Изобретение относится к области органической химии, к способу получения новых биологически активных веществ класса 1-замещенных 4,4,4-трихлорбутан-1,3-дионов (β-дикетоны), их таутомерам, а именно к 4,4,4-трихлор-1-(4-хлорфенил)бутан-1,3-диону (1) и его таутомерам, общей формулы: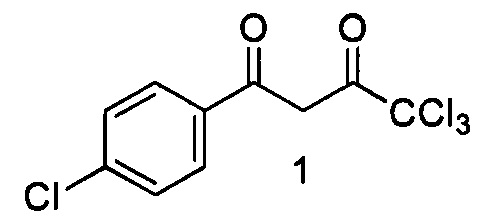
обладающему противогрибковой активностью, что позволяет предположить его использование в медицине в качестве противогрибкового средства.
В литературе описаны несколько подходов к синтезу β-дикетонов, содержащих трихлорметильный заместитель.
Так, при взаимодействии ацетофенонов с трихлорацетонитрилом в присутствии фенилэтиламиномагнийбромида образуются (β-аминовинил)кетоны, гидролиз которых приводит к образованию β-дикетонов - структурных аналогов соединения (1) [В.Я. Сосновских, И.С. Овсянников ЖОрХ. 1990, 26 (10), 2086-2091]. К недостаткам этого метода можно отнести умеренные выходы конечно продукта (71-73%), а также высокую токсичность трихлорацетонитрила (очень ядовит, сильно раздражает слизистые верхних дыхательных путей и глубокие дыхательные пути).
Еще один способ синтеза аналогов соединения (1) конденсация триметилсилиловых эфиров с хлорангидридом трихлоруксусной кислоты. Данный метод дает умеренные выходы конечных продуктов реакции (41-67%) [S. Murai, Y. Kuroki, K. Hasegawa, S. Tsutsumi J.C.S. Chem.Comm. 1972, 946-947].
В литературе описан метод синтеза полигалогензамещенных β-дикетонов из ацеталей ацетофенонов [Flores, A.C.F. Synthesis and structure of novel 1-aryl-4,4,4-trichloro-1,3-butanediones / A.C.F. Flores, M.J. Martins, L.M. Frigo // Synth. Commun. - 2012. - №.42. - Р. 727-737].
Из уровня техники известен способ получения 4,4,4-трихлор-1-(4-хлорфенил)бутан-1,3-диона (патент РФ 2582236, МПК С07С 49/807, А61К 31/122, А61Р 31/04, А61Р 29/00, опубл. 20.04.2016), заключающийся в осуществлении взаимодействия предварительно охлажденного до 0°С 4-хлор-1-(1,1-диэтокси)бензола, пиридина с хлорангидридом трихлоруксусной кислоты в среде абсолютного хлороформа при перемешивании в течение 10-12 часов, последующем добавлении 2М серной кислоты и дальнейшем перемешивании при 80°С в течение 2 часов. Полученную смесь разделяют на делительной воронке, органический слой сушат сульфатом натрия, после удаления растворителя выпавший продукт перекристаллизовывают из гексана.
Недостатками данного способа являются недостаточная чистота конечного продукта, низкий выход конечного продукта. Указанные недостатки устраняются в заявляемом изобретении за счет дополнительных технологических приемов.
Нами были проведены оптимизация условий реакции синтеза и очистки соединения (1) (табл. 1).
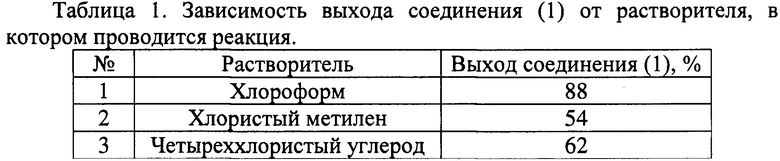
Как следует из данных таблицы, максимальный выход соединения (1) достигнут при использовании в качестве растворителя хлороформа.
При попытках очистки и перекристаллизации конечного продукта (1) нами было установлено, что предварительно осушенную сульфатом натрия реакционную массу нужно пропускать через слой силикагеля среднего размера для избавления от смолянистых примесей. Подбор растворителей для перекристаллизации показал, что самым удобным является этанол (по данным ГЖХ).
Заявляемое соединение (1) синтезируют взаимодействием 4-хлор-1-(1,1-диэтокси)бензола и хлорангидридом трихлоруксусной кислоты в присутствии пиридина в среде безводного хлороформа, с последующим кислотным гидролизом:
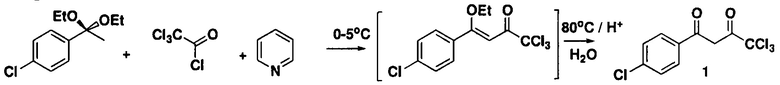
Технический результат заключается в том, что использование заявляемого способа позволяет получать целевой продукт, который не содержит нежелательных примесей и не требует дополнительной очистки, а также позволяет получать целевой продукт с высоким выходом.
Изобретение иллюстрируется следующими примерами:
Пример 1. В плоскодонную колбу объемом 500 мл помещают 22.85 г (0.1 моль) свежеперегнанного 1-(1,1-диэтоксиэтил)4-хлорбензола и 16 мл (0.2 моль) пиридина в 40 мл безводного хлороформа, полученную смесь охлаждают до 0°С. Затем при интенсивном перемешивании на магнитной мешалке добавляют охлажденный до 0°С раствор 20.63 мл (0.2 моль) хлорангидрида трихлоруксусной кислоты в 30 мл безводного хлороформа. Реакционную смесь оставляют на магнитной мешалке при комнатной температуре в течение 10 ч (без доступа влаги), по истечению времени добавляют 130 мл 2М серной кислоты и продолжают перемешивание еще 2 часа при 80°С. После охлаждения смесь переносят в делительную воронку объемом 500 мл, отделяют водный слой и промывают органический слой дистиллированной водой до нейтральной реакции среды (рН среды определяют по универсальной индикаторной бумаге). Промытый органический слой сушат над безводным сульфатом натрия в течение суток. По истечению времени отфильтровывают сульфат натрия, полученный фильтрат пропускают через слой силикагеля (70-100 мкм, 15 г) и упаривают. Образовавшийся осадок соединения (1) дважды перекристаллизовывают из этанола. Выход 88%, точка плавления 76°С (по литературным данным т.пл 69-71°С). ИК-спектр (ФСМ-1202, вазелиновое масло, ν, см-1): 3208, 1642, 1598, 1562 (O=С-С=С). ЯМР 1Н спектр (Bruker Avance III HD 400 [рабочая частота 400 МГц] в CDCl3, внутренний стандарт - ТМС, δ, м.д.): 6.79 (1Н, с, СН), 7.50 (2Н, д, HAr, J 8 Гц), 7.89 (2Н, д, HAr, J 8 Гц), 14.22 (1Н, с, ОН). ЯМР 13С спектр (Bruker Avance III HD 400 [рабочая частота 100 МГц] в CDCl3, внутренний стандарт - ТМС, δ, м.д.): 76.4, 89.2, 127.9, 128.7, 130.4, 139.2, 178.8, 185.7. Найдено, %: С 40.10, Н 2.00. C10H6Cl4O2. Вычислено, %: С 82.04, Н 2.02. Однозначно структура соединения (1) была подтверждена методом РСА.
Полученное соединение (1) представляет собой бесцветное кристаллическое вещество, растворимое в хлороформе, ацетоне, практически не растворимое в воде.
Предлагаемый способ получения соединения (1) имеет следующие преимущества относительно известных способов:
1. Использование данного способа позволяет получать целевой продукт с выходом 88%;
2. Целевой продукт не содержит нежелательных примесей и не требует дополнительной очистки, что важно для использования его в качестве фармацевтической субстанции.
Пример 2. Противомикробная активность соединения (1).
Изучение противогрибковой активности in vitro проводили методом двукратных серийных разведений в жидкой питательной среде [Руководство по экспериментальному (доклиническому) изучению новых фармакологических веществ / Хабриев Р.У. - М.: Медицина, 2005. - 832 с]. Противогрибковые свойства заявляемого соединения (1) изучали на пяти коллекционных тест-культурах, полученных в ФГБОУ ВО СЗМУ им. Мечникова Минздрава России, лаборатория «Российская коллекция патогенных грибов»:
С. albicans, PКПГY 1353/1277;
С. parapsilosis, РКПГY 1579/296;
С. krusei, РКПГY 1472/310;
С. glabrata, РКПГY 1485/47;
С. kefir, РКПГY 1559/577.
Культуры выращивали в пробирках на скошенной агаризированной среде Сабуро. Для приготовления рабочей взвеси микробов тестируемые штаммы разводили в стерильном 0,9% физиологическом растворе до мутности эквивалентной 5,0 по стандарту Mac-Farland с использованием денситометра. После ряда разведений плотность суспензии в пробирке соответствовала 5×105 клеток/мл, далее приготовленные суспензии спор грибов исследуемых штаммов вносили в разведения заявленного соединения по 200 мкл.
В качестве стандартной навески изучаемого соединения (1) брали 10 мг. Растворяли в 1 мл ДМСО, а затем добавляли 9 мл дистиллированной воды, далее разводили раствор еще в 10 раз, таким образом, в первой пробирке содержалось 100,0 мкг/мл испытуемого вещества. После чего готовили ряд серийных последовательных разведений с двукратно уменьшающей концентрацией. Максимально испытанная концентрация соответствовала 100,0 мкг/мл, минимальная - 0,05 мкг/мл.
Учет результатов контрольных и опытных пробирок учитывали после 24 часового (ингибирующее действие) и 7-суточного (фунгицидное действие) термостатирования при температуре 37±1°С.
За действующую дозу принимали минимальную ингибирующую концентрацию (МИК) препарата, которая задерживает рост дрожжевых культур. Эталоном сравнения служил известный в медицинской практике противогрибковый препарат флуконазол, производителя ООО «Озон», Самарская область, г. Жигулевск.
Результаты исследований по заявленному соединению (1) изложены в таблице 2.
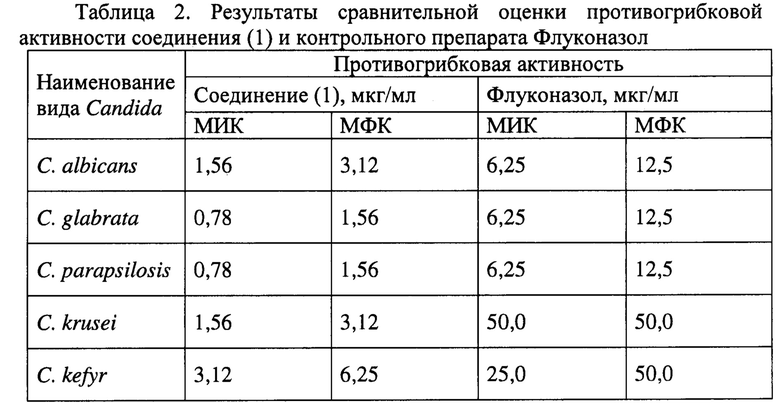
Примечание: "-" - отсутствие противогрибкового действия в испытанных концентрациях.
Результаты, представленные в таблице 2, демонстрируют достаточно высокую активность изучаемого соединения (1) в отношении коллекционных штаммов грибов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРИМЕНЕНИЕ 1-(4-БРОМФЕНИЛ)-4,4,4-ТРИХЛОРБУТАН-1,3-ДИОНА В КАЧЕСТВЕ СРЕДСТВА, ОБЛАДАЮЩЕГО ПРОТИВОГРИБКОВОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ ASPERGILLUS FUMIGATUS И ASPERGILLUS NIGER | 2022 |
|
RU2798469C1 |
| 4,4,4-ТРИХЛОР-1-(4-ХЛОРФЕНИЛ)БУТАН-1,3-ДИОН, ОБЛАДАЮЩИЙ АНАЛЬГЕТИЧЕСКОЙ И ПРОТИВОМИКРОБНОЙ АКТИВНОСТЯМИ | 2015 |
|
RU2582236C1 |
| НАТРИЕВЫЕ СОЛИ 1-АРИЛ-4,4,4-ТРИФТОРБУТАН-1,3-ДИОНОВ, ПРОЯВЛЯЮЩИЕ ПРОТИВОМИКРОБНУЮ АКТИВНОСТЬ | 2024 |
|
RU2829962C1 |
| СЕРЕБРЯНЫЕ СОЛИ 1-(4-ГАЛОГЕНФЕНИЛ)-4,4,4-ТРИФТОРБУТАН-1,3-ДИОНОВ, ПРОЯВЛЯЮЩИЕ ПРОТИВОМИКРОБНУЮ АКТИВНОСТЬ | 2023 |
|
RU2798433C1 |
| Способ получения непредельных @ -дикетонов | 1989 |
|
SU1726472A1 |
| СПОСОБ ПОЛУЧЕНИЯ НЕСИММЕТРИЧНЫХ 1,2-ДИТИЕНИЛЗАМЕЩЕННЫХ ЦИКЛОПЕНТЕНОВ | 2009 |
|
RU2421453C9 |
| ПРИМЕНЕНИЕ 1-(4-БРОМФЕНИЛ)-4,4,4-ТРИФТОРБУТАН-1,3-ДИОНА В КАЧЕСТВЕ БИОПРОТЕКТОРА В СОСТАВЕ ТЕХНИЧЕСКОЙ СМАЗКИ ОТ ВОЗДЕЙСТВИЯ МИКРОМИЦЕТОВ НА СТАЛЬ | 2022 |
|
RU2815904C2 |
| 3-ФЕНОКСИФЕНИЛСОДЕРЖАЩИЕ 1,3-ДИКЕТОНЫ В КАЧЕСТВЕ ИСХОДНЫХ СОЕДИНЕНИЙ ДЛЯ ПОЛУЧЕНИЯ ИХ ХЕЛАТНЫХ КОМПЛЕКСОВ С ИОНАМИ МЕДИ (II) И СПОСОБ ПОЛУЧЕНИЯ 3-ФЕНОКСИФЕНИЛСОДЕРЖАЩИХ 1,3-ДИКЕТОНОВ | 2012 |
|
RU2475473C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-(3-ФЕНОКСИФЕНИЛ)БУТАН-1,3-ДИОНА | 2013 |
|
RU2529029C1 |
| Способ получения ацилированных дикетоновых соединений | 1985 |
|
SU1697591A3 |
Настоящее изобретение относится к способу получения 4,4,4-трихлор-1-(4-хлорфенил)бутан-1,3-диона, формулы:
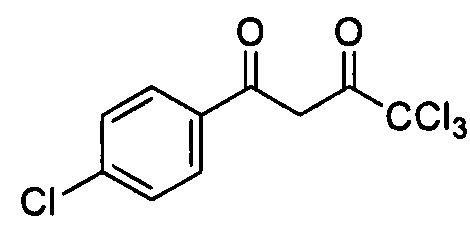 , обладающего противокандидозной активностью. Способ заключается в том, что 4-хлор-1-(1,1-диэтокси)бензол в присутствии пиридина в среде безводного хлороформа охлаждают до 0°С, добавляют при перемешивании раствор хлорангидрида трихлоруксусной кислоты, реакционную смесь выдерживают при перемешивании 10 часов, по истечении времени добавляют 2М серную кислоту и осуществляют перемешивание в течение 2 часов при температуре 80°С, полученную смесь разделяют на делительной воронке, органический слой сушат сульфатом натрия, после удаления растворителя выпавший продукт перекристаллизовывают, с последующим выделением целевого продукта. При этом раствор хлорангидрида трихлоруксусной кислоты перед добавлением в реакционную смесь предварительно охлаждают до 0°С; выдерживание реакционной смеси при перемешивании в течение 10 часов осуществляют при комнатной температуре, без доступа влаги; перед переносом в делительную воронку смесь охлаждают; отделенный органический слой промывают дистиллированной водой до нейтральной реакции среды; фильтрат, полученный после осушения промытого органического слоя над безводным сульфатом натрия, пропускают через слой силикагеля; упаривают; образовавшийся осадок соединения дважды перекристаллизовывают из этанола. Предлагаемый способ позволяет получить целевой продукт, не содержащий нежелательные примеси, с выходом 88%. 4 з.п. ф-лы, 2 табл., 2 пр.
, обладающего противокандидозной активностью. Способ заключается в том, что 4-хлор-1-(1,1-диэтокси)бензол в присутствии пиридина в среде безводного хлороформа охлаждают до 0°С, добавляют при перемешивании раствор хлорангидрида трихлоруксусной кислоты, реакционную смесь выдерживают при перемешивании 10 часов, по истечении времени добавляют 2М серную кислоту и осуществляют перемешивание в течение 2 часов при температуре 80°С, полученную смесь разделяют на делительной воронке, органический слой сушат сульфатом натрия, после удаления растворителя выпавший продукт перекристаллизовывают, с последующим выделением целевого продукта. При этом раствор хлорангидрида трихлоруксусной кислоты перед добавлением в реакционную смесь предварительно охлаждают до 0°С; выдерживание реакционной смеси при перемешивании в течение 10 часов осуществляют при комнатной температуре, без доступа влаги; перед переносом в делительную воронку смесь охлаждают; отделенный органический слой промывают дистиллированной водой до нейтральной реакции среды; фильтрат, полученный после осушения промытого органического слоя над безводным сульфатом натрия, пропускают через слой силикагеля; упаривают; образовавшийся осадок соединения дважды перекристаллизовывают из этанола. Предлагаемый способ позволяет получить целевой продукт, не содержащий нежелательные примеси, с выходом 88%. 4 з.п. ф-лы, 2 табл., 2 пр.
1. Способ получения 4,4,4-трихлор-1-(4-хлорфенил)бутан-1,3-диона, формулы:
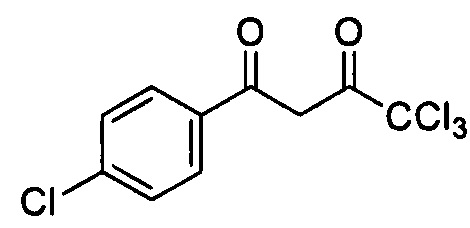
заключающийся в том, что 4-хлор-1-(1,1-диэтокси)бензол в присутствии пиридина в среде безводного хлороформа охлаждают до 0°С, добавляют при перемешивании раствор хлорангидрида трихлоруксусной кислоты, реакционную смесь выдерживают при перемешивании 10 часов, по истечении времени добавляют 2М серную кислоту и осуществляют перемешивание в течение 2 часов при температуре 80°С, полученную смесь разделяют на делительной воронке, органический слой сушат сульфатом натрия, после удаления растворителя выпавший продукт перекристаллизовывают, с последующим выделением целевого продукта, отличающийся тем, что раствор хлорангидрида трихлоруксусной кислоты перед добавлением в реакционную смесь предварительно охлаждают до 0°С; выдерживание реакционной смеси при перемешивании в течение 10 часов осуществляют при комнатной температуре, без доступа влаги; перед переносом в делительную воронку смесь охлаждают; отделенный органический слой промывают дистиллированной водой до нейтральной реакции среды; фильтрат, полученный после осушения промытого органического слоя над безводным сульфатом натрия, пропускают через слой силикагеля; упаривают; образовавшийся осадок соединения дважды перекристаллизовывают из этанола.
2. Способ по п. 1, отличающийся тем, что добавление хлорангидрида трихлоруксусной кислоты осуществляют при интенсивном перемешивании.
3. Способ по п. 2, отличающийся тем, что перемешивание осуществляют на магнитной мешалке.
4. Способ по п. 1, отличающийся тем, что осушение промытого органического слоя над безводным сульфатом натрия осуществляют в течение суток.
5. Способ по п. 1, отличающийся тем, что высушенный органический слой пропускают через слой силикагеля с размерами частиц 70-100 мкм.
| 4,4,4-ТРИХЛОР-1-(4-ХЛОРФЕНИЛ)БУТАН-1,3-ДИОН, ОБЛАДАЮЩИЙ АНАЛЬГЕТИЧЕСКОЙ И ПРОТИВОМИКРОБНОЙ АКТИВНОСТЯМИ | 2015 |
|
RU2582236C1 |
| Н.Ю | |||
| Лисовенко и др | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Вестник Пермского университета, 2016, вып.1(21), стр | |||
| Приспособление для записи звуковых явлений на светочувствительной поверхности | 1919 |
|
SU101A1 |
| A.F.C | |||
| Florens et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Synthetic Communications, 2012, 42(5), 727-737 | |||
| US 3636214 A1, 18.01.1972. | |||

