ОБЛАСТЬ ИЗОБРЕТЕНИЯ
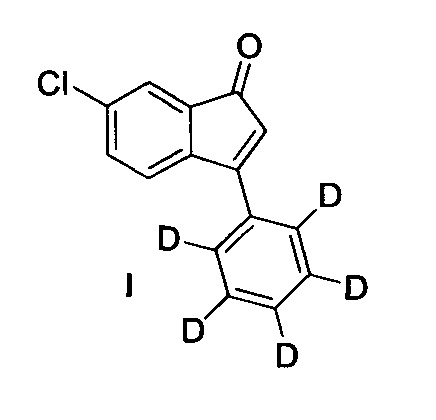
Настоящее изобретение относится к 6-хлор-3-(фенил-d5)-инден-1-ону, применению данного соединения, а также к способам получения данного соединения.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Дейтерированные 1-пиперазино-3-фенилинданы для лечения шизофрении были раскрыты в публикации патентной заявки США №13/527364. В публикации патентной заявки США №13/527364 также раскрыто, как определенные дейтерированные 1-пиперазино-3-фенилинданы можно получать с использованием 6-хлор-3-(фенил-d5)-индан-1-она. Однако раскрытые пути синтеза как рацемического, так и энантиомерно чистого 6-хлор-3-(фенил-d5)-индан-1-она построены на неэкономичном использовании больших загрузок либо хирального родиевого катализатора, либо хирального палладиевого катализатора. Поэтому требуются новые способы синтеза рацемического и энантиомерно чистого 6-хлор-3-(фенил-d5)-индан-1-она, которые и описаны в данном документе, причем в них используется 6-хлор-3-(фенил-d5)-инден-1-он (I) в качестве промежуточного соединения.
В публикации Clark, W. М. et al, Organic Letter, 1999, Vol. 1, No. 11, pp. 1839-1842 авторы описывают попытку получения 3-арилинденонов, таких как 6-хлор-3-(фенил-d5)-индан-1-он (I), имеющих электроноакцепторные группы (Cl, Br, NO2) в С(5)- или С(6)-положении в инденоновом кольце, используя реакцию Сузуки, но им не удалось получить значимые количества требуемого продукта. В отличие от этого, настоящее изобретение описывает удачное получение таких 3-арилинденонов, например, 6-хлор-3-(фенил-d5)-индан-1-она (I), с помощью реакции Сузуки.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
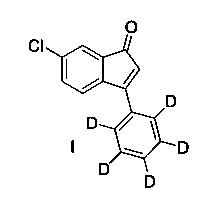
В настоящем изобретении раскрыто соединение 6-хлор-3-(фенил-d5)-инден-1-он (I)
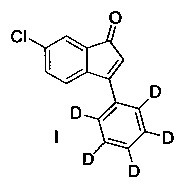
и пути синтеза для получения (I). В дополнительном аспекте в настоящем изобретении раскрыто применение 6-хлор-3-(фенил-d5)-инден-1-она (I) для получения рацемического 6-хлор-3-(фенил-d5)-индан-1-она (VIII) или (S)-6-хлор-3-(фенил-d5)-индан-1-она (IX).
Кроме того, в настоящем изобретении раскрыто применение 6-хлор-3-(фенил-d5)-инден-1-она (I) для получения 4-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазина и его фармацевтически приемлемых солей.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Перечень соединений
(I): 6-хлор-3-(фенил-d5)-инден-1-он
(II): 6-хлор-1-инданон
(III): 3-бром-6-хлоринден-1-он
(IV): 6-хлор-3-(фенил-d5)-1Н-инден
(V): 5-хлор-1-инданон
(VI): (±)-6-хлор-3-(фенил-d5)-1Н-инден-1-ол
(VIa): 6-хлор-3-(фенил-d5)-1H-инден-1-ол
(VII): (S)-6-хлор-3-(фенил-d5)-1Н-инден-1-ол
(VIII): (±)-6-хлор-3-(фенил-d5)-индан-1-он
(VIIIa): 6-хлор-3-(фенил-d5)-индан-1-он
(IX): (S)-6-хлор-3-(фенил-d5)-индан-1-он
(X): (±)-цис-6-хлор-3-(фенил-d5)-индан-1-ол
(Ха): (1S,3S)-6-хлор-3-(фенил-d5)-индан-1-ол
(Xb): 6-хлор-3-(фенил-d5)-индан-1-ол
(XI): (±)-цис-3,5-дихлор-1-(фенил-d5)-индан
(XIa): (1S,3S)-3,5-дихлор-1-(фенил-d5)-индан
(XIb): 3,5-дихлор-1-(фенил-d5)-индан
(XII): (±)-транс-1-(6-хлор-3-(фенил-d5)-индан-1-ил)-3,3-диметилпиперазина малеат
(XIIa): 1-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-3,3-диметилпиперазина малеат
(XIIb): Фармацевтически приемлемая соль 1-(6-хлор-3-(фенил-d5)-индан-1-ил)-3,3-диметилпиперазина
(XIII): (±)-транс-4-(6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазина сукцинат
(XIIIa): 4-(6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазин
(XIV): 4-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазина L-(+)-тартрат
(XV): 4-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазина фумарат
(XVa): Фармацевтически приемлемая соль 4-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазина
(XVI): 1(d3),2,2-триметилпиперазина бис-2,2,2-трифторацетат
(XVII): 2,2-диметилпиперазин
(XVIII): трет-бутил 3,3-диметилпиперазин-1-карбоксилата геми-D,L-тартрат
(XIX): (E)-1-(6-хлор-3-фенил(d5)-1Н-инден-1-илиденметил)-N,N-диметиламин
Настоящее изобретение предлагает соединение (I)
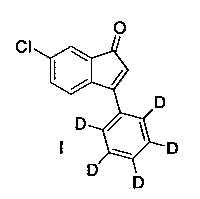
и пути синтеза для получения данного соединения, а также применение данного соединения для получения 4-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазина фармацевтически приемлемых солей 4-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазина.
В настоящем изобретении раскрывается, как осуществлять синтез соединения (I) с помощью первой стадии, включающей синтез соединения (III), за которой следует вторая стадия, в ходе которой (III) взаимодействует с арилбороновой кислотой или ее сложным эфиром эфиром, например, 4,4,5,5-тетраметил-2-d5-фенил-[1,3,2]диоксабороланом, в присутствии подходящего катализатора, например, ацетата палладия(II), и основания, например, фосфата калия.
В настоящем изобретении дополнительно раскрывается, как осуществлять синтез соединения (I), исходя из соединения (IV), путем окисления енамина (XIX), например, в присутствии периодата.
В дополнительном аспекте настоящего изобретения раскрыто применение соединения (I) для получения соединения (VIII) или (IX) любым из следующих путей.
(1) Восстановлением с последующей перегруппировкой с целью получения соединения (VIII) (путь В и D2 в Схеме 1, ниже)
(2) Энантиоселективным восстановлением с последующей перегруппировкой с целью получения соединения (IX) (путь С и Е1 в Схеме 1, ниже)
(3) Гидрированием с целью получения соединения (VIII) (путь D1 в Схеме 1, ниже)
(4) Органокаталитическое гидрирование с асимметричным переносом водорода с целью получения соединения (IX) (путь Е2 в Схеме 1, ниже)
(5) Асимметричное гидрирование с целью получения соединения (IX) (путь Е3 в Схеме 1, ниже).
Эти пути синтеза изобретения можно обобщить следующим образом:
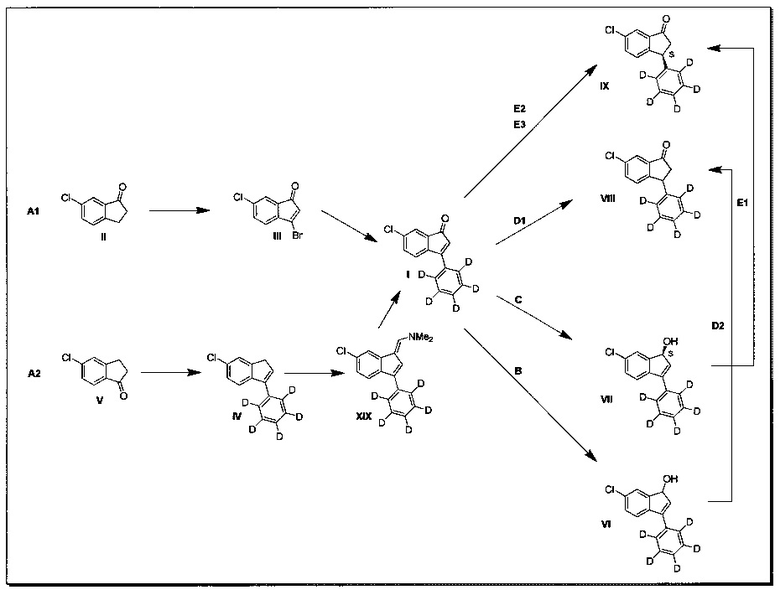
Схема 1: Получение соединений (I), (VI)/(VIa), (VII), (VIII)/(VIIIa) и (IX)
В дополнительном аспекте настоящего изобретения раскрыто применение соединения (VIII), полученного как описано выше, для приготовления соединения (XIV) через промежуточные соединения (X), (XI), (XII) и (XIII) (путь А и В в Схеме 2).
В дополнительном аспекте настоящего изобретения раскрыто применение соединения (IX), полученного как описано выше, для приготовления соединения (XV) или соединения (XIV) через промежуточные соединения (Ха) и (XIa) (путь С в Схеме 2).
В дополнительном аспекте настоящего изобретения раскрыто применение соединения (IX), полученного как описано выше, для приготовления соединения (XV) или соединения (XIV) через промежуточные соединения (Ха), (XIa) и (XIIa) (путь А и В в Схеме 2).
В другом аспекте настоящего изобретения раскрыто получение соединения (XVI) из соединения (XVII) через промежуточное соединение (XVIII) (Схема 2).
Данные пути синтеза изобретения можно обобщить следующим образом:
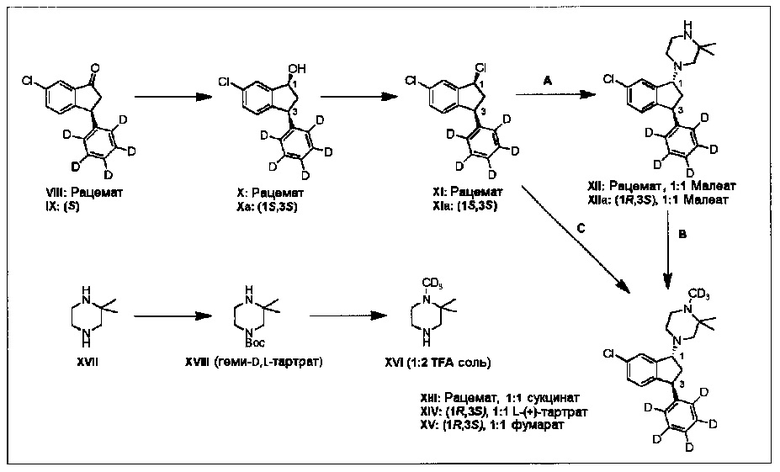
Схема 2: Получение соединений (XIII), (XIV), (XV) и (XVI)
Настоящее изобретение будет проиллюстрировано в следующих неограничивающих примерах.
Варианты осуществления согласно настоящему изобретению
Если не указано иначе, ссылка на любое из соединений в вариантах осуществления ниже охватывает энантиомерно чистую форму соединения или смеси энантиомеров в любом соотношении. Например, соединение (VIIIa) 6-хлор-3-(фенил-d5)-индан-1-он относится к рацемической смеси (VIIIa), т.е. (±)-6-хлор-3-(фенил-d5)-индан-1-ону, а также к энантиомерным изомерам (VIIIa) в любом соотношении.
В первом варианте осуществления (Е1) настоящее изобретение относится к соединению, имеющему структуру (I) (которое также упоминается, как соединение формулы (I) или соединение (I))
В (Е2) соединение (I) из (Е1) получают реакцией 3-бром-6-хлоринден-1-она (III) с бороновой кислотой фенила-с-5 или сложным эфиром бороновой кислоты фенила-d5.
В дополнительном варианте осуществления (Е3) варианта (Е2) соединение (III) получают, используя (II) в качестве исходного материала.
В дополнительном варианте осуществления (Е4) варианта (Е3) синтез соединения (I) включает следующие стадии:
1. Бромирование соединения (II), например, путем введения 2,2'-азо-бис-изобутиронитрила и N-бромсукцинимида в раствор, содержащий 6-хлор-1-инданон (II).
2. Отщепление, опосредованное основанием, путем введения основания, например, триэтиламина, в раствор, полученный в стадии 1, с целью получения 3-бром-6-хлоринден-1-она (III).
3. 3-бром-6-хлоринден-1-он (III), полученный в стадии 2, по желанию отделяют и приводят во взаимодействие с фенил-d5 бороновой кислотой или эфиром, например, 4,4,5,5-тетраметил-2-d5-фенил-[1,3,2]диоксабороланом, в присутствии подходящего катализатора и основания с целью получения соединения (I).
В одном варианте осуществления (Е5) варианта (Е1) синтез соединения (I) включает следующие стадии:
1. Синтез 6-хлор-3-(фенил-d5)-1Н-индена (IV) путем взаимодействия органометаллических частиц (полученных из моногалогенированного бензола-d5) и 5-хлор-1-инданона (V) с последующей дегидратацией.
2. Преобразование 6-хлор-3-(фенил-d5)-1Н-индена (IV) в соединение (XIX) и его последующее окислительное расщепление с получением соединения (I).
В варианте осуществления (Е6) варианта осуществления (Е5) соединение (I) получают способом, включающим:
1. Синтез 6-хлор-3-(фенил-d5)-1Н-индена (IV) реакцией Гриньяра с участием бромбензола-d5, магния и 5-хлор-1-инданона (V) с последующей дегидратацией.
2. Взаимодействие 6-хлор-3-(фенил-d5)-1Н-индена (IV) с 1,1-диметокси-N,N-диметилметанамином с последующим окислительным расщеплением образовавшегося соединения (XIX) с целью получения соединения (I).
В дополнительном варианте осуществления (Е7) вариантов осуществления (Е5) и (Е6) окислительное расщепление в синтезе соединения (I) проводят, используя окислитель, выбранный из группы, состоящей из метапериодата натрия, метапериодата калия, озона, дихромата калия, дихромата натрия, синглетного кислорода и m-хлорпербензойной кислоты.
В определенном варианте осуществления (Е8) варианта осуществления (Е7) окислительное расщепление проводят, используя метапериодат натрия.
В варианте осуществления (Е9) соединение (I), полученное в (Е1), восстанавливают с целью получения (VIa), в частности, (±)-6-хлор-3-(фенил-d5)-1Н-инден-1-ола (VI).
В дополнительном варианте осуществления (Е10) варианта осуществления (Е9) восстановление происходит в присутствии восстановителей, выбранных из группы, состоящей из борогидрида натрия, борогидрида магния, борогидрида кальция, борогидрида лития, триацетоксиборогидрида натрия, триацетоксиборогидрида лития, алюмогидрида лития, бис(2-метоксиэтокси)алюмодигидрида натрия, диизобутилалюмогидрида и триэтилборогидрида лития.
В определенном варианте осуществления (Е11) варианта осуществления (Е10) восстановление происходит в присутствии диизобутилалюмогидрида.
В варианте осуществления (Е12) соединение (VIa), полученное в (Е9), преобразуют в (VIIIa), в частности, в (±)-6-хлор-3-(фенил-d5)-индан-1-он (VIII), путем перегруппировки в присутствии основания.
В варианте осуществления (Е13) соединение (I), полученное в (Е1), преобразуют в (S)-6-хлор-3-(фенил-d5)-1H-инден-1-ол (VII) путем энантиоселективного восстановления.
В определенном варианте осуществления (Е14) варианта осуществления (Е13) энантиоселективное восстановление происходит в присутствии энантиоселективного катализатора, а восстановители выбирают из группы, состоящей из энантиомерно чистого 2-метил-CBS-оксазаборолидина, о-толил-CBS-оксазаборолидина, 2-бутил-CBS-оксазаборолидина, Alpine-Borane® и В-хлордиизопинокамфеилборана.
В определенном варианте осуществления (Е15) варианта осуществления (Е14) энантиоселективное восстановление происходит в присутствии энантиомерно чистого 2-метил-CBS-оксазаборолидина.
В варианте осуществления (Е16) вариантов осуществления (Е13)-(Е15) соединение (VII) преобразуют в (S)-6-хлор-3-(фенил-d5)-индан-1-он (IX) путем перегруппировки в присутствии основания.
В дополнительном варианте осуществления (Е17) любого из (Е12) и (Е16) опосредованная основанием перегруппировка происходит в присутствии подходящего основания, выбранного из группы, состоящей из 1,4-диазабицикло[2.2.2]октана, бис(триметилсилил)амида калия и бис(триметилсилил)амида лития.
В определенном варианте осуществления (Е18) варианта осуществления (Е17) опосредованная основанием перегруппировка происходит в присутствии 1,4-диазабицикло[2.2.2]октана.
В варианте осуществления (Е19) соединение (I), полученное в (Е1), преобразуют с целью получения (VIIIa), в частности, (±)-6-хлор-3-(фенил-d5)-индан-1-она (VIII), путем гидрирования в присутствии подходящего катализатора в подходящем растворителе.
В определенном варианте осуществления (Е20) варианта осуществления (Е19) соединение (I) преобразуют в соединение (VIII) в присутствии хлорида трис(трифенилфосфин)родия(I).
В определенном варианте осуществления (Е21) варианта осуществления (Е19) растворитель представляет собой этилацетат.
В варианте осуществления (Е22) соединение (I), полученное в (Е1), преобразуют в (S)-6-хлор-3-(фенил-d5)-индан-1-он (IX), путем асимметричного гидрирования в присутствии подходящего катализатора в подходящем растворителе.
В дополнительном варианте осуществления (Е23) варианта осуществления (Е22) асимметричное гидрирование соединения (I) проводят в присутствии соли родия.
В дополнительном варианте осуществления (Е24) любого из (Е22) и (Е23) асимметричное гидрирование соединения (I) проводят в присутствии хирального фосфинового лиганда.
В определенном варианте осуществления (Е25) варианта осуществления (Е23) соль родия выбирают из группы, состоящей из трифторметансульфоната бис(норборнадиен)родия(I), тетрафторбората бис(норборнадиен)родия(I), трифторметансульфоната бис(1,5-циклооктадиен)родия(I), тетрафторбората бис(норборнадиен)родия(I) и тетракис[(бис(3,5-трифторметил)фенил]бората бис(1,5-циклооктадиен)родия(I).
В определенном варианте осуществления (Е26) варианта осуществления (Е24) хиральный фосфиновый лиганд выбирают из группы, состоящей из (R)-(-)-5,5'-бис[ди(3,5-ди-трет-бутил-4-метоксифенил)фосфино]-4,4,-би-1,3-бензодиоксола ((R)-DTBM-SEGPHOS), (S)-(+)-4,12-бис(дифенилфосфино)-[2.2]-парациклофана ((S)-Phanephos) и (S)-(+)-4,12-бис[ди(3,5-ксилил)фосфино]-[2.2]-парациклофана ((S)-DM-Phanephos).
В определенном варианте осуществления (Е27) варианта осуществления (Е22) растворитель представляет собой этилацетат.
В варианте осуществления (Е28) соединение (VIII) любого из вариантов осуществления (Е12) и (Е19) преобразуют в (Xb), в частности, в (±)-цис-6-хлор-3-(фенил-d5)-индан-1-ол (X).
В варианте осуществления (Е29) любого из вариантов осуществления (Е16)-(Е18) и (Е22)-(Е27) (S)-6-хлор-3-(фенил-d5)-индан-1-он (IX) преобразуют в (1S,3S)-цис-6-хлор-3-(фенил-d5)-индан-1-ол (Ха).
В варианте осуществления (Е30) варианта осуществления (Е28) (Xb) преобразуют в (XIb) хлорированием, в частности, (±)-цис-6-хлор-3-(фенил-d5)-индан-1-ол (X) преобразуют в (±)-цис-3,5-дихлор-1-(фенил-d5)-индан (XI) путем хлорирования.
В варианте осуществления (Е31) варианта осуществления (Е29) (1S,3S)-6-хлор-3-(фенил-d5)-индан-1-ол (Ха) преобразуют в (1S,3S)-3,5-дихлор-1-(фенил-d5)-индан (XIa) путем хлорирования.
В дополнительном варианте осуществления (Е32) любого из (Е30) и (Е31) хлорирование происходит в присутствии реагента, выбранного из группы, состоящей из тионилхлорида, оксихлорида фосфора и пентахлорида фосфора.
В определенном варианте осуществления (Е33) варианта осуществления (Е32) хлорирование происходит в присутствии тионилхлорида.
В варианте осуществления (Е34) варианта осуществления (Е30) 3,5-дихлор-1-(фенил-d5)-индан (XIb) преобразуют в (XIIb) путем нуклеофильного замещения, используя 2,2-диметилпиперазин или соединение, которое впоследствии можно преобразовать в 3,3-диметилпиперазиновый фрагмент в (XIIb); в частности, (XI) преобразуют в (XII) нуклеофильным замещением, используя 2,2-диметилпиперазин или соединение, которое впоследствии можно преобразовать в 3,3-диметилпиперазиновый фрагмент в (XII). В варианте осуществления (Е35) варианта осуществления (Е31) (1S,3S)-3,5-дихлор-1-(фенил-d5)-индан (XIa) преобразуют в соединение (XIIa) нуклеофильным замещением, используя 2,2-диметилпиперазин или соединение, которое впоследствии можно преобразовать в 3,3-диметилпиперазиновый фрагмент соединения (XIIa). В дополнительном варианте осуществления (Е36) любого из (Е34) и (Е35) нуклеофильное замещение проводят, используя 2,2-диметилпиперазин, в присутствии основания.
В определенном варианте осуществления (Е37) варианта осуществления (Е36) основание представляет собой карбонат, например, карбонат калия.
В варианте осуществления (Е38) соединение (XII) преобразуют в соединение (XIII) путем алкилирования.
В варианте осуществления (Е39) соединение (XIIa) преобразуют в соединение (XVa), такое как (XIV) или (XV), путем алкилирования.
В дополнительном варианте осуществления (Е40) любого из (Е38) и (Е39) алкилирование проводят в присутствии активного донора метил-d3 и основания.
В определенном варианте осуществления (Е41) варианта осуществления (Е40) активный донор метила выбирают из группы, состоящей из иодида метила-d3, бромида метила-d3 и диметилсульфата-d6.
В определенном варианте осуществления (Е42) любого из (Е40) и (Е41) активный донор метила представляет собой метилиодид-d3.
В определенном варианте осуществления (Е43) варианта осуществления (Е40) основание выбирают из группы, состоящей из гидроксида натрия и калия, карбоната натрия и калия и трет-бутоксида натрия и калия.
В определенном варианте осуществления (Е44) любого из (Е40) и (Е43) основание представляет собой гидроксид калия. В варианте осуществления (Е45) (1S,3S)-3,5-дихлор-1-(фенил-d5)-индан (XIa) из (Е31) преобразуют в соединение (XIV) или (XV) путем нуклеофильного замещения, используя соединение (XVI) или соединение, которое впоследствии можно преобразовать в 1(d3),2,2-триметилпиперазиновый фрагмент соединения (XIV) или соединения (XV).
В дополнительном варианте осуществления (Е46) варианта осуществления (Е45) нуклеофильное замещение проводят, используя соединение (XVI) в присутствии основания.
В определенном варианте осуществления (Е47) варианта осуществления (Е46) основание выбирают из группы, состоящей из гидроксида натрия и калия, карбоната натрия и калия и трет-бутоксида натрия и калия.
В определенном варианте осуществления (Е48) варианта осуществления (Е47) основание представляет собой карбонат калия.
В определенном варианте осуществления (Е49) варианта осуществления (Е46) соединение (XVI) получают из соединения (XVII) через промежуточное соединение (XVIII).
В варианте осуществления (Е50) любого из предшествующих вариантов осуществления реакции проводят, используя соединения, содержащие водород (Н) вместо дейтерия (D), таким образом предоставляя соответствующие недейтерированные соединения.
В варианте осуществления (Е51) любого из вариантов осуществления (Е34), (Е35), (Е38), (Е39), (Е45) и (Е50) реакции могут проводиться в целях получения любой другой фармацевтически приемлемой соли соединений (XII), (XIIa), (XIII), (XIV) и (XV).
Определения
Энантиомерный избыток определяется как абсолютная разница между мольными фракциями каждого энантиомера.
Энантиомерный избыток, выраженный в процентах, вычисляют как
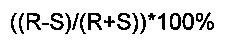
где R и S представляют собой соответствующие мольные фракции энантиомеров в смеси, таким образом, что R+S=1.
Настоящее изобретение будет проиллюстрировано в следующих неограничивающих примерах.
Соединения, описанные в данном документе, предназначены обозначать любую форму соединения, такую как свободное основание, его фармацевтически приемлемые соли, например, фармацевтически приемлемые соли присоединения кислот, такие как сукцинат, тартрат, в частности, L(+)-тартрат, и малонаты, гидраты или сольваты свободного основания или его солей, а также безводные формы, аморфные формы, кристаллические формы и растворы.
Фармацевтически приемлемые соли соединений настоящего изобретения включают фармацевтически приемлемые соли присоединения кислот. Соли присоединения кислот включают соли неорганических кислот, а также органических кислот. Типичные примеры подходящих неорганических кислот включают хлористоводородную, бромистоводородную, йодистоводородную, фосфорную, серную, сульфаминовую, азотную кислоты и т.п. Типичные примеры подходящих органических кислот включают муравьиную, уксусную, трихлоруксусную, трифторуксусную, пропионовую, бензойную, коричную, лимонную, фумаровую, гликолевую, итаконовую, молочную, метансульфоновую, малеиновую, яблочную, малоновую, миндальную, оксалиновую, пикриновую, пировиноградную, салициловую, янтарную, метансульфоновую, этансульфоновую, винную, аскорбиновую, памовую, бисметиленсалициловую, этандисульфоновую, глюконовую, цитраконовую, аспарагиновую, стеариновую, пальмитиновую, EDTA, гликолевую, п-аминобензойную, глутаминовую, бензолсульфоновую, п-толуолсульфоновую кислоты, теофиллин-уксусные кислоты, а также 8-галогентеофиллины, например, 8-бромтеофиллин и т.п.
Если не указано иначе, ссылка на любое из соединений, раскрытых в данной заявке, охватывает энантиомерно чистую форму соединения, а также смеси энантиомеров в любом соотношении.
Экспериментальная часть
Общая экспериментальная часть
Все реакции проводили в атмосфере азота, если не указано иначе. За ходом реакций следили, используя тонкослойную хроматографию (ТСХ) и/или ЖХ-МС. Все реагенты приобретали и использовали без дополнительной очистки. Пятна визуализировали с помощью ультрафиолетового (УФ) излучения (254 нм) или путем выдерживания в 5%-ном (вес/вес.) растворе фосфомолибденовой кислоты (ФМК) в этаноле или в щелочном водном перманганате калия (KMnO4) с последующим нагреванием. Колоночный хроматографический анализ проводили, используя силикагель Merck С60 (40-63 мкм, 230-240 меш). Спектры ЯМР записывали при 250, 500 или 600 МГц (1Н ЯМР) и калибровали относительно пика остаточного растворителя. Для данных ЯМР использовали следующие аббревиатуры: s - синглет; d - дублет; t - триплет; m - мультиплет. Константы взаимодействия округляли до ближайших 0,5 Гц. Энантиомерный избыток определяли методом хиральной ВЭЖХ.
Разделение рацемических соединений можно проводить, как описано, например, в публикациях WO 12/093165 и WO 11/003423.
Метод ЖХ-МС
Колонка Acquity UPLC ВЕН С18 1,7 мкм; 2,1 × 50 мм, работающая при 60°С с расходом 1,2 мл/минуту бинарного градиента, состоящего из воды + 0,1% муравьиной кислоты (А) и ацетонитрила + 5% воды + 0,1% муравьиной кислоты (В). УФ детектирование при 254 нм.
Метод хиральной ВЭЖХ
Колонка Phenomenex Lux 5μ Cellulose-2; 250 × 4,6 мм, работающая при 30°С с расходом 0,5 или 1,0 мл/минуту смеси н-гексан : изопропанол : диэтиламин, 90:10:0.1. УФ детектирование при 220 нм.
Методы ВЭЖХ
Метод 1: Колонка Chromolith Performance Rp-18e 2 μ; 100 × 4,6 мм, работающая при 30°С с расходом 2,0 мл/минуту смеси вода:триэтиламин:ацетонитрил, 1000:5.5:1000 со значением рН 3, полученным с помощью H3PO4. УФ детектирование при 254 нм.
Метод 2: Колонка Agilent Zorbax SB-Phenyl 3.5 μ; 150 × 4,6 мм, работающая при 40°С с расходом 1,0 мл/минуту. УФ детектирование при 220 нм. Подвижная фаза А: вода + трифторуксусная кислота = 1000 + 0,5 мл; подвижная фаза В: ацетонитрил + трифторуксусная кислота = 1000 + 0,5 мл. Градиент: 0 мин: 90% А, 10% В, 20 мин: 5% А, 95% В, 25 мин: 5% А, 95% В, 25,1 мин: 90% А, 10% В, 30 мин: 90% А, 10% В.
Метод 3: Колонка Phenomenex Luna С18 3.0 μ; 150 × 4,6 мм, работающая при 40°С с расходом 1,0 мл/минуту. УФ детектирование при 220 нм. Подвижная фаза А: 25 мМ фосфатный буфер рН 7.4 : ацетонитрил = 40:60; подвижная фаза В: вода : ацетонитрил = 10:90. Градиент: 0 мин: 100% А, 0% В, 32 мин: 100% А, 0% В, 35 мин: 50% А, 50% В, 37 мин: 50% А, 50% В, 39 мин: 100% А, 0% В, 40 мин: 100% А, 0% В.
Метод ГХ
Rtx-5 amine 0.5 μ; 30 m × 0,25 мм с расходом гелия 1 мл/минуту. Детектирование ПИД (пламенно-ионизационный детектор) (250°С). Градиент: 0 мин: 50°С; 9 мин: 140°С; 11 мин: 140°С; 21 мин: 240°С; 23 мин 240°С; 26 мин: 300°С; 28 мин: 300°С.
Синтез соединений данного изобретения
А. Синтез 6-хлор-3-(фенил-d5)-инден-1-она (I)

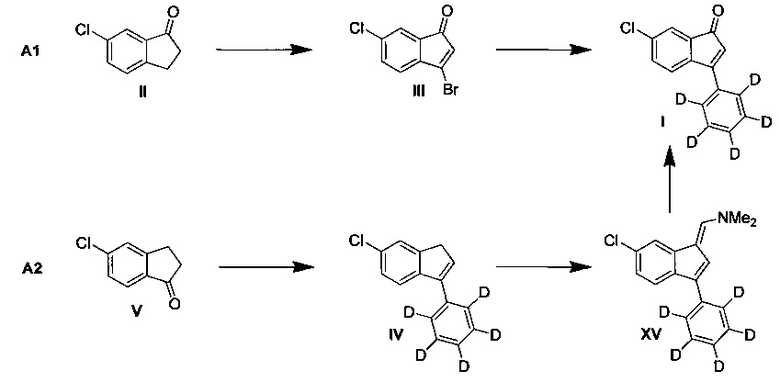
А1. С помощью реакции Сузуки (Схема 3, путь А1):
Синтез 3-бром-6-хлоринден-1-она (III)
К раствору 6-хлор-1-инданона (II) (100,0 г, 600,2 ммоль) в 1,2-дихлорэтане (1,00 л) добавляли 2,2'-азо-бис-изобутиронитрил (9,86 г, 60,0 ммоль), а затем - N-бромсукцинимид (224,3 г, 1,26 моль). Реакционную смесь быстро нагревали до кипячения с обратным холодильником. Через 30 минут после начала кипячения с обратным холодильником вводили дополнительное количество 2,2'-азо-бис-изобутиронитрила (9,86 г, 60,0 ммоль). Реакционную смесь выдерживали при кипячении с обратным холодильником в течение 4,5 часа. Затем смесь перемешивали при комнатной температуре в течение ночи. Смесь охлаждали до 0°С, и по каплям добавляли триэтиламин (126 мл, 904 ммоль). Смесь перемешивали 1 час при 0°С, а затем давали нагреться до комнатной температуры. Добавляли воду (1,0 л). Смесь интенсивно перемешивали 15 минут. Перемешивание прекращали и водный слой удаляли отсасыванием. Добавляли свежую воду (1,0 л) и смесь перемешивали 15 минут. Водный слой затем отсасывали. Органическую фазу дополнительно встряхивали с соляным раствором (500 мл) в разделительной воронке.
Органический слой отделяли и перемешивали с MgSO4 и активированным углем в течение 30 мин. Смесь фильтровали через слой целита. Фильтрат выпаривали досуха в вакууме. Таким образом получали неочищенный 3-бром-6-хлоринден-1-он (III) (190 г) в виде твердого вещества, которое затем использовали в следующей стадии без дополнительной очистки.
Синтез 6-хлор-3-(фенил-d5)-инден-1-она (I)
К сырому 3-бром-6-хлоринден-1-ону (III), полученному выше, добавляли ацетат палладия (5,78 г, 25,8 ммоль), трифенилфосфин (13,5 г, 51,5 ммоль) и 4,4,5,5-тетраметил-2-d5-фенил-[1,3,2]диоксаборолан (116 г, 566 ммоль), а затем - ТГФ (1,50 л) при комнатной температуре. Добавляли воду (750 мл) и фосфат калия (115 г, 541 ммоль). Реакционную смесь интенсивно перемешивали при комнатной температуре в течение 2 часов. Образовался темный, почти черный раствор. Добавляли гептан (0,70 л). Затем органическую фазу промывали водой (1,0 л) и соляным раствором (0,5 л), сушили над MgSO4, фильтровали и выпаривали до сухого состояния в вакууме. В результате получали сырой 6-хлор-3-(фенил-d5)-инден-1-он (I) в виде темного твердого вещества. Сырой 6-хлор-3-(фенил-d5)-инден-1-он (I) растворяли в смеси гептан-EtOAc (2:1) и раствор фильтровали через силикагель. Фильтрат выпаривали досуха в вакууме. Остаток повторно осаждали из гептана, растворяя в кипящем гептане, фильтруя в горячем состоянии и позволяя медленно охладиться до комнатной температуры, в результате получая 6-хлор-3-(фенил-d5)-инден-1-он (I) (75,8 г, 52%) в виде темно-оранжевого твердого вещества, имеющего чистоту 95% согласно результатам анализа ЖХ-МС.
Аналитические данные для 6-хлор-3-(фенил-d5)-инден-1-она (I):
1Н ЯМР (600 МГц, CDCl3 (дейтерированный хлороформ)) δН 6,04 (s, 1Н), 7,32 (d, J=8,0 Гц, 1Н), 7,37 (dd, J=2,0, 8,0 Гц, 1Н), 7,49 (dd, J=0,5, 2,0 Гц, 1Н); 13С ЯМР (150 МГц, CDCl3) δC 122,7, 123,1, 123,5, 127,2 (t, J=23,5 Гц), 128,7 (t, J=23,5 Гц), 130,6 (t, J=23,5 Гц), 132,2, 132,6, 134,2, 134,4 (t, J=23,5 Гц), 135,7, 142,1, 162,8; ЖХ-МС (APPI-фотоионизация при атмосферном давлении): масса/заряд рассчетн. для C15H5D5ClO [М+Н]+ 246,1, эксперим. 246,1.
А2. Путем окисления (Схема 3, путь А2):
Пример 1:
Синтез 6-хлор-3-(фенил-d5)-1Н-индена (IV)
К суспензии магния (4,43 г, 182 ммоль) в ТГФ (15,0 мл) добавляли Red-Al (бис(2-метоксиэтокси)алюмогидрид натрия) (0,50 мл, 1,67 ммоль, 65 масс. % в толуоле). Небольшое количество (примерно 5 мл) раствора бромбензол-d5 (29,3 г, 181 ммоль) в ТГФ (100 мл) добавляли при комнатной температуре. Смесь осторожно нагревали (40-50°С), что приводило к инициированию реакции. Начало реакции определяли по экзотермическому эффекту и оставшийся раствор бромбензола-d5 добавляли по каплям так, чтобы поддерживать устойчивое кипение с обратным холодильником, вся процедура заняла 35 минут. После этого смесь нагревали при кипячении с обратным холодильником в течение 1,5 ч. Полученную смесь охлаждали до комнатной температуры и раствор декантировали (используя иглу), отделяя избыток магния. К этому раствору добавляли раствор 5-хлор-1-инданона (V) (20,0 г, 120,0 ммоль) в ТГФ (100 мл) за период, равный 30 мин, что позволяло поддерживать температуру ниже 50°С (нагрев или охлаждение не применяли). По завершении добавления реакционную смесь перемешивали 1 ч (нагрев или охлаждение не применяли). Очень медленно и осторожно прибавляли концентрированную серную кислоту (13,3 мл, 96 масс. %), при этом температуру реакционной смеси поддерживали ниже 50°С. По завершении добавления добавляли воду (125 мл). Большую часть ТГФ удаляли выпариванием в вакууме. Оставшуюся водную смесь дважды экстрагировали, используя гептан (2×100 мл). Объединенные экстракты промывали насыщенным водн. раствором NaHCO3 (100 мл), водой (2×100 мл) и соляным раствором (100 мл). Органическую фазу интенсивно перемешивали с MgSO4 и активированным углем в течение 20 мин и фильтровали через слой целита. Фильтрат выпаривали досуха. Остаток выпаривали в вакууме вместе с этанолом до сухого состояния с целью удалить большую часть гептана путем азеотропной дистилляции. В результате получали неочищенный 6-хлор-3-(фенил-d5)-1Н-инден (IV) (26,7 г) в виде твердого вещества. Сырой продукт повторно осаждали из этанола, растворяя в минимальном количестве кипящего этанола и медленно охлаждая до 5°С при перемешивании, в результате получая 6-хлор-3-(фенил-d5)-1H-инден (IV) (20,5 г, 74%) в виде желтоватого твердого вещества, имеющего чистоту 99% согласно результатам анализа ЖХ-МС.
Аналитические данные для 6-хлор-3-(фенил-d5)-1H-индена (IV):
1Н ЯМР (600 МГц, CDCl3) δH 3,49 (d, J=2,0 Гц, 2Н), 6,57 (t, J=2,0 Гц, 1Н), 7,29 (dd, J=2,0, 8,0 Гц, 1Н), 7,48 (d, J=8,0 Гц, 1Н), 7,50 (m, 1Н); 13С ЯМР (150 МГц, CDCl3) δC 38,1, 121,2, 124,6, 126,5, 127,3 (t, J=24,0 Гц), 127,4 (t, J=24,0 Гц), 128,3 (t, J=24,0 Гц), 131,1, 131,2, 135,6, 142,6, 144,7, 146,6; ЖХ-МС (APPI): масса/заряд рассчетн. для C15H6D5Cl (М+) 231,1, эксперим. 231,1.
Синтез 6-хлор-3-(фенил-d5)-инден-1-она (I)
К раствору 6-хлор-3-(фенил-d5)-1Н-индена (3,00 г, 12,9 ммоль) в ТГФ (30,0 мл) добавляли 1,1-диметокси-N,N-диметилметанамин (4,30 мл, 32,4 ммоль) при комнатной температуре. Смесь нагревали при 45°С в течение 2,5 ч. Добавляли воду (15,0 мл), а затем - метапериодад натрия (8,31 г, 38,8 ммоль). Смесь нагревали далее при 60°С при интенсивном перемешивании в течение 1,5 ч. Смесь фильтровали через слой целита. Осадок на фильтре тщательно промывали дихлорметаном. Объединенные фильтраты промывали соляным раствором, сушили над MgSO4, фильтровали и выпаривали в вакууме до сухого состояния. Остаток очищали колоночной хроматографией, элюируя смесью гептан-EtOAc (20:1) и получая 6-хлор-3-(фенил-d5)-инден-1-он (I) (2,86 г, 90%) в виде желто-оранжевого твердого вещества, имеющего чистоту 97% согласно результатам анализа ЖХ-МС.
Аналитические данные (ЯМР и ЖХ-МС) соединения (I) были такими же, как и те, что приведены выше.
Пример 2:
Синтез (Е)-1-(6-хлор-3-фенил(d5)-1Н-инден-1-илиденметил)-N,N-диметиламина (XIX)
Магниевую стружку (5,60 кг, 230 моль) суспендировали в 2-МеТГФ (21,3 л). Изопропилмагний хлорид (25 мл, 50,0 ммоль, 2 М) в ТГФ добавляли к магниевой стружке и суспензию магниевой стружки нагревали до кипения с обратным холодильником при перемешивании. Раствор бромбензола-d5 (34,23 кг, 211 моль) в 2-МеТГФ (79,6 л) добавляли к магниевой стружке за период, равный 1 ч 3 мин. Добавляли 2-МеТГФ (10,5 л) и реакционную смесь кипятили с обратным холодильником в течение 38 мин. Реакционную смесь охлаждали до 22°С перед тем, как добавляли раствор 5-хлор-1-инданона (V) (32,5 кг, 195 моль), растворенный в 2-МеТГФ (198 л), за период, равный 42 минутам, при максимальной температуре, равной 44°С. Добавляли 2-МеТГФ (10,5 л) и реакционную смесь перемешивали в течение ночи. К реакционной смеси добавляли водн. раствор соляной кислоты (80 л, 15 масс. %) и реакционную смесь перемешивали в течение 2 ч 46 мин. Фазы разделяли и органическую фазу промывали водн. раствором NaCl (40 л, 15 масс. %). Фазы разделяли и объем органической фазы уменьшали до 170 л путем дистилляции. Реакционную смесь охлаждали до 30°С, а затем добавляли 1,1-диметокси-N,N-диметилметанамин (31,0 кг, 260 моль). Реакционную смесь перемешивали в течение ночи и затем охлаждали до 6°С.Образовавшийся осадок отфильтровывали и дважды промывали гептаном (2х 38 л). Полученный твердый продукт сушили в вакуумной печи при 50°С в течение двух дней, получая (E)-1-(6-хлор-3-фенил(d5)-1Н-инден-1-илиден)-N,N-диметилметанамин (XIX) (48,0 кг, 86%) с чистотой >99% согласно результатам анализа ВЭЖХ (метод 1).
Аналитические данные для (Е)-1-(6-хлор-3-фенил(d5)-1Н-инден-1-илиденметил)-N,N-диметиламина (XIX):
1Н ЯМР (250 МГц, CDCl3) δH 3,26 (s, 6Н), 7,11 (s, 1Н), 7,12 (dd, J=2,0, 8,5 Гц, 1Н), 7,37 (в, 1Н), 7,58 (d, J=2,0 Гц, 1Н), 7,66 (d, J=8,0 Гц, 1Н).
Синтез 6-хлор-3-(фенил-d5)-инден-1-она (I)
Смесь (E)-1-(6-хлор-3-фенил-1Н-инден-1-илиденметил)-N,N-диметиламина (XIX) (803 г, 2,80 моль), метапериодата натрия (1,80 кг, 8,40 моль), ТГФ (3,9 л) и воды (3,9 л) перемешивали при 30°С. Через 48 мин в результате экзотермического эффекта реакционная смесь нагрелась до 36°С - максимальной температуры, достигнутой в ходе реакции. Реакционную смесь перемешивали в течение ночи при 30°С и затем охлаждали до 21°С. Добавляли толуол (280 мл), метансульфокислоту (546 мл) и гептан (4,2 л) и реакционную смесь нагревали до 29°С. Фазы разделяли и органическую фазу промывали водой (2×4 л). К органической фазе добавляли гептан (4 л) и объем органической фазы уменьшали до 3 л путем дистилляции в вакууме (макс. 45°С). Добавляли ТГФ (280 мл) и гептан (4 л) и реакционную смесь перемешивали в течение ночи. Реакционную смесь охлаждали до 5°С в течение 2 ч, прежде чем образовавшийся осадок отфильтровывали и промывали гептаном (2,5 л). Твердый продукт сушили в вакуумной печи при 40°С в течение ночи, получая 6-хлор-3-(фенил-d5)-инден-1-он (I) (508 г, 74%) с чистотой >99% согласно результатам анализа ВЭЖХ (метод 1). Аналитические данные (ЯМР и ЖХ-МС) соединения (I) были такими же, как и те, что приведены выше.
В. Синтез (±)-6-хлор-3-(фенил-d5)-1Н-инден-1-ола (VI)
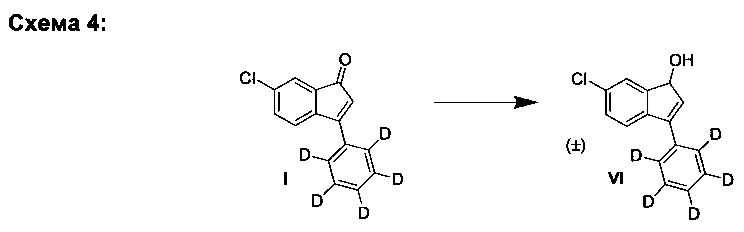
К раствору 6-хлор-3-(фенил-d5)-инден-1-она (I) (1,00 г, 4,07 ммоль) в ТГФ (10,0 мл) за период, равный 45 мин, добавляли диизобутилалюмогидрид в ТГФ (5,70 мл, 5,70 ммоль, 1,0 М) при -10°С при перемешивании. Полученную реакционную смесь перемешивали 30 мин при -10°С. Добавляли метанол (3,0 мл) при -10°С и охлаждение удаляли. Через 5 минут добавляли насыщенный водн. раствор тартрата калия-натрия (10 мл). Полученную смесь перемешивали 15 мин и добавляли насыщенный водн. раствор NH4Cl (5 мл), а затем - дихлорметан (30 мл). Органический слой отделяли и промывали соляным раствором. Органический слой сушили над MgSO4, фильтровали и выпаривали в вакууме до сухого состояния.
Остаток очищали колоночной хроматографией, элюируя смесью гептан-EtOAc (4:1) и получая (±)-6-хлор-3-(фенил-d5)-1Н-инден-1-ол (VI) (907 мг, 90%) в виде грязно-белого твердого вещества, имеющего чистоту 98% согласно результатам анализа ЖХ-МС.
Аналитические данные для (±)-6-хлор-3-(фенил-d5)-1Н-инден-1-ола (VI):
1Н ЯМР (600 МГц, CDCl3) δH 5,18 (dd, J=2,0, 7,0 Гц, 1Н), 5,76 (d, J=7,0 Гц, 1Н), 6,55 (d, J=2,0 Гц, 1Н), 7,36 (dd, J=2,0, 8,0 Гц, 1Н), 7,42 (d, J=8,0 Гц, 1Н), 7,53 (m, 1Н); 13С ЯМР (150 МГц, CDCl3) δС 74,8, 121,7, 124,3, 127,0 (t, J=24,0 Гц), 127,8, 128,1 (t, J=24,0 Гц), 128,6 (t, J=24,0 Гц), 131,1, 134,1, 136,5, 140,2, 142,4, 150,5; ЖХ-МС (APPI): масса/заряд рассчетн. для C15H7D5ClO [М+Н]+ 248,1, эксперим. 248,2.
C. Синтез (S)-6-хлор-3-(фенил-d5)-1Н-инден-1-ола (VII)
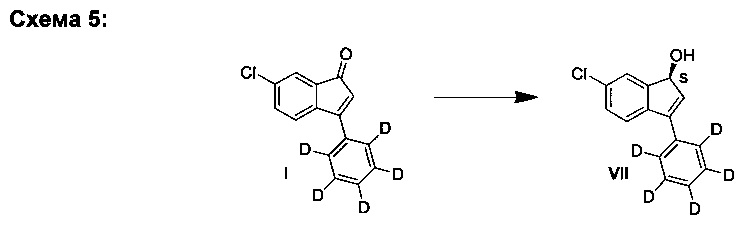
К раствору (R)-(+)-2-метил-CBS-оксазаборолидина в ТГФ (61 мкл, 61 мкмоль, 1,0 М) добавляли ТГФ (4,0 мл), а затем - раствор комплекса боран-ТГФ в ТГФ (1,34 мл, 1,34 ммоль, 1,0 М). Полученный раствор охлаждали до -10°С и медленно добавляли раствор 6-хлор-3-(фенил-d5)-инден-1-она (I) (300 мг, 1,22 ммоль) в ТГФ (4,0 мл) за период, равный 1,5 ч. Реакционную смесь перемешивали еще 45 мин при -10°С. Чтобы погасить реакцию, добавляли метанол (5 мл) и смеси давали нагреться до комнатной температуры. Смесь выпаривали вместе с силикагелем. Полученный материал загружали в силикагельную колонку и элюированием смесью гептан-EtOAc (4:1) получали (S)-6-хлор-3-(фенил-d5)-1Н-инден-1-ол (VII) (243 мг, 80%) в виде белого твердого вещества, имеющего 97% э.и. согласно данным анализа методом хиральной ВЭЖХ.
Аналитические данные (ЯМР и ЖХ-МС) соединения (VII) были такими же, как и те, что приведены выше для соединения (VI).
D. Синтез (±)-6-хлор-3-(фенил-d5)-индан-1-она (VIII)
D1. Путем гидрирования:
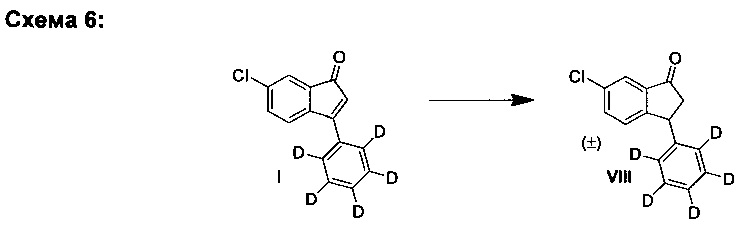
Общий способ:
К твердой смеси 6-хлор-3-(фенил-d5)-инден-1-она (I) (200 мг, 0,814 ммоль) и хлорида трис(трифенилфосфин)родия(I) (7,5 мг, 8,1 мкмоль) добавляли растворитель (3,0 мл, подробности см. в Таблице 1). Полученный раствор гидрировали при 4 бар газа водорода в течение 22 ч при комнатной температуре. Реакционную смесь выпаривали на силикагель, загружали в силикагельную колонку и элюированием смесью гептан-EtOAc (20:1) получали (±)-6-хлор-3-(фенил-d5)-индан-1-он (VIII). Полученное соединение (VIII) анализировали, используя ЖХ-МС, подробности см. в Таблице 1.

Пример:
К твердой смеси 6-хлор-3-(фенил-d5)-инден-1-она (I) (200 мг, 0,814 ммоль) и хлорида трис(трифенилфосфин)родия(I) (7,5 мг, 8,1 мкмоль) добавляли EtOAc (3,0 мл). Полученный раствор гидрировали при 4 бар газа водорода в течение 22 ч при комнатной температуре. Реакционную смесь выпаривали на силикагель, загружали в силикагельную колонку и элюированием смесью гептан-EtOAc (20:1) получали (±)-6-хлор-3-(фенил-d5)-индан-1-он (VIII) (164 мг, 81%).
Аналитические данные для (±)-6-хлор-3-(фенил-d5)-инден-1-она (VIII):
1Н ЯМР (500 МГц, CDCl3) δH 2,72 (dd, 1Н, J=4,0, 19,5 Гц), 3,27 (dd, 1Н, J=8,0, 19,5 Гц), 4,55 (dd, 1Н, J=4,0, 8,0 Гц), 7,21 (d, 1Н; J=8,0 Гц), 7,52 (dd, 1Н, J=2,0, 8,0 Гц), 7,77 (d, 1Н, J=2,0 Гц); 13С ЯМР (125 МГц, CDCl3) δC 44,0, 47,2, 123,2, 126,8 (t, J=24,0 Гц), 127,3 (t, J=24,0 Гц), 128,7 (t, J=24,0 Гц), 134,4, 135,1, 138,2, 142,9, 156,0, 206,4; ЖХ-МС (APPI): масса/заряд рассчетн. для C15H7D5ClO [М+Н]+ 248,1, эксперим. 247,6.
D2. Путем перегруппировки:
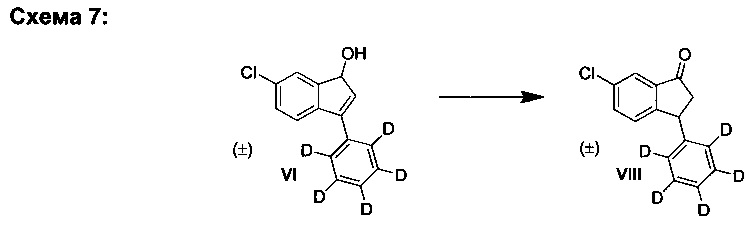
К раствору (±)-6-хлор-3-(фенил-d5)-1Н-инден-1-ола (VI) (200 мг, 0,807 ммоль) и DABCO (1,4-диазабицикло[2.2.2]октана) (45,3 мг, 0,404 ммоль) в ТГФ (3,0 мл) добавляли триэтиламин (281 мкп, 2,02 ммоль) при комнатной температуре. Реакционную смесь нагревали при 60°С в течение 1 ч. Реакционную смесь охлаждали и выпаривали вместе с силикагелем. Полученный материал загружали в силикагельную колонку и элюированием смесью гептан-EtOAc (10:1) получали (±)-6-хлор-3-(фенил-d5)-индан-1-он (VIII) (188 мг, 94%).
Аналитические данные (ЯМР и ЖХ-МС) соединения (VIII) были такими же, как и те, что приведены выше.
Е. Синтез (S)-6-хлор-3-(фенил-d5)-индан-1-она (IX)
Е1. Путем перегруппировки:
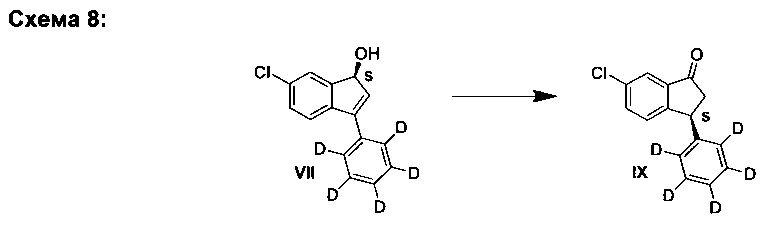
К раствору (S)-6-хлор-3-(фенил-d5)-1Н-инден-1-ола (VII) (200 мг, 0,807 ммоль, 97% э.и.) и DABCO (45,3 мг, 0,404 ммоль) в ТГФ (3,0 мл) добавляли триэтиламин (281 мкл, 2,02 ммоль) при комнатной температуре. Реакционную смесь нагревали при 60°С в течение 1 ч. Реакционную смесь охлаждали и выпаривали вместе с силикагелем. Полученный материал загружали в силикагельную колонку и элюированием смесью гептан-EtOAc (10:1) получали (S)-6-хлор-3-(фенил-d5)-индан-1-он (IX) (188 мг, 94%), имеющий 80% э.и. согласно данным анализа методом хиральной ВЭЖХ.
Аналитические данные (ЯМР и ЖХ-МС) соединения (IX) были такими же, как и те, что приведены выше для соединения (VIII).
Е2. Путем органокаталитического гидрирования с асимметричным переносом водорода
Общий способ:
К раствору 6-хлор-3-(фенил-d5)-инден-1-она (I) (300 мг) в растворителе добавляли катализатор и восстановитель при комнатной температуре или при 60°С (подробности см. в Таблице 2). Реакционную смесь перемешивали в течение 10-24 ч. Реакционную смесь выпаривали на силикагель, загружали в силикагельную колонку и элюированием смесью гептан-EtOAc (20:1) получали (S)-6-хлор-3-(фенил-d5)-индан-1-он (IX).
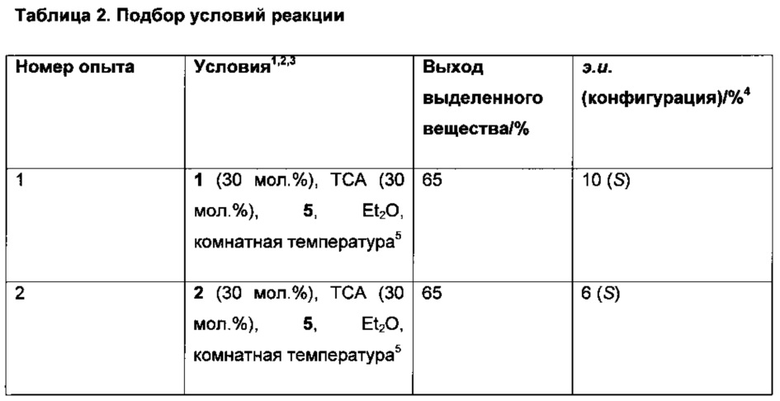

2. Катализаторы:
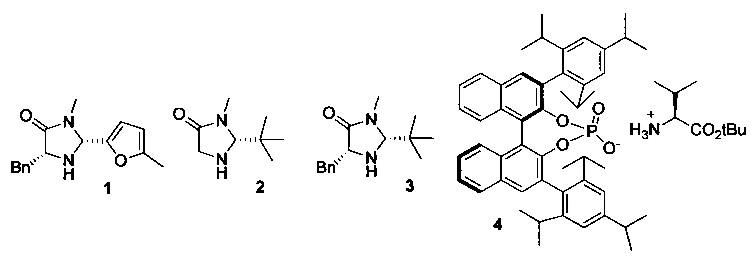
3. Восстановители:
4. Из анализа реакционной смеси методом хиральной ВЭЖХ.
5. Jamison В. Turtle et al, J. Am. Chem. Soc. 2006, 128, 12662-12663.
6. Nolwenn J. A. Martin et al, J. Am. Chem. Soc. 2006, 128, 13368-13369.
Пример:
Катализатор 4 изготавливали путем смешивания эквимолярных количеств (R)-TRIP и трет-бутилового эфира L-валина в Et2O. Образовавшийся осадок отфильтровывали и сушили в вакууме, получая катализатор 4.
К твердой смеси 6-хлор-3-(фенил-d5)-инден-1-она (I) (300 мг, 1,22 ммоль), восстановителя 6 (402 мг, 1,59 ммоль) и катализатора 4 (57 мг, 0,0610 ммоль) при комнатной температуре добавляли Bu2O. Реакционную смесь нагревали при 60°С в течение 10 ч. Реакционную смесь выпаривали вместе с силикагелем, загружали в силикагельную колонку и элюированием смесью гептан-EtOAc (от 20:1 до 10:1) получали (S)-6-хлор-3-(фенил-d5)-индан-1-он (IX) (287 мг, 95%), имеющий 46% э.и. согласно данным анализа методом хиральной ВЭЖХ.
Аналитические данные (ЯМР и ЖХ-МС) соединения (IX) были такими же, как и те, что приведены выше для соединения (VIII).
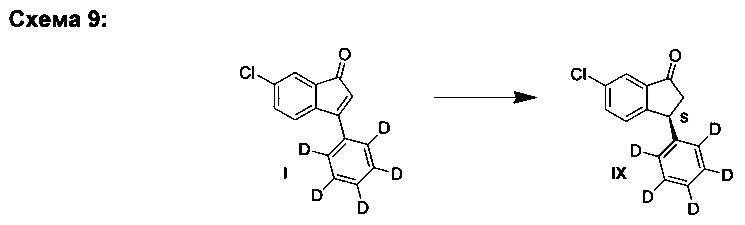
Е3. Путем асимметричного гидрирования (Схема 9):
Общий способ:
К твердой смеси металлического соединения-предшественника и лиганда, или катализатора, добавляли растворитель. Реакционную смесь интенсивно перемешивали при комнатной температуре в течение 30 мин, после чего добавляли раствор 6-хлор-3-(фенил-d5)-инден-1-она (I) в растворителе (подробности см. в Таблицах 3-6). Полученную смесь гидрировали при 4 бар газа водорода, перемешивая в течение 18-70 ч при комнатной температуре. Реакционную смесь непосредственно анализировали, используя ЖХ-МС и хиральную ВЭЖХ. Продукт - (S)-6-хлор-3-(фенил-d5)-индан-1-он (IX) - можно выделять выпариванием реакционной смеси в вакууме и очисткой с помощью колоночной хроматографии, элюируя смесью гептан-EtOAc (20:1), или перекристаллизацией из этанола.
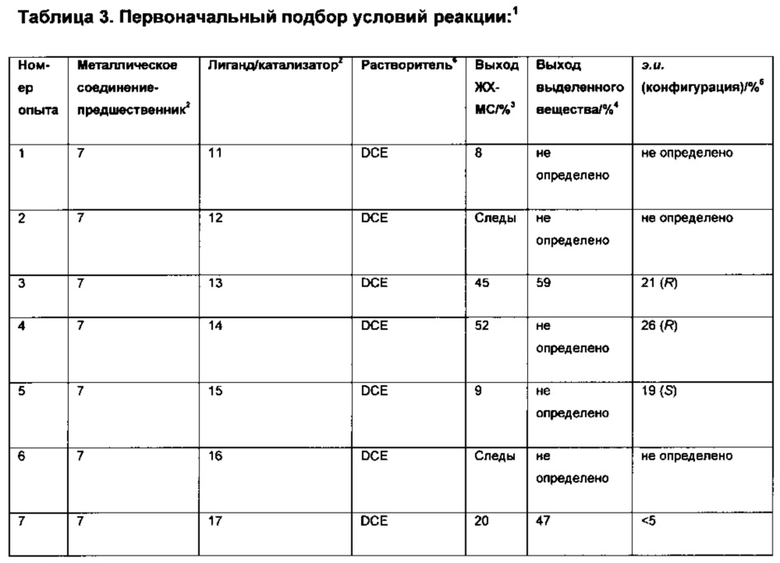
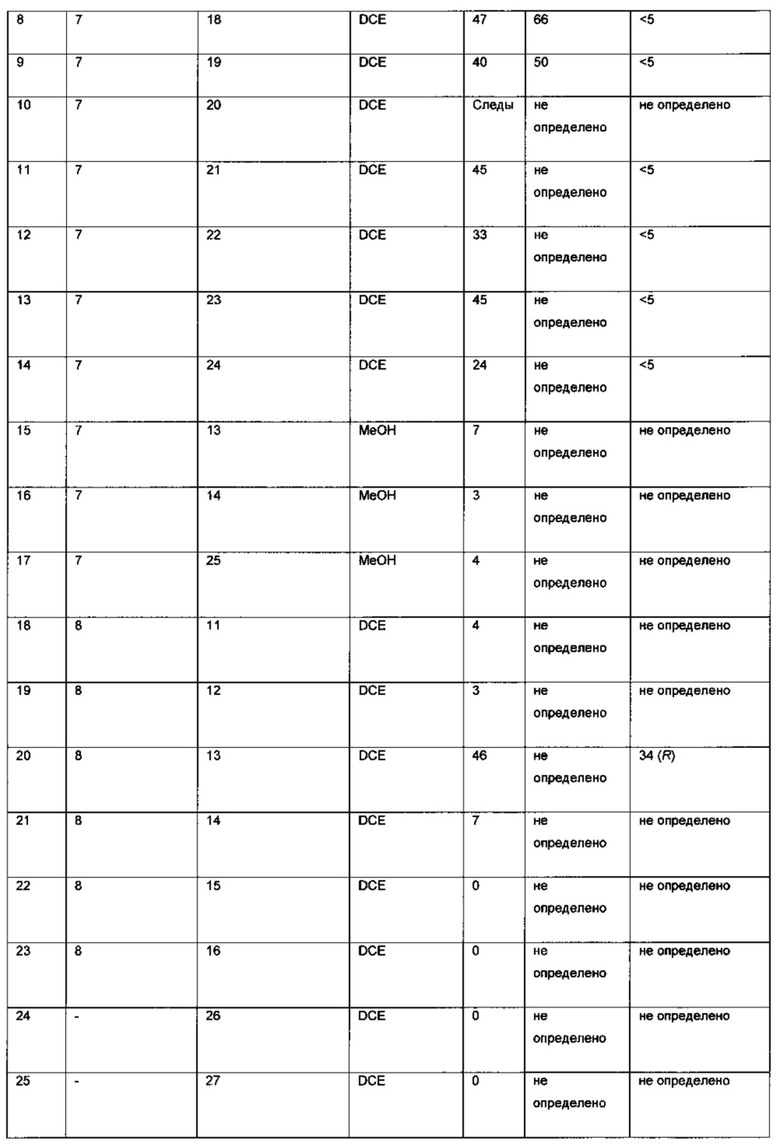
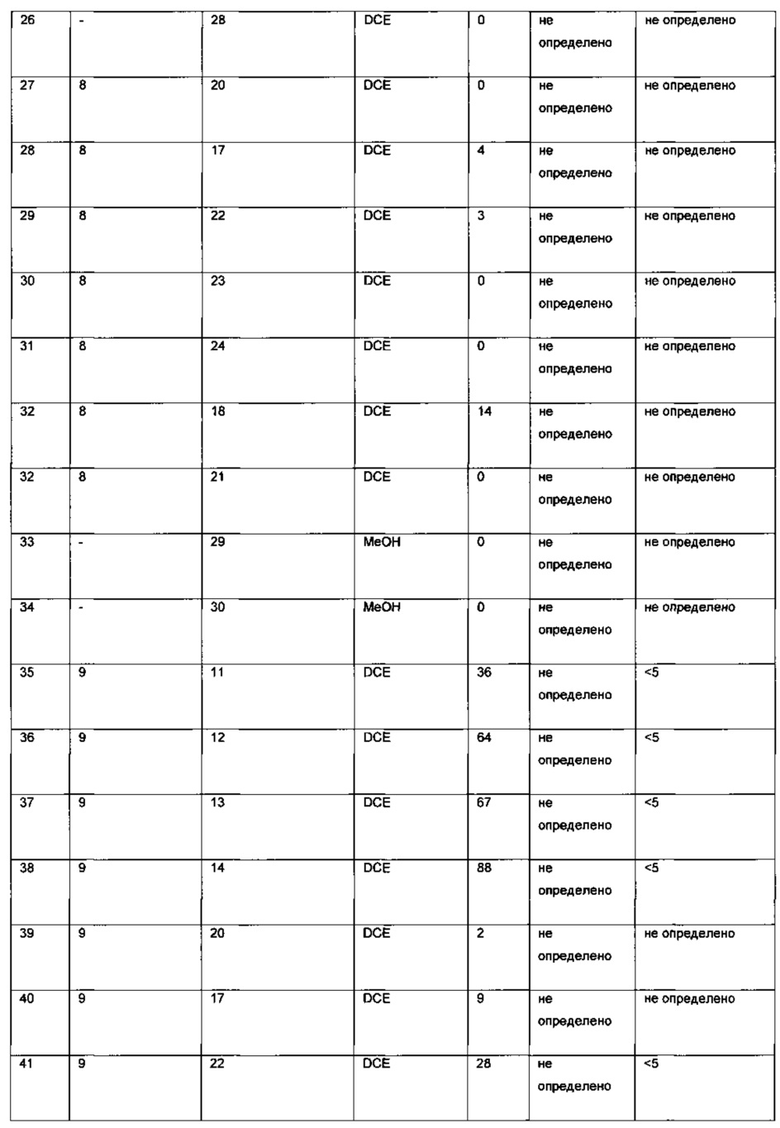
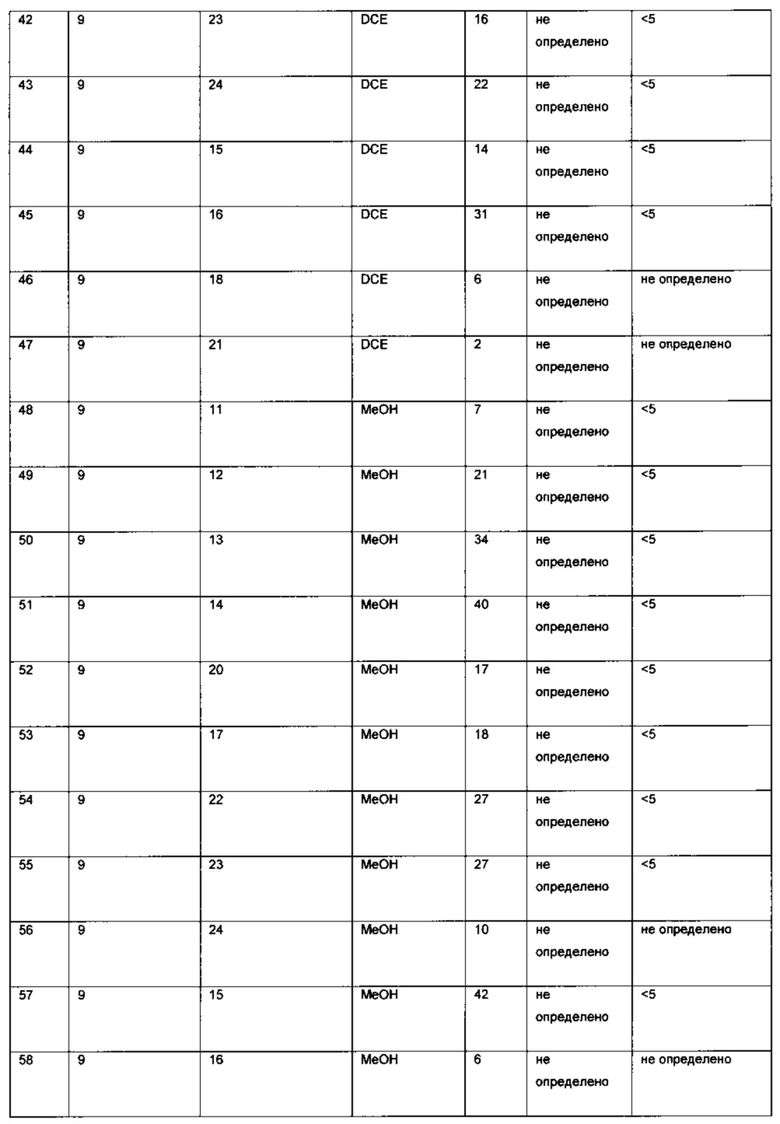
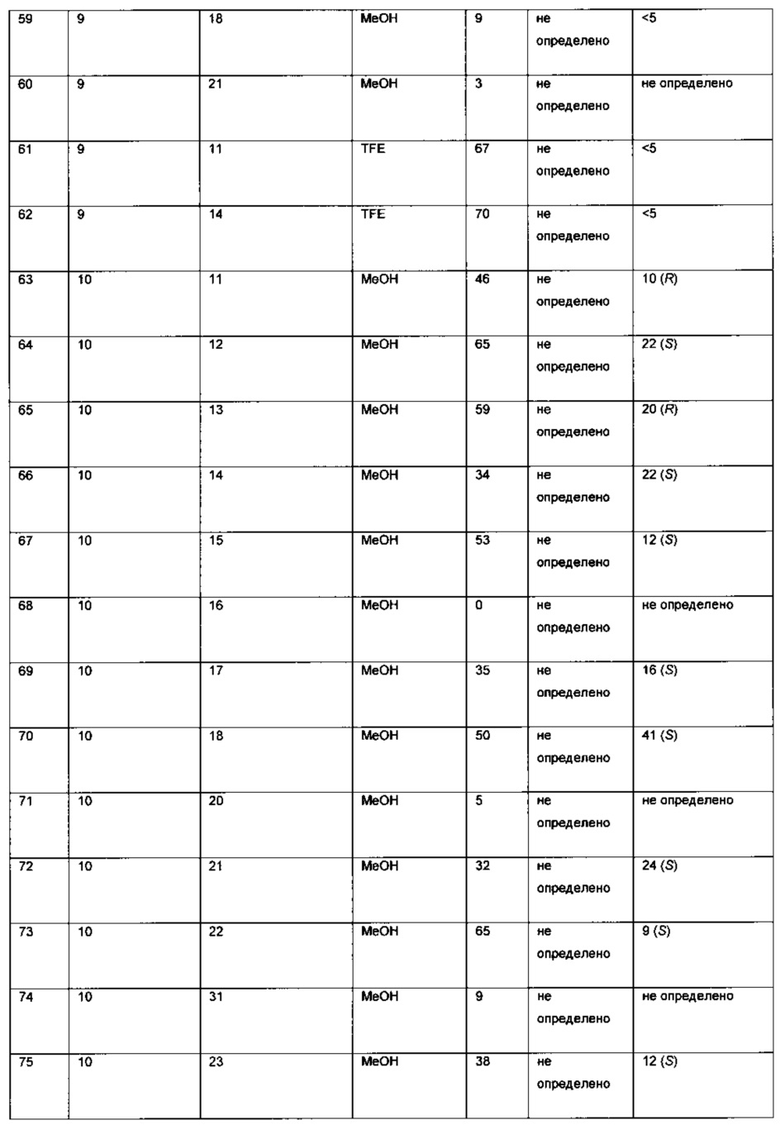
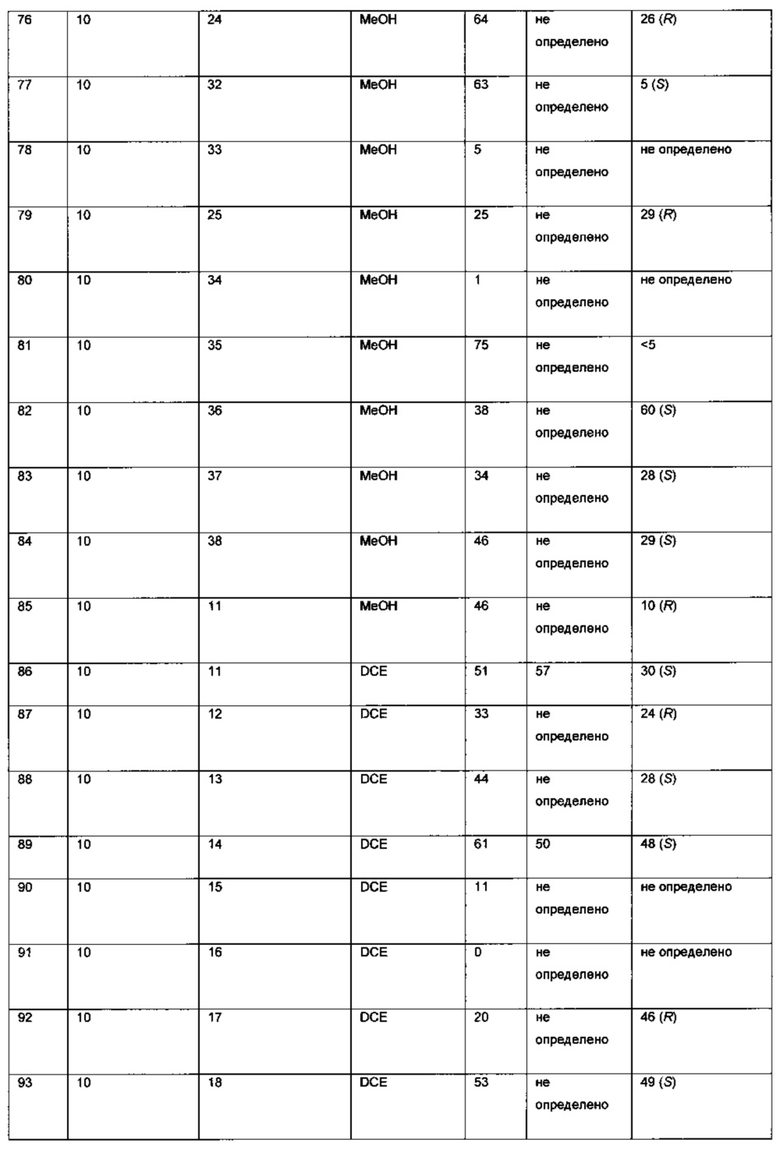
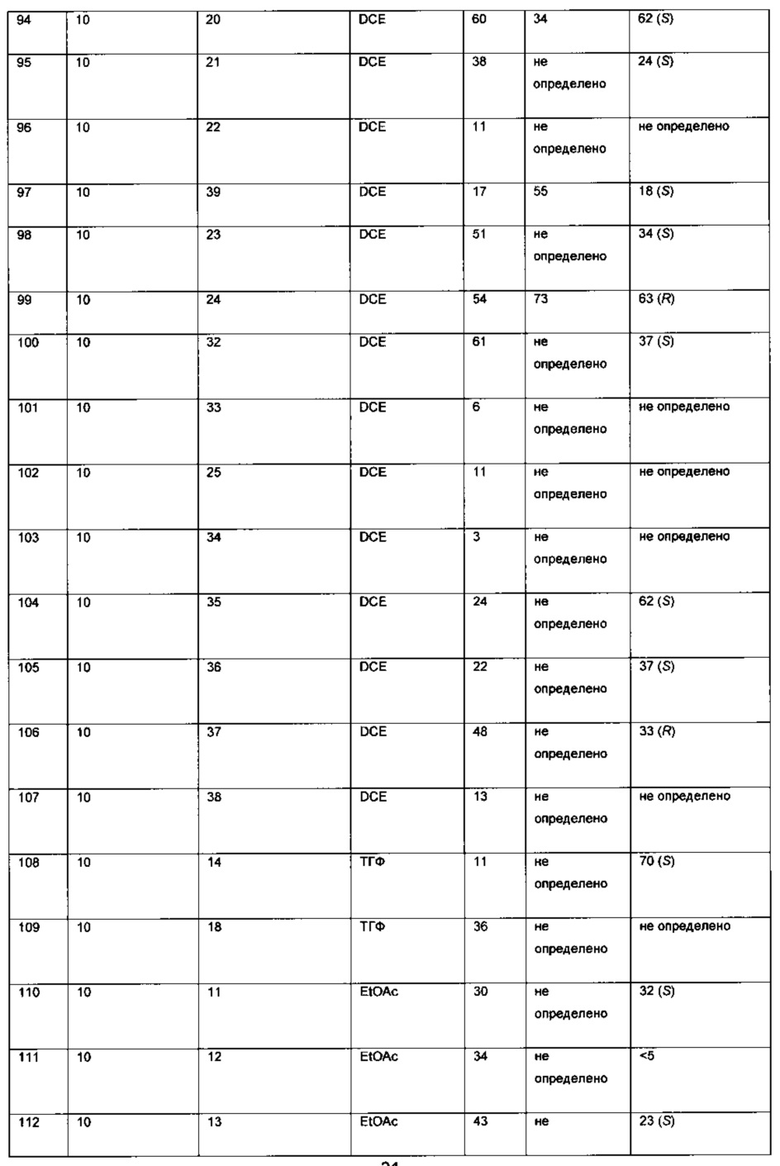



Оптимизация с использованием четырех ведущих лигандов:

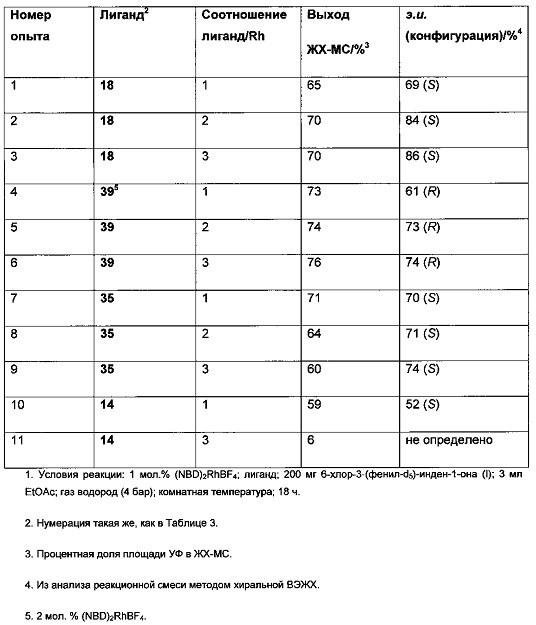
Оптимизация с использованием (S)-Phanephos:
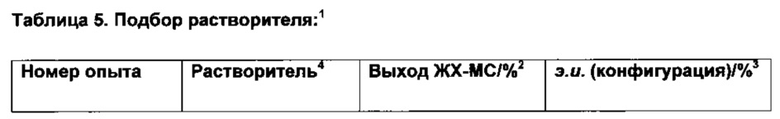


Пример 1:
К твердой смеси (NBD)2RhBF4 (0,8 мг, 2 мкмоль) и (S)-Phanephos (3,5 мг, 6,1 мкмоль) добавляли EtOAc (обезгаженный, 4,0 мл). Реакционную смесь интенсивно перемешивали в течение 30 мин, после чего к мутному раствору добавляли раствор 6-хлор-3-(фенил-d5)-инден-1-она (I) (1,00 г, 4,07 ммоль) в EtOAc (обезгаженный, 3,0 мл). Полученную смесь гидрировали при 4 бар газа водорода, перемешивая в течение 18 ч. Результаты анализа реакционной смеси методом хиральной ВЭЖХ показали образование (S)-6-хлор-3-(фенил-d5)-индан-1-она (IX), имеющего 86% э.и. Реакционную смесь выпаривали до сухого состояния в вакууме и остаток повторно растворяли в минимальном количестве кипящего этанола, раствору дали медленно охладиться до комнатной температуры. Образовавшийся осадок отфильтровывали из раствора и сушили в вакууме, получая (S)-6-хлор-3-(фенил-d5)-индан-1-он (IX) (712 мг, 71%) в виде грязно-белого порошка, имеющего 98% э.и. согласно результатам анализа методом хиральной ВЭЖХ. Дополнительный выход продукта можно получить, охлаждая фильтрат в морозильной камере (-5°С) и выделяя (S)-6-хлор-3-(фенил-d5)-индан-1-он (IX) (64 мг, 6%), имеющий 93% э.и. согласно результатам анализа методом хиральной ВЭЖХ.
Аналитические данные (ЯМР и ЖХ-МС) соединения (IX) были такими же, как и те, что приведены выше для соединения (VIII).
Пример 2:
К твердой смеси комплекса [(S)-Phanephos][NBD]RhBF4 (17 мг, 20 мкмоль) и 6-хлор-3-(фенил-d5)-инден-1-она (I) (10,0 г, 40,7 ммоль) добавляли EtOAc (обезгаженный, 100 мл). Смесь гидрировали при 4 бар газа водорода, перемешивая в течение 2 ч. Результаты анализа реакционной смеси методом хиральной ВЭЖХ показали образование (S)-6-хлор-3-(фенил-d5)-индан-1-она (IX), имеющего 89% э.и. Реакционную смесь перемешивали с активированным углем (1 г) в течение 1 часа и фильтровали через целит. Фильтрат выпаривали до сухого состояния в вакууме и остаток повторно растворяли в минимальном количестве кипящего этанола, раствору дали медленно охладиться до комнатной температуры. Образовавшийся осадок отфильтровывали из раствора и сушили в вакууме, получая (S)-6-хлор-3-(фенил-d5)-индан-1-он (IX) (7,1 г, 70%), имеющего 99% э.и. согласно результатам анализа методом хиральной ВЭЖХ.
Аналитические данные (ЯМР и ЖХ-МС) соединения (IX) были такими же, как и те, что приведены выше для соединения (VIII).
Пример 3:
К твердой смеси (NBD)2RhBF4 (435 мг, 1,16 ммоль) и (S)-Phanephos (1,31 г, 2,27 ммоль) добавляли EtOAc (обезгаженный, 300 мл). Смесь интенсивно перемешивали в течение 30 мин и вносили в суспензию 6-хлор-3-(фенил-d5)-инден-1-она (I) (400 г, 1,63 моль) в EtOAc (обезгаженный, 2,7 л). Смесь переносили в 25-литровый автоклав и гидрировали при 4 бар газа водорода в течение 22 ч при к.т. Затем реакционную смесь смешивали с активированным углем (56 г) и перемешивали 1 ч, фильтровали через Arbocel ВС 200®, используя дополнительное количество EtOAc (200 мл). Фильтрат выпаривали до сухого состояния в вакууме и добавляли этанол (1,2 л). Смесь нагревали до 80°С до образования гомогенного раствора, которому затем дали медленно охладиться до к.т. при перемешивании, а полученную суспензию дополнительно охладили на ледяной бане и отфильтровали. Осадок промывали охлажденным на ледяной бане этанолом (200 мл), сушили в вакууме при 50°С в течение одного дня, получая (S)-6-хлор-3-(фенил-d5)-индан-1-он (IX) (339 г, 84%) в виде твердого вещества, имеющего 99% э.и. согласно результатам анализа методом хиральной ВЭЖХ и чистоту >99% согласно результатам анализа методом ЖХ-МС.
Аналитические данные (ЯМР и ЖХ-МС) соединения (IX) были такими же, как и те, что приведены выше для соединения (VIII).
F. Синтез (±)-цис-6-хлор-3-(фенил-d5)-индан-1-ола (X)
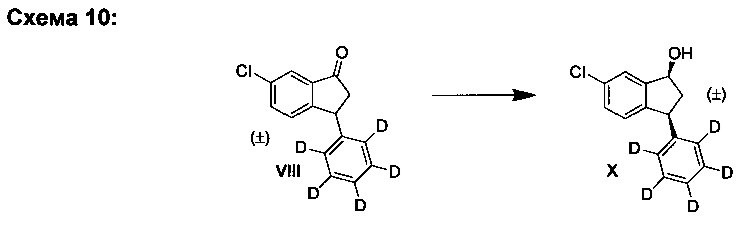
К суспензии борогидрида натрия (443 мг, 11,7 ммоль) в IPA (10,0 мл) добавляли раствор (±)-6-хлор-3-(фенил-d5)-индан-1-она (VIII) (1,45 г, 5,85 ммоль) в IPA (10,0 мл) и ТГФ (5,0 мл) при -10°С. Смеси дали медленно нагреться до к.т. в течение ночи. К реакционной смеси осторожно добавляли водн. раствор HCl (10 мл, 4 М), охлаждая при этом реакционную смесь на ледяной бане, поддерживая температуру при комнатной температуре или ниже. Полученную смесь концентрировали выпариванием в вакууме и добавляли воду (20 мл). Водную смесь трижды экстрагировали, используя EtOAc (3×30 мл). Объединенные экстракты промывали соляным раствором (20 мл), сушили над MgSO4 и фильтровали. Фильтрат выпаривали вместе с силикагелем. Полученный материал загружали в силикагельную колонку и элюированием смесью гептан-EtOAc (4:1) получали (±)-цис-6-хлор-3-(фенил-d5)-индан-1-ол (X) (1,43 г, 98%) в виде грязно-белого твердого вещества, имеющего соотношение цис : транс форм, равное 97:3 согласно анализу методом 1Н ЯМР, и чистоту 97% согласно анализу методом ЖХ-МС.
Аналитические данные для (±)-цис-6-хлор-3-(фенил-d5)-индан-1-ола (X):
1Н ЯМР (600 МГц, CDCl3) δH 1,96 (ddd, J=8,0, 13,0 Гц, 1Н), 2,06 (d, J=8,0 Гц, 1Н), 3,03 (dt, J=8,0, 13,0 Гц, 1Н), 4,14 (t, J=8,0 Гц, 1Н), 5,25 (q, J=8,0 Гц, 1Н), 6,86 (dd, J=1,0, 8,0 Гц, 1Н), 7,20 (dd, J=2,0, 8,0 Гц, 1Н), 7,45 (d, J=2,0 Гц, 1Н); 13С ЯМР (150 МГц, CDCl3) δC 47,3, 47,8, 74,7, 124,1, 126,3, 126,4, 128,0, 128,5, 129,3, 133,2, 143,6, 144,1, 147,2; ЖХ-МС (APPI): масса/заряд рассчетн. для C15H9D5ClO [М+Н]+ 250,1, эксперим. 250,0.
G. Синтез (±)-транс-1-(6-хлор-3-(фенил-d5)-индан-1-ил)-3,3-диметилпиперазина малеата (XII), используя (±)-цис-3,5-дихлор-1-(фенил-d5)-индан (XI)
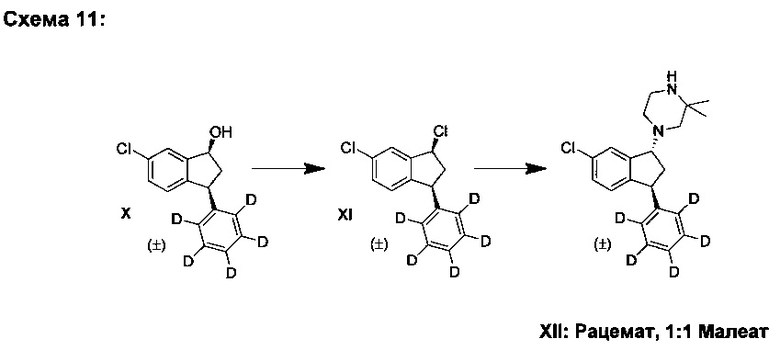
Тионилхлорид (2,01 кг, 16,9 моль) и ТГФ (7,2 кг) смешивали и смесь охлаждали до 10-15°С. Медленно добавляли раствор (±)-цис-6-хлор-3-(фенил-d5)-индан-1-ола (X) (2,76 кг, 11,1 моль) в ТГФ (7,2 кг) и по завершении добавляли ТГФ (5,9 кг). Реакционную смесь перемешивали при 15°С в течение приблизительно 90 ч. Воду (16,7 кг) охлаждали до 11°С и медленно прибавляли к реакционной смеси, а затем медленно добавляли водн. раствор NaOH (7,8 кг, 27,7 масс. %), затем - EtOAc (10 кг). Смесь перемешивали 20-40 мин. Фазы разделяли и проводили дистилляцию органической фазы, пока ее объем не уменьшился до примерно 6 л. Добавляли MIBK (16 кг) и проводили дистилляцию, пока объем не уменьшился до примерно 8 л, получая при этом раствор соединения (XI). Добавляли карбонат калия (1,58 кг, 11,4 моль), 2,2-диметилпиперазин (1,69 кг, 14,8 моль) и MIBK (13,6 кг). Реакционную смесь перемешивали при 90-95°С в течение 35 ч. После охлаждения до комнатной температуры добавляли воду (11 кг) и смесь перемешивали 30-60 мин. Фазы разделяли. К органической фазе добавляли воду (13,7 кг) и смесь медленно перемешивали в течение 30-60 мин. Фазы разделяли и органическую фазу отфильтровывали без использования специальных фильтрующих материалов. Добавляли MIBK (5 кг), воду (7,8 кг) и водн. раствор соляной кислоты (5,9 кг, 36 масс. %) и смесь перемешивали при 50°С в течение 30-60 мин. Фазы разделяли, к водной фазе прибавляли MIBK (8 кг) и смесь охлаждали до 10-15°С. Смесь MIBK (3,5 кг) и водн. раствора NH3 (7,8 кг, 25 масс. %) медленно добавляли к смеси и реакционную смесь перемешивали при 20-25°С в течение 60-90 мин. Фазы разделяли и органическую фазу промывали водой (10,5 кг). Органическую фазу подвергали дистилляции, пока ее объем не уменьшился до 8 л. Добавляли малеиновую кислоту (1,19 кг, 10,3 моль) и MIBK (9 кг) и реакционную смесь затем нагревали до 75-80°С. После охлаждения до 10-15°С осадок отфильтровывали и промывали, используя MIBK (10 кг). Твердый продукт сушили в вакуумной печи при 50°С в течение примерно 20 ч, получая (±)-транс-1-(6-хлор-3-(фенил-d5)-индан-1-ил)-3,3-диметилпиперазина малеат (XII) (3,47 кг, 68%).
Аналитические данные для (±)-транс-1-(6-хлор-3-(фенил-d5)-индан-1-ил)-3,3-диметилпиперазина малеата (XII):
1Н ЯМР (250 МГц, DMSO-d6) δH 1,31 (s, 3Н), 1,33 (s, 3Н), 2,12 (ddd, J=6,0, 8,0, 14,0 Гц, 1Н), 2,31 (d, J=12,0 Гц, 1Н), 2,58-2,50 (m, 3Н), 2,77 (bs, 1Н), 3,16 (bs, 2Н), 3,37 (bs, 1Н), 4,48 (dd, J=6,0, 8,5 Гц, 1Н), 4,56 (dd, J=5,0, 8,0 Гц, 1Н), 6,04 (s, 2Н, малеиновая кислота), 6,98 (d, J=8,0 Гц, 1Н), 7,29 (dd, J=2,0, 8,0 Гц, 1Н), 7,39 (d, J=1,5 Гц, 1Н), 8,60 (bs, 2Н).
Н. Синтез (±)-транс-1-(6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазина сукцината (XIII)

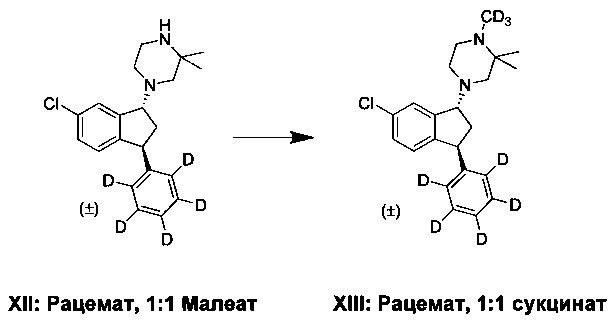
Соединение (XII) (1,1 кг, 2,38 моль), ТВМЕ (11 л), воду (1,8 л) и водн. раствор NH3 (1 л, 25 масс. %) перемешивали в течение 1-2 ч. Фазы разделяли и органическую фазу промывали водой (2×2 л). К органической фазе добавляли водн. раствор KOH (254 г, 3,85 моль, 85 масс. %) и воду (1,5 л), затем вводили метилиодид-d3 (450 г, 3,11 моль). Реакционную смесь перемешивали при 20-25°С в течение 16-24 ч. Добавляли воду (2 л) и выпадающий в осадок побочный продукт отфильтровывали. К фильтрату добавляли воду (0,8 л) и водн. раствор NH3 (0,2 л, 25 масс. %) и смесь перемешивали в течение 20-40 мин. Фазы разделяли и органическую фазу промывали водой (2 л). Фазы разделяли и к органической фазе добавляли ацетилхлорид (38 г, 0,48 моль), которую перемешивали 20-40 мин. Добавляли воду (0,8 л) и водн. раствор NH3 (0,2 л, 25 масс. %) и смесь перемешивали в течение 20-40 мин. Фазы разделяли и органическую фазу промывали водой (2 л). Органическую фазу выпаривали досуха. Янтарную кислоту (225 г, 1,91 моль) и ацетон добавляли таким образом, чтобы общий объем реакционной смеси составлял 6-6,5 л. Реакционную смесь нагревали до кипения с обратным холодильником, а затем охлаждали до 5-10°С. Осадок отфильтровывали и промывали ацетоном (1 л). Твердый продукт сушили в вакуумной печи при 50°С в течение более чем 16 ч, получая соединение (XIII) (630 г, 55%).
Аналитические данные для (±)-транс-1-(6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазина сукцината (XIII):
1Н ЯМР (600 МГц, DMSO-d6) δH 1,02 (s, 3Н), 1,04 (s, 3Н), 2,02 (ddd, J=6,0, 8,0, 14,0 Гц, 1Н), 2,13 (d, J=11,0 Гц, 1Н), 2,31 (bs, 1Н), 2,37 (s, 4Н, янтарная кислота), 2,46-2,41 (m, 1Н), 2,65-2,56 (m, 4Н), 4,46 (dd, J=6,0, 9,0 Гц, 1Н), 4,46 (dd, J=5,0, 8,0 Гц, 1Н), 6,95 (d, J=8,0 Гц, 1Н), 7,26 (dd, J=2,0, 8,0 Гц, 1Н), 7,33 (d, J=2,0 Гц, 1Н).
I. Синтез 4-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазина L-(+)-тартрата (XIV) путем разделения

Соединение (XIII) (1,00 кг, 2,08 моль), EtOAc (8 л), воду (2 л) и водн. раствор NH3 (1 л, 25 масс. %) перемешивали в течение 0,5-1 ч. Фазы разделяли и органическую фазу промывали водой (2 л). Органическую фазу подвергали дистилляции, пока ее объем не уменьшился до примерно 1,5 л. Добавляли ацетон (10 л) и L-(+)-винную кислоту (312 г, 2,08 моль). Смесь нагревали до кипения с обратным холодильником, а затем охлаждали до 5-10°С. Осадок отфильтровывали и промывали ацетоном (1,2 л). Сырой осадок на фильтре смешивали с этанолом (11 л). Смесь нагревали до кипения с обратным холодильником, а затем охлаждали до 5-10°С. Осадок отфильтровывали и промывали абсолютным этанолом (1,2 л). Твердый продукт сушили в вакуумной печи при 50°С в течение более чем 16 ч, получая соединение (XIV) (395 г, 37%).
Аналитические данные для 4-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазина L-(+)-тартрата (XIV):
1Н ЯМР (600 МГц, DMSO-d6) δH 1,18 (s, 3Н), 1,21 (s, 3Н), 2,04 (ddd, J=6,0, 8,0, 14,0 Гц, 1Н), 2,31 (d, J=12,0 Гц, 1Н), 2,61-2,50 (m, 3Н), 2,77 (bs, 1Н), 2,95 (bs, 1Н), 4,07 (s, 2Н, тартрат), 4,45 (dd, J=6,0, 8,5 Гц, 1Н), 4,50 (dd, J=5,0, 8,0 Гц, 1Н), 6,96 (d, J=8,0 Гц, 1Н), 7,27 (d, J=8,0 Гц, 1Н), 7,36 (s, 1Н).
J. Синтез (1S,3S)-6-хлор-3-(фенил-d5)-индан-1-ола (Ха)
Борогидрид натрия (67 г, 1,77 моль) растворяли в IPA (2,1 л) и раствор охлаждали до -10°С. (S)-6-хлор-3-(фенил-d5)-индан-1-он (IX) (438 г, 1,77 моль) растворяли в ТГФ (2,3 л) и IPA (0,4 л). Раствор (S)-6-хлор-3-(фенил-d5)-индан-1-она (IX) добавляли к раствору борогидрида натрия за период, равный 2 ч 24 мин при максимальной температуре в ходе добавления, равной -4°С. Реакционную смесь перемешивали в течение ночи когда температура достигла комнатной температуры. Реакционную смесь охлаждали до -2°С и добавляли водн. раствор HCl (1,55 л, 4 М) за период, равный 1 ч 35 мин. Объем реакционной смеси уменьшали путем дистилляции в вакууме до примерно 2,5 л. Добавляли воду (2,5 л) и толуол (4 л) и реакционную смесь перемешивали при 45°С в течение 15 мин. Фазы разделяли и органическую фазу промывали водн. раствором NaCl (3 л, 5 масс. %). Фазы разделяли и органическую фазу подвергали дистилляции в вакууме (макс. 70°С), пока ее объем не уменьшился до примерно 1,4 л. Органическую фазу прибавляли к гептану (12,5 л) за период, равный 9 мин. Температуру реакции снижали до -5°С и образовавшийся осадок отфильтровывали через 1 ч 20 мин. Осадок промывали гептаном (1 л) и затем сушили в вакуумной печи при 40°С в течение ночи, получая (1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ол (Ха) (377 г, 86%) в виде грязно-белого твердого вещества, имеющего чистоту 99,5% согласно результатам анализа ВЭЖХ (метод 2).
Аналитические данные (ЯМР и ЖХ-МС) соединения (Ха) были такими же, как и те, что приведены выше для соединения (X).
K. Синтез 4-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазина L-(+)-тартрата (XIV), используя (1S,3S)-3,5-дихлор-1-(фенил-d5)-индан (XIa)
Охлажденный на ледяной бане раствор соединения (Ха) (25 г, 100 ммоль) в 2-МеТГФ (80 мл) добавляли к охлажденному на ледяной бане раствору тионилхлорида (11,0 мл, 152 ммоль) в 2-МеТГФ (60 мл) за период, равный 10 мин, причем максимальная температура составляла 1°С. Реакционную смесь перемешивали на протяжении ночи при комнатной температуре, затем охлаждали до 2°С перед тем как добавляли воду (180 мл) за период, равный 25 мин, поддерживая температуру ниже 18°С. Значение рН доводили до 7 добавлением водн. раствора NH3 (34 мл, 25 масс. %) и после этого фазы разделяли. Органическую фазу выпаривали в вакууме и полученное масло десорбировали один раз, используя MIBK (50 мл), получая неочищенное соединение (XIa). Добавляли MIBK (160 мл), карбоната калия (42,8 г, 310 ммоль) и соединение (XVI) (43,1 г, 120 ммоль) и реакционную смесь нагревали при 90°С в течение 24 ч. Реакционную смесь охлаждали до комнатной температуры и затем добавляли воду (300 мл). Реакционную смесь перемешивали 15 мин, фазы разделяли и органическую фазу промывали водой (300 мл). Фазы разделяли и к органической фазе добавляли ацетилхлорид (1,0 мл). Реакционную смесь перемешивали 3 ч, затем добавляли воду (20 мл) и водн. раствор NH3 (6 мл, 25 масс. %). Фазы разделяли и органическую фазу промывали водой (130 мл). Органическую фазу фильтровали через Arbocel ВС-200, а затем добавляли водн. раствор HCl (240 мл, 1,08 моль, 4,5 М). Температуру реакции повышали до 50°С, фазы разделяли и к водной фазе добавляли MIBK (300 мл), а затем - водн. раствор NH3 (180 мл, 25 масс. %). Фазы разделяли и органическую фазу промывали водой (300 мл), а затем уменьшали ее объем путем выпаривания в вакууме. Полученное масло десорбировали, используя ацетон (100 мл). Затем масло растворяли в этаноле (300 мл) и добавляли L-(+)-винную кислоту (15,0 г, 100 ммоль). Смесь нагревали до кипения с обратным холодильником, а затем охлаждали до комнатной температуры. Полученный осадок отфильтровывали и твердое вещество промывали ацетоном (50 мл). Твердый продукт сушили в вакуумной печи при 50°С в течение ночи, получая соединение (XIV) (29,8 г, 58%), имеющее чистоту 97,8% согласно результатам анализа ВЭЖХ (метод 3).
Аналитические данные (ЯМР и ЖХ-МС) соединения (XIV) были такими же, как и те, что приведены выше.
L. Синтез 4-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазина фумарата (XV), используя (1R,3S)-3,5-дихлор-1-(фенил-d5)-индан (XIa)

Охлажденный на ледяной бане раствор соединения (Ха) (23,7 г, 94,9 ммоль) в 2-МеТГФ (80 мл) добавляли к охлажденному на ледяной бане раствору тионилхлорида (10,3 мл, 142 ммоль) в 2-МеТГФ (60 мл) за период, равный 10 мин, причем максимальная температура составляла 1°С. Реакционную смесь перемешивали на протяжении ночи при комнатной температуре, затем охлаждали до 3°С перед тем как добавляли воду (180 мл) за период, равный 25 мин, поддерживая температуру ниже 16°С. Значение рН доводили до 7 добавлением водн. раствора NH3 (35 мл, 25 масс. %) и после этого фазы разделяли. Органическую фазу выпаривали в вакууме и полученное масло десорбировали один раз, используя MIBK (50 мл), получая неочищенное соединение (XIa). Добавляли MIBK (160 мл), карбоната калия (40,7 г, 295 ммоль) и соединение (XVI) (40,9 г, 114 ммоль) и реакционную смесь нагревали при 80°С в течение 68 ч. Реакционную смесь охлаждали до 39°С и затем прибавляли воду (270 мл). Реакционную смесь перемешивали 15 мин, фазы разделяли и органическую фазу промывали водой (270 мл). Фазы разделяли и к органической фазе добавляли ацетилхлорид (0,9 мл). Реакционную смесь перемешивали 72 ч, затем добавляли воду (25 мл) и водн. раствор NH3 (7 мл, 25 масс. %). Фазы разделяли и органическую фазу промывали водн. раствором NaCl (100 мл, 7,5 масс. %), а затем водой (100 мл). Органическую фазу фильтровали через Arbocel ВС-200, а затем добавляли водн. раствор HCl (250 мл, 1,0 моль, 4,0 М). Температуру реакции повышали до 55°С, фазы разделяли и к водной фазе добавляли MIBK (300 мл), а затем - водн. раствор NH3 (100 мл, 25 масс. %). Фазы разделяли и органическую фазу промывали водой (300 мл), а затем уменьшали ее объем путем выпаривания в вакууме. Полученное масло десорбировали, используя ацетон (200 мл), а затем эту операцию повторяли с этанолом (200 мл). Затем масло растворяли в этаноле (200 мл) и добавляли фумаровую кислоту (9,75 г, 84,0 ммоль). Реакционную смесь нагревали до 55°С, а затем охлаждали до комнатной температуры. Полученный осадок отфильтровывали и твердое вещество дважды промывали этанолом (2×25 мл). Твердый продукт сушили в вакуумной печи при 50°С в течение двух дней, получая соединение (XV) (26,4 г, 58%), имеющее чистоту 99,2% согласно результатам анализа ВЭЖХ (метод 3).
Аналитические данные для 4-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазина фумарата (XV):
1Н ЯМР (250 МГц, CDCl3) δH 1,14 (s, 3Н), 1,16 (s, 3Н), 2,04 (ddd, J=6,0, 8,0, 13,5 Гц, 1Н), 2,26 (d, J=12,0 Гц, 1Н), 2,73-2,40 (m, 3Н), 2,86-2,75 (m, 1Н), 2,92-2,86 (m, 2Н), 4,52-4,41 (m, 2Н), 6,53 (s, 2Н, фумарат), 6,95 (d, J=8,0 Гц, 1Н), 7,26 (dd, J=2,5, 8,0 Гц, 1Н), 7,34 (d, J=2,5 Гц, 1Н).
М. Синтез трет-бутил 3,3-диметилпиперазин-1-карбоксилата геми-D,L-тартрата (XVIII)
2,2-диметилпиперазин (11,5 кг, 101 моль) растворяли в этаноле (48,5 л) и раствор охлаждали до примерно 9°С. Ди-трет-бутил дикарбонат (21,9 кг, 100 моль) растворяли в этаноле (41,7 л). Раствор ди-трет-бутил дикарбоната прибавляли к раствору диметилпиперазина за период, равный 2 ч 30 мин, поддерживая температуру реакции ниже 15°С. Добавляли этанол (12,4 л) и раствор перемешивали на протяжении ночи при температуре 12-25°С. Реакционную смесь нагревали до кипения и отгоняли 75 л. К реакционной смеси добавляли этанол (76 л) и раствор нагревали до 52°С и переносили в суспензию D,L-винной кислоты (7,5 кг, 50,0 моль) в этаноле (25,2 л), и нагревали до 51°С. Добавляли этанол (25,3 л) и реакционную смесь выдерживали при 20°С в течение ночи. Осадок отфильтровывали и промывали этанолом (28,1 л). Твердый продукт сушили в вакуумной печи при 50°С в течение ночи, получая соединение (XVIII) (27,1 кг, 93%), имеющее чистоту 99% согласно результатам анализа методом ГХ.
Аналитические данные для mpem-бутил 3,3-диметилпиперазин-1-карбоксилата геми-D,L-тартрата (XVIII):
1Н ЯМР (250 МГц, CDCl3) δH 1,35 (s, 6Н), 1,46 (s, 9Н), 3,10 (bs, 2Н), 3,42 (bs, 2Н), 3,63 (bs, 2Н), 4,29 (s, 1Н, тартрат), 7,60 м.д. (bs, 3Н); 13С ЯМР (62,5 МГц, CDCl3) δC 22,3, 28,3, 39,0, 40,8, 50,2, 51,8, 53,6, 73,6 (тартрат), 80,6, 154,2, 178,3 (тартрат).
N. Синтез 1(d3),2,2-триметилпиперазина бис-2,2,2-трифторацетата (XVI)
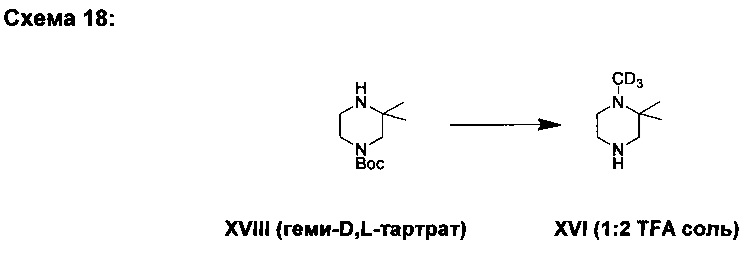
Соединение (XVIII) (23,0 кг, 79,5 моль) суспендировали в толуоле (133 л), добавляли воду (85,2 л) и водн. раствор NaOH (14,1 кг, 27,7 масс. %) и реакционную смесь перемешивали 1 ч. После разделения фаз органическую фазу прибавляли к карбонату калия (11,1 кг, 80,3 моль). Добавляли N-метилпирролидин (7,0 кг). Иодометан-d3 (12,7 кг, 87,6 моль) растворяли в толуоле (11,5 л) и затем прибавляли к реакционной смеси, в последующем добавляли толуол (11,5 л). Реакционную смесь перемешивали при 23°С в течение ночи. После того, как по данным контроля за процессом в реакции осталось 5,7% соединения (XVIII), добавляли иодометан-d3 (0,9 кг, 6,21 моль) и толуол (12,7 л) и реакционную смесь перемешивали в течение ночи при 23°С. Добавляли воду (85 л) и водн. раствор NH3 (3,5 кг, 25 масс. %) и реакционную смесь перемешивали 40 мин. Фазы разделяли и объем органической фазы уменьшали до примерно 20 л путем дистилляции в вакууме. Температуру реакции понижали до 0°С и добавляли трифторуксусную кислоту (38,0 кг, 333 моль) за период, равный 36 мин. Реакционную смесь перемешивали при 39°С в течение ночи, а затем охлаждали до 13°С. Добавляли диэтиловый эфир (77,1 л) и реакционную смесь перемешивали при температуре примерно 22°С на протяжении ночи. Температуру реакции понижали до 8°С, перемешивали 3,5 ч и затем фильтровали. Осадок на фильтре промывали диэтиловым эфиром (44,9 л) и затем дополнительным количеством диэтилового эфира (30,8 л). Полученный твердый продукт сушили в вакуумной печи при 50°С в течение ночи, получая соединение (XVI) (23,4 кг, 82%), имеющее чистоту 93,2% согласно результатам анализа методом ГХ.
Аналитические данные для 1(d3),2,2-триметилпиперазина бис-2,2,2-трифторацетата (XVI):
1Н ЯМР (250 МГц, D2O) δH 1,35 (s, 6Н), 3,16 (d, J=14,5 Гц, 1Н), 3,31-3,21 (m, 1Н), 3,58-3,35 м.д. (m, 4Н); 13С ЯМР (62,5 МГц, D2O) δС 17,0, 23,8, 37,0, 41,9, 47,3, 51,7, 60,8, 117,6 (q, J=36 Гц, TFA), 164,0 (q, J=291 Гц, TFA).
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ С КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ | 2019 |
|
RU2783414C2 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНИЛПИПЕРИДИНА В КАЧЕСТВЕ АНТАГОНИСТОВ ХЕМОКИНОВОГО РЕЦЕПТОРА | 2005 |
|
RU2369604C2 |
| 2-АМИНОБЕНЗОКСАЗОЛКАРБОКСАМИДЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ 5-НТ3 | 2007 |
|
RU2448105C2 |
| ПРОИЗВОДНЫЕ 1,3-ДИОКСОИНДЕНА, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ АНТИВИРУСНАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2012 |
|
RU2566761C2 |
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2001 |
|
RU2265011C2 |
| 3-ФЕНИЛСУЛЬФОНИЛ-8-ПИПЕРАЗИН-1-ИЛ-ХИНОЛИНЫ, ОБЛАДАЮЩИЕ АФФИННОСТЬЮ К 5-НТ6 РЕЦЕПТОРУ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 2003 |
|
RU2309154C9 |
| СОЕДИНЕНИЯ ПИРИМИДИНА И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ РАКА | 2019 |
|
RU2807277C2 |
| ФЕНОКСИПИРИДИНИЛАМИДНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ PDE4-ОПОСРЕДОВАННЫХ БОЛЕЗНЕННЫХ СОСТОЯНИЙ | 2009 |
|
RU2509077C2 |
| ПРОИЗВОДНЫЕ СПИРОПИПЕРИДИНА, ЛЕКАРСТВЕННОЕ СРЕДСТВО, СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ | 1999 |
|
RU2184735C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ С ИХ ИСПОЛЬЗОВАНИЕМ | 2001 |
|
RU2265598C2 |
Изобретение относится к 6-хлор-3-(фенил-d5)-инден-1-ону (I), к способам получения соединения (I) и к использованию соединения (I) для получения фармацевтически приемлемых солей дейтерированных 1-пиперазино-3-фенилинданов, применяемых для лечения шизофрении. 11 н. и 7 з.п. ф-лы, 6 табл., 7 пр.
1. Соединение, имеющее структурную формулу (I)
2. Способ получения соединения (I), где соединение (I) получают реакцией 3-бром-6-хлоринден-1-она с фенил-d5 бороновой кислотой или ее сложным эфиром.
3. Способ по п. 2, где фенил-d5 бороновая кислота или ее сложный эфир представляет собой 4,4,5,5-тетраметил-2-d5-фенил-[1,3,2]диоксаборолан.
4. Способ по п. 2, включающий следующие стадии:
a) 2,2'-азо-бис-изобутиронитрил и N-бромсукцинимид вводят в раствор, содержащий 6-хлор-1-инданон;
b) триэтиламин вводят в раствор, полученный в стадии а), с целью получения 3-бром-6-хлоринден-1-она;
c) 3-бром-6-хлоринден-1-он отделяют и вводят в реакцию с 4,4,5,5-тетраметил-2-d5-фенил-[1,3,2]диоксабороланом в присутствии подходящего катализатора и основания с целью получения соединения (I).
5. Способ по п. 4, где стадию с) проводят в присутствии ацетата палладия и трифенилфосфина.
6. Способ получения соединения (I), включающий стадии:
1. Синтеза 6-хлор-3-(фенил-d5)-1Н-индена (IV) путем взаимодействия органометаллических частиц, полученных из моногалогенированного бензола-d5 и 5-хлор-1-инданона (V) с последующей дегидратацией;
2. Преобразования 6-хлор-3-(фенил-d5)-1H-индена (IV) в (E)-1-(6-хлор-3-фенил(d5)-1Н-инден-1-илиденметил)-N,N-диметиламин (XIX) и его последующего окислительного расщепления с целью получения соединения (I).
7. Способ по п. 6, включающий стадии:
a) синтеза 6-хлор-3-(фенил-d5)-1Н-индена (IV) реакцией Гриньяра с участием бромбензола-d5, магния и 5-хлор-1-инданона с последующей дегидратацией;
b) взаимодействия 6-хлор-3-(фенил-d5)-1H-индена, полученного в стадии а), с 1,1-диметокси-N,N-диметилметанамином с последующим окислительным расщеплением образовавшегося енаминного промежуточного соединения (XIX) с целью получения соединения (I).
8. Способ по п. 6 или 7, где окислительное расщепление в стадии b) проводят, используя окислитель, выбранный из группы, состоящей из метапериодата натрия, метапериодата калия, озона, дихромата калия, дихромата натрия, синглетного кислорода и m-хлорпербензойной кислоты.
9. Способ получения 6-хлор-3-(фенил-d5)-индан-1-она (VIIIa), включающий стадии:
a) восстановления соединения (I) по п. 1 с целью получения 6-хлор-3-(фенил-d5)-1Н-инден-1-ола (VIa);
b) преобразования соединения (VIa) в 6-хлор-3-(фенил-d5)-индан-1-он (VIIIa) путем перегруппировки в присутствии основания.
10. Способ получения (S)-6-хлор-3-(фенил-d5)-индан-1-она (IX), включающий стадии:
а) преобразования соединения (I) по п. 1 в (S)-6-хлор-3-(фенил-d5)-1Н-инден-1-ол (VII) путем энантиоселективного восстановления в присутствии энантиоселективного катализатора и восстановителя;
b) преобразования соединения (VII) в (S)-6-хлор-3-(фенил-d5)-индан-1-он (IX) путем перегруппировки в присутствии основания.
11. Способ получения 6-хлор-3-(фенил-d5)-индан-1-она (VIIIa), включающий преобразование соединения (I) по п. 1 с целью получения 6-хлор-3-(фенил-d5)-индан-1-она (VIIIa) путем гидрирования в присутствии подходящего катализатора, такого как хлорид трис(трифенилфосфин)родия(I), в подходящем растворителе, таком как этилацетат.
12. Способ получения (S)-6-хлор-3-(фенил-d5)-индан-1-она (IX), включающий преобразование соединения (I) по п. 1 с целью получения (S)-6-хлор-3-(фенил-d5)-индан-1-она (IX) путем асимметричного гидрирования в присутствии подходящего катализатора, такого как соль родия, и хирального фосфинового лиганда, и подходящего растворителя, такого как этилацетат.
13. Способ получения 3,5-дихлор-1-(фенил-d5)-индана, (±)-цис-3,5-дихлор-1-(фенил-d5)-индана или (1S,3S)-3,5-дихлор-1-(фенил-d5)-индана, причем способ включает следующие стадии:
a) восстановление (±)-6-хлор-3-(фенил-d5)-индан-1-она (VIII), 6-хлор-3-(фенил-d5)-индан-1-она (VIIIa) или (S)-6-хлор-3-(фенил-d5)-индан-1-она (IX) с целью получения соответствующего инданола: (±)-цис-6-хлор-3-(фенил-d5)-индан-1-ола (X), 6-хлор-3-(фенил-d5)-индан-1-ола (Xb) или (1S,3S)-6-хлор-3-(фенил-d5)-индан-1-ола (Ха) в присутствии подходящего восстановителя;
b) хлорирование любого из соединений, полученных в стадии а), с целью получения соответствующего хлорированного инданового соединения (±)-цис-3,5-дихлор-1-(фенил-d5)-индана (XI), 3,5-дихлор-1-(фенил-d5)-индана (XIb) или (1S,3S)-3,5-дихлор-1-(фенил-d5)-индана (XIa).
14. Способ по п. 13, в котором восстановитель на стадии а) представляет собой NaBH4.
15. Способ по п. 13, в котором хлорирование на стадии б) проводят в присутствии тионилхлорида.
16. Способ получения фармацевтически приемлемой соли (±)-транс-1-(6-хлор-3-(фенил-d5)-индан-1-ил)-3,3-диметилпиперазина, 1-(6-хлор-3-(фенил-d5)-индан-1-ил)-3,3-диметилпиперазина или 1-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-3,3-диметилпиперазина, где (±)-цис-3,5-дихлор-1-(фенил-d5)-индан (XI), 3,5-дихлор-1-(фенил-d5)-индан (XIb) или (1S,3S)-3,5-дихлор-1-(фенил-d5)-индан (XIa), как получено в п. 13, преобразуют в фармацевтически приемлемую соль (±)-транс-1-(6-хлор-3-(фенил-d5)-индан-1-ил)-3,3-диметилпиперазина, 1-(6-хлор-3-(фенил-d5)-индан-1-ил)-3,3-диметилпиперазина или 1-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-3,3-диметилпиперазина, причем способ включает следующие стадии:
a) взаимодействие 2,2-диметилпиперазина или соединения, которое впоследствии можно преобразовать в 3,3-диметилпиперазиновый фрагмент соединения (±)-транс-1-(6-хлор-3-(фенил-d5)-индан-1-ил)-3,3-диметилпиперазина, 1-(6-хлор-3-(фенил-d5)-индан-1-ил)-3,3-диметилпиперазина или 1-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-3,3-диметилпиперазина;
b) образование и необязательно осаждение фармацевтически приемлемой соли путем введения соответствующей кислоты.
17. Способ получения фармацевтически приемлемой соли 4-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазина, где фармацевтически приемлемую соль (±)-транс-1-(6-хлор-3-(фенил-d5)-индан-1-ил)-3,3-диметилпиперазина, 1-(6-хлор-3-(фенил-d5)-индан-1-ил)-3,3-диметилпиперазина или 1-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-3,3-диметилпиперазина, полученную в п. 16, преобразуют в фармацевтически приемлемую соль 4-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазина, причем способ включает следующие стадии:
a) алкилирование в присутствии активного донора метила-d3, такого как метилиодид-d3, и основания, такого как гидроксид калия, и
b) получение и необязательно осаждение фармацевтически приемлемой соли путем введения соответствующей кислоты.
18. Способ получения фармацевтически приемлемой соли 4-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазина, где 3,5-дихлор-1-(фенил-d5)-индан, (±)-цис-3,5-дихлор-1-(фенил-d5)-индан или (1S,3S)-3,5-дихлор-1-(фенил-d5)-индан, полученный в п. 13, преобразуют в фармацевтически приемлемую соль 4-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазина, причем способ включает следующие стадии:
a) нуклеофильное замещение 3,5-дихлор-1-(фенил-d5)-индана с использованием 1(d3),2,2-триметилпиперазина бис-2,2,2-трифторацетата или соединения, которое впоследствии можно преобразовать в 1(d3),2,2-триметилпиперазиновый фрагмент 4-((1R,3S)-6-хлор-3-(фенил-d5)-индан-1-ил)-1(d3),2,2-триметилпиперазина;
b) получение и необязательно осаждение фармацевтически приемлемой соли путем введения соответствующей кислоты.
| W.M.Clark et al., A Highly Enantioselective Conjugate Reduction of 3-Arylinden-1-ones Using Bakers’ Yeast for the Preparation of (S)-3-Arylindan-1-ones | |||
| Organic Letters, 1999, 1(11), 1839-1842 | |||
| WO 2005016901 A1, 24.02.2005 | |||
| НОВЫЕ ДИЗАМЕЩЕННЫЕ ФЕНИЛПИПЕРИДИНЫ/ПИПЕРАЗИНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ ДОПАМИНОВОЙ НЕЙРОТРАНСМИССИИ | 2005 |
|
RU2366654C2 |

