ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к арил-замещенным карбоксамидным производным, которые обладают блокирующими активностями кальциевых каналов Т-типа или потенциалзависимых натриевых каналов в качестве тетродотоксин-чувствительных (TTX-S) блокаторов, таких как NaV1.3 и NaV1.7, и которые являются полезными в лечении или профилактике расстройств и заболеваний, в которые вовлечены кальциевые каналы Т-типа или потенциалзависимые натриевые каналы. Настоящее изобретение также относится к фармацевтическим композициям, содержащим эти соединения, и применению этих соединений и композиций в профилактике или лечении таких заболеваний, в которые вовлечены кальциевые каналы Т-типа или потенциалзависимые натриевые каналы.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Кальциевые каналы плазматических мембран являются членами многообразного суперсемейства белков потенциалзависимых каналов. Кальциевые каналы представляют собой распространенные на мембране, включающие множество субэлементов, белки, которые обеспечивают контролируемое поступление в клетки ионов Ca2+ из внеклеточной жидкости. Возбудимые клетки во всем разнообразии видов животных и, по меньшей мере, в некоторых бактериальных, грибковых и растительных клетках имеют один или несколько видов кальциевых каналов. Почти все "возбудимые" клетки у животных, такие как нейроны центральной нервной системы (ЦНС), периферические нервные клетки и клетки мышц, в том числе и скелетных мышц, сердечных мышц и венозных и артериальных гладких мышц, имеют потенциалзависимые кальциевые каналы.
Различные типы кальциевых каналов были идентифицированы в клетках млекопитающих из различных тканей, в том числе скелетных мышц, сердечной мышцы, легких, гладких мышц и мозга. Основной тип этого семейства представляет собой кальциевые каналы L-типа, функция которых ингибируется известными классами блокаторов кальциевых каналов (дигидропиридины, такие как нифедипин, фенилалкиламины, такие как верапамил, и бензотиазепины, такие как дилтиазем). Дополнительные классы кальциевых каналов плазменных мембран известны как T-тип, N-тип, P-тип, Q-тип и R-тип.
Кальциевые каналы "T-типа" (или "активируемые низким напряжением") названы так потому, что их открывание является менее протяженным (T=транзиторный (временный)), чем более протяженное (L=долго действующие) открывание кальциевых каналов L-типа. Каналы L, N, P и Q-типа активируются при более положительных потенциалах (активируемые высоким напряжением) и демонстрируют различную кинетику и потенциалзависимые свойства.
Кальциевые каналы Т-типа вовлечены в патологии, связанные с различными заболеваниями и расстройствами, включая эпилепсию, эссенциальный тремор, боль, нейропатическую боль, шизофрению, болезнь Паркинсона, депрессию, тревогу, расстройства сна, нарушения сна, бессонницу, психоз, сердечную аритмию, гипертензию, рак, диабет, бесплодие и сексуальную дисфункцию (J Neuroscience, 14, 5485 (1994); Drugs Future 30(6), 573-580 (2005); EMBO J, 24, 315-324 (2005); Drug Discovery Today, 11, 5/6,245-253 (2006); Neuropharmacology 53, 308-317(2007) и J. Biol. Chem., 283(15), 10162-10173 (2008)).
С другой стороны, блокаторы потенциалзависимых натриевых каналов, таких как TTX-S каналы, также имеют различные терапевтические применения.
Канал NaV1.3 крысы и канал NaV1.3 человека были клонированы в 1988 и 1998/2000 годах, соответственно (FEBS Lett. 228 (1), 187-194, 1988; J. Mol. Neurosci., 10 (1), 67-70, 1998; Eur. J. Neurosci. 12 (12), 4281-4289, 2000). Канал NaV1.3 ранее был известен как тип III натриевого канала головного мозга. NaV1.3 присутствует в относительно высоких уровнях в нервной системе эмбриона крысы, но практически не поддается обнаружению у взрослых крыс. NaV1.3 активируется после аксотомии при лигировании спинномозгового нерва (SNL), хронической компрессии (CCI) и диабетической невропатии (J Neurophysiol 82, 2776-2785, 1999. J. A. Black et al.; Ann Neurol 52, 786-792, 2002. M.J. Cranner et al.; Pain 83, 591-600, 1999. S. Dib-Hajj et al.; J Biol Chem 279, 29341-29350, 2004. S. Hong et al.; Mol Brain Res 95, 153-161, 2001. C.H. Kim et al.). Повышающее регулирование канала NaV1.3 способствует быстрому репраймингу тока натрия в малом дорсальном корешковом ганглии (DRG) (J Neurophysiol 82, 2776-2785, 1999. J.A. Black et al.). Эти наблюдения позволяют предположить, что NaV1.3 может вносить ключевой вклад в повышенную возбудимость нейронов.
В целях подтверждения вклада, вносимого натриевым каналом NaV1.3 в болевые состояния, были использованы специфические антисмысловые олигонуклеотиды (ASO) в моделях боли у животных. Обработка ASO натриевых каналов NaV1.3 значительно ослабляла связанное с болью поведение после операции CCI (J. Neurosci. 24, 4832-4839, 2004, Hains, B.C. et al.). Эти открытия говорят о том, что антагонист натриевого канала NaV1.3 является полезным для лечения состояний невропатической боли.
Оказалось, что канал NaV1.7 является лучшей 'подтвержденной' связанной с болью мишенью. Самые потрясающие открытия в отношении NaV1.7 связаны с генетическими исследованиями человека. Cox et al. (Nature 444, 894-898, 2006) обнаружили мутации SCN9A, которые вызывали потерю функции NaV1.7 в трех семьях из Пакистана. Их наблюдения говорят о том, что потеря функции NaV1.7 связана с врожденной неспособностью испытывать боль, это является дополнительным подтверждением, указывающим на то, что канал NaV1.7 принимает существенно важное участие в ноцицепции человека.
В отличие от этого, были также описаны мутации “приобретения новой функции”, которые приводят к усилению боли, например, первичному ограниченному отеку в одном случае и пароксизмальному сильному болевому расстройству в другом. Эти мутации “приобретения новой функции” у пациентов приводили к различным типам изменений механизма управления пропускания в NaV1.7 натриевых токах, и, что примечательно, различным степеням эффективности лекарственных средств, специфическим образом блокирующих натриевые каналы. Вывод из этих данных следует, что селективный блокатор NaV1.7 может быть эффективным средством для лечения боли у человека.
Местный анестетик лидокаин и летучий анестетик галотан, как известно, действуют на оба ТТХ-R и ТТХ-S натриевых канала с плохой селективностью и низкой активностью (диапазон значений IC50 от 50 мМ до 10 мМ). Эти анестетики при высокой системной концентрации могут вызвать разрушительные побочные эффекты, например, паралич и остановку сердца. Однако системное введение лидокаина при низких концентрациях является эффективным для лечения хронической боли (Trends in Pharm. Sci 22, 27-31, 2001, Baker, M.D. et al.). У крыс, применение очень низкой дозы ТТХ для DRG поврежденного сегмента L5 спинномозгового нерва значительно снижает механическое аллодиническое поведение (Brain Res 871, 98-103, 2000, Lyu, Y.S. et al.). Это говорит о том, что ТТХ-S подтипы натриевых каналов играют важную роль в поддержании аллодинического поведения в животной модели невропатической боли.
Канал NaV1.5 является также членом TTX-резистентных натриевых каналов. Канал NaV1.5 почти исключительно экспрессируется в сердечной ткани, и было показано, что он лежит в основе различных сердечных аритмий и нарушений сердечной проводимости.
В частности, арил-замещенные карбоксамидные производные по настоящему изобретению являются селективными в отношении каналов TTX-S в сравнении с каналами NaV1.5, приводя к улучшению профиля побочных эффектов.
Арил-замещенные карбоксамидные производные поэтому являются полезными для лечения широкого спектра заболеваний, в частности, таких заболеваний, как боль, острая боль, хроническая боль, невропатическая боль, боль при воспалении, висцеральная боль, ноцицептивная боль, в том числе послеоперационная боль, и боль смешанного типа, включая боль внутренних органов, желудочно-кишечного тракта, структур черепа, опорно-двигательного аппарата, позвоночника, мочеполовой системы, сердечно-сосудистой системы и ЦНС, в том числе раковая боль, спинная и орофациальная боль.
Другие состояния, которые можно лечить при помощи пиколинамидных производных по настоящему изобретению, включают рассеянный склероз, нейродегенеративные заболевания, синдром раздраженного кишечника, остеоартрит, ревматоидный артрит, невропатологические расстройства, функциональные расстройства кишечника, воспалительные заболевания кишечника; боль, связанную с дисменореей; тазовую боль, цистит, панкреатит, мигрень, кластерные и тензионные головные боли, диабетическую невропатию, периферическую невропатическую боль, пояснично-крестцовый радикулит; болезнь Крона, вызванную фибромиалгией; эпилепсию или эпилептические состояния, биполярную депрессию, тахиаритмию, расстройство настроения, биполярное расстройство, психические расстройства, такие как тревога и депрессия, миотонию, аритмию, нарушения движений, нейроэндокринные расстройства, атаксию, недержание, висцеральную боль, невралгию тройничного нерва, герпетическую невралгию, общую невралгию, постгерпетическую невралгию, радикулярную боль, пояснично-крестцовый радикулит, боль в спине, головную или шейную боль, сильную или некупируемую боль, приступ неконтролируемой боли, послеоперационную боль, инсульт, раковую боль, эпилепсию и каузалгию.
WO 2007120729, WO 2009054982, WO 2009054983 и WO 2009054984 раскрывают ряд гетероциклических амидных соединений, которые являются блокаторами кальциевых каналов Т-типа.
Соединения по настоящему изобретению, однако, структурно отличаются от известных соединений, раскрытых в приведенных выше документах предшествующего уровня техники, наличием уникального спейсера между карбонильной группой и концевой арильной группой. В частности, раскрытые соединения предшествующего уровня техники включают введение только одного атома углерода в качестве спейсера между карбонильной группой и гетероарилом, тогда как соединения по настоящему изобретению характеризуются введением различных уникальных спейсеров между карбонильной группой и концевой арильной группой.
WO 2003037274 раскрывает производные пиразола в качестве блокаторов натриевых каналов. Кроме того, WO 2002091830 раскрывает пиридинил-конденсированные бициклические амиды в качестве фунгицидов.
Новые соединения с трифторэтокси- или метоксигруппой на пиридиновом кольце или пиразиновом кольце и алкильной боковой цепью являются полезными для лечения состояния или расстройства, в котором участвуют потенциалзависимые натриевые каналы.
С другой стороны, циклопропанкарбоксамид наряду с трифторэтокси- или метоксигруппой на пиридиновом кольце или пиразиновом кольце является важным для лечения состояния или расстройства, в котором участвуют кальциевые каналы Т-типа. Такие соединения имеют преимущество перед соединениями, раскрытыми в WO 2007120729, WO 2009054982, WO 2009054983 и WO 2009054984 с точки зрения метаболизма.
Приведенные выше статьи, однако, никогда не раскрывали потенциалзависимые натриевые каналы. Поэтому арил-замещенные карбоксамидные производные по настоящему изобретению обеспечивают первые сведения о блокировании не только кальциевых каналов Т-типа, но также потенциалзависимых натриевых каналов.
Целью настоящего изобретения является обеспечение новых блокаторов кальциевых каналов Т-типа или блокаторов TTX-S, которые являются хорошими потенциальными лекарственными средствами. Предпочтительные соединения должны сильно связываться с каналами TTX-S (NaV1.3 и NaV1.7), в то же время, демонстрируя низкую аффинность в отношении других натриевых каналов, в частности, канала NaV1.5. Они должны хорошо абсорбироваться из желудочно-кишечного тракта, быть метаболически стабильными и обладать подходящими фармакокинетическими свойствами. Например, соединения по настоящему изобретению обладают отличными метаболическими свойствами по сравнению с соединениями, раскрытыми в WO 2007120729, WO 2009054982, WO 2009054983 и WO 2009054984. Они должны быть нетоксичными и демонстрировать мало побочных эффектов. Более того, идеальное лекарственное средство-кандидат должно существовать в физической форме, которая является стабильной, негигроскопичной и легко составляется в композиции.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение направлено на арил-замещенные карбоксамидные производные, которые являются блокаторами кальциевых каналов Т-типа или потенциалзависимых натриевых каналов, и которые являются полезными в лечении или профилактике неврологических и психических расстройств и заболеваний, в которых участвуют кальциевые каналы Т-типа или потенциалзависимые натриевые каналы. Настоящее изобретение также направлено на фармацевтические композиции, содержащие эти соединения, и применение этих соединений и композиций в профилактике или лечении таких заболеваний, в которых участвуют кальциевые каналы Т-типа или потенциалзависимые натриевые каналы. Нет необходимости говорить, что кальциевые каналы Т-типа или потенциалзависимые натриевые каналы действительно охватывают кальциевые каналы Т-типа и потенциалзависимые натриевые каналы.
ОПИСАНИЕ ВАРИАНТОВ ВОПЛОЩЕНИЯ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Настоящее изобретение обеспечивает применение соединения следующей формулы (I) для получения лекарственного средства для лечения состояния или расстройства, в котором участвуют кальциевые каналы Т-типа или потенциалзависимые натриевые каналы:
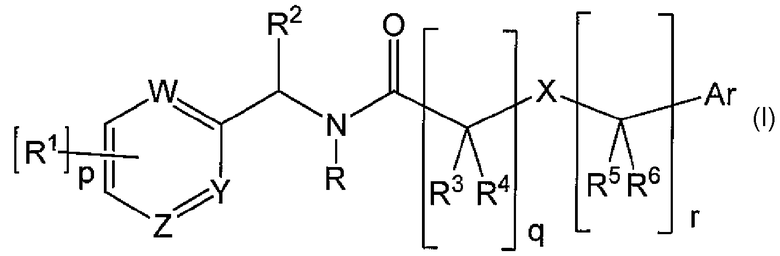
где:
R представляет собой водород или C1-6 алкил, который может быть замещен одним или несколькими заместителями, независимо выбранными из R7;
R1 независимо выбран из группы, включающей:
(1) водород, (2) галоген, (3) гидроксил, (4) -On-C1-6 алкил, где алкил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (5) -On-C3-6 циклоалкил, где циклоалкил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (6) C2-4 алкенил, где алкенил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (7) -On-фенил или -On-нафтил, где фенил или нафтил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (8) -On-гетероциклическую группу, где гетероциклическая группа является незамещенной или замещена одним или несколькими заместителями, независимо выбранными из R7, (9) -(C=O)-NR9R10, (10) -NR9R10, (11) -S(O)2-NR9R10, (12) -NR9-S(O)2R10, (13) -S(O)t-R9, где t имеет значение 0, 1 или 2, (14) -NR9(C=O)R10, (15) -CN и (16) -NO2;
где n имеет значение 0 или 1, когда n имеет значение 0, вместо On присутствует химическая связь;
p имеет значение 1, 2, 3 или 4; когда p имеет значение два или более чем два, R1 могут быть одинаковыми или отличными друг от друга;
R2 выбран из группы, включающей:
(1) водород, (2) C1-6 алкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (3) C3-6 циклоалкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (4) C2-6 алкенил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (5) C2-6 алкинил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (6) фенил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (7) -(C=O)-NR9R10 и (8) -(C=O)-O-C1-6 алкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7;
или R2 образует 5-7-членное кольцо с заместителем R1, который может содержать атом азота, атом кислорода, атом серы или двойную связь, где 5-7-членное кольцо необязательно замещено 1-6 заместителями, независимо выбранными из группы, включающей: (1) водород, (2) гидроксил, (3) галоген, (4) C1-6 алкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (5) C3-6 циклоалкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (6) -O-C1-6 алкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, и (7) -O-C3-6 циклоалкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7;
X представляет собой химическую связь, -C=C-, -циклоалкилен-, -циклоалкилен-C1-4-алкилен-O-, атом кислорода, атом серы или атом азота; когда X представляет собой -C=C-, -циклоалкилен-, -циклоалкилен-C1-4-алкилен-O- или атом азота, указанный заместитель X может содержать заместитель, независимо выбранный из определений для R9 и R10;
W, Y и Z независимо выбраны из атома азота и атома углерода, которые независимо необязательно замещены R1;
по меньшей мере один из W, Y и Z представляет собой азот, и W, Y и Z не могут одновременно представлять собой углерод;
R3, R4, R5 и R6 независимо выбраны из группы, включающей:
(1) водород, (2) гидроксил, (3) галоген, (4) C1-6 алкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (5) C3-6 циклоалкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (6) -O-C1-6 алкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (7) -O-C3-6 циклоалкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, и (8) -NR7R8;
или R3 и R4 и атом углерода, к которому они присоединены, образуют оксогруппу;
или R3 и R4 и атом углерода, к которому они присоединены, образуют C3-6 циклоалкильное кольцо, которое является незамещенным или замещено R7;
или R5 и R6 и атом углерода, к которому они присоединены, образуют оксогруппу;
или R5 и R6 и атом углерода, к которому они присоединены, образуют C3-6 циклоалкильное кольцо, которое является незамещенным или замещено R7;
q имеет значение 0, 1, 2, 3 или 4; когда q имеет значение один или более чем один, R3 и R4 могут быть одинаковыми или отличными друг от друга;
r имеет значение 0, 1, 2, 3 или 4; когда r имеет значение один или более чем один, R5 и R6 могут быть одинаковыми или отличными друг от друга;
когда (i) q имеет значение 1 и r имеет значение 0, или (ii) q имеет значение 0 и r имеет значение 1, X не является химической связью;
R7 выбран из группы, включающей:
(1) водород, (2) галоген, (3) гидроксил, (4) -(C=O)m-Ol-C1-6 алкил, где алкил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R8, (5) -Ol-(C1-3)перфторалкил, (6) -(C=O)m-Ol-C3-6 циклоалкил, где циклоалкил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R8, (7) -(C=O)m-C2-4 алкенил, где алкенил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R8, (8) -(C=O)m-Ol-фенил или -(C=O)m-Ol-нафтил, где фенил или нафтил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R8, (9) -(C=O)m-Ol-гетероциклическую группу, где гетероциклическая группа является незамещенной или замещена одним или несколькими заместителями, независимо выбранными из R8, (10) -(C=O)-NR9R10, (11) -NR9R10, (12) -S(O)2-NR9R10, (13) -S(O)t-R9, где t имеет значение 0, 1 или 2, (14) -CO2H, (15) -CN и (16) -NO2;
где l имеет значение 0 или 1, и m имеет значение 0 или 1; когда l имеет значение 0 или m имеет значение 0, присутствует химическая связь вместо (C=O)m или Ol, и когда l имеет значение 0 и m имеет значение 0, присутствует химическая связь вместо (C=O)m-Ol;
R8 независимо выбран из группы, включающей:
(1) водород, (2) гидроксил, (3) галоген, (4) C1-6 алкил, (5) -C3-6 циклоалкил, (6) -O-C1-6 алкил, (7) -O(C=O)-C1-6 алкил, (8) -NH-C1-6 алкил, (9) фенил, (10) гетероциклическую группу и (11) -CN;
R9 и R10 представляют собой, независимо, водород или C1-6 алкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из галогена, гидроксила и -O-C1-6 алкила; или R9 образует 4-7-членное кольцо вместе с R10, который может содержать атом азота, атом кислорода, атом серы или двойную связь, где 4-7-членное кольцо необязательно замещено 1-6 заместителями, независимо выбранными из группы, включающей: (1) водород, (2) гидроксил, (3) галоген, (4) C1-6 алкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R8, (5) C3-6 циклоалкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R8, (6) -O-C1-6 алкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R8, и (7) -O-C3-6 циклоалкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R8;
Ar представляет собой арил, который необязательно замещен 1-5 заместителями, независимо выбранными из группы, включающей:
(1) галоген, (2) гидроксил, (3) -On-фенил или -On-нафтил, где фенил или нафтил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (4) -On-гетероциклическую группу, где гетероциклическая группа является незамещенной или замещена одним или несколькими заместителями, независимо выбранными из R7, (5) -On-C1-6 алкил, где алкил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (6) -On-C3-6 циклоалкил, где циклоалкил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (7) -C2-4 алкенил, где алкенил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (8) -(C=O)-NR9R10, (9) -NR9R10, (10) -S(O)2-NR9R10, (11) -NR9-S(O)2R10, (12) -S(O)t-R9, где t имеет значение 0, 1 или 2, (13) -NR9(C=O)R10, (14) -CN и (15) -NO2;
где n имеет значение 0 или 1, когда n имеет значение 0, химическая связь присутствует вместо On;
или его фармацевтически приемлемой соли.
Настоящее изобретение обеспечивает соединения формулы (II)
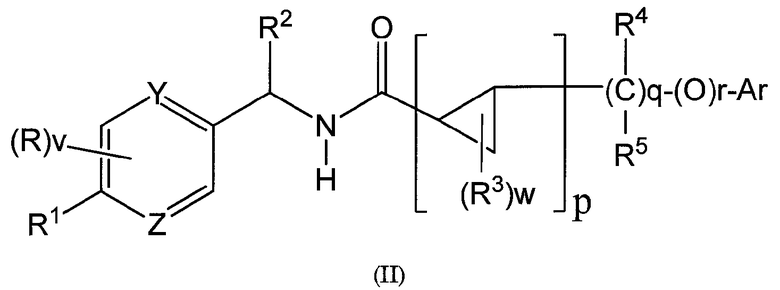
где
R представляет собой галоген или C1-6 алкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из галогена, гидроксила и -O-C1-6 алкила;
v имеет значение 0, 1, 2 или 3; когда v имеет значение два или более чем два, R могут быть одинаковыми или отличными друг от друга;
R1 представляет собой -OCH2CF3 или -OCH3;
R2 представляет собой C1-6 алкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из галогена, гидроксила и -O-C1-6 алкила;
R3 независимо выбран из группы, включающей:
(1) галоген, (2) C1-6 алкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R6, (3) C3-6 циклоалкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R6, (4) -O-C1-6 алкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R6, (5) -O-C3-6 циклоалкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R6 и (6) -NR7R8.
Предпочтительный R3 независимо выбран из группы, включающей:
(1) галоген, (2) C1-6 алкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из галогена;
w имеет значение 0, 1, 2, 3 или 4; когда w имеет значение два или более чем два, R3 могут быть одинаковыми или отличными друг от друга;
R4 и R5 представляют собой, независимо, водород, галоген или C1-6 алкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из галогена, гидроксила и -O-C1-6 алкила;
Предпочтительные R4 и R5 представляют собой, независимо, водород, галоген или C1-6 алкил, который является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из галогена;
R6 независимо выбран из группы, включающей:
(1) водород, (2) гидроксил, (3) галоген, (4) -OlR7, (5) -CN, (6) -(C=O)-NR7R8, (7) -NR7R8, (8) -S(O)2-NR7R8, (9) -S(O)t-R7, где t имеет значение 0, 1 или 2, (10) -CN и (11) -NO2;
где l имеет значение 0 или 1; когда l имеет значение 0, вместо Ol присутствует химическая связь;
R7 и R8 представляют собой, независимо, водород, C1-6 алкил или C3-8 циклоалкил, которые являются незамещенными или замещены одним или несколькими заместителями, независимо выбранными из галогена, гидроксила и -O-C1-6 алкила; или R7 образует 4-7-членное кольцо вместе с R8, который может содержать атом азота или атом кислорода, где 4-7-членное кольцо необязательно замещено 1-6 заместителями, независимо выбранными из группы, включающей: (1) водород, (2) гидроксил, (3) галоген, (4) C1-6 алкил и (5) -O-C1-6 алкил;
p, q и r представляют собой, независимо, 0 или 1; когда p имеет значение 0, оба q и r имеют значение 1, или оба q и r имеют значение 0.
Y и Z независимо выбраны из атома азота и атома углерода; Y и Z не могут одновременно представлять собой атом углерода;
когда p имеет значение 0, Ar выбран из группы, включающей фенил, индолил и хинолинил; где Ar необязательно замещен 1-5 заместителями, независимо выбранными из группы, включающей:
(1) галоген, (2) гидроксил, (3) -O-фенил или -O-нафтил, где фенил или нафтил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R6, (4) -On-гетероциклическую группу, где гетероциклическая группа является незамещенной или замещена одним или несколькими заместителями, независимо выбранными из R6, (5) -On-C1-6 алкил, где алкил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R6, (6) -On-C3-6 циклоалкил, где циклоалкил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R6, (7) -NR7R8, (8) -S(O)2-NR7R8, (9) -S(O)t-R7, где t имеет значение 0, 1 или 2, (10) -NR7SO2R8, (11) -(C=O)-NR7R8, (12) -NR7(C=O)R8, (13) -CN и (14) -NO2;
где предпочтительный Ar необязательно замещен 1-5 заместителями, независимо выбранными из группы, включающей:
(1) галоген, (2) гидроксил, (3) -O-фенил, где фенил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из галогена, метила, трифторметила и трифторметокси, (4) -On-гетероциклическую группу, где гетероциклическая группа является незамещенной или замещена одним или несколькими заместителями, независимо выбранными из галогена, метила, трифторметила и трифторметокси, (5) -On-C1-6 алкил, где алкил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из галогена, (6) -On-C3-6 циклоалкил, где циклоалкил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из галогена и (7) -CN;
где n имеет значение 0 или 1, когда n имеет значение 0, вместо On присутствует химическая связь;
когда p имеет значение 1, Ar представляет собой арил, который необязательно замещен 1-5 заместителями, независимо выбранными из группы, включающей:
(1) галоген, (2) гидроксил, (3) -On-гетероциклическую группу, где гетероциклическая группа является незамещенной или замещена одним или несколькими заместителями, независимо выбранными из R6, (4) -On-C1-6 алкил, где алкил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R6, (5) -On-C3-6 циклоалкил, где циклоалкил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R6, (6) -NR7R8, (7) -S(O)2-NR7R8, (8) -S(O)t-R7, где t имеет значение 0, 1 или 2, (9) -NR7SO2R8, (10) -(C=O)-NR7R8, (11) -NR7(C=O)R8, (12) -CN и (13) -NO2;
когда p имеет значение 1, предпочтительный Ar представляет собой арил, который необязательно замещен 1-5 заместителями, независимо выбранными из группы, включающей:
(1) галоген, (2) гидроксил, (3) -On-C1-6 алкил, где алкил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из галогена, (4) -On-C3-6 циклоалкил, где циклоалкил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из галогена, и (5) -CN;
где n имеет значение 0 или 1, когда n имеет значение 0, вместо On присутствует химическая связь;
или их фармацевтически приемлемые соли.
Подходящие соединения по настоящему изобретению представляют собой:
(R)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-2-(4-(трифторметил)фенокси)ацетамид;
(R)-3,5-дихлор-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
(R)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)хинолин-2-карбоксамид;
(1R,2R)-2-метил-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-2-(4-(трифторметил)фенил)циклопропанкарбоксамид;
(R)-4-трет-бутил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
(R)-N-(1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)-2-(4-(трифторметил)фенокси)ацетамид;
(R)-2-(пара-толилокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)ацетамид;
(R)-4-(2,2,2-трифторэтокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
(R)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-2-карбоксамид;
(R)-1-метил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-2-карбоксамид;
(R)-2-(2,4-дихлорфенокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)ацетамид;
(R)-2-(4-бромфенокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)ацетамид;
(R)-3-(3-фторфенил)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)пропанамид;
(R)-3-метил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензофуран-2-карбоксамид;
(R)-5-трет-бутил-2-метил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)фуран-3-карбоксамид;
(R)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-3-(трифторметил)бензамид;
(R)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-4-(трифторметил)бензамид;
(R)-5-фенил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-2-(трифторметил)фуран-3-карбоксамид;
(R)-3-фтор-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-5-(трифторметил)бензамид;
(R)-3-фтор-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-4-(трифторметил)бензамид;
(R)-4-фтор-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-3-(трифторметил)бензамид;
(R)-2-метил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-6-(трифторметил)-2H-индазол-3-карбоксамид;
(R)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-5-(трифторметил)пиколинамид;
(R)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-3-карбоксамид;
(R)-1-метил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-3-карбоксамид;
(R)-3-(1H-индол-1-ил)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)пропанамид;
(R)-1-метил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-5-(трифторметил)-1H-индол-2-карбоксамид;
(R)-N-(1-(5-(циклопропилметокси)пиридин-2-ил)этил)-5-фтор-1H-индол-2-карбоксамид;
(R,E)-N-(1-(5-(циклопропилметокси)пиридин-2-ил)этил)-3-(4-(трифторметил)фенил)акриламид;
(R,E)-N-(1-(5-(бензилокси)пиридин-2-ил)этил)-3-(4-(трифторметил)фенил)акриламид;
(R)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-2-(4-(трифторметил)фенил)тиазол-4-карбоксамид;
(R)-3-(6-фтор-1H-индол-1-ил)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)пропанамид;
(R)-N-(1-(5-(циклопропилметокси)пиридин-2-ил)этил)-3-(6-фтор-1H-индол-1-ил)пропанамид;
(R)-N-(1-(5-(2-фторбензилокси)пиридин-2-ил)этил)-2-(4-(трифторметил)фенокси)ацетамид;
(R)-5-(2,2,2-трифторэтокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)пиколинамид;
(R)-N-(1-(5-(пиридин-2-илметокси)пиридин-2-ил)этил)-2-(4-(трифторметил)фенокси)ацетамид;
N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1,2,3,4-тетрагидронафталин-2-карбоксамид;
(R,E)-3-(1H-индол-3-ил)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)акриламид;
(1R,2R)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)-2-(4-(трифторметил)фенил)циклопропанкарбоксамид;
(R)-N-(1-(5-((1-метилциклопропил)метокси)пиридин-2-ил)этил)-2-(4-(трифторметил)фенокси)ацетамид;
транс-2-(7-фтор-1H-индол-3-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(R)-3-хлор-4-метил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
(R)-4-трет-бутил-N-(1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)бензамид;
(R)-3-хлор-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
(R)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)хиноксалин-2-карбоксамид;
(R)-4-метокси-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)хинолин-2-карбоксамид;
(R)-5-изобутил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)изоксазол-3-карбоксамид;
(R)-3-(2-метилтиазол-4-ил)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
(R)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензо[b]тиофен-2-карбоксамид;
(R)-3-(бензилокси)-4-метокси-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
(R)-3-фенокси-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
(1S*,2S*)-2-(1H-индол-3-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-(1H-индол-3-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(R)-5-хлор-1-метил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-3-карбоксамид;
(R)-5-метокси-1-метил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-3-карбоксамид;
(R)-1,6-диметил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-3-карбоксамид;
(R)-6-фтор-1-метил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-3-карбоксамид;
(R)-5-метил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-3-карбоксамид;
(R)-5-фтор-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-3-карбоксамид;
(R)-5-хлор-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-3-карбоксамид;
(R)-6-хлор-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-3-карбоксамид;
транс-2-(1H-индол-6-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
(R)-1,5-диметил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-2-карбоксамид;
(R)-5-фтор-1-метил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-2-карбоксамид;
(R)-5-хлор-1-метил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-2-карбоксамид;
(R)-6-фтор-1-метил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-2-карбоксамид;
(R)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-4-(трифторметокси)бензамид;
(R)-5-фенил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)изоксазол-3-карбоксамид;
(R)-5-бром-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-2-карбоксамид;
(R)-6-хлор-1-метил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-2-карбоксамид;
(R)-1-метил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-5-(трифторметокси)-1H-индол-2-карбоксамид;
(R)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-3-(трифторметокси)бензамид;
транс-2-(хинолин-7-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
транс-2-(хинолин-7-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)циклопропанкарбоксамид;
транс-2-(изохинолин-3-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
транс-2-(хинолин-3-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)циклопропанкарбоксамид;
транс-2-((4-хлорфенокси)метил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)циклопропанкарбоксамид;
транс-2-(2-фтор-5-метоксифенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
транс-2-((1H-индол-1-ил)метил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(R)-6-фтор-1-метил-N-(1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)-1H-индол-2-карбоксамид;
транс-2-(2,5-дифторфенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
транс-N-((R)-1-(5-(циклопропилметокси)пиридин-2-ил)этил)-2-(2,5-дифторфенил)циклопропанкарбоксамид;
транс-2-(2,5-дифторфенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)циклопропанкарбоксамид;
транс-N-((R)-1-(5-(циклопропилметокси)пиридин-2-ил)этил)-2-(1H-индол-4-ил)циклопропанкарбоксамид;
транс-2-(4-метокси-3-метилфенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-(1H-индол-6-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
транс-2-(хинолин-6-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
транс-2-(5-фтор-1H-индол-2-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
транс-2-(хинолин-3-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
транс-2-(1H-индол-4-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(8-хлорхинолин-2-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)циклопропанкарбоксамид;
(R)-5-метокси-N-(1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)-1H-индол-2-карбоксамид;
(R)-N-(1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)-4-(трифторметокси)бензамид;
(R)-3-фенокси-N-(1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)бензамид;
(R)-6-метокси-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)хинолин-2-карбоксамид;
(1S*,2S*)-2-(1H-индол-2-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-(1H-индол-2-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(1H-индол-2-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)циклопропанкарбоксамид;
транс-2-(1-метил-1H-индазол-6-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-(4-(бензилокси)фенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(R,E)-3-(хинолин-2-ил)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)акриламид;
(1S*,2S*)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-2-(2,4,6-трифторфенил)циклопропанкарбоксамид;
(1S*,2S*)-2-(3,5-дифторфенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(3-метоксифенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(4-метоксифенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-(2-хлор-4-фторфенил)-N-((R)-1-(5-(циклопропилметокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-N-((R)-1-(5-(циклопропилметокси)пиридин-2-ил)этил)-2-(2-фтор-4-метоксифенил)циклопропанкарбоксамид;
(1S*,2S*)-N-((R)-1-(5-(циклопропилметокси)пиридин-2-ил)этил)-2-(2,4,6-трифторфенил)циклопропанкарбоксамид;
(1S*,2S*)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)-2-(2,4,6-трифторфенил)циклопропанкарбоксамид;
(1R*,2R*)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)-2-(2,4,6-трифторфенил)циклопропанкарбоксамид;
(1S*,2S*)-2-(1H-индол-4-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-фенил-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-фенил-N-((R)-1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-фенил-N-((R)-1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-фенил-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-фенил-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(1H-бензо[d]имидазол-2-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(1H-бензо[d]имидазол-2-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-(1H-бензо[d]имидазол-2-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(феноксиметил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-((3-фторфенокси)метил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-((3-цианофенокси)метил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-((4-фторфенокси)метил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-((4-цианофенокси)метил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-(феноксиметил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-((3-фторфенокси)метил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-((3-цианофенокси)метил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-((4-фторфенокси)метил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-((4-цианофенокси)метил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(4-((3-метилоксетан-3-ил)метокси)фенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-N-((R)-1-(5-(циклопропилметокси)пиридин-2-ил)этил)-2-(1H-индол-7-ил)циклопропанкарбоксамид;
(1S*,2S*)-2-(феноксиметил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-(хинолин-2-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
4-(бензилокси)-3-метокси-N-((6-(трифторметил)пиридин-3-ил)метил)бензамид;
2-(4-(трифторметил)фенокси)-N-((6-(трифторметил)пиридин-3-ил)метил)ацетамид;
(R)-N-(1-(6-метил-3-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-2-(4-(трифторметил)фенокси)ацетамид;
(R)-5-фтор-N-(1-(6-метил-3-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-2-карбоксамид;
(S)-4-изопропил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
(S)-2-(4-хлорфенокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)ацетамид;
(S)-4-(2,2,2-трифторэтокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
(1S*,2S*)-2-(4-(бензилокси)фенил)-N-((S)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-(4-(бензилокси)фенил)-N-((S)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(2-фтор-4-метоксифенил)-N-((S)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-(2-фтор-4-метоксифенил)-N-((S)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(2-хлор-4-фторфенил)-N-((S)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-(2-хлор-4-фторфенил)-N-((S)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-фенил-N-((S)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-фенил-N-((S)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
трет-бутил ((R)-1-оксо-1-(((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)амино)-3-(2-(трифторметокси)фенил)пропан-2-ил)карбамат;
трет-бутил ((R)-1-оксо-1-(((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)амино)-3-(2-(трифторметил)фенил)пропан-2-ил)карбамат;
(R)-N-(1-(5-метоксипиридин-2-ил)этил)-3-феноксибензамид;
(R)-2-гидрокси-4-фенил-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бутанамид;
трет-бутил ((S)-1-(4-хлорфенил)-3-оксо-3-(((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)амино)пропил)карбамат;
трет-бутил ((R)-1-(4-хлорфенил)-3-оксо-3-(((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)амино)пропил)карбамат;
трет-бутил ((R)-3-(4-хлорфенил)-1-оксо-1-(((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)амино)пропан-2-ил)карбамат;
трет-бутил ((S)-3-(2-хлорфенил)-1-оксо-1-(((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)амино)пропан-2-ил)карбамат;
трет-бутил ((S)-3-(2-фторфенил)-1-оксо-1-(((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)амино)пропан-2-ил)карбамат;
(R)-2-(2-хлорфенокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)ацетамид;
(R)-2-(3-хлорфенокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)ацетамид;
(R)-2-(2-хлорфенокси)-2-метил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)пропанамид;
(R)-2-(2,3-дихлорфенокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)ацетамид;
(R)-2-(o-толилокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)ацетамид;
(R)-2-(м-толилокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)ацетамид;
(R)-2-(2,4-диметилфенокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)ацетамид;
(R)-2-(2-хлор-6-метилфенокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)ацетамид;
(R)-2-(4-(трет-бутил)фенокси)-N-(1-(5-метоксипиридин-2-ил)этил)ацетамид;
(R)-2-амино-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-3-(2-(трифторметил)фенил)пропанамид;
изобутил ((R)-1-оксо-1-(((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)амино)-3-(2-(трифторметил)фенил)пропан-2-ил)карбамат;
этил ((R)-1-оксо-1-(((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)амино)-3-(2-(трифторметил)фенил)пропан-2-ил)карбамат;
N-((5-(2,2,2-трифторэтокси)пиридин-2-ил)метил)-3-(трифторметокси)бензамид;
4-(2,2,2-трифторэтокси)-N-((5-(2,2,2-трифторэтокси)пиридин-2-ил)метил)бензамид;
6-фтор-1-метил-N-((5-(2,2,2-трифторэтокси)пиридин-2-ил)метил)-1H-индол-2-карбоксамид;
3-(2,2,2-трифторэтокси)-N-((5-(2,2,2-трифторэтокси)пиридин-2-ил)метил)бензамид;
(R)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-2-(3-(трифторметил)фенокси)ацетамид;
(R)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-2-(2-(трифторметокси)фенокси)ацетамид;
(R)-N-(1-(5-метоксипиридин-2-ил)этил)-2-(3-(трифторметил)фенокси)ацетамид;
(R)-3-(2,2,2-трифторэтокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
(R)-N-(1-(5-метоксипиридин-2-ил)этил)-2-(4-(трифторметил)фенил)тиазол-4-карбоксамид;
(R)-N-(1-(5-метоксипиридин-2-ил)этил)-1-метил-5-(трифторметокси)-1H-индол-2-карбоксамид;
(R)-2-(4-хлорфенокси)-N-(1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)ацетамид;
(R)-5-(2,2,2-трифторэтокси)-N-(1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)пиколинамид;
(R)-N-(1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)-3-(трифторметокси)бензамид;
(R)-4-фтор-3-фенокси-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
4-(трет-бутил)-N-((6-метоксипиридин-3-ил)метил)бензамид;
N-((6-метоксипиридин-3-ил)метил)-2-(4-(трифторметил)фенокси)ацетамид;
4-(трет-бутил)-N-((5-метоксипиридин-2-ил)метил)бензамид;
(S)-4-(трет-бутил)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
(S)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-3-(трифторметокси)бензамид;
(S)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-4-(трифторметокси)бензамид;
(S)-3-(2,2,2-трифторэтокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
(R)-N-(1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)-4-(трифторметокси)бензамид;
(R)-4-(2,2,2-трифторэтокси)-N-(1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)бензамид;
(R)-3-(2,2,2-трифторэтокси)-N-(1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)бензамид;
4-(трет-бутил)-N-((5-(трифторметил)пиридин-2-ил)метил)бензамид;
3-(трифторметокси)-N-((5-(трифторметил)пиридин-2-ил)метил)бензамид;
4-(трифторметокси)-N-((5-(трифторметил)пиридин-2-ил)метил)бензамид;
4-(2,2,2-трифторэтокси)-N-((5-(трифторметил)пиридин-2-ил)метил)бензамид;
3-(2,2,2-трифторэтокси)-N-((5-(трифторметил)пиридин-2-ил)метил)бензамид;
4-(трет-бутил)-N-((6-(пиперидин-1-ил)пиридин-3-ил)метил)бензамид;
N-((6-(пиперидин-1-ил)пиридин-3-ил)метил)-3-(трифторметокси)бензамид;
N-((6-(пиперидин-1-ил)пиридин-3-ил)метил)-4-(2,2,2-трифторэтокси)бензамид;
4-(трет-бутил)-N-((6-(пирролидин-1-ил)пиридин-3-ил)метил)бензамид;
N-((6-(пирролидин-1-ил)пиридин-3-ил)метил)-3-(трифторметокси)бензамид;
N-((6-(пирролидин-1-ил)пиридин-3-ил)метил)-4-(2,2,2-трифторэтокси)бензамид;
4-(трет-бутил)-N-((6-(трифторметил)пиридин-3-ил)метил)бензамид;
3-(трифторметокси)-N-((6-(трифторметил)пиридин-3-ил)метил)бензамид;
4-(трет-бутил)-N-((6-(пирролидин-1-ил)пиридин-2-ил)метил)бензамид;
N-((6-(пирролидин-1-ил)пиридин-2-ил)метил)-3-(трифторметокси)бензамид;
N-((6-(пирролидин-1-ил)пиридин-2-ил)метил)-4-(2,2,2-трифторэтокси)бензамид;
(R)-4-хлор-2-метокси-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
(R)-4-(2-цианопропан-2-ил)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
(R)-3-хлор-4-метокси-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
(R)-6-метокси-1-метил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-2-карбоксамид;
(R)-N-(1-(6-метил-3-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-3-(трифторметокси)бензамид;
(R)-N-(1-(6-метил-3-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-4-(2,2,2-трифторэтокси)бензамид;
(S)-2-(3-хлорфенокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)ацетамид;
2-(3-хлорфенокси)-N-((5-(2,2,2-трифторэтокси)пиридин-2-ил)метил)ацетамид;
(R)-2-(3-хлорфенокси)-N-(1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)ацетамид;
(R)-4-этил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
(R)-3-фтор-4-метил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
(R)-5-хлор-2-метокси-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)бензамид;
(R)-N-(1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)хиноксалин-2-карбоксамид;
(R)-N-(1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)-5-(трифторметил)пиколинамид;
и их соли.
Также, настоящее изобретение обеспечивает применение соединения формулы (I) или его фармацевтически приемлемой соли, которые описаны в настоящей заявке, для получения лекарственного средства для лечения состояния или расстройства, опосредованного кальциевыми каналами Т-типа или потенциалзависимыми натриевыми каналами; в частности, блокирующей активностью кальциевых каналов Т-типа или блокирующей активностью потенциалзависимых натриевых каналов. Для того, чтобы применять соединения формулы (I) и их фармацевтически приемлемые соли в терапии, их, как правило, формируют в фармацевтическую композицию в соответствии со стандартной фармацевтической практикой. Настоящее изобретение также обеспечивает фармацевтическую композицию, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель или эксципиент.
Предпочтительно, настоящее изобретение, также обеспечивает применение соединения формулы (I) или его фармацевтически приемлемой соли, описанных в настоящей заявке, для получения лекарственного средства для лечения заболеваний, выбранных из заболеваний, связанных с кальциевыми каналами Т-типа, или заболеваний, связанных с потенциалзависимыми натриевыми каналами.
Также, настоящее изобретение обеспечивает применение соединения формулы (I) или его фармацевтически приемлемой соли, описанных в настоящей заявке, для получения лекарственного средства для лечения состояния или расстройства, в котором участвуют потенциалзависимые натриевые каналы, как описано в формуле (I) в настоящей заявке, где, когда Y представляет собой атом азота, и в то же время (i) q имеет значение 1 и r имеет значение 0, или (ii) q имеет значение 0 и r имеет значение 1, тогда X может быть химической связью;
или, как описано в формуле (I) в настоящей заявке, где, когда Y представляет собой атом углерода, Z представляет собой атом азота, W представляет собой атом азота, и в то же время (i) q имеет значение 1 и r имеет значение 0, или (ii) q имеет значение 0 и r имеет значение 1, тогда Х может быть химической связью;
определение других дескрипторов, такое же, как описано в настоящей заявке.
Также, настоящее изобретение обеспечивает фармацевтическую композицию, содержащую соединение формулы (I) или его фармацевтически приемлемую соль, описанные в настоящей заявке, вместе с фармацевтически приемлемым носителем для указанного соединения.
Также, настоящее изобретение обеспечивает фармацевтическую композицию, содержащую соединение формулы (I) или его фармацевтически приемлемую соль, описанные в настоящей заявке, вместе с фармацевтически приемлемым носителем для указанного соединения и другим фармакологически активным веществом.
Также, настоящее изобретение обеспечивает способ получения фармацевтической композиции, который включает смешивание соединения формулы (I) или его фармацевтически приемлемой соли и фармацевтически приемлемого носителя или эксципиента.
Также, настоящее изобретение обеспечивает промежуточное соединение для способа получения соединения формулы (I) или его фармацевтически приемлемой соли.
Кроме того, настоящее изобретение обеспечивает способ лечения состояния или расстройства, опосредованного активностью кальциевых каналов Т-типа или активностью потенциалзависимых натриевых каналов у млекопитающего, который включает введение млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, которые описаны в настоящей заявке.
В следующем аспекте, настоящее изобретение обеспечивает способ получения фармацевтической композиции, который включает смешивание соединения формулы (I) или его фармацевтически приемлемой соли и фармацевтически приемлемого носителя или эксципиента.
Примеры состояний или расстройств, опосредованных активностью кальциевых каналов Т-типа или активностью потенциалзависимых натриевых каналов включают, но не ограничиваются этим, заболевания, связанные с кальциевыми каналами Т-типа или заболевания, связанные с потенциалзависимыми натриевыми каналами. Соединения по настоящему изобретению демонстрируют блокирующую активность кальциевых каналов Т-типа или блокирующую активность потенциалзависимых натриевых каналов. Соединения по настоящему изобретению могут демонстрировать меньшую токсичность, хорошую абсорбцию, распределение, хорошую растворимость, меньшую белок-связывающую активность по сравнению с другими кальциевыми каналами Т-типа или потенциалзависимыми натриевыми каналами, меньшее межлекарственное взаимодействие, хорошую метаболическую стабильность, меньшую ингибирующую активность в отношении HERG канала и уменьшенную продолжительность QT.
Как будет понятно специалисту в данной области, термины "галоген" или "гало", используемые в настоящей заявке, предназначены для включения фтора, хлора, брома и йода.
Аналогичным образом, C1-6, как это используется в термине C1-6 алкил, означает группу, содержащую 1, 2, 3, 4, 5 или 6 атомов углерода в линейном или разветвленном расположении, таким образом, C1-8 алкил конкретно включает метил, этил, н-пропил, изо-пропил, н-бутил, изобутил, трет-бутил, пентил и гексил. Аналогичным образом, C2-6 алкенил означает группу, содержащую 2, 3, 4, 5 или 6 атомов углерода, которая включает, по меньшей мере, одну двойную связь, которая может быть в E- или Z- конфигурации. Группа, которая обозначается как независимо замещенная заместителями, может быть независимо замещена множеством таких заместителей.
Термин "алкенил", используемый в настоящей заявке, означает углеводородный радикал, содержащий, по меньшей мере, одну двойную связь, включая, но не ограничиваясь этим, этенил, пропенил, 1-бутенил, 2-бутенил и подобные.
Термин "циклоалкил", используемый в настоящей заявке, означает моно- или бициклическое кольцо, включая, но не ограничиваясь этим, группы циклопропила, циклобутила, циклопентила, циклогексила, циклогептила, норборанила и адамантила и подобные.
Термин "арил", используемый в настоящей заявке, означает моно- или би-карбоциклическое или моно- или би-гетероциклическое кольцо, которое может содержать 0-4 гетероатома, выбранных из O, N и S, включая, но не ограничиваясь этим, фенил, фурил, тиенил, оксазолил, тетразолил, тиадиазолил, пиридил, пиримидинил, пирролил, тиофенил, пиразинил, пиридазинил, изооксазолил, изотиазолил, триазолил, фуразанил, нафтил, тетрагидронафтил, инданил, бензофуранил, изобензофуранил, бензотиофенил, индолил, изоиндолил, бензохазолил, бензотиазолил, индазолил, бензоимидазолил, бензотриазолил, имидазопиридинил, пиразолопиримидинил, хинолил, изохинолил, циннолинил, нафтиридинил, фталазинил, хиназолинил, хиноксалинил, триазолопиримидинил и указанные кольца, которые являются полностью или частично насыщенными, такие как пиридин-2-онил, пиперидинил, пирролидинил, тетрагидронафталенил и подобные.
Термин "гетероциклическая группа", используемый в настоящей заявке, включает как ненасыщенные, так и насыщенные гетероциклические группы, где ненасыщенные гетероциклические группы (то есть "гетероарил") включают бензоимидазолил, бензимидазолонил, бензофуранил, бензофуразанил, бензопиразолил, бензотриазолил, бензотиофенил, бензоксазолил, карбазолил, карболинил, циннолинил, фуранил, имидазолил, индолинил, индолил, индолазинил, индазолил, изобензофуранил, изоиндолил, изохинолил, изотиазолил, изоксазолил, нафтпиридинил, оксадиазолил, оксазолил, оксазолин, изоксазолин, оксетанил, пиразинил, пиразолил, пиридазинил, пиридопиридинил, пиридазинил, пиридил, пиримидинил, пирролил, хиназолинил, хинолил, хиноксалинил, тетразолил, тетразолопиридил, тиадиазолил, тиазолил, тиенил, триазолил и их N-оксиды, и где насыщенные гетероциклические группы включают азетидинил, 1,4-диоксанил, гексагидроазепинил, пиперазинил, пиперидинил, пиридин-2-онил, пирролидинил, морфолинил, тетрагидрофуранил, тиоморфолинил и тетрагидротиенил и их N-оксиды и S-оксиды.
Термин "C0", используемый в настоящей заявке, означает простую связь.
Термин "защитная группа", используемый в настоящей заявке, означает гидрокси- или амино-защитную группу, которые выбирают из типичных гидрокси- или аминозащитных групп, описанных в Protective Groups in Organic Synthesis edited by T. W. Greene et al. (John Wiley & Sons, 1991);
Термин "лечение" и "терапия", используемый в настоящей заявке, относится к лечебному, паллиативному и профилактическому лечению, включая реверсирование, облегчение, ингибирование развития или предотвращения расстройства или состояния, к которому этот термин применяется, или одного или нескольких симптомов такого расстройства или состояния.
Используемая в настоящей заявке форма единственного числа охватывает также форму множественного числа объекта, если не указано иное.
В объем "соединений по настоящему изобретению" включены все соли, сольваты, гидраты, комплексы, полиморфы, пролекарства, радиоактивно меченные производные, стереоизомеры и оптические изомеры соединений формулы (I).
Соединения формулы (I) могут образовывать их кислотно-аддитивные соли. Должно быть понятно, что для использования в медицине соли соединений формулы (I) должны быть фармацевтически приемлемыми. Подходящие фармацевтически приемлемые соли будут очевидны специалистам в данной области и включают соли, описанные в J. Pharm. Sci, 1977, 66, 1-19, такие как кислотно-аддитивные соли, образованные с неорганическими кислотами, например хлористоводородной, бромистоводородной, серной, азотной или фосфорной кислотами; и органическими кислотами, например янтарной, малеиновой, муравьиной, уксусной, трифторуксусной, пропионовой, фумаровой, лимонной, винной, бензойной, пара-толуолсульфоновой, метансульфоновой или нафталинсульфоновой кислотой. Некоторые из соединений формулы (I) могут образовывать кислотно-аддитивные соли с одним или несколькими эквивалентами кислоты. Настоящее изобретение включает во всем его объеме все возможные стехиометрические и нестехиометрические формы. Кроме того, некоторые соединения, содержащие кислотные функциональные группы, такие как карбокси, могут быть выделены в форме их неорганической соли, в которой противоион может быть выбран из натрия, калия, лития, кальция, магния и подобных, а также из органических оснований.
Соединения формулы (I) и их соли могут быть получены в кристаллической или некристаллической форме, и, в случае кристаллической формы, необязательно могут быть гидратированы или сольватированы. Настоящее изобретение включает в полном объеме стехиометрические гидраты или сольваты, а также соединения, содержащие различные количества воды и/или растворителя.
Соли и сольваты, содержащие фармацевтически неприемлемые противоионы или ассоциированные растворители, входят в объем настоящего изобретения, например, для использования в качестве промежуточных соединений в получении других соединений формулы (I) и их фармацевтически приемлемых солей.
Соединения формулы (I) могут содержать полиморфы в кристаллической форме, которые входят в объем настоящего изобретения.
Кроме того, соединения формулы (I) можно вводить в качестве пролекарств. Используемый в настоящей заявке термин "пролекарство" соединения формулы (I) представляет собой функциональное производное соединения, которое после введения пациенту, в конечном счете, высвобождает in vivo соединение формулы (I). Введение соединения формулы (I) в качестве пролекарства может позволить специалисту в данной области сделать одно или несколько из следующих: (a) модифицировать начало действия соединения in vivo; (b) модифицировать продолжительность действия соединения in vivo; (c) модифицировать доставку или распределение соединения in vivo; (d) модифицировать растворимость соединения in vivo; и (e) преодолеть побочные эффекты или другие трудности, возникающие с соединением. Типичные функциональные производные, используемые для получения пролекарств, включают модификации соединения, которые химически или ферментативно расщепляются in vivo. Такие модификации, которые включают получение фосфатов, амидов, сложных эфиров, тиоэфиров, карбонатов и карбаматов, хорошо известны специалистам в данной области.
В некоторых из соединений формулы (I), могут содержаться несколько хиральных атомов углерода. В таких случаях соединения формулы (I) существуют в виде стереоизомеров. Настоящее изобретение распространяется на все оптические изомеры, такие как стереоизомерные формы соединений формулы (I), включая энантиомеры, диастереоизомеры и их смеси, такие как рацематы. Различные стереоизомерные формы могут быть разделены или отделены друг от друга при помощи обычных способов, или любой данный изомер может быть получен путем обычного стереоселективного или асимметричного синтеза.
Некоторые из соединений в настоящей заявке могут существовать в различных таутомерных формах, и должно быть понятно, что настоящее изобретение охватывает все такие таутомерные формы.
Настоящее изобретение также включает изотопно-меченные соединения, которые идентичны тем, что описаны в настоящей заявке, но при этом один или более атомов замещены атомом, имеющим атомную массу или массовое число, отличное от атомной массы или массового числа, обычно встречающихся в природе. Примеры изотопов, которые могут быть включены в соединения по настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора, йода и хлора, такие как 3H, 11C, 14C, 18F, 123I и 125I. Соединения по настоящему изобретению, которые содержат вышеуказанные изотопы и/или другие изотопы других атомов, включены в объем настоящего изобретения. Изотопно-меченные соединения по настоящему изобретению, например, те, в которые включены радиоактивные изотопы, такие как 3H, 14C, являются полезными в анализах распределения в тканях лекарственного средства и/или субстрата. Особенно предпочтительными изотопами являются тритий, то есть 3H, и углерод-14, то есть 14C, благодаря простоте их получения и способности к обнаружению. Изотопы 11C и 18F особенно полезны в PET (позитронная эмиссионная томография), и изотопы 125I особенно полезны в SPECT (однофотонная эмиссионная компьютерная томография), все они полезны в томографии головного мозга. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, то есть 2Н, может обеспечить определенные терапевтические преимущества, вытекающие из большей метаболической стабильности, например увеличение периода полураспада in vivo или сокращение необходимой дозировки, и, следовательно, может быть предпочтительным в некоторых случаях. Изотопно-меченные соединения по настоящему изобретению, как правило, можно получить путем осуществления процедур, раскрытых ниже на Схемах и/или в примерах, затем используя легко доступный изотопно-меченный регент вместо немеченного изотопом реагента.
Активности и эффективности соединений по настоящему изобретению в отношении кальциевых каналов Т-типа или потенциалзависимых натриевых каналов могут быть определены при помощи хорошо известной в данной области методики, включая "анализ притока Ca2+", "электрофизиологический анализ для Ca2+ T-типа", "FRET анализ для Navs" и "электрофизиологический анализ для Navs", как описано в настоящей заявке. Соединения формулы (I) продемонстрировали блокирующую активность кальциевых каналов Т-типа в анализах, описанные в настоящей заявке.
Характерную блокирующую активность кальциевых каналов Т-типа или блокирующую активность потенциалзависимых натриевых каналов соединения, которое может быть использовано в настоящем изобретении, можно определить при помощи этих анализов. В частности, соединения следующих примеров обладали активностью блокирования кальциевых каналов T-типа или потенциалзависимых натриевых каналов в вышеуказанных анализах, как правило, со значением IC50 менее чем около 10 мкМ, предпочтительно менее чем около 1 мкМ, более предпочтительно менее чем около 0,3 мкМ. Некоторые из соединений по настоящему изобретению обладали активностью блокирования кальциевых каналов T-типа или потенциалзависимых натриевых каналов в вышеуказанных анализах с IC50 менее чем около 1 мкМ. Такой результат свидетельствует о характерной для соединений активности при использовании в качестве блокаторов активности кальциевых каналов Т-типа или активности потенциалзависимых натриевых каналов.
Что касается других соединений, раскрытых в известном уровне техники, соединения настоящего изобретения проявляют неожиданные свойства, например, что касается продолжительности действия и/или метаболизма, такие как увеличение метаболической стабильности, повышение оральной биодоступности или абсорбции, и/или снижение межлекарственных взаимодействий.
Кальциевые каналы Т-типа вовлечены в широкий спектр биологических функций. Это предполагает возможную роль этих рецепторов в различных патологических процессах у человека или других видов. Соединения по настоящему изобретению применимы для лечения, предотвращения, облегчения, контроля или снижения риска различных неврологических и психических расстройств, связанных с кальциевыми каналами, включая одно или несколько из следующих состояний или заболеваний: двигательные расстройства, включая акинез и акинетически-ригидные синдромы (включая болезнь Паркинсона, медикаментозный паркинсонизм, постэнцефалитический паркинсонизм, прогрессирующий супрануклеарный паралич, множественную системную атрофию, кортико-базальную дегенерацию, комплекс паркинсонизм-ALS деменция и кальцификацию базальных ядер головного мозга), синдром хронической усталости, усталость, включая усталость Паркинсонового типа, усталость при рассеянном склерозе; усталость, вызванная расстройством сна или расстройством суточного ритма, медикаментозно-индуцированный паркинсонизм (такой, как индуцированный нейролептиками паркинсонизм; злокачественный нейролептический синдром; индуцированная нейролептиками острая дистония; индуцированная нейролептиками острая акатизия; индуцированная нейролептиками поздняя дискинезия и медикаментозно-индуцированный постуральный тремор), синдром Жиль де ля Туретта, судороги, эпилепсия и дискинезия [включая тремор (такой как тремор в покое, эссенциальный тремор, постуральный тремор и интенционное дрожание), хорея (такая как хорея Сиденгама, болезнь Гентингтона, доброкачественная наследственная хорея, нейроакантоцитоз, симптоматическая хорея; хорея, обусловленная действием лекарственного средства, и гемибаллизм), миоклонус (включая генерализованный миоклонус и очаговый миоклонус), тики (включая простые тики, сложные тики и симптоматические тики), синдром беспокойных ног и дистония (включая генерализованную дистонию, такую как идиопатическая дистония; дистония, обусловленная действием лекарственного средства; симптоматическая дистония и приступообразная дистония и фокальная дистония, такая как тонический блефароспазм, оромандибулярная дистония, спастическая дисфония, спастическая кривошея, осевая дистония, дистонический графоспазм и гемиплегическая дистония); болезнь сердца, нарушения сердечного ритма и аритмии, инфаркт миокарда, застойная сердечная недостаточность, ишемическая болезнь сердца, внезапная смерть, инсульт, сексуальные и репродуктивные дисфункции, такие как нарушение фертильности, бесплодие, заболевания или расстройства, при которых в головном мозге происходит аномальная осцилляторная активность, включая депрессию, мигрень, невропатическую боль, болезнь Паркинсона, психоз и шизофрению, а так же заболевания или расстройства, при которых происходит аномальное сочетание активности, в частности через таламус; усиление когнитивной функции, улучшение памяти, увеличение сохранения данных в памяти; улучшение способности к обучению; повышение иммунного ответа; повышение иммунной функции; приливы; ночные приливы; увеличение продолжительности жизни; шизофрения; связанные с мышцами расстройства, которые контролируются ритмами возбуждения/релаксации, задаваемыми нервной системой, такие как сердечный ритм и другие расстройства сердечно-сосудистой системы; состояния, связанные с пролиферацией клеток, такие как расширение кровеносных сосудов или сужение сосудов и артериальное давление; рак; сердечная аритмия; гипертензия; застойная сердечная недостаточность; состояния половой/мочевыделительной системы; расстройства половой функции и фертильности; адекватность функции почек; чувствительность к анестетикам; расстройства сна, нарушения сна, включая повышение качества сна, улучшение качества сна, повышение эффективности сна, способствующие поддержанию сна; увеличение значения, которое рассчитывается, исходя из времени, в течение которого субъект спит, деленное на время, в течение которого субъект пытается заснуть; улучшение засыпания; уменьшение латентности сна или начала сна (время, которое требуется, чтобы заснуть); уменьшение трудностей с засыпанием, повышение непрерывности сна; уменьшение числа пробуждений во время сна; снижение прерывистых пробуждений во время сна; уменьшение случаев ночного пробуждения, снижение времени бодрствования после начала наступления сна, повышение общего количества сна; уменьшения фрагментации сна, изменение времени, частоты или продолжительности приступов быстрого сна; изменение времени, частоты или длительности медленной волны (т.е. стадии 3 или 4) периодов сна; увеличение количества и процента стадии 2 сна; активирование медленного сна; усиление EEG-дельта активности во время сна; увеличение количества Дельта сна в начале периода сна, увеличение фазы быстрого сна в конце периода сна; снижение ночного пробуждения, особенно ранних утренних пробуждений; увеличение дневного бодрствования; снижение дневной сонливости; лечение или снижение избыточной сонливости в дневное время; повышение удовлетворенности интенсивностью сна; усиление поддержания сна; идиопатическая бессонница; проблемы со сном; бессонница, повышенная сонливость, идиопатическая гиперсомния, повторяемая гиперсомния, внутренняя гиперсомния, нарколепсия, прерванный сон, приступы апноэ во сне, синдром обструктивного апноэ во сне, бессонница, ночной миоклонус, прерывания фазы быстрого сна, суточный ритм, нарушения сна вахтовиков, диссомния, ночной ужас, бессонницы, вызванные депрессией, эмоциональные расстройства/расстройства настроения, болезнь Альцгеймера или когнитивные расстройства, а также хождение во сне и энурез и расстройства сна, которые сопровождают старение; вечерняя спутанность Альцгеймерового типа; состояния, связанные с суточным ритмом, а также психические и физические расстройства, связанные с поездками через несколько часовых поясов и с постоянными сдвигами графиков работы, состояния, вызванные приемом лекарственных средств, которые в качестве побочного эффекта вызывают сокращение в фазе быстрого сна; фибромиалгия; синдромы, которые проявляются не-восстанавливающимся сном и болью в мышцах, или синдром апноэ во сне, связанный с нарушениями дыхания во время сна; состояния, которые являются результатом снижения качества сна; расстройства настроения, такие как депрессия или, более конкретно, депрессивные расстройства, например, одиночные эпизодические или повторяющиеся сильные депрессивные расстройства и дистимические расстройства, или биполярные расстройства, например, биполярное расстройство I, биполярное расстройство II и циклотимическое расстройства, расстройства настроения из-за общего состояния здоровья и расстройства настроения, вызванные приемом психоактивных веществ; тревожные расстройства, включая острые стрессовые расстройства, агорафобия, генерализованное тревожное расстройство, обсессивно-компульсивные расстройства, паническая атака, панические расстройства, посттравматические стрессовые расстройства; тревожное расстройство, вызванное разлукой; социофобии, специфические фобии; тревожные расстройства, вызванные приемом психоактивных веществ, и тревога из-за общего состояния здоровья; острые неврологические и психические расстройства, такие как церебральный дефицит после сердечного шунтирования и трансплантации, инсульт, ишемический инсульт, ишемия головного мозга, травма спинного мозга, травма головы, перинатальная гипоксия, остановка сердца, гипогликемическое повреждение нейронов; Хорея Гентингтона; амиотрофический боковой склероз; рассеянный склероз; поражение зрения; ретинопатия; когнитивные расстройства; идиопатическая и обусловленная действием лекарственного средства болезнь Паркинсона; мышечные спазмы и расстройства, связанные с мышечной спастичностью, включая треморы, эпилепсию, судороги; когнитивные расстройства, включая слабоумие (связанные с болезнью Альцгеймера, ишемией, травмами, сосудистыми проблемами или инсультом, ВИЧ-инфекцией, болезнью Паркинсона, болезнью Гентингтона, болезнью Пика, болезнью Крейтцфельдта-Якоба, перинатальной гипоксией, другими общими медицинскими состояниями или злоупотреблением психоактивными веществами); бред, амнестические расстройства или снижение когнитивных способностей, связанное со старением; шизофрения или психоз включая шизофрению (параниодную, гебефрению, кататоническую или недифференцированную), шизофреноформные расстройства, шизоаффективные расстройства, бредовые расстройства, краткие психотические расстройства, индуцированные психотические расстройства, психотические расстройства вследствие общего заболевания и психотические расстройства, вызванные приемом психоактивных веществ; расстройства, связанные с приемом психоактивных веществ, и вызванное привыканием поведение (включая бред, вызванный приемом психоактивных веществ, персистирующее слабоумие, персистирующие амнестические расстройства, психотические расстройства или тревожные расстройства; толерантность, зависимость или синдром отмены, включая спирт, амфетамины, марихуану, кокаин, галлюциногены, ингалянты, никотин, опиоиды, фенциклидин, седативные препараты, снотворные препараты или транквилизаторы); синдром дефицита внимания с гиперактивностью (ADHD); расстройство поведения; мигрень (включая головную боль при мигрени); недержание мочи; гиперактивность мочевого пузыря (DAB); побудительное недержание мочи (UUI); симптомы, связанные с нижними мочевыводящими путями (LUTS); толерантность к психоактивным веществам, синдром отмены психоактивных веществ (включая, такие вещества, как опиаты, никотин, табачные изделия, спирт, бензодиазепины, кокаин, седативные препараты, снотворные вещества и т.д.); психоз; шизофрения; тревога (включая генерализованное тревожное расстройство, паническое расстройство и обсессивно-компульсивное расстройство); расстройства настроения (включая депрессию, манию, биполярные расстройства); невралгия тройничного нерва; потеря слуха, шум в ушах; нейрональные повреждения, включая глазные повреждения; ретинопатия, дегенерация желтого пятна глаза, рвота, отек мозга; боль, включая острую боль, хроническую боль, тяжелую боль, неутихающую боль, боль при воспалении, хроническую боль при воспалении, диабетическую невропатию, хроническую невропатическую боль, посттравматическую боль, боль в костях и суставах (остеоартрит), повторяющуюся боль при движении, зубную боль, раковую боль, миофасциальный болевой синдром (мышечные травмы, фибромиалгия), периоперационную боль (общая хирургическая, гинекологическая), хроническую боль, невропатическую боль, посттравматическую боль, невралгию тройничного нерва, мигрень и головную боль при мигрени.
Таким образом, в вариантах воплощения настоящего изобретения обеспечиваются способы для: лечения, контроля, улучшения состояния или снижения риска эпилепсии, включая малый эпилептический припадок; лечения или контроля болезни Паркинсона; лечения эссенциального тремора; лечения или контроля боли, включая невропатическую боль; повышения качества сна, увеличения содержания сна; увеличения фазы быстрого сна, увеличения фазы медленного сна, снижения фрагментации сна; лечения бессонницы; повышения познавательной способности, улучшения сохранения данных в памяти; лечения или контроля депрессии; лечения или контроля психоза; или лечения, контроля, улучшения состояния или снижения риска шизофрении у млекопитающего пациента, нуждающегося в этом, которые включают введение пациенту терапевтически эффективного количества соединения по настоящему изобретению. Соединения по настоящему изобретению являются так же полезными в способе профилактики, лечения, контроля, улучшения состояния или снижения риска заболеваний, расстройств и состояний, указанных в настоящей заявке.
По аналогии с кальциевыми каналами Т-типа, тетродотоксин-чувствительные (TTX-S) потенциалзависимые натриевые каналы, такие как NaV1.3 и NaV1.7, также вовлечены во множество различных биологических функций. Это говорит о потенциальной роли этих рецепторов в различных патологических процессах у человека или других видов. Соединения по настоящему изобретению полезны для лечения, профилактики, облегчения, контроля или снижения риска различных неврологических и психических расстройств, связанных с TTX-S натриевыми каналами, включая одно или несколько из следующих состояний или заболеваний: боль, острая боль, хроническая боль, невропатическая боль, боль при воспалении, висцеральная боль, ноцицептивная боль, рассеянный склероз, нейродегенеративные расстройства, синдром раздраженного кишечника, остеоартрит, ревматоидный артрит, невропатологические расстройства, функциональные расстройства кишечника, воспалительные заболевания кишечника; боль, связанная с дисменореей; тазовая боль, цистит, панкреатит, мигрень, кластерные и тензионные головные боли, диабетическая невропатия, периферическая невропатическая боль, пояснично-крестцовый радикулит; болезнь Крона, вызванная фибромиалгией; эпилепсия или эпилептические состояния, биполярная депрессия, тахиаритмия, расстройство настроения, биполярное расстройство, психические расстройства, такие как тревога и депрессия, миотония, аритмия, двигательные расстройства, нейроэндокринные расстройства, атаксия, недержание, висцеральная боль, невралгия тройничного нерва, герпетическая невралгия, общая невралгия, постгерпетическая невралгия, корешковая боль, пояснично-крестцовый радикулит, боль в спине, головная или шейная боль, сильная или некупируемая боль, приступ неконтролируемой боли, послеоперационная боль, инсульт, раковая боль, эпилепсия и каузалгия.
Доза активного ингредиента в композиции по настоящему изобретению может варьировать, однако необходимо, чтобы количеств активного ингредиента было таким, чтобы можно было получить подходящую дозированную форму. Активный ингредиент можно вводить пациентам (животным и человеку), нуждающимся в таком лечении, в дозах, которые будут обеспечивать оптимальную фармацевтическую эффективность.
Выбранная доза зависит от желаемого терапевтического эффекта, пути введения и от продолжительности лечения. Доза для разных пациентов будет разной, в зависимости от природы и тяжести заболевания, массы тела пациента, специального режима питания пациента в этот период, одновременно принимаемого лечения и других факторов, которые должны быть известны специалистам.
Как правило, уровни доз в пределах от 0,0001 до 20 мг/кг массы тела в день вводят пациенту, например, человеку и пожилым людям для получения эффективной блокады T-типа кальциевого канала. Диапазон доз, как правило, составляет от около 0,5 мг до 1,0 г в расчете на пациента в день, это количество можно вводить в виде единой или множества доз.
В одном варианте воплощения, диапазон доз составляет от около 0,5 мг до 500 мг в расчете на пациента в день; в другом варианте воплощения от около 0,5 мг до 200 мг в расчете на пациента в день; в другом варианте воплощения от около 1 мг до 100 мг в расчете на пациента в день; и в другом варианте воплощения от около 5 мг до 50 мг в расчете на пациента в день; еще в одном варианте воплощения от около 1 мг до 30 мг в расчете на пациента в день. Фармацевтические композиции по настоящему изобретению могут обеспечиваться в твердой дозированной форме, например, включающей от около 0,5 мг до 500 мг активного ингредиента или включающей от около 1 мг до 250 мг активного ингредиента. Фармацевтическая композиция может обеспечиваться в твердой дозированной форме, включающей около 1 мг, 5 мг, 10 мг, 25 мг, 50 мг, 100 мг, 200 мг или 250 мг активного ингредиента. Для перорального введения, композиция может обеспечиваться в форме таблеток, содержащих от 1,0 до 1000 миллиграмм активного ингредиента, например, 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, 600, 750, 800, 900 и 1000 миллиграмм активного ингредиента для симптоматического регулирования дозы для пациента, подлежащего лечению. Соединения можно вводить по схеме от 1 до 4 раз в день, например, один или два раза в день.
Соединения по настоящему изобретению можно применять в комбинации с одним или несколькими другими лекарственными средствами для лечения, профилактики, контроля, облегчения или снижения риска заболеваний или состояний, для которых соединения по настоящему изобретению или другие лекарственные средства могут быть полезными, при этом комбинация таких лекарственных средств является более безопасной или более эффективной, чем любое из этих средств, используемое отдельно. Такое другое лекарственное средство(средства) можно вводить, с использованием обычных для таких средств пути введения и количества, одновременно или последовательно с соединением по настоящему изобретению. Когда соединение по настоящему изобретению используют одновременно с одним или несколькими другими лекарственными средствами, предусматривается фармацевтическая композиция в единичной дозированной форме, содержащая такие другие лекарственные средства и соединение по настоящему изобретению. Однако комбинированная терапия также может включать терапии, в которых соединение по настоящему изобретению и одно или несколько других лекарственных средств вводят по разным перекрывающимся схемам. Также предусматривается, что при использовании в комбинации с одним или несколькими другими активными ингредиентами соединения по настоящему изобретению и другие активные ингредиенты можно использовать в меньших дозах, чем дозы при их отдельном использовании.
Соответственно, фармацевтические композиции по настоящему изобретению включают такие композиции, которые содержат один или несколько других активных ингредиентов, в дополнение к соединению по настоящему изобретению. Указанные выше комбинации включают комбинации соединения по настоящему изобретению не только с одним другим активным соединением, но также с двумя или более другими активными соединениями.
Подобным образом, соединения по настоящему изобретению можно использовать в комбинации с другими лекарственными средствами, которые используют для профилактики, лечения, контроля, облегчения или снижения риска заболеваний или состояний, для которых соединения по настоящему изобретению являются полезными. Такие другие лекарственные средства можно вводить, с использованием обычных для таких средств пути введения и количества, одновременно или последовательно с соединением по настоящему изобретению. Когда соединение по настоящему изобретению используют одновременно с одним или несколькими другими лекарственными средствами, предусматривается фармацевтическая композиция, содержащая такие другие лекарственные средства в дополнение к соединению по настоящему изобретению. Соответственно, фармацевтические композиции по настоящему изобретению включают такие, которые также содержат один или несколько других активных ингредиентов в дополнение к соединению по настоящему изобретению.
Массовое отношение соединения по настоящему изобретению к второму активному ингредиенту может варьировать и зависит от эффективной дозы каждого ингредиента. Как правило, используют эффективную дозу каждого. Так, например, когда соединение по настоящему изобретению используют в комбинации с другим средством, массовое отношение соединения по настоящему изобретению к другому средству, как правило, находится в пределах от около 1000:1 до около 1:1000, включая от около 200:1 до около 1:200. Комбинации соединения по настоящему изобретению и других активных ингредиентов, в основном также находятся в указанных пределах, но в каждом случае следует использовать эффективную дозу каждого активного ингредиента. В таких комбинациях соединение по настоящему изобретению и другие активные средства можно вводить по отдельности или вместе. Кроме того, введение одного элемента можно осуществлять до, одновременно или после введения другого средства(средств).
Блокатор кальциевых каналов Т-типа или блокатор потенциалзависимых натриевых каналов можно с пользой сочетать с таким же или другим фармакологически активным соединением или двумя или более такими же или другими фармакологически активными соединениями, особенно для лечения воспалительных заболеваний, боли и урологических заболеваний или расстройств. Например, блокатор кальциевых каналов Т-типа или блокатор потенциалзависимых натриевых каналов, в частности, соединение формулы (I) или его фармацевтически приемлемая соль или сольват, определенные выше, можно вводить одновременно, последовательно или отдельно, в сочетании с одним или несколькими средствами, выбранными из следующих:
- опиоидный анальгетик, например морфин, героин, гидроморфон, оксиморфон, леворфанол, леваллорфан, метадон, меперидин, фентанил, кокаин, кодеин, дигидрокодеин, оксикодон, гидрокодон, пропоксифен, налмефен, налорфин, налоксон, налтрексон, бепренорфин, буторфанол, налбуфин или пентазоцин;
- нестероидное противовоспалительное лекарственное средство (NSAID), например аспирин, диклофенак, дифлусинал, эдотолак, фенбуфен, фенопрофен, флуфенисал, флурбипрофен, ибупрофен, индометацин, кетопрофен, кеторолак, меклофенамовая кислота, мефенамовая кислота, мелоксикам, набуметон, напроксен, нимесулид, нитрофлурбипрофен, олсалазин, оксапрозин, фенилбутазон, пироксикам, сульфасалазин, сулиндак, толметин или зомепирак;
- седативное средство на основе барбитурата, например, амобарбитал, апробарбитал, бутабарбитал, бутабитал, мефобарбитал, метарбитал, метогекситал, пентобарбитал, фенобартитал, секобарбитал, талбутал, теамилал или тиопентал;
- бензодиазепин, обладающий седативным действием, например, хлордиазепоксид, клоразепат, диазепам, флуразепам, лоразепам, оксазепам, темазепам или триазолам;
- антагонист H1, обладающий седативным действием, например, дифенгидрамин, пириламин, прометазин, хлорфенирамин или хлорциклизин;
- седативное средство, такое как глутетимид, мепробамат, метагуалон или дихлоралфеназон;
- скелетно-мышечный релаксант, например, баклофен, карисопродол, хлорзоксазон, циклобензаприн, метокарбамол или орфенадин;
- антагонист NMDA рецептора, например, декстрометорфан ((+)-3-гидрокси-N-метилморфинан) или его метаболит декстрорфан ((+)-3-гидрокси-N-метилморфинан), кетамин, мемантин, пирролохинолин хинин, цис-4-(фосфонометил)-2-пиперидинкарбоновая кислота, будипин, EN-3231 (MorphiDex (зарегистрированная торговая марка), комбинированный препарат морфина и декстрометорфана), топирамат, нерамексан или перзинфотел, в том числе антагонист NR2B, например, ифенпродил, траксопродил или (-)-(R)-6-{2-[4-(3-фторфенил)-4-гидрокси-1-пиперидинил]-1-гидроксиэтил-3,4-дигидро-2(1H)-хинолинон;
- альфа-адренергетическое средство, например, доксазосин, тамсулосин, клонидин, гуанфацин, дексметатомидин, модафинил или 4-амино-6,7-диметокси-2-(5-метан-сульфонамидо-1,2,3,4-тетрагидроизохинол-2-ил)-5-(2-пиридил)хиназолин;
- трициклический антидепрессант, например, десипрамин, имипрамин, амитриптилин или нортриптилин;
- противосудорожное средство, например, карбамазепин, ламотригин, топиратмат или валпроат;
- антагонист тахикинина (NK), в частности, антагонист NK-3, NK-2 или NK-1, например, (альфа-R,9R)-7-[3,5-бис(трифторметил)бензил]-8,9,10,11-тетрагидро-9-метил-5-(4-метилфенил)-7H-[1,4]диазоцино[2,1-g][1,7]-нафтиридин-6-13-дион (TAK-637), 5-[[(2R,3S)-2-[(1R)-1-[3,5-бис(трифторметил)фенил]этокси-3-(4-фторфенил)-4-морфолинил]-метил]-1,2-дигидро-3H-1,2,4-триазол-3-он (MK-869), апрепитант, ланепитант, дапитант или 3-[[2-метокси-5-(трифторметокси)фенил]-метиламино]-2-фенилпиперидин (2S,3S);
- антагонист мускаринового рецептора, например, оксибутинин, толтеродин, пропиверин, тропсиум хлорид, дарифенацин, солифенацин, темиверин и ипратропиум;
- селективный ингибитор COX-2, например, целекоксиб, рофекоксиб, парекоксиб, валдекоксиб, деракоксиб, эторикоксиб или лумиракоксиб;
- анальгетик на основе каменноугольной смолы, в частности парацетамол;
- нейролептическое средство, такое как дроперидол, хлорпромазин, галоперидол, перфеназин, тиоридазин, мезоридазин, трифлуоперазин, флуфеназин, клозапин, оланзапин, рисперидон, зипразидон, кветиапин, сертиндол, арипипразол, сонепипразол, блонансерин, илоперидон, пероспирон, раклоприд, зотепин, бифепрунокс, асенапин, лурасидон, амисулприд, балаперидон, палиндрон, эпливансерин, осанетант, римонабант, меклинертант, Miraxion (зарегистрированная торговая марка) или саризотан;
- агонист ваниллоидного рецептора (например, ресинфератоксин) или антагонист ваниллоидного рецептора (например, капсазепин);
- бета-адренергетическое средство, такое как пропранолол;
- местный анестетик, такой как мексилетин;
- кортикостероид, такой как дексаметазон;
- агонист или антагонист 5-HT рецептора, в частности, агонист 5-HT1B/1D, такой как элетриптан, суматриптан, наратриптан, золмитриптан или ризатриптан;
- антагонист 5-HT2A рецептора, такой как R(+)-альфа-(2,3-диметокси-фенил)-1-[2-(4-фторфенилэтил)]-4-пиперидинметанол (MDL-100907);
- холинергический (никотиновый) анальгетик, такой как испрониклин (TC-1734), (E)-N-метил-4-(3-пиридинил)-3-бутен-1-амин (RJR-2403), (R)-5-(2-азетидинилметокси)-2-хлорпиридин (ABT-594) или никотин;
- Tramadol (зарегистрированная торговая марка);
- ингибитор PDEV, такой как 5-[2-этокси-5-(4-метил-1-пиперазинил-сульфонил)фенил]-1-метил-3-н-пропил-1,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он (силденафил), (6R,12aR)-2,3,6,7,12,12a-гексагидро-2-метил-6-(3,4-метилендиоксифенил)-пиразино[2',1':6,1]-пиридо[3,4-b]индол-1,4-дион (IC-351 или тадалафил), 2-[2-этокси-5-(4-этил-пиперазин-1-ил-1-сульфонил)-фенил]-5-метил-7-пропил-3H-имидазо[5,1-f][1,2,4]триазин-4-он (варденафил), 5-(5-ацетил-2-бутокси-3-пиридинил)-3-этил-2-(1-этил-3-азетидинил)-2,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он, 5-(5-ацетил-2-пропокси-3-пиридинил)-3-этил-2-(1-изопропил-3-азетидинил)-2,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он, 5-[2-этокси-5-(4-этилпиперазин-1-илсульфонил)пиридин-3-ил]-3-этил-2-[2-метоксиэтил]-2,6-дигидро-7H-пиразоло[4,3-d]пиримидин-7-он, 4-[(3-хлор-4-метоксибензил)амино]-2-[(2S)-2-(гидроксиметил)пирролидин-1-ил]-N-(пиримидин-2-илметил)пиримидин-5-карбоксамид, 3-(1-метил-7-оксо-3-пропил-6,7-дигидро-1H-пиразоло[4,3-d]пиримидин-5-ил)-N-[2-(1-метилпирролидин-2-ил)этил]-4-пропоксибензолсульфонамид;
- альфа-2-дельта лиганд, такой как габапентин, прегабалин, 3-метилгабапентин, (1 альфа,3 альфа,5 альфа)(3-амино-метил-бицикло[3.2,0]гепт-3-ил)-уксусная кислота, (3S,5R)-3 аминометил-5 метил-гептановая кислота, (3S,5R)-3 амино-5 метил-гептановая кислота, (3S,5R)-3 амино-5 метил-октановая кислота, (2S,4S)-4-(3-хлорфенокси)пролин, (2S,4S)-4-(3-фторбензил)пролин, [(1R,5R,6S)-6-(аминометил)бицикло[3.2,0]гепт-6-ил]уксусная кислота, 3-(1-аминометил-циклогексилметил)-4H-[1,2,4]оксадиазол-5-он, C-[1-(1H-тетразол-5-илметил)циклогептил]метиламин, (3S,4S)-(1-аминометил-3,4-диметил-циклопентил)уксусная кислота, (3S,5R)-3 аминометил-5 метил-октановая кислота, (3S,5R)-3 амино-5 метил-нонановая кислота, (3S,5R)-3 амино-5 метил-октановая кислота, (3R,4R,5R)-3-амино-4,5-диметил-гептановая кислота и (3R,4R,5R)-3-амино-4,5-диметил-октановая кислота;
- каннабиноид;
- антагонист метаботропного рецептора глутамата подтипа 1 (mGluR1);
- ингибитор повторного поглощения серотонина, такой как сертралин, сертралиновый метаболит деметилсертралин, флуоксетин, норфлуоксетин (флуоксетин десметил метаболит), флувоксамин, пароксетин, циталопрам, циталопрамовый метаболит десметилциталопрам, эсциталопрам, d,l-фенфлурамин, фемоксетин, ифоксетин, цианодотиепин, литоксетин, дапоксетин, нефазодон, церикламин и тразодон;
- ингибитор повторного поглощения норадреналина (норэпинефрина), такой как мапротилин, лофепрамин, миртазепин, оксапротилин, фезоламин, томоксетин, миансерин, бупропирон, бупропироновый метаболит гидроксибупропирон, номифенсин и вилоксазин (Vivalan®), особенно селективный ингибитор повторного поглощения норадреналина, такой как ребоксетин, в частности (S,S)-ребоксетин;
- обладающий двойным действием ингибитор повторного поглощения серотонина-норадреналина, такой как венлафаксин, венлафаксиновый метаболит O-десметилвенлафаксин, кломипрамин, кломипраминовый метаболит десметилкломипрамин, дулоксетин, милнаципран и имипрамин;
- ингибитор индуцируемой синтазы оксида азота (iNOS), такой как S-[2-[(1-иминоэтил)амино]этил]-L-гомоцистеин, S-[2-[(1-иминоэтил)-амино]этил]-4,4-диоксо-L-цистеин, S-[2-[(1-иминоэтил)амино]этил]-2-метил-L-цистеин, (2S,5Z)-2-амино-2-метил-7-[(1-иминоэтил)амино]-5-гептеновая кислота, 2-[[(1R,3S)-3-амино-4-гидрокси-1-(5-тиазолил)-бутил]тио]-5-хлор-3-пиридинкарбонитрил; 2-[[(1R,3S)-3-амино-4-гидрокси-1-(5-тиазолил)бутил]тио]-4-хлорбензонитрил, (2S,4R)-2-амино-4-[[2-хлор-5-(трифторметил)фенил]тио]-5-тиазолбутанол, 2-[[(1R,3S)-3-амино-4-гидрокси-1-(5-тиазолил)бутил]тио]-6-(трифторметил)-3 пиридинкарбонитрил, 2-[[(1R,3S)-3-амино-4-гидрокси-1-(5-тиазолил)бутил]тио]-5-хлорбензонитрил, N-[4-[2-(3-хлорбензиламино)этил]фенил]тиофен-2-карбоксамидин или гуанидиноэтилдисульфид;
- ингибитор ацетилхолинэстеразы, такой как допенезил;
- антагонист простагландина E2 подтипа 4 (EP4, такой как N-[({2-[4-(2-этил-4,6-диметил-1H-имидазо[4,5-c]пиридин-1-ил)фенил]этил}амино)-карбонил]-4-метилбензолсульфонамид или 4-[(1S)-1-({[5-хлор-2-(3-фторфенокси)пиридин-3-ил]карбонил}амино)этил]бензойная кислота;
- антагонист лейкотриена B4; такой как
1-(3-бифенил-4-илметил-4-гидрокси-хроман-7-ил)-циклопентанкарбоновая кислота (CP-105696), 5-[2-(2-карбоксиэтил)-3-[6-(4-метоксифенил)-5E-гексенил]оксифенокси]-валериановая кислота (ONO-4057) или DPC-11870,
- ингибитор липоксигеназы, такой как зилеутон, 6-[(3-фтор-5-[4-метокси-3,4,5,6-тетрагидро-2H-пиран-4-ил])фенокси-метил]-1-метил-2-хинолон (ZD-2138) или 2,3,5-триметил-6-(3-пиридилметил),1,4-бензохинон (CV-6504);
- блокатор натриевых каналов, такой как лидокаин;
- блокатор кальциевых каналов, такой как зиконидин, зонисамид, мибефраздил;
- антагонист 5-HT3, такой как ондасетрон;
и их фармацевтически приемлемые соли и сольваты.
Такие комбинации могут дать существенные преимущества, включая синергетическую активность, в терапии.
Фармацевтическая композиция по настоящему изобретению, которую можно получить путем смешивания, подходяще при температуре окружающей среды и атмосферном давлении, обычно адаптирована для орального, парентерального или ректального введения, и, как таковая, может быть в форме таблеток, капсул, оральных жидких препаратов, порошков, гранул, лепешек, реструктурируемых порошков, растворов или суспензий для инъекций или инфузий, или суппозиториев. Орально вводимые композиции, как правило, являются предпочтительными. Таблетки и капсулы для орального введения могут быть в единичной дозированной форме и могут содержать традиционные эксципиенты, такие как связующие (например, пре-желатинизированный кукурузный крахмал, поливинилпирролидон или гидроксипропилметилцелюлоза); наполнители (например, лактоза, микрокристаллическая целюлоза или гидрофосфат кальция); лубриканты для таблетирования (например, стеарат магния, тальк или диоксид кремния); дезинтегранты (например, картофельный крахмал или натрийкрахмалгликолят); и приемлемые смачивающие вещества (например, лаурилсульфат натрия). На таблетки может быть нанесено покрытие в соответствии со способами, хорошо известными в обычной фармацевтической практике.
Оральные жидкие препараты могут быть в форме, например, водной или масляной суспензии, растворов, эмульсий, сиропов или эликсиров, или могут быть в форме сухого продукта для восстановления водой или другим подходящим носителем перед применением. Такие жидкие препараты могут содержать традиционные добавки, такие как суспендирующие вещества (например, сироп сорбита, производные целлюлозы или гидрогенизированные пищевые жиры), эмульгаторы (например, лецитин или аравийская камедь), не-водные носители (которые могут включать пищевые масла например, миндальное масло, сложные эфиры масел, этиловый спирт или фракционированные растительные масла), консерванты (например, метил- или пропил-п-гидроксибензоаты или сорбиновая кислота) и, если желательно, традиционные отдушки или красители, буферные соли и подсластители, если это является подходящим. Препараты для орального введения могут быть подходящим образом сформулированы с получением контролируемого высвобождения активного соединения или его фармацевтически приемлемой соли.
Для парентерального введения, жидкие единичные дозированные формы получают с использованием соединения формулы (I) или его фармацевтически приемлемой соли и стерильного носителя. Препараты для инъекций могут быть представлены в единичной дозированной форме, например, в ампулах или в виде множественных доз, с использованием соединения формулы (I) или его фармацевтически приемлемой соли и стерильного носителя, необязательно с добавлением консерванта. Композиции могут быть в таких формах как суспензии, растворы или эмульсии в масляных или водных носителях, и могут содержать вещества для формулирования, такие как суспендирующие, стабилизирующие и/или диспергирующие вещества. Альтернативно, активный ингредиент может быть в форме порошка для восстановления подходящим носителем, например, стерильной апирогенной водой, перед применением. Соединение, в зависимости от используемого носителя и концентрации, может быть либо суспендировано или растворено в носителе. При получении растворов, соединение может быть растворено для инъекций, и его можно стерилизовать через фильтр перед заполнением в подходящих флакон или ампулу и запаивания. Преимущество получают, когда адъюванты, такие как местный анестетик, консерванты и буферные вещества растворены в носителе. Для повышения стабильности, композицию можно заморозить после заполнения флакона и удалить воду в условиях вакуума. Парентеральные суспензии получают по существу таким же способом, за исключением того, что соединение суспендируют в носителе вместо его растворения, и стерилизацию нельзя осуществлять путем фильтрования. Соединение можно стерилизовать путем воздействия этиленоксида до суспендирования в стерильном носителе. Преимущество получают при включение поверхностно-активного вещества или смачивающего вещества в композицию для облегчения однородного распределения соединения.
Лосьоны могут быть сформулированы с водной или масляной основой и, помимо этого, содержат один или несколько эмульгаторов, стабилизаторов, диспергирующих веществ, суспендирующих веществ, загустителей или красителей. Капли могут быть сформулированы с водной или не-водной основой и также включают одно или несколько диспергирующих веществ, стабилизаторов, солюбилизирующих веществ или суспендирующих веществ. Они также могут содержать консервант.
Соединения формулы (I) или их фармацевтически приемлемые соли также могут быть сформулированы в композиции для ректального введения, такие как суппозитории или удерживаемые клизмы, например, содержащие традиционные основы для суппозиториев, такие как масло какао или другие глицериды.
Соединения формулы (I) или их фармацевтически приемлемые соли также могут быть сформулированы в виде депо препаратов. Такие долгодействующие препараты можно вводить путем имплантации (например, подкожно или внутримышечно) или путем внутримышечной инъекции. Так, например, соединения формулы (I) или их фармацевтически приемлемые соли могут быть сформулированы с подходящими полимерными или гидрофобными веществами (например, в виде эмульсии в приемлемом масле) или ионообменными смолами, или в виде медленно растворимых производных, например, в виде медленно растворимой соли.
Для интраназального введения, соединения формулы (I) или их фармацевтически приемлемые соли могут быть сформулированы в виде растворов для введения через подходящее мерное или однодозовое устройство или, альтернативно, в виде порошкообразной смеси с подходящим носителем для введения с использованием подходящего устройства доставки. Таким образом, соединения формулы (I) или их фармацевтически приемлемые соли могут быть сформулированы для орального, буккального, парентерального, местного (включая глазное и назальное), депо или ректального введение или в форме, подходящей для введения путем ингаляции или инсуффляции (либо через рот, либо через нос). Соединения формулы (I) и их фармацевтически приемлемые соли могут быть сформулированы для местного введения в форме мазей, кремов, гелей, лосьонов, пессариев, аэрозолей или капель (например, глазных, ушных или назальных капель). Мази и кремы, например, могут быть сформулированы с водной или масляной основой с добавлением подходящих загустителей и/или гелеобразующих веществ. Мази для глазного введения можно получить стерильным образом, используя стерилизованные компоненты.
Общие способы синтеза
По всему тексту настоящей заявки используются следующие сокращения, которые имеют следующие значения:
Термин "основание" также не предполагает никаких конкретных ограничений на природу используемых оснований, и любое основание, обычно используемое в реакциях этого типа, может также быть использовано в настоящей заявке. Примеры таких оснований включают: гидроксиды щелочных металлов, такие как гидроксид лития, гидроксид натрия, гидроксид калия и гидроксид бария; гидриды щелочных металлов, такие как гидрид лития, гидрид натрия и гидрид калия; алкоксиды щелочных металлов, такие как метоксид натрия, этоксид натрия и трет-бутоксид калия; карбонаты щелочных металлов, такие как карбонат лития, карбонат натрия, карбонат калия и карбонат цезия; гидрокарбонаты щелочных металлов, такие как гидрокарбонат лития, гидрокарбонат натрия и гидрокарбонат калия; амины, такие как N-метилморфолин, триэтиламин, трипропиламин, трибутиламин, диизопропилэтиламин, N-метилпиперидин, пиридин, 4-пирролидинопиридин, пиколин, 2,6-ди(трет-бутил)-4-метилпиридин, хинолин, N,N-диметиланилин, N,N-диэтиланилин, 1,5-диазабицикло[4.3,0]нон-5-ен (DBN), 1,4-диазабицикло[2.2.2]октан (DABCO), 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), лутидин и колидин; амиды щелочных металлов, такие как амид лития, амид натрия, амид калия, диизопропиламид лития, диизопропиламид калия, диизопропиламид натрия, бис(триметилсилил)амид лития и бис(триметилсилил)амид калия. Из них предпочтительными являются триэтиламин, диизопропилэтиламин, DBU, DBN, DABCO, пиридин, лутидин, колидин, карбонат натрия, гидрокарбонат натрия, гидроксид натрия, карбонат калия, гидрокарбонат калия, гидроксид калия, гидроксид бария и карбонат цезия.
Реакции, как правило, и предпочтительно, осуществляют в присутствии инертного растворителя. Не существует никаких конкретных ограничений на природу применяемого растворителя, при условии, что он не оказывает негативного влияния на реакцию или участвующие в ней реагенты, и что он может растворять реагенты, по крайней мере, до некоторой степени. Примеры подходящих растворителей включают, но не ограничиваются этим: галогенированные углеводороды, такие как дихлорметан, хлороформ, четыреххлористый углерод и дихлорэтан; эфиры, такие как диэтиловый эфир, диизопропиловый эфир, ТГФ и диоксан; ароматические углеводороды, такие как бензол, толуол и нитробензол; амиды, такие как, ДМФА, N,N-диметилацетамид и триамид гексаметилфосфорной кислоты; амины, такие как N-метилморфолин, триэтиламин, трипропиламин, трибутиламин, диизопропилэтиламин, N-метилпиперидин, пиридин, 4-пирролидинопиридин, N,N-диметиланилин и N,N-диэтиланилин; спирты, такие как метанол, этанол, пропанол, изопропанол и бутанол; нитрилы, такие как ацетонитрил и бензонитрил; сульфоксиды, такие как диметилсульфоксид (ДМСО) и сульфолан; кетоны, такие как ацетон и диэтилкетон. Из этих растворителей предпочтительными являются, включая, но не ограничивается этим, ДМФА, ДМСО, ТГФ, диэтилэфир, диизопропилэфир, диметоксиэтан, ацетонитрил, дихлорметан, дихлорэтан и хлороформ.
Примеры
Настоящее изобретение проиллюстрировано следующими не-ограничивающими примерами, в которых, если не указано иное: все реагенты являются коммерчески доступными, все процедуры осуществляли при комнатной температуре или температуре окружающей среды, то есть в диапазоне от 18 до 25ºC; выпаривание растворителя осуществляли с использованием роторного испарителя, при пониженном давлении с температурой бани до около 60ºC; реакции отслеживали при помощи тонкослойной хроматографии (ТСХ), и время реакции указано только для иллюстрации; структуру и чистоту всех выделенных соединений подтверждали, по меньшей мере, одним из следующих методов: ТСХ (пластины для ТСХ с предварительно нанесенным силикагелем 60 F254 Merck или пластины для ТСХВД с предварительно нанесенным покрытием NH2 F254 Merck), масс-спектрометрия или ядерный магнитный резонанс (ЯМР). Выход продукта приведен только для иллюстрации. Колоночную флэш-хроматографию осуществляли, используя Merck силикагель 60 (230-400 меш ASTM) или Fuji Silysia Chromatorex (зарегистрированная торговая марка) DU3050 (амино-типа, 30-50 мкм) или диоксид кремния Biotage (32-63 мкм, KP-Sil) или амино-связанный диоксид кремния Biotage (35-75 мкм, KP-NH) или Hi-Flash ColumnTM (40 мкм, силикагель). Данные масс-спектрометрии низкого разрешения (ESI) были получены с использованием следующего устройства и условий: Устройство: система ВЭЖХ Waters Alliance на ZQ или ZMD масс-спектрометре и УФ-детектор. Данные ЯМР определяли при 270 МГц (JEOL JNM-LA 270 спектрометр) или 300 МГц (JEOL JNM-LA300), используя дейтерированный хлороформ (99,8% D) или диметилсульфоксид (99,9% D) в качестве растворителя, если не указано иное, относительно тетраметилсилана (TMS) в качестве внутреннего стандарта, миллионных долях (м.д.); обычные используемые сокращения следующие: с - синглет, д - дублет, т - триплет, кв - квартет, м - мультиплет, шир. - широкий и так далее. Химические символы имеют свои обычные значения;
мкм (микрометр(ы)), мкл (микролитр(микролитры)), мкг (микрограмм(микрограммы)), М (моль(моли) на литр), л (литр(литры)), мл (миллилитр(миллилитры)), г(грамм(граммы)), мг (миллиграмм(миллиграммы)), моль(моли), ммоль(миллимоль(миллимоли)).
Способы очистки:
Ахиральная обращенно-фазовая ВЭЖХ:
Оборудование: Waters MS-trigger AutopurificationTM System (2525 Модуль бинарного насоса, 2767 Автоматическая система пробоотборника, 2996 PDA детектор и ZQ2000 масс-спектрометр).
Колонка: XBridgeTM Prep C 18 5 мкм, 19×50 мм.
Температура колонки: температура окружающей среды (комнатная температура).
Скорость потока: 20 мл/мин.
Подвижная фаза А: метанол или ацетонитрил/0,05% (об/об) водный раствор муравьиной кислоты.
Подвижная фаза В: метанол или ацетонитрил/0,05% (об/об) водный раствор аммиака.
Элюирование: оптимизированная программа градиента с выбранными подвижными фазами.
Время выполнения программы: 7 минут.
ЖХСД (жидкостная хроматография среднего давления):
Оборудование: Biotage SP System.
Колонка: Hi-FlashTM с силикагелем 40 мкм, 60Å.
Температура колонки: комнатная температура.
Растворители:
менее полярный растворитель: гексан;
высокополярный растворитель: этилацетат.
Хиральная ВЭЖХ с нормальной фазой:
Оборудование: Shimadzu Preparative-HPLC System.
Колонка: DAICEL Chiralpac AD-H, 20×250 мм.
DAICEL Chiralpac AS-H, 20×250 мм.
DAICEL Chiralpac OJ-H, 20×250 мм.
DAICEL Chiralpac OD-H, 20×250 мм.
Температура колонки: 40°C.
Растворители:
А1: н-гексан;
В1: этанол или 2-пропанол.
Элюирование: оптимизированные изократические условия с выбранной колонкой и подвижными фазами.
Способ оценки чистоты:
Способ A:
Оборудование: Waters Acquity Ultra Performance LC на TUV детекторе и ZQ масс-спектрометре.
Колонка: XTerra MS C18 3,5 мкм, 2,1×30 мм.
Температура колонки: 45°С.
Растворители:
А1: ацетонитрил;
В1: 5 мМ водный раствор ацетат аммония.
Способ B:
Ахиральная обращенно-фазовая жидкостная хроматография сверхвысокого давления (ЖХСВД):
Оборудование: Waters Acquity Ultra Performance LC (UPLCTM) c детектором TUV и масс-спектрометром ZQ2000.
Колонка: Waters Acquity UPLCTM C18, 2,1×100 мм, 1,7 мкм.
Температура колонки: 60°С.
Скорость потока: 0,7 мл/мин.
Растворители:
А1: 10 мМ водный раствор ацетат аммония;
В1: ацетонитрил.
Программа элюирования
Все арил-замещенные карбоксамидные производные формулы (I) могут быть получены при помощи процедур, описанных в общих способах, представленных ниже, или при помощи конкретных способов, описанных в разделе “Примеры” и разделе “Получения”, или путем стандартной модификации таких способов. Настоящее изобретение также включает любой один или несколько из этих способов получения арил-замещенных карбоксамидных производных формулы (I), в дополнение к любым новым промежуточным соединениям, используемым в них.
В следующих общих способах, Ar, W, X, Y, Z, R1, R2, R3, R4, R5, R6, p, q и r имеют значения, определенные выше для арил-замещенных карбоксамидных производных формулы (I), если не указано иное.
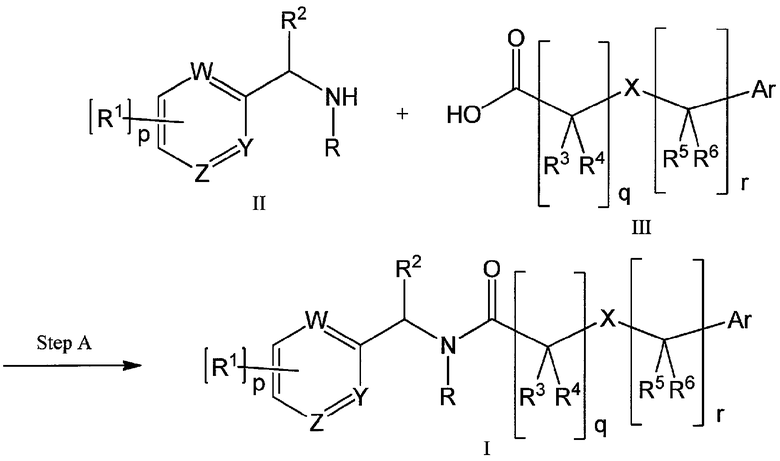
На стадии A, соединение формулы (I) может быть получено из соединения формулы (III) путем амидирования с использованием соединения формулы (II) с использованием подходящего агента конденсации, такого как EDC, предпочтительно в присутствии основания, такого как сочетание триметиламина и HOBT, в подходящем растворителе, таком как дихлорметан, при температуре от 5 до 40ºC в течение 5-20 часов.
Для получения некоторых других соединений формулы (I), будут использовать подходящую реакцию преобразования заместителей.
Например, алкил-замещенные производные могут быть получены из соединения соответствующего галогенида путем реакции сочетания с подходящей бороновой кислотой, с использованием подходящего катализатора, такого как тетракистрифенилфосфинпалладий, в присутствии основания, такого как фосфат калия, и подходящего растворителя, такого как диоксан, при температуре от 5 до 90ºC в течение 12-24 часов; циклопропановые производные могут быть получены из соединения соответствующего альфа, бета-ненасыщенного амида путем реакции циклизации с подходящим алкилдийодидом, с использованием подходящего реагента, такого как диэтилцинк, в подходящем растворителе, таком как дихлорметан, при температуре от 5 до 90ºC в течение 12-24 часов, или путем реакции циклизации с подходящим триалкилсульфоксонийгалогенидом, таким как триметилсульфоксониййодид, и подходящим основанием, таким как гидрид натрия, в подходящем растворителе, таком как ДМСО, при температуре от 5 до 90ºC в течение 1-24 часов; гидроксильные производные могут быть получены из соединения соответствующего бензилокси-производного путем реакции гидрирования с подходящим палладиевым катализатором, таким как гидроксилпалладий, в подходящем растворителе, таком как этанол, в атмосфере водорода; эфирные производные могут быть получены из соединения соответствующего гидроксильного производного путем реакции алкилирования с алкильным спиртом в присутствии агента конденсации, такого как ди-трет-бутилазодикарбоксилат и трифенилфосфин, и основания, такого как N-N-диизопропилэтиламин, и подходящего растворителя, такого как тетрагидрофуран, или с алкилгалогенидом в присутствии основания, такого как карбонат калия, и подходящего растворителя, такого как диметилформамид; N-алкилированные производные могут быть получены из соединения соответствующего NH-амидного производного путем реакции алкилирования с подходящим алкилгалогенидом, с использованием основания, такого как гидрид натрия, в подходящем растворителе, таком как диметилформамид.
Все исходные вещества в следующих общих способах синтеза могут быть коммерчески доступными, или их можно получить обычными способами, известными специалистам в данной области, если не указано иное в части, относящейся к синтезу промежуточных соединений.
Синтез промежуточных соединений
Аминовое промежуточное соединение-1
(R)-1-(5-(циклопропилметокси)пиридин-2-ил)этанамин, соль 2HCl
Стадия 1: 5-(циклопропилметокси)-2-метилпиридин
К раствору 6-метилпиридин-3-ола (5,0 г, 46 ммоль) в ДМФА (45 мл) при комнатной температуре добавляли карбонат цезия (16,5 г, 53 ммоль) и (бромметил)циклопропан (7,1 г, 53 ммоль). После перемешивания при комнатной температуре в течение 18 часов смесь выливали в H2O и водную фазу экстрагировали этилацетатом (два раза). Объединенные органические слои сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (3:1 (об/об)), с получением 3,9 г (52% выход) указанного в заголовке соединения в виде желтого масла:
1H-ЯМР (300 МГц, CDCl3) δ 8,20 (1H, д, J=2,9 Гц), 7,04-7,14 (2H, м), 3,82 (2H, д, J=6,6 Гц), 2,49 (3H, с), 1,21 -1,34 (1H, м), 0,67 (2H, кв, J=7,3 Гц), 0,37 (2H, кв, J=5,9 Гц). LCMS (способ A) m/z, M+1 obs 164,3, tR=2,07 мин.
Стадия 2: (5-(циклопропилметокси)пиридин-2-ил)метанол
К раствору 5-(циклопропилметокси)-2-метилпиридина (3,9 г, 24 ммоль) в дихлорметане (50 мл) при комнатной температуре добавляли 3-хлорбензопероксикислоту (7,6 г, 32 ммоль). После перемешивания при комнатной температуре в течение 1 часа, смесь выливали в насыщенный водный раствор бикарбоната натрия. Органическую фазу экстрагировали дихлорметаном (два раза). Объединенные органические слои сушили над сульфатом магния и концентрировали в вакууме. Остаток растворяли в уксусном ангидриде (50 мл) и смесь перемешивали при 100ºC в течение 2 часов. Половину растворителя удаляли при пониженном давлении. Остаток растворяли в метаноле (50 мл). К смеси осторожно добавляли карбонат калия (20 г, 143 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа. Смесь выливали в H2O и водную фазу экстрагировали этилацетатом (два раза). Объединенные органические слои сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (1:1 (об/об)), с получением 4,5 г (количественный выход) указанного в заголовке соединения в виде коричневого масла:
1H-ЯМР (300 МГц, CDCl3) δ 8,25 (1H, д, J=2,9 Гц), 7,21 (1H, дд, J=8,8, 2,9 Гц), 7,17 (1H, д, J=8,8 Гц), 4,70 (2H, с), 3,85 (2H, 6, J=7,4 Гц), 1,28 (1H, м), 0,75-0,63 (2H, м), 0,40-0,28 (2H, м). LCMS (способ A) m/z M+1 obs 180,3, tR=2,09 мин.
Стадия 3: (R,E)-N-((5-(циклопропилметокси)пиридин-2-ил)метилен)-2-метилпропан-2-сульфинамид
К раствору (5-(циклопропилметокси)пиридин-2-ил)метанола (4,5 г, 25 ммоль) в дихлорметане (50 мл), добавляли 15% водный раствор бромида калия (20 мл) с последующим добавлением насыщенного раствора бикарбоната (20 мл). Двухфазную смесь охлаждали в ледяной бане и добавляли TEMPO (200 мг, 1,3 ммоль). После перемешивания в течение 10 минут по каплям добавляли 5% раствор гипохлорит натрия (30 мл). Реакционную смесь перемешивали в течение 10 минут. Раствор выливали в делительную воронку и органический слой сушили над сульфатом магния и концентрировали в вакууме. Остаток растворяли в дихлорметане (50 мл). К смеси добавляли сульфат меди(II) (10,1 г, 63 ммоль) с последующим добавлением (R)-(+)-2-метил-2-пропансульфинамида (3,1 г, 25 ммоль), соответственно, и смесь перемешивали в течение 18 часов при комнатной температуре. Реакционную смесь фильтровали и фильтрат концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (1:1 (об/об)), с получением 6,2 г (87% выход) указанного в заголовке соединения в виде твердого вещества с чешуйчатой структурой:
1H-ЯМР (300 МГц, CDCl3) δ 8,63 (1H, с), 8,42 (1H, д, J=2,2 Гц), 7,96 (1H, д, J=8,8 Гц), 7,24-7,28 (1H, м), 3,92 (2H, д, J=6,6 Гц), 1,27 (10H, м), 0,67-0,73 (2H, м), 0,39-0,42 (2H, м). LCMS (способ A) m/z M+1 obs 281,2, tR=2, 98 мин.
Стадия 4: (R)-N-((R)-1-(5-(циклопропилметокси)пиридин-2-ил)этил)-2-метилпропан-2-сульфинамид
Раствор (R,E)-N-((5-(циклопропилметокси)пиридин-2-ил)метилен)-2-метилпропан-2-сульфинамида (6,2 г, 22 ммоль) растворяли в дихлорметане (110 мл). К смеси при -78ºC по каплям добавляли метилмагнийбромид (44 мл, 44 ммоль, 1,0M раствор в ТГФ). Смесь перемешивали в течение 1 часа при -78ºC. Смесь выливали в насыщенный водный раствор хлорида аммония и водную фазу экстрагировали этилацетатом (два раза). Объединенные органические слои сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (1:1 (об/об)), с получением 3,2 г (49% выход) указанного в заголовке соединения в виде белого твердого вещества:
1H-ЯМР (300 МГц, CDCl3) δ 8,24 (1H, д, J=2,2 Гц), 7,15-7,23 (2H, м), 4,51-4,57 (2H, м), 3,83 (2H, д, J=6,6 Гц), 1,49 (3H, д, J=6,6 Гц), 1,25 (10H, м), 0,59-0,75 (2H, м) 0,34-0,44 (2H, м), LCMS (способ A) m/z M+1 obs 297,3, tR=2,81 мин.
Стадия 5: (R)-1-(5-(циклопропилметокси)пиридин-2-ил)этанамин, соль 2HCl
(R)-N-((R)-1-(5-(циклопропилметокси)пиридин-2-ил)этил)-2-метилпропан-2-сульфинамид (3,2 г, 10,9 ммоль) растворяли в растворе 10 н. HCl/MeOH (50 мл). Смесь перемешивали при комнатной температуре в течение 3 часов. Смесь концентрировали при помощи N2-потока с получением белого осадка. Твердое вещество собирали при помощи фильтрации и промывали диизопропиловым эфиром с получением 3,2 г (49% выход) указанного в заголовке соединения в виде белого твердого вещества:
1H-ЯМР (300 МГц, ДМСО-d6) δ 8,67 (3H, шир.с), 8,41 (1H, д, J=2,2 Гц), 7,70-7,55 (2H, м), 4,56 (1H, м), 4,01 (2H, д, J=7,3 Гц), 1,57 (3H, д, J=6,6 Гц), 1,33 (1H, м), 0,70-0,60 (2H, м), 0,45-0,35 (2H, м). LCMS (способ A) m/z M+1 obs 193,3, tR=1,90 мин.
Аминовое промежуточное соединение-2
(R)-1-(5-(бензилокси)пиридин-2-ил)этанамин, соль 2HCl
Стадия 1: (R,E)-N-((5-(бензилокси)пиридин-2-ил)метилен)-2-метилпропан-2-сульфинамид
Получали, как на стадии 3 получения аминового промежуточного соединения-1, из (5-(бензилокси)пиридин-2-ил)метанола.
1H-ЯМР (300 МГц, CDCl3) δ 8,64 (1H, с), 8,49 (1H, д, J=2,94 Гц), 7,97 (1H, д, J=8,1 Гц), 7,31-7,45 (6H, м), 5,19 (2H, с), 1,27 (9H, с). LCMS (способ A) m/z M+1 obs 317,2, tR=3,15 мин.
Стадия 2: (R)-N-((R)-1-(5-(бензилокси)пиридин-2-ил)этил)-2-метилпропан-2-сульфинамид
Получали, как на стадии 4 получения аминового промежуточного соединения-1, из (R,E)-N-((5-(бензилокси)пиридин-2-ил)метилен)-2-метилпропан-2-сульфинамида.
1H-ЯМР (300 МГц, CDCl3) δ 8,31 (1H, д, J=2,2 Гц), 7,50-7,30 (5H, м), 7,23 (2H, 6,J=2,2 Гц), 5,09 (2H, с), 4,57 (2H, м), 1,49 (3H, д, J=6,6 Гц), 1,25 (9H, с). LCMS (способ A) m/z M+1 obs 333,2, tR=2,97 мин.
Стадия 3: (R)-1-(5-(бензилокси)пиридин-2-ил)этанамин, соль 2HCl
Получали, как на стадии 5 получения аминового промежуточного соединения-1, из (R)-N-((R)-1-(5-(бензилокси)пиридин-2-ил)этил)-2-метилпропан-2-сульфинамида.
1H-ЯМР (300 МГц, ДМСО-d6) δ 7,50 (2H, шир.с), 8,38 (1H, д, J=2,9 Гц), 7,65-6,25 (7H, м), 6,01 (2H, шир.с), 5,22 (2H, с), 4,45 (1H, м), 1,46 (3H, д, J=6,6 Гц). LCMS (способ A) m/z M+1 obs 229,3, tR=2,24 мин.
Аминовое промежуточное соединение-3
(R)-1-(5-(2-фторбензилокси)пиридин-2-ил)этанамин, соль 2HCl
Стадия 1: 5-(2-фторбензилокси)пиколинонитрил
К смеси 2-бром-5-(2-фторбензилокси)пиридина (1,5 г, 5,3 ммоль) и цианида цинка (0,81 г, 6,9 ммоль) в ДМФА (20 мл) добавляли тетракис(трифенилфосфин)палладий(0) (0,61 г, 0,53 ммоль) при комнатной температуре. После перемешивания при 60ºC в течение 4 часов к смеси добавляли насыщенный водный раствор бикарбоната натрия. Смесь фильтровали через слой целита. Фильтрат экстрагировали этилацетатом, сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (2:1 (об/об)), с получением 0,69 г (57% выход) указанного в заголовке соединения в виде светло-желтого твердого вещества:
1H-ЯМР (300 МГц, CDCl3) δ 8,45 (1H, д, J=2,9 Гц), 7,65 (1H, д, J=8,7 Гц), 7,50-7,28 (3H, м), 7,24-7,05 (2H, м), 5,24 (2H, с). LCMS (способ A) m/z M+1 obs 229,3, tR=2,94 мин.
Стадия 2: (R,E)-N-((5-(2-фторбензилокси)пиридин-2-ил)метилен)-2-метилпропан-2-сульфинамид
К раствору реагента 5-(2-фторбензилокси)пиколинонитрила (690 мг, 3,0 ммоль) в дихлорметане (20 мл) при -78ºC добавляли DIBAL-H (3,7 мл, 3,6 ммоль, 0,99 M). После перемешивания при -78ºC в течение 4 часов к смеси добавляли метанол (2 мл). При комнатной температуре к смеси добавляли 1 н. раствор хлористоводородной кислоты (0,5 мл). Смесь перемешивали при комнатной температуре в течение 1 часа. К смеси добавляли насыщенный водный раствор бикарбоната натрия до достижения нейтрального рН. Органический слой экстрагировали при помощи дихлорметана, сушили над сульфатом натрия и концентрировали в вакууме. Остаток растворяли в дихлорметане (20 мл). К смеси добавляли сульфат меди(II) (1,2 г, 7,6 ммоль) с последующим добавлением (R)-(+)-2-метил-2-пропансульфинамида (370 мг, 3,0 ммоль), соответственно, и смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь фильтровали через слой целита и фильтрат концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (2:1 (об/об)), с получением 360 мг (36% выход) указанного в заголовке соединения в виде бесцветного масла:
1H-ЯМР (300 МГц, CDCl3) δ 8,64 (1H, с), 8,49 (1H, д, J=2,9 Гц), 7,98 (1H, д, J=8,8 Гц), 7,49 (1H, тд, J=7,3,1,5 Гц), 7,40-7,30 (2H, м), 7,23-7,05 (2H, м), 5,26 (2H, с), 1,27 (9H, с). LCMS (способ A) m/z, M+1 obs 335,3, tR=3,14 мин.
Стадия 3: (R)-N-((R)-1-(5-(2-фторбензилокси)пиридин-2-ил)этил)-2-метилпропан-2-сульфинамид
Получали, как на стадии 4 получения аминового промежуточного соединения-1, из (R,E)-N-((5-(2-фторбензилокси)пиридин-2-ил)метилен)-2-метилпропан-2-сульфинамида.
1H-ЯМР (300 МГц, CDCl3) δ 8,32 (1H, д, J=1,4 Гц), 7,48 (1H, т, J=7,3 Гц), 7,40-7,05 (5H, м), 5,16 (2H, с), 4,60-4,50 (2H, м), 1,49 (3H, д, J=5,9 Гц), 1,25 (9H, с). LCMS (способ A) m/z M+1 obs 351,3, tR=2,97 мин.
Стадия 4: (R)-1-(5-(2-фторбензилокси)пиридин-2-ил)этанамин, соль 2HCl
Получали, как на стадии 5 получения аминового промежуточного соединения-1, из (R)-N-((R)-1-(5-(2-фторбензилокси)пиридин-2-ил)этил)-2-метилпропан-2-сульфинамида.
1H-ЯМР (300 МГц, ДМСО-d6) δ 8,97 (2H, шир.с), 8,40 (1H, м), 7,87 (1H, д, J=8,8 Гц), 7,64 (1H, д, J=8,8 Гц), 7,55-7,35 (2H, м), 7,30-7,08 (2H, м), 5,24 (2H, с), 4,78 (1H, м), 1,76 (3H, д, J=6,6 Гц). LCMS (способ A) m/z M+1 obs 247,3, tR=2,34 мин.
Аминовое промежуточное соединение-4
(R)-1-(6-метил-5-(2,2,2-трифторэтокси)пиридин-2-ил)этанамин
Стадия 1: 2,6-диметил-3-(2,2,2-трифторэтокси)пиридин
Получали, как на стадии 1 получения аминового промежуточного соединения-1, из 2,6-диметилпиридин-3-ола и 2,2,2-трифторэтилтрифторметансульфоната.
К суспензии 2,6-диметилпиридин-3-ола (5,0 г, 41 ммоль) и карбоната цезия (15 г, 47 ммоль) в ДМФА (50 мл) по каплям добавляли 2,2,2-трифторэтилтрифторметансульфонат (11 мл, 47 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После перемешивания при комнатной температуре в течение 18 часов смесь выливали в H2O и водную фазу экстрагировали этилацетатом (два раза). Объединенные органические слои сушили над сульфатом магния и концентрировали в вакууме с получением 8,3 г (количественный выход) указанного в заголовке соединения в виде коричневого масла:
1H-ЯМР (300 МГц, CDCl3) δ 7,01 (1H, д, J=8,0 Гц), 6,95 (1H, д, J=8,0 Гц), 4,33 (2H, кв, J=8,0 Гц), 2,48 (6H, с). LCMS (способ A) m/z M+1 obs 206,2, tR=2,58 мин.
Стадия 2: (6-метил-5-(2,2,2-трифторэтокси)пиридин-2-ил)метанол
Получали, как на стадии 2 получения аминового промежуточного соединения-1, из 2,6-диметил-3-(2,2,2-трифторэтокси)пиридина в качестве побочного продукта.
1H-ЯМР (300 МГц, CDCl3) δ 7,12 (1H, 6, J=8,0 Гц), 7,08 (1H, д, J=8,0 Гц), 4,68 (2H, с), 4,37 (2H, кв, J=8,0 Гц), 2,52 (3H, с), 2,05 (1H, шир.с) (побочный продукт).
Стадия 3: (R,E)-2-метил-N-((6-метил-5-(2,2,2-трифторэтокси)пиридин-2-ил)метилен)пропан-2-сульфинамид
Получали, как на стадии 3 получения аминового промежуточного соединения-1, из (6-метил-5-(2,2,2-трифторэтокси)пиридин-2-ил)метанола.
1H-ЯМР (300 МГц, CDCl3) δ 8,63 (1H, с), 7,89 (1H, д, J=8,1 Гц), 7,16 (1H, д, J=8,1 Гц), 4,44 (2H, кв, J=8,1 Гц), 2,58 (3H, с), 1,27 (9H, с). LCMS (способ A) m/z M+1 obs 323,2, tR=3,00 мин.
Стадия 4: (R)-2-метил-N-((R)-1-(6-метил-5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)пропан-2-сульфинамид
Получали, как на стадии 4 получения аминового промежуточного соединения-1, из (R,E)-2-метил-N-((6-метил-5-(2,2,2-трифторэтокси)пиридин-2-ил)метилен)пропан-2-сульфинамида.
1H-ЯМР (300 МГц, CDCl3) δ 7,11 (1H, д, J=8,0 Гц), 7,05 (1H, д, J=8,0 Гц), 4,79 (1H, д, J=5,1 Гц), 4,55 (1H, м), 4,33 (2H, кв, J=8,1 Гц), 2,42 (3H, с), 1,48 (3H, д, J=6,6 Гц), 1,25 (9H, с).
Стадия 5: (R)-1-(6-метил-5-(2,2,2-трифторэтокси)пиридин-2-ил)этанамин, соль 2 HCl
Получали, как на стадии 5 получения аминового промежуточного соединения-1, из (R)-2-метил-N-((R)-1-(6-метил-5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)пропан-2-сульфинамида.
LCMS (способ A) m/z M+1 obs 235,3, tR=2,24 мин.
Аминовое промежуточное соединение-5
(R)-1-(6-метил-3-(2,2,2-трифторэтокси)пиридин-2-ил)этанамин
Стадия 1: (6-метил-3-(2,2,2-трифторэтокси)пиридин-2-ил)метанол
Получали, как на стадии 2 получения аминового промежуточного соединения-1, из 2,6-диметил-3-(2,2,2-трифторэтокси)пиридина в качестве основного продукта.
1H-ЯМР (300 МГц, CDCl3) δ 7,03-7,09 (2H, м), 4,74 (2H, д, J=4,4 Гц), 4,36 (2H, кв, J=8,1 Гц), 2,52 (3H, с), 1,64 (1H, шир.с). LCMS (способ A) m/z M+1 obs 222,3, tR=2,17 мин.
Стадия 2: (R,E)-2-метил-N-((6-метил-3-(2,2,2-трифторэтокси)пиридин-2-ил)метилен)пропан-2-сульфинамид
Получали, как на стадии 3 получения аминового промежуточного соединения-1, из (6-метил-3-(2,2,2-трифторэтокси)пиридин-2-ил)метанола.
1H-ЯМР (300 МГц, CDCl3) δ 8,95 (1H, с), 7,28 (2H, с), 4,44 (2H, м), 2,60 (3H, с), 1,27 (9H, с). LCMS (способ A) m/z M+1 obs 323,2, tR=2,84 мин.
Стадия 3: (R)-2-метил-N-((R)-1-(6-метил-3-(2,2,2-трифторэтокси)пиридин-2-ил)этил)пропан-2-сульфинамид
Получали, как на стадии 4 получения аминового промежуточного соединения-1, из (R,E)-2-метил-N-((6-метил-3-(2,2,2-трифторэтокси)пиридин-2-ил)метилен)пропан-2-сульфинамида.
1H-ЯМР (300 МГц, CDCl3) δ 7,04 (1H, д, J=8,1 Гц), 6,99 (1H, д, J=8,1 Гц), 5,21 (1H, д, J=7,3 Гц), 4,86(1 H, м), 4,38 (2H, кв, J=8,0 Гц), 2,48 (3H, с), 1,41 (3H, д, J=6,6 Гц), 1,26 (9H, с).
Стадия 4: (R)-1-(6-метил-3-(2,2,2-трифторэтокси)пиридин-2-ил)этанамин, соль 2HCl
Получали, как на стадии 5 получения аминового промежуточного соединения-1, из (R)-2-метил-N-((R)-1-(6-метил-3-(2,2,2-трифторэтокси)пиридин-2-ил)этил)пропан-2-сульфинамида.
1H-ЯМР (300 МГц, CDCl3-ДМСО-d6) δ 8,43 (3H, шир.с), 7,60 (1H, д, J=8,8 Гц), 7,32 (1H, д, J=8,1 Гц), 4,91 (2H, кв, J=8,8 Гц), 4,54 (1H, м), 2,48 (3H, с), 1,41 (3H, д, J=6,6 Гц).
Аминовое промежуточное соединение-6
(R)-1-(6-(2-фторбензилокси)пиридин-3-ил)этанамин
Стадия 1: (R,E)-N-((6-(2-фторбензилокси)пиридин-3-ил)метилен)-2-метилпропан-2-сульфинамид
К суспензии гидрида натрия (640 мг, 16 ммоль, 60%) в ДМФА (20 мл) при 0ºC добавляли (2-фторфенил)метанол (1,9 г, 15 ммоль). После перемешивания при комнатной температуре в течение 30 минут к смеси добавляли 6-хлорникотинонитрил (2,6 г, 19 ммоль). Смесь перемешивали при комнатной температуре в течение 14 часов. Смесь выливали в насыщенный водный раствор хлорида аммония и водную фазу экстрагировали этилацетатом (два раза). Объединенные органические слои сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (19:1 (об/об)), с получением промежуточного соединения. Промежуточное соединение растворяли в дихлорметане (30 мл). К смеси добавляли при -78ºC DIBAL-H (6,3 мл, 6,4 ммоль, 0,99 M). После перемешивания при -78ºC в течение 4 часов к смеси добавляли метанол (2 мл). При комнатной температуре к смеси добавляли 1 н. раствор хлористоводородной кислоты (0,5 мл). Смесь перемешивали при комнатной температуре в течение 1 часа. К смеси добавляли насыщенный водный раствор бикарбоната натрия до достижения нейтрального рН. Органический слой экстрагировали дихлорметаном, сушили над сульфатом натрия и концентрировали в вакууме. Остаток растворяли в дихлорметане (20 мл). К смеси добавляли, соответственно, сульфат меди(II) (2,3 г, 14 ммоль) с последующим добавлением (R)-(+)-2-метил-2-пропансульфинамида (700 мг, 5,8 ммоль) и смесь перемешивали в течение 18 часов при комнатной температуре. Реакционную смесь фильтровали через слой целита и фильтрат концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (4:1 (об/об)), с получением 155 мг (3% выход) указанного в заголовке соединения в виде белого твердого вещества:
1H-ЯМР (300 МГц, CDCl3) δ 8,55-8,62 (2H, м), 8,14 (1H, дд, J=8,0 Гц), 7,30-7,53 (2H, м), 7,07-7,18 (1H, м), 6,89 (1H, д, J=8,8 Гц), 5,53 (2H, с), 1,26 (9H, с).
Стадия 2: (R)-N-((R)-1-(6-(2-фторбензилокси)пиридин-3-ил)этил)-2-метилпропан-2-сульфинамид
Получали, как на стадии 4 получения аминового промежуточного соединения-1, из (R,E)-N-((6-(2-фторбензилокси)пиридин-3-ил)метилен)-2-метилпропан-2-сульфинамида.
1H-ЯМР (300 МГц, CDCl3) δ 8,14 (1H, с), 7,51-7,58 (2H, м), 7,29-7,33 (1H, м), 7,06-7,18 (2H, м), 6,80 (1H, д, J=8,0 Гц), 5,44 (2H, с), 4,55-4,59 (2H, м), 1,54 (3H, д, J=6,6 Гц), 1,20 (9H, с).
Стадия 3: (R)-1-(6-(2-фторбензилокси)пиридин-3-ил)этанамин, соль 2HCl
Получали, как на стадии 5 получения аминового промежуточного соединения-1, из (R)-N-((R)-1-(6-(2-фторбензилокси)пиридин-3-ил)этил)-2-метилпропан-2-сульфинамида.
1H-ЯМР (300 МГц, CDCl3-ДМСО-d6) δ 8,60 (2H, шир.с), 8,31 (1H, д, J=3,0 Гц), 7,96 (1H, дд, J=2,2 Гц), 7,51-7,61 (1H, м), 7,38-7,45 (1H, м), 7,20-7,27 (2H, м), 6,76 (1H, д, J=8,0 Гц), 6,49 (2H, м), 5,41 (2H, с), 4,40-4,44 (1H, м), 1,53 (3H, д, J=7,3 Гц). LCMS (способ A) m/z, M+1 obs 247,3, tR=2,44 мин.
Аминовое промежуточное соединение-7
(R)-1-(5-((1-метилциклопропил)метокси)пиридин-2-ил)этанамин, соль 2HCl
Стадия 1: 2-метил-5-((1-метилциклопропил)метокси)пиридин
К раствору 6-метилпиридин-3-ола (0,5 г, 4,6 ммоль) в толуоле (6 мл) добавляли (1-метилциклопропил)метанол (0,59 г, 6,9 ммоль) и перемешивали в атмосфере азота. К раствору добавляли цианометилентри-н-бутилфосфоран (CMBP, 2,5 мл, 9,53 ммоль) и перемешивали при 100ºC в течение 3 часов. Реакционную смесь упаривали. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (2:1 (об/об)), с получением 820 мг (количественный выход) указанного в заголовке соединения в виде коричневого масла:
1H-ЯМР (300 МГц, CDCl3) δ 8,18 (1H, д, J=2,9 Гц), 7,03-7,13 (2H, м), 3,74 (2H, с), 2,48 (3H, с) 1,24 (3H, с), 0,31-0,56 (4H, м). LCMS (способ A) m/z, M+1 obs 178,3, tR=2,54 мин.
Стадия 2: (5-((1-метилциклопропил)метокси)пиридин-2-ил)метанол
Получали, как на стадии 2 получения аминового промежуточного соединения-1, из 2-метил-5-((1-метилциклопропил)метокси)пиридина.
1H-ЯМР (300 МГц, CDCl3) δ 8,24 (1H, д, J=1,4 Гц), 7,16-7,27 (2H, м), 4,70 (2H, с), 3,78 (2H, с), 2,83 (1H, шир.с), 1,25 (3H, с), 0,45-0,58 (4H, м). LCMS (способ A) m/z M+1 obs 194,32, tR=2,37 мин.
Стадия 3: (R,E)-2-метил-N-((5-((1-метилциклопропил)метокси)пиридин-2-ил)метилен)пропан-2-сульфинамид
Получали, как на стадии 3 получения аминового промежуточного соединения-1, из (5-((1-метилциклопропил)метокси)пиридин-2-ил)метанола.
1H-ЯМР (300 МГц, CDCl3) δ 8,64 (1H, с), 8,42 (1H, д, J=2,9 Гц), 7,96 (1H, д, J=8,8 Гц), 7,24 (1H, д, J=2,9 Гц), 3,85 (2H, с), 1,28 (9H, с), 1,24 (3H, с), 0,51-0,59 (4H, м). LCMS (способ A) m/z M+1 obs 295,3, tR=3,14 мин.
Стадия 4: (R)-2-метил-N-((R)-1-(5-((1-метилциклопропил)метокси)пиридин-2-ил)этил)пропан-2-сульфинамид
Получали, как на стадии 4 получения аминового промежуточного соединения-1, из (R,E)-2-метил-N-((5-((1-метилциклопропил)метокси)пиридин-2-ил)метилен)пропан-2-сульфинамида.
1H-ЯМР (300 МГц, CDCl3) δ 8,23 (1H, д, J=2,2 Гц), 7,13-7,22 (2H, м), 4,52-4,56 (2H, м), 3,75 (2H, с), 1,49 (3H, д, J=6,6 Гц), 1,25 (9H, с), 1,23 (3H, с) 0,44-0,56 (4H, м) . LCMS (способ A) m/z M+1 obs 311,3, tR=2,95 мин.
Стадия 5: (R)-1-(5-((1-метилциклопропил)метокси)пиридин-2-ил)этанамин, соль 2HCl
Получали, как на стадии 5 получения аминового промежуточного соединения-1, из (R)-2-метил-N-((R)-1-(5-((1-метилциклопропил)метокси)пиридин-2-ил)этил)пропан-2-сульфинамида.
1H-ЯМР (300 МГц, CDCl3-ДМСО-d6) δ 8,52 (2H, шир.с), 8,34 (1H, с), 7,52 (2H, с), 5,80 (2H, шир.с), 4,48 (1H, м), 3,88 (2H, с), 1,49 (3H, д, J=6,6 Гц), 1,19 (3H, с), 0,41-0,56 (4H, м). LCMS (способ A) m/z M+1 obs 207,3, tR=2,07 мин.
Аминовое промежуточное соединение-8
3-(2,2,2-трифторэтокси)-5,6,7,8-тетрагидрохинолин-8-амин
Стадия 1: 3-(2,2,2-трифторэтокси)хинолин
Получали, как на стадии 1 получения аминового промежуточного соединения-4, из хинолин-3-ола.
1H-ЯМР (300 МГц, CDCl3) δ 8,77 (1H, д, J=2,9 Гц), 8,08 (1H, д, J=8,0 Гц), 7,75 (1H, д, J=8,1 Гц), 7,67-7,50 (2H, м), 7,45 (1H, д, J=2,9 Гц), 4,50 (2H, кв, J=8,0 Гц). LCMS (способ A) m/z M+1 obs 228,3, tR=2,90 мин.
Стадия 2: 3-(2,2,2-трифторэтокси)-5,6,7,8-тетрагидрохинолин
Смесь 3-(2,2,2-трифторэтокси)хинолина (1,13 г, 5,0 ммоль) и оксида платины(IV) (50 мг) в ТФА (8 мл) перемешивали при комнатной температуре в течение 12 часов в атмосфере водорода (1 атмосфера). Затем смесь фильтровали через слой целита и фильтрат концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (10:1 - 7:1), с получением 495 мг (43% выход) указанного в заголовке соединения в виде бесцветного масла:
1H-ЯМР (300 МГц, CDCl3): δ 8,12 (1H, д, J=2,9 Гц), 6,96 (1H, д, J=2,9 Гц), 4,36 (2H, кв, J=8,1 Гц), 2,87 (2H, т, J=6,6 Гц), 2,77 (2H, т, J=6,6 Гц), 1,93-1,75 (4H, м). LCMS (способ A) m/z M+1 obs 232,3, tR=2,84 мин.
Стадия 3: 3-(2,2,2-трифторэтокси)-5,6,7,8-тетрагидрохинолин 1-оксид
Смесь 3-(2,2,2-трифторэтокси)-5,6,7,8-тетрагидрохинолина (495 мг, 2,1 ммоль) и 3-хлорпербензойной кислоты (приблизительно 75%, 739 мг, 3,2 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 1,5 часа. Затем смесь выливали в насыщенный водный раствор бикарбоната натрия (50 мл) и водную фазу экстрагировали дихлорметаном. Органический слой сушили над сульфатом магния и концентрировали в вакууме с получением 740 мг неочищенного указанного в заголовке соединения. Это соединение использовали на следующей стадии без дополнительной очистки:
LCMS (способ A) m/z M+1 obs 248,3, tR=2,52 мин.
Стадия 4: 3-(2,2,2-трифторэтокси)-5,6,7,8-тетрагидрохинолин-8-ол
Смесь 3-(2,2,2-трифторэтокси)-5,6,7,8-тетрагидрохинолин 1-оксида (530 мг, 2,1 ммоль) и уксусного ангидрида (3 мл) перемешивали при 100ºC в течение 2 часов. После охлаждения до комнатной температуры уксусный ангидрид удаляли в вакууме. К остатку добавляли метанол (5 мл) и карбонат калия (1,77 г, 13 ммоль) и смесь перемешивали при комнатной температуре в течение 20 часов. Затем метанол выпаривали в вакууме. К остатку добавляли этилацетат и смесь фильтровали через слой целита. Фильтрат концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (1:1-1:2), с получением 193 мг (36% выход) указанного в заголовке соединения в виде белого твердого вещества:
1H-ЯМР (300 МГц, CDCl3): δ 8,17 (1H, д, J=2,9 Гц), 7,00 (1H, д, J=2,9 Гц), 4,69 (1H, шир.т, J=5,9 Гц), 4,38 (2H, кв, J=8,1 Гц), 3,64 (1H, с), 2,85-2,75 (2H, м), 2,31-2,20 (1H, м), 2,05-1,94 (1H, м), 1,88-1,75 (2H, м). LCMS (способ A) m/z M+1 obs 248,2, tR=2,52 мин.
Стадия 5: 3-(2,2,2-трифторэтокси)-6,7-дигидрохинолин-8(5H)-он
Смесь 3-(2,2,2-трифторэтокси)-5,6,7,8-тетрагидрохинолин-8-ола (193 мг, 0,78 ммоль) и оксида марганца (IV) (543 мг, 6,3 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 3 часов. Затем смесь фильтровали через слой целита и фильтрат концентрировали в вакууме. Оставшееся твердое вещество промывали диэтиловым эфиром с получением 155 мг (81% выход) указанного в заголовке соединения в виде бледно-желтого твердого вещества:
1H-ЯМР (300 МГц, CDCl3): δ 8,44 (1H, д, J=2,9 Гц), 7,11 (1H, д, J=2,9 Гц), 4,48 (2H, кв, J=8,1 Гц), 3,03 (2H, т, J=5,9 Гц), 2,79 (2H, т, J=5,9 Гц), 2,20 (2H, квинтет, J=5,9 Гц). LCMS (способ A) m/z M+1 obs 246,3, tR=2,48 мин.
Стадия 6: оксим 3-(2,2,2-трифторэтокси)-6,7-дигидрохинолин-8(5H)-она
Смесь 3-(2,2,2-трифторэтокси)-6,7-дигидрохинолин-8(5H)-она (155 мг, 0,63 ммоль), гидрохлорида гидроксиламина (88 мг, 1,3 ммоль) и ацетата натрия (104 мг, 1,3 ммоль) в смеси этанол-вода (3:1, 4 мл) кипятили с обратным холодильником при перемешивании в течение 2 часов. После охлаждения до комнатной температуры смесь выливали в воду и водный слой экстрагировали дихлорметаном (два раза). Объединенные органические слои сушили над сульфатом магния и концентрировали в вакууме с получением 167 мг неочищенного указанного в заголовке соединения в виде коричневого твердого вещества. Это вещество использовали на следующей стадии без дополнительной очистки:
1H-ЯМР (300 МГц, ДМСО-d6): δ 8,25 (1H, д, J=2,9 Гц), 7,37 (1H, д, J=2,9 Гц), 4,88 (2H, кв, J=8,8 Гц), 2,79-2,68 (2H, м), 1,95-1,75 (4H, м) (сигнал не обнаружен из-за OH). LCMS (способ A) m/z M+1 obs 261,3, tR=2,62 мин.
Стадия 7: 3-(2,2,2-трифторэтокси)-5,6,7,8-тетрагидрохинолин-8-амин
Смесь оксима 3-(2,2,2-трифторэтокси)-6,7-дигидрохинолин-8(5H)-она (167 мг) и 10% палладия на углероде (100 мг) в метаноле (7 мл) перемешивали при комнатной температуре в течение 24 часов в атмосфере водорода (4 атмосферы). Затем смесь фильтровали через слой целита и фильтрат концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на NH-геле, элюируя смесью гексан/этилацетат (1:1-0:1), с получением 68 мг (43% выход) указанного в заголовке соединения в виде светло-коричневого масла:
1H-ЯМР (300 МГц, CDCl3): δ 8,18 (1H, с), 7,96 (1H, с), 4,37 (2H, кв, J=8,1 Гц), 4,03-3,95 (1H, м), 2,90-2,68 (2H, м), 2,24-2,13 (1H, м), 2,03-1,90 (1H, м), 1,85-1,66 (2H, м) (сигнал не обнаружен из-за NH2). LCMS (способ A) m/z M+1 obs 247,3, tR=2,14 мин.
Аминовое промежуточное соединение-9
(R)-1-(3-фтор-5-(2,2,2-трифторэтокси)пиридин-2-ил)этанамин, соль 2HCl
Стадия 1: 3-фтор-5-(2,2,2-трифторэтокси)пиколинонитрил
К раствору 2,2,2-трифторэтанола (0,257 мл, 3,57 ммоль) в триамиде N,N,N',N',N'',N''-гексаметилфосфорной кислоты (6 мл) при 0ºC добавляли 60% гидрид натрия (0,219 г, 5,71 ммоль) и перемешивали в течение 1 часа. Затем к реакционной смеси добавляли 3,5-дифторпиколинонитрил (1,0 г, 7,1 ммоль) в триамиде N,N,N',N',N'',N''-гексаметилфосфорной кислоты (4 мл) и перемешивали при комнатной температуре в течение 20 часов. Затем в реакционную смесь добавляли 2,2,2-трифторэтанол (0,257 мл, 3,57 ммоль) и 60% гидрид натрия (0,22 г, 5,7 ммоль) и перемешивали при комнатной температуре в течение 3 часов. После завершения реакции смесь выливали в воду и водную фазу экстрагировали этилацетатом. Органические слои сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (6:1-4:1), с получением 398 мг (25% выход) указанного в заголовке соединения в виде маслянистого раствора:
1H-ЯМР (300 МГц, CDCl3) δ 8,31 (1H, д, J=2,6 Гц), 7,15 (1H, дд, J=9,5, 2,6 Гц), 4,50 (2H, кв, J=7,7 Гц).
Стадия 2: (R,E)-N-((3-фтор-5-(2,2,2-трифторэтокси)пиридин-2-ил)метилен)-2-метилпропан-2-сульфинамид
Получали, как на стадии 2 получения аминового промежуточного соединения-3, из 3-фтор-5-(2,2,2-трифторэтокси)пиколинонитрила.
1H-ЯМР (300 МГц, CDCl3) δ 8,83 (1H, с) 8,39 (1H, д, J=2,2 Гц), 7,11 (1H, дд, J=2,2 Гц), 4,89 (2H, кв, J=7,4 Гц), 1,30 (9H, с). LCMS (способ A) m/z M+1 obs 327,2, tR=2,94 мин.
Стадия 3: (R)-N-((R)-1-(3-фтор-5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-2-метилпропан-2-сульфинамид
Получали, как на стадии 4 получения аминового промежуточного соединения-1, из (R,E)-N-((3-фтор-5-(2,2,2-трифторэтокси)пиридин-2-ил)метилен)-2-метилпропан-2-сульфинамида.
1H-ЯМР (300 МГц, CDCl3) δ 8,14 (1H, с), 7,02 (1H, дд, J=1,5, 2,2 Гц), 4,70-4,88 (2H, м), 4,38 (2H, кв, J=6,6 Гц), 1,45 (3H, д, J=6,6 Гц), 1,25 (9H, с). LCMS (способ A) m/z M+1 obs 343,2, tR=2,92 мин.
Стадия 4: (R)-1-(3-фтор-5-(2,2,2-трифторэтокси)пиридин-2-ил)этанамин, соль 2HCl
Получали, как на стадии 5 получения аминового промежуточного соединения-1, из (R)-N-((R)-1-(3-фтор-5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-2-метилпропан-2-сульфинамида.
LCMS (способ A) m/z M+1 obs 222,3, tR=2,00 мин.
Аминовое промежуточное соединение-10
дигидрохлорид (R)-1-(3-метил-5-(2,2,2-трифторэтокси)пиридин-2-ил)этанамина
Стадия 1: 3-метил-5-(2,2,2-трифторэтокси)пиколинонитрил
Получали, как на стадии 1 получения аминового промежуточного соединения-4, из коммерчески доступного 5-гидрокси-3-метилпиколинонитрила:
1H-ЯМР (300 МГц, CDCl3) δ 8,28 (1H, д, J=2,9 Гц), 7,18 (1H, д, J=2,9 Гц), 4,46 (2H, кв, J=7,4 Гц), 2,58 (3H, с). LCMS (способ A) m/z M+1 obs 217,3, tR=2,79 мин.
Стадия 2: (R,E)-2-метил-N-((3-метил-5-(2,2,2-трифторэтокси)пиридин-2-ил)метилен)пропан-2-сульфинамид
Получали, как на стадии 2 получения аминового промежуточного соединения-3, из 3-метил-5-(2,2,2-трифторэтокси)пиколинонитрила.
LCMS (способ A) m/z M+1 obs 323,3, tR=3,02 мин.
Стадия 3: (R)-2-метил-N-((R)-1-(3-метил-5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)пропан-2-сульфинамид
Получали, как на стадии 4 получения аминового промежуточного соединения-1, из (R,E)-2-метил-N-((3-метил-5-(2,2,2-трифторэтокси)пиридин-2-ил)метилен)пропан-2-сульфинамида:
1H-ЯМР (270 МГц, CDCl3): δ 8,13 (1H, д,J==3,3 Гц), 7,05 (1H, д, J=3,3 Гц), 4,88 (1H, д, J=7,2 Гц), 4,69 (1H, квинтет, J=6,5 Гц), 4,38 (2H, кв, J=7,9 Гц), 2,38 (3H, с), 1,39 (3H, д, J=6,6 Гц), 1,25 (9H, с). LCMS (способ A) m/z M+1 obs 339,3, tR=2,95 мин.
Стадия 4: дигидрохлорид (R)-1-(3-метил-5-(2,2,2-трифторэтокси)пиридин-2-ил)этанамина
Получали, как на стадии 5 получения аминового промежуточного соединения-1, из (R)-2-метил-N-((R)-1-(3-метил-5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)пропан-2-сульфинамида:
LCMS (способ A) m/z M+1 obs 235,3, tR=2,20 мин.
Аминовое промежуточное соединение-11
(S)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этанамин дигидрохлорид
Указанное в заголовке соединение получали в соответствии с процедурой, аналогичной той, которую использовали для получения (R)-изомера, используя (S)-(-)-2-метил-2-пропансульфинамид:
[α]D 22-16,7° (c=1,61, MeOH).
Промежуточное карбоновокислотное соединение-1
1-метил-6-(трифторметил)-1H-индазол-3-карбоновая кислота
Стадия 1: метил 1-метил-6-(трифторметил)-1H-индазол-3-карбоксилат
К раствору метил 6-(трифторметил)-1H-индазол-3-карбоксилата (300 мг, 1,2 ммоль) в ацетонитриле (5 мл) при комнатной температуре добавляли карбонат калия (1,0 г, 7,4 ммоль) и йодометан (350 мг, 2,5 ммоль), соответственно. Смесь перемешивали при комнатной температуре в течение 4 часов. Твердое вещество удаляли путем фильтрования и промывали ацетонитрилом. Фильтрат концентрировали в вакууме. Фильтрат отфильтровывали и затем концентрировали при пониженном давлении, остаток очищали при помощи колоночной хроматографии на силикагеле и элюировали смесью гексан/этилацетат = 4/1 с получением 239 мг (75% выход, основной продукт) указанного в заголовке соединения в виде белого твердого вещества.
1H-ЯМР (300 МГц, CDCl3) δ 8,36 (1H, д, J=8,0 Гц), 7,79 (1H, с), 7,55 (1H, д, J=8,0 Гц), 4,24 (3H, с), 4,06 (3H, с). LCMS (способ A) m/z M+1 obs 259,1; tR=3,15 мин.
Стадия 2: 1-метил-6-(трифторметил)-1H-индазол-3-карбоновая кислота
К раствору метил 1-метил-6-(трифторметил)-1H-индазол-3-карбоксилата (50 мг, 0,19 ммоль) в тетрагидрофуране (2 мл) при комнатной температуре добавляли 2 н. раствор гидроксида натрия (0,2 мл, 4,0 ммоль). Смесь кипятили с обратным холодильником при 90ºC при перемешивании в течение 3 часов. После охлаждения до комнатной температуры к смеси добавляли 2 н. раствор хлористоводородной кислоты до достижения рН 4,0. Органический слой экстрагировали этилацетатом, промывали насыщенным солевым раствором и сушили над сульфатом магния. После фильтрования для удаления растворителя и сульфата магния растворитель удаляли при пониженном давлении с получением 47 мг (количественный выход) указанного в заголовке соединения в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки;
LCMS (способ А) m/z, M+1 obs 245,0; tR=2,57 мин.
Промежуточное карбоновокислотное соединение-2
2-метил-6-(трифторметил)-2H-индазол-3-карбоновая кислота
Стадия 1: метил 2-метил-6-(трифторметил)-2H-индазол-3-карбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-1, в качестве второстепенного продукта.
1H-ЯМР (300 МГц, CDCl3) δ 8,13 (1H, д, J=8,8 Гц), 8,10 (1H, с), 7,45 (1H, д, J=8,8 Гц), 4,56 (3H, с), 4,06 (3H, с). LCMS (способ A) m/z M+1 obs 259,1, tR=2,99 мин.
Стадия 2: 2-метил-6-(трифторметил)-2H-индазол-3-карбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-1, из метил 2-метил-6-(трифторметил)-2H-индазол-3-карбоксилата.
LCMS (способ A) m/z M+1 obs 245,0, tR=2,52 мин.
Промежуточное карбоновокислотное соединение-3
1-метил-6-(трифторметил)-1H-индол-2-карбоновая кислота
Стадия 1: этил 1-метил-6-(трифторметил)-1H-индол-2-карбоксилат
Смесь этил 6-(трифторметил)-1H-индол-2-карбоксилата (100 мг, 0,39 ммоль), йодометана (36 микро л, 0,58 ммоль) и карбоната калия (134 мг, 0,97 ммоль) в ДМФА перемешивали при комнатной температуре в течение 7 часов. Затем смесь выливали в воду и водный слой экстрагировали дихлорметаном (три раза). Объединенные органические слои сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (20:1-10:1), с получением 95,3 мг (90% выход) указанного в заголовке соединения в виде белого твердого вещества:
1H-ЯМР (300 МГц, CDCl3): δ 7,98 (1H, с), 7,56 (1H, д, J=8,8 Гц), 7,47 (1H, д, J=8,8 Гц), 7,37 (1H, с), 4,40 (2H, кв, J=7,4 Гц), 4,11 (3H, с), 1,43 (3H, т, J=7,4 Гц). LCMS (способ A) m/z M+1 obs 272,1, tR=3,45 мин.
Стадия 2: 1-метил-6-(трифторметил)-1H-индол-2-карбоновая кислота
Смесь этил 1-метил-6-(трифторметил)-1H-индол-2-карбоксилата (90 мг, 0,33 ммоль) и 2 моль/л водного раствора гидроксида натрия (0,42 мл, 0,83 ммоль) в метаноле (2 мл) перемешивали при комнатной температуре в течение 2 часов. Затем добавляли 2 моль/л хлористоводородной кислоты и образовавшийся осадок собирали путем фильтрования с получением 75,6 мг (94% выход) указанного в заголовке соединения в виде белого твердого вещества:
1H-ЯМР (300 МГц, ДМСО-d6): δ 13,23 (1H, шир.), 8,12 (1H, с), 7,80 (1H, д, J=8,8 Гц), 7,60 (1H, д, J=8,8 Гц), 7,39 (1H, с), 4,08 (3H, с). LCMS (способ A) m/z M-1 obs 242,1, tR=2,88 мин.
Промежуточное карбоновокислотное соединение-4
1-метил-6-(трифторметил)-1H-индол-3-карбоновая кислота
Стадия 1: 2,2,2-трифтор-1-[6-(трифторметил)-1H-индол-3-ил]этанон
К раствору 6-(трифторметил)-1H-индола (460 мг, 2,5 ммоль) в тетрагидрофуране (5 мл) при 0ºC добавляли трифторуксусный ангидрид (0,52 мл, 3,7 ммоль) и полученную смесь перемешивали при такой же температуре в течение 1 часа и при комнатной температуре в течение 1 часа. Затем смесь выливали в воду и образовавшийся осадок собирали путем фильтрования с получением 583 мг (83% выход) указанного в заголовке соединения в виде светло-коричневого твердого вещества:
1H-ЯМР (300 МГц, ДМСО-d6) δ 13,04 (1H, шир.), 8,72 (1H, с), 8,37 (1H, д, J=8,8 Гц), 7,93 (1H, с), 7,66 (1H, д, J=8,1 Гц). LCMS (способ A) m/z M-1 obs 280,0, tR=3,20 мин.
Стадия 2: 2,2,2-трифтор-1-(1-метил-6-(трифторметил)-1H-индол-3-ил)этанон
К смеси 2,2,2-трифтор-1-[6-(трифторметил)-1H-индол-3-ил]этанона (200 мг, 0,71 ммоль) и карбоната калия (246 мг, 1,8 ммоль) в ДМФА (2 мл) при комнатной температуре добавляли йодометан (0,067 мл, 1,1 ммоль). После перемешивания при той же температуре в течение 2 часов смесь выливали в воду и водную фазу экстрагировали при помощи смеси EtOAc (этилацетат)-гексан (2:1, два раза). Объединенные органические слои сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (4:1 (об/об)), с получением 195 мг (93% выход) указанного в заголовке соединения в виде светло-коричневого твердого вещества:
1H-ЯМР (300 МГц, CDCl3) δ 8,51 (1H, д, J=8,0 Гц), 8,04 (1H, с), 7,70 (1H, с), 7,64 (1H, д, J=8,1 Гц), 3,99 (3H, с). LCMS (способ A) m/z M+1 obs 296,0, tR=3,32 мин.
Стадия 3: 1-метил-6-(трифторметил)-1H-индол-3-карбоновая кислота
Смесь 2,2,2-трифтор-1-(1-метил-6-(трифторметил)-1H-индол-3-ил)этанона (195 мг, 0,66 ммоль) и 20% водного раствора гидроксида натрия (5 мл) кипятили с обратным холодильником при перемешивании в течение 10 часов. После охлаждения до комнатной температуры смесь выливали в 1 M раствор хлористоводородной кислоты и водную фазу экстрагировали этилацетатом (два раза). Объединенные органические слои сушили над сульфатом магния и концентрировали в вакууме. Оставшееся твердое вещество промывали 2-пропанолом с получением 107 мг (67% выход) указанного в заголовке соединения в виде светло-оранжевого твердого вещества:
1H-ЯМР (300 МГц, ДМСО-d6) δ 12,25 (1H, с), 8,26 (1H, с), 8,18 (1H, д, J=8,8 Гц), 7,98 (1H, с), 7,50 (1H, д, J=8,6 Гц), 3,94 (3H, с). LCMS (способ A) m/z M-1 obs 242,1, tR=2,84 мин.
Промежуточное карбоновокислотное соединение-5
5-(2,2,2-трифторэтокси)пиколиновая кислота
Стадия 1: этил 5-(2,2,2-трифторэтокси)пиколинат
Получали, как на стадии 1 получения аминового промежуточного соединения-4, из этил 5-гидроксипиколината (EP1748048):
1H-ЯМР (300 МГц, CDCl3): δ 8,47 (1H, д, J=2,9 Гц), 8,15 (1H, д, J=8,8 Гц), 7,32 (1H, дд, J=2,9 и 8,8 Гц), 4,52-5,52 (4H, м), 1,44 (3H, т, J=7,2 Гц). LCMS (способ A) m/z M+1 obs 250,3, tR=2,72 мин.
Стадия 2: 5-(2,2,2-трифторэтокси)пиколиновая кислота
Смесь этил 5-(2,2,2-трифторэтокси)пиколината (253 мг, 1,0 ммоль) и 2 моль/л водного раствора гидроксида натрия (1,0 мл, 2,0 ммоль) в метаноле (5 мл) перемешивали при комнатной температуре в течение 4 часов. Затем метанол удаляли в вакууме. К остатку добавляли воду (2 мл) и 2 моль/л хлористоводородной кислоты (pH~4). Образовавшийся осадок собирали путем фильтрования с получением 118 мг (52% выход) указанного в заголовке соединения в виде серого твердого вещества:
1H-ЯМР (300 МГц, CDCl3): δ 8,49 (1H, д, J=2,9 Гц), 8,06 (1H, д, J=8,8 Гц), 7,66 (1H, дд, J=2,9 и 8,8 Гц), 4,99 (2H, кв, J=8,8 Гц) (сигнал не обнаружен из-за COOH). LCMS (способ A) m/z M+1 obs 222,3, tR=1,59 мин.
Промежуточное карбоновокислотное соединение-6
транс-2-(1-метил-1H-индол-3-ил)циклопропанкарбоновая кислота
Стадия 1: этил транс-2-(1-метил-1H-индол-3-ил)циклопропанкарбоксилат
К суспензии гидрида натрия (приблизительно 60%, 21 мг, 0,52 ммоль) в ДМСО (1 мл) добавляли йодид триметилсульфоксония (115 мг, 0,52 ммоль) и смесь перемешивали при комнатной температуре в течение 20 минут. Затем к смеси добавляли этил (E)-3-(1-метил-1H-индол-3-ил)акрилат (Synlett, (9), 1319-1322 (2006)) (100 мг, 0,44 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа и при 60ºC в течение 20 часов. После охлаждения до комнатной температуры смесь выливали в воду (30 мл) и водную фазу экстрагировали этилацетатом (два раза). Объединенные органические слои сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (7:1), с получением 23 мг (21% выход) указанного в заголовке соединения в виде светло-желтого масла:
1H-ЯМР (300 МГц, CDCl3): δ 7,65 (1H, д, J=8,0 Гц), 7,30-7,19 (2H, м), 7,16-7,07 (1H, м), 6,79 (1H, с), 4,20 (2H, кв, J=8,0 Гц), 3,71 (3H, с), 2,65-2,55 (1H, м), 1,91-1,82 (1H, м), 1,61-1,51 (1H, м), 1,30 (3H,т, J=8,0 Гц), 1,35-1,25 (1H, м). LCMS (способ A) m/z M+1 obs 244,4, tR=3,22 мин.
Стадия 2: транс-2-(1-метил-1H-индол-3-ил)циклопропанкарбоновая кислота
Смесь этил транс-2-(1-метил-1H-индол-3-ил)циклопропанкарбоксилата (20 мг, 0,082 ммоль) и 2 моль/л водного раствора гидроксида натрия (0,20 мл, 0,40 ммоль) в метаноле (3 мл) перемешивали при 60ºC в течение 3 часов. После охлаждения до комнатной температуры добавляли 2 моль/л хлористоводородной кислоты (0,20 мл, 0,40 ммоль) и растворитель удаляли в вакууме. К остатку добавляли ТГФ (2 мл) и отфильтровывали. Фильтрат концентрировали в вакууме с получением 25 мг указанного в заголовке соединения в виде светло-желтого масла. Это соединение использовали на следующей стадии без дополнительной очистки:
1H-ЯМР (300 МГц, CDCl3): δ 7,68 (1H, д, J=8,8 Гц), 7,31-7,20 (2H, м), 7,18-7,09 (1H, м), 6,82 (1H, с), 3,73 (3H, с), 2,75-2,64 (1H, м), 1,90-1,80 (1H, м), 1,69-1,60 (1H, м), 1,45-1,35 (1H, м). LCMS (способ A) m/z M+1 obs 216,4, tR=2,72 мин.
Промежуточное карбоновокислотное соединение-7
транс-2-(7-фтор-1H-индол-3-ил)циклопропанкарбоновая кислота
Стадия 1: этил (E)-3-(7-фтор-1-тозил-1H-индол-3-ил)акрилат
К суспензии гидрида натрия (приблизительно 60%, 240 мг, 6,3 ммоль) в ТГФ (10 мл) при 0ºC добавляли по каплям раствор триэтилфосфоноацетата (1,33 г, 5,9 ммоль) в ТГФ (5 мл). После перемешивания при комнатной температуре в течение 0,5 часа к смеси при 0ºC добавляли раствор 7-фтор-1-тозил-1H-индол-3-карбальдегида (J. Med. Chem., 48 (19), 6023-6034 (2005)) (1,10 г, 3,48 ммоль) в ТГФ (5 мл). Полученную смесь перемешивали при 0ºC в течение 0,5 часа и при комнатной температуре в течение 19 часов. Смесь выливали в воду, экстрагировали дихлорметаном, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Оставшееся твердое вещество промывали этилацетатом с получением 864 мг (65% выход) указанного в заголовке соединения в виде белого твердого вещества:
1H-ЯМР (270 МГц, CDCl3): δ 8,03 (1H, с), 7,85-7,75 (3H, м), 7,57 (1H, д, J=7,6 Гц), 7,31-7,15 (3H, м), 6,99 (1H, дд, J=8,2 и 12,2 Гц), 6,50 (1H, 6, J=16,1 Гц), 4,27 (2H, кв, J=7,2 Гц), 2,37 (3H, с), 1,33 (3H, т, J=7,2 Гц).
Стадия 2: этил транс-2-(7-фтор-1-тозил-1H-индол-3-ил) циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из этил (E)-3-(7-фтор-1-тозил-1H-индол-3-ил)акрилата:
1H-ЯМР (270 МГц, CDCl3): δ 7,78 (2H, д, J=7,6 Гц), 7,45 (1H, с), 7,34 (1H, д, J=6,9 Гц), 7,28-7,22 (2H, м), 7,13 (1H, дт, J=4,3 и 7,9 Гц), 6,94 (1H, дд, J=7,9 и 12,2 Гц), 4,19 (2H, кв, J=7,3 Гц), 2,51-2,42 (1H, м), 2,36 (3H, с), 1,92-1,84 (1H, м), 1,61-1,52 (1H, м), 1,30 (3H, т, J=7,3 Гц), 1,33-1,20 (1H, м).
Стадия 3: транс-2-(7-фтор-1H-индол-3-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил транс-2-(7-фтор-1-тозил-1H-индол-3-ил)циклопропанкарбоксилата:
1H-ЯМР (300 МГц, CDCl3): δ 8,16 (1H, шир.с), 7,44 (1H, д, J=8,0 Гц), 7,1-6,8 (3H, м), 2,71-2,59 (1H, м), 1,95-1,85 (1H, м), 1,70-1,60 (1H, м), 1,48-1,35 (1H, м) (сигнал не обнаружен из-за COOH). LCMS (способ A) m/z M+1 obs 220,3, tR=2,57 мин.
Промежуточное карбоновокислотное соединение-8
транс-2-(5-фтор-1H-индол-3-ил)циклопропанкарбоновая кислота
Стадия 1: этил (E)-3-(5-фтор-1-тозил-1H-индол-3-ил) акрилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-7, из 5-фтор-1-тозил-1H-индол-3-карбальдегида (J. Med. Chem., 41 (25), 4995-5001 (1998)):
1H-ЯМР (270 МГц, CDCl3): δ 7,95 (1H, дд, J=4,6 и 9,2 Гц), 7,86 (1H, с), 7,79-7,68 (3H, м), 7,44 (1H, дд, J=2,6 и 8,6 Гц), 7,29-7,24 (2H, м), 7,11 (1H, дт, J=2,6 и 8,6 Гц), 6,43 (1H, д, J=16,5 Гц), 4,27 (2H, кв, J=7,2 Гц), 2,37 (3H, с), 1,35 (3H, т, J=7,2 Гц). LCMS (способ A) m/z M+1 obs 388,2, tR=3,52 мин.
Стадия 2: этил транс-2-(5-фтор-1-тозил-1H-индол-3-ил) циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из этил (E)-3-(5-фтор-1-тозил-1H-индол-3-ил)акрилата:
1H-ЯМР (300 МГц, CDCl3): δ 7,93-7,86 (1H, м), 7,71 (2H, д, J=9,5 Гц), 7,30-7,17 (4H, м), 7,09-7,00 (1H, м), 4,20 (2H, кв, J=7,3 Гц), 2,46-2,35 (1H, м), 2,35 (3H, с), 1,88-1,80 (1H, м), 1,63-1,55 (1H, м), 1,31 (3H, т, J=7,3 Гц), 1,30-1,20 (1H, м). LCMS (способ A) m/z M+1 obs 402,3, tR=3,54 мин.
Стадия 3: транс-2-(5-фтор-1H-индол-3-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил транс-2-(5-фтор-1-тозил-1H-индол-3-ил)циклопропанкарбоксилата:
1H-ЯМР (300 МГц, ДМСО-d6): δ 11,00 (1H, шир.с), 7,36-7,22 (3H, м), 6,98-6,88 (1H, м), 2,43-2,33 (1H, м), 1,75-1,67 (1H, м), 1,42-1,28 (2H, м) (сигнал не обнаружен из-за COOH). LCMS (способ A) m/z M+1 obs 220,3, tR=2,59 мин.
Промежуточное карбоновокислотное соединение - 9
транс-2-(1H-индол-6-ил)циклопропанкарбоновая кислота
Стадия 1: этил (E)-3-(1-тозил-1H-индол-6-ил)акрилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-7, из 1-тозил-1H-индол-6-карбальдегида.
1H-ЯМР (300 МГц, CDCl3) δ 8,13 (1H, с), 7,90-7,75 (3H, м), 7,62 (1H, д, J=3,7 Гц), 7,52 (1H, д, J=8,8 Гц), 7,43 (1H, д, J=8,8 Гц), 7,24 (2H, д, J=8,1 Гц), 6,66 (1H, д, J=3,7 Гц), 6,49 (1H, д, J=16,1 Гц), 4,29 (2H, кв, J=7,3 Гц), 2,35 (3H, с), 1,37 (3H, т, J=7,3 Гц). LCMS (способ A) m/z M+1 obs 370,2, tR=3,44 мин.
Стадия 2: этил транс-2-(1-тозил-1H-индол-6-ил) циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из этил (E)-3-(1-тозил-1H-индол-6-ил)акрилата.
1H-ЯМР (300 МГц, CDCl3) δ 7,76 (2H, д, J=8,1 Гц), 7,76 (1H, с), 7,53 (1H, д, J=3,7 Гц), 7,43 (1H, д, J=8,1 Гц), 7,25 (2H, д, J=8,1 Гц), 6,97 (1H, дд, J=8,1, 1,5 Гц), 6,62 (1H, д, J=3,7 Гц), 4,21 (2H, кв, J=7,3 Гц), 2,67 (1H, м), 2,37 (3H, с), 1,95 (1H, м), 1,67 (1H, м), 1,39 (1H, м), 1,32 (3H, т, J=7,3 Гц). LCMS (способ A) m/z M+1 obs 384, tR=3,44 мин.
Стадия 3: транс-2-(1H-индол-6-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил транс-2-(1-тозил-1H-индол-6-ил)циклопропанкарбоксилата.
1H-ЯМР (300 МГц, ДМСО-d6) δ 11,00 (1H, с), 7,44 (1H, д, J=8,0 Гц), 7,29 (1H, т, J=2,2 Гц), 7,18 (1H, с), 6,79 (1H, 6, J=8,1 Гц), 6,37 (1H, м), 2,50 (1H, м), 1,78 (1H, м), 1,48-1,30 (2H, м). LCMS (способ A) m/z M-1 obs 200,3, tR=2,52 мин.
Промежуточное карбоновокислотное соединение-10
транс-2-(5-циано-1H-индол-3-ил)циклопропанкарбоновая кислота
Стадия 1: этил (E)-3-(5-циано-1-тозил-1H-индол-3-ил)акрилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-7, из 3-формил-1-тозил-1H-индол-5-карбонитрила.
1H-ЯМР (300 МГц, CDCl3) δ 8,14 (1H, с), 8,11 (1H, д, J=8,8 Гц), 7,94 (1H, с), 7,81 (2H, д, J=8,1 Гц), 7,74 (1H, д, J=16,1 Гц), 7,63 (1H, дд, J=8,8, 1,5 Гц), 7,30 (2H, д, J=8,0 Гц), 6,49 (1H, д, J=16,1 Гц), 4,29 (2H, кв, J=6,6 Гц), 2,39 (3H, с), 1,36 (3H, т, J=6,6 Гц). LCMS (способ A) m/z M+1 obs 395,2, tR=3,40 мин.
Стадия 2: транс-2-(5-циано-1H-индол-3-ил)циклопропанкарбоновая кислота
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения 6 и на стадии 2 получения промежуточного карбоновокислотного соединения 6, из этил (E)-3-(5-циано-1-тозил-1H-индол-3-ил)акрилата.
LCMS (способ A) m/z M+1 obs 227,3, tR=2,39 мин.
Промежуточное карбоновокислотное соединение-11
транс-2-(1H-индол-7-ил)циклопропанкарбоновая кислота
Стадия 1: 1-тозил-1H-индол-7-карбальдегид
К суспензии гидрида натрия (240 мг, 5,9 ммоль) в ТГФ (10 мл) при комнатной температуре добавляли 1H-индол-7-карбальдегид (570 мг, 3,9 ммоль). После перемешивания при комнатной температуре к смеси в течение 20 минут добавляли 4-метилбензол-1-сульфонилхлорид (1,1 г, 5,9 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа. Смесь выливали в насыщенный водный раствор хлорида аммония и водную фазу экстрагировали этилацетатом (два раза). Объединенные органические слои сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (4:1 (об/об)), с получением 1,0 г (89% выход) указанного в заголовке соединения в виде белого твердого вещества:
1H-ЯМР (300 МГц, CDCl3) δ 10,73 (1H, с), 7,82 (1H, д, J=8,8 Гц), 7,75-7,65 (2H, м), 7,47 (2H, д, J=8,8 Гц), 7,38 (1H, т, J=8,3 Гц), 7,17 (2H, д, J=8,8 Гц), 6,79 (1H, д, J=3,7 Гц), 2,34 (3H, с). LCMS (способ A) m/z M+1 obs 300,2, tR=3,15 мин.
Стадия 2: этил (E)-3-(1-тозил-1H-индол-7-ил)акрилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения 7, из 1-тозил-1H-индол-7-карбальдегида.
1H-ЯМР (300 МГц, CDCl3) δ 8,64 (1H, д, J=15,4 Гц), 7,84 (1H, д, J=3,7 Гц), 7,65-7,55 (3H, м), 7,34 (1H, д, J=7,3 Гц), 7,26-7,10 (3H, м), 6,72 (1H, д, J=4,4 Гц), 6,10 (1H, д, J=15,4 Гц), 4,32 (2H, кв, J=7,3 Гц), 2,34 (3H, с), 1,41 (3H, т, J=7,3 Гц). LCMS (способ A) m/z M+1 obs 370,3, tR=3,40 мин.
Стадия 3: этил транс-2-(1-тозил-1H-индол-7-ил)циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из (E)-этил 3-(1-тозил-1H-индол-7-ил)акрилата.
1H-ЯМР (300 МГц, CDCl3) δ 7,75 (1H, д, J=3,7 Гц), 7,54 (2H, д, J=8,1 Гц), 7,38 (1H, д, J=8,9 Гц), 7,21-7,10 (3H, м), 6,91 (1H, д, J=8,0 Гц), 6,67 (1H, д, J=3,7 Гц), 4,29-4,19 (2H, м), 3,17 (1H, м), 2,34 (3H, с), 1,92 (1H, м), 1,48 (1H, м), 1,33 (3H, т, J=6,6 Гц), 1,24 (1H, м). LCMS (способ A) m/z M+1 obs 384,2, tR=3,42 мин.
Стадия 4: транс-2-(1H-индол-7-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения 6, из этил транс-2-(1-тозил-1H-индол-7-ил)циклопропанкарбоксилата.
1H-ЯМР(300 МГц, ДМСО-d6) δ 11,31 (1H, с), 7,39 (1H, д, J=8,1 Гц), 7,33 (1H, т, J=1,5 Гц), 6,91 (1H, т, J=7,3 Гц), 6,68 (1H, д, J=7,3 Гц), 6,44 (1H, т, J=1,5 Гц), 2,79 (1H, м), 1,88 (1H, м), 1,51 (1H, м), 1,34 (1H, м). LCMS (способ A) m/z M-1 obs 200,3, tR=2,62 мин.
Промежуточное карбоновокислотное соединение-12
транс-2-(1H-индол-2-ил)циклопропанкарбоновая кислота
Стадия 1: этил (E)-3-(1-тозил-1H-индол-2-ил)акрилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-7, из 1-тозил-1H-индол-2-карбальдегида (Heterocycles, 76(2), 1155-1170; 2008).
1H-ЯМР (300 МГц, CDCl3) δ 8,37 (1H, д, J=16,1 Гц), 8,22 (1H, д, J=8,4 Гц), 7,62 (2H, д, J=8,4 Гц), 7,48 (1H, д, J=8,1 Гц), 7,36 (1H, дт, J=7,3, 1,1 Гц), 7,26 (1H, м), 7,16 (2H, д, J=8,1 Гц), 6,96 (1H, с), 6,36 (1H, д, J=16,1 Гц), 4,30 (2H, кв, J=7,3 Гц), 2,32 (3H, с), 1,37 (3H, т, J=7,3 Гц).
Стадия 2: этил транс-2-(1-тозил-1H-индол-2-ил)циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из этил (E)-3-(1-тозил-1H-индол-2-ил)акрилата:
1H-ЯМР (300 МГц, CDCl3) δ 8,20 (1H, д, J=8,1 Гц), 7,73 (2H, д, J=8,1 Гц), 7,42-7,19 (5H, м), 6,28 (1H, с), 4,28-4,11 (2H, м), 2,93 (1H, м), 2,34 (3H, с), 1,82 (1H, м), 1,62 (1H, м), 1,35-1,22 (4H, м).
Стадия 3: транс-2-(1H-индол-2-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил транс-2-(1-тозил-1H-индол-2-ил)циклопропанкарбоксилата:
1H-ЯМР(300 МГц, CDCl3) δ 8,04 (1H, с), 7,49 (1H, д, J=7,3 Гц), 7,23 (1H, д, J=8,8 Гц), 7,10 (2H, м), 6,14 (1H, с), 2,60 (1H, м), 1,92 (1H, м), 1,62 (1H, м), 1,41 (1H, м). LCMS (способ A) m/z M+1 obs 202,3, tR=2,59 мин.
Промежуточное карбоновокислотное соединение-13
транс-2-(5-фтор-1H-индол-2-ил)циклопропанкарбоновая кислота
Стадия 1: фтор-N-метокси-N-метил-1H-индол-2-карбоксамид
Гидрохлорид N,O-диметилгидроксиламина (1,089 г, 11,16 ммоль) и триэтиламин (3,92 мл, 27,9 ммоль) добавляли к раствору 5-фтор-1H-индол-2-карбоновой кислоты (2,0 г, 11,16 ммоль) в дихлорметане(30 мл) и перемешивали при комнатной температуре в течение 5 минут. Затем добавляли гидрохлорид1-этил-3-(3-диметиламинопропил)карбодиимида (2,140 г, 11,16 ммоль) и перемешивали в течение 20 часов. После завершения реакции растворитель удаляли. Остаток суспендировали в минимальном объеме ацетона и нерастворившееся белое твердое вещество удаляли путем фильтрования. После концентрирования в вакууме смесь выливали в насыщенный водный раствор бикарбоната натрия и водную фазу экстрагировали этилацетатом. Органический слой сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (2:1), с получением 690 мг (28% выход) указанного в заголовке соединения в виде белого кристалла:
1Н-ЯМР (300 МГц, ДМСО-d6) δ 11,6 (1H, с), 7,44 (1H, м), 7,38 (1H, д, J=2,6 Гц), 7,12 (1H, д, J=1,5 Гц), 7,06 (1H, дт, J=9,5, 2,6 Гц), 3,78 (3H, с), 3,32 (3H, с).
Стадия 2: 5-фтор-1H-индол-2-карбальдегид
Алюмогидрид лития (0,094 г, 2,488 ммоль) при 0ºC добавляли к раствору 5-фтор-N-метокси-N-метил-1H-индол-2-карбоксамида (0,691 г, 3,11 ммоль) в тетрагидрофуране (10 мл) и перемешивали в течение 1 часа. Реакционную смесь охлаждали до 0ºC и к реакционной смеси добавляли по каплям 25% раствор аммиака до тех пор, пока цвет алюмогидрида лития не изменялся от серого до белого. Затем к реакционной смеси добавляли дихлорметан и церит и перемешивали в течение 30 минут. Смесь фильтровали через слой целита и концентрировали в вакууме с получением 523 мг неочищенного указанного в заголовке соединения. Это соединение использовали на следующей стадии без дополнительной очистки:
1H-ЯМР (300 МГц, CDCl3) δ 9,85 (1H, с), 9,13 (1H, шир.с), 7,42-7,36 (2H, м), 7,24 (1H, д, J=1,1 Гц), 7,16 (1H, дт, J=9,2, 2,6 Гц).
Стадия 3: 5-фтор-1-тозил-1H-индол-2-карбальдегид
К раствору 5-фтор-1H-индол-2-карбальдегида (0,523 г, 3,21 ммоль) в дихлорметане (10 мл) добавляли пара-толуолсульфонилхлорид (2,445 г, 12,82 ммоль), N,N-диметил-4-аминопиридин (0,196 г, 1,603 ммоль) и триэтиламин (2,253 мл, 16,03 ммоль) и перемешивали при комнатной температуре в течение 20 часов. После завершения реакции смесь выливали в насыщенный водный раствор бикарбоната натрия и водную фазу экстрагировали этилацетатом. Органический слой сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (10:1), с получением 823 мг (81% выход) указанного в заголовке соединения в виде белого кристалла:
1H-ЯМР (300 МГц, CDCl3) δ 10,5 (1H, с), 8,20 (1H, дд, J=10,0, 4,2 Гц), 7,62 (2H, д, J=8,4 Гц), 7,40 (1H, с), 7,29-7,19 (4H, м), 2,34 (3H, с).
Стадия 4: этил (E)-3-(5-фтор-1-тозил-1H-индол-2-ил)акрилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-7, из 5-фтор-1-тозил-1H-индол-2-карбальдегида.
1H-ЯМР (300 МГц, CDCl3) δ 8,33 (1H, д, J=16,1 Гц), 8,17 (1H, дд, J=9,2, 4,4 Гц), 7,59 (2H, д, J=8,4 Гц), 7,19-7,05 (4H, м), 6,90 (1H, с), 6,35 (1H, д, J=16,1 Гц), 4,30 (2H, кв, J=7,3 Гц), 2,33 (3H, с), 1,37 (3H, т, J=7,3 Гц).
Стадия 5: этил транс-2-(5-фтор-1-тозил-1H-индол-2-ил)циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из этил (E)-3-(5-фтор-1-тозил-1H-индол-2-ил)акрилата.
1H-ЯМР (300 МГц, CDCl3) δ 8,14 (1H, дд, J=8,8, 4,4 Гц), 7,69 (2H, д, J=8,1 Гц), 7,21 (2H, д, J=8,1 Гц), 7,07-6,98 (2H, м), 6,23 (1H, с), 4,23 (2H, м), 2,91 (1H, м), 2,35 (3H, с), 1,82 (1H, м), 1,62 (1H, м), 1,32 (3H, т, J=7,3 Гц), 1,28 (1H, м).
Стадия 6: транс-2-(5-фтор-1H-индол-2-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил транс-2-(5-фтор-1-тозил-1H-индол-2-ил)циклопропанкарбоксилата.
1H-ЯМР (300 МГц, ДМСО-d6) δ 11,1 (1H, с), 7,21 (1H, дд, J=8,8, 4,8 Гц), 7,10 (1H, дд, J=10,3, 2,6 Гц), 6,81 (1H, дт, J=8,8, 2,6 Гц), 6,18 (1H, д, J=1,8 Гц), 2,48 (1H, м), 1,87 (1H, м), 1,42 (2H, м). LCMS (способ A) m/z: M+1 obs 220,3, tR=2,64 мин.
Промежуточное карбоновокислотное соединение-14
транс-2-(4-фтор-1H-индол-3-ил)циклопропанкарбоновая кислота
Стадия 1: этил (E)-3-(4-фтор-1-тозил-1H-индол-3-ил)акрилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-7, из 4-фтор-1-тозил-1H-индол-3-карбальдегида.
1H-ЯМР (270 МГц, CDCl3): δ 7,88-7,72 (5H, м), 7,32-7,20 (3H, м), 6,95 (1H, дд, J=8,2 и 10,9 Гц), 6,48 (1H, д, J=16,1 Гц), 4,24 (2H, кв, J=7,2 Гц), 2,35 (3H, с), 1,32 (3H, т, J=7,2 Гц).
Стадия 2: этил транс-2-(4-фтор-1-тозил-1H-индол-3-ил) циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из этил (E)-3-(4-фтор-1-тозил-1H-индол-3-ил)акрилата.
1H-ЯМР (300 МГц, CDCl3): δ 7,77-7,70 (3H, м), 7,6-7,3 (4H, м), 6,95-6,86 (1H, м), 4,18 (2H, кв, J=7,3 Гц), 2,72-2,62 (1H, м), 2,36 (3H, с), 1,87-1,79 (1H, м), 1,65-1,55 (1H, м), 1,35-1,25 (1H, м), 1,29 (3H, т, J=7,3 Гц).
Стадия 3: транс-2-(4-фтор-1H-индол-3-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил транс-2-(4-фтор-1-тозил-1H-индол-3-ил)циклопропанкарбоксилата.
LCMS (способ A) m/z M-1 obs 218,3, tR=2,52 мин.
Промежуточное карбоновокислотное соединение-15
транс-2-(хинолин-2-ил)циклопропанкарбоновая кислота
Стадия 1: этил транс-2-(хинолин-2-ил)циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из (E)-этил 3-(хинолин-2-ил)акрилата.
1H-ЯМР (300 МГц, CDCl3): δ 8,04 (1H, д, J=8,8 Гц), 7,93 (1H, д, J=8,1 Гц), 7,75 (1H, д, J=8,1 Гц), 7,65 (1H, т, J=8,1 Гц), 7,46 (1H, т, J=8,1 Гц), 7,33 (1H, д, J=8,8 Гц), 4,18 (2H, д, J=7,3 Гц), 2,80-2,73 (1H, м), 2,47-2,40 (1H, м), 1,82-1,75 (1H, м), 1,71-1,64 (1H, м), 1,29 (3H, т, J=7,3 Гц). LCMS (способ A) m/z M+1 obs 242,2, tR=3,09 мин.
Стадия 2: транс-2-(хинолин-2-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил транс-2-(хинолин-2-ил)циклопропанкарбоксилата.
1H-ЯМР (300 МГц, ДМСО-d6): δ 8,27 (1H, д, J=8,8 Гц), 7,94 (1H, д, J=8,0 Гц), 7,87 (1H, д, J=8,0 Гц), 7,70 (1H, т, J=8,0 Гц), 7,58 (1H, д, J=8,8 Гц), 7,52 (1H, т, J=8,0 Гц), 2,75 (1H, шир.), 2,20 (1H, шир.), 1,68-1,48 (2H, м) (сигнал не обнаружен из-за COOH). LCMS (способ A) m/z M-1 obs 212,2, tR=2,27 мин.
Промежуточное карбоновокислотное соединение-16
транс-2-(1H-индазол-3-ил)циклопропанкарбоновая кислота
Стадия 1: метил (E)-3-(1-тозил-1H-индазол-3-ил)акрилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-11, из метил (E)-3-(1H-индазол-3-ил)акрилата.
1H-ЯМР (300 МГц, CDCl3) δ 8,25 (1H, д, J=8,8 Гц), 7,93-7,84 (4H, м), 7,60 (1H, т, J=7,7 Гц), 7,41 (1H, т, J=7,7 Гц), 7,30-7,24 (2H, м), 6,87 (1H, д, J=16,8 Гц), 3,84 (3H, с), 2,37 (3H, с). LCMS (способ A) m/z M+1 obs 357,2, tR=3,32 мин.
Стадия 2: метил транс-2-(1-тозил-1H-индазол-3-ил)циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из метил (E)-3-(1-тозил-1H-индазол-3-ил)акрилата.
1H-ЯМР (300 МГц, CDCl3) δ 8,16 (1H, д, J=8,0 Гц), 7,79 (2H, д, J=8,0 Гц), 7,69 (1H, д, J=8,0 Гц), 7,55 (1H, т, J=8,0 Гц), 7,33 (1H, т, J=8,0 Гц), 7,22 (2H, д, J=8,0 Гц), 3,73 (3H, с), 2,80-2,70 (1H, м), 2,35 (3H, с), 2,33-2,27 (1H, м), 1,72-1,62 (2H, м). LCMS (способ A) m/z M+1 obs 371,2, tR=3,25 мин.
Стадия 3: транс-2-(1H-индазол-3-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из метил транс-2-(1-тозил-1H-индазол-3-ил)циклопропанкарбоксилата.
1H-ЯМР (300 МГц, ДМСО-d6) δ 12,75 (1H, шир.), 7,78 (1H, д, J=8,0 Гц), 7,46 (1H, д, J=8,0 Гц), 7,33 (1H, т, J=8,0 Гц), 7,09 (1H, т, J=8,0 Гц), 2,77-2,65 (1H, м), 2,08-2,00 (1H, м), 1,58-1,47 (2H, м). LCMS (способ A) m/z M-1 obs 201,3, tR=2,29 мин.
Промежуточное карбоновокислотное соединение-17
транс-2-(хинолин-7-ил)циклопропанкарбоновая кислота
Стадия 1: этил (E)-3-(хинолин-7-ил)акрилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-7, из хинолин-7-карбальдегида.
1H-ЯМР (300 МГц, CDCl3) δ 8,95 (1H, д, J=4,4 Гц), 8,20 (1H, с), 8,15 (1H, д, J=8,8 Гц), 7,87 (1H, д, J=16,1 Гц), 7,83 (1H, д, J=8,8 Гц), 7,73 (1H, д, J=8,8 Гц), 7,42 (1H, дд, J=4,4 и 8,0 Гц), 6,62 (1Н, д, J=16,1 Гц), 4,31 (2H, кв, J=7,3 Гц), 1,37 (3H, т, J=7,3 Гц). LCMS (способ A) m/z M+1 obs 228,3, tR=2,82 мин.
Стадия 2: этил транс-2-(хинолин-7-ил)циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из этил (E)-3-(хинолин-7-ил)акрилата.
1H-ЯМР (300 МГц, CDCl3) δ 8,91-8,85 (1H, м), 8,11 (1H, д, J=8,8 Гц), 7,80 (1H, с), 7,74 (1H, д, J=8,8 Гц), 7,40-7,30 (2H, м), 4,19 (2H, кв, J=8,0 Гц), 2,76-2,67 (1H, м), 2,10-2,02 (1H, м), 1,76-1,68 (1H, м), 1,54-1,45 (1H, м), 1,30 (3H, т, J=8,0 Гц). LCMS (способ A) m/z M+1 obs 242,3, tR=2,79 мин.
Стадия 3: транс-2-(хинолин-7-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил транс-2-(хинолин-7-ил)циклопропанкарбоксилата.
1H-ЯМР (300 МГц, ДМСО-d6) δ 8,86 (1H, д, J=4,4Гц), 8,31 (1H, д, J=8,8 Гц), 7,90 (1H, д, J=9,5 Гц), 7,81 (1H, с), 7,46 (1H, дд, J=4,4 и 8,8 Гц), 7,40 (1H, д, J=9,5 Гц), 2,66-2,58 (1H, м), 2,03-1,95 (1H, м), 1,57-1,50 (2H, м) (сигнал не обнаружен из-за COOH). LCMS (способ A) m/z M-1 obs 212,3, tR=2,13 мин.
Промежуточное карбоновокислотное соединение-18
транс-2-(1-метил-1H-индол-6-ил)циклопропанкарбоновая кислота
Стадия 1: этил (E)-3-(1-метил-1H-индол-6-ил)акрилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-7, из 1-метил-1H-индол-6-карбальдегида.
1H-ЯМР (300 МГц, CDCl3) δ 7,85 (1H, д, J=15,4 Гц), 7,60 (1H, д, J=8,0 Гц), 7,47 (1H, с), 7,34 (1H, д, J=8,0 Гц), 7,13 (1H, д, J=2,9 Гц), 6,49 (1H, д, J=2,9 Гц), 6,47 (1H, д, J=15,4 Гц), 4,27 (2H, кв, J=7,3 Гц), 3,82 (3H, с), 1,35 (3H, т, J=7,3 Гц). LCMS (способ A) m/z M+1 obs 230,3, tR=3,15 мин.
Стадия 2: этил транс-2-(1-метил-1H-индол-6-ил)циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из этил (E)-3-(1-метил-1H-индол-6-ил)акрилата.
1H-ЯМР (300 МГц, CDCl3) δ 7,52 (1H, д, J=8,1 Гц), 7,08 (1H, с), 7,00 (1H, д, J=2,9 Гц), 6,86 (1H, д, J=8,1 Гц), 6,43 (1H, д, J=2,9 Гц), 4,18 (2H, кв, J=7,4 Гц), 3,76 (3H, с), 2,72-2,63 (1H, м), 1,99-1,90 (1H, м), 1,66-1,59 (1H, м), 1,44-1,35 (1H, м), 1,29 (3H, т, J=7,4 Гц). LCMS (способ A) m/z M+1 obs 244,3, tR=3,17 мин.
Стадия 3: транс-2-(1-метил-1H-индол-6-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил транс-2-(1-метил-1H-индол-6-ил)циклопропанкарбоксилата.
1H-ЯМР (300 МГц, ДМСО-d6) δ 12,2 (1H, шир.), 7,42 (1H, д, J=8,1 Гц), 7,26-7,20 (2H, м), 6,82 (1H, д, J=8,0 Гц), 6,35 (1H, д, J=2,9 Гц), 3,75 (3H, с), 2,50-2,44 (1H, м), 1,85-1,77 (1H, м), 1,47-1,38 (2H, м). LCMS (способ A) m/z M-1 obs 214,2, tR=2,67 мин.
Промежуточное карбоновокислотное соединение-19
транс-2-(6-фтор-1H-индол-3-ил)циклопропанкарбоновая кислота
Стадия 1: этил (E)-3-(6-фтор-1-тозил-1H-индол-3-ил)акрилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-7, из 6-фтор-1-тозил-1H-индол-3-карбальдегида.
1H-ЯМР (300 МГц, CDCl3) δ 7,82-7,70 (6H, м), 7,34-7,25 (2H, м), 7,08 (1H, т, J=8,8 Гц), 6,48 (1H, д, J=16,1 Гц), 4,27 (2H, кв, J=7,3 Гц), 2,38 (3H, с), 1,34 (3H, т, J=7,3 Гц). LCMS (способ A) m/z M+1 obs 388,2, tR=3,57 мин.
Стадия 2: этил транс-2-(6-фтор-1-тозил-1H-индол-3-ил)циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из этил (E)-3-(6-фтор-1-тозил-1H-индол-3-ил)акрилата.
1H-ЯМР (300 МГц, CDCl3) δ 7,73 (2H, д, J=8,0 Гц), 7,68 (1H, дд, J=2,2 и 9,5 Гц), 7,47 (1H, дд, J=5,1 и 8,8 Гц), 7,28-7,22 (2H, м), 7,00 (1H, дт, J=2,2 и 8,8 Гц), 4,19 (2H, кв, J=7,3 Гц), 2,50-2,40 (1H, м), 2,36 (3H, с), 1,87-1,80 (1H, м), 1,61-1,53 (1H, м), 1,31 (3H, т, J=7,3 Гц), 1,28-1,21 (1H, м). LCMS (способ A) m/z M+1 obs 402,2, tR=3,48 мин.
Стадия 3: транс-2-(6-фтор-1H-индол-3-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил транс-2-(6-фтор-1-тозил-1H-индол-3-ил)циклопропанкарбоксилата.
LCMS (способ A) m/z M-1 obs 218,3, tR=2,54 мин.
Промежуточное карбоновокислотное соединение-20
транс-2-((4-хлорфенокси)метил)циклопропанкарбоновая кислота
Стадия 1: этил транс-2-((4-хлорфенокси)метил)циклопропанкарбоксилат
К суспензии гидрида натрия (60%, 650 мг, 16,3 ммоль) в толуоле (25 мл) при 0ºC добавляли по каплям раствор триэтилфосфоноацетата (3,64 г, 16,3 ммоль) в толуоле (5 мл). После перемешивания при комнатной температуре в течение 10 минут, добавляли 2-((4-хлорфенокси)метил)оксиран (1,50 г, 8,1 ммоль) и смесь кипятили с обратным холодильником при перемешивании в течение 1 дня. После охлаждения до комнатной температуры смесь выливали в насыщенный солевой раствор и водный слой экстрагировали при помощи EtOAc два раза. Объединенный органический слой сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (10:1-5:1), с получением 1,25 г (60%) указанного в заголовке соединения в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3) δ 7,22 (2H, д, J=8,8 Гц), 6,80 (2H, д, J=8,8 Гц), 4,15 (2H, кв, J=7,3 Гц), 3,92 (1H, дд, J=6,6 и 10,2 Гц), 3,83 (1H, дд, J=6,6 и 10,2 Гц), 1,93-1,82 (1H, м), 1,71-1,65 (1H, м), 1,27 (3H, т, J=7,3 Гц), 1,01-0,93 (1H, м), 0,90-0,76 (1H, м). LCMS (способ A) m/z M+1 obs 255,2, tR=3,25 мин.
Стадия 2: транс-2-((4-хлорфенокси)метил) циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из транс-2-((4-хлорфенокси)метил)циклопропанкарбоновой кислоты.
1H-ЯМР (300 МГц, CDCl3) δ 7,22 (2Н, J=8,8 Гц), 6,80 (2H, д, J=8,8 Гц), 3,96 (1H, дд, J=5,9 и 10,3 Гц), 8,81 (1H, дд, J=6,6 и 10,3 Гц), 2,00-1,90 (1H, м), 1,75-1,68 (1H, м), 1,41-1,32 (1H, м), 1,12-1,05 (1H, м) (сигнал не обнаружен из-за COOH). LCMS (способ A) m/z M-1 obs 225,2, tR=2,80 мин.
Промежуточное карбоновокислотное соединение-21
транс-2-(изохинолин-3-ил)циклопропанкарбоновая кислота
Стадия 1: этил (E)-3-(изохинолин-3-ил)акрилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-7, из изохинолин-3-карбальдегида.
1H-ЯМР (300 МГц, CDCl3) δ 9,25 (1H, с), 7,99 (1H, д, J=8,0 Гц), 7,85 (1H, д, J=7,3 Гц), 7,83 (1H, д, J=15,4 Гц), 7,75-7,62 (3H, м), 7,19 (1H, д, J=15,4 Гц), 4,29 (2H, кв, J=6,6 Гц), 1,35 (3H, т, J=6,6 Гц). LCMS (способ A) m/z M+1 obs 228,2, tR=2,99 мин.
Стадия 2: этил транс-2-(изохинолин-3-ил)циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из этил (E)-3-(изохинолин-3-ил)акрилата.
1H-ЯМР (300 МГц, CDCl3) δ 9,11 (1H, с), 7,92 (1H, д, J=8,0 Гц), 7,75 (1H, д, J=7,4 Гц), 7,66 (1H, м), 7,58 (1H, с), 7,53 (1H, м), 4,19 (2H, кв, J=6,6 Гц), 2,76 (1H, м), 2,33 (1H, м), 1,77-1,63 (2H, м), 1,29 (3H, т, J=6,6 Гц).
Стадия 3: транс-2-(изохинолин-3-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил транс-2-(изохинолин-3-ил)циклопропанкарбоксилата.
LCMS (способ A) m/z M+1 obs 214,3, tR=2,40 мин.
Промежуточное карбоновокислотное соединение-22
транс-2-(хинолин-3-ил)циклопропанкарбоновая кислота
Стадия 1: этил транс-2-(хинолин-3-ил)циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из этил (E)-3-(хинолин-3-ил)акрилата.
1H-ЯМР (300 МГц, CDCl3) δ 8,77 (1H, д, J=2,2 Гц), 8,08 (1H, д, J=8,8 Гц), 7,80 (1H, д, J=2,2 Гц), 7,75 (1H, дд, J=8,0, 1,5 Гц), 7,68 (1H, тд, J=6,6, 1,5 Гц), 7,54 (1H, м), 4,22 (2H, кв, J=7,3 Гц), 2,73 (1H, м), 2,07 (1H, м), 1,75 (1H, м), 1,46 (1H, м), 1,31 (3H, т, J=7,3 Гц). LCMS (способ A) m/z M+1 obs 242,3, tR=2,85 мин.
Стадия 2: транс-2-(хинолин-3-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил транс-2-(хинолин-3-ил)циклопропанкарбоксилата.
1H-ЯМР (300 МГц, ДМСО-d6) δ 8,83 (1H, д, J=2,2 Гц), 8,10 (1H, д, J=2,2 Гц), 7,99 (1H, д, J=8,0 Гц), 7,89 (1H, д, J=7,3 Гц), 7,71 (1H, м), 7,59 (1H, т, J=8,1 Гц), 2,63 (1H, м), 2,05 (1H, м), 1,55 (2H, т, J=6,6 Гц). LCMS (способ A) m/z M+1 obs 214,3, tR=2,30 мин.
Промежуточное карбоновокислотное соединение-23
2-(3-(дифторметокси)фенил)циклопропанкарбоновая кислота
Стадия 1: этил (E)-3-(3-(дифторметокси)фенил)акрилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-7, из 3-(дифторметокси)бензальдегида.
1H-ЯМР (300 МГц, CDCl3) δ 7,95 (1H, д, J=16,1 Гц), 7,62 (1H, дд, J=7,7, 1,8 Гц), 7,38 (1H, дт, J=7,7, 1,5 Гц), 7,26-7,16 (2H, м), 6,56 (1H, т, J=73 Гц), 6,48 (1H, д, J=16,1 Гц), 4,27 (2H, кв, J=7,0 Гц), 1,34 (3H, т, J=7,0 Гц).
Стадия 2: этил транс-2-(3-(дифторметокси)фенил)циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из этил (E)-3-(3-(дифторметокси)фенил)акрилата.
1H-ЯМР (300 МГц, CDCl3) δ 7,25-7,09 (3H, м), 6,97 (1H, дд, J=7,7, 1,8 Гц), 6,52 (1H, т, J=74 Гц), 4,19 (2H, м), 2,71 (1H, м), 1,83 (1H, м), 1,61 (1H, м), 1,31 (1H, м), 1,28 (3H, т, J=7,0 Гц).
Стадия 3: транс-2-(3-(дифторметокси)фенил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил транс-2-(3-(дифторметокси)фенил)циклопропанкарбоксилата.
LCMS (способ A) m/z M-1 ObS 228,2, tR=2,66 мин.
Промежуточное карбоновокислотное соединение-24
транс-2-(2-фтор-5-метоксифенил)циклопропанкарбоновая кислота
Стадия 1: этил транс-2-(2-фтор-5-метоксифенил) циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из этил (E)-3-(2-фтор-5-метоксифенил)акрилата.
1H-ЯМР (270 МГц, CDCl3) δ 6,93 (1H, т, J=9,2 Гц), 6,66 (1H, дт, J=8,9, 3,3 Гц), 6,45 (1H, дд, J=5,9, 3,0 Гц), 4,17 (2H, кв, J=7,3 Гц), 3,75 (3H, с), 2,62 (1H, м), 1,93 (1H, м), 1,58 (1H, м), 1,33 (1H, м), 1,28 (3H, т, J=7,3 Гц).
Стадия 2: транс-2-(2-фтор-5-метоксифенил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил транс-2-(2-фтор-5-метоксифенил)циклопропанкарбоксилата.
LCMS (способ A) m/z: M-1 obs 209,2, tR=2,59 мин.
Промежуточное карбоновокислотное соединение-25
транс-2-((1H-индол-1-ил)метил)циклопропанкарбоновая кислота
Стадия 1: этил транс-2-((1H-индол-1-ил)метил)циклопропанкарбоксилат
К суспензии гидрида натрия (60%, 55 мг, 1,4 ммоль) в ДМФА (5 мл) добавляли индол (135 мг, 1,2 ммоль). После перемешивания при комнатной температуре в течение 10 минут добавляли этил 2-(((метилсульфонил)окси)метил) циклопропанкарбоксилат (307 мг, 1,4 ммоль). После перемешивания при комнатной температуре в течение 6 часов, смесь выливали в воду и водный слой экстрагировали при помощи EtOAc два раза. Объединенный органический слой сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат (10:1-5:1), с получением 153 мг (54%) указанного в заголовке соединения в виде светло-коричневого масла;
1H-ЯМР (300 МГц, CDCl3) δ 7,67-7,60 (1H, м), 7,42-7,09 (4H, м), 6,57-6,50 (1H, м), 4,22-4,02 (4H, м), 1,96-1,86 (1H, м), 1,69-1,62 (1H, м), 1,31-1,25 (1H, м), 1,24 (3H, т, J=7,3 Гц), 0,95-0,87 (1H, м). LCMS (способ A) m/z M+1 obs 244,3, tR=3,17 мин.
Стадия 2: транс-2-((1H-индол-1-ил)метил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил транс-2-((1H-индол-1-ил)метил)циклопропанкарбоксилата.
1H-ЯМР (300 МГц, CDCl3) δ 7,66-7,61 (1H, м), 7,43-7,08 (4H, м), 6,58-6,50 (1H, м), 4,20-4,06 (2H, м), 2,00-1,91 (1H, м), 1,70-1,62 (1H, м), 1,36-1,27 (1H, м), 1,01-0,94 (1H, м) (сигнал не обнаружен из-за COOH). LCMS (способ A) m/z M-1 obs 214,3, tR=2,72 мин.
Промежуточное карбоновокислотное соединение-26
транс-2-(1-((3-метилоксетан-3-ил)метил)-1H-индол-6-ил)циклопропанкарбоновая кислота
Стадия 1: 1-((3-метилоксетан-3-ил)метил)-1H-индол-6-карбальдегид
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-11, из 1H-индол-6-карбальдегида.
1H-ЯМР (300 МГц, CDCl3) δ 10,06 (1H, с), 7,93 (1H, с), 7,73 (1H, д, J=8,0 Гц), 7,64 (1H, д, J=8,0 Гц), 7,30 (1H, д, J=3,7 Гц), 6,63 (1H, д, J=3,7 Гц), 4,67 (2H, д, J=6,6 Гц), 4,45 (2H, с), 4,42 (2H, д, J=6,6 Гц), 1,31 (3H, с). LCMS (способ A) m/z M+1 obs 230,2, tR=2,62 мин.
Стадия 2: этил (E)-3-(1-((3-метилоксетан-3-ил)метил)-1H-индол-6-ил)акрилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-7, из 1-((3-метилоксетан-3-ил)метил)-1H-индол-6-карбальдегида.
1H-ЯМР (300 МГц, CDCl3) δ 7,83 (1H, д, J=16,1 Гц), 7,62 (1H, д, J=8,1 Гц), 7,50 (1H, с), 7,36 (1H, д, J=8,1 Гц), 7,15 (1H, д, J=3,0 Гц), 6,55 (1H, д, J=3,0 Гц), 6,47 (1H, д, J=16,1 Гц), 4,68 (2H, д, J=5,9 Гц), 4,43 (2H, д, J=5,9 Гц), 4,40 (2H, с), 4,29 (2H, кв, J=7,4 Гц), 1,36 (3H, т, J=7,4 Гц), 1,32 (3H, с). LCMS (способ A) m/z M+1 obs 300,2, tR=3,09 мин.
Стадия 3: этил транс-2-(1-((3-метилоксетан-3-ил)метил)-1H-индол-6-ил)циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из этил (E)-3-(1-((3-метилоксетан-3-ил)метил)-1H-индол-6-ил)акрилата.
1H-ЯМР (300 МГц, CDCl3) δ 7,52 (1H, д, J=8,1 Гц), 7,13 (1H, с), 7,01 (1H, д, J=2,9 Гц), 6,83 (1H, д, J=8,1 Гц), 6,48 (1H, д, J=3,0 Гц), 4,67 (2H, д, J=6,6 Гц), 4,41 (2H, д, J=6,6 Гц), 4,34 (2H, с), 4,18 (2H, кв, J=7,3 Гц), 2,67 (1H, м), 1,93 (1H, м), 1,63 (1H, м), 1,40-1,25 (7H, м). LCMS (способ A) m/z M+1 obs 314,2, tR=3,10 мин.
Стадия 4: транс-2-(1-((3-метилоксетан-3-ил)метил)-1H-индол-6-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил транс-2-(1-((3-метилоксетан-3-ил)метил)-1H-индол-6-ил)циклопропанкарбоксилата.
1H-ЯМР (300 МГц, CDCl3) δ 7,54 (1H, д, J=8,1 Гц), 7,17 (1H, с), 7,03 (1H, д, J=3,7 Гц), 6,85 (1H, д, J=8,1 Гц), 6,50 (1H, д, J=3,6 Гц), 4,69 (2H, д, J=6,6 Гц), 4,43 (2H, д, J=5,9 Гц), 4,35 (2H, с), 2,76 (1H, м), 1,96 (1H, м), 1,72 (1H, м), 1,49 (1H, м), 1,32 (3H, с).
Промежуточное карбоновокислотное соединение-27
транс-2-(2-(изопропиламино)пиридин-4-ил)циклопропанкарбоновая кислота
Стадия 1: этил транс-2-(2-хлорпиридин-4-ил)циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из (E)-этил 3-(2-хлорпиридин-4-ил)акрилата.
1H-ЯМР (300 МГц, CDCl3) δ 8,25 (1H, д, J=5,1 Гц), 7,04 (1H, с), 6,91 (1H, дд, J=5,1, 1,4 Гц), 4,18 (2H, кв, J=6,6 Гц), 2,46 (1H, м), 2,00 (1H, м), 1,71 (1H, м), 1,36 (1H, м), 1,29 (3H, т, 6,6 Гц). LCMS (способ A) m/z M+1 obs 226,2, tR =2,82 мин.
Стадия 2: этил транс-2-(2-(изопропиламино)пиридин-4-ил)циклопропанкарбоксилат
К раствору этил транс-2-(2-хлорпиридин-4-ил)циклопропанкарбоксилата (250 мг, 1,1 ммоль) и изопропиламина (393 мг, 6,7 ммоль) в диоксане (5 мл) при комнатной температуре добавляли карбонат цезия (1,1 г, 3,3 ммоль), Xantophos (224 мг, 0,4 ммоль) и ацетат палладия (50 мг, 0,2 ммоль), соответственно. Смесь герметично закрывали и перемешивали при 100ºC в течение 14 часов. Фильтрат отфильтровывали и затем концентрировали при пониженном давлении, остаток очищали при помощи колоночной хроматографии на силикагеле и элюировали смесью гексан/этилацетат 6/1 с получением 100 мг (36% выход) указанного в заголовке соединения в виде бесцветного масла;
1H-ЯМР (300 МГц, CDCl3) δ 7,93 (1H, д, J=5,9 Гц), 6,18 (1H, д, J=5,9 Гц), 6,11 (1H, с), 4,32 (1H, шир.д, J=7,3 Гц), 4,16 (2H, кв, J=7,3 Гц), 3,87 (1H, м), 2,36 (1H, м), 1,93 (1H, м), 1,60 (1H, м), 1,33-1,18 (10H, м). LCMS (способ A) m/z M+1 obs 249,3, tR=2,04 мин.
Стадия 3: транс-2-(2-(изопропиламино)пиридин-4-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил транс-2-(2-(изопропиламино)пиридин-4-ил)циклопропанкарбоксилата.
LCMS (способ A) m/z M+1 obs 221,3, tR=0,82 мин.
Промежуточное карбоновокислотное соединение-28
2-(1H-индол-4-ил)циклопропанкарбоновая кислота
Стадия 1: этил 2-(1-тозил-1H-индол-4-ил)циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из (E)-этил 3-(1-тозил-1H-индол-4-ил)акрилата.
1H-ЯМР (300 МГц, CDCl3) δ 7,86 (1H, д, J=8,8 Гц), 7,76 (2H, д, J=8,1 Гц), 7,59 (1H, д, J=4,4 Гц), 7,26-7,19 (3H, м), 6,85-6,80 (2H, м), 4,19 (2H, кв, J=6,6 Гц), 2,73 (1H, м), 2,34 (3H, с), 1,94 (1H, м), 1,64 (1H, м), 1,36 (1H, м), 1,29 (3H, т, J=7,3 Гц). LCMS (способ A) m/z M+1 obs 384,2, tR=3,47 мин.
Стадия 2: 2-(1H-индол-4-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил 2-(1-тозил-1H-индол-4-ил)циклопропанкарбоксилата.
1H-ЯМР (300 МГц, CDCl3) δ 8,25 (1H, шир.с), 7,33-7,24 (2H, м), 7,14 (1H, т, J=7,3 Гц), 6,80 (1H, д, J=7,3 Гц), 6,72 (1H, м), 2,98 (1H, м), 2,05 (1H, м), 1,75 (1H, м), 1,58 (1H, м). LCMS (способ A) m/z M+1 obs 202,2, tR=2,38 мин.
Промежуточное карбоновокислотное соединение-29
2-(8-хлорхинолин-2-ил)циклопропанкарбоновая кислота
Стадия 1: (E)-этил 3-(8-хлорхинолин-2-ил)акрилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-7, из 8-хлорхинолин-2-карбальдегида.
1H-ЯМР (300 МГц, CDCl3) δ 8,23 (1H, д, J=8,8 Гц), 7,96 (1H, д, J=15,4 Гц), 7,88 (1H, дд, J=7,3, 1,4 Гц), 7,77 (1H, д, J=7,3 Гц), 7,69 (1H, д, J=8,8 Гц), 7,50 (1H, т, J=8,1 Гц), 7,13 (1H, д, J=15,4 Гц), 4,34 (2H, кв, J=6,6 Гц), 1,40 (3H, т, J=6,6 Гц). LCMS (способ A) m/z M+1 obs 262,1, tR=3,24 мин.
Стадия 2: этил 2-(8-хлорхинолин-2-ил)циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из (E)-этил 3-(8-хлорхинолин-2-ил)акрилата.
LCMS (способ A) m/z M+1 obs 276,1, tR=3,40 мин.
Стадия 3: 2-(8-хлорхинолин-2-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил 2-(8-хлорхинолин-2-ил)циклопропанкарбоксилата.
LCMS (способ A) m/z M+1 obs 248,2, tR=2,82 мин.
Промежуточное карбоновокислотное соединение-30
2-(1-метил-1H-индазол-6-ил)циклопропанкарбоновая кислота
Стадия 1: (E)-этил 3-(1-метил-1H-индазол-6-ил)акрилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-7, из 1-метил-1H-индазол-6-карбальдегида.
1H-ЯМР (270 МГц, CDCl3) δ 7,97 (1H, с), 7,82 (1H, д, J=16,1 Гц), 7,71 (1H, д, J=8,2 Гц), 7,50 (1H, с), 7,35 ( 1H, дд, J=8,6, 1,0 Гц), 6,53 (1H, 6, J=16,1 Гц), 4,28 (2H, кв, J=6,9 Гц), 4,09 (3H, с), 1,35 (3H, т, J=6,9 Гц). LCMS (способ A) m/z: M+1 obs 231,2, tR=2,88 мин.
Стадия 2: этил 2-(1-метил-1H-индазол-6-ил)циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из (E)-этил 3-(1-метил-1H-индазол-6-ил)акрилата.
1H-ЯМР (270 МГц, CDCl3) δ 7,91 (1H, с), 7,61 (1H, д, J=8,6 Гц), 7,13 (1H, с), 6,87 (1H, дд, J=8,6, 1,3 Гц), 4,18 (2H, кв, J=7,3 Гц), 4,04 (3H, с), 2,67 (1H, м), 1,98 (1H, м), 1,66 (1H, м), 1,41 (1H, м), 1,29 (3H, т, J=7,3 Гц). LCMS (способ A) m/z: M+1 obs 245,3, tR=2,95 мин.
Стадия 3: 2-(1-метил-1H-индазол-6-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил 2-(1-метил-1H-индазол-6-ил)циклопропанкарбоксилата.
1H-ЯМР (300 МГц, ДМСО-d6) δ 7,94 (1H, с), 7,62 (1H, д, J=8,1 Гц), 7,42 (1H, с), 6,92 (1H, д, J=8,4 Гц), 3,98 (3H, с), 2,49 (1H, м), 1,90 (1H, м), 1,48-1,43 (2H, м). LCMS (способ A) m/z: M+1 obs 217,2, tR=2,37 мин.
Промежуточное карбоновокислотное соединение-31
2-(1H-индол-5-ил)циклопропанкарбоновая кислота
Стадия 1: (E)-этил 3-(1-тозил-1H-индол-5-ил)акрилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-7, из 1-тозил-1H-индол-5-карбальдегида.
1H-ЯМР (270 МГц, CDCl3) δ 7,98 (1H, д, J=8,6 Гц), 7,76 (2H, д, J=8,6 Гц), 7,74 (1H, д, J=15,8 Гц), 7,66 (1H, с), 7,58 (1H, д, J=3,9 Гц), 7,50 (1H, д, J=8,6 Гц), 7,23 (2H, д, J=8,6 Гц), 6,66 (1H, д, J=3,3 Гц), 6,41 (1H, д, J=15,8 Гц), 4,26 (2H, кв, J=7,2 Гц), 2,34 (3H, с), 1,33 (3H, т, J=7,2 Гц). LCMS (способ A) m/z M+1 obs 370,2, tR=3,45 мин.
Стадия 2: этил 2-(1-тозил-1H-индол-5-ил)циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из (E)-этил 3-(1-тозил-1H-индол-5-ил)акрилата.
1H-ЯМР (300 МГц, CDCl3) δ 7,88 (1H, д, J=8,8 Гц), 7,74 (2H, д, J=8,1 Гц), 7,53 (1H, д, J=3,7 Гц), 7,25 (1H, д, J=2,2 Гц), 7,21 (2H, д, J=8,1 Гц), 7,05 (1H, дд, J=8,8, 2,2 Гц), 6,58 (1H, д, J=3,7 Гц), 4,18 (2H, кв, J=7,3 Гц), 2,57 (1H, м), 2,34 (3H, с), 1,88 (1H, м), 1,61 (1H, м), 1,32 (1H, м), 1,27 (3H, т, J=7,3 Гц). LCMS (способ A) m/z M+1 obs 384,3, tR=3,45 мин.
Стадия 3: 2-(1H-индол-5-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил 2-(1-тозил-1H-индол-5-ил)циклопропанкарбоксилата.
1H-ЯМР (300 МГц, ДМСО-d6) δ 12,2 (1H, шир.с), 11,1 (1H, с), 7,36-7,30 (3H, м), 6,92 (1H, д, J=8,1 Гц), 6,39 (1H, с), 2,49 (1H, м), 1,77 (1H, м), 1,49-1,37 (2H, м).
Промежуточное карбоновокислотное соединение-32
2-(3-(бензилокси)фенил)циклопропанкарбоновая кислота
Стадия 1: этил 2-(3-(бензилокси)фенил) циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из (E)-этил 3-(3-(бензилокси)фенил)акрилата.
1H-ЯМР (300 МГц, CDCl3) δ 7,44-7,32 (5H, м), 7,19 (1H, м), 6,81 (1H, м), 6,72-6,69 (2H, м), 5,04 (2H, с), 4,16 (2H, кв, J=7,3 Гц), 2,48 (1H, м), 1,89 (1H, м), 1,58 (1H, м), 1,30 (1H, м), 1,27 (3H, т, J=7,3 Гц).
Стадия 2: 2-(3-(бензилокси)фенил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил 2-(3-(бензилокси)фенил)циклопропанкарбоксилата.
LCMS (способ A) m/z M-1 obs 267,2, tR=3,03 мин.
Промежуточное карбоновокислотное соединение-33
2-(2-хлор-4-фторфенил)циклопропанкарбоновая кислота
Стадия 1: (E)-этил 3-(2-хлор-4-фторфенил)акрилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-7, из 2-хлор-4-фторбензальдегида.
1H-ЯМР (300 МГц, CDCl3) δ 8,02 (1H, д, J=16,1 Гц), 7,62 (1H, дд, J=8,8, 6,6 Гц), 7,18 (1H, дд, J=7,3, 1,5 Гц), 7,02 (1H, м), 6,38 (1H, д, J=16,1 Гц), 4,28 (2H, кв, J=7,3 Гц), 1,35 (3H, т, J=7,3 Гц). LCMS (способ A) m/z M-1 obs 229,2, tR=3,22 мин.
Стадия 2: этил 2-(2-хлор-4-фторфенил)циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из (E)-этил 3-(2-хлор-4-фторфенил)акрилата.
1H-ЯМР (300 МГц, CDCl3) δ 7,12 (1H, дд, J=8,8, 2,9 Гц), 6,99 (1H, м), 6,89 (1H, м), 4,20 (2H, кв, J=7,3 Гц), 2,66 (1H, м), 1,77 (1H, м), 1,61 (1H, м), 1,29 (3H, т, J=7,3 Гц), 1,29 (1H, м). LCMS (способ A) m/z M-1 obs 243,2, tR=3,27 мин.
Стадия 3: 2-(2-хлор-4-фторфенил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил 2-(2-хлор-4-фторфенил)циклопропанкарбоксилата.
1H-ЯМР (300 МГц, ДМСО-d6) S 7,44 (1H, д, J=8,8 Гц), 7,25-7,10 (2H, м), 2,48 (1H, м), 1,70 (1H,м), 1,45-1,35 (2H, м). LCMS (способ A) m/z M-1 obs 213,2, tR=2,72 мин.
Промежуточное карбоновокислотное соединение-34
2-(2-фтор-4-метоксифенил)циклопропанкарбоновая кислота
Стадия 1: этил 2-(2-фтор-4-метоксифенил) циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из (E)-этил 3-(2-фтор-4-метоксифенил)акрилата.
1H-ЯМР (300 МГц, CDCl3) δ 6,90 (1H, т, J=6,6 Гц), 6,65-6,55 (2H, м), 4,17 (2H, кв, J=7,3 Гц), 3,77 (3H, с), 2,57 (1H, м), 1,86 (1H, м), 1,54 (1H, м), 1,28 (3H, т, J=7,3 Гц), 1,28 (1H, м). LCMS (способ A) m/z: M+1 obs 239,3, tR=3,13 мин.
Стадия 2: 2-(2-фтор-4-метоксифенил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил 2-(2-фтор-4-метоксифенил)циклопропанкарбоксилата.
1H-ЯМР (300 МГц, ДМСО-d6) δ 7,01 (1H, т, J=8,8 Гц), 6,79 1H, м), 6,69 (1H, м), 3,72 (3H, с), 2,34 (1H, м), 1,71 (1H, м), 1,40-1,30 (2H, м). LCMS (способ A) m/z: M-1 obs 209,2 tR=2,60 мин.
Промежуточное карбоновокислотное соединение-35
2-(2,4,6-трифторфенил)циклопропанкарбоновая кислота
Стадия 1: (E)-этил 3-(2,4,6-трифторфенил)акрилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-7, из 2,4,6-трифторбензальдегида.
1H-ЯМР (300 МГц, CDCl3) δ 7,69 (1H, д, J=16,8 Гц), 6,77-6,65 (3H, м), 4,28 (2H, кв, J=7,3 Гц), 1,35 (3H, т, J=7,3 Гц). LCMS (способ A) m/z: M+1 obs 231,2, tR=3,18 мин.
Стадия 2: этил 2-(2,4,6-трифторфенил)циклопропанкарбоксилат
Получали, как на стадии 1 получения промежуточного карбоновокислотного соединения-6, из (E)-этил 3-(2,4,6-трифторфенил)акрилата.
1H-ЯМР (300 МГц, CDCl3) δ 6,66-6,50 (2H, м), 4,18 (2H, кв, J=7,3 Гц), 2,40 (1H, м), 2,07 (1H, м), 1,58-1,44 (2H, м), 1,30 (3H, т, J=7,3 Гц). LCMS (способ A) m/z: M+1 obs 245,2 tR=3,23 мин.
Стадия 3: 2-(2,4,6-трифторфенил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил 2-(2,4,6-трифторфенил)циклопропанкарбоксилата.
1H-ЯМР (300 МГц, ДМСО-d6) δ 7,18-7,10 (2H, м), 2,17 (1H, м), 1,88 (1H, м), 1,45-1,30 (2H, м). LCMS (способ A) m/z M-1 obs 215,2 tR=2,65 мин.
Промежуточное карбоновокислотное соединение-36
2-(5-циано-1H-бензо[d]имидазол-2-ил)циклопропанкарбоновая кислота
Стадия 1: этил 2-(5-циано-1H-бензо[d]имидазол-2-ил)циклопропанкарбоксилат
К смеси 3,4-диаминобензонитрила (326 мг, 2,45 ммоль), транс-2-(этоксикарбонил)циклопропанкарбоновой кислоты (323 мг, 2,04 ммоль) и триэтиламина (1,44 мл, 10,2 ммоль) в ДМФА (10 мл) добавляли HBTU (1,01 г, 2,66 ммоль). После перемешивания при комнатной температуре в течение 3 часов смесь выливали в воду и водную фазу экстрагировали при помощи EtOAc два раза. Объединенный органический слой сушили над сульфатом натрия и концентрировали в вакууме. К остатку добавляли уксусную кислоту (10 мл) и смесь перемешивали при 90ºC в течение 12 часов. После охлаждения до комнатной температуры растворитель удаляли в вакууме. Остаток выливали в насыщенный водный раствор бикарбоната натрия и водный слой экстрагировали при помощи EtOAc два раза. Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью гексан/этилацетат, с получением 210 мг (40%) указанного в заголовке соединения в виде белого аморфного вещества:
1H-ЯМР (300 МГц, CDCl3): δ 9,65 (1H, м), 7,73-7,68 (1H, м), 7,53-7,43 (2H, м), 4,20 (2H, кв, J=7,3 Гц), 2,66-2,57 (1H, м), 2,51-2,41 (1H, м), 1,88-1,80 (1H, м), 1,80-1,70 (1H, м), 1,30 (3H, т, J=7,3 Гц). LCMS (способ A) m/z M+1 obs 256,2, tR=2,61 мин.
Стадия 2: 2-(5-циано-1H-бензо[d]имидазол-2-ил)циклопропанкарбоновая кислота
Получали, как на стадии 2 получения промежуточного карбоновокислотного соединения-6, из этил 2-(5-циано-1H-бензо[d]имидазол-2-ил)циклопропанкарбоксилата.
1H-ЯМР (300 МГц, CDCl3) δ 8,01 (1H, с), 7,63 (1H, д, J=8,1 Гц), 7,53 (1H, д, J=8,1 Гц), 2,63-2,55 (1H, м), 2,20-2,12 (1H, м), 1,65-1,51 (2H, м) (сигнал не обнаружен из-за NH и COOH). LCMS (способ A) m/z M+1 obs 228,2, tR=1,88 мин.
Промежуточное карбоновокислотное соединение-37
4-((1H-имидазол-1-ил)метил)-1H-индол-2-карбоновая кислота
Стадия 1: метил 3-(2-метил-6-нитрофенил)пропеноат
2-Бром-3-нитротолуол (0,5 г, 23 ммоль), метилакрилат (0,39 г, 46 ммоль), ацетат палладия (29 мг, 1,3 ммоль), трифенилфосфин (0,06 г, 0,23 ммоль) и TEA (0,4 мл) объединяли в плотно закрытой пробирке и нагревали до 95ºC в течение 24 часов. Остаток растворяли в MeOH, растворитель удаляли и неочищенный продукт очищали при помощи колоночной хроматографии (EtOAc:гексан=7,5:92,5) с получением 0,024 г (48% выход) указанного в заголовке соединения в виде желтого масла:
1H-ЯМР (300 МГц, CDCl3) δ 7,86 (1H, д, J=16,4 Гц), 7,75 (1H, д, J=8,0 Гц), 7,46 (1H, д, J=7,6 Гц), 7,35 (1H, кв, J=8,0 Гц, 7,6 Гц), 3,79 (3H, с), 2,37 (3H, с).
Стадия 2: метил 4-метилиндол-2-карбоксилат
Метил 3-(2-метил-6-нитрофенил)пропеноат (0,24 г, 1,1 ммоль) растворяли в триэтилфосфит (1 мл) и нагревали при кипячении с обратным холодильником в течение 20 часов. Растворитель удаляли в вакууме и неочищенный продукт очищали при помощи хроматографии на силикагеле (смесь EtOAc:гексан=8:92) с получением 0,15 г (71% выход) указанного в заголовке соединения в виде бледно-желтого твердого вещества:
1H-ЯМР (300 МГц, CDCl3) δ 8,82 (1H, шир.с), 7,25-7,18 (3H, м), 6,92 (1H, д, J=6,0 Гц), 3,93 (3H, с), 2,54 (3H, с)
Стадия 3: метил 1-трет-бутоксикарбонил-4-метилиндол-2-карбоксилат
Ди-трет-бутилдикарбонат (0,35g, 1,6 ммоль) и DMAP (0,015 г, 0,12 ммоль) добавляли к раствору метил 4-метилиндол-2-карбоксилата (0,15 г, 0,8 ммоль) в ацетонитриле (7,5 мл). Полученную смесь перемешивали при комнатной температуре в течение 16 часов и растворитель выпаривали в вакууме. Остаток распределяли между этилацетатом (7,5 мл) и водой (7,5 мл). Водный слой затем экстрагировали при помощи этилацетата (2x7,5 мл) и органические экстракты объединяли, промывали насыщенным раствором бикарбоната натрия, сушили над сульфатом натрия, выпаривали в вакууме и очищали при помощи колоночной хроматографии на силикагеле (смесь EtOAc:гексан=5:95) с получением 0,13 г (59% выход) указанного в заголовке соединения в виде светло-желтого масла:
1H-ЯМР (300 МГц, CDCl3) δ 7,88 (1H, д, J=8,4 Гц), 7,31-7,03 (3H, м), 3,90 (3H, с), 2,50 (3H, с), 1,60 (9H, с).
Стадия 4: метил 4-бромметил-1-трет-бутоксикарбонилиндол-2-карбоксилат
Раствор метил 1-трет-бутоксикарбонил-4-метилиндол-2-карбоксилата (0,13 г, 0,48 ммоль), NBS (0,087 г, 0,48 ммоль) и AIBN (4 мг, 0,024 ммоль) в четыреххлористом углероде (1,9 мл) нагревали до температуры кипения с обратным холодильником в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры, фильтровали и промывали раствором четыреххлористого углерода. Фильтрат выпаривали с получением желтого масла, которое очищали при помощи хроматографии на силикагеле (смесь EtOAc:гексан=8:92) с получением 0,12 г (72% выход) указанного в заголовке соединения в виде бледно-желтого твердого вещества:
1H-ЯМР (300 МГц, CDCl3) δ 8,06 (1H, д, J=8,4Гц), 7,38-7,27 (3H, м), 4,73 (2H, с), 3,94 (3H, с).
Стадия 5: 1-трет-бутиловый эфир, 2-метиловый эфир 4-имидазол-1-илметил-индол-1,2-дикарбоновой кислоты
Раствор метил 4-бромметил-1-трет-бутоксикарбонилиндол-2-карбоксилата (0,92 г, 2,4 ммоль) и имидазола (0,82 г, 12 ммоль) перемешивали при 90ºC в ацетонитриле (14 мл) в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры и упаривали досуха. Остаток очищали при помощи колоночной хроматографии на силикагеле (смесь MeOH:ДХМ=8:92) с получением 0,413 г (46% выход) указанного в заголовке соединения в виде белого твердого вещества:
1H-ЯМР (300 МГц, CDCl3) δ 8,10 (1H, д, J=8,4 Гц), 7,59 (1H, с), 7,39 (1H, кв, J=7,6 Гц, 8,4 Гц), 7,08 (1H, с), 7,04 (1H, д, J=7,6 Гц), 6,94 (1H, с), 6,88 (1H, с), 5,33 (2H, с), 3,91 (3H, с), 1,62 (9H, с).
Стадия 6: 4-((1H-имидазол-1-ил)метил)-1H-индол-2-карбоновая кислота
Смесь 1-трет-бутилового эфира, 2-метилового эфира 4-имидазол-1-илметил-индол-1,2-дикарбоновой кислоты (350 мг, 0,99 ммоль) и 2 н. водного раствора гидроксида натрия (1 мл, 2 ммоль) в ТГФ (5 мл) кипятили с обратным холодильником при 80ºC при перемешивании в течение 2 дней. Добавляли 2 н. раствор хлористоводородной кислоты до достижения уровня pH 7,0. Смесь концентрировали в вакууме. Полученный осадок собирали путем фильтрования и промывали дихлорметаном, метанолом, H2O и этилацетатом с получением 63 мг (27% выход) указанного в заголовке соединения в виде белого твердого вещества:
1H-ЯМР (300 МГц, ДМСО-d6) δ 7,82 (1H, с), 6,72 (1H, д, J=8,8 Гц), 6,65 (1H, с), 6,55 (1H, с), 6,50 (1H, т, J=7,3 Гц), 6,36 (1H, с), 6,29 (1H, д, J=6,6 Гц), 4,86 (2H, с).
Пример 1
(R)-5-трет-бутил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)изоксазол-3-карбоксамид
К суспензии (R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этанамина в форме соли 2HCl (18 мг, 0,06 ммоль) и 5-трет-бутилизоксазол-3-карбоновой кислоты (10 мг, 0,06 ммоль) в дихлорметане (2 мл) добавляли триэтиламин (19 мг, 0,18 ммоль), EDC (19 мг, 0,1 ммоль) и HOBT (9,4 мг, 0,06 ммоль), соответственно. Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Растворитель выпаривали при помощи N2-потока. Полученный остаток растворяли в этилацетате и к смеси добавляли воду. Органический слой затем промывали насыщенным солевым раствором и сушили над сульфатом натрия. После фильтрования для отделения растворителя и сульфата натрия, растворитель удаляли при пониженном давлении с получением остатка. Остаток разбавляли метанолом и наносили на сильный катионообменный картридж (BondElute (зарегистрированная торговая марка) SCX, 1 г/6 мл, Varian Inc.) и твердофазную матрицу промывали метанолом (6 мл). Неочищенную смесь элюировали в пробирку для сбора фильтрата при помощи 1 моль/л раствора аммиака в метаноле (6 мл) и концентрировали в вакууме. Остаток очищали при помощи препаративной LC-MS с получением 17 мг, (75% выход) указанного в заголовке соединения.
При помощи способа, аналогичного описанному в примере 1, за исключением того, что реагирующее вещество было другим, были также получены следующие соединения примеров 2-27, 30-80, 82-241, 243-254, 258-291, 307-313, 315-423 и 426-464 (также смотрите таблицу 1). Для реакций использовали коммерчески доступные вещества, в иных случаях указанные в частях, относящихся к получению промежуточных соединений.
Пример 28
(R)-2-(4-бромфенокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)ацетамид
К суспензии (R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этанамина в форме соли 2HCl (173 мг, 0,79 ммоль) и 2-(4-бромфенокси)уксусной кислоты (200 мг, 0,87 ммоль) в дихлорметане (5 мл) добавляли триэтиламин (400 мг, 3,9 ммоль), EDC (180 мг, 0,94 ммоль) и HOBT (60 мг, 0,39 ммоль), соответственно. Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. К смеси добавляли насыщенный водный раствор гидрохлорида аммиака. Органический слой экстрагировали этилацетатом, промывали насыщенным солевым раствором и сушили над сульфатом натрия. После фильтрования для отделения растворителя и сульфата натрия растворитель удаляли при пониженном давлении с получением остатка, который очищали при помощи колоночной хроматографии на силикагеле и элюировали смесью гексан/этилацетат 2/1 (об/об) с получением 276 мг (81% выход) указанного в заголовке соединения в виде бесцветного масла;
1H-ЯМР (300 МГц, CDCl3) δ 8,30 (1H, м), 7,66 (1H, шир.д, J=8,1 Гц), 7,41 (2H, д, J=8,8 Гц), 7,26-7,24 (2H, м), 6,84 (2H, д, J=8,8 Гц), 5,22 (1H, м), 4,48 (2H, квАВ, J=14,6 Гц), 4,40 (2H, квАВ, J=8,1 Гц), 1,49 (3H, д, J=7,3 Гц). LCMS (способ A) m/z M+1 434,8; tR=3,15 мин.
Пример 29
(R)-2-(4-циклопропилфенокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)ацетамид
К раствору (R)-2-(4-бромфенокси)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)ацетамида (100 мг, 0,23 ммоль) и циклопропилбороновой кислоты (26 мг, 0,30 ммоль) в диоксане (2 мл) при комнатной температуре добавляли 1,27 M раствор фосфата калия (0,36 мл) и тетракистрифенилфосфинпалладий (13 мг, 0,012 ммоль). Смесь перемешивали при 120ºC, используя микроволновую печь, в течение 2 часов. Смесь сушили над сульфатом магния. После фильтрования для отделения растворителя и сульфата натрия растворитель удаляли при пониженном давлении с получением остатка. Остаток разбавляли метанолом и наносили на сильный катионообменный картридж (BondElute (зарегистрированная торговая марка) SCX, 1 г/6 мл, Varian Inc.) и твердофазную матрицу промывали метанолом (6 мл). Неочищенную смесь элюировали в пробирку для сбора фильтрата при помощи 1 моль/л раствора аммиака в метаноле (6 мл) и концентрировали в вакууме. Остаток очищали при помощи препаративной LC-MS с получением 6,5 мг, (7% выход) указанного в заголовке соединения.
Пример 81
(R)-N-(1-(5-(пиридин-2-илметокси)пиридин-2-ил)этил)-2-(4-(трифторметил)фенокси)ацетамид
Стадия 1: (R)-N-(1-(5-гидроксипиридин-2-ил)этил)-2-(4-(трифторметил)фенокси)ацетамид
Смесь (R)-N-(1-(5-(бензилокси)пиридин-2-ил)этил)-2-(4-(трифторметил)фенокси)ацетамида (пример 63, 550 мг, 1,3 ммоль) и гидроксида палладия на углероде (20 масс.%, 50 мг) в метаноле (30 мл) гидрировали в течение 6 часов. Смесь фильтровали через слой целита, промывали метанолом, фильтрат концентрировали с получением 410 мг (94% выход) (R)-N-(1-(5-гидроксипиридин-2-ил)этил)-2-(4-(трифторметил)фенокси) ацетамида в виде белого кристаллического твердого вещества;
1H-ЯМР (300 МГц, CDCl3) δ 8,19 (1H, д, J=2,9 Гц), 7,77 (1H, д, J=7,3 Гц), 7,58 (2H, д, J=8,1 Гц), 7,14 (1H, дд, J=8,8 Гц, 2,9 Гц), 7,08-7,02 (3H, м), 5,20-5,10 (1H, м), 4,58 (1H, д, J=13,9 Гц), 4,51 (1H, д, J=13,9 Гц), 1,48 (3H, д, J=6,6 Гц). LCMS (способ A) m/z M+1 obs 341.
Стадия 2: (R)-N-(1-(5-(пиридин-2-илметокси)пиридин-2-ил)этил)-2-(4-(трифторметил)фенокси)ацетамид
Смесь (R)-N-(1-(5-гидроксипиридин-2-ил)этил)-2-(4-(трифторметил)фенокси)ацетамида (30 мг, 0,088 ммоль), 2-(бромметил)пиридингидробромида (22 мг, 0,088 ммоль) и карбоната цезия (115 мг, 0,35 ммоль) в ДМФА (3 мл) нагревали при 90ºC в течение ночи. После охлаждения смесь фильтровали через слой целита, промывали дихлорметаном, фильтрат концентрировали и остаток очищали при помощи SCX картриджа с получением 34 мг (89% выход) (R)-N-(1-(5-(пиридин-2-илметокси)пиридин-2-ил)этил)-2-(4-(трифторметил)фенокси)ацетамида в виде прозрачного бесцветного масла;
LCMS (способ A) m/z M+1 obs 432, M-1 obs 430.
Альтернативный маршрут для смеси примеров 133 и 134
транс-2-(1H-индол-3-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид
Стадия 1: (R,E)-3-(1H-индол-3-ил)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)акриламид
К суспензии (R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этанамина в форме соли 2HCl (1,1 г, 3,8 ммоль) и (E)-3-(1-(трет-бутоксикарбонил)-1H-индол-3-ил)акриловой кислоты (1,0 г, 3,5 ммоль) в дихлорметане (8 мл) добавляли триэтиламин (1,8 г, 17 ммоль), EDC (800 мг, 4,2 ммоль) и HOBT (270 мг, 1,7 ммоль), соответственно. Реакционную смесь перемешивали при комнатной температуре в течение 6 часов. К смеси добавляли насыщенный водный раствор бикарбоната натрия. Органический слой экстрагировали этилацетатом, промывали насыщенным солевым раствором и сушили над сульфатом натрия. После фильтрования для отделения растворителя и сульфата натрия, растворитель удаляли при пониженном давлении с получением остатка, который очищали при помощи колоночной хроматографии на силикагеле и элюировали смесью гексан/этилацетат 2/1 (об/об), с получением 900 мг (53% выход) указанного в заголовке соединения в виде желтого твердого вещества;
1H-ЯМР (300 МГц, CDCl3) δ 8,31 (1H, д, J=1,5 Гц), 8,18 (1H, д, J=8,0 Гц), 7,90-7,70 (3H, м), 7,40-7,20 (4H, м), 6,93 (1H, д, J=8,0 Гц), 6,59 (1H, д, J=16,1 Гц), 5,30 (1H, м), 4,39 (2H, кв, J=8,0 Гц), 1,08 (9H, с), 1,53 (3H, д, J=6,6 Гц). LCMS (способ A) m/z M+1 490,3; tR=3,44 мин.
Стадия 2: транс-2-(1H-индол-3-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид
К раствору (R,E)-3-(1H-индол-3-ил)-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)акриламида (600 мг, 1,3 ммоль) в дихлорметане (10 мл) при комнатной температуре добавляли этилцинк (4,1 мл, 4,1 ммоль, 1,0 M). После перемешивания при комнатной температуре в течение 3 минут к смеси добавляли дийодометан (1,8 г, 6,7 ммоль). Смесь кипятили с обратным холодильником при 55ºC при перемешивании в течение 18 часов. К смеси добавляли насыщенный водный раствор гидрохлорида аммиака. Органический слой экстрагировали этилацетатом, промывали насыщенным солевым раствором, сушили над сульфатом натрия. После фильтрования для отделения растворителя и сульфата натрия, растворитель удаляли при пониженном давлении с получением остатка, который очищали при помощи колоночной хроматографии на силикагеле и элюировали смесью гексан/этилацетат 1/1 (об/об) и препаративной LC-MS с получением 14 мг, (3% выход) указанного в заголовке соединения в виде белого твердого вещества (2:1 смесь диастереомеров).
1H-ЯМР (600 МГц, CDCl3) δ 8,28 (1H, с), 7,99 (1H, шир.с), 7,60 (1H, м), 7,34 (1H, м), 7,26-7,22 (2H, м), 7,18 (1H, м), 7,08 (1H, м), 6,95-6,86 (2H, м),5,21 (1H, м), 4,40 (2H, кв, J=7,9 Гц), 2,54 (1H, м), 1,68 (1H, м), 1,60 (1H, м), 1,48 (3H, д, J=6,8 Гц), 1,29 (1H, м). LCMS (способ А) m/z, M+1 404,3; tR=2,98 мин.
Пример 242
(1S*,2S*)-2-(4-гидроксифенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид
Смесь гидроксида палладия (20 масс.% нагрузка) на углероде (63 мг) и (1S*,2S*)-2-(4-(бензилокси)фенил)-N-(R-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамида (631 мг, 1,341 ммоль) в метаноле (30 мл) перемешивали в течение 4 часов при комнатной температуре в атмосфере H2. Смесь фильтровали через слой целита, промывали этилацетатом, фильтрат концентрировали с получением 485 мг (95% выход) указанного в заголовке соединения в виде белого аморфного вещества. 8 мг Остаток очищали при помощи препаративной LC-MS с получением 4,8 мг указанного в заголовке соединения.
Пример 255
(1S*,2S*)-2-(3-гидроксифенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид
Получали как в примере 242 из (1S*,2S*)-2-(3-(бензилокси)фенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамида. Остаток очищали при помощи препаративной LC-MS с получением 5,4 мг указанного в заголовке соединения.
Пример 256
(1S*,2S*)-2-(4-(2-(4,4-дифторпиперидин-1-ил)-2-оксоэтокси)фенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид
Стадия 1: трет-бутил 2-(4-((1S*,2S*)-2-(((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)карбамоил)циклопропил)фенокси)ацетат
Смесь трет-бутил 2-бромацетата (0,063 мл, 0,434 ммоль), карбоната калия (109 мг, 0,789 ммоль) и (1S*,2S*)-2-(4-гидроксифенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамида (150 мг, 0,394 ммоль) в дихлорметане (4 мл) кипятили с обратным холодильником при перемешивании в течение 3 часов. После охлаждения до комнатной температуры смесь выливали в воду и водный слой экстрагировали этилацетатом, сушили над сульфатом магния и концентрировали в вакууме. Остаток перекристаллизовывали из смеси тетрагидрофуран/гексан с получением 137 мг (70% выход) указанного в заголовке соединения в виде белого кристалла:
1H-ЯМР (300 МГц, ДМСО-d6) δ 8,55(1H, д, J=8,1 Гц), 8,31 (1H, д, J=2,6 Гц), 7,49 (1H, дд, J=8,8, 2,9 Гц), 7,28 (1H, д, J=8,4 Гц), 7,03 (2H, д, J=8,4 Гц), 6,78 (2H, д, J=8,4 Гц), 4,95 (1H, т, J=7,3 Гц), 4,84 (2H, кв, J=8,8 Гц), 4,59 (2H, с), 2,17 (1H, м), 1,89 (1H, м), 1,41 (9H, с), 1,32 (2H, д, J=6,6 Гц), 1,22 (1H, м), 1,09 (1H, м). LCMS (способ A) m/z: M+1 obs 495,1, tR=3,25 мин.
Стадия 2: 2-(4-((1S*,2S*)-2-(((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)карбамоил)циклопропил) фенокси)уксусная кислота
Смесь трифторуксусной кислоты (0,213 мл, 2,77 ммоль) и трет-бутил 2-(4-((1S*,2S*)-2-(((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)карбамоил)циклопропил) фенокси)ацетата (137 мг, 0,277 ммоль) в дихлорметане (5 мл) кипятили с обратным холодильником при перемешивании в течение 6 часов. Избыток трифторуксусной кислоты и дихлорметана удаляли при пониженном давлении с получением 200 мг указанного в заголовке соединения в виде белого твердого вещества. Это соединение использовали на следующей стадии без дополнительной очистки:
LCMS (способ A) m/z: M+1 obs 439,0, tR=2,54 мин.
Стадия 3: (1S*,2S*)-2-(4-(2-(4,4-дифторпиперидин-1-ил)-2-оксоэтокси)фенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид
Смесь HBTU (28 мг, 0,075 ммоль), триэтиламина (0,03 мл, 0,25 ммоль), 4,4-дифторпиперидингидрохлорида (9,5 мг, 0,060 ммоль) и 2-(4-((1S*,2S*)-2-(((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)карбамоил)циклопропил) фенокси)уксусной кислоты (28 мг, 0,050 ммоль) перемешивали в течение 4 часов при комнатной температуре. Смесь выливали в 2 моль/л хлористоводородной кислоты и водный слой экстрагировали этилацетатом, сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали при помощи препаративной LC-MS с получением 13,7 мг (50% выход) указанного в заголовке соединения.
Пример 257
(1S*,2S*)-2-(3-(2-(4,4-дифторпиперидин-1-ил)-2-оксоэтокси)фенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид
Стадия 1: трет-бутил 2-(3-((1S*,2S*)-2-(((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)карбамоил)циклопропил)фенокси) ацетат
Получали, как на стадии 1 примера 256, из (1S*,2S*)-2-(3-гидроксифенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамида.
1H-ЯМР (300 МГц, ДМСО-d6) δ 8,56 (1H, д, J=7,7 Гц), 8,31 (1H, д, J=2,9 Гц), 7,49 (1H, дд, J=8,4, 2,9 Гц), 7,29 (1H, д, J=8,8 Гц), 7,16 (1H, т, J=7,7 Гц), 6,72-6,65 (3H, м), 4,95 (1H, т, J=7,3 Гц), 4,84 (2H, кв, J=8,8 Гц), 4,61 (2H, с), 2,19 (1H, м), 1,98 (1H, м), 1,41 (9H, с), 1,32 (2H, д, J=7,0 Гц), 1,25 (1H, м), 1,15 (1H, м). LCMS (способ A) m/z: M+1 obs 495,1, tR=3,28 мин.
Стадия 2: 2-(3-((1S*,2S*)-2-(((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)карбамоил)циклопропил)фенокси) уксусная кислота
Получали, как на стадии 2 примера 256, из трет-бутил 2-(3-((1S*,2S*)-2-(((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)карбамоил)циклопропил)фенокси)ацетата.
LCMS (способ A) m/z: M+1 obs 439,0, tR=2,61 мин.
Стадия 3: (1S*,2S*)-2-(3-(2-(4,4-дифторпиперидин-1-ил)-2-оксоэтокси)фенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид
Получали, как на стадии 3 примера 256, из 2-(3-((1S*,2S*)-2-(((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)карбамоил) циклопропил)фенокси)уксусной кислоты. Остаток очищали при помощи препаративной LC-MS с получением 11,4 мг указанного в заголовке соединения.
Пример 292
(R)-4-(трет-бутил)-N-(1-(5-гидроксипиридин-2-ил)этил)бензамид
Получали, как в примере 242, из (R)-N-(1-(5-(бензилокси)пиридин-2-ил)этил)-4-(трет-бутил)бензамида (пример 313).
1H-ЯМР (300 МГц, ДМСО-d6) δ 9,74 (1H, с), 8,60 (1H, д, J=8,1 Гц), 8,04 (1H, д, J=2,9 Гц), 7,81 (2H, д, J=8,4 Гц), 7,45 (2H, д, J=8,1 Гц), 7,19 (1H, д, J=8,4 Гц), 7,10 (1H, дд, J=8,4, 2,9 Гц), 5,11 (1H, квинтет, J=7,0 Гц), 1,43 (3H, д, J=7,0 Гц), 1,28 (9H, с). LCMS (способ A) m/z: M+1 obs 299,2, tR=3,21 мин.
Пример 293
(1S*,2S*)-2-(феноксиметил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид
Стадия 1: (1R*,2R*)-2-(гидроксиметил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид и (1S*,2S*)-2-(гидроксиметил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид
К смеси дигидрохлорида (R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этанамина (997 мг, 3,40 ммоль), транс-2-(гидроксиметил)циклопропанкарбоновой кислоты (329 мг, 2,83 ммоль) и триэтиламина (1,99 мл, 14,2 ммоль) в ацетонитриле добавляли HBTU. После перемешивания при комнатной температуре в течение 5 часов, смесь выливали в воду и водный слой экстрагировали дихлорметаном три раза. Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюируя смесью дихлорметан/метанол (20:1), с получением 263 мг (29%) верхнего пятна (условно отнесенного к (1R*,2R*)-2-(гидроксиметил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамиду) в виде бесцветного масла и 292 мг нижнего пятна (условно отнесенного к (1S*,2S*)-2-(гидроксиметил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамиду) в виде кристалла.
(1R*,2R*)-2-(гидроксиметил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид)
1H-ЯМР (300 МГц, CDCl3): δ 8,26 (1H, с), 7,27-7,21 (2H, м), 6,91 (1H, д, J=7,3 Гц), 5,11 (квинтет, J=6,6 Гц), 3,66 (1H, дд, J=5,9 и 11,0 Гц), 3,41 (1H, дд, J=7,3 и 11,0 Гц), 1,70-1,60 (2H, м), 1,45 (3H, д, J=6,6 Гц), 1,27-1,19 (1H, м), 0,81 -0,73 (1H, м) (сигнал не обнаружен из-за OH). LCMS (способ A) m/z, M+1 obs 319,1, tR=2,40 мин.
(1S*,2S*)-2-(гидроксиметил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид)
1H-ЯМР (300 МГц, CDCl3): δ 8,29 (1H, с), 7,27-7,20 (2H, м), 6,89 (1H, д, J=6,6 Гц), 5,11 (1H, квинтет, J=6,6 Гц), 4,40 (2H, кв, J=8,0 Гц), 3,66 (1H, дд, J=5,9 и 11,7 Гц), 3,49 (1H, дд, J=6,6 и 11,7 Гц), 1,77-1,65 (2H, м), 1,20-1,12 (1H, м), 0,77-0,70 (1H, м) (сигнал не обнаружен из-за OH). LCMS (способ A) m/z M+1 obs 319,1, tR=2,37 мин.
Стадия 2: (1S*,2S*)-2-(феноксиметил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид
К смеси (1S*,2S*)-2-(гидроксиметил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамида (30 мг, 0,094 ммоль) и фенола (16,0 мг, 0,17 ммоль) в тетрагидрофуране (1 мл) последовательно добавляли трифенилфосфин (45 мг, 0,17 ммоль) и ди-трет-бутилазодикарбоксилат (28,2 мг, 0,12 ммоль). После перемешивания при комнатной температуре в течение 1 дня, смесь выливали в воду и водный слой экстрагировали дихлорметаном три раза. Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали при помощи препаративной LC-MS с получением 12,1 мг (33% выход) указанного в заголовке соединения.
При помощи способа, аналогичного описанному в примере 293, за исключением того, что реагирующее вещество было другим, были также получены следующие соединения примеров 294-302 (так же смотрите таблицу 3). Для реакций использовали коммерчески доступные вещества, в иных случаях указанные в частях, относящихся к получению промежуточных соединений.
Пример 303
(1S*,2S*)-2-(3-((3-метилоксетан-3-ил)метокси)фенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид
Смесь (1S*,2S*)-2-(3-гидроксифенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамида (15 мг, 0,039 ммоль), 3-(хлорметил)-3-метилоксетана (24 мг, 0,197 ммоль) и карбоната калия (27 мг, 0,197 ммоль) в ДМФА (2 мл) нагревали при 70ºC при перемешивании в течение 15 часов. Смесь выливали в воду и водный слой экстрагировали этилацетатом, сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали при помощи препаративной LC-MS с получением 8,9 мг (49 % выход) указанного в заголовке соединения.
Пример 304
(1S*,2S*)-2-(4-((3-метилоксетан-3-ил)метокси)фенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид
Получали, как в примере 303, из (1S*,2S*)-2-(4-гидроксифенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамида. Остаток очищали при помощи препаративной LC-MS с получением 8 мг указанного в заголовке соединения.
Пример 305
(1S*,2S*)-2-(4-(пиридин-2-илметокси)фенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид
Смесь (1S*,2S*)-2-(4-гидроксифенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамида (15 мг, 0,039 ммоль), 2-(бромметил)пиридингидробромида (100 мг, 0,394 ммоль) и карбоната калия (27 мг, 0,197 ммоль) в ДМФА (2 мл) нагревали при 70ºC при перемешивании в течение 2 дней. Смесь выливали в воду и водный слой экстрагировали этилацетатом, сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали при помощи препаративной LC-MS с получением 5 мг (27% выход) указанного в заголовке соединения.
Пример 306
(1S*,2S*)-2-(3-(пиридин-2-илметокси)фенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид
Получали, как в примере 305, из (1S*,2S*)-2-(3-гидроксифенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамида. Остаток очищали при помощи препаративной LC-MS с получением 12 мг указанного в заголовке соединения.
Пример 314
(R)-4-трет-бутил-N-(1-(5-(пиридин-2-илметокси)пиридин-2-ил)этил)бензамид
Получали, как в примере 81 и примере 305, из (R)-N-(1-(5-(бензилокси)пиридин-2-ил)этил)-4-трет-бутилбензамида (пример 313).
Пример 424
(R)-6-фтор-N,1-диметил-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-2-карбоксамид
К перемешиваемому раствору (R)-6-фтор-N-(1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-1H-индол-2-карбоксамида (пример 48, 18 мг, 0,046 ммоль) в ДМФА (1 мл) при комнатной температуре добавляли гидрид натрия (60%, 1,6 мг, 0,068 ммоль). Через 20 минут добавляли йодометан (0,0034 мл, 0,055 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа. Смесь выливали в воду и экстрагировали при помощи этилацетата, сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток разбавляли метанолом и наносили на сильный катионообменный картридж (BondElute (зарегистрированная торговая марка) SCX, 1 г/6 мл, Varian Inc.) и твердофазную матрицу промывали метанолом (6 мл). Неочищенную смесь элюировали в пробирку для сбора фильтрата при помощи 1 моль/л раствора аммиака в метаноле (6 мл) и концентрировали в вакууме. Остаток очищали при помощи препаративной LC-MS с получением 9,4 мг (50% выход) указанного в заголовке соединения.
Пример 425
(1S*,2S*)-N-метил-2-(хинолин-2-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)циклопропанкарбоксамид
Стадия 1: (1S*,2S*)-2-(хинолин-2-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)циклопропанкарбоксамид
Получали, как в примере 1, из 2-(хинолин-2-ил)циклопропанкарбоновой кислоты (очищали при помощи хиральной ВЭЖХ).
1H-ЯМР (300 МГц, CDCl3) δ 8,25 (1H, с), 8,07 (1H, с), 8,00 (1H, д, J=8,0 Гц), 7,90 (1H, д, J=8,0 Гц), 7,74 (1H, д, J=8,0 Гц), 7,65 (1H, т, J=8,0 Гц), 7,45 (1H, т, J=8,0 Гц), 7,31 (1H, д, J=8,0 Гц), 6,58 (1H, д, J=8,0 Гц), 5,27 (1H, квинтет, J=7,3 Гц), 4,80-4,67 (2H, м), 2,73-2,66 (1H, м), 2,35-2,27 (1H, м), 1,73-1,66 (2H, м), 1,50 (3H, д, J=7,3 Гц).
Стадия 2: (1S*,2S*)-N-метил-2-(хинолин-2-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)циклопропанкарбоксамид
Получали, как в примере 424, из (1S*,2S*)-2-(хинолин-2-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)циклопропанкарбоксамида. Остаток очищали при помощи препаративной LC-MS с получением 11 мг указанного в заголовке соединения.
Пример 426
(1R*,2R*)-N-метил-2-(хинолин-2-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)циклопропанкарбоксамид
Получали, как в примере 424, из (1R*,2R*)-2-(хинолин-2-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиразин-2-ил)этил)циклопропанкарбоксамида (пример 222). Остаток очищали при помощи препаративной LC-MS с получением 2,3 мг указанного в заголовке соединения.
Условия анализа контроля качества (способ B), используемые амин/карбоновая кислота, способ очистки и данные спектров описаны для примеров 1-464 ниже в таблице 3 и таблице 4.
Таблица 3
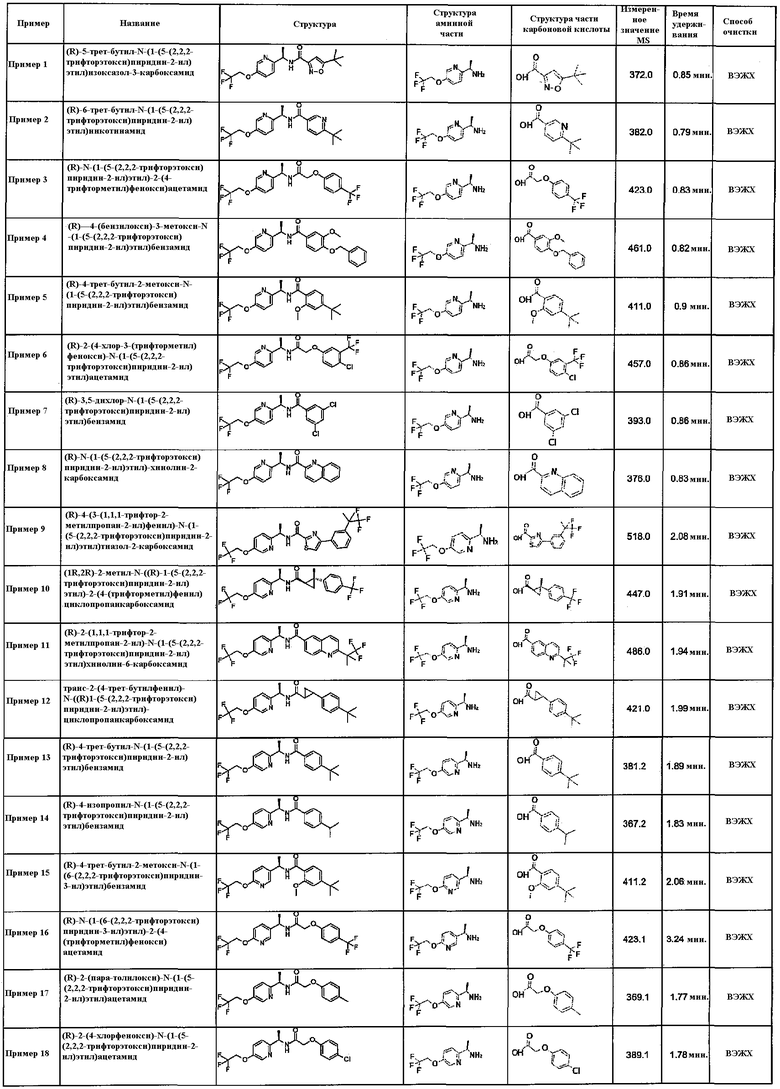
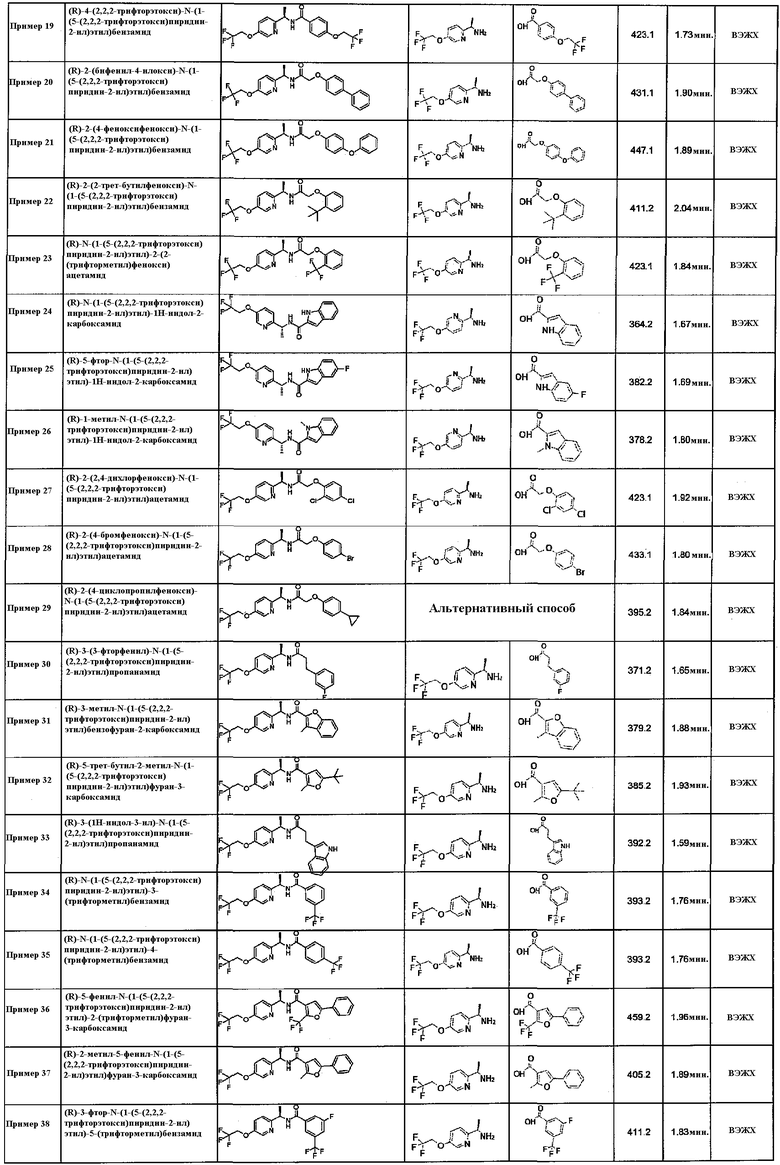
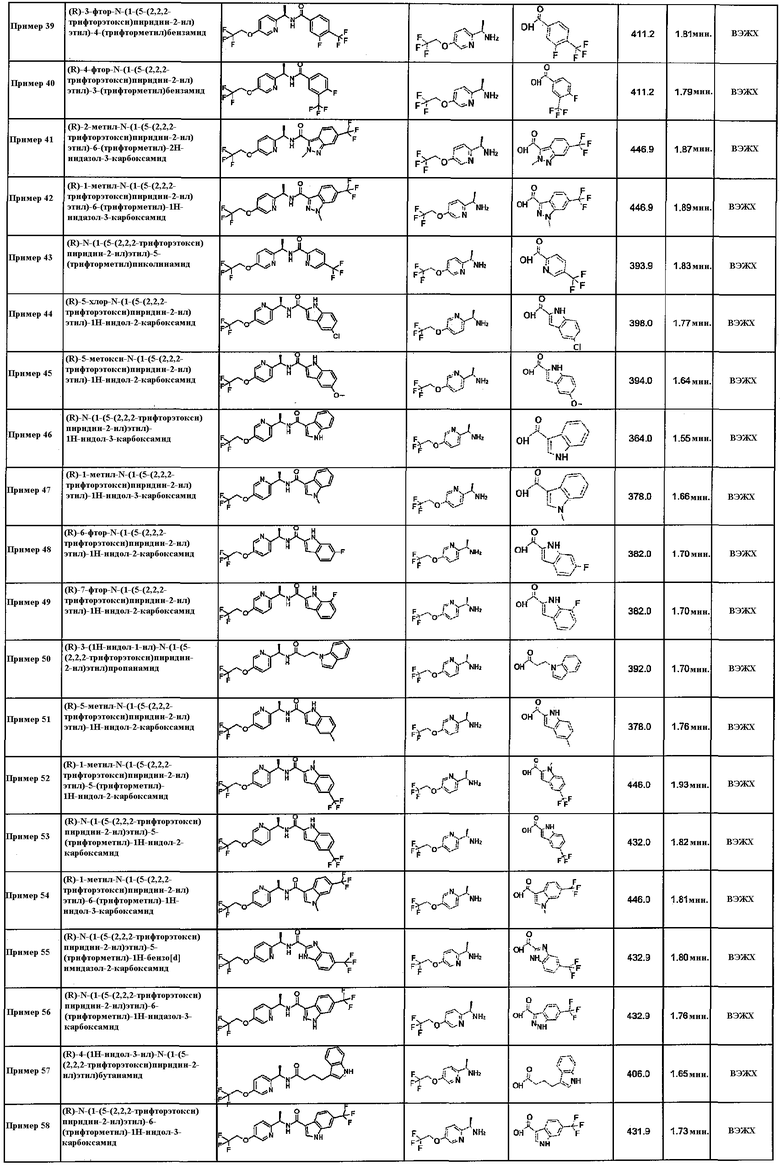
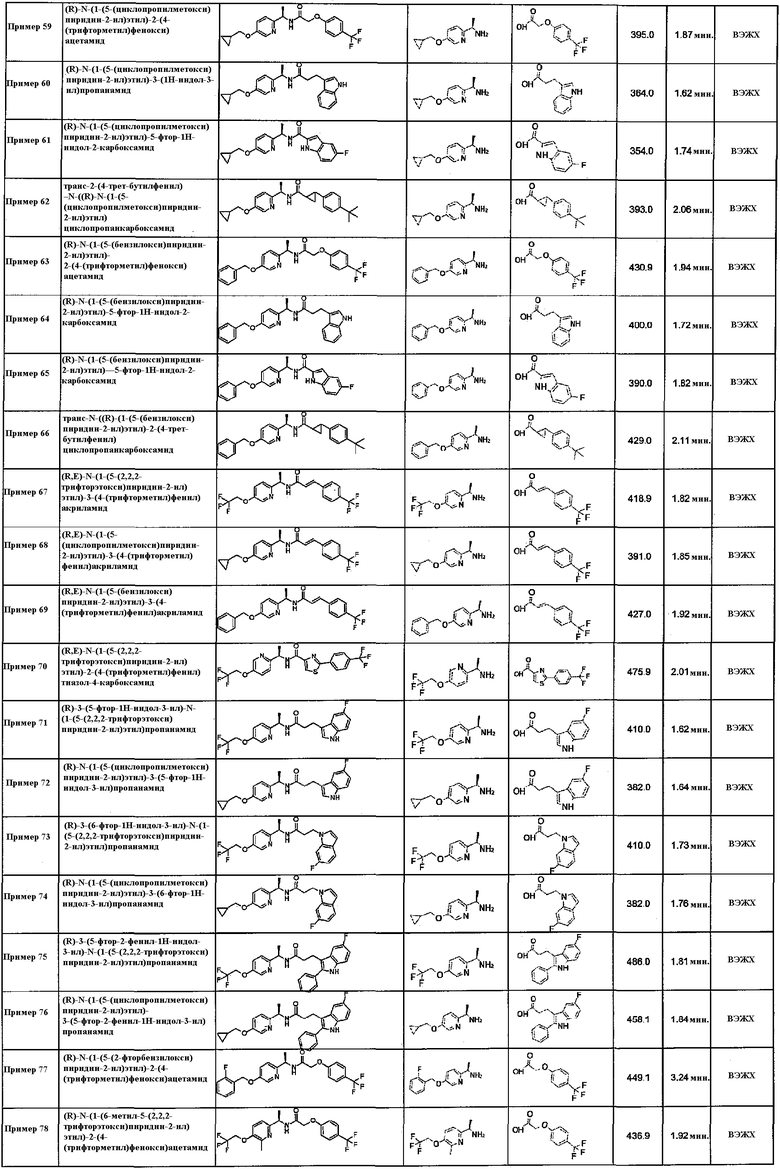
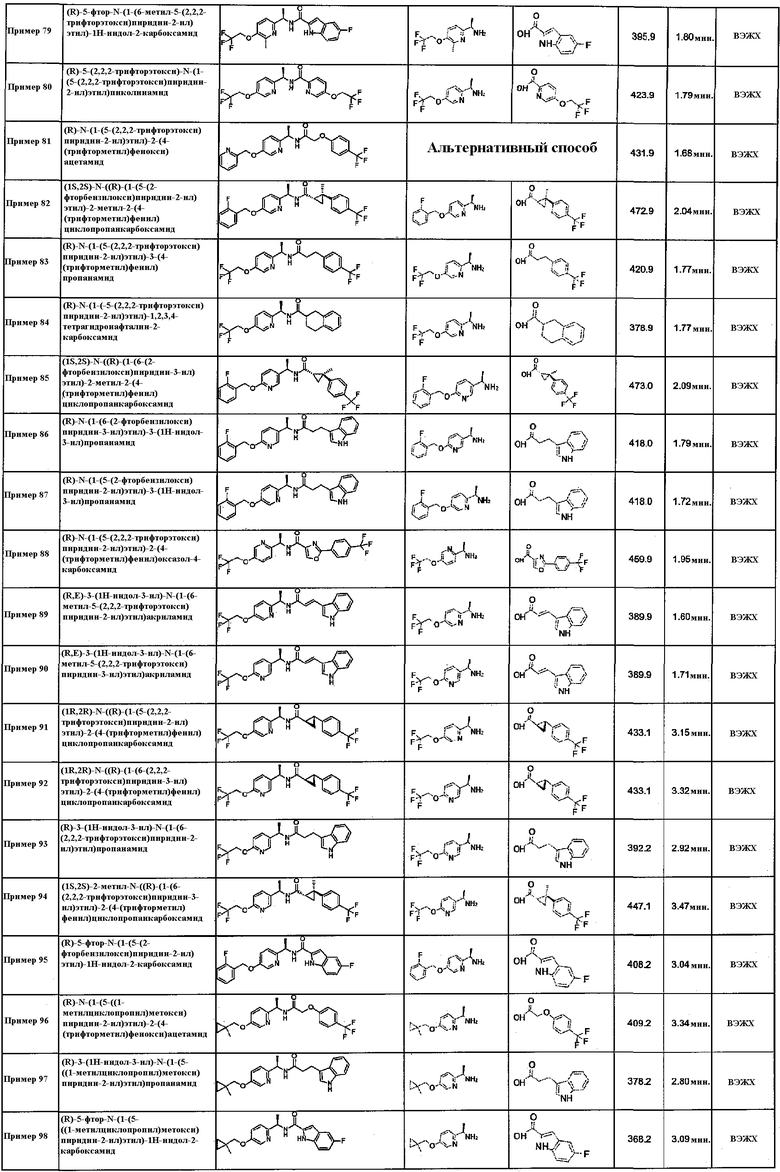
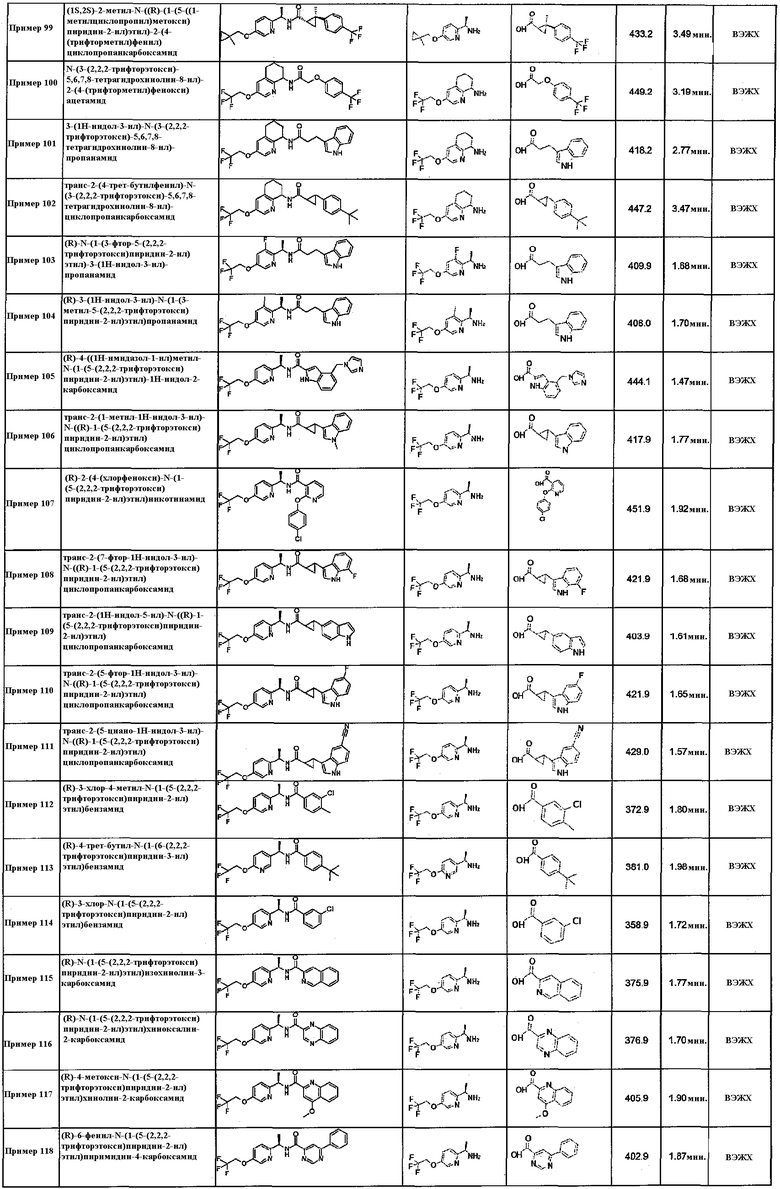
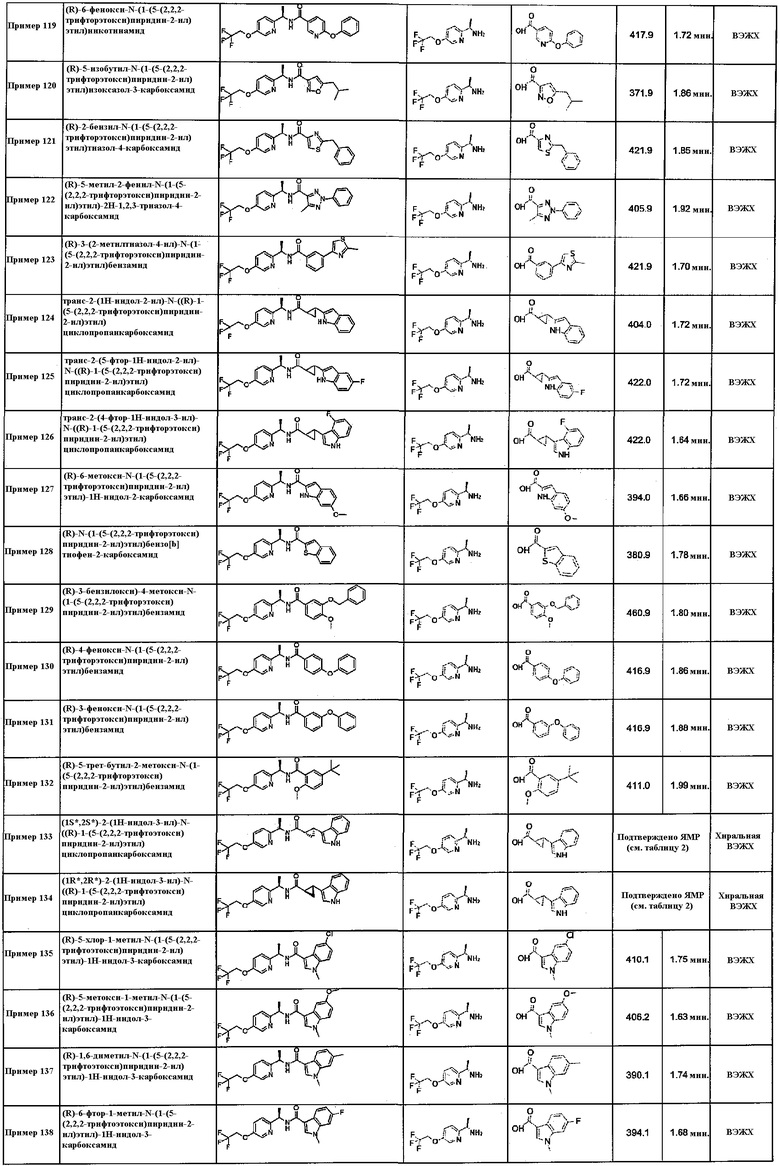
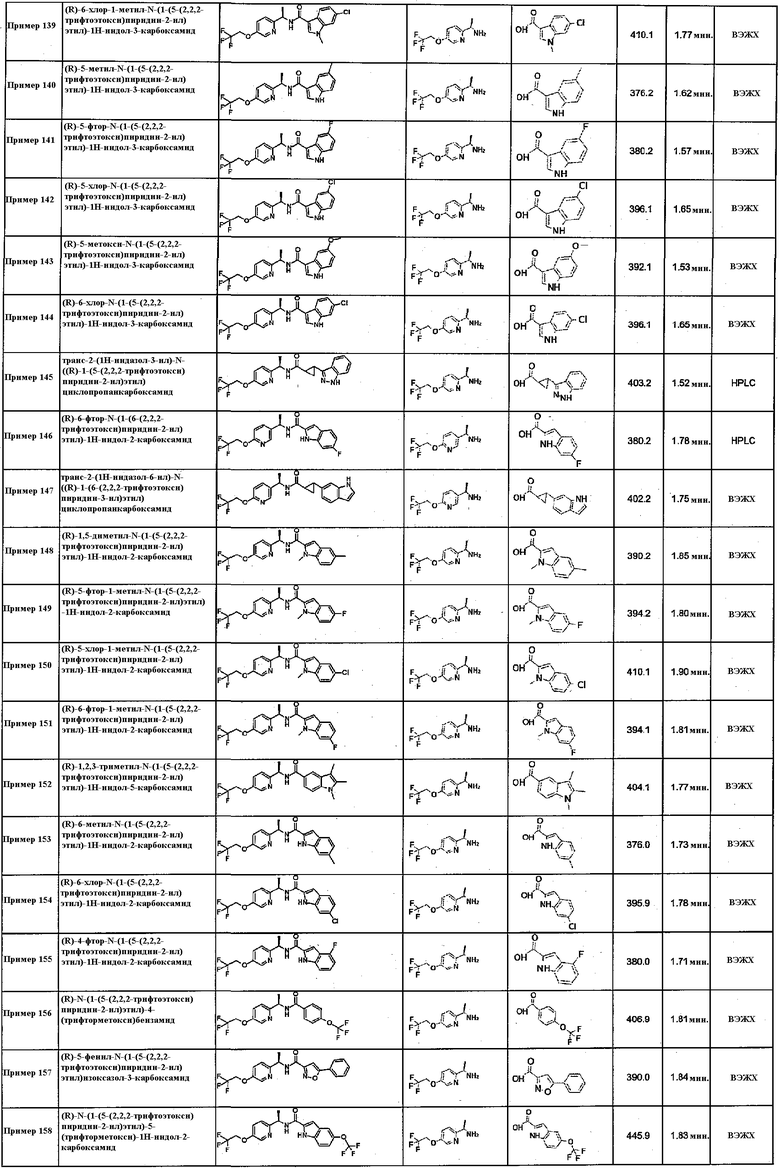
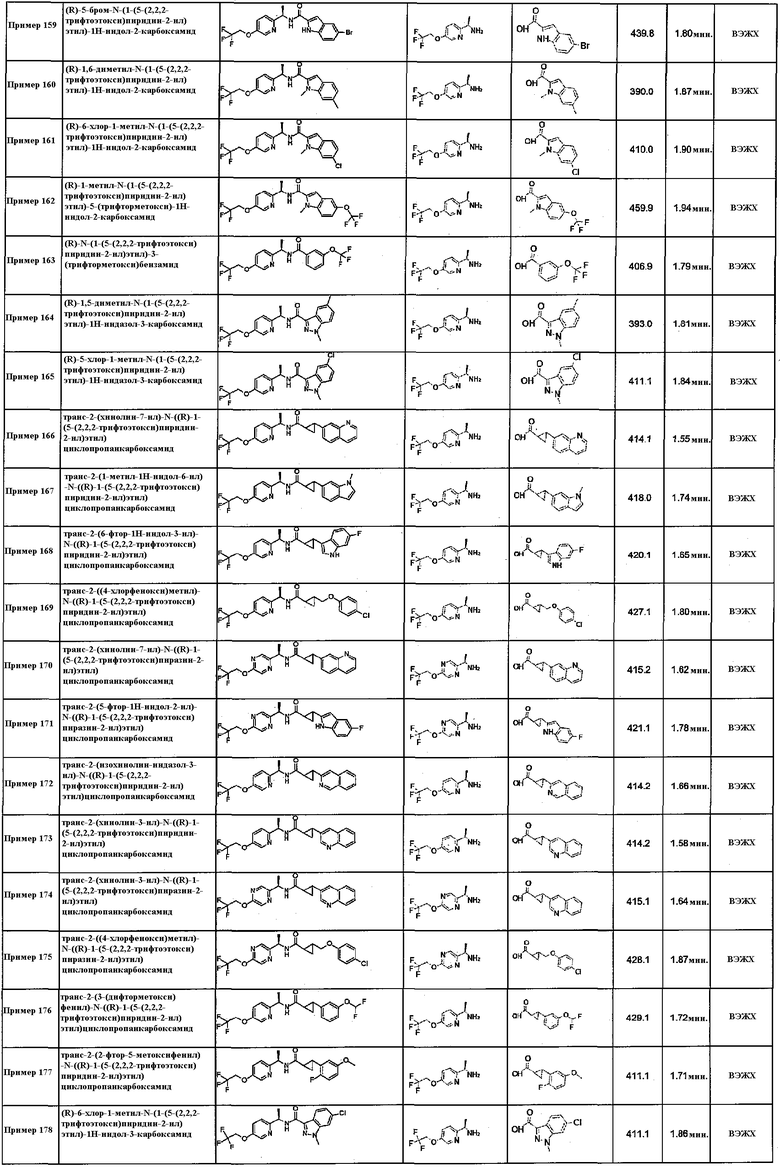
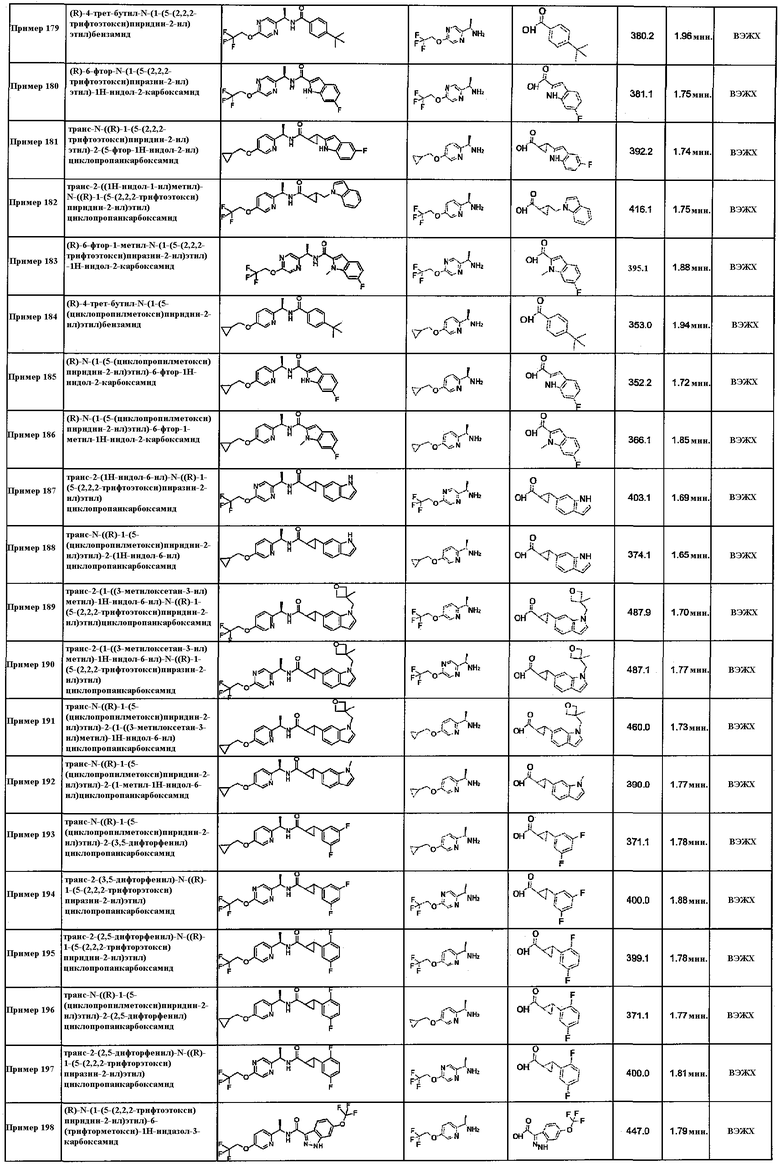
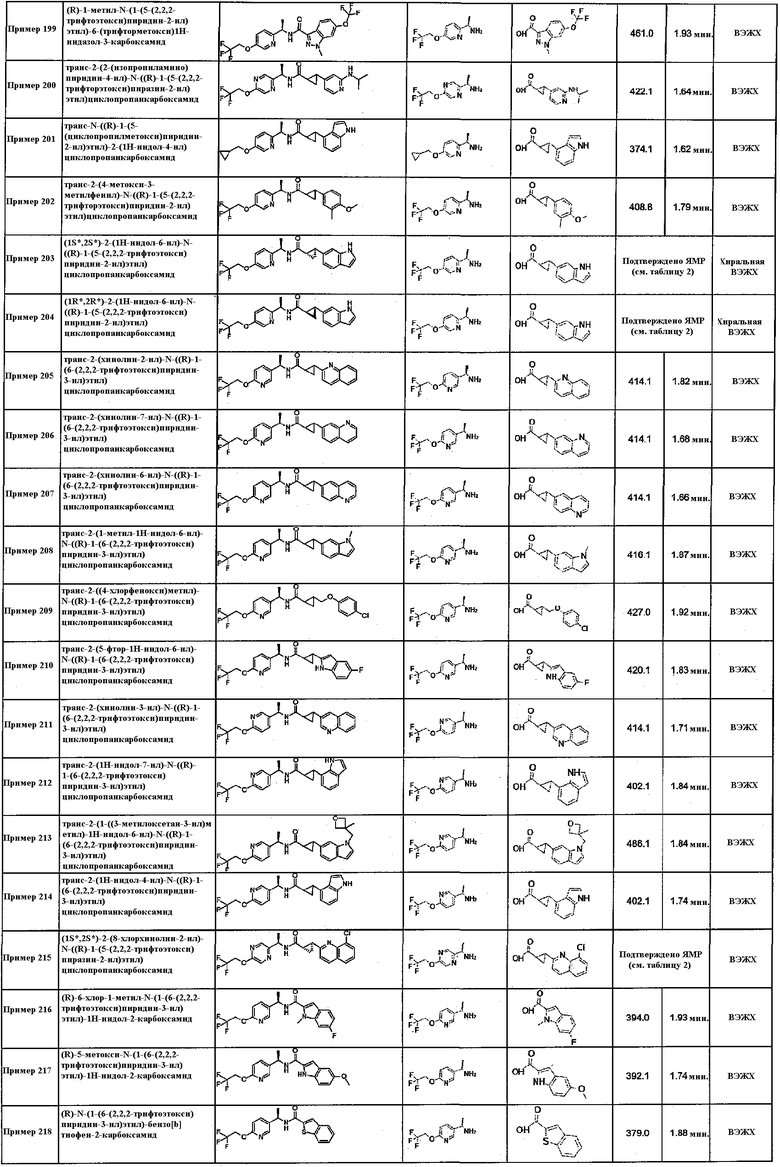
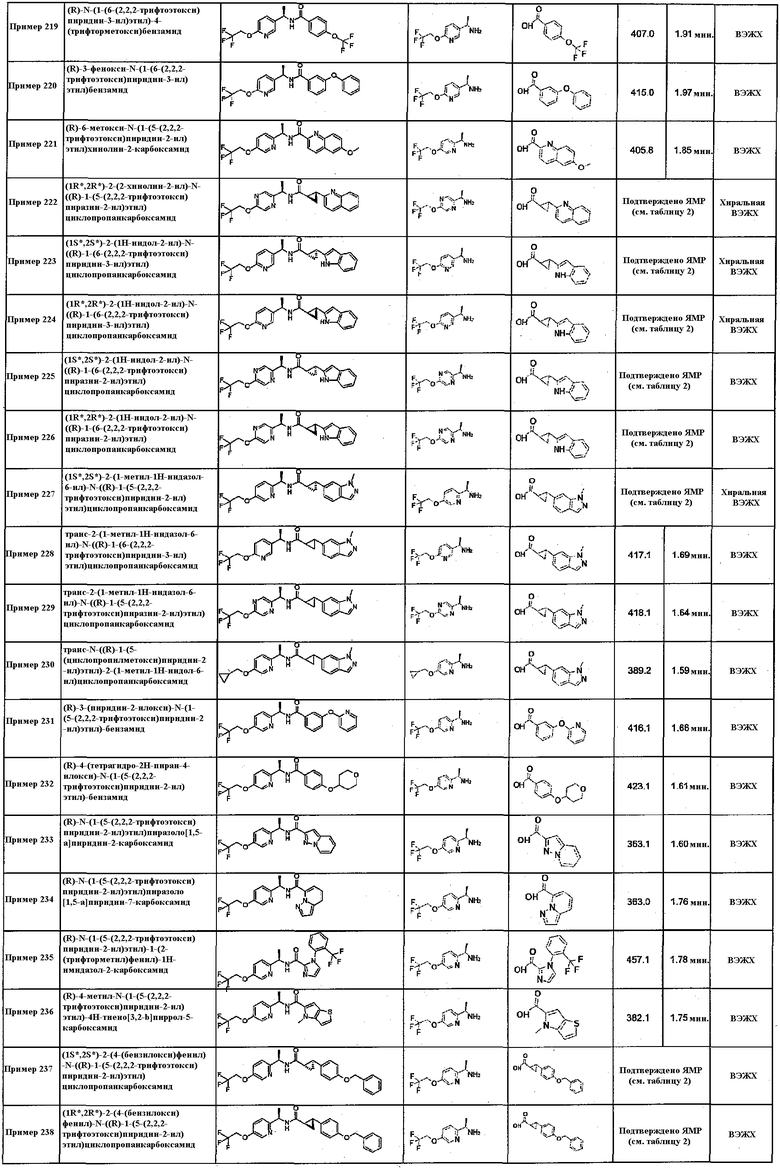
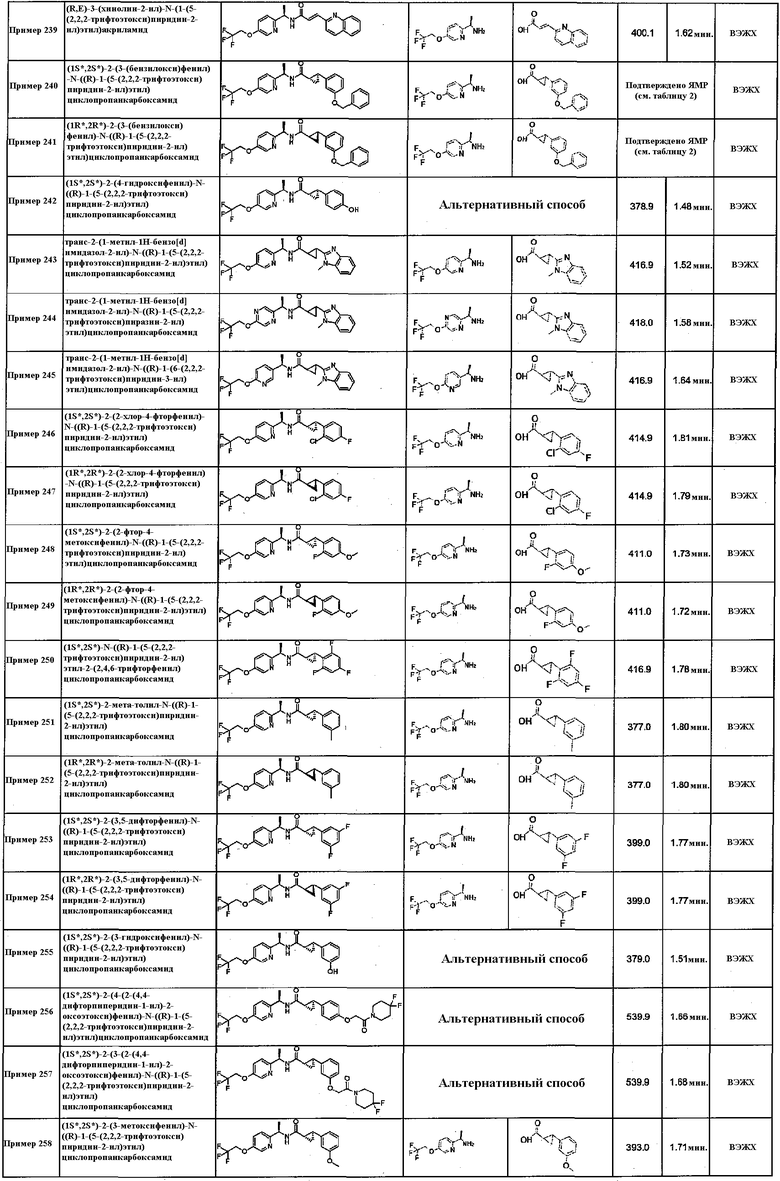
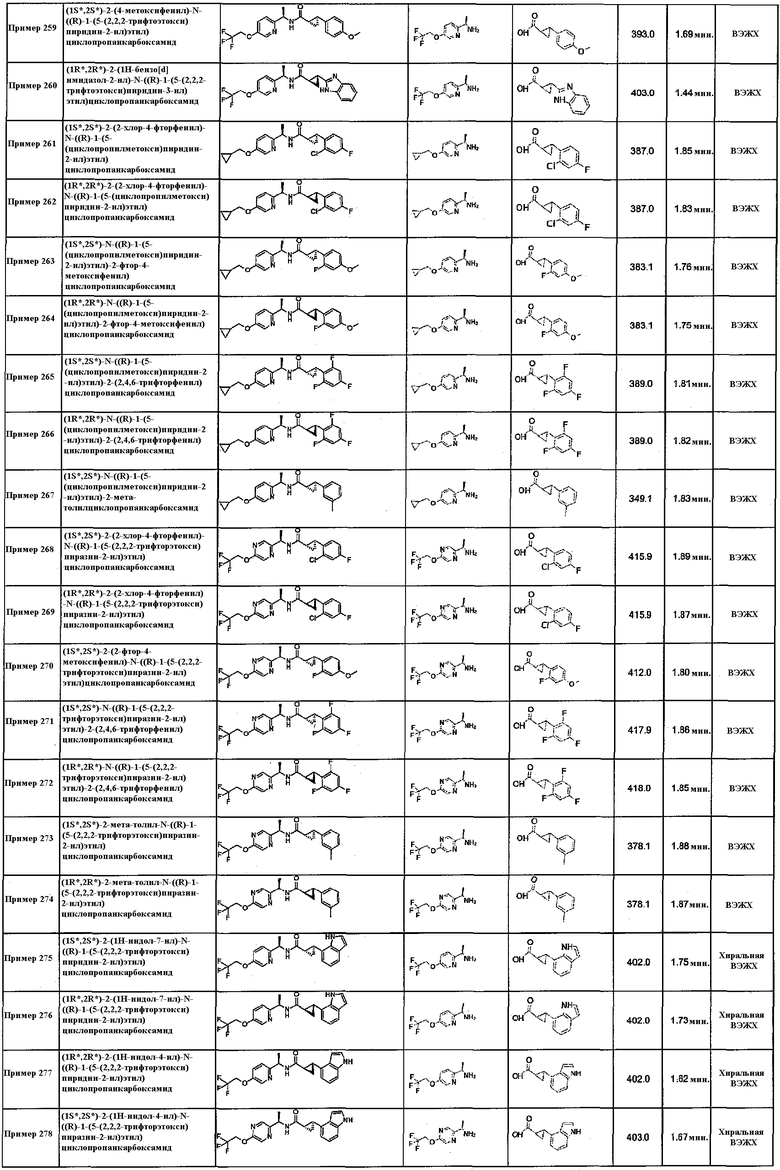
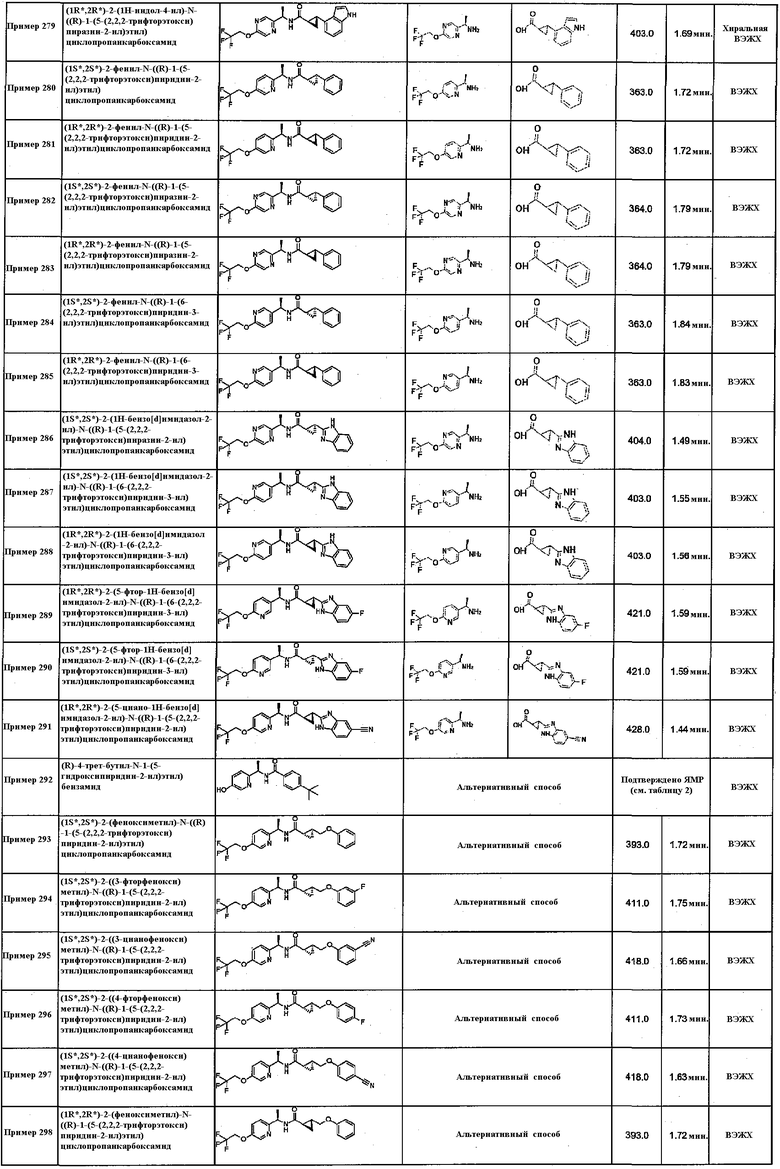

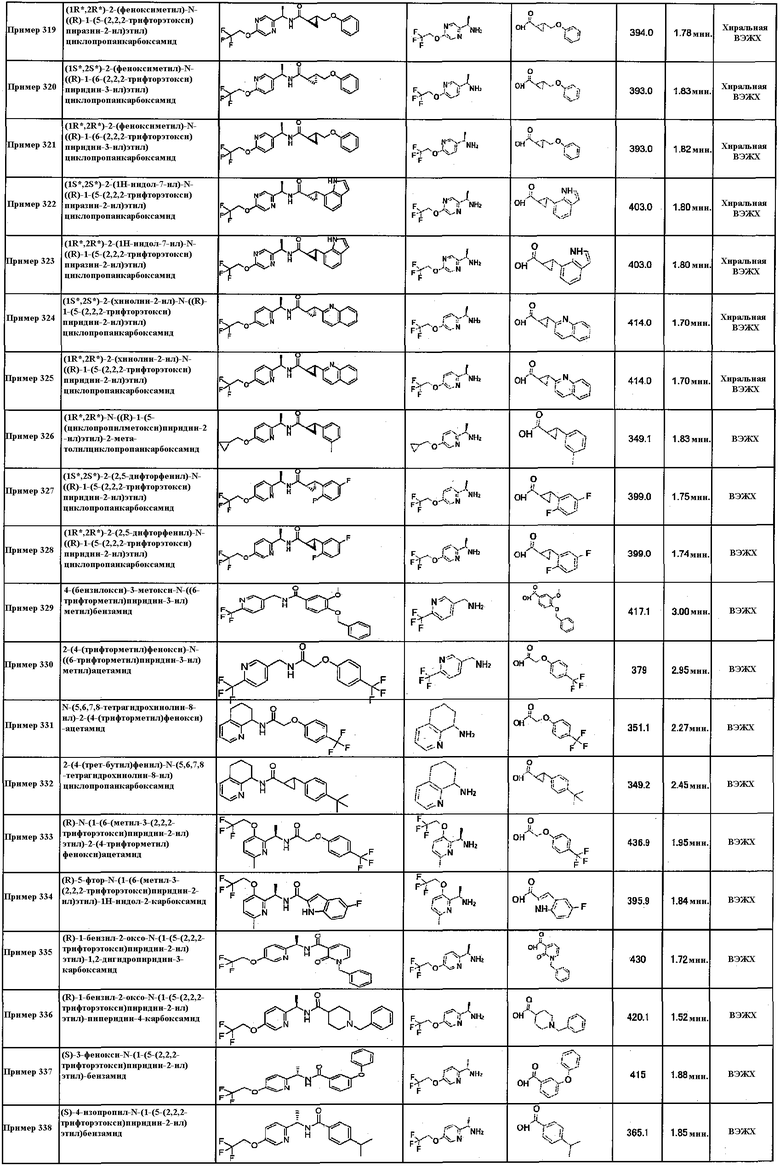
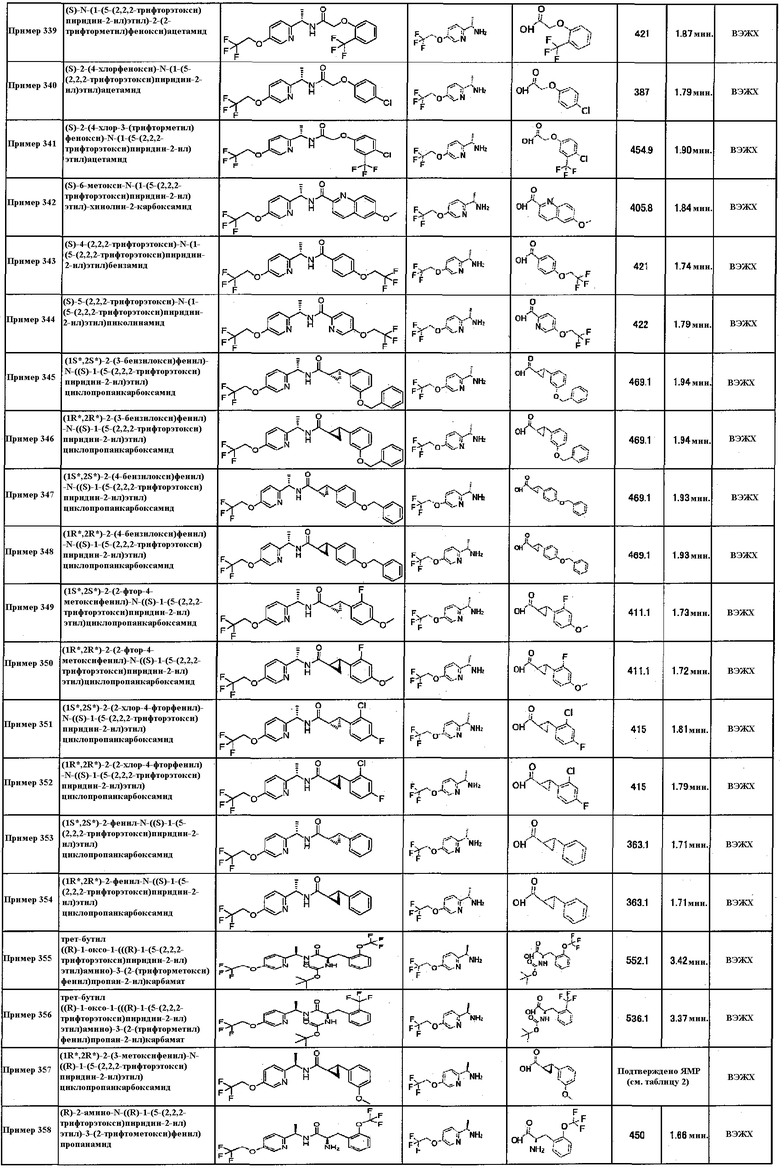
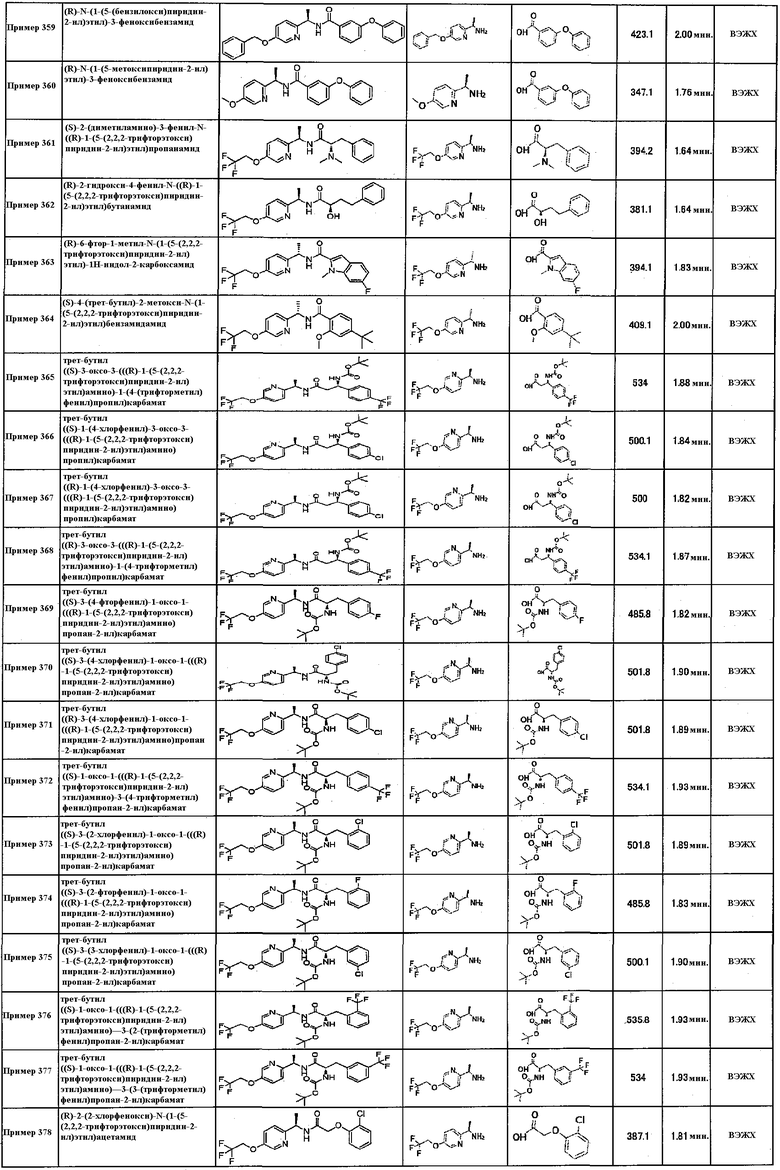
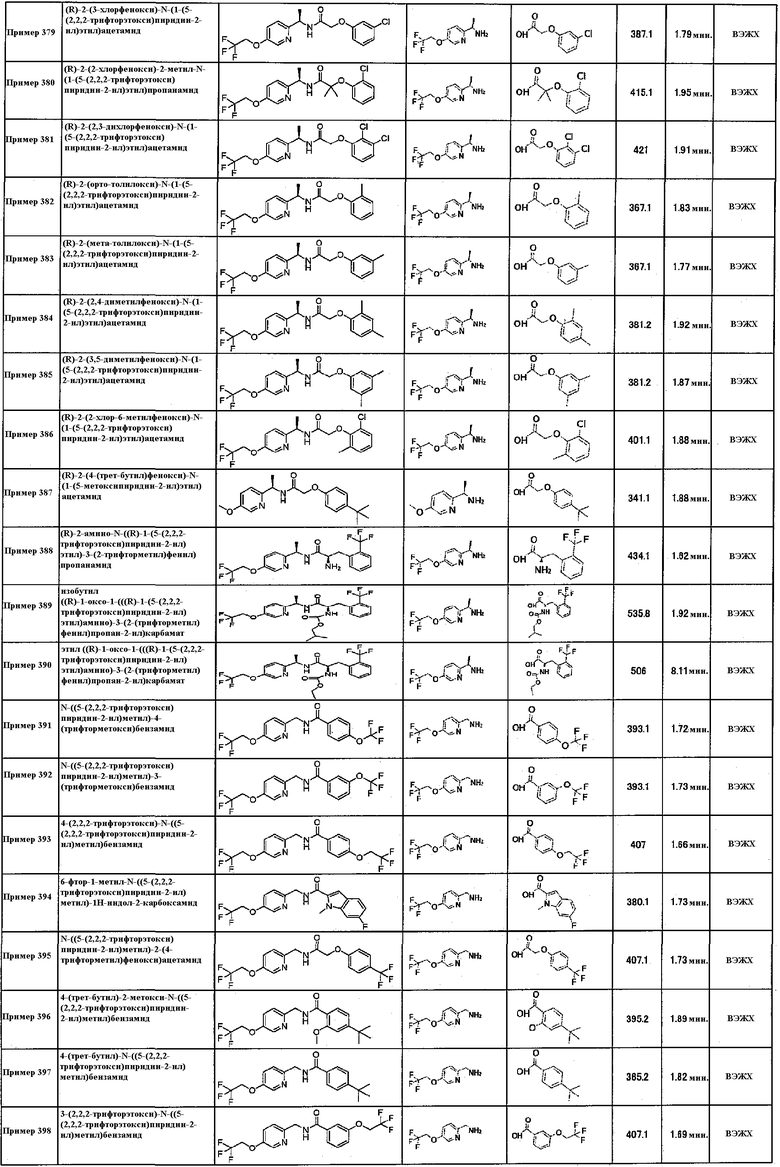
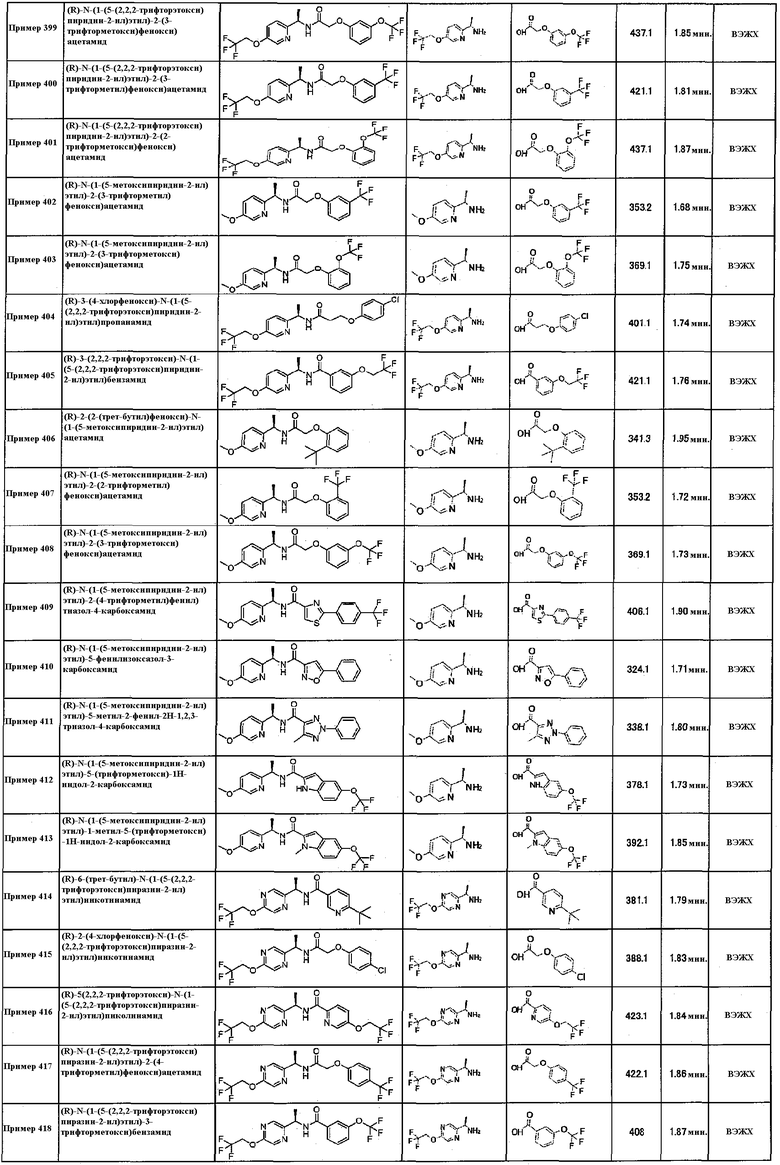
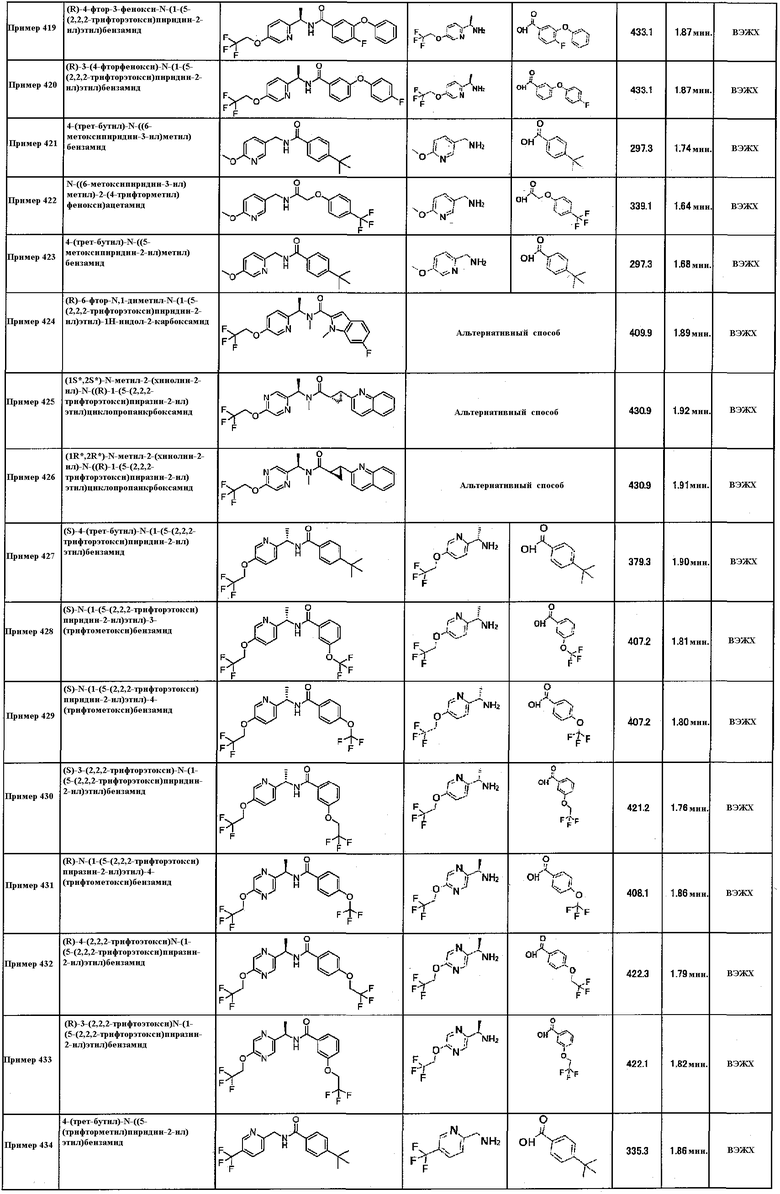
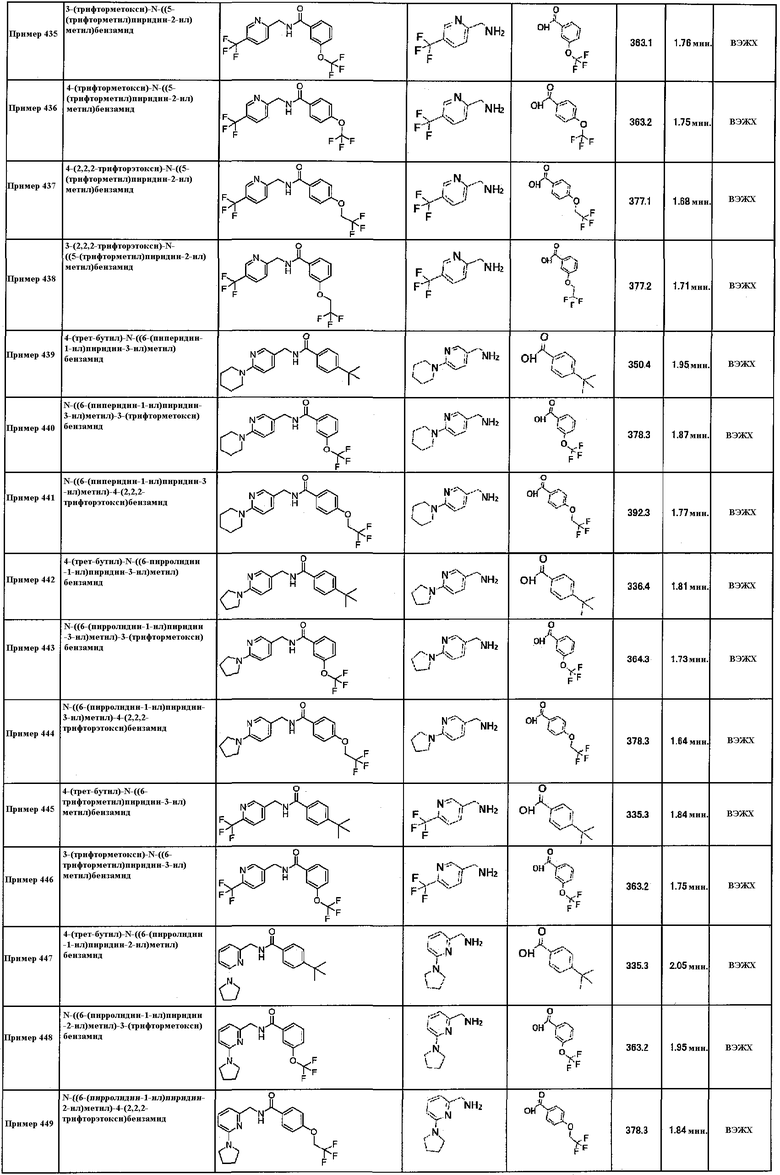
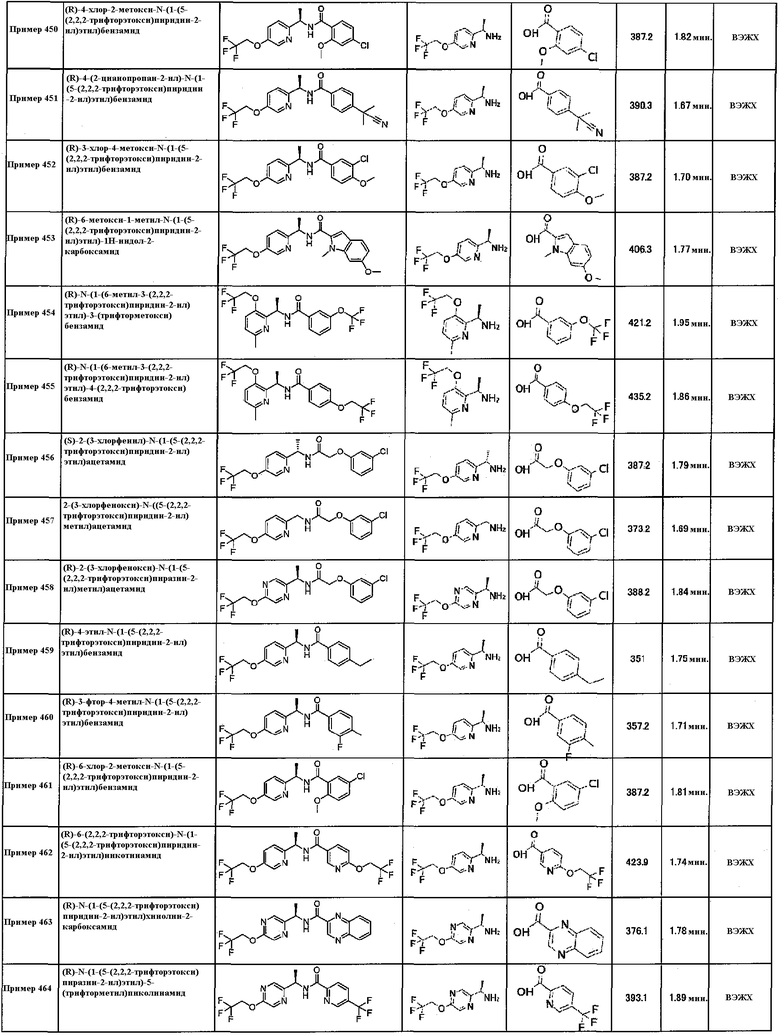
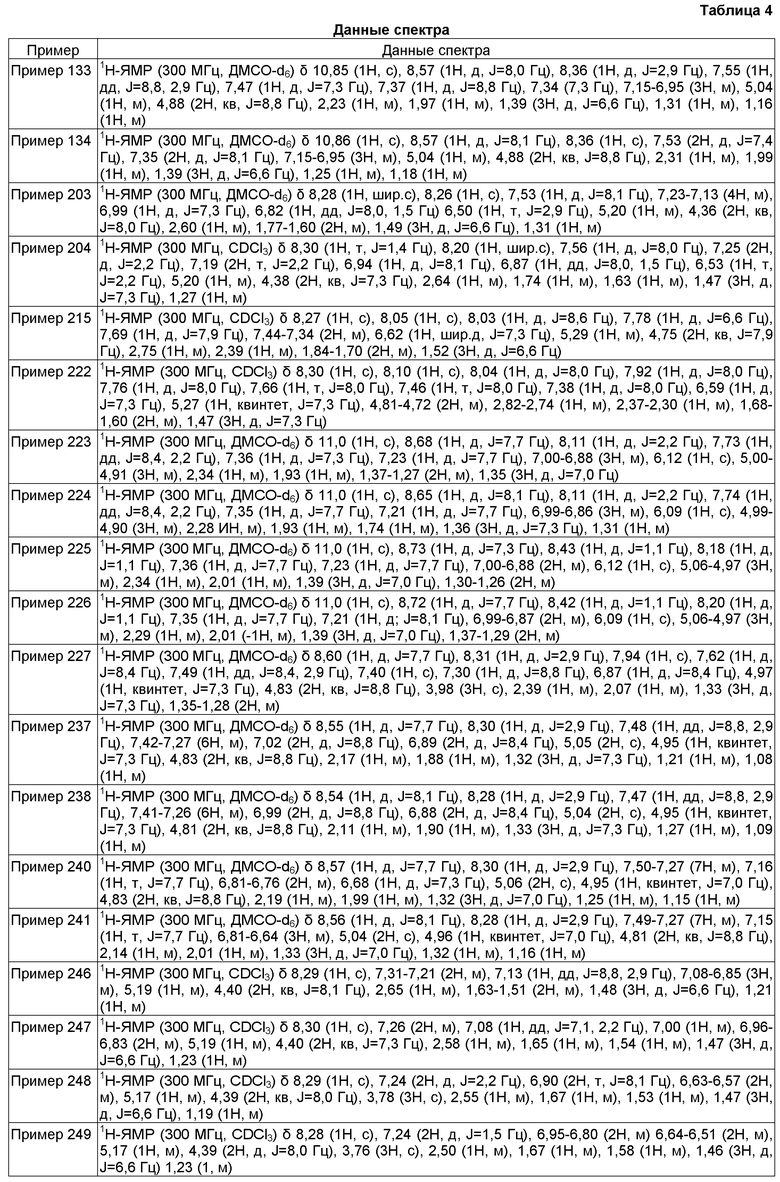
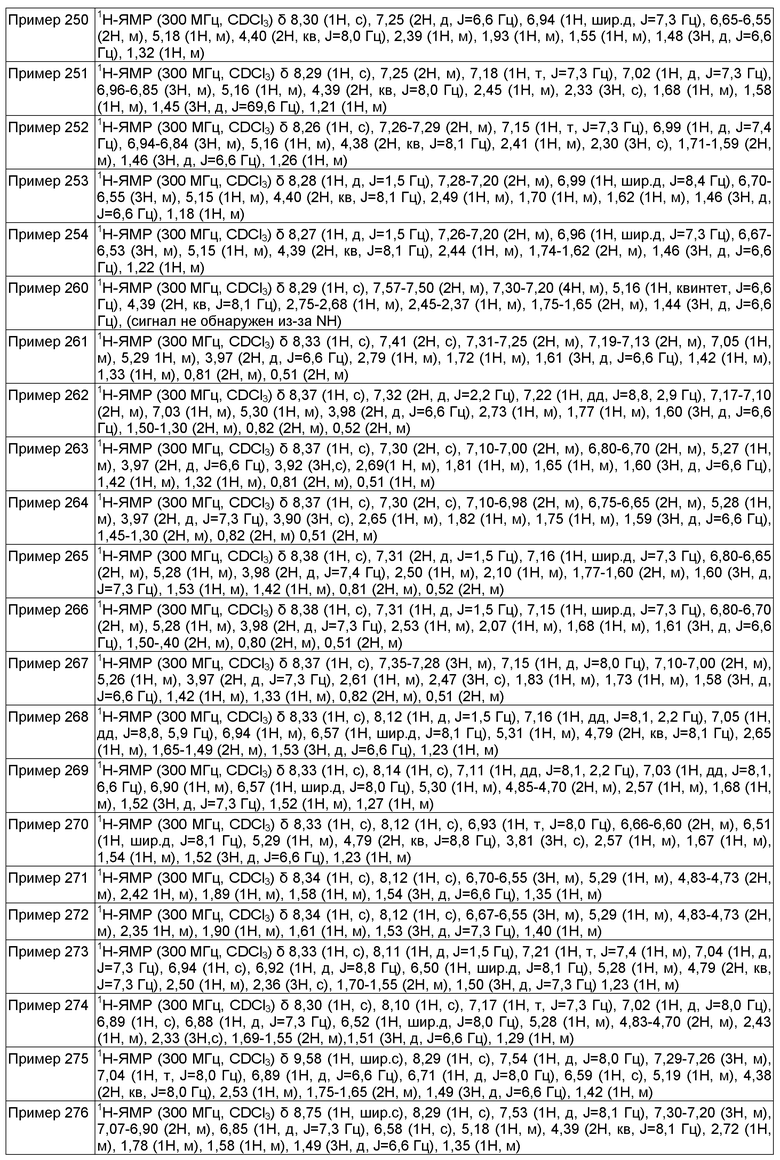
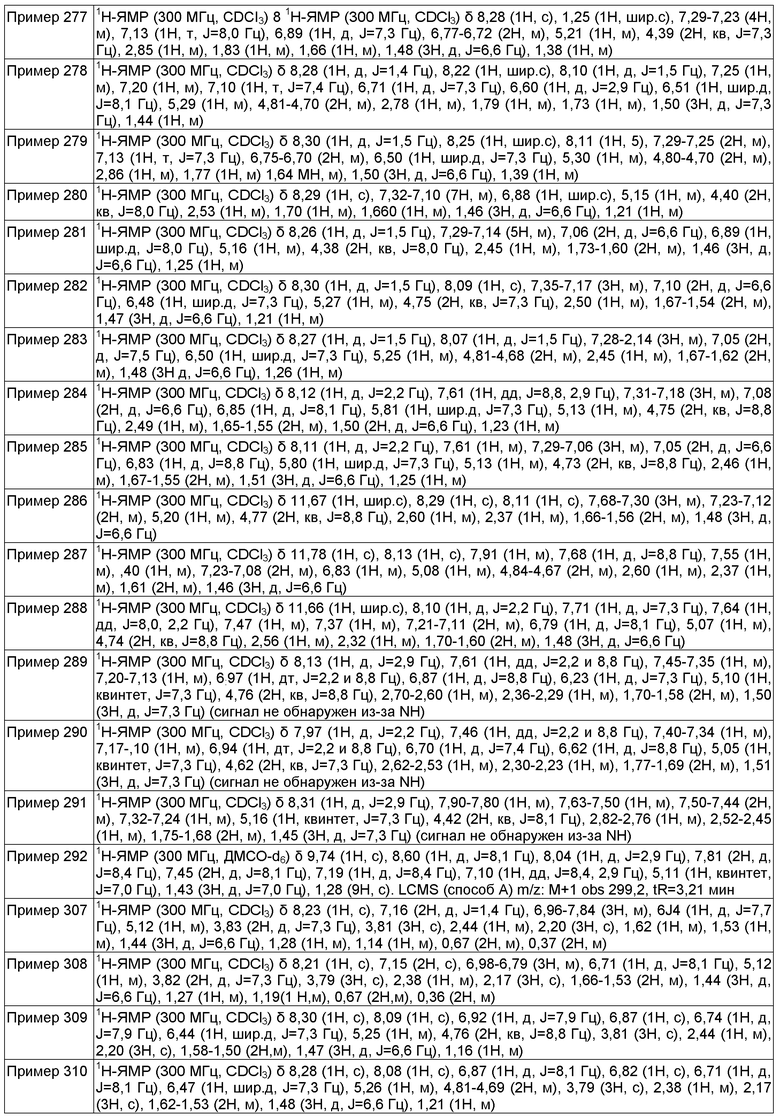
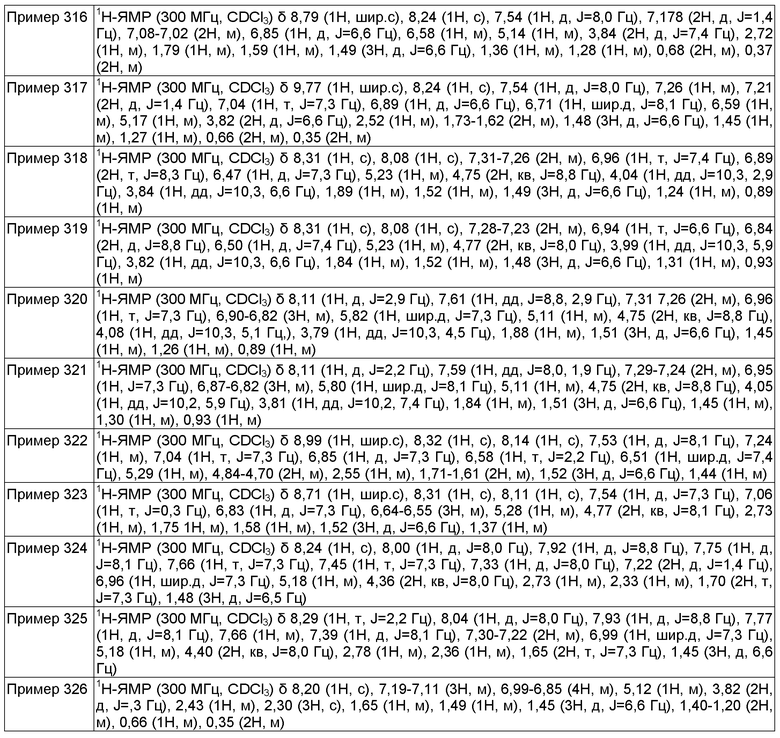
Фармакологические анализы
In vitro анализы активности в отношении T-типа Ca 2+ каналов человека
Активность соединений в отношении кальциевых каналов T-типа определяли методом, хорошо известными из уровня техники, включая "Анализ притока Ca2+" и "Анализ блокатора T-типа Ca2+ методом фиксации потенциала".
Анализ притока Ca 2+
Ингибирование активности T-типа кальциевых каналов определяли с использованием клеточного флуоресцентного анализа притока Ca2+, в котором ионофор калия добавляли для снижения мембранного потенциала покоя, и использовали стимуляцию высоким уровнем внеклеточного K+ для модулирования мембранного потенциала клетки. Изменения сигнала флуоресценции отслеживали методом визуального наблюдения клеток с использованием Hamamatsu Photonics's Functional Drug Screening System (FDSS).
Поддержание клеток
HEK 293 клетки, экспрессирующие человеческий T-типа канал альфа-1H (CaV 3,2), поддерживали в DMEM, дополненной 10% термоактивированной FBS, 100 единиц/мл Пенициллина, 100 мкг/мл стрептомицина, 150 мкг/мл зеоцина, 300 мкг/мл генетицина. Клетки выращивали в 5% CO2 увлажненном инкубаторе при 37ºC.
Протокол анализа
День 1:
1.Клетки собирали и высевали в покрытые поли-D-лизином 384-луночные планшеты с темными стенками и прозрачным дном при плотности 10000 клеток/лунка за 24 часа до анализа.
2.Инкубировали при 37ºC в 5% CO2.
День 2:
1.Каждую лунку промывали буфером для анализа (смотрите ниже) и оставляли 20 мкл с использованием устройства для промывки планшетов Elx-405 Select CW (BIO-TEK).
2. Добавляли в каждую лунку 20 мкл буфера для аналитза, содержащего 6 мкМ Fluo4-AM (molecular Probes) и 0,0005% Pluronic F-127.
3. Инкубировали планшет при 37ºC в течение 1 часа.
4. Каждую лунку промывали буфером для анализа (смотрите ниже) и оставляли 20 мкл с использованием устройства для промывки планшетов Elx-405 Select CW (BIO-TEK).
5. Добавляли в каждую лунку 10 мкл раствора соединения при помощи FDSS6000 и оставляли планшет на 4,5 мин и затем добавляли 10 мкл раствора ионофора калия.
6. Добавляли 20 мкл деполяризующего раствора с высоким содержанием К+ (смотрите ниже) и отслеживали изменения сигнала флуоресценции.
IC50 значения для соединений по настоящему изобретению определяли на основании 7 точек кривой доза-ответ. Кривые строили с использованием среднего значения от двух лунок для каждой точки данных. В конце рассчитывали значения IC50 с использованием кривой наиболее подходящей дозы, определенной при помощи XLfit.
Буфер для анализа (рН 7,4, отрегулирован при помощи HCl)
Деполяризующий раствор с высоким содержианием K+
Электрофизиологический анализ T-типа Ca 2+
В типичном эксперименте регистрировали функцию ионных каналов из HEK 293 клеток, экспрессирующих человеческий T-типа канал альфа-1H (CaV 3,2), для определения активности соединений в блокировани тока кальция, опосредованного T-типа каналом. Клетки, экспрессирующие каналы T-типа, выращивали в питательной среде, которая включала: DMEM, 10% термоинактивированную FBS, 100 единиц/мл пенициллина, 100 мкг/мл стрептомицина, 150 мкг/мл зеоцина, 300 мкг/мл генетицина. Экспрессирующие Ca канал T-типа клетки HEK293 were разделяли при помощи 0,05% трипсина-ЭДТА и затем высевали на предметном стекле в течение 24 часов.
Стеклянные пипетки вытягивали за кончик с диаметром 1-2 микрометра на устройстве для вытягивания пипеток. Пипетки заполняли внутриклеточным раствором и внутри по всей длине протягивали хлорированную серебряную проволоку, которую затем соединяли с головкой усилителеля фиксации потенциала. Раствор для внеклеточной регистрации состоял из (мМ): 150 мМ NMDG, 2 мМ CaCl2, 10 мМ HEPES, 10 мМ глюкозы, pH 7,4. Внутренний раствор состоял из (мМ): 110 CsF, 10 EGTA, 10 HEPES, 3 ATP-Mg, 0,6 GTP pH 7,2; при введении кончика пипетки в ванночку, отмечали последовательное сопротивление (приемлемый диапазон в пределах 1-4 мегаОм). Контактную разность потенциалов между растворами в пипетке и в ванночке устанавливали на нуль на усилителе. Клетки затем разделяли на отельные пэтчи, и после компенсации на последовательное сопротивление (≥80%) применяли протокол электрического напряжения, при этом регистрируя ответный Ca2+ ток для целой клетки. Протоколы электрического напряжения: (1) -80 мВ фиксирующий потенциал каждые 30 секунд импульс до -20 мВ в течение 100 мсек; эффективность лекарственного средства в ингибировании тока, опосредованного таким каналом, измеряли непосредственно из данных уменьшения максимальной амплитуды тока, инициированного изменением напряжения от -80 мВ до -20 мВ; (2) -140 мВ фиксирующий потенциал каждые 30 секунд импульс до -20 мВ в течение 100 мсек; эффективность лекарственного средства в ингибировани тока, опосредованного этим каналом, измеряли непосредственно из данных уменьшения максимальной амплитуды тока, инициированного изменением напряжения от -140 мВ до -20 мВ. Разницу в блокировании при двух фиксирующих потенциалах использовали для определения эффекта лекарственного средства при отличающихся уровнях инактивации, индуцируемых уровнем потенциала состояние покоя клетки. После получения контрольной базовой линии токов кальция, вводили внеклеточные растворы, содержащие увеличивающиеся концентрации испытываемого соединения. После достижения устойчивого ингибирования при данной концентрации соединения применяли бóльшую концентрацию соединения. % ингибирования максимального направленного внутрь контрольного Ca2+ тока в ходе стадии деполяризации до -20 мВ использовали для построения графика как функцию от концентрации соединения.
(3) Нормализованную кривую инактивации в устойчивом состоянии строили с использованием 5 сек (для носителя) или 60 сек (для лекарственных средств) кондиционирующего импульса до различных потенциалов, после которого немедленно следовал испытываемый импульс до -20 мВ. Максимальные токи наносили на график как функцию от максимального тока при кондиционирующих потенциалах в пределах от -140 мВ до -20 мВ. V1/2 или k значения определяли из подборов Больцмана. Аффинность лекарственных средств к Ca каналам T-типа в состоянии покоя (Кпокоя или Kr) оценивали при помощи 30 мсек испытываемого импульса от отрицательного фиксирующего потенциала -140 мВ, где по существу все каналы находятся в состоянии покоя. Значение Kr рассчитывали с использованием традиционной 1:1 модели связывания:
Кпокоя (Kr)={[лекарственное средство]Iмакс, лекарственное средство/(Iмакс, контроль-Iмакс, лекарственное средство)},
где Кпокоя (=Kr) представляет собой константу диссоциации для состояния покоя и [лекарственное средство] представляет собой концентрацию соединения. Imax,контроль и Imax,лекарственное средство представляют собой максимальные токи в отсутствие и в присутствии соединения, соответственно.
Аффинность лекарственного средства к T-типа Ca каналам в инактивированном состоянии (Kинакт. или Ki) оценивали на основании сдвига кривой доступности для соединения. Взаимодействие соединения с каналом в инактивированном состоянии оценивали, как предложено Bean et al (1983 Journal of general pharmacology 81, 613-), путем подгонки экспериментальных точек индуцированных соединением сдвигов средней точки потенциала инвактивации в устойчивом состоянии к уравнению:
Kинтакт.(Ki)={[лекарственное средство]/((1+[лекарственное средство]/Kr)*эксп/(ΔV/k)-1)},
где Kинтакт.(Ki) представляет собой константу диссоциации инактивированного состояния. ΔV представляет собой индуцированный соединением сдвиг напряжения от полумаксимального напряжения кривой Больцмана, и k представляет собой фактор наклона кривой соединения.
Все примеры по настоящему изобретению имеют IC50≤1 мкМ в анализе притока Ca2+ или IC50≤3 мкМ в FRET анализах Nav1.3 или FRET анализах Nav1.7.
В частности, пример 3, пример 33, пример 57, пример 104, пример 106, пример 108, пример 110, пример 111, пример 124, пример 125, пример 133, пример 134, пример 147, пример 167, пример 168, пример 169, пример 171, пример 172, пример 181, пример 182, пример 190, пример 193, пример 194, пример 202, пример 203, пример 204, пример 205, пример 206, пример 207, пример 208, пример 210, пример 211, пример 212, пример 213, пример 222, пример 223, пример 224, пример 225, пример 226, пример 227, пример 228, пример 229, пример 237, пример 240, пример 243, пример 244, пример 245, пример 246, пример 248, пример 250, пример 251, пример 253, пример 258, пример 259, пример 260, пример 261, пример 263, пример 266, пример 267, пример 268, пример 270, пример 271, пример 273, пример 275, пример 276, пример 277, пример 279, пример 280, пример 282, пример 284, пример 286, пример 287, пример 289, пример 294, пример 296, пример 305, пример 306, пример 307, пример 309, пример 310, пример 316, пример 317, пример 318, пример 320, пример 322, пример 323, пример 325, пример 327 и пример 346 по настоящему изобретению имеют IC50≤0,3 мкМ в анализе притока Ca2+.
In vitro анализы активности в отношении потенциалзависимых натриевых каналов человека
Активность соединений в отношении потенциалзависимых натриевых каналов определяли методом, хорошо известными из уровня техники.
Способность арил-замещенных карбоксамидных производных формулы (I) ингибировать каналы NaV1.3, Nav1.7 и Nav1.5 измеряли при помощи FRET анализа и электрофизиологического анализа, описанных ниже.
FRET анализ для Navs
Этот скрининговый анализ используют для определения эффектов соединений на человеческие каналы NaV1.3, человеческие каналы Nav1.7 и человеческие каналы Nav1.5 методом визуального наблюдения клеток с использованием Hamamatsu Photonics's Functiall Drug Screening System (FDSS). Этот эксперимент основан на FRET (Передача Энергии Резонансной Флуоресценции), и использовали две флуоресцентные молекулы. Первая молекула, оксонол (DiSBAC2(3)), представляет собой высоко флуоресцентный отрицательно заряженный и гидрофобный ион, который "чувствует" транс-мембранный электрический потенциал. В ответ на изменения мембранного потенциала он может быстро перераспределяться между двумя сайтами связывания на противоположных сторонах плазматической мембраны. Потенциалзависимое перераспределение преобразовывают в логометрические данные флуоресценции через вторую флуоресцентную молекулу (Кумарин (CC2-DMPE)), которая связывается специфическим образом с одной стороной плазменной мембраны и функционирует как FRET партнер для подвижного потенциал-чувствительного иона. Чтобы этот анализ работал, каналы необходимо фармакологически поддерживать в открытом состоянии. Это достигается путем обработки клеток вератридином.
Поддержание клеток:
Каждые клетки HEK293, экспрессирующие человеческие каналы NaV1.3, и клетки HEK293, экспрессирующие человеческие каналы NaV1.5, выращивали в T225 колбах, в 5% CO2 увлажненном инкубаторе до около 80% слияния. Состав среды включал модифицированную Дульбекко среду Игла (с высоким содержанием глюкозы), 10% фетальную телячью сыворотку (FCS), 100 единиц/мл Пенициллина, 100 мкг/мл Стрептомицина и 500 мкг/мл генетицина.
Клетки CHO, экспрессирующие человеческие каналы NaV1.7, выращивали в T225 колбах, в 5% CO2 увлажненном инкубаторе до около 80% слияния. Состав среды включал HAM/F12 с Glutamax I, 10% фетальную телячью сыворотку (FCS), 100 единиц/мл Пенициллина и 100 мкг/мл гигромицина.
Протокол:
- Высевали каждую из клеточных линий (1,5×104 клеток/лунка) в покрытые поли-D-лизином 384-луночные планшеты до начала эксперимента.
- Инкубировали при 37ºC в 5% CO2 в течение 24 часов.
- Каждую лунку промывали буфером #1 (140 мМ NaCl, 4,5 мМ KCl, 10 мМ D-глюкозы, 2 мМ CaCl2, 1 мМ MgCl2, 10 HEPES, pH 7,4 доведенный при помощи NaOH) два раза с использованием устройства для промывки планшетов.
- Добавляли 1-й нагружающий раствор, содержащий 5 мкм CC-2-DMPE и 0,02% Pluronic F-127 в буфере #1.
- Инкубировали планшет при комнатной температуре в темноте в течение 0,5 часа.
- Каждую лунку промывали буфером #2 (160 мМ холина, 10 мМ D-глюкозы, 0,1 мМ CaCl2, 1 мМ MgCl2, 10 HEPES, pH 7,4 доведенный при помощи KOH) два раза с использованием устройства для промывки планшетов.
- Добавляли 2-й нагружающий раствор, содержащий 15 мкм DISBAC2(3), 0,5 мМ VABSC-1, 10 мМ вератридина и 0,004% Pluronic F-127 в буфере #2.
- Добавляли растворы соединений в аналитический планшет и оставляли планшет на 30 минут в темноте при комнатной температуре.
- Осуществляли измерения методом FDSS.
Данные анализировали и представляли как нормализованные отношения интенсивностей, измеренных в 465 нм и 575 нм каналах. Расчет этих отношений осуществляли следующим способом:
"FI465B" = среднее значение интенсивности флуоресценции в качестве базовой линии (до добавления лиганда Na+) при 465 нм
"FI575B" = среднее значение интенсивности флуоресценции в качестве базовой линии (до добавления лиганда Na+) при 575 нм
"FI465Max" = максимальная интенсивность флуоресценции при 465 нм после стимуляции Na+
"FI575Min" = минимальная интенсивность флуоресценции при 575 нм после стимуляции Na+
"FR" = отношение флуоресценции = (FI465Max/FI575Min)-(FI465B/FI575B)

Этот анализ осуществляли с использованием специальной компьютерной программы, разработанной для FDSS данных. Значения отношения флуоресценции наносили на график с использованием XLfit для определения IC50 значения для каждого соединения.
Электрофизиологический анализ для Navs
Метод пэтч-кламп регистрации в конфигурации “целая клетка” использовали для оценки эффективности или селективности блокатора Na каналов на экспрессирующие человеческий Nav1.3 (hSCN3A) клетки HEK293 или экспрессирующие человеческий Nav1.7 (hSCN9A) клетки CHO. Экспрессирующие человеческий Nav1.3 клетки HEK293 выращивали в питательной среде, которая включала: DMEM, 10% термоинактивированную FBS (Hyclone Laboratories Inc), 100 мкг/мл пенициллина/100 Ед/мл стрептомицина, 150 мкг/мл зеоцина, 3 мкг/мл генетицина. Экспрессирующие человеческий Nav1.7 клетки CHO выращивали в питательной среде, которая включала: HAM/F-12, 9% термоинактивированную FBS (Hyclone Laboratories Inc), 100 мкг/мл пенициллина/100 Ед/мл стрептомицина, 100 мкг/мл гигромицина.
Экспрессирующие Na канал клетки диссоциировали при помощи 0,05% трипсина-ЭДТА и затем высевали на предметное стекло в течение 24-48 часов.
Стеклянные пипетки вытягивали за кончик с диаметром 1-2 микрометра на устройстве для вытягивания пипеток. Пипетки заполняли внутриклеточным раствором и внутри по всей длине протягивали хлорированную серебряную проволоку, которую затем соединяли с головкой усилителя фиксации потенциала (Axon Instruments или HEKA electronik). Раствор для внеклеточной регистрации состоял из (мМ): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 Глюкозы, pH 7,4 доведенный при помощи NaOH. Внутренний раствор состоял из (мМ): 120 CsF, 15 NaCl, 10 EGTA, 10 HEPES, pH 7,2 доведенный при помощи CsOH; При введении кончика пипетки в ванночку, отмечали сопротивление пипетки (приемлемый диапазон в пределах 1-3 МОм). Контактную разность потенциалов между растворами в пипетке и в ванночке устанавливали на нуль на усилителе. После установления конфигурации “целая клетка”, приблизительно 10 минут отводили для уравновешивания раствора в пипетке в клетке до начала регистрации. Токи подвергали низкочастотной фильтрации в пределах 2-5 кГц и осуществляли отбор проб цифровым методом при 10 кГц. Последовательное сопротивление компенсировали (>80%) и непрерывно контролировали.
Нормализованную кривую инактивации в устойчивом состоянии строили с использованием 5 сек (для носителя) или 60 сек (для лекарственных средств) кондиционирующего импульса до различных потенциалов, после которого немедленно следовал испытываемый импульс до 0 мВ. Максимальные токи наносили на график как функцию от максимального тока при кондиционирующих потенциалах в пределах от -120 мВ до -40 мВ. V1/2 или k значения определяли из подборов Больцмана. Аффинность лекарственных средств к Na каналам в состоянии покоя (Кпокоя или Kr) оценивали при помощи 30 мс испытываемого импульса от отрицательного фиксирующего потенциала -140 мВ, где по существу все каналы находятся в состоянии покоя. Значение Kr рассчитывали с использованием традиционной 1:1 модели связывания:
Кпокоя (Kr)={[лекарственное средство]Iмакс, лекарственное средство/(Iмакс, контроль-Iмакс, лекарственное средство)},
где Кпокоя (=Kr) представляет собой константу диссоциации для состояния покоя, и [лекарственное средство] представляет собой концентрацию соединения. «Iмакс, контроль» и «Iмакс, лекарственное средство» представляют собой максимальные токи в отсутствие и в присутствии соединения, соответственно.
Аффинность лекарственного средства к инактивированному состоянию Na каналов (Kinact или Ki) оценивали из сдвига кривой доступности для соединения. Взаимодействие соединения с каналом в инактивированном состоянии оценивали при помощи следующего уравнения:
Kинтакт.(Ki)={[лекарственное средство]/((1+[лекарственное средство]/Kr)*эксп/(-ΔV/k)-1)},
где Kинтакт. (=Ki) представляет собой константу диссоциации инактивированного состояния. ΔV представляет собой индуцированный соединением сдвиг напряжения от полумаксимального напряжения кривой Больцмана, и k представляет собой угол наклона кривой в присутствии соединения.
Соединения примеров были испытаны в описанном выше анализе. Результаты следующие:
Все примеры по настоящему изобретению имеют IC50≤1 мкМ в анализе притока Ca2+ или IC50≤3 мкМ в Nav1.3 FRET или Nav1.7 FRET анализах.
В частности, пример 1, пример 4, пример 5, пример 6, пример 9, пример 10, пример 11, пример 12, пример 13, пример 14, пример 15, пример 16, пример 18, пример 20, пример 21, пример 22, пример 23, пример 28, пример 29, пример 32, пример 36, пример 37, пример 41, пример 42, пример 44, пример 45, пример 48, пример 51, пример 52, пример 53, пример 54, пример 56, пример 59, пример 62, пример 63, пример 64, пример 65, пример 66, пример 67, пример 68, пример 69, пример 70, пример 74, пример 75, пример 76, пример 77, пример 78, пример 80, пример 82, пример 85, пример 86, пример 87, пример 88, пример 89, пример 90, пример 91, пример 92, пример 93, пример 94, пример 95, пример 99, пример 102, пример 103, пример 113, пример 130, пример 131, пример 138, пример 143, пример 146, пример 150, пример 151, пример 152, пример 154, пример 156, пример 157, пример 158, пример 161, пример 162, пример 175, пример 184, пример 192, пример 195, пример 196, пример 197, пример 201, пример 209, пример 214, пример 220, пример 238, пример 241, пример 269, пример 274, пример 285, пример 308, пример 313, пример 314, пример 315, пример 321, пример 324, пример 326, пример 328, пример 332, пример 333, пример 337, пример 338, пример 339, пример 341, пример 345, пример 359, пример 377, пример 424, пример 3, пример 57, пример 104, пример 124, пример 125, пример 147, пример 169, пример 182, пример 194, пример 202, пример 204, пример 205, пример 206, пример 208, пример 210, пример 211, пример 212, пример 213, пример 226, пример 240, пример 246, пример 248, пример 251, пример 253, пример 261, пример 267, пример 268, пример 273, пример 279, пример 294, пример 306, пример 307, пример 310, пример 316, пример 317, пример 318, пример 320, пример 322, пример 323, пример 325, пример 327, пример 346, пример 329, пример 347, пример 355, пример 386, пример 396, пример 397, пример 399, пример 400, пример 413, пример 415, пример 417, пример 419, пример 420, пример 427, пример 431, пример 432, пример 434, пример 439, пример 440, пример 441, пример 442, пример 443, пример 444, пример 447, пример 448, пример 449, пример 454, пример 455, пример 456 и пример 458 по настоящему изобретению имеют IC50=<1,0 мкМ в Nav1.3 FRET или Nav1.7 FRET анализах.
In vivo анализ
Хроническим констрикционным поражением (CCI)-индуцированная статическая аллодиния
Самцов крыс Sprague-Dawley с массой тела 210-240 г закупали у Charles River Japan (Kanagawa, Japan). Животных размещали группами по два животных в условиях 12-часового цикла свет/темнота (свет с 07:00) с доступом к пище и воде ad libitum. CCI осуществляли в соответствии со способом Bennett GJ and Xie YK (Pain 1988, 33: 87-107). Животных анестезировали при помощи интраперитонеальной инъекции пентобарбитала натрия. Левый общий седалищный нерв обнажали на уровне середины бедра и накладывали вокруг него свободно четыре лигатуры с использованием 4-0 шелковой нити (Ethicon Inc, Brussels, Belgium) приблизительно на расстоянии 1 мм. Разрез зашивали и крысе давали восстановиться. Осуществляли ложную операцию таким же способом, за исключением лигирования седалищного нерва. Через 2-3 недели оценивали статическую аллодинию с использованием волосков фон Фрея (VFHs; North Coast Medical Inc., San Jose, CA), как описано Field MJ et al. (Pain 1999, 83: 303-311). Животных помещали в клетки с решетчатым полом и давали акклиматизироваться, по меньшей мере, в течение 30 минут до начала эксперимента. VFH в восходящим по силе порядке (0,16, 0,4, 0,6, 1, 1,4, 2, 4, 6, 8, 10, 15 и 26 г) прилагали к плантарной поверхности задней лапы. Каждый VFH прилагали к ипсилатеральной лапе в течение 6 секунд или до ответной реакции отдергивания лапы. После ответной реакции отдергивания, лапу снова испытывали, начиная со следующего нисходящего VFH до тех пор, пока не происходило никакой ответной реакции. Наименьшее количество силы, необходимое, чтобы вызвать ответную реакцию, определяли как порог отдергивания лапы (PWT) в граммах. Животных с PWT <2 г отбирали для оценки и рандомизированно распределяли почти поровну по всем группам. Соединения по настоящему изобретению или их носители вводили системно.
Все испытываемые соединения по настоящему изобретению показали сильную активность в этой модели.
Индуцированная полным адъювантом Фрейнда (CFA) термическая гипералгезия
Самцов крыс Sprague-Dawley с массой тела 200-250 г закупали у Charles River Japan (Kanagawa, Japan). Животных размещали в условиях 12-часового цикла свет/темнота (свет с 07:00) с доступом к пище и воде ad libitum. CFA-индуцированную термическую гипералгезию оценивали с использованием аппарата для плантарного исследования (Ugo Basile, Verse, Italy), как описано Hargreaves K et al. (Pain 1988, 32: 77-88). Животных помещали в аппарат, состоящий из индивидуальной клетки для испытания на приподнятом стеклянном столе и давали акклиматизироваться, по меньшей мере, в течение 10 минут. Когда животные привыкали к условиям, под столом устанавливали мобильный источник излучения тепла и термостимуляцию прилагали к плантарной поверхности правой задней лапы. Латентность, с которой животное убирало лапу, определяли как латентность отдергивания лапы (PWL) в секундах. CFA подготавливали при концентрации 200 мкг/100 мкл Mycobacterium tuberculosis H37 RA (Difco Laboratories Inc.) в жидком парафине и вводили путем инъекции в плантарную поверхность правой задней лапы. PWL измеряли до и через 2 дня после CFA инъекции. Животных, показывающих снижение PWL в день 2, отбирали для оценки и рандомизированно распределяли почти поровну по всем группам. Соединения по настоящему изобретению или их носители вводили системно. PWL измеряли в подходящее время после введения соединения.
Анализ метаболической стабильности
Период полужизни в микросомах печени человека (HLM)
Испытываемые соединения (1 мкМ) инкубировали с 3,3 мМ MgCl2 и 0,78 мг/мл HLM (HL101) в 100 мМ калий-фосфатного буфера (pH 7,4) при 37ºC в 96-луночном планшете с глубокими лунками. Реакционную смесь разделяли на две группы, не-P450 и P450 группа. NADPH добавляли только к реакционной смеси P450 группы. (систему генерирования NADPH также использовали вместо NADPH). Аликвоту образцов P450 группы собирали в точках времени 0, 10, 30 и 60 мин, где точка времени 0 мин указывала время, когда NADPH добавляли к реакционной смеси P450 группы. Аликвоту образцов не-P450 группы собирали в точках времени -10 и 65 мин. Собранные аликвоты экстрагировали при помощи раствора ацетонитрила, содержащего внутренний стандарт. Осажденный белок центрифугировали (2000 об/мин, 15 мин). Концентрацию соединения LC/MS/MS.
Значение периода полужизни получали путем построения графика с использованием натурального логарифма отношения площадей пиков соединений/внутреннего стандарта против времени. Угол наклона кривой лучшей подборки через точки дает скорость метаболизма (k). Это значение преобразовывали в значение периода полужизни с использованием следующих уравнений: Период полужизни = ln 2/k
Анализ межлекарственного взаимодействия
Этот способ по существу включает определение процента ингибирования образования метаболитов из зондов (Tacrine (Sigma A3773-1G) 2 мкМ, Декстрометорфана (Sigma D-9684) 5 мкМ, диклофенака (Sigma D-6899-10G) 5 мкМ и мидазолама (ULTRAFINE UC-429) 2 мкМ) при 3 мкМ каждого соединения.
Более конкретно, анализ осуществляли следующим способом. Соединения (60 мкМ, 10 мкл) предварительно инкубировали в 170 мкл смеси, включающей микросомы печени человека, 100 мМ калий-фосфатного буфера и зонды в качестве субстрата, в течение 5 мин. Реакцию начинали путем добавления NADPH (10 мМ, 20 мкл) (также использовали систему генерирования NADPH, которая состоит из 0,5 мМ NADP, 10 мМ MgCl2, 6,2 мМ DL-изолимонной кислоты и 0,5 Ед/мл изолимонной дегидрогеназы). Аналитический планшет инкубировали при 37ºC. К раствору инкубата добавляли ацетонитрил в подходящее время (например, 8 мин).
Концентрацию метаболитов в супернатанте измеряли с использованием системы LC/MS/MS.
Степень взаимодействия между лекарственными средствами интерпретировали на основании % метаболитов в присутствии или в отсутствие испытываемого соединения.
Анализ связывания человеческого дофетилида
Трансфицированные человеческим HERG клетки HEK293S клетки получали и выращивали в лаборатории. Собранные клетки суспендировали в 50 мМ Tris-HCl, 10 мМ KCl, 1 мМ MgCl2, Complete (Roche) (pH 7,4 при 4ºC) и гомогенизировали с использованием ручного устройства для разрыва клеток Polytron PT 1300 D, установленного на 15000 об/мин, в течение 20 сек на льду. Гомогенаты центрифугировали при 48000×g при 4ºC в течение 20 минут. Осадки после центрифугирования затем ресуспендировали, гомогенизировали и центрифугировали таким же способом. Конечные осадки после центрифугирования ресуспендировали в подходящем объеме 50 мМ Tris-HCl, 10 мМ KCl, 1 мМ MgCl2, Complete (pH 7,4 при 4ºC), гомогенизировали, делили на аликвоты и хранили при -80ºC до использования. Аликвоту мембранных фракций использовали для определения концентрации белка при помощи BCA набора для анализа белка (PIERCE) и ARVOsx планшет-ридера (Wallac). Анализы связывания осуществляли в общем объеме 30 мкл в 384-луночных планшетах. Активность измеряли при помощи PHERAstar (BMG LABTECH) с использованием метода поляризации флуоресценции. 10 мкл испытываемых соединений инкубировали с 10 мкл флуоресцентного лиганда (6 нМ Cy3B-меченного производного дофетилида) и 10 мкл мембранного гомогената (6 мкг белка) в течение 120 минут при комнатной температуре. Неспецифическое связывани определяли при помощи 10 мкМ E4031 при конечной концентрации. Значения IC50 рассчитывали с использованием односайтовых моделей доза-ответ, логистической модели с 4 параметрами (XLfit).
Все публикации, включая, но не ограничиваясь этим, изданные патенты, патентные заявки и журнальные статьи, на которые ссылаются в настоящей заявке, включены каждая в настоящую заявку посредством ссылки во всей их полноте. Хотя настоящее изобретение было описано выше со ссылкой на раскрываемые варианты воплощения, специалисты в данной области легко поймут, что конкретные подробно описанные эксперименты являются только иллюстрацией настоящего изобретения. Должно быть понятно, что возможны различные модификации без отступления от сути настоящего изобретения. Соответственно, настоящее изобретение ограничено только следующей формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ БЛОКАТОРОВ TTX-S | 2013 |
|
RU2652117C2 |
| ПРОИЗВОДНЫЕ АРИЛАМИДОВ В КАЧЕСТВЕ БЛОКАТОРОВ TTX-S | 2011 |
|
RU2535671C1 |
| ПРОИЗВОДНЫЕ ПИРРОЛОПИРИДИНОНА В КАЧЕСТВЕ TTX-S БЛОКАТОРОВ | 2013 |
|
RU2646754C2 |
| ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ СПИРОПИПЕРИДИНИЛА И ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2021 |
|
RU2839891C1 |
| ПРОИЗВОДНЫЕ АМИДА В КАЧЕСТВЕ БЛОКАТОРОВ Nav1.7 И Nav1.8 | 2018 |
|
RU2768149C2 |
| ПОЗИТИВНЫЕ АЛЛОСТЕРИЧЕСКИЕ МОДУЛЯТОРЫ НИКОТИНОВОГО РЕЦЕПТОРА АЦЕТИЛХОЛИНА | 2012 |
|
RU2604737C2 |
| N-(2-ЦИАНОГЕТЕРОЦИКЛИЛ)ПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2669922C2 |
| АМИДОПРОИЗВОДНЫЕ КАК БЛОКАТОРЫ TTX-S | 2013 |
|
RU2632899C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗО[1,2-b]ПИРИДАЗИНЫ, ЗАМЕЩЕННЫЕ ИМИДАЗО[1,5-b]ПИРИДАЗИНЫ, РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ПАТОЛОГИЧЕСКИХ СОСТОЯНИЙ | 2017 |
|
RU2773333C2 |
| ИНГИБИТОРЫ FXIA, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В ФАРМАЦЕВТИКЕ | 2020 |
|
RU2802878C1 |
Изобретение относится к арил-замещенным карбоксамидным производным формулы (I) или их фармацевтически приемлемым солям, где в формуле (I) R представляет собой водород; R1 независимо выбран из группы, состоящей из: (1) водорода, (2) галогена, (3) гидроксила, (4) -On-C1-6 алкила, где алкил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (5) -On-гетероциклической группы, выбранной из пиперидинила, пирролидинила, тетрагидропиранила, тетрагидрофуранила и оксетанила; n имеет значение 0 или 1, когда n имеет значение 0, вместо On присутствует химическая связь; р имеет значение 1 или 2; когда р имеет значение два, R1 могут быть одинаковыми или отличными друг от друга; R2 представляет собой C1-6 алкил, который является незамещенным или замещенным одним или несколькими заместителями, независимо выбранными из R7; или R2 вместе с R1 образует С3-С6 циклоалкил; X представляет собой 1,2-С3 циклоалкилен; W, Y и Z независимо выбраны из атома азота и атома углерода; по меньшей мере, один из W, Y и Z представляет собой азот и W, Y и Z, в одно и то же время, не являются углеродом; R3, R4, R5 и R6 являются такими, как указано в формуле изобретения; Ar означает арил, который представляет собой моно- или би-карбоциклическое или моно- или би-гетероциклическое кольцо, содержащее 0-3 гетероатома, выбранных из О, N и S, включая фенил, фурил, оксазолил, тиазолил, имидозолил, пиридил, пиперидинил, пиримидинил, изооксазолил, триазолил, тетрагидронафтил, бензофуранил, бензотиофенил, индолил, бензоимидазолил, хинолил, изохинолил, хиноксалинил, пиразоло [1,5-а] пиридил, тиено [3,2-b] пирролил, где арил необязательно замещен 1-3 заместителями, указанными в формуле изобретения. Также изобретение относится к соединениям формулы (II) и к конкретным арил-замещенным карбоксамидным производным, указанным в п.3 формулы изобретения, фармацевтической композиции, способу лечения и применению. Технический результат - арил-замещенные карбоксамидные производные, обладающие блокирующей активностью в отношении кальциевых каналов Т-типа или потенциалзависимых натриевых каналов. 7 н. и 3. з.п. ф-лы, 6 табл., 464 пр.


1. Соединение формулы (I)
где:
R представляет собой водород;
R1 независимо выбран из группы, состоящей из:
(1) водорода, (2) галогена, (3) гидроксила, (4) -On-C1-6 алкила, где алкил является незамещенным или замещен одним или несколькими заместителями, независимо выбранными из R7, (5) -On-гетероциклической группы, выбранной из пиперидинила, пирролидинила, тетрагидропиранила, тетрагидрофуранила и оксетанила;
где n имеет значение 0 или 1, когда n имеет значение 0, вместо On присутствует химическая связь;
р имеет значение 1 или 2; когда р имеет значение два, R1 могут быть одинаковыми или отличными друг от друга;
R2 представляет собой C1-6 алкил, который является незамещенным или замещенным одним или несколькими заместителями, независимо выбранными из R7;
или R2 вместе с R1 образует С3-С6 циклоалкил;
X представляет собой 1,2-С3 циклоалкилен;
W, Y и Z независимо выбраны из атома азота и атома углерода;
по меньшей мере, один из W, Y и Z представляет собой азот и W, Y и Z, в одно и то же время, не являются углеродом;
R3, R4, R5 и R6 независимо выбраны из группы, состоящей из:
(1) водорода, (2) гидроксила, (3) C1-6 алкила и (4) -NR7R8;
q имеет значение 0;
r имеет значение 0;
R7 выбран из группы, состоящей из:
(1) водорода, (2) галогена, (3) гидроксила, (4) -(С=O)m-О1-C1-6 алкила, (5) -О1-(C1-3) перфторалкила, (6) -(С=O)m-O1-C1-6 циклоалкила, где циклоалкил является незамещенным или замещен одним R8, (7) -(С=O)m-O1-фенила, где фенил является незамещенным или замещен одним R8, (8) -(С=O)m-O1-гетероциклической группы, выбранной из пиридила, имидазолила и оксетанила, где гетероциклическая группа является незамещенной или замещена одним R8;
где l имеет значение 0 или 1 и m имеет значение 0 или 1; когда l имеет значение 0 или m имеет значение 0, вместо (С=O)m или O1 присутствует химическая связь, и когда l имеет значение 0 и m имеет значение 0, вместо (C=O)m-O1 присутствует химическая связь;
R8 независимо выбран из группы, состоящей из:
(1) водорода, (2) галогена, (3) C1-6 алкила;
R9 и R10 представляют собой, независимо, водород или C1-6 алкил, или R9 образует 6-членное насыщенное моноциклическое кольцо вместе с R10, которое содержит атом азота, и замещено 2 заместителями, независимо выбранными из атомов галогена;
Ar означает арил, который представляет собой моно- или би-карбоциклическое или моно- или би-гетероциклическое кольцо, содержащее 0-3 гетероатома, выбранных из О, N и S, включая фенил, фурил, оксазолил, тиазолил, имидозолил, пиридил, пиперидинил, пиримидинил, изооксазолил, триазолил, тетрагидронафтил, бензофуранил, бензотиофенил, индолил, бензоимидазолил, хинолил, изохинолил, хиноксалинил, пиразоло [1,5-а] пиридил, тиено [3,2-b] пирролил, где арил необязательно замещен 1-3 заместителями, независимо выбранными из группы, состоящей из:
(1) галогена, (2) гидроксила, (3) -On-C1-6 алкила, где алкил является незамещенным или замещен 1-3 заместителями, независимо выбранными из R7, (4) -On-С3-6 циклоалкила, (5) -NR9R10, (6) -CN;
где n имеет значение 0 или 1; когда n имеет значение 0, вместо On присутствует химическая связь;
или его фармацевтически приемлемая соль.
2. Соединение формулы (II)
где
R представляет собой C1-6 алкил
v имеет значение 0 или 1;
R1 представляет собой -OCH2CF3 или -ОСН3;
R2 представляет собой C1-6 алкил;
R3 представляет собой C1-6 алкил;
w имеет значение 0 или 1;
R4 и R5 представляют собой, независимо, водород или С1-6 алкил;
R6 представляет собой галоген;
где l имеет значение 0 или 1; когда l имеет значение 0, вместо O1 присутствует химическая связь;
R7 и R8 представляют собой, независимо, водород или C1-6 алкил;
p представляет собой 1, q и r представляют собой, независимо, 0 или 1;
Y и Z независимо выбраны из атома азота и атома углерода; Y и Z, в одно то же самое время, не являются атомом углерода;
Ar означает арил, который представляет собой моно- или би-карбоциклическое или моно- или би-гетероциклическое кольцо, содержащее 1-2 атома N, включая, фенил, индолил, бензоимидазолил, хинолил, изохинолил, индазолил, где арил необязательно замещен 1-3 заместителями, независимо выбранными из группы, состоящей из:
(1) галогена, (2) гидроксила, (3) -On-C1-6 алкила, где алкил является незамещенным или замещен 1-3 заместителями, независимо выбранными из R6, (4) -NR7R8 и (5) -CN;
где n имеет значение 0 или 1; когда n имеет значение 0, вместо On присутствует химическая связь;
или его фармацевтически приемлемая соль.
3. Соединение, выбранное из следующих соединений:
(1R*,2R*)-2-м-толил-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-(5-циано-1Н-бензо[d]имидазол-2-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
транс-2-(изохинолин-3-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(2,5-дифторфенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
транс-2-((4-хлорфенокси)метил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-N-((R)-1-(5-(циклопропилметокси)пиридин-2-ил)этил)-2-(2-фтор-4-метоксифенил)циклопропанкарбоксамид;
(1R*,2R*)-N-((R)-1-(5-(циклопропилметокси)пиридин-2-ил)этил)-2-(1Н-индол-7-ил)циклопропанкарбоксамид;
транс-N-((R)-1-(5-(циклопропилметокси)пиридин-2-ил)этил)-2-(3,5-дифторфенил)циклопропанкарбоксамид;
(1S*,2S*)-2-(1Н-индол-6-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-фенил-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)-2-(2,4,6-трифторфенил)циклопропанкарбоксамид;
(1R*,2R*)-2-(1Н-индол-4-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
транс-2-(1-метил-1Н-бензо[d]имидазол-2-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-((3-фторфенокси)метил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(1Н-индол-7-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(2-хлор-4-фторфенил)-N-((R)-1-(5-(циклопропилметокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(2-фтор-4-метоксифенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(4-метоксифенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-N-((R)-1-(5-(циклопропилметокси)пиридин-2-ил)этил)-2-(2,4,6-трифторфенил)циклопропанкарбоксамид;
(1R*,2R*)-2-(1Н-бензо[d]имидазол-2-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
транс-2-(1-метил-1Н-индол-6-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-((4-фторфенокси)метил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(3,5-дифторфенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
транс-2-(5-фтор-1Н-индол-3-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
транс-2-((1Н-индол-1-ил)метил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
транс-2-(7-фтор-1Н-индол-3-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(1-метил-1Н-индазол-6-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
транс-2-(5-фтор-1Н-индол-2-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(2-хлор-4-фторфенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(3-метоксифенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-(хинолин-2-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
транс-2-(1Н-индол-2-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
транс-N-((R)-1-(5-(циклопропилметокси)пиридин-2-ил)этил)-2-(5-фтор-1Н-индол-2-ил)циклопропанкарбоксамид;
(1S*,2S*)-N-((R)-1-(5-(циклопропилметокси)пиридин-2-ил)этил)-2-(4-метокси-3-метилфенил)циклопропанкарбоксамид;
транс-2-(4-метокси-3-метилфенил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-N-((R)-1-(5-(циклопропилметокси)пиридин-2-ил)этил)-2-(1Н-индол-7-ил)циклопропанкарбоксамид;
(1S*,2S*)-N-((R)-1-(5-(циклопропилметокси)пиридин-2-ил)этил)-2-м-толил-циклопропанкарбоксамид;
(1S*,2S*)-2-м-толил-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
транс-2-(6-фтор-1Н-индол-3-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-(1H-индол-6-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-(1H-индол-7-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-(1Н-индол-3-ил)-N-((R)-1-(5-(2,2,2-трифторэтокси)пиридин-2-ил)этил)циклопропанкарбоксамид;
транс-2-(хинолин-3-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
транс-2-(хинолин-7-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
транс-2-(хинолин-2-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-(1Н-индол-2-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(феноксиметил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
транс-2-(хинолин-6-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
транс-2-(1-метил-1Н-бензо[d]имидазол-2-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-фенил-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
транс-2-(1-метил-1Н-индазол-6-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
транс-2-(1Н-индол-6-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
(1S*,2S*)-2-(1Н-бензо[d]имидазол-2-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
(1R*,2R*)-2-(5-фтор-1Н-бензо[d]имидазол-2-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
транс-2-(1-метил-1Н-индол-6-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
транс-2-(5-фтор-1Н-индол-2-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
транс-2-(1Н-индол-7-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид; и
(1S*,2S*)-2-(1Н-индол-2-ил)-N-((R)-1-(6-(2,2,2-трифторэтокси)пиридин-3-ил)этил)циклопропанкарбоксамид;
и их фармацевтически приемлемых солей.
4. Фармацевтическая композиция, обладающая блокирующей активностью в отношении кальциевых каналов Т-типа или потенциалзависимых натриевых каналов, содержащая в качестве активного ингредиента соединение или его фармацевтически приемлемую соль по любому из пп. 1-3 в эффективной дозировке и фармацевтически приемлемый носитель.
5. Способ лечения состояния или расстройства, в котором участвуют кальциевые каналы Т-типа или потенциалзависимые натриевые каналы, у млекопитающего, в том числе человека, который включает введение млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения или его фармацевтически приемлемой соли, по любому из пп. 1-3.
6. Соединение или его фармацевтически приемлемая соль по п. 1 для применения в лечении состояния или расстройства, в котором участвуют кальциевые каналы Т-типа или потенциалзависимые натриевые каналы.
7. Соединение или его фармацевтически приемлемая соль по п. 2 для применения в лечении состояния или расстройства, в котором участвуют кальциевые каналы Т-типа или потенциалзависимые натриевые каналы.
8. Соединение или его фармацевтически приемлемая соль по п. 3 для применения в лечении состояния или расстройства, в котором участвуют кальциевые каналы Т-типа или потенциалзависимые натриевые каналы.
9. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-3 для получения лекарственного средства для лечения состояния или расстройства, в котором участвуют кальциевые каналы Т-типа или потенциалзависимые натриевые каналы.
10. Применение соединения формулы (I) или его фармацевтически приемлемой соли по п. 1 для получения лекарственного средства для лечения состояния или расстройства, в котором участвуют потенциалзависимые натриевые каналы, как описано в формуле (I) в п. 1, где, когда Y представляет собой атом азота и в то же время (i) q имеет значение 1 и r имеет значение 0, или (ii) q имеет значение 0 и r имеет значение 1, тогда X может быть химической связью;
или, как описано в формуле (I) в п. 1, где, когда Y представляет собой атом углерода, Z представляет собой атом азота, W представляет собой атом азота и в то же время (i) q имеет значение 1 и r имеет значение 0, или (ii) q имеет значение 0 и r имеет значение 1, тогда X может быть химической связью;
определение других переменных такое же, как в п. 1.

