ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к производным 1,2,4-оксадиазола, применимым в качестве ингибиторов гистондезацетилазы 6. Настоящее изобретение также относится к фармацевтическим композициям, содержащим эти соединения, и к их применению в терапии.
УРОВЕНЬ ТЕХНИКИ
Гистондезацетилазы (HDACs) являются частью большого семейства ферментов, которые катализируют отщепление ацетильной группы от гистонов и негистонных белков. HDACs играют критически важную роль в многочисленных биологических процессах, преимущественно вследствие их репрессивного влияния на транскрипцию. У людей имеются четыре класса HDACs, которые включают всего 18 белков: в класс I HDACs входят HDAC1, HDAC2, HDAC3 и HDAC8; в класс II HDACs входят HDAC4, HDAC5, HDAC6, HDAC7, HDAC9 и HDAC10; в класс III HDACs входят Sir2-подобные белки SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6 и SIRT7; и в класс IV HDACs входит HDAC11. Ферменты класса II дополнительно разделяют на два подкласса, класс IIa (HDAC4, HDAC5, HDAC7 и HDAC9) и класс IIb (HDAC6 и HDAC10).
Гистондезацетилаза 6 (HDAC6) катализирует преимущественно дезацетилирование негистонных субстратов, такие как альфа-тубулин, белок теплового шока (Hsp)90 и кортактин.
Сообщали, что активность HDAC6 проявляется в разных патологических состояниях, включая рак, неврологические, цилиопатические, инфекционные, сердечно-сосудистые, инфекционные и иммунные и воспалительные заболевания, которые подробнее рассмотрены ниже. Таким образом, ингибиторы HDAC6 проявились для использования в привлекательном терапевтическом подходе для лечения широкого спектра заболеваний.
Многие из исследуемых в настоящее время ингибиторов HDAC являются pan-HDAC ингибиторами, которые являются неселективными агонистами разных изоформ HDAC. Известно, что pan-HDAC ингибиторы характеризуются значительными побочными эффектами; в частности, токсические побочные эффекты были связаны с ингибированием некоторых изоформ HDAC класса I, в особенности HDAC1 и HDAC2.
Желательно выявить ингибиторы HDAC, которые ингибируют одну или большее количество, но не все изоформы HDAC и, в частности, соединения, которые ингибируют HDAC6, но не ингибируют HDAC1 или HDAC2 или ингибируют их в намного меньшей степени.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Одним объектом настоящего изобретения является соединение формулы (I), описанное ниже, или его соль:
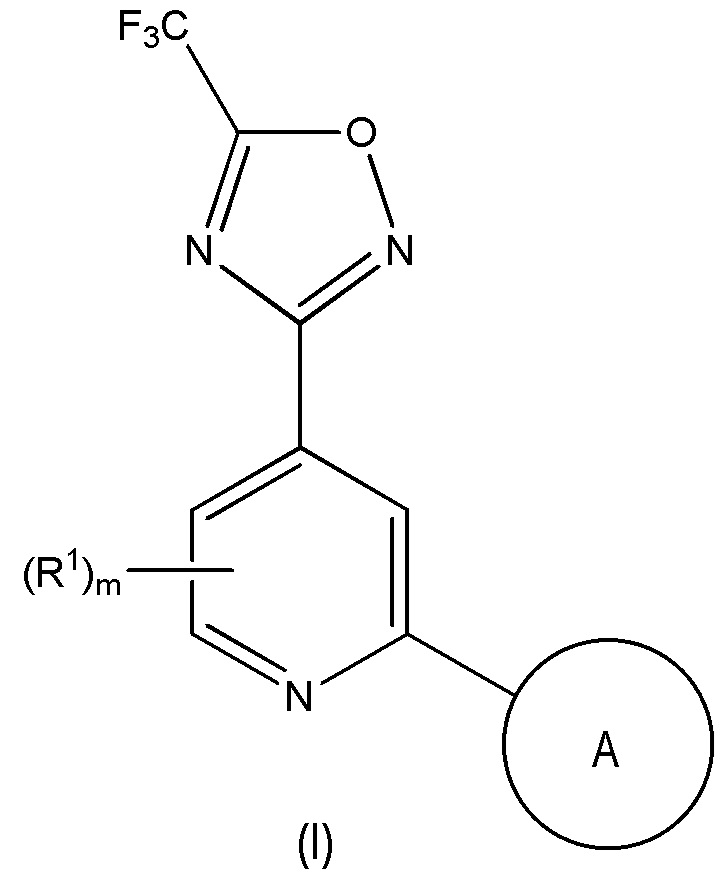
в которой
m равно 0, 1 или 2;
каждый R1 независимо выбран из галогена, метила и трифторметила;
A выбран из:
5- или 6-членного моноциклического или 9- или 10-членного бициклического гетероарильного кольца, которое является полностью ароматическим, и
9- или 10-членного бициклическго гетероарильного кольца, состоящего из 5- или 6-членного моноциклического гетероарильного кольца, конденсированное с насыщенным или частично ненасыщенным карбоциклическим или гетероциклическим кольцом, где 9- или 10-членное бициклическое гетероарильное кольцо связано с остальной частью молекулы через 5- или 6-членное моноциклическое гетероарильное кольцо,
где A содержит один кольцевой атом N в положении рядом с кольцевым атомом, через который кольцо A присоединено к остальной части молекулы, где A необязательно содержит от 1 до 3 дополнительных кольцевых гетероатомов, независимо выбранных из N, O и S, и
где A необязательно замещен одним или двумя R2 и дополнительно A необязательно замещен одним R3;
каждый R2 независимо выбран из галогена, C1-6 алкила, C1-6 галогеналкила, C3-7 циклоалкила и -(C1-6 алкилен)-OR4;
R3 выбран из: -L1-R5, -L2-OR6, -L3-NR7R8, -L4-CONR9R10, -L5-NR11COR12, -Y-L6-OR6 и -Y-L7-NR7R8;
L1, L2, L3, L4 и L5 каждый независимо выбраны из связи и C1-6 алкилена;
L6 и L7 каждый независимо выбраны из C2-6 алкилена;
каждый Y независимо выбран из -O-, -NR13-, -CONR14- и -NR15CO-;
каждый R4 независимо выбран из водорода, C1-6 алкила, C1-6 галогеналкила, C3-7 циклоалкила и C3-7 циклоалкил-C1-6 алкила;
каждый R5 независимо выбран из карбоциклила, арила, гетероциклила и гетероарила, где карбоциклил, арил, гетероциклил и гетероарил каждый необязательно замещены одним или большим количеством R16;
R6 и R12 каждый независимо выбраны из водорода, C1-6 алкила, C1-6 галогеналкила и -L1-R5;
R7 и R8 каждый независимо выбраны из водорода, C1-6 алкила, C1-6 галогеналкила, -(C1-6 алкилен)-OR4 и -L1-R5;
R9 и R10 каждый независимо выбраны из водорода, C1-6 алкила, C1-6 галогеналкила, -(C1-6 алкилен)-OR4 и -L1-R5, или R9 и R10 вместе с атомом N, к которому они присоединены, образуют насыщенное 4-12-членное гетероциклическое кольцо, необязательно содержащее один дополнительный гетероатом, выбранный из N, O и S, где указанное гетероциклическое кольцо необязательно замещено одним или большим количеством R16;
R11, R13, R14 и R15 каждый независимо выбраны из водорода, C1-6 алкила, C1-6 галогеналкила, C3-7 циклоалкила, C3-7 циклоалкил-C1-6 алкила и -(C1-6 алкилен)-OR4;
каждый R16 независимо выбран из C1-6 алкила, C1-6 галогеналкила, галогена, C1-6 алкокси, C1-6 галогеналкокси, -OH, -NR17R18, -COR19, -CN, -L8-карбоциклила, -L8-арила, -L8-гетероциклила и -L8-гетероарила, где карбоциклил в -L8-карбоциклил, арил в -L8-арил, гетероциклил в -L8-гетероциклил и гетероарил в -L8-гетероарил каждый необязательно замещены одним или большим количеством R20;
каждый L8 независимо выбран из связи и C1-6 алкилена;
R17 и R18 каждый независимо выбраны из водорода, C1-6 алкила, C1-6 галогеналкила, C3-7 циклоалкила и C3-7 циклоалкил-C1-6 алкила;
R19 выбран из водорода, C1-6 алкила и C1-6 галогеналкила; и
каждый R20 независимо выбран из C1-6 алкила, C1-6 галогеналкила, галогена, C1-6 алкокси, C1-6 галогеналкокси, -OH, -NR17R18, -COR19 и -CN.
Соединения формулы (I), описанные в настоящем изобретении, являются ингибиторами HDACs, в частности, HDAC6. Эти соединения и фармацевтические композиции, содержащие эти соединения, применимы для лечения заболеваний, связанных с HDAC6. Например, заболеванием является рак, аутоиммунное или воспалительное заболевание, отторжение трансплантата, цилиопатия, заболевание нервной системы, психическое нарушение или нарушение поведения, сердечно-сосудистое заболевание, атрофия мышц или кахексия.
Настоящее изобретение также относится к фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения в качестве лекарственного средства.
Настоящее изобретение также относится к соединению формулы (I) или его фармацевтически приемлемой соли или фармацевтической композиции, содержащей указанное соединение и фармацевтически приемлемый носитель, для применения для лечения заболевания, связанного с HDAC6.
Настоящее изобретение также относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения заболевания, связанного с HDAC6.
Настоящее изобретение также относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для лечения заболевания, связанного с HDAC6.
Настоящее изобретение также относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения в качестве ингибитора HDAC6.
Настоящее изобретение также относится к способу лечения заболевания, связанного с HDAC6, включающему введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли нуждающемуся в нем пациенту.
Настоящее изобретение также относится к способу ингибирования активности HDAC6, включающему введение пациенту, нуждающемуся в таком лечении, количества соединения формулы (I) или его фармацевтически приемлемой соли, достаточного для ингибирования активности HDAC6.
Настоящее изобретение также относится к соединению формулы (I) или его фармацевтически приемлемой соли или фармацевтической композиции, содержащей указанное соединение и фармацевтически приемлемый носитель, для применения для лечения заболевания, выбранного из рака, аутоиммунного или воспалительного заболевания, отторжения трансплантата, цилиопатии, заболевания нервной системы, психического нарушения или нарушения поведения, инфекционного заболевания, сердечно-сосудистого заболевания, атрофии мышц и кахексии.
Настоящее изобретение также относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения заболевания, выбранного из рака, аутоиммунного или воспалительного заболевания, отторжения трансплантата, цилиопатии, заболевания нервной системы, психического нарушения или нарушения поведения, инфекционного заболевания, сердечно-сосудистого заболевания, атрофии мышц и кахексии.
Настоящее изобретение также относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для лечения заболевания, выбранного из рака, аутоиммунного или воспалительного заболевания, отторжения трансплантата, цилиопатии, заболевания нервной системы, психического нарушения или нарушения поведения, инфекционного заболевания, сердечно-сосудистого заболевания, атрофии мышц и кахексии.
Настоящее изобретение также относится к способу лечения заболевания, выбранного из рака, аутоиммунного или воспалительного заболевания, отторжения трансплантата, цилиопатии, заболевания нервной системы, психического нарушения или нарушения поведения, инфекционного заболевания, сердечно-сосудистого заболевания, атрофии мышц и кахексии, включающему введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли нуждающемуся в нем пациенту.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединению формулы (I) или его соли:

в которой
m равно 0, 1 или 2;
каждый R1 независимо выбран из галогена, метила и трифторметила;
A выбран из:
5- или 6-членного моноциклического или 9- или 10-членного бициклического гетероарильного кольца, которое является полностью ароматическим, и
9- или 10-членное бициклического гетероарильного кольца, состоящего из 5- или 6-членного моноциклического гетероарильного кольца, конденсированного с насыщенным или частично ненасыщенным карбоциклическим или гетероциклическим кольцом, где 9- или 10-членное бициклическое гетероарильное кольцо связано с остальной частью молекулы через 5- или 6-членное моноциклическое гетероарильное кольцо,
где A содержит один кольцевой атом N в положении рядом с кольцевым атомом, через который кольцо A присоединено к остальной части молекулы, где A необязательно содержит от 1 до 3 дополнительных кольцевых гетероатомов, независимо выбранных из N, O и S, и
где A необязательно замещен одним или двумя R2 и дополнительно A необязательно замещен одним R3;
каждый R2 независимо выбран из галогена, C1-6 алкила, C1-6 галогеналкила, C3-7 циклоалкила и -(C1-6 алкилен)-OR4;
R3 выбран из: -L1-R5, -L2-OR6, -L3-NR7R8, -L4-CONR9R10, -L5-NR11COR12, -Y-L6-OR6 и -Y-L7-NR7R8;
L1, L2, L3, L4 и L5 каждый независимо выбраны из связи и C1-6 алкилена;
L6 и L7 каждый независимо выбраны из C2-6 алкилена;
каждый Y независимо выбран из -O-, -NR13-, -CONR14- и -NR15CO-;
каждый R4 независимо выбран из водорода, C1-6 алкила, C1-6 галогеналкила, C3-7 циклоалкила и C3-7 циклоалкил-C1-6 алкила;
каждый R5 независимо выбран из группы, включающей карбоциклил, арил, гетероциклил и гетероарил, где карбоциклил, арил, гетероциклил и гетероарил каждый необязательно замещены одним или большим количеством R16;
R6 и R12 каждый независимо выбраны из водорода, C1-6 алкила, C1-6 галогеналкила и -L1-R5;
R7 и R8 каждый независимо выбраны из водорода, C1-6 алкила, C1-6 галогеналкила, -(C1-6 алкилен)-OR4 и -L1-R5;
R9 и R10 каждый независимо выбраны из водорода, C1-6 алкила, C1-6 галогеналкила, -(C1-6 алкилен)-OR4 и -L1-R5, или R9 и R10 вместе с атомом N, к которому они присоединены, образуют насыщенное 4-12-членное гетероциклическое кольцо, необязательно содержащее один дополнительный гетероатом, выбранный из N, O и S, где указанное гетероциклическое кольцо необязательно замещено одним или большим количеством R16;
R11, R13, R14 и R15 каждый независимо выбраны из водорода, C1-6 алкила, C1-6 галогеналкила, C3-7 циклоалкила, C3-7 циклоалкил-C1-6 алкила и -(C1-6 алкилен)-OR4;
каждый R16 независимо выбран из C1-6 алкила, C1-6 галогеналкила, галогена, C1-6 алкокси, C1-6 галогеналкокси, -OH, -NR17R18, -COR19, -CN, -L8-карбоциклила, -L8-арила, -L8-гетероциклила и -L8-гетероарила, где карбоциклил в -L8-карбоциклил, арил в -L8-арил, гетероциклил в -L8-гетероциклил и гетероарил в -L8-гетероарил каждый необязательно замещены одним или большим количеством R20;
каждый L8 независимо выбран из связи и C1-6 алкилена;
R17 и R18 каждый независимо выбраны из водорода, C1-6 алкила, C1-6 галогеналкила, C3-7 циклоалкила и C3-7 циклоалкил-C1-6 алкила;
R19 выбран из водорода, C1-6 алкила и C1-6 галогеналкила; и
каждый R20 независимо выбран из C1-6 алкила, C1-6 галогеналкила, галогена, C1-6 алкокси, C1-6 галогеналкокси, -OH, -NR17R18, -COR19 и -CN.
Варианты осуществления настоящего изобретения описаны в последующих параграфах. Каждый из вариантов осуществления, описанный ниже, можно объединить с любым другим вариантом осуществления, описанным в настоящем изобретении, который не согласуется с вариантом осуществления, с которые он объединяется.
Комбинациями заместителей и переменных, приведенными в настоящем изобретении, являются только такие, которые приводят к образованию стабильных соединений. Термин "стабильный" при использовании в настоящем изобретении означает соединения, которые обладают стабильностью, достаточной для обеспечения изготовления и которые сохраняют целостность соединения в течение периода времени, достаточного для его использования в целях, подробно описанных в настоящем изобретении (например, для терапевтического введения субъекту).
Кроме того, каждый из вариантов осуществления, описанный в настоящем изобретении, включает в свой объем соли (например, фармацевтически приемлемые соли) соединений, описанных в настоящем изобретении. Соответственно, выражение "или его соль" (также включая "или его фармацевтически приемлемая соль") подразумевается в описании всех соединений, описанных в настоящем изобретении. Настоящее изобретение, в частности, также относится ко всем соединениям, описанным в настоящем изобретении, в несолевой форме.
В соединении формулы (I) каждый R1 независимо выбран из галогена, метила и трифторметила, и предпочтительно, если каждый R1 представляет собой фтор. Следует понимать, что каждый заместитель R1 может находится у любого доступного кольцевого атома C пиридинового кольца, к которому присоединен R1.
Предпочтительно, если в соединении формулы (I) m равно 0.
В соединении формулы (I) A представляет собой циклическую группу, выбранную из:
5- или 6-членного моноциклического или 9- или 10-членного бициклического гетероарильного кольца, которое является полностью ароматическим, и
9- или 10-членного бициклического гетероарильного кольца, состоящего из 5- или 6-членного моноциклического гетероарильного кольца, конденсированного с насыщенным или частично ненасыщенным карбоциклическим или гетероциклическим кольцом, где 9- или 10-членное бициклическое гетероарильное кольцо связано с остальной частью молекулы через 5- или 6-членное моноциклическое гетероарильное кольцо,
где A содержит один кольцевой атом N в положении рядом с кольцевым атомом, через который кольцо A присоединено к остальной части молекулы, где A необязательно содержит от 1 до 3 дополнительных кольцевых гетероатомов, независимо выбранных из N, O и S (и где все остальные кольцевые атомы A являются атомами углерода) и где A необязательно замещен одним или двумя R2 и дополнительно A необязательно замещен одним R3. Следует понимать, что кольцо A может быть присоединено к остальной части молекулы через кольцевой атом C или кольцевой атом N кольца A.
Следует понимать, что каждый заместитель R2 или R3, который присоединен к кольцу A, может находиться у любого доступного кольцевого атома. В частности, любой заместитель R2 или R3 (если содержится) может быть присоединен к кольцевому атому C или кольцевому атому N кольца A. Также следует понимать, что, если кольцо A является бициклическим кольцом, заместитель (заместители) R2 и/или R3 может быть присоединен к любому доступному кольцевому атому (например, к любому доступному кольцевому атому C или к любому доступному кольцевому атому N) любого из колец, образующих бициклическую кольцевую группу A. Например, если кольцо A является бициклическим кольцом, один или два необязательных заместителя R2 (если содержатся) и необязательный заместитель R3 (если содержится) могут быть присоединены к кольцу, которое не содержит кольцевой атом, через который A присоединено к остальной части молекулы, или указанный необязательный заместитель (заместители) может быть присоединен к кольцу, которое кольцевой атом, через который A присоединено к остальной части молекулы, или соответствующие необязательные заместители могут быть присоединены к обеим кольцам, образующих бициклическую кольцевую группу A. Кроме того, следует понимать, что один или два необязательных заместителя R2 (если содержатся) и необязательный заместитель R3 (если содержится) обычно присоединены к разным кольцевым атомам A. Присоединение двух из этих необязательных заместителей к одному и тому же кольцевому атому A возможно только если соответствующий кольцевой атом обладает достаточным количеством положений присоединения. Например, если A представляет собой 2,3-дигидро-1H-пирроло[3,2-c]пиридин-6-ильное кольцо (которое состоит из пиридинового кольца, сконденсированного с пирролидиновым кольцом), кольцевой атом -CH2- пирролидинового фрагмента 2,3-дигидро-1H-пирроло[3,2-c]пиридин-6-ильного кольца может быть замещен двумя из указанных выше необязательных заместителей (например, двумя заместителями R2, что дает кольцевой атом -C(R2)(R2)-, или одним заместителем R2 и одним заместителем R3, что дает кольцевой атом -C(R2)(R3)-).
Неограничивающие примеры циклических групп A включают группы, приведенные ниже в таблице 1, и любую их таутомерную форму:
Таблица 1:


где любая из указанных групп A, приведенных в таблице 1, необязательно замещена одним или двумя R2 и дополнительно любая из указанных A необязательно замещена одним R3.
В некоторых вариантах осуществления в соединении формулы (I) A выбран из групп, приведенных в таблице 1 (включая любую их таутомерную форму), где A необязательно замещен одним или двумя R2 и дополнительно A необязательно замещен одним R3.
Предпочтительно, если в соединении формулы (I) A выбран из:
5- или 6-членного моноциклического или 9- или 10-членного бициклического гетероарильного кольца, которое является полностью ароматическим, и
9- или 10-членного бициклического гетероарильного кольца, состоящего из 5- или 6-членного моноциклического гетероарильного кольца, конденсированного с насыщенным или частично ненасыщенным карбоциклическим или гетероциклическим кольцом, где 9- или 10-членное бициклическое гетероарильное кольцо связано с остальной частью молекулы через 5- или 6-членное моноциклическое гетероарильное кольцо,
где A содержит один кольцевой атом N в положении рядом с кольцевым атомом, через который кольцо A присоединено к остальной части молекулы, где A необязательно содержит от 1 до 3, предпочтительно 1 или 2, дополнительных кольцевых атомов N (и где все остальные кольцевые атомы A являются атомами углерода) и где A необязательно замещен одним или двумя R2 и дополнительно A необязательно замещен одним R3.
Более предпочтительно, если в соединении формулы (I) A представляет собой 5- или 6-членное моноциклическое или 9- или 10-членное бициклическое гетероарильное кольцо, которое является полностью ароматическим, где A содержит один кольцевой атом N в положении рядом с кольцевым атомом, через который кольцо A присоединено к остальной части молекулы, где A необязательно содержит от 1 до 3, предпочтительно 1 или 2, дополнительных кольцевых атомов N (и где все остальные кольцевые атомы A являются атомами углерода) и где A необязательно замещен одним или двумя, предпочтительно одним R2 и дополнительно A необязательно замещен одним R3.
Еще более предпочтительно, если в соединении формулы (I) A выбран из циклических групп, приведенных ниже:
 ,
,
где A необязательно замещен одним или двумя, предпочтительно одним R2 и дополнительно A необязательно замещен одним R3.
Особенно предпочтительно, если в соединении формулы (I) A выбран из циклических групп, приведенных ниже:
 ,
,
где A необязательно замещен одним или двумя, предпочтительно одним R2 и дополнительно A необязательно замещен одним R3.
В некоторых вариантах осуществления в соединении формулы (I) A является замещенным, т. е. содержится по меньшей мере один из необязательных заместителей R2 и/или R3. Указанный заместитель (заместители) может быть присоединен к любому доступному кольцевому атому A (включая любой доступный кольцевой атом N), как указано выше. В некоторых вариантах осуществления в соединении формулы (I) A замещено одним R3 и дополнительно необязательно замещено одним или двумя (предпочтительно одним) R2.
В некоторых других вариантах осуществления, в соединении формулы (I) A является незамещенным (т. е. A не содержит какой-либо необязательный заместитель R2 или R3).
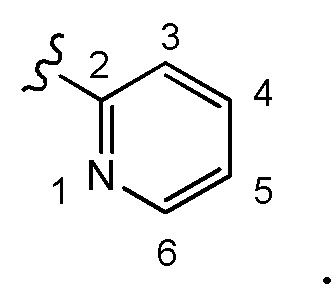
В некоторых предпочтительных вариантах осуществления в соединении формулы (I) A представляет собой
 , где A необязательно замещен одним или двумя, предпочтительно одним R2 и дополнительно A необязательно замещен одним R3, где предпочтительно, если A замещено одним R3 и дополнительно необязательно замещено одним или двумя, предпочтительно одним R2. В некоторых вариантах осуществления A замещено одним R3 и не содержит необязательный заместитель (заместители) R2. Предпочтительно, если заместитель R3, если содержится, находится у кольцевого атома C в положении 4 или 5 пиридильного кольца A в соответствии с нумерацией, приведенной ниже в химической структуре:
, где A необязательно замещен одним или двумя, предпочтительно одним R2 и дополнительно A необязательно замещен одним R3, где предпочтительно, если A замещено одним R3 и дополнительно необязательно замещено одним или двумя, предпочтительно одним R2. В некоторых вариантах осуществления A замещено одним R3 и не содержит необязательный заместитель (заместители) R2. Предпочтительно, если заместитель R3, если содержится, находится у кольцевого атома C в положении 4 или 5 пиридильного кольца A в соответствии с нумерацией, приведенной ниже в химической структуре:
В некоторых других предпочтительных вариантах осуществления в соединении формулы (I) A представляет собой
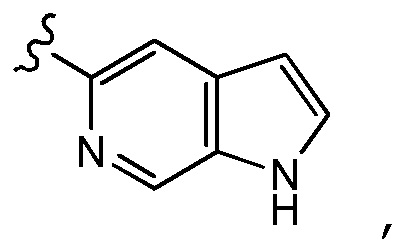 где A необязательно замещен одним или двумя, предпочтительно одним R2 и дополнительно A необязательно замещен одним R3. Предпочтительно, если один или два необязательных заместителя R2 (если содержится) и необязательный заместитель R3 (если содержится) присоединены к пиррольному кольцу, образующему часть кольца A. В некоторых вариантах осуществления A является незамещенным. Более предпочтительно, если A замещено одним R3 и дополнительно необязательно замещено одним или двумя, предпочтительно одним R2, где заместитель R3 и один или два необязательных заместителя R2 (если содержится) предпочтительно присоединены к пиррольному кольцу, образующему часть кольца A.
где A необязательно замещен одним или двумя, предпочтительно одним R2 и дополнительно A необязательно замещен одним R3. Предпочтительно, если один или два необязательных заместителя R2 (если содержится) и необязательный заместитель R3 (если содержится) присоединены к пиррольному кольцу, образующему часть кольца A. В некоторых вариантах осуществления A является незамещенным. Более предпочтительно, если A замещено одним R3 и дополнительно необязательно замещено одним или двумя, предпочтительно одним R2, где заместитель R3 и один или два необязательных заместителя R2 (если содержится) предпочтительно присоединены к пиррольному кольцу, образующему часть кольца A.
В некоторых других предпочтительных вариантах осуществления в соединении формулы (I) A представляет собой
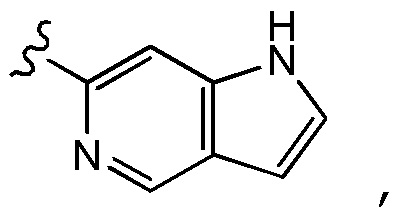 где A необязательно замещен одним или двумя, предпочтительно одним R2 и дополнительно A необязательно замещен одним R3. Предпочтительно, если один или два необязательных заместителя R2 (если содержится) и необязательный заместитель R3 (если содержится) присоединены к пиррольному кольцу, образующему часть кольца A. В некоторых предпочтительных вариантах осуществления A является незамещенным. В некоторых других предпочтительных вариантах осуществления A замещено одним R3 и дополнительно необязательно замещено одним или двумя, предпочтительно одним R2, где заместитель R3 и один или два необязательных заместителя R2 (если содержится) предпочтительно присоединены к пиррольному кольцу, образующему часть кольца A.
где A необязательно замещен одним или двумя, предпочтительно одним R2 и дополнительно A необязательно замещен одним R3. Предпочтительно, если один или два необязательных заместителя R2 (если содержится) и необязательный заместитель R3 (если содержится) присоединены к пиррольному кольцу, образующему часть кольца A. В некоторых предпочтительных вариантах осуществления A является незамещенным. В некоторых других предпочтительных вариантах осуществления A замещено одним R3 и дополнительно необязательно замещено одним или двумя, предпочтительно одним R2, где заместитель R3 и один или два необязательных заместителя R2 (если содержится) предпочтительно присоединены к пиррольному кольцу, образующему часть кольца A.
В некоторых других предпочтительных вариантах осуществления в соединении формулы (I) A представляет собой
 где A необязательно замещен одним или двумя, предпочтительно одним R2 и дополнительно A необязательно замещен одним R3.
где A необязательно замещен одним или двумя, предпочтительно одним R2 и дополнительно A необязательно замещен одним R3.
В некоторых других предпочтительных вариантах осуществления в соединении формулы (I) A представляет собой
 , где A необязательно замещен одним или двумя, предпочтительно одним R2 и дополнительно A необязательно замещен одним R3.
, где A необязательно замещен одним или двумя, предпочтительно одним R2 и дополнительно A необязательно замещен одним R3.
В некоторых вариантах осуществления в соединении формулы (I) каждый R2 независимо выбран из галогена, C1-4 алкила, C1-4 галогеналкила, C3-7 циклоалкила и -(C1-4 алкилен)-OR4.
Предпочтительно, если в соединении формулы (I) каждый R2 независимо выбран из C1-4 алкила, C1-4 галогеналкила и -(C1-4 алкилен)-OR4. В некоторых вариантах осуществления каждый R2 независимо выбран из C1-4 алкила и -(C2-4 алкилен)-OR4. В некоторых вариантах осуществления один R2 выбран из метила, этила, пропила и бутила (например, н-бутил). В некоторых вариантах осуществления один R2 представляет собой -CH2CH2-OCH3.
В некоторых вариантах осуществления в соединении формулы (I) R3 выбран из -L1-R5, -L2-OR6, -L3-NR7R8, -CONR9R10, -NR11COR12 и -Y-L7-NR7R8, где предпочтительно, если Y выбран из -O- и -NR13-.
В некоторых предпочтительных вариантах осуществления в соединении формулы (I) R3 представляет собой -L1-R5, где предпочтительно, если R5 выбран из гетероциклила и гетероарила, где гетероциклил и гетероарил каждый необязательно замещены одним или большим количеством R16.
В некоторых предпочтительных вариантах осуществления в соединении формулы (I) R3 представляет собой -CONR9R10 или -NR11COR12.
В некоторых предпочтительных вариантах осуществления в соединении формулы (I) R3 представляет собой -Y-L7-NR7R8, где Y выбран из -O- и -NR13-.
В некоторых предпочтительных вариантах осуществления в соединении формулы (I) R3 представляет собой -OR6, где R6 представляет собой -L1-R5, где L1 в указанном -L1-R5 предпочтительно представляет собой C1-6 алкилен, более предпочтительно C1-4 алкилен, и R5 в указанном -L1-R5 предпочтительно выбран из гетероциклила и гетероарила, где гетероциклил и гетероарил каждый необязательно замещены одним или большим количеством R16, или R3 представляет собой -NR7R8, где один из R7 или R8 представляет собой -L1-R5, где L1 в указанном -L1-R5 предпочтительно представляет собой C1-6 алкилен, более предпочтительно C1-4 алкилен, и R5 в указанном -L1-R5 предпочтительно выбран из гетероциклила и гетероарила, где гетероциклил и гетероарил каждый необязательно замещены одним или большим количеством R16.
В некоторых предпочтительных вариантах осуществления в соединении формулы (I) R3 представляет собой -L2-OR6 или -L3-NR7R8, где L2 и L3 каждый независимо выбраны из C1-6 алкилена, предпочтительно C1-4 алкилена.
В некоторых вариантах осуществления в соединении формулы (I) R11, R13, R14 и R15 каждый независимо выбраны из водорода, C1-6 алкила и C1-6 галогеналкила.
В некоторых вариантах осуществления в соединении формулы (I) каждый R16 независимо выбран из C1-6 алкила, C1-6 галогеналкила, галогена, C1-6 алкокси, C1-6 галогеналкокси, -OH, -NR17R18, -COR19, -CN и C3-7 циклоалкила.
Предпочтительный вариант осуществления относится к соединению формулы (I) или его соли, где:
A представляет собой циклическую группу, выбранную из:
5- или 6-членного моноциклического или 9- или 10-членного бициклического гетероарильного кольца, которое является полностью ароматическим, и
9- или 10-членного бициклического гетероарильного кольца, состоящего из 5- или 6-членного моноциклического гетероарильного кольца, конденсированного с насыщенным или частично ненасыщенным карбоциклическим или гетероциклическим кольцом, где 9- или 10-членное бициклическое гетероарильное кольцо связано с остальной частью молекулы через 5- или 6-членное моноциклическое гетероарильное кольцо,
и предпочтительно A представляет собой 5- или 6-членное моноциклическое или 9- или 10-членное бициклическое гетероарильное кольцо, которое является полностью ароматическим,
где A содержит один кольцевой атом N в положении рядом с кольцевым атомом, через который кольцо A присоединено к остальной части молекулы, где A необязательно содержит от 1 до 3, предпочтительно 1 или 2, дополнительных кольцевых атомов N (и где все остальные кольцевые атомы A являются атомами углерода) и где A необязательно замещен одним или двумя R2 и дополнительно A необязательно замещен одним R3;
R3, если содержится, выбран из -L1-R5, -L2-OR6, -L3-NR7R8, -CONR9R10, -NR11COR12 и -Y-L7-NR7R8, где предпочтительно, если Y выбран из -O- и -NR13-; и
где предпочтительно, если m равно 0.
Более предпочтительный вариант осуществления относится к соединению формулы (I) или его соли, где:
A выбран из циклических групп, приведенных ниже:
и предпочтительно, если A выбран из циклических групп, приведенных ниже:
 ,
,
где A необязательно замещен одним или двумя, предпочтительно одним R2 и дополнительно A необязательно замещен одним R3;
R3, если содержится, выбран из -L1-R5, -L2-OR6, -L3-NR7R8, -CONR9R10, -NR11COR12 и -Y-L7-NR7R8, где предпочтительно, если Y выбран из -O- и -NR13-; и
где предпочтительно, если m равно 0.
В особенно предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы (I) или его соли, описывающемуся формулой (IIa) или (IIb):
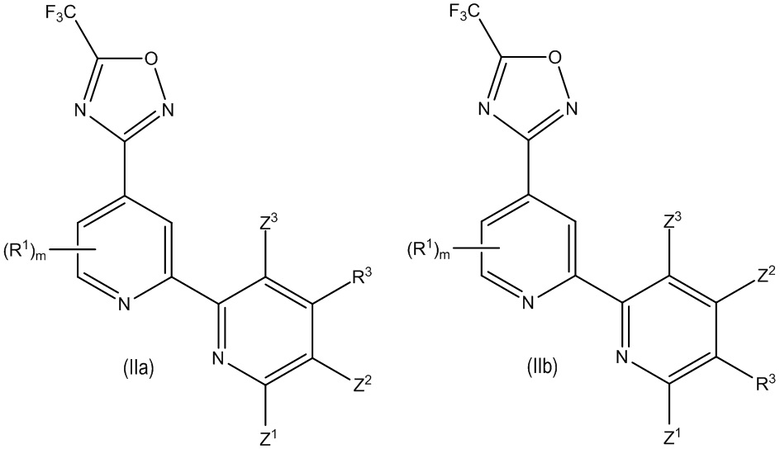
где один из Z1, Z2 и Z3 представляет собой H и остальные независимо выбраны из H и R2, и предпочтительно, если каждый Z1, Z2 и Z3 представляют собой H; и
где предпочтительно, если R3 выбран из -L1-R5, -L2-OR6, -L3-NR7R8, -CONR9R10, -NR11COR12 и -Y-L7-NR7R8, и более предпочтительно, если R3 выбран из -OR6, -NR7R8, -NR11COR12 и -Y-L7-NR7R8, где предпочтительно, если Y выбран из -O- и -NR13-; и
где предпочтительно, если m равно 0. В некоторых предпочтительных вариантах осуществления в соединении формулы (IIa) или (IIb) R3 представляет собой -N R11COR12. В некоторых других предпочтительных вариантах осуществления в соединении формулы (IIa) или (IIb) R3 представляет собой -Y-L7-NR7R8 и Y выбран из -O- и -NR13-. В некоторых других предпочтительных вариантах осуществления в соединении формулы (IIa) или (IIb) R3 представляет собой -OR6, где R6 представляет собой -L1-R5, где L1 в указанном -L1-R5 предпочтительно представляет собой C1-4 алкилен и R5 в указанном -L1-R5 предпочтительно выбран из гетероциклила и гетероарила, где гетероциклил и гетероарил каждый необязательно замещены одним или большим количеством R16, или R3 представляет собой -N R7R8, где один из R7 или R8 представляет собой -L1-R5, где L1 в указанном -L1-R5 предпочтительно представляет собой C1-4 алкилен и R5 в указанном -L1-R5 предпочтительно выбран из гетероциклила и гетероарила, где гетероциклил и гетероарил каждый необязательно замещены одним или большим количеством R16.
В другом особенно предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы (I) или его соли, описывающемуся формулой (IIIa) или (IIIb):
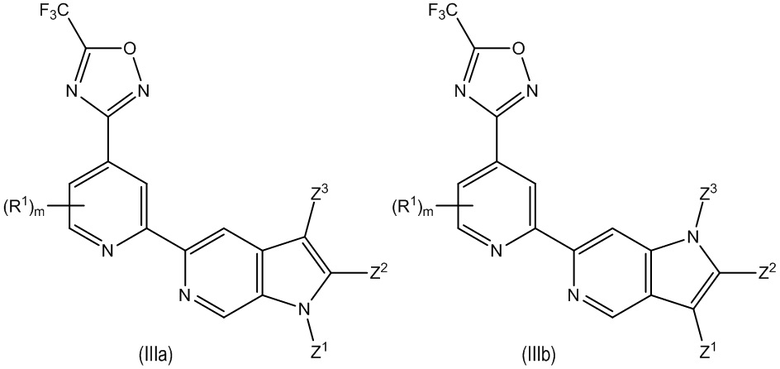
где один из Z1, Z2 и Z3 представляет собой R3 или H, предпочтительно R3 и остальные независимо выбраны из H и R2;
где предпочтительно, если R3, если содержится, выбран из группы, включающей -L1-R5, -L2-OR6, -L3-NR7R8, -CONR9R10, -NR11COR12 и -Y-L7-NR7R8, где предпочтительно, если Y выбран из -O- и -NR13-, и более предпочтительно, если R3 выбран из -L1-R5, -L2-OR6, -L3-NR7R8 и -CONR9R10, где предпочтительно, если L2 и L3 каждый независимо выбраны из C1-4 алкилена; и
где предпочтительно, если m равно 0. В некоторых предпочтительных вариантах осуществления соединение формулы (I) представляет собой соединение формулы (IIIa). В некоторых предпочтительных вариантах осуществления соединение формулы (I) представляет собой соединение формулы (IIIb). В некоторых предпочтительных вариантах осуществления в соединении формулы (IIIa) или (IIIb) один из Z1, Z2 и Z3, предпочтительно Z2, представляет собой R3 и остальные независимо выбраны из H и R2, и R3 представляет собой -CONR9R10. В некоторых других предпочтительных вариантах осуществления в соединении формулы (IIIa) или (IIIb) один из Z1, Z2 и Z3 представляет собой R3 и остальные независимо выбраны из H и R2, и R3 представляет собой -L1-R5, где предпочтительно, если R5 в указанном -L1-R5 выбран из гетероциклила и гетероарила, где гетероциклил и гетероарил каждый необязательно замещены одним или большим количеством R16. В некоторых других предпочтительных вариантах осуществления в соединении формулы (IIIa) или (IIIb) один из Z1, Z2 и Z3 представляет собой R3 и остальные независимо выбраны из H и R2, и R3 представляет собой -L2-OR6 или -L3-NR7R8 где L2 и L3 каждый независимо выбраны из C1-4 алкилена.
В другом особенно предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы (I) или его соли, описывающемуся формулой (IVa):

где один из Z1, Z2, Z3 и Z4 выбран из R2, R3 и H, и остальные независимо выбраны из H и R2, при условии, что только до двух из Z1, Z2, Z3 и Z4 представляют собой R2; и
где предпочтительно, если m равно 0. В некоторых предпочтительных вариантах осуществления в соединении формулы (IVa) Z4 выбран из R2, R3 и H, и Z1, Z2 и Z3 независимо выбраны из H и R2 при условии, что только до двух из Z1, Z2, Z3 и Z4 представляют собой R2 и где предпочтительно, если Z3 представляет собой H. В некоторых предпочтительных вариантах осуществления в соединении формулы (IVa) Z4 выбран из R2, R3 и H, и Z1, Z2 и Z3 представляют собой H.
В другом особенно предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы (I) или его соли, описывающемуся формулой (IVa-1):

где один из Z1, Z2 и Z3 представляет собой R3 или H и остальные независимо выбраны из H и R2, и предпочтительно, если каждый Z1, Z2 и Z3 представляют собой H; и
где предпочтительно, если m равно 0.
В другом особенно предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы (I) или его соли, описывающемуся формулой (IVb):
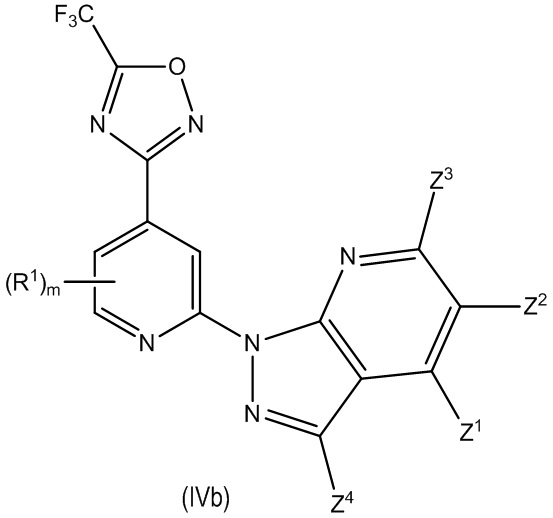
где один из Z1, Z2, Z3 и Z4 выбран из R2, R3 и H, и остальные независимо выбраны из H и R2, при условии, что только до двух из Z1, Z2, Z3 и Z4 представляют собой R2; и
где предпочтительно, если m равно 0.
В другом особенно предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы (I) или его соли, описывающемуся формулой (IVb-1):
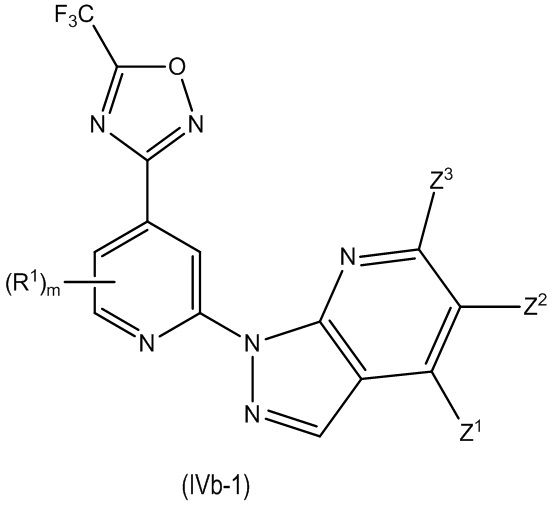
где один из Z1, Z2 и Z3 представляет собой R3 или H и остальные независимо выбраны из H и R2, и
где предпочтительно, если m равно 0.
В некоторых вариантах осуществления настоящее изобретение относится к соединению формулы (I) или его соли, выбранному из:
3-(2-(1-Бутил-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-Пропил-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
1-Бутил-N,N-диметил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-2-карбоксамида,
N,N-Диэтил-3-((4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)окси)пропан-1-амина,
1-Бутил-N-этил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-2-карбоксамида,
4-(3-((4'-(5-(Трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)окси)пропил)морфолина,
3-(5'-(3-(4,4-Дифторпиперидин-1-ил)пропокси)-[2,2'-бипиридин]-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(3-(Пиперидин-1-илметил)-1-пропил-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
4-((1-Пропил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-3-ил)метил)морфолина,
N-Бутил-3-метокси-N-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)пропанамида,
N-(Циклопропилметил)-N-метил-4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-амина,
N1,N1-Диэтил-N3-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)пропан-1,3-диамина,
N-(3-(4,4-Дифторпиперидин-1-ил)пропил)-N-метил-4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-амина,
N,N-Диэтил-3-(2-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)пиримидин-5-илокси)пропан-1-амина,
N1,N1-Диэтил-N3-метил-N3-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-2,2'-бипиридин-5-ил)пропан-1,3-диамина,
3-(2-(1-(Тетрагидро-2H-пиран-4-ил)-1H-пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
N-Этил-N-фенэтил-3-((4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)окси)пропан-1-амина,
2-Фенил-N-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)ацетамида,
3-(2-(1-((Тетрагидро-2H-пиран-4-ил)метил)-1H-пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(4'-(2-(4,4-Дифторпиперидин-1-ил)этокси)-[2,2'-бипиридин]-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
4-(2-((4'-(5-(Трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-ил)окси)этил)морфолина,
N,N,1-Триметил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-2-карбоксамида,
3-(2-(1-Пропил-3-(1H-пиразол-4-ил)-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
1-Бутил-N,N-диэтил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-2-карбоксамида,
3-(2-(1-(2-Метоксиэтил)-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазолаа,
2-(4,4-Дифторпиперидин-1-ил)-N-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-ил)ацетамид,
N-(2-(4,4-Дифторпиперидин-1-ил)этил)-4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-амина,
3-(2-(3-(Пиперидин-1-илметил)-1H-пиразоло[3,4-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-(2-(4,4-Дифторпиперидин-1-ил)этил)-1H-пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
1-Метил-N-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)пиперидин-4-карбоксамида,
3-Фенил-N-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-ил)пропанамида,
2-Циклобутил-N-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-ил)ацетамида,
N-(Пиперидин-3-ил)-4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-карбоксамида,
3-(5'-(3-(1H-Пиразол-1-ил)пропокси)-[2,2'-бипиридин]-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
(1-Пропил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-3-ил)метанола,
3-(2-(3-(Метоксиметил)-1-пропил-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
4-((1-Пропил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-3-ил)метил)морфолина,
3-(2-(1H-Пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-(Пиридин-4-илметил)-1H-пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
N-((1-Метилпиперидин-4-ил)метил)-4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-карбоксамида,
N-((1-Метилпиперидин-4-ил)метил)-4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-карбоксамида,
3-(2-(1-(1-(2,2,2-Трифторэтил)пиперидин-4-ил)-1H-пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-Метил-3-(1H-пиразол-4-ил)-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-Бутил-3-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
N-Метил-3-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-2,2'-бипиридин-5-илокси)пропан-1-амина,
1-(1-Бутил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-3-ил)-N,N-диметилметанамина,
3-(2-(1H-Пиразоло[4,3-b]пиридин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
N-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-ил)пиперидин-3-карбоксамида,
1-(2-Метоксиэтил)-N,N-диметил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-2-карбоксамида,
3-(2-(1-(2-Метоксиэтил)-3-(1H-пиразол-4-ил)-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-Метил-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-(2-Метоксиэтил)-1H-пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-(Пиридин-3-илметил)-1H-пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-(Пиридин-2-илметил)-1H-пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
2-(Метил(3-((4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)окси)пропил)амино)этан-1-ола,
3-(2-(1-(2-Метоксиэтил)-1H-пиразоло[3,4-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(2-(2-Метоксиэтил)-2H-пиразоло[3,4-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1H-Пиразоло[3,4-b]пиридин-1-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
N,N-Диметил-3-((4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)окси)пропан-1-амина,
3-(2-(1-Метил-1H-пиразоло[4,3-b]пиридин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-(2-Метоксиэтил)-1H-пиразоло[4,3-b]пиридин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-Этил-1H-пиразоло[4,3-b]пиридин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-(2-(1-Метил-1H-имидазол-2-ил)этил)-1H-пиразоло[4,3-b]пиридин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола и
3-(2-(1-((1-Метил-1H-пиразол-4-ил)метил)-1H-пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
или его соль.
Другие примеры соединений формулы (I), предлагаемых в настоящем изобретении, включают соединения, приведенные ниже, и их соли:

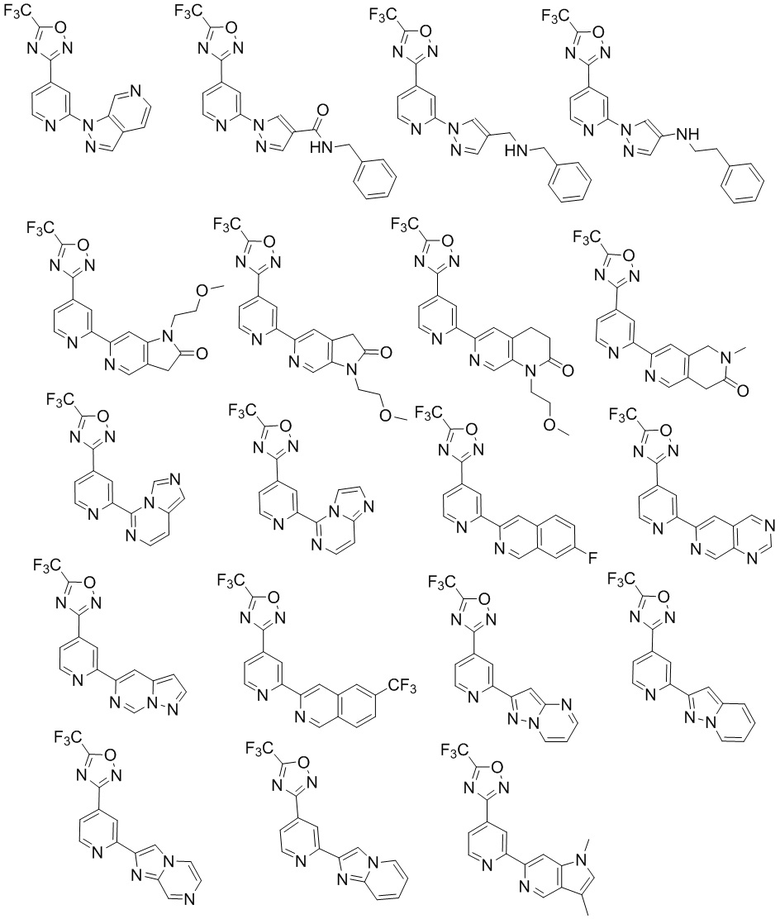
В особенно предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы (I) или его соли, выбранному из
N,N-Диэтил-3-((4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)окси)пропан-1-амина,
1-Бутил-N,N-диметил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-2-карбоксамида,
N,N,1-Триметил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-2-карбоксамида,
3-(2-(1-Пропил-3-(1H-пиразол-4-ил)-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
2-(4,4-Дифторпиперидин-1-ил)-N-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-ил)ацетамида,
3-(2-(1-(2-Метоксиэтил)-1H-пиразоло[4,3-b]пиридин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-Этил-1H-пиразоло[4,3-b]пиридин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-(2-(1-Метил-1H-имидазол-2-ил)этил)-1H-пиразоло[4,3-b]пиридин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1H-Пиразоло[4,3-b]пиридин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола и
3-(2-(1-Метил-1H-пиразоло[4,3-b]пиридин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
или его соль.
Настоящее изобретение также относится к любому отдельному соединению или любой подгруппе соединений, указанных в приведенных выше перечнях, и их соли.
Кроме того, настоящее изобретение также относится к соединению формулы (I) или его соли, описанной и определенной в настоящем изобретении (включая любое из предпочтительных определений/вариантов осуществления, описанных выше), где предпочтительно, если следующие соединения исключены из формулы (I):
3-(2-(тиазол-4-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(2-метилтиазол-4-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
N,N-диметил-4-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)тиазол-2-карбоксамид,
3-(2-(оксазол-4-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(2-метилоксазол-4-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
N,N-диметил-4-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)оксазол-2-карбоксамид,
3-(2-(1H-имидазол-4-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(1,2-диметил-1H-имидазол-4-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
N,N,1-триметил-4-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-имидазол-2-карбоксамид,
3-(2-(1H-пиррол-2-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(5-метил-1H-пиррол-2-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
N,N-диметил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пиррол-2-карбоксамид,
3-(2-(1,2,4-тиадиазол-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(3-метил-1,2,4-тиадиазол-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
N,N-диметил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1,2,4-тиадиазол-3-карбоксамид,
3-(2-(1,2,4-оксадиазол-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-метил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1,2,4-оксадиазол,
N,N-диметил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1,2,4-оксадиазол-3-карбоксамид,
3-(2-(1H-1,2,4-триазол-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(3-метил-1H-1,2,4-триазол-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
N,N-диметил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-1,2,4-триазол-3-карбоксамид,
3-(2-(1H-пиразол-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(3-метил-1H-пиразол-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
N,N-диметил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пиразол-3-карбоксамид,
3-(2-(1H-1,2,3-триазол-4-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(1-метил-1H-1,2,3-триазол-4-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
N,N-диметил-4-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-1,2,3-триазол-1-карбоксамид,
3-(2-(4H-1,2,4-триазол-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(5-метил-4H-1,2,4-триазол-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
N,N-диметил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-4H-1,2,4-триазол-3-карбоксамид,
3-(2-(1,3,4-оксадиазол-2-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(5-метил-1,3,4-оксадиазол-2-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
N,N-диметил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1,3,4-оксадиазол-2-карбоксамид,
3-(2-(1,3,4-тиадиазол-2-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(5-метил-1,3,4-тиадиазол-2-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
N,N-диметил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1,3,4-тиадиазол-2-карбоксамид,
3-(2-(2H-1,2,3-триазол-4-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(2-метил-2H-1,2,3-триазол-4-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
N,N-диметил-4-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-2H-1,2,3-триазол-2-карбоксамид,
3-(2-(2H-тетразол-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(2-метил-2H-тетразол-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
N,N-диметил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-2H-тетразол-2-карбоксамид,
3-(2-(1,3,5-триазин-2-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(4-метил-1,3,5-триазин-2-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
N,N-диметил-4-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1,3,5-триазин-2-карбоксамид,
3-(2-(пиридазин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(6-метилпиридазин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
N,N-диметил-6-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)пиридазин-3-карбоксамид,
3-(2-(пиримидин-4-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(2-метилпиримидин-4-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
N,N-диметил-4-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)пиримидин-2-карбоксамид,
3-(2-(пиразин-2-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(5-метилпиразин-2-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
N,N-диметил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)пиразин-2-карбоксамид,
3-(2-(индолизин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(1H-индол-2-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(1-метил-1H-бензо[d]имидазол-2-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(1-метил-1H-индазол-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(бензо[d]изотиазол-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)бензо[d]изоксазол,
3-(2-(изохинолин-1-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол,
3-(2-(1,8-нафтиридин-2-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол и
3-(2-(фталазин-1-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол.
Соответственно, предпочтительно, если исключены соединения, указанные в предыдущем предложении, а также их соли и таутомеры.
Определения конкретных терминов, использующихся в описании и формуле изобретения, приведены ниже. Все остальные технические и научные термины, использующиеся в настоящем изобретении и не определенные ниже, обладают такими же значениями, какие обычно известны специалисту с общей подготовкой в области техники, к которой относится настоящее изобретение. В случае противоречия определяющим является настоящее описание, включая определения.
В случае противоречия между химическими структурами и названиями соединений, раскрытых в настоящем изобретении, определяющими являются химические структуры.
В разных разделах настоящего описания заместители соединений, предлагаемых в настоящем изобретении, раскрыты в виде групп или диапазонов значений. Следует особо отметить, что настоящее изобретение включает все и каждую субкомбинацию представителей таких групп и диапазонов значений.
В разных разделах настоящего описания описаны разные арильные, гетероарильные, карбоциклильные и гетероциклильные группы. Если не указано иное, эти кольца могут быть присоединены к остальной части молекулы по любому элементу кольца, если это допускает валентность. Например, термин "пиридил" (или пиридинил) может означать пиридин-2-ильное, пиридин-3-ильное или пиридин-4-ильное кольцо и термин "пиперидинил" может означать пиперидин-1-ильное, пиперидин-2-ильное, пиперидин-3-ильное или пиперидин-4-ильное кольцо.
Термин "n-членное", где n является целым числом, указывает количество образующих кольцо атомов в кольцевой системе, в которой количество образующих кольцо атомов равно n. Например, фенил является примером 6-членного арила, циклопропил является примером 3-членного карбоциклила, пиразолил является примером 5-членного гетероарила, хинолинил является примером 10-членного гетероарила, пиперидинил является примером 6-членного гетероциклила и декагидрохинолинил является примером 10-членного гетероциклила.
Термин "Cy-z", где y и z являются целым числами, при использовании в комбинации с химической группой, означает диапазон количеств атомов углерода в химической группе, где y и z являются граничными значениями, которые включены в диапазон. Примеры включают C1-6, C2-6, C3-7 и т. п.
Термин "Cy-z алкил" означает насыщенную линейную или разветвленную ациклическую углеводородную группу, содержащую от y до z атомов углерода. Таким образом, C1-6 алкил представляет собой алкил, содержащий от 1 до 6 атомов углерода. Примеры C1-6 алкила включают, но не ограничиваются только ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, втор-пентил, неопентил, н-гексил или втор-гексил.
Термин "Cy-z алкоксигруппа" означает Cy-z алкильную группу (определенную выше), ковалентно связанную с атомом кислорода, т. е. группу формулы -O-алкил, где алкильная группа содержит от y до z атомов углерода. Таким образом, термин C1-6 алкоксигруппа означает алкоксигруппу, где алкильный фрагмент содержит от 1 до 6 атомов углерода. Примеры C1-6 алкоксигруппы включают, но не ограничиваются только ими, метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропоксигруппу, н-бутоксигруппу, изобутоксигруппу, втор-бутоксигруппу, трет-бутоксигруппу, н-пентоксигруппу или н-гексилоксигруппу.
Термин "Cy-z алкилен" означает насыщенную линейную или разветвленную двухвалентную ациклическую углеводородную группу, содержащую от y до z атомов углерода. Так, например, C1-6 алкилен представляет собой алкилен, содержащий от 1 до 6 атомов углерода. Предпочтительно, если указанные алкиленовые группы представляют собой полиметиленовые группы, т. е. (CH2)x, где x означает количество звеньев CH2 в соответствующей алкиленовой группе, равное от 1 до 6. Примеры C1-6 алкилена включают, но не ограничиваются только ими, метилен, этилен, пропилен, н-бутилен, н-пентилен или н-гексилен.
Термин "арил", если не указано иное, означает 6-18-членную углеводородную кольцевую систему, которая содержит только атомы водорода и углерода и которая является моноциклической или полициклической (например, конденсированной, мостиковой или спиросочлененной кольцевой системой), где по меньшей мере одно из колец в кольцевой системе является ароматическим. Таким образом, арил при использовании в настоящем изобретении включает полностью ароматические углеводородные кольцевые системы, т. е. в которых все кольца в системе являются ароматическими, такие как фенил, нафтил или антрацил, а также кольцевые системы, в которых ароматическое углеводородное кольцо (например, фенил) сконденсировано с одним или большим количеством неароматических (т. е. насыщенных или частично ненасыщенных) углеводородных колец, таких как инданил, инденил, 1-оксо-2,3-дигидро-1H-инденил, тетрагидронафтил, флуоренил и т. п. В некоторых вариантах осуществления положение присоединения находится в ароматическом углеводородном кольце. В некоторых вариантах осуществления арильная группа содержит от 6 до 10 атомов углерода. В некоторых вариантах осуществления арильная группа представляет собой полностью ароматическую углеводородную кольцевую систему. Предпочтительно, если арильная группа представляет собой фенил. Арильные группы необязательно могут быть замещенными, как указано в других частях описания, и заместитель (заместители) может находиться в любом доступном положении в кольцевой системе.
Термин "связь" означает ординарную связь, если специально не указано иное.
Термин "карбоциклил", если не указано иное, означает 3-18-членную неароматическую углеводородную кольцевую систему, которая содержит только атомы водорода и углерода и которая является моноциклической или полициклической (например, конденсированной, мостиковой или спиросочлененной кольцевой системой). Каждое из колец в кольцевой системе является полностью насыщенным или частично ненасыщенным, т. е. ни одно из колец не является ароматическим. Один или большее количество кольцевых атомов углерода карбоциклильной группы необязательно могут быть окислены с образованием группы CO. В некоторых вариантах осуществления карбоциклил содержит от 3 до 10 атомов углерода. В некоторых вариантах осуществления карбоциклил представляет собой полностью насыщенную углеводородную кольцевую систему, т. е. она не содержит каких-либо кратных связей; полностью насыщенный карбоциклил в настоящем изобретении также называют "циклоалкилом". Примеры карбоциклила включают, но не ограничиваются только ими, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклопентенил, циклогексенил, циклогептенил, циклооктенил, адамантил, бицикло[2.2.1]гептанил, бицикло[2.2.2]октанил, декалинил и т. п. Предпочтительно, если карбоциклил представляет собой C3-7 циклоалкил. Карбоциклильные группы необязательно могут быть замещенными, как указано в других частях описания, и заместитель (заместители) может находиться в любом доступном положении в кольцевой системе.
Термин "C3-7 циклоалкил" означает моноциклический циклоалкил, содержащий от 3 до 7 образующих кольцо атомов углерода и включает циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Циклоалкильные группы необязательно могут быть замещенными, как указано в других частях описания, и заместитель (заместители) могут находиться в любом доступном положении в кольцевой системе.
"Галоген" означает бром, хлор, фтор или йод. Предпочтительно, если галоген означает фтор.
Термин "Cy-z галогеналкил" означает алкильную группу, содержащую от y до z атомов углерода, определенную в настоящем изобретении, которая один или большее количество раз замещена одним или большим количеством галогенов, которые могут быть одинаковыми или разными. Соответственно, C1-6 галогеналкил представляет собой C1-6 алкил, который один или большее количество раз замещен одним или большим количеством галогенов. Галогеналкильные группы включают пергалогеналкильные группы, т. е. алкильные группы, в которых все атомы водорода замещены галогеном. Примеры галогеналкильных групп включают, но не ограничиваются только ими, фторметил, дифторметил, трифторметил, 2,2,2-трифторэтил, 1-фтор-2-фторэтил, пентафторэтил, 3,3,3-трифторпропил, гептафторпропил, 4,4,4-трифторбутилхлорметил, дихлорметил, трихлорметилдифторхлорметил, дихлорфторметил, 1,2-дихлорэтил, 3,3-дихлорпропил и т. п. В некоторых вариантах осуществления галогеналкил представляет собой фторалкил, т. е. алкильную группу, которая один или большее количество раз замещена одним или большим количеством атомов фтора.
Термин "Cy-z галогеналкоксигруппа" означает галогеналкильную группу, содержащую от y до z атомов углерода, определенную в настоящем изобретении, ковалентно связанную с атомом кислорода, т. е. группу формулы -O-Cy-z галогеналкил. Таким образом, C1-6 галогеналкоксигруппа означает галогеналкоксигруппу, где галогеналкильный фрагмент содержит от 1 до 6 атомов C. Примеры галогеналкоксигрупп включают, но не ограничиваются только ими, трифторметоксигруппу, 2-фторэтоксигруппу, пентафторэтоксигруппу, 3-хлорпропоксигруппу, 3-фторпропоксигруппу, гептафторпропоксигруппу и т. п.
Термин "гетероарил", если не указано иное, означает 5-18-членную гетероциклическую кольцевую систему, которая является моноциклической или полициклической (например, конденсированной, мостиковой или спиросочлененной кольцевой системой) и которая в дополнение к атомам C включает от 1 до 6 гетероатомов, независимо выбранных из N, O и S, где по меньшей мере одно из колец в кольцевой системе является ароматическим и содержит по меньшей мере один из гетероатомов. Таким образом, при использовании в настоящем изобретении гетероарил включает полностью ароматические кольцевые системы, т. е. в которых все кольца в системе являются ароматическими, такие как имидазолил, пиридил, хинолил, пиридо[2,3-d]пиримидинил и т. п. и группы, в которых гетероароматическое кольцо (кольца) сконденсировано с одним или большим количеством неароматических (т. е. насыщенных или частично ненасыщенных) карбоциклических или гетероциклических колец, таких как 5,6,7,8-тетрагидрохинолин, 1,2,3,4-тетрагидро-1,8-нафтиридин и т.п. Гетероатом(ы) необязательно могут быть окислены. Аналогичным образом, если гетероарил содержит гетероароматическое кольцо, сконденсированное с одним или большим количеством неароматических карбоциклических или гетероциклических колец, один или большее количество кольцевых атомов углерода в неароматическом карбоциклическом или гетероциклическом кольце необязательно может быть окислен с образованием группы CO. Гетероарильная группа может быть присоединена к остальной части молекулы через любой атом C или N, если это приводит к стабильной структуре. В некоторых вариантах осуществления положение присоединения находится в гетероароматическом кольце. В некоторых вариантах осуществления гетероарильная группа содержит от 1 до 4 гетероатомов. В некоторых вариантах осуществления гетероарильная группа содержит от 1 до 3 гетероатомов. В некоторых вариантах осуществления гетероарил является 5-6-членным моноциклическим или 9-10-членным бициклическим. В некоторых вариантах осуществления гетероарил является 5-6-членным моноциклическим. В некоторых вариантах осуществления гетероарильная группа является полностью ароматической. Неограничивающие примеры гетероарильных групп включают пиридил, пиразинил, пиримидинил, пиридазинил, фурил, тиенил, пирролил, имидазолил, пиразолил, оксазолил, тиазолил, изоксазолил, изотиазолил, триазолил, оксадиазолил, тиадиазолил, тетразолил, триазин, хинолинил, изохинолинил, хиназолинил, хиноксалинил, нафтиридинил, фталазинил, индолил, бензимидазолил, бензофуранил, бензотиенил, бензотиазолил, бензоксазолил, циннолинил, индазолил, индолизинил, изоиндолил, птеридинил, пуринил, фуропиридинил, акридинил, феназинил, 5,6,7,8-тетрагидрохинолин, 1,2,3,4-тетрагидро-1,8-нафтиридин и т.п. Гетероарильные группы необязательно могут быть замещенными, как указано в других частях описания, и заместитель (заместители) может находиться в любом доступном положении в кольцевой системе.
Термин "гетероциклил", если не указано иное, означает 3-18-членную частично ненасыщенную или полностью насыщенную гетероциклическую кольцевую систему, которая является моноциклической или полициклической (например, конденсированной, мостиковой или спиросочлененной кольцевой системой) и которая в дополнение к атомам C включает от 1 до 6 гетероатомов, независимо выбранных из N, O и S. Атомы азота или серы необязательно могут быть окислены (например, -N=O, -S(=O)-, или -S(=O)2-) и дополнительно один или большее количество кольцевых атомов углерода гетероциклила необязательно могут быть окислены с образованием группы CO. "Гетероциклил" при использовании в настоящем изобретении также включает группы, в которых частично ненасыщенное или полностью насыщенное гетероциклическое кольцо сконденсировано с одним или большим количеством фенильных колец, как в соединениях 1,2,3,4-тетрагидрохинолинил, бензодиоксолил, карбазолил или фталимидил. Гетероциклил может быть присоединен к остальной части молекулы через любой кольцевой атом C или N, если это приводит к стабильной структуре. В некоторых вариантах осуществления гетероциклил является 3-7-членным моноциклическим. Примеры гетероциклилльной группы включают, но не ограничиваются только ими, пирролидинил, 2-оксопирролидинил, тетрагидрофуранил, тетрагидротиенил, тетрагидропиранил, дигидропиранил, тетрагидротиопиранил, пиперидинил, морфолинил, тиоморфолинил, пиперазинил, гомопиперазинил, азетидинил, оксетанил, гомопиперидинил, оксепанил, тиепанил, 2H-пиранил, 4H-пиранил, диоксанил, 1,3-диоксоланил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил, пирролинил, пиразолинил, пиразолидинил, имидазолинил, имидазолидинил, оксазолидинил, индолинил, 1-оксоизоиндолинил, декагидрохинолинил, 1,2,3,4-тетрагидрохинолинил, 6-азабицикло[3.3.1]гептанил, 8-азабицикло[3.2.1]октанил, 3-азаспиро[5.5]ундеканил, 7-азаспиро[3.5]нонанил, карбазолил, фталимидил, тетрагидротиопиранил 1,1-диоксид, 2-азаспиро[4.5]деканил, 2,3-дигидроспиро[инден-1,4´-пиперидинил] и т.п. Гетероциклильные группы необязательно могут быть замещенными, как указано в других частях описания, и заместитель (заместители) может находиться в любом доступном положении в кольцевой системе.
Термин "необязательно замещенный" означает незамещенный или замещенный. При использовании в настоящем изобретении термин "замещенный" означает, что атом водорода удален и заменен одновалентным заместителем. Следует понимать, что замещение у данного атома ограничено его валентностью. Если не приведены другие определения (или нет ограничения по валентности), группа, которая является необязательно замещенной "одним или большим количеством" заместителей может быть незамещенной или может, например, содержать 1, 2 или 3 (предпочтительно 1 или 2) заместителя.
Термин "CO" при использовании в настоящем изобретении означает карбонильную группу.
Термин "частично ненасыщенное" при использовании в настоящем изобретении применительно к кольцу означает кольцо, которое включает по меньшей мере одну двойную связь между кольцевыми атомами, но не является полностью ненасыщенным (т. е. ароматическим).
Термин "насыщенное" используется взаимозаменяемым образом с термином "полностью насыщенное" и при использовании в настоящем изобретении применительно к кольцу означает кольцо, которое не содержит кратных связей.
Термин "полностью ароматическое" используется взаимозаменяемым образом с термином "ароматическое" и при использовании в настоящем изобретении применительно к кольцу означает кольцо, которое является полностью ненасыщенным.
Волнистая линия  в химических формулах указывает положение присоединения к остальной части молекулы.
в химических формулах указывает положение присоединения к остальной части молекулы.
Для соединений, предлагаемых в настоящем изобретении, в которых переменная содержится больше одного раза, каждая переменная может означать другой фрагмент, независимо выбранный из группы, определяющей переменную. Например, если структура описана, как содержащая две группы R, которые одновременно содержатся в одном и том же соединении, эти две группы R могут представлять собой разные фрагменты, независимо выбранные из группы, определяющей указанный R.
Соединения, предлагаемые в настоящем изобретении, могут содержать один или большее количество асимметрических центров и поэтому могут образовывать стереоизомеры. Включены все стереоизомеры, такие как энантиомеры, диастереоизомеры и их смеси, если не указано иное. Соединения, предлагаемые в настоящем изобретении, которые содержат асимметрично замещенные атомы углерода, можно выделить в виде оптически активной формы или рацемических смесей. Методики получения оптически активных форм из оптически неактивных исходных веществ известны в данной области техники и включают, например, разделение рацемических смесей или стереоселективный синтез.
Соединения, предлагаемые в настоящем изобретении, в некоторых вариантах осуществления могут существовать в виде таутомеров. Следует понимать, что, если соединения обладают таутомерными формами, все таутомерные формы входят в объем настоящего изобретения. "Таутомер" означает молекулу, в которой возможно смещение протона от одного атома к другому атому той же молекулы. Примеры включают кето-енольные пары и циклические формы, в которых протон может занимать два или большее количество положений в гетероциклической системе, как, например, в 1H- и 3H-имидазоле. Таутомерные формы могут находиться в равновесии или быть стерически заблокированными в одной форме путем подходящего замещения.
Соединения, предлагаемые в настоящем изобретении, включают немеченые формы соединений формулы (I), а также их изотопно меченые формы. Изотопно меченные формы соединений являются соединениями, которые различаются только заменой одного или большего количества атомов на соответствующую изотопно обогащенную форму. Примеры изотопов, которые можно ввести в соединения, предлагаемые в настоящем изобретении, включают, например, изотопы водорода, углерода, азота, кислорода, фтора, хлора и йода, такие как 2H, 3H, 11C, 13C, 14C, 15N, 18O, 17O, 35S, 18F, 36Cl и 125I. Такие изотопно меченные соединения применимы, например, в качестве зондов в биологических анализах, в качестве средств для анализа или в качестве терапевтических средств.
"Полиморфы" или "кристаллические формы" означают кристаллические структуры, в которых соединение (или его соль или сольват) может кристаллизоваться в разных кристаллических упаковках, и все они обладают одинаковыми элементными составами. Разные кристаллические формы обычно, в частности, обладают разными рентгенограммами, инфракрасными спектрами, спектрами комбинационного рассеяния, температурами плавления, спектрами дифференциальной сканирующей калориметрии (DSC), формами кристалла, растворимостями и/или стабильностями. Если соединения, предлагаемые в настоящем изобретении, существуют в разных твердых формах, все их формы, включая аморфные формы и кристаллические формы, входят в объем настоящего изобретения.
Термины "соединение, предлагаемое в настоящем изобретении", "соединение, описанное в настоящем изобретении" и т. п. включают соединение формулы (I) (включая все и каждые подроды соединений формулы (I), описанных в настоящем изобретении и в формуле изобретения, а также соединения, описанные в примерах), включая все их стереоизомеры, таутомеры, изотопно меченные формы и полиморфы.
Настоящее изобретение также включает соли соединений, предлагаемых в настоящем изобретении. Предпочтительно, если указанные соли являются фармацевтически приемлемыми солями. При использовании в настоящем изобретении "фармацевтически приемлемая соль" означает соль, которая сохраняет биологическую эффективность и свойства исходного соединения (т. е. свободной кислоты или свободного основания в соответствующих случаях) и которая не является биологически или в ином отношении нежелательной. Фармацевтически приемлемые соли включают соли, образованные с неорганическими или органическими основаниями, и соли, образованные с неорганическими и органическими кислотами. Фармацевтически приемлемые соли хорошо известны в данной области техники. Типичные фармацевтически приемлемые соли включают соли, полученные по реакции соединений, предлагаемых в настоящем изобретении, с неорганической или органической кислотой, такие как гидрохлориды, гидробромиды, сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, фосфаты, моногидрофосфаты, дигидрофосфаты, метафосфаты, пирофосфаты, хлориды, бромиды, йодиды, нитраты, ацетаты, галогенацетаты (например, трифторацетаты), пропионаты, деканоаты, каприлаты, акрилаты, формиаты, изобутираты, капроаты, гептаноаты, пропиолаты, оксалаты, малонаты, сукцинаты, субераты, себацинаты, фумараты, малеаты, бутин-1,4 диоаты, гексин-1,6-диоаты, бензоаты, хлорбензоаты, метилбензоаты, динитробензоаты, гидроксибензоаты, метоксибензоаты, фталаты, сульфонаты, ксилолсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, гамма-гидроксибутираты, гликоляты, тартраты, метансульфонаты, этансульфонаты, пропансульфонаты, бензолсульфонаты, толуолсульфонаты, трифторметансульфонаты, нафталин-1-сульфонаты, нафталин-2-сульфонаты, манделаты, пируваты, стеараты, аскорбаты или салицилаты. Если соединения, предлагаемые в настоящем изобретении, содержат кислотный фрагмент, их подходящие фармацевтически приемлемые соли могут включать соли щелочных металлов, например, соли натрия или калия; соли щелочноземельных металлов, например, соли кальция или магния; и соли, образованные с подходящими органическими лигандами, такими как аммиак, алкиламины, гидроксиалкиламины, лизин, аргинин, N-метилглюкамин, прокаин и т. п. Фармацевтически приемлемые соли, предлагаемые в настоящем изобретении, можно получить из исходных соединений, которые содержат основной или кислотный фрагмент, по обычным химическим методикам. Например, такие соли можно получить по реакции форм свободной кислоты или основания этих соединений со стехиометрическим количеством подходящего основания или кислоты в подходящем растворителе.
Дополнительно соединения, предлагаемые в настоящем изобретении, или их соли, могут находиться в гидратированной или негидратированной (безводной) форме или в форме сольватов с молекулами других растворителей. "Сольват" при использовании в настоящем изобретении означает форму с присоединением растворителя, которая содержит стехиометрические или нестехиометрические количества растворителя. Некоторые соединения склонны захватывать молекулы растворителя в кристаллическом твердом состоянии в постоянном молярном отношении и таким образом образовывать сольват. Если растворителем является вода, то образовавшийся сольват является гидратом. Неограничивающие примеры сольватов включают гидраты и сольваты со спиртами (также называющиеся алкоголятами), такими как этанол (этаноляты). Если соединения, предлагаемые в настоящем изобретении (или их соли), существуют, как сольваты, все их сольваты входят в объем настоящего изобретения, предпочтительно фармацевтически приемлемые сольваты. При использовании в настоящем изобретении "фармацевтически приемлемый сольват" означает сольват, образовавшийся с фармацевтически приемлемым растворителем. Фармацевтически приемлемые растворители хорошо известны в данной области техники и включают растворители, такие как вода и этанол.
Соединения, предлагаемые в настоящем изобретении, включая их соли, можно получить по многим путям синтеза, включая общие пути синтеза, описанные ниже, начиная с имеющихся в продаже исходных веществ, соединений, известных из литературы, или с легко получаемых промежуточных продуктов, по стандартным методикам синтеза и процедурам. Стандартные методики синтеза и процедуры для получения органических соединений и преобразования функциональных групп и операций с ними известны в данной области техники и приведены в стандартных пособиях, таких как Smith M.B., "March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure", 7th Edition, Wiley, 2013; Greene TW and Wuts PGM "Greene’s Protective Groups in Organic Synthesis", 4th edition, Wiley, 2006)
Схемы реакций, описанные ниже, являются только иллюстративными для методик получения соединений, предлагаемых в настоящем изобретении. Другие пути, известные специалисту с общей подготовкой в данной области техники, а также другие реагенты и промежуточные продукты, также можно использовать для получения соединений формулы (I).
В некоторых методиках, описанных ниже, может быть необходимой или желательной защита реакционноспособных или нестабильных групп обычными защитными группами. Природа этих защитных групп и методики их введения и удаления хорошо известны в данной области техники (см., например, Greene TW and Wuts PGM, cited supra). Если содержится защитная группа, потребуется стадия удаления защитной группы, которую можно провести при стандартных условиях, хорошо известных в данной области техники, таких как описанные в указанной выше литературе.
Если не указано иное, в методиках, описанных ниже, значения разных заместителей в каждом синтетическом промежуточном продукте и в каждом соединении формулы (I) являются такими, как указано ниже.
В целом соединения формулы (I) можно получить по методике, представленной ниже на схеме 1.
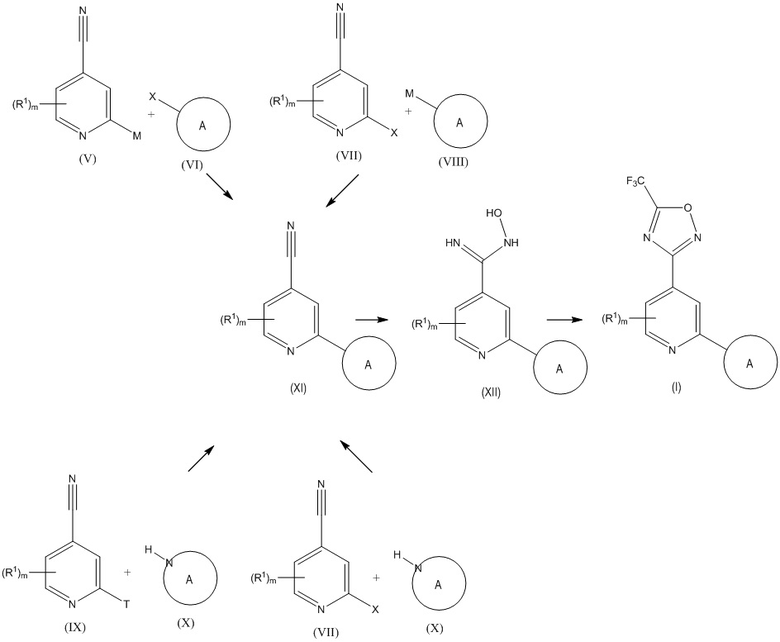
Схема 1
где R1, m и кольцо A обладают такими же значениями, как описанные для соединения формулы (I), и M, T и X обладают значениями, определенными ниже.
Кольцо A может быть присоединено к остальной части молекулы через кольцевой атом C или кольцевой атом N кольца A. Если кольцо A присоединено через кольцевой атом C, соединение формулы (XI) получают по реакции перекрестного сочетания металлоорганических соединений (V) или (VIII) с галогенидом (VI) или (VII).
Для сочетания соединения формулы (V) с соединением формулы (VI) или соединения формулы (VII) с соединением формулы (VIII) можно использовать разные реакции перекрестного сочетания, включая: перекрестное сочетание по Сузуки, в котором M означает бороновую кислоту или производное бороновой кислоты и X означает Cl, Br или I; реакцию Стилле, в которой M означает триалкилстаннильную группу и X означает Cl, Br или I; сочетание по Негиши, в котором M означает галогенид цинка и X означает трифлат, Cl, Br или I; и сочетание по Хиияма, в котором M означает триалкилсилильную группу и X означает Cl, Br или I.
Если соединения формулы (XI) получают с помощью перекрестного сочетания по Судзуки с промежуточными продуктами, указанными на схеме 1, реакцию можно провести с использованием подходящей комбинации Pd/лиганд, такой как XPhos и Pd2(dba)3 или Pd(PPh3)4, в присутствии подходящей соли Cu, такой как Cu(OAc)2 или CuI, в подходящем растворителе, таком как тетрагидрофуран или диметилформамид, с использованием подходящего основания, такого как карбонат калия. Температура, при которой проводят реакцию, обычно может меняться от комнатной температуры до 120°C и продолжительность реакций обычно равна от 1 ч до 48 ч. Примеры производного бороновой кислоты включают, в частности, диэтил-, диметилпроизводные N-метилиминодиуксусной кислоты (MIDA) и производное 2,2'-(фенилазандиил)бис(этан-1-ола).
Если соединения формулы (XI) получают с помощью перекрестного сочетания по Стилле с промежуточными продуктами, указанными на схеме 1, реакцию можно провести с использованием подходящей комбинации Pd/лиганд, такой как Pd(PPh3)4, Pd(PPh3)Cl2 или Pd(dppb)Cl2, в присутствии подходящей соли Cu, такой как CuI или CuO, в присутствии или при отсутствии CsF, в подходящем растворителе, таком как тетрагидрофуран, диоксан или диметилформамид. Температура, при которой проводят реакцию, обычно может меняться от комнатной температуры до 120°C и продолжительность реакций обычно равна от 1 ч до 48 ч. Использующимся оловоорганическим соединением может быть производное триметилстаннила. Также можно использовать межмолекулярную реакцию Стилле-Келли, в которой оба реагента являются галогенсодержащими производными и их обрабатывают с помощью (Bu3Sn)2, Et4NI и комбинации Pd/лиганд.
Если соединения формулы (XI) получают с помощью перекрестного сочетания по Негиши с промежуточными продуктами, указанными на схеме 1, реакцию можно провести с использованием подходящей комбинации Pd/лиганд, такой как PPh3 и Pd2(dba)3, XPhos и Pd2(dba)3, RuPhos и Pd2(dba)3 или Pd(PPh3)4, в подходящем растворителе, таком как тетрагидрофуран, диоксан или диметилформамид. Температура, при которой проводят реакцию, обычно может меняться от комнатной температуры до 120°C и продолжительность реакций обычно равна от 1 ч до 48 ч.
Если соединения формулы (XI) получают с помощью перекрестного сочетания по Хиияма с промежуточными продуктами, указанными на схеме 1, реакцию можно провести с использованием подходящей комбинации Pd/лиганд, такой как PdCl2(PPh3)2 и PPh3 или Pd(OAc)2 и ди(1-адамантил)-н-бутилфосфин в присутствии подходящей соли Cu, такой как CuI или CuBr, в присутствии или при отсутствии тетрабутиламмонийфторида в подходящем растворителе, таком как тетрагидрофуран, диоксан или диметилформамид. Температура, при которой проводят реакцию, обычно может меняться от комнатной температуры до 120°C и продолжительность реакций обычно равна от 1 ч до 48 ч. Если кольцо A присоединено через кольцевой атом N, соединение формулы (XI) получают по реакции соединения формулы (IX) и соединения формулы (X) (где кольцевой атом N в A, который образует связь с остальной частью молекулы, находится в форме NH) по реакции перекрестного сочетания или, альтернативно, между соединением формулы (VII) и соединением формулы (X) по реакции замещения. В случае реакции перекрестного сочетания для сочетания соединения формулы (IX) с соединением формулы (X) можно использовать реакцию Чана-Лэма. Эта реакция обеспечивает образование связи углерод-гетероатом путем окислительного сочетания бороновых кислот, станнанов или силоксанов с содержащими группу N-H соединениями в присутствии воздуха. T в соединениях формулы (IX) означает производное бора, олова или кремния. Реакцию инициируют стехиометрическим или каталитическим количеством соли меди, такой как Cu(OAc)2 или CuI, в присутствии или при отсутствии основания, такого как NEt3, пиридин или DMAP, в подходящем растворителе, таком как CH2Cl2, пиридин или DMSO, в присутствии воздуха. Температура, при которой проводят реакцию, обычно может меняться от комнатной температуры до 120°C и продолжительность реакций обычно равна от 1 ч до 48 ч. Альтернативно, соединение формулы (XI) можно получить по реакции замещения между соединением формулы (VII) и соединением формулы (X), которую можно провести в присутствии основания, такого как CsCO3, tBuOK, K2CO3, NaH или пиридин, и в присутствии или при отсутствии производного меди, такого как Cu2O или CuI, в подходящем растворителе, таком как DMF, DMSO или THF. Температура, при которой проводят реакцию, обычно может меняться от комнатной температуры до 120°C и продолжительность реакций обычно равна от 1 ч до 48 ч.
Соединения формулы (I) получают в две стадии из цианопроизводного формулы (XI). На первой стадии цианопроизводное формулы (XI) превращают в N’-гидроксиимидамид формулы (XII) по реакции с гидроксиламином с последующей конденсацией (XII) с трифторуксусным ангидридом и получают соединения формулы (I). Добавление гидроксиламина к цианопроизводным (XI) проводят в подходящем растворителе, таком как EtOH или MeOH, и в присутствии основания, в этом случае используют хлоргидрат гидроксиламина. Температура, при которой проводят реакцию, обычно может меняться от комнатной температуры до 60°C и продолжительность реакций обычно равна от 1 ч до 48 ч. Конденсация N’-гидроксиимидамидов (XII) с трифторуксусным ангидридом в подходящем растворителе, таком как CH2Cl2 или THF, дает соответствующий N’-трифторацетоксиимидамид, который в этой же реакционной среде или после добавления трифторуксусной кислоты дает 1,2,4-оксадиазолы формулы (I).
Альтернативно, соединения формулы (I) также можно получить в три стадии путем получения 1,2,4-оксадиазола из цианопроизводного (VII) до проведения реакции перекрестного сочетания или замещения с подходящими производными кольца A. Реакция цианопроизводного (VII) с гидроксиламином дает промежуточный продукт (XIII), который затем конденсируют с трифторуксусным ангидридом и получают соединения формулы (XIV), которые затем вводят в реакции перекрестного сочетания или замещения с подходящим производным кольца A и получают соединения формулы (I), указанные на схеме 2. Если реакцию перекрестного сочетания используют для превращения соединения (XIV) в соединение формулы (I), металлоорганические соединения можно получить из галогенпроизводного формулы (XIV) или из подходящих производных кольца A, как на схеме 1. Реакции, приведенные на схеме 2, можно провести при таких же условиях, как указанные выше для схемы 1.

Схема 2
где R1, m и кольцо A обладают такими же значениями, как описанные для соединения формулы (I) и X обладает значением, определенным выше.
Соединения формулы (V), (VI), (VII), (VIII), (IX) и (X) имеются в продаже или их можно получить по стандартным методикам, хорошо известным специалистам в области органической химии.
Металлоорганические производные формулы (V), (VIII), (IX) и образованные из соединения формулы (XIV) можно получить из соединений формулы (VII) и (XIV) путем переметаллирования по стандартным методикам получения реагентов для сочетаний по Сузуки, Стилле, Хиияма, Негиши и Чана-Лэма, хорошо известны специалистам в области органической химии. Например, боронатные производные N-метилиминодиуксусной кислоты можно получить по реакции соединений формулы (VII) и (XIV) с nBuLi в присутствии B(OiPr)3 при -78°C с последующим добавлением N-метилиминодиуксусной кислоты; производные триметилолова можно получить по реакции соединений формулы (VII) и (XIV) с гексаметилдиоловом и Pd (PPh3)4 в толуоле при 110°C в течение 16 ч; цинкорганические производные можно получить из соединений формулы (VII) и (XIV) путем обработки с помощью Zn в THF при комнатной температуре в течение от 1 до 6 ч; и триметилсилильные производные можно получить по реакции соединений формулы (VII) и (XIV) с nBuLi в присутствии триметилсилилхлорида при -78°C в THF.
Введение заместителей R2 и R3 в кольцо A, а также превращения заместителей R2 и R3 кольца A можно провести по стандартным методикам, хорошо известным специалистам в области органической химии. Указанные стандартные методики включают, например: замещение первичных или вторичных аминов путем обработки алкилирующим реагентом при стандартных условиях; или путем восстановительного аминирования, т. е. путем обработки альдегидом или кетоном в присутствии восстановительного реагента, такого как цианоборогидрид натрия или триацетоксиборогидрид натрия; превращения амина в амид с помощью активирующих реагентов, таких как дициклогексилкарбодиимид (DCC), 1-гидроксибензотриазол (HOBT), N-гидроксисукцинимид (HOSu), 1-этил-3-(3’-диметиламино)карбодиимид (EDC) в присутствии основания, такого как, диизопропилэтиламин, пиридин, триэтиламин или N-метилморфолин, в растворителе, таком как диметоксиэтан, N,N-диметилформамид, тетрагидрофуран, дихлорметан или диоксан, или, альтернативно, по реакции с хлорангидридами кислот в присутствии подходящего основания; алкилирования амида путем обработки алкилирующим реагентом в щелочной среде; превращения спирта в простой эфир при стандартных условиях; частичного или полного окисления спирта с получением кетона при стандартных условиях окисления; восстановления кетона путем обработки с помощью восстановительным реагентом, таким как борогидрид натрия; превращения спирта в галогенид по реакции с SOCI2, PBr3, тетрабутиламмонийбромидом в присутствии P2O5 или PCl3; превращения галогенида в амин по реакции с амином, необязательно в присутствии подходящего растворителя, и предпочтительно с нагреванием; и превращения первичного амида в соединение, содержащее группу -CN, при стандартных условиях.
Аналогичным образом, любое из ароматических колец соединений, предлагаемых в настоящем изобретении, можно ввести в реакции электрофильного ароматического замещения или нуклеофильного ароматического замещения, подробно описанные в литературе.
Некоторые из этих реакций взаимопревращения подробнее описаны в примерах. Как должны понимать специалисты в данной области техники, эти реакции взаимопревращения можно провести с соединением формулы (I) и таким образом получить другие соединения формулы (I), а также провести с любым подходящим промежуточным продуктом.
Соли соединения формулы (I) можно получить при окончательном выделении и очистке соединений, предлагаемых в настоящем изобретении, или можно получить путем обработки соединения формулы (I) достаточным количеством желательной кислоты (или основания) и обычным образом получить соль.
Если методики получения соединений, предлагаемых в настоящем изобретении, дают к смеси стереоизомеров, отдельные стереоизомеры соединения формулы (I) можно получить, например, путем разделения, исходя из соединения формулы (I), полученного в виде смеси стереоизомеров, с использованием хорошо известных методик, таких как образование пар диастереоизомеров путем получения соли с оптически активной кислотой с последующей фракционной кристаллизацией и выделением свободного основания, или с помощью хиральной препаративной хроматографии. Альтернативно, можно получить оптически чистые или энантиомерно обогащенные синтетические промежуточные продукты, которые затем можно использовать без обработки на следующих стадиях, на разных стадиях методик синтеза, описанных выше, с использованием любой известной методики хирального разделения. Альтернативно, можно получить оптически чистые или энантиомерно обогащенные конечные соединения (или синтетические промежуточные продукты) с помощью хиральной хроматографии.
Соединения, предлагаемые в настоящем изобретении, ингибируют активность гистондезацетилаз. В частности, установлено, что соединения, предлагаемые в настоящем изобретении, являются активными ингибиторами HDAC6.
Активность соединений, предлагаемых в настоящем изобретении, как ингибиторов HDAC6 можно определить с использованием например, исследований in vitro, описанных в разделе примеров. В частности, в примере 8 описана методика определения ингибирующей активности по отношению к HDAC6. С использованием анализа, описанного в примере 8, показано, что соединения, предлагаемые в настоящем изобретении, являются активными ингибиторами HDAC6. Также показано, что соединения, предлагаемые в настоящем изобретении, ингибируют активность HDAC6 в клетках, о чем свидетельствуют результаты, описанные в примере 9. Селективность по отношению к HDAC6 сравнительно с селективностью по отношению к другим изоформам HDAC можно определить по методикам, хорошо известным в данной области техники, например, с помощью анализов in vitro, сходных с описанным в примере 8, с использованием соответствующей представляющей интерес изоформы HDAC. Показано, что соединения, предлагаемые в настоящем изобретении, обладают селективностью по отношению к HDAC6, более значительной, чем по отношению к HDAC2, о чем свидетельствуют результаты, описанные в примере 8 для HDAC2 с использованием типичных соединений, предлагаемых в настоящем изобретении.
HDAC6 является классом IIb HDAC, которые могут дезацетилировать субстраты, такие как тубулин, белок теплового шока (Hsp)90 и кортактин. HDAC6 локализован в цитозоле и содержит два каталитических домена и C-концевой цинк-пальцевый домен, который может связывать свободный убиквитин, а также моно- и полиубиквитинированные белки (Li et al, FEBS J. 2013 Feb;280(3):775-93). Связывающий убиквитин домен в HDAC6 связывается с несколькими белками, участвующими в контроле убиквитина, системы протеасомы, образования агресомы и аутофагии. Дополнительно, способность HDAC6 дезацетилировать альфа-тубулин влияет на опосредуемые микротрубочками процессы, такие как миграция клеток, образование иммунного синапса, вирусная инфекция, разложение неправильно свернутых белков и образование стрессовой гранулы. Также было показано, что HDAC6 дезацетилирует Hsp90 и модулирует его шапероновую активность, тем самым модулирует разные связанные с Hsp90 пути передачи сигналов в клетках, такие как контроль связанного со стрессом ответа.
Во многих исследованиях описана роль HDAC6 при раке. Например, было показано, что ингибирование HDAC6 уменьшает рост множественной миеломы в доклинических моделях и усиливает воздействие ингибиторов протеасомы и иммуномодулирующих лекарственных средств на основе талидомида, использующихся в качестве стандартов лечения (Santo et al, Blood. 2012 Mar 15;119(11):2579-89; North et al, PLoS One. 2017 Mar 6;12(3):e0173507). Также было показано, что ингибирование HDAC6 усиливает воздействие других стандартных лечебных лекарственных средств, таких как паклитаксел при раке яичников, поджелудочной железы и молочной железы (Huang et al, Oncotarget 2017 Jan 10;8(2):2694-2707). Антипролиферативную активность ингибиторов HDAC6 также наблюдали для клеток рака предстательной железы и меланомы (Li et al, Eur J Med Chem. 2015 Jul 15;100:270-6; Seidel et al, Biochem Pharmacol. 2016 Jan 1;99:31-52). Кроме того, in vivo эффективность ингибиторов HDAC6 описана для колоректального рака, воспалительного рака молочной железы, лейкоза, лимфомы и моделей ксеротрансплантата мутанта ARID1A яичников (Yang et al, J Med Chem. 2016 Feb 25;59(4):1455-70; Putcha et al, Breast Cancer Res. 2015 Dec 8;17(1):149; Bitler et al, Nat Cell Biol. 2017 Aug;19(8):962-973). Аналогичным образом, нокаутный HDAC6 снижает пролиферацию клеток лейомиомы матки и рака желудка, а сверхэкспрессирование HDAC6 стимулирует пролиферацию и стимулирует лекарственную резистентность клеток немелкоклеточного рака легких и клеток глиобластомы (Wei et al, Reprod Sci. 2011 Aug;18(8):755-62; Park et al, Cancer Lett. 2014 Nov 1;354(1):97-106; Wang et al, Oncol Rep. 2016 Jul;36(1):589-97; Wang et al, Cancer Lett. 2016 Aug 28;379(1):134-42). Кроме того, показано, что ингибиторы HDAC6 обладают противораковой активностью благодаря стимулированию иммунного ответа на опухоли в моделях меланомы и немелкоклеточного рака легких при использовании по отдельности или в комбинации с ингибиторами иммунных конечных точек или эпигенетическими модуляторами (Knox et al, Abstract 4055, AACR Annual Meeting 2017; April 1-5, 2017; Washington, DC; Woan et al, Mol Oncol. 2015 Aug;9(7):1447-1457; Tavares et al, ACS Med Chem Lett. 2017 Sep 5;8(10):1031-1036; Adeegbe et al, Cancer Discov. 2017 Aug;7(8):852-867).
Многократно показано, что HDAC6 играет роль в воспалительных и аутоиммунных заболеваниях. Мыши с нокаутом HDAC6 обладают увеличенным количеством регуляторных T-клеток в кровотоке (Tregs), которые являются ключевыми для поддержания иммунного гомеостаза. Аналогичным образом, специфические ингибиторы HDAC6 стимулируют супрессивную активность Treg в моделях воспалительной болезни кишечника и реакции "трансплантат против хозяина" (de Zoeten et al, Mol Cell Biol. 2011 May;31(10):2066-78). Было показано, что ингибиторы HDAC6 обладают модифицирующей заболевание активностью в моделях воспаления, ревматоидного артрита и системной красной волчанки (Vishwakarma et al, Int Immunopharmacol. 2013 May;16(1):72-8; Regna et al, Clin Immunol. 2016 Jan;162:58-73). У мышей с отсутствием HDAC6 обнаруживается ослабление аутофагии, что уменьшает дисфункцию ресничек, связанную с хроническим обструктивным заболеванием легких (COPD) (Lam et al, J Clin Invest. 2013 Dec;123(12):5212-30).
Также было показано, что ингибиторы HDAC6 эффективны для лечения цилиопатий. Цилиопатии являются генетическими заболеваниями, связанными с дефектами в цилиарной структуре или нарушениями функции и включают, в частности, поликистозное заболевание почек, поликистозное заболевание печени, синдром Барде-Бидля и дегенерацию сетчатки. В модели поликистозного заболевания почек ингибиторы HDAC6 предупреждают образование кисты и улучшают функцию почек (Cebotaru et al, Kidney Int. 2016 Jul;90(1):90-9). Аналогичным образом, в модели поликистозного заболевания печени фармакологическое ингибирование HDAC6 уменьшает пролиферацию кистозных холангиоцитов и уменьшает развитие кисты печени и фиброза (Gradilone et al, Am J Pathol. 2014 Mar;184(3):600-8).
Также было показано, что HDAC6 играет важную роль в заболеваниях нервной системы. В частности, ингибиторы HDAC6 проявили эффективность в моделях периферической невропатии, такой как болезнь Шарко-Мари-Тута и вызванная химиотерапией периферическая невропатия (Benoy et al, Neurotherapeutics. 2017 Apr;14(2):417-428; Krukowski et al, Pain. 2017 Jun;158(6):1126-1137). Кроме того, в нейронной культуре, взятой у пациентов с боковым амиотрофическим склерозом, обработка ингибиторами HDAC6 восстановила ее дефектный фенотип (Guo et al, Nat Commun. 2017 Oct 11;8(1):861).
Также было показано, что ингибиторы HDAC6 эффективны для лечения некоторых других заболеваний нервной системы. Например, было показано, что уменьшение содержания или ингибирование HDAC6 восстанавливает память и улучшает познавательную способность в моделях болезни Альцгеймера на мышах (Govindarajan et al, EMBO Mol Med. 2013 Jan;5(1):52-63). Утрата или ингибирование HDAC6 подавляет накопление невритных белков тау, таким образом ингибирование HDAC6 можно применять для лечения не только болезни Альцгеймера, но и других 4-повторных таупатий человека, таких как кортикобазальная дегенерация и прогрессирующий супрануклеарный паралич (Tseng et al, Cell Rep. 2017 Aug 29;20(9):2169-2183). Кроме того, в модели болезни Гентингтона ингибирование HDAC6 уменьшает чувствительность нейронов к мутантному гентингтину, что свидетельствует о нейропротективном действии ингибиторов HDAC6 при HD (Guedes-Dias et al, Biochim Biophys Acta. 2015 Nov;1852(11):2484-93).
Также было показано, что HDAC6 играет роль при психических нарушениях и нарушениях поведения, таких как депрессия. Например, ингибиторы HDAC6 стимулируют исследовательское поведение мышей и оказывают благоприятное влияние на результаты анксиолитических тестов и тестов социального взаимодействия (Jochems et al, Neuropsychopharmacology. 2014 Jan;39(2):389-400).
Кроме того, в некоторых публикациях подчеркнута важная роль HDAC6 при инфекционных заболеваниях. Применение ингибиторов HDAC6 уменьшает репликацию вирусов, таких как вирус японского энцефалита (JEV), вирус гепатита C (HCV) и вирус бешенства (Lu et al, Int J Mol Sci. 2017 May 1;18(5); Zan et al, Front Cell Infect Microbiol. 2017 Apr 26;7:146; Ai et al, J Med Chem. 2015 Jan 22;58(2):785-800). Также было показано, что HDAC6 облегчает вход в клетку вирусов гриппа A и регулирование переключения цикл лизиса вирусов-латентность для других вирусов (Banerjee et al, Science 2014 Oct 24;346(6208):473-7). Например, было показано, что HDAC6 участвует с поддержании латентности HIV, таким образом ингибирование HDAC6 может промотировать удаление вирусов из организма (Huo et al, J Biol Chem. 2011 Mar 18;286(11):9280-6). Кроме того, селективные ингибиторы HDAC6 увеличивают продолжительность жизни и удаление бактерий в моделях сепсиса (Zhao et al, J Trauma Acute Care Surg. 2016 Jan;80(1):34-40).
В некоторых публикациях также была показана роль HDAC6 при сердечно-сосудистых заболеваниях. Мыши с нокаутом HDAC6 характеризуются улучшенным состоянием сердца в моделях сердечной недостаточности на мышах. Кроме того, мыши с отсутствием HDAC6 устойчивы к скелетной мышечной слабости, которая считается опасным для жизни осложнением при застойной сердечной недостаточности (Demos-Davies et al, Am J Physiol Heart Circ Physiol. 2014 Jul 15;307(2):H252-8). Показано, что фармакологическое ингибирование HDAC6 защищает от ремоделирования предсердий в связи с фибрилляцией предсердий (Zhang et al, Circulation. 2014 Jan 21;129(3):346-58). Активность HDAC6 последовательно увеличивалась при стрессе миокарда, что свидетельствует о роли ингибиторов HDAC6 в миокардиопатиях (Lemon et al, J Mol Cell Cardiol. 2011 Jul;51(1):41-50). Также было показано, что селективное ингибирование HDAC6 улучшает выживаемость в модели геморрагического шока на грызунах (Chang et al, J Trauma Acute Care Surg. 2015 Dec;79(6):905-10). Кроме того, ингибирование of HDAC6 улучшает стабильную легочную артериальную гипертензию в экспериментальных моделях и оказывает нейропротективное воздействие в моделях ишемии головного мозга (Boucherat et al, Sci Rep. 2017 Jul 3;7(1):4546; Liesz et al, J Neurosci. 2013 Oct 30;33(44):17350-62).
Таким образом, соединения, предлагаемые в настоящем изобретении, предположительно применимы для лечения заболеваний, связанных с HDACs, в частности HDAC6. Примеры заболеваний, связанных с HDAC6, включают, без наложения ограничений, заболевания, приведенные ниже:
Раковые заболевания, такие как: рак легких, рак толстой кишки, рак молочной железы, рак предстательной железы, рак печени, рак головного мозга и другие неоплазии CNS, рак почки, рак яичников, рак желудка, рак кожи, рак кости, рак желудка, рак поджелудочной железы, рак сердца, глиома, глиобластома, рак пищевода, печеночно-клеточная карцинома, рак костей и суставов, папиллярная карцинома почки, плоскоклеточная карцинома головы и шеи, саркомы, мезотелиома, лейкозы, лимфомы и миеломы;
Аутоиммунные или воспалительные заболевания, такие как: ревматоидный артрит, остеоартрит, ревматоидный спондилит, псориатический артрит, инфекционный артрит, прогрессирующий хронический артрит, деформирующий артрит, травматический артрит, подагрический артрит, синдром Рейтера, полихондрия, острый синовит и спондилит, псориаз, постишемическое перфузионное поражение, воспалительная болезнь кишечника (например, язвенный колит или болезнь Крона), экзема, ишемическое/реперфузионное поражение, гломерулонефрит, гемолитическая анемия, апластическая анемия, идиопатическая тромбоцитопения, нейтропения, хронический тиреоидит, болезнь Грейвса, диабет типа I, склеродермия, диабет, гепатит, первичный биллиарный цирроз, синдром системного воспалительного ответа, послеоперационное или посттравматическое воспаление, злокачественная миастения, пузырчатка, алкогольное заболевание печени, муковисцидоз, рассеянный склероз (MS), болезнь Аддисона, болезнь Кастлемена, нодозный полиартериит, системная красная волчанка, атопический дерматит, контактный дерматит, хроническая почечная недостаточность, синдром Стивенса-Джонсона, идиопатическое спру, саркоидоз, синдром Гийена-Барре, увеит, конъюнктивит, кератоконъюнктивит, отит среднего уха, периодонтальное заболевание, интерстициальный фиброз легких, острый респираторный дистресс-синдром, астма, бронхит, ринит, синусит, панкреатит, воспалительное заболевание кости, менингит, цистит, фаринголарингит, пневмокониоз, синдром легочной недостаточности, эмфизема легких, хроническое обструктивное заболевание легких (COPD), фиброз легких, силикоз, хроническое воспалительное заболевание легких или перитониальный фиброз;
Отторжение трансплантата, в том числе реакция "хозяин против трансплантата", реакция "трансплантант против хозяина" и отторжение аллотрансплантата;
Инфекционные заболевания, в том числе грипп, вирусный энцефалит, HIV, гепатит вирусного происхождения, пневмония и сепсис;
Цилиопатии, такие как поликистозное заболевание почек, поликистозное заболевание почек, синдром Альстрема, синдром Барде-Бидля, некоторые формы дегенерации сетчатки, синдром Жубера, синдром Меккель-Грубера, нефронофтиз, рото-лице-пальцевой синдром 1, синдром Сениора-Локена, первичная цилиарная дискинезия (синдром Картагенера) или торакоасфиктическая дистрофия (синдром Жена), синдром Мардена-Уолкера или изомерия;
Заболевания нервной системы, такие как болезнь Вильсона, прионное заболевание, болезнь Паркинсона, болезнь Гентингтона, боковой амиотрофический склероз, амилоидоз, болезнь Альцгеймера, болезнь Александера, болезнь Пика, спинальная мышечная дистрофия, слабоумие с тельцами Леви, вызванная химиотерапией когнитивная дисфункция, синдромы митохондриальной энцефалопатии нарушения моторики кишечника, болезнь моторного нейрогенеза (MND), синдромы атаксии, в том числе атаксия Фридрейха и спинально-церебеллярная атаксия (SCA), повреждение спинного мозга, оливомостомозжечковая атрофия, мультисистемная атрофия, прогрессирующий супрануклеарный паралич, синуклеопатии, синдром Дауна, кортикобазальная дегенерация, прогрессирующая семейная миоклоническая эпилепсия, стрионигральная дегенерация, торсионная дистония, семейный тремор, синдром Туретта, синдром Шай-Драгера и болезнь Галлервордена-Шпатца, а также периферические невропатии, такие как болезнь Шарко-Мари-Тута, периферическая невропатия, вызванная химиотерапевтическими средствами (например, химиотерапевтическими средствами на основе платины, таксан, винкристин, бортезомиб и т. п.) и т. п.;
Психические нарушения и нарушения поведения, в том числе психотические расстройства и расстройства шизофренического спектра, такие как шизотипическое (личностное) нарушение, бредовое расстройство, кратковременное психотическое расстройство, шизофреноподобное расстройство, шизофрения, шизоаффективное расстройство, психотическое расстройство, вызванное веществом/лекарством, и психотическое расстройство, обусловленное другим патологическим состоянием; биполярные расстройства, такие как биполярное расстройство I, биполярное расстройство II, циклотимическое нарушение, биполярное и родственное расстройство, вызванное веществом/лекарством; депрессивные нарушения, такие как деструктивное расстройство, дисрегуляции настроения, большая депрессия, одиночные и рецидивирующие приступы, устойчивое депрессивное нарушение (дистимия), предменструальное дисфорическое нарушение, депрессивное нарушение, вызванное веществом/лекарством, и депрессивное нарушение, обусловленное другим патологическим состоянием; тревожные нарушения, такие как вызванное разлукой тревожное нарушение, селективный мутизм, специфическая фобия, состояние социальной тревоги (социофобия), паническое расстройство, агорафобия, генерализованное тревожное нарушение и т. п.;
Сердечно-сосудистые заболевания, такие как сердечная недостаточность, миокардиопатия, фибрилляция предсердий, легочная артериальная гипертензия, геморрагический шок, инсульт, ишемическое заболевание сердца, миокардит и клапанный порок;
атрофия мышц; и
кахексия.
Для применений и способов лечения, описанных в настоящем изобретении, можно использовать любые из соединений, предлагаемых в настоящем изобретении, включая любой из их вариантов осуществления.
Соответственно, настоящее изобретение также относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения в качестве лекарственного средства.
Настоящее изобретение также относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения для лечения заболевания, связанного с HDAC6.
Настоящее изобретение также относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения заболевания, связанного с HDAC6.
Настоящее изобретение также относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для лечения заболевания, связанного с HDAC6.
Настоящее изобретение также относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения в качестве ингибитора HDAC6.
Настоящее изобретение также относится к способу лечения заболевания, связанного с HDAC6, включающему введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли нуждающемуся в нем пациенту.
Настоящее изобретение также относится к способу ингибирования активности HDAC6, включающему введение пациенту, нуждающемуся в таком лечении, количества соединения формулы (I) или его фармацевтически приемлемой соли, достаточного для ингибирования активности HDAC6.
Настоящее изобретение также относится к соединению формулы (I) или его фармацевтически приемлемой соли для применения для лечения заболевания, выбранного из рака, аутоиммунного или воспалительного заболевания, отторжения трансплантата, цилиопатии, заболевания нервной системы, психического нарушения или нарушения поведения, инфекционного заболевания, сердечно-сосудистого заболевания, атрофии мышц и кахексии.
Настоящее изобретение также относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения заболевания, выбранного из рака, аутоиммунного или воспалительного заболевания, отторжения трансплантата, цилиопатии, заболевания нервной системы, психического нарушения или нарушения поведения, инфекционного заболевания, сердечно-сосудистого заболевания, атрофии мышц и кахексии.
Настоящее изобретение также относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для лечения заболевания, выбранного из рака, аутоиммунного или воспалительного заболевания, отторжения трансплантата, цилиопатии, заболевания нервной системы, психического нарушения или нарушения поведения, инфекционного заболевания, сердечно-сосудистого заболевания, атрофии мышц и кахексии.
Настоящее изобретение также относится к способу лечения заболевания, выбранного из рака, аутоиммунного или воспалительного заболевания, отторжения трансплантата, цилиопатии, заболевания нервной системы, психического нарушения или нарушения поведения, инфекционного заболевания, сердечно-сосудистого заболевания, атрофии мышц и кахексии, включающему введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли нуждающемуся в нем пациенту.
Настоящее изобретение также относится к способу ингибирования активности HDAC6 в образце (например, биологическом образце), включающему взаимодействие указанного образца (например, указанного биологического образца) с соединением формулы (I) или его фармацевтически приемлемой солью.
Настоящее изобретение также относится к применению соединения формулы (I) или его фармацевтически приемлемой соли в качестве ингибитора HDAC6 в исследованиях, предпочтительно в качестве средства исследования соединения для ингибирования HDAC6. Соответственно, настоящее изобретение относится к применению in vitro соединения формулы (I) или его фармацевтически приемлемой соли в качестве ингибитора HDAC6 и, в частности, к применению in vitro соединения формулы (I) или его фармацевтически приемлемой соли в качестве средства исследования соединения, действующего в качестве ингибитора HDAC6. Настоящее изобретение также относится к способу, предпочтительно к способу in vitro ингибирования HDAC6, способ включает нанесение соединения формулы (I) или его фармацевтически приемлемой соли на образец (например, биологический образец). Следует понимать, что термин "in vitro" в этом специфическом контексте используется в смысле "за пределами живого человека или тела животного", что включает, в частности, эксперименты, проводимые с клетками, клеточными или субклеточными экстрактами и/или биологическими молекулами в искусственной среде, такой как водный раствор или культуральная среда, которые можно провести, например, в колбе, пробирке чашке Петри, планшете для микротитрования и т. п.
Если не указано иное, любое описание способа лечения включает применение соединений для проведения такого лечения, как описанное в настоящем изобретении, а также применение соединений для приготовления лекарственного средства для лечения такого патологического состояния.
Любое указание на соединение формулы (I) в настоящем изобретении включает указание на любое из соединений формулы (IIa), (IIb), (IIIa), (IIIb), (IVa), (IVa-1), (IVb) и (IVb-1), и на любые их варианты осуществления, описанные в настоящем изобретении.
Термин "заболевание, связанное с HDAC6" и т. п. означает любое заболевание или патологическое состояние в котором играет роль HDAC6, и/или заболевание или патологическое состояние, связанное с экспрессией или активностью HDAC6, и/или заболевания или патологические состояния на протекание которых можно повлиять путем модулирования HDAC6. Заболевания, связанные с HDAC6, включают, без наложения ограничений, заболевания и патологические состояния, описанные в настоящем изобретении. Предпочтительно, если заболевание, связанное с HDAC, представляет собой заболевание, выбранное из рака, аутоиммунного или воспалительного заболевания, отторжения трансплантата, цилиопатии, заболевания нервной системы, психического нарушения или нарушения поведения, инфекционного заболевания, сердечно-сосудистого заболевания, атрофии мышц и кахексии.
При использовании в настоящем изобретении термин "субъект" или "пациент" или "индивидуум" означает любых животных, включая млекопитающих, предпочтительно мышей, крыс, других грызунов, кроликов, собак, кошек, свиней, крупный рогатый скот, овец, лошадей или приматов и наиболее предпочтительно людей (например, мужчин или женщин).
При использовании в настоящем изобретении термин "биологический образец" включает, без наложения ограничений, клетки, клеточные культуры или их экстракты; биоптат, взятый у животного, например, человека, или их экстракты; и кровь, слюну, мочу, фекалии или любые другие жидкости организма, или их экстракты.
При использовании в настоящем изобретении термин "терапевтически эффективное количество" количество активного соединения, которое вызывает биологический или медицинский ответ, который ожидают от субъекта (предпочтительно человека). Соответственно, терапевтически эффективное количество соединения может представлять собой количество, которое достаточно для лечения заболевания или нарушения, задержки начала или прогрессирования заболевания или нарушения, и/или смягчения одного или большего количества симптомов заболевания или нарушения при введении субъекту, страдающему от указанного заболевания или нарушения. Точное эффективное количество для субъекта зависит от множества факторов, таких как масса тела субъекта, размер и состояние здоровья, природа и тяжесть патологического состояния, подвергающегося лечению, и лекарственного средства или комбинации лекарственных средств, выбранных для введения. Терапевтически эффективные количества для данной ситуации можно определить с помощью стандартных экспериментов, которые находятся в пределах компетенции и мнения клинициста.
Для любого соединения терапевтически эффективное количество можно определить в начале с помощью анализов in vitro, например, анализов на основе клеточных культур или посредством моделей на животных, например, мышах, крысах или собаках. Модель на животных также можно использовать для определения подходящего диапазона концентрации и пути введения. Такую информацию можно использовать для определения применимых доз и путей введения для людей. Терапевтическую эффективность и токсичность можно определить по стандартным методикам с использованием клеточных культур или экспериментальных животных, например, можно определить значения ED50 и LD50 и отношение токсического эффекта к терапевтическому эффекту, также известное, как терапевтический индекс, можно рассчитать и использовать для определения доз, подходящих для применения для людей.
При использовании в настоящем изобретении если не указано иное, термин "лечить" и "лечение" применительно к заболеванию, нарушению или патологическому состоянию означает лечение и уход за пациентом для борьбы с заболеванием, нарушением или патологическим состоянием, такое как обращение, облегчение протекания, подавление процесса или предупреждение заболевания, нарушения или патологического состояния, к которым применим такой термин, или одного или большего количества симптомов такого заболевания, нарушения или патологического состояния и включает введение соединения, предлагаемого в настоящем изобретении (или его фармацевтически приемлемой соли) для предупреждения появления симптомов или осложнений, или смягчения симптомов или осложнений, или устранения заболевания, патологического состояния или нарушения. Предпочтительно, если лечение является излечивающим или улучшающим состояние.
Если это допустимо и соединение, предлагаемое в настоящем изобретении, можно вводить для применения в терапии непосредственно без обработки, его обычно вводят в форме фармацевтической композиции. Эти композиции содержат соединение, предлагаемое в настоящем изобретении (или его фармацевтически приемлемую соль) в качестве активного фармацевтического ингредиента вместе с одним или большим количеством фармацевтически приемлемых носителей. Для задач настоящего изобретения носитель является подходящим для применения в фармацевтических композициях, описанных в настоящем изобретении, если он совместим с другими ингредиентами композиции и не вреден для реципиента композиции. "Фармацевтически приемлемый носитель" включает не-API (API означает активный фармацевтический ингредиент) вещества, такие как разрыхлители, связующие, наполнители, смазывающие вещества и т. п., использующиеся для приготовления фармацевтических продуктов и считающиеся безопасными для введения субъектам (в особенности людям) в соответствии с установленными государственными стандартами, включая приведенные в United States Food and Drug Administration (Управление по контролю за качеством пищевых продуктов медикаментов и косметических средств США) и European Medical Agency (Европейское агентство лекарственных средств). Фармацевтически приемлемые носители хорошо известны специалистам в данной области техники и их выбирают на основе выбранного типа препарата и пути введения в соответствии со стандартной фармацевтической практикой, как это описано, например, в Remington: The Science and Practice of Pharmacy 22nd edition, edited by Loyd V Allen Jr, Pharmaceutical Press, Philadelphia, 2012).
Соответственно, настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (I) (включая любой его подрод формулы (IIa), (IIb), (IIIa), (IIIb), (IVa), (IVa-1), (IVb) и (IVb-1), и любые его варианты осуществления, описанные в настоящем изобретении) или его фармацевтически приемлемые соли и один или большее количество фармацевтически приемлемых носителей.
Фармацевтические композиции можно получить хорошо известным в фармацевтике образом и можно вводить разными путями, например, пероральным, парентеральным, легочным или местным путем. Парентеральное введение включает внутривенное, внутриартериальное, подкожное, внутрибрюшинное или внутримышечное. Парентеральное введение можно провести в виде одной болюсной дозы или можно провести, например, путем непрерывного вливания с помощью насоса. Легочное введение включает например, введение путем ингаляции или вдувания порошков или аэрозолей, включая проводимое с помощью небулайзера. Местное введение включает чрескожное, эпидермальное, глазное и в слизистые оболочку, включая интраназальную, вагинальную и ректальную доставку.
Композиции можно приготовить для быстрого (немедленного), замедленного отложенного высвобождения активного ингредиента после введения пациенту путем использования методик, известных в данной области техники.
Примеры фармацевтически приемлемых инертных наполнителей включают лактозу, декстрозу, сахарозу, сорбит, маннит, крахмалы, камедь акации, фосфат кальция, альгинаты, трагакантовую камедь, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду и метилцеллюлозу. Фармацевтические композиции могут дополнительно включать другие фармацевтически приемлемые инертные наполнители, включая: смазывающие агенты, такие как тальк, стеарат магния и минеральное масло; смачивающие агенты; эмульгирующие и суспендирующие агенты; консервирующие агенты, такие как метил- и пропилгидроксибензоаты; подсластители; вкусовые агенты; и окрашивающие агенты.
Подходящие пероральные дозированные формы включают, например, таблетки, пилюли, саше или капсулы из твердого или мягкого желатина или любого другого подходящего материала. Например, активное соединение можно включить в препарат, который содержит фармацевтически приемлемые носители, такие как связующие (например, желатин, целлюлоза, трагакантовая камедь), инертные наполнители (например, крахмал, лактоза), смазывающие вещества (например, стеарат магния, диоксид кремния), разрыхляющие агенты (например, альгинат, Primogel, кукурузный крахмал) и подслащивающие или вкусовые агенты (например, глюкоза, сахароза, сахарин, метилсалицилат и мята). Затем их можно спрессовать в таблетки или поместить в капсулы с использованием обычных методик. На капсулы и таблетки также можно нанести различные покрытия, известные в данной области техники, для изменения аромата, вкуса, цвета и формы капсулы и таблетки. Кроме того, в капсулы также можно включать жидкие носители, такие как нелетучее масло. Пероральные препараты также могут находиться в форме суспензий, растворов, сиропов и т. п. При желании можно добавить обычные агенты, для изменения аромата, вкуса, цвета и т. п.
Фармацевтические композиции, подходящие для парентерального введения включают стерильные водные растворы или суспензии, или, альтернативно, их можно приготовить в лиофилизированной форме для проводимого перед использованием приготовления раствора или суспензии с помощью стерильного водного носителя. В таких препаратах можно использовать разбавители или фармацевтически приемлемые носители, такие как стерильная вода и физиологический солевой буфер. Можно включать другие обычные растворители, pH буферы, стабилизаторы, антибактериальные средства, поверхностно-активные вещества и антиоксиданты. Например, применимые компоненты включают хлорид натрия, ацетатный, цитратный или фосфатный буферы, глицерин, декстрозу, нелетучие масла, метилпарабены, полиэтиленгликоль, пропиленгликоль, бисульфат натрия, бензиловый спирт, аскорбиновую кислоту и т. п. Парентеральные препараты можно хранить в любых обычных контейнерах, таких как флаконы и ампулы.
Композиции для введения путем ингаляции или вдувания включают растворы и суспензии в фармацевтически приемлемых водных или органических растворителях, или их смеси и порошки. Жидкие или твердые композиции могут включать подходящие фармацевтически приемлемые инертные наполнители, описанные выше. Такие композиции можно вводить пероральным или респираторным назальным путем для оказания местного или системного воздействия. Композиции можно распылять путем использования подходящего газа. Распыленные растворы можно вдыхать прямо из распыляющего устройства или распыляющее устройство можно прикрепить к лицевой маске или дыхательной камере. Растворы, суспензии и порошкообразные композиции можно вводить перорально или назально из устройств, которые доставляют препарат подходящим образом.
Фармацевтические композиции для местного введения могут включать чрескожные пластыри, мази, примочки, кремы, гели, капли, суппозитории, спреи, жидкости и порошки. Местные препараты могут содержать один или большее количество обычных носителей. Например, мази могут содержать воду и один или большее количество гидрофобных носителей, выбранных из жидкогоа парафин, алкилового эфира полиоксиэтилена, пропиленгликоля, белого вазелина и т. п. Носители композиции кремов могут быть на водной основе в комбинации с глицерином и одним или большим количеством других компонентов, таких как цетилстеариловый спирт, глицеринмоностеарат и т. п. Гели можно приготовить с использованием изопропилового спирта и воды, предпочтительно в комбинации с другими инертными наполнителями, такими как глицерин, гидроксиэтилцеллюлоза и т. п.
Фармацевтические композиции, такие как пероральные и парентеральные композиции, можно приготовить в виде разовых дозированных форм для облегчения введения и обеспечения равномерности дозы. При использовании в настоящем изобретении "разовые дозированные формы" означают представляют собой отдельные порции, применимые в качестве разовых доз для введения субъектам, каждая порция содержит заданное количество активного ингредиента, рассчитанное для оказания желательного терапевтического воздействия, вместе с одним или большим количеством подходящих фармацевтических носителей.
Для применения в терапии композиции следует вводить таким образом, который подходит для подвергающегося лечению заболевания, что определяет специалист по медицине. Подходящую дозу и подходящую длительность и частоту введения, в частности, определяют такие факторы, как состояние пациента, тип и тяжесть заболевания, конкретная форма активного ингредиента, методика введения. Обычно подходящую дозу и режим введения обеспечивает фармацевтическая композиция в количестве, достаточном для обеспечения терапевтического преимущества, например, улучшенного клинического результата, такого как более частая полная или частичная ремиссия, или более длительная проходящая без заболевания или общая продолжительность жизни, или уменьшение тяжести симптомов, или любое другое объективно выявляемое улучшение, установленное клиницистом. Эффективные дозы обычно можно оценить или экстраполировать с помощью экспериментальных моделей, таких как зависимости доза-ответ, полученные с помощью систем оценки in vitro или посредством моделей на животных.
Фармацевтические композиции, предлагаемые в настоящем изобретении, можно поместить в контейнер, упаковку или дозатор вместе с инструкциями для введения.
Соединения, предлагаемые в настоящем изобретении, можно вводить в виде одного активного средства или также можно использовать или вводить в комбинации с одним или большим количеством дополнительных терапевтически активных средств, например, лекарственных средств, применимых для лечения заболевания, выбранного из раковых заболеваний, аутоиммунных или воспалительных заболеваний, отторжения трансплантата, цилиопатии, заболеваний нервной системы, психических нарушений или нарушений поведения, инфекционных заболеваний, сердечно-сосудистых заболеваний, атрофии мышц и кахексии. Комбинированная терапия включает введение одного фармацевтического дозированного препарата, который содержит соединение, предлагаемое в настоящем изобретении, и один или большее количество дополнительных терапевтически активных средств, а также введение соединения, предлагаемого в настоящем изобретении, и каждого дополнительного терапевтически активного средства в своем собственном фармацевтическом дозированном препарате для раздельного введения. При раздельном введении введение может быть одновременным, последовательным или раздельным, и соединение, предлагаемое в настоящем изобретении, и дополнительное терапевтическое средство (средства) можно вводить по одному введения пути или с использованием разных путей введения, например, одно соединение можно вводить перорально и другое внутривенно. Кроме того, как показано выше, соединения, предлагаемые в настоящем изобретении, также можно использовать в монотерапии, в особенности при монотерапевтическом лечении заболевания, выбранного из рака, аутоиммунного или воспалительного заболевания, отторжения трансплантата, цилиопатии, заболевания нервной системы, психического нарушения или нарушения поведения, инфекционного заболевания, сердечно-сосудистого заболевания, атрофии мышц и кахексии.
ПРИМЕРЫ
В примерах использованы следующие аббревиатуры:
AcN: ацетонитрил
AcOH: уксусная кислота
Boc: трет-бутилоксикарбонил
n-BuOH: н-бутанол
DCM: дихлорметан
DIAD: диизопропилазодикарбоксилат
DIPEA: N,N-диизопропилэтиламин,
DMF: N,N-диметилформамид
EtOAc: этилацетат
EtOH: этанол
FA: муравьиная кислота
HPLC: высокоэффективная жидкостная хроматография
LC-MS: жидкостная хроматография-масс-спектроскопия
MeI: йодметан
MeOH: метанол
PPh3 : трифенилфосфин
Pd(PPh3)4 : тетракис(трифенилфосфин)палладий(0)
Pd(PPh3)2Cl2 : Бис(трифенилфосфин)паладийхлорид
pTSA: моногидрат п-толуолсульфоновой кислоты
КТ (или КТ): комнатная температура
Rt: время удерживания,
TBAB: тетрабутиламмонийбромид
TEA: триэтиламин
TFA: трифторуксусная кислота
THF: тетрагидрофуран
THP: тетрагидропиран
T3P: раствор пропилфосфонового ангидрида ≥50 мас.% в этилацетате
Для исследования с помощью LC-MS использовали следующие методики:
Методика 1: колонка: KINETEX-1,7u XB-C18 100A (50 мм×2,1 мм, 1,7 мкм); подвижная: фаза: A: 0,05% муравьиная кислота в воде B: 0,05% муравьиная кислота в ацетонитриле; градиентный режим: время/%A: 0/97, 0,3/97,3,2/2,4,8/2,5/97,5,10/97 температура колонки: 35°C; скорость потока: 0,6 мл/мин
Методика 2: колонка: Aquity UPLC BEH C18 (50 мм×2,1 мм, 1,7 мкм); подвижная: фаза: B: 0,1% муравьиная кислота в воде A: 0,1% муравьиная кислота в ацетонитриле; градиентный режим: время/% B: 0/97, 0,3/97, 3,2/2, 4/2, 4,01/97; температура колонки: 35°C; скорость потока: 0,6 мл/мин;
Методика 3: колонка: Aquity UPLC BEH C18 (50 мм×2,1 мм, 1,7 мкм); подвижная: фаза: B: 0,1% муравьиная кислота в воде, A: 0,1% муравьиная кислота в ацетонитриле; градиентный режим: время/% B: 0/97, 0,3/97, 3,0/2, 4,5/2, 4,51/97; температура колонки: 35°C; скорость потока: 0,6 мл/мин.
Методика 4: колонка: Aquity UPLC BEH C18 (50 мм×2,1 мм, 1,7 мкм); подвижная: фаза: B: 0,1% муравьиная кислота в воде A: 0,1% муравьиная кислота в ацетонитриле; градиентный режим: время/% B: 0/97,0,3/97,2,2/2, 3,30/2,4,5/2,4,51/97; температура колонки: 35°C; скорость потока: 0,6 мл/мин;
Методика 5: колонка - AQUITY UPLC BEH C18 (50 мм×2,1 мм, 1,7 мкм); подвижная: фаза:-A: 0,1%FA в воде, B: 0,1% FA в ацетонитриле: T%A: 0/90, 1/10, 2,20/10, 2,30/90,2,60./90
скорость потока: 0,8 мл/мин, температура: 50°C.
Методика 6: колонка - AQUITY UPLC BEH C18 (50 мм×2,1 мм, 1,7 мкм); подвижная: фаза-A: 0,1%FA в воде, B: 0,1% FA в ацетонитриле T%A:0/95,0,3/95,2,0/5,3,5/5,3,6/95,4,2/95
скорость потока: 0,6 мл/мин, температура: 40°C
Методика 7: колонка: Aquity UPLC BEH C18 (50 мм×2,1 мм, 1,7 мкм); подвижная: фаза: A: 0,05% муравьиная кислота в ацетонитриле, B: 0,05% муравьиная кислота в воде; градиентный режим: время/% B: 0/97, 0,3/97, 3,2/2, 3,8/2, 4,3/97, 4,5/97; температура колонки: 35°C; скорость потока: 0,6 мл/мин
Методика 8: колонка: Aquity UPLC BEH C18 (50 мм×2,1 мм, 1,7 мкм); подвижная: фаза: A: 0,1% муравьиная кислота в воде, B: 0,1% муравьиная кислота в ацетонитриле; градиентный режим: время/% B: 0/97, 0,3/97, 2,7/2, 3,5/2, 3,51/97, температура колонки: 35°C; скорость потока: 0,6 мл/мин;
Методика 9: колонка - AQUITY UPLC BEH C18 (50 мм×2,1 мм, 1,7 мкм); подвижная: фаза-A: 0,1%FA в воде, B: 0,1% FA в ацетонитриле; T%A: 0/97 0,3/97,3,0/2,4,0/2,4,2/97,4,50/97
скорость потока: 0,6 мл/мин, температура: 35°C
Методика 10: колонка: Aquity UPLC BEH C18 (50 мм×2,1 мм, 1,7 мкм); подвижная: фаза: A: 0,1% муравьиная кислота в воде, B: 0,1% муравьиная кислота в ацетонитриле; градиентный режим: время/% A: 0/97, 0,3/97, 3,0/2, 4,0/2, 4,3/97, 4,50/97; температура колонки: 35°C; скорость потока: 0,6 мл/мин
Методика 11: колонка: Aquity UPLC BEH C18 (50 мм×2,1 мм, 1,7 мкм); подвижная: фаза: A: 0,1% муравьиная кислота в воде, B: 0,1% муравьиная кислота в ацетонитриле; градиентный режим: время/% B: 0/5, 0,3/95, 2,0/95, 3,7/95, 4,2/5, 5,7/5; температура колонки: 40°C; скорость потока: 0,5 мл/мин
Методика 12: колонка - AQUITY UPLC BEH C18 (50 мм×2,1 мм, 1,7 мкм); подвижная: фаза-A: 0,1%FA в воде, B: 0,1% FA в ацетонитриле; T%A: 0/98 0,2/98,1,8/2,2,4/2,2,60/98,3,0/98. скорость потока: 0,8 мл/мин, температура: 50°
Методика 13: колонка: YMC TRAIT C18; подвижная: фаза: A: Ацетонитрил, B: 0,01 M бикарбонат аммония в воде; градиентный режим: A=40%, B=60%. Скорость потока: 25,0 мл/мин.
Методика 14: колонка: XBridge BEH C18 (50 мм×2,1 мм, 2,5 мкм); подвижная: фаза: A: 0,01 M формиат аммония в воде; B: AcN; градиентный режим: время/% B: 0/5, 3/100, 3,5/100, 3,8/5, 4,3/5; скорость потока: 0,7 мл/мин. температура: 40°C
Методика 15: колонка: XBridge BEH C18 (50 мм×3,0 мм, 2,5 мкм); подвижная: фаза: A: 0,01 M формиат аммония в воде: AcN(95:5), B: 0,01 M формиат аммония в воде:AcN(5:95); градиентный режим: время/% B: 0/2, 4/98, 4,5/98, 5/2, 5,5/2, 6,5/2; скорость потока: 1,0 мл/мин
Методика 16: колонка: XBridge BEH C18 (50 мм×3,0 мм, 2,5 мкм); подвижная: фаза: A: 0,01 M формиат аммония в воде: AcN(95:5), B: 0,01 M формиат аммония в воде:AcN(5:95); градиентный режим: время/% B: 0/2,2/2,7/98,7,5/98,8,5/2,10/2; скорость потока: 1,0 мл/мин;
Методика 17: колонка: Aquity UPLC BEH C18 (50 мм×2,1 мм, 1,7 мкм); подвижная: фаза: A: 0,01 M бикарбонат аммония в воде, B: Ацетонитрил; градиентный режим: время/% B: 0/3; 1,0/3; 7,0/100; 7,5/100; 9,0/3; 10,0/3. температура колонки: 35°C; скорость потока: 0,5 мл/мин;
Методика 18: колонка: XBridge BEH C18 (50 мм×2,10 мм, 2,5 мкм); подвижная: фаза: A: 0,01 M формиат аммония в воде: AcN(95:5),B: Ацетонитрил; градиентный режим: время/% B: 0/5; 1,0/5; 7/100; 7,5/100; 9/5,10/5. температура колонки: 40°C; скорость потока: 0,7 мл/мин;
Методика 19 : колонка: XBridge BEH C18 (50 мм×3,0 мм, 2,5 мкм); подвижная: фаза A: 0,01 M формиат аммония в воде: AcN(95:5), B: 0,01 M формиат аммония в воде:AcN(5:95); градиентный режим: время/% B: 0/2,1/2,4/98,4,5/98,5,5/2,6,5/2; скорость потока: 1,0 мл/мин;
Методика 20: колонка: Aquity UPLC BEH C18 (50 мм×2,1 мм, 1,7 мкм); подвижная: фаза: A: 0,1% муравьиная кислота в воде, B: 0,1% муравьиная кислота в ацетонитриле; градиентный режим: время/% A: 0/97, 1,0/97, 7,0/0, 7,5/0,9,0/97; температура колонки: 35°C; скорость потока: 0,5 мл/мин
Методика 21: колонка: Aquity UPLC BEH C18 (50 мм×2,1 мм, 1,7 мкм); подвижная: фаза: A: 0,1% муравьиная кислота в воде, B: 0,1% муравьиная кислота в ацетонитриле; градиентный режим: время/% A: 0/5, 0,1/5, 2,7/100, 3,5/100, 3,8/5;4,3/5 температура колонки: 40°C; скорость потока: 0,7 мл/мин
Методика 22 : колонка: XBridge BEH C18 (50 мм×2,1 мм, 2,5 мкм); подвижная: фаза A: 0,01 M формиат аммония в воде: AcN(95:5), B: 0,01 M формиат аммония в воде:AcN(5:95); градиентный режим: время/% B: 0/2,1/2,7/100,7,5/100,9/2,10/2; скорость потока: 0,7 мл/мин;
Методика 23 : колонка: Aquity UPLC BEH C18 (50 мм×2,1 мм, 2,5 мкм); подвижная: фаза A: 0,01 M ацетат аммония в воде; B: AcN; градиентный режим: время/% B: 0/5,0,2/5,7/100,8/100,8,5/5,11/5; температура колонки: 40°C; скорость потока: 0,5 мл/мин;
Методика 24: колонка: Luna Omega 3 мкм PS C18 100A; подвижная: фаза A: 0,01 M формиат аммония в воде:AcN (95:5); B: 0,01 M формиат аммония в воде:AcN (5:95); градиентный режим: время/% B: 0/2,1/2,4/98,4,5/98,5,5/2,6,5/2; скорость потока: 1,0 мл/мин;
Методика 25: колонка - AQUITY UPLC BEH C18 (50 мм×2,1 мм, 1,7 мкм); подвижная: фаза-A: 0,1%FA в воде, B: 0,1% FA в ацетонитриле T%A:0/95,0,3/95,2,0/5,3,5/5,3,6/95,4,4/95
скорость потока: 0,6 мл/мин, температура: 40°C
Методика 26: колонка: Aquity UPLC BEH C18 (50 мм×2,1 мм, 1,7 мкм); подвижная: фаза A: 0,01 M ацетат аммония в воде; B: AcN; градиентный режим: время/% A: 0/5, 0,1/5, 2,4/100, 3,8/100,4,0/5;4,5/5 температура колонки: 50°C; скорость потока: 0,5 мл/мин
Методика 27: колонка: Luna Omega 3 мкм PS C18 100A); подвижная: фаза A: 0,01 M формиат аммония в воде: AcN(95:5), B: 0,01 M формиат аммония в воде:AcN(5:95); градиентный режим: время/% B: 0/2,1/2,7/100,7,5/100,9/2,10/2; скорость потока: 1,0 мл/мин.
ССЫЛОЧНЫЙ ПРИМЕР 1
2-(Триметилстаннил)изоникотинонитрил
При перемешивании к раствору 2-бромизоникотинонитрила (2 г, 10,92 ммоля) в толуоле (20 мл) добавляли гексаметилдиолово (4,6 г, 14,20 ммоля) и Pd(PPh3)4 (1,2 г, 1,09 ммоля) при КТ. Полученный раствор дегазировали азотом в течение 10 мин и нагревали при 110°C в течение 16 ч. Реакционную смесь выпаривали при пониженном давлении и неочищенное соединение очищали с помощью колоночной флэш-хроматографии на нейтральном оксиде алюминия с использованием 50% EtOAc в петролейном эфире и получали искомое соединение (1,5 г, 51,7%).
LC-MS (методика 1): Rt=1,93 мин; m/z=269,08 (M+H+)
ССЫЛОЧНЫЙ ПРИМЕР 2
5-Бром-1-бутил-1H-пирроло[2,3-c]пиридин
При перемешивании к суспензии 60% NaH (0,146 г, 6,091 ммоля) в DMF (20 мл) при 0°C добавляли 5-бром-1H-пирроло[2,3-c]пиридин (0,8 г, 4,06 ммоля) и перемешивали в течение 15 мин. Затем при 0°C к реакционной смеси добавляли 1-бромбутан (0,66 г, 4,873 ммоля). Полученной смеси давали нагреться до КТ и перемешивали в течение 16 ч. Реакцию останавливали водой и экстрагировали с помощью EtOAc и органический слой сушили над безводным Na2SO4 и его концентрировали при пониженном давлении. Неочищенное соединение очищали с помощью колоночной флэш-хроматографии с использованием 10% EtOAc в петролейном эфире в качестве элюента и получали искомое соединение (0,78 г, 67%).
LC-MS (методика 2): Rt =2,27 мин; m/z=253,17 (M+H+).
По методике, аналогичной описанной в ссылочном примере 2, но с использованием в каждом случае соответствующих исходных веществ, получали следующие соединения:
(*)добавляли 0,02 экв. NaI.
ССЫЛОЧНЫЙ ПРИМЕР 3
5-Бром-1-бутил-N,N-диметил-1H-пирроло[2,3-c]пиридин-2-карбоксамид
Стадия a. 5-Бром-N,N-диметил-1H-пирроло[2,3-c]пиридин-2-карбоксамид
При перемешивании к раствору 5-бром-1H-пирроло[2,3-c]пиридин-2-карбоновой кислоты (500 мг, 2,07 ммоля) в DMF при 0°C добавляли диметиламингидрохлорид (168 мг, 2,07 ммоля), TEA (1,49 г, 10,37 ммоля) и T3P (1,97 г, 6,21 ммоля). Полученную смесь перемешивали при КТ в течение 16 ч. Реакционную смесь разбавляли водой и экстрагировали с помощью EtOAc. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенное соединение очищали с помощью колоночной хроматографии на силикагеле и элюировали с помощью 5% MeOH в DCM и получали искомое соединение (450 мг, 78%).
LC-MS (методика 4): Rt=1,75 мин; m/z=270,15 (M+H++2).
Стадия b. 5-Бром-1-бутил-N,N-диметил-1H-пирроло[2,3-c]пиридин-2-карбоксамид
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия a (1,4 г, 5,24 ммоля) в DMF при 0°C добавляли 60% NaH (1,04 г, 26,21 ммоля). Реакционную смесь перемешивали в течение 15 мин при такой же температуре и затем добавляли бутилйодид (1,92 г, 10,48 ммоля). Суспензию перемешивали при КТ в течение 16 ч. Реакционную смесь охлаждали до 0°C, реакцию останавливали водой и экстрагировали с помощью EtOAc. Отделенный органический слой сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенное соединение очищали с помощью колоночной хроматографии на силикагеле и элюировали с помощью 5% MeOH в DCM и получали искомое соединение (1,0 г, 59,8%)
LC-MS (методика 5): Rt=1,10 мин; m/z=325,36 (M+H+).
По методике, аналогичной описанной в ссылочном примере 3, но с использованием в каждом случае соответствующего исходного вещества, получали следующие соединения:
ССЫЛОЧНЫЙ ПРИМЕР 4
3-((6-Бромпиридин-3-ил)окси)-N,N-диэтилпропан-1-амин
При перемешивании к раствору 6-бромпиридин-3-ола (5,0 г, 28,73 ммоля) в THF (50 мл) при 0°C добавляли PPh3 (15,0 г, 57,47 ммоля), DIAD (8,7 г, 43,10 ммоля) и 3-(диэтиламино)пропан-1-ол (5,64 г, 43,10 ммоля). Реакционную смесь перемешивали при КТ в течение 16 ч. Реакционную смесь разбавляли водой (100 мл) и экстрагировали с помощью EtOAc (2×100 мл). Объединенный органический слой сушили над безводным Na2SO4, фильтровали и фильтрат концентрировали. Неочищенное соединение очищали с помощью колоночной флэш-хроматографии с использованием 100-200 силикагеля и элюировали с помощью 40% EtOAc/петролейный эфир и получали искомое соединение (3,1 г, 38%) в виде смолообразной жидкости.
LC-MS (методика 7): Rt=1,50 мин; m/z=287,18 (M+H+).
ССЫЛОЧНЫЙ ПРИМЕР 5
1-(5-Бром-1-бутил-1H-пирроло[2,3-c]пиридин-3-ил)-N,N-диметилметанамин
При перемешивании к раствору соединения ссылочного примера 2 (500 мг, 1,97 ммоля) в n-BuOH (20 мл) добавляли 37% водный раствор формальдегида (37%) (0,78 мл, 9,88 ммоля) и диметиламингидрохлорид (805 мг, 9,88 ммоля). Реакционную смесь перемешивали при 120°C в течение 16 ч и затем ее охлаждали до КТ. Органический растворитель выпаривали при пониженном давлении и получали остаток, который разбавляли 1 н. водным раствором NaOH и экстрагировали с помощью 10% MeOH/DCM (2×50 мл). Неочищенный остаток выпаривали и получали неочищенное соединение, которое очищали с помощью колоночной хроматографии на 230-400 диоксиде кремния с использованием 85% EtOAc/петролейный эфир вместе с 0,5 мл TEA и получали 400 мг (65%) искомого соединения в виде смолообразного твердого вещества.
LC-MS (методика 5): Rt=0,77 мин; m/z=312,12 (M+H++2).
По методике, аналогичной описанной в ссылочном примере 5, но с использованием в каждом случае соответствующих исходных веществ, получали следующие соединения:
ССЫЛОЧНЫЙ ПРИМЕР 6
2-Бром-5-(3-(4,4-дифторпиперидин-1-ил)пропокси)пиридин
Стадия a. 2-Бром-5-(3-хлорпропокси)пиридин
При перемешивании к раствору 6-бромпиридин-3-ола (3 г, 17,2 ммоля) в DMF (50 мл) добавляли K2CO3 (7,1 г, 51,6 ммоля) и перемешивали при КТ в течение 15 мин. Его охлаждали до 0°C и добавляли 1-бром-3-хлорпропан (4 г, 25,8 ммоля). Полученную смесь перемешивали при КТ в течение 16 ч. Реакционную смесь разбавляли охлажденной льдом водой и экстрагировали с помощью EtOAc (3×100 мл), промывали водой (2×80 мл) и затем рассолом (50 мл). Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении и получали искомое соединение (2,8 г, 64%) в виде желтой смолообразной жидкости.
LC-MS (методика 6): Rt=2,04 мин; m/z=250,01 (M+H+).
Стадия b. 2-Бром-5-(3-(4,4-дифторпиперидин-1-ил)пропокси)пиридин
К раствору соединения, полученного в предыдущем разделе, стадия a (2 г, 7,98 ммоля) в ацетонитриле при КТ добавляли 4,4-дифторпиперидингидрохлорид (1,88 г, 11,97 ммоля), K2CO3 (3,3 г, 23,95 ммоля) и NaI (1,19 г, 7,98 ммоля). Полученную смесь перемешивали при 70°C в течение 24 ч и затем ее охлаждали до КТ. Реакционную смесь выливали в воду со льдом (30 мл) и экстрагировали с помощью EtOAc (2×80 мл): Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток промывали н-пентаном и сушили и получали 1,5 г (56%) искомого соединения в виде смолообразной жидкости.
LC-MS (методика 5): Rt=0,66 мин; m/z=335,15 (M+H+).
По методике, аналогичной описанной в ссылочном примере 6, но с использованием соответствующего исходного вещества, получали следующее соединение:
ССЫЛОЧНЫЙ ПРИМЕР 7
N-(6-Бромпиридин-3-ил)-N-бутил-3-метоксипропанамид
Стадия a. 6-Бром-N-бутилпиридин-3-амин (2) (C2134-130):
При перемешивании к раствору 6-бромпиридин-3-амина (10 г, 57 ммоля) в MeOH (100 мл) добавляли масляный альдегид (4,9 г, 69,36 ммоля) и перемешивали в течение 16 ч при КТ. Его охлаждали до 0°C и добавляли NaBH3CN (7,2 г, 115,6 ммоля). Полученную смесь перемешивали при КТ в течение 16 ч. Реакционную смесь выливали в холодную воду (100 мл), экстрагировали с помощью EtOAc (2×200 мл) и промывали рассолом (150 мл). Органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении и получали неочищенное соединение, которое очищали с помощью колоночной хроматографии с использованием 13% EtOAc/петролейный эфир и получали 7 г (52%) искомого соединения.
LC-MS (методика 4): Rt=2,31 мин; m/z=229,16 (M+H+).
Стадия b. N-(6-Бромпиридин-3-ил)-N-бутил-3-метоксипропанамид
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия a (1500 мг, 6,54 ммоля, 1,0 экв.) в DCM при 0°C добавляли 3-метоксипропановую кислоту (1020 мг, 9,84 ммоля), TEA (3,30 г, 32,75 ммоля) и T3P (8,3 г, 26,18 ммоля). Реакционную смесь перемешивали при КТ в течение 16 ч. Реакционную смесь разбавляли водой и экстрагировали с помощью EtOAc. Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенное соединение очищали с помощью колоночной хроматографии на силикагеле и элюировали с помощью 30% EtOAc в петролейном эфире и получали (1450 мг, 70%) искомое соединение.
LC-MS (методика 10): Rt=2,40 мин; m/z=315,12 (M+H+).
ССЫЛОЧНЫЙ ПРИМЕР 8
6-Бром-N-(циклопропилметил)-N-метилпиридин-3-амин
При перемешивании к раствору 6-бром-N-метилпиридин-3-амина (1200 мг, 6,44 ммоля) в DMF при 0°C добавляли 60% NaH (1030 мг, 25,76 ммоля) и перемешивали при КТ в течение 15 мин. Его охлаждали при 0°C и добавляли (бромметил)циклопропан (1738 мг, 12,88 ммоля). Его перемешивали при КТ в течение 1 ч. Реакционную смесь охлаждали до 0°C, реакцию останавливали водой и экстрагировали с помощью EtOAc. Отделенный органический слой промывали водой и рассолом и сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенное соединение очищали с помощью колоночной хроматографии и элюировали с помощью 15% EtOAc в петролейном эфире и получали искомое соединение (1,0 г, 64,2%) в виде желтого смолообразного вещества;
LC-MS (методика 5): Rt=1,20 мин; m/z=240,97 (M+H+).
ССЫЛОЧНЫЙ ПРИМЕР 9
Трет-бутил-(6-бромпиридин-3-ил)(3-(диэтиламино)пропил)карбамат
Стадия a. 3-Хлор-N,N-диэтилпропан-1-амингидрохлорид
При перемешивании к раствору 3-(диэтиламино)пропан-1-ола (1 г, 7,63 ммоля) в DCM (10 мл) при 0°C добавляли SOCl2 (1,1 мл, 15,26 ммоля) и полученную смесь перемешивали при КТ в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении и получали искомое соединение (1 г, 88%).
Стадия b. Трет-бутил-(6-бромпиридин-3-ил)(3-(диэтиламино)пропил)карбамат
При перемешивании к суспензии 60% NaH (0,15 г, 6,598 ммоля) в DMF (50 мл) при 0°C добавляли раствор трет-бутил-(6-бромпиридин-3-ил)карбамата (1,2 г, 4,399 ммоля) в DMF (10 мл). Полученную смесь перемешивали в течение 15 мин и затем к реакционной смеси при 0°C добавляли соединение (0,785 г, 5,274 ммоля), полученное в предыдущем разделе, стадия a. Полученную смесь перемешивали при КТ в течение 16 ч. Реакцию останавливали охлажденной льдом водой и экстрагировали с помощью EtOAc. Органический слой концентрировали при пониженном давлении и полученное неочищенное соединение очищали с помощью колоночной флэш-хроматографии с использованием 100% EtOAc в качестве элюента и получали искомое соединение (0,9 г, 53)
LC-MS (методика 13): Rt=1,55 мин; m/z=388,09 (M+H++2).
ССЫЛОЧНЫЙ ПРИМЕР 10
6-Бром-N-(3-(4,4-дифторпиперидин-1-ил)пропил)-N-метилпиридин-3-амин
Стадия a. Трет-бутил-(6-бромпиридин-3-ил)(3-хлорпропил)карбамат
При перемешивании к раствору трет-бутил 6-бромпиридин-3-илкарбамата (2,0 г, 7,352 ммоля) в DMF (20 мл) при 0°C добавляли 60%NaH (0,529 г, 22,05 ммоля) и перемешивали при КТ в течение 15 мин. Затем добавляли 1-бром-3-хлорпропан (2,308 г, 14,705 ммоля) и полученную смесь перемешивали при КТ в течение 16 ч. Реакционную смесь охлаждали до 0°C, реакцию останавливали водой со льдом и экстрагировали с помощью EtOAc. Органические слои промывали водой и рассолом, сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенное соединение очищали с помощью колоночной хроматографии на силикагеле и элюировали с помощью 8% EtOAc в петролейном эфире и получали искомое соединение (2,0 г, LCMS~75%) виде коричневого смолообразного вещества;
LC-MS (методика 5): Rt=1,31 мин; m/z=349,20 (M+H+).
Стадия b Трет-бутил-(6-бромпиридин-3-ил)(3-(4,4-дифторпиперидин-1-ил)пропил)карбамат.
При перемешивании к раствору 4,4-дифторпиперидингидрохлорида (1,815 г, 11,49 ммоля) в AcN (30 мл) добавляли NaI (1,027 г, 6,896 ммоля) и K2CO3 (2,379 г, 17,24 ммоля). Реакционную смесь перемешивали в течение 10 мин, добавляли соединение, полученное в предыдущем разделе, стадия a (2,0 г, 5,747 ммоля) и нагревали при 90°C в течение 16 ч. Реакционную смесь фильтровали через слой целита и ее промывали с помощью 10% MeOH в DCM. Фильтрованный раствор концентрировали при пониженном давлении и неочищенное соединение очищали с помощью колоночной хроматографии на силикагеле с использованием 25% EtOAc в петролейном эфире в качестве элюента и получали искомое соединение (1,1 г, 44%) в виде почти белого твердого вещества;
LC-MS (методика 5): Rt=0,86 мин; m/z=434,35 (M+H+).
Стадия c. 6-Бром-N-(3-(4,4-дифторпиперидин-1-ил)пропил)пиридин-3-амин
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия b (1,4 г, 3,22 ммоля) в DCM (10 мл) при 0°C добавляли TFA (3,0 мл) и его перемешивали при КТ в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении. Неочищенный остаток разбавляли водой, подщелачивали до pH 8 с помощью NaHCO3 и экстрагировали с помощью EtOAc. Органические слои промывали водой и рассолом. Органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении и получали искомое соединение (1,01 г, 94%) в виде почти белого твердого вещества;
LC-MS (методика 6): Rt=1,40 мин; m/z=334,11 (M+H+).
Стадия d. 6-Бром-N-(3-(4,4-дифторпиперидин-1-ил)пропил)-N-метилпиридин-3-амин
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия c (3,0 г, 9,0 ммоля) в муравьиной кислоте (40 мл) добавляли параформальдегид (2,70 г, 90,09 ммоля) и нагревали при 100°C в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении: Неочищенный остаток растворяли в воде и подщелачивали до pH~8 насыщенным водным раствором NaHCO3 и экстрагировали с помощью EtOAc (3×50 мл). Отделенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении Неочищенное соединение очищали с помощью колоночной хроматографии на силикагеле и элюировали с помощью 20% EtOAc в петролейном эфире и получали искомое соединение (2,5 г, 79%) в виде почти белого твердого вещества.
LC-MS (методика 6): Rt=1,55 мин; m/z=348,13 (M+H+).
ССЫЛОЧНЫЙ ПРИМЕР 11
3-(2-Бромпиримидин-5-илокси)-N,N-диэтилпропан-1-амин
Стадия a. 2-Бромпиримидин-5-ол
При перемешивании к раствору 2-хлорпиримидин-5-ола (2 г, 15,3 ммоля) в AcOH (6 мл) при 0°C добавляли HBr (47% водный раствор) (6 мл) и его перемешивали при КТ в течение 15 мин. Полученную смесь перемешивали при 100°C в течение 24 ч. Растворители выпаривали в вакууме и затем выливали в охлажденную льдом воду и экстрагировали с помощью EtOAc (2×100 мл). Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении и получали неочищенный остаток, который очищали с помощью колоночной хроматографии на 100-200 диоксиде кремния с использованием 30% EtOAc/петролейный эфир и получали искомое соединение (1,5 г, 55%) в виде почти белого твердого вещества.
LC-MS (методика 6): Rt=0,94 мин; m/z=175,03 (M+H+).
Стадия b. 2-Бром-5-(3-хлорпропокси)пиримидин
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия a (1,5 г, 8,6 ммоля) в DMF добавляли K2CO3 (3,5 г, 25,8 ммоля) и 1-бром-3-хлорпропан (2) (2 г, 12,9 ммоля). Полученную смесь перемешивали при КТ в течение ночи. Реакционную смесь разбавляли водой, экстрагировали с помощью EtOAc (2×100 мл). Органический слой сушили над Na2SO4, и выпаривали при пониженном давлении и получали 1,8 г (83%) неочищенного соединения, которое использовали на следующей стадии без какой-либо дополнительной очистки.
LC-MS (методика 6): Rt=1,89 мин; m/z=251,08 (M+H+).
Стадия c. 3-(2-Бромпиримидин-5-илокси)-N,N-диэтилпропан-1-амин
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия b (1,8 г, 7,2 ммоля) в AcN добавляли K2CO3 (2,9 г, 21,6 ммоля) и NaI (1,07 г, 7,2 ммоля). Полученную смесь перемешивали при КТ в течение 15 мин и диэтиламингидрохлорид (2,6 г, 36 ммоля) добавляли при КТ. Ее перемешивали при 70°C в течение 16 ч. Реакционную смесь разбавляли охлажденной льдом водой и экстрагировали с помощью EtOAc (3×100 мл). Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении и получали искомое соединение (1,2 г, 58%) виде коричневой смолообразной жидкости.
LC-MS (методика 6): Rt=1,08 мин; m/z=288,18 (M+H+).
ССЫЛОЧНЫЙ ПРИМЕР 12
N1-(6-Бромпиридин-3-ил)-N3,N3-диэтил-N1-метилпропан-1,3-диамин
Стадия a. N1-(6-Бромпиридин-3-ил)-N3,N3-диэтилпропан-1,3-диамин
При перемешивании к раствору соединения ссылочного примера 9 (1,2 г, 3,1 ммоля) в DCM (20 мл) добавляли TFA (3,5 мл) и перемешивали при КТ в течение 8 ч. Реакционную смесь концентрировали досуха и получали неочищенный остаток, который очищали с помощью колоночной хроматографии и элюировали с помощью 10%MeOH/DCM и получали 0,9 г (100%) искомого соединения.
LC-MS (методика 6): Rt=1,27 мин; m/z=286,13 (M+H+).
Стадия b. N1-(6-Бромпиридин-3-ил)-N3,N3-диэтил-N1-метилпропан-1,3-диамин
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия a (800 мг, 2,795 ммоля) в муравьиной кислоте (10 мл) при 0°C медленно добавляли параформальдегид (839 мг, 27,95 ммоля) и перемешивали при 95°C в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении и остаток растворяли в воде. Водный слой подщелачивали насыщенным водным раствором NaOH и концентрировали при пониженном давлении. Полученный неочищенный остаток очищали с помощью колоночной хроматографии и элюировали с помощью 10%MeOH/DCM и получали искомое соединение (700 мг, 67,3%).
LC-MS (методика 6): Rt=1,37 мин; m/z=300,16 (M+H+).
ССЫЛОЧНЫЙ ПРИМЕР 13
3-(6-Бромпиридин-3-илокси)-N-метилпропан-1-амин
В герметизированной пробирке при перемешивании к раствору соединения, полученного в ссылочном примере 6, стадия a (1 г, 4 ммоля) в AcN при 0°C добавляли K2CO3 (1,6 г, 12 ммоля) и NaI (300 мг, 2 ммоля) и перемешивали при КТ в течение 15 мин. Затем при 0°C добавляли 33% раствор метиламина в EtOH, (0,6 мл, 6 ммоля). Полученную смесь перемешивали при 60°C в течение 16 ч. Ее разбавляли охлажденной льдом водой и экстрагировали с помощью EtOAc. Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении и получали неочищенный остаток, который очищали с помощью колоночной хроматографии на 230-400 диоксиде кремния с использованием 2% MeOH/DCM в качестве элюента и получали искомое соединение (750 мг, 76%) в виде смолообразной жидкости.
LC-MS (методика 14): Rt=1,07 мин; m/z=245,01 (M+H+).
ССЫЛОЧНЫЙ ПРИМЕР 14
6-Бром-1-(тетрагидро-2H-пиран-4-ил)-1H-пирроло[3,2-c]пиридин
При перемешивании к раствору 6-бром-1H-пирроло[3,2-c]пиридина (1 г, 3,36 ммоля) в DMF (20 мл) при 0°C добавляли тетрагидро-2H-пиран-4-илметансульфонат (1,51 г, 8,38 ммоля), и Cs2CO3 (5,46 г, 16,8 ммоля). Полученную смесь перемешивали при 100°C в течение 16 ч. Реакционную смесь разбавляли с помощью EtOAc и промывали водой. Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток очищали с помощью колоночной хроматографии и элюировали с помощью 10%MeOH в DCM и получали искомое соединение (0,33 г, 35%) в виде почти белого твердого вещества.
LC-MS (методика 6): Rt=1,73 мин; m/z=281,07 (M+H+).
По методике, аналогичной описанной в ссылочном примере 14, но с использованием соответствующего исходного вещества, получали следующее соединение:
ССЫЛОЧНЫЙ ПРИМЕР 15
3-((6-Бромпиридин-3-ил)окси)-N-этил-N-фенэтилпропан-1-амин
Стадия a. 3-((6-Бромпиридин-3-ил)окси)пропан-1-ол
При перемешивании к суспензии 6-бромпиридин-3-ола (5,0, 28,73 ммоля) в DMF (40 мл) при КТ добавляли K2CO3 (11,90 г, 86,20 ммоля) и 3-бромпропанол (4,39 г, 31,60 ммоля) и перемешивали в течение 16 ч. Реакцию останавливали охлажденной льдом водой (150 мл) и экстрагировали этилацетатом (2×60 мл). Объединенные органические слои промывали водой (100 мл), затем рассолом (100 мл) и сушили над безводным Na2SO4 и фильтровали. Фильтрованный раствор концентрировали при пониженном давлении и полученное неочищенное соединение очищали с помощью колоночной флэш-хроматографии с использованием 30% этилацетата в петролейном эфире в качестве элюента и получали искомое соединение (3,50 г, 53%) в виде бесцветной жидкости.
LC-MS (методика 5): Rt=0,82 мин; m/z=232,05 (M+H+).
Стадия b. 2-Бром-5-(3-бромпропокси)пиридин
К раствору соединения, полученного в предыдущем разделе, стадия a (3,50 г, 15,08 ммоля) в DCM (50 мл) при 0°C добавляли PPh3 (11,0 г, 30,17 ммоля) и CBr4 (9,98 г, 30,17 ммоля). Полученную смесь перемешивали при комнатной температуре в течение 6 ч. Растворитель удаляли при пониженном давлении и неочищенный остаток очищали на колонке с силикагелем с использованием 10% EtOAc в петролейном эфире в качестве элюента и получали искомое соединение (2,50 г, 56%) в виде бесцветной жидкости.
LC-MS (методика 5): Rt=1,19 мин; m/z=293,99 (M+H+).
Стадия c. 3-((6-Бромпиридин-3-ил)окси)-N-фенэтилпропан-1-амин
К раствору соединения, полученного в предыдущем разделе, стадия b (2,50 г, 8,47 ммоля) и 2-фенилэтанамина (1,54 г, 12,71 ммоля) в AcN (50 мл) добавляли NaI (1,27 г, 8,47 ммоля) и K2CO3 (3,50 г, 25,42 ммоля). Полученную смесь нагревали при 60°C в течение 16 ч. Реакционную смесь быстро охлаждали, фильтровали и фильтрат концентрировали при пониженном давлении: Неочищенный остаток очищали с помощью колоночной хроматографии с обращенной фазой на колонке Grace с использованием 35% AcN в 0,1% водном растворе муравьиной кислоты в качестве элюента и получали искомое соединение (1,50 г, 53%) в виде бесцветной жидкости.
LC-MS (методика 5): Rt=0,81 мин; m/z=335,20 (M+H+).
Стадия d. 3-((6-Бромпиридин-3-ил)окси)-N-этил-N-фенэтилпропан-1-амин
К раствору соединения, полученного в предыдущем разделе, стадия c (1,50 г, 4,47 ммоля) в DMF (20 мл) добавляли K2CO3 (1,85 г, 13,43 ммоля) и этилйодид (1,04 г, 6,71 ммоля) и перемешивали при комнатной температуре в течение 16 ч. Реакцию останавливали охлажденной льдом водой (150 мл) и экстрагировали этилацетатом (2×60 мл). Объединенные органические слои промывали водой (100 мл), затем рассолом (100 мл) и сушили над безводным Na2SO4 и фильтровали. Фильтрованный раствор концентрировали при пониженном давлении и полученное неочищенное соединение очищали на колонке Grace с обращенной фазой с использованием 40% ацетонитрила в 0,1% водном растворе муравьиной кислоты в качестве элюента и получали искомое соединение (1,10 г, 68%) в виде бесцветной жидкости.
LC-MS (методика 6): Rt=1,62 мин; m/z=363,29 (M+H+).
ССЫЛОЧНЫЙ ПРИМЕР 16
N-(6-Бромпиридин-3-ил)-2-фенилацетамид
При перемешивании к раствору 6-бромпиридин-3-амина (2,0 г, 11,56 ммоля) в DCM (50 мл) добавляли 2-фенилуксусную кислоту (2,04 г, 15,02 ммоля) и TEA (4,60 г, 46,24 ммоля), затем добавляли T3P (9,19 г, 28,90 ммоля) при 0°C. Полученную смесь перемешивали при КТ в течение 16 ч. Реакционную смесь промывали насыщенным водным раствором NaHCO3 (100 мл). Органический слой сушили над безводным Na2SO4 и фильтровали. Фильтрованный раствор концентрировали при пониженном давлении и полученное неочищенное соединение очищали с помощью колоночной флэш-хроматографии с использованием 25% этилацетата в петролейном эфире в качестве элюента и получали искомое соединение (2,30 г, 68%) в виде почти белого твердого вещества.
LC-MS (методика 1): Rt=2,46 мин; m/z=291,08 (M+H+).
По методике, аналогичной описанной в ссылочном примере 16, но с использованием в каждом случае соответствующего исходного вещества, получали следующее соединение:
(*) с использованием THF вместо DCM.
(**) с использованием DMF вместо DCM
ССЫЛОЧНЫЙ ПРИМЕР 17
2-Бром-4-(2-(4,4-дифторпиперидин-1-ил)этокси)пиридин
Стадия a. 2-((2-Бромпиридин-4-ил)окси)этан-1-ол
При перемешивании к раствору 2-бромпиридин-4-ола (2,0 г, 11,5 ммоля) в DMF при КТ добавляли K2CO3 (3,96 г, 28,75 ммоля). При 0°C медленно добавляли 2-бромэтанол (2,15 г,17,25 ммоля). Затем его перемешивали при 70°C в течение 12 ч. Реакцию останавливали охлажденной льдом водой и экстрагировали с помощью DCM. Органический слой промывали насыщенным водным раствором NaHCO3, затем рассолом и сушили над безводным Na2SO4 и фильтровали. Фильтрованный раствор концентрировали при пониженном давлении и полученное неочищенное соединение очищали с помощью колоночной флэш-хроматографии на (100-200) силикагеле с использованием 20% EtOAc/петролейный эфир в качестве элюента и получали искомое соединение (1,2 г, 47,8%) в виде бледно-желтой жидкости.
LC-MS (методика 14): Rt=1,26 мин; m/z=220,0 (M+H++2).
Стадия b. 2-Бром-4-(2-бромэтокси)пиридин
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия a (1,1 г, 5,0 ммоля) в DCM при 0°C добавляли PPh3 (1,57 г, 1,2 экв.) и CBr4 (3,31 г, 10 ммоля). Его перемешивали при КТ в течение 12 ч. Реакцию останавливали охлажденной льдом водой и экстрагировали с помощью DCM. Органический слой промывали насыщенным водным раствором NaHCO3, затем рассолом и сушили над безводным Na2SO4 и фильтровали. Фильтрованный раствор концентрировали при пониженном давлении и полученное неочищенное соединение очищали с помощью колоночной флэш-хроматографии на (230-400) силикагеле с использованием 10% EtOAc/петролейный эфир в качестве элюента и получали искомое соединение (0,9 г, 64%) в виде бледно-желтой жидкости.
LC-MS (методика 14): Rt=2,18 мин; m/z=281,9 (M+H+).
Стадия c. 2-Бром-4-(2-(4,4-дифторпиперидин-1-ил)этокси)пиридин
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия b (400 мг, 1,42 ммоля) в AcN, K2CO3 (0,58 г, 4,26 ммоля) при КТ добавляли NaI (0,1 г, 0,71 ммоля) и дифторпиперидингидрохлорид (0,25 г, 2,13 ммоля) и затем нагревали при 80°C в течение 12 ч. Органические растворители выпаривали досуха. Полученный неочищенный остаток растворяли в воде и DCM. Объединенные органические слои промывали водой, затем рассолом и сушили над безводным Na2SO4 и фильтровали. Фильтрованный раствор концентрировали при пониженном давлении и полученное неочищенное соединение очищали с помощью колоночной флэш-хроматографии на (230-400) силикагеле с использованием 50% EtOAc/петролейный эфир в качестве элюента и получали искомое соединение (0,350 г, 76,7%) в виде бледно-желтой жидкости.
LC-MS (методика 15): Rt=2,91 мин; m/z=321,30 (M+H+).
По методике, аналогичной описанной в ссылочном примере 17, но с использованием соответствующего исходного вещества, получали следующее соединение:
ССЫЛОЧНЫЙ ПРИМЕР 18
4-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразол
При перемешивании к раствору 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (2 г, 10,3 ммоля) в DMF (30 мл) добавляли (2-(хлорметокси)этил)триметилсилан (2 г, 12,3 ммоля), и Cs2CO3 (10 г, 30,9 ммоля). Полученную смесь перемешивали при КТ в течение 3 ч. Растворители выпаривали и неочищенный остаток разбавляли охлажденной льдом водой и экстрагировали с помощью EtOAc. Объединенные органические слои сушили над безводным Na2SO4 и фильтровали. Фильтрованный раствор концентрировали при пониженном давлении и полученное неочищенное соединение очищали с помощью колоночной флэш-хроматографии с использованием 20-30% EtOAc/петролейный эфир и получали искомое соединение (1,5 г, 44%) в виде бледно-желтого смолообразного вещества.
LC-MS (методика 14): Rt=3,08 мин; m/z=325,2 (M+H+).
СЫЛОЧНЫЙ ПРИМЕР 19
5-Бром-1-пропил-3-(1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-ил)-1H-пирроло[2,3-c]пиридин
Стадия a. 5-Бром-3-йод-1H-пирроло[2,3-c]пиридин
При перемешивании к раствору 5-бром-1H-пирроло[2,3-c]пиридина (1 г, 5,1 ммоля) в DMF добавляли KOH (0,86 г, 15,3 ммоля). Его перемешивали при КТ в течение 20 мин и затем добавляли йод (1,54 г, 6,12 ммоля). Полученную смесь перемешивали при КТ в течение 16 ч. Реакционную смесь выпаривали в вакууме и получали неочищенный остаток. К полученному неочищенному остатку добавляли колотый лед и осажденное твердое вещество отфильтровывали, сушили и промывали диэтиловым эфиром и получали 1 г (60%) неочищенного соединения, которое использовали на следующей стадии без какой-либо дополнительной очистки.
LC-MS (методика 6): Rt=1,94 мин; m/z=323,01 (M+H+).
Стадия b. 5-Бром-3-йод-1-пропил-1H-пирроло[2,3-c]пиридин
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия a (1 г, 3,1 ммоля) в DMF при 0°C добавляли NaH (60%) (0,371g, 9,2 ммоля). Через 15 мин медленно добавляли бромпропан (0,45 г, 3,72 ммоля) и перемешивали при КТ в течение 3 ч. К реакционной смеси добавляли колотый лед и осадившееся твердое вещество отфильтровывали, сушили и промывали диэтиловым эфиром и н-пентаном и получали 900 мг (79%) неочищенного соединения, которое использовали на следующей стадии без какой-либо дополнительной очистки.
LC-MS (методика 6): Rt=2,28 мин; m/z=365,01 (M+H+).
Стадия c. 5-Бром-1-пропил-3-(1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-ил)-1H-пирроло[2,3-c]пиридин
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия b (500 мг, 1,4 ммоля) в смеси DMF/вода 1:1 добавляли соединение ссылочного примера 18 (0,545 г, 1,7 ммоля) и Cs2CO3 (1,36 г, 4,2 ммоля). Полученный раствор дегазировали азотом в течение 15 мин, затем добавляли PdCl2dppf (0,1 г, 0,14 ммоля) и перемешивали при 90°C в течение 16 ч. Реакционную смесь выпаривали в вакууме и получали неочищенный остаток, который разбавляли холодной водой и экстрагировали с помощью EtOAc. Объединенные органические слои сушили над безводным Na2SO4 и фильтровали. Фильтрованный раствор концентрировали при пониженном давлении и полученное неочищенное соединение очищали с помощью колоночной флэш-хроматографии с использованием 20-30% EtOAc/петролейный эфир и получали искомое соединение (380 мг, 63%) в виде бледно-коричневого смолообразного вещества.
LC-MS (методика 6): Rt=2,39 мин; m/z=435,34 (M+H+).
По методике, аналогичной описанной в ссылочном примере 19, но с использованием соответствующего исходного вещества, получали следующее соединение:
ССЫЛОЧНЫЙ ПРИМЕР 20
N-(2-Бромпиридин-4-ил)-2-(4,4-дифторпиперидин-1-ил)ацетамид
Стадия a. N-(2-Бромпиридин-4-ил)-2-хлорацетамид
При перемешивании к раствору 2-бромпиридин-4-амина (2 г, 11,5 ммоля) в DCM (40 мл) при 0°C добавляли DIPEA (4 мл, 23 ммоля) и хлорацетилхлорид (1,85 мл, 23 ммоля). Его перемешивали при КТ в течение 3 ч. Реакционную смесь разбавляли с помощью EtOAc и промывали водой. Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенное соединение очищали с помощью колоночной хроматографии и элюировали с помощью 20% EtOAc в петролейном эфире и получали искомое соединение (2,2 г, 76,5%) в виде желтоватого твердого вещества.
LC-MS (методика 6): Rt=1,70 мин; m/z=249,02 (M+H+).
Стадия b. N-(2-Бромпиридин-4-ил)-2-(4,4-дифторпиперидин-1-ил)ацетамид
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия a (2,1 г, 8,4 ммоля) в AcN (50 мл) при 0°C добавляли K2CO3 (3,5 г, 25,2 ммоля), NaI (1,62 г, 10,9 ммоля) и 4,4-дифторпиперидингидрохлорид (1,52 г, 12,6 ммоля). Его перемешивали при КТ в течение 16 ч. Реакционную смесь фильтровали через слой целита и промывали с помощью EtOAc (50 мл). Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении и получали неочищенное соединение, которое использовали на следующей стадии без какой-либо дополнительной очистки (1,5 г, 53,4%)
LC-MS (методика 6): Rt=1,42 мин; m/z=336,16 (M+H++2).
ССЫЛОЧНЫЙ ПРИМЕР 21
2-Бром-N-(2-(4,4-дифторпиперидин-1-ил)этил)пиридин-4-амин
При перемешивании к раствору соединения ссылочного примера 20 (1,0 г, 2,97 ммоля) в THF (40 мл) при 0°C добавляли LiAlH4 (0,135 г, 3,56 ммоля). Его перемешивали при КТ в течение 3 ч. Реакционную смесь выливали в воду с колотым льдом и разбавляли с помощью EtOAc. Полученную смесь фильтровали через слой целита и концентрировали при пониженном давлении и получали неочищенное соединение, которое использовали на следующей стадии без какой-либо дополнительной очистки (0,5 г, 52,6%)
LC-MS (методика 19): Rt=3,44 мин; m/z=320,31 (M+H+).
ССЫЛОЧНЫЙ ПРИМЕР 22
5-Бром-3-(пиперидин-1-илметил)-1H-пиразоло[3,4-c]пиридин
По методике, аналогичной описанной в ссылочном примере 5, но с использованием 5-бром-1H-пиразоло[3,4-c]пиридина вместо соединения ссылочного примера 2, получали искомое соединение.
LC-MS (методика 6): Rt=1,12 мин; m/z=295,18 (M+H+).
ССЫЛОЧНЫЙ ПРИМЕР 23
6-Бром-1-(2-(4,4-дифторпиперидин-1-ил)этил)-1H-пирроло[3,2-c]пиридин
Стадия a. 6-Бром-1-(2-бромэтил)-1H-пирроло[3,2-c]пиридин
При перемешивании к 40% водному раствору NaOH (15 мл) добавляли 6-бром-1H-пирроло[3,2-c]пиридин (1 г, 5,07 ммоля), TBAB (163 мг, 0,50 ммоля) и 1,2-дибромэтан (15 мл). Полученную смесь перемешивали при 80°C в течение 12 ч. Реакцию останавливали водой и экстрагировали EtOAc. Органический слой сушили над безводным Na2SO4, концентрировали при пониженном давлении и получали неочищенное соединение, которое использовали на следующей стадии без какой-либо дополнительной очистки (1,8 г).
LC-MS (методика 6): Rt=1,77 мин; m/z=304,80 (M+H+).
Стадия b. 6-Бром-1-(2-(4,4-дифторпиперидин-1-ил)этил)-1H-пирроло[3,2-c]пиридин
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия a (1 г, 3,2 ммоля) в AcN (50 мл) добавляли 4,4-дифторпиперидингидрохлорид (780 мг, 5 ммоля), K2CO3 (1,3 г, 9,6 ммоля). Полученную суспензию перемешивали при 70°C в течение 16 ч. Реакционную смесь выливали в холодную воду и экстрагировали с помощью EtOAc (2×80 мл). Органический слой сушили и концентрировали и получали неочищенное соединение, которое очищали с помощью колоночной флэш-хроматографии на (230-400) силикагеле с использованием 28% EtOAc/петролейный эфир в качестве элюента и получали искомое соединение (650 мг, 59%) в виде почти белого твердого вещества.
LC-MS (методика 6): Rt=1,26 мин; m/z=344,26 (M+H+).
ССЫЛОЧНЫЙ ПРИМЕР 24
N-(2-Бромпиридин-4-ил)-3-фенилпропанамид
При перемешивании к раствору 2-бромпиридин-4-амина (500 мг, 2,9 ммоля) в THF (20 мл) добавляли 3-фенилпропаноилхлорид (585 мг, 3,5 ммоля) и DIPEA (1,5 мл, 8,7 ммоля) и его перемешивали при КТ в течение 16 ч. Реакционную смесь выливали в воду со льдом (50 мл) и экстрагировали с помощью EtOAc (2×100 мл).Органический слой сушили над безводным Na2SO4, фильтровали и получали неочищенный остаток, который растирали с н-пентаном и сушили и получали искомое соединение (880, 36%) в виде смолообразного твердого вещества.
LC-MS (методика 14): Rt=2,27 мин; m/z=305,0 (M+H+).
ССЫЛОЧНЫЙ ПРИМЕР 25
6-Бром-1-(пиридин-4-илметил)-1H-пирроло[3,2-c]пиридин
При перемешивании к раствору 6-бром-1H-пирроло[3,2-c]пиридина (1 г, 5,07 ммоля) в DMF (25 мл) добавляли Cs2CO3 (4,95 г, 15,21 ммоля) и 4-(бромметил)пиридингидробромид (1,92 г, 7,60 ммоля). Полученную суспензию перемешивали при КТ и затем нагревали при 50°C в течение 16 ч. Реакционную смесь выливали в воду со льдом и экстрагировали с помощью EtOAc (2×120 мл). Органический слой промывали водой и рассолом, сушили над безводным Na2SO4 и фильтровали. Полученный раствор концентрировали при пониженном давлении и получали неочищенный остаток, который очищали с помощью колоночной флэш-хроматографии на (230-400) силикагеле с использованием 70% EtOAc/петролейный эфир в качестве элюента и получали искомое соединение (800 мг, 57,7%) искомого соединения в виде светло-коричневого твердого вещества.
LC-MS (методика 15): Rt=2,33 мин; m/z=288,27 (M+H+).
По методике, аналогичной описанной в ссылочном примере 25, но с использованием в каждом случае соответствующих исходных веществ, получали следующие соединения:
ССЫЛОЧНЫЙ ПРИМЕР 26
3-Бром-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразоло[4,3-b]пиридин
При перемешивании к раствору 3-бром-1H-пиразоло[4,3-b]пиридина (200 мг, 1,01 ммоля) в DMF (5 мл) при КТ добавляли 3,4-дигидро-2H-пиран (127 мг, 1,51 ммоля) и каталитическое количество pTSA. Полученный раствор перемешивали при 85°C в течение 24 ч. Реакционную смесь охлаждали до КТ, разбавляли водой и экстрагировали с помощью EtOAc. Органический слой промывали водой и рассолом, сушили над безводным Na2SO4 и фильтровали. Полученный раствор концентрировали при пониженном давлении и получали неочищенный остаток, который очищали с помощью колоночной флэш-хроматографии и получали искомое соединение (150 мг, 52,8%).
LC-MS (методика 1): Rt=2,24 мин; m/z=282,30 (M+H+).
ССЫЛОЧНЫЙ ПРИМЕР 27
5-(3-(1H-Пиразол-1-ил)пропокси)-2-бромпиридин
При перемешивании к раствору 1H-пиразола (378 мг, 5,55 ммоля) в DMF (10 мл) при 0°C добавляли 60%NaH (666 мг, 16,6 ммоля) и его перемешивали при КТ в течение 15 мин. Затем добавляли (медленно) соединение, полученное в ссылочном примере 6, раздел a, (1,25 г, 6,1 ммоля). Его перемешивали при КТ в течение 3 ч. Его очищали с помощью колоночной хроматографии на силикагеле с использованием 30-80% EtOAc/петролейный эфир и получали искомое соединение (1,2 г, 70%) в виде почти белого твердого вещества.
LC-MS (методика 14): Rt=2,09 мин; m/z=282,0 (M+H+).
ССЫЛОЧНЫЙ ПРИМЕР 28
6-Бром-1-(1-(2,2,2-трифторэтил)пиперидин-4-ил)-1H-пирроло[3,2-c]пиридин
Стадия a. Трет-бутил-4-(6-бром-1H-пирроло[3,2-c]пиридин-1-ил)пиперидин-1-карбоксилат
При перемешивании к раствору трет-бутил-4-(метилсульфонилокси)пиперидин-1-карбоксилата в DMF (30 мл) при КТ добавляли Cs2CO3 (3,08 г. 9,5 ммоля) и 6-бром-1H-пирроло[3,2-c]пиридин (372 мг, 1,9 ммоля). Реакционную смесь перемешивали при 90°C в течение 12 ч. Реакцию останавливали водой со льдом и экстрагировали с помощью DCM. Органический слой сушили над безводным Na2SO4 и его концентрировали при пониженном давлении. Неочищенное соединение очищали с помощью колоночной флэш-хроматографии с использованием 20% EtOAc в петролейном эфире в качестве элюента и получали искомое соединение (0,60 г, 44%).
LC-MS (методика 14): Rt=2,65 мин; m/z=380,0 (M+H+).
Стадия b. 6-Бром-1-(пиперидин-4-ил)-1H-пирроло[3,2-c]пиридингидрохлорид
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия a (0,6 г, 1,6 ммоля) в диоксане (25 мл) добавляли 4M HCl в диоксане (0,2 мл). Полученную смесь перемешивали при КТ в течение 3 ч. Органические растворители удаляли в вакууме и неочищенный остаток растирали с диэтиловым эфиром. Его фильтровали при пониженном давлении и получали 400 мг (89%) искомого соединения
LC-MS (методика 14): Rt=1,23 мин; m/z=280,0 (M+H+).
Стадия c. 6-Bbromo-1-(1-(2,2,2-трифторэтил)пиперидин-4-ил)-1H-пирроло[3,2-c]пиридин
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия b, (200 мг, 0,7 ммоля) в ацетоне (15 мл) при КТ добавляли K2CO3 (0,483 мг, 3,5 ммоля) и 2,2,2-трифторэтилтрифторметансульфонат (450 мг, 1,4 ммоля). Его перемешивали при 55°C в течение ночи. Органические растворители удаляли в вакууме. Неочищенный остаток экстрагировали с помощью DCM/вода. Затем объединяли органический слой концентрировали при пониженном давлении и получали искомое соединение (150 мг. 58%) в виде почти белого твердого вещества.
LC-MS (методика 6): Rt=2,07 мин; m/z=362,24 (M+H+).
ССЫЛОЧНЫЙ ПРИМЕР 29
5-Бром-1-бутил-3-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)-1H-пирроло[2,3-c]пиридин
Стадия a. 5-Бром-3-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)-1H-пирроло[2,3-c]пиридин
При перемешивании к раствору 5-бром-1H-пирроло[2,3-c]пиридина (1,0 г, 5,07 ммоля) в MeOH (20 мл) добавляли порошкообразный KOH (1,13 г, 20,28 ммоля). Полученную смесь перемешивали при КТ в течение 10 мин. Затем добавляли 1-метилпиперидин-4-он (1,14 г, 10,14 ммоля) и ее кипятили с обратным холодильником в течение 16 ч. Реакцию останавливали водой и экстрагировали с помощью 10% MeOH в DMC. Отделенный органический слой сушили над безводным Na2SO4, фильтровали и концентрировали при пониженном давлении и получали 800 мг (54%) искомого соединения.
LC-MS (методика 6): Rt=0,95 мин; m/z=290,20 (M-H+).
Стадия b. 5-Бром-1-бутил-3-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)-1H-пирроло[2,3-c]пиридин
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия a (800 мг, 2,74 ммоля) в DMF (10 мл) при 0°C добавляли NaH (60%) (325 мг, 8,22 ммоля) и его перемешивали в течение 15 мин. Затем добавляли йодбутан (1,0 г, 5,48 ммоля) и реакционную смесь перемешивали при КТ в течение 16 ч. Реакцию останавливали водой и экстрагировали с помощью EtOAc и органический слой сушили над безводным Na2SO4 и его концентрировали при пониженном давлении. Неочищенное соединение очищали с помощью колоночной флэш-хроматографии с использованием 25-30% EtOAc в петролейном эфире в качестве элюента и получали искомое соединение (400 мг, 42%).
LC-MS (методика 6): Rt=1,66 мин; m/z=348,29 (M+H+).
ССЫЛОЧНЫЙ ПРИМЕР 30
N,N-Диметил-3-(6-(триметилстаннил)пиридин-3-илокси)пропан-1-амин)
При перемешивании к раствору соединения ссылочного примера 6b (500 мг, 1,92 ммоля) в толуоле (20 мл) при КТ добавляли гексаметилдиолово (695 мг, 2,12 ммоля) и Pd(PPh3)4 (223 мг, 0,192 ммоля). Полученный раствор дегазировали азотом в течение 10 мин и нагревали при 110°C в течение 16 ч. Реакционную смесь выпаривали при пониженном давлении и неочищенное соединение очищали с помощью колоночной флэш-хроматографии на нейтральном оксиде алюминия с использованием 10% EtOAc в петролейном эфире и получали искомое соединение (390 мг, 58%).
LC-MS (методика 21): Rt=2,02 мин; m/z=345,0 (M+H+)
ССЫЛОЧНЫЙ ПРИМЕР 31
3-(2-Бромпиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол
Стадия a. 2-Бром-N-гидроксиизоникотинимидамид
При перемешивании к раствору 2-бромизоникотинонитрила (3 г, 16,4 ммоля) в EtOH/MeOH (1:1) (50 мл) добавляли 50% раствор NH2OH.HCl (2,2 г, 32,8 ммоля) в NaHCO3 (2,7 г, 32,8 ммоля). Полученный раствор нагревали при 60°C в течение 3 ч. Реакционную смесь выпаривали при пониженном давлении и выливали в воду со льдом (20 мл) и экстрагировали с помощью EtOAc (2×150 мл). Органический слой сушили над безводным Na2SO4 и фильтровали. и получали неочищенное соединение, которое использовали на следующей стадии без какой-либо дополнительной очистки.
LC-MS (методика 21): Rt=0,97 мин; m/z=214,90 (M+H+)
Стадия b. 3-(2-Бромпиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия a (3,4 мг, 15,7 ммоля) в THF (20 мл) добавляли трифторуксусный ангидрид (4,94 г, 23,5 ммоля). Полученный раствор нагревали при 50°C в течение 2 ч. Реакционную смесь выпаривали в вакууме, выливали в воду со льдом (30 мл) и экстрагировали с помощью EtOAc (2×50 мл). Органический слой сушили над безводным Na2SO4, фильтровали и фильтрованный раствор концентрировали и получали неочищенный остаток, который очищали с помощью колоночной флэш-хроматографии на диоксиде кремния с использованием 25% EtOAc в петролейном эфире и получали искомое соединение (3,2 г, 69%) в виде почти белого твердого вещества.
LC-MS (методика 15): Rt =4,15 мин; m/z=293,51 (M+H+).
ССЫЛОЧНЫЙ ПРИМЕР 32
3-Бром-1-(2-метоксиэтил)-1H-пиразоло[4,3-b]пиридин
При перемешивании к раствору 3-бром-1H-пиразоло[4,3-b]пиридин (500 мг, 2,52 ммоля) в DMF при 0°C добавляли NaH (60%) (201 мг, 5,04 ммоля) и его перемешивали в течение 15 мин. Затем добавляли 1-бром-2-метоксиэтан (420 мг, 3,02 ммоля) и реакционную смесь перемешивали при КТ в течение 2 ч. Реакцию останавливали водой и экстрагировали с помощью EtOAc. Органический слой сушили над безводным Na2SO4 и его концентрировали при пониженном давлении. Неочищенное соединение очищали с помощью колоночной флэш-хроматографии на 230-400 диоксиде кремния с использованием 45% EtOAc в петролейном эфире в качестве элюента и получали искомое соединение (430 мг, 66%) в виде смолообразного твердого вещества.
LC-MS (методика 24): Rt=2,63 мин; m/z=258,01 (M+2H+).
По методике, аналогичной описанной в ссылочном примере 32, но с использованием в каждом случае соответствующих исходных веществ, получали следующие соединения:
ССЫЛОЧНЫЙ ПРИМЕР 33
3-Бром-1-(2-(1-метил-1H-имидазол-2-ил)этил)-1H-пиразоло[4,3-b]пиридин
При перемешивании к раствору 3-бром-1H-пиразоло[4,3-b]пиридина (800 мг, 4,03 ммоля) в ацетонитриле (10 мл) при КТ добавляли Cs2CO3 (3940 мг, 12,09 ммоля), 2-(2-хлорэтил)-1-метил-1H-имидазол (640 мг, 4,43 ммоля) и NaI (303 мг, 2,02 ммоля). Реакционную смесь нагревали при 90°C в течение ночи. Неочищенную реакционную смесь фильтровали через слой целита, промывали с помощью EtOAc (50 мл), и раствор выпаривали досуха. Неочищенное соединение очищали дважды с помощью колоночной хроматографии с обращенной фазой с использованием 10 мМ ацетат аммония/ацетонитрил и получали и получали искомое соединение (430 мг, 35%) в виде смолообразного твердого вещества.
LC-MS (методика 26): Rt=1,99 мин; m/z=306,0 (M+H+).
ССЫЛОЧНЫЙ ПРИМЕР 34
1-Метил-3-(триметилстаннил)-1H-пиразоло[4,3-b]пиридин
По методике, аналогичной описанной в ссылочном примере 30, но с использованием соединения ссылочного примера 32b вместо соединения ссылочного примера 6b, получали искомое соединение.
ПРИМЕР 1
3-(2-(1-Бутил-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол

Стадия a. 2-(1-Бутил-1H-пирроло[2,3-c]пиридин-5-ил)изоникотинонитрил
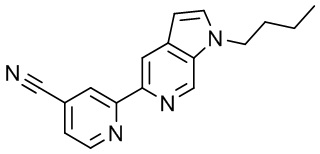
При перемешивании к раствору соединения ссылочного примера 2 (800 мг, 3,16 ммоля) в 1,4-диоксане (5 мл) добавляли соединение ссылочного примера 1 (928 мг, 3,47 ммоля), CsF (948 мг, 6,32 ммоля) и CuI (119 мг, 0,63 ммоля). Полученный раствор дегазировали азотом в течение 5 мин и затем добавляли Pd (PPh3)4 (358 мг, 0,31 экв.). Реакционную смесь повторно дегазировали в течение еще 5 мин и затем нагревали при 110°C в течение 16 ч. Неочищенную реакционную смесь фильтровали через слой целита, промывали с помощью EtOAc (50 мл) и фильтрованный раствор выпаривали досуха. Неочищенное соединение очищали с помощью колоночной флэш-хроматографии на (230-400) силикагеле с использованием 20% EtOAc/петролейный эфир и получали искомое соединение (500 мг, 57%) в виде светло-желтого твердого вещества.
LC-MS (методика 6): Rt =1,66 мин; m/z=277,54 (M+H+).
Стадия b. 2-(1-Бутил-1H-пирроло[2,3-c]пиридин-5-ил)-N-гидроксиизоникотинимидамид
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия a (300 мг, 1,08 ммоля) в EtOH/MeOH (1:1) (10 мл), 50% водный раствор NH2OH.HCl (300 мг, 2,16 ммоля) добавляли. Полученный раствор нагревали при 60°C в течение 5 ч. Реакционную смесь выпаривали в вакууме, выливали в воду со льдом (20 мл) и экстрагировали с помощью EtOAc (2×80 мл). Органический слой сушили над безводным Na2SO4, фильтровали и фильтрованный раствор концентрировали и получали искомое соединение (320 мг, 95%), в виде смолообразного твердого вещества.
LC-MS (методика 6): Rt =1,47 мин; m/z=310,27 (M+H+).
Стадия c. 3-(2-(1-Бутил-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия b (300 мг, 0,97 ммоля) в THF (10 мл), трифторуксусный ангидрид (243 мг, 1,16 ммоля) добавляли. Полученный раствор нагревали при 50°C в течение 4 ч. Реакционную смесь выпаривали в вакууме, выливали в воду со льдом (20 мл) и экстрагировали с помощью EtOAc (2×80 мл). Органический слой сушили над безводным Na2SO4, фильтровали и фильтрованный раствор концентрировали и получали неочищенный остаток, который очищали с помощью препаративной HPLC и получали искомое соединение (70 мг, 18,6%) в виде почти белого твердого вещества.
LC-MS (методика 6): Rt =2,00 мин; m/z=388,28 (M+H+).
Условия проведения препаративной HPLC: колонка/размеры : PRONTOSIL C18 (20×250 мм), 10,0мкм, подвижная: фаза : 0,1% FA в воде : Ацетонитрил (A:B); градиентный режим (время/%B) : 0/20, 1/20,10/80,10,1/98,14/98,14,1/20,17/20., Скорость потока : 20 мл/мин.
По методике, аналогичной описанной в примере 1, но с использованием в каждом случае соответствующих исходных веществ, получали следующие соединения:
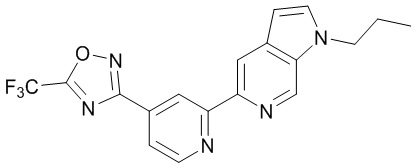





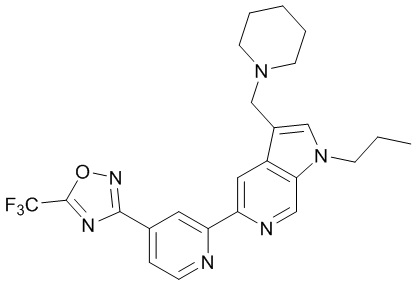


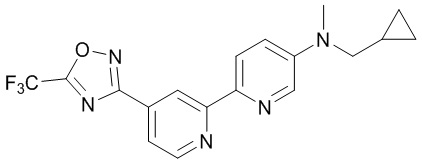
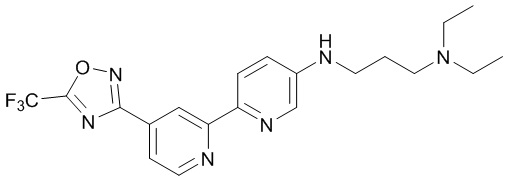
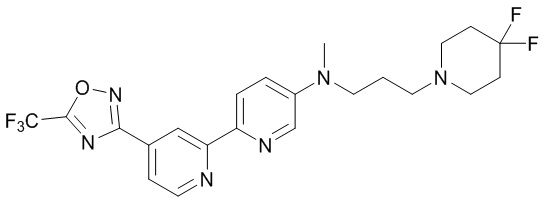

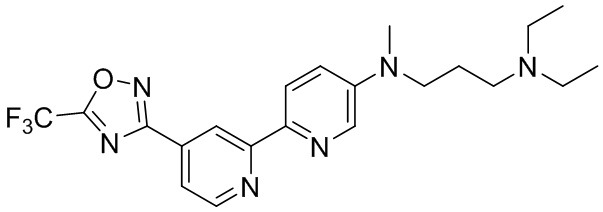
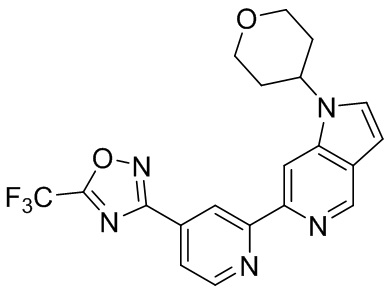


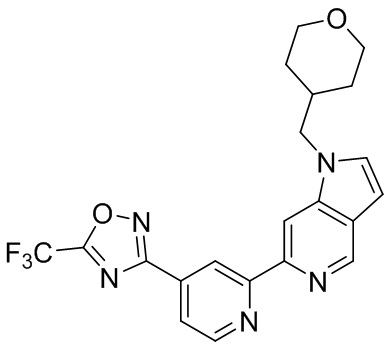

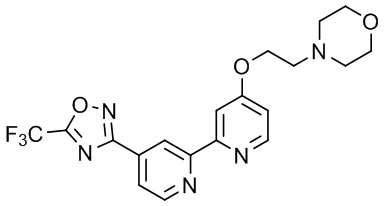
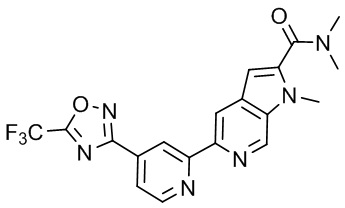




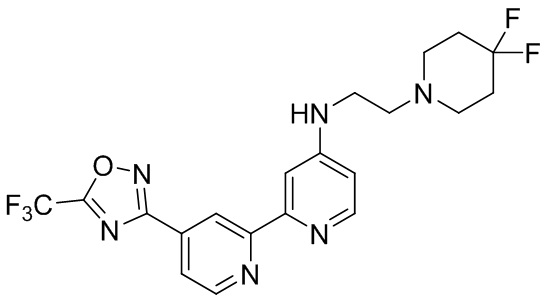


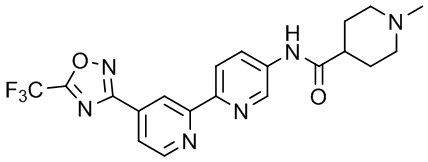

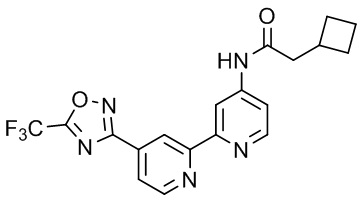
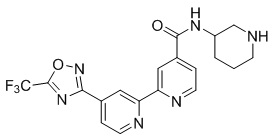
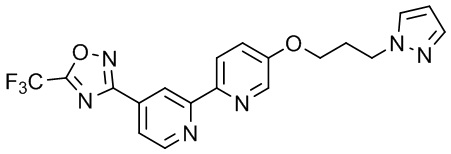
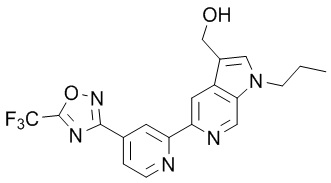

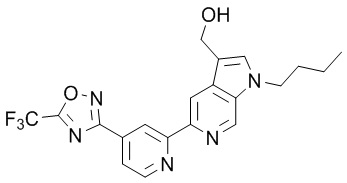


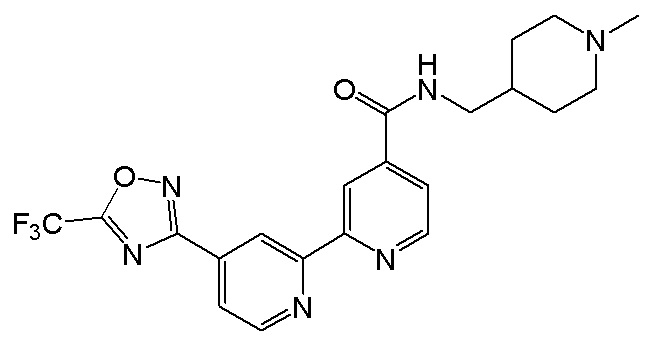

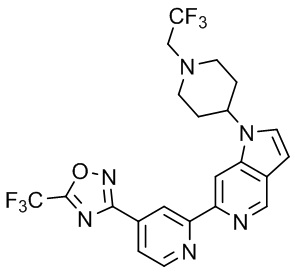


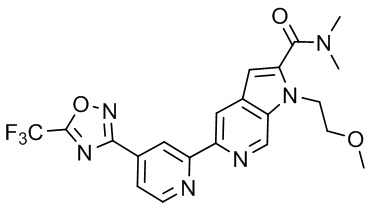

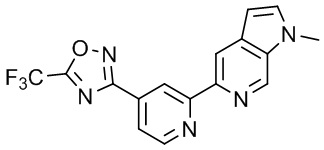
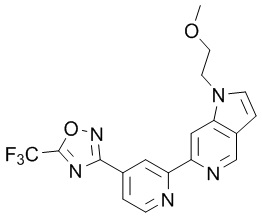
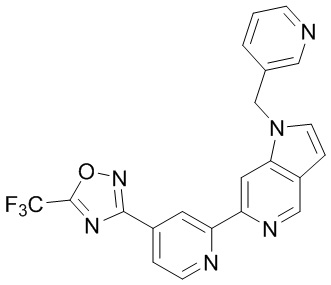

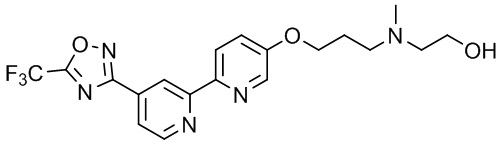


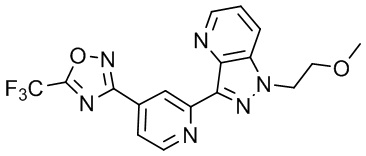



(*) TFA добавляли для завершения удаления защитной группы Boc
(**) получали с использованием 0,7 экв. TFA и 0,3 экв. муравьиной кислоты
ПРИМЕР 2
N-Метил-3-((4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)окси)пропан-1-амин (в виде формиата)
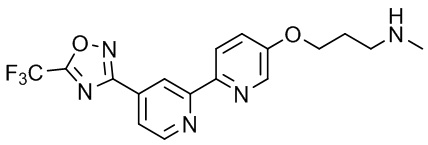
Стадия a. 5'-(3-(Метиламино)пропокси)-[2,2'-бипиридин]-4-карбонитрил
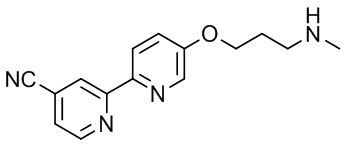
При перемешивании к раствору соединения ссылочного примера 13 (250 мг, 1,01 ммоля) в 1,4-диоксане (20 мл) добавляли соединение ссылочного примера 1 (16) (299 мг, 1,12 ммоля) CsF (308 мг, 2,03 ммоля) и CuI (38 мг, 0,20 ммоля) добавляли. Полученный раствор дегазировали азотом в течение 15 мин, затем добавляли Pd(PPh3)4 (117 мг, 0,1 ммоля) и нагревали при 110°C в течение 16 ч. Реакционную смесь фильтровали через слой целита, промывали с помощью EtOAc (50 мл). Фильтрованный раствор выпаривали досуха. Неочищенный остаток очищали с помощью колоночной хроматографии на 230-400 диоксиде кремния с использованием 90% EtOAc/петролейный эфир в качестве элюента и получали искомое соединение (180 мг, 65%) в виде смолообразного твердого вещества.
LC-MS (методика 14): Rt =1,45 мин; m/z=269,1 (M+H+).
Стадия b. Трет-бутил-(3-((4'-циано-[2,2'-бипиридин]-5-ил)окси)пропил)(метил)карбамат

При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия a (180 мг, 0,67 ммоля) в ацетонитриле (10 мл) добавляли Boc ангидрид (219 мг, 1,0 ммоля) и TEA (0,27 мл, 2,01 ммоля). Полученную смесь перемешивали КТ в течение 16 ч. Реакционную смесь выпаривали в вакууме и получали неочищенное соединение, которое использовали на следующей стадии без какой-либо дополнительной очистки (200 мг, 80%)
LC-MS (методика 15): Rt =3,75 мин; m/z=369,08 (M+H+).
Стадия c. Трет-бутил-(3-((4'-(N-гидроксикарбамимидоил)-[2,2'-бипиридин]-5-ил)окси)пропил)(метил)карбамат
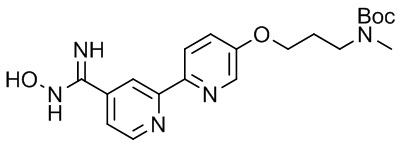
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия b (200 мг, 0,54 ммоля) в EtOH/MeOH (4:1) добавляли 50% водный раствор NH2OH.HCl (75 мг, 1,08 ммоля) и NaHCO3 (91 мг, 1,08 ммоля). Полученную смесь перемешивали при 50°C в течение 3 ч. Реакционную смесь концентрировали и получали неочищенный остаток, который разбавляли с помощью EtOH и фильтровали через воронку Бюхнера. Фильтрованный раствор концентрировали досуха и получали искомое соединение (170 мг (78%) в виде в виде светло-коричневого смолообразного твердого вещества, которое использовали на следующей стадии без очистки.
LC-MS (методика 15): Rt =2,89 мин; m/z=402,21 (M+H+).
Стадия d. N-Метил-3-((4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)окси)пропан-1-амин формиат
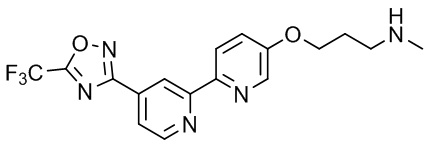
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия c (150 мг, 0,37 ммоля) в THF добавляли трифторуксусный ангидрид (0,1 мл, 0,56 ммоля). Полученную смесь нагревали при 70°C и добавляли TFA (0,5 мл, 0,64 ммоля). Ее перемешивали при 50°C в течение 2 ч. Растворители выпаривали в вакууме и неочищенный остаток выливали в воду со льдом и экстрагировали с помощью EtOAc. Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенное соединение очищали с помощью препаративной HPLC и получали искомое соединение (65 мг, 45%) в виде розового твердого вещества.
LC-MS (методика 15): Rt=2,79 мин; m/z=380,15 (M+H+).
Условия проведения препаративной HPLC: колонка/размеры: SYNERGY POLAR C18(21,2×250 мм), 5 мкм подвижная: фаза : 0,1% FA : Ацетонитрил (A:B) градиентный режим (время/%B) : 0/20, 1/20,7/50,10/50,10,1/98,13/98,13,120,16/20. Скорость потока: 20 мл/мин.
ПРИМЕР 3
1-(1-Бутил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-3-ил)-N,N-диметилметанамин

При перемешивании к раствору соединения примера 1 (400 мг,1 ммоля) в н-бутаноле добавляли HCHO (300 мг, 10 ммоль) и диметиламинхлорид (815 мг 10 ммоля). Полученную смесь перемешивали при 120°C в течение 16 ч. Реакционную смесь концентрировали и получали неочищенное соединение, которое очищали с помощью препаративной HPLC и получали искомое соединение (38 мг, 8,6%) в виде липкого розового твердого вещества.
LC-MS (методика 15): Rt=3,37 мин; m/z=445,39 (M+H+).
Условия проведения препаративной HPLC: колонка/размеры: XSELECT C18 (19×250×5 мкм) подвижная: фаза A: 0,1% FA в воде; подвижная: фаза B : Ацетонитрил (org) градиентный режим (время/%B) : 0/10,1/10,5/30,9/30,9,1/100,11/100,11,1/10,13/10. Скорость потока : 18 мл/мин Растворимость : Метанол+ THF.
ПРИМЕР 4
3-(2-(1H-Пиразоло[4,3-b]пиридин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол

Стадия a. 2-(1-(Тетрагидро-2H-пиран-2-ил)-1H-пиразоло[4,3-b]пиридин-3-ил)изоникотинонитрил

При перемешивании к раствору соединения ссылочного примера 26 (900 мг, 3,19 ммоля) в толуоле (25 мл) добавляли соединение ссылочного примера 1 (1100 мг, 4,15 ммоля) и дегазировали азотом в течение 10 мин. Затем добавляли PdCl2(PPh3)2 (112 мг, 0,16 ммоля) и полученный раствор дегазировали в течение еще 5 мин и нагревали при 100°C в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении и получали неочищенный остаток, который очищали с помощью колоночной хроматографии и получали искомое соединение (800 мг, 82,2%).
LC-MS (методика 1): Rt=2,44 мин; m/z=306,24 (M+H+).
Стадия b. 2-(1H-Пиразоло[4,3-b]пиридин-3-ил)изоникотинонитрил

При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия a (800 мг, 2,62 ммоля) в DCM (15 мл) добавляли TFA (10 мл) и его перемешивали при КТ в течение 48 ч. Реакционную смесь концентрировали при пониженном давлении и получали неочищенный остаток, который очищали с помощью колоночной хроматографии и получали искомое соединение (400 мг, 69,0%).
LC-MS (методика 1): Rt=1,85 мин; m/z=222,13 (M+H+).
Стадия c. N-гидрокси-2-(1H-пиразоло[4,3-b]пиридин-3-ил)изоникотинимидамид

По методике, аналогичной описанной в примере 1, раздел b, но с использованием соединения, полученного в предыдущем разделе, стадия b, вместо 2-(1-бутил-1H-пирроло[2,3-c]пиридин-5-ил)изоникотинонитрила, получали искомое соединение.
LC-MS (методика 14): Rt=1,25 мин; m/z=255,1 (M+H+).
Стадия d. 3-(2-(1H-пиразоло[4,3-b]пиридин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол
По методике, аналогичной описанной в примере 1, раздел c, но с использованием соединения, полученного в предыдущем разделе, стадия c вместо 2-(1-бутил-1H-пирроло[2,3-c]пиридин-5-ил)-N-гидроксиизоникотинимидамида, получали искомое соединение.
LC-MS (методика 6): Rt=1,98 мин; m/z=333,20 (M+H+).
ПРИМЕР 5
N-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-ил)пиперидин-3-карбоксамид (в виде гемиформиата)
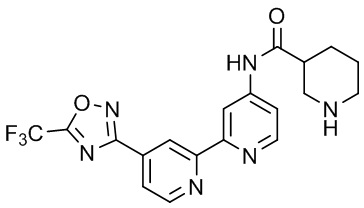
Стадия a. трет-бутил-3-((4'-циано-[2,2'-бипиридин]-4-ил)карбамоил)пиперидин-1-карбоксилат
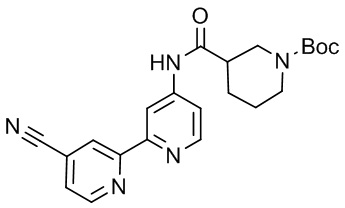
При перемешивании к раствору соединения ссылочного примера 16c (3 г, 7,81 ммоля) в толуоле (30 мл) добавляли 2-бромизоникотинонитрил (2,14 г, 11,71 ммоля) и гексаметилдиолово (2,8 мл, 13,67 ммоля). Полученный раствор дегазировали аргоном в течение 10 мин, затем добавляли Pd(PPh3)4 (902 мг, 0,781 ммоля). Полученную смесь дегазировали в течение 5 мин и нагревали при 110°C в течение 16 ч. Реакционную смесь разбавляли водой и экстрагировали с помощью EtOAc (2×20 мл). Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении и получали неочищенный остаток, который очищали с помощью колоночной хроматографии с использованием 10% EtOAc в петролейном эфире и получали искомое соединение (800 мг, 25,1%)
LC-MS (методика 1): Rt=2,62 мин; m/z=408,31 (M+H+).
Стадия b. N-(4'-циано-[2,2'-бипиридин]-4-ил)пиперидин-3-карбоксамидгидрохлорид
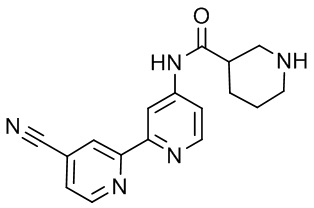
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия a (200 мг, 0,486 ммоля) в DCM (2 мл) при 0°C добавляли 2M раствор HCl в диэтиловом эфире (3 мл). Реакционную смесь перемешивали при КТ в течение 2 ч. Полученную смесь концентрировали и неочищенный остаток растирали с н-пентаном и диэтиловым эфиром. Ее фильтровали при пониженном давлении и получали 800 мг (54%) искомого соединения
LC-MS (методика 1): Rt=1,54 мин; m/z=308,29 (M+H+).
Стадия c. N-(4'-(N-гидроксикарбамимидоил)-[2,2'-бипиридин]-4-ил)пиперидин-3-карбоксамидгидрохлорид
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия b (50 мг, 0,16 ммоля) в EtOH (6 мл) добавляли гидроксиламингидрохлорид (22 мг, 0,32 ммоля) и NaHCO3 (27 мг, 0,32 ммоля) в воде (2 мл). Реакционную смесь перемешивали при 70°C в течение 6 ч. Полученный раствор концентрировали досуха и неочищенный остаток разбавляли с помощью EtOH и фильтровали через воронку Бюхнера для удаления NaHCO3. Фильтрованный раствор концентрировали и получали искомое соединение (45 мг, 82%), которое использовали на следующей стадии без дополнительной очистки.
LC-MS (методика 14): Rt=1,26 мин; m/z=341,1 (M+H+).
Стадия d. 1-(2,2,2-трифторацетил)-N-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-ил)пиперидин-3-карбоксамид

При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия c (40 мг, 0,11 ммоля) в THF (5 мл) добавляли трифторуксусный ангидрид (0,1 мл). Полученную смесь перемешивали при 70°C в течение 3 ч. Реакционную смесь концентрировали при пониженном давлении и получали неочищенный остаток, который очищали с помощью препаративной HPLC и получали искомое соединение (16 мг, 28%) в виде почти белого твердого вещества.
LC-MS (методика 14): Rt=2,82 мин; m/z=515,1 (M+H+).
Условия проведения препаративной HPLC: колонка/размеры Prontosil C18 (20×250 мм), 10 мкм подвижная: фаза : 0,1% Ацетат аммония в воде : Ацетонитрил (A:B) градиентный режим (время/%B) : 0/20,1/20, 5/70,15/700,15,1/100 Скорость потока : 20 мл/мин, растворимость : ACN+THF+ВОДА
Стадия e. N-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-ил)пиперидин-3-карбоксамидгемиформиат

При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия c (16 мг, 0,03 ммоля) в MeOH (5 мл) добавляли K2CO3 (4,7 мг, 0,034 ммоля). Полученную смесь перемешивали при КТ в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении и получали неочищенный остаток, который очищали с помощью препаративной HPLC и получали искомое соединение (6 мг, 46%) в виде почти белого твердого вещества.
LC-MS (методика 6): Rt=1,84 мин; m/z=419,34 (M+H+).
Условия проведения препаративной HPLC: колонка/размеры : SUN FIRE C18 (19×150 мм, 5 мкм) подвижная: фаза A : 0,1% FA в воде; подвижная: фаза B : Ацетонитрил (org) градиентный режим (время/%B) : 0/10,1/10,5/25,8/25,8,1/98,10/98,10,1/10,12/10. Скорость потока : 18 мл/мин Растворимость:Ацетонитрил+ THF
ПРИМЕР 6
3-(2-(1H-Пиразоло[3,4-b]пиридин-1-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол
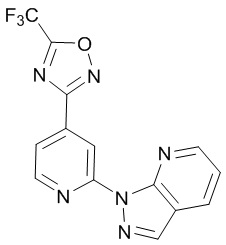
Стадия a. 2-(1H-пиразоло[3,4-b]пиридин-1-ил)изоникотинонитрил

При перемешивании к раствору 2-бромизоникотинонитрила (500 мг, 2,7 ммоля) в толуоле (25 мл) добавляли 1H-пиразоло[3,4-b]пиридин (0,6432 г, 5,4 ммоля) и K2CO3 (1,86 г, 13,5 ммоля), затем CuI (0,256 г, 1,35 ммоля). Полученный раствор дегазировали азотом в течение 15 мин и добавляли транс-N,N-диметилциклогексан-1,2-диамин (0,192 г, 1,35 ммоля,). Полученную реакционную смесь нагревали при 110°C в течение 16 ч. Реакционную смесь выпаривали в вакууме и получали неочищенный остаток, который очищали с помощью препаративной HPLC и получали искомое соединение (300 мг, 29,3%)
LC-MS (методика 6): Rt =1,626 мин; m/z=222,01 (M+H+).
Стадия b. N-гидрокси-2-(1H-пиразоло[3,4-b]пиридин-1-ил)изоникотинимидамид
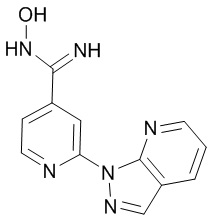
При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия a (300 мг, 1,0 ммоля) в EtOH (5 мл) добавляли 50% водный раствор NH2OH.HCl (179 мг, 2,1 ммоля) и Na2CO3 (296 мг, 2,8 ммоля) в H2O (5 мл). Полученный раствор перемешивали при КТ в течение 18 ч. Реакционную смесь выпаривали в вакууме и получали неочищенное соединение, которое использовали на следующей стадии без какой-либо дополнительной очистки.
LC-MS (методика 6): Rt =1,16 мин; m/z=255,12 (M+H+).
Стадия c. 3-(2-(1H-Пиразоло[3,4-b]пиридин-1-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол

При перемешивании к раствору соединения, полученного в предыдущем разделе, стадия b (300 мг, 1,2 ммоля) в THF (10 мл) добавляли трифторуксусный ангидрид (252 мг, 1,2 ммоля). Полученный раствор нагревали при 70°C в течение 3 ч. Реакционную смесь выпаривали в вакууме и получали неочищенный остаток, который очищали с помощью препаративной HPLC и получали искомое соединение (115 мг, 29,3%) в виде почти белого твердого вещества.
LC-MS (методика 20): Rt =4,79 мин; m/z=333,11 (M+H+).
ПРИМЕР 7
N,N-Диметил-3-((4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)окси)пропан-1-амин
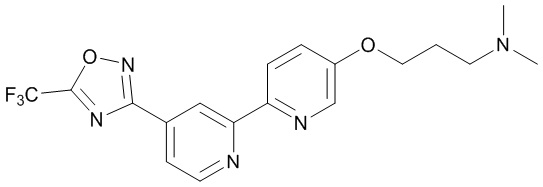
При перемешивании к раствору соединения ссылочного примера 31 (300 мг, 1,0 ммоля) в толуоле (3 мл) добавляли соединение ссылочного примера 30 (928 мг, 3,47 ммоля). Полученный раствор дегазировали азотом в течение 15 мин и затем Pd (PPh3)4 (115 мг, 0,1 ммоля) добавляли. Реакционную смесь повторно дегазировали в течение еще 5 мин и затем нагревали при 110°C в течение 16 ч. Неочищенную реакционную смесь фильтровали через слой целита, промывали с помощью EtOAc (50 мл) и фильтрованный раствор выпаривали досуха. Неочищенное соединение очищали с помощью препаративной HPLC и получали искомое соединение (65 мг, 16%) в виде светло-розового твердого вещества.
LC-MS (методика 16): Rt =4,49 мин; m/z=394,29 (M+H+).
По методике, аналогичной описанной в примере 7, но с использованием соответствующего исходного вещества, получали следующее соединение:
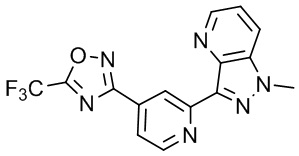
ПРИМЕР 8
Исследование ингибирования фермента HDAC6 и HDAC2
Методика:
Флуориметрический анализ использовали для исследования активности соединений, предлагаемых в настоящем изобретении, по отношению к HDAC6 и HDAC2. Эталонный рекомбинантный белок и количества, использованные в каждом анализе, приведены в таблице ниже.
Соединения растворяли в DMSO. Серийные разведения готовили в DMSO и затем разводили 1:10 в буфере для анализа HDAC (забуференный с помощью Tris раствор, BPS Bioscience Inc. #50031). 5 мкл Разведения соединения добавляли к 50 мкл реакционной смеси, так что конечная концентрация DMSO равнялась 1%.
Соединения предварительно инкубировали (эксперимент проводили дважды) при КТ в течение 3 ч в смеси, содержащей буфер для анализа HDAC, 5 мкг BSA и ркомбинантный фермент HDAC (см. таблицу выше). Ферментативные реакции инициировали путем добавления флуорогенного ацетилированного пептидного субстрата на основе гистонного белка (BPS Bioscience Inc. #50037) до конечной концентрации, равной 10 мкМ. Ферментативная реакция протекала в течение 30 мин при 37°C. Затем добавляли 50 мкл 2× проявителя HDAC, который обладал пептидазной активностью и содержал трихостатин A (BPS Bioscience Inc. #50030), и планшет инкубировали при КТ в течение еще 15 мин. Интенсивность флуоресценции измеряли при возбуждении с длиной волны 360 нм и испускании при длине волны 460 нм с использованием считывающего устройства для микропланшетов Tecan Infinite M1000.
Трихостатин A (TSA, Selleckchem #S1045) использовали в качестве эталонного ингибитора.
Данные по интенсивности флуоресценции анализировали с использованием компьютерного программного обеспечения Graphpad Prism (GraphPad Software, San Diego, CA). Равную 100% активность определяли, как интенсивность флуоресценции (Ft) при отсутствии соединения. Равную 0% активность определяли, как интенсивность флуоресценции (Fb) при отсутствии фермента. Выраженную в процентах активность в присутствии каждого соединения рассчитывали по следующему уравнению: %активность = (F-Fb)/(Ft-Fb), где F= интенсивность флуоресценции в присутствии соединения.
Результаты:
Результаты, полученные в указанных выше исследованиях, для соединений, предлагаемых в настоящем изобретении, приведены ниже в таблице:
NT: Не исследовали.
ПРИМЕР 9
Исследование in vitro на основе клеток
Методика:
Для определения активности HDAC6 по отношению к клеткам после обработки ингибиторами HDAC6, степени ацетилирование альфа-тубулина (специфического субстрата HDAC6) измеряли с помощью вестерн-блоттинга. Для этого клетки MOLP8 высевали в 6-луночные планшеты при плотности клеток, равной 500000 клеток/лунка, и обрабатывали выбранными соединениями при 5 и 1 мкМ в течение 18 ч при 37°C и 5% CO2 во влажном инкубаторе для клеточных культур. Последовательно собирали пеллеты клеток и экстракты цельного белка, полученные с использованием буфера RIPA (SIGMA) с добавлением 1× ингибиторов протеазы (complete mini, Roche). Концентрацию белка определяли с помощью реагента Брэдфорда (Bio-Rad) в соответствии с инструкциями изготовителя и 7 мкг общего белка вносили в предварительно отлитые гели 10% NuPAGE Novex (Life Technologies). Гели вводили в реакцию в буфере MOPS-SDS (Life Technologies) и белки переносили с использованием iBlot 2 Dry Blotting System (Life Technologies). Затем блоты промывали в дистиллированной воде и окрашивали раствором Ponceau S (SIGMA). Затем блоты промывали в дистиллированной воде для удаления избытка Ponceau и сканировали профессиональным сканером Epson Perfection V600 Photo. Затем блоты обесцвечивали и блокировали в 5% молоко/PBS-Tween 0,1% в течение 1 ч при комнатной температуре, затем инкубировали с первичными антителами к анти-ацетилальфа-тубулину (SIGMA cat. #T7451, разведение 1:10000) и анти-бета-актину (SIGMA, cat. #A5316, разведение 1:2000) в 5% молоко/PBS-T 0,1% в течение ночи при 4°C на платформе для встряхивания. После инкубации блоты промывали 3 раза по 5 мин каждая в PBS-Tween 0,5% и инкубировали с антимышиными HRP-сопряженными вторичными антителами (Jackson Immuno Research, cat.# 115-035-068) при разведении 1:8000 в 5% молоко/PBS-Tween 0,1%, 1 ч при комнатной температуре на платформе для встряхивания. После 3 промывок по 5 мин каждая с помощью PBS-Tween 0,5% и 1 промывки в PBS 1×, блоты проявляли с помощью ECL Plus (GE Healthcare) и хемилюминесцентную реакцию визуализировали с помощью системы визуализации G:Box Chemi XRQ (Syngene). Изображения WB и Ponceau анализировали с помощью программного обеспечения ImageJ, интенсивности полос WB нормированы на содержание общего белка или бета-актина определяли относительно ACY-1215 1 мкМ (эквивалентно 100%). ACY-1215 является ингибитором HDAC6 и также известен, как риколиностат, и обладает химическим названием 2-(дифениламино)-N-[7-(гидроксиамино)-7-оксогептил]-5-пиримидинкарбоксамид. Интенсивности полос в процентах классифицировали следующим образом:
Результаты:
Результаты, полученные в указанных выше исследованиях, для соединений, предлагаемых в настоящем изобретении, приведены ниже в таблице:
Данные, приведенные в примерах 8 и 9, показывают, что соединения формулы (I) обладают высокой ингибирующей активностью по отношению к HDAC6, включая активность в клетках. Кроме того, соединения, предлагаемые в настоящем изобретении, обладают селективностью по отношению к HDAC6, более высокой, чем по отношению к HDAC2, что основано на данных, полученных для типичных соединений, предлагаемых в настоящем изобретении, по отношению к HDAC2, как показано в примере 8.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ, ГЕТЕРОБИЦИКЛО-ЗАМЕЩЕННЫЕ-[1,2,4]ТРИАЗОЛО[1,5c]ХИНАЗОЛИН-5-АМИНА, ОБЛАДАЮЩИЕ СВОЙСТВАМИ А2А АНТАГОНИСТОВ | 2013 |
|
RU2671628C2 |
| НОВЫЕ ЗАМЕЩЕННЫЕ БИАРИЛОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ИНДОЛАМИН-2,3-ДИОКСИГЕНАЗЫ (IDO) | 2018 |
|
RU2786586C2 |
| ОКТАГИДРО КОНДЕНСИРОВАННЫЕ АЗАДЕКАЛИНОВЫЕ МОДУЛЯТОРЫ ГЛЮКОКОРТИКОИДНОГО РЕЦЕПТОРА | 2014 |
|
RU2674983C1 |
| ПИПЕРИДИНИЛПИРАЗОЛОПИРИМИДИНОНЫ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2713937C2 |
| ИНГИБИТОРЫ КАТЕХОЛ-О-МЕТИЛТРАНСФЕРАЗЫ И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ ПСИХОТИЧЕСКИХ РАССТРОЙСТВ | 2011 |
|
RU2586974C2 |
| КОНДЕНСИРОВАННЫЕ С ГЕТЕРОАРИЛКЕТОНАМИ АЗАДЕКАЛИНЫ - МОДУЛЯТОРЫ ГЛЮКОКОРТИКОИДНЫХ РЕЦЕПТОРОВ | 2013 |
|
RU2639867C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ АРИЛСУЛЬФОНЫ, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, КОТОРЫЕ ОТВЕЧАЮТ НА МОДУЛИРОВАНИЕ 5HT РЕЦЕПТОРОВ | 2007 |
|
RU2451012C2 |
| ПРОИЗВОДНЫЕ 1,3,4-ОКСАДИАЗОЛА, КАК ИНГИБИТОР ГИСТОНДЕЗАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2019 |
|
RU2763936C1 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОАРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ СТИМУЛЯТОРОВ CFTR | 2017 |
|
RU2753056C2 |
Группа изобретений относится к области медицины, а именно к терапии, и предназначена для лечения заболеваний, связанных с HDAC6. Представлены производные 1,2,4-оксадиазола формулы (I) и их применение для лечения заболеваний, связанных с HDAC6 и выбранных из рака, аутоиммунного или воспалительного заболевания, отторжения трансплантата, цилиопатии, заболевания нервной системы, психического нарушения или нарушения поведения, инфекционного заболевания, сердечно-сосудистого заболевания, атрофии мышц и кахексии, а именно из периферической невропатии и болезни Шарко-Мари-Тута. В другом воплощении обеспечивается фармацевтическая композиция, содержащая данные соединения. Также обеспечивается in vitro способ ингибирования HDAC6 и in vitro применение производных 1,2,4-оксадиазола формулы (I) в качестве ингибитора HDAC6. Группа изобретений обеспечивает новые производные 1,2,4-оксадиазола формулы (I), обладающие ингибирующей активностью по отношению к гистондезацетилазе 6 (HDAC6). 8 н. и 24 з.п. ф-лы, 1 табл., 9 пр.

1. Соединение формулы (I) или его фармацевтически приемлемая соль
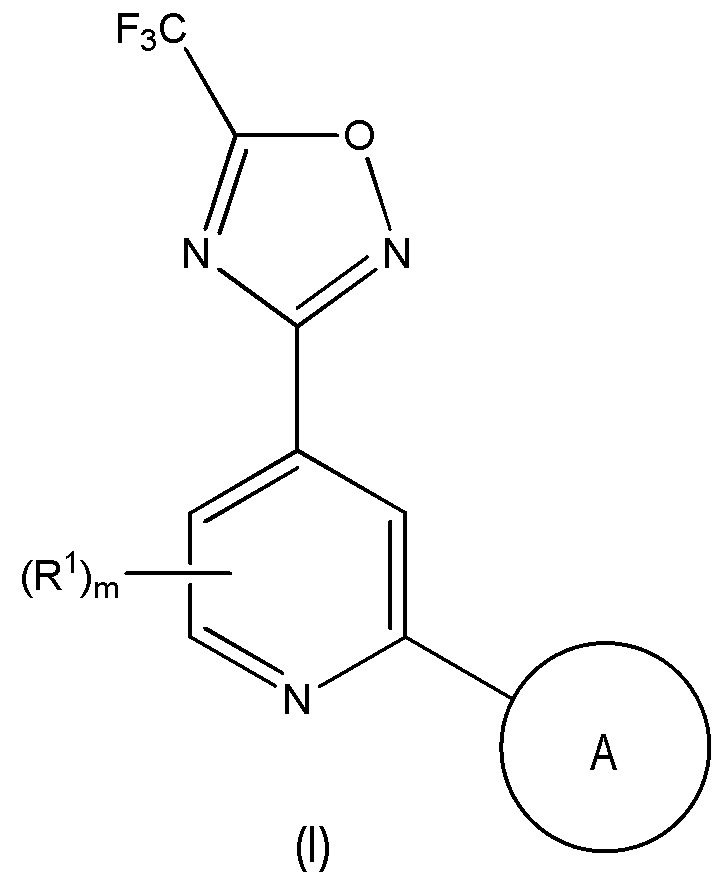
в которой
m равно 0;
A представляет собой:
5- или 6-членное моноциклическое или 9- или 10-членное бициклическое гетероарильное кольцо, которое является полностью ароматическим,
где A содержит один кольцевой атом N в положении рядом с кольцевым атомом, через который кольцо A присоединено к остальной части молекулы, где A необязательно содержит от 1 или 2 дополнительных кольцевых атомов N и
где A необязательно замещен одним или двумя R2 и дополнительно A необязательно замещен одним R3;
каждый R2 независимо выбран из C1-6 алкила, C1-6 галогеналкила и -(C1-6 алкилен)-OR4;
R3 выбран из -L1-R5, -L2-OR6, -L3-NR7R8, -L4-CONR9R10, -L5-NR11COR12, -Y-L6-OR6 и -Y-L7-NR7R8;
L1, L2, L3, L4 и L5, каждый независимо, выбраны из связи и C1-6 алкилена;
L6 и L7, каждый независимо, выбраны из C2-6 алкилена;
каждый Y независимо выбран из -O-, -NR13-, -CONR14- и -NR15CO-;
каждый R4 независимо выбран из водорода, C1-6 алкила и C1-6 галогеналкила;
каждый R5 независимо выбран из моноциклического C3-10 карбоциклила, моноциклического C6-10 арила, 3-7-членного моноциклического гетероциклила, содержащего 1 или 2 гетероатома, независимо выбранных из N, O и S, и 5-6-членного моноциклического гетероарила, содержащего 1-3 гетероатома, независимо выбранных из N, O и S, где карбоциклил, арил, гетероциклил и гетероарил, каждый необязательно, замещены одним, двумя или тремя R16;
R6 и R12, каждый независимо, выбраны из водорода, C1-6 алкила, C1-6 галогеналкила и -L1-R5;
R7 и R8, каждый независимо, выбраны из водорода, C1-6 алкила, C1-6 галогеналкила, -(C1-6 алкилен)-OR4 и -L1-R5;
R9 и R10, каждый независимо, выбраны из водорода, C1-6 алкила, C1-6 галогеналкила, -(C1-6 алкилен)-OR4 и -L1-R5;
R11, R13, R14 и R15, каждый независимо, выбраны из водорода, C1-6 алкила или C1-6 галогеналкила; и
каждый R16 независимо выбран из C1-6 алкила, C1-6 галогеналкила, галогена и C1-6 алкокси.
2. Соединение по п.1, в котором A выбран из циклических групп, приведенных ниже:
 ,
,
где A необязательно замещен одним или двумя R2 и дополнительно A необязательно замещен одним R3.
3. Соединение по п.1, в котором A выбран из циклических групп, приведенных ниже:
 ,
,
где A необязательно замещен одним или двумя R2 и дополнительно A необязательно замещен одним R3.
4. Соединение по п.1, где соединение описывается формулой (IIa) или (IIb), или его фармацевтически приемлемая соль
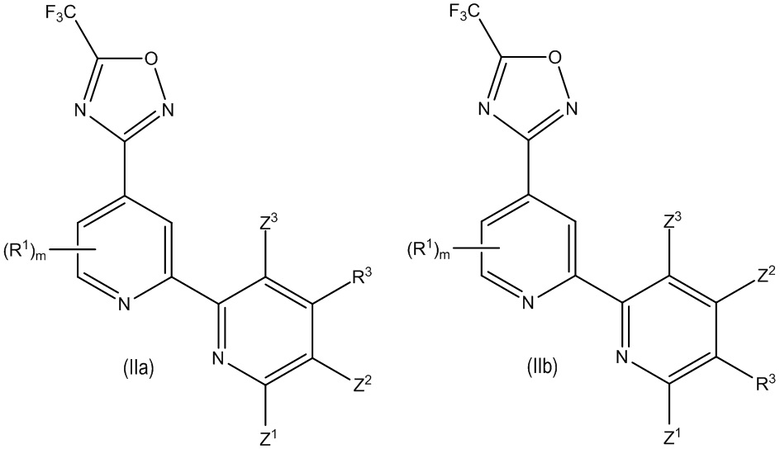
в которой один из Z1, Z2 и Z3 представляет собой H и остальные независимо выбраны из H и R2.
5. Соединение по п.1, где соединение описывается формулой (IIIa) или (IIIb), или его фармацевтически приемлемая соль

в которой один из Z1, Z2 и Z3 представляет собой R3 или H и остальные независимо выбраны из H и R2.
6. Соединение по п.1, где соединение описывается формулой (IIIa), или его фармацевтически приемлемая соль
в которой один из Z1, Z2 и Z3 представляет собой R3 или H и остальные независимо выбраны из H и R2.
7. Соединение по п.5 или 6, где Z2 представляет собой R3, где Z1 и Z3 независимо выбраны из H и R2 и где R3 представляет собой -CONR9R10.
8. Соединение по п.1, где соединение описывается формулой (IVa), или его фармацевтически приемлемая соль
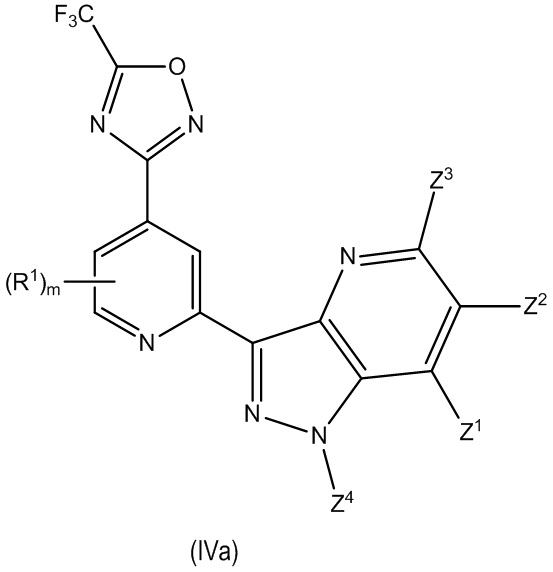
в которой один из Z1, Z2, Z3 и Z4 выбран из R2, R3 и H и остальные независимо выбраны из H и R2 при условии, что только до двух из Z1, Z2, Z3 и Z4 представляют собой R2.
9. Соединение по п.1, где соединение описывается формулой (IVb), или его фармацевтически приемлемая соль
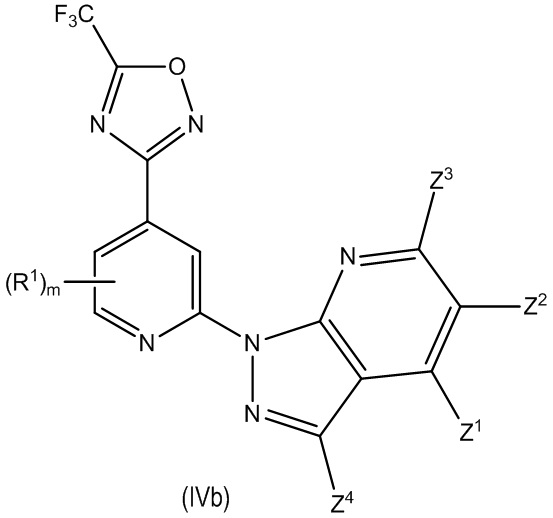
в которой один из Z1, Z2, Z3 и Z4 выбран из R2, R3 и H и остальные независимо выбраны из H и R2 при условии, что только до двух из Z1, Z2, Z3 и Z4 представляют собой R2.
10. Соединение по любому из пп.1-9, в котором R3 выбран из -L1-R5, -L2-OR6, -L3-NR7R8, -CONR9R10, -NR11COR12 и -Y-L7-NR7R8.
11. Соединение по любому из пп.1-10, в котором R3 представляет собой -L1-R5.
12. Соединение по любому из пп.1-10, в котором R3 представляет собой -CONR9R10 или -NR11COR12.
13. Соединение по любому из пп.1-10, в котором R3 представляет собой -Y-L7-NR7R8 и Y выбран из -O- и -NR13-.
14. Соединение по любому из пп.1-10, в котором R3 представляет собой -OR6, где R6 представляет собой -L1-R5, или R3 представляет собой -NR7R8, где один из R7 или R8 представляет собой -L1-R5.
15. Соединение по любому из пп.1-10, в котором R3 представляет собой -L2-OR6 или -L3-NR7R8, где L2 и L3 каждый независимо выбраны из C1-6 алкилена.
16. Соединение по любому из пп.1-15, в котором каждый R2 независимо выбран из C1-4 алкила, C1-4 галогеналкила и -(C1-4 алкилен)-OR4.
17. Соединение по п.1, которым является соединение, выбранное из
3-(2-(1-Бутил-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-Пропил-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
1-Бутил-N,N-диметил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-2-карбоксамида,
N,N-Диэтил-3-((4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)окси)пропан-1-амина,
1-Бутил-N-этил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-2-карбоксамида,
4-(3-((4'-(5-(Трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)окси)пропил)морфолина,
3-(5'-(3-(4,4-Дифторпиперидин-1-ил)пропокси)-[2,2'-бипиридин]-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(3-(Пиперидин-1-илметил)-1-пропил-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
4-((1-Пропил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-3-ил)метил)морфолина,
N-Бутил-3-метокси-N-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)пропанамида,
N-(Циклопропилметил)-N-метил-4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-амина,
N1,N1-Диэтил-N3-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)пропан-1,3-диамина,
N-(3-(4,4-Дифторпиперидин-1-ил)пропил)-N-метил-4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-амина,
N,N-Диэтил-3-(2-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)пиримидин-5-илокси)пропан-1-амина,
N1,N1-Диэтил-N3-метил-N3-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-2,2'-бипиридин-5-ил)пропан-1,3-диамина,
3-(2-(1-(Тетрагидро-2H-пиран-4-ил)-1H-пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
N-Этил-N-фенэтил-3-((4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)окси)пропан-1-амина,
2-Фенил-N-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)ацетамида,
3-(2-(1-((Тетрагидро-2H-пиран-4-ил)метил)-1H-пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(4'-(2-(4,4-Дифторпиперидин-1-ил)этокси)-[2,2'-бипиридин]-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
4-(2-((4'-(5-(Трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-ил)окси)этил)морфолина,
N,N,1-Триметил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-2-карбоксамида,
3-(2-(1-Пропил-3-(1H-пиразол-4-ил)-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
1-Бутил-N,N-диэтил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-2-карбоксамида,
3-(2-(1-(2-Метоксиэтил)-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
2-(4,4-Дифторпиперидин-1-ил)-N-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-ил)ацетамида,
N-(2-(4,4-Дифторпиперидин-1-ил)этил)-4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-амина,
3-(2-(3-(Пиперидин-1-илметил)-1H-пиразоло[3,4-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-(2-(4,4-Дифторпиперидин-1-ил)этил)-1H-пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
1-Метил-N-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)пиперидин-4-карбоксамида,
3-Фенил-N-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-ил)пропанамида,
2-Циклобутил-N-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-ил)ацетамида,
N-(Пиперидин-3-ил)-4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-карбоксамида,
3-(5'-(3-(1H-Пиразол-1-ил)пропокси)-[2,2'-бипиридин]-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
(1-Пропил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-3-ил)метанола,
3-(2-(3-(Метоксиметил)-1-пропил-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
(1-Бутил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-3-ил)метанола,
3-(2-(1H-Пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-(Пиридин-4-илметил)-1H-пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
N-((1-Метилпиперидин-4-ил)метил)-4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-карбоксамида,
N-((1-Метилпиперидин-4-ил)метил)-4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-карбоксамида,
3-(2-(1-(1-(2,2,2-Трифторэтил)пиперидин-4-ил)-1H-пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-Метил-3-(1H-пиразол-4-ил)-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-Бутил-3-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
N-Метил-3-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-2,2'-бипиридин-5-илокси)пропан-1-амина,
1-(1-Бутил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-3-ил)-N,N-диметилметанамина,
3-(2-(1H-Пиразоло[4,3-b]пиридин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
N-(4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-4-ил)пиперидин-3-карбоксамида,
1-(2-Метоксиэтил)-N,N-диметил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-2-карбоксамида,
3-(2-(1-(2-Метоксиэтил)-3-(1H-пиразол-4-ил)-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-Метил-1H-пирроло[2,3-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-(2-Метоксиэтил)-1H-пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-(Пиридин-3-илметил)-1H-пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-(Пиридин-2-илметил)-1H-пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
2-(Метил(3-((4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)окси)пропил)амино)этан-1-ола,
3-(2-(1-(2-Метоксиэтил)-1H-пиразоло[3,4-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(2-(2-Метоксиэтил)-2H-пиразоло[3,4-c]пиридин-5-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1H-Пиразоло[3,4-b]пиридин-1-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
N,N-Диметил-3-((4'-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)-[2,2'-бипиридин]-5-ил)окси)пропан-1-амина,
3-(2-(1-Метил-1H-пиразоло[4,3-b]пиридин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-(2-Метоксиэтил)-1H-пиразоло[4,3-b]пиридин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-Этил-1H-пиразоло[4,3-b]пиридин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
3-(2-(1-(2-(1-Метил-1H-имидазол-2-ил)этил)-1H-пиразоло[4,3-b]пиридин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола и
3-(2-(1-((1-Метил-1H-пиразол-4-ил)метил)-1H-пирроло[3,2-c]пиридин-6-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазола,
или его фармацевтически приемлемая соль.
18. Соединение по п.1, которое представляет собой N,N,1-триметил-5-(4-(5-(трифторметил)-1,2,4-оксадиазол-3-ил)пиридин-2-ил)-1H-пирроло[2,3-c]пиридин-2-карбоксамид, или его фармацевтически приемлемая соль.
19. Соединение по п.1, которое представляет собой 3-(2-(1-(2-метоксиэтил)-1H-пиразоло[4,3-b]пиридин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол, или его фармацевтически приемлемая соль.
20. Соединение по п.1, которое представляет собой 3-(2-(1H-пиразоло[4,3-b]пиридин-3-ил)пиридин-4-ил)-5-(трифторметил)-1,2,4-оксадиазол, или его фармацевтически приемлемая соль.
21. Фармацевтическая композиция, обладающая активностью ингибирования HDAC6, которая содержит терапевтически эффективное количество соединения по любому из пп.1-20 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
22. Соединение по любому из пп.1-20 или его фармацевтически приемлемая соль для применения в качестве лекарственного средства.
23. Соединение по любому из пп.1-20 или его фармацевтически приемлемая соль или фармацевтическая композиция по п.21 для применения в лечении заболевания, связанного с HDAC6.
24. Соединение по любому из пп.1-20 или его фармацевтически приемлемая соль или фармацевтическая композиция по п.21 для применения для лечения заболевания, выбранного из рака, аутоиммунного или воспалительного заболевания, отторжения трансплантата, цилиопатии, заболевания нервной системы, психического нарушения или нарушения поведения, инфекционного заболевания, сердечно-сосудистого заболевания, атрофии мышц и кахексии.
25. Соединение по любому из пп.1-20 или его фармацевтически приемлемая соль или фармацевтическая композиция по п.21 для применения для лечения периферической невропатии.
26. Соединение по любому из пп.1-20 или его фармацевтически приемлемая соль или фармацевтическая композиция по п.21 для применения для лечения болезни Шарко-Мари-Тута.
27. Применение соединения по любому из пп.1-20 или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения заболевания, связанного с HDAC6.
28. Применение соединения по любому из пп.1-20 или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения заболевания, выбранного из рака, аутоиммунного или воспалительного заболевания, отторжения трансплантата, цилиопатии, заболевания нервной системы, психического нарушения или нарушения поведения, инфекционного заболевания, сердечно-сосудистого заболевания, атрофии мышц и кахексии.
29. Применение соединения по любому из пп.1-20 или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения периферической невропатии.
30. Применение соединения по любому из пп.1-20 или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения болезни Шарко-Мари-Тута.
31. In vitro способ ингибирования HDAC6, включающий нанесение на образец соединения по любому из пп.1-20 или его фармацевтически приемлемой соли.
32. In vitro применение соединения по любому из пп.1-20 или его фармацевтически приемлемой соли в качестве ингибитора HDAC6.
| WO 2013080120 A1, 06.06.2013 | |||
| EP 1870401 A1, 26.12.2007 | |||
| EA 201390711 A1, 31.03.2014 | |||
| BERGE S | |||
| M | |||
| et al | |||
| Pharmaceutical Salts | |||
| Journal of Pharmaceutical Sciences, 1977, Vol | |||
| Приспособление для соединения пучка кисти с трубкою или втулкою, служащей для прикрепления ручки | 1915 |
|
SU66A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| БУДЧАНОВ Ю | |||
| И | |||
| Трансплантационная иммунология | |||
| Учебно-методическое пособие для студентов, 2012, С | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

