Область техники
Изобретение относится к области полимерных композиционных материалов (ПКМ), а именно к аппретированию углеродных волокон с целью повышения их прочности, а также улучшения совместимости последних со связующим и повышения физико-механических характеристик получаемых из волокон композиционных материалов, которые могут быть использованы в химической, нефтяной и металлургической промышленности, авиатехнике для создания изделий и элементов конструкций, подвергающихся воздействию повышенных температур.
Уровень техники
Углепластики, обладающие комплексом ценных свойств - низкой плотностью, высоким модулем упругости, прочностью, высокой износостойкостью, низким коэффициентом линейного температурного расширения (КЛТР), нашли широкое применение в авиакосмической технике, автомобилестроении, при производстве спортивного оборудования, промышленных товаров - там, где требуется высокая прочность материала в сочетании с низким весом. К недостаткам углепластиков как на основе термореактивного, так и термопластичного связующих относится низкая прочность при сдвиге, что связано с недостаточной адгезией полимерных, особенно термопластичных, связующих к поверхности углеродных волокон (УВ). Адгезия на границе раздела «УВ-полимерная матрица» определяется наличием механических связей вследствие проникновения полимера в шероховатости поверхности УВ, химическими связями между поверхностью УВ и полимерной матрицей и взаимодействием, обусловленным действием сил Ван-дер-Ваальса.
Необходимое для обеспечения хорошей адгезии возникновение химических связей в системе «УВ-полимер» может быть обеспечено формированием на поверхности УВ химически активных функциональных групп, способных реагировать с атомами и группами в цепочках полимера. С этой целью, а также для сохранения УВ, сразу после его изготовления, на волокно наносят замасливатели (аппреты), в качестве которых обычно используют водные эмульсии полимеров или олигомеров, или их растворы в органических растворителях. Большинство применяемых в Российской Федерации и за рубежом аппретов для углеродного волокна представляют собой эпоксидные смолы, обладающие низкой термической устойчивостью (до 200°С), которая отрицательно проявляется при высокотемпературных испытаниях углепластиков. Полимерный композиционный материал (ПКМ) получают посредством нанесения на аппретированные волокна связующего, в качестве которого могут быть использованы фталонитрильные матрицы, с последующим отверждением при температурах до 375°С. Распространенные эпоксидные аппреты разлагаются уже на этом этапе, в результате чего в материале (ПКМ) образуются микропоры на границе волокно-матрица, что приводит к ухудшению механических свойств композита, особенно при повышенных температурах, когда падает модуль упругости матрицы.
Аппрет служит для улучшения технологических характеристик волокон (https://en.wikipedia.org/wiki/Sizing), основная его функция - предотвращать распушение и истирание углеродного волокна при намотке его на бобины и во время текстильной обработки. Аппретирование углеродного волокна позволяет наматывать его на бобины малого диаметра без разрушения. Известен целый ряд способов аппретирования углеродных волокон с использованием различных полимеров и олигомеров. Аппретирование осуществляется путем пропускания углеродного волокна через раствор или дисперсию аппретирующего состава. Однако не все аппреты отвечают предъявляемым требованиям, а именно, обеспечивают хорошую смачиваемость углеродного волокна, образуя нелипкую пленку, обеспечивающую сохранность волокон и жгутов в процессе их дальнейшей обработки, снижают напряжение в зоне контакта связующего и углеродного волокна.
Известен способ получения полимерного композиционного материала (RU 2536969, 27.12.2014), включающий аппретирование углеродных лент или волокон путем нанесения аппретирующего материала из раствора с последующей сушкой и прессованием. Композиционный материал, армированный углеродными наполнителями, получают предварительной обработкой углеродной ленты (волокна) аппретирующим составом - полигидроксиэфиром на основе бисфенола А с молекулярной массой 50-60 тыс. в виде 3-7% растворов в легколетучих органических растворителях (хлороформ, тетрагидрофуран, промышленный растворитель 646 и др.). Такая обработка термопластичным аппретом -полигидроксиэфиром, повышает смачиваемость наполнителя полисульфоном, позволяет снизить температуру прессования на 30-40°С, а также многократно проводить при необходимости термообработку без изменения свойств аппрета. Углеродный наполнитель покрывают аппретирующим составом, затем высушивают до постоянного веса и собирают пакет, чередуя наполнитель с пленкой полисульфона, и прессуют на гидропрессе при давлении 1,0-2,0 МПа при температуре 250-260°С в течение 30-40 минут. Однако получить материал из углеродного волокна, аппретированного полигидроксиэфиром, сохраняющего механические свойства при температурах до 400°С не представляется возможным.
Из уровня техники известно углеродное волокно, имеющее устойчивое к разложению при температуре 400°С покрытие, образованное аморфным преполимером, полученным частичной полимеризацией бис-фталонитрильного мономера в присутствии ароматического амина (US 5389441, 14.02.1995). Для нанесения покрытия на волокна фталонитрильный мономер сплавляют с ароматическим диамином при температуре не менее 200°С в течение не менее 1 минуты, после чего полученный преполимер растворяют в органическом растворителе (дихлорметан, диметилсульфоксид и др.). В полученном растворе вымачивают неаппретированный жгут углеродного волокна или ткань, а затем углеродный материал высушивают от растворителя с получением аппретированного материала. Таким образом, для нанесения на волокно используют преполимер, чтобы придать пластичность наносимому покрытию (аппрету). Однако использование в процессе аппретирования органических растворителей затрудняет непрерывность процесса получения аппретированного волокна, так как нахождение легко воспламеняющихся жидкостей (ЛВЖ) в одном помещении с печью графитации недопустимо по правилам пожарной безопасности, и кроме того отходы от использования растворителей требуют утилизации.
Наиболее близким к заявляемому является способ изготовления углеродного волокна, включающий нанесение аппрета на углеродное волокно путем пропускания его через ванну, содержащую аммонийную соль полиамидокислоты с последующим нагреванием в диапазоне от 200°С до 450°С до образования покрытия (US 20140343218, 20.11.2014). Однако недостатком указанного способа является то, что для образования термостойкого полиимидного покрытия на волокне проводят стадию нагревания волокна с нанесенной солью полиамидокислоты, при которой происходит газовыделение, способное нарушить равномерность нанесения аппрета. При этом выделяющиеся газы могут быть токсичными (например, в случае использования аммонийных солей полиамидокислот).
Таким образом, из уровня техники не выявлено источников информации, из которых было бы известно аппретирование углеродных волокон фталонитрильными мономерами.
В заявляемом изобретении предлагается использовать низкоплавкие фталонитрильные мономеры в качестве аппрета для углеродных волокон для получения из них ПКМ с улучшенными механическими свойствами, в частности, сохраняющими стабильность при температурах до 450°С.
Раскрытие изобретения
Задачей заявляемого изобретения является разработка термостойкого аппрета и создание углеродного термостойкого аппретированного волокна, позволяющего получать из него прочный и стабильный при высоких температурах ПКМ (до 450°С).
Технический результат, достигаемый при использовании заявляемого изобретения, заключается в разработке соединений, обеспечивающих возможность использования их в составе для получения термостойкого (до 450°С) аппретирующего покрытия для углеродных волокон. Кроме того, технический результат также заключается в разработке способа аппретирования углеродного волокна, имеющего упрощенную, по сравнению с аналогами, технологию, меньшее количество технологических операций, необходимых для нанесения аппрета. ПКМ, полученные из аппретированных фталонитрилами волокон и с использованием в качестве связующего фталонитрильных матриц, обеспечивает повышение прочности получаемого из них ПКМ при межслоевом сдвиге и сдвиге в плоскости не менее, чем на 10%, по сравнению с аналогичными материалами, полученными из волокон, аппретированных эпоксидными смолами.
Следует отметить, что заявляемый состав для получения аппрета состоит из фталонитрильных мономеров (одного или смеси нескольких) и не требует при его нанесении на волокно использования ароматического амина или другого инициатора полимеризации (дифенолы, кислоты Льюиса). За счет этого значительно упрощается способ аппретирования углеродного волокна. При этом после пропитки такого аппретированного волокна связующим, например, фталонитрильным, происходит сополимеризация аппрета со связующим (так как связующее содержит инициатор полимеризации, например, ароматический диамин). В результате образуется прочная и термостойкая граница волокно-матрица, обеспечивающая сохранение механических свойств ПКМ при повышенных температурах.
Поставленная задача решается тем, что состав для получения аппретированного углеродного волокна включает, по меньшей мере, одно соединение формулы:
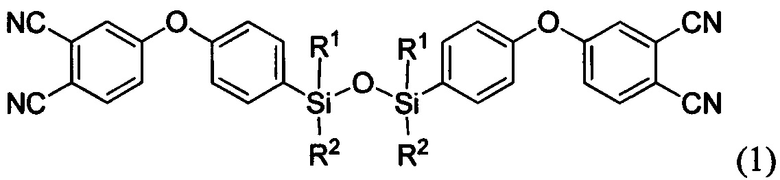
где R1=R2 и представляет собой метил или фенил, или в случае, когда R1 представляет собой метил, то R2 - фенил;
и/или, по меньшей мере, одно соединение формулы:
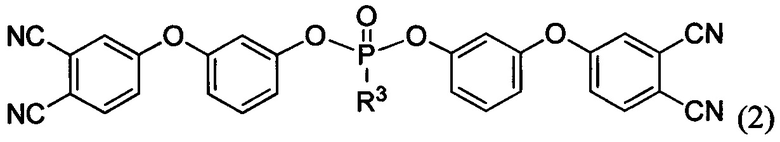
где R3 выбирают из группы, включающей арил, арилокси или алкилокси заместители.
Задача также решается посредством создания аппретированного углеродного волокна, характеризующегося наличием слоя аппрета указанного выше состава. При этом аппретированное волокно характеризуется содержанием аппрета в количестве 0,41-4,6 мас. % от общей массы аппретированного волокна. Более предпочтительно содержание аппрета составляет 1-2 мас. % от общей массы аппретированного волокна. Волокно также имеет краевой угол смачивания от 32 до 80 град, в легкоплавком фталонитрильном связующем (например, PN-3M), при этом оно сохраняет стабильность при температуре не менее 450°С.
Заявляется также способ получения аппретированного углеродного волокна,
характеризующийся нанесением на углеродное волокно описанного выше состава посредством контакта водной дисперсии мономеров указанного состава с углеродным волокном, после чего волокно сушат до достижения постоянного веса. Водная дисперсия мономеров имеет концентрацию от 0,4 до 2 мас. %. Мономеры для реализации заявленного способа выбирают с температурой стеклования не более 60°С. Нанесение состава на волокно осуществляют посредством погружения волокна в пропиточную ванну или его протягивания через ванну с обеспечением времени контакта волокна с водной дисперсией мономеров не менее 10 с, предпочтительно в течение 15-30 с. Сушку осуществляют при температуре от 100 до 400°С в течение 10-300 с, предпочтительно при температуре от 250 до 300°С в течение 20-200 с. Водную дисперсию получают посредством смешения мономеров с органическим растворителем, выбранным из группы: метанол, этанол, изопропанол, тетрагидрофуран, ацетон, с последующим добавлением воды и удалением органического растворителя с получением водной дисперсии с содержанием твердой фазы не более 20 масс. %. Состав дополнительно может содержать неионогенные поверхностно-активные вещества (ПАВ), выбранные из группы: ди- или триэтаноламин, полиэтиленгликоль в количестве до 5 мас. %, при этом ПАВ вносят в раствор до добавления воды.
В другом варианте реализации изобретения водную дисперсию можно получить расплавлением мономера с последующим его диспергированием в воде посредством ультразвукового воздействия с частотой 15-44 кГц с добавлением неионогенных ПАВ, выбранных из группы: ди- или триэтаноламин, полиэтиленгликоль, в количестве до 5 мас. %.
В одном из вариантов осуществления способа получения аппретированного углеродного волокна на водную дисперсию воздействую ультразвуком частотой 15-44 кГц в процессе ее нанесения на углеродные волокна. В качестве углеродных волокон могут быть использованы волокна с линейной плотностью от 0,06 до 4,0 г/м. В результате применения заявляемого способа получают волокно с содержанием аппрета 0,41-4,6 мас. % от общей массы аппретированного волокна, предпочтительно - волокно с содержанием аппрета 1-2 мас. % от общей массы аппретированного волокна, а также с краевым углом смачивания от 32 до 80 град в легкоплавком фталонитрильном связующем, например PN-3M, сохраняющего стабильность при температуре не менее 400°С.
Поставленная задача решается также тем, что полимерный композиционный материал в виде ленты, или полотна, или ткани, содержит заявляемое аппретированное волокно и связующее, в качестве которого может быть использовано фталонитрильное связующее.
Краткое описание чертежей
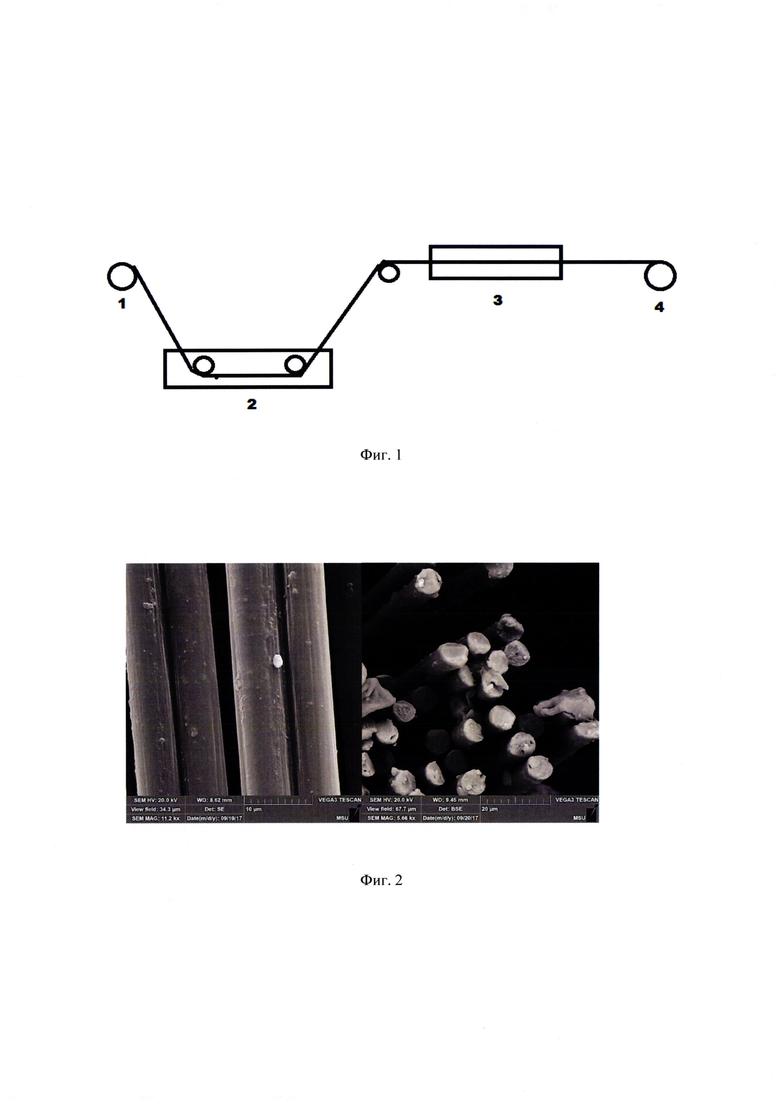
На фиг. 1 представлена возможная схема осуществления способа аппретирования углеродного волокна, где 1 - подающая бобина, 2 - пропиточная ванна, 3 - сушильная печь, 4 - приемная бобина.
На фиг. 2 представлена фотография углеродного волокна с нанесенным аппретом из примера 28.
На фиг. 3 представлена схема синтеза мономеров (9) и (10).
Термины и определения
Ниже приведены основные термины и определения, используемые при описании осуществления изобретения.
Углеродное волокно (УВ) - материал, состоящий из тонких нитей диаметром от 5 до 15 мкм, образованных, преимущественно, атомами углерода.
Аппрет - вещества/композиции/соединения, наносимые на поверхность материалов, придающие материалам определенные свойства.
Углеродные волокна могут иметь разнообразную текстильную форму, определяемую чаще всего формой исходного сырья (непрерывные или штапельные нити, жгуты, ленты и др.). Возможна также переработка углеродного волокна в тканые и нетканые материалы с использованием обычного текстильного оборудования.
Углепластики - композиционные полимерные материалы, армированные наполнителями из углеродных волокон в виде нитей, ленты, ткани. Углепластики характеризуются низкой плотностью, высокой механической прочностью, вибропрочностью, повышенной химический стойкостью, практически нулевым коэффициентом линейного расширения.
Осуществление изобретения
Круг фталонитрилов, пригодных для аппретирования, ограничивается аморфными бис-фталонитрилами с температурами стеклования не выше 60°С, чтобы при комнатной температуре аппрет оставался эластичным и не разрушался при технологических деформациях (то есть не крошился и не осыпался). Выбор класса фталонитрилов обусловлен тем, что фталонитрилы являются наиболее термостойкими матрицами для ПКМ из известных, с возможностью их эксплуатации при температурах выше 300°С. Большинство классических аппретов разрушаются при таких температурах (например, наиболее часто используемый эпоксидный), в связи с разрушением границы волокно-матрица в ПКМ и образованием пористости. Чтобы этого избежать, предлагается использовать аппреты на основе фталонитрильных мономеров, так как при нанесении на волокно связующего фталонитрильной природы, содержащего инициатор полимеризации, аппрет будет полимеризоваться вместе с матрицей, встраиваясь в ее структуру. При этом полимер, образованный из аппрета, должен выдерживать высокие температуры (те же, что и матрица). В связи с перечисленными выше требованиями были выявлены и разработаны соединения фталонитрилов и их смеси для использования в качестве аппретов (см. соединения, представленные формулами 1 и 2).
где R1=R2 и представляет собой метил или фенил, или в случае, когда R1 представляет собой метил, то R2 - фенил; и фосфорсодержащие фталонитрильные мономеры:
где R3 выбирают из группы, включающей арил, арилокси или алкилокси заместители.
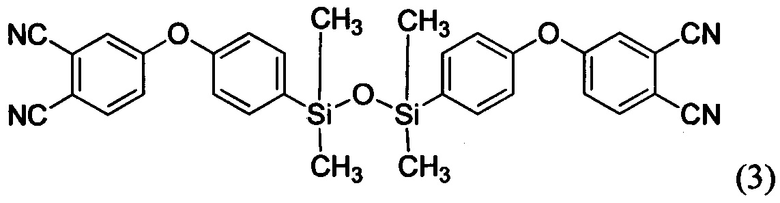
При этом следует отметить, что соединение формулы (3) и способ получения данного соединения известно из источника Dzhevakov Р.В. et al. Synthesis and polymerization of disiloxane Si-O-Si-linked phthalonitrile monomer // Mendeleev Commun. 2016. Vol. 26, №6. P. 527-529.
Ниже приведены конкретные примеры фосфорсодержащих фталонитрильных мономеров, демонстрирующие достижение заявляемого результата.
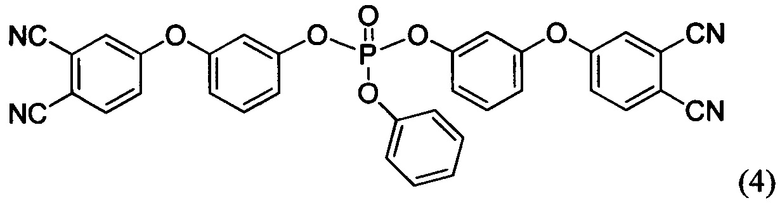
Соединения формул (4) и (5), а также способ их получения известны из источника Bulgakov В.А. et al. Low-melting phthalonitrile thermosetting monomers with siloxane - and phosphate bridges // Eur. Polym. J. 2016. Vol. 84. P. 205-217.
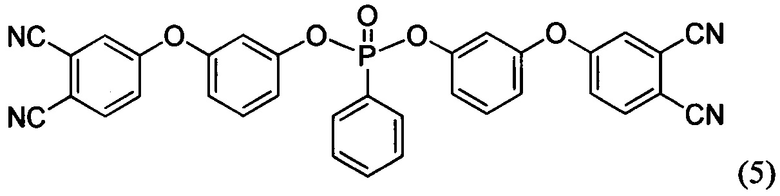
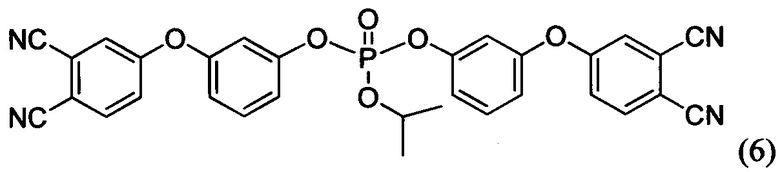
Формулы соединений (6), (7) и (8) раскрыты в статье Bulgakov В.А., Babkin A.V., Dzhevakov Р.В., Bogolyubov A.A., Sulimov A.V., Kepman A.V., Kolyagin Yu G., Guseva D.V., Rudyak V.Yu, Chertovich A.V. Low-melting phthalonitrile thermosetting monomers with siloxane - and phosphate bridges в журнале European Polymer Journal, 2016, №84, c. 205-217, а также в тезисах Сулимов А.В., Булгаков Б.А., Бабкин А.В. Полимерные матрицы на основе легкоплавких фосфорсодержащих фталонитрильных мономеров описаны в сборнике VII Всероссийская Каргинская конференция «Полимеры - 2017» (ссылка не полная…).
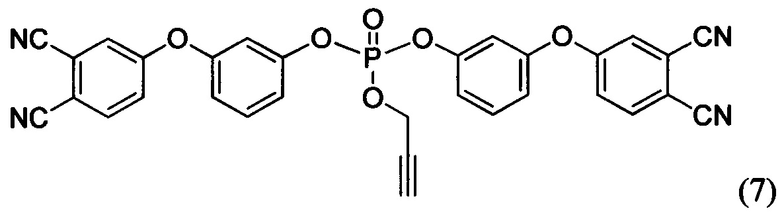
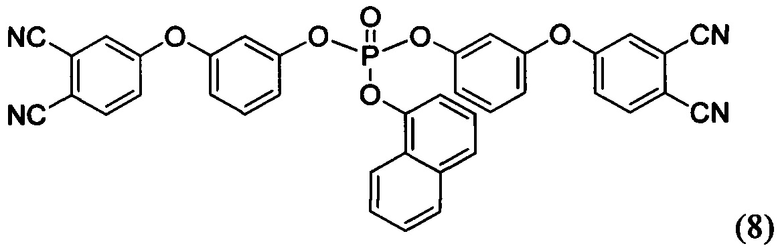
Способ получения указанных мономеров приведен в примерах и заключается во взаимодействии 4-(3-гидроксифенокси)фталонитрила с дихлорангидридом алкил- или арилфосфорной кислоты в присутствии основания. Более подробно синтез раскрыт при описании примеров №№14-16 осуществления настоящего изобретения.
Соединения формул (9) и (10) были синтезированы впервые
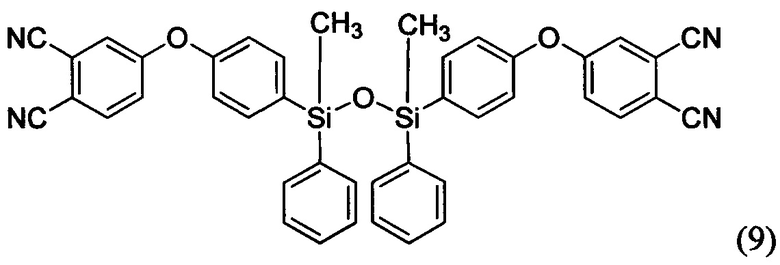
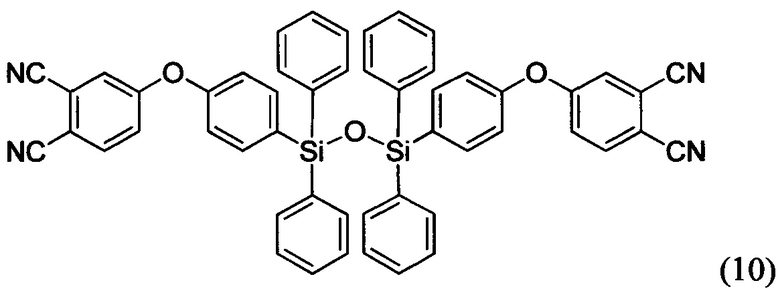
Способ синтеза мономеров 9 и 10 заключается в реакции нуклеофильного замещения между бис-фенолом, соединенным дисилоксановым мостиком (Si-O-Si), и 4-нитрофталонитрилом в апротонном полярном растворителе в присутствии основания. Бис-фенол, содержащий дисилоксановый мостик, может быть синтезирован различными способами (например, см. фиг. 3). В качестве апротонного полярного растворителя предпочтительно использовать диметилацетамид, ацетонитрил, этилацетат, диметилформамид, тетригидрофуран и другие. В качестве оснований предпочтительно использовать карбонат калия (поташ), карбонат натрия, гидрокарбонат натрия, гидроксид калия, гидроксид натрия, оксид магния, оксид кальция, фосфат калия и др.
Характеристики полимеров, получаемых при полимеризации фталонитрильных мономеров, которые использованы в качестве аппретов важны с точки зрения нижней границы ТТД (температуры тепловой деформации) - не менее 400°С, и температуры начала критического разложения (T5%) - не менее 500°С.
Таблица 1. Свойства фталонитрильных мономеров, пригодных для использования в качестве аппретов
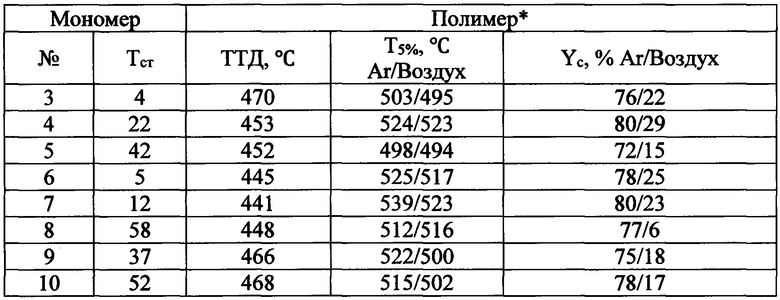
* - получен нагреванием мономера в присутствии ароматического амина Аппрет может наноситься на любые углеродные волокна независимо от их происхождения. В качестве углеродного волокна для использования подойдут коммерчески доступные углеродные волокна, имеющие высокую прочность на растяжение и высокий модуль упругости. В частности, могут быть использованы углеродные волокна, полученные из вискозы, или PAN (полиакрилонитрил, ПАН), или каменноугольного пека. Для реализации заявленного способа аппретирования углеродных волокон могут быть использованы волокна, которые предпочтительно имеют линейную плотность от 0,06 до 4,0 г/м, а также число монофиламентов от 1000 до 48000. Модуль упругости при растяжении составляет 200 ГПа или выше, предпочтительно 240 ГПа или выше. Если прочность нити и модуль углеродного волокна ниже 3,0 ГПа и 200 ГПа, соответственно, трудно получить желаемые механические свойства изготавливаемого из него ПКМ.
Аппретирующий состав по настоящему изобретению можно наносить любыми известными из уровня техники способами, например, путем осуществления контакта нитей с валиковым или ленточным аппликатором, путем распыления или другими способами. Наиболее предпочтительным является формирование аппретирующего слоя на поверхности волокна посредством погружения волокна в емкость с пропитывающим (аппретирующим) составом или его протягиванием через раствор мономеров, содержащийся в ванне, с обеспечением времени контакта аппетирующего раствора с волокном в течение 10-30 с, предпочтительно 15-25 с.
Мономеры формул 3-10 можно использовать в качестве аппрета для получения высокотемпературных ПКМ, так как они относятся к классу фталонитрилов и при отверждении с аминами образуют сшитый термостойкий полимер, а также являются аморфными стеклами с температурами стеклования от 0 до 60°С (табл. 1). В отличие от заявляемого состава, использование эпоксидного аппрета приводит к ухудшению механических свойств ПКМ с фталонитрильными матрицами, так как при отверждении матрицы при температуре выше 300°С происходит разложение эпоксидного аппрета, за счет чего нарушается целостность границы фаз, а, следовательно, и ее прочность.
Нанесение на поверхность углеродных волокон составов, включающих одно или несколько мономеров формул 1 и 2, осуществляют из их водной дисперсии, характеризующейся ее высокой гидролитической устойчивостью в водной среде. Кремнийсодержащие мономеры формул 3, 9 и 10 не содержат чувствительных к гидролизу фрагментов (последовательность «C-Si-O-Si-С» состоит из химически прочных связей). Фосфорсодержащие мономеры формул 4-8 устойчивы к гидролизу в кислой и нейтральной среде [Belsky K.S. et al. Hydrolysis rate constants and activation parameters for phosphate - and phosphonate-bridged phthalonitrile monomers under acid, neutral and alkali conditions // Data Br. 2017. Vol. 13. P. 10-17.].
Аппретирование углеродных волокон проводится путем пропускания волокон через ванну с водной дисперсией (эмульсией или суспензией) мономеров формул 1 и 2 или их смесей. Мономеры, выбранные из соединений формул 1 и 2 могут быть использованы как в виде отдельных соединений, так и в виде смесей соединений. Для образования водной дисперсии возможно использование смесей любых мономеров в любом соотношении, однако предпочтительнее использование комбинаций фосфорсодержащих мономеров с кремниевыми с соотношением мономеров от 1:100 до 100:1.
Водные дисперсии фталонитрильных мономеров для нанесения (аппретирования) углеродного волокна можно получать любыми известными способами, обеспечивающими получение устойчивой водной дисперсии (эмульсии или суспензии), характеризующейся равномерным распределением мономеров во всем объеме, например, следующими способами.
1. Смешением растворов мономеров из группы 3-10 в органическом растворителе (метанол, этанол, изопропанол, тетрагидрофуран, ацетон) с водой с последующим удалением органического растворителя для получения водных дисперсий с содержанием твердой фазы до 20 мас. %. Кроме того, могут применяться поверхностно-активные вещества (ПАВ) для увеличения устойчивости эмульсии во времени. Важно, чтобы ПАВ были выбраны из группы неионогенных ПАВ, например, ди- или триэтаноламин, полиэтиленгликоль (ПЭГ-400). Концентрация ПАВ - до 5 мас. %. Добавление ПАВ необходимо производить непосредственно в органическую фазу перед смешением раствора фталонитрильного мономера и воды.
2. Диспергированием расплава мономера в воде ультразвуком с использованием неионогенных ПАВ частотой 15-44 кГц при непрерывном воздействии в процессе нанесения на волокно.
Для равномерного нанесения аппрета на волокно при непрерывном методе необходимо, чтобы волокно находилось в пропиточной ванне не менее 10 секунд. После прохождения волокна через ванну с эмульсией мономера (массовая доля твердого вещества в эмульсии от 0.1 до 5 мас. %) жгут попадает в сушильную печь. Время нахождения материала в печи может составлять от 10 до 300 секунд при температурах от 200 до 350°С. Скорость прохождения волокна должна быть подобрана в зависимости от используемого оборудования и толщины используемого волокна для обеспечения необходимого целевого количества нанесенного аппрета. После сушки волокно наматывают на бобину. Произведенное таким образом волокно можно использовать для получения композиционных материалов, эксплуатируемых при повышенных температурах.
Углеродные волокна, аппретированные фталонитрилом не заламываются и не распушаются. Кроме того, использование таких волокон при создании ПКМ способствует улучшению механических свойств ПКМ, полученных с использованием фталонитрильных связующих, например, методом вакуумной инфузии.
Получение ПКМ из УВ, аппретированного фталонитрильным мономером, может осуществляться по препреговой технологии (аналогично S.B. Sastri, J.P. Armistead, Т.М. Keller, Phthalonitrile-carbon fiber composites, Polym. Compos. 17 (1996) 816-822. doi:10.1002/pc. 10674 и L. Zong, C. Liu, S. Zhang, J. Wang, X. Jian, Enhanced thermal properties of phthalonitrile networks by cooperating phenyl-s-triazine moieties in backbones, Polymer (Guildf). 77 (2015) 177-188. doi:10.1016/j.polymer.2015.09.035) или по инжекционным техникам, таким как вакуумная инфузия или инжекция в форму (аналогично В. Bulgakov, A. Sulimov, A. Babkin, I. Timoshkin, A. Solopchenko, A. Kepman, V. Avdeev, Phthalonitrile-carbon fiber composites produced by vacuum infusion process, J. Compos. Mater. (2017). doi:10.1177/0021998317699452 и В.A. Bulgakov, A.V.. Sulimov, A.V.. Babkin, D.V.. Afanasiev, A.V. Solopchenko, E.S.. Afanaseva, A.V.. Kepman, V.V. Avdeev, Flame-retardant carbon fiber reinforced phthalonitrile composite for high-temperature applications obtained by resin transfer molding, Mendeleev Commun. 3 (2017) 257-259. doi:10.1016/j.mencom.2017.05.013). Причем аппретированное УВ может быть использовано для текстильной переработки в ткань или однонаправленную ленту. Отверждение и пост-отверждение образца осуществляется согласно режиму, подходящему для используемого связующего. Для изготовления ПКМ в качестве связующих могут быть использованы термореактивные смолы, а именно эпоксидные, полиэфирные, фенольные, кремнийорганические и поли-имидные смолы. Для получения термостабильных ПКМ при температурах до 450°С, более предпочтительно использовать фталонитрильные и поли-имидные связующие.
Ниже представлено более детальное описание заявляемого способа, которое не ограничивает объем притязаний заявляемого изобретения, а демонстрирует возможность осуществления изобретения с достижением заявляемого технического результата.
Синтез прекурсоров для использования в синтезе мономеров.
Пример 1. Синтез 1,3-диметил-1,3-дифенил-1,3-дихлордисилоксана
Раствор воды (3,6 г, 0,2 моль) в сухом тетрагидрофуране (70 мл) прибавляли к раствору метилфенилдихлорсилана (79,7 г, 0,4 моль) в сухом тетрагидрофуране (70 мл) при интенсивном перемешивании при температуре 0°С. Полученный раствор кипятили с обратным холодильником в течение 3 часов. Реакционную смесь упаривали досуха. Остаток перегоняли в вакууме, собирая фракцию, кипящую 125°С (1,22 мм рт.ст.). Получали продукт 1,3-диметил-1,3-дифенил-1,3-дихлордисилоксан, представляющий собой бесцветную жидкость. Выход составлял 41,9 г (32%).
Пример 2. Синтез диметоксиметилфенилсилана
В двугорлой кругло донной колбе объемом 100 мл, снабженной обратным холодильником, растворяли метилфенилдихлорсилан (4,2 г, 0,022 моль) в 10 мл сухого толуола. К полученному раствору при -5°С по каплям прибавляли смесь метанола (4,22 г, 0,132 моль) и пиридина (3,48 г, 0,044 моль). Затем реакционную смесь кипятили в течение 5 часов, после чего выливали в воду. Продукт экстрагировали диэтиловым эфиром. Полученную органическую фазу высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали методом колоночной хроматографии на силикагеле с использованием в качестве элюента смесей дихлорметан - петролейный эфир. Полученный продукт представлял собой белое кристаллическое вещество, его выход составил 3,84 г (96%).
Пример 3. Синтез метоксиметилфенил(4-тетрагидро-2Н-пиран-2-илокси)фенил)силана
В двугорлую круглодонную колбу емкостью 100 мл, снабженную обратным холодильником, капельной воронкой и механической мешалкой помещали магниевые стружки (0,53 г, 0,022 моль) и 10 мл тетрагидрофурана. К полученной смеси прибавляли раствор 5,14 г (0,02 моль) 2-(4-бромфенокси)тетрагидропирана в 10 мл тетрагидрофурана из капельной воронки с такой скоростью, чтобы реакционная смесь слегка кипела. По окончанию прибавления смесь кипятили еще в течение 30 минут. Полученный раствор 4-(тетрагидропиран-2-илокси)фенил)магний бромида медленно прибавляли к раствору диметоксиметилфенилсилана (3,64 г, 0,02 моль) в 30 мл тетрагидрофурана при температуре 0°С в течение 15 минут. Итоговую смесь перемешивали в течение часа при комнатной температуре, после чего выливали в ледяную воду. Водную фазу экстрагировали дизтиловым эфиром. Полученную органическую вытяжку промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали методом колоночной хроматографии на силикагеле с использованием в качестве элюента смесей дихлорметан - петролейный эфир. Получали 5,79 г (84%) продукта в виде белого кристаллического вещества.
Пример 4. Синтез метилфенил(4-(тетрагидро-2Н-пиран-2-илокси)фенил)силанола
В двугорлую круглодонную колбу емкостью 100 мл, снабженную обратным холодильником, капельной воронкой и механической мешалкой помещали магниевые стружки (1,12 г, 0,05 моль) и 20 мл тетрагидрофурана. К полученной смеси прибавляли раствор 9,90 г (0,04 моль) 2-(4-бромфенокси)тетрагидропирана в 20 мл тетрагидрофурана из капельной воронки с такой скоростью, чтобы реакционная смесь слегка кипела. По окончанию прибавления смесь кипятили еще в течение 30 минут. Полученный раствор 4-(тетрагидропиран-2-илокси)фенил)магний бромида медленно прибавляли к смеси метилфенилдихлорсилана (7,64 г, 0,04 моль), тризтиламина (7,79 г, 0,08 моль) и 30 мл тетрагидрофурана при температуре 0°С в течение 15 минут. Итоговую смесь перемешивали в течение часа при комнатной температуре после чего выливали в ледяную воду и аккуратно нейтрализовали до нейтрального рН. Продукт экстрагировали диэтиловым эфиром. Полученный раствор промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и упаривали досуха. Продукт промывали (2×30 мл) гексаном и высушивали в вакууме. Получали 11,95 г (95%) продукта в виде бесцветного твердого вещества.
Пример 5. Синтез 1,3-бис(4-(тетрагидро-2Н-пиран-2-илокси)фенил)-1,3-диметил-1,3-дифенилдисилоксана
Вариант 1. В трехгорлую круглодонную колбу емкостью 250 мл, снабженную обратным холодильником, капельной воронкой и механической мешалкой помещали магниевые стружки (2,6 г, 0,09 моль) и 60 мл тетрагидрофурана. К полученной смеси прибавляли раствор 22,3 г (0,08 моль) 2-(4-бромфенокси)тетрагидропирана в 60 мл тетрагидрофурана из капельной воронки с такой скоростью, чтобы реакционная смесь слегка кипела. По окончанию прибавления смесь кипятили еще в течение 30 минут. Полученный раствор 4-(тетрагидропиран-2-илокси)фенил)магний бромида медленно прибавляли к раствору 1,3-диметил-1,3-дифенил-1,3-дихлордисилоксана (15,0 г, 0,03 моль) и триэтиламина (6,7 г, 0,06 моль) в тетрагидрофуране (70 мл) при температуре 0°С в течение 40 минут. Итоговую смесь перемешивали в течение часа при комнатной температуре, после чего выливали в ледяную воду и аккуратно нейтрализовали до нейтрального рН. Продукт экстрагировали диэтиловым эфиром. Полученный раствор промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и упаривали досуха. Продукт промывали (2×30 мл) гексаном и высушивали в вакууме. Получали 16,2 г (89%) продукта в виде бесцветного твердого вещества.
Вариант 2. В двугорлой круглодонной колбе, снабженной обратным холодильником растворяли метилфенил(4-(тетрагидро-2Н-пиран-2-илокси)фенил)силанол (0,94 г, 3 ммоль) в 30 мл диоксана. К раствору приливали раствор гидроксида натрия (0,24 г, 6 ммоль) в 13 мл воды. Полученную смесь кипятили при перемешивании в течение 5 часов, затем выливали в воду. Продукт экстрагировали диэтиловым эфиром. Органическую вытяжку высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Полученный продукт представлял собой бесцветное твердое вещество, его выход составлял 0,31 г (30%).
Вариант 3. В двугорлой круглодонной колбе, снабженной обратным холодильником растворяли метилфенил(4-(тетрагидро-2Н-пиран-2-илокси)фенил)силанол (0,94 г, 3 ммоль) в 30 мл диоксана. К раствору приливали раствор гидроксида натрия (0,48 г, 12 ммоль) в 13 мл воды. Полученную смесь кипятили при перемешивании в течение 5 часов, затем выливали в воду. Продукт экстрагировали диэтиловым эфиром. Полученную органическую вытяжку высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Получали 0,04 г (4%) продукта в виде бесцветного твердого вещества.
Вариант 4. В двугорлой круглодонной колбе, снабженной обратным холодильником растворяли метилфенил(4-(тетрагидро-2Н-пиран-2-илокси)фенил)силанол (0,94 г, 3 ммоль) в 30 мл диоксана. К раствору прибавляли раствор фторида цезия (0,23 г, 1,5 ммоль) в 13 мл воды. Полученную смесь кипятили при перемешивании в течение 5 часов, после чего вылили в воду. Продукт экстрагировали диэтиловым эфиром. Органическую вытяжку высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Получали 0,15 г (15%) продукт в виде бесцветного твердого вещества.
Вариант 5. В двугорлой круглодонной колбе, снабженной обратным холодильником растворяли метилфенил(4-(тетрагидро-2Н-пиран-2-илокси)фенил)силанол (0,94 г, 3 ммоль) в 50 мл сухого диоксана. К раствору прибавляли гидрид натрия (0,14 г, 1,5 ммоль). Полученную смесь кипятили при перемешивании в течение 5 часов, после чего выливали в воду. Продукт экстрагировали диэтиловым эфиром. Полученную органическую вытяжку высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Получали 0,72 г (81%) продукта в виде бесцветного твердого вещества.
Вариант 6. В стеклянной виале с завинчивающейся крышкой объемом 4 мл смешавали метилфенил(4-(тетрагидро-2Н-пиран-2-илокси)фенил)силанол (0,22 г, 0,7 ммоль) и гидрид натрия (13 мг, 0,3 ммоль). Смесь нагревали при температуре 200°С в течение 30 минут. Полученную смесь растворяли в воде, продукт экстрагировали диэтиловым эфиром, после чего раствор сушили над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Получали 0,21 г (91%) продукта в виде бесцветного твердого вещества.
Вариант 7. В стеклянной виале с завинчивающейся крышкой объемом 4 мл смешивали метилфенил(4-(тетрагидро-2Н-пиран-2-илокси)фенил)силанола (0,22 г, 0,7 ммоль) и гидроксида натрия (27 мг, 0,7 ммоль). Смесь нагревали при температуре 200°С в течение 30 минут. Полученную смесь растворяли в воде, продукт экстрагировали диэтиловым эфиром, после чего раствор сушили над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Получали 0,168 г (77%) продукта в виде бесцветного твердого вещества.
Вариант 8. В двугорлую круглодонную колбу емкостью 100 мл, снабженную обратным холодильником, капельной воронкой и механической мешалкой помещали магниевые стружки (1,12 г, 0,05 моль) и 20 мл тетрагидрофурана. К полученной смеси прибавляли раствор 9,90 г (0,04 моль) 2-(4-бромфенокси)тетрагидропирана в 20 мл тетрагидрофурана из капельной воронки с такой скоростью, чтобы реакционная смесь слегка кипела. По окончанию прибавления смесь кипятили еще в течение 30 минут. Полученный раствор 4-(тетрагидропиран-2-илокси)фенил)магний бромида медленно прибавляли к раствору метилфенилдихлорсилана (7,64 г, 0,04 моль) в 30 мл тетрагидрофурана при температуре 0°С в течение 15 минут. Затем к раствору приливали раствор метилфенил(4-(тетрогидро-2Н-пиран-2-илокси)фенил)силанола (12,58 г, 0,04 моль) и сухого пиридина (6,33 г, 0,08 моль) в 50 мл тетрагидрофурана. Итоговую смесь перемешивали в течение часа при комнатной температуре, после чего выливали в ледяную воду и аккуратно нейтрализовали до нейтрального рН. Продукт экстрагировали диэтиловым эфиром. Полученный раствор промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали методом колоночной хроматографии на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента и высушивали в вакууме. Получали 11,95 г (95%) продукта в виде бесцветного твердого вещества.
Пример 6. Синтез 1,3-бис(4-гидроксифенил)-1,3-диметил-1,3-дифенилдисилокеана.
Вариант 1. Метилфенил(4-(тетрагидропиран-2-илокси)фенил)силанол (428,0 мг, 1,36 ммоль) растворяли в 20 мл толуола в круглодонной колбе емкостью 50 мл и прибавляли 4-толуолсульфокислоту (10,8 мг, 0,03 ммоль) при комнатной температуре, затем реакционную смесь доводили до кипения. После остывания к смеси приливали 50 мл воды. Продукт экстрагировали диэтиловым эфиром. Полученный раствор высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейнй эфир - дихлорметан в качестве элюента. Получали 378,9 мг (63%) продукта в виде бесцветного твердого вещества.
Вариант 2. Метилфенил(4-(тетрагидропиран-2-илокси)фенил)силанол (428,0 мг, 1,36 ммоль) растворяли в 20 мл тетрагидрофурана в круглодонной колбе емкостью 50 мл и прибавляли 1,1 мг кристаллического йода при комнатной температуре, после чего смесь перемешивали в течение 2 часов. Затем к смеси приливали 50 мл воды. Продукт экстрагировали диэтиловым эфиром. Полученный раствор высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Получали 162 мг (27%) продукта в виде бесцветного твердого вещества.
Вариант 3. Метилфенил(4-(тетрагидропиран-2-илокси)фенил)силанол (3,0 г, 9,5 ммоль) растворяли в 40 мл этанола в круглодонной колбе емкостью 100 мл и прибавляли 0,5 мл 5%-ной соляной кислоты при комнатной температуре. После перемешивания в течение 1 часа прибавляли 100 мл ледяной воды. Продукт экстрагировали диэтиловым, эфиром. Полученный раствор высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Полученный продукт представлял собой бесцветное твердое вещество, его выход составлял 0,27 г (36%).
Вариант 4. В сухой стеклянной виале с завинчивающейся крышкой объемом 20 мл смешивали метоксиметилфенил(4-тетрагидро-2Н-пиран-2-илокси)фенил)силана (0,689 г, 2 ммоль), сухого ацетонитрила (10 мл), поташа (0,276 г, 2 ммоль) и тетраборфтората триэтилоксония (0,380 г, 2 ммоль). Смесь перемешивали в течение 90 минут при температуре 100°С. Реакционную смесь охлаждали до комнатной температуры, растворяли в воде - дихлорметане. Органическую фазу отделяли, водную экстрагировали дихлорметаном (3×30 мл). Объединенную органическую вытяжку промывали водой, насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали методом колоночной хроматографии на силикагеле с использованием в качестве элюента смесей дихлорметан - петролейный эфир. Получили 0,175 г (35%) продукта в виде белого кристаллического вещества.
Вариант 5. 1,3-бис(4-(тетрагидро-2Н-пиран-2-илокси)фенил)-1,3-диметил-1,3-дифенилдисилоксан (5,5 г, 9 ммоль) растворяли в 10 мл метанола в круглодонной колбе емкостью 50 мл и прибавляли хлорид алюминия гексагидрат (44 мг, 0,18 ммоль) при комнатной температуре. После перемешивания в течение 1 часа прибавляли 100 мл ледяной воды. Продукт экстрагировали диэтиловым эфиром. Полученный раствор высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Получали желтоватое твердое вещество с выходом 3,51 г (88%).
Пример 7. Синтез 1,3-бис(4-(3,4-динитрилфенилокси))-1,3-диметил-1,3-дифенилдисилоксана (мономер формулы (9))
1,3-Бис(4-гидроксифенил)-1,3-диметил-1,3-дифенилдисилоксан (2,21 г, 5 ммоль) растворяли в 20 мл сухого диметилацетамида. К раствору прибавляли 1,47 г (10 ммоль) тонко растертого поташа. К полученной смеси прибавляли раствор 4-нитрофталонитрила (1,85 г, 10 ммоль) в 10 мл сухого диметилацетамида. Реакционную смесь перемешивали при 70°С в течение суток (контроль за ходом реакции осуществляли методом тонкослойной хроматографии) после чего выливали в 100 мл холодной воды. Продукт экстрагировали дихлорметаном. Органическую фазу промывали водой (5×50 мл), насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Получали продукт в виде желтоватого стеклообразного вещества с выход 2,08 г (60%).
Пример 8. Синтез 1,1,3,3-тетрафенил-1,3-дихлордисилоксана
Раствор воды (3,6 г, 0,2 моль) в тетрагидрофуране (70 мл) прибавляли по каплям к раствору дифенилдихлорсилана (100 г, 0,4 моль) в тетрагидрофуране (70 мл) при интенсивном перемешивании при температуре 0°С. Полученный раствор кипятили с обратным холодильником в течение 3 часов. Реакционную смесь упаривали досуха. Остаток перегоняли в вакууме, собирая фракцию, кипящую 228-233°С (1,07 мм рт.ст.). Получали 67,3 г (37%) продукта в виде бесцветной жидкости.
Пример 9. Синтез диметоксидифенилсилана
В двугорлой круглодонной колбе объемом 100 мл, снабженной обратным холодильником, растворяли дифенил дихлорсилан (5,57 г, 0,022 моль) в 10 мл сухого толуола. К полученному раствору при -5°С по каплям прибавляли смесь метанола (4,22 г, 0,132 моль) и сухого пиридина (3,48 г, 0,044 моль). Реакционную смесь кипятили в течение 5 часов, после чего выливали в воду. Продукт экстрагировали диэтиловым эфиром, затем раствор высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали методом колоночной хроматографии на силикагеле с использованием в качестве элюента смесей дихлорметан - петролейный эфир. Получали 4,98 г (93%) продукта в виде белого кристаллического вещества.
Пример 10. Синтез метоксидифенил(4-тетрагидро-2Н-пиран-2-илокси)фенил)силана
В двугорлую круглодонную колбу емкостью 100 мл, снабженную обратным холодильником, капельной воронкой и механической мешалкой помещали магниевые стружки (0,53 г, 0,022 моль) и 10 мл тетрагидрофурана. К полученной смеси прибавляли раствор 5,14 г (0,02 моль) 2-(4-бромфенокси)тетрагидропирана в 10 мл тетрагидрофурана из капельной воронки с такой скоростью, чтобы реакционная смесь слегка кипела. По окончанию прибавления смесь кипятили еще в течение 30 минут. Полученный раствор 4-(тетрагидропиран-2-илокси)фенил)магний бромида медленно прибавляли к раствору диметоксидифенилсилана (4,89 г, 0,02 моль) в 30 мл тетрагидрофурана при температуре 0°С в течение 15 минут. Итоговую смесь перемешивали в течение часа при комнатной температуре, после чего выливали в ледяную воду. Органическую часть экстрагировали диэтиловым эфиром. Полученный раствор промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали методом колоночной хроматографии на силикагеле с использованием в качестве элюента смесей дихлорметан - петролейный эфир. Получали 6,65 г (85%) продукта в виде белого кристаллического вещества.
Пример 11. Синтез дифенил(4-(тетрагидро-2Н-пиран-2-илокси)фенил)силанола
В двугорлую круглодонную колбу емкостью 100 мл, снабженную обратным холодильником, капельной воронкой и механической мешалкой помещали магниевые стружки (1,12 г, 0,05 моль) и 20 мл тетрагидрофурана. К полученной смеси прибавляли раствор 9,90 г (0,04 моль) 2-(4-бромфенокси)тетрагидропирана в 20 мл тетрагидрофурана из капельной воронки с такой скоростью, чтобы реакционная смесь слегка кипела. По окончанию прибавления смесь кипятили еще в течение 30 минут. Полученный раствор 4-(тетрагидропиран-2-илокси)фенил)магний бромида медленно прибавляли к смеси дифенилдихлорсилана (10,12 г, 0,04 моль), триэтиламина (7,79 г, 0,08 моль) и 30 мл тетрагидрофурана при температуре 0°С в течение 15 минут. Итоговую смесь перемешивали в течение часа при комнатной температуре, после чего выливали в ледяную воду и аккуратно нейтрализовали до нейтрального рН. Продукт экстрагировали диэтиловым эфиром. Полученный раствор промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и упаривали досуха. Продукт промывали (2×30 мл) гексаном и высушивали в вакууме. Получали 14,29 г (95%) продукта в виде бесцветного твердого вещества.
Пример 12. Синтез 1,3-бис(4-(тетрагидро-2Н-пиран-2-илокси)фенил)-1,1,3,3-тетрафенилдилисилоксана
Вариант 1. В двугорлой круглодонной колбе, снабженной обратным холодильником растворяли дифенил(4-(тетрагидро-2Н-пиран-2-илокси)фенил)силанол (1,13 г, 3 ммоль) в 30 мл диоксана. Затем к раствору приливали раствор гидроксида натрия (0,24 г, 6 ммоль) в 13 мл воды. Полученную смесь кипятили при перемешивали в течение 5 часов, после чего выливали в воду. Продукт экстрагировали диэтиловым эфиром. Полученный раствор высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Получали 0,044 г (4%) продукта в виде бесцветного твердого вещества.
Вариант 2. В двугорлой круглодонной колбе, снабженной обратным холодильником растворяли дифенил(4-(тетрагидро-2Н-пиран-2-илокси)фенил)силанол (1,13 г, 3 ммоль) в 30 мл диоксана. После к раствору приливали раствор гидроксида натрия (0,48 г, 6 ммоль) в 13 мл воды. Полученную смесь кипятили при перемешивании в течение 5 часов, после чего выливали в воду. Продукт экстрагировали диэтиловым эфиром. Полученный раствор высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Получали 0,053 г (5%) продукта в виде бесцветного твердого вещества.
Вариант 3. В двугорлой круглодонной колбе, снабженной обратным холодильником растворяли дифенил(4-(тетрагидро-2Н-пиран-2-илокси)фенил)силанол (1,13 г, 3 ммоль) в 30 мл диоксана. После к раствору приливали раствор фторида цезия (0,23 г, 1,5 ммоль) в 13 мл воды. Полученную смесь кипятили при перемешивали в течение 5 часов, после чего выливали в воду. Продукт экстрагировали диэтиловым эфиром. Полученный раствор высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Получали 0,019 г (2%) продукта в виде бесцветного твердого вещества.
Вариант 4. В двугорлой круглодонной колбе, снабженной обратным холодильником растворяли дифенил(4-(тетрагидро-2Н-пиран-2-илокси)фенил)силанол (1,13 г, 3 ммоль) в 50 мл сухого диоксана. После к раствору добавляли гидрид натрия (0,14 г, 1,5 ммоль). Полученную смесь кипятили при перемешивали в течение 5 часов, после чего выливали в воду. Продукт экстрагировали диэтиловым эфиром. Полученный раствор высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Получали 0,074 г (7%) продукта в виде бесцветного твердого вещества.
Вариант 5. В стеклянной виале с завинчивающейся крышкой объемом 4 мл смешивали дифенил(4-(тетрагидро-2Н-пиран-2-илокси)фенил)силанол (0,25 г, 0,7 ммоль) и гидрид натрия (13 мг, 0,3 ммоль). Смесь нагревали при температуре 200°С в течение 30 минут. Полученную смесь растворяли в воде, продукт экстрагировали диэтиловым эфиром, после чего раствор высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Получали продукт в виде бесцветного твердого вещества с выходом 0,024 г (11%).
Вариант 6. В стеклянной виале с завинчивающейся крышкой объемом 4 мл смешивали дифенил(4-(тетрагидро-2Н-пиран-2-илокси)фенил)силанол (0,25 г, 0,7 ммоль) и гидроксид натрия (27 мг, 0,7 ммоль). Смесь нагревали при температуре 200°С в течение 30 минут. Полученную смесь растворяли в воде, продукт экстрагировали диэтиловым эфиром, после чего раствор высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Получали 0,019 г (9%) продукта в виде бесцветного твердого вещества.
Вариант 7. Синтез 1,3-бис(4-(тетрагадро-2Н-пиран-2-илокси)фенил)-1,1,3,3-дифенилдисилоксана
В трехгорбую круглодонную колбу емкостью 250 мл, снабженную обратным холодильником, капельной воронкой и механической мешалкой помещали магниевые стружки (2,55 г, 0,09 моль) и 60 мл тетрагидрофурана. К полученной смеси прибавляли раствор 22,27 г (0,08 моль) 2-(4-бромфенокси)тетрагидропирана в 60 мл тетрагидрофурана из капельной воронки с такой скоростью, чтобы реакционная смесь слегка кипела. По окончанию прибавления смесь кипятили еще в течение 30 минут. Полученный раствор 4-(тетрагидропиран-2-илокси)фенил)магний бромида медленно прибавляли к раствору 1,1,3,3-тетрафенил-1,3-дихлордисилоксана (13,54 г, 0,03 моль) и триэтиламина (6,72 г, 0,06 моль) в тетрагидрофуране (70 мл) при температуре 0°С в течение 40 минут. Итоговую смесь перемешивали в течение часа при комнатной температуре, после чего выливали в ледяную воду и аккуратно нейтрализовали до нейтрального рН. Продукт экстрагировали диэтиловым эфиром. Полученный раствор промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и упаривали досуха. Продукт промывали (2×30 мл) гексаном и высушивали в вакууме. Получали 3,31 г (15%) продукта в виде бесцветного твердого вещества.
Вариант 8. 1,3-бис(4-(тетрагидро-2Н-пиран-2-илокси)фенил)-1,1,3,3-тетрафенилдисилоксана
В двугорлую круглодонную колбу емкостью 250 мл, снабженную обратным холодильником, капельной воронкой и механической мешалкой помещали магниевые стружки (1,12 г, 0,05 моль) и 20 мл тетрагидрофурана. К полученной смеси прибавляли раствор 9,90 г (0,04 моль) 2-(4-бромфенокси)тетрагидропирана в 20 мл тетрагидрофурана из капельной воронки с такой скоростью, чтобы реакционная смесь слегка кипела. По окончанию прибавления смесь кипятили еще в течение 30 минут. Полученный раствор 4-(тетрагидропиран-2-илокси)фенил)магний бромида медленно прибавляли к раствору дифенилдихлорсилана (10,12 г, 0,04 моль) в 30 мл тетрагидрофурана при температуре 0°С в течение 15 минут. Затем к раствору приливали раствор дифенил(4-(тетрагидро-2Н-пиран-2-илокси)фенил)силанола (15,06 г, 0,04 моль) и сухого пиридина (6,33 г, 0,08 моль) в 50 мл тетрагидрофурана. Итоговую смесь перемешивали в течение часа при комнатной температуре, после чего выливали в ледяную воду и аккуратно нейтрализовали до нейтрального рН. Продукт экстрагировали диэтиловым эфиром. Полученный раствор промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали методом колоночной хроматографии на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента и высушивали в вакууме. Получали 4,41 г (15%) продукта в виде бесцветного твердого вещества.
Пример 12. Синтез 1,3-бис(4-гидроксифенил)-1,1,3,3-тетрафенилдисилоксана
Вариант 1. Дифенил(4-(тетрагидропиран-2-илокси)фенил)силанол (0,512 г, 1,36 ммоль) растворяли в 20 мл сухого толуола в круглодонной колбе емкостью 50 мл и прибавляли 4-толуолсульфокислоту (10,8 мг, 0,03 ммоль) при комнатной температуре, затем реакционную смесь доводили до кипения. После остывания к смеси приливали 50 мл воды. Продукт экстрагировали диэтиловым эфиром. Полученный раствор высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Получали 0,054 г (7%) продукта в виде желтоватого твердого вещества.
Вариант 2. Дифенил(4-(тетрагидропиран-2-илокси)фенил)силанол (0,512 г, 1,36 ммоль) растворяли в 20 мл тетрагидрофурана в круглодонной колбе емкостью 50 мл и прибавляли 1,5 мг кристаллического йода при комнатной температуре, после чего смесь перемешивали в течение 2 часов. Затем к смеси приливали 50 мл воды. Продукт экстрагировали диэтиловым эфиром. Полученный раствор высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Получали 0,046 г (6%) продукта в виде желтоватого твердого вещества.
Вариант 3. Дифенил(4-(тетрагидропиран-2-илокси)фенил)силанол (3 г, 8 ммоль) растворяли в 40 мл этанола в круглодонной колбе емкостью 100 мл и прибавляли 0,5 мл 5%-ной соляной кислоты при комнатной температуре. После перемешивания в течение 1 часа прибавляли 100 мл ледяной воды. Продукт экстрагировали диэтиловым эфиром. Полученный раствор высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Получали продукт в виде желтоватого твердого вещества с выходом 0,015 г (2%).
Вариант 4. В сухой стеклянной виале с завинчивающейся крышкой объемом 20 мл смешивали метоксидифенил(4-тетрагидро-2Н-пиран-2-илокси)фенил)силан (0,781 г, 2 ммоль), сухой ацетонитрил (10 мл), поташ (0,276 г, 2 ммоль) и тетраборфторат триэтилоксония (0,380 г, 2 ммоль). Смесь перемешивали в течение 90 минут при температуре 100°С. Реакционную смесь охлаждали до комнатной температуры, растворяли в воде - дихлорметане. Органическую фазу отделяли, водную экстрагировали дихлорметаном (3×30 мл). Объединенную органическую вытяжку промывали водой, насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали методом колоночной хроматографии на силикагеле с использованием в качестве элюента смесей дихлорметан - петролейный эфир. Получали 0,045 г (8%) продукта в виде бесцветного кристаллического вещества.
Вариант 5. 1,3-Бис(4-(тетрагидро-2Н-пиран-2-илокси)фенил)-1,1,3,3-тетрафенил дисилоксан (6,62 г, 9 ммоль) растворяли в 10 мл метанола в круглодонной колбе емкостью 50 мл и прибавляли хлорид алюминия гексагидрат (44 мг, 0,18 ммоль) при комнатной температуре. После перемешивания в течение 1 часа прибавляли 100 мл ледяной воды. Продукт экстрагировали диэтиловым эфиром. Полученный раствор высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Получали продукт в виде желтоватого твердого вещества с выходом 4,36 г (87%).
Синтез мономеров формул 6, 7, 8, 10
Пример 13. Синтез 1,3-бис(4-(3,4-динитрилфенилокси))-1,1,3,3-дифенилдисилоксана (мономер формулы (10))
1,3-Бис(4-гадроксифенил)-1,1,3,3-дифенилдисилоксан (2,84 г, 5 ммоль) растворяли в 20 мл сухого диметилацетамида. К раствору прибавляли 1,47 г (10 ммоль) тонко растертого поташа. К полученной смеси прибавляли раствор 4-нитрофталонитрила (1,85 г, 10 ммоль) в 10 мл сухого диметилацетамида. Реакционную смесь перемешивали при 70°С в течение суток (контроль за ходом реакции осуществляли методом тонкослойной хроматографии) после чего выливали в 100 мл холодной воды. Продукт экстрагировали дихлорметаном. Органическую фазу промывали водой (5×50 мл), насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и упаривали досуха. Продукт очищали колоночной хроматографией на силикагеле с использованием смесей петролейный эфир - дихлорметан в качестве элюента. Получали 2,51 г (61%) продукта в виде желтоватого стеклообразного вещества.
Пример 14. Синтез бис-(3-(3,4-дицианофенокси)фенил)-нафталин-2-ил фосфата (мономер формулы (8))
В круглодонную трехгорлую колбу емкостью 250 мл, снабженную обратным холодильником добавляли 60 мл сухого толуола, 4-(3-гидроксифенокси)фталонитрил (10 г, 0.04 моль) и KBr (0.5 г, 0.004 моль), после чего при перемешивании через септу добавляли сухой пиридин (3.35 г, 0.04 моль). Затем через септу добавляли нафталин-1-ил дихлорфосфат (5.22 г, 0.02 моль), нагревали до 130°С и перемешивали 24 часа. Полноту протекания реакции проверяли методом тонкослойной хроматографии. Реакционную смесь отфильтровывали на стеклянном фильтре от гидрохлорида пиридина, после чего продукт очищали флеш-хроматографией на силикагеле (элюент - хлористый метилен). После удаления растворителя выделяли желтовато-белое стеклообразное вещество. Выход 10.8 г (84%).
Методика синтеза аналогична синтезу бис-(3-(3,4-дицианофенокси)фенил)фенил фосфата. Продукт представляет собой светло-коричневое стеклообразное вещество. Выход 6,5 г (93%).
Пример 15. Синтез изо-пропил бис-(3-(3,4-дицианофенокси)фенил) фосфата (мономер формулы (6))
В круглодонную трехгорлую колбу емкостью 250 мл, снабженную капельной воронкой, добавляли суспензию гидрида натрия (0.61 г, 0.025 моль) в ТГФ (30 мл), после чего при перемешивании на магнитной мешалке по каплям добавляли раствор 4-(3-гидроксифенокси)фталонитрила (6 г, 0.025 моль) в ТГФ (50 мл). Следует отметить, что образование фенолята происходит с выделением тепла и реакционная смесь вспенивается, в связи с чем необходимо соблюдать скорость прикапывания. После полного добавления фталонитрильного прекурсора, бутил дихлорфосфат (2.26 г, 0.0127 моль) добавляли по каплям через септу, и реакционную смесь оставляли перемешиваться на ночь. Далее реакционную смесь выливали в 500 мл воды, и экстрагировали хлористым метиленом, контролируя наличие искомого вещества в экстракте методом тонкослойной хроматографии. Хлористый метилен упаривали на роторном испарителе, и выделяли конечный продукт методом колоночной хроматографии, используя хлористый метилен в качестве элюента. Выход 6.7 г (89%).
Пример 16. Синтез бис-(3-(3,4-дицианофенокси)фенил)проп-2-инил фосфата (мономер формулы (7)
Методика синтеза аналогична синтезу изо-пропил бис-(3-(3,4-дицианофенокси)фенил) фосфата (мономер 4). Выход 6.68 г (88%).
Приготовление водных дисперсий фталонитрильных мономеров
Пример 17.
К расплаву мономера 3 (10 г), нагретому до 40°С при перемешивании на механической мешалке добавляли 89 мл воды, также предварительно нагретой до 40°С. Скорость вращения мешалки увеличили до 600 об/мин, добавляли 1 г диэтаноламина. Через 30 минут перемешивание выключили и получали устойчивую водную эмульсию фталонитрила 3 с концентрацией твердой фазы 10%.
Пример 18.
К раствору мономера 9 (10 г) в 100 мл изопропилового спирта добавляли 50 мл воды при интенсивном перемешивании. В смесь добавляли 0.5 мас. % ПАВ (полисорбат-20), после чего в ультрозвуковой бане упаривали изопропанол. Получали устойчивую эмульсию мономера 9 с массовым содержанием 20%.
Пример 19.
В колбу помещали 10 г мономера 3 и 2 г мономера 4. Смесь сплавляли при перемешивании при 50°С, добавляли 3 г диэтиленгликоля, после чего при перемешивании добавляли 100 мл воды, предварительно нагретой до 50°С. Получали устойчивую эмульсию смешанного аппретирующего агента (из двух мономеров 3 и 4) с массовой долей 10.4%.
Эксперименты проводились и для других смесей мономеров, взятых в различных сочетаниях из диапазона значения от 1:100 до 100:1, при этом были получены аппреты, которые характеризовались устойчивостью в водной дисперсии и стабильностью при нагреве до 450°С.
Пример 20.
В колбу помещали 10 г мономера 8 и растворяли его в ТГФ. В раствор добавляли 0.5 г диэтаноламина, после чего при перемешивании добавляли 100 мл воды, предварительно нагретой до 50°С. Получали устойчивую эмульсию мономера 8 с массовой долей 9,9%
Для нанесения аппрета на волокно полученные концентрированные эмульсии (Примеры №№17-19) разбавляли до концентрации от 0.1 до 5 мас. %, после чего помещали в пропиточную емкость (ванну (фиг. 1). Нить с подающей бобины 1 пропускали через ванну 2, наполненную эмульсией фталонитрильного аппрета. После этого нить направляли в сушильную печь 3, откуда она попадала на приемную бобину 4.
Приготовление аппретированных волокон
Пример 21.
Для экспериментов по аппретированию использовали волокно марки TohoTenax 3k НТА40 (4 ГПа/240 ГПа), предварительно отмытое от аппрета путем многократного пропускания через ванну, наполненную ацетоном. После каждой промывки волокно помещали в сушильный шкаф на 110°С и грели до постоянной массы. Промывку повторяли до тех пор, пока масса волокна до и после промывки не совпадала. По разности масс между исходным и отмытым волокном определяли содержание аппрета.
Пример 22.
Использовали эмульсию, полученную в Примере 17, разбавленную до концентрации 1.1%. Углеродное волокно протягивали через ванну с эмульсией при температуре 25°С. Пропиточная ванна была снабжена ультразвуком с частотой 35 кГц и мощностью генератора 50 Вт. Сушили волокно при температуре 250°С для удаления воды в течение 2 минут. Полученное волокно содержало 1.05% аппрета по массе. Содержание нанесенного аппрета определяли по разности масс до и после нанесения.
Пример 23.
Использовали эмульсию, полученную в примере 17, разбавленную до концентрации 0.5%. Углеродное волокно протягивали через ванну с эмульсией при температуре 25°С, после чего пропускали через воздушную печь при температуре 300°С в течение 1,5 минут. Полученное волокно содержало 0.41% аппрета по массе. Содержание нанесенного аппрета определяли по разности масс до и после нанесения.
Пример 24.
Использовали эмульсию, полученную в примере 17, разбавленную до концентрации 2.1%. Углеродное волокно протягивали через ванну с эмульсией при температуре 30°С, после чего пропускали через воздушную печь при температуре 350°С в течение 10 секунд. Полученное волокно содержало 2.03% аппрета по массе. Содержание нанесенного аппрета определяли по разности масс до и после нанесения.
Пример 25.
Использовали эмульсию, полученную в примере 17, разбавленную до концентрации 0.1%. Углеродное волокно протягивали через ванну с эмульсией при температуре 30°С, после чего пропускали через воздушную печь при температуре 200°С в течение 5 минут. Полученное волокно содержало 0.08% аппрета по массе. Волокна, полученные в данном примере, распушались и заламывались. Содержание нанесенного аппрета определяли по разности масс до и после нанесения.
Пример 26.
Использовали эмульсию, полученную в примере 17, разбавленную до концентрации 5%. Углеродное волокно протягивали через ванну с эмульсией при температуре 30°С, после чего пропускали через воздушную печь при температуре 350°С в течение 20 секунд. Полученное волокно содержало 4.6% аппрета по массе. Содержание нанесенного аппрета определяли по разности масс до и после нанесения. Жгуты волокна, полученные в данном примере, слипались между собой, а излишки связующего налипали на поверхности, с которыми соприкасался жгут.
Пример 27.
Использовали эмульсию, полученную в примере 18, разбавленную до концентрации 1.6%. Углеродное волокно протягивали через ванну с эмульсией при температуре 30°С, после чего пропускали через воздушную печь при температуре 350°С в течение 10 секунд. Полученное волокно содержало 1.55% аппрета по массе. Содержание нанесенного аппрета определяли по разности масс до и после нанесения.
Пример 28.
Использовали эмульсию, полученную в Примере 18, разбавленную до концентрации 1.1%. Углеродное волокно протягивали через ванну с эмульсией при температуре 40°С. Пропиточная ванна была снабжена ультразвуком с частотой 35 кГц и мощностью генератора 50 Вт. Сушили волокно при температуре 250°С для удаления воды в течение 2 минут (фиг. 2). Полученное волокно содержало 1.07% аппрета по массе. Содержание нанесенного аппрета определяли по разности масс до и после нанесения.
Пример 29.
Использовали эмульсию, полученную в примере 18, разбавленную до концентрации 1.6%. Углеродное волокно протягивали через ванну с эмульсией при температуре 35°С, после чего пропускали через воздушную печь при температуре 300°С в течение 60 секунд. Полученное волокно содержало 1.59% аппрета по массе. Содержание нанесенного аппрета определяли по разности масс до и после нанесения.
Пример 30.
Использовали эмульсию, полученную в примере 18, разбавленную до концентрации 0.5%. Углеродное волокно протягивали через ванну с эмульсией при температуре 30°С после чего пропускали через воздушную печь при температуре 300°С в течение 1,5 минут. Полученное волокно содержало 0.43% аппрета по массе. Содержание нанесенного аппрета определяли по разности масс до и после нанесения.
Пример 31.
Использовали эмульсию, полученную в примере 18, разбавленную до концентрации 2.2%. Углеродное волокно протягивали через ванну с эмульсией при температуре 25°С, после чего пропускали через воздушную печь при температуре 350°С в течение 20 секунд. Полученное волокно содержало 2.12% аппрета по массе. Содержание нанесенного аппрета определяли по разности масс до и после нанесения.
Пример 32.
Использовали эмульсию, полученную в Примере 19, разбавленную до концентрации 1.1%. Углеродное волокно протягивали через ванну с эмульсией при температуре 40°С. Сушили волокно при температуре 300°С для удаления воды в течение 1 минуты. Полученное волокно содержало 1.02% аппрета по массе. Содержание нанесенного аппрета определяли по разности масс до и после нанесения.
Пример 33.
Использовали эмульсию, полученную в Примере 20, разбавленную до концентрации 1.2%. Углеродное волокно протягивали через ванну с эмульсией при температуре 40°С. Сушили волокно при температуре 300°С для удаления воды в течение 1 минуты. Полученное волокно содержало 0.98% аппрета по массе. Содержание нанесенного аппрета определяли по разности масс до и после нанесения.
Для проверки формирования на поверхности аппретированного волокна химически активных функциональных групп, способных реагировать с полимером - связующим, используемым при производстве ПКМ, были проведены измерения краевых углов смачивания полученного аппретированного волокна и волокна, аппретированного эпоксидной смолой. Определяли поверхностное натяжения связующего PN-3M, для этого кольцо Дю Нуи помещали в расплав связующего (Т=165°С), находящийся в измерительной чаше тензиометра. В результате проведенных испытаний среднее поверхностное натяжение расплава связующего PN-3M составило 41,444 мН/м.
Измерение краевых углов смачивания для образцов волокна, полученных в примерах №№21-33 проводилось аналогично измерению поверхностного натяжения для связующего PN-3M, однако вместо кольца Дю Нуи в расплав связующего опускали образец углеродного волокна (жгута) с нанесенным аппретом. Результаты измерений представлены в табл. 2.
Таблица 2. Значения краевых углов смачивания углеродного волокна с различными аппретами
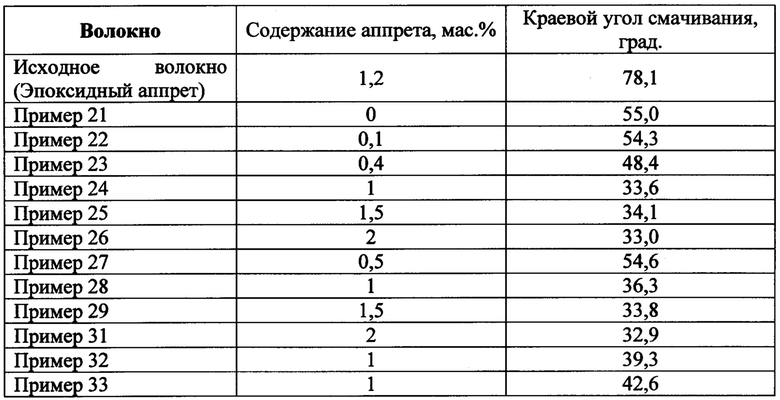
Равномерность нанесения аппретов подтверждали методом растровой электронной микроскопии. Из данных, представленных в таблице 2 видно, что количество аппрета менее 0.4 мас. % не приводит к достижению технического результата, а при количествах выше 2 мас. % не происходит существенного улучшения смачивания волокна. Кроме того, можно отметить, что в случае фталонитрильного связующего PN-3M при использовании волокна с эпоксидным аппретом смачивание жгута связующим хуже, чем при использовании неаппретированного волокна.
Таким образом, оптимальным содержанием аппрета на волокне является количество от 1 до 2 мас. % от массы аппретированного волокна.
Из аппретированных волокон была изготовлена ткань саржевого плетения 2*2 (аналогично ткани 22502 производства ЗАО «ИНУМиТ», http://inumit.ru/rus/produkciya-i-uslugi/ugleplastiki/uglerodnye-tkani/).
Приготовление полимерных композиционных материалов
Пример 34.
Получали ПКМ методом вакуумной инфузии, используя в качестве матрицы фталонитрильное связующее PN-3M (ООО «Итекма»). Для получения образцов ПКМ ткань нарезали на квадраты 30×30 см, укладывали по формуле [12]0, собирали вакуумный пакет и вели пропитку при 160°С в течение 15 минут. Образцы отверждали по программе отверждения, рекомендованной для связующего (http://itecma.ru/upload/iblock/211/211040069a575ba6597adfaef165bff1.pdf).
Для ПКМ исследовали прочности при межслоевом сдвиге (то) методом короткой балки согласно методике из ASTM D2344.
Прочность при сдвиге в плоскости листа определяли по методу Иосипеску в соответствии со стандартом ASTM D5379 (τ12). Полученные результаты представлены в табл. 3.
Таблица 3. Прочности межслоевых сдвигов (τ13), и сдвигов в плоскости листа (τ12), полученные для образцов ПКМ с различными аппретами. 
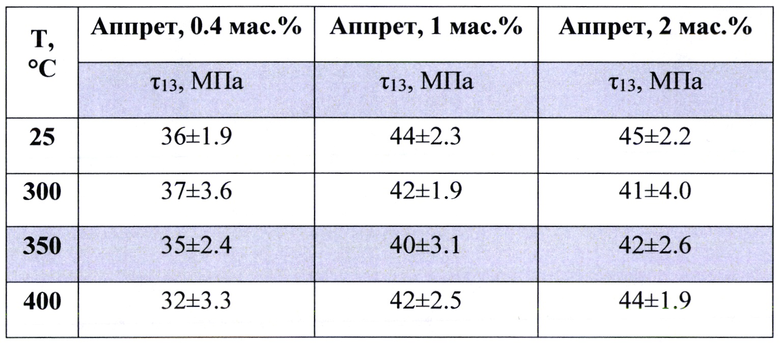
Аппрет целесообразно использовать в количестве не менее 0.4 мас. % и не более 2 мас. % (табл. 4). При нанесении аппрета в меньших количествах, чем 0.4 мас. % не наблюдается замасливания, то есть волокно продолжается распушаться и заламываться, краевой угол смачивания практически не меняется (табл. 2). При увеличении содержания аппрета выше 2 мас. % не наблюдается усиления эффекта увеличения прочности межфазной границы (прочность межслоевого сдвига).
Таблица 4. Влияние содержания аппрета на величину прочности межслоевого сдвига ПКМ.
Таким образом, материал, полученный из заявляемого углеродного волокна, аппретированного фталонитрильными мономерами, сохраняет свои механические свойства при высоких температурах (до 450°С), при этом улучшается совместимость (адгезия) аппретированного волокна со связующими, за счет чего повышаются физико-механические характеристики композиционных материалов.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДИФИЦИРОВАННЫЙ КРЕМНИЙОРГАНИЧЕСКИМИ ФРАГМЕНТАМИ ФТАЛОНИТРИЛЬНЫЙ МОНОМЕР, СПОСОБ ЕГО ПОЛУЧЕНИЯ, СВЯЗУЮЩЕЕ НА ЕГО ОСНОВЕ И ПРЕПРЕГ | 2014 |
|
RU2580927C1 |
| МОДИФИЦИРОВАННЫЙ ФОСФОРОРГАНИЧЕСКИМИ ФРАГМЕНТАМИ МОНОМЕР ФТАЛОНИТРИЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, СВЯЗУЮЩЕЕ НА ЕГО ОСНОВЕ И ПРЕПРЕГ | 2016 |
|
RU2638307C1 |
| Полифениленсульфидные композиционные материалы с аппретированными стекловолокнами и способ их получения | 2021 |
|
RU2767551C1 |
| Полифениленсульфидные композиционные материалы с аппретированными углеродными волокнами и способ их получения | 2021 |
|
RU2767562C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИРИМИДИНА | 1990 |
|
RU2019543C1 |
| СПОСОБ ОБРАБОТКИ УГЛЕРОДНОГО ВОЛОКНА АППРЕТИРУЮЩИМ СОСТАВОМ, СОВМЕСТИМЫМ С ВЫСОКОТЕМПЕРАТУРНЫМИ ТЕРМОПЛАСТИЧНЫМИ СВЯЗУЮЩИМИ | 2023 |
|
RU2815005C1 |
| ИНГИБИТОРЫ FXIA, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В ФАРМАЦЕВТИКЕ | 2020 |
|
RU2802878C1 |
| Способ получения производных 2-фенил-3-ароилбензотиофена или их солей | 1976 |
|
SU701539A3 |
| ПРОИЗВОДНЫЕ 4-ГИДРОКСИ-1,2,3,4-ТЕТРАГИДРОНАФТАЛИН-1-ИЛ-МОЧЕВИНЫ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ, СРЕДИ ПРОЧЕГО, ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНОГО ТРАКТА | 2011 |
|
RU2586333C1 |
| ФЕНОКСИПИРИДИНИЛАМИДНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ PDE4-ОПОСРЕДОВАННЫХ БОЛЕЗНЕННЫХ СОСТОЯНИЙ | 2009 |
|
RU2509077C2 |

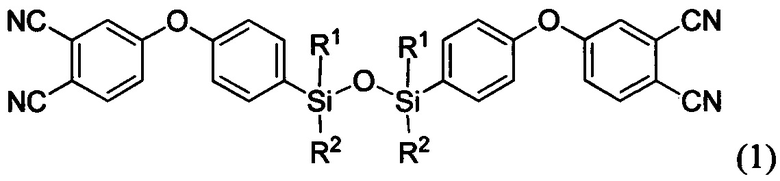
Изобретение относится к области полимерных композиционных материалов (ПКМ), а именно к аппретированию углеродных волокон, предназначенных для получения материалов, которые могут быть использованы в химической, нефтяной и металлургической промышленности, авиатехнике для создания изделий и элементов конструкций, подвергающихся воздействию повышенных температур. Агент для аппретирования углеродного волокна представляет собой по меньшей мере одно соединение формулы
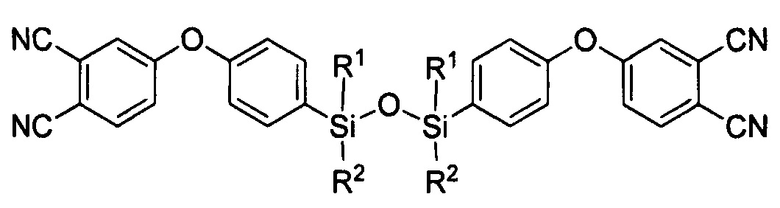
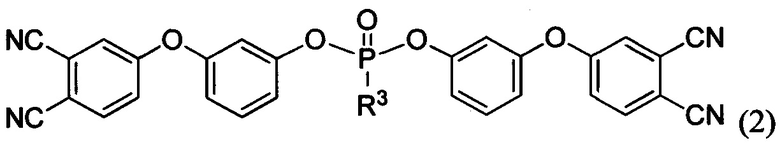
где R1=R2 и представляет собой метил или фенил или в случае, когда R1 представляет собой метил, то R2 - фенил; и/или по меньшей мере одно соединение формулы
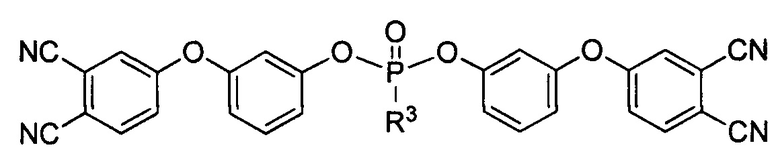
где R3 выбирают из группы, включающей арил, арилокси или алкилокси заместители. Изобретение также относится к аппретированному углеродному волокну, покрытому слоем указанного агента и полимерному композиционному материалу в виде ленты, или полотна, или ткани, который содержит аппретированное углеродное волокно и фталонитрильное связующее. Технический результат - повышение прочности, а также улучшение совместимости волокон со связующим и повышение физико-механических характеристик композиционных материалов. Фталонитрильные мономеры для аппретирования углеродных волокон за счет улучшения совместимости (адгезии) аппретированного волокна со связующими позволяют получить материал, сохраняющий свои механические свойства при высоких температурах (до 450°С). 4 н. и 19 з.п. ф-лы, 3 ил., 4 табл., 33 пр.
1. Агент для аппретирования углеродного волокна, характеризующийся тем, что представляет собой по меньшей мере одно соединение формулы
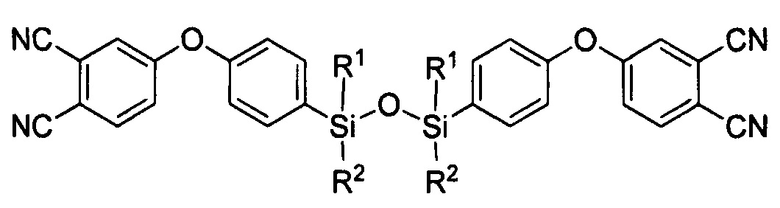
где R1=R2 и представляет собой метил или фенил или в случае, когда R1 представляет собой метил, то R2 - фенил;
и/или по меньшей мере одно соединение формулы
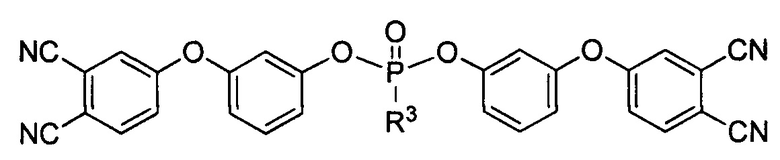
где R3 выбирают из группы, включающей арил, арилокси или алкилокси заместители.
2. Аппретированное углеродное волокно, характеризующееся тем, что волокно покрыто слоем агента по п. 1, формирующего аппретирующее покрытие на углеродных волокнах.
3. Волокно по п. 2, характеризующееся тем, что содержание аппрета составляет 0, 41-4,6 мас.% от общей массы аппретированного волокна.
4. Волокно по п. 3, характеризующееся тем, что содержание аппрета составляет 1-2 мас.% от общей массы аппретированного волокна.
5. Волокно по п. 2, характеризующееся тем, что оно имеет краевой угол смачивания во фталонитрильных связующих от 32 до 55 град.
6. Волокно по п. 2, характеризующееся тем, что оно сохраняет стабильность при температуре не менее 400°С.
7. Способ получения аппретированного углеродного волокна по п. 2, характеризующийся нанесением на углеродное волокно агента по п. 1 посредством контакта водной дисперсии мономеров по п. 1 с углеродным волокном, после чего волокно сушат до достижения постоянной массы.
8. Способ по п. 7, характеризующийся тем, что водная дисперсия мономеров имеет концентрацию от 0,4 до 2 мас.%.
9. Способ по п. 7, характеризующийся тем, что мономеры выбирают с температурой стеклования не более 60°С.
10. Способ по п. 7, характеризующийся тем, что нанесение состава на волокно осуществляют посредством погружения волокна в пропиточную ванну или его протягивания через ванну с обеспечением времени контакта волокна с водной дисперсией мономеров не менее 10 с.
11. Способ по п. 10, характеризующийся тем, что время контакта волокна с водной дисперсией мономеров составляет 15-30 с.
12. Способ по п. 7, характеризующийся тем, что сушку осуществляют при температуре от 100 до 400°С в течение 10-300 с.
13. Способ по п. 12, характеризующийся тем, что сушку осуществляют при температуре от 250 до 300°С в течение 20-200 с.
14. Способ по п. 7, характеризующийся тем, что водную дисперсию получают посредством смешения мономеров с органическим растворителем, выбранным из группы: метанол, этанол, изопропанол, тетрагидрофуран, ацетон, с последующим добавлением воды и удалением органического растворителя с получением водной дисперсии с содержанием твердой фазы не более 20 мас.%.
15. Способ по п. 7, характеризующийся тем, что водную дисперсию получают расплавлением мономера с последующим его диспергированием в воде посредством ультразвукового воздействия с частотой 15-44 кГц с добавлением неионогенных ПАВ, выбранных из группы: ди- или триэтаноламин, полиэтиленгликоль в количестве не более 5 мас.%.
16. Способ по п. 14, характеризующийся тем, что состав дополнительно содержит неионогенные ПАВ, выбранные из группы: ди- или триэтаноламин, полиэтиленгликоль в количестве не более 5 мас.%, при этом ПАВ вносят в раствор до добавления воды.
17. Способ по п. 7, характеризующийся тем, что в процессе контакта углеродного волокна с водной дисперсией проводят обработку ультразвуком с частотой 15-44 кГц.
18. Способ по п. 7, характеризующийся тем, что в качестве углеродных волокон используют волокна с линейной плотностью от 0,06 до 4,0 г/м.
19. Способ по п. 7, характеризующийся тем, что получают волокно с содержанием аппрета 0,41-4,6 мас.% от общей массы аппретированного волокна.
20. Способ по п. 7, характеризующийся тем, что получают волокно с содержанием аппрета 1-2 мас.% от общей массы аппретированного волокна.
21. Способ по п. 7, характеризующийся тем, что получают волокно с краевым углом смачивания от 32 до 55 град.
22. Способ по п. 7, характеризующийся тем, что получают волокно, сохраняющее стабильность при температуре не менее 400°С.
23. Полимерный композиционный материал в виде ленты, или полотна, или ткани, характеризующийся тем, что содержит аппретированное волокно по п. 2 и фталонитрильное связующее.
| US 20140343218 A1, 20.11.2014 | |||
| US 5389441 A, 14.02.1995 | |||
| WO 2000000350 A1, 06.01.2000 | |||
| ПОЛИМЕРНОЕ СВЯЗУЮЩЕЕ И ПРЕПРЕГ НА ЕГО ОСНОВЕ | 2012 |
|
RU2510408C1 |
| ПОЛИМЕРНЫЙ КОМПОЗИЦИОННЫЙ МАТЕРИАЛ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2536969C2 |

