В данном изобретении предложены соединения, которые представляют собой агонисты мускаринового М1 рецептора и/или М4 рецептора, и которые пригодны для лечения опосредованных мускариновыми М1/М4 рецепторами заболеваний. Также предложены фармацевтические композиции, содержащие соединения, и использование соединений в терапевтических целях.
Уровень техники
Мускариновые ацетилхолиновые рецепторы (mAChR) являются членами суперсемейства связанных с G-белками рецепторов, которые опосредуют действие нейромедиатора ацетилхолина как в центральной, так и в периферической нервной системе. Пять подтипов mAChR клонировано от M1 до М5. M1 mAChR экспрессируется преимущественно постсинаптически в коре головного мозга, гиппокампе, стриатуме и таламусе; М2 mAChR преимущественно локализируются в стволе головного мозга и таламусе, хотя также и в коре головного мозга, гиппокампе и стриатуме, где они расположены в холинергических терминальных синаптических окончаниях (Langmead et al, 2008 Br J Pharmacol). Тем не менее, M2 mAChR также экспрессируются периферически в сердечной мышечной ткани (где они опосредуют вагальную иннервацию сердца) и в гладких мышцах и экзокринных железах. М3 mAChR экспрессируются на относительно низком уровне в ЦНС, но значительно экспрессируются в гладких мышцах и железистых тканях, таких как потовые и слюнные железы (Langmead et al, 2008 Br J Pharmacol).
В центральной нервной системе мускариновые рецепторы, особенно M1 mAChR, играют существенно важную роль в опосредовании высших когнитивных функций. Заболевания, связанные с когнитивными нарушениями, такие как болезнь Альцгеймера, сопровождаются потерей холинергических нейронов в базальном отделе переднего мозга (Whitehouse et al, 1982 Science). При шизофрении, которая также включает когнитивное нарушение в качестве важной составляющей клинической картины, плотность mAChR снижена в префронтальной коре, гиппокампе и дорсолатеральных отделах стриатума субъектов, больных шизофренией (Dean et al, 2002 Mol Psychiatry). Более того, в животных моделях блокирование или повреждение центральных холинергических путей приводит к углублению когнитивных расстройств и неселективные антагонисты mAChR показали способность индуцировать психотомиметические эффекты у психиатрических пациентов. Заместительная холинергическая терапия значительно основывается на использовании ингибиторов ацетилхолинэстеразы для предотвращения расщепления эндогенного ацетилхолина. Данные соединения показали эффективность против симптоматического снижения когнитивных способностей в клинике, но приводит к ограничивающим дозу побочным эффектам, обусловленным стимуляцией периферических М2 и М3 mAChR, включая расстройство моторики желудочно-кишечного тракта, брадикардию, тошноту и рвоту (http://www.druqs.com/pro/donepezil.html; http://www.druqs.com/pro/rivastiqmine.html).
Дальнейшие попытки исследований были направлены на идентификацию прямых агонистов M1 mAChR с целью стимуляции селективного улучшения когнитивной функции с благоприятным набором побочных эффектов. Такие попытки привели к идентификации ряда агонистов, представленных на примерах соединений, таких как ксаномелин, AF267B, сабкомелин, миламелин и цевимелин. Большинство данных соединений показали высокую эффективность в профилактических модельных исследованиях как на грызунах, так и/или на нечеловекообразных приматах. Миламелин показал эффективность против вызванных скополамином дефицтов в работе и пространственной памяти у грызунов; сабкомелин показал эффективность в задании различения визуального объекта у мартышек и ксаномелин обратил вызванный антагонистом mAChR дефицит когнитивной деятельности в реакции пассивного избегания.
Болезнь Альцгеймера (AD) представляет собой наиболее часто встречающееся нейродегенеративное заболевание (26,6 миллионов людей по всему миру в 2006 г.), которое поражает пожилых людей, приводя к глубокой потере памяти и когнитивной дисфункции. Этиология заболевания является комплексной, но характеризуется двумя отличительными патологиями мозга: агрегатами амилоидных бляшек, в основном состоящих из амилоид-β пептида (Аβ), и нейрофибриллярными клубками, образованными гиперфосфорилированными тау-белками. Считается, что аккумуляция Aβ играет главную роль в развитии AD и, в связи с этим, общепризнанные терапии для лечения AD нацелены на ингибирование образования Aβ. Аβ образуется в результате протеолитического расщепления мембранной связи белка-предшественника амилоида (АРР). АРР образуется двумя путями, неамилоидогенетическим и амилоидогенетическим. Расщепление АРР γ-секретазой является общим для двух путей, но в первом АРР расщепляется α-секретазой с образованием растворимого АРРα. При этом, в амилоидогенетическом пути АРР расщепляется β-секретазой с образованием растворимого АРРβ и также Аβ. Исследования In vitro показали, что агонисты mAChR могут увеличивать образование АРР по отношению к растворимому, неамилоидогенетическому пути. Исследования In vivo показали что агонист mAChR, AF267B, изменил болезнеобразную патологию у 3×TgAD трансгенной мыши, модели различных составляющих болезни Альцгеймера (Caccamo et al., 2006 Neuron). Агонист mAChR цевимелин показал небольшое, но значительное уменьшение уровней спинномозговой жидкости Aβ у пациентов с болезнью Альцгеймера, демонстрируя таким образом потенциальную эффективность в изменении заболевания (Nitsch et al, 2000 Neurol).
Доклинические исследования позволяют утверждать, что агонисты mAChR показывают необычное антипсихотик-подобное действие в ряде случаев доклинических исследований. Агонист mAChR ксаномелин обращает ряд опосредованных дофамином поведений, включая вызванную амфетамином двигательную активность у крыс, вызванное апоморфином вскарабкивание у мышей, вызванное агонистом дофамина вращение у односторонне пораженных 6-OH-DA крыс и вызванное амфетамином двигательное беспокойство у обезьян (без предрасположенности к EPS). Он также показал способность ингибировать А10, но не A9, дофамин-опосредованное возбуждение клеток и условно-рефлекторное избегание, и вызывает c-fos экспрессию в префронтальной коре и прилежащем ядре, но не полосатом теле у крыс. Указанные данные также свидетельствуют об атипическом антипсихотик-подобном действии (Mirza et al, 1999 CNS Drug Rev). Мускариновые рецепторы также вовлечены в нейробиологию наркотической зависимости. Подкрепляющий эффект кокаина и других вызывающих наркотическую зависимость веществ опосредуется мезолимбической дофаминовой системой, где поведенческие и нейрохимические исследования показали, что холинергические подтипы мускариновых рецепторов играют важные роли в регуляции дофаминергической нейропередачи. Например, мыши М(4) (-/-) показали значительно повышенное мотивированное вознаграждением поведение в результате действия кокаина (Schmidt et al Psychopharmacology (2011) Aug; 216 (3): 367-78). Более того, ксаномелин показал способность блокировать эффекты кокаина на данных моделях.
Мускариновые рецепторы также вовлечены в контроль движения и потенциально могут быть представлены в новых способах лечения двигательных расстройств, таких как болезнь Паркинсона, СДВГ, болезнь Хантингтона, синдром Туретта, и других синдромов, связанных с дофаминергической дисфункцией в качестве основного фактора патогенеза, вызывающего заболевание.
Ксаномелин, самбкомелин, миламелин и цевимелин прошли различные этапы клинических исследований для лечения болезни Альцгеймера и/или шизофрении. Клинические исследования (Фаза II) ксаномелина показали его эффективность против различных видов когнитивных симптомов, включая нарушение поведения и галлюцинации, связанные с болезнью Альцгеймера (Bodick et al, 1997 Arch Neurol). Данное соединение также оценили в небольшом исследовании (Фаза II) шизофреников, где оно привело к значительному уменьшению положительных и отрицательных симптомов по сравнению с плацебо-контролем (Shekhar et al, 2008 Am J Psych). Тем не менее, во всех клинических исследованиях ксаномелин и другие родственные агонисты mAChR проявили неприемлемый уровень безопасности в отношении холинергических побочных эффектов, включая тошноту, боль в нижней части живота, диарею, диафорез (повышенное потоотделение), гиперсаливацию (повышенное слюноотделение), обморок и брадикардию.
Мускариновые рецепторы вовлечены в процесс формирования центральной и периферической боли. Выделяют три различных типа боли: острая, воспалительная и невропатическая. Острая боль служит в качестве важной защитной функции при поддержании организма в безопасности от стимулов, которые могут вызывать разрушение ткани; тем не менее, лечение послеоперационной боли является необходимым. Воспалительная боль может возникать в результате большого числа причин, включая повреждение ткани, аутоимунную реакцию и инвазию патогенов, и вызвана действием воспалительных медиаторов, таких как нейропептиды и простагландины, которые приводят к нейрональному воспалению и боли. Невропатическая боль связана с отклоняющимися от нормы болезненными ощущениями к неболевым стимулам. Невропатическая боль связана с рядом разных заболеваний/травм, таких как повреждение спинного мозга, рассеянный склероз, диабеты (диабетическая невропатия), вирусные инфекции (такие как ВИЧ или герпес). Она также обычно встречается при раке как результат заболевания, так и побочный эффект химиотерапии. В ряде болевых состояний активация мускариновых рецепторов привела к анальгезии путем активации рецепторов в спинном мозге и высших болевых центрах мозга. Повышение ингибиторами ацетилхолинэстеразы эндогенных уровней ацетилхолина, непосредственная активация агонистами мускариновых рецепторов или аллостерические модуляторы проявили аналгетическую активность. Напротив, блокирование агонистами мускариновых рецепторов или использование нокаутированных мышей повышает чувствительность к боли. Доказательство роли рецептора М1 в боли рассмотрено в обзоре D. F. Fiorino и М. Garcia-Guzman, 2012.
Недавно было открыто небольшое число соединений, показавших улучшенную селективность для подтипа mAChR относительно экспрессируемых периферически mAChR подтипов (Bridges et al, 2008 Bioorg Med Chem Lett; Johnson et al, 2010 Bioorg Med Chem Lett, Budzik et al, 2010 ACS Med Chem Lett). Несмотря на повышенные уровни селективности в отношении М3 mAChR подтипа, некоторые из данных соединений сохраняют значительную активность в качестве агонистов обоих данных подтипов и М2 mAChR подтипа. В данном документе мы описываем серию соединений, неожиданно показавших высокие уровни селективности по отношению к M1 и/или М4 mAChR по сравнению с М2 и М3 подтипами рецептора.
Описание Фигур
Описание фигур может быть найдено в экспериментальных секциях B и C.
Фигура 1 иллюстрирует найденную для Примера 1-33 Изомера 2 способность обращать вызванную скополамином амнезию дозозависимым способом, с ЭД50 приблизительно 10 мг/кг (п/о). Эффект от 30 мг/кг оказался схожим с эффектом, вызванным ингибитором холинэстеразы донепезилом (0,1 мг/кг, и/п), который служил в качестве положительного контроля.
Фигура 2 иллюстрирует эффект нового тестируемого соединения на d-амфетамин-индуцированную гиперактивность у крыс. Антипсихотически-подобное поведение крыс оценивали путем ингибирования гиперактивности (или гиперлокомоции), вызванной d-амфетамином. Представлены данные для Примеров 1-21 Изомера 2, 1-32 Изомера 2, 1-33 Изомера 2, 2-7 Изомера 2 и 2-17 Изомера 2.
Подробное описание сущности изобретения
В данном изобретении предложены соединения, имеющие активность как агонисты мускаринового М1 и/или М4 рецептора. В частности, в изобретении предложены соединения, которые проявляют селективность к М1 рецептору и/или М4 рецептору по отношению к М2 и М3 подтипам рецептора.
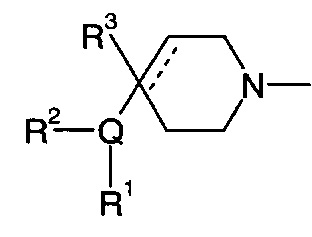
Соответственно, в одном варианте реализации изобретения (Вариант реализации изобретения 1.1), в изобретении предложено соединение формулы (1):
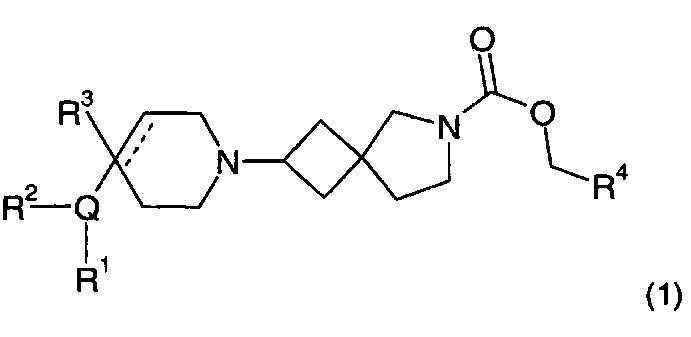
или его соль, где
Q представляет собой пяти- или шестичленное моноциклическое гетероциклическое кольцо, содержащее 1, 2, 3 или 4 члена гетероатомного кольца, выбранных из N, О и S;
R1 выбран из водорода; фтора; хлора; брома; циано; оксо; гидрокси; OR5; NR5R6; COR5; COOR5; OCOR5; NR7COR5; CONR5R6; NR7CONR5R6; NR7COOR5; OCONR5R6; SR5; SOR5 и SO2R5; C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомомами фтора и причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из О, N и S, и их окисленных форм; и необязательно замещенного 5- или 6-членного кольца, содержащего 0, 1, 2 или 3 гетероатома, выбранных из О, N и S и их окисленных форм;
R2 выбран из водорода; фтора; хлора; брома; циано; гидрокси; метокси; OR5; NR5R6; COR5; COOR5; OCOR5; NR7COR5; CONR5R6; NR7CONR5R6; NR7COOR5; OCONR5R6; SR5; SOR5 и SO2R5; C1-6 неароматической углеводородной группы; или R1 и R2 могут быть соединены вместе с образованием 6-членного конденсированного ароматического кольца;
R3 выбран из водорода; фтора; циано; гидрокси; амино; и C1-9 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и причем один, два или три, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S и их окисленных форм;
R4 представляет собой водород или C1-6 неароматическую углеводородную группу, которая необязательно замещена от одного до шести атомомами фтора и, причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из О, N и S и их окисленных форм;
R5, R6 и R7 являются одинаковыми или разными и каждый независимо выбран из водорода, неароматической C1-4 углеводородной группы, необязательно замещенной одним или более атомами фтора; или группы формулы CH2N(Ra)COORb;
Ra выбран из водорода и неароматической C1-4 углеводородной группы;
Rb представляет собой неароматическую C1-4 углеводородную группу, которая необязательно замещена одной или более группами, выбранными из фтора; хлора; брома; циано; гидрокси; метокси; амино; или циклоалкильной, гетероциклоалкильной, арильной или гетероарильной группой;
и пунктирная линия означает необязательную вторую углерод-углеродную связь, при условии, что если присутствует вторая углерод-углеродная связь, то R3 отсутствует.
Соответственно, в одном варианте реализации изобретения (Вариант реализации изобретения 1.1а) в изобретении предложено соединение формулы (1а):
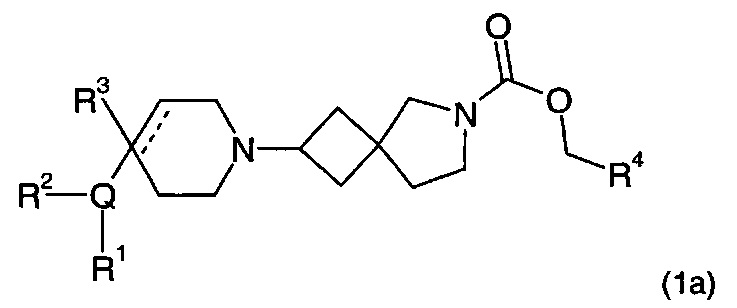
или его соль, где
Q представляет собой пяти- или шести-, или семичленное моноциклическое гетероциклическое кольцо, содержащее 1, 2, 3 или 4 членов гетероатомного кольца, выбранных из N, О и S;
R1 выбран из водорода; фтора; хлора; брома; циано; оксо; гидрокси; OR5; NR5R6; COR5; COOR5; OCOR5; NR7COR5; CONR5R6; NR7CONR5R6; NR7COOR5; OCONR5R6; SR5; SOR5 и SO2R5; C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомомами фтора и, причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S и их окисленных форм; и необязательно замещенного 5- или 6-членного кольца, содержащего 0, 1, 2 или 3 гетероатома, выбранных из O, N и S и их окисленных форм;
R2 выбран из водорода; фтора; хлора; брома; циано; гидрокси; метокси; OR5; NR5R6; COR5; COOR5; OCOR5; NR7COR5; CONR5R6; NR7CONR5R6; NR7COOR5; OCONR5R6; SR5; SOR5 и SO2R5; C1-6 неароматической углеводородной группы; или R1 и R2 могут быть соединены вместе с образованием 6-членного конденсированного ароматического кольца;
R3 выбран из водорода; фтора; циано; гидрокси; амино; и C1-9 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и причем один, два или три, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S и их окисленных форм;
R4 представляет собой водород или C1-6 неароматическую углеводородную группу, которая необязательно замещена от одного до шести атомомами фтора и причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S и их окисленных форм;
R5, R6 и R7 являются одинаковыми или разными и каждый независимо выбран из водорода, неароматической С1-4 углеводородной группы, необязательно замещенной одним или более атомами фтора; или группы формулы CH2N(Ra)COORb;
Ra выбран из водорода и неароматической C1-4 углеводородной группы;
Rb представляет собой неароматическую С1-4 углеводородную группу, которая необязательно замещена одной или более группами, выбранными из фтора; хлора; брома; циано; гидрокси; метокси; амино; или циклоалкильной, гетероциклоалкильной, арильной или гетероарильной группой;
и пунктирная линия означает необязательную вторую углерод-углеродную связь, при условии, что если присутствует вторая углерод-углеродная связь, то R3 отсутствует.
Соответственно, в одном варианте реализации изобретения (Вариант реализации изобретения 1.1b), в изобретении предложено соединение формулы (1b):
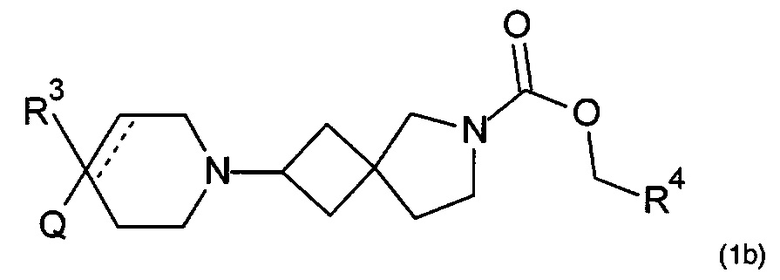
или его соль, где
Q представляет собой необязательно замещенное пяти- или шести- или семичленное гетероциклическое кольцо, содержащее 1, 2, 3 или 4 члена гетероатомного кольца, выбранных из N, O и S;
R3 выбран из водорода; фтора; циано; гидрокси; амино; и C1-9 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и причем один, два или три, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S и их окисленных форм;
R4 представляет собой водород или C1-6 неароматическую углеводородную группу, которая необязательно замещена от одного до шести атомомами фтора и причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S и их окисленных форм; и пунктирная линия означает необязательную вторую углерод-углеродную связь, при условии, что если присутствует вторая углерод-углеродная связь, то R3 отсутствует.
Частные случаи соединений формулы (1), (1а) или (1b) находятся в соответствии с определениями в изложенных ниже Вариантах реализации изобретения от 1.2 до 1.180.
1.2 Соединение по Варианту реализации изобретения 1.1, отличающееся тем, что Q представляет собой ароматическое или ненасыщенное гетероциклическое кольцо.
1.3 Соединение по Варианту реализации изобретения 1.2, отличающееся тем, что Q представляет собой ароматическое гетероциклическое кольцо.
1.4 Соединение по Варианту реализации изобретения 1.3, отличающееся тем, что Q представляет собой гетероциклическое кольцо, содержащее азот-содержащий кольцевой член и необязательно один или два дополнительных кольцевых члена, выбранных из O, N и S.
1.5 Соединение по Варианту реализации изобретения 1.4, отличающееся тем, что Q представляет собой ароматическое гетероциклическое кольцо, содержащее азотсодержащий кольцевой член и необязательно один дополнительный кольцевой член, выбранный из O, N и S.
1.6 Соединение по Варианту реализации изобретения 1.5, отличающееся тем, что Q представляет собой ароматическое гетероциклическое кольцо, содержащее один или два азот-содержащих кольцевых члена.
1.7 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.6, отличающееся тем, что Q представляет собой пятичленное гетероциклическое кольцо, соединенное со смежным шестичленным кольцом с помощью атома углерода, указанного пятичленного гетероциклического кольца.
1.8 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.6, отличающееся тем, что Q представляет собой пятичленное гетероциклическое кольцо, соединенное со смежным шестичленным кольцом с помощью атома азота указанного пятичленного гетероциклического кольца.
1.9 Соединение по Варианту реализации изобретения 1.1, отличающееся тем, что Q выбран из 1-пирролила, 2-имидазолила, 1-пиразолила, 3-пиразолила, 5-пиразолила, 2-тиазолила, 2-оксазолила, триазолила, тетразолила, тиадиазолила, оксадиазолила и их таутомерных форм.
1.10 Соединение по Варианту реализации изобретения 1.6, отличающееся тем, что Q представляет собой пиррольное кольцо.
1.11 Соединение по Варианту реализации изобретения 1.6, отличающееся тем, что Q представляет собой имидазольное кольцо.
1.12 Соединение по Варианту реализации изобретения 1.6, отличающееся тем, что Q представляет собой пиразольное кольцо.
1.13 Соединение по Варианту реализации изобретения 1. 6, отличающееся тем, что Q выбран из 1-пиразолила, 3-пиразолила, 5-пиразолила и их таутомерных форм.
1.14 Соединение по Варианту реализации изобретения 1.1, отличающееся тем, что Q представляет собой шестичленное кольцо, содержащее один или более атомов азота.
1.15 Соединение по Варианту реализации изобретения 1.14, отличающееся тем, что Q представляет собой радикал пиридинового, пиразинового или 2-оксо-3N (3-пиперидин-2-онового) кольца, содержащего 0-2 C-C ненасыщенных связей.
1.16 Соединение по Варианту реализации изобретения 1.1, отличающееся тем, что Q представляет собой 5, 6 или 7-членное ненасыщенное гетероциклическое кольцо.
1.17 Соединение по Варианту реализации изобретения 1.16, отличающееся тем, что Q представляет собой 5-пирролидинил.
1.18 Соединение по Варианту реализации изобретения 1.1, отличающееся тем, что Q является бициклическим; имеющим дополнительное кольцо, присоединенное к Q.
1.19 Соединение по Варианту реализации изобретения 1.1b, отличающееся тем, что Q имеет один или более заместителей, например один, два или три заместителя, которые могут быть выбраны из одного R1 и/или R2, причем R1 и R2 могут быть одинаковыми или разными. Дополнительные заместители для Q могут включать (L)-R10, (L)-R11 и (L)-R12, где L представляет собой связь или группу CH2; R10, R11 и R12 независимо выбраны из водорода; фтора; хлора; брома; циано; оксо; гидрокси; OR15; NR15R16; COR15; CSR15; COOR15; COSR15; OCOR15; NR17COR15; CONR15R16; CSNR15R16; NR17CONR15R16; R17COOR15; OCONR15R16; SR15; SOR15 и SO2R15; C1-6 неароматическую углеводородную группу, которая необязательно замещена от одного до шести атомомами фтора и причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S, и их окисленных форм; и необязательно замещенного 5- или 6-членного кольца, содержащего 0, 1, 2 или 3 гетероатома, выбранных из O, N и S и их окисленных форм;
при этом необязательные заместители для необязательно замещенного 5- или 6-членного кольца выбраны из группы R8, содержащей водород; фтор; хлор; бром; циано; оксо; гидрокси; OR5; NR5R6; COR5; COOR5; OCOR5; NR7COR5; CONR5R6; NR7CONR5R6; NR7COOR5; OCONR5R6; SR5; SOR5 и SO2R5; и C1-6 неароматическую углеводородную группу, которая необязательно замещена от одного до шести атомомами фтора и причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S, и их окисленных форм;
при этом R15, R16 и R17 являются одинаковыми или разными или могут быть соединены вместе с образованием кольца, и каждый независимо выбран из водорода, неароматической C1-6 углеводородной группы, необязательно замещенной одним или более атомами фтора и причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S, и их окисленных форм; или группы формулы CH2N(Ra)COORb; или группы формулы (L)-R18, где L представляет собой связь или группу CH2 и R18 представляет собой необязательно замещенное 5- или 6-членное кольцо, содержащее 0, 1, 2 или 3 гетероатома, выбранных из O, N и S, и их окисленных форм;
при этом необязательные заместители для необязательно замещенного 5- или 6-членного кольца выбраны из группы R8.
1.20 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.19, отличающееся тем, что R1 выбран из водорода; фтора; хлора; брома; циано; оксо; гидрокси; OR5; NR5R6; COR5, COOR5; OCOR5; NR7COR5; CONR5R6; NR7CONR5R6; NR7COOR5; OCONR5R6; SR5; SOR5 и SO2R5; неароматической углеводородной группы, которая необязательно замещена от одного до шести атомомами фтора и причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S и их окисленных форм; и необязательно замещенного 5- или 6-членного кольца, содержащего 0, 1, 2 или 3 гетероатома, выбранных из O, N и S, и их окисленных форм;
при этом необязательные заместители для необязательно замещенного 5- или 6-членного кольца выбраны из группы R8, содержащей водород; фтор; хлор; бром; циано; оксо; гидрокси; OR5; NR5R6; COR5; COOR5; OCOR5; NR7COR5; CONR5R6; NR7CONR5R6; NR7COOR5; OCONR5R6; SR5; SOR5 и SO2R5; и C1-6 неароматическую углеводородную группу, которая необязательно замещена от одного до шести атомомами фтора и, причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из О, N и S, и их окисленных форм.
1.21 Соединение по Варианту реализации изобретения 1.20, отличающееся тем, что R1 выбран из водорода; фтора; хлора; брома; циано; оксо; гидрокси; OR5; NR5R6; COR5; COOR5; OCOR5; NR7COR5; CONR5R6; NR7CONR5R6; NR7COOR5; OCONR5R6; SR5; SOR5 и SO2R5; C1-5 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомомами фтора и причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S и их окисленных форм; и необязательно замещенного 5- или 6-членного кольца, содержащего 0, 1 или 2 гетероатома, выбранных из O, N и S, и их окисленных форм;
при этом необязательные заместители для необязательно замещенного 5- или 6-членного кольца выбраны из группы R8, содержащей фтор; хлор; бром; циано; оксо; гидрокси; OR5; NR5R6; COR5; COOR5; OCOR5; NR7COR5; CONR5R6; NR7CONR5R6; NR7COOR5; OCONR5R6; SR5; SOR5 и SO2R5; и С1-4 неароматическую углеводородную группу, которая необязательно замещена от одного до шести атомомами фтора и причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S, и их окисленных форм.
1.22 Соединение по Варианту реализации изобретения 1.21, отличающееся тем, что R1 выбран из водорода; фтора; хлора; брома; циано; оксо; гидрокси; OR5; NR5R6; COR5; COOR5; OCOR5; NR7COR5; CONR5R6; NR7CONR5R6; NR7COOR5; OCONR5R6; SR5; SOR5 и SO2R5; C1-4 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомомами фтора и причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S, и их окисленных форм; и необязательно замещенное 5- или 6-членное арильное или гетероарильное кольцо, содержащее 0, 1 или 2 гетероатомов, выбранных из O, N и S, и их окисленных форм;
при этом необязательные заместители для необязательно замещенного радикала 5- или 6-членного арильного или гетероарильного кольца выбраны из группы R8, содержащей фтор; хлор; бром; циано; оксо; гидрокси; OR5; NR5R6; COR5; COOR5; OCOR5; NR7COR5; CONR5R6; NR7CONR5R6; NR7COOR5; OCONR5R6; SR5; SOR5 и SO2R5; и C1-4 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомомами фтора и, причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S и их окисленных форм.
1.23 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.19, отличающееся тем, что R1 выбран из водорода; фтора; хлора; циано; оксо; гидрокси; OR5; NR5R6; COR5; COOR5; OCOR5; NR7COR5; CONR5R6; NR7CONR5R6; NR7COOR5; OCONR5R6; SO2R5; C1-4 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомомами фтора и причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S и их окисленных форм; и необязательно замещенного 5- или 6-членного кольца, содержащего 0, 1, 2 или 3 гетероатома, выбранных из O, N и S, и их окисленных форм, при этом необязательные заместители для необязательно замещенного 5- или 6-членного кольца выбраны из группы R8, содержащей фтор; хлор; бром; циано; оксо; гидрокси; OR5; NR5R6; COR5; COOR5; OCOR5; NR7COR5; CONR5R6; NR7CONR5R6; NR7COOR5; OCONR5R6; SR5; SOR5 и SO2R5; и C1-4 неароматическую углеводородную группу, которая необязательно замещена от одного до шести атомомами фтора и причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S, и их окисленных форм.
1.24 Соединение по Варианту реализации изобретения 1.23, отличающееся тем, что R1 выбран из водорода; фтора; хлора; циано; гидрокси; OR5; NR5R6; COR5; COOR5; OCOR5; NR7COR5; CONR5R6; NR7CONR5R6; NR7COOR5; OCONR5R6; SO2R5; и C1-4 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомомами фтора и причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S, и их окисленных форм.
1.25 Соединение по Варианту реализации изобретения 1.24, отличающееся тем, что R1 выбран из водорода; фтора; хлора; циано; гидрокси; OR5; NR5R6; COR5; COOR5; OCOR5; NR7COR5; CONR5R6; NR7CONR5R6; NR7COOR5; SO2R5; и C1-4 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора.
1.26 Соединение по Варианту реализации изобретения 1.25, отличающееся тем, что R1 выбран из водорода; фтора; хлора; циано; NR5R6; COR5; COOR5 и C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора.
1.27 Соединение по Варианту реализации изобретения 1.26, отличающееся тем, что R1 выбран из водорода; фтора; хлора; циано; NH2, COR5; COOR5 и C1-4 насыщенной неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора.
1.28 Соединение по Варианту реализации изобретения 1.27, отличающееся тем, что R1 выбран из водорода; COR5; COOR5; CONR5R6 и C1-4 алкильной группы.
1.29 Соединение по Варианту реализации изобретения 1.28, отличающееся тем, что R1 выбран из водорода; COR5; COOR5 и C1-3 алкильной группы.
1.30 Соединение по Варианту реализации изобретения 1.29, отличающееся тем, что R1 выбран из водорода; метила; этила и COOR5.
1.31 Соединение по Варианту реализации изобретения 1.30, отличающееся тем, что R1 представляет собой водород.
1.32 Соединение по Варианту реализации изобретения 1.30, отличающееся тем, что R1 представляет собой метил или этил.
1.33 Соединение по Варианту реализации изобретения от 1.20 до 1.30, отличающееся тем, что R1 представляет собой СООМе; COOEt; СОМе; COEt; CONH2; CF3; CONHMe; CON(Me)2; COCF3; СО-циклопропил; СО-циклобутил; CONHEt; СОН; NH2; OMe;
1.34 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.33, отличающееся тем, что R2 выбран из водорода; фтора; хлора; брома; циано; гидрокси; метокси; и C1-6 неароматической углеводородной группы; или соединен вместе с R1 с образованием 6-членного конденсированного ароматического кольца.
1.35 Соединение по Варианту реализации изобретения 1.34, отличающееся тем, что R2 выбран из водорода; фтора; гидрокси; метокси; и C1-6 неароматической углеводородной группы.
1.36 Соединение по Варианту реализации изобретения 1.35, отличающееся тем, что R2 выбран из водорода; фтора; метокси; и C1-4 насыщенной углеводородной группы.
1.37 Соединение по Варианту реализации изобретения 1.36, отличающееся тем, что R2 выбран из водорода; фтора; метокси; и C1-4 алкильной группы.
1.38 Соединение по Варианту реализации изобретения 1.37, отличающееся тем, что R2 выбран из водорода и C1-3 алкильной группы.
1.39 Соединение по Варианту реализации изобретения 1.38, отличающееся тем, что R2 выбран из водорода и метила.
1.40 Соединение по Варианту реализации изобретения 1.34, отличающееся тем, что R2 соединен вместе с R1 с образованием 6-членного конденсированного ароматического кольца, которое может представлять собой арил или гетероарил.
1.41 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.40, отличающееся тем, что пунктирная линия представляет собой вторую углерод-углеродную связь и R3 отсутствует.
1.42 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.40, отличающееся тем, что R3 присутствует и необязательная вторая углерод-углеродная связь отсутствует.
1.43 Соединение по Варианту реализации изобретения 1.42, отличающееся тем, что R3 выбран из водорода; фтора; циано; гидрокси; амино; и C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомомами фтора и причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S и их окисленных форм.
1.44 Соединение по Варианту реализации изобретения 1.43, отличающееся тем, что R3 выбран из водорода; фтора; циано; гидрокси; амино; и C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора и при этому один, но не все, атом углерода углеводородной группы может быть необязательно замещен гетероатомом, выбранным из O, N и S, и их окисленных форм.
1.45 Соединение по Варианту реализации изобретения 1.44, отличающееся тем, что R3 выбран из водорода; фтора; циано; гидрокси; амино; C1-4 алкила и С1-4 алкокси, при этом каждый C1-4 алкил и С1-4 алкокси необязательно замещен от одного до шести атомами фтора.
1.46 Соединение по Варианту реализации изобретения 1.45, отличающееся тем, что R3 выбран из водорода; фтора; гидрокси и метокси.
1.47 Соединение по Варианту реализации изобретения 1.46, отличающееся тем, что R3 представляет собой водород.
1.48 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.47, отличающееся тем, что R4 представляет собой водород или ациклическую C1-6 углеводородную группу.
1.49 Соединение по Варианту реализации изобретения 1.48, отличающееся тем, что R4 представляет собой водород или ациклическую С1-3 углеводородную группу.
1.50 Соединение по Варианту реализации изобретения 1.49, отличающееся тем, что R4 представляет собой водород или С1-3 алкильную группу или С2-3 алкинильную группу.
1.51 Соединение по Варианту реализации изобретения 1.50, отличающееся тем, что R4 выбран из водорода, метила, этила, этинила и 1-пропинила.
1.52 Соединение по Варианту реализации изобретения 1.51, отличающееся тем, что R4 выбран из водорода и метила.
1.53 Соединение по Варианту реализации изобретения 1.52, отличающееся тем, что R4 представляет собой метил.
1.54 Соединение по любому из предшествующих Вариантов реализации изобретения, отличающееся тем, что R5, если присутствует, представляет собой неароматическую C1-4 углеводородную группу, необязательно замещенную одним или более атомами фтора; или группу формулы CH2N(Ra)COORb.
1.55 Соединение по Варианту реализации изобретения 1.54, отличающееся тем, что неароматическая С1-4 углеводородная группа является насыщенной С1-4 углеводородной группой.
1.56 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.53, отличающееся тем, что R5, если присутствует, представляет собой водород.
1.57 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.53, отличающееся тем, что R5, если присутствует, выбран из водорода и насыщенной C1-4 углеводородной группы.
1.58 Соединение по Варианту реализации изобретения 1.55 или Варианту реализации изобретения 1.56, отличающееся тем, что насыщенная С1-4 углеводородная группа представляет собой С1-4 алкильную группу.
1.59 Соединение по Варианту реализации изобретения 1.58, отличающееся тем, что насыщенная С1-4 углеводородная группа представляет собой С1-3 алкильную группу.
1.60 Соединение по Варианту реализации изобретения 1.59, отличающееся тем, что C1-3 алкильная группа выбрана из метила, этила и изопропила.
1.61 Соединение по Варианту реализации изобретения 1.60, отличающееся тем, что C1-3 алкильная группа представляет собой этил.
1.62 Соединение по любому из предшествующих Вариантов реализации изобретения, отличающееся тем, что R6, если присутствует, представляет собой неароматическую C1-4 углеводородную группу.
1.63 Соединение по Варианту реализации изобретения 1.62, отличающееся тем, что неароматическая C1-4 углеводородная группа представляет собой насыщенную C1-4 углеводородную группу.
1.64 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.61, отличающееся тем, что R6, если присутствует, представляет собой водород.
1.65 Соединение по Варианту реализации изобретения 1.63, отличающееся тем, что насыщенная C1-4 углеводородная группа представляет собой C1-3 алкильную группу.
1.66 Соединение по Варианту реализации изобретения 1.65, отличающееся тем, что C1-3 алкильная группа выбрана из метила, этила и изопропила.
1.67 Соединение по любому из предшествующих Вариантов реализации изобретения, отличающееся тем, что R7, если присутствует, представляет собой неароматическую C1-4 углеводородную группу.
1.68 Соединение по Варианту реализации изобретения 1.67, отличающееся тем, что неароматическая C1-4 углеводородная группа представляет собой насыщенную C1-4 углеводородную группу.
1.69 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.66, отличающееся тем, что R7, если присутствует, представляет собой водород.
1.70 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.66, отличающееся тем, что R7, если присутствует, выбран из водорода и насыщенной C1-4 углеводородной группы.
1.71 Соединение по Варианту реализации изобретения 1.68 или Варианту реализации изобретения 1.70, отличающееся тем, что насыщенная C1-4 углеводородная группа представляет собой С1-4 алкильную группу.
1.72 Соединение по Варианту реализации изобретения 1.71, отличающееся тем, что насыщенная C1-4 углеводородная группа представляет собой C1-3 алкильную группу.
1.73 Соединение по Варианту реализации изобретения 1.72, отличающееся тем, что C1-3 алкильная группа выбрана из метила, этила и изопропила.
1.74 Соединение по любому из предшествующих Вариантов реализации изобретения, отличающееся тем, что когда R1 представляет собой необязательно замещенное 5- или 6-членное кольцо, то он выбран из ароматических колец, содержащих 0, 1 или 2 или 3 гетероатома, выбранных из O, N и S, и их окисленных форм.
1.75 Соединение по Варианту реализации изобретения 1.74, отличающееся тем, что ароматическое кольцо является карбоциклическим.
1.76 Соединение по Варианту реализации изобретения 1.74, отличающееся тем, что ароматическое кольцо является гетероциклическим.
1.77 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.73, отличающееся тем, что когда R1 представляет собой необязательно замещенное 5- или 6-членное кольцо, то он выбран из неароматических колец, содержащих 0, 1 или 2, или 3 гетероатома, выбранных из O, N и S, и их окисленных форм.
1.78 Соединение по Варианту реализации изобретения 1.77, отличающееся тем, что неароматическое кольцо является карбоциклическим.
1.79 Соединение по Варианту реализации изобретения 1.77, отличающееся тем, что неароматическое кольцо является гетероциклическим.
1.80 Соединение по любому из Вариантов реализации изобретения от 1.74 до 1.79, отличающееся тем, что кольцо представляет собой 5-членное кольцо.
1.81 Соединение по любому из Вариантов реализации изобретения от 1.74 до 1.79, отличающееся тем, что кольцо представляет собой 6-членное кольцо.
1.82 Соединение по любому из предшествующих Вариантов реализации изобретения, отличающееся тем, что когда R1 представляет собой необязательно замещенное 5- или 6-членное кольцо, то оно замещено 0, 1, 2 или 3 заместителями R8.
1.83 Соединение по Варианту реализации изобретения 1.82, отличающееся тем, что присутствуют 0, 1 или 2 заместителя R8.
1.84 Соединение по Варианту реализации изобретения 1.83 отличающееся тем, что присутствует 0 заместителей R8.
1.85 Соединение по Варианту реализации изобретения 1.82, отличающееся тем, что присутствует 1 заместитель R8.
1.86 Соединение по Варианту реализации изобретения 1.82, отличающееся тем, что присутствуют 2 заместителя R8.
1.87 Соединение по любому из Вариантов реализации изобретения 1.81, 1.82, 1.83, 1.85 и 1.86, отличающееся тем, что R8, если присутствует, выбран из фтора; циано; оксо; гидрокси; OR5; NR5R6; COR5; COOR5; OCOR5; NR7COR5; CONR5R6; SR5; SOR5 и SO2R5; и C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомомами фтора и причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S, и их окисленных форм.
1.88 Соединение по Варианту реализации изобретения 1.87, отличающееся тем, что R8 выбран из фтора; циано; оксо; гидрокси; OR5; NR5R6; COR5; COOR5; OCOR5 и SO2R5; и C1-4 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомомами фтора и причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S, и их окисленных форм.
1.89 Соединение по Варианту реализации изобретения 1.88, отличающееся тем, что R8 выбран из фтора; циано; оксо; гидрокси; OR5; NR5R6; и C1-4 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомами фтора.
1.90 Соединение по Варианту реализации изобретения 1.89, отличающееся тем, что R8 выбран из циано; оксо; гидрокси; OR5; NR5R6; и C1-4 алкила.
1.91 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.40 и от 1.42 до 1.53, отличающееся тем, что фрагмент:
 или
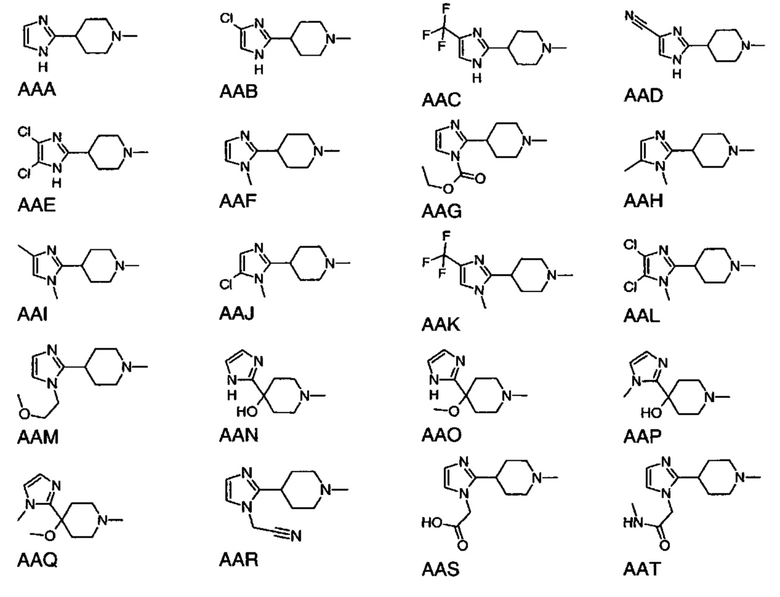
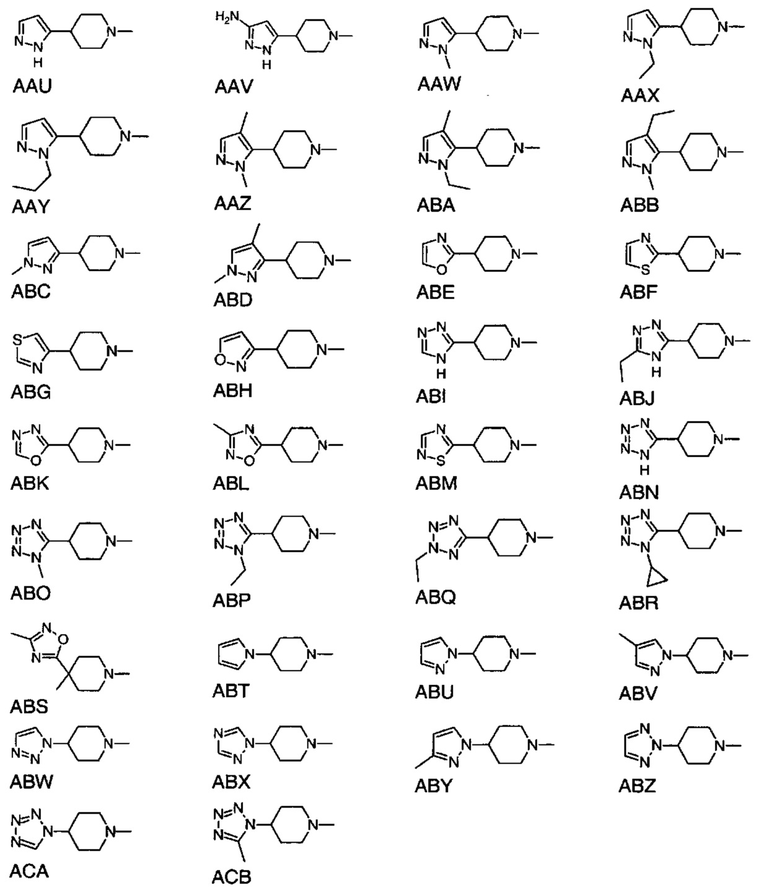
или 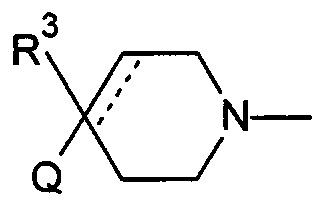
выбран из групп от AAA до АСВ ниже:
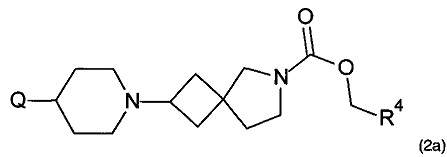
1.92 Соединение формулы (2) или формулы 2а:
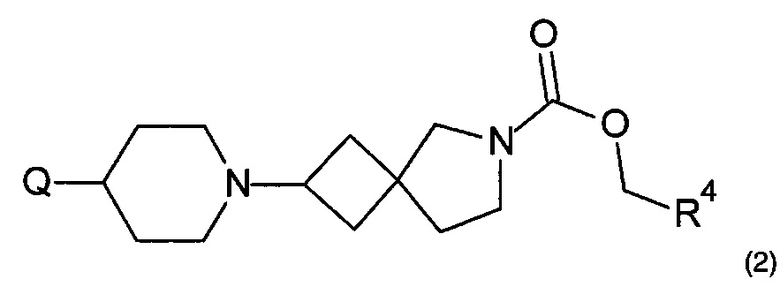 ,
,
где Q представляет собой необязательно замещенный радикал 5 или 6-членного гетероциклического или гетероарильного кольца, имеющий один или более атомов азота, и R4 находится в соответствии с определениями по любому из Вариантов реализации изобретения от 1.48 до 1.53; или
 ,
,
где Q представляет собой необязательно замещенный радикал 5, 6 или 7-членного гетероциклического или гетероарильного кольца, имеющий один или более атомов азота, и R4 находится в соответствии с определениями по любому из Вариантов реализации изобретения от 1.48 до 1.53.
1.93 Соединение согласно формуле (2) или формуле (2а), отличающееся тем, что Q имеет один или более заместителей, например один, два или три заместителя, которые выбраны из (L)-R10, (L)-R11 и (L)-R12, где L представляет собой связь или группу CH2; R10, R11 и R12 независимо выбраны из водорода; фтора; хлора; брома; циано; оксо; гидрокси; OR15; NR15R16; COR15; CSR15; COOR15; COSR15; OCOR15; NR17COR15; CONR15R16; CSNR15R16; NR17CONR15R16; R17COOR15; OCONR15R16; SR15; SOR15 и SO2R15; C1-6 неароматической углеводородной группы, которая необязательно замещена от одного до шести атомомами фтора и причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S, и их окисленных форм; и необязательно замещенного 5- или 6-членного кольца, содержащего 0, 1, 2 или 3 гетероатома, выбранных из O, N и S, и их окисленных форм;
при этом необязательные заместители для необязательно замещенного 5- или 6-членного кольца выбраны из группы R8, состоящей из водорода; фтора; хлора; брома; циано; оксо; гидрокси; OR5; NR5R6; COR5; COOR5; OCOR5; NR7COR5; CONR5R6; NR7CONR5R6; NR7COOR5; OCONR5R6; SR5; SOR5 и SO2R5; и C1-6 неароматическую углеводородную группу, которая необязательно замещена от одного до шести атомомами фтора и причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S, и их окисленных форм;
при этом R15, R16 и R17 являются одинаковыми или разными, или могут быть соединены вместе с образованием кольца, и каждый независимо выбран из водорода, неароматической C1-6 углеводородной группы, необязательно замещенной одним или более атомами фтора и причем один или два, но не все, атомы углерода углеводородной группы могут быть необязательно заменены гетероатомом, выбранным из O, N и S, и их окисленных форм; или группы формулы CH2N(Ra)COORb; или группы формулы (L)-R18, где L представляет собой связь или группу CH2 и R18 представляет собой необязательно замещенное 5- или 6-членное кольцо, содержащее 0, 1, 2 или 3 гетероатома, выбранных из O, N и S, и их окисленных форм;
при этом необязательные заместители для необязательно замещенного 5- или 6-членного кольца выбраны из группы R8.
1.94 Соединение по Вариантам реализации изобретения от 1.1 до 1.93, имеющее формулу (3):
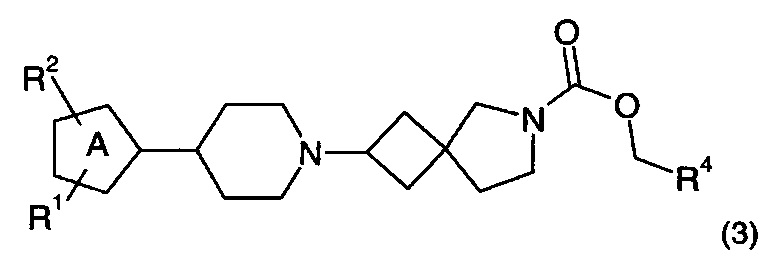
где R1, R2 и R4 находятся в соответствии с определениями по любому из Вариантов реализации изобретения от 1.1 до 1.40 и от 1.42 до 1.90 и кольцо A представляет собой пятичленное гетероциклическое или гетероарильное кольцо, содержащее один или два азот-содержащих кольцевых члена.
1.95 Соединение по Варианту реализации изобретения 1.94, отличающееся тем, что кольцо A представляет собой гетероарильное кольцо, содержащее два азот-содержащих кольцевых члена.
1.96 Соединение по Варианту реализации изобретения 1.95, отличающееся тем, что кольцо A представляет собой имидазольное кольцо.
1.97 Соединение по Варианту реализации изобретения 1.96, имеющее формулу (4):
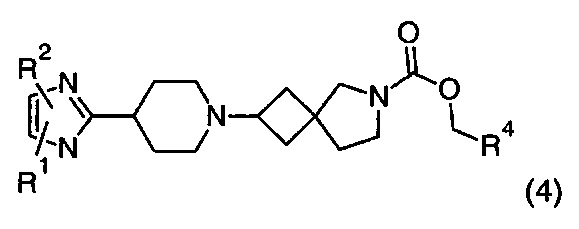
где R1, R2 и R4 находятся в соответствии с определениями по любому из Вариантов реализации изобретения от 1.1 до 1.40 и от 1.42 до 1.90.
1.98 Соединение по Варианту реализации изобретения 1.95, отличающееся тем, что кольцо A представляет собой пиразольное кольцо.
1.99 Соединение по Варианту реализации изобретения 1.98, имеющее формулу (5):
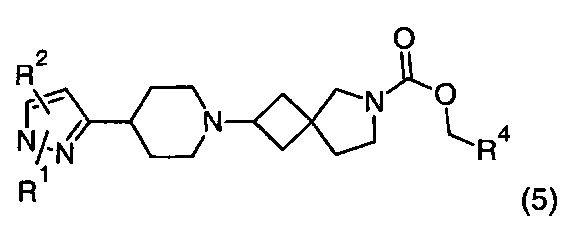
где R1, R2 и R4 находятся в соответствии с определениями по любому из Вариантов реализации изобретения от 1.1 до 1.40 и от 1.42 до 1.90.
1.100 Соединение по Варианту реализации изобретения 1.98, имеющее формулу (6):
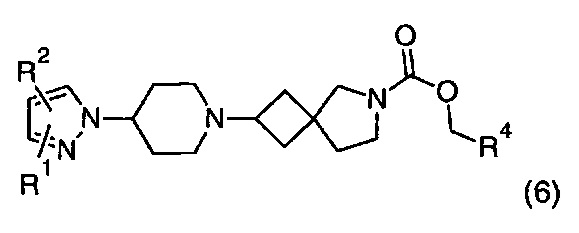
где R1, R2 и R4 находятся в соответствии с определениями по любому из Вариантов реализации изобретения от 1.1 до 1.40 и от 1.42 до 1.90.
1.101 Соединение по Варианту реализации изобретения 1.94, отличающееся тем, что кольцо A представляет собой 5-членное пятичленное гетероциклическое кольцо, содержащее один атом азота.
1.102 Соединение по Варианту реализации изобретения 1.101, имеющее формулу (7):
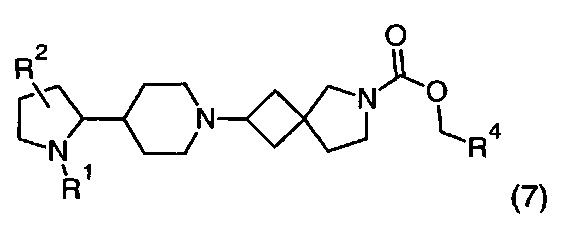
где R1, R2 и R4 находятся в соответствии с определениями по любому из Вариантов реализации изобретения от 1.1 до 1.40 и от 1.42 до 1.90.
1.103 Соединение по Варианту реализации изобретения 1.101, отличающееся тем, что фрагмент:
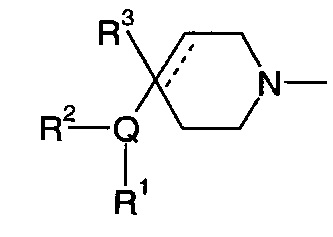 или
или 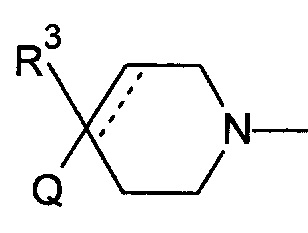
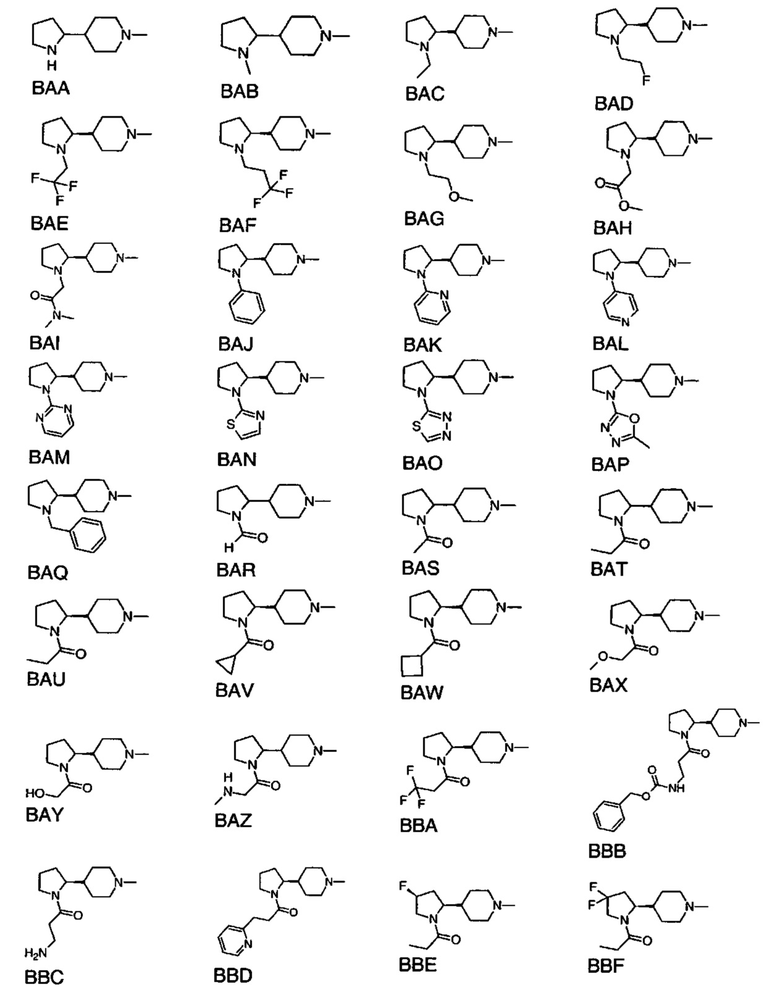
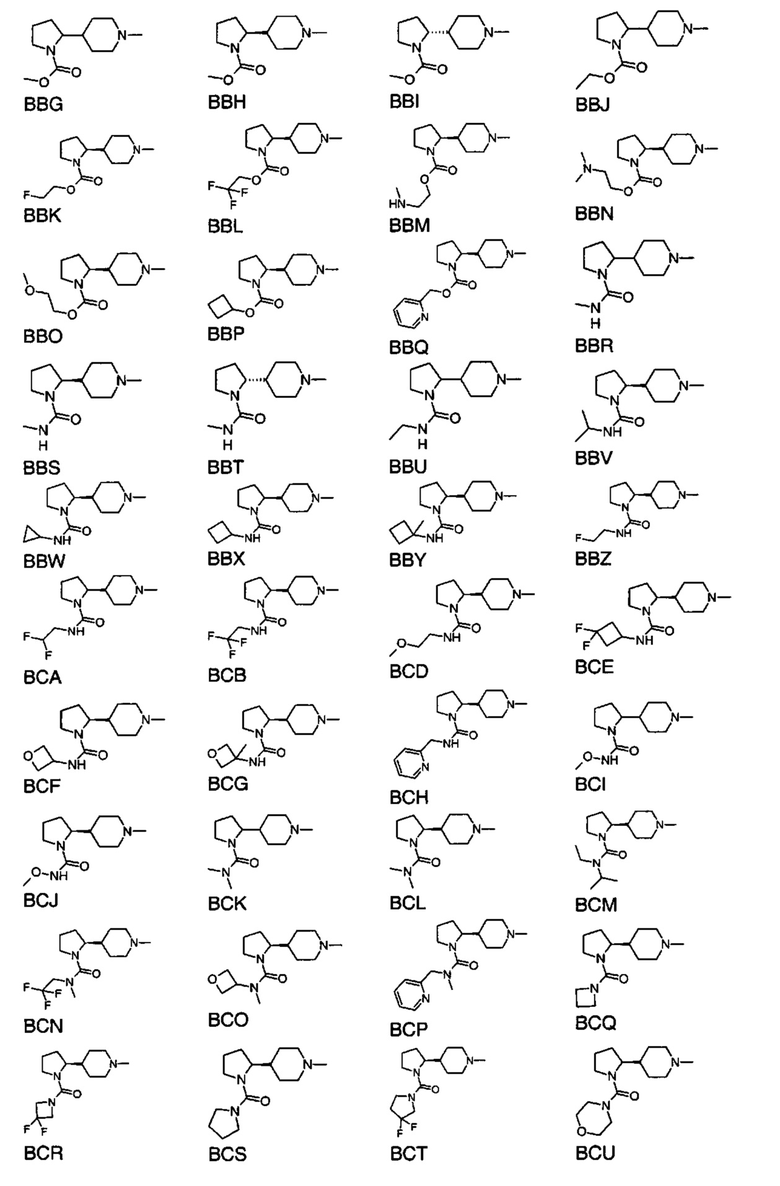
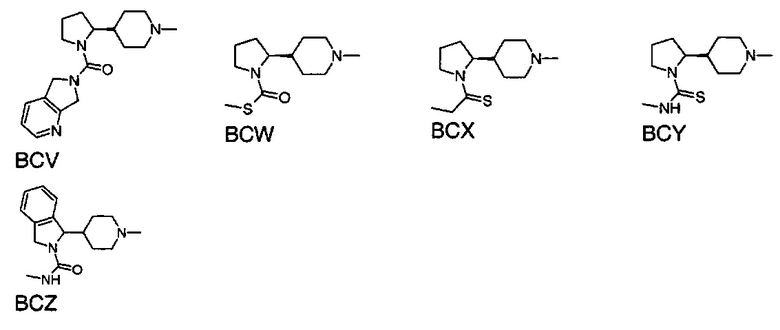
выбран из групп от ВАА до BCZ ниже:
1.104 Соединение по Варианту реализации изобретения 1.101, имеющее формулу (8):
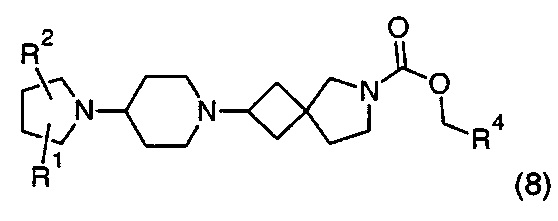
где R1, R2 и R4 находятся в соответствии с определениями по любому из Вариантов реализации изобретения от 1.1 до 1.40 и от 1.42 до 1.90.
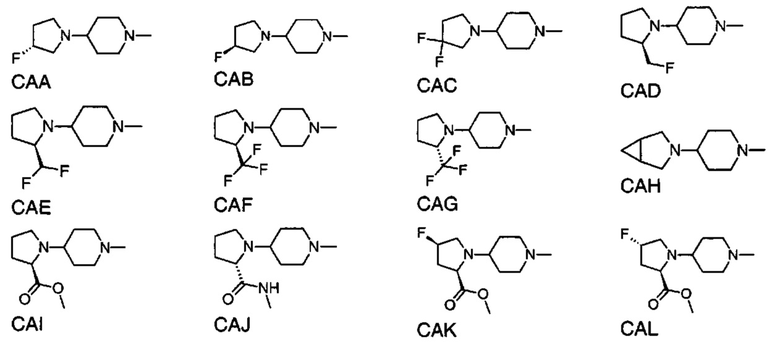
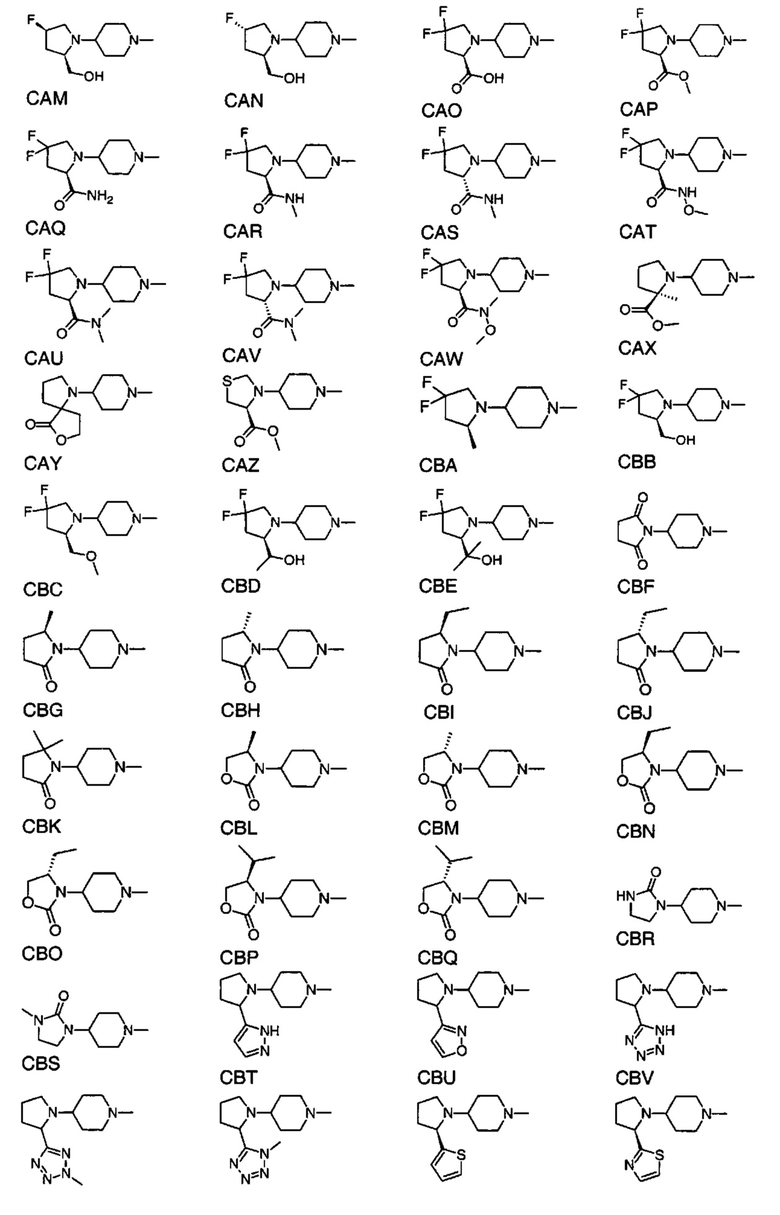
1.105 Соединение по Варианту реализации изобретения 1.101, отличающееся тем, что фрагмент:
 или
или 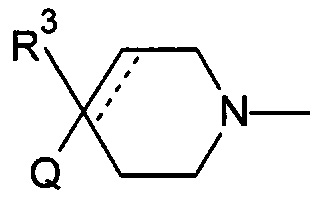
выбран из групп от САА до СВХ ниже:

1.106 Соединение по Варианту реализации изобретения 1.1, отличающееся тем, что Q представляет собой шестичленное моноциклическое гетероциклическое кольцо, содержащее 1, 2, 3 или 4 члена гетероатомного кольца, выбранных из N, O и S.
1.107 Соединение по Варианту реализации изобретения 1.106, отличающееся тем, что фрагмент:
 или
или 
выбран из групп от DAA до DBG ниже:
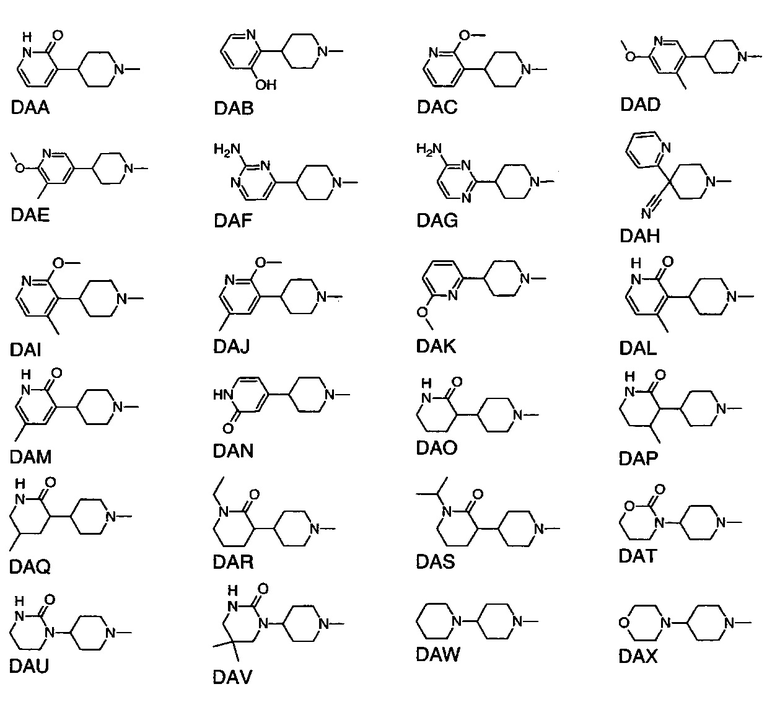
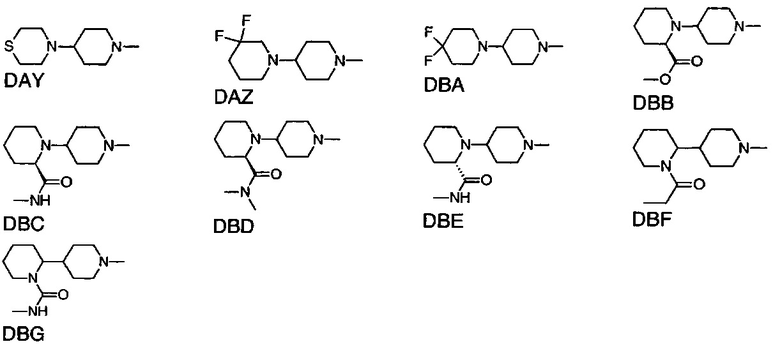
1.108 Соединение по Варианту реализации изобретения 1.1, отличающееся тем, что Q представляет собой семичленное моноциклическое гетероциклическое кольцо, содержащее 1, 2, 3 или 4 члена гетероатомного кольца, выбранных из N, O и S.
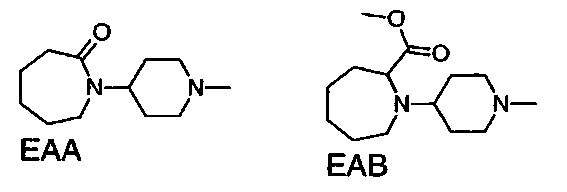
1.109 Соединение по Варианту реализации изобретения 1.108, отличающееся тем, что фрагмент:
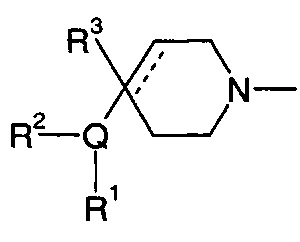 или
или 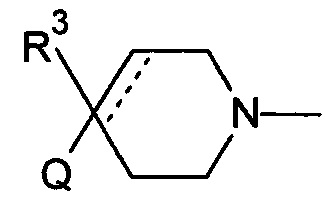
выбран из групп от ЕАА до ЕАВ ниже:
1.110 Соединение по Варианту реализации изобретения 1.1, который находится в соответствии с определениями в любом из Примеров от 1-1 до 1-73, от 2-1 до 2-138, от 3-1 до 3-16, от 4-1 до 4-20 или от 5-1 до 5-2.
1.111 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.110, имеющее молекулярную массу менее чем 550.
1.112 Соединение по Варианту реализации изобретения 1.111, имеющее молекулярную массу менее чем 500.
1.113 Соединение по Варианту реализации изобретения 1.112, имеющее молекулярную массу равную или менее чем 450.
1.114 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.113, которое находится в форме соли.
1.115 Соединение по Варианту реализации изобретения 1.114, отличающееся тем, что соль представляет собой кислотно-аддитивную соль.
1.116 Соединение по Варианту реализации изобретения 1.115 или Варианту реализации изобретения 1.115, отличающееся тем, что соль представляет собой фармацевтически приемлемую соль.
Определения
В данной заявке использованы следующие определения, если не указано иное.
Термин «лечение», в отношении применений соединений формулы (1), (1а) или (1b), использован для описания любой формы воздействия, когда соединение вводится субъекту, страдающему от или с риском страдания от, или с потенциальным риском страдания от указанного заболевания или нарушения. Поэтому термин «лечение» охватывает как профилактическое лечение, так и лечение, когда проявляются измеримые и поддающиеся обнаружению симптомы заболевания или нарушения.
Термином «эффективное терапевтическое количество» в контексте данного документа (например, в отношении способов лечения заболевания или патологического состояния) называют количество соединения, которое является эффективным для получения желаемого терапевтического эффекта. Например, если патологическое состояние представляет собой боль, то эффективное терапевтическое количество представляет собой количество достаточное для обеспечения желаемого уровня облегчения боли. Желаемый уровень облегчения боли может представлять собой, например, полное устранение боли или уменьшение тяжести боли.
Термином «неароматическая углеводородная группа» как в случае «C1-10 неароматической углеводородной группы» или «ациклической C1-5 неароматической углеводородной группы» называют группу, состоящую из атомов углерода и водорода и не содержащую ароматических колец. Углеводородная группа может быть полностью насыщенной или может содержать одну или более двойных углерод-углеродных связей или тройных углерод-углеродных связей, или сочетание двойных и тройных связей. Углеводородная группа может быть группой с линейной или разветвленной цепью или может состоять из или содержать циклическую группу. Поэтому термин неароматический углеводород включает алкил, алкенил, алкинил, циклоалкил, циклоалкенил, циклоалкилалкил, циклоалкенилаклил и др.
Термины «алкил», «алкенил», «алкинил», «циклоалкил» арил, гетероарил и «циклоалкенил» использованы в принятом значении (например, в соответствии с определениями в IUPAC Gold Book), если не указано иное.
Термином «насыщенная углеводородная группа», как в случае «С1-4 насыщенной углеводородной группы», называют углеводородную группу, не содержащую двойных или тройных углерод-углеродных связей. Насыщенная углеводородная группа может, таким образом, представлять собой алкильную группу, циклоалкильную группу, циклоалкилалкильную группу, алкилциклоалкильную группу или алкилциклоалкилалкильную группу. Примеры C1-4 насыщенных углеводородных групп включают C1-4 алкильные группы, циклопропил, циклобутил и циклопропилметил.
Термин «циклоалкил» в контексте данного документа, когда позволяет указанное число атомов углерода, включает как моноциклические циклоалкильные группы, такие как циклопропил, циклобутил, циклопентил, циклогексил и циклогептил, так и бицилкические, и трициклические группы. Бициклические циклоалкильные группы включают мостиковые кольцевые системы, такие как бициклогептан, бициклооктан и адамантан.
В определениях R1, R2, R3 и R4 выше, где указано, один или два, но не все, атомы углерода неароматической углеводородной группы необязательно могут быть заменены на гетероатом, выбранный из O, N и S и (в случае R1 и R4) их окисленных форм. Понятно, что когда атом углерода заменен на гетероатом, более низкие валентности гетероатомов по сравнению с углеродом означают, что меньшее число атомов будет связано с гетероатомом, чем могло бы быть связано с атомом углерода, который бы был заменен. Поэтому, например, замена атома углерода (четырехвалентный) в группе CH2 кислородом (двухвалентный) будет означать, что полученная молекула будет содержать на два атома водорода меньше и замена атома углерода (четырехвалентный) в группе CH2 азотом (трехвалентный) будет означать, что полученная молекула будет содержать на один атом водорода меньше.
Примеры гетероатомных замен для атомов углерода включают замену атома углерода в цепи -CH2-CH2-CH2- на кислород или серу с получением как -CH2-O-CH2-, так и тиоэфира -CH2-S-CH2-, замену атома углерода в группе CH2-С≡С-Н на азот с получением нитрильной (циано) группы CH2-C≡N, замену атома углерода в группе -CH2-CH2-CH2- на C=O с получением кетона -CH2-С(O)-CH2-, замену атома углерода в группе -CH2-CH2-CH2- на S=O или SO2 с получением сульфоксида -CH2-S(O)-CH2- или сульфона -CH2-S(O)2-CH2-, замену атома углерода в цепи -CH2-CH2-CH2- на C(O)NH с получением амида -CH2-CH2-C(O)-NH-, замену атома углерода в цепи -CH2-CH2-CH2- азотом с получением амина -CH2-NH-CH2-, и замену атома углерода в цепи -CH2-CH2-CH2- на C(O)O с получением сложного эфира (или карбоновой кислоты) -CH2-CH2-С(O)-O-. В каждой из таких замен по меньшей мере один атом углерода углеводородной группы должен остаться.
Соли
Большинство соединений формулы (1), (1а) или (1b) может существовать в форме солей, например кислотно-аддитивных солей или, в некоторых случаях, солей органических или неорганических оснований, таких как карбоксилатные, сульфонатные и фосфатные соли. Все такие соли находятся в пределах описания данного изобретения, и ссылки на соединения формулы (1), (1а) или (1b) соединения в форме солей в соответствии с определениями в Вариантах реализации изобретения от 1.114 до 1.116.
В основном соли представляют собой кислотно-аддитивные соли.
Соли по данному изобретению могут быть синтезированы из исходного соединения, которое содержит основный или кислотный фрагмент, с помощью принятых способов, таких как способы, описанные в Pharmaceutical Salts: Properties, Selection, and Use, P. Heinrich Stahl (Editor), Camille G. Wermuth (Editor), ISBN: 3-90639-026-8, Hardcover, 388 страниц, Август 2002. Обычно такие соли могут быть получены с помощью приведения в контакт свободных кислотных или основных форм данных соединений с подходящим основанием или кислотой в воде или органическом растворителе, или смеси обоих; обычно используют неводную среду, такую как эфир, этилацетат, этанол, изопропанол или ацетонитрил.
Кислотно-аддитивные соли (в соответствии с определением в Варианте реализации изобретения 1.120) могут быть образованы с широким разнообразием кислот, как неорганических, так и органических. Примеры кислотно-аддитивных солей, находящиеся в пределах Варианта реализации изобретения 1.120, включают моно- или дисоли, образованные с кислотой, выбранные из группы, состоящей из уксусной, 2,2-дихлоруксусной, адипиновой, альгиновой, аскорбиновой (например, L-аскорбиновой, L-аспарагиновой, бензолсульфоновой, бензойной, 4-ацетамидобензойной, масляной, (+) камфорной, камфорсульфоновой, (+)-(1S)-камфор-10-сульфоновой, каприновой, капроновой, каприловой, коричной, лимонной, цикламовой, додецилсерной, этан-1,2-дисульфоновой, этансульфоновой, 2-гидроксиэтансульфоновой, муравьиной, фумаровой, слизевой, гентизиновой, глюкогептоновой, D-глюконовой, глюкуроновой (например, D-глюкуроновой), глутаминовой (например, L-глутаминовой), α-оксоглутаровой, гликолевой, гиппуровой, галогенводородных кислот (например, бромистоводородной, соляной, иодистоводородной), изэтионовой, молочной (например, (+)-L-молочной, (±)-DL-молочной), лактобионовой, малеиновой, яблочной, (-)-L-яблочной, малоновой, (±)-DL-миндальной, метансульфоновой, нафталин-2-сульфоновой, нафталин-1,5-дисульфоновой, 1-гидрокси-2-нафтойной, никотиновой, азотной, олеиновой, оротовой, щавелевой, пальмитиновой, памовой, фосфорной, пропионовой, пировиноградной, L-пироглутаминовой, салициловой, 4-амино-салициловой, себациновой, стеариновой, янтарной, серной, дигалловой, (+)-L-винной, тиоциановой, р-толуолсульфоновой, ундеценовой и валериановой кислот, также как и ацилированных аминокислот и катионообменных смол.
Если соединения формулы (1), (1а) или (1b) содержат функциональную амино-группу, они могут образовывать четвертичные аммонийные соли, например, по реакции с алкилирующим агентом, согласно способам, известным специалисту в данной области техники. Такие четвертичные аммонийные соединения находятся в пределах объема формулы (1), (1а) или (1b).
Соединения по данному изобретению могут существовать в виде моно- или дисолей, в зависимости от pKa кислоты из которой образуется соль.
Солевые формы соединений по данному изобретению обычно представляют собой фармацевтически приемлемые соли, и примеры фармацевтически приемлемых солей рассмотрены в Berge et al.., 1977, "Pharmaceutically Acceptable Salts," J. Pharm. Sci., Том 66, с. 1-19. Тем не менее, соли, которые не являются фармацевтически приемлемыми, могут быть получены в качестве форм промежуточных соединений, которые затем могут быть превращены в фармацевтически приемлемые соли. Такие формы фармацевтически неприемлемых солей, которые могут быть пригодны, например, при очистке или разделении соединений по данному изобретению, также являются частью данного изобретения.
Стереоизомеры
Стереоизомеры представляют собой изомерные молекулы, имеющие одинаковую молекулярную формулу и последовательность связанных атомов, но отличающиеся только трехмерной ориентацией их связей в пространстве. Стереоизомеры могут быть, например, геометрическими изомерами или оптическими изомерами.
Геометрические Изомеры
В геометрических изомерах изомерия обусловлена различными положениями атома или группы относительно двойной связи, как в цис и транс (Z и Е) изомерии относительно двойной углерод-углеродной связи или цис и транс изомерах относительно амидной связи, или син и анти изомерии относительно двойной связи углерод-азот (например, в оксиме), или ротационной изомерии относительно связи, вокруг которой ограничено вращение, или цис и транс изомерии относительно кольца, такого как циклоалкановое кольцо.
Соответственно, в другом варианте реализации изобретения (Вариант реализации изобретения 1.121) в изобретении предложен геометрический изомер соединения по любому из Вариантов реализации изобретения от 1.1 до 1.116.
Оптические Изомеры
Если соединения данной формулы содержат один или более хиральных центров и могут существовать в форме двух или более оптических изомеров, то ссылки на соединения включают все их оптически изомерные формы (например, энантиомеры, эпимеры и диастереомеры), как и индивидуальные оптические изомеры, или смеси (например, рацемические смеси) так и два или более оптических изомеров, если контекст описания не требует другого.
Соответственно, в другом варианте реализации изобретения (Вариант реализации изобретения 1.132) в изобретении предложено соединение по любому из Вариантов реализации изобретения от 1.1 до 1.121, которое содержит хиральный центр.
Оптические изомеры могут быть охарактеризованы и идентифицированы по их оптической активности (например, как + и - изомеры, или d и I изомеры) или они могут быть охарактеризованы в терминах их абсолютной стереохимической конфигурации, используя «R и S» номенклатуру, предложенную Каном, Ингольдом и Прелогом, см. Advanced Organic Chemistry by Jerry March, 4th Edition, John Wiley & Sons, Нью-Йорк, 1992, с. 109-114, и также см. Cahn, Ingold & Prelog, Angew. Chem. Int. Ed. Engl., 1966, 5, 385-415. Оптические изомеры могут быть разделены с помощью ряда техник, включая хиральную хроматографию (хроматографию на хиральной фазе) и таких техник, которые хорошо известны специалисту в данной области техники. В качестве альтернативы хиральной хроматографии, оптические изомеры могут быть разделены путем образования диастереомерных солей с хиральными кислотами, такими как (+)-винная кислота, (-)-пироглутаминовая кислота, (-)-ди-толуоил-Ьвинная кислота, (+)-миндальная кислота, (-)-яблочная кислота, и (-)-камфорсульфоновая, разделяя диастереомеры избирательной кристаллизацией, и затем разложив соли с получением индивидуальных энантиомеров в форме свободных оснований.
Если соединения по данному изобретению существуют в виде двух или более форм оптических изомеров, один энантиомер из пары энантиомеров может показать преимущества относительно другого энантиомера, например, в терминах биологической активности. Поэтому, при определенных обстоятельствах, может быть желательно использование в качестве лекарственного средства только одного из пары энантиомеров или только одного из множества диастереомеров.
Соответственно, в другом варианте реализации изобретения (Вариант реализации изобретения 1.133), в изобретении предложены композиции, содержащие соединение согласно Варианту реализации изобретения 1.132, имеющее один или более хиральных центров, в котором по меньшей мере 55% (например, по меньшей мере 60%, 65%, 70%, 75%, 80%, 85%, 90% или 95%) соединения по Варианту реализации изобретения 1.108 присутствует в виде одного оптического изомера (например, энантиомера или диастереомера).
В одном общем варианте реализации изобретения (Вариант реализации изобретения 1.134), 99% или более (например, существенная часть) общего количества соединения (или соединения к использованию) соединения по Варианту реализации изобретения 1.132 присутствует в форме одного оптического изомера.
Например, в одном варианте реализации изобретения (Вариант реализации изобретения 1.135) соединение присутствует в виде одного энантиомера.
В другом варианте реализации изобретения (Вариант реализации изобретения 1.136), соединение присутствует в виде одного диастереомера.
В изобретении также предложены смеси оптических изомеров, которые могут быть рацемическими или нерацемическими. Так, в изобретении предложено:
1.137 Соединение по Варианту реализации изобретения 1.132, которое находится в форме рацемической смеси оптических изомеров.
1.138 Соединение по Варианту реализации изобретения 1.132, которое находится в форме нерацемической смеси оптических изомеров.
Изотопы
Соединения по данному изобретению в соответствии с определениями по любому из Вариантов реализации изобретения от 1.1 до 1.138 могут содержать одно или более изотопных замещений, и ссылка на определенный элемент находится в пределах объема всех изтопов данного элемента. Например, ссылка на водород находится в пределах всего объема его изотопов 1Н, 2Н (D) и 3Н (Т). Аналогично, ссылки на углерод или кислород находятся в пределах объема, соответственно 12С, 13С и 14С, и 16О, и 18O.
Аналогичным образом, ссылка на конкретную функциональную группу также находится в пределах объема ее изотопных вариаций, если в контексте данного описания не указано иного. Например, ссылка на алкильную группу, такую как этильная группа также включает вариации, в которых один или более атомов водорода в группе находится в форме дейтериевого или тритиевого изотопа, например, как в этильной группе, в которой все пять атомов водорода находятся в форме дейтериевого изотопа (пердейтероэтильная группа).
Изотопы могут быть радиоактивными или нерадиоактивными. В одном варианте реализации данного изобретения (Вариант реализации изобретения 1.140) соединение по любому из Вариантов реализации изобретения от 1.1 до 1.138 не содержит радиоактивных изотопов. Такие соединения являются предпочтительными для использования в терапевтических целях. В другом варианте реализации изобретения (Вариант реализации изобретения 1.141),тем не менее, соединение по любому из Вариантов реализации изобретения от 1.1 до 1.138 может содержать один или более радиоактивных изотопов. Соединения, содержащие такие радиоактивные изотопы, могут быть пригодными в диагностических целях.
Сольваты
Соединения формулы (1), (1а) или (1b) в соответствии с определениями по любому из Вариантов реализации изобретения от 1.1 до 1.141 могут образовывать сольваты. Предпочтительные сольваты представляют собой сольваты, образованные путем введения в находящуюся в твердом состоянии (например, кристаллическую структуру) структуру соединений по данному изобретению молекулы нетоксичного фармацевтически приемлемого растворителя (далее именуемого как сольватирующий растворитель). Примеры таких растворителей включают воду, спирты (такие как этанол, изопропанол и бутанол) и диметилсульфоксид. Сольваты могут быть получены путем перекристаллизации соединений по данному изобретению из растворителя или смеси растворителей, содержащей сольватирующий растворитель. Образуются или нет сольваты в любом отдельно взятом случае может определяться с помощью анализа кристаллов соединений, используя хорошо известные и стандартные техники, такие как термический гравиметрический анализ (TGE), дифференциальная сканирующая калориметрия (DSC) и рентгеновская кристаллография. Сольваты могут представлять собой стехиометрические или нестехиометрические сольваты. Конкретные предпочтительные сольваты представляют собой гидраты и примеры гидратов включают гемигидраты, моногидраты и дигидраты.
Соответственно, в дополнительных вариантах реализации изобретения 1.150 и 1.151 в изобретении предложено:
1.151 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.141 в форме сольвата.
1.152 Соединение по Варианту реализации изобретения 1.151, отличающееся тем, что сольват представляет собой гидрат.
Для более подробного рассмотрения сольватов и способов, использованных для их получения и характеризации, см. Bryn et al, Solid-State Chemistry of Drugs, Second Edition, опубликована SSCI, Inc of West Lafayette, Индиана, США, 1999, ISBN 0-967-06710-3.
Альтернативно, более вероятно, чем в гидратной, соединение по данному изобретению может находится в безводной форме. Поэтому, в другом варианте реализации изобретения (Вариант реализации изобретения 1.153) в изобретении предложено соединение в соответствии с определениями по любому из Вариантов реализации изобретения от 1.1 до 1.141 в безводной форме (например, безводной кристаллической форме).
Кристаллические и аморфные Формы
Соединения по любому из Вариантов реализации изобретения от 1.1 до 1.153 может существовать в кристаллической или некристаллической (например, аморфной) форме. Существует или нет соединение в кристаллической форме может быть легко определено с помощью стандартных техник, таких как порошковая рентгеновская дифракция (XRPD). Кристаллы и их кристаллические структуры могут быть охарактеризованы, используя ряд техник, включая рентгеновскую кристаллографию монокристалла, порошковую рентгеновскую дифракцию (XRPD), дифференциальную сканирующую калориметрию (DSC) и инфракрасную спектроскопию, например ИК-спектроскопию с Фурье-преобразованием (FTIR). Поведение кристаллов в условиях с переменной влажностью может анализироваться путем исследований гравиметрической сорбции пара и также с помощью XRPD. Определение кристаллической структуры соединений может быть выполнено с помощью рентгеновской кристаллографии, которая может быть проведена в соответствии с традиционно принятыми способами, такими как те, которые описаны в данном документе и описаны в Fundamentals of Crystallography, С. Giacovazzo, Н.L. Monaco, D. Viterbo, F. Scordari, G. Gilli, G. Zanotti и M. Catti, (International Union of Crystallography / Oxford University Press, 1992 ISBN 0-19-855578-4 (p/b), 0-19-85579-2 (h/b)). Данная техника включает анализ и интерпретацию рентгеновской дифракции монокристалла. В аморфном теле обычно существующие в кристаллической форме, трехмерные структуры не существуют и положения молекул относительно друг друга в аморфном теле являются исключительно случайными, см, например, Hancock et al J. Pharm. Sci. (1997), 86, 1).
Соответственно, в дополнительных вариантах реализации изобретения, в изобретении предложено:
1.160 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.153 в кристаллической форме.
1.161 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.153, которое находится:
(а) от 50% до 100% в кристаллической форме, и более конкретно находится по меньшей мере 50% в кристаллической форме или по меньшей мере 60% в кристаллической форме, или по меньшей мере 70% в кристаллической форме, или по меньшей мере 80% в кристаллической форме, или по меньшей мере 90% в кристаллической форме, или по меньшей мере 95% в кристаллической форме, или по меньшей мере 98% в кристаллической форме, или по меньшей мере 99% в кристаллической форме, или по меньшей мере 99,5% в кристаллической форме, или по меньшей мере 99,9% в кристаллической форме, например 100% в кристаллической форме.
1.162 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.153, которое находится в аморфной форме.
Пролекарства
Соединения формулы (1), (1а) или (1b) в соответствии с определениями по любому из Вариантов реализации изобретения от 1.1 до 1.162 могут быть представлены в форме пролекарства. Под «пролекарствами» подразумевают, например, любое соединение, которое превращается in vivo в биологически активное соединение формулы (1), (1а) или (1b), в соответствии с определениями по любому из Вариантов реализации изобретения от 1.1 до 1.162.
Например, некоторые пролекарства представляют собой сложные эфиры активного соединения (например, физиологически приемлемый метаболически разлагаемый сложный эфир). Во время метаболизма сложноэфирная группа (-C(=O)OR) расщепляется с образованием активного лекарства. Такие сложные эфиры могут быть образованы путем эстерификации, например, любой гидроксильной группы, присутствующей в родительском соединении, где необходимо, предшествуя защите любой другой реакционноспособной группы, присутствующей в родительском соединении, с последующей депротекцией, если это необходимо.
Также некоторые пролекарства активируются под воздействием ферментов с образованием активного соединения или соединения, которое в условиях дополнительной химической реакции дает активное соединение (например, как в ADEPT, GDEPT, LIDEPT, и т.д.). Например, пролекарство может представлять собой производное сахара или другого гликозидного конъюгата, или может представлять собой сложноэфирное производное аминокислоты.
Соответственно, в другом варианте реализации изобретения (Вариант реализации изобретения 1.170) в изобретении предложено пролекарства соединения в соответствии с определениями по любому из Вариантов реализации изобретения от 1.1 до 1.170, причем соединение содержит функциональную группу, способную превращаться в физиологических условиях с образованием гидроксильной группы или амино-группы.
Комплексы и клатраты
Также формулой (1), (1а) или (1b) в Вариантах реализации изобретения от 1.1 до 1.170 охвачены комплексы (например, комплексы включения или клатраты с соединениями, такими как циклодекстрины или комплексы с металлами) соединений по Вариантам реализации изобретения от 1.1 до 1.170.
Соответственно, в другом варианте реализации изобретения (Вариант реализации изобретения 1.180) в изобретении предложено соединение по любому из Вариантам реализации изобретения от 1.1 до 1.170 в форме комплекса или клатрата.
Биологическая активность и использование в терапевтических целях
Соединения по данному изобретению обладают активностью в качестве агонистов рецептора М1. Мускариновая активность соединений может быть определена, используя исследование Фосфо-ERK1/2, описанное ниже в Примере А.
Значительное преимущество соединений по данному изобретению состоит в том, что они являются высоко селективными для рецептора М1 относительно М2 и М3 подтипов рецепторов. Соединения по данному изобретению не являются агонистами М2 и М3 подтипов рецепторов. Например, при этом соединения по данному изобретению обычно имеют значения рЕС50 по меньшей мере 6 (предпочтительно по меньшей мере 6,5) и значения Emax более чем 80 (предпочтительно более, чем 95) относительно рецептора М1 в функциональном исследовании, описанном в Примере А, они могут иметь значения рЕС50 менее чем 5 и значения Emax менее чем 20%, когда тестировались относительно подтипов М2 и М3 в функциональном исследовании Примера А.
Некоторые соединения по данному изобретению также являются высоко селективными для рецептора М4 относительно рецептора М1. Примеры таких соединений включают соединение из Примеров 1-6, 1-9, 1-21 и 2-17.
Другие соединения по данному изобретению обладают активностью по отношению к обоим рецепторам М1 и М4. Примеры таких соединений включают соединения из Примеров от 1-1 до 1-4 и от 1-8 до 1-10 и 2-116.
Соответственно, в Вариантах реализации изобретения от 2.1 до 2.9 в изобретении предложены:
2.1 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.180 для применения в медицине.
2.2 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.180 для применения в качестве агониста мускариновых рецепторов М1 и/или М4.
2.3 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.180, которое представляет собой агонист мускаринового рецептора М1, имеющее рЕС50 в диапазоне от 6,0 до 8,1 и Еmax, равный по меньшей мере 90 относительно рецептора М1 в исследовании Примера A данного описания или в прилагаемом к нему полностью аналогичном исследовании.
2.4 Соединение по Варианту реализации изобретения 2.3, которое представляет собой агонист мускаринового рецептора М1, имеющее рЕС50 в диапазоне от 6,5 до 7,5.
2.5 Соединение по Варианту реализации изобретения 2.3 или Варианту реализации изобретения 2.4, имеющее Еmax, равный по меньшей мере 95 относительно рецептора М1.
2.6 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.180, которое представляет собой агонист мускаринового рецептора М4, имеющее рЕС50 в диапазоне от 6,0 до 9,0 и Еmax по меньшей мере 90 относительно рецептора М4 в исследовании Примера A данного описания или в прилагаемом к нему полностью аналогичном исследовании.
2.7 Соединение по Варианту реализации изобретения 2.6, которое представляет собой агонист мускаринового рецептора М4, имеющее рЕС50 в диапазоне от 6,5 до 9,0.
2.8 Соединение по Варианту реализации изобретения 2.6 или Варианту реализации изобретения 2.7, имеющее Еmax, равный по меньшей мере 95 относительно рецептора М4.
2.9 Соединение по любому из Вариантов реализации изобретения от 2.3 до 2.8, которое является селективным для рецепторов М1 и/или М4 по сравнению с мускариновыми рецепторами М2 и М3.
2.10 Соединение по Варианту реализации изобретения 2.9, которое является селективным для рецептора М1 по сравнению с мускариновыми рецепторами М2 и М3.
2.11 Соединение по Варианту реализации изобретения 2.9, которое является селективным для рецептора М4 по сравнению с мускариновыми рецепторами М2 и М3.
2.12 Соединение по любому из Вариантов реализации изобретения от 2.3 до 2.5, которое является селективным для рецептора М1 по сравнению с мускариновыми рецепторами М2, М3 и М4.
2.13 Соединение по любому из Вариантов реализации изобретения от 2.6 до 2.8, которое является селективным для рецептора М4 по сравнению с мускариновыми рецепторами М1, М2 и М3.
2.14 Соединение по любому из Вариантов реализации изобретения от 2.3 до 2.8, которое является селективным для рецепторов М1 и М4 по сравнению с мускариновыми рецепторами М2 и М3.
2.15 Соединение по любому из Вариантов реализации изобретения от 2.3 до 2.14, которое имеет рЕС50 менее чем 5 и Еmax менее чем 50 относительно подтипов М2 и М3 мускариновых рецепторов.
2.16 Соединение по Варианту реализации изобретения 2.15, которые имеют рЕС50 менее чем 4,5 и/или Еmax менее чем 30 относительно подтипов М2 и М3 мускариновых рецепторов.
2.17 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.180 и по Вариантам реализации изобретения от 2.3 до 2.16 для применения при лечении болезни или паталогического состояния, опосредованного мускариновым М1 рецептором.
Ввиду своей активности в качестве агонистов мускариновых М1 и/или М4 рецепторов, соединения по данному изобретению могут быть применены при лечении болезни Альцгеймера, шизофрении и других психических расстройств, когнитивных расстройств и других болезней, опосредованных мускариновыми М1 и/или М4 рецепторами и могут быть также применены при лечении различных типов боли.
Соответственно, в Вариантах реализации изобретения от 2.18 до 2.34, в изобретении предложено:
2.18 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.180 для применения при лечении когнитивного расстройства или психического расстройства.
2.19 Соединение для применения по Варианту реализации изобретения 2.18, отличающееся тем, что когнитивное расстройство или психическое расстройство включает, вызвано или связано с паталогическим состоянием, выбранным из когнитивного нарушения, умеренного когнитивного нарушения, лобно-височной деменции, мультиинфарктной деменции, деменции с тельцами Леви, пресенильной деменции, сенильной деменции, атаксии Фридрейха, синдрома Дауна, болезни Гентингтона, гиперкинезии, мании, синдрома Туретта, болезни Альцгеймера, прогрессирующего супрануклеарного пареза взора, ухудшения когнитивных функций, включая нарушение внимания, ориентации, способности к обучению, памяти (т.е. расстройство памяти, амнезию, амнестические расстройства, синдром транзиторной глобальной амнезии и связанные с возрастом нарушения памяти) и речевую функцию; когнитивного нарушения как результата инсульта, болезни Гентингтона, болезни Пика, СПИД-ассоциированной деменции или других состояний деменции, таких как мультиинфарктная деменция, алкогольная деменция, гипотиреозная деменция и деменция, асоциированная с другими дегенеративными заболеваниями, такими как атрофия мозжечка и боковой амиотрофический склероз; других острых или подострых патологических состояний, которые могут обусловить снижение когнитивных способностей, таких как делирий или депрессия (псевдодементные состояния) травма, травма головы, связанное с возрастом снижение когнитивных способностей, инсульт, нейродегенерация, обусловленные действием наркотиков состояния, нейротоксичные агенты, возрастные когнитивные нарушения, связанные с аутизмом когнитивные нарушения, синдром Дауна, относящееся к психозу когнитивное расстройство и когнитивные нарушения после электросудорожной терапии; вызванных злоупотреблением наркотиков когнитивных нарушений или отмены наркотического средства, включая никотин, каннабис, амфетамин, кокаин, синдрома дефицита внимания с гиперактивностью (СДВГ) и дискинетических расстройств, таких как болезнь Паркинсона, вызванный нейролептиками паркинсонизм и поздняя дискинезия, шизофрения, шизофреноформные расстройства, психотическая депрессия, мания, острая мания, параноидальные, галлюциногенные и бредовые расстройства, личностные расстройства, синдромы навязчивых состояний, шизотипические расстройства, бредовые расстройства, психоз благодаря злокачественной опухоли, нарушение обмена веществ, эндокринное заболевание или нарколепсия, психоз благодаря злоупотреблению наркотиками или отмене накотического средства, биполярные эффектные расстройства, эпилепсия и шизоаффектное расстройство.
2.20 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.180 для применения при лечении болезни Альцгеймера.
2.21 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.180 для применения при лечении шизофрении.
2.22 Способ лечения когнитивного расстройства у субъекта (например, млекопитающего больного, такого как человек, например, человек, нуждающийся в таком лечении), который включает введение терапевтически эффективной дозы соединения по любому из Вариантов реализации изобретения от 1.1 до 1.180.
2.23 Способ по Варианту реализации изобретения 2.20, отличающийся тем, что когнитивное расстройство включает, вызвано или связано с паталогическим состоянием в соответствии с определением в Варианте реализации изобретения 2.19.
2.24 Способ по Варианту реализации изобретения 2.23, отличающийся тем, что когнитивное расстройство вызвано или связано с болезнью Альцгеймера.
2.25 Способ по Варианту реализации изобретения 2.24, отличающийся тем, что когнитивное расстройство представляет собой шизофрению.
2.26 Применение соединения по любому из Вариантов реализации изобретения от 1.1 до 1.180 для изготовления лекарственных средств для лечения когнитивного расстройства.
2.27 Применение по Варианту реализации изобретения 2.26, отличающееся тем, что когнитивное расстройство включает, вызвано или связано с паталогическим состоянием в соответствии с определением в Варианте реализации изобретения 2.11.
2.28 Применение по Варианту реализации изобретения 2.27, отличающееся тем, что когнитивное расстройство вызвано или связано с болезнью Альцгеймера.
2.29 Применение по Варианту реализации изобретения 2.29, отличающееся тем, что когнитивное расстройство представляет собой шизофрению.
2.30 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.180 для лечения или уменьшения тяжести острой, хронической, невропатической или воспалительной боли, артрита, мигрени, кластерной головной боли, тригеминальной невралгии, невралгии при опоясывающем лишае, общей невропатологии, висцеральной боли, остеоартритической боли, постгерпетической невралгии, диабетической невропатии, радикулита, ишиаса, боли в пояснице, боли в голове и шее, острой и неукротимой боли, ноцицептивной боли, прорыва боли, послеоперационной боли или раковой боли.
2.31 Способ лечения или уменьшения тяжести острой, хронической, невропатической или воспалительной боли, артрита, мигрени, кластерной головной боли, тригеминальной невралгии, невралгии при опоясывающем лишае, общей невропатологии, висцеральной боли, остеоартритической боли, постгерпетической невралгии, диабетической невропатии, радикулита, ишиаса, боли в пояснице, боли в голове и шее, острой и неукротимой боли, ноцицептивной боли, прорыва боли, послеоперационной боли или раковой боли, включающий введение терапевтически эффективной дозы соединения по любому из Вариантов реализации изобретения от 1.1 до 1.180.
2.32 Соединение по любому из Вариантов реализации изобретения от 1.1 до 1.180 для лечения заболеваний периферической нервной системы, таких как уменьшение внутриглазного давления при глаукоме, и лечения сухости глаз и сухости во рту, включая синдром Гужеро-Шегрена.
2.33 Способ лечения заболеваний периферической нервной системы, таких как уменьшение внутриглазного давления при глаукоме, и лечения сухости глаз и сухости во рту, включая синдром Гужеро-Шегрена, включающий введение терапевтически эффективной дозы соединения по любому из Вариантов реализации изобретения от 1.1 до 1.180.
2.34 Применение соединения по любому из Вариантов реализации изобретения от 1.1 до 1.180 для изготовления лекарственных средств для лечения или уменьшения тяжести острой, хронической, невропатической или воспалительной боли, артрита, мигрени, кластерной головной боли, тригеминальной невралгии, невралгии при опоясывающем лишае, общей невропатологии, висцеральной боли, остеоартритической боли, постгерпетической невралгии, диабетической невропатии, радикулита, ишиаса, боли в пояснице, боли в голове и шее, острой и неукротимой боли, ноцицептивной боли, прорыва боли, послеоперационной боли или раковой боли или для лечения заболеваний периферической нервной системы, таких как уменьшение внутриглазного давления при глаукоме, и лечения сухости глаз и сухости во рту, включая синдром Гужеро-Шегрена.
2.35 Применение соединения по любому из Вариантов реализации изобретения от 1.1 до 1.180 для лечения зависимости.
2.36 Применение соединения по любому из Вариантов реализации изобретения от 1.1 до 1.180 для лечения двигательных расстройств, таких как болезнь Паркинсона, СДВГ, болезнь Хантингтона, синдром Туретта и другие синдромы, связанные с допаминергической дисфункцией в качестве основного фактора патогенеза, вызывающего заболевание.
Способы получения соединений формулы (1), (1а) или (1b)
Соединения формулы (1), (1а) или (1b) могут быть получены в соответствии с синтетическими способами, известными специалисту в данной области техники и согласно настоящему описанию.
Соответственно, в другом варианте реализации изобретения (Вариант реализации изобретения 3.1), в изобретении предложен процесс получения соединения в соответствии с определениями по любому из Вариантов реализации изобретения от 1.1 до 1.180, в которых процесс включает:
(A) приведение соединения формулы (10) в контакт
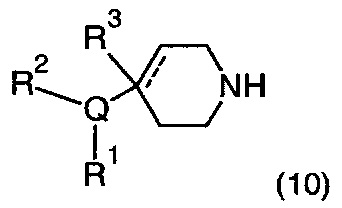
с соединением формулы (11):
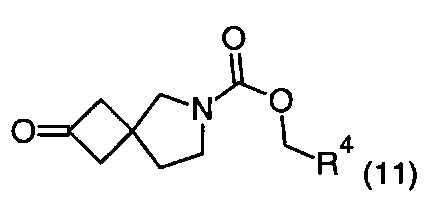
в условиях восстановительного аминирования; причем R1, R2, R3, R4 и Q находятся в соответствии с определениями по любому из Вариантов реализации изобретения от 1.1 до 1.180; или
(B) приведение соединения формулы (12) в контакт:
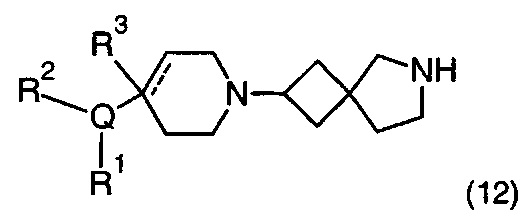
с соединением формулы Cl-C(=O)-CH2-R4, в присутствии основания; или
(C) приведение соединения формулы (10) в контакт
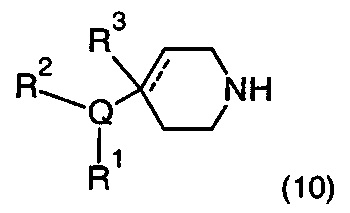
с соединением формулы (13):
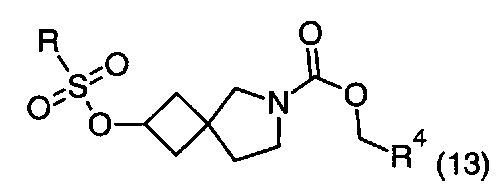
в условиях реакции нуклеофильного замещения; причем R1, R2, R3, R4 и Q находятся в соответствии с определениями по любому из Вариантов реализации изобретения от 1.1 до 1.180; и необязательно:
(D) превращение соединения формулы (1), (1а) или (1b) в другое соединение формулы (1), (1а) или (1b).
В варианте (А) процесса пиперидиновый гетероцикл (10) приводят в контакт с замещенным кетоном (11) в условиях восстановительного аминирования. Реакцию восстановительного аминирования обычно проводят при температуре окружающей среды, используя борогидридный восстанавливающий агент, такой как триацетоксиборогидрид натрия в растворителе, таком как дихлорметан или дихлорэтан, содержащем уксусную кислоту.
В варианте (С) процесса пиперидиновый гетероцикл (10) приводят в контакт с сульфоновым эфиром (13, R = метил, трифторметил или 4-метилфенил) в условиях реакции нуклеофильного замещения, которую обычно проводят при небольшом нагревании (например, до температуры от около 40°C до около 70°C) как в сухую, без растворителя, так и в подходящем растворителе, таком как тетрагидрофуран, ацетонитрил или диметилацетамид.
Промежуточные соединения формулы (12) могут быть получены серией реакций, проиллюстрированной ниже в Схеме 1.
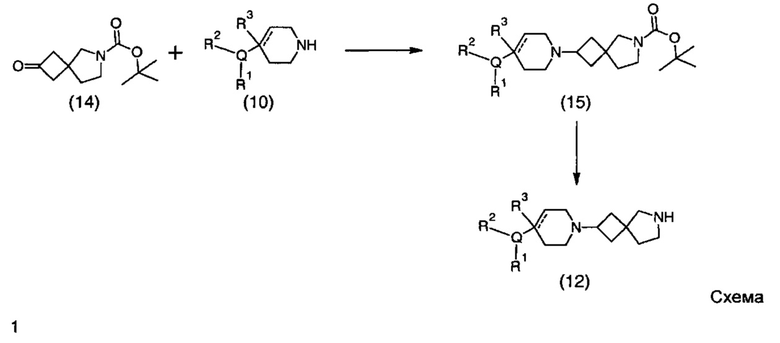
В реакционной Схеме 1 пиперидиновый гетероцикл (10) приводят в контакт с Boc-защищенным спирокетоном (14) в условиях восстановительного аминирования. Реакцию восстановительного аминирования обычно проводят при небольшом нагревании (например, до температуры от около 40°C до около 70°C) в присутствии как цианоборогидрида натрия, так и в комбинации с хлоридом цинка или триацетоксиборогидрида натрия в комбинации с изопропоксидом титана в растворителе, таком как дихлорметан или дихлорэтан, содержащем уксусную кислоту, с получением промежуточного пиперидинового соединения (15), в котором затем проводят депротекцию с помощью удаления Boc-группы путем обработки кислотой (например, трифторуксусной кислотой в дихлорметане) с получением соединения (12).
Соединения формулы (12) также могут быть получены последовательностью реакций, проиллюстрированной ниже в Схеме 2.
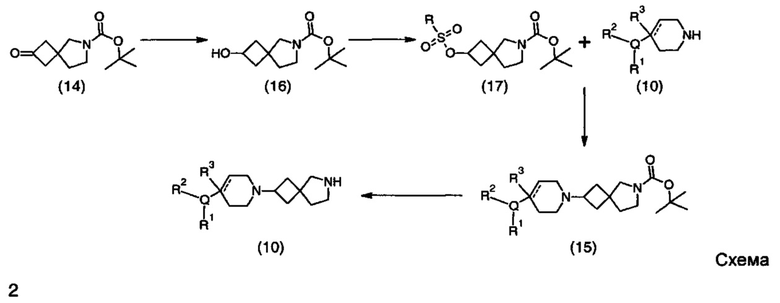
В Схеме 2 Boc-защищенный спирокетон (14) восстанавливают до спирта (16), используя борогидрид натрия в метаноле. Затем спирт (16) активируют в виде сульфонового эфира (17, R = метил, трифторметил или 4-метилфенил), используя соответствующий сульфонилхлорид в дихлорметане в присутствии третичного амина, такого как триэтиламин или N,N-диизопропилэтиламин. Сульфоновый эфир (17) приводят в контакт с пиперидиновым гетероциклом (10) в условиях реакции нуклеофильного замещения, которую обычно проводят при небольшом нагревании (например, до температуры от около 40°C до около 70°C) как в сухую, без растворителя, так и в подходящем растворителе, таком как тетрагидрофуран, ацетонитрил или диметилацетамид, с получением соединения (15), в котором затем проводят депротекцию с помощью удаления Вос-группы путем обработки кислотой (например, трифторуксусной кислотой в дихлорметане) с получением соединения (12).
Образовавшись, соединение формулы (1), (1а) или (1b), или его защищенная производная, может быть превращено в другое соединение формулы (1), (1а) или (1b) с помощью способов, известных специалисту в данной области техники. Примеры синтетических способов превращения одной функциональной группы в другую функциональную группу изложены в стандартных текстах, таких как Advanced Organic Chemistry and Organic Syntheses (см. ссылки выше) или Fiesers' Reagents for Organic Synthesis, тома 1-17, John Wiley, изданные Mary Fieser (ISBN: 0-471-58283-2). Примеры данных превращений включают образование амидной связи, образование мочевины, образование карбамата, реакции алкилирования, реакцию N-арилирования и образование C-C связи путем реакции кросс-сочетания.
В большинстве реакций, описанных выше, защита одной или нескольких групп может быть необходима для предотвращения реакции, имеющей место по нежелательному положению в молекуле. Примеры защитных групп и способов протекции и депротекции функциональных групп, могут быть найдены в Protective Groups in Organic Synthesis (Т. Greene и P. Wuts; 3rd Edition; John Wiley and Sons, 1999).
Соединения, полученные вышеизложенными способами, могут быть изолированы или очищены с помощью любого разнообразия способов, известных специалисту в данной области техники, и примеры таких способов включают перекристаллизацию и хроматографические методы, такие как колоночная хроматография (например, флеш-хроматография) и ВЭЖХ.
Фармацевтические композиции
До тех пор, пока это возможно для активного соединения, предпочтительно представлять его в виде фармацевтической композиции (например, лекарственной формы).
Соответственно, в другом варианте реализации изобретения (Вариант реализации изобретения 4.1) предложена фармацевтическая композиция, содержащая по меньшей мере одно соединение формулы (1), (1а) или (1b) в соответствии с определениями по любому из Вариантов реализации изобретения от 1.1 до 1.180 вместе с по меньшей мере одним фармацевтически приемлемым вспомогательным веществом.
В одном варианте реализации изобретения (Вариант реализации изобретения 4.2), композиция представляет собой таблеточную композицию.
В другом варианте реализации изобретения (Вариант реализации изобретения 4.3) композиция представляет собой композицию в форме капсулы.
Фармацевтически приемлемое вспомогательное вещество(-а) может быть выбрано из, например, веществ-носителей (например, твердого, жидкого или полутвердого вещества-носителя), вспомогательных лекарственных веществ, разбавителей (например, твердых разбавителей, таких как наполнители или объемообразующие агенты; и жидких разбавителей, таких как растворители и сорастворители), гранулирующих агентов, связывающих веществ, агентов для повышения текучести, покрывающих агентов, контролирующих высвобождение веществ (например, полимеров или восков, отсрачивающих или замедляющих высвобождение лекарственных средств), связывающих веществ, разрыхлителей, буферных веществ, смазывающих веществ, консервантов, антигрибковых и антибактериальных средств, антиоксидантов, буферных веществ, регулирующих тоничность веществ, загустителей, вкусовых добавок, подсластителей, пигментов, смягчителей, веществ, корригирующих вкус и запах, стабилизаторов или любых других вспомогательных веществ, традиционно используемых в фармацевтических композициях.
Термином «фармацевтически приемлемый» в контексте данного документа называют соединения, материалы, композиции, и/или лекарственные формы, которые находятся в пределах известных медицинских представлений, пригодные для приведения в контакт с тканями испытуемого (например, испытуемого человека) без излишней токсичности, раздражения, аллергической реакции или другой проблемы, или осложнения, соразмерной с соотношением рациональное преимущество/риск. Каждое вспомогательное вещество должно быть «приемлемым» в смысле сочетаемости с другими ингредиентами лекарственной формы.
Фармацевтические композиции, содержащие соединения формулы (1), (1а) или (1b) приготовлены согласно известным техникам, см, например, Remington's Pharmaceutical Sciences, Mack Publishing Company, Истон, Пенсильвания, США.
Фармацевтические композиции могут находиться в любой форме, подходящей для перорального, парентерального, местного, внутриносового, внутрибронхиального, сублингвального, глазного, ушного, ректального, интравагинального или внутрикожного введения.
Фармацевтические лекарственные формы, подходящие для перорального введения включают таблетки (покрытые или непокрытые оболочкой), капсулы (с твердой или мягкой оболочкой), капсуловидные таблетки, пилюли, пастилки, сиропы, растворы, порошки, гранулы, эликсиры и суспензии, сублингвальные таблетки, пластинки или пластыри, такие как трансбуккальные пластыри.
Таблеточные композиции могут содержать лекарственную форму действующего вещества в сочетании с инертным разбавителем или носителем, таким как сахар или сахарный спирт, например; лактоза, сахароза, сорбит или маннит; и/или разбавитель несахарного происхождения, такой как карбонат натрия, фосфат кальция, карбонат кальция или целлюлоза или ее производные, такие как микрокристаллическая целлюлоза (МКЦ), метилцеллюлоза, этилцеллюлоза, гидроксипропилметилцеллюлоза и крахмалы, такие как кукурузный крахмал. Также таблетки могут содержать такие стандартные ингредиенты, как связывающие и гранулирующие агенты, такие как поливинилпирролидон, разрыхлители (например, поддающиеся разбуханию сшитые полимеры, такие как сшитая карбоксиметилцеллюлоза), смазывающие вещества (например, стеараты), консерванты (например, парабены), антиоксиданты (например, ВНТ), буферные вещества (например фосфатные или цитратные буферы), и выделяющие газ агенты, такие как цитратные/гидрокарбонатные смеси. Такие вспомогательные вещества хорошо известны и не нуждаются в подробном обсуждении в данном документе.
Таблетки могут быть созданы для высвобождения лекарства как в случае приведения в контакт с желудочным соком (таблетки с быстрым высвобождением), так и высвобождать его контролируемым способом (таблетки с контролируемым высвобождением) в течение продолжительного периода времени или в специфической области ЖКТ.
Фармацевтические композиции обычно содержат от приблизительно 1% (в весовом отношении) до приблизительно 95%, предпочтительно % (в весовом отношении) действующего вещества и от 99% (в весовом отношении) до 5% (в весовом отношении) фармацевтически приемлемого вспомогательного вещества (например, в значении данного термина, приведенном выше) или комбинации таких вспомогательных веществ. Предпочтительно, композиции содержат от приблизительно 20% (в весовом отношении) до приблизительно 90% (в весовом отношении) действующего вещества и от 80% (в весовом отношении) до 10% фармацевтического вспомогательного вещества или комбинации вспомогательных веществ. Фармацевтические композиции содержат приблизительно от 1% до приблизительно 95%, предпочтительно от приблизительно 20% до приблизительно 90% действующего вещества. Фармацевтические композиции по данному изобретению могут находиться, например, в форме унифицированной дозы, такой как ампулы, флаконы, суппозитории, предварительно заполненный шприц, драже, порошки, таблетки или капсулы.
Таблетки и капсулы могут содержать, например, 0-20% разрыхлителей, 0-5% смягчителей, 0-5% агентов для повышения текучести и/или 0-99% (в весовом отношении) наполнителей и/или объемообразующих агентов (в зависимости от дозы лекарства). Также они могут содержать 0-10% (в весовом отношении) полимерных связующих веществ, 0-5% (в весовом отношении) антиоксидантов, 0-5% (в весовом отношении) пигментов. Таблетки с замедленным высвобождением обычно могут в дополнение содержать 0-99% (в весовом отношении) контролирующих высвобождение (например, замедляющих) полимеров (в зависимости от дозы). Пленочные оболочки таблетки или капсулы обычно содержат 0-10% (в весовом отношении) полимеров, 0-3% (в весовом отношении) пигментов и/или 0-2% (в весовом отношении) смягчителей.
Парентеральные лекарственные формы обычно содержат 0-20% (в весовом отношении) буферов, 0-50% (в весовом отношении) сорастворителей и/или 0-99% (в весовом отношении) воды для инъекций (воды д/и) (в зависимости от дозы и лиофилизированности). Лекарственные формы для внутримышечных инъекций замедленного всасывания также могут содержать 0-99% (в весовом отношении) масел.
Фармацевтические лекарственные формы могут быть предоставлены больному в «упаковках для пациента», содержащих целый курс лечения в единичной упаковке, обычно в блистерной упаковке.
Соединения формулы (1), (1а) или (1b) обычно будут представлены в единичной лекарственной форме и, соответственно, обычно будут содержать достаточное количество соединения для обеспечения желаемого уровня биологической активности. Например, лекарственная форма может содержать от 1 нанограмма до 2 грамм действующего вещества, например, от 1 нанограмма до 2 миллиграммов действующего вещества. В пределах этих диапазонов конкретные поддиапазоны соединения представляют собой от 0,1 миллиграмма до 2 грамм действующего вещества (более типично от 10 миллиграмм до 1 грамма, например, от 50 миллиграмм до 500 миллиграмм) или от 1 микрограмма до 20 миллиграмм (например, от 1 микрограмма до 10 миллиграмм, например, от 0,1 миллиграмма до 2 миллиграмм действующего вещества).
Для пероральных композиций единичная лекарственная форма может содержать от 1 миллиграмма до 2 грамм, более типично от 10 миллиграмм до 1 грамма, например, от 50 миллиграмм до 1 грамма, например, от 100 миллиграмм до 1 грамма активного соединения.
Активное соединение будет введено нуждающемся в нем больному (например, больному человеку или животному) в количестве, достаточном для достижения желаемого терапевтического эффекта (эффективное количество). Точное количество введенного соединения может быть определено лечащим врачом согласно стандартным методикам.
ПРИМЕРЫ
Изобретение будет далее проиллюстрировано, но не ограничено, путем ссылки на конкретные варианты реализации изобретения, описанные в следующих примерах.
ПРИМЕРЫ ОТ 1-1 ДО 5-2
Соединения, полученные из Примеров от 1-1 до 5-2, проиллюстрированы ниже в Таблице 1. Их ЯМР и ЖХ/МС свойства и использованные для их получения способы описаны в Таблице 3.
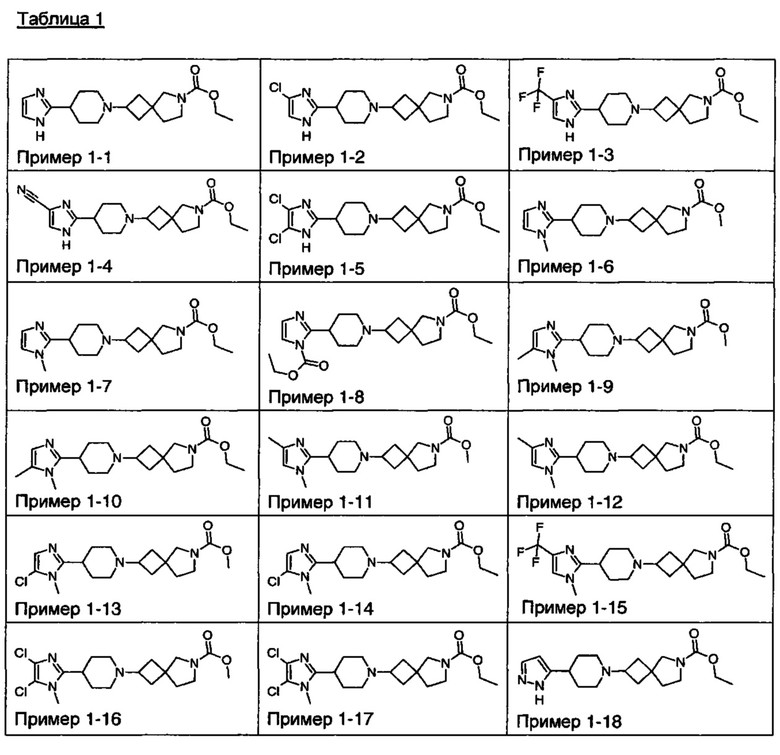
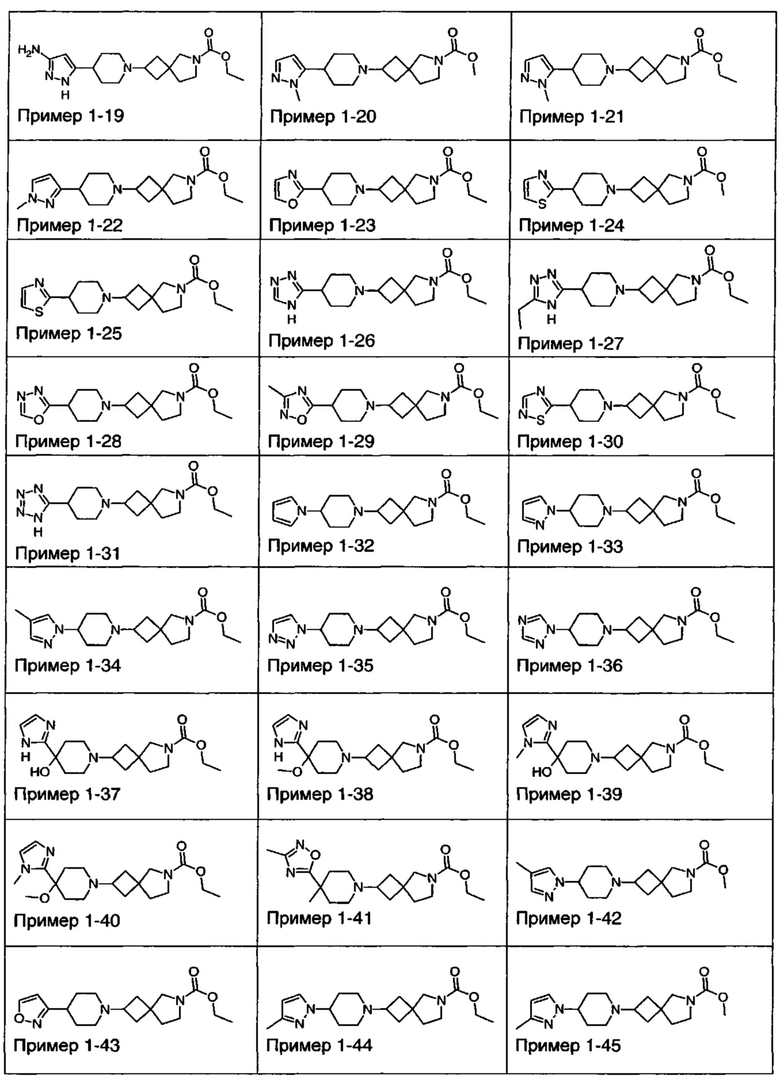
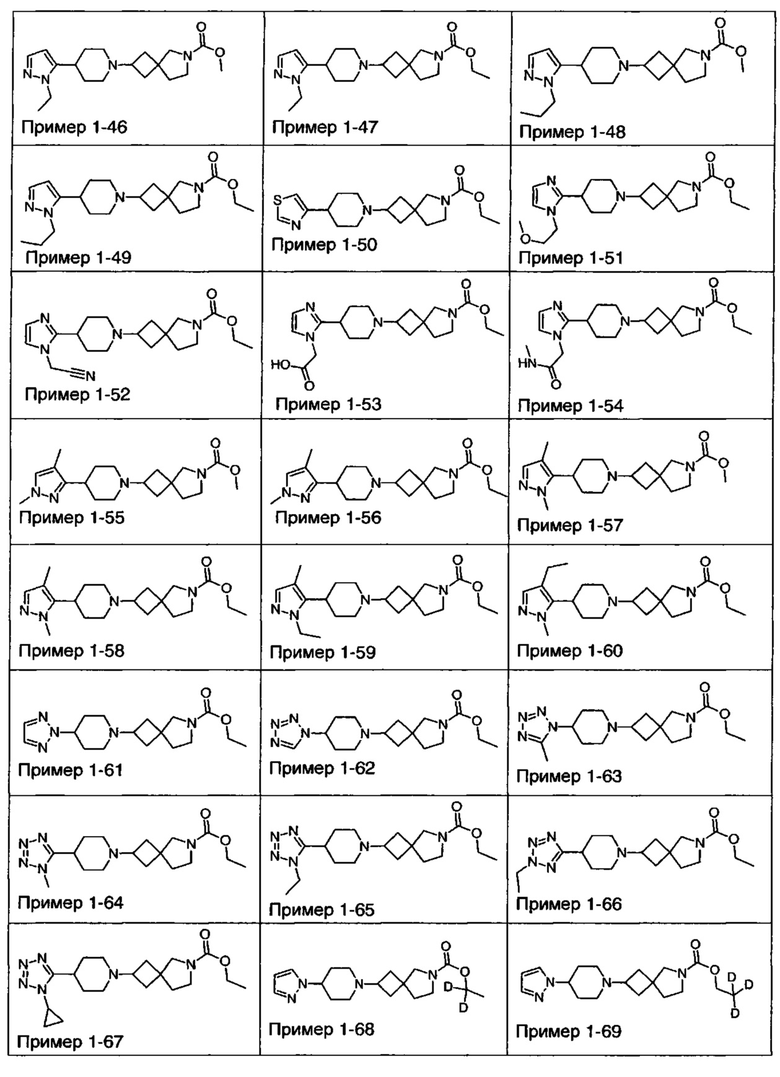
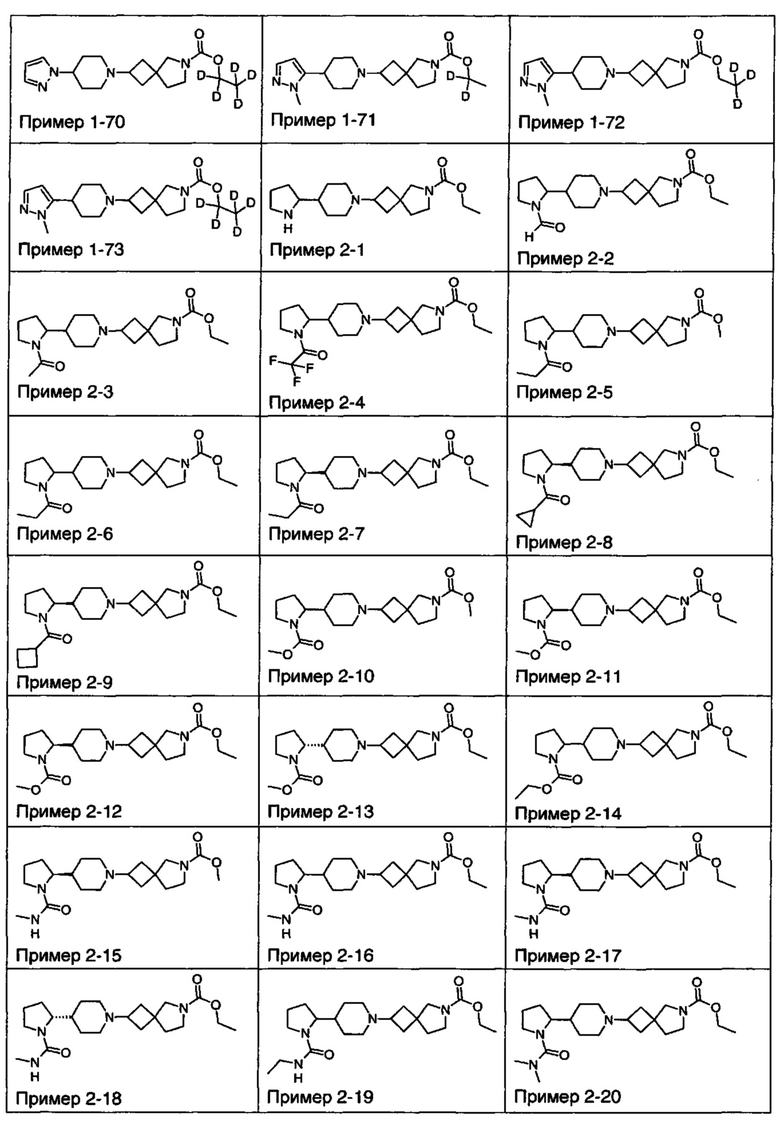
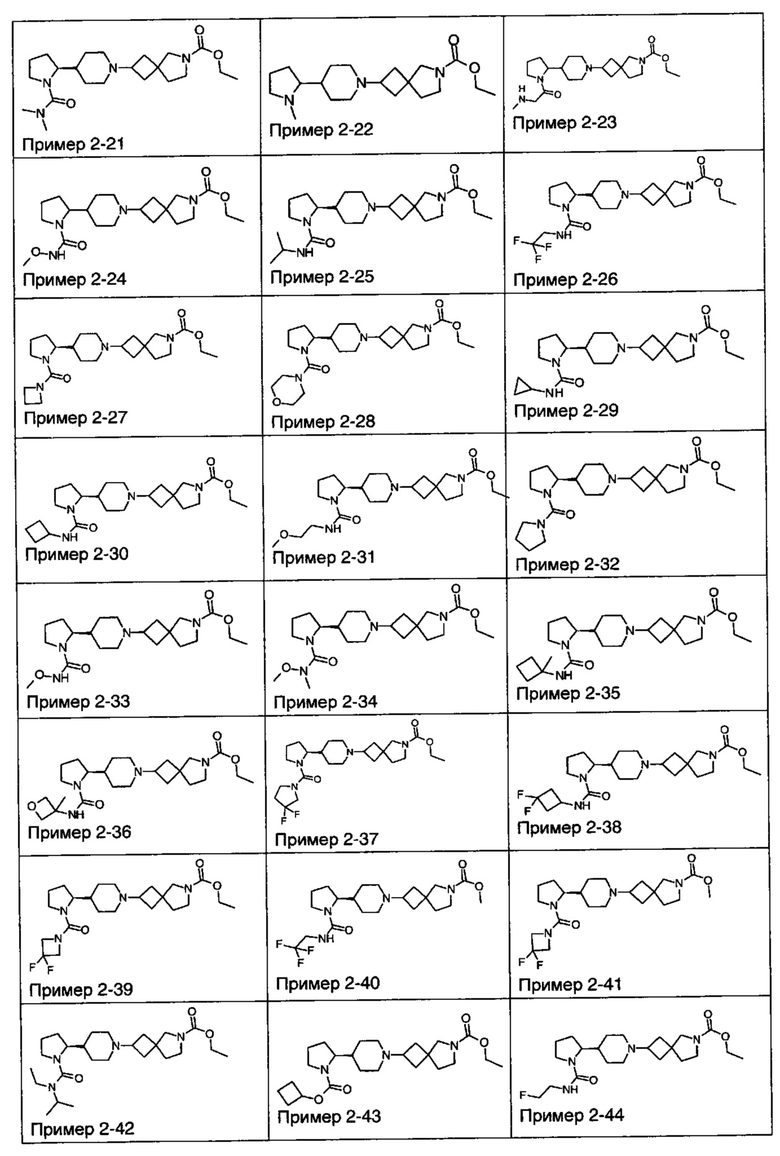
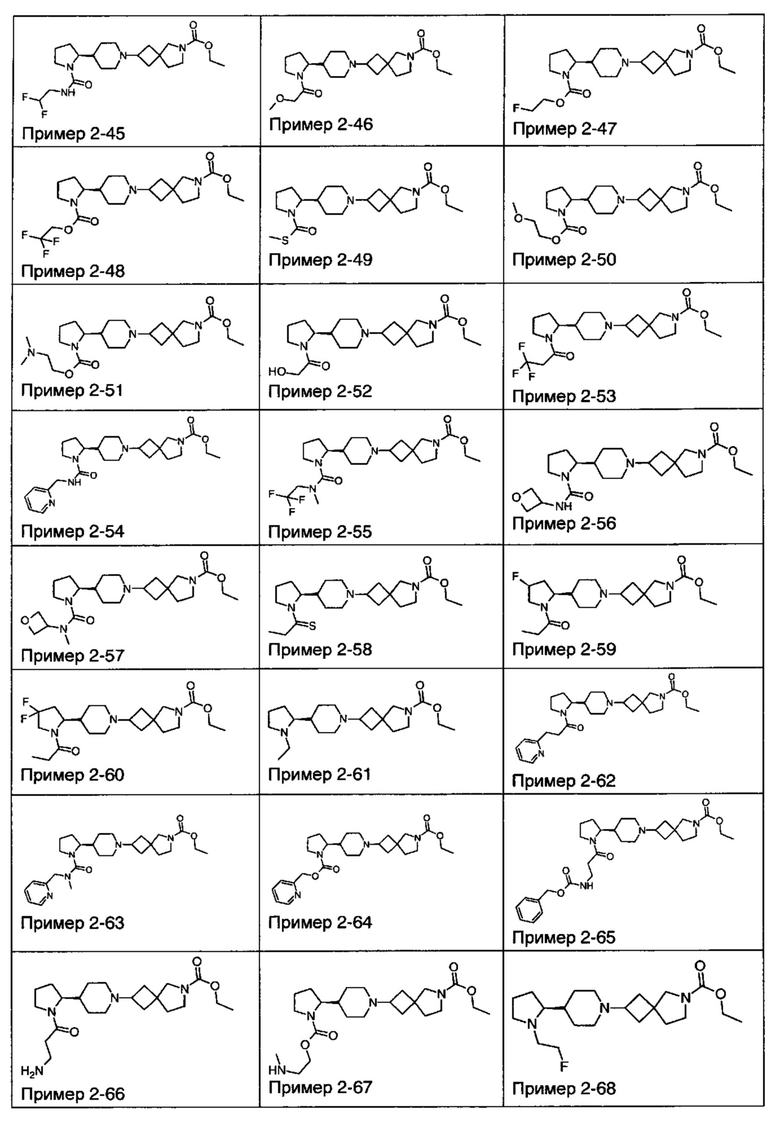
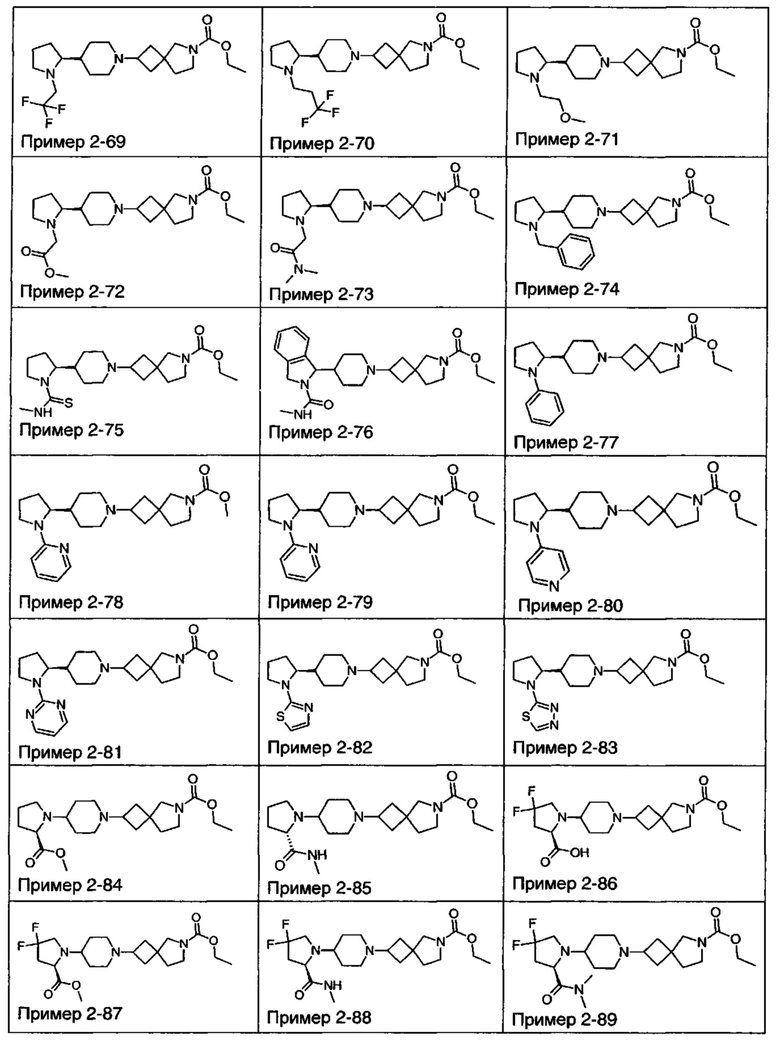
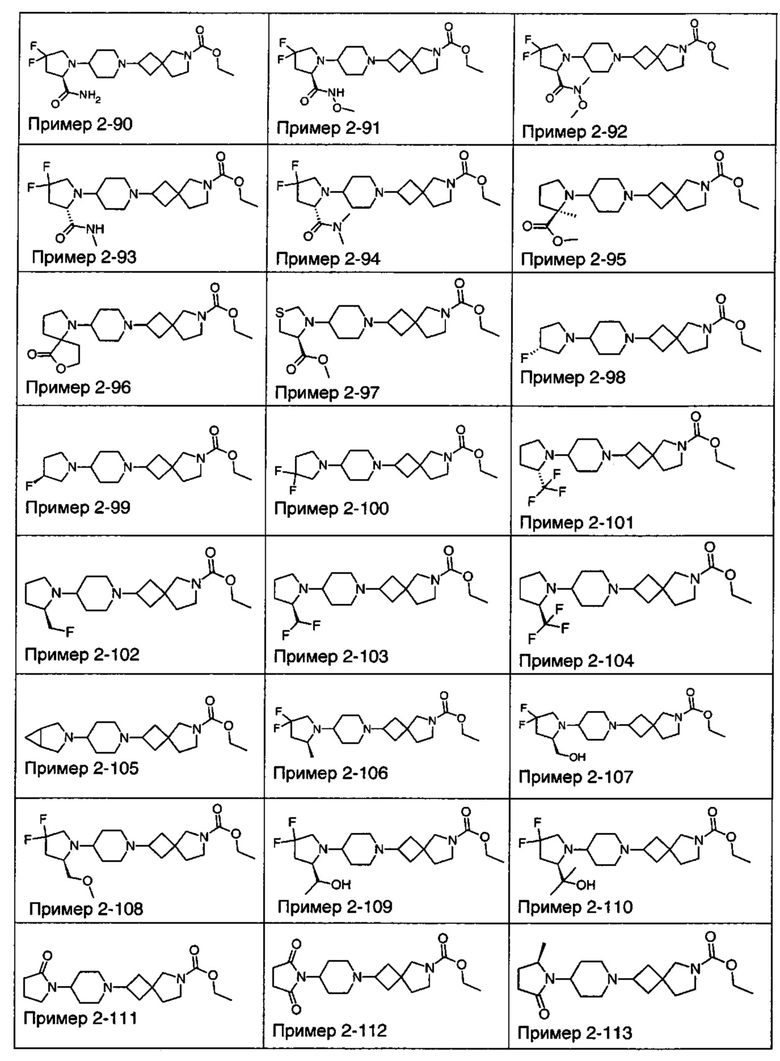
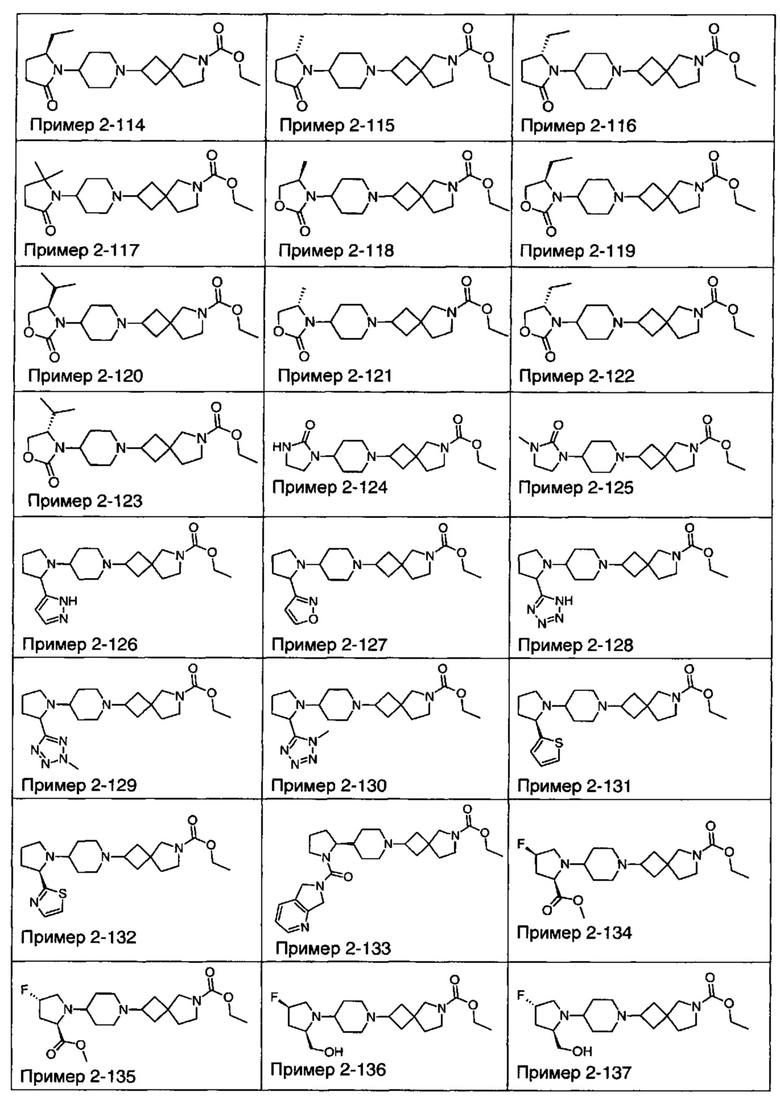
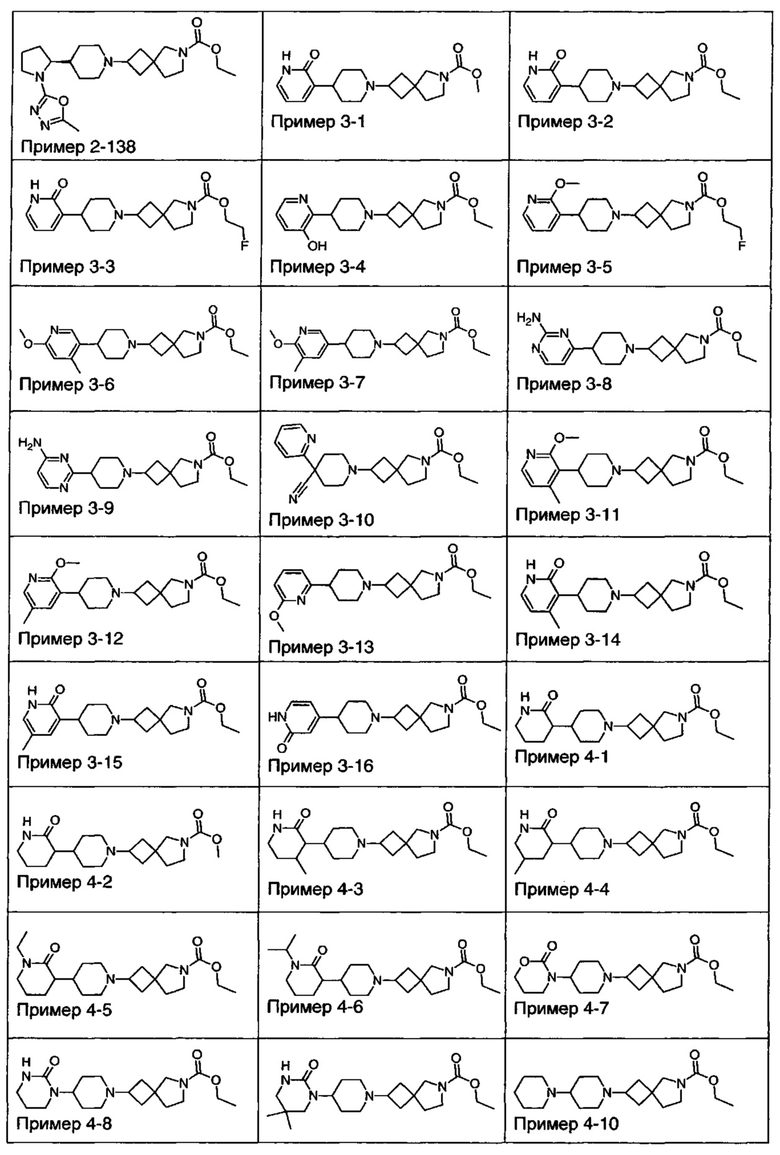
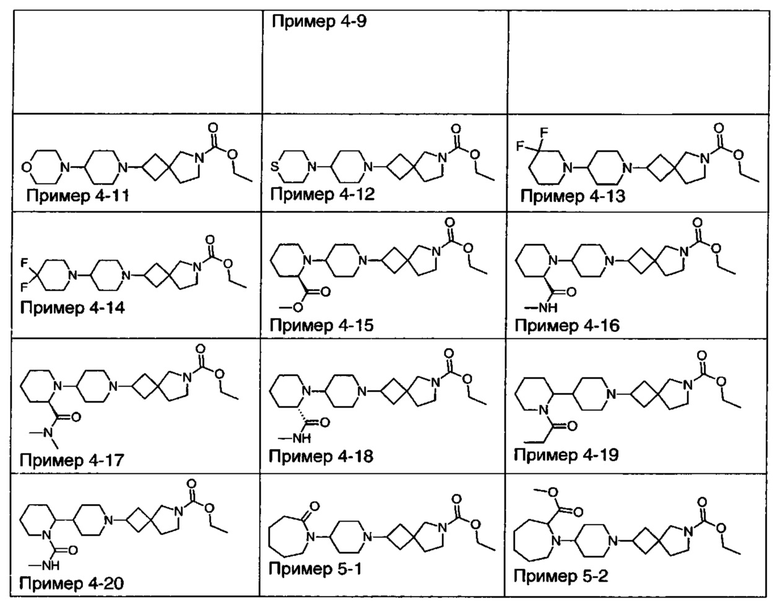
Общие методы синтеза
Где не указано никаких препаративных путей получения, соответствующее промежуточное соединение является коммерчески доступным. Коммерческие реагенты были использованы без дополнительной очистки. Комнатной температурой (комн. темп.) называют температуру приблизительно 20-27°C. 1Н ЯМР спектры записаны при 400 МГц либо на Bruker, либо на Jeol instrument. Значения химических сдвигов выражены в миллионных долях (м.д.), т.е. (δ:)-значениях. Следующие сокращения использованы для обозначения мультиплетности сигналов ЯМР: с = синглет, уш = уширеный, д = дублет т = триплет, к = квартет, квинт = квинтет, тд = триплет дублетов, тт = триплет триплетов, кд = квартет дублетов, ддд = дублет дублетов дублетов, ддт = дублет дублетов триплетов, м = мультиплет. Константы спин-спинового взаимодействия перечислены в виде значений J, измеренных в Гц. Результаты ЯМР-спектроскопии и масс-спектрометрии были откорректированы с учетом фонового шума. Хроматография относится к колоночной хроматографии, выполненной с использованием 60-120 меш силикагеля и проведенной под давлением азота (флеш-хроматография). ТСХ для контроля течения реакции относится к ТСХ, проведенной с использованием специальной подвижной фазы и силикагеля F254 (Merck) в качестве стационарной фазы. Реакции под действием микроволнового излучения выполнены на Biotage Initiator или СЕМ Discover микроволновом реакторе.
ЖХ/МС эксперименты обычно проводили, используя условия электроспрея, как указано для каждого соединения, в следующих условиях:
ЖХ/МС Способы A и B
Инструменты: Waters Alliance 2795, Waters 2996 PDA детектор, Micromass ZQ; Колонка: Waters X-Bridge C-18, 2,5 микрон, 2,1×20 мм или Phenomenex Gemini-NX C-18, 3 микрон, 2,0×30 мм; Градиент [время (мин)/растворитель D в С (%)]: Способ A: 0,00/2, 0,10/2, 2,50/95, 3,50/95, 3,55/2, 4,00/2 или Способ B: 0,00/2, 0,10/2, 8,40/95, 9,40/95, 9,50/2, 10,00/2; Растворители: растворитель C=2,5 л H2O + 2,5 мл раствора аммиака; растворитель D=2,5 л MeCN + 135 мл H2O + 2,5 мл раствора аммиака); объем введенной пробы 3 μL; УФ детектирование от 230 до 400 нм; температура колонки 45°C; Скорость потока 1,5 мл/мин.
ЖХ/МС Способ C
Инструменты: Agilent 1260 Infinity LC с Фотодиодной матрицей, одноквадрупольный масс-спектрометр Agilent 6120 B с источником API-ES; Колонка: Phenomenex Gemini-NX C-18, 3 микрон, 2,0×30 мм; Градиент [время (мин)/растворитель В в А (%)]: Способ: 0,00/5, 2,00/95, 2,50/95, 2,60/5, 3,00/5; Растворители: растворитель A = 2,5 л H2O + 2,5 мл (28% NH3 в H2O); растворитель B = 2,5 л MeCN + 129 мл H2O + 2,7 мл (28% NH3 в H2O); объем введенной пробы 0,5 μL; УФ детектирование от 190 до 400 нм; температура колонки 40°C; Скорость потока 1,5 мл/мин.
ЖХ/МС Способы D и E
Инструменты: HP 1100 с G1315A DAD, Micromass ZQ; Колонка: Waters X-Bridge C-18, 2,5 микрон, 2,1×20 мм или Phenomenex Gemini-NX C-18, 3 микрон, 2,0×30 мм; Градиент [время (мин)/растворитель D в С (%)]: Способ D: 0,00/2, 0,10/2, 2,50/95, 3,50/95, 3,55/2, 4,00/2 или Способ Е: 0,00/2, 0,10/2, 8,40/95, 9,40/95, 9,50/2, 10,00/2; Растворители: растворитель C = 2,5 л H2O + 2,5 мл раствора 28% аммиака в H2O; растворитель D = 2,5 л MeCN + 135 мл H2O + 2,5 мл раствора 28% аммиака в H2O); объем введенной пробы 1 мкл; УФ детектирование от 230 до 400 нм; масс-детекция от 130 до 800 а.е.м. (+ve и -ve электроспрей); температура колонки 45°C; Скорость потока 1,5 мл/мин.
ЖХ/МС Способ F:
Инструменты: Waters Acquity Н Class, Фотодиодная матрица, SQ Детектор; Колонка: ВЕН С18, 1,7 микрон, 2,1×50 мм; Градиент [время (мин)/растворитель B в А (%)]: 0,00/5, 0,40/5, 0,8/35, 1,20/55, 2,50/100, 3,30/100 4,00/5; Растворители: растворитель А=5 мМ ацетата аммония и 0,1% муравьиной кислоты в H2O; растворитель B = 0,1% муравьиной кислоты в MeCN; объем введенной пробы 2 μL; УФ детектирование от 200 до 400 нм; масс-детекция 100 до 1200 а.е.м. (+ve электроспрей); колонка при температуре окружающей среды; Скорость потока 0,5 мл/мин.
ЖХ/МС Способ G:
Инструменты: Waters 2695, Фотодиодная матрица, ZQ-2000 Детектор; Колонка: X-Bridge С18, 5 микрон, 150×4,6 мм; Градиент [время (мин)/растворитель B в A (%)]: 0,00/10, 5,00/90, 7,00/100, 11,00/100, 11,01/10 12,00/10; Растворители: растворитель A = 0,1% аммиака в H2O; растворитель B = 0,1% аммиака в MeCN; объем введенной пробы 10 мкл; УФ детектирование от 200 до 400 нм; масс-детекция от 60 до 1000 а.е.м. (+ve электроспрей); колонка при температуре окружающей среды; Скорость потока 1,0 мл/мин.
ЖХ/МС Способ Н:
Инструменты: Waters 2695, Фотодиодная матрица, ZQ-2000 Детектор; Колонка: X-Bridge С18, 5 микрон, 150×4,6 мм; Градиент [время (мин)/растворитель B в A (%)]: 0,00/100, 7,00/50, 9,00/0, 11,00/0, 11,01/100, 12,00/100; Растворители: растворитель A = 0,1% аммиака в H2O; растворитель B = 0,1% аммиака в MeCN; объем введенной пробы 10 мкл; УФ детектирование от 200 до 400 нм; масс-детекция от 60 до 1000 а.е.м. (+ve электроспрей); колонка при температуре окружающей среды; Скорость потока 1,0 мл/мин.
ЖХ/МС Способ I:
Инструменты: Waters 2695, Фотодиодная матрица, ZQ-2000 Детектор; Колонка: X-Bridge С18, 3,5 микрон, 150×4,6 мм; Градиент [время (мин)/растворитель B в A (%)]: 0,00/5, 5,00/90, 5,80/95, 10/95; Растворители: растворитель A = 0,1% аммиака в H2O; растворитель B = 0,1% аммиака в MeCN; объем введенной пробы 10 мкл; УФ детектирование от 200 до 400 нм; масс-детекция от 60 до 1000 а.е.м. (+ve электроспрей); колонка при температуре окружающей среды; Скорость потока 1,0 мл/мин.
ЖХ/МС Способ J:
Инструменты: Waters 2695, Фотодиодная матрица, ZQ-2000 Детектор; Колонка: X-Bridge С18, 5 микрон, 150×4,6 мм; Градиент [время (мин)/растворитель B в A (%)]: 0,01/10, 5,00/90, 7,00/100, 11,00/100, 11,01/10, 12,00/10; Растворители: растворитель A = 20 мМ ацетата аммония в H2O; растворитель B = МеОН; объем введенной пробы 10 мкл; УФ детектирование от 200 до 400 нм; масс-детекция от 60 до 1000 а.е.м. (+ve электроспрей); колонка при температуре окружающей среды; Скорость потока 1,0 мл/мин.
ЖХ/МС Способ K:
Инструменты: Waters 2695, Фотодиодная матрица, ZQ-2000 Детектор; Колонка: X-Bridge С18, 3,5 микрон, 50×4,6 мм; Градиент [время (мин)/растворитель B в A (%)]: 0,01/0, 0,20/0, 5,00/90, 5,80/95, 7,20/95, 7,21/100, 10,00/100; Растворители: растворитель A=0,1% аммиака в H2O; растворитель B = 0,1% аммиака в MeCN; объем введенной пробы 10 мкл; УФ детектирование от 200 до 400 нм; Масс-детекция от 60 до 1000 а.е.м. (+ve электроспрей); колонка при температуре окружающей среды; Скорость потока 1,0 мл/мин.
ЖХ/МС Способ L
Инструменты: Waters Acquity UPLC, Waters 3100 PDA Детектор, SQD; Колонка: Acquity ВЕН C-18, 1,7 микрон, 2,1×100 мм; Градиент [время (мин)/растворитель B в A (%)]: 0,00/2, 2,00/2, 7,00/50, 8,50/80, 9,50/2, 10 0/2; Растворители: растворитель A = 5 мМ ацетата аммония в воде; растворитель B = ацетонитрил; объем введенной пробы 1 мкл; Длина волны детектирования 214 нм; Температура колонки 30°C; Скорость потока 0,3 мл в мин.
ЖХ/МС Способ М
Инструменты: Agilent 1260 Infinity series UHPLC; ELSD: Agilent 1260 infinity; Колонка: Acquity C-18, 1,7 микрон, 2,1×50 мм; Градиент [время (мин)/растворитель B в A (%)]: 0,00/10, 1,00/10, 2,00/15, 4,50/55, 6,00/90, 8,00/90, 9,00/10, 10,00/10; Растворители: А=5 мМ ацетата аммония в воде, В = ацетонитрил; объем введенной пробы: 1 мкл; Детектирование с помощью ELSD; Температура колонки: 40°C; Скорость потока: 0,6 мл в мин.
ЖХ/МС Способ N
Инструменты: Waters Acquity UPLC, Waters 3100 PDA Детектор, SQD; Колонка: Acquity ВЕН C-18, 1,7 микрон, 2,1×100 мм; Градиент [время (мин)/растворитель B в A (%)]: 0,00/2, 0,50/2, 1,50/20, 4,00/92, 5,00/92, 5,50/50, 6,00/2; Растворители: растворитель A = 5 мМ ацетата аммония в воде; растворитель B = ацетонитрил; объем введенной пробы 1 мкл; Длина волны детектирования 214 нм; Температура колонки 35°C; Скорость потока 0,6 мл в мин.
ЖХ/МС Способ O
Инструменты: Waters Acquity UPLC, Waters 3100 PDA Детектор, SQD; Колонка: Acquity HSS-T3, 1,8 микрон, 2,1×100 мм; Градиент [время (мин)/растворитель B в A (%)]: 0,00/10, 1,00/10, 2,00/15, 4,50/55, 6,00/90, 8,00/90, 9,00/10, 10,00/10; Растворители: растворитель A = 0,1% трифторуксусной кислоты в воде; растворитель B=ацетонитрил; объем введенной пробы 1 мкл; Длина волны детектирования 214 нм; Температура колонки 30°C; Скорость потока 0,3 мл в мин.
Данные ЖХ/МС в экспериментальной части даны в формате: Масса иона, время удерживания, УФ-активность.
Аббревиатуры
АсОН = уксусная кислота
CDI = 1,1'-Карбонилдиимидазол
д = день (дней)
DAST = Трифторид диэтиламиносеры
DCE = дихлорэтан
ДХМ = дихлорметан
DIPEA = диизопропилэтиламин
DIAD = диизопропилазодикарбоксилат
ДМФА = диметилформамид
DMP = периодинан Десса-Мартина
ДМСО = диметилсульфоксид
ES = электроспрей ионизация
EtOAc = этилацетат
ч = час (ы)
HATU = 1-[Бис(диметиламино)метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиний-3-оксид гексафторфосфат
ВЭЖХ = высокоэффективная жидкостная хроматография
ЖХ = жидкостная хроматография
LiAlH4/LАН = алюмогидрид лития
МеСМ = ацетонитрил
МеОН = метанол
min = минута (-ы)
МС = масс-спектрометрия
Et3N = триэтиламин
ЯМР = ядерный магнитный резонанс
комн. темп. = комнатная температура
насыщ. = насыщенный
р-р. = раствор
STAB = триацетоксиборогидрид натрия
ТГФ = тетрагидрофуран
ТСХ = тонкослойная хроматография
Префиксы н-, втор-, и-, т- и трет- имеют свои обычные значения: нормальный, вторичный, изо-, и третичный.
Синтез промежуточных соединений:
Метод получения промежуточного соединения 2, этил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилата
6-Boc-2-оксо-6-азаспиро[3.4]октан (3,37 г, 15 ммоль) порциями прибавили к хлороводороду (4 М раствор в диоксане, 50 мл, 210 ммоль). Осторожно: бурное выделение газа. Через 24 ч реакционную смесь концентрировали в вакууме и твердый остаток растворили в смеси Et3N (4,18 мл, 30 ммоль) и ДХМ (66 мл). По завершении растворения раствор немедленно охладили до 0°C, затем по каплям прибавляли этилхлорформиат (1,57 мл, 16,5 ммоль). Через 18 ч смесь вылили в дихлорметан (100 мл) и NaHCO3 (водн.) (100 мл) и экстрагировали (2×100 мл). Органические фазы собрали, промыли насыщенным водным раствором хлорида натрия (20 мл), сушили над MgSO4, затем остаток после выпаривания очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 100 г, 40-63 мкм, 60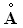 , 50 мл в мин, градиент от 0% до 4% МеОН в ДХМ) с получением промежуточного соединения 2, этил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилата в виде бесцветного масла (2,47 г, 83%). Данные для указанного в заголовке соединения приведены в Таблице 2.
, 50 мл в мин, градиент от 0% до 4% МеОН в ДХМ) с получением промежуточного соединения 2, этил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилата в виде бесцветного масла (2,47 г, 83%). Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 3, метил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилата
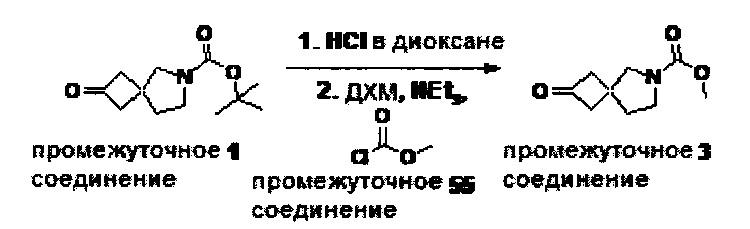
6-Вос-2-оксо-6-азаспиро[3.4]октан (5,00 г, 22,2 ммоль) по порциям прибавляли к хлороводороду (4 М раствор в диоксане, 45 мл, 180 ммоль) в дихлорметане (5 мл). Осторожно: бурное выделение газа. Через 2 ч реакционную смесь концентрировали в вакууме и 1,29 г твердого остатка растворили в смеси триэтиламина (2,23 мл, 16,0 ммоль) и дихлорметана (10 мл). По завершении растворения раствор немедленно охладили до 0°C, затем прибавляли по каплям метилхлорформиат (0,68 мл, 8,83 ммоль). Через 3 ч смесь вылили в дихлорметан (50 мл), промыли NaHCO3 (водн.) (2×50 мл) и экстрагировали ДХМ (50 мл). Органические фазы объединили, промыли насыщенным водным раствором хлорида натрия (50 мл), пропустили через картридж фазного сепаратора Biotage, растворитель удалили в вакууме и остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 50 г, 40-63 мкм, 60, 40 мл в мин, градиент от 0% до 10% МеОН в ДХМ) с получением промежуточного соединения 3, метил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилата в виде оранжевого масла (0,93 г, 66%). Данные для указанного в заголовке соединения приведены в Таблице 2
Метод получения промежуточного соединения 4, 2-фторэтил 2-оксо-6-азаспиро [3.4]октан-6-карбоксилата
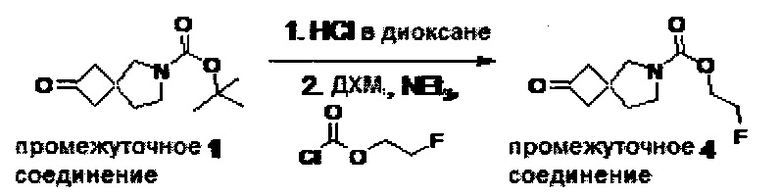
трет-Бутил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (5 г, 22,19 ммоль) перемешивали в растворе HCl в 1,4-иоксане (25 мл) в течение 10 ч при комн. темп. Реакционную смесь концентрировали в вакууме и растерли с ацетоном (3×50 мл) с получением 6-азаспиро[3.4]октан-2-она (2,77 г, 55,4%) в виде коричневой смолы. Остаток растворили в сухом ДХМ (20 мл) и прибавили Et3N (0,7 мл, 4,8 ммоль). Реакционную смесь охладили до 0°C и прибавили 2-фторэтилкарбонохлоридат (0,45 г, 3,6 ммоль). Реакционную смесь перемешивали при 30°C в течение 5 ч, затем разбавили водой (50 мл), экстрагировали ДХМ (2×100 мл), органические фазы объединили, сушили (Na2SO4), и растворитель удалили в вакууме. Остаток очистили с помощью колоночной хроматографии (нормально-фазовая, силикагель 60-120 меш, от 0 до 10% EtOAc в гексане) с получением промежуточного соединения 4, 2-фторэтил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилата (0,2 г, 38,8%) в виде коричневой смолы. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточных соединений 20 и 21, 4-(5-хлор-1-метил-1Н-имидазол-2-ил)пиперидин трифторацетата и 4-(4,5-дихлор-1-метил-1Н-имидазол-2-ил)пиперидин трифторацетата соответственно
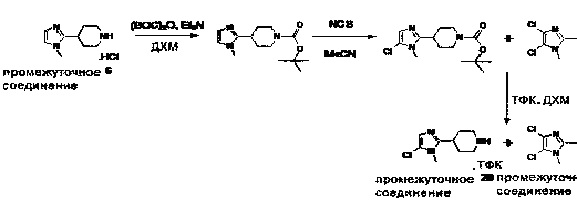
4-(1-Метилимидазол-2-ил)пиперидин гидрохлорид (1 г, 4,96 ммоль) суспендировали в смеси безводного ДХМ (20 мл) и Et3N (2,1 мл, 15,1 ммоль) и охладили на ледяной бане. (ВОС)2O (1,19 г, 5,45 ммоль) прибавляли по порциям в течение 5 мин, смесь нагрели до комн. темп. и перемешивали в течение 48 ч. Смесь разбавили ДХМ, промыли насыщенным водным NaHCO3 (x2) и насыщенным водным раствором хлорида натрия (х1), затем пропустили через фазный сепаратор и концентрировали в вакууме с получением трет-бутил-4-(1-метил-1Н-имидазол-2-ил)пиперидин-1-карбоксилата (1,34 г, колич.) в виде твердого вещества.
ЖХ/МС (Способ С): m/z 266 (М+Н)+ (ES+), за 1,43 мин, активно в ультрафиолетовом свете.
трет-Бутил-4-(1-метил-1Н-имидазол-2-ил)пиперидин-1-карбоксилат (0,250 г, 0,942 ммоль) растворили в MeCN (7,5 мл), обработали NCS (0,314 г, 2,35 ммоль) и перемешивали при комн. темп. в течение ночи. Реакционную смесь концентрировали на флэш силикагеле (~15 мл) в вакууме. Полученный порошок очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 50 г, 40-63 мкм, 60, 40 мл в мин, градиент от 2% до 10% Растворитель А в ДХМ за 15 объемов колонки, где растворитель А представляет собой 10% (7 М NH3/MeOH) в МеОН]) с получением смеси, содержащей трет-бутил-4-(5-хлор-1-метил-1Н-имидазол-2-ил)пиперидин-1-карбоксилат и трет-бутил-4-(4,5-дихлор-1-метил-1Н-имидазол-2-ил)пиперидин-1-карбоксилат и сукцинимид (0,495 г).
ЖХ/МС (Способ С): монохлор: m/z 300/302 (М+Н)+ (ES+), при 1,68 мин, активно в ультрафиолетовом свете; дихлор: m/z 334/336/338 (М+Н)+ (ES+), при 1,87 мин, активно в ультрафиолетовом свете. Соотношение монохлор; дихлор ~16:1 согласно ЖХ-УФ. Смесь, содержащую трет-бутил-4-(5-хлор-1-метил-1Н-имидазол-2-ил)пиперидин-1-карбоксилат и трет-бутил-4-(4,5-дихлор-1-метил-1Н-имидазол-2-ил)пиперидин-1-карбоксилат и сукцинимид (0,495 г) растворили в ДХМ (5 мл), обработали ТФК (5 мл) и перемешивали при комн. темп. в течение 6 ч. Реакционную смесь концентрировали в вакууме и остаток подвергли азеотропной перегонке с толуолом (х2) с получением сырой смеси промежуточного соединения 20, 4-(5-хлор-1-метил-1Н-имидазол-2-ил)пиперидин трифторацетата и промежуточного соединения 21, 4-(4,5-дихлор-1-метил-1Н-имидазол-2-ил)пиперидин трифторацетата, смешанных с сукцинимидом. Предполагали количественный выход. Использовали без дополнительной очистки. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточных соединений 22 и 25, 4-(5-хлор-1Н-имидазол-2-ил)пиперидин дигидробромида и 4-(4,5-дихлор-1Н-имидазол-2-ил)пиперидин дигидробромида соответственно
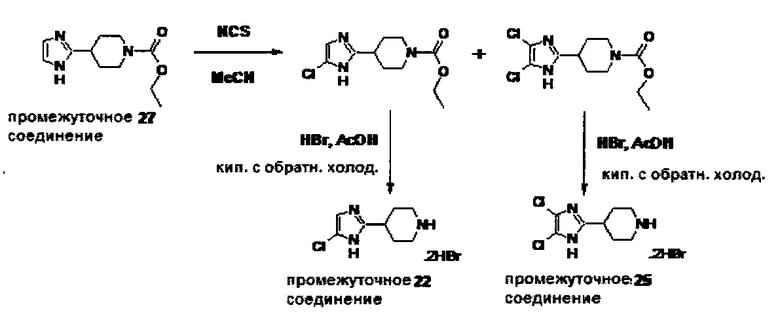
Этил-4-(1Н-имидазол-2-ил)пиперидин-1-карбоксилат (0,40 г, 1,79 ммоль) растворили в MeCN (12 мл), обработали NCS (0,360 г, 2,70 ммоль) и перемешивали при комн. темп. в течение 5,5 ч. Реакционную смесь концентрировали на силикагеле для флеш-хроматографии (~10 мл) в вакууме. Полученный порошок очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 50 г, 40-63 мкм, 60, 40 мл в мин, градиент от 0% до 5% Растворителя A в ДХМ за 15 объемов колонки, затем 5% Растворителя A в ДХМ за 5 объемов колонки, где растворитель A представляет собой 10% (7 М NH3/MeOH) в МеОН]) с получением разделенного этил-4-(5-хлор-1Н-имидазол-2-ил)пиперидин-1-карбоксилата и этил-4-(4,5-дихлор-1Н-имидазол-2-ил)пиперидин-1-карбоксилата, обоих смешанных с сукцинимидом. Каждый растворили в ДХМ, промыли H2O (х3), пропустили через фазный сепаратор и концентрировали в вакууме для удаления сукцинимида.
Этил-4-(5-хлор-1Н-имидазол-2-ил)пиперидин-1-карбоксилат (0,12 г, 26%), ЖХ/МС (Способ С): m/z 258/260 (М+Н)+ (ES+), при 1,34 мин, активно в ультрафиолетовом свете. Этил-4-(4,5-дихлор-1Н-имидазол-2-ил)пиперидин-1-карбоксилат (0,24 г, 45%), ЖХ/МС (Способ С): m/z 292/294/296 (М+Н)+ (ES+), при 1,24 мин, активно в ультрафиолетовом свете.
Этил-4-(5-хлор-1Н-имидазол-2-ил)пиперидин-1-карбоксилат (0,12 г, 0,47 ммоль) растворили в АсОН (2 мл), обработали 48% водным HBr (2 мл) и нагревали при кипячении с обратным холодильником при ~120° в течение 2 ч. Реакционную смесь концентрировали в вакууме, остаток подвергли азеотропной перегонке с толуолом (х1) и концентрировали в вакууме для получения твердого вещества. Выход промежуточного соединения 22, 4-(5-хлор-1Н-имидазол-2-ил)пиперидин дигидробромидной соли приняли за количественный. Немедленно использовали.
Этил-4-(4,5-дихлор-1Н-имидазол-2-ил)пиперидин-1-карбоксилат (0,24 г, 0,82 ммоль) растворили в АсОН (2 мл), обработали 48% водным HBr (2 мл) и нагревали ~120°C в течение 2 ч. Реакционную смесь концентрировали в вакууме, остаток подвергли азеотропной перегонке с толуолом (х1) и концентрировали в вакууме для получения твердого вещества. Выход промежуточного соединения 25, 4-(4,5-дихлор-1Н-имидазол-2-ил)пиперидин дигидробромида приняли за количественный. Немедленно использовали. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 46, трет-бутил-4-(4-(трифторметил)-1Н-имидазол-2-ил)пиперидин-1-карбоксилата и промежуточного соединения 33, 4-[4-(трифторметил)-1Н-имидазол-2-ил]пиперидина
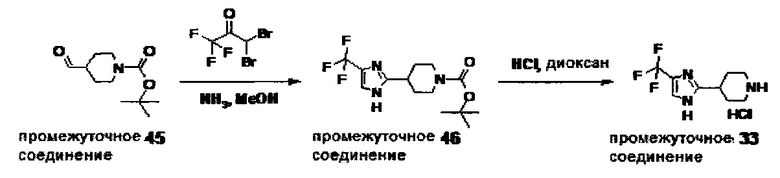
трет-Бутил-4-формилпиперидин-1-карбоксилат (2,0 г, 9,4 ммоль) растворили в МеОН (10 мл) с последующим прибавлением в течение 30 мин охлажденного до 0°C 7 М метанольного раствора аммиака, с последующим прибавлением 3,3-дибром-1,1,1-трифторпропан-2-она (5,07 г, 18,5 ммоль) порциями. Полученную реакционную смесь перемешивали при 25°C в течение 2 ч, растворители удалили в вакууме и остаток разделили между H2O (80 мл) и EtOAc (50 мл), водную фазу экстрагировали EtOAc (2×50 мл), органические фазы объединили, сушили (Na2SO4), растворитель удалили в вакууме и остаток очистили с помощью колоночной хроматографии (активированный оксид алюминия при 0,5% МеОН в ДХМ) с получением промежуточного соединения 46, трет-бутил-4-(4-(трифторметил)-1Н-имидазол-2-ил)пиперидин-1-карбоксилата (1,80 г, 60%) в виде белого твердого вещества.
Данные для указанного в заголовке соединения приведены в Таблице 2.
трет-Бутил-4-(4-(трифторметил)-1Н-имидазол-2-ил)пиперидин-1-карбоксилат (1 г, 3,13 ммоль) растворили в 1,4-диоксане (5 мл) и с последующим прибавлением по каплям HCl в 1,4-диоксане (20 мл, 3 М р-р.). Полученную реакционную смесь перемешивали при 30°C в течение 16 ч, растворители удалили в вакууме и остаток очистили путем растирания с диэтиловым эфиром (3×5 мл) с получением промежуточного соединения 33, 4-(4-(трифторметил)-1Н-имидазол-2-ил)пиперидин гидрохлорида (650 мг, 95%) в виде белого твердого вещества. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 37, 4-[1-метил-4-(трифторметил)-1Н-имидазол-2-ил]пиперидин гидрохлоридной соли
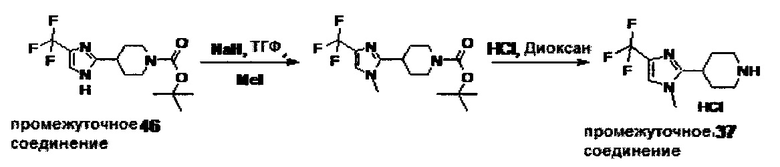
трет-Бутил-4-(4-(трифторметил)-1Н-имидазол-2-ил)пиперидин-1-карбоксилат (200 мг, 0,63 ммоль) растворили в ТГФ (5 мл) и прибавили при 0°C 60% гидрид натрия (74 мг, 1,88 ммоль). Реакционную смесь перемешивали при 0°C в течение 10 мин, затем прибавили метилиодид (0,06 мл, 0,96 ммоль) и перемешивали полученную реакционную смесь при 25°C в течение 2 ч. Реакционную смесь разделили между H2O (60 мл) и EtOAc (45 мл), водную фазу дополнительно экстрагировали EtOAc (2×45 мл), органические фазы объединили, сушили (Na2SO4) и растворители удалили в вакууме. Остаток очистили с помощью колоночной хроматографии (нормально-фазовая на силикагеле, размер в меш: 60-120, от 0% до 2,0% до 3,5% МеОН в ДХМ) с получением трет-бутил-4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)пиперидин-1-карбоксилата (190 мг, 91%) в виде желтой смолы.
ЖХ/МС (Способ F): m/z 334 (М+Н)+ (ES+), при 2,31 мин, активно в ультрафиолетовом свете
трет-Бутил-4-(1-метил-4-(трифторметил)-1Н-имидазол-2-ил)пиперидин-1-карбоксилат (200 мг, 0,6 ммоль) растворили в 1,4-диоксане (5 мл) с последующим прибавлением по каплям HCl в 1,4-диоксане (20 мл, 4 М р-р). Полученную реакционную смесь перемешивали при 30°C в течение 16 ч, растворители удалили в вакууме и остаток очистили путем растирания с диэтиловым эфиром (3×5 мл) с получением промежуточного соединения 37, 4-[1-метил-4-(трифторметил)-1Н-имидазол-2-ил]пиперидин гидрохлоридной соли (140 мг, 86,8%) в виде белого твердого вещества. Данные для указанного в заголовке соединения приведены в Таблице 2
Метод получения промежуточного соединения 43, 4-(1,3,4-оксадиазол-2-ил)пиперидина
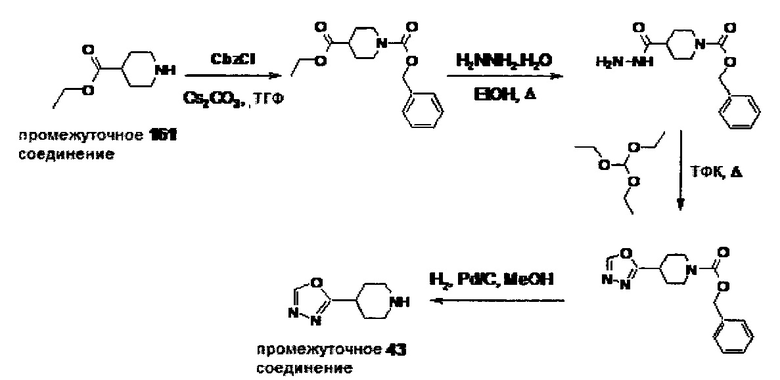
Этилпиперидин-4-карбоксилат (3,0 г, 19,1 ммоль) растворили в ТГФ (15 мл) и прибавили Cs2CO3 (7,4 г, 22,9 ммоль) при 0°C. Полученную реакционную смесь перемешивали при 0-5°C в течение 10 мин, затем по каплям прибавляли бензилхлорформиат (3,2 г, 19,1 ммоль), и перемешивали реакционную смесь при комнатной температуре в течение 18 ч. Реакционную смесь разделили между H2O (100 мл) и EtOAc (200 мл), водную фазу экстрагировали EtOAc (2×200 мл), органические фазы объединили, сушили (Na2SO4) и растворитель удалили в вакууме. Остаток очистили с помощью колоночной хроматографии (нормально-фазовая на силикагеле, размер в меш: 60-120, от 0% до 10% EtOAc в гексане) с получением 1-бензил 4-этилпиперидин-1,4-дикарбоксилата (4,2 г, 76,4%) в виде желтого твердого вещества.
ЖХ/МС (Способ F): m/z 292 (М+Н)+ (ES+), при 2,35 мин, активно в ультрафиолетовом свете
1-Бензил-4-этилпиперидин-1,4-дикарбоксилат (4,2 г, 14,43 ммоль) растворили в EtOH (10 мл), прибавили гидразин моногидрат (10 мл, 5,41 ммоль) и полученную реакционную смесь перемешивали при 90°C в течение ночи. Растворители удалили в вакууме и сырой продукт растерли с пентаном и гексаном с получением бензил-4-(гидразинилкарбонил)пиперидин-1-карбоксилата (3,8 г, 95%) в виде белого твердого вещества.
ЖХ/МС (Способ Н): m/z 278 (М+Н)+ (ES+), при 1,76 мин, активно в ультрафиолетовом свете
Бензил-4-(гидразинилкарбонил)пиперидин-1-карбоксилат (0,5 г, 1,79 ммоль), растворили в триэтилортоформиате (8 мл) и затем прибавили ТФК (0,1 мл). Полученную реакционную смесь перемешивали при 80°C в течение ночи. Реакционную смесь разделили между H2O (50 мл) и EtOAc (100 мл), водную фазу экстрагировали EtOAc (2×100 мл), органические фазы объединили, сушили (Na2SO4) и растворитель удалили в вакууме. Остаток очистили с помощью колоночной хроматографии (нормально-фазовая на силикагеле, размер в меш: 60-120, от 50% до 60% EtOAc в гексане) с получением бензил-4-(1,3,4-оксадиазол-2-ил)пиперидин-1-карбоксилата (0,19 г, 38%) в виде желтого твердого вещества.
ЖХ/МС (Способ Н): m/z 288 (М+Н)+ (ES+), при 2,03 мин, активно в ультрафиолетовом свете
Бензил-4-(1,3,4-оксадиазол-2-ил)пиперидин-1-карбоксилат (0,15 г, 0,52 ммоль) растворили в МеОН (10 мл). Катализатор сухого 10% Pd на угле (20 мг) прибавили и реакционную смесь продули Н2 при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 12 ч в атмосфере Н2. Реакционную массу фильтровали через целит и растворитель удалили в вакууме с получением промежуточного соединения 43, 4-(1,3,4-оксадиазол-2-ил)пиперидина (0,078 г, 99%) в виде бесцветной смолы. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 44, 4-(3-метил-1,2,4-оксадиазол-5-ил)пиперидина
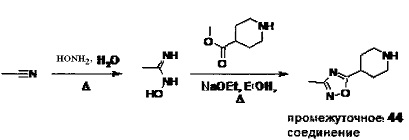
Ацетонитрил (40,0 мл) и 50% гидроксиламина в воде (4,20 мл) нагревали при кипячении с обратным холодильником при 90°C в течение 24 ч. Реакционную смесь охладили до 0°C и фильтровали. Остаток сушили с получением (Z)-N-гидроксиэтанимидамида (2,1 г, >100%) в виде белого кристаллического вещества.
ЖХ/МС (Способ Н): m/z 7 4 (М+Н)+ (ES+), при 1,86 мин, не активно в ультрафиолетовом свете
(Z)-N-гидроксиэтанимидамид (0,50 г, 6,75 ммоль) и метилпиперидин-4-карбоксилат (1,17 г, 7,42 ммоль) растворили в этаноле (20 мл). 21% раствор этоксида натрия в этаноле (0,92 мл, 13,4 ммоль) по каплям прибавляли и перемешивали реакционную смесь в течение 30 мин при комн. темп. и затем при 100°C в течение 16 ч. Растворители удалили в вакууме и остаток очистили с помощью колоночной хроматографии (нормально-фазовая на силикагеле, от 0 до 12% метанола в ДХМ) с получением промежуточного соединения 44, 4-(3-метил-1,2,4-оксадиазол-5-ил)пиперидина (380 мг, 34%) в виде желтой смолы. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 47, 4-(1H-имидазол-2-ил)пиперидин-4-ол гидрохлорида
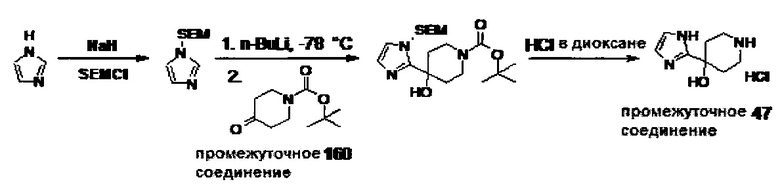
1Н-Имидазол (8,0 г, 117,5 ммоль) растворили в ДМФА (100 мл), прибавили гидрид натрия (4,7 г, 117,5 ммоль, 60% в минеральном масле) при комнатной температуре. Реакционную смесь перемешивали при комн. темп. в течение 2 ч. К реакционной смеси по каплям прибавляли 2-(триметилсилил)этоксиметилхлорид (20,5 г, 123,38 ммоль) при комн. темп., после прибавления реакционную смесь перемешивали при комн. темп. в течение 16 ч. Реакционную смесь вылили в ледяную воду (1000 мл) и экстрагировали EtOAc (500 мл), водную фазу дополнительно экстрагировали EtOAc (2×500 мл), органические фазы объединили, сушили (Na2SO4) и растворители удалили в вакууме. Остаток очистили с помощью колоночной хроматографии (нормальная фаза - силикагель, от 0 до 1% метанол в ДХМ) с получением 1-((2-(триметилсилил)этокси)метил)-1Н-имидазола (16,2 г, 68,6%) в виде светло-зеленой смолы.
ЖХ/МС (Способ F): m/z 199 (М+Н)+ (ES+), при 1,73 мин, активно в ультрафиолетовом свете
1-((2-(триметилсилил)этокси)метил)-1Н-имидазол (5,0 г, 25,0 ммоль) растворили в ТГФ (50 мл) и охладили до -78°C. По каплям прибавляли n-BuLi (19,0 мл, 30 ммоль, 1,6 М в гексане) при -78°C, затем реакционную смесь перемешивали при -78°C в течение 1 ч. К реакционной смеси при -78°C по каплям прибавляли раствор трет-бутил-4-оксопиперидин-1-карбоксилата (5,53 г, 27 ммоль) в ТГФ (10 мл). Реакционную смесь оставили нагреваться до комн. темп. в течение 2 ч. Реакционную смесь погасили насыщенным раствором NH4Cl (100 мл), экстрагировали EtOAc (50 мл), водную фазу дополнительно экстрагировали EtOAc (2×50 мл), органические фазы объединили, сушили (Na2SO4), и растворители удалили в вакууме. Остаток очистили с помощью колоночной хроматографии (нормальная фаза - силикагель, от 0 до 20% EtOAc в гексане) с получением трет-бутил-4-гидрокси-4-(1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-2-ил)пиперидин-1-карбоксилата (8 г, 80,0%) в виде светло-желтой смолы.
ЖХ/МС (Способ F): m/z 398 (М+Н)+ (ES+), при 2,16 мин, активно в ультрафиолетовом свете
трет-Бутил-4-гидрокси-4-(1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-2-ил)пиперидин-1-карбоксилат (2,0 г, 5,0 ммоль) растворили в 4 М HCl в 1,4-диоксане (20 мл), реакционную смесь перемешивали при комн. темп. в течение 10 ч. Растворители удалили в вакууме, и остаток растерли с ацетоном (3×20 мл) с получением промежуточного соединения 47, 4-(1Н-имидазол-2-ил)пиперидин-4-ола (0,5 г, 60,2%) в виде коричневой смолы. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 48, 4-(1H-имидазол-2-ил)-4-метоксипиперидин гидрохлоридной соли
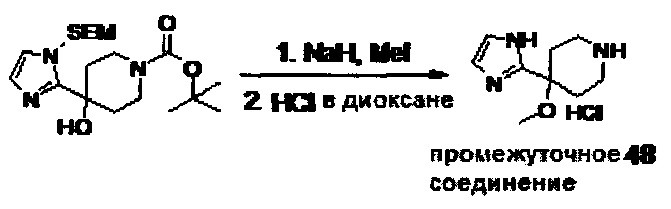
трет-Бутил-4-гидрокси-4-(1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-2-ил)пиперидин-1-карбоксилат (2,0 г, 5,0 ммоль) растворили в ДМФА (20 мл). Раствор охладили до 0°C в атмосфере N2 и прибавили NaH (0,24 г, 10,0 ммоль). Реакционную смесь перемешивали при 0°C в течение 30 мин, затем прибавили метилиодид (1,07 г, 7,5 ммоль) и перемешивали реакционную смесь при комн. темп. в течение 2 ч. Реакционную смесь разделили между водой (50 мл) и EtOAc (50 мл), водную фазу дополнительно экстрагировали EtOAc (3×100 мл), органические фазы объединили, сушили (Na2SO4), и растворители удалили в вакууме. Остаток очистили с помощью колоночной хроматографии (нормально-фазовая, силикагель 60-120 меш, от 0 до 10% EtOAc в гексане) с получением трет-бутил-4-метокси-4-(1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-2-ил)пиперидин-1-карбоксилата (1,5 г, 75,0%) в виде желтой смолы.
трет-бутил-4-метокси-4-(1-((2-(триметилсилил)этокси)метил)-1Н-имидазол-2-ил)пиперидин-1-карбоксилат (1,5 г, 3,6 ммоль) растворили в 4 М HCl в 1,4-диоксане (20 мл), реакционную смесь перемешивали при комн. темп. в течение 10 ч. Растворители удалили в вакууме, и остаток растерли с ацетоном (3×20 мл) с получением промежуточного соединения 48, 4-(1Н-имидазол-2-ил)-4-метоксипиперидина (0,5 г, 76,0%) в виде коричневого твердого вещества. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 49, 4-(1-метил-1Н-имидазол-2-ил)пиперидин-4-ол гидрохлоридной соли
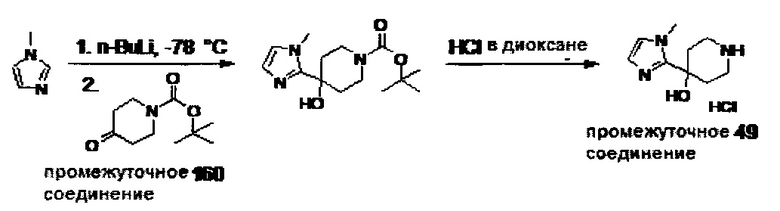
1-Метилимидазол (6,0 г, 73,0 ммоль) растворили в ТГФ (100 мл) при комн. темп. и охладили реакционную смесь до -78°C в атмосфере азота, медленно прибавили n-BuLi в гексане (45,4 мл, 73,0 ммоль). Реакционную смесь постепенно нагрели до 40°C и перемешивали в течение 4 ч, затем охладили до -78°C. Прибавили трет-бутил 4-оксопиперидин-1-карбоксилат (14,56 г, 73,0 ммоль) в ТГФ (100 мл). Реакционную смесь постепенно нагрели до 40°C и перемешивали в течение 10 ч, затем погасили водой (50 мл). Реакционную смесь разделили между EtOAc (200 мл) и водой (150 мл), водную фазу экстрагировали EtOAc (2×200 мл) и органические фазы объединили и сушили (Na2SO4). Растворители удалили в вакууме и остаток промыли метанолом с получением трет-бутил-4-гидрокси-4-(1-метил-1H-имидазол-2-ил)пиперидин-1-карбоксилата (14,0 г, 68,1%) в виде твердого вещества, которое использовали в сыром виде в последующей реакции.
ЖХ/МС (Способ F): m/z 282 (М+Н)+ (ES+), при 2,05 мин, активно в ультрафиолетовом свете
трет-бутил-4-гидрокси-4-(1-метил-1Н-пиррол-2-ил)пиперидин-1-карбоксилат (0,5 г, 1,7 ммоль) растворили в 1,4-диоксане (30 мл) при комн. темп. и охладили реакционную смесь до 0°C в атмосфере азота, медленно прибавили HCl в диоксане (15 мл, 4 М р-р). Реакционную смесь перемешивали при комн. темп. в течение 6 ч, растворители удалили в вакууме, и остаток очистили растиранием в пентане (10 мл) и диэтиловом эфире (10 мл) с получением промежуточного соединения 49, 4-(1-метил-1Н-имидазол-2-ил)пиперидин-4-ола (0,2 г, 62,5%) в виде коричневого твердого вещества. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 50, 4-метокси-4-(1-метил-1H-имидазол-2-ил)пиперидин гидрохлоридной соли
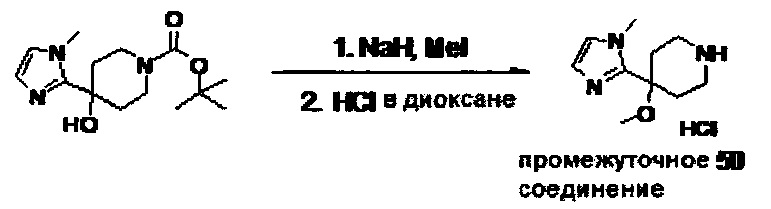
трет-Бутил-4-гидрокси-4-(1-метил-1H-имидазол-2-ил)пиперидин-1-карбоксилат (3,0 г, 10,6 ммоль) растворили в ДМФА (50 мл) при комн. темп. и реакционную смесь охладили до 0°C в атмосфере азота, прибавили NaH (0,64 г, 16,0 ммоль, 60% дисперсия в масле). Реакционную смесь перемешивали при 0°C в течение 1 ч и затем прибавляли по каплям метилиодид (1,8 г, 128 ммоль). Реакционную смесь нагрели до комн. темп. и перемешивали в течение 10 ч, затем погасили водой (50 мл). Реакционную смесь экстрагировали EtOAc (3×200 мл) и органические фазы объединили и сушили (Na2SO4). Растворители удалили в вакууме и остаток очистили с помощью колоночной хроматографии (нормально-фазовая, силикагель, 60-120 меш, градиент от 0% до 50% EtOAc в гексане) с получением трет-бутил-4-метокси-4-(1-метил-1Н-имидазол-2-ил)пиперидин-1-карбоксилата (1,3 г, 41,3%) в виде бледно-желтого твердого вещества.
ЖХ/МС (Способ F): m/z 296 (М+Н)+ (ES+), при 2,36 мин, активно в ультрафиолетовом свете
трет-Бутил-4-метокси-4-(1-метил-1H-имидазол-2-ил)пиперидин-1-карбоксилат (1,3 г, 3,3 ммоль) растворили в 1,4-диоксане (30 мл) при комн. темп. и реакционную смесь охладили до 0°C в атмосфере азота, медленно прибавили HCl в диоксане (15 мл, 4 М р-р). Реакционную смесь перемешивали при комн. темп. в течение 6 ч, растворители удалили в вакууме и остаток очистили растиранием в пентане (10 мл) и диэтиловом эфире (10 мл) с получением промежуточного соединения 50, 4-метокси-4-(1-метил-1H-имидазол-2-ил)пиперидина (0,80 г, 94,1%) в виде белого твердого вещества с металлическим оттенком. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 111, бензил-4-[(2S,4R)-1-(трет-бутоксикарбонил)-4-гидроксипирролидин-2-ил]пиперидин-1-карбоксилата
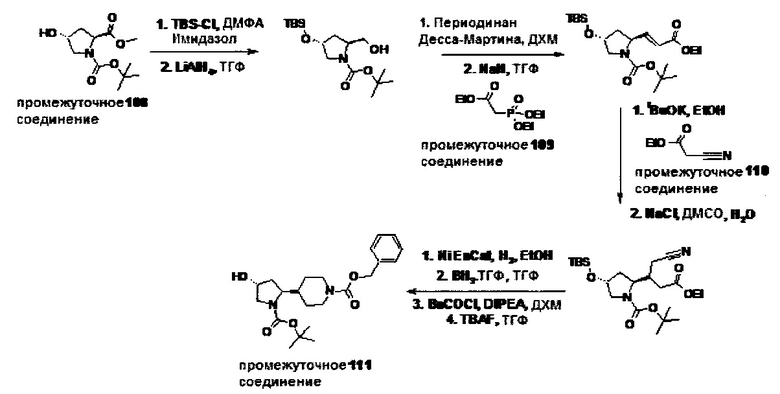
Метиловый эфир (2S,4R)-1-Boc-4-гидроксипирролидин-2-карбоновой кислоты (25 г, 101,93 ммоль) и имидазол (34,687 г, 509,5 ммоль) растворили в ДМФА (100 мл) и охладили реакционную смесь до 0°C. Затем прибавили трет-бутилдиметилсилилхлорид (36,86 г, 244,56 ммоль), нагрели реакционную смесь до комн. темп. и перемешивали в течение 18 ч. Летучие компоненты удалили на роторном испарителе и разбавили реакционную смесь ДХМ (250 мл). Смесь промыли H2O (2×250 мл), объединенные водные фазы промыли ДХМ (250 мл), объединенные органические фазы промыли насыщенным NH4Cl(водн.) (250 мл) и насыщенным водным раствором хлорида натрия (250 мл) и пропустили через фазный сепаратор Biotage. Летучие компоненты удалили в вакууме с получением (2S,4R)-1-Вос-4-трет-бутилдиметилсилилового эфира метилового эфира пирролидин-2-карбоновой кислоты (35,812 г, 99%).
ЖХ/МС (Способ D): m/z 260 (М+Н-Вос)+ (ES+), при 2,64 мин, не активно в ультрафиолетовом свете
(2S,4R)-1-Вос-4-трет-бутилдиметилсилиловый эфир метилового эфира пирролидин-2-карбоновой кислоты (42,7 г, 118,76 ммоль) растворили в ТГФ (100 мл) и охладили реакционную смесь до 0°C. Затем прибавили алюмогидрид лития (120 мл в 1,0 М растворе в ТГФ, 120,0 ммоль) и перемешивали реакционную смесь при 0°C в течение 1 ч. Реакционную смесь погасили H2O (4,5 мл), раствором 15% NaOH (4,5 мл) и H2O (13,5 мл) и фильтровали через слой целита. Летучие компоненты удалили в вакууме с получением (2S,4R)-1-Вос-4-трет-бутилдиметилсилилового эфира пирролидин-2-гидроксиметила (30,320 г, 77%).
ЖХ/МС (Способ D): m/z 232 (М+Н-Вос)+ (ES+), при 2,00 мин, не активно в ультрафиолетовом свете
(2S,4R)-1-Вос-4-трет-бутилдиметилсилиловый эфир пирролидин-2-гидроксиметила (10,050 г, 30,362 ммоль) растворили в ДХМ (100 мл) и прибавили периодинан Десса-Мартина (15,371 г, 36,253 ммоль). Реакционную смесь перемешивали при комн. темп. в течение 2 часов, затем летучие компоненты удалили на роторном испарителе и сырой продукт загрузили непосредственно в колонку Biotage SNAP (100 г) для очистки (градиент от 10% до 50% EtOAc в н-гексане) с получением (2S,4R)-1-Boc-4-тpeт-бутилдиметилсилилового эфира пирролидин-2-карбальдегида (2,150 г, 22%). К суспензии гидрида натрия (135 мг 60% дисперсии в минеральном масле, 3,338 ммоль) в ТГФ (8 мл) при 0°C прибавили триэтилфосфоноацетат (0,665 мл, 3,338 ммоль). Через 10 мин прибавили (2S,4R)-1-Вос-4-трет-бутилдиметилсилиловый эфир пирролидин-2-карбальдегида (1,002 г, 3,034 ммоль) в ТГФ (2 мл) и перемешивали реакционную смесь при 0°C в течение 30 мин. Летучие компоненты удалили на роторном испарителе и разбавили реакционную смесь ДХМ (20 мл). Смесь промыли H2O (2×20 мл), объединенные водные фазы промыли ДХМ (20 мл), объединенные органические фазы промыли насыщенным водным раствором хлорида натрия (20 мл) и пропустили через фазный сепаратор Biotage. Летучие компоненты удалили в вакууме и сырую смесь загрузили в колонку Biotage SNAP (100 г) и очистили с помощью колоночной хроматографии (от 0 до 30% EtOAc в гексане) с получением трет-бутил-(2S,4R)-4-{[трет-бутил(диметил)силил]окси)-2-[(1Е)-3-этокси-3-оксопроп-1-ен-1-ил]пирролидин-1-карбоксилата в виде бесцветного масла (545 мг, 45%).
ЖХ/МС (Способ D): m/z 300 (М+Н-Вос)+ (ES+), при 2,85 мин, не активно в ультрафиолетовом свете.
К раствору трет-бутоксида калия (421 мг, 3,753 ммоль) и этилцианоацетат (0,399 мл, 3,753 ммоль) в EtOH (5 мл) прибавили трет-бутил-(2S,4R)-4-{[трет-бутил(диметил)силил]окси)-2-[(1Е)-3-этокси-3-оксопроп-1-ен-1-ил]пирролидин-1-карбоксилат (500 мг, 1,251 ммоль) и перемешивали реакционную смесь при 78°C в течение 18 ч. Прибавили АсОН (0,200 мл) и удалили летучие компоненты на роторном испарителе. Реакционную смесь разбавили ДХМ (50 мл) и промыли H2O (2×50 мл), объединенные водные фазы промыли ДХМ (50 мл), объединенные органические фазы промыли насыщенным водным раствором хлорида натрия (50 мл) и пропустили через фазный сепаратор Biotage. Летучие компоненты удалили в вакууме и сырую смесь загрузили в колонку Biotage SNAP (100 г) и очистили с помощью колоночной хроматографии (от 0 до 30% EtOAc в гексане) с получением диэтил-3-[(4R)-1-(трет-бутоксикарбонил)-4-{[трет-бутил(диметил)силил]окси)пирролидин-2-ил]-2-цианопентандиоата в виде желтого масла (567 мг, 89%).
К раствору хлорида натрия (71 мг, 1,212 ммоль) и ДМСО (0,040 мл, 2,204 ммоль) в H2O (3 мл) прибавили диэтил-3-[(4R)-1-(трет-бутоксикарбонил)-4-{[трет-бутил(диметил)силил]окси)пирролидин-2-ил]-2-цианопентандиоат (565 мг, 1,102 ммоль) и перемешивали реакционную смесь при 145°C в течение 2 ч. Прибавили ледяную воду (50 мл) с последующим прибавлением EtOAc (50 мл) и промыли органическую фазу H2O (2×50 мл). Объединенные органические фазы промыли насыщенным водным раствором хлорида натрия (50 мл) и пропустили через фазный сепаратор Biotage. Летучие компоненты удалили в вакууме и сырую смесь загрузили в колонку Biotage SNAP (50 г) и очистили с помощью колоночной хроматографии (от 0 до 30% EtOAc в гексане) с получением трет-бутил-(4R)-4-{[трет-бутил(диметил)силил]окси)-2-(1-циано-4-этокси-4-оксобутан-2-ил)пирролидин-1-карбоксилата в виде желтого масла (351 мг, 72%).
ЖХ/МС (Способ D): m/z 341 (М+Н-Вос)+ (ES+), при 2,77 мин, не активно в ультрафиолетовом свете
В колбу, содержащую NiEnCat™ (65 г влажный, ~0,25 экв.) прибавили трет-бутил-(4Н)-4-{[трет-бутил(диметил)силил]окси)-2-(1-циано-4-этокси-4-оксобутан-2-ил)пирролидин-1-карбоксилата (8,700 г, 19,7 ммоль) в EtOH (75 мл) и перемешивали реакционную смесь при 78°C в атмосфере водорода из баллона в течение 96 ч. Реакционную смесь отфильтровали через слой целита и удалили летучие компоненты в вакууме, сырую смесь загрузили в колонку Biotage SNAP (340 г) и очистили с помощью колоночной хроматографии (от 2,5 до 10% МеОН в ДХМ) с получением трет-бутил-(4R)-4-{[трет-бутил(диметил)силил]окси)-2-(2-оксопиперидин-4-ил)пирролидин-1-карбоксилата в виде желтого масла (4,665 г, 59%).
ЖХ/МС (Способ D): m/z 399 (М+Н)+ (ES+), при 1,90 мин, не активно в ультрафиолетовом свете
К раствору трет-бутил (4R)-4-{[трет-бутил(диметил)силил]окси)-2-(2-оксопиперидин-4-ил)пирролидин-1-карбоксилата (1,850 г, 4,648 ммоль) ТГФ (30 мл) прибавили боран:тетрагидрофуран (9,3 мл 1,0 М раствора в ТГФ, 9,300 ммоль) при 0°C и перемешивали реакционную смесь при 60°C в течение 30 мин. Реакционную смесь охладили до комн. темп. и погасили МеОН (10 мл) и удалили летучие компоненты на роторном испарителе. Реакционную смесь разбавили ДХМ (100 мл) и промыли 1 М NaOH(водн.) (2×100 мл), объединенные водные фазы промыли ДХМ (100 мл), объединенные органические фазы промыли насыщенным водным раствором хлорида натрия (250 мл) и пропустили через фазный сепаратор Biotage. Летучие компоненты удалили в вакууме с получением трет-бутил-(2S,4R)-4-{[трет-бутил(диметил)силил]окси)-2-(пиперидин-4-ил)пирролидин-1-карбоксилата в виде желтого масла (1,830 г, >99%).
ЖХ/МС (Способ D): m/z 285 (М+Н-Вос)+ (ES+), при 3,00 мин, не активно в ультрафиолетовом свете
К раствору трет-бутил-(2S,4R)-4-{[трет-бутил(диметил)силил]окси)-2-(пиперидин-4-ил)пирролидин-1-карбоксилата (1,830 г, 4,766 ммоль) в ДХМ (20 мл) прибавили диизопропилэтиламин (1,814 мл, 10,484 ммоль) и бензилхлорформиат (0,816 мл, 5,719 ммоль) при 0°C. Реакционную смесь нагрели до комн. темп. и перемешивали в течение 18 ч. Реакционную смесь разбавили ДХМ (100 мл) и промыли 1 М NaОН(водн.) (2×100 мл), объединенные водные фазы промыли ДХМ (100 мл), объединенные органические фазы промыли насыщенным водным раствором хлорида натрия (250 мл) и пропустили через фазный сепаратор Biotage. Летучие компоненты удалили в вакууме и сырую смесь загрузили в колонку Biotage SNAP (100 г), и очистили с помощью колоночной хроматографии (от 10 до 40% EtOAc в гексане) с получением бензил-4-[(2S,4R)-1-(трет-бутоксикарбонил)-4-{[трет-бутил(диметил)силил]окси)пирролидин-2-ил]пиперидин-1-карбоксилата в виде бесцветного масла (700 мг, 28%).
К раствору бензил-4-[(2S,4R)-1-(трет-бутоксикарбонил)-4-{[трет-бутил(диметил)силил]окси)пирролидин-2-ил]пиперидин-1-карбоксилата (0,780 г, 1,504 ммоль) в ТГФ (5 мл) прибавили фторид тетрабутиламмония (1,800 мл 1,0 М раствора в ТГФ, 1,800 ммоль) и перемешивали реакционную смесь при комн. темп. в течение 1 ч. Реакционную смесь разбавили ДХМ (100 мл) и промыли H2O (2×100 мл), объединенные водные фазы промыли ДХМ (100 мл), объединенные органические фазы промыли насыщенным водным раствором хлорида натрия (250 мл) и пропустили через фазный сепаратор Biotage. Летучие компоненты удалили в вакууме с получением промежуточного соединения 111, бензил-4-[(2S,4R)-1-(трет-бутоксикарбонил)-4-гидроксипирролидин-2-ил]пиперидин-1-карбоксилата в виде бесцветного масла (500 мг, 82%). Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 112, трет-бутил (2S,4R)-4-фтор-2-(пиперидин-4-ил)пирролидин-1-карбоксилата
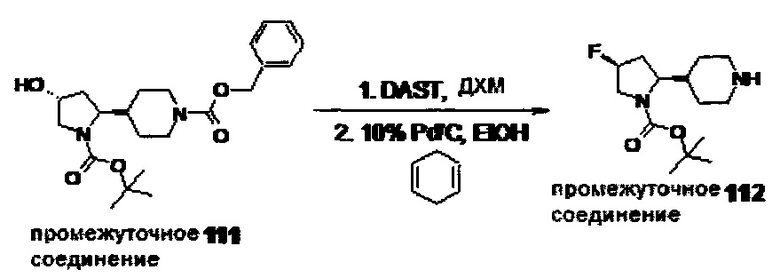
Бензил-4-[(2S,4R)-1-(трет-бутоксикарбонил)-4-гидроксипирролидин-2-ил]пиперидин-1-карбоксилат (100 мг, 0,247 ммоль) растворили в ДХМ (1 мл) при -40°C и прибавили DAST (0,049 мл, 0,371 ммоль). Реакционную смесь нагрели до комн. темп. и перемешивали в течение 2 ч. Реакционную смесь разбавили ДХМ (25 мл) и промыли насыщенным NaНСO3(водн.) (2×25 мл), объединенные водные фазы промыли ДХМ (25 мл), объединенные органические фазы промыли насыщенным водным раствором хлорида натрия (25 мл) и пропустили через фазный сепаратор Biotage. Летучие компоненты удалили в вакууме с получением бензил-4-[(2S,4S)-1-(трет-бутоксикарбонил)-4-фторпирролидин-2-ил]пиперидин-1-карбоксилата (0,090 г, 90%).
ЖХ/МС (Способ D): m/z 307 (М+Н-Вос)+ (ES+), при 2,31 мин, не активно в ультрафиолетовом свете
К раствору бензил-4-[(2S,4S)-1-(трет-бутоксикарбонил)-4-фторпирролидин-2-ил]пиперидин-1-карбоксилата (0,090 г, 0,221 ммоль) в EtOH (2 мл) прибавили 10% Pd/C (10 мг) и 1,4-циклогексадиена (0,147 мл, 1,530 ммоль) и перемешивали реакционную смесь при 70°C в течение 1 ч. Отфильтровали реакционную смесь через слой целита и удалили летучие компоненты в вакууме с получением промежуточного соединения 112, трет-бутил-(2S,4S)-4-фтор-2-(пиперидин-4-ил)пирролидин-1-карбоксилата (55 мг, 92%). Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 113, трет-бутил-(2S)-4,4-дифтор-2-(пиперидин-4-ил)пирролидин-1-карбоксилата
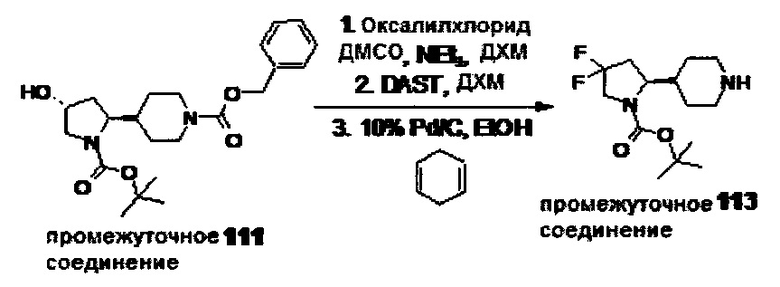
Оксалилхлорид (0,065 мл, 0,741 ммоль) растворили в ДХМ (1 мл) при -78°C и прибавили ДМСО (0,100 мл). Через 5 минут при -78°C прибавили бензил-4-[(2S,4R)-1-(трет-бутоксикарбонил)-4-гидроксипирролидин-2-ил]пиперидин-1-карбоксилат (200 мг, 0,494 ммоль) в ДХМ (2 мл) с последующим прибавлением триэтиламина (0,345 мл, 2,47 ммоль) через дополнительные 5 мин при -78°C. Реакционную смесь нагрели до комн. темп. и перемешивали в течение 30 мин. Реакционную смесь разбавили ДХМ (25 мл) и промыли насыщенным NaНСO3(водн.) (2×25 мл), объединенные водные фазы промыли ДХМ (25 мл), объединенные органические фазы промыли насыщенным водным раствором хлорида натрия (25 мл) и пропустили через фазный сепаратор Biotage. Летучие компоненты удалили в вакууме с получением бензил-4-[(2S)-1-(трет-бутоксикарбонил)-4-оксопирролидин-2-ил]пиперидин-1-карбоксилата (0,170 г, 85%).
ЖХ/МС (Способ D): m/z 303 (М+Н-Вос)+ (ES+), при 2,15 мин, не активно в ультрафиолетовом свете
бензил-4-[(2S)-1-(трет-бутоксикарбонил)-4-оксопирролидин-2-ил]пиперидин-1-карбоксилат (170 мг, 0,422 ммоль) растворили в ДХМ (1 мл) при -78°C и прибавили DAST (0,167 мл, 1,267 ммоль). Реакционную смесь нагрели до комн. темп. и перемешивали в течение 18 ч. Реакционную смесь разбавили ДХМ (25 мл) и промыли насыщенным NaHCO3(водн.) (2×25 мл), объединенные водные фазы промыли ДХМ (25 мл), объединенные органические фазы промыли насыщенным водным раствором хлорида натрия (25 мл) и пропустили через фазный сепаратор Biotage. Летучие компоненты удалили в вакууме с получением бензил-4-[(2S)-1-(трет-бутоксикарбонил)-4,4-дифторпирролидин-2-ил]пиперидин-1-карбоксилата (0,070 г, 39%).
ЖХ/МС (Способ D): m/z 325 (М+Н-Вос)+ (ES+), при 2,41 мин, не активно в ультрафиолетовом свете
К раствору бензил-4-[(2S)-1-(трет-бутоксикарбонил)-4,4-дифторпирролидин-2-ил]пиперидин-1-карбоксилата (0,067 г, 0,158 ммоль) в EtOH (2 мл) прибавили 10% Pd/C (10 мг) и 1,4-циклогексадиен (0,105 мл, 1,105 ммоль) и перемешивали реакционную смесь при 70°C в течение 1 ч. Реакционную смесь отфильтровали через слой целита и летучие компоненты удалили в вакууме с получением промежуточного соединения 113, трет-бутил-(2S)-4,4-дифтор-2-(пиперидин-4-ил)пирролидин-1-карбоксилата (30 мг, 65%). Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 125, трет-бутил-(2S)-4,4-дифтор-2-метилпирролидин-1-карбоксилата
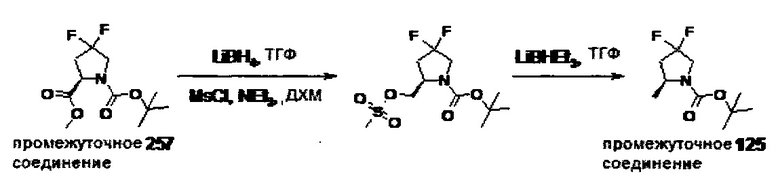
К 1-трет-бутил-2-метил (2R)-4,4-дифторпирролидин-1,2-дикарбоксилату (2 г, 7,5 ммоль) в ТГФ (20 мл) прибавили раствор борогидрида лития в ТГФ (2,0 М, 7,5 мл, 15 ммоль) при 0°C и нагрели реакционную смесь до комн. темп., и перемешивали в течение 2 ч. Реакционную смесь погасили путем прибавления по порциям насыщенного водного NaHCO3, и как только выделения газа прекратилось, смесь концентрировали для удаления ТГФ. Водную смесь разделили между насыщенным водным NaHCO3 и ДХМ (х2), органические фазы пропустили через фазный сепаратор и концентрировали для получения сырого трет-бутил-(2R)-4,4-дифтор-2-(гидроксиметил)пирролидин-1-карбоксилата (1,98 г, >100%) в виде масла.
ЖХ/МС (Способ С): m/z 260 (M+Na)+ (ES+), при 1,09 мин, не активно в ультрафиолетовом свете.
К раствору трет-бутил-(2R)-4,4-дифтор-2-(гидроксиметил)пирролидин-1-карбоксилата (1 г, 4,2 ммоль) в ДХМ (10 мл) и триэтиламина (1,5 мл, 11 ммоль) при 0°C порциями прибавили MsCl (0,42 мл, 5,4 ммоль). Смесь перемешивали при 0°C в течение 100 мин, затем разделили между ледяным насыщенным водным NaHCO3 и ледяным ДХМ (х2), органические фазы пропустили через фазный сепаратор и концентрировали для получения сырого трет-бутил-(2R)-4,4-дифтор-2-{[(метилсульфонил)окси]метил}пирролидин-1-карбоксилата (1,55 г, >100%) в виде масла.
ЖХ/МС (Способ С): m/z 338 (M+Na)+ (ES+), при 1,28 мин, не активно в ультрафиолетовом свете.
К раствору трет-бутил-(2R)-4,4-дифтор-2-{[(метилсульфонил)окси]метил}пирролидин-1-карбоксилата (1,55 г, 4,9 ммоль) в ТГФ (15 мл) при 0°C порциями прибавили раствор LiBHEt3 в ТГФ (1,0 М, 9,8 мл, 9,8 ммоль) в течение 10 мин. Затем смесь перемешивали в течение 3 дней, оставив охлаждающую баню нагреваться до комнатной температуры. Смесь снова охладили до 0°C, погасили путем прибавления H2O, затем концентрировали для удаления ТГФ. Водную смесь разделили между насыщенным водным NaHCO3 и ДХМ (х2), органические фазы пропустили через фазный сепаратор и концентрировали для получения сырого промежуточного соединения 125, трет-бутил-(2S)-4,4-дифтор-2-метилпирролидин-1-карбоксилата (0,89 г, 82%) в виде масла. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 126, третбутил-(2R)-4,4-дифтор-2-(гидроксиметил)пирролидин-1-карбоксилата
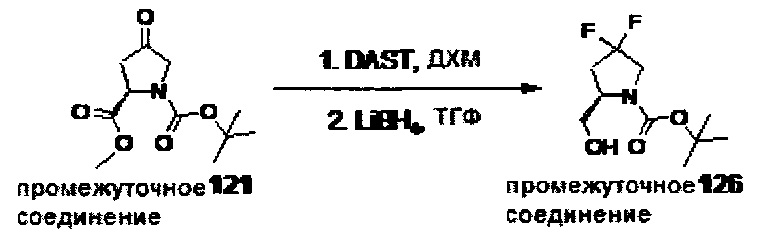
(R)-1-трет-Бутил-2-метил 4-оксопирролидин-1,2-дикарбоксилат (1,00 г, 4,111 ммоль) растворили в ДХМ (10 мл) при -78°C и прибавили DAST (1,629 мл, 12,332 ммоль). Реакционную смесь нагрели до комн. темп. и перемешивали в течение 2 ч. Реакционную смесь разбавили ДХМ (100 мл) и промыли насыщенным NaHCO3(водн.) (2×100 мл), объединенные водные фазы промыли ДХМ (100 мл), объединенные органические фазы промыли насыщенным водным раствором хлорида натрия (25 мл) и пропустили через сепаратор Biotage Phase. Растворитель удалили в вакууме с получением оранжевого масла (0,957 г, 90%).
К (R)-1-трет-Бутил 2-метил 4,4-дифторпирролидин-1,2-дикарбоксилату (500 мг, 1,885 ммоль) в ТГФ (5 мл) прибавили борогидрид лития в виде 2,0 М раствора в ТГФ (1,90 мл, 3,80 ммоль) при 0°C и реакционную смесь нагрели до комн. темп. и перемешивали в течение 1 ч. Растворители удалили е вакууме и реакционную смесь разбавили ДХМ (50 мл) и промыли насыщенным NaHCO3(водн.) (2×50 мл), объединенные водные фазы промыли ДХМ (50 мл), объединенные органические фазы промыли насыщенным водным раствором хлорида натрия (50 мл) и пропустили через фазный сепаратор Biotage. Летучие компоненты удалили в вакууме с получением промежуточного соединения 126, трет-бутил-(2R)-4,4-дифтор-2-(гидроксиметил)пирролидин-1-карбоксилата (452 мг, 92%).
Метод получения промежуточного соединения 132, 3-(пиперидин-4-ил)-1,3-оксазинан-2-он гидрохлорида
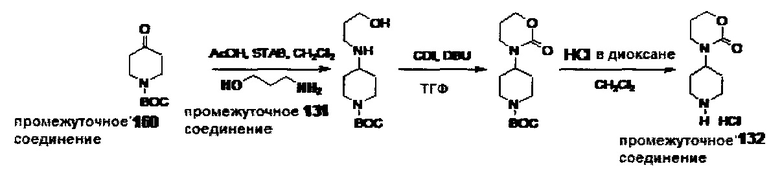
трет-Бутил-4-оксопиперидин-1-карбоксилат (0,796 г, 4,00 ммоль) и 3-аминопропан-1-ола (0,330 г, 4,4 ммоль) смешали в CH2Cl2 (20 мл) при комн. темп., прибавили АсОН (0,68 мл, 12,0 ммоль) и перемешивали в течение 3 ч. Прибавили STAB (2,34 г, 10,0 ммоль) и перемешивали реакционную смесь в атмосфере азота при комн. темп. в течение ночи. Реакционную смесь погасили путем прибавления NaHCO3 (насыщ. водн.) (40 мл), экстрагировали CH2Cl2 (4×45 мл) и объединенные органические фазы промыли насыщенным водным раствором хлорида натрия, затем сушили над MgSO4 и фильтровали. Растворители удалили в вакууме с получением сырого трет-бутил 4-[(3-гидроксипропил)амино]пиперидин-1-карбоксилата (1,03 г, 4,00 ммоль), который использовали без очистки.
ЖХ/МС (Способ В): m/z 259 (М+Н)+ (ES+), при 0,24 мин, не активно в ультрафиолетовом свете.
трет-Бутил 4-[(3-гидроксипропил)амино]пиперидин-1-карбоксилат (1,03 г, 4,00 ммоль), CDI (1,36 г, 8,4 ммоль) и ДБУ (0,24 мл, 1,60 ммоль) растворили в ТГФ (40 мл), смесь нагрели до кипения и выдержали в течение 72 ч. Растворители удалили в вакууме и остаток очистили с помощью колоночной хроматографии [картридж Biotage SNAP KP-sil 25 г, 40-63 мкм, 60, 50 мл в мин, градиент от 0% до 10% МеОН в ДХМ]) с получением трет-бутил-4-(2-оксо-1,3-оксазинан-3-ил)пиперидин-1-карбоксилата (0,60 г, 53%) в виде бесцветного масла.
ЖХ/МС (Способ В): m/z 307 (M+Na)+ (ES+), при 0,16 мин, не активно в ультрафиолетовом свете.
трет-Бутил-4-(2-оксо-1,3-оксазинан-3-ил)пиперидин-1-карбоксилат (0,60 г, 2,11 ммоль) растворили в CH2Cl2 (21 мл), прибавили 4 М хлористого водорода в диоксане (2,64 мл, 10,5 ммоль) и реакционную смесь перемешивали при комн. темп. в течение ночи. Осадок собрали фильтрованием, промыли CH2Cl2 (2×20 мл) и сушили с получением промежуточного соединения 132, 3-(пиперидин-4-ил)-1,3-оксазинан-2-он гидрохлорида (0,352 г, 76%) в виде бесцветного твердого вещества. Данные для указанного в заголовке соединения приведены в Таблице 2
Метод получения промежуточного соединения 151, этил-2-(4-оксопиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат гидрохлорида
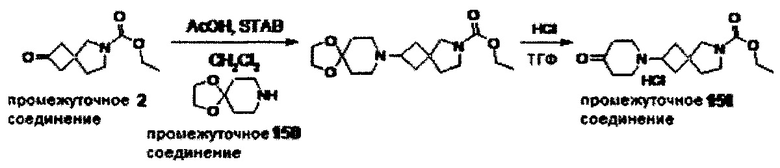
Этил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (0,985 г, 5,00 ммоль) и 1,4-диокса-8-азаспиро[4.5]декан (0,715 г, 5,00 ммоль) смешали в CH2Cl2 (50 мл) при комн. темп., прибавили АсОН (0,31 мл, 5,50 ммоль) и перемешивали в течение 3 ч. Прибавили STAB (2,65 г, 12,5 ммоль) и реакционную смесь перемешивали в атмосфере азота при комн. темп. в течение ночи. Реакционную смесь погасили путем прибавления NaHCO3 (насыщ. водн.) (40 мл), экстрагировали CH2Cl2 (4×45 мл) и объединенные органические фазы промыли насыщенным водным раствором хлорида натрия, затем сушили над MgSO4 и фильтровали. Растворители удалили в вакууме с получением сырого этил-2-(1,4-диокса-8-азаспиро[4.5]дек-8-ил)-6-азаспиро[3.4]октан-6-карбоксилата в виде смеси диастереомеров, которую использовали без дополнительной очистки.
ЖХ/МС (Способ D): m/z 325 (М+Н)+ (ES+), при 1,11 мин и 1,16 мин, не активно в ультрафиолетовом свете.
Сырой этил-2-(1,4-диокса-8-азаспиро[4.5]дек-8-ил)-6-азаспиро[3.4]октан-6-карбоксилат (1,62 г, 5,00 ммоль) растворили в ТГФ (10 мл), прибавили воду (10 мл) и концентрированную соляную кислоту (10 мл) и смесь перемешивали при комн. темп. в течение ночи. Растворители удалили в вакууме и остаток растерли в Et2O с получением промежуточного соединения 151, этил-2-(4-оксопиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат гидрохлорида (1,30 г, 82%) в виде бесцветного твердого вещества. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 164, 4-(1,3-тиазол-4-ил)пиперидин гидробромида
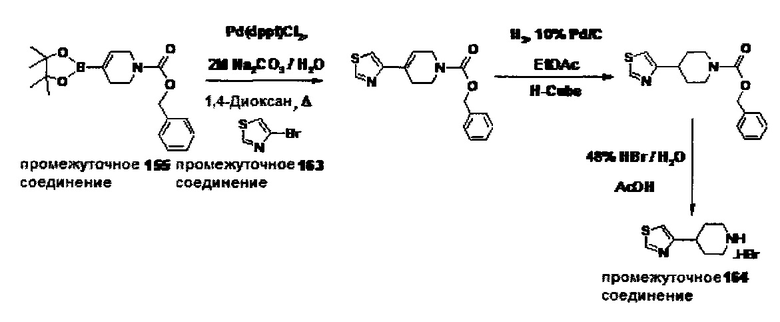
Водный раствор карбоната натрия (2 М) и 1,4-диоксан оба дегазировали, пропуская в жидкости в течение 15 мин струи азота через трубку с пористой стеклянной пластинкой. Бензил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2Н)-карбоксилат (250 мг, 0,73 ммоль), 4-бром-1,3-тиазол (119 мг, 0,73 ммоль), [1,1'-Бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (32 мг, 0,044 ммоль), дегазированный водный раствор карбоната натрия (2 М, 1,1 мл, 2,2 ммоль) и дегазированный 1,4-диоксан (3 мл) поместили в продутую азотом пробирку, герметично закрыли и нагревали под давлением при 90°C в течение 2,5 ч. Охлажденную реакционную смесь разбавили H2O и экстрагировали EtOAc. Органическую фазу пропустили через фазный сепаратор и концентрировали на силикагеле для флеш-хроматографии (15 мл). Полученный порошок очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 50 г, 40-63 мкм, 60], 40 мл в мин, 65% Et2O в изогексане, изократичный режим) с получением бензил 4-(1,3-тиазол-4-ил)-3,6-дигидропиридин-1(2Н)-карбоксилата (173 мг, 79%).
ЖХ/МС (Способ С): m/z 301 (М+Н)+ (ES+), при 1,46 мин, активно в ультрафиолетовом свете.
Раствор бензил-4-(1,3-тиазол-4-ил)-3,6-дигидропиридин-1(2Н)-карбоксилата (150 мг, 0,50 ммоль) в EtOAc (10 мл) гидрировали на катализаторе 10% палладия на угле при давлении в 100 бар и при 50°C при скорости потока 1 мл/мин, используя H-Cube аппарат. Раствор концентрировали с получением бензил-4-(1,3-тиазол-4-ил)пиперидин-1-карбоксилата (143 мг, 95%).
ЖХ/МС (Способ A): m/z 303 (М+Н)+ (ES+), при 1,92 мин, активно в ультрафиолетовом свете.
Раствор бензил-4-(1,3-тиазол-4-ил)пиперидин-1-карбоксилата (127 мг, 0,42 ммоль) в АсОН (1 мл) и 48% водный HBr (1 мл) перемешивали при комн. темп. в течение ночи. Затем смесь концентрировали и остаток подвергли азеотропной перегонке с толуолом с получением промежуточного соединения 164, 4-(1,3-тиазол-4-ил)пиперидин гидробромида (160 мг, >100%). Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 172, (1R,5S)-3-фенил-2,4-диокса-3-борабицикло[3.3.1]нонан-7-она
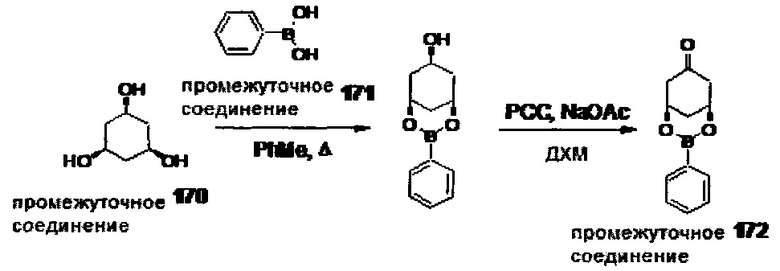
(1S,3S,5S)-Циклогексан-1,3,5-триол (1,0 г, 6,0 ммоль) и фенилбороновую кислоту (0,72 г, 6,0 ммоль) растворили в толуоле (35 мл) и нагревали при кипячении с обратным холодильником при 120°C в течение16 ч. Реакционную смесь концентрировали с получением сырого (1R,5S,7R)-3-фенил-2,4-диокса-3-борабицикло[3.3.1]нонан-7-ола (1,43 г, 87%) в виде твердого вещества, которое немедленно использовали. (1R,5S,7R)-3-фенил-2,4-диокса-3-борабицикло[3.3.1]нонан-7-ол (1,4 г, 6,4 ммоль) растворили в ДХМ (50 мл). Прибавили ацетат натрия (1,31 г, 16 ммоль) и пиридиния хлорохромат (12,9 г, 11 ммоль) и реакционную смесь перемешивали в течение 16 ч. Реакционную смесь фильтровали через целит и фильтрат концентрировали с получением сырого продукта, который перекристаллизовали из ДХМ : гексан (1:4) с получением промежуточного соединения 172, (1R,5S)-3-фенил-2,4-диокса-3-борабицикло[3.3.1]нонан-7-она (0,65 г, 38%) в виде твердого вещества. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 174, 4-[(2R)-4,4-дифтор-2-(метоксиметил)пирролидин-1-ил]пиперидин трифторацетатной соли
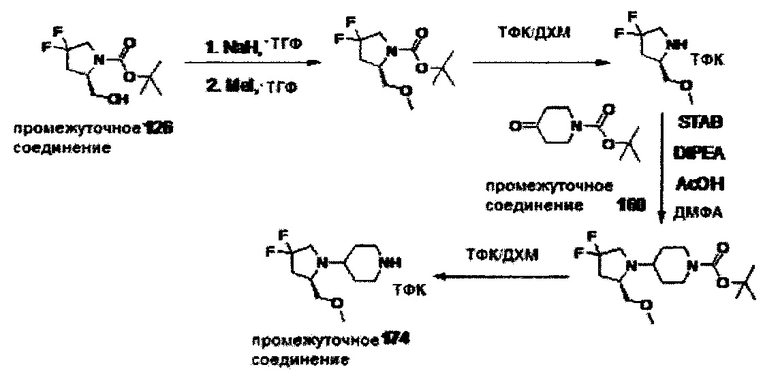
Раствор трет-бутил-(2R)-4,4-дифтор-2-(гидроксиметил)пирролидин-1-карбоксилат (150 мг, 0,63 ммоль) в ТГФ (5 мл) охладили в ледяной воде и обработали 60% суспензией гидрида натрия в минеральном масле (30 мг, 0,75 ммоль). Смесь перемешивали, охлаждая льдом в течение 30 мин, затем при комн. темп. в течение 1,5 ч до прибавления метилиодида (0,118 мл, 1,9 ммоль) и перемешивания при комн. темп. в течение ночи. Смесь погасили каплей H2O, затем концентрировали для удаления ТГФ. Остаток разделили между насыщ. водным NaHCO3 и ДХМ (х2), органическую фазу пропустили через фазный сепаратор и концентрировали с получением сырого трет-бутил-(2R)-4,4-дифтор-2-(метоксиметил)пирролидин-1-карбоксилата (110 мг, 69%) в виде масла.
ЖХ/МС (Способ С): m/z 274 (M+Na)+ (ES+), при 1,35 мин, не активно в ультрафиолетовом свете.
Раствор сырого трет-бутил-(2R)-4,4-дифтор-2-(метоксиметил)пирролидин-1-карбоксилата (110 мг, 0,44 ммоль) в ДХМ (2 мл) и ТФК (2 мл) перемешивали при комн. темп. в течение 40 мин, затем разбавили толуолом и концентрировали. Остаток подвергли азеотропной перегонке с толуолом (х2) с получением сырой (2R)-4,4-дифтор-2-(метоксиметил)пирролидин трифторацетатной соли в виде масла (172 мг, >100%).
ЖХ/МС (Способ С): m/z 152 (М+Н)+ (ES+), при 0,73 мин, не активно в ультрафиолетовом свете.
Сырую (2R)-4,4-дифтор-2-(метоксиметил)пирролидин трифторацетатную соль (172 мг, приблизительно 0,44 ммоль) растворили в ДМФА (5 мл). К раствору в указанном порядке прибавили DIPEA (0,38 мл, 2,2 ммоль), АсОН (0,038 мл, 0,66 ммоль), трет-бутил-4-оксопиперидин-1-карбоксилат (0,087 г, 0,44 ммоль) и STAB (0,278 г, 1,3 ммоль). Смесь перемешивали при комн. темп. в течение 2 дней, затем концентрировали для удаления ДМФА. Остаток разделили между насыщ. водным NaHCO3 и ДХМ (х2) и органическую фазу пропустили через фазный сепаратор и концентрировали с получением сырого трет-бутил-4-[(2R)-4,4-дифтор-2-(метоксиметил)пирролидин-1-ил]пиперидин-1-карбоксилата (0,241 г, >100%) в виде масла.
ЖХ/МС (Способ С): m/z 335 (М+Н)+ (ES+), при 1,43 мин, не активно в ультрафиолетовом свете.
Раствор сырого трет-бутил-4-[(2R)-4,4-дифтор-2-(метоксиметил)пирролидин-1-ил]пиперидин-1-карбоксилата (0,241 г, приблизительно 0,44 ммоль) в ДХМ (2 мл) и ТФК (2 мл) перемешивали при комн. темп. в течение 45 мин, затем разбавили толуолом и концентрировали. Остаток подвергли азеотропной перегонке с толуолом (х2) с получением сырого промежуточного соединения 174, 4-[(2R)-4,4-дифтор-2-(метоксиметил)пирролидин-1-ил]пиперидин трифторацетатной соли в виде масла. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 179,1-[(2R)-4,4-дифтор-1-(пиперидин-4-ил)пирролидин-2-ил]этанол трифторацетатной соли
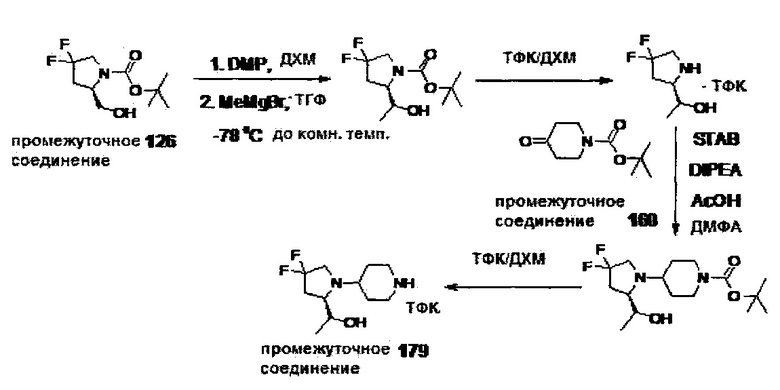
Раствор трет-бутил (2R)-4,4-дифтор-2-(гидроксиметил)пирролидин-1-карбоксилата (150 мг, 0,63 ммоль) в ДХМ (5 мл) охладили в ледяной воде и обработали периодинаном Десса-Мартина (402 мг, 0,95 ммоль). Охлаждающую баню убрали и смесь перемешивали при комн. темп. в течение 3 ч. Прибавили насыщ. водный NaHCO3 (5 мл), насыщ. водный раствор тиосульфата натрия (5 мл) и EtOAc (10 мл) и смесь интенсивно перемешивали в течение 30 мин. Фазы разделили и водную фазу повторно экстрагировали EtOAc. Объединенные органические фазы пропустили через фазный сепаратор и концентрировали с получением сырого альдегида, который немедленно растворили в ТГФ (5 мл), охладили до -78°C и обработали метилмагнийбромидом в эфире (3 М, 0,42 мл, 1,3 ммоль). Смесь убрали с охлаждающей бани, перемешивали в течение 2,75 ч и затем погасили путем прибавления насыщ. водного раствора NH4Cl. Смесь концентрировали для удаления ТГФ и затем разделили между насыщ. NH4Cl и ДХМ (х2). Органические фазы пропустили через фазный сепаратор и концентрировали на силикагеле для флеш-хроматографии (10 мл). Полученный порошок очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 25 г, 40-63 мкм, 60], 30 мл в мин, от 20 до 50% EtOAc в изогексане, с получением трет-бутил (2R)-4,4-дифтор-2-(1-гидроксиэтил)пирролидин-1-карбоксилата (0,106 г, 67%) в виде масла.
ЖХ/МС (Способ С): m/z 152 (М-ВОС+Н)+, 196 (M-tBu+H)+ (ES+), при 1,24 мин, не активно в ультрафиолетовом свете.
Раствор трет-бутил (2R)-4,4-дифтор-2-(1-гидроксиэтил)пирролидин-1-карбоксилата (102 мг, 0,41 ммоль) в ДХМ (2 мл) и ТФК (2 мл) перемешивали при комн. темп. в течение 30 мин, затем разбавили толуолом и концентрировали. Остаток подвергли азеотропной перегонке с толуолом с получением сырого 1-[(2R)-4,4-дифторпирролидин-2-ил]этанол трифторацетатной соли в виде смолы. Немедленно использовали.
ЖХ/МС (Способ С): m/z 152 (М+Н)+ (ES+), при 0,27 мин, не активно в ультрафиолетовом свете.
Указанную выше сырую 1-[(2R)-4,4-дифторпирролидин-2-ил]этанол трифторацетатную соль (приблизительно 0,41 ммоль) растворили в ДМФА (5 мл). К раствору в указанном порядке прибавили DIPEA (0,38 мл, 2,0 ммоль), АсОН (0,035 мл, 0,61 ммоль), трет-бутил-4-оксопиперидин-1-карбоксилат (0,081 г, 0,41 ммоль) и STAB (0,258 г, 1,2 ммоль). Смесь перемешивали при комн. темп. в течение 3 дней, затем концентрировали для удаления ДМФА. Остаток подвергли азеотропной перегонке с толуолом, растворили в МеОН и концентрировали на силикагеле для флеш-хроматографии (5 мл). Полученный порошок очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 25 г, 40-63 мкм, 60], 30 мл в мин, от 0 до 15% Растворителя А в ДХМ, где Растворитель А представляет собой 10% (7 М NH3/MeOH) в МеОН) с получением трет-бутил 4-[(2R)-4,4-дифтор-2-(1-гидроксиэтил)пирролидин-1-ил]пиперидин-1-карбоксилата (0,301 г, >100%) в виде масла.
ЖХ/МС (Способ С): m/z 335 (М+Н)+ (ES+), при 1,41 мин, не активно в ультрафиолетовом свете.
Раствор трет-бутил 4-[(2R)-4,4-дифтор-2-(1-гидроксиэтил)пирролидин-1-ил]пиперидин-1-карбоксилат (0,301 г, приблизительно 0,41 ммоль) в ДХМ (2 мл) и ТФК (2 мл) перемешивали при комн. темп. в течение 30 мин, затем разбавили толуолом и концентрировали. Остаток подвергли азеотропной перегонке с толуолом с получением сырого промежуточного соединения 179, 1-[(2R)-4,4-дифтор-1-(пиперидин-4-ил)пирролидин-2-ил]этанол трифторацетатной соли в виде масла (0,553 г, >100%). Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 215, 4-(1Н-тетразол-1-ил)пиперидин гидрохлоридной соли
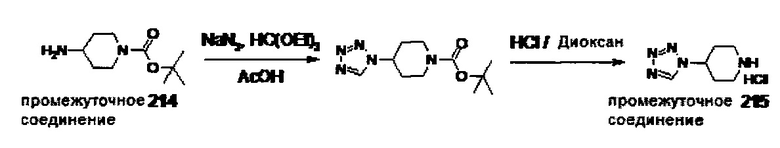
Триэтилортоформиат (3,5 г, 23 ммоль), трет-бутил-4-аминопиперидин-1-карбоксилат (0,80 г, 3,9 ммоль) и азид натрия (1,52 г, 23 ммоль) растворили в уксусной кислоте (50 мл). Полученную реакционную смесь перемешивали при 100°C в течение 6 ч, затем охладили до комн. темп. Летучие компоненты удалили путем концентрирования и остаток разделили между H2O (100 мл) и этилацетатом (150 мл). Водную фазу дополнительно экстрагировали этилацетатом (2×100 мл), объединенные органические фазы сушили (Na2SO4), и растворитель удалили путем концентрирования с получением сырого продукта, который растерли с диэтиловым эфиром с получением трет-бутил-4-(1Н-тетразол-1-ил)пиперидин-1-карбоксилата (0,51 г, 9%) в виде твердого вещества.
ЖХ/МС (Способ F): m/z 254 (М+Н)+ (ES+), при 1,92 мин, малоактивно в ультрафиолетовом свете.
трет-Бутил-4-(1Н-тетразол-1-ил)пиперидин-1-карбоксилат (0,51 г, 2,0 ммоль) растворили в 1,4-диоксане (10 мл). Прибавляли по каплям раствор HCl в 1,4-диоксане (4 М, 5 мл, 20 ммоль) и полученную смесь перемешивали при комн. темп. в течение 16 ч. Растворители удалили путем концентрирования и остаток очистили путем растирания с диэтиловым эфиром (3×10 мл) с получением промежуточного соединения 215, 4-(1Н-тетразол-1-ил)пиперидин гидрохлорида (0,30 г, 97%) в виде твердого вещества. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 218, 4-(1-циклопропил-1Н-тетразол-5-ил)пиперидин гидрохлоридной соли
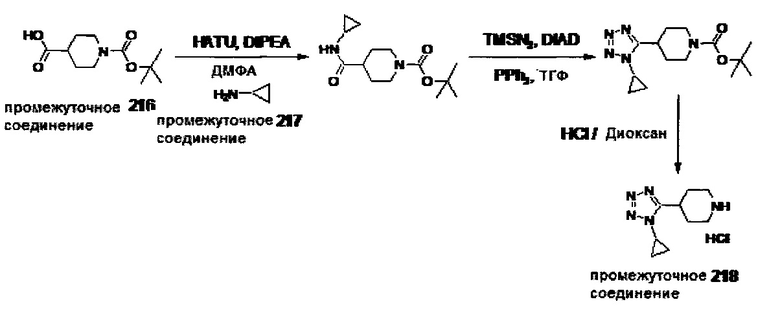
1-(трет-Бутоксикарбонил) пиперидин-4-карбоновая кислота (2,0 г, 8,7 ммоль) и циклопропиламин (0,6 мл, 8,7 мл) растворили в ДМФА (45 мл). Прибавили HATU (3,3 г, 8,7 ммоль) при комнатной температуре с последующим прибавлением DIPEA (3,1 мл, 17 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь разбавили холодной водой (250 мл) и экстрагировали EtOAc (3×100 мл). Объединенные органические фазы сушили (Na2SO4) и концентрировали с получением сырого продукта, который очистили с помощью колоночной хроматографии (Нормально-фазовой, Нейтральный силикагель, 60-120 меш, от 0 до 35% EtOAC в гексанах) с получением трет-бутил-4-(циклопропилкарбамоил)пиперидин-1-карбоксилата (1,5 г, 64%) в виде твердого вещества.
ЖХ/МС (Способ F): m/z 269 (М+Н)+ (ES+), при 1,80 мин, малоактивно в ультрафиолетовом свете.
трет-Бутил-4-(циклопропилкарбамоил)пиперидин-1-карбоксилат (1,5 г, 5,6 ммоль) и трифенилфосфин (2,9 г, 11 ммоль) растворили в ТГФ (160 мл). DIAD (2,26 г, 11 ммоль) прибавили при комнатной температуре в течение 15 мин. Прибавили триметилсилилазид (1,3 г, 11 ммоль) и реакционную смесь оставили перемешиваться при комнатной температуре в течение 24 ч. Реакционную смесь разбавили водой (250 мл) и экстрагировали EtOAc (3×100 мл). Объединенные органические фазы сушили (Na2SO4) и концентрировали с получением сырого продукта, который очистили с помощью колоночной хроматографии (Нормально-фазовая, Нейтральный силикагель, 60-120 меш, от 0 до 30% EtOAC в гексане) с получением трет-бутил-4-(1-циклопропил-1Н-тетразол-5-ил)пиперидин-1-карбоксилата (400 мг, 24%) в виде твердого вещества.
ЖХ/МС (Способ F): m/z 294 (М+Н)+ (ES+), при 1,96 мин, малоактивно в ультрафиолетовом свете.
трет-Бутил 4-(1-циклопропил-1Н-тетразол-5-ил)пиперидин-1-карбоксилат (400 мг, 1,4 ммоль) растворили в диоксане (5 мл). Раствор HCl в диоксане (4 М, 5 мл, 20 ммоль) прибавили при 0°C и смесь перемешивали при комнатной температуре в течение 5 ч. Растворитель удалили путем концентрирования и остаток растерли с диэтиловым эфиром (10 мл) с получением промежуточного соединения 218 4-(1-циклопропил-1Н-тетразол-5-ил)пиперидин гидрохлорида (260 мг, 98%) в виде твердого вещества. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 193, 1-(пиперидин-4-ил)пирролидин-2,5-дион трифторацетатной соли
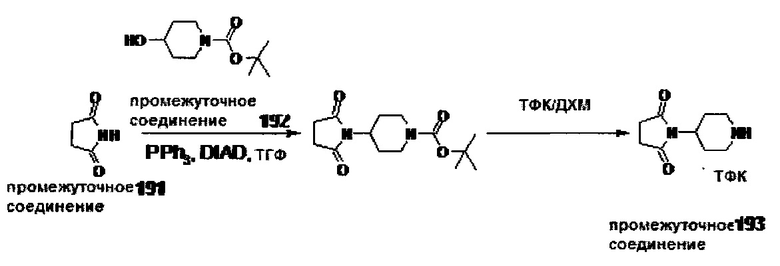
Сукцинимид (0,099 г, 1,0 ммоль), трет-бутил-4-гидроксипиперидин-1-карбоксилат (0,221 г, 1,10 ммоль и трифенилфосфин (0,314 г, 1,20 ммоль) растворили в ТГФ (5 мл), затем обработали диизопропилазодикарбоксилатом (0,236 мл, 1,20 ммоль) и перемешивали при комн. темп. в течение ночи. Реакционную смесь концентрировали на силикагеле для флеш-хроматографии (5 мл). Полученный порошок очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 25 г, 40-63 мкм, 60], 30 мл в мин, от 20% до 100% EtOAc в изогексане) с получением трет-бутил-4-(2,5-диоксопирролидин-1-ил)пиперидин-1-карбоксилата (0,253 г, 90%) в виде твердого вещества.
ЖХ/МС (Способ С): m/z 305 (M+Na)+ (ES+), при 1,11 мин, не активно в ультрафиолетовом свете
Раствор трет-бутил-4-(2,5-диоксопирролидин-1-ил)пиперидин-1-карбоксилата (0,141 г, 0,50 ммоль) в ДХМ (3 мл) и ТФК (3 мл) перемешивали при комн. темп. в течение 30 мин, затем разбавили толуолом и концентрировали. Остаток подвергли азеотропной перегонке с толуолом с получением промежуточного соединения 193, 1-(пиперидин-4-ил)пирролидин-2,5-дион трифторацетатной соли в виде смолы. Немедленно использовали. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 229, этил-2-([2,4'-бипиперидин]-1'-ил)-6-азаспиро[3.4]октан-6-карбоксилата
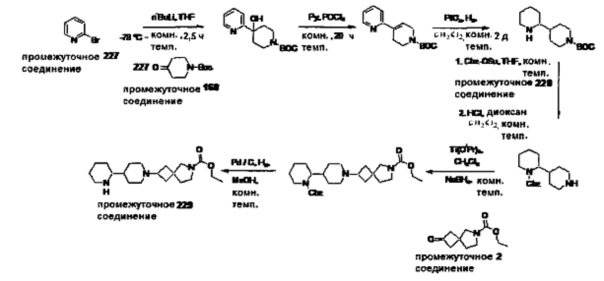
К раствору 2-бромпиридина (10,0 г, 63,3 ммоль) в сухом ТГФ (60 мл) медленно прибавили n-BuLi (79,1 мл, 2,5 М в гексане, 126 ммоль) при -78°C. После перемешивания при данной температуре в течение 30 мин медленно прибавили трет-бутил 4-оксопиперидин-1-карбоксилат (13,8 г, 69,6 ммоль) в ТГФ (40 мл). Температуру реакции постепенно подняли до комнатной температуры и перемешивали в течение 2 ч. После охлаждения до 0°C реакционную смесь осторожно погасили ледяной водой (50 мл). После удаления летучих компонентов водн. фазу экстрагировали этилацетатом (3×50 мл). Органические фазы объединили и промыли насыщенным водным раствором хлорида натрия, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очистили с помощью колоночной флеш-хроматографии [нормально-фазовая, силикагель (100-200 меш), градиент от 0% до 30% этилацетата в гексане] с получением трет-бутил 4-гидрокси-4-(пиридин-2-ил)пиперидин-1 -карбоксилата (11,4 г, 65%) в виде желтого масла.
1Н-ЯМР (400 МГц; CDCl3) δ: 1,48 (с, 9Н), 1,56-1,63 (м, 2Н), 1,90-2,0 (м, 2Н), 3,25-3,46 (м, 2Н), 4,05-4,22 (м, 2Н), 5,29 (уш. с., 1Н), 7,20-7,25 (м, 1Н), 7,32 (д, J=8,0, Гц, 1Н), 7,73 (дт, J=1,6, 8,4 Гц, 1Н), 8,53 (д, J=4,8 Гц, 1Н).
К раствору трет-бутил 4-гидрокси-4-(пиридин-2-ил)пиперидин-1-карбоксилат (11,4 г, 41,0 ммоль) в пиридине (100 мл), POCl3 (5,7 мл, 61,5 ммоль) прибавили и перемешивали при комнатной температуре в течение 20 ч. После удаления пиридина в вакууме реакционную смесь погасили водн. NaOH (10%, 30 мл) и экстрагировали хлороформом (2×30 мл). Органические фазы объединили, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очистили с помощью колоночной флеш-хроматографии [нормально-фазовая, силикагель (100-200 меш), градиент от 0% до 30% этилацетата в гексане] с получением трет-бутил-3',6'-дигидро-[2,4'-бипиридин]-1'(2'Н)-карбоксилат (2,3 г, 21%) в виде желтого масла.
1Н-ЯМР (400 МГц; CDCl3) δ: 1,48 (с, 9Н), 2,61-2,70 (м, 2Н), 3,60-3,70 (м, 2Н), 4,10-4,19 (м, 2Н), 6,58-6,62 (м, 1Н), 7,11-7,19 (м, 1Н), 7,36 (д, J=7,88 Гц, 1Н), 7,62-7,66 (м, 1Н), 8,55 (д, J=4,4 Гц, 1Н).
К раствору трет-бутил-3',6'-дигидро-[2,4'-бипиридин]-1'(2'Н)-карбоксилата (2,3 г, 8,84 ммоль) в CH2Cl2 (20 мл) прибавили PtO2 (200 мг, 0,88 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 дней в атмосфере Н2. Реакционную смесь отфильтровали через слой целита, промыли МеОН и концентрировали в вакууме. Остаток очистили с помощью колоночной флеш-хроматографии [нормально-фазовая, силикагель (100-200 меш), градиент от 0% до 15% МеОН в ДХМ, содержащего 0,1% водн. аммиака] с получением трет-бутил-[2,4'-бипиперидин]-1'-карбоксилата (1,05 г, 44%) в виде бесцветного масла.
1Н-ЯМР (400 МГц; CDCl3) δ: 1,30-1,40 (м, 1Н), 1,48 (с, 9Н), 1,60-1,91 (м, 8Н), 2,45-2,55 (м, 2Н), 2,58-2,75 (м, 4Н), 3,24-3,31 (м, 1Н), 4,14-4,24 (м, 2Н).
К раствору трет-бутил-[2,4'-бипиперидин]-1'-карбоксилата (1,05 г, 3,91 ммоль) в ТГФ (5 мл) прибавили Cbz-OSu (975 мг, 3,91 ммоль) в ТГФ (5 мл) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь погасили водой (20 мл) и водн. фазу экстрагировали этилацетатом (2×20 мл). Органические фазы объединили и промыли насыщенным водным раствором хлорида натрия, сушили (Na2SО4), фильтровали и концентрировали в вакууме. Остаток очистили с помощью колоночной флеш-хроматографии [нормально-фазовая, силикагель (100-200 меш), градиент от 0% до 20% этилацетата в гексане] с получением 1-бензил-1'-(трет-бутил)-[2,4'-бипиперидин]-1,1'-дикарбоксилата (840 мг, 53%) в виде бесцветного масла.
1Н-ЯМР (400 МГц; CDCl3) δ: 1,44 (с, 9Н), 1,50-1,63 (м, 2Н), 1,68-1,92 (м, 4Н), 1,95-2,14 (м, 2Н), 2,55-2,80 (м, 2Н), 2,81-2,95 (м, 4Н), 2,98-3,10 (м, 1Н), 3,75-4,24 (м, 3Н), 5,11 (с, 2Н), 7,34-7,37 (м, 5Н).]
К раствору 1-бензил-1'-(трет-бутил)-[2,4'-бипиперидин]-1,1'-дикарбоксилата (840 мг, 2,1 ммоль) в CH2Cl2 (10 мл) медленно прибавили HCl в диоксане (10 мл, 4 М) при 0°C и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь погасили насыщенным NaHCO3 (20 мл) и водн. фазу экстрагировали CH2Cl2 (2×20 мл). Органические фазы объединили, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очистили с помощью колоночной флеш-хроматографии [нормально-фазовая, силикагель (100-200 меш), градиент от 0% до 15% МеОН в ДХМ, содержащего 0,1% водн. аммиака] с получением бензил-[2,4'-бипиперидин]-1-карбоксилата (570 мг, 90%) в виде бесцветного липкого твердого вещества.
1Н-ЯМР (400 МГц; CDCl3) δ: 1,35-1,70 (м, 6Н), 1,71-1,98 (м, 4Н), 2,46-2,63 (м, 2Н), 2,68-2,73 (м, 1Н), 3,03-3,18 (м, 1Н), 3,65-3,80 (м, 2Н), 3,86-4,16 (м, 2Н), 5,11 (с, 2Н), 7,34-7,37 (м, 5Н).
К раствору бензил-[2,4'-бипиперидин]-1-карбоксилата (520 мг, 1,72 ммоль) и этил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилата (305 мг, 1,55 ммоль) в CH2Cl2 (15 мл) прибавили Ti(OiPr)4 (1,6 мл, 5,16 ммоль) и перемешивали при 0°C в течение 40 мин. К данной реакционной смеси прибавили NaBH4 (1,1 г, 5,16 ммоль) и перемешивание продолжили при этой температуре в течение 2 ч. Реакционную смесь погасили водой (20 мл) и водн. фазу экстрагировали CH2Cl2 (2×20 мл). Органические фазы объединили, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очистили с помощью препаративной ВЭЖХ (обращенно-фазовая, X BRIDGE, С-18, 19×250 мм, 5 мк, градиент от 68% до 90% ACN в воде, содержащей 0,1% NH4OH, 214 нм, комн. темп.: 7,45 мин для изомера-1 и 8,37 мин для изомера-2 с получением этил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилата изомера 1, (120 мг, 15%) и этил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилата изомера-2, (160 мг, 19%) в виде бесцветных липких твердых веществ.
Изомер-1:
ЖХ/МС (Способ L): m/z 484 (М+Н)+ (ES+), при 5,70 мин, активно в ультрафиолетовом свете.
1Н-ЯМР (400 МГц; CDCl3) δ: 1,10-1,32 (м, 6Н), 1,36-1,95 (м, 14Н), 2,00-2,18 (м, 2Н), 2,52-3,05 (м, 4Н), 3,20-3,45 (м, 4Н), 3,87-4,18 (м, 4Н), 5,11 (с, 2Н), 7,30-7,35 (м, 5Н).
Изомер-2:
ЖХ/МС (Способ L): m/z 484 (M+H)+ (ES+), при 5,81 мин, активно в ультрафиолетовом свете.
1Н-ЯМР (400 МГц; CDCl3) δ: 1,10-1,32 (м, 6Н), 1,35-1,53 (м, 5Н), 1,62-1,80 (м, 5Н), 1,81-1,97 (м, 4Н), 2,00-2,18 (м, 2Н), 2,52-3,00 (м, 4Н), 3,18-3,52 (м, 4Н), 3,88-4,20 (м, 4Н), 5,11 (с, 2Н), 7,32-7,37 (м, 5Н).
К раствору смеси изомеров этил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилата (1,0 г, 2,06 ммоль) в МеОН (20 мл) прибавили 10% Pd на угле (320 мг, 50% влажный) и реакционную смесь перемешивали при комн. темп. в течение 16 ч в атмосфере Н2. Реакционную смесь отфильтровали через слой целита, промыли МеОН и концентрировали в вакууме, и растерли с пентаном для получения промежуточного соединения 229, этил-2-([2,4'-бипиперидин]-1'-ил)-6-азаспиро[3.4]октан-6-карбоксилата (445 мг, 92%) в виде бесцветной жидкости. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 243, трет-бутил-1-(пиперидин-4-ил)-1,3-дигидро-2Н-изоиндол-2-карбоксилата
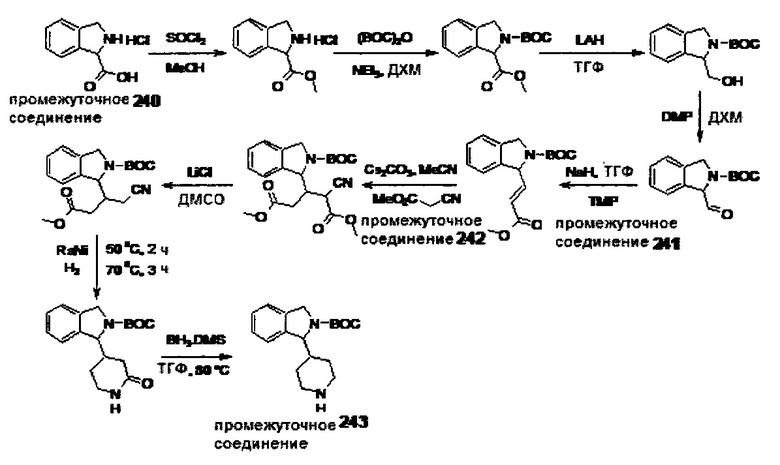
К раствору гидрохлорида изоиндолин-1-карбоновой кислоты (5,0 г, 25,0 ммоль) в метаноле (60 мл), медленно прибавили SOCl2 (2,7 мл, 37,5 ммоль) при 0°C и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. По окончании реакционную смесь концентрировали в вакууме. Остаток растерли с диэтиловым эфиром с получением метилизоиндолин-1-карбоксилат гидрохлорида (4,9 г, 92%) в виде белого твердого вещества с металлическим оттенком. Остаток использовали для следующей стадии без дополнительной очистки.
1Н-ЯМР (400 МГц; ДМСО-d6) δ: 3,81 (с, 3Н), 4,52-4,63 (м, 2Н), 5,70 (с, 1Н), 7,44-7,50 (м, 4Н), 9,77 (уш. с., 2Н).
К раствору метилизоиндолин-1-карбоксилат гидрохлорида (4,9 г, 23,0 ммоль) в ДХМ (50 мл) последовательно прибавили Et3N (9,9 мл, 69,0 ммоль) и (Вос)2O (8,0 мл, 34,0 ммоль) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 12 ч. Реакционную смесь погасили водой (20 мл). После отделения органической фазы, водн. фазу экстрагировали CH2Cl2 (3×15 мл). Органические фазы объединили и промыли насыщенным водным раствором хлорида натрия, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Сырой остаток очистили с помощью колоночной флеш-хроматографии [нормально-фазовая, силикагель (100-200 меш), градиент от 10% до 30% этилацетата в гексане] с получением 2-(трет-бутил) 1-метил изоиндолин-1,2-дикарбоксилата (6,5 г, 90%) в виде бесцветной жидкости.
1Н-ЯМР (400 МГц; CDCl3) δ: 1,52 (с, 9Н), 3,75 (с, 3Н), 4,65-4,85 (м, 2Н), 5,45 (с, 1Н), 7,25-7,43 (м, 4Н).
К раствору 2-(трет-бутил) 1-метил изоиндолин-1,2-дикарбоксилата (6,5 г, 23,0 ммоль) в ТГФ (60 мл) медленно прибавили LAH (2 М, 11,5 мл, 23,0 ммоль) при 0°C и перемешивали в течение 30 мин. По окончании реакционную смесь погасили насыщ. водн. Na2SO4 (20 мл). Реакционную смесь отфильтровали через слой целита и промыли этилацетатом (100 мл), сушили (Na2SO4) и концентрировали в вакууме с получением трет-бутил 1-(гидроксиметил)изоиндолин-2-карбоксилата (5,2 г, 91%) в виде белого твердого вещества с металлическим оттенком. Сырой остаток использовали для следующей стадии без дополнительной очистки.
1Н-ЯМР (400 МГц; CDCl3) δ: 1,52 (с, 9Н), 3,70-3,78 (м, 1Н), 3,98-4,03 (м, 1Н), 4,60-4,69 (м, 1Н), 4,70-4,85 (м, 2Н), 5,22 (уш. с., 1Н), 7,25-7,40 (м, 4Н).
К раствору трет-бутил 1-(гидроксиметил)изоиндолин-2-карбоксилата (5,2 г, 20,0 ммоль) в ДХМ (100 мл) порциями прибавили периодинан Десса-Мартина (27 г, 62,0 ммоль) при 0°C и перемешивали при комнатной температуре в течение 48 ч. По окончании реакционную смесь отфильтровали через слой целита и промыли диэтиловым эфиром (3×20 мл). Фильтрат промыли насыщ. водн. раствором NaНСO3, насыщенным водным раствором хлорида натрия, сушили (Na2SO4) и концентрировали в вакууме с получением трет-бутил-1-формилизоиндолин-2-карбоксилата (4,5 г, 88%) в виде коричневой жидкости. Сырой остаток использовали для следующей стадии без дополнительной очистки.
1Н-ЯМР (400 МГц; CDCl3) δ: 1,48 (с, 9Н), 4,65-4,90 (м, 2Н), 5,29-5,35 (с, 1Н), 7,25-7,35 (м, 4Н), 9,51 (с, 1Н).
К раствору NaH (874 мг, 18,2 ммоль) в ТГФ прибавили триметилфосфоноацетат (3,3 мл, 18,2 ммоль) при -78°C. После перемешивания в течение 1 ч при -78°C медленно прибавили трет-бутил 1-формилизоиндолин-2-карбоксилат (4,5 г, 18,2 ммоль) и реакционную смесь оставили при 0°C. По окончании реакционную смесь погасили насыщ. водн. раствором NH4Cl (10 мл) и водную фазу экстрагировали этилацетатом (3×20 мл). Органические фазы объединили, сушили (Na2SO4) и концентрировали в вакууме с получением трет-бутил (E)-1-(3-метокси-3-оксопроп-1-ен-1-ил)изоиндолин-2-карбоксилата (5,2 г, 92%) в виде коричневой жидкости. Остаток использовали для следующей стадии без дополнительной очистки.
МС (ESI +ve): 304
К раствору трет-бутил (E)-1-(3-метокси-3-оксопроп-1-ен-1-ил)изоиндолин-2-карбоксилата (5,2 г, 17,2 ммоль) в MeCN (20 мл) порциями прибавили Cs2CO3 (11,1 г, 34,4 ммоль) при комнатной температуре. После перемешивания в течение 20 мин медленно прибавили метилцианоацетат (3,0 мл, 34,4 ммоль) и реакционную смесь перемешивали при 70°C в течение 16 ч. По окончании реакционную смесь отфильтровали через слой целита и основательно промыли гексаном (3×20 мл). Фильтрат концентрировали в вакууме с получением диметил 3-(2-(трет-бутоксикарбонил)изоиндолин-1-ил)-2-цианопентандиоата) (5,5 г, сырой) в виде липкого твердого вещества. Сырой остаток использовали для следующей стадии без дополнительной очистки.
МС (ESI +ve): 403
К раствору диметил 3-(2-(трет-бутоксикарбонил)изоиндолин-1-ил)-2-цианопентандиоата) (1,6 г, сырой) в ДМСО (15 мл), прибавили LiCl (500 мг, 11,7 ммоль) с последующим прибавлением воды (0,1 мл, кат.) и перемешивали реакционную смесь при 135°C в течение 16 ч. По окончании реакционую смесь погасили водой (20 мл) и водн. фазу экстрагировали диэтиловым эфиром (3×20 мл). Органические фазы объединили, сушили (Na2SO4) и концентрировали в вакууме с получением трет-бутил 1-(1-циано-4-метокси-4-оксобутан-2-ил)изоиндолин-2-карбоксилата (1,5 г, сырой) в виде коричневого полутвердого вещества. Сырой остаток использовали для следующей стадии без дополнительной очистки.
МС (ESI +ve): 345
К раствору трет-бутил 1-(1-циано-4-метокси-4-оксобутан-2-ил)изоиндолин-2-карбоксилата (300 мг, 0,8 ммоль) в МеОН (30 мл) прибавили никель Ренея (0,30 г, влажный) и нагревали реакционную смесь до 50°C в течение 2 ч в атмосфере Н2 при 50 фунт на кв. дюйм. Затем температуру реакции повысили до 70°C и перемешивали в течение 3 ч. По окончании реакционую смесь отфильтровали через слой целита, промыли МеОН (25 мл) и концентрировали в вакууме. Остаток растерли с диэтиловым эфиром (30 мл) с получением трет-бутил-1-(2-оксопиперидин-4-ил)изоиндолин-2-карбоксилата (0,21 г, 76%) в виде коричневого твердого вещества.
МС (ESI +ve): 317
К раствору трет-бутил 1-(2-оксопиперидин-4-ил)изоиндолин-2-карбоксилата (210 мг, 0,60 ммоль) в ТГФ (5 мл) медленно прибавили BH3-DMS (0,5 мл, 6,60 ммоль) при 0°C и реакционную смесь перемешивали при 78°C в течение 8 ч. После охлаждения при 0°C реакционную массу погасили метанолом (0,5 мл) с последующим гашением водой (1 мл). К сырой реакционной массе прибавили 5% МеОН/ДХМ (30 мл) и отфильтровали. Фильтрат концентрировали в вакууме. Остаток растерли с диэтиловым эфиром (20 мл) с получением трет-бутил 1-(пиперидин-4-ил)изоиндолин-2-карбоксилата, промежуточного соединения 243 (200 мг, 99%) в виде коричневого твердого вещества. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 247, 4-(2Н-1,2,3-триазол-2-ил)пиперидина 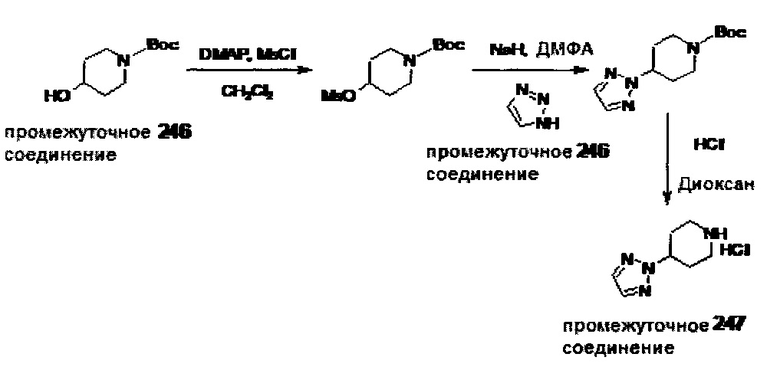
трет-Бутил-4-гидроксипиперидин-1-карбоксилат (0,500 г, 2,4 ммоль) растворили в CH2Cl2, затем по каплям прибавили DMAP (0,302 г, 2,4 ммоль) и метансульфонилхлорид (0,284 г, 2,48 ммоль) при 0°С. Полученную реакционную смесь перемешивали при комн. темп. в течение 6 ч, затем разделили между H2O (70 мл) и CH2Cl2 (70 мл), водную фазу дополнительно экстрагировали CH2Cl2 (2×70 мл), органические фазы объединили, сушили (Na2SO4), фильтровали и растворитель удалили в вакууме с получением сырого трет-бутил 4-((метилсульфонил)окси)пиперидин-1-карбоксилата (0,520 г, 75,0%) в виде белого твердого вещества, которое непосредственно использовали без дополнительной очистки.
1Н-ЯМР (400 МГц; ДМСО) δ: 1,23 (д, J=9,38 Гц, 2Н) 1,54-1,69 (м, 4Н) 1,86-1,96 (м, 2Н) 2,35 (с, 1Н) 2,85-3,00 (м, 2Н) 3,18 (д, J=5,42 Гц, 5Н) 3,54-3,67 (м, 4Н) 4,83 (с, 1Н).
1H-1,2,3-Триазол (0,098 г, 1,4 ммоль) растворили в ДМФА (5 мл) прибавили NaH (0,037 г, 1,5 ммоль) и перемешивали при 0°С в течение 30 мин. Прибавили трет-бутил-4-((метилсульфонил)окси)пиперидин-1-карбоксилат (0,400 г, 1,4 ммоль) и перемешивали при 150°C в течение 1 ч. Реакционную смесь разделили между H2O (50 мл) и EtOAc (50 мл), водную фазу дополнительно экстрагировали EtOAc (2×50 мл), органические фазы объединили, сушили (Na2SO4), фильтровали и растворитель удалили в вакууме с получением сырого трет-бутил-4-(2Н-1,2,3-триазол-2-ил) пиперидин-1-карбоксилата (0,350 г, 97,0%) в виде бесцветной смолы, которую непосредственно использовали без дополнительной очистки.
ЖХ/МС (Способ F): m/z 253 (M+H)+ (ES+), при 1,95 мин, активно в ультрафиолетовом свете. трет-Бутил-4-(2H-1,2,3-триазол-2-ил)пиперидин-1-карбоксилат (0,500 г, 1,9 ммоль) растворили в 1,4-диоксане (10 мл) с последующим прибавлением по каплям HCl в 1,4-диоксане (5 мл, 4 М). Полученную реакционную смесь перемешивали при 25°С в течение 16 ч, растворители удалили в вакууме и остаток очистили путем растирания с диэтиловым эфиром (3×10 мл) с получением 4-(2Н-1,2,3-триазол-2-ил)пиперидин гидрохлорида, промежуточного соединения 247, (0,290 г, 96,3%) в виде светлого белого твердого вещества. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 255, 4-(5-метил-1Н-тетразол-1-ил)пиперидин гидрохлорида
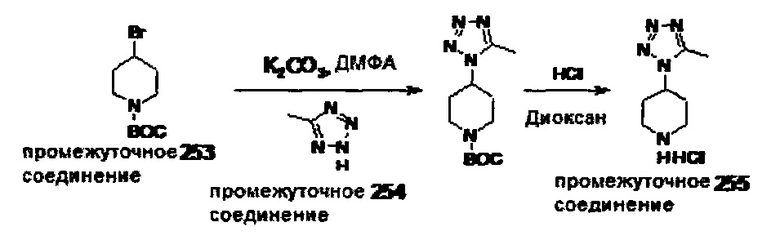
5-Метил-2Н-тетразол (0,500 г, 5,9 ммоль) и трет-бутил 4-бромпиперидин-1-карбоксилат (1,29 г, 4,8 ммоль) растворили в ДМФА. Прибавили K2CO3 (1,64 г, 11,8 ммоль), полученную реакционную смесь перемешивали при 100°C в течение 6 ч, затем разделили между H2O (100 мл) и этилацетатом (150 мл). Водную фазу дополнительно экстрагировали этилацетатом (2×100 мл), органические фазы объединили, сушили (Na2SO4), фильтровали и растворитель удалили в вакууме. Остаток очистили с помощью combi-flash колоночной хроматографии (нормально-фазовая, нейтральный силикагель, 60-120 меш, от 10 до 20% EtOAc в гексане) с получением трет-бутил 4-(5-метил-1Н-тетразол-1-ил)пиперидин-1-карбоксилата (0,280 г, 34,6%) в виде белого твердого вещества.
1Н-ЯМР (400 МГц, ДМСО) δ: 1,43 (с, 9Н), 1,73-1,88 (м, 2Н), 2,01 (уш. с., 2Н), 2,68-2,75 (м, 3Н), 2,88-2,91 (м, 2Н), 4,03-4,10 (м, 2Н), 4,60-4,70 (м, 1Н).
трет-Бутил 4-(5-метил-1Н-тетразол-1-ил) пиперидин-1-карбоксилат (0,280 г, 1,04 ммоль) растворили в 1,4-диоксане (10 мл) с последующим прибавлением по каплям HCl в 1,4-диоксане (5 мл, 4М). Полученную реакционную смесь перемешивали при 25°C в течение 16 ч, растворители удалили в вакууме и остаток очистили путем растирания с диэтиловым эфиром (3×10 мл) с получением 4-(5-метил-1Н-тетразол-1-ил)пиперидин гидрохлорида, промежуточного соединения 255 (0,170 г, 97,6%) в виде белого твердого вещества. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 258, дающий (R)-2-(4,4-дифтор-1-(пиперидин-4-ил)пирролидин-2-ил)пропан-2-ола гидрохлорид
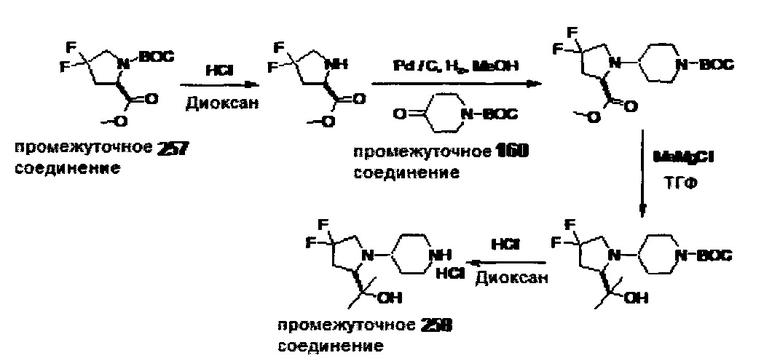
К раствору 1-(трет-бутил) 2-метил (R)-4,4-дифторпирролидин-1,2-дикарбоксилат (500 мг, 1,89 ммоль) в диоксане (15 мл) медленно прибавили HCl в диоксане(4 М, 15 мл) при 0°C и перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь концентрировали в вакууме и остаток растерли с гексаном (10 мл). Данный остаток подщелочили водн. насыщ. NaHCO3 (10 мл) и концентрировали. К сырой реакционной массе прибавили CDM (30 мл) и отфильтровали. Фильтрат концентрировали в вакууме с получением метил (R)-4,4-дифторпирролидин-2-карбоксилата (2, 320 мг, 84%) в виде коричневой жидкости. Данный сырой остаток использовали для следующей стадии без дополнительной очистки.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,40-1,51 (м, 1Н), 2,58-2,84 (м, 3Н), 3,52-3,62 (м, 1Н), 3,84 (с, 3Н), 4,40-4,52 (м, 1Н).
К раствору метил-(R)-4,4-дифторпирролидин-2-карбоксилата (200 мг, 1,21 ммоль) и трет-бутил 4-оксопиперидин-1-карбоксилата (240 мг, 1,21 ммоль) в метаноле (20 мл) прибавили 10% палладии на угле (300 мг, 50% влажный) и реакционную смесь перемешивали в атмосфере Н2 (1 атм) при комнатной температуре в течение 24 ч. По окончании реакционую смесь отфильтровали через слой целита, основательно промыли метанолом и концентрировали в вакууме с получением трет-бутил-(R)-4-(4,4-дифтор-2-(метоксикарбонил)пирролидин-1-ил)пиперидин-1-карбоксилата (400 мг, 95%) в виде бесцветной жидкости.
1Н-ЯМР (400 МГц, CDCl3) δ: 1,32-1,45 (м, 1Н), 1,45 (с, 9Н), 1,61-1,80 (м, 4Н), 2,39-2,49 (м, 1Н), 2,50-2,83 (м, 2Н), 3,19 (с, 3Н), 3,35-3,49 (м, 2Н), 3,61-3,82 (м, 3Н), 3,94-4,05 (м, 1Н). К раствору трет-бутил (R)-4-(4,4-дифтор-2-(метоксикарбонил)пирролидин-1-ил)пиперидин-1-карбоксилата (375 мг, 1,07 ммоль) в ТГФ (10 мл) медленно прибавили MeMgBr (3 М, 1,07 мл, 3,21 ммоль) при 0°C и перемешивали в течение 4 ч при комнатной температуре. По окончании реакционую смесь погасили насыщ. водн. раствором NH4Cl (10 мл) и водную фазу экстрагировали этилацетатом (3×10 мл). Органические фазы объединили, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Сырой остаток очистили с помощью колоночной флеш-хроматографии [нормально-фазовая, силикагель (100-200 меш), градиент от 10% до 30% этилацетата в гексане] с получением трет-бутил (R)-4-(4,4-дифтор-2-(2-гидроксипропан-2-ил)пирролидин-1-ил)пиперидин-1-карбоксилата (240 мг, 64%) в виде бесцветной жидкости.
1H-ЯМР (400 МГц, CDCl3) δ: 1,11 (с, 3Н), 1,21 (с, 3Н), 1,21-1,40 (м, 2Н), 1,46 (с, 9Н), 1,61-1,80 (м, 2Н), 2,15-2,30 (м, 3Н), 2,50-2,83 (м, 3Н), 3,02-3,23 (м, 2Н), 4,09-4,30 (м, 2Н). O-H не наблюдались.
К раствору (R)-4-(4,4-дифтор-2-(2-гидроксипропан-2-ил)пиррол идин-1-ил)пиперидин-1-карбоксилата (240 мг, 0,69 ммоль) в диоксане (10 мл) медленно прибавили HCl в диоксане (4 М, 10 мл) при 0°C и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали в вакууме и остаток растерли с гексаном (10 мл). К сырой реакционной массе прибавили CH2Cl2 (30 мл) и фильтровали. Фильтрат концентрировали в вакууме с получением (R)-2-(4,4-дифтор-1-(пиперидин-4-ил)пирролидин-2-ил)пропан-2-ол гидрохлорида, промежуточного соединения 258 (150 мг, 87%) в виде коричневой жидкости. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 282, трет-бутил-(2R)-2-(диметилкарбамоил)пиперидин-1-карбоксилата
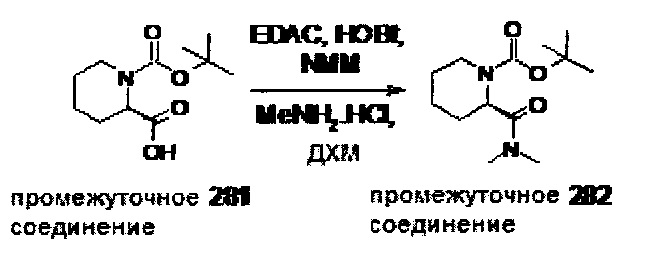
(R)-(трет-бутоксикарбонил)пиперидин-2-карбоновую кислоту (0,500 г, 2,18 ммоль) растворили в безводном ДХМ (8 мл) и охладили реакционную смесь до 0°C в атмосфере азота. Прибавили 1-этил-3-(3-диметиламинопропил)-карбодиимид HCl (0,628 г, 3,275 ммоль), гидроксибензотриазол (0,334 г, 2,183 ммоль), N-метилморфолин (1,104 г, 10,915 ммоль) и диметиламина гидрохлорид (0,356 г, 4,36 ммоль) и реакционную смесь перемешивали при комн. темп. в атмосфере азота в течение ночи. Реакционную смесь разбавили ДХМ (20 мл) и промыли насыщ. NaHCO3 (водн.) (20 мл) и насыщ. NaCl (водн.) (20 мл). Органическую фазу пропустили через картридж фазного сепаратора Biotage и растворители удалили в вакууме. Остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 25 г 40-63 мкм, 60 , 25 мл в мин, градиент от 0% до 10% МеОН / ДХМ]) с получением трет-бутил-(2R)-2-(диметилкарбамоил)пиперидин-1-карбоксилата, промежуточного соединения 282, (0,241 г, 43%) в виде масла янтарного цвета. Данные для указанного в заголовке соединения приведены в Таблице 2.
, 25 мл в мин, градиент от 0% до 10% МеОН / ДХМ]) с получением трет-бутил-(2R)-2-(диметилкарбамоил)пиперидин-1-карбоксилата, промежуточного соединения 282, (0,241 г, 43%) в виде масла янтарного цвета. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 295, трет-бутил-(2R)-2-(фторметил)пирролидин-1-карбоксилата

(2R)-(+)-1-Вос-2-пирролидинметанол (0,300 г, 1,49 ммоль) растворили в ДХМ (8 мл) и охладили до -78°C в атмосфере азота. К реакционной смеси прибавили по каплям N,N-трифторид диэтиламиносеры (0,360 г 2,24 ммоль), реакционную смесь перемешивали при -78°C в атмосфере азота в течение 4 ч и затем нагрели до комн. темп. в течение ночи. Реакционную смесь погасили путем прибавления насыщ. NaHCO3 (водн.) (20 мл) и экстрагировали ДХМ (2×15 мл), органические фазы объединили и сушили, пропуская через картридж фазного сепаратора Biotage, и удалили растворители в вакууме. Остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 10 г 40-63 мкм, 60, 12 мл в мин, градиент от 0% до 4% МеОН / ДХМ]) с получением трет-бутил (2R)-2-(фторметил)пирролидин-1-карбоксилата, промежуточного соединения 295, (0,104 г, 34%), в виде масла янтарного цвета. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 285, метил-(4S)-1,3-тиазолидин-4-карбоксилат гидрохлорида
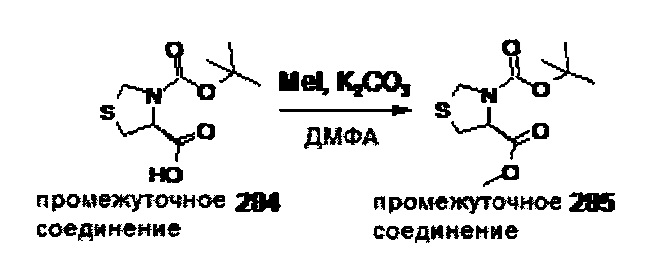
(S)-3-Вос-тиазолидин-4-карбоновую кислоту (1,00 г, 4,29 ммоль) растворили в безводном ДМФА (4 мл), прибавили карбонат калия (2,372 г, 17,16 ммоль) и подметан (0,730 г, 5,14 ммоль). Реакционную смесь перемешивали при комн. темп. в атмосфере азота в течение ночи. Растворители удалили в вакууме и остаток растворили в EtOAc (40 мл) и промыли водой (3×20 мл) и насыщ. NaCl (водн.) (20 мл), сушили (MgSO4). Растворители удалили в вакууме с получением 3-трет-бутил-4-метил-(4S)-1,3-тиазолидин-3,4-дикарбоксилата, промежуточного соединения 285, (0,812 г, 77%) в виде бледно-желтого масла. Данные для указанного в заголовке соединения приведены в Таблице 2.
Метод получения промежуточного соединения 297, трет-бутил (2R)-2-(дифторметил)пирролидин-1-карбоксилата
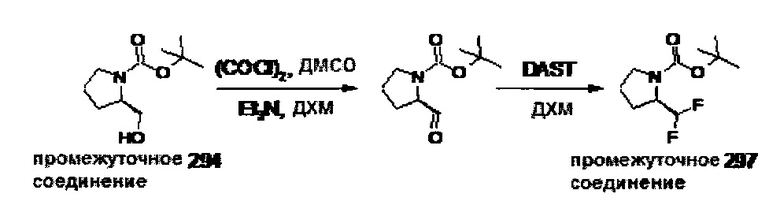
ДМСО (0,698 г, 8,94 ммоль) по каплям прибавляли к раствору оксалилхлорида (0,566 г, 2,93 ммоль) в безводном ДХМ (12 мл) при -78°C в атмосфере азота. Реакционную смесь перемешивали при -78°C в атмосфере азота в течение 15 мин, затем по каплям прибавляли раствор (2R)-(+)-1-Вос-2-пирролидинметанола (0,600 г, 2,98 ммоль) в безводном ДХМ (4 мл). Реакционную смесь перемешивали при -78°C в атмосфере азота в течение 15 мин, затем Et3N (1,06 г, 11,92 ммоль) прибавили и реакционную смесь перемешивали при 0°C в атмосфере азота в течение 1 ч. Реакционную смесь погасили насыщ. NaHCO3 (водн.) (20 мл) и экстрагировали ДХМ (2×20 мл), органические фазы объединили и сушили, пропуская через картридж фазного сепаратора Biotage и удалили растворители в вакууме. Остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 10 г 40-63 мкм, 60, 12 мл в мин, градиент 0% до 4% МеОН / ДХМ]) с получением трет-бутил(2R)-2-формилпирролидин-1-карбоксилата (0,435 г, 73%).
трет-бутил(2R)-2-формилпирролидин-1-карбоксилат (0,435 г, 2,19 ммоль) растворили в безводном ДХМ (8 мл) и охладили до -78°C в атмосфере азота. К смеси прибавили по каплям трифторид N,N-диэтиламиносеры (0,528 г, 3,28 ммоль), реакционную смесь перемешивали при -78°C в атмосфере азота в течение 3 ч и затем нагрели до комн. темп. в течение ночи. Реакционную смесь погасили путем прибавления насыщ. NaHCO3 (водн.) (20 мл) и экстрагировали ДХМ (2×15 мл), органические фазы объединили и сушили, пропуская через картридж фазного сепаратора Biotage, и удалили растворители в вакууме. Остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 10 г 40-63 мкм, 60, 12 мл в мин, градиент от 0% до 4% МеОН / ДХМ]) с получением трет-бутил(2R)-2-(дифторметил)пирролидин-1-карбоксилата, промежуточного соединения 297, (0,217 г, 45%) в виде масла янтарного цвета. Данные для указанного в заголовке соединения приведены в Таблице 2.
Общие методы синтеза промежуточных соединений:
Путь 1
Типовый метод получения пиперидинов реализуется путем реакции Сузуки, гидрирования и Вос-депротекции, как проиллюстрировано на примере получения промежуточного соединения 30, 5-(пиперидин-4-ил)-1,2,4-тиадиазола
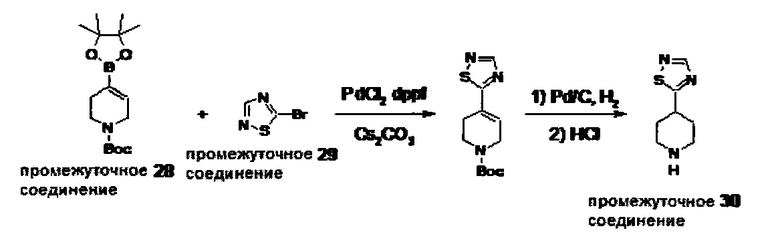
2-5-Бром-1,2,4-тиадиазол (108 мг, 0,65 ммоль), трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2Н)-карбоксилат (200 мг, 0,65 ммоль) и Cs2CO3 (632 мг, 1,94 ммоль) растворили в диоксан: вода (10: 2 мл). Реакционную смесь дегазировали в течение 30 мин с последующим прибавлением PdCl2dppf (24 мг, 0,03 ммоль), затем перемешивали в течение 16 ч при 90°C. Реакционную смесь разделили между H2O (80 мл) и EtOAc (50 мл), водную фазу дополнительно экстрагировали EtOAc (2×50 мл), органические фазы объединили, сушили (Na2SO4), растворители удалили в вакууме и остаток очистили с помощью колоночной хроматографии (нормально-фазовая, силикагель, размер в меш: 60-120, от 16% до 20% EtOAc в гексане) с получением трет-бутил 4-(1,2,4-тиадиазол-5-ил)-3,6-дигидропиридин-1(2Н)-карбоксилата (158 мг, 92,0%) в виде белого твердого вещества с металлическим оттенком.
ЖХ/МС (Способ F): m/z 212 (М+Н-56)+ (ES+), при 2,37 мин, активно в ультрафиолетовом свете
Прибавили трет-бутил-4-(1,2,4-тиадиазол-5-ил)-3,6-дигидропиридин-1(2Н)-карбоксилат (200 мг, 0,74 ммоль) растворили в МеОН (15 мл) и 10% Pd/C (20 мг). Реакционную смесь продули Н2 и перемешивали при 25°C в течение 8 ч под давлением Н2. Реакционную смесь отфильтровали через целит, остаток промыли МеОН и растворители удалили в вакууме, и остаток очистили с помощью колоночной хроматографии (нормально-фазовая, силикагель, размер в меш: 60-120, от 20% до 24% EtOAc в гексане) с получением трет-бутил 4-(1,2,4-тиадиазол-5-ил)пиперидин-1-карбоксилата (150 мг, 74,6%) в виде темно-зеленой смолы.
ЖХ/МС (Способ F): m/z 214 (М+Н)+ (ES+), при 2,14 мин, активно в ультрафиолетовом свете
трет-Бутил 4-(1,2,4-тиадиазол-5-ил)пиперидин-1-карбоксилат (150 мг, 0,56 ммоль) растворили в 1,4-диоксане (5 мл), по каплям прибавляли HCl в диоксане (10 мл, 3,0 М р-р.) и реакционную смесь перемешивали при 30°C в течение 16 ч. Растворители удалили в вакууме и остаток очистили путем растирания с диэтиловым эфиром (3×3 мл) с получением промежуточного соединения 30, 5-(пиперидин-4-ил)-1,2,4-тиадиазола (102 мг, 89,5%) в виде темно-зеленой смолы. Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 2
Метод получения промежуточного соединения 34, 4-(1,5-диметил-1Н-имидазол-2-ил)-1,2,3,6-тетрагидропиридина
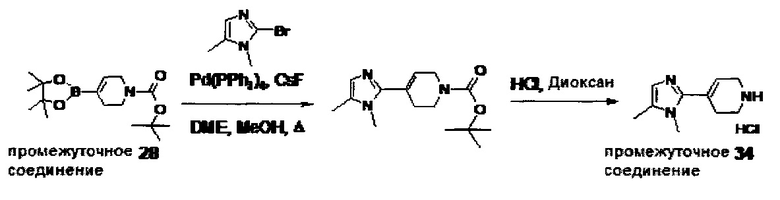
трет-Бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2Н)-карбоксилат (2,0 г, 6,55 ммоль), 2-бром-1,5-диметил-1Н-имидазол (1,13 г, 6,45 ммоль) и CsF (2,9 г, 1,85 ммоль) растворили в DME : МеОН (2:1, 30 мл). Реакционную смесь дегазировали в течение 5 мин, затем прибавили Pd(PPh3)4 (73 мг, 0,064 ммоль) и полученную реакционную смесь перемешивали в течение 5 ч при 100°C. Реакционную смесь разделили между H2O (100 мл) и EtOAc (100 мл), водную фазу дополнительно экстрагировали EtOAc (2×100 мл), органические фазы объединили, сушили (Na2SO4) и растворители удалили в вакууме. Остаток очистили с помощью колоночной хроматографии (нормально-фазовая, силикагель, размер в меш: 60-120, от 13% до 17% этилацетата в гексане) с получением трет-бутил 4-(1,5-диметил-1Н-имидазол-2-ил)-3,6-дигидропиридин-1(2Н)-карбоксилата (1 г, 55%) в виде желтой смолы.
ЖХ/МС (Способ F): m/z 278 (М+Н)+ (ES+), при 1,70 мин, активно в ультрафиолетовом свете
трет-Бутил 4-(1,5-диметил-1Н-имидазол-2-ил)-3,6-дигидропиридин-1(2Н)-карбоксилат (1,0 г, 3,61 ммоль) растворили в 1,4-диоксане (20 мл) с последующим прибавлением по каплям HCl в 1,4-диоксане (20 мл, 3 М р-р). Полученную реакционную смесь перемешивали при 30°C в течение 16 ч, растворители удалили в вакууме и остаток очистили путем растирания с диэтиловым эфиром (3×5 мл) с получением промежуточного соединения 34, 4-(1,5-диметил-1Н-имидазол-2-ил)-1,2,3,6-тетрагидропиридин гидрохлорида (0,5 г, 65%) в виде белого твердого вещества. Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 3
Типовый метод получения пиперидинов реализуется путем реакции Сузуки, гидрирования и Вос-депротекции, как проиллюстрировано на примере получения промежуточного соединения 65,3-(пиперидин-4-ил)пиридин-2(1Н)-он гидрохлорида
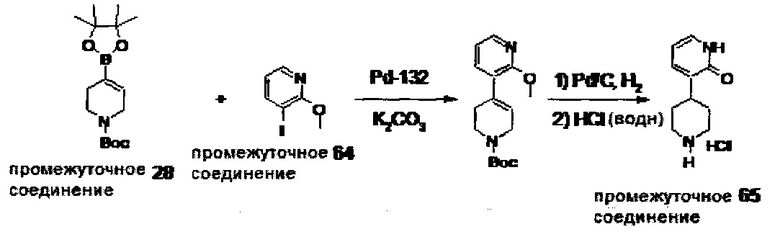
трет-Бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2Н)-карбоксилат (2,5 г, 10,0 ммоль), 3-иод-2-метоксипиридин (8,21 г, 26,0 ммоль) и K2CO3 (4,3 г, 31,8 ммоль) растворили в 1,4-диоксане (10 мл) и воде (5 мл). Реакционную смесь дегазировали, используя N2 в течение 15 мин; прибавили Pd-132 (0,376 г, 0,53 ммоль) и перемешивали реакционную смесь при 80°C в течение 2 ч. Реакционную смесь разбавили водой (50 мл), экстрагировали EtOAc (2×100 мл), органические фазы объединили, сушили (Na2SO4), растворитель удалили в вакууме и сырой продукт очистили с помощью колоночной хроматографии (нормально-фазовая, силикагель 60-120 меш, от 0 до 20% EtOAc в гексане) с получением трет-бутил-2-метокси-3',6'-дигидро-[3,4'-бипиридин]-1'(2'Н)-карбоксилата (2,0 г, 69,0%) в виде белого твердого вещества с металлическим оттенком.
ЖХ/МС (Способ F): m/z 291 (М+Н)+ (ES+), при 2,39 мин, активно в ультрафиолетовом свете
трет-Бутил 2-метокси-3',6'-дигидро-[3,4'-бипиридин]-1'(2'Н)-карбоксилат (1,89 г, 6,51 ммоль) растворили в МеОН (10 мл) и прибавили 10% Pd/C (0,2 г). Реакционную смесь продули Н2 и перемешивали при комн. темп. в течение 12 ч в атмосфере Н2. Реакционную смесь отфильтровали через целит и растворители удалили в вакууме с получением трет-бутил 4-(2-метоксипиридин-3-ил) пиперидин-1-карбоксилата (0,91 г, 47,9%) в виде бесцветной смолы.
ЖХ/МС (Способ F): m/z 293 (М+Н)+ (ES+), при 2,50 мин, активно в ультрафиолетовом свете
трет-Бутил 4-(2-метоксипиридин-3-ил)пиперидин-1-карбоксилат (0,200 г, 0,6 ммоль) растворили в 1,4-диоксане (4,0 мл) и воде (2,0 мл) и прибавили конц. НCl, реакционную смесь перемешивали в течение 10 ч при 100°C. Растворители удалили в вакууме и остаток растерли с ацетоном (3×10 мл) с получением промежуточного соединения 65, 3-(пиперидин-4-ил)пиридин-2(1H)-он гидрохлорида (0,100 г, 82,6%) в виде коричневого твердого вещества. Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 4
Типовым метод получения пиперидинов реализуется путем реакции Сузуки, гидрирования и Вос-депротекции, как проиллюстрировано на примере получения промежуточного соединения 66, 2-метокси-3-(пиперидин-4-ил)пиридин гидрохлорида
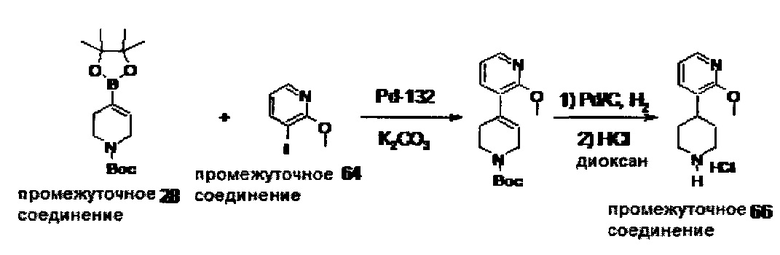
трет-Бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2Н)-карбоксилат (2,5 г, 10,0 ммоль), 3-иод-2-метоксипиридин (8,21 г, 26,0 ммоль) и K2CO3 (4,3 г, 31,8 ммоль) растворили в 1,4-диоксане (10 мл) и воде (5 мл). Реакционную смесь дегазировали, используя N2 в течение 15 мин; прибавили Pd-132 (0,376 г, 0,53 ммоль) и перемешивали реакционную смесь при 80°C в течение 2 ч. Реакционную смесь разбавили водой (50 мл), экстрагировали EtOAc (2×100 мл), органические фазы объединили, сушили (Na2SO4), растворитель удалили в вакууме и сырой продукт очистили с помощью колоночной хроматографии (нормально-фазовая, силикагель 60-120 меш, от 0 до 20% EtOAc в гексане) с получением трет-бутил-2-метокси-3',6'-дигидро-[3,4'-бипиридин]-1'(2'Н)-карбоксилата (2,0 г, 69,0%) в виде белого твердого вещества с металлическим оттенком.
ЖХ/МС (Способ F): m/z 291 (М+Н)+ (ES+), при 2,39 мин, активно в ультрафиолетовом свете
трет-Бутил 2-метокси-3',6'-дигидро-[3,4,-бипиридин]-1'(2'Н)-карбоксилат (1,89 г, 6,51 ммоль) растворили в МеОН (10 мл) и прибавили 10% Pd/C (0,2 г). Реакционную смесь продули Н2 и перемешивали при комн. темп. в течение 12 ч в атмосфере Н2. Реакционную смесь отфильтровали через целит и растворители удалили в вакууме с получением трет-бутил 4-(2-метоксипиридин-3-ил) пиперидин-1-карбоксилата (0,91 г, 47,9%) в виде бесцветной смолы.
ЖХ/МС (Способ F): m/z 293 (М+Н)+ (ES+), при 2,50 мин, активно в ультрафиолетовом свете
трет-Бутил 4-(2-метоксипиридин-3-ил)пиперидин-1-карбоксилат (0,8 г, 2,7 ммоль) перемешивали в HCl в 1,4-диоксане (4,0 мл, 4,0 М р-р.) в течение 10 ч при комн. темп. Растворители удалили в вакууме и остаток растерли в ацетоне (3×10 мл) с получением промежуточного соединения 66, 2-метокси-3-(пиперидин-4-ил)пиридин гидрохлорида (0,135 г, 25,7%) в виде белого твердого вещества. Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 5
Типовый метод получения пиперидинов реализуется путем гидрирования, как проиллюстрировано на примере получения промежуточного соединения 69, 3,4'-бипиперидин-2-она
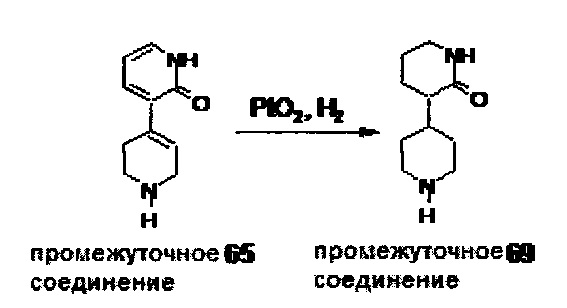
3-(Пиперидин-4-ил)-1,6-дигидропиридин-2-ол (0,5 г, 2,8 ммоль) растворили в МеОН (10 мл) и прибавили PtO2 (0,2 г). Реакционную смесь продули Н2 и перемешивали при комн. темп. в течение 12 ч в атмосфере Н2. Реакционную смесь отфильтровали через целит и растворители удалили в вакууме с получением промежуточного соединения 69, 3,4'-бипиперидин-2-она (0,4 г, 78,3%) в виде коричневой смолы. Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 6
Типовым метод получения пирролидинов реализуется путем восстановительного аминирования и Вос-депротекции, как проиллюстрировано на примере получения промежуточного соединения 127, смеси диастереомеров этил-2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
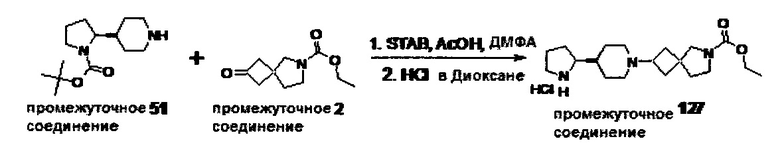
(S)-трет-Бутил 2-(пиперидин-4-ил)пирролидин-1-карбоксилат (1,24 г, 6,29 ммоль) и этил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (1,60 г, 6,29 ммоль) растворили в ДМФА (15 мл) при комн. темп. и прибавили уксусную кислоту (0,54 мл, 9,44 ммоль). Реакционную смесь перемешивали при комн. темп. в течение 3 ч. Затем прибавили STAB (2,67 г, 12,6 ммоль) и реакционную смесь перемешивали в атмосфере азота при комн. темп. в течение ночи. Растворители удалили в вакууме, и остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 340 г, 40-63 мкм, 60, 80 мл в мин, градиент от 0% до 10% 7N NH3 в МеОН в ДХМ]) с получением неразделимой смеси изомеров этил-2-{4-[(2S)-1-(трет-бутоксикарбонил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (2,46 г, 90%) в виде желтого твердого вещества.
ЖХ/МС (Способ D): m/z 436 (М+Н)+ (ES+), при 2,36 мин, не активно в ультрафиолетовом свете.
Смесь диастереомеров этил-2-{4-[(2S)-1-(трет-бутоксикарбонил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (0,6 г, 1,4 ммоль) растворили в 1,4-диоксане (10 мл) и обработали, прикапывая HCl в 1,4-диоксане (4 М, 15 мл, 60 ммоль). Полученную реакционную смесь перемешивали при 25°C в течение 16 ч, растворители удалили и остаток очистили путем растирания с диэтиловым эфиром (3×10 мл) с получением смеси диастереомеров этил-2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, промежуточного соединения 127 в виде твердого вещества (0,45 г, 97%). Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 7
Типовый метод получения пиперидинов реализуется путем восстановительного аминирования, Вос-депротекции, образования мочевины и гидрирования, как проиллюстрировано на примере получения промежуточного соединения 137, 1-(пиперидин-4-ил)тетрагидропиримидин-2(1Н)-она
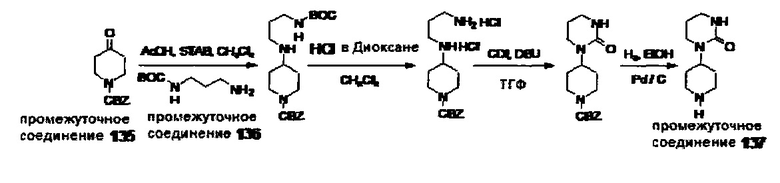 Бензил-4-оксопиперидин-1-карбоксилат (0,932 г, 4,00 ммоль) и трет-бутил-(3-аминопропил)карбамат (0,766 г, 4,4 ммоль) смешали в CH2Cl2 (20 мл) при комн. темп., прибавили АсОН (0,68 мл, 12,0 ммоль) и перемешивали в течение 3 ч. Прибавили STAB (2,59 г, 12,0 ммоль) и реакционную смесь перемешивали в атмосфере азота при комн. темп. в течение ночи. Реакционную смесь погасили путем прибавления NaHCO3 (насыщ. водн.) (40 мл), экстрагировали CH2Cl2 (4×45 мл) и объединенные органические фазы промыли насыщенным водным раствором хлорида натрия, затем сушили над MgSO4 и фильтровали. Растворители удалили в вакууме и остаток очистили с помощью колоночной хроматографии [картридж Biotage SNAP KP-sil 25 г, 40-63 мкм, 60, 50 мл в мин, градиент от 0% до 10% МеОН в ДХМ]) с получением бензил-4-({3-[(трет-бутоксикарбонил)амино]пропил}амино)пиперидин-1-карбоксилата (1,54 г, 98%) в виде бесцветного масла.
Бензил-4-оксопиперидин-1-карбоксилат (0,932 г, 4,00 ммоль) и трет-бутил-(3-аминопропил)карбамат (0,766 г, 4,4 ммоль) смешали в CH2Cl2 (20 мл) при комн. темп., прибавили АсОН (0,68 мл, 12,0 ммоль) и перемешивали в течение 3 ч. Прибавили STAB (2,59 г, 12,0 ммоль) и реакционную смесь перемешивали в атмосфере азота при комн. темп. в течение ночи. Реакционную смесь погасили путем прибавления NaHCO3 (насыщ. водн.) (40 мл), экстрагировали CH2Cl2 (4×45 мл) и объединенные органические фазы промыли насыщенным водным раствором хлорида натрия, затем сушили над MgSO4 и фильтровали. Растворители удалили в вакууме и остаток очистили с помощью колоночной хроматографии [картридж Biotage SNAP KP-sil 25 г, 40-63 мкм, 60, 50 мл в мин, градиент от 0% до 10% МеОН в ДХМ]) с получением бензил-4-({3-[(трет-бутоксикарбонил)амино]пропил}амино)пиперидин-1-карбоксилата (1,54 г, 98%) в виде бесцветного масла.
ЖХ/МС (Способ В): m/z 392 (М+Н)+ (ES+), при 1,73 мин, активно в ультрафиолетовом свете.
Бензил-4-({3-[(трет-бутоксикарбонил)амино]пропил}амино)пиперидин-1-карбоксилат (1,54 г, 3,92 ммоль) растворили в CH2Cl2 (19,5 мл), прибавили 4 М хлороводород в диоксане (4,90 мл, 19,6 ммоль) и перемешивали реакционную смесь при комн. темп. в течение ночи. Растворители удалили в вакууме, остаток промыли CH2Cl2 (2×20 мл) и сушили с получением сырого бензил-4-[(3-аминопропил)амино]пиперидин-1-карбоксилат дигидрохлорида (1,41 г, 99%) в виде белого твердого вещества с металлическим оттенком.
ЖХ/МС (Способ В): m/z 292 (М+Н)+ (ES+), при 1,46 мин, активно в ультрафиолетовом свете.
Сырой бензил-4-[(3-аминопропил)амино]пиперидин-1-карбоксилат дигидрохлорид (1,41 г, 3,88 ммоль), CDI (0,778 г, 4,80 ммоль) и пиридин (0,24 мл, 12,0 ммоль) растворили в ТГФ (39 мл), смесь нагрели до кипения и выдержали в течение 18 ч. Растворители удалили в вакууме и остаток очистили с помощью колоночной хроматографии [картридж Biotage SNAP KP-sil 50 г, 40-63 мкм, 60, 50 мл в мин, градиент от 0% до 10% МеОН в ДХМ]) с получением бензил-4-(2-оксотетрагидропиримидин-1(2Н)-ил)пиперидин-1-карбоксилата (0,82 г, 65%) в виде бесцветного твердого вещества.
ЖХ/МС (Способ В): m/z 318 (М+Н)+ (ES+), при 2,62 мин, активно в ультрафиолетовом свете.
Бензил-4-(2-оксотетрагидропиримидин-1(2Н)-ил)пиперидин-1-карбоксилат (0,82 г, 2,59 ммоль) растворили в EtOH (100 мл) и пропустили через 10% Pd/С картридж, используя реактор H-Cube при 50°C, 40 Бар Н2 при 1 мл/мин. Элюированный раствор концентрировали в вакууме с получением промежуточного соединения 137, 1-(пиперидин-4-ил)тетрагидропиримидин-2(1Н)-она (0,470 г, 99%) в виде бесцветного твердого вещества. Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 8
Типовый метод получения пиперидинов реализуется путем восстановительного аминирования и Вос-депротекции, как проиллюстрировано на примере получения промежуточного соединения 139, (2S)-N-метил-1-(пиперидин-4-ил)пирролидин-2-карбоксамида
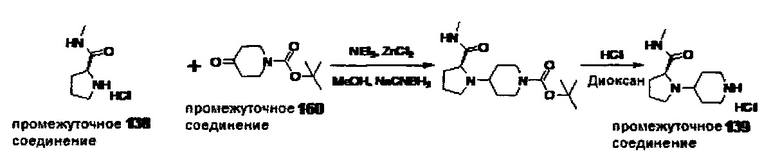
(S)-N-метилпирролидин-2-карбоксамид (0,5 г, 3,8 ммоль), NEt3 (1,5 мл, 11,0 ммоль), трет-бутил-4-оксопиперидин-1-карбоксилат (0,38 г, 3,9 ммоль) и ZnCl2 (0,15 г, 4,5 ммоль) растворили в МеОН (15 мл) в атмосфере азота и перемешивали в течение 1 ч при 50-60°C. Прибавили по порциям NaCNBH3 (0,16 г, 0,67 ммоль) при 0-10°C и перемешивали смесь в течение 3 ч при комнатной температуре. Реакционную смесь разделили между EtOAc (2×100 мл) и водой (50 мл), органические фазы объединили, сушили (Na2SO4), фильтровали, растворитель удалили в вакууме и сырой продукт очистили с помощью колоночной хроматографии (нормально-фазовая, силикагель, 0 до 20% EtOAc в гексане) с получением трет-бутил-(S)-4-(2-(метилкарбамоил) пирролидин-1-ил) пиперидин-1-карбоксилата (0,3 г, 25,0%) в виде светло-коричневого твердого вещества.
ТСХ наблюдения: Значение RF: 0,5 (ЭА: Геке, 5:5).
ЖХ/МС (Способ G): m/z 312 (М+Н)+ (ES+), при 1,61 мин, не активно в ультрафиолетовом свете.
трет-Бутил (S)-4-(2-(метилкарбамоил) пирролидин-1-ил) пиперидин-1-карбоксилат (0,3 г, 0,96 ммоль) перемешивали в растворе HCl в 1,4-диоксане (5,00 мл) в течение 10 ч при комнатной температуре. Реакционную смесь концентрировали под высоким давлением и затерли в ацетоне (3×10 мл) с получением промежуточного соединения 139, (S)-N-метил-1-(пиперидин-4-ил)пирролидин-2-карбоксамид дигидрохлорида (0,135 г, 67,16%) в виде бесцветного твердого вещества. Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 9
Общий метод получения пиперидинов, содержащих N-замещенный циклический амин в 4-положении, реализуется путем восстановительного алкилирования и депротекции, как проиллюстрировано на примере получения промежуточного соединения 181, 4-[2-(1Н-пиразол-5-ил)пирролидин-1-ил]пиперидин трифторацетатной соли
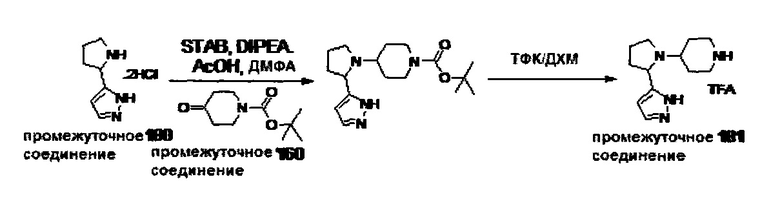
5-(Пирролидин-2-ил)-1Н-пиразол-1-ил дигидрохлорид (0,105 г, 0,50 ммоль) растворили в ДМФА (5 мл). К раствору в указанном порядке прибавили DIPEA (0,435 мл, 2,5 ммоль), АсОН (0,043 мл, 0,75 ммоль), трет-бутил-4-оксопиперидин-1-карбоксилат (0,100 г, 0,50 ммоль) и STAB (0,318 г, 1,50 ммоль). Смесь перемешивали при комн. темп. в течение 2 дней, затем концентрировали для удаления ДМФА. Остаток разделили между насыщ. водным NaHCO3 и ДХМ (х2) и органическую фазу пропустили через фазный сепаратор и концентрировали с получением сырого трет-бутил-4-[2-(1Н-пиразол-5-ил)пирролидин-1-ил]пиперидин-1-карбоксилата (0,271 г, >100%) в виде масла.
ЖХ/МС (Способ С): m/z 321 (М+Н)+ (ES+), при 1,18 мин, активно в ультрафиолетовом свете
Раствор сырого трет-бутил-4-[2-(1Н-пиразол-5-ил)пирролидин-1-ил]пиперидин-1-карбоксилата (0,271 г, предположительно 0,50 ммоль) в ДХМ (3 мл) и ТФК (3 мл) перемешивали при комн. темп. в течение 110 мин, затем разбавили толуолом и концентрировали. Остаток подвергли азеотропной перегонке с толуолом с получением сырого промежуточного соединения 181, 4-[2-(1Н-пиразол-5-ил)пирролидин-1-ил]пиперидин трифторацетатной соли (0,598 г, >100%) в виде масла. Немедленно использовали. Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 10
Общий метод получения пирролидинона или оксадиазолона, содержащих пиперидины, реализуется путем катализируемого медью кросс-сочетания с пиридином с последующим гидрированием, как проиллюстрировано на примере получения промежуточного соединения 184, 5-метил-1-(пиперидин-4-ил)пирролидин-2-он ацетатной соли
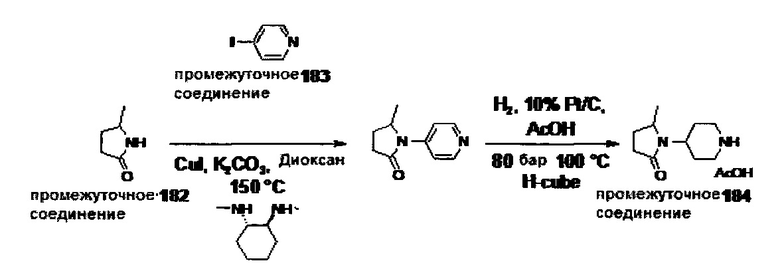
Смесь 5-метилпирролидин-2-она (0,050 г, 0,50 ммоль), 4-иодпиридина (0,103 г, 0,50 ммоль), (транс)-N,N'-диметилциклогексан-1,2-диамина (0,016 мл, 0,10 ммоль), CuI (0,019 г, 0,10 ммоль) и K2CO3 (0,209 г, 1,5 ммоль) в диоксане (2 мл) герметично закрыли в продутой азотом стеклянной пробирке и нагревали при перемешивании при 150°C в течение ночи. Охлажденную реакционную смесь концентрировали на силикагеле для флеш-хроматографии (5 мл). Полученный порошок очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 25 г, 40-63 мкм, 60], 30 мл в мин, от 0 до 5% Растворителя А в ДХМ, где Растворитель А представляет собой 10% (7 М NHa/MeOH) в МеОН) с получением 5-метил-1-(пиридин-4-ил)пирролидин-2-она (0,088 г, 99%) в виде масла.
ЖХ/МС (Способ С): m/z 177 (М+Н)+ (ES+), при 0,69 мин, активно в ультрафиолетовом свете
5-Метил-1-(пиридин-4-ил)пирролидин-2-он (0,080 г, 0,45 ммоль) растворили в АсОН (8 мл) и гидрировали на 10% Pt/C катализаторе под давлением 80 бар и 100°C при скорости потока 1 мл/мин, используя H-Cube. Затем раствор концентрировали и остаток подвергли азеотропной перегонке толуолом (х2) для получения сырого промежуточного соединения 184, 5-метил-1-(пиперидин-4-ил)пирролидин-2-он ацетатной соли (0,166 г, >100%) в виде масла. Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 11
Типовый метод получения пиперидинов реализуется путем катализируемого медью кросс-сочетания с пиридином с последующим гидрированием, как проиллюстрировано на примере получения промежуточного соединения 199, (5R)-5-метил-1-(пиперидин-4-ил)пирролидин-2-он ацетатной соли и промежуточного соединения 200, (5R)-5-этил-1-(пиперидин-4-ил)пирролидин-2-он ацетатной соли
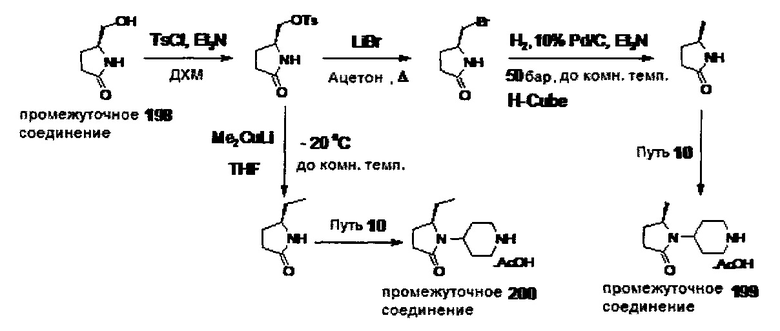
промежуточного соединения 199, (5R)-5-метил-1-(пиперидин-4-ил)пирролидин-2-он ацетатной соли:
К раствору (5S)-5-(гидроксиметил)пирролидин-2-он (2,0 г, 17 ммоль) и 4-метилбензолсульфонилхлорида (5,3 г, 28 ммоль) в ДХМ (24 мл) прибавили триэтиламин (12 мл, 86 ммоль). Полученную смесь перемешивали при комн. темп. в течение ночи, затем концентрировали. Остаток растворили в ДХМ и промыли 1 М водным HCl (х3) и насыщенным водным раствором хлорида натрия (х1), затем пропустили через фазный сепаратор и концентрировали с получением коричневого твердого вещества. Твердое вещество перекристаллизовали из ДХМ/изогексан с получением желто-коричневого твердого вещества, которое изолировали с помощью фильтрации, промыли смесью ДХМ/изогексан и сушили на воздухе с получением [(2S)-5-оксопирролидин-2-ил]метил 4-метилбензолсульфоната (3,13 г, 67%).
ЖХ/МС (Способ С): m/z 270 (М+Н)+ (ES+), при 0,97 мин, активно в ультрафиолетовом свете
Смесь [(2S)-5-оксопирролидин-2-ил]метил-4-метилбензолсульфоната (0,50 г, 1,9 ммоль) и бромида лития (0,484 г, 5,6 ммоль) в ацетоне (5 мл) нагревали до кипения под N2 в течение ночи, затем оставили охлаждаться. Растворитель удалили с помощью концентрирования, остаток разделили между ДХМ и H2O и фазы разделили. Водную фазу экстрагировали ДХМ (х3), затем органические фазы пропустили через фазный сепаратор и концентрировали с получением (5S)-5-(бромметил)пирролидин-2-она (0,284 г, 86%) в виде смолы.
ЖХ/МС (Способ С): m/z 178/180 (М+Н)+ (ES+), при 0,37 мин, малоактивно в ультрафиолетовом свете
Раствор (5S)-5-(бромметил)пирролидин-2-он (0,284 г, 1,6 ммоль) в триэтиламине (0,267 мл, 1,9 ммоль) и этаноле (32 мл) гидрировали на 10% Pd/C катализаторе под давлением 50 бар и при комн. темп, при скорости потока 1 мл/мин, используя H-Cube. Раствор концентрировали с получением сырого (5R)-5-метилпирролидин-2-она (0,445 г, >100%) в виде липкого твердого вещества.
ЖХ/МС (Способ С): m/z 100 (М+Н)+ (ES+), при 0,34 мин, малоактивно в ультрафиолетовом свете
Сырой (5R)-5-метилпирролидин-2-он (0,445 г, предположительно 1,5 ммоль) вводили в реакцию согласно Пути 10 (кросс-сочетание с промежуточным соединением 183) с получением сырого промежуточного соединения 199, (5R)-5-метил-1-(пиперидин-4-ил)пирролидин-2-он ацетатной соли (0,125 г, 46%) в виде масла. Данные для указанного в заголовке соединения приведены в Таблице 2.
Промежуточное соединение 200, (5R)-5-этил-1-(пиперидин-4-ил)пирролидин-2-он ацетатной соли:
При перемешивании быстро прибавили метиллитий (1,5 М в эфире, 7,4 мл, 11 ммоль) к суспензии иодида меди (1,06 г, 5,6 ммоль) в ТГФ (6 мл), предварительно охлажденной в ледяной воде под N2. Бледно-коричневый раствор перемешивали в ледяной воде в течение 45 мин, затем охладили до -20°C. Раствор [(2S)-5-оксопирролидин-2-ил]метил 4-метилбензолсульфоната (0,50 г, 1,9 ммоль) в ТГФ (6 мл) порциями прибавляли в течение 2 мин и перемешивали полученный раствор при -20°C в течение 45 мин, затем в ледяной воде в течение ночи, оставив охлаждающую баню медленно нагреваться до комнатной температуры. Смесь погасили насыщенным водным NH4Cl (15 мл) и перемешивали в течение нескольких часов. Двухфазную смесь экстрагировали эфиром (х3), органические фазы промыли насыщенным водным раствором хлорида натрия, пропустили через фазный сепаратор и концентрировали с получением сырого (5R)-5-этилпирролидин-2-она (0,124 г, 59%) в виде масла.
ЖХ/МС (Способ С): m/z 114 (М+Н)+ (ES+), при 0,50 мин, малоактивно в ультрафиолетовом свете
Сырой (5R)-5-этилпирролидин-2-он (0,124 г, 1,10 ммоль) вводили в реакцию согласно Пути 10 (кросс-сочетание с промежуточным соединением 183) с получением сырого промежуточного соединения 200, (5R)-5-этил-1-(пиперидин-4-ил)пирролидин-2-он ацетатной соли (0,156 г, 72%) в виде смолы. Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 12
Типовый метод получения пиперидинов реализуется путем образования карбамата, катализируемым медью кросс-сочетанием с пиридином с последующим гидрированием, как проиллюстрировано на примере получения промежуточного соединения 205, (4R)-4-метил-3-(пиперидин-4-ил)-1,3-оксазолидин-2-он ацетатной соли
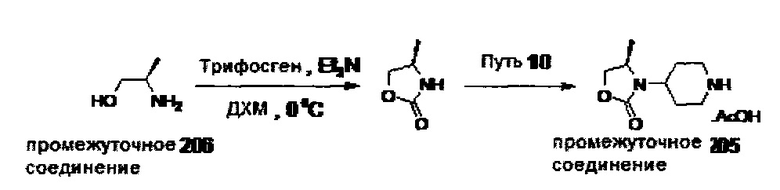
Раствор трифосгена (0,297 г, 1,0 ммоль) в ДХМ (5 мл) порциями прибавляли в течение 1 ч к раствору (2R)-2-аминопропан-1-ола (0,156 мл, 2,0 ммоль) и триэтиламина (0,56 мл, 4,0 ммоль) в ДХМ (5 мл), предварительно охлажденному в ледяной воде. Смесь перемешивали в ледяной воде в течение дополнительных 2 ч, затем прибавили эфир (6 мл). Густую суспензию отфильтровали через стеклянный фильтр, еще промыли твердое вещество эфиром (6 мл). Фильтрат концентрировали на силикагеле для флеш-хроматографии (5 мл) и полученный порошок очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 25 г, 40-63 мкм, 60], 30 мл в мин, 100% EtOAc) с получением
(4R)-4-метил-1,3-оксазолидин-2-она (192 мг, 95%) в виде твердого вещества.
ЖХ/МС (Способ С): m/z 102 (М+Н)+ (ES+), при 0,14 мин, не активно в ультрафиолетовом свете
(4R)-4-Метил-1,3-оксазолидин-2-он (0,188 г, 1,9 ммоль) вводили в реакцию согласно Пути 10 (кросс-сочетание с промежуточным соединением 183) с получением сырого промежуточного соединения 205, (4R)-4-метил-3-(пиперидин-4-ил)-1,3-оксазолидин-2-он ацетатной соли (0,343 г, 100%) в виде твердого вещества. Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 13
Типовый метод получения пиперидинов реализуется путем восстановительных аминирований, как проиллюстрировано на примере получения промежуточного соединения 159, трет-бутил-4-[(2R)-2-(метоксикарбонил)пирролидин-1-ил]пиперидин-1-карбоксилата 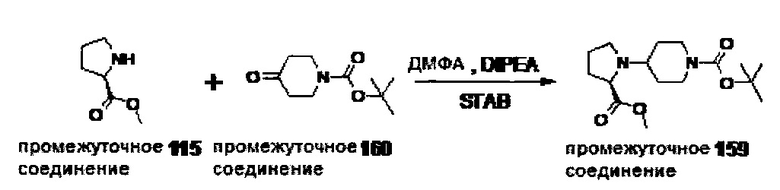
Гидрохлорид D-пролинметилового эфира (0,200 г, 1,208 ммоль) и 1-Вос-4-пиперидинон (0,24 г, 1,208 ммоль) растворили в ДМФА (2 мл) при комн. темп. и прибавили диизопропилэтиламин (0,209 мл, 1,208 ммоль). Реакционную смесь перемешивали при комн. темп. в течение 3 ч. Затем прибавили STAB (0,512 г, 2,416 ммоль) и перемешивали реакционную смесь в течение ночи в атмосфере азота при комн. темп. Растворители удалили в вакууме и остаток разделили между H2O (15 мл) и EtOAc (25 мл), водную фазу экстрагировали EtOAc (2×25 мл), органические фазы объединили, сушили над Na2SO4 и растворитель удалили в вакууме с получением трет-бутил-4-[(2R)-2-(метоксикарбонил)пирролидин-1-ил]пиперидин-1-карбоксилата, промежуточного соединения 159, в виде белого твердого вещества (393 мг, >99%). Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 14
Типовым метод получения пиперидинов реализуется путем восстановительных аминирований, как проиллюстрировано на примере получения промежуточного соединения 271, трет-бутил-3,3-дифтор-1,4'-бипиперидин-1'-карбоксилата
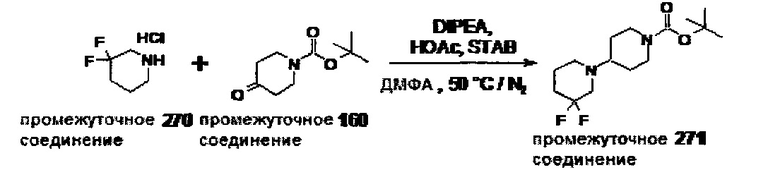
3,3-дифторпиперидин. HCl (0,30 г, 1,90 ммоль) и 1-Вос-4-пиперидинон (0,379 г, 1,90 ммоль) растворили в ДМФА (8 мл) при комн. темп. и прибавили диизопропилэтиламин (0,246 г, 1,90 ммоль). Реакционную смесь перемешивали при 50°C в атмосфере азота в течение 2 ч. Реакционную смесь охладили до комн. темп., затем прибавили ледяную уксусную кислоту (0,114 г, 1,90 ммоль) и STAB (1,01 г, 4,76 ммоль) и перемешивали реакционную смесь в течение ночи при 50°C в атмосфере азота. К охлажденной реакционной смеси прибавили воду (2 мл) и удалили растворители в вакууме. Остаток разбавили насыщ. NaHCO3 (водн.) (10 мл) и экстрагировали ДХМ (2×10 мл). Объединенные органические фазы пропустили через картридж фазного сепаратора Biotage для осушения и удалили растворители в вакууме. Остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 25 г 40-63 мкм, 60, 25 мл в мин, градиент от 0% до 10% МеОН / ДХМ]) с получением трет-бутил 3,3-дифтор-1,4'-бипиперидин-1'-карбоксилата, промежуточного соединения 271, (0,347 г, 60%) в виде масла янтарного цвета. Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 15
Типовым метод получения пиперидинов реализуется путем образования тетразола с последующим алкилированием, как проиллюстрировано на примере получения промежуточного соединения 195, 4-(1-метил-1Н-тетразол-5-ил)пиперидин гидрохлоридной соли
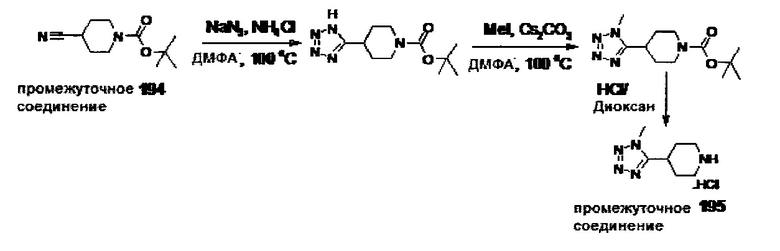
трет-Бутил 4-цианопиперидин-1-карбоксилат (2,1 г, 10 ммоль), азид натрия (1,95 г, 30 ммоль) и хлорид аммония (1,6 г, 30 ммоль) растворили в ДМФА (20 мл). Реакционную смесь перемешивали при 100°C в течение 24 ч, затем разбавили водой (250 мл) и экстрагировали EtOAc (3×100 мл). Объединенные органические фазы сушили (Na2SO4) и концентрировали с получением сырого продукта, который очистили с помощью колоночной хроматографии (нормально-фазовая, нейтральный силикагель, 60-120 меш, от 0 до 5% МеоН в ДХМ) с получением трет-бутил 4-(1Н-тетразол-5-ил)пиперидин-1-карбоксилата (1,25 г, 50%) в виде твердого вещества.
ЖХ/МС (Способ F): m/z 198 (M-tBu+H)+ (ES+), при 1,69 мин, не активно в ультрафиолетовом свете
трет-Бутил 4-(1Н-тетразол-5-ил)пиперидин-1-карбоксилат (1,2 г, 4,7 ммоль), иодметан (2,0 г, 14 ммоль) и Cs2CO3 (9,6 г, 28 ммоль) растворили в сухом ДМФА (36 мл). Реакционную смесь перемешивали при 100°C в течение 2 ч, затем разбавили водой (250 мл) и экстрагировали EtOAc (3×100 мл). Объединенные органические фазы сушили (Na2SO4) и концентрировали с получением сырого продукта, который очистили с помощью колоночной хроматографии (нормально-фазовая, нейтральный силикагель, 60-120 меш, от 0 до 35% EtOAc в гексане и затем от 45 до 60% EtOAc в гексане для разделения двух региоизомеров. Требуемый региоизомер, трет-бутил 4-(1-метил-1Н-тетразол-5-ил)пиперидин-1-карбоксилат (0,160 г, 13%) элюировали вторым из колонки и получили в виде твердого вещества.
ЖХ/МС (Способ F): m/z 212 (M-tBu+H)+ (ES+), при 1,79 мин, не активно в ультрафиолетовом свете
трет-Бутил 4-(1-метил-1Н-тетразол-5-ил)пиперидин-1-карбоксилат (0,160 г, 0,60 ммоль) растворили в диоксане (3 мл). Прибавили HCl в диоксане (4 М, 3 мл, 12 ммоль) при 0°C и перемешивали смесь при комнатной температуре в течение 5 ч. Растворитель удалили и смесь растерли с диэтиловым эфиром (5 мл) с получением 4-(1-метил-1Н-тетразол-5-ил)пиперидин гидрохлоридной соли, промежуточного соединения 195, (0,130 г, >100%) в виде твердого вещества. Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 16
Типовый метод получения пиперидинов реализуется путем восстановительного аминирования, образования амида и Вос-депротекции, как проиллюстрировано на примере получения промежуточного соединения 223, (2R)-N-метил-1,4'-бипиперидин-2-карбоксамида
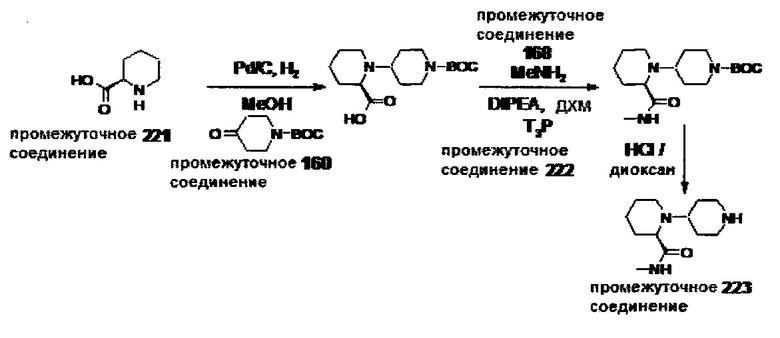
К раствору R-пипеколиновой кислоты (1 г, 7,75 ммоль) и трет-бутил-4-оксопиперидин-1-карбоксилата (2,31 г, 11,6 ммоль) в МеОН (40 мл) прибавили 10% Pd на угле (1 г, 50% влажный) и перемешивали реакционную смесь при комнатной температуре под Н2 (1 атм) в течение 48 ч. Реакционную смесь отфильтровали через слой целита и фильтрат выпарили в вакууме. Сырой остаток затерли в ДХМ (50 мл) с получением (R)-1'-(трет-бутоксикарбонил)-[1,4'-бипиперидин]-2-карбоновой кислоты (1,2 г, 50%) в виде белого твердого вещества. Данный сырой остаток использовали для следующей стадии без дополнительной очистки.
1Н-ЯМР (400 МГц; CDCl3) δ: 1,46 (с, 9Н), 1,50-1,59 (м, 1Н), 1,75-1,91 (м, 4Н), 1,93-2,05 (м, 2Н), 2,10-2,19 (м, 2Н), 2,35-2,41 (м, 1Н), 2,51-2,69 (м, 3Н), 3,41-3,49 (м, 1Н), 3,55-3,61 (м, 1Н), 3,70-3,79 (м, 1Н), 4,25-4,36 (м, 2Н).
К раствору (R)-1'-(трет-бутоксикарбонил)-[1,4'-бипиперидин]-2-карбоновой кислоты (1,0 г, 3,20 ммоль) и MeNH2 (2 M в ТГФ, 3,2 мл, 6,41 ммоль) в ДХМ (20 мл), прибавили DIPEA (1,75 мл, 9,60 ммоль) при 0°C. После перемешивания в течение 10 мин прибавляли 1-пропанфосфоновый ангидрид [50% раствор в этилацетате (4,07 мл, 6,41 ммоль)] и перемешивали при комнатной температуре в течение 3 ч. По окончании реакционую смесь погасили насыщенным водн. NaHCO3 и экстрагировали ДХМ (3×30 мл). Органические фазы объединили и промыли насыщенным водным раствором хлорида натрия, сушили (Na2SO4) и концентрировали в вакууме с получением трет-бутил (R)-2-(метилкарбамоил)-[1,4'-бипиперидин]-1'-карбоксилата (4,1 г, 97%) в виде бесцветной смолообразной жидкости. Данный сырой остаток использовали для следующей стадии без дополнительной очистки.
1Н-ЯМР (400 МГц, ДМСО) δ: 1,46 (с, 9Н), 1,61-1,80 (м, 4Н), 1,91-2,08 (м, 4Н), 2,25-2,33 (м, 2Н), 2,61-2,71 (м, 4Н), 2,82 (д, J=4,8 Гц, 3Н), 3,32-3,45 (м, 2Н), 4,25-4,36 (м, 2Н), 6,85 (уш. с., 1Н).
К раствору трет-бутил (R)-2-(метилкарбамоил)-[1,4'-бипиперидин]-1'-карбоксилата (700 мг, 2,15 ммоль) в диоксане (10 мл) медленно прибавили HCl в диоксане (4 М, 10 мл) при 0°C и перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь концентрировали в вакууме. Полученное подщелочили водн. насыщ. NaHCO3 (10 мл) и концентрировали. К сырой реакционной массе прибавили 5% МеОН/ ДХМ (30 мл), перемешивали в течение 10 мин и фильтровали. Фильтрат концентрировали в вакууме с получением промежуточного соединения 223, (R)-N-метил-[1,4'-бипиперидин]-2-карбоксамида (400 мг, 83%) в виде коричневой смолообразной жидкости. Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 17
Типовым метод получения иодпиразолов реализуется путем реакции Зандмейера, как проиллюстрировано на примере получения промежуточного соединения 250, 4-этил-5-иод-1-метил-1Н-пиразола

1-Этил-4-метил-1Н пиразоламин (0,5 г, 3,932 ммоль) растворили в дииодметане (9,0 мл) при 0-5°C в атмосфере азота с последующим прибавлением по каплям изоамилнитрита и перемешивали смесь в течение 2 ч при 80°C, затем 2 ч при комнатной температуре. Реакционную смесь разделили между Н2О (100 мл) и EtOAc (250 мл), водную фазу дополнительно экстрагировали EtOAc (2×250 мл), объединенные органические фазы сушили (Na2SO4), фильтровали и удалили растворитель в вакууме. Остаток очистили с помощью колоночной хроматографии (нормально-фазовая, нейтральный силикагель, 60-120 меш, от 30 до 50% этилацетата в гексане) с получением 4-этил-5-иод-1-метил-1H-пиразола, промежуточного соединения 250 (0,5 г, 53,23%) в виде светло-желтой смолы. Данные для указанного в заголовке соединения приведены в Таблице 2.
Путь 18
Типовый метод получения активированных карбаматов реализуется путем депротекции, образования карбамата с последующим восстановительным аминированием, как проиллюстрировано на примере получения промежуточного соединения 302, трет-бутил-(2R)-2-(дифторметил)пирролидин-1-карбоксилата
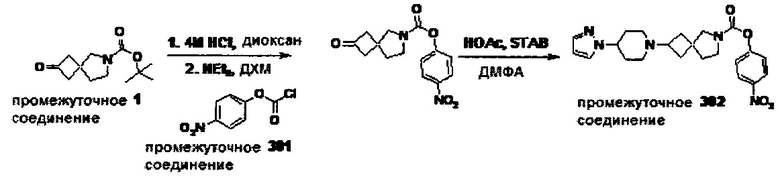
6-Вос-2-оксо-6-аза-спиро[3.4]октан (4,00 г, 0,017 моль) растворили в 4 М HCl в диоксане (25 мл) и перемешивали при комн. темп. в атмосфере азота в течение ночи. Растворители удалили в вакууме с получением белого твердого вещества с металлическим оттенком, которое суспендировали в ДХМ (40 мл), реакционную смесь охладили до 0°C в атмосфере азота. Прибавили Et3N (3,60 г, 0,036 моль) и 4-нитрофенилхлорформиат (3,767 г, 0,0187 моль) и перемешивали реакционную смесь при комн. темп. в течение ночи. Реакционную смесь погасили насыщ. NaHCO3 (водн.) (30 мл) и экстрагировали ДХМ (3×20 мл). Органические фазы объединили и сушили, пропуская через картридж фазного сепаратора Biotage и удалили растворители в вакууме. Остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 50 г 40-63 мкм, 60, 50 мл в мин, градиент от 0% до 6% МеОН / ДХМ]) с получением 4-нитрофенил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилата в виде желтого твердого вещества (1,40 г, 27%).
ЖХ/МС (Способ С): m/z 291 (М+Н)+ (ES+) при 1,167 мин
4-нитрофенил 2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (0,700 г, 2,41 ммоль) растворили в ДМФА (15 мл). Прибавили 4-(1Н-пиразол-1-ил)пиперидин (0,365 г, 2,41 ммоль), ледяную уксусную кислоту (0,144 г, 2,41 ммоль) и STAB (1,535 г, 7,24 ммоль), перемешивали реакционную смесь при 50°C в атмосфере азота в течение ночи. Реакционную смесь погасили водой (2 мл) и удалили растворители в вакууме. Остаток разделили между ДХМ (20 мл) и насыщ. NaHCO3 (водн.) (20 мл), водную фазу экстрагировали ДХМ (2×20 мл), органические фазы объединили и сушили, пропуская через картридж фазного сепаратора Biotage и удалили растворители в вакууме. Остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 25 г 40-63 мкм, 60, 25 мл в мин, градиент от 0% до 10% МеОН / ДХМ]) с получением 4-нитрофенил 2-[4-(1Н-пиразол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата, промежуточного соединения 302, (0,738 г, 72%). Данные для указанного в заголовке соединения приведены в Таблице 2.
Общие методы синтеза Примеров:
Путь а
Типовым метод получения пиперидинов реализуется путем восстановительного аминирования триацетоксиборогидридом натрия как проиллюстрировано на примере получения Примера 1-1, этил-2-[4-(1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата
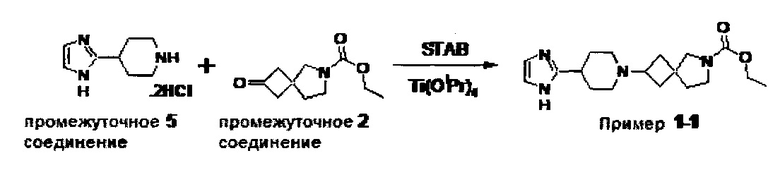
4-(1Н-Имидазол-2-ил)пиперидин дигидрохлорид (1,43 г, 7,1 ммоль) и этил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (1,60 г, 7,1 ммоль) растворили в ДХМ (60 мл) при комн. темп. и прибавили изопропоксид титана (2,31 мл, 7,81 ммоль). Реакционную смесь перемешивали при комн. темп. в течение 1 ч. Реакционную смесь охладили до -5°C, затем прибавили STAB (3,01 г, 14,2 ммоль) и уксусную кислоту (350 мкл, 4,26 ммоль) и перемешивали реакционную смесь в течение ночи в атмосфере азота при нагревании до комн. темп.. Реакционную смесь погасили путем прибавления NaHCO3 (насыщ. водн.) (10 мл) и разбавили ДХМ, затем отфильтровали через слой целита. Фазы разделили и водную фазу экстрагировали ДХМ. Объединенные фазы ДХМ промыли насыщенным водным раствором хлорида натрия, затем сушили над MgSO4. Растворители удалили в вакууме и остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 50 г, 40-63 мкм, 60, 50 мл в мин, градиент от 1% до 10% МеОН в ДХМ с 0,5% NEt3]) с получением неразделимой смеси диастереомеров этил-2-[4-(1H-имидазол-2-ил)пиперидин]-6-азаспиро[3.4]октан-6-карбоксилата (2,645 г, 98,3%) в виде белого твердого вещества. Препаративную ВЭЖХ использовали для разделения диастереомеров, используя колонку Phenomenex Gemini-N С18, 150×21 мм, элюируя от 28 до 38% MeCN/H2O при 18 мл/мин и собирая фракции при 218 нм с получением изомера 1 этил-2-[4-(1Н-имидазол-2-ил)пиперидин]-6-азаспиро[3.4]октан-6-карбоксилата (0,338 г, 14%) в виде бесцветного твердого вещества и изомера 2 этил-2-[4-(1Н-имидазол-2-ил)пиперидин]-6-азаспиро[3.4]октан-6-карбоксилата (0,369 г, 16%) в виде бесцветного твердого вещества. Данные для изомера 2 приведены в Таблице 3.
Путь b
Типовым метод получения пиперидинов реализуется путем восстановительного аминирования цианоборогидридом натрия и хлоридом цинка, как проиллюстрировано на примере получения Примера 1-3, этил-2-(4-(4-(трифторметил)-1Н-имидазол-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата
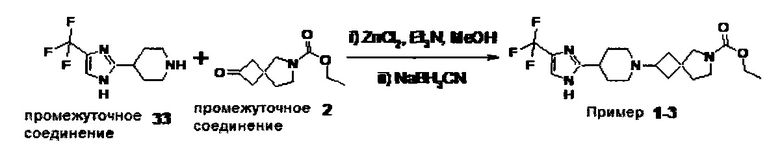
4-(4-(Трифторметил)-1Н-имидазол-2-ил)пиперидин (100 мг, 0,46 ммоль), этил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (89 мг, 0,46 ммоль), ZnCl2 (2 мг, 0,01 ммоль) и триэтиламин (0,3 мл, 2,28 ммоль) растворили в МеОН (5 мл) и перемешивали реакционную смесь при 50°C в течение 2 ч. Реакционную смесь охладили до 0°C, и порциями прибавили NaBH3CN (114 мг, 1,83 ммоль). Полученную реакционную смесь перемешивали при 25°C в течение 7 ч и удалили растворители в вакууме. Остаток разделили между H2O (50 мл) и EtOAc (35 мл), водную фазу экстрагировали EtOAc (2×35 мл), органические фазы объединили, сушили (Na2SO4) и растворитель удалили в вакууме. Остаток очистили с помощью препаративной ВЭЖХ [обращенно-фазовая (X-BRIDGE, С-18, 250×19 мм, 5 мкм, 18 мл в мин, градиент 28,0% (в течение 40,0 мин), 100% (в течение 3,0 мин), затем 28,0% (в течение 5,0 мин), 0,1% NH3 в MeCN/вода] с получением этил-2-(4-(4-(трифторметил)-1Н-имидазол-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 1-3 Изомера 1, (15 мг, 8,24%) в виде желтого твердого вещества и этил-2-(4-(4-(трифторметил)-1Н-имидазол-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 1-3 Изомера 2, (12 мг, 6,6%) в виде желтого твердого вещества. Данные для Изомера 2 приведены в Таблице 3.
Путь c
Типовый метод превращения трифторметил-замещенных имидазолов в циано-замещенные имидазолы, как проиллюстрировано на примере получения Примера 1-4, этил-2-[4-(4-циано-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата
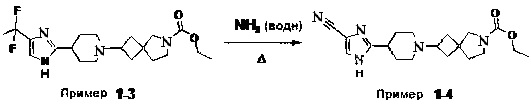
Этил-2-(4-(4-(трифторметил)-1Н-имидазол-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат (200 мг, 0,50 ммоль) растворили в растворе NH3 (20 мл) и перемешивали при 60°C в течение 8 ч. Растворители удалили в вакууме и остаток разделили между H2O (60 мл) и EtOAc (40 мл), водную фазу экстрагировали EtOAc (2×40 мл),органические фазы объединили, сушили (Na2SO4). Растворитель удалили в вакууме и остаток очистили с помощью препаративной ВЭЖХ [обращенно-фазовая (DURASHELL, С-18, 250×21,2 мм, 5 мкм, 22 мл в мин, градиент 25,0% (в течение 30,0 мин), 100% (в течение 3,0 мин), затем 25,0% (в течение 7,0 мин), 0,1% NH3 в MeCN/вода] с получением этил-2-(4-(4-циано-1Н-имидазол-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата, Примера 1-4 Изомера 1, (26 мг, 14,6%) в виде желтого твердого вещества и этил-2-[4-(4-циано-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата, Примера 1-4 Изомера 2, (25 мг, 14,06%) в виде желтого твердого вещества. Данные для изомера 2 приведены в Таблице 3.
Путь d
Типовый метод получения пиперидинов реализуется путем восстановительного аминирования триацетоксиборогидридом натрия, Вос-депротекции и образования этилкарбамата, как проиллюстрировано на примере получения Примера 1-7, этил-2-[4-(1-метил-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата
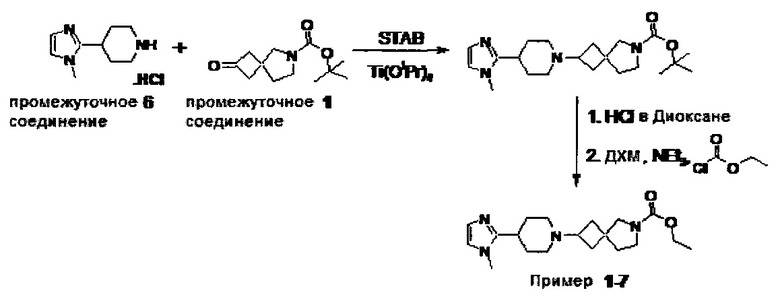
4-(1-Метилимидазол-2-ил)пиперидин гидрохлорид (0,244 г, 1,21 ммоль) и 6-Вос-2-оксо-6-азаспиро[3,4]октан (0,273 г, 1,21 ммоль) растворили в ДХМ (10 мл) при комн. темп. и прибавили изопропоксид титана (0,4 мл, 2,42 ммоль). Реакционную смесь перемешивали при комн. темп. в течение 1 ч. Реакционную смесь охладили до -5°C, затем прибавили STAB (0,513 г, 2,42 ммоль) и уксусную кислоту (27 мкл, 480 мкмоль) и перемешивали реакционную смесь в течение ночи в атмосфере азота при нагревании до комн. темп.. Реакционную смесь погасили путем прибавления NaHCO3 (насыщ. водн.) (10 мл) и разбавили ДХМ, затем отфильтровали через слой целита. Фазы разделили и водную фазу экстрагировали ДХМ. Объединенные фазы ДХМ промыли насыщенным водным раствором хлорида натрия, затем сушили над MgSO4. Растворители удалили в вакууме и остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 25 г, 40-63 мкм, 60, 50 мл в мин, градиент от 1% до 10% МеОН в ДХМ]) с получением неразделимой смеси изомеров трет-бутил 2-[4-(1-метил-1Н-имидазол-2-ил)пиперидин]-6-азаспиро[3.4]октан-6-карбоксилата (0,330 г, 72%) в виде желтой смолы.
ЖХ/МС (Способ A): m/z 374 (М+Н)+ (ES+), при 1,68 мин, не активно в ультрафиолетовом свете.
Трет-бутил 2-[4-(1-метил-1Н-имидазол-2-ил)пиперидин]-6-азаспиро[3.4]октан-6-карбоксилат (0,326 г, 0,87 ммоль) растворили в 4 М растворе хлороводорода в диоксане (1,2 мл, 5,2 ммоль). Реакционную смесь перемешивали при комн. темп. в течение 18 ч. Затем летучие компоненты удалили в вакууме и остаток растворили в ДХМ (17 мл) и триэтиламине (0,49 мл, 3,49 ммоль). Прибавляли по каплям этилхлорформиат (125 мкл, 1,31 ммоль) и перемешивали раствор при комн. темп. в течение 18 ч. Затем смесь вылили в NaHCO3 (водн.) (75 мл) и ДХМ (75 мл), экстрагировали (2×75 мл), и промыли объединенные экстракты ДХМ насыщенным водным раствором хлорида натрия (20 мл), затем сушили над MgSO4. После концентрирования остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 25 г, 40-63 мкм, 60, 50 мл в мин, градиент от 1% до 10% МеОН в ДХМ]) с получением этил-2-[4-(1-метил-1Н-имидазол-2-ил)пиперидин]-6-азаспиро[3.4]октан-6-карбоксилата в виде коричневого масла и смеси диастереомеров (0,25 г, 83%). Препаративную ВЭЖХ использовали для разделения диастереомеров, используя колонку Phenomenex Gemini-N С18, 150×21 мм, элюируя от 38 до 48% MeCN/H2O при 18 мл/мин и собирая фракции при 218 нм с получением этил-2-[4-(1-метил-1Н-имидазол-2-ил)пиперидин]-6-азаспиро[3.4]октан-6-карбоксилата, Примера 1-7 Изомера 1, (0,044 г, 15%) в виде бесцветного масла и этил-2-[4-(1-метил-1Н-имидазол-2-ил)пиперидин]-6-азаспиро[3.4]октан-6-карбоксилата, Примера 1-7 Изомера 2, (0,031 г, 10%) в виде бесцветного масла. Данные для Изомера 2 приведены в Таблице 3.
Путь е
Типовый метод гидрирования соединений, содержащих 3,6-дигидропиридин-1(2Н)-ил с получением соединений, содержащих пиперидинил, как проиллюстрировано на примере получения Примера 1-9, метил-2-[4-(1,5-диметил-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата,

Метил-2-(4-(1,5-диметил-1Н-имидазол-2-ил)-3,6-дигидропиридин-1(2Н)-ил)-6-азаспиро[3.4]октан-6-карбоксилат (102 мг, 0,29 ммоль) [синтезировали путем d и растворили промежуточные соединения 3 и 34] в МеОН (10 мл), и прибавили 10% Pd/C (25 мг). Реакционную смесь продули Н2, затем перемешивали при 25°C в течение 20 ч под балонным Н2. Реакционную смесь отфильтровали через целит и промыли МеОН, растворители из фильтрата удалили в вакууме, и остаток очистили с помощью препаративной ВЭЖХ (X Bridge, С-18, 150×30 мм, 5 мкм, 40 мл в мин, градиент 30% (в течение 12,00 мин), 100% (в течение 14,00 мин), затем 30% (в течение 14,01 мин), 0,1% аммиака в ацетонитрил/вода] с получением метил-2-[4-(1,5-диметил-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата, Примера 1-9 Изомера 1, (5,6 мг, 5,8%) в виде бесцветной смолы и метил-2-[4-(1,5-диметил-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата, Примера 1-9 Изомера 2, (11,6 мг, 11,7%) в виде бесцветной смолы. Данные для изомера 2 приведены в Таблице 3.
Путь f
Типовый метод получения пиперидинов реализуется путем восстановительного аминирования триацетоксиборогидридом натрия, Вос-депротекции и образования этилкарбамата, как проиллюстрировано на примере получения Примера 1-36, этил-2-[4-(1Н-1,2,4-триазол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата
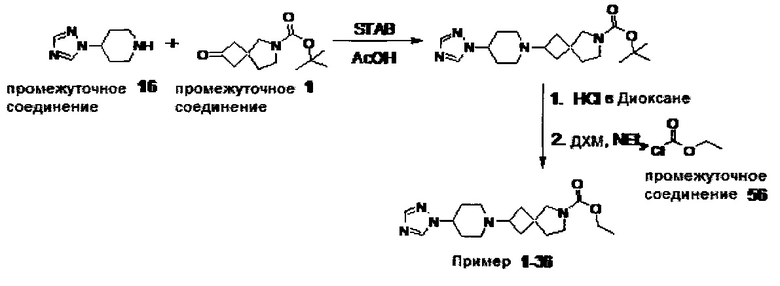
4-(1H-1,2,4-Триазол-1-ил)пиперидин (0,152 г, 1,0 ммоль) и 6-Вос-2-оксо-6-азаспиро[3,4]октан (0,222 г, 1,05 ммоль) растворили в ДХМ (10 мл) под N2 при комн. темп. и прибавили уксусную кислоту (0,13 мл, 2,22 ммоль). Реакционную смесь перемешивали при комн. темп. в течение 2 ч, прибавили STAB (0,53 г, 2,50 ммоль) и перемешивали реакционную смесь в течение ночи при комн. темп. Реакционную смесь погасили путем прибавления NaHCO3 (насыщ. водн.) (30 мл), экстрагировали ДХМ (4×25 мл) и объединенные фазы ДХМ пропустили через фазный сепаратор Biotage. Растворители удалили в вакууме с получением сырой смеси диастереомеров трет-бутил-2-[4-(1Н-1,2,4-триазол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата, который использовали без очистки.
ЖХ/МС (Способ С): m/z 362 (М+Н)+ (ES+), при 1,58 мин и 1,61 мин, не активны в ультрафиолетовом свете.
Сырой трет-бутил 2-[4-(1Н-1,2,4-триазол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат (предположительно 1,0 ммоль) растворили в 4 М растворе хлороводорода в диоксане (1,2 мл, 5,2 ммоль) и перемешивали реакционную смесь при комн. темп. в течение ночи. Летучие компоненты удалили в вакууме и остаток растворили в ДХМ (10 мл) и прибавили NEt3 (0,70 мл, 5,0 ммоль). Прибавляли по каплям этилхлорформиат (0,14 мл, 1,5 ммоль) и перемешивали раствор при комн. темп. в течение ночи. Смесь вылили в NaHCO3 (водн.) (40 мл), экстрагировали ДХМ (4×40 мл), и пропустили объединенные фазы ДХМ через фазный сепаратор Biotage. Растворители удалили в вакууме и остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 25 г, 40-63 мкм, 60, 40 мл в мин, градиент от 0% до 10% МеОН в ДХМ) с получением неразделимой смеси диастереомеров этил-2-[4-(1Н-1,2,4-триазол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата. Препаративную ВЭЖХ использовали для разделения диастереомеров, используя колонку Phenomenex Gemini-NX 5 мкм С18 110A Axia, 100×30 мм, элюируя от 25 до 55% MeCN/Растворитель В в течение 14,4 при 30 мл/мин [где Растворитель В представляет собой 0,2% (28% NH3/Н2О) в H2O] и собирая фракции при 210 нм с получением этил-2-[4-(1Н-1,2,4-триазол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата, Примера 1-36 Изомера 1, (0,026 г, 8%) в виде бесцветного твердого вещества и этил-2-[4-(1Н-1,2,4-триазол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата, Примера 1-36 Изомера 2, (0,026 г, 8%) в виде бесцветного твердого вещества. Данные для изомера 2 приведены в Таблице 3.
Путь g
Типовый метод алкилирования имидазола, включающий соединения с использованием гидрида натрия в ДМФА, как проиллюстрировано на примере получения Примера 1-51, этил-2-{4-[1-(2-метоксиэтил)-1Н-имидазол-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
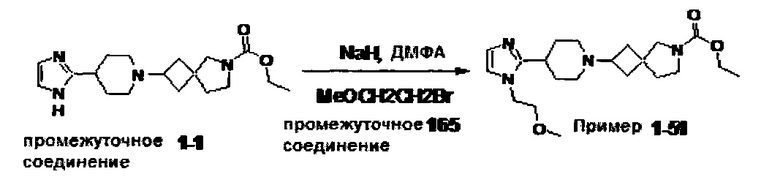
Смесь диастереомеров этил-2-[4-(1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата (150 мг, 0,45 ммоль) растворили в безводном ДМФА (3 мл), обработали суспензией 60% гидрида натрия в минеральном масле (27 мг, 0,68 ммоль) и перемешивали при комн. темп. в течение 2 ч. Прибавили 2-бромэтилметиловый эфир (0,051 мл, 0,54 ммоль) и перемешивали смесь при комн. темп. в течение ночи. Смесь концентрировали для удаления ДМФА. Остаток растворили в МеОН и концентрировали на силикагеле для флеш-хроматографии (5 мл). Полученный порошок очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 25 г, 40-63 мкм, 60], 30 мл в мин, от 0 до 20% Растворителя А в ДХМ, где Растворитель А представляет собой 10% (7 М NH3/MeOH) в МеОН) с получением смеси диастереомеров этил-2-{4-[1-(2-метоксиэтил)-1Н-имидазол-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (159 мг, 90%). Данную смесь растворили в МеОН и очистили раствор с помощью препаративной обратно-фазовой ВЭЖХ, используя колонку Phenomenex Gemini-NX 5 мкм С18 110А Axia, 100×30 мм, элюируя от 15 до 45% MeCN/Растворитель B в течение 14,4 мин при 30 мл/мин [где растворитель B представляет собой 0,2% (28% NH3/H2O) в H2O] и собирая фракции при 210 нм с получением этил-2-{4-[1-(2-метоксиэтил)-1Н-имидазол-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, Примера 1-51 Изомера 1, (54 мг, 31%) и этил-2-{4-[1-(2-метоксиэтил)-1Н-имидазол-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, Примера 1-51 Изомера 2, (27 мг, 15%).
Данные для Изомера 2 приведены в Таблице 3.
Путь h
Типовый метод алкилирования имидазола, включающий соединения с использованием карбоната калия в ДМФА, как проиллюстрировано на примере получения Примера 1-52, этил-2-{4-[1-(цианометил)-1Н-имидазол-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
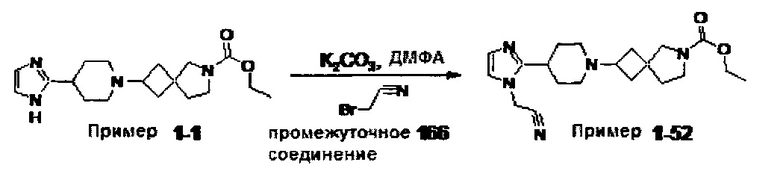
Смесь диастереомеров этил-2-[4-(1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата (150 мг, 0,45 ммоль) растворили в безводном ДМФА (3 мл). Прибавили карбонат калия (187 мг, 1,4 ммоль) и бромацетонитрил (0,114 мл, 1,6 ммоль) и перемешивали смесь при комн. темп. в течение двух ночей. Смесь концентрировали для удаления ДМФА. Остаток растворили в МеОН и концентрировали на силикагеле для флеш-хроматографии (5 мл). Полученный порошок очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 25 г, 40-63 мкм, 60], 30 мл в мин, от 0 до 20% Растворителя А в ДХМ, где Растворитель А представляет собой 10% (7 М NH3/MeOH) в МеОН) с получением смеси диастереомеров этил-2-{4-[1-(цианометил)-1Н-имидазол-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (91 мг, 54%). Данную смесь растворили в МеОН и раствор очистили с помощью препаративной обратно-фазовой ВЭЖХ, используя колонку Phenomenex Gemini-NX 5 мкм С18 110A Axia, 100×30 мм, элюируя от 15 до 45% MeCN/Растворитель B в течение 14,4 мин при 30 мл/мин [где растворитель B представляет собой 0,2% (28% NH3/H2O) в H2O] и собирая фракции при 210 нм с получением этил-2-{4-[1-(цианометил)-1Н-имидазол-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, Примера 1-52 Изомера 1, (8 мг, 5%) и этил-2-{4-[1-(цианометил)-1Н-имидазол-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, Примера 1-52 Изомера 2, (5 мг, 3%). Данные для Изомера 2 приведены в Таблице 3.
Путь i
Метод получения Примера 1-53, (2-{1-[6-(этоксикарбонил)-6-азаспиро[3.4]окт-2-ил]пиперидин-4-ил}-1Н-имидазол-1-ил)уксусной кислоты и Примера 1-54, этил-2-(4-{1-[2-(метиламино)-2-оксоэтил]-1Н-имидазол-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата
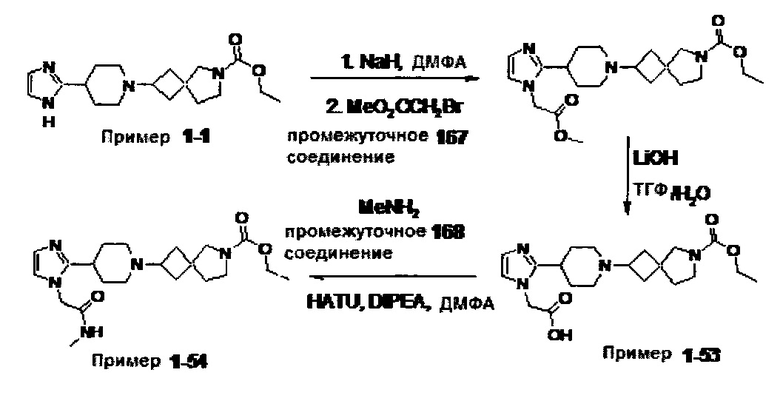
Смесь диастереомеров этил-2-[4-(1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата (500 мг, 1,5 ммоль) привели в контакт с 60% дисперсией гидрида натрия в минеральном масле (90 мг, 2,3 ммоль) и метилбромацетатом (0,171 мл, 1,8 ммоль) в ДМФА (10 мл), используя способ Пути g с получением смеси диастереомеров этил-2-{4-[1-(2-метокси-2-оксоэтил)-1Н-имидазол-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (393 мг, 65%).
ЖХ/МС (Способ С): m/z 405 (М+Н)+ (ES+), при 1,12 и 1,17 мин, малоактивны в ультрафиолетовом свете.
Смесь диастереомеров этил-2-{4-[1-(2-метокси-2-оксоэтил)-1Н-имидазол-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (180 мг, 0,45 ммоль) перемешивали с моногидратом гидроксида лития (75 мг, 1,8 ммоль) в ТГФ (4 мл) и H2O (1 мл) при комн. темп. в течение 5 дней. Смесь концентрировали для удаления ТГФ, подкислили 1 М водным HCl и концентрировали для получения сырой смеси диастереомеров (2-{1-[6-(этоксикарбонил)-6-азаспиро[3.4]окт-2-ил]пиперидин-4-ил}-1Н-имидазол-1-ил)уксусной кислоты (0,4 г, >100%). Приблизительно 0,2 г данной смеси растворили в МеОН и раствор очистили с помощью препаративной обратно-фазовой ВЭЖХ, используя колонку Phenomenex Gemini-NX 5 мкм С18 110А Axia, 100×30 мм, элюируя от 5 до 15% MeCN/Растворитель B в течение 14,4 мин при 30 мл/мин [где растворитель B представляет собой 0,2% (28% NH3/H2O) в H2O] и собирая фракции при 210 нм с получением (2-{1-[6-(этоксикарбонил)-6-азаспиро[3.4]окт-2-ил]пиперидин-4-ил}-1Н-имидазол-1-ил)уксусной кислоты, Примера 1-53 Изомера 1, (30 мг, 17%) и (2-{1-[6-(этоксикарбонил)-6-азаспиро[3.4]окт-2-ил]пиперидин-4-ил}-1Н-имидазол-1-ил)уксусной кислоты, Примера 1-53 Изомера 2, (22 мг, 13%).
Данные для изомера 2 приведены в Таблице 3.
Оставшуюся сырую смесь диастереомеров (2-{1-[6-(этоксикарбонил)-6-азаспиро[3.4]окт-2-ил]пиперидин-4-ил}-1Н-имидазол-1-ил)уксусной кислоты, Примера 1-53, (0,2 г, приблизительно 0,22 ммоль) растворили в ДМФА (3 мл) и обработали диизопропилэтиламином (0,155 мл, 0,89 ммоль) и раствором метиламина в метаноле (2 М, 0,33 мл, 0,66 ммоль). Затем прибавили HATU (0,127 г, 0,33 ммоль) и смесь перемешивали при комн. темп. в течение ночи. Смесь концентрировали для удаления ДМФА, остаток растворили в смеси ДХМ и МеОН и концентрировали на силикагеле для флеш-хроматографии (10 мл). Полученный порошок очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 25 г, 40-63 мкм, 60], 30 мл в мин, от 0 до 20% Растворителя А в ДХМ, где Растворитель А представляет собой 10% (7 М NH3/MeOH) в МеОН) с получением смеси диастереомеров этил-2-(4-{1-[2-(метиламино)-2-оксоэтил]-1Н-имидазол-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата. Данную смесь растворили в МеОН и очистили раствор с помощью препаративной обратно-фазовой ВЭЖХ используя колонку Phenomenex Gemini-NX 5 мкм С18 110A Axia, 100×30 мм, элюируя от 15 до 45% MeCN/Растворитель В в течение 14.4 мин при 30 мл/мин [где растворитель В представляет собой 0,2% (28% NH3/H2O) в H2O] и собирая фракции при 210 нм с получением этил-2-(4-{1-[2-(метиламино)-2-оксоэтил]-1Н-имидазол-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата, Примера 1-54 Изомера 1, (9 мг, 4%) и этил-2-(4-{1-[2-(метиламино)-2-оксоэтил]-1Н-имидазол-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата, Примера 1-54 Изомера 2, (6 мг, 3%). Данные для Изомера 2 приведены в Таблице 3.
Путь j
Типовый метод получения пиперидинов реализуется путем образования карбамата, как проиллюстрировано на примере получения Примера 1-70, этил-2-{4-[(2R)-4,4-дифтор-2-(метилкарбамоил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата

4-нитрофенил-2-[4-(1H-пиразол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат (0,125 g, 0,294 ммоль) суспендировали в безводном ТГФ (4 мл) и обрабатывали ультразвуком для растворения. Прибавили гидрид натрия, 60% дисперсию в минеральном масле, (0,026 г, 0,647 ммоль) и перемешивали реакционную смесь при комн. темп. в атмосфере азота в течение 10 мин. Прибавили этанол-1,1-2,2,2-d5 (0,150 г, 2,94 ммоль) и перемешивали реакционную смесь при комн. темп. в атмосфере азота в течение ночи. К реакционной смеси прибавили воду (1 мл) и в вакууме удалили растворители. Остаток разделили между ДХМ (20 мл) и насыщ. NaHCO3 (водн.) (10 мл), водную фазу экстрагировали ДХМ (2×10 мл). Органические фазы объединили и сушили, пропуская через картридж фазного сепаратора Biotage. Растворители удалили в вакууме, и остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 10 г 40-63 мкм, 60, 12 мл в мин, градиент 0% до 10% МеОН / ДХМ]). Остаток дополнительно очистили с помощью препаративной обратно-фазовой ВЭЖХ (колонка Phenomenex Gemini-NX 5 мкм С18 110A Axia, 100×30 мм, элюируя 20 до 50% MeCN/Растворитель В в течение 14,4 мин при 30 мл/мин [где растворитель В представляет собой 0,2% (28% NH3/H2O) в H2O] и собирая фракции при 210 нм) с получением (2Н5)этил-2-[4-(1Н-пиразол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата, Примера 1-70 Изомера 1, (0,017 г, 17%) в виде белого твердого вещества и (2Н5)этил-2-[4-(1H-пиразол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата, Примера 1-70 Изомера 2, (0,013 г, 13%) в виде белого твердого вещества. Данные для изомера 2 приведены в Таблице 3.
Путь k
Типовый метод получения пиперидинов реализуется путем образования формамида, как проиллюстрировано на примере получения Примера 2-2, этил-2-[4-(1-формилпирролидин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата
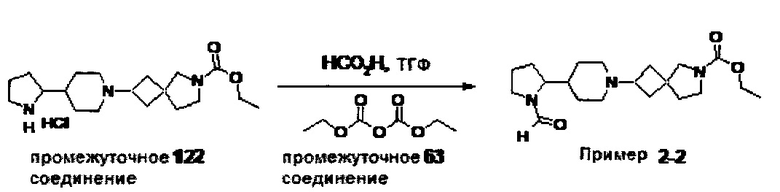
Смесь муравьиной кислоты (2 мл) и уксусного ангидрида (0,1 мл, 1,43 ммоль) перемешивали при 60°C в течение 1 ч, затем реакционную смесь охладили до 0°C, и прибавляли по каплям смесь диастереомеров этил-2-(4-(пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата. HCl (100 мг, 0,30 ммоль) в ТГФ (2 мл). Полученную реакционную смесь перемешивали при 60°C в течение 8 ч, довели до основного pH, затем разделили реакционную смесь между H2O (40 мл) и EtOAc (25 мл). Водную фазу дополнительно экстрагировали EtOAc (2×25 мл) и органические фазы объединили и сушили над Na2SO4. Растворители удалили в вакууме и остаток очистили с помощью препаративной ВЭЖХ (X Bridge, С-18, 150×30 мм, 5 мкм, 40 мл в мин, градиент 30% (в течение 12,00 мин), 100% (в течение 14,00 мин), затем 30% (в течение 14,01 мин), 0,1% аммиак в ацетонитрил/вода] с получением этил-2-[4-(1-формилпирролидин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-2 Изомера 1 (14,6 мг, 13,0%) в виде желтой смолы и этил-2-[4-(1-формилпирролидин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-2 Изомера 2 (12,5 мг, 11,1%) в виде желтой смолы. Данные для Изомера 2 приведены в Таблице 3.
Путь L
Типовый метод получения пиперидинов реализуется путем образования амида, как проиллюстрировано на примере получения Примера 2-4, этил-2-{4-[1-(трифторацетил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
Этил-2-(4-(пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат (50 мг, 0,15 ммоль) и NEt3 (0,06 мл, 0,45 ммоль) растворили в ТГФ (3 мл) при комн. темп. По каплям прибавляли этил-2,2,2-трифторацетат (0,03 мг, 0,22 ммоль) и перемешивали полученную реакционную смесь при комн. темп. в течение 8 ч. Реакционную смесь разделили между H2O (40 мл) и EtOAc (25 мл), водную фазу дополнительно экстрагировали EtOAc (2×25 мл), органические фазы объединили и сушили над Na2SO4. Растворители удалили в вакууме и остаток очистили с помощью препаративной ВЭЖХ (X Bridge, С-18, 150×30 мм, 5 мкм, 40 мл в мин, градиент 30% (в течение 12,00 мин), 100% (в течение 14,00 мин), затем 30% (в течение 14,01 мин), 0,1% аммиак в ацетонитрил/вода] с получением этил-2-{4-[1-(трифторацетил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-4 Изомера-1 (5,5 мг, 8,0%) в виде желтой смолы и этил-2-{4-[1-(трифторацетил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-4 Изомера-2 (6,2 мг, 9,7%) в виде желтой смолы. Данные для Изомера 2 приведены в Таблице 3.
Путь m
Типовый метод получения пиперидинов реализуется путем образования амида/карбамата/мочевины, как проиллюстрировано на примере получения Примера 2-17, этил-2-{4-[(2S)-1-(метилкарбамоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
Смесь диастереомеров этил-2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата. HCl (2,10 г, 5,65 ммоль) растворили в ДХМ (20 мл) и триэтиламине (1,54 мл, 11,1 ммоль). Метиламиноформилхлорид (620 мг, 6,63 ммоль) прибавили и перемешивали раствор при комн. темп. в течение 2 ч. Затем смесь вылили в 1 М NaOH (водн.) (50 мл), экстрагировали ДХМ (2×50 мл), и промыли объединенные экстракты ДХМ насыщенным водным раствором хлорида натрия (50 мл), затем пропустили через фазный сепаратор Biotage и концентрировали в вакууме с получением этил-2-{4-[(2S)-1-(метилкарбамоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата в виде желтого твердого вещества и смеси диастереомеров (1,79 г, 82%). Препаративную ВЭЖХ использовали для разделения диастереомеров, используя колонку Phenomenex Gemini-NX С18, 100×30 мм, элюируя от 25 до 35% MeCN/0,2% аммиака в H2O (об.) при 18 мл/мин и собирая фракции при 210 нм с получением Примера 2-17 Изомера 1, этил-2-{4-[(2S)-1-(метилкарбамоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (0,78 г, 36%) в виде бесцветного масла и Примера 2-17 Изомера 2, этил-2-{4-[(2S)-1-(метилкарбамоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (0,67 г, 31%) в виде бесцветного масла. Данные для Изомера 2 приведены в Таблице 3.
Путь n
Типовый метод получения пиперидинов реализуется путем образования мочевины/карбамата, как проиллюстрировано на примере получения Примера 2-19, этил-2-{4-[1-(этилкарбамоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
Смесь диастереомеров этил-2-(4-(пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата. HCl (100 мг, 0,30 ммоль), диэтиламина (0,3 мл, 0,60 ммоль) и NEt3 (0,1 мл, 0,90 ммоль) растворили в DCE (5 мл) при комн. темп. Прибавили CDI (145 мг, 0,60 ммоль) и перемешивали реакционную смесь при комн. темп. в течение 15 ч. Реакционную смесь разделили между H2O (40 мл) и EtOAc (25 мл), водную фазу дополнительно экстрагировали EtOAc (2×25 мл), органические фазы объединили, сушили (Na2SO4), растворители удалили в вакууме и остаток очистили с помощью препаративной ВЭЖХ [обращенно-фазовая ВЭЖХ (X-BRIDGE, С-18, 250×19 мм, 5 мкм, 15 мл в мин, градиент от 30,0% до 38,0% (в течение 25,0 мин), 100,0% (в течение 3,0 мин), затем 30,0% (в течение 2,0 мин), 0,1% NH3 в MeCN/вода] с получением этил-2-(4-(1-(этилкарбамоил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2 19 изомера-1, (7,5 мг, 6,20%) в виде желтой смолы и этил-2-(4-(1-(этилкарбамоил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-19 изомера 2, (8,1 мг, 6,60%) в виде желтой смолы. Данные для Изомера 2 приведены в Таблице 3.
Путь о
Типовый метод получения пиперидинов реализуется путем алкилирования, как проиллюстрировано на примере получения Примера 2-22, этил-2-[4-(1-метилпирролидин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата
Смесь диастереомеров этил-2-(4-(пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата. HCl (200 мг, 0,60 ммоль) и формальдегид (40% р-р, 1,01 мл, 3,60 ммоль) растворили в H2O (2 мл) при 25°C. Прибавляли по каплям муравьиную кислоту (0,303 мл, 0,90 ммоль) и перемешивали полученную смесь при 70°C в течение 14 ч. Реакционную смесь погасили раствором NaHCO3(5 мл), затем реакционную смесь разделили между H2O (50 мл) и EtOAc (35 мл). Водную фазу дополнительно экстрагировали EtOAc (2×35 мл), органические фазы объединили и сушили над Na2SO4. Растворители удалили в вакууме и остаток очистили с помощью препаративной ВЭЖХ [обращенно-фазовая ВЭЖХ (X-Bridge, C-18, 250×19,0 мм, 5 мкм, 14 мл в мин, градиент 37% (в течение 28,0 мин), 100% (в течение 4,0 мин), затем 37% (в течение 3,0 мин), 0,1% NH3 в MeCN/вода] с получением этил-2-(4-(1-метилпирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-22 изомера 1 (12 мг, 5,80%) в виде желтой смолы и этил-2-(4-(1-метилпирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-22 изомера 2 (11 мг, 5,30%) в виде желтой смолы. Данные для Изомера 2 приведены в Таблице 3.
Путь p
Типовым метод получения пиперидинов реализуется путем образования амида, как проиллюстрировано на примере получения Примера 2-23, этил-2-{4-[1-(N-метилглицил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
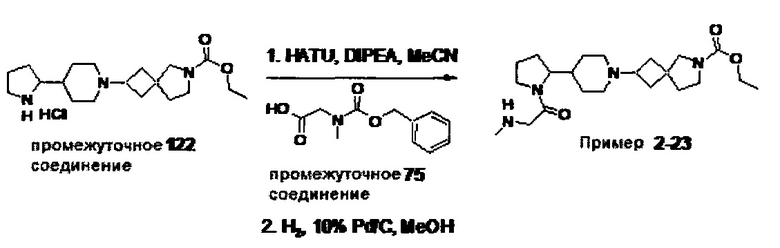
N-[(бензилокси)карбонил]-N-метилглицин (73 мг, 0,33 ммоль) растворили в ацетонитриле (5 мл) с последующим прибавлением HATU (170 мг, 0,45 ммоль) и DIPEA (0,2 мл, 0,90 ммоль). Реакционную смесь перемешивали при 0°C в течение 10 минут, с последующим прибавлением смеси диастереомеров этил-2-(4-(пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата. HCl (100 мг, 0,30 ммоль) и перемешивали полученную реакционную смесь при 25°C в течение 3 ч. Реакционную смесь разделили между H2O (50 мл) и EtOAc (35 мл), водную фазу дополнительно экстрагировали EtOAc (2×35 мл), органические фазы объединили, сушили над Na2SO4 и удалили растворители в вакууме. В конце остаток очистили с помощью колоночной хроматографии (нормально-фазовая на активированном оксиде алюминия, от 0,5% до 1,0% МеОН в ДХМ) с получением этил-2-(4-(1-(N-((бензилокси)карбонил)-N-метилглицил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата (130 мг, 80,74%) в виде коричневой смолы. Этил-2-(4-(1-(N-((бензилокси)карбонил)-N-метилглицил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат (130 мг, 0,24 ммоль) растворили в МеОН (10 мл) с последующим прибавлением Pd/C (сухая подложка, 13 мг). Затем реакционную смесь продули Н2 и полученную реакционную смесь перемешивали при 25°C в течение 10 ч. Реакционную смесь фильтровали через слой целита и промыли метанолом, затем фильтрат сушили над Na2SO4 и удалили растворители в вакууме. Остаток очистили с помощью препаративной ВЭЖХ [обращенно-фазовая ВЭЖХ (X-BRIDGE, С-18, 250×19 мм, 5 мкм, 15 мл в мин, градиент от 20,0% до 35,0% (в течение 30,0 мин), 100,0% (в течение 3,0 мин), затем 20,0% (в течение 2,0 мин), 0,1% NH3 в MeCN/вода] с получением этил-2-(4-(1-(метилглицил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-23 Изомера 1, (9,0 мг, 9,27%) в виде желтой смолы и этил-2-(4-(1-(этилкарбамоил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-23 Изомера 2, (8,0 мг, 8,50%) в виде желтой смолы. Данные для Изомера 2 приведены в Таблице 3.
Путь q
Типовый метод получения пиперидинов реализуется путем образования мочевины, как проиллюстрировано на примере получения Примера 2-27, этил-2-{4-[(2S)-1-(азетидин-1-илкарбонил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
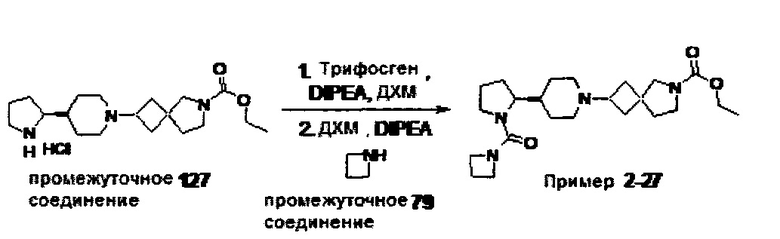
Смесь диастереомеров этил-2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата. HCl (100 мг, 0,291 ммоль) растворили в смеси ДХМ (5 мл) идиизопропилэтиламина (0,099 мл, 0,58 ммоль). Трифосген (88 мг, 0,291 ммоль) прибавили при 0°C и раствор нагрели до комн. темп. и перемешивали в течение 1 ч. Затем смесь разбавили ДХМ (50 мл) и промыли H2O (70 мл). Водную фазу экстрагировали ДХМ (2×50 мл), и объединенные экстракты ДХМ сушили Na2SO4 и концентрировали в вакууме. Остаток растворили в ДХМ (5 мл) и прибавили азетидин (0,020 мл, 0,291 ммоль) и диизопропилэтиламин (0,256 мл, 1,48 ммоль). Реакционную смесь перемешивали при комн. темп. в течение 1 ч. Затем смесь разбавили ДХМ (50 мл) и промыли H2O (70 мл). Водную фазу экстрагировали ДХМ (2x 50 мл), и объединенные экстракты ДХМ сушили Na2SO4 и концентрировали в вакууме с получением этил-2-{4-[(2S)-1-(азетидин-1-илкарбонил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата в виде желтого твердого вещества и смеси диастереомеров. Остаток очистили с помощью препаративной ВЭЖХ [обращенно-фазовая ВЭЖХ (CHIRALPAK AD-Н, С-18, 250×20 мм, 5 мкм, 18,0 мл в мин, градиент от 0% до 50% (в течение 15,0 мин), 0,1% аммиака в ацетонитриле и 0,1% аммиака в H2O с получением этил (S)-2-(4-(1-(азетидин-1-карбонил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-27 Изомера 1 (20 мг, 16,12%) в виде бесцветной смолы, и Примера 2-27 Изомера 2 (20 мг, 16,12%) в виде бесцветной смолы. Данные для Изомера 2 приведены в Таблице 3.
Путь r
Типовый метод получения пиперидинов реализуется путем образования мочевины и дегидратации, как проиллюстрировано на примере получения Примера 2-42, этил-2-(4-{(2S)-1-[этил(пропан-2-ил)карбамоил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата и Примера 2-138, этил-2-{4-[(2S)-1-(5-метил-1,3,4-оксадиазол-2-ил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
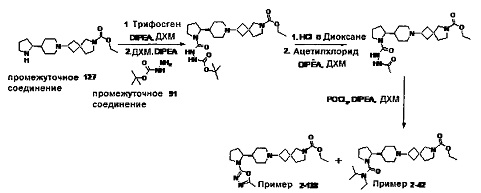
Смесь диастереомеров этил-2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата. HCl (164 мг, 0,403 ммоль) растворили в смеси ДХМ (2 мл) и диизопропилэтиламина (0,209 мл, 1,21 ммоль). Трифосген (43 мг, 0,145 ммоль) прибавили при 0°C и нагрели раствор до комн. темп., и перемешивали в течение 18 ч. К данной смеси прибавили трет-бутилкарбазат (108 мг, 0,82 ммоль) и диизопропилэтиламин (0,142 мл, 0,82 ммоль) и реакционную смесь перемешивали при комн. темп. в течение 18 ч. Затем смесь разбавили ДХМ (20 мл) и промыли насыщенным NaHCO3 (водн.) (2×20 мл). Водные фазы экстрагировали ДХМ (20 мл), и объединенные экстракты ДХМ промыли насыщенным водным раствором хлорида натрия (50 мл), затем пропустили через фазный сепаратор Biotage и концентрировали в вакууме с получением этил-2-{4-[(2S)-1-(трет-бутил карбазоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата в виде желтого масла и смеси диастереомеров (192 мг, 97%).
ЖХ/МС (Способ D): m/z 494 (М+Н)+ (ES+), при 1,83 и 1,87 мин, не активно в ультрафиолетовом свете.
Сырой продукт растворили в 4 М хлорида водорода в диоксане (2,0 мл, 8,0 ммоль) и ДХМ (1 мл). Реакционную смесь перемешивали при комн. темп. в течение 1 ч. Затем летучие компоненты удалили в вакууме до того, как реакционную смесь растворили в ДХМ (2 мл) и диизопропилэтиламине (0,142 мл, 0,82 ммоль). Прибавили ацетилхлорид (0,031 мл, 0,428 ммоль) при 0°C, и раствор нагрели до комн. темп. и перемешивали в течение 2 ч. Летучие компоненты удалили в вакууме и использовали в следующей стадии без дополнительной очистки. Остаток растворили в толуоле (2 мл) и диизопропилэтиламине (0,135 мл, 0,78 ммоль) и охладили до 0°C. Прибавили оксихлорид фосфора (0,182 мл, 1,945 ммоль) и реакционную смесь перемешивали при 110°C в течение 30 минут до того, как реакционную смесь охладили до комн. темп. и погасили ледяной водой (20 мл). Затем смесь разбавили ДХМ (20 мл) и промыли 1 М NaOH(BOAH.) (2×20 мл). Водные фазы экстрагировали ДХМ (3×20 мл), и объединенные экстракты ДХМ промыли насыщенным водным раствором хлорида натрия (50 мл), затем пропустили через фазный сепаратор Biotage и концентрировали в вакууме. Остаток очистили с помощью препаративной ВЭЖХ, используя колонку Phenomenex Gemini-NX С18, 100×30 мм, элюируя от 25 до 45% MeCN/0,2% аммиака в H2O (об.) при 18 мл/мин и собирая фракции при 210 нм с получением Примера 2-42 Изомера 1, этил-2-(4-{(2S)-1-[этил(пропан-2-ил)карбамоил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата (1,7 мг, 1%) в виде бесцветного масла, Примера 2-42 Изомера 2, этил-2-(4-{(2S)-1-[этил(пропан-2-ил)карбамоил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата (1,6 мг, 1%) в виде бесцветного масла, Примера 2-138 Изомера 1, этил-2-{4-[(2S)-1-(5-метил-1,3,4-оксадиазол-2-ил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (3,9 мг, 2,5%) в виде бесцветного масла и Примера 2-138 Изомера 2, этил-2-{4-[(2S)-1-(5-метил-1,3,4-оксадиазол-2-ил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (3,0 мг, 2%) в виде бесцветного масла. Данные для Изомеров 2 приведены в Таблице 3.
Путь s
Типовый метод получения пиперидинов реализуется путем образования карбамата, как проиллюстрировано на примере получения Примера 2-47, этил-2-(4-{(2S)-1-[(2-фторэтокси)карбонил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата
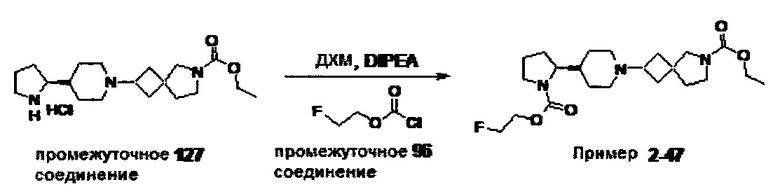
Смесь диастереомеров этил-2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата. HCl (0,15 г, 0,44 ммоль) и диизопропилэтиламин (0,152 мл, 0,89 ммоль) растворили в ДХМ (5 мл), затем реакционную смесь охладили до 0°C. 2-фторэтилхлорформиат (0,062 г, 0,492 ммоль) прибавили и перемешивали полученную реакционную смесь при 25°C в течение 16 ч. Реакционную смесь разделили между H2O (70 мл) и ДХМ (50 мл), водную фазу дополнительно экстрагировали ДХМ (2×50 мл), органические фазы объединили, сушили над Na2SO4, и удалили растворитель в вакууме. Остаток очистили с помощью препаративной ВЭЖХ [обращенно-фазовая ВЭЖХ (CHIRALPAK AD-H, С-18, 250×20 мм, 5 мкм, 18,0 мл в мин, градиент от 0% до 35% (в течение 52 мин), 0,1% аммиака в ацетонитриле и 0,1% аммиака в воде с получением этил-2-(4-{(2S)-1-[(2-фторэтокси)карбонил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-47 Изомера-1 (17 мг, 8,9%) в виде желтой смолы, и этил-2-(4-{(2S)-1-[(2-фторэтокси)карбонил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-47 Изомера-2 (19 мг, 10%) в виде желтой смолы. Данные для Изомера 2 приведены в Таблице 3.
Путь t
Типовый метод получения пиперидинов реализуется путем образования амида, как проиллюстрировано на примере получения Примера 2-52, этил-2-{4-[(2S)-1-(гидроксиацетил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
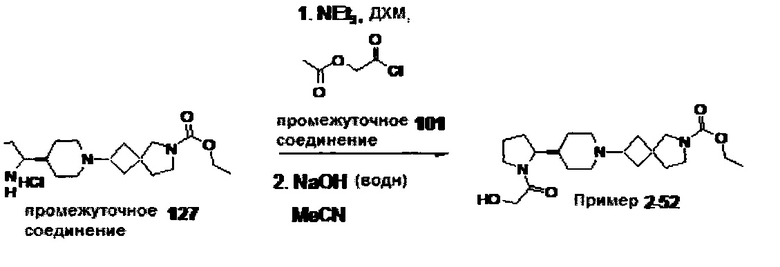
Смесь диастереомеров этил-2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата. HCl (0,2 г, 0,541 ммоль) и триэтиламина (0,152 мл, 1,1 ммоль) растворили в ДХМ (5 мл), затем реакционную смесь охладили до 0°C и прибавили ацетоксиацетилхлорид (0,080 г, 0,591 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Летучие компоненты удалили в вакууме, затем остаток растворили в ацетонитриле (25 мл) и 20% растворе NaOH в воде (10 мл) и перемешивали его при комнатной температуре в течение 1 ч. Реакционную смесь разделили между H2O (70 мл) и ДХМ (50 мл), водную фазу дополнительно экстрагировали ДХМ (2×50 мл), органические фазы объединили, сушили над Na2SO4, растворитель удалили под вакуумом и остаток очистили с помощью препаративной ВЭЖХ [обращенно-фазовая ВЭЖХ (CHIRALPAK AD-H, С-18, 250×20 мм, 5 мкм, 18,0 мл в мин, градиент от 0% до 30% (в течение 35,0 мин), 0,1% аммиака в ацетонитриле и 0,1% аммиака в воде с получением этил(S)-2-(4-(1-(2-гидроксиацетил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-52 Изомера 1 (8 мг, 8,3%) в виде бесцветной смолы, и этил(S)-2-(4-(1-(2-гидроксиацетил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-52 Изомера 2 (12 мг, 12,24%) в виде бесцветной смолы. Данные для Изомера 2 приведены в Таблице 3.
Путь u
Типовый метод получения пиперидинов реализуется путем образования амида, как проиллюстрировано на примере получения Примера 2-53, этил-2-{4-[(2S)-1-(3,3,3-трифторпропаноил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
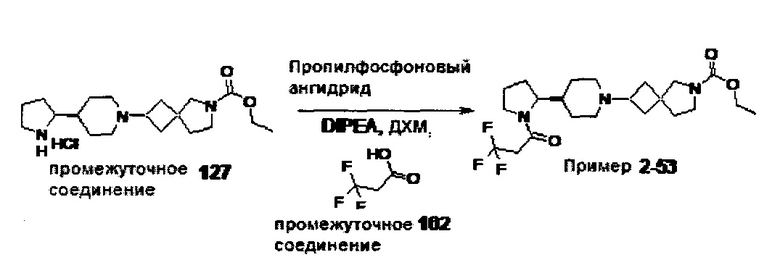
(Смесь диастереомеров этил-2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата. HCl (0,12 г, 0,36 ммоль) и диизопропилэтиламин (0,123 мл, 0,71 ммоль) растворили в ДХМ (10 мл) с последующим прибавлением трифторпропионовой кислоты (0,045 г, 0,394 ммоль). Реакционную смесь охладили до 0°C, и прибавили пропилфосфоновый ангидрид (0,140 г, 0,462 ммоль 50% EtOAc раствор) и перемешивали полученную реакционную смесь при 25°C в течение 2 ч. Реакционную смесь разделили между H2O (20 мл) и ДХМ (50 мл), водную фазу дополнительно экстрагировали ДХМ (2×50 мл), органические фазы объединили, сушили над Na2SO4, и растворитель удалили в вакууме. Остаток очистили с помощью препаративной ВЭЖХ [обращенно-фазовая ВЭЖХ (CHIRALPAK AD-H, С-18, 250×20 мм, 5 мкм, 18,0 мл в мин, градиент от 0% до 30% (в течение 27,0 мин), 0,1% аммиака в ацетонитриле и 0,1% аммиака в воде с получением этил(S)-2-(4-(1-(3,3,3-трифторпропаноил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-53 Изомера 1 (9 мг, 6,0%) в виде бесцветной смолы, и этил(S)-2-(4-(1-(3,3,3-трифторпропаноил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-53 Изомера 2 (9 мг, 6,0%) в виде бесцветной смолы. Данные для Изомера 2 приведены в Таблице 3.
Путь v
Типовый метод получения тиоамидов реализуется путем использования реагента Лавессона, как проиллюстрировано на примере получения Примера 2-58, этил-2-{4-[(2S)-1-пропантиоилпирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
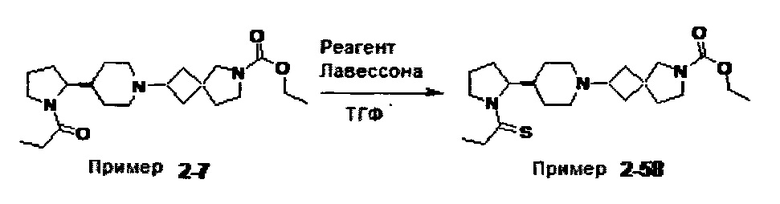
Этил-2-{4-[(2S)-1-пропаноилпирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат (0,341 г, 0,87 ммоль) растворили в ТГФ (4 мл) с последующим прибавлением реагента Лавессона (0,265 г, 0,65 ммоль). Реакционную смесь перемешивали при 70°C в течение 24 часов. Летучие компоненты удалили в вакууме и разделили реакционную смесь между 1 М NaOH (водн.) (50 мл) и ДХМ (30 мл), водную фазу дополнительно экстрагировали ДХМ (2×50 мл), органические фазы объединили, промыли 5% пиросульфита натрия (водн.), сушили над Na2SO4, и удалили растворители в вакууме. Остаток очистили с помощью препаративной ВЭЖХ, используя колонку Phenomenex Gemini-NX С18, 100×30 мм, элюируя от 25 до 45% MeCN/0,2% аммиака в H2O (об.) при 18 мл/мин и собирали фракции при 210 нм с получением Примера 2-58 Изомера 1 этил-2-{4-[(2S)-1-пропантиоилпирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (14,1 мг, 4%) в виде желтого масла и Примера 2-58 Изомера 2, этил-2-{4-[(2S)-1-пропантиоилпирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (7,6 мг, 2%) в виде желтого масла. Данные для Изомера 2 приведены в Таблице 3.
Путь w
Типовый метод получения пиперидинов реализуется путем алкилирования, как проиллюстрировано на примере получения Примера 2-61, этил-2-{4-[(2S)-1-этилпирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
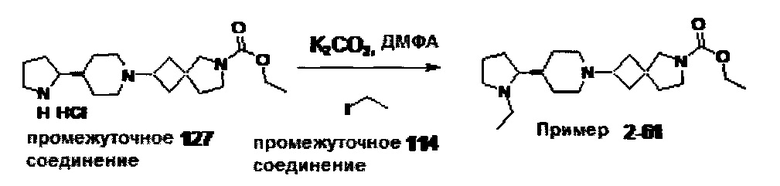
Смесь диастереомеров этил-2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата. HCl (0,100 г, 0,29 ммоль) и карбонат калия (0,123 мг, 0,89 ммоль) растворили в ДМФА (5 мл) и перемешивали реакционную смесь при 60°C в течение 2 часов. Затем прибавили иодэтан (0,049 г, 0,31 ммоль и перемешивали реакционную смесь при 100°C в течение 62 часов. Реакционную смесь разделили между H2O (70 мл) и EtOAc (50 мл), водную фазу дополнительно экстрагировали EtOAc (2×50 мл), органические фазы объединили, сушили над Na2SO4, и растворитель удалили в вакууме. Остаток очистили с помощью препаративной ВЭЖХ (X Bridge, С-18, 150×19 мм, 5 мкм, 20 мл в мин, градиент 35% (в течение 0,01 мин), 100% (в течение 25,01 мин), затем 35% (в течение 30,00 мин), 0,1% аммиака в ацетонитриле/воде] с получением этил(S)-2-(4-(1-этилпирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-61 Изомера 1 (43 мг, 38,8%) в виде бесцветной смолы, и этил(S)-2-(4-(1-этилпирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-61 Изомера 2 (26 мг, 23,1%) в виде бесцветной смолы. Данные для Изомера 2 приведены в Таблице 3.
Путь x
Типовый метод получения пиперидинов реализуется путем s образования амида, как проиллюстрировано на примере получения Примера 2-62, этил-2-(4-{(2S)-1-[3-(пиридин-2-ил)пропаноил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата
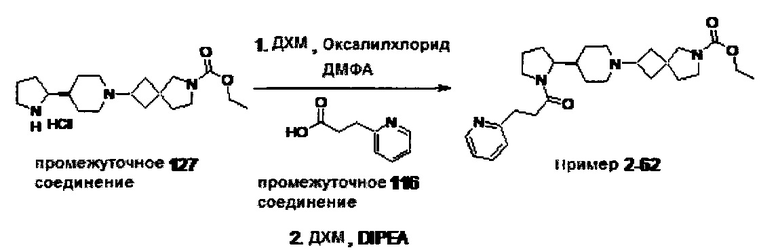
К раствору оксалил хлорида (0,065 мл, 0,768 ммоль) в ДХМ (2 мл) при 0°C прибавили 2-пиридинпропионовую кислоту (106 мг, 0,704 ммоль) и ДМФА (1 каплю). Смесь диастереомеров этил-2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата. HCl (262 мг, 0,640 ммоль) растворили в ДХМ (1 мл) и добавили диизопропилэтиламин (0,355 мл, 2,049 ммоль) к раствору, который перемешивали при комн. темп. в течение 2 ч. Затем смесь вылили в 1 М NaOH (водн.) (50 мл), экстрагировали ДХМ (2×50 мл), и объединенные ДХМ экстракты промыли насыщенным водным раствором хлорида натрия (50 мл), затем пропустили через фазный сепаратор Biotage и концентрировали в вакууме для получения этил-2-(4-{(2S)-1-[3-(пиридин-2-ил)пропаноил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата в виде черного масла и смеси диастереомеров (0,245 г, 82%). Препаративную ВЭЖХ использовали для разделения диастереомеров, используя колонку Phenomenex Gemini-NX С18, 100×30 мм, элюируя от 25 до 45% MeCN/0,2% аммиака в H2O (об.) при 18 мл/мин и собирая фракции при 210 нм с получением Примера 2-62 Изомера 1, этил 2-(4-{(2S)-1-[3-(пиридин-2-ил)пропаноил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата (0,042 г, 14%) в виде бесцветного масла и Примера 2-62 Изомера 2, этил 2-(4-{(2S)-1-[3-(пиридин-2-ил)пропаноил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата (0,030 г, 10%) в виде бесцветного масла. Данные для 2 приведены в Таблице 3.
Путь y
Типовый метод получения пиперидинов реализуется путем образования амида и CBZ-депротекции, как проиллюстрировано на примере получения Примера 2-65, этил-2-{4-[(2S)-1-{N-[(бензилокси)карбонил]-β-аланил}пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата и Примера 2-66, этил-2-{4-[(2S)-1-(β-аланил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
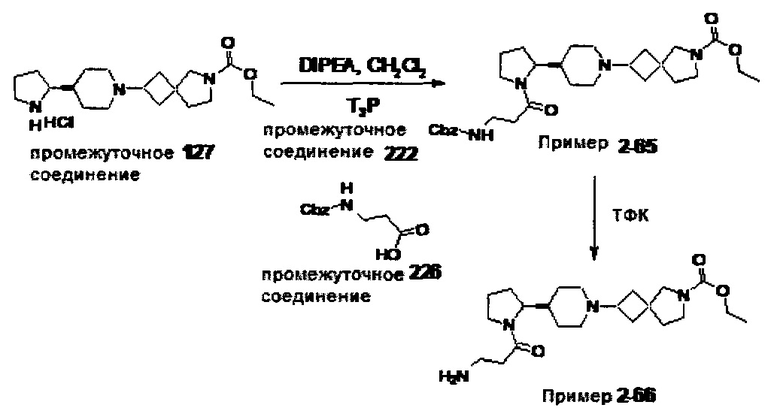
Смесь диастереомеров этил-2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата. НСl (100 мг, 0,298 ммоль) и DIPEA (0,102 мл, 0,597 ммоль) растворили в CH2Cl2 (5 мл), охладили до 0°C и прибавили Z-p-ala-OH (0,066 г, 0,298 ммоль) с последующим прибавлением пропилфосфонового ангидрида (0,123 г, 0,388 ммоль, 50% в этилацетате). Полученную реакционную смесь перемешивали при 25°C в течение 3 ч, разделили между H2O (70 мл) и CH2Cl2 (50 мл) и дополнительно экстрагировали водную фазу CH2Cl2 (2×50 мл). Объединенные органические фазы сушили (Na2SO4), фильтровали и удалили растворитель в вакууме с получением этил-2-{4-[(2S)-1-{N-[(бензилокси)карбонил]-β-аланил}пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (100 мг, 71,4%) в виде бесцветной смолы, который использовали непосредственно без очистки для синтеза Примера 2-66.
ЖХ/МС (Способ I): m/z 541 (М+Н)+ (ES+) при 4,38 и 4,51 мин, не активно в ультрафиолетовом свете.
Остаток может быть очищен с помощью препаративной ВЭЖХ [обращенно-фазовая ВЭЖХ (CHIRALPAK AD-H, С-18, 250×20 мм, 5 мкм, 18,0 мл в мин, градиент от 0% до 50% (в течение15,0 мин), 0,1% аммиака в ацетонитриле и 0,1% аммиака в воде с получением этил-2-{4-[(2S)-1-{N-[(бензилокси)карбонил]-β-аланил}пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-65 Изомера 1 (20 мг, 12,5%) в виде бесцветной смолы, и этил-2-{4-[(2S)-1-{N-[(бензилокси)карбонил]-β-аланил}пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]окган-6-карбоксилата Примера 2-65 Изомера 2 (20 мг, 12,5%) в виде бесцветной смолы. Данные для изомера 2 приведены в Таблице 3.
Смесь диастереомеров этил-2-{4-[(2S)-1-{N-[(бензилокси)карбонил]-(3-аланил}пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (100 мг, 0,185 ммоль) растворили в ТФК (2,0 мл). Полученный раствор перемешивали при 80°C в течение 3 ч, концентрировали в вакууме и остаток очистили с помощью препаративной ВЭЖХ [обращенно-фазовая ВЭЖХ (CHIRALPAK AD-Н, С-18, 250×19 мм, 5 мкм, 13,0 мл в мин, градиент от 0% до 30% (в течение 30,0 мин), 0,1% аммиака в ацетонитриле и 0,1% аммиака в воде с получением этил (S)-2-(4-(1-(3-аминопропаноил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4] октан-6-карбоксилата Примера 2-66 Изомера 1 (2 мг, 2,63%) в виде бесцветной смолы и этил(S)-2-(4-(1-(3-аминопропаноил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-66 Изомера 2 (3 мг, 4,0%) в виде бесцветной смолы. Данные для изомера 2 приведены в Таблице 3.
Путь z
Типовым метод получения пиперидинов реализуется путем алкилирования, как проиллюстрировано на примере получения Примера 2-68, этил-2-{4-[(2S)-1-(2-фторэтил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
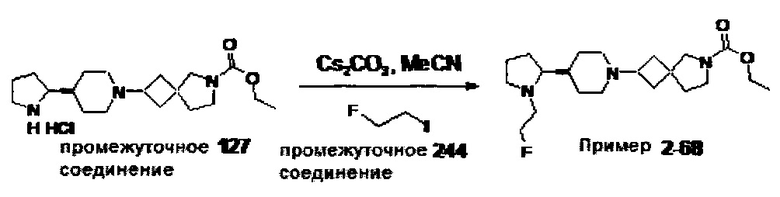
Смесь диастереомеров этил-2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата. HCl (100 мг, 0,27 ммоль) растворили в MeCN (5 мл), прибавили CS2CO3 (290 мг, 0,89 ммоль) с последующим прибавлением 2-иод-1-фторэтан (56 мг, 0,32 ммоль) и перемешивали реакционную смесь при 50°C в течение 16 ч. Реакционную смесь разделили между H2O (70 мл) и этилацетатом (50 мл), водную фазу дополнительно экстрагировали этилацетатом (2×50 мл), органические фазы объединили, сушили (Na2SO4), фильтровали и удалили растворитель в вакууме. Остаток очистили с помощью препаративной ВЭЖХ [обращенно-фазовая ВЭЖХ (CHIRALPAK AD-Н, С-18, 250×19 мм, 5 мкм, 15,0 мл в мин, градиент от 0% до 40% (в течение19,0 мин), 0,1% аммиака в ацетонитриле и 0,1% аммиака в воде] с получением этил(S)-2-(4-(1-(2-фторэтил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-68 Изомера 1 (25 мг, 25%) в виде желтоватой смолы и этил(S)-2-(4-(1-(2-фторэтил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-68 Изомера 2 (20 мг, 20,3%) в виде желтоватой смолы. Данные для изомера 2 приведены в Таблице 3.
Путь аа
Типовый метод получения пиперидинов реализуется путем алкилирования, как проиллюстрировано на примере получения Примера 2-69, этил-2-{4-[(2S)-1-(2,2,2-трифторэтил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
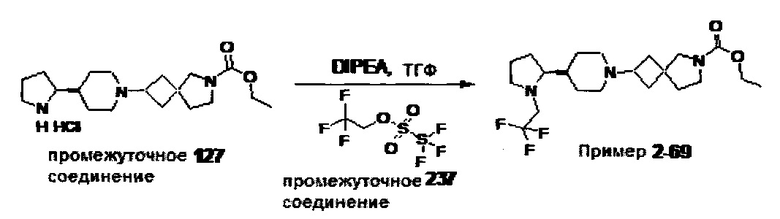
Смесь диастереомеров этил-2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата. HCl (0,100 г, 0,29 ммоль) и DIPEA (0,112 г, 0,87 ммоль) растворили в ТГФ (5 мл) и перемешивали при 60°C в течение 2 ч. По каплям прибавляли 2,2,2-Трифторэтилтрифторметансульфонат (0,067 г, 0,29 ммоль) при 0°C и перемешивали полученную реакционную смесь при комнатной температуре в течение 24 ч. Реакционную смесь разделили между H2O (70 мл) и EtOAc (50 мл), водную фазу дополнительно экстрагировали EtOAc (2×50 мл), органические фазы объединили, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очистили с помощью препаративной ВЭЖХ (X Bridge, С-18, 250×19 мм, 5 мкм, 12 мл в мин, градиент 45% (в течение 0,01 мин), 100% (в течение 30,00 мин), затем 45% (в течение 32,00 мин), 0,1% аммиака в ацетонитрил/вода с получением этил (S)-2-(4-(1-(2,2,2-трифторэтил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-69 Изомера 1 (0,003 г, 2,4%) в виде бесцветной смолы и этил (S)-2-(4-(1-(2,2,2-трифторэтил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-69 Изомера 2 (0,002 мг, 1,6%) в виде бесцветной смолы. Данные для изомера 2 приведены в Таблице 3.
Путь ab
Типовый метод получения пиперидинов реализуется путем алкилирования, как проиллюстрировано на примере получения Примера 2-70, этил 2-{4-[(2S)-1-(3,3,3-трифторпропил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
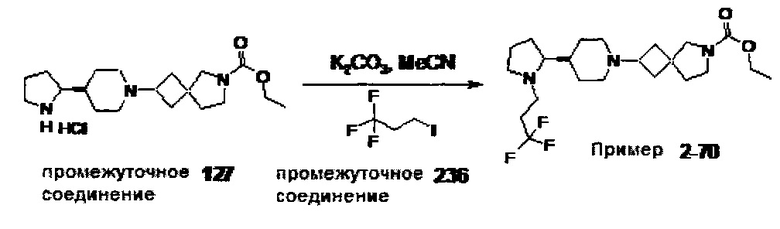
Смесь диастереомеров этил-2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата. HCl (0,100 г, 0,29 ммоль) и K2CO3 (0,123 г, 0,89 ммоль) растворили в MeCN (5 мл) и перемешивали реакционную смесь при 60°C в течение 2 ч. По каплям прибавляли 1,1,1-трифтор-3-иодпропан (0,066 г, 0,29 ммоль) при 0°C и перемешивали полученную смесь при комнатной температуре в течение 8 ч. Реакционную смесь разделили между H2O (70 мл) и EtOAc (50 мл), водную фазу дополнительно экстрагировали EtOAc (2×50 мл), органические фазы объединили, сушили (Na2SO4), фильтровали и растворитель удалили в вакууме. Остаток очистили с помощью препаративной ВЭЖХ (X Bridge, С-18, 250×19 мм, 5 мкм, 15 мл в мин, градиент 60% (в течение 0,01 мин), 100% (в течение 14,01 мин), затем 60% (в течение 23,00 мин), 0,1% аммиака в ацетонитрил/вода с получением этил (S)-2-(4-(1-(3,3,3-трифторпропил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-70 Изомера 1 (0,005 г, 3,9%) в виде бесцветной смолы, и этил-(S)-2-(4-(1-(3,3,3-трифторпропил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-70 Изомера 2 (0,005 мг, 3,9%) в виде бесцветной смолы. Данные для Изомера 1 и Изомера 2 приведены в Таблице 3.
Путь ас
Типовый метод получения пиперидинов реализуется путем алкилирования, как проиллюстрировано на примере получения Примера 2-72, этил (S)-2-(4-(1-(2-метокси-2-оксоэтил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата
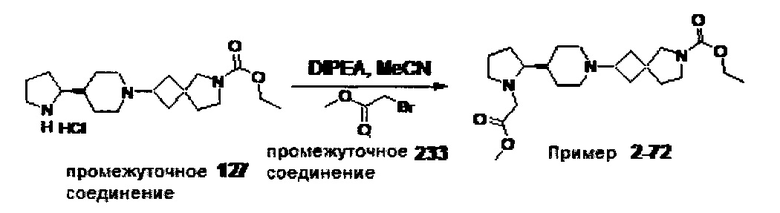
Смесь диастереомеров этил 2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата. HCl (0,100 г, 0,29 ммоль) и DIPEA (0,14 мл, 0,87 ммоль) растворили в MeCN (5 мл) и реакционную смесь перемешивали при комн. темп. в течение 1 ч. По каплям прибавляли метилбромацетат (0,044 г, 0,29 ммоль) и перемешивали полученную реакционную смесь при 100°C в течение 3 ч. Реакционную смесь разделили между H2O (70 мл) и EtOAc (50 мл), водную фазу дополнительно экстрагировали EtOAc (2×50 мл), органические фазы объединили, сушили (Na2SO4), фильтровали и растворитель удалили в вакууме. Остаток очистили с помощью препаративной ВЭЖХ (X Bridge, С-18, 250×19 мм, 5 мкм, 15 мл в мин, градиент 48% (в течение 0,01 мин), 100% (в течение 11,1 мин), 48% (в течение 48,00 мин), 0,1% аммиака в ацетонитрил/вода] с получением этил (S)-2-(4-(1-(2-метокси-2-оксоэтил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-72 Изомера 1 (0,012 г, 9,9%) в виде бесцветной смолы, и этил-(S)-2-(4-(1-(2-метокси-2-оксоэтил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-72 Изомера 2 (0,013 мг, 10,7%) в виде бесцветной смолы. Данные для Изомера 2 приведены в Таблице 3.
Путь ad
Типовый метод получения пиперидинов реализуется путем алкилирования, как проиллюстрировано на примере получения Примера 2-72, этил-2-(4-{(2S)-1-[2-(диметиламино)-2-оксоэтил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата
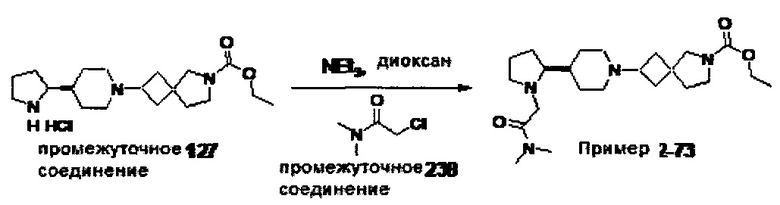
Смесь диастереомеров этил 2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата.HCl (0,100 г, 0,29 ммоль) и NEt3 (0,087 г, 0,85 ммоль) растворили в диоксане (5 мл) и перемешивали реакционную смесь при 60°C в течение 30 мин. По каплям прибавляли 2-хлор-N,N-диметилацетамид (0,036 г, 0,29 ммоль) при 0°C и перемешивали полученную смесь при комнатной температуре в течение 12 ч. Реакционную смесь разделили между H2O (70 мл) и EtOAc (50 мл), водную фазу дополнительно экстрагировали EtOAc (2×50 мл), органические фазы объединили, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очистили с помощью препаративной ВЭЖХ (X Bridge, С-18, 250×19 мм, 5 мкм, 14 мл в мин, градиент 20% (в течение 0,01 мин), 40% (в течение 36,00 мин), 100% (в течение 44,00 мин), затем 20% (в течение 52,00 мин), 0,1% аммиака в ацетонитрил/вода] с получением этил (S)-2-(4-(1-(2-(диметиламино)-2-оксоэтил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]окган-6-карбоксилата Примера 2-72 Изомера 1 (0,002 г, 1,6%) в виде желтой смолы и этил (S)-2-(4-(1-(2-(диметиламино)-2-оксоэтил)пирролидин-2-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-72 Изомера 2 (0,002 мг, 1,6%) в виде желтой смолы. Данные для Изомера 2 приведены в Таблице 3.
Путь ае
Типовым метод получения пиперидинов реализуется путем восстановительного аминирования, Вос-депротекции и образования мочевины/амида, как проиллюстрировано на примере получения Примера 2-76, этил-2-{4-[2-(метилкарбамоил)-2,3-дигидро-1Н-изоиндол-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
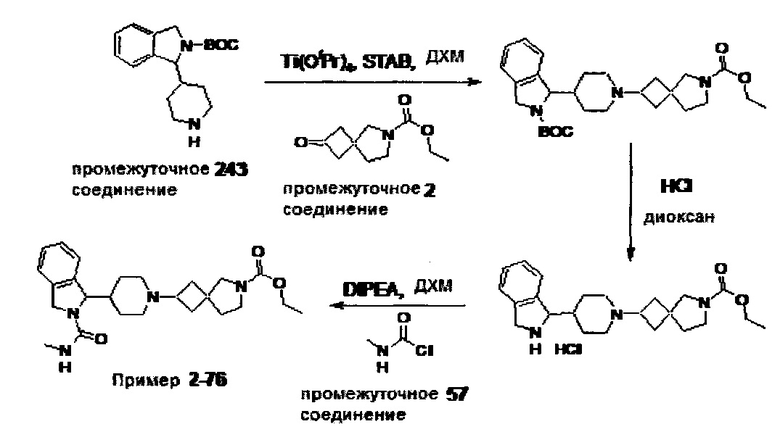
К раствору трет-бутил-1-(пиперидин-4-ил)изоиндолин-2-карбоксилата (135 мг, 0,45 ммоль) и этил-3-оксо-8-азабицикло[3.2.1]октан-8-карбоксилата (88 мг, 0,890 ммоль) в ДХМ (5 мл) прибавили Ti(OiPr)4 (0,40 мл, 1,34 ммоль) при 0°C и перемешивали реакционную смесь в течение 1 ч. К реакционной смеси порциями прибавили Na(OAc)3BH (283 мг, 1,34 ммоль) и перемешивали при 0°C в течение 2 ч. По окончании реакционую смесь погасили водн. насыщ. NaHCO3 и экстрагировали ДХМ (3×30 мл). Органические фазы объединили и промыли насыщенным водным раствором хлорида натрия, сушили (Na2SO4) и концентрировали в вакууме. Остаток очистили с помощью колоночной флеш-хроматографии [нормально-фазовая, силикагель (100-200 меш), градиент от 5% до 10% метанол в ДХМ] с получением трет-бутил 1-(1-(6-(этоксикарбонил)-6-азаспиро[3.4]октан-2-ил)пиперидин-4-ил)изоиндолин-2-карбоксилата (35 мг, 75%) в виде бесцветной жидкости.
МС (ESI +ve): 484
К раствору трет-бутил 1-(1-(6-(этоксикарбонил)-6-азаспиро[3.4]октан-2-ил)пиперидин-4-ил)изоиндолин-2-карбоксилата (290 мг, 0,61 ммоль) в 1,4-диоксане (10 мл) медленно прибавили HCl в диоксане (4 М, 5 мл) при 0°C и перемешивали смесь при комнатной температуре в течение 5 ч. Растворитель выпарили в вакууме. Твердый остаток растерли с диэтиловым эфиром с получением этил 2-(4-(изоиндолин-1-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат гидрохлорида (250 мг, сырой) в виде белого твердого вещества с металлическим оттенком.
МС (ESI +ve): 384
1Н-ЯМР (400 МГц; ДМСО-d6) δ: 1,15 (т, J=6,9 Гц, 3Н), 1,16-1,26 (м, 1Н), 1,70-1,90 (м, 5Н), 1,95-2,28 (м, 5Н), 3,49-3,72 (м, 4Н), 3,60-3,72 (м, 4Н), 3,98-4,15 (м, 2Н), 4,13 (к, J=6,9 Гц, 2Н), 4,45-4,59 (м, 2Н), 7,37-7,49 (м, 5Н), 9,54, 10,19 (2 уш.с., 2Н).
К раствору этил 2-(4-(изоиндолин-1-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат гидрохлорида (240 мг, 0,40 ммоль) в ДХМ (5 мл) прибавили DIPEA (0,43 мл, 2,38 ммоль) при 0°C. К данной реакционной смеси прибавили метилкарбамоилхлорид (67 мг, 0,72 ммоль) и перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь погасили водой (10 мл) и водн. фазу экстрагировали ДХМ (2×20 мл). Органические фазы объединили, сушили (Na2SO4) и концентрировали в вакууме. Остаток очистили с помощью колоночной флеш-хроматографии [нормально-фазовая, силикагель (100-200 меш), градиент от 5% до 10% метанол в ДХМ] с получением этил 2-(4-(2-(метилкарбамоил)-изоиндолин-1-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата в виде смеси диастереомеров (130 мг, 48%) в виде смолообразной жидкости.
ЖХ/МС (Способ М): m/z 441 (M+H)+ (ES+), при 1,97 и 1,99 мин, активны в ультрафиолетовом свете.
1H-ЯМР (400 МГц; ДМСО-d6): δ: 1,15 (т, J=6,9 Гц, 3Н), 1,16-1,26 (м, 4 Н), 1,49-1,90 (м, 5Н), 1,91-2,01 (м, 2Н), 2,62 (д, J=3,9 Гц, 3 Н), 2,70-2,90 (м, 2Н), 3,09-3,25 (м, 4Н), 3,97 (к, J=6,8 Гц, 2Н), 4,45-4,59 (м, 2Н), 5,01-5,09 (м, 1Н), 6,27 (уш. с., 1Н), 7,22-7,32 (м, 4Н).
Разделение диастереомеров осуществляли с использованием препаративной ВЭЖХ (73,0 мг введено, полупрепаративная ВЭЖХ система Gilson, включая двухпоршневые насосы 331 и 332, детектор на диодной матрице 171 и систему дозирования жидкостей GX-271, растворители: водный = вода + 0,2% аммиака (28% раствор аммиака) и органический = ацетонитрил, градиент: 20-50% органического в водном, скорость потока: 30 мл/мин, колонка: Gemini-NX, С18.5 мк, 100×30 мм), давая этил 2-(4-(2-(метилкарбамоил)-изоиндолин-1-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат Пример 2-76 Изомер 1 (8,99 мг, 12,3%) в виде бесцветной смолы и этил 2-(4-(2-(метилкарбамоил)-изоиндолин-1-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат Пример 2-76 Изомер 2 (10,9 мг, 14,9%) в виде бесцветной смолы. Данные для Изомера 1 и Изомера 2 приведены в Таблице 3.
Путь af
Типовый метод арилирования пирролидина, как проиллюстрировано на примере получения Примера 2-77, этил 2-{4-[(2S)-1-фенилпирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
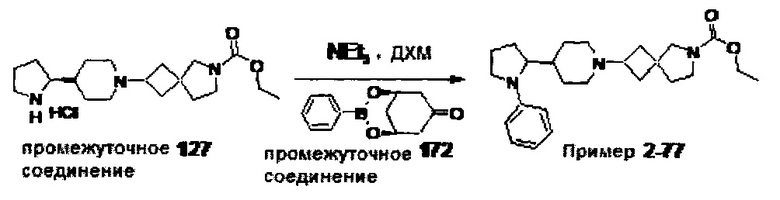
Смесь диастереомеров этил 2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата.HCl (0,100 г, 0,27 ммоль) растворили в ДХМ (5 мл) и прибавили триэтиламин (54 мг, 0,54 ммоль) с последующим прибавлением (1R,5S)-3-фенил-2,4-диокса-3-борабицикло[3.3.1]нонан-7-она (см. J. Luo et al. Tetrahedron Letters 54 (2013), 4505-4508, 58 мг, 0,53 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, затем разделили между H2O (70 мл) и ДХМ (100 мл). Водную фазу дополнительно экстрагировали ДХМ (2×50 мл) и органические фазы объединили, сушили (Na2SO4), растворитель удалили и остаток очистили с помощью препаративной ВЭЖХ [обращенно-фазовая ВЭЖХ (CHIRALPAK AD-H, С-18, 250×19 мм, 5 мкм, 15,0 мл в мин, градиент от 0% до 30% (в течение 21,0 мин), 0,1% аммиака в ацетонитриле и 0,1% аммиака в воде с получением этил 2-{4-[(2S)-1-фенилпирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-77 Изомера 1 (10 мг, 13%) в виде смолы, и этил 2-{4-[(2S)-1-фенилпирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-77 Изомера 2 (10 мг, 13%) в виде смолы. Данные для изомера 2 приведены в Таблице 3.
Путь ag
Типовый метод арилирования гетероциклами пирролидин-содержащих соединений, используя карбонат цезия и иодид меди в ДМФА, как проиллюстрировано на примере получения Примера 2-78, метил 2-{4-[(2S)-1-(пиридин-2-ил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
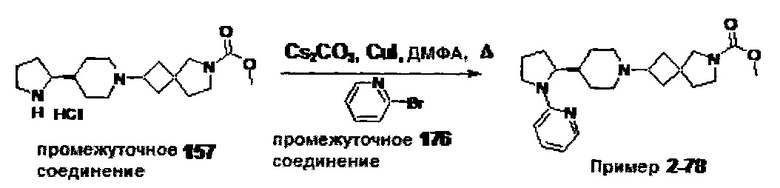
Смесь диастереомеров метил 2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата. HCl (0,120 г, 0,37 ммоль), Cs2CO3 (0,361 г, 1,1 ммоль) и CuI (0,105 г, 0,50 ммоль) растворили в ДМФА (5 мл) и перемешивали при комн. темп. в течение 30 мин. Затем прибавили 2-бромпиридин (0,058 г, 0,37 ммоль) и перемешивали полученную смесь при 100°C в течение 18 ч. Реакционную смесь разделили между H2O (70 мл) и EtOAc (50 мл). Водную фазу дополнительно экстрагировали EtOAc (2×50 мл). Органические фазы объединили, сушили (Na2SO4), растворитель удалили с помощью концентрирования и остаток очистили с помощью препаративной ВЭЖХ (X Bridge, С-18, 150×19 мм, 5 мкм, 13 мл в мин, градиент от 40% до 100% (в течение 20 мин), 0,1% аммиак в ацетонитрил/вода] с получением метил 2-{4-[(2S)-1-(пиридин-2-ил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-78 Изомера 1 (0,011 г, 2%) в виде смолы, и метил 2-{4-[(2S)-1-(пиридин-2-ил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-78 Изомера 2 (0,09 мг, 2%) в виде смолы. Данные для изомера 2 приведены в Таблице 3.
Путь ah
Типовый метод арилирования гетероциклами пирролидин-содержащих соединений, используя карбонат натрия в этаноле, как проиллюстрировано на примере получения Примера 2-81, этил 2-{4-[(2S)-1-(пиримидин-2-ил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
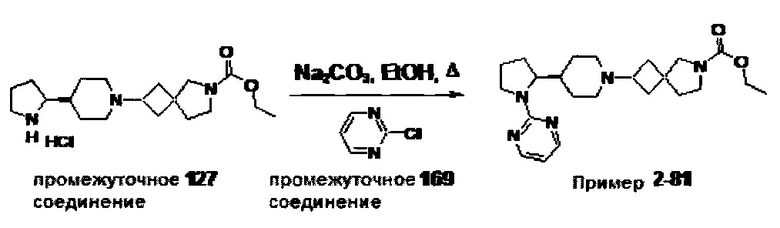
Смесь диастереомеров этил 2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата. HCl (0,100 г, 0,29 ммоль) и Na2CO3 (0,092 г, 0,87 ммоль) растворили в этаноле (10 мл) и перемешивали при комн. темп. в течение 30 мин. Затем прибавили 2-хлорпиримидин (0,034 г, 0,29 ммоль) при 0°C. Полученную реакционную смесь перемешивали при 80°C в течение 6 ч. Реакционную смесь концентрировали и прибавили дихлорметан. Смесь отфильтровали и фильтрат концентрировали и очистили с помощью препаративной ВЭЖХ (X Bridge, С-18, 150×19 мм, 5 мкм, 15 мл в мин, градиент 38% (в течение 0,01 мин), 42% (в течение 15,00 мин), 100% (в течение 19,00 мин), затем 38% (в течение 23,00 мин), 0,1% аммиак в ацетонитрил/вода] с получением этил-2-{4-[(2S)-1-(пиримидин-2-ил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-81 Изомера 1 (0,031 г, 25%) в виде смолы, и этил 2-{4-[(2S)-1-(пиримидин-2-ил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-81 Изомера 2 (0,017 мг, 14%) в виде смолы. Данные для изомера 2 приведены в Таблице 3.
Путь ai
Типовым метод арилирования гетероциклами пирролидин-содержащих соединений, используя карбонат цезия в ДМФА, как проиллюстрировано на примере получения Примера 2-82, этил-2-{4-[(2S)-1-(1,3-тиазол-2-ил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
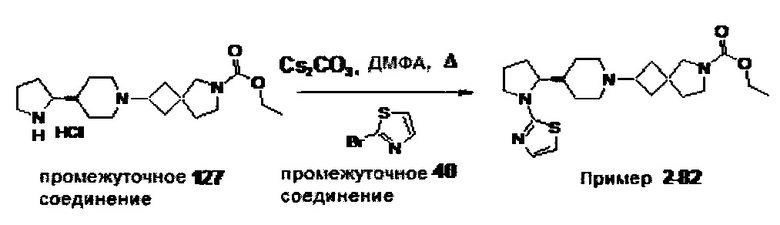
Смесь диастереомеров этил-2-{4-[(2S)-пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата. HCl, промежуточного соединения 127, (100 мг, 0,27 ммоль) растворили в ДМФА (5 мл) и прибавили CS2CO3 (260 мг, 0,81 ммоль), с последующим прибавлением 2-броитиазола (58 мг, 0,29 ммоль). Реакционную смесь перемешивали при 90°C в течение 16 ч. Реакционную смесь разделили между H2O (70 мл) и EtOAc (50 мл). Водную фазу дополнительно экстрагировали EtOAc (2×50 мл). Органические фазы объединили, сушили (Na2SO4), растворитель удалили путем концентрирования и остаток очистили с помощью препаративной ВЭЖХ [обращенно-фазовая ВЭЖХ (CHIRALPAK AD-H, С-18, 250×19 мм, 5 мкм, 15,0 мл в мин, градиент от 0% до 30% (в течение 21,0 мин), 0,1% аммиака в ацетонитриле и 0,1% аммиака в воде с получением этил-2-{4-[(2S)-1-(1,3-тиазол-2-ил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-82 Изомера 1 (20 мг, 18%) в виде бесцветной смолы, и этил 2-{4-[(2S)-1-(1,3-тиазол-2-ил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-82 Изомера 2 (6 мг, 6%) в виде бесцветной смолы. Данные для Изомера 1 и Изомера 2 приведены в Таблице 3.
Путь aj
Типовый метод получения пиперидинов реализуется путем депротекции и восстановительных аминирований, как проиллюстрировано на примере получения Примера 2-84, этил 2-{4-[(2R)-2-(метоксикарбонил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
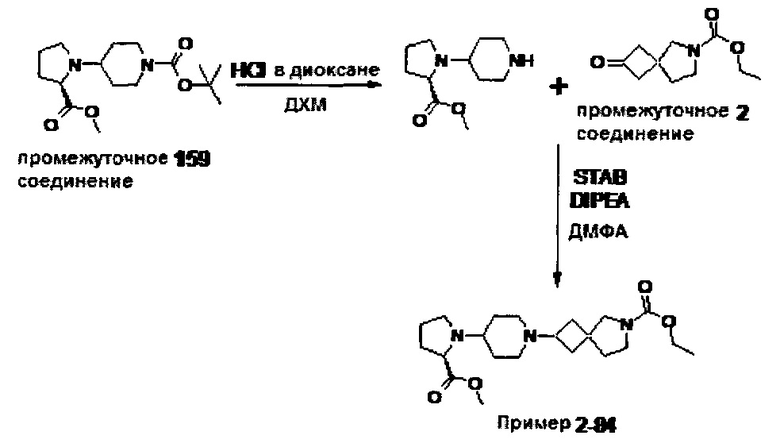
трет-Бутил 4-[(2R)-2-(метоксикарбонил)пирролидин-1-ил]пиперидин-1-карбоксилат (0,396 г, 1,26 ммоль) растворили в ДХМ (1 мл), с последующим прибавлением по каплям HCl в диоксане (3 мл, 4,0 М р-р.). Полученную реакционную смесь перемешивали при комн. темп. в течение 1 ч, растворители удалили в вакууме и остаток использовали в следующей стадии без дополнительной очистки.
Метил 1-пиперидин-4-ил-D-пролинат. HCl (0,358 г, 1,26 ммоль) и этил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (0,266 г, 1,26 ммоль) растворили в ДМФА (4 мл) при комн. темп. и прибавили DIPEA (0,435 мл, 2,510 ммоль). Реакционную смесь перемешивали при комн. темп. в течение 3 ч. Затем прибавили STAB (0,533 г, 2,518 ммоль) и перемешивали реакционную смесь в атмосфере азота при комн. темп. в течение ночи. Растворители удалили в вакууме и препаративную ВЭЖХ использовали для разделения диастереомеров, используя колонку Phenomenex Gemini-NX С18, 100×30 мм, элюируя от 25 до 45% MeCN/0,2% аммиака в H2O (об.) при 18 мл/мин и собирая фракции при 210 нм с получением Примера 2-84 Изомера 1 этил 2-{4-[(2R)-2-(метоксикарбонил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (18,4 мг, 4%) в виде бесцветного масла и Примера 2-84 Изомера 2, этил 2-{4-[(2R)-2-(метоксикарбонил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (13,9 мг, 3%) в виде бесцветного масла. Данные для Изомера 2 приведены в Таблице 3.
Путь ak
Типовый метод получения пиперидинов реализуется путем восстановительного аминирования, как проиллюстрировано на примере получения Примера 2-85, этил 2-{4-[(2S)-2-(метилкарбамоил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
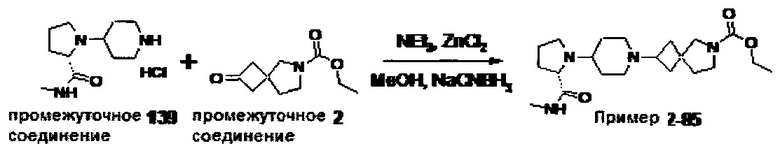
(S)-N-метил-1-(пиперидин-4-ил)пирролидин-2-карбоксамид дигидрохлорид (0,2 г, 0,94 ммоль), NEt3 (0,75 мл, 5,0 ммоль), 6-(этоксикарбонил)-2-оксо-6-азаспиро[3.4]октан-8-илий (0,188 г, 0,93 ммоль) и ZnCl2 (30 мг, 0,02 ммоль) растворили в МеОН (15,00 мл) в атмосфере азота и перемешивали в течение 1 ч при 50-60°C. Порциями прибавили NaCNBH3 (0,069 г, 1,0 ммоль) при 0-10°C и перемешивали реакционную смесь в течение 3 ч при комнатной температуре. Реакционную смесь разделили между EtOAc (2×50 мл) и водой (30 мл), органические фазы объединили, сушили (Na2SO4), фильтровали и растворитель удалили в вакууме и сырой продукт очистили с помощью препаративной ВЭЖХ [обращенно-фазовая ВЭЖХ (X-Bridge PREP С18, 250×19 мм, 5 мкм, 15 мл в мин, градиент от 30% до 100% (в течение 22 мин), затем 100% (2 мин), 0,1% NH3 в ацетонитриле с получением Примера 2-85 Изомера 1, этил (S)-2-(4-(2-(метилкарбамоил)пирролидин-1-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата (0,09 г, 24,32%) в виде белого твердого вещества и Примера 2-85 Изомера 2, этил (S)-2-(4-(2-(метилкарбамоил)пирролидин-1-ил)пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилата (0,089 г, 24,10%) в виде белого твердого вещества. Данные для изомера 2 приведены в Таблице 3.
Путь ap
Типовый метод получения пиперидинов реализуется путем депротекции и восстановительного аминирования триацетоксиборогидридом натрия, как проиллюстрировано на примере получения Примера 2-87, этил 2-{4-[(2R)-4,4-дифтор-2-(метоксикарбонил)пирролидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
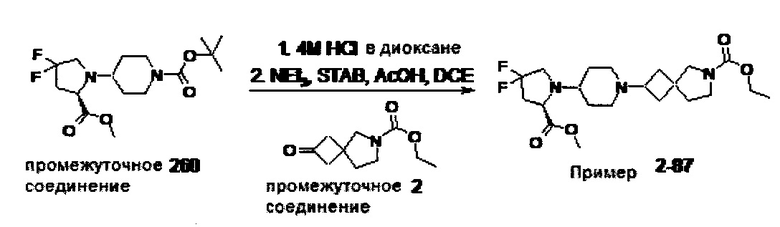
трет-бутил 4-[(2R)-4,4-дифтор-2-(метоксикарбонил)пирролидин-1-ил]пиперидин-1-карбоксилат (0,36 г, 1,03 ммоль) растворили в 4,0 М HCl в диоксане (10 мл) и перемешивали реакционную смесь при комн. темп. в течение 6 ч. Растворители удалили в вакууме и остаток использовали в следующей стадии без дополнительной очистки. Сырую реакционную смесь и этил 2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (0,243 г, 1,233 ммоль) растворили в DCE (10 мл) при комн. темп. и прибавили Et3N (0,249 г, 2,47 ммоль). Реакционную смесь перемешивали при 50С в атмосфере азота в течение 2 ч. Реакционную смесь охладили до комн. темп., прибавили ледяную уксусную кислоту (0,114 г, 1,90 ммоль) и STAB (0,784 г, 3,69 ммоль) и перемешивали реакционную смесь в течение ночи при 50°C в атмосфере азота. К охлажденной реакционной смеси прибавили воду (2 мл) и удалили растворители в вакууме. Остаток разделили между ДХМ (15 мл) и насыщ. NaHCO3 (водн.) (15 мл), водную фазу промыли ДХМ (2×15 мл). Органические фазы объединили и сушили, пропуская через картридж фазного сепаратора Biotage. Растворители удалили в вакууме, и остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 10 г 40-63 мкм, 60 , 12 мл в мин, градиент от 1% до 10% МеОН/ДХМ]). Остаток дополнительно очистили с помощью препаративной обратно-фазовой ВЭЖХ (колонка Phenomenex Gemini-NX 5 мкм С18 110А Axia, 100×30 мм, элюируя от 20 до 50% MeCN/Растворитель B в течение 14,4 мин при 30 мл/мин [где растворитель B представляет собой 0,2% (28% NH3/H2O) в H2O] и собирая фракции при 210 нм) с получением этил 2-{4-[(2R)-4,4-дифтор-2-(метоксикарбонил)пирролидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-87 Изомера 1 (0,020 г, 4,5%) и этил 2-{4-[(2R)-4,4-дифтор-2-(метоксикарбонил)пирролидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-87 Изомера 2 (0,020 г, 4,5%). Данные для изомера 2 приведены в Таблице 3.
, 12 мл в мин, градиент от 1% до 10% МеОН/ДХМ]). Остаток дополнительно очистили с помощью препаративной обратно-фазовой ВЭЖХ (колонка Phenomenex Gemini-NX 5 мкм С18 110А Axia, 100×30 мм, элюируя от 20 до 50% MeCN/Растворитель B в течение 14,4 мин при 30 мл/мин [где растворитель B представляет собой 0,2% (28% NH3/H2O) в H2O] и собирая фракции при 210 нм) с получением этил 2-{4-[(2R)-4,4-дифтор-2-(метоксикарбонил)пирролидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-87 Изомера 1 (0,020 г, 4,5%) и этил 2-{4-[(2R)-4,4-дифтор-2-(метоксикарбонил)пирролидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-87 Изомера 2 (0,020 г, 4,5%). Данные для изомера 2 приведены в Таблице 3.
Путь am
Типовый метод получения пиперидинов реализуется путем депротекции и образования амида, как проиллюстрировано на примере получения Примера 2-88, этил-2-{4-[(2R)-4,4-дифтор-2-(метилкарбамоил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата

Смесь диастереомеров этил 2-{4-[(2R)-4,4-дифтор-2-(метоксикарбонил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-87, (0,400 г, 0,932 ммоль) растворили в ТГФ (8 мл) и прибавили 1 М LiOH (водн.) (1,9 мл), реакционную смесь перемешивали при комн. темп. в течение ночи. Реакционную смесь нейтрализовали, используя 2,0 М раствор HCl и удалили растворители в вакууме. Остаток подвергли азеотропной перегонке с толуолом с получением 1-{1-[6-(этоксикарбонил)-6-азаспиро[3.4]окт-2-ил]пиперидин-4-ил}-4,4-дифтор-D-пролин (0,440 г, 100%) в виде желтого стеклообразного вещества.
ЖХ/МС (Способ С): m/z 416 (М+Н)+ (ES+) при 0,71 min, не активно в ультрафиолетовом свете
1-{1-[6-(этоксикарбонил)-6-азаспиро[3.4]окт-2-ил]пиперидин-4-ил}-4,4-дифтор-D-пролин (0,193 г, 0,466 ммоль) растворили в безводном ДМФА (5 мл) и прибавили HATU (0,533 г, 1,398 ммоль), 2,0 М раствор метиламина в ТГФ (2,3 мл, 2,33 ммоль) и DIPEA (0,301 г, 2,33 ммоль), реакционную смесь перемешивали в течение ночи при комн. темп. Растворители удалили в вакууме, и остаток разделили между ДХМ (20 мл) и насыщ. NaHCO3 (водн.) (20 мл), водную фазу экстрагировали ДХМ (2×15 мл). Органические фазы объединили, промыли насыщенным водным раствором хлорида натрия (20 мл) и сушили, пропуская через картридж фазного сепаратора Biotage. Растворители удалили в вакууме, и остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 10 г 40-63 мкм, 60, 12 мл в мин, градиент от 0% до 10% МеОН / ДХМ]). Остаток дополнительно очистили с помощью препаративной обратно-фазовой ВЭЖХ (колонка Phenomenex Gemini-NX 5 мкм С18 110А Axia, 100×30 мм, элюируя от 20 до 50% MeCN/Растворитель B в течение 14,4 мин при 30 мл/мин [где растворитель B представляет собой 0,2% (28% NH3/H2O) в H2O] и собирая фракции при 210 нм) с получением этил-2-{4-[(2R)-4,4-дифтор-2-(метилкарбамоил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-88 Изомера 1, (0,038 г, 18%) в виде бесцветного масла и этил-2-{4-[(2R)-4,4-дифтор-2-(метилкарбамоил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-88 Изомера 2, (0,037 г, 18%) в виде бесцветного масла. Данные для изомера 2 приведены в Таблице 3.
Путь an
Типовым метод получения пиперидинов реализуется путем образования амида, как проиллюстрировано на примере получения Примера 2-90, этил-2-{4-[(2R)-2-карбамоил-4,4-дифторпирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
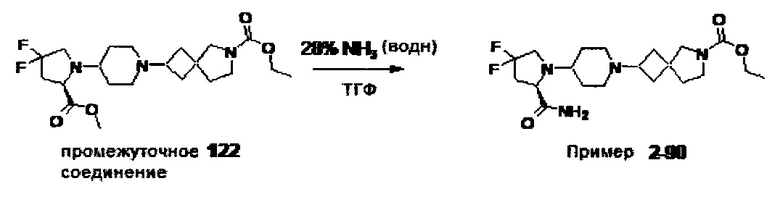
Смесь диастереомеров этил 2-{4-[(2R)-2-метоксикарбонил-4,4-дифторпирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (456 мг, 1,062 ммоль) растворили в ТГФ (1 мл) и 28% раствор NH3 (9 мл) при 60°C и перемешивали реакционную смесь в течение 18 ч. Реакционную смесь нейтрализовали с помощью 1 М HCl(водн.) разбавленным ДХМ (25 мл) и промыли H2O (2×25 мл), промыли объединенные водные фазы ДХМ (25 мл), объединенные органические фазы промыли насыщенным водным раствором хлорида натрия (25 мл) и пропустили через фазный сепаратор Biotage. Летучие компоненты удалили в вакууме с получением оранжевого масла (0,290 г, 65%). Препаративную ВЭЖХ использовали для разделения диастереомеров, используя колонку Phenomenex Gemini-NX С18, 100×30 мм, элюируя от 20 до 45% MeCN/0,2% аммиака в H2O (об.) при 18 мл/мин и собирая фракции при 210 нм с получением Примера 2-90 Изомера 1 этил-2-{4-[(2R)-2-карбамоил-4,4-дифторпирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (16,8 мг, 4%) в виде бесцветного масла и Примера 2-90 Изомера 2, этил-2-{4-[(2R)-2-карбамоил-4,4-дифторпирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (12,4 мг, 3%) в виде бесцветного масла. Данные для Изомера 2 приведены в Таблице 3.
Путь ао
Типовый метод получения пиперидинов реализуется путем гидролиза и образования амида, как проиллюстрировано на примере получения Примера 2-91, этил-2-{4-[(2R)-4,4-дифтор-2-(метоксикарбамоил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
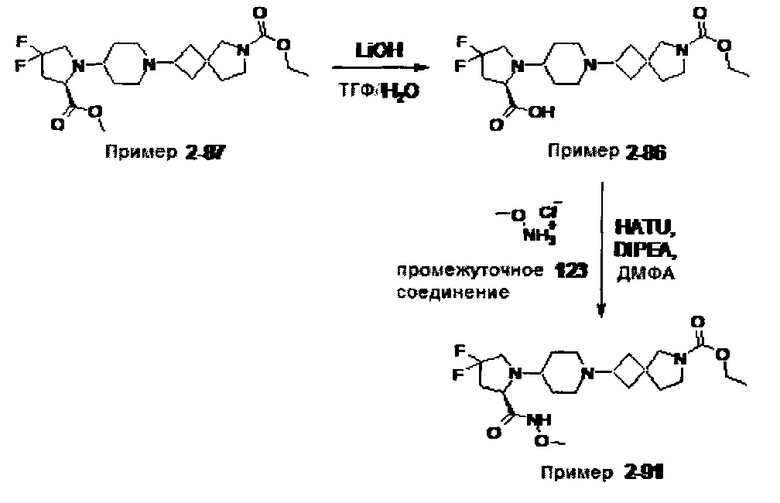
Смесь диастереомеров этил 2-{4-[(2R)-2-метоксикарбонил-4,4-дифторпирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (230 мг, 0,536 ммоль) растворили в ТГФ (6,5 мл) и 1,0 М раствора LiOH (1,1 мл, 1,1 ммоль) при комн. темп. и перемешивали реакционную смесь в течение 18 ч. Летучие компоненты удалили в вакууме и соединение использовали без дополнительной очистки. 1-{1-[6-(этоксикарбонил)-6-азаспиро[3.4]окт-2-ил]пиперидин-4-ил}-4,4-дифтор-D-пролин (125 мг, 0,300 ммоль) растворили в ДМФА (1 мл) с последующим прибавлением HATU (228 мг, 0,60 ммоль) и DIPEA (0,260 мл, 1,50 ммоль). Реакционную смесь перемешивали при 0°C в течение 10 минут с последующим прибавлением метоксиамин гидрохлорида (25 мг, 0,30 ммоль) и перемешивали полученную реакционную смесь при комн. темп. в течение 18 ч. Реакционную смесь концентрировали в вакууме, затем разделили между насыщенным NaHCO3(водн.) (50 мл) и ДХМ (50 мл), водную фазу дополнительно экстрагировали ДХМ (2×50 мл), органические фазы объединили, промыли насыщенным водным раствором хлорида натрия (50 мл) и пропустили через фазный сепаратор Biotage. Летучие компоненты удалили в вакууме с получением оранжевого масла (0,102 г, 77%). Препаративную ВЭЖХ использовали для разделения диастереомеров, используя колонку Phenomenex Gemini-NX С18, 100×30 мм, элюируя от 20 до 50% MeCN/0,2% аммиака в H2O (об.) при 18 мл/мин и собирая фракции при 210 нм с получением Примера 2-91 Изомера 1 этил-2-{4-[(2R)-4,4-дифтор-2-(метоксикарбамоил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (12,2 мг, 4%) в виде бесцветного масла и Примера 2-91 Изомера 2, этил 2-{4-[(2R)-4,4-дифтор-2-(метоксикарбамоил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (7,2 мг, 3%) в виде бесцветного масла. Данные для Изомера 2 приведены в Таблице 3.
Путь ар
Типовый метод получения пиперидинов реализуется путем депротекции и восстановительного аминирования триацетоксиборогидридом натрия, как проиллюстрировано на примере получения Примера 2-111, этил 2-[4-(2-оксопирролидин-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата
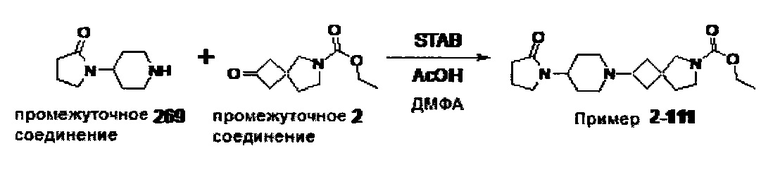
1-Пиперидин-4-ил-пирролидин-2-он (0,200 г, 1,19 ммоль) и этил 2-оксо-6-азаспиро [3.4]октан-6-карбоксилат (0,212 г, 1,14 ммоль) растворили в ДМФА (6 мл) при комн. темп., реакционную смесь перемешивали при 40°C в атмосфере азота в течение 3 ч. Реакционную смесь охладили до комн. темп., прибавили STAB (0,630 г, 2,97 ммоль) и ледяную уксусную кислоту (0,071 г, 1,189 ммоль) и перемешивали реакционную смесь в течение ночи при 40°C в атмосфере азота. К охлажденной реакционной смеси прибавили воду (2 мл) и удалили растворители в вакууме. Остаток разделили между ДХМ (15 мл) и насыщ. NaHCO3 (водн.) (15 мл), водную фазу промыли ДХМ (2×15 мл). Органические фазы объединили и сушили, пропуская через картридж фазного сепаратора Biotage. Растворители удалили в вакууме и остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 10 г 40-63 мкм, 60, 12 мл в мин, градиент от 1% до 10% МеОН/ДХМ]). Остаток дополнительно очистили с помощью препаративной обратно-фазовой ВЭЖХ (колонка Phenomenex Gemini-NX 5 мкм С18 110А Axia, 100×30 мм, элюируя от 20 до 35% MeCN/Растворитель В в течение 14,4 мин при 30 мл/мин [где растворитель В представляет собой 0,2% (28% NH3/H2O) в H2O] и собирая фракции при 210 нм) с получением этил 2-[4-(2-оксопирролидин-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-111 Изомера 1, (0,008 г, 2%) в виде бесцветного масла и этил 2-[4-(2-оксопирролидин-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-111 Изомера 2, (0,009 г, 2%) в виде бесцветного масла. Данные для Изомера 2 приведены в Таблице 3.
Если использованный в восстановительном аминировании амин содержит вторую аминогруппу, которая защищена (стандартными амино-защитными группами, такими как ВОС или Cbz). Тогда стандартные способы депротекции могут быть использованы для удаления данных защитных групп после того, как выполнено восстановительное аминирование, чтобы позволить дополнительную функционализацию целевого соединения.
Путь aq
Типовым метод получения пиперидинов реализуется путем восстановительного аминирования, Boc депротекции и образования мочевины, как проиллюстрировано на примере получения Примера 2-124, этил 2-[4-(2-оксо-1,2-дигидропиридин-4-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата
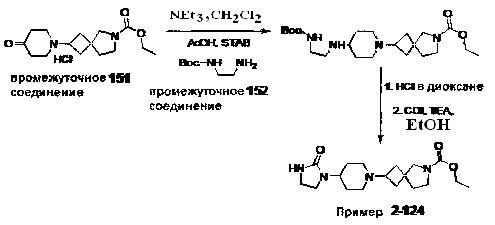
Этил 2-(4-оксопиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат гидрохлорид (0,316 г, 1,00 ммоль) и трет-бутил-(2-аминоэтил)карбамат (0,320 г, 2,00 ммоль) растворили в ДХМ (10 мл) под N2 при комн. темп., NEt3 (0,15 мл, 1,10 ммоль) прибавили и реакционную смесь перемешивали при комн. темп. в течение 0,5 ч. Прибавили уксусную кислоту (0,13 мл, 2,20 ммоль), перемешивали реакционную смесь при комн. темп. в течение 2 ч, прибавили STAB (0,530 г, 2,50 ммоль) и перемешивали реакционную смесь в течение ночи. Реакционную смесь погасили путем прибавления NaHCO3 (насыщ. водн.) (30 мл), экстрагировали ДХМ (4×25 мл), объединенные фазы ДХМ пропустили через фазный сепаратор Biotage и концентрировали в вакууме с получением сырого этил 2-[4-({2-[(трет-бутоксикарбонил)амино]этил}амино)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата в виде смеси диастереомеров, которую использовали без какой-либо дополнительной очистки.
ЖХ/МС (Способ D): m/z 425 (М+Н)+ (ES+), при 1,30 и 1,35 мин, не активно в ультрафиолетовом свете.
Сырой этил 2-[4-({2-[(трет-бутоксикарбонил)амино]этил}амино)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат (0,424 г, 1,00 ммоль) растворили в CH2Cl2 (10 мл), прибавили 4 М хлороводород в диоксане (1,25 мл, 5,0 ммоль) и реакционную смесь перемешивали при комн. темп. в течение ночи. Летучие компоненты удалили в вакууме, остаток растворили в EtOH (10 мл), прибавили NEt3 (1,40 мл, 10,0 ммоль) и CDI (0,244 г, 1,50 ммоль) и нагрели смесь до кипения и выдержали в течение ночи. Растворитель удалили в вакууме, остаток разделили между CH2Cl2 (20 мл) и водой (20 мл), и водную фазу экстрагировали CH2Cl2 (4×20 мл). Объединенные органические фазы концентрировали в вакууме с получением сырого этил-2-[4-(2-оксоимидазолидин-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата в виде смеси диастереомеров. Препаративную ВЭЖХ использовали для разделения диастереомеров, используя колонку Phenomenex Gemini-N С18, 150×21 мм, элюируя от 25 до 45% MeCN в 0,2% NH3/H2O при 18 мл/мин и собирая фракции при 210 нм с получением этил 2-[4-(2-оксо-1,2-дигидропиридин-4-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-124 Изомера 1, (0,008 г, 2,3%) в виде бесцветного твердого вещества и этил-2-[4-(2-оксо-1,2-дигидропиридин-4-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата Примера 2-124 Изомера 2, (0,008 г, 2,3%) в виде бесцветного твердого вещества. Данные для Изомера 2 приведены в Таблице 3.
Путь ar
Типовый метод получения пиперидинов реализуется путем восстановления сложного эфира, как проиллюстрировано на примере получения Примера 2-136, этил-2-{4-[(2R,4R)-4-фтор-2-(гидроксиметил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата
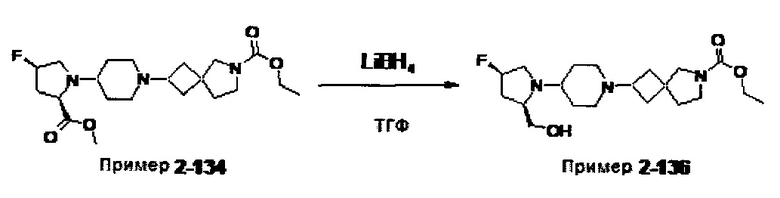
Смесь диастереомеров этил-2-{4-[(2R,4R)-4-фтор-2-(метоксикарбонил) пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата (0,140 г, 0,341 ммоль) растворили в безводном ТГФ (10 мл) и охладили до 0°C в атмосфере азота. К реакционной смеси по каплям прибавляли 2,0 М раствор борогидрида лития в ТГФ (1,02 мл, 1,023 ммоль) и затем реакционную смесь оставили нагреваться до комн. темп. в течение ночи. Реакционную смесь погасили насыщ. NaHCO3 (водн.) (15 мл) и затем экстрагировали EtOAc (2×15 мл), органические фазы объединили и сушили (MgSO4). Растворители удалили в вакууме, и остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 10 г 40-63 мкм, 60, 12 мл в мин, градиент от 0% до 10% МеОН / ДХМ]). Остаток дополнительно очистили с помощью препаративной обратно-фазовой ВЭЖХ (колонка Phenomenex Gemini-NX 5 мкм С18 110A Axia, 100×30 мм, элюируя от 20 до 50% MeCN/Растворитель B в течение 14,4 мин при 30 мл/мин [где растворитель B представляет собой 0,2% (28% NH3/H2O) в H2O] и собирая фракции при 210 нм) с получением этил-2-{4-[(2R,4R)-4-фтор-2-(гидроксиметил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилата, Примера 2-136 Изомера 1, (2,99 мг, 0,23%) в виде белого твердого вещества и этил 2-{4-[(2R,4R)-4-фтор-2-(гидроксиметил)пирролидин-1-илъпиперидин-1-ил}-6-азоспиро[3.4]октан-6-карбоксилата, Примера 2-136 Изомера 2, (3,10 мг, 0,24%) в виде белого твердого вещества. Данные для изомера 2 приведены в Таблице 3.
Путь as
Типовый метод получения пиперидинов реализуется путем восстановительного аминирования триацетоксиборогидридом натрия в ДМФА, как проиллюстрировано на примере получения Примера 3-4, этил 2-[4-(3-гидроксипиридин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата
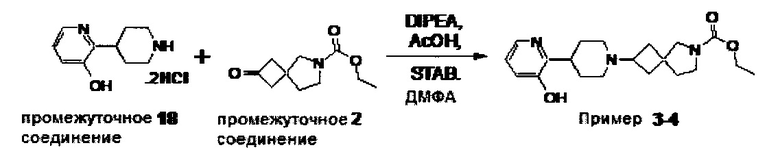
2-(Пиперидин-4-ил)пиридин-3-ола дигидрохлорид (0,20 г, 0,8 ммоль) и этил 2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (0,157 г, 0,8 ммоль) смешали в ДМФА (8 мл) при комн. темп.. Прибавили DIPEA (0,28 мл, 1,6 ммоль) и АсОН (0,07 мл, 1,2 ммоль) с последующим прибавлением STAB (0,34 г, 1,6 ммоль). Реакционную смесь перемешивали в атмосфере азота при комн. темп. в течение ночи, затем погасили путем прибавления небольшого количества МеОН и концентрировали в вакууме для удаления всех растворителей. Остаток растворили в смеси МеОН и ДХМ и концентрировали в вакууме на силикагеле для флеш-хроматографии (~10 мл). Полученный порошок очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 25 г, 40-63 мкм, 60, 30 мл в мин, градиент от 0% до 15% Растворителя A в ДХМ в течение 15 объемов колонки, где растворитель A представляет собой 10% (7 М NH3/MeOH) в МеОН]) с получением сырой смеси диастереомеров (0,258 г). Данную смесь растворили в МеОН, прибавили небольшое количество 28% NH3/H2O (~0,1 мл), и очистили раствор с помощью препаративной обратно-фазовой ВЭЖХ, используя колонку Phenomenex Gemini-NX 5 мкм С18 110А Axia, 100×30 мм, элюируя от 15 до 25% MeCN/Растворитель B в течение 14,4 мин при 30 мл/мин [где растворитель B представляет собой 0,2% (28% NH3/H2O) в H2O] и собирая фракции при 230 нм с получением этил 2-[4-(3-гидроксипиридин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата, Примера 3-4 Изомера 1, (0,034 г, 12%) и этил 2-[4-(3-гидроксипиридин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата, Примера 3-4 Изомера 2, (0,052 г, 18%). Данные для изомера 2 приведены в Таблице 3.
Путь at
Типовый метод получения пиперидинов реализуется путем восстановительного аминирования триацетоксиборогидридом натрия, как проиллюстрировано на примере получения Примера 3-10, этил-2-[4-циано-(пиридин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата
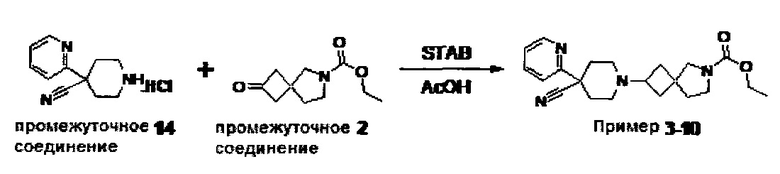
4-(Пиридин-2-ил)пиперидин-4-карбонитрила гидрохлорид (0,187 г, 1,0 ммоль) и этил 2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (0,197 г, 1,0 ммоль) растворили в ДХМ (10 мл) под N2 при комн. темп. и прибавили NEt3 (0,15 мл, 1,1 ммоль). Реакционную смесь перемешивали при комн. темп. в течение 1 ч, прибавили уксусную кислоту (0,13 мл, 2,2 ммоль) и перемешивали реакционную смесь при комн. темп. в течение 3 ч. прибавили STAB (0,636 г, 3,0 ммоль) и перемешивали реакционную смесь в течение ночи. Реакционную смесь погасили путем прибавления NaHCO3 (насыщ. водн.) (30 мл), экстрагировали ДХМ (4×25 мл) и объединенные фазы ДХМ пропустили через фазный сепаратор Biotage. Растворители удалили в вакууме, и остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 25 г, 40-63 мкм, 60, 40 мл в мин, градиент от 0% до 10% МеОН в ДХМ) с получением неразделимой смеси диастереомеров этил 2-[4-циано-(пиридин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата. Препаративную ВЭЖХ использовали для разделения диастереомеров, используя колонку Phenomenex Gemini-N С18, 150×21 мм, элюируя от 25 до 65% МеОН/H2O при 18 мл/мин и собирая фракции при 210 нм с получением этил-2-[4-циано-(пиридин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата, Примера 3-10 Изомера 1, (0,012 г, 3%) в виде бесцветного твердого вещества и этил 2-[4-циано-(пиридин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата, Примера 3-10 Изомера 2, (0,014 г, 4%) в виде бесцветного твердого вещества. Данные для обоих Изомеров приведены в Таблице 3.
Путь au
Типовый метод получения пиперидинов реализуется путем алкилирования, как проиллюстрировано на примере получения Примера 4-5, этил-2-(1-этил-2-оксо-3,4'-бипиперидин-1'-ил)-6-азаспиро[3.4]октан-6-карбоксилата
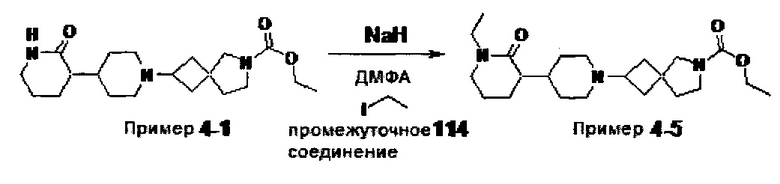
Этил-2-(2-оксо-[3,4'-бипиперидин]-1'-ил)-6-азаспиро[3.4]октан-6-карбоксилат, Пример 4-1, (0,2 г, 0,55 ммоль), растворили в ДМФА (3 мл) и охладили до 0-5°C. Прибавили гидрид натрия (0,080 г, 1,6 ммоль) и иодэтан (0,139 г, 0,8 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь погасили водой (50 мл), экстрагировали EtOAc (3×30 мл), и объединенные органические фазы сушили над (Na2SO4), фильтровали и концентрировали в вакууме с получением сырого этил 2-(1-этил-2-оксо-[3,4'-бипиперидин-1'-ил)-6-азаспиро[3.4]октан-6-карбоксилата в виде смеси диастереомеров. Сырой продукт очистили с помощью препаративной ВЭЖХ [X-Bridge С18 (150 X 19 мм, 5 мкм, 17 мл в мин, градиент от 27% до 100% (в течение 30 мин), затем 100% (4 мин), 0,1% NH3 в ацетонитриле с получением этил 2-(1-этил-2-оксо-[3,4'-бипиперидин]-1'-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 4-5 Изомера-1 (0,011 г, 5,11%) в виде бесцветной смолы и этил 2-(1-этил-2-оксо-[3,4'-бипиперидин]-1'-ил)-6-азаспиро[3.4]октан-6-карбоксилата Примера 4-5 Изомера-2 (0,012 г, 5,80%) в виде бесцветной смолы. Данные для Изомера 2 приведены в Таблице 3.
Путь av
Типовый метод получения пиперидинов реализуется путем восстановительного аминирования триацетоксиборогидридом натрия, как проиллюстрировано на примере получения Примера 4-8, этил 2-[4-(2-оксотетрагидропиримидин-1(2Н)-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата
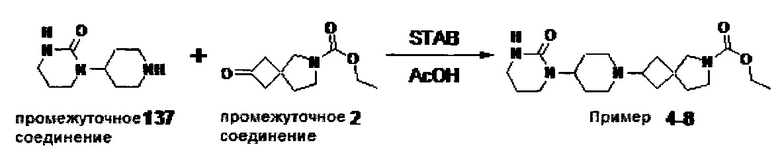
1-(Пиперидин-4-ил)тетрагидропиримидин-2(1H)-он (0,183 г, 1,0 ммоль) и этил 2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (0,197 г, 1,0 ммоль) растворили в ДХМ (10 мл) под N2 при комн. темп., прибавили уксусную кислоту (0,13 мл, 2,2 ммоль) и перемешивали реакционную смесь при комн. темп. в течение 3 ч. Прибавили STAB (0,530 г, 2,5 ммоль) и перемешивали реакционную смесь в течение ночи. Реакционную смесь погасили путем прибавления NaHCO3 (насыщ. водн.) (30 мл), экстрагировали ДХМ (4×25 мл) и объединенные фазы ДХМ пропустили через фазный сепаратор Biotage. Растворители удалили в вакууме и остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 25 г, 40-63 мкм, 60, 40 мл в мин, градиент от 0% до 10% МеОН в ДХМ) с получением неразделимой смеси диастереомеров этил 2-[4-(2-оксотетрагидропиримидин-1(2H)-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата. Препаративную ВЭЖХ использовали для разделения диастереомеров, используя колонку Phenomenex Gemini-N С18, 150×21 мм, элюируя от 15 до 30% MeCN в 0,2% NH3/H2O при 18 мл/мин и собирая фракции при 210 нм с получением этил 2-[4-(2-оксотетрагидропиримидин-1(2H)-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата, Примера 4-8 Изомера 1, (0,028 г, 7,7%) в виде бесцветного твердого вещества и этил 2-[4-(2-оксотетрагидропиримидин-1(2Н)-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата Примера 4-8 Изомера 2, (0,025 г, 6,9%) в виде бесцветного твердого вещества.
Данные для обоих Изомеров приведены в Таблице 3.
Путь aw
Типовый метод получения пиперидинов реализуется путем депротекции и восстановительного аминирования триацетоксиборогидридом натрия, как проиллюстрировано на примере получения Примера 4-13, этил 2-(3,3-дифтор-1,4'-бипиперидин-1'-ил)-6-азаспиро[3.4]октан-6-карбоксилата
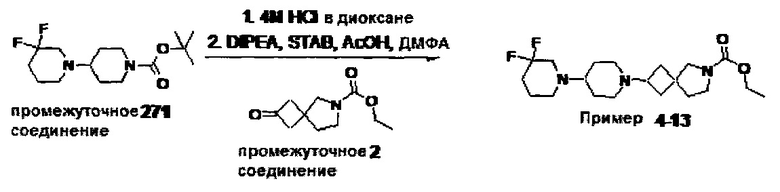
трет-Бутил 3,3-дифтор-1,4'-бипиперидин-1'-карбоксилат (0,347 г, 1,14 ммоль) растворили в 4,0 М HCl в диоксане (5 мл) и перемешивали реакционную смесь при комн. темп. в течение ночи. Растворители удалили в вакууме и остаток использовали в следующей стадии без дополнительной очистки. Сырую реакционную смесь и этил-2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (0,212 г, 1,14 ммоль) растворили в ДМФА (6 мл) при комн. темп. и прибавили DIPEA (0,295 г, 2,28 ммоль). Реакционную смесь перемешивали при 50°C в атмосфере азота в течение 2 ч. Реакционную смесь охладили до комн. темп., прибавили ледяную уксусную кислоту (0,068 г, 1,14 ммоль) и STAB (0,604 г, 2,85 ммоль) и перемешивали реакционную смесь в течение ночи при 50°C в атмосфере азота. К охлажденной реакционной смеси прибавили воду (2 мл) и удалили растворители в вакууме. Остаток разделили между ДХМ (15 мл) и насыщ. NaHCO3 (водн.) (15 мл), водную фазу промыли ДХМ (2×15 мл). Органические фазы объединили и сушили, пропуская через картридж фазного сепаратора Biotage. Растворители удалили в вакууме и остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 10 г 40-63 мкм, 60, 12 мл в мин, градиент от 1% до 10% МеОН/ДХМ]). Остаток дополнительно очистили с помощью препаративной обратно-фазовой ВЭЖХ (колонка Phenomenex Gemini-NX 5 мкм С18 110A Axia, 100×30 мм, элюируя от 30 до 60% MeCN/Растворитель B в течение 14,4 мин при 30 мл/мин [где растворитель B представляет собой 0,2% (28% NH3/H2O) в H2O] и собирая фракции при 210 нм) с получением этил-2-(3,3-дифтор-1,4'-бипиперидин-1'-ил)-6-азаспиро[3.4]октан-6-карбоксилата, Примера 4-13 Изомера 1, (0,011 г, 2,6%) в виде бесцветного масла и этил-2-(3,3-дифтор-1,4'-бипиперидин-1'-ил)-6-азаспиро[3.4]октан-6-карбоксилата, Примера 4-13 Изомера 2, (0,005 г, 1,3%) в виде бесцветного масла. Данные для изомера 2 приведены в Таблице 3.
Путь ax
Типовый метод получения пиперидинов реализуется путем восстановительного аминирования, как проиллюстрировано на примере получения Примера 4-16, этил 2-[(2R)-2-(метилкарбамоил)-1,4'-бипиперидин-1'-ил]-6-азаспиро[3.4]октан-6-карбоксилата
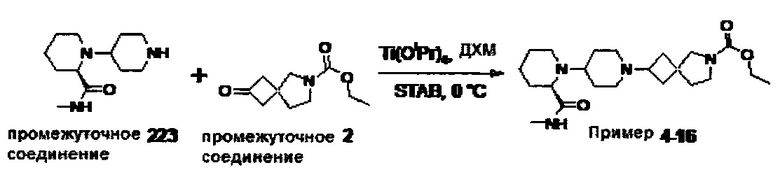
К раствору (R)-N-метил-[1,4'-бипиперидин]-2-карбоксамида (200 мг, 0,890 ммоль) и этил 3-оксо-8-азабицикло[3.2.1]октан-8-карбоксилата (175 мг, 0,890 ммоль) в ДХМ (7,5 мл), прибавили Ti(OiPr)4 (0,80 мл, 2,67 ммоль) при 0°C и перемешивали реакционную смесь в течение 1 ч. К реакционной смеси порциями прибавили Na(OAc)3BH (562 мг, 2,67 ммоль) и перемешивали при 0°C в течение 2 ч. По окончании реакционую смесь погасили водн. насыщ. NaHCO3 и экстрагировали ДХМ (3×30 мл). Органические фазы объединили и промыли насыщенным водным раствором хлорида натрия, сушили (Na2SO4) и концентрировали в вакууме. Остаток очистили с помощью препаративной-HPLC (обращенно-фазовая, X BRIDGE, С-18, 19×250 мм, 5 мк, градиент от 10% до 90% ACN в воде, содержащей 5 мМ NH4OAc, с получением 25 мг (7%) этил 2-[(2R)-2-(метилкарбамоил)-1,4'-бипиперидин-1'-ил]-6-азаспиро[3.4]октан-6-карбоксилата Примера 4-16 Изомера-1 и 25 мг (7%) этил-2-[(2R)-2-(метилкарбамоил)-1,4'-бипиперидин-1'-ил]-6-азаспиро[3.4]октан-6-карбоксилата Примера 4-16 Изомера-2 в виде бесцветного полутвердого вещества. Данные для Изомера 2 приведены в Таблице 3.
Путь ay
Типовый метод получения пиперидинов реализуется путем алкилирования, циклизации и восстановительных аминирований триацетоксиборогидридом натрия, как проиллюстрировано на примере получения Примера 5-1, этил 2-[4-(2-оксоазепан-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата
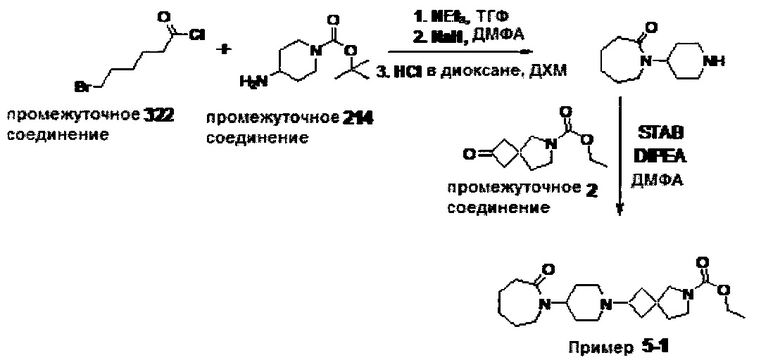
К раствору 4-Амино1-Вос-пиперидина (200 мг, 1,0 ммоль) в ТГФ (2 мл) прибавили триэтиламин (0,153 мл, 1,1 ммоль) и 6-бромгексаноилхлорид (0,168 мл, 1,098 ммоль) и мутную суспензию перемешивали при комн. темп. в течение 2 ч. Растворители удалили в вакууме, и остаток разделили между H2O (15 мл) и EtOAc (25 мл), водную фазу экстрагировали EtOAc (2×25 мл), органические фазы объединили, сушили над Na2SO4 и растворитель удалили в вакууме с получением трет-бутил-4-[(6-бромгексаноил)амино]пиперидин-1-карбоксилата (378 мг, >99%) в виде оранжевого масла.
трет-бутил 4-[(6-бромгексаноил)амино]пиперидин-1-карбоксилат (378 мг, 1,0 ммоль) растворили в ДМФА (25 мл) и прибавили гидрид натрия (48 мг, 1,2 ммоль). Реакционную смесь перемешивали при 80°C в течение 1 ч, растворитель удалили в вакууме и остаток очистили с помощью колоночной хроматографии (нормально-фазовая, [картридж Biotage SNAP KP-sil 10 г, 40-63 мкм, 60, 25 мл в мин, от 1% до 10% МеОН в ДХМ]) с получением трет-бутил 4-(2-оксоазепан-1-ил)пиперидин-1-карбоксилата (178 мг, 60%). Остаток растворили в ДХМ (1 мл), с последующим прибавлением по каплям HCl в диоксане (3 мл, 4,0 М р-р.). Полученную реакционную смесь перемешивали при комн. темп. в течение 1 ч, растворители удалили в вакууме и остаток использовали в следующей стадии без дополнительной очистки. 1-(пиперидин-4-ил)азепан-2-он. HCl (0,182 г, 0,738 ммоль) и этил 2-оксо-6-азаспиро[3.4]октан-6-карбоксилат (0,155 г, 0,785 ммоль) растворили в ДМФА (2 мл) при комн. темп. и прибавили DIPEA (0,136 мл, 0,790 ммоль). Реакционную смесь перемешивали при комн. темп. в течение 3 ч. Затем прибавили STAB (0,332 г, 1,569 ммоль) и перемешивали реакционную смесь в течение ночи в атмосфере азота при комн. темп. Растворители удалили в вакууме и остаток очистили с помощью препаративной обратно-фазовой ВЭЖХ (колонка Phenomenex Gemini-NX 5 мкм С18 110A Axia, 100×30 мм, элюируя от 25 до 45% MeCN/Растворитель B в течение 14,4 мин при 30 мл/мин [где растворитель B представляет собой 0,2% (28% NH3/H2O) в H2O] и собирая фракции при 210 нм) с получением этил-2-[4-(2-оксоазепан-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата, Примера 5-1 Изомера 1 (6,2 мг, 2%) в виде бесцветного масла и этил 2-[4-(2-оксоазепан-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилата, Примера 5-1 Изомера 2 (3,9 мг, 1%) в виде бесцветного масла. Данные для Изомера 2 приведены в Таблице 3.
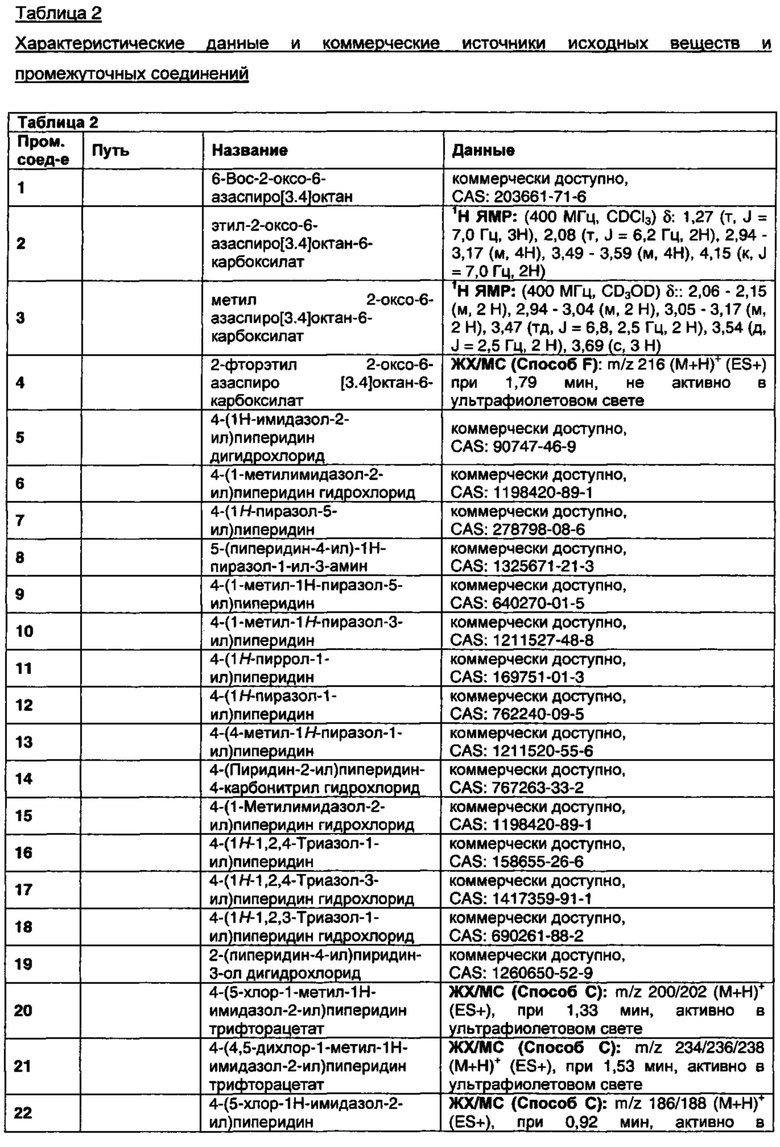
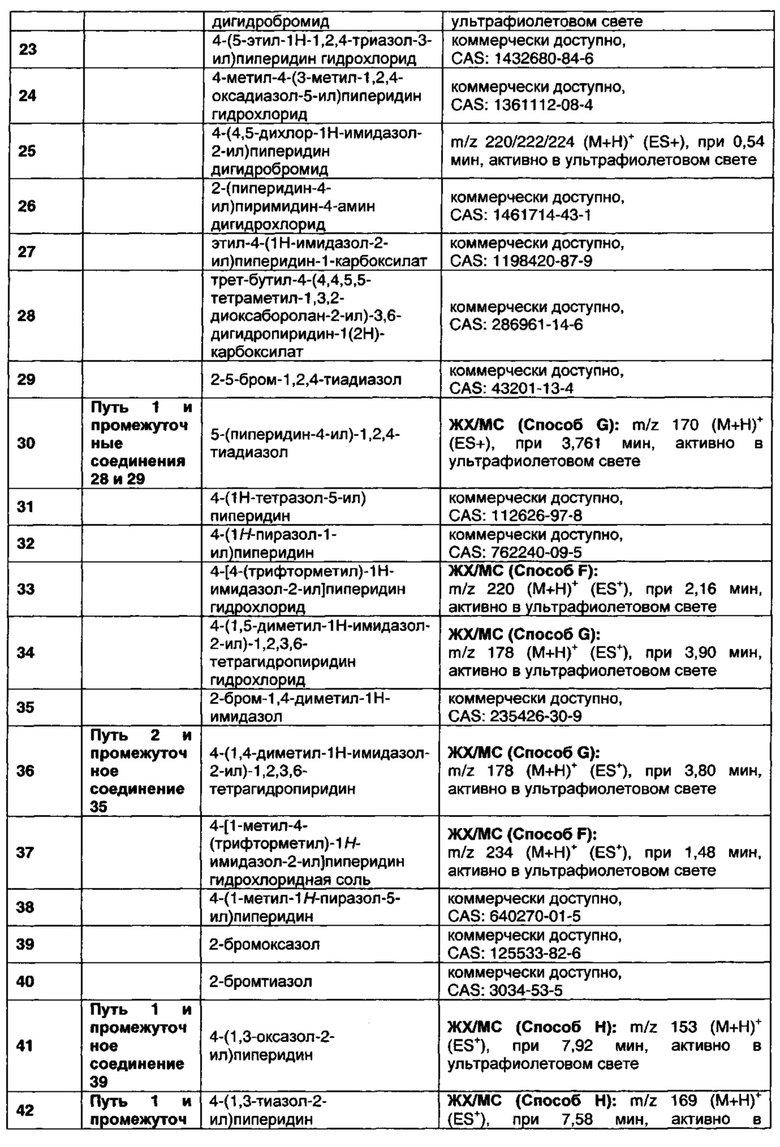
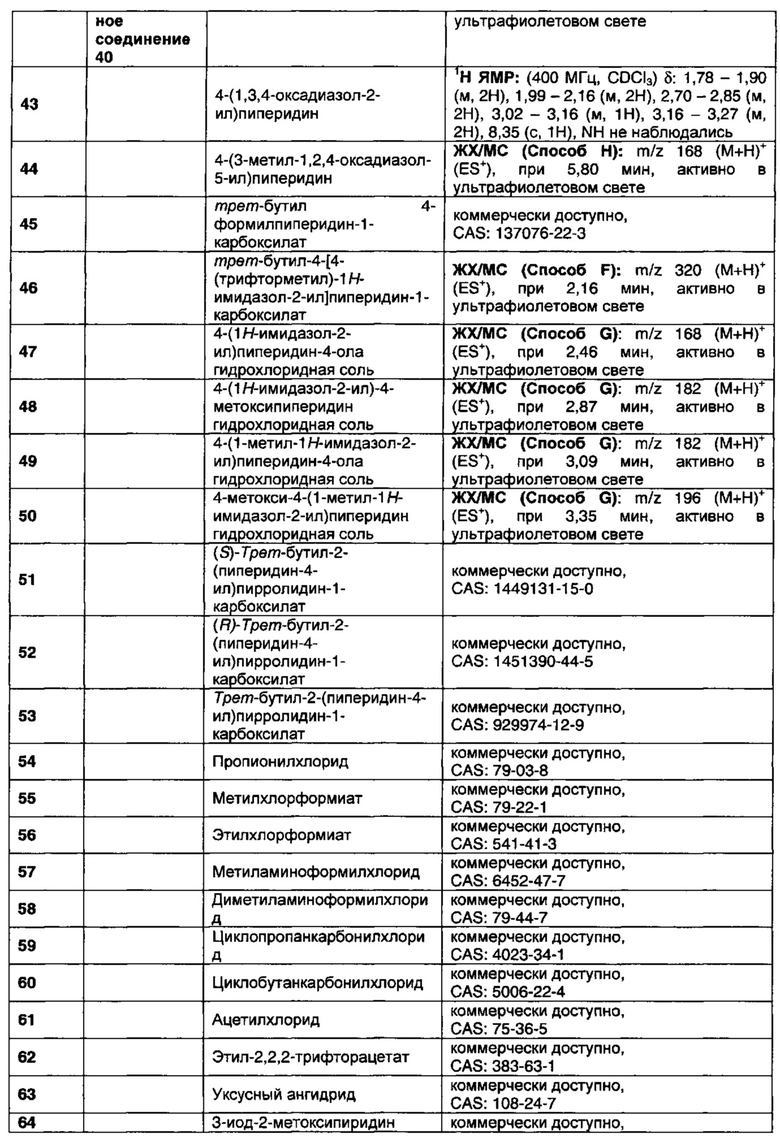
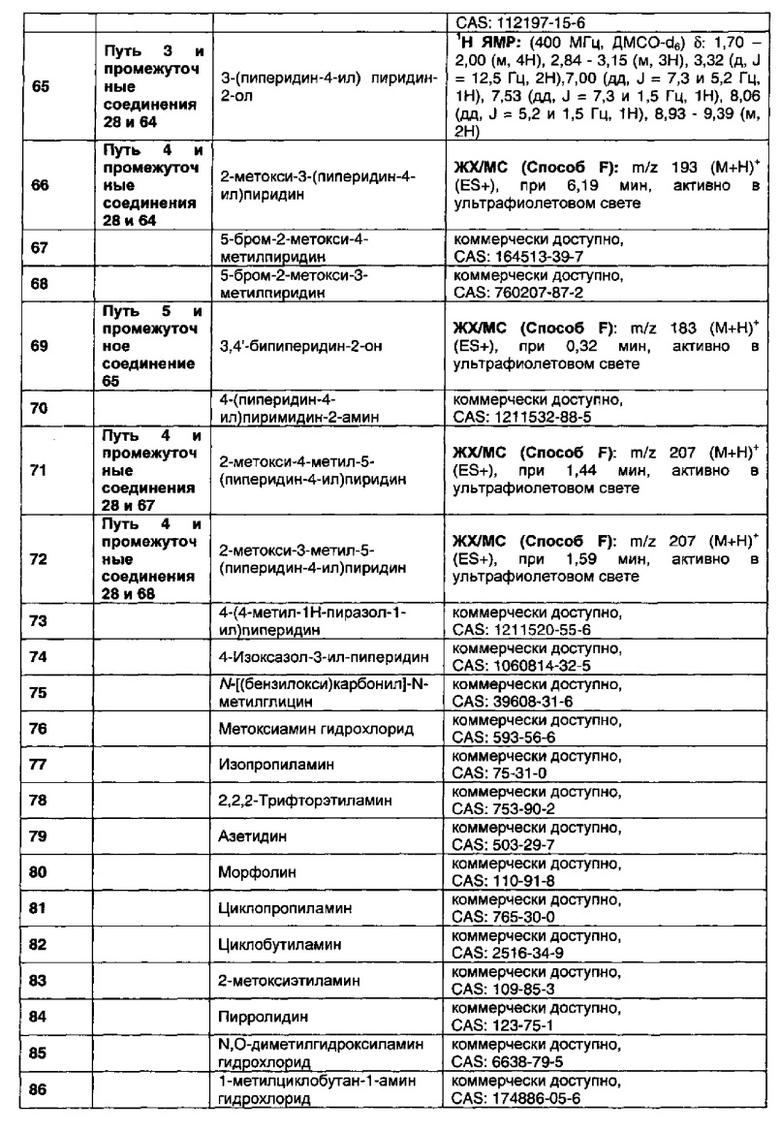
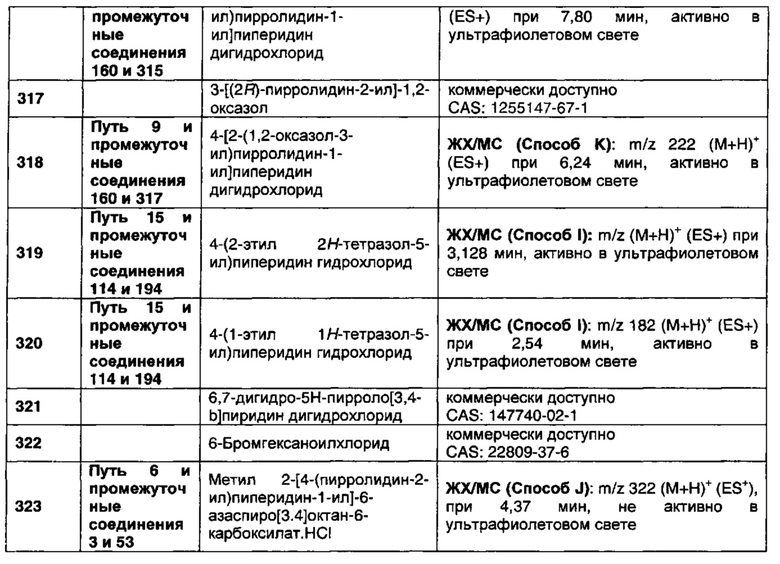
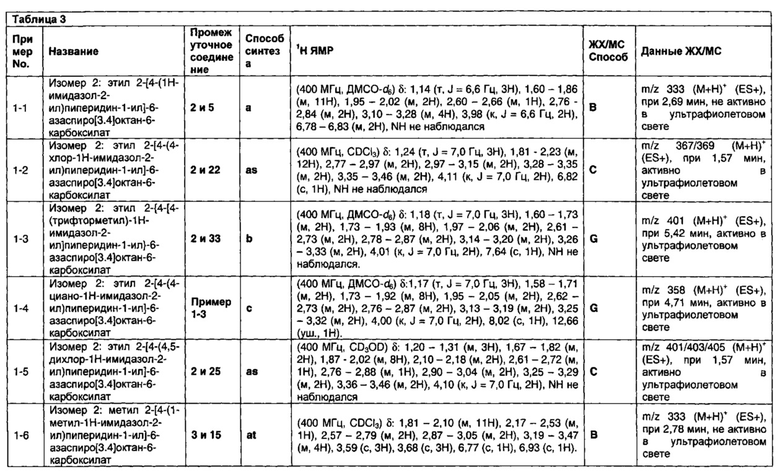
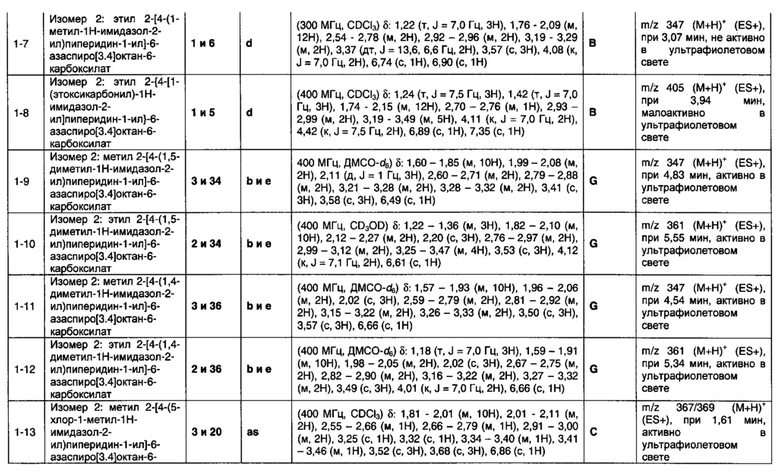
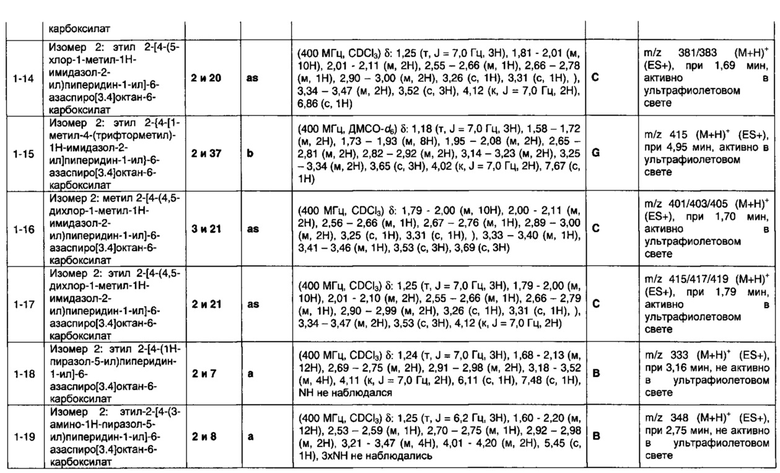
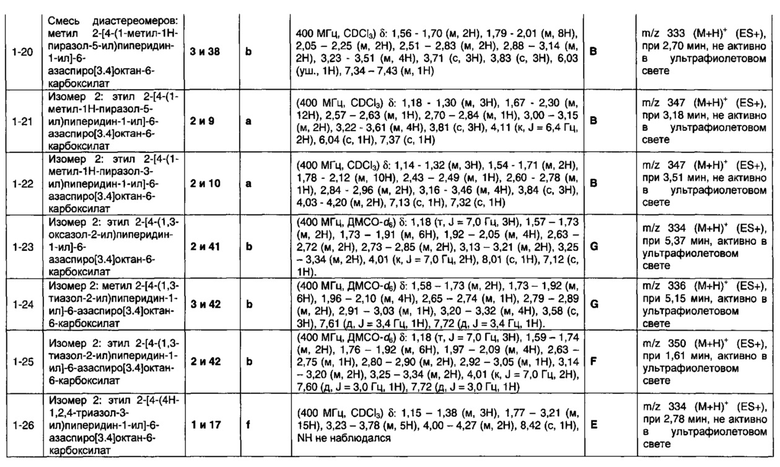
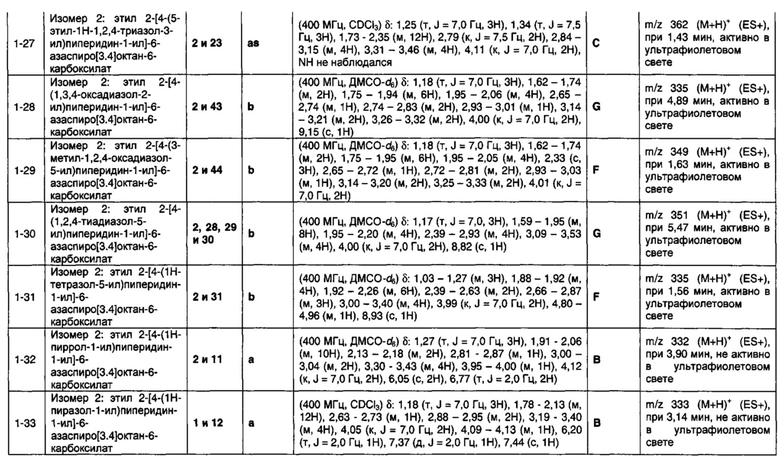
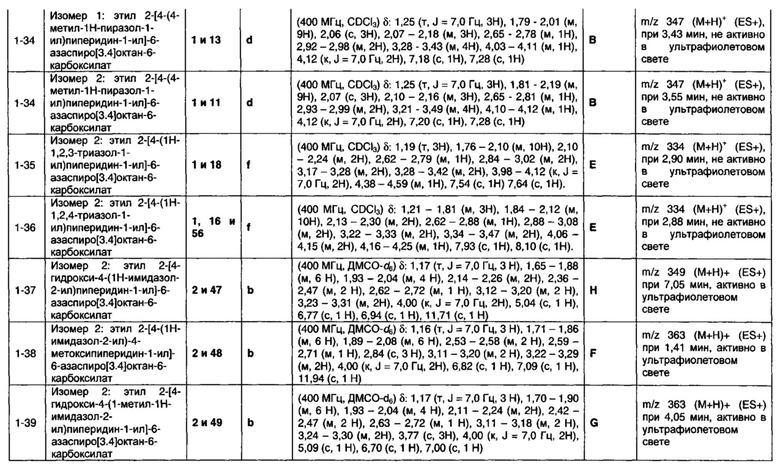
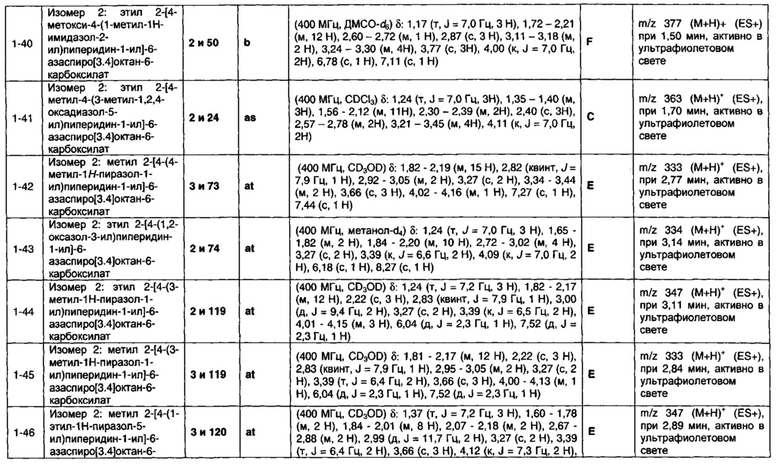
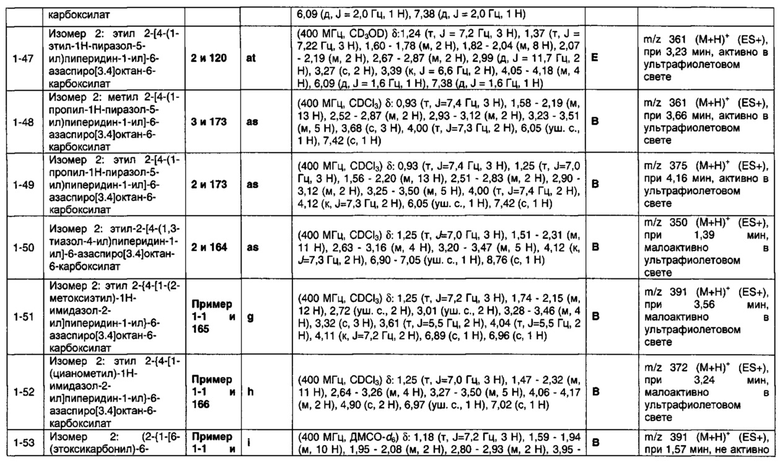
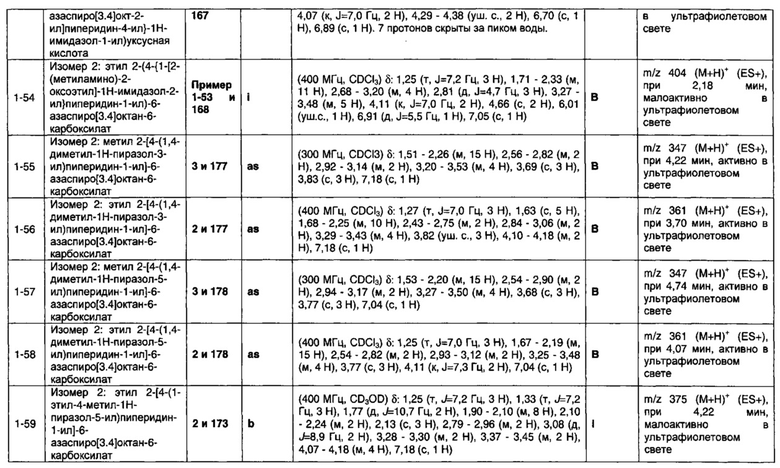
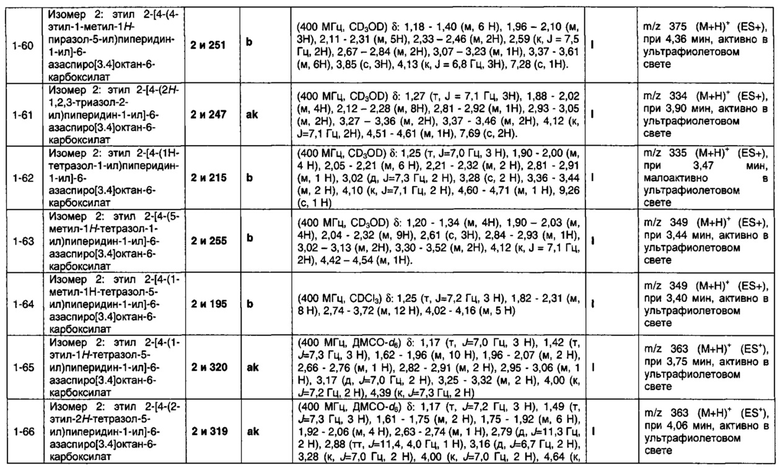
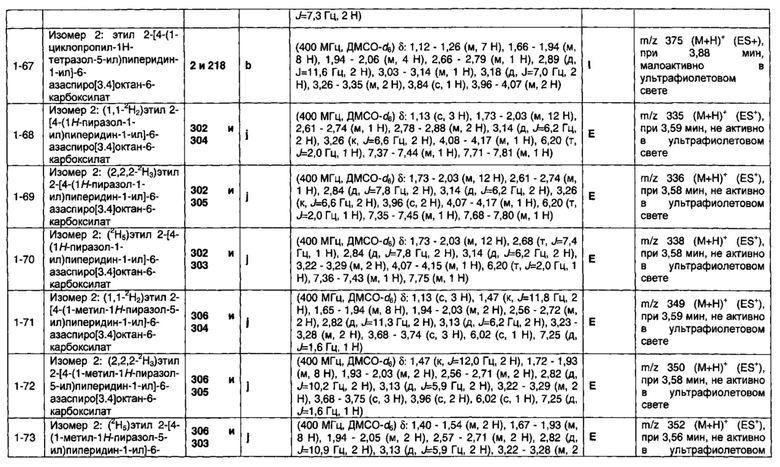
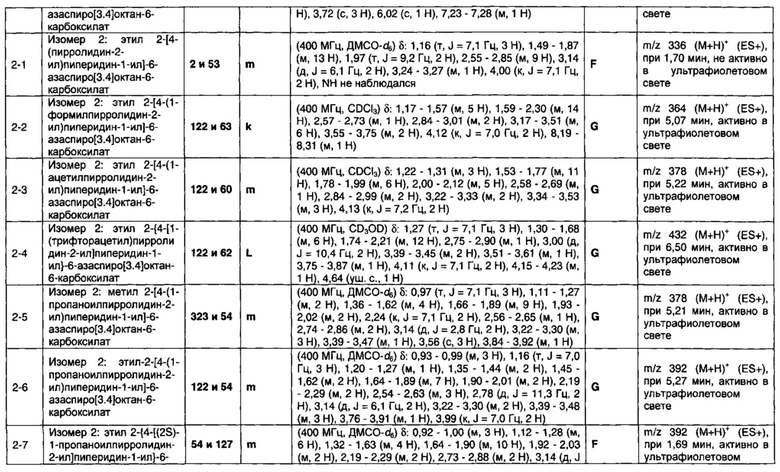
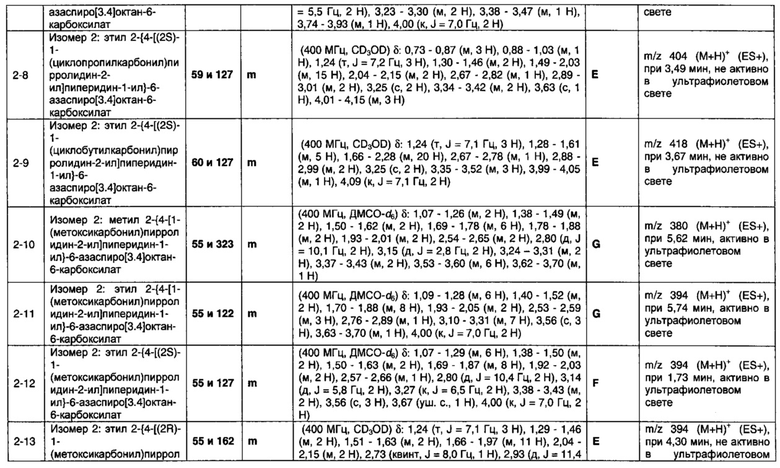
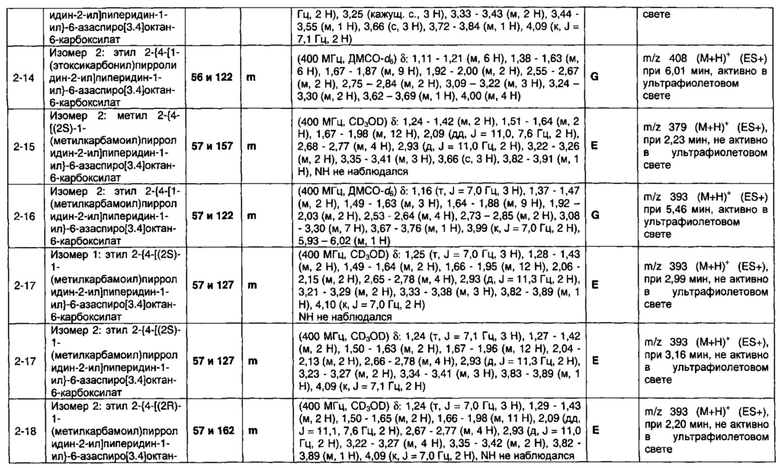
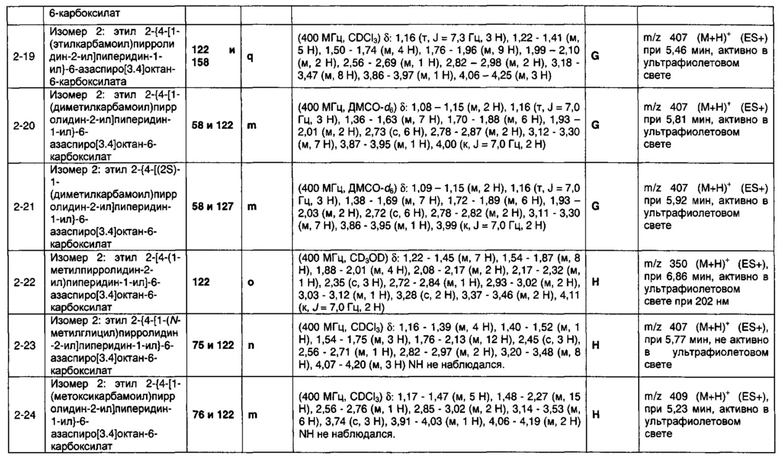
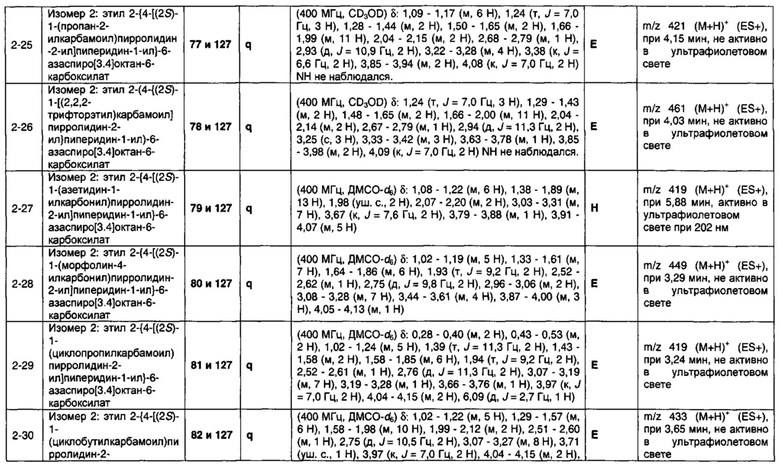
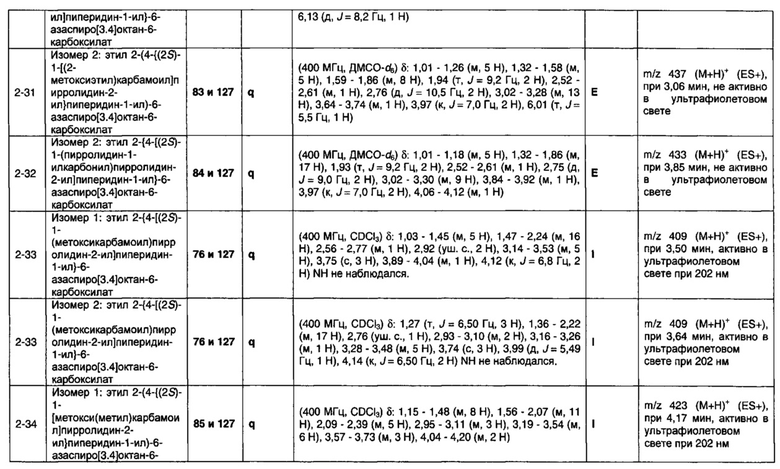
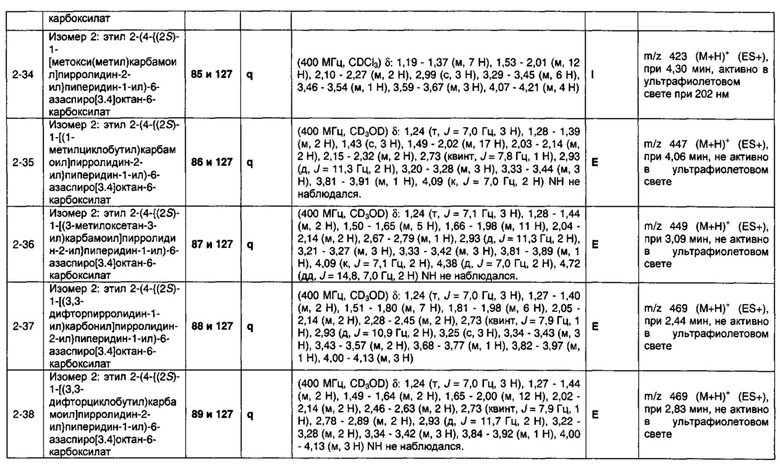
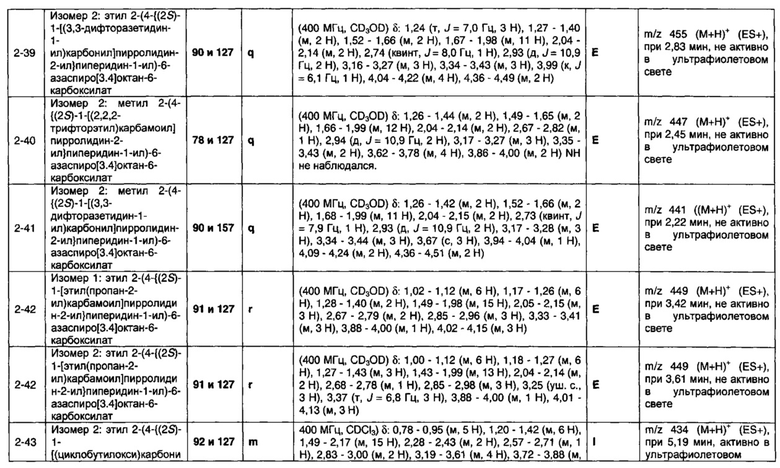
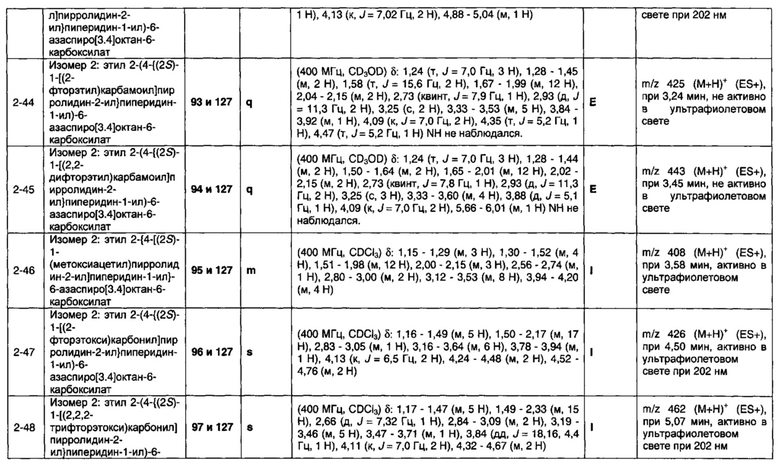
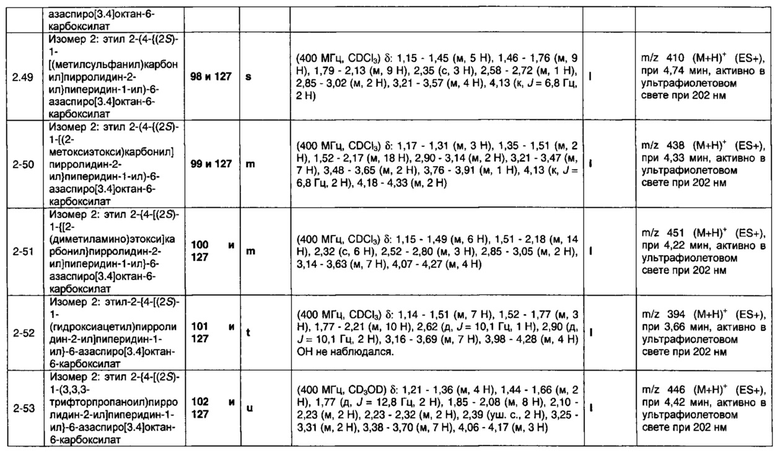
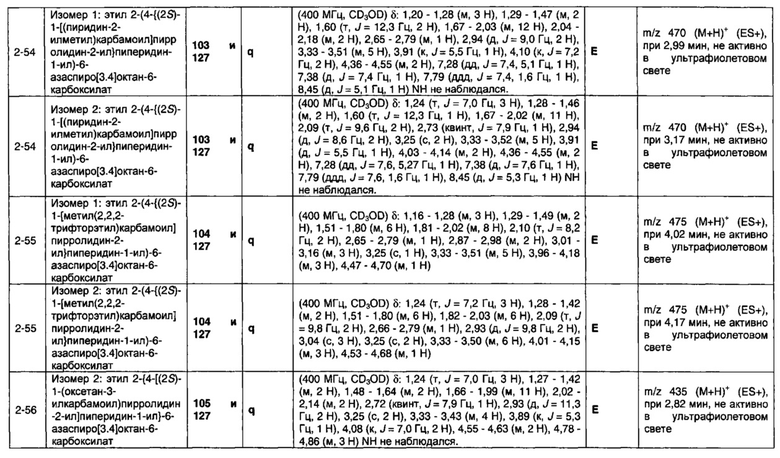
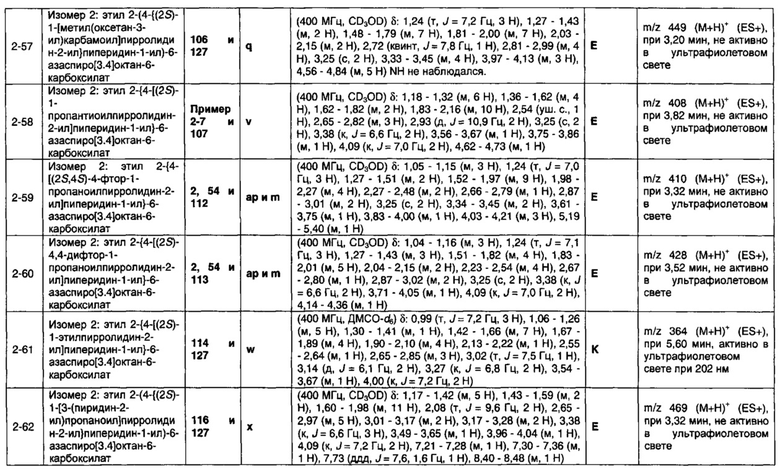
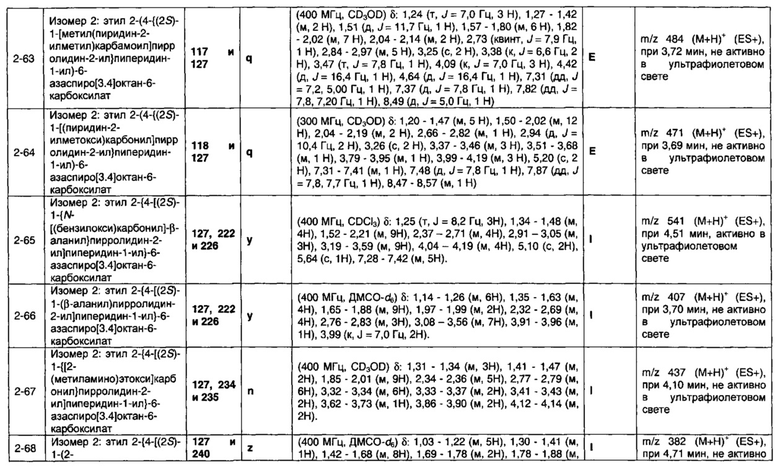
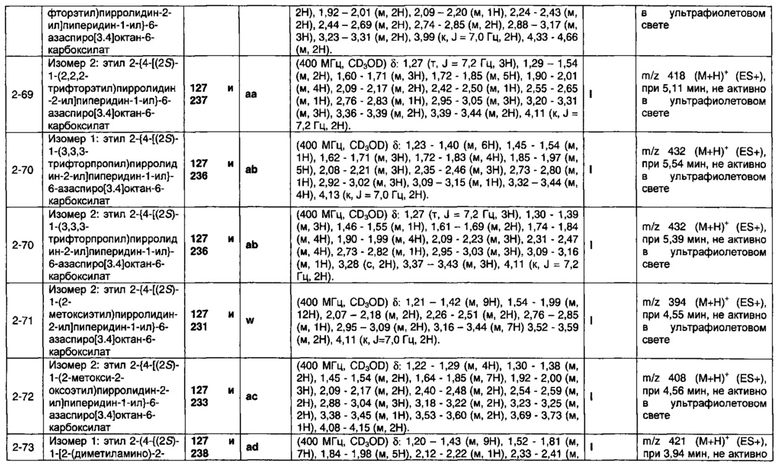
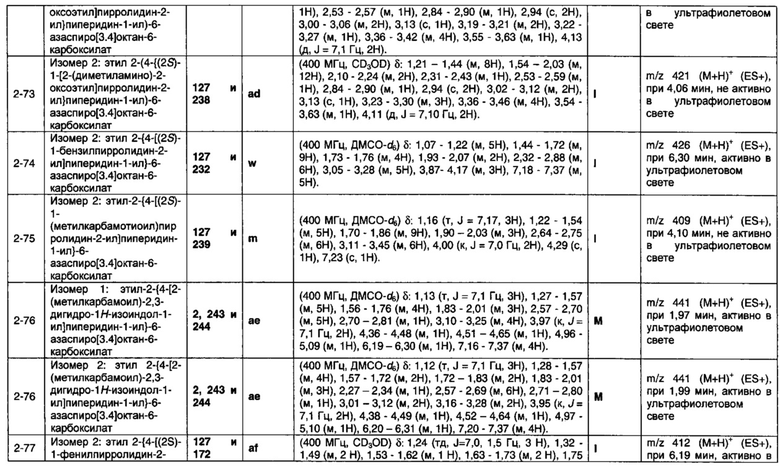
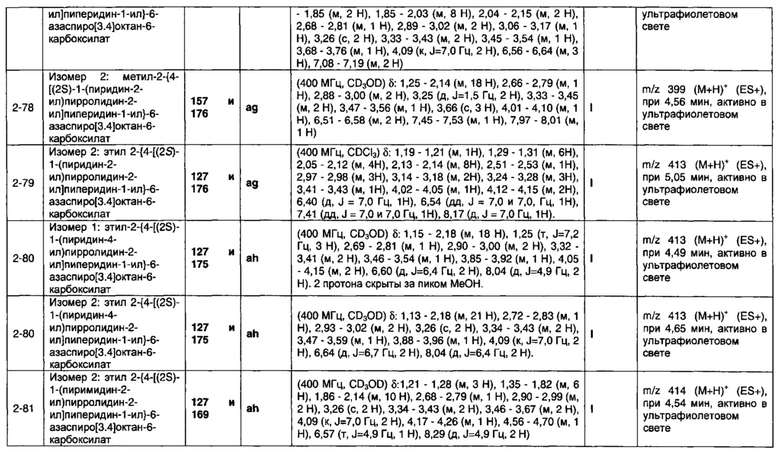
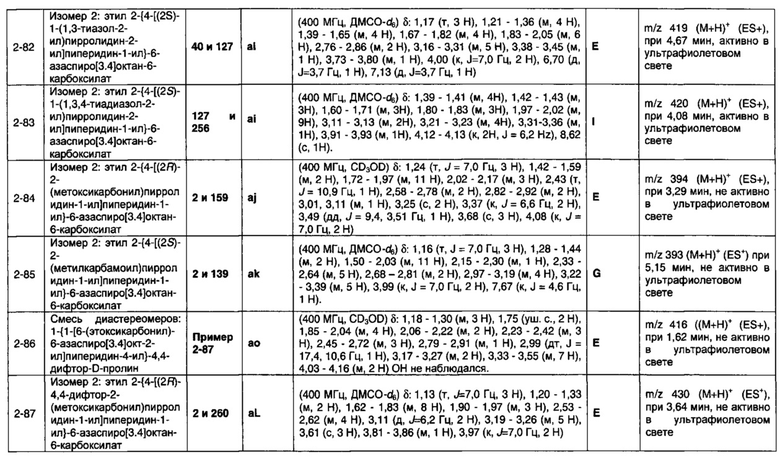
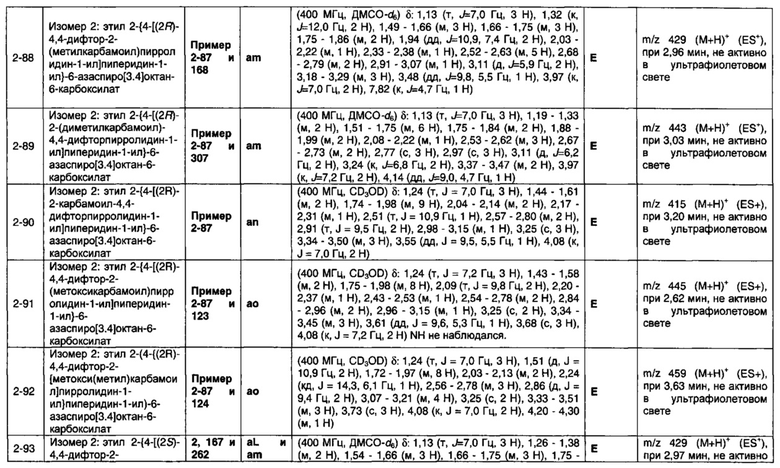
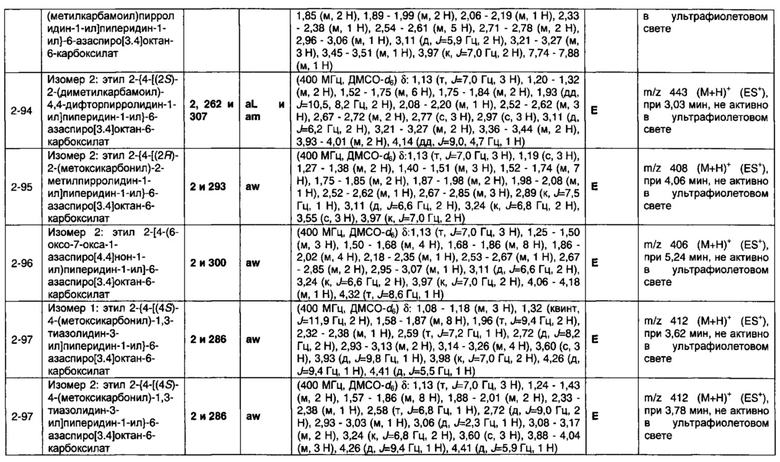
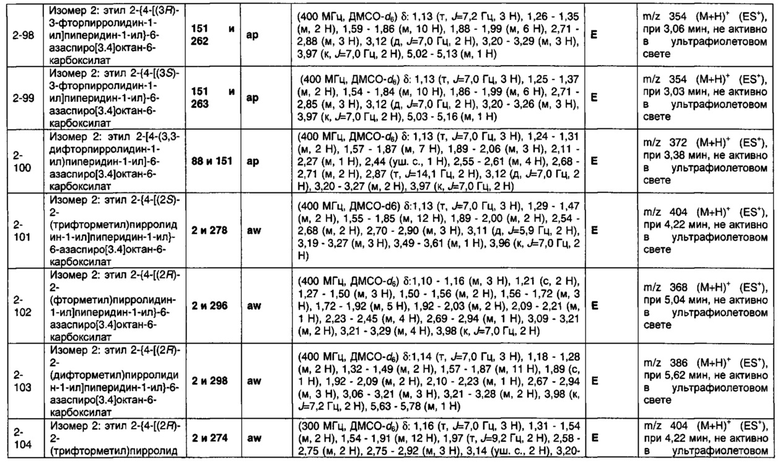
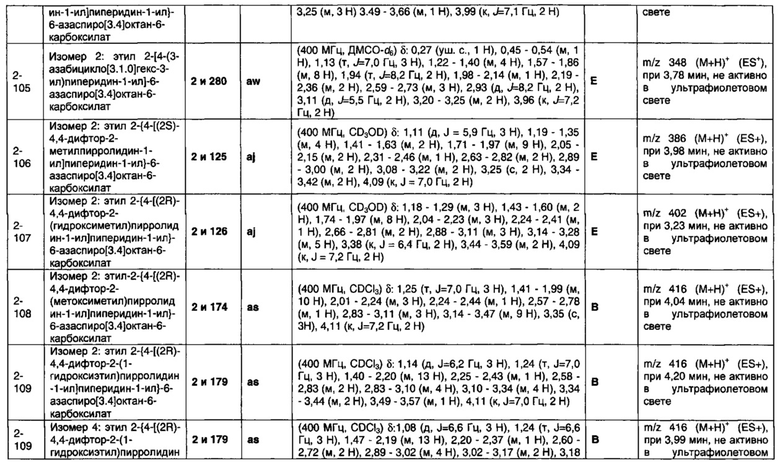
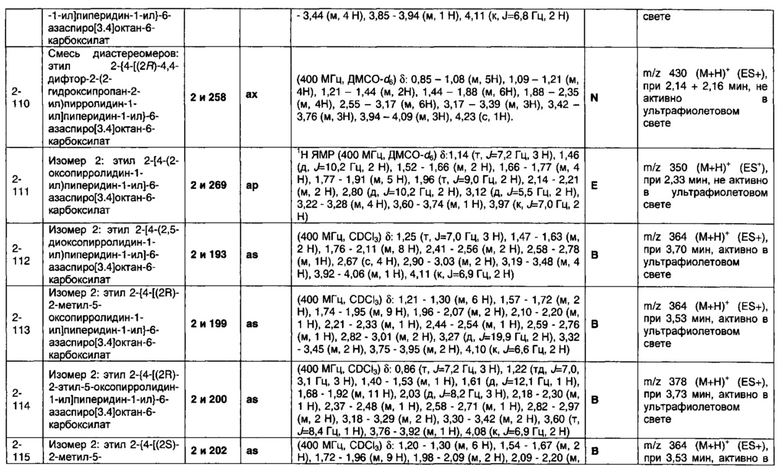
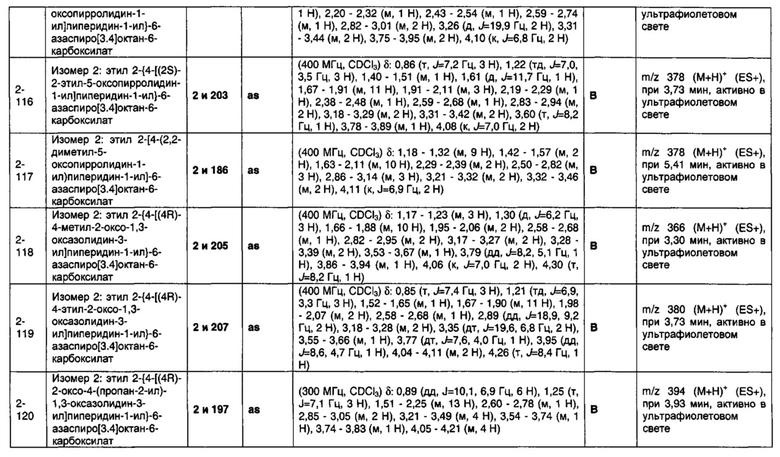
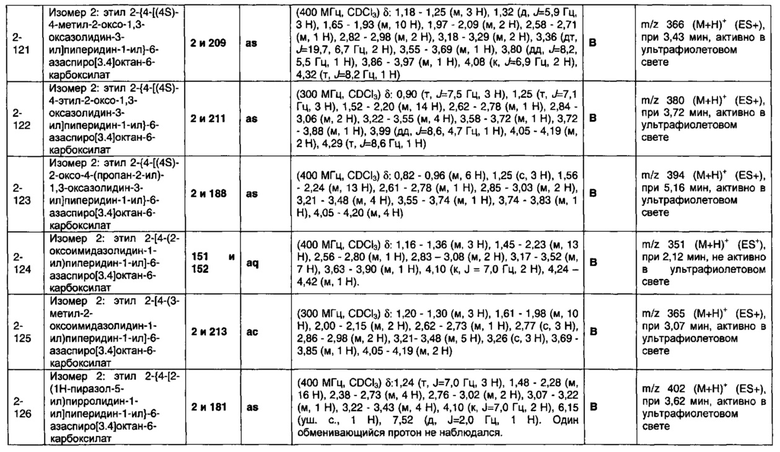
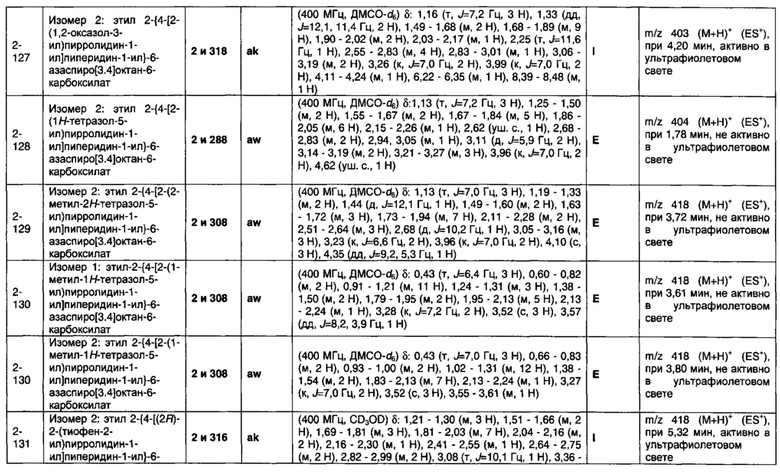
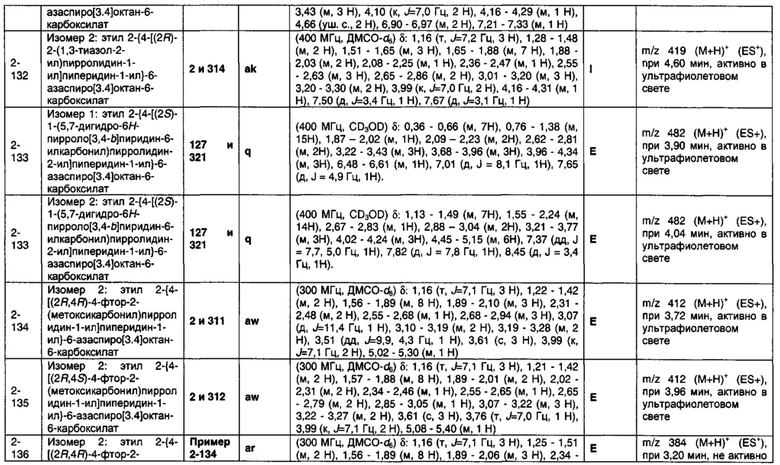
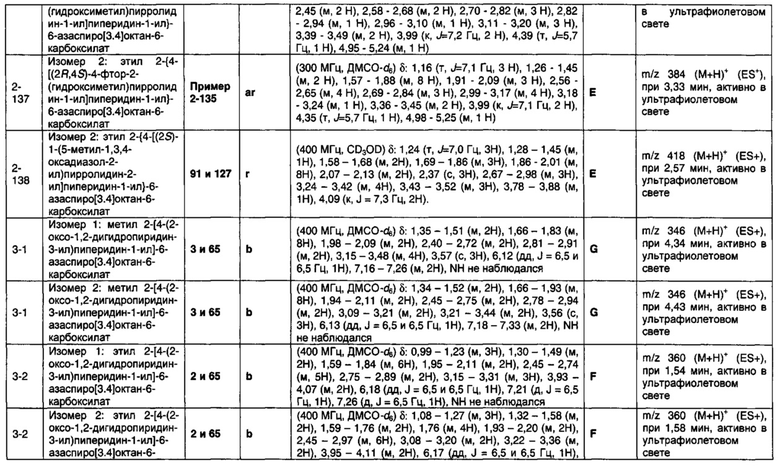
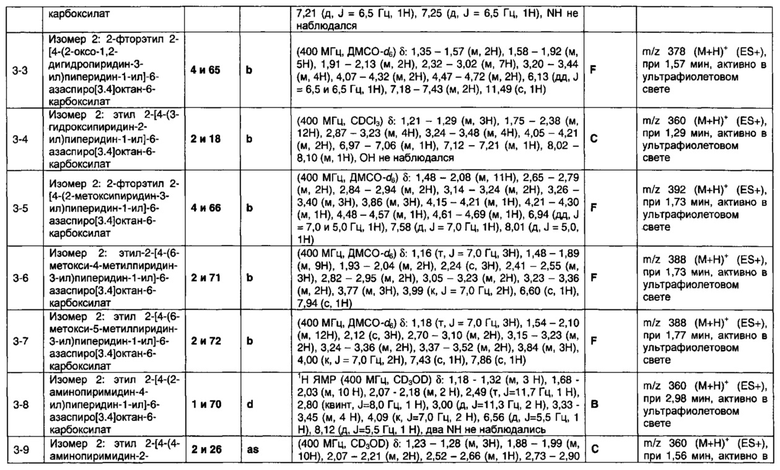
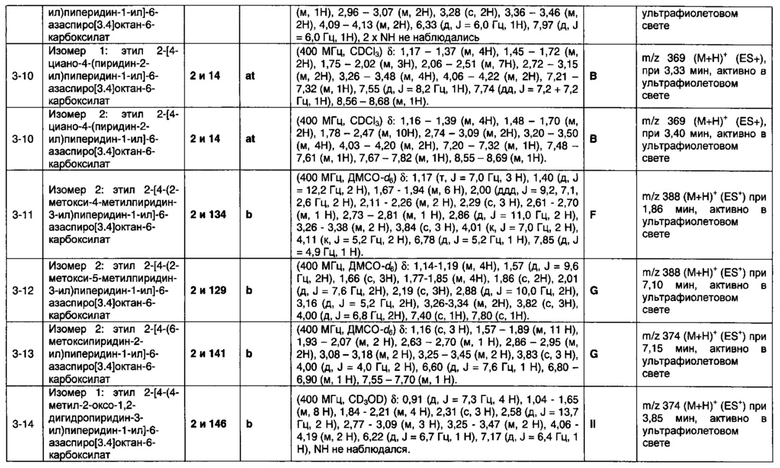
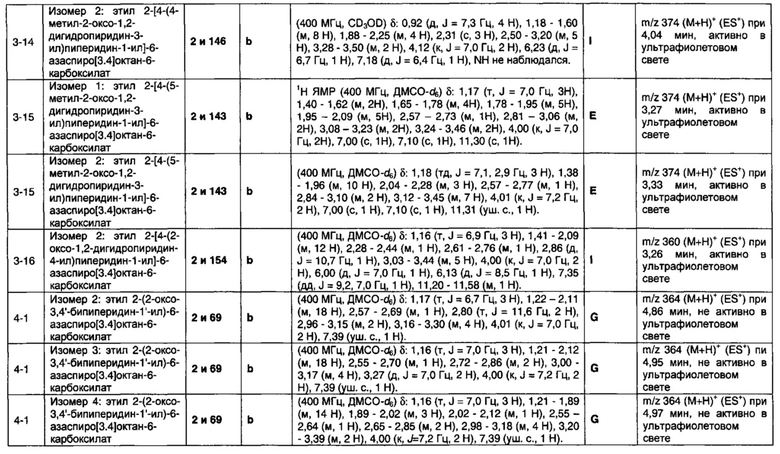
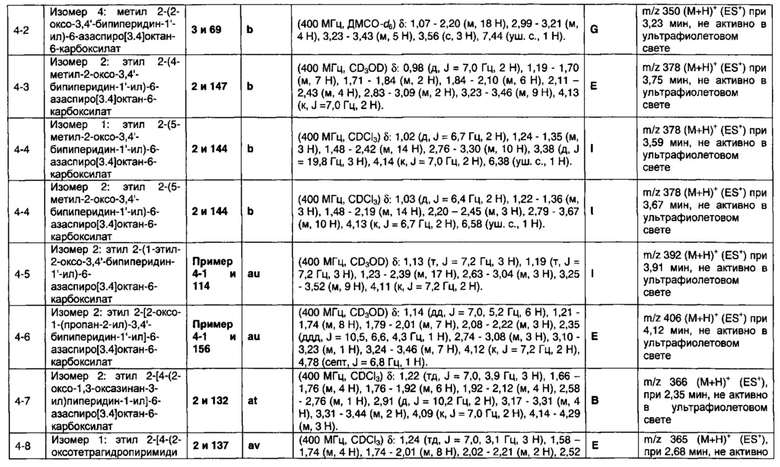
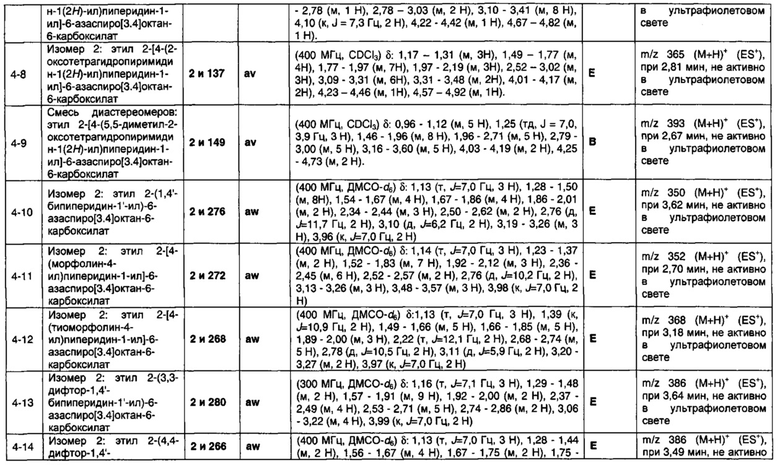
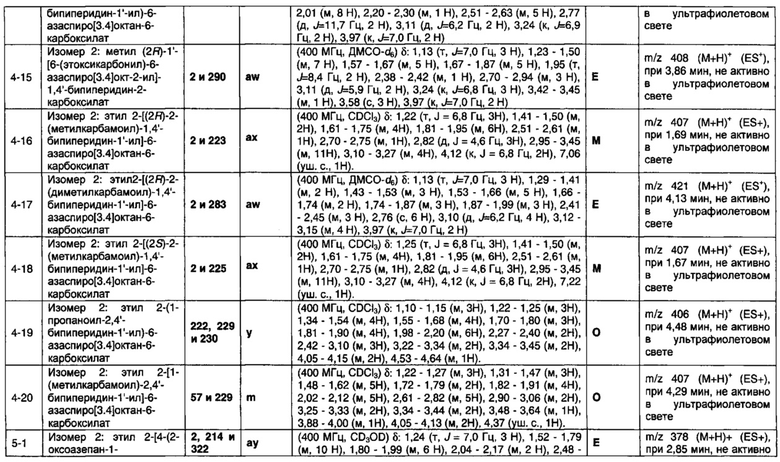

БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ ПРИМЕРА
Исследования cboccbo-ERK1/2
Функциональные исследования выполняли, используя Alphascreen Surefire исследования фосфо-ERK1/2 (Crouch & Osmond, Comb. Chem. High Throughput Screen, 2008). Фосфорилирование ERK1/2 представляет собой нисходящий сигнальный каскад для активации связанных с Gq/11 и Gi/o белками рецепторов, делая его более подходящим для оценки M1, М3 (Gq/11 связанный) и М2, М4 рецепторов (Gi/o связанный), чем использование различных форматов исследований разных подтипов рецепторов. Стабильно экспрессирующие человеческий мускариновый М1, М2, М3 или М4 рецептор клетки СНО высевали (25000 / лунка) в 96-луночные планшеты для выращивания живой ткани в альфа-MEM + 10% диализированная ФБС. После прикрепления к поверхности клетки оставили культивироваться в условиях сывороточного голодания в течение ночи. Стимуляцию агонистом выполнили путем прибавления 5 мкл агониста к клеткам в течение 5 мин (37°C). Питательную среду удалили и прибавили 50 мкл лизирующего буфера. Через 15 мин 4 мкл образца перенесли в 384-луночный планшет и прибавили 7 мкл детектирующей смеси. Планшеты инкубировали в течение 2 ч с легким перемешиванием в темноте и затем считывали на планшет-ридере PHERAstar.
Значения рЕС50 и Еmax рассчитывали, исходя из полученных для каждого подтипа рецептора данных.
Результаты приведены ниже в Таблице 4.
Для каждого примера существуют два диастереомера, которые были разделены, если не указано иное, и установлены на основании времени удерживания на аналитическом пути ЖХ/МС. В большинстве примеров изомер 1 не активен. Аналитические данные для активных изомеров приведены в Таблице 3. Данные для нескольких малоактивных соединений включены в Таблицу 4 для отображения предпочтительности абсолютной стереохимической конфигурации.
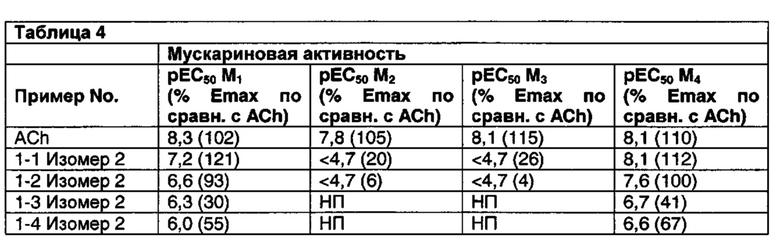
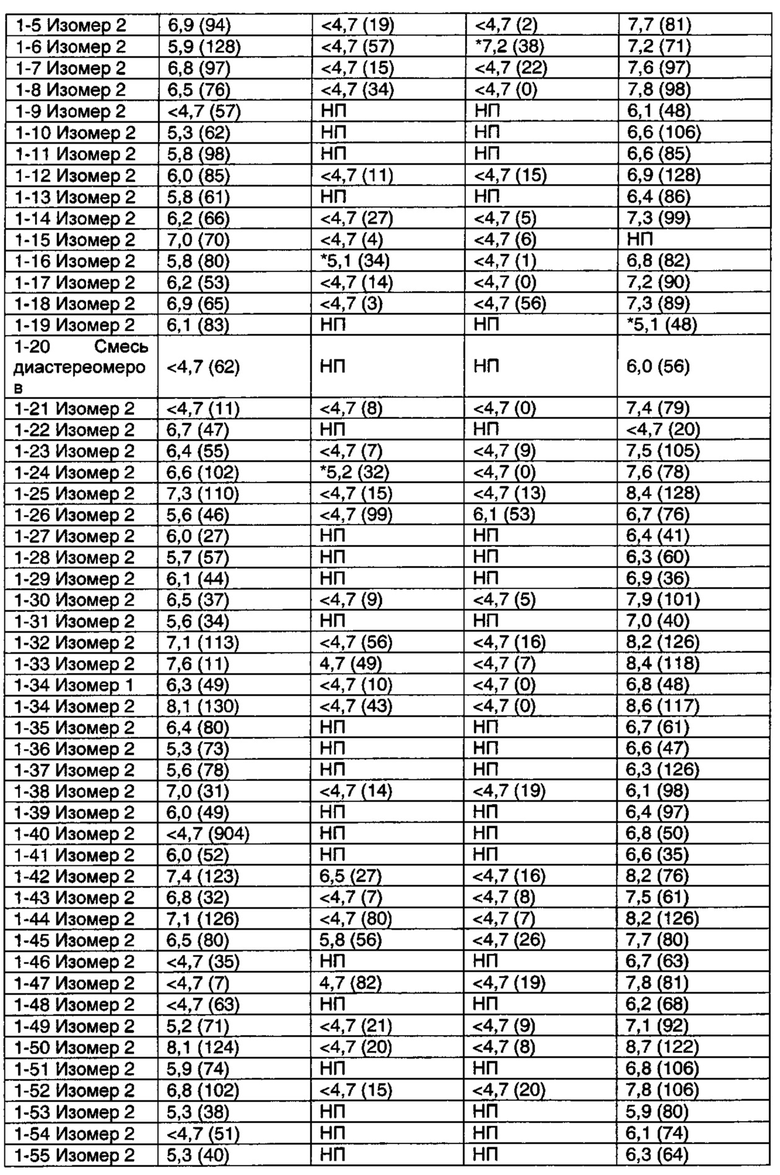
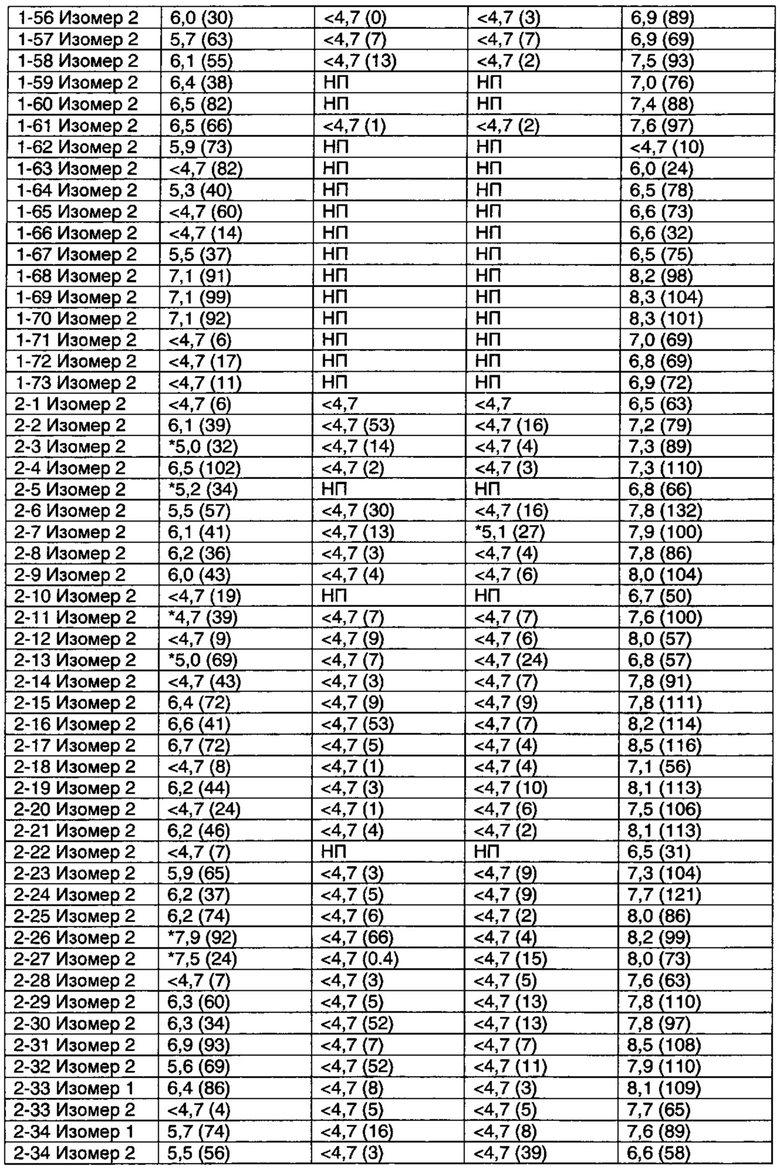
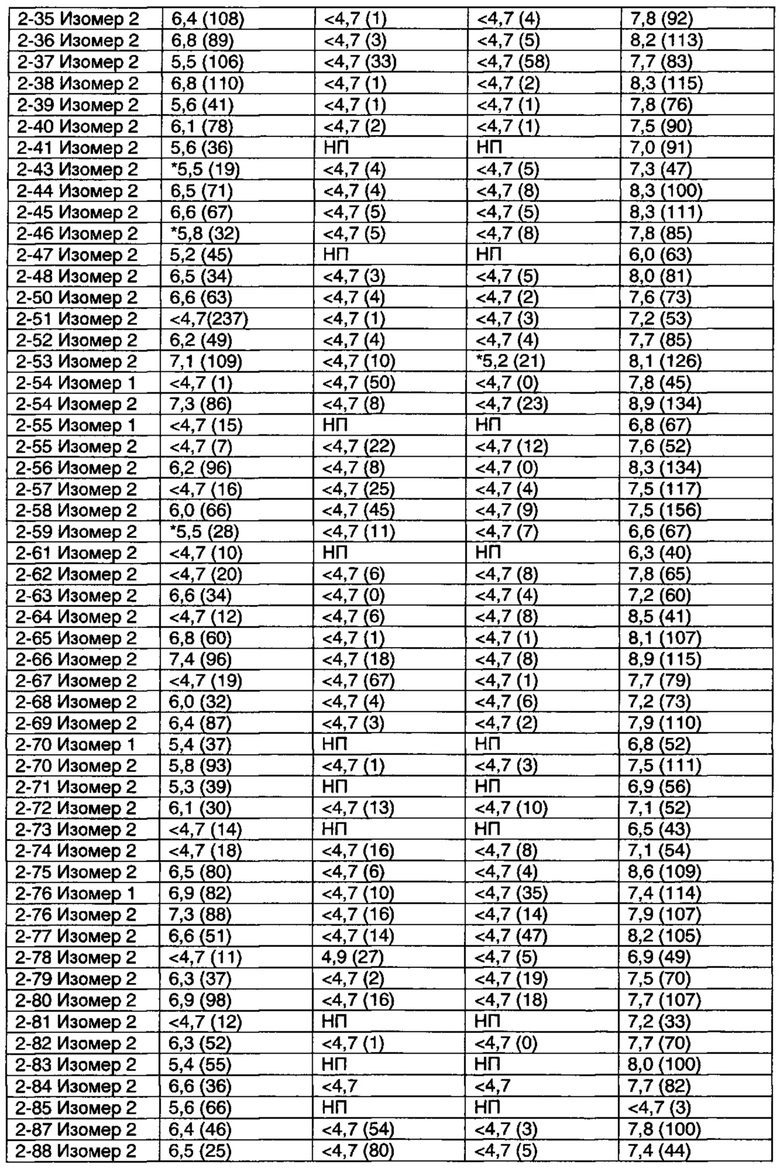
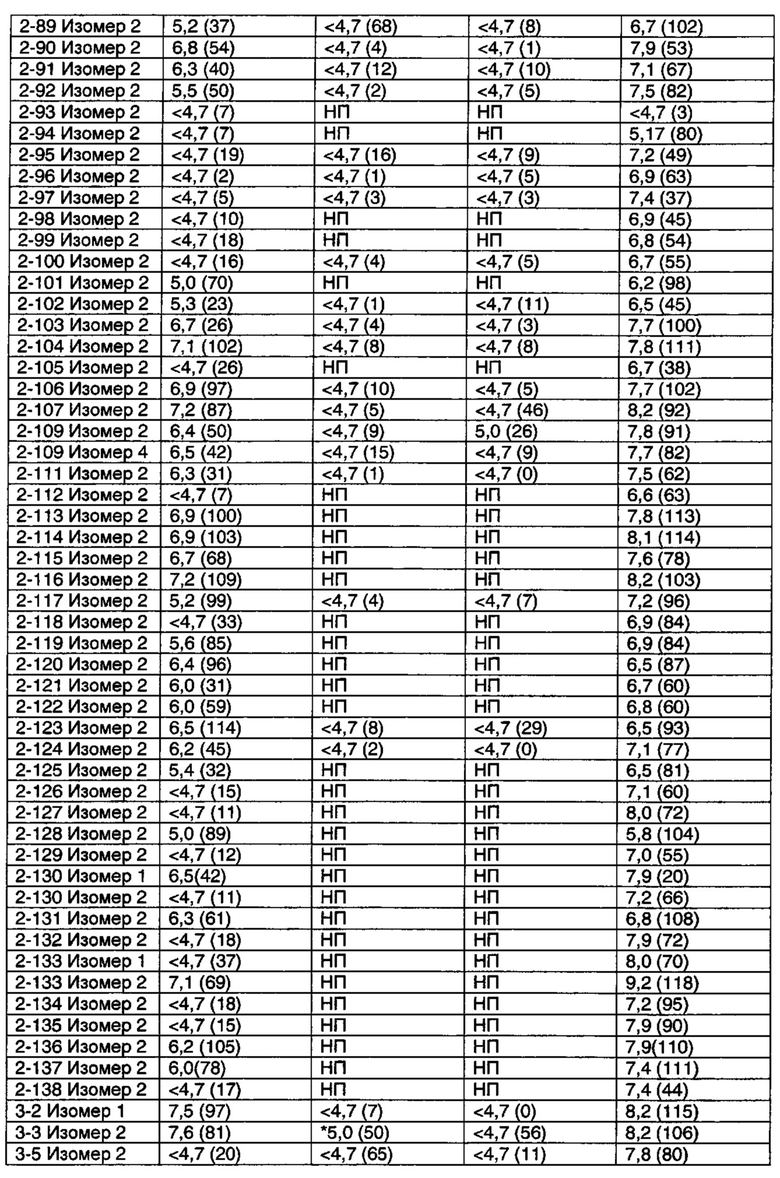
ПРИМЕР B
Пассивное избегание
Исследования проводили согласно ранее описанному Foley et al., (2004) Neuropsychopharmacology. В задании пассивного избегания введение скополамина (1 мг/кг, и/п) вызывало у животных потерю памяти на 6 часов последующих обучений. Диапазон доз свободных оснований в 3, 10, и 30 мг/кг (п/о) вводили путем принудительного кормления за 90 минут до изучения периода обучения. Было обнаружено, что Пример 1-33 Изомер 2 способен обращать вызванную скополамином амнезию в доза-зависимый способ, с ЭД50 приблизительно 10 мг/кг (п/о). Эффект от 30 мг/кг оказался схожим с эффектом, вызванным ингибитором холинэстеразы донепезилом (0,1 мг/кг, и/п), который служил в качестве положительного контроля (Фигура 1).
ПРИМЕР C
Эффект нового тестируемого соединения и ксаномелина на d-амфетамин-индуцированную гиперактивность у крыс
Цель данного исследования изучить эффект нового тестируемого соединения на d-амфетамин-индуцированную гиперактивность у крыс. Шизофрения представляет собой сложное многофакторное заболевание, которое не может быть представлено в полной мере одной экспериментальной методикой. Антипсихотически-подобное поведение крыс оценивали путем ингибирования гиперактивности (или гиперлокомоции), вызванной d-амфетамином. Данная методика чувствительна к клинически значимым антагонистам дофаминовых рецепторов и, таким образом, считается подходящей для сравнения агонистов мускариновых рецепторов, которые оказывают влияние на дофаминергический путь. Выявившую ранее способность значительно уменьшать индуцированную d-амфетамином гиперактивность, дозу ксаномелина использовали в качестве положительного контроля. Статистический анализ обычно включал трехсторонний анализ ковариаций или устойчивой регрессии с лечением, день и подачу питания в качестве факторов и активность за 30 минут до лечения как ковариацию, с последующими подходящими тестами множественных сравнений. Значение А P<0,05 считали статистически значимым и соответственно обозначили на всех последующих фигурах. Данные для Примеров 1-21 Изомера 2, 1-32 Изомера 2, 1-33 Изомера 2, 2-7 Изомера 2 и 2-17 Изомера 2 проиллюстрированы на Фигуре 2.
ПРИМЕР D
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
(i) Таблеточные композиции
Таблеточную композицию, содержащую соединение формулы (1), (1а) или (1b), получают смешиванием 50 мг соединения с 197 мг лактозы (BP) в качестве разбавителя и 3 мг стеарата магния в качестве смазывающего вещества и, сжимая известным способом в форме таблетки.
(ii) Композиция в капсуле
Композицию в капсуле получают смешиванием 100 мг соединения формулы (1), (1а) или (1b) с 100 мг лактозы и необязательно 1% по массе стеарата магния и полученной смесью заполняют стандартную светонепроницаемую твердую желатиновую капсулу.
Эквиваленты
Вышеизложенные примеры представлены в целях иллюстрации изобретения и не должны подразумевать наложение каких-либо ограничений на объем изобретения. Очевидно, что большое число модификаций и изменений может быть осуществлено в специфических вариантах реализации изобретения, описанных выше, и проиллюстрированных в примерах, без отхождения от принципов, лежащих в основе данного изобретения. Все такие модификации и изменения осуществляются с целью быть охваченными данной заявкой.
| название | год | авторы | номер документа |
|---|---|---|---|
| Бициклические азотсодержащие соединения как агонисты М1 мускариновых рецепторов | 2019 |
|
RU2811601C1 |
| АГОНИСТЫ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2016 |
|
RU2737158C2 |
| ОКСАДИАЗОЛЫ В КАЧЕСТВЕ АГОНИСТОВ МУСКАРИНОВОГО РЕЦЕПТОРА M1 И/ИЛИ M4 | 2019 |
|
RU2817018C2 |
| СПИРО-КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ПИПЕРИДИНА ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КАЛИЕВОГО КАНАЛА НАРУЖНОГО МЕДУЛЛЯРНОГО СЛОЯ | 2013 |
|
RU2642066C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПРОСТОГО ЭФИРА ИЗОКСАЗОЛИЛА В КАЧЕСТВЕ ПАМ ГАМК A АЛЬФА5 | 2019 |
|
RU2800160C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2019 |
|
RU2821941C2 |
| КЛАСС БИФУНКЦИОНАЛЬНЫХ ХИМЕРНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ДЛЯ НАПРАВЛЕННОГО РАЗРУШЕНИЯ АНДРОГЕННЫХ РЕЦЕПТОРОВ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2825000C2 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2019 |
|
RU2809257C2 |
| СПИРО-АМИНОСОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ НАРУШЕНИЙ СНА И ЛЕКАРСТВЕННОГО ПРИВЫКАНИЯ | 2010 |
|
RU2562609C2 |
| ПРОИЗВОДНЫЕ АЗАСПИРОАЛКАНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕАЗ | 2004 |
|
RU2379303C2 |
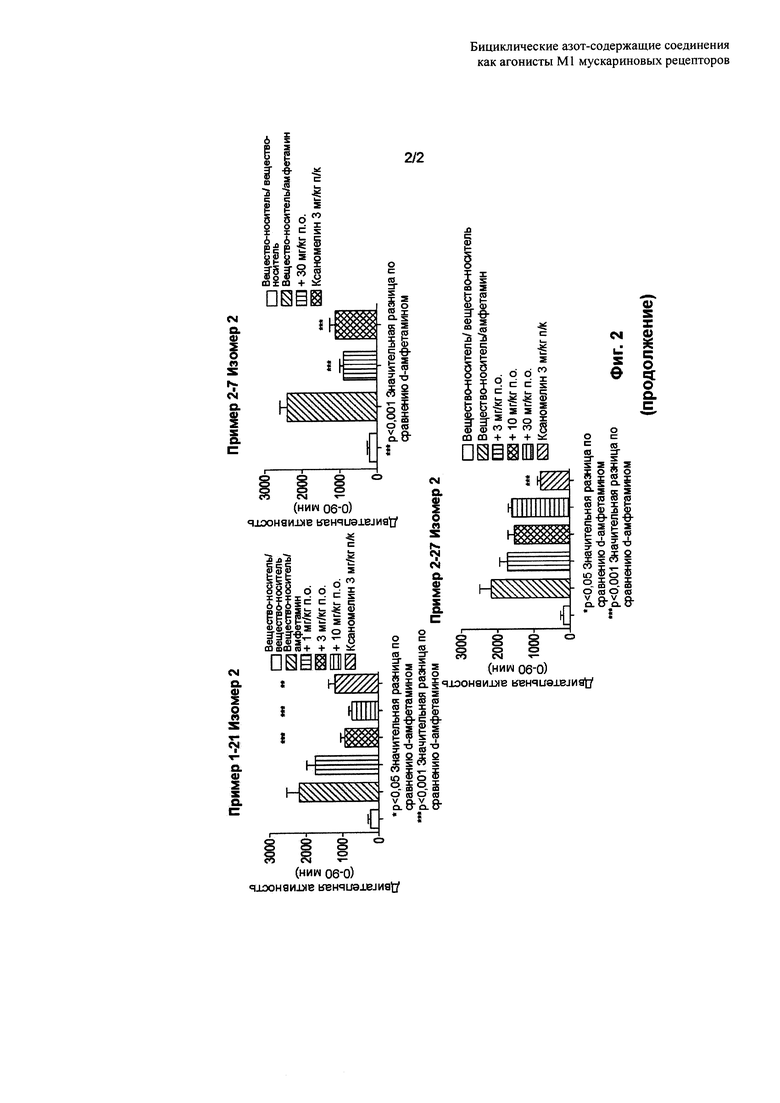
Изобретение относится к соединению формулы (1b)
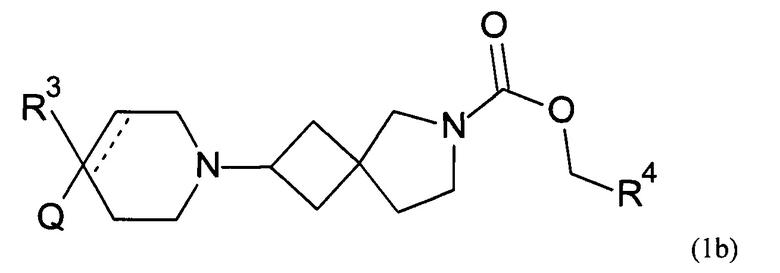
или его фармацевтически приемлемым солям, где Q представляет собой пяти-, или шести-, или семичленное гетероциклическое кольцо, содержащее 1, 2, 3 или 4 члена гетероатомного кольца, выбранных из N, О и S, где указанное гетероциклическое кольцо необязательно замещено одним, двумя или тремя заместителями, которые могут быть одинаковыми или разными и выбраны из (L)-R10, где L представляет собой связь или группу СН2 и R10 независимо выбран из водорода, фтора, хлора, брома, циано, оксо, гидрокси, OR15, NR15R16, COR15, CSR15, COOR15, COSR15, CONR15R16, CSNR15R16, SR15, SOR15, SO2R15, С1-6алкильной, С3-6циклоалкильной, С4-6циклоалкилалкильной, С4-6алкилциклоалкильной или С5-6алкилциклоалкилалкильной группы, которая необязательно замещена от одного до шести атомами фтора и в которой один или два, но не все, атомы углерода могут быть необязательно заменены гетероатомом, выбранным из О, N и S; и 5- или 6-членной арильной или гетероарильной группы, содержащей 0, 1, 2, 3 или 4 гетероатома, выбранных из О, N и S, которая необязательно замещена C1-6алкильной, С3-6циклоалкильной, С4-6циклоалкилалкильной, С4-6алкилциклоалкильной или С5-6алкилциклоалкилалкильной группой; где R15 и R16 являются одинаковыми или разными или могут быть соединены вместе с образованием кольца и каждый независимо выбран из водорода, C1-6алкильной, С3-6циклоалкильной, С4-6циклоалкилалкильной, С4-6алкилциклоалкильной или С5-6алкилциклоалкилалкильной группы, которая необязательно замещена 1-3 атомами фтора и в которой один или два, но не все, атомы углерода могут быть необязательно заменены гетероатомом, выбранным из О, N и S; или группы формулы -CH2CH2NHC(О)OCH2-фенил; или 5,7-дигидро-6H-пирроло[3,4-b]пиридин-6-ильной группы; или группы формулы (L)-R18, где L представляет собой связь или группу СН2 либо СН2СН2 и R18 представляет собой 5- или 6-членное кольцо, содержащее 0, 1, 2 или 3 гетероатома, выбранных из О, N и S, которое необязательно замещено одним или двумя атомами фтора, хлора или брома; R3 выбран из водорода, циано, гидрокси, амино и С1-9алкильной, С3-9циклоалкильной, С4-9циклоалкилалкильной, С4-9алкилциклоалкильной или С5-9алкилциклоалкилалкильной группы, в которой один атом углерода может быть необязательно заменен О; и R4 представляет собой водород или С1-6алкильную, С3-6циклоалкильную, С4-6циклоалкилалкильную, С4-6алкилциклоалкильную или С5-6алкилциклоалкилалкильную группу, которая необязательно замещена одним атомом фтора, которые представляют собой агонисты мускаринового М1 рецептора и/или М4 рецептора и которые пригодны для лечения опосредованных мускариновыми М1/М4 рецепторами заболеваний. 2 н. и 17 з.п. ф-лы, 2 ил., 4 табл.
1. Соединение формулы (1b)
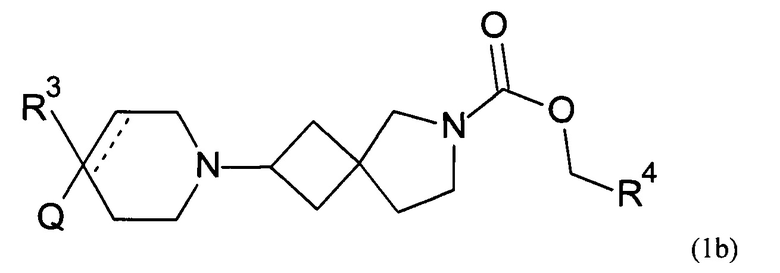
или его фармацевтически приемлемая соль,
где Q представляет собой пяти- или шести-, или семичленное гетероциклическое кольцо, содержащее 1, 2, 3 или 4 члена гетероатомного кольца, выбранных из N, О и S;
где указанное гетероциклическое кольцо необязательно замещено одним, двумя или тремя заместителями, которые могут быть одинаковыми или разными и выбраны из (L)-R10, где L представляет собой связь или группу СН2 и R10 независимо выбран из водорода, фтора, хлора, брома, циано, оксо, гидрокси, OR15, NR15R16, COR15, CSR15, COOR15, COSR15, CONR15R16, CSNR15R16, SR15, SOR15, SO2R15, С1-6алкильной, С3-6циклоалкильной, С4-6циклоалкилалкильной, С4-6алкилциклоалкильной или С5-6алкилциклоалкилалкильной группы, которая необязательно замещена 1-6 атомами фтора и в которой один или два, но не все, атомы углерода могут быть необязательно заменены гетероатомом, выбранным из О, N и S; и 5- или 6-членной арильной или гетероарильной группы, содержащей 0, 1, 2, 3 или 4 гетероатома, выбранных из О, N и S, которая необязательно замещена C1-6алкильной, С3-6циклоалкильной, С4-6циклоалкилалкильной, С4-6алкилциклоалкильной или С5-6алкилциклоалкилалкильной группой;
где R15 и R16 являются одинаковыми или разными или могут быть соединены вместе с образованием кольца и каждый независимо выбран из водорода, C1-6алкильной, С3-6циклоалкильной, С4-6циклоалкилалкильной, С4-6алкилциклоалкильной или С5-6алкилциклоалкилалкильной группы, которая необязательно замещена 1-3 атомами фтора и в которой один или два, но не все, атомы углерода могут быть необязательно заменены гетероатомом, выбранным из О, N и S; или группы формулы -CH2CH2NHC(О)OCH2-фенил; или 5,7-дигидро-6H-пирроло[3,4-b]пиридин-6-ильной группы; или группы формулы (L)-R18, где L представляет собой связь или группу СН2 либо СН2СН2 и R18 представляет собой 5- или 6-членное кольцо, содержащее 0, 1, 2 или 3 гетероатома, выбранных из О, N и S, которое необязательно замещено одним или двумя атомами фтора, хлора или брома;
R3 выбран из водорода, циано, гидрокси, амино и С1-9алкильной, С3-9циклоалкильной, С4-9циклоалкилалкильной, С4-9алкилциклоалкильной или С5-9алкилциклоалкилалкильной группы, в которой один атом углерода может быть необязательно заменен О; и
R4 представляет собой водород или С1-6алкильную, С3-6циклоалкильную, С4-6циклоалкилалкильную, С4-6алкилциклоалкильную или С5-6алкилциклоалкилалкильную группу, которая необязательно замещена одним атомом фтора.
2. Соединение по п. 1, отличающееся тем, что Q представляет собой ароматическое пяти-, или шести-, или семичленное гетероциклическое кольцо, содержащее 1, 2, 3 или 4 члена гетероатомного кольца, выбранных из N, О и S.
3. Соединение по п. 2, отличающееся тем, что Q представляет собой ароматическое пяти-, или шести-, или семичленное гетероциклическое кольцо, содержащее один или два атома азота.
4. Соединение по п. 3, отличающееся тем, что Q представляет собой кольцо (i) имидазола или (ii) пиразола.
5. Соединение по п. 1, отличающееся тем, что Q представляет собой кольцо (i) пиперидин-2-она или (ii) пирролидина.
6. Соединение по п. 1, отличающееся тем, что Q представляет собой 5, 6 или 7-членное ненасыщенное гетероциклическое кольцо, содержащее 1, 2, 3 или 4 члена гетероатомного кольца, выбранных из N, О и S.
7. Соединение по п. 1, отличающееся тем, что фрагмент
выбран из групп от AAA до АСВ, от ВАА до BCY, от САА до CBZ, от DAA до DBG или от ЕАА до ЕАВ:
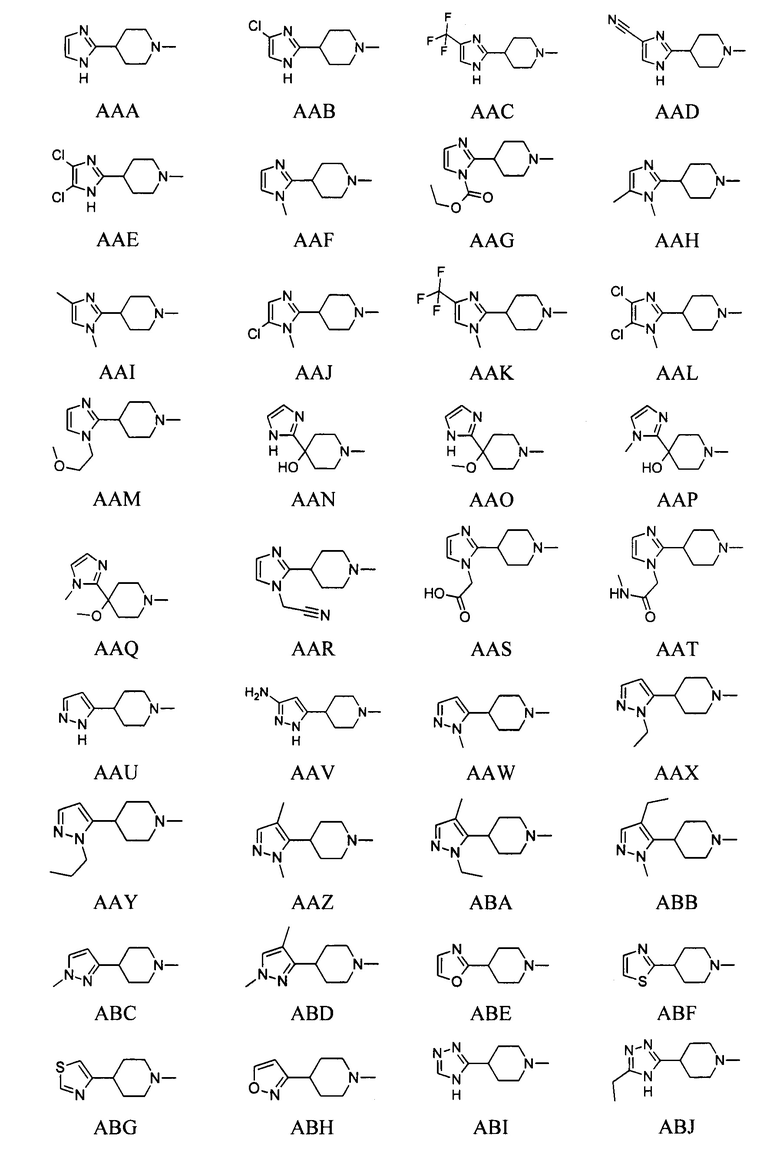
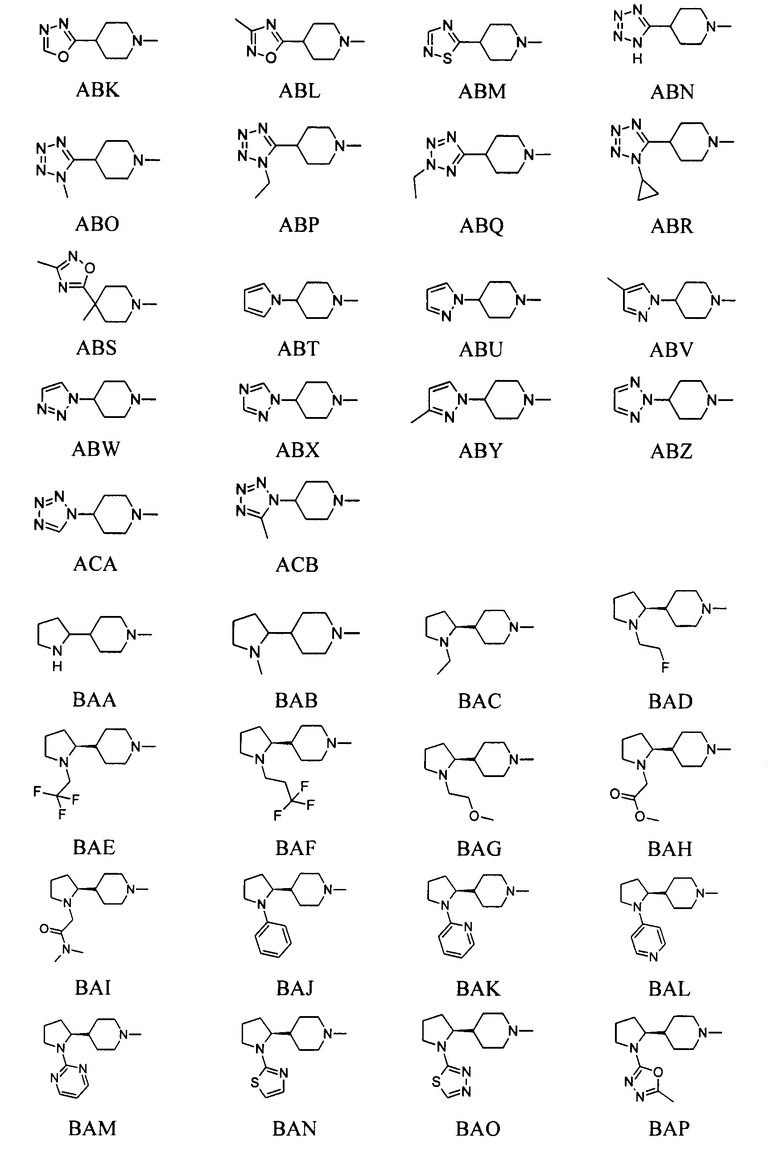
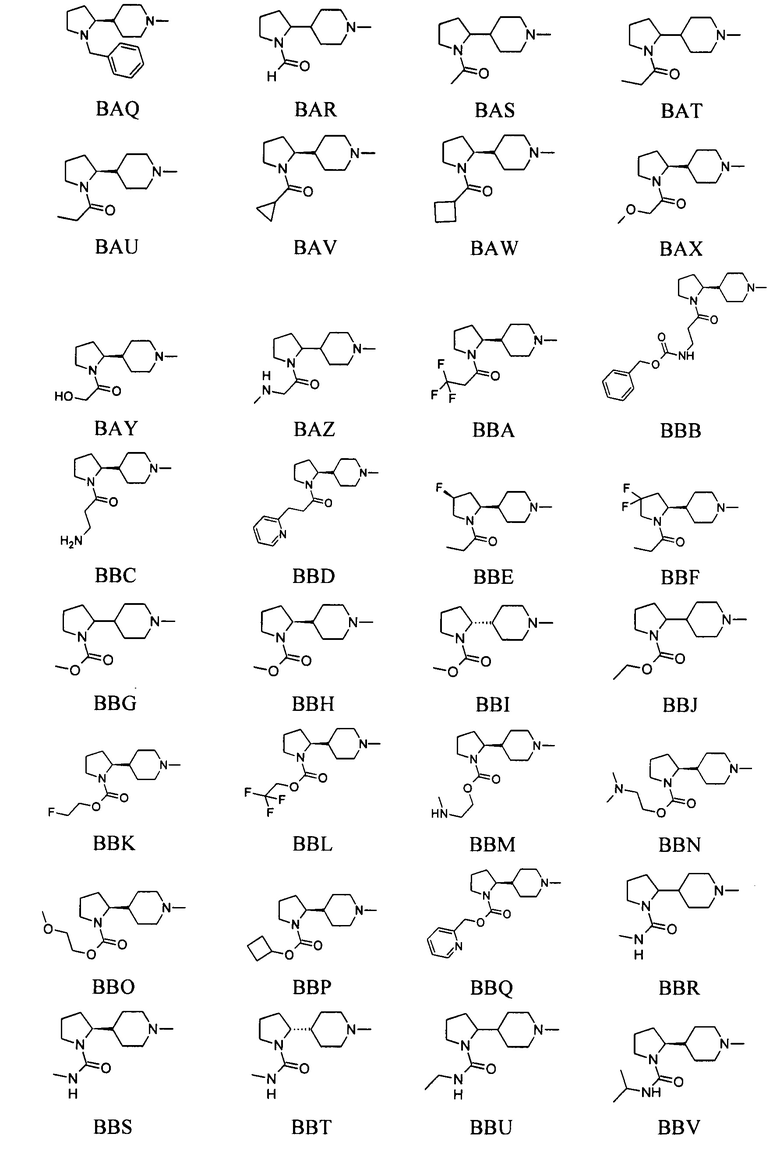
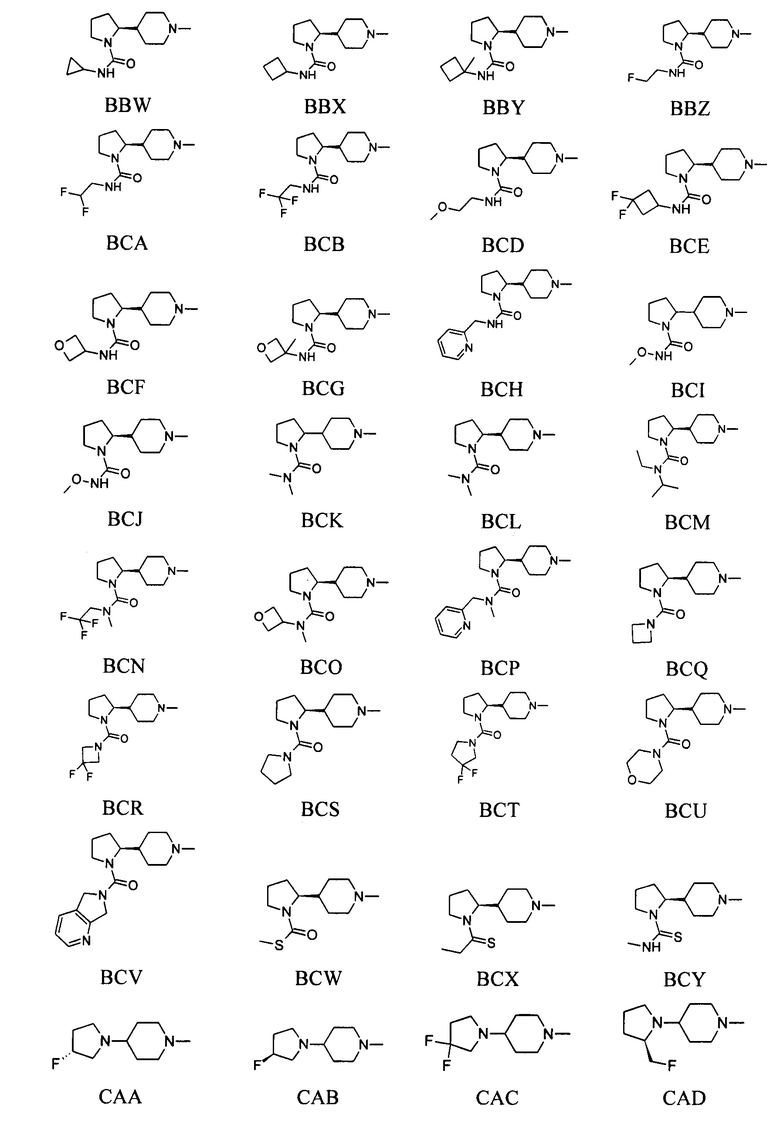
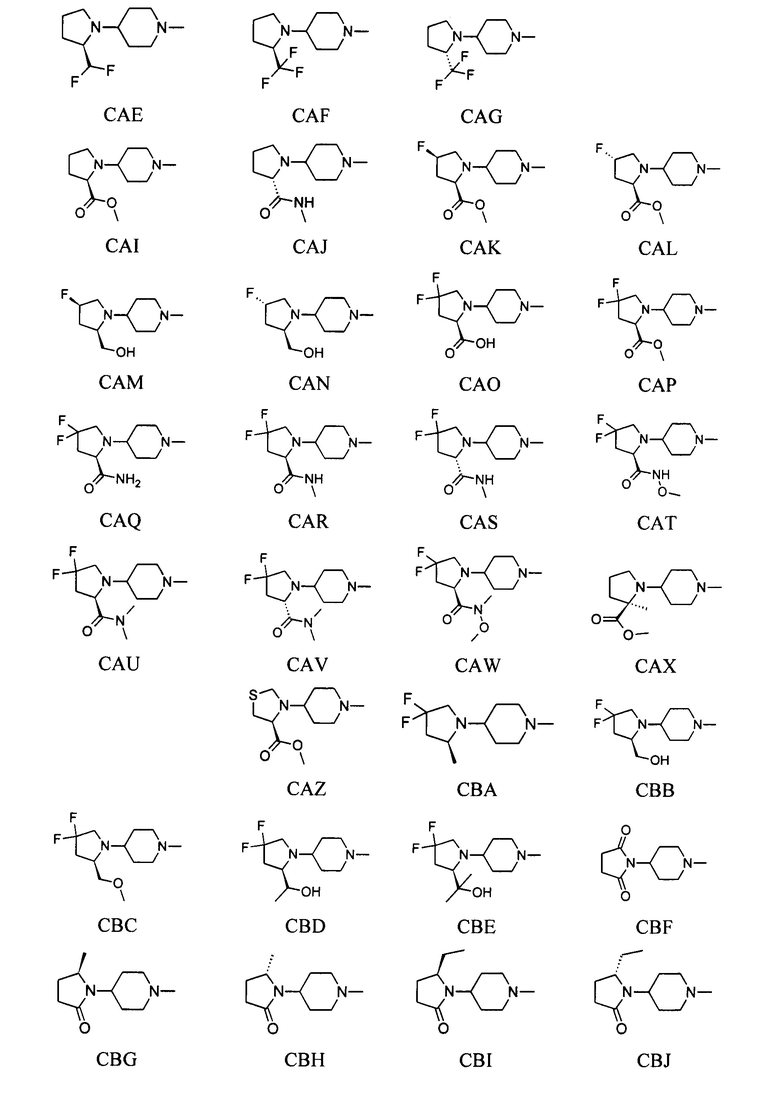
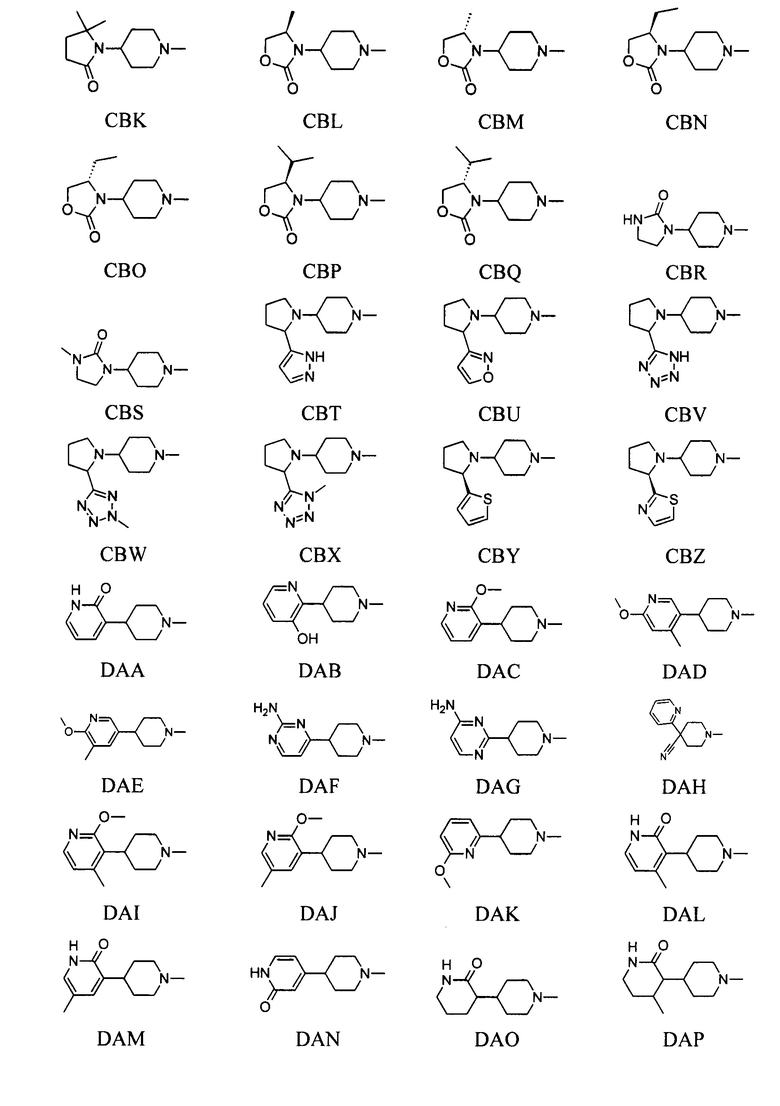
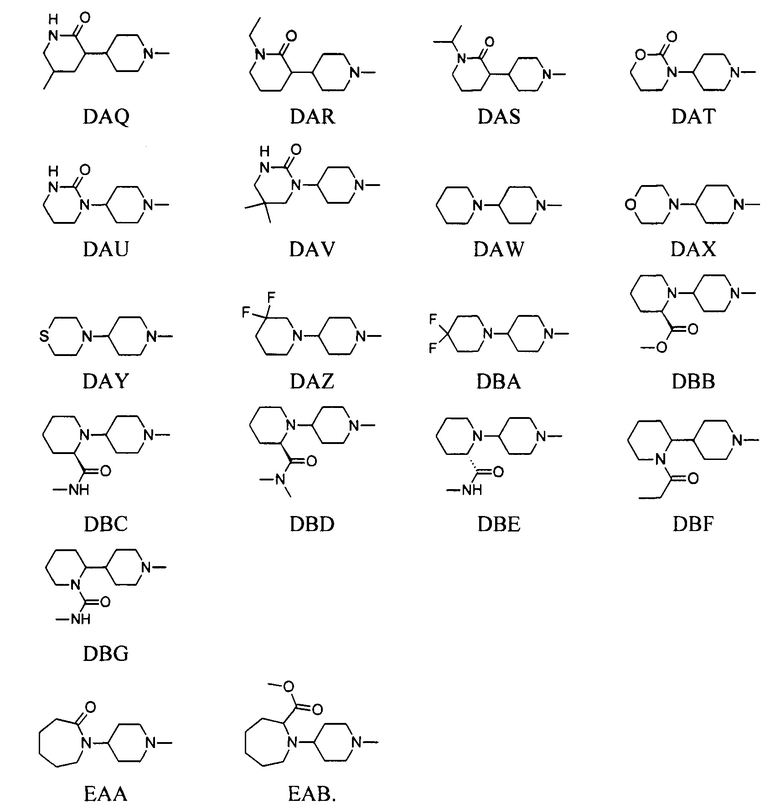
8. Соединение по п. 1, отличающееся тем, что R10 выбран из водорода, фтора, циано, гидрокси, OR15, NR15R16, COR15, COOR15, CONR15R16 и С1-4алкильной группы, которая необязательно замещена 1-3 атомами фтора.
9. Соединение по п. 8, отличающееся тем, что R10 выбран из водорода, NH2, COR15, COOR15 и С1-4алкильной группы, которая необязательно замещена 1-3 атомами фтора, и при этом R15 выбран из С1-4алкила.
10. Соединение по п. 8, отличающееся тем, что R10 выбран из водорода, метила, этила, СООМе, COOEt, СОМе, COEt, CF3, CONHMe, CON(Me)2, COCF3, CO-циклопропила, СО-циклобутила, CONHEt, СОН и NH2.
11. Соединение по п. 1, отличающееся тем, что R3 выбран из водорода, фтора, гидроксила, метокси и циано.
12. Соединение по п. 11, отличающееся тем, что R3 представляет собой водород.
13. Соединение по п. 1, отличающееся тем, что R4 выбран из водорода и метила.
14. Соединение по п. 1, которое представляет собой
Этил-2-[4-(1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(4-хлор-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[4-(трифторметил)-1Н-имидазол-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(4-циано-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(4,5-дихлор-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-[4-(1-метил-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1-метил-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[1-(этоксикарбонил)-1Н-имидазол-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-[4-(1,5-диметил-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1,5-диметил-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-[4-(1,4-диметил-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1,4-диметил-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-[4-(5-хлор-1-метил-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(5-хлор-1-метил-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[1-метил-4-(трифторметил)-1Н-имидазол-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-[4-(4,5-дихлор-1-метил-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(4,5-дихлор-1-метил-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1Н-пиразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(3-амино-1Н-пиразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-[4-(1-метил-1Н-пиразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1-метил-1Н-пиразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1-метил-1Н-пиразол-3-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1,3-оксазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-[4-(1,3-тиазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1,3-тиазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(4Н-1,2,4-триазол-3-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(5-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1,3,4-оксадиазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(3-метил-1,2,4-оксадиазол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1,2,4-тиадиазол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1Н-тетразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1Н-пиррол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1Н-пиразол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(4-метил-1Н-пиразол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1Н-1,2,3-триазол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1Н-1,2,4-триазол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-гидрокси-4-(1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1Н-имидазол-2-ил)-4-метоксипиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-гидрокси-4-(1-метил-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-метокси-4-(1-метил-1Н-имидазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-метил-4-(3-метил-1,2,4-оксадиазол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-[4-(4-метил-1H-пиразол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1,2-оксазол-3-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(3-метил-1Н-пиразол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-[4-(3-метил-1Н-пиразол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-[4-(1-этил-1Н-пиразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1-этил-1Н-пиразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-[4-(1-пропил-1Н-пиразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1-пропил-1Н-пиразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1,3-тиазол-4-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[1-(2-метоксиэтил)-1Н-имидазол-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[1-(цианометил)-1Н-имидазол-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
(2-{1-[6-(этоксикарбонил)-6-азаспиро[3.4]окт-2-ил]пиперидин-4-ил}-1Н-имидазол-1-ил)уксусная кислота,
Этил-2-(4-{1-[2-(метиламино)-2-оксоэтил]-1Н-имидазол-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-[4-(1,4-диметил-1Н-пиразол-3-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1,4-диметил-1Н-пиразол-3-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-[4-(1,4-диметил-1Н-пиразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1,4-диметил-1Н-пиразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1-этил-4-метил-1Н-пиразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(4-этил-1-метил-1H-пиразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(2H-1,2,3-триазол-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1Н-тетразол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(5-метил-1H-тетразол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1-метил-1Н-тетразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1-этил-1H-тетразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(2-этил-2H-тетразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1-циклопропил-1Н-тетразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
(1,1-2Н2)Этил-2-[4-(1H-пиразол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
(2,2,2-2Н3)Этил-2-[4-(1H-пиразол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
(2Н5)Этил-2-[4-(1H-пиразол-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
(1,1-2Н2)Этил-2-[4-(1-метил-1H-пиразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
(2,2,2-2Н3)Этил-2-[4-(1-метил-1H-пиразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
(2Н5)Этил-2-[4-(1-метил-1H-пиразол-5-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(пирролидин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1-формилпирролидин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1-ацетилпирролидин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[1-(трифторацетил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-[4-(1-пропаноилпирролидин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1-пропаноилпирролидин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-[(2S)-1-пропаноилпирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(циклопропилкарбонил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро [3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(циклобутилкарбонил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-{4-[1-(метоксикарбонил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[1-(метоксикарбонил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(метоксикарбонил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R)-1-(метоксикарбонил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[1-(этоксикарбонил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-{4-[(2S)-1-(метилкарбамоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[1-(метилкарбамоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(метилкарбамоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R)-1-(метилкарбамоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[1-(этилкарбамоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[1-(диметилкарбамоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(диметилкарбамоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(1-метилпирролидин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[1-(N-метилглицил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[1-(метоксикарбамоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(пропан-2-илкарбамоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[(2,2,2-трифторэтил)карбамоил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(азетидин-1-илкарбонил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(морфолин-4-илкарбонил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(циклопропилкарбамоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(циклобутилкарбамоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[(2-метоксиэтил)карбамоил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(пирролидин-1-илкарбонил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-[(2S)-1-(метоксикарбамоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[метокси(метил)карбамоил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[(1-метилциклобутил)карбамоил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[(3-метилоксетан-3-ил)карбамоил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[(3,3-дифторпирролидин-1-ил)карбонил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[(3,3-дифторциклобутил)карбамоил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[(3,3-дифторазетидин-1-ил)карбонил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-(4-{(2S)-1-[(2,2,2-трифторэтил)карбамоил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-(4-{(2S)-1-[(3,3-дифторазетидин-1-ил)карбонил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[этил(пропан-2-ил)карбамоил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[(циклобутилокси)карбонил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[(2-фторэтил)карбамоил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро [3.4] октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[(2,2-дифторэтил)карбамоил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(метоксиацетил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[(2-фторэтокси)карбонил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[(2,2,2-трифторэтокси)карбонил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[(метилсульфанил)карбонил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[(2-метоксиэтокси)карбонил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-{[2-(диметиламино)этокси]карбонил}пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-[(2S)-1-(гидроксиацетил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(3,3,3-трифторпропаноил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[(пиридин-2-илметил)карбамоил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[метил(2,2,2-трифторэтил)карбамоил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(оксетан-3-илкарбамоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[метил(оксетан-3-ил)карбамоил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-пропантиоилпирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S,4S)-4-фтор-1-пропаноилпирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-4,4-дифтор-1-пропаноилпирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-этилпирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[3-(пиридин-2-ил)пропаноил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[метил(пиридин-2-илметил)карбамоил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[(пиридин-2-илметокси)карбонил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4] октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-{N-[(бензилокси)карбонил]-β-аланил}пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(β-аланил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-{[2-(метиламино)этокси]карбонил}пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(2-фторэтил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(2,2,2-трифторэтил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(3,3,3-трифторпропил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(2-метоксиэтил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(2-метокси-2-оксоэтил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2S)-1-[2-(диметиламино)-2-оксоэтил]пирролидин-2-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-бензилпирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(метилкарбамотиоил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-фенилпирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-{4-[(2S)-1-(пиридин-2-ил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(пиридин-2-ил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(пиридин-4-ил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(пиримидин-2-ил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(1,3-тиазол-2-ил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(1,3,4-тиадиазол-2-ил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R)-2-(метоксикарбонил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S-2-(метилкарбамоил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
1-{1-[6-(этоксикарбонил)-6-азаспиро[3.4]окт-2-ил]пиперидин-4-ил}-4,4-дифтор-D-пролин,
Этил-2-{4-[(2R)-4,4-дифтор-2-(метоксикарбонил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R)-4,4-дифтор-2-(метилкарбамоил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R)-2-(диметилкарбамоил)-4,4-дифторпирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R)-2-карбамоил-4,4-дифторпирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R)-4,4-дифтор-2-(метоксикарбамоил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-{(2R)-4,4-дифтор-2-[метокси(метил)карбамоил] пирролидин-1-ил}пиперидин-1-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-4,4-дифтор-2-(метилкарбамоил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-2-(диметилкарбамоил)-4,4-дифторпирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R)-2-(метоксикарбонил)-2-метилпирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(4S)-4-(метоксикарбонил)-1,3-тиазолидин-3-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(3R)-3-фторпирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(3S)-3-фторпирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(3,3-дифторпирролидин-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-2-(трифторметил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R)-2-(фторметил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R)-2-(дифторметил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R)-2-(трифторметил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(3-азабицикло[3.1.0]гекс-3-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-4,4-дифтор-2-метилпирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R)-4,4-дифтор-2-(гидроксиметил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R)-4,4-дифтор-2-(метоксиметил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R)-4,4-дифтор-2-(1-гидроксиэтил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R)-4,4-дифтор-2-(2-гидроксипропан-2-ил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(2-оксопирролидин-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(2,5-диоксопирролидин-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R)-2-метил-5-оксопирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R)-2-этил-5-оксопирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-2-метил-5-оксопирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-2-этил-5-оксопирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(2,2-диметил-5-оксопирролидин-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(4R)-4-метил-2-оксо-1,3-оксазолидин-3-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(4R)-4-этил-2-оксо-1,3-оксазолидин-3-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(4R)-2-оксо-4-(пропан-2-ил)-1,3-оксазолидин-3-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(4S)-4-метил-2-оксо-1,3-оксазолидин-3-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(4S)-4-этил-2-оксо-1,3-оксазолидин-3-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(4S)-2-оксо-4-(пропан-2-ил)-1,3-оксазолидин-3-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(2-оксоимидазолидин-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(3-метил-2-оксоимидазолидин-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[2-(1Н-пиразол-5-ил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[2-(1,2-оксазол-3-ил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[2-(1H-тетразол-5-ил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[2-(2-метил-2H-тетразол-5-ил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[2-(1-метил-1H-тетразол-5-ил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R)-2-(тиофен-2-ил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R)-2-(1,3-тиазол-2-ил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(5,7-дигидро-6H-пирроло[3,4-b]пиридин-6-илкарбонил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R,4R)-4-фтор-2-(метоксикарбонил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R,4S)-4-фтор-2-(метоксикарбонил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R,4R)-4-фтор-2-(гидроксиметил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2R,4S)-4-фтор-2-(гидроксиметил)пирролидин-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[(2S)-1-(5-метил-1,3,4-оксадиазол-2-ил)пирролидин-2-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-[4-(2-оксо-1,2-дигидропиридин-3-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(2-оксо-1,2-дигидропиридин-3-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
2-Фторэтил-2-[4-(2-оксо-1,2-дигидропиридин-3-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(3-гидроксипиридин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
2-Фторэтил-2-[4-(2-метоксипиридин-3-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(6-метокси-4-метилпиридин-3-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(6-метокси-5-метилпиридин-3-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(2-аминопиримидин-4-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(4-аминопиримидин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-циано-4-(пиридин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(2-метокси-4-метилпиридин-3-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(2-метокси-5-метилпиридин-3-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(6-метоксипиридин-2-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(4-метил-2-оксо-1,2-дигидропиридин-3-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(5-метил-2-оксо-1,2-дигидропиридин-3-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(2-оксо-1,2-дигидропиридин-4-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(2-оксо-3,4'-бипиперидин-1'-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-2-(2-оксо-3,4'-бипиперидин-1'-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4-метил-2-оксо-3,4'-бипиперидин-1'-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(5-метил-2-оксо-3,4'-бипиперидин-1'-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(1-этил-2-оксо-3,4'-бипиперидин-1'-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[2-оксо-1-(пропан-2-ил)-3,4'-бипиперидин-1'-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(2-оксо-1,3-оксазинан-3-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(2-оксотетрагидропиримидин-1(2H)-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(5,5-диметил-2-оксотетрагидропиримидин-1(2H)-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(1,4'-бипиперидин-1'-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(морфолин-4-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(тиоморфолин-4-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(3,3-дифтор-1,4'-бипиперидин-1'-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(4,4-дифтор-1,4'-бипиперидин-1'-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Метил-(2R)-1'-[6-(этоксикарбонил)-6-азаспиро[3.4]окт-2-ил]-1,4'-бипиперидин-2-карбоксилат,
Этил-2-[(2R)-2-(метилкарбамоил)-1,4'-бипиперидин-1'-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[(2R)-2-(диметилкарбамоил)-1,4'-бипиперидин-1'-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[(2S)-2-(метилкарбамоил)-1,4'-бипиперидин-1'-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-(1-пропаноил-2,4'-бипиперидин-1'-ил)-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[1-(метилкарбамоил)-2,4'-бипиперидин-1'-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-[4-(2-оксоазепан-1-ил)пиперидин-1-ил]-6-азаспиро[3.4]октан-6-карбоксилат,
Этил-2-{4-[2-(метоксикарбонил)азепан-1-ил]пиперидин-1-ил}-6-азаспиро[3.4]октан-6-карбоксилат.
15. Соединение по п. 1 или его фармацевтически приемлемая соль, обладающие активностью агониста мускаринового M1 рецептора и/или М4 рецептора.
16. Фармацевтическая композиция, содержащая эффективное количество соединения по п. 15 или его фармацевтически приемлемой соли и фармацевтически приемлемое вспомогательное вещество.
17. Соединение по п. 1 или его фармацевтически приемлемая соль для применения при лечении когнитивного расстройства или психического расстройства или для лечения или уменьшения тяжести острой, хронической, невропатической или воспалительной боли.
18. Соединение по п. 1 или его фармацевтически приемлемая соль, отличающиеся тем, что проявляют селективность для рецептора M1 и/или рецептора M1 и М4 относительно М2 и М3 подтипов рецептора для применения при лечении болезни Альцгеймера, деменции с тельцами Леви и других когнитивных расстройств, или для лечения или уменьшения тяжести острой, хронической, невропатической или воспалительной боли, или для лечения зависимости, или для лечения двигательных расстройств.
19. Соединение по п. 1 или его фармацевтически приемлемая соль, отличающиеся тем, что показывают селективность для рецептора М4 относительно M1, М2 и М3 подтипов рецептора, для применения в лечении шизофрении или других психических расстройств, или для лечения или уменьшения тяжести острой, хронической, невропатической или воспалительной боли, или для лечения зависимости, или для лечения двигательных расстройств.
| WO 2013072705 A1, 23.05.2013 | |||
| WO 2007100670 A1, 07.09.2007 | |||
| RU 2010110036 A, 20.10.2011 | |||
| WO 2005117883 A1, 15.12.2005. |

