ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ НАСТОЯЩЕЕ ИЗОБРЕТЕНИЕ
[0001] Настоящее изобретение относится к новой соли тиофенового соединения, замещенного α-галогеном, пригодной в качестве лекарственного средства. Соль тиофенового соединения, замещенного α-галогеном, настоящего изобретения обладает антагонистическим действием относительно рецептора лизофосфатидной кислоты (LPA) и, следовательно, является пригодной для предотвращения и/или лечения заболеваний, вызванных LPA.
УРОВЕНЬ ТЕХНИКИ [0002] Лизофосфатидная кислота (LPA) представляет собой физиологически активный фосфолипид, который присутствует в живом организме. Связываясь со специфическими рецепторами, сопряженными с G-белком (LPA1, LPA2, LPA3, LPA4, LPA5 и LPA6), LPA передает сигналы в клетки и регулирует пролиферацию, дифференциацию, выживание, миграцию, адгезию, инфильтрацию и морфогенез клеток. Кроме того, известно, что LPA вовлечена в заболевания, сопровождаемые фиброзом в различных органах.
[0003] Сообщалось, что в печени, LPA стимулирует пролиферацию или сокращение звездчатых клеток, которые играют важную роль в процессе фиброза печени и стимулируют миграцию миофибробластов (смотри непатентные документы 1, 2 и 3).
Сообщалось, что в почках, продуцирование LPA или экспрессия LPA1 усиливается у мышей с односторонним лигированием мочеточника в качестве животных моделей фиброза почек, и что фиброз почек подавляется дефицитом LPA1 или введением LPA рецепторного антагониста (смотри непатентные документы 4 и 5).
[0004] Что касается легких, сообщалось, что жидкость бронхоальвеолярного лаважа пациентов с идиопатическим фиброзирующим альвеолитом содержала повышенную концентрацию LPA, и что LPA1 представлял собой самый экспрессируемый рецептор в фибробластах, играющих важную роль в процессе фиброза легких, и LPA вызывала миграцию фибробластов. Кроме того, сообщалось, что дефицит LPA1 или введение LPA рецепторного антагониста подавляло фиброз у мышей, которым вводили трахеально блеомицин, в качестве животных моделей фиброза легких (смотри непатентные документы 6 и 7).
[0005] Что касается кожи, сообщалось, что фиброз кожи подавляется дефицитом LPA1 или введением LPA рецепторного антагониста мышам, которым вводили подкожно блеомицин, в качестве моделей склеродермы (смотри непатентный документ 8).
[0006] Также известно, что LPA вовлечена в иммунологические или воспалительные заболевания. Сообщалось, что LPA стимулирует миграцию моноцитов человека и вовлечена в пролиферацию или инфильтрацию T-клеток. Кроме того, сообщалось, что синовиальные клетки пациентов с ревматоидным артритом экспрессируют LPA рецепторы и мигрируют или продуцируют IL-6 и IL-8 стимулированием LPA, и что данное действие ингибировалось LPA рецепторным антагонистом (смотри непатентные документы 9, 10 и 11).
[0007] Кроме того, сообщалось, что LPA и LPA1 участвуют в развитии нейропатической боли (смотри патентный документ 12), что LPA вызывала сокращение выделенных образцов уретры и простаты и повышение внутриуретрального давления, и она, таким образом, участвует в урологических заболеваниях (смотри патентный документ 1), и что LPA участвовала в раковых заболеваниях стимулированием инфильтрации раковых клеток, стимулированием пролиферации раковых клеток яичника или стимулированием пролиферации раковых клеток простаты (смотри непатентные документы 13, 14 и 15).
[0008] На основании данных сообщений, лекарственное средство, которое проявляет антагонизм относительно LPA рецепторов (в частности, LPA1 рецептора) считают пригодным для предотвращения и/или лечения заболеваний, сопровождающих фиброз, иммунологических или воспалительных заболеваний, заболеваний центральной или периферической нервной системы, урологических заболеваний и раковых заболеваний и т.д.
[0009] С другой стороны, патентный документы 2-23 и непатентные документы 5, 7, 8 и 16 описывают производные ([1,1ʹ-бифенил]-4-ил)уксусной кислоты, патентный документ 17 описывает производные (2ʹ-метокси-[1,1ʹ-бифенил]-4-ил)уксусной кислоты, и патентный документ 19 описывает 3-хлоризотиазоловые производные в качестве соединения, обладающего антагонистической функцией относительно LPA рецептором, отсутствует описание соединений согласно настоящему изобретению.
ДОКУМЕНТЫ ПРЕДШЕСТВУЮЩЕГО УРОВНЯ ТЕХНИКИ
Патентные документы
[0010] Патентный документ 1: WO 2002/062389
Патентный документ 2: WO 2010/077882
Патентный документ 3: WO 2010/077883
Патентный документ 4: WO 2010/141761
Патентный документ 5: WO 2010/141768
Патентный документ 6: WO 2011/017350
Патентный документ 7: WO 2011/041461
Патентный документ 8: WO 2011/041462
Патентный документ 9: WO 2011/041694
Патентный документ 10: WO 2011/041729
Патентный документ 11: WO 2011/091167
Патентный документ 12: WO 2011/159632
Патентный документ 13: WO 2011/159633
Патентный документ 14: WO 2011/159635
Патентный документ 15: WO 2012/078593
Патентный документ 16: WO 2012/078805
Патентный документ 17: WO 2012/138648
Патентный документ 18: WO 2012/138797
Патентный документ 19: WO 2013/025733
Патентный документ 20: WO 2013/085824
Патентный документ 21: WO 2013/189862
Патентный документ 22: WO 2013/189864
Патентный документ 23: WO 2013/189865
Непатентные документы
[0011] Непатентный документ 1: Biochemical and Biophysical Research Communications, 248(1998) 436-440
Непатентный документ 2: Biochemical and Biophysical Research Communications, 277 (2000) 72-78
Непатентный документ 3: Journal of Biomedical Science, 10 (2003) 352-358
Непатентный документ 4: Journal of the American Society of Nephrology, 18 (2007) 3110-3118
Непатентный документ 5: The Journal of Pharmacology and Experimental Therapeutics, 336 (2011) 693-700
Непатентный документ 6: Nature Medicine, 14 (2008) 45-54
Непатентный документ 7: British Journal of Pharmacology, 160 (2010) 1699-1713
Непатентный документ 8: Arthritis & Rheumatism, 63(2011) 1405-1415
Непатентный документ 9: Journal of Biological Chemistry, 270 (1995) 25549-25556
Непатентный документ 10: Biochimica et Biophysica Acta, 1582 (2002) 168-174
Непатентный документ 11: Molecular Pharmacology, 73 (2008) 587-600
Непатентный документ 12: Nature Medicine, 10 (2004) 712-718
Непатентный документ 13: Biochemical and Biophysical Research Communications, 193 (1993) 497-503
Непатентный документ 14: Biochemical Journal, 309 (1995) 933-940
Непатентный документ 15: The Journal of Urology, 163 (2000) 1027-1032
Непатентный документ 16: Journal of Medicinal Chemistry, 55 (2012) 7920-7939
ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Проблемы, которые будет решать настоящее изобретение
[0012] Изобретатели настоящего изобретения провели исследования на различных солях замещенных галогеном гетероциклических соединениях для того, чтобы разработать превосходное лекарственное средство для лечения или предотвращения заболеваний, сопровождающих фиброз, иммунологических или воспалительных заболеваний, заболеваний центральной или периферической нервной системы, урологических заболеваний и раковых заболеваний и т.д. Как результат, изобретатели настоящего изобретения обнаружили, что новая соль тиофенового соединения, замещенного α-галогеном, имеющая специфическую структуру, показывает превосходное LPA рецепторное антагонистическое действие и является пригодным в качестве лекарственного средства (в частности для предотвращения и/или лечения заболеваний, сопровождающих фиброз, иммунологических или воспалительных заболеваний, заболеваний центральной или периферической нервной системы, урологических заболеваний и раковых заболеваний). Настоящее изобретение завершено на основе данной находки.
[0013] Настоящее изобретение относится к новой соли тиофенового соединения, замещенного α-галогеном, которая обладает мощным антагонистическим действием относительно LPA рецептора и является пригодной в качестве лекарственного средства для лечения и/или предотвращения (предпочтительно, лекарственного средства для лечения) в частности заболеваний, с последующим фиброзом, иммунологических или воспалительных заболеваний, заболеваний центральной или периферической нервной системы, урологических заболеваний и раковых заболеваний.
Способы решения данных проблем
[0014] Настоящее изобретение относится к:
[0015] (1) соли, представленной общей формулой (I):
[0016] [Chem. 1]

[0017] (где
R представляет собой атом водорода или метокси группу,
X представляет собой атом галогена,
A выбран из группы, состоящей из:
[0018] [Chem. 2]
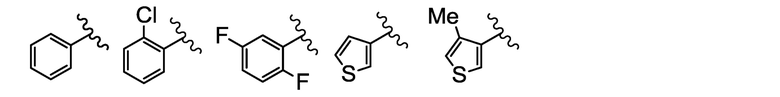
M представляет собой щелочной или щелочноземельный металл, и
n равен 1, когда M представляет собой щелочной металл, и равен 2, когда M представляет собой щелочноземельный металл).
[0019] (2) соли по (1), где щелочной металл или щелочноземельный металл представляет собой натрий, калий или кальций.
[0020] (3) соли (R)-1-[4ʹ-(5-хлор-3-{[(1-фенилэтокси)карбонил]амино}тиофен-2-ил)-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил]циклопропанкарбоновой кислоты с щелочным или щелочноземельным металлом.
[0021] (4) соли по (3), где щелочной металл или щелочноземельный металл представляет собой натрий, калий или кальций.
[0022](5) соли (R)-1-{4ʹ-[5-хлор-3-({[1-(2,5-дифторфенил)этокси]карбонил}амино)тиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты с щелочным или щелочноземельным металлом.
[0023] (6) соли по (5), где щелочной металл или щелочноземельный металл представляет собой натрий, калий или кальций.
[0024] (7) соли (R)-1-{4ʹ-[3-({[1-(2-хлорфенил)этокси]карбонил}амино)-5-фтортиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты с щелочным или щелочноземельным металлом.
[0025] (8) соли по (7), где щелочной металл или щелочноземельный металл представляет собой натрий, калий или кальций.
[0026](9) соли (R)-1-{4ʹ-[5-хлор-3-({[1-(тиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты с щелочным или щелочноземельным металлом.
[0027](10) соли по (9), где щелочной металл или щелочноземельный металл представляет собой натрий, калий или кальций.
[0028](11) соли (R)-1-{4ʹ-[5-фтор-3-({[1-(4-метилтиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты с щелочным или щелочноземельным металлом.
[0029](12) соли по (11), где щелочной металл или щелочноземельный металл представляет собой натрий, калий или кальций.
[0030] (13) LPA рецепторному антагонисту, содержащему соль по любому из (1)-(12) в качестве активного ингредиента.
[0031] (14) фармацевтической композиции, содержащей соль по любому из (1)-(12) в качестве активного ингредиента.
[0032] (15) фармацевтической композиции по (14) для лечения или предотвращения заболевания, сопровождающего фиброз, иммунологического или воспалительного заболевания, заболевания центральной или периферической нервной системы, урологического заболевания или ракового заболевания.
[0033] Конкретные примеры соединений, представленных общей формулой (I), настоящего изобретения включают соединения, описанные в таблице 1 ниже. В таблице 1, OMe представляет собой метокси группу, и ʺрацемическоеʺ и ʺ(R)-ʺ представляют собой конфигурацию атома углерода, помеченного ʺ*ʺ в общей формуле (I) ниже.
[0034] [Chem. 3]

[0035] [Таблица 1]
Эффекты настоящего изобретения
[0036] Соли тиофенового соединения, замещенного α-галогеном, настоящего изобретения, которые представлены общей формулой (I), обладают мощным LPA рецепторным антагонистическим действием и, следовательно, являются пригодными в качестве лекарственных средств для предотвращения и/или лечения заболеваний, сопровождающих фиброз, иммунологических или воспалительных заболеваний, заболеваний центральной или периферической нервной системы, урологических заболеваний и раковых заболеваний.
СПОСОБЫ ОСУЩЕСТВЛЕНИЯ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
[0037] Предпочтительные варианты осуществления каждой замещающей группы в солях, представленных общей формулой (I), будут описаны ниже.
[0038] Примеры ʺатомов галогенаʺ, представленных X, включают атом фтора, атом хлора, атом брома и атом йода.
[0039] Предпочтительно, ʺатом галогенаʺ, представленный X, представляет собой атом фтора, атом хлора или атом брома, и более предпочтительно представляет собой атом фтора или атом хлора.
[0040] Примеры ʺщелочных металловʺ, представленных M, включают литий, натрий, калий, рубидий и цезий, причем натрий и калий являются предпочтительными.
[0041] Примеры ʺщелочноземельных металловʺ, представленных M, включают магний, кальций, стронций и барий, причем кальций является предпочтительным.
[0042] В случае, когда соли настоящего изобретения, представленные общей формулой (I), имеют оптические изомеры, геометрические изомеры и изомеры вращения, данные изомеры включены в объем настоящего изобретения. Кроме того, в случае, когда имеется протонная таутомерия, данные таутомеры также включены в объем настоящего изобретения.
[0043] В общей формуле (I), группа, представленная:
[0044] [Chem. 4]

[0045] предпочтительно представляет собой следующую группу:
[0046] [Chem. 5]

[0047] Соли настоящего изобретения, представленные общей формулой (I), могут образовывать гидраты или сольваты, каждый и смесь которых также включены в объем настоящего изобретения.
[0048] Один или более типов атомов, образующих соли настоящего изобретения, представленные формулой (I), могут содержать атомные изотопы в неприродном соотношении. Примеры атомных изотопов включают дейтерий (2H), тритий (3H), углерод-14 (14C), фтор-18 (18F), серу-35 (35S) и йод-125 (125I). Данные соединения являются пригодными в качестве терапевтических или превентативных лекарственных средств, реагентов для исследований, таких как реагенты для анализа, и диагностических агентов, таких как in vivo диагностические визуализирующие агенты. Все изотопные варианты солей настоящего изобретения, представленных формулой (I), включены в объем настоящего изобретения, независимо от того, являются ли они радиоактивными или нет.
[0049] Общий способ получения соединения настоящего изобретения показан ниже. Каждый конкретный способ получения соединения настоящего изобретения будет описан отдельно и подробно в примерах ниже.
[0050] [Chem. 6]
[0051] [Chem. 7]

[0052] [Chem. 8]

(В формулах, R, X, A, M и n являются такими же, как определено в настоящем изобретении выше, Mʹ и nʹ имеют такие же значения, как M и n, соответственно, Z- представляет собой анион, такой как гидроксид, галоген или ацетат, и R1 представляет собой защитную группу карбокси группы, такую как алкильная группа, которая удаляется гидролизом.)
[0053] Соли настоящего изобретения, представленные общей формулой (I), можно получить любой из стадий 1-3, описанных выше.
[0054] В реакции на каждой из стадий 10-18, описанных ниже, любой растворитель можно применять без ограничений при условии, что растворитель не ингибирует реакцию и может растворять часть исходного материала. Примеры растворителей включают алифатические углеводороды, такие как гексан, пентан, гептан, петролейный эфир и циклогексан; ароматические углеводороды, такие как бензол, толуол, ксилол и этилбензол; галогенированные углеводороды, такие как дихлорметан, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол и дихлорбензол; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, 2-метилтетрагидрофуран, 1,4-диоксан, диметоксиэтан и диметиловый эфир диэтиленгликоля; кетоны, такие как ацетон, метилэтилкетон, метилизобутилкетон и циклогексанон; сложные эфиры, такие как метилацетат, этилацетат, пропилацетат, изопропилацетат и бутилацетат; нитрилы, такие как ацетонитрил, пропионитрил, бутиронитрил и изобутиронитрил; карбоновые кислоты, такие как уксусная кислота и пропионовая кислота; спирты, такие как метанол, этанол, 1-пропанол, 2-пропанол, 1-бутанол, 2-бутанол, 2-метил-1-пропанол, 2-метил-2-пропанола и 1,2-пропандиола; амиды, такие как формамид, N,N-диметилформамид, N,N-диметилацетамид, 1-метил-2-пирролидон, диметилимидазолон и гексаметилфосфортриамид; сульфоксиды, такие как диметилсульфоксид; сульфоны, такие как сульфолан; воду; и их смешанные растворители.
[0055] В реакции на каждой из стадий 10-18, описанных ниже, температура реакции изменяется в зависимости от условий, таких как растворители, исходные соединения и реагенты, и продолжительность реакции изменяется в зависимости от условий, таких как растворители, исходные соединения, реагенты и температура реакции.
[0056] Стадия 1: соединение (1) может реагировать в растворителе для реакции, применяя гидроксид щелочного металла или щелочноземельного металла, давая соль общей формулы (I).
Растворитель для реакции предпочтительно представляет собой воду или смесь вода/органический растворитель, и более предпочтительно представляет собой воду, смесь ацетонитрил/вода или смесь ацетонитрил/тетрагидрофуран/вода.
[0057] Стадия 2: соединение (Iʹ) и соединение (2) можно подвергать катионному обмену в растворителе для реакции, получая соль общей формулы (I).
Растворитель для реакции предпочтительно представляет собой воду или смесь вода/органический растворитель, и более предпочтительно представляет собой воду или смесь ацетонитрил/вода.
[0058] Стадия 3: соединение (3) можно гидролизовать в растворителе для реакции, применяя гидроксид щелочного металла или щелочноземельного металла, получая соль общей формулы (I).
Растворитель для реакции предпочтительно представляет собой воду или смесь вода/органический растворитель, и более предпочтительно представляет собой смесь 2-пропанол/вода или смесь 2-пропанол/тетрагидрофуран/вода.
[0059] Общий способ получения синтетического промежуточного соединения настоящего изобретения будет описан ниже. Каждый конкретный способ получения синтетического промежуточного соединения настоящего изобретения будет описан отдельно и подробно в примерах далее.
В способах получения, показанных ниже, R, X, A, M, Mʹ, n, nʹ, Z- и R1 имеют те же значения, как описано выше. L, La и M представляют собой заместители, необходимые для реакции конденсации и, например, в случае, когда L или La представляет собой атом хлора, атом брома, атом йода или трифторметансульфонилокси группу и т.д., M представляет собой бороновую кислоту, боронатный эфир или триалкилолово и т.д., и в случае, когда L или La представляет собой бороновую кислоту, боронатный эфир или триалкилолово и т.д., M представляет собой атом хлора, атом брома, атом йода или трифторметансульфонилокси группу и т.д.
[0060] [Chem. 9]
[0061] Стадия 4: Согласно способу, описанному, например, в Tetrahedron, 64 (2008), стр. 9733-9737 или Bioorganic and Medicinal Chemistry Letters, 21 (2011), стр. 528-530, соединение (4) можно галогенировать галогенирующим агентом в растворителе для реакции, получая соединение (5).
Предпочтительные примеры растворителей для реакции включают алифатические углеводороды, галогенированные углеводороды, эфиры, нитрилы, карбоновые кислоты, амиды, сульфоксиды, воду и их смешанный растворитель. N,N-Диметилформамид является более предпочтительным.
Примеры галогенирующих агентов включают йод, N-йодсукцинимид, бром, N-бромсукцинимид, 1,2-дибромэтан, 1,2-дибром-1,1,2,2-тетрафторэтан, хлор, N-хлорсукцинимид, дифторид ксенона, N-фторбензолсульфонимид и N-фтор-Nʹ-(хлорметил)триэтилендиамин бис(тетрафторборат).
Альтернативно, соединение (4) можно превратить в анион в растворителе для реакции, применяя основание, и затем обрабатывая галогенирующим агентом, получая соединение (5) согласно способу, описанному, например, в Tetrahedron Letters, 51 (2010), стр. 4526-4529 или Journal of Medicinal Chemistry, 54 (2011), стр. 2687-2700.
Предпочтительные примеры растворителей для реакции включают алифатические углеводороды, ароматические углеводороды, галогенированные углеводороды, эфиры и их смешанный растворитель. Эфиры, алифатические углеводороды и их смешанный растворитель являются более предпочтительными.
Примеры оснований включают алкиллитии, такие как н-бутиллитий, втор-бутиллитий и трет-бутиллитий; амиды лития, такие как диизопропиламид лития и 2,2,6,6-тетраметилпиперидин лития; реагенты Гриньяра, такие как этилмагнийбромид, этилмагнийхлорид, изопропилмагнийхлорид и фенилмагнийхлорид; амиды магния, такие как диизопропиламидмагнийхлорид и 2,2,6,6-тетраметилпиперидинмагнийхлорид; и дисилазановые основания, такие как 1,1,1,3,3,3-гексаметилдисилазан лития и 1,1,1,3,3,3-гексаметилдисилазан калия.
Примеры галогенирующих агентов включают йод, N-йодсукцинимид, бром, N-бромсукцинимид, тетрабромид углерода, 1,2-дибромэтан, 1,2-дибром-1,1,2,2-тетрафторэтан, хлор, N-хлорсукцинимид, тетрахлорид углерода, дифторид ксенона, N-фторбензолсульфонимид и N-фтор-Nʹ-(хлорметил)триэтилендиамин бис(тетрафторборат).
[0062] [Chem. 10]

[0063] Стадия 5: согласно способу, описанному, например, в Tetrahedron, 58 (2002), стр. 9633-9695, соединение (6) и соединение (7) может реагировать в растворителе для реакции в присутствии катализатора конденсации, лиганда и/или основания, давая соединение (8).
Предпочтительные примеры растворителей для реакции включают алифатические углеводороды, ароматические углеводороды, эфиры, кетоны, сложные эфиры, нитрилы, спирты, амиды, сульфоксиды, сульфоны, воду и их смешанный растворитель. Смесь 1,4-диоксан/вода является более предпочтительной.
Примеры катализаторов для конденсации включают палладиевые катализаторы, такие как тетракис(трифенилфосфин)палладий (0), продукт присоединения хлористого метилена и дихлорида [1,1ʹ-бис(дифенилфосфино)ферроцен]палладия (II), дихлорид бис(трифенилфосфин)палладия (II), трис(дибензилиденацетон)дипалладий (0) и ацетат палладия (II); и никелевые катализаторы, такие как дихлорид бис(трифенилфосфин)никеля (II).
Примеры лигандов, иногда присутствующих в самих катализаторах для конденсации, включают трифенилфосфин, [1,1ʹ-бис(дифенилосфино)ферроцен], дибензилиденацетон, трифениларсин, три(o-толил)фосфин, три-трет-бутилфосфин и трициклогексилфосфин.
Примеры оснований включают фторидные соли, такие как фторид калия и фторид цезия; карбонатные соли, такие как гидрокарбонат натрия, карбонат натрия, карбонат калия, карбонат цезия и карбонат таллия; гидроксиды металлов, такие как гидроксид натрия, гидроксид калия, гидроксид бария и гидроксид таллия; фосфатные соли, такие как фосфат калия; и органические основания, такие как триэтиламин и диизопропилэтиламин. Карбонат натрия является предпочтительным.
[0064] [Chem. 11]
[0065] Стадия 6: согласно способу, описанному, например, в Journal of the American Chemical Society, 129 (2007), стр. 4595-4605, соединение (8) может реагировать в растворителе для реакции в присутствии палладиевого катализатора, лиганда, реагента на основе бороновой кислоты и/или основания, давая соединение (9). Например, L представляет собой атом хлора, атом брома, атом йода или трифторметансульфонилокси группу, и M представляет собой бороновую кислоту или боронатный эфир.
Предпочтительные примеры растворителей для реакции включают алифатические углеводороды, ароматические углеводороды, эфиры, кетоны, сложные эфиры, нитрилы, спирты, амиды, сульфоксиды, сульфоны, воду и их смешанный растворитель. 1,4-Диоксан является более предпочтительным.
Примеры палладиевых катализаторов включают тетракис(трифенилфосфин)палладий (0), продукт присоединения хлористого метилена к дихлориду [1,1ʹ-бис(дифенилосфино)ферроцен]палладия (II), дихлорид бис(трифенилфосфин)палладия (II), трис(дибензилиденацетон)дипалладий (0) и ацетат палладия (II). продукт присоединения хлористого метилена к дихлориду [1,1ʹ-бис(дифенилосфино)ферроцен]палладия (II) и ацетат палладия (II) являются предпочтительными.
Примеры лигандов, иногда присутствующих в самих катализаторах для конденсации, включают трифенилфосфин, [1,1ʹ-бис(дифенилосфино)ферроцен], дибензилиденацетон, трифениларсин, три(o-толил)фосфин, три-трет-бутилфосфин и трициклогексилфосфин. Трициклогексилфосфин является предпочтительным.
Примеры реагентов на основе бороновой кислоты включают 4,4,4ʹ,4ʹ,5,5,5ʹ,5ʹ-октаметил-2,2ʹ-бис(1,3,2-диоксаборолан) и 4,4,5,5-тетраметил-1,3,2-диоксаборолан.
Примеры оснований включают ацетат калия и ацетат натрия.
[0066] Альтернативно, согласно способу, описанному, например, в Angewandte Chemie-International Edition, 45 (2006), стр. 1404-1408, соединение (8), в котором L представляет собой атом хлора, атом брома или атом йода, можно обрабатывать в растворителе для реакции так, чтобы подвергать галогеновую группу L обмену галогена на металл, применяя основание, и затем продукт можно обрабатывать реагентом на основе бороновой кислоты, получая соединение (9).
Предпочтительные примеры растворителей для реакции включают алифатические углеводороды, ароматические углеводороды, галогенированные углеводороды, эфиры, амиды, сульфоксиды, сульфоны и их смешанный растворитель. Эфиры, алифатические углеводороды и их смешанный растворитель являются более предпочтительными.
Примеры оснований включают алкиллитии, такие как н-бутиллитий, втор-бутиллитий и трет-бутиллитий; и реагенты Гриньяра, такие как этилмагнийбромид, этилмагнийхлорид, изопропилмагнийхлорид и фенилмагнийхлорид.
Примеры реагентов на основе бороновой кислоты включают триметилборат, триизопропилборат, тригесадецилборат и 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолан.
[0067] [Chem. 12]

[0068] [Chem. 13]

[0069] Стадия 7 и стадия 8: соединение (5) или соединение (11) и соединение (9) может реагировать способом, аналогичным стадии 5, давая соединение (10) или соединение (12), соответственно.
[0070] [Chem. 14]

[0071] Стадия 9: соединение (12) можно обрабатывать способом, аналогичным способу 4, получая соединение (13).
[0072] [Chem. 15]
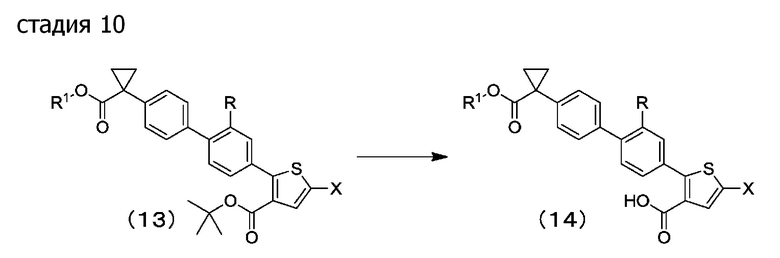
[0073] Стадия 10: в растворителе для реакции или без растворителя, кислота действует на соединение (13), деблокируя соединение, посредством этого давая соединение (14).
Предпочтительные примеры растворителей для реакции включают алифатические углеводороды, ароматические углеводороды, галогенированные углеводороды, эфиры, кетоны, сложные эфиры, нитрилы, карбоновые кислоты, спирты, амиды, сульфоксиды, сульфоны, воду и их смешанный растворитель. Хлористый метилен является более предпочтительным.
Примеры кислот включают неорганические кислоты, такие как хлористоводородная кислота и серная кислота; органические кислоты, такие как уксусная кислота, трифторуксусная кислота и трихлоруксусная кислота; и сульфокислоты, такие как метансульфокислота, бензолсульфокислота и п-толуолсульфокислота, причем трифторуксусная кислота является предпочтительной.
[0074] [Chem. 16]

[0075] Стадия 11: в растворителе для реакции, соединение (10) гидролизуют в присутствии кислоты или основания, давая соединение (15).
Предпочтительные примеры растворителей для реакции включают алифатические углеводороды, ароматические углеводороды, галогенированные углеводороды, эфиры, кетоны, нитрилы, карбоновые кислоты, спирты, амиды, сульфоксиды, сульфоны, воду и их смешанный растворитель. Смесь этанол/тетрагидрофуран/вода и смесь 2-пропанол/тетрагидрофуран/вода являются более предпочтительными.
Примеры кислот и оснований включают неорганические кислоты, такие как хлористоводородная кислота и серная кислота; органические кислоты, такие как уксусная кислота и трифторуксусная кислота; сульфокислоты, такие как метансульфокислота, бензолсульфокислота и п-толуолсульфокислота; гидроксиды щелочных металлов, такие как гидроксид литий, гидроксид натрия и гидроксид калия; и карбонаты щелочных металлов, такие как карбонат калия и карбонат натрия. Применение основания является предпочтительным, и применение гидроксида лития или гидроксида натрия является более предпочтительным.
[0076] [Chem. 17]
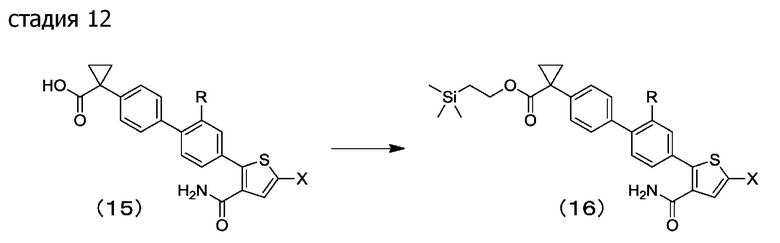
[0077] Стадия 12: в растворителе для реакции соединение (15) можно конденсировать с триметилсилилэтанолом, получая конденсирующий агент в присутствии или отсутствии основания и в присутствии или отсутствии добавки, посредством этого получая соединение (16).
Предпочтительные примеры растворителей для реакции включают алифатические углеводороды, ароматические углеводороды, галогенированные углеводороды, эфиры, кетоны, сложные эфиры, нитрилы, амиды, сульфоксиды, сульфоны и их смешанный растворитель. N,N-диметилформамид является более предпочтительным.
Примеры оснований включают карбонатные соли, такие как гидрокарбонат натрия, карбонат натрия, карбонат калия, карбонат цезия и карбонат таллия; пиридины, такие как пиридин, 2,6-лутидин и 4-пиколин; и органические основания, такие как триэтиламин и диизопропилэтиламин. Диизопропилэтиламин является предпочтительным.
Примеры конденсирующих реагентов включают карбодиимидные конденсирующие реагенты, такие как N,Nʹ-дициклогексилкарбодиимид, N,Nʹ-диизопропилкарбодиимид, гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида и мето-п-толуолсульфонатная соль N-циклогексил-Nʹ-(2-морфолиноэтил)карбодиимида; имидазольные конденсирующие реагенты, такие как N,Nʹ-карбонилдиимидазол; триазиновые конденсирующие реагенты, такие как хлорид 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния и трифторметансульфонат (4,6-диметокси-1,3,5-триазин-2-ил)-(2-октокси-2-оксоэтил)диметиламмония; фосфониевые конденсирующие реагенты, такие как гексафторфосфатная соль 1H-бензотриазол-1-илокситрис(диметиламино)фосфония, гексафторфосфатная соль 1H-бензотриазол-1-илокситрипирролидинофосфония и гексафторфосфатная соль хлортрипирролидинофосфония; и урониевые конденсирующие реагенты, такие как гексафторфосфатная соль ({[(1-циано-2-этокси-2-оксоэтилиден)амино]окси}-4 морфолинометилен)диметиламмония, гексафторфосфатная соль O-(бензотриазол-1-ил)-N,N,Nʹ,Nʹ-тетраметилурония, гексафторфосфатная соль O-(7-азабензотриазол-1-ил)-N,N,Nʹ,Nʹ-тетраметилурония, тетрафторборатная соль O-(N-сукцинимидил)-N,N,Nʹ,Nʹ-тетраметилурония и тетрафторборатная соль O-(3,4-дигидро-4-оксо-1,2,3-бензотриазин-3-ил)-N,N-Nʹ,Nʹ-тетраметилурония, причем гексафторфосфатная соль O-(бензотриазол-1-ил)-N,N,Nʹ,Nʹ-тетраметилурония является предпочтительной.
Примеры добавок включают бензотриазолы, такие как 1-гидроксибензотриазол и 1-гидроксиазабензотриазол; пиридины, такие как N,N-диметиламинопиридин; и их комбинации. N,N-диметиламинопиридин является предпочтительным.
[0078] [Chem. 18]
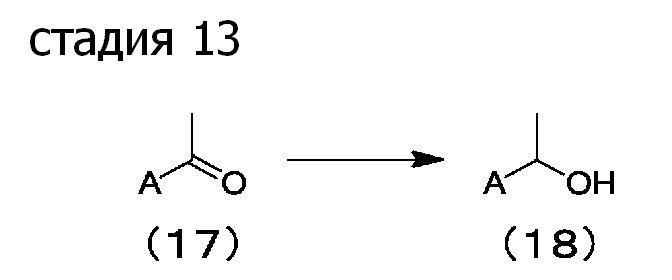
[0079] Стадия 13: Согласно способу, описанному, например, в Tetrahedron Letters, 47 (2006), стр. 5261-5264, соединение (17) можно восстановить восстанавливающим агентом в растворителе для реакции, получая соединение (18).
Предпочтительные примеры растворителей для реакции включают алифатические углеводороды, ароматические углеводороды, галогенированные углеводороды, эфиры, сложные эфиры, нитрилы, спирты, амиды, сульфоксиды, сульфоны, воду и их смешанный растворитель.
Примеры восстанавливающих агентов включают боргидриды, такие как боргидрид лития, боргидрид натрия, боргидрид калия и триметоксиборгидрид натрия; и гидриды алюминия, такие как алюмогидрид лития, алюмогидрид натрия, диизобутилалюмогидрид и триметоксиалюмогидрид литий.
[0080] Оптически активное соединение (18) можно получить, применяя (R)- или (S)-5,5-дифенил-2-метил-3,4-пропано-1,3,2-оксазаборолидин и т.д., согласно способу, описанному, например, в Journal of Organic Chemistry, 56 (1991), стр. 763-769.
[0081] Оптическую чистоту соединения (18), полученного способом выше, можно увеличить известным способом, таким как применяя фермент или разделяющий агент или комбинацию данных способов.
[0082] [Chem. 19]
[0083] [Chem. 20]
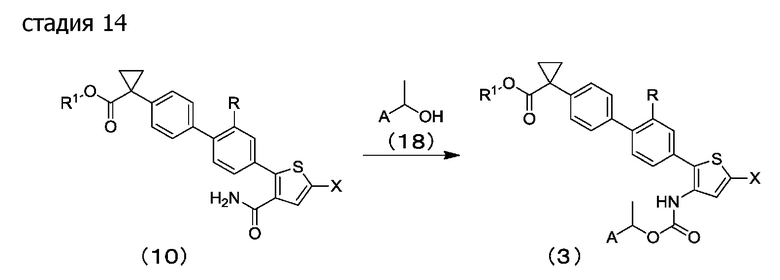
[0084] Стадия 14 и стадия 15: в растворителе для реакции или без растворителя соединение (10) или соединение (16) подвергают перегруппировке Гофмана в присутствии или отсутствии основания, применяя соединение (18) и окисляющий агент согласно способу, описанному, например, в Organic Synthesis, 66 (1988), стр. 132-137, посредством этого получая соединение (3) или соединение (19), соответственно.
Предпочтительные примеры растворителей для реакции включают алифатические углеводороды, ароматические углеводороды, галогенированные углеводороды, эфиры, кетоны, сложные эфиры, нитрилы, амиды, сульфоксиды, сульфоны и их смешанный растворитель. Толуол является более предпочтительным.
Примеры оснований включают органические амины, такие как триэтиламин и диизопропилэтиламин; и пиридины, такие как пиридин, 2,6-лутидин и 4-пиколин. Пиридин является предпочтительным.
Примеры окисляющих агентов включают высоковалентные йодные соединения, такие как [бис(ацетокси)йод]бензол, [бис(трифторацетокси)йод]бензол и йодозилбензол, причем [бис(трифторацетокси)йод]бензол является предпочтительным.
[0085] [Chem. 21]

[0086] Стадия 16: в растворителе для реакции или без растворителя соединение (14) можно подвергать перегруппировке Курциуса, применяя соединение (18), дифенилфосфорилазид и основание согласно способу, описанному, например, в Journal of the American Chemical Society, 94 (1972), стр. 6203-6205, посредством этого получая соединение (3).
Предпочтительные примеры растворителей для реакции включают алифатические углеводороды, ароматические углеводороды, галогенированные углеводороды, эфиры, кетоны, сложные эфиры, нитрилы, амиды, сульфоксиды, сульфоны, воду и их смешанный растворитель. Толуол является более предпочтительным.
Примеры оснований включают органические амины, такие как триэтиламин и диизопропилэтиламин, причем триэтиламин является предпочтительным.
[0087] [Chem. 22]

[0088] Стадия 17: соединение (3) можно обработать способом, аналогичным способу 11, получая соединение (1).
[0089] [Chem. 23]
[0090] Стадия 18: в растворителе для реакции, соединение (19) можно деблокировать деблокирующим агентом, получая соединение (1).
Предпочтительные примеры растворителей для реакции включают алифатические углеводороды, ароматические углеводороды, галогенированные углеводороды, эфиры, кетоны, сложные эфиры, нитрилы, карбоновые кислоты, спирты, амиды, сульфоксиды, сульфоны, воду и их смешанный растворитель. N,N-Диметилформамид является более предпочтительным.
Примеры деблокирующих агентов включают фтороводородную кислоту; неорганические фторидные соли, такие как фторид калия; органические гидрофторидные соли, такие как гидрофторидная соль пиридина, гидрофторидная соль триэтиламина и гидрофторидная соль 1-гексадекана; фториды аммония, такие как фторид тетраэтиламмония и фторид тетрабутиламмония; и дифтортриметилсиликатные соли, такие как дифтортриметилсиликат трис(диметиламино)сульфония. Фторид тетрабутиламмония является предпочтительным.
[0091] Соединение формулы (Iʹ) можно получить обработкой соединения формулы (1) согласно стадии 1, применяя Mʹnʹ+(OH-)nʹ.
[0092] Целевое соединение, полученное в каждой стадии, можно выделить из жидкости реакционной смеси стандартным способом. В случае, когда, например, целевое соединение осаждается полностью или частично, выпадает или кристаллизуется в жидкости реакционной смеси, твердый остаток, содержащий целевое соединение, можно получить фильтрованием жидкости реакционной смеси. Когда целевое соединение растворяется полностью или частично в жидкости реакционной смеси, целевое соединение можно получить удалением растворителя (например, лиофилизацией) непосредственно или после удаления нерастворимого материала фильтрованием. Альтернативно, целевое соединение можно получить подходящей нейтрализацией жидкости реакционной смеси, удаляя любой нерастворимый материал фильтрованием, добавляя смешиваемый с водой органический растворитель, такой как этилацетат, промывая смешанную жидкость водой, суспендируя органическую фазу, содержащую целевое соединение, сушкой органической фазы осушителем, таким как безводный сульфат магния или безводный сульфат натрия, и упариванием растворителя отгонкой.
[0093] При необходимости, полученное целевое соединение можно выделить и очистить подходящей комбинацией стандартных способов, таких как промывка водой, органическим растворителем или смесью данных растворителей; перекристаллизация; переосаждение; и способами, обычно применяемыми для выделения и очистки органических соединений (например, способами адсорбционной колоночной хроматографии, применяя носитель, такой как силикагель или оксид алюминия; способами ионообменной хроматографии; способами хроматографии с нормальной или обратной фазой (предпочтительно, высокоэффективная жидкостная хроматография), применяя силикагель или алкилированный силикагель; и способы хроматографии с нормальной или обратной фазой (предпочтительно, высокоэффективная жидкостная хроматография), применяя наполнитель, в котором зафиксированы оптически активные молекулы или в котором силикагель покрыт оптически активными молекулами).
[0094] Когда соли настоящего изобретения, представленные общей формулой (I), применяют в качестве лекарственных средств, сами соли (в качестве ингредиента) можно вводить как есть или можно вводить перорально или парентерально (таким как внутривенное введение, внутримышечное введение, внутрибрюшинное введение, подкожное введение, введение через трахею или внутрикожное введение и подкожное введение) в формах, таких как таблетки, капсулы, порошки, сиропы, гранулы, высокодисперсные гранулы, пилюли, суспензии, эмульсии, препараты для впитывания через кожу, суппозитории, мази, лосьоны, ингалянты и продукты для инъекции, которые получают смешением солей с подходящими фармакологически приемлемыми вспомогательными веществами, разбавителями и т.д.
[0095] Данные препараты получают известными способами, применяя добавки, такие как вспомогательные вещества, смазывающие вещества, связующие вещества, разрыхлители, эмульгаторы, стабилизаторы, ароматизаторы и разбавители.
[0096] Примеры вспомогательных веществ включают органические вспомогательные вещества и неорганические вспомогательные вещества. Примеры органических вспомогательных веществ включают сахарные производные, такие как лактоза, сахароза, глюкоза, маннит и сорбит; производные крахмала, такие как кукурузный крахмал, картофельный крахмал, α-крахмал и декстрин; производные целлюлозы, такие как кристаллическая целлюлоза; гуммиарабик; декстран; и пуллулан. Примеры неорганических вспомогательных веществ включают легкую безводную кремниевую кислоту; и сульфатные соли, такие как сульфат кальция.
[0097] Примеры смазывающих веществ включают стеариновую кислоту; соль металла стеариновой кислоты, такую как стеарат кальция и стеарат магния; тальк; коллоидный диоксид кремния; воски, такие как пчелиный воск и спермацетовый воск; борную кислоту; адипиновую кислоту; сульфатные соли, такие как сульфат натрия; гликоль; фумаровую кислоту; бензоат натрия; D,L-лейцин; лаурилсульфат натрия; кремневые кислоты, такие как кремниевый ангидрид и гидрат кремниевой кислоты; и производные крахмала, перечисленные в качестве вспомогательных веществ выше.
[0098] Примеры связующих веществ включают гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, поливинилпирролидон, макрогель и соединения, перечисленные в качестве вспомогательных веществ выше.
[0099] Примеры разрыхлителей включают производные целлюлозы, такие как гидроксипропилцеллюлозу с низкой степенью замещения, карбоксиметилцеллюлозу, карбоксиметилцеллюлозу кальция и карбоксиметилцеллюлозу кальция с внутренними сшивками; сшитый поливинилпирролидон; и химически модифицированный крахмал или производные целлюлозы, такие как карбоксиметилкрахмал и карбоксиметилкрахмал натрия.
[0100] Примеры эмульгаторов включают глины, такие как бентонит и пчелиная смола; анионные поверхностно-активные вещества, такие как лаурилсульфат натрия; катионные поверхностно-активные вещества, такие как хлорид бензалкония; и неионные поверхностно-активные вещества, такие как полиоксиэтиленалкиловый эфир, сложный эфир жирной кислоты и полиоксиэтиленсорбитана, сложный эфир сахарозы и жирных кислот.
[0101] Примеры стабилизаторов включают п-гидроксибензоатные сложные эфиры, такие как метилпарабен и пропилпарабен; спирты, такие как хлорбутанол, бензиловый спирт и фенилэтиловый спирт; хлорид бензалкония; фенолы, такие как фенол и крезол; тимеросал; уксусный ангидрид; и сорбиновую кислоту.
[0102] Примеры ароматизаторов включают подсластители, такие как сахарин натрия и аспартам; подкислители, такие как лимонная кислота, яблочная кислота и винная кислота; и ароматизаторы, такие как ментол, экстракт лимона и экстракт апельсина.
[0103] Разбавители представляют собой соединения, обычно применяемые для разбавления. Их примеры включают лактозу, маннитол, глюкозу, сахарозу, сульфат кальция, гидроксипропилцеллюлозу, микрокристаллическую целлюлозу, воду, этанол, полиэтиленгликоль, пропиленгликоль, глицерин, крахмал, поливинилпирролидон и их смеси.
[0104] Доза солей настоящего изобретения, представленных общей формулой (I), может изменяться в зависимости от условий, таких как симптомы, возраст и вес тела пациентов. В случае перорального введения, наименьшая и наибольшая дозы на введение могут составлять 0,001 мг/кг (предпочтительно 0,01 мг/кг) и 20 мг/кг (предпочтительно 10 мг/кг), соответственно. В случае парентерального введения, наименьшая и наибольшая дозы на введение могут составлять 0,0001 мг/кг (предпочтительно 0,0005 мг/кг) и 10 мг/кг (предпочтительно 5 мг/кг), соответственно. В обоих случаях, количество введений для взрослых может составлять 1-6 в день, в зависимости от симптомов.
ПРИМЕРЫ
[0105] Настоящее изобретение будет описано более подробно в настоящем изобретении ниже, представлением примеров (примеры 1-15), сравнительных примеров (сравнительные примеры 1-32), примеров испытаний (примеры испытаний 1-8) и примеров получения (1-3). Данные примеры служат только для того, чтобы способствовать лучшему пониманию настоящего изобретения и не предполагаются ограничивающими объем настоящего изобретения.
[0106] Из свойств в примерах и сравнительных примерах, величины Rf предсталвяют собой величины, измеренные тонкослойной хроматографией (Merck Co., пластинки с силикагелем для ТСХ 60F254 (торговая марка)). Проявляющие растворители (и их объемное соотношение) описаны в скобках.
[0107] Термин COOH колонка в колоночной хроматографии на силикагеле обозначает Chromatorex (зарегистрированная торговая марка) Q-PACK COOH колонку с силикагелем Fuji Silysia Chemical Ltd.
[0108] Применяли ультрачистую воду Wako Pure Chemical Industries, Ltd. (214-01301).
[0109] В случае, когда получали ряд масс-спектральных величин благодаря наличию изотопов, описывали только наименьшую величину m/z. DUIS в масс-спектроскопии представляет собой смешанный режим ионизации ESI и APCI.
[0110] В химических структурах, Me обозначает метильную группу, если не указано иначе.
[0111] (Пример 1)
(R)-1-[4ʹ-(5-Хлор-3-{[(1-фенилэтокси)карбонил]амино}тиофен 2-ил)-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил]циклопропанкарбоксилат натрия (соединение No. I-2)
[Chem. 24]

[0112] В бане со льдом и при перемешивании 2,00 мл (2,00 ммоль) 1N водного раствора гидроксида натрия добавляли к суспензии в ацетонитриле (80 мл) 1,10 г (2,00 ммоль) (R)-1-[4ʹ-(5-хлор-3-{[(1-фенилэтокси)карбонил]амино}тиофен-2-ил)-2ʹ-метокси[1,1ʹ-бифенил]-4-ил]циклопропанкарбоновой кислоты, полученной аналогично сравнительному примеру 29. После этого, добавляли ультрачистую воду (6 мл), и смесь обрабатывали ультразвуком, получая однородный раствор, который, затем, перемешивали при комнатной температуре в течение 3 часов. К жидкости реакционной смеси дополнительно добавляли небольшое количество ультрачистой воды. Растворитель удаляли лиофилизацией, и остаток сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 1,08 г (1,89 ммоль, выход 95%), в виде белого твердого остатка.
Масс-спектр (ESI+, m/z): 570 [M+1]+.
1H-ЯМР-спектр (400 МГц, DMSO-d6) δ: 9,44 (1H, уш с), 7,43-7,22 (10H, м), 7,21-7,14 (2H, м), 7,08 (1H, дд, J=7,8, 1,4 Гц), 5,75 (1H, кв, J=6,1 Гц), 3,73 (3H, с), 1,56-1,38 (3H, м), 1,16 (2H, дд, J=5,6, 2,6 Гц), 0,66 (2H, дд, J=5,7, 2,7 Гц).
[0113] (Пример 2)
(R)-1-[4ʹ-(5-Хлор-3-{[(1-фенилэтокси)карбонил]амино}тиофен-2-ил)-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил]циклопропанкарбоксилат калия (соединение No. I-4)
[Chem. 25]

[0114] При перемешивании 0,500 мл (0,500 ммоль) 1N водного раствора гидроксида калия добавляли к суспензии в ацетонитриле (20 мл) и ультрачистой воде (1,5 мл) 275 мг (0,501 ммоль) (R)-1-[4ʹ-(5-хлор-3-{[(1-фенилэтокси)карбонил]амино}тиофен-2-ил)-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил]циклопропанкарбоновой кислоты, полученной аналогично сравнительному примеру 29, посредством этого получая однородный раствор. После этого, добавляли ультрачистую воду (6 мл), и смесь обрабатывали ультразвуком. Полученную в результате жидкость реакционной смеси выдерживали при комнатной температуре в течение 30 минут. Дополнительно добавляли небольшое количество ультрачистой воды. Растворитель удаляли лиофилизацией, и остаток сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 235 мг (0,401 ммоль, выход 80%) в виде белого твердого остатка.
Масс-спектр (ESI+, m/z): 586 [M+1]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 9,44 (1H, уш с), 7,43-7,14 (12H, м), 7,08 (1H, дд, J=7,8, 1,4 Гц), 5,75 (1H, кв, J=6,2 Гц), 3,73 (3H, с), 1,54-1,39 (3H, м), 1,11 (2H, дд, J=5,8, 2,7 Гц), 0,60 (2H, дд, J=5,8, 2,6 Гц).
[0115] (Пример 3)
(R)-1-[4ʹ-(5-Хлор-3-{[(1-фенилэтокси)карбонил]амино}тиофен-2-ил)-2ʹ-метокси[1,1ʹ-бифенил]-4-ил]циклопропанкарбоксилат 1/2 кальция (соединение No. I-6)
[Chem. 26]

[0116] 0,180 мл (0,090 ммоль) 0,5 M водного раствора ацетата кальция добавляли к раствору в ультрачистой воде (25 мл) 101 мг (0,177 ммоль) (R)-1-[4ʹ-(5-хлор-3-{[(1-фенилэтокси)карбонил]амино}тиофен-2-ил)-2ʹ-метокси[1,1ʹ-бифенил]-4-ил]циклопропанкарбоксилата натрия, полученного в примере 1. Смесь перемешивали при комнатной температуре в течение 2 дней. Полученную в результате суспензию фильтровали через мембранный фильтр (Millipore). Остаток промывали ультрачистой водой и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 40,4 мг (0,071 ммоль, выход 40%), в виде белого твердого остатка.
Масс-спектр (ESI+, m/z): 1133 [2M+1]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 9,42 (1H, уш с), 7,41-7,25 (10H, м), 7,19-7,15 (2H, м), 7,07 (1H, дд, J=7,9, 1,3 Гц), 5,75 (1H, кв, J=6,2 Гц), 3,72 (3H, с), 1,54-1,38 (3H, м), 1,38-1,22 (2H, м), 0,90-0,80 (2H, м).
[0117] (Пример 4)
(R)-1-{4ʹ-[5-хлор-3-({[1-(2,5-дифторфенил)этокси]карбонил}амино)тиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоксилат натрия (соединение No. I-8)
[Chem. 27]

[0118] В бане со льдом и при перемешивании, 1,00 мл (1,00 ммоль) 1N водного раствора гидроксида натрия добавляли к суспензии в ацетонитриле (40 мл) и ультрачистой воде (3 мл) 584 мг (1,00 ммоль) (R)-1-{4ʹ-[5-хлор-3-({[1-(2,5-дифторфенил)этокси]карбонил}амино)тиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты, полученной аналогично сравнительному примеру 32, посредством этого получая однородный раствор. Раствор перемешивали при данной температуре в течение 30 минут. К жидкости реакционной смеси дополнительно добавляли небольшое количество ультрачистой воды. Растворитель удаляли лиофилизацией, и остаток сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 534 мг (0,882 ммоль, выход 88%), в виде белого твердого остатка.
Масс-спектр (ESI+, m/z): 606 [M+1]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 9,56 (1H, уш с), 7,36-7,14 (10H, м), 7,08 (1H, дд, J=7,8, 1,4 Гц), 5,91 (1H, кв, J=6,6 Гц), 3,75 (3H, с), 1,61-1,36 (3H, м), 1,16 (2H, дд, J=5,6, 2,8 Гц), 0,66 (2H, дд, J=5,6, 2,6 Гц).
[0119] (Пример 5)
(R)-1-{4ʹ-[5-Хлор-3-({[1-(2,5-дифторфенил)этокси]карбонил}амино)тиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоксилат калия (соединение No. I-10)
[Chem. 28]

[0120] В бане со льдом и при перемешивании 0,500 мл (0,500 ммоль) 1N водного раствора гидроксида калия добавляли к однородному раствору в ацетонитриле (20 мл) и ультрачистой воде (1,5 мл) 292 мг (0,500 ммоль) (R)-1-{4ʹ-[5-хлор-3-({[1-(2,5-дифторфенил)этокси]карбонил}амино)тиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты, полученной аналогично сравнительному примеру 32. Смесь перемешивали при данной температуре в течение 1 часа. К жидкости реакционной смеси дополнительно добавляли небольшое количество ультрачистой воды. Растворитель удаляли лиофилизацией, и остаток сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 276 мг (0,444 ммоль, выход 89%), в виде белого твердого остатка.
Масс-спектр (ESI+, m/z): 622 [M+1]+.
1H-ЯМР-спектр (400МГц,DMSO-d6) δ: 9,56 (1H, уш с), 7,33-7,14 (10H, м), 7,08 (1H, дд, J=7,8, 1,1 Гц), 5,91 (1H, кв, J=6,1 Гц), 3,75 (3H, с), 1,58-1,40 (3H, м), 1,12 (2H, дд, J=5,7, 2,7 Гц), 0,61 (2H, дд, J=5,6, 2,5 Гц).
[0121] (Пример 6)
(R)-1-{4ʹ-[5-Хлор-3-({[1-(2,5-дифторфенил)этокси]карбонил}амино)тиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоксилат 1/2 кальция (соединение No. I-12)
[Chem. 29]
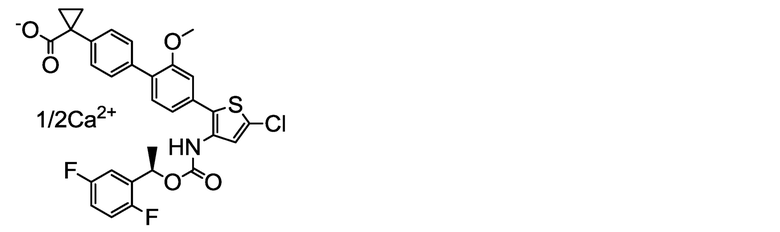
[0122] 0,170 мл (0,085 ммоль) 0,5 M водного раствора ацетата кальция добавляли к однородному раствору в ультрачистой воде (25 мл) 100 мг (0,165 ммоль) (R)-1-{4ʹ-[5-хлор-3-({[1-(2,5-дифторфенил)этокси]карбонил}амино)тиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоксилата натрия, полученного в примере 4. Смесь перемешивали при комнатной температуре в течение 2 дней. Полученную в результате суспензию фильтровали через мембранный фильтр (Millipore). Остаток промывали ультрачистой водой и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 47,8 мг (0,079 ммоль, выход 48%) в виде светло-желтого твердого остатка.
Масс-спектр (ESI+, m/z): 1205 [2M+1]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 9,55 (1H, уш с), 7,38-7,14 (10H, м), 7,07 (1H, дд, J=7,8, 1,6 Гц), 5,90 (1H, кв, J=6,4 Гц), 3,75 (3H, с), 1,60-1,40 (3H, м), 1,40-1,20 (2H, м), 0,91-0,78 (2H, м).
[0123] (Пример 7)
(R)-1-{4ʹ-[3-({[1-(2-Хлорфенил)этокси]карбонил}амино)-5-фтортиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоксилат натрия (соединение No. I-14)
[Chem. 30]

[0124] В бане со льдом и при перемешивании 2,00 мл (2,00 ммоль) 1N водного раствора гидроксида натрия добавляли к суспензии в ацетонитриле (80 мл) и ультрачистой воде (6 мл) 1,13 г (2,00 ммоль) (R)-1-{4ʹ-[3-({[1-(2-хлорфенил)этокси]карбонил}амино)-5-фтортиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты, полученной аналогично сравнительному примеру 31. Смесь обрабатывали ультразвуком, получая однородный раствор, который, затем, выдерживали при данной температуре в течение 3 часов. К жидкости реакционной смеси дополнительно добавляли небольшое количество ультрачистой воды. Растворитель удаляли лиофилизацией, и остаток сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 1,15 г (1,96 ммоль, выход 98%), в виде белого твердого остатка.
Масс-спектр (ESI+, m/z): 588 [M+1]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 9,55 (1H, уш с), 7,62-7,22 (9H, м), 7,14-7,20 (1H, м), 7,07 (1H, дд, J=7,8, 1,2 Гц), 6,84 (1H, д, J=2,5 Гц), 6,00 (1H, кв, J=5,6 Гц), 3,76 (3H, с), 1,59-1,35 (3H, м), 1,16 (2H, дд, J=5,7, 2,7 Гц), 0,66 (2H, дд, J=5,6, 2,6 Гц)
[0125] (Пример 8)
(R)-1-{4ʹ-[3-({[1-(2-Хлорфенил)этокси]карбонил}амино)-5-фтортиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоксилат калия (соединение No. I-16)
[Chem. 31]

[0126] При комнатной температуре 0,500 мл (0,500 ммоль) 1N водного раствора гидроксида калия добавляли к суспензии в ацетонитриле (20 мл) и ультрачистой воде (1,5 мл) 284 мг (0,501 ммоль) (R)-1-{4ʹ-[3-({[1-(2-хлорфенил)этокси]карбонил}амино)-5-фтортиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты, полученной аналогично сравнительному примеру 31. Смесь обрабатывали ультразвуком, получая однородный раствор, который, затем, выдерживали при комнатной температуре в течение 1 часа. К жидкости реакционной смеси дополнительно добавляли небольшое количество ультрачистой воды. Растворитель удаляли лиофилизацией, и остаток сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 270 мг (0,447 ммоль, выход 89%) в виде белого твердого остатка.
Масс-спектр (ESI+, m/z): 604 [M+1]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 9,56 (1H, уш с), 7,60-7,21 (9H, м), 7,19-7,14 (1H, м), 7,07 (1H, дд, J=7,9, 1,4 Гц), 6,84 (1H, д, J=2,4 Гц), 6,00 (1H, кв, J=6,4 Гц), 3,76 (3H, с), 1,55-1,37 (3H, м), 1,12 (2H, дд, J=5,8, 2,7 Гц), 0,61 (2H, дд, J=5,8, 2,6 Гц).
[0127] (Пример 9)
(R)-1-{4ʹ-[3-({[1-(2-Хлорфенил)этокси]карбонил}амино)-5-фтортиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоксилат 1/2 кальция (соединение No. I-18)
[Chem. 32]

[0128] При комнатной температуре и при перемешивании, 0,500 мл (0,500 ммоль) 1N водного раствора гидроксида натрия добавляли к суспензии в ацетонитриле (20 мл) и ультрачистой воде (1,5 мл) 282 мг (0,498 ммоль) (R)-1-{4ʹ-[3-({[1-(2-хлорфенил)этокси]карбонил}амино)-5-фтортиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты, полученной аналогично сравнительному примеру 31. Смесь обрабатывали ультразвуком, получая однородный раствор. Затем, 0,500 мл (0,085 ммоль) 0,5 M водного раствора ацетата кальция добавляли к жидкости реакционной смеси, и перемешивание осуществляли при комнатной температуре в течение 1 часа. Ацетонитрил отгоняли из жидкости реакционной смеси, и добавляли ультрачистую воду. Полученную в результате смесь перемешивали при комнатной температуре в течение 18 часов. Полученную в результате суспензию фильтровали, и остаток промывали ультрачистой водой и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 213 мг (0,365 ммоль, выход 73%), в виде белого твердого остатка.
Масс-спектр (ESI+, m/z): 1169 [2M+1]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 9,54 (1H, уш с), 7,58-7,25 (9H, м), 7,17 (1H, д, J=1,3 Гц), 7,07 (1H, дд, J=7,8, 1,1 Гц), 6,84 (1H, д, J=2,5 Гц), 6,00 (1H, кв, J=6,5 Гц), 3,75 (3H, с), 1,57-1,38 (3H, м), 1,38-1,18 (2H, м), 0,90-0,79 (2H, м).
[0129] (Пример 10)
(R)-1-{4ʹ-[5-Хлор-3-({[1-(тиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоксилат натрия (соединение No. I-20)
[Chem. 33]

[0130] При комнатной температуре и при перемешивании 2,00 мл (2,00 ммоль) 1N водного раствора гидроксида натрия добавляли к однородному раствору в ацетонитриле (80 мл) 1,05 г (2,00 ммоль) (R)-1-{4ʹ-[5-хлор-3-({[1-(тиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты, полученной аналогично сравнительному примеру 28. Смесь обрабатывали ультразвуком и перемешивали при данной температуре в течение 4 часов. Полученную в результате суспензию фильтровали, и остаток промывали маточным раствором и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 1,06 г (1,94 ммоль, выход 97%), в виде белого твердого остатка.
Масс-спектр (ESI+, m/z): 546 [M+1]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 9,34 (1H, уш с), 7,70-7,65 (2H, м), 7,57-7,40 (6H, м), 7,35-7,30 (2H, м), 7,23-7,16 (1H, м), 7,16-7,08 (1H, м), 5,82 (1H, кв, J=6,4 Гц), 1,61-1,40 (3H, м), 1,18 (2H, дд, J=5,8, 2,8 Гц), 0,68 (2H, дд, J=5,7, 2,7 Гц).
[0131] (Пример 11)
(R)-1-{4ʹ-[5-Хлор-3-({[1-(тиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоксилат калия (соединение No. I-22)
[Chem. 34]

[0132] При комнатной температуре и при перемешивании 0,500 мл (0,500 ммоль) 1N водного раствора гидроксида калия добавляли к однородному раствору в ацетонитриле (20 мл) 262 мг (0,500 ммоль) (R)-1-{4ʹ-[5-хлор-3-({[1-(тиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты, полученной аналогично сравнительному примеру 28. Смесь обрабатывали ультразвуком и перемешивали при комнатной температуре в течение 3,5 часов. Полученную в результате суспензию фильтровали через мембранный фильтр (Millipore). Остаток промывали маточным раствором и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 220 мг (0,392 ммоль, выход 78%), в виде белого твердого остатка.
Масс-спектр (ESI+, m/z): 562 [M+1]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 9,34 (1H, уш с), 7,71-7,64 (2H, м), 7,58-7,39 (6H, м), 7,33-7,27 (2H, м), 7,24-7,16 (1H, м), 7,16-7,07 (1H, м), 5,82 (1H, кв, J=6,5 Гц), 1,61-1,41 (3H, м), 1,13 (2H, дд, J=5,8, 2,7 Гц), 0,62 (2H, дд, J=5,8, 2,7 Гц).
[0133] (Пример 12)
(R)-1-{4ʹ-[5-Хлор-3-({[1-(тиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоксилат 1/2 кальция (соединение No. I-24)
[Chem. 35]

[0134] 0,190 мл (0,095 ммоль) 0,5 M водного раствора ацетата кальция добавляли к однородному раствору в ультрачистой воде (20 мл) и ацетонитриле (5 мл) 104 мг (0,190 ммоль) (R)-1-{4ʹ-[5-хлор-3-({[1-(тиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоксилата натрия, полученного в примере 10. Смесь перемешивали при комнатной температуре в течение 2 дней. Полученную в результате суспензию фильтровали через мембранный фильтр (Millipore). Остаток промывали небольшим количеством ацетонитрила и ультрачистой воды и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 47,8 мг (0,079 ммоль, выход 48%) в виде белого твердого остатка.
Масс-спектр (ESI+, m/z): 1085 [2M+1]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 9,32 (1H, уш с), 7,74-7,64 (2H, м), 7,59-7,49 (5H, м), 7,47-7,40 (1H, м), 7,40-7,34 (2H, м), 7,22-7,16 (1H, м), 7,16-7,08 (1H, м), 5,82 (1H, кв, J=6,4 Гц), 1,62-1,40 (3H, м), 1,40-1,20 (2H, м), 0,90-0,78 (2H, м).
[0135] (Пример 13)
(R)-1-{4ʹ-[5-фтор-3-({[1-(4-Метилтиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоксилат натрия (соединение No. I-26)
[Chem. 36]
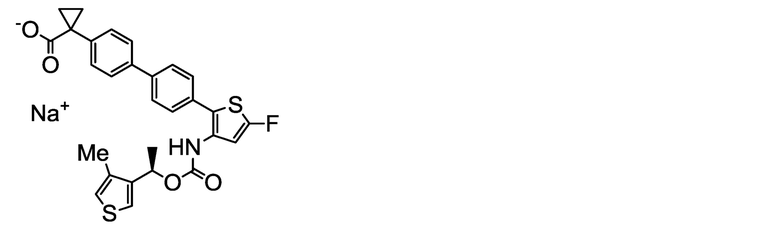
[0136] В бане со льдом и при перемешивании 2,00 мл (2,00 ммоль) 1N водного раствора гидроксида натрия добавляли к суспензии в ацетонитриле (80 мл) и ультрачистой воде (6 мл) 1,04 г (2,00 ммоль) (R)-1-{4ʹ-[5-фтор-3-({[1-(4-метилтиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты, полученной аналогично сравнительному примеру 30. Ацетонитрил (80 мл) и тетрагидрофуран (50 мл) дополнительно добавляли к жидкости реакционной смеси, и полученную в результате смесь обрабатывали ультразвуком при комнатной температуре в течение 30 минут и перемешивали при данной температуре в течение 25 часов. Полученную в результате суспензию фильтровали, и остаток сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 760 мг (1,40 ммоль, выход 70%) в виде белого твердого остатка.
Масс-спектр (ESI+, m/z): 544 [M+1]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 9,33 (1H, уш с), 7,69-7,63 (2H, м), 7,54-7,39 (5H, м), 7,35-7,29 (2H, м), 7,16 (1H, д, J=1,9 Гц), 6,83 (1H, уш с), 5,74 (1H, кв, J=6,5 Гц), 2,17 (3H, с), 1,59-1,43 (3H, м), 1,18 (2H, дд, J=5,8, 2,8 Гц), 0,68 (2H, дд, J=5,8, 2,8 Гц).
[0137] (Пример 14)
(R)-1-{4ʹ-[5-Фтор-3-({[1-(4-метилтиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоксилат калия (соединение No. I-28)
[Chem. 37]

[0138] При комнатной температуре и при перемешивании 0,500 мл (0,500 ммоль) 1N водного раствора гидроксида калия добавляли к однородному раствору в ацетонитриле (20 мл) и тетрагидрофуране (5 мл) 260 мг (0,499 ммоль) (R)-1-{4ʹ-[5-фтор-3-({[1-(4-метилтиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты, полученной аналогично сравнительному примеру 30. Смесь обрабатывали ультразвуком и перемешивали при комнатной температуре в течение 2 часов. Полученную в результате суспензию фильтровали через мембранный фильтр (Millipore). Остаток промывали небольшим количеством ацетонитрила и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 126 мг (0,226 ммоль, выход 45%), в виде белого твердого остатка.
Масс-спектр (ESI+, m/z): 560 [M+1]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 9,34 (1H, уш с), 7,69-7,63 (2H, м), 7,56-7,41 (5H, м), 7,34-7,27 (2H, м), 7,16 (1H, д, J=2,0 Гц), 6,83 (1H, уш с), 5,74 (1H, кв, J=6,5 Гц), 2,17 (3H, уш с), 1,57-1,45 (3H, м), 1,14 (2H, дд, J=5,9, 2,8 Гц), 0,63 (2H, дд, J=5,8, 2,6 Гц).
[0139] (Пример 15)
(R)-1-{4ʹ-[5-Фтор-3-({[1-(4-метилтиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоксилат 1/2 кальция (соединение No. I-30)
[Chem. 38]

[0140] 0,190 мл (0,095 ммоль) 0,5 M водного раствора ацетата кальция добавляли к однородному раствору в ультрачистой воде (25 мл) 103 мг (0,190 ммоль) R)-1-{4ʹ-[5-фтор-3-({[1-(4-метилтиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоксилата натрия (полученного аналогично примеру 13. Смесь перемешивали при комнатной температуре в течение 2 дней. Полученную в результате суспензию фильтровали через мембранный фильтр (Millipore). Остаток промывали ультрачистой водой и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 27,7 мг (0,051 ммоль, выход 27%), в виде белого твердого остатка.
Масс-спектр (ESI+, m/z): 1081 [2M+1]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 9,32 (1H, уш с), 7,70-7,63 (2H, м), 7,57-7,31 (8H, м), 7,14 (1H, д, J=1,9 Гц), 6,82 (1H, уш с), 5,74 (1H, кв, J=6,5 Гц), 2,17 (3H, с), 1,61-1,41 (3H, м), 1,41-1,20 (2H, м), 0,91-0,78 (2H, м).
[0141] [Сравнительные примеры]
(Сравнительный пример 1)
трет-Бутиловый эфир 2-бромтиофен-3-карбоновой кислоты
[Chem. 39]

[0142] В атмосфере азота 7,6 мл (87 ммоль) оксалилхлорида добавляли по каплям к раствору в хлористом метилене (70 мл) 15 г (72 ммоль) 2-бромтиофен-3-карбоновой кислоты (Aldrich) и 0,60 мл (7,8 ммоль) N,N-диметилформамида при комнатной температуре при перемешивании. Смесь перемешивали при данной температуре в течение 15 часов. После завершения реакции, жидкость реакционной смеси концентрировали при пониженном давлении. 2-Метил-2-пропанол (70 мл), 65 мл (372 ммоль) N,N-диизопропилэтиламина и 0,90 г (7,4 ммоль) N,N-диметиламинопиридина последовательно добавляли к остатку. В атмосфере азота, полученную в результате смесь перемешивали в течение 2 часов при нагревании при 80°C. После завершения реакции, жидкость реакционной смеси концентрировали при пониженном давлении. Добавляли к остатку воду, и смесь экстрагировали толуолом. Органическую фазу промывали насыщенным соляным раствором, сушили безводным сульфатом магния, и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=100:0-90:10 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая заявленное в заголовке соединение, весящее 12 г (32 ммоль (чистота 71% масс.), выход 45%), в виде светло-желтого масла.
Масс-спектр (EI, m/z): 262 [M]+.
1H-ЯМР-спектр (400МГц, CDCl3) δ: 7,32 (1H, д, J=5,8 Гц), 7,18 (1H, д, J=5,8 Гц), 1,59 (9H, с).
[0143] Заявленное в заголовке соединение также получали следующим способом.
[0144] В атмосфере аргона, 1,80 г (9,70 ммоль) п-толуолсульфонилхлорида добавляли маленькими порциями к раствору в пиридине (9,6 мл) 1,005 г (4,85 ммоль) 2-бромтиофен-3-карбоновой кислоты (Aldrich) в бане со льдом при перемешивании. Затем, добавляли 0,46 мл (4,8 ммоль) 2-метил-2-пропанола. В бане со льдом, смесь перемешивали в течение 2 часов. Перемешивание дополнительно проводили при комнатной температуре в течение 1 часа. После этого, добавляли 0,47 мл (5,0 ммоль) 2-метил-2-пропанола, и смесь перемешивали при комнатной температуре в течение 27 часов. После завершения реакции, жидкость реакционной смеси концентрировали при пониженном давлении. Добавляли этилацетат и насыщенный водный раствор гидрокарбоната натрия, осуществляя разделение жидкостей. Органическую фазу промывали насыщенным водным раствором гидрокарбоната натрия, и затем насыщенным соляным раствором. Затем, органическую фазу промывали 5% масс. водным раствором гидросульфата калия и снова промывали насыщенным соляным раствором. Органическую фазу сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=99:1-94:6 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая заявленное в заголовке соединение, весящее 1,22 г (4,64 ммоль, выход 96%), в виде светло-желтого масла.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 7,63 (1H, д, J=5,8 Гц), 7,28 (1H, д, J=5,8 Гц), 1,53 (9H, с).
[0145] (Сравнительный пример 2)
2-Бром-5-хлортиофен-3-карбоксамид
[Chem. 40]

[0146] В атмосфере аргона и при перемешивании 8,70 г (65,2 ммоль) N-хлорсукцинимида добавляли к раствору в N,N-диметилформамиде (50 мл) 4,48 г (21,7 ммоль) 2-бромтиофен-3-карбоксамида (полученного согласно WO 10/036497). Смесь перемешивали в течение 3 часов при нагревании при 60°C. После завершения реакции, 50 мл воды и 100 мл этилацетата добавляли к ней в бане со льдом. При перемешивании, добавляли 6,80 г (65,3 ммоль) гидросульфита натрия. Смесь перемешивали при комнатной температуре в течение 15 минут. После этого, добавляли воду, осуществляя разделение жидкостей. Органическую фазу промывали два раза 50 мл насыщенного водного раствора гидрокарбоната натрия. Затем, органическую фазу промывали насыщенным соляным раствором и сушили безводным сульфатом магния. Растворитель концентрировали при пониженном давлении до приблизительно половины объема. Добавляли к полученной в результате суспензии гексан, и смесь обрабатывали ультразвуком. Затем, твердый остаток собирали фильтрованием, промывали гексаном и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 3,64 г (15,1 ммоль, выход 70%), в виде белого твердого остатка.
Масс-спектр (DUIS+, m/z): 240 [M+1]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 7,75 (1H, уш с), 7,58 (1H, уш с), 7,33 (1H, с).
[0147] Заявленное в заголовке соединение также получали следующим способом.
[0148] В атмосфере азота, 0,90 г (6,7 ммоль) N-хлорсукцинимида добавляли к раствору в N,N-диметилформамиде (16 мл) 1,0 г (4,8 ммоль) 2-бромтиофен-3-карбоновой кислоты (Aldrich) при комнатной температуре при перемешивании. Смесь перемешивали в течение 1 часа при нагревании при 80°C. После завершения реакции, жидкость реакционной смеси охлаждали. Добавляли воду, и жидкость подкисляли добавлением 2N хлористоводородной кислоты. Смесь экстрагировали этилацетатом. Органическую фазу промывали последовательно водным раствором гидросульфита натрия и насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток растворяли в хлористом метилене (15 мл), и 0,80 мл (9,1 ммоль) оксалилхлорида добавляли по каплям к раствору в атмосфере азота при 0°C при перемешивании. Температуру повышали до комнатной температуры, и смесь перемешивали в течение 30 минут. Затем, 3,7 мл (48 ммоль) 28% масс. водного аммиака добавляли по каплям при комнатной температуре при перемешивании, и смесь перемешивали при комнатной температуре в течение 1 часа. После завершения реакции, добавляли к жидкости реакционной смеси воду, и смесь экстрагировали этилацетатом. Органическую фазу промывали насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=69:31-48:52 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 0,62 г (2,6 ммоль, выход 53%), в виде белого твердого остатка.
[0149] (Сравнительный пример 3)
4-Бром-1-йод-2-метоксибензол
[Chem. 41]

[0150] В бане со льдом и при перемешивании 0,75 г (11 ммоль) нитрита натрия добавляли к раствору в уксусной кислоте (15 мл) и концентрированной хлористоводородной кислоте (1 мл) 2,0 г (9,0 ммоль) 4-бром-2-метоксианилина (Tokyo Chemical Industry Co., Ltd.) таким способом, чтобы температура внутри колбы не превышала 10°C. Смесь перемешивали при комнатной температуре в течение 30 минут. Затем, жидкость реакционной смеси добавляли по каплям к водному раствору 1,0 г (30 ммоль) йодида калия в 48% масс. водного раствора бромистоводородной кислоты (30 мл) при комнатной температуре при перемешивании. Полученную в результате смесь перемешивали при данной температуре в течение 1 часа. После завершения реакции, жидкость реакционной смеси добавляли маленькими порциями к смеси водного раствора карбоната натрия и хлористого метилена. После подтверждения основности водной среды, жидкость отделяли. Органическую фазу промывали последовательно 10% масс. водным раствором гидросульфита натрия, насыщенным водным раствором гидрокарбоната натрия и насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=100:0-91:9 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая заявленное в заголовке соединение, весящее 2,2 г (7,2 ммоль, выход 73%), в виде оранжевого твердого остатка.
Масс-спектр (EI, m/z): 312 [M]+.
1H-ЯМР-спектр (400МГц, CDCl3) δ: 7,61 (1H, д, J=8,3 Гц), 6,94 (1H, д, J=2,1 Гц), 6,87 (1H, дд, J=8,2, 2,1 Гц), 3,88 (3H, с).
[0151] (Сравнительный пример 4)
Этиловый эфир 1-(4ʹ-бром-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил)циклопропанкарбоновой кислоты
[Chem. 42]

[0152] Раствор в 1,4-диоксане (15 мл) и воде (10 мл) 1,2 г (3,8 ммоль) 4-бром-1-йод-2-метоксибензола, полученного аналогично сравнительному примеру 3, 1,1 г (3,5 ммоль) этилового эфира 1-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]циклопропанкарбоновой кислоты (полученного согласно способу, описанному в WO 12/078593) и 1,1 г (10 ммоль) карбоната натрия дегазировали и продували азотом. Затем, добавляли 0,10 г (0,12 ммоль) продукта присоединения хлористого метилена к дихлориду [1,1ʹ-бис(дифенилосфино)ферроцен]палладия (II). Смесь перемешивали в атмосфере азота в течение 1,5 часов при нагревании при 80°C. После завершения реакции, добавляли к жидкости реакционной смеси воду, и смесь экстрагировали этилацетатом. Органическую фазу промывали насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=94:6-75:25 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 0,72 г (1,9 ммоль, выход 55%), в виде белого твердого остатка.
Масс-спектр (EI, m/z): 374 [M]+.
1H-ЯМР-спектр (400МГц, CDCl3) δ: 7,45-7,41 (2H, м), 7,39-7,35 (2H, м), 7,19 (1H, д, J=8,0 Гц), 7,15 (1H, дд, J=8,0, 1,8 Гц), 7,10 (1H, д, J=1,8 Гц), 4,12 (2H, кв, J=7,1 Гц), 3,81 (3H, с), 1,61 (2H, дд, J=7,0, 4,0 Гц), 1,22 (2H, дд, J=7,0, 4,0 Гц), 1,19 (3H, т, J=7,1 Гц).
[0153] (Сравнительный пример 5)
Этиловый эфир 1-(4ʹ-хлор-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил)циклопропанкарбоновой кислоты
[Chem. 43]

[0154] Раствор в 1,4-диоксане (20 мл) и воде (20 мл) 2,0 г (9,0 ммоль) 1-бром-4-хлор-2-метоксибензола (Tokyo Chemical Industry Co., Ltd.), 2,6 г этилового эфира (8,2 ммоль) 1-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]циклопропанкарбоновой кислоты (полученного согласно способу, описанному в WO 12/078593) и 2,7 г (25 ммоль) карбоната натрия дегазировали и продували азотом. Затем, добавляли 0,21 г (0,25 ммоль) продукта присоединения хлористого метилена к дихлориду [1,1ʹ-бис(дифенилосфино)ферроцен]палладия (II). Смесь перемешивали в атмосфере азота в течение 2 часов при нагревании при 80°C. После завершения реакции, добавляли к жидкости реакционной смеси воду, и смесь экстрагировали этилацетатом. Органическую фазу промывали насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле, и фракцию, имеющую Rf=0,5 (проявляющий растворитель: гексан:этилацетат=90:10 (об./об.)), концентрировали при пониженном давлении и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 2,46 г (7,4 ммоль, выход 90%), в виде белого твердого остатка.
Масс-спектр (EI, m/z): 330 [M]+.
1H-ЯМР-спектр (400МГц, CDCl3) δ: 7,45-7,41 (2H, м), 7,39-7,35 (2H, м), 7,25 (1H, д, J=8,2 Гц), 7,00 (1H, дд, J=8,2, 2,0 Гц), 6,96 (1H, д, J=2,0 Гц), 4,12 (2H, кв, J=7,1 Гц), 3,81 (3H, с), 1,61 (2H, дд, J=6,9, 3,9 Гц), 1,22 (2H, дд, J=7,0, 4,0 Гц), 1,19 (3H, т, J=7,1 Гц).
[0155] (Сравнительный пример 6)
Этиловый эфир 1-[2ʹ-метокси-4ʹ-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-[1,1ʹ-бифенил]-4-ил]циклопропанкарбоновой кислоты
[Chem. 44]

[0156] Раствор в 1,4-диоксане (10 мл) 0,72 г (1,9 ммоль) этилового эфира 1-(4ʹ-бром-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил)циклопропанкарбоновой кислоты, полученного аналогично сравнительному примеру 4, 0,60 г (2,4 ммоль) 4,4,4ʹ,4ʹ,5,5,5ʹ,5ʹ-октаметил-2,2ʹ-би(1,3,2-диоксаборолана) и 0,30 г (3,1 ммоль) ацетата калия дегазировали и продували азотом. Затем, добавляли 0,10 г (0,12 ммоль) продукта присоединения хлористого метилена к дихлориду [1,1ʹ-бис(дифенилосфино)ферроцен]палладия (II). В атмосфере азота, смесь перемешивали в течение 3 часов при кипячении с обратным холодильником. После завершения реакции, добавляли к жидкости реакционной смеси воду, и смесь экстрагировали толуолом. Органическую фазу промывали насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=92:8-79:21 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 0,81 г (1,9 ммоль, количественный выход), в виде светло-желтого твердого остатка.
Масс-спектр (EI, m/z): 422 [M]+.
1H-ЯМР-спектр (400МГц,CDCl3) δ: 7,52-7,46 (3H, м), 7,40-7,33 (4H, м), 4,12 (2H, кв, J=7,1 Гц), 3,86 (3H, с), 1,60 (2H, дд, J=6,9, 3,9 Гц), 1,36 (12H, с), 1,22 (2H, дд, J=7,0, 4,0 Гц), 1,19 (3H, т, J=7,1 Гц).
[0157] Заявленное в заголовке соединение также получали следующим способом.
[0158] Раствор в 1,4-диоксане (30 мл) 2,46 г (7,43 ммоль) этилового эфира 1-(4ʹ-хлор-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил)циклопропанкарбоновой кислоты, полученного аналогично сравнительному примеру 5, 2,43 г (9,57 ммоль) 4,4,4ʹ,4ʹ,5,5,5ʹ,5ʹ-октаметил-2,2ʹ-би(1,3,2-диоксаборолана) и 1,1 г (11 ммоль) ацетата калия дегазировали и продували азотом. Затем, добавляли 0,30 г (0,37 ммоль) продукта присоединения хлористого метилена к дихлориду [1,1ʹ-бис(дифенилосфино)ферроцен]палладия (II) и 0,30 г (1,1 ммоль) трициклогексилфосфина. Смесь перемешивали в атмосфере азота в течение 24 часов при кипячении с обратным холодильником. Затем, добавляли к жидкости реакционной смеси 0,15 г (0,18 ммоль) продукта присоединения хлористого метилена к дихлориду [1,1ʹ-бис(дифенилосфино)ферроцен]палладия (II) и 0,15 г (0,54 ммоль) трициклогексилфосфина, и смесь перемешивали в атмосфере азота в течение 5 часов при кипячении с обратным холодильником. После завершения реакции, жидкость реакционной смеси охлаждали до комнатной температуры. Добавляли толуол, и нерастворимый материал отфильтровывали. Фильтрат промывали последовательно насыщенным водным раствором гидрокарбоната натрия и насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=92:8-79:21 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении. К остатку добавляли гексан. Твердый остаток собирали фильтрованием, промывали гексаном и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 1,89 г (4,5 ммоль, выход 60%), в виде белого твердого остатка.
[0159] (Сравнительный пример 7)
Этиловый эфир 1-[4ʹ-(3-карбамоил-5-хлортиофен-2-ил)-[1,1ʹ-бифенил]-4-ил]циклопропанкарбоновой кислоты
[Chem. 45]

[0160] Раствор в 1,4-диоксане (15 мл) и воде (5 мл) 486 мг (2,02 ммоль) 2-бром-5-хлортиофен-3-карбоксамида, полученного аналогично сравнительному примеру 2, 877 мг (2,23 ммоль) этилового эфира 1-[4ʹ-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-[1,1ʹ-бифенил]-4-ил]циклопропанкарбоновой кислоты (полученного согласно способу, описанному в WO 12/078593) и 658 мг (6,21 ммоль) карбоната натрия дегазировали замораживанием в бане с сухим льдом и ацетоном и продували аргоном. Затем, добавляли 230 мг (0,199 ммоль) тетракис(трифенилфосфин)палладия (0), и смесь перемешивали в течение 3 часов при нагревании при 90°C. После завершения реакции, жидкость реакционной смеси охлаждали. Добавляли этилацетат и воду, осуществляя разделение жидкостей. Органическую фазу сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=64:36-43:57 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении. Добавляли к остатку раствор гексан-этилацетат (2:1 (об./об.)), и выпавший твердый остаток собирали фильтрованием и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 705 мг (1,66 ммоль, выход 82%), в виде белого твердого остатка.
Масс-спектр (EI, m/z): 425 [M]+.
1H-ЯМР-спектр (400МГц, CDCl3) δ: 7,69-7,65 (2H, м), 7,58-7,53 (4H, м), 7,46-7,42 (2H, м), 7,33 (1H, с), 5,44 (2H, уш с), 4,12 (2H, кв, J=7,1 Гц), 1,65 (2H, дд, J=7,0, 4,0 Гц), 1,23 (2H, дд, J=7,0, 4,0 Гц), 1,19 (3H, т, J=7,1 Гц).
[0161] (Сравнительный пример 8)
Этиловый эфир 1-[4ʹ-(3-карбамоил-5-хлортиофен-2-ил)-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил]циклопропанкарбоновой кислоты
[Chem. 46]

[0162] Раствор в 1,4-диоксане (30 мл) и воде (10 мл) 2,0 г (4,7 ммоль) этилового эфира 1-[2ʹ-метокси-4ʹ-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-[1,1ʹ-бифенил]-4-ил]циклопропанкарбоновой кислоты, полученного аналогично сравнительному примеру 6, 1,25 г (5,2 ммоль) 2-бром-5-хлортиофен-3-карбоксамида, полученного аналогично сравнительному примеру 2, и 1,5 г (14 ммоль) карбоната натрия дегазировали. Затем, добавляли 0,30 г (0,26 ммоль) тетракис(трифенилфосфин)палладия (0). Смесь перемешивали в атмосфере азота в течение 4,5 часов при нагревании при 90°C. После завершения реакции, жидкость реакционной смеси охлаждали. Добавляли воду, и смесь экстрагировали этилацетатом. Органическую фазу промывали насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. К остатку добавляли этилацетат. Твердый остаток собирали фильтрованием, промывали небольшим количеством этилацетата и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 1,53 г (3,4 ммоль, выход 71%), в виде белого твердого остатка.
Масс-спектр (EI, m/z): 455 [M]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 7,73 (1H, уш с), 7,50 (1H, уш с), 7,48-7,43 (2H, м), 7,39-7,33 (3H, м), 7,32 (1H, с), 7,25 (1H, д, J=1,6 Гц), 7,13 (1H, дд, J=7,8, 1,6 Гц), 4,05 (2H, кв, J=7,1 Гц), 3,79 (3H, с), 1,51 (2H, дд, J=6,8, 4,0 Гц), 1,23 (2H, дд, J=7,0, 4,0 Гц), 1,12 (3H, т, J=7,1 Гц).
[0163] (Сравнительный пример 9)
трет-Бутиловый эфир 2-{4ʹ-[1-(этоксикарбонил)циклопропил]-[1,1ʹ-бифенил]-4-ил}тиофен-3-карбоновой кислоты
[Chem. 47]

[0164] Раствор в 1,4-диоксане (15 мл) и воде (5 мл) 0,80 г (2,0 ммоль) этилового эфира 1-[4ʹ-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-[1,1ʹ-бифенил]-4-ил]циклопропанкарбоновой кислоты (полученного согласно способу, описанному в WO 12/078593), 0,50 г (1,9 ммоль) трет-бутилового эфира 2-бромтиофен-3-карбоновой кислоты, полученного аналогично сравнительному примеру 1, и 0,61 г (5,8 ммоль) карбоната натрия дегазировали. Затем, добавляли 0,10 г (0,089 ммоль) тетракис(трифенилфосфин)палладия (0). Смесь перемешивали в атмосфере азота в течение 14,5 часов при нагревании при 90°C. После завершения реакции, жидкость реакционной смеси охлаждали. Добавляли воду, и смесь экстрагировали этилацетатом. Органическую фазу промывали насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле, и фракцию, имеющую Rf=0,41 (проявляющий растворитель: гексан:этилацетат=90:10 (об./об.)), концентрировали при пониженном давлении и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 0,58 г (1,3 ммоль, выход 68%), в виде белого твердого остатка.
Масс-спектр (EI, m/z): 448 [M]+.
1H-ЯМР-спектр (400МГц, CDCl3) δ: 7,64-7,60 (2H, м), 7,59-7,55 (2H, м), 7,55-7,51 (2H, м), 7,48 (1H, д, J=5,4 Гц), 7,45-7,41 (2H, м), 7,23 (1H, д, J=5,3 Гц), 4,13 (2H, кв, J=7,1 Гц), 1,64 (2H, дд, J=6,9, 3,9 Гц), 1,38 (9H, с), 1,23 (2H, дд, J=7,2, 4,1 Гц), 1,20 (3H, т, J=7,2 Гц).
[0165] (Сравнительный пример 10)
трет-Бутиловый эфир 2-{4ʹ-[1-(этоксикарбонил)циклопропил]-2-метокси-[1,1ʹ-бифенил]-4-ил}тиофен-3-карбоновой кислоты
[Chem. 48]

[0166] 2,96 г (27,9 ммоль) карбоната натрия добавляли к раствору в 1,4-диоксане (23 мл) и воде (23 мл) 3,81 г (9,02 ммоль) этилового эфира 1-[2ʹ-метокси-4ʹ-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-[1,1ʹ-бифенил]-4-ил]циклопропанкарбоновой кислоты, полученного аналогично сравнительному примеру 6, и 2,7 г (7,3 ммоль (чистота 71% масс.)) трет-бутилового эфира 2-бромтиофен-3-карбоновой кислоты, полученного в сравнительном примере 1. Смесь дегазировали. Затем, добавляли 540 мг (0,467 ммоль) тетракис(трифенилфосфин)палладия (0). Полученную в результате смесь перемешивали в атмосфере азота в течение 7 часов при нагревании при 90°C. После завершения реакции, жидкость реакционной смеси охлаждали. Добавляли воду, и смесь экстрагировали этилацетатом. Органическую фазу промывали насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. К остатку добавляли этилацетат. Полученный в результате нерастворимый материал удаляли фильтрованием и промывали смесью гексан-этилацетат (1:2 (об./об.)). Затем, маточный раствор и промывки объединяли и концентрировали при пониженном давлении. Концентрат растворяли в этилацетате, и добавляли гексан до того, как раствор становился мутным. Выпавший твердый остаток отфильтровывали, промывали гексаном и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 3,09 г (5,55 ммоль (чистота 86% масс.), выход 61%), в виде белого твердого остатка.
Масс-спектр (EI, m/z): 478 [M]+.
1H-ЯМР-спектр (400МГц, CDCl3) δ: 7,53-7,48 (2H, м), 7,47 (1H, д, J=5,4 Гц), 7,41-7,36 (2H, м), 7,34 (1H, д, J=7,8 Гц), 7,23 (1H, д, J=5,4 Гц), 7,12 (1H, дд, J=7,7, 1,6 Гц), 7,07 (1H, д, J=1,6 Гц), 4,13 (2H, кв, J=7,1 Гц), 3,83 (3H, с), 1,62 (2H, дд, J=6,8, 4,0 Гц), 1,40 (9H, с), 1,23 (2H, дд, J=6,8, 3,8 Гц), 1,20 (3H, т, J=7,1 Гц).
[0167] (Сравнительный пример 11)
трет-Бутиловый эфир 2-{4ʹ-[1-(этоксикарбонил)циклопропил]-[1,1ʹ-бифенил]-4-ил}-5-фтортиофен-3-карбоновой кислоты
[Chem. 49]

[0168] В атмосфере аргона 11 мл (12,0 ммоль) 1,09 M раствора в тетрагидрофуране и гексане диизопропиламида лития (Kanto Chemical Co., Inc.) добавляли по каплям в течение 5 минут к раствору в сухом тетрагидрофуране (60 мл) 4,50 г (10,0 ммоль) трет-бутилового эфира 2-{4ʹ-[1-(этоксикарбонил)циклопропил]-[1,1ʹ-бифенил]-4-ил}тиофен-3-карбоновой кислоты, полученного аналогично сравнительному примеру 9, при охлаждении системы до -70°C или ниже в бане с сухим льдом и ацетоном. Смесь перемешивали при данной температуре в течение 30 минут. Затем, при охлаждении системы до -65°C или ниже, раствор в тетрагидрофуране (15 мл) 4,75 г (15,1 ммоль) N-фторбензолсульфонимида добавляли по каплям в течение 5 минут, и смесь перемешивали при данной температуре в течение 30 минут. Затем, температуру постепенно повышали, и реакцию прекращали при -45°C добавлением 40 мл насыщенного водного раствора хлорида аммония. Температуру повышали до комнатной температуры, и смесь экстрагировали этилацетатом. Органическую фазу сушили безводным сульфатом магния и концентрировали при пониженном давлении. К остатку добавляли хлористый метилен. Нерастворимый материал отфильтровывали, и фильтрат концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=100:0-79:21 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 2,23 г (4,78 ммоль, выход 48%), в виде белого твердого остатка.
Масс-спектр (CI, m/z): 467 [M+1]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 7,78-7,72 (2H, м), 7,68-7,63 (2H, м), 7,56-7,50 (2H, м), 7,46-7,41 (2H, м), 7,03 (1H, д, J=2,4 Гц), 4,05 (2H, кв, J=7,1 Гц), 1,52 (2H, дд, J=6,9, 3,9 Гц), 1,31 (9H, с), 1,24 (2H, дд, J=7,1, 4,1 Гц), 1,12 (3H, т, J=7,1 Гц).
[0169] (Сравнительный пример 12)
трет-Бутиловый эфир 2-{4ʹ-[1-(этоксикарбонил)циклопропил]-2-метокси-[1,1ʹ-бифенил]-4-ил}-5-фтортиофен-3-карбоновой кислоты
[Chem. 50]
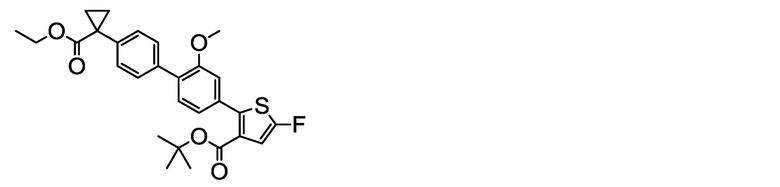
[0170] В атмосфере аргона, 6,56 мл (7,22 ммоль) 1,1 M раствор диизопропиламида литий в тетрагидрофуране добавляли по каплям к раствору в тетрагидрофуране (37 мл) 2,88 г (5,17 ммоль (чистота 86% масс.)) трет-бутилового эфира 2-{4ʹ-[1-(этоксикарбонил)циклопропил]-2-метокси-[1,1ʹ-бифенил]-4-ил}тиофен-3-карбоновой кислоты, полученного в сравнительном примере 10, при -78°C при перемешивании. Смесь перемешивали при данной температуре в течение 30 минут. Затем, добавляли по каплям раствор в тетрагидрофуране (9,5 мл) 2,85 г (9,04 ммоль) N-фторбензолсульфонимида, и смесь перемешивали при данной температуре в течение 30 минут. После завершения реакции, добавляли к жидкости реакционной смеси насыщенный водный раствор хлорида аммония и этилацетат. Органическую фазу отделяли. Органическую фазу промывали насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:дихлорэтан=100:0-30:70 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении. К остатку добавляли гексан, и смесь нагревали, получая раствор, который обрабатывали ультразвуком. Выпавший твердый остаток собирали фильтрованием, промывали гексаном и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 496 мг (1,00 ммоль, выход 20%) в виде белого твердого остатка.
Масс-спектр (DUIS+, m/z): 497 [M+1]+.
1H-ЯМР-спектр (400МГц, CDCl3) δ: 7,52-7,46 (2H, м), 7,41-7,36 (2H, м), 7,33 (1H, д, J=7,8 Гц), 7,08 (1H, дд, J=7,8, 1,6 Гц), 7,03 (1H, д, J=1,5 Гц), 6,84 (1H, д, J=2,3 Гц), 4,13 (2H, кв, J=7,1 Гц), 3,82 (3H, с), 1,62 (2H, дд, J=6,9, 3,9 Гц), 1,37 (9H, с), 1,23 (2H, дд, J=6,5, 3,5 Гц), 1,20 (3H, т, J=7,2 Гц).
[0171] (Сравнительный пример 13)
2-{4ʹ-[1-(Этоксикарбонил)циклопропил]-[1,1ʹ-бифенил]-4-ил}-5 фтортиофен-3-карбоновая кислота
[Chem. 51]
[0172] В атмосфере аргона 5,0 мл (65 ммоль) трифторуксусной кислоты добавляли к раствору в хлористом метилене (20 мл) 2,18 г (4,67 ммоль) трет-бутилового эфира 2-{4ʹ-[1-(этоксикарбонил)циклопропил]-[1,1ʹ-бифенил]-4-ил}-5-фтортиофен-3 карбоновой кислоты, полученного аналогично сравнительному примеру 11, в бане со льдом при перемешивании. Смесь перемешивали при данной температуре в течение 1 часа и дополнительно при комнатной температуре в течение 2 часов. После завершения реакции, смесь концентрировали при пониженном давлении. Остаток промывали последовательно диэтиловым эфиром и гексаном, и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 1,87 г (4,56 ммоль, выход 98%), в виде белого твердого остатка.
Масс-спектр (CI, m/z): 411 [M+1]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 12,92 (1H, с), 7,74-7,70 (2H, м), 7,68-7,64 (2H, м), 7,60-7,55 (2H, м), 7,46-7,41 (2H, м), 7,05 (1H, д, J=2,4 Гц), 4,05 (2H, кв, J=7,1 Гц), 1,52 (2H, дд, J=6,9, 3,9 Гц), 1,24 (2H, дд, J=7,0, 4,1 Гц), 1,11 (3H, т, J=7,1 Гц).
[0173] (Сравнительный пример 14)
2-{4ʹ-[1-(Этоксикарбонил)циклопропил]-2-метокси-[1,1ʹ-бифенил]-4-ил}-5 фтортиофен-3-карбоновая кислота
[Chem. 52]
[0174] В атмосфере аргона при комнатной температуре 1,1 мл (14 ммоль) трифторуксусной кислоты добавляли к раствору в хлористом метилене (4,4 мл) 491 мг (0,989 ммоль) трет-бутилового эфира 2-{4ʹ-[1-(этоксикарбонил)циклопропил]-2-метокси-[1,1ʹ-бифенил]-4-ил}-5-фтортиофен-3-карбоновой кислоты, полученного аналогично сравнительному примеру 12. Смесь перемешивали при комнатной температуре в течение 3 часов. После завершения реакции, жидкость реакционной смеси концентрировали при пониженном давлении. Добавляли хлористый метилен, и смесь концентрировали при пониженном давлении. К остатку добавляли гексан, и смесь концентрировали при пониженном давлении и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 436 мг (0,99 ммоль, количественный выход), в виде белого твердого остатка.
Масс-спектр (EI, m/z): 440 [M]+.
1H-ЯМР спектр (400МГц, CDCl3) δ: 7,53-7,48 (2H, м), 7,40-7,37 (2H, м), 7,34 (1H, д, J=7,8 Гц), 7,14 (1H, д, J=1,6 Гц), 7,13-7,10 (1H, м), 6,94 (1H, д, J=2,3 Гц), 4,12 (2H, кв, J=7,1 Гц), 3,82 (3H, s), 1,62 (2H, дд, J=6,9, 3,9 Гц), 1,23 (2H, дд, J=7,0, 4,0 Гц), 1,20 (3H, т, J=7,1 Гц).
[0175] (Сравнительный пример 15)
1-[4ʹ-(3-Карбамоил-5-хлортиофен-2-ил)-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил]циклопропанкарбоновая кислота
[Chem. 53]

[0176] При комнатной температуре и при перемешивании 6,6 мл (6,6 ммоль) 1N водного раствора гидроксида натрия добавляли к суспензии в этаноле (10 мл) и тетрагидрофуране (10 мл) 1,00 г (2,19 ммоль) этилового эфира 1-[4ʹ-(3-карбамоил-5-хлортиофен-2-ил)-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил]циклопропанкарбоновой кислоты, полученного аналогично сравнительному примеру 8. Смесь перемешивали при данной температуре в течение 4 дней. После завершения реакции, жидкость реакционной смеси нейтрализовали добавлением 6,6 мл (6,6 ммоль) 1N хлористоводородной кислоты. Добавляли воду, и выпавший твердый остаток отфильтровывали через мембранный фильтр (Millipore), промывали водой, и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 884 мг (2,07 ммоль, выход 94%), в виде белого твердого остатка.
Масс-спектр (DUIS+, m/z): 428 [M+1]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 12,35 (1H, уш с), 7,75 (1H, уш с), 7,52 (1H, уш с), 7,45-7,41 (2H, м), 7,38-7,35 (2H, м), 7,33 (1H, д, J=7,9 Гц), 7,32 (1H, с), 7,24 (1H, д, J=1,8 Гц), 7,13 (1H, дд, J=7,8, 1,6 Гц), 3,79 (3H, с), 1,47 (2H, дд, J=6,7, 3,8 Гц), 1,18 (2H, дд, J=6,9, 3,9 Гц).
[0177] (Сравнительный пример 16)
2-(Триметилсилил)этиловый эфир 1-[4ʹ-(3-карбамоил-5-хлортиофен-2-ил)-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил]циклопропанкарбоновой кислоты
[Chem. 54]
[0178] Толуол добавляли к 0,80 г (1,9 ммоль) 1-[4ʹ-(3-карбамоил-5-хлортиофен-2-ил)-2ʹ-метокси-[1,1ʹ-бифенил]-4 ил]циклопропанкарбоновой кислоты, полученной в сравнительном примере 15. После проведения азеотропного осушения, атмосферу заменяли на аргон. Затем, добавляли N,N-диметилформамид (10 мл), 23,0 мг (0,188 ммоль) N,N-диметиламинопиридина, 0,42 мл (2,8 ммоль) триметилсилилэтанола и 0,98 мл (5,6 ммоль) N,N-диизопропилэтиламина. Затем, в бане со льдом добавляли 1,06 г (2,81 ммоль) гексафторфосфатной соли о-(бензотриазол-1-ил)-N,N,Nʹ,Nʹ-тетраметилурония. Смесь перемешивали при комнатной температуре в течение 18 часов. Затем, добавляли к жидкости реакционной смеси 64,6 мг (0,529 ммоль) N,N-диметиламинопиридина, и смесь перемешивали при данной температуре в течение 1 дня. После завершения реакции, добавляли к жидкости реакционной смеси воду и этилацетат. Экстракцию проводили два раза этилацетатом. Органическую фазу сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=71:29-50:50 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 889 мг (1,68 ммоль, выход 90%), в виде светло-желтого твердого остатка.
Масс-спектр (DUIS+, m/z): 528 [M+1]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 7,75 (1H, уш с), 7,52 (1H, уш с), 7,46-7,42 (2H, м), 7,38-7,34 (2H, м), 7,33 (1H, д, J=7,8 Гц), 7,32 (1H, с), 7,24 (1H, д, J=1,6 Гц), 7,13 (1H, дд, J=7,8, 1,7 Гц), 4,14-4,07 (2H, м), 3,78 (3H, с), 1,49 (2H, дд, J=6,8, 3,8 Гц), 1,22 (2H, дд, J=7,0, 4,1 Гц), 0,91-0,84 (2H, м), -0,05 (9H, с).
[0179] (Сравнительный пример 17)
1-(4-Метилтиофен-3-ил)этанон
[Chem. 55]

[0180] В атмосфере аргона при -78°C 4,0 мл (6,4 ммоль) 1,6 M раствора в гексане н-бутиллития добавляли по каплям к раствору в диэтиловом эфире (23 мл) 1,0 г (5,3 ммоль) 3-бром-4-метилтиофена (Tokyo Chemical Industry Co., Ltd.). Смесь перемешивали при данной температуре в течение 15 минут. Затем, добавляли по каплям при -78°C раствор в диэтиловом эфире (1 мл) 0,70 мл (6,9 ммоль) N-метокси-N-метилацетамида, и смесь перемешивали при данной температуре в течение 15 минут и при комнатной температуре в течение 23 часов. После завершения реакции добавляли к жидкости реакционной смеси насыщенный водный раствор хлорида аммония, и полученную в результате смесь экстрагировали этилацетатом. Органическую фазу промывали водой, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=80:20 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая заявленное в заголовке соединение, весящее 552 мг (3,94 ммоль, выход 75%), в виде светло-желтого масла.
Масс-спектр (CI, m/z): 141 [M+1]+.
1H-ЯМР-спектр (400МГц, CDCl3) δ: 7,99 (1H, д, J=3,1 Гц), 6,91 (1H, dq, J=3,1, 1,0 Гц), 2,53 (3H, с), 2,46 (3H, д, J=1,0 Гц).
[0181] Сравнительный пример 18)
(RS)-1-(2,5-дифторфенил)этанол
[Chem. 56]

[0182] При перемешивании 10 г (260 ммоль) боргидрида натрия добавляли к раствору в этаноле (200 мл) 39,0 г (250 ммоль) 1-(4-фтор-2-метилфенил)этанона (комбинация продуктов Wako Pure Chemical Industries, Ltd. и Tokyo Chemical Industry Co., Ltd.). Смесь перемешивали при комнатной температуре в течение 30 минут. После завершения реакции, жидкость реакционной смеси концентрировали при пониженном давлении. Добавляли воду, и полученную в результате смесь экстрагировали этилацетатом. Органическую фазу промывали насыщенным соляным раствором, сушили безводным сульфатом магния, и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=90:10-69:31 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая заявленное в заголовке соединение, весящее 41,5 г (243 ммоль, выход 97%), в виде бесцветного масла.
1H-ЯМР-спектр (400МГц, CDCl3) δ: 7,25-7,19 (1H, м), 7,01-6,87 (2H, м), 5,22-5,14 (1H, м), 1,88 (1H, д, J=4,3 Гц), 1,50 (3H, д, J=6,4 Гц).
[0183] (Сравнительный пример 19)
(R)-1-(тиофен-3-ил)этанол
[Chem. 57]

[0184] Согласно способу, описанному в Journal of Organic Chemistry, 72 (2007) стр. 1639-1644, 0,446 г (1,61 ммоль) (S)-5,5-дифенил-2-метил-3,4-пропано-1,3,2-оксазаборолидина (Aldrich) добавляли к раствору в тетрагидрофуране (100 мл) 2,023 г (16,03 ммоль) 1-(тиофен-3-ил)этанона (Aldrich), высушенного над молекулярными ситами 4A 1/16 (торговое название, Wako Pure Chemical Industries, Ltd.) в атмосфере аргона при комнатной температуре при перемешивании. Затем, при контролировании температуры в районе -30°C в бане с сухим льдом и этанолом и при перемешивании, 19,0 мл (17,1 ммоль) добавляли по каплям в течение 1 часа 0,9 M боран-тетрагидрофураный комплекс (Tokyo Chemical Industry Co., Ltd.). Смесь перемешивали при приблизительно -30°C в течение 1 часа. После завершения реакции, добавляли 50 мл воды, и затем добавляли 100 мл этилацетата и 5 мл 1N хлористоводородной кислоты, осуществляя разделение жидкостей. Органическую фазу промывали 50 мл насыщенного соляного раствора, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=95:5-74:26 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая заявленное в заголовке соединение, весящее 1,81 г (14,1 ммоль, выход 88%, оптическая чистота 82,9% ee), в виде бесцветного масла.
Условия анализа на оптическую чистоту
Колонка: CHIRALCEL OJ-RH (торговое название, Daicel Corporation)
Размер: 0,46 см вн.д. x 25 см дл.
Подвижная фаза: 0,03% об. водный раствор трифторуксусной кислоты/ацетонитрил=75/25 (об./об.)
Скорость потока: 1,0 мл/мин.
Температура: 40°C
Длина волны: 254 нм
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 7,44 (1H, дд, J=5,0, 3,0 Гц), 7,25 (1H, ддд, J=3,0, 1,2, 0,9 Гц), 7,07 (1H, дд, J=5,0, 1,2 Гц), 5,11 (1H, д, J=4,8 Гц), 4,80-4,71 (1H, м), 1,34 (3H, д, J=6,4 Гц).
[0185] Заявленное в заголовке соединение можно получить с повышенной оптической чистотой следующим способом.
[0186] При комнатной температуре и при перемешивании 13 г липазы PS Amano SD (Wako Pure Chemical Industries, Ltd.) добавляли к раствору в диизопропиловом эфире (200 мл) 13,1 г (102 ммоль, оптическая чистота 69% ee) (R)-1-(тиофен-3-ил)этанола, полученного аналогично сравнительному примеру 19, и 15,0 мл (163 ммоль) винилацетата. Жидкость реакционной смеси перемешивали при 45°C в течение 6,5 часов и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=80:20 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая (R)-1-(тиофен-3-ил)этиловый эфир уксусной кислоты, весящий 11,8 г (67 ммоль, выход 65%, оптическая чистота > 99% ee) в виде светло-желтого масла.
Условия анализа на оптическую чистоту
Колонка: CHIRALPAK IA (торговое название, Daicel Corporation)
Размер: 0,46 см I.D. x 25 см л.
Подвижная фаза: гексан:2-пропанол=99,5:0,5 (об./об.)
Скорость потока: 1,0 мл/мин.
Температура: 40°C
Длина волны: 254 нм
1H-ЯМР-спектр (400МГц, CDCl3) δ: 7,30 (1H, дд, J=5,0, 2,9 Гц), 7,25-7,22 (1H, м), 7,09 (1H, дд, J=5,0, 1,3 Гц), 6,00 (1H, кв, J=6,6 Гц), 2,07 (3H, с), 1,56 (3H, д, J=6,5 Гц).
[0187] В потоке азота 2,50 г (104 ммоль) гидроксида лития добавляли к раствору в этаноле (100 мл) и воде (10 мл) 11,8 г полученного выше (R)-1-(тиофен-3-ил)этилового эфира уксусной кислоты (67,1 ммоль, оптическая чистота >99% ee) при комнатной температуре при перемешивании. Смесь перемешивали при данной температуре в течение 1,5 часов. После завершения реакции, жидкость реакционной смеси концентрировали при пониженном давлении, удаляя этанол. К остатку добавляли воду, и смесь экстрагировали этилацетатом. Органическую фазу промывали насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=95:5-74:26 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая заявленное в заголовке соединение, весящее 8,1 г (63 ммоль, выход 95%, оптическая чистота >99,0% ee), в виде светло-желтого масла.
[0188] (Сравнительный пример 20)
(R)-1-(4-метилтиофен-3-ил)этанол
[Chem. 58]

[0189] В атмосфере аргона при -30°C--27°C 3,4 мл (3,1 ммоль) 0,9 M боран-тетрагидрофуранового комплекса добавляли по каплям к раствору в тетрагидрофуране (1,0 мл) 78 мг (0,28 ммоль) (S)-5,5-дифенил-2-метил-3,4-пропано-1,3,2-оксазаборолидина (Aldrich). Смесь перемешивали при данной температуре в течение 30 минут. Затем, раствор в тетрагидрофуране (20 мл) 406 мг (2,90 ммоль) 1-(4-метилтиофен-3-ил)этанона, полученного аналогично сравнительному примеру 17, добавляли по каплям при -30°C--27°C, и смесь перемешивали при данной температуре в течение 1 часа. После завершения реакции, добавляли к жидкости реакционной смеси воду и 1N хлористоводородную кислоту, и смесь экстрагировали этилацетатом. Органическую фазу промывали насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=95:5-70:30 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая заявленное в заголовке соединение, весящее 387 мг (2,72 ммоль, выход 94%), в виде бесцветного масла.
Масс-спектр (EI, m/z): 142 [M]+.
1H-ЯМР-спектр (400МГц, CDCl3) δ: 7,23-7,21 (1H, м), 6,92 (1H, дкв, J=3,1, 0,9 Гц), 4,92 (1H, квдд, J=6,4, 4,8, 0,8 Гц), 2,27 (3H, д, J=0,9 Гц), 1,63 (1H, д, J=4,6 Гц), 1,53 (3H, д, J=6,4 Гц).
[0190] Заявленное в заголовке соединение можно получить с повышенной оптической чистотой следующим способом.
[0191] При комнатной температуре и при перемешивании 6,7 г липазы PS Amano SD (Wako Pure Chemical Industries, Ltd.) добавляли к раствору в диизопропиловом эфире (67 мл) 13,3 г (94 ммоль, оптическая чистота 90% ee) (R)-1-(4-метилтиофен-3-ил)этанола, полученного аналогично сравнительному примеру 20, и 16,3 мл (163 ммоль) винилацетата. Смесь перемешивали при 45°C в течение 25 часов. Полученную жидкость реакционной смеси фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=90:10 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая (R)-1-(4-метилтиофен-3-ил)этиловый эфир уксусной кислоты, весящий 10,3 г (56,0 ммоль, выход 60%, оптическая чистота > 99% ee), в виде светло-желтого масла.
Условия анализа на оптическую чистоту
Колонка: CHIRALPAK IA (торговое название, Daicel Corporation)
Размер: 0,46 см I.D. x 25 см л.
Подвижная фаза: гексан:2-пропанол=99,5:0,5 (об./об.)
Скорость потока: 1,0 мл/мин.
Температура: 40°C
Длина волны: 254 нм
1H-ЯМР-спектр (400МГц, CDCl3) δ: 7,23 (1H, д, J=3,3 Гц), 6,94-6,90 (1H, м), 5,94 (1H, дкв, J=6,5, 0,9 Гц), 2,23 (3H, д, J=1,0 Гц), 2,07 (3H, с), 1,55 (3H, д, J=6,5 Гц).
[0192] В потоке азота 2,0 г (84 ммоль) гидроксида лития добавляли к раствору в этаноле (50 мл) и воде (5 мл) 10,3 г (56,0 ммоль) (R)-1-(4-метилтиофен-3-ил)этилового эфира уксусной кислоты, полученного выше, при комнатной температуре при перемешивании. Смесь перемешивали при данной температуре в течение 1,5 часов. После завершения реакции, жидкость реакционной смеси концентрировали при пониженном давлении, удаляя этанол. К остатку добавляли воду, и смесь экстрагировали этилацетатом. Органическую фазу промывали насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=95:5-70:30 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая заявленное в заголовке соединение, весящее 7,8 г (55 ммоль, выход 98%, оптическая чистота 99,5% ee), в виде бесцветного масла.
Условия анализа на оптическую чистоту
Колонка: CHIRALPAK IA (торговое название, Daicel Corporation)
Размер: 0,46 см I.D. x 25 см л.
Подвижная фаза: жидкость A:жидкость B=95:1 (об./об.)
Жидкость A: гексан:2-пропанол=99,5:0,5 (об./об.)
Жидкость B: 2-пропанол
Скорость потока: 1,0 мл/мин.
Температура: 40°C
Длина волны: 254 нм
[0193] (Сравнительный пример 21)
(R)-1-(2,5-Дифторфенил)этиловый эфир уксусной кислоты
[Chem. 59]

[0194] При комнатной температуре и при перемешивании 33 г липазы PS Amano SD (Wako Pure Chemical Industries, Ltd.) добавляли к раствору в диизопропиловом эфире (200 мл) 39,3 г (249 ммоль) (RS)-1-(2,5-дифторфенил)этанола, полученного аналогично сравнительному примеру 18, и 45,0 мл (450 ммоль) винилацетата. Смесь перемешивали при 45°C в течение 46,5 часов. Полученную жидкость реакционной смеси фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=90:10 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая заявленное в заголовке соединение, весящее 20,1 г (100 ммоль, выход 40%, оптическая чистота 97% ee), в виде светло-желтого масла.
Условия анализа на оптическую чистоту
Колонка: CHIRALPAK IA (торговое название, Daicel Corporation)
Размер: 0,46 см I.D. x 25 см л.
Подвижная фаза: гексан:2-пропанол=99,5:0,5 (об./об.)
Скорость потока: 1,0 мл/мин.
Температура: 40°C
Длина волны: 254 нм
1H-ЯМР-спектр (400МГц, CDCl3) δ: 7,11-7,05 (1H, м), 7,03-6,90 (2H, м), 6,09 (1H, кв, J=6,6 Гц), 2,11 (3H, с), 1,52 (3H, д, J=6,5 Гц).
[0195] (Соединение, полученное в данной реакции (оптическая чистота 97% ee), гидролизовали тем же способом, как в сравнительном примере 22 (оптическая чистота 92,2% ee), и продукт обрабатывали в тех же условиях, как в реакции выше, посредством этого повышая оптическую чистоту.)
При комнатной температуре и при перемешивании, 16 г липазы PS Amano SD (Wako Pure Chemical Industries, Ltd.) добавляли к раствору в диизопропиловом эфире (70 мл) 15,8 г (100 ммоль) (R)-1-(2,5-дифторфенил)этанола (оптическая чистота 92,2% ee) и 20,0 мл (200 ммоль) винилацетата. Смесь перемешивали при 45°C в течение 64,5 часов. Полученную жидкость реакционной смеси фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=90:10 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая заявленное в заголовке соединение, весящее 19,2 г (96 ммоль, выход 96%, оптическая чистота 99,7% ee), в виде светло-желтого масла.
[0196] (Сравнительный пример 22)
(R)-1-(2,5-дифторфенил)этанол
[Chem. 60]

[0197] В потоке азота 3,5 г (150 ммоль) гидроксида лития добавляли к раствору в этаноле (100 мл) и воде (10 мл) 19,2 г (96 ммоль, оптическая чистота 99,7% ee) (R)-1-(2,5-дифторфенил)этилового эфира уксусной кислоты, полученного в сравнительном примере 21, при комнатной температуре при перемешивании. Смесь перемешивали при данной температуре в течение 1,5 часов. После завершения реакции, жидкость реакционной смеси концентрировали при пониженном давлении, удаляя этанол. Добавляли к остатку воду, и смесь экстрагировали этилацетатом. Органическую фазу промывали насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=90:10-69:31 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая заявленное в заголовке соединение, весящее 13,5 г (85,6 ммоль, выход 89%, оптическая чистота 99,0% ee), в виде светло-желтого масла.
Условия анализа на оптическую чистоту
Колонка: CHIRALPAK IA (торговое название, Daicel Corporation)
Размер: 0,46 см I.D. x 25 см д.
Подвижная фаза: жидкость A:жидкость B=99:1 (об./об.)
Жидкость A: гексан:2-пропанол=99,5:0,5 (об./об.)
Жидкость B: 2-пропанол
Скорость потока: 1,0 мл/мин.
Температура: 40°C
Длина волны: 254 нм
1H-ЯМР-спектр (400МГц, CDCl3) δ: 7,25-7,19 (1H, м), 7,01-6,87 (2H, м), 5,22-5,14 (1H, м), 1,88 (1H, д, J=4,3 Гц), 1,50 (3H, д, J=6,4 Гц).
[0198] (Сравнительный пример 23)
Этиловый эфир (R)-1-{4ʹ-[5-хлор-3-({[1-(тиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты
[Chem. 61]
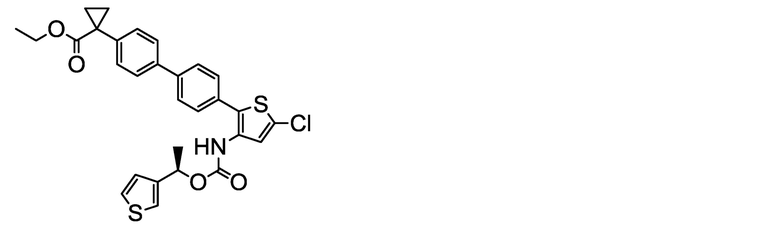
[0199] В атмосфере аргона 340 мг (0,791 ммоль) [бис(трифторацетокси)йод]бензола добавляли к раствору в толуоле (6 мл) 300 мг (0,704 ммоль) этилового эфира 1-[4ʹ-(3-карбамоил-5-хлортиофен-2-ил)-[1,1ʹ-бифенил]-4-ил]циклопропанкарбоновой кислоты, полученного аналогично сравнительному примеру 7, и 0,53 мл (6,6 ммоль) пиридина. Смесь перемешивали при комнатной температуре в течение 30 минут. Затем, 105 мг (0,819 ммоль) (R)-1-(тиофен-3-ил)этанола, полученного аналогично сравнительному примеру 19, добавляли в атмосфере аргона при комнатной температуре, и смесь перемешивали в течение 2 часов при нагревании при 70°C. После завершения реакции, добавляли к жидкости реакционной смеси этилацетат и воду, и органическую фазу отделяли. Органическую фазу промывали насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=80:20-30:70 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая заявленное в заголовке соединение, весящее 306 мг (0,55 ммоль, выход 79%), в виде коричневого масла.
Масс-спектр (DUIS-, m/z): 550 [M-1]-.
1H-ЯМР-спектр (400МГц, CDCl3) δ: 7,69-7,63 (2H, м), 7,63-7,51 (3H, м), 7,48-7,40 (4H, м), 7,31 (1H, дд, J=5,0, 2,9 Гц), 7,28-7,26 (1H, м), 7,11 (1H, дд, J=5,0, 1,3 Гц), 6,72 (1H, с), 5,99 (1H, кв, J=6,6 Гц), 4,13 (2H, кв, J=7,1 Гц), 1,64 (2H, дд, J=7,0, 4,0 Гц), 1,62 (3H, д, J=6,5 Гц), 1,23 (2H, дд, J=7,0, 4,0 Гц), 1,19 (3H, т, J=7,1 Гц).
[0200] (Сравнительный пример 24)
Этиловый эфир (R)-1-[4ʹ-(5-хлор-3-{[(1-фенилэтокси)карбонил]амино}тиофен-2-ил)-2ʹ-метокси[1,1ʹ-бифенил]-4-ил]циклопропанкарбоновой кислоты
[Chem. 62]
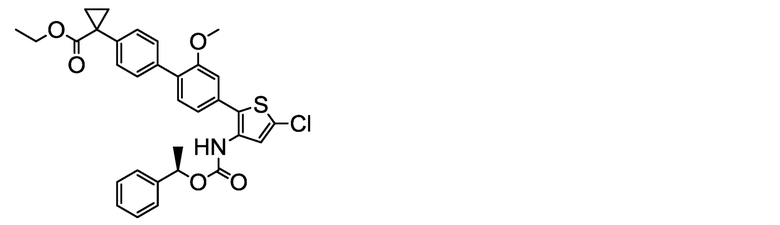
[0201] В атмосфере азота при комнатной температуре 2,4 г (5,6 ммоль) [бис(трифторацетокси)йод]бензола добавляли к раствору в толуоле (20 мл) 2,0 г (4,4 ммоль) этилового эфира 1-[4ʹ-(3-карбамоил-5-хлортиофен-2-ил)-2ʹ-метокси-[1,1ʹ-бифенил]-4 ил]циклопропанкарбоновой кислоты, полученного аналогично сравнительному примеру 8, 0,80 г (6,6 ммоль) (R)-1-фенилэтанола (Tokyo Chemical Industry Co., Ltd.) и 1,2 мл (15 ммоль) пиридина. Смесь перемешивали в течение 1,5 часов при нагревании при 60°C. После завершения реакции, жидкость реакционной смеси концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=99:1-80:20 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая заявленное в заголовке соединение, весящее 1,46 г (4,0 ммоль (чистота 72% масс.), выход 42%), в виде оранжевого масла.
Масс-спектр (CI, m/z): 575 [M]+.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 9,39 (1H, уш с), 7,40-7,26 (10H, м), 7,23-7,16 (2H, м), 7,10 (1H, дд, J=7,8, 1,6 Гц), 5,75 (1H, кв, J=6,4 Гц), 4,06 (2H, кв, J=7,1 Гц), 3,73 (3H, с), 1,56-1,41 (5H, м), 1,23 (2H, дд, J=7,0, 4,0 Гц), 1,13 (3H, т, J=7,0 Гц).
[0202] (Сравнительный пример 25)
2-(Триметилсилил)этиловый эфир (R)-1-{4ʹ-[5-хлор-3-({[1-(2,5-дифторфенил)этокси]карбонил}амино)тиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты
[Chem. 63]

[0203] В атмосфере аргона при комнатной температуре 874 мг (2,03 ммоль) [бис(трифторацетокси)йод]бензола добавляли к раствору в толуоле (10 мл) 886 мг (1,68 ммоль) 2-(триметилсилил)этилового эфира 1-[4ʹ-(3-карбамоил-5-хлортиофен-2-ил)-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил]циклопропанкарбоновой кислоты, полученного аналогично сравнительному примеру 16, и 0,65 мл (8,0 ммоль) пиридина. Смесь перемешивали в течение 5 минут. После этого, добавляли 413 мг (2,61 ммоль) (R)-1-(2,5-дифторфенил)этанола (Enamine). Смесь перемешивали в течение 1 часа при нагревании при температуре бани 70°C. После завершения реакции, добавляли к жидкости реакционной смеси воду и этилацетат, и смесь экстрагировали этилацетатом. Органическую фазу сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=99:1-94:6 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая заявленное в заголовке соединение, весящее 1,16 г (1,46 ммоль (чистота 86% масс.), выход 87%), в виде коричневого масла (полутвердое).
Масс-спектр (DUIS-, m/z): 682 [M-1]-.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 9,55 (1H, уш с), 7,45-7,40 (2H, м), 7,39-7,17 (8H, м), 7,10 (1H, дд, J=7,9, 1,5 Гц), 5,91 (1H, кв, J=6,3 Гц), 4,14-4,07 (2H, м), 3,76 (3H, с), 1,55-1,42 (3H, м), 1,49 (2H, дд, J=6,8, 3,9 Гц), 1,23 (2H, дд, J=7,0, 4,0 Гц), 0,91-0,84 (2H, м), -0,05 (9H, с).
[0204] (Сравнительный пример 26)
Этиловый эфир (R)-1-{4ʹ-[5-фтор-3-({[1-(4-метилтиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты
[Chem. 64]
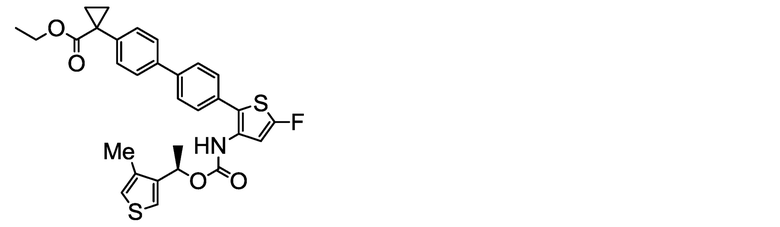
[0205] В атмосфере аргона 0,24 мл (1,7 ммоль) триэтиламина и 0,29 мл (1,4 ммоль) дифенилфосфорилазида добавляли к раствору в толуоле (10 мл) 456 мг (1,11 ммоль) 2-{4ʹ-[1-(этоксикарбонил)циклопропил]-[1,1ʹ-бифенил]-4-ил}-5-фтортиофен-3 карбоновой кислоты, полученной аналогично сравнительному примеру 13. Смесь перемешивали при комнатной температуре в течение 30 минут. Затем, добавляли раствор в толуоле (1 мл) 190 мг (1,34 ммоль) (R)-1-(4-метилтиофен-3-ил)этанола, который получали аналогично сравнительному примеру 20 и сушили молекулярными ситами 4Ǻ (порошок) (торговое название, NACALAI TESQUE, INC.) (0,3 г). Полученную в результате смесь перемешивали в течение 2 часов при нагревании при 70°C. После завершения реакции, добавляли к жидкости реакционной смеси этилацетат и насыщенный водный раствор хлорида аммония, и органическую фазу отделяли. Органическую фазу промывали насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=80:20 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая заявленное в заголовке соединение, весящее 570 мг (1,04 ммоль, выход 93%), в виде бесцветного масла.
Масс-спектр (DUIS-, m/z): 548 [M-1]-.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 9,31 (1H, уш с), 7,74-7,68 (2H, м), 7,66-7,61 (2H, м), 7,57-7,40 (5H, м), 7,17-7,13 (1H, м), 6,83 (1H, уш с), 5,74 (1H, кв, J=6,5 Гц), 4,05 (2H, кв, J=7,2 Гц), 2,17 (3H, уш с), 1,60-1,43 (3H, м), 1,51 (2H, дд, J=6,8, 4,0 Гц), 1,23 (2H, дд, J=7,1, 4,1 Гц), 1,11 (3H, т, J=7,1 Гц).
[0206] (Сравнительный пример 27)
Этиловый эфир (R)-1-{4ʹ-[3-({[1-(2-хлорфенил)этокси]карбонил}амино)-5-фтортиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты
[Chem. 65]
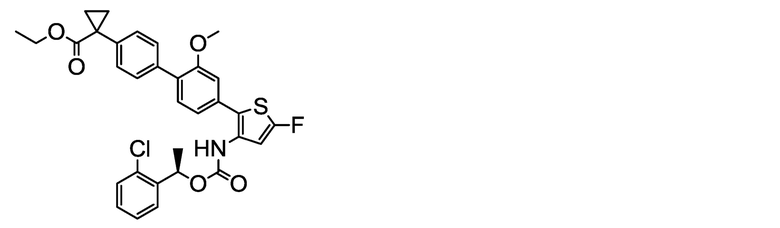
[0207] В атмосфере аргона 0,040 мл (0,29 ммоль) триэтиламина и 0,045 мл (0,21 ммоль) дифенилфосфорилазида добавляли к раствору в толуоле (4,0 мл) 72 мг (0,16 ммоль) 2-{4ʹ-[1-(этоксикарбонил)циклопропил]-2-метокси-[1,1ʹ-бифенил]-4-ил}-5 фтортиофен-3-карбоновой кислоты, полученной аналогично сравнительному примеру 14. Смесь перемешивали при комнатной температуре в течение 30 минут. Затем, добавляли 35 мг (0,22 ммоль) (R)-1-(2-хлорфенил)этанола (Shanghai AoBo Bio-pharm). Смесь перемешивали в течение 2 часов при нагревании при 70°C. После завершения реакции, добавляли к жидкости реакционной смеси этилацетат и воду, и органическую фазу отделяли. Органическую фазу промывали насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=93:7-72:28 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая заявленное в заголовке соединение, весящее 84 мг (0,061 ммоль (чистота 43% масс.), выход 37%), в виде бесцветного масла.
Масс-спектр (EI, m/z): 593 [M]+.
1H-ЯМР-спектр (400МГц, CDCl3) δ: 7,52-7,48 (2H, м), 7,43-7,17 (9H, м), 7,06 (1H, дд, J=7,8, 1,6 Гц), 6,97 (1H, д, J=1,6 Гц), 6,23 (1H, кв, J=6,7 Гц), 4,13 (2H, кв, J=7,1 Гц), 3,82 (3H, с), 1,63 (2H, дд, J=6,9, 3,9 Гц), 1,57 (3H, д, J=6,7 Гц), 1,24 (2H, дд, J=7,0, 4,0 Гц), 1,20 (3H, т, J=7,2 Гц).
[0208] (Сравнительный пример 28)
(R)-1-{4ʹ-[5-хлор-3-({[1-(тиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновая кислота
[Chem. 66]
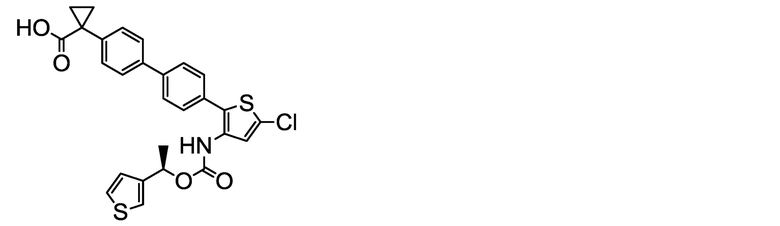
[0209] 2,0 мл (4,0 ммоль) 2N водного раствора гидроксида натрия добавляли к раствору в 2-пропаноле (4 мл) 304 мг этилового эфира (0,551 ммоль) (R)-1-{4ʹ-[5-хлор-3-({[1-(тиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты, полученного аналогично сравнительному примеру 23. Смесь перемешивали при комнатной температуре в течение 42,5 часов. После завершения реакции, жидкость реакционной смеси подкисляли добавлением 2N хлористоводородной кислоты и экстрагировали этилацетатом. Органическую фазу промывали последовательно водой и насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (COOH колонка, растворитель для элюирования: гексан:этилацетат=70:30-10:90 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении. Добавляли к остатку гексан (10 мл) и этилацетат (3 мл). Выпавший белый осадок отфильтровывали и промывали смесью гексан-этилацетат (3:1 (об./об.)). Маточный раствор и промывки концентрировали при пониженном давлении, получая заявленное в заголовке соединение, весящее 65 мг (0,55 ммоль, выход 23%), в виде белого твердого остатка.
Масс-спектр (DUIS-, m/z): 522 [M-1]-.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 12,37 (1H, уш с), 9,33 (1H, уш с), 7,74-7,68 (2H, м), 7,65-7,60 (2H, м), 7,58-7,50 (3H, м), 7,48-7,37 (3H, м), 7,25-7,07 (2H, м), 5,82 (1H, кв, J=6,4 Гц), 1,56-1,44 (3H, м), 1,48 (2H, дд, J=6,7, 3,8 Гц), 1,19-1,16 (2H, м).
[0210] (Сравнительный пример 29)
(R)-1-[4ʹ-(5-хлор-3-{[(1-фенилэтокси)карбонил]амино}тиофен-2-ил)-2ʹ метокси-[1,1ʹ-бифенил]-4-ил]циклопропанкарбоновая кислота
[Chem. 67]

[0211] 8,0 мл (16,0 ммоль) 2N водного раствора гидроксида натрия добавляли к раствору в 2-пропаноле (30 мл) 1,46 г (4,0 ммоль (чистота 72% масс.)) этилового эфира (R)-1-[4ʹ-(5-хлор-3-{[(1-фенилэтокси)карбонил]амино}тиофен-2-ил)-2ʹ-метокси [1,1ʹ-бифенил]-4-ил]циклопропанкарбоновой кислоты, полученной в сравнительном примере 24. Смесь перемешивали при комнатной температуре в течение 110 часов. После завершения реакции, жидкость реакционной смеси подкисляли добавлением 2N хлористоводородной кислоты и экстрагировали хлористым метиленом. Органическую фазу промывали насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=80:20-0:100 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении. Остаток растворяли в небольшом количестве этанола. Добавляли к раствору воду, осаждая твердый остаток. Твердый остаток собирали фильтрованием, промывали водой и сушили нагревом в вакууме, получая заявленное в заголовке соединение, весящее 588 мг (1,07 ммоль, выход 58%), в виде светло-красного твердого остатка.
Масс-спектр (DUIS-, m/z): 546 [M-1]-.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 12,34 (1H, уш с), 9,40 (1H, уш с), 7,45-7,25 (10H, м), 7,21-7,16 (2H, м), 7,09 (1H, дд, J=7,9, 1,6 Гц), 5,75 (1H, кв, J=6,4 Гц), 3,73 (3H, с), 1,54-1,41 (5H, м), 1,18-1,12 (2H, м).
[0212] (Сравнительный пример 30)
(R)-1-{4ʹ-[5-Фтор-3-({[1-(4-метилтиофен-3 ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновая кислота
[Chem. 68]

[0213] 4,0 мл (8,0 ммоль) 2N водного раствора гидроксида натрия добавляли к раствору в 2-пропаноле (12 мл) 565 мг (1,03 ммоль) этилового эфира (R)-1-{4ʹ-[5-фтор-3-({[1-(4-метилтиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислота, полученного аналогично сравнительному примеру 26. Смесь перемешивали при комнатной температуре в течение 91 часов. После завершения реакции, жидкость реакционной смеси подкисляли добавлением 1N хлористоводородной кислоты и экстрагировали хлористым метиленом. Органическую фазу промывали последовательно водой и насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=50:50 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении. Затем, добавляли 6 мл гексана и 12 мл этилацетата, и смесь нагревали при 50°C и охлаждали. Выпавший твердый остаток отфильтровывали и промывали смесью гексан-этилацетат (50:50 (об./об.)). Маточный раствор и промывки концентрировали при пониженном давлении. К остатку добавляли 8 мл ацетонитрила, 4 мл воды и 3 мл тетрагидрофурана. Лиофилизация смеси приводила в результате к заявленному в заголовке соединению, весящему 193 мг (0,37 ммоль, выход 36%), в виде белого твердого остатка.
Масс-спектр (DUIS-, m/z): 520 [M-1]-.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 12,38 (1H, уш с), 9,33 (1H, уш с), 7,73-7,67 (2H, м), 7,65-7,59 (2H, м), 7,57-7,50 (2H, м), 7,49-7,38 (3H, м), 7,19-7,12 (1H, м), 6,83 (1H, уш с), 5,74 (1H, кв, J=6,4 Гц), 2,17 (3H, уш с), 1,59-1,44 (3H, м), 1,48 (2H, дд, J=6,7, 3,8 Гц), 1,18 (2H, дд, J=6,9, 3,9 Гц).
[0214] (Сравнительный пример 31)
(R)-1-{4ʹ-[3-({[1-(2-хлорфенил)этокси]карбонил}амино)-5-фтортиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновая кислота
[Chem. 69]

[0215] 1,5 мл (3,0 ммоль) 2N водного раствора гидроксида натрия добавляли к раствору в 2-пропаноле (3,0 мл) 80 мг (0,058 ммоль (чистота 43% масс.)) этилового эфира (R)-1-{4ʹ-[3-({[1-(2-хлорфенил)этокси]карбонил}амино)-5-фтортиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты, полученного в сравнительном примере 27. Смесь перемешивали при комнатной температуре в течение 23 часов. После завершения реакции, жидкость реакционной смеси подкисляли добавлением 2N хлористоводородной кислоты и экстрагировали хлористым метиленом. Органическую фазу промывали последовательно водой и насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле (COOH колонка, растворитель для элюирования: гексан:этилацетат=80:20-20:80 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении, получая заявленное в заголовке соединение, весящее 13 мг (0,023 ммоль, выход 40%), в виде белого твердого остатка.
Масс-спектр (DUIS-, m/z): 564 [M-1]-.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 12,35 (1H, уш с), 9,55 (1H, уш с), 7,60-7,28 (9H, м), 7,18 (1H, д, J=1,5 Гц), 7,09 (1H, дд, J=7,8, 1,4 Гц), 6,84 (1H, д, J=2,5 Гц), 6,00 (1H, кв, J=6,1 Гц), 3,77 (3H, с), 1,55-1,39 (5H, м), 1,21-1,10 (2H, м).
[0216] (Сравнительный пример 32)
(R)-1-{4ʹ-[5-хлор-3-({[1-(2,5 дифторфенил)этокси]карбонил}амино)тиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновая кислота
[Chem. 70]

[0217] В атмосфере аргона при комнатной температуре 9,0 мл (9,0 ммоль) 1 M тетрагидрофуранового раствора фторида тетрабутиламмония добавляли к раствору в диметилформамиде (90 мл) 3,00 г (4,38 ммоль) 2-(триметилсилил)этилового эфира (R)-1-{4ʹ-[5-хлор-3-({[1-(2,5-дифторфенил)этокси]карбонил}амино)тиофен-2-ил]-2ʹ-метокси-[1,1ʹ-бифенил]-4-ил}циклопропанкарбоновой кислоты, полученного аналогично сравнительному примеру 25. Смесь перемешивали при данной температуре в течение 2 часов. После завершения реакции, этилацетат и воду добавляли к жидкости реакционной смеси, и pH доводили до приблизительно 3 0,5N хлористоводородной кислотой. Жидкость реакционной смеси разделялась на фазы. Органическую фазу промывали последовательно водой и насыщенным соляным раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Добавляли к остатку 2-пропанол (40 мл) и воду (40 мл), и смесь обрабатывали ультразвуком. Выпавший твердый остаток отфильтровывали и промывали водой. Таким образом, получали неочищенное заявленное в заголовке соединение, весящее 2,86 г. Неочищенное соединение подвергали колоночной хроматографии на силикагеле (растворитель для элюирования: гексан:этилацетат=70:30-10:90 (об./об.)), и фракцию, содержащую целевое соединение, концентрировали при пониженном давлении. Добавляли к остатку гептан, и выпавший твердый остаток отфильтровывали. Таким образом, получали заявленное в заголовке соединение, весящее 1,62 г (2,77 ммоль, выход 63%), в виде белого твердого остатка.
Масс-спектр (DUIS-, m/z): 582 [M-1]-.
1H-ЯМР-спектр (400МГц, DMSO-d6) δ: 12,35 (1H, уш с), 9,54 (1H, уш с), 7,44-7,39 (2H, м), 7,39-7,19 (7H, м), 7,18 (1H, д, J=1,6 Гц), 7,10 (1H, дд, J=7,9, 1,6 Гц), 5,91 (1H, кв, J=6,5 Гц), 3,76 (3H, с), 1,56-1,43 (3H, м), 1,47 (2H, дд, J=6,7, 3,7 Гц), 1,18 (2H, дд, J=6,8, 4,0 Гц).
[0218] [Пример испытания 1]
Испытание на связывание GTPγS с LPA1 рецептором
5 мкг мембранной фракции RH 7777 клеток, экспрессирующих человеческий LPA1 рецептор (A324, ChanTest), суспендировали в рабочем буферном растворе (20 мМ HEPES, 100 мМ NaCl, 10 мМ MgCl2, 10 мкМ GDP, 5 мкг сапонин, 0,2% BSA, 0,1 нМ [35S]GTPγS (NEG030X, Perkin Elmer), pH 7,4). Каждое из испытуемых соединений, растворенных в DMSO при различных концентрациях, добавляли к суспензии. После предварительного выдерживания при 30°C в течение 15 минут, добавляли LPA (L7260, Sigma, конечная концентрация 100 нМ), и суспензии выдерживали при 30°C в течение 30 минут. Мембранные фракции собирали на фильтр из стекловолокна (GF/B, Whatman), применяя коллектор клеток (M30, Brandel), и промывали 10 мМ фосфатным буфером (pH 7,4). Радиоактивность мембранных фракций измеряли жидкостно-сцинтилляционным анализатором (2900TR, Packard), и концентрацию (IC50) испытуемого соединения, требуемую для 50% ингибирования связывания LPA1 рецептора и [35S]GTPγS, определяли нелинейным регрессионным анализом, применяя EXSAS (версия 7.6.0, Arm Systex).
[0219] [Пример испытания 2]
Испытание на миграцию клеток
Испытание на миграцию клеток осуществляли, применяя Chemo-Tx (зарегистрированная торговая марка) (116-8, Neuro Probe). A2058 клетки меланомы человека (полученные у европейской коллекции культивируемых клеток) выращивали в бессывороточной EMEM среде в течение 24 часов, и суспендировали в DMEM среде, содержащей 0,1% BSA, получая клеточную суспензию. Каждое из испытуемых соединений, растворенных в DMSO при различных концентрациях, добавляли к клеточной суспензии, и суспензии выращивали при 37°C в течение 15 минут (конечная концентрация в DMSO 0,5%). LPA, растворенную в DMEM среде, содержащей 0,1% BSA (конечная концентрация 100 нМ), добавляли в Chemo-Tx 96 луночный планшет, и Chemo-Tx фильтр, покрытый 0,001% фибронектином с обеих сторон, помещали на планшет. Суспензии культивируемых клеток (25000 клеток) добавляли на верхнюю поверхность фильтра и дополнительно культивировали при 37°C в течение 3 часов. После этого, клетки на верхней поверхности фильтра удаляли. После того, как с верхней поверхности фильтра удаляли клетки и сушили, клетки, которые мигрировали к нижней поверхности фильтра, прокрашивали красителем Дифф-Квик (16920, Sysmex). Измеряли поглощение фильтра (570 нм), и концентрацию (IC50) испытуемого соединения, требуемую для 50% ингибирования клеточной миграционной активности LPA, определяли нелинейным регрессионным анализом, применяя EXSAS (версия 7.6.0, Arm Systex).
[0220] В данном испытании, соединения настоящего изобретения показали превосходную активность. Например, величины IC50 соединений примеров 1-15 были не больше чем 200 нМ.
[0221] [Пример испытания 3]
Испытание на высвобождение гистамина, вызванное LPA, на мышах
Испытание на высвобождение гистамина, вызванное LPA, на мышах осуществляли согласно способу Swaney et al. (The Journal of Pharmacology and Experimental Therapeutics, 336 (2011), стр. 693-700). Испытуемое соединение суспендировали в 0,5% растворе метилцеллюлозы (133-14255, Wako Pure Chemical Industries, Ltd.) и перорально вводили мужским особям CD1 мышей (вес тела 30-40 г, поставляемым Charles River Laboratories Japan) при дозе 10 мл/кг. Через 4 часа после введения, LPA (857130P, Avanti), растворенную в PBS, содержащем 0,1% BSA, вводили через хвостовую вену (300 мкг/мышь). Сразу же после этого, каждую из мышей анестезировали изофлураном, и кровь собирали из вены через 2 минуты после введения LPA. Кровь помещали в пробирки, содержащие EDTA, и центрифугировали при 4°C, 2000 x g в течение 10 минут, получая плазму.
Концентрацию гистамина в плазме измеряли EIA набором (62HTMPEB, Cisbio Bioassays).
Степень ингибирования (%) в группе с введением 0,5% раствора метилцеллюлозы рассчитывали для каждого индивида, исходя из концентрации гистамина в плазме у мыши, которой вводили испытуемое соединение, и долю индивидов, которые показали степень ингибирования 80% или более, выражали в виде коэффициента эффективности (%).
[0222] В данном испытании, соединения настоящего изобретения показали превосходную активность. Например, соединения примеров 1-15 достигали коэффициента эффективности 50% или более при дозе 10 мг/кг.
[0223] [Пример испытания 4]
Модели фиброза легкого, вызванного блеомицином
Гидрохлорид блеомицина (Nippon Kayaku Co., Ltd.) вводили мышам, получая модели фиброза легкого. Испытуемое соединение вводили перорально каждый день со дня, в который начинали введение блеомицина. В 3-42 дни после обработки блеомицином, жидкость бронхоальвеолярного лаважа (BALF) или легкие собирали при анестезии изофлураном. BALF центрифугировали при 800 x g в течение 10 минут, получая супернатанты. Супернатанты анализировали набором для определения белка DC (500-0114, Biorad), определяя количества белка, и анализировали набором для опредедления растворимого коллагена Sircol (S111, Biocolor), определяя количества коллагена. Затем, биомаркеры воспаления и фиброза в супернатантах измеряли ELISA способом. В отношении легких, после измерения их сырой массы, количества гидроксипролина в тканях измеряли модификацией способа Воесснера (Archives of Biochemistry and Biophysics, 93 (1961), стр. 440-447). Часть легких фиксировали в 10% формалиновом нейтральном буферном растворе и наблюдали за гистологическими изменениями. Результаты анализировали статистически, применяя EXSAS (версия 7.6.0, Arm Systex).
[0224] [Пример испытания 5]
Модели фиброза почки с односторонней обструкцией мочеточника (ООМ)
Брюшную полость мышей, анестезированных изофлураном, разрезали. Левый мочеточник лигировали шелковой нитью, получая модель ООМ. Испытуемое соединение вводили перорально каждый день со дня, в который получали ООМ модели. На 8 день, 14 день или 21 день после получения модели ООМ, почки собирали и измеряли их влажную массу. РНК экстрагировали из части почек, и степени экспрессии генов, являющихся маркерами фиброза, измеряли способом количественного ПЦР. Затем, измеряли количества гидроксипролина или коллагена в почечной ткани. Результаты анализировали статистически, применяя EXSAS.
[0225] [Пример испытания 6]
Модели фиброза печени, вызванного тетрахлоридом углерода (CCl4)
Разбавленный CCl4 (035-01273, Wako Pure Chemical Industries, Ltd.) вводили мышам два раза в неделю, получая модель фиброза печени. Испытуемое соединение вводили перорально каждый день со дня, в которой начинали введение CCl4. В 3-28 дни после начала введения CCl4, печень собирали при анестезии изофлураном, и измеряли их влажную массу. РНК экстрагировали из частей печени, и степени экспрессии генов, являющихся маркерами фиброза, измеряли способом количественного ПЦР. Затем, измеряли количества гидроксипролина или коллагена в тканях печени. Части печени фиксировали в 10% формалиновом нейтральном буферном растворе и наблюдали за гистологическими изменениями. Результаты анализировали статистически, применяя EXSAS.
[0226] [Пример испытания 7]
Модель неалкогольного стеатогепатита (NASH) на крысах
NASH модели получали кормлением крыс пищей, лишенной метионина/холина (MCD). Крысам обеспечивали свободный доступ к нормальной пище или MCD пище в течение 20 недель. Испытуемое соединение вводили перорально каждый день со дня, в который начинали кормление MCD пищей. После разведения в течение 20 недель, печени собирали при анестезии изофлураном, и измеряли их влажную массу. РНК экстрагировали из частей печени, и степени экспрессии генов, являющихся маркерами фиброза, измеряли способом количественного ПЦР. Затем, измеряли количества гидроксипролина или коллагена в тканях печени. Части печени фиксировали в 10% формалиновом нейтральном буферном растворе и наблюдали за гистологическими изменениями. Результаты анализировали статистически, применяя EXSAS.
[0227] [Пример испытания 8]
Модель неалкогольного стеатогепатита (NASH) на мышах
STAM (загестрированная торговая марка) мышей (доступных у Stelic Institute & Co., Inc.) применяли в качестве NASH моделей. STAM (загестрированная торговая марка) мышей получали подкожным введением 200 мкг стрептозотоцина (Sigma Aldrich) один раз в спину 2-дневным мужским особям мышей и кормили мышей пищей с высоким содержанием жиров (High Fat Diet 32, CLEA Japan, Inc.) через 4 недели после рождения (Medical Molecular Morphology, 46 (2013) стр. 141-152).
Испытуемое соединение вводили перорально каждый день через 5 или 6 недель после рождения. В возрасте 9 или 10 недель, кровь и печень собирали при анестезии. Кровь подвергали биохимическим испытаниям. После измерения влажной массы печени, РНК экстрагировали из частей печени, и степень экспрессии генов, являющихся маркером воспаления и фиброза, измеряли способом количественного ПЦР. Затем, измеряли количества гидроксипролина или коллагена в тканях печени. Парафиновые срезы или замороженные срезы получали из частей печени и подвергали гистологическим испытаниям, определяя баллы активности NAFLD, участки фиброза или участки воспаления. Результаты анализировали статистически, применяя EXSUS (версия 8.0, CAC EXICARE CORPORATION) или Prism 4 (GraphPad Software, Inc.).
[0228] [Пример испытания 9]
Модели неалкогольного стеатогепатита (NASH) на мышах
NASH модели получали выращиванием мышей на пище с высоким содержанием жиров и 0,1% метионином и несодержащей холин (A06071302, Research Diets, Inc.) (International Journal of Experimental Pathology, 94 (2013) стр. 93-103).
Испытуемое соединение вводили перорально каждый день со дня, в который начинали кормление CDAHFD. Через 8-12 недель печень собирали при анестезии изофлураном и измеряли их влажную массу. РНК экстрагировали из частей печени, и степень экспрессии генов, являющихся маркером воспаления и фиброза, измеряли способом количественного ПЦР. Затем, измеряли количества гидроксипролина или коллагена в тканях печени. Части печени фиксировали в 10% формалиновом нейтральном буферном растворе и наблюдали за гистологическими изменениями. Результаты анализировали статистически, применяя EXSUS (версия 8.0, CAC EXICARE CORPORATION).
[0229] [Пример испытания 10]
Фармакокинетические исследования на обезьянах
Одноразовый катетер вставляли через носовую полость в желудок макаки-крабоеда, лишенного питания с вечера дня перед испытанием. Через шприц-тюбик перорально вводили суспензию 0,5% метилцеллюлозы или раствор, содержащий испытуемое соединение при 10 мг/2 мл один раз при дозе 2 мл/кг. Через шприц-тюбик образцы крови отбирали из бедренной вены перед введением и через 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 8 часов, 12 часов и 24 часа после введения. EDTA-2K добавляли к образцам крови, и образцы центрифугировали (4°C, 1710 x g, 3000 об/мин, 15 минут), получая плазму. Плазму освобождали от белков добавлением ацетонитрила (смесь 50 мкл плазмы+200 мкл ацетонитрила), и смеси фильтровали (PTFE, 0,2 мкм). Фильтраты анализировали LC-MS/MS (3200 QTrap, AB SCIEX; и LC-20A или LC-30A серии, Shimadzu Corporation), определяя концентрации испытуемого соединения в плазме. AUC (площадь под кривой зависимости концентрации в плазме) рассчитывали Phoenix WinNonlin (CERTARA), исходя из изменений концентрации в плазме.
[0230] Из результатов примеров испытаний 2 и 3, соли тиофенового соединения, замещенного α-галогеном, настоящего изобретения обладают LPA рецепторный антагонистическим действием и являются в частности пригодными в качестве лекарственных средств для лечения и/или предотвращения (предпочтительно, лекарственных средств для лечения) заболеваний, сопровождающих фиброз, иммунологических или воспалительных заболеваний, заболеваний центральной или периферической нервной системы, урологических заболеваний и раковых заболеваний.
[0231][Пример получения 1] твердые капсулы
Стандартные твердые желатиновые капсулы, состоящие из двух частей, загружали порошком (100 мг) соли соединения примера, лактозой (150 мг), целлюлозой (50 мг) и стеаратом магния (6 мг), получая твердые капсулы, которые промывали, и затем сушили.
[0232] [Пример получения 2] мягкие капсулы
Смесь усвояемого масла, такого как соевое масло или оливковое масло, и соли соединения примера вводили в желатин, получая мягкие капсулы, содержащие 100 мг активного ингредиента, и мягкие капсулы промывали, и затем сушили.
[0233][Пример получения 3] таблетки
Согласно способу, известному в фармацевтической области техники, таблетки получали, применяя соль соединения (100 мг) примера, коллоидный диоксид кремния (0,2 мг), стеарат магния (0,2 мг), микрокристаллическую целлюлозу (0,2 мг), крахмал (0,2 мг) и лактозу (98,8 мг). Таблетки можно покрывать, при необходимости.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
[0234] Соли тиофенового соединения, замещенного α-галогеном, настоящего изобретения, представленные общей формулой (I), обладают большим LPA рецепторным антагонистическим действием и превосходными свойствами, такими как долгосрочные лечебные эффекты и растворимость, и являются пригодными в качестве лекарственных средств (лекарственных средств для лечения и/или предотвращения заболеваний, сопровождающих фиброз, иммунологических или воспалительных заболеваний, заболеваний центральной или периферической нервной системы, урологических заболеваний и раковых заболеваний).
| название | год | авторы | номер документа |
|---|---|---|---|
| ГАЛОГЕНЗАМЕЩЕННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2013 |
|
RU2649398C2 |
| ГАЛОГЕНЗАМЕЩЕННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2013 |
|
RU2756506C2 |
| АНТАГОНИСТЫ РЕЦЕПТОРА СОМАТОСТАТИНА ПОДТИПА 5 (SSTR5) | 2014 |
|
RU2671958C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2681849C2 |
| ТРИАЗОЛОПИРИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ ДЛЯ ЛЕЧЕНИЯ КОЖНЫХ ЗАБОЛЕВАНИЙ | 2009 |
|
RU2544011C2 |
| N-(2-ЦИАНОГЕТЕРОЦИКЛИЛ)ПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2669922C2 |
| ПРОИЗВОДНЫЕ 1,2-ДИ(ЦИКЛО)ЗАМЕЩЕННОГО БЕНЗОЛА | 2004 |
|
RU2340602C2 |
| АРИЛ- И ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ТЕТРАГИДРОИЗОХИНОЛИНЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ БЛОКИРОВАНИЯ ОБРАТНОГО ЗАХВАТА НОРЭПИНЕФРИНА, ДОПАМИНА И СЕРОТОНИНА | 2005 |
|
RU2388751C2 |
| АРИЛ-ЗАМЕЩЕННЫЕ КАРБОКСАМИДНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ БЛОКАТОРОВ КАЛЬЦИЕВЫХ ИЛИ НАТРИЕВЫХ КАНАЛОВ | 2010 |
|
RU2575168C2 |
| ПРОИЗВОДНЫЕ ЦИКЛОПРОПИЛАМИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ H-ГИСТАМИНОВОГО РЕЦЕПТОРА | 2007 |
|
RU2449989C2 |
Изобретение относится к солям тиофенового соединения, замещенного α-галогеном, общей формулы(I):

(где R представляет собой атом водорода или метокси группу; X представляет собой атом галогена; A выбран из группы, состоящей из:
 ;
;
M представляет собой щелочной или щелочноземельный металл; и n равен 1, когда M представляет собой щелочной металл, и равен 2, когда M представляет собой щелочноземельный металл), которое обладает сильным LPA рецепторным антагонистическим действием. 8 н. и 7 з.п. ф-лы, 1 табл., 45 пр.
1. Соль, представленная общей формулой (I):
[Chem. 71]

(где
R представляет собой атом водорода или метокси группу,
X представляет собой атом галогена,
A выбран из группы, состоящей из:
[Chem. 72]

M представляет собой щелочной металл или щелочноземельный металл, и
n равен 1, когда M представляет собой щелочной металл, и равен 2, когда M представляет собой щелочноземельный металл).
2. Соль по п.1, где щелочной металл или щелочноземельный металл представляет собой натрий, калий или кальций.
3. Соль (R)-1-[4'-(5-хлор-3-{[(1-фенилэтокси)карбонил]амино}тиофен-2-ил)-2'-метокси[1,1'-бифенил]-4-ил]циклопропанкарбоновой кислоты с щелочным металлом или щелочноземельным металлом.
4. Соль по п.3, где щелочной металл или щелочноземельный металл представляет собой натрий, калий или кальций.
5. Соль (R)-1-{4'-[5-хлор-3-({[1-(2,5-дифторфенил)этокси]карбонил}амино)тиофен-2-ил]-2'-метокси-[1,1'-бифенил]-4-ил}циклопропанкарбоновой кислоты с щелочным металлом или щелочноземельным металлом.
6. Соль по п.5, где щелочной металл или щелочноземельный металл представляет собой натрий, калий или кальций.
7. Соль (R)-1-{4'-[3-({[1-(2-хлорфенил)этокси]карбонил}амино)-5-фтортиофен-2-ил]-2'-метокси-[1,1'-бифенил]-4-ил}циклопропанкарбоновой кислоты с щелочным металлом или щелочноземельным металлом.
8. Соль по п.7, где щелочной металл или щелочноземельный металл представляет собой натрий, калий или кальций.
9. Соль (R)-1-{4'-[5-хлор-3-({[1-(тиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1'-бифенил]-4-ил}циклопропанкарбоновой кислоты с щелочным металлом или щелочноземельным металлом.
10. Соль по п.9, где щелочной металл или щелочноземельный металл представляет собой натрий, калий или кальций.
11. Соль (R)-1-{4'-[5-фтор-3-({[1-(4-метилтиофен-3-ил)этокси]карбонил}амино)тиофен-2-ил]-[1,1'-бифенил]-4-ил}циклопропанкарбоновой кислоты с щелочным металлом или щелочноземельным металлом.
12. Соль по п.11, где щелочной металл или щелочноземельный металл представляет собой натрий, калий или кальций.
13. Антагонист LPA рецептора, содержащий соль по любому из пп. 1-12 в качестве активного ингредиента.
14. Фармацевтическая композиция, обладающая антагонистическим действием относительно LPA рецептора, содержащая эффективное количество соли по любому из пп. 1-12 в качестве активного ингредиента.
15. Фармацевтическая композиция по п.14 для лечения или предотвращения заболевания, сопровождающего фиброз, иммунологического или воспалительного заболевания, заболевания центральной или периферической нервной системы, урологического заболевания или ракового заболевания.
| EA 201100691 A1, 28.02.2012 | |||
| WO 2012078593 A2, 14.06.2012 | |||
| WO 2012138797 A1, 11.10.2012. |

