ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Изобретение относится к соединениям, которые модулируют активность или связываются с хемокиновыми рецепторами, такими как CCR5. В некоторых вариантах осуществления соединения селективны в отношении CCR5. Соединения могут использоваться, например, для лечения заболеваний, ассоциированных с экспрессией или активностью хемокинового рецептора, таких как воспалительные заболевания, иммунные заболевания и вирусные инфекции.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Миграция и доставка лейкоцитов из кровеносных сосудов в поврежденные ткани вовлечена в инициацию нормальных воспалительных реакций, противоборствующих развитию заболевания. Данный процесс, так же известный как рекрутирование лейкоцитов, также относится к началу и прогрессу угрожающего жизни воспаления, а также повреждающих аутоиммунных заболеваний. В результате патология данных заболеваний является следствием атаки защитных сил иммунной системы организма на нормальные ткани. В соответствии с этим, профилактика и блокирование рекрутирования лейкоцитов в ткани-мишени при воспалительном и аутоиммунном заболевании является очень эффективным подходом терапевтического вмешательства.
Другие классы лейкоцитарных клеток, которые вовлечены в клеточные иммунные ответы, включают моноциты, лимфоциты, нейтрофилы, эозинофилы и базофилы. В большинстве случаев лимфоциты представляют собой класс лейкоцитов, который инициирует, координирует и поддерживает хронические воспалительные реакции, и требуется блокада данных клеток от проникновения в участки воспаления. Лимфоциты привлекают в данные участки ткани моноциты, которые, вместе с лимфоцитами, отвечают за большую часть повреждений ткани, которые происходят при воспалительном заболевании. Инфильтрация лимфоцитами и/или моноцитами, как известно, приводит к различным хроническим аутоиммунным заболеваниям и также к отторжению трансплантированного органа. Данные заболевания включают в качестве не ограничивающих примеров ревматоидный артрит, хронический контактный дерматит, воспалительное заболевание кишечника, волчанку, системную красную волчанку, рассеянный склероз, атеросклероз, псориаз, саркоидоз, идиопатический фиброз легких, дерматомиозит, пемфигоид кожи и связанные заболевания (например, вызванные Pemphigus vulgaris, P. foliacious, P. erythematosis), гломерулонефриты, васкулиты, гепатит, диабет, отторжение аллотрансплантата и болезнь «трансплантат против хозяина».
Процесс, посредством которого лейкоциты покидают кровяное русло, накапливаются в участках воспаления и инициируют заболевание, как полагают, имеет, по меньшей мере, три стадии, описанные как (1) прокатывание, (2) активация/плотная адгезия и (3) миграция через эндотелий [Springer, T. A., Nature 346: 425-433 (1990); Lawrence and Springer, Cell 65: 859-873 (1991); Butcher, E. C., Cell 67: 1033-1036 (1991)]. Вторая стадия опосредуется на молекулярном уровне рецепторами хемоаттрактантов. Рецепторы хемоаттрактантов на поверхности лейкоцитов затем связываются с цитокинами-хемоаттрактантами, которые секретируются клетками в участке повреждения или инфекции. Связывание рецепторов активирует лейкоциты, повышает адгезивность молекул адгезии, которые опосредуют трансэндотелиальную миграцию и способствуют прямой миграции клеток в направлении источника цитокина-хемоаттрактанта.
Цитокины хемотаксиса (лейкоцитарные хемоаттрактанты/активирующие факторы) так же известные как хемокины, так же известные как интеркрины и SIS-цитокины, представляют собой группу воспалительных/иммуномодулирующих полипептидных факторов с молекулярной массой 6-15 кДа, которые высвобождаются различными клетками, такими как макрофаги, моноциты, эозинофилы, нейтрофилы, фибробласты, эндотелиальные клетки сосудов, гладкомышечные клетки и тучные клетки в участках воспаления (обзор в Luster, New Eng. J Med., 338, 436-445 (1998) и Rollins, Blood, 90, 909-928 (1997)). Также хемокины описаны в Oppenheim, J. J. et al., Annu. Rev. Immunol., 9: 617-648 (1991); Schall and Bacon, Curr. Opin. Immunol., 6: 865-873 (1994); Baggiolini, M., et al., and Adv. Immunol., 55: 97-179 (1994). Хемокины имеют способность стимулировать направленную миграцию клеток, процесс, известный как хемотаксис. Каждый хемокин содержит четыре остатка цистеина (C) и два внутренних дисульфидных мостика. Хемокины могут быть сгруппированы на два надсемейства на основе того, прилегают ли два амино-концевых цистеиновых остатка непосредственно друг к другу (семейство CC) или же они разделены одной аминокислотой (семейство CXC). Данные отличия коррелируют с организацией данных двух надсемейств в отдельные генные кластеры. В каждом генном кластере хемокины обычно характеризуются сходством последовательности от 25 до 60%. CXC-хемокины, такие как интерлейкин-8 (IL-8), нейтрофилактивирующий белок-2 (NAP-2) и белок с активностью по стимуляции роста меланомы (MGSA) обладают хемотаксисом преимущественно для нейтрофилов и Т-лимфоцитов, в то время как CC-хемокины, такие как RANTES, MIP-1α, MIP-1β; белки хемотаксиса моноцитов (MCP-1, MCP-2, MCP-3, MCP-4, и MCP-5) и эотаксины (-1 и -2), помимо других клеточных типов, характеризуются хемотаксисом в отношении макрофагов, Т-лимфоцитов, эозинофилов, дендритных клеток и базофилов. Также существуют хемокины лимфотактин-1, лимфотактин-2 (оба C-хемокины) и фракталкин (CXXXC-хемокин), которые не относятся к главным хемокиновым надсемействам.
MCP-1 (так же известный как MCAF (сокращение от фактора активации и хемотаксиса макрофагов) или JE) представляет собой CC-хемокин, продуцируемый моноцитами/макрофагами, гладкомышечными клетками, фибробластами и клетками эндотелия сосудов, и вызывают миграцию клеток и адгезию клеток моноцитов (см., например, Valente, A. J., et al., Biochemistry, 1988, 27, 4162; Matsushima, K., et al., J. Exp. Med., 1989, 169, 1485; Yoshimura, T., et al., J. Immunol., 1989, 142, 1956; Rollins, B. J., et al., Proc. Natl. Acad. Sci. USA, 1988, 85, 3738; Rollins, B. J., et al., Blood, 1991, 78, 1112; Jiang, Y., et al., J. Immunol., 1992, 148, 2423; Vaddi, K., et al., J. Immunol., 1994, 153, 4721), Т-лифмоцитов памяти (см., например, Carr, M. W., et al., Proc. Natl. Acad. Sci. USA, 1994, 91, 3652), Т-лимфоцитов (см., например, Loetscher, P., et al., FASEB J., 1994, 8, 1055) и клеток-естественных киллеров (см., например, Loetscher, P., et al., J. Immunol., 1996, 156, 322; Allavena, P., et al., Eur. J. Immunol., 1994, 24, 3233), а также опосредуют высвобождение гистамина базофилами (см., например, Alam, R., et al., J. Clin. Invest., 1992, 89, 723; Bischoff, S. C., et al., J. Exp. Med., 1992, 175, 1271; Kuna, P., et al., J. Exp. Med., 1992, 175, 489). Кроме того, о высоком уровне экспрессии MCP-1 сообщали при заболеваниях, где, как полагают, накопление моноцитов/макрофагов и/или Т-клеток важно при инициации или прогрессии заболеваний, таких как атеросклероз (см., например, Hayes, I. M., et al., Arterioscler. Thromb. Vasc. Biol., 1998, 18, 397; Takeya, M. et al., Hum. Pathol., 1993, 24, 534; Yla-Herttuala, S., et al., Proc. Natl. Acad. Sci. USA, 1991, 88, 5252; Nelken, N. A., J. Clin. Invest., 1991, 88, 1121), ревматоидный артрит (см., например, Koch, A. E., et al., J. Clin. Invest., 1992, 90, 772; Akahoshi, T., et al., Arthritis Rheum., 1993, 36, 762; Robinson, E., et al., Clin. Exp. Immunol., 101, 398), нефрит (см., например, Noris, M., et al., Lab. Invest., 1995, 73, 804; Wada, T., at al., Kidney Int., 1996, 49, 761; Gesualdo, L., et al., Kidney Int., 1997, 51, 155), нефропатия (см., например, Saitoh, A., et al., J. Clin. Lab. Anal., 1998, 12, 1; Yokoyama, H., et al., J. Leukoc. Biol., 1998, 63, 493), фиброз легких, саркоидоз легких (см., например, Sugiyama, Y., et al., Internal Medicine, 1997, 36, 856), астма (см., например, Karina, M., et al., J. Invest. Allergol. Clin. Immunol., 1997, 7, 254; Stephene, T. H., Am. J. Respir. Crit. Care Med., 1997, 156, 1377; Sousa, A. R., et al., Am. J. Respir. Cell Mol. Biol., 1994, 10, 142), рассеянный склероз (см., например, McManus, C., et al., J. Neuroimmunol., 1998, 86, 20), псориаз (см., например, Gillitzer, R., et al., J. Invest. Dermatol., 1993, 101, 127), воспалительное заболевание толстой кишки (см., например, Grimm, M. C., et al., J. Leukoc. Biol., 1996, 59, 804; Reinecker, H. C., et al., Gastroenterology, 1995, 106, 40), миокардит (см., например, Seino, Y., et al., Cytokine, 1995, 7, 301), эндометриоз (см., например, Jolicoeur, C., et al., Am. J. Pathol., 1998, 152, 125), внутрибрюшинная адгезия (см., например, Zeyneloglu, H. B., et al., Human Reproduction, 1998, 13, 1194), застойная сердечная недостаточность (см., например, Aurust, P., et al., Circulation, 1998, 97, 1136), хроническое заболевание печени (см., например, Marra, F., et al., Am. J. Pathol., 1998, 152, 423), вирусный менингит (см., например, Lahrtz, F., et al., Eur. J. Immunol., 1997, 27, 2484), болезнь Кавасаки (см., например, Wong, M.; et al., J. Rheumatol., 1997, 24, 1179) и сепсис (см., например, Salkowski, C. A.; et al., Infect. Immun., 1998, 66, 3569). Более того, антитело против MCP-1, как сообщалось, характеризуется ингибиторным действием или терапевтическим действием на животных-моделях ревматоидного артрита (см., например, Schimmer, R. C., et al., J. Immunol., 1998, 160, 1466; Schrier, D. J., J. Leukoc. Biol., 1998, 63, 359; Ogata, H., et al., J. Pathol., 1997, 182, 106), рассеянного склероза (см., например, Karpus, W. J., et al., J. Leukoc. Biol., 1997, 62, 681), нефрита (см., например, Lloyd, C. M., et al., J. Exp. Med., 1997, 185, 1371; Wada, T., et al., FASEB J., 1996, 10, 1418), астмы (см., например, Gonzalo, J.-A., et al., J. Exp. Med., 1998, 188, 157; Lukacs, N. W., J. Immunol., 1997, 158, 4398), атеросклероза (см., например, Guzman, L. A., et al., Circulation, 1993, 88 (suppl.), I-371), гиперчувствительности отложенного типа (см., например, Rand, M. L., et al., Am. J. Pathol., 1996, 148, 855), легочной гипертензии (см., например, Kimura, H., et al., Lab. Invest., 1998, 78, 571) и внутрибрюшинной адгезии (см., например, Zeyneloglu, H. B., et al., Am. J. Obstet. Gynecol., 1998, 179, 438). Также сообщалось, что пептидный антагонист MCP-1, MCP-1(9-76), ингибирует артрит у экспериментальных мышей (см. Gong, J.-H., J. Exp., 4ed., 1997, 186, 131), также исследования в дефицитных по MCP-1 мышах показали, что MCP-1 является существенным для рекрутирования моноцитов in vivo (см. Lu, B., et al., J. Exp. Med., 1998, 187, 601; Gu, L., et al., Moll. Cell, 1998, 2, 275).
Литература указывает на то, что хемокины, такие как MCP-1 и MIP-1α привлекают моноциты и лимфоциты в участки патологии и опосредуют их активацию, и полагают, что они тесно вовлечены в инициацию, прогрессию и поддержание заболеваний с непосредственным участием моноцитов и лимфоцитов, таких как атеросклероз, повторный стеноз, ревматоидный артрит, псориаз, астма, язвенный колит, нефрит (нефропатия), рассеянный склероз, легочный фиброз, миокардит, гепатит, панкреатит, саркоидоз, болезнь Крона, эндометриоз, застойная сердечная недостаточность, вирусный менингит, инсульт, невропатия, болезнь Кавасаки и сепсис (см., например, Rovin, B. H., et al., Am. J. Kidney. Dis., 1998, 31, 1065; Lloyd, C., et al., Curr. Opin. Nephrol. Hypertens., 1998, 7, 281; Conti, P., et al., Allergy and Asthma Proc., 1998, 19, 121; Ransohoff, R. M., et al., Trends Neurosci., 1998, 21, 154; MacDermott, R. P., et al., Inflammatory Bowel Diseases, 1998, 4, 54).
Хемокины связываются со специфичными рецепторами клеточной поверхности, принадлежащими к семейству связанных с G-белком белков с семью трансмембранными доменами (обзор в Horuk, Trends Pharm. Sci., 15, 159-165 (1994)), которые называются «хемокиновыми рецепторами». После связывания соответствующего им лиганда хемокиновые рецепторы трансдуцируют внутриклеточный сигнал через ассоциированные тримерные G-белки, что приводит, среди прочих реакций, к быстрому повышению внутриклеточной концентрации кальция, изменениям в форме клетки, повышенной экспрессии молекул клеточной адгезии, дегрануляции и осуществления миграции клеток.
Гены, кодирующие рецепторы конкретных хемокинов, клонировали, и известно, что данные рецепторы представляют собой связанные с G-белком рецепторы с семью трансмембранными доменами, присутствующие на различных популяциях лейкоцитов. До настоящего времени идентифицировали, по меньшей мере, пять CXC-хемокиновых рецепторов (CXCR1-CXCR5) и восемь CC-хемокиновых рецепторов (CCR1-CCR8). Например, IL-8 представляет собой лиганд CXCR1 и CXCR2, MIP-1α представляет собой таковой для CCR1 и CCR5, а MCP-1 является лигандом для CCR2A и CCR2B (для ссылки, см., например, Holmes, W. E., et al., Science 1991, 253, 1278-1280; Murphy P. M., et al., Science, 253, 1280-1283; Neote, K. et al, Cell, 1993, 72, 415-425; Charo, I. F., et al., Proc. Natl. Acad. Sci. USA, 1994, 91, 2752-2756; Yamagami, S., et al., Biochem. Biophys. Res. Commun., 1994, 202, 1156-1162; Combadier, C., et al., The Journal of Biological Chemistry, 1995, 270, 16491-16494, Power, C. A., et al., J. Biol. Chem., 1995, 270, 19495-19500; Samson, M., et al., Biochemistry, 1996, 35, 3362-3367; Murphy, P. M., Annual Review of Immunology, 1994, 12, 592-633). Сообщалось, что воспаление легких и образование гранулемы подавляются у CCR1-дефицитных мышей (см. Gao, J.-L., et al., J. Exp. Med., 1997, 185, 1959; Gerard, C., et al., J. Clin. Invest., 1997, 100, 2022), и что рекрутирование макрофагов и образование атеросклеротических повреждений снижалось у дефицитных по CCR2 мышей (см. Boring, L., et al., Nature, 1998, 394, 894; Kuziel, W. A., et al., Proc. Natl. Acad. Sci., USA, 1997, 94, 12053; Kurihara, T., et al., J. Exp. Med., 1997, 186, 1757; Boring, L., et al., J. Clin. Invest., 1997, 100, 2552).
Хемокиновые рецепторы также известны как сорецепторы для проникновения вирусов, что приводит к вирусной инфекции, такой как, например, инфекция ВИЧ. Обратная транскрипция и процессинг белка представляют собой классические стадии вирусного жизненного цикла, который призван блокировать противоретровирусные терапевтические средства. Хотя многие новые лекарственные средства, которые, как полагают, блокируют проникновение вирусов, остаются перспективными, в настоящее время не имеется средства, к которому ВИЧ-1 не способен обрести устойчивости. Многочисленные раунды репликации вируса требуются для генерирования генетического разнообразия, которое образует основу устойчивости. Комбинированная терапия, при которой репликация максимально подавлена, остается краеугольным камнем лечения ингибиторами проникновения, а также другими средствами. Направленное действие на множественные стадии в процессе проникновения вируса, как полагают, имеет потенциал в плане синергии (Starr-Spires et al., Clin. Lab. Med., 2002, 22 (3), 681.)
Проникновение ВИЧ-1 в CD4(+) клетки требует последовательных взаимодействий вирусных оболочечных гликопротеинов с CD4 и сорецептором, таким как хемокиновые рецепторы CCR5 и CXCR4. Внушающий доверие подход блокирования данного процесса представляет собой применение низкомолекулярных антагонистов функции сорецептора. Молекула TAK-779 представляет собой один из таких антагонистов CCR5, которые предотвращают инфекцию ВИЧ-1. TAK-779 ингибирует репликацию ВИЧ-1 на стадии слияния мембраны за счет блокирования взаимодействия гликопротеина клеточной поверхности gp120 с CCR5. Участок связывания TAK-779 на CCR5 расположен вблизи внеклеточной поверхности рецептора в полости, образованной между трансмембранными спиралями 1, 2, 3 и 7 (Dragic et al., Proc. Natl. Acad. Sci. USA, 2000, 97(10), 5639).
Хемокиновые рецепторы CXCR4 и CCR5, как полагают, используются в качестве сорецепторов штаммами ВИЧ-1, тропными в отношении Т-клеток (X4) и макрофагов (R5), соответственно, для проникновения в их клетки хозяина. Размножение штаммов R5 ВИЧ-1 на CD4 лимфоцитах и макрофагах требует экспрессии сорецептора CCR5 на клеточной поверхности. Субъекты, лишенные CCR5 (гомозиготный фенотип CCR5 дельта 32), фенотипически нормальны и устойчивы к инфекции ВИЧ-1. Проникновение вируса может ингибироваться естественными лигандами CXCR4 (CXC-хемокин SDF-1) и CCR5 (CC-хемокины RANTES, MIP-1-альфа и MIP-1-бета). Первое непептидное соединение, которое взаимодействует с CCR5, но не с CXCR4, представляет собой производное четвертичной соли аммония, называемое TAK-779, которое также имеет мощную, но изменчивую активность против ВИЧ (De Clercq et al., Antivir. Chem. Chemother. 2001, 12 Suppl. 1, 19.
SCH-C (SCH 351125) представляет собой другой низкомолекулярный ингибитор проникновения ВИЧ-1 через сорецептор CCR5. SCH-C, соединения оксима-пиперидина, представляет собой специфичный антагонист CCR5, определенный в множественных анализах связывания рецептора и трансдукции сигнала. Данное соединение специфично ингибирует инфекцию ВИЧ-1, опосредованную CCR5 в клетках астроглиомы U-87, но не имеет эффекта в отношении инфекции экспрессирующих CXCR4 клеток (Strizki et al, Proc. Natl. Acad Sci. USA, 2001, 98(22), 12718 или Tremblay et al., Antimicrobial Agents and Chemotherapy, 2002, 46(5), 1336).
AD101, химически сходный с SCH-C, также ингибирует проникновение вируса иммунодефицита человека типа 1 (ВИЧ-1) через человеческий CCR5. Обнаружено, что AD101 ингибирует проникновение ВИЧ-1 через CCR5 макаки-резуса, тогда как SCH-C этого не делает. Среди восьми остатков, которые отличаются в человеческом сорецепторе от белка макаки, только один, метионин-198, вносит вклад в интенсивность ингибирования CCR5 макаки за счет SCH-C. Положение 198 находится в трансмембранной (ТМ) спирали 5 CCR5 и не расположено в ранее выявленных участках связывания AD101 и SCH-C, которые затрагивают остатки ТМ спиралей 1, 2, 3, и 7. Основываясь на исследованиях аминокислотных замен в CCR5, предполагалось, что область CCR5 вблизи остатка 198 может воздействовать на конформационное состояние данного рецептора (Billick et al., 2004, J. Virol., 78(8), 4134).
Соответственно, лекарственные средства, которые ингибируют связывание хемокинов с соответствующими им рецепторами, могут использоваться в качестве фармацевтических средств, которые ингибируют действие хемокинов на клетки-мишени и/или блокируют проникновение вирусов в клетки, экспрессирующие данные рецепторы. Идентификация соединений, которые модулируют активность хемокиновых рецепторов или блокируют связывание вирусных белков, представляет отличный подход к конструированию лекарств для разработки фармакологических средств для лечения воспалительных заболеваний, вирусных инфекций и других заболеваний, ассоциированных с активацией хемокиновых рецепторов. Соединения по настоящему изобретению могут удовлетворить эти и другие потребности.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям формулы I:
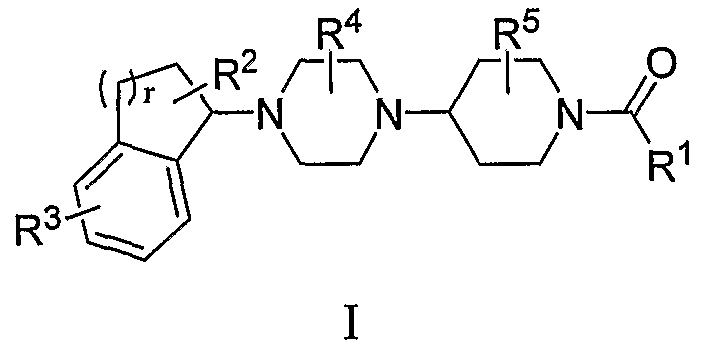
или к их фармацевтически приемлемой соли или пролекарству, где заместители определены здесь.
Настоящее изобретение далее относится к композициям, содержащим соединения формулы I и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к способам модулирования активности хемокинового рецептора, включающему взаимодействие хемокинового рецептора с соединением формулы I.
Настоящее изобретение далее относится к способам лечения заболевания, ассоциированного с экспрессией или активностью хемокинового рецептора у пациента, включающему введение указанному пациенту терапевтически эффективного количества соединения формулы I.
Настоящее изобретение далее относится к способам лечения заболевания или состояния, выбранного из воспалительного заболевания, иммунного нарушения, и вирусной инфекции у пациента, включающему в себя введение указанному пациенту терапевтически эффективного количества соединения формулы I.
Настоящее изобретение далее относится к способам лечения ВИЧ-инфекции у пациента, включающему введение указанному пациенту терапевтически эффективного количества соединения формулы I.
Настоящее изобретение далее относится к применению соединения формулы I в лечении.
Настоящее изобретение далее относится к применению соединения формулы I для получения лекарственного средства для применения в лечении.
ПОДРОБНОЕ ОПИСАНИЕ
Среди прочего, настоящее соединение относится к соединениям формулы I:
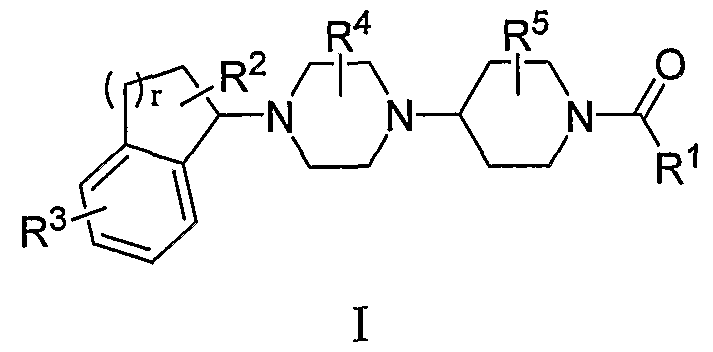
или к их фармацевтически приемлемой соли или пролекарству, где:
R1 представляет собой гетероарил, необязательно замещенный одним или несколькими R6;
R2 представляет собой H, галоген, циано, нитро, C1-C6 алкил, C1-C6 галогеналкил, C2-C6 алкенил, C2-C6 алкинил, арил, гетероарил, C3-C7 циклоалкил, гетероциклоалкил, SOR7,SO2R7, COR8, OR9, SR9, COOR9, NR10R11 или NR10COR8;
R3 представляет собой F, Cl, Br, I, C1-C4 галогеналкил, C1-C4 галогеналкокси или гетероарил;
R4 представляет собой H, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил или C1-C6 галогеналкил;
R5 представляет собой H, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил или C1-C6 галогеналкил;
R6 представляет собой H, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 галогеналкил, C1-C6 алкокси, C1-C6 галогеналкокси, амино, (C1-C6 алкил)амино или ди(C1-C6 алкил)амино;
R7 представляет собой H, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 галогеналкил, арил, гетероарил, C3-C7 циклоалкил, гетероциклоалкил, арилалкил, гетероарилалкил, (C3-C7 циклоалкил)алкил, гетероциклоалкилалкил или NR12R13;
R8 представляет собой H, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 галогеналкил, арил, гетероарил, C3-C7 циклоалкил, гетероциклоалкил, арилалкил, гетероарилалкил, (C3-C7 циклоалкил)алкил, гетероциклоалкилалкил, или NR12R13;
R9 представляет собой H, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 галогеналкил, алкоксиалкил, галогеналкоксиалкил, арилоксиалкил, гетероарилоксиалкил, циклоалкилоксиалкил, гетероциклоалкилоксиалкил, арил, гетероарил, C3-C7 циклоалкил, гетероциклоалкил, арилалкил, гетероарилалкил; (C3-C7 циклоалкил)алкил или гетероциклоалкилалкил;
R10 и R11 оба независимо представляют собой H, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 галогеналкил, арил, гетероарил, C3-C7 циклоалкил, гетероциклоалкил, арилалкил, гетероарилалкил; (C3-C7 циклоалкил)алкил или гетероциклоалкилалкил;
или R10 и R11 вместе с атомом азота, к которому они присоединены, образуют 3-, 4-, 5-, 6- или 7-членную гетероциклоалкильную группу;
R12 и R13 оба независимо представляют собой H, C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 галогеналкил, арил, гетероарил, C3-C7 циклоалкил, гетероциклоалкил, арилалкил, гетероарилалкил; (C3-C7 циклоалкил)алкил или гетероциклоалкилалкил;
или R12 и R13 вместе с атомом азота, к которому они присоединены, образуют 3-, 4-, 5-, 6- или 7-членную гетероциклоалкильную группу; и
r представляет собой 1, 2 или 3.
В некоторых вариантах осуществления R1 представляет собой 5-, 6-, 9- или 10-членную гетероарильную группу, содержащую, по меньшей мере, один образующий кольцо атом N, где указанная 5-, 6-, 9- или 10-членная гетероарильная группа необязательно замещена 1, 2, 3 или 4 группами R6.
В некоторых вариантах осуществления R1 представляет собой 9- или 10-членную гетероарильную группу, содержащую, по меньшей мере, один образующий кольцо атом N, где указанная 6-членная гетероарильная группа необязательно замещена 1, 2, 3 или 4 группами R6.
В некоторых вариантах осуществления R1 представляет собой 5- или 6-членную гетероарильную группу, содержащую, по меньшей мере, один образующий кольцо атом N, где указанная 5-членная гетероарильная группа необязательно замещена 1, 2, 3 или 4 группами R6.
В некоторых вариантах осуществления R1 представляет собой 6-членную гетероарильную группу, содержащую, по меньшей мере, один образующий кольцо атом N, где указанная 6-членная гетероарильная группа необязательно замещена 1, 2, 3 или 4 группами R6.
В некоторых вариантах осуществления R1 представляет собой 5-членную гетероарильную группу, содержащую, по меньшей мере, один образующий кольцо атом N, где указанная 5-членная гетероарильная группа необязательно замещена 1, 2, 3 или 4 группами R6.
В некоторых вариантах осуществления R1 представляет собой хинолинил, изохинолинил, нафтиридинил, индолил, индазолил, пиридил, пиримидинил, N-оксопиридил, N-оксопиримидинил, изоксазол, пиразол, пирролил, имидазолил, оксазолил или тиазолил, причем каждый из них необязательно замещен 1, 2, 3 или 4 группами R6.
В некоторых вариантах осуществления R1 представляет собой хинолинил, изохинолинил, нафтиридинил, пиридил, пиримидинил, N-оксопиридил, изоксазол или пиразол, причем каждый из них необязательно замещен 1, 2, 3 или 4 группами R6.
В некоторых вариантах осуществления R1 представляет собой пиридил, пиримидинил, N-оксопиридил, N-оксопиримидинил, изоксазол, пиразол, пирролил, имидазолил, оксазолил или тиазолил, причем каждый из них необязательно замещен 1, 2, 3 или 4 группами R6.
В некоторых вариантах осуществления R1 представляет собой пиридил, пиримидинил, N-оксопиридил, изоксазол или пиразол, причем каждый из них необязательно замещен 1, 2, 3 или 4 группами R6.
В некоторых вариантах осуществления R1 представляет собой:
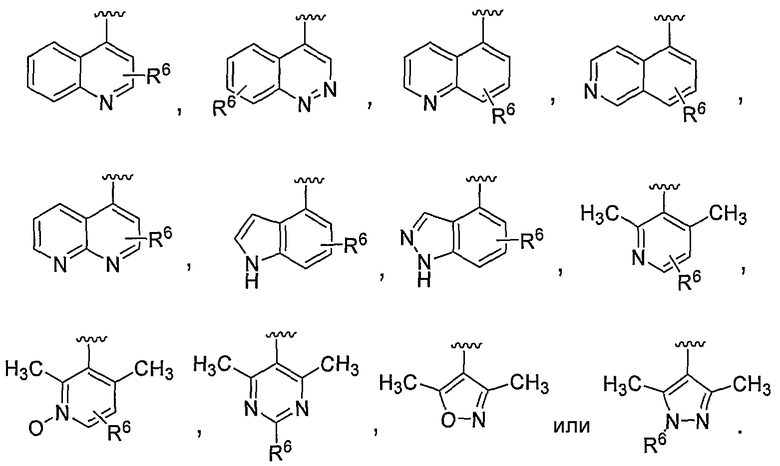
В некоторых вариантах осуществления R1 представляет собой:
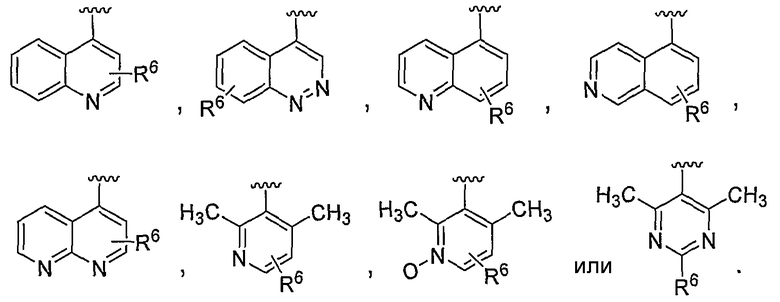
В некоторых вариантах осуществления R1 представляет собой:
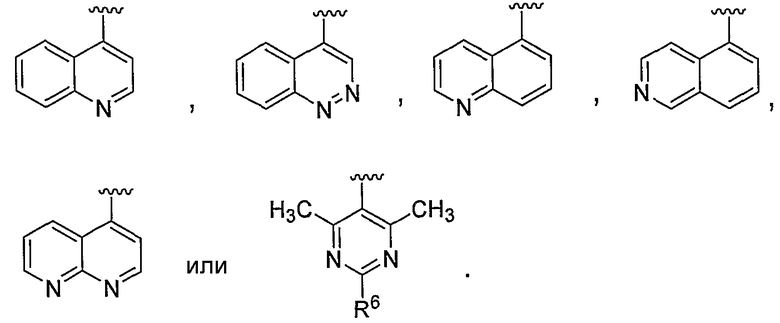
В некоторых вариантах осуществления R1 представляет собой:
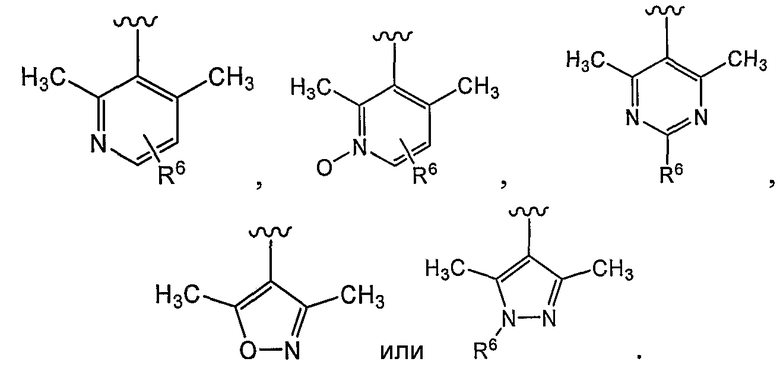
В некоторых вариантах осуществления R1 представляет собой:
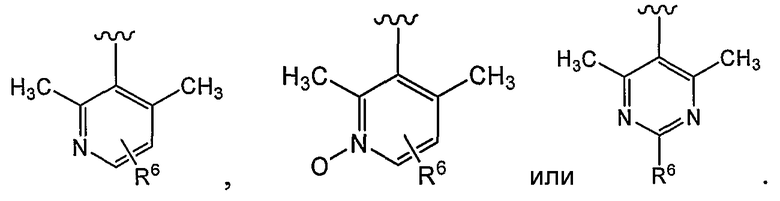
В некоторых вариантах осуществления R1 представляет собой:

В некоторых вариантах осуществления R2 представляет собой H, C1-C6 алкил, C1-C6 галогеналкил, OR9, SR9 или NR10R11.
В некоторых вариантах осуществления R2 представляет собой H или OR9.
В некоторых вариантах осуществления R3 представляет собой F, Br, CF3, или 6- или 5-членный гетероарил.
В некоторых вариантах осуществления R3 представляет собой F, Br, CF3, OCF3, тиазолил, пиримидинил, пиридил.
В некоторых вариантах осуществления R3 представляет собой F, Br или CF3.
В некоторых вариантах осуществления R4 представляет собой C1-C6 алкил.
В некоторых вариантах осуществления R4 представляет собой метил.
В некоторых вариантах осуществления R5 представляет собой C1-C6 алкил.
В некоторых вариантах осуществления R5 представляет собой метил.
В некоторых вариантах осуществления r представляет собой 1.
В некоторых вариантах осуществления r представляет собой 2.
В некоторых вариантах осуществления соединения по изобретению имеют формулу IIa или IIb:

В некоторых вариантах осуществления соединений, имеющих формулу IIa или IIb, R1 представляет собой:
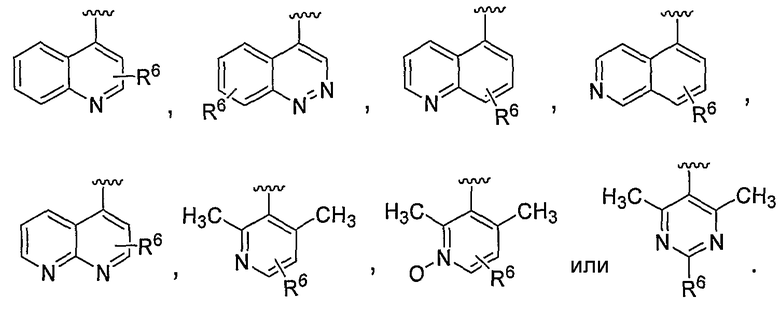
В некоторых вариантах осуществления соединений, имеющих формулу IIa или IIb, R1 представляет собой:
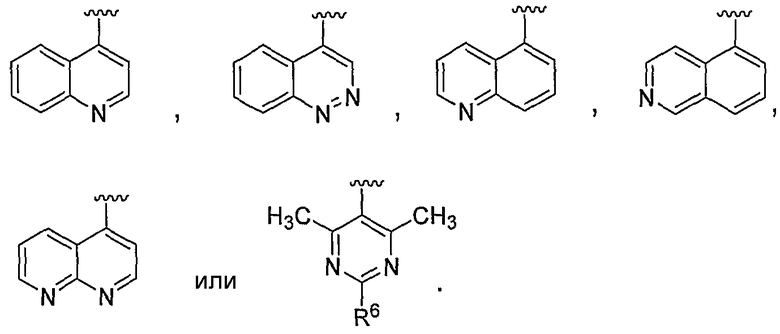
В некоторых вариантах осуществления соединений, имеющих формулу IIa или IIb, R1 представляет собой:
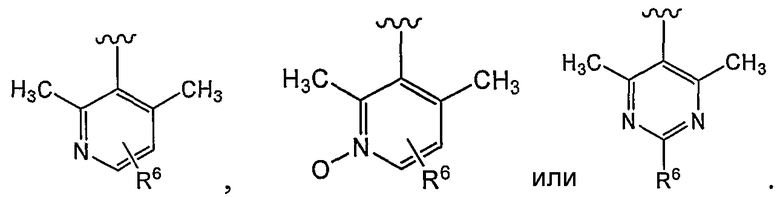
В некоторых вариантах осуществления соединений, имеющих формулу IIa или IIb, R1 представляет собой:
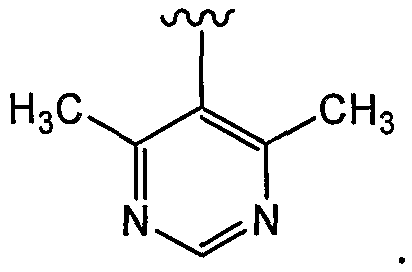
В некоторых вариантах осуществления соединений, имеющих формулу IIa или IIb, R2 представляет собой H, C1-C6 алкил, C1-C6 галогеналкил, OR9, SR9 или NR10R11.
В некоторых вариантах осуществления соединений, имеющих формулу IIa или IIb, R2 представляет собой H или OR9.
В некоторых вариантах осуществления соединений, имеющих формулу IIa или IIb, R3 представляет собой F, Br, CF3, 5- или 6-членный гетероарил.
В некоторых вариантах осуществления соединений, имеющих формулу IIa или IIb, R3 представляет собой F, Br или CF3.
Понятно, что некоторые характеристики изобретения, которые для ясности описаны в контексте отдельных вариантов осуществления, могут также предоставляться в комбинации в одном варианте осуществления. Наоборот, различные характеристики изобретения, которые для краткости описаны в контексте одного варианта осуществления, могут также предоставлять отдельно или в любой подходящей субкомбинации.
Используемый здесь термин «алкил», как подразумевается, относится к насыщенной углеводородной группе, цепь которой неразветвлена или разветвлена. Типовые алкильные группы включают метил (Me), этил (Et), пропил (например, н-пропил и изопропил), бутил (например, н-бутил, изобутил, втор-бутил, трет-бутил), пентил (например, н-пентил, изопентил, неопентил) и тому подобное. Алкильная группа может содержать от 1 примерно до 20, от 2 примерно до 20, от 1 примерно до 10, от 1 примерно до 8, от 1 примерно до 6, от 1 примерно до 4, или от 1 примерно до 3 атомов углерода.
Используемый здесь термин «алкенил» относится к алькильной группе, имеющей одну или несколько двойных углерод-углеродных связей. Типовые алкенильные группы включают этенил, пропенил, бутенил, пентенил, гексенил, бутадиенил, пентадиенил, гексадиенил и тому подобное.
Используемый здесь термин «алкинил» относится к алкильной группе, имеющей одну или несколько тройных углерод-углеродных связей. Типовые алкинильные группы включают этинил, пропинил, бутинил, пентинил и тому подобное.
Используемый здесь термин «галогеналкил» относится к алкильной группе, имеющей один или несколько галогеновых заместителей. Типовые галогеналкильные группы включают CF3, C2F5, CHF2, CCl3, CHCl2, C2Cl5 и тому подобное. Алкильная группа, в которой все атомы водорода замещены галогеновыми атомами, может обозначаться как «пергалогеналкил». Типовые пергалогеналкильные группы включают CF3 и C2F5.
Используемый здесь термин «арил» относится к моноциклическим или полициклическим ароматическим углеводородам, таким как, например, фенил, нафтил, антраценил, фенантренил, инданил, инденил и тому подобное. В некоторых вариантах осуществления арильные группы имеют от 6 примерно до 18 углеродных атомов.
Используемый здесь термин «циклоалкил» относится к неароматическим циклическим углеводородам, включая циклические алкильные, алкенильные и алкинильные группы. Циклоалкильные группы могут включать би- или полициклические кольцевые системы и могут необязательно содержать ненасыщенные связи. Типовые циклоалкильные группы включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклопентенил, циклогексенил, циклогексадиенил, циклогептатриенил, норборнил, норпинил, норкарнил, адамантил и тому подобное. Также в определение циклоалкила включены радикалы, которые имеют одно или несколько ароматических колец, конденсированных (т.е. имеющих общую связь) с циклоалкильным кольцом, например, бензопроизводные циклопентана (инданил), циклогексана (тетрагидронафтил) и тому подобное. Циклоалкильные группы могут иметь примерно от 3 до 20, 3 до 12 или 3 до 7 углеродных атомов.
Используемые здесь «гетероарильные» группы представляют собой моноциклические или полициклические ароматические углеводороды, имеющие, по меньшей мере, один гетероатомный член кольца, такой как сера, кислород или азот. Гетероарильные группы включают в качестве не ограничивающих примеров пиридил, N-оксопиридил, пиримидинил, N-оксопиримидинил, пиразинил, пиридазинил, триазинил, нафтиридинил, фурил, хинолил, изохинолил, тиенил, имидазолил, тиазолил, индолил, пирролил, оксазолил, бензофурил, бензотиенил, бензтиазолил, изоксазолил, пиразолил, триазолил, тетразолил, индазолил, 1,2,4-тиадиазолил, изотиазолил, бензотиенил, пуринил, карбазолил, бензимидазолил, 2,3-дигидробензофуранил, 2,3-дигидробензотиенил, 2,3-дигидробензотиенил-S-оксид, 2,3-дигидробензотиенил-S-диоксид и тому подобное. В некоторых вариантах осуществления гетероарильные группы могут иметь от 1 примерно до 20 углеродных атомов и в дальнейших осуществлениях примерно от 3 до 20 углеродных атомов. В некоторых вариантах осуществления гетероарильные группы имеют от 1 примерно до 4, от 1 примерно до 3 или от 1 до 2 гетероатомов. В некоторых вариантах осуществления гетероарильная группа имеет от 5 до 50, от 5 до 20, от 5 до 14 или от 5 до 7 членов кольца. В некоторых вариантах осуществления гетероарильная группа представляет собой 5-, 6-, 9- или 10-членную группу. В некоторых вариантах осуществления гетероарильная группа содержит, по меньшей мере, один кольцеобразующий атом N.
Используемый здесь термин «гетероциклоалкил» относится к неароматическому циклическому углеводороду, включая циклические алкильные, алкенильные и алкинильные группы, в котором один или несколько кольцеобразующих атомов замещены гетероатомом, таким как атом О, N или S. Типовые гетероциклоалкильные группы включают пиперидинил, пиролидинил, морфолин, тетрагидрофуранил и тому подобное. Также в определение гетероциклоалкила включены радикалы, которые имеют одно или несколько ароматических колец, конденсированных (т.е. имеющих общую связь) с неароматическим гетероциклическим кольцом, например, фталимидил, нафталимидил, пиромеллитиновый диимидил, фталанил, и бензо-производные насыщенных гетероциклов, таких как индоленовая и изоиндоленовая группы. В некоторых вариантах осуществления гетероциклоалкильная группа имеет от 3 до 20, от 3 до 14 или от 3 до 7 членов кольца.
Используемый здесь термин «галоген» или «галогено» включает в себя фтор, хлор, бром и йод.
Используемый здесь термин «алкокси» относится к -O-алкильной группе. Типовые алкоксигруппы включают метокси, этокси, пропокси (например, н-пропокси и изопропокси), трет-бутокси и тому подобное. «Галогеналкокси» относится к -O-галогеналкильной группе.
Используемый здесь термин «арилалкил» относится к алкильной группе, замещенной, по меньшей мере, одной арильной группой. Типовой арилалкильной группой является бензил.
Используемый здесь термин «циклоалкилалкил» относится к алкильной группе, замещенной, по меньшей мере, одной циклоалкильной группой.
Используемый здесь термин «гетероарилалкил» относится к алкильной группе, замещенной, по меньшей мере, одной гетероарильной группой.
Используемый здесь термин «гетероциклоалкилалкил» относится к алкильной группе, замещенной, по меньшей мере, одной гетероциклоалкильной группой.
Используемый здесь термин «арилокси» относится к -О-арилу.
Используемый здесь термин «гетероарилокси» относится к -O-гетероарилу.
Используемый здесь термин «циклоалкилокси» относится к -О-циклоалкилу.
Используемый здесь термин «гетероциклоалкилокси» относится к -О-гетероциклоалкилу.
Используемый здесь термин «алкоксиалкил» относится к алкильной группе, замещенной, по меньшей мере, одной алкоксигруппой. Типовые алкоксиалкильные группы включают метоксиметил, метоксиэтил, метоксипропил и тому подобное.
Используемый здесь термин «галогеналкоксиалкил» относится к алкильной группе, замещенной, по меньшей мере, одной галогеналкоксигруппой.
Используемый здесь термин «арилалкоксиалкил» относится к алкильной группе, замещенной, по меньшей мере, одной арилоксигруппой.
Используемый здесь термин «циклоалкилоксиалкил» относится к алкильной группе, замещенной, по меньшей мере, одной циклоалкилоксигруппой.
Используемый здесь термин «гетероарилоксиалкил» относится к алкильной группе, замещенной, по меньшей мере, одной гетероарилоксигруппой.
Используемый здесь термин «гетероциклоалкоксиалкил» относится к алкильной группе, замещенной, по меньшей мере, одной гетероциклоалкилоксигруппой.
Используемый здесь термин «амино» относится к NH2. Сходным образом, термин «алкиламино» относится к аминогруппе, замещенной алкильной группой, а термин «диалкиламино» относится к аминогруппе, замещенной двумя алкильными группами.
Используемый здесь термин «замещенный» указывает на то, что, по меньшей мере, один атом водорода химической группы замещен на не относящийся к водороду радикал. Когда приведенная здесь химическая группа «замещена», она может иметь до полного набора заместителей, при обеспечении того, что полученное в результате соединение является стабильным или имеет стабильную структуру; например, метильная группа может быть замещена 1, 2, или 3 заместителями, метиленовая группа может быть замещена 1 или 2 заместителями, фенильная группа может замещаться 1, 2, 3, 4 или 5 заместителями и тому подобное.
Описанные здесь соединения могут быть асимметричными (например, иметь один или несколько стереоцентров). Подразумеваются все стереоизомеры, такие как энантиомеры и диастереоизомеры, кроме указанных иначе случаев. Соединения по настоящему изобретению, которые содержат асимметрично замещенные атомы углерода, могут выделяться в оптически активной форме или в виде рацемата. Способы получения оптически активных форм из оптически активных исходных материалов, например, путем разрешения рацемических смесей или путем стереоселективного синтеза, известны в данной области. Многие геометрические изомеры олефинов, двойных связей C=N и тому подобного также могут присутствовать в описанных здесь соединениях, и все такие стабильные изомеры относятся к настоящему изобретению. Цис- и транс-геометрические изомеры соединений по настоящему изобретению описаны и могут выделяться в виде смеси изомеров или в виде отдельных изомерных форм.
Разрешение рацемических смесей соединений может проводиться любыми из многочисленных способов, известных в данной области. Типовой способ включает в себя фракционную перекристаллизацию с использованием «разрешающей хиральность кислоты», которая является оптически активной солеобразующей органической кислотой. Подходящие разрешающие агенты для способов фракционной перекристаллизации представляют собой, например, оптически активные кислоты, такие как D- и L-формы винной кислоты, диацетилвинной кислоты, дибензоилвинной кислоты, миндальной кислоты, яблочной кислоты, молочной кислоты или различных оптически активных камфорсульфоновых кислот, таких как β-камфорсульфоновая кислота. Другие разрешающие средства, подходящие для способов фракционной перекристаллизации, включают стереоизомерно чистые формы α-метилбензиламина (например, S- и R-формы, или диастереомерно чистые формы), 2-фенилглицинол, норэфедрин, эфедрин, н-метилэфедрин, циклогексилэтиламин, 1,2-диаминоциклогексан и тому подобное.
Разрешение рацемических смесей также может проводиться элюцией на колонке, забитой оптически активным разрешающим агентом (например, динитробензоилфенилглицином). Подходящая композиция элюирующего растворителя может определяться специалистом в данной области.
Соединения по изобретению также могут включать таутомерные формы, такие как кето-енольные таутомеры. Таутомерные формы могут быть в равновесии или стерически заблокированы в одной форме подходящей заменой.
Соединения по изобретению также включают гидраты и сольваты.
Соединения по изобретению могут также включать все изотопы атомов, встречающихся в виде промежуточных соединений или конечных соединений. Изотопы включают те атомы, которые имеют то же атомное число, но другие значения массы. Например, изотопы водорода включают тритий и дейтерий.
Выражение «фармацевтически приемлемый» применяется здесь для обозначения тех соединений, материалов, композиций, и/или дозированных форм, которые в рамках обоснованных медицинских представлений подходят для применения в контакте с тканями человеческих существ и животных без избыточной токсичности, ирритации, аллергической реакции, или других проблем или осложнений, соизмеримо с обоснованным отношением польза/риск.
Настоящее изобретение также относится к фармацевтически приемлемым солям описанных здесь соединений. Используемый здесь термин «фармацевтически приемлемые соли» относится к производным описанных соединений, где исходное соединение модифицировано преобразованием существующего кислотного или основного остатка в форму его соли. Примеры фармацевтически приемлемых солей включают в качестве не ограничивающих примеров минеральные или органические соли основных остатков, таких как амины; щелочные или органические соли кислых остатков, таких как карбоновые кислоты; и тому подобное. Фармацевтически приемлемые соли по настоящему изобретению включают общепринятые нетоксичные соли или четвертичные аммонийные соли исходного образованного соединения, например, из нетоксичных неорганических или органических кислот. Фармацевтически приемлемые соли по настоящему изобретению могут синтезироваться из исходного соединения, которое содержит основной или кислый радикал, общепринятыми химическими методами. В общем, такие соли могут быть получены путем взаимодействия форм данных соединений в виде свободной кислоты или основания со стехиометрическим количеством подходящего основания или кислоты в воде или в органическом растворителе или в их смеси; в основном, предпочтительны неводные среды, такие как простой эфир, этилацетат, этанол, изпропанол или ацетонитрил. Списки подходящих солей находятся в Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, Pa., 1985, p. 1418 и в Journal of Pharmaceutical Science, 66, 2 (1977), причем каждый из данных источников включен сюда полностью в качестве ссылки.
Настоящее изобретение также включает в себя пролекарства описанных здесь соединений. Используемый здесь термин «пролекарства» относится к любым ковалентно связанным носителям, которые высвобождают активное конечное лекарство при введении субъекту-млекопитающему. Пролекарства могут быть получены модификацией функциональных групп, присутствующих в соединениях таким путем, что модификации отщепляются, путем рутинных манипуляций или in vivo, от конечных соединений. Пролекарства включают соединения, в которых гидроксильные, амино-, сульфгидрильные или карбоксильные группы связаны с любой группой, которая при введении субъекту-млекопитающему отщепляется с образованием свободной гидроксильной, амино-, сульфгидрильной или карбоксильной группы, соответственно. Не ограничивающие примеры пролекарственных средств включают ацетатные, формиатные и бензоатные производные функциональных спиртовых и аминогрупп в соединениях по изобретению. Получение и применение пролекарств обсуждается в T. Higuchi and V. Stella, "Pro-drugs as Novel Delivery Systems, " Vol. 14 of the A.C.S. Symposium Series, и в Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987, причем оба данных источника включены сюда полностью в качестве ссылки.
Синтез
Соединения по изобретению, включая их соли, гидраты и сольваты, могут быть получены с использованием известных способов органического синтеза и могут синтезироваться по любому из возможных многочисленных путей синтеза.
Взаимодействия для получения соединений по изобретению могут проводиться в подходящих растворителях, которые могут быть легко выбраны специалистом в области органического синтеза. Подходящие растворители, по существу, могут не взаимодействовать с исходными веществами (реагентами), промежуточными продуктами или продуктами при температурах проведения взаимодействий, например, при температурах, которые могут меняться от температуры плавления до температуры кипения растворителя. Данное взаимодействие может проводиться в одном растворителе или в смеси более одного растворителя. В зависимости от конкретной стадии взаимодействия, могут быть выбраны подходящие растворители для конкретной стадии взаимодействия.
Получение соединений по изобретению может включать защиту или снятие защиты с различных химических групп. Потребность в защите и снятии защиты и выбор подходящих защитных групп может легко определяться специалистом в данной области. Сведения о химии защитных групп могут быть найдены, например, в T. W. Green and P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd. Ed., Wiley & Sons, Inc., New York (1999), причем данный источник включен сюда полностью в качестве ссылки.
Мониторинг взаимодействий может проводиться любым подходящим способом, известным в данной области. Например, мониторинг образования продукта может проводиться посредством спектроскопии, например, путем спектроскопии ядерного магнитного резонанса (например, 1H или 13C), инфракрасной спектроскопии, спектрофотометрии (например, УФ-видимой), или масс-спектрометрии, или путем хроматографии, такой как высокоэффективная жидкостная хроматография (ВЭЖХ) или тонкослойная хроматография.
Типовые пути синтеза соединений по изобретению предоставлены ниже на схемах 1-5, где составляющие члены изображенных формул определены в данном описании.
Схема 1
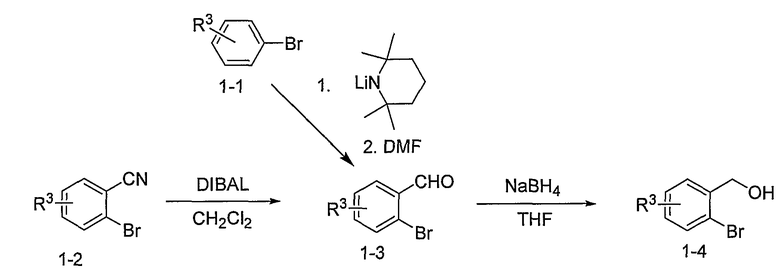
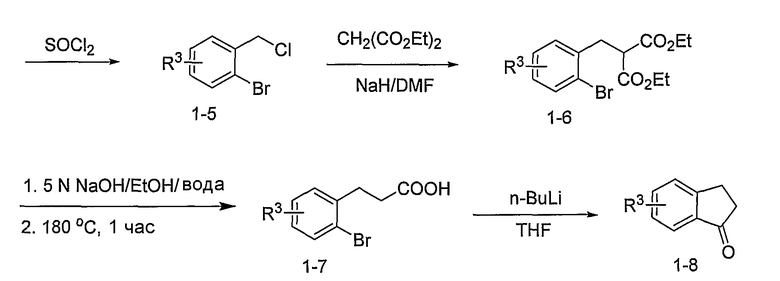
Промежуточные инданоны формулы 1-8 могут синтезироваться с использованием процедур, показанных на схеме 1. Например, бензальдегид 1-3 может образоваться депротонированием бромбензола (1-1) сильным основанием, таким как 2,2,6,6-тетраметилпиперидин/н-бутиллитий с последующим гашением, например, за счет DMF. Альтернативно, бензальдегид 1-3 может образоваться восстановлением бензонитрила (1-2) с использованием подходящего восстанавливающего агента, такого как гидрид диизобутилалюминия (DIBAL). После восстановления альдегида до спирта с использованием следующего восстанавливающего агента, такого как борогидрид натрия, полученный спирт 1-4 может преобразовываться в хлорид обработкой подходящим хлорирующим агентом, таким как тионилхлорид. Замещение хлорида 1-5 диэтилмалонатом с использованием подходящего основания (например, гидрида натрия) приводит к получению сложного диэфира 1-6. Омыление сложного диэфира с использованием основания, такого как гидроксид натрия, с последующим декарбоксилированием, дает монокарбоновую кислоту 1-7. Обработка 1-7 подходящим циклизующим агентом, таким как н-бутиллитий, приводит к получению циклического продукта индан-1-она 1-8.
Схема 2
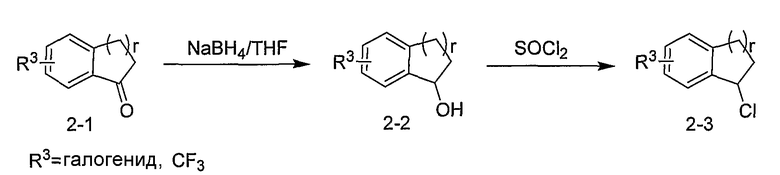
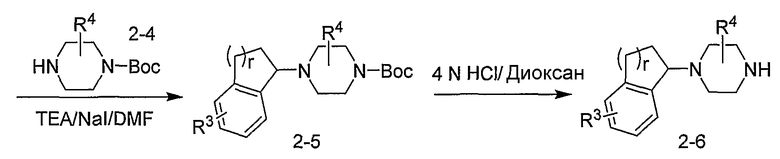
Промежуточные продукты формулы 2-6 могут синтезироваться с использованием способов, показанных на схеме 2. Кетоновое производное формулы 2-1 может подвергаться восстановлению с использованием подходящего восстанавливающего агента, такого как борогидрид натрия, с получением спирта 2-2. После преобразования спирта в хлорид с использованием подходящего хлорирующего агента, такого как SOCl2, хлорид 2-3 взаимодействует с производным пиперазина формулы 2-4 с получением 2-5. Удаление защитной группы Boc с использованием кислоты, такой как 4 Н HCl в диоксане приводит к получению промежуточных продуктов формулы 2-6.
Схема 3
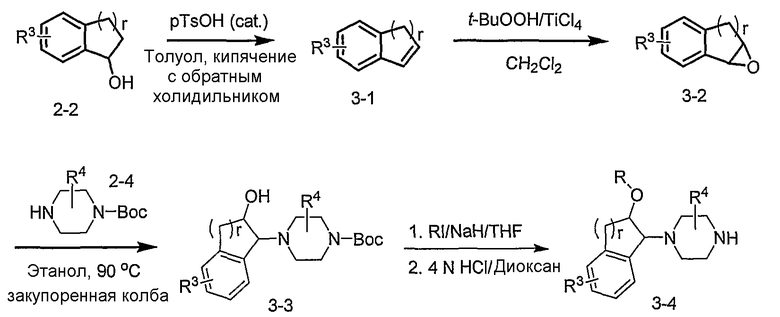
Промежуточные продукты формулы 3-4 могут быть получены с использованием последовательности действий, показанной на схеме 3. Промежуточный спирт 2-2 подвергают дегидрированию в подходящих условиях (например, р-TsOH, кипячение в толуоле с обратным холодильником) с получением индена 3-1. Эпоксидирование с использованием подходящего окислителя, такого как трет-бутилгидропероксид дает эпоксид 3-2. Открытие кольца эпоксида пиперазиновым производным формулы 2-4 предоставляет 3-3. Алкилирование спирта в 3-3 алкилирующим агентом, таким как алкилйодид (RI) с последующим удалением Boc с использованием кислоты, дает промежуточные продукты формулы 3-4 (в которой R представляет собой алкильную группу).
Схема 4

Соединения формулы 4-4 могут быть получены процедурами, описанными на схеме 4. Взаимодействие производного пиперазина формулы 4-1 с защищенным пиперидином формулы 4-2 (Pr представляет собой защитную группу аминогруппы, такую как Boc) предоставляет производное 4-3. После удаления защитной группы аминогруппы (Pr) с использованием подходящего реагента (например, кислоты, такой как 4 Н HCl в диоксане), полученный свободный амин может быть присоединен к карбоновой кислоте с использованием подходящего агента присоединения, такого как BOP, с получением соединений формулы 4-4.
Схема 5
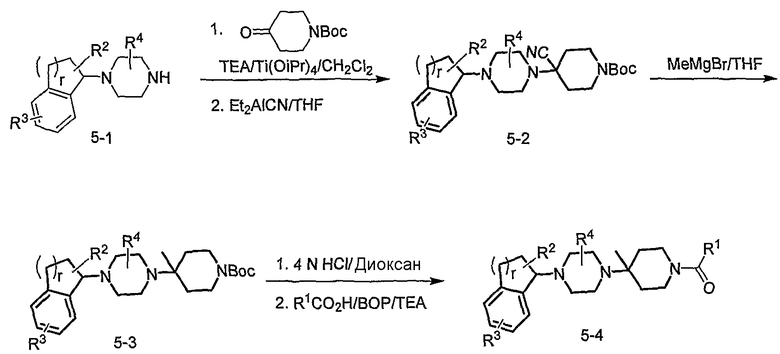
Соединения формулы 5-4 могут быть получены с использованием процедур, описанных на схеме 5. Взаимодействие производного пиперазина формулы 5-1 с трет-бутил-4-оксо-1-пиперидинкарбоксилатом с последующей обработкой цианидом диэтилалюминия приводит к образованию цианопроизводного 5-2. Замещение цианового остатка бромидом метилмагния дает 5-3. После удаления Boc-группы с использованием кислоты, такой как 4 Н HCl в диоксане, полученный в результате амин может присоединяться к карбоновой кислоте с использованием агента присоединения, такого как BOP, с получением соединений формулы 5-4.
Схема 6
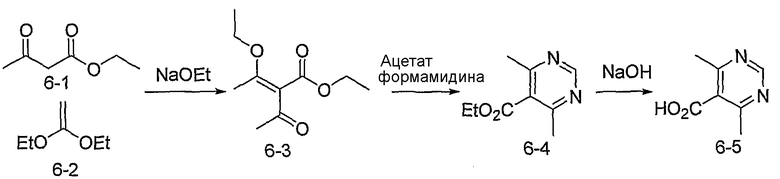
4,6-Диметилпиримидин-5-карбоновые кислоты (6-5) могут быть получены с использованием процедур, показанных на схеме 6. Взаимодействие этилацетоацетата с диэтилацеталем кетена в присутствии основания, такого как этоксид натрия, дает промежуточный продукт 6-3. Циклизация 6-3 ацетатом формамидина предоставляет этиловый сложный эфир 6-4, который омыляется с получением карбоновой кислоты 6-5.
Схема 7
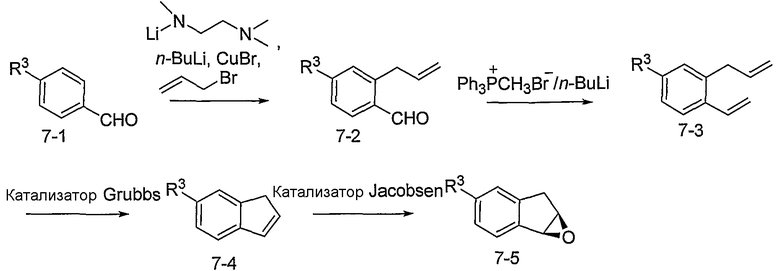
Альтернативно, соединения формулы I могут синтезироваться с использованием процедур, показанных на схемах 7-9. Обработка литием производного бензальдегида 7-1 посредством н-бутиллития в присутствии N,N,N'-триметилэтан-1,2-диамина с последующим гашением аллилбромидом предоставляет производное аллила 7-2. После преобразования альдегида в олефин обработкой Ph3PCH3Br/н-BuLi, 7-3 циклизуют с использованием катализатора Grubbs с получением производного индена 7-4. Асимметричное эпоксидирование с использованием катализатора Jacobsen приводит к получению эпоксида 7-5.
Схема 8
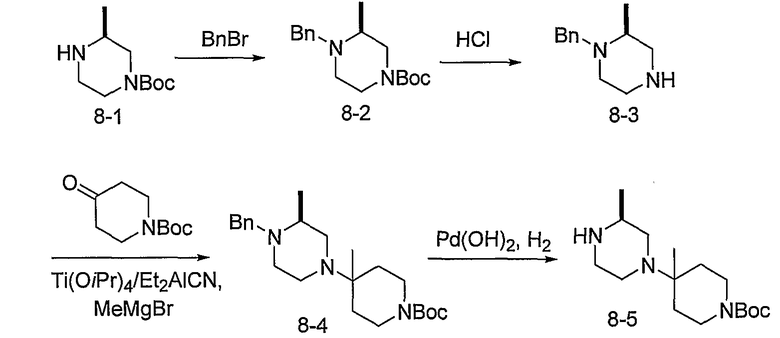
Алкилирование 4-Boc-2-метилпиперазина 8-1 бензилбромидом с последующим удалением Boc с использованием кислоты, такой как HCl, предоставляет 8-3. Промежуточный продукт 8-3 может преобразовываться в 8-4 с использованием способа, описанного на схеме 5. Удаление бензильной группы в 8-4 гидрированием с использованием катализатора Pd(OH)2 приводит к получению промежуточного продукта 8-5.
Схема 9
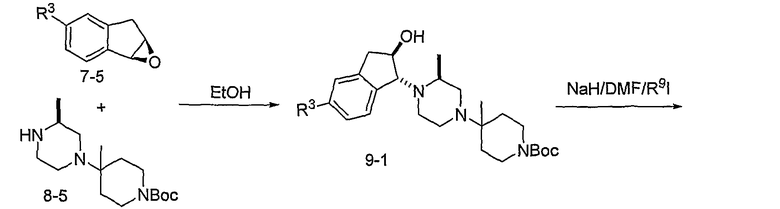
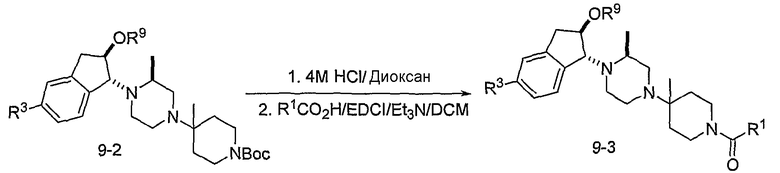
Промежуточные продукты 7-5 и 8-5 могут конденсироваться при повышенной температуре в таком растворителе, как этанол, с образованием промежуточного продукта 9-1, как показано на схеме 9. Алкилирование полученного спирта R9I может проводиться с использованием основания, такого как гидрид натрия. После удаления Boc-группы в 9-2, присоединения полученного в результате амина к RCO2H с использованием агента присоединения, такого как EDCI, предоставляет соединения формулы 9-3.
Схема 10
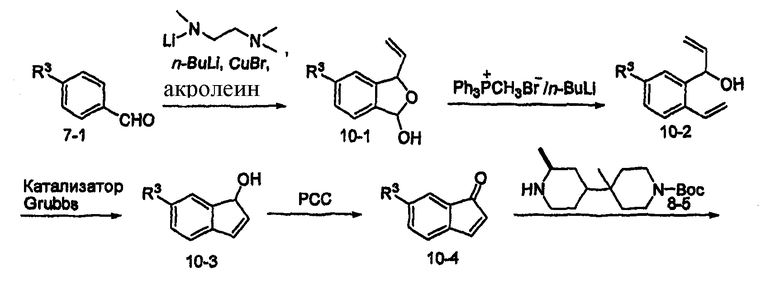
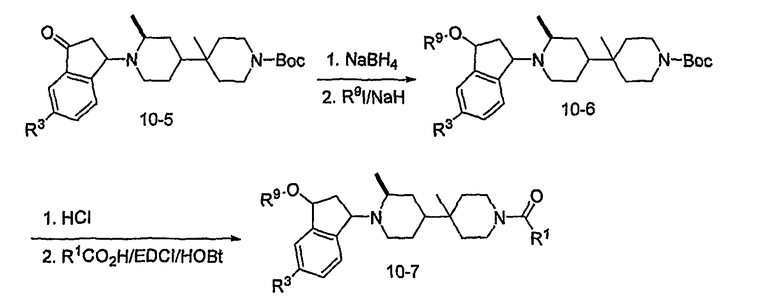
Альтернативно, соединения формулы I могут быть получены, как показано на схеме 10. Производное бензальдегида 7-1 может алкилироваться обработкой н-бутиллитием в присутствии N,N,N'-триметилэтан-1,2-диамина с последующим тушением акролеином. Полученный в результате полуацеталь 10-1 может преобразовываться в олефин обработкой Ph3PCH3Br/н-бутиллитием. Циклизация с использованием катализатора Grubbs дает производное 3-гидроксииндена 10-3, которое может подвергаться окислению с использованием окислителя, такого как хлорхромат пиридиния (PCC). Присоединение по Michael промежуточного продукта 8-5 к полученному в результате кетону 10-4 дает промежуточный продукт 10-5. После восстановления кетона до спирта алкилирование R9I может проводиться с использованием основания, такого как гидрид натрия. Удаление Boc с последующим присоединением к R1CО2H с использованием агента присоединения, такого как EDCI/HOBt, дает соединения формулы 10-7.
Способы
В некоторых вариантах осуществления, соединения по изобретению могут изменять активность одного или более хемокиновых рецепторов. Термин «изменять» относится к способности увеличивать или уменьшать активность рецептора. Соответственно, соединения по изобретению могут использоваться в способах изменения хемокиновых рецепторов путем контакта рецептора с одним или более соединениями или смесями, описанными здесь. В некоторых вариантах осуществления, соединения по настоящему изобретению могут действовать как ингибиторы хемокиновых рецепторов. В других вариантах осуществления, соединения по изобретению могут применяться для изменения активности хемокиновых рецепторов индивидуума при необходимости изменения рецептора путем введения изменяющего количества соединения формулы I.
В некоторых вариантах осуществления, соединения по изобретению могут связываться с хемокиновым рецептором таким образом, что блокируют или ингибируют связывание эндогенных и других лигандов хемокиновых рецепторов. В некоторых вариантах осуществления, соединения по изобретению могут блокировать или ингибировать связывание экзогенных лигандов, включающих в себя вирусные белки, вовлеченные в проникновение вируса в клетки, экспрессирующие хемокиновые рецепторы. Соответственно, соединения по изобретению могут блокировать проникновение вируса и ингибировать вирусную инфекцию. В некоторых вариантах осуществления, соединения по изобретению могут ингибировать инфицирование вирусом иммунодефицита человека (ВИЧ) путем, например, блокирования взаимодействия хемокиновых рецепторов (например, CCR5) с гликопротеином120 (gp120) ВИЧ.
Хемокиновые рецепторы, с которыми настоящие соединения связываются и/или изменяют их, включают любые хемокиновые рецепторы. В некоторых вариантах осуществления, хемокиновые рецепторы принадлежат семейству хемокиновых рецепторов СС, включая в себя, например, CCR1, CCR2, CCR3, CCR4, CCR5, CCR6, CCR7 и CCR8. В некоторых вариантах осуществления, хемокиновым рецептором является CCR2. В некоторых вариантах осуществления, хемокиновым рецептором является CCR5.
Соединения по изобретению могут быть избирательными. Под термином «избирательный» подразумевается, что соединение связывается или ингибирует один хемокиновый рецептор с большим сродством или силой, соответственно, в сравнении с, по меньшей мере, одним другим хемокиновым рецептором.
Соединения по изобретению могут избирательно связываться с CCR5, что означает, что соединения по изобретению могут связываться с CCR5 с большим сродством, чем с другим хемокиновым рецептором, таким как, по меньшей мере, один из CCR1, CCR2, CCR3, CCR4, CCR6, CCR7 и CCR8. В некоторых вариантах осуществления, соединения по изобретению обладают связывающей избирательностью к ССR5, большей, чем к ССR2. В некоторых вариантах осуществления, соединения по изобретению обладают связывающей избирательностью к ССR5, большей, чем к ССR1. В некоторых вариантах осуществления, соединения по изобретению обладают связывающей избирательностью к ССR5, большей, чем к любым другим ССR. Избирательность может быть, по меньшей мере, примерно, 10-кратной, по меньшей мере, примерно, 20-кратной, по меньшей мере, примерно, 50-кратной, по меньшей мере, примерно, 100-кратной, по меньшей мере, примерно, 200-кратной, по меньшей мере, примерно, 500-кратной или, по меньшей мере, примерно, 1000-кратной. В некоторых вариантах осуществления, соединения по изобретению обладают связывающим сродством к CCR5, которое, по меньшей мере, примерно, 10-кратно, по меньшей мере, примерно, 20-кратно, по меньшей мере, примерно, 50-кратно, по меньшей мере, примерно, 100-кратно, по меньшей мере, примерно, 200-кратно, по меньшей мере, примерно, 500-кратно или, по меньшей мере, примерно, 1000-кратно превышает связывающее сродство к CCR1, CCR2 или к любому другому хемокиновому рецептору. Связывающее сродство может быть измерено в соответствии с рутинными способами, известными в науке, такими, как анализы, приведенные здесь.
В некоторых вариантах осуществления, соединения по изобретению могут быть избирательными ингибиторами CCR5, то есть соединения по изобретению могут ингибировать активность CCR5 более сильно, чем, по меньшей мере, одного другого хемокинового рецептора, такого как, например, CCR1, CCR2, CCR3, CCR4, CCR6, CCR7 и CCR8. В некоторых вариантах осуществления, соединения по изобретению обладают ингибирующей избирательностью к ССR5, большей, чем к ССR2. В некоторых вариантах осуществления, соединения по изобретению обладают ингибирующей избирательностью к ССR5, большей, чем к ССR1. В некоторых вариантах осуществления, соединения по изобретению обладают ингибирующей избирательностью к ССR5, большей, чем к любым другим ССR. Избирательность может быть, по меньшей мере, примерно, 10-кратной, по меньшей мере, примерно, 20-кратной, по меньшей мере, примерно, 50-кратной, по меньшей мере, примерно, 100-кратной, по меньшей мере, примерно, 200-кратной, по меньшей мере, примерно, 500-кратной или, по меньшей мере, примерно, 1000-кратной. В некоторых вариантах осуществления, соединения по изобретению обладают ингибирующим сродством к CCR5, которое, по меньшей мере, примерно, 10-кратно, по меньшей мере, примерно, 20-кратно, по меньшей мере, примерно, 50-кратно, по меньшей мере, примерно, 100-кратно, по меньшей мере, примерно, 200-кратно, по меньшей мере, примерно, 500-кратно или, по меньшей мере, примерно, 1000-кратно превышает связывающее сродство к CCR1, CCR2 или к любому другому хемокиновому рецептору. Ингибирующая способность может быть измерена в соответствии с рутинными способами, известными в науке, такими, как анализы, приведенные здесь.
Другой аспект настоящего изобретения относится к способам лечения заболеваний или нарушений, связанных с хемокиновыми рецепторами, у индивидуума (например, пациента) путем введения индивидууму, при необходимости такого лечения, терапевтически эффективного количества или дозы соединения по настоящему изобретению, или его фармацевтического препарата. Заболевания, связанные с хемокиновыми рецепторами, могут включать любое заболевание, нарушение или условие, которое прямо опосредованно связано с экспрессией или активностью хемокиновых рецепторов. Заболевания, связанные с хемокиновыми рецепторами, могут также включать любое заболевание, нарушение или условие, которое может быть предотвращено, излечено или течение которого может быть облегчено путем изменения активности хемокиновых рецепторов. Заболевания, связанные с хемокиновыми рецепторами, могут, кроме того, включать любое заболевание, нарушение или условие, характеризующееся связыванием инфекционного агента, такого как вирус или вирусный белок, с хемокиновыми рецепторами. В некоторых вариантах осуществления, заболевание, связанное с хемокиновыми рецепторами, является CCR5-связанным заболеванием, таким как инфекция ВИЧ.
Примеры заболеваний, нарушений или условий, связанных с хемокиновыми рецепторами, включают воспаление или воспалительные заболевания, иммунные нарушения и вирусные инфекции. Примеры воспалительных заболеваний включают заболевания, имеющие воспалительный компонент, такие как астма, аллергический ринит, рестеноз, атеросклероз, множественный склероз, болезнь Крона, язвенный колит, гиперчувствительные заболевания легких, аллергический пневмонит, эозинофильная пневмония, гиперчувствительность замедленного типа, астма, интерстициальная болезнь легких (ИБЛ) (например, идиопатический легочный фиброз или ИБЛ, связанная с ревматоидным артритом, системной красной волчанкой, анкилозирующим спондилитом, системным склерозом, синдромом Шегрена, полимиозитом или дерматомиозитом) и подобные заболевания. Примеры иммунных нарушений включают ревматоидный артрит, псориатический артрит, системную красную волчанку, злокачественную миастению, юношеский диабет; гломерулонефрит, аутоиммунный тиреоидит, реакцию отторжения трансплантата, включающую в себя отторжение аллотрансплантата и болезнь трансплантат-против-хозяина. Пример вирусной инфекции включает в себя инфекцию ВИЧ.
Используемый здесь термин «взаимодействие» относится к совместной доставке указанных агентов в системе in vitro или in vivo. Например, «взаимодействие» хемокиновых рецепторов с соединением по изобретению включает в себя введение соединения по настоящему изобретению индивидууму или пациенту, например, человеку, имеющему хемокиновые рецепторы, так же, как и, например, введение соединения по изобретению в образец, содержащий клеточный или очищенный препарат, содержащий хемокиновые рецепторы.
Используемые здесь термины «индивидуум» или «пациент», применяемые попеременно, относятся к любому животному, включая в себя млекопитающих, преимущественно мышей, крыс, других грызунов, кроликов, собак, кошек, свиней, коров, овец, лошадей или приматов, а наиболее предпочтительно - людей.
Используемая здесь фраза «терапевтически эффективное количество» относится к количеству активного соединения или фармацевтического агента, которое вызывает биологический или медицинский ответ в ткани, системе, животном, индивидууме или человеке, ожидаемый исследователем, ветеринаром, врачом или другим клиницистом, включающий в себя одно или более из следующего:
(1) предотвращение заболевания; например, предотвращение заболевания, условия или нарушения у индивидуума, который может быть предрасположен к заболеванию, условию или нарушению, но еще не подвергся ему, или не проявляет патологических или симптоматических признаков болезни;
(2) подавление заболевания; например, подавление заболевания, условия или нарушения у индивидуума, который подвергся или проявляет патологические или симптоматические признаки заболевания, условия или нарушения (например, прекращение дальнейшего развития патологии и/или симптоматологии), такое как стабилизация вирусной нагрузки в случае вирусной инфекции; и
(3) облегчение течения заболевания; например, облегчение течения заболевания условия или нарушения у индивидуума, который подвергся или проявляет патологические или симптоматические признаки заболевания, условия или нарушения (например, обратное развитие патологии и/или симптоматологии), такое как снижение вирусной нагрузки в случае вирусной инфекции.
Один или более других фармацевтических агентов, таких как, например, антивирусные агенты, антитела, антивоспалительные агенты и/или иммуносупрессоры могут быть использованы в комбинации с соединением по настоящему изобретению для лечения заболеваний, нарушений или условий, связанных с хемокиновыми рецепторами. Агенты могут комбинироваться с настоящими соединениями в единой форме дозирования или могут вводиться одновременно или отдельно как особые формы дозирования.
Предположительно, антивирусные агенты, пригодные для использования в комбинации с соединениями по настоящему изобретению, могут содержать нуклеозидные и нуклеотидные ингибиторы обратной транскриптазы (NRTIs), ненуклеозидные ингибиторы обратной транскриптазы (NNRTIs), ингибиторы протеаз и другие антивирусные лекарства.
Пример приемлемых NRTIs включает в себя зидовудин (AZT); диданозин (ddl); зальцитабин (ddC); ставудин (d4T); ламивудин (3TC); абакавир (1592U89); дипивоксил адефовира [bis(POM)-PMEA]; лобукавир (BMS-180194); BCH-10652; эмитрицитабин [(-)-FTC]; бета-1-FD4 (также называемый бета-1-D4C и бета-1-2',3'-диклеокси-5-фтороцитидин); DAPD, ((-)-бета-D-2,6,-диаминопурин диоксолан) и лоденозин (FddA).
Типичные приемлемые NNRTIs включают невирапин (BI-RG-587); делавирадин (BHAP, U-90152); эфавиренц (DMP-266); PNU-142721; AG-1549; MKC-442 (1-(этокси-метил)-5-(1-метилэтил)-6-(фенилметил)-(2,4(1H,3H)-пиримидин недион) и (+)-каланолид A (NSC-675451) и B.
Типичные приемлемые ингибиторы протеаз включают саквинавир (Ro 31-8959); ритонавир (ABT-538); индинавир (MK-639); нелфнавир (AG-1343); ампренавир (141W94); лазинавир (BMS-234475); DMP-450; BMS-2322623; ABT-378 и AG-1549.
Другие антивирусные агенты включают гидроксимочевину, рибавирин, IL-2, IL-12, пентафусид и Yissum Project No. 11607.
В некоторых вариантах осуществления, противовоспалительные и обезболивающие агенты, которые предположительно могут быть использованы в комбинации с соединением по настоящему изобретению, могут содержать, например, опиатный агонист, ингибитор липоксигеназы, такой как ингибитор липоксигеназы-5, ингибитор циклооксигеназы, такой как ингибитор циклооксигеназы-2, ингибитор интерлейкина, такой как ингибитор интерлейкина-I, антагонист NNMA, ингибитор оксида азота или ингибитор синтеза оксида азота, нестероидный противовоспалительный агент или цитокин-супрессорный противовоспалительный агент, например, такой, как ацетаминофен, аспирин, кодеин, фентанил, ибупрофен, индометацин, кеторолак, морфин, напроксен, фенацетин, пироксикам, стероидные обезболивающие, суфентанил, сунлиндак, тенидап и подобные агенты. Сходным образом, необходимые соединения могут вводиться с обезболивающим; с потенцирующим агентом, таким как кофеин, Н2-антагонист, симетикон, гидрохлорид алюминия или магния; с противоотечным агентом, таким как фенилэфрин, фенилпропаноламин, псевдофедрин, оксиметазолин, эпинефрин, нафазолин, ксилометазолин, пропилгекседфин или лево-дезоксиэфедрин; с противокашлевым агентом, таким как кодеин, гидрокодон, карамифен, карбетапентан или декстраметорфан; с диуретиком; и с седативным или неседативным антигистаминовым агентом.
В некоторых вариантах осуществления, фармацевтические агенты, которые предположительно могут быть использованы в комбинации с соединением по настоящему изобретению, могут содержать (а) антагонисты VLA-4, такие как описанные в патентах США 5510332, W095/15973, W096/01644, W096/06108, W096/20216, W096/229661, W096/31206, W096/4078, W097/030941, W097/022897, WO98/426567, W098/53814, W098/53817, W098/538185, W098/54207 и W098/58902; (b) стероиды, такие как беклометазон, метилпреднизолон, бетаметазон, преднизон, дексаметазон и гидрокортизон; (c) иммуносупрессанты, такие как циклоспорин, такролимус, рапамицин и другие иммуносупрессанты типа FK506; (d) антигистаминовые агенты (антагонисты НI-гистамина), такие как бромофенирамин, хлорфенирамин, дексхлорфенирамин, трипролидин, клемастин, дифенгидрамин, дифенилпиралин, трипеленнамин, гидроксизин, метдилазин, прометазин, тримепразин, азатадин, ципрогептадин, антазолин, пириламин фенирамина, астемизол, терфенадин, лоратадин, цетиризин, фексофенадин, десербоэтоксилоратадин и подобные агенты; (е) нестероидные антиастматические агенты, такие как тербуталин, метапротеренол, фенотерол, изоэтаин, албутерол, битолтерол, пирбутерол, теофиллин, кромолин натрия, атропин, бромид ипратропиума, антагонисты лейкотриенов (например, зафирлукаст, монтелукаст, пранлукаст, иралукаст, побилукаст, SKB-106203), ингибиторы биосинтеза лейкотриенов (например, зилейтон, BAY-1005); (f) нестероидные противовоспалительные агенты (NSAIDs), такие как производные пропионовой кислоты (например, альминопрофен, беноксапрофен, буклоксовая кислота, карпрофен, фенбуфен, фенопрофен, флюпрофен, флюрбипрофен, ибупрофен, индопрофен, кетопрофен, миропрофен, напроксен, оксапрозин, пирпрофен, пранопрофен, супрофен, тиапрофеновая кислота и тиоксапрофен), производные уксусной кислоты (например, индометацин, ацеметацин, алклофенак, клиданак, диклофенак, фенклофенак, фенклозовая кислота, фентиазак, фурофенак, ибуфенак, изоксепак, окспинак, сулиндак, тиопинак, толметин, зидометацин и зомепирак), производные фенамовой кислоты (флуфенамовая кислота, меклофенамовая кислота, мефенамовая кислота, нифлумовая кислота и толфенамовая кислота), производные бифенилкарбоксиловой кислоты (например, дифлюнизал и флюфенизал), оксикамы (изоксикам, пироксикам, судоксикам и теноксикам), салицилаты (ацетилсалициловая кислота, сульфазалазин) и пиразолоны (апазон, безпиперилон, фепразон, мофебутазон, оксифенбутазон, фенилбутазон); (g) ингибиторы циклооксигеназы-2 (COX-2); (h) ингибиторы фосфодиэстеразы типа IV (PDE-IV); (i) другие антагонисты хемокиновых рецепторов, особенно CXCR-4, CCRI, CCR2, CCR3 и CCR5; (j) агенты, снижающие уровень холестерина, такие как ингибиторы ГМГ-КоА редуктазы (ловастатин, симвастатин и правастатин, флювастатин, аторвастатин и другие статины), секвестранты (холестирамин и холестипол), никотиновая кислота, производные фенофиброевой кислоты (гемфиброзил, клофибрат, фенофибрат и бензафибрат) и пробукол; (k) антидиабетические агенты, такие как инсулин, сульфонилмочевина, бигуаниды (метформин), ингибиторы U-глюкозидазы (акарбоза) и орлитазоны (троглитазон и пиоглитазон); (l) препараты бета-интерферона (интерферон бета-lo., интерферон бета-lP); (m) другие соединения, такие как аминосалициловые кислоты, антиметаболиты, такие как азатиоприн и 6-меркаптопурин, и цитотоксичные агенты раковой химиотерапии. Весовое соотношение соединения по настоящему изобретению к второму активному ингредиенту может различаться и зависит от эффективной дозы каждого ингредиента.
Фармацевтические прописи и формы дозирования
Соединения формулы I, когда используются в качестве фармацевтических препаратов, могут вводиться в форме фармацевтических смесей. Эти смеси готовят способом, хорошо известным в фармацевтической науке, и вводят различными путями, в зависимости от того, местное или системное требуется лечение и от области лечения. Введение может быть местное (включающее в себя внутриглазное и в слизистые оболочки, включая интраназальное, вагинальное и ректальное введение), пульмонарное (например, путем ингаляции или инсуффляции порошков или аэрозолей, включающим в себя небулайзер; интратрахеальное, интраназальное, эпидермальное и трансдермальное), глазное, оральное и парентеральное. Способы глазного введения могут включать местное введение (глазные капли), субконъюнктивальные, периокулярные инъекции или инъекции в стекловидное тело, или введение через баллонный катетер, или хирургическое введение в конъюнктивальный мешок. Парентеральное введение включает в себя внутривенные, внутриартериальные, подкожные, интраперитонеальные или внутримышечные инъекции или инфузии; или интракраниальные, например, интратекальные или интравентрикулярные, введения. Парентеральное введение может быть в форме единичной болюсной дозы или может проводиться, например, с помощью постоянного перфузионного насоса. Фармацевтические смеси и прописи для местного введения могут включать трансдермальные пластыри, мази, лосьоны, кремы, гели, капли, свечи, спреи, растворы и порошки. Общепринятые фармацевтические носители на водной, порошковой или масляной основе, загустители и подобные вещества могут быть необходимы или желательны.
Настоящее изобретение также включает в себя фармацевтические смеси, которые содержат, в качестве активных ингредиентов, одно или более соединений формулы I, указанных выше, в комбинации с одним или более фармацевтически пригодным носителем. При получении смеси по изобретению активный ингредиент обычно смешивают с наполнителем, разводят наполнителем или помещают в такой носитель в форме, например, капсулы, пакета, бумаги или другого контейнера. Когда наполнитель используют в качестве разбавителя, он может быть плотным, полуплотным или жидким, который действует как транспортное средство, носитель или среда для активного ингредиента. Таким образом, смеси могут быть в форме таблеток, пилюль, порошков, ромбов, пакетов, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей (на твердой или жидкой основе), мазей, содержащих, например, до 10% по весу активного компонента, мягких или твердых желатиновых капсул, свечей, стерильных растворов для инъекций и стерильно упакованных порошков.
При приготовлении прописи активное соединение обрабатывают для получения частиц нужного размера перед комбинацией с другими ингредиентами. Если активное соединение преимущественно нерастворимое, его обрабатывают для получения частиц размером менее 200 меш. Если активное соединение преимущественно водорастворимое, размер частиц корректируют путем обработки для достижения по возможности однотипного распределения в прописи, например, около 40 меш.
Некоторые примеры приемлемых наполнителей включают лактозу, декстрозу, сахарозу, сорбит, маннит, крахмал, аравийскую камедь, фосфат кальция, альгинаты, трагакант, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду, сироп и метилцеллюлозу. Пропись может дополнительно включать: лубрикантные агенты, такие как тальк, стеарат магния и минеральное масло; увлажняющие агенты; эмульгирующие и суспендирующие агенты; и ароматизирующие агенты. Смесь по изобретению составляют таким образом, чтобы вызвать быстрое, длительное или отсроченное высвобождение активного ингредиента после введения пациенту путем использования способов, известных в науке.
Смесь может быть изготовлена в виде стандартной лекарственной формы, когда каждая доза содержит от, примерно, 5 до, примерно, 100 мг, чаще - от, примерно, 10 до, примерно, 30 мг активного ингредиента. Термин «стандартная лекарственная форма» относится к физически обособленной дозе, пригодной для отдельного дозирования человеческим субъектам и другим млекопитающим, при этом каждая доза содержит определенное количество активного материала, рассчитанное для вызывания желаемого терапевтического эффекта, в сочетании с приемлемым фармацевтическим наполнителем.
Активное соединение может быть эффективным в широком диапазоне дозировок и обычно вводится в фармацевтически эффективном количестве. Должно быть понятно, однако, что количество вводимого соединения должно быть определено врачом, в соответствии с конкретными обстоятельствами, включающими в себя состояние, которое подвергается лечению, выбранный способ введения, конкретное вводимое соединение, возраст, вес и индивидуальный ответ пациента, тяжесть симптомов пациента и тому подобное.
Для получения плотных составов, таких как таблетки, основные активные компоненты смешивают с фармацевтическим наполнителем для формирования плотной предварительной смеси, содержащей гомогенную смесь соединения по настоящему изобретению. Когда предварительную смесь оценивают как гомогенную, активный ингредиент обычно равномерно распределен по всей смеси, так что смесь может быть легко разделена на равно эффективные стандартные лекарственные формы, такие как таблетки, пилюли и капсулы. Эту плотную предварительную смесь затем разделяют на стандартные лекарственные формы описанных выше типов, содержащие от, например, 0,1 до, например, 500 мг активного ингредиента по настоящему изобретению.
Таблетки или пилюли по настоящему изобретению покрывают или иным образом обрабатывают для того, чтобы данная форма дозирования могла проявить преимущества продленного действия. Например, таблетки или пилюли могут содержать внутренний и внешний компоненты дозы, при этом последний представляет собой оболочку определенного шаблона. Эти два компонента могут быть разделены внутренним слоем, который служит для предохранения от разрушения в желудке и позволяет внутреннему компоненту попасть в интактном состоянии в двенадцатиперстную кишку или высвободиться через определенное время. Различные материалы могут применяться для создания таких внутренних слоев или покрытий, такие материалы включают большое число полимерных кислот и смесей полимерных кислот с такими материалами, как шеллак, цетиловый спирт и ацетат целлюлозы.
Жидкие формы, в которые соединения и смеси по настоящему изобретению могут включаться для введения внутрь или через инъекцию, включают водные растворы, ароматизированные сиропы, водные или масляные суспензии, ароматизированные эмульсии со съедобными маслами, такими как хлопковое масло, горчичное масло, кокосовое масло или арахисовое масло, а также эликсиры и сходные фармацевтические носители.
Смеси для ингаляции или инсуффляции включают растворы и суспензии в фармацевтически приемлемых, водных или органических, растворителях или их смеси, и порошки. Жидкие или плотные смеси могут содержать приемлемые фармацевтически пригодные наполнители, как описано выше. В некоторых вариантах осуществления, смеси вводят оральным или назальным респираторным путем для местного или системного эффекта. Смеси распыляют при помощи инертных газов. Распыленные растворы вдыхают непосредственно из аппарата распыления, или аппарат распыления подключают к лицевой маске, или к аппарату http://www.multitran.ru/c/m.exe?t=2591461_2_1. Растворы, суспензии или порошковые смеси вводят орально или назально с помощью оборудования, обеспечивающего доставку препарата надлежащим способом.
Количество соединения или смеси, вводимое пациенту, будет варьировать в зависимости от того, что вводится, цели введения, например, профилактики или лечения, состояния пациента, способа введения и подобных факторов. При терапевтическом применении смеси вводят пациенту, уже страдающему заболеванием, в количестве, достаточном для лечения или, по меньшей мере, частичного уменьшения симптомов заболевания и его осложнений. Эффективность доз будет зависеть от условий заболевания, так же как заключение лечащего врача зависит от таких факторов, как тяжесть заболевания, возраст, вес, общее состояние пациента и тому подобного.
Смеси, вводимые пациенту, могут быть в форме фармацевтических смесей, описанных выше. Эти смеси стерилизуют общепринятыми способами стерилизации или путем фильтрации. Водные растворы упаковывают для использования либо в исходном виде, либо лиофилизируют, лиофилизованные препараты перед введением смешивают со стерильным водным носителем. Уровень рН препаратов соединения обычно бывает между 3 и 11, более предпочтительно - от 5 до 9, и наиболее предпочтительно - от 7 до 8. Должно быть понятно, что использование определенных наполнителей, носителей или стабилизаторов будет сказываться на образовании фармацевтических солей.
Терапевтическая доза соединения по настоящему изобретению будет варьировать в зависимости от, например, конкретной цели, с которой предпринимается лечение, способа введения соединения, здоровья и состояния пациента и заключения назначающего лечение врача. Пропорции или концентрации соединения по изобретению в фармацевтической смеси могут варьировать в зависимости от большого числа факторов, включающих в себя дозировку, химические характеристики (например, гидрофобность) и путь введения. Например, соединение по изобретению применяют для парентерального введения в виде раствора в водном физиологическом буфере, содержащем от, примерно, 0,1 до, примерно, 10% вес/объем соединения. Некоторые типичные величины дозировок находятся в пределах от, примерно, 1 мкг/кг до, примерно, 1 г/кг веса тела в день. В некоторых вариантах осуществления, величины дозировок находятся в пределах от, примерно, 0,01 мг/кг до, примерно, 100 мг/кг веса тела в день. Дозировка зависит от таких особенностей, как тип и степень прогрессирования заболевания или нарушения, общее состояние здоровья конкретного пациента и пути введения. Эффективные дозы могут быть экстраполированы с кривых доза-ответ, полученных in vitro, или с тестовых систем животных моделей.
Соединения по изобретению также могут быть изготовлены в комбинации с одним или более активными ингредиентами, которые могут включать любой фармацевтический агент, такой как антивирусные агенты, антитела, иммуносупрессанты, противовоспалительные агенты и подобные агенты. В некоторых вариантах осуществления, соединения по изобретению могут быть изготовлены в комбинации с одним или более антивирусными агентами, включающими в себя ингибиторы протеаз и другие агенты, используемые в анти-ВИЧ терапии.
Меченые соединения и способы анализа
Другой аспект по настоящему изобретению относится к радиоактивно меченым соединениям формулы I, которые будут полезны не только в радиодиагностике, но и в исследованиях, как in vitro, так и in vivo, по локализации и подсчету хемокиновых рецепторов в образцах ткани, включающих в себя ткани человека, и для идентификации лигандов хемокиновых рецепторов путем ингибирования связывания радиоактивно меченых соединений.
Далее, настоящее изобретение включает в себя изотопно-меченые соединения формулы I. «Изотопно» или «радиоактивно меченое» соединение является соединением по изобретению, в котором один или более атомов заменены или замещены атомом, имеющим атомарную массу или атомарное число, отличное от атомарной массы или атомарного числа, обнаруживаемого в природе (то есть обычно встречающегося). Подходящие радионуклиды, которые могут быть введены в соединения по настоящему изобретению, включают, но не ограничиваются ими, 2Н (также обозначаемый как D, дейтерий), 3Н (также обозначаемый как Т, тритий), 11С, 13С, 14С, 13N, 15N, 15O, 17O, 18O, 18F, 35S, 36Cl, 82Br, 75Br, 76Br, 77Br, 123I, 124I, 125I и 131I. Введение того или иного радионуклида в исходные радиоактивно меченые соединения зависит от особенностей применения этого радиоактивно меченого соединения. Например, для исследований по мечению и конкурентному связыванию хемокиновых рецепторов in vitro, наиболее полезны соединения, содержащие 3Н, 14С, 82Br, 125I, 131I, 35S. Для применения в радиодиагностике наиболее полезны соединения, содержащие 11С, 18F, 125I, 123I, 124I, 131I, 75Br, 76Br или 77Br.
Понятно, что «радиоактивно меченым» или «меченым соединением» является соединение, содержащее, по меньшей мере, один радионуклид. В некоторых вариантах осуществления, радионуклид выбирают из группы, состоящей из 3Н, 14С, 125I, 35S и 82Br.
Искусственные способы введения радиоизотопов в органические соединения применимы для соединений по изобретению и хорошо известны в науке.
Радиоактивно меченое соединение по изобретению используют в скрининговых исследованиях для идентификации/оценки соединений. В целом, вновь синтезированное или идентифицированное соединение (например, тестовое соединение) может быть оценено на способность уменьшать связывание радиоактивно меченого соединения по изобретению с хемокиновыми рецепторами. Соответственно, способность тестового соединения конкурировать с радиоактивно меченым соединением за связывание с хемокиновыми рецепторами напрямую коррелирует с его связывающей способностью.
Наборы
Настоящее изобретение также включает в себя фармацевтические наборы, применимые, например, в лечении или предотвращении хемокин-ассоциированных заболеваний или нарушений, таких как инфекция ВИЧ, которые включают один или более контейнеров, содержащих фармацевтические смеси терапевтически эффективных количеств соединения формулы I. Эти наборы могут также включать, при необходимости, один или более различных общепринятых компонентов фармацевтических наборов, таких как, например, контейнеры с одним или более фармацевтически приемлемыми носителями, дополнительные контейнеры, и так далее, что очевидно специалисту в данной области науки. Инструкции, как в виде вкладышей, так и в виде наклеек, указывающие количества компонентов для введения, рекомендации по введению и/или рекомендации по смешиванию компонентов также могут быть включены в набор.
Изобретение будет описано в больших деталях на специальных примерах. Следующие примеры использованы для иллюстративных целей и не являются ограничениями изобретения каким-либо способом. Специалист в данной области науки легко обнаружит многие некритичные параметры, которые могут быть изменены или модифицированы с получением существенно схожих результатов.
Примеры
Пример 1
5-({4-[(3S)-4-(5-Бром-2,3-дигидро-1H-инден-1-ил)-3-метилпиперазин-1-ил]-4-метилпиперидин-1-ил}карбонил)-4,6-диметилпиримидин
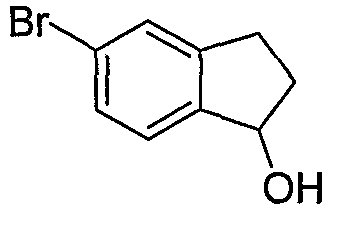
Стадия A
5-Бром-1-инданол
К раствору 5-бром-1-инданона (2,0 г, 9,5 ммоль) в THF (20 мл) добавляли NaBH4 (0,5 г, 12,8 ммоль). После перемешивания при комнатной температуре в течение ночи раствор гасили добавлением воды. Полученный в результате раствор экстрагировали дважды EtOAc. Объединенные слои EtOAc сушили над Na2SО4 и концентрировали в вакууме с получением 2,0 г указанного в заголовке соединения в виде твердого вещества. MS раcсчетная для C9H9BrO: (M+H)+ 212,9; обнаружено 194,9 (M+H-H2O)+, 197,0 (M+H-H2O)+.
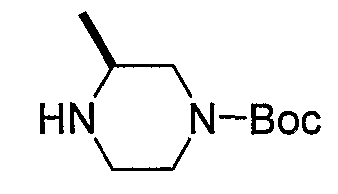
Стадия B
трет-Бутил-(3S)-3-метилпиперазин-1-карбоксилат
К раствору (2S)-2-метилпиперазина (20,0 г, 0,200 моль) в метиленхлориде (300 мл) и триэтиламине (20,4 г, 0,202 моль) добавляли по каплям раствор ди-трет-бутилдикарбоната (44,0 г, 0,202 моль) в CH2Cl2 (100 мл) в течение 5 часов. Смесь промывали водой, солевым раствором и затем сушили над MgSО4 и концентрировали. Колоночная хроматография на силикагеле (10-20% MeOH в EtOAc) давала 32,0 г (80%) указанного в заголовке соединения в виде масла.
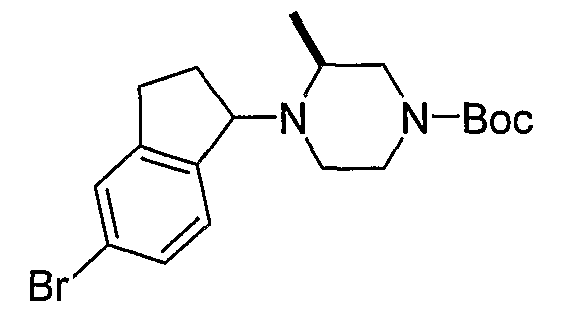
Стадия C
трет-Бутил-(3S)-4-(5-бром-2,3-дигидро-1H-инден-1-ил)-3-метилпиперазин-1-карбоксилат
5-Бром-1-инданол (1,0 г, 4,7 ммоль) стадии А растворяли в тионилхлориде (10 мл). После перемешивания при комнатной температуре в течение 2 часов раствор концентрировали в вакууме. Остаток растворяли в DMF (10 мл). К смеси добавляли трет-бутил-(3S)-3-метилпиперазин-1-карбоксилат (0,94 г, 4,7 ммоль), NaI (2 г, 13 ммоль) и триэтиламин (1,5 мл, 10 ммоль). Полученный в результате раствор перемешивали при 70°C в течение ночи. После охлаждения при комнатной температуре добавляли воду. Раствор дважды экстрагировали EtOAc. Объединенные слои EtOAc промывали солевым раствором, сушили над MgSО4 и концентрировали. Колоночная хроматография на силикагеле (50% EtOAc в гексане) предоставляла два изомера. Изомер 1 (быстро двигавшийся изомер): 0,36 г; MS расчетная для C19H27BrN2О2 (M+H)+ 395; обнаруженная 395,1, 397,0. Изомер 2 (медленно двигавшийся изомер): 0,33 г; MS обнаруженная 395,1, 397,0.
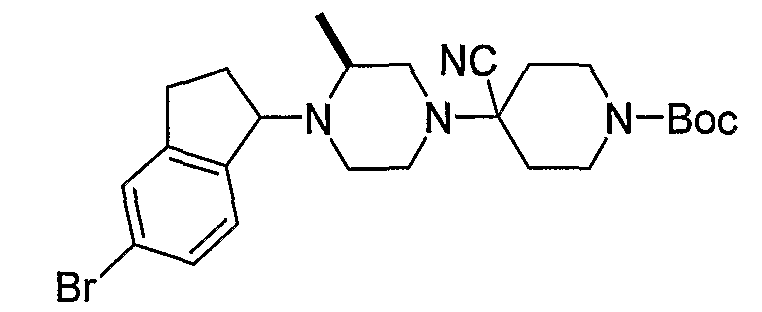
Стадия D
трет-Бутил-4-[(3S)-4-(5-бром-2,3-дигидро-1H-инден-1-ил)-3-метилпиперазин-1-ил]-4-цианопиперидин-1-карбоксилат
Изомер 1 со стадии C (0,33 г, 0,83 ммоль) растворяли в 4 Н HCl в диоксане (4 мл). После перемешивания при комнатной температуре в течение 2 ч раствор концентрировали. Остаток растворяли в CH2Cl2 (5 мл). К нему добавляли трет-бутил-4-оксо-1-пиперидинкарбоксилат (0,17 г, 0,85 ммоль), Ti(Oi-Pr)4 (0,87 мл) и триэтиламин (0,6 мл). Смесь перемешивали при комнатной температуре в течение ночи, и летучие вещества удаляли в вакууме. Остаток растворяли в THF (5 мл). К смеси добавляли 1,0 M раствор цианида диэтилалюминия (1 мл). Полученный в результате раствор перемешивали при 30°C в течение 5 ч и концентрировали для обеспечения неочищенного указанного в заголовке соединения (0,32 г), которое использовали для следующей реакции без очистки. MS расчетная для C25H35BrN4О2: (M+H)+ 503; обнаружено 503,1, 505,1.
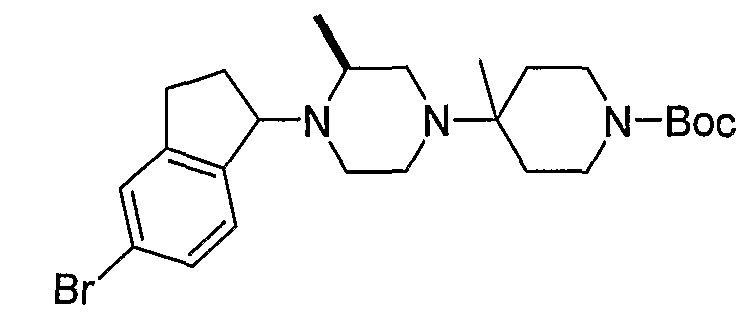
Стадия E
трет-Бутил-4-[(3S)-4-(5-бром-2,3-дигидро-1H-инден-1-ил)-3-метилпиперазин-1-ил]-4-метилпиперидин-1-карбоксилат
К раствору трет-бутил-4-[(3S)-4-(5-бром-2,3-дигидро-1H-инден-1-ил)-3-метилпиперазин-1-ил]-4-цианопиперидин-1-карбоксилата (0,32 г, 0,64 ммоль) в THF (2 мл) добавляли 3 M раствора бромида метилмагния (1,1 мл, 3,3 ммоль). После перемешивания при комнатной температуре в течение ночи раствор концентрировали. Очистка на силикагеле (2:1 гексан/EtOAc) дала указанное в заголовке соединение (0,25 г). MS расчетная для С25H37BrN3О2:(M+H)+ 491; обнаружено 491,2, 494,2.
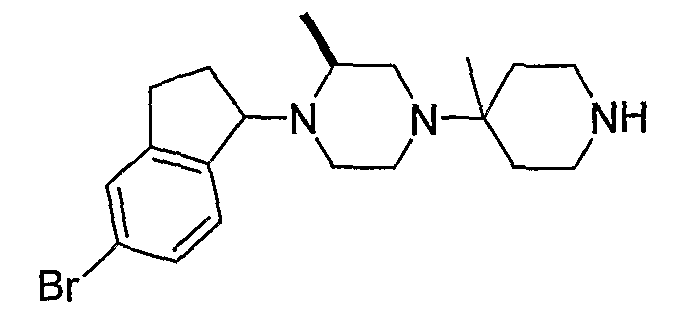
Стадия F
(2S)-1-(5-Бром-2,3-дигидро-1H-инден-1-ил)-2-метил-4-(4-метилпиперидин-4-ил)пиперазин
Промежуточный продукт, полученный на стадии E (0,23 г) растворяли в растворе 4 Н HCl в диоксане (3 мл). После перемешивания при комнатной температуре в течение 2 ч, раствор концентрировали для предоставления указанного в заголовке соединения в виде соли тригидрохлорида (0,23 г). MS расчетная для C20H30BrN3: (M+H)+ 392; обнаружено 392,2, 394,2.
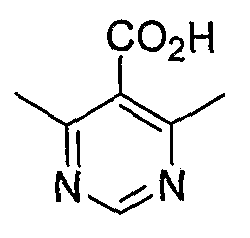
Стадия G
4,6-Диметилпиримидин-5-карбоновая кислота
Этил-2-ацeтил-3-этоксибут-2-еноат. Колбу с 4 горлышками объемом 5 л, снабженную механической мешалкой, термолункой, конденсером, воронкой для добавления и выходом N2, заряжали этилацетоацетатом (493,1 г, 483 мл, 3,7931 моль, 1,0 экв.) и этоксидом натрия (3,1 г, 0,046 моль, 1,2 молярных %). Диэтилацеталь кетена (880,0 г, 1000 мл, 7,5862 моль, 2,0 экв.) добавляли в течение 1 ч при поддержании температуры взаимодействия ниже 22°C за счет внешнего охлаждения. По завершении добавления реакционную смесь нагревали при 85°C±5°C в течение 7,5 ч. Желто-коричневую реакционную смесь охлаждали до комнатной температуры и перемешивали в течение ночи. Большую часть низкокипящих компонентов [EtOH, EtOAc, Me(OEt)3] упаривали на роторном испарителе (температура бани ~65°C). Оставшееся желто-оранжевое масло перегоняли, собирая фракцию с т.к. 100-107°C (1,8-2,1 торр) с получением 675,2 г (89%) продукта в виде желтой жидкости.
Этил-4,6-диметилпиримидин-5-карбоксилат. Этил-2-ацетил-3-этоксибут-2-еноат (10,7 г, 0,0537 моль), ацетата формамидина (5,6 г, 0,054 моль) и этоксид натрия (2,7 M в этаноле, 20,0 мл) смешивали в этаноле (30 мл), и смесь перемешивали при 90°C в течение 4 ч. Реакционную смесь охлаждали, гасили водой и концентрировали. Неочищенный материал очищали тонкослойной хроматографией на силикагеле, элюируя 10%, 50% этилацетат/гексан с получением требуемого продукта (7,4 г, 76%) в виде желтого масла. MS (EI) 181,1 (M+1). 1H ЯМР (300 МГц, CDCl3) δ (м.д.) 8,97 (с, 1H), 4,44 (кв., 2H), 2,56 (с, 6H), 1,42 (т, 3H).
4,6-Диметилпиримидин-5-карбоновая кислота. Этил-4,6-диметилпиримидин-5-карбоксилат (10,9 г, 0,0605 моль) смешивали с раствором гидроксида натрия (4,0 г, 0,10 моль) в воде (70 мл). Смесь перемешивали при комнатной температуре в течение ночи. Водную реакционную смесь подкисляли концентрированной соляной кислотой и затем концентрировали до сухости. К данному остатку добавляли ацетон (100 мл). Нерастворимый хлорид натрия фильтровали и промывали метанолом (100 мл). Фильтрат концентрировали до сухости. Остаток промывали ACN с получением 8,5 г (92%) продукта в виде твердого вещества. MS (EI) 153,1 (M+1). 1H ЯМР (400 МГц, CD3ОD) δ (м.д.) 8,89 (с, 1H), 2,56 (с, 6H).
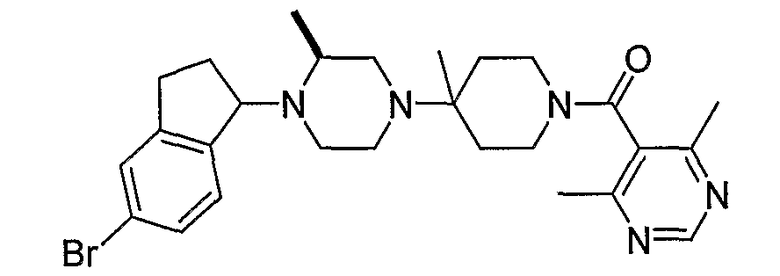
Стадия H
5-({4-[(3S)-4-(5-Бром-2,3-дигидро-1H-инден-1-ил)-3-метилпиперазин-1-ил]-4-метилпиперидин-1-ил}карбонил)-4,6-диметилпиримидин
К раствору тригидрохлорида (2S)-1-(5-бром-2,3-дигидро-1H-инден-1-ил)-2-метил-4-(4-метилпиперидин-4-ил)пиперазина (30 мг, 0,06 ммоль) и 4,6-диметилпиримидин-5-карбоновой кислоты (9 мг, 0,06 ммоль) в DMF (2 мл) добавляли бензотриазол-1-илокситрис(диметиламино)фосфония гексафторфосфат (30 мг, 0,06 ммоль) с последующим добавлением триэтиламина (30 мг, 0,3 ммоль). После перемешивания при комнатной температуре в течение 5 ч смесь разбавляли EtOAc и раствором Na2CО3 в воде. Органический слой отделяли, промывали несколько раз водой, сушили над Na2SО4 и концентрировали. Очистка путем обращенно-фазовой ВЭЖХ и лиофилизации давала конечный продукт в виде соли ТФУ (20 мг). MS расчетная для C27H36BrN5О: (M+H)+ 526; обнаружено 526,1, 528,1.
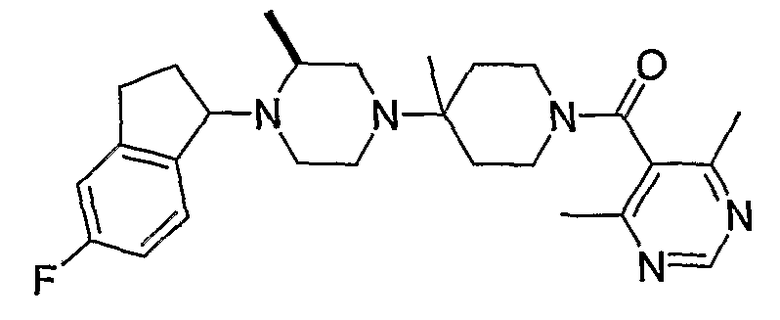
Пример 2
5-({4-[(3S)-4-(5-Фтор-2,3-дигидро-1H-инден-1-ил)-3-метилпиперазин-1-ил]-4-метилпиперидин-1-ил}карбонил)-4,6-диметилпиримидин
Данное соединение получали, по существу, как описано в примере 1 с использованием подходящих исходных материалов. MS расчетная для C27H36FN5О: (M+H)+ 466; обнаружено 466,2.
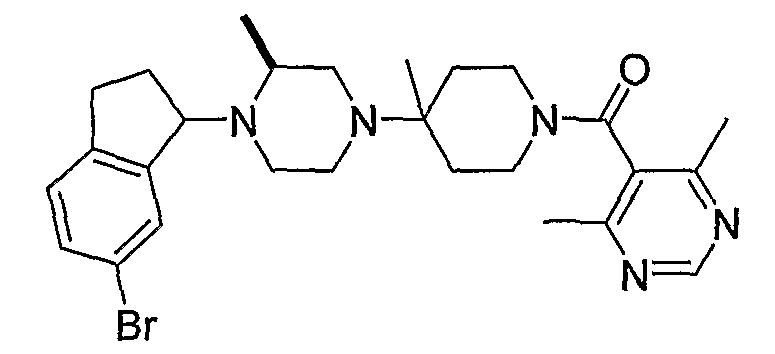
Пример 3
5-({4-[(3S-4-(6-Бром-2,3-дигидро-1H-инден-1-ил)-3-метилпиперазин-1-ил]-4-метилпиперидин-1-ил}карбонил)-4,6-диметилпиримидин
Данное соединение получали, по существу, как описано в примере 1 с использованием подходящих исходных материалов. MS расчетная для C27H36BrN5О: (M+H)+ 526; обнаружено 526,1, 528,1.
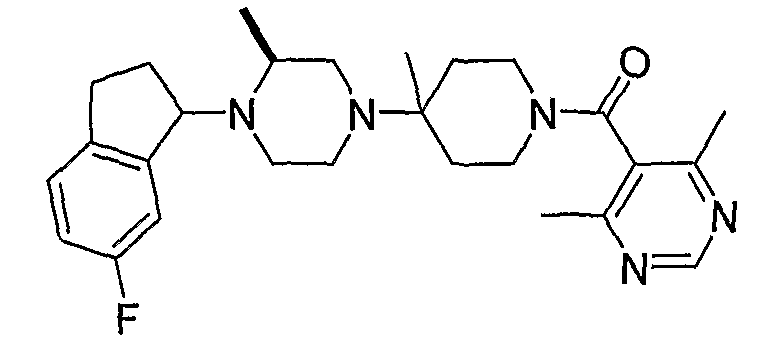
Пример 4
5-({4-[(3S)-4-(6-Фтор-2,3-дигидро-1H-инден-1-ил)-3-метилпиперазин-1-ил]-4-метилпиперидин-1-ил}карбонил)-4,6-диметилпиримидин
Данное соединение получали, по существу, как описано в примере 1 с использованием подходящих исходных материалов. MS расчетная для C27H36FN5O: (M+H)+ 466; обнаружено 466,2.
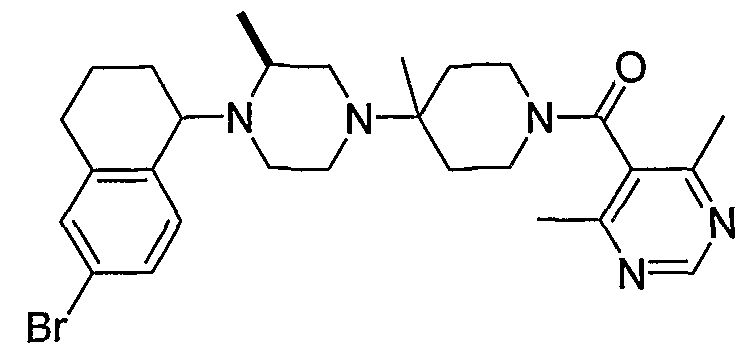
Пример 5
5-({4-[(3S-4-(6-Бром-1,2,3,4-тетрагидронафталин-1-ил)-3-метилпиперазин-1-ил]-4-метилпиперидин-1-ил}карбонил)-4,6-диметилпиримидин
Данное соединение получали, по существу, как описано в примере 1 с использованием подходящих исходных материалов. MS расчетная для C28H38BrN5O: (M+H)+ 540; обнаружено 540,2, 542,1.
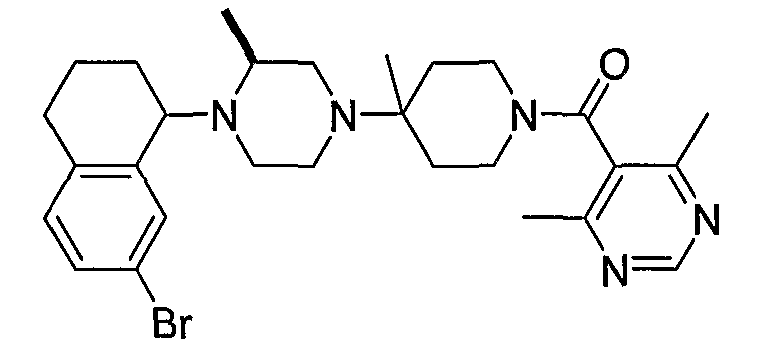
Пример 6
5-({4-[(3S-4-(7-Бром-1,2,3,4-тетрагидронафталин-1-ил)-3-метилпиперазин-1-ил]-4-метилпиперидин-1-ил}карбонил)-4,6-диметилпиримидин
Данное соединение получали, по существу, как описано в примере 1 с использованием подходящих исходных материалов. MS расчетная для C28H38BrN5О: (M+H)+ 540; обнаружено 540,2, 542,1.
Пример 7
4,6-Диметил-5-[(4-метил-4-{(3S)-3-метил-4-[6-(трифторметил)-2,3-дигидро-1H-инден-1-ил]пиперазин-1-ил}пиперидин-1-ил)карбонил]пиримидин
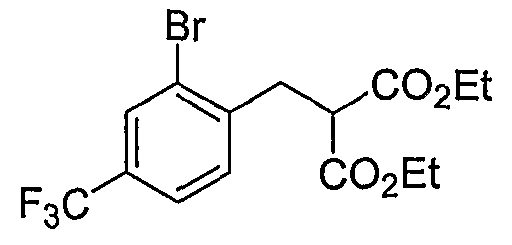
Стадия A
Диэтил-[2-бром-4-(трифторметил)бензил]малонат
В суспензию гидрида натрия (1,4 г, 58 ммоль) в DMF (37 мл) при 10°C по каплям добавляли этилмалонат (14 г, 88 ммоль). После добавления смесь перемешивали в течение 1 ч при комнатной температуре. К ней медленно добавляли раствор 2-бром-1-(хлорметила)-4-(трифторметил)бензола (10,0 г, 36,6 ммоль) в DMF (20 мл). После перемешивания при комнатной температуре в течение ночи смесь выливали в ледяную воду (300 мл). Полученный в результате раствор экстрагировали дважды эфиром. Объединенные экстракты промывали водой и солевым раствором, сушили над MgSО4 и концентрировали. Колоночная хроматография на силикагеле (5-10% EtOAc в гексане) приводила к получению указанного в заголовке соединения в виде масла (13,5 г, 93%). MS расчетная для C15H16BrF3O4: (M+H)+ 397; обнаружено 397,0, 399,0.
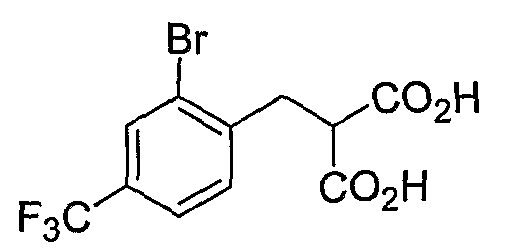
Стадия B
2-Бром-4-(трифторметил)бензил]малоновая кислота
К раствору диэтил-[2-бром-4-(трифторметил)бензил]малоната (13,5 г, 34 ммоль) в этаноле (60 мл) и воде (28 мл) добавляли 5 M раствора гидроксида натрия в воде (20 мл). После перемешивания при комнатной температуре в течение ночи этанол удаляли в вакууме. Водный раствор разбавляли добавлением большего количества воды и дважды экстрагировали эфиром. Полученный водный слой подкисляли до pH 3 концентрированной HCl и экстрагировали эфиром 3 раза. Объединенные эфирные слои промывали водой и солевым раствором, сушили над MgSО4 и концентрировали с получением указанного в заголовке соединения в виде белого твердого вещества (9,8 г, 84%). MS расчетная для C11H8BrF3O4: (M+H)+ 341; обнаружено 363 (M+Na)+.
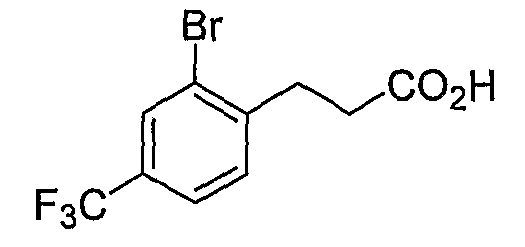
Стадия C
3-[2-Бром-4-(трифторметил)фенил]пропановая кислота
[2-Бром-4-(трифторметил)бензил]малоновую кислоту (9,80 г, 28,7 ммоль) в круглодонной колбе нагревали до 180°C, и продолжали нагревание при 180°C в течение 1,5 ч. После охлаждения до комнатной температуры жидкость растворяли в эфире. Полученный раствор сушили над MgSО4 и концентрировали. Твердое вещество промывали гексаном с образованием указанного в заголовке соединения (7,20 г, 85%). MS расчетная для C10H8BrF3O2: (M+H)+ 297; обнаружено 297,0, 299,0.
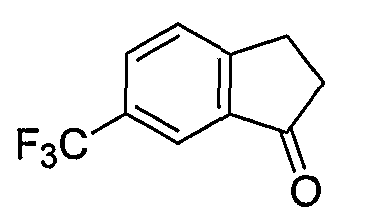
Стадия D
6-(Трифторметил)индан-1-он
К раствору 3-[2-бром-4-(трифторметил)фенил]пропановой кислоты (6,40 г, 21,5 ммоль) в THF (300 мл) и гексана (80 мл) при -78°C добавляли 2,5 M раствор н-бутиллития в гексане (19 мл). После перемешивания в течение 15 мин смесь выливали в раствор 2 Н HCl (150 мл). Два слоя разделяли, и водный слой экстрагировали эфиром. Объединенные органические слои промывали раствором NaHCО3, водой, солевым раствором, сушили над MgSО4 и концентрировали. Колоночная хроматография на силикагеле (10-20% EtOAC в гексане) приводила к получению указанного в заголовке соединения (2,2 г, 51%) в виде масла. MS расчетная для C10H7F3О: (M+H)+ 201; обнаружено 201,0.
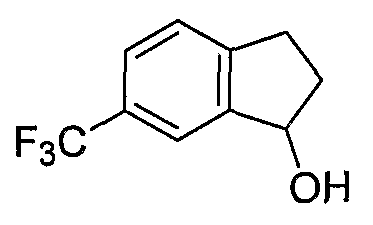
Стадия E
6-(Трифторметил)индан-1-ол
К раствору 6-(трифторметил)индан-1-она (2,20 г, 11 ммоль) в THF (30 мл) добавляли борогидрид натрия (0,50 г, 13 ммоль). После перемешивания в течение 30 мин медленно добавляли метанол (10 мл). Перемешивание продолжали в течение 2 ч. Реакционную смесь гасили добавлением водного раствора хлорида аммония. Два слоя разделяли и водный слой экстрагировали эфиром. Объединенные органические слои промывали водой, солевым раствором, сушили над MgSО4 и концентрировали. Колоночная хроматография на силикагеле (10-15% EtOAC в гексане) приводила к получению указанного в заголовке соединения (1,52 г, 68%) в виде масла. MS расчетная для C10H9F3О: (M+H)+ 203; обнаружено 185,0 (M-H2O+1)+.
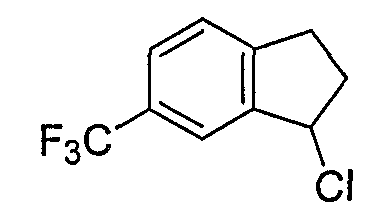
Стадия F
1-Хлор-6-(трифторметил)индан
6-(Трифторметил)индан-1-ол (1,52 г, 7,5 ммоль) растворяли в тионилхлориде (15 мл). После перемешивания при комнатной температуре в течение 2 ч раствор концентрировали в вакууме с предоставлением указанного в заголовке соединения (1,5 г, 90%).
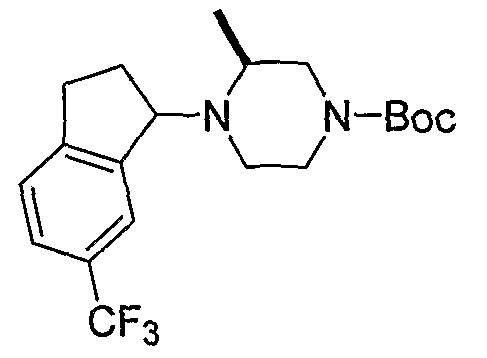
Стадия G
трет-Бутил-(3S)-3-Метил-4-[6-(трифторметил)-2,3-дигидро-1H-инден-1-ил]пиперазин-1-карбоксилат
Раствор 1-хлор-6-(трифторметил)индана (1,52 г, 6,89 ммоль), трет-бутил-(3S)-3-метилпиперазин-1-карбоксилата (2,1 г, 10 ммоль), йодида натрия (3 г, 20 ммоль) и триэтиламина (3 г, 30 ммоль) в DMF (20 мл) перемешивали при 60°C в течение ночи. После охлаждения до комнатной температуры добавляли воду. Полученный в результате раствор экстрагировали дважды EtOAc. Объединенные экстракты промывали водой и солевым раствором, сушили над MgSО4 и концентрировали. Колоночная хроматография на силикагеле (10%-30% EtOAc в гексане) давала два изомера. Изомер 1: 0,52 г (коричневое масло). MS расчетная для C20H27F3N2О2: (M+H)+ 385; обнаружено 385,2. Изомер 2: 0,41 г (коричневое масло). MS: (M+H)+ 385,2.
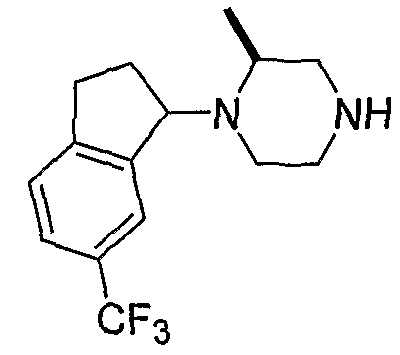
Стадия H
(2S)-2-Метил-1-[6-(трифторметил)-2,3-дигидро-1H-инден-1-ил]пиперазин
трет-Бутил-(3S)-3-метил-4-[6-(трифторметил)-2,3-дигидро-1H-инден-1-ил]пиперазин-1-карбоксилат (изомер 1 со стадии G, 0,52 г, 1,4 ммоль) растворяли в 4 M растворе HCl в 1,4-диоксане (10 мл). После перемешивания при комнатной температуре в течение 2 ч раствор концентрировали в вакууме с предоставлением указанного в заголовке соединения в виде соли дигидрохлорида (0,48 г, 100%). MS расчетная для C15H19F3N2: (M+H)+ 285; обнаружено 285,1.
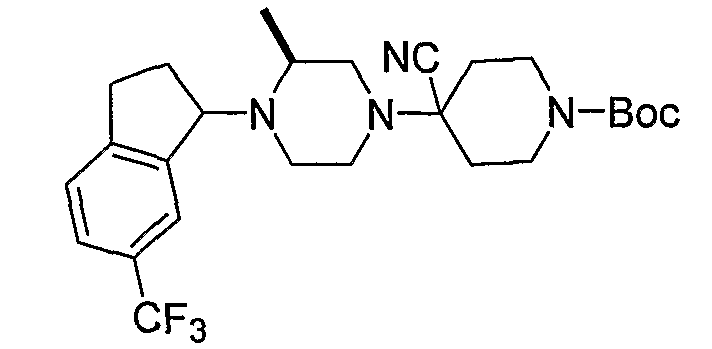
Стадия I
трет-Бутил-4-циано-4-{(3S)-3-метил-4-[6-(трифторметил)-2,3-дигидро-1H-инден-1-ил]пиперазин-1-ил}пиперидин-1-карбоксилат
Дигидрохлорид (2S)-2-метил-1-[6-(трифторметил)-2,3-дигидро-1H-инден-1-ил]пиперазина (0,38 г, 1,3 ммоль) растворяли в дихлорметане. Раствор промывали насыщенным раствором NaHCО3, сушили над MgSО4 и концентрировали. Остаток растворяли в дихлорметане (20 мл). К нему добавляли трет-бутил-4-оксо-1-пиперидинкарбоксилат (0,32 г, 1,6 ммоль) и тетраизопропоксид титана (0,8 г, 3 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и концентрировали в вакууме. Остаток растворяли в THF (20 мл). Добавляли цианид диэтилалюминия (0,18 г, 1,6 ммоль). После перемешивания при комнатной температуре в течение 5 ч раствор гасили добавлением воды (3 мл). Полученный в результате раствор фильтровали через целит, и целит промывали несколько раз дихлорметаном. Фильтрат сушили над MgSО4 и концентрировали с получением указанного в заголовке соединения (0,72 г, 98%) в виде коричневого вязкого масла. MS расчетная для C26H35F3N4О2: (M+H)+ 493; обнаружено 493,2.
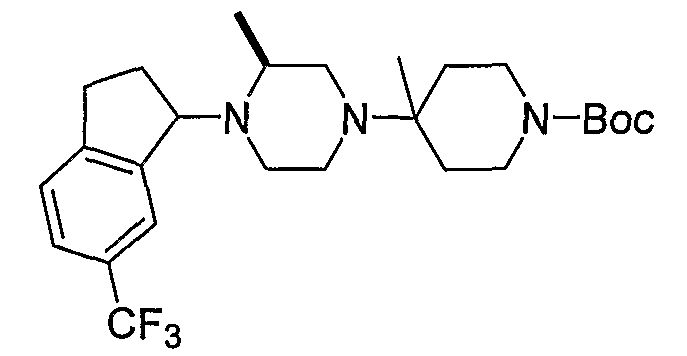
Стадия J
трет-Бутил-4-метил-4-{(3S)-3-метил-4-[6-(трифторметил)-2,3-дигидро-1H-инден-1-ил]пиперазин-1-ил}пиперидин-1-карбоксилат
К раствору трет-бутил-4-циано-4-{(3S)-3-метил-4-[6-(трифторметил)-2,3-дигидро-1H-инден-1-ил]пиперазин-1-ил}пиперидин-1-карбоксилата (0,72 г, 1,3 ммоль) в THF (20 мл) добавляли 3 M раствор бромида метилмагния в эфире (4,0 мл). После перемешивания при комнатной температуре в течение ночи реакцию гасили добавлением воды. Полученный в результате раствор экстрагировали дважды EtOAc. Объединенные слои EtOAc сушили и концентрировали. Колоночная хроматография на силикагеле (20-30% EtOAc в гексане) предоставляла указанное в заголовке соединение (0,32 г, 50%) в виде вязкого масла. MS расчетная для C26H35F3N3О2:(M+H)+ 482; обнаружено 482,3.
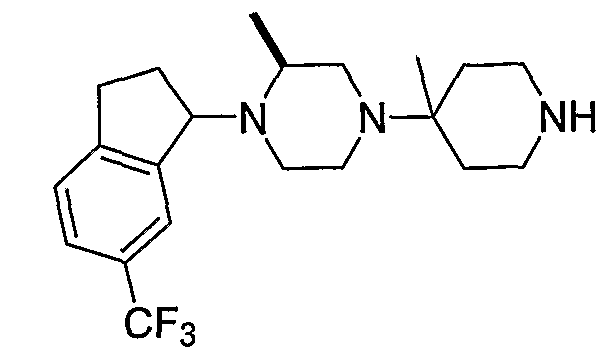
Стадия K
(2S)-2-Метил-4-(4-метилпиперидин-4-ил)-1-[6-(трифторметил)-2,3-дигидро-1H-инден-1-ил]пиперазин
Трет-бутил-4-метил-4-{(3S)-3-метил-4-[6-(трифторметил)-2,3-дигидро-1H-инден-1-ил]пиперазин-1-ил}пиперидин-1-карбоксилат (0,32 г, 0,6 ммоль) растворяли в 4 M растворе HCl в диоксане (8,0 мл). После перемешивания при комнатной температуре в течение 2 ч раствор концентрировали с получением указанного в заголовке соединения (0,35 г) в виде соли тригидрохлорид. MS расчетная для C21H30F3N3: (M+H)+ 382; обнаружено 382,2.
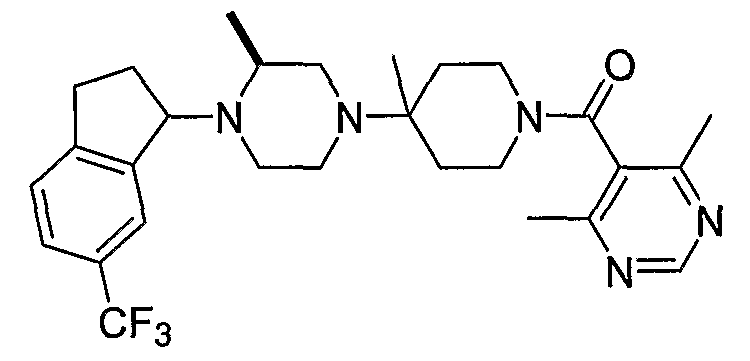
Стадия L
4,6-Диметил-5-[(4-метил-4-{(3S)-3-метил-4-[6-(трифторметил)-2,3-дигидро-1H-инден-1-ил]пиперазин-1-ил}пиперидин-1-ил)карбонил]пиримидин
К раствору тригидрохлорида (2S)-2-метил-4-(4-метилпиперидин-4-ил)-1-[6-(трифторметил)-2,3-дигидро-1H-инден-1-ил]пиперазина (100 мг, 0,183 ммоль), 4,6-диметилпиримидин-5-карбоновой кислоты (67 мг, 0,22 ммоль) в DMF (5 мл) добавляли гексафторфосфат бензотриазол-1-илокси-трис(диметиламино)фосфония (97 мг, 0,22 ммоль) и затем триэтиламин (90 мг, 0,9 ммоль). После перемешивания при комнатной температуре в течение ночи добавляли раствор NaHCО3 в воде. Полученный в результате раствор экстрагировали дважды EtOAc. Объединенные слои EtOAc промывали солевым раствором, сушили над MgSO4 и концентрировали. Колоночная хроматография на силикагеле (10-20% MeOH в EtOAc) приводила к получению указанного в заголовке соединения (60 мг) в виде масла. MS расчетная для C28H36F3N5О: (M+H)+ 516; обнаружено 516,2.
Пример 8
4,6-Диметил-5-[(4-метил-4-{(3S)-3-метил-4-[5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]пиперазин-1-ил}пиперидин-1-ил)карбонил]пиримидин
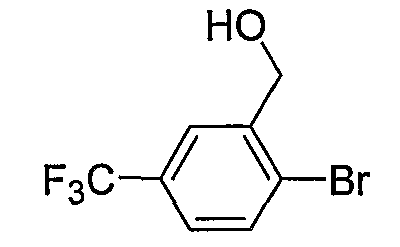
Стадия A
[2-Бром-5-(трифторметил)фенил]метанол
К раствору 2-бром-5-(трифторметил)бензонитрила (10,0 г, 40 ммоль) в дихлорметане (100 мл) по каплям добавляли 1,0 M раствор гидрида диизобутилалюминия в гексане (48 мл). Полученный в результате раствор перемешивали в азоте при комнатной температуре в течение 1 ч и затем разбавляли добавлением эфира (100 мл). После нагревания на ледяной бане осторожно добавляли 3 Н раствор HCl, и смесь интенсивно встряхивали при комнатной температуре в течение 15 минут. Органический слой промывали солевым раствором, сушили (MgSО4) и упаривали. Полученное в результате масло очищали тонкослойной хроматографией (5% EtOAc/гексан) с получением 5 г 2-бром-5-трифторметилбензальдегида. 1H-ЯМР (CDCl3) δ 10,39 (с, 1H), 8,18 (д, J=2 Гц, 1H), 7,82 (д, J=8,8 Гц, 1H), 7,70 (дд, J=8,5 Гц, 2 Гц, 1H).
К смеси 2-бром-5-(трифторметил)бензальдегида (5 г, 20 ммоль) в THF (20 мл) при 0°C добавляли борогидрид натрия (0,8 г, 20 ммоль). Полученную в результате смесь перемешивали при 0°C до комнатной температуры в течение 1 ч. Реакцию гасили добавлением водного раствора NaHCО3. Полученный в результате раствор экстрагировали дважды EtOAc. Объединенные экстракты промывали солевым раствором, сушили (MgSО4), фильтровали и концентрировали с получением требуемого спирта в виде белого твердого вещества (4,4 г). 1H-ЯМР (CDCl3) δ 7,81 (с, 1H), 7,66 (д, J=8,3 Гц, 1H), 7,42 (д, J=8,3 Гц, 2 Гц, 1H), 4,81 (д, J=6,3 Гц, 2H), 2,03 (м, 1Н).
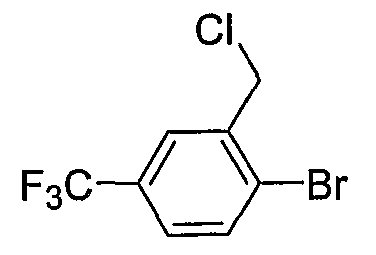
Стадия B
1-Бром-2-(хлорметил)-4-(трифторметил)бензол
К [2-бром-5-(трифторметил)фенил]метанолу (4,4 г, 17 ммоль) добавляли тионилхлорид (5 мл), полученную смесь перемешивали при комнатной температуре в течение 1 ч. Упаривание в вакууме давало неочищенный продукт в виде масла. 1H-ЯМР (CDCl3) δ 7,73 (д, J=8,3 Гц 1Н), 7,73 (д, J=2,0 Гц, 1H), 7,53 (дд, J=8,5, 2,2 Гц, 1H), 5,66 (д, J=12,7 Гц, 1H), 5,46 (д, J=12,2 Гц).
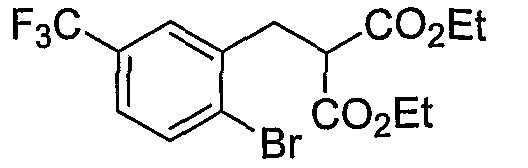
Стадия C
Диэтил-[2-бром-5-(трифторметил)бензил]малонат
К раствору этилмалоната (23 г, 140 ммоль) в DMF (70 мл) при 0ºC добавляли гидрид натрия (3,9 г, 60% в минеральном масле, 97 ммоль), и полученную смесь перемешивали при комнатной температуре в течение 30 мин. К смеси добавляли раствор 1-бром-2-(хлорметил)-4-(трифторметил)бензола (16 г, 60 ммоль) в DMF (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч и гасили ледяной водой. Полученный в результате раствор экстрагировали дважды EtOAc. Экстракты промывали солевым раствором, сушили (MgSО4), фильтровали и концентрировали. Неочищенный материал очищали тонкослойной хроматографией на силикагеле с элюцией от 3% до 5% EtOAc/гексан с получением требуемого продукта (15,2 г, 64%) в виде масла. LC/MS расчетная для C15H16BrF3O4:(M+H)+ 397; обнаружено 397,1/399,1.
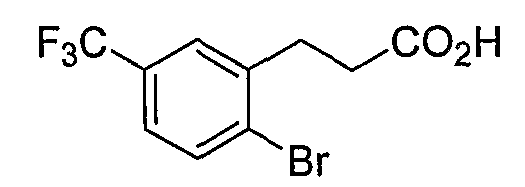
Стадия D
3-[2-бром-5-(трифторметил)фенил]пропановая кислота
К раствору диэтил[2-бром-5-(трифторметил)бензил]малоната (22,9 г, 57,6 ммоль) в этаноле (100 мл) и воде (50 мл) добавляли 5 M раствор гидроксида натрия в воде (30 мл). Смесь нагревали до кипячения с обратным холодильником в течение 2 ч. Этанол удаляли путем упаривания. Водный слой экстрагировали эфиром и затем подкисляли концентрированной HCl до pH 5, причем в это время осаждалось большое количество белого твердого вещества. Твердое вещество собирали фильтрацией. Фильтрат экстрагировали дважды этилацетатом, и экстракты промывали солевым раствором, сушили (MgSО4) и концентрировали с получением белого твердого вещества.
Объединенное твердое вещество декарбоксилировали нагреванием на масляной бане до 180°C примерно в течение 1 ч. Полученное в результате желтое масло охлаждали и помещали в вакуум с получением требуемой монокислоты (11,5 г, 67%). LC/MS расчетная для C10H8BrF3O2: (M+H)+ 297; обнаружено 297,1/299,1.
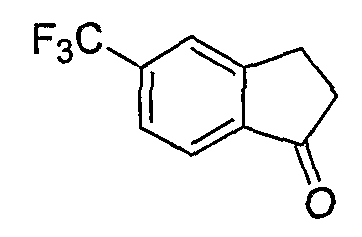
Стадия E
5-(Трифторметил)индан-1-он
К раствору 3-[2-бром-5-(трифторметил)фенил]пропановой кислоты (2,8 г, 9,4 ммоль) в THF (100 мл) и гексане (20 мл) при -78°C по каплям добавляли 2,5 M раствор н-бутиллития в гексане (8,3 мл). После окончания добавления реакцию гасили насыщенным NH4Cl. Полученный в результате раствор экстрагировали дважды этилацетатом. Экстракты промывали насыщенным NaHCО3, солевым раствором, сушили над MgSО4 и концентрировали. Неочищенный материал очищали тонкослойной хроматографией на силикагеле с элюцией 10-20% EtOAc/гексан с получением требуемого продукта в виде белого твердого вещества (0,7 г, 37%). LC/MS расчетная для C10H7F3O: (M+H)+ 201; обнаружено 201,1.
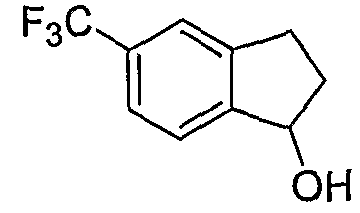
Стадия F
5-(Трифторметил)индан-1-ол
К раствору 5-(трифторметил)индан-1-она (0,7 г, 3 ммоль) в THF (5 мл), охлажденного на ледяной бане, добавляли борогидрид натрия (0,1 г, 3 ммоль) с последующим добавлением MeOH (1 мл). После перемешивания в течение 30 мин, реакцию гасили водным NaHCО3. Полученный в результате раствор экстрагировали дважды EtOAc. Экстракты промывали солевым раствором, сушили (MgSО4), фильтровали и концентрировали. Неочищенный материал очищали тонкослойной хроматографией на силикагеле с элюцией 20% EtOAc/гексан с получением требуемого продукта (0,65 г, 92%) в виде масла. LC/MS расчетная для C10H9F3О: (M+H)+ 203; обнаружено 185,1 (M+H-H2О)+.
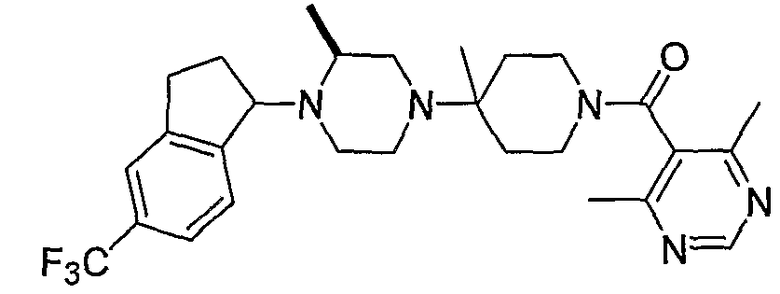
Стадия G
4,6-Диметил-5-[(4-метил-4-{(3S)-3-метил-4-[5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]пиперазин-1-ил}пиперидин-1-ил)карбонил]пиримидин
Исходя из 5-(трифторметил)индан-1-ола, указанное в заголовке соединение получали по процедурам, описанным для примера 7. MS расчетная для C28H36F3N5О: (M+H)+ 516; обнаружено 516,2.
Пример 9
1-((2S)-4-{1-[(4,6-Диметилпиримидинил-5-ил)карбонил]-4-метилпиперидин-4-ил}-2-метилпиперазин-1-ил)-5-(трифторметил)индан-2-ол
Стадия A
6-(Трифторметил)-1H-инден
Смесь 5-(трифторметил)индан-1-ола (1,6 г, 7,9 ммоль) и п-толуолсульфоновой кислоты (0,02 г, 0,1 ммоль) в толуоле (20 мл) кипятили с обратным холодильником через ловушку Дина-Старка примерно в течение 3 ч. Раствор концентрировали в вакууме, и остаток очищали путем тонкослойной хроматографии на силикагеле, элюируя 5% EtOAc/гексан с получением требуемого продукта в виде масла (1,4 г, 96%).
Стадия B
4-(Трифторметил)-6,6a-дигидро-1aH-инденo[l,2-b]оксирен
К раствору 6-(трифторметил)-1H-индена (1,1 г, 6 ммоль) в безводном дихлорметане (80 мл) при -78°C добавляли 5,5 M раствор трет-бутилгидропероксида в н-декане (1,3 мл) с последующим добавлением тетрахлорида титана (0,79 мл, 7,2 ммоль). После перемешивания при -78°C в течение 1 ч, полученный в результате коричневый раствор гасили смесью Et2О/насыщенного раствора Na2SО3. Смесь перемешивали при комнатной температуре в течение 1 ч с получением бесцветного раствора. Органический слой разделяли и промывали солевым раствором, сушили над MgSО4 и концентрировали с получением неочищенного продукта (1,2 г) в виде твердого вещества. LC/MS расчетная для C10H7F3O:(M+H)+ 201; обнаружено 201,0.
Стадия C
трет-Бутил-(3S)-4-[2-гидрокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-карбоксилат
Смесь 4-(трифторметил)-6,6a-дигидро-1aH-инденo[1,2-b]оксирена (1,2 г, 6,0 ммоль) и трет-бутил-(3S)-3-метилпиперазин-1-карбоксилата (1,4 г, 7,2 моль) в этаноле (20 мл) кипятили с обратным холодильником в течение ночи. Добавляли еще 1 грамм трет-бутил-(3S)-3-метилпиперазин-1-карбоксилата. Смесь переносили в закупоренный флакон и нагревали до 95°C в течение 2 суток. Растворитель концентрировали и остаток очищали тонкослойной хроматографией с элюцией 25% EtOAc/гексан, с последующей элюцией 5% MeOH/EtOAc + 0,5% концентрированного NH4ОH. Выделяли два изомера. Изомер 1 (быстро двигающийся изомер): 0,45 г; MS расчетная для C20H27F3N2О3 (M+H)+ 401; обнаружено 401,1. Изомер 2 (медленно двигающийся изомер): 0,38 г; MS обнаруженная 401,1.
Стадия D
1-((2S)-4-{1-[(4,6-Диметилпиримидин-5-ил)карбонил]-4-метилпиперидин-4-ил}-2-метилпиперазин-1-ил)-5-(трифторметил)индан-2-ол
Исходя из трет-бутил-(3S)-4-[2-гидрокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-карбоксилата (изомер 1 со стадии C) указанное в заголовке соединение получали с использованием процедур, сходных с описанными в примере 7. MS расчетная для C28H36F3N5O2: (M+H)+ 532; обнаружено 532.
Пример 10
5-[(4-{(3S)-4-[2-Метокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидин
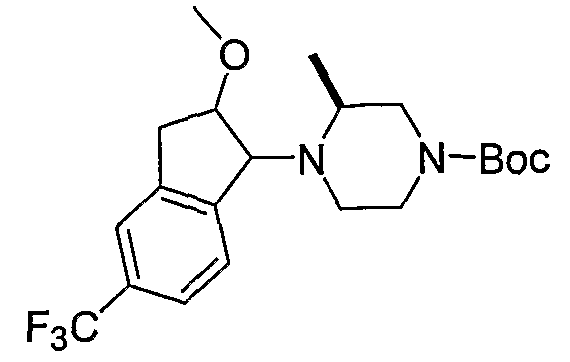
Стадия A
трет-Бутил (3S)-4-[2-метокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-карбоксилат
К раствору трет-бутил-(3S)-4-[2-гидрокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-карбоксилата (изомер 1 со стадии C в примере 9) (50 мг, 0,1 ммоль) в THF (2 мл) при 0°C добавляли NaH (8 мг, 60% в масле, 0,2 ммоль). После перемешивания в течение 10 мин добавляли MeI (28 мг, 0,2 ммоль). Смесь перемешивали при температуре окружающей среды в течение 1 ч и гасили водным NH4Cl. Полученный в результате раствор экстрагировали дважды EtOAc. Экстракты промывали солевым раствором, сушили (MgSО4), фильтровали и концентрировали с получением неочищенного продукта. LC/MS расчетная для C21H29F3N2О3: (M+H)+ 415; обнаружено 415,2.
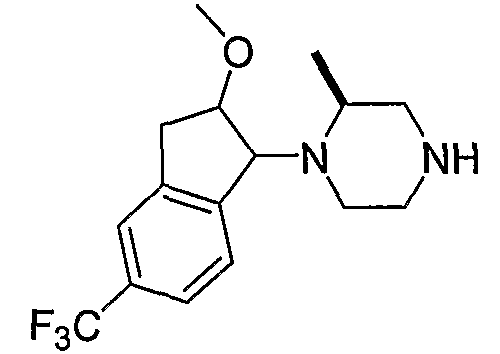
Стадия B
(2S)-1-[2-Метокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-2-метилпиперазин
К трет-бутил-(3S)-4-[2-метокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-карбоксилату (51,8 мг, 0,125 ммоль) добавляли 4,0 M раствор хлороводорода в диоксане (2 мл). Смесь перемешивали при комнатной температуре в течение 1 ч и концентрировали в вакууме с получением указанного в заголовке соединения в виде соли дигидрохлорида. LC/MS расчетная для C16H22ClF3N2О: (M+H)+ 351; обнаружено 351,1.
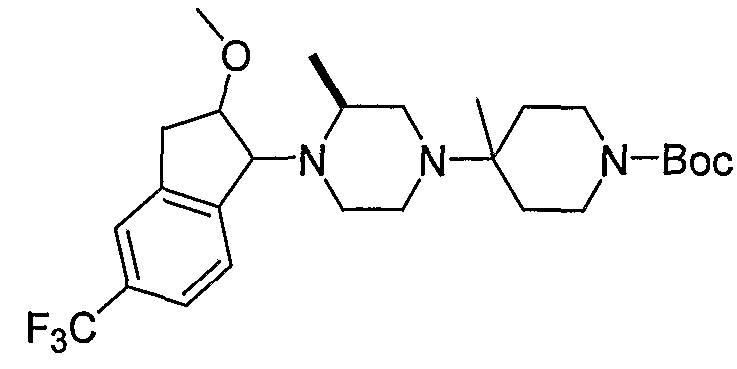
Стадия C
трет-Бутил-4-{(3S)-4-[2-метокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-карбоксилат
К смеси (2S)-1-[2-метокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-2-метилпиперазин гидрохлорида (44 мг, 0,12 ммоль) и трет-бутил-4-оксо-1-пиперидинкарбоксилата (25 мг, 0,12 ммоль) в метиленхлориде (2 мл) добавляли триэтиламин (0,07 мл, 0,5 ммоль), с последующим добавлением тетраизопропоксида титана (0,037 мл, 0,12 ммоль). Полученную в результате смесь перемешивали при комнатной температуре в течение ночи и концентрировали с предоставлением неочищенного энамина.
Неочищенный энамин растворяли в THF и обрабатывали 1,0 M цианида диэтилалюминия в толуоле (0,15 мл). Смесь перемешивали при комнатной температуре в течение ночи и гасили добавлением водного NaHCО3 и EtOAc. Полученный в результате раствор фильтровали через целит. Фильтрат разделяли, и промывали органический слой солевым раствором, сушили (MgSО4), фильтровали и концентрировали с получением неочищенного цианосоединения. LC/MS: 523,2 (M+H)+.
Неочищенное цианосоединение растворяли в THF и обрабатывали 3,0 M раствором бромида метилмагния в простом эфире (0,2 мл) при 0°C. Смесь перемешивали при комнатной температуре в течение 4 ч. Добавляли еще 0,2 мл раствора бромида метилмагния, и смесь перемешивали при комнатной температуре в течение 2 суток. Реакцию гасили водным NaHCО3 и дважды экстрагировали EtOAc. Экстракты промывали солевым раствором, сушили (MgSО4), фильтровали и концентрировали. Неочищенный материал очищали тонкослойной хроматографией на силикагеле с элюцией 25% и затем 50% EtOAc/гексаном с получением указанного в заголовке соединения (25 мг). MS расчетная для C27H40F3N3О3: (M+H)+ 512; обнаружено 512,2.
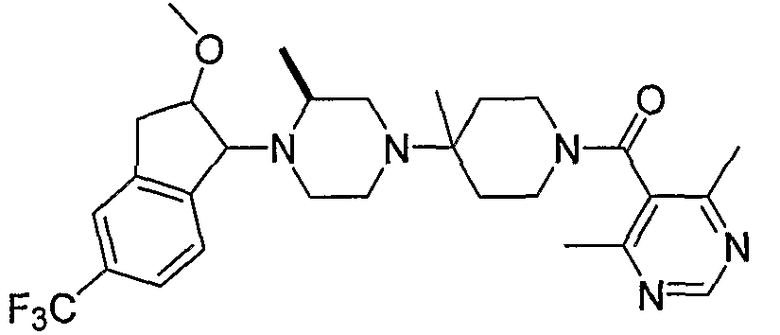
Стадия D
5-[(4-{(3S)-4-[2-Метокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил)-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидин
К трет-бутил-4-{(3S)-4-[2-метокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-карбоксилату (0,02 г, 0,04 ммоль) добавляли 4,0 M раствор HCl в 1,4-диоксане (2,0 мл). Полученную в результате смесь перемешивали при комнатной температуре в течение 1 ч, концентрировали и упаривали в вакууме до сухости.
К указанному выше гидрохлориду амина добавляли DMF (2,0 мл), 4,6-диметилпиримидин-5-карбоновую кислоту (0,01 г, 0,08 ммоль), гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония (0,026 г, 0,059 ммоль) и триэтиламин (0,02 мл, 0,1 ммоль). После перемешивания при комнатной температуре в течение ночи реакционную смесь гасили водным NaHCО3 и экстрагировали дважды EtOAc. Экстракты промывали солевым раствором, сушили (MgSО4), фильтровали и концентрировали. Неочищенный материал очищали путем обращено-фазовой ВЭЖХ (10-80% ацетонитрил-вода с 0,05% TFA, 30 мин, 10 мл/мин) с получением указанного в заголовке соединения (25 мг) в виде бис-соли TFA. LC/MS расчетная для C29H38F3N5О2: (M+H)+ 546; обнаружено 546,2.
Пример 11
5-[(4-(3S)-4-[(1R,2R)-2-Этокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидин дигидрохлорид
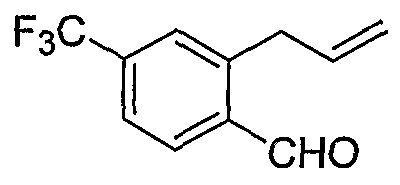
Стадия A
2-Аллил-4-(трифторметил)бензальдегид
В высушенный в термостате 5 литровый флакон с 4 горлышками вокруг дна, оснащенный подвесной мешалкой, азотным шприцем, воронкой для добавления на 500 мл, воронкой для добавления на 250 мл и термометром, заряжали тетрагидрофуран (1400 мл) и N,N,N'-триметилэтан-1,2-диамин (105 мл, 0,788 моль). Раствор охлаждали до -40°C (сухой лед/ MeCN баня). н-Бутиллитий (1,6 M в гексане, 510 мл) добавляли в 500 мл воронку для добавления и затем добавляли к вышеуказанному раствору в течение 40 минут (от -40°С до -35°C). Бесцветный раствор приобретал светло-желтый цвет. Затем холодную баню удаляли и реакционную смесь перемешивали в течение 30 минут, пока она не нагревалась до температуры в -10°C. Реакцию охлаждали до -40°С, до -45°С и п-трифторметилбензальдегид (77 мл, 0,56 М) добавляли через шприц в 250 мл воронку для добавления. Альдегид добавляли по каплям в течение 15 минут, пока температура реакции оставалась на уровне от -40°C до -35°C. В процессе добавления реакция приобретала коричневый цвет. Реакцию перемешивали при температуре от -50°C до -40°C в течение 30 минут. Второе добавление 1,6 M н-бутиллития в гексане (400 мл) проводили через 50 минут через воронку для добавления. Реакционную смесь оставляли до повышения температуры до -25°C и выдерживали при от -20°C до -30°C в течение 3 часов, после чего бромид меди (I) (108 г, 0,738 моль) добавляли непосредственно через воронку для порошка. Холодную баню удаляли, и реакции позволяли нагреться при перемешивании в течение дополнительных 90 минут. Реакцию охлаждали до -30°C до -25°C и раствор бромида аллила (78 мл, 0,90 моль) в тетрагидрофуране (240 мл) добавляли по каплям в течение 40 минут через 250 мл воронку для добавления порционно. После перемешивания в течение 2 часов, реакцию гасили добавлением метанола (100,0 мл). Холодную баню удаляли, и смесь перемешивали в течение 5 минут перед добавлением 6,00 M раствора соляной кислоты (300,0 мл) (pH~7). Смесь дополнительно перемешивали в течение 15 минут, после чего пропускали через целитную подушку, и целитную подушку промывали эфиром. Насыщенный хлорид аммония (400 мл) добавляли и собирали органическую фазу. Водную фазу затем экстрагировали 300 мл эфира. Объединенную органическую фазу разбавляли 400 мл насыщенного хлорида аммония (400 мл), 1 M бикарбоната натрия (3×400 мл) и солевым раствором (400 мл) и высушивали над сульфатом магния. После удаления высушивающего агента фильтрацией, фильтрат концентрировали с использованием ротационного испарителя (сокр. Rotovap) до получения коричневой жидкости. Дальнейшее очищение проводили путем дистилляции до получения 105,4 г (85%) продукта. 1H-ЯМР (400 МГц, CDCl3) δ (м.д.) 10,22 (с, 1Н), 8,02 (д, 1H), 7,80 (д, 1H), 7,74 (с, 1H), 6,02 (м, 1H), 5,06 (м, 1H), 4,96 (м, 1Н), 3,89 (д, 2H).
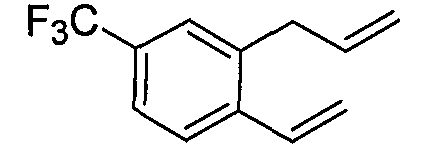
Стадия B
2-Аллил-4-(трифторметил}-1-винилбензол
Бромид трифенилметилфосфония (182 г, 0,508 М) суспендировали в диэтилэфире (900 мл, 8 моль) и охлаждали до 0°C. н-Бутиллитий (1,60 M в гексане, 289 мл) быстро добавляли через шприц и получившуюся смесь нагревали до комнатной температуры и перемешивали в течение ночи (18 ч). Перемешивание прекращали для осаждения сухого осадка и переносили верхний светлый раствор через канюлю к раствору 2-аллил-4-(трифторметил)бензальдегида (99,0 г, 0,462 моль) в метиленхлориде (900 мл), который перемешивали при 0°C. После добавления, ледяную баню удаляли и смесь нагревали кипячением с обратным холодильником в течение примерно 30 часов. После охлаждения до комнатной температуры оранжевый раствор концентрировали, пока не оставалось небольшое количество растворителя. Силикагель добавляли к раствору при помешивании до образования слегка вязкой массы. Пентан (500 мл) добавляли для растворения более плотных сгустков. Смесь пропускали через силикагель в стеклянной пористой вакуумной воронке с использованием пентана (1,5 л). Раствор пентана собирали в 3 л флакон с круглым дном. Почти бесцветную жидкость затем концентрировали до получения чистого диена (78 г, 79,3%); 1H-ЯМР (400 МГц, CDCl3) δ (м.д.) 7,58 (д, 1H), 7,45 (д, 1H), 7,41 (с, 1Н), 6,95 (дд, 1Н), 5,95 (м, 1H), 5,72 (д, 1H), 5,41 (д, 1H), 5,11 (д, 1H), 5,00 (д, 1Н), 3,48 (д, 2H).
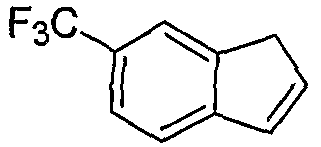
Стадия C
6-(Трифторметил)-1H-инден
Хлорид метилена (0,6 л) добавляли в 1 л флакон, содержащий 2-аллил-4-(трифторметил)-1-винилбензол (80,0 г, 0,377 моль). Дихлорид бис(трициклогексилфосфин)бензилидина рутения(IV) (катализатор Grubbs, 1-ое поколение) (3,1 г, 0,0038 моль) добавляли в раствор и полученный в результате раствор кипятили с обратным холодильником в течение ночи (18 ч). Растворитель упаривали до получения темного масла, которое пропускали через силикагелевую подушку с использованием пентана. После осторожного удаления пентана, собирали 57 г (82,8%) чистого продукта в виде светло-коричневого масла. 1H-ЯМР (400 МГц, CDCl3) δ (м.д.) 7,72 (с, 1H), 7,55 (д, 1H), 7,48 (д, 1Н), 6,93 (м, 1Н), 6,74 (м, 1H), 3,46 (ушир.с, 1H).
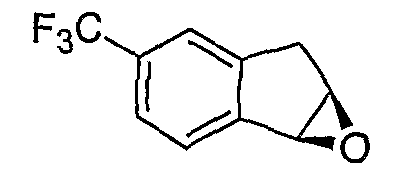
Стадия D
4-(Трифторметил)-6,6a-дигидро-1aH-индено[1,2-b]оксирен
К раствору 2 M гипохлорита натрия в воде (200 мл) при 0°C добавляли водный гидроксид натрия (1 M, 40 мл), N-оксид 4-(3-фенилпропил)пиридин (6,0 г, 0,03 М) и раствор хлорида (S,S)-(+)-N,N'-бис(3,5-ди-трет-бутилсалицилиден)-1,2-циклогександиамино-марганца(III) (4,13 г, 0,00651 моль) в дихлорметане (700 мл). Полученный коричневый раствор встряхивали в течение 15 мин при 0°C. К холодному раствору добавляли раствор 6-(трифторметил)-1H-индена (51 г, 0,24 моль) в дихлорметане (700 мл) с одновременным добавлением водного гипохлорита натрия (2 M, 200 мл). Реакцию проводили при 0°C, и коричневый раствор сохранял свой цвет при добавлении индена. Через 4 часа органическую фазу собирали и высушивали над сульфатом натрия. Смесь пропускали через силикагель с использованием пентана. После удаления растворителя получали 42 г (88%, 84% ee) эпоксида. 1H-ЯМР (400 МГц, CDCl3) δ (м.д.) 7,72 (с, 1H), 7,55 (д, 1H), 7,48 (д, 1Н), 6,93 (м, 1Н), 6,74 (м, 1H), 3,46 (ушир.с, 1H).
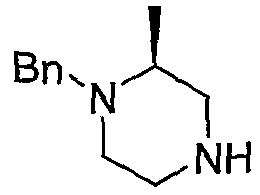
Стадия E
(2S)-1-Бензил-2-метилпиперазин
трет-Бутил-(3S)-3-метилпиперазин-1-карбоксилат (380,0 г, 1,897 М) и бромид бензила (248 мл, 2,09 М) смешивали в ацетонитриле (440 мл). Триэтиламин (300,0 мл, 2,152 моль) осторожно добавляли, и смесь кипятили с обратным холодильником в течение ночи. После охлаждения смеси до комнатной температуры, сухой осадок отфильтровывали. Фильтрат концентрировали. Остаток соединяли с сухим осадком и растворяли в хлориде метилена. Раствор хлорида метилена промывали 1 N NaOH и высушивали над сульфатом магния. После удаления растворителя остаток непосредственно обрабатывали 6 N HCl при 0°C и, спустя 3 часа, раствор защелачивали медленным добавлением сухого гидроксида натрия. Полученную смесь экстрагировали хлоридом метилена, и экстракт высушивали над сульфатом магния. После удаления растворителя получали 330 г (91,4%) продукта. Продукт использовали непосредственно в следующей стадии. 1H-ЯМР (400 МГц, CDCl3) δ (м.д.) 7,30 (м, 5H), 4,05 (д, 1H), 3,15 (д, 1Н), 2,92 (м, 1Н), 2,83 (м, 2H), 2,67 (м, 1Н), 2,60 (м, 1Н), 2,38 (м, 1Н), 2,36 (ушир.с, 2H), 2,06 (м, 1Н), 1,14 (д, 3Н). MS (EI) 191,1 (М+1).
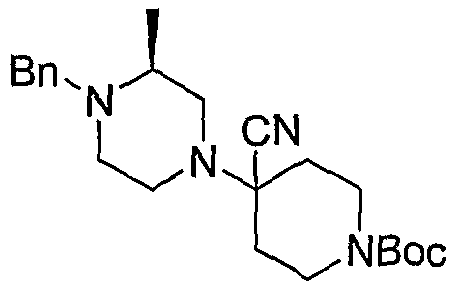
Стадия F
трет-Бутил-4-[(3S)-4-Бензил-3-метилпиперазин-1-ил]-4-цианопиперидин-1-карбоксилат
В 5 л флаконе (2S)-1-бензил-2-метилпиперазин (260,0 г, 1,366 М), дихлорометан (1000 мл), трет-бутил-4-оксо-1-пиперидинкарбоксилат (272 г, 1,37 моль) и тетраизопропоксид титана (480,0 мл, 1,626 моль) смешивали и смесь перемешивали при комнатной температуре в течение 20 часов. Смесь охлаждали до 0°C и по каплям добавляли цианид диэтилалюминия в толуоле (1,0 M, 1600 мл). Полученную смесь перемешивали при комнатной температуре в течение 20 часов. Содержимое реакции затем разделяли на два 5 л флакона, в каждый флакон добавляли 1 л этилацетата, 500 г бикарбоната натрия, 150 г целита, после чего их охлаждали -40°C с использованием сухого льда/ацетонитрила. Затем в каждый флакон медленно добавляли 200 мл насыщенного водного сульфата натрия при энергичном перемешивании. После медленного нагревания реакционной смеси до комнатной температуры и перемешивания в течение 4 часов, в каждый флакон добавляли 1 л метанола. После перемешивания в течение ночи, реакционную смесь фильтровали через тонкий слой песка. Сухой остаток переносили обратно в 5 л флакон и перемешивали с 3 л метанола в течение 5 часов, и нерастворимый сухой осадок отфильтровывали. Объединенные фильтраты концентрировали до сухого состояния и добавляли 3 л хлорида метилена. Нерастворимый сухой осадок отфильтровывали. Фильтрат высушивали над сульфатом магния, растворитель удаляли до получения 484 г (88,9%) продукта в виде светло-желтого сухого осадка. Неочищенный продукт тщательно очищали и использовали непосредственно в следующей стадии. 1H-ЯМР (400 МГц, CDCl3) δ (м.д.) 7,32 (м, 5H), 4,04 (д, 1H), 3,95 (ушир.с, 2Н), 3,15 (д, 1Н), 3,15 (ушир.с, 2Н), 2,82 (м, 2H), 2,73 (м, 2Н), 2,44 (м, 2Н), 2,25 (т, 1Н), 2,15 (м, 1H), 2,05 (м, 1Н), 1,66 (м, 1Н), 1,46 (с, 9Н), 1,17 (д, 3Н). MS (EI) 399,2 (М+1).
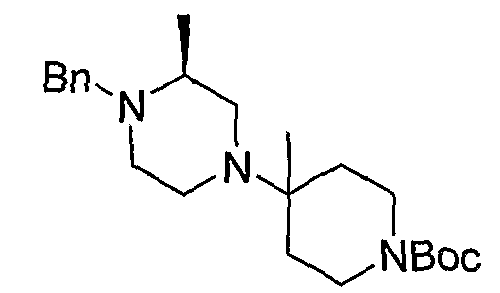
Стадия G
трет-бутил-4-[(3S)-4-Бензил-3-метилпиперазин-1-ил]-4-метилпиперидин-1-карбоксилат
Раствор трет-бутил-4-[(3S)-4-бензил-3-метилпиперазин-1-ил]-4-цианопиперидин-1-карбоксилата (242 г, 0,605 моль) в тетрагидрофуране (1,5 л) в 5 л флаконе охлаждали до -40°C с использованием сухого льда/ацетонитрила. Медленно добавляли бромид метилмагния (3,0 M в тетрагидрофуране, 800 мл). После добавления, реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение ночи. После охлаждения до -40°C с использованием сухого льда/ацетонитрила, целит (200 г), и затем осторожно добавляли этилацетат (500 мл). После добавления смесь перемешивали в течение 4 часов, пока температура медленно нарастала до комнатной температуры. Реакционную смесь снова охлаждали до -40°C и добавляли воду (200 мл), а затем метанол (1,5 л). После перемешивания при комнатной температуре в течение ночи, смесь фильтровали через тонкий слой песка. Сухой остаток переносили обратно в 5 л флакон и перемешивали с метанолом (2 л) в течение 5 часов. Нерастворимый сухой осадок отфильтровывали. Объединенные фильтраты концентрировали до сухого состояния. Добавляли хлорид метилена (3,5 л). Нерастворимый сухой осадок отфильтровывали. Фильтрат высушивали над сульфатом магния. После удаления растворителя получали 436 г (92,6%) продукта в виде белого трудно растворимого сухого осадка. Неочищенный продукт тщательно очищали и использовали непосредственно в следующей стадии. MS (EI) 388,3 (M+1).
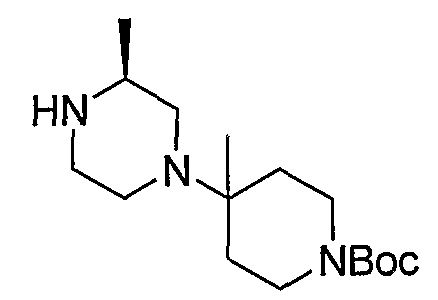
Стадия H
трет-Бутил-4-метил-4-[(3S)-3-метилпиперазин-1-ил]пиперидин-1-карбоксилат
Раствор трет-бутил-4-[(3S)-4-бензил-3-метилпиперазин-1-ил]-4-метилпиперидин-1-карбоксилата (45,5 г, 0,118 моль) в метаноле (320 мл) и уксусная кислота (35 мл, ~5 эквивалентов) в 2,25 л сосуде Парра насыщали H2 до 60 фунт/дюйм2 и смесь встряхивали в течение 18 часов. Реакционную смесь фильтровали через целитную подушку, и подушку промывали метанолом. Фильтрат концентрировали в вакууме. Остаточное масло растворяли в DCM (500 мл) и промывали водным гидроксидом натрия (300 мл). Водную фазу обращенно экстрагировали хлоридом метилена (200 мл). Смешанный органический раствор промывали солевым раствором (500 мл), высушивали над сульфатом натрия и растворитель удаляли в вакууме до получения 35 г (100%) продукта в виде бледно-желтого вязкого масла, которое медленно кристаллизовалось. 1H-ЯМР (400 МГц, CDCl3) δ (м.д.) 3,44 (м, 2H), 3,36 (м, 2Н), 2,97 (дт, 1Н), 2,84 (дд, 1H), 2,78 (м, 1Н), 2,71 (ушир.д, 2Н), 2,16 (дт, 1Н), 1,81 (т, 2H), 1,76 (м, 1Н), 1,45 (с, 9Н), 1,34 (м, 3Н), 1,03 (д, 3Н), 0,90 (с, 3Н); MS (EI) 298,2 (М+1).
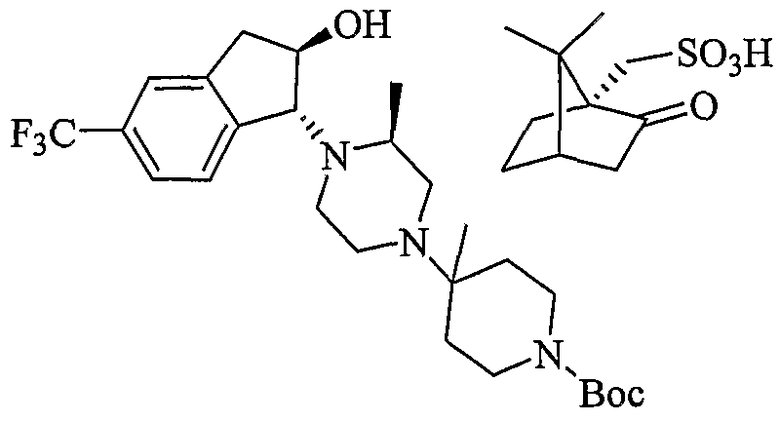
Стадия I
Соль трет-бутил-4-(3S)-4-[((1R,2R)-2-гидрокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил-4-метилпиперидин-1-карбоксилат [(1R)-7,7-Диметил-2-оксобицикло[2.2.1]гепт-1-ил]метансульфоновой кислоты
Смесь (1aS,6aR)-4-(трифторметил)-6,6a-дигидро-1aH-инденo[1,2-b]оксирена (118,4 г, 0,5917 М) и трет-бутил-4-метил-4-[(3S)-3-метилпиперазин-1-ил]пиперидин-1-карбоксилата (160,00 г, 0,53793 моль) в этаноле (100 мл) нагревали до 70°C (температура масляной бани) в течение двух дней. Смесь концентрировали и остаток пропускали через пластинку силикагеля с использованием этилацетата, содержащего 1% триэтиламина. После удаления растворителя получали 267,67 г вспененного сухого остатка. К этому остатку добавляли ацетонитрил (500 мл) и смесь перемешивали в течение 30 минут. К указанной выше смеси сразу добавляли сухую [(1R)-7,7-диметил-2-оксобицикло[2.2.1]гепт-1-ил]метансульфоновую кислоту (124,96 г, 0,53793 моль). Раствор медленно становился прозрачным, а затем выпадал белый осадок. После перемешивания в течение ночи, осадок собирали путем фильтрации, промывали ацетонитрилом и высушивали до получения 232 г продукта. Фильтрат нейтрализовали и концентрировали, кроме того, проводили тот же процесс формирования соли с остатком, для получения дополнительных 74 г продукта. Общий выход составлял 70%. MS (EI) 498,2 (M+1).
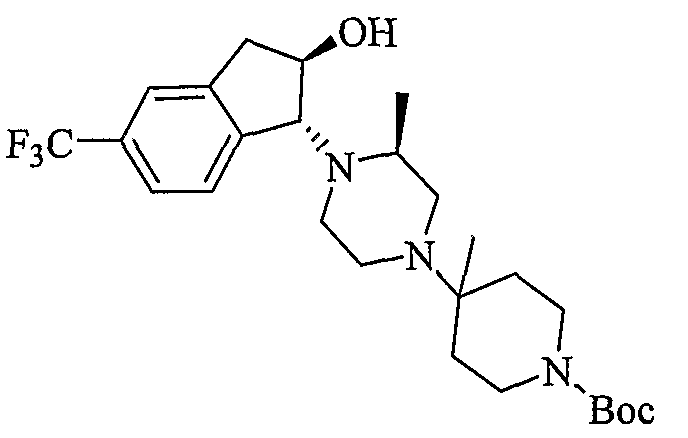
Стадия J
трет-Бутил-4-(3S)-4-((1R,2R)-2-гидрокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил-4-метилпиперидин-1-карбоксилат
Соль трет-бутил-4-(3S)-4-[(1R,2R)-2-гидрокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил-4-метилпиперидин-1-карбоксилат [(1R)-7,7-диметил-2-оксобицикло[2.2.1]гепт-1-ил]метансульфоновой кислоты (512 г, 0,701 моль) растворяли в 1 M водного гидроксида натрия (1 л), и раствор экстрагировали хлоридом метилена (2×2 л). Объединенные органические слои высушивали над сульфатом магния и концентрировали. Остаток затем высушивали в высоком вакууме до получения грязно-белого вспененного осадка (346 г, 99,1%). MS (EI) 498,2 (M+1).
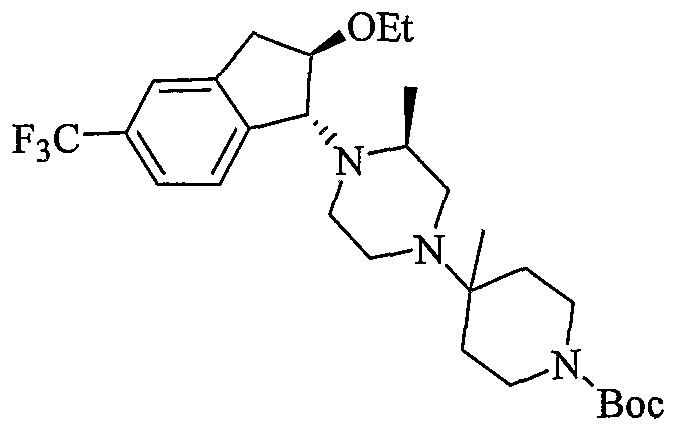
Стадия K
трет-Бутил-4-(3S)-4-[2-Этокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил-4-метилпиперидин-1-карбоксилат
Гидрид натрия (5,225 г, 0,1306 М) смешивали с сухим DMF (150 мл) при 0°C. Раствор трет-бутил-4-(3S)-4-[2-гидрокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил-4-метилпиперидин-1-карбоксилата (50,0 г, 0,1005 М) в DMF (100 мл) добавляли по каплям при 0°C в течение 20 мин. После добавления смесь перемешивали в течение еще 20 мин перед добавлением йодоэтана (12,06 мл, 0,1507 М) одной порцией. После перемешивания в течение 1 ч, содержимое реакции осторожно вливали в 500 мл ледяной воды. Смесь экстрагировали хлоридом метилена (500 мл ×3). Смешанный органический слой промывали солевым раствором и высушивали над сульфатом магния. После удаления растворителя осадок растворяли в хлориде метилена и пропускали через пластинку силикагеля с этилацетат/гексан/триэтиламином 50:50:1 (предварительно пластинку насыщали той же системой растворителей). После удаления растворителя получали 48,4 г (91,6%) продукта в виде вязкого осадка, MS (EI) 526,2 (M+1).
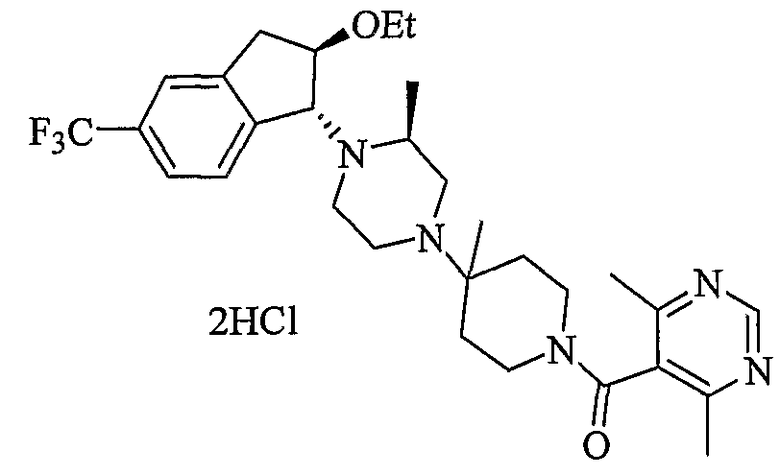
Стадия L
Дигидрохлорид 5-[(4-(3S)-4-[(1R,2R)-2-этокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидин
трет-Бутил-4-(3S)-4-[2-этокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил-4-метилпиперидин-1-карбоксилат (48,4 г, 0,0921 М) обрабатывали 4,0 M раствором хлорида водорода в 1,4-диоксане (230 мл) при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали до сухого состояния, и остаток затем высушивали в высоком вакууме. Полученный гидрохлорид амина смешивали с 4,6-диметилпиримидин-5-карбоновой кислотой (16,8 г, 0,110 моль) в хлориде метилена (100 мл), и затем добавляли 1-гидроксибензотриазол (16,80 г, 0,1243 М), гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида (25,00 г, 0,1304 М) и триэтиламин (65,0 мл, 0,466 моль). Полученную реакционную смесь перемешивали при комнатной температуре в течение ночи, после чего разбавляли хлоридом метилена и промывали водным гидроксидом натрия (1 M) и солевым раствором. Органический слой собирали и высушивали над сульфатом магния. После удаления растворителя осадок растворяли в хлориде метилена (50 мл) и пропускали через пластинку силикагеля с этилацетатом, содержащим 1% триэтиламина. Раствор концентрировали и осадок растворяли в 900 мл ацетата изопропила. К этому раствору медленно добавляли 185 мл 1,0 N HCl в ацетате изопропила. Смесь медленно становилась мутной, и ее перемешивали в течение ночи. Образовавшийся белый осадок собирали, промывали 40 мл ацетата изопропила. Массу высушивали в токе воздуха до получения 47,0 г (80,7%) продукта. MS (EI) 560,3 (M+1).
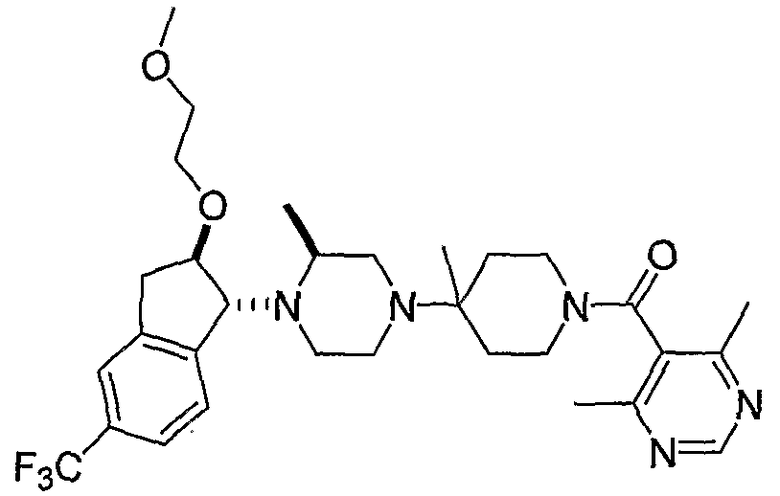
Пример 12
5-[(4-{(3S)-4-[(1R,2R)-2-(2-Метоксиэтокси)-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидин
Это соединение получали в основном так же, как описано в Примере 11, с использованием соответствующих исходных материалов. MS(M+H)+ 590.
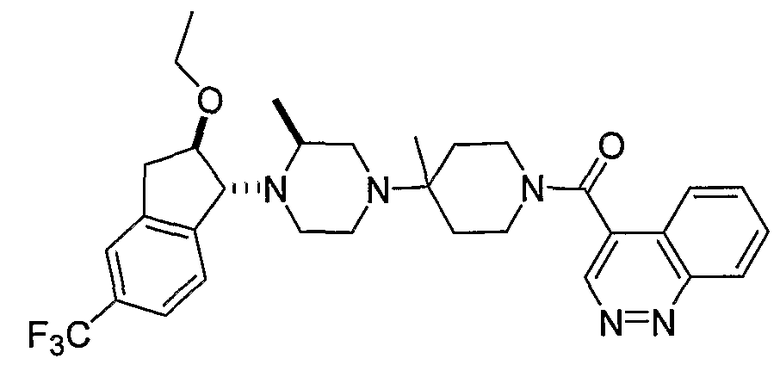
Пример 13
4-[(4-{(3S)-4-[(1S,2R)-2-Этокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]циннолин
Это соединение получали в основном так же, как описано в Примере 11, с использованием соответствующих исходных материалов. MS(M+H)+ 582,4.
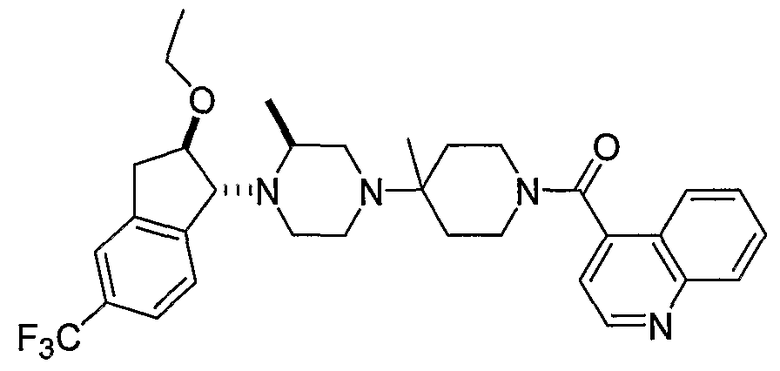
Пример 14
4-[(4-{(3S)-4-[(1R,2R)-2-Этокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]хинолин
Это соединение получали в основном так же, как описано в Примере 11, с использованием соответствующих исходных материалов. MS(M+H)+ 581,4.
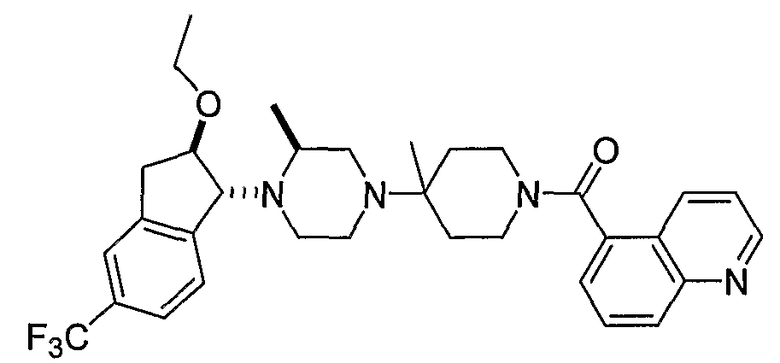
Пример 15
5-[(4-{(3S)-4-[(1R,2R)-2-Этокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]хинолин
Это соединение получали в основном так же, как описано в Примере 11, с использованием соответствующих исходных материалов. MS(M+H)+ 581,4.
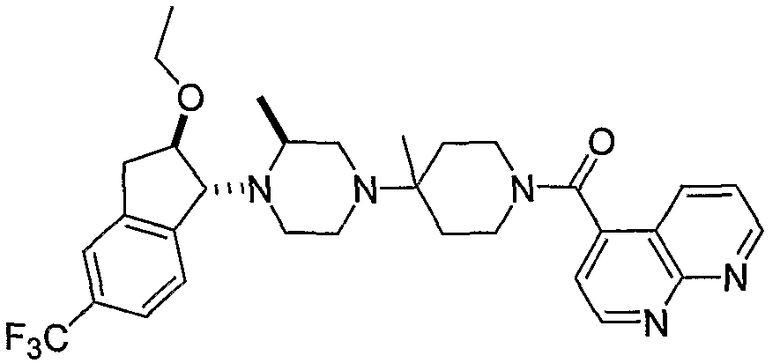
Пример 16
4-[(4-{(3S)-4-[(1R,2R)-2-Этокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-1,8-нафтиридин
Это соединение получали в основном так же, как описано в Примере 11, с использованием соответствующих исходных материалов. MS (M+H)+ 582,4.
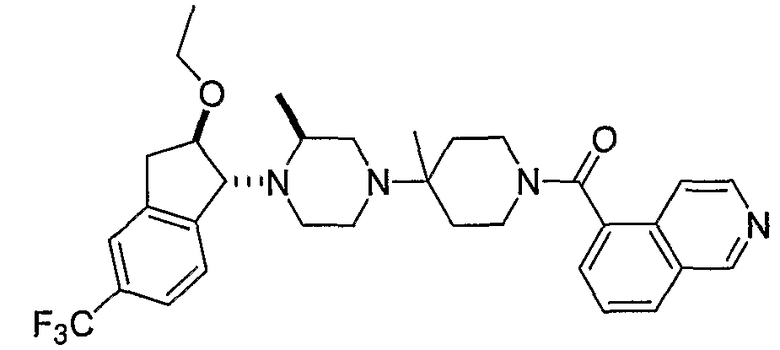
Пример 17
5-[(4-{(3S)-4-[(1R,2R)-2-Этокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]изохинолин
Это соединение получали в основном так же, как описано в Примере 11, с использованием соответствующих исходных материалов. MS (M+H)+ 581,4.
Пример 18
5-[(4-{(3S)-4-[(1R,2R)-5-Бром-2-этокси-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидин
Стадия A
6-Бром-1H-инден
Раствор 5-броминдан-1-ола (5,00 г, 23,5 ммоль) и моногидрата п-толуолсульфоновой кислоты (100 мг, 0,5 ммоль) в бензоле (150,00 мл) кипятили с обратным холодильником в течение 2 часов, постоянно удаляя воду из реакционной смеси ловушкой Дина-Старка. После охлаждения до комнатной температуры, раствор бензола промывали водой, высушивали над обезвоженным Na2SO4 и концентрировали при низком давлении. Очищением осадка тонкослойной хроматографией (гексан) получали чистый 5-броминден (4,0 г, 87%).
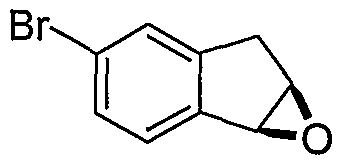
Стадия B
4-Бром-6,6a-дигидро-1aH-инденo[1,2-b]оксирен
К раствору N-оксид 4-(3-фенилпропил)пиридина (68,88 мг, 0,323 ммоль) в хлориде метилена (6,00 мл) добавляли хлорид (S,S)-(+)-N,N'-би(3,5-ди-трет-бутилсалицилиден)-1,2-циклогександиамино-марганца(III) (58,62 мг, 0,0923 мМ) и 2,0 M гипохлорита натрия в воде (4,00 мл) при 0°C. Полученную коричневую суспензию перемешивали при 0°C в течение 15 мин. К охлажденной суспензии добавляли раствор 6-бром-1H-индена (900 мг, 4,6141 ммоль) в хлориде метилена (6,00 мл) при 0°C с одновременным добавлением 2,0 M гипохлорита натрия в воде (4,00 мл) при 0°C. Реакцию проводили при 0°C в течение 1 ч. Реакции позволяли нагреться до комнатной температуры и затем перемешивали при комнатной температуре в течение ночи. Реакционную смесь переливали в солевой раствор и затем экстрагировали хлоридом метилена (4×40 мл). Объединенные экстракты высушивали над обезвоженным Na2SO4, фильтровали и упаривали при низком давлении. Неочищенный продукт использовали непосредственно в следующей реакции. Соотношение двух диастереомеров было 8/1.
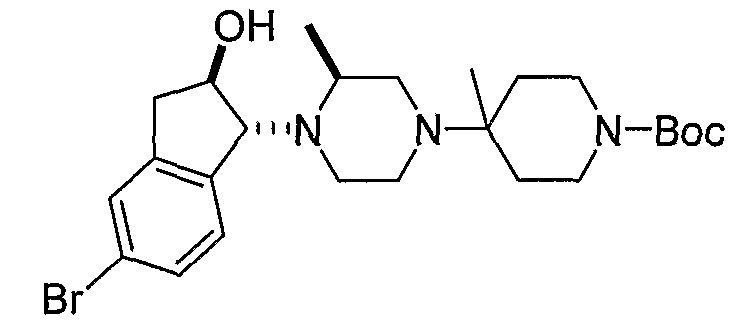
Стадия C
трет-Бутил-4-{(3S)-4-[(1R,2R)-5-бром-2-гидрокси-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-карбоксилат
К раствору 4-бром-6,6a-дигидро-1aH-инденo[1,2-b]оксирена (247,9 мг, 1,1746 мМ) в этаноле (10,00 мл) добавляли трет-бутил-4-метил-4-[(3S)-3-метилпиперазин-1-ил]пиперидин-1-карбоксилат (349,36 мг, 1,1746 ммоль). Реакционную смесь кипятили с обратным холодильником в течение ночи. После охлаждения смесь упаривали при низком давлении. Очищением на силикагеле с 50% ацетатом этила (1% NH4OH) и гексаном получали требуемое соединение в виде продукта с высокой подвижностью (295 мг; 49,4%).
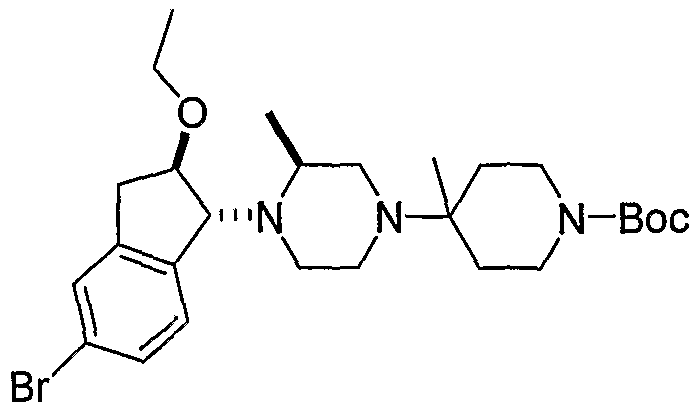
Стадия D
трет-Бутил-4-{(3S)-4-[(1R,2R)-5-Бром-2-этокси-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-карбоксилат
К раствору трет-бутил-4-{(3S)-4-[(1R,2R)-5-бром-2-гидрокси-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-карбоксилата (564 мг, 1,1 ммоль) в тетрагидрофуране (20,00 мл) добавляли гидрид натрия (448 мг, 17,75 ммоль) при комнатной температуре. После перемешивания в течение 10 мин добавляли йодоэтан (1,42 мл, 17,75 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи и гасили насыщенным водным раствором хлорида аммония (20 мл). Органический слой отделяли и экстрагировали водную фазу ацетатом этила (3×30 мл). Объединенные экстракты промывали солевым раствором, высушивали над обезвоженным Na2SO4, фильтровали и упаривали при низком давлении до получения неочищенного продукта (488 мг, 82%). LC-MS[M+1]=536,3, 538,8.
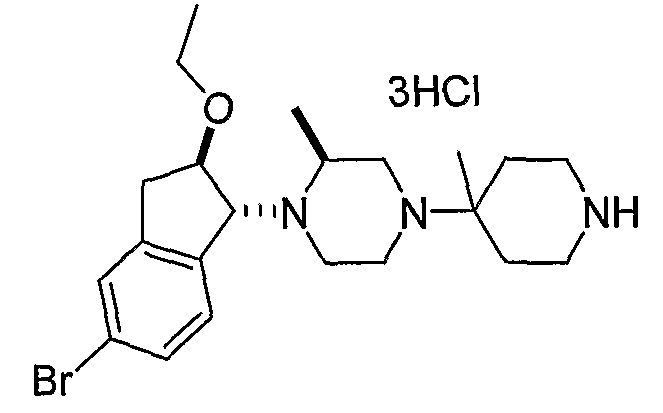
Стадия E
Тригидрохлорид (2S)-1-[(1R,2R)-5-бром-2-этокси-2,3-дигидро-1H-инден-1-ил]-2-метил-4-(4-метилпиперидин-4-ил)пиперазин
К раствору трет-бутил-4-{(3S)-4-[(1R,2R)-5-бром-2-этокси-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-карбоксилата (50 мг, 0,093 ммоль) в тетрагидрофуране (3 мл) добавляли 4,00 M раствор хлорида водорода в 1,4-диоксане (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Упариванием при низком давлении получали требуемый, лишенный Boc, продукт.
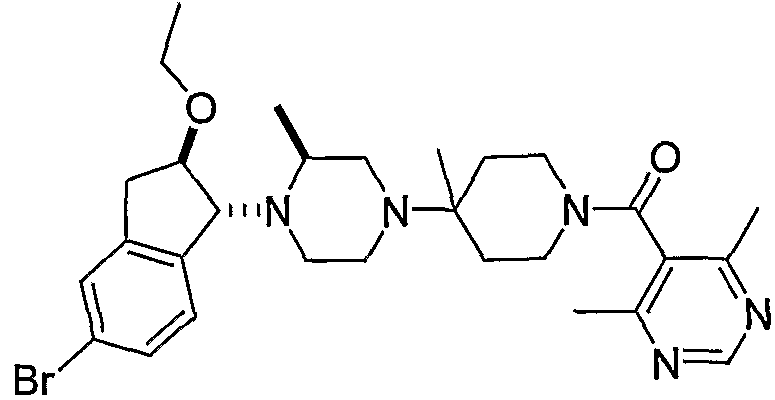
Стадия F
5-[(4-{(3S)-4-[(1S,2R)-5-Бром-2-этокси-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил)-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидин
К кашицеобразному тригидрохлориду (2S)-1-[(1R,2R)-5-бром-2-этокси-2,3-дигидро-1H-инден-1-ил]-2-метил-4-(4-метилпиперидин-4-ил)пиперазина (50 мг, 0,0916 ммоль) в хлориде метилена (6 мл) добавляли 4,6-диметилпиримидин-5-карбоновую кислоту (27,88 мг, 0,183 ммоль), гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида (21,07 мг, 0,11 ммоль), 1-гидроксибензотриазол (13,62 мг, 0,101 ммоль) и триэтиламин (0,0894 мл, 0,641 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. С помощью прямой хроматографии на силикагеле с 10% MeOH/EtOAc (1% NH4OH) получали требуемый продукт (42 мг, 80%). LC-MS[M+1]=570,10; 572,05.
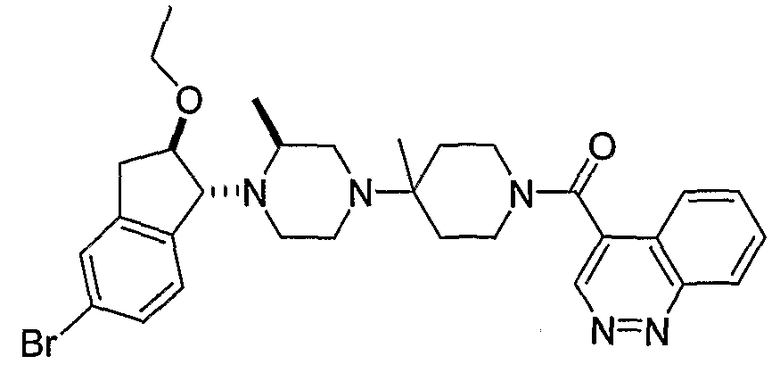
Пример 19
4-[(4-{(3S)-4-[(1R,2R)-5-бром-2-этокси-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил)-4-метилпиперидин-1-ил)карбонил]циннолин
Это соединение получали в основном так же, как описано в Примере 18, с использованием соответствующих исходных материалов. MS(M+H)+ 592,3, 594,3.
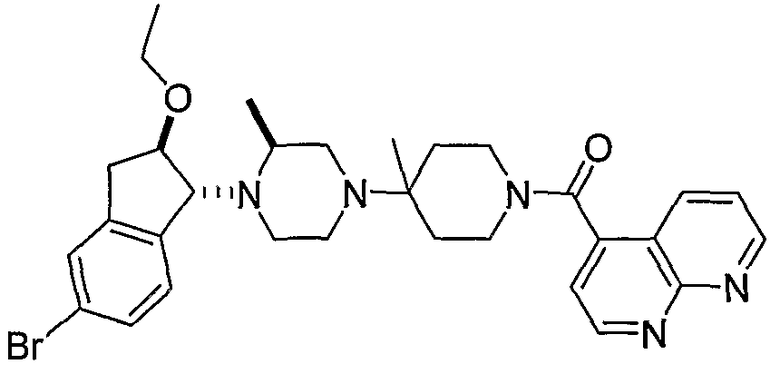
Пример 20
4-[(4-{(3S)-4-[(1R,2R)-5-Бром-2-этокси-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-1,8-нафтиридин
Это соединение получали в основном так же, как описано в Примере 18, с использованием соответствующих исходных материалов. MS(M+H)+ 592,3, 594,3.
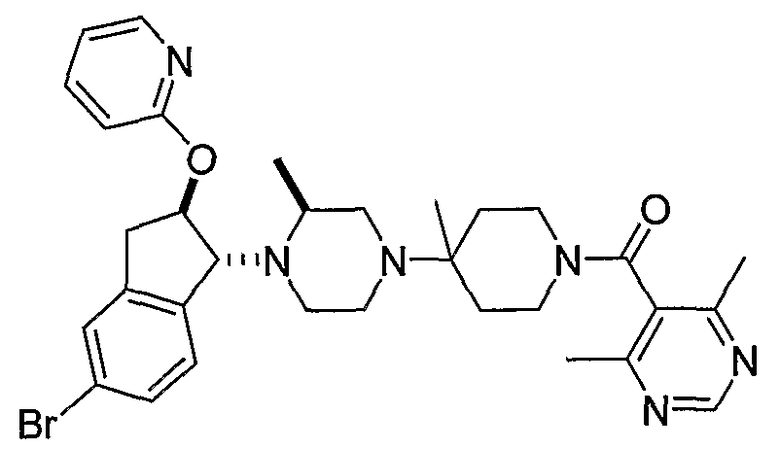
Пример 21
5-[(4-{(3S)-4-[(1R,2R)-5-Бром-2-(пиридин-2-илокси)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидин
Это соединение получали в основном так же, как описано в Примере 18, с использованием соответствующих исходных материалов. MS:(M+H)+ 619,3, 621,3.
Пример 22
5-[(4-{(3S)-4-[(1R,2R)-2-Этокси-5-(1,3-тиазол-2-ил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидин
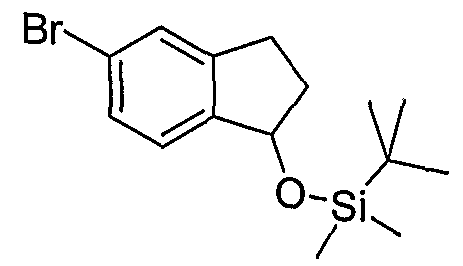
Стадия A
[(5-Бром-2,3-дигидро-1H-инден-1-ил)окси](трет-бутил)диметилсилан
К раствору 5-броминдан-1-ола (4,00 г, 18,8 ммоль) в N,N-диметилформамиде (25,00 мл) добавляли триэтиламин (5,23 мл, 37,5 ммоль), хлорид трет-бутилдиметилсилила (4,244 г, 28,16 ммоль) и 4-диметиламинопиридин (115 мг, 0,939 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Смесь разводили эфиром (100 мл) и гасили водой. Водную фазу экстрагировали эфиром (4×40 мл). Объединенные экстракты промывали солевым раствором, высушивали над обезвоженным Na2SO4, фильтровали и упаривали при низком давлении. С помощью хроматографии на силикагеле с 5% EtOAc/гексан получали требуемый продукт (6,05 г, 98%).
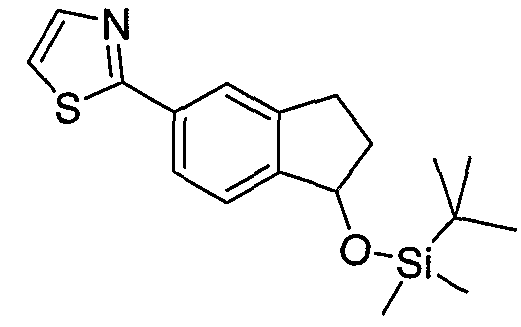
Стадия B
2-(1-{[трет-Бутил(диметил)силил]окси}-2,3-дигидро-1H-инден-5-ил)-1,3-тиазол
К суспензии цинка (899 мг, 13,75 ммоль) в тетрагидрофуране (1,60 мл) после перемешивания добавляли 1,2-дибромэтан (0,118 мл, 1,37 ммоль). Суспензию нагревали в струе горячего воздуха до прекращения испарения этилена. Добавляли хлортриметилсилан (0,0698 мл, 0,55 ммоль) и раствор 2-бромтиазола (0,413 мл, 4,58 ммоль) в тетрагидрофуране. Через 15 мин добавляли [(5-бром-2,3-дигидро-1H-инден-1-ил)окси](трет-бутил)диметилсилан (1,0 г, 3,055 ммоль) и тетракис(трифенилфосфин)палладий(0) (70,6 мг, 0,0611 ммоль), разведенный в тетрагидрофуране (8,00 мл). Смесь перемешивали в течение 24 ч при кипячении с обратным холодильником и гасили 15 мл солевого раствора. Органический слой отделяли и экстрагировали водную фазу хлоридом метилена (25 мл ×3). Смешанные экстракты промывали солевым раствором, высушивали над обезвоженным Na2SO4 и упаривали при низком давлении. С помощью хроматографии на силикагеле с 2,5% EtOAc/гексан получали требуемый парный продукт (800 мг, 79%).
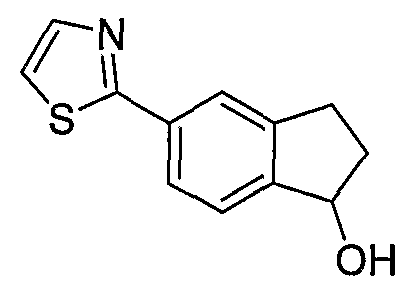
Стадия C
5-(1,3-Тиазол-2-ил)индан-1-ол
К раствору 2-(1-{[трет-бутил(диметил)силил]окси}-2,3-дигидро-1H-инден-5-ил)-1,3-тиазола (630 мг, 1,9 ммоль) в тетрагидрофуране (10,00 мл) добавляли 1,00 M раствор фторида тетрабутиламмония, тригидрата в тетрагидрофуране (1,90 мл) при 0°C. Ледяную баню удаляли и реакционную среду перемешивали при комнатной температуре в течение 1 ч. Смесь разводили эфиром, промывали насыщенным водным раствором NaCl, высушивали над обезвоженным MgSO4, фильтровали и упаривали при низком давлении до получения требуемого продукта (410 мг, 99%).
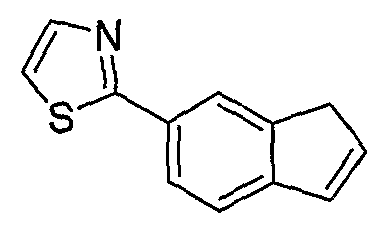
Стадия D
2-(1H-Инден-6-ил)-1,3-тиазол
К раствору 5-(1,3-тиазол-2-ил)индан-1-ола (900,00 мг, 0,0041420 моль) в тетрагидрофуране (20,00 мл) добавляли 1,0 M раствор хлорида водорода в воде (20,00 мл). Реакционную смесь кипятили с обратным холодильником в течение ночи. После охлаждения до комнатной температуры реакцию гасили 30 мл водного раствора 1 N NaOH. Органический слой отделяли и водную фазу экстрагировали EtOAc (3×30 мл). Объединенные экстракты промывали солевым раствором, высушивали над обезвоженным MgSO4, фильтровали и упаривали при низком давлении до получения требуемого продукта (381 мг, 46%). LC-MS [M+1]=200,2.
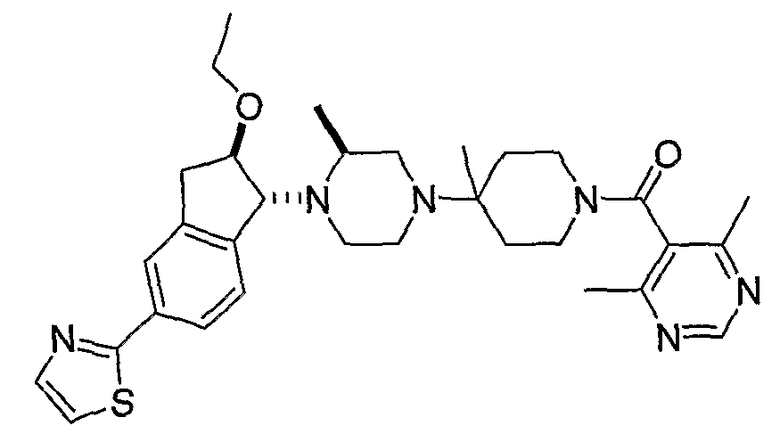
Стадия E
5-[(4-{(3S)-4-((1R,2R)-2-Этокси-5-(1,3-тиазол-2-ил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидин
Начиная с середины стадии D, указанное в заголовке соединение получали с использованием способов, аналогичных описанным в Примере 18. MS (M+H) 575,2.
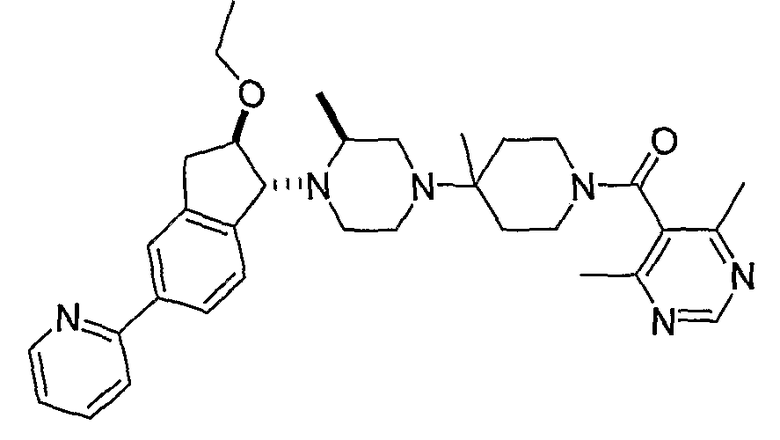
Пример 23
5-[(4-{(3S)-4-[(1R,2R)-2-Этокси-5-пиридин-2-ил-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидин
Указанное в заголовке соединение получали способом, аналогичным указаному в Примере 22. MS (M+H) 569,3.
Пример 24
5-[(4-{(3S)-4-[3-Метокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидин
Стадия A
5-(Трифторметил)-3-винил-1,3-дигидро-2-бензофуран-1-ол
К раствору N,N,N'-триметил-1,2-этандиамина (4,76 мл, 37,4 ммоль) в тетрагидрофуране (150,00 мл) добавляли 1,6 M раствор н-бутиллития в гексане (25,7 мл) при -40°C. Бесцветный раствор приобретал светло-желтый цвет. Затем холодную баню удаляли и реакционную смесь перемешивали в течение 30 минут, пока она не нагревалась до температуры в -15°C. Реакцию повторно охлаждали до -40°C и добавляли 4-трифторметилбензальдегид (5,00 мл, 37,4 ммоль). Реакцию перемешивали при -50°C в течение 35 минут, после чего повторно добавляли 1,60 M раствор н-бутиллития в гексане. Реакционную смесь оставляли до повышения температуры до -25°C и выдерживали при -25°C в течение 2 часов, добавляя в это время акролеин (2,75 мл, 41,2 ммоль). Реакционную смесь перемешивали в течение ночи и гасили добавлением 30 мл водного раствора 6 N HCl. Органический слой отделяли и экстрагировали водную фазу EtOAc дважды (2×30 мл). Объединенные экстракты промывали солевым раствором, высушивали над Na2SO4, упаривали в вакууме и очищали тонкослойной хроматографией до получения требуемого продукта (2,4 г, 28% выход). LC-MS[M+1]=231,2.
Стадия B
1-[5-(Трифторметил)-2-винилфенил]проп-2-ен-1-ол
Бромид трифенилметилфосфония (1,71 г, 4,78 ммоль) растворяли в эфире (20,00 мл). После охлаждения до 0°C, 1,60 M раствора н-бутиллития в гексане (2,72 мл) быстро добавляли через шприц и полученную оранжевую смесь нагревали до комнатной температуры и перемешивали в течение ночи. Перемешивание останавливали, чтобы позволить осадку осесть, и затем результат переносили канюлей в раствор 5-(трифторметил)-3-винил-1,3-дигидро-2-бензофуран-1-ола (1,00 г, 4,34 ммоль) в эфире (10,00 мл) при перемешивании при 0°C. После добавления, ледяную баню удаляли и смесь кипятили с обратным холодильником в течение ночи. Реакции позволяли остыть, затем отфильтровывали осадок и промывали небольшим количеством эфира. Большую часть растворителей упаривали, неочищенный продукт помещали на силикагель и элюировали смесью гексан/EtOAc (10:1) до получения требуемого продукта (0,81 г, 82%), LC-MS [M+1]=229,2.
Стадия C
6-(Трифторметил)-1H-инден-1-ол
К раствору 1-[5-(трифторметил)-2-винилфенил]проп-2-ен-1-ола (462 мг, 2,02 ммоль) в хлориде метилена (20 мл) в азоте добавляли бензилиден-би(трициклогексилфосфин)дихлоррутений (70 мг, 0,08 ммоль). Темную смесь перемешивали при 25°C в течение 30 мин и концентрировали. Остаток очищали тонкослойной хроматографией до получения требуемого продукта (278,1 мг, 68%). LC-MS [M+1]=279,2.
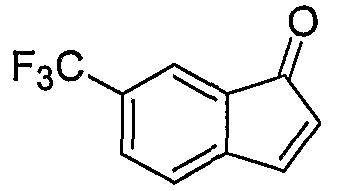
Стадия D
6-(Трифторметил)-1H-инден-1-он
К раствору хлорхромата пиридиния (1,08 г, 5,0 ммоль) в хлориде метилена (15 мл) добавляли по каплям раствор 6-(трифторметил)-1H-инден-1-ола (500 мг, 2,498 ммоль) в хлориде метилена (10 мл). После перемешивания в течение 14 ч, добавляли эфир (30 мл), и реакционную смесь фильтровали через силикагель. Фильтрат концентрировали в вакууме. Неочищенный материал хроматографировали (10:1 гексан/EtOAc) до получения 200 мг (40%) продукта. LC-MS [M+1]=199,2.
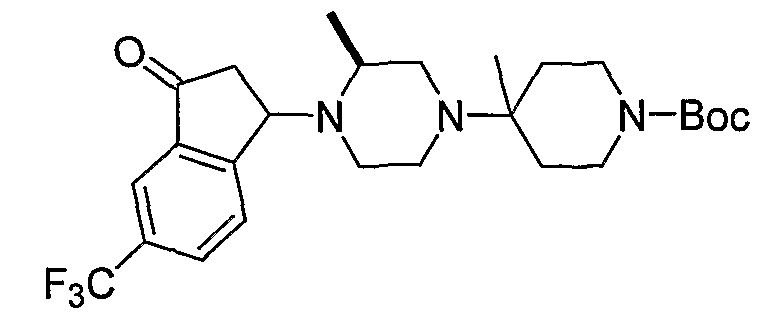
Стадия E
трет-Бутил-4-метил-4-{3-метил-4-[3-оксо-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]пиперазин-1-ил}пиперидин-1-карбоксилат
Раствор 6-(трифторметил)-1H-инден-1-она (100 мг, 0,505 ммоль) и трет-бутил-4-метил-4-(3-метилпиперазин-1-ил)пиперидин-1-карбоксилата (420 мг, 1,412 мМ) в тетрахлориде углерода (8,00 мл) нагревали до 60°C в течение 18 ч при перемешивании. После испарения растворителя остаток очищали на силикагеле до получения двух диастереомеров (соотношение 3/1). Получали 180 мг (72%). LC-MS [M+1]=496,4.
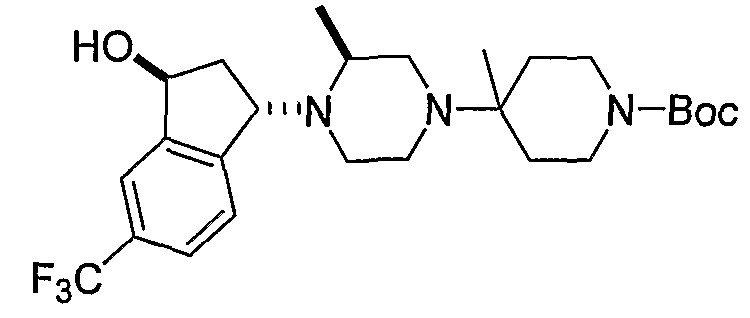
Стадия F
трет-Бутил-4-{(3S)-4-[3-гидрокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-карбоксилат
К раствору трет-бутил-4-метил-4-{3-метил-4-[3-оксо-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]пиперазин-1-ил}пиперидин-1-карбоксилата (100 мг, 0,202 ммоль) в этаноле (7 мл) добавляли борогидрид натрия (57 мг, 1,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Растворитель испаряли в вакууме, и остаток гасили 10 мл 1 N водного раствора NaOH и 10 мл EtOAc. Органическую фазу отделяли и водный слой экстрагировали EtOAc дважды (2×15 мл). Объединенные экстракты промывали солевым раствором, высушивали над Na2SO4 и упаривали при низком давлении до получения требуемого продукта, который непосредственно использовали в следующей стадии (93 мг, 92%). LC-MS [M+1]=498,4.
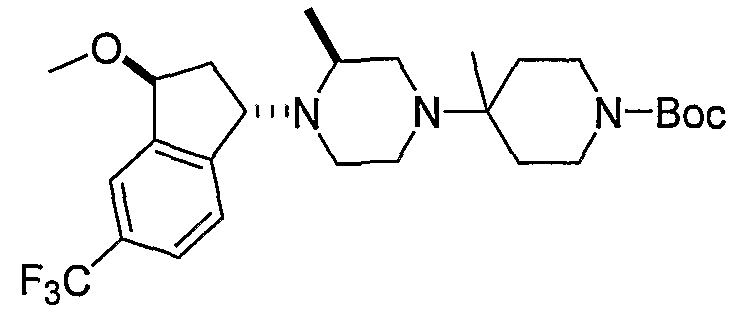
Стадия G
трет-Бутил-4-{(3S)-4-[3-метокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-карбоксилат
К суспензии гидрида натрия (150 мг, 3,75 ммоль) в тетрагидрофуране (15 мл) добавляли раствор трет-бутил-4-{4-[3-гидрокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-карбоксилата (92 мг, 0,185 ммоль) в тетрагидрофуране (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, после чего добавляли йодид метила (0,50 мл, 8,05 ммоль). Реакцию постоянно перемешивали при комнатной температуре в течение ночи и гасили добавлением 10 мл воды и 10 мл EtOAc. Органическую фазу отделяли и водный слой экстрагировали EtOAc дважды (2×15 мл). Объединенные экстракты промывали солевым раствором, высушивали над Na2SO4 и упаривали при низком давлении до получения неочищенного продукта (81 мг, 85%), который непосредственно использовали в следующей стадии. LC-MS[M+1]=511,3.
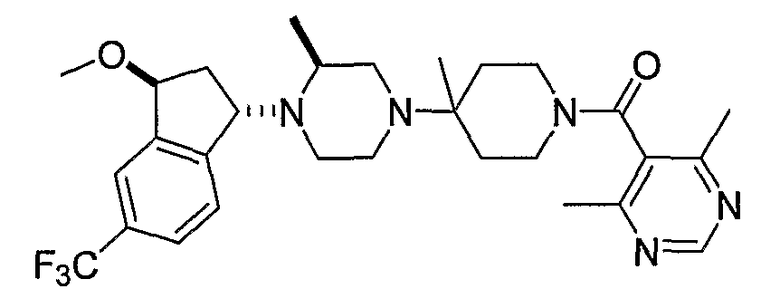
Стадия H
5-[(4-{(3S)-4-[3-Метокси-5-(трифторoметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидин
трет-Бутил-4-{4-[3-метокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-карбоксилат (85 мг, 0,166 ммоль) растворяли в 4,00 M растворе хлорида водорода в 1,4-диоксане (2 мл). Смесь перемешивали в течение 1 ч, концентрировали до сухого состояния и упаривали в вакууме.
К кашицеобразной 4,6-диметилпиримидин-5-карбоновой кислоте (50,6 мг, 0,333 мМ) в ацетонитриле (4 мл) при 0°C добавляли каплю DMF (используемого в качестве катализатора) и затем добавляли хлорид оксалила (0,028 мл, 0,333 ммоль). Полученную массу перемешивали при комнатной температуре в течение 2 ч. К реакционной смеси добавляли раствор указанного выше гидрохлорида амина в ацетонитриле (4 мл) в присутствии триэтиламина (0,139 мл, 0,998 ммоль) при 0°C. Полученную массу выдерживали при 45-50°C в течение 6 ч и при 80°C в течение 3 ч. С помощью прямой хроматографии на силикагеле получали требуемый продукт (71 мг, 78%). LC-MS [M+1]=546,3.
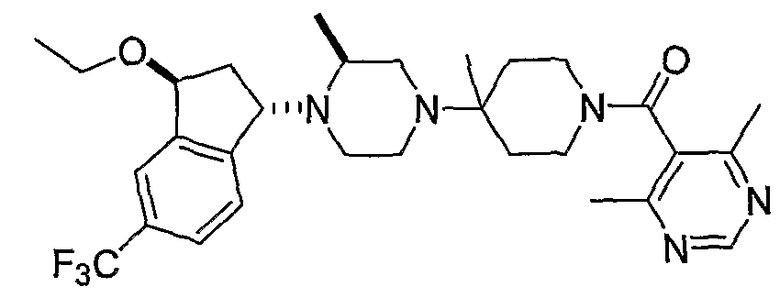
Пример 24
5-[(4-{(3S)-4-[3-Этокси-5-(трифторметил)-2,3-дигидро-1H-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидин
Указанное в заголовке соединение получали способом, аналогичным способу в Примере 23. MS (M+H) 560,2.
Пример A
Экспрессия CCR5
Лейкофорез (Biological Specialty, Colmar, PA) получали от нормальных, не получавших лечения доноров и изолировали мононуклеарные клетки периферической крови (PBMCs) с помощью центрифугирования в градиенте плотности. Затем моноциты изолировали центрифужным промыванием. После промывания моноциты ресуспендировали из расчета 106 клеток/мл в RPMI (Invitrogen, Carlsbad, CA) с добавлением 10% FBS (Hyclone, Logan, UT) и 10-20 нг/мл рекомбинантного человеческого IL-10 (R&D systems, Minneapolis, MN), и инкубировали в этой среде при 37°C с 5% CO2 в течение 24-48 часов. Экспрессию CCR5 в моноцитах, обработанных IL-10, затем верифицировали путем окрашивания клеток PE-конъюгированными антителами против человеческого CCR5 (PharMingen, San Diego, CA), с последующим FACS анализом с помощью FACSCalibur (BD Biosciences, Bedford, MA).
Пример B
Анализ связывания CCR5
В 96-луночном фильтрационном планшете MultiScreenТМ (Millipore Systems, Billerica, MA), 3×105 моноцитов, обработанных IL-10 в 150 мкл RPMI (Invitrogen, Carlsbad, CA) с 20 мМ HEPES (Invitrogen, Carlsbad, CA) и 0,3% BSA (Sigma, St Louis, MO), инкубировали при комнатной температуре в течение 1 часа с 0,2 нМ 125I-MIP-1β (Perkin Elmer, Boston, MA) и сериями концентраций соединения по изобретению. Неспецифическое связывание определяли путем инкубирования клеток с 0,3 мкМ MIP-1β (R&D Systems, Minneapolis, MN). Реакцию связывания прекращали сбором клеток на фильтре плашки в вакуумной установке (Millipore Systems, Billerica, MA). Затем фильтр промывали 5 раз RPMI (Invitrogen, Carlsbad, CA), с добавлением 20 мМ HEPES (Invitrogen, Carlsbad, CA), 0,3% BSA (Sigma, St Louis, MO) и 0,4 M NaCl, в вакуумной установке, высушивали в токе воздуха и счищали с планшета. Фильтрационные чашки, соотносящиеся с лунками на фильтрационном планшете, обрабатывали с использованием Millipore Punch System (Millipore Systems, Billerica, MA). Количество связавшейся радиоактивности на каждой фильтрационной чашке определяли подсчетом на счетчике гамма-излучения. Специфическое связывание определяли как общее связывание минус неспецифическое связывание. Данные по связыванию оценивали с помощью Prism (GraphPad Software, San Diego, CA). Было обнаружено, что соединения по изобретению обладают связывающими свойствами около 1 мкМ или менее, в соответствии с данным исследованием.
Пример C
Исследование проникновения ВИЧ-1
Репортерные вирионы ВИЧ-1 с нарушенной репликацией получали котрансфицированием плазмиды, кодирующей штамм NL4-3 ВИЧ-1 (который модифицировали мутацией оболочечного гена и внедрением люциферазной репортерной плазмиды), вместе с плазмидой, кодирующей один из нескольких оболочечных генов ВИЧ-1, как описано, например, Connor et al, Virology, 206 (1995), 935-944. После трансфицирования двух плазмид преципитацией фосфатом кальция, вирусные супернатанты собирали на 3 день и определяли функциональный вирусный титр. Этот материал использовали затем для инфицирования клеток U87, стабильно экспрессирующих CD4 и хемокиновые рецепторы CCR5, которые преинкубировали с или без тестируемого соединения. Инфекционные агенты выдерживали в течение 2 часов при 37°C, клетки отмывали и среду заменяли на свежую среду, содержащую соединение. Клетки инкубировали в течение 3 суток, лизировали и определяли активность люциферазы. Результаты представляли как концентрацию соединения, необходимую для ингибирования 50% люциферазной активности в контрольных культурах.
Пример D
Исследование репликации ВИЧ-1 на MT-4 клетках
Исследования ингибирования репликации ВИЧ-1 NL4.3 (или IIIB) могут проводится, как описано ранее (Bridger, et al., J. Med. Chem. 42:3971-3981 (1999); De Clercq, et al., Proc. Natl. Acad. Sci. 89:5286-5290 (1992); De Clercq, et al., Antimicrob. Agents Chemother. 38:668-674 (1994); Bridger, et al., J. Med. Chem. 38:366-378 (1995)). В целом, определение анти-ВИЧ активности и цитотоксичности проводили параллельно и на основе жизнеспособности клеток MT-4, которые инфицировали ВИЧ в присутствии различных концентраций тестируемого соединения. После пролиферации клеток MT-4 в течение 5 суток, количество жизнеспособных клеток подсчитывали с помощью основанного на тетразоле колориметрического способа с применением бромида 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразола (MTT) в 96-луночных микрокюветах. Результаты оценивали по достижении значений EC50, которые представляют концентрации, необходимые для защиты 50% инфицированных вирусом клеток от вирусной цитопатогенности.
Пример E
Исследование ингибирования/связывания хемокиновых рецепторов
Способность соединения по изобретению быть антагонистом функции хемокиновых рецепторов (например, CCR2) может определяться с использованием приемлемых способов скрининга (например, высокопроизводительных анализов). Например, агент можно тестировать с помощью анализа внеклеточного окисления, анализа кальциевого потока, анализа связывания лиганда или анализа хемотаксиса (см., например, Hesselgesser et al., J. Biol. Chem. 273(25):15687-15692 (1998); WO 00/05265 и WO 98/02151, каждый из которых включен здесь в качестве ссылки).
Примером такого анализа может быть использование хемокинового рецептора, изолированного или полученного рекомбинантно, который имеет, по меньшей мере, одно свойство, активность или функциональную характеристику хемокинового рецептора млекопитающих. Специфичным свойством может быть способность к связыванию (например, с лигандом или ингибитором), сигнальная активность (например, активация белка G млекопитающих, индукция быстрого и преходящего увеличения концентрации свободного цитозольного кальция [Ca++]), функция клеточного ответа (например, стимуляция хемотаксиса или выделение медиаторов воспаления лейкоцитами) и тому подобное.
В одном варианте осуществления, смесь, содержащая хемокиновые рецепторы или их разновидности, выдерживали в условиях, способствующих связыванию. Рецепторы контактировали с тестируемым соединением, и связывание определяли и измеряли.
В другом варианте осуществления, исследование основано на анализе клеток, где используемые клетки стабильно или временно трансфицированы вектором или экспрессирующей кассетой, содержащей последовательность нуклеиновых кислот, кодирующую рецептор. Клетки выращивали в условиях, способствующих экспрессии рецептора, и обеспечивали контакт с агентом в условиях, способствующих связыванию. Связывание выявляли с использованием стандартных способов. Например, степень связывания может быть определена по отношению к соответствующему контролю. Таким образом, клеточная фракция, такая как мембранная фракция, содержащая рецептор, может быть использована в цельных клетках или их фрагментах.
Определение связывания или образования комплексов между соединениями по изобретению и хемокиновыми рецепторами может проводиться напрямую или косвенно. Например, соединение может быть помечено приемлемой меткой (например, флюоресцентной меткой, меткой, изотопной меткой, ферментной меткой и подобными метками) и связывание может быть определено путем выявления метки. Специфичное и/или конкурентное связывание может быть определено конкурентными или замещающими исследованиями с использованием не меченных агентов или лигандов как конкурентов.
Активность тестируемых агентов в качестве антагонистов оценивали как концентрацию ингибитора, необходимую для 50% ингибирования (величина IC50) специфичного связывания в используемых анализах рецепторного связывания, например, 125I-меченного MCP-1, как лиганда, и мононуклеарных клеток периферической крови (PBMCs), полученных из нормальной цельной человеческой крови с помощью центрифугирования в градиенте плотности. Специфичное связывание обычно определяют как общее связывание (например, общее количество импульсов в минуту на фильтрах) минус неспецифичное связывание. Неспецифичное связывание определяют как количество импульсов в минуту, которое продолжает определяться в присутствии избытка не меченного конкурента (например, MCP-1).
PBMCs человека, описанные выше, использовали в соответствующих исследованиях связывания. Например, от 200000 до 500000 клеток инкубировали с 125I-меченными MCP-l, от 0,1 до 0,2 нМ, с или без не меченного конкурента (10 нМ MCP-1), или с разными концентрациями тестируемого соединения. 125I-меченные MCP-1 могут быть получены приемлемыми способами или коммерчески приобретены (Perkin Elmer, Boston MA). Реакции связывания проводили в связывающем буфере, от 50 до 250 мкл, состоящем из 1M HEPES pH 7,2, и 0,1% BSA (альбумин бычьей сыворотки), в течение 30 минут при комнатной температуре. Реакции связывания прекращали удалением мембран быстрой фильтрацией через фильтры из стекловолокна (Perkin Elmer), которые предварительно обрабатывали 0,3% полиэтиленимином или солевым фосфатным буфером (PBS). Фильтры промывали примерно 600 мкл связывающего буфера, содержащего 0,5 M NaCl или PBS, затем высушивали, и количество связавшейся радиоактивности определяли подсчетом в гамма-камере (Perkin Elmer).
Способность соединений по изобретению быть антагонистами функции хемокиновых рецепторов может быть также определена исследованием хемотаксиса лейкоцитов, с применением соответствующих клеток. Приемлемые клетки включают, например, линии клеток, рекомбинантные клетки или изолированные клетки, которые экспрессируют хемокиновые рецепторы (например, CCR2) и подвержены хемотаксису, индуцированному лигандами хемокиновых рецепторов (например, MCP-1). В исследовании применяли мононуклеарные клетки периферической крови человека, в модификации Boyden Chamber (Neuro Probe). 500000 клеток в среде DMEM, свободной от сыворотки (In Vitrogen), инкубировали с или без ингибиторов и нагревали до 37°C. Камеру хемотаксиса (Neuro Probe) также предварительно нагревали. 400 мкл нагретых 10 нМ MCP-1 добавляли в нижнюю камеру всех лунок с ожидаемым отрицательным контролем, в который добавляли DMEM. Восьмимикронный мембранный фильтр (Neuro Probe) помещали сверху и закрывали крышку камеры. Затем клетки добавляли в отверстия крышки камеры, которые были связаны с лунками камеры под мембраной фильтра. Камеру инкубировали при 37°C, 5% CO2 в течение 30 минут. Затем клетки аспирировали, открывали крышку камеры и фильтр аккуратно удаляли. Верхнюю часть фильтра промывали 3 раза PBS, а нижнюю часть оставляли нетронутой. Фильтр высушивали в токе воздуха и окрашивали краской по Райту-Гимзе (Sigma). Фильтры изучали с помощью микроскопии. Лунки с отрицательным контролем служили фоном и их вычитали из всех значений. Антагонистическую активность определяли путем сравнения числа клеток, мигрировавших в нижнюю камеру в лунках, содержавших антагонист, с числом клеток, мигрировавших в нижнюю камеру в контрольных лунках с MCP-1.
Соединения по настоящему изобретению признавали активными, если они имели значения IC50 в пределах от, примерно, 0,01 до, примерно, 500 нМ в анализе связывания, приведенном выше. В анализе хемотаксиса активные соединения имели значения IC50 в пределах от, примерно, 1 до, примерно, 3000 нМ.
Различные модификации изобретения, в дополнение к описанным здесь, могут проводиться специалистами в этой области науки, исходя из вышеприведенного описания. Все такие модификации также должны быть включены в общую формулу изобретения. Каждая ссылка, упомянутая в настоящей заявке, в том числе все патенты, публикации и книги, включена сюда полностью в качестве ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| СП0СОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ ДИГИДРОИНДЕНАМИДА, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИИ, СОДЕРЖАЩИЕ ДАННЫЕ СОЕДИНЕНИЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗЫ | 2009 |
|
RU2528408C2 |
| МОДУЛЯТОРЫ CCR2 | 2016 |
|
RU2726206C2 |
| РАСТВОРИМЫЕ С5аR АНТАГОНИСТЫ | 2017 |
|
RU2748260C2 |
| ПРОИЗВОДНЫЕ 3-АМИНОПИРРОЛИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРОВ ХЕМОКИНОВ | 2003 |
|
RU2355679C2 |
| ПРОИЗВОДНЫЕ 2,3-ДИГИДРО-1Н-ИНДЕН-1-ИЛ-2,7-ДИАЗАСПИРО[3.5]НОНАНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ ИЛИ ОБРАТНЫХ АГОНИСТОВ ГРЕЛИНОВОГО РЕЦЕПТОРА | 2011 |
|
RU2524341C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ДИГИДРОПИРИМИДИНКАРБОКСАМИДА В КАЧЕСТВЕ ИНГИБИТОРОВ RHO-КИНАЗЫ | 2017 |
|
RU2778478C2 |
| БЕНЗОКСАЗИНИЛ-АМИДОЦИКЛОПЕНТИЛ-ГЕТЕРОЦИКЛИЧЕСКИЕ МОДУЛЯТОРЫ ХЕМОКИНОВЫХ РЕЦЕПТОРОВ | 2004 |
|
RU2301802C2 |
| ПРОИЗВОДНЫЕ АЗАСПИРОАЛКАНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕАЗ | 2004 |
|
RU2379303C2 |
| МОДУЛЯТОРЫ ЯДЕРНЫХ РЕЦЕПТОРОВ | 2016 |
|
RU2715897C2 |
| Бициклические гетероциклические соединения и их применения в терапии | 2012 |
|
RU2662827C2 |
Изобретение относится к соединениям формулы I, обладающим активностью в отношении хемокинового рецептора, которые могут применяться для лечения иммунных и вирусных заболеваний
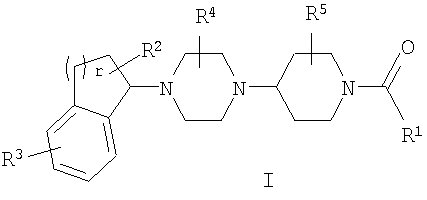
где R1 представляет собой гетероарил; R2 представляет собой Н, гетероарил, OR9 или SR9; R3 представляет собой F, Cl, Br, I, C1-C4 галогеналкил или гетероарил; R4 представляет собой C1-С6 алкил; R5 представляет собой C1-С6 алкил; R9 представляет собой Н, C1-C6 алкил, алкоксиалкил или гетероарил; r представляет собой 1 или 2. Технический результат - получение новых биологически активных соединений. 9 н. и 23 з.п. ф-лы.
1. Соединение формулы I:
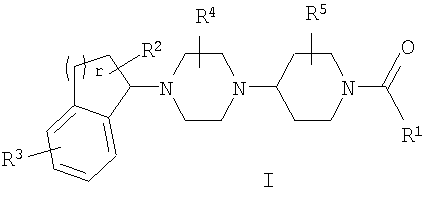
или его фармацевтически приемлемая соль, в котором:
R1 представляет собой гетероарил, необязательно замещенный одним или несколькими R6;
R2 представляет собой Н, гетероарил, OR9 или SR9;
R3 представляет собой F, Cl, Br, I, C1-C4 галогеналкил или гетероарил;
R4 представляет собой C1-С6 алкил;
R5 представляет собой C1-С6 алкил;
R6 представляет собой Н или C1-С6 алкил;
R9 представляет собой Н, C1-С6 алкил, алкоксиалкил или гетероарил;
r представляет собой 1 или 2,
где гетероарил представляет собой 5- или 6-членный моноциклический или 10-членный бициклический ароматический углеводород, имеющий 1-2 гетероатома, выбранных из серы, кислорода или азота.
2. Соединение по п.1, или его фармацевтически приемлемая соль, где R1 представляет собой 5- или 6-членную гетероарильную группу, содержащую по меньшей мере один образующий кольцо атом N, где указанная гетероарильная группа необязательно замещена 1, 2, 3 или 4 группами R6.
3. Соединение по п.1, или его фармацевтически приемлемая соль, где R1 представляет собой:
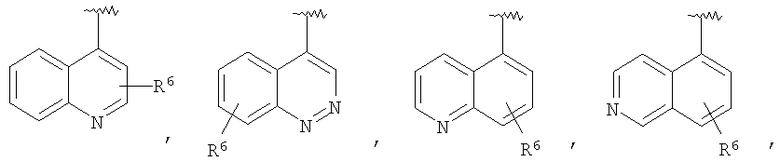
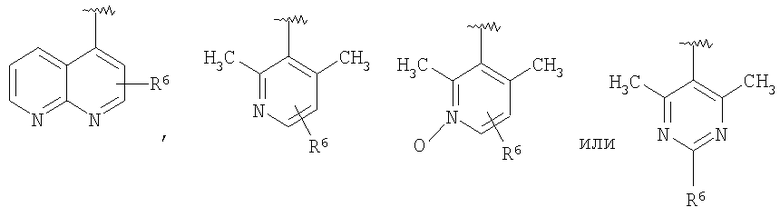
4. Соединение по п.1, или его фармацевтически приемлемая соль, где R1 представляет собой:
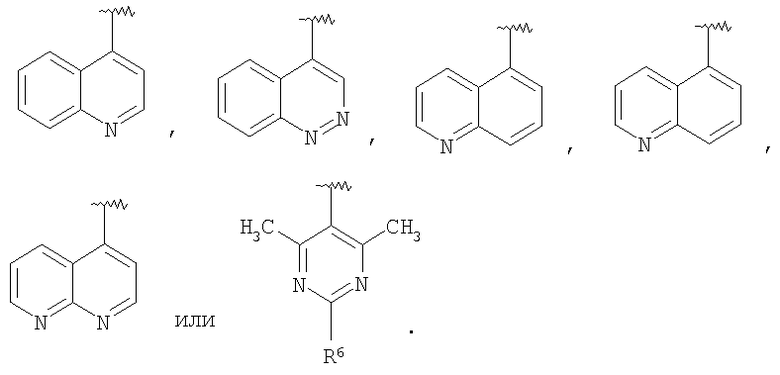
5. Соединение по п.1, или его фармацевтически приемлемая соль, где R2 представляет собой Н или OR9.
6. Соединение по п.1, или его фармацевтически приемлемая соль, где R3 представляет собой F, Вr, СF3, или 6- или 5-членный гетероарил.
7. Соединение по п.1, или его фармацевтически приемлемая соль, где R4 представляет собой метил.
8. Соединение по п.1, или его фармацевтически приемлемая соль, где R5 представляет собой метил.
9. Соединение по п.1, или его фармацевтически приемлемая соль, имеющее формулу IIа:
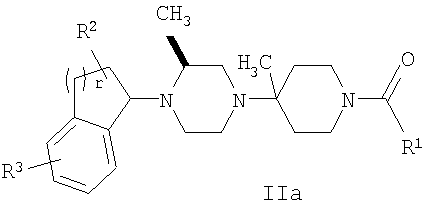
10. Соединение по п.9, или его фармацевтически приемлемая соль, где R1 представляется собой:
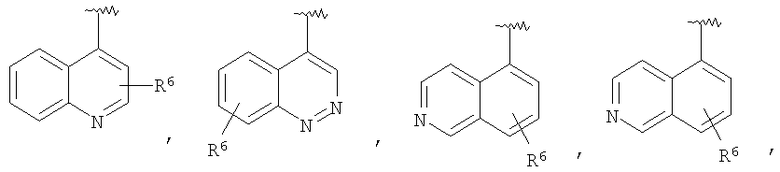
11. Соединение по п.10, или его фармацевтически приемлемая соль, где R1 представляется собой:
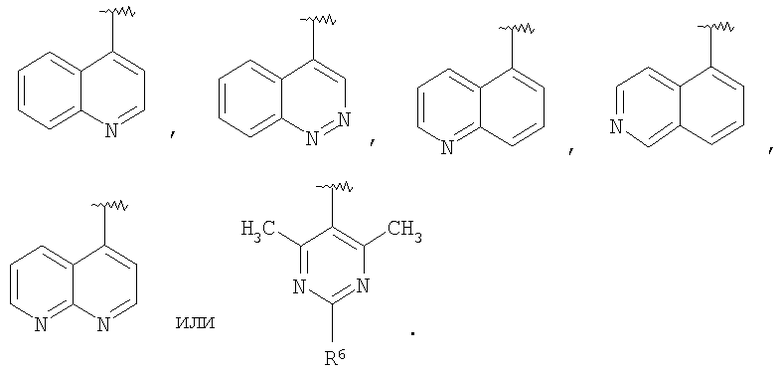
12. Соединение по п.1, или его фармацевтически приемлемая соль, в качестве агента, избирательно связывающегося с CCR5, или ингибитора CCR5.
13. Соединение по п.1, выбранное из:
5-({4-[(3S)-4-(5-бром-2,3-дигидро-1Н-инден-1-ил)-3-метилпиперазин-1-ил]-4-метилпиперидин-1-ил}карбонил)-4,6-диметилпиримидина;
5-({4-[(3S)-4-(5-фтор-2,3-дигидро-1H-инден-1-ил)-3-метилпиперазин-1-ил]-4-метилпиперидин-1-ил}карбонил)-4,6-диметилпиримидина;
5-({4-[(3S)-4-(6-бром-2,3-дигидро-1Н-инден-1-ил)-3-метилпиперазин-1-ил]-4-метилпиперидин-1-ил}карбонил)-4,6-диметилпиримидина;
5-({4-[(3S)-4-(6-фтор-2,3-дигидро-1Н-инден-1-ил)-3-метилпиперазин-1-ил]-4-метилпиперидин-1-ил}карбонил)-4,6-диметилпиримидина;
5-({4-[(3S)-4-(6-бром-1,2,3,4-тетрагидронафталин-1-ил)-3-метилпиперазин-1-ил]-4-метилпиперидин-1-ил}карбонил)-4,6-диметилпиримидина;
5-({4-[(3S)-4-(7-бром-1,2,3,4-тетрагидронафталин-1-ил)-3-метилпиперазин-1-ил]-4-метилпиперидин-1-ил}карбонил)-4,6-диметилпиримидина;
4,6-диметил-5-[(4-метил-4-{(3S)-3-метил-4-[6-(трифторметил)-2,3-дигидро-1Н-инден-1-ил]пиперазин-1-ил}пиперидин-1-ил)карбонил]пиримидина;
4,6-диметил-5-[(4-метил-4-{(3S)-3-метил-4-[5-(трифторметил)-2,3-дигидро-1Н-инден-1-ил]пиперазин-1-ил}пиперидин-1-ил)карбонил]пиримидина;
1-((2S)-4-{1-[(4,6-диметилпиримидин-5-ил)карбонил]-4-метилпиперидин-4-ил}-2-метилпиперазин-1-ил)-5-(трифторметил)индан-2-ола;
5-[(4-{(3S)-4-[2-метокси-5-(трифторметил)-2,3-дигидро-1Н-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидина;
дигидрохлорида 5-[(4-(3S)-4-[(1R,2R)-2-этокси-5-(трифторметил)-2,3-дигидро-1Н-инден-1-ил]-3-метилпиперазин-1-ил-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидина;
5-[(4-{(3S)-4-[(1R,2R)-2-(2-метоксиэтокси)-5-(трифторметил)-2,3-дигидро-1Н-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидина;
4-[(4-{(3S)-4-[(1S,2R)-2-этокси-5-(трифторметил)-2,3-дигидро-1Н-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]циннолина;
4-[(4-{(3S)-4-[(1R,2R)-2-этокси-5-(трифторметил)-2,3-дигидро-1Н-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]хинолина;
5-[(4-{(3S)-4-[(1R,2R)-2-этокси-5-(трифторметил)-2,3-дигидро-1Н-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]хинолина;
4-[(4-{(3S)-4-[(1R,2R)-2-этокси-5-(трифторметил)-2,3-дигидро-1Н-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-1,8-нафтиридина;
5-[(4-{(3S)-4-[(1R,2R)-2-этокси-5-(трифторметил)-2,3-дигидро-1Н-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]изохинолина;
5-[(4-{(3S)-4-[(1R,2R)-5-бром-2-этокси-2,3-дигидро-1Н-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидина;
4-[(4-{(3S)-4-[(1R,2R)-5-бром-2-этокси-2,3-дигидро-1Н-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]циннолина;
4-[(4-{(3S)-4-[(1R,2R)-5-бром-2-этокси-2,3-дигидро-1Н-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-1,8-нафтиридина;
5-[(4-{(3S)-4-[(1R,2R)-5-бром-2-(пиридин-2-илокси)-2,3-дигидро-1Н-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидина;
5-[(4-{(3S)-4-[(1R,2R)-2-этокси-5-(1,3-тиазол-2-ил)-2,3-дигидро-1Н-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидина;
5-[(4-{(3S)-4-[(1R,2R)-2-этокси-5-пиридин-2-ил-2,3-дигидро-1Н-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидина; и
5-[(4-{(3S)-4-[3-метокси-5-(трифторметил)-2,3-дигидро-1Н-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидина;
или его фармацевтически приемлемая соль.
14. Соединение, представляющее собой 5-[(4-(3S)-4-[(1R,2R)-2-этокси-5-(трифторметил)-2,3-дигидро-1Н-инден-1-ил]-3-метилпиперазин-1-ил-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидин или его фармацевтически приемлемую соль.
15. Соединение по п.14, представляющее собой 5-[(4-(3S)-4-[(1R,2R)-2-этокси-5-(трифторметил)-2,3-дигидро-1Н-инден-1-ил]-3-метилпиперазин-1-ил-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидиндигидрохлорид.
16. Соединение, представляющее собой 5-[(4-{(3S)-4-[(1R,2R)-2-этокси-5-(1,3-тиазол-2-ил)-2,3-дигидро-1Н-инден-1-ил]-3-метилпиперазин-1-ил}-4-метилпиперидин-1-ил)карбонил]-4,6-диметилпиримидин или его фармацевтически приемлемую соль.
17. Соединение формулы I:
или его фармацевтически приемлемая соль, в котором:
R1 представляет собой гетероарил, необязательно замещенный одним или несколькими R6;
R2 представляет собой Н, гетероарил, OR9 или SR9;
R3 представляет собой F, Cl, Br, I, C1-C4 галогеналкил;
R4 представляет собой C1-С6 алкил;
R5 представляет собой C1-C6 алкил;
R6 представляет собой Н или C1-С6 алкил;
R9 представляет собой Н, С1-С6 алкил, алкоксиалкил или гетероарил;
r представляет собой 1 или 2;
где гетероарил представляет собой 5- или 6-членный моноциклический или 10-членный бициклический ароматический углеводород, имеющий 1-2 гетероатомов, выбранных из серы, кислорода или азота.
18. Фармацевтическая композиция для модулирования активности или связывания с хемокиновым рецептором, содержащая соединение по любому из пп.1-17 или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель.
19. Способ модулирования активности хемокинового рецептора, включающий взаимодействие указанного хемокинового рецептора с соединением по любому из пп.1-17 или его фармацевтически приемлемой солью.
20. Способ по п.19, в котором указанный хемокиновый рецептор представляет собой CCR5.
21. Способ по п.19, в котором указанное модулирование соответствует ингибированию.
22. Способ по п.19, в котором указанное соединение представляет собой избирательный ингибитор CCR5.
23. Способ по п.19, в котором указанное соединение представляет собой агент, избирательно связывающийся с CCR5.
24. Способ лечения заболевания, ассоциированного с экспрессией или активностью хемокинового рецептора у пациента, включающий введение указанному пациенту терапевтически эффективного количества соединения по любому из пп.1-17.
25. Способ по п.24, в котором указанный хемокиновый рецептор представляет собой CCR5.
26. Способ по п.25, в котором указанное соединение представляет собой избирательный ингибитор CCR5 или агент, избирательно связывающийся с CCR5.
27. Способ лечения заболевания или состояния, выбранного из воспалительного заболевания, иммунного нарушения и вирусной инфекции у пациента, включающий введение указанному пациенту терапевтически эффективного количества соединения по любому из пп.1-17 или его фармацевтически приемлемой соли.
28. Способ по п.27, в котором указанное заболевание или состояние представляет собой воспалительное заболевание.
29. Способ по п.28, в котором указанное заболевание или состояние представляет собой иммунное нарушение.
30. Способ по п.28, в котором указанное заболевание или состояние представляет собой вирусную инфекцию.
31. Способ по п.30, в котором указанная вирусная инфекция представляет собой инфекцию ВИЧ.
32. Способ лечения инфекции ВИЧ у пациента, включающий введение указанному пациенту терапевтически эффективного количества соединения по любому из пп.1-17 или его фармацевтически приемлемой соли.
33. Способ по п.32, дополнительно включающий одновременное или последовательное введение, по меньшей мере, одного противовирусного агента.
| ПРОИЗВОДНЫЕ N-ФЕНИЛ-2-ПИРИМИДИНАМИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ИНГИБИРОВАНИЯ (ЛЕЧЕНИЯ) ОПУХОЛИ | 1994 |
|
RU2135491C1 |
| US 5935958 A, 10.08.1999 | |||
| WO 9800412, 08.01.1998. | |||

