Настоящее изобретение относится к функционализованным азокарбонилом силанам, их получению и их применению.
В DE 102010003387.1 описан способ получения кремнийсодержащих азодикарбамидов взаимодействием соединений формулы
R3-X1-C(O)-N=N-C(O)-X1-R4 с соединениями формулы (R1)3-a(R2)aSi-RI-NH2.
Из DE 2704506 известны соединения общей формулы
Y-X-CO-N=N-CO-X1-Z и их применение в резиновых смесях с наполнителем.
Помимо этого из US 2009/0234066 А1 известны соединения общей формулы A-CO-N=N-CO-Z-G, которые совместно с серусодержащими силанами используются в изопреновом каучуке.
Из US 2009/0186961 А1 известны соединения общей формулы A-CO-N=N-CO-Z-G, которые совместно с аппретами используются в изопреновом каучуке.
В US 2009/0216000 и US 2011/282040 описаны способы получения кремнийорганических соединений, содержащих структурный фрагмент -CO-N=N-CO-.
Помимо этого в DE 2434426 описан 1,2,4-триазолин-3,5-дион.
В ЕР 2508559 описаны резиновые смеси, содержащие определенный каучук, оксидный наполнитель и азодикарбамид формулы
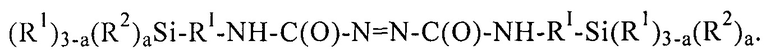
Недостаток известных резиновых смесей, содержащих силаны, состоит в их неудовлетворительном сопротивлении раздиру.
В основу настоящего изобретения была положена задача предложить силан, который при его включении в состав резиновой смеси обеспечивал бы улучшение ее характеристик сопротивления раздиру.
Объектом настоящего изобретения являются функционализованные азокарбонилом силаны общей формулы I
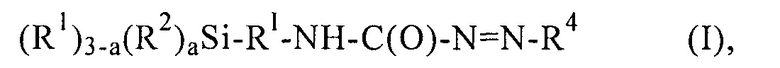
где
R1 в каждом случае независимо обозначает замещенные либо незамещенные алкильные группы с C1-C18, предпочтительно с C1-C10, особенно предпочтительно с С1-С6, наиболее предпочтительно с С1, циклоалкильные группы с С5-С18, предпочтительно с С6, или арильные группы с С6-С18, предпочтительно фенил,
R2 в каждом случае независимо обозначает -ОН, замещенную либо незамещенную алкоксигруппу с C1-C18, предпочтительно СН3-О-, С2Н5-O-, С3Н7-О-, С12Н25-О-, C14H29-O-, С16Н33-О- или С18Н37-О-, особенно предпочтительно С2Н5-О-, циклоалкоксигруппу с С5-С18 или группу простого алкилового полиэфира O(CH2-CH2-O)n-R3 или O(CH(CH3)-CH2-O)n-R3, где n в среднем составляет от 1 до 18, a R3 в каждом случае независимо представляет собой разветвленную либо неразветвленную, насыщенную либо ненасыщенную одновалентную углеводородную цепь с С1-С32,
RI обозначает разветвленную либо неразветвленную, насыщенную либо ненасыщенную алифатическую, ароматическую либо смешанно алифатическую/ароматическую двухвалентную углеводородную группу с С1-С30, предпочтительно с С1-С20, более предпочтительно с С1-С10, особенно предпочтительно с С1-С7, которая необязательно может быть замещена F-, Cl-, Br-, I-, -CN или HS-,
а обозначает число 1, 2 или 3,
R4 обозначает замещенный либо незамещенный арил, предпочтительно фенил, галогензамещенный фенил, например хлорфенил, бромфенил или иодфенил, толил, алкоксифенил, например метоксифенил, о-, м- или n-нитрофенил, или замещенный либо незамещенный алкил, предпочтительно метил, этил, пропил, бутил, изобутил, трет-бутил, нитрометил, нитроэтил, нитропропил, нитробутил или нитроизобутил.
В предпочтительном варианте R2 может представлять собой этоксигруппу, а индекс а обозначает 3.
В более предпочтительном варианте R4 может представлять собой фенил, n-нитрофенил или трет-бутил.
В предпочтительном варианте RI может представлять собой -СН2-, -СН2СН2-, -СН2СН2СН2-, -СН2СН2СН2СН2-, -СН(СН3)-, -СН2СН(СН3)-, -СН(СН3)СН2-, -С(СН3)2-, -СН(С2Н5)-, -СН2СН2СН(СН3), -СН(СН3)СН2СН2-, -СН2СН(СН3)СН2-, -СН2СН2СН2СН2СН2-, -СН2СН2СН2СН2СН2СН2-, -СН2СН2СН2СН2СН2СН2СН2-, -СН2СН2СН2СН2СН2СН2СН2СН2-, -СН2СН2СН2СН2СН2СН2СН2СН2СН2-, -СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2-, -СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2-, -СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2-, -СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2-, -СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2СН2-, 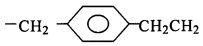 - или -СН2-СН2-С6Н4-СН2-.
- или -СН2-СН2-С6Н4-СН2-.
Функционализованные азокарбонилом силаны общей формулы I в предпочтительном варианте могут представлять собой
(CH3CH2O-)3Si-СН2-NH-СО-N=N-фенил,
(CH3CH2O-)3Si-(СН2)2-NH-СО-N=N-фенил,
(CH3CH2O-)3Si-(CH2)3-NH-CO-N=N-фенил, (CH3O-)3Si-CH2-NH-CO-N=N-фенил,
(CH3O-)3Si-(СН2)2-NH-СО-N=N-фенил, (CH3O-)3Si-(CH2)3-NH-CO-N=N-фенил,
(CH3CH2O-)2Si(-O(CH2-CH2-O)5-C13H27)-CH2-NH-CO-N=N-фенил,
(CH3CH2O-)2Si(-O(СН2-СН2-O)5-С13Н27)-(СН2)2-NH-СО-N=N-фенил,
(CH3CH2O-)2Si(-O(СН2-СН2-O)5-С13Н27)-(СН2)3-NH-СО-N=N-фенил,
(CH3CH2O-)Si(-O(СН2-СН2-O)5-С13Н27)2-СН2-NH-СО-N=N-фенил,
(CH3CH2O-)Si(-O(СН2-СН2-O)5-С13Н27)2-(СН2)2-NH-СО-N=N-фенил,
(CH3CH2O-)Si(-O(СН2-СН2-O)5-С13Н27)2-(СН2)3-NH-СО-N=N-фенил,
(CH3CH2O-)3Si-СН2-NH-СО-N=N-фенил,
(CH3CH2O-)3Si-(СН2)2-NH-СО-N=N-(n-нитрофенил),
(CH3CH2O-)3Si-(СН2)3-NH-СО-N=N-(n-нитрофенил),
(CH3O-)3Si-СН2-NH-СО-N=N-(n-нитрофенил),
(CH3O-)3Si-(СН2)2-NH-СО-N=N-(n-нитрофенил),
(CH3O-)3Si-(СН2)3-NH-СО-N=N-(n-нитрофенил),
(CH3CH2O-)2Si(-O(СН2-СН2-O)5-С13Н27)-СН2-NH-СО-N=N-(n-нитрофенил),
(CH3CH2O-)2Si(-O(СН2-СН2-O)5-С13Н27)-(СН2)2-NH-СО-N=N-(n-нитрофенил),
(CH3CH2O-)2Si(-O(СН2-СН2-O)5-C13H27)-(СН2)3-NH-СО-N=N-(n-нитрофенил),
(CH3CH2O-)Si(-O(СН2-СН2-O)5-С13Н27)2-СН2-NH-СО-N=N-(n-нитрофенил),
(CH3CH2O-)Si(-O(СН2-СН2-O)5-С13Н27)2-(СН2)2-NH-СО-N=N-(n-нитрофенил),
(CH3CH2O-)Si(-O(СН2-СН2-O)5-С13Н27)2-(СН2)3-NH-СО-N=N-(n-нитрофенил),
(CH3CH2O-)3Si-CH2-NH-CO-N=N-CH3, (CH3CH2O-)3Si-(CH2)2-NH-CO-N=N-CH3,
(CH3CH2O-)3Si-(CH2)3-NH-CO-N=N-CH3, (CH3O-)3Si-CH2-NH-CO-N=N-CH3,
(CH3O-)3Si-(CH2)2-NH-CO-N=N-CH3, (CH3O-)3Si-(CH2)3-NH-CO-N=N-CH3,
(CH3CH2O-)3Si-CH2-NH-CO-N=N-CH2CH3,
(CH3CH2O-)3Si-(CH2)2-NH-CO-N=N-CH2CH3,
(CH3CH2O-)3Si-(CH2)3-NH-CO-N=N-CH2CH3,
(CH3O-)3Si-CH2-NH-CO-N=N-CH2CH3, (CH3O-)3Si-(CH2)2-NH-CO-N=N-CH2CH3,
(CH3O-)3Si-(CH2)3-NH-CO-N=N-CH2CH3,
(CH3CH2O-)3Si-CH2-NH-CO-N=N-CH2CH2CH3,
(CH3CH2O-)3Si-(CH2)2-NH-CO-N=N-CH2CH2CH3,
(CH3CH2O-)3Si-(CH2)3-NH-CO-N=N-CH2CH2CH3,
(CH3O-)3Si-CH2-NH-CO-N=N-CH2CH2CH3,
(CH3O-)3Si-(CH2)2-NH-CO-N=N-CH2CH2CH3,
(CH3O-)3Si-(CH2)3-NH-CO-N=N-CH2CH2CH3,
(CH3CH2O-)3Si-CH2-NH-CO-N=N-CH2CH2CH2CH3,
(CH3CH2O-)3Si-(CH2)2-NH-CO-N=N-CH2CH2CH2CH3,
(CH3CH2O-)3Si-(CH2)3-NH-CO-N=N-CH2CH2CH2CH3,
(CH3O-)3Si-CH2-NH-CO-N=N-CH2CH2CH2CH3,
(CH3O-)3Si-(CH2)2-NH-CO-N=N-CH2CH2CH2CH3,
(CH3O-)3Si-(CH2)3-NH-CO-N=N-CH2CH2CH2CH3,
(CH3CH2O-)3Si-CH2-NH-CO-N=N-C(CH3)3,
(CH3CH2O-)3Si-(CH2)2-NH-CO-N=N-C(CH3)3,
(CH3CH2O-)3Si-(CH2)3-NH-CO-N=N-C(CH3)3,
(CH3O-)3Si-CH2-NH-CO-N=N-C(CH3)3, (CH3O-)3Si-(CH2)2-NH-CO-N=N-C(CH3)3
или (CH3O-)3Si-(CH2)3-NH-CO-N=N-C(CH3)3.
Функционализованные азокарбонилом силаны общей формулы I могут представлять собой смесь из функционализованных азокарбонилом силанов общей формулы I и дополнительно олигомеров, получаемых путем гидролиза или конденсации функционализованных азокарбонилом силанов общей формулы I.
Объектом изобретения является также первый способ получения предлагаемых в изобретении функционализованных азокарбонилом силанов общей формулы I
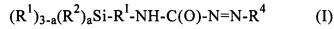 ,
,
отличающийся тем, что на первой стадии гидразин формулы II
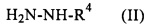
подвергают взаимодействию с изоцианатосиланом общей формулы III
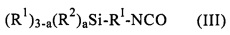
и на второй стадии полученный на первой стадии продукт окисляют окислителем, при этом R1, R2, R4, RI и а имеют указанные выше значения.
Первую стадию первого способа можно проводить в атмосфере инертного газа, например азота или аргона.
Первую стадию первого способа можно проводить при температуре в пределах от -50 до 50°С, предпочтительно от -10 до 25°С, особенно предпочтительно от 0 до 15°С.
Первую стадию первого способа можно проводить в течение 5-500 мин, предпочтительно 60-300 мин.
Первую стадию первого способа можно проводить в растворителе, например дихлорметане, этилацетате, пентане или воде, либо в отсутствие растворителя.
На первой стадии первого способа для образования гидразина формулы II in situ можно использовать HCl-соль гидразина формулы IV
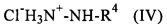
совместно с основанием, например пиридином или NaOH.
Вторую стадию первого способа можно проводить в атмосфере инертного газа, например азота или аргона.
Вторую стадию первого способа можно проводить при температуре в пределах от -50 до 50°С, предпочтительно от -10 до 25°С, особенно предпочтительно от 0 до 25°С.
Вторую стадию первого способа можно проводить в течение 5-300 мин, предпочтительно 60-210 мин.
Вторую стадию первого способа можно проводить в растворителе, например дихлорметане, этилацетате, пентане или воде, либо в отсутствие растворителя.
В качестве окислителя на второй стадии первого способа можно использовать NaOCl, бром, N-бромсукцинимид, надуксусную кислоту, 1,3-дибром-5,5-диметилгидантоин или (мета)периодат тетрабутиламмония.
Окисление на второй стадии первого способа можно проводить в присутствии основания, например карбоната натрия, пиридина или имидазола, или в присутствии буферного раствора.
Объектом изобретения является также второй способ получения предлагаемых в изобретении функционализованных азокарбонилом силанов общей формулы I
,
отличающийся тем, что на первой стадии гидразин формулы II
подвергают взаимодействию с ацилгалогенидом общей формулы V
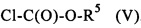 ,
,
на второй стадии полученный на первой стадии продукт окисляют окислителем и на третьей стадии полученный на второй стадии продукт подвергают взаимодействию с аминосиланом общей формулы (VI)
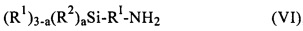 ,
,
при этом R1, R2, R4, RI и а имеют указанные выше значения, a R5 обозначает арил или С1-С30алкил, предпочтительно СН3, СН2СН3, СН2СН2СН3 или СН(СН3)2.
Первую стадию второго способа можно проводить в атмосфере инертного газа, например азота или аргона.
Первую стадию второго способа можно проводить при температуре в пределах от -50 до 50°С, предпочтительно от -10 до 25°С, особенно предпочтительно от -5 до 15°С.
Первую стадию второго способа можно проводить в течение 5-300 мин, предпочтительно 30-180 мин.
Первую стадию второго способа можно проводить в растворителе, например ацетонитриле, дихлорметане, этилацетате, пентане или воде, либо в отсутствие растворителя.
Первую стадию второго способа можно проводить в присутствии основания, например пиридина, имидазола или карбоната натрия.
На первой стадии второго способа для образования гидразина формулы II in situ можно использовать HCl-соль гидразина формулы IV
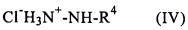
совместно с основанием, например пиридином или NaOH.
Вторую стадию второго способа можно проводить в атмосфере инертного газа, например азота или аргона.
Вторую стадию второго способа можно проводить при температуре в пределах от -25 до 50°С, предпочтительно от -10 до 25°С, особенно предпочтительно от 0 до 20°С.
Вторую стадию второго способа можно проводить в течение 5-300 мин, предпочтительно 60-210 мин.
Вторую стадию второго способа можно проводить в растворителе, например дихлорметане, пентане, этилацетате, этаноле, уксусной кислоте или воде, либо в отсутствие растворителя.
В качестве окислителя на второй стадии второго способа можно использовать бром, N-бромсукцинимид, надуксусную кислоту, пероксимоносульфат калия, NaOCl, 1,3-дибром-5,5-диметилгидантоин или (мета)периодат тетрабутиламмония.
Окисление на второй стадии второго способа можно проводить в присутствии основания, предпочтительно карбоната натрия, пиридина или имидазола.
Третью стадию второго способа можно проводить в атмосфере инертного газа, например азота или аргона.
Третью стадию второго способа можно проводить при температуре в пределах от -25 до 50°С, предпочтительно от -10 до 25°С, особенно предпочтительно от -5 до 15°С.
Третью стадию второго способа можно проводить в течение 5-300 мин, предпочтительно 30-200 мин.
Третью стадию второго способа можно проводить в растворителе, например ацетонитриле, дихлорметане, этилацетате, пентане или воде, либо в отсутствие растворителя.
Объектом изобретения являются также резиновые смеси, отличающиеся тем, что они содержат
(А) по меньшей мере один каучук, выбранный из группы, включающей сополимер этилена, пропилена и диенового мономера (СКЭПТ), сополимер этилена с пропиленом (СКЭП), хлоропреновый каучук (ХК), хлорполиэтилен (ХП), хлорированный сополимер изобутена и изопрена (хлорбутилкаучук) (СКИИХ), хлорсульфированный полиэтилен (ХСПЭ), сополимер этилена с винилацетатом (ЕАМ), сополимер алкилакрилата (САА), полиэфироуретан со сложноэфирными группами (AU), полиэфироуретан с простыми эфирными группами (EU), бромированный сополимер изобутена и изопрена (бромбутилкаучук) (СКИИБ), полихлортрифторэтилен (CFM), сополимер изобутена и изопрена (бутилкаучук, СКИИ), изобутеновый каучук (IM), полиизопрен (СКИ), термопластичный полиэфироуретан со сложноэфирными группами (YAU), термопластичный полиэфироуретан с простыми эфирными группами (YEU), кремнийорганический каучук с метальными группами в полимерной цепи (MQ), гидрированный бутадиен-нитрильный каучук (СКНГ), бутадиен-нитрильный каучук (СКН) и карбоксилатный бутадиен-нитрильный каучук (СКНК), предпочтительно сополимер этилена, пропилена и диенового мономера (СКЭПТ),
(Б) по меньшей мере один оксидный наполнитель и
(В) по меньшей мере один функционализованный азокарбонилом силан общей формулы I
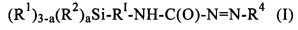
где R1, R2, R4, RI и а имеют указанные выше значения.
Функционализованные азокарбонилом силаны общей формулы I можно добавлять в процесс смешения в чистом виде или же можно добавлять в абсорбированном на инертном органическом или неорганическом носителе виде или в предварительно прореагировавшем с органическим или неорганическим носителем виде. В качестве примера предпочтительных носителей можно назвать осажденный или коллоидный диоксид кремния, воски, термопласты, природные или синтетические силикаты, природные или синтетические оксиды, такие как оксид алюминия, или технический углерод различных типов. Функционализованные азокарбонилом силаны общей формулы I можно также добавлять в процесс смешения в предварительно прореагировавшем с применяемым оксидным наполнителем виде.
В качестве примера предпочтительных восков можно назвать воски с температурой плавления, интервалом температур плавления или интервалом температур размягчения от 50 до 200°С, предпочтительно от 70 до 180°С, особенно предпочтительно от 90 до 150°С, наиболее предпочтительно от 100 до 120°С. Применяемые воски могут представлять собой олефиновые воски. Применяемые воски могут содержать насыщенные и ненасыщенные углеводородные цепи. Применяемые воски могут содержать полимеры или олигомеры, предпочтительно бутадиен-стирольный каучук эмульсионной полимеризации (Э-СКС) и/или бутадиен-стирольный каучук, полученный полимеризацией в растворе (Р-СКС). Применяемые воски могут содержать алканы с длинной цепью и/или карбоновые кислоты с длинной цепью. Применяемые воски могут содержать сополимер этилена с винилацетатом и/или поливиниловые спирты.
Функционализованные азокарбонилом силаны общей формулы I можно добавлять в процесс смешения в виде физической смеси с органическим веществом или в виде физической смеси со смесью органических веществ. Такое органическое вещество может представлять собой или такая смесь органических веществ может содержать полимеры или олигомеры. Подобные полимеры или олигомеры могут представлять собой гетероатомсодержащие полимеры или олигомеры, например, сополимер этилена с виниловым спиртом и/или поливиниловые спирты.
В предлагаемых в изобретениях резиновых смесях можно использовать следующие оксидные наполнители:
- аморфный диоксид кремния, получаемый, например, путем осаждения растворов силикатов (осажденный диоксид кремния) или путем пламенного гидролиза галогенидов кремния (коллоидный диоксид кремния). Удельная поверхность аморфного диоксида кремния (удельная поверхность, определяемая методом Брунауэра-Эммета-Теллера по адсорбции азота (БЭТ-поверхность)) может составлять от 5 до 1000 м2/г, предпочтительно от 20 до 400 м2/г, а размер его первичных частиц может составлять от 10 до 400 нм. Диоксид кремния при необходимости может быть также представлен в виде смешанного оксида с другими оксидами металлов, такими как оксиды Al, оксиды Mg, оксиды Ca, оксиды Ba, оксиды Zn и оксиды титана;
- синтетические силикаты, такие как силикат алюминия или силикаты щелочноземельных металлов, такие как силикат магния или силикат кальция. БЭТ-поверхность синтетических силикатов может составлять от 20 до 400 м2/г, а диаметр их первичных частиц может составлять от 10 до 400 нм;
- синтетические или природные оксиды алюминия и синтетические или природные гидроксиды алюминия;
- природные силикаты, такие как каолин и диоксид кремния других встречающихся в природе типов;
- стекловолокно и стекловолокнистые продукты (стекловолокнистые маты, стекложгуты) или стеклянные микрошарики.
В некоторых случаях может оказаться предпочтительным использовать аморфный диоксид кремния, полученный осаждением растворов силикатов (осажденный диоксид кремния) с БЭТ-поверхностью от 20 до 400 м2/г. Количества, в которых можно использовать аморфный диоксид кремния, составляют от 5 до 150 мас. частей в каждом случае в пересчете на 100 частей каучука.
Указанные выше наполнители можно использовать индивидуально или в смеси. В одном из особенно предпочтительных вариантов резиновые смеси могут содержать оксидные наполнители в количестве от 10 до 150 мас. частей, при необходимости совместно с техническим углеродом в количестве от 0 до 100 мас. частей, а также функционализованные азокарбонилом силаны общей формулы I в количестве от 1 до 20 мас. частей, в каждом случае в пересчете на 100 мас. частей каучука.
К числу дополнительных наполнителей, которые можно использовать в предлагаемых в изобретении резиновых смесях, относятся технический углерод (сажа) различных сортов, такой как пламенная сажа, печная сажа, газовая сажа или термическая сажа, либо синтетические или природные карбонаты кальция, такие как осажденный карбонат кальция. БЭТ-поверхность технического углерода подобных сортов может составлять от 20 до 200 м2/г. При необходимости технический углерод может также содержать гетероатомы, такие как Si.
Предпочтительным материалом для приготовления предлагаемых в изобретении резиновых смесей является сополимер этилена с пропиленом (СКЭП) и диеновым мономером (СКЭПТ), каковой сополимер может содержать третий мономер (тройной этилен-пропиленовый сополимер). Предлагаемые в изобретении резиновые смеси дополнительно могут содержать натуральный каучук или синтетические каучуки. Предпочтительные для применения в этих целях синтетические каучуки описаны, например, у W. Hofmann в справочнике Kautschuktechnologie, изд-во Genter Verlag, Stuttgart, 1980. К ним относятся, в частности, полибутадиен (СКД), полиизопрен (СКИ), сополимеры стирола и бутадиена (СКС), например бутадиен-стирольный каучук эмульсионной полимеризации (Э-СКС) или бутадиен-стирольный каучук, получаемый полимеризацией в растворе (Р-СКС), с содержанием стирола от 1 до 60 мас. %, предпочтительно от 2 до 50 мас. %, особенно предпочтительно от 10 до 40 мас. %, наиболее предпочтительно от 15 до 35 мас. %, хлоропрен (ХП), сополимеры изобутилена и изопрена (СКИИ), сополимеры бутадиена и акрилонитрила (СКН) с содержанием акрилонитрила от 5 до 60 мас. %, предпочтительно от 10 до 50 мас. %, особенно предпочтительно от 10 до 45 мас. %, наиболее предпочтительно от 19 до 45 мас. %, частично либо полностью гидрированный бутадиен-нитрильный каучук (СКНГ), вышеназванные каучуки, дополнительно содержащие функциональные группы, такие, например, как карбоксигруппы, силанольные группы или эпоксигруппы, например эпоксидированный натуральный каучук, функционализованный карбоксигруппами СКН или функционализованный силанольными группами (-SiOH), соответственно силилалкоксигруппами (-Si-OR) СКС, или смесь указанных каучуков.
Предлагаемые в изобретении резиновые смеси могут содержать дополнительные ингредиенты, такие как ускорители реакции, антиоксиданты (противостарители), термостабилизаторы, светостабилизаторы, антиозонанты, технологические добавки, пластификаторы, вещества для повышения клейкости, порообразователи, красители, пигменты, воски, разбавители, органические кислоты, ингибиторы, оксиды металлов, а также активаторы, такие как триэтаноламин или гексантриол.
Предлагаемая в изобретении резиновая смесь может дополнительно содержать другие силаны. К таким дополнительным силанам, которые можно добавлять к предлагаемым в изобретении резиновым смесям, относятся меркаптоорганилсиланы, содержащие этоксисилильные группы, и/или тиоцианатоорганилсиланы, содержащие этоксисилильные группы, и/или блокированные меркаптоорганилсиланы, содержащие этоксисилильные группы, и/или полисульфидные алкоксисиланы, содержащие этоксисилильные группы.
К дополнительным силанам, которые можно добавлять к предлагаемым в изобретении резиновым смесям, относятся, в частности, меркаптоорганил(алкоксисиланы) с C8H17-O-, С10Н21-О-, С12Н25-О-, С14Н29-О-, С16Н33-О- или С18Н37-О-группой у атома кремния.
К дополнительным силанам, которые можно добавлять к предлагаемым в изобретении резиновым смесям, относятся, в частности, тиоцианатоорганил(алкоксисиланы) с C8H17-O-, С10Н21-О-, С12Н25-О-, С14Н29-О-, С16Н33-О- или C18H37-O-группой у атома кремния.
К дополнительным силанам, которые можно добавлять к предлагаемым в изобретении резиновым смесям, относятся, в частности, блокированные меркаптоорганил(алкоксисиланы) с C8H17-O-, С10Н21-О-, С12Н25-О-, C14H29-O-, С16Н33-О- или С18Н37-O-группой у атома кремния.
К дополнительным силанам, которые можно добавлять к предлагаемым в изобретении резиновым смесям, относятся, в частности, блокированные меркаптоорганил(алкоксисиланы) с бифункциональными спиртами (диолами) у атома кремния (например, продукт NXT LowV или NXT Ultra-LowV фирмы General Electric).
К дополнительным силанам, которые можно добавлять к предлагаемым в изобретении резиновым смесям, относятся, в частности, полисульфидные алкоксисиланы с С8Н17-О-, С10Н21-О-, С12Н25-О-, C14H29-O-, С16Н33-О- или С18Н37-O-группой у атома кремния.
К дополнительным силанам, которые можно добавлять к предлагаемым в изобретении резиновым смесям, относятся, в частности, полисульфидные алкоксисиланы формулы
EtO-Si(Me)2-CH2-CH2-CH2-S2-CH2-CH2-CH2-Si(Me)2(OEt),
EtO-Si(Me)2-CH2-CH2-CH2-S3-CH2-CH2-CH2-Si(Me)2(OEt) или
EtO-Si(Me)2-CH2-CH2-CH2-S4-CH2-CH2-CH2-Si(Me)2(OEt).
К дополнительным силанам, которые можно добавлять к предлагаемым в изобретении резиновым смесям, относятся, в частности, 3-меркаптопропил(триэтоксисилан) (например, продукт Si 263 фирмы Evonik Industries AG), 3-тиоцианатопропил(триэтоксисилан) (например, продукт Si 264 фирмы Evonik Industries AG), бис-(триэтоксисилилпропил)полисульфид (например, продукт Si 69 фирмы Evonik Industries AG), а также бис-(триэтоксисилилпропил)дисульфид (например, продукт Si 266 фирмы Evonik Industries AG).
К дополнительным силанам, которые можно добавлять к предлагаемым в изобретении резиновым смесям, относятся, в частности, меркаптоорганилсиланы, содержащие алкилполиэфироспирты (например, продукт Si 363 фирмы Evonik Industries AG), и/или тиоцианатоорганилсиланы, содержащие алкилполиэфироспирты, и/или блокированные меркаптоорганилсиланы, содержащие алкилполиэфироспирты, и/или полисульфидные силаны, содержащие алкилполиэфироспирты.
С учетом экономических аспектов или с учетом особенностей технологии переработки каучука может оказаться целесообразным минимизировать необходимость в применении дополнительных силанов или их требуемое количество.
Указанные выше дополнительные ингредиенты резиновых смесей можно применять в обычных количествах, зависящих помимо прочего от назначения резиновой смеси. Как правило, такие количества в зависимости от применяемой технологической добавки составляют от 0,001 до 50 мас. %, предпочтительно от 0,001 до 30 мас. %, особенно предпочтительно от 0,01 до 30 мас. %, наиболее предпочтительно от 0,1 до 30 мас. %, в пересчете на массу каучука (част./100 част, каучука).
Предлагаемые в изобретении резиновые смеси могут представлять собой вулканизуемые серой резиновые смеси.
Предлагаемые в изобретении резиновые смеси могут представлять собой сшиваемые пероксидом резиновые смеси.
В качестве сшивающих агентов можно использовать серу или соединения-доноры серы. Серу можно использовать в количестве от 0,1 до 10 мас. %, предпочтительно от 0,1 до 5 мас. %, в пересчете на массу каучука.
Функционализованные азокарбонилом силаны общей формулы I можно использовать в качестве усилителя адгезии между неорганическими материалами (например, стеклянными шариками, стеклянной крошкой, стеклянными поверхностями, стекловолокнами, металлами, оксидными наполнителями, диоксидом кремния) и органическими полимерами (например, термореактопластами, термопластами, эластомерами) либо в качестве сшивающего агента и модификатора поверхности для оксидных поверхностей. Функционализованные азокарбонилом силаны общей формулы I можно далее использовать в качестве аппретов в наполненных резиновых смесях, в качестве примера которых можно назвать резиновые смеси для изготовления уплотнений.
С учетом экономических аспектов или с учетом особенностей технологии переработки каучука может оказаться целесообразным минимизировать необходимость в применении дополнительных ингредиентов резиновых смесей или требуемое количество таких ингредиентов резиновых смесей.
Предлагаемые в изобретении резиновые смеси могут, кроме того, содержать ускорители вулканизации.
Ускорители вулканизации можно использовать в количестве от 0,1 до 10 мас. %, предпочтительно от 0,1 до 5 мас. %, в пересчете на массу применяемого каучука.
Предлагаемые в изобретении резиновые смеси могут также содержать
(Г) тиурамсульфид в качестве ускорителя и/или карбамат в качестве ускорителя и/или меркаптобензотриазол и/или дитиофосфат и/или соответствующие цинковые соли,
(Д) при необходимости азотсодержащий соактиватор и
(Е) при необходимости другие ингредиенты.
Объектом изобретения является далее способ приготовления предлагаемых в изобретении резиновых смесей, отличающийся тем, что между собой смешивают по меньшей мере один каучук, выбранный из группы, включающей сополимер этилена, пропилена и диенового мономера (СКЭПТ), сополимер этилена с пропиленом (СКЭП), хлоропреновый каучук (ХК), хлорполиэтилен (ХП), хлорированный сополимер изобутена и изопрена (хлорбутилкаучук) (СКИИХ), хлорсульфированный полиэтилен (ХСПЭ), сополимер этилена с винилацетатом (ЕАМ), сополимер алкилакрилата (АХП), полиэфироуретан со сложноэфирными группами (AU), полиэфироуретан с простыми эфирными группами (EU), бромированный сополимер изобутена и изопрена (бромбутилкаучук) (СКИИБ), полихлортрифторэтилен (CFM), сополимер изобутена и изопрена (бутилкаучук, СКИИ), изобутеновый каучук (IM), полиизопрен (СКИ), термопластичный полиэфироуретан со сложноэфирными группами (YAU), термопластичный полиэфироуретан с простыми эфирными группами (YEU), кремнийорганический каучук с метальными группами в полимерной цепи (MQ), гидрированный бутадиен-нитрильный каучук (СКНГ), бутадиен-нитрильный каучук (СКН) и карбоксилатный бутадиен-нитрильный каучук (СКНК), предпочтительно сополимер этилена, пропилена и диенового мономера (СКЭПТ), по меньшей мере один оксидный наполнитель и по меньшей мере один функционализованный азокарбонилом силан общей формулы I.
Предлагаемый в изобретении способ можно проводить при температуре выше 25°С. Предлагаемый в изобретении способ можно, в частности, проводить при температуре в пределах от 80 до 220°С, предпочтительно от 100 до 200°С, особенно предпочтительно от 110 до 180°С.
Предлагаемый в изобретении способ можно проводить в непрерывном или периодическом режиме.
Функционализованные азокарбонилом силаны общей формулы I, а также наполнители можно добавлять при температуре смеси в пределах от 100 до 220°С. Однако их можно добавлять и при более низкой температуре, составляющей от 40 до 100°С, например, совместно с дополнительными ингредиентами резиновых смесей.
Процесс смешения каучуков с наполнителем, с возможно используемыми дополнительными ингредиентами резиновых смесей и с функционализованными азокарбонилом силанами общей формулы I можно проводить в обычных смесительных устройствах, таких как вальцы, резиносмесители закрытого типа и шнековые смесители. Обычно такие резиновые смеси можно приготавливать в резиносмесителях закрытого типа, при этом сначала на одной либо нескольких последовательных термомеханических стадиях смешения каучуки, наполнитель, функционализованные азокарбонилом силаны общей формулы I и другие дополнительные ингредиенты резиновых смесей смешивают при температуре в интервале от 100 до 180°С. При этом последовательность и момент добавления отдельных компонентов могут оказывать решающее влияние на свойства получаемой резиновой смеси. Затем с полученной таким путем резиновой смесью можно смешивать сшивающие агенты, обычно в смесителе закрытого типа либо на вальцах при температуре в интервале от 40 до 110°С, с получением невулканизованной резиновой смеси, так называемой сырой смеси, которую подвергают дальнейшей переработке на последующих технологических стадиях, таких, например, как формование и вулканизация.
Вулканизацию предлагаемых в изобретении резиновых смесей можно проводить при температуре в интервале от 80 до 220°С, предпочтительно от 130 до 190°С, при необходимости под давлением в пределах от 10 до 200 бар.
Предлагаемые в изобретении резиновые смеси могут использоваться для изготовления уплотнений, вибраторов, желобков для подъемных оконных стекол, радиаторов, садовых шлангов и шлангов для бытовой техники, труб, прокладок, приводных ремней, электроизоляции, диффузоров громкоговорителей, изделий, используемых в соединителях электрических кабелей, профилей, наружных оболочек электрических проводов, кровельных мембран, геомембран, резинотехнических изделий, эластификаторов для пластмасс, термопластов, вулканизатов и многих других продуктов. Предлагаемые в изобретении резиновые смеси могут использоваться для изготовления предохраняющих от атмосферных осадков уплотнений для любых транспортных средств. К таким уплотнениям относятся уплотнители дверей, уплотнители стекол, уплотнители крышек багажных отсеков и уплотнители крышек моторных отсеков. Предлагаемые в изобретении резиновые смеси могут использоваться для изготовления шлангов и трубок систем циркуляционного охлаждения на автомобилях. Помимо этого предлагаемые в изобретении резиновые смеси могут использоваться для изготовления трубок для подачи наддувочного воздуха в компрессорные двигатели внутреннего сгорания.
Объектом изобретения являются далее формованные изделия, изготавливаемые из предлагаемой в изобретении резиновой смеси путем вулканизации.
Преимущество предлагаемых в изобретении резиновых смесей состоит в наличии у них улучшенных характеристик сопротивления раздиру.
Примеры
В резиновых смесях использовали следующие соединения:
- 3-изоцианатопропил(триэтоксисилан) фирмы ABCR;
- этилхлорформиат и изопропилхлорформиат фирмы Isochem;
- 3-аминопропил(триэтоксисилан) фирмы Evonik Industries AG;
- фенилгидразин, раствор гипохлорита натрия (13%-ный), бром,
N-бромсукцинимид, пиридин, этанол, пентан, этилацетат, трет-бутилметиловый эфир, дихлорметан, Oxone®, раствор надуксусной кислоты (39%-ный в уксусной кислоте), 4-нитрофенилгидразин, гидроксид натрия, гидрохлорид трет-бутилгидразина фирмы Aldrich;
- Lutensol ТО 5 (этоксилированный изотридеканол) фирмы BASF;
- ацетонитрил и силикагель фирмы Merck.
Пример 1: Получение (EtO)3Si-(CH2)3-NH-C(=O)-N=N-фенила
а) Получение (EtO)3Si-(CH2)3-NH-C(=O)-NH-NH-фенила
178,5 г (1,6 моля) фенилгидразина в атмосфере аргона растворяли в 2000 мл этилацетата и охлаждали до 10°С. Далее к перемешиваемому раствору в течение 120 мин добавляли 416,6 г (1,6 моля) 3-изоцианатопропил(триэтоксисилана) таким образом, чтобы температура оставалась в пределах от 5 до 15°С. После этого удаляли охлаждающую баню, суспензии давали нагреться до комнатной температуры и перемешивали в течение 150 мин. Затем смесь концентрировали под пониженным давлением при 40°С. Далее добавляли 2000 мл пентана. Выпавшее в осадок твердое вещество отфильтровывали, промывали пентаном и сушили под вакуумом. Полученный таким путем продукт представлял собой белое твердое вещество (492,22 г, 87%) с чистотой по данным ЯМР-спектроскопии более 95 мол. %.
б) Получение (EtO)3Si-(CH2)3-NH-C(=O)-N=N-фенила
В атмосфере аргона 178 г (0,5 моля) продукта из примера 1а смешивали с 79,1 г (1 моль) пиридина и 650 мл дихлорметана и перемешивали при 0°С. Далее в течение 120 мин добавляли раствор 79,9 г (0,5 моля) брома в 150 мл дихлорметана, поддерживая температуру в пределах от 0 до 10°С. После этого удаляли охлаждающую баню и смесь перемешивали еще в течение 150 мин. Затем на роторном испарителе при 40°С под вакуумом удаляли летучие соединения. Далее добавляли 300 мл трет-бутилметилового эфира и отфильтровывали осадок. Фильтрат концентрировали под пониженным давлением (до 0,2 мбара), получая продукт в виде красного масла (171 г, 97%) с чистотой по данным ЯМР-спектроскопии более 90 мол. %.
Пример 2: Получение (EtO)3Si-(CH2)3-NH-C(=O)-N=N-фенила
a) Получение (EtO)3Si-(CH2)3-NH-C(=O)NH-NH-фенила
Раствор 5,2 г (50 ммолей) фенилгидразина в 40 мл воды охлаждали в атмосфере аргона до 5°С. Далее в течение 120 мин добавляли 12,3 г (50 ммолей) изоцианатопропил(триэтоксисилана), поддерживая температуру в пределах от 0 до 10°С. Выпавшее в осадок твердое вещество собирали путем фильтрации, промывали водой и сушили под вакуумом. Полученный таким путем продукт (15,5 г, 87%) представлял собой белое твердое вещество с чистотой более 85 мол. % (по данным ЯМР-спектроскопии).
б) Получение (EtO)3Si-(СН2)3-NH-С(=O)-N=N-фенила
В колбу в атмосфере аргона добавляли навеску продукта из примера 2а массой 17,8 г (50 ммолей) и навеску карбоната натрия массой 5,3 г (50 ммолей). Далее добавляли 50 мл дихлорметана, смесь перемешивали и охлаждали до 5°С. Затем в течение 60 мин малыми порциями добавляли в общей сложности 9,0 г (50 ммолей) N-бромсукцинимида, поддерживая температуру в пределах от 0 до 10°С.Реакционную смесь перемешивали в течение 120 мин при комнатной температуре. После этого удаляли растворитель под пониженным давлением и остаток растворяли в смеси из 20 мл дихлорметана и 80 мл пентана. Твердые вещества удаляли путем фильтрации. Фильтрат концентрировали под пониженным давлением (до 0,2 мбара). Полученный таким путем продукт представлял собой красное масло (12,1 г, 69%) с чистотой более 70 мол. % (по данным ЯМР-спектроскопии). Другие 20 мол. % приходились на димеризованные или олигомеризованные структуры целевого соединения.
Пример 3: Получение (EtO)3Si-(СН2)3-NH-С(=O)-N=N-фенила
54,1 г (0,5 моля) фенилгидразина в атмосфере аргона растворяли в 1000 мл этилацетата и охлаждали до 10°С. Далее к перемешиваемому раствору в течение 120 мин добавляли 123,7 г (0,5 моля) 3-изоцианатопропил(триэтоксисилана), поддерживая температуру в пределах от 5 до 15°С. Суспензию перемешивали в течение еще 150 мин, после чего добавляли 40,0 г (0,5 моля) пиридина. Затем в течение 30 мин малыми порциями добавляли в общей сложности 89,9 г (0,5 моля) N-бромсукцинимида, поддерживая температуру в пределах от 5 до 15°С. После этого удаляли охлаждающую баню, суспензии давали нагреться до комнатной температуры и перемешивали в течение 120 мин. Растворитель удаляли под пониженным давлением и остаток растворяли в смеси из 200 мл дихлорметана и 800 мл пентана. Твердые вещества удаляли путем фильтрации. Фильтрат концентрировали под пониженным давлением (до 0,2 мбара). Полученный таким путем продукт представлял собой красное масло (169 г, 96%) с чистотой более 90 мол. % (по данным ЯМР-спектроскопии).
Пример 4: Получение (EtO)3Si-(CH2)3-NH-C(=O)-N=N-фенила
а) Получение (EtO)3Si-(CH2)3-NH-C(=O)-NH-NH-фенила
178,5 г (1,6 моля) фенилгидразина в атмосфере аргона растворяли в 2000 мл этилацетата и охлаждали до 10°С. Далее к перемешиваемому раствору в течение 120 мин добавляли 416,6 г (1,6 моля) 3-изоцианатопропил(триэтоксисилана), поддерживая температуру в пределах от 5 до 15°С. После этого удаляли охлаждающую баню, суспензии давали нагреться до комнатной температуры и перемешивали в течение 150 мин. Растворитель удаляли под пониженным давлением и концентрат обрабатывали 2000 мл пентана. Затем отфильтровывали осадок, промывали его пентаном и сушили под вакуумом с получением целевого соединения в виде белого твердого вещества (492,2 г, 87%) с чистотой более 95 мол. %.
б) Получение (EtO)3Si-(CH2)3-NH-C(=O)-N=N-фенила
17,8 г (50 ммолей) продукта из примера 4а, 80 мл толуола, 15 г 0,05-молярного буферного раствора (цитрат натрия в лимонной кислоте, рН 5), 0,2 г (2,5 ммоля) пиридина и 0,3 г (2,5 ммоля) бромида натрия перемешивали в колбе в атмосфере аргона и охлаждали до 2°С. Далее в течение 120 мин добавляли 35,5 г (62 ммоля) раствора гипохлорита натрия (13%-ного), поддерживая температуру в пределах от 0 до 10°С. Реакционную смесь перемешивали в течение 90 мин при комнатной температуре. Фазы разделяли и водную фазу экстрагировали толуолом. Объединенные органические фазы сушили над сульфатом магния и фильтровали. Фильтрат концентрировали под пониженным давлением (до 0,2 мбара). Полученный таким путем продукт представлял собой красное масло (14,4 г, 81%) с чистотой более 64 мол. % (по данным ЯМР-спектроскопии). 12 мол. % приходились на димеризованные или олигомеризованные структуры целевого соединения, а около 20% приходились на непрореагировавший исходный материал и его димеризованные и олигомеризованные структуры.
Пример 5: Получение (EtO)3Si-(CH2)3-NH-C(=O)-N=N-фенила
a) Получение EtO-С(=O)-NH-NH-фенила (аналогично J. Chem. Soc. (С), 1970, 26)
Раствор 324 г (3 моля) фенилгидразина и 240 г (3 моля) пиридина в 1800 мл воды при перемешивании охлаждали до 2°С. Далее при интенсивном перемешивании по каплям добавляли 336 г (3 моля) этилхлорформиата, поддерживая температуру в пределах от 0 до 15°С. По завершении такой процедуры добавления суспензию перемешивали в течение 60 мин при комнатной температуре. Выпавшее в осадок твердое вещество собирали путем фильтрации, промывали водой и сушили под вакуумом (до 0,2 мбара, 55°С). Полученный таким путем продукт (464 г, 86%) представлял собой грязно-белое твердое вещество с чистотой более 95 мол. % (по данным ЯМР-спектроскопии) и температурой плавления 72°С.
б) Получение EtO-С(=O)-N=N-фенила
К перемешиваемому раствору 4,5 г (25 ммолей) 1-этоксикарбонил-2-фенилгидразина в 50 мл 80%-ной уксусной кислоты в течение 15 мин дозировали 6,1 г 39%-ного раствора надуксусной кислоты в уксусной кислоте, поддерживая температуру в пределах от 5 до 15°С. Смеси при ее перемешивании давали нагреться до комнатной температуры в течение последующих 120 мин. После этого на роторном испарителе под пониженным давлением удаляли летучие соединения. Остаток растворяли в 100 мл этилацетата, промывали водой (3 раза, порциями по 100 мл), сушили над сульфатом магния и фильтровали. После этого растворитель удаляли под пониженным давлением (до 0,2 мбара) с получением целевого соединения в виде красной жидкости (3,9 г, 88%) с чистотой более 95 мол. %.
в) Получение (EtO)3Si-(CH2)3-NH-C(=O)-N=N-фенила
В атмосфере аргона 2,2 г (10 ммолей) 3-аминопропил(триэтоксисилана) растворяли в 50 мл пентана и при перемешивании охлаждали до 5°С. Далее в течение 15 мин при температуре в пределах от 5 до 15°С добавляли 1,9 г (10 ммолей) продукта из примера 56. После этого удаляли охлаждающую баню и смесь перемешивали в течение 120 мин. Затем удаляли пентан и этанол под пониженным давлением (до 0,2 мбара). Полученный таким путем продукт (3,5 г, 98%) представлял собой красное масло с чистотой более 85 мол. %.
Пример 6: Получение (EtO)3Si-(CH2)3-NH-C(=O)-N=N-фенила
а) Получение EtO-С(=O)-NH-NH-фенила (аналогично J. Chem. Soc. (С), 1970. 26)
324 г (3 моля) фенилгидразина и 240 г (3 моля) пиридина при перемешивании растворяли в 1800 мл воды. Раствор охлаждали до 2°С, после чего в течение 60 мин по каплям добавляли 336 г (3 моля) этилхлорформиата, поддерживая температуру в пределах от 0 до 15°С. По завершении такой процедуры добавления суспензию перемешивали в течение 60 мин при комнатной температуре. Выпавшее в осадок твердое вещество собирали путем фильтрации, промывали водой и сушили под вакуумом (до 0,2 мбара, 55°С). Полученный таким путем продукт (464 г, 86%) представлял собой грязно-белое твердое вещество с чистотой более 95 мол. % (по данным ЯМР-спектроскопии) и температурой плавления 72°С.
б) Получение EtO-С(=O)-N=N-фенила
4,5 г (25 ммолей) продукта из примера 6а добавляли к смеси из 75 мл воды и 75 мл этанола. Смесь охлаждали до 10°С и перемешивали. Далее в течение 30 мин порциями добавляли 15,4 г (25 ммолей) продукта Oxone®, поддерживая температуру в пределах от 10 до 20°С. Смеси при ее перемешивании давали нагреться до комнатной температуры в течение последующих 180 мин. Затем смесь концентрировали под пониженным давлением с получением остатка объемом приблизительно 60 мл, который экстрагировали петролейным эфиром (3 раза, порциями по 50 мл). Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния и фильтровали. После этого растворитель удаляли под пониженным давлением (до 0,2 мбара) с получением целевого соединения в виде красной жидкости (3,8 г, 86%) с чистотой более 95 мол. %.
в) Получение (EtO)3Si-(CH2)3-NH-C(=O)-N=N-фенила
В атмосфере аргона 2,2 г (10 ммолей) 3-аминопропил(триэтоксисилана) при перемешивании растворяли в 50 мл пентана и охлаждали до 5°С. Далее в течение 15 мин порциями добавляли 1,9 г (10 ммолей) этил-2-фенилдиазенкарбоксилата, поддерживая температуру в пределах от 5 до 15°С. Смеси при ее перемешивании давали нагреться до комнатной температуры в течение последующих 120 мин. После этого на роторном испарителе под пониженным давлением (до 0,2 мбара) удаляли летучие соединения. Полученный таким путем продукт представлял собой красное масло (3,5 г, 98%) с чистотой более 85 мол. % (по данным ЯМР-спектроскопии).
Пример 7: Получение (EtO)3Si-(CH2)3-NH-C(=O)-N=N-фенила
а) Получение EtO-С(=O)-NH-NH-фенила (аналогично J. Chem. Soc. (С) 1970. 26)
324 г (3 моля) фенилгидразина и 240 г (3 моля) пиридина при перемешивании растворяли в 1800 мл воды. Раствор охлаждали до 2°С, после чего в течение 60 мин по каплям добавляли 336 г (3 моля) этилхлорформиата, поддерживая температуру в пределах от 0 до 15°С. По завершении такой процедуры добавления суспензию перемешивали в течение 60 мин при комнатной температуре. Выпавшее в осадок твердое вещество собирали путем фильтрации, промывали водой и сушили под вакуумом (до 0,2 мбара, 55°С). Полученный таким путем продукт (464 г, 86%) представлял собой грязно-белое твердое вещество с чистотой более 95 мол. % (по данным ЯМР-спектроскопии) и температурой плавления 72°С.
б) Получение EtO-С(=O)-N=N-фенила
К перемешиваемому раствору 108 г (0,6 моля) этил-2-фенилгидразинкарбоксилата в 900 мл этилацетата добавляли раствор 6,2 г (60 ммолей) бромида натрия в 20 мл воды. Смесь охлаждали до 10°С. Далее в течение 30 мин добавляли 429 г (0,75 ммоля) раствора гипохлорита натрия (13%-ного), поддерживая температуру в пределах от 10 до 20°С. Реакционную смесь перемешивали в течение 120 мин при комнатной температуре. Фазы разделяли, органическую фазу промывали насыщенным раствором хлорида натрия, сушили над сульфатом магния и фильтровали. Фильтрат концентрировали под пониженным давлением (до 0,2 мбара). Полученный таким путем продукт представлял собой красное масло (106 г, 99%) с чистотой более 95 мол. % (по данным ЯМР-спектроскопии).
в) Получение (EtO)3Si-(CH2)3-NH-C(=O)-N=N-фенила
В атмосфере аргона 122,5 г (0,55 моля) 3-аминопропил(триэтоксисилана) при перемешивании растворяли в 200 мл пентана и охлаждали до 5°С. Далее в течение 60 мин порциями добавляли 98,6 г (0,55 моля) этил-2-фенилдиазенкарбоксилата, поддерживая температуру в пределах от 5 до 15°С. Смеси при ее перемешивании давали нагреться до комнатной температуры в течение последующих 120 мин. После этого на роторном испарителе под пониженным давлением (до 0,2 мбара) удаляли летучие соединения.
Полученный таким путем продукт представлял собой красное масло (194,9 г, 99%) с чистотой более 85 мол. % (по данным ЯМР-спектроскопии).
Пример 8: Получение (EtO)3Si-(СН2)3-NH-С(=О)-N=N-фенила
а) Получение изо-PrO-С(=О)-NH-NH-фенила (аналогично DE 2246282)
324 г (3 моля) фенилгидразина и 238 г (3 моля) пиридина при перемешивании растворяли в 1800 мл воды. Раствор охлаждали до 2°С, после чего в течение 60 мин по каплям добавляли 368 г (3 моля) изопропилхлорформиата, поддерживая температуру в пределах от 0 до 15°С. По завершении такой процедуры добавления суспензию перемешивали в течение 60 мин при комнатной температуре. Выпавшее в осадок твердое вещество собирали путем фильтрации, промывали водой и сушили под вакуумом (до 0,2 мбара, 55°С). Полученный таким путем продукт (582 г, 98%) представлял собой грязно-белое твердое вещество с чистотой более 90 мол. % (по данным ЯМР-спектроскопии).
б) Получение изо-PrO-С(=O)-N=N-фенила
4,9 г (25 ммолей) изопропил-2-фенилгидразинкарбоксилата и 2,0 г (25 ммолей) пиридина растворяли в 50 мл дихлорметана. Перемешиваемый раствор охлаждали до 5°С. Далее порциями добавляли 3,8 г (13 ммолей) 1,3-дибром-5,5-диметилгидантоина, поддерживая температуру в пределах от 0 до 10°С. Реакционную смесь перемешивали в течение 120 мин при комнатной температуре. Затем под пониженным давлением удаляли летучие соединения. Остаток растворяли в 50 мл пентана. Осадок удаляли путем фильтрации. Фильтрат концентрировали под пониженным давлением (до 0,2 мбара). Полученный таким путем продукт представлял собой красное масло (3,0 г, 63%) с чистотой более 95 мол. % (по данным ЯМР-спектроскопии).
в) Получение (EtO)3Si-(CH2)3-NH-C(=O)-N=N-фенила
В атмосфере аргона 3,3 г (15 ммолей) 3-аминопропил(триэтоксисилана) при перемешивании растворяли в 20 мл дихлорметана и раствор охлаждали до 5°С. Далее в течение 20 мин порциями добавляли 2,9 г (15 ммолей) изопропил-2-фенилдиазенкарбоксилата, поддерживая температуру в пределах от 5 до 15°С. Смеси при ее перемешивании давали нагреться до комнатной температуры в течение последующих 120 мин. После этого на роторном испарителе под пониженным давлением (до 0,2 мбара) удаляли летучие соединения. Полученный таким путем продукт представлял собой красное масло (5,4 г, 98%) с чистотой более 70 мол. % (по данным ЯМР-спектроскопии).
Пример 9: Получение (EtO)3Si-(CH2)3-NH-C(=O)-N=N-фенил-n-NO2
а) Получение (EtO)3Si-(CH2)3-NH-C(=O)-NH-NH-фенил-n-NO2
10,3 г (65 молей) 4-нитрофенилгидразина в атмосфере аргона растворяли в 50 мл этилацетата и охлаждали до 10°С. Далее к перемешиваемому раствору добавляли 17,0 г (65 ммолей) 3-изоцианатопропил(триэтоксисилана), поддерживая температуру в пределах от 5 до 15°С. Затем удаляли охлаждающую баню, смеси давали нагреться до комнатной температуры и перемешивали в течение 140 мин. После этого растворитель удаляли под пониженным давлением и концентрат обрабатывали 100 мл пентана. Осадок отфильтровывали, промывали пентаном и сушили под вакуумом с получением целевого соединения в виде красно-коричневого твердого вещества (16,7 г, 61%) с чистотой более 95 мол. %.
б) Получение (EtO)3Si-(CH2)3-NH-C(=O)-N=N-фенил-n-NO2
10,0 г (25 ммолей) продукта из примера 9а и 2,0 г (25 ммолей) пиридина в атмосфере аргона растворяли в 50 мл этилацетата. Перемешиваемый раствор охлаждали до 15°С.Далее в течение 25 мин малыми порциями добавляли в общей сложности 4,5 г (25 ммолей) N-бромсукцинимида, поддерживая температуру в пределах от 10 до 20°С. Реакционную смесь перемешивали в течение 120 мин при комнатной температуре. После этого растворитель удаляли под пониженным давлением и остаток растворяли в 100 мл трет-бутилметилового эфира. Твердые вещества удаляли путем фильтрации. Фильтрат концентрировали под пониженным давлением (до 0,2 мбара). Полученный таким путем продукт представлял собой красное твердое вещество (9,9 г, 82%) с чистотой более 85 мол. % (по данным ЯМР-спектроскопии).
Пример 10: Получение (EtO)2(C13H27[OC2H4]5O)Si-(CH2)3-NH-C(=O)-N=N-фенила
а) Получение (EtO)2(C13H27[OC2H4]5O)Si-(CH2)3-NH2 с переэтерифицированным по одной эфирной группе АМЕО
В 2-литровую четырехгорлую колбу с дистилляционным мостом, мешалкой kpg и термометром при комнатной температуре в атмосфере азота добавляли навеску 3-аминопропил(триэтоксисилана) (АМЕО) массой 400 г (1,8 моля) и навеску этоксилированного спирта C13H27[OC2H4]nOH, где n составляет в среднем 5 (продукт Lutensol ТО 5 фирмы BASF), массой 759 г (1,8 моля). Смесь при перемешивании нагревали до 140°С. Образующийся этанол непрерывно удаляли при атмосферном давлении. Через 1 ч давление постепенно снижали до 15 мбар в течение 5,5 ч. Полученный таким путем продукт (1,05 кг, 98%) представлял собой желтоватое масло со средней степенью переэтерификации, равной 1 по данным ЯМР-спектроскопии.
б) Получение (EtO)2(C13H27[OC2H4]5O)Si-(CH2)3-NH-C(=O)-N=N-фенила
8,16 г (13,7 ммоля) продукта из примера 10а, а именно (EtO)2(C13H27[OC2H4]5O)Si-(CH2)3-NH2, в атмосфере аргона при перемешивании растворяли в 10 мл пентана и раствор охлаждали до 5°С. Далее в течение 15-30 мин порциями добавляли 2,44 г (13,7 ммоля) этил-2-фенилдиазенкарбоксилата, поддерживая температуру в пределах от 0 до 15°С. Смеси при ее перемешивании давали нагреться до комнатной температуры в течение последующих 180 мин. После этого на роторном испарителе под пониженным давлением (до 0,2 мбара) удаляли летучие соединения. Полученный таким путем продукт представлял собой красное масло (9,8 г, 98%) с чистотой более 90 мол. % (по данным ЯМР-спектроскопии).
Пример 11: Получение (EtO)3Si-(CH2)3-NH-C(=O)-N=N-C(CH3)3
а) Получение EtO-С(=O)-NH-NH-трет-бутила (аналогично М.С. Chaco, N. Rabjohn. J. Org. Chem., 1962, 27(8). cc. 2765-2767)
К перемешиваемому раствору 74,8 г (0,6 моля) гидрохлорида трет-бутилгидразина в 60 мл воды осторожно добавляли раствор 24,0 г (0,6 моля) гидроксида натрия в 60 мл воды. Далее добавляли 47,5 г (0,6 моля) пиридина и смесь охлаждали до 0°С. Затем при этой температуре в течение 10 мин добавляли 65,13 г (0,6 моля) этилхлорформиата. После этого удаляли охлаждающую баню и смесь перемешивали при комнатной температуре в течение последующих 180 мин. Далее добавляли 100 мл дихлорметана, фазы разделяли и водную фазу экстрагировали дихлорметаном (60 мл, 3 раза). Объединенные органические фазы сушили над сульфатом магния и фильтровали. После этого под пониженным давлением удаляли органические летучие соединения. Полученный таким путем продукт представлял собой бесцветную жидкость (88,6 г, 93%) с чистотой более 85 мол. % (по данным ЯМР-спектроскопии).
б) Получение EtO-C(=O)-N=N-трет-бутила (аналогично М.С. Chaco, N. Rabjohn, J. Org. Chem., 1962, 27(8), cc. 2765-2767)
6,01 г (37,5 ммоля) этил-2-трет-бутилгидразинкарбоксилата и 3,27 г (41,25 ммоля) пиридина в атмосфере аргона растворяли в 100 мл этилацетата. Перемешиваемый раствор охлаждали до 0°С. Далее в течение 15 мин порциями добавляли 6,66 г (37,5 ммоля) N-бромсукцинимида, поддерживая температуру в пределах от 0 до 10°С. Смеси при ее перемешивании давали нагреться до комнатной температуры в течение последующих 135 мин. После этого на роторном испарителе под пониженным давлением удаляли летучие соединения. Затем добавляли гексан и осадок удаляли путем фильтрации. Сырой продукт дистиллировали под пониженным давлением (30°С, 3 мбара) с получением указанного в заголовке соединения в виде желтой жидкости (5,28 г, 89%) с чистотой более 95 мол. %.
в) Получение (EtO)3Si-(CH2)3-NH-C(=O)-N=N-C(CH3)3
В атмосфере аргона 21,0 г (95 ммолей) 3-аминопропил(триэтоксисилана) растворяли в 20 мл пентана и при перемешивании охлаждали до 5°С. Далее в течение 15 мин при температуре в пределах от -5 до 15°С добавляли 15,0 г (95 ммолей) этил-2-трет-бутилдиазенкарбоксилата. После этого удаляли охлаждающую баню и смесь перемешивали в течение 180 мин. Затем под пониженным давлением (до 0,2 мбара) удаляли пентан и этанол. Полученный таким путем продукт (28,7 г, 91%) представлял собой желтое масло с чистотой более 95 мол. %.
Пример 12: Резиновые смеси
Основная рецептура резиновых смесей приведена ниже в таблице 1. При этом величина "част./100 част, каучука" представляет собой массовую долю соответствующего компонента в пересчете на 100 частей используемого сырого каучука.
Общий способ приготовления резиновых смесей и получения их вулканизатов описан в справочнике "Rubber Technology Handbook", W. Hofmann, изд-во Hanser Verlag, 1994.
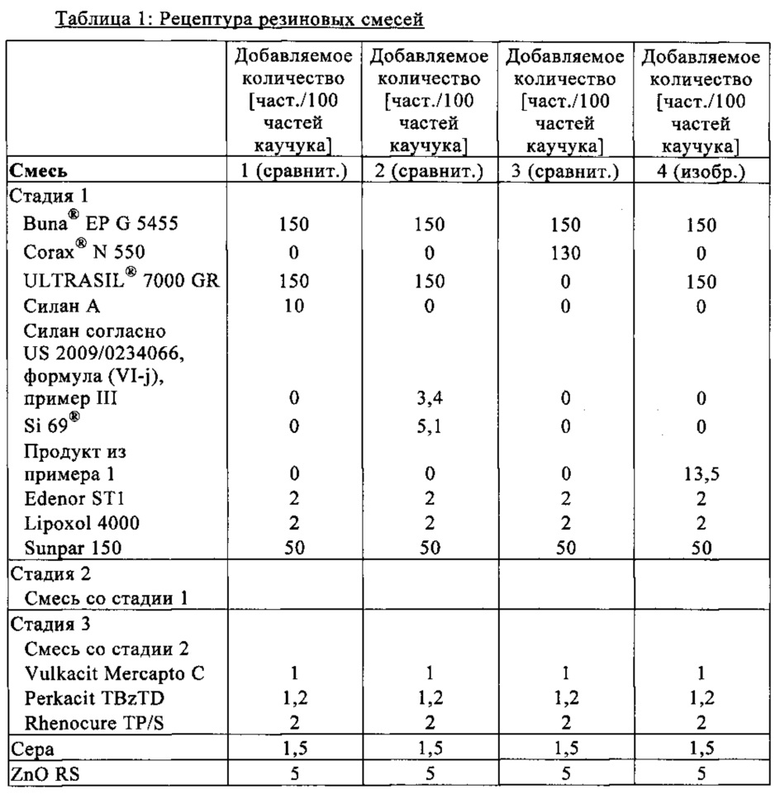
Полимер Buna® ЕР G 5455 представляет собой тройной этилен-пропиленовый сополимер со средней ненасыщенностью (содержание этилиденнорборнена составляет 4,3), содержащий парафиновое масло в количестве 50 част./100 част, каучука и выпускаемый фирмой Lanxess. Его вязкость по Муни (UML (1+4) 125°С) составляет 46.
Продукт ULTRASIL® 7000 GR представляет собой высоко дисперсный диоксид кремния, выпускаемый фирмой Evonik Industries AG и имеющий БЭТ-поверхность 170 м2/г.
Аппрет Si 69 представляет собой бис-(триэтоксисилилпропил)полисульфид, выпускаемый фирмой Evonik Industries AG.
Силан А представляет собой (CH3CH2O)3Si-(CH2)3-NH-C(O)-N=N-C(O)-NH-(CH2)3-Si(OCH2CH3)3 и описан в ЕР 2508559.
Продукт Edenor ST1 представляет собой стеариновую кислоту, выпускаемую фирмой KemCare.
Продукт Lipoxol 4000 представляет собой полиэтиленгликоль 4000, выпускаемый фирмой Sasol, продукт Sunpar 150 представляет собой парафиновое масло, выпускаемое фирмой Holly Corporation, продукт Vulkacit Mercapto С представляет собой 2-меркаптобензотиазол (МБТ), выпускаемый фирмой Lanxess, продукт Perkacit TBzTD (тетрабензилтиурамдисульфид) выпускается фирмой Flexsys N.V., продукт Rhenocure TP/S представляет собой диалкилдитиофосфат цинка (67%), связанный с диоксидом кремния (33%), и выпускается фирмой RheinChemie.
Резиновые смеси приготавливают в резиносмесителе закрытого типа, используя оборудование и условия, указанные ниже в таблице 2.


Резинотехнические свойства исследуют по методам, представленным в таблице 3.
Определение коэффициента диспергирования
Коэффициент диспергирования можно определять топографическим методом, описанным у A. Wehmeier в "Entwicklung eines Verfahrens zur Charakterisierung der Füllstoffdispersion in Gummimischungen mittels einer Oberflächentopographie", диссертация, защищенная в 1998 г. при Мюнстерском техническом университете, г. Штайнфурт, химико-технологический факультет, и в "Filler dispersion Analysis by Topography Measurements", фирма Degussa AG, Applied Technology Advanced Fillers, технический отчет TR 820 (Technical Report TR 820). Альтернативно этому коэффициент диспергирования можно также определять DIAS-методом (оптически), разработанным Немецким институтом технологии резины ("Deutsches Institut für Kautschuktechnologie") в г. Ганновер, Германия (см. Н. Geisler, DIK aktuell, 1-е изд., 1997, и Medalia, Rubber Age, апрель 1965). Максимально достижимая степень диспергирования составляет 100%, а наихудшая соответственно будет теоретически равняться 0%. Диоксиды кремния, у которых коэффициент диспергирования составляет не менее 90%, классифицируют как высокодиспергируемые (ВД).
Коэффициент диспергирования (КД) можно вычислить на основании результатов анализа топографии поверхности по следующей формуле:
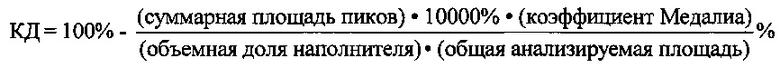 ,
,
где
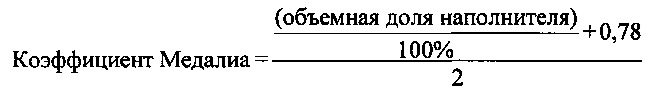 .
.
В приведенных выше формулах коэффициент диспергирования указывается в %, суммарная площадь пиков (мера шероховатости) указывается в мм2, объемная доля наполнителя указывается в %, а общая анализируемая площадь указывается в мм2.
Результаты исследования свойств резиновых смесей представлены ниже в таблице 4.
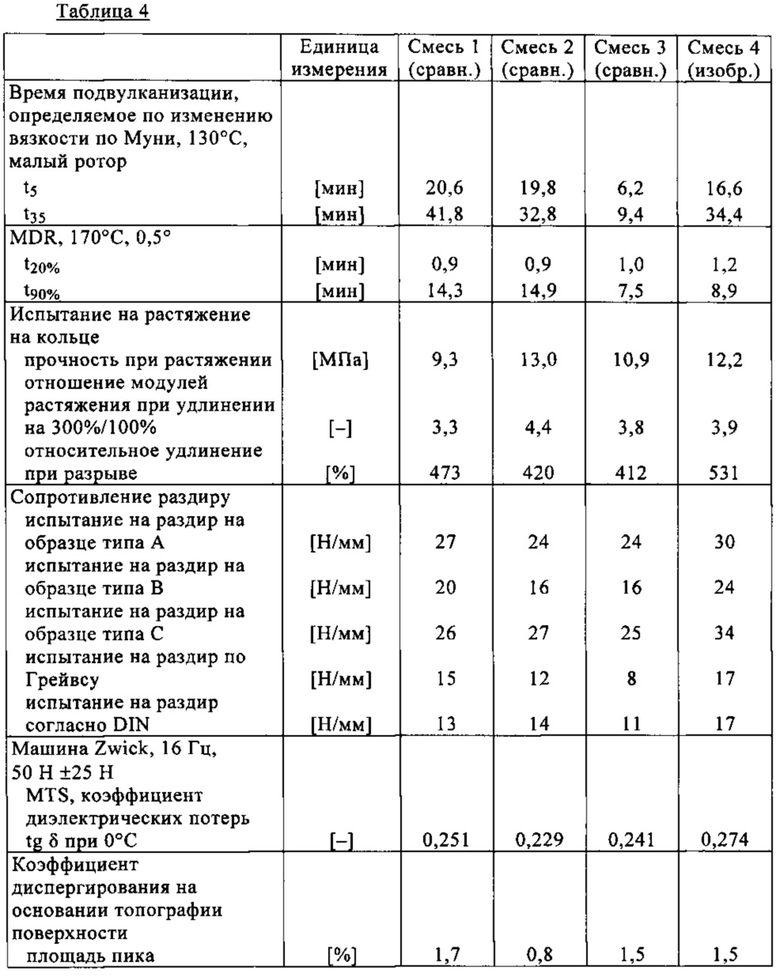
Как следует из приведенных в таблице 4 данных, смесь 4, содержащая функционализованный азокарбонилом силан общей формулы I, обладает явно улучшенными показателями сопротивления раздиру.
Пример 13: Получение (EtO)3Si-(CH2)3-NH-C(=O)-N=N-фенил-n-NO2
а) Получение EtO-С(=O)-NH-NH-фенил-n-NO2 (аналогично Р. Urankar, М. Steinbücher, J. Kosjek, J. Kosmrlj, Tetrahedron, 2010, 66, cc. 2602-2613)
Раствор 130 г (~70%, 0,59 моля) 4-нитрофенилгидразина и 79,1 г (1 моль) пиридина в 500 мл ацетонитрила при перемешивании охлаждали до 2°С. Далее при интенсивном перемешивании по каплям добавляли 79,0 г (0,73 моля) этилхлорформиата, поддерживая температуру в пределах от 0 до 15°С. По завершении такой процедуры добавления суспензию перемешивали в течение 120 мин при комнатной температуре. После этого под пониженным давлением удаляли большую часть летучих соединений. Затем добавляли воду (500 мл), выпавшее в осадок твердое вещество собирали путем фильтрации, промывали водой и сушили под вакуумом. Полученный таким путем продукт (88,2 г, 88%) представлял собой коричневое твердое вещество с чистотой более 95%.
б) Получение EtO-С(=O)-N=N-фенил-n-NO2 (аналогично P. Urankar, М. Steinbücher, J. Kosjek, J. Kosmrlj, Tetrahedron, 2010, 66, cc. 2602-2613)
144 г (0,64 моля) этил-2-(4-нитрофенил)гидразинкарбоксилата растворяли в 400 мл дихлорметана. Далее добавляли 55,7 г (0,70 моля) пиридина и смесь при перемешивании охлаждали до 5°С. Затем порциями добавляли 115,0 г (0,64 моля) N-бромсукцинимида, поддерживая температуру в пределах от 0 до 10°С. Смесь перемешивали в течение последующих 90 мин при комнатной температуре. После этого смесь промывали насыщенным раствором NH4Cl (200 мл), раствором NaHCO3 (200 мл) и водой (200 мл). Затем под пониженным давлением частично удаляли органические летучие соединения и смесь фильтровали через силикагель (циклогексан/этилацетат в соотношении 2:1). В завершение под вакуумом (до 0,02 мбара) удаляли растворитель с получением целевого соединения в виде коричневого твердого вещества (139,5 г, 0,63 моля, 98%) с чистотой более 95%.
в) Получение (EtO)3Si-(CH2)3-NH-C(=O)-N=N-фенил-n-NO2
В атмосфере аргона 2,98 г (13,4 ммоля) 3-аминопропил(триэтоксисилана) растворяли в 30 мл ацетонитрила и при перемешивании охлаждали до -15°С. Далее в течение 30 мин при температуре в пределах от -5 до 15°С добавляли раствор 3,00 г (13,4 ммоля) этил-2-(4-нитрофенил)диазенкарбоксилата в ацетонитриле (10 мл). После этого удаляли охлаждающую баню и смесь перемешивали в течение 180 мин. Затем под пониженным давлением (до 0,2 мбара) удаляли летучие соединения. Полученный таким путем продукт (5,23 г, 98%) представлял собой твердое вещество с цветом от красного до коричневого (чистота более 95 мол. %).
Пример 14: Резиновые смеси
Основная рецептура резиновых смесей приведена ниже в таблице 5. При этом величина "част./100 част, каучука" представляет собой массовую долю соответствующего компонента в пересчете на 100 частей используемого сырого каучука.
Общий способ приготовления резиновых смесей и получения их вулканизатов описан в справочнике "Rubber Technology Handbook", W. Hofmann, изд-во Hanser Verlag, 1994.
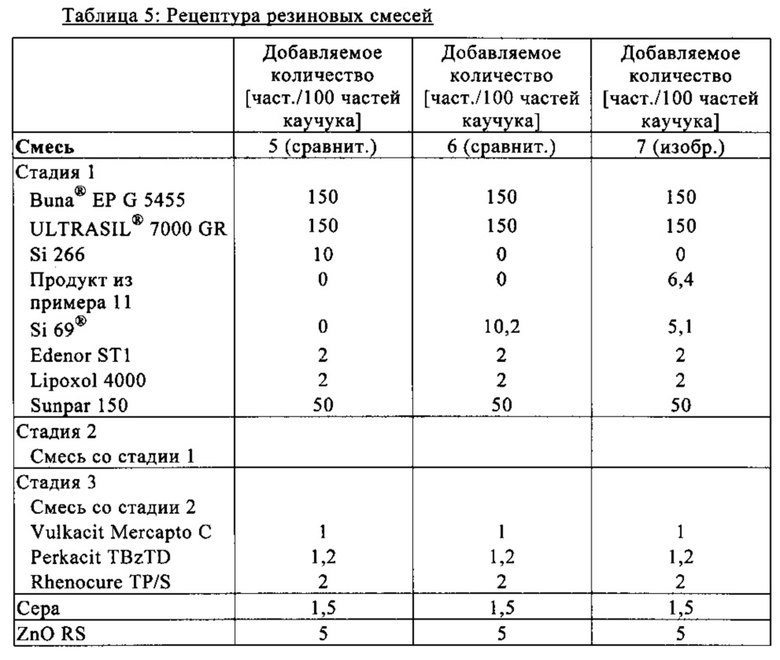
Аппрет Si 266 представляет собой бис-(триэтоксисилилпропил)дисульфид, выпускаемый фирмой Evonik Industries AG.
Резиновые смеси приготавливают в резиносмесителе закрытого типа, используя оборудование и условия, указанные ниже в таблице 6.


Резинотехнические свойства исследуют по методам, представленным в таблице 3.
Результаты исследования свойств резиновых смесей представлены ниже в таблице 7 (вулканизация при 165°С в течение 20 мин).
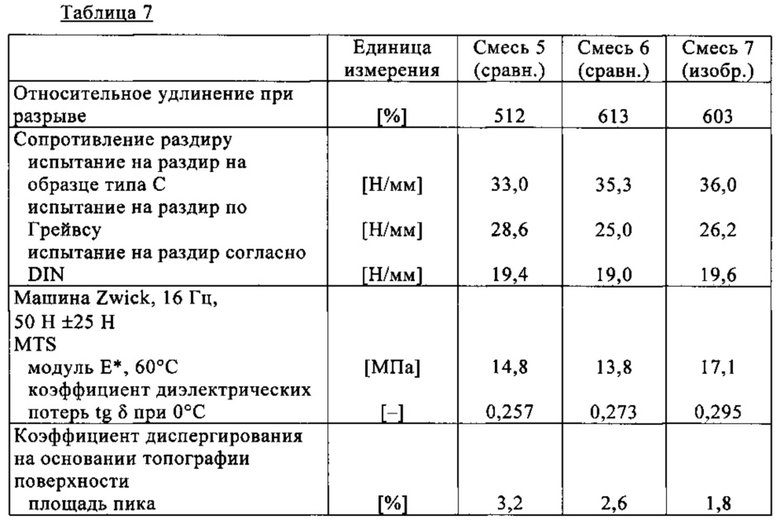
Как следует из приведенных в таблице 7 данных, смесь 7, содержащая предлагаемый в изобретении силан, обладает явно улучшенными показателями сопротивления раздиру, лучшими показателями сопротивления проскальзыванию на мокрой дороге, улучшенными показателями динамической жесткости и лучшей степенью диспергирования наполнителя.
| название | год | авторы | номер документа |
|---|---|---|---|
| РЕЗИНОВЫЕ СМЕСИ | 2012 |
|
RU2612148C2 |
| Карбамидсодержащие меркаптосиланы, способ их получения и их применение | 2015 |
|
RU2678701C2 |
| КРЕМНИЙСОДЕРЖАЩИЕ АЗОДИКАРБОКСАМИДЫ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2016 |
|
RU2727928C2 |
| КАРБАМИДСОДЕРЖАЩИЕ СИЛАНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2678320C2 |
| КАРБАМИДСОДЕРЖАЩИЕ СИЛАНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2677482C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАРБАМИДСОДЕРЖАЩИХ СИЛАНОВ | 2015 |
|
RU2679800C2 |
| РЕЗИНОВЫЕ СМЕСИ | 2016 |
|
RU2734414C2 |
| СПОСОБ ПОЛУЧЕНИЯ КРЕМНИЙСОДЕРЖАЩИХ АЗОДИКАРБАМИДОВ | 2011 |
|
RU2559876C2 |
| РЕЗИНОВЫЕ СМЕСИ | 2012 |
|
RU2619696C2 |
| БЕНЗОТИАЗОЛСОДЕРЖАЩИЕ СИЛАНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2762110C2 |
Изобретение относится к функционализованным азокарбонилом силанам общей формулы (I) (R1)3-a(R2)aSi-RI-NH-C(O)-N=N-R4. Функционализованные азокарбонилом силаны получают способом, при осуществлении которого на первой стадии гидразин формулы H2N-NH-R4 подвергают взаимодействию с изоцианатосиланом общей формулы (R1)3-a(R2)aSi-RI-NCO и на второй стадии полученный на первой стадии продукт окисляют окислителем или на первой стадии гидразин формулы H2N-NH-R4 подвергают взаимодействию с ацилгалогенидом общей формулы Cl-C(O)-O-R5, на второй стадии полученный на первой стадии продукт окисляют окислителем и на третьей стадии полученный на второй стадии продукт подвергают взаимодействию с аминосиланом общей формулы (R1)3-a(R2)aSi-RI-NH2. Изобретение позволяет улучшить сопротивление раздиру резин, содержащих силаны. 7 н. и 8 з.п. ф-лы, 7 табл.
1. Функционализованные азокарбонилом силаны общей формулы I
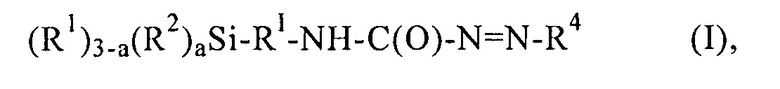
где R1 в каждом случае независимо обозначает незамещенные алкильные группы с C1-C18,
R2 в каждом случае независимо обозначает незамещенную алкоксигруппу с C1-C18 или группу простого алкилового полиэфира O(CH2-CH2-O)n-R3 или O(CH(CH3)-CH2-O)n-R3, где n в среднем составляет от 1 до 18, а
R3 в каждом случае независимо представляет собой разветвленную либо неразветвленную, насыщенную одновалентную углеводородную цепь с С1-С32,
RI обозначает разветвленную либо неразветвленную, насыщенную алифатическую двухвалентную углеводородную группу с С1-С30,
а обозначает число 1, 2 или 3,
R4 обозначает замещенный либо незамещенный фенил или замещенный либо незамещенный алкил.
2. Функционализованные азокарбонилом силаны общей формулы I по п. 1, отличающиеся тем, что R2 представляет собой этоксигруппу, а индекс обозначает 3.
3. Функционализованные азокарбонилом силаны общей формулы I по п. 1 или 2, отличающиеся тем, что R1 представляет собой -СН2СН2СН2-.
4. Функционализованные азокарбонилом силаны общей формулы I по одному из пп. 1-3, отличающиеся тем, что R4 представляет собой фенил, нитрофенил или трет-бутил.
5. Функционализованные азокарбонилом силаны общей формулы I по п. 1, отличающиеся тем, что они представляют собой (CH3CH2O)3Si-(CH2)3-NH-C(O)-N=N-фенил.
6. Функционализованные азокарбонилом силаны общей формулы I по п. 1, отличающиеся тем, что они представляют собой (CH3CH2O)3Si-(CH2)3-NH-C(O)-N=N-C(CH3)3.
7. Функционализованные азокарбонилом силаны общей формулы I по п. 1, отличающиеся тем, что они представляют собой (CH3CH2O)3Si-(СН2)3-NH-С(O)-N=N-(n-нитрофенил).
8. Способ получения функционализованных азокарбонилом силанов общей формулы I по одному из пп. 1-7, отличающийся тем, что на первой стадии гидразин формулы II
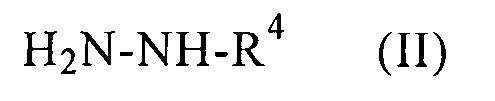
подвергают взаимодействию с изоцианатосиланом общей формулы III
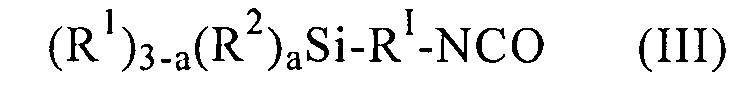
и на второй стадии полученный на первой стадии продукт окисляют окислителем, при этом R1, R2, R4, RI и а имеют указанные в п. 1 значения.
9. Способ по п. 8, отличающийся тем, что в качестве окислителя на второй стадии используют NaOCl, бром, N-бромсукцинимид, надуксусную кислоту, 1,3-дибром-5,5-диметилгидантоин или (мета)периодат тетрабутиламмония.
10. Способ получения функционализованных азокарбонилом силанов общей формулы I по одному из пп. 1-7, отличающийся тем, что на первой стадии гидразин формулы II
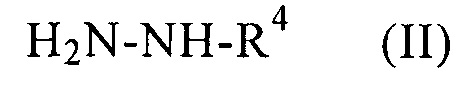
подвергают взаимодействию с ацилгалогенидом общей формулы V
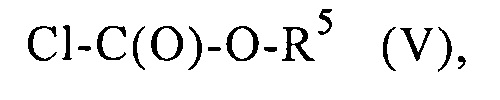
на второй стадии полученный на первой стадии продукт окисляют окислителем и на третьей стадии полученный на второй стадии продукт подвергают взаимодействию с аминосиланом общей формулы (VI)
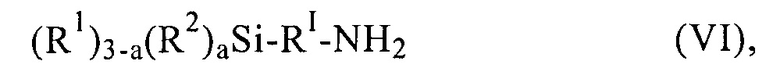
при этом R1, R2, R4, RI и а имеют указанные в п. 1 значения, a R5 обозначает арил или C1-С30алкил.
11. Способ по п. 10, отличающийся тем, что в качестве окислителя на второй стадии используют бром, N-бромсукцинимид, надуксусную кислоту, пероксимоносульфат калия, NaOCl, 1,3-дибром-5,5-диметилгидантоин или (мета)периодат тетрабутиламмония.
12. Резиновая смесь, отличающаяся тем, что она содержит
(A) по меньшей мере один каучук, выбранный из группы, включающей сополимер этилена, пропилена и диенового мономера (СКЭПТ), сополимер этилена с пропиленом (СКЭП), хлоропреновый каучук (ХК), хлорполиэтилен (ХП), хлорированный сополимер изобутена и изопрена (хлорбутилкаучук) (СКИИХ), хлорсульфированный полиэтилен (ХСПЭ), сополимер этилена с винилацетатом (ЕАМ), сополимер алкилакрилата (САА), полиэфироуретан со сложноэфирными группами (AU), полиэфироуретан с простыми эфирными группами (EU), бромированный сополимер изобутена и изопрена (бромбутилкаучук) (СКИИБ), полихлортрифторэтилен (CFM), сополимер изобутена и изопрена (бутилкаучук, СКИИ), изобутеновый каучук (IM), полиизопрен (СКИ), термопластичный полиэфироуретан со сложноэфирными группами (YAU), термопластичный полиэфироуретан с простыми эфирными группами (YEU), кремнийорганический каучук с метильными группами в полимерной цепи (MQ), гидрированный бутадиен-нитрильный каучук (СКНГ), бутадиен-нитрильный каучук (СКН) и карбоксилатный бутадиен-нитрильный каучук (СКНК), предпочтительно сополимер этилена, пропилена и диенового мономера (СКЭПТ),
(Б) по меньшей мере один оксидный наполнитель и
(B) по меньшей мере один функционализованный азокарбонилом силан общей формулы I по одному из пп. 1-7.
13. Способ приготовления резиновой смеси по п. 12, отличающийся тем, что между собой смешивают по меньшей мере один каучук, выбранный из группы, включающей сополимер этилена, пропилена и диенового мономера (СКЭПТ), сополимер этилена с пропиленом (СКЭП), хлоропреновый каучук (ХК), хлорполиэтилен (ХП), хлорированный сополимер изобутена и изопрена (хлорбутилкаучук) (СКИИХ), хлорсульфированный полиэтилен (ХСПЭ), сополимер этилена с винилацетатом (ЕАМ), сополимер алкилакрилата (АХП), полиэфироуретан со сложноэфирными группами (AU), полиэфироуретан с простыми эфирными группами (EU), бромированный сополимер изобутена и изопрена (бромбутилкаучук) (СКИИБ), полихлортрифторэтилен (CFM), сополимер изобутена и изопрена (бутилкаучук, СКИИ), изобутеновый каучук (IM), полиизопрен (СКИ), термопластичный полиэфироуретан со сложноэфирными группами (YAU), термопластичный полиэфироуретан с простыми эфирными группами (YEU), кремнийорганический каучук с метальными группами в полимерной цепи (MQ), гидрированный бутадиен-нитрильный каучук (СКНГ), бутадиен-нитрильный каучук (СКН) и карбоксилатный бутадиен-нитрильный каучук (СКНК), предпочтительно сополимер этилена, пропилена и диенового мономера (СКЭПТ), по меньшей мере один оксидный наполнитель и по меньшей мере один функционализованный азокарбонилом силан общей формулы I.
14. Применение резиновой смеси по п. 12 для изготовления формованных изделий.
15. Применение резиновой смеси по п. 12 в таких изделиях как, уплотнения, предохраняющие от атмосферных осадков уплотнения, уплотнители дверей, уплотнители стекол, уплотнители крышек багажных отсеков, уплотнители крышек моторных отсеков, вибраторы, желобки для подъемных оконных стекол, радиаторы, гибкие трубопроводы, садовые шланги и шланги для бытовой техники, трубки, прокладки, приводные ремни, электроизоляция, диффузоры громкоговорителей, изделия, используемые в соединителях электрических кабелей, профили, наружные оболочки электрических проводов, кровельные мембраны, геомембраны, пневматические рессоры, покрытия для различных валков, конвейерные ленты, резинотехнические изделия, эластификаторы для пластмасс, термопласты, трубки систем циркуляционного охлаждения и трубки для подачи наддувочного воздуха в компрессорные двигатели внутреннего сгорания.
| СПОСОБ ОБЗОРА ПРОСТРАНСТВА РАДИОЛОКАЦИОННОЙ СТАНЦИЕЙ | 2012 |
|
RU2508559C2 |
| US 4952641 A1, 28.08.1990 | |||
| US 4118367 A1, 03.10.1978 | |||
| RU 2002120188 A, 10.01.2004. | |||

