Область техники
Настоящее изобретение относится к новым способам получения производных триазина, пиримидина и пиридина, к их промежуточным соединениям и к получению промежуточных соединений.
Уровень техники
В WO 2010/052569 описаны некоторые производные триазина, пиримидина и пиридина, обладающие ингибирующими свойствами в отношении PI3K и mTOR, их применение в качестве лекарственных средств и способы их получения. Описанные способы получения подходят для надежного получения описанных соединений, но только в лабораторных масштабах.
Одно конкретное соединение триазина, описанное в WO 2010/052569, представляет собой двойной ингибитор фосфатидилинозитол 3-киназы/mTOR 5-(4,6-диморфолино-1,3,5-триазин-2-ил)-4-(трифторметил)пиридин-2-амин 1.

Остается потребность в новых твердых формах соединения 1, подходящих в качестве активных ингредиентов для лекарственных средств, поскольку указанное соединение до сих пор было доступно только в виде масла с низкой чистотой.
Биарильные структуры, такие как соединение 1, часто получают с использованием реакций кросс-сочетания. Среди них, реакция Сузуки обычно является предпочтительной вследствие доступности и стабильности борорганических реагентов. Тем не менее, борорганические реагенты, содержащие свободные амины, представляют собой особую проблему для кросс-сочетания, поскольку они способны отравлять катализатор на основе палладия и, следовательно, снижать выход целевого продукта. Кроме того, наличие свободного амина, как правило, приводит к маслянистым реагентам, которые не так легко выделить в чистом виде, и, прежде всего, с которыми трудно работать в случае крупномасштабных синтезов.
Сущность изобретения
В настоящем изобретении предложены улучшенные способы получения производных триазинов, пиримидинов и пиридинов формулы (II), новые промежуточные соединения, подходящие для таких способов и способам получения таких промежуточных соединений.
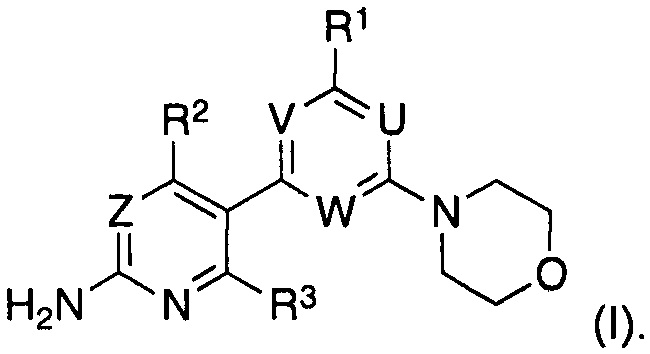
Таким образом, в одном аспекте, изобретение относится к способу получения соединения формулы (I)

или его стереомера, таутомера или соли, где,
U представляет собой CRU или N, где RU выбран из группы, состоящей из водорода, циано, галогена, метила и трифторметила;
V представляет собой CRV или N, где RV выбран из группы, состоящей из водорода, циано, галогена, метила и трифторметила;
W представляет собой CRW или N, где RW выбран из группы, состоящей из водорода, циано, галогена, метила и трифторметила;
при условии, что по меньшей мере один из U, V и W представляет собой N;
Z представляет собой CRZ или N, где RZ выбран из группы, состоящей из водорода, циано, галогена, метила и трифторметила;
R1 выбран из группы, состоящей из водорода, галогена и -N(RT)RS, где RT и RS представляют собой водород или C1-C7-алкил, или где RT и RS вместе с атомом азота, к которому они присоединены, образуют а C3-C8 моно- или бициклическое гетероциклическое кольцо, необязательно содержащее один или более дополнительных атомов в кольце, выбранных из N, O или S, где указанное гетероциклическое кольцо необязательно замещено одной или более группами, независимо выбранными из C1-C7-алкила или C3-C7-циклоалкила;
R2 выбран из группы, состоящей из водорода, циано, галогена, метила и трифторметила; и
R3 представляет собой водород или галоген,
характеризующийся тем, что соединение формулы (II)

где
Y2B представляет собой остаток ациклической борной кислоты, ациклического сложного эфира борной кислоты или циклического сложного эфира борной кислоты, и R2 и R3 являются такими, как определено для соединения формулы (I);
R4 представляет собой водород, C1-C7-алкил или C5-C7-циклоалкил;
R5 и R6 представляют собой C1-C7-алкил, или R5 и R6 вместе представляют собой C4-C6-циклоалкил;
и скрещенная двойная связь между N и C(R4)N указывает на цис и/или транс двойную связь;
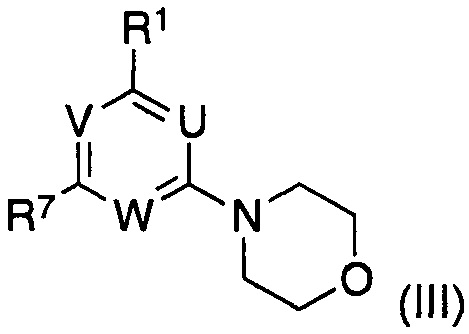
подвергают взаимодействию с соединением формулы (III)
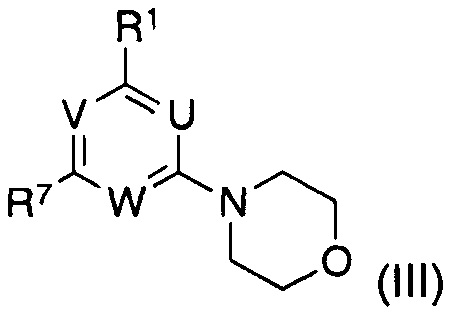
в котором указанные группы U, V, W и R1 являются такими, как определено выше; и
R7 представляет собой галоген;
в водном органическом растворителе или несмешивающейся смеси органический растворитель - вода при температурах от 0°C до температуры кипения растворителя или смеси растворителя в присутствии катализатора на основе фосфина Pd(0) или Pd(II) и основания;
и полученный формамидин формулы (IV)

где заместители имеют значения, как определено выше,
гидролизуют, in situ или после выделения, в водном растворе кислоты или основания.
В другом аспекте настоящее изобретение относится к соединениям формулы (II) как таковым, где заместители имеют значения, как указано выше.
В другом аспекте настоящее изобретение относится к способу получения соединения формулы (II), где заместители имеют значения, как указано выше, характеризующийся тем, что соединение формулы (V)
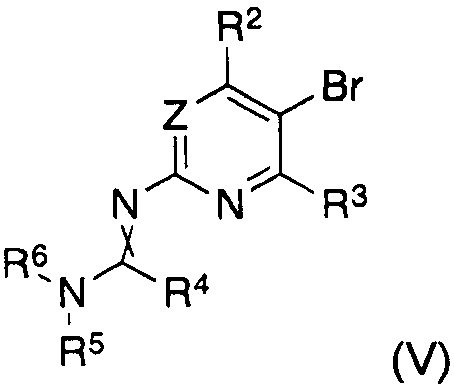
в котором указанные группы R2-R6 и группа Z являются такими, как определено выше, обрабатывают металлоорганическим соединением в органическом растворителе при температурах от -80°C до температуры кипения растворителя и, после завершения реакции обмена бром-металл, дополнительно подвергают взаимодействию с борорганическим реагентом формулы (VI)
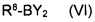
где R8 представляет собой уходящую группу и Y является таким, как определено выше.
В другом аспекте настоящее изобретение относится к соединениям формулы (V) как таковым, где заместители имеют значения, как указано выше.
В другом аспекте настоящее изобретение относится к способу получения соединения формулы (V), где заместители имеют значения, как указано выше, характеризующийся тем, что соединение формулы (VII)

в котором указанные группы R2, R3 и Z являются такими, как определено выше, галогенируют бромом, бромидом меди (II), бромоксоном (bromoxone) или N-галогенимидом, в инертном органическом растворителе, экстрагируют водным основанием, и подвергают взаимодействию с соединением формулы (VIII)

где R4, R5 и R6 в формуле (VIII) являются такими, как определено выше, и R9 представляет собой C1-C4-алкил или C5-C7-циклоалкил, предпочтительно метил, этил или трет-бутил.
В другом аспекте настоящее изобретение относится к способу получения соли присоединения кислоты формулы (Ia)

где
U, V, W, Z и R1-R3 являются такими, как определено для соединения формулы (I), и HX представляет собой протонную кислоту,
характеризующийся тем, что свободное основание формулы (I) обрабатывают протонной кислотой HX, необязательно в подходящем растворителе, и полученную соль присоединения кислоты очищают с помощью осаждения из растворителя или перекристаллизации.
Кроме того, настоящее изобретение относится к соединению 5-(4,6-диморфолино-1,3,5-триазин-2-ил)-4-(трифторметил)пиридин-2-амин 1

высокой чистоты, предпочтительно в виде кристаллического твердого вещества, его гидратам, солям и гидратам и сольватам его солей, и способам образования такого специфического твердого вещества, предпочтительно в кристаллических формах.
Краткое описание чертежей
На Фигуре 1 показаны спектры рамановского рассеяния до и после динамической сорбции паров (DVS) 5-(4,6-диморфолино-1,3,5-триазин-2-ил)-4-(трифторметил)пиридин-2-амина 1.
Подробное описание изобретения
В настоящем изобретении предложен существенно улучшенный способ синтеза соединений формулы (I)
По сравнению со способами, описанными в уровне техники, например, в публикации WO 2010/052569, настоящий способ позволяет получить соединения с более высоким выходом и более высокой чистотой и не требует опасных химических веществ. Кроме того, описанный способ можно масштабировать и он прост для осуществления. Например, не требуется продолжительная очистка хроматографическими методами.
Центральным аспектом настоящего изобретения является защита свободной аминной функциональной группы в борном реагенте для использования в реакции Сузуки. Группа амидина является ценной альтернативой, обеспечивая стабильное и легкодоступное кристаллическое вещество, которое можно успешно использовать в реакции Сузуки с высоким выходом. Амидинная защитная группа может быть впоследствии удалена с помощью простой кислотной или основной обработки, с получением требуемого аддукта свободного амина. Данная новая стратегия позволяет получить требуемые соединения с более высоким выходом и более высокой чистотой. Соединение 1, например, может быть получено в кг масштабе с отличным выходом и чистотой.
В частности изобретение относится к способу получения соединения формулы (I), его стереомеру, таутомеру или соли, где
U представляет собой CRU или N, где RU выбран из группы, состоящей из водорода, циано, галогена, метила и трифторметила, предпочтительно водорода;
V представляет собой CRV или N, где RV выбран из группы, состоящей из водорода, циано, галогена, метила и трифторметила, предпочтительно водорода;
W представляет собой CRW или N, где RW выбран из группы, состоящей из водорода, циано, галогена, метила и трифторметила, предпочтительно водорода;
при условии, что по меньшей мере один из U, V и W представляет собой N;
Z представляет собой CRZ или N, где RZ выбран из группы, состоящей из водорода, циано, галогена, метила и трифторметила, предпочтительно водорода;
R1 выбран из группы, состоящей из водорода, галогена, предпочтительно хлора, и -N(RT)RS, где RT и RS независимо друг от друга представляют собой водород или C1-C7-алкил, предпочтительно метил или этил, или
где RT и RS вместе с атомом азота, к которому они присоединены, образуют C3-C8 моно- или бициклическое гетероциклическое кольцо, необязательно содержащее один или более дополнительных атомов в кольце, выбранных из N, O или S, где указанное гетероциклическое кольцо необязательно замещено одной или более группами, независимо выбранными из C1-C7-алкила или C3-C7-циклоалкила, предпочтительно метила;
предпочтительно где RT и RS вместе с атомом азота, к которому они присоединены, образуют C4-C6-гетероциклическое кольцо, содержащее один дополнительный атом в кольце, выбранный из O, N или S, такой как в
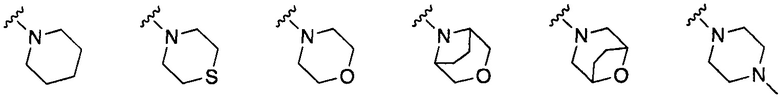


более предпочтительно, где RT и RS вместе с атомом азота, к которому они присоединены, образуют морфолино;
R2 выбран из группы, состоящей из водорода, циано, галогена, метила и трифторметила, предпочтительно водорода или трифторметила и более предпочтительно трифторметила;
и R3 представляет собой водород или галоген, предпочтительно водород;
характеризующийся тем, что соединение формулы (II)
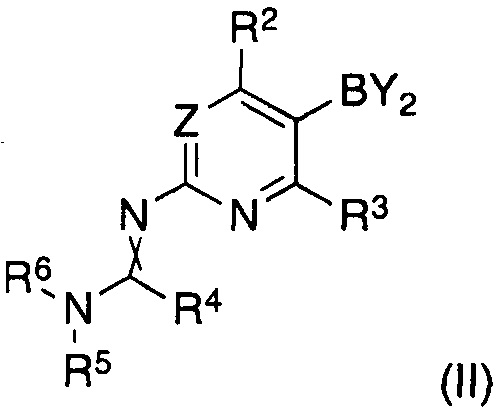
где
Y2B представляет собой остаток борной кислоты, ациклического сложного эфира борной кислоты или циклического сложного эфира борной кислоты, предпочтительно циклический сложный эфир борной кислоты, в частности пинаколатоборонат, и R2 и R3 являются такими, как определено для соединения формулы (I);
R4 представляет собой водород, C1-C7-алкил или C5-C7-циклоалкил, предпочтительно водород или C1-C7-алкил, в частности водород;
R5 и R6 представляют собой C1-C7-алкил, предпочтительно C1-C4-алкил, в частности метил, или R5 и R6 вместе представляют собой C4-C6-алкилен, в частности бутилен;
и скрещенная двойная связь между N и C(R4)N указывает на цис и/или транс двойную связь;
подвергают взаимодействию с соединением формулы (III)
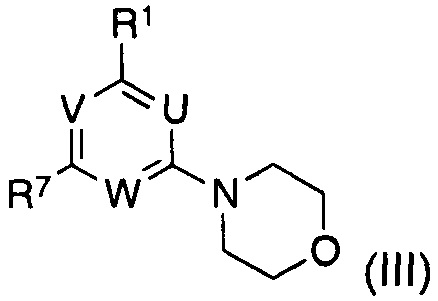
в котором указанные группы U, V, W и R1 являются такими, как определено выше; и
R7 представляет собой галоген, предпочтительно бром или хлор, в частности хлор;
в водном органическом растворителе или несмешивающейся смеси органический растворитель - вода при температурах от 0°C до температуры кипения растворителя или смеси растворителя в присутствии катализатора на основе фосфина Pd(0) или Pd(II) и основания.
и полученный формамидин формулы (IV)
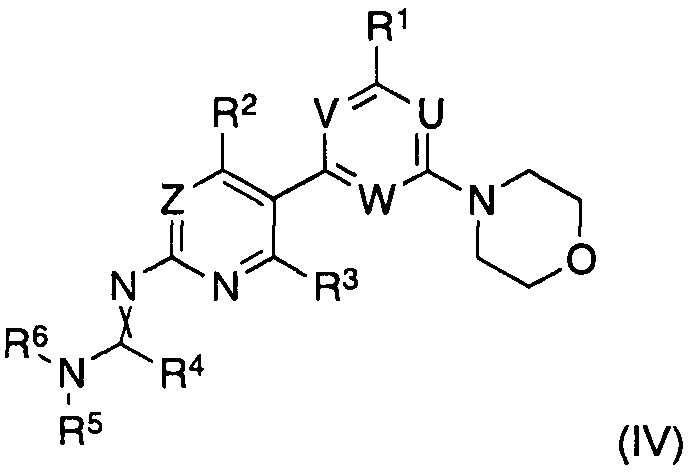
где заместители имеют значения, как определено выше,
гидролизуют, in situ или после выделения, в водном растворе кислоты или основания.
Было обнаружено, что значительно увеличенные выходы могут быть достигнуты с использованием формамидино защищенных борорганических реагентов формулы (II), по сравнению с известным способом с использованием свободных аминов. Такие амины вызывают побочные реакции, в частности реакции нуклеофильного замещения в ароматическом кольце с участием указанной функциональной группы амина, что таким образом приводит к смеси продуктов, снижающих выход и требующих хроматографической очистки. Кроме того, получение указанных выше борорганических реагентов с незащищенной функциональной группой амина, например, как описано в публикации WO 2007/084786, сопровождается образованием трудноотделимых прото-деборилированых побочных продуктов, в которых борный заместитель заменен водородом. Получение N-ацетил защищенных борорганических реагентов, таких как описанные в публикации WO 2012/044727, требует дополнительных стадий и криогенных условий (-78°C). Для сравнения, борорганические реагенты, описанные в настоящем документе, могут быть получены в чистом виде и с хорошим выходом без дополнительных стадий и при температуре не ниже 0°C. Кроме того, выделение описанных в настоящем документе борорганических реагентов в чистой твердой форме, хотя и легко достижимо, не является обязательным условием для успешной реакции Сузуки. Описанные в настоящем документе борорганические реагенты могут быть использованы in situ, таким образом, сокращая дальнейшие стадии получения соединений формулы (I).
Таким образом, согласно другому варианту реализации способа получения соединения формулы (I) согласно настоящему изобретению, указанное соединение формулы (II) получают in situ до указанной реакции с указанным соединением формулы (III), где указанное получение in situ осуществляют путем обработки соединения формулы (V)
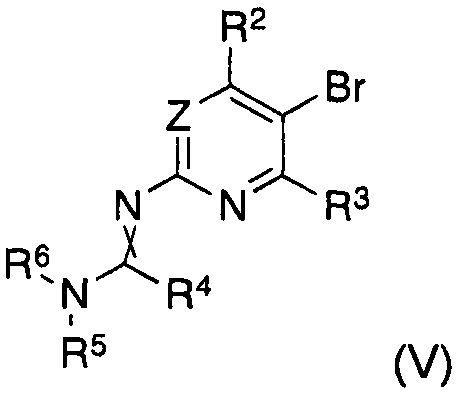
в котором указанные группы R2-R6 и группа Z являются такими, как указано выше, металлоорганическим соединением в органическом растворителе при температурах от -80°C до температуры кипения растворителя и, после завершения реакции обмена бром-металл, дополнительно подвергают взаимодействию с борорганическим реагентом формулы (VI)
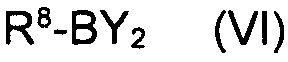
где R8 представляет собой уходящую группу и Y является таким, как указано выше.
После данного способа и метода, соответственно, получения соединения формулы (I) согласно настоящему изобретению, можно необязательно осуществлять одну или более реакций образования солей с применением протонной кислоты НХ как описано выше или ниже в настоящем документе.
Алкил представляет собой, например, C1-C7-алкил, такой как C1-C4-алкил, н-пентил, 1-этилпропил, н-гексил, 2-гексил, изогексил или н-гептил, в частности, C1-C4-алкил, такой как метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил или трет-бутил, в частности метил или этил.
Галоген представляет собой фтор, хлор, бром или йод, предпочтительно фтор и хлор, в частности хлор.
В остатке -BY2 ациклического сложного эфира борной кислоты, сложноэфирные заместители представляют собой, например, C1-C7-алкокси, такой как метокси, этокси, изо-пропокси. В остатке -BY2 предпочтительного циклического сложного эфира борной кислоты, диол, формирующий циклический сложный эфир, предпочтительно представляет собой 1,2-гликоль, например, 1,2-этандиол, 1,2-пропандиол, 2,3-бутандиол, или, в частности, пинакол (2,3-диметилбутан-2,3-диол). Кроме того, рассматриваются также циклические сложные эфиры 1,3-гликоля, например, 1,3-пропандиола или 2,2-диметил-1,3-пропандиола, также циклические сложные эфиры, дополнительно содержащие азот, такие как циклические сложные эфиры необязательно замещенного диэтаноламина, например N-фенилдиэтаноламина (N,N-ди(2-гидроксиэтил)анилина) или N-метиламиндиуксусной кислоты. Уходящая группа R8 предпочтительно представляет собой дополнительную группу Y, например, C1-C7-алкокси. В случае циклического бороната, в котором две группы Y представляют собой диол, уходящая группа может также представлять собой C1-C7-алкокси, в частности, изопропокси.

Согласно предпочтительному варианту реализации настоящего изобретения, в указанном соединении формулы (I), U представляет собой CRU или N, где RU представляет собой водород; V представляет собой CRV или N, где RV представляет собой водород; W представляет собой CRW или N, где RW представляет собой водород; Z представляет собой CRZ или N, где RZ представляет собой водород; R1 представляет собой морфолино; R2 представляет собой трифторметил; и R3 представляет собой водород.
Согласно дополнительному предпочтительному варианту реализации настоящего изобретения, в указанном соединении формулы (II) Y2B представляет собой циклический сложный эфир борной кислоты; R4 представляет собой водород; и R5 и R6 представляют собой метил.
Растворители для применения в реакции борорганического реагента (II) с соединением формулы (III) представляют собой, например, водный тетрагидрофуран, водный диоксан или смесь толуол - вода. Если требуемое основание добавляют в водном растворе, тогда полученная смесь растворителя является гомогенной (тетрагидрофуран, диоксан) или гетерогенной (толуол).
Рассматриваемые катализаторы палладия(0) и палладия(II) представляют собой тетракистрифенил-фосфин палладий(0), Pd(dppf)Cl2, Pd(dppf)Cl2⋅CH2Cl2, бис(трифенилфосфин)-палладий(II) дихлорид, хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил) [2-(2'-амино-1,1'-бифенил)]-палладий(II), смеси триарилфосфина, например, трифенилфосфина, тритолилфосфина или трифурилфосфина, или диалкиларилфосфина, например 2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил и соли палладия, такой как ацетат палладия или дихлорид палладия, в соотношении 1-5:1, предпочтительно в соотношении 3-5:1, в частности 3:1. Согласно предпочтительному варианту реализации, указанные катализаторы на основе фосфина Pd(0) или Pd(II), которые применяют в способе согласно изобретению для получения указанного соединения формулы (I), представляют собой смесь трифенилфосфина и ацетата палладия в соотношении 3:1.
Катализатор может быть использован в присутствии основания, например, карбоната калия, ацетата калия или трехосновного фосфата калия, предпочтительно, карбоната калия, в частности, карбоната калия, предпочтительно в присутствии водного основания, например раствора карбоната калия в воде, ацетата калия или трехосновного фосфата калия, предпочтительно раствора карбоната калия в воде, в частности, 10% (масс./об.) раствора карбоната калия в воде.
Реакцию проводят в растворителе, таком как тетрагидрофуран, диоксан или толуол при температуре между 0°C и температурой кипения растворителя в течение от 1 до 48 ч, предпочтительно между 20°C и температурой кипения растворителя в течение от 1 до 24 ч, более предпочтительно между 40°C и температурой кипения растворителя в течение от 1 до 12 ч, особенно предпочтительно при температуре кипения растворителя в течение от 2 до 4 ч.
В частности реакцию можно проводить в смеси тетрагидрофурана и воды при температуре между 0°C и температурой кипения смеси растворителя в течение от 1 до 48 ч, предпочтительно между 20°C и температурой кипения смеси растворителя в течение от 1 до 24 ч, более предпочтительно между 20 и 65°C в течение от 1 до 12 ч, особенно предпочтительно между 55 и 60°C в течение от 2 до 4 ч.
Альтернативно реакцию можно проводить в смеси диоксана и воды при температуре между 0°C и температурой кипения смеси растворителя в течение от 1 до 48 ч, предпочтительно между 20°C и температурой кипения смеси растворителя в течение от 1 до 36 ч, более предпочтительно между 80 и 105°C в течение от 1 до 24 ч, особенно предпочтительно между 95 и 105°C в течение 24 ч.
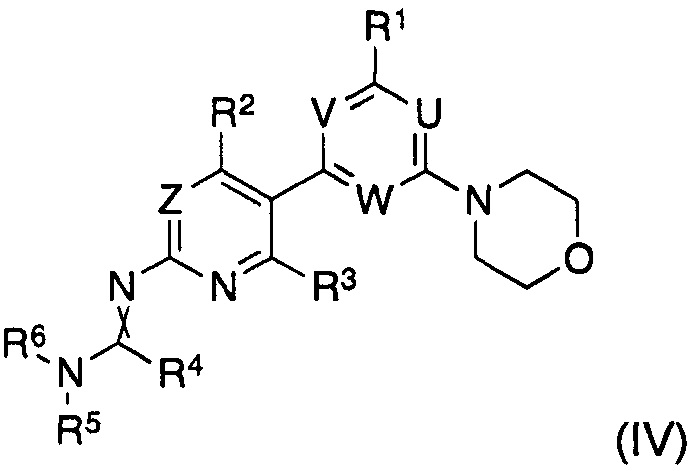
Соединение формулы (IV), полученное в такой реакции, затем подвергают взаимодействию in situ или после выделения с подходящим основанием, таким как гидроксид щелочного металла, предпочтительно гидроксид натрия, или подходящей кислотой, такой как соляная кислота при концентрации 1-10 М, предпочтительно 2-8 М, в частности, 4-5 М, с получением, после нейтрализации, соединения формулы (I).

Согласно другому варианту реализации способа получения соединения формулы (I) согласно настоящему изобретению, в указанном соединении формулы (I), формулы (II), формулы (III) и формулы (IV), где это применимо, U представляет собой CRU или N, где RU представляет собой водород; V представляет собой CRV или N, где RV представляет собой водород; W представляет собой CRW или N, где RW представляет собой водород; Z представляет собой CRZ или N, где RZ представляет собой водород; R1 представляет собой галоген, предпочтительно хлор; R2 представляет собой трифторметил; и R3 представляет собой водород; и где дополнительно указанный полученный формамидин формулы (IV) подвергают взаимодействию с морфолином до указанной гидролизации, где предпочтительно указанный формамидин формулы (IV) выделяют для указанной реакции с указанным морфолином. Кроме того, предпочтительно, в указанном соединении формулы (II) Y2B представляет собой циклический сложный эфир борной кислоты; R4 представляет собой водород; и R5 и R6 представляют собой метил.
Согласно предпочтительному варианту реализации способа получения соединения формулы (I) согласно настоящему изобретению, в указанном соединении формулы (I), формулы (II), формулы (III) и формулы (IV), где это применимо, U представляет собой N; V представляет собой N; W представляет собой N; Z представляет собой CRZ, где RZ представляет собой водород; R1 представляет собой галоген, предпочтительно хлор или морфолино; R2 представляет собой трифторметил; и R3 представляет собой водород.
Согласно другому очень предпочтительному варианту реализации способа получения соединения формулы (I) согласно настоящему изобретению, в указанном соединении формулы (I), формулы (II), формулы (III) и формулы (IV), где это применимо, U представляет собой N; V представляет собой N; W представляет собой N; Z представляет собой CRZ, где RZ представляет собой водород; R1 представляет собой морфолино; R2 представляет собой трифторметил; и R3 представляет собой водород.
Согласно другому очень предпочтительному варианту реализации способа получения соединения формулы (I) согласно настоящему изобретению, в указанном соединении формулы (I), формулы (II), формулы (III) и формулы (IV), где это применимо, U представляет собой N; V представляет собой N; W представляет собой N; Z представляет собой CRZ, где RZ представляет собой водород; R1 представляет собой морфолино; R2 представляет собой трифторметил; и R3 представляет собой водород, и в указанном соединении формулы (II) Y2B представляет собой циклический сложный эфир борной кислоты; R4 представляет собой водород; и предпочтительно R5 и R6 представляют собой метил. Предпочтительно, указанный R7 представляет собой хлор.
Согласно другому очень предпочтительному варианту реализации способа получения соединения формулы (I) согласно настоящему изобретению, в указанном соединении формулы (I), формулы (II), формулы (III) и формулы (IV), где это применимо, U представляет собой N; V представляет собой N; W представляет собой N; Z представляет собой CRZ, где RZ представляет собой водород; R1 представляет собой морфолино; R2 представляет собой трифторметил; и R3 представляет собой водород, и в указанном соединении формулы (II) Y2B представляет собой циклический сложный эфир борной кислоты, и предпочтительно, пинаколатоборонат; R4 представляет собой водород; и R5 и R6 представляют собой метил. Предпочтительно, указанный R7 представляет собой хлор.
Согласно другому очень предпочтительному варианту реализации способа получения соединения формулы (I) согласно настоящему изобретению, в указанном соединении формулы (I), формулы (II), формулы (III) и формулы (IV), где это применимо, U представляет собой N; V представляет собой N; W представляет собой N; Z представляет собой CRZ, где RZ представляет собой водород; R1 представляет собой морфолино; R2 представляет собой трифторметил; и R3 представляет собой водород, и в указанном соединении формулы (II) Y2B представляет собой пинаколатоборонат; R4 представляет собой водород; и R5 и R6 представляют собой метил. Предпочтительно, указанный R7 представляет собой хлор.
Согласно другому аспекту, изобретение относится к способу получения соли присоединения кислоты формулы (Ia)

где
U, V, W, Z и R1-R3 являются такими, как определено для соединения формулы (I), и HX представляет собой протонную кислоту,
характеризующийся тем, что свободное основание формулы (I) обрабатывают протонной кислотой HX, необязательно в подходящем растворителе, и полученную соль присоединения кислоты очищают с помощью осаждения из растворителя или перекристаллизации.
Превращение свободных соединений в их соответствующие соли хорошо известно в области органической химии. Основные соединения, такие как в настоящем изобретении, могут быть превращены в соответствующие соли путем добавления кислотных соединений (HX), например, растворенных в органической или водной среде, в виде газа или в веществе. Указанная реакция еще не применялась с использованием конкретных исходных веществ, описанных в настоящем документе, и, таким образом, образует новый и неочевидный способ.
Указанную стадию предпочтительно применяют для получения фармацевтически приемлемых солей присоединения кислоты из соединений формулы (I). Предпочтительные фармацевтически приемлемые соли присоединения кислоты образуют из следующих протонных кислот HX: I) неорганические кислоты, в частности, выбранные из группы, состоящей из соляной кислоты, бороводородной кислоты (англ. hydroboric acid), азотной кислоты, серной кислоты и фосфорной кислоты, II) органические кислоты, в частности, выбранные из группы, состоящей из муравьиной кислоты, уксусной кислоты, трифторуксусной кислоты, фумаровой кислоты, винной кислоты, щавелевой кислоты, малеиновой кислоты, лимонной кислоты, метансульфоновой кислоты, янтарной кислоты, яблочной кислоты, бензолсульфоновой кислоты и п-толуолсульфоновой кислоты, и III) кислые аминокислоты, в частности, выбранные из группы, состоящей из аспарагиновой кислоты и глутаминовой кислоты. Особенно предпочтительной HX кислотой является соляная кислота. Еще одной предпочтительной HX кислотой является метансульфоновая кислоты.
Согласно другому аспекту изобретение относится к соединениям формулы (II), как таковым,
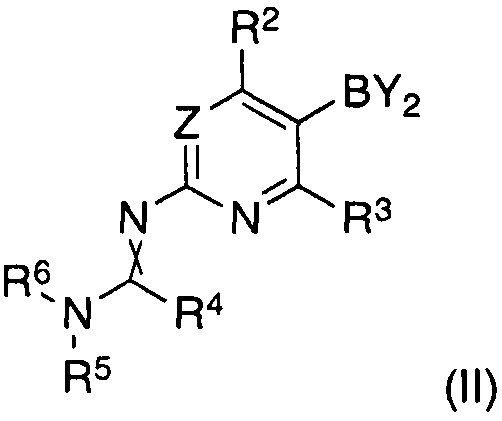
где
Y2B представляет собой остаток борной кислоты, ациклического сложного эфира борной кислоты или циклического сложного эфира борной кислоты, предпочтительно циклического сложного эфира борной кислоты, в частности пинаколатобороната;
R2 представляет собой водород, циано, галоген, метил или трифторметил, предпочтительно водород или трифторметил, в частности трифторметил;
R3 представляет собой водород или галоген, предпочтительно водород;
R4 представляет собой водород, C1-C7-алкил или C5-C7-циклоалкил, предпочтительно водород или C1-C7-алкил, в частности водород;
R5 и R6 представляют собой C1-C7-алкил, предпочтительно C1-C4-алкил, в частности метил, или R5 и R6 вместе представляют собой C4-C6-алкилен, в частности бутилен;
и скрещенная двойная связь между N и C(R4)N указывает на цис и/или транс двойную связь.
Согласно очень предпочтительному варианту реализации соединения согласно настоящему изобретению формулы (II) указанный Z представляет собой CRZ, где RZ представляет собой водород; R3 представляет собой водород, Y2B представляет собой циклический сложный эфир борной кислоты, и предпочтительно пинаколатоборонат; R4 представляет собой водород; и R5 и R6 представляют собой метил.
Согласно очень предпочтительному варианту реализации соединения согласно настоящему изобретению формулы (II), указанный Z представляет собой CRZ, где RZ представляет собой водород; R3 представляет собой водород, Y2B представляет собой пинаколатоборонат; R4 представляет собой водород; и R5 и R6 представляют собой метил.
Согласно другому аспекту изобретение относится к способу получения соединения формулы (II), где заместители имеют значения, как указано выше,
характеризующийся тем, что соединение формулы (V)
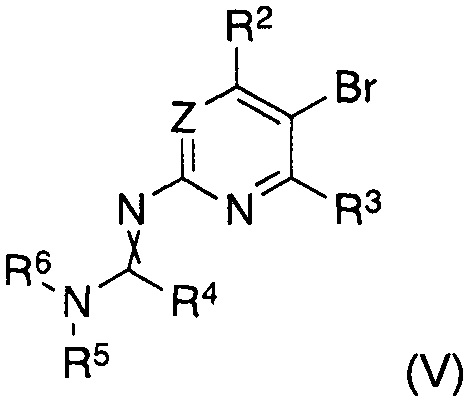
в котором указанные группы R2-R6 и группа Z являются такими, как определено выше, обрабатывают металлоорганическим соединением в органическом растворителе при температурах от -80°C до температуры кипения растворителя и, после завершения реакции обмена бром-металл, дополнительно подвергают взаимодействию с органоборатом (VI)

где R8 представляет собой уходящую группу и Y является таким, как определено выше.
Согласно очень предпочтительному варианту реализации способа получения соединения формулы (II) согласно настоящему изобретению, в указанном соединении формулы (II), (V) и (VI), где это применимо, указанный Z представляет собой CRZ, где RZ представляет собой водород; R3 представляет собой водород, Y2B представляет собой циклический сложный эфир борной кислоты, и предпочтительно пинаколатоборонат; R4 представляет собой водород; и R5 и R6 представляют собой метил.
Согласно другому очень предпочтительному варианту реализации способа получения соединения формулы (II) согласно настоящему изобретению, в указанном соединении формулы (II), (V) и (VI), где это применимо, указанный Z представляет собой CRZ, где RZ представляет собой водород; R3 представляет собой водород, Y2B представляет собой циклический сложный эфир борной кислоты, и предпочтительно пинаколатоборонат; R4 представляет собой водород; и R5 и R6 представляют собой метил.
При получении соединения формулы (II), соединение формулы (V)

подвергают обмену бром-метал способами, известными в данной области, такими как, взаимодействие с металлоорганическими соединениями, предпочтительно литийорганическими и магнийорганическими соединениями, в частности хлоридом изопропилмагния в подходящем растворителе, таком как тетрагидрофуран или 2-метил-тетрагидрофуран при температурах от -80°C до температуры кипения растворителя, предпочтительно между -20°C и температурой кипения растворителя, в частности между 0°C и 65°C. После завершения обмена бром-метал полученное металлоорганическое соединение подвергают взаимодействию in situ с соответствующим борным реагентом R8-BY2 формулы (VI), где Y является таким, как определено выше и уходящая группа R8 выбрана из галогена или C1-C4 алкокси, предпочтительно из метокси или изопропокси, более предпочтительно изопропокси, в частности с 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксабороланом, с получением предпочтительного соединения формулы (II). Соединение формулы (II) можно дополнительно использовать in situ или выделять в чистой форме.
Согласно другому аспекту изобретение относится к соединениям формулы (V)
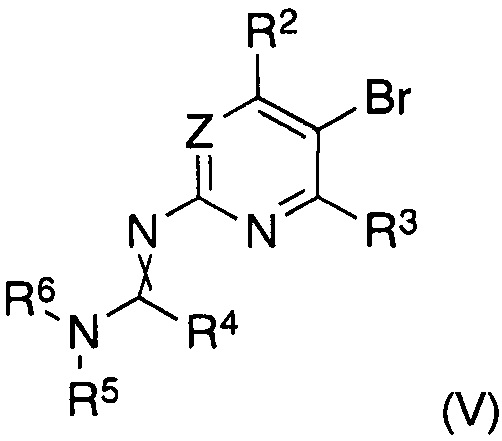
как таковым, где
Z представляет собой CRZ или N, где RZ выбран из группы, состоящей из водорода, циано, галогена, метила и трифторметила;
R2 представляет собой водород, циано, галоген, метил или трифторметил, предпочтительно водород или трифторметил, в частности трифторметил;
R3 представляет собой водород или галоген, предпочтительно водород;
R4 представляет собой водород, C1-C7-алкил или C5-C7-циклоалкил, предпочтительно водород или C1-C7-алкил, в частности водород;
R5 и R6 представляют собой C1-C7-алкил, предпочтительно C1-C4-алкил, в частности метил, или R5 и R6 вместе представляют собой C4-C6-алкилен, в частности бутилен;
и скрещенная двойная связь между N и C(R4)N указывает на цис и/или транс двойную связь.
Согласно очень предпочтительному варианту реализации соединения согласно настоящему изобретению формулы (V), указанный Z представляет собой CRZ, где RZ представляет собой водород; R2 представляет собой трифторметил; R3 представляет собой водород; R4 представляет собой водород; и R5 и R6 представляют собой метил.
Согласно другому аспекту изобретение относится к способу получения соединения формулы (V). Соединениям формулы (V) получают из соединения формулы (VII)

в котором указанные группы R2, R3 и Z являются такими, как определено выше.
Соединения формулы (VII) галогенируют галогенирующими агентами, такими как бром, бромид меди (II), бромоксон ((1R,5S,6R)-3-бром-5-гидрокси-7-оксабицикло[4,1.0]гепт-3-ен-2-он) или N-галогенимид, предпочтительно N-галогенсукцинимиды, в частности, N-бромсукцинимид, в инертном органическом растворителе, таком как сложноэфирный, простой эфирный или нитрильный растворитель, предпочтительно C1-C4-алкиловые эфиры уксусной кислоты, C1-C4-алкилнитрилы или циклические простые эфиры, в частности этиловый эфир уксусной кислоты, ацетонитрил или 2-метилтетрагидрофуран. Затем реакционную смесь экстрагируют подходящим водным основанием, таким как карбонат металла, предпочтительно карбонат щелочного металла, в частности растворами карбоната натрия и калия в воде. Полученное промежуточное соединение дополнительно подвергают взаимодействию, либо in situ, либо после выделения промежуточного соединения, с соединением формулы (VIII)

где R4, R5 и R6 в формуле (VIII) являются такими, как определено выше, и R9 представляет собой C1-C4-алкил или C5-C7-циклоалкил, предпочтительно C1-C4-алкил, в частности метил, этил или трет-бутил.
Согласно очень предпочтительному варианту реализации способа получения соединения формулы (V) согласно настоящему изобретению, в указанном соединении формулы (VII) и (VIII), где это применимо, указанный Z представляет собой CRZ, где RZ представляет собой водород; R2 представляет собой трифторметил; R3 представляет собой водород; R4 представляет собой водород; и R5 и R6 представляют собой метил.
Соединения формулы (III)
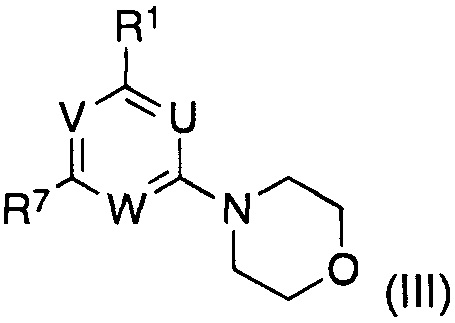
где указанные группы U, V, W и R1 являются такими, как определено выше, R7 представляет собой галоген, предпочтительно бром или хлор, в частности хлор, получают из соединений формулы (IX), где U, V, W и R7 являются такими, как определено выше, способами нуклеофильного замещения в ароматическом кольце, известными в данной области.
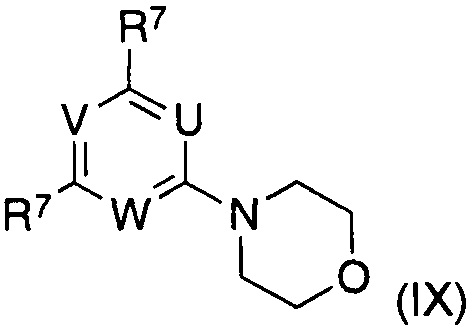
Соединения формулы (IX) получают из соответствующих соединений формулы (X)

где R7 представляет собой галоген, посредством взаимодействия с морфолином в подходящих органических растворителях, таких как алканы, галогеналканы, сложные эфиры, простые эфиры, нитрилы, предпочтительно галогеналканы, в частности дихлорметан. В частности, ситуация, при которой R1 в соединении формулы (III) представляет собой морфолин, указанное соединение получают непосредственно из соединения формулы (X) посредством взаимодействия с избытком морфолина в указанных подходящих органических растворителях, например, дихлорметане.
Кроме того, изобретение относится к соединению 5-(4,6-диморфолино-1,3,5-триазин-2-ил)-4-(трифторметил)пиридин-2-амин 1
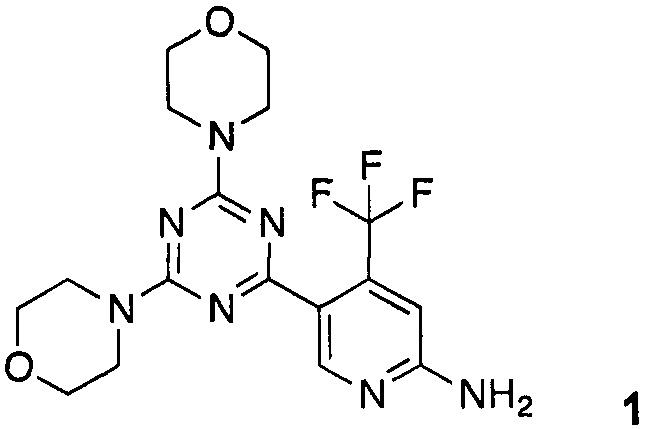
в твердом виде с высокой степенью чистоты, предпочтительно с более чем 99% чистотой, более предпочтительно, с более чем 99,5% чистотой, например 99,7% чистотой, и плавлении при 219-220°C.
В других аспектах, изобретение относится к специфически высокочистым твердым, предпочтительно кристаллическим, формам соединения формулы 1, его гидратам, его солям и гидратам и сольватам его солей, и способам образования таких специфических твердых, предпочтительно кристаллических, форм.
Примеры
экв = эквиваленты
ТСХ = тонкослойная хроматография
Пример 1а: Получение 4-(4-хлор-6-морфолино-1,3,5-триазин-2-ил)морфолина

Смесь морфолина (2,83 кг, 2,84 л, 32,5 моль, 4 экв.), воды (6,75 л) и дихлорметана (5 л) охлаждали до 5°C. К полученной двухфазной смеси добавляли раствор хлорангидрида циануровой кислоты (1,50 кг, 8,13 моль, 1 экв.) в дихлорметане (4,5 л) с такой скоростью, что температура не превышала 10°C (са. 3 ч), и перемешивание продолжали в течение 15 мин при 5°C. Фазы разделяли и органическую фазу промывали дважды водой (2×15 л). Объем органической фазы понижали на половину путем выпаривания при пониженном давлении (700 мбар) с использованием роторного испарителя. Замену растворителя осуществляли путем постепенного добавления изомеров гептана (15 л) и выпаривания в общей сложности 14,5 л растворителя при пониженном давлении (150 мбар) с использованием роторного испарителя. Полученную суспензию белого цвета охлаждали до 20°C и перемешивали при указанной температуре в течение 18 ч. Продукт собирали фильтрованием, промывали изомерами гептана (1,4 л) и сушили при 35°C при пониженном давлении (<50 мбар) до постоянной массы с получением указанного в заголовке соединения в виде белого твердого вещества (2,297 кг, 7,97 моль, 98% выход).
1H ЯМР (400 МГц, CDCl3, δ): 3,78 (m, 8Н), 3,70 (m, 8Н).
MS m/z: 287,6 [M+H]+.

Пример 1b: Получение 4-(4-хлор-6-морфолино-1,3,5-триазин-2-ил)морфолина
Получение, описанное в Примере 1b, было осуществлено в модифицированном виде, по сравнению с процедурой, описанной в Примере 1а, что привело к дальнейшему улучшению чистоты.
К раствору морфолина (9,21 кг, 9,20 л, 106 моль, 4 экв.) в воде (21,9 л) добавляли дихлорметан (14,6 л) и смесь охлаждали до 0°C. К полученной двухфазной смеси добавляли раствор хлорангидрида циануровой кислоты (4,87 кг, 26,4 моль, 1 экв.) в дихлорметане (34,1 л) при постоянной скорости в течение 3 ч (так, чтобы температура не превышала 5°C) и перемешивание продолжали в течение 15 мин при 5°C. Фазы разделяли и органическую фазу три раза промывали водой (3×29,2 л) и сушили над безводным сульфатом натрия (3 кг). Твердые вещества удаляли фильтрованием и фильтрационный осадок промывали дихлорметаном (9,7 л). К объединенным фильтратам добавляли гептан (39 л) и объем (102 л) понижали до 68 L путем выпаривания при пониженном давлении (са 700 мбар). К полученной смеси добавляли гептан (14,6 л) и полученную суспензию белого цвета охлаждали до 3°C в течение 1,5 ч. Продукт собирали фильтрованием, промывали гептаном (2×14,6 л) и сушили при 40°C при пониженном давлении (<50 мбар) до постоянной массы с получением указанного в заголовке соединения в виде белого твердого вещества (7,35 кг, 25,72 моль, 97% нескорректированный выход, чистота 97% а/а). При втором прогоне, начиная с 4,40 кг хлорангидрид циануровой кислоты, указанное в заголовке соединение получали в виде белого твердого вещества с использованием той же процедуры (6,57 кг, 22,99 моль, 94% нескорректированный выход, чистота 97% а/а). Две партии указанного в заголовке соединения совместно суспендировали в гептане (110 л) и перемешивали при 21°C в течение 18 ч. Твердое вещество собирали фильтрованием и фильтрационный осадок промывали гептаном (10 л) и сушили до постоянной массы при 40°C в вакууме с получением 13,6 кг (98% извлечение, чистота 98,3% а/а). Указанное вещество разделяли на две равные партии для перекристаллизации. К перемешанному раствору полученного выше указанного в заголовке соединения (6,8 кг) в дихлорметане (20 л) при 40°C добавляли гептан (60 л) в течение 1,5 ч при 40°C и перемешивание продолжали в течение 0,75 ч. Полученную суспензию охлаждали до 0°C в течение 8 ч. Твердое вещество собирали фильтрованием промывали смесью гептана и дихлорметана (99:1, 7 л) и сушили до постоянной массы в вакууме при 40°C с получением 6,13 кг и 6,03 кг. Обе партии объединяли с получением 12,16 кг (84% всего, чистота 99,9% а/а) указанного в заголовке соединения.
Пример 2а: Получение N'-[5-бром-4-(трифторметил)-2-пиридил]-N,N-диметил-формамидин
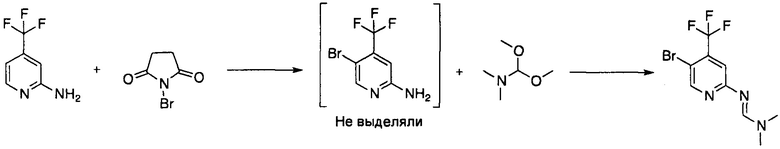
К раствору 4-(трифторметил)пиридин-2-амина (1,39 кг, 8,59 моль, 1 экв.) в 2-метил-тетрагидрофуране (16,8 л) добавляли N-бромсукцинимид (1,528 кг, 8,59 моль, 1 экв.) в 10 равных порциях в течение 50 мин при температуре 0°C, и перемешивание продолжали при указанной температуре в течение дополнительных 2 ч. К полученной оранжевой суспензии добавляли 8% водный раствор карбоната натрия (14 л). Фазы разделяли и водную фазу экстрагировали 2-метилтетрагидрофураном (2,4 л). Объединенные органические фазы смешивали с 5% водным раствором хлорида натрия (5,6 л) и фазы разделяли. Органическую фазу азеотропно перегоняли с 2-метилтетрагидрофураном (2×9 л) и объем коричневого раствора понижали до 18 л путем выпаривания при пониженном давлении с использованием роторного испарителя. К полученному раствору добавляли при 35°C 1,1-диметокси-N,N'-диметилметанамин (1,275 кг, 1,52 л, 10,7 моль, 1,25 экв. и смесь нагревали до 60°C в течение 2,5 ч. Смесь охлаждали до комнатной температуры и растворитель заменяли на изомеры гептана посредством азеотропной перегонки (4 раза) при пониженном давлении с использованием изомеров гептана (4×9 л). Изомеры гептана добавляли до достижения объема 28 ч. Темно-коричневый осадок удаляли фильтрованием, полученный коричневый маточный раствор промывали дважды водой (2×14 л). Объем органической фазы понижали до 7 л путем выпаривания при пониженном давлении, в результате чего образовалась суспензия. Указанную суспензию перемешивали при 20°C в течение 1 ч и затем охлаждали до 0°C. Твердое вещество собирали фильтрованием, промывали холодными изомерами гептана (2 л) и сушили до постоянной массы при пониженном давлении (<50 мбар) с получением указанного в заголовке соединения в виде оранжевого твердого вещества (1,85 кг, 6,20 моль, 73% выход).
1H ЯМР (400 МГц, CDCl3, δ): 8,47 (s, 1Н, пиридил-H), 8,40 (s, 1Н, пиридил-H), 7,20 (s, 1H, CH(N(Me)2), 3,12 (s, 3H, CH3), 3,10 (s, 3H, CH3);
19F ЯМР (100,6 МГц, CDCl3, δ): -65,01.
MS m/z: 296,0 [M(79Br)+H]+, 298,0 [M(81Br)+H]+.
Пример 2b: Получение N'-[5-бром-4-(трифторметил)-2-пиридил]-N,N-диметил-формамидина
Получение, описанное в Примере 2b, было осуществлено в модифицированном виде, по сравнению с процедурой, описанной в Примере 2а, что привело к дальнейшему улучшению чистоты.
К раствору 4-(трифторметил)пиридин-2-амина (6,50 кг, 40,1 моль, 1 экв.) в 2-метилтетрагидрофуране (78 л) добавляли при 0°C в десяти равных порциях N-бромсукцинимид (7,49 кг, 42,1 моль, 1,05 экв.) в течение 50 мин. Полученную суспензию перемешивали при 0°C в течение 1 ч. Твердые вещества удаляли фильтрованием. Фильтрационный осадок промывали 2-метил-тетрагидрофураном (10 л). Объединенные фильтраты смешивали с 4% водным раствором карбоната натрия (65 л). Фазы разделяли и органическую фазу экстрагировали 2N соляной кислотой (3×20 л и 3×10 л). Объединенные водные экстракты смешивали с 2-метил-тетрагидрофураном (5 л), деионизированной водой (3 л) и водным раствором гидроксида натрия (30% масс./об., 19 л) с такой скоростью, что температура не превышала 20°C. pH полученной двухфазной смеси доводили до 8 путем добавления насыщенного водного раствора бикарбоната натрия (8% масс./об., 3,4 л). Фазы разделяли и слегка мутную коричневую органическую фазу сушили над безводным сульфатом натрия (3,2 кг). Твердые вещества удаляли фильтрованием и фильтрационный осадок промывали 2-метил-тетрагидрофураном (5 л). Растворитель выпаривали при пониженном давлении. Полученное коричневое масло (8,35 кг) растворяли в 2-метилтетрагидрофуране (94 л) при 60°C и 1,1-диметокси-N,N'-диметилметанамин (6,8 л, 51,2 моль, 1,28 Экв.) добавляли в течение 10 мин. Полученный коричневый раствор перемешивали при 60°C в течение 3 ч охлаждали до 20°C и смешивали с насыщенным водным раствором бикарбоната натрия (8% масс./об., 33 л). Фазы разделяли и органическую фазу промывали дважды водным раствором хлорида натрия (5% масс./об., 32 л). Органическую фазу сушили над безводным сульфатом натрия (3,2 кг). Твердые вещества удаляли фильтрованием и фильтрационный осадок промывали 2-метил-тетрагидрофураном (11 л). Объединенные фильтраты смешивали с гептаном (39 л) и смесь концентрировали при пониженном давлении. Остаток смешивали с гептаном (39 л) и смесь концентрировали при пониженном давлении в результате чего образовалась суспензия. Суспензию перемешивали при 0°C. Твердое вещество собирали фильтрованием и промывали гептаном (8 л) с получением указанного в заголовке соединения (8,20 кг, 27,7 моль, 69%, чистота 98,0% а/а) в виде желто-коричневатого твердого вещества.
Пример 3a: Получение (E)-N,N-диметил-N'-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-4-(трифторметил)пиридин-2-ил)формамидина
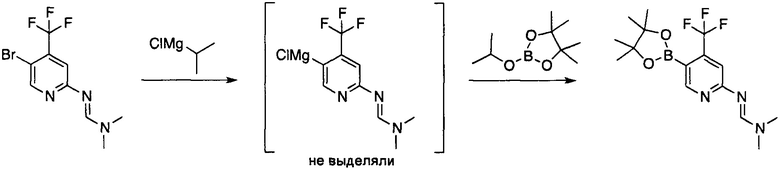
К 2 М раствору хлорида изопропилмагния (29,3 мл, 1,15 экв.) в ТГФ (60 мл) медленно добавляли при 0°C раствор N'-[5-бром-4-(трифторметил)-2-пиридил]-N,N-диметил-формамидина (Пример 2, 15,06 г, 50,9 ммоль, 1 экв.) в ТГФ (50 мл) в течение 5 мин. Смесь перемешивали при 0°C в течение 45 мин и при комнатной температуре в течение 15 мин. Контроль по ТСХ подтвердил полный обмен бром-магний. К полученной суспензии добавляли 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолан (13,4 мл, 1,3 экв.) и смесь перемешивали при 60°C в течение 3 ч. Полученный темный раствор охлаждали до 0°C и гасили путем добавления 15% водного раствора NH4Cl (210 мл). Слои разделяли и водный слой дополнительно экстрагировали ТРФ (100 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и растворитель удаляли при пониженном давлении с использованием роторного испарителя. Добавляли гептан (200 мл) и полученный раствор промывали насыщенным водным раствором NaHCO3 (100 мл). Органический слой сушили над безводным Na2SO4, фильтровали и объем растворителя понижали до 100 мл с использованием роторного испарителя. Полученный оранжевый раствор охлаждали до -20°C в течение 18 ч. Желто-оранжевые кристаллы указанного в заголовке соединения (11,45 г, 66% выход) собирали фильтрованием. Маточный раствор концентрировали и подвергали второй перекристаллизации из гептана с получением дополнительных желто-оранжевых кристаллов указанного в заголовке соединения (1,18 г, 7% выход). Объединенный выход реакции составил 73%.
1H ЯМР (CDCl3, 400 МГц, δ): 8,70 (s, 1Н), 8,61 (s, 1Н), 7,17 (s, 1Н), 3,13 (s, 3H), 3,11 (s, 3H), 1,35 (s, 12Н).
19F ЯМР (CDCl3, 376 МГц, δ): -62,7 (s, 3F).
MS m/z: 344,8 [M+H]+.
Пример 3b: Получение (E)-N,N-диметил-N'-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-4-(трифторметил)пиридин-2-ил)формамидина
Получение, описанное в Пример 3b было осуществлено в модифицированном виде, по сравнению с процедурой, описанной в Примере 3a что привело к дальнейшему улучшению чистоты.
2М раствор хлорида изопропилмагния в тетрагидрофуране (17,46 кг, 33,8 моль, 1,25 Экв.) добавляли к тетрагидрофурану (32 л) при 0°C в течение 50 мин. К полученному раствору добавляли раствор N'-[5-бром-4-(трифторметил)-2-пиридил]-N,N-диметил-формамидина (8,0 кг, 27 моль, 1 Экв.) в тетрагидрофуране (28 л) в течение 30 мин при 0--4°C. Полученную оранжевую суспензию перемешивали при 0°C в течение 16 мин и затем нагревали до 20°C в течение 35 мин, и данной температуре перемешивание продолжали в течение 18 мин. К полученной оранжевой суспензии добавляли 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолан (6,84 кг, 7,48 л, 36,7 моль, 1,36 экв.) в течение 8 мин. Полученную смесь нагревали до 55°C в течение 50 мин и оставляли перемешиваться при указанной температуре в течение 4,5 ч. Полученный раствор охлаждали до 0°C и ледяной 15% водный раствор хлорида аммония (64 л) добавляли в течение 17 мин поддерживая температуру между 1-8°C. Двухфазную смесь перемешивали в течение 36 мин, фазы разделяли и органическую фазу промывали 3× водным раствором хлорида натрия (13% масс./об., 3×40 л). Растворитель выпаривали при пониженном давлении при 40°C. Полученный остаток растворяли в гептане (102 л), промывали насыщенным водным раствором бикарбоната натрия (8% масс./об., 55 л) и сушили над безводным сульфатом натрия (3,2 кг). Твердые вещества удаляли фильтрованием и фильтрационный осадок промывали гептаном (11 л). Объединенные фильтраты концентрировали до объема са 32 л в результате чего образовалась желто-коричневатая суспензия. Указанную суспензию охлаждали до -20°C и перемешивали при указанной температуре в течение 3 ч. Твердое вещество собирали фильтрованием и промывали дважды холодным гептаном (2×6 л) и сушили до постоянной массы при пониженном давлении при 40°C с получением указанного в заголовке соединения (6,01 кг, 17,5 моль, 65%, чистота 99% а/а).
Пример 4a: Получение 5-(4,6-диморфолино-1,3,5-триазин-2-ил)-4-(трифторметил)пиридин-2-амина 1
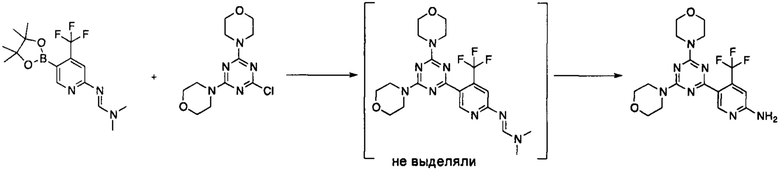
Смесь ацетата палладия (18 мг, 0,08 ммоль, 0,04 экв.) и трифенилфосфина (63 мг, 0,24 ммоль, 0,12 экв.) в тетрагидрофуране (6,25 мл) перемешивали при комнатной температуре в течение 1 ч. К полученному раствору добавляли раствор 4-(4-хлор-6-морфолино-1,3,5-триазин-2-ил)морфолина (Пример 1, 572 мг, 2 ммоль, 1 экв.) и (E)-N,N-диметил-N'-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-4-(трифторметил)пиридин-2-ил)формамидина (Пример 3, 823 мг, 2,4 ммоль, 1,2 экв.) в тетрагидрофуране (5 мл) и раствор карбоната калия (828 мг, 6 ммоль, 3 экв.) в воде (2,5 мл). Полученную смесь нагревали до 55°C и перемешивали при указанной температуре. Реакцию контролировали с помощью ТСХ с использованием этилацетата в качестве элюента, и было показано полное превращение через 2 ч. Смесь охлаждали до комнатной температуры и осторожно добавляли 5 М раствор HCl в диоксане (4 мл) (выделение CO2). Смесь перемешивали при 55°C в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли 5 М водным раствором HCl (20 мл) и этилацетатом (5 мл). Фазы разделяли. pH водной фазы доводили до 7,0 путем добавления 2 М водного раствора гидроксида натрия и экстрагировали этилацетатом (2×50 мл). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с использованием роторного испарителя. Остаток очищали с помощью флэш-хроматографии на силикагеле (50 г) с использованием первой смеси 1:2 этилацетата и циклогексана, и затем чистым этилацетатом в качестве элюента. Фракции продукта объединяли и выпаривали с получением указанного в заголовке соединения в виде беловатого порошка (707 мг, 1,71 ммоль, 86% выход).
Пример 4b: Получение 5-(4,6-диморфолино-1,3,5-триазин-2-ил)-4-(трифторметил)пиридин-2-амина 1
Получение, описанное в Примере 4b, было осуществлено в модифицированном виде, по сравнению с процедурой, описанной в Примере 4а, что позволяет избежать описанной в нем хроматографической очистки.
К суспензии Pd(OAc)2 (0,131 кг, 0,58 моль, 0,04 Экв.) в тетрагидрофуране (35 л) добавляли трифенилфосфин (0,452 кг, 1,72 моль, 0,12 Экв.) и смесь перемешивали в инертных условиях при 20°C в течение 23 мин с получением раствора катализатора. Параллельно, двухфазную смесь, полученную из раствора карбоната калия (6,047 кг, 43,57 моль, 3,2 Экв.) в воде (15 л) и N,N-диметил-N'-[5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-4-(трифторметил)-2-пиридил]формамидина (5,00 кг, 14,6 моль, 1,07 Экв.) и 4-(4-хлор-6-морфолино-1,3,5-триазин-2-ил)морфолина (3,916 кг, 13,7 моль, 1,0 Экв.) в тетрагидрофуране (30 л) нагревали до 44°C. К полученной смеси добавляли раствор катализатора в течение 10 мин и полученную смесь нагревали до 56°C в течение 24 мин и оставляли перемешиваться при указанной температуре в течение 2 ч. Смесь охлаждали до 24°C и фазы разделяли. К органической фазе добавляли 5 N водную соляную кислоту (35 л) за 16 мин и смесь нагревали до 54°C в течение 2 ч и оставляли перемешиваться при указанной температуре в течение 13 ч. Смесь концентрировали до 30 л посредством выпаривания при пониженном давлении при 55°C в течение 1,75 ч. К оставшейся смеси добавляли 30 л 2-метил-тетрагидрофурана и смесь снова концентрировали до 30 л посредством выпаривания при пониженном давлении при 55°C в течение 53 мин. К оставшейся смеси добавляли 30 л 2-метил-тетрагидрофурана и смесь снова концентрировали до 30 л посредством выпаривания при пониженном давлении при 55°C в течение 49 мин. Полученный раствор охлаждали до 27°C в течение 57 мин и разбавляли 2-метил-тетрагидрофураном (40 л) и водой (20 л) и смесь перемешивали при 25°C в течение 1 ч. Некоторое количество твердого вещества удаляли фильтрованием. Фазы фильтрата разделяли и водную фазу смешивали с 2-метилтетрагидрофураном (40 л). pH полученной смеси доводили до 8 путем добавления 4 М водного раствора гидроксида натрия (35,7 кг) в течение 50 мин при 20°C и pH стабилизировали при 8,0 путем добавления 8% водного раствора бикарбоната натрия (12 кг) и смесь перемешивали в течение 0,5 ч. Фазы разделяли, органическую фазу нагревали до 60°C, добавляли Si-Thiol (Silicycle 0,59 кг) и смесь перемешивали при 60°C в течение 1 ч. Твердые вещества удаляли фильтрованием и фильтрационный осадок промывали 2-метил-тетрагидрофураном (5 л). К объединенным фильтратам добавляли Si-Thiol (Silicycle 0,59 кг) и смесь перемешивали при 60°C в течение 1 ч. Твердые вещества удаляли фильтрованием и фильтрационный осадок промывали 2-метил-тетрагидрофураном (5 л). К объединенным фильтратам добавляли Si-Thiol (Silicycle 0,59 kg) и смесь перемешивали при 60°C в течение 1 ч. Твердые вещества удаляли фильтрованием и фильтрационный осадок промывали 2-метил-тетрагидрофураном (5 л). Объединенные фильтраты концентрировали до 40 л посредством выпаривания при пониженном давлении. К полученному темно-коричневому раствору добавляли гептан (35 л) при 54°C в течение 20 мин и смесь концентрировали до 30 л посредством выпаривания при пониженном давлении при 60°C и смесь разбавляли гептаном (35 л) и смесь концентрировали снова до 25 л посредством выпаривания при пониженном давлении при 60°C. Полученную густую суспензию охлаждали до 25°C и перемешивали при указанной температуре в течение 14 ч. Твердое вещество собирали фильтрованием, промывали гептаном (15 л) и сушили при пониженном давлении при 60°C с получением неочищенного указанного в заголовке соединения (5,096 кг 12,39 моль, 90%, чистота 99,4% а/а). В результате второго идентичного прогона получали 5,287 кг (12,85 моль, 94%, чистота 99,3% а/а) неочищенного указанного в заголовке соединения. Суспензию 10,305 кг неочищенного продукта в этаноле (72 л) нагревали до 75°C в течение 20 мин. К полученной жидкой суспензии добавляли воду (72 л) 5 порциями (10 л, 11 л, 11 л, 20 л, 20 л) (прозрачный раствор получали после добавления первых двух порций). Смесь концентрировали путем выпаривания при пониженном давлении до объема 56 л. Полученную густую суспензию охлаждали до 20°C и перемешивали при указанной температуре в течение 15 ч. Твердое вещество собирали фильтрованием промывали дважды смесью 1:1 этанола и воды (2×20 л) и сушили до постоянной массы при пониженном давлении при 60°C в течение 2 дней с получением указанного в заголовке соединения (9,835 кг, 23,91 моль, 87% всего, чистота 99,9% а/а) в виде белого твердого вещества, которое плавилось при 220°C.
Пример 5: Альтернативное получение 5-(4,6-диморфолино-1,3,5-триазин-2-ил)-4-(трифторметил)пиридин-2-амина 1
В отличие от примеров 3 и 4 получение соединения 1 проводили без выделения промежуточных соединений, начиная с N'-[5-бром-4-(трифторметил)-2-пиридил]-N,N-диметил-формамидина.

К раствору N'-[5-бром-4-(трифторметил)-2-пиридил]-N,N-диметил-формамидина (Пример 2, 3,40 кг, 11,49 моль, 1 экв.) в тетрагидрофуране (24 л) добавляли по каплям в течение 1,2 ч 20% раствор хлорида изопропилмагния в тетрагидрофуране (2,95 л, 13,4 моль, 1,20 экв.) при температуре 0-4°C. Полученную суспензию перемешивали при 2°C в течение 20 мин, затем нагревали до 20°C в течение а периода 40 мин и перемешивали при указанной температуре в течение 7 мин. К полученной суспензии добавляли 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолан (2,78 кг, 3,00 л, 14,9 моль, 1,3 экв.) в течение 5 мин при 20°C. Суспензию затем нагревали до 54°C в течение периода в 20 мин и перемешивали при указанной температуре в течение 1,5 ч. Полученный темно-коричневый раствор охлаждали до 21°C в течение 0,5 ч и добавляли к холодному (5°C) 15% водному раствору хлорида аммония (20 л) в течение 10 мин при температуре, не превышающей 15°C. Полученную смесь перемешивали в течение 10 мин при 13°C. Фазы разделяли и органическую фазу промывали 3 раза 13% водным раствором хлорида натрия (3×34 л). Количественный анализ 1H ЯМР органической фазы выявил 7,3 моль (64% выход) бороната в 27,3 кг раствора. Указанный раствор хранили в течение 5 дней до проведения сочетания Сузуки без какого-либо разложения, как определено по результатам ВЭЖХ. С целью снижения риска раствор бороната разделяли на два идентичных прогона. К раствору бороната (13,6 кг, 3,64 моль, 1 экв.) добавляли 4-(4-хлор-6-морфолино-1,3,5-триазин-2-ил)морфолин (Пример 1, 1,064 кг, 3,73 моль, 1,02 экв.) и тетрагидрофуран (3,5 л) и смесь нагревали до 40°C с получением однородного раствора. К указанному раствору добавляли раствор, полученный путем смешивания ацетата палладия (0,051 кг, 0,23 моль, 0,04 экв.), трифенилфосфина (0,178 кг, 0,69 моль, 0,12 экв.) и тетрагидрофурана (10 л) при 20°C в течение 30 мин в инертных условиях. К полученной смеси добавляли раствор карбонат калия (2,38 кг, 27,22 моль, 3 экв.) в воде (5,1 л) и тетрагидрофуран (0,5 л). Полученную смесь нагревали до 55°C в течение 20 мин и перемешивали при указанной температуре в течение 3,5 ч. Реакционную смесь охлаждали до 24°C и фазы разделяли. К органической фазе добавляли 16% водный раствор соляной кислотой (4,6 л), смесь нагревали до 55°C в течение 1,3 ч и перемешивали при указанной температуре в течение 14 ч. Указанные два идентичных прогона (28 л каждый) объединяли для дальнейшей процедуры выделения. Полученный раствор концентрировали путем выпаривания на роторном испарителе при 55°C в течение 1,5 ч. К полученному раствору дважды добавляли 2-метил-тетрагидрофуран (21 л) с последующим повторным выпариванием на роторном испарителе при 55°C в течение 0,75 ч. Полученный раствор охлаждали до 27°C, смешивали с 2-метил-тетрагидрофураном (27 л) и высокочистой водой (13 л). Полученную смесь пропускали через пресс-фильтр, в результате чего удаляли и выбрасывали небольшое количество твердого вещества. Фазы фильтрата разделяли и водную фазу смешивали с 2-метилтетрагидрофураном (27 л). pH полученной смеси доводили до 8 посредством добавления по каплям 4 М раствора гидроксида натрия в воде (10 кг) в течение 1,3 ч при 20°C. Фазы разделяли и органическую фазу нагревали до 55°C, смешивали с Si-Thiol (Silicycle продукт No R51030B, 0,57 кг) и перемешивали при 60°C в течение 1 ч. Горячую суспензию фильтровали и твердые вещества промывали 2-метилтетрагидрофураном (3,5 л). Фильтрат снова смешивали со свежим Si-Thiol (0,57 кг) и полученную смесь перемешивали при 60°C в течение дополнительных 1 ч. Полученный раствор концентрировали путем выпаривания 28 л растворителя при пониженном давлении с использованием роторного испарителя. К полученному темно-коричневому раствору добавляли изомеры гептана (23 л). Полученную суспензию концентрировали путем выпаривания 23 л растворителя при пониженном давлении с использованием роторного испарителя при 60°C. К полученной густой суспензии снова добавляли изомеры гептана (23 л) и указанную смесь снова концентрировали путем выпаривания 23 л растворителя при пониженном давлении с использованием роторного испарителя при 60°C. Полученную густую суспензию разбавляли изомерами гептана (10 л), смесь охлаждали до 20°C и перемешивали при указанной температуре в течение 1 ч. Твердое вещество собирали фильтрованием, промывали изомерами гептана и сушили на роторном испарителе при 60°C в течение 1 ч с получением указанного в заголовке соединения в виде коричневого твердого вещества с 98,4% чистотой (1,977 кг, 4,81 моль, 42%). Суспензию 1,95 кг указанного вещества в этаноле (20 л) нагревали до 71°C в течение 0,5 ч и перемешивали при указанной температуре в течение 20 мин. К полученной суспензии добавляли высокочистую воду в течение периода в 20 мин. Полученный темно-коричневый раствор концентрировали с удалением 14 л на роторном испарителе при 75°C в течение 2,5 ч. Полученную суспензию охлаждали до 20°C и перемешивали при указанной температуре в течение 15 ч. Твердое вещество собирали фильтрованием, промывали дважды смесью этанола (3,4 л) и высокочистой воды (3,4 л) и сушили на роторном испарителе при 60°C в течение 24 ч с получением указанного в заголовке соединения в виде беловатого твердого вещества с 99,7% чистотой (1,683 кг, 4,09 моль, 36% выход), плавящегося при 219-220°C.


1H-ЯМР (600 МГц, ДМСО-d6, δ): 8,62 (s, 1Н, о-пиридил-H), 6,98 (s, 2Н, NH2), 6,83 (s, 1Н, m-пиридил-H), 3,76 (m, 8Н, морфолин), 3,63 (m, 8Н, морфолин).
13C-ЯМР (150 МГц, ДМСО-d6, δ): 169,5 (s, триазин), 164,1 (s, триазин), 161,2 (s, о-пиридин), 152,6 (s, о-пиридин), 136,5, 136,2, 136,0, 135,8 (q, р-пиридин), 125,8, 124,0, 122,2, 120,4 (q, CF3), 118,7 (s, m-пиридин), 104,8, 104,7, 104,7, 104,6 (q, m-пиридин), 66,0 (s, морфолин), 43,2 (s, морфолин).
MS (ESI+) m/z: 412,2 [M+H]+. MS (ESI-) m/z: 410,4 [M-H]- и 456,4 [M+HCOO]-.
FT-рамановский спектр до и после динамической сорбции паров (DVS) для 5-(4,6-диморфолино-1,3,5-триазин-2-ил)-4-(трифторметил)пиридин-2-амина 1 см. на Фигуре 1.

Пример 6: Получение 5-(4,6-диморфолино-1,3,5-триазин-2-ил)-4-(трифторметил)пиридин-2-амина гидрохлорида

5-(4,6-диморфолино-1,3,5-триазин-2-ил)-4-(трифторметил)пиридин-2-амин 1 (12 г, 29,2 ммоль, 1 экв.) загружали в 1 л круглодонную колбу и растворяли в ацетоне (400 мл). Затем добавляли 5 М раствор HCl в изопропаноле (8,76 мл, 43,8 ммоль, 1,5 экв.) и белый осадок образовался в течение нескольких минут. Гетерогенную реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Полученную суспензию фильтровали с получением требуемого продукта в виде белого твердого вещества (11,5 г, 88%).
1H ЯМР (ДМСО-d6, 400 МГц, δ): 8,60 (s, 1Н), 7,28 (s, 1Н), 3,74-3,76 (m, 8Н), 3,61-3,64 (m, 8Н).
19F ЯМР (ДМСО-d6, 376 МГц, δ): -59,3 (s, 3F).
Анал. Вычислено для C17H21ClF3N7O2: C, 45,59; H, 4,73; N, 21,89. Найдено: C, 45,49; H, 4,83; N, 21,55.
Пример 7а: Получение 5-(4,6-диморфолино-1,3,5-триазин-2-ил)-4-(трифторметил)пиридин-2-амина гидрохлорида моногидрата

5-(4,6-диморфолино-1,3,5-триазин-2-ил)-4-(трифторметил)пиридин-2-амин 1 (12 г, 29,2 ммоль, 1 экв.) загружали в 1 л круглодонную колбу и растворяли в ацетоне (400 мл). Затем, добавляли 12 М раствор HCl в воде (3,65 мл, 43,8 ммоль, 1,5 экв.) и а белый осадок образовался в течение нескольких минут. Гетерогенную реакционную смесь перемешивали в течение 15 ч при комнатной температуре. Полученную суспензию фильтровали с получением требуемого продукта в виде белого твердого вещества (11,4 г, 88%).
1H ЯМР (ДМСО-d6, 400 МГц, δ): 8,59 (s, 1Н), 7,21 (s, 1Н), 3,74-3,76 (m, 8Н), 3,61-3,64 (m, 8Н).
19F ЯМР (ДМСО-d6, 376 МГц, δ): -59,2 (s, 3F).
Анал. Вычислено для C17H23ClF3N7O3: C, 43,83; H, 4,98; N, 21,05. Найдено: C, 43,89; H, 4,83; N, 21,24.
Пример 7b: Получение 5-(4,6-диморфолино-1,3,5-триазин-2-ил)-4-(трифторметил)пиридин-2-амина метансульфоната
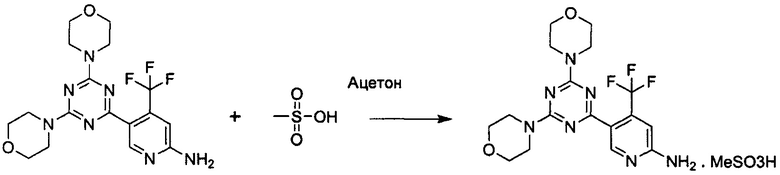
К раствору 5-(4,6-диморфолино-1,3,5-триазин-2-ил)-4-(трифторметил)пиридин-2-амина 1 (411 мг, 1 ммоль, 1 Экв.) в ацетоне (13,7 мл) добавляли раствор метансульфоновой кислоты (65 мкл, 1 ммоль, 1 экв.) в ацетоне (0,65 мл). Белый осадок образовался в течение нескольких минут. Гетерогенную реакционную смесь перемешивали в течение 16 ч при комнатной температуре. Полученную суспензию фильтровали с получением указанного в заголовке соединения в виде белого твердого вещества, плавящегося при 265°C (460 мг, 91% выход, чистота 99,2% а/а).
1Н ЯМР (ДМСО-d6, 400 МГц, δ): 8,58 (s, 1Н), 8,50-7,90 (bs, 3H), 7,18 (s, 1Н), 3,78-3,73 (m, 8Н), 3,65-3,60 (m, 8Н), 2,40 (s, 3H).
19F ЯМР (ДМСО-d6, 376 МГц, δ): -59,3 (s, 3F).
Анал. Вычислено для C18H24F3N7O5S: C, 42,51; H, 5,03; N, 20,01. Найдено: C, 42,60; H, 4,77; N, 19,32.
Пример 8: Получение 4,4'-(6-хлорпиримидин-2,4-диил)диморфолин и 4,4'-(2-хлорпиримидин-4,6-диил)диморфолина

2,4,6-трихлорпиримидин (11,2 г, 61 ммоль, 1 экв.), N,N-диизопропилэтиламин (23,3 мл, 134,2 ммоль, 2,2 экв.) и морфолин (11,7 мл, 134,2 ммоль, 2,2 экв.) загружали в колбу и растворяли в этаноле (120 мл). Колбу снабжали обратным холодильником и помещали на масляную баню, предварительно нагретую до 100°C. Реакционную смесь перемешивали при указанной температуре в течение 18 ч. После чего реакционную смесь охлаждали до к.т. и летучие вещества удаляли при пониженном давлении с использованием роторного испарителя. Полученную смесь растворяли в дихлорметане (100 мл) и промывали дважды водным раствором NaHSO4 (2×80 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с использованием роторного испарителя. Продукты A1 и A2 выделяли с помощью флэш-хроматографии на силикагеле с использованием первой смеси 3:1 циклогексана и этилацетата и затем смеси 1:1 циклогексана и этилацетата в качестве элюента. Фракции продукта объединяли и выпаривали с получением А1 в виде белого порошка (13,8 г, 80%) и А2 в виде белого порошка (2,2 г, 13% выход).
4,4'-(6-хлорпиримидин-2,4-диил)диморфолин A1:
1H ЯМР (CDCl3, 400 МГц, δ): 5,85 (s, 1Н), 3,71-3,75 (m, 12Н), 3,52-3,55 (m, 4Н).
MS m/z: 285,42 [M+H]+.
4,4'-(2-хлорпиримидин-4,6-диил)диморфолин А2:
1H ЯМР (CDCl3, 400 МГц, δ): 5,38 (s, 1Н), 3,73-3,76 (m, 8Н), 3,52-3,54 (m, 8Н).
MS m/z: 285,24 [M+H]+.
Пример 9: Получение 5-(2,6-диморфолинопиримидин-4-ил)-4-(трифторметил)пиридин-2-амина 2

Смесь ацетата палладия (2,2 мг, 0,001 ммоль. 0,04 экв.) и трифенилфосфина (7,6 мг, 0,03 ммоль, 0,12 экв.) в тетрагидрофуране (0,8 мл) перемешивали при комнатной температуре в течение 1 ч. Полученный раствор добавляли в колбу, содержащую раствор 4,4'-(6-хлорпиримидин-2,4-диил)диморфолина (Пример 8, 69 мг, 0,24 ммоль, 1 экв.) и (E)-N,N-диметил-N'-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-4-(трифторметил)пиридин-2-ил)формамидина (Пример 3,100 мг, 0,29 ммоль, 1,2 экв.) в тетрагидрофуране (0,6 мл) и раствор карбоната калия (101 мг, 0,73 ммоль, 3 экв.) в воде (0,3 мл) и полученную смесь нагревали до 55°C. Реакцию контролировали с помощью ТСХ с использованием этилацетата в качестве элюента, и было показано полное превращение через 2 ч. Смесь охлаждали до комнатной температуры, и осторожно добавляли 5 М раствор HCl в диоксане (0,5 мл) (выделение CO2), и смесь перемешивали при 55°C в течение 18 ч. Смесь охлаждали до комнатной температуры и разбавляли 5 М водным раствором HCl (2 мл) и этилацетата (1 мл). Фазы разделяли. pH водной фазы доводили до 7,0 путем добавления а 2 М водного раствора гидроксида натрия и экстрагировали этилацетатом (2×10 мл). Объединенные органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с использованием роторного испарителя. Остаток очищали с помощью флэш-хроматографии на силикагеле (5 г) с использованием первой смеси 1:2 этилацетата и циклогексана и затем чистым этилацетатом в качестве элюента. Фракции продукта объединяли и выпаривали с получением указанного в заголовке соединения в виде беловатого порошка (82 мг, 0,2 ммоль, 82% выход).
1H ЯМР (CDCl3, 400 МГц, δ): 8,27 (s, 1Н), 6,78 (s, 1Н), 5,97 (s, 1Н), 4,79 (s, 2Н), 3,77 (m, 8Н), 3,60 (m, 8Н).
19F ЯМР (CDCl3, 376 МГц, δ): -59,7 (s, 3F).
MS m/z: 411,25 [M+H]+.
Пример 10: Получение 5-(4,6-диморфолинопиримидин-2-ил)-4-(трифторметил)пиридин-2-амина 3

Смесь ацетата палладия (9 мг, 0,04 ммоль. 0,04 экв.) и трифенилфосфина (31 мг, 0,12 ммоль, 0,12 экв.) в тетрагидрофуране (3,1 мл) перемешивали при комнатной температуре в течение 1 ч. Полученный раствор добавляли к раствору 4,4'-(2-хлорпиримидин-4,6-диил)диморфолина (Пример 8, 285 мг, 1 ммоль, 1 экв.) и (E)-N,N-диметил-N'-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-4-(трифторметил)пиридин-2-ил)формамидина (Пример 3, 411 мг, 1,2 ммоль, 1,2 экв.) в тетрагидрофуране (2,5 мл) и раствору карбоната калия (414 мг, 3 ммоль, 3 экв.) в воде (1,25 мл) и полученную смесь нагревали до 55°C. Реакцию контролировали с помощью ТСХ с использованием этилацетата в качестве элюента, и было показано полное превращение через 2 ч. Смесь охлаждали до комнатной температуры и осторожно добавляли 5 М раствор HCl в диоксане (2 мл) (выделение CO2), и смесь перемешивали при 55°C в течение 18 ч. Смесь охлаждали до комнатной температуры и разбавляли 5 М водным раствором HCl (20 мл) и этилацетата (5 мл). Фазы разделяли. pH водной фазы доводили до 7,0 путем добавления а 2 М водного раствора гидроксида натрия и экстрагировали этилацетатом (2×50 мл). Объединенные органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с использованием роторного испарителя. Остаток очищали с помощью флэш-хроматографии на силикагеле (10 г) с использованием первой смеси 1:2 этилацетата и циклогексана и затем чистым этилацетатом в качестве элюента. Фракции продукта объединяли и выпаривали с получением указанного в заголовке соединения в виде беловатого порошка (235 мг, 0,57 ммоль, 57% выход).
1H ЯМР (CDCl3, 400 МГц, δ): 8,86 (s, 1Н), 6,77 (s, 1Н), 5,51 (s, 1Н), 4,78 (s, 2Н), 3,78 (m, 8Н), 3,59 (m, 8Н).
19F ЯМР (CDCl3, 376 МГц, δ): -59,9 (s, 3F).
MS m/z: 411,25 [M+H]+.
Пример 11: Получение 4,4'-(6-хлорпиридин-2,4-диил)диморфолина и 4,4'-(4-хлорпиридин-2,6-диил)диморфолина

К смеси 2,4,6-трихлорпиридина (5 г, 27,5 ммоль. 1 экв.), морфолина (7,2 мл, 82,3 ммоль, 3 экв.), трет-бутоксида натрия (7,9 г, 82,3 ммоль, 3 экв.), (2-бифенил)ди-трет-бутилфосфина (408 мг, 2,7 ммоль, 0,05 экв.) в тетрагидрофуране (80 мл) добавляли Pd(dppf)Cl2 (от Combi-blocks, номер продукта: ОТ-0746), 1 г, 2,7 ммоль, 0,05 экв.). Смесь перемешивали при 80°C в течение 4 ч. Смесь охлаждали до комнатной температуры и выливали в насыщенный раствор NH4Cl (100 мл). Фазы разделяли и водную фазу экстрагировали этилацетатом (2×100 мл). Объединенные органические экстракты сушили над безводным сульфатом натрия, фильтровали и выпаривали при пониженном давлении с использованием роторного испарителя. Продукты B1 и B2 выделяли с помощью флэш-хроматографии на силикагеле с использованием первой смеси 1:4,5 этилацетата и циклогексана и затем смеси 1:1 этилацетата и циклогексана в качестве элюента. Фракции продукта объединяли и выпаривали с получением В1 в виде беловатого порошка (2,45 г, 8,6 ммоль, 31%) и В2 в виде беловатого порошка (2,2 г, 7,8 ммоль, 28% выход).
4,4'-(6-хлорпиридин-2,4-диил)диморфолин) В1:
1H ЯМР (CDCl3, 400 МГц, δ): 6,19 (s, 1Н), 5,77 (s, 1Н), 3,80 (m, 8Н), 3,45 (m, 4Н), 3,24 (m, 4Н).
MS m/z: 283,67 [M+H]+.
4,4'-(4-хлорпиридин-2,6-диил)диморфолин В2:
1H ЯМР (CDCl3, 400 МГц, δ): 6,00 (s, 1Н), 3,78 (m, 8Н), 3,45 (m, 8Н).
MS m/z: 283,56 [M+H]+.
Пример 12: Получение 5-(4,6-диморфолино-2-пиридил)-4-(трифторметил)пиридин-2-амина 4

4,4'-(6-Хлорпиридин-2,4-диил)диморфолин (Пример 11, 281 мг, 1 ммоль, 1 экв.), (E)-N,N-диметил-N'-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-4-(трифторметил)пиридин-2-ил)формамидин (Пример 3, 412 мг, 1,2 ммоль, 1,2 экв.), трехосновный фосфат калия (424 мг, 2 ммоль, 2 экв.) и хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил) [2-(2'-амино-1,1'-бифенил)]-палладия(II) (Sigma-Aldrich (номер продукта: 741825), 39,4 мг, 0,05 ммоль, 0,05 экв.) загружали в колбу. Сосуд помещали в вакуум и затем повторно наполняли азотом. Указанную операцию повторяли три раза и добавляли диоксан (10 мл) с последующим добавлением деионизированной воды (5 мл). Затем колбу помещали на масляную баню, предварительно нагретую до 100°C и перемешивали в течение 24 ч. После чего реакционную смесь охлаждали до комнатной температуры, гасили солевым раствором (20 мл) и экстрагировали этилацетатом (3×40 мл). Органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха при пониженном давлении с использованием роторного испарителя. Неочищенную смесь очищали с помощью флэш-хроматографии на силикагеле (этилацетат, Rf=0,2) с получением указанного в заголовке соединения в виде белой - бледно-желтой пены (360 мг, 88% выход).
1H ЯМР (CDCl3, 400 МГц, δ): 8,26 (в, 1Н), 6,77 (s, 1Н), 6,31-6,31 (d, JHH=1,9 Hz, 1Н), 5,93-5,93 (d, JHH=1,9 Hz, 1H), 4,73 (brs, 2H), 3,79-3,85 (m, 8H), 3,48-3,51 (m, 4H), 3,26-3,29 (m, 4H).
19F ЯМР (CDCl3, 376 МГц, δ): -59,8 (s, 3F).
MS m/z: 410 [M+H]+.
Пример 13: Получение 5-(2,6-диморфолино-4-пиридил)-4-(трифторметил)пиридин-2-амина 5

4,4'-(4-Хлорпиридин-2,6-диил)диморфолин (Пример 11, 140,5 мг, 0,5 ммоль, 1 экв.), (E)-N,N-диметил-N'-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-4-(трифторметил)пиридин-2-ил)формамидин (Пример 3, 206 мг, 0,6 ммоль, 1,2 экв.), трехосновный фосфат калия (212 мг, 1 ммоль, 2 экв.) и хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]-палладия(II) (Sigma-Aldrich (номер продукта: 741825), 19,7 мг, 0,025 ммоль, 0,05 экв.) загружали в колбу. Сосуд помещали в вакуум и затем повторно наполняли азотом. Указанную операцию повторяли три раза и добавляли диоксан (5 мл) с последующим добавлением деионизированной воды (2,5 мл). Колбу затем помещали на масляную баню, предварительно нагретую до 100°C, и перемешивали в течение 24 ч. После чего реакционную смесь охлаждали до комнатной температуры, гасили солевым раствором (10 мл) и экстрагировали этилацетатом (3×20 мл). Органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха при пониженном давлении с использованием роторного испарителя. Неочищенную смесь очищали с помощью флэш-хроматографии на силикагеле (циклогексан / этилацетат : 1/1, Rf=0,2) с получением указанного в заголовке соединения в виде белой - бледно-желтой пены (166 мг, 81% выход).
1H ЯМР (CDCl3, 400 МГц, δ): 8,06 (s, 1Н), 6,77 (s, 1Н), 5,95 (s, 2Н), 4,73 (brs, 2Н), 3,80-3,82 (m, 8Н), 3,47-3,49 (m, 8Н).
19F ЯМР (CDCl3, 376 МГц, δ): -59,9 (s, 3F).
MS m/z: 410 [M+H]+.
Пример 14: Получение 4-(4,6-дихлор-1,3,5-триазин-2-ил)морфолина

К раствору хлорангидрид циануровой кислоты (10,0 г, 54,2 ммоль, 1,0 экв.) в дихлорметане (200 мл) по каплям добавляли морфолин (9,49 мл, 108,4 ммоль, 2,0 экв.) при -10°C. Реакционную смесь перемешивали при указанной температуре в течение 6 ч, разбавляли дихлорметаном (200 мл) и смешивали с насыщенным водным раствором NaHSO4 (50 мл). Фазы разделяли. Органическую фазу последовательно промывали насыщенным водным раствором NaHSO4 (2×50 мл), сушили над безводным Na2SO4, фильтровали и выпаривали при пониженном давлении с получением чистого указанного в заголовке соединение в виде белого твердого вещества (11,7 г, 92% выход).
1H ЯМР (CDCl3, 400 МГц, δ): 3,88 (t, J=4,9 Hz, 4Н), 3,75 (t, J=4,8 Hz, 4H).
MS m/z: 258,6 [M+Na]+.
Пример 15: Получение (E)-N'-(5-(4-хлор-6-морфолино-1,3,5-триазин-2-ил)-4-(трифторметил)пиридин-2-ил)-N,N-диметилформамидина

Смесь ацетата палладия (4,5 мг, 0,02 ммоль, 0,04 экв.) и трифенилфосфина (15,5 мг, 0,06 ммоль, 0,12 экв.) в тетрагидрофуране (1,2 мл) перемешивали при комнатной температуре в течение 1 ч. Полученный раствор добавляли в колбу, содержащую раствор 4-(4,6-дихлор-1,3,5-триазин-2-ил)морфолина (Пример 14, 176 мг, 0,75 ммоль, 1,5 экв.) и (E)-N,N-диметил-N'-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-4-(трифторметил)пиридин-2-ил)формамидина (Пример 3, 171 мг, 0,5 ммоль, 1 экв.) в тетрагидрофуране (1 мл) и раствор карбонат калия (138 мг, 1 ммоль, 2 экв.) в воде (1 мл), и полученную смесь нагревали до 75°C. Реакцию контролировали с помощью ТСХ с использованием этилацетата в качестве элюента, и было показано полное превращение через 2 ч. После чего реакционную смесь охлаждали до комнатной температуры, гасили солевым раствором (10 мл) и экстрагировали этилацетатом (3×20 мл). Органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали досуха при пониженном давлении с использованием роторного испарителя. Неочищенную смесь очищали с помощью флэш-хроматографии на силикагеле (циклогексан / этилацетат 1:3, Rf=0,5) с получением указанного в заголовке соединения в виде белой - бледно-желтой пены (104 мг, 50% выход).
1H ЯМР (CDCl3, 400 МГц, δ): 8,90 (s, 1Н), 8,68 (s, 1Н), 7,25 (s, 1Н), 3,88 (m, 8Н), 3,77 (m, 8Н), 3,24 (s, 3H), 3,22 (s, 3H).
19F ЯМР (CDCl3, 376 МГц, δ): -59,8 (s, 3F).
MS m/z: 415,84 [M+H]+.
Пример 16: Получение (E)-N'-(5-(4,6-диморфолино-1,3,5-триазин-2-ил)-4-(трифторметил)пиридин-2-ил)-N,N-диметилформимидамида

К раствору (E)-N'-(5-(4-хлор-6-морфолино-1,3,5-триазин-2-ил)-4-(трифторметил)пиридин-2-ил)-N,N-диметилформамидина (Пример 15, 207 мг, 0,5 ммоль, 1,0 экв.) в диметилформамиде (2 мл) добавляли морфолин (44 мкл, 0,5 ммоль, 1 экв.) и карбонат калия (69 мг, 0,5 ммоль, 1 экв.). Реакционную смесь помещали на масляную баню, предварительно нагретую до 70°C, и перемешивали при указанной температуре в течение 15 ч. Затем реакционную смесь охлаждали до комнатной температуры, выливали в насыщенный водный раствор NH4Cl (75 мл) и экстрагировали этилацетатом (2×20 мл). Органический слой сушили над безводным Na2SO4, фильтровали и выпаривали при пониженном давлении с использованием роторного испарителя. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием первой смеси 1:2 циклогексан и этилацетат и затем смеси 1:10 циклогексана и этилацетата в качестве элюента. Фракции продукта объединяли и выпаривали с получением указанного в заголовке соединения в виде белого твердого вещества (184 мг, 79% выход).
1H ЯМР (CDCl3, 400 МГц, δ): 8,81 (s, 1Н), 8,56 (s, 1Н), 7,23 (s, 1Н), 3,84 (brs, 8Н), 3,71-3,74 (m, 8Н), 3,13 (s, 3H), 3,12 (s, 3H).
19F ЯМР (CDCl3, 376 МГц, δ): -59,7 (s, 3F).
MS m/z: 467,09 [M+H]+.
Пример 17: Получение 5-(4,6-диморфолино-1,3,5-триазин-2-ил)-4-(трифторметил)-пиридин-2-амина 1
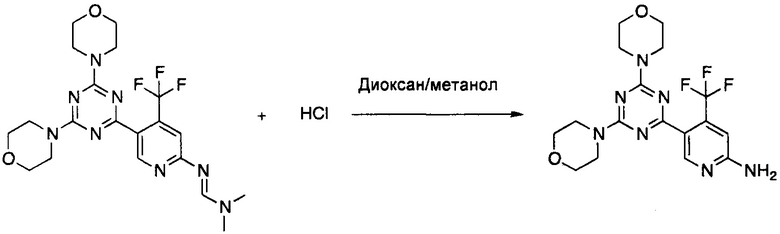
К раствору (E)-N'-(5-(4,6-диморфолино-1,3,5-триазин-2-ил)-4-(трифторметил)пиридин-2-ил)-N,N-диметилформимидамида (Пример 16, 121 мг, 0,26 ммоль, 1,0 экв.) в метаноле (2 мл) добавляли 4 М раствор HCl в диоксане (4 мл, 16 ммоль, 62 экв.). Реакционную смесь помещали на масляную баню, предварительно нагретую при 90°C, и перемешивали при указанной температуре в течение 4 ч. Затем реакционную смесь охлаждали до комнатной температуры, выливали в 2 М водный раствор NaOH (50 мл) и экстрагировали этилацетатом (2×30 мл). Органический слой сушили над безводным Na2SO4, фильтровали и выпаривали при пониженном давлении с использованием роторного испарителя. Остаток очищали с помощью флэш-хроматографии на силикагеле с использованием смеси 1:3 циклогексана и этилацетата в качестве элюента. Фракции продукта объединяли и выпаривали с получением указанного в заголовке соединения в виде белого твердого вещества (77 мг, 72% выход).
| название | год | авторы | номер документа |
|---|---|---|---|
| ТРИАЗИНОВЫЕ, ПИРИМИДИНОВЫЕ И ПИРИДИНОВЫЕ АНАЛОГИ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ТЕРАПЕВТИЧЕСКИХ АГЕНТОВ И ДИАГНОСТИЧЕСКИХ ПРОБ | 2009 |
|
RU2537945C2 |
| ДИФТОРМЕТИЛАМИНОПИРИДИНЫ И ДИФТОРМЕТИЛАМИНОПИРИМИДИНЫ | 2015 |
|
RU2712091C2 |
| ЛЕЧЕНИЕ НЕВРОЛОГИЧЕСКИХ РАССТРОЙСТВ | 2017 |
|
RU2765868C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СПОСОБ ПОЛУЧЕНИЯ 5-(2,6-ДИ-4-МОРФОЛИНИЛ-4-ПИРИМИДИНИЛ)-4-ТРИФТОРМЕТИЛПИРИДИН-2-АМИНА | 2013 |
|
RU2646760C2 |
| ПРОТИВОГЕЛЬМИНТНЫЕ ДЕПСИПЕПТИДНЫЕ СОЕДИНЕНИЯ | 2016 |
|
RU2707298C2 |
| ИНГИБИТОРЫ GCN2 И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2811403C2 |
| НОВЫЕ СОЕДИНЕНИЯ, ИХ ИЗОМЕР ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ В КАЧЕСТВЕ АНТАГОНИСТА ВАНИЛОИДНОГО РЕЦЕПТОРА И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2007 |
|
RU2448108C2 |
| ХИНОЛИНКАРБОКСАМИДНЫЕ И ХИНОЛИНКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ mGLuR2-НЕГАТИВНЫХ АЛЛОСТЕРИЧЕСКИХ МОДУЛЯТОРОВ, КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2610262C2 |
| Соединения, активные по отношению к бромодоменам | 2015 |
|
RU2743074C2 |
| ПРОИЗВОДНЫЕ 1,2,4-ОКСАДИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ГИСТОНДЕЗАЦЕТИЛАЗЫ 6 | 2018 |
|
RU2797613C2 |

Изобретение относится к способу получения производных триазина, пиримидина и пиридина формулы (I) путем конденсации соответствующего галоген-триазина, пиримидина или пиридина посредством реакции сочетания Сузуки с пиридил- или пиримидинил-бораном, где аминофункциональную группу защищают в виде формамидина. Кроме того, изобретение относится к подходящим промежуточным соединениям и способам получения таких промежуточных соединений. Технический результат: разработан новый способ получения соединений формулы (I), позволяющий получать целевые соединения с высоким выходом и чистотой. 5 н. и 14 з.п. ф-лы, 3 табл., 17 пр., 1 ил.
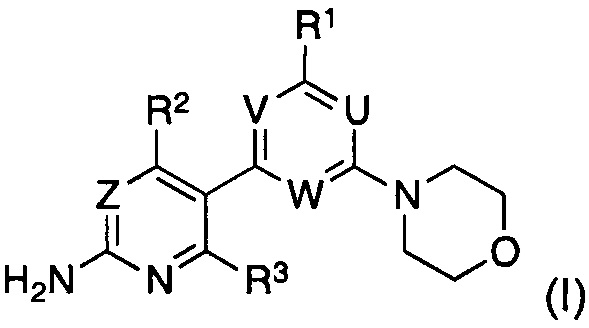
1. Способ получения соединения формулы (I)
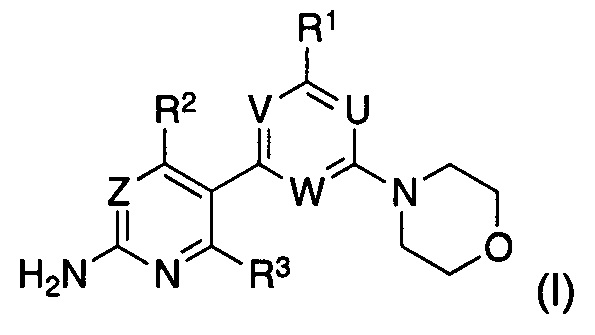
или его стереомера, таутомера или соли,
где U представляет собой CRU или N, где RU выбран из группы, состоящей из водорода, циано, галогена, метила и трифторметила;
V представляет собой CRV или N, где RV выбран из группы, состоящей из водорода, циано, галогена, метила и трифторметила;
W представляет собой CRW или N, где RW выбран из группы, состоящей из водорода, циано, галогена, метила и трифторметила;
при условии, что по меньшей мере один из U, V и W представляет собой N;
Z представляет собой CRZ или N, где RZ выбран из группы, состоящей из водорода, циано, галогена, метила и трифторметила;
R1 выбран из группы, состоящей из водорода, галогена и -N(RT)RS, где RT и RS представляют собой водород или С1-С7алкил или где RT и RS вместе с атомом азота, к которому они присоединены, образуют С3-C8 моно- или бициклическое гетероциклическое кольцо, необязательно содержащее один или более дополнительных атомов в кольце, выбранных из N, О или S, где указанное гетероциклическое кольцо необязательно замещено одной или более группами, независимо выбранными из С1-С7алкила или С3-С7циклоалкила;
R2 выбран из группы, состоящей из водорода, циано, галогена, метила и трифторметила; и
R3 представляет собой водород или галоген, характеризующийся тем, что соединение формулы (II)
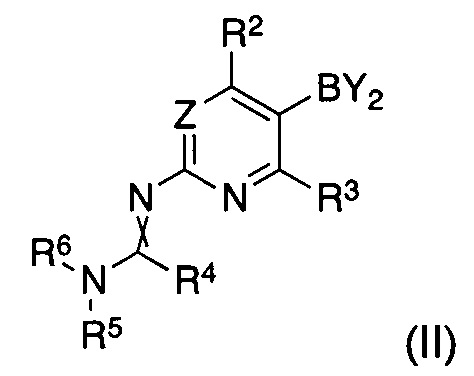
,
где Y2B представляет собой остаток ациклической борной кислоты, ациклического сложного эфира борной кислоты или циклического сложного эфира борной кислоты и R2 и R3 являются такими, как определено для соединения формулы (I);
R4 представляет собой водород, С1-С7алкил или С5-С7-циклоалкил;
R5 и R6 представляют собой С1-С7-алкил или R5 и R6 вместе представляют собой С4-С6-циклоалкил
и скрещенная двойная связь между N и C(R4)N указывает на цис и/или транс двойную связь;
подвергают взаимодействию с соединением формулы (III)
 ,
,
в котором указанные группы U, V, W и R1 являются такими, как определено выше; и
R7 представляет собой галоген;
в водном органическом растворителе или несмешивающейся смеси органический растворитель - вода при температурах от 0°С до температуры кипения растворителя или смеси растворителя в присутствии катализатора на основе фосфина Pd(0) или Pd(II) и основания;
и полученный формамидин формулы (IV)
 ,
,
где заместители имеют значения, как определено выше,
гидролизуют in situ или после выделения в водном растворе кислоты или основания.
2. Способ по п. 1, отличающийся тем, что в соединении формулы (I)
U представляет собой CRU или N, где RU представляет собой водород;
V представляет собой CRV или N, где RV представляет собой водород;
W представляет собой CRW или N, где RW представляет собой водород;
Z представляет собой CRZ или N, где RZ представляет собой водород;
R1 представляет собой морфолино;
R2 представляет собой трифторметил
и R3 представляет собой водород.
3. Способ по п. 1 или 2, отличающийся тем, что в соединении формулы (I)
U представляет собой N;
V представляет собой N;
W представляет собой N;
Z представляет собой CRZ, где RZ представляет собой водород;
R1 представляет собой морфолино;
R2 представляет собой трифторметил и
R3 представляет собой водород.
4. Способ по любому из предшествующих пунктов, отличающийся тем, что в соединении формулы (II)
Y2B представляет собой циклический сложный эфир борной кислоты;
R4 представляет собой водород и
R5 и R6 представляют собой метил.
5. Способ по п. 1, отличающийся тем, что в соединении формулы (I)
U представляет собой CRU или N, где RU представляет собой водород;
V представляет собой CRV или N, где RV представляет собой водород;
W представляет собой CRW или N, где RW представляет собой водород;
Z представляет собой CRZ или N, где RZ представляет собой водород;
R1 представляет собой галоген;
R2 представляет собой трифторметил и
R3 представляет собой водород
и где дополнительно указанный полученный формамидин формулы (IV) подвергают взаимодействию с морфолином до указанной гидролизации.
6. Способ по п. 1, отличающийся тем, что в соединении формулы (I)
U представляет собой CRU или N, где RU представляет собой водород;
V представляет собой CRV или N, где RV представляет собой водород;
W представляет собой CRW или N, где RW представляет собой водород;
Z представляет собой CRZ или N, где RZ представляет собой водород;
R1 представляет собой хлор;
R2 представляет собой трифторметил и
R3 представляет собой водород
и где дополнительно указанный полученный формамидин формулы (IV) подвергают взаимодействию с морфолином до указанной гидролизации.
7. Способ по п. 1, отличающийся тем, что в соединении формулы (I)
U представляет собой N;
V представляет собой N;
W представляет собой N;
Z представляет собой CRZ, где RZ представляет собой водород;
R1 представляет собой галоген или морфолино;
R2 представляет собой трифторметил и
R3 представляет собой водород.
8. Способ по п. 1, отличающийся тем, что в соединении формулы (I)
U представляет собой N;
V представляет собой N;
W представляет собой N;
Z представляет собой CRZ, где RZ представляет собой водород;
R1 представляет собой хлор или морфолино;
R2 представляет собой трифторметил и
R3 представляет собой водород.
9. Способ по п. 1, отличающийся тем, что в соединении формулы (I)
U представляет собой N;
V представляет собой N;
W представляет собой N;
Z представляет собой CRZ, где RZ представляет собой водород;
R1 представляет собой морфолино;
R2 представляет собой трифторметил и
R3 представляет собой водород.
10. Способ по п. 9, отличающийся тем, что в соединении формулы (II)
Y2B представляет собой циклический сложный эфир борной кислоты;
R4 представляет собой водород и
R5 и R6 представляют собой метил.
11. Способ по п. 9, отличающийся тем, что в соединении формулы (II)
Y2B представляет собой пинаколатоборонат;
R4 представляет собой водород и
R5 и R6 представляют собой метил.
12. Способ по п. 9, отличающийся тем, что в соединении формулы (II)
Y2B представляет собой пинаколатоборонат;
R4 представляет собой водород;
R5 и R6 представляют собой метил и
R7 представляет собой хлор.
13. Способ по пп. 1 или 2, отличающийся тем, что органический растворитель выбран из группы, состоящей из тетрагидрофурана, диоксана и толуола.
14. Способ по пп. 1 или 2, отличающийся тем, что катализатор на основе фосфина Pd представляет собой смесь трифенилфосфина и ацетата палладия(II) или дихлорида палладия.
15. Соединение формулы (II)
 ,
,
где Y2B представляет собой остаток борной кислоты, ациклического сложного эфира борной кислоты или циклического сложного эфира борной кислоты;
Z представляет собой CRZ или N, где RZ выбран из группы, состоящей из водорода, циано, галогена, метила и трифторметила;
R2 выбран из группы, состоящей из водорода, циано, галогена, метила и трифторметила;
R3 представляет собой водород или галоген;
R4 представляет собой водород, С1-С7-алкил или С5-С7-циклоалкил;
R5 и R6 представляют собой С1-С7-алкил или R5 и R6 вместе представляют собой С4-С6-циклоалкил
и скрещенная двойная связь между N и C(R4)N указывает на цис и/или транс двойную связь.
16. Соединение формулы (II) по п. 15, отличающееся тем, что в соединении формулы (II)
Y2B представляет собой пинаколатоборонат;
R2 представляет собой трифторметил;
R3 представляет собой водород;
R4 представляет собой водород;
R5 и R6 представляют собой метил.
17. Способ получения соединения формулы (II) по п. 15,
характеризующийся тем, что соединение формулы (V)
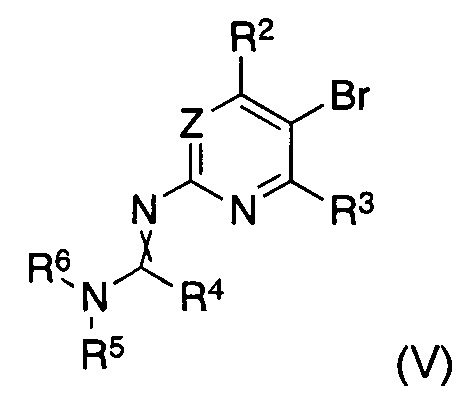 ,
,
в котором указанные группы R2-R6 и группа Z являются такими, как определено для соединения (II) по п. 15, обрабатывают металлоорганическим соединением в органическом растворителе при температурах от -80°С до температуры кипения растворителя и после завершения реакции обмена бром-металл дополнительно подвергают взаимодействию с борорганическим реагентом формулы (VI)
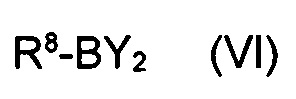 ,
,
где R8 представляет собой уходящую группу и Y является таким, как определено для соединения (II) по п. 15.
18. Соединение формулы (V)
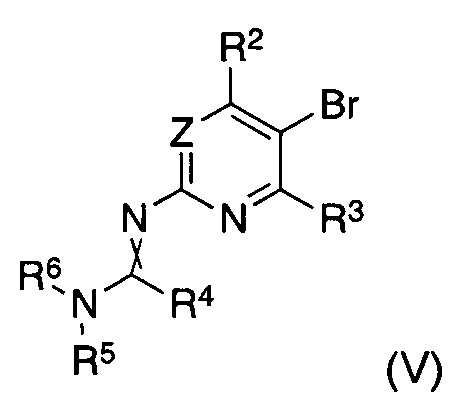 ,
,
где Z представляет собой CRZ, где RZ представляет собой водород;
R2 представляет собой трифторметил;
R3 представляет собой водород;
R4 представляет собой водород;
R5 и R6 представляют собой метил
и скрещенная двойная связь между N и C(R4)N указывает на цис и/или транс двойную связь.
19. Способ получения соединения формулы (V) по п. 18,
характеризующийся тем, что соединение формулы (VII)
 ,
,
в котором указанные группы R2, R3 и Z являются такими, как определено для соединения (V) по п. 18, галогенируют бромом, бромидом меди (II), бромоксоном или N-галогенимидом в инертном органическом растворителе, экстрагируют водным основанием и подвергают взаимодействию с соединением формулы (VIII)
 ,
,
где R4, R5 и R6 в формуле (VIII) являются такими, как определено для соединения (V) по п. 18, и R9 представляет собой С1-С4-алкил или С5-С7-циклоалкил.
| WO 2012109423 A1, 16.08.2012 | |||
| Gomez-Garcia, Omar et al, Tertiary formylated amines by microwave irradiation of N,N-dimethyl-N′-(2-pyridyl)formamidines with methyl vinyl ketone, Heterocycl | |||
| Commun., 20(1), 21-23, 2014 | |||
| Gomez-Garcia, Omar, Reaction of Tosylmethyl Isocyanide with N-Heteroaryl Formamidines: an Alternative Approach to the Synthesis of N-Heteroaryl Tosylimidazoles, Bulletin of the Korean Chemical Society, pp.2807-2810, 2013 | |||
| US 2011224438 A1, 15.09.2011 | |||
| WO 2012044727 A2, 05.04.2012 | |||
| RU 2011114559 A, 20.12.2012. |

