Настоящее изобретение относится к новым соединениям и способам получения ингибиторов деубиквитилирующих ферментов (DUBs). В частности, настоящее изобретение относится к ингибированию убиквитин-С-концевой гидролазы L1 (UCHL1). Настоящее изобретение также относится к применению ингибиторов DUB для лечения рака и в способах скрининга.
Уровень техники
Указание или обсуждение в настоящем описании очевидно ранее опубликованных документов не должно непременно толковаться как признание таких документов частью уровня техники или общих знаний.
Убиквитин представляет собой небольшой белок, состоящий из 76 аминокислот, который является важным для регуляции функции белков в клетке. Убиквитилирование и деубиквитилирование - это опосредованные ферментами процессы, посредством которых убиквитин ковалентно связывается или отщепляется от целевого белка. Эти процессы задействованы в регуляции большого количества клеточных функций, включая протекание клеточного цикла, апоптоз, модификацию рецепторов на поверхности клетки, регуляцию транскрипции ДНК и восстановление ДНК. Таким образом, система убиквитина участвует в патогенезе многочисленных болезненных состояний, включая воспаление, вирусную инфекцию, метаболическую дисфункцию, расстройства ЦНС и онкогенез (Clague et al., Physiol Rev 93:1289-1315, 2013). Был идентифицирован ряд убиквитиноподобных (Ubls) молекул, которые регулируют функции белков в клетках подобным убиквитину образом.
Убиквитин и подобные убиквитину молекулы отщепляются от белков посредством ферментов, называемых изопептидазами или деубиквитинирующими ферментами (DUBs), и в клетках человека существует приблизительно 95 DUBs, разделяемых на подсемейства на основании гомологии последовательностей: убиквитин-С-концевые гидролазы (UCHs), убиквитинспецифичные протеазы (USPs), протеазы опухолей яичников (OTUs), протеазы домена Мачадо-Джозефа (MJDs), металлопротеазы JAB1/MPN/MOV34 (JAMMs) или сентринспецифичные протеазы (SENPs). DUBs могут перерабатывать убиквитин или убиквитиноподобные аддукты. Ряд DUBs связаны с различными заболеваниями, включая рак, воспаление, нейродегенеративные заболевания и инфекции (Kim КН et al., Curr Pharm Des. 2013; 19(22):4039-52; Nicholson В et al., J Biomol Screen. 2014 Mar 14:19(7); 989-999; Ristic G et al., Front Mol Neurosci. 2014 Aug 19; 7:72; Ashida H et al., Nat Rev Microbiol. 2014 June; 12(6):399-413). Семейство убиквитин-С-концевых гидролаз (UCH), состоящее из UCHL1, UCHL3, UCHL5 и ВАР1, представляет собой цистеинпротеазы, которые действуют через тиольный активный участок. При этом UCHs должны быть задействованы в переработке и рециклинге убиквитина и предпочтительно расщеплять небольшие белковые субстраты. UCHL1 представляет собой белок, состоящий из 223 аминокислот, экспрессия которого в норме ограничена мозгом, периферической нервной системой, яичниками и семенниками у млекопитающих. Однако, как сообщается, UCHL1 повышающе регулируется при некоторых патологических состояниях, включая опухолевые ткани, хроническую обструктивную болезнь легких (ХОБЛ), инсульт, болезнь Паркинсона, болезнь Альцгеймера, невропатическую боль или лизосомные болезни накопления. Кроме того, UCHL1 функционирует как онкоген при прогрессировании большого числа раковых заболеваний, включая рак груди, лимфому, колоректальный рак, остеосаркому, карциному поджелудочной железы и немелкоклеточную карциному легких, и находится в обратной корреляции с выживанием пациента (Hurst-Kennedy et al., Biochem Res Int, 2012; Hussain et al., Leukemia 24:1641-1655, 2010). Фармакологическое ингибирование UCHL1 могло бы, соответственно, стать новым способом лечения патологий, таких как раковые заболевания.
Убиквитин-протеасомная система привлекла к себе внимание в качестве мишени для лечения рака после одобрения ингибитора протеасом бортезомиба (Велкейд (Velcade®)) для лечения множественной миеломы. Продолжительность лечения бортезомибом ограничена его токсичностью и устойчивостью к данному лекарственному средству. Тем не менее, предполагается, что терапевтические стратегии, которые имеют своей целью конкретные аспекты убиквитин-протеасомного пути выше по пути относительно протеасом, такие как DUBs, обеспечивают лучшую переносимость (Bedford et al., Nature Rev 10:29-46, 2011). На сегодняшний день, несмотря на имеющиеся отдельные публикации об ингибиторах (обзор см. в источнике: Lill and Wertz, Trends in Pharmaceutical Sciences, 35 (4), 2014), отсутствуют сообщения об ингибиторах DUB, успешно внедренных в клиническую практику. Таким образом, существует потребность в соединениях и фармацевтических композициях для ингибирования DUBs, таких как UCHL1, USP6 или USP30, для лечения рака или других состояний, при которых наблюдается активность DUB. Альтернативные DUBs, которые также были предложены в качестве потенциальных терапевтических агентов для лечения рака, включают USP1, USP2, USP4, USP6, USP7, USP8, USP9x, USP10, USP11, USP13, USP14, USP17 и USP28. Также показания, при которых ингибирование DUB может оказаться предпочтительным, включают расстройства ЦНС (например, USP30, USP14) или воспаление (например, А20, CYLD).
Краткое описание изобретения
В соответствии с первым аспектом настоящего изобретения предложено соединение формулы (I):
Соединение, имеющее формулу (I)
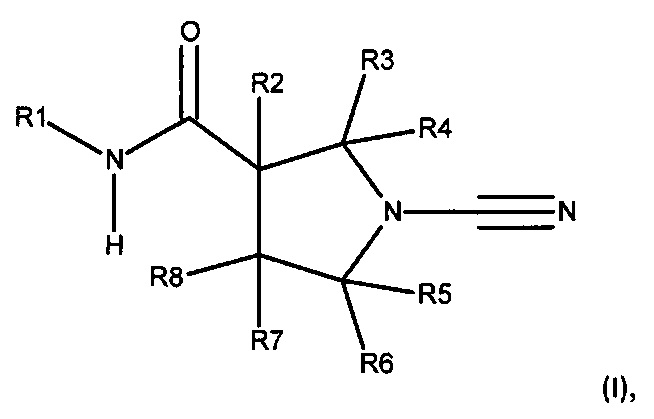
или его фармацевтически приемлемая соль, где:
R1 представляет собой 5-10-членное гетероарильное кольцо, которое может быть необязательно замещенным; и
каждый из R2, R3, R4, R5, R6 и R8 независимо представляет собой атом водорода, циано, необязательно замещенный C1-С6 алкил, необязательно замещенную C1-С6 алкоксигруппу, одну или более спироциклических групп, где R3 связан с R4, R5 связан с R6, или R8 связан с R7, или R2 связан с R8 с образованием необязательно замещенного С3-С4 циклоалкильного кольца, или R6 связан с R7 с образованием необязательно замещенного С3-С4 циклоалкильного кольца; и
R7 представляет собой атом водорода, атом фтора, циано, необязательно замещенный C1-С3 алкил, необязательно замещенную C1-С3 алкоксигруппу, или необязательно замещенный арил, или 5-6-членное гетероарильное кольцо, или связан с R8 с образованием спироциклической группы, или связан с R6 с образованием необязательно замещенного С3-С4 циклоалкильного кольца.
R1 может представлять собой 5-10-членное гетероарильное кольцо, замещенное одним или более из Q1-(R9)n, где:
n равен 0 или 1
Q1 представляет собой атом водорода, атом галогена, циано, ковалентную связь, -NR10-, -CONR10-, -NR10CO-, атом кислорода, оксо, нитро, -S(O)m-, C1-С6 алкокси, C1-С6 галогеналкокси, C1-С6 гидроксиалкил, -СО-, -SO2R11, -NR11R12, -NR11COR12, -NR10CONR11R12, -CONR11R12, -CO2 R11, -NR11CO2 R12, -SO2NR11R12, -CONR11, -C(O)R11, -NR11SO2R12, NR11SO2NR13R14 и SO2NR11 или необязательно замещенный C1-С6 алкилен, -С2-С6 алкенилен или -C1-С6 алкильную группу;
m равен 0, 1 или 2;
каждый из R10, R11 и R12 независимо представляет собой атом водорода, или необязательно замещенный C1-С6 алкил, или необязательно замещенную C1-С6 алкиленовую группу.
Когда n равен 1, R9 представляет собой необязательно замещенный 3-10-членный гетероциклил, гетероарил, арил или циклоалкильное кольцо (когда n равен 0, Q присутствует и R9 отсутствует).
R9 может быть необязательно замещен одним или более заместителями, выбранными из галогена, необязательно замещенного C1-С6 галогеналкила, необязательно замещенного C1-С6 алкокси, C1-С6 галогеналкокси, необязательно замещенного C1-С6 алкила, необязательно замещенного С2-С6 алкенила, необязательно замещенного С2-С6 алкинила, C1-С6 гидроксиалкила, оксо, циано, нитро, необязательно замещенного гетероциклина, необязательно замещенного циклоалкила, необязательно замещенного гетероарила, необязательно замещенного арила, -Q2-NR13CONR14R15, -Q2-NR13R14, -Q2-NR13COR14, -Q2-COR13, -Q2-SO2R13, -Q2-CONR13, Q2-CONR13R14, -Q2-CO2R13, -Q2-SO2NR13R14, -Q2-NR13SO2R14 и -Q2-NR13SO2NR14R15; где
Q2 представляет собой ковалентную связь, или C1-С6 алкилен, или С2-С6 алкениленовую группу; и
каждый из R13, R14 и R15 независимо представляет собой водород, или необязательно замещенный C1-С6 алкил, или необязательно замещенный гетероциклил, или необязательно замещенный гетероарил, или необязательно замещенный арил, или необязательно замещенный циклоалкил.
Краткое описание чертежей
На фигуре 1 представлено изображение FLAG-UCHL1, очищенной от клеток млекопитающих. FLAG-очишенный белок или указанные концентрации бычьего сывороточного альбумина выделяли посредством электрофореза в полиакриламидном геле с применением додецилсульфата натрия (SDS-PAGE) и окрашивали красителем Imperial Protein Stain (Pierce Biotechnology).
Фигура 2 представляет собой график, на котором представлена протеолитическая активность очищенной FLAG-UCHL1, определенная с применением флуоресцентного поляризационного анализа. Различные указанные объемы очищенной UCHL1 инкубировали с TAMRA-меченным пептидом, связанным с убиквитином посредством изопептидной связи.
Подробное описание настоящего изобретения
Когда указывается, что какая-либо группа соединений формулы (I) является необязательно замещенной, такая группа может быть замещенной или незамещенной. Замещение может осуществляться одним или более конкретными заместителями, которые могут быть одинаковыми или различными. Будет очевидно, что количество и природа заместителей будут выбраны таким образом, чтобы избежать каких-либо пространственно нежелательных комбинаций.
В контексте настоящего описания, если не указано иное, алкильная, алкенильная или алкинильная группа заместителя или алкильный, алкенильный фрагмент в группе заместителя могут быть линейными или разветвленными. Алкильные и алкенильные цепи также могут содержать внутри цепи гетероатомы, такие как кислород.
Сх-Су алкил относится к насыщенной алифатической углеводородной группе, содержащей х-у атомов углерода, которая может быть линейной или разветвленной. Например, С1-С6 алкил содержит от 1 до 6 атомов углерода. «Разветвленный» означает, что в группе присутствует по меньшей мере одно место ответвления от атома углерода. Например, трет-бутил и изопропил представляют собой разветвленные группы. Примеры C1-С6 алкильных групп включают метил, этил, пропил, 2-метил-1-пропил, 2-метил-2-пропил, 2-метил-1-бутил, 3-метил-1-бутил, 2-метил-3-бутил, 2,2-диметил-1-пропил, 2-метилпентил, 3-метил-1-пентил, 4-метил-1-пентил, 2-метил-2-пентил, 3-метил-2-пентил, 4-метил-2-пентил, 2,2-диметил-1-бутил, 3,3-диметил-1-бутил, 2-этил-1-бутил, н-бутил, изобутил, трет-бутил, н-пентил, изопентил, неопентил и н-гексил.
Сх-Су алкиленовая группа или фрагмент могут быть линейными или разветвленными и они относятся к двухвалентной углеводородной группе, содержащей на один меньше атомов водорода, чем Сх-Су алкил, определение которого представлено выше. Примеры C1-С6 алкиленовой группы включают метилен, этилен, н-пропилен, н-бутилен, метилметилен и диметилметилен.
С2-С6 алкенил относится к радикалу с линейной или разветвленной углеводородной цепью, содержащему по меньшей мере два атома углерода и по меньшей мере одну двойную связь. Примеры алкенильной группы включают этенил, пропенил, 2-пропенил, 1-бутенил, 2-бутенил, 1-гексенил, 2-метил-1-пропенил, 1,2-бутадиенил, 1,3-пентадиенил, 1,4-пентадиенил и 1-гексадиенил.
С2-С6 алкинил относится к радикалу с линейной или разветвленной углеводородной цепью, содержащему по меньшей мере два атома углерода и по меньшей мере одну тройную связь. Примеры алкинильных групп включают этинил, пропинил, 2-пропинил, 1-бутинил, 2-бутинил и 1-гексинил.
С1-С6 алкокси относится к группе или части группы, содержащей -O-Сх-Су алкильную группу согласно определению Сх-Су алкила, приведенному выше. Примеры C1-С6алкокси включают метокси, этокси, пропокси, изопропокси, бутокси, пентокси и гексокси.
С1-С6 галогеналкил и С1-С6 галогеналкокси относятся к Сх-Су алкильной группе, определение которой приведено выше, в которой по меньшей мере один атом водорода заменен атомом галогена. Примеры C1-С6 галогеналкильных групп включают фторметил, дифторметил, трифторметил, трифторэтил, пентафторэтил, фторметокси, дифторметокси и трифторметокси.
С1-С6 гидроксиалкил относится к Сх-Су алкильной группе, определение которой приведено выше, в которой по меньшей мере один атом водорода заменен гидрокси (-ОН) группой. Примеры гидрокси-С1-6 алкильных групп включают гидроксиметил, гидроксиэтил, дигидроксиэтил, гидроксипропил и гидроксиизопропил.
Термин «галоген» или «гало» относится к атомам хлора, брома, фтора или иода.
Во избежание сомнений следует понимать, что 3-10-членное гетероарильное, арильное, циклоалкильное или гетероциклическое кольцо, определенное в соответствии с R1 или R9, не включает каких-либо нестабильных кольцевых структур или каких-либо связей О-О, O-S или S-S и что заместитель, в случае его наличия, может быть присоединен к любому подходящему атому кольца, который может представлять собой атом углерода или гетероатом. Замещение в кольце может также включать изменение атома кольца в положении замещения. Например, замещение по фенильному кольцу может включать изменение атома кольца в положении замещения с углерода на азот с получением пиридинового кольца. Указанное 3-10-членное гетероарильное, арильное, циклоалкильное или гетероциклическое кольцо может быть моноциклическим или бициклическим.
«Сх-Су циклоалкил» относится к циклической неароматической углеводородной группе, содержащей х-у атомов углерода. Например, С3-C8 циклоалкил относится к углеводородному кольцу, содержащему 3-8 атомов углерода. Примерами С3-С8 циклоалкила являются циклопропил, циклобутил, циклопентил, циклогексил, циклопентил, циклогексил, циклогептил и циклооктил.
«Арильная» группа/фрагмент относится к любой моноциклической или бициклической углеводородной группе, содержащей по меньшей мере одну ароматическую группу, например, содержащую вплоть до 12 атомов углерода в качестве членов колец. Примеры арильных групп включают фенил, нафтил, тетрагидронафтил и бифенил.
«Гетероарильные» группы могут быть моноциклическими или бициклическими. Бициклические кольца могут представлять собой конденсированные ароматические кольца, где оба кольца являются ароматическими или могут представлять собой конденсированные кольца, где одно из колец не является ароматическим. В случае R1 кольцо, присоединенное к амидному азоту, представляет собой ароматическое кольцо, которое может быть конденсировано с другим ароматическим или неароматическим кольцом. Гетероарильные кольца содержат 1, 2 или 3 гетероатома, выбранных из кислорода, серы и азота. Когда гетероатом представляет собой азот, он может быть окислен. Примеры гетероарильных групп включают пиридинил, пиразинил, пиримидинил, пиридазинил, фурил, тиофенил, пирролил, оксазолил, тиазолил, пиразолил, триазолил, тетразолил, индолил, индолизинил, изоиндолил, индолинил, пуринил, фуразанил, имидазолил, индазолил, изотиазолил, изоксазолил, оксадиазолил, оксазинанил, тетразолил, тиадиазолил, бензофуранил, изобензофуранил, бензотиофенил, изобензотиофенил, бензимидазолил, бензотиазолил, нафтиридинил, птеридинил, пиразинил, 4Н-хинолизинил, хинолинил, изохинолинил, циннолинил, фталазинил, хиназолинил, имидазопиридинил, пиразолопиридинил, тиазолопиридинил, индолинил, изоиндолинил, триазинил, пиридазинил и хиноксалинил.
«Гетероциклильные» группы также могут быть моноциклическими или могут содержать 2 или более конденсированных колец, которые могут быть насыщенными или частично ненасыщенными и содержать 1, 2 или 3 гетероатома, выбранных из кислорода, серы и азота. Примеры гетероциклильных групп включают азетидинил, пирролидинил, пиперидинил, азепанил, диазепанил, дигидрофуранил (например, 2,3-дигидрофуранил, 2,5-дигидрофуранил), 4,5-дигидро-1Н-малеимидо, диоксоланил, морфолинил, оксазолидинил, пиперазинил, тетрагидрофуранил, тиоморфолинил, дигидропиранил (например, 3,4-дигидропиранил, 3,6-дигидропиранил), диоксанил, гексагидропиримидинил, пиразолинил, пиразолидинил, пиридазинил, 4Н-хинолизинил, хинуклинил, тетрагидропиранил, тетрагидропиридинил, тетрагидропиримидинил, тетрагидротиофенил, тетраметиленсульфоксид, тиазолидинил, гидантоинил, бензопиранил, тетрагидротиазолопиридинил, тетрагидрохинолинил, тетрагидроизохинолинил, тетрагидропиразолопиразинил и тетрагидротиазолоазепинил.
Термин «необязательно замещенный» в отношении любой группы означает, что указанная группа может, если необходимо, быть замещенной одним или более заместителями, которые могут быть одинаковыми или различными. Необязательные заместители в определениях R2, R3, R4, R5, R6 и R8 могут включать галоген, дейтеро, С1-3 алкил, С1-3 алкокси, циано, амино, нитро или SF5 (известный миметик NO2). Примеры подходящих заместителей для всех остальных «замещенных» и «необязательно замещенных» фрагментов, включая кольца R1 и R9, могут включать галоген, дейтеро, С1-3 алкил, гидрокси, С1-3 алкокси, циано, амино, нитро, оксо, арил, гетероарил, гетероциклил, С3-С6 циклоалкил, С1-3 алкиламино, С2-6 алкениламино, ди-С1-3 алкиламино, С1-3 ациламино, ди-С1-3 ациламино, карбокси, С1-3 алкоксикарбонил, карбамоил, моно-С1-3 карбамоил, ди-С1-3 карбамоил или любой из вышеуказанных фрагментов, в которых углеводородный фрагмент сам замещен галогеном. В группах, содержащих атом кислорода, таких как гидрокси и алкокси, указанный атом кислорода может быть заменен серой с получением групп, таких как тио (SH) и тиоалкил (S-алкил). Необязательные заместители, таким образом, включают группы, такие как S-метил. В тиоалкильных группах атом серы может быть дополнительно окислен с получением сульфоксида или сульфона, и, таким образом, необязательные заместители, соответственно, включают группы, такие как S(O)-алкил и S(O)2-алкил.
Замещенные группы, таким образом, включают, например, CN, CFH2, CF2H, CF3, OCF3, CH2NH2, СН2ОН, CH2CN, CH2SCH3, СН2ОСН3, ОМе, OEt, OPr, Me, Et, t-Bu, -ОСН2О-, СО2Ме, С(O)Ме, i-Pr, SCF3, SO2Me, NMe2 и т.п. В случае арильных групп замещения может быть в форме колец от соседних атомов углерода в арильном кольце, примерами являются циклические ацетали, такие как О-CH2-О.
Термин «лечить», или «осуществление лечения», или «лечение» включает профилактику и означает подавление, облегчение симптомов, устранение причины симптомов либо временно, либо постоянно, или предотвращение или задержку возникновения симптомов указанного расстройства или состояния. Соединения согласно настоящему изобретению являются подходящими для применения для лечения людей и отличных от человека животных.
Доза соединения представляет собой количество, эффективное для предотвращения возникновения симптомов расстройства или для лечения некоторых симптомов расстройства, которым страдает пациент. Под «эффективным количеством», или «терапевтически эффективным количеством», или «эффективной дозой» понимают количество, достаточное для достижения необходимых фармакологических или терапевтических эффектов с обеспечением, тем самым, эффективного предотвращения или лечения расстройства. Предотвращение расстройства проявляется в задержке наступления симптомов расстройства до значимой с медицинской точки зрения степени. Лечение расстройства проявляется в уменьшении симптомов, связанных с расстройством, или подавлении повторного возникновения симптомов расстройства.
Фармацевтически приемлемые соли соединений согласно настоящему изобретению включают, но не ограничиваются ими, соли присоединения (например, фосфаты, нитраты, сульфаты, бораты, ацетаты, малеаты, цитраты, фумараты, сукцинаты, метансульфонаты, бензоаты, салицилаты и гидрогалогениды), соли полученные из неорганических оснований (такие как соли лития, калия и натрия), соли аминокислот (таких как глицин, аланин, валин, лейцин, изолейцин, цистеин, метионин и пролин), органических оснований (таких как триэтиламин, гидроксид, холин, тиамин и N-N'-диацетилэтилендиамин). Другие фармацевтически приемлемые соли включают соли аммония, соли замещенного аммония и соли алюминия. Другие фармацевтически приемлемые соли включают соли четвертичного аммония и соединений формулы (I).
Общие способы получения солей хорошо известны специалисту в данной области техники. Такие соли могут быть образованы стандартными способами, например, путем реакции свободной кислоты или свободного основания соединения с одним или более эквивалентами соответствующей кислоты или основания, необязательно в растворителе или в среде, в которой соль нерастворима, с последующим удалением указанного растворителя или указанной среды с применением стандартных методик (например, под вакуумом, путем сублимационной сушки или путем фильтрования). Соли также могут быть получены путем обмера противоиона соединения в форме соли на другой противоион, например, с применением подходящей ионообменной смолы.
В тех случаях, когда соединения согласно настоящему изобретению существуют в различных энантиомерных и/или диастереоизомерных формах, настоящее изобретение относится к таким соединениям, полученным в виде изомерных смесей или рацематов, присутствующих в оптически чистой форме или в виде смесей с другими изомерами. Энантиомеры различаются исключительно по их способности вращать плоскость плоскополяризованного света в равных количествах в противоположных направлениях и обозначаются как (+) / (S) или (-) / (R) формы, соответственно. Отдельные энантиомеры или изомеры могут быть получены способами, известными в данной области техники, такими как оптическое разделение продуктов или промежуточных соединений (например, хиральное хроматографическое разделение, например, хиральная ВЭЖХ, или с помощью энантиомерного синтеза). Аналогичным образом, когда соединения согласно настоящему изобретению существуют в виде альтернативных таутомерных форм, например, кето/енол, амид/имидная кислота, настоящее изобретение относится к индивидуальным таутомерам в выделенном виде и к смесям таутомеров во всех пропорциях.
В настоящее описание включено соединение в соответствии с формулой (II):
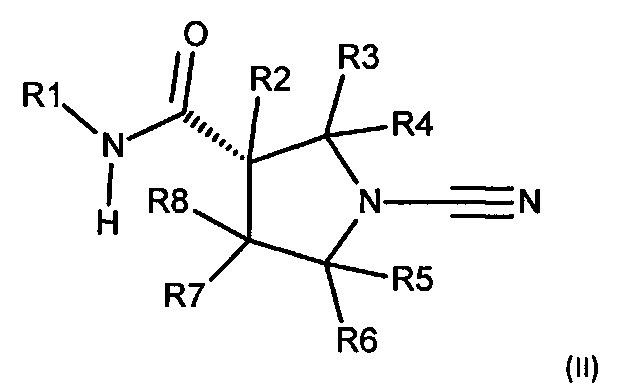
или его фармацевтически приемлемая соль, где:
R1 представляет собой 5-10-членное гетероарильное кольцо, которое может быть необязательно замещенным; и
каждый из R2, R3, R4, R5, R6 и R8 независимо представляет собой атом водорода, циано, необязательно замещенный C1-С6 алкил, необязательно замещенную C1-С6 алкоксигруппу, одну или более спироциклических групп, где R3 связан с R4, R5 связан с R6, или R8 связан с R7, или R2 связан с R8 с образованием необязательно замещенного С3-С4 циклоалкильного кольца, или R6 связан с R7 с образованием необязательно замещенного С3-С4 циклоалкильного кольца; и
R7 представляет собой атом водорода, атом фтора, циано, необязательно замещенный С1-С3 алкил, необязательно замещенную C1-С3 алкоксигруппу или необязательно замещенное арильное или гетероарильное кольцо, или связан с R8 с образованием спироциклической группы, или связан с R6 с образованием необязательно замещенного С3-С4 циклоалкильного кольца.
В одном из вариантов реализации R1 представляет собой 5-10-членное гетероарильное кольцо, которое может быть необязательно замещенным; и
каждый из R2, R3, R4, R5, R6 и R8 независимо представляет собой атом водорода, циано, необязательно замещенный C1-С3 алкил, необязательно замещенную С1-С3 алкоксигруппу, одну или более спироциклических групп, где R3 связан с R4, R5 связан с R6, или R8 связан с R7, или R2 связан с R8 с образованием С3-С4 циклоалкильного кольца; и
R7 представляет собой атом водорода, циано, необязательно замещенный C1-С3 алкил, необязательно замещенную C1-С3 алкоксигруппу или необязательно замещенное арильное или гетероарильное кольцо или связан с R8 с образованием спироциклической группы.
Когда R3 и R4 различны, группа R3 может быть цис или транс в отношении амида. Соединения предпочтительно представляют собой цис, когда R4 представляет собой Н. Когда R3 = Me и R2 = R4 = Н, соединение может быть представлено формулой (III):
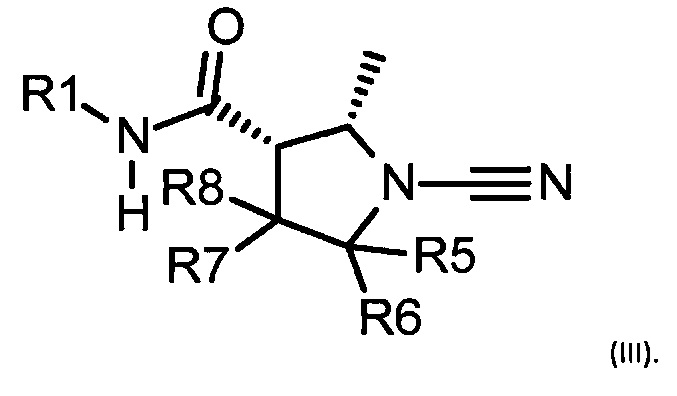
Изотопы
Соединения, описанные в настоящем документе, могут содержать одну или более изотопных замен, и ссылка на конкретный элемент охватывает все изотопы элемента. Например, ссылка на водород включает в себя 1Н, 2Н (D), и 3Н (Т). Аналогичным образом, ссылки на углерод и кислород охватывают, соответственно, 12С, 13С и 14С и 16O и 18O. Примеры изотопов включают 2Н, 3Н, 11С, 13С, 14С, 36Cl, 18F, 123I, 125I, 13N, 15N, 15O, 17O, 18O, 32P и 35S.
Аналогичным способом, ссылка на конкретную функциональную группу также включает в себя изотопные вариации, если контекстом не предусмотрено иное. Например, ссылка на алкильную группу, такую как этильная группа, также охватывает варианты, в которых один или более атомов водорода в группе находятся в форме дейтериевого или тритиевого изотопа, примером является этильная группа, в которой все пять атомов водорода находятся в дейтериевой изотопной форме (пердейтероэтильная группа).
Изотопы могут быть радиоактивными или нерадиоактивными. В одном из вариантов реализации соединения не содержат радиоактивных изотопов. Такие соединения являются предпочтительными для применения в терапии. Тем не менее, в другом варианте реализации соединения могут содержать один или более радиоактивных изотопов. Соединения, содержащие такие радиоактивные изотопы могут быть подходящими для применения в контексте диагностики.
Некоторые изотопно-меченные соединения формулы (I), например, соединения, содержащие радиоактивный изотоп, являются подходящие для применения в исследованиях лекарственных средств и/или распределения субстрата в тканях. Радиоактивные изотопы, такие как 3Н и 14С, являются особенно подходящими для данной цели ввиду легкости их включения и наличия готовых средств детектирования. Замена более тяжелыми изотопами, такими как 2Н, может обеспечить некоторые терапевтические преимущества, являющиеся следствием более высокой стабильности к метаболизму, например, повышенное время полувыведения in vivo или сниженные требования к дозировкам, и, следовательно, может быть предпочтительной в некоторых случаях. Замена позитрон-излучающими изотопами, такими как 11C, 18F, 15O и 13N, может быть подходящей для применения в исследованиях посредством позитронно-эмиссионной томографии (ПЭТ) для оценки занятости рецепторов. Изотопно-меченные соединения формулы (I) могут быть в целом получены традиционными способами, известными в данной области техники, или посредством процессов, аналогичных описанных в сопроводительных примерах и способах получения с применением соответствующего изотопно-меченного реагента вместо ранее применяемого немеченного реагента.
Кристаллические и аморфные формы
Соединения формулы (I) могут существовать в кристаллической или аморфной форме, и некоторые из кристаллических форм могут существовать в качестве полиморфов, которые включены в объем настоящего изобретения. Полиморфные формы соединений формулы (I) могут быть охарактеризованы и их можно отличить, применяя ряд обычных аналитических методик, включая, но не ограничиваясь ими, инфракрасную спектроскопию, рамановскую спектроскопию, рентгеновскую порошковую дифракцию, дифференциальную сканирующую калориметрию, термогравиметрический анализ и твердофазный ядерный магнитный резонанс.
Соответственно, в других вариантах реализации настоящего изобретения предложено соединение в соответствии с любыми описанными вариантами реализации в кристаллической форме. Соединение может быть на 50%-100% кристаллическим, и в частности по меньшей мере на 50% кристаллическим, или по меньшей мере на 60% кристаллическим, или по меньшей мере на 70% кристаллическим, или по меньшей мере на 80% кристаллическим, или по меньшей мере на 90% кристаллическим, или по меньшей мере на 95% кристаллическим, или по меньшей мере на 98% кристаллическим, или по меньшей мере на 99% кристаллическим, или по меньшей мере на 99,5% кристаллическим, или по меньшей мере на 99,9% кристаллическим, например 100% кристаллическим. Соединение может, в качестве альтернативы, находиться в аморфной форме.
Настоящее изобретение, описанное в настоящем документе, относится ко всем кристаллическим формам, сольватам и гидратам любых раскрытых соединений, неважно каким образом полученным. В тех случаях, когда любые соединения, раскрытые в настоящем описании, имеют кислотные или основные центры, такие как карбоксилаты или аминогруппы, в настоящее описание включены все солевые формы таких соединений. В случае фармацевтического применения должно быть показано, что соль представляет собой фармацевтически приемлемую соль.
Настоящее изобретение относится к любым сольватам соединений и их солям. Предпочтительные сольваты представляют собой сольваты, образованные путем включения в твердую структуру (например, кристаллическую структуру) соединений согласно настоящему изобретению молекул нетоксичного фармацевтически приемлемого растворителя (называемого далее сольватирующим растворителем). Примеры таких растворителей включают воду, спирты (такие как этанол, изопропанол и бутанол) и диметилсульфоксид. Сольваты могут быть получены путем перекристаллизации соединений согласно настоящему изобретению с растворителем или смесью растворителей, содержащей сольватирующий растворитель. Образуется ли сольват в любом конкретном случае может быть определено путем анализа кристаллов соединения с применением хорошо известных и стандартных методик, таких как термогравиметрический анализ (ТГА), дифференциальная сканирующая калориметрия (ДСК) и рентгеновская кристаллография.
Сольваты могут представлять собой стехиометрические и нестехиометрические сольваты. Отдельные сольваты могут представлять собой гидраты, и примеры гидратов включают полугидраты, моногидраты и дигидраты. Более подробное обсуждение сольватов и способов, применяемых для их получения и определения их характеристик, см. в источнике Bryn et al., Solid-State Chemistry of Drugs, Second Edition, published by SSCI, Inc of West Lafayette, IN, USA, 1999, ISBN 0-967-06710-3.
Настоящее изобретение относится к фармацевтически функциональным производным соединений, определенных в настоящем описании, включая сложноэфирные производные и/или производные, которые имеют или обеспечивают ту же биологическую функцию и/или активность, что и соответствующее соединение согласно настоящему изобретению. Таким образом, для целей настоящего изобретения данный термин также включает пролекарства соединений, определенных в настоящем описании.
Термин «пролекарство» соответствующего соединения включает любое соединение, которое после перорального или парентерального введения метаболизируется in vivo с получением соответствующего соединения в экспериментально детектируемом количестве и в течение определенного времени (например, в течение интервала дозирования от 6 до 24 часов (то есть при введении один-четыре раза в сутки).
Пролекарства соединений могут быть получены путем модификации функциональных групп, присутствующих в соединении таким образом, что эти модификации могут быть отщеплены in vivo, когда такое пролекарство вводят субъекту, представляющему собой млекопитающее. Такие модификации, как правило, получают путем синтеза исходного соединения с пролекарственным заместителем. Пролекарства включают соединения, в которых гидроксильная, амино, сульфгидрильная, карбоксильная или карбонильная группа в соединении связана с любой группой, которая может быть отщеплена in vivo для регенерации свободной гидроксильной, амино, сульфгидрильной, карбоксильной или карбонильной группы, соответственно.
Примеры пролекарств включают, но не ограничиваются ими, сложные эфиры и карбаматы гидроксильных функциональных групп, сложноэфирных групп карбоксильных функциональных групп, N-ацильных производных и N-оснований Манниха. Общая информация о пролекарствах может быть найдена, например, в источнике Bundegaard, Н. "Design of Prodrugs" p. 1-92, Elsevier, New York-Oxford (1985).
Соединения согласно настоящему изобретению могут метаболизироваться in vivo. Метаболиты соединений формулы (I) также входят в объем настоящего изобретения. Термин «метаболиты» относится ко всем молекулам, полученным из любых соединений в соответствии с настоящим изобретением в клетке или организме, предпочтительно, млекопитающего. Предпочтительно указанный термин относится к молекулам, которые отличаются от любой молекулы, присутствующей в любой такой клетке или организме в физиологических условиях.
Лечение, определенное в настоящем описании, может быть применено в качестве единственной терапии или может включать, помимо соединений согласно настоящему изобретению, традиционное хирургическое лечение, или радиотерапию, или химиотерапию. Кроме того, соединения формулы (I) также могут быть применены в комбинации с существующими терапевтическими агентами для лечения рака, включая терапевтические агенты, представляющие собой малые молекулы, или терапевтические агенты на основе антител.
Настоящее описание включает соединение, имеющее формулу (I):
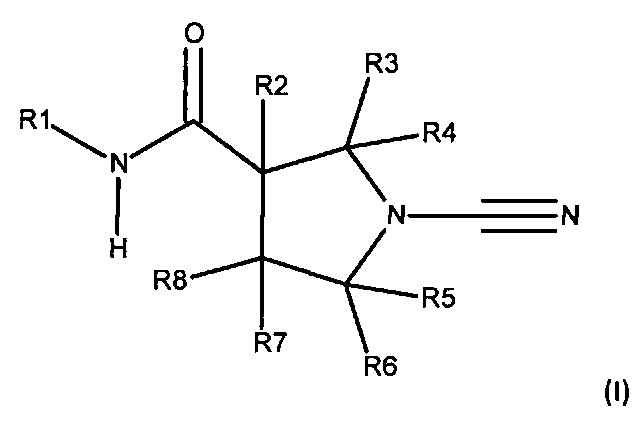
или его фармацевтически приемлемую соль, где:
R1 представляет собой 5-10-членное гетероарильное кольцо, которое может быть необязательно замещенным;
каждый из R2, R3, R4, R5, R6 и R8 независимо представляет собой атом водорода, циано, необязательно замещенный C1-С6 алкил, необязательно замещенную C1-С6 алкоксигруппу, одну или более спироциклических групп, где R3 связан с R4, R5 связан с R6, или R8 связан с R7, или R2 связан с R8 с образованием необязательно замещенного С3-С4 циклоалкильного кольца, R6 связан с R7 с образованием необязательно замещенного С3-С4 циклоалкильного кольца; и
R7 представляет собой атом водорода, атом фтора, циано, необязательно замещенный C1-С3 алкил, необязательно замещенную C1-С3 алкоксигруппу или необязательно замещенное арильное или гетероарильное кольцо, или связан с R8 с образованием спироциклической группы, или связан с R6 с образованием необязательно замещенного С3-С4 циклоалкильного кольца.
В одном из вариантов реализации R1 представляет собой 5-10-членное гетероарильное кольцо, которое может быть необязательно замещенным;
каждый из R2, R3, R4, R5, R6 и R8 независимо представляет собой атом водорода, циано, необязательно замещенный C1-С3 алкил, необязательно замещенную С1-С3 алкоксигруппу, одну или более спироциклических групп, где R3 связан с R4, R5 связан с R6, или R8 связан с R7, или R2 связан с R8 с образованием С3-С4 циклоалкильного кольца; и
R7 представляет собой атом водорода, циано, необязательно замещенный C1-С3 алкил, необязательно замещенную C1-С3 алкоксигруппу или необязательно замещенное арильное или гетероарильное кольцо или связан с R8 с образованием спироциклической группы.
В формуле (I), определение которой представлено в настоящем описании, гетероарильное кольцо R1 может быть присоединено непосредственно к амидному атому азота с образованием N-арильной связи. Арильное кольцо может быть моноциклическим или бициклическим. Если кольцо является бициклическим, второе кольцо может быть ароматическим или может быть частично насыщенным, и, таким образом, не каждый атом в 5-10-членном гетероарильном кольце должен входить в арильную систему, должно быть по меньшей мере одно гетероарильное кольцо на 5-10 атомов.
В одном из вариантов реализации R1 представляет собой 5-10-членное (например, 5, 6, 7, 8, 9 или 10-членное) гетероарильное кольцо, которое может быть необязательно замещено одним или более (например, одним, двумя, тремя или четырьмя) из -Q1-(R9)n.
В другом варианте реализации R1 представляет собой 5- или 6-членное гетероарильное кольцо, которое может быть необязательно замещено одним или более (например, одним, двумя, тремя или четырьмя) из -Q1-(R9)n.
В дополнительном варианте реализации R1 представляет собой 9-членное бициклическое гетероарильное кольцо, которое может быть необязательно замещено одним или более (например, одним, двумя, тремя или четырьмя) из -Q1-(R9)n.
Гетероарильное кольцо R1 может быть моноциклическим или бициклическим и содержать один или более (например, 1, 2 или 3) гетероатомов, независимо выбранных из азота, кислорода и серы.
В одном из вариантов реализации необязательно замещенное 5-10-членное гетероарильное кольцо R1 выбрано из тиазолила, пиридинила, изоксазолила, тиадиазолила, индазолила, имидазолила, бензимидазолила, бензотиазолила, пиразолила, пиридазинила, пиримидинила, нафтиридинила, изохинолинила, пиразинила, тетрагидротиазолопиридинила, имидазопиридинила, триазолила, пиразолопиридинила, тетрагидропиразолопиразинила, тетрагидротиазолоазепинила, тиазолопиридинила.
Указанное 5-10-членное гетероарильное кольцо R1 может быть выбрано из тиазолила, пиридинила, изоксазолила, тиадиазолила, индазолила, имидазолила, бензимидазолила, бензотиазолила, пиразолила, пиридазинила, пиримидинила, нафтиридинила, изохинолинила, пиразинила, тетрагидротиазолопиридинила, имидазопиридинила, триазолила и пиразолопиридинила.
В дополнительном варианте реализации 5-10-членное гетероарильное кольцо R1 выбрано из тиазолила, имидазолила, бензотиазолила, пиразолила, имидазопиридинила и тетрагидротиазолопиридинила, пиридинила, тетрагидропиразолопиразинила, пиримидинила, изохинолинила, пиразолопиридинила, пиридазинила, тетрагидротиазолоазептина, фенила, тиазолопиридинила и индазолила.
Указанное 5-10-членное гетероарильное кольцо R1 может быть выбрано из тиазолила, имидазолила, бензотиазолила, пиразолила, имидазопиридинила и тетрагидротиазолопиридинила.
В дополнительном варианте реализации 5-10-членное гетероарильное кольцо R1 выбрано из тиазолила и имидазолила.
Типичные примеры 5-10-членного гетероарильного кольца R1 включают тиазол-2-ил, тиазол-5-ил, пиридин-2-ил, пиридин-3-ил, тиадиазол-2-ил, изоксазол-5-ил, индазол-5-ил, бензоимидазол-5-ил, бензотиазол-2-ил, пиразол-5-ил, пиразол-3-ил, пиримидин-5-ил, пиримидин-4-ил пиридазин-3-ил, имидазол-4-ил, 1,8-нафтиридин-2-ил, имидазо[1,2-а]пиридин-2-ил, имидазо[1,2-а]пиридин-6-ил, имидазо[1,2-а]пиридин-5-ил, пиразин-2-ил, тиазоло[5,4-с]пиридин-2-ил, имидазо-4-ил, 1,2,3-триазол-4-ил, 4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил, 5,6,7,8-тетрагидро-4Н-тиазоло[4,5-d]азепин-2-ил, тиазоло[4,5-b]пиридин-2-ил, тиазоло[4,5-с]пиридин-2-ил, тетрагидропиразоло[1,5-а]пиразин-2-ил, пиразоло[1,5-а]пиридин-2-ил, хинолин-3-ил, изохинолин-3-ил, бензо[d]тиазол-2-ил.
Группа R1 может быть дополнительно замещенной. R1 может представлять собой 5-10-членное гетероарильное кольцо, замещенное одним или более из Q1-(R9)n, где
n равен 0 или 1;
Q1 представляет собой атом водорода, атом галогена, циано, ковалентную связь, -NR10-, -CONR10-, -NR10CO-, атом кислорода, оксо, нитро, -S(O)m-, C1-С6 алкокси, C1-С6 галогеналкокси, C1-С6 гидроксиалкил, -СО-, -SO2R11, -NR11R12, -NR11COR12, -NR10CONR11R12, -CONR11R12, -CO2 R11, -NR11CO2R12, -SO2NR11R12, -CONR11, -C(O)R11, -NR11SO2R12, NR11SO2NR13R14 и SO2NR11 или необязательно замещенный C1-С6алкилен, -С2-С6 алкенилен или -C1-С6 алкильную группу;
m равен 0, 1 или 2;
каждый из R10, R11 и R12 независимо представляет собой атом водорода, или необязательно замещенный C1-С6 алкил, или необязательно замещенную C1-С6 алкиленовую группу.
Когда n равен 1, R9 представляет собой необязательно замещенный 3-10-членный гетероциклил, гетероарил, арил или циклоалкильное кольцо (когда n равен 0, Q присутствует и R9 отсутствует).
R9 может быть необязательно замещен одним или более заместителями, выбранными из галогена, необязательно замещенного C1-С6 галогеналкила, необязательно замещенного C1-С6 алкокси, C1-С6 галогеналкокси, необязательно замещенного C1-С6 алкила, необязательно замещенного С2-С6 алкенила, необязательно замещенного С2-С6 алкинила, C1-С6 гидроксиалкила, оксо, циано, нитро, необязательно замещенного гетероциклила, необязательно замещенного циклоалкила, необязательно замещенного гетероарила, необязательно замещенного арила, -Q2-NR13CONR14R15, -Q2-NR13R14, -Q2-NR13COR14, -Q2-COR13, -Q2-SO2R13, -Q2-CONR13, Q2-CONR13R14; -Q2-CO2R13, Q2-SO2NR13R14, -Q2-NR13SO2R14 и -Q-NR13SO2NR14R15; где
Q2 представляет собой ковалентную связь, или C1-С6 алкилен, или С2-С6 алкениленовую группу; и
каждый из R13, R14 и R15 независимо представляет собой водород или необязательно замещенный C1-С6 алкил, или необязательно замещенный гетероциклил, или необязательно замещенный гетероарил, или необязательно замещенный арил, или необязательно замещенный циклоалкил.
В качестве альтернативы R9 может быть замещен дополнительно необязательно замещенными 3-10-членными гетероциклильными, гетероарильными, арильными или циклоалкильными кольцами, присоединенными либо непосредственно, либо посредством связующей группы. Связующая группа может представлять собой кислород, карбонил, необязательно замещенный C1-С6 алкилен или цепь необязательно замещенного C1-С6 алкиленокси. Связующая группа может представлять собой кислород, -СО-, C1-С6 алкиленовую группу или группу C1-С6 алкиленокси. В одном из вариантов реализации связующая группа может представлять собой карбонил или алкиленовую цепь, например -СО- или C1-С6 алкиленовую группу.
В одном из вариантов реализации R2 представляет собой C1-С6 алкил. В одном из вариантов реализации R2 представляет собой С1-С4 алкил. В одном из вариантов реализации R2 представляет собой C1-С3 алкил. В одном из вариантов реализации R2 представляет собой С1-С2 алкил. В одном из вариантов реализации R2 представляет собой C1-С3 алкокси. В одном из вариантов реализации R2 представляет собой С1-С2 алкокси. В одном из вариантов реализации R2 представляет собой циано. В одном из вариантов реализации R2 представляет собой метил. В одном из вариантов реализации R2 представляет собой замещенный метил. В одном из вариантов реализации R2 представляет собой СН2Х, где X представляет собой ОМе, F или Cl. В одном из вариантов реализации R2 представляет собой метокси. В одном из вариантов реализации R2 представляет собой C1-С6 алкил, С1-С4 алкил, C1-С3 алкил или С1-С2 алкил, С1-С3 алкокси, С1-С2 алкокси, циано, метил или замещенный метил и каждый из R3, R4, R5, R6, R7 и R8 независимо представляет собой атом водорода.
В другом варианте реализации R3 представляет собой C1-С6 алкил. В другом варианте реализации R3 представляет собой С1-С4 алкил. В другом варианте реализации R3 представляет собой C1-С3 алкил. В другом варианте реализации R3 представляет собой С1-С2 алкил (например, метил или этил). В другом варианте реализации R3 представляет собой C1-С6 алкил, С1-С4 алкил, C1-С3 алкил или С1-С2 алкил (например, метил или этил) и каждый из R1, R2, R4, R5, R6, R7 и R8 независимо представляет собой атом водорода.
В альтернативном варианте реализации R3 связан с R4 с образованием спироциклической группы. Указанная спироциклическая группа может быть образована из атомов углерода или может содержать один или более гетероатомов. Спироциклическая группа может содержать 3, 4, 5 или 6 атомов, то есть представлять собой С3-С6 спироциклическую группу. Спироциклические группы могут быть необязательно дополнительно замещенными.
В альтернативном варианте реализации R5 связан с R6 с образованием спироциклической группы. Указанная спироциклическая группа может быть образована из атомов углерода или может содержать один или более гетероатомов. Спироциклическая группа может содержать 3, 4, 5 или 6 атомов, то есть представлять собой С3-С6 спироциклическую группу. Спироциклические группы могут быть необязательно дополнительно замещенными.
В дополнительном варианте реализации R2 представляет собой метил или метокси. В одном из вариантов реализации R2 представляет собой метил или метокси и каждый из R3, R4, R5, R6, R7 и R8 независимо представляет собой атом водорода. В дополнительном варианте реализации R2 представляет собой метил. В одном из вариантов реализации R2 представляет собой метил и каждый из R3, R4, R5, R6, R7 и R8 независимо представляет собой атом водорода.
В дополнительном варианте реализации, когда R2 не является водородом, R3 представляет собой водород. Если R3 не является водородом, R2 представляет собой водород, то есть либо R2, либо R3 должен быть Н.
В дополнительном варианте реализации R2 связан с R8 с образованием С3-С4 циклоалкильного кольца. Циклоалкильное кольцо может быть циклопропильным или циклобутильным.
В дополнительном варианте реализации R7 представляет собой атом водорода, атом фтора, циано, необязательно замещенный C1-С3 алкил, необязательно замещенную C1-С3 алкоксигруппу или необязательно замещенное арильное или гетероарильное кольцо. В еще одном дополнительном варианте реализации R7 представляет собой атом водорода, циано, необязательно замещенный С1-С3 алкил, необязательно замещенную C1-С3 алкоксигруппу или необязательно замещенное арильное или гетероарильное кольцо. R7 может представлять собой Н, метил или замещенный метил. R7 может быть метилом. R7 может быть CF3. R7 может представлять собой необязательно замещенное арильное или гетероарильное кольцо. R7 может представлять собой необязательно замещенное фенильное, пиридильное или пиримидильное кольцо. R7 может представлять собой необязательно замещенное фенильное или пиридильное кольцо.
В альтернативном варианте реализации R7 связан с R8 с образованием спироциклической группы. Спироциклическая группа может быть образована из атомов углерода, или может содержать один или более гетероатомов. Спироциклическая группа может содержать 3, 4, 5 или 6 атомов. Спироциклические группы могут быть необязательно дополнительно замещенными.
В альтернативном варианте реализации R7 связан с R6 с образованием С3-С4 циклоалкильного кольца, то есть циклопропила или циклобутила. В одном из вариантов реализации R7 и R6 совместно образуют циклопропильное кольцо.
С3-С4 циклоалкильные кольца, раскрытые в настоящем описании, по отношению к R2, R6, R7 и R8 могут быть необязательно замещены галогеном, дейтеро, С1-3 алкилом, С1-3 алкокси, циано, амино, нитро или SF5.
Q1 представляет собой атом водорода, атом галогена, циано, ковалентную связь, -NR10-, -CONR10-, -NR10CO-, атом кислорода, оксо, нитро, -S(O)m-, C1-С6 алкокси, C1-С6 галогеналкокси, C1-С6 гидроксиалкил, -СО-, -SO2R11, -NR11R12, -NR11COR12, -NR10CONR11R12, -CONR11R12, -CO2 R11, -NR11CO2R12, -SO2NR11R12, -CONR11, -C(O)R11, -NR11SO2R12, NR11SO2NR13R14 и SO2NR11 или необязательно замещенный C1-С6алкилен, -С2-С6 алкенилен или -С1-С6 алкильную группу;
В одном из вариантов реализации Q1 представляет собой атом водорода, атом галогена, циано, ковалентную связь, -NR10 -, -CONR10-, -NR10CO-, атом кислорода, -S(O)m-, С1-С6 алкокси, C1-С6 галогеналкокси, C1-С6 гидроксиалкил, -СО-, -SO2R11, -NR11R12, -NR11COR12, -NR10CONR11R12, -CONR11R12, -CO2R11, -NR11CO2R12, -SO2NR11R12, -CONR11, -C(O)R11 и -NR11SO2R12 или необязательно замещенный C1-С6алкилен, -С2-С6 алкенилен или -C1-С6 алкильную группу. Например, Q1 может быть выбран из атома галогена, атома водорода, ковалентной связи, NR10 C1-С6 алкилена или С2-С6 алкениленовой группы, которая может быть необязательно замещена гидрокси, атомом галогена, C1-С6 алкилом, C1-С6 галогеналкилом, C1-С6 алкокси, C1-С6 галогеналкокси, C1-С6 гидроксиалкилом, -COR11, -SO2R11, -NR11R12, -NR11COR12, -CONR11R12, -CO2R11, -NR11CO2R12, -SO2NR11R12 -C(O)N, -С(О) и -NR11SO2R12.
В другом варианте реализации Q1 может быть выбран из атома водорода, ковалентной связи, NR10 или C1-С6 алкилена, - С1-С4 алкилена, С1-С2 алкилена, С2-С6 алкенилена или С2-С4 алкениленовой группы, которая может быть необязательно замещена гидрокси, атомом галогена (например, фтором, хлором или бромом), C1-С6 алкилом, С1-С4 алкилом, С1-С2 алкилом, C1-С6 галогеналкилом, С1-С4 галогеналкилом, С1-С2 галогеналкилом, C1-С6 алкокси, С1-С4 алкокси, С1-С2 алкокси, С1-С6 галогеналкокси, С1-С4 галогеналкокси, C1-С2 галогеналкокси, -C1-С6 гидроксиалкилом, С1-С4 гидроксиалкилом, С1-С2 гидроксиалкилом, -COR11, -SO2R11, -NR11R12, -NR11COR12, -CONR11R12, -CO2R11, -NR11CO2R12, -SO2NR11R12 и -NR11SO2R12.
В дополнительном варианте реализации Q1 может быть выбран из ковалентной связи, NR10 (например, метиламино), -С1-С4 алкилена (например, метилена или этилена) или С2-С4 алкениленовой (например, винильной) группы, которая может быть необязательно замещена гидрокси, атомом галогена (например, фтором, бромом или хлором), С1-С4 алкилом (например, пропилом, изобутилом или трет бутилом), С1-С2 алкилом (например, метилом или этилом), С1-С2 галогеналкилом (например, трифторметилом), С1-С2 алкокси (например, метокси или метоксиметилом), С1-С2 галогеналкокси (например, трифторметокси), С1-С2 гидроксиалкилом (например, гидроксиметилом или гидроксиэтилом), -COR11 (например, ацетилом), -SO2R11 (например, метилсульфонилом) -NR11R12 (например, амино или N,N-диметиламино), -NR11COR12 (например, N-ацетилом), -CONR11R12 (например, амидо), -CO2R11 (например, метоксикарбонилом или этоксикарбонилом), -NR11CO2 R12, -SO2NR11R12 (например, диметиламиносульфонилом) и -NR11SO2R12.
В дополнительном варианте реализации Q1 может быть выбран из атома галогена (например, брома, хлора или фтора), С1-С4 алкила (например, пропила или трет-бутила), С1-С2 алкила (например, метила или этила), С1-С2 галогеналкила (например, трифторметила), С1-С2 алкокси (например, метокси), -COR11 (например, ацетила), -SO2R11 (например, метилсульфонила), циано, CONR11R12 и С1-С2 галогеналкокси (например, трифторметокси).
В дополнительном варианте реализации Q1 может быть выбран из атома галогена (например, брома или хлора), С1-С4 алкила (например, пропила или трет бутила), С1-С2 алкила (например, метила или этила), С1-С2 галогеналкила (например, трифторметила), С1-С2 алкокси (например, метокси), -COR11(например, ацетила) и -SO2R11 (например, метилсульфонила).
В одном из вариантов реализации Q2 может быть выбран из атома водорода, ковалентной связи или необязательно замещенного C1-С6 алкилена (например, С1-С3 алкилена, С1-С4 алкилена, С1-С2 алкилена, С2-С6 алкенилена, С2-С4 алкенилена).
В другом варианте реализации Q2 может быть выбран из атома водорода, ковалентной связи, С1-С2 алкилена (например, метилена, этилена), С2-С4 алкенилена (например, этенилена).
В дополнительном варианте реализации Q2 выбран из Н, ковалентной связи, метилена и этилена.
Когда n равен 0, Q1 может быть выбран из атома водорода, атома галогена, необязательно замещенного C1-С6алкила, COR11, -SO2R11, -C(O)NR11R12, необязательно замещенного C1-С6 алкокси, C1-С6 галогеналкила и C1-С6 галогеналкокси, где R11 и R12 такие, как определено выше.
Необязательно, когда n равен 0, Q1 может быть выбран из атома водорода, атома галогена, C1-С6алкила, COR11 или -SO2R11, где R11 такой, как определено выше.
В качестве альтернативы, когда n равен 1, Q1 может быть выбран из ковалентной связи, -СО-, C1-С6 алкилена или С2-С6 алкениленовой группы, которая может быть необязательно замещена гидрокси и -NR10.
В одном из вариантов реализации n равен 1
В одном из вариантов реализации R9 представляет собой 3-10-членное (например, 3, 4, 5, 6, 7, 8, 9 или 10-членное) гетероциклильное, гетероарильное или циклоалкильное кольцо, которое может быть необязательно замещенным одним или более заместителями, выбранными из галогена, необязательно замещенного C1-С6 галогеналкила, необязательно замещенного C1-С6 алкокси, C1-С6 галогеналкокси, необязательно замещенного C1-С6 алкила, необязательно замещенного С2-С6 алкенила, С2-С6 алкинила, C1-С6 гидроксиалкила, оксо, циано, нитро, гетероциклила, -Q2-NR13CONR14R15, -Q2-NR13R14, -Q2-NR13COR14, -Q2-COR13, -Q2-SO2R13, -Q2-CONR13R14, -Q2-CO2R13, -Q2-SO2NR13R14, -Q2-NR13SO2R14 и -Q2-NR13SO2R14R15.
В одном из вариантов реализации R9 представляет собой 3-10-членное (например, 3, 4, 5, 6, 7, 8, 9 или 10-членное) гетероциклильное, гетероарильное или циклоалкильное кольцо, которое может быть необязательно замещено одним или более (например, одним, двумя или тремя) заместителями, выбранными из атома галогена (например, фтора, брома или хлора), С1-С6 галогеналкила, С1-С4 галогеналкила, С1-С2 галогеналкила, C1-С6 алкокси, С1-С4 алкокси, С1-С2 алкокси, C1-С6 галогеналкокси, С1-С4 галогеналкокси, С1-С2 галогеналкокси, C1-С6 алкила, С1-С4 алкила, С1-С2 алкила, С2-С6 алкенила, С2-С4 алкенила, С1-С6 гидроксиалкила, оксо, циано, -Q2-NR13CONR14, -Q2-NR13R14, -Q2-NR13SO2R14, -Q2-NR13COR14, -Q2-COR13, -Q2-SO2R13, -Q2-CONR13, -Q2-CO2R13, -Q2-SO2NR13R14 и -Q2-NR13SO2R14.
В другом варианте реализации R9 представляет собой 3-10-членное (например, 3, 4, 5, 6, 7, 8, 9 или 10-членное) гетероциклильное, гетероарильное, арильное или циклоалкильное кольцо, которое может быть необязательно замещено одним или более (например, одним, двумя или тремя) заместителями, выбранными из атома галогена (например, фтора, брома или хлора), С1-С2 алкокси (например, метокси, метоксиметила), С1-С4 алкила (например, метила, этила, пропила, трет бутила), оксо, циано, -Q2-NR13CONR14 (например, ацетамидо, ацетамидометила), -Q2-NR13R14 (например, амино), -Q2-NR13SO2R14 (например, метилсульфониламино), -Q2-SO2R13 (например, метилсульфонила) или дополнительно необязательно замещенного 3-10-членного гетероциклильного, гетероарилного, арильного или циклоалкильного кольца.
В другом варианте реализации R9 представляет собой 3-10-членное (например, 3, 4, 5, 6, 7, 8, 9 или 10-членное) гетероциклильное, арильное, гетероарильное или циклоалкильное кольцо, такое как морфолинил, пиперидинил, пирролидинил, диазепанил, пиперазинил, пиридазинил, пиразинил, пиразолил, циклопропил, циклогексил, циклопентил, пиридинил, имидазолил, индолинил, изоиндолинил, пиримидинил, изоксазолил, дигидроинденил, дигидроизохинолинил, тетрагидропиранил, фенил, оксадиазолил, триазолил, изохинолинил, индазолил, пиразолопиридинил, пиразолопиримидинил, имидазолпиридинил, имидазопиримидинил, имидазопиразинил, оксазолил и хинолинил, которое может быть необязательно замещенным. R9 может быть необязательно замещен одним или более (например, одним, двумя или тремя) заместителями, выбранными из атома галогена (например, фтора, брома или хлора), необязательно замещенного C1-С3алкокси (например, метокси, метоксиметила), необязательно замещенного С1-С4алкила (например, метила, этила, пропила, трет бутила), оксо, циано, -Q2-NR13CONR14 (например, ацетамидо, ацетамидометила), -Q2-NR13R14 (например, амино), -Q2-NR13SO2R14 (например, метилсульфониламино), -Q2-SO2R13 (например, метилсульфонила), -Q2-SO2NR13R14 и -Q2-CONR13R14.
В еще одном варианте реализации R9 представляет собой 3-10-членное (например, 3, 4, 5, 6, 7, 8, 9 или 10-членное) гетероциклильное, арильное, гетероарильное или циклоалкильное кольцо, такое как морфолинил, пиперидинил, пирролидинил, диазепанил, пиперазинил, пиридазинил, пиразинил, пиразолил, циклопропил, циклогексил, циклопентил, пиридинил, имидазолил, индолинил, изоиндолинил, пиримидинил, изоксазолил, дигидроинденил, дигидроизохинолинил, тетрагидропиранил, фенил, оксадиазолил и триазолил, которое может быть необязательно замещенным. R9 может быть необязательно замещен одним или более (например, одним, двумя или тремя) заместителями, выбранными из атома галогена (например, фтора, брома или хлора), С1-С2алкокси (например, метокси, метоксиметилом), С1-С4алкилом (например, метилом, этилом, пропилом, трет бутилом), оксо, циано, -Q2-NR13CONR14 (например, ацетамидо, ацетамидометилом), -Q2-NR13R14 (например, амино), -Q2-NR13SO2R14 (например, метилсульфониламино), -Q2-SO2R13 (например, метилсульфонилом).
В другом варианте реализации R9 представляет собой 3-10-членное (например, 3, 4, 5, 6, 7, 8, 9 или 10-членное) гетероциклильное, арильное, гетероарильное или циклоалкильное кольцо, такое как фенил, морфолинил, пиперидинил, пирролидинил, диазепанил, пиперазинил, циклопропил, циклогексил, циклопентил, пиридинил, имидазолил, индолинил, пиримидинил, изоксазолил, хинолинил, триазолил, изохинолинил, индазолил, пиразолопиридинил, пиразолопиримидинил, имидазолпиридинил, имидазопиримидинил, имидазопиразинил, оксазолил и хинолинил, которое может быть необязательно замещено одним или более (например, одним, двумя или тремя) заместителями, выбранными из атома галогена, C1-С6алкокси, C1-С6алкила, оксо, циано, -Q2-NR13CONR14, -Q2-NR13R14, -Q2-NR13SO2R14, -Q2-SO2NR13R14 и -Q2-CONR13R14
В другом варианте реализации R9 представляет собой 3-10-членное (например, 3, 4, 5, 6, 7, 8, 9 или 10-членное) гетероциклильное, арильное, гетероарильное или циклоалкильное кольцо, такое как фенил, морфолинил, пиперидинил, пирролидинил, диазепанил, пиперазинил, циклопропил, циклогексил, циклопентил, пиридинил, имидазолил, индолинил, пиримидинил, изоксазолил, хинолинил, триазолил, которое может быть необязательно замещено одним или более (например, одним, двумя или тремя) заместителями, выбранными из атома галогена, С1-С6алкокси, C1-С6алкил, оксо, циано, -Q2-NR13CONR14, -Q2-NR13R14 и -Q2-NR13SO2R14.
В дополнительном варианте реализации R9 представляет собой необязательно замещенный 5 или 6-членный моноциклический гетероциклил, арил, гетероарил или циклоалкильное кольцо. В другом варианте реализации R9 представляет собой необязательно замещенный 9 или 10-членный бициклический гетероциклил, арил, гетероарил или циклоалкильное кольцо.
В еще одном дополнительном варианте реализации R9 представляет собой необязательно замещенный 6-членный гетероциклил, арил, гетероарил или циклоалкильное кольцо.
Типичные примеры 3-10-членного гетероциклила, арила, гетероарила или циклоалкильного кольца R9 включают пиперидин-1-ил, индолин-1-ил, индолин-2-ил, пиперазин-1-ил, пирролидин-1-ил, 3,4-дигидроизохинолин-2(1Н)-ил, фенил, пиридин-2,3,4-ил, имидазол-1-ил, изоксазол-4-ил, пиримидин-4-ил, 1Н-1,2,3-триазол-2-ил, тиазол-2-ил, тиазолил, циклогексил циклопропил, индазол-5-ил, индозол-4-ил, пиразоло[1,5-а]пиримидин-7-ил, имидазо[1,2-а]пиримидин-5-ил, имидазо[1,2-а]пиридин-5-ил, имидазо[1,2-а]пиразин-6-ил, имидазо[1,2-а]пиримидин-6-ил, оксазол-5-ил, пиразол-4-ил, изоксазол-4-ил, имидазол-4-ил и хинолин-4-ил.
В некоторых вариантах реализации R9 выбран из замещенного или незамещенного фенила, морфолинила, изоксазолила, пиридинила, пиперазинила, циклопропила, индолинила, пирролидинила, изохинолинила, индазолила, пиразолопиридинила, пиразолопиримидинила, имидазолпиридинила, имидазопиримидинила, имидазопиразинила, оксазолила и хинолинила.
В некоторых вариантах реализации R9 выбран из замещенного или незамещенного фенила, морфолинила, изоксазолила, пиридинила, пиперазинила, циклопропила, индолинила и пирролидинила.
В некоторых вариантах реализации R9 представляет собой замещенный или незамещенный фенил.
В одном из вариантов реализации m равен 1 или 2, предпочтительно 2.
В другом варианте реализации каждый из R10, R11 и R12 независимо представляет собой водород или необязательно замещенный C1-С6 алкил. C1-С6 алкил может быть замещен одним или более галогеном, например фтором.
В другом варианте реализации каждый из R10, R11 и R12 независимо представляет собой водород, C1-С6 алкил, С1-С4 алкил или С1-С2 алкил.
В другом варианте реализации каждый из R10, R11 и R12 независимо представляет собой водород или С1-С2 алкил (например, метил или этил).
Каждый из R10, R11 и R12 может независимо представлять собой необязательно замещенную C1-С6 алкиленовую группу, которая служит в качестве связующего фрагмента для связи с дополнительным кольцом.
Каждый из R13, R14 и R15 независимо представляет собой водород, или необязательно замещенный C1-С6 алкил, или необязательно замещенный гетероциклил, или необязательно замещенный гетероарил, или необязательно замещенный арил, или необязательно замещенный циклоалкил. Каждый из R13, R14 и R15 может независимо представлять собой водород или C1-С3 алкил.
В дополнительном варианте реализации n равен 0 и R1 может быть необязательно замещен одним или более (например, одним, двумя, тремя или четырьмя) Q1 заместителями, независимо выбранными из атома галогена (например, брома, хлора или фтора), С1-С4 алкила (например, пропила или трет бутила), С1-С2 алкила (например, метила или этила), С1-С2 галогеналкила (например, трифторметила), С1-С2 алкокси (например, метокси), -COR11 (например, ацетила), -SO2R11 (например, метилсульфонила), циано, CONR11R12 и С1-С2 галогеналкокси.
В дополнительном варианте реализации n равен 0 и R1 может быть необязательно замещен одним или более (например, одним, двумя, тремя или четырьмя) Q1 заместителями, независимо выбранными из атома галогена (например, брома или хлора), С1-С4 алкила (например, пропила или трет бутила), С1-С2 алкила (например, метила или этила), С1-С2 галогеналкила (например, трифторметила), С1-С2 алкокси (например, метокси), -COR11 (например, ацетила) и -SO2R11 (например, метилсульфонила).
В одном из вариантов реализации n равен 0 и R1 представляет собой 5 или 6 членное гетероарильное кольцо, которое может быть необязательно замещено одним или более (например, одним, двумя, тремя или четырьмя) Q1 заместителями, независимо выбранными из атома галогена (например, фтора, брома или хлора), С1-С4 алкила (например, пропила, изобутила или трет бутила), С1-С2 алкила (например, метила или этила), С1-С2 галогеналкила (например, трифторметила), С1-С2 алкокси (например, метокси или метоксиметила), С1-С2 галогеналкокси (например, трифторметокси), С1-С2 гидроксиалкила (например, гидроксиметила или гидроксиэтила), -COR11 (например, ацетила), -SO2R11 (например, метилсульфонила), -NR11R12 (например, амино или N,N-диметиламино), -NR11COR12 (например, N-ацетила), -CONR11R12 (например, амидо), -CO2R11 (например, метоксикарбонила или этоксикарбонила), -NR11CO2R12, -SO2NR11R12 (например, диметиламиносульфонила) и -NR11SO2R12;
В другом варианте реализации n равен 0 и R1 представляет собой 9 членное гетероарильное кольцо, которое может быть необязательно замещено одним или более (например, одним, двумя, тремя или четырьмя) Q1 заместителями, независимо выбранными из атома галогена (например, фтора, брома или хлора), С1-С4 алкила (например, пропила, изобутила или трет бутила), С1-С2 алкила (например, метила или этила), С1-С2 галогеналкила (например, трифторметила), С1-С2 алкокси (например, метокси или метоксиметила), С1-С2 галогеналкокси (например, трифторметокси), С1-С2 гидроксиалкила (например, гидроксиметила или гидроксиэтила), -COR11 (например, ацетила), -SO2R11 (например, метилсульфонила) -NR11R12 (например, амино или N,N-диметиламино), -NR11COR12 (например, N-ацетила), -CONR11R12 (например, амидо) -CO2R11 (например, метоксикарбонила или этоксикарбонила), -NR11CO2R12, -SO2NR11R12 (например, диметиламиносульфонила) и -NR11SO2R12;
В другом варианте реализации n равен 0 и R1 представляет собой 9 членное гетероарильное кольцо, которое может быть необязательно замещено одним или более (например, одним, двумя, тремя или четырьмя) Q1 заместителями, независимо выбранными из атома галогена (например, фтора, брома или хлора), циано, С1-С4 алкила (например, пропила, изобутила или трет бутила), С1-С2 алкила (например, метила или этила), С1-С2 галогеналкила (например, трифторметила), С1-С2 алкокси (например, метокси или метоксиметила), С1-С2 галогеналкокси (например, трифторметокси), С1-С2 гидроксиалкила (например, гидроксиметила или гидроксиэтила), -COR11 (например, ацетила), -SO2R11 (например, метилсульфонила), -NR11R12 (например, амино или N,N-диметиламино), -NR11COR12 (например, N-ацетила), -CONR11R12 (например, амидо) -CO2R11 (например, метоксикарбонила или этоксикарбонила), -NR11CO2 R12, -SO2NR11R12 (например, диметиламиносульфонила) и -NR11SO2R12;
Соединения согласно настоящему описанию могут иметь атом азота в ортио-положении по отношению к атому углерода, присоединенному к амидному азоту. В таких случаях R1 имеет орто-атом с образованием соединения, включающего фрагмент N-C-NH-CO; представленного формулой (IV):
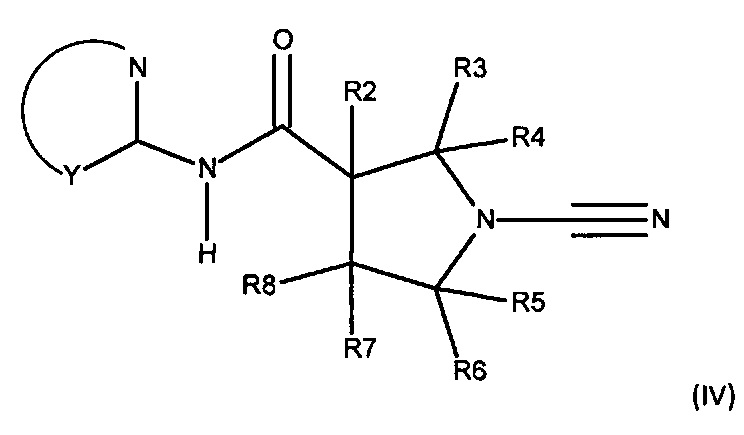
или его фармацевтически приемлемой соли, где:
Y представляет собой остальные атомы 5- или 6-членного гетероарильного кольца, которое может быть необязательно замещенным или конденсированным с дополнительным кольцом, которое может быть дополнительно необязательно замещенным;
каждый из R2, R3, R4, R5, R6 и R8 независимо представляет собой атом водорода, циано, необязательно замещенный C1-С6 алкил, необязательно замещенную С1-С6 алкоксигруппу, одну или более спироциклических групп, где R3 связан с R4, R5 связан с R6, или R8 связан с R7, или R2 связан с R8 с образованием необязательно замещенного С3-С4 циклоалкильного кольца, или R6 связан с R7 с образованием необязательно замещенного С3-С4 циклоалкильного кольца; и
R7 представляет собой атом водорода, атом фтора, циано, необязательно замещенный C1-С3 алкил, необязательно замещенную C1-С3 алкоксигруппу или необязательно замещенное арильное или гетероарильное кольцо, или связан с R8 с образованием спироциклической группы, или связан с R6 с образованием необязательно замещенного С3-С4 циклоалкильного кольца.
В одном из вариантов реализации Y представляет собой остальные атомы 5 или 6 членного гетероарильного кольца, которое может быть необязательно замещенным или конденсированным с дополнительным кольцом, которое может быть дополнительно необязательно замещенным;
каждый из R2, R3, R4, R5, R6 и R8 независимо представляет собой атом водорода, циано, необязательно замещенный C1-С6 алкил, необязательно замещенную C1-С6 алкоксигруппу, одну или более спироциклических групп, где R3 связан с R4, R5 связан с R6, или R8 связан с R7, или R2 связан с R8 с образованием С3-С4 циклоалкильного кольца; и
R7 представляет собой атом водорода, циано, необязательно замещенный C1-С3 алкил, необязательно замещенную C1-С3 алкоксигруппу или необязательно замещенное арильное или гетероарильное кольцо или связан с R8 с образованием спироциклической группы.
Фрагмент, обозначенный символом Y, может быть замещенным в соответствии с Q1-(R9)n, как определено в настоящем описании.
Примеры R1 включают значения, приведенные ниже:
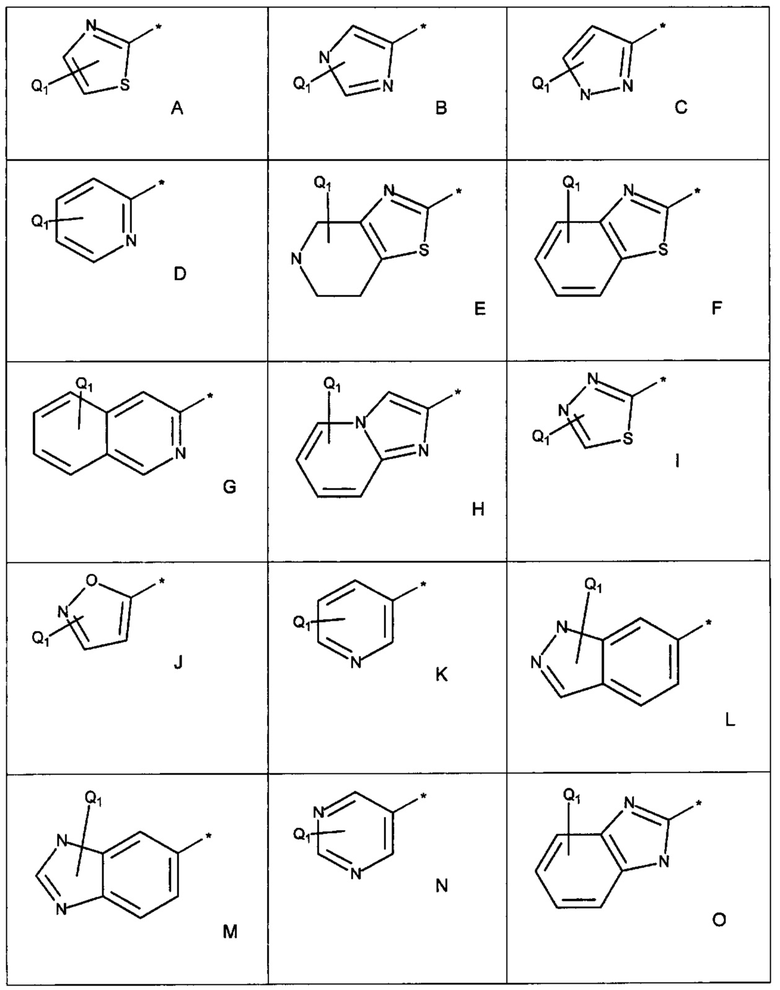
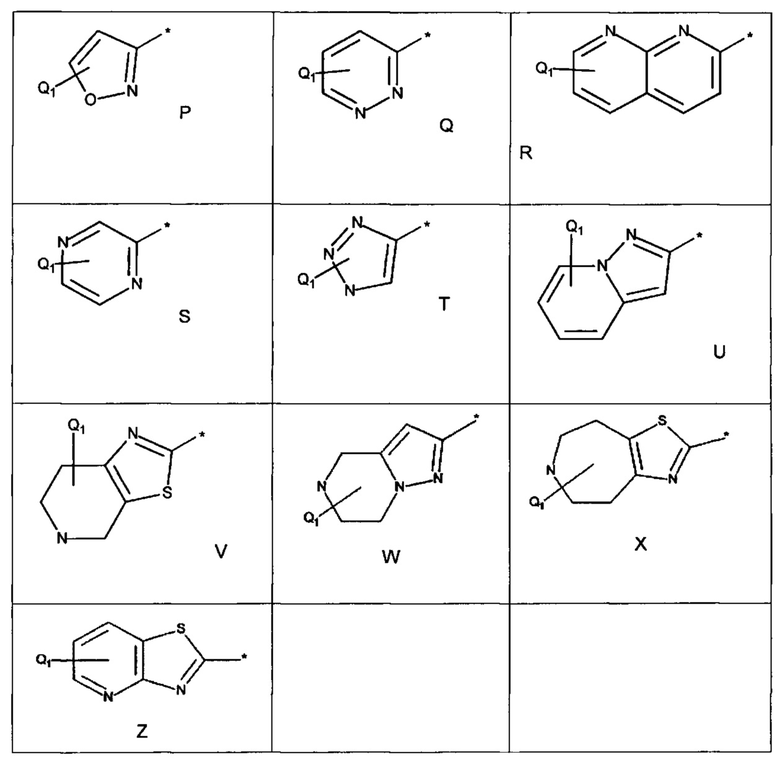
Примеры соединений согласно настоящему изобретению включают:
(S)-1-циано-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамид
1-циано-N-(5-(3-фторфенил)тиазол-2-ил)пирролидин-3-карбоксамид
1-циано-N-(5-(2-фторфенил)тиазол-2-ил)пирролидин-3-карбоксамид
N-(5-(4-хлорфенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
N-(5-(3-хлорфенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
N-(5-(2-хлорфенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
1-циано-N-(5-метилтиазол-2-ил)пирролидин-3-карбоксамид
N-(5-(трет-бутил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
1-циано-N-(4-фенилтиазол-2-ил)пирролидин-3-карбоксамид
1-циано-N-(2-фенилтиазол-5-ил)пирролидин-3-карбоксамид
1-циано-N-(5-фенил-1,3,4-тиадиазол-2-ил)пирролидин-3-карбоксамид
1-циано-N-(5-этил-1,3,4-тиадиазол-2-ил)пирролидин-3-карбоксамид
1-циано-N-(3-фенилизоксазол-5-ил)пирролидин-3-карбоксамид
1-циано-N-(3-(4-метоксифенил)изоксазол-5-ил)пирролидин-3-карбоксамид
1-циано-N-(5-фенилизоксазол-3-ил)пирролидин-3-карбоксамид
N-(5-(трет-бутил)изоксазол-3-ил)-1-цианопирролидин-3-карбоксамид
N-(3-(трет-бутил)-1Н-пиразол-5-ил)-1-цианопирролидин-3-карбоксамид
N-(бензо [d]тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
1-циано-N-(6-метилбензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
1-циано-N-(6-(трифторметил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
1-циано-N-(6-метоксибензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
N-(6-бромбензо[d]тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
N-(1Н-бензо[d]имидазол-2-ил)-1-цианопирролидин-3-карбоксамид
1-циано-N-(пиридин-2-ил)пирролидин-3-карбоксамид
N-(5-хлорпиридин-2-ил)-1-цианопирролидин-3-карбоксамид
1-циано-N-(5-метилпиридин-2-ил)пирролидин-3-карбоксамид
1-циано-N-(5-метоксипиридин-2-ил)пирролидин-3-карбоксамид
1-циано-N-(5-морфолинопиридин-2-ил)пирролидин-3-карбоксамид
1-циано-N-(5-(пиперидин-1-ил)пиридин-2-ил)пирролидин-3-карбоксамид
N-(5-(1Н-имидазол-1-ил)пиридин-2-ил)-1-цианопирролидин-3-карбоксамид
1-циано-N-(4-фенилпиридин-2-ил)пирролидин-3-карбоксамид
N-([2,3'-бипиридин]-6'-ил)-1-цианопирролидин-3-карбоксамид
N-([3,3'-бипиридин]-6-ил)-1-цианопирролидин-3-карбоксамид
N-([3,4'-бипиридин]-6-ил)-1-цианопирролидин-3-карбоксамид
1-циано-N-(6-фенилпиридин-3-ил)пирролидин-3-карбоксамид
1-циано-N-(6-фенилпиридазин-3-ил)пирролидин-3-карбоксамид
1-циано-N-(2-фенилпиримидин-5-ил)пирролидин-3-карбоксамид
1-циано-N-(5-циклогексилпиридин-2-ил)пирролидин-3-карбоксамид
N-(1-бензил-1Н-индазол-5-ил)-1-цианопирролидин-3-карбоксамид
1-циано-N-(1-пропил-1Н-бензо[d]имидазол-5-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-фенил-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-фенил-1Н-пиразол-3-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразол-3-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-фенил-1Н-1,2,3-триазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-фенил-1Н-пиразол-3-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(4-метилбензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(7-метилбензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
(S)-N-(4-бромбензо[d]тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-N-(7-бромимидазо[1,2-а]пиридин-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-N-(6-бромимидазо[1,2-а]пиридин-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(пиразоло[1,5-а]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(4-метоксипиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-фенилпиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-фенилпиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1,8-нафтиридин-2-ил)пирролидин-3-карбоксамид
(S)-N-(5-бензилтиазол-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(изохинолин-3-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(тетрагидро-2Н-пиран-4-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-метил-5-фенил-1Н-пиразол-3-ил)пирролидин-3-карбоксамид
1-циано-N-(5-(4-метоксипиперидин-1-ил)пиридин-2-ил)пирролидин-3-карбоксамид
(S)-N-(6-(1Н-1,2,3-триазол-1-ил)бензо[d]тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-N-(6-(2Н-1,2,3-триазол-2-ил)бензо[d]тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(5-(4-(метилкарбамоил)фенил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(4-метил-5-(морфолинометил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(4-фторфенил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(3,4-дифторфенил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(4-(трифторметил)фенил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(пиридин-4-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(пиридин-2-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(R)-1-циано-N-(5-фенилпиридин-2-ил)пирролидин-3-карбоксамид
(2S,3S)-1-циано-2-метил-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамид
(2S,3S)-1-циано-N-(5-(4-фторфенил)тиазол-2-ил)-2-метилпирролидин-3-карбоксамид
(2S,3S)-1-циано-2-метил-N-(1-фенил-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(2S,3S)-1-циано-2-метил-N-(5-фенил-1Н-пиразол-3-ил)пирролидин-3-карбоксамид
(2S,3S)-1-циано-2-метил-N-(5-(тетрагидро-2Н-пиран-4-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(2S,3S)-N-(5-(2-хлорфенил)тиазол-2-ил)-1-циано-2-метилпирролидин-3-карбоксамид
1-циано-3-метил-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамид
1-циано-3-метил-N-(1-фенил-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
1-циано-3-(метоксиметил)-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамид
1,3-дициано-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамид
(3S,4S)-1-циано-4-метил-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамид
(3S,4S)-1-циано-4-метил-N-(5-метилтиазол-2-ил)пирролидин-3-карбоксамид
(3S,4S)-N-(5-(2-хлорфенил)тиазол-2-ил)-1-циано-4-метилпирролидин-3-карбоксамид
(3S,4S)-1-циано-4-метил-N-(1-фенил-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(3S,4S)-1-циано-4-этил-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамид
(3S,4S)-1-циано-4-этил-N-фенил-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
1-циано-5-метил-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамид
1-циано-5-метил-N-(1-фенил-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
1-циано-5-метил-N-(5-фенил-1Н-пиразол-3-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-морфолинотиазол-2-ил)пирролидин-3-карбоксамид
1-циано-N-(5-(4-метилпиперазин-1-ил)тиазол-2-ил)пирролидин-3-карбоксамид
N-(5-(2-(ацетамидометил)пиперидин-1-ил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
1-циано-N-(5-(метил(фенил)амино)тиазол-2-ил)пирролидин-3-карбоксамид
1-циано-N-(5-(индолин-1-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(пиперидин-1-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(изоиндолин-2-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(3,4-дигидроизохинолин-2(1Н)-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-((R)-2-(метоксиметил)пирролидин-1-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-((S)-2-(метоксиметил)пирролидин-1-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(5-оксо-1,4-диазепан-1-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(R)-1-циано-N-(5-морфолинотиазол-2-ил)пирролидин-3-карбоксамид
(R)-1-циано-N-(5-(пиперидин-1-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(3S,4S)-1-циано-4-метил-N-(5-(пиперидин-1-ил)тиазол-2-ил)пирролидин-3-карбоксамид
1-циано-5-метил-N-(5-(пиперидин-1-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(пирролидин-1-ил)пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(4-(пирролидин-1-ил)пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(пирролидин-1-ил)пиразин-2-ил)пирролидин-3-карбоксамид
N-(5-(2-аминофенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
N-(5-(3-аминофенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
N-(5-(4-аминофенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
1-циано-N-(5-(пиридин-3-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(Е)-1-циано-N-(5-(2-циклопропилвинил)таазол-2-ил)пирролидин-3-карбоксамид
N-(5-(4-ацетамидофенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
N-(5-(2-ацетамидофенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
1-циано-N-(5-(3-(метилсульфонамидо)фенил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(3-цианофенил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(4-цианофенил)тиазол-2-ил)пирролидин-3-карбоксамид
1-циано-N-(5-(3,5-диметилизоксазол-4-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(4-метоксифенил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-фенилбензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(1-метил-1Н-пиразол-5-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(1-метил-1Н-пиразол-4-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(3,5-диметил-1Н-пиразол-4-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(5-метилизоксазол-4-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(7-(3,5-диметилизоксазол-4-ил)имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-циклопропилимидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамид
1-циано-N-(6-циклопропилбензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(3,6-диметоксипиридазин-4-ил)имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(3,6-диметоксипиридазин-4-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
(2S,3S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)бензо[d]тиазол-2-ил)-2-метилпирро лидин-3-карбоксамид
1-циано-N-(5-(п-толил)пиридин-2-ил)пирролидин-3-карбоксамид
1-циано-N-(5-(м-толил)пиридин-2-ил)пирролидин-3-карбоксамид
1-циано-N-(5-(о-толил)пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)-5-метилимидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)-7-метилбензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(7-метил-6-(1-метил-1Н-пиразол-4-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
1-циано-N-(5-(морфолинометил)тиазол-2-ил)пирролидин-3-карбоксамид
1-циано-N-(5-(пирролидин-1-илметил)тиазол-2-ил)пирролидин-3-карбоксамид
(3S)-1-циано-N-(5-((2,6-диметилморфолино)метил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(((R)-2-(метоксиметил)пирролидин-1-ил)метил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(((S)-2-(метоксиметил)пирролидин-1-ил)метил)тиазол-2-ил)пирролидин-3-карбоксамид
(2S,3S)-1-циано-N-(5-(((R)-2-(метоксиметил)пирролидин-1-ил)метил)тиазол-2-ил)-2-метилпирролидин-3-карбоксамид
(S)-N-(1-бензил-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(1-фенетил-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-изобутил-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(2S,3S)-N-(1-бензил-1Н-имидазол-4-ил)-1-циано-2-метилпирролидин-3-карбоксамид
(S)-1-циано-N-(1-(4-фторфенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(3-фторфенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(2-фторфенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(4-(трифторметил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(3-(трифторметил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(4-(метилкарбамоил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(3-(метилкарбамоил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(2-(метилкарбамоил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(4-((2-метоксиэтил)карбамоил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(4-метоксифенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(2-метоксифенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(тетрагидро-2Н-пиран-4-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-циклогексил-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(3S)-1-циано-N-(1-(2,3-дигидро-1Н-инден-1-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(2,3-дигидро-1Н-инден-2-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-((тетрагидро-2Н-пиран-4-ил)метил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-((S)-1-фенилэтил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-((R)-1-фенилэтил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(пиридин-2-илметил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(пиридин-3-илметил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(пиридин-4-илметил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-((3,5-диметилизоксазол-4-ил)метил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(2S,3S)-1-циано-2-метил-N-(1-((S)-1-фенилэтил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(3S,4S)-1-циано-4-метил-N-(1-((S)-1-фенилэтил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(4-метилпиридин-2-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(6-метилпиридин-2-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(R)-1-циано-N-(1-(2-метилпиримидин-4-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(4-цианофенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-N-(1-бензил-2-метил-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(1-(2-(3,5-диметилизоксазол-4-ил)этил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(3-цианофенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(пиридин-3-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(пиридин-4-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(3-метоксифенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(3S)-N-(1-(1-бензоилпиперидин-3-ил)-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамид
(3S)-N-(1-(1-бензоилпирролидин-3-ил)-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамид
(3S)-N-(1-(1-бензилпиперидин-3-ил)-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамид
(3S)-1-циано-N-(1-(1-метилпиперидин-3-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(3S)-1-циано-N-(1-(1-метилпирролидин-3-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-N-(5-ацетил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(5-изобутирил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-N-(5-бензоил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(5-(2-метоксибензоил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-пиколиноил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3 -карбоксамид
(S)-1-циано-N-(5-(1-метил-1Н-пиразол-3-карбонил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(1-метил-2-оксо-1,2-дигидропиридин-3-карбонил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-никотиноил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(диметилглицил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид
метил (S)-2-(1-цианопирролидин-3-карбоксамидо)-6,7-дигидротиазоло[5,4-с]пиридин-5 (4Н)-карбоксилат
2-метоксиэтил (S)-2-(1-цианопирролидин-3-карбоксамидо)-6,7-дигидротиазоло[5,4-с]пиридин-5(4Н)-карбоксилат
(S)-1-циано-N-(5-метил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-N-(5-бензил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(5-(метилсульфонил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(изопропилсульфонил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(фенилсульфонил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(4-этинилфенил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(3-этинилфенил)тиазол-2-ил)пирролидин-3-карбоксамид
(3S)-1-циано-N-(5-(N-(1-фенилэтил)сульфамоил)пиридан-2-ил)пирролидин-3-карбоксамид
(3S)-1-циано-N-(5-(N-метил-N-(1-фенилэтил)сульфамоил)пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(5-метил-1,2,4-оксадиазол-3-ил)имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамид
(3S,4S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)имидазо[1,2-а]пиридин-2-ил)-4-(трифторметил)пирролидин-3-карбоксамид
(S)-1-циано-N-(4-(1-метил-1Н-пиразол-4-ил)пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(N,N-диметилсульфамоил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(пиридазин-4-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(4-циано-3-фторфенил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-N-(5-(1Н-индазол-7-ил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-N-(5-(3-(1Н-имидазол-1-ил)фенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(5-(4-(метилсульфонамидо)фенил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-N-(5-(3-(1Н-пиразол-1-ил)фенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(5-(4-циано-3-(трифторметокси)фенил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(4-циано-3-метоксифенил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(4-сульфамоилфенил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-N-(5-(1Н-индазол-6-ил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-N-(5-(1Н-индазол-5-ил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-6-(2-(1-цианопирролидин-3-карбоксамидо)тиазол-5-ил)-N-метилпиколинамид
(S)-6-(2-(1-цианопирролидин-3-карбоксамидо)тиазол-5-ил)-N-этилпиколинамид
(S)-1-циано-N-(5-(1-метил-1Н-индазол-5-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(1-(2-метоксиэтил)-1Н-индазол-5-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(2-оксоиндолин-7-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(3-метил-1Н-индазол-5-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-N-(5-(1Н-индазол-4-ил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(5-(4-фтор-3-(метилкарбамоил)фенил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-N-(5-(3-карбамоилфенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(5-(3-(метилкарбамоил)фенил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(3-(этилкарбамоил)фенил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(3-(диметилкарбамоил)фенил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-N-(5-(3-карбамоил-4-фторфенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-N-(5-(3-карбамоил-4-хлорфенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-N-(5-(4-хлор-3-(метилкарбамоил)фенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-N-(5-(4-хлор-3-(проп-2-ин-1-илкарбамоил)фенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(5-(изопропилсульфонил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-фенил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-ил)пирролидин-3-карбоксамид
(S)-N-(5-бензил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(1-(4-циано-3-(2-метоксиэтокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(2-циано-5-(2-метоксиэтокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(4-циано-3-(3-морфолинопропокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(2-циано-5-(3-морфолинопропокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(4-циано-3-(((S)-тетрагидрофуран-3-ил)окси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(2-циано-5-(((S)-тетрагидрофуран-3-ил)окси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(4-циано-3-(оксетан-3-илокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(2-циано-5-(оксетан-3-илокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(4-(пиразоло[1,5-а]пиримидин-7-ил)пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(4-(имидазо[1,2-а]пиримидин-5-ил)пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(4-(имидазо[1,2-а]пиридин-5-ил)пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(4-(имидазо[1,2-а]пиразин-6-ил)пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(4-(имидазо[1,2-а]пиримидин-6-ил)пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1,7-нафтиридин-6-ил)пирролидин-3-карбоксамид
(S)-N-(6-(3-хлорфенил)пиримидин-4-ил)-1-цианопирролидин-3-карбоксамид
(S)-N-(2'-амино-[4,4'-бипиридин]-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(2'-(метиламино)-[4,4'-бипиридин]-2-ил)пирролидин-3-карбоксамид
(S)-3-(1-цианопирролидин-3-карбоксамидо)-N-метилизохинолин-6-карбоксамид
(S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)изохинолин-3-ил)пирролидин-3-карбоксамид
(S)-N-(4-(1Н-индазол-4-ил)пиридин-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(6-этинилизохинолин-3-ил)пирролидин-3-карбоксамид
(2S,3S)-1-циано-N-(6-этинилизохинолин-3-ил)-2-метилпирролидин-3-карбоксамид
(S)-3-(1-цианопирролидин-3-карбоксамидо)изохинолин-6-карбоксамид
(2S,3S)-N-(6-(1Н-пиразол-4-ил)изохинолин-3-ил)-1-циано-2-метилпирролидин-3-карбоксамид
(S)-1-циано-N-(пиразоло[1,5-а]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(4-фенилпиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(пиридин-3-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(пиридин-3-ил)тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(трифторметил)имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-метил-5-(м-толил)-1Н-пиразол-3-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(хинолин-3-ил)пирролидин-3-карбоксамид
(S)-N-(4-(трет-бутил)пиридин-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(6-(пирвдин-4-ил)пиримидин-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-фенилпиридазин-3-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(оксазол-5-ил)имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(изопропилсульфонил)-5,6,7,8-тетрагидро-4Н-тиазоло[4,5-d]азепин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(4-(1-(2-метоксиэтил)-1Н-пиразол-4-ил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
3-циано-N-(5-фенилтиазол-2-ил)-3-азабицикло[3.1.0]гексан-1-карбоксамид
2-циано-N-(5-фенилтиазол-2-ил)-2-азабицикло[3.1.0]гексан-4-карбоксамид
(3S,4S)-1-циано-N-(5-фенилтиазол-2-ил)-4-(трифторметил)пирролидин-3-карбоксамид
(3S,4R)-1-циано-N-(5-фенилтиазол-2-ил)-4-(пиридин-3-ил)пирролидин-3-карбоксамид
(3S,4R)-1-циано-N-(5-фенилтиазол-2-ил)-4-(пиримидин-5-ил)пирролидин-3-карбоксамид
1-циано-3-метокси-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)тиазоло[4,5-b]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(5-(3,5-диметилизоксазол-4-ил)тиазоло[5,4-b]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(2,6-диметилфенил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(1,3-диметил-1Н-пиразол-4-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(5-(трифторметил)-1Н-пиразол-4-ил)изохинолин-3-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(4-(3-(5-метилизоксазол-4-ил)фенил)пиридин-2-ил)пирролидин-3-карбоксамид
(S)-N-(4-(1Н-пиразол-4-ил)пиридин-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-N-(6-(1Н-пиразол-4-ил)бензо[d]тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
(S)-N-(1-(4-(1Н-пиразол-4-ил)фенил)-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(4-(5-метил-1Н-пиразол-4-ил)пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)тиазоло[4,5-с]пиридин-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(2'-(5-метилизоксазол-4-ил)-[4,4'-бипиридин]-2-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(1,3-диметил-1Н-индазол-5-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(3-метил-1Н-индазол-6-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-N-(1-(1Н-индазол-5-ил)-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамид
(S)-N-(1-(1Н-индазол-6-ил)-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(1-(4-фтор-3-метоксифенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-N-(1-(1Н-индазол-4-ил)-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамид
(S)-1-циано-N-(1-(4-циано-3-циклопропилфенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(хинолин-4-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-метил-N-(4-(4-(метилкарбамоил)фенил)тиазол-2-ил)пирролидин-2-карбоксамид
(S)-1-циано-N-(1-(4-циано-3-метоксифенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(4-циано-3-метилфенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(4-циано-3-(трифторметил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(S)-1-циано-N-(1-(4-циано-3-(трифторметокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
Необходимо отметить, что каждое из вышеперечисленных химических соединений представляет собой конкретный и независимый аспект настоящего изобретения.
В соответствии с дополнительным аспектом настоящего изобретения предложен способ получения соединения формулы (I) или его фармацевтически приемлемой соли, включающий стадии проведения реакции кислоты формулы (V) с соединением R1-NH2 с образованием амида:
где R2-R8 имеют значения, определенные в любом другом месте, a Pg представляет собой аминозащитную группу. Указанная защитная группа может представлять собой ВОС, но не ограничивается им. Специалисту в данной области техники очевидна необходимость объединения или добавления такой защитной химической группы. После сочетания R1-NH2 с образованием амида защитная группа может быть удалена с получением свободного амина в соответствии с формулой (VI), который может быть затем обработан цианоген бромидом с образованием соединений в соответствии с формулой (I).
В соответствии с дополнительным аспектом настоящего изобретения предложен способ получения соединения формулы (I) или его фармацевтически приемлемой соли, включающий стадии проведения реакции амина формулы (VI) с цианоген бромидом с получением соединения N-CN:
В соответствии с дополнительным аспектом настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I).
Фармацевтические композиции согласно настоящему изобретению содержат любые соединения формулы (I) согласно настоящему изобретению, объединенные с любым фармацевтически приемлемым носителем, адъювантом или переносящей средой.
Примеры фармацевтически приемлемых носителей известны специалистам в данной области техники и включают, но не ограничиваются ими, консервирующие агенты, наполнители, дезинтегрирующие агенты, смачивающие агенты, эмульгирующие агенты, суспендирующие агенты, подслащивающие агенты, ароматизирующие агенты (flavouring agents), отдушки (perfuming agents), антибактериальные агенты, противогрибковые агенты, смазывающие агенты и диспергирующие агенты в зависимости от вида пути введения и лекарственных форм. Композиции могут быть представлены в форме, например, таблеток, капсул, порошков, гранул, эликсиров, таблеток для рассасывания (lozenges), суппозиториев, сиропов и жидких препаратов, включая суспензии и растворы. Термин «фармацевтическая композиция» в контексте настоящего изобретения означает композицию, содержащую активный агент и дополнительно содержащую один или более фармацевтически приемлемых носителей. Композиция может дополнительно содержать ингредиенты, выбранные из, например, разбавителей, адъювантов, вспомогательных веществ, переносящих сред, консервирующих агентов, наполнителей, дезинтегрирующих агентов, смачивающих агентов, эмульгирующих агентов, суспендирующих агентов, подслащивающих агентов, ароматизирующих агентов, отдушек, антибактериальных агентов, противогрибковых агентов, смазывающих агентов и диспергирующих агентов, в зависимости от вида пути введения и лекарственных форм.
В соответствии с дополнительным аспектом настоящего изобретения предложено соединение формулы (I) или содержащая его фармацевтическая композиция для применения для терапии. В частности, соединения согласно настоящему изобретению в соответствии с формулой (I) применяют при лечении рака и, в частности, при лечении раковых заболеваний, связанных с деубиквитилирующей (DUB) активностью. Соединения согласно настоящему изобретению могут быть подходящими для применения против DUB фермента, включая, но не ограничиваясь ими, UCHL1, USP6 или USP30.
Соединения формулы (I), описанные в настоящем документе, могут быть применены для получения лекарственного средства для лечения рака, связанного с DUB активностью.
UCHL1 сверхэкспрессирована в большом количестве типов опухолей. В другом аспекте настоящего изобретения предложен способ лечения или предотвращения рака, связанного с активностью UCHL1, причем указанный способ включает введение фармацевтически эффективного количества соединения формулы (I) содержащей его фармацевтической композиции индивидууму, страдающему раком, связанным с активностью UCHL1.
Соединения или композиции в соответствии с формулой (I) могут быть применены для лечения рака. Указания на «рак» или «опухоль» включают, но не ограничиваются ими, рак груди, яичников, предстательной железы, легкого, почки, желудка, прямой кишки, яичек, головы и шеи, поджелудочной железы, головного мозга, меланомы, рака кости или другие раковые заболевания органов и тканей и раковые заболевание клеток крови, таких как лимфомы и лейкозы. Конкретные раковые заболевания включают карциному груди, лимфому, множественную миелому, колоректальный рак, остеосаркому, карциному поджелудочной железы и немелкоклеточную карциному легкого.
Соединения или композиции в соответствии с формулой (I) могут быть применены для лечения дополнительных заболеваний, связанных с активностью UCHL1. Например, заболевание, связанное с активностью UCHL1, может быть выбрано из нейродегенеративных расстройств (таких как болезнь Альцгеймера, болезнь Паркинсона), ХОБЛ, воспаления, вирусных инфекций, включая ближневосточный респираторный синдром (БВРС) или тяжелый острый респираторный синдром (ТОРС), бактериальных инфекций, включая туберкулез, или метаболических расстройств.
Соединения формулы (I) или их фармацевтическая композиция, описанные в настоящем описании, могут быть объединены с одним или более дополнительными агентами. Соединения могут быть объединены с дополнительным противоопухолевым терапевтическим агентом, например, химиотерапевтическими лекарственными средствами или ингибиторами других регуляторных белков. В одном из вариантов реализации указанный дополнительный противоопухолевый терапевтический агент выбран из ингибиторов PARP (поли-(АДФ-рибоза)-полимеразы), ингибитора BRCA2 и ингибитора ATM. В других варианте реализации ингибитор PARP представляет собой ингибирующую молекулу РНК (англ.: inhibitory RNA, RNAi) (PARPi). В дополнительном варианте реализации ингибиторы PARP могут быть выбраны из одного или более из Инипариба (BSI 201), Олапариба (AZD-2281), Рукапариба (AG014699, PF-01367338) и Велипариба (АВТ-888), МК-4827, СЕР-9722, Е716 (GPI-221016), LT-673, MP-124, NMC-P118. В дополнительном варианте реализации дополнительный противоопухолевый агент представляет собой химиотерапевтический агент. Химиотерапевтические агенты могут быть выбраны из олапариба, митомицина С, цисплатина, карбоплатина, оксалиплатина, ионизирующего излучения (ИИ), камптотецина, иринотекана, топотекана, темозоломида, таксанов, 5-фторпиримидинов, гемцитабина и доксорубицина.
Фармацевтические композиции согласно настоящему изобретению могут быть введены любым эффективным образом, подходящим для нацеливания на раковые клетки, например, перорально в любой перорально приемлемой лекарственной форме, включая, но не ограничиваясь ими, таблетки, драже, порошки, эликсиры, сиропы, жидкие препараты, включая суспензии, спреи, ингаляторы, таблетки, таблетки для рассасывания (lozenges), эмульсии, растворы, саше, гранулы и капсулы. Такие лекарственные формы получают в соответствии с методиками, известными в области получения фармацевтических составов. В случае формы спреев или препаратов для ингаляций фармацевтические композиции могут быть введены назально. Подходящие для данной цели составы известны специалистам в данной области техники.
Фармацевтические композиции согласно настоящему изобретению могут быть введены путем инъекции и могут быть представлены в форме стерильного жидкого препарата для инъекций, включая липосомные препараты.
Фармацевтические композиции согласно настоящему изобретению также могут быть представлены в форме суппозиториев для ректального введения. Их готовят таким образом, что фармацевтическая композиция является твердой при комнатной температуре и жидкой при температуре тела, позволяя высвобождаться активному соединению.
Дозировки могут варьироваться в зависимости от требований пациента, тяжести состояния, подвергаемого лечению, и применяемого соединения. Подходящая дозировка для конкретной ситуации определяется специалистом в данной области техники. В целом, лечение начинают с небольших доз, которые меньше оптимальной дозы соединения. После этого дозу увеличивают небольшими интервалами до достижения оптимального эффекта в конкретных условиях.
Величина эффективной дозы соединения будет, несомненно, меняться в зависимости от природы и тяжести подлежащего лечению состояния и в зависимости от конкретного соединения и пути его введения. Выбор подходящих дозировок находится в пределах компетенции специалиста в данной области и не требует излишних затрат. Диапазон суточных доз составляет от примерно 10 мкг до примерно 100 мг на кг массы тела человека или отличного от человека животного и, в целом, может составлять примерно от 10 мкг до 30 мг на кг массы тела на дозу. Указанная выше доза может быть введена один-три раза в сутки.
Способы синтеза
Соединения согласно настоящему изобретению могут быть получены с применением различных способов синтеза. Иллюстративные пути синтеза некоторых соединений согласно настоящему изобретению приведены ниже. Примеры соединений согласно настоящему изобретению могут быть синтезированы в соответствии с общими способами синтеза, описанными ниже, и проиллюстрированы более конкретно следующими схемами. Поскольку указанные схемы являются иллюстрацией, не следует считать, что настоящее изобретение ограничено представленными химическими реакциями и условиями. Получение различных исходных веществ, применяемых в указанных схемах, находится в пределах компетенции специалистов в данной области техники. Специалистам в данной области техники будет очевидно, что в соответствующих случаях отдельные превращения в схеме могут быть выполнены в различном порядке. На следующих схемах описаны общие способы синтеза, согласно которым могут быть получены промежуточные и целевые соединения согласно настоящему изобретению. Дополнительные иллюстративные соединения и их стереоизомеры, рацемические смеси, диастереомеры и энантиомеры могут быть синтезированы с применением промежуточных соединений, полученных в соответствие с общими схемами, и других материалов, соединений и реагентов, известных специалистам в данной области техники. Предполагается, что все такие соединения, их стереоизомеры, рацемические смеси, диастереомеры и энантиомеры включены в объем настоящего изобретения.
Все соединения были охарактеризованы посредством жидкостной хроматографии-масс-спектроскопии (ЖХМС) и 1Н ЯМР.
Сокращения:
водн. Водный
Ar Арил
ВОС Трет-бутоксикарбонил
ушир. Уширенный (сигнал ЯМР)
d Дублет (сигнал ЯМР)
dba Дибензилиденацетон
ДХЭ 1,2-Дихлорэтан
ДХМ Дихлорметан
DIAD Диизопропилазодикарбоксилат
DIPEA Диизопропилэтиламин
ДМАА Диметилацетамид
DMAP Диметиламинопиридин
ДМФА Диметилформамид
ДМСО Диметилсульфоксид
dppf 1,1'-Бис(дифенилфосфино)ферроцен
EDC 1-Этил-3-(3-диметиламинопропил)карбодиимид
ЭР Электрораспыление
EtOAc Этилацетат
EtOH Этанол
МК Муравьиная кислота
Fmoc Флуоренилметилоксикарбонил
ч Час (-ы)
HATU (1-[Бис(диметиламино)метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиний-3-оксида гексафторфосфат)
HBTU О-Бензотриазол-N,N,N'N'-тетраметилурония гексафторфосфат
HPLC Высокоэффективная жидкостная хроматография
HOAt 1-Гидрокси-7-азабензотриазол
IPA Изопропиловый спирт
LDA Диизопропиламид лития
LiHMDS Гексаметилдисилазид лития
m Мультиплет (сигнал ЯМР)
MeCN Ацетонитрил
МеОН Метанол
n-BuLi н-Бутиллитий
к.т. Комнатная температура
s Синглет (сигнал ЯМР)
t Триплет (сигнал ЯМР)
Т3Р 2,4,6-Трипропил-1,3,5,2,4,6-триоксатрифосфоринан-2,4,6-триоксид
TBAI Тетрабутиламмония иодид
ТЭА Триэтиламин
ТФУ Трифторуксусная кислота
ТФУА Трифторуксусный ангидрид
ТГФ Тетрагидрофуран
X-phos 2-Дициклогексилфосфино-2',4',6'-триизопропилбифенил
Методы анализа:

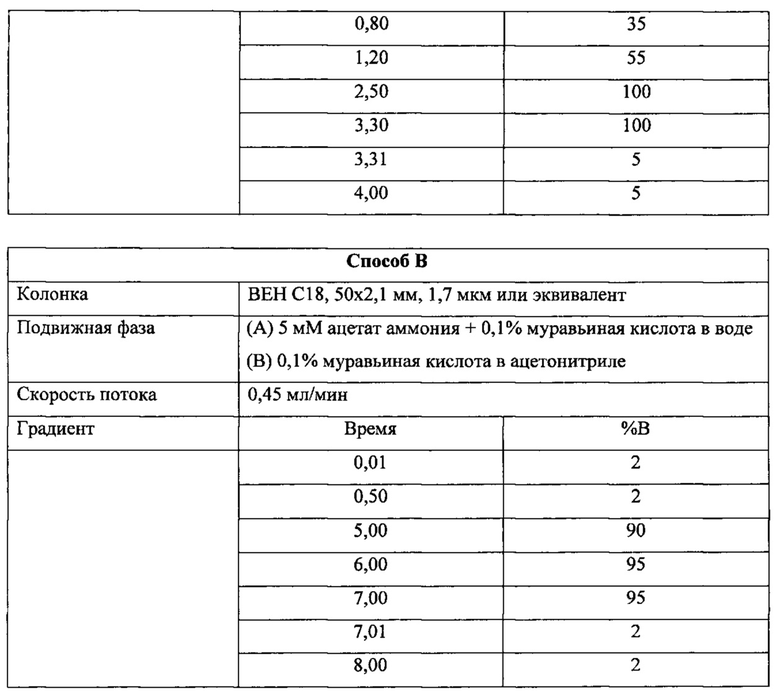

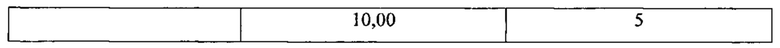
Схемы синтеза:
Схема 1
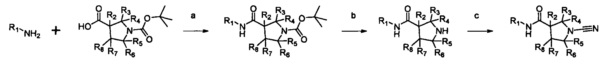
Реагенты и условия: а) Т3Р (50% в EtOAc), DIPEA, ТГФ, 0°С, к.т. 1 ч или POCl3, пиридин, ДХМ, от 0°С до к. т. 1 ч; b) 4М HCl в EtOAc, от 0°С до к. т. 4 ч или ТФУ, ДХМ, от 0°С до к. т. 1 ч; с) цианоген бромид, К2СО3, ДХМ, от 0°С до к. т. 16 ч.
Схема 2
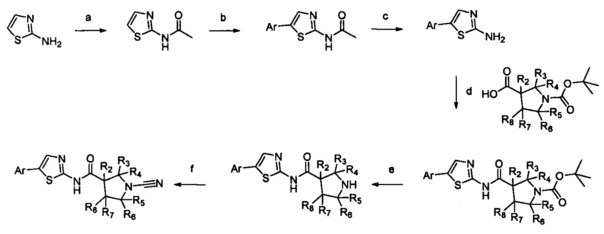
Реагенты и условия: а) Ac2O, ТЭА, ТГФ, 0°С, к. т. 2 ч; b) ArBr, К3РО4, Pd(OAc)2, трициклогексилфосфин, ДМФА, к. т., 140°С микроволновое излучение 1,5 ч; с) 1,4-диоксан, конц. HCl, 100°С 4 ч; d) Т3Р (50% в EtOAc), DIPEA, ТГФ, 0°С, к. т. 1 ч; е) 4М HCl в EtOAc, 0°С, к. т. 4 ч или ТФУ, ДХМ, 0°С, к. т. 1 ч; f) цианоген бромид, К2СО3, ДХМ, 0°С, к.т. 16 ч.
Схема 3
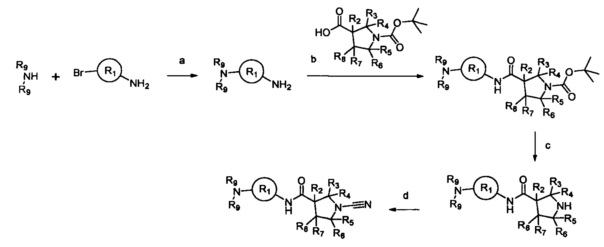
Реагенты и условия: a) Cs2CO3, MeCN, к. т. 2 ч; b) Т3Р (50% в EtOAc), DIPEA, ТГФ, 0°С 1 ч, затем при комнатной температуре 1 ч; b) 4М HCl в EtOAc, 0°С, к. т. 4 ч или ТФУ, ДХМ, 0°С, к. т. 1 ч; с) цианоген бромид, K2CO3, ДХМ, 0°С, к. т. 16 ч.
Схема 4
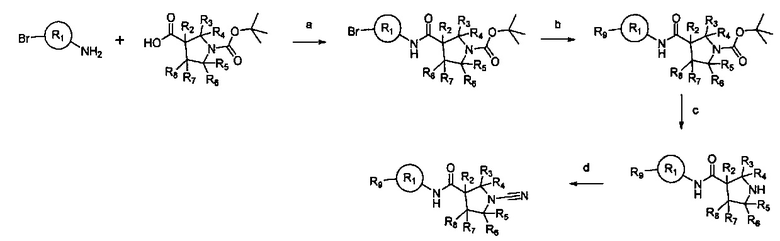
Реагенты и условия: а) Т3Р (50% в EtOAc), ТЭА, ТГФ, к. т. 24 ч; b) R9B(OH)2 или боронатный сложный эфир, Na2CO3, Pd(PPh3)4, 1,4-диоксан, вода, 110°С, 24 ч или трифторборатная соль калия, К3РО4, Pd(dppf)Cl2.ДХМ, толуол, вода, 100°С, плотно закрытая пробирка, 18 ч; с) 4М HCl в 1,4-диоксан, 0°С, к. т. 24 ч или ТФУ, ДХМ, 0°С, к. т. 1 ч; d) цианоген бромид, ТЭА, ДХМ, 0°С, к. т. 30 мин.
Схема 5
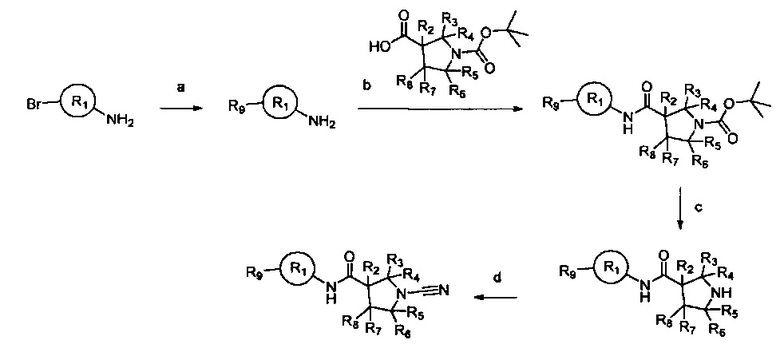
Реагенты и условия: a) R9B(OH)2 или боронатный сложный эфир, Na2CO3, Pd(PPh3)4, этанол, толуол, вода, микроволновое излучение, 100°С, 3 ч; b) HBTU, DIPEA, ДХМ, к. т., 2,5 ч или POCl3, пиридин, ДХМ, от 0°С до к. т. 2 ч; с) ТФУ, ДХМ, 0°С, к. т. 1 ч; d) цианоген бромид, К2СО3, ТГФ, 0°С 10 мин.
Схема 6
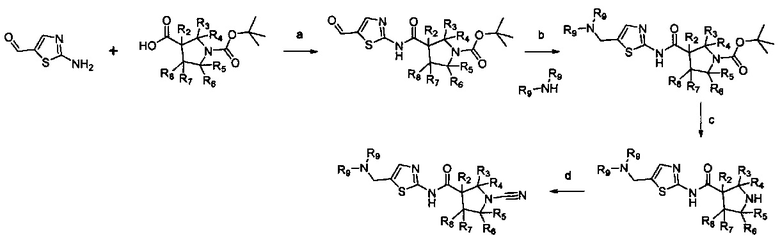
Реагенты и условия: a) HOAt, DIPEA, EDC.HCl, ДМФА, к. т. 3 ч; b) NaBH(OAc)3, ДХЭ, к. т. 3 ч; с) ТФУ, 0°С, к. т. 40 мин; d) цианоген бромид, К2СО3, ТГФ, 0°С, к. т. 2 ч.
Схема 7
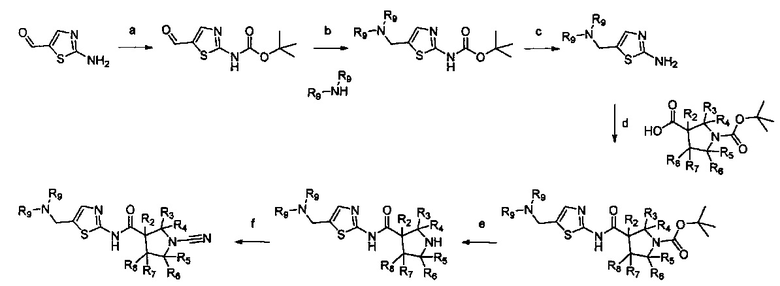
Реагенты и условия: а) (ВОС)2О, DMAP, ТГФ, 0°С, к. т. 1 ч; b) NaBH(OAc)3, МеОН, к. т., 80°С 2 ч; с) ТФУ, ДХМ, к. т. 1 ч; d) Т3Р (50% в EtOAc), DIPEA, ТГФ, к. т. 1 ч; е) ТФУ, ДХМ, к. т. 1 ч; f) цианоген бромид, K2CO3, ТГФ, 0°С, к. т. 1 ч.
Схема 8
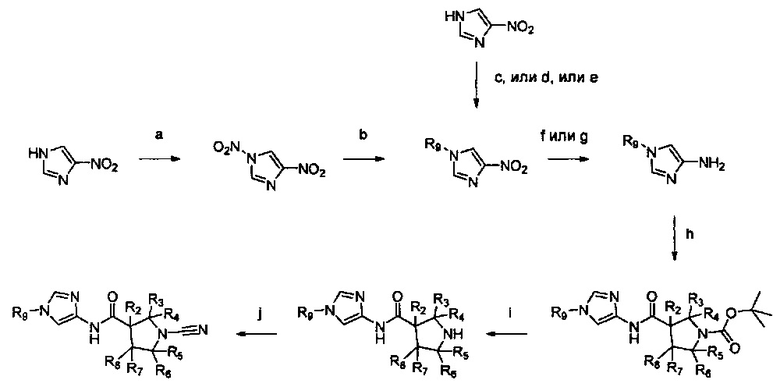
Реагенты и условия: а) дымящая HNO3, уксусная кислота, уксусный ангидрид, 0°С, к. т. 2 ч; b) R9NH2, МеОН: вода (1:1), к. т. 2 ч; с) ArX или R9Br, КОН, ДМСО, к. т. 3 ч; d) ArX, KI, К2СО3, ДМФА, 100°С 18 ч; е) R9B(OH)2, CuCl2, NaOH, МеОН, O2, 80°С 18 ч; f) порошок Fe (или Zn), NH4Cl, МеОН: вода (1:1), 60-80°С 2 ч; g) 10% Pd/C, Н2, МеОН, к. т. 2 ч; h) Т3Р (50% в EtOAc), ТЭА, ТГФ, 0°С, к. т. 2 ч; i) 4М HCl в 1,4-диоксане, ДХМ, 0°С 1 ч; j) цианоген бромид, K2CO3, ДМФА, 0°С, к. т. 30 мин.
Схема 9
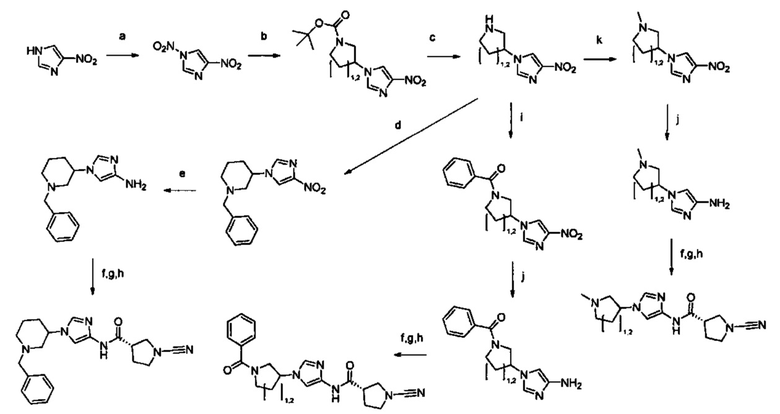
Реагенты и условия: а) дымящая HNO3, уксусная кислота, уксусный ангидрид, 0°С, к. т. 2 ч; b) 3-амино-1-ВОС-пирролидин или 3-амино-1-ВОС-пиперидин, МеОН, вода, к. т. 16 ч; с) ТФУ, ДХМ, к. т. 10 мин; d) бензилбромид, К2СО3, ТГФ, 80°С 16 ч; е) Fe, NH4Cl, ТГФ: вода (1:1), 80°С 0,5 ч; f) Т3Р (50% в EtOAc), DIPEA, ТГФ, 0°С, к. т. 1 ч; g) 4М HCl в EtOAc, 0°С, к. т. 4 ч; h) цианоген бромид, К2СО3, ДХМ, 0°С, к. т. 16 ч; i) бензойная кислота, Т3Р (50% в EtOAc), ТЭА, ТГФ, 0°С, к. т. 16 ч j) 10% Pd/C, Н2, МеОН, к. т. 30 мин k) 37% водный раствор формальдегида, NaCNBH3, МеОН, АсОН, к. т. 16 ч.
Схема 10
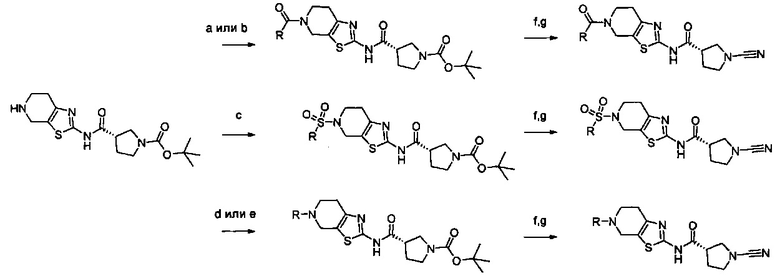
Реагенты и условия: a) RCOCl, ТЭА, ДХМ, 0°С, к. т. 1 ч; b) RCO2H, Т3Р (50% в EtOAc), DIPEA, ТГФ, 0°С, к. т.; с) сульфонилхлорид, ТЭА, ДХМ, 0°С, к. т. 1 ч; d) 37% водн. формальдегид, уксусная кислота, NaCNBH3, МеОН, 0°С 0,5 ч, затем при комнатной температуре 3 ч; е) бензальдегид, уксусная кислота, NaCNBH3, МеОН, 0°С 0,5 ч, затем при комнатной температуре 3 ч f) ТФУ, ДХМ, к. т. 3 ч; g) цианоген бромид, ТЭА, ДХМ, 0°С 30 мин.
Схема 11
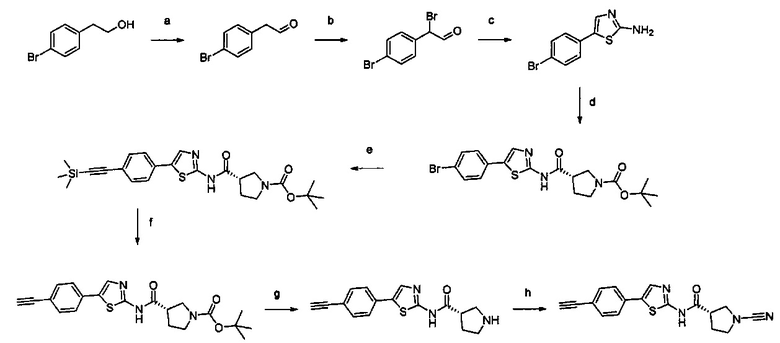
Реагенты и условия: а) периодинан Десса Мартина, ДХМ, к. т. 16 ч; b) бром, ДХМ, 0°С, к. т. 2 ч; с) тиомочевина, этанол, 90°С 6 ч; d) Т3Р (50% в EtOAc), ТЭА, ТГФ, 0°С, к. т. 1 ч; е) триметилсилилацетилен, CuI, Pd(PPh3)2Cl2, DIPEA, 110°С, 16 ч; f) 5М водный КОН, МеОН, 0°С, к. т. 30 мин; g) ТФУ, ДХМ, 0°С, к. т. 2 ч; h) цианоген бромид, К2СО3, ДМФА, 0°С, к. т. 1 ч.
Схема 12
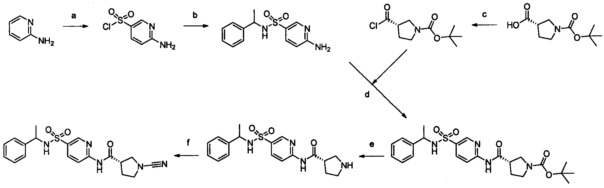
Реагенты и условия: а) хлорсульфоновая кислота, 150°С, 2 ч; b) 1-фенилэтан-1-амин, ТЭА, ТГФ, от 0°С до к. т., 30 мин; с) ДХМ, (COCl)2, ДМФА, пиридин, от 0°С до к. т., 1 ч; d) ТЭА, ДМФА, от 0°С до к. т., 16 ч; е) ДХМ, ТФУ, к. т., 15 мин f) цианоген бромид, ТЭА, ТГФ, 0°С, к. т. 30 мин.
Схема 13
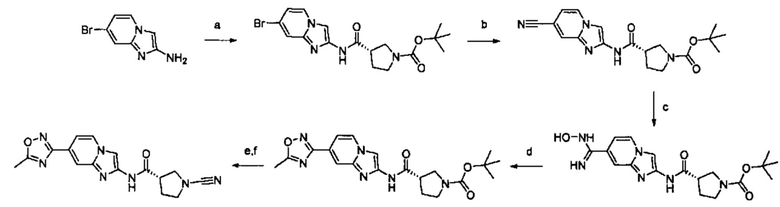
Реагенты и условия: а) Т3Р (50% в EtOAc), DIPEA, ТГФ, 0°С, 1 ч; b) Zn(CN)2, Pd(dba)2, 1,1'бис(дифенилфосфино)ферроцен, ТЭА, ДМФА, 100°С 24 ч; с) гидроксиламина гидрохлорид, ТЭА, IPА, 70°С 3 ч; d) N,N-циметилацетамид-диметилацеталь, 70°С 1 ч; е) ДХМ, ТФУ, к. т. 1 ч; f) цианоген бромид, К2СО3, ТГФ, 0°С 30 мин, затем при к. т. 1 ч.
Схема 14
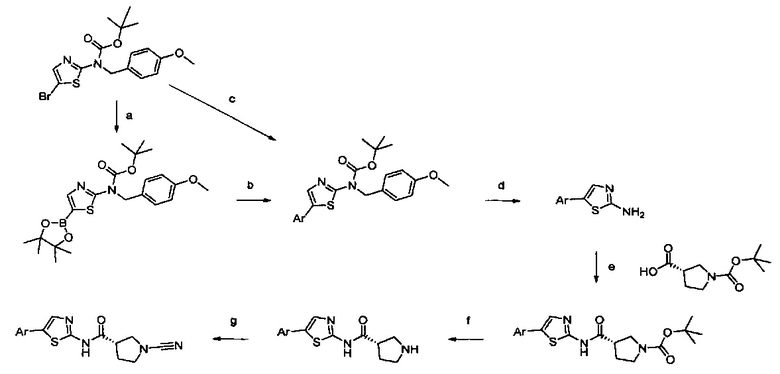
Реагенты и условия: a) n-BuLi, 4,4,5,5-тетраметил-1,3,2-диоксаборолан, ТГФ, -78°С до 0°С, 30 мин; b) арилгалогенид, PdCl2(dppf), Na2CO3, толуол: вода (5:1), к. т. затем 100°С, 1 ч; с) арилбороновая кислота / боронат, PdCl2(dppf), Cs2CO3, 1,4-диоксан: вода (9:1), к. т. затем 80°С, 2 ч; d) ТФУ, 80°С, 8 ч; е) HATU, DIPEA, ТГФ, к. т., 2 ч; f) ТФУ, ДХМ, 0°С затем к. т., 4 ч; g) цианоген бромид, K2СО3, ТГФ: ДМФА (1:1), 0°С затем к. т., 30 мин.
Схема 15
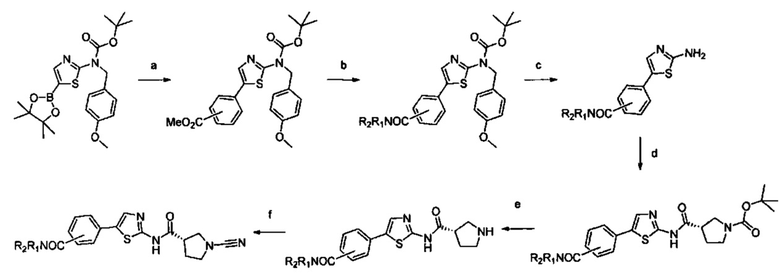
Реагенты и условия: а) Арилгалогенид, PdCl2(dppf), Na2CO3, толуол: вода (5:1), к. т. затем 100°С, 1 ч; b) R1R2NH, триазабициклодецен, ТГФ, от 0°С до к. т., 4 ч; с) ТФУ, 80°С, 8 ч; d) (3S)-ВОС-1-пирролидин-3-карбоновая кислота, HATU, DIPEA, ТГФ, к. т., 2 ч; е) ТФУ, ДХМ, от 0°С до к. т., 4 ч; f) цианоген бромид, K2СО3, ТГФ: ДМФА (1:1), 0°С затем к. т., 30 мин.
Схема 16
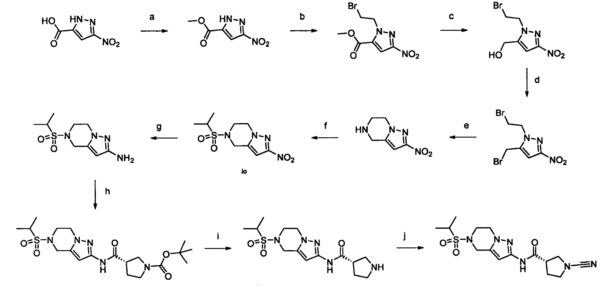
Реагенты и условия: a) SOCl2, МеОН, ДМФА, 70°С, 4 ч; b) 1,2-дибромэтан, K2СО3, ацетон, 60°С, 4 ч; с) LiBH4 (3М в ТГФ), ТГФ, 0°С затем к. т., 2 ч; d) PBr3, хлороформ, 0°С затем к. т., 2 ч; е) водный раствор аммиака, ТГФ, к. т., 72 ч; f) изопропилсульфонилхлорид, ТЭА, ДХМ, к. т., 4 ч; g) Н2, Pd/C (10% сух.), МеОН, к. т., 3 ч; h) Т3Р (50% в EtOAc), DIPEA, ТГФ, к. т. 1 ч; i) ТФУ, ДХМ, к. т., 30 мин; j) цианоген бромид, К2СО3, ТГФ, к. т., 30 мин.
Схема 17
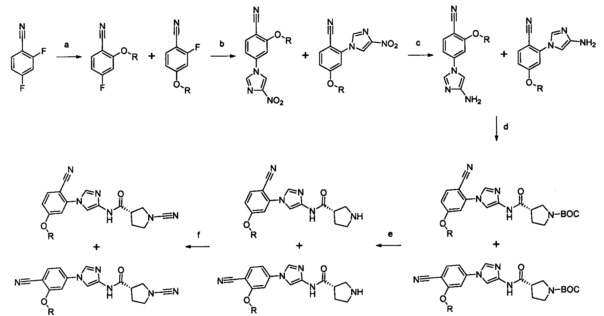
Реагенты и условия: a) ROH, NaH, ТГФ, 1,4-диоксан, от к. т. до 70°С, 16 ч; b) 4-нитро-1H-имидазол, K2СО3, KI, ДМФА, 130°С, 16 ч; с) Н2, 10% Pd/C (50% влажн.), ТГФ, к. т., 2 ч; d) Т3Р (50% в EtOAc), DIPEA, ТГФ, к. т., 1 ч; е) ТФУ, ДХМ, к. т., 30 мин f) цианоген бромид, К2СО3, ТГФ, к. т., 30 мин.
Схема 18
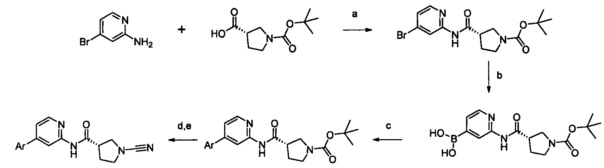
Реагенты и условия: а) POCl3, пиридин, от 0°С до к. т., 1 ч; b) биспинаколатодибор, Pd2(dba)3, X-Phos, СН3СО2K, 1,4-диоксан, 110°С, 2 ч; с) Ar-Hal, Pd(dppi)Cl2, K2СО3, ДМФА, 80°С, 2 ч; d) ДХМ, ТФУ, к. т., 1 ч; е) цианоген бромид, K2СО3, ТГФ, к. т. 1 ч.
Схема 19
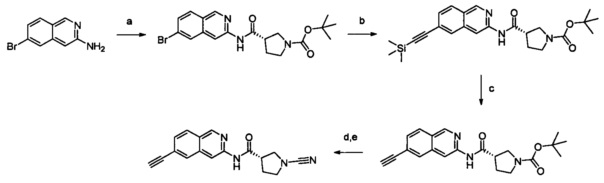
Реагенты и условия: а) (3S)-ВОС-1-пирролидин-3-карбоновая кислота, POCl3, пиридин, ДХМ, 0°С, 30 мин; b) триметилсилилацетилен, CuI, PdCl2(PPh3)2, диизопропиламин, 120°С 30 мин; с) K2СО3, МеОН, от 0°С до к. т., 1 ч; d) ТФУ, ДХМ, от 0°С до к. т., 1 ч; е) цианоген бромид, K2СО3, ТГФ, от 0°С до к. т., 1 ч.
Схема 20
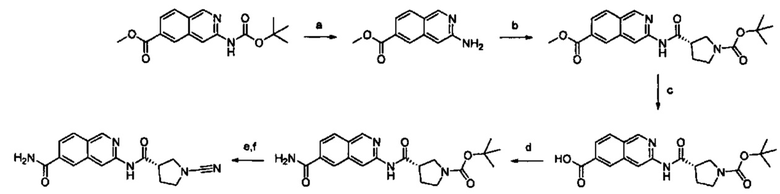
Реагенты и условия: а) ТФУ, ДХМ, от 0°С до к. т., 3 ч; b) (3S)-ВОС-1-пирролидин-3-карбоновая кислота, POCl3, пиридин, ДХМ, 0°С, 30 мин; с) LiOH, МеОН, вода, 60°С, 3 ч; d) NH4HCO3, HATU, DIPEA, ТГФ, к. т., 42 ч; е) ТФУ, ДХМ, от 0°С до к. т., 2 ч; f) цианоген бромид, K2СО3, ТГФ, от 0°С до к. т., 30 мин.
Промежуточное соединение 1 ((2S,3S)-1-[(трет-бутокси)карбонил]-2-метилпирролидин-3-карбоновая кислота)
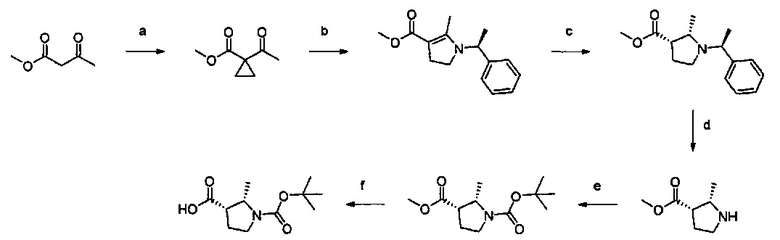
Реагенты и условия: а) K2CO3, ацетон, 1,2-дибромэтан, 70°С 24 ч; b) (S)-1-фенилэтан-1-амин, толуол, 110°С 22 ч; с) NaBH(ОАс)3, уксусная кислота, MeCN, 0°С 3 часа d) 10% Pd/C, Н2 425 psi (примерно 2,9 МПа), МеОН, автоклав, к. т. 16 ч; е) ВОС-ангидрид, DMAP, ТГФ, 0°С, к. т. 16 ч; f) LiOH, вода: МеОН (1:1), от 0°С до 20°С 6 ч.
Стадия а. К раствору метилацетоацетата (1,72 моль) и 1,2-дибромэтана (1,90 моль) в ацетоне (2000 мл) добавляли K2СО3 (2,59 моль) при комнатной температуре. Реакционную смесь нагревали при 70°С в течение 24 часов. Полученной реакционной смеси давали возможность остыть до к. т. и фильтровали через Celite Hyflo. Остаток на целите промывали ацетоном (2×100 мл). Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью колоночной хроматографии (4% EtOAc в гексане), получая метил-1-ацетилциклопропан-1-карбоксилат (0,70 моль). МС: ЭР+ 143,14.
Стадия b. Раствор метил-1-ацетилциклопропан-1-карбоксилата (0,70 моль) и (S)-1-фенилэтан-1-амина (0,84 моль) в толуоле (1000 мл) вносили в стеклянный аппарат Дина-Старка. Реакционную смесь нагревали при температуре (130-140°С) для удаления воды путем азеотропной перегонки. Процесс продолжали в течение 22 часов. Реакционной смеси давали возможность остыть до к. т. и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (2% EtOAc в гексане), получая метил-(S)-2-метил-1-(1-фенилэтил)-4,5-дигидро-1Н-пиррол-3-карбоксилат (0,37 моль). МС: ЭР+ 246,33; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 7,36-7,39 (m, 2 Н), 7,26-7,29 (m, 3 Н), 4,95 (q, J=7,01 Гц, 1 Н), 3,50 (s, 3 Н), 3,41-3,49 (m, 1 Н), 3,03-3,10 (m, 1 Н), 2,55-2,57 (m, 2 Н), 2,25 (s, 3 Н), 1,48 (d, J=7,01 Гц, 3 Н).
Стадия с. К раствору (S)-2-метил-1-(1-фенилэтил)-4,5-дигидро-1Н-пиррол-3-карбоксилата (375 ммоль) в MeCN (1000 мл) добавляли уксусную кислоту (300 мл) при 0°С. Добавляли триацетоксиборогидрид натрия (751 ммоль) при 0°С равными частями. Реакционную смесь перемешивали при 0°С в течение 3 ч и полученную реакционную смесь перегоняли при пониженном давлении для удаления большей части MeCN. Полученную смесь вливали в ледяную воду (500 мл) и нейтрализовали твердым Na2CO3. Полученную смесь подвергали экстракции EtOAc (3×200 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (2-10% EtOAc в гексане), получая (210 ммоль) указанного в заголовке соединения в виде смеси диастереомеров. Указанную диастереомерную смесь далее кристаллизовали из гексана (350 мл) при -70°С, получая метил-(2S,3S)-2-метил-1-((S)-1-фенилэтил)пирролидин-3-карбоксилат (137 ммоль). МС: ЭР+ 248,15; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 7,28-7,36 (m, 4 Н), 7,19-7,24 (m, 1 Н), 3,59 (s, 3 Н), 3,55-3,58 (m, 1 Н), 3,31-3,39 (m, 1 Н), 3,03-3,10 (m, 1 Н), 2,56-2,62 (m, 1 Н), 2,40-2,46 (m, 1 Н), 1,93-2,04 (m, 1 Н), 1,76-1,84 (m, 1 Н), 1,25 (d, J=6,71 Гц, 3 Н), 0,71 (d, J=6,40 Гц, 3 Н).
Стадия d. Раствор метил-(2S,3S)-2-метил-1-((S)-1-фенилэтил)пирролидин-3-карбоксилата (137 ммоль) в МеОН (700 мл) вносили в резервуар автоклава при комнатной температуре в атмосфере азота. К реакционной смеси добавляли 10% Pd/C (0,3% масс./масс.) в атмосфере азота. Полученную реакционную смесь перемешивали в автоклаве при комнатной температуре при давлении Н2 425 psi (примерно 2,9 МПа) в течение 16 ч. Полученную реакционную смесь тщательно фильтровали через Celite Hyflo и концентрировали при пониженном давлении с получением метил-(2S,3S)-2-метилпирролидин-3-карбоксилата (количественный выход). Данный материал сразу же применяли на следующей стадии. МС: ЭР+ 144,05; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 3,59 (s, 3 H), 3,18-3,25 (m, 1 H), 2,96-3,02 (m, 1 H), 2,82-2,88 (m, 1 H), 2,61-2,66 (m, 1 H), 1,80-1,97 (m, 2 H), 0,94 (d, J=6,714 Гц, 3 H).
Стадия е. К перемешиваемому раствору метил-(2S,3S)-2-метилпирролидин-3-карбоксилата (139 ммоль) в ТГФ (200 мл) добавляли DMAP (8,1 ммоль) и ВОС-ангидрид (751 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 16 ч. Полученную реакционную смесь вливали в воду (100 мл) и подвергали экстракции посредством ДХМ (2×200 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (10% EtOAc в гексане), получая 1-(трет-бутил)-3-метил-(2S,3S)-2-метилпирролидин-1,3-дикарбоксилат (111 ммоль). МС: ЭР+ 188 (М-56) 144 (М-100); 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 3,98-4,03 (m, 1 Н), 3,64 (s, 3 Н), 3,30-3,41 (m, 1 Н), 3,12-3,29 (m, 2 Н), 2,06-2,14 (m, 1 Н), 1,91-1,99 (m, 1 Н), 1,40 (s, 9 Н), 0,95 (d, J=6,40 Гц, 3 Н).
Стадия f. Раствор 1-(трет-бутил) 3-метил-(2S,3S)-2-метилпирролидин-1,3-дикарбоксилата (111 ммоль) в смеси МеОН : вода (1:1, 400 мл) перемешивали при 0°С в течение 10 мин. Твердый LiOH (166 ммоль) добавляли порциями к реакционной смеси при 0°С. Реакционную смесь перемешивали при 20°С в течение 6 часов. Полученную реакционную смесь подвергали экстракции EtOAc (2×100 мл). Водный слой охлаждали до 0°С и нейтрализовали путем медленного добавления 1 н. HCl. Полученную смесь подвергали экстракции EtOAc (3×200 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая указанное в заголовке соединение (87,33 ммоль). МС: ЭР- 228; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,43 (ушир. s, 1 Н), 3,97-4,02 (m, 1 Н), 3,30-3,35 (m, 1Н) 3,06-3,18 (m, 2 Н), 2,01-2,10 (m, 1 Н), 1,85-1,93 (m, 1 Н), 1,40 (s, 9 Н), 1,00 (d, J=5,79 Гц, 3 Н).
Промежуточное соединение 2 (1-[(трет-бутокси)карбонил]-(±)-транс-4-метилпирролидин-3-карбоновая кислота)
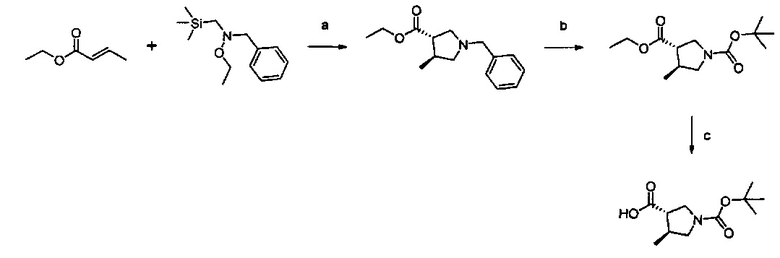
Реагенты и условия: а) ТФУ, толуол, 50°С, 16 ч; b) 20% Pd(ОН)2 на угле, полиметилгидросилоксан, ВОС-ангидрид, этанол, 0°С, к. т. 1,5 ч; с) водный раствор NaOH, вода:ТГФ (1:1), 0°С, к. т. 16 ч.
Стадия а. Раствор этилкротоната (17,5 ммоль) и N-бензил-O-этил-N-((триметилсилил)метил)гидроксиламина (19,2 ммоль) в толуоле (40 мл) перемешивали при комнатной температуре в течение 5 мин. К полученной реакционной смеси по каплям добавляли ТФУ (17,5 ммоль) при комнатной температуре. Реакционную смесь затем нагревали при 50°С в течение 16 часов. Полученную реакционную смесь вливали в воду (100 мл) и подщелачивали твердым NaHCO3. Полученную смесь подвергали экстракции EtOAc (2×180 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (0-9% EtOAc в гексане), получая этил-(±)-транс-1-бензил-4-метилпирролидин-3-карбоксилат (9,0 ммоль). МС: ЭР+ 248,33; 1H ЯМР (400 МГц, CDCl3) δ ppm 7,24-7,36 (m, 5 Н), 4,13 (q, J=8,0, 5,2 Гц, 2 Н), 3,67 (d, J=13 Гц, 1 Н), 3,58 (d, J=13 Гц, 1 Н), 2,77-2,91 (m, 3 Н), 2,47-2,59 (m, 2 Н), 2,21-2,26 (m, 1 Н), 1,27 (t, J=7,2 Гц, 3 Н), 1,16 (d, J=6,71 Гц, 3 Н).
Стадия b. К раствору этил-(±)-транс-1-бензил-4-метилпирролидин-3-карбоксилата (10 ммоль) в этаноле (30 мл) добавляли полиметилгидросилоксан (1,0 масс./масс.), 20% Pd(OH)2 на угле (0,5 масс./масс.) и ВОС-ангидрид (20 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часа. Полученную реакционную смесь тщательно фильтровали через Celite Hyflo и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (0-10% EtOAc в гексане), получая 1-трет-бутил-3-этил-(±)-4-метилпирролидин-1,3-дикарбоксилат (8,5 ммоль). МС: ЭР+ 202,2 (М-56).
Стадия с. Раствор 1-трет-бутил-3-этил-(±)-4-метилпирролидин-1,3-дикарбоксилата (8,5 ммоль) в ТГФ (15 мл) перемешивали при 0°С в течение 5 минут. К полученной реакционной смеси по каплям добавляли раствор NaOH (34,0 ммоль) в воде (15 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Полученную реакционную смесь вливали в воду (200 мл) и подкисляли до рН 4,0 разбавленной HCl. Полученную смесь подвергали экстракции EtOAc (2×150 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 1-[(трет-бутокси)карбонил]-(±)-транс-4-метилпирролидин-3-карбоновую кислоту (7,1 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР- 228,28; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,51 (ушир. s, 1 Н), 3,47-3,56 (m, 2 Н), 3,28-3,34 (m, 1 Н), 2,78-2,86 (m, 1 Н), 2,58-2,64 (m, 1H), 2,27-2,34 (m, 1 Н), 1,38 (s, 9 Н), 1,04 (d, J=4,8 Гц, 3H).
Промежуточное соединение 3 5-(тетрагидро-2Н-пиран-4-ил)тиазол-2-амин
Реагенты и условия: а) п-толуолсульфоновой кислоты моногидрат, пирролидин, циклогексан 80°С, 3 ч, далее порошок S, цианамид, МеОН, 0°С, к. т. 16 ч.
Стадия а. К перемешиваемому раствору 2-(тетрагидро-2Н-пиран-4-ил) ацетальдегида (4,7 ммоль), пирролидина (5,6 ммоль) и п-толуолсульфоновой кислоты моногидрата (0,5 ммоль) в циклогексане (20 мл) добавляли высушенные в печи молекулярные сита. Реакционную смесь нагревали при 80°С в течение 3 часов. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток повторно растворяли в МеОН (3 мл) и охлаждали до 0°С. К этому раствору добавляли порошок серы (4,7 ммоль) и раствор цианамида (4,7 ммоль) в МеОН (0,5 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Полученную реакционную смесь концентрировали при пониженном давлении, вливали в воду (50 мл) и подвергали экстракции посредством EtOAc (3×50 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (2-3% МеОН в ДХМ), получая 5-(тетрагидро-2Н-пиран-4-ил)тиазол-2-амин (1,6 ммоль). МС: ЭР+ 185,14.
Промежуточное соединение 4 1-метил-5-фенил-1Н-пиразол-3-амин
Реагенты и условия: а) Бром, МеОН, 0°С, к. т. 16 ч; b) метилгидразин, МеОН, 75°С, 16 ч.
Стадия а. Раствор циннамонитрила (7,7 ммоль) в МеОН (10 мл) перемешивали при 0°С в течение 5 минут. К реакционной смеси добавляли бром (15,5 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Полученную реакционную смесь вливали в воду (50 мл) и подщелачивали твердым NaHCO3. Полученную смесь подвергали экстракции EtOAc (3×50 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (0-4% EtOAc в гексане), получая 2,3-дибром-3-фенилпропаннитрил (1,66 ммоль). Данный материал сразу же применяли на следующей стадии без дополнительной очистки.
Стадия b. К раствору 2,3-дибром-3-фенилпропаннитрила (1,6 ммоль) в МеОН (10 мл) добавляли метилгидразин (1,6 ммоль) при комнатной температуре. Реакционную смесь нагревали при 75°С в течение 16 ч. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (2-3% МеОН в ДХМ), получая 1-метил-5-фенил-1Н-пиразол-3-амин (количественный выход). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 174,24
Промежуточное соединение 5 5-(4-метоксипиперидин-1-ил)пиридин-2-амин
Реагенты и условия: а) 4-метоксипиперидин HCl, K2CO3, TBAI, ДМСО, 80°С 15 ч; b) 10% Pd/C, Н2, МеОН, к. т. 1 ч.
Стадия а. Раствор 5-бром-2-нитропиридина (2,0 ммоль) и 4-метоксипиперидина HCl (4,0 ммоль) в ДМСО (10 мл) перемешивали при комнатной температуре в течение 5 мин. Затем к реакционной смеси добавляли K2CO3 (4,0 ммоль) и TBAI (0,2 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 15 ч.
Полученную реакционную смесь вливали в воду (200 мл) и подвергали экстракции посредством EtOAc (2×150 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (15% EtOAc в гексане), получая 5-(4-метоксипиперидин-1-ил)-2-нитропиридин (1,6 ммоль). МС: ЭР+ 238,35; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 8,27 (d, J=3,05 Гц, 1 Н), 8,14 (d, J=9,16 Гц, 1 Н), 7,49 (dd, J=9,16, 3,05 Гц, 1 Н), 3,75-3,84 (m, 2 Н), 3,46-3,48 (m, 1 Н), 3,30-3,35 (m, 2 Н), 3,28 (s, 3 Н), 1,90-1,95 (m, 2 Н), 1,48-1,53 (m, 2 Н).
Стадия b. К раствору 5-(4-метоксипиперидин-1-ил)-2-нитропиридина (1,6 ммоль) в МеОН (20 мл) добавляли 10% Pd/C (0,25% масс./масс.) при комнатной температуре. Реакционную смесь продували газообразным Н2 при комнатной температуре в течение 1 часа. Полученную реакционную смесь тщательно фильтровали через Celite Hyflo и концентрировали при пониженном давлении с получением 5-(4-метоксипиперидин-1-ил)пиридин-2-амина (1,55 ммоль). МС: ЭР+ 208,08; 1Н ЯМР (400 МГц, ДMCO-d6) δ ppm 7,61 (d, J=3,05 Гц, 1 Н), 7,16 (dd, J=8,85, 2,75 Гц, 1 Н), 6,38 (d, J=8,85 Гц, 1 Н), 5,40 (ушир. s, 2 Н), 3,22-3,31 (m, 4 Н), 3,15-3,22 (m, 2 Н), 2,61-2,71 (m, 2 Н), 1,88-1,97 (m, 2 Н), 1,46-1,58 (m, 2 Н).
Промежуточное соединение 6 5-циклогексилпиридин-2-амин
Реагенты и условия: a) ZnCl2 (0,7М в ТГФ), циклогексилмагния бромид (1M в ТГФ), 2-амино-5-бромпиридин, Pd(dppf)Cl2.CH2Cl2, 1,4-диоксан, 0°С, затем при к. т. до кипения в течение 20 ч.
Стадия а. К раствору 2-амино-5-бромпиридина (2,8 ммоль) в 1,4-диоксане (5 мл) добавляли Pd(dppf)Cl2.CH2Cl2 (0,06 ммоль) при 0°С. Полученную реакционную смесь оставляли перемешиваться при 0°С. В это время готовили свежий раствор Гриньяра путем добавления циклогексилмагния бромида (1М в ТГФ) (11,5 ммоль) к ZnCl2 (0,7М в ТГФ) (5,8 ммоль) при 0°С. Полученный раствор Гриньяра разбавляли путем добавления по каплям 1,4-диоксана (10 мл) при 0°С. Полученный свежеприготовленный раствор Гриньяра по каплям добавляли к реакционной смеси, поддерживая температуру при 0°С. Полученной реакционной смеси затем давали достичь к. т. и затем кипятили с обратным холодильником в течение 20 ч. Полученную реакционную смесь охлаждали и затем вливали в насыщенный раствор NaHCO3 (25 мл) и подвергали экстракции посредством EtOAc (3×50 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (40-50% EtOAc в гексане), получая 5-циклогексилпиридин-2-амин (2,3 ммоль). МС: ЭР+ 177,19.
Промежуточные соединения 7 и 8 6-(1Н-1,2,3-триазол-1-ил)бензо[d]тиазол-2-амин и 6-(2Н-1,2,3-триазол-2-ил)бензо[d]тиазол-2-амин
Реагенты и условия: а) 1,2,3-триазол, Cs2CO3, ДМФА, 70°С 3 ч; b) Fe, уксусная кислота, вода, этанол, 80°С 1 ч; с) тиоцианат аммония, бром, уксусная кислота, 0°С, к. т. 3 ч.
Стадия а. Раствор 1,2,3-триазола (14,17 ммоль) и Cs2CO3 (35,5 ммоль) в ДМФА (20 мл) перемешивали при комнатной температуре в течение 30 мин. К реакционной смеси добавляли 1-фтор-4-нитробензол (14,17 ммоль) при комнатной температуре и затем перемешивали при 70°С в течение 3 часов. Полученную реакционную смесь вливали в охлажденный солевой раствор (50 мл). Полученный осадок белого цвета собирали путем фильтрования при пониженном давлении и промывали охлажденной водой (2×10 мл). Полученной твердое вещество представляло собой смесь региоизомеров, и их дополнительно разделяли посредством колоночной хроматографии (0-9% EtOAc в гексане), получая 1-(4-нитрофенил)-1Н-1,2,3-триазол (4,3 ммоль) МС: ЭР+ 191,34; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 9,08 (d, J=1,22 Гц, 1 Н), 8,45-8,49 (m, 2 Н), 8,24-8,30 (m, 2 Н), 8,09 (d, J=1,22 Гц, 1 Н) и 2-(4-нитрофенил)-2Н-1,2,3-триазол (5,8 ммоль) МС: ЭР+ 191,3; 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 8,43-8,46 (m, 2 Н), 8,27-8,30 (m, 4 Н).
Стадия b. К раствору 1-(4-нитрофенил)-1Н-1,2,3-триазола (5,78 ммоль) в этаноле (8 мл) и воде (4 мл) добавляли порошок Fe (34,73 ммоль) и уксусную кислоту (34,74 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 80°С в течение 1 часа. Полученной реакционной смеси давали возможность остыть до к. т. и вливали в смесь ДХМ (20 мл) и воды (10 мл). Полученную эмульсию фильтровали через Celite Hyflo. Органический слой отделяли от полученного фильтрата и водный слой снова подвергали экстракции посредством ДХМ (10 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 4-(1Н-1,2,3-триазол-1-ил)анилин (5,6 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 161,10.
С 2-(4-нитрофенил)-2Н-1,2,3-триазолом проводили аналогичные процессы с получением 4-(2Н-1,2,3-триазол-2-ил)анилина. МС: ЭР+ 161,10.
Стадия с. К раствору 4-(1Н-1,2,3-триазол-1-ил)анилина (3,43 ммоль) в уксусной кислоте (5 мл) добавляли тиоцианат аммония (8,59 ммоль) при комнатной температуре и реакционную смесь затем охлаждали до 10°С. К вышеуказанной реакционной смеси по каплям добавляли бром (3,43 ммоль) в уксусной кислоте (2 мл) при 10°С. При добавлении брома наблюдали получение густой массы. Реакционную смесь затем тщательно перемешивали при к. т. в течение 3 ч. Полученную реакционную смесь вливали в охлажденную воду (30 мл) и нейтрализовали, применяя гидроксид аммония. Полученный осадок желтого цвета собирали путем фильтрования при пониженном давлении, промывали охлажденной водой (2×10 мл), МеОН (2×5 мл) и сушили с получением 6-(1Н-1,2,3-триазол-1-ил)бензо[d]тиазол-2-амина (2,85 ммоль). МС: ЭР+ 218,13; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 8,73 (d, J=0,92 Гц, 1 Н), 8,23 (d, J=2,14 Гц, 1 Н), 7,95 (d, J=0,92 Гц, 1 Н), 7,75 (s, 2 Н), 7,71 (dd, J=8,55, 2,44 Гц, 1 Н), 7,48 (d, J=8,55 Гц, 1Н).
С 4-(2Н-1,2,3-триазол-2-ил)анилином проводили аналогичные процессы с получением 6-(2Н-1,2,3-триазол-2-ил)бензо[d]тиазол-2-амина. МС: ЭР+ 218,13; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 8,35 (s, 1 Н), 8,08-8,11 (m, 2 Н), 7,88 (d, J=9,16 Гц, 1 Н), 7,69 (ушир. s, 2 Н), 7,45 (d, J=8,85 Гц, 1 Н).
Промежуточное соединение 9 4-(2-аминотиазол-5-ил)-N-метилбензамид
Реагенты и условия: а) метил-4-бромбензоат, КОАс, Pd(PPh3)4, ДМАА, 130°С, 16 ч; b) триметилалюминий (2M в толуоле), метиламин, DIPEA, толуол, 0°С 15 мин, затем при 100°С 16 ч; с) ТФУ, ДХМ, к. т., 48 ч.
Стадия а. Готовили раствор N-(2,4-диметоксибензил)тиазол-2-амина (7,9 ммоль), метил-4-бромбензоата (11,9 ммоль) и КОАс (23,97 ммоль) в ДМАА (50 мл) в стеклянной пробирке. Через полученную реакционную смесь пропускали поток газообразного азота при комнатной температуре в течение 30 мин. К реакционной смеси добавляли Pd(PPh3)4 (0,79 моль) и стеклянную пробирку плотно закрывали. Закрытую пробирку нагревали при 140°С (наружная температура) в течение 16 часов. Полученную реакционную смесь вливали в воду (100 мл) и подвергали экстракции посредством EtOAc (2×50 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (25% EtOAc в гексане), получая метил-4-(2-((2,4-диметоксибензил)-амино)тиазол-5-ил)бензоат (3,9 ммоль). МС: ЭР+ 385,68.
Стадия b. К раствору метил-4-(2-((2,4-диметоксибензил)амино)тиазол-5-ил)бензоата (1,8 ммоль) и DIPEA (0,9 ммоль) в толуоле (15 мл) добавляли триметилалюминий (2 М в толуоле) (9,1 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 15 мин. К реакционной смеси добавляли метиламин (2,18 ммоль) и затем нагревали при 100°С в течение 16 часов. Полученную реакционную смесь концентрировали при пониженном давлении и полученный остаток вливали в насыщенный раствор NaHCO3 (30 мл). Полученную смесь подвергали экстракции EtOAc (2×20 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 4-(2-((2,4-диметоксибензил)амино)тиазол-5-ил)-N-метилбензамид (0,86 ммоль). МС: ЭР+ 384,38.
Стадия с. К раствору 4-(2-((2,4-диметоксибензил)амино)тиазол-5-ил)-N-метилбензамида (0,80 ммоль) в ДХМ (5 мл) добавляли ТФУ (3 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 48 часов. Полученную реакционную смесь вливали в воду (10 мл) и подвергали экстракции посредством EtOAc (2×10 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая соль ТФУ и 4-(2-аминотиазол-5-ил)-N-метилбензамида (0,63 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 234,35 Промежуточное соединение 10 5-(2-хлорфенил)тиазол-2-амин
Синтез проводили с помощью методики, аналогичной методике, описанной для Промежуточного соединения 9, применяя 1-бром-2-хлорбензол (номер в CAS 694-80-4) на стадии а. МС: ЭР+ 211,13; 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 7,55 (dd, J=7,6 Гц, 1,6 Гц, 1 Н), 7,50 (dd, J=6,0 Гц, 1,2 Гц, 1 Н), 7,36 (s, 1 Н), 7,33 (dd, J=6,0 Гц, 1,2 Гц, 1 Н), 7,25-7,29 (m, 1 Н), 7,21 (ушир. s, 2 Н).
Промежуточное соединение 11 1-(трет-бутоксикарбонил)-3-(метоксиметил)пирролидин-3-карбоновая кислота
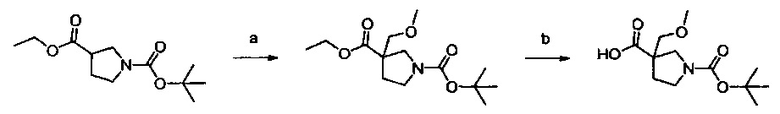
Реагенты и условия: а) бромметилметиловый эфир, свежеприготовленный LDA, ТГФ, -78°С 45 мин, к. т. 16 ч; b) LiOH, ТГФ, вода, к. т. 16 ч.
Стадия а. Раствор 1-(трет-бутил) 3-этил-пирролидин-1,3-дикарбоксилата (2,0 ммоль) в ТГФ (5 мл) охлаждали до -78°С. К полученной реакционной смеси по каплям добавляли свежеприготовленный LDA (2,2 ммоль) при -78°С. Реакционную смесь перемешивали при -78°С в течение 45 мин. К реакционной смеси по каплям добавляли бромметилметиловый эфир (2,2 ммоль) при -78°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Полученную реакционную смесь вливали в насыщенный раствор NH4Cl (10 мл) и подвергали экстракции посредством EtOAc (3×20 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 1-(трет-бутил) 3-этил-3-(метоксиметил)пирролидин-1,3-дикарбоксилат (количественный выход). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 288,70
Стадия b. К раствору 1-(трет-бутил)-3-этил-3-(метоксиметил)пирролидин-1,3-дикарбоксилата (2,1 ммоль) в смеси ТГФ: вода (3:1, 10 мл) добавляли LiOH (8,3 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Полученную реакционную смесь разбавляли водой (10 мл) и доводили до рН 3, применяя насыщенную водным раствором лимонной кислоты. Полученную смесь подвергали экстракции ДХМ (3×20 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 1-(трет-бутоксикарбонил)-3-(метоксиметил)-пирролидин-3-карбоновую кислоту (0,96 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР- 257,40
Промежуточное соединение 12 1-(трет-бутоксикарбонил)-3-цианопирролидин-3-карбоновая кислота
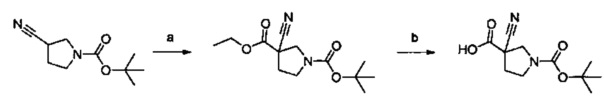
Реагенты и условия: а) этилхлорформиат, LiHMDS, ТГФ, -78°С 30 мин, к. т., 2 ч; b) LiOH, ТГФ, вода, к. т. 16 ч.
Стадия а. Раствор трет-бутил-3-цианопирролидин-1-карбоксилата (2,8 ммоль) в ТГФ (5 мл) охлаждали до -78°С. К полученной реакционной смеси по каплям добавляли LiHMDS (1М в гексане) (7,0 ммоль) при -78°С. Реакционную смесь перемешивали при -78°С в течение 30 мин. К реакционной смеси по каплям добавляли этилхлорформиат (8,4 ммоль) при -78°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Полученную реакционную смесь повторно охлаждали до -78°С и гасили насыщенным раствором NH4Cl (10 мл). Полученную смесь подвергали экстракции EtOAc (3×20 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 1-(трет-бутил) 3-этил-3-цианопирролидин-1,3-дикарбоксилат (количественный выход). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 269,60
Стадия b. К раствору 1-(трет-бутил)-3-этил-3-цианопирролидин-1,3-дикарбоксилата (1,8 ммоль) в смеси ТГФ: вода (3:1, 13 мл) добавляли LiOH (5,6 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Полученную реакционную смесь разбавляли водой (10 мл) и доводили до рН 3, применяя насыщенную водным раствором лимонной кислоты. Полученную смесь подвергали экстракции EtOAc (3×20 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 1-(трет-бутоксикарбонил)-3-цианопирролидин-3-карбоновую кислоту (0,8 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР- 239,50.
Промежуточное соединение 13 трет-бутил-(4-метил-5-(морфолинометил)тиазол-2-ил)карбамат
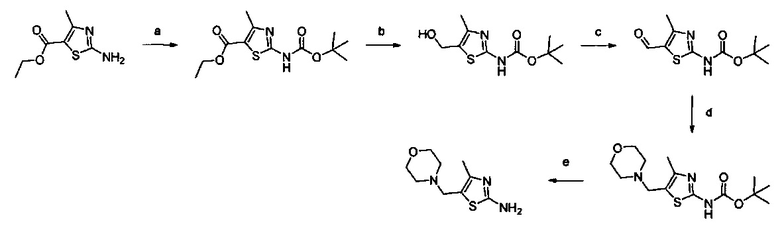
Реагенты и условия: а) (ВОС)2O, DMAP, ТГФ, к. т. 18 ч; b) 2,0 М LiAlH4 в ТГФ, ТГФ, 0°С, к. т. 2 ч; с) периодинан Десса-Мартина, ДХМ, к. т., 24 ч; d) морфолин, ДХЭ, 35°С 3 ч, NaBH(OAc)3, 60°С 5 ч; е) ТФУ, ДХМ, к. т. 3 ч.
Стадия а. К раствору этил-2-амино-4-метилтиазол-5-карбоксилата (10,75 ммоль) в ТГФ (15 мл) добавляли (ВОС)2О (12,90 ммоль), DMAP (1,07 ммоль) и ТЭА (16,13 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Полученную реакционную смесь разбавляли посредством EtOAc (180 мл) и промывали 1М HCl (100 мл) и солевым раствором (100 мл). Органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая этил-2-((трет-бутоксикарбонил)амино)-4-метилтиазол-5-карбоксилат (8,39 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки.
Стадия b. К раствору этил-2-((трет-бутоксикарбонил) амино)-4-метилтиазол-5-карбоксилата (8,39 ммоль) в ТГФ (15 мл) добавляли LiAlH4 (2,0М в ТГФ) (10,06 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Полученную реакционную смесь вливали в воду (100 мл). Полученную смесь фильтровали через Celite Hyflo и промывали EtOAc (50 мл). Отделяли органический слой от фильтрата и водный слой дополнительно подвергали экстракции посредством EtOAc (3×150 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая трет-бутил-(5-(гидроксиметил)-4-метилтиазол-2-ил)карбамат (7,17 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 245,33.
Стадия с. К раствору трет-бутил-(5-(гидроксиметил)-4-метилтиазол-2-ил)карбамата (7,17 ммоль) в ДХМ (80 мл) добавляли периодинан Десса-Мартина (7,05 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Полученную реакционную смесь вливали в воду (100 мл). Полученную смесь нейтрализовали путем добавления водного раствора насыщенного NaHCO3. Полученную смесь подвергали экстракции ДХМ (2×100 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая трет-бутил-(5-формил-4-метилтиазол-2-ил)карбамат (4,00 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 243,48.
Стадия d. К раствору трет-бутил-(5-формил-4-метилтиазол-2-ил)карбамата (4,00 ммоль) в ДХЭ (5 мл) добавляли морфолин (8,01 ммоль) при комнатной температуре. Реакционную смесь нагревали при 35°С в течение 3 часов. К реакционной смеси добавляли NaBH(OAc)3 (12,02 ммоль) при 35°С. Реакционную смесь затем нагревали при 60°С в течение 5 часов. Полученной реакционной смеси давали возможность остыть до к. т., вливали в ледяную воду (100 мл) и подвергали экстракции посредством ДХМ (3×100 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (5% МеОН в ДХМ), получая трет-бутил-(4-метил-5-(морфолинометил)тиазол-2-ил)карбамата (2,55 ммоль). МС: ЭР+ 314,43.
Стадия е. К раствору трет-бутил-(4-метил-5-(морфолинометил)тиазол-2-ил)карбамата (2,55 ммоль) в ДХМ (10 мл) добавляли ТФУ (25,5 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток растирали с диэтиловым эфиром (2×5 мл), получая соль ТФУ и 4-метил-5-(морфолинометил) тиазол-2-амина (1,69 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 214,15
Промежуточное соединение 14 трет-бутил-(S)-3-((4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)карбамоил)пирролидин-1-карбоксилат
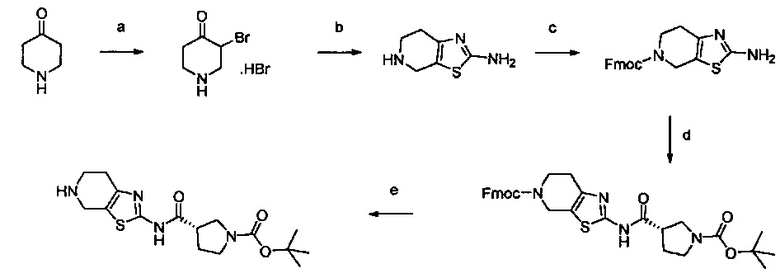
Реагенты и условия: а) 30% HBr в АсОН, 4,8М бром в АсОН, АсОН, к. т. 1 ч; b) тиомочевина, этанол, 80°С 16 ч; с) FMoc-Cl, K2СО3, 1,4-диоксан, вода, 0°С, к. т. 3 ч; d) Т3Р (50% в EtOAc), DIPEA, ТГФ, к. т. 3 ч; е) пиперидин, МеОН, к. т. 2 ч.
Стадия а. К раствору пиперидин-4-он гидрохлорида (37,31 ммоль) в уксусной кислоте (30 мл) добавляли 30% HBr в уксусной кислоте (0,47 мл) и 4,8 М бром в уксусной кислоте (3,0 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Полученную реакционную смесь концентрировали при пониженном давлении с получением остатка, который суспендировали в ацетоне (100 мл) и нагревали с обратным холодильником в течение 1 часа. Полученный твердый осадок собирали путем фильтрования при пониженном давлении, промывали ацетоном (30 мл) и сушили с получением 3-бромпиперидин-4-она гидробромида (31,28 ммоль). Материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 178,0, 180,0
Стадия b. К раствору 3-бромпиперидин-4-она гидробромида (31,39 ммоль) в этаноле (100 мл) добавляли тиомочевину (32,0 ммоль) и реакционную смесь нагревали при 80°С в течение 16 часов. Полученной реакционной смеси давали возможность остыть до к. т. Наблюдали выпадение осадка в реакционной смеси. Полученный осадок собирали путем фильтрования при пониженном давлении, промывали этанолом (50 мл) и сушили с получением 4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-амина гидробромида (количественный выход). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 156,19.
Стадия с. К раствору 4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-амина гидробромида (31,77 ммоль) в 1,4-диоксане (47 мл) добавляли воду (75 мл), K2СО3 (63,5 ммоль) и Fmoc-Cl (47,0 ммоль) при 0°С. Полученной реакционной смеси давали возможность нагреться до к. т. и перемешивали ее в течение 3 ч. Полученную реакционную смесь вливали в воду (500 мл) и подвергали экстракции посредством ДХМ (3×100 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (2% МеОН в ДХМ), получая (9Н-флуорен-9-ил)метил-2-амино-6,7-дигидротиазоло[5,4-с]пиридин-5(4Н)-карбоксилат (11,93 ммоль). МС: ЭР+ 378,54; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 7,85-7,90 (m, 2 Н), 7,63-7,65 (m, 2 Н), 7,40-7,44 (m, 2 Н), 7,30-7,34 (m, 2 Н), 6,83 (ушир. s, 2 Н), 4,31-4,42 (m, 5 Н), 3,47-3,60 (m, 2 Н), 2,29-2,43 (m, 2 Н).
Стадия d. К раствору (3S)-ВОС-1-пирролидин-3-карбоновой кислоты (8,37 ммоль) в ТГФ (20 мл) добавляли Т3Р (50% в EtOAc) (25,11 ммоль) при комнатной температуре. Полученную реакционную смесь перемешивали в течение 20 мин. К реакционной смеси добавляли (9Н-флуорен-9-ил)метил-2-амино-6,7-дигидротиазоло[5,4-с]пиридин-5(4Н)-карбоксилат (10,04 ммоль) и DIPEA (25,11 ммоль) и перемешивали при комнатной температуре в течение 3 ч. Полученную реакционную смесь вливали в воду (100 мл) и проводили экстракцию посредством EtOAc (3×100 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (2-3% МеОН в ДХМ), получая (9Н-флуорен-9-ил)метил-(S)-2-(1-(трет-бутоксикарбонил)пирролидин-3-карбоксамидо)-6,7-дигидротиазоло[5,4-с]пиридин-5(4Н)-карбоксилат (3,74 ммоль). МС: ЭР+ 575,74.
Стадия е. К раствору (9Н-флуорен-9-ил)метил-(S)-2-(1-(трет-бутоксикарбонил)пирролидин-3-карбоксамидо)-6,7-дигидротиазоло[5,4-с]пиридин-5(4Н)-карбоксилата (3,7 ммоль) в МеОН (10 мл) добавляли пиперидин (14,9 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (4% МеОН в ДХМ), получая трет-бутил-(S)-3-((4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)карбамоил)пирролидин-1-карбоксилат (0,77 ммоль). МС: ЭР+ 353,43.
Промежуточное соединение 15 6-бром-5-метилбензо[d]тиазол-2-амин
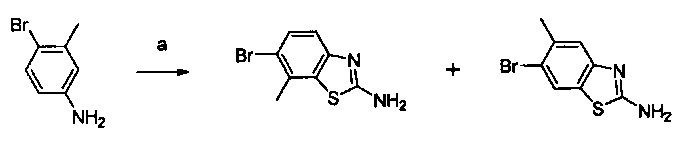
Реагенты и условия: а) тиоцианат аммония, АсОН, 10°С, к. т. 1,5 ч, далее бром в АсОН, 10°С 1,5 ч, далее NaOH, вода, рН=9.
Стадия а. К раствору 4-бром-3-метил-анилина (26,88 ммоль) в ледяной уксусной кислоте (77 мл) добавляли тиоцианат аммония (53,68 ммоль) при комнатной температуре. Полученную реакционную смесь оставляли перемешиваться до практически прозрачного состояния. Реакционную смесь охлаждали до 10°С и в нее по каплям добавляли раствор брома (26,88 ммоль) в ледяной уксусной кислоте (2,5 мл) при 10°С. Полученную реакционную смесь перемешивали при комнатной температуре в течение 1,5 ч. Полученные осадки собирали путем фильтрования и промывали холодной уксусной кислотой. Полученный остаток грязно-белого цвета вносили в воду и доводили рН до значения 9 посредством 1М раствора NaOH. Полученное твердое вещество собирали путем фильтрования, промывали водой (100 мл) и сушили при пониженном давлении, получая 14,46 ммоль смеси, содержащей 70% 6-бром-7-метил-бензотиазол-2-иламина. МС: ЭР+ 243,1 (М) 245,1 (М+2); 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 7,61 (ушир. s, 2Н), 7,40, (d, J=8,8 Гц, 1H), 7,10 (d, J=8,8 Гц, 1Н), 2,41 (s, 3Н) и 30% 6-бром-5-метил-бензотиазол-2-иламина. МС: ЭР+ 243,10; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 7,88 (s, 1H), 7,57, (ушир. s, 2Н), 7,31 (s, 1H), 2,34 (s, 3Н).
Промежуточное соединение 16 (6-бром-5-метилимидазо[1,2-а]пиридин-2-амин)
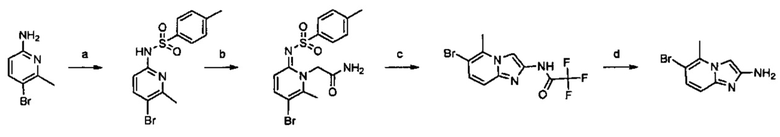
Реагенты и условия: а) пиридин, 4-толуолсульфонилхлорид, 110°С 3 ч; b) DIPEA, ДМФА, 2-бромацетамид, 80°С 48 ч; с) ТФУА, ДХМ, 80°С 30 мин; d) LiOH.H2O, ТГФ, вода, к. т. 48 ч.
Стадия а. К 4-толуолсульфонилхлориду (32,08 ммоль) медленно добавляли раствор 5-бром-6-метилпиридин-2-амина (26,73 ммоль) в пиридине (60 мл) при 0°С. Реакционную смесь нагревали при 110°С в течение 3 ч. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток вливали в воду (300 мл) и перемешивали в течение 1 ч при комнатной температуре. Полученные твердые вещества собирали путем фильтрования и сушили с получением N-(5-бром-6-метилпиридин-2-ил)-4-метилбензолсульфонамида (13,19 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 341,3, 343,28; 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 11,14 (s, 1 Н), 7,82 (t, J=7,6 Гц, 3 Н), 7,81 (d, J=8,0, 2 Н), 6,84 (d, J=8,4 Гц, 1 Н), 2,39 (s, 3 Н), 2,35 (s, 3 Н).
Стадия b. К раствору N-(5-бром-6-метилпиридин-2-ил)-4-метилбензолсульфонамида (13,19 ммоль) в ДМФА (36 мл) в стеклянной пробирке добавляли 2-бромацетамид (15,81 ммоль) и DIPEA (15,81 ммоль) при комнатной температуре. Реакционную смесь нагревали при 80°С в течение 48 часов. Полученной реакционной смеси давали возможность остыть до к. т. и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (2% МеОН в ДХМ), получая (Z)-2-(5-бром-6-метил-2-(тозилимино) пиридин-1(2Н)-ил) ацетамид (2,26 ммоль). МС: ЭР+ 398,0, 400,5.
Стадия с. К раствору (Z)-2-(5-бром-6-метил-2-(тозилимино)пиридин-1(2Н)-ил) ацетамида (2,26 ммоль) в ДХМ (9 мл) добавляли ТФУА (4,5 мл) при комнатной температуре. Реакционную смесь нагревали при 80°С в течение 30 мин. Полученной реакционной смеси давали возможность остыть до к. т. и вливали в ледяную воду (150 мл). Полученную смесь нейтрализовали путем добавления водного раствора насыщенного NaHCO3. Полученную смесь подвергали экстракции ДХМ (3×50 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (1% МеОН в ДХМ), получая N-(6-бром-5-метилимидазо[1,2-а]пиридин-2-ил)-2,2,2-трифторацетамид (1,86 ммоль). МС: ЭР+ 322,40, 324,38; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,57 (ушир. s, 1 Н), 8,05 (s, 1 Н), 7,52 (d, J=9,6, 1 Н), 7,43 (d, J=9,6 Гц, 1 Н), 2,75 (s, 3 Н).
Стадия d. К раствору N-(6-бром-5-метилимидазо[1,2-а]пиридин-2-ил)-2,2,2-трифторацетамида (3,11 ммоль) в смеси ТГФ: вода (3:1, 13 мл) порциями добавляли твердый LiOH.H2O (12,44 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 48 ч. Полученную реакционную смесь вливали в воду (100 мл) и подвергали экстракции посредством EtOAc (3×50 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (7% МеОН в ДХМ), получая 6-бром-5-метилимидазо[1,2-а]пиридин-2-амин (2,47 ммоль). МС: ЭР+ 226,0, 227,90; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 7,20 (d, J=9,20 Гц, 1 H), 7,06 (d, J=9,20 Гц, 1 H), 6,83 (s, 1 H), 5,22 (ушир. s, 2 H), 2,59 (s, 3 H).
Промежуточное соединение 17 (1-[(трет-бутокси)карбонил]-(±)-транс-4-этилпирролидин-3-карбоновая кислота)
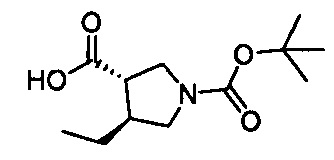
Синтез проводили с помощью методики, аналогичной методике, описанной для Промежуточного соединения 2, применяя этил-(2Е)-пент-2-эноат на стадии а. МС: ЭР+ (М-56) 188,4, (М-100) 142,2; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 1,27 (ушир. s, 1Н), 3,47-3,55 (m, 2Н), 2,85-2,93 (m, 1Н), 2,64-2,70 (m, 1H), 2,27-2,23 (m, 1H), 1,49-1,56 (m, 1H), 1,39 (s, 9Н), 1,27-1,34 (m, 2Н), 0,86 (t, J=7,2 Гц, 3Н).
Промежуточное соединение 18 1-(2-метоксиэтил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ип)-1Н-индазол
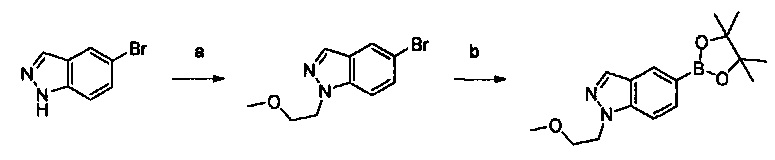
Реагенты и условия: а) 1-бром-2-метоксиэтан, Cs2CO3, ДМСО, к. т., 30 мин b) бис(пинаколато)дибор, Pd(dppf)Cl2.ДХМ, КОАс, ДМФА, 80°С, 2 ч.
Стадия а. К раствору 5-бром-1Н-индазола (2,00 г, 10,15 ммоль) и 1-бром-2-метоксиэтана (1,69 г, 12,19 ммоль) в ДМСО (20 мл) добавляли Cs2CO3 (8,22 г, 25,25 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 0,5 ч. Полученную реакционную смесь разбавляли водой (50 мл) и подвергали экстракции посредством EtOAc (2×50 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (20% EtOAc в гексане), получая 5-бром-1-(2-метоксиэтил)-1Н-индазол (1,20 г, 4,72 ммоль). ЖХМС: Способ А, 2,28 мин, МС: ЭР+ 255,16.
Стадия b. К раствору 5-бром-1-(2-метоксиэтил)-1Н-индазола (0,30 г, 1,181 ммоль) в ДМФА добавляли бис(пинаколато)дибор (0,59 г, 2,36 ммоль) и ацетат калия (0,57 г, 5,90 ммоль) при комнатной температуре. Полученную реакционную смесь дегазировали в течение 30 мин. К реакционной смеси добавляли комплекс Pd(dppf)Cl2.ДХМ (0,14 г, 0,17 ммоль) при комнатной температуре. Реакционную смесь нагревали при 80°С в течение 2 часов. Полученную реакционную смесь разбавляли водой (30 мл) и подвергали экстракции посредством EtOAc (3×30 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток перемешивали в гексане (30 мл) в течение 10 мин и декантировали гексановый слой (этот процесс повторяли еще два раза). Объединенный гексановый слой концентрировали при пониженном давлении, получая 1-(2-метоксиэтил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индазол (0,41 г, количественный выход). ЖХМС: Способ А, 2,35 мин, МС: ЭР+ 303,37.
Промежуточное соединение 19 Региоизомерная смесь трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индазол-1-карбоксилата и трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2Н-индазол-2-карбоксилата
Реагенты и условия: а) ВОС-ангидрид, DMAP, ТЭА, ДХМ, к. т., 3 ч; b) бис(пинаколато)дибор, Pd(dppf)Cl2, ДМФА, 110°С, 1 ч.
Стадия а. К перемешиваемому раствору 4-бром-1Н-индазола (3,00 г, 15,22 ммоль) в ДХМ (30 мл) добавляли DMAP (0,18 г, 1,52 ммоль), ТЭА (1,84 г, 18,27 ммоль) и ВОС-ангидрид (3,7 мл, 16,75 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, вливали в воду (100 мл) и подвергали экстракции посредством ДХМ (3×30 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (20% EtOAc в гексане), получая смесь региоизомеров с приблизительным соотношением 60:40 трет-бутил-4-бром-1Н-индазол-1-карбоксилата и трет-бутил-4-бром-2Н-индазол-2-карбоксилата (3,2 г, 10,77 ммоль). ЖХМС: Способ А, 2,50 мин, 2,61 мин, МС: ЭР+ 297,14.
Стадия b. Полученную выше региоизомерную смесь трет-бутил-4-бром-1Н-индазол-1-карбоксилата и трет-бутил-4-бром-2Н-индазол-2-карбоксилата (0,77 г, 2,59 ммоль), бис(пинаколато)дибор (1,31 г, 5,18 ммоль) и ацетат калия (1,27 г, 12,96 ммоль) в ДМФА (10 мл) дегазировали в течение 30 мин при комнатной температуре. К реакционной смеси добавляли Pd(dppf)Cl2 (0,09 г, 0,13 ммоль) при комнатной температуре. Реакционную смесь нагревали при 110°С в течение 2 часов. Полученную реакционную смесь вливали в воду (50 мл) и подвергали экстракции посредством EtOAc (3×30 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (20% EtOAc в гексане), получая региоизомерную смесь трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индазол-1-карбоксилата и трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2Н-индазол-2-карбоксилата (0,45 г, 1,30 ммоль). ЖХМС: Способ А, 3,03 мин, МС: ЭР+ 345,64.
Промежуточное соединение 20 трет-бутил-(5-бромтиазол-2-ил)(4-метоксибензил)карбамат
Реагенты и условия: а) (ВОС)2O, DMAP, ТГФ, к. т., 6 ч; b) PPh3, DIAD, n-метоксибензиловый спирт, ТГФ, от 0°С до к. т., 2 ч.
Стадия а. К раствору 2-амино-5-бромтиазола (73,06 ммоль) и DMAP (3,6 ммоль) в ТГФ (130 мл) добавляли (ВОС)2О (73,06 ммоль) при комнатной температуре. Полученную реакционную смесь перемешивали при комнатной температуре в течение 6 ч. Избыток ТГФ удаляли при пониженном давлении и полученный остаток очищали с помощью колоночной хроматографии (0-5% EtOAc в гексане), получая трет-бутил-(5-бромтиазол-2-ил)карбамат (52,1 ммоль). МС: ЭР+ 279,08; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 11,75 (s, 1H), 7,44 (s, 1Н), 1,48 (s, 9Н).
Стадия b. Смесь трет-бутил-(5-бромтиазол-2-ил)карбамата (3,6 ммоль), PPh3 (7,9 ммоль) и n-метоксибензилового спирта (7,2 ммоль) в ТГФ (10 мл) охлаждали при 0°С. К реакционной смеси по каплям добавляли DIAD (7,9 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 10 мин и затем при комнатной температуре в течение 2 ч. Полученную реакционную смесь концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии (0-5% EtOAc в гексане), получая трет-бутил-(5-бромтиазол-2-ил)(4-метоксибензил)карбамат (2,76 ммоль). МС: ЭР-56 343,1; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 7,57 (s, 1Н), 7,22 (d, J=8,4 Гц, 2Н), 6,89 (d, J=8,4 Гц, 2Н), 5,13 (s, 2Н), 3,74 (s, 3Н), 1,50 (s, 9Н).
Промежуточное соединение 21 5-(4-фторфенил)-4-метилтиазол-2-амин
Реагенты и условия: а) тиомочевина, Cu(ОАс)2, I2, метанол, 70°С, 8 ч.
Стадия а. К перемешиваемому раствору 4-фторфенилацетона (3,28 ммоль) в метаноле (5 мл) добавляли тиомочевину (4,92 ммоль) и Cu(ОАс)2 (0,33 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. К реакционной смеси добавляли иод (3,28 ммоль) при комнатной температуре и нагревали до 70°С в течение 8 часов. Полученную реакционную смесь охлаждали до к. т. и концентрировали при пониженном давлении, удаляя избыток метанола, к полученной концентрированной смеси добавляли насыщенный раствор Na2S2O3 (30 мл). Полученную смесь подвергали экстракции EtOAc (2×15 мл). Объединенную органическую фазу собирали, промывали солевым раствором (30 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток растирали с диэтиловым эфиром (2×10 мл), получая 5-(4-фторфенил)-4-метилтиазол-2-амин (2,45 ммоль). МС: ЭР+ 209,18; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 7,34 - 7,37 (m, 2 Н), 7,21 (t, J=9,2 Гц, 2 Н), 7,01 (s, 2 Н), 2,15 (s, 3 Н).
Промежуточное соединение 22 5-(1-фенилэтил) тиазол-2-амин
Реагенты и условия: а) Br2, ДХМ, от 0°С до к. т., 16 ч; b) тиомочевина, этанол, кипячение с обратным холодильником, 16 ч.
Стадия а. К раствору 3-фенилбутаналя (2,0 ммоль) в ДХМ (5 мл) по каплям добавляли бром (2,0 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Полученную реакционную смесь концентрировали при пониженном давлении, получая 2-бром-3-фенилбутанал. Данный материал сразу же применяли на следующей стадии без какой-либо дополнительной обработки.
Стадия b. К раствору 2-бром-3-фенилбутаналя (1,76 ммоль) в EtOH (7 мл) добавляли тиомочевину (3,5 ммоль) при комнатной температуре. Реакционную смесь нагревали при 80°С в течение 16 часов. Полученную реакционную смесь охлаждали до к. т. и концентрировали при пониженном давлении, к полученной концентрированной смеси добавляли воду (10 мл). Полученную смесь подвергали экстракции EtOAc (3×20 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 5-(1-фенилэтил)-тиазол-2-амин (1,71 ммоль). Данный материал применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 205,28.
Промежуточное соединение 23 5-(4-фторбензил)тиазол-2-амин
Синтез проводили с помощью методики, аналогичной методике, описанной для Промежуточного соединения 22, применяя 3-(4-фторфенил)пропиональдегид на стадии а. МС: ЭР+ 209,18; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 7,22-7,31 (m, 2 Н), 7,09-7,15 (m, 2 Н), 6,72 (ушир. s, 2 Н), 6,69 (s, 1 Н), 3,92 (s, 2 Н).
Промежуточное соединение 24 трет-бутил-(2'-амино-[4,4'-бипиридин]-2-ил)карбамат
Реагенты и условия: а) пинаколовый эфир 2-аминопиридин-4-бороновой кислоты, PdCl2(dppf), К3РО4, 1,4-диоксан, вода, 110°С (плотно закрытая пробирка), 2 ч.
Стадия а. Смесь трет-бутил-(4-бромпиридин-2-ил)карбамата (0,19 г, 0,68 ммоль) в смеси 1,4-диоксан: вода (8:2) (10 мл) получали в стеклянном флаконе. Реакционную смесь дегазировали в течение 15 мин. К реакционной смеси добавляли пинаколовый эфир 2-аминопиридин-4-бороновой кислоты (0,15 г, 0,68 ммоль) и К3РО4 (0,43 г, 2,04 ммоль) при комнатной температуре. Реакционную смесь снова дегазировали в течение 15 мин. Pd(dppf)Cl2 (0,024 г, 0,034 ммоль). Стеклянный флакон плотно закрывали и подвергали нагреванию при 110°С (наружная температура) в течение 2 часов. Полученную реакционную смесь объединяли с еще одной партией, полученной в том же масштабе идентичным способом, вливали в воду (50 мл) и подвергали экстракции посредством EtOAc (2×30 мл). Объединенную органическую фазу промывали солевым раствором (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая трет-бутил-(2'-амино-[4,4'-бипиридин]-2-ил)карбамат (0,50 г, количественный выход). ЖХМС: Способ А, 1,71 мин, МС: ЭР+ 287,48.
Промежуточное соединение 25 трет-бутил-(2'-амино-[4,4'-бипиридин]-2-ил) (метил)карбамат
Реагенты и условия: a) NaH, MeI, ДМФА, от 0°С до к. т. 1 ч; b) пинаколовый эфир 2-аминопиридин-4-бороновой кислоты, PdCl2(dppf), К3РО4, 1,4-диоксан, вода, 110°С (плотно закрытая пробирка), 3 ч.
Стадия а. К раствору трет-бутил-(4-бромпиридин-2-ил)карбамата (0,25 г, 0,915 ммоль) в ДМФА (5 мл) добавляли NaH (60% в масле) (0,073 г, 1,83 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 15 мин. К реакционной смеси добавляли метилиодид (0,26 г, 1,37 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, вливали в ледяную воду (20 мл) и подвергали экстракции посредством EtOAc (2×15 мл). Органический слой промывали солевым раствором (20 мл). Органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая трет-бутил-(4-бромпиридин-2-ил)(метил)карбамат (0,25 г, 0,87 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. ЖХМС: Способ А, 2,50 мин, МС: ЭР+ 230,9; 232,9 (М-56).
Стадия b. Методику, аналогичную описанной для Промежуточного соединения 24, применяли с получением указанного в заголовке соединения ЖХМС: Способ А, 1,71 мин, МС:ЭР+ 301,37
Промежуточное соединение 26 соль трифторуксусной кислоты и 3-амино-N-метилизохинолин-6-карбоксамида
Реагенты и условия: a) LiHMDS, ТГФ -40°С до 0°С, 30 мин, затем ВОС-ангидрид, от 0°С до к. т., 1 ч; b) Pd(dppf)Cl2.ДХМ, NaOAc, МеОН, СО (25 кг/см2), 85°С, 2 суток; с) NaOH, вода, МеОН, 70°С, 4 ч; d) метиламин (2М в ТГФ), HATU, DIPEA, ТГФ, к. т. 18 ч; е) ТФУ, ДХМ, к. т., 2 ч.
Стадия a. LiHMDS (1М в гексане) (18 мл, 18,02 ммоль) добавляли по каплям к перемешиваемому раствору 6-бромизохинолин-3-амина (2,00 г, 9,01 ммоль) в ТГФ (40 мл) при -40°С. Полученную реакционную смесь перемешивали при 0°С температуре в течение 30 мин. Раствор ВОС-ангидрида (1,96 г, 9,01 ммоль) в ТГФ (10 мл) при 0°С добавляли по каплям к реакционной смеси. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и затем вливали в раствор NH4Cl (80 мл) и подвергали экстракции посредством EtOAc (3×80 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая трет-бутил-(6-бромизохинолин-3-ил)карбамат (2,87 г, 8,91 ммоль). ЖХМС: Способ А, 2,64 мин, МС: ЭР+ 323,19.
Стадия b. Смесь трет-бутил-(6-бромизохинолин-3-ил)карбамата (2,80 г, 8,69 ммоль), натрий ацетата (3,56 г, 43,48 ммоль) и комплекс Pd(dppf)Cl2.ДХМ (3,55 г, 4,35 ммоль) в МеОН (60 мл) вносили в резервуар, работающий под давлением. Полученную реакционную смесь перемешивали при 85°С под давлением СО (25 кг/см2) в течение 48 часов. Полученную реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении. Остаток суспендировали в ДХМ и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали с помощью хроматографии (1% МеОН в ДХМ), получая метил-3-((трет-бутоксикарбонил)амино)изохинолин-6-карбоксилат (2,40 г, 7,95 ммоль). ЖХМС: Способ А, 2,43 мин, МС: ЭР+ 303.
Стадия с. Раствор NaOH (0,70 г, 17,38 ммоль) в 50 мл воды добавляли к раствору метил-3-((трет-бутоксикарбонил)амино)изохинолин-6-карбоксилата (1,75 г, 5,79 ммоль) в метаноле (50 мл) при комнатной температуре. Реакционную смесь нагревали при 70°С в течение 4 часов. Полученной реакционной смеси давали возможность остыть до к. т. и подкисляли путем медленного добавления раствора лимонной кислоты при непрерывном перемешивании. Полученную смесь подвергали экстракции EtOAc (3×100 мл). Объединенную органическую фазу собирали сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 3-((трет-бутоксикарбонил)амино)изохинолин-6-карбоновую кислоту (1,80 г, количественный выход). ЖХМС: Способ А, 2,12 мин, МС: ЭР+ 289,33.
Стадия d. Смесь 3-((трет-бутоксикарбонил)амино)изохинолин-6-карбоновой кислоты (1,80 г, 6,25 ммоль), HATU (3,56 г, 9,37 ммоль) и DIPEA (2,15 мл, 12,50 ммоль) в ТГФ (50 мл) получали при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 0,5 ч. К реакционной смеси добавляли метиламин (2М в ТГФ) (6,25 мл, 12,50 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Полученную реакционную смесь вливали в насыщенный раствор NaHCO3 (100 мл) и подвергали экстракции посредством EtOAc (3×100 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (3% МеОН в ДХМ), получая трет-бутил-(6-(метилкарбамоил)изохинолин-3-ил)карбамат (1,35 г, 4,48 ммоль). ЖХМС: Способ А, 2,01 мин, МС: ЭР+ 302,38.
Стадия е. К раствору трет-бутил-(6-(метилкарбамоил)изохинолин-3-ил)карбамата (1,30 г, 4,318 ммоль) в ДХМ (50 мл) добавляли ТФУ (13 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Полученную реакционную смесь концентрировали при пониженном давлении, получая соль ТФУ и 3-амино-N-метилизохинолин-6-карбоксамида (2,0 г, количественный выход). ЖХМС: Способ А, 0,89 мин, МС: ЭР+ 202,13.
Промежуточное соединение 27 трет-бутил-(2S,3S)-3-((6-(1Н-пиразол-4-ил)изохинолин-3-ил)карбамот)-2-метилпирролидин-1-карбоксилат
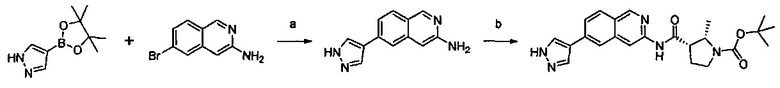
Реагенты и условия: a) Pd(dppf)Cl2, Cs2CO3, ДМФА, вода, плотно закрытая пробирка, микроволновое излучение, 150°С 1 ч; b) Промежуточное соединение 1, POCl3, пиридин, ДХМ, от 0°С до к. т., 30 мин.
Стадия а. В стеклянном флаконе для обработки микроволновым излучением готовили раствор 6-бромизохинолин-3-амина (0,20 г, 0,89 ммоль) и 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразола (0,19 г, 0,98 ммоль) в смеси ДМФА: вода (4:1, 5 мл). К полученной реакционной смеси добавили Cs2CO3 (0,87 г, 2,68 ммоль) при комнатной температуре. Реакционную смесь дегазировали при комнатной температуре в течение 15 мин, после чего добавляли Pd(dppi)Cl2 (0,06 г, 0,09 ммоль) и стеклянный флакон плотно закрывали. Реакционную смесь подвергали нагреванию посредством микроволнового излучения при 150°С в течение 1 ч. Полученную реакционную смесь вливали в воду (50 мл) и подвергали экстракции посредством EtOAc (3×50 мл). Объединенный органический слой промывали солевым раствором (50 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (2,2% МеОН в ДХМ), получая 6-(1Н-пиразол-4-ил)изохинолин-3-амин (0,11 г, 0,52 ммоль). ЖХМС: Способ А, 1,47 мин, МС: ЭР+ 211,18.
Стадия b. К раствору 6-(1Н-пиразол-4-ил)изохинолин-3-амина (0,09 г, 0,42 ммоль) и Промежуточного соединения 1 (0,10 г, 0,42 ммоль) в ДХМ (2 мл) добавляли пиридин (0,9 мл г) при 0°С. К реакционной смеси по каплям добавляли POCl3 (0,08 г, 0,51 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 15 мин и затем при комнатной температуре в течение 30 мин. Полученную реакционную смесь разбавляли посредством холодной воды (30 мл) и подвергали экстракции посредством EtOAc (3×30 мл). Объединенный органический слой промывали насыщенным водным раствором лимонной кислоты (30 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (1,2% МеОН в ДХМ), получая трет-бутил-(2S,3S)-3-((6-(1Н-пиразол-4-ил)изохинолин-3-ил)карбамоил)-2-метилпирролидин-1-карбоксилат (0,045 г, 0,10 ммоль). ЖХМС: Способ А, 1,96 мин, МС: ЭР+ 422,52.
Промежуточное соединение 28 6-(оксазол-5-ил)имидазо[1,2-а]пиридин-2-амин
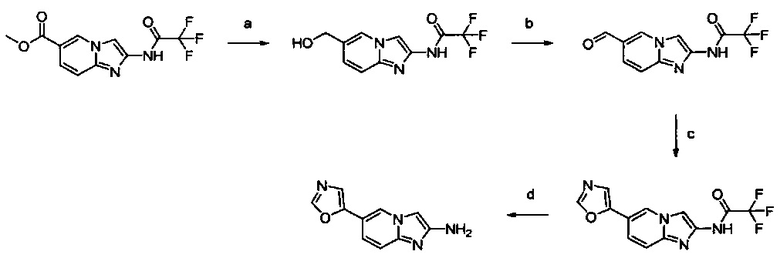
Реагенты и условия: a) LiAlH4, ТГФ, 0°С, 1 ч b) периодинан Десса-Мартина, ДХМ, к. т., 2 суток; с) 4-толуолсульфонилметилизоцианид, K2СО3, МеОН, 70°С, 3 ч; d) NaOH, вода, к. т., 2 ч.
Стадия а. К суспензии порошка LiAlH4 (0,165 г, 4,35 ммоль) в ТГФ (10 мл) добавляли раствор метил-2-(2,2,2-трифторацетамидо)имидазо[1,2-а]пиридин-6-карбоксилата (0,5 г, 1,74 ммоль) в ТГФ (5 мл) по каплям при 0°С в атмосфере азота. Реакционную смесь перемешивали при 0°С в атмосфере азота в течение 1 часа. Полученную реакционную смесь объединяли с другой партией, полученной в том же масштабе идентичным способом, и гасили посредством EtOAc (100 мл). Полученный органический раствор промывали водой (50 мл). Полученную эмульсию фильтровали через Celite Hyflo. Отделяли органическую фазу от фильтрата. Полученную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток растирали с н-пентаном (2×10 мл) и сушили с получением 2,2,2-трифтор-N-(6-(гидроксиметил)имидазо[1,2-а]пиридин-2-ил)ацетамида (0,55 г, 2,12 ммоль). ЖХМС: Способ А, 1,57 мин, МС: ЭР+ 260,21.
Стадия b. К раствору 2,2,2-трифтор-N-(6-(гидроксиметил)имидазо[1,2-а]пиридин-2-ил)ацетамида (0,55 г, 2,12 ммоль) в ДХМ (20 мл) добавляли периодинан Десса-Мартина (3,6 г, 8,49 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 суток. Полученную реакционную смесь объединяли с другой партией, полученной идентичным способом в масштабе 0,25 г. Полученную реакционную смесь тщательно фильтровали через Celite Hyflo. Полученный фильтрат вливали в воду (200 мл), подщелачивали твердым NaHCO3 и подвергали экстракции посредством EtOAc (2×100 мл). Объединенную органическую фазу собирали сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток растирали с н-гексаном (3×7 мл) и сушили, получая 2,2,2-трифтор-N-(6-формилимидазо[1,2-а]пиридин-2-ил)ацетамид (0,75 г, 2,92 ммоль). ЖХМС: Способ А, 1,85 мин, МС: ЭР+ 258,36.
Стадия с. Смесь 2,2,2-трифтор-N-(6-формилимидазо[1,2-а]пиридин-2-ил)ацетамида (0,75 г, 2,92 ммоль), 4-толуолсульфонилметил изоцианида (0,569 г, 2,92 ммоль) и К2СО3 (0,805 г, 5,83 ммоль) в МеОН (10 мл) нагревали до 70°С в течение 3 ч. Полученную реакционную смесь вливали в воду (150 мл) и подвергали экстракции посредством EtOAc (3×70 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 2,2,2-трифтор-N-(6-(оксазол-5-ил)имидазо[1,2-а]пиридин-2-ил)ацетамид (0,55 г, 1,86 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. ЖХМС: Способ А, 1,88 мин, МС: ЭР+ 297,38.
Стадия d. К раствору 2,2,2-трифтор-N-(6-(оксазол-5-ил)имидазо[1,2-а]пиридин-2-ил)ацетамида (0,25 г, 0,844 ммоль) в воде добавляли NaOH (0,13 г, 3,375 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Полученную реакционную смесь вливали в воду (120 мл) и подвергали экстракции посредством EtOAc (2×20 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 6-(оксазол-5-ил)имидазо[1,2-а]пиридин-2-амин (0,17 г, 0,84 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки.
Промежуточное соединение 29 6-(изопропилсульфонил)-5,6,7,8-тетрагидро-4Н-тиазоло[4,5-d]азепин-2-амин
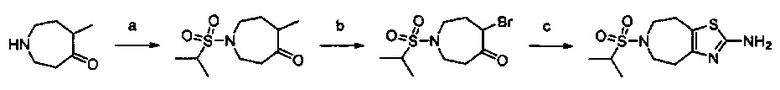
Реагенты и условия: а) изопропилсульфонилхлорид, DIPEA, ДХМ, к. т., 2 ч; b) Br2, СНCl3, от 0°С до к. т., 1 ч; с) тиомочевина, этанол, 90°С, 2 ч.
Стадия а. К раствору 4-пергидроазепинона гидрохлорида (1 г, 6,68 ммоль) в ДХМ (10 мл) добавляли DIPEA (3,45 г, 26,73 ммоль) при комнатной температуре в течение 10 мин. К реакционной смеси добавляли изопропилсульфонилхлорид (1,14 г, 8,02 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Полученную реакционную смесь вливали в воду (100 мл) и подвергали экстракции посредством ДХМ (3×40 мл). Органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 1-(изопропилсульфонил)азепан-4-она (0,54 г, 2,46 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. ЖХМС: Способ А, 1,54 мин, МС: ЭР+ 220,23.
Стадия b. К раствору 1-(изопропилсульфонил)азепан-4-она (0,54 г, 2,46 ммоль) в CHCl3 (10,8 мл) добавляли бром (0,29 г, 3,6 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь вливали в раствор метабисульфита натрия (60 мл) и подвергали экстракции посредством ДХМ (3×30 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 5-бром-1-(изопропилсульфонил)азепан-4-он (0,6 г, 2,02 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки.
Стадия с. К раствору 5-бром-1-(изопропилсульфонил)азепан-4-она (0,6 г, 2,02 ммоль) в этаноле (9 мл) добавляли тиомочевину (0,76 г, 10,10 ммоль) при комнатной температуре. Реакционную смесь нагревали при 90°С в течение 2 ч. Полученную реакционную смесь вливали в 1М раствор HCl (50 мл) и подвергали экстракции посредством ДХМ (2×20 мл). Водный слой нейтрализовали твердым Na2CO3 с доведением рН до 8 и подвергали экстракции посредством ДХМ (3×30 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (2% МеОН в ДХМ), получая 6-(изопропилсульфонил)-5,6,7,8-тетрагидро-4Н-тиазоло[4,5-d]азепин-2-амин (0,18 г, 0,67 ммоль). ЖХМС: Способ С, 3,03 мин, МС: ЭР+ 276,05
Промежуточное соединение 30 1-(4-(1-(2-метоксиэтил)-1Н-пиразол-4-ил)фенил)-1H-имидазол-4-амин
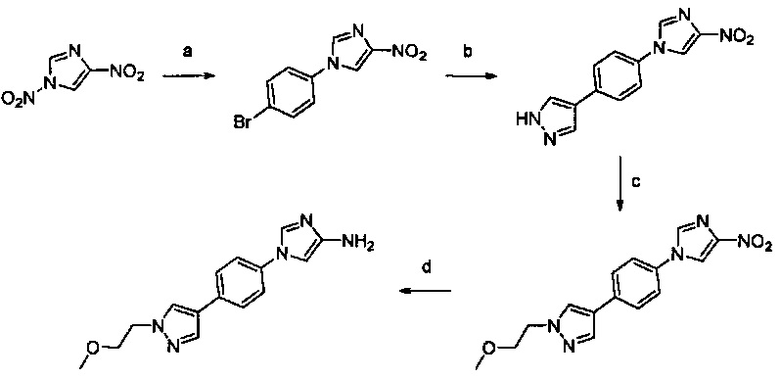
Реагенты и условия: а) 4-броманилин, МеОН, к. т., 16 ч; b) 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол, Pd(dppf)Cl2.ДХМ, ДМФА, вода, 150°С, 1 ч; с) 1-бром-2-метоксиэтан, K2CO3, ДМФА, 100°С, 16 ч; d) Н2, 10% Pd/C, ТГФ, к. т., 4 ч.
Стадия а. К раствору 1,4-динитро-1H-имидазола (2,5 г, 15,80 ммоль) в МеОН (40 мл) добавляли 4-броманилин (2,70 г, 15,80 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Полученный твердый осадок собирали путем фильтрования при пониженном давлении и сушили, получая 1-(4-бромфенил)-4-нитро-1Н-имидазол (3,5 г, 13,11 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. ЖХМС: Способ А, 2,11 мин, МС: ЭР+ 268,20.
Стадия b. Раствор 1-(4-бромфенил)-4-нитро-1Н-имидазола (0,30 г, 1,12 ммоль) и 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразола (0,26 г, 1,35 ммоль) в смеси ДМФА: вода (9:1, 10 мл) получали в стеклянном флаконе для обработки микроволновым излучением. К полученной реакционной смеси добавляли Na2CO3 (0,23 г, 2,24 ммоль) при комнатной температуре. Реакционную смесь дегазировали в течение 30 мин при комнатной температуре. К реакционной смеси добавляли комплекс Pd(dppf)Cl2.ДХМ (0,10 г, 0,11 ммоль) при комнатной температуре. Реакционную смесь подвергали нагреванию посредством микроволнового излучения при 150°С в течение 1 ч. Полученную реакционную смесь вливали в воду (20 мл) и подвергали экстракции посредством EtOAc (3×20 мл). Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт растирали с н-пентаном (30 мл), получая 4-(4-(4-нитро-1Н-имидазол-1-ил)фенил)-1Н-пиразол (0,25 г, 0,98 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. ЖХМС: Способ А, 1,78 мин, МС: ЭР+ 256,41.
Стадия с. К раствору (4-(4-(4-нитро-1Н-имидазол-1-ил)фенил)-1Н-пиразола (0,23 г, 0,90 ммоль) в ДМФА (6 мл) добавляли К2СО3 (0,37 г, 2,70 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 5 мин и обрабатывали 1-бром-2-метоксиэтаном (0,19 г, 1,35 ммоль). Реакционную смесь нагревали при 100°С в течение 16 часов. Полученной реакционной смеси позволяли остыть до к. т., вливали в воду (15 мл) и подвергали экстракции посредством EtOAc (3×10 мл). Объединенную органическую фазу собирали, промывали водой (2×10 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 1-(2-метоксиэтил)-4-(4-(4-нитро-1Н-имидазол-1-ил)фенил)-1Н-пиразол (0,25 г, 0,79 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. ЖХМС: Способ А, 1,91 мин, МС: ЭР+ 314,59.
Стадия d. К раствору 1-(2-метоксиэтил)-4-(4-(4-нитро-1Н-имидазол-1-ил)фенил)-1Н-пиразола (0,25 г, 0,79 ммоль) в ТГФ (5 мл) добавляли 10% Pd/C (0,05 г) при комнатной температуре. Реакционную смесь продували газообразным Н2 при комнатной температуре в течение 2 ч. Полученную реакционную смесь тщательно фильтровали через Celite Hyflo. Полученный фильтрат непосредственно применяли в следующей реакции сочетания кислоты с амином. ЖХМС: Способ А, 1,41 мин, МС: ЭР+ 284,39.
Промежуточное соединение 31 (3S,4R)-1-(трет-бутоксикарбонил)-4-(пиридин-3-ил)пирролидин-3-карбоновая кислота
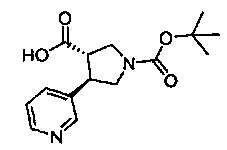
Синтез проводили с помощью методики, аналогичной методике, описанной для Промежуточного соединения 2, применяя этил-транс-3-(3-пиридил)акрилат на стадии а. ЖХМС: Способ А, 1,51 мин, МС: ЭР+ 293,28; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,5 (ушир. s, 1 Н), 8,55 (d, J=2 Гц, 1 H), 8,47 (dd, J=1,6 Гц, 4,8 Гц, 1 Н), 7,84 (d, J=7,6 Гц, 1 Н), 7,37-7,40 (m, 1 Н), 3,72-3,80 (m, 2 Н), 3,48-3,61 (m, 1 Н), 3,36-3,45 (m, 1 Н), 3,23-3,28 (m, 1 Н), 2,64-2,78 (m, 1 Н), 1,38 (s, 9 Н).
Промежуточное соединение 32 (3S,4R)-1-(трет-бутоксикарбонил)-4-(пиримидин-5-ил)пирролидин-3-карбоновая кислота
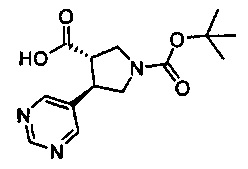
Синтез проводили с помощью методики, аналогичной методике, описанной для Промежуточного соединения 2, применяя этиловый эфир 3-пиримидин-5-ил-акриловой кислоты на стадии а. ЖХМС: Способ А, 1,65 мин, МС: ЭР+ 294,23; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,47 (ушир. s, 1 Н), 9,08 (s, 1 Н), 8,83 (s, 2 Н), 3,78-3,83 (m, 1 Н), 3,59-3,53 (m, 2 Н), 3,51-3,46 (m, 2 Н), 2,78-2,64 (m, 1 Н), 1,43 (s, 9 Н).
Промежуточное соединение 33 1-(трет-бутоксикарбонил)-3-метоксипирролидин-3-карбоновая кислота

Реагенты и условия: а) 4М HCl в 1,4-диоксан, МеОН, от 0°С до к. т. 1 ч; b) ВОС-ангидрид, NaHCO3, EtOAc, к. т. 8 ч; с) Mel, Cs2CO3, MeCN, 80°С 20 ч; d) LiOH.H2O, МеОН, к. т. 2 ч.
Стадия а. К раствору трет-бутил-3-циано-3-гидроксипирролидин-1-карбоксилата (0,60 г, 2,83 ммоль) в МеОН (15 мл) добавляли 4М HCl в 1,4-диоксан (6 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь концентрировали при пониженном давлении, получая метил-3-гидроксипирролидин-3-карбоксилатную соль HCl (0,55 г, количественный выход). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. ЖХМС: Способ А, 0,25 мин, МС: ЭР+ 146,08.
Стадия b. К суспензии метил-3-гидроксипирролидин-3-карбоксилатной соли HCl (0,55 г, 3,03 ммоль) в насыщенном растворе NaHCO3 (2 мл) и EtOAc (5 мл) добавляли ВОС-ангидрид (1,32 г, 6,07 ммоль) и перемешивали при комнатной температуре в течение 8 ч. Полученную реакционную смесь вливали в воду (15 мл) и подвергали экстракции посредством EtOAc (3×10 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 1-(трет-бутил)-3-метил-3-гидроксипирролидин-1,3-дикарбоксилат (0,29 г, 1,18 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. ЖХМС: Способ А, 1,88 мин, МС: ЭР+ 246,30.
Стадия с. К раствору метил-1-(трет-бутил)-3-метил-3-гидроксипирролидин-1,3-дикарбоксилата (0,27 г, 1,10 ммоль) в MeCN (10 мл) добавляли Cs2CO3 (1,79 г, 5,51 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 5 мин. К реакционной смеси добавляли CH3I (0,78 г, 5,51 ммоль) при комнатной температуре. Реакционную смесь нагревали при 80°С в течение 20 часов. Полученную реакционную смесь вливали в холодную воду (15 мл) и подвергали экстракции посредством EtOAc (3×10 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (10% EtOAc в гексане), получая 1-(трет-бутил)-3-метил-3-метоксипирролидин-1,3-дикарбоксилат (0,23 г, 0,88 ммоль). ЖХМС: Способ А, 2,13 мин, МС: ЭР+ 260,30.
Стадия d. К раствору 1-(трет-бутил)-3-метил-3-метоксипирролидин-1,3-дикарбоксилата (0,23 г, 0,88 ммоль) в МеОН (10 мл) добавляли LiOH.H2O (0,15 г, 3,55 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Полученную реакционную смесь разбавляли водой (10 мл) и подвергали экстракции посредством EtOAc (2×10 мл). Водный слой подкисляли до рН 6, применяя 10% водный раствор лимонной кислоты. Полученную смесь подвергали экстракции смесью МеОН: EtOAc (1:9, 7×10 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 1-(трет-бутоксикарбонил)-3-метоксипирролидин-3-карбоновую кислоту (0,20 г, 0,81 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. ЖХМС: Способ А, 1,85 мин, МС: ЭР- 244,20.
Промежуточное соединение 34 2'-хлор-[4,4'-бипиридин]-2-амин
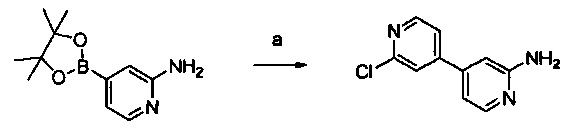
Реагенты и условия: а) 2-хлор-4-иодпиридин, PdCl2(dppf), K3РО4, 1,4-диоксан, вода, 100°С, 16 ч
Стадия а. Смесь 2-хлор-4-иодпиридина (0,75 г, 3,13 ммоль), пинаколового эфира 2-аминопиридин-4-бороновой кислоты (0,76 г, 3,45 ммоль) и K3PO4 (1,3 г, 6,26 ммоль) получали в смеси 1,4-диоксан: вода (2:1, 3 мл) при комнатной температуре. Реакционную смесь дегазировали в течение 15 мин. К реакционной смеси добавляли PdCl2(dppf) (0,11 г, 0,16 ммоль) при комнатной температуре. Реакционную смесь нагревали при 100°С в течение 16 часов. Полученной реакционной смеси давали возможность остыть до к. т., вливали в солевой раствор (100 мл) и подвергали экстракции посредством EtOAc (3×75 мл). Объединенную органическую фазу промывали дименирализованной (ДМ) водой (75 мл). Отделяли органическую фазу, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (75% EtOAc в гексане), получая 2'-хлор-[4,4'-бипиридин]-2-амин (0,3 г, 1,46 ммоль). ЖХМС: Способ А, 1,49 мин, МС: ЭР+ 206,13.
Промежуточное соединение 35 4-амино-2-циклопропилбензонитрил
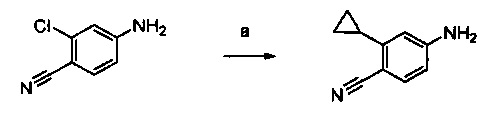
Реагенты и условия: а) циклопропилбороновая кислота, Pd(OAc)2, трициклогексилфосфен, К3РО4, толуол, вода, 80°С, 15 ч.
Стадия а. Раствор 4-амино-2-хлорбензонитрила (0,46 г, 3,00 ммоль) и циклопропилбороновой кислоты (0,39 г, 4,50 ммоль) в смеси толуол: вода (6:1, 7 мл) получали в стеклянном флаконе при к. т. К реакционной смеси добавляли K3РО4 (2,55 г, 12,01 ммоль) и трициклогексилфосфин (0,34 г, 1,20 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь продували азотом в течение 15 мин. К реакционной смеси добавляли Pd(OAc)2 (0,13 г, 0,6 ммоль) при комнатной температуре в атмосфере азота. Стеклянный флакон плотно закрывали и реакционную смесь нагревали при 80°С в течение 15 ч. Полученную реакционную смесь вливали в воду (200 мл) и подвергали экстракции посредством EtOAc (3×100 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (25% EtOAc в гексане), получая 4-амино-2-циклопропилбензонитрил (0,34 г, 2,15 ммоль). ЖХМС: Способ А, 1,90 мин, МС: ЭР+ 159,3.
Пример 1 (S)-1-циано-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 1)
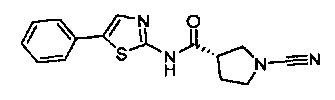
Стадия а. Раствор 5-фенилтиазол-2-амина (2,6 ммоль) и (3S)-ВОС-1-пирролидин-3-карбоновой кислоты (2,9 ммоль) в ТГФ перемешивали при 0°С в течение 5 минут. К этому раствору добавляли Т3Р (50% в EtOAc) (3,9 ммоль) и DIPEA (5,2 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь вливали в воду (40 мл) и подвергали экстракции посредством ДХМ (40 мл). Органическую фазу собирали и промывали 1М раствором NaOH (30 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (2-3% МеОН в ДХМ), получая трет-бутил-(S)-3-((5-фенилтиазол-2-ил)карбамоил)пирролидин-1-карбоксилат (1,2 ммоль). МС: ЭР+ 374,13; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,37 (ушир. s, 1 Н), 7,89 (s, 1 Н), 7,60-7,62 (m, 2 Н), 7,40-7,43 (m, 2 Н), 7,28-7,32 (m, 1 Н), 3,51-3,53 (m, 1 Н), 3,36-3,43 (m, 2 Н), 3,22-3,31 (m, 2 Н), 2,12-2,18 (m, 1 Н), 2,01-2,09 (m, 1 Н), 1,37- 1,43 (m, 9 Н).
Стадия b. К раствору трет-бутил-(S)-3-((5-фенилтиазол-2-ил)карбамоил)пирролидин-1-карбоксилата (1,2 ммоль) в ДХМ (10 мл) добавляли 4М HCl в EtOAc (40 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток растирали с диэтиловым эфиром (10 мл), получая (S)-N-(5-фенилтиазол-2-ил) пирролидин-3-карбоксамид гидрохлорид (1,28 ммоль). МС: ЭР+ 274,13; 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 12,55 (ушир. s, 1 Н), 9,58 (ушир. s, 1 Н), 9,34 (ушир. s, 1 Н), 7,91 (s, 1 Н), 7,60-7,62 (m, 2 Н), 7,40-7,44 (m, 2 Н), 7,29-7,33 (m, 1 Н), 3,38-3,44 (m, 3 Н), 3,19-3,24 (m, 2 Н), 2,25-2,33 (m, 1 Н), 2,08-2,12 (m, 1 Н).
Стадия с. К раствору (S)-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамида гидрохлорида (1,2 ммоль) в ДХМ (5 мл) добавляли K2СО3 (2,4 ммоль) и цианоген бромид (2,4 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Полученную реакционную смесь вливали в воду (40 мл) и подвергали экстракции посредством ДХМ (50 мл). Органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (1-1,5% МеОН в ДХМ), получая указанное в заголовке соединение (0,77 ммоль). МС: ЭР+ 299,0; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,43 (s, 1 Н), 7,90 (s, 1 Н), 7,61-7,63 (m, 2 Н), 7,40-7,44 (m, 2 Н), 7,29-7,33 (m, 1 Н), 3,63-3,66 (m, 1 Н), 3,53-3,61 (m, 1 Н), 3,42-3,49 (m, 2 Н), 3,33-3,40 (m, 1 Н), 2,16-2,23 (m, 1 Н), 2,06-2,12 (m, 1 Н).
Соединения, приведенные в таблице 1, синтезировали с помощью методики, аналогичной методике, описанной для Примера 1, применяя 1-ВОС-пирролидин-3-карбоновую кислоту.
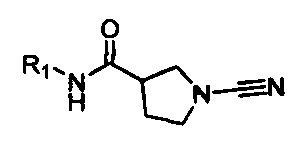
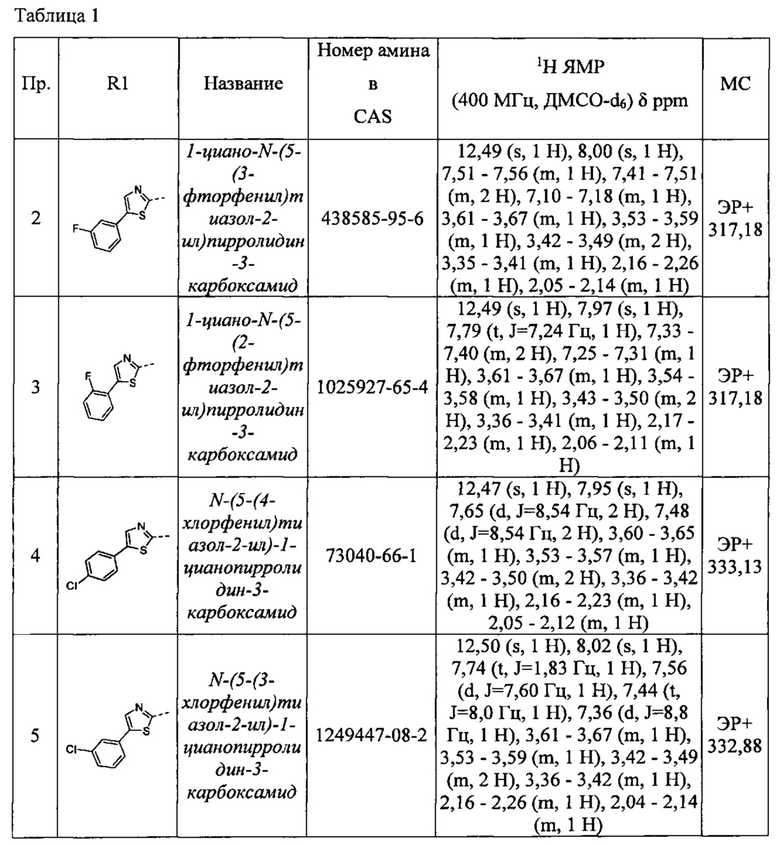
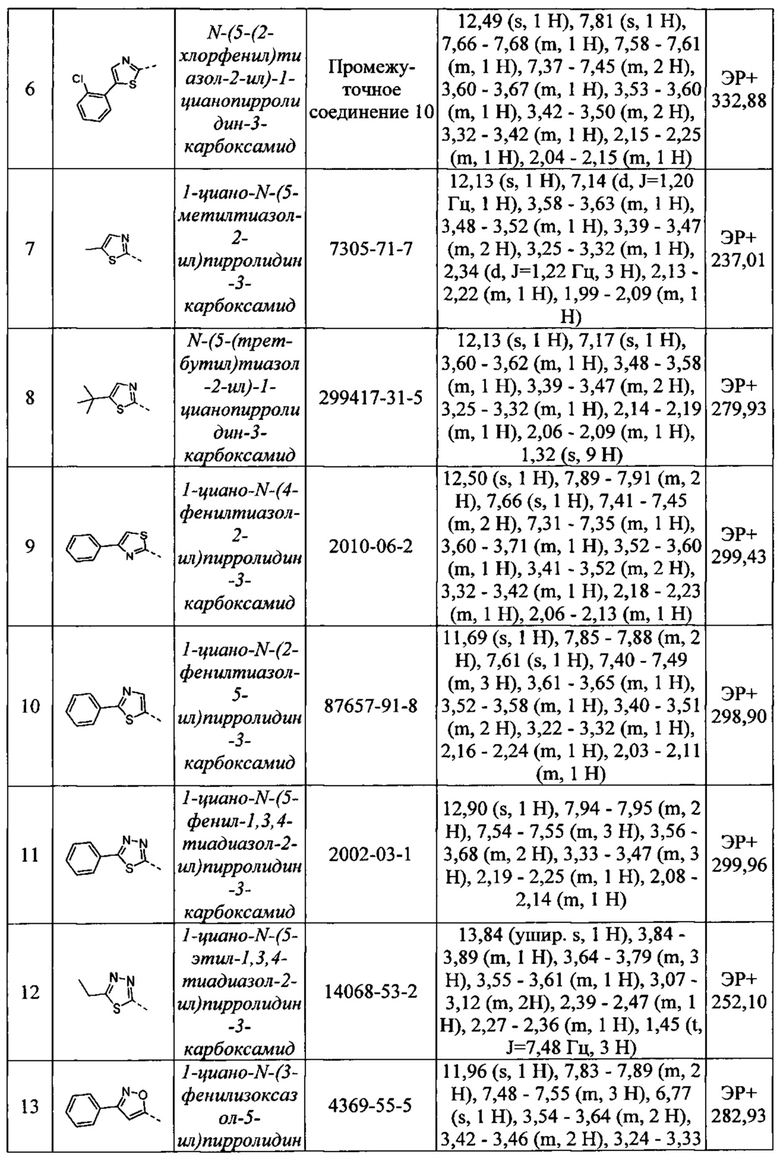
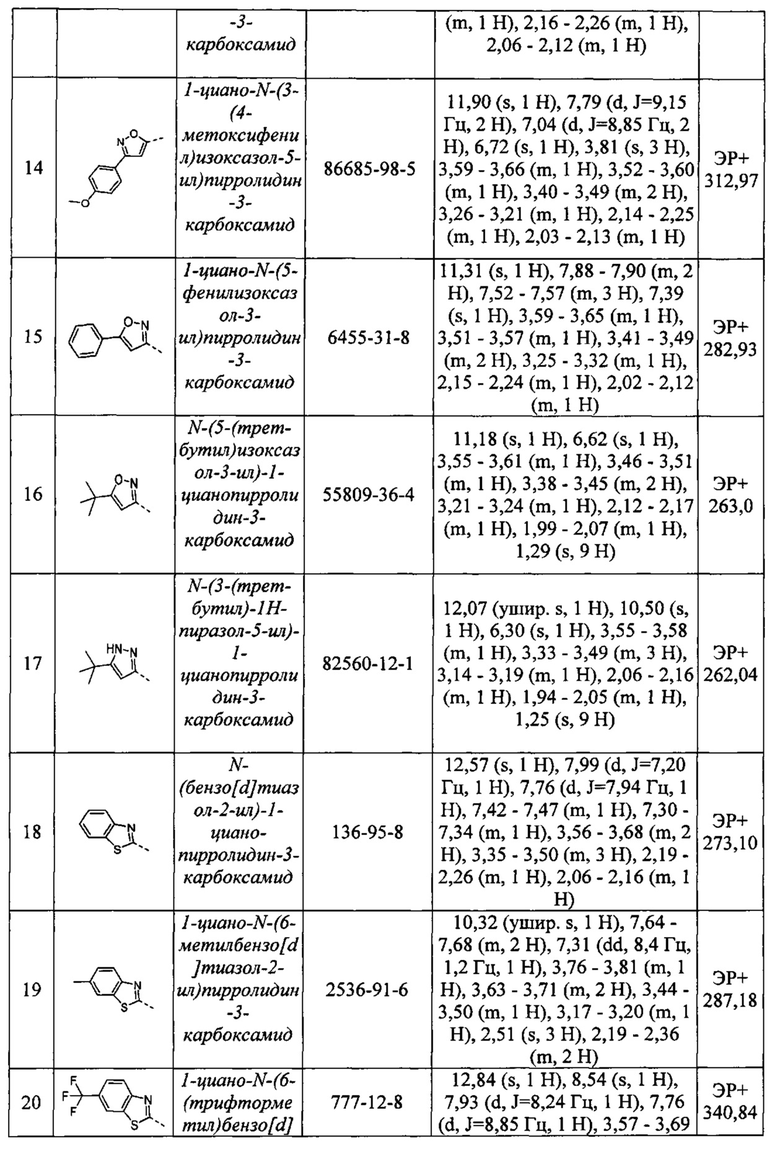
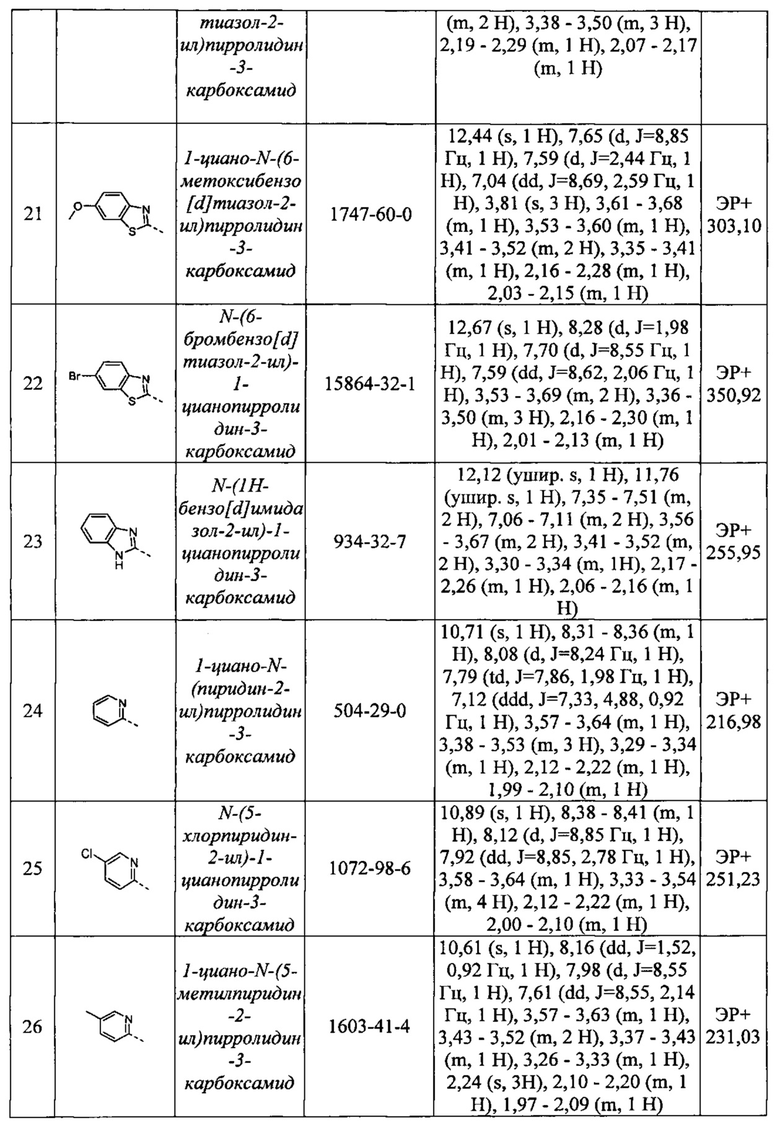
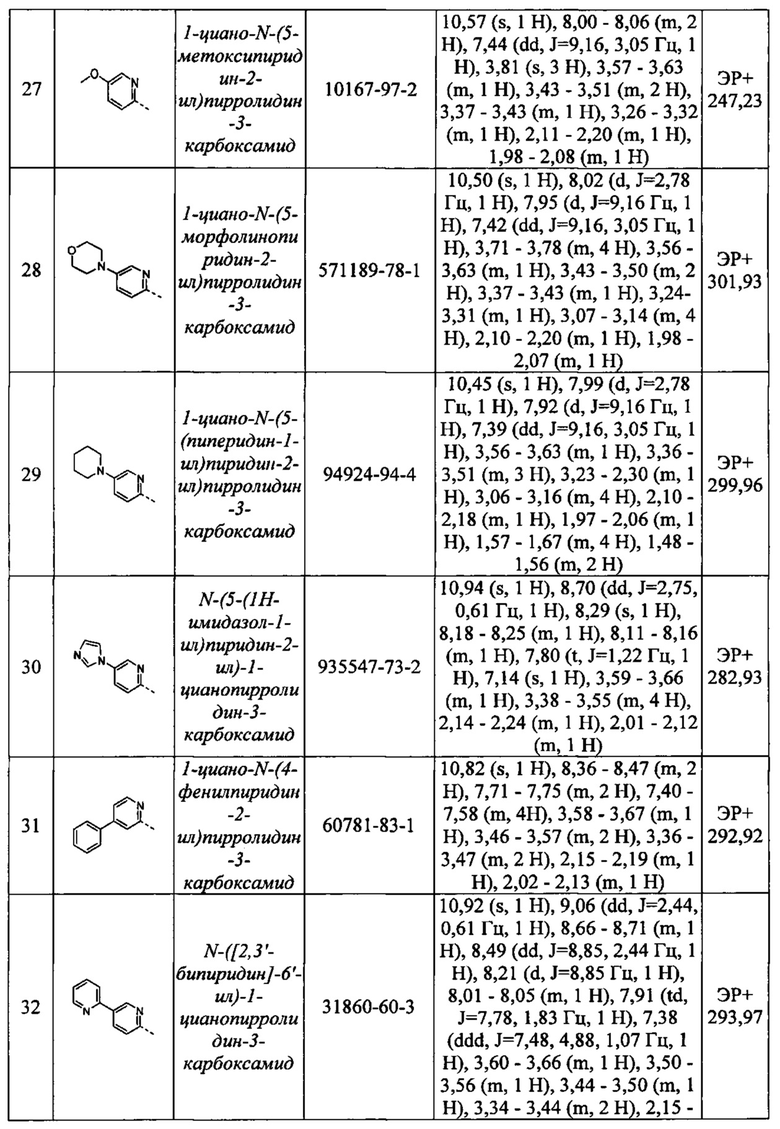
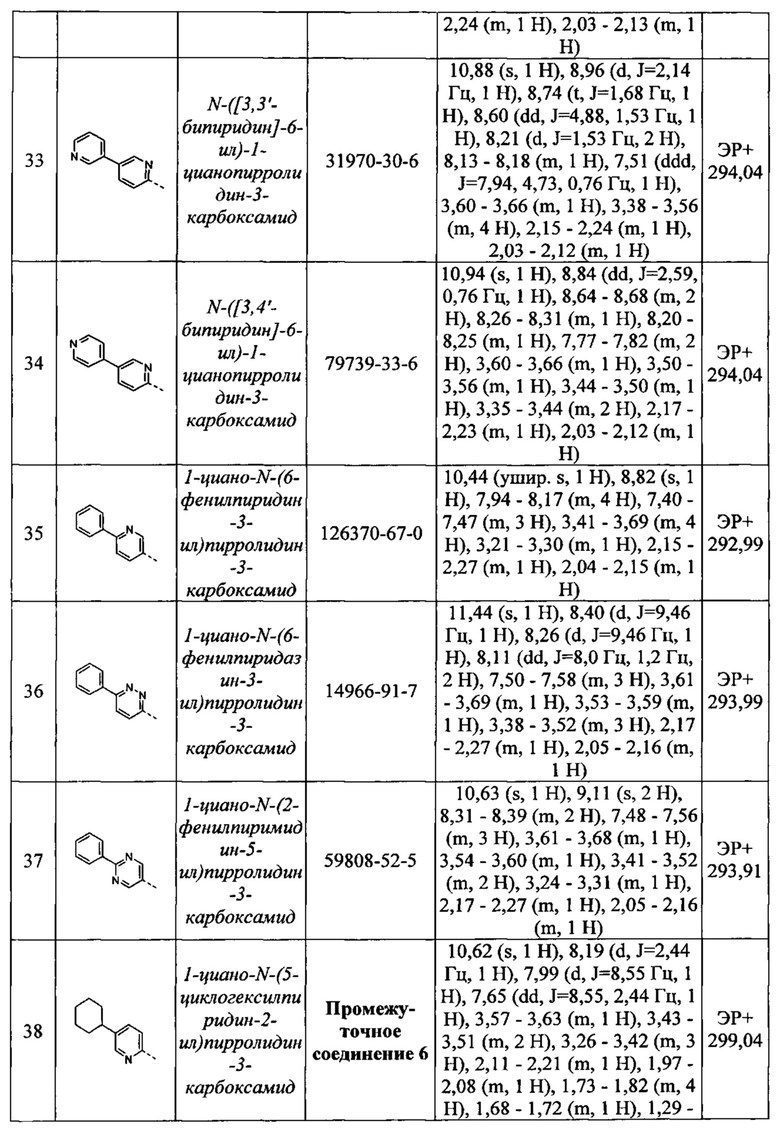
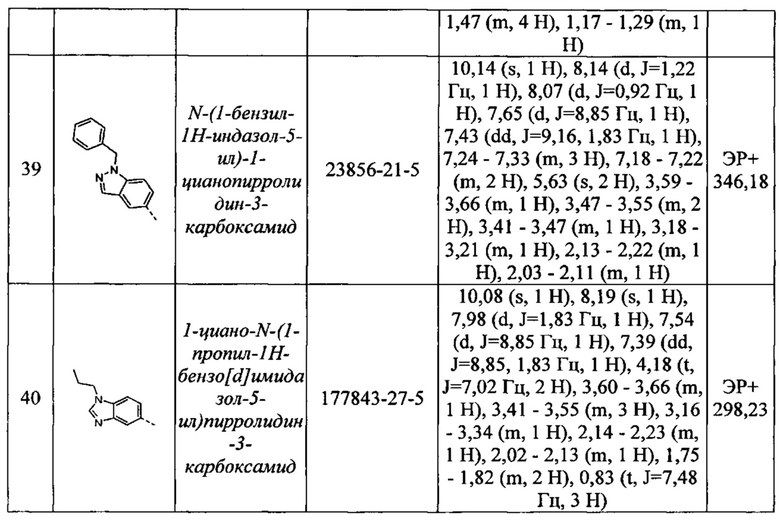
Соединения, приведенные в таблице 2, синтезировали с помощью методики, аналогичной методике, описанной для Примера 1, применяя (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту.
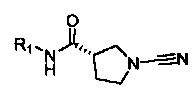
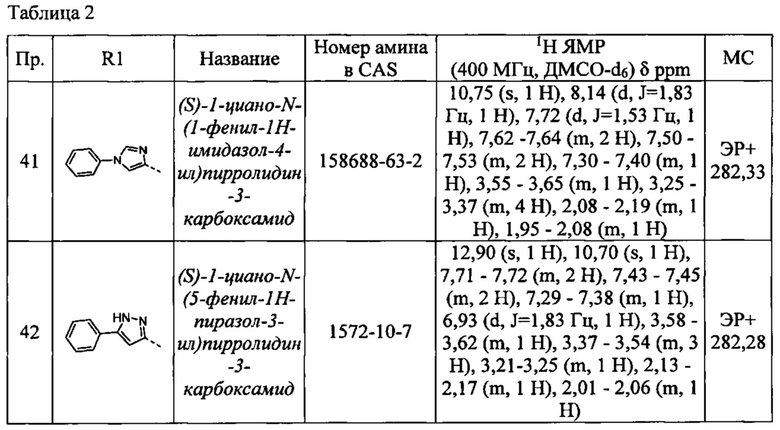
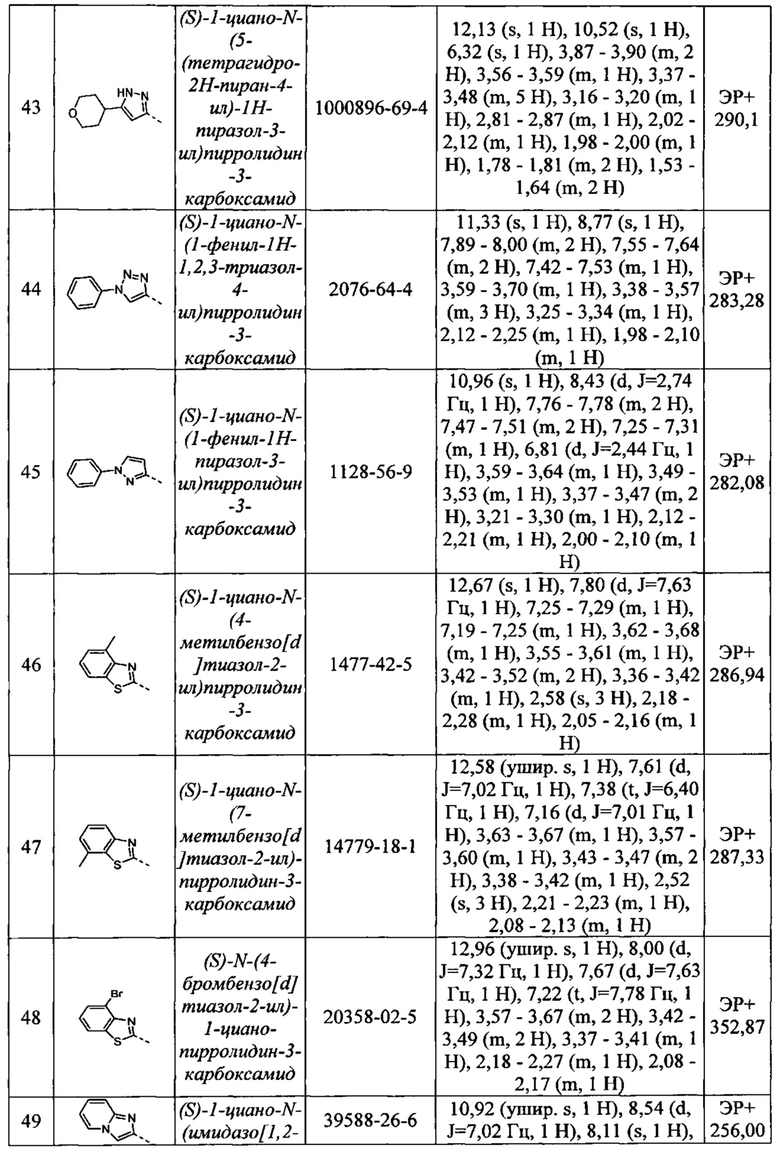
Пример 66 (S)-1-циано-N-(5-(4-фторфенил)тиазол-2-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 2)
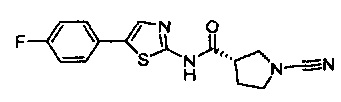
Стадия а. К раствору 2-аминотиазола (35 ммоль) в ТГФ (35 мл) добавляли ТЭА (52 ммоль) при комнатной температуре. Полученную реакционную смесь охлаждали до 0°С. Уксусный ангидрид (52 ммоль) добавляли к реакционной смеси. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. В реакционной смеси наблюдали выпадение осадка. Полученный осадок отфильтровывали при пониженном давлении, промывали ледяной водой (2×20 мл), сушили под вакуумом, получая N-(тиазол-2-ил)ацетамид (16,9 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 143,19; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,09 (ушир. s, 1 Н), 7,45 (d, J=3,60 Гц, 1 Н), 7,18 (d, J=3,60 Гц, 1 Н), 2,14 (s, 3 Н).
Стадия b. Раствор N-(тиазол-2-ил)ацетамида (7,9 ммоль) и 1-бром-4-фторбензола (11,8 ммоль) в ДМФА (10 мл) перемешивали при комнатной температуре в атмосфере азота в стеклянной пробирке для обработки микроволновым излучением в течение 5 минут. К реакционной смеси добавляли K3РО4 (9,4 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь продували азотом в течение 30 мин. К реакционной смеси добавляли Pd(OAc)2 (0,5 ммоль) и трициклогексилфосфин (0,4 ммоль) при комнатной температуре в атмосфере азота и стеклянную пробирку плотно закрывали. Закрытую пробирку подвергали действию микроволнового излучения при 140°С в течение 1,5 ч. Полученную реакционную смесь вливали в воду (30 мл) и подвергали экстракции посредством EtOAc (3×30 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (70-80% EtOAc в гексане), получая N-(5-(4-фторфенил)тиазол-2-ил)ацетамид (1,3 ммоль). МС: ЭР+ 237,28; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,19 (s, 1 Н), 7,83 (s, 1 Н), 7,64-7,67 (m, 2 Н), 7,23-7,28 (m, 2 Н), 2,16 (s, 3 Н).
Стадия с. К раствору N-(5-(4-фторфенил) тиазол-2-ил)ацетамида (1,3 ммоль) в 1,4-диоксане (6 мл) добавляли концентрированную HCl (24 мл) при комнатной температуре. Реакционную смесь нагревали при 100°С в течение 4 часов. Полученную реакционную смесь вливали в воду (30 мл) и подщелачивали водным 1М раствором NaOH. Полученный осадок отфильтровывали при пониженном давлении, промывали ледяной водой (3×10 мл) и сушили под вакуумом, получая 5-(4-фторфенил)тиазол-2-амин (0,7 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 195,18; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 7,43-7,46 (m, 2 Н), 7,35 (s, 1 Н), 7,17-7,29 (m, 2 Н), 7,15 (ушир. s, 2 Н).
Стадии d-f. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для Примера 1, применяя (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту. МС: ЭР+ 316,91; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,43 (ушир. s, 1 Н), 7,87 (s, 1 H), 7,64-7,68 (m, 2 H), 7,25-7,29 (m, 2 H), 3,61-3,65 (m, 1 H), 3,53-3,57 (m, 1 H), 3,40-3,50 (m, 3 H), 2,16-2,33 (m, 1 H), 2,03-2,12 (m, 1 H).
Пример 67 (S)-1-циано-N-(5-(3,4-дифторфенил)тиазол-2-ил)пирролидин-3-карбоксамид
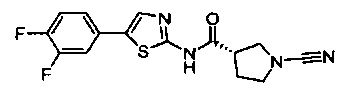
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 66, применяя 1-бром-3,4-дифторбензол. МС: ЭР+ 334,99; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,48 (s, 1 Н), 7,95 (s, 1 Н), 7,77-7,82 (m, 1 Н), 7,43-7,52 (m, 2 Н), 3,61-3,65 (m, 1 Н), 3,53-3,57 (m, 1 Н), 3,37-3,50 (m, 2 Н), 3,25-3,29 (m, 1 Н), 2,16-2,25 (m, 1 Н), 2,04-2,12 (m, 1 Н).
Пример 68 (S)-1-циано-N-(5-(4-(трифторметил)фенил)тиазол-2-ил)пирролидин-3-карбоксамид
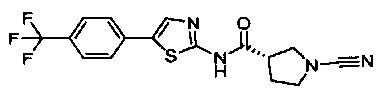
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 66, применяя 1-бром-4-(трифторметил)бензол. МС: ЭР+ 366,87; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,55 (ушир. s, 1 Н), 8,08-8,16 (m, 1 Н), 7,82-7,90 (m, 2 Н), 7,74-7,80 (m, 2 Н), 3,61-3,65 (m, 1 Н), 3,57-3,58 (m, 1 Н), 3,33-3,47 (m, 3 Н), 2,16-2,30 (m, 1Н), 2,04-2,15 (m,1Н).
Пример 69 (S)-1-циано-N-(5-(пиридин-4-ил)тиазол-2-ил)пирролидин-3-карбоксамид
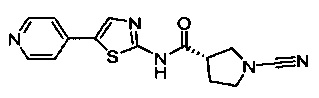
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 66, применяя 4-бромпиридина гидрохлорид. МС: ЭР+ 300,07; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,68 (s, 1 Н), 8,61 (d, J=6,0 Гц, 2 Н), 8,32 (s, 1 Н), 7,74 (d, J=6,0 Гц, 2 Н), 3,59-3,66 (m, 1 Н), 3,55-3,57 (m, 1 Н), 3,43-3,50 (m, 2 Н), 3,34-3,41 (m, 1 Н), 2,19-2,24 (m, 1 Н), 2,07-2,12 (m, 1 Н).
Пример 70 (S)-1-циано-N-(5-(пиридин-2-ил)тиазол-2-ил)пирролидин-3-карбоксамид
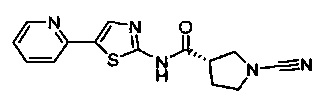
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 66, применяя 2-бромпиридин. МС: ЭР+ 300,05; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,42 (s, 1 Н), 8,52 (d, J=4,4 Гц, 1Н), 8,19 (s, 1 Н), 7,91-7,95 (m, 1 Н), 7,80-7,84 (m, 1 Н), 7,24-7,27 (m, 1 Н), 3,62-3,66 (m, 1 Н), 3,54-3,58 (m, 1 Н), 3,40-3,51 (m, 3 Н), 2,17-2,28 (m, 1 Н), 2,04-2,13 (m, 1 Н).
Пример 71 (R)-1-циано-N-(5-фенилпиридин-2-ил)пирролидин-3-карбоксамид
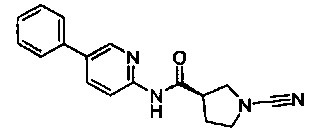
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 1, применяя (R)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту и 2-амино-5-фенилпиридин. МС: ЭР+ 292,99; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,83 (s, 1 Н), 8,67 (d, J=1,67 Гц, 1 Н), 8,10-8,20 (m, 2 Н), 7,70-7,75 (m, 2 Н), 7,49 (t, J=7,6 Гц, 2 Н), 7,39 (t, J=7,6 Гц, 1 Н), 3,59-3,67 (m, 1 Н), 3,32-3,55 (m, 4 Н), 2,14-2,24 (m, 1 Н), 2,02-2,12 (m, 1 Н).
Пример 72 (2S,3S)-1-циано-2-метил-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамид
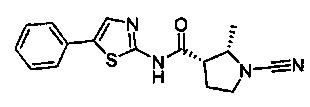
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 1, применяя Промежуточное соединение 1 и 2-амино-5-фенилтиазол. Очистка с помощью препаративной ВЭЖХ; подвижная фаза: (А) 100% н-гексан (В) 100% IPА, колонка: YMC PACKSIL, 250×20 мм, 5 мкм, скорость потока: 18 мл/мин. МС: ЭР+ 312,90; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,41 (ушир. s, 1 Н), 7,90 (s, 1 Н), 7,60-7,63 (m, 2 Н), 7,39-7,45 (m, 2 Н), 7,27-7,34 (m, 1 Н), 3,96-4,04 (m, 1 Н), 3,60-3,68 (m, 1 Н), 3,40-3,46 (m, 1 Н), 3,28-3,34 (m, 1 Н), 2,17-2,24 (m, 1 Н), 2,03-2,10 (m, 1 Н), 1,10 (d, J=6,41 Гц, 3 Н).
Пример 73 (2S,3S)-1-циано-N-(5-(4-фторфенил)тиазол-2-ил)-2-метилпирролидин-3-карбоксамид
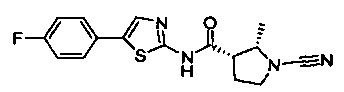
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 66, применяя Промежуточное соединение 1. МС: ЭР+ 331,05; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,42 (s, 1 Н), 7,87 (s, 1 Н), 7,65-7,69 (m, 2 Н), 7,25-7,29 (m, 2 Н), 3,98-4,02 (m, 1 Н), 3,61-3,66 (m, 1 Н), 3,40-3,46 (m, 1 Н), 3,28-3,32 (m, 1 Н), 2,16-2,23 (m, 1 Н), 2,06-2,09 (m, 1 Н), 1,10 (d, J=6,71 Гц, 3 Н).
Пример 74 (2S,3S)-1-циано-2-метил-N-(1-фенил-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
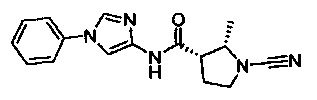
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 1, применяя Промежуточное соединение 1 и 4-амино-1-фенилимидазол. Очистка с помощью препаративной ВЭЖХ; подвижная фаза: (А) 100% н-гексан (В) 100% IPА, колонка: YMC PACKSIL, 250×20 мм, 5 мкм, скорость потока: 20 мл/мин. МС: ЭР+ 296,09; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,69 (s, 1 Н), 8,14 (d, J=1,83 Гц, 1 H), 7,73 (d, J=1,83 Гц, 1 H), 7,62-7,66 (m, 2 Н), 7,50-7,54 (m, 2 Н), 7,33-7,39 (m, 1 Н), 3,90-3,98 (m, 1 Н), 3,59-3,68 (m, 1 Н), 3,34-3,43 (m, 1 Н), 3,18-3,23 (m, 1 Н), 2,09-2,21 (m, 1 Н), 1,95-2,05 (m, 1 Н), 1,11 (d, J=6,71 Гц, 3 H).
Пример 75 (2S,3S)-1-циано-2-метил-N-(5-фенил-1Н-пиразол-3-ил)пирролидин-3-карбоксамид
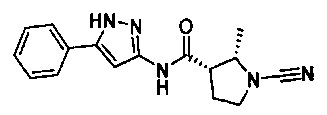
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 1, применяя Промежуточное соединение 1 и 5-амино-3-фенилпиразол. Очистка с помощью препаративной ВЭЖХ; подвижная фаза: (А) 100% н-гексан (В) 100% IPА, колонка: YMC PACKSIL, 250×20 мм, 5 мкм, скорость потока: 20 мл/мин. МС: ЭР+ 296,47; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,89 (ушир. s, 1 Н), 10,64 (ушир. s, 1 Н), 7,70-7,74 (m, 2 Н), 7,41-7,47 (m, 2 Н), 7,31-7,37 (m, 1 Н), 6,94 (ушир. s, 1 Н), 3,93-3,98 (m, 1 Н), 3,59-3,67 (m, 1 Н), 3,38-3,44 (m, 1 Н), 3,16-3,21 (m, 1 Н), 2,12-2,20 (m, 1 Н), 1,97-2,04 (m, 1 Н), 1,13 (d, J=6,40 Гц, 3 Н).
Пример 76 (2S,3S)-1-циано-2-метил-N-(5-(тетрагидро-2Н-пиран-4-ил)тиазол-2-ил)пирролидин-3-карбоксамид
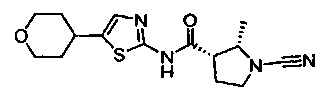
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 1, применяя Промежуточное соединение 1 и Промежуточное соединение 3. МС: ЭР+ 321,18; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,19 (ушир. s, 1 Н), 7,19 (s, 1 Н), 3,93-3,98 (m, 1 Н), 3,86-3,92 (m, 2 Н), 3,58-3,64 (m, 1 Н), 3,34-3,44 (m, 3 Н), 3,17-3,27 (m, 1 Н), 2,99-3,05 (m, 1 Н), 2,12-2,19 (m, 1 Н), 1,99-2,06 (m, 1 Н), 1,83-1,86 (m, 2 Н), 1,56-1,86 (m, 2 Н), 1,05 (d, J=6,4 Гц, 3 Н).
Пример 77 (2S,3S)-N-(5-(2-хлорфенил)тиазол-2-ил)-1-циано-2-метилпирролидин-3-карбоксамид
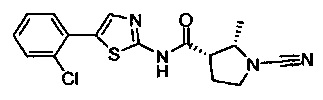
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 1, применяя Промежуточное соединение 1 и 5-(2-хлорфенил)тиазол-2-амин. МС: ЭР+ 347,63; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,43 (ушир. s, 1 Н), 7,28 (s, 1 Н), 7,67 (dd, J=7,2 Гц, 1,6 Гц, 1 Н), 7,59 (dd, J=8,0 Гц, 2,0 Гц, 1 Н), 7,37-7,44 (m, 2 Н), 3,97-4,03 (m, 1 Н), 3,61-3,66 (m, 1 Н), 3,40-3,46 (m, 1 Н), 3,29-3,32 (m, 1 Н), 2,16-2,24 (m, 1 Н), 2,02-2,09 (m, 1 Н), 1,10 (d, J=6,4 Гц, 3 Н).
Пример 78 1-циано-3-метил-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамид
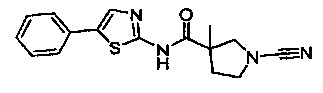
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 1, применяя 1-(трет-бутоксикарбонил)-3-метилпирролидин-3-карбоновую кислоту и 2-амино-5-фенилтиазол. МС: ЭР+ 312,9; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,30 (s, 1 Н), 7,93 (s, 1 Н), 7,61-7,63 (m, 2 Н), 7,44-7,40 (m, 2 Н), 7,29-7,33 (m, 1 Н), 3,86-3,89 (m, 1 Н), 3,48-3,52 (m, 1 Н), 3,37-3,41 (m, 1 Н), 3,29-3,33 (m, 1 Н), 2,38-2,45 (m, 1 Н), 1,92-1,99 (m, 1 Н), 1,42 (s, 3 Н).
Пример 79 1-циано-3-метил-N-(1-фенил-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
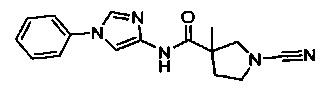
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 1, применяя 1-(трет-бутоксикарбонил)-3-метилпирролидин-3-карбоновую кислоту и 4-амино-1-фенилимидазол. МС: ЭР+ 296,53; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,52 (s, 1 Н), 8,16 (d, J=1,6 Гц, 1 Н), 7,73 (d, J=l,6 Гц, 1 Н), 7,63-7,65 (m, 2 Н), 7,50-7,54 (m, 2 Н), 7,34-7,38 (m, 1 Н), 3,85 (d, J=9,16 Гц, 1 Н), 3,36-3,60 (m, 2 Н), 3,25 (d, J=9,16 Гц, 1 Н), 2,37-2,42 (m, 1Н), 1,89-1,94 (m, 1 Н), 1,38 (s, 3 Н).
Пример 80 1-циано-3-(метоксиметил)-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамид
(получали в соответствии со Схемой 1)
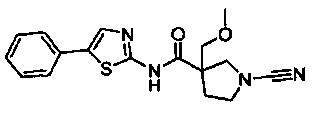
Стадия а. К раствору Промежуточного соединения 11 (1,0 ммоль) в ДМФА (5 мл) добавляли HATU (1,5 ммоль) и 5-фенилтиазол-2-амин (1,0 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 10 мин. ТЭА (2,9 ммоль) добавляли по каплям к реакционной смеси. Реакционную смесь перемешивали при комнатной температуре в течение 6 ч. Полученную реакционную смесь вливали в воду (20 мл) и подвергали экстракции посредством EtOAc (3×20 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая трет-бутил-3-(метоксиметил)-3-((5-фенилтиазол-2-ил)карбамоил)пирролидин-1-карбоксилат (0,5 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 418,48
Стадии b-с. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для Примера 1 в соответствии со стадиями b и с Схемы 1. МС: ЭР+ 343,33; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,54 (ушир. s, 1 Н), 7,87 (s, 1 Н), 7,59-7,61 (m, 2 Н), 7,39-7,43 (m, 2 Н), 7,27-7,30 (m, 1 Н), 3,92 (d, J=9,60 Гц, 1 Н), 3,80 (d, J=9,60 Гц, 1 Н), 3,62 (d, J=9,60 Гц, 1 Н), 3,45-3,52 (m, 1 Н), 3,32-3,41 (m, 2 Н), 3,25 (s, 3 Н), 2,30-2,36 (m, 1 Н), 1,98-2,06 (m, 1 Н).
Пример 81 1,3-дициано-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамид
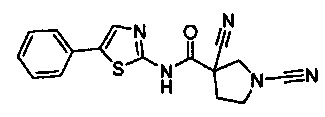
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 1, применяя 2-амино-5-метилтиазол и Промежуточное соединение 12. МС: ЭР+ 324,48; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 13,49 (s, 1 Н), 7,96 (s, 1 Н), 7,61-7,63 (m, 2 Н), 7,40-7,44 (m, 2 Н), 7,31-7,33 (m, 1 Н), 3,93-4,00 (m, 2 Н), 3,59-3,63 (m, 2 Н), 2,57-2,67 (m, 2 Н).
Пример 82 (3S,4S)-1-циано-4-метил-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамид
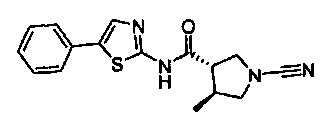
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 1, применяя Промежуточное соединение 2 и 2-амино-5-фенилтиазол. Очистка с помощью препаративной хиральной ВЭЖХ; подвижная фаза: (А) 70-50% н-гексан (В) 30-50% IPA, колонка: Chiralpak IC, 250×10 мм, 5 мкм, скорость потока: 8 мл/мин. МС: ЭР+ 313,23; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,48 (ушир. s, 1 Н), 7,91 (s, 1 Н), 7,61-7,63 (m, 2 Н), 7,39-7,44 (m, 2 Н), 7,29-7,35 (m, 1 Н), 3,73-3,77 (m, 1 Н), 3,59-3,63 (m, 1 Н), 3,52-3,56 (m, 1 Н), 3,05-3,10 (m, 1 Н), 2,94-2,99 (m, 1 Н), 2,46-2,47 (m, 1 Н), 1,06 (d, J=6,71 Гц, 3 Н).
Пример 83 (3S,4S)-1-циано-4-метил-N-(5-метилтиазол-2-ил)пирролидин-3-карбоксамид
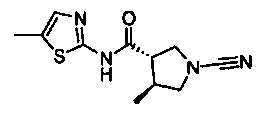
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 1, применяя Промежуточное соединение 2 и 2-амино-5-метилтиазол. Очистка с помощью препаративной хиральной ВЭЖХ; подвижная фаза: (А) 70-50% н-гексан (В) 30-50% IPA, колонка: Chiralpak IC, 250×10 мм, 5 мкм, скорость потока: 8 мл/мин. МС: ЭР+ 251,17; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,20 (s, 1 Н), 7,16 (d, J=1,2 Гц, 1 Н), 3,69-3,74 (m, 1 Н), 3,57-3,61 (m, 1 Н), 3,47-3,52 (m, 1 Н), 3,03-3,07 (m, 1 Н), 2,87-2,95 (m, 1 Н), 2,40-2,48 (m, 1 Н), 2,35 (s, 3 Н), 1,03 (d, J=6,71 Гц, 3 Н).
Пример 84 (±)-транс-N-(5-(2-хлорфенил)тиазол-2-ил)-1-циано-4-метилпирролидин-3-карбоксамид
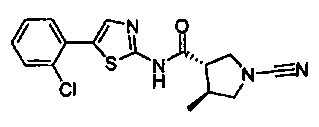
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 1, применяя Промежуточное соединение 2 и Промежуточное соединение 10. МС: ЭР+ 347,07; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,57 (s, 1 Н), 7,82 (s, 1 Н), 7,67 (dd, J=7,2, 1,6 Гц, 1 Н), 7,59 (dd, J=7,6, 2,0 Гц, 1 Н), 7,37-7,45 (m, 2 Н), 3,73-3,77 (m, 1 Н), 3,59-3,63 (m, 1Н) 3,53-3,57 (m, 1 H), 3,06-3,10 (m, 1 H), 2,94-3,01 (m, 1 H), 2,46-2,49 (m, 1H), 1,06 (d, J=6,71 Гц, 3 H).
Пример 85 (±)-1-циано-4-метил-N-(1-фенил-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
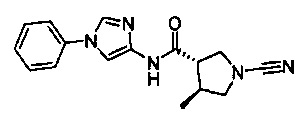
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 1, применяя Промежуточное соединение 2 и 4-амино-1-фенилимидазол. МС: ЭР+ 296,38; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,80 (s, 1 Н), 8,14 (d, J=1,52 Гц, 1 H), 7,74 (d, J=1,52 Гц, 1 H), 7,64 (d, J=8,0 Гц, 2H), 7,51 (t, J=7,6 Гц, 2H), 7,33-7,39 (m, 1 Н), 3,67-3,72 (m, 1 Н), 3,60-3,64 (m, 1 Н), 3,45-3,50 (m, 1 Н), 3,01-3,06 (m, 1 Н), 2,85-2,91 (m, 1 Н), 2,40-2,45 (m, 1 Н), 1,03 (d, J=6,71 Гц, 3 H).
Пример 86 (±)-1-циано-4-этил-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамид
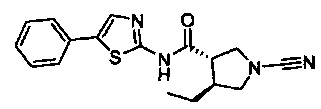
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 1, применяя Промежуточное соединение 17 и 2-амино-5-фенилтиазол. МС: ЭР+ 327,43; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,51 (s, 1H), 7,91 (s, 1H), 7,61-7,64 (m, 2H), 7,40-7,45 (m, 2H), 7,30-7,34 (m, 1H) 3,72-3,76 (m, 1H), 3,62-3,66 (m, 1H), 3,49-3,53 (m, 1H), 3,13-3,17 (m, 1H), 3,01-3,05 (m, 1H), 2,38-2,41 (m, 1H), 1,46-1,51 (m, 1H), 1,36-1,42 (m, 1H), 0,87 (t, J=7,6 Гц, 3H).
Пример 87 (±)-1-циано-4-этил-N-(1-фенил-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
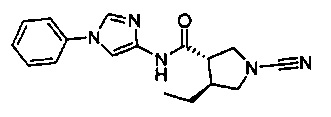
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 1, применяя Промежуточное соединение 17 и 1-фенил-1Н-имидазол-4-амин. МС: ЭР+ 310,13; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,82 (s, 1 Н), 8,14 (d, J=1,6 Гц, 1 Н), 7,74 (d, J=1,6 Гц, 1 Н), 7,62-7,74 (m, 2 Н), 7,49-7,53 (m, 2 Н), 7,34-7,39 (m, 1 Н), 3,63-3,70 (m, 2 Н), 3,40-3,50 (m, 1 Н), 3,07-3,12 (m, 1 Н), 2,91-2,97 (m, 1 Н), 2,30-2,35 (m, 1 Н), 1,44-1,49 (m, 1 Н), 1,29-1,38 (m, 1 Н), 0,86 (d, J=7,6 Гц, 3 Н).
Пример 88 1-циано-5-метил-N-(5-фенттиазол-2-ил)пирролидин-3-карбоксамид
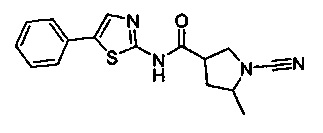
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 1, применяя 1-(трет-бутоксикарбонил)-5-метилпирролидин-3-карбоновую кислоту (полученную в соответствии со способом, описанным в WO 2010059658) и 2-амино-5-фенилтиазол. Соединение представляло собой смесь диастереомеров 86:14, указанные ниже пики ЯМР-спектра относятся только к основному диастереомеру. МС: ЭР+ 313,13; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,41 (s, 1 Н), 7,90 (s, 1 Н), 7,61-7,63 (m, 2 Н), 7,40-7,44 (m, 2 Н), 7,29-7,33 (m, 1 Н), 3,61-3,72 (m, 3 Н), 3,33-3,42 (m, 1 Н), 2,34-2,43 (m, 1 Н), 1,63-1,72 (m, 1 Н), 1,27 (d, J=6,10 Гц, 3 Н).
Пример 89 1-циано-5-метил-N-(1-фенил-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
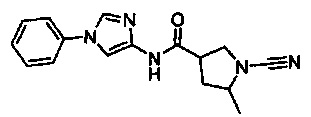
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 1, применяя 1-(трет-бутоксикарбонил)-5-метилпирролидин-3-карбоновую кислоту (полученную в соответствии со способом, описанном в WO 2010059658) и 4-амино-1-фенилимидазол. Соединение представляло собой смесь диастереомеров 89:11, указанные ниже пики ЯМР-спектра относятся только к основному диастереомеру. МС: ЭР+ 296,10; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,73 (s, 1 Н), 8,13 (d, J=1,52 Гц, 1 Н), 7,70 (d, J=1,52 Гц, 1 Н), 7,61-7,65 (m, 2 Н), 7,48-7,54 (m, 2 Н), 7,33-7,39 (m, 1 Н), 3,54-3,71 (m, 3 H), 3,23-3,33 (m, 1 H), 2,28-2,32 (m, 1 H), 1,59-1,67 (m, 1 H), 1,20-1,27 (m, 3Н).
Пример 90 1-циано-5-метил-N-(5-фенил-1Н-пиразол-3-ил)пирролидин-3-карбоксамид
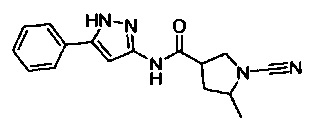
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 1, применяя 1-(трет-бутоксикарбонил)-5-метилпирролидин-3-карбоновую кислоту (полученную в соответствии со способом, описанном в WO 2010059658) и 5-амино-3-фенилпиразол. Соединение представляло собой смесь диастереомеров 88:12, указанные ниже пики ЯМР-спектра относятся только к основному диастереомеру. МС: ЭР+ 296,08; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,89 (ушир. s, 1 Н), 10,67 (s, 1 Н), 7,70-7,72 (m, 2 Н), 7,42-7,47 (m, 2 Н), 7,31-7,37 (m, 1 Н), 6,88 (ушир. s, 1 Н), 3,54-3,73 (m, 3 Н), 3,20-3,32 (m, 1 Н), 2,29-2,37 (m, 1 Н), 1,60-1,68 (m, 1 Н), 1,27 (d, J=6,40 Гц, 3 Н).
Пример 91 (S)-1-циано-N-(5-морфолинотиазол-2-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 3)
Стадия а. К раствору морфолина (11,17 ммоль) в MeCN (15 мл) добавляли Cs2CO3 (16,75 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 5 минут. К реакционной смеси добавляли 5-бромтиазол-2-амин (5,58 ммоль) и перемешивали при комнатной температуре в течение 2 ч. Полученную реакционную смесь вливали в воду (100 мл) и подвергали экстракции посредством EtOAc (3×50 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (2% МеОН в ДХМ), получая 5-морфолинотиазол-2-амин (1,83 ммоль). МС: ЭР+ 186,05; 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 6,49 (s, 2 Н), 6,28 (s, 1 Н), 3,64-3,67 (m, 4Н), 2,79-2,80 (m, 4 Н).
Стадии b-d. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для Примера 1, применяя (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту. МС: ЭР+ 308,33; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 11,97 (s, 1 Н), 6,69 (s, 1 Н), 3,70-3,73 (m, 4 Н), 3,57-3,63 (m, 1 Н), 3,39-3,50 (m, 3 Н), 3,24-3,33 (m, 1 Н), 2,97-3,00 (m, 4 Н), 2,10-2,21 (m, 1 Н), 1,97-2,10 (m, 1 Н).
Соединения, приведенные в таблице 3, синтезировали с помощью методики, аналогичной методике, описанной для Примера 91, применяя 1-ВОС-пирролидин-3-карбоновую кислоту.
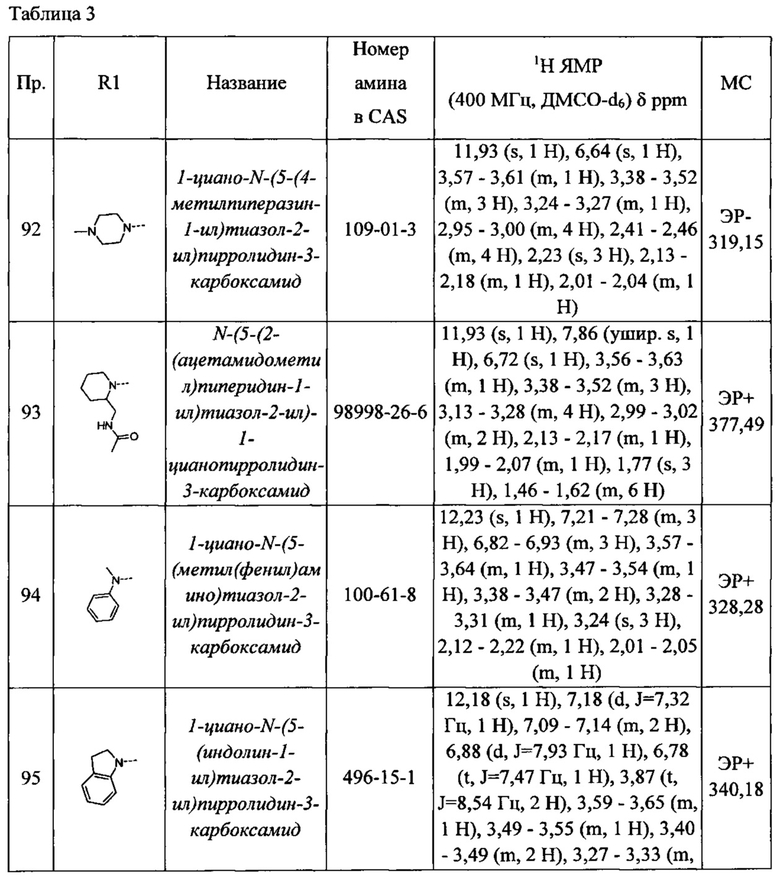
Соединения, приведенные в таблице 4, синтезировали с помощью методики, аналогичной методике, описанной для Примера 91, применяя (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту.
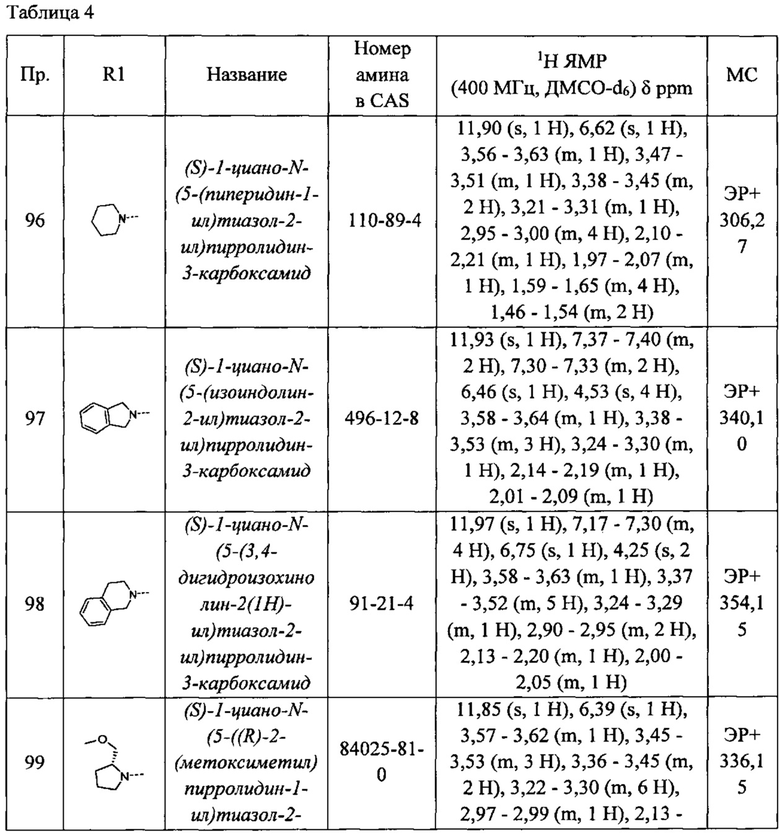
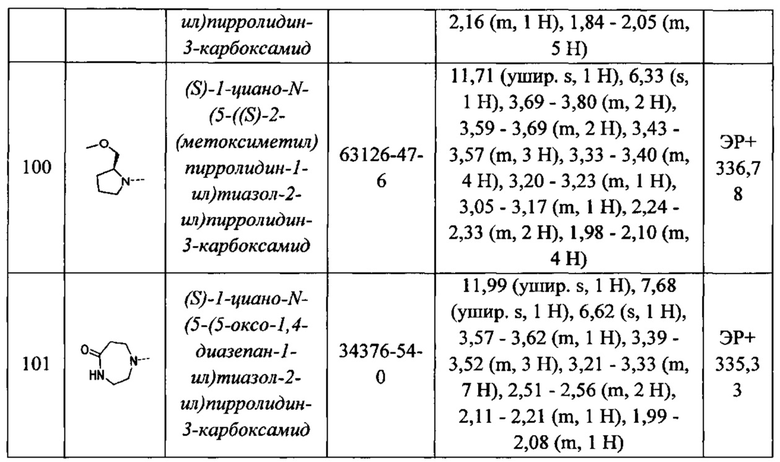
Пример 102 (R)-1-циано-N-(5-морфолинотиазол-2-ил)пирролидин-3-карбоксамид
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 91, применяя морфолин и (R)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту. МС: ЭР+ 308,33; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 11,97 (s, 1 Н), 6,69 (s, 1 Н), 3,70-3,73 (m, 4 Н), 3,57-3,63 (m, 1 Н), 3,39-3,50 (m, 3 Н), 3,24-3,33 (m, 1 Н), 2,97-3,00 (m, 4 Н), 2,10-2,21 (m, 1 Н), 1,97-2,10 (m, 1 Н).
Пример 103 (R)-1-циано-N-(5-(пиперидин-1-ил)тиазол-2-ил)пирролидин-3-карбоксамид
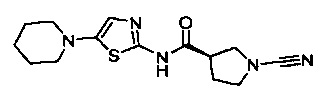
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 91, применяя пиперидин и (R)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту. МС: ЭР+ 305,9; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 11,89 (s, 1 Н), 6,61 (s, 1 Н), 3,57-3,61 (m, 1 Н), 3,37-3,50 (m, 3 Н), 3,23-3,34 (m, 1 Н), 2,95-3,00 (m, 4 Н), 2,09-2,21 (m, 1 Н), 1,97-2,08 (m, 1 Н), 1,57-1,69 (m, 4 Н), 1,50 (m, 2 Н).
Пример 104 (±)-транс-1-циано-4-метил-N-(5-(пиперидин-1-ил)тиазол-2-ил)пирролидин-3-карбоксамид
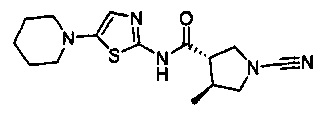
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 91, применяя пиперидин и Промежуточное соединение 2. МС: ЭР+ 320,21; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 11,99 (s, 1 Н), 6,62 (s, 1 Н), 3,67-3,72 (m, 1 Н), 3,57-3,61 (m, 1 Н), 3,45-3,49 (m, 1 Н), 3,02-3,06 (m, 1 Н), 2,96-3,00 (m, 4 Н), 2,84-2,90 (m, 1 Н), 2,40-2,46 (m, 1 Н), 1,59-1,64 (m, 4 Н), 1,49-1,52 (m, 2 Н), 1,01 (d, J=6,71 Гц, 3 Н).
Пример 105 1-циано-5-метил-N-(5-(пиперидин-1-ил)тиазол-2-ил)пирролидин-3-карбоксамид
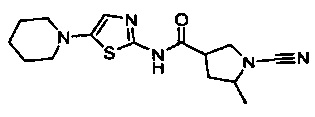
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 91, применяя пиперидин и 1-(трет-бутоксикарбонил)-5-метилпирролидин-3-карбоновую кислоту (полученную в соответствии со способом, описанном в WO 2010059658). МС: ЭР+ 320,38; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 11,89 (s, 1 Н), 6,62 (s, 1 Н), 3,67-3,71 (m, 1 Н), 3,54-3,63 (m, 2 Н), 3,23-3,32 (m, 1 Н), 2,95-3,00 (m, 4 Н), 2,29-2,35 (m, 1 Н), 1,56-1,66 (m, 5 Н), 1,49-1,52 (m, 2 Н), 1,25 (d, J=6,10 Гц, 3 H).
Пример 106 (S)-1-циано-N-(6-(пирролидин-1-ил)пиридин-2-ил)пирролидин-3-карбоксамид
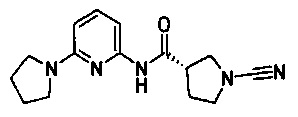
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 91, применяя 2-амино-6-бромпиридин, пирролидин и (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту. МС: ЭР+ 286,38; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,07 (s, 1 Н), 7,44 (t, J=7,93 Гц, 1 Н), 7,27 (d, J=8,24 Гц, 1 Н), 6,16 (d, J=8,24 Гц, 1 Н), 3,56-3,62 (m, 1 Н), 3,42-3,48 (m, 2 Н), 3,31-3,41 (m, 6 Н), 2,10-2,19 (m, 1 Н), 1,98-2,06 (m, 1 Н), 1,90-1,97 (m, 4 Н).
Пример 107 (S)-1-циано-N-(4-(пирролидин-1-ил)пиридин-2-ил)пирролидин-3-карбоксамид
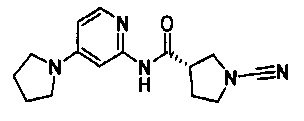
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 91, применяя 2-амино-4-бромпиридин, пирролидин и (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту. МС: ЭР+ 286,33; 1Н ЯМР (400 МГц, CDCl3) δ ppm 7,77 (d, J=6,40 Гц, 1 Н), 7,41 (s, 1 Н), 6,27 (dd, J=6,4, 2,4 Гц, 1 Н), 3,59-3,77 (m, 3 Н), 3,45-3,54 (m, 1 Н), 3,40-3,48 (m, 4 Н), 3,23-3,26 (m, 1 Н), 2,24-2,33 (m, 2Н), 2,06-2,11 (m, 4Н).
Пример 108 (S)-1-циано-N-(5-(пирролидин-1-ил)пиразин-2-ил)пирролидин-3-карбоксамид
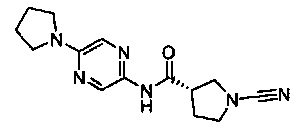
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 91, применяя 2-амино-5-бромпиразин, пирролидин и (S)-1-трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту. МС: ЭР+ 287,48; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,42 (s, 1 Н), 8,73 (s, 1 Н), 7,72 (d, J=1,52 Гц, 1 Н), 3,56-3,65 (m, 1 Н), 3,45-3,52 (m, 2 Н), 3,40-3,43 (m, 5 Н), 3,25-3,29 (m, 1 Н), 2,13-2,16 (m, 1 Н), 2,01-2,06 (m, 1 Н), 1,93-1,96 (m, 4Н).
Пример 109 N-(5-(2-аминофенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид (получали в соответствии со Схемой 4)
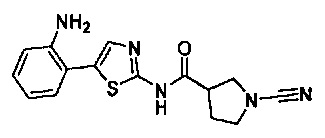
Стадия а. Раствор 5-бромтиазол-2-амина (39,3 ммоль) и ВОС-1-пирролидин-3-карбоновой кислоты (32,5 ммоль) в ТГФ (50 мл) перемешивали при комнатной температуре в течение 5 мин. К полученной реакционной смеси добавляли Т3Р (50% в EtOAc) (65,1 ммоль) и ТЭА (97,6 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Полученную реакционную смесь вливали в воду (100 мл) и подвергали экстракции посредством EtOAc (3×100 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (45-50% EtOAc в гексане), получая трет-бутил-3-((5-бромтиазол-2-ил)карбамоил)-пирролидин-1-карбоксилат (12,88 ммоль). МС: ЭР+ 376,23; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,54 (s, 1 Н), 7,58 (s, 1 Н), 3,50-3,54 (m, 1 Н), 3,36-3,40 (m, 2 Н), 3,24-3,34 (m, 2 Н), 2,12-2,15 (m, 1 Н), 2,00-2,05 (m, 1Н),1,4 (s, 9Н).
Стадия b. Раствор трет-бутил-3-((5-бромтиазол-2-ил)карбамоил)пирролидин-1-карбоксилата (0,26 ммоль) и 2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (0,26 ммоль) в смеси 1,4-диоксан: вода (1:1, 1 мл) перемешивали при комнатной температуре в стеклянной пробирке в течение 5 мин. К реакционной смеси добавляли Na2CO3 (1,06 ммоль) и дегазировали в течение 30 мин. Pd(PPh3)4 (0,026 моль) добавляли и стеклянную пробирку плотно закрывали. Реакционную смесь нагревали при 110°С (наружная температура) в течение 24 ч. Полученную реакционную смесь вливали в воду (150 мл) и подвергали экстракции посредством EtOAc (3×50 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (45-50% EtOAc в гексане), получая трет-бутил-3-((5-(2-аминофенил)тиазол-2-ил)карбамоил)пирролидин-1-карбоксилат (количественный выход). МС: ЭР+ 389,30
Стадия с. К раствору трет-бутил-3-((5-(2-аминофенил)тиазол-2-ил)карбамоил)пирролидин-1-карбоксилата (0,38 ммоль) в ДХМ (2 мл) добавляли 4М HCl в 1,4-диоксане (1,54 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 24 ч. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток растирали с диэтиловым эфиром (10 мл), получая N-(5-(2-аминофенил)тиазол-2-ил)пирролидин-3-карбоксамид гидрохлорид (0,34 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 289,23
Стадия d. К раствору N-(5-(2-аминофенил)тиазол-2-ил)пирролидин-3-карбоксамида гидрохлорида (0,34 ммоль) в ДХМ (2 мл) добавляли ТЭА (1,38 ммоль) и цианоген бромид (0,5 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Полученную реакционную смесь вливали в воду (20 мл) и подвергали экстракции посредством ДХМ (3×20 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (3-4% МеОН в ДХМ), получая указанное в заголовке соединение (0,05 ммоль). МС: ЭР+ 314,16; 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 12,35 (s, 1 Н), 7,56 (s, 1 Н), 7,13 (dd, J=7,63, 1,52 Гц, 1 H), 7,03-7,08 (m, 1 Н), 6,79 (d, J=7,2 Гц, 1 H), 6,59-6,64 (m, 1 Н), 5,09 (ушир. s, 2 Н), 3,61-3,66 (m, 1 Н), 3,52-3,56 (m, 1 Н), 3,40-3,48 (m, 2 Н), 3,33-3,38 (m, 1Н), 2,18-2,22 (m, 1 Н), 2,04-2,09 (m, 1 Н).
Соединения, приведенные в таблице 5, синтезировали с помощью методики, аналогичной методике, описанной для Примера 109, применяя 1-ВОС-пирролидин-3-карбоновую кислоту.
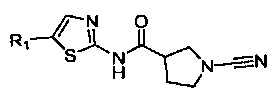
Пример 117 (S)-1-циано-N-(5-(3-цианофенил)тиазол-2-ил)пирролидин-3-карбоксамид
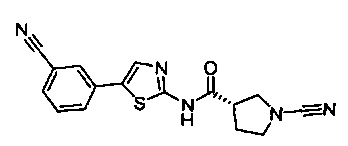
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 109, применяя (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту и 3-цианофенилбороновую кислоту на стадии b. МС: ЭР+ 324,33; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,49 (s, 1 Н), 8,12-8,22 (m, 1 Н), 8,08 (s, 1 Н), 7,88-7,96 (m, 1 Н), 7,72-7,80 (m, 1 Н), 7,57-7,66 (m, 1 Н), 3,58-3,66 (m, 1 Н), 3,50-3,57 (m, 1 Н), 3,34-3,48 (m, 3 Н), 2,16-2,30 (m, 1 Н), 2,00-2,15 (m, 1 Н).
Пример 118 (S)-1-циано-N-(5-(4-цианофенил)тиазол-2-ил)пирролидин-3-карбоксамид
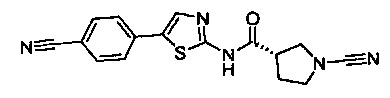
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 109, применяя (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту и 4-цианофенилбороновую кислоту. МС: ЭР+ 324,48; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,60 (s, 1 Н), 8,15 (s, 1 Н), 7,86-7,88 (m, 2 Н), 7,81-7,84 (m, 2 Н), 3,61-3,66 (m, 1 Н), 3,54-3,59 (m, 1 Н), 3,40-3,49 (m, 2 Н), 3,36-3,40 (m, 1 Н), 2,17-2,23 (m, 1 Н), 2,06-2,13 (m, 1 Н).
Пример 119 1-циано-N-(5-(3,5-диметилизоксазол-4-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 109, применяя 2-амино-5-бромбензотиазол на стадии а и 3,5-диметилизоксазол-4-бороновую кислоту на стадии b. МС: ЭР+ 367,92; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,61 (s, 1 Н), 8,09 (d, J=8,24 Гц, 1 Н), 7,75 (s, 1 Н), 7,33 (d, J=7,94 Гц, 1 Н), 3,53-3,72 (m, 2 Н), 3,39-3,49 (m, 3 Н), 2,44 (s, 3 Н), 2,21-2,27 (m, 4 Н), 2,05-2,19 (m, 1 Н).
Соединения, приведенные в таблице 6, синтезировали с помощью методики, аналогичной методике, описанной для Примера 109, применяя (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту и 2-амино-6-бромбензотиазол.
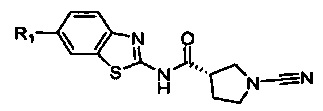
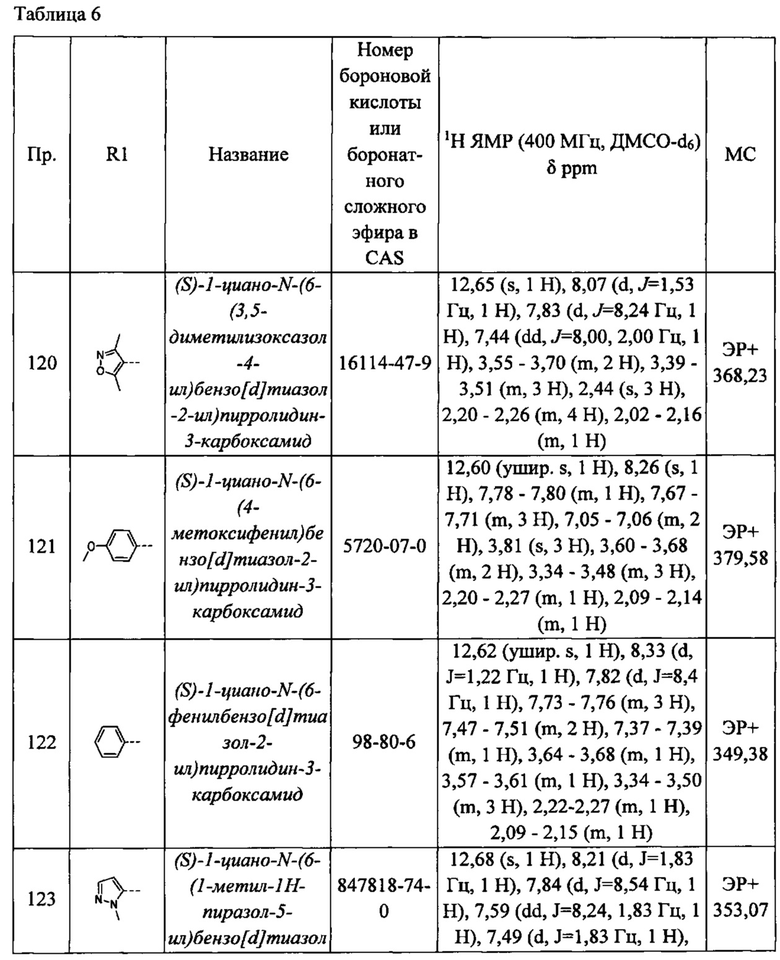
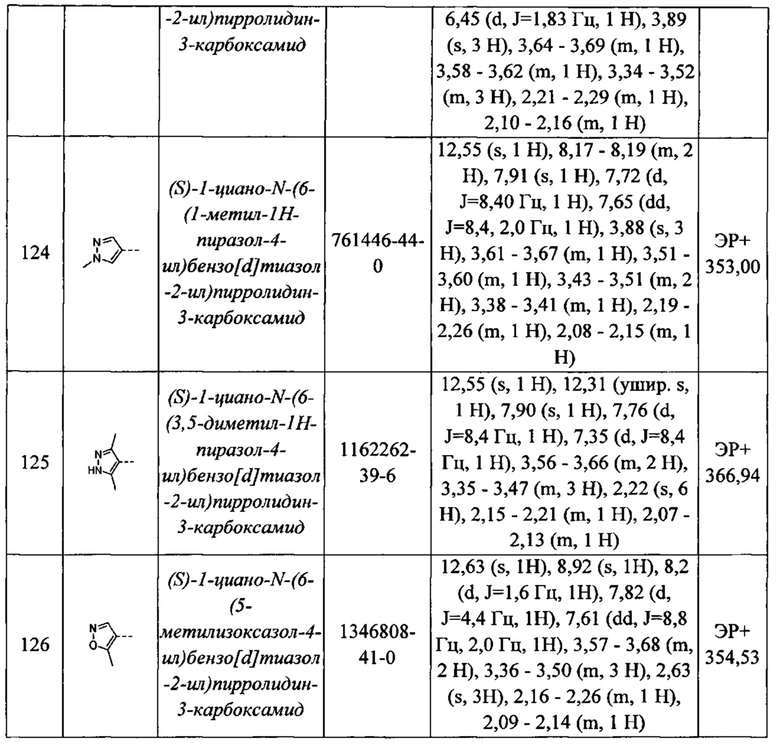
Пример 127 (S)-1-циано-N-(7-(3,5-диметилизоксазол-4-ил)имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамид
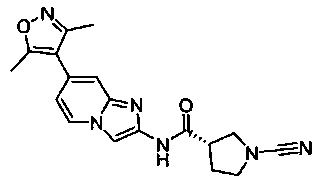
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 109, применяя (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту, 2-амино-7-бромимидазо[1,2-а]пиридин (полученный в соответствии со способом, описанном в WO 2005089763) и 3,5-диметилизоксазол-4-бороновую кислоту. МС: ЭР+ 351,08; 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 10,94 (s, 1 Н), 8,62 (d, J=7,02 Гц, 1 Н), 8,16 (s, 1 Н), 7,45 (s, 1 Н), 6,94 (dd, J=7,02, 1,83 Гц, 1 H), 3,61-3,66 (m, 1 Н), 3,44-3,54 (m, 2 Н), 3,40-3,43 (m, 1 Н), 3,30-3,33 (m, 1 Н), 2,47 (s, 3 Н), 2,29 (s, 3 Н), 2,16-2,21 (m, 1 Н), 2,05-2,10 (m, 1 Н).
Пример 128 (S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамид
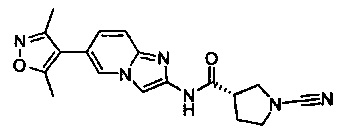
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 109, применяя (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту, 2-амино-6-бромимидазо[1,2-а]пиридин (полученный в соответствии со способом, описанным в WO 2012174312) и 3,5-диметилизоксазол-4-бороновую кислоту. МС: ЭР+ 351,09; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 11,00 (ушир. s, 1 Н), 8,66 (s, 1 Н), 8,15 (s, 1 Н), 7,52 (d, J=9,20 Гц, 1 Н), 7,26 (dd, J=9,16, 1,53 Гц, 1 Н), 3,49-3,52 (m, 1 Н), 3,44-3,48 (m, 1 Н), 3,38-3,42 (m, 2 Н), 3,28-3,31 (m, 1 Н), 2,43 (s, 3 Н), 2,20 (s, 3 Н), 2,14-2,22 (m, 1 Н), 2,01-2,10 (m, 1 Н).
Пример 129 (S)-1-циано-N-(6-циклопропилимидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 4)
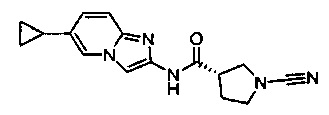
Стадия b. Раствор трет-бутил-(S)-3-((6-бромимидазо[1,2-а]пиридин-2-ил)карбамоил)пирролидин-1-карбоксилата (0,98 ммоль) [получали с помощью методики, аналогичной методике, описанной для Примера 109, применяя (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту и 2-амино-7-бромимидазо[1,2-а]пиридин (полученный в соответствии со способом, описанном в WO 2005089763) на стадии а] и циклопропилтрифторборат калия (1,96 ммоль) в смеси толуол: вода (8:1, 9 мл) смешивали в стеклянной пробирке. К реакционной смеси добавляли K3РО4 (1,96 ммоль) при комнатной температуре и дегазировали в течение 10 мин. К реакционной смеси добавляли Pd(dppf)Cl2.CH2Cl2 (0,098 ммоль) при комнатной температуре и стеклянную пробирку плотно закрывали и нагревали до 100°С в течение 18 часов. Полученную реакционную смесь вливали в воду (100 мл) и подвергали экстракции посредством EtOAc (2×100 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (4% МеОН в ДХМ), получая трет-бутил-(S)-3-((6-циклопропилимидазо[1,2-а]пиридин-2-ил)карбамоил) пирролидин-1-карбоксилат (0,45 ммоль). МС: ЭР+ 371,5
Стадии c-d. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для стадий c и d Примера 109. МС: ЭР+ 296,38; 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 10,87 (s, 1 Н), 8,37 (d, J=0,9 Гц, 1 Н), 7,99 (s, 1 Н), 7,32 (d, J=9,6 Гц, 1 Н), 6,97 (dd, J=9,6, 2,0 Гц, 1 Н), 3,59-3,61 (m, 1 Н), 3,40-3,51 (m, 2 Н), 3,26-3,34 (m, 2 Н), 2,13-2,19 (m, 1 Н), 2,01-2,08 (m, 1 Н), 1,90-1,94 (m, 1 Н), 0,88-0,95 (m, 2 Н), 0,66-0,70 (m, 2 Н).
Пример 130 1-циано-N-(6-циклопропилбензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
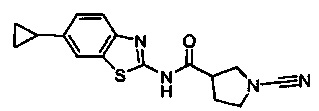
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 129, применяя 2-амино-6-бромбензотиазол и калий циклопропилтрифторборат. МС: ЭР+ 312,90; 1Н ЯМР (400 МГц, CD3OD) δ ppm 7,63 (d, J=8,40 Гц, 1 H), 7,59 (d, J=1,60 Гц, 1 Н), 7,19 (dd, J=8,40, 1,60 Гц, 1 Н), 3,70-3,74 (m, 2 Н), 3,50-3,64 (m, 3 Н), 2,18-2,39 (m, 2 Н), 2,00-2,10 (m, 1 Н), 0,97-1,06 (m, 2 Н), 0,71-0,78 (m, 2 Н).
Пример 131 (S)-1-циано-N-(6-(3,6-диметоксипиридазин-4-ил)имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 4)
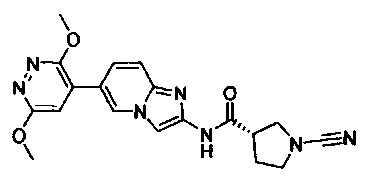
Стадия b. Раствор трет-бутил-(3S)-3-[(6-бромимидазо[1,2-а]пиридин-2-ил)карбамоил] пирролидин-1-карбоксилата (0,61 ммоль) [получали с помощью методики, аналогичной методике, описанной для Примера 109, применяя (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту и 2-амино-7-бромимидазо[1,2-а]пиридин (полученный в соответствии со способом, описанном в WO 2005089763) на стадии а] и 3,6-диметоксилпиридазин-4-бороновую кислоту (1,22 ммоль) в смеси 1,4-диоксан: вода (4:1, 5 мл) перемешивали при комнатной температуре в стеклянной пробирке в течение 5 минут. К реакционной смеси добавляли CsF (1,83 ммоль) при комнатной температуре и дегазировали в течение 30 мин. К реакционной смеси добавляли Pd(dppf)Cl2.CH2Cl2 (0,06 моль) при комнатной температуре и стеклянную пробирку плотно закрывали. Полученную реакционную смесь нагревали при 100°С в течение 2 часов. Полученную реакционную смесь вливали в воду (20 мл) и подвергали экстракции посредством EtOAc (3×20 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (100% ДХМ), получая трет-бутил-(S)-3-((6-(3,6-диметоксипиридазин-4-ил)имидазо[1,2-а]пиридин-2-ил)карбамоил) пирролидин-1-карбоксилат (0,44 ммоль). МС: ЭР+ 469,91. 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 10,94 (ушир. s, 1Н) 9,01 (s, 1Н), 8,17 (s, 1Н), 7,50-7,57 (m, 2Н), 7,39 (s, 1H), 4,02 (s, 3Н), 4,06 (s, 3Н), 3,51-3,53 (m, 1H), 3,35-3,43 (m, 2Н), 3,23-3,28 (m, 2Н), 2,09-2,11 (m, 1H), 2,03-2,04 (m, 1Н), 1,41 (s, 9Н).
Стадии c-d. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для стадий c и d Примера 109. МС: ЭР+ 394,11; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 11,01 (s, 1 Н), 9,02 (s, 1 Н), 8,19 (s, 1 Н), 7,49-7,57 (m, 2 Н), 7,40 (s, 1 Н), 4,04 (s, 3 Н), 4.00 (s, 3 Н), 3,59-3,69 (m, 1 Н), 3,46-3,54 (m, 2 Н), 3,40-3,44 (m, 2 Н), 2,14-2,21 (m, 1Н), 1,99-2,10 (m,1Н).
Пример 132 (S)-1-циано-N-(6-(3,6-диметоксипиридазин-4-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
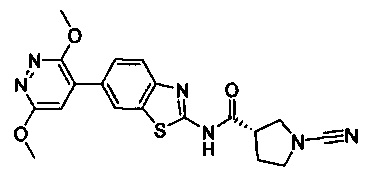
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 131, применяя 2-амино-5-бромбензотиазол и 3,6-диметоксилпиридазин-4-бороновую кислоту. МС: ЭР+ 411,06; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,69 (s, 1 Н), 8,34 (d, J=1,52 Гц, 1 Н), 7,83 (d, J=8,4 Гц, 1 Н), 7,75 (dd, J=2,0, 8,8 Гц, 1 Н), 7,31 (s, 1 Н), 4.01 (s, 3 Н), 4,00 (s, 3 Н) 3,57-3,68 (m, 2 Н), 3,37-3,51 (m, 3 Н), 2,19-2,33 (m, 1 Н), 2,07-2,16 (m, 1 Н).
Пример 133 (2S,3S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)бензо[d]тиазол-2-ил)-2-метил-пирролидин-3-карбоксамид
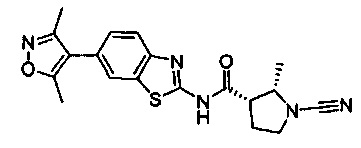
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 109, применяя Промежуточное соединение 1, 2-амино-6-бромбензотиазол и 3,5-диметилизоксазол-4-бороновую кислоту. Очистка с помощью препаративной ВЭЖХ; подвижная фаза: (А) 100% н-гексан (В) 50% IPA/МеОН, колонка: YMC PACKSIL, 250×20 мм, 5 мкм, скорость потока: 15 мл/мин. МС: ЭР+ 382,10; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,64 (s, 1 Н), 8,05 (d, J=1,52 Гц, 1 Н), 7,83 (d, J=8,24 Гц, 1 Н), 7,44 (dd, J=8,4, 2,0 Гц, 1 Н), 4,01-4,06 (m, 1 Н), 3,61-3,67 (m, 1 Н), 3,41-3,48 (m, 1 Н), 3,34-3,47 (m, 1 Н), 2,43 (s, 3 Н), 2,26 (s, 3 Н), 2,20-2,24 (m, 1 Н), 2,05-2,14 (m, 1 Н), 1,14 (d, J=6,71 Гц, 3 Н).
Пример 134 1-циано-N-(5-(n-толил)пиридин-2-ил)пирролидин-3-карбоксамид
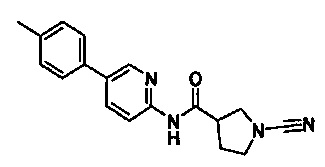
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 109, применяя 2-амино-5-бром-пиридин и 4-метилфенилбороновую кислоту. МС: ЭР+ 307,23; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,82 (s, 1 Н), 8,64 (d, J=2,0 Гц, 1 Н), 8,13-8,20 (m, 1 Н), 8,07-8,12 (m, 1 Н), 7,62 (d, J=8,24 Гц, 2 Н), 7,29 (d, J=7,94 Гц, 2 Н), 3,59-3,66 (m, 1 Н), 3,38-3,55 (m, 4 Н), 2,35 (s, 3 Н), 2,14-2,23 (m, 1 Н), 2,01-2,11 (m, 1 Н).
Пример 135 1-циано-N-(5-(м-толил)пиридин-2-ил)пирролидин-3-карбоксамид
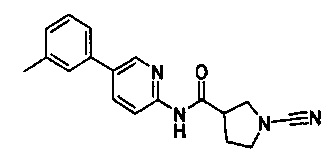
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 109, применяя 2-амино-5-бром-пиридин и 3-метилфенилбороновую кислоту. МС: ЭР+ 307,23; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,84 (s, 1 Н), 8,66 (dd, J=2,44, 0,61 Гц, 1 Н), 8,14-8,20 (m, 1 Н), 8,08-8,13 (m, 1 Н), 7,49-7,55 (m, 2 Н), 7,37 (t, J=7,63 Гц, 1 Н), 7,21 (d, J=7,33 Гц, 1 H), 3,59-3,66 (m, 1 Н), 3,38-3,55 (m, 4 Н), 2,38 (s, 3 Н), 2,16-2,21 (m, 1 Н), 2,02-2,12 (m, 1 Н).
Пример 136 1-циано-N-(5-(о-толил)пиридин-2-ил)пирролидин-3-карбоксамид
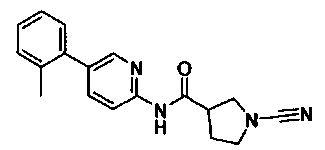
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 109, применяя 2-амино-5-бром-пиридин и 2-метилфенилбороновую кислоту. МС: ЭР+ 307,23; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,81 (s, 1 Н), 8,31 (d, J=2,44 Гц, 1 Н), 8,16 (d, J=8,55 Гц, 1 Н), 7,82 (dd, J=8,55, 2,44 Гц, 1 Н), 7,28-7,38 (m, 3 Н), 7,23-7,27 (m, 1 Н), 3,59-3,67 (m, 1 Н), 3,36-3,56 (m, 4 Н), 2,25 (s, 3 Н), 2,14-2,23 (m, 1 Н), 2,03-2,13 (m, 1 Н).
Пример 137 (S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)-5-метилимидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамид
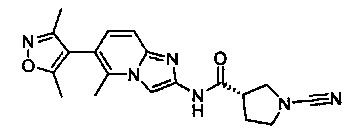
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 109, применяя (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту, Промежуточное соединение 16 и 3,5-диметилизоксазол-4-бороновую кислоту. МС: ЭР+ 365,23; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 11,02 (s, 1 Н), 7,95 (s, 1 Н), 7,44 (d, J=9,2, 1 H), 7,15 (d, J=9,2, 1 H), 3,62-3,66 (m, 1 H), 3,42-3,54 (m, 3 H), 3,30-3,34 (m, 1 H), 2,41 (s, 3 H), 2,27 (s, 3 H), 2,15-2,23 (m, 1 H), 2,09 (s, 3 H), 2,02-2,08 (m, 1 H).
Пример 138 (S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)-7-метилбензо[d]тиазол-2-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 5)
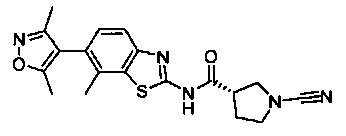
Стадия а. Раствор Промежуточного соединения 15 (1,23 ммоль) и 3,5-диметилизоксазол-4-бороновой кислоты (2,46 ммоль) в этанол: толуол: вода (1:2:1, 10 мл) перемешивали при комнатной температуре в стеклянной пробирке в течение 5 минут. К реакционной смеси добавляли Na2CO3 (2,46 ммоль) и продували с помощью азота в течение 20 мин. К реакционной смеси добавляли Pd(PPh3)4 (0,12 ммоль) и стеклянную пробирку плотно закрывали. Полученную реакционную смесь нагревали при 100°С (наружная температура) в течение 3 ч. Полученную реакционную смесь вливали в воду (100 мл) и подвергали экстракции посредством EtOAc (2×80 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (40% EtOAc в гексане), получая 75:25 смесь 6-(3,5-диметил-1,2-оксазол-4-ил)-7-метил-1,3-бензотиазол-2-амина & 6-(3,5-диметил-1,2-оксазол-4-ил)-5-метил-1,3-бензотиазол-2-амина (0,54 ммоль). МС: ЭР+ 260,33.
Стадия b. К раствору (3S)-ВОС-1-пирролидин-3-карбоновой кислоты (0,97 ммоль) и DIPEA (1,62 ммоль) в ДХМ (5 мл) добавляли HBTU (1,2 ммоль) при комнатной температуре и реакционную смесь перемешивали в течение 30 мин. К полученной реакционной смеси по каплям добавляли раствор 75:25 смеси 6-(3,5-диметил-1,2-оксазол-4-ил)-7-метил-1,3-бензотиазол-2-амина с 6-(3,5-диметил-1,2-оксазол-4-ил)-5-метил-1,3-бензотиазол-2-амином (0,81 ммоль) в ДХМ (5 мл) и перемешивали при комнатной температуре в течение 2 ч. Полученную реакционную смесь вливали в воду (100 мл) и подщелачивали, применяя твердый NaHCO3. Полученную смесь подвергали экстракции EtOAc (2×80 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (38% EtOAc в гексане), получая смесь 80:20 трет-бутил-(S)-3-((6-(3,5-диметилизоксазол-4-ил)-7-метилбензо[d]тиазол-2-ил)карбамоил)пирролидин-1-карбоксилата и трет-бутил-(S)-3-((6-(3,5-диметилизоксазол-4-ил)-5-метилбензо[d]тиазол-2-ил)карбамоил)пирролидин-1-карбоксилата (1,24 ммоль). МС: ЭР+ 456,91.
Стадия с. К раствору 80:20 смеси трет-бутил-(S)-3-((6-(3,5-диметилизоксазол-4-ил)-7-метилбензо[d]тиазол-2-ил)карбамоил)пирролидин-1-карбоксилата & трет-бутил-(S)-3-((6-(3,5-диметилизоксазол-4-ил)-5-метилбензо[d]тиазол-2-ил)карбамоил)пирролидин-1-карбоксилата (0,43 ммоль) в ДХМ (5 мл) добавляли ТФУ (1 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток подвергали азеотропной перегонке с применением ДХМ, получая смесь 80:20 (S)-N-(6-(3,5-диметилизоксазол-4-ил)-7-метилбензо[d]тиазол-2-ил)пирролидин-3-карбоксамида и соли ТФУ и (S)-N-(6-(3,5-диметилизоксазол-4-ил)-5-метилбензо[d]тиазол-2-ил)пирролидин-3-карбоксамида (количественный выход). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 357,48.
Стадия d. К раствору смеси 80:20 (S)-N-(6-(3,5-диметилизоксазол-4-ил)-7-метилбензо[d]тиазол-2-ил)пирролидин-3-карбоксамида и соли ТФУ и (S)-N-(6-(3,5-диметилизоксазол-4-ил)-5-метилбензо[d]тиазол-2-ил)пирролидин-3-карбоксамида (0,71 ммоль) в ТГФ (5 мл) добавляли K2СО3 (7,1 ммоль) при комнатной температуре. К реакционной смеси добавляли цианоген бромид (0,56 ммоль) при 0°С и перемешивали в течение 10 мин. Полученную реакционную смесь вливали в воду (150 мл) и подвергали экстракции посредством EtOAc (2×80 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (72% EtOAc в гексане), получая смесь 70:30 (3S)-1-циано-N-[6-(3,5-диметил-1,2-оксазол-4-ил)-7-метил-1,3-бензотиазол-2-ил] пирролидин-3-карбоксамида и (3S)-1-циано-N-[6-(3,5-диметил-1,2-оксазол-4-ил)-5-метил-1,3-бензотиазол-2-ил]пирролидин-3-карбоксамида (0,17 ммоль). Региоизомеры разделяли с помощью препаративной хиральной ВЭЖХ; подвижная фаза: (А) 75-70% 10 мМ водн. NH4OAc (В) 25-30% MeCN, колонка: Sunfire С18, 250×19 мм, 5 мкм, скорость потока: 19 мл/мин, получая указанное в заголовке соединение (0,057 ммоль). МС: ЭР+ 382,43; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,64 (ушир. s, 1Н), 7,60 (d, J=8,0 Гц, 1H), 7,22 (d, J=8,4 Гц, 1H), 3,57-3,66 (m, 2Н), 3,42-3,47 (m, 2Н), 3,32-3,36 (m, 1Н), 2,28 (s, 3Н), 2,23 (s, 3Н), 2,18-2,22 (m, 1H), 2,10-2,13 (m, 1Н), 2,05 (s, 3Н).
Пример 139 (S)-1-циано-N-(7-метил-6-(1-метил-1H-пиразол-4-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамид
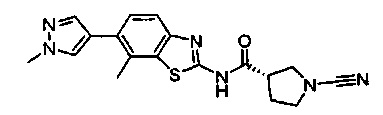
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 138, применяя пинаколовый эфир 1-метил-1Н-пиразол-4-бороновой кислоты. МС: ЭР+ 366,3; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,56 (s, 1H), 7,97 (s, 1H), 7,68 (s, 1H), 7,61 (d, J=8,4 Гц, 1H), 7,44 (d, J=8,4 Гц, 1Н), 3,90 (s, 3Н), 3,63-3,68 (m, 1H), 3,56-3,60 (m, 1H), 3,43-3,49 (m, 2H), 3,37-3,42 (m, 1H), 2,55 (s, 3H), 2,21-2,28 (m, 1H), 2,07-2,19 (m, 1H).
Пример 140 1-циано-N-(5-(морфолинометил)тиазол-2-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 6)
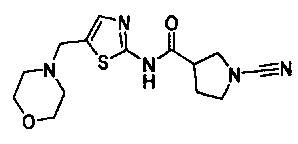
Стадия а. Раствор 2-амино-5-формилтиазола (15,6 ммоль), 1-(трет-бутоксикарбонил)пирролидин-3-карбоновой кислоты (19,5 ммоль) и HOAt (18,7 ммоль) в ДМФА (20 мл) перемешивали при комнатной температуре в течение 5 мин. К реакционной смеси добавляли DIPEA (21,8 ммоль) и EDC.HCl (18,7 ммоль) и перемешивали в течение дополнительных 3 ч при комнатной температуре. Полученную реакционную смесь вливали в воду (200 мл) и подвергали экстракции посредством EtOAc (2×50 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (60% EtOAc в гексане), получая трет-бутил-3-((5-формилтиазол-2-ил)карбамоил)пирролидин-1-карбоксилат (4,6 ммоль). МС: ЭР+ 326,20; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,91 (s, 1 Н), 9,97 (s, 1 Н), 8,44 (s, 1 Н), 3,63-4,02 (m, 1 Н), 3,27-3,36 (m, 4 Н), 2,11-2,22 (m, 1 Н), 1,99-2,10 (m, 1 Н), 1,45 (s, 9Н).
Стадия b. К раствору 3-((5-формилтиазол-2-ил)карбамоил)пирролидин-1-карбоксилата (1,5 ммоль) и морфолина (0,8 ммоль) в ДХЭ (10 мл) добавляли триацетоксиборогидрид натрия (1,5 ммоль) при комнатной температуре и перемешивали в течение 3 ч. Полученную реакционную смесь вливали в воду (50 мл) и подвергали экстракции посредством EtOAc (2×25 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (25% EtOAc в гексане), получая трет-бутил-3-((5-(морфолинометил)тиазол-2-ил)карбамоил) пирролидин-1-карбоксилат (1,2 ммоль). МС: ЭР+ 397,33; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,16 (s, 1 Н), 7,30 (s, 1 Н), 3,49-3,62 (m, 6 Н), 3,35-3,39 (s, 3 Н), 3,19-3,31 (m, 2 Н), 2,31-2,37 (m, 4 Н), 2,05-2,15 (m, 1 Н), 1,92-2,01 (m, 1 Н), 1,40 (s, 9Н).
Стадия с. К раствору трет-бутил-3-((5-(морфолинометил)тиазол-2-ил)карбамоил)пирролидин-1-карбоксилата (1,2 ммоль) в ДХМ (4 мл) добавляли ТФУ (0,6 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 40 мин. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток подвергали азеотропной перегонке с применением ДХМ, получая соль ТФУ и N-(5-(морфолинометил)тиазол-2-ил)пирролидин-3-карбоксамида (1,0 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки.
Стадия d. К раствору соли ТФУ и N-(5-(морфолинометил)тиазол-2-ил)пирролидин-3-карбоксамида (1,0 ммоль) в ТГФ (4 мл) добавляли K2СО3 (3,0 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 30 мин. К реакционной смеси добавляли цианоген бромид (1,1 ммоль) при 0°С. Реакционную смесь затем перемешивали при комнатной температуре в течение 2 ч. Полученную реакционную смесь вливали в воду (30 мл) и подвергали экстракции посредством EtOAc (20 мл). Органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (7-8% МеОН в ДХМ), получая указанное в заголовке соединение (0,08 ммоль). МС: ЭР+ 322,23; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,20 (ушир. s, 1 Н), 7,30 (s, 1 Н), 3,56-3,63 (m, 3 Н), 3,46-3,60 (m, 4 Н), 3,40-3,44 (m, 2 Н), 3,20-3,30 (m, 2 Н), 2,30-2,48 (m, 4 Н), 2,15-2,18 (m, 1 Н), 2,02- 2,07 (m, 1 Н).
Пример 141 1-циано-N-(5-(пирролидин-1-илметил)тиазол-2-ил)пирролидин-3-карбоксамид
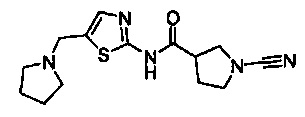
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 140, применяя пирролидин. МС: ЭР+ 306,38; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,29 (s, 1 Н), 7,29 (s, 1 Н), 3,72 (s, 2 Н), 3,59-3,63 (m, 1 Н), 3,49-3,53 (m, 1 Н), 3,40-3,46 (m, 2 Н), 3,29-3,34 (m, 1 Н), 2,45 (m, 4 Н), 2,12-2,23 (m, 1 Н), 2,01-2,12 (m, 1 Н), 1,69 (m, 4 Н).
Пример 142 (3S)-1-циано-N-(5-((2,6-диметилморфолино)метил)тиазол-2-ил)пирролидин-3-карбоксамид
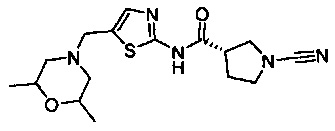
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 140, применяя (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту и 2,6-диметилморфолин. МС: ЭР+ 350,43; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,22 (ушир. s, 1 Н), 7,30 (s, 1 Н), 3,62 (m, 4 Н), 3,28-3,55 (m, 5 Н), 2,67-2,70 (m, 2 Н), 2,11-2,27 (m, 1 Н), 2,05-2,07 (m, 1 Н), 1,64 (t, J=10,84 Гц, 2 Н), 1,03 (d, J=6,41 Гц, 6 Н).
Пример 143 (S)-1-циано-N-(5-(((R)-2-(метоксиметил)пирролидин-1-ил)метил)тиазол-2-ил)пирролидин-3-карбоксамид
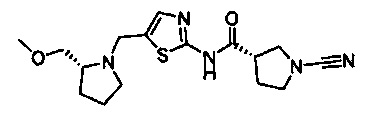
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 140, применяя (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту и (R)-2-(метоксиметил)пирролидин. МС: ЭР+ 349,98; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,19 (s, 1 Н), 7,27 (s, 1 Н), 4,10 (d, J=14,04 Гц, 1 Н), 3,50-3,67 (m, 2 Н), 3,42-3,46 (m, 1 Н), 3,37-3,45 (m, 2 Н), 3,20-3,32 (m, 6 Н), 2,79-2,87 (m, 1 Н), 2,65-2,74 (m, 1 Н), 2,09-2,27 (m, 2 Н), 1,97-2,10 (m, 1 Н), 1,77-1,87 (m, 1 Н), 1,56-1,68 (m, 2Н), 1,48 (m, 1 Н).
Пример 144 (S)-1-циано-N-(5-(((S)-2-(метоксиметил)пирролидин-1-ил)метил)тиазол-2-ил)пирролидин-3-карбоксамид
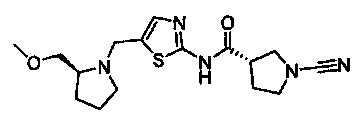
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 140, применяя (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту и (S)-2-(метоксиметил)пирролидин. МС: ЭР+ 349,98; 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 12,19 (s, 1 Н), 7,27 (s, 1 Н), 4,10 (d, J=14,04 Гц, 1 Н), 3,59-3,68 (m, 2 Н), 3,49-3,53 (m, 1 Н), 3,37-3,47 (m, 3 Н), 3,20-3,31 (m, 5 Н), 2,73-2,89 (m, 1 Н), 2,60-2,75 (m, 1 Н), 2,08-2,27 (m, 2 Н), 1,99-2,08 (m, 1 Н), 1,75-1,87 (m, 1 Н), 1,54-1,71 (m, 2 Н), 1,45-1,48 (m, 1 Н).
Пример 145 (2S,3S)-1-циано-N-(5-(((R)-2-(метоксиметил)пирролидин-1-ил)метил)тиазол-2-ил)-2-метилпирролидин-3-карбоксамид (получали в соответствии со Схемой 7)
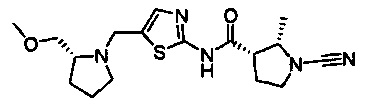
Стадия а. К раствору 2-аминотиазол-5-карбальдегида (3,9 ммоль) в ТГФ (10 мл) добавляли DMAP (5,85 ммоль) и (ВОС)2О (5,85 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь вливали в воду (50 мл) и подвергали экстракции посредством EtOAc (3×100 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (40% EtOAc в гексане), получая трет-бутил-(5-формилтиазол-2-ил) карбамат (2,63 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 229,05.
Стадия b. К раствору трет-бутил-(5-формилтиазол-2-ил) карбамата (2,19 ммоль) и (R)-2-(метоксиметил) пирролидина (4,83 ммоль) в МеОН (5 мл) добавляли натрий триацетоксиборогидрид (3,28 ммоль) при комнатной температуре. Реакционную смесь нагревали при 80°С в течение 2 часов. Полученную реакционную смесь вливали в 1М водный раствор HCl (50 мл) и подвергали экстракции посредством EtOAc (2×25 мл). Органический слой отбрасывали. Водный слой подщелачивали твердым Na2CO3. Полученную смесь подвергали экстракции EtOAc (2×50 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая трет-бутил-(R)-(5-((2-(метоксиметил)пирролидин-1-ил) метил) тиазол-2-ил) карбамат (1,56 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 328,48; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 11,29 (s, 1 Н), 7,14 (s, 1 Н), 4,06 (d, J=14,4 Гц, 1 Н), 3,62 (d, J=14,4 Гц, 1 Н), 3,34-3,38 (m, 1 Н), 3,23-3,29 (m, 3 Н), 3,19-3,22 (m, 1 Н), 2,82-2,86 (m, 1 Н), 2,67-2,70 (m, 1 Н), 2,18-2,24 (m, 1 Н), 1,77-1,85 (m, 1 Н), 1,57-1,63 (m, 3 Н), 1,47 (s, 9H).
Стадия с. К раствору трет-бутил-(R)-(5-((2-(метоксиметил)пирролидин-1-ил)метил) тиазол-2-ил)карбамата (1,56 ммоль) в ДХМ (6 мл) добавляли ТФУ (2,5 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток нейтрализовали водным раствором насыщенного NaHCO3. Полученную смесь подвергали экстракции ДХМ (2×100 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая (R)-5-((2-(метоксиметил) пирролидин-1-ил) метил)тиазол-2-амин (количественный выход). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 228,04.
Стадия d. К раствору (2S,3S)-1-(трет-бутоксикарбонил)-2-метилпирролидин-3-карбоновой кислоты (1,74 ммоль) в ТГФ (6 мл) добавляли Т3Р (50% в EtOAc) (5,24 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 30 мин и затем охлаждали до 0°С. (R)-5-((2-(метоксиметил) пирролидин-1-ил)метил)тиазол-2-амин (1,57 ммоль) и DIPEA (5,24 ммоль) добавляли и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь вливали в насыщенный NaHCO3 (25 мл) и подвергали экстракции посредством EtOAc (3×50 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая трет-бутил-(2S,3S)-3-((5-(((R)-2-(метоксиметил)пирролидин-1-ил)метил)тиазол-2-ил) карбамоил)-2-метилпирролидин-1-карбоксилат (0,52 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 439,25; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,09 (ушир. s, 1 Н), 7,26 (s, 1 Н), 4,18-4,22 (m, 1 Н), 4,08-4,11 (m, 1 Н), 3,63-3,66 (m, 1 Н), 3,34-3,39 (m, 2 Н), 3,26 (s, 3 Н), 3,16-3,22 (m, 3 Н), 2,81-2,93 (m, 1 Н), 2,67-2,69 (m, 1 Н), 2,19-2,25 (m, 2 Н), 1,90-1,93 (m, 1 Н), 1,80-1,85 (m, 1 Н), 1,58-1,63 (m, 2 Н), 1,47-1,50 (m, 1 Н), 1,47 (s, 9 Н), 0,90 (d, J=5,2 Гц, 3 Н).
Стадия е. К раствору трет-бутил-(2S,3S)-3-((5-(((R)-2-(метоксиметил)пирролидин-1-ил)метил)тиазол-2-ил) карбамоил)-2-метилпирролидин-1-карбоксилата (0,52 ммоль) в ДХМ (10 мл) добавляли ТФУ (1,0 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток растирали с диэтиловым эфиром (10 мл), получая соль ТФУ и (2S,3S)-N-(5-(((R)-2-(метоксиметил)пирролидин-1-ил)метил)тиазол-2-ил)-2-метилпирролидин-3-карбоксамида (0,409 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 339,10.
Стадия f. К раствору соли ТФУ и (2S,3S)-N-(5-(((R)-2-(метоксиметил)пирролидин-1-ил)метил)тиазол-2-ил)-2-метилпирролидин-3-карбоксамида (0,409 ммоль) в ТГФ (6 мл) добавляли K2СО3 (1,2 ммоль) при 0°С. Реакционную смесь перемешивали при 0° С в течение 30 мин. К реакционной смеси добавляли цианоген бромид (0,60 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Полученную реакционную смесь вливали в воду (30 мл). Полученный твердый осадок собирали путем фильтрования при пониженном давлении и сушили, получая указанное в заголовке соединение (0,09 ммоль). МС: ЭР+ 364,15; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,16 (ушир. s, 1 Н), 7,27 (s, 1 Н), 4,08-4,12 (m, 1 Н), 3,97-4,01 (m, 1 Н), 3,63-3,67 (m, 2 Н), 3,35-3,42 (m, 4 Н), 3,26 (s, 3 Н), 2,83-2,87 (m, 1 Н), 2,62-2,69 (m, 1 Н), 2,15-2,28 (m, 2 Н), 2,03-2,11 (m, 1 Н), 1,75-1,83 (m, 1 Н), 1,55-1,70 (m, 2 Н), 1,49-1,51 (m, 1 Н), 0,87 (d, J=5,2 Гц, 3 Н).
Пример 146 (S)-N-(1-бензил-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамид (получали в соответствии со Схемой 8, стадии с, f, h, i, j)
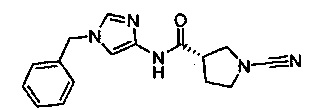
Стадия с. К раствору 4-нитро-1Н-имидазола (4,42 ммоль) в ДМСО (5 мл) добавляли пеллеты KОН (6,6 ммоль) при комнатной температуре. Реакционную смесь охлаждали до 0°С и обрабатывали бензилбромидом (5,31 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Полученную реакционную смесь вливали в воду (50 мл) и подвергали экстракции посредством EtOAc (3×50 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (1,5-2,5% МеОН в ДХМ), получая 1-бензил-4-нитро-1Н-имидазол (3,2 ммоль). 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 8,50 (d, J=1,2 Гц, 1 Н), 8,01 (d, J=1,2 Гц, 1 Н), 7,34-7,43 (m, 5 Н), 5,31 (s, 2 Н).
Стадия f. Раствор 1-бензил-4-нитро-1Н-имидазола (2,46 ммоль) в МеОН: вода (1:1, 12 мл) перемешивали при комнатной температуре в течение 5 мин. К реакционной смеси добавляли порошок Fe (12,3 ммоль) и NH4Cl (4,9 ммоль). Реакционную смесь нагревали при 80°С в течение 2 ч. Полученной реакционной смеси давали возможность остыть до к. т. и вливали ее в воду (30 мл). Полученную смесь фильтровали через Celite Hyflo и промывали посредством EtOAc (20 мл). Отделяли органический слой и водный слой подвергали экстракции EtOAc (3×20 мл). Объединенную органическую фазу собирали, промывали насыщенным раствором NaHCO3 (20 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 1-бензил-1Н-имидазол-4-амин (1,4 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 174,24.
Стадия h. Раствор 1-бензил- 1Н-имидазол-4-амина (1,4 ммоль) и (3S)-BOC-1-пирролидин-3-карбоновой кислоты (1,4 ммоль) в ТГФ (3 мл) перемешивали при 0°С в течение 5 мин. К полученной реакционной смеси добавляли Т3Р (50% в EtOAc) (2,1 ммоль) и ТЭА (4,3 ммоль) и перемешивали при комнатной температуре в течение 2 ч. Полученную реакционную смесь вливали в воду (40 мл) и подвергали экстракции посредством EtOAc (3×20 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (2-3% МеОН в ДХМ), получая трет-бутил-(S)-3-((1-бензил-1Н-имидазол-4-ил)карбамоил)пирролидин-1-карбоксилат (0,80 ммоль). МС: ЭР+ 371,33
Стадия i. К раствору трет-бутил-(S)-3-((1-бензил-1Н-имидазол-4-ил)карбамоил)пирролидин-1-карбоксилата (0,80 ммоль) в ДХМ (6 мл) добавляли 4М HCl в 1,4-диоксане (24 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток растирали в диэтиловом эфире (10 мл), получая (S)-N-(1-бензил-1Н-имидазол-4-ил)пирролидин-3-карбоксамид гидрохлорид (0,65 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 271,33.
Стадия j. К раствору (S)-N-(1-бензил-1Н-имидазол-4-ил)пирролидин-3-карбоксамида гидрохлорида (0,65 ммоль) в ДМФА (4 мл) добавляли K2СО3 (1,95 ммоль) и цианоген бромид (0,78 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Полученную реакционную смесь вливали в воду (40 мл) и подвергали экстракции посредством EtOAc (3×20 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (2,5-3,5% МеОН в ДХМ), получая указанное в заголовке соединение (0,14 ммоль). МС: ЭР+ 296,01; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,53 (s, 1 Н), 7,60 (d, J=1,2 Гц, 1 Н), 7,35-7,38 (m, 2 Н), 7,27-7,33 (m, 3 Н), 7,21 (d, J=1,2 Гц, 1 Н), 5,14 (s, 2 Н), 3,52-3,56 (m, 1 Н), 3,34-3,46 (m, 3Н), 3,15-3,21 (m, 1 Н), 2,04-2,10 (m, 1 Н), 1,93-2,00 (m, 1 Н).
Пример 147 (S)-1-циано-N-(1-фенетил-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
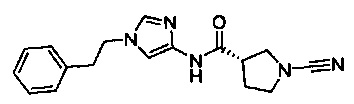
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 146, применяя (2-бромэтил)бензол на стадии с. МС: ЭР+ 310,01; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,47 (s, 1 Н), 7,27-7,30 (m, 4 Н), 7,19-7,23 (m, 3 Н), 4,16 (t, J=7,32 Гц, 2 Н), 3,53-3,60 (m, 1 Н), 3,36-3,48 (m, 3 Н), 3,15-3,23 (m, 1 Н), 3,01 (t, J=7,32 Гц, 2 Н), 2,06-2,15 (m, 1 Н), 1,97-2,05 (m, 1 Н).
Пример 148 (S)-1-циано-N-(1-изобутил-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
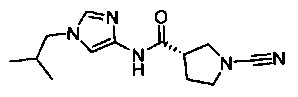
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 146, применяя 1-бром-2-метилпропан на стадии с. МС: ЭР+ 262,04; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,51 (s, 1 Н), 7,41 (d, J=1,52 Гц, 1 Н), 7,20 (d, J=1,52 Гц, 1 Н), 3,72 (d, J=7,32 Гц, 2 Н), 3,54-3,58 (m, 1 Н), 3,36-3,49 (m, 3 Н), 3,17-3,21 (m, 1 Н), 2,05-2,15 (m, 1 Н), 1,92-2,04 (m, 2 Н), 0,82 (d, J=6,71 Гц, 6 Н).
Пример 149 (2S,3S)-N-(1-бензил-1Н-имидазол-4-ил)-1-циано-2-метилпирролидин-3-карбоксамид
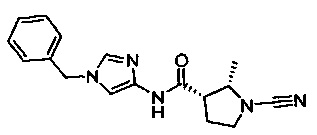
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 146, применяя бензилбромид и Промежуточное соединение 1. Очистка с помощью препаративной ВЭЖХ; подвижная фаза: (А) 100% н-гексан (В) 50% IPA / МеОН, колонка: YMC PACKSIL, 250×20 мм, 5 мкм, скорость потока: 20 мл/мин. МС: ЭР+ 310,15; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,48 (s, 1 Н), 7,60 (d, J=1,60 Гц, 1 Н), 7,35-7,39 (m, 2 Н), 7,29-7,33 (m, 3 Н), 7,22 (d, J=1,60 Гц, 1 Н), 5,13 (s, 2 Н), 3,87-3,90 (m, 1 Н), 3,55-3,59 (m, 1 Н), 3,36-3,39 (m, 1 Н), 3,12-3,14 (m, 1 Н), 2,06-2,09 (m, 1 Н), 1,92-1,96 (m, 1 Н), 1,04 (d, J=6,40 Гц, 3 Н).
Пример 150 (S)-1-циано-N-(1-(4-фторфенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 8, стадии a, b, g, h, i, j)
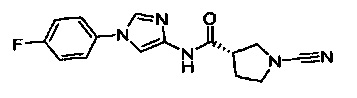
Стадия а. Раствор 4-нитро-1Н-имидазола (8,84 ммоль) в уксусной кислоте (18 мл) охлаждали до 0°С. К реакционной смеси по каплям добавляли дымящую HNO3 (4,3 мл). После завершения добавления к реакционной смеси по каплям добавляли уксусный ангидрид (12 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Полученную реакционную смесь медленно вливали в ледяную воду (100 мл) и подвергали экстракции посредством EtOAc (3×50 мл). Объединенную органическую фазу медленно промывали насыщенным раствором K2СО3 (50 мл) (экзотермический процесс), солевым раствором (50 мл) сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 1,4-динитро-1Н-имидазол (8,8 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3) δ ppm 8,53 (d, J=1,5 Гц, 1 Н), 8,40 (d, J=1,5 Гц, 1 Н).
Стадия b. К раствору 1,4-динитро-1Н-имидазола (1,25 ммоль) в МеОН : вода (1:1, 8 мл) добавляли 4-фторанилин (1,32 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Полученный твердый осадок собирали путем фильтрования при пониженном давлении и сушили, получая 1-(4-фторфенил)-4-нитро-1Н-имидазол (0,96 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 208,23.
Стадия g. К раствору 1-(4-фторфенил)-4-нитро-1Н-имидазола (0,97 ммоль) в МеОН (5 мл) добавляли 10% Pd/C (0,5% масс./масс.) при комнатной температуре. Реакционную смесь продували газообразным H2 в течение 2 часов при комнатной температуре. Полученную реакционную смесь тщательно фильтровали через Celite Hyfio и концентрировали при пониженном давлении с получением 1-(4-фторфенил)-1Н-имидазол-4-амина (0,90 ммоль). Данный материал сразу же применяли на следующей стадии без дополнительной очистки. МС: ЭР+: 177,96
Стадии h-j. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для Примера 146 в соответствии со стадиями h, i и j Схемы 8. МС: ЭР+ 300,23; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,73 (s, 1 Н), 8,08 (d, J=1,2 Гц, 1 Н), 7,66-7,70 (m, 3 Н), 7,33-7,38 (m, 2 Н), 3,58-3,62 (m, 1 Н), 3,44-3,50 (m, 2 Н), 3,37-3,43 (m, 1 Н), 3,21-3,28 (m, 1 Н), 2,10-2,18 (m, 1 Н), 1,98-2,07 (m, 1 Н).
Пример 151 (S)-1-циано-N-(1-(3-фторфенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
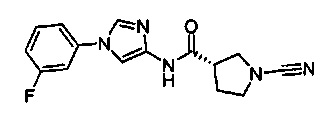
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя 3-фторанилин на стадии b. МС: ЭР+ 300,28; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,75 (s, 1 Н), 8,21 (d, J=1,52 Гц, 1 Н), 7,78 (d, J=1,52 Гц, 1 Н), 7,62-7,67 (m, 1 Н), 7,50-7,59 (m, 2 Н), 7,16-7,23 (m, 1 Н), 3,58-3,63 (m, 1 Н), 3,37-3,51 (m, 3 Н), 3,23-3,34 (m, 1 Н), 2,10-2,20 (m, 1 Н), 1,98-2,08 (m, 1 Н).
Пример 152 (S)-1-циано-N-(1-(2-фторфенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
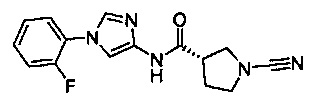
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя 2-фторанилин на стадии b. МС: ЭР+ 300,23; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,75 (ушир. s, 1 Н), 8,22 (s, 1 Н), 7,78 (s, 1 Н), 7,63-7,65 (m, 1 Н), 7,50-7,58 (m, 2 Н), 7,19-7,20 (m, 1 Н), 3,58-3,62 (m, 1 Н), 3,29-3,58 (m, 3 Н), 3,22-3,29 (m, 1 Н), 2,10-2,19 (m, 1 Н), 1,98-2,08 (m, 1 Н).
Пример 153 (S)-1-циано-N-(1-(4-(трифторметил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
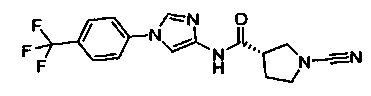
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя 4-(трифторметил)анилин. МС: ЭР+ 350,1; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,77 (s, 1 Н), 8,31 (d, J=1,6 Гц, 1 Н), 7,84-7,92 (m, 5 Н), 3,58-3,63 (m, 1 Н), 3,38-3,50 (m, 3 Н), 3,22-3,50 (m, 1 Н), 2,11-2,19 (m, 1 Н), 1,99-2,08 (m, 1 Н).
Пример 154 (S)-1-циано-N-(1-(3-(трифторметил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
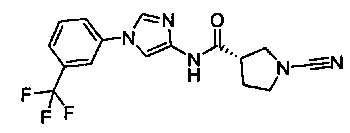
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя 3-(трифторметил)анилин. МС: ЭР+ 350,13; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,76 (s, 1 Н), 8,30 (d, J=1,6 Гц, 1 Н), 8,35-8,47 (m, 1 Н), 7,98 (d, J=7,2 Гц, 1 Н), 7,86 (d, J=1,2 Гц, 1 Н), 7,69-7,76 (m, 2 Н), 3,58-3,63 (m, 1 Н), 3,38-3,50 (m, 3 Н), 3,24-3,29 (m, 1 Н), 2,11-2,17 (m, 1 Н), 2,01-2,07 (m, 1 Н).
Пример 155 (S)-1-циано-N-(1-(4-(метилкарбамоил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
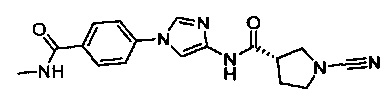
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя 4-амино-N-метилбензамид. МС: ЭР+ 339,23; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,77 (s, 1 Н), 8,53 (d, J=4,8 Гц, 1 Н), 8,25 (d, J=1,6 Гц, 1 Н), 7,96 (d, J=8,8 Гц, 2 Н), 7,81 (d, J=1,6 Гц, 1 Н), 7,76 (d, J=8,8 Гц, 2 Н), 3,58-3,62 (m, 1 Н), 3,44-3,49 (m, 2 Н), 3,37-3,41 (m, 1 Н), 3,24-3,27 (m, 1 Н), 2,79 (d, J=4,8 Гц, 3 Н), 2,12-2,17 (m, 1 Н), 2,00-2,05 (m, 1 Н).
Пример 156 (S)-1-циано-N-(1-(3-(метилкарбамоил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
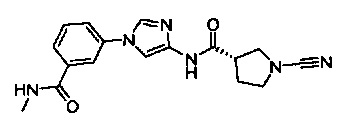
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя 3-амино-N-метилбензамид. МС: ЭР+ 339,18; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,76 (s, 1 Н), 8,65 (d, J=4,8 Гц, 1 Н), 8,20 (d, J=1,6 Гц, 1 Н), 8,02 (s, 1 Н), 7,85 (d, J=1,6 Гц, 1 Н), 7,79-7,83 (m, 2 Н), 7,59 (t, J=8,0 Гц, 1 Н), 3,59-3,63 (m, 1 Н), 3,37-3,50 (m, 3 Н), 3,16-3,28 (m, 1 Н), 2,81 (d, 4,4 Гц, 3 Н), 2,13-2,18 (m, 1 Н), 2,01-2,06 (m, 1 Н).
Пример 157 (S)-1-циано-N-(1-(2-(метилкарбамоил)фенил)-1H-имидазол-4-ил)пирролидин-3-карбоксамид
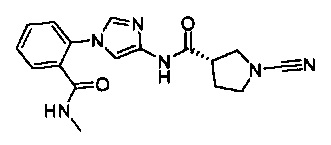
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя 2-амино-N-метилбензамид. МС: ЭР+ 339,18; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,67 (s, 1 Н), 8,36-8,39 (m, 1 Н), 7,62 (d, J=1,6 Гц, 1 Н), 7,55-7,59 (m, 1 Н), 7,46-7,50 (m, 3 Н), 7,41 (d, J=1,6 Гц, 1 Н), 3,57-3,61 (m, 1 Н), 3,36-3,49 (m, 3 Н), 3,20-3,27 (m, 1 Н), 2,64 (d, J=4,8 Гц, 3 Н), 2,09-2,18 (m, 1 Н), 1,97-2,06 (m, 1 Н).
Пример 158 (S)-1-циано-N-(1-(4-((2-метоксиэтил)карбамоил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
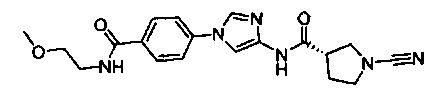
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя 4-амино-N-(2-метоксиэтил)бензамид. МС: ЭР+ 383,28; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,77 (s, 1 Н), 8,63 (t, J=4,4 Гц, 1 Н), 8,27 (d, J=1,6 Гц, 1 Н), 7,98 (d, J=8,8 Гц, 2 Н), 7,81 (d, J=1,6 Гц, 1 Н), 7,76 (d, J=8,8 Гц, 2 Н), 3,58-3,62 (m, 1 Н), 3,42-3,50 (m, 5 Н), 3,37-3,39 (m, 2 Н), 3. 27 (s, 3 Н), 3,22-3,26 (m, 1 Н), 2,11-2,17 (m, 1 Н), 1,98-2,05 (m, 1 Н).
Пример 159 (S)-1-циано-N-(1-(4-метоксифенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
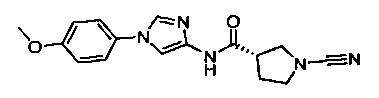
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя 4-метоксианилин. МС: ЭР+ 312,18; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,70 (s, 1 Н), 7,99 (d, J=1,6 Гц, 1 Н), 7,62 (d, J=1,2 Гц, 1 Н), 7,54 (d, J=8,8 Гц, 2 Н), 7,06 (d, J=9,2 Гц, 2 Н), 3,80 (s, 3 Н), 3,57-3,62 (m, 1 Н), 3,44-3,48 (m, 2 Н), 3,37-3,42 (m, 1 Н), 3,22-3,26 (m, 1 Н), 2,11-2,16 (m, 1 Н), 1,99-2,04 (m, 1 Н).
Пример 160 (S)-1-циано-N-(1-(2-метоксифенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
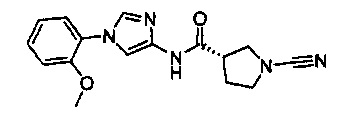
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя 2-метоксианилин. МС: ЭР+ 312,18; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,68 (s, 1 Н), 7,75 (d, J=1,6 Гц, 1 Н), 7,39-7,45 (m, 3 Н), 7,25 (d, J=8,0 Гц, 1 Н), 7.05-7,07(m, 1 H), 3,83 (s, 3 H), 3,57-3,61 (m, 1 H), 3,44-3,49 (m, 2 H), 3,34-3,42 (m, 1 H), 3,22-3,26 (m, 1 H), 2,09-2,17 (m, 1 H), 1,99-2,06 (m, 1 H).
Пример 161 (S)-1-циано-N-(1-(тетрагидро-2Н-пиран-4-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
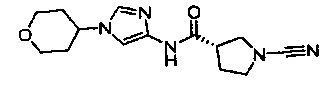
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя 4-амино-тетрагидропиран. МС: ЭР+ 290,53; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,51 (s, 1 Н), 7,56 (d, J=1,60 Гц, 1 Н), 7,29 (d, J=1,60 Гц, 1 Н), 4,21-4,27 (m, 1 Н), 3,93-3,97 (m, 2 Н), 3,54-3,58 (m, 1 Н), 3,37-3,48 (m, 5 Н), 3,17-3,21 (m, 1 Н), 2,06-2,12 (m, 1 Н), 1,82-2,03 (m, 5 Н).
Пример 162 (S)-1-циано-N-(1-циклогексил-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
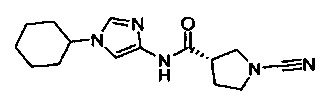
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя циклогексиламин. МС: ЭР+ 288,15; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,51 (s, 1 Н), 7,53 (d, J=1,2 Гц, 1 Н), 7,28 (d, J=1,6 Гц, 1 Н), 3,94-3,98 (m, 1 Н), 3,54-3,58 (m, 1 Н), 3,36-3,48 (m, 3 Н), 3,17-3,21 (m, 1 Н), 2,08-2,12 (m, 1 Н), 1,93-2,01 (m, 3 Н), 1,75-1,85 (m, 2 Н), 1,56-1,66 (m, 4 Н), 1,31-1,41 (m, 2 Н).
Пример 163 (S)-1-циано-N-(1-циклогексил-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
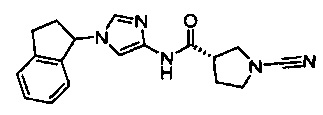
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя (±)-1-аминоиндан. МС: ЭР+ 322,58; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,54 (s, 1 Н), 7,59 (d, J=1,2 Гц, 1 Н), 7,37 (d, J=7,6 Гц, 1 Н), 7,31 (t, J=7,2 Гц, 1 Н), 7,22 (t, J=7,2 Гц, 1 Н), 7,07 (d, J=7,6 Гц, 1 Н), 7,00 (d, J=1,2 Гц, 1 Н), 5,79 (t, J=6,8 Гц, 1 Н), 3,51-3,56 (m, 1 Н), 3,36-3,43 (m, 3 Н), 3,15-3,18 (m, 1 Н), 3,07-3,13 (m, 1 Н), 2,89-2,97 (m, 1 Н), 2,60-2,67 (m, 1 Н), 2,13-2,19 (m, 1 Н), 2,05-2,10 (m, 1 Н), 1,92-1,98 (m, 1Н).
Пример 164 (S)-1-циано-N-(1-(2,3-дигидро-1Н-инден-2-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
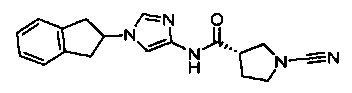
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя 2,3-дигидро-1Н-инден-2-амин. МС: ЭР+ 322,58; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,51 (s, 1 Н), 7,54 (d, J=1,60 Гц, 1 Н), 7,30 (dd, J=5,20, 3,20 Гц, 2 Н), 7,23 (dd, J=5,20, 2,00 Гц, 2 Н), 7,10 (d, J=1,60 Гц, 1 Н), 5,01-5,10 (m, 1 Н), 3,51-3,57 (m, 1 Н), 3,36-3,47 (m, 5 Н), 3,08-3,18 (m, 3 Н), 2,05-2,10 (m, 1 Н), 1,93-1,99 (m, 1 Н).
Пример 165 (S)-1-циано-N-(1-((тетрагидро-2Н-пиран-4-ил)метил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
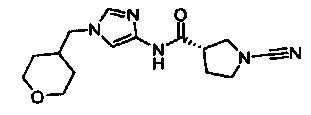
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя 4-(аминометил)тетрагидропиран. МС: ЭР+ 304,25; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,52 (s, 1 Н), 7,43 (s, 1 Н), 7,23 (s, 1 Н), 3,81-3,83 (m, 4 Н), 3,54-3,58 (m, 1 Н), 3,43-3,46 (m, 2 Н), 3,17-3,26 (m, 4 Н), 2,08-2,11 (m, 1 Н), 1,96-2,01 (m, 1 Н), 1,89-1,90 (m, 1 Н), 1,36-1,39 (m, 2 Н), 1,13-1,24 (m, 2 Н).
Пример 166 (S)-1-циано-N-(1-((S)-1-фенилэтил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
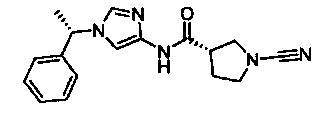
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя (S)-(-)-1-метилбензиламин на стадии b. Стадию восстановления осуществляли, как описано в Примере 146 (Fe, NH4Cl, МеОН, Н2О, 80°С). МС: ЭР+ 310,10; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,54 (s, 1 Н), 7,66 (d, J=1,22 Гц, 1 H), 7,34-7,39 (m, 2 Н), 7,28-7,32 (m, 3 Н), 7,22 (d, J=1,52 Гц, 1 H), 5,47 (q, J=6,91 Гц, 1 H), 3,51-3,57 (m, 1 Н), 3,35-3,48 (m, 3 Н), 3,13-3,21 (m, 1 Н), 2,04-2,13 (m, 1 Н), 1,92-2,02 (m, 1 Н), 1,77 (d, J=7,01 Гц, 3Н).
Пример 167 (S)-1-циано-N-(1-((R)-1-фенилэтил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
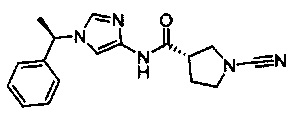
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя (R)-(-)-1-метилбензиламин. МС: ЭР+ 310,23; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,54 (s, 1 Н), 7,66 (d, J=1,22 Гц, 1 Н), 7,34-7,39 (m, 2 Н), 7,28-7,32 (m, 3 Н), 7,22 (d, J=1,52 Гц, 1 Н), 5,47 (q, J=6,91 Гц, 1 Н), 3,51-3,57 (m, 1 Н), 3,35-3,48 (m, 3 Н), 3,13-3,21 (m, 1 Н), 2,04-2,13 (m, 1 Н), 1,92-2,02 (m, 1 Н), 1,77 (d, J=7,01 Гц, 3 Н).
Пример 168 (S)-1-циано-N-(1-(пиридин-2-илметил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
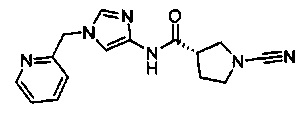
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя 2-(хлорметил)-пиридин гидрохлорид. Стадию восстановления осуществляли, как описано в Примере 146 (10% Pd/C, МеОН, Н2, rt). МС: ЭР+ 297,53; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,54 (s, 1 Н), 8,54 (dd, J=4,52, 1,12 Гц, 1 Н), 7,78-7,83 (m, 1 Н), 7,59 (d, J=1,20 Гц, 1 Н), 7,32-7,35 (m, 1 Н), 7,21-7,25 (m, 2 Н), 5,24 (s, 2 Н), 3,53-3,57 (m, 1 Н), 3,37-3,50 (m, 3 Н), 3,15-3,22 (m, 1 Н), 2,05-2,13 (m, 1 Н), 1,94-2,01 (m, 1 Н).
Пример 169 (S)-1-циано-N-(1-(пиридин-3-илметил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
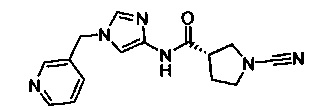
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 146, применяя 3-(хлорметил)-пиридина гидрохлорид. Стадию восстановления осуществляли, как описано в Примере 150 (10% Pd/C, МеОН, 1H, rt). МС: ЭР+ 297,43; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,56 (s, 1 Н), 8,53-8,57 (m, 2 Н), 7,65-7,71 (m, 2 Н), 7,39-7,42 (m, 1 Н), 7,26 (s, 1 Н), 5,20 (s, 2 Н), 3,52-3,56 (m, 1 Н), 3,34-3,46 (m, 3 Н), 3,15-3,19 (m, 1 Н), 2,04-2,10 (m, 1 Н), 1,93-2,00 (m, 1 Н).
Пример 170 (S)-1-циано-N-(1-(пиридин-4-илметил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
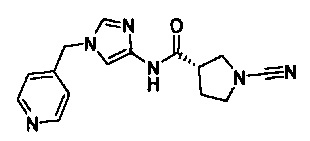
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 146, применяя 4-(хлорметил)-пиридин гидрохлорид. Стадию восстановления осуществляли, как описано в Примере 150 (10% Pd/C, МеОН, Н2, rt). МС: ЭР+ 297,48; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,59 (s, 1 Н), 8,55 (dd, J=4,40, 1,60 Гц, 2 Н), 7,63 (d, J=1,20 Гц, 1 Н), 7,25 (d, J=1,20 Гц, 1 Н), 7,18 (dd, J=4,36, 1,32 Гц, 2 Н), 5,23 (s, 2 Н), 3,53-3,58 (m, 1 Н), 3,37-3,47 (m, 3 Н), 3,17-3,20 (m, 1 Н), 2,05-2,12 (m, 1 Н), 1,95-2,01 (m, 1Н).
Пример 171 (S)-1-циано-N-(1-((3,5-диметилизоксазол-4-ил)метил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
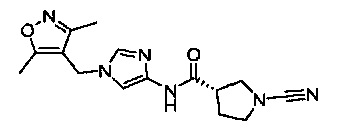
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 146, применяя 4-(хлорметил)-3,5-диметилизоксазол. Стадию восстановления осуществляли, как описано в Примере 150 (10% Pd/C, МеОН, Н2, rt). МС: ЭР+ 315,38; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,55 (s, 1 Н), 7,54 (d, J=1,00 Гц, 1 Н), 7,13 (d, J=1,00 Гц, 1 Н), 5,00 (s, 2 Н), 3,53-3,57 (m, 1 Н), 3,37-3,46 (m, 3 Н), 3,16-3,21 (m, 1 Н), 2,39 (s, 3 Н), 2,08 (s, 3 Н), 2,06-2,16 (m, 1 Н), 1,92-2,00 (m, 1 Н).
Пример 172 (2S,3S)-1-циано-2-метил-N-(1-((S)-1-фенилэтил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
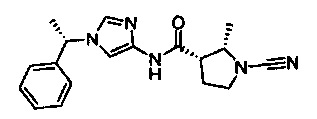
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя (S)-(-)-1-метил-бензиламин и Промежуточное соединение 1. Стадию восстановления осуществляли, как описано в Примере 146 (Fe, NH4Cl, МеОН, H2O, 80°С). МС: ЭР+ 324,38; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,47 (s, 1 Н), 7,65 (s, 1 Н), 7,23-7,63 (m, 5 Н), 7,25 (s, 1 Н), 5,47-5,48 (m, 1 Н), 3,86-3,90 (m, 1 Н), 3,55-3,62 (m, 1 Н), 3,36-3,42 (m, 1 Н), 3,13-3,16 (m, 1 Н), 2,07-2,09 (m, 1 Н), 1,91-1,99 (m, 1 Н), 1,78 (d, J=7,01 Гц, 3 Н), 1,06 (d, J=6,71 Гц, 3 Н).
Пример 173 (3S,4S)-1-циано-4-метил-N-(1-((S)-1-фенилэтил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
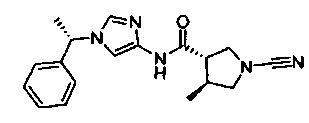
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 150, применяя (S)-(-)-1-метилбензиламин и Промежуточное соединение 2. Стадию восстановления осуществляли, как описано в Примере 146 (Fe, NH4Cl, МеОН, H2O, 80°С). МС: ЭР+ 324,43; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,59 (s, 1 Н), 7,67 (s, 1 Н), 7,28-7,38 (m, 5 Н), 7,24-7,25 (m, 1 Н), 5,46-5,48 (m, 1 Н), 3,55-3,65 (m, 2 Н), 3,36-3,40 (m, 1 Н), 2,97-3,01 (m, 1 Н), 2,76-2,83 (m, 1 Н), 2,33-2,38 (m, 1 Н), 1,78 (d, J=7,02 Гц, 3 Н), 0,98 (d, J=6,10 Гц, 3 Н).
Пример 174 (S)-1-циано-N-(1-(4-метилпиридин-2-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 8, стадии d, g, h, i, j)
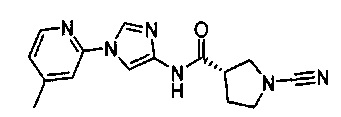
Стадия d. Раствор 2-фтор-4-метилпиридина (9,0 ммоль), K2СО3 (27,0 ммоль) и KI (9,0 ммоль) в ДМФА (10 мл) перемешивали при комнатной температуре в течение 5 мин. К реакционной смеси добавляли 4-нитроимидазол (9,0 ммоль) и затем нагревали при 100°С в течение 72 часов. Полученную реакционную смесь вливали в воду (50 мл) и подвергали экстракции посредством EtOAc (3×100 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (90-100% EtOAc в гексане), получая 4-метил-2-(4-нитро-1Н-имидазол-1-ил)пиридин (1,34 ммоль). МС: ЭР+ 205,23; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 9,07 (d, J=1,2 Гц, 1 Н), 8,69 (d, J=1,2 Гц, 1 Н), 8,44 (d, J=5,2 Гц, 1 Н), 7,93 (s, 1 Н), 7,38 (d, J=5,2 Гц, 1 Н), 2,44 (s, 3 Н).
Стадия g. К раствору 4-метил-2-(4-нитро-1Н-имидазол-1-ил) пиридина (2,4 ммоль) в МеОН (15 мл) добавляли 10% Pd/C (0,10% масс./масс.) при комнатной температуре. Реакционную смесь продували газообразным Н2 в течение 2 часов при комнатной температуре. Полученную реакционную смесь тщательно фильтровали через Celite Hyflo и концентрировали при пониженном давлении с получением 1-(4-метилпиридин-2-ил)-1Н-имидазол-4-амина (2,4 ммоль). МС: ЭР+ 175,1
Стадии h-j. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для Примера 146 в соответствии со стадиями h, i и j Схемы 8. МС: ЭР+ 297,0; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,76 (s, 1 Н), 8,40 (d, J=1,53 Гц, 1 Н), 8,34 (d, J=5,19 Гц, 1 Н), 7,99 (d, J=1,53 Гц, 1 Н), 7,69 (s, 1 Н), 7,20 (d, J=4,88 Гц, 1 Н), 3,57-3,64 (m, 1 Н), 3,38-3,52 (m, 3 Н), 3,24-3,27 (m, 1 Н), 2,41 (s, 3 Н), 2,10-2,21 (m, 1 Н), 1,98-2,08 (m, 1 Н).
Пример 175 (S)-1-циано-N-(1-(6-метилпиридин-2-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
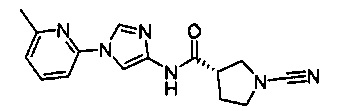
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 174, применяя 2-фтор-6-метилпиридин на стадии d. МС: ЭР+ 297,0; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,76 (s, 1 Н), 8,40 (d, J=1,53 Гц, 1 H), 7,99 (d, J=1,53 Гц, 1 H), 7,86 (t, J=7,94 Гц, 1 H), 7,60 (d, J=7,94 Гц, 1 H), 7,22 (d, J=7,63 Гц, 1 H), 3,57-3,63 (m, 1 Н), 3,44-3,51 (m, 2 Н), 3,37-3,44 (m, 1 Н), 3,23-3,27 (m, 1 Н), 2,51 (s, 3 Н), 2,10-2,20 (m, 1 Н), 1,98-2,08 (m, 1 Н).
Пример 176 (S)-1-циано-N-(1-(2-метилпиримидин-4-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
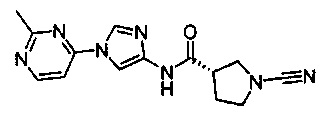
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 174, применяя 4-хлор-2-метилпиримидин на стадии d. МС: ЭР+ 297,98; 1Н ЯМР (400 МГц, ДMCO-d6) δ ppm 10,87 (s, 1 Н), 8,77 (d, J=5,80 Гц, 1 H), 8,60 (d, J=1,53 Гц, 1 H), 8,06 (d, J=1,53 Гц, 1 H), 7,77 (d, J=5,49 Гц, 1 H), 3,57-3,63 (m, 1 Н), 3,37-3,51 (m, 3 Н), 3,22-3,31 (m, 1 Н), 2,64 (s, 3Н), 2,11-2,20 (m, 1 Н), 1,99-2,08 (m, 1 Н).
Пример 177 (S)-1-циано-N-(1-(4-цианофенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
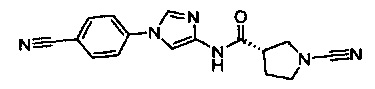
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 174, применяя 4-фторбензонитрил на стадии d. МС: ЭР+ 307,43; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,81 (s, 1 Н), 8,35 (d, J=1,83 Гц, 1 Н), 7,97-8,04 (m, 2 Н), 7,88-7,93 (m, 2 Н), 7,87 (d, J=1,83 Гц, 1 Н), 3,58-3,62 (m, 1 Н), 3,37-3,54 (m, 3 Н), 3,21-3,30 (m, 1 Н), 2,09-2,21 (m, 1 Н), 1,96-2,08 (m, 1 Н).
Пример 178 (S)-N-(1-бензил-2-метил-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамид
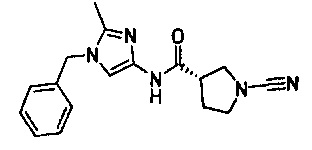
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 174, применяя 2-метил-4(5)-нитроимидазол и бензилхлорид. МС: ЭР+ 310,47; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,43 (s, 1 Н), 7,35-7,38 (m, 2 Н), 7,27-7,31 (m, 1 Н), 7,17-7,18 (m, 2 Н), 7,14 (s, 1 Н), 5,09 (s, 2 Н), 3,53-3,55 (m, 1 Н), 3,35-3,46 (m, 3 Н), 3,12-3,20 (m, 1 Н), 2,22 (s, 3 Н), 2,06-2,13 (m, 1 Н), 1,91-1,99 (m, 1 Н).
Пример 179 (S)-1-циано-N-(1-(2-(3,5-диметилизоксазол-4-ил)этил)-1H-имидазол-4-ил)пирролидин-3-карбоксамид
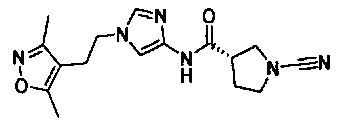
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 174, применяя 4-(2-хлор-этил)-3,5-диметил-изоксазол. МС: ЭР+ 329,43; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,47 (s, 1 Н), 7,30 (s, 1 Н), 7,25 (s, 1 Н), 4,0 (t, J=8,0 Гц, 2 Н), 3,55-3,59 (m, 1 Н), 3,38-3,46 (m, 3 Н), 3,15-3,22 (m, 1 Н), 2,71 (t, J=6,8 Гц, 2 Н), 2,08-2,15 (m, 1 Н), 2,09 (s, 3 Н), 2,05 (s, 3 Н), 1,97-2,02 (m, 1 Н).
Пример 180 (S)-1-циано-N-(1-(3-цианофенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 8, стадии d, f, h, i, j)
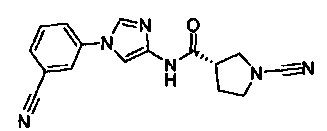
Стадия f. К раствору 3-(4-нитро-1Н-имидазол-1-ил) бензонитрила [полученного с помощью методики, аналогичной методике, описанной для Примера 174, применяя 3-фторбензонитрил на стадии d] (0,65 ммоль) в МеОН (8 мл) добавляли цинковую пыль (1,96 ммоль) и насыщенный раствор NH4Cl (0,5 мл) при комнатной температуре. Реакционную смесь перемешивали при 60°С в течение 1 часа. Полученной реакционной смеси давали возможность остыть до к. т. и вливали в воду (50 мл) и подвергали экстракции посредством EtOAc (2×50 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 3-(4-амино-1H-имидазол-1-ил) бензонитрил (0,46 ммоль). Данный материал сразу же применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 185,29
Стадии h-j. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для Примера 146 в соответствии со стадиями h, i и j Схемы 8. МС: ЭР+ 307,28; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,76 (s, 1 Н), 8,27 (m, 2 Н), 8,02 (dd, J=8,00 Гц, 1,2 Гц, 1 Н), 7,87 (d, J=1,53 Гц, 1 Н), 7,81 (d, J=7,94 Гц, 1 Н), 7,66-7,75 (m, 1 Н), 3,58-3,61 (m, 1 Н), 3,44-3,50 (m, 2 Н), 3,38-3,40 (m, 1 Н), 3,23-3,29 (m, 1 Н), 2,09-2,21 (m, 1 Н), 1,97-2,08 (m, 1 Н).
Пример 181 (S)-1-циано-N-(1-(пиридин-3-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(получали в соответствии со Схемой 8, стадии е, g, h, i, j)
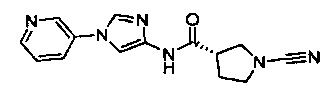
Стадия е. К раствору 4-нитроимидазола (17,6 ммоль), пиридин-3-бороновой кислоты (35,3 ммоль) и CuCl2 в МеОН (40 мл) добавляли NaOH (35,3 ммоль) при комнатной температуре и реакционную смесь перемешивали в течение 5 минут. В реакционную смесь вводили поток газообразного О2 с низкой скоростью. Реакционную смесь нагревали при 80°С в течение 18 часов, продолжая продувку газообразным О2 во время протекания. Полученной реакционной смеси давали возможность остыть до к. т. и продувку газообразным О2 прекращали. Реакционную смесь затем концентрировали при пониженном давлении. Полученный неочищенного материал вливали в воду (100 мл) и подвергали экстракции посредством EtOAc (2×100 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 3-(4-нитро-1Н-имидазол-1-ил)пиридин (3,89 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 191,1
Стадия g. К раствору 3-(4-нитро-1Н-имидазол-1-ил)пиридина (3,68 ммоль) в МеОН : ТГФ (1:1, 10 мл) добавляли 10% Pd/C (0,25% масс./масс.) при комнатной температуре. Реакционную смесь продували газообразным Н2 в течение 2 часов при комнатной температуре. Полученную реакционную смесь тщательно фильтровали через Celite Hyflo и концентрировали при пониженном давлении с получением 1-(пиридин-3-ил)-1Н-имидазол-4-амина (3,12 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки.
Стадии h-j. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для Примера 146 в соответствии со стадиями h, i и j Схемы 8. МС: ЭР+ 283,48; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,78 (s, 1 Н), 8,94 (d, J=2,78 Гц, 1 Н), 8,56 (dd, J=4,58, 1,22 Гц, 1 Н), 8,24 (d, J=1,53 Гц, 1 Н), 8,09-8,12 (m, 1 Н), 7,81 (d, J=1,53 Гц, 1 Н), 7,53-7,57 (m, 1 Н), 3,56-3,65 (m, 1 Н), 3,36-3,52 (m, 3 Н), 3,22-3,31 (m, 1 Н), 2,11-2,19 (m, 1 Н), 1,99-2,07 (m, 1 Н).
Пример 182 (S)-1-циано-N-(1-(пиридин-4-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
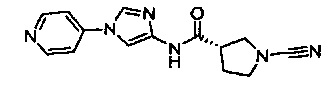
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 181, применяя пиридин-4-бороновую кислоту. МС: ЭР+ 283,43; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,83 (s, 1 Н), 8,63-8,65 (m, 2 Н), 8,45 (d, J=1,6 Гц, 1 Н), 7,91 (d, J=1,6 Гц, 1 Н), 7,75-7,77 (m, 2 Н), 3,59-3,63 (m, 1 Н), 3,42-3,51 (m, 2 Н), 3,35-3,40 (m, 1 Н), 3,21-3,31 (m, 1 Н), 2,09-2,18 (m, 1 Н), 1,99-2,07 (m, 1 Н).
Пример 183 (S)-1-циано-N-(1-(3-метоксифенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
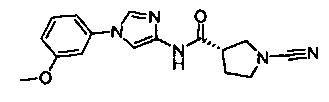
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 181, применяя 3-метокси-фенилбороновую кислоту. МС: ЭР+ 312,43; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,73 (s, 1Н), 8,15 (d, J=1,6 Гц, 1Н), 7,72 (d, J=1,6 Гц, 1Н), 7,38-7,43 (m, 1Н), 7,17-7,19 (m, 2Н), 6.92 (dd, J=6,0 Гц, 2,0 Гц, 1 Н), 3,84 (s, 3Н), 3,58-3,61 (m, 1 Н), 3,45-3,50 (m, 2 Н), 3,37-3,44 (m, 1 Н), 3,21-3,28 (m, 1Н), 2,07-2,16 (m, 1 Н), 1,98-2,05 (m, 1 Н).
Пример 184 (3S)-N-(1-(1-бензоилпиперидин-3-ил)-1H-имидазол-4-ил)-1-цианопирролидин-3-карбоксамид (получали в соответствии со Схемой 9)
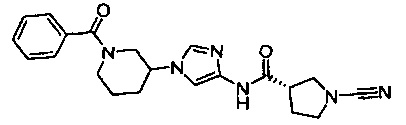
Стадия b. К раствору 1,4-динитро-1Н-имидазола (15,82 ммоль) [получали, применяя методику, описанную, например, 150 на стадии а] в МеОН : вода (1:1, 40 мл) добавляли 1-ВОС-3-аминопиперидин (15,82 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Полученную реакционную смесь подвергали выпариванию при пониженном давлении для удаления большей части МеОН. Полученную смесь разбавляли водой (100 мл) и подвергали экстракции посредством EtOAc (2×60 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (60% EtOAc в гексане), получая трет-бутил-3-(4-нитро-1Н-имидазол-1-ил)пиперидин-1-карбоксилат (9,12 ммоль). МС: ЭР+ 297,43; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 8,55 (d, J=1,2 Гц, 1Н) 8,00 (d, J=1,2 Гц, 1H), 4,26-4,33 (m, 1H), 4,00-4,05 (m, 1Н), 3,71-3,76 (m, 1Н), 2,91-2,98 (m, 1H), 2,07-2,11 (m, 1H), 1,99-2,02 (m, 1H), 1,68-1,74 (m, 1Н), 1,44-1,50 (m, 2Н), 1,40 (s, 9Н).
Стадия с. К раствору трет-бутил-3-(4-нитро-1Н-имидазол-1-ил)пиперидин-1-карбоксилата (9,12 ммоль) в ДХМ (30 мл) добавляли ТФУ (10 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток подвергали азеотропной перегонке с применением ДХМ и дополнительно растирали с н-гексаном с получением соли ТФУ и 3-(4-нитро-1Н-имидазол-1-ил)пиперидина (количественный выход). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 197,10; 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 9,35 (ушир. s, 1Н), 9,03 (ушир. s, 1Н), 8,61 (d, J=1,4 Гц, 1Н), 8,01 (d, J=1,4 Гц, 1H), 4,51-4,59 (m, 1H), 3,59-3,62 (m, 1Н), 3,30-3,40 (m, 2Н), 2,79-2,89 (m, 1Н), 2,15-2,18 (m, 1H), 1,95-2,08 (m, 2Н), 1,69-1,80 (m, 1H).
Стадия i. К раствору бензойной кислоты (2,04 ммоль) и ТЭА (3,07 ммоль) в сухом ТГФ (5 мл) добавляли Т3Р (50% в EtOAc) (4,5 ммоль) при комнатной температуре. Раствор соли ТФУ и 3-(4-нитро-1Н-имидазол-1-ил)пиперидина (1,63 ммоль) в ТГФ (2 мл) добавляли по каплям и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь вливали в воду (200 мл), подщелачивали твердым NaHCO3 и подвергали экстракции посредством EtOAc (2×100 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая [3-(4-нитро-1Н-имидазол-1-ил)пиперидин-1-ил] (фенил)метанон (количественный выход). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 301,38.
Стадия j. К раствору [3-(4-нитро-1Н-имидазол-1-ил)пиперидин-1-ил](фенил)метанона (1,0 ммоль) в МеОН (20 мл) добавляли 10% Pd/C (0,3 масс./масс.) при комнатной температуре. Реакционную смесь продували газообразным Н2 в течение 30 мин при комнатной температуре. Полученную реакционную смесь тщательно фильтровали через Celite Hyflo и концентрировали при пониженном давлении с получением [3-(4-амино-1Н-имидазол-1-ил)пиперидин-1-ил](фенил)метанона (количественный выход). Данный материал сразу же применяли на следующей стадии без дополнительной очистки.
Стадии f-h. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для Примера 1, применяя (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту. МС: ЭР+ 393,19; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,58 (ушир. s, 1 Н), 7,61 (ушир. s, 1 Н), 7,40-7,46 (m, 5 Н), 7,24 (ушир. s, 1 Н), 4,35-4,47 (m, 1 Н), 4,22-4,27 (m, 1 Н), 3,54-3,64 (m, 2 Н), 3,37-3,50 (m, 3 Н), 3,19-3,30 (m, 2 Н), 2,89-2,98 (m, 1 Н), 1,90-2,2 (m, 4 Н), 1,45-1,75 (m, 2Н).
Пример 185 (3S)-N-(1-(1-бензоилпирролидин-3-ил)-1H-имидазол-4-ил)-1-цианопирролидин-3-карбоксамид
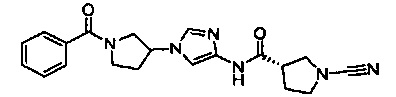
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 184, применяя 1-ВОС-3-аминопирролидин. МС: ЭР+ 379,58; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,58 (ушир. s, 1Н), 7,62 (s, 1Н), 7,43-7,54 (m, 5Н), 7,28 (s, 1Н), 4,88-4,93 (m, 1H), 4,79-4,83 (m, 1H), 3,95-3,99 (m, 1H), 3,84-3,89 (m, 1Н), 3,69-3,72 (m, 1Н), 3,52-3,62 (m, 3Н), 3,42-3,46 (m, 2Н), 3,17-3,22 (m, 2Н), 2,13-2,16 (m, 1Н), 2,07-2,10 (m, 1Н).
Пример 186 (3S)-N-(1-(1-бензилпиперидин-3-ил)-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамид (получали в соответствии со Схемой 9)
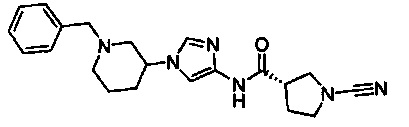
Стадия d. К раствору соли ТФУ и 3-(4-нитро-1Н-имидазол-1-ил)пиперидина (полученному в Примере 184) (2,25 ммоль) и K2СО3 (6,77 ммоль) в ТГФ (8 мл) добавляли бензилбромид (1,8 ммоль) при комнатной температуре. Реакционную смесь нагревали до 80°С в течение 16 часов. Полученную реакционную смесь вливали в воду (70 мл) и подвергали экстракции посредством EtOAc (2×50 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 1-бензил-3-(4-нитро-1Н-имидазол-1-ил)пиперидина (количественный выход). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 287,43.
Стадия е. Раствор 1-бензил-3-(4-нитро-1Н-имидазол-1-ил)пиперидина (1,39 ммоль) в ТГФ: вода (1:1, 10 мл) перемешивали при комнатной температуре в течение 5 мин. Добавляли порошок Fe (13,98 ммоль) и NH4Cl (13,98 ммоль) и нагревали реакционную смесь при 80°С в течение 0,5 часа. Полученной реакционной смеси давали возможность остыть до к. т. и фильтровали через Celite Hyflo. Фильтрат вливали в воду (20 мл) и подвергали экстракции посредством EtOAc (2×5 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и полученный раствор сразу же применяли на следующей стадии без упаривания.
Стадии f-h. Указанное в заголовке соединение синтезировали из предыдущего промежуточного соединения с помощью методики, аналогичной методике, описанной для Примера 1, применяя (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновую кислоту. МС: ЭР+ 379,24; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,50 (s, 1 Н), 7,55 (s, 1 Н), 7,24-7,33 (m, 6 Н), 4,15-4,25 (m, 1 Н), 3,52-3,57 (m, 3 Н), 3,38-3,50 (m, 2 Н), 3,16-3,22 (m, 1 Н), 2,86-2,89 (m, 1 Н), 2,66-2,73 (m, 1 Н), 2,25-2,35 (m, 1 Н), 2,11-2,17 (m, 2 Н), 1,93-2,07 (m, 2 Н), 1,60-1,68 (m, 2 Н), 1,54-1,57 (m, 2Н).
Пример 187 (3S)-1-циано-N-(1-(1-метилпиперидин-3-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 9)
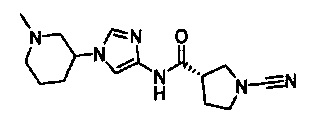
Стадия k. К раствору соли 3-(4-нитро-1Н-имидазол-1-ил)пиперидина, образованной с ТФУ (полученной в Примере 184) (2,58 ммоль) и 37% водного раствора формальдегида (10 мл) в МеОН (20 мл) добавляли цианоборогидрид натрия (3,87 ммоль) при комнатной температуре. К полученной реакционной смеси по каплям добавляли уксусную кислоту (6,19 ммоль) и перемешивали при комнатной температуре в течение 16 часов. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток вливали в воду (100 мл), подщелачивали твердым NaHCO3 и подвергали экстракции посредством EtOAc (2×100 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 1-метил-3-(4-нитро-1Н-имидазол-1-ил)пиперидин (количественный выход). МС: ЭР+ 211,04; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 8,56 (s, 1 Н), 8,01 (s, 1 Н), 4,33-4,40 (m, 1 Н), 2,86-2,89 (m, 1 Н), 2,51-2,57 (m, 1 Н), 2,20-2,36 (m, 1 Н), 2,22 (s, 3 Н), 2,05-2,10 (m, 1 Н), 1,95-1,99 (m, 1 Н), 1,67-1,79 (m, 2 Н), 1,54-1,60 (m, 1Н).
Стадия j. К раствору 1-метил-3-(4-нитро-1Н-имидазол-1-ил)пиперидина (1,90 ммоль) в МеОН (50 мл) добавляли 10% Pd/C (0,25 масс./масс.) при комнатной температуре. Реакционную смесь продували газообразным H2 при комнатной температуре в течение 0,5 часа. Полученную реакционную смесь тщательно фильтровали через Celite Hyflo и концентрировали при пониженном давлении с получением 1-(1-метилпиперидин-3-ил)-1Н-имидазол-4-амина (количественный выход). Данный материал сразу же применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 378,98; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,46 (ушир. s, 1 Н), 7,56 (s, 1 Н), 7,29 (s, 1 Н), 4,10-4,21 (m, 1 Н), 3,40-3,52 (m, 2Н), 3,18-3,38 (m, 4Н), 3,07-3,19 (m, 1Н), 2,80-2,91 (m, 1 Н), 2,59-2,68 (m, 1 Н), 2,20 (S, 3 Н), 2,08-2,18 (m, 2 Н), 1,87-1,90 (m, 2 Н), 1,51-1,73 (m, 2 Н), 1,39 (s, 9Н).
Стадии f-h. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для Примера 1, с применением (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновой кислоты. МС: ЭР+ 303,04; 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 10,58 (ушир. s, 1 Н), 7,58 (s, 1 Н), 7,37 (s, 1 Н), 4,57-4,62 (m, 1 Н), 3,51-3,60 (m, 1 Н), 3,34-3,50 (m, 4 Н), 3,20-3,26 (m, 2 Н), 3,01-3,14 (m, 2 Н), 2,50 (s, 3 Н), 2,07-2,17 (m, 2 Н), 1,95-2,07 (m, 2 Н), 1,70-1,80 (m, 2 Н).
Пример 188 (3S)-1-циано-N-(1-(1-метилпирролидин-3-ил)-1H-имидазол-4-ил)пирролидин-3-карбоксамид
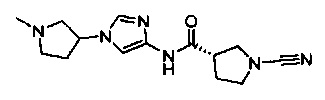
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 187, применяя 1-ВОС-3-аминопирролидин. МС: ЭР+ 289,18; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,52 (s, 1 Н), 7,50 (s, 1 Н), 7,33 (s, 1 Н), 4,71-4,82 (m, 1 Н), 3,54-3,58 (m, 1Н), 3,42-3,46 (m, 2 Н), 3,17-3,22 (m, 1 Н), 2,87-2,88 (m, 1 Н), 2,67-2,70 (m, 1 Н), 2,52-2,61 (m, 1 Н), 2,33-2,41 (m, 2 Н), 2,28 (s, 3 Н), 2,23-2,26 (m, 1Н), 2,06-2,12 (m, 1 Н), 1,96-2,02 (m, 1 Н), 1,82-1,84 (m, 1 Н).
Пример 189 (S)-N-(5-ацетил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)-1-цианопирролидин-3-карбоксамид (получали в соответствии со Схемой 10)
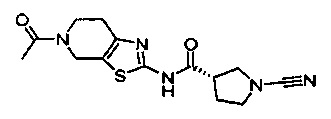
Стадия а. К раствору Промежуточного соединения 14 (0,7 ммоль) в ДХМ (2 мл) добавляли ТЭА (1,42 ммоль) и ацетилхлорид (0,85 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Полученную реакционную смесь вливали в ледяную воду (30 мл) и подвергали экстракции посредством ДХМ (3×30 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (2% МеОН в ДХМ), получая трет-бутил-(S)-3-((5-ацетил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)карбамоил)пирролидин-1-карбоксилат (0,55 ммоль). МС: ЭР+ 395,60.
Стадия f. 4М HCl в 1,4-диоксан (5 мл) добавляли к трет-бутил-(S)-3-((5-ацетил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)карбамоил) пирролидин-1-карбоксилату (0,55 ммоль) и перемешивали при комнатной температуре в течение 1 часа. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток нейтрализовали водным раствором насыщенного NaHCO3. Полученную смесь подвергали экстракции ДХМ (3×100 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток растирали с диэтиловым эфиром (10 мл), получая (S)-N-(5-ацетил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид (0,49 ммоль). Материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 295,28.
Стадия g. К раствору (S)-N-(5-ацетил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил) пирролидин-3-карбоксамида (0,5 ммоль) в CHCl3 (5 мл) добавляли DIPEA (1,5 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 15 мин. Цианоген бромид (0,7 ммоль) добавляли и реакционную смесь перемешивали при 0°С в течение 30 мин. Полученную реакционную смесь вливали в ледяную воду (50 мл) и подвергали экстракции посредством ДХМ (2×30 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (2% МеОН в ДХМ). Материал, полученный после хроматографии, дополнительно растирали с диэтиловым эфиром (5 мл), получая указанное в заголовке соединение (0,13 ммоль). МС: ЭР+ 320,3; 1Н ЯМР (проводили при 80°С, 400 МГц, ДМСО-d6) δ ppm 11,99 (ушир. s, 1 Н), 4,62 (s, 2 Н), 3,72-3,85 (m, 2 Н), 3,60-3,65 (m, 1 Н), 3,49-3,56 (m, 1 Н), 3,38-3,48 (m, 2 Н), 3,28-3,37 (m, 1 Н), 2,60-2,80 (m, 2 Н), 2,16-2,28 (m, 1 Н), 2,05-2,10 (m, 4Н).
Пример 190 (S)-1-циано-N-(5-изобутирил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид
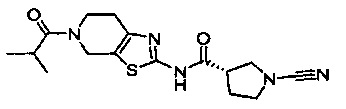
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 189, применяя 2-метилпропаноилхлорид. МС: ЭР+ 348,48; 1Н ЯМР (проводили при 80°С, 400 МГц, ДМСО-d6) δ ppm 12,00 (ушир. s, 1 Н), 4,65 (s, 2 Н), 3,79-3,82 (m, 2 Н), 3,60-3,64 (m, 1 Н), 3,51-3,54 (m, 1 Н), 3,40-3,45 (m, 2 Н), 3,33-3,38 (m, 1 Н), 2,95-2,98 (m, 1 Н), 2,67-2,72 (m, 2 Н), 2,16-2,23 (m, 1 Н), 2,05-2,12 (m, 1 Н), 1,04 (d, J=6,40 Гц, 6 Н).
Пример 191 (S)-N-(5-бензоил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)-1-цианопирролидин-3-карбоксамид
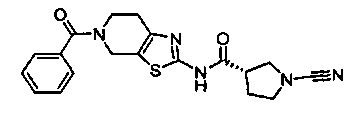
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 189, применяя бензоилхлорид. МС: ЭР+ 382,38; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,27 (ушир. s, 1 H), 7,41-7,62 (m, 5 H), 4,76 (s, 2 H), 3,92-3,94 (m, 1 H), 3,34-3,61 (m, 6 H), 2,66-2,73 (m, 2 H), 2,21-2,28 (m, 1 H), 2,04-2,11 (m, 1 H).
Пример 192 (S)-1-циано-N-(5-(2-метоксибензоил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид
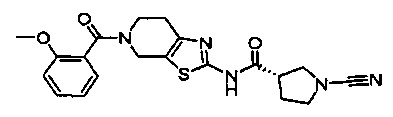
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 189, применяя 2-метоксибензоилхлорид. МС: ЭР+ 412,43; 1Н ЯМР (проводили при 80°С, 400 МГц, ДМСО-d6) δ ppm 12,03 (ушир. s, 1 Н), 7,42 (t, J=7,6 Гц, 1 Н), 7,11-7,23 (m, 2 Н), 7,01-7,04 (m, 1 Н), 4,78 (s, 2 Н), 4,29-4,39 (m, 1 Н), 3,94-4,08 (m, 1 Н), 3,81 (s, 3 Н), 3,60-3,62 (m, 1 Н), 3,38-3,47 (m, 3 Н), 3,31-3,36 (m, 1 Н), 2,73-2,79 (m, 1 Н), 2,58-2,65 (m, 1 Н), 2,19-2,22 (m, 1 Н), 2,06-2,11 (m, 1 Н).
Пример 193 (S)-1-циано-N-(5-пиколиноил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 10)
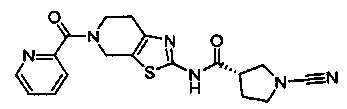
Стадия b. К раствору 2-пиридинкарбоновой кислоты (0,406 ммоль) в ТГФ (2 мл) добавляли Т3Р (50% в EtOAc) (13,95 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 15 мин. Промежуточное соединение 14 (0,41 ммоль) и DIPEA (1,23 ммоль) добавляли к реакционной смеси. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Полученную реакционную смесь вливали в воду (50 мл) и подвергали экстракции посредством EtOAc (3×50 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (4% МеОН в ДХМ), получая трет-бутил-(S)-3-((5-пиколиноил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил) карбамоил) пирролидин-1-карбоксилат (0,32 ммоль). МС: ЭР+ 458,66.
Стадия f. К раствору трет-бутил-(S)-3-((5-пиколиноил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)карбамоил)пирролидин-1-карбоксилата (0,32 ммоль) в ДХМ (4 мл) добавляли ТФУ (1,31 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток нейтрализовали водным раствором насыщенного NaHCO3 и подвергали экстракции посредством ДХМ (3×50 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая (S)-N-(5-пиколиноил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил) пирролидин-3-карбоксамид (0,31 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 358,43.
Стадия g. К раствору (S)-N-(5-пиколиноил-4,5,6,7-тетрагидротиазоло [5,4-с]пиридин-2-ил)пирролидин-3-карбоксамида (0,31 ммоль) в ТГФ (6 мл) добавляли K2СО3 (0,92 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 30 мин. Добавляли цианоген бромид (0,46 ммоль) и реакционную смесь затем перемешивали при комнатной температуре в течение 30 мин. Полученную реакционную смесь вливали в ледяную воду (100 мл) подвергали экстракции посредством EtOAc (3×50 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (4% МеОН в ДХМ), получая указанное в заголовке соединение (0,14 ммоль). МС: ЭР+ 383,63; 1H ЯМР (проводили при 80°С, 400 МГц, ДМСО-d6) δ ppm 11,99 (ушир. s, 1 Н), 8,62 (d, J=4,58 Гц, 1 Н), 7,94 (t, J=7,48 Гц, 1 Н), 7,62 (d, J=7,33 Гц, 1 Н), 7,49-7,52 (m, 1 Н), 4,80 (s, 2 Н), 3,97-4,07 (m, 1 Н), 3,88-3,94 (m, 1 Н), 3,60-3,65 (m, 1 Н), 3,47-3,53 (m, 1 Н), 3,37-3,45 (m, 2 Н), 3,31-3,35 (m, 1 Н), 2,71-2,81 (m, 2 Н), 2,15-2,22 (m, 1 Н), 2,04-2,13 (m, 1 Н).
Пример 194 (S)-1-циано-N-(5-(1-метил-1H-пиразол-3-карбонил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-гил)пирролидин-3-карбоксамид
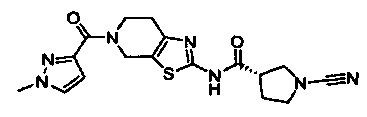
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 193, применяя 1-метил-1Н-пиразол-3-карбоновую кислоту. МС: ЭР+ 386,43; 1Н ЯМР (проводили при 80°С, 400 МГц, ДМСО-d6) δ ppm 11,98 (ушир. s, 1 Н), 7,74 (s, 1 Н), 6,57 (s, 1 Н), 4,89 (s, 2 Н), 4,06-4,19 (m, 2 Н), 3,93 (s, 3 Н), 3,60-3,64 (m, 1 Н), 3,51-3,55 (m, 1 Н), 3,43-3,49 (m, 2 Н), 3,31-3,37 (m, 1 Н), 2,74-2,77 (m, 2 Н), 2,19-2,25 (m, 1 Н), 2,04-2,11 (m, 1 Н).
Пример 195 (S)-1-циано-N-(5-(1-метил-2-оксо-1,2-дигидропиридин-3-карбонил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид
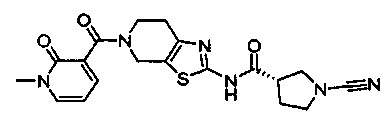
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 193, применяя 1-метил-2-оксо-1,2-дигидропиридин-3-карбоновую кислоту. МС: ЭР+ 413,15; 1Н ЯМР (проводили при 80°С, 400 МГц, ДМСО-d6) δ ppm 11,99 (ушир. s, 1 Н), 7,78 (dd, J=6,71, 2,14 Гц, 1 Н), 7,47-7,49 (m, 1 Н), 6,28 (t, J=6,714 Гц, 1 Н), 4,71 (s, 2 Н), 3,72-3,79 (m, 1 Н), 3,60-3,67 (m, 1 Н), 3,50-3,54 (m, 2 Н), 3,47 (s, 3 Н), 3,38-3,45 (m, 2 Н), 3,29-3,36 (m, 1 Н), 2,70-2,73 (m, 2 Н), 2,17-2,23 (m, 1 Н), 2,07-2,13 (m, 1Н).
Пример 196 (S)-1-циано-N-(5-никотиноил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид
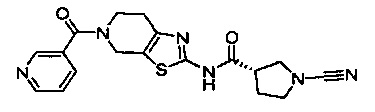
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 193, применяя никотиновую кислоту. МС: ЭР+ 383,20; 1Н ЯМР (проводили при 80°С, 400 МГц, ДМСО-d6) δ ppm 12,00 (ушир. s, 1 Н), 8,66-8,69 (m, 2 Н), 7,87 (d, J=8,0 Гц, 1 Н), 7,48-7,51 (m, 1 Н), 4,72 (s, 2 Н), 3,73-3,89 (m, 1 Н), 3,60-3,65 (m, 1 Н), 3,51-3,55 (m, 2 Н), 3,41-3,48 (m, 2 Н), 3,31-3,37 (m, 1 Н), 2,75-2,77 (m, 2 Н), 2,19-2,25 (m, 1 Н), 2,04-2,11 (m, 1 Н).
Пример 197 (S)-1-циано-N-(5-(диметилглицил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид
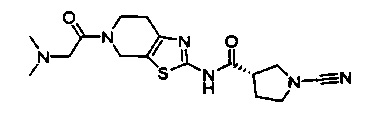
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 193, применяя N,N-диметилглицин. МС: ЭР+ 363,23; 1Н ЯМР (400 МГц, CD3OD) δ ppm 4,72-4,77 (m, 2 Н), 3,86-3,98 (m, 2 Н), 3,61-3,65 (m, 2 Н), 3,51-3,62 (m, 2 Н), 3,32-3,42 (m, 2 Н), 2,72-2,84 (m, 2 Н), 2,38 (s, 3Н), 2,32 (s, 3Н), 2,15-2,30 (m, 3 Н).
Пример 198 метил (S)-2-(1-цианопирролидин-3-карбоксамидо)-6,7-дигидротиазоло[5,4-с]пиридин-5(4Н)-карбоксилат
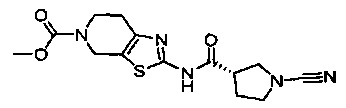
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 189, применяя метилхлорформиат. МС: ЭР+ 336,43; 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 12,24 (ушир. s, 1 Н), 4,55 (s, 2 Н), 3,67-3,70 (m, 2 Н), 3,64 (s, 3 Н), 3,59-3,61 (m, 1 Н), 3,48-3,53 (m, 1 Н), 3,38-3,46 (m, 2 Н), 3,28-3,32 (m, 1 Н), 2,65-2,68 (m, 2 Н), 2,08-2,20 (m, 1 Н), 2,01-2,08 (m, 1 Н).
Пример 199 2-метоксиэтил (S)-2-(1-цианопирролидин-3-карбоксамидо)-6,7-дигидротиазоло[5,4-с]пиридин-5(4Н)-карбоксилат
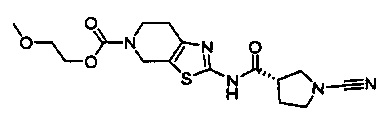
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 189, применяя 2-метоксиэтилхлорформиат. МС: ЭР+ 380,38; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,24 (s, 1 Н), 4,55 (s, 2 Н), 4,15-4,17 (m, 2 Н), 3,68-3,70 (m, 2 Н), 3,59-3,63 (m, 1 Н), 3,51-3,54 (m, 2 Н), 3,38-3,45 (m, 3 Н), 3,29-3,31 (m, 1 Н), 3,26 (s, 3 Н), 2,65-2,67 (m, 2 Н), 2,14-2,22 (m, 1 Н), 1,99-2,08 (m, 1 Н).
Пример 200 (S)-1-циано-N-(5-метил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 10)
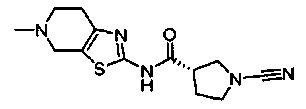
Стадия d. К раствору Промежуточного соединения 14 (0,56 ммоль) в МеОН (5 мл) добавляли 37% водный раствор формальдегида (1,70 ммоль) и уксусную кислоту (3 капли) при 0°С. Реакционную смесь перемешивали при 0°С в течение 30 мин, к реакционной смеси добавляли NaCNBH3 (1,12 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Полученную реакционную смесь вливали в ледяную воду (10 мл) и подвергали экстракции посредством ДХМ (3×25 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (3% МеОН в ДХМ), получая трет-бутил-(S)-3-((5-метил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)карбамоил) пирролидин-1-карбоксилат (0,35 ммоль). МС: ЭР+ 367,3.
Стадия f. К раствору трет-бутил-(S)-3-((5-метил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)карбамоил)пирролидин-1-карбоксилата (0,35 ммоль) в ДХМ (10 мл) добавляли ТФУ (1,4 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток растирали с диэтиловым эфиром (10 мл), получая соль ТФУ и (S)-N-(5-метил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамида (0,21 ммоль). Материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 267,0.
Стадия g. К раствору соли ТФУ и (S)-N-(5-метил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамида (0,21 ммоль) в ДХМ (2 мл) добавляли ТЭА (0,63 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 10 мин. К реакционной смеси добавляли цианоген бромид (0,24 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 30 мин. Полученную реакционную смесь вливали в ледяную воду (10 мл) и подвергали экстракции посредством ДХМ (3×25 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (5% МеОН в ДХМ) с получением указанного в заголовке соединения (0,07 ммоль). МС: ЭР+ 292,08; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,20 (ушир. s, 1 Н), 3,55-3,68 (m, 3 Н), 3,48-3,54 (m, 1 Н), 3,39-3,47 (m, 2 Н), 3,25-3,31 (m, 1 Н), 2,79 (m, 2 Н), 2,68 (m, 2 Н), 2,43 (s, 3 Н), 2,16-2,20 (m, 1 Н), 2,00-2,08 (m, 1 Н).
Пример 201 (S)-N-(5-бензил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)-1-цианопирролидин-3-карбоксамид (получали в соответствии со Схемой 10)
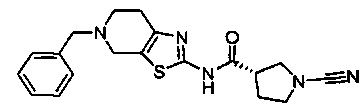
Стадия е. К раствору Промежуточного соединения 14 (0,85 ммоль) в МеОН (5 мл) добавляли бензальдегид (1,70 ммоль) и уксусную кислоту (3 капли) при 0°С. Реакционную смесь перемешивали при 0°С в течение 30 мин. К реакционной смеси добавляли NaCNBH3 (1,70 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Полученную реакционную смесь вливали в ледяную воду (10 мл) и подвергали экстракции посредством ДХМ (3×25 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (2% МеОН в ДХМ), получая трет-бутил-(S)-3-((5-бензил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)карбамоил)пирролидин-1-карбоксилат (0,58 ммоль). МС: ЭР+ 443,4.
Стадии f-g. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для Примера 200 в соответствии со стадиями d и е Схемы 8. МС: ЭР+ 368,38; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,16 (ушир. s, 1 Н), 7,34-7,46 (m, 4 Н), 7,24-7,31 (m, 1 Н), 3,69 (s, 2 Н), 3,47-3,65 (m, 4 Н), 3,37-3,43 (m, 2 Н), 3,25-3,33 (m, 1 Н), 2,73-2,79 (m, 2 Н), 2,63-2,67 (m, 2 Н), 2,13-2,22 (m, 1 Н), 1,99-2,07 (m, 1 Н).
Пример 202 (S)-1-циано-N-(5-(метилсульфонил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 10)
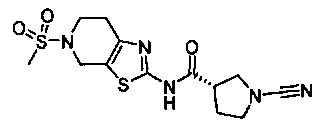
Стадия с. К раствору Промежуточного соединения 14 (0,56 ммоль) в ДХМ (2 мл) добавляли ТЭА (1,13 ммоль) и метансульфонилхлорид (0,84 ммоль) при 0°С.Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь вливали в ледяную воду (10 мл) и проводили экстракцию посредством ДХМ (3×20 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (2% МеОН в ДХМ), получая трет-бутил-(S)-3-((5-(метилсульфонил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)карбамоил)-пирролидин-1-карбоксилат (0,43 ммоль). МС: ЭР+ 431,38.
Стадия f. 4М HCl в 1,4-диоксане (4 ммоль) добавляли к трет-бутил-(S)-3-((5-(метилсульфонил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)карбамоил) пирролидин-1-карбоксилату (0,43 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток растирали с диэтиловым эфиром (10 мл), получая (S)-N-(5-(метилсульфонил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид гидрохлорид (0,36 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 331,28.
Стадия g. К раствору (S)-N-(5-(метилсульфонил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамида гидрохлорида (0,35 ммоль) в CHCl3 (4 мл) добавляли DIPEA (1,1 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 10 мин. Цианоген бромид (0,53 ммоль) добавляли и реакционную смесь перемешивали при 0°С в течение 30 мин. Полученную реакционную смесь вливали в ледяную воду (10 мл) и подвергали экстракции посредством ДХМ (3×25 мл). Органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (2% МеОН в ДХМ), получая указанное в заголовке соединение (0,26 ммоль). МС: ЭР+ 356,39; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,28 (s, 1 Н), 4,40 (s, 2 Н), 3,59-3,64 (m, 1 Н), 3,47-3,55 (m, 3 Н), 3,38-3,47 (m, 2 Н), 3,26-3,33 (m, 1 Н), 2,96 (s, 3 Н), 2,75-2,78 (m, 2 Н), 2,14-2,23 (m, 1 Н), 2,00-2,10 (m, 1 Н).
Пример 203 (S)-1-циано-N-(5-(изопропилсульфонил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид
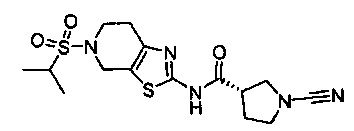
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 202, применяя изопропилсульфонилхлорид. МС: ЭР+ 384,28; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,26 (s, 1 Н), 4,47 (s, 2 Н), 3,59-3,63 (m, 3 Н), 3,44-3,53 (m, 1 Н), 3,37-3,42 (m, 3 Н), 3,26-3,31 (m, 1 Н), 2,67-2,71 (m, 2 Н), 2,14-2,17 (m, 1 Н), 2,00-2,08 (m, 1 Н), 1,22 (d, J=6,4 Гц, 6 Н).
Пример 204 (S)-1-циано-N-(5-(фенилсулъфонил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид
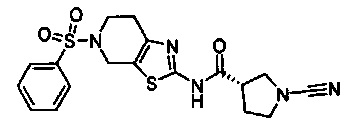
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 202, применяя бензолсульфонилхлорид. МС: ЭР+ 418,58; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,22 (ушир. s, 1 Н), 7,81-7,83 (m, 2 Н), 7,68-7,72 (m, 1 Н), 7,59-7,63 (m, 2 H), 4,31 (s, 2 H), 3,60-3,62 (m, 1 H), 3,47-3,52 (m, 1 H), 3,35-3,45 (m, 4 H), 3,24-3,29 (m, 1 H), 2,61-2,67 (m, 2 H), 2,12-2,21 (m, 1 H), 1,98-2,07 (m, 1 H).
Пример 205 (S)-1-циано-N-(5-(4-этинилфенил)тиазол-2-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 11)
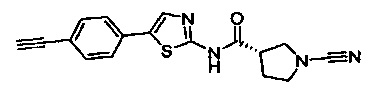
Стадия а. К раствору 2-(4-бромфенил)этан-1-ола (4,97 ммоль) в ДХМ (13 мл) добавляли периодинан Десса-Мартина (6,2 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь частично упаривали и полученный остаток фильтровали через Celite Hyflo. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (9-10% EtOAc в гексане), получая 2-(4-бромфенил)-ацетальдегид (3,51 ммоль). МС: ЭР- 197,10; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 9,68 (s, 1 Н), 7,54 (d, J=2,0 Гц, 2 Н), 7,21 (d, J=8,4 Гц, 2 Н), 3,79 (d, J=0,8 Гц, 2 Н).
Стадия b. К раствору 2-(4-бромфенил)ацетальдегида (3,5 ммоль) в ДХМ (13 мл) по каплям добавляли бром (3,5 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Полученную реакционную смесь затем вливали в насыщенный раствор NaHCO3 (20 мл) и подвергали экстракции посредством ДХМ (2×40 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 2-бром-2-(4-бромфенил)ацетальдегид. Данный материал сразу же применяли на следующей стадии без какой-либо дополнительной обработки.
Стадия с. Тиомочевину (7,0 ммоль) добавляли к раствору 2-бром-2-(4-бромфенил) ацетальдегида в EtOH (8 мл) при комнатной температуре. Реакционную смесь нагревали при 90°С в течение 6 ч. Полученной реакционной смеси давали возможность остыть до к. т. и упаривали ее при пониженном давлении. Полученный остаток распределяли между насыщенным NaHCO3 (20 мл) и ДХМ (30 мл). Органический слой отделяли и полученный водный слой дополнительно подвергали экстракции посредством ДХМ (2×30 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 5-(4-бромфенил)тиазол-2-амин (2,37 ммоль). МС: ЭР+ 255,1, 257,08; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 7,49-7,52 (m, 2 Н), 7,46 (s, 1 Н), 7,36-7,38 (m, 2 Н), 7,23 (ушир. s, 2 Н).
Стадия d. К раствору (3S)-ВОС-1-пирролидин-3-карбоновой кислоты (0,78 ммоль) в ТГФ (2 мл) добавляли Т3Р (50% в EtOAc) (1,17 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 15 мин. 5-(4-бромфенил) тиазол-2-амин (0,78 ммоль) и ТЭА (2,3 ммоль) затем добавляли к реакционной смеси. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь вливали в воду (50 мл) и подвергали экстракции посредством EtOAc (3×30 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (1-2% МеОН в ДХМ), получая трет-бутил-(S)-3-((5-(4-бромфенил)тиазол-2-ил)карбамоил)пирролидин-1-карбоксилат (0,37 ммоль). МС: ЭР+ 452,34, 454,28
Стадия е. Трет-бутил-(S)-3-((5-(4-бромфенил)тиазол-2-ил)карбамоил)пирролидин-1-карбоксилат (0,88 ммоль), Pd(PPh3)2Cl2 (0,13 мл) и CuI (0,05 ммоль) смешивали в высушенной в печи стеклянной пробирке в атмосфере азота. Стеклянную пробирку медленно заполняли азотом и сразу же плотно закрывали. К реакционной смеси добавляли триметилсилилацетилен (4,42 ммоль) и DIPEA (10 мл) с помощью шприца. В закрытую пробирку снова направляли потом азота с низкой скоростью. Указанную плотно закрытую стеклянную пробирку затем подвергали нагреванию при 110°С (наружная температура) в течение 16 часов. Полученную реакционную смесь вливали в воду (30 мл) и подвергали экстракции посредством EtOAc (2×50 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (30-40% EtOAc в гексане), получая трет-бутил-(S)-3-((5-(4-((триметилсилил)этинил)фенил)тиазол-2-ил)карбамоил)пирролидин-1-карбоксилат (0,63 ммоль). МС: ЭР+ 470,53.
Стадия f. Раствор трет-бутил-(S)-3-((5-(4-((триметилсилил)этинил)фенил)тиазол-2-ил)карбамоил)пирролидин-1-карбоксилата (0,63 ммоль) в МеОН (10 мл) перемешивали при 0°С в течение 5 минут. К реакционной смеси добавляли 5М водный раствор КОН (7 мл) добавляли при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Полученную реакционную смесь вливали в воду (40 мл) и подвергали экстракции посредством EtOAc (2×50 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (60-70% EtOAc в гексане), получая трет-бутил-(S)-3-((5-(4-этинилфенил) тиазол-2-ил) карбамоил) пирролидин-1-карбоксилат (0,1 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной обработки.
Стадия g. К раствору трет-бутил-(S)-3-((5-(4-этинилфенил) тиазол-2-ил) карбамоил) пирролидин-1-карбоксилата (0,08 ммоль) в ДХМ (2 мл) добавляли ТФУ (0,3 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток растирали с диэтиловым эфиром (5 мл), получая соль ТФУ и (S)-N-(5-(4-этинилфенил) тиазол-2-ил)пирролидин-3-карбоксамида (0,067 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 298,38.
Стадия h. К раствору соли ТФУ и (S)-N-(4-этинилфенил) тиазол-2-ил)пирролидин-3-карбоксамида (0,12 ммоль) в ДХМ (2 мл) добавляли K2СО3 (0,36 ммоль) при комнатной температуре и перемешивали в течение 15 мин. К реакционной смеси добавляли цианоген бромид (0,14 ммоль) при 0°С. Реакционную смесь затем перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь вливали в воду (20 мл) и подвергали экстракции посредством EtOAc (2×20 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (1,5-2,5% МеОН в ДХМ), получая указанное в заголовке соединение (0,01 ммоль). МС: ЭР+ 322,96; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,49 (s, 1 Н), 7,99 (s, 1 Н), 7,64 (d, J=8,4 Гц, 2 Н), 7,51 (d, J=8,4 Гц, 2 Н), 4,29 (s, 1 Н), 3,61-3,66 (m, 1 Н), 3,53-3,58 (m, 1 Н), 3,40-3,48 (m, 3 Н), 2,16-2,23 (m, 1 Н), 2,06-2,10 (m, 1 Н).
Пример 206 (S)-1-циано-N-(5-(3-этинилфенил)тиазол-2-ил)пирролидин-3-карбоксамид
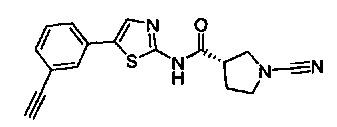
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 205, применяя 3-бромфенетиловый спирт на стадии а. МС: ЭР+ 323,06; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,48 (s, 1 Н), 7,99 (s, 1 Н), 7,61-7,66 (m, 2 Н), 7,49-7,53 (m, 1 Н), 7,39-7,46 (m, 1 Н), 4,29 (s, 1 Н), 3,61-3,67 (m, 1 Н), 3,53-3,59 (m, 1 Н), 3,42-3,50 (m, 2 Н), 3,36-3,42 (m, 1 Н), 2,16-2,26 (m, 1 Н), 2,08-2,12 (m, 1 Н).
Пример 207 (3S)-1-циано-N-(5-(N-(1-фенилэтил)сулъфамоил)пиридин-2-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 12)
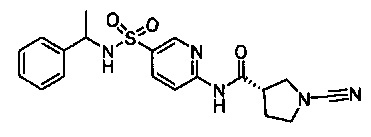
Стадия а. К 2-аминопиридину (10,6 ммоль) медленно добавляли хлорсульфоновую кислоту (89,2 ммоль) в атмосфере азота при 0°С. По завершении добавления реакционной смеси давали возможность нагреться до к. т. и затем нагревали при 150°С в течение 2 часов. Полученной реакционной смеси давали возможность остыть до к. т., вливали в ледяную воду (100 мл) и нейтрализовали путем порционного добавления твердого NaHCO3. Полученную в результате суспензию подвергали экстракции EtOAc (2×50 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный материал кристаллизовали из смеси диэтиловый эфир: гептан, получая 6-аминопиридин-3-сульфонилхлорид (5,75 ммоль). МС: ЭР+ 193,19; 1Н ЯМР (400 МГц, CDCl3) δ ppm 8,71 (d, J=2,75 Гц, 1 Н), 7,98 (dd, J=9,00, 2,59 Гц, 1 Н), 6,59 (dd, J=9,16, 0,61 Гц, 1 Н), 5,41 (ушир. s, 2 Н).
Стадия b. К раствору 1-фенилэтан-1-амина (2,08 ммоль) в ТГФ (5 мл) добавляли ТЭА (2,5 ммоль) при 0°С и реакционную смесь перемешивали в течение 5 мин при 0°С. К реакционной смеси добавляли 6-аминопиридин-3-сульфонилхлорид (2,08 ммоль) при 0°С и затем перемешивали при комнатной температуре в течение 30 мин. Полученную реакционную смесь вливали в воду (50 мл) и подвергали экстракции посредством EtOAc (2×30 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (25% EtOAc в гексане), получая 6-амино-N-(1-фенилэтил)пиридин-3-сульфонамид (1,62 ммоль). МС: ЭР+ 278,33; 1Н ЯМР (400 МГц, ДМСО6) δ ppm 8,12 (dd, J=2,44, 0,61 Гц, 1 Н), 7,94 (d, J=8,24 Гц, 1 Н), 7,49 (dd, J=8,85, 2,44 Гц, 1 Н), 7,18-7,25 (m, 4 Н), 7,13-7,17 (m, 1 Н), 6,75 (ушир. s, 2 Н), 6,35 (dd, J=8,85, 0,61 Гц, 1 Н), 4,25-4,30 (m, 1 Н), 1,22 (d, J=7,02 Гц, 3 Н).
Стадия с. Смесь безводного ДХМ (5 мл) и безводного ДМФА (1,39 ммоль) вносили в стеклянную пробирку в атмосфере азота при 0°С. К реакционной смеси добавляли оксалилхлорид (1,39 ммоль) в атмосфере азота при 0°С. К реакционной смеси добавляли безводный пиридин (1,39 ммоль) в атмосфере азота при 0°С. К реакционной смеси по каплям добавляли раствор (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновой кислоты (1,39 ммоль) в ДХМ (5 мл) в атмосфере азота при 0°С. Полученную реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение 1 ч, в результате чего получали необходимый хлорангидрид кислоты. Одновременно с этим в другой стеклянной пробирке получали раствор 6-амино-N-(1-фенилэтил)пиридин-3-сульфонамида в ДМФА (3 мл) в атмосфере азота. ТЭА (1,65 ммоль) добавляли к реакционной смеси, находящейся во второй стеклянной пробирке, в атмосфере азота при 0°С. После этого раствор хлорангидрида из первой стеклянной пробирки аккуратно набирали в шприц и по каплям добавляли к реакционной смеси, находящейся во второй стеклянной пробирке, в атмосфере азота при 0°С. Конечную реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Полученную реакционную смесь концентрировали при пониженном давлении и полученный неочищенный материал подвергали азеотропной перегонке с применением ДХМ (2×50 мл). Полученный остаток очищали с помощью колоночной хроматографии (4% МеОН в ДХМ), получая трет-бутил-(3S)-3-((5-(N-(1-фенилэтил)-сульфамоил)пиридин-2-ил)карбамоил)пирролидин-1-карбоксилат (0,46 ммоль). МС: ЭР+ 475,71
Стадия d. К раствору трет-бутил-(3S)-3-((5-(N-(1-фенилэтил)сульфамоил)пиридин-2-ил)карбамоил)пирролидин-1-карбоксилата (0,46 ммоль) в ДХМ (3 мл) добавляли ТФУ (2,5 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 15 мин. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток подвергали азеотропной перегонке с применением ДХМ (2×15 мл), получая соль ТФУ и (3S)-N-(5-(N-(1-фенилэтил)сульфамоил)пиридин-2-ил)пирролидин-3-карбоксамида (0,37 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки.
Стадия е. К раствору соли ТФУ и (3S)-N-(5-(N-(1-фенилэтил)сульфамоил)пиридин-2-ил)пирролидин-3-карбоксамида (0,37 ммоль) в ТГФ (3 мл) добавляли ТЭА (1,49 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 5 минут. К реакционной смеси добавляли цианоген бромид (0,44 ммоль) при 0°С. Реакционную смесь затем перемешивали при комнатной температуре в течение 30 мин. Полученную реакционную смесь вливали в воду (30 мл) и подвергали экстракции посредством EtOAc (2×20 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (10% МеОН в ДХМ), получая указанное в заголовке соединение (0,04 ммоль). МС: ЭР+ 399,93; 1Н ЯМР (400 МГц, ДМСО6) δ ppm 11,07 (ушир. s, 1 Н), 8,43-8,45 (m, 1 Н), 8,33-8,35 (m, 1 Н), 8,07-8,09 (m, 1 Н), 7,93-7,97 (m, 1 Н), 7,09-7,21 (m, 5 Н), 4,35-4,47 (m, 1 Н), 3,56-3,63 (m, 1 Н), 3,35-3,53 (m, 4 Н), 2,14-2,20 (m, 1 Н), 2,06-2,12 (m, 1 Н), 1,25-1,27 (m, 3 Н).
Пример 208 (3S)-1-циано-N-(5-(N-метил-N-(1-фенилэтил)сульфамоил)пиридин-2-ил)пирролидин-3-карбоксамид
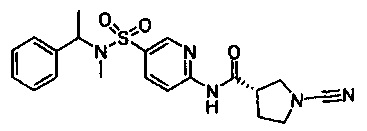
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 207, применяя метил(1-фенилэтил)амин на стадии b. МС: ЭР+ 414,68; 1Н ЯМР (400 МГц, CD3OD) δ ppm 8,72-8,73 (m, 1 Н), 8,33-8,35 (m, 1 Н), 8,16-8,19 (m, 1 Н), 7,24-7,37 (m, 5 Н), 5,29-5,32 (m, 1 Н), 3,69-3,71 (m, 2 Н), 3,51-3,60 (m, 2 Н), 3,32-3,39 (m, 1 Н), 2,65 (s, 3Н), 2,21-2,32 (m, 2 Н), 1,38-1,40 (m, 3 Н).
Пример 209 (S)-1-циано-N-(6-(5-метил-1,2,4-оксадиазол-3-ил)имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 13)
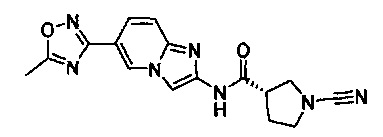
Стадия а. К раствору (3S)-ВОС-1-пирролидин-3-карбоновой кислоты (4,65 ммоль) в ТГФ (20 мл) добавляли Т3Р (50% в EtOAc) (13,95 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 15 мин. К реакционной смеси добавляли 6-бромимидазо[1,2-а]пиридин-2-амин (4,65 ммоль) и DIPEA (13,95 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь вливали в воду (50 мл) и подвергали экстракции посредством EtOAc (3×100 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая трет-бутил-(S)-3-((6-бромимидазо[1,2-а]пиридин-2-ил)карбамоил)пирролидин-1-карбоксилат (3,80 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 409,50, 411,50.
Стадия b. Раствор трет-бутил-(S)-3-((6-бромимидазо[1,2-а]пиридин-2-ил)карбамоил)пирролидин-1-карбоксилат (2,45 ммоль) получали в ДМФА (20 мл) в стеклянной пробирке. К реакционной смеси добавляли Zn(CN)2 (7,35 ммоль) и ТЭА (4,90 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь продували азотом gas при комнатной температуре в течение 15 мин. К реакционной смеси добавляли Pd(dba)2 (0,24 моль) и 1,1'-бис(дифенилфосфино)ферроцен (0,49 ммоль) при комнатной температуре в атмосфере азота и стеклянную пробирку плотно закрывали. Стеклянную пробирку подвергали нагреванию при 100°С (наружная температура) в течение 24 ч. Полученной реакционной смеси давали возможность остыть до к. т. и вливали в воду (50 мл) и подвергали экстракции посредством EtOAc (3×75 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (чистый ДХМ), получая трет-бутил-(S)-3-((6-цианоимидазо [1,2-а] пиридин-2-ил) карбамоил) пирролидин-1-карбоксилат (2,25 ммоль). МС: ЭР+ 356,53.
Стадия с. К раствору (трет-бутил-(S)-3-((6-цианоимидазо[1,2-а]пиридин-2-ил)карбамоил)пирролидин-1-карбоксилата (2,25 ммоль) в IPA (30 мл) добавляли гидроксиламин гидрохлорид (13,5 ммоль) и ТЭА (15,7 ммоль) при комнатной температуре. Реакционную смесь нагревали при 70°С в течение 3 ч. Полученной реакционной смеси позволяли остыть до к. т. и вливали в воду (50 мл) и подвергали экстракции посредством EtOAc (3×75 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая трет-бутил-(S)-3-((6-(N-гидроксикарбамимидоил)имидазо[1,2-а]пиридин-2-ил)карбамоил)пирролидин-1-карбоксилат (1,28 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 389,48.
Стадия d. Раствор трет-бутил-(S)-3-((6-(N-гидроксикарбамимидоил)имидазо[1,2-а]пиридин-2-ил)карбамоил)пирролидин-1-карбоксилата (1,28 ммоль) в диметилацетамид-диметилацетале (20 мл) нагревали при 100°С в течение 1 часа. Полученной реакционной смеси позволяли остыть до к. т. и вливали в воду (25 мл) и подвергали экстракции посредством EtOAc (3×25 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (100% ДХМ), получая трет-бутил-(S)-3-((6-(5-метил-1,2,4-оксадиазол-3-ил) имидазо[1,2-а]пиридин-2-ил)карбамоил) пирролидин-1-карбоксилат (0,70 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 413,54.
Стадия е. К раствору трет-бутил-(S)-3-((6-(5-метил-1,2,4-оксадиазол-3-ил) имидазо[1,2-а] пиридин-2-ил)карбамоил) пирролидин-1-карбоксилат (0,70 ммоль) в ДХМ (6 мл) добавляли ТФУ (1,2 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток растирали с диэтиловым эфиром (10 мл), получая соль ТФУ и (S)-N-(6-(5-метил-1,2,4-оксадиазол-3-ил)имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамида (0,69 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 313,53.
Стадия f. К раствору соли ТФУ и (S)-N-(6-(5-метил-1,2,4-оксадиазол-3-ил)имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамида (0,69 ммоль) в ТГФ (6 мл) добавляли К2СО3 (2,03 ммоль) при 0°С. Полученную реакционную смесь перемешивали при 0°С в течение 30 мин. К реакционной смеси добавляли цианоген бромид (1,03 ммоль) при 0°С. Реакционную смесь затем перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь вливали в воду (30 мл). Полученный твердый осадок собирали путем фильтрования при пониженном давлении и сушили, получая указанное в заголовке соединение (0,16 ммоль). МС: ЭР+ 338,10; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 11,06 (ушир. s, 1 Н), 9,32 (s, 1 Н), 8,33 (s, 1 Н), 7,72 (dd, J=9,16, 1,53 Гц, 1 Н), 7,58 (d, J=9,16 Гц, 1 Н), 3,61-3,65 (m, 1 Н), 3,41-3,54 (m, 3 Н), 3,28-3,32 (m, 1 Н), 2,69 (s, 3 Н), 2,14-2,22 (m, 1 Н), 2,01-2,10 (m, 1 Н).
Пример 210 (±)-транс-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)имидазо[1,2-а]пиридин-2-ил)-4-(трифторметил)пирролидин-3-карбоксамид
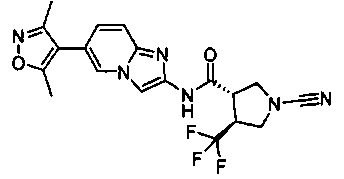
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 109, применяя транс-1-трет-бутиловый эфир (±)[4-(трифторметил)пирролидин]-1,3-дикарбоновой кислоты, 2-амино-6-бром-имидазо[1,2-а]пиридин (полученный в соответствии со способом, описанном в WO 2012174312) и 3,5-диметилизоксазол-4-бороновую кислоту. ЖХМС: Способ В, 3,57 мин, МС: ЭР+ 419,31.
Пример 211 (S)-1-циано-N-(4-(1-метил-1Н-пиразол-4-ил)пиридин-2-ил)пирролидин-3-карбоксамид
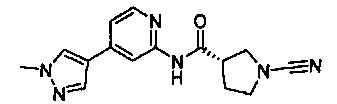
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 138, применяя 2-амино-4-бромпиридин и пинаколовый эфир 1-метил-1Н-пиразол-4-бороновой кислоты. ЖХМС: Способ С, 2,96 мин, МС: ЭР+ 297,02.
Пример 212 (S)-1-циано-N-(5-(N,N-диметилсульфамоил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 10, стадии с, f, g)
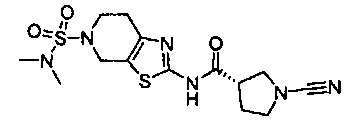
Стадия с. К раствору Промежуточного соединения 14 (0,48 ммоль) в ДХМ (3 мл) добавляли ТЭА (1,44 ммоль) и N,N-диметиламиносульфонилхлорид (0,57 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Полученную реакционную смесь вливали в воду (150 мл) и подвергали экстракции посредством EtOAc (3×50 мл). Объединенную органическую фазу собирали, промывали насыщенным раствором NaHCO3 (50 мл), 10% лимонной кислотой (50 мл), солевым раствором (50 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая трет-бутил-(S)-3-((5-(N,N-диметилсульфамоил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)карбамоил)пирролидин-1-карбоксилат (0,39 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 460,17.
Стадия f, g. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для стадий f и g Примера 202. ЖХМС: Способ В, время удерживания 3,15 мин, МС: ЭР+ 385,14.
Пример 213 (S)-1-циано-N-(5-(пиридазин-4-ил)тиазол-2-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 14, стадии a, b, d-g)
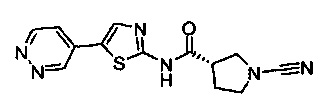
Стадия а. Раствор Промежуточного соединения 20 (2,5 ммоль) в сухом ТГФ (15 мл) охлаждали при -78°С. К полученной реакционной смеси по каплям добавляли 2,4М n-BuLi в гексане (2,5 ммоль) при -78°С. Реакционную смесь перемешивали при -78°С в течение 20 мин. К полученной реакционной смеси добавляли 4,4,5,5-тетраметил-1,3,2-диоксаборолан (3,0 ммоль) при -78°С. Реакционную смесь перемешивали при -78°С в течение 30 мин. Полученной реакционной смеси давали возможность нагреться до 0°С и гасили путем добавление насыщенного раствора хлорида аммония (100 мл). Полученную смесь подвергали экстракции EtOAc (2×100 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением трет-бутил-(4-метоксибензил)-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол-2-ил)карбамата (количественный выход). Данный материал сразу же применяли на следующей стадии без какой-либо очистки.
Стадия b. В стеклянной закрытой пробирке суспензию 4-бромпиридазина гидробромида (1,25 ммоль), трет-бутил-(4-метоксибензил)-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)тиазол-2-ил)карбамата (2,18 ммоль) и Na2CO3 (6,25 ммоль) в смеси толуол: вода (15 мл: 3 мл) дегазировали азотом в течение 30 мин при комнатной температуре. В полученную реакционную смесь добавляли PdCl2(dppf) (0,12 ммоль) при комнатной температуре, и стеклянную пробирку запаивали. Реакционную смесь нагревали при 100°С (внешняя температура) в течение 1 ч. Полученную реакционную смесь охлаждали до к. т., вливали в насыщенный раствор NaHCO3 (300 мл) и подвергали экстракции посредством EtOAc (3×150 мл). Объединенную органическую фазу промывали солевым раствором (250 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (2,5% МеОН в ДХМ), получая трет-бутил-(4-метоксибензил)(5-(пиридазин-4-ил)тиазол-2-ил)карбамата (1,13 ммоль). МС: ЭР+ 399,35; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 9,58-9,60 (m, 1 Н), 9,15 (dd, J=1,2, 5,6 Гц, 1H), 8,38 (s, 1H), 7,82-7,84 (m, 1H), 7,26 (d, J=8,4 Гц, 2H), 6,90 (d, J=8,8 Гц, 2H), 5,22 (s, 2H), 3,72 (s, 3H), 1,5 (s, 9H).
Стадия d. Раствор трет-бутил-(4-метоксибензил)(5-(пиридазин-4-ил)тиазол-2-ил)карбамата (0,75 ммоль) в ТФУ (15 мл) нагревали при 80°С в течение 8 часов. Полученную реакционную смесь концентрировали при пониженном давлении и к ней добавляли воду (75 мл). Полученную смесь подвергали экстракции EtOAc (3×50 мл). Полученный водный слой подщелачивали, применяя твердый NaHCO3, и подвергали экстракции посредством EtOAc (15×50 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 5-(пиридазин-4-ил)тиазол-2-амин (0,67 ммоль). Данный материал применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 179,01.
Стадия е. К раствору (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновой кислоты (1,17 ммоль) в ТГФ (7 мл) добавляли HATU (1,18 ммоль) и DIPEA (2,35 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. К реакционной смеси добавляли 5-(пиридазин-4-ил)тиазол-2-амин (0,78 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Полученную реакционную смесь вливали в насыщенный раствор NaHCO3 (100 мл) и подвергали экстракции посредством EtOAc (3×50 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (3,5% МеОН в ДХМ), получая трет-бутил-(S)-3-((5-(пиридазин-4-ил)тиазол-2-ил)карбамоил)пирролидин-1-карбоксилат (0,46 ммоль). МС: ЭР+ 376,29; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,68(s, 1Н), 9,58-9,60 (m, 1 Н), 9,16 (dd, J=1,2, 5,6 Гц, 1H), 8,48 (s, 1H), 7,81-7,83 (m, 1H), 3,53-3,54 (m, 1Н), 3,28-3,44 (m, 4Н), 2,15-2,17 (m, 1H), 2,01-2,09 (m, 1Н), 1,45 (s, 9Н).
Стадия f, g. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для стадий c и d Примера 140. ЖХМС: Способ С, 1,87 мин, МС: ЭР+ 300,88; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,78(s, 1Н), 9,58-9,60 (m, 1 Н), 9,16 (d, J=5,6, 1H), 8,41 (s, 1H), 7,82-7,84 (m, 1H), 3,62-3,66 (m, 1H), 3,55-3,59 (m, 1H), 3,41-3,47 (m, 2H), 3,37-3,39 (m, 1H), 2,17-2,26 (m, 1H), 2,05-2,13 (m, 1H).
Соединения, приведенные в таблице 7, синтезировали с помощью методики, аналогичной методике, описанной для Примера 213.
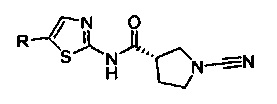

Пример 227 (S)-1-циано-N-(5-(1-(2-метоксиэтил)-1H-индазол-5-ил)тиазол-2-ил)пирролидии-3-карбоксамид (получали в соответствии со Схемой 14, стадии c-g)
Стадия с. Получали смесь Промежуточного соединения 20 (0,50 г, 1,25 ммоль), Промежуточного соединения 18 (0,38 г, 1,25 ммоль) и Cs2CO3 (0,77 г, 2,37 ммоль) в смеси 1,4-диоксан: вода (9:1) (5 мл) в стеклянном флаконе. Реакционную смесь дегазировали в течение 30 мин. К реакционной смеси добавляли Pd(dppf)Cl2 (0,03 г, 0,03 ммоль) при комнатной температуре. Стеклянный флакон плотно закрывали и подвергали нагреванию при 80°С (наружная температура) в течение 2 часов. Полученную реакционную смесь разбавляли водой (50 мл) и подвергали экстракции посредством ДХМ (3×30 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (25% EtOAc в гексане), получая трет-бутил-(4-метоксибензил)(5-(1-(2-метоксиэтил)-1Н-индазол-5-ил)тиазол-2-ил)карбамата (0,37 г, 0,74 ммоль). ЖХМС: Способ А, 2,93 мин, МС: ЭР+ 495,6.
Стадия d-g. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для стадий d-g of Пример 213. ЖХМС: Способ С, 3,24 мин, МС: ЭР+ 397,02
Пример 228 (S)-1-циано-N-(5-(2-оксоиндолин-7-ил)тиазол-2-ил)пирролидин-3-карбоксамид
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 227, применяя веществом с номером в CAS 1150271-45-6 на стадии с. ЖХМС: Способ С, 2,82 мин, МС: ЭР+ 353,90
Пример 229 (S)-1-циано-N-(5-(3-метил-1Н-индазол-5-ил)тиазол-2-ил)пирролидин-3-карбоксамид
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 227, применяя веществом с номером в CAS 864771-17-5 на стадии с. ЖХМС: Способ В, 3,31 мин, МС: ЭР+ 353,57
Пример 230 (S)-N-(5-(1Н-индазол-4-ил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамид
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 227, применяя Промежуточное соединение 19 на стадии с. ЖХМС: Способ А, 1,82 мин, МС: ЭР+ 339,54
Пример 231 (S)-1-циано-N-(5-(4-фтор-3-(метилкарбамоил)фенил)тиазол-2-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 15)
Стадия а. Данную стадию осуществляли с помощью методики, аналогичной методике, описанной для стадии а Примера 213. ЖХМС: Способ А, 3,21 мин, МС: ЭР+ 473,40
Стадия b. К раствору метил-5-(2-((трет-бутоксикарбонил)(4-метоксибензил)амино)тиазол-5-ил)-2-фторбензоата (0,23 г, 0,48 ммоль), триазабициклодецен (0,13 г, 0,97 ммоль) в ТГФ (5 мл) добавляли CH3NH2 (2М в ТГФ) (1,21 мл, 2,43 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Полученную реакционную смесь вливали в воду (25 мл) и подвергали экстракции посредством EtOAc (3×25 мл). Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (35% EtOAc в гексане), получая трет-бутил-(5-(4-фтор-3-(метилкарбамоил)фенил)тиазол-2-ил)(4-метоксибензил)карбамат (0,13 г, 0,27 ммоль). ЖХМС: Способ А, 2,87 мин, МС: ЭР+ 472,45
Стадии c-f. Указанное в заголовке соединение синтезировали из описанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для стадий b-е Примера 213. ЖХМС: Способ А, 1,81 мин, МС: ЭР+ 374,35
Соединения, приведенные в таблице 8, синтезировали с помощью методики, аналогичной методике, описанной для Примера 231.
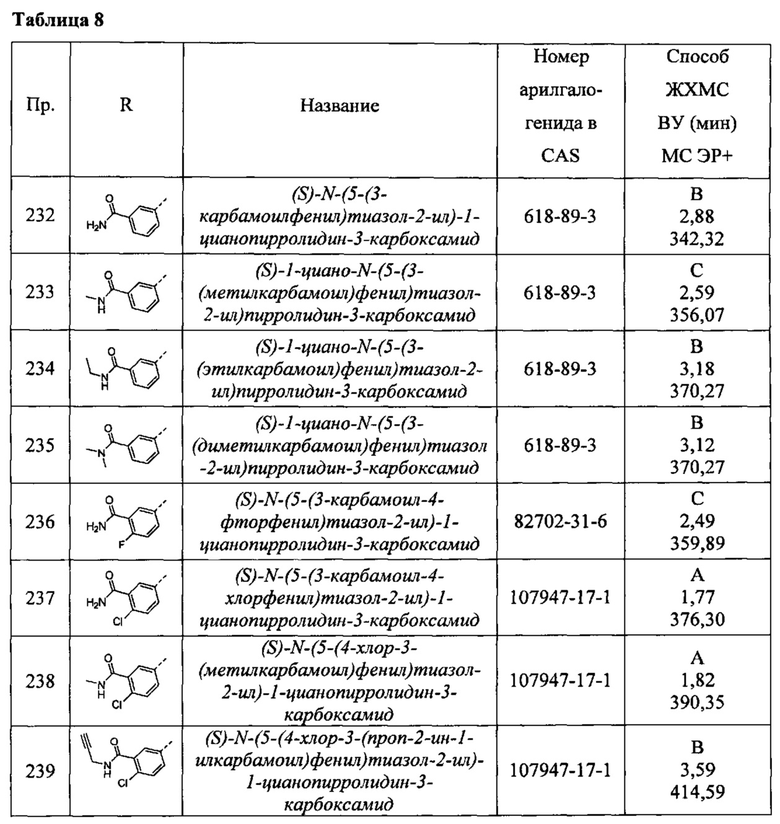
Пример 240 (S)-1-циано-N-(5-(изопропилсульфонил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-ил)пирролидин-3-карбоксамид
(получали в соответствии со Схемой 16, стадии a-j)
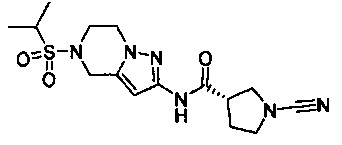
Стадия а. Раствор 3-нитро-1Н-пиразол-5-карбоновой кислоты (63,66 ммоль) в МеОН (100 мл) и каталитическое количество ДМФА (0,05 мл) перемешивали при 0°С в течение 10 мин. К полученной реакционной смеси по каплям добавляли SOCl2 (165,52 ммоль) при 0°С. Реакционной смеси давали возможность нагреться до к. т. и затем нагревали при 70°С в течение 4 часов. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток подвергали азеотропной перегонке с применением МеОН (100 мл), получая метил-3-нитро-1Н-пиразол-5 -карбоксилат (количественный выход). МС: ЭР- 170,12.
Стадия b. Раствор метил-3-нитро-1Н-пиразол-5-карбоксилата (65,27 ммоль) и K2СО3 (326,4 ммоль) в ацетоне (335 мл) перемешивали при комнатной температуре в течение 20 мин. К реакционной смеси добавляли 1,2-дибромэтан (195,81 ммоль) при комнатной температуре и затем перемешивали при 60°С в течение 1 часа. Реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (9-10% EtOAc в гексане), получая метил-1-(2-бромэтил)-3-нитро-1Н-пиразол-5-карбоксилат (41,10 ммоль) МС: ЭР+ 278,05 (М) 280 (М+2); 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 7,63 (s, 1 Н), 5,01-5,05 (m, 2 Н), 3,92-3,96 (m, 2 Н), 3,90 (s, 3 Н).
Стадия с. К раствору метил-1-(2-бромэтил)-3-нитро-1Н-пиразол-5-карбоксилата (41,11 ммоль) в ТГФ (171 мл) добавляли LiBH4 (3,0М в ТГФ) (82,21 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. К полученной реакционной смеси добавляли EtOAc (125 мл), а затем добавляли воду (120 мл). Отделяли органический слой и водный слой дополнительно подвергали экстракции посредством EtOAc (3×60 мл). Объединенную органическую фазу собирали, промывали солевым раствором (60 мл) сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением (1-(2-бромэтил)-3-нитро-1Н-пиразол-5-ил)метанола (39,46 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. МС: ЭР+ 250 (М) 252 (М+2); 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 6,99 (s, 1 Н), 5,68 (t, J=5,2 Гц, 1 Н), 4,61-4,67 (m, 4 Н), 3,91 (t, J=6,4 Гц, 2 Н).
Стадия d. Раствор (1-(2-бромэтил)-3-нитро-1Н-пиразол-5-ил)метанола (39,43 ммоль) в хлороформе (394,4 мл) добавляли PBr3 (39,43 ммоль) по каплям при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Полученную реакционную смесь охлаждали до 0°С и добавляли ДХМ (1000 мл). Смесь подщелачивали насыщенным раствором NaHCO3 (400 мл). Отделяли органический слой и водный слой дополнительно подвергали экстракции посредством ДХМ (3×400 мл). Объединенную органическую фазу собирали, промывали солевым раствором (650 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (15% EtOAc в гексане), получая 1-(2-бромэтил)-5-(бромметил)-3-нитро-1Н-пиразол (18,27 ммоль). 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 7,22 (s, 1 Н), 4,91 (s, 2 Н), 4,71 (t, J=6,0 Гц, 2 Н), 3,93 (t, J=6,0 Гц, 2 Н).
Стадия е. К раствору 1-(2-бромэтил)-5-(бромметил)-3-нитро-1Н-пиразола (18,27 ммоль), водный раствор аммиака (257 мл) в ТГФ (68 мл) перемешивали при комнатной температуре в течение 72 ч. Реакционную смесь концентрировали при пониженном давлении. Полученный остаток растворяли в ТГФ (50 мл) и насыщенный раствор К2СО3 (5 мл) добавляли (для нейтрализации остаточного HBr). Смесь концентрировали при пониженном давлении и полученный в результате остаток очищали с помощью колоночной хроматографии (4% МеОН в ДХМ), получая 2-нитро-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин (13,54 ммоль). МС: ЭР+ 169,04.
Стадия f. К раствору 2-нитро-4,5,6,7-тетрагидропиразоло[1,5-а]пиразина (1,78 ммоль) в ДХМ (9 мл) добавляли ТЭА (3,56 ммоль) и смесь перемешивали при комнатной температуре в течение 15 мин. Метансульфонилхлорид (1,96 ммоль) добавляли при комнатной температуре и реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (40% EtOAc в гексане), получая 5-(изопропилсульфонил)-2-нитро-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин (1,51 ммоль). МС: ЭР+ 275,5.
Стадия g. К раствору 5-(изопропилсульфонил)-2-нитро-4,5,6,7-тетрагидропиразоло[1,5-а]пиразина (1,45 ммоль) в МеОН (16 мл) добавляли 10% Pd/C (0,5% масс./масс.) при к. т. Реакционную смесь продували газообразным H2 при к. т. в течение 3 ч. Полученную реакционную смесь тщательно фильтровали через Celite Hyflo и концентрировали при пониженном давлении с получением 5-(изопропилсульфонил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-амина (1,27 ммоль). Данный материал сразу же применяли на следующей стадии. МС: ЭР+ 245,2.
Стадия h. К раствору (3S)-ВОС-1-пирролидин-3-карбоновой кислоты (1,47 ммоль) в ТГФ (9 мл) добавляли Т3Р (50% в EtOAc) (3,65 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 10 мин. К реакционной смеси добавляли 5-(изопропилсульфонил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-амин (1,22 ммоль) и DIPEA (3,68 ммоль) и перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь вливали в воду (100 мл) и подвергали экстракции посредством EtOAc (3×100 мл). Объединенную органическую фазу промывали насыщенным NaHCO3 (100 мл) и солевым раствором (100 мл), собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (3% МеОН в ДХМ), получая трет-бутил-(S)-3-((5-(изопропилсульфонил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-ил)карбамоил)пирролидин-1-карбоксилат (0,9 ммоль). МС: ЭР+ 442,4.
Стадия i, j. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для стадий c и d Примера 140. ЖХМС: Способ С, 2,554 мин, МС: ЭР+ 367,15; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,63 (s, 1 Н), 6,39 (s, 1 Н), 4,53 (s, 2 Н), 4,00 (t, J=5,2 Гц, 2 Н), 3,79 (t, J=5,2 Гц, 2 Н), 3,55-3,59 (m, 1 Н), 3,37-3,50 (m, 4Н), 3,67-3,33 (m, 1 Н), 2,10-2,15 (m, 1 Н), 1,96-2,01 (m, 1 Н), 1,23 (d, J=6,8 Гц, 6 Н).
Пример 241 (S)-1-циано-N-(5-фенил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-ил)пирролидин-3-карбоксамид
(получали в соответствии со Схемой 16, стадии а-е и g-j).
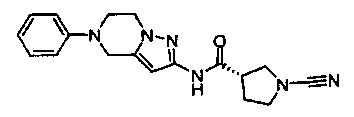
Стадии а-е осуществляли, как описано в Примере 240.
Стадия f. Раствор 2-нитро-4,5,6,7-тетрагидропиразоло[1,5-а]пиразина (2,97 ммоль), иодбензола (3,56 ммоль), Xantphos (0,44 ммоль), Cs2CO3 (8,92 ммоль) в 1,4-диоксане (15 мл) перемешивали при комнатной температуре в атмосфере азота в 25 мл стеклянном флаконе. Реакционную смесь продували азотом в течение 30 мин. К реакционной смеси добавляли Pd(OAc)2 (0,29 ммоль) при комнатной температуре в атмосфере азота и стеклянный флакон плотно закрывали. Закрытый флакон подвергали нагреванию при 100°С в течение 2 часов. Полученную реакционную смесь охлаждали и вливали в воду (100 мл) и подвергали экстракции посредством EtOAc (3×100 мл). Объединенную органическую фазу промывали солевым раствором (100 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (20% EtOAc в гексане), получая 2-нитро-5-фенил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин (количественный выход). МС: ЭР+ 245,43.
Стадии g-j. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для Примера 240 в соответствии со стадиями g-j Схемы 16. ЖХМС: Способ В, 3,511 мин, МС: ЭР+ 337,78; 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 10,6 (s, 1 Н), 7,26 (t, J=8,4 Гц, 2 Н), 7,06 (d, J=8,0 Гц, 2 Н), 6,83 (t, J=7,6 Гц, 1 Н), 6,41 (s, 1 Н), 4,22 (s, 2 Н), 4,04 (t, J=5,6 Гц, 2 Н), 3,77 (t, J=5,4 Гц, 2 Н), 3,57 (t, J=7,6 Гц, 1 Н), 3,72-3,47 (m, 3 Н), 3,15-3,22 (m, 1 Н), 2,10-2,17 (m, 1 Н), 1,91-2,02 (m, 1 Н).
Пример 242 (S)-N-(5-бензил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-ил)-1-цианопирролидин-3-карбоксамид
(получали в соответствии со Схемой 16, стадии а-е и g-j).
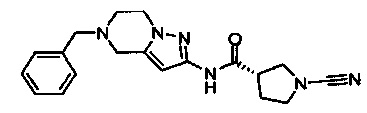
Стадии а-е проводили, как описано в Примере 240.
Стадия f. К раствору 2-нитро-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин (2,08 ммоль) в МеОН (14 мл) добавляли бензальдегид (4,16 ммоль) и уксусную кислоту (5 капли) при 0°С. Реакционную смесь перемешивали при 0°С в течение 1 ч. К реакционной смеси добавляли NaCNBH3 (4,16 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь вливали в воду (100 мл) и подвергали экстракции посредством EtOAc (3×100 мл). Объединенную органическую фазу промывали солевым раствором (100 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая 5-бензил-2-нитро-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин (количественный выход). МС: ЭР+ 259,48.
Стадии g-j. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для Примера 240 в соответствии со стадиями g-j Схемы 16. ЖХМС: Способ С, 3,52 мин, МС: ЭР+ 351,23; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,55 (s, 1 Н), 7,28-7,36 (m, 5 Н), 6,25 (s, 1 Н), 3,91 (t, J=5,6 Гц, 2 Н), 3,67 (s, 2 Н), 3,57 (s, 2 Н), 3,54 (s, 1 Н), 3,38-3,47 (m, 3 Н), 3,15-3,19 (m, 1 Н), 2,87 (t, J=5,2 Гц, 2 Н), 2,09-2,14 (m, 1 Н), 1,95-2,00 (m, 1 Н).
Пример 243 (S)-1-циано-N-(1-(4-циано-3-(2-метоксиэтокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
Пример 244 (S)-1-циано-N-(1-(2-циано-5-(2-метоксиэтокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
(получали в соответствии со Схемой 17)
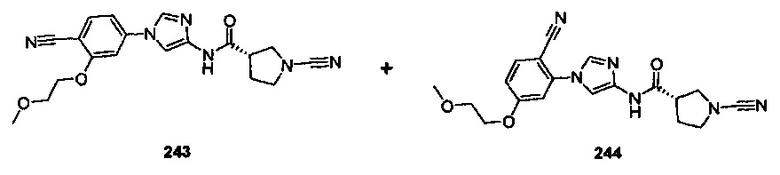
Стадия а. К раствору 2-метоксиэтанола (1,08 г, 14,19 ммоль) в ТГФ (20 мл) добавляли NaH (60% в минеральное масло) (0,56 г, 14,91 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 6 ч. Реакционную смесь охлаждали до 0°С и раствор 2,4-дифторбензонитрил (2,0 г, 14,19 ммоль) в диоксан (20 мл) добавляли по каплям. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Полученную реакционную смесь разбавляли водой (100 мл) и подвергали экстракции посредством EtOAc (3×100 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (10% EtOAc в гексане), получая региоизомерную смесь 2-фтор-4-(2-метоксиэтокси)бензонитрила и 4-фтор-2-(2-метоксиэтокси)бензонитрила (1,5 г, 7,69 ммоль). МС: ЭР+ 213,20.
Стадия b. К раствору 2-фтор-4-(2-метоксиэтокси)бензонитрила и 4-фтор-2-(2-метоксиэтокси)бензонитрила (0,7 г, 3,59 ммоль) в ДМФА (10 мл) добавляли К2СО3 (1,48 г, 10,77 ммоль) при комнатной температуре в атмосфере азота в пробирке для обработки микроволновым излучением. К реакционной смеси добавляли KI (0,06 г, 0,36 ммоль) при комнатной температуре. Реакционную смесь нагревали при 140°С в течение 2 часов в микроволновой печи. Полученную реакционную смесь вливали в воду (50 мл) и подвергали экстракции посредством EtOAc (2×100 мл). Объединенную органическую фазу промывали солевым раствором (50 мл). Органическую фазу собирали сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (30-40% EtOAc в гексане), получая региоизомерную смесь 2-(2-метоксиэтокси)-4-(4-нитро-1Н-имидазол-1-ил)бензонитрила и 4-(2-метоксиэтокси)-2-(4-нитро-1Н-имидазол-1-ил)бензонитрила (0,55 г, 1,91 ммоль). МС: ЭР+ 289,20
Стадия с. К раствору 2-(2-метоксиэтокси)-4-(4-нитро-1Н-имидазол-1-ил)бензонитрила и 4-(2-метоксиэтокси)-2-(4-нитро-1Н-имидазол-1-ил)бензонитрила (0,5 г, 1,74 ммоль) в ТГФ (30 мл) добавляли 10% сухого Pd/C (0,5 г) при комнатной температуре. Реакционную смесь продували газообразным Н2 при комнатной температуре в течение 1 ч. Полученную реакционную смесь тщательно фильтровали через целитчерез Celite Hyflo. Фильтрат, содержащий региоизомерную смесь 4-(4-амино-1Н-имидазол-1-ил)-2-(2-метоксиэтокси) бензонитрила и 2-(4-амино-1Н-имидазол-1-ил)-4-(2-метоксиэтокси) бензонитрила, сразу же применяли на следующей стадии без перегонки.
Стадия d. К раствору (S)-1-(трет-бутоксикарбонил)пирролидин-3-карбоновой кислоты (0,42 г, 1,95 ммоль) в ТГФ (20 мл) добавляли HATU (0,89 г, 2,34 ммоль) и DIPEA (0,68 мл, 3,91 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. По каплям при комнатной температуре добавляли полученный на стадии с фильтрат, содержащий региоизомерную смесь 4-(4-амино-1Н-имидазол-1-ил)-2-(2-метоксиэтокси)бензонитрила и 2-(4-амино-1Н-имидазол-1-ил)-4-(2-метоксиэтокси)бензонитрила (0,4 г, 1,56 ммоль) в ТГФ (40 мл), и проводили перемешивание в течение 2 ч. Полученную реакционную смесь вливали в насыщенный раствор NaHCO3 (50 мл) и подвергали экстракции посредством EtOAc (3×50 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая региоизомерную смесь трет-бутил-(S)-3-((1-(4-циано-3-(2-метоксиэтокси)фенил)-1Н-имидазол-4-ил)карбамоил)пирролидин-1-карбоксилата и трет-бутил-(S)-3-((1-(2-циано-5-(2-метоксиэтокси)фенил)-1Н-имидазол-4-ил)карбамоил)пирролидин-1-карбоксилата (0,81 г, 1,78 ммоль). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. ЖХМС: Способ В, 4,02 мин, 4,09 мин, МС: ЭР+ 456,83
Стадия е, f. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для стадий c и d Примера 140, получая региоизомерную смесь (S)-1-циано-N-(1-(4-циано-3-(2-метоксиэтокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида и (S)-1-циано-N-(1-(2-циано-5-(2-метоксиэтокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида (0,47 г, 1,24 ммоль). ЖХМС: Способ С, 3,44 мин, 3,51 мин, МС: ЭР+ 381,05
Региоизомеры отделяли с помощью препаративной ВЭЖХ; подвижная фаза: (А) 100% вода (В) 100% ацетонитрил, колонка: Sunfire С18, 250×19 мм, 5 мкм, скорость потока: 15 мл/мин; с получением (S)-1-циано-N-(1-(4-циано-3-(2-метоксиэтокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида ЖХМС: Способ С, 3,44 мин, МС: ЭР+ 381,05; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,79 (s, 1 Н), 8,36 (d, J=1,6 Гц, 1 Н), 7,92 (d, J=1,6 Гц, 1 H), 7,85 (d, J=8,4 Гц, 1 H), 7,50 (d, J=1,6, 1 H), 7,41 (dd, J=2,0 Гц, 8,4 Гц, 1 H), 4,43 (t, J=4,4 Гц, 2 H), 3,75 (t, J=4,4 Гц, 2 H), 3,58-3,63 (m, 1 H), 3,46-3,50 (m, 2 H), 3,38-3,44 (m, 1 H), 3,33 (s, 3 H), 3,24-3,29 (m, 1 H), 2,13-2.17 (m, 1 H), 2,00-2,07 (m, 1 H) и (S)-1-циано-N-(1-(2-циано-5-(2-метоксиэтокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида ЖХМС: Способ С, 3,35 мин, МС: ЭР+ 381,05; 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 10,81 (s, 1 Н), 8,01 (d, J=1,6 Гц, 1 H), 7,94 (d, J=8,8 Гц, 1 H), 7,69 (d, J=1,6 Гц, 1 H), 7,28 (d, J-2,4, 1 H), 7,18 (dd, J=2,4 Гц, 8,4 Гц, 1 H), 4,28-4,31 (m, 2 Н), 3,67-3,70 (m, 2 Н), 3,58-3,67 (m, 1 Н), 3,46-3,50 (m, 2 Н), 3,37-3,44 (m, 1 Н), 3,34 (s, 3 Н), 3,24-3,17 (m, 1 Н), 2,13-2.17 (m, 1 Н), 2,00-2,07 (m, 1 Н).
Пример 245 (S)-1-циано-N-(1-(4-циано-3-(3-морфолинопропокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
Пример 246 (S)-1-циано-N-(1-(2-циано-5-(3-морфолинопропокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
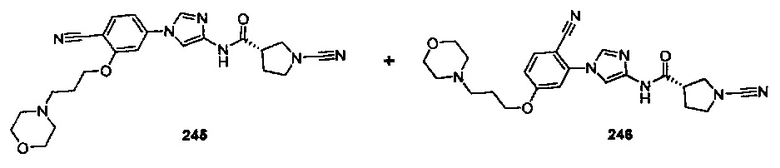
Указанное в заголовке соединение синтезировали в виде смеси региоизомеров с помощью методики, аналогичной методике, описанной для Примера 243/244, и указанную смесь разделяли после конечной стадии с помощью препаративной ВЭЖХ; подвижная фаза: (А) 0,1% МК в воде; (В) ацетонитрил: метанол (50:50), колонка: Sunfire С18, 250×19 мм, 5 мкм, скорость потока: 15 мл/мин с получением (S)-1-циано-N-(1-(4-циано-3-(3-морфолинопропокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида
ЖХМС: Способ В, 2,79 мин, МС: ЭР+ 450,94; 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 10,82(s, 1 Н), 8,36(d, J=1,6 Гц, 1 Н), 7,91(d, J=1,2 Гц, 1 Н), 7,84(d, J=8,4 Гц, 1 Н), 7,48(d, J=2,0 Гц, 1 Н), 7,40(dd, J=2,0, 8,4 Гц, 1 Н), 4,32(t, J=6,0 Гц, 2 Н), 3,58-3,62 (m, 5 Н), 3,40-3,49 (m, 4 Н), 3,23-3,35 (m, 2 Н), 2,43 (ушир. s, 4 Н), 2,12-2,17 (m, 1 Н), 1,99-2,05 (m, 1 Н), 1,94-1,97 (m, 2 Н), и (S)-1-циано-N-(1-(2-циано-5-(3-морфолинопропокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид.
ЖХМС: Способ В, 2,58 мин, МС: ЭР+ 450,74; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,79(s, 1 Н), 8,01 (d, J=1,2 Гц, 1 Н), 7,93(d, J=8,8 Гц, 1 Н), 7,68(d, J=1,2 Гц, 1 Н), 7,25(d, J=2,4 Гц, 1 Н), 7,16(dd, J=2,4, 8,8 Гц, 1 Н), 4,19(t, J=6,4 Гц, 2 Н), 3,55 - 3,58 (m, 5 Н), 3,41-3,49 (m, 4 H), 3,22-3,35 (m, 2 H), 2,36 (ушир. s, 4 Н), 2,12-2,17 (m, 1 Н), 2,00-2,07 (m, 1 Н), 1,87-1,93 (m,2H).
Пример 247 (S)-1-циано-N-(1-(4-циано-3-(((S)-тетрагидрофуран-3-ил)окси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
Пример 248 (S)-1-циано-N-(1-(2-циано-5-(((S)-тетрагидрофуран-3-ил)окси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
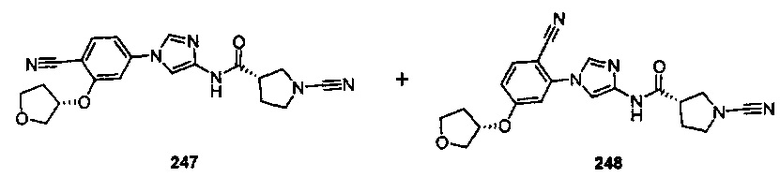
Указанное в заголовке соединение синтезировали в виде смеси региоизомеров с помощью методики, аналогичной методике, описанной для Примера 243/244, и указанную смесь разделяли после конечной стадии с помощью препаративной ВЭЖХ; подвижная фаза: (А) 100% гексан (В) IPA: метанол (50:50), колонка: YMC PACKSIL, 250×20 мм, 5 мкм, скорость потока: 15 мл/мин с получением (S)-1-циано-N-(1-(4-циано-3-(((S)-тетрагидрофуран-3-ил)окси)фенил)-1H-имидазол-4-ил)пирролидин-3-карбоксамида, ЖХМС: Способ В, 3,20 мин, МС: ЭР+ 393,59; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,79 (s, 1 Н), 8,36(d, J=1,6 Гц, 1 H), 7,92(d, J=1,6 Гц, 1 H), 7,86(d, J=8,4 Гц, 1 H), 7,48(d, J=1,6 Гц, 1 H), 7,42(dd, J=2,0, 8,4 Гц, 1 H), 5,48(s, 1 Н), 3,86-3,96 (m, 3 Н), 3,77-3,83 (m, 1 Н), 3,58-3,62 (m, 1 Н), 3,40-3,49 (m, 3 Н), 3,24-3,29 (m, 1 Н), 2,28-2,32 (m, 1 Н), 2,12-2,17 (m, 1 Н), 2,05-2,07 (m, 2 Н), и (S)-1-циано-N-(1-(2-циано-5-(((S)-тетрагидрофуран-3-ил)окси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида, ЖХМС: Способ В, 3,38 мин, МС: ЭР+ 393,19; 1H ЯМР (400 МГц, ДМСО-d6) δ ppm 10,81 (s, 1 Н), 8,00(d, J=2,0 Гц, 1 Н), 7,95(d, J=8,4 Гц, 1 Н), 7,67(d, J=2,0 Гц, 1 Н), 7,25(d, J=2,4 Гц, 1 Н), 7,17(dd, J=2,4, 8,4 Гц, 1 Н), 5,28(t, J=4,0 Гц, 1 Н), 3,83-3,91 (m, 3 Н), 3,76-3,79 (m, 1 Н), 3,58-3,62 (m, 1 Н), 3,24-3,49 (m, 4 Н), 2,27-2,33 (m, 1 Н), 2,14-2,18 (m, 1 Н), 1,98-2,05 (m, 1 Н), 1,88-1,92 (m, 1 Н).
Пример 249 (S)-1-циано-N-(1-(4-циано-3-(оксетан-3-илокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
Пример 250 (S)-1-циано-N-(1-(2-циано-5-(оксетан-3-илокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамид
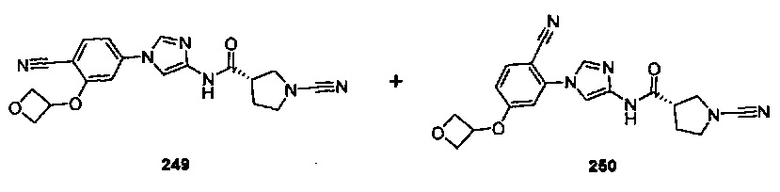
Указанное в заголовке соединение синтезировали в виде смеси региоизомеров с помощью методики, аналогичной методике, описанной для Примера 243/244 и отделяли после конечной стадии с помощью препаративной ВЭЖХ; подвижная фаза: (А) 100% вода (В) 100% ацетонитрил, колонка: X-bridge С18, 150 х 19 мм, 5 мкм, скорость потока: 13 мл/мин, с получением (S)-1-циано-N-(1-(4-циано-3-(оксетан-3-илокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида, ЖХМС: Способ В, 3,31 мин, МС: ЭР+ 379,26; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,79 (s, 1 Н), 8,34(s, 1 Н), 7,92(s, 1 Н), 7,90(s, 1 Н), 7,45(d, J=8,0 Гц, 1 Н), 7,11(s, 1 Н), 5,67(t, J=4,8 Гц, 1 Н), 5,00 (t, J=6,8 Гц, 2 Н), 4,60 (t, J=6,4 Гц, 2 Н), 3,58-3,62 (m, 1 Н), 3,4-3,49 (m, 3 Н), 3,23-3,27 (m, 1 Н), 2,12-2,16 (m, 1 Н), 2,03-2,05 (m, 1 Н), и (S)-1-циано-N-(1-(2-циано-5-(оксетан-3-илокси)фенил)-1H-имидазол-4-ил)пирролидин-3-карбоксамида, ЖХМС: Способ В, 3,24 мин, МС: ЭР+ 379,26; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,81 (s, 1 Н), 7,96-7,99 (m, 2 Н), 7,67(d, J=1,6 Гц, 1 Н), 7,13(d, J=2,4 Гц, 1 Н), 7,07(dd, J=2,4 Гц, 8,4 Гц, 1 Н), 5,5(t, J=5,2 Гц, 1 Н), 4,96 (t, J=6,8 Гц, 2 Н), 4,55-4,58 (m, 2 Н), 3,58-3,62 (m, 1 Н), 3,39-3,49 (m, 3 Н), 3,24-3,29 (m, 1 Н), 2,12-2,16 (m, 1 Н), 2,00-2,05 (m, 1 Н).
Пример 251 (S)-1-циано-N-(4-(пиразоло[1,5-а]пиримидин-7-ил)пиридин-2-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 18)
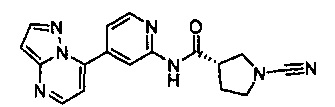
Стадия а. Раствор 2-амино-4-бромпиридина (2,50 г, 14,45 ммоль) и (S)-1-N-BOC-бета-пролина (3,72 г, 17,34 ммоль) в пиридине (50 мл) перемешивали при комнатной температуре в течение 15 мин, к реакционной смеси по каплям добавляли оксихлорид фосфора (2,67 мл, 28,90 ммоль) при 0°С и затем проводили перемешивание при комнатной температуре в течение 1 ч. Полученную реакционную смесь вливали в воду (100 мл) и подвергали экстракции посредством EtOAc (3×30 мл). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (20% EtOAc в гексане), получая трет-бутил-(S)-3-((4-бромпиридин-2-ил)карбамоил)пирролидин-1-карбоксилат (5,0 г, 13,55 ммоль). ЖХМС: Способ А, 2,39 мин, МС: ЭР+ 370,1, 372,1.
Стадия b. К раствору трет-бутил-(S)-3-((4-бромпиридин-2-ил)карбамоил)пирролидин-1-карбоксилата (5,0 г, 13,50 ммоль) в 1,4-диоксане добавляли бис(пинаколато)дибор (5,14 г, 20,26 ммоль) и ацетат калия (2,64 г, 27,01 ммоль) при комнатной температуре. Реакционную смесь дегазировали в течение 30 мин при комнатной температуре. К реакционной смеси добавляли Pd2(dba)3 (0,61 г, 0,67 ммоль) и X-phos (0,64 г, 1,35 ммоль) при комнатной температуре. Реакционную смесь нагревали при 110°С в течение 2 часов. Полученную реакционную смесь фильтровали через целит и промывали МеОН (100 мл). Органическую фазу концентрировали при пониженном давлении, получая трет-бутил-(S)-3-((4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)карбамоил)пирролидин-1-карбоксилат (11,7 г, количественный выход). Данный материал непосредственно применяли на следующей стадии без дополнительной очистки. ЖХМС: Способ А, 1,71 мин, МС: ЭР+ 336,5; 280,38
Стадия с. Получали раствор 7-хлорпиразоло[1,5-а]пиримидина (0,18 г, 1,17 ммоль), трет-бутил-(S)-3-((4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)карбамоил)пирролидин-1-карбоксилата (2,43 г, 5,86 ммоль) и К2СО3 (0,48 г, 3,52 ммоль) в ДМФА (10 мл) в стеклянном флаконе. Полученную реакционную смесь дегазировали в течение 20 мин. Добавляли Pd(dppf)Cl2 (0,06 г, 0,093 ммоль) и реакционную смесь нагревали при 80°С в течение 2 ч. Полученную реакционную смесь охлаждали до к. т. и вливали в воду (50 мл). Полученную смесь подвергали экстракции EtOAc (2×50 мл). Объединенную органическую фазу промывали водой (100 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (60% EtOAc в гексане), получая трет-бутил-(S)-3-((4-(пиразоло[1,5-а]пиримидин-7-ил)пиридин-2-ил)карбамоил)пирролидин-1-карбоксилат (0,28 г, 0,68 ммоль). ЖХМС: Способ А, 2,14 мин, МС: ЭР+ 409,28.
Стадии d,e. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для Примера 193, стадий f, g. ЖХМС: Способ В, 3,23 мин, МС: ЭР+ 334,37; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,92 (s, 1 Н), 9,28 (dd, J=7,6 Гц, 0,8 Гц, 1 Н), 8,91(s, 1 Н), 8,52 (d, J=4,8 Гц, 1 Н), 8,33 (d, J=2,4 Гц, 1 Н), 7,88 (dd, J=5,2 Гц, 1,6 Гц, 1 Н), 7,67 (d, J=7,2 Гц, 1 Н), 6,92 (dd, J=2,4 Гц, 0,8 Гц, 1 Н), 3,61-3,65 (m, 1 Н), 3,53-3,57 (m, 1 Н), 3,37-3,50 (m, 3 Н), 2,18-2,21 (m, 1 Н), 2,09-2,12 (m, 1 Н).
Соединения, приведенные в таблице 9, синтезировали с помощью методики, аналогичной методике, описанной для Примера 251.

Пример 256 (S)-1-циано-N-(1,7-нафтиридин-6-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 1)
Стадия а. К раствору 1,7-нафтиридин-6-амина (0,22 г, 0,98 ммоль) и (3S)-BOC-1-пирролидин-3-карбоновой кислоты (0,21 г, 0,98 ммоль) в ДХМ (10 мл) добавляли пиридин (0,95 мл, 11,78 ммоль) при 0°С. К реакционной смеси по каплям добавляли оксихлорид фосфора (0,75 мл, 7,86 ммоль) при 0°С и затем перемешивали при комнатной температуре в течение 20 мин. Полученную реакционную смесь вливали в насыщенный раствор NaHCO3 (50 мл) и подвергали экстракции посредством ДХМ (3×50 мл). Объединенную органическую фазу собирали, промывали солевым раствором (25 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (70-80% EtOAc в н-гексане), получая трет-бутил-(S)-3-((1,7-нафтиридин-6-ил)карбамоил)пирролидин-1-карбоксилат (0,12 г, 0,35 ммоль). ЖХМС: Способ А, 1,90 мин, МС: ЭР+ 343,34.
Стадия b, с. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для стадий c и d Примера 140. ЖХМС: Способ В, 2,72 мин, МС: ЭР+ 268,11
Соединения, приведенные в таблице 10, синтезировали с помощью методики, аналогичной методике, описанной для Примера 256.
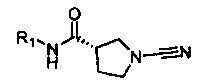
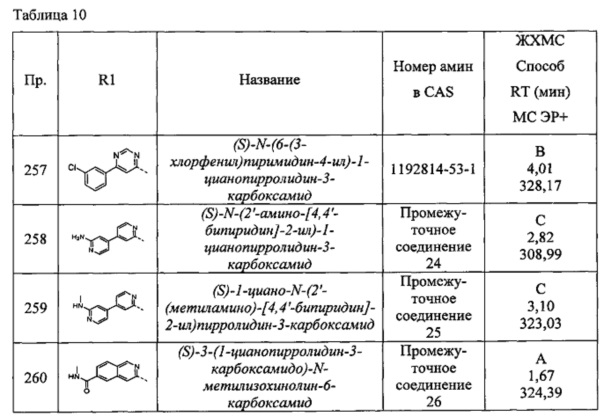
Пример 261 (S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)изохинолин-3-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 5)
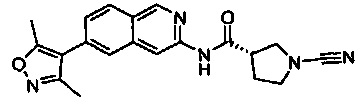
Стадия а. Данную стадию проводили с помощью методики, аналогичной методике, описанной для Примера 138, применяя 6-бромизохинолин-3-амин и пинаколовый эфир 3,5-диметилизоксазол-4-бороновой кислоты. ЖХМС: Способ А, 1,69 мин, МС: ЭР+ 240,15.
Стадия b. К раствору 6-(3,5-диметилизоксазол-4-ил)изохинолин-3-амина (0,30 г, 1,25 ммоль) и (3S)-ВОС-1-пирролидин-3-карбоновой кислоты (0,30 г, 1,38 ммоль) в ДХМ (15 мл) добавляли пиридин (1,10 мл, 13,81 ммоль) при 0°С. Реакционную смесь перемешивали в течение 20 мин при 0°С. К реакционной смеси по каплям добавляли оксихлорид фосфора (1,20 мл, 12,547 ммоль) при 0°С и затем перемешивали при комнатной температуре в течение 2 ч. Полученную реакционную смесь вливали в воду (30 мл) и подвергали экстракции посредством ДХМ (3×25 мл). Объединенную органическую фазу собирали, промывали 10% раствором лимонной кислоты (25 мл) затем промывали насыщенным раствором NaHCO3 (2×30 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (1% МеОН в ДХМ), получая трет-бутил-(S)-3-((6-(3,5-диметилизоксазол-4-ил)изохинолин-3-ил)карбамоил)пирролидин-1-карбоксилат (0,20 г, 0,46 ммоль). ЖХМС: Способ А, 2,32 мин, МС: ЭР+437,46.
Стадия с, d. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для стадий c и d Примера 138. ЖХМС: Способ В, 4,62 мин, МС: ЭР+ 362,34.
Пример 262 (S)-N-(4-(1Н-индазол-4-ил)пиридин-2-ил)-1-цианопирролидин-3-карбоксамид
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 261, применяя 1-ВОС-4-бром-1Н-индазол и пинаколовый эфир 2-аминопиридин-4-бороновой кислоты. ЖХМС: Способ С, 3,30 мин, МС: ЭР+ 332,94.
Пример 263 (S)-1-циано-N-(6-этинилизохинолин-3-ил)пирролидин-3-карбоксамид (получали в соответствии со Схемой 19)
Стадия а. К раствору 6-бромизохинолин-3-амина (0,40 г, 1,79 ммоль) и (3S)-BOC-1-пирролидин-3-карбоновой кислоты (0,42 г, 1,97 ммоль) в ДХМ (5 мл) добавляли пиридин (4 мл) при 0°С. К реакционной смеси по каплям добавляли POCl3 (0,34 г, 2,15 ммоль) при 0°С и перемешивали в течение 30 мин. Полученную реакционную смесь разбавляли водой (100 мл) и подвергали экстракции посредством EtOAc (3×100 мл). Объединенный органический слой промывали 10% водным раствором лимонной кислоты (2×50 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (2% МеОН в ДХМ), получая трет-бутил-(S)-3-((6-бромизохинолин-3-ил)карбамоил)пирролидин-1-карбоксилат (0,39 г, 0,92 ммоль). ЖХМС: Способ А, 2,63 мин, МС: ЭР+ 420,28; 422,3
Стадия b. Раствор трет-бутил-(S)-3-((6-бромизохинолин-3-ил)карбамоил)пирролидин-1-карбоксилата (0,25 г, 0,59 ммоль) и CuI (0,005 г, 0,03 ммоль) в диизопропиламине (5 мл) дегазировали при комнатной температуре в течение 15 мин. К реакционной смеси добавляли Pd(PPh3)2Cl2 (0,06 г, 0,09 ммоль) и триметилсилилацетилен (0,29 г, 2,99 ммоль) при комнатной температуре. Реакционную смесь нагревали при 120°С в течение 30 мин. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (20-25% EtOAc в гексане), получая трет-бутил-(S)-3-((6-((триметилсилил)этинил)изохинолин-3-ил)карбамоил)пирролидин-1-карбоксилат (0,31 г, количественный выход). ЖХМС: Способ А, 2,90 мин, МС: ЭР+ 438,72.
Стадия с. Раствор трет-бутил-(S)-3-((6-((триметилсилил)этинил)изохинолин-3-ил)карбамоил)пирролидин-1-карбоксилат (0,30 г, 0,68 ммоль) в МеОН (5 мл) добавляли К2СО3 (0,24 г, 1,71 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Полученную реакционную смесь фильтровали для удаления непрореагировавшего К2СО3, промывали МеОН (10 мл). Объединенный фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью флэш-хроматографии (25-30% EtOAc в гексане), получая трет-бутил-(S)-3-((6-этинилизохинолин-3-ил)карбамоил)пирролидин-1-карбоксилат (0,24 г, 0,65 ммоль). ЖХМС: Способ А, 2,46 мин, МС: ЭР+ 366,28.
Стадии d, e. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для стадий f и g Примера 193. ЖХМС: Способ С, 4,07 мин, МС: ЭР+ 291,10; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,89 (s, 1 Н), 9,17 (s, 1 Н), 8,48 (s, 1 Н), 8,05-8,08 (m, 2 Н), 7,53 (dd, J=8,40 Гц, 1,20 Гц, 1 Н), 4,47 (s, 1 Н), 3,62-3,66 (m, 1 Н), 3,48-3,56 (m, 2 Н), 3,34-3,47 (m, 2 Н), 2,16-2,24 (m, 1 Н), 2,04-2,13 (m, 1 Н).
Пример 264 (2S,3S)-1-циано-N-(6-этинилизохинолин-3-ил)-2-метилпирролидин-3-карбоксамид
Синтез проводили с помощью методики, аналогичной методике, описанной для Примера 263, применяя Промежуточное соединение 1. ЖХМС: Способ С, 4,22 мин, МС: ЭР+ 305,14; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 10,87 (s, 1 Н), 9,17 (s, 1 Н), 8,49 (s, 1 Н), 8,05-8,09 (m, 2 Н), 7,53 (dd, J=8,40 Гц, 1,20 Гц, 1 Н), 4,48 (s, 1 Н), 3,97-4,04 (m, 1 Н), 3,62-3,68 (m, 1 Н), 3,36-3,45 (m, 2 Н), 2,17-2,25 (m, 1 Н), 1,99-2,07 (m, 1 Н), 1,15 (d, J=6,40 Гц, 3 Н).
Пример 265 (S)-3-(1-цианопирролидин-3-карбоксамидо)изохинолин-6-карбоксамид (получали в соответствии со Схемой 20)
Стадия а. К раствору метил-3-((трет-бутоксикарбонил)амино)изохинолин-6-карбоксилата (полученного в соответствии со стадиями а-b способа, описанного для синтеза Промежуточного соединения 26, 0,36 г, 1,19 ммоль) в ДХМ (15 мл) добавляли ТФУ (3,6 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Полученную реакционную смесь концентрировали при пониженном давлении. Полученный остаток растирали Et2O (10 мл) и сушили с получением соли ТФУ и метил-3-аминоизохинолин-6-карбоксилата (0,37 г, 1,15 ммоль). ЖХМС: Способ А, 1,66 мин, МС: ЭР+ 203,18. Данный материал непосредственно применяли на следующей стадии без дополнительной очистки.
Стадия b. К раствору соли ТФУ и метил-3-аминоизохинолин-6-карбоксилата (0,35 г, 1,11 ммоль) и (3S)-ВОС-1-пирролидин-3-карбоксилата в ДХМ (20 мл) добавляли пиридин (1,34 мл, 16,61 ммоль) при 0°С. К реакционной смеси по каплям добавляли оксихлорид фосфора (1,06 мл, 11,07 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 20 мин. Полученную реакционную смесь вливали в насыщенный раствор NaHCO3 (50 мл) и подвергали экстракции посредством ДХМ (2×50 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (2% МеОН в ДХМ), получая метил (S)-3-(1-(трет-бутоксикарбонил)пирролидин-3-карбоксамидо)изохинолин-6-карбоксилат (0,27 г, 0,68 ммоль). ЖХМС: Способ А, 2,26 мин, МС: ЭР+ 400,42.
Стадия с. К раствору метил-(S)-3-(1-(трет-бутоксикарбонил)пирролидин-3-карбоксамидо)изохинолин-6-карбоксилата (0,25 г, 0,63 ммоль) в МеОН (10 мл) добавляли раствор LiOH (0,26 г, 6,26 ммоль) в 5 мл воды при комнатной температуре. Реакционную смесь нагревали при 60°С в течение 3 ч. Полученной реакционной смеси давали возможность остыть до к. т. Полученную реакционную смесь концентрировали при пониженном давлении, удаляя органический растворитель. Полученный водный слой вливали в раствор лимонной кислоты (50 мл) при постоянном перемешивании. Полученную смесь подвергали экстракции EtOAc (3×50 мл). Объединенную органическую фазу собирали сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая (S)-3-(1-(трет-бутоксикарбонил)пирролидин-3-карбоксамидо)изохинолин-6-карбоновую кислоту (0,21 г, 0,54 ммоль). ЖХМС: Способ А, 2,04 мин, МС: ЭР+ 386,5.
Стадия d. Получали смесь (S)-3-(1-(трет-бутоксикарбонил)пирролидин-3-карбоксамидо)изохинолин-6-карбоновой кислоты (0,2 г, 0,52 ммоль), HATU (0,30 г, 0,78 ммоль) и DIPEA (0,18 мл, 1,04 ммоль) в ТГФ (5 мл) при 0°С. Данную реакционную смесь перемешивали при комнатной температуре в течение 0,5 ч. К реакционной смеси добавляли бикарбонат аммония (0,08 г, 1,04 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 42 ч. Полученную реакционную смесь вливали в насыщенный раствор NaHCO3 (40 мл) и подвергали экстракции посредством EtOAc (2×50 мл). Объединенную органическую фазу собирали, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии (3% МеОН в ДХМ), получая трет-бутил-(S)-3-((6-карбамоилизохинолин-3-ил)карбамоил)пирролидин-1-карбоксилат (0,065 г, 0,17 ммоль). ЖХМС: Способ А, 1,92 мин, МС: ЭР+ 385,55.
Стадии е, f. Указанное в заголовке соединение синтезировали из указанного выше промежуточного соединения с помощью методики, аналогичной методике, описанной для стадий f и g Примера 193. ЖХМС: Способ В, 2,73 мин, МС: ЭР+ 310,5.
Пример 266 (2S,3S)-N-(6-(1Н-пиразол-4-ил)изохинолин-3-ил)-1-циано-2-метилпирролидин-3-карбоксамид
Синтез проводили с помощью методики, аналогичной методике, описанной для стадий f и g Примера 193, применяя Промежуточное соединение 27. ЖХМС: Способ С, 3,29 мин, МС: ЭР+ 347,58; 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 13,12 (ушир. s, 1 Н), 10,75 (s, 1 Н), 9,05 (s, 1 Н), 8,43-8,47 (m, 2 Н), 8,13-8,16 (m, 2 Н), 8,02 (d, J=8,40 Гц, 1 Н), 7,83 (d, J=8,40 Гц, 1 Н), 3,97-4,04 (m, 1 Н), 3,63-3,68 (m, 1 Н), 3,34-3,45 (m, 2 Н), 2,18-2,27 (m, 1 Н), 1,99-2,07 (m, 1 Н), 1,16 (d, J=6,40 Гц, 3 Н).
Соединения, приведенные в таблице 11, синтезировали с помощью методики, аналогичной методике, описанной для Примера 1.
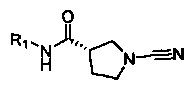
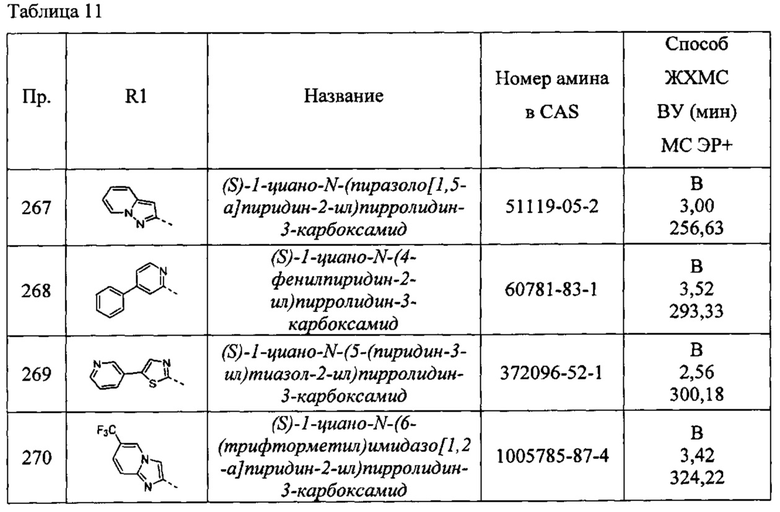
Соединения, приведенные в таблице 12, синтезировали с помощью методики, аналогичной методике, описанной для Примера 1, применяя 2-амино-5-фенилтиазол.
Соединения, приведенные в таблице 13, синтезировали с помощью методики, аналогичной методике, описанной для Примера 109.

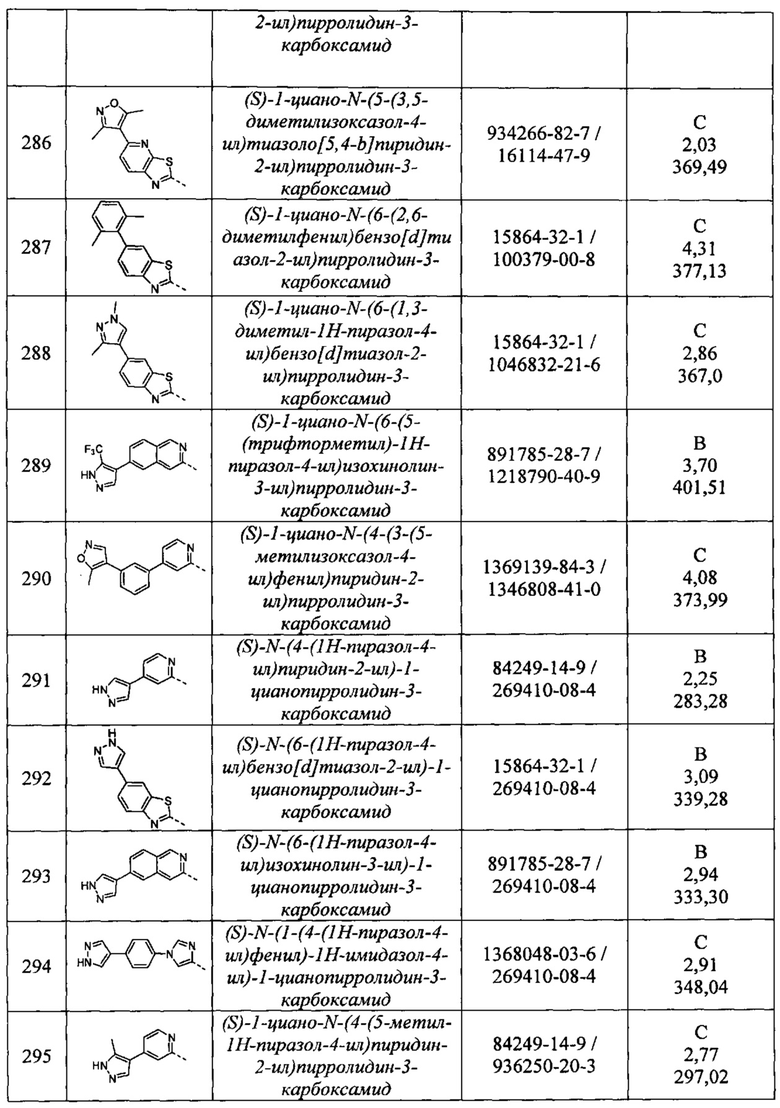

Соединения, приведенные в таблице 14, синтезировали с помощью методики, аналогичной методике, описанной для Примера 150.
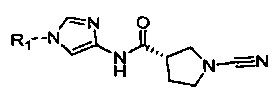

Соединения, приведенные в таблице 15, синтезировали с помощью методики, аналогичной методике, описанной для Примера 174.
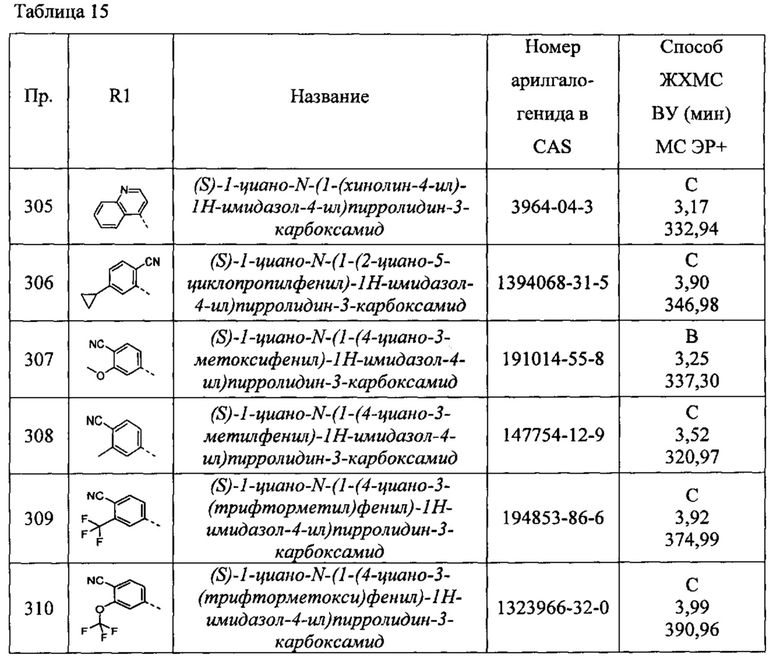
Биологическая активность соединений согласно настоящему изобретению
Сокращения:
TAMRA карбокситетраметилродамин
ПЦР полимеразная цепная реакция
ФСБ фосфатно-солевой буферный раствор
ЭДТА этилендиаминтетрауксусная кислота
Tris 2-амино-2-(гидроксиметил)-1,3-пропандиол
NP-40 Нонидет Р-40, октилфеноксиполиэтоксиэтанол
БСА бычий сывороточный альбумин
Анализ ингибирования UCHL1 in vitro
Экспрессия и очистка UCHL1
Конструкцию UCHL1 амплифицировали посредством ПЦР и клонировали в вектор pFLAG-CMV-6a (Sigma-Aldrich) с N-концевой меткой FLAG. Клетки НЕК293Т трансфицировали FLAG-UCHL1, применяя реагент для трансфекции TransIT-LT1 (Mirus) в соответствии с инструкциями производителя. Клетки собирали через 40 часов после трансфекции. Клетки однократно промывали посредством ФСБ и счищали в лизирующий буфер (50 мМ Tris, рН 7,5, 150 мМ NaCl, 3 мМ ЭДТА, 0,5% NP40, 10% глицерола, 5 мМ бета-меркаптоэтанола, ингибиторы протеазы (complete mini, Roche) и ингибиторы фосфатазы (PhosSTOP mini, Roche). Лизаты инкубировали в течение 30 мин на льду и центрифугировали при 1200 об/мин в течение 10 мин при 4°С. Растворимый супернатант добавляли к смоле с аффинностью к FLAG (аффинный гель EZview Rad ANTI-FLAG M2, Sigma-Aldrich), уравновешенной буфером с низким содержанием соли (20 мМ Tris, рН 7,5, 150 мМ NaCl, 0,5 мМ ЭДТА, 5 мМ бета-меркаптоэтанола), и инкубировали при 4°С в течение 3 часов при вращении. Смолу отделяли центрифугированием при 2000 об/мин в течение 2 мин и полученную надосадочную жидкость удаляли. Смолу промывали два раза буфером с низким содержанием соли и один раз буфером с высоким содержанием соли (20 мМ Tris, рН 7,5, 500 мМ NaCl, 0,5 мМ ЭДТА, 5 мМ бета-меркаптоэтанола, ингибиторы протеазы (complete mini, Roche) и ингибиторы фосфатазы (PhosSTOP mini, Roche). Для элюирования связанной UCHL1 к указанной смоле добавляли элюирующий буфер (10 мМ Tris, рН 7,5, 150 мМ NaCl, 0,5 мМ ЭДТА, 10% глицерина, 0,5% NP40,5 мМ бета-меркаптоэтанола, 0,15 мг/мл 3Х FLAG пептида (Sigma-Aldrich)) и проводили инкубирование при 4°С в течение 2,5 часов при вращении. Смолу отделяли центрифугированием при 4000 об/мин в течение 30 секунд и супернатант, содержащий очищенный комплекс FLAG-UCHL1, удаляли и хранили при -80°С.
Биохимический кинетический анализ UCHL1
Реакции проводили в двух повторностях в черных 384-луночных планшетах (малого объема, Greiner 784076) при конечном объеме реакционной среды 21 мкл. UCHL1 разводили в реакционном буфере (20 мМ Tris, рН 7,5, 100 мМ NaCl, 0,05% Tween 20, 0,5 мг/мл БСА, 5 мМ бета-меркаптоэтанола) до эквивалента 0, 0,01, 0,05, 0,1, 0,5, и 1 мкл на лунку. Буфер оптимизировали в отношении температуры, рН, восстанавливающего агента, соли, времени инкубирования и детергента. Реакции инициировали путем добавления 50 нМ TAMRA-меченного пептида, связанного с убиквитином посредством изопептидной связи, в качестве субстрата для поляризации флуоресценции. Реакционные смеси инкубировали при комнатной температуре и проводили считывание каждые 2 мин в течение 120 мин. Считывание осуществляли на приборе Pherastar Plus (ВМГ Labtech). λ возбуждения 540 нм; λ эмиссии 590 нм.
Биохимический анализ IC50 для UCHL1
В 96-луночном полипропиленовом планшете с V-образным дном (Greiner #651201) готовили разведения в 21 раз до конечной концентрации (от 2100 мкМ до конечной концентрации 100 мкМ) в 50% ДМСО. Типичные серии разведения с получением 8 точек содержали конечные концентрации 100, 30, 10, 3, 1, 0,3, 0,1, 0,03 мкМ. Реакции осуществляли в двух повторностях в черных 384-луночных планшетах (малого объема, Greiner 784076) при конечном объеме реакционной среды 21 мкл. В планшет добавляли либо 1 мкл 50% ДМСО, либо разбавленное соединение. UCHL1 разводили в реакционном буфере (20 мМ Tris, рН 7,5, 100 мМ NaCl, 0,05% Tween 20, 0,5 мг/мл БСА, 5 мМ бета-меркаптоэтанола) до эквивалента 0,05 мкл на лунку и 10 мкл разбавленной UCHL1 добавляли к соединению. Фермент и соединение инкубировали в течение 30 мин при комнатной температуре. Реакции инициировали путем добавления 50 нМ TAMRA-меченного пептида, связанного с убиквитином посредством изопептидной связи, в качестве субстрата для поляризации флуоресценции. Считывание реакционных смесей проводили сразу же после добавления субстрата и после инкубирования в течение 2 ч при комнатной температуре. Считывание проводили на приборе Pherastar Plus (ВМГ Labtech). λ возбуждения 540 нм; λ эмиссии 590 нм.
Активность иллюстративных соединений в биохимическом анализе IC50 UCHL1
Диапазоны
А<1 мкМ;
1<В<10 мкМ;
10<С<30 мкМ
| название | год | авторы | номер документа |
|---|---|---|---|
| Новые соединения | 2016 |
|
RU2734256C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ МОДУЛЯЦИИ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ, СПОСОБ ПОЛУЧЕНИЯ ЭТИХ СОЕДИНЕНИЙ | 2000 |
|
RU2255937C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2015 |
|
RU2733400C2 |
| Бициклические азотсодержащие соединения как агонисты М1 мускариновых рецепторов | 2015 |
|
RU2685230C2 |
| N-ацил-N'-(пиридин-2-ил) карбамиды и их аналоги, проявляющие противораковую и антипролиферативную активность | 2014 |
|
RU2769607C2 |
| Бициклические азотсодержащие соединения как агонисты М1 мускариновых рецепторов | 2019 |
|
RU2811601C1 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3K И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2600927C2 |
| ПРОИЗВОДНЫЕ 3-АМИНОПИРРОЛИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРОВ ХЕМОКИНОВ | 2003 |
|
RU2355679C2 |
| 6,7-ДИГИДРО-5H-ПИРИДО[2,3-C]ПИРИДАЗИНОВЫЕ ПРОИЗВОДНЫЕ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛКОВ BCLXL И ПРОАПОПТОТИЧЕСКИХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ | 2020 |
|
RU2832191C2 |
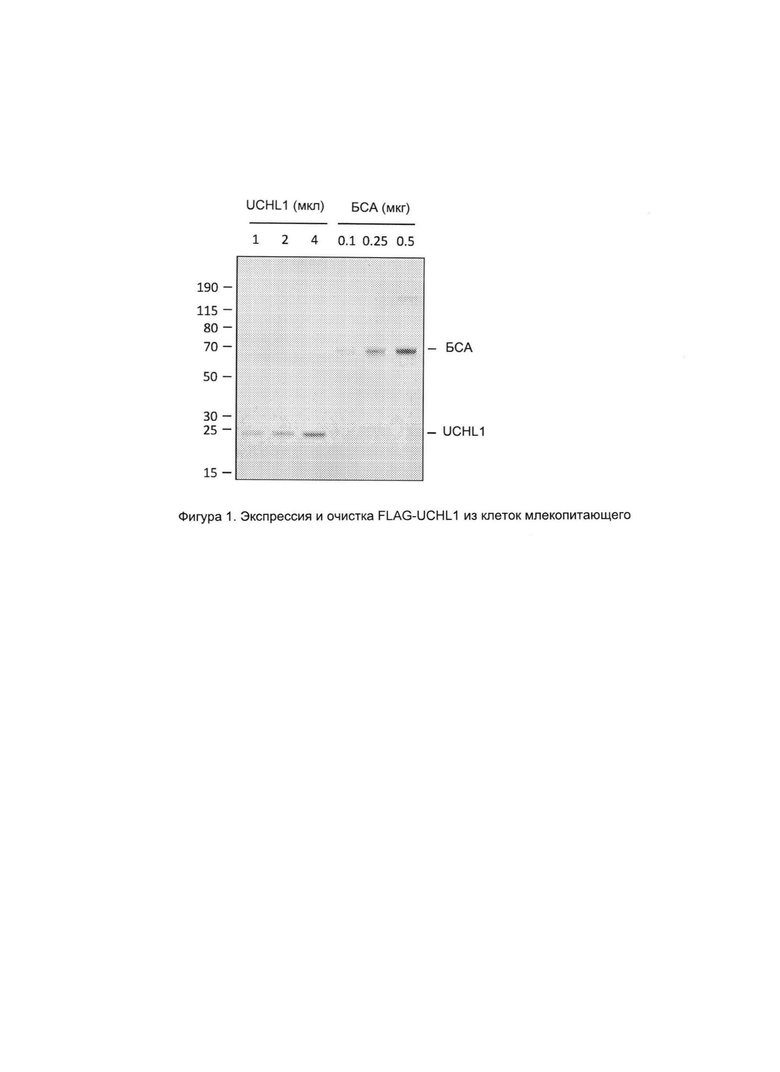
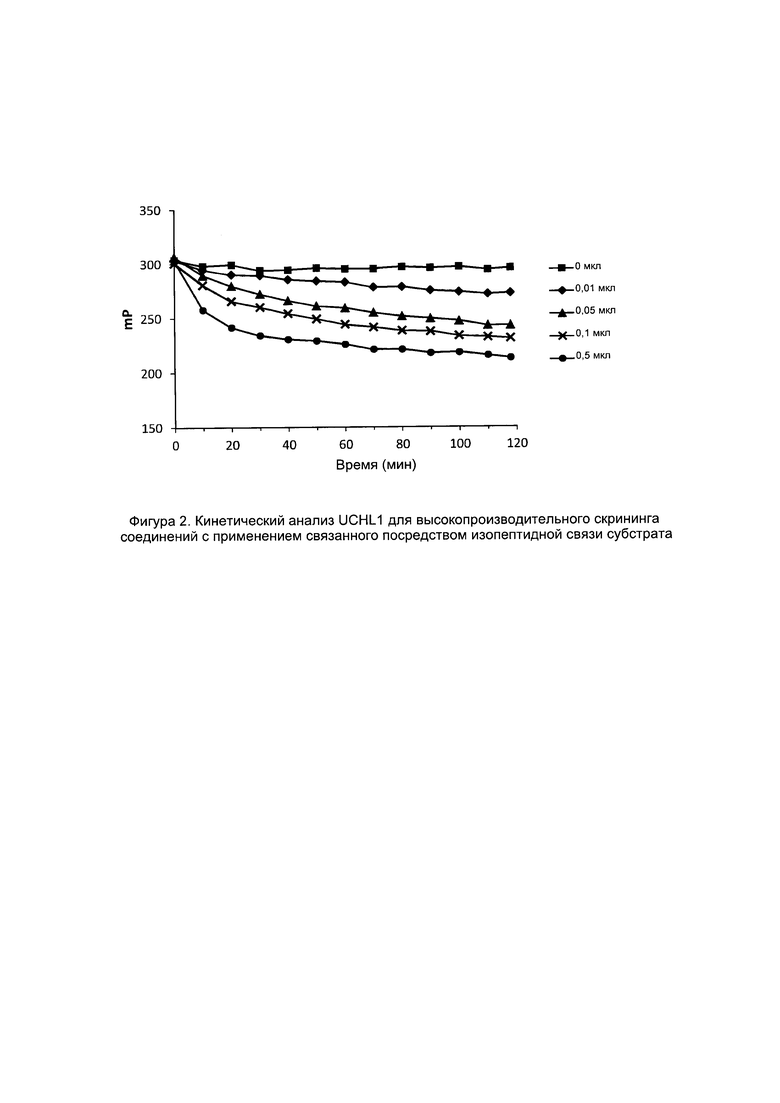
Настоящее изобретение относится к соединениям формулы (I), где R1 - R8 такие, как определено в настоящем описании, или их фармацевтически приемлемым солям, в качестве ингибиторов убиквитин-C-концевой гидролазы L1 (UCHL1). Настоящее изобретение также относится к применению соединений для лечения рака, который связан с активностью UCHL1 и других состояний, связанных с UCHL1. 2 н. и 16 з.п. ф-лы, 2 ил., 15 табл., 266 пр.
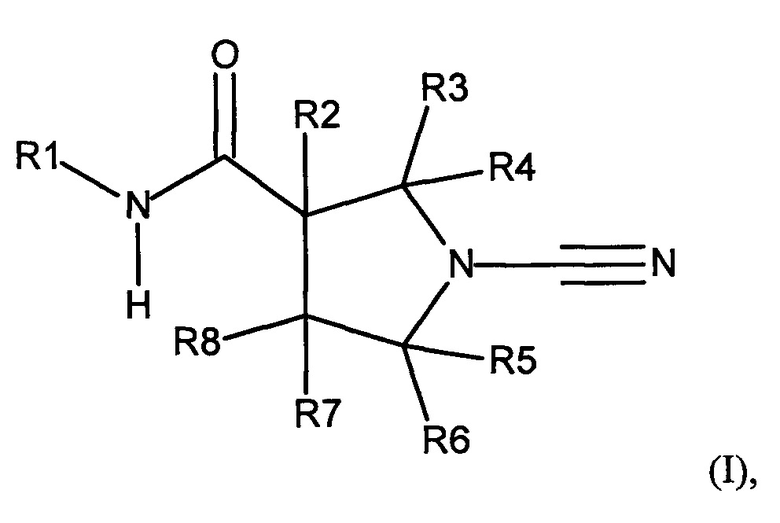
1. Соединение формулы (I):
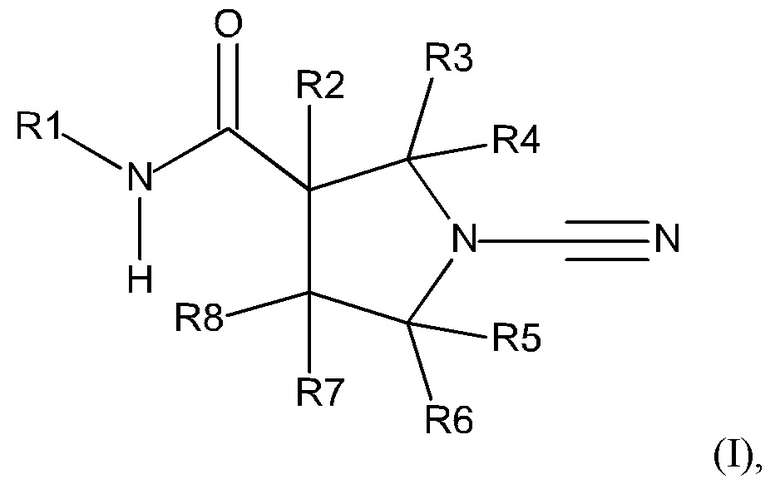
его таутомер или фармацевтически приемлемая соль указанного соединения или таутомера, где:
R1 представляет собой 5-10-членное моноциклическое или бициклическое гетероарильное кольцо, которое содержит от одного до трех гетероатомов, независимо выбранных из азота, кислорода и серы;
R2 выбран из водорода, циано, C1-С3 алкила и С1-С3 алкокси, где алкильная группа необязательно замещена С1-С3 алкокси; и R8 представляет собой водород; или R2 связан с R8 с образованием С3-С4 циклоалкильного кольца;
R3 выбран из водорода и С1-С3 алкила;
R4 представляет собой водород;
R5 выбран из водорода и С1-С3 алкила;
R6 представляет собой водород; и R7 выбран из водорода, С1-С3 алкила, CF3, фенила, пиридила и пиримидила; или R6 связан с R7 с образованием С3-С4 циклоалкильного кольца;
где R1 может быть необязательно замещено от одного до четырех Q1-(R9)n;
n равен 0 или 1;
Q1 представляет собой атом водорода, атом галогена, циано, ковалентную связь, -NR10-, атом кислорода, оксо, нитро, -S(O)m-, C1-С6 алкокси, C1-С6 галогеналкокси, -СО-, -SO2R11, -NR11R12, -CONR11R12, -CO2R11, -SO2NR11R12, -C(O)R11, C1-C6 алкилен, -C2-C6 алкенилен, -C1-С6 алкильную или C1-C2 галогеналкильную группу;
m равен 0, 1 или 2;
R10 выбран из водорода и С1-С4 алкила;
каждый из R11 и R12 независимо представляет собой атом водорода, C1-С6 алкил или C1-С6 алкиленовую группу, где алкил R11 необязательно замещен ОСН3 или N(CH3)2;
R9 представляет собой 3-10-членное моноциклическое или бициклическое гетероциклильное, гетероарильное, арильное или циклоалкильное кольцо; где гетероциклильное и гетероарильное кольца содержат от одного до трех гетероатомов, независимо выбранных из азота, кислорода и серы;
где R9 может быть необязательно замещено от одного до трех заместителей, выбранных из галогена, C1-С6 галогеналкила, C1-С6 алкокси, C1-С6 галогеналкокси, C1-С6 алкила, С2-С6 алкинила, оксо, циано, нитро, -Q2-NR13R14, -Q2-NR13COR14, -Q2-COR13, -Q2-SO2R13, -Q2-CONR13, Q2-CONR13R14, -Q2-CO2R13, -Q2-SO2NR13R14 и -Q2-NR13SO2R14; или R9 может быть замещено 3-10-членным моноциклическим гетероциклильным, гетероарильным, арильным или циклоалкильным кольцом, присоединенным либо непосредственно, либо посредством связующей группы, выбранной из кислорода, карбонила, C1-С6 алкилена и C1-С6 алкиленокси; где гетероциклильное и гетероарильное кольца содержат от одного до трех гетероатомов, независимо выбранных из азота, кислорода и серы; и где указанные алкильная, алкокси и гетероарильная группы могут быть необязательно замещены С1-С3 алкилом, С1-С3 алкокси и С1-С3 алкокси-С1-С3 алкилом;
Q2 представляет собой ковалентную связь или C1-С6 алкилен; и
каждый из R13 и R14 независимо представляет собой водород или C1-С6 алкил, где указанная алкильная группа может быть необязательно замещена галогеном или C1-С3 алкокси.
2. Соединение по п. 1, отличающееся тем, что гетероарильное кольцо R1 представляет собой 5-6-членное гетероарильное кольцо или 9-10-членное бициклическое гетероарильное кольцо, где R1 может быть необязательно замещено как определено в п. 1.
3. Соединение по п. 2, отличающееся тем, что кольцо R1 выбрано из тиазолила, пиридинила, изоксазолила, тиадиазолила, индазолила, имидазолила, бензотиазолила, бензоимидазолила, пиразолила, пиридазинила, пиримидинила, нафтиридинила, пиразинила, изохинолинила, хинолинила, тетрагидротиазолопиридинила, имидазопиридинила, триазолила, пиразолопиридинила, тетрагидропиразоло[1,5-а]пиразинила, тетрагидротиазолоазепинила и тиазолопиридинила, где R1 может быть необязательно замещено как определено в п. 1.
4. Соединение по любому из пп. 1-3, отличающееся тем, что для каждой группировки Q1-(R9)n, когда n равен 0, каждый Q1 заместитель может быть независимо выбран из атома галогена, С1-С4 алкила, С1-С2 галогеналкила, С1-С2 алкокси, -COR11, -SO2R11, циано, C(O)NR11R12 и С1-С2 галогеналкокси, и
тем, что для каждой группировки Q1-(R9)n, когда n равен 1, каждый Q1 заместитель может быть независимо выбран из ковалентной связи, -СО-, C1-С6 алкилена, С2-С6 алкениленовой группы и -NR10-.
5. Соединение по любому из пп. 1-4, отличающееся тем, что R2 представляет собой водород, циано, метил или метокси, где метильная группа необязательно замещена метокси.
6. Соединение по любому из пп. 1-5, отличающееся тем, что R7 представляет собой водород, метил, CF3, фенил, пиридил или пиримидил.
7. Соединение по любому из пп. 1-6, отличающееся тем, что каждый из R2, R3, R4, R5, R6, R7 и R8 независимо представляет собой водород.
8. Соединение по любому из пп. 1-7, отличающееся тем, что R3 представляет собой метил.
9. Соединение по любому из пп. 1-8, отличающееся тем, что кольцо R9 представляет собой 5-6-членное моноциклическое или 9-10-членное бициклическое гетероциклильное, арильное, гетероарильное или циклоалкильное кольцо, где R9 может быть необязательно замещено как определено в п. 1.
10. Соединение по п. 9, отличающееся тем, что кольцо R9 выбрано из морфолинила, пиперидинила, пирролидинила, диазепанила, пиперазинила, пиридазинила, пиразинила, пиразолила, циклопропила, циклогексила, циклопентила, пиридинила, имидазолила, индолинила, изоиндолинила, пиримидинила, изоксазолила, дигидроинденила, дигидроизохинолинила, тетрагидропиранила, фенила, оксадиазолила, триазолила, изохинолинила, индазолила, пиразолопиридинила, пиразолопиримидинила, имидазолпиридинила, имидазопиримидинила, имидазопиразинила, оксазолила и хинолинила, где R9 может быть необязательно замещено как определено в п. 1.
11. Соединение по любому из пп. 1-10, имеющее формулу (IV):
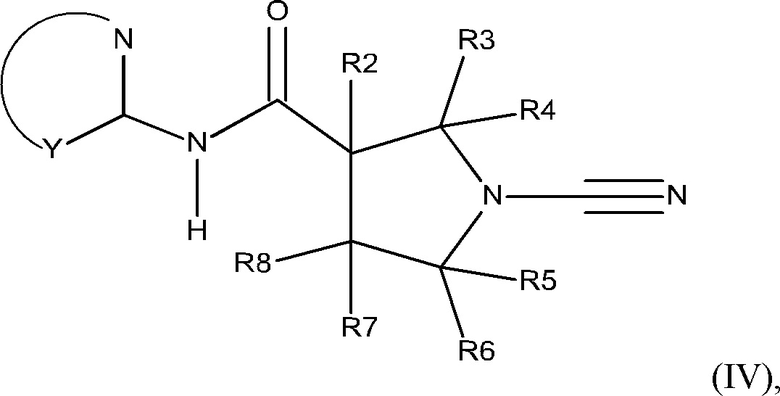
его таутомер или его фармацевтически приемлемая соль указанного соединения или таутомера, где:
Y представляет собой остальные атомы 5-10-членного моноциклического или бициклического гетероарильного кольца, которое содержит от одного до трех гетероатомов, независимо выбранных из азота, кислорода и серы, которое может быть необязательно замещенным, где указанные необязательные заместители являются такими, как определено по любому из пп. 1-10.
12. Соединение по п. 1, которое выбрано из группы, состоящей из:
(S)-1-циано-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамида;
1-циано-N-(5-(3-фторфенил)тиазол-2-ил)пирролидин-3-карбоксамида;
1-циано-N-(5-(2-фторфенил)тиазол-2-ил)пирролидин-3-карбоксамида;
N-(5-(4-хлорфенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
N-(5-(3-хлорфенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
N-(5-(2-хлорфенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
1-циано-N-(5-метилтиазол-2-ил)пирролидин-3-карбоксамида;
N-(5-(трет-бутил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
1-циано-N-(4-фенилтиазол-2-ил)пирролидин-3-карбоксамида;
1-циано-N-(2-фенилтиазол-5-ил)пирролидин-3-карбоксамида;
1-циано-N-(5-фенил-1,3,4-тиадиазол-2-ил)пирролидин-3-карбоксамида;
1-циано-N-(5-этил-1,3,4-тиадиазол-2-ил)пирролидин-3-карбоксамида;
1-циано-N-(3-фенилизоксазол-5-ил)пирролидин-3-карбоксамида;
1-циано-N-(3-(4-метоксифенил)изоксазол-5-ил)пирролидин-3-карбоксамида;
1-циано-N-(5-фенилизоксазол-3-ил)пирролидин-3-карбоксамида;
N-(5-(трет-бутил)изоксазол-3-ил)-1-цианопирролидин-3-карбоксамида;
N-(3-(трет-бутил)-1Н-пиразол-5-ил)-1-цианопирролидин-3-карбоксамида;
N-(бензо[d]тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
1-циано-N-(6-метилбензо[d]тиазол-2-ил)пирролидин-3-карбоксамида;
1-циано-N-(6-(трифторметил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамида;
1-циано-N-(6-метоксибензо[d]тиазол-2-ил)пирролидин-3-карбоксамида;
N-(6-бромбензо[d]тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
N-(1Н-бензо[d]имидазол-2-ил)-1-цианопирролидин-3-карбоксамида;
1-циано-N-(пиридин-2-ил)пирролидин-3-карбоксамида;
N-(5-хлорпиридин-2-ил)-1-цианопирролидин-3-карбоксамида;
1-циано-N-(5-метилпиридин-2-ил)пирролидин-3-карбоксамида;
1-циано-N-(5-метоксипиридин-2-ил)пирролидин-3-карбоксамида;
1-циано-N-(5-морфолинопиридин-2-ил)пирролидин-3-карбоксамида;
1-циано-N-(5-(пиперидин-1-ил)пиридин-2-ил)пирролидин-3-карбоксамида;
N-(5-(1Н-имидазол-1-ил)пиридин-2-ил)-1-цианопирролидин-3-карбоксамида;
1-циано-N-(4-фенилпиридин-2-ил)пирролидин-3-карбоксамида;
N-([2,3'-бипиридин]-6'-ил)-1-цианопирролидин-3-карбоксамида;
N-([3,3'-бипиридин]-6-ил)-1-цианопирролидин-3-карбоксамида;
N-([3,4'-бипиридин]-6-ил)-1-цианопирролидин-3-карбоксамида;
1-циано-N-(6-фенилпиридин-3-ил)пирролидин-3-карбоксамида;
1-циано-N-(6-фенилпиридазин-3-ил)пирролидин-3-карбоксамида;
1-циано-N-(2-фенилпиримидин-5-ил)пирролидин-3-карбоксамида;
1-циано-N-(5-циклогексилпиридин-2-ил)пирролидин-3-карбоксамида;
N-(1-бензил-1H-индазол-5-ил)-1-цианопирролидин-3-карбоксамида
1-циано-N-(1-пропил-1Н-бензо[d]имидазол-5-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-фенил-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-фенил-1Н-пиразол-3-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразол-3-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-фенил-1Н-1,2,3-триазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-фенил-1Н-пиразол-3-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(4-метилбензо[d]тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(7-метилбензо[d]тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-N-(4-бромбензо[d]тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-N-(7-бромимидазо[1,2-а]пиридин-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-N-(6-бромимидазо[1,2-а]пиридин-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(пиразоло[1,5-а]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(4-метоксипиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-фенилпиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-фенилпиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1,8-нафтиридин-2-ил)пирролидин-3-карбоксамида;
(S)-N-(5-бензилтиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(изохинолин-3-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(тетрагидро-2Н-пиран-4-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-метил-5-фенил-1Н-пиразол-3-ил)пирролидин-3-карбоксамида;
1-циано-N-(5-(4-метоксипиперидин-1-ил)пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-N-(6-(1H-1,2,3-триазол-1-ил)бензо[d]тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-N-(6-(2H-1,2,3-триазол-2-ил)бензо[d]тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(4-(метилкарбамоил)фенил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(4-метил-5-(морфолинометил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(4-фторфенил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(3,4-дифторфенил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(4-(трифторметил)фенил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(пиридин-4-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(пиридин-2-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(R)-1-циано-N-(5-фенилпиридин-2-ил)пирролидин-3-карбоксамида;
(2S,3S)-1-циано-2-метил-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамида;
(2S,3S)-1-циано-N-(5-(4-фторфенил)тиазол-2-ил)-2-метилпирролидин-3-карбоксамида;
(2S,3S)-1-циано-2-метил-N-(1-фенил-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(2S,3S)-1-циано-2-метил-N-(5-фенил-1H-пиразол-3-ил)пирролидин-3-карбоксамида;
(2S,3S)-1-циано-2-метил-N-(5-(тетрагидро-2Н-пиран-4-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(2S,3S)-N-(5-(2-хлорфенил)тиазол-2-ил)-1-циано-2-метилпирролидин-3-карбоксамида;
1-циано-3-метил-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамида;
1-циано-3-метил-N-(1-фенил-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
1-циано-3-(метоксиметил)-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамида;
1,3-дициано-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамида;
(3S,4S)-1-циано-4-метил-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамида;
(3S,4S)-1-циано-4-метил-N-(5-метилтиазол-2-ил)пирролидин-3-карбоксамида;
(3S,4S)-N-(5-(2-хлорфенил)тиазол-2-ил)-1-циано-4-метилпирролидин-3-карбоксамида;
(3S,4S)-1-циано-4-метил-N-(1-фенил-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(3S,4S)-1-циано-4-этил-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамида;
(3S,4S)-1-циано-4-этил-N-(1-фенил-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
1-циано-5-метил-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамида;
1-циано-5-метил-N-(1-фенил-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
1-циано-5-метил-N-(5-фенил-1Н-пиразол-3-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-морфолинотиазол-2-ил)пирролидин-3-карбоксамида;
1-циано-N-(5-(4-метилпиперазин-1-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
N-(5-(2-(ацетамидометил)пиперидин-1-ил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
1-циано-N-(5-(метил(фенил)амино)тиазол-2-ил)пирролидин-3-карбоксамида;
1-циано-N-(5-(индолин-1-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(пиперидин-1-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(изоиндолин-2-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(3,4-дигидроизохинолин-2(1Н)-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-((R)-2-(метоксиметил)пирролидин-1-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-((S)-2-(метоксиметил)пирролидин-1-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(5-оксо-1,4-диазепан-1-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(R)-1-циано-N-(5-морфолинотиазол-2-ил)пирролидин-3-карбоксамида;
(R)-1-циано-N-(5-(пиперидин-1-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(3S,4S)-1-циано-4-метил-N-(5-(пиперидин-1-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
1-циано-5-метил-N-(5-(пиперидин-1-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(пирролидин-1-ил)пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(4-(пирролидин-1-ил)пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(пирролидин-1-ил)пиразин-2-ил)пирролидин-3-карбоксамида;
N-(5-(2-аминофенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
N-(5-(3-аминофенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
N-(5-(4-аминофенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
1-циано-N-(5-(пиридин-3-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(Е)-1-циано-N-(5-(2-циклопропилвинил)тиазол-2-ил)пирролидин-3-карбоксамида;
N-(5-(4-ацетамидофенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
N-(5-(2-ацетамидофенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
1-циано-N-(5-(3-(метилсульфонамидо)фенил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(3-цианофенил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(4-цианофенил)тиазол-2-ил)пирролидин-3-карбоксамида;
1-циано-N-(5-(3,5-диметилизоксазол-4-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(4-метоксифенил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-фенилбензо[d]тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(1-метил-1Н-пиразол-5-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(1-метил-1Н-пиразол-4-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(3,5-диметил-1Н-пиразол-4-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(5-метилизоксазол-4-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(7-(3,5-диметилизоксазол-4-ил)имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-циклопропилимидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамида;
1-циано-N-(6-циклопропилбензо[d]тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(3,6-диметоксипиридазин-4-ил)имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(3,6-диметоксипиридазин-4-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамида;
(2S,3S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)бензо[d]тиазол-2-ил)-2-метилпирролидин-3-карбоксамида;
1-циано-N-(5-(п-толил)пиридин-2-ил)пирролидин-3-карбоксамида;
1-циано-N-(5-(м-толил)пиридин-2-ил)пирролидин-3-карбоксамида;
1-циано-N-(5-(о-толил)пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)-5-метилимидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)-7-метилбензо[d]тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(7-метил-6-(1-метил-1Н-пиразол-4-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамида;
1-циано-N-(5-(морфолинометил)тиазол-2-ил)пирролидин-3-карбоксамида;
1-циано-N-(5-(пирролидин-1-илметил)тиазол-2-ил)пирролидин-3-карбоксамида;
(3S)-1-циано-N-(5-((2,6-диметилморфолино)метил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(((R)-2-(метоксиметил)пирролидин-1-ил)метил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(((S)-2-(метоксиметил)пирролидин-1-ил)метил)тиазол-2-ил)пирролидин-3-карбоксамида;
(2S,3S)-1-циано-N-(5-(((R)-2-(метоксиметил)пирролидин-1-ил)метил)тиазол-2-ил)-2-метилпирролидин-3-карбоксамида;
(S)-N-(1-бензил-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(1-фенетил-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-изобутил-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(2S,3S)-N-(1-бензил-1Н-имидазол-4-ил)-1-циано-2-метилпирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(4-фторфенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(3-фторфенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(2-фторфенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(4-(трифторметил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(3-(трифторметил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(4-(метилкарбамоил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(3-(метилкарбамоил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(2-(метилкарбамоил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(4-((2-метоксиэтил)карбамоил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(4-метоксифенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(2-метоксифенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(тетрагидро-2Н-пиран-4-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-циклогексил-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(3S)-1-циано-N-(1-(2,3-дигидро-1Н-инден-1-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(2,3-дигидро-1Н-инден-2-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-((тетрагидро-2Н-пиран-4-ил)метил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-((S)-1-фенилэтил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-((R)-1-фенилэтил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(пиридин-2-илметил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(пиридин-3-илметил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(пиридин-4-илметил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-((3,5-диметилизоксазол-4-ил)метил)-1H-имидазол-4-ил)пирролидин-3-карбоксамида;
(2S,3S)-1-циано-2-метил-N-(1-((S)-1-фенилэтил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(3S,4S)-1-циано-4-метил-N-(1-((S)-1-фенилэтил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(4-метилпиридин-2-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(6-метилпиридин-2-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(R)-1-циано-N-(1-(2-метилпиримидин-4-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(4-цианофенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-N-(1-бензил-2-метил-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(2-(3,5-диметилизоксазол-4-ил)этил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(3-цианофенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(пиридин-3-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(пиридин-4-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(3-метоксифенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(3S)-N-(1-(1-бензоилпиперидин-3-ил)-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамида;
(3S)-N-(1-(1-бензоилпирролидин-3-ил)-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамида;
(3S)-N-(1-(1-бензилпиперидин-3-ил)-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамида;
(3S)-1-циано-N-(1-(1-метилпиперидин-3-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(3S)-1-циано-N-(1-(1-метилпирролидин-3-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-N-(5-ацетил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(5-изобутирил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-N-(5-бензоил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(2-метоксибензоил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-пиколиноил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(1-метил-1Н-пиразол-3-карбонил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(1-метил-2-оксо-1,2-дигидропиридин-3-карбонил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-никотиноил-4,5,6,7-тетрагидротиазоло[5,4-с] пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(диметилглицил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамида;
метил-(S)-2-(1-цианопирролидин-3-карбоксамидо)-6,7-дигидротиазоло[5,4-с]пиридин-5(4Н)-карбоксилата;
2-метоксиэтил-(S)-2-(1-цианопирролидин-3-карбоксамидо)-6,7-дигидротиазоло[5,4-с]пиридин-5(4Н)-карбоксилата;
(S)-1-циано-N-(5-метил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-N-(5-бензил-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(метилсульфонил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(изопропилсульфонил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(фенилсульфонил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(4-этинилфенил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(3-этинилфенил)тиазол-2-ил)пирролидин-3-карбоксамида;
(3S)-1-циано-N-(5-(N-(1-фенилэтил)сульфамоил)пиридин-2-ил)пирролидин-3-карбоксамида;
(3S)-1-циано-N-(5-(N-метил-N-(1-фенилэтил)сульфамоил)пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(5-метил-1,2,4-оксадиазол-3-ил)имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамида;
(3S,4S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)имидазо[1,2-а]пиридин-2-ил)-4-(трифторметил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(4-(1-метил-1Н-пиразол-4-ил)пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(N,N-диметилсульфамоил)-4,5,6,7-тетрагидротиазоло[5,4-с]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(пиридазин-4-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(4-циано-3-фторфенил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-N-(5-(1Н-индазол-7-ил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-N-(5-(3-(1Н-имидазол-1-ил)фенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(4-(метилсульфонамидо)фенил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-N-(5-(3-(1Н-пиразол-1-ил)фенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(4-циано-3-(трифторметокси)фенил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(4-циано-3-метоксифенил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(4-сульфамоилфенил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-N-(5-(1Н-индазол-6-ил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-N-(5-(1H-индазол-5-ил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-6-(2-(1-цианопирролидин-3-карбоксамидо)тиазол-5-ил)-N-метилпиколинамида;
(S)-6-(2-(1-цианопирролидин-3-карбоксамидо)тиазол-5-ил)-N-этилпиколинамида;
(S)-1-циано-N-(5-(1-метил-1Н-индазол-5-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(1-(2-метоксиэтил)-1Н-индазол-5-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(2-оксоиндолин-7-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(3-метил-1Н-индазол-5-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-N-(5-(1Н-индазол-4-ил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(4-фтор-3-(метилкарбамоил)фенил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-N-(5-(3-карбамоилфенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(3-(метилкарбамоил)фенил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(3-(этилкарбамоил)фенил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(3-(диметилкарбамоил)фенил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-N-(5-(3-карбамоил-4-фторфенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-N-(5-(3-карбамоил-4-хлорфенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-N-(5-(4-хлор-3-(метилкарбамоил)фенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-N-(5-(4-хлор-3-(проп-2-ин-1-илкарбамоил)фенил)тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(изопропилсульфонил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-фенил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-ил)пирролидин-3-карбоксамида;
(S)-N-(5-бензил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(4-циано-3-(2-метоксиэтокси)фенил)-1H-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(2-циано-5-(2-метоксиэтокси)фенил)-1H-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(4-циано-3-(3-морфолинопропокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(2-циано-5-(3-морфолинопропокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(4-циано-3-(((S)-тетрагидрофуран-3-ил)окси)фенил)-1H-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(2-циано-5-(((S)-тетрагидрофуран-3-ил)окси)фенил)-1H-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(4-циано-3-(оксетан-3-илокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(2-циано-5-(оксетан-3-илокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(4-(пиразоло[1,5-а]пиримидин-7-ил)пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(4-(имидазо[1,2-а]пиримидин-5-ил)пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(4-(имидазо[1,2-а]пиридин-5-ил)пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(4-(имидазо[1,2-а]пиразин-6-ил)пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(4-(имидазо[1,2-а]пиримидин-6-ил)пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1,7-нафтиридин-6-ил)пирролидин-3-карбоксамида;
(S)-N-(6-(3-хлорфенил)пиримидин-4-ил)-1-цианопирролидин-3-карбоксамида;
(S)-N-(2'-амино-[4,4'-бипиридин]-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(2'-(метиламино)-[4,4'-бипиридин]-2-ил)пирролидин-3-карбоксамида;
(S)-3-(1-цианопирролидин-3-карбоксамидо)-N-метилизохинолин-6-карбоксамида;
(S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)изохинолин-3-ил)пирролидин-3-карбоксамида;
(S)-N-(4-(1H-индазол-4-ил)пиридин-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(6-этинилизохинолин-3-ил)пирролидин-3-карбоксамида;
(2S,3S)-1-циано-N-(6-этинилизохинолин-3-ил)-2-метилпирролидин-3-карбоксамида;
(S)-3-(1-цианопирролидин-3-карбоксамидо)изохинолин-6-карбоксамида;
(2S,3S)-N-(6-(1Н-пиразол-4-ил)изохинолин-3-ил)-1-циано-2-метилпирролидин-3-карбоксамида;
(S)-1-циано-N-(пиразоло[1,5-а]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(4-фенилпиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(пиридин-3-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(пиридин-3-ил)тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(трифторметил)имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-метил-5-(м-толил)-1Н-пиразол-3-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(хинолин-3-ил)пирролидин-3-карбоксамида;
(S)-N-(4-(трет-бутил)пиридин-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(пиридин-4-ил)пиримидин-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-фенилпиридазин-3-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(оксазол-5-ил)имидазо[1,2-а]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(изопропилсульфонил)-5,6,7,8-тетрагидро-4Н-тиазоло[4,5-d]азепин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(4-(1-(2-метоксиэтил)-1Н-пиразол-4-ил)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
3-циано-N-(5-фенилтиазол-2-ил)-3-азабицикло[3.1.0]гексан-1-карбоксамида;
2-циано-N-(5-фенилтиазол-2-ил)-2-азабицикло[3.1.0]гексан-4-карбоксамида;
(3S,4S)-1-циано-N-(5-фенилтиазол-2-ил)-4-(трифторметил)пирролидин-3-карбоксамида;
(3S,4R)-1-циано-N-(5-фенилтиазол-2-ил)-4-(пиридин-3-ил)пирролидин-3-карбоксамида;
(3S,4R)-1-циано-N-(5-фенилтиазол-2-ил)-4-(пиримидин-5-ил)пирролидин-3-карбоксамида;
1-циано-3-метокси-N-(5-фенилтиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)тиазоло[4,5-b]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(5-(3,5-диметилизоксазол-4-ил)тиазоло[5,4-b]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(2,6-диметилфенил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(1,3-диметил-1Н-пиразол-4-ил)бензо[d]тиазол-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(5-(трифторметил)-1Н-пиразол-4-ил)изохинолин-3-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(4-(3-(5-метилизоксазол-4-ил)фенил)пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-N-(4-(1H-пиразол-4-ил)пиридин-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-N-(6-(1Н-пиразол-4-ил)бензо[d]тиазол-2-ил)-1-цианопирролидин-3-карбоксамида;
(S)-N-(1-(4-(1Н-пиразол-4-ил)фенил)-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(4-(5-метил-1Н-пиразол-4-ил)пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(6-(3,5-диметилизоксазол-4-ил)тиазоло[4,5-с]пиридин-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(2'-(5-метилизоксазол-4-ил)-[4,4'-бипиридин]-2-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(1,3-диметил-1Н-индазол-5-ил)-1H-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(3-метил-1Н-индазол-6-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-N-(1-(1Н-индазол-5-ил)-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамида;
(S)-N-(1-(1Н-индазол-6-ил)-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(4-фтор-3-метоксифенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-N-(1-(1Н-индазол-4-ил)-1Н-имидазол-4-ил)-1-цианопирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(4-циано-3-циклопропилфенил)-1H-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(хинолин-4-ил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-метил-N-(4-(4-(метилкарбамоил)фенил)тиазол-2-ил)пирролидин-2-карбоксамида;
(S)-1-циано-N-(1-(4-циано-3-метоксифенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(4-циано-3-метилфенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида;
(S)-1-циано-N-(1-(4-циано-3-(трифторметил)фенил)-1H-имидазол-4-ил)пирролидин-3-карбоксамида и
(S)-1-циано-N-(1-(4-циано-3-(трифторметокси)фенил)-1Н-имидазол-4-ил)пирролидин-3-карбоксамида,
его таутомер или фармацевтически приемлемая соль указанного соединения или таутомера.
13. Соединение по любому из пп. 1-12, его таутомер или фармацевтически приемлемая соль указанного соединения или таутомера для применения в качестве ингибиторов убиквитин-С-концевой гидролазы L1 (UCHL1).
14. Соединение по любому из пп. 1-12, его таутомер или фармацевтически приемлемая соль указанного соединения или таутомера для применения для лечения рака, который связан с активностью UCHL1.
15. Соединение по п. 14, где рак выбран из рака груди, яичников, предстательной железы, легкого, почки, желудка, прямой кишки, яичек, головы и шеи, поджелудочной железы, головного мозга, меланомы, рака кости, раковых заболеваний тканевых органов, раковых заболеваний клеток крови, лимфомы, лейкоза, множественной миеломы, колоректального рака, остеосаркомы, карциномы поджелудочной железы и немелкоклеточной карциномы легкого.
16. Соединение по любому из пп. 1-12 для применения для лечения заболевания, связанного с активностью UCHL1, которое выбрано из нейродегенеративных расстройств, хронической обструктивной болезни легких (ХОБЛ), воспаления, вирусной инфекции, ближневосточного респираторного синдрома (БВРС), тяжелого острого респираторного синдрома (ТОРС), бактериальной инфекции, туберкулеза и метаболических расстройств.
17. Соединение по п. 16, где нейродегенеративное расстройство выбрано из болезни Альцгеймера и болезни Паркинсона.
18. Фармацевтическая композиция, обладающая активностью ингибиторов UCHL1, содержащая эффективное количество соединения формулы (I) как определено по любому из пп. 1-12, его таутомера или фармацевтически приемлемой соли указанного соединения или таутомера совместно с одним или более фармацевтически приемлемыми вспомогательными веществами.
| WO 2001077073 A1, 18.10.2001 | |||
| WO 1995008534 A1, 30.03.1995 | |||
| US 20120077806 A1, 29.03.2012 | |||
| Формовочная машина со сквозной протяжной доской для формовки двойных шестерен | 1929 |
|
SU26409A1 |

