до 1:1. Мембранное покрытие в основном включает от приблизительно 5% до приблизительно 60%, предпочтительно от приблизительно 10% до приблизительно 50% по массе покрытых гранул. В соответствии с уже другим вариантом осуществления, IR гранулы могут просто покрываться комбинацией водонерастворимого полимера и кишечнорастворимого полимера в вышеупомянутых количествах.
Единичная капсула или обычная лекарственная форма таблетки согласно настоящему изобретению могут включать TPR гранулы, одну или в комбинации с IR гранулами, в то время как единичная ОРТ может включать TPR гранулы, одну или в комбинации с гранулами быстрого высвобождения (IR) с исправленным вкусом. IR гранулы без наличия мембраны исправленного вкуса будут обеспечивать быстрое высвобождение слабоосновного лекарственного средства в гастроинтеральном тракте в течение приблизительно 60 минут, предпочтительно в течение 30 минут после орального приема. Если они с исправленным вкусом, то эти гранулы проявляют исправление вкуса в полости рта и в основном завершает высвобождение слабоосновного лекарственного средства в гастроинтеральном тракте в течение приблизительно 2 часов, предпочтительно в течение одного часа после орального приема. TPR гранулы будут высвобождать слабоосновное лекарственное средство в течение периода до приблизительно 4-20 часов в гастроинтеральном тракте после времени задержки приблизительно 1-10 часов после орального приема.
Настоящее изобретение также обеспечивает способ для получения фармацевтической лекарственной формы, состоящей из множества частиц, имеющей одно или более объединения гранул с регулируемым по времени, пульсирующим высвобождением одного или более слабоосновных активных средств, включающих SR покрытые ядра, содержащие органическую кислоту, то есть хорошо регулируемые во времени серии импульсов так, что активные средства и кислота, депонируемая в хорошо отделенные/изолированные слои, не контактируют друг с другом для образования дополнительных кислотных композиций до тех пор, пока лекарственная форма не проконтактирует со средой растворения или жидкостями организма после орального приема. Полученная таким образом лекарственная форма показывает сложные профили высвобождения активного средства и кислоты, которые сопоставимы, а конкретнее, профиль высвобождения кислоты медленнее, чем профиль высвобождения лекарственного средства так, чтобы никакое нерастворенное лекарственное средство не осталось после в лекарственной форме, чтобы обеспечить отсутствие солюбилизирующей органической кислоты.
В соответствии с одним вариантом осуществления настоящего изобретения способ может включать стадии:
a) обеспечение ядра частицы, содержащего органическую кислоту (т.е. кристаллическая органическая кислота с желаемым распределением размера частицы или частица, включающая инертную частицу (например, сахарная сфера, сфера целлюлозы, сфера диоксида кремния), покрытую органической кислотой из полимерного связующего раствора);
b) покрытие ядра частицы, содержащего органическую кислоту, SR покрывающей мембраной, состоящей из водонерастворимого полимера, такого как EC-10 (этилцеллюлоза со средней вязкостью 10 cps), одного или в комбинации с водорастворимым полимером (например, повидоном или ПЭГ 400) или кишечнорастворимым полимером, таким как фталат гидроксипропилметилцеллюлозы (например, HP-55);
c) использование слоя слабоосновного лекарственного средства на SR покрытое ядро частицы, содержащей органическую кислоту, для образования IR гранулы;
d) нанесение барьера, покрывающего мембрану, на IR гранулу с раствором водонерастворимого полимера, одного или в комбинации с водорастворимым полимером;
e) использование мембраны с покрытием с задержкой времени на SR гранулу с раствором водонерастворимого полимера в комбинации с кишечнорастворимым при массовом соотношении от приблизительно 9:1 до 1:4 для образования лекарственной частицы с регулируемым по времени пульсирующим высвобождением (TPR гранула).
В соответствии с некоторыми вариантами осуществления настоящего изобретения способ может включать стадии:
i. IR гранулы, исправляющие вкус накапливанием растворителя с водонерастворимым полимером (например, этилцеллюлозой со средней вязкостью 100 cps), одним или в комбинации с растворимым в желудке порообразующим веществом (например, карбонатом кальция) в соответствии с описанием заявки на патент США № 11/213266, находящейся на стадии рассмотрения, поданной 26 августа 2005 г. (публикация США 2006/0105038 от 18 мая 2006 г.) или покрытием псевдоожиженным слоем с водонерастворимым полимером (например, этилцеллюлозой со средней вязкостью 10 cps), одним или в комбинации с растворимым в желудке полимером (например, эудрагит E100 или EPO) в соответствии с описанием заявки на патент США № 11/248596, находящейся на стадии рассмотрения, поданной 12 октября 2005 г. (публикация США № 2006/0078614 от 13 апреля 2006 г.) или растворимым в желудке порообразующим веществом (например, карбонатом кальция) в соответствии с описанием заявки на патент № США 11/256653, находящейся на стадии рассмотрения, поданной 21 октября 2005 г. (публикация США 2006/0105039 от 18 мая 2006 г.), содержание заявок, изложенных в этом абзаце, включены в настоящее изобретение ссылкой;
ii. Гранулирование порошковой смеси сахарного спирта, такого как маннитол, или сахарида, такого как лактоза и кросповидон, например, с использованием описания заявки на патент США № 10/827106, находящейся на стадии рассмотрения, поданной 19 апреля 2004 г. (публикация США № 2005/0232988 от 20 октября 2005 г.), содержание которой включено в настоящее изобретение ссылкой для получения быстродиспергирующих микрогранул;
iii. Смешивание одного или более объединений TPR гранул из стадии (e) самих по себе или в комбинации с IR гранулами с исправленным вкусом из стадии (i), и/или SR гранулами из стадии (d) в желаемом соотношении для обеспечения желаемого одноразового суточного профиля плазмы, быстродиспергирующих микрогранул из стадии (ii) и других фармацевтически приемлемых наполнителей; и
iv. Прессование смеси из стадии (iii) в орально распадающиеся таблетки, включающие требуемую дозу, одно или более слабоосновные лекарственные средства, которые распадались бы быстро при контакте со слюной в полости рта, образуя однородную, легкую для глотания суспензию и показывая профиль плазмы, подходящий для двухразового или одноразового суточного режима дозирования с уменьшенной частотой возникновения побочных явлений, включая несоблюдение.
Водная или фармацевтически приемлемая среда растворителя может использоваться для приготовления частиц ядра, основанных на покрытых инертных частицах. Тип инертного связывающего вещества, который используется, чтобы связать водорастворимую органическую кислоту или слабоосновное лекарственное средство с инертной частицей или с SR покрытым ядром, содержащим кислоту, не критичен, но обычно могут использоваться водорастворимые или растворимые в спирте связывающие вещества, такие как поливинилпирролидон (ПВП или повидон) или гидроксипропилметилцеллюлоза. Связывающее вещество может использоваться при любой концентрации, способной к применению к инертной частице. Как правило, связывающее вещество используется при концентрации от приблизительно 0,5 до 10% по массе. Органическая кислота или слабоосновное лекарственное средство предпочтительно могут присутствовать в этом покрывающем образовании в форме раствора или суспензии. Содержание твердых частиц композиции с нанесенным лекарственным средством может изменяться в зависимости от применения, но в основном изменяется от приблизительно 5 до 30% по массе в зависимости от вязкости покрывающего образования и/или растворимости лекарственного средства.
В соответствии с другими вариантами осуществления ядра, содержащие органическую кислоту, могут быть приготовлены ротогранулированием или гранулированием с последующей экструзией-сферонизацией или таблетированием в микротаблетки. Органическая кислота, связывающее вещество и произвольно другие фармацевтически приемлемые наполнители (например, разбавители/наполнители) могут быть смешаны вместе в грануляторе с высоким сдвигом или грануляторе псевдоожижженного слоя, таком как Glatt GPCG гранулятор, и гранулированы для образования агломератов. Влажная масса может быть экструдирована и сферонизирована для получения сферических частиц (драже). Смесь, включающая частицы кислоты, связывающее вещество и произвольный наполнитель/разбавитель или гранулы, содержащие лекарственное средство, может быть также спрессована в микротаблетки (приблизительно 1-1,5 мм в диаметре) для получения драже, содержащие органическую кислоту. В этих вариантах осуществления содержание кислоты могло быть столь же высоко как 95%, по массе на основе общей массы гранулированного, экструдированного или спрессованного ядра. Эти ядра, содержащие кислоту, покрываются SR мембраной до наслоения лекарственного средства и последующего покрытия функциональными полимерами.
Индивидуальные полимерные покрытия на ядрах, содержащих кислоту, и IR гранулах будут изменяться от приблизительно 5 до 50% по массе в зависимости от относительной растворимости органической кислоты в активном веществе, природы активного вещества, композиции барьерного покрытия и регулируемого времени задержки. В одном варианте осуществления кислотные ядра можно обеспечить барьерным покрытием пластифицированным водонерастворимым полимером, таким как этилцеллюлоза (ЕС-10), при приблизительно 5-50% по массе, чтобы поддержать высвобождение кислоты приблизительно свыше 5-20 часов. В некоторых других вариантах осуществления кислотные ядра могут обеспечиваться барьерным покрытием пластифицированной этилцеллюлозой и фталатом (HP-55) гидроксипропилметилцеллюлозы (гипромеллозой) при приблизительно 10-50% по массе, в то время как IR гранулы покрываются этилцеллюлозой (ЕС-10) при 5-20% по массе, чтобы достигнуть высвобождения лекарственного средства, синхронизированного с высвобождением кислоты. В уже другом варианте осуществления настоящего изобретения IR гранулы не могут быть обеспечены любым барьерным покрытием, а внешнее покрытие с задержкой времени EC-10/HP-55/пластификатор при приблизительно 45,5/40/14,5 для увеличения массы приблизительно 30-50% по массе контролирует высвобождение лекарственного средства после задержки времени. Композиция мембранного слоя и индивидуальных масс полимеров представляет собой важные факторы, которые нужно рассмотреть для достижения желаемого профиля высвобождения лекарственного средства/кислоты и времени задержки до заметного высвобождения лекарственного средства.
Профили высвобождения лекарственного средства/кислоты из IR гранул, гранул, покрытых барьером/SR, и TPR гранул могут быть определены согласно следующей методике.
Исследование растворения IR гранул, с исправленным вкусом или нет, проводится с USP установкой 1 (барабан при 100 оборотах в минуту) или установкой 2 (мешалки с лопастью при 50 оборотах в минуту) в 900 мл 0,1 н. HCl при 37°C, в то время как исследование растворения SR и TPR гранул проводится в USP аппарате с использованием среды растворения с двумя стадиями (сначала 2 часа в 700 мл 0,1 н. HCl при 37°C с последующим исследованием растворения при pH=6,8, полученного добавлением 200 мл pH модификатора). Высвобождение лекарственного средства/кислоты со временем определяют ВЭЖХ на образцах, собранных выбранных интервалах.
Есть случаи, в которых начало высвобождения лекарственного средства должно начинаться несколькими часами позже орального приема, чтобы обеспечить адекватную концентрацию в плазме, чтобы быть подходящей для двухразового или одноразового суточного режима дозирования, в зависимости от элиминационного периода полувыведения активного вещества. В соответствии с определенными аспектами изобретения высвобождение лекарственного средства может быть задержано приблизительно до 8-10 часов после орального приема.
Единственный расчетный профиль замедленного высвобождения свыше нескольких часов после орального приема, с или без быстрого пульсирующего высвобождения, обеспечивается в соответствии с некоторыми вариантами осуществления настоящего изобретения.
Водная или фармацевтически приемлемая среда растворителя может использоваться для приготовления внутренних слоев частицы, содержащих органическую кислоту, или IR гранул, содержащих лекарственное средство, наслоением лекарственного средства на инертные ядра, такие как сахарные сферы, или на SR покрытые ядра, содержащие кислоту. Тип инертного связывающего вещества, которое используется, чтобы связать водорастворимую органическую кислоту с инертной частицей или слабоосновным лекарственным средством на SR покрытых кислотных ядрах, не критичен, но обычно используются связывающие вещества, растворимые в воде или спирте и/или растворимые в ацетоне. Типичные примеры связывающих веществ включают, но не ограничиваются, поливинилпирролидон (ПВП), гидроксипропилметилцеллюлозу (ГПМЦ), гидроксипропилцеллюлозу, карбоксиалкилцеллюлозы, полиэтиленоксид, полисахариды, такие как декстран, кукурузный крахмал, который может быть растворен или диспергирован в воде, спирте, ацетоне или их смесях. Связывающие вещества в основном используются при концентрации от приблизительно 0,5 до 10% по массе.
Типичные инертные частицы, применяемые к слою кислоты или фармацевтическому активному веществу, включают сахарные сферы, сферы целлюлозы и сферы диоксида кремния с подходящим распределением размера частицы (например, 20-25 меш сахарных сфер для создания покрытых гранул для объединения в образование капсулы и 60-80 меш сахарных сфер для создания покрытых гранул для объединения в ОРТ образование).
Примеры слабоосновных, азот-(N)-содержащих терапевтических средств, имеющих pKa в диапазоне от приблизительно 5 до 14, включают, но не ограничиваются, болеутоляющие, противосудорожные, антидиабетические средства, антибактериальные средства, противоопухолевые, антипаркинсонические средства, антиревматические средства, сердечно-сосудистые средства, ЦНС (центральная нервная система) стимулянты, агонисты допаминового рецептора, противорвотные средства, желудочно-кишечные средства, психотерапевтические средства, опиоидные агонисты, опиоидные антагонисты, антиэпилептические лекарственные средства, гистаминовые Н2 антагонисты, антиастматические средства и релаксанты скелетных мышц.
Типичные фармацевтически приемлемые органические кислоты, которые увеличивают растворимость фармацевтического активного вещества, включают лимонную кислоту, фумаровую кислоту, яблочную кислоту, винную кислоту, янтарную кислоту, щавелевую кислоту, аспарагиновую кислоту, глутаминовую кислоту и т.п. Отношение органической кислоты к фармацевтическому активному веществу в основном изменяется от приблизительно 5:1 до 1:10, а конкретнее от приблизительно 3:1 до 1:3 по массе в некоторых вариантах осуществления настоящего изобретения.
Типичные примеры водонерастворимых полимеров, полезных в изобретении, включают этилцеллюлозу, ацетат поливинила (например, Kollicoat SR# 30D от BASF), ацетат целлюлозы, ацетат бутират целлюлозы, нейтральные сополимеры, основанные на этилакрилате и метилметакрилате, сополимеры сложных эфиров акриловой и метакриловой кислоты с группами четвертичного аммония, такими как эудрагит NE, RS и RS30D, RL или RL30D и т.п. Типичные примеры водорастворимых полимеров, полезных в изобретении, включают поливинилпирролидон (ПВП), гидроксипропилметилцеллюлозу (ГПМЦ), гидроксипропилцеллюлозу (ГПЦ), полиэтиленгликоль и т.п.
Типичные примеры кишечнорастворимых полимеров, полезных в изобретении, включают сложные эфиры целлюлозы и ее производных (фталат ацетата целлюлозы, фталат гидроксипропилметилцеллюлозы, сукцинат ацетата гидроксипропилметилцеллюлозы), фталат ацетата поливинила, pH-чувствительные сополимеры метакриловой кислоты - метакрилата и шеллак. Эти полимеры могут использоваться в качестве сухого порошка или водной дисперсии. Некоторыми коммерчески доступными материалами, которые могут использоваться, являются сополимеры метакриловой кислоты, реализованные под торговой маркой эудрагит (L100, S100, L30D), изготовленные Rohm Pharma, Cellacefate (фталат ацетата целлюлозы) от Eastman Chemical Co., Aquateric (водная дисперсия фталата ацетата целлюлозы) от корпорации FMC и Aqoat (водная дисперсия сукцината ацетата гидроксипропилметилцеллюлозы) от Shin Etsu K.K.
Кишечнорастворимые, водонерастворимые и водорастворимые полимеры, используемые при образовании мембран, обычно пластифицированы. Типичные примеры пластификаторов, которые могут применяться к пластификатору, мембранам, включают триацетилглицерин, трибутилцитрат, триэтилцитрат, ацетилтри-н-бутилцитрат, диэтилфталат, касторовое масло, дибутилсебацинат, моно- и диацилглицериды (коммерчески доступные как Myvacet 9-45), и т.п. или их смеси. Пластификатор при использовании может включать от приблизительно 3 до 30% по массе и более в основном от приблизительно 10 до 25% по массе на основе полимера. Тип пластификатора и его содержания зависит от полимера или полимеров и природы системы покрытия (например, водная или на основе растворителя, раствор или на основе дисперсии и общей массы твердых частиц).
Вообще, желательно заполнить поверхность частиц слоистым лекарственным средством, перед применением покрытий с мембранным барьером или отделить различные мембранные слои использованием тонкой пленки гидроксипропилметилцеллюлозы (ГПМЦ) (например, Pharmacoat® 603 или Opadry® Clear). В то время как используется в основном ГПМЦ, также могут использоваться другие заполнители, такие как гидроксипропилцеллюлоза (ГПЦ), или с более низкой вязкостью этилцеллюлоза.
Активные фармацевтические ингредиенты, подходящие для объединения в них систем с регулируемым по времени, пульсирующим высвобождением, включают слабоосновные активные фармацевтические ингредиенты, производные или их соли, которые являются азот-(N)-содержащими биоактивными фрагментами, имеющими pKa в диапазоне от приблизительно 5 до 14, растворимость не больше, чем 200 мкг/мл при pH 6,8, и отношение оптимальной самой высокой дозы к растворимости при pH 6,8 не меньше, чем приблизительно 100. Лекарственное вещество может быть выбрано из группы фармацевтически приемлемых химических объектов с доказанной фармакологической активностью на людях.
Определенные примеры слабоосновных, азот-(N)-содержащих терапевтических средств включают без ограничения, оланзапин, производное пиперазина, предназначенное для лечения шизофрении, ондансетрон или гидрохлорид ондансетрона, селективный антагонист серотонинового 5-HT3 рецептора, предназначенного для предотвращения тошноты и рвоты, связанного с химиотерапией или постоперационной хирургией, дипиридамол, дипиримидиновое производное, предназначенное для предотвращения постоперационных тромбоэмболических осложнений замены сердечного клапана, карведилол, бета-адренергическое блокирующее средство, предназначенное для лечения паралича сердца ишемического или кардиомиопатического происхождения, ламотригин, триазиновое производное, предназначенное для терапии эпилепсии у взрослых и детей, оланзапин или его фармацевтически приемлемая соль, психотропное средство, предназначенное для лечения шизофрении, кветиапин, производное пиперазина, предназначенное для лечения биполярных расстройств.
Мембранные покрытия могут применяться к внутреннему слою с использованием любого из способов покрытия, обычно используемых в фармацевтической промышленности, но покрытие псевдоожиженным слоем особенно полезно. Настоящее изобретение относится к формам многократного приема лекарственного средства, то есть лекарственным продуктам в виде лекарственных форм, состоящих из множества частиц, в качестве твердых желатиновых капсул или обычных и орально распадающихся спрессованных таблеток с использованием ротационного таблетного пресса, включающих одно или более объединений гранул для орального приема, чтобы обеспечить расчетные ФК параметры у пациентов, нуждающихся в лечении. Обычные таблетки быстро диспергируют при попадании в желудок, в то время как ОРТ быстро распадаются при контакте со слюной в полости рта, образуя однородную, легкую для проглатывания суспензию покрытых гранул для легкого проглатывания. Одно или более покрытых объединений гранул могут быть спрессованы вместе с соответствующими наполнителями таблетки (например, связывающее вещество, разбавитель/наполнитель, и дезинтегрирующее вещество для обычных таблеток, в то время как быстро диспергирующее гранулирование может заменять комбинацию связывающего вещества-разбавителя/наполнителя в ОРТ). Кроме того, прессование в ОРТ может быть выполнено с использованием таблетного пресса, оборудованного внешней системой смазывания, чтобы смазать удары и штампы до прессования.
Следующие неограничивающие примеры иллюстрируют лекарственные формы в виде капсулы, включающие один или более импульсов, причем каждый с предопределенным началом запаздывания и общим количеством in vitro параметра высвобождения лекарственного средства или последующего in vivo параметра концентрации в плазме при оральном приеме лекарственной формы должен подражать желаемому параметру для достижения максимальной терапевтической эффективности, чтобы увеличить соблюдение больным режима и схемы лечения и качество жизни. Такие формы дозировки, при введении «правильное время» или как рекомендуется врачом, позволили бы поддержать концентрацию лекарственного средства в плазме на уровне, потенциально выгодном в уменьшении возникновения побочных эффектов, связанных с Cmax или Cmin.
Пример 1
A. SR гранулы фумаровой кислоты
40-80 меш кристаллов фумаровой кислоты (3750 г) загрузили в устройство для нанесения покрытий с псевдоожиженным слоем, Glatt GPCG 5, оборудованным 9" вставкой Wurster распыления дна, 10" длина колонки и трубкой на 16 мм. Эти кристаллы кислоты покрыли раствором (при 6% твердых частиц) 250 г этилцеллюлозы (Ethocel Premium 10 cps, упомянутой далее как ЕС-10) и 166,7 г полиэтиленгликоля (ПЭГ 400) при соотношении 60/40, растворенным в смеси 98/2 ацетон/вода (6528,3 г) для увеличения массы до 10% по массе. Условия обработки были следующие: давление воздуха распыления: 2,0 бара; диаметр сопла: 1,00 мм; нижняя тарелка распределения: B; интервал распыления/встряхивания: 30 с/3 с; температуру продукта поддерживали при 35±1°C; объем входного воздуха: 145-175 кубических футов в минуту (cfm), а скорость распыления увеличивали от приблизительно 8 до 30 г/мин.
Кристаллы фумаровой кислоты также покрыли, как описано выше, с использованием различных соотношений этилцеллюлозы и ПЭГ. А конкретнее, кристаллы кислоты покрыли раствором ЕС-10 (Ethocel Premium 10 cps)/ПЭГ 400 в соотношении или 75/25 или 67,5/32,5 для увеличения массы до 10% по массе в каждом случае. Фиг.4 показывает профили высвобождения фумаровой кислоты из ее кристаллов, покрытых при различных отношениях EC-10/ПЭГ.
B. Кристаллы винной кислоты, покрытые барьером
60-100 меш кристаллы винной кислоты (900 г) загрузили в устройство для нанесения покрытий с псевдоожиженным слоем, Glatt GPCG 1, оборудованным 6" вставкой Wurster распыления дна, 6" длина колонки, и 1 см от дна. Эти кристаллы кислоты покрывали раствором (при 6% твердых частиц) 202,5 г этилцеллюлозы (Ethocel Premium 10 cps) и 22,5 г триэтилцитрата (ТЕС) для увеличения по массе 20%. Условия обработки были следующие: давление воздуха распыления: 1,5 бара; диаметр сопла: 1,00 мм; нижняя тарелка распределения: B; температуру продукта поддерживали при 33±1°C; скорость входного воздуха: 4-5 м/с и скорость распыления увеличивали от приблизительно 5 до 8 г/минут. После покрытия гранулы высушили в блоке в течение 10 минут, чтобы убрать избыток остаточного растворителя. Высвобождение винной кислоты было слишком быстрым. 20% SR покрытых кристаллов высвободили 67% винной кислоты в течение часа при исследовании растворения в 0,1 н. HCl. Эти покрытые кристаллы покрывали EC-10/HP-55/ТЕС при отношении 60/25/15, растворенного в смеси 95/5 ацетон/вода для увеличения массы 20%. Высвобождение винной кислоты в 2- и 4-часовых точках времени равнялось соответственно 66% и 93% при исследовании методикой двухстадийного растворения.
Пример 2
A. Ядра, содержащие фумаровую кислоту
Гидроксипропилцеллюлозу (Klucel LF, 33,3 г) медленно добавляли к 90/10 денатурированному SD 3C 190 крепости спирту с водой при 4% твердых частиц при строгом перемешивании для растворения и затем медленно добавляли фумаровую кислоту (300 г) для растворения. Glatt GPCG 3, оборудованный 6" вставкой Wurster распыления дна, 8" колонну разделения загрузили 866,7 г 25-30 меш сахарных сфер. Сахарные сферы наслаивали раствором фумаровой кислоты при поддержании температуры продукта при приблизительно 33-34°C и скорости входного воздуха приблизительно 3,5-4,5 м/с. Кислотные ядра высушили в блоке в течение 10 минут, чтобы убрать остаточный растворитель/влажность и просеяли через 20-30 меш.
B. Ядра, SR покрытые фумаровой кислотой
Кислотные ядра (1080 г) из вышеупомянутого пункта покрыли раствором (при 7,5% твердых частиц) 108 г этилцеллюлозы (ЕС-10) и 12 г триэтилцитрата (ТЕС), растворенного при соотношении 90/10 в 95/5 растворе ацетон/вода для увеличения массы 10% по массе.
C. IR гранулы дипиридамола, включающие SR покрытые кислотные ядра
Дипиридамол (225 г) медленно добавляли к водному раствору поливинилпирролидона повидон K-29/32 (25 г) для растворения лекарств. SR покрытые кислотные ядра покрывали в Glatt GPCG 3, причем раствор лекарственного средства и гранулы, покрытые лекарственным средством, обеспечивали защитным изолирующим слоем Opadry Clear (увеличение массы приблизительно на 2%) для образования IR гранул с загрузкой лекарственного средства 17,29% по массе.
D. SR гранулы дипиридамола
Вышеупомянутые IR гранулы дипиридамола (1080 г) покрывали барьером (SR покрытым) распылением раствора (7,5% твердых частиц) 90/10 EC-10/TЕС (триэтилцитрат) при 5-10% по массе и высушивали в Glatt в течение 10 минут, чтобы убрать избыток остаточного растворителя. Высушенные гранулы просеяли, чтобы отбросить любое образование дублей.
E. TPR гранулы дипиридамола
SR гранулы дипиридамола (1080 г) с покрытием на 7% из примера 2D далее покрывали мембраной покрытия с задержкой времени EC-10/HP-55 (фталат гидросипропилметилцеллюлозы)/TЕС (триэтилцитрат) при соотношении 50/35/15 для увеличения массы приблизительно на 20%.
TPR гранулы высушили в Glatt при той же самой температуре, чтобы удалить остаточный растворитель, и просеяли. Фиг.5 показывает профили высвобождения дипиридамола из TPR гранулы дипиридамола.
Пример 3
А. Ядра, содержащие фумаровую кислоту
Гидроксипропилцеллюлоза (Klucel LF, 20 г) медленно добавили к 90/10 денатурированному 3D ЗС 190 крепости спирта с водой при 4% твердых частиц при строгом перемешивании для растворения, и затем медленно добавляли фумаровую кислоту (200 г) для растворения. Glatt GPCG 3 загрузили 780 г 25-30 меш сахарных сфер, сахарные сферы наслоили раствором фумаровой кислоты, как описано в примере 1. Кислотные ядра высушили в блоке в течение 10 минут, чтобы убрать остаточный растворитель/влажность, и просеяли через 20-30 меш.
В. Ядра, SR покрытые фумаровой кислотой
Вышеупомянутые кислотные ядра (900 г) покрывали раствором (при 7,5% твердых частиц) 90 г этилцеллюлозы (ЕС-10) и 10 г триэтилцитрата (ТЕС), растворенного при соотношении 90/10, в 95/5 смеси ацетон/вода для увеличения массы 10% по массе.
С.IR гранулы ламотригина
Ламотригин (162 г) медленно добавляли к водному раствору Klucel LF (13 г) для растворения лекарственного средства. Вышеупомянутые SR покрытые кислотные ядра (900 г) покрывали в Glatt GPCG 3 с раствором лекарственного средства, а гранулы, покрытые лекарственным средством, обеспечивали защитным изолирующим слоем Opadry Clear (увеличение массы приблизительно на 2%) и высушивали в Glatt для получения IR гранул.
D. SR гранулы ламотригина
IR гранулы ламотригина покрывали барьером, распыляя раствор (7,5% твердых частиц) 70/30 EC-10/ТЕС при 3-5% по массе, и высушивали в Glatt GPCG 3 при той же самой температуре в течение 10 минут, чтобы удалить избыток остаточного растворителя. Высушенные гранулы просеяли, чтобы отбросить любое образование дублей.
E. TPR гранулы ламотригина
SR гранулы ламотригина при покрытии на 5% далее покрывали мембраной покрытия с задержкой времени EC-10/HP-55/ТЕС при соотношении 42,5/42,5/15 для увеличения массы приблизительно 10-15%. TPR гранулы высушивали в Glatt, чтобы удалить остаточный растворитель, и просеивали через 20 меш сита.
F. MR капсулы ламотригина, 50 мг:
Твердые желатиновые капсулы заполняли IR гранулами, SR гранулами (покрытие на 3%) и TPR гранулами (покрытие на 10%) при соотношении 35/40/25.
Пример 4
A. Кристаллы винной кислоты, покрытым барьером
60-100 меш кристаллов винной кислоты (900 г) покрывали в Glatt GPCG 3 с раствором фумаровой кислоты (90 г) и 10 г Klucel LF при соотношении 90/10, растворенной в денатурированном SD 3C 190 крепости спирта в воде при 4% твердых частиц, и далее покрывали с EC-10/HP-55/ТЕС при соотношении 65/20/15, растворенного в 95/5 ацетон/вода при 7,5% твердых частиц для увеличения 30% по массе, как описано в вышеупомянутых примерах. Покрытые кристаллы высушивали и просеивали, чтобы отбросить образование дублей.
B. IR гранулы ламотригина
Ламотригин (540 г) медленно добавляли к водному раствору Klucel LF (60 г), чтобы диспергировать лекарственное средство гомогенно. Вышеупомянутые SR покрытые кислотные ядра (900 г) покрывали в Glatt GPCG 3 лекарственной суспензией, а покрытые лекарственным средством гранулы обеспечивали защитным изолирующим слоем Opadry Clear для увеличения массы на 2% и высушивали в Glatt для получения IR гранул.
C. SR гранулы ламотригина
IR гранулы ламотригина (800 г) покрывались SR распылением раствора (7,5% твердых частиц) 85/15 EC-10/TЕС для увеличения массы на 5-10%. Гранулы SR, высушенные в Glatt при той же самой температуре в течение 10 минут, чтобы удалить избыток остаточного растворителя, просеивали, чтобы отбросить любое образование дублей.
D. TPR гранулы ламотригина
Гранулы SR ламотригина далее покрывали мембраной покрытия с задержкой времени ЕС-10/HP-55/ТЕС при соотношении 45/40/15 для увеличения массы приблизительно на 10-20%.
E. MR капсулы ламотригина, 50 мг:
Твердые желатиновые капсулы заполняли IR гранулами, гранулами SR (покрытие на 10% или на 5%) и TPR гранулами (покрытие на 20% или на 10%) при соотношении 35/40/25.
Пример 5
А. Ядра, содержащие фумаровую кислоту
Гидроксипропилцеллюлозу (Klucel LF, 33,3 г) медленно добавляли к 90/10 190 крепости спирта в воде при 4% твердых частиц, при строгом перемешивании для растворения и затем медленно добавляли фумаровую кислоту (300 г) для растворения. Glatt GPCG 3, оборудованный 6” вставкой Wurster распыления дна, 8” колонку разделения загружали с 866,7 г 60-80 меш сахарных сфер. Сахарные сферы наслаивали раствором фумаровой кислоты при поддержании температуры продукта при приблизительно 33-34°С и скорости входного воздуха приблизительно 3,5-4,5 м/с. Кислотные ядра высушивали в блоке в течение 10 минут, чтобы удалить остаточный растворитель/влажность, и просеивали через 40-80 меш.
В. Ядра, SR покрытые фумаровой кислотой
Вышеупомянутые кислотные ядра (800 г) покрывали раствором (при 7,5% твердых частиц) 180 г этилцелюллозы (ЕС-10) и 20 г триэтилцитрата (ТЕС) при соотношении 90/10, растворенного в 95/5 ацетон/вода для увеличения массы от 10% до 20%.
С.IR гранулы карведилола
Гидроксипропилцеллюлозу (Klucel LF, 77,8 г) медленно добавляли к очищенной воде (при 6% твердых частиц) при строгом перемешивании для растворения, а карведилол (700 г) медленно добавляли при перемешивании, чтобы диспергировать лекарственное средство гомогенно. Вышеупомянутые покрытые SR кислотные ядра (900 г) покрывали в Glatt GPCG 3 с дисперсией лекарственного средства, а покрытые лекарственным средством гранулы обеспечивали защитным изолирующим слоем Opadry Clear (34,2 г приблизительно для увеличения массы на 2%) и высушивали в Glatt для получения IR гранул.
D. SR гранулы карведилола
IR гранулы карведилола (1080 г) покрывали барьером (покрытым SR) распылением раствора (7,5% твердых частиц) 90/10 EC-10/TЕС при 5% по массе и высушивали в Glatt при той же самой температуре в течение 10 минут, чтобы удалить избыток остаточного растворителя. Высушенные гранулы просеивали, чтобы отбросить любое образование дублей.
E. РПВП гранулы карведилола
Гранулы SR карведилола далее покрывали мембраной покрытия с задержкой времени EC-10/HP-55/TEC при соотношении 50/35/15 для увеличения массы приблизительно на 10-20% по массе. TPR гранулы высушивали в Glatt, чтобы удалить остаточный растворитель, и просеивали через 20 меш. Фиг.6 показывает профили высвобождения TPR гранул карведилола.
F. IR гранулы с замаскированным вкусом
IR гранулы, полученные выше, покрывались 50/50 EC-10/эудрагит E100, растворенным в 48,5/24/27,5 ацетон/IPA/вода для увеличения массы приблизительно на 10-20% по массе.
G. Быстродиспергируемые микрогранулы
Быстродиспергируемые микрогранулы, включающие сахарный спирт, такой как маннитол, и дезинтегрирующее вещество кросповидон приготовили после методики, описанной в заявке на патент США № 2005/0232988, находящейся на стадии рассмотрения, поданной 20 октября 2005 г., содержание которой включено в настоящее изобретение ссылкой. D-маннитол (152 кг) со средним размером частицы приблизительно 20 мкм или меньше (Pearlitol 25 от Roquette, Франция) смешали с 8 кг кросс-связанного повидона (Кросповидон XL-10 от ISP) в грануляторе с высоким сдвигом (GMX 600 от Vector) и гранулировали с очищенной водой (приблизительно 32 кг) и мокро измельчили с использованием Comil от Quadro и высушили в лотках для LOD (потеря при высыхании) меньше, чем приблизительно 0,8%. Высушенные гранулы просеивали и материал большого размера измельчили для получения быстродиспергирующих микрогранул со средним размером частицы в диапазоне приблизительно 175-300 мкм.
H. CR ОДТ карведилола
Быстродиспергирующие микрогранулы смешали с TPR гранулами, гранулами SR, IR гранулами с замаскированным вкусом и другие фармацевтические приемлемые ингредиенты, такие как ароматические добавки, подсластители и дополнительное дезинтегрирующее вещество при соотношении быстродиспергирующих микрогранул к многослойным гранулам карведилола 2:1, в V-смесителе с двойным корпусом в течение достаточного времени, чтобы получить гомогенно распределенную смесь для прессования. Таблетки, включающие гранулы с замаскированным вкусом, гранулы SR и TPR гранулы при соотношении 35/40/25 как карведилол спрессовали с использованием таблетного пресса масштабного производства, оборудованного внешней системой смазывания при средней твердости приблизительно 5-7 kP. Таким образом, полученные МВ ОДТ карведилола, в количестве 50 мг, быстро распались в полости рта, создавая однородную, легкую для проглатывания суспензию, включающую покрытые гранулы карведилола, которые обеспечивают расчетные параметры, подходящие для одноразового суточного режима дозирования.
Пример 6
A. Ядра, содержащие фумаровую кислоту
Гидроксипропилцеллюлозу (Klucel LF, 53,6 г) медленно добавляли к 90/10 190 крепости спирта в воде при 4% твердых частиц при строгом перемешивании для растворения, и затем медленно добавили фумаровую кислоту (482,1 г) для растворения. Glatt GPCG 5, оборудованный 9" вставкой Wurster распыления дна, 10" колонку разделения загрузили 3750 г 25-30 меш сахарных сфер. Сахарные сферы наслаивали раствором фумаровой кислоты при поддержании температуры продукта при приблизительно 33-35°C и скорости распыления 8-60 мл/мин. Кислотные ядра высушили в блоке в течение 10 минут, чтобы удалить остаточный растворитель/влажность и просеяли через 40-80 меш.
B. Ядра SR, покрытые фумаровой кислотой
Вышеупомянутые кислотные ядра (3750 г) покрывали раствором (при 7,5% твердых частиц) 177,6 г этилцеллюлозы (ЕС-10) и 19,7 г триэтилцитрата (TEC) при соотношении 90/10, растворенного в смеси 95/5 ацетон/вода для увеличения массы 5% по массе после методик, описанных выше.
C. IR гранулы дигидрата гидрохлорида ондансетрона
Гидроксипропилцеллюлозу (Klucel LF, 77,8 г) медленно добавили к 50/50 190 крепости спирта в воде (4247,4 г спирта + 4247,4 г воды при 5% твердых частиц) при строгом перемешивании для растворения, и медленно добавили гидрохлорид ондансетрона (402,8 г) при перемешивании для растворения лекарственного средства. SR покрытые кислотные ядра (3500 г) покрывали в Glatt GPCG 5 с раствором лекарственного средства, а гранулы, слоистые лекарственные средства, обеспечивали защитным изолирующим слоем Pharmacoat 603 (80,5 г для увеличения массы приблизительно на 2%) и высушили в Glatt для получения IR гранулы (размер пробы: 4028 г),
D. SR гранулы гидрохлорида ондансетрона
IR гранулы гидрохлорида ондансетрона (3500 г) покрывали барьером (SR покрытым) распылением раствора (7,5% твердых частиц) 90/10 EC-10/TEC при 5% по массе и высушивали в Glatt при той же самой температуре в течение 10 минут, чтобы удалить избыток остаточного растворителя. Высушенные гранулы просеивали, чтобы исключить любое образование дублей.
E. TPR гранулы гидрохлорида ондансетрона
SR гранулы гидрохлорида ондансетрона далее покрывали мембраной покрытия с задержкой времени EC-10/HP-55/TEC при соотношении 60,5/25/14,5 для увеличения массы с 20% до 45% по массе. TPR гранулы высушивали в Glatt, чтобы удалить остаточный растворитель, и просеивали через 30 меш. TPR гранулы, расфасованные при введении в герметично закрытые HDPE сосуды, поместили на основании неизменных ICH норм. Фиг. 7 демонстрирует профили высвобождения лекарственного средства, полученные от TPR гранул на основе ускоренной стабильности (то есть в 40°C/75%RH) в течение до 6 месяцев. Таблица 2 показывает, что образование настоящего изобретения физически и химически стабильно.
(Лот# 1117-NHV-099)
Пример 7 (сравнительный)
A. IR гранулы карведилола, наслоенные на сахарных сферах
Гидроксипропилцеллюлозу (Klucel LF, 77,8 г) медленно добавляли к 90/10 190 крепости спирта в воде (11667 г спирта + 1296 г воды при 6% твердых частиц) при строгом перемешивании для растворения, и медленно добавляли карведилол (700 г) при перемешивании для растворения лекарственного средства. 25-30 меш сахарных сфер (900 г) покрывали в Glatt GPCG 3 с раствором лекарственного средства, и гранулы, покрытые лекарственным средством, обеспечивали защитным изолирующим слоем Opadry Clear (34,2 г для увеличения массы приблизительно на 2%) и высушивали в Glatt для получения IR гранул (размер пробы: 1712 г).
B. SR гранулы карведилола
IR гранулы карведилола (800 г) покрывали барьером (покрытым SR) распылением раствора (при 6% твердых частиц) 80/10 EC-10/TEC при 15% по массе и высушивали в Glatt в течение 10 минут, чтобы удалить избыток остаточного растворителя. Собранные образцы при 5%, 7,5% и 10% покрытия. Высушенные гранулы просеивали, чтобы исключить любое образование дублей. Исследование растворения 5% и 10% покрытых SR гранул, чтобы показать воздействие объединение ядра органической кислоты.
C. Рентгеновская дифракция порошка
Образцы порошка для рентгена получили для фумаровой кислоты, карведилола, SR гранул, покрытых фумаровой кислотой, IR гранул карведилола, SR гранул, и TPR гранул примера 6. Анализ этих образцов рентгена показал что, карведилол присутствует в IR и TPR гранулах в первоначальном прозрачном состоянии, а не соли муравьиной кислоты.
Пример 8
A. Ядра, содержащие фумаровую кислоту
Ядра, содержащие фумаровую кислоту (при загрузке фумаровой кислоты 5,4% по массе), приготовили в соответствии с методикой, описанной выше.
B. SR покрытые ядра, содержащие фумаровую кислоту
Вышеупомянутые ядра, содержащие фумаровую кислоту (3750 г), покрывали раствором ЕС-10 и либо ПЭГ 400 (B.1) при соотношении 60/40, либо TEC (B.2) при соотношении 90/10 в качестве пластификатора, растворенного в смеси 98/2 ацетон/вода (при 6% твердых частиц) для увеличения массы 10%.
C. IR гранулы гидрохлорида ондансетрона
Вышеупомянутые IR гранулы гидрохлорида ондансетрона от B.1 и B.2 приготовили, как описано в примере 3C. Гранулы, покрытые лекарственным средством, обеспечивали защитным изолирующим слоем с Pharmacoat 603 (гидропропилметилцеллюлоза 2910; 3 cps) для увеличения массы 2%.
D. SR гранулы гидрохлорида ондансетрона
IR гранулы гидрохлорида ондансетрона (1080 г) покрывали барьером (покрытый SR) распылением раствора EC-10 и либо ПЭГ 400 (D.1) при отношении 60/40 либо TEC (D.2) при соотношении 90/10 в качестве пластификатора, растворенного в смеси 98/2 ацетон/вода (при 7,5% твердых частиц) для увеличения массы на 10% и высушивали в Glatt при той же самой температуре в течение 10 минут, чтобы удалить избыток остаточного растворителя. Высушенные гранулы просеивали, чтобы исключить любое образование дублей.
E. TPR гранулы гидрохлорида ондансетрона
Вышеупомянутые SR гранулы гидрохлорида ондансетрона из D.1 и D.2 далее покрывали мембраной покрытия с задержкой времени EC-10/HP-55/TEC при трех отношениях 45,5/40/14,5 (E.1-лот# 1084-066), 50,5/35/14,5 (E.2-лот# 1117-025) и 60,5/25/14,5 (E.3 - лот# 1117-044), растворенной в смеси 90/10 ацетон/вода (при 7,5% твердых частиц) для увеличения до 50% по массе. TPR гранулы высушивали в Glatt, чтобы удалить остаточный растворитель, и высушивали через 18 меш сит. Фиг.8 показывает профили высвобождения для гидрохлорида ондансетрона из TPR гранул, покрытых EC-10/HP-55/TEC при трех различных соотношениях (E.1, E.2 и E.3). А конкретнее, фиг.8 показывает профили высвобождения для следующих образований.
1) Лот# 1084-066 TPR гранул - покрытие EC-10/HP-55/TEC при соотношении 45,5/40/14,5 при 50% по массе, используемое на IR гранулах, покрытых 60/40 ЕС-10/PEG 400 при 10%, в то время как IR гранулы (5% лекарственного средства, покрытого 90/10 ондансетрон/ПВП), включает ядра, содержащие фумаровую кислоту (4% покрытую на сахарные сферы из кислоты/Klucel), покрытые 60/40 EC-10/ПЭГ 400 при 10%.
2) Лот# 1117-025 TPR гранул - покрытие EC-10/HP-55/TEC при соотношении 50,5/35/14,5 при 50%, по массе, используемое на IR гранулах, покрытых 90/10 ЕС-10/TEC при 10%, в то время как IR гранулы (6% лекарственного средства, покрытого 90/10 ондансетрон/Klucel LF) включают ядра, содержащие фумаровую кислоту (покрытую на сахарные сферы из кислоты/ПВП), покрытые 90/10 EC-10/TEC при 10%.
3) Лот# 1117-044 TPR гранул - покрытие EC-10/HP-55/TEC при соотношении 60,5/25/14,5 при 50% по массе, используемые на IR гранулах, покрытых 90/10 ЕС-10/TEC при 10%, в то время как IR гранулы (6% лекарственного средства, покрытого 90/10 ондансетрон/Klucel LF, включает ядра, содержащие фумаровую кислоту (покрытую на сахарные сферы из кислоты/ПВП), покрытые 90/10 EC-10/TEC при 10%.
Пример 9
A. Доказательство концепции исследуемых образований
IR гранулы гидрохлорида ондансетрона (PE364EA0001) и TPR гранулы (лот# PE366EA0001 с покрытием с задержкой времени 30%, лот# PE367EA0001 с покрытием с задержкой времени 45%, и лот# PE368EA0001 с покрытием с задержкой времени 50%) инкапсулировали при соотношении 35%/65% в твердые желатиновые капсулы для получения МВ (модифицированного высвобождения) капсул, 16 мг (лот# PF380EA0001, лот# PF381EA0001, и лот# PF382EA0001) QD (один раз в сутки) для экспериментального изучения биодоступности на людях по сравнению с коммерческим зофраном® 8 мг (как ондансетрон), дозированным BID (два раза в день).
B. Доказательство концепции ФК изучения на человеке
Провели перекрестное пробное исследование 4-х групп пациентов для ДК (доказательства концепции), которое включало 12 мужчин - уроженцев Кавказа, здоровых, в возрасте от 18 до 55 лет, с периодом выведения 7 дней. Каждому добровольцу давали дозу 250 мл разового исследуемого образования (16 мг) в дистиллированной минеральной воде А (PF380EA0001), B (PF381EA0001) или C (PF382EA0001 примера 4) в 8:00 или два зофрана® (8 мг) в 8:00 и 16:30 после голодания накануне вечером (по меньшей мере, 12 часов и завтрак принимался в 11:00). Были взяты образцы крови в 0 (преддоза), 20 мин, 40 мин, 1 ч, 1,5 ч, 2 ч, 3 ч, 4 ч, 6 ч, 8,5 ч (до 2-й дозы), 9 ч 10 мин, 9,5 ч, 10 ч, 10,5 ч, 11,5 ч, 12,5 ч, 14,5 ч, 17 ч, 20 ч, 22 ч, 24 ч и 36 ч. Фиг.9 показывает профили зависимости от времени достигнутой средней концентрации в плазме. Фигура показывает, что профили плазмы исследуемых образований А (PE280EA0001), B (PE281EA0001) и C (PE282EA0001) - представляют собой характеристики образований с замедленным высвобождением, то есть очевидный период полувыведения значительно длиннее, чем у зофрана, AUC или Cmax исследуемых образований существенно не отклоняется от таких же параметров зофрана (то есть AUC в пределах ±25% и Cmax приблизительно 70% зофрана). Фактическая Cmax для 8 мг зофрана была 30 нг/мл по сравнению с предсказанными 24 нг/мл, в то время как фактическая Cmax для IR компонента была приблизительно 24 нг/мл при нормализации. Приблизительно 70% 8 мг зофрана BID (2 раза в день), было поглощено в течение 24 часов. Исследуемые образования от А до C показали ожидаемую тенденцию постдозирования до точки пересечения при приблизительно 15-16 ч. Затем формула C продолжала показывать более низкую концентрацию в плазме на профиле вопреки предсказанному поведению.
Пример 10
A. SR покрытые кристаллы винной кислоты
60-100 меш кристаллов винной кислоты покрывали барьером, как описано в примере 2B.
B. IR гранулы карведилола (загрузка лекарственного средства: 40,9% мас./мас.)
IR гранулы приготавливали, как описано в примере 5.
C. SR гранулы карведилола
IR гранулы, полученные выше, покрыли SR с 90/10 EC-10/TEC для увеличения массы 5-10%.
D. TPR гранулы карведилола
На покрытые SR гранулы карведилола с 5% покрытием наносили покрытие с задержкой времени EC-10/HP-55/TEC при соотношении 50/35/15 для увеличения массы до приблизительно 30% по массе.
E. CR капсулы карведилола, 50 мг
Твердые желатиновые капсулы наполняли IR гранулами, SR гранулами (5% или 10% покрытия) и TPR гранулами (покрытие на 30%) при соотношении 35/40/25.
Из этих демонстраций очевидно, что объединение органической кислоты, в качестве солюбилизатора для слабоосновных лекарственных средств, показывающих профили растворимости, зависимые от pH (то есть показывают уменьшение в растворимости при кишечном pH 6,8 примерно на 2 порядка величины по сравнению с его максимальной растворимостью в жидкости ЖК) и функциональном покрытии кислоты до применения активного фармацевтического ингредиента существенно воздействует на задержку времени, желаемого, но завершенного профиля высвобождения лекарственного средства, до того как закончится буферный раствор. Кроме того, активный фармацевтический ингредиент остается в неизменной форме в твердой лекарственной форме, до тех пор, пока он не высвободится для поглощения в ЖК тракте.
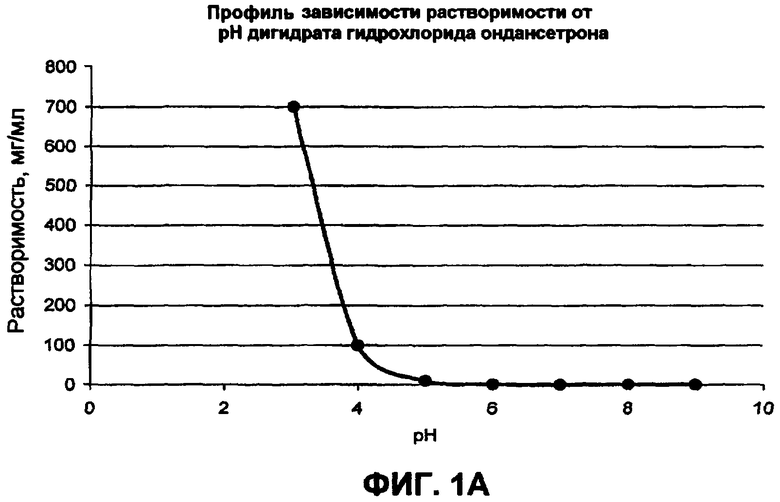
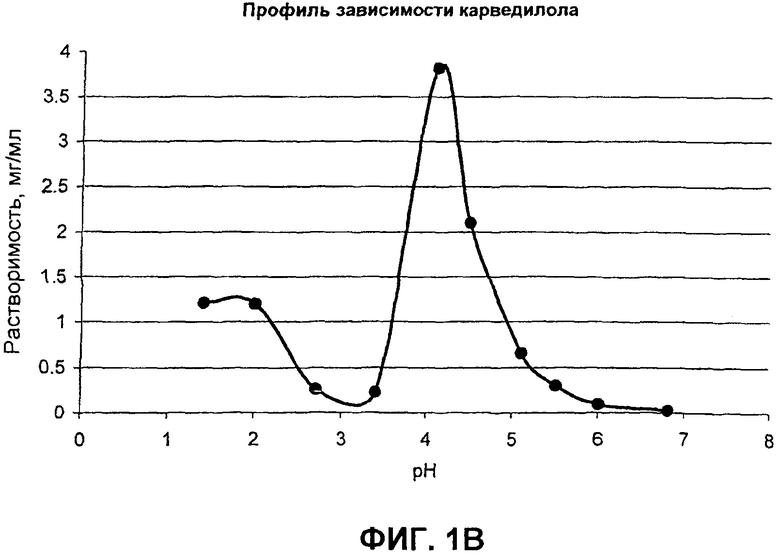
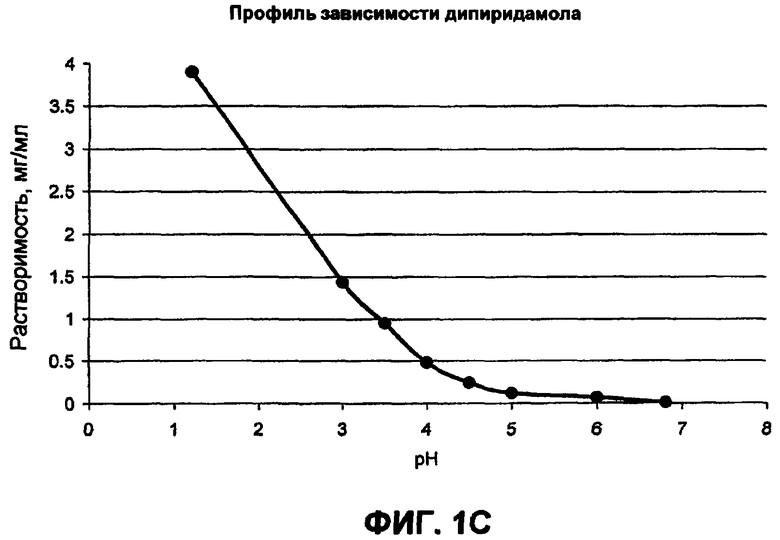
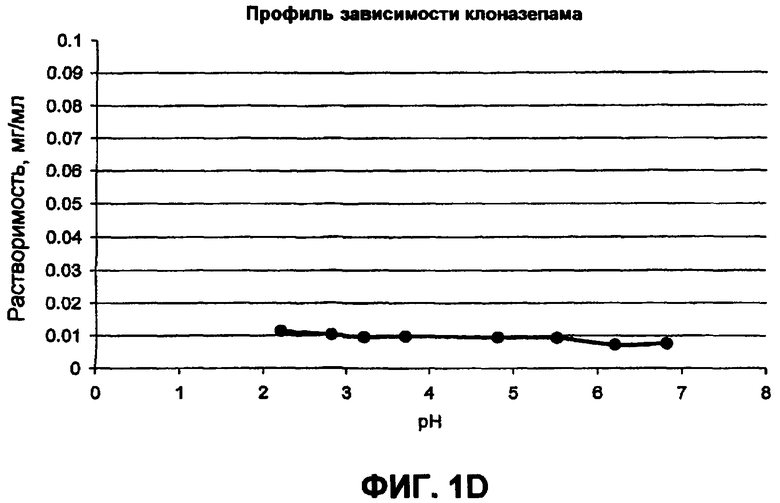
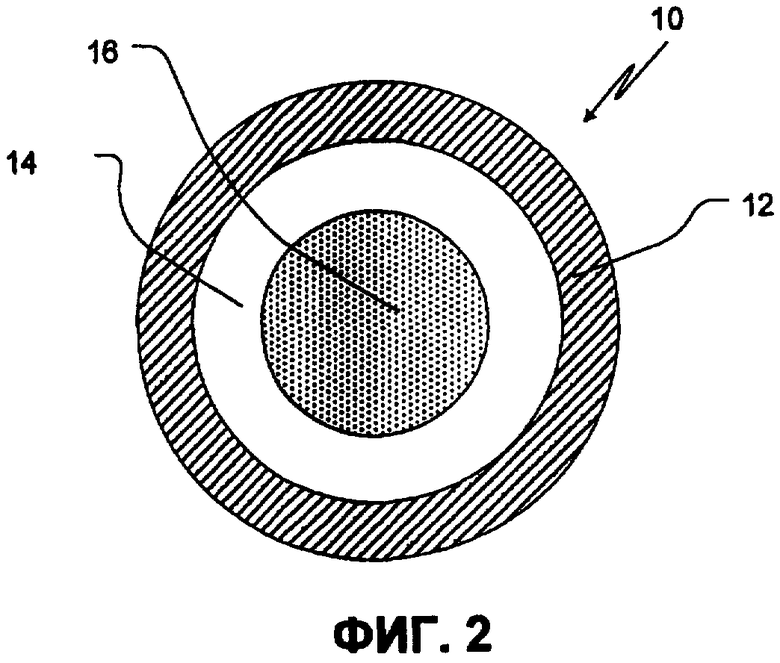
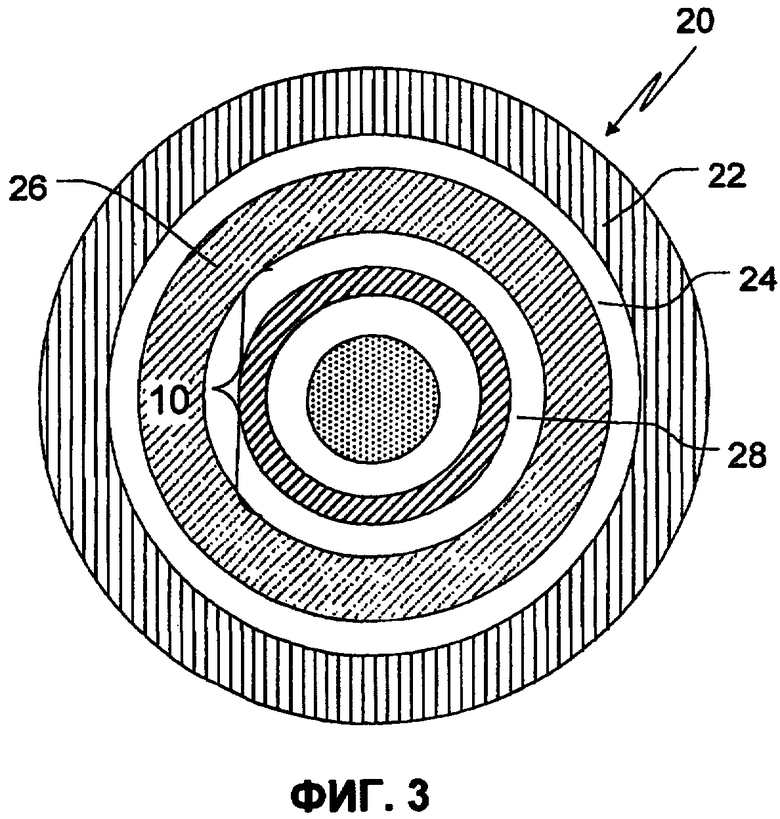
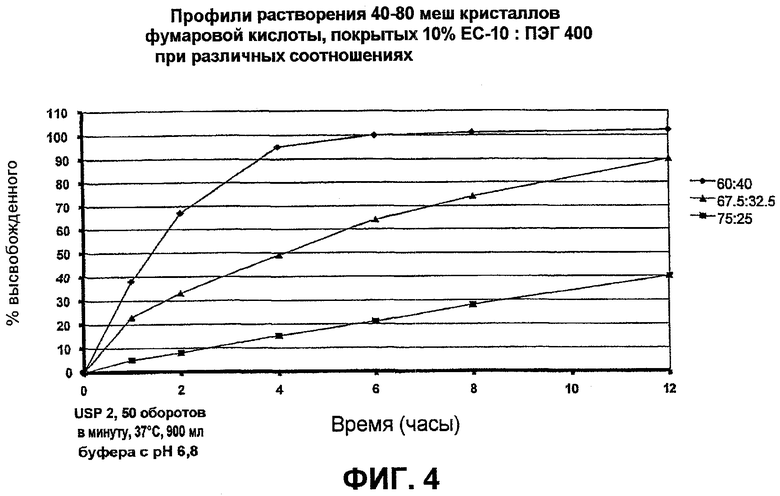
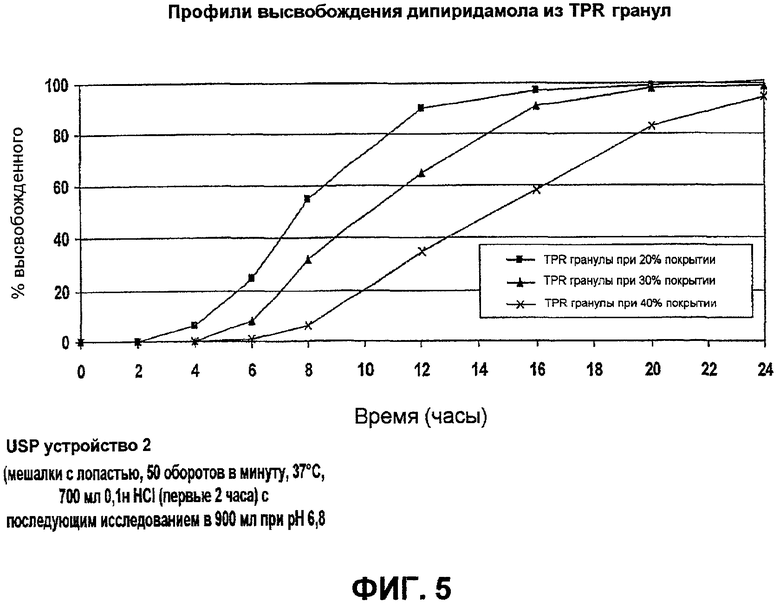
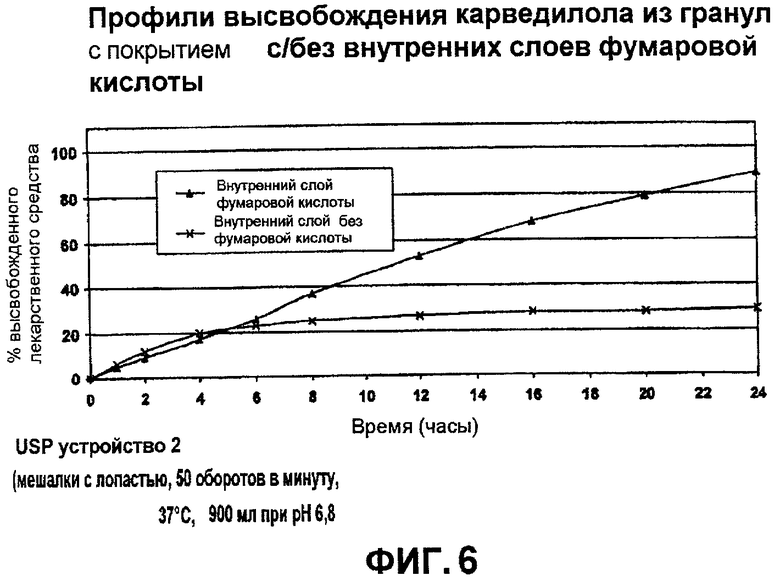
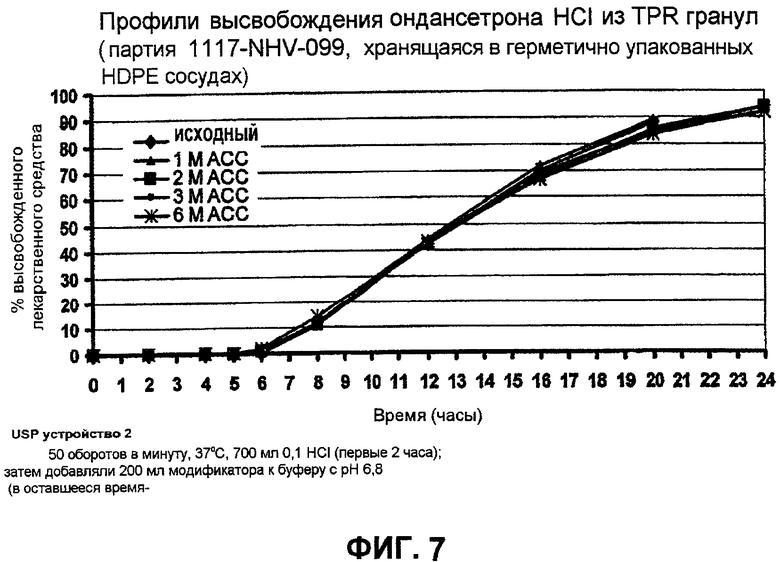
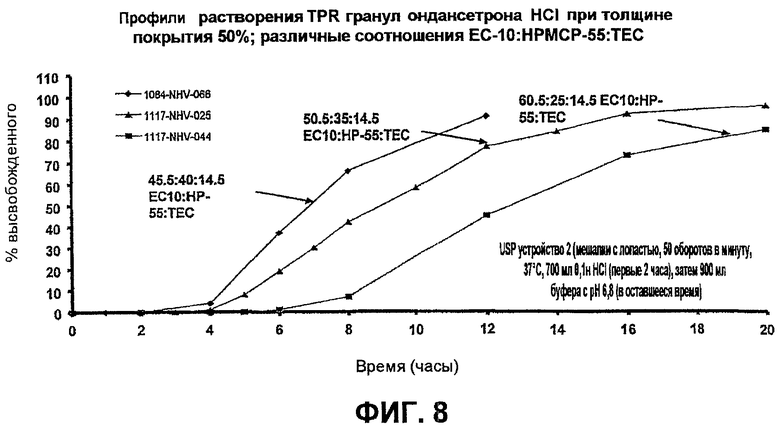
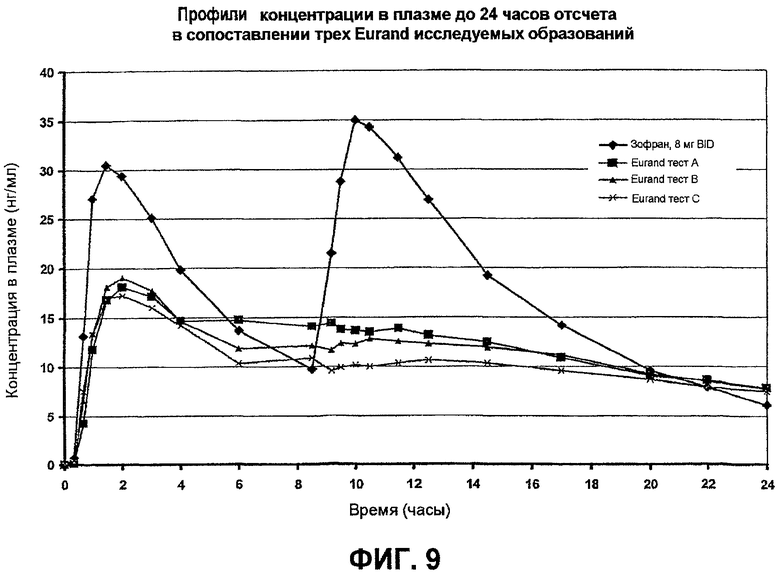
Изобретение относится к области медицины, а именно к химико-фармацевтической промышленности, в частности к лекарственной форме, такой как капсула, обычная или орально распадающаяся таблетка, способная к доставке азот-(N)-содержащих терапевтических средств, имеющих рКа в диапазоне от приблизительно 5 до 14 в организме в форме с замедленным высвобождением для того, чтобы быть подходящей для двухразового или одноразового суточного режима дозирования, включает, по меньшей мере, одну органическую кислоту, которая солюбилизирует лекарственное средство до его высвобождения в неблагоприятную окружающую среду кишечника, в которой указанное слабоосновное лекарственное средство практически нерастворимо. Единичная лекарственная форма, составленная из множества многослойных частичек, изготавливается таким способом, чтобы вышеупомянутое слабоосновное лекарственное средство и вышеупомянутая органическая кислота не вступали в близкий контакт с образованием in situ кислотных продуктов присоединения, гарантируя то, что кислота не закончится до завершения высвобождения лекарственнрго средства. 2 н. и 37 з.п. ф-лы, 2 табл., 9 ил.
1. Фармацевтическая лекарственная форма, состоящая из множества частиц, включающая одну или более популяций гранул с замедленным высвобождением (SR) и/или одну или более популяций гранул с пульсирующим регулируемым высвобождением (TPR), по меньшей мере, одного слабоосновного лекарственного средства, в которой слабоосновное лекарственное средство включает фармацевтически приемлемое азот-(N)-содержащее терапевтическое средство, или его фармацевтически приемлемую соль, имеющее рКа в диапазоне от приблизительно 5 до 14, и растворимость не более чем приблизительно 200 мкг/мл при рН 6,8, в которой SR гранулы включают частицы ядра из органической кислоты, покрытые SR слоем; TPR гранулы включают частицы ядра из органической кислоты, покрытые слоем, обеспечивающим высвобождение с задержкой по времени, где частицы ядра из органической кислоты, включают по меньшей мере одну фармацевтически приемлемую органическую кислоту и слабоосновное лекарственное средство, где слабоосновное лекарственное средство расположено поверх органической кислоты и где мембрана с замедленным высвобождением, включающая водонерастворимый полимер необязательно в комбинации с водорастворимым полимером или кишечнорастворимым полимером расположена между слабоосновным лекарственным средством и органической кислотой.
2. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.1, в которой соотношение оптимальной максимальной дозы слабоосновного лекарственного средства к растворимости при рН 6,8 не меньше, чем приблизительно 100, и указанное слабоосновное лекарственное средство практически нерастворимо.
3. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.1, в которой
a) указанные TPR гранулы включают внешнее обеспечивающее задержку во времени покрытие, включающее нерастворимый в воде полимер в комбинации с кишечнорастворимым полимером, расположенное на указанных SR гранулах, указанное покрытие обеспечивает время задержки от приблизительно 2 до приблизительно 7 ч перед началом высвобождения слабоосновного лекарственного средства;
b) указанные SR гранулы включают SR (защитное) покрытие, расположенное на указанных частицах ядра, из органической кислоты, указанное SR покрытие включает нерастворимый в воде полимер необязательно в комбинации с водорастворимым полимером.
4. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.1 в форме орально распадающейся таблетки (ОРТ).
5. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.1, в которой
а) указанные TPR гранулы включают внешнее покрытие, задерживающее время высвобождения, включающее нерастворимый в воде полимер в комбинации с кишечнорастворимым полимером, расположенное на частицах ядра из органической кислоты, указанное задерживающее высвобождение покрытие обеспечивает время задержки от приблизительно 2 до приблизительно 7 ч до начала высвобождения слабоосновного лекарственного средства.
6. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.1, включающая популяцию гранул быстрого высвобождения (IR гранул), первую TPR популяцию гранул и SR популяцию гранул, или IR популяцию гранул, первую TPR популяцию гранул и вторую TPR популяцию гранул, в которой соотношение IR популяции гранул к первой TPR популяции гранул, к SR популяции гранул или соотношение IR популяции гранул к первой TPR популяции гранул ко второй TPR популяции гранул варьирует от приблизительно 10:90:0 до приблизительно 40:10:50.
7. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.1, в которой указанное слабоосновное лекарственное средство выбирают из группы, состоящей из болеутоляющих средств, противосудорожных средств, антидиабетических средств, противоинфекционных средств, противоопухолевых средств, антипаркинсонических средств, противоревматических средств, сердечно-сосудистых средств, стимуляторов ЦНС (центральной нервной системы), агонистов допаминового рецептора, противорвотных средств, желудочно-кишечных средств, психотерапевтических средств, опиоидных агонистов, опиоидных антагонистов, антиэпилептических лекарственных средств, гистаминовых Нз антагонистов, антиастматических средств и релаксантов скелетных мышц.
8. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.7, в которой указанное слабоосновное лекарственное средство выбирают из группы, состоящей из оланзапина, ондансетрона, гидрохлорида ондансетрона, дипиридамола, карведилола, ламотригина, оланзапина, кветиапина, их фармацевтически приемлемых солей и их комбинаций.
9. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.1, в которой органическую кислоту выбирают из группы, состоящей из лимонной кислоты, фумаровой кислоты, яблочной кислоты, малеиновой кислоты, винной кислоты, янтарной кислоты, щавелевой кислоты, аспарагиновой кислоты, глутаминовой кислоты и их смесей.
10. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.1, в которой соотношение слабоосновного лекарственного средства к органической кислоте изменяется от приблизительно 5:1 до 1:10 по весу.
11. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.3, в которой указанные частицы ядра из органической кислоты представлены в форме:
i. кристалла органической кислоты;
ii. инертной частицы, покрытой органической кислотой и полимерным связующим веществом, или
iii. гранулы или микротаблетки, включающей органическую кислоту, полимерное связующее вещество и разбавитель/наполнитель.
12. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.11, в которой частицы ядра из органической кислоты включают слой лекарственного средства, включающий слабоосновное лекарственное средство и полимерное связующее вещество при соотношении лекарственного средства и связующего вещества от приблизительно 85:15 до приблизительно 99:1, и указанное полимерное связующее вещество выбрано из группы, состоящей из поливинилпирролидона, метилцеллюлозы, гидроксипропилцеллюлозы, гидроксипропилметилцеллюлозы, кукурузного крахмала, прежелатинированного крахмала и их смесей.
13. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.3, в которой мембрана с замедленным высвобождением включает один нерастворимый в воде полимер или нерастворимый в воде полимер в комбинации с водорастворимым полимером при соотношении водонерастворимый полимер/водорастворимый полимер от приблизительно 9:1 до 5:5, и в которой указанная мембрана с замедленным высвобождением нанесена для увеличения веса от приблизительно 1,5% до 20% по массе от массы SR-покрытых частиц ядра из органической кислоты.
14. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.13, в которой указанный нерастворимый в воде полимер выбран из группы, состоящей из этилцеллюлозы, ацетата целлюлозы, ацетата бутирата целлюлозы, поливинилацетата, нейтральных сополимеров метакриловой кислоты-метилметакрилата и их смесей.
15. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.13, в которой указанный растворимый в воде полимер выбран из группы, состоящей из метилцеллюлозы, гидроксипропилметилцеллюлозы, гидроксипропилцеллюлозы, поливинилпирролидона и полиэтиленгликоля и их смесей.
16. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.3, в которой указанное покрытие, задерживающее высвобождение, включает нерастворимый в воде полимер в комбинации с кишечнорастворимым полимером в соотношении от приблизительно 9:1 до 1:3 соответственно для увеличения массы приблизительно от 10% до 60% по массе от массы сухой ТPR гранулы.
17. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.16, в которой кишечнорастворимый полимер, выбран из группы, состоящей из ацетатфталата целлюлозы, фталата гидроксипропилметилцеллюлозы, сукцината гидроксипропилметилцеллюлозы, фталата поливинилацетата, рН-чувствительных сополимеров метакриловой кислоты-метилметакрилата, шеллака, их производных и их смесей.
18. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.3, в которой по меньшей мере одно из внутренних защитных покрытий и внешнее задерживающее высвобождение покрытие дополнительно включает пластификатор, выбранный из группы, состоящей из триацетина, трибутилцитрата, триэтилцитрата, ацетил три-н-бутилцитрата, диэтилфталата, дибутилсебацината, полиэтиленгликоля, полипропиленгликоля, касторового масла, ацетилированных моно- и диглицеридов и их смесей.
19. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.3, дополнительно включающая IR гранулы, которые высвобождают не меньше, чем приблизительно 50% слабоосновного лекарственного средства, содержащегося в указанных IR гранулах в течение первого часа после орального приема лекарственной формы.
20. Фармацевтическая лекарственная форма, состоящая из множества частиц по п.3, дополнительно включающая IR гранулы, которые включают указанное слабоосновное лекарственное средство и связующий полимер, нанесенный на инертное ядро.
21. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.1, включающая одну или более популяций TPR гранул, где указанное слабоосновное лекарственное средство включает карведилол или его фармацевтически приемлемую соль и органической кислотой является винная кислота, каждая популяция TPR гранул включает покрытие, задерживающее высвобождение, расположенное на SR гранулах, где задерживающий высвобождение слой включает этилцеллюлозу и фталат гидроксипропилметилцеллюлозы в соотношении от приблизительно 9:1 до приблизительно 1:3 для увеличения массы до 50%, и каждая популяция TPR гранул показывает при оральном приеме лекарственной формы заранее определенное время задержки и различающиеся характеристики высвобождения.
22. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.1, включающая одну или более популяций TPR гранул, где указанное слабоосновное лекарственное средство включает ондансетрон или его фармацевтически приемлемую соль, и органической кислотой является фумаровая кислота, каждая TPR популяция гранул включает покрытие, задерживающее высвобождение, расположенное на SR гранулах, где покрытие задерживающее высвобождение включает этилцеллюлозу и фталат гидроксипропилметилцеллюлозы в соотношении от приблизительно 9:1 до приблизительно 1:3 для увеличения массы от 5% до 60%, и каждая популяция TPR гранул показывает при оральном приеме лекарственной формы заранее определенное время задержки и различающиеся характеристики высвобождения.
23. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.1 в форме орально распадающейся таблетки, включающая SR популяцию гранул и/или одну или две популяции TPR гранул, в которой каждая SR или TPR популяция гранул включает соответственно внешнее SR покрытие или TPR покрытие, расположенное на частицах ядра из органической кислоты, включающая ядра фумаровой кислоты, покрытые мембраной с замедленным высвобождением, дополнительно покрытых слабоосновным лекарственным средством.
24. Способ изготовления лекарственной формы, состоящей из множества частиц по п.1, включающий
а. изготовление частиц ядра, включающих органическую кислоту, включающих по меньшей мере одну фармацевтически приемлемую органическую кислоту;
b. изготовление частиц ядра, содержащих органическую кислоту, покрытых мембраной с замедленным высвобождением, мембрана с замедленным высвобождением включает один нерастворимый в воде полимер, или водонерастворимый полимер в комбинации с растворимым в воде полимером или кишечнорастворимым полимером в соотношении от приблизительно 95/5 до приблизительно 50/50 для увеличения массы приблизительно на 20%;
с. изготовление частиц ядра из органической кислоты путем нанесения раствора, включающего слабоосновное лекарственное средство, или его фармацевтически приемлемую соль, и полимерное связующее и необязательно нанесение защитного герметичного слоя, содержащего растворимый в воде полимер, на частицы из органической кислоты, покрытые мембраной с замедленным высвобождением;
d. изготовление SR гранул путем нанесения SR (защитного) покрытия нерастворимого в воде полимера, одного, или нерастворимого в воде полимера в комбинации с растворимым в воде полимером в соотношении от приблизительно 95:5 до приблизительно 50:50 на частицы ядра из органической кислоты для увеличения массы приблизительно от 1,5% до 20% сухой массы покрытых SR гранул;
е. изготовление TPR гранул путем нанесения внешнего задерживающего высвобождение покрытия, включающего нерастворимый в воде полимер в комбинации с кишечнорастворимым полимером в соотношении к SR гранулам от приблизительно 9:1 до 1:3 для увеличения массы приблизительно от 10% до 60% от общей сухой массы TPR гранул; и
f. заполнение в капсулу или прессование в таблетку или орально распадающуюся таблетку, SR гранул и/или одной или более TPR популяций гранул в количествах, достаточных для обеспечения фармакокинетических профилей, пригодных для одноразового суточного режима дозирования для пациентов, нуждающихся в таком лечении.
25. Способ по п.24, в котором каждое из указанных покрытий или стадий нанесения включает покрытие или нанесение из раствора в фармацевтически приемлемой системе растворителей или из водной дисперсии.
26. Способ по п.24, в котором стадия (f) является стадией прессования в орально распадающуюся таблетку, и указанный способ дополнительно включает
i. маскировку вкуса SR гранулы и/или одной или более TPR популяций гранул коацервацией раствора либо покрытием в псевдоожиженном слое до прессования.
27. Способ по п.24, в котором указанная стадия прессования в орально распадающиеся таблетки включает прессование на таблеточном прессе, оборудованном внешней смазочной системой, чтобы смазывать матрицы и штампы до сжатия.
28. Способ п.24, в котором фармацевтическая лекарственная форма, состоящая из множества частиц, включает терапевтически эффективные количества SR гранул и/или одну или более популяций TPR гранул, где каждая из SR гранул и/или одна или более популяций TPR гранул показывает различающиеся характеристики высвобождения и предварительно определенное время задержки высвобождения.
29. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.1, в которой фармацевтически приемлемая органическая кислота не убывает из вышеупомянутой лекарственной формы до полного высвобождения слабоосновного лекарственного средства, при исследовании растворения методикой растворения согласно Фармакопее США (USP), с использованием двухстадийной среды растворения (сначала 2 ч в 0,1 н НСl с последующим исследованием в буфере при рН 6,8).
30. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.1, дополнительно включающая гранулы с быстрым высвобождением (IR).
31. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.2, в которой указанная лекарственная форма показывает фармакокинетический профиль через 24 ч, после приема дозы, подходящей для одноразового суточного режима дозирования пациентов, нуждающихся в этом.
32. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.1, в которой соотношение слабоосновного лекарственного средства к органической кислоте находится в диапазоне от приблизительно 5:1 до приблизительно 1:10.
33. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.4, в которой указанные ОРТ дополнительно включают быстродиспергирующиеся микрогранулы, где указанные быстрорастворяющиеся микрогранулы включают дезинтегратор и сахарный спирт или сахарид или их сочетание, и каждый из дезинтегратора и сахарного спирта или сахарида имеет средний размер частицы не больше, чем приблизительно 30 мкм.
34. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.33, в которой быстродиспергирующиеся микрогранулы имеют средний размер частицы не больше, чем 400 мкм.
35. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.33, в которой указанные ОРТ имеют хрупкость меньше, чем 1% по массе, и время дезинтеграции приблизительно 60 с или меньше при контакте со слюной.
36. Фармацевтическая лекарственная форма, состоящая из множества частиц, по п.3, включающая TPR гранулы, в которой задерживающее высвобождение покрытие включает этилцеллюлозу и фталат гидроксипропилметилцеллюлозы.
37. Способ по п.26, дополнительно включающий
ii. обеспечение спрессованного покрытия на указанных SR гранулах и/или одной или более популяций TPR гранул, где указанное спрессованное покрытие включает пластифицированный полимер, и посредством чего в течение прессования разрушение покрытия устраняется/минимизируется.
38. Способ по п.26, дополнительно включающий
h. гранулирование сахарного спирта или сахарида или их комбинации и дезинтегранта, причем каждое из которых имеет средний размер частицы не больше, чем приблизительно 30 мкм, для изготовления быстро диспергирующихся микрогранул;
iv. смешивание SR гранул и/или одной или более популяций TPR гранул с быстро диспергирующимися микрогранулами; и
v. прессование смеси со стадии (iv) в орально распадающиеся таблетки (ОРТ).
39. Способ по п.38, в котором соотношение SR гранул и/или одной или более TPR гранул к быстро распадающимся микрогранулам находится в соотношении от приблизительно 1:6 до приблизительно 1:2.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Промышленная технология лекарств./Под ред.проф | |||
| В.И.Чуешова | |||
| - Харьков, 2002, т.2, с.383-392, Глава 15. | |||

