Область техники, к которой относится изобретение
Настоящее изобретение относится к области химических лекарственных средств, и в нём описана серия селективных ингибиторов Фактора XIa (FXIa для краткости). Настоящее изобретение касается также фармацевтических композиций, содержащих эти соединения, и их применения в лекарственных средствах для лечения заболеваний, таких как тромбоэмболия.
Предшествующий уровень техники
Сердечнососудистые и цереброваскулярные заболевания, такие как цереброваскулярная болезнь, инфаркт мозга, инфаркт миокарда, ишемическая болезнь сердца и артериосклероз, являются причиной смерти примерно 12 миллионов человек в мире каждый год, что составляет почти ¼ от общего числа смертей в мире, и они становятся угрозой номер один для здоровья людей. Более 2.6 миллионов человек умирают от сердечнососудистых заболеваний в Китае каждый год, и 75% выживающих пациентов становятся недееспособными, из которых более 40% получают тяжелую инвалидность. Тромбоз, вызванный сердечнососудистыми и цереброваскулярными заболеваниями, диабетом и осложнениями после них, стал проблемой, требующей немедленного решения.
Процесс свертывания крови человека состоит из внутреннего пути, внешнего пути и общего пути (Annu.Rev.Med.2011.62:41–57), и представляет собой цепную реакцию, в которой процесс непрерывно усиливается и расширяется путем последовательной активации различных зимогенов. Коагуляционный каскад инициируется эндогенным путем (известен также как внутренний путь активации свертывания крови) и экзогенным путем (известен также как внешний путь активации свертывания крови), в ходе которых генерируется FXa, и затем вырабатывается тромбин (FIIa) через общий путь, и наконец образуется фибрин.
Внутренний путь означает процесс, в котором фактор XII активируется с образованием комплекса XIa-VIIIa-Ca2+-PL и активирует фактор X, в то время как внешний путь активации свертывания крови означает процесс, в котором выделяется тканевый фактор (TF), образуется комплекс TF-VIIa-Ca2+ и затем активируется фактор X. Общий путь означает процесс комбинации этих двух путей в один после образования фактора Xa, активации протромбина и наконец – генерирования фибрина, где FXI необходим для запуска эндогенного пути, и он играет ключевую роль для амплификации коагуляционного каскада, тромбин активирует обратную связь от FXI, и активированный FXI (FXIa) в свою очередь способствует выработке тромбина, тем самым амплифицируя реакцию коагуляционного каскада. Поэтому антагонисты FXI активно разрабатывались для лечения различного рода тромбов.
Традиционные антикоагулирующие лекарственные средства, такие как варфарин, гепарин, низкомолекулярный гепарин (НМГ), а также новые лекарственные средства, выпущенные в последние годы, такие как ингибиторы FXa (ривароксабан, апиксабан и т.д.) и ингибиторы тромбина (дабигатран этексилат, гирудин и т.д.) – все они эффективно уменьшают тромбоз и занимают объемный рынок сердечнососудистых и цереброваскулярных средств благодаря своей хорошей эффективности. Однако все более и более значимыми становятся их побочные эффекты. Среди них одной из наиболее серьезных проблем является “риск кровотечений” (N Engl J Med 1991; 325: 153-8, Blood. 2003; 101: 4783-4788).
В ходе исследований было показано, что в моделях тромбоза ингибирование фактора FXIa эффективно подавляло образование тромбов, но при более тяжелых стадиях тромбоза эффект FXIa был минимальный (Blood. 2010; 116(19): 3981-3989). Клиническая статистика показывает, что повышение уровня FXIa повышает преобладание венозного тромбоэмболизма (ВТЭ) (Blood 2009;114:2878-2883), а у пациентов с сильным дефицитом FXIa снижается риск тромбоза глубоких вен (ТГВ) (Thromb Haemost 2011;105:269–273).
FXIa в настоящее время является набирающей популярность мишенью для подавления тромбоза, и соединения с FXIa ингибирующей активностью раскрыты в патентных документах WO9630396, WO9941276, WO2013093484, WO2004002405, WO2013056060, WO2017005725, WO2017/023992, WO2018041122 и т.д. Из их числа только антисмысловой олигонуклеотид BAY-2306001 от Bayer перешел в Фазу II клинических испытаний.
Соединения по настоящему изобретению имеют высокую активность. В частности, соединение по настоящему изобретению демонстрирует прекрасное антикоагулирующее действие на кровь человека, обладает хорошей фармакокинетикой и может применяться для эффективного лечения и/или предотвращения сердечнососудистых и цереброваскулярных заболеваний и симптомов тромбоза.
Краткое описание изобретения
В настоящем изобретении описана серия оксопиридазинамидных производных, способы их получения и применение в фармацевтике.
В частности, в настоящем изобретении описано соединение формулы (I) или его стереоизомер, таутомер, фармацевтически приемлемая соль, где все переменные имеют значения, указанные в настоящем тексте.
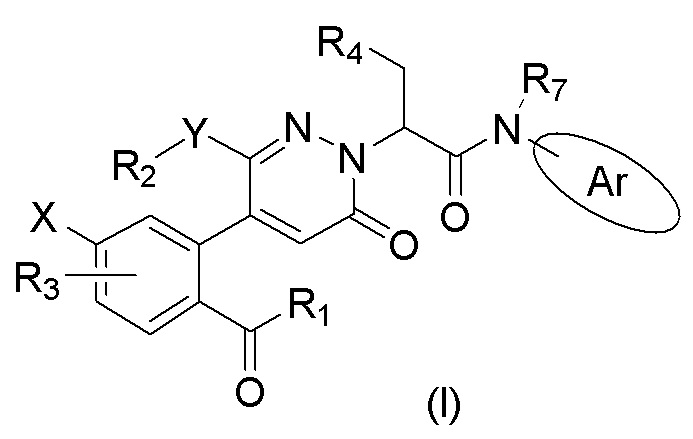
Эти соединения являются селективными ингибиторами фактора XIa (FXIa). Настоящее изобретение касается также фармацевтических композиций, содержащих эти соединения, и применения этих соединений в лекарственных средствах для лечения заболеваний, таких как тромбоэмболия.
В частности, в настоящем изобретении описаны следующие технические решения:
Соединение формулы (I) или его стереоизомер, таутомер, фармацевтически приемлемая соль,
где:
R1 выбран из группы, состоящей из алкила, галогеналкила, алкокси-группы, алкоксиалкила, гидроксиалкила;
X выбран из группы, состоящей из галогена, алкокси-группы и галогеналкила;
R3 представляет собой атом водорода или галоген;
Y выбран из группы, состоящей из атома кислорода, атома азота и связи;
R2 выбран из группы, состоящей из атома водорода, бензольного кольца, алкила, алкокси-группы, алкоксиалкила, гидроксиалкила, галогеналкила, гетероциклоалкила и циклоалкилметилена;
R4 выбран из группы, состоящей из алкила, бензила и арила или гетероарила, имеющих один заместитель R6, где R6 выбран из группы, состоящей из алкила, галогена, циано-группы, замещенной или незамещенной амидо-группы, замещенного или незамещенного оксопиперазинила, и замещенного или незамещенного 2-пиперидинонила, где замещенная амидо-группа, замещенный оксопиперазинил и замещенный 2-пиперидинонил имеют заместитель, выбранный из группы, состоящей из алкила, циклоалкила и алкоксиалкила;
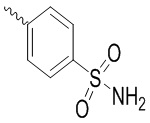
Ar выбран из группы, состоящей из бензольного кольца и индола, имеющего один или два заместителя R5, индазола, хиноксалина, бензимидазола, индолин-2-она, изохинолин-1(2H)-она и 3,4-дигидрохинолин-2(1H)-она, где R5 выбран из группы, состоящей из атома водорода, галогена, алкокси-группы, гидроксила, карбоксила, сульфо-группы, сульфонамидо-группы и амидной группы; и
R7 представляет собой атом водорода или алкил.
В предпочтительном варианте осуществления настоящего изобретения, алкил представляет собой C1-4 алкил, где C1-4 алкил выбран из группы, состоящей из метила, этила, пропила, изопропила, н-бутила, изо-бутила, втор-бутила и трет-бутила.
В предпочтительном варианте осуществления настоящего изобретения, алкокси-группа представляет собой C1-4 алкокси-группу, где C1-4 алкокси-группа выбрана из группы, состоящей из метокси-группы, этокси-группы, пропокси-группы, изопропокси-группы, н-бутокси-группы, изобутокси-группы, втор-бутокси-группы и трет-бутокси-группы.
В предпочтительном варианте осуществления настоящего изобретения, алкоксиалкил представляет собой C1-4 алкокси C1-4 алкил, где C1-4 алкокси C1-4 алкил выбран из группы, состоящей из метоксиметила, метоксиэтила, метоксипропила, метоксибутила, этоксиметила, этоксиэтила, этоксипропила, этоксибутила, пропоксиметила, пропоксиэтила, пропоксипропила, пропоксибутила, бутоксиметила, бутоксиэтила, бутоксипропила и бутоксибутила и т.п.
В предпочтительном варианте осуществления настоящего изобретения, галоген выбран из группы, состоящей из фтора, хлора, брома и иода. Термин «галогеналкил» означает, что один или больше атомов водорода в алкиле заменены на галоген, и термин «гидроксиалкил» означает, что один или больше атомов водорода в алкиле заменены на гидроксил. Термин «гетероциклоалкил» означает, что один или больше атомов водорода в алкиле заменены на гетероциклическое кольцо. Термин «циклоалкилметилен» означает, что один или больше атомов водорода в метиле заменены на циклоалкил.
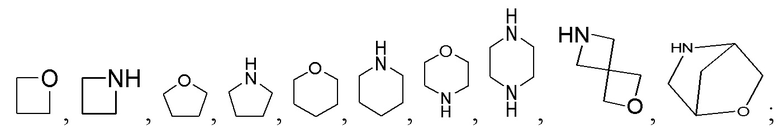
В предпочтительном варианте осуществления настоящего изобретения, гетероциклоалкил представляет собой 4-10-членный гетероциклоалкил, где 4-10-членный гетероциклоалкил выбран из группы, состоящей из
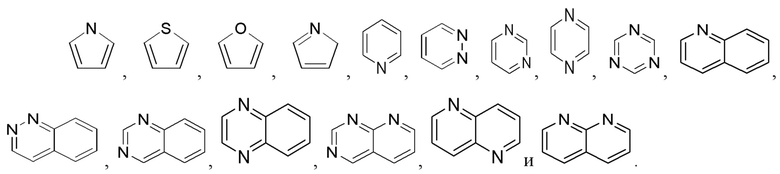
арил представляет собой фенил; гетероарил представляет собой 5-12-членный гетероарил, где 5-12-членный гетероарил выбран из группы, состоящей из
В предпочтительном варианте осуществления настоящего изобретения, циклоалкил представляет собой C3-6 циклоалкил, где C3-6 циклоалкил выбран из группы, состоящей из циклопропила, циклобутила, циклопентила и циклогексила.
В предпочтительном варианте осуществления настоящего изобретения, R1 выбран из группы, состоящей из метила, этила, гидроксиметила, дифторметила, фторметила и метоксиметила;
X выбран из группы, состоящей из хлора, фтора и трифторметила;
R3 представляет собой атом водорода;
Y представляет собой связь, и R2 представляет собой атом водорода или  , или Y представляет собой атом кислорода, и R2 выбран из группы, состоящей из атома водорода, метила, этила, фенила, гидроксиэтила, циклопропилметила, метоксиэтила, изопропила, дифторметила,
, или Y представляет собой атом кислорода, и R2 выбран из группы, состоящей из атома водорода, метила, этила, фенила, гидроксиэтила, циклопропилметила, метоксиэтила, изопропила, дифторметила, 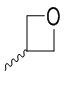 и CF3CH2-;
и CF3CH2-;
R4 выбран из группы, состоящей из фенила, 4-фторфенила, 4-бромфенила, 3-метилфенила, 4-метилфенила, бензила, изопропила, 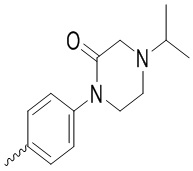 ,
, 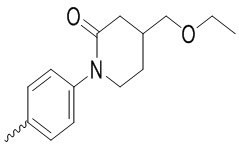 ,
, 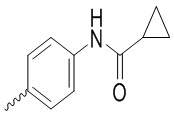 ,
, 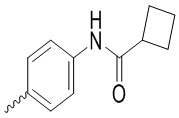 и
и 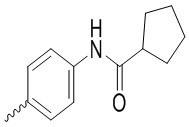 ;
;
Ar выбран из группы, состоящей из 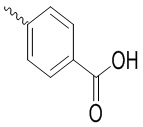 ,
,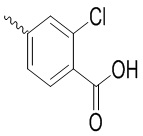 ,
,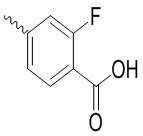 ,
,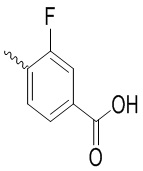 ,
,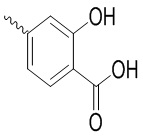 ,
,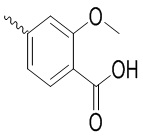 ,
,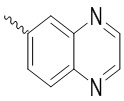 ,
,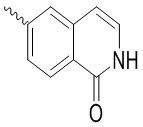 ,
,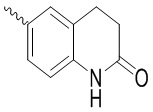 ,
,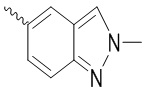 ,
,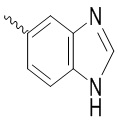 ,
,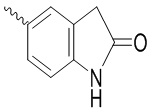 ,
,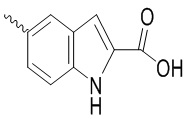 ,
,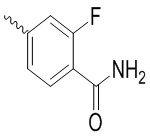 ,
, и
и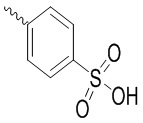 ;
;
R7 представляет собой атом водорода или метил.
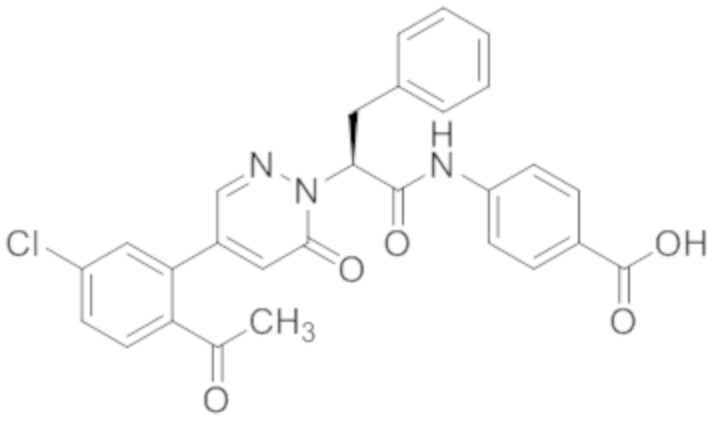
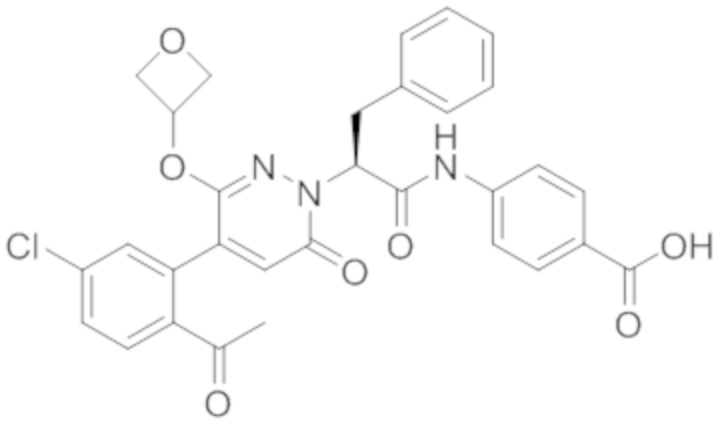
В предпочтительном варианте осуществления настоящего изобретения, соединение или его фармацевтически приемлемая соль выбраны из следующих соединений:
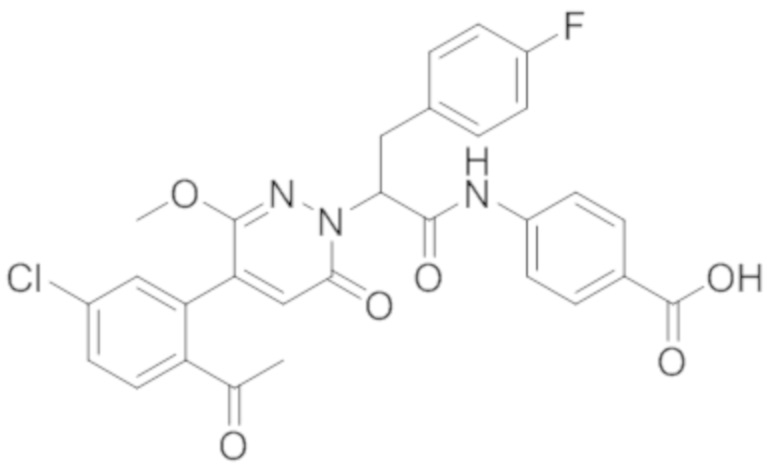
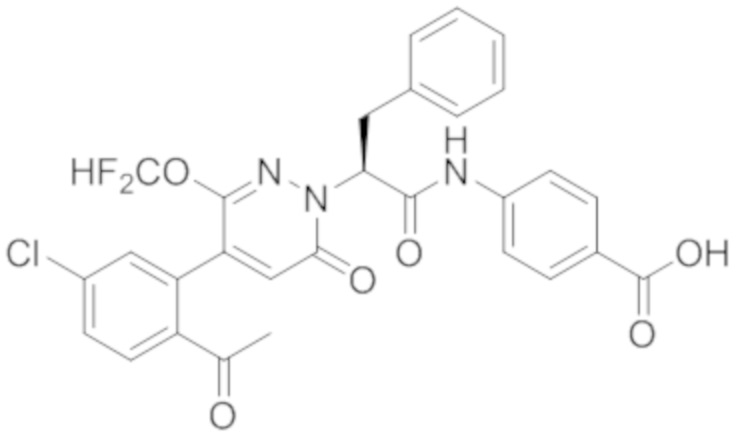
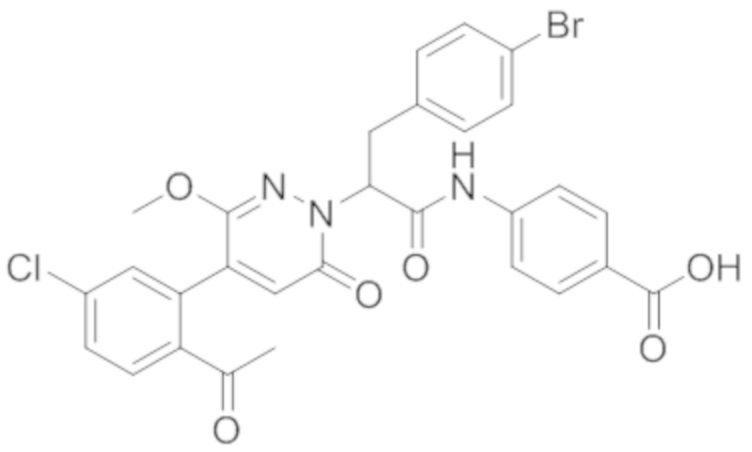
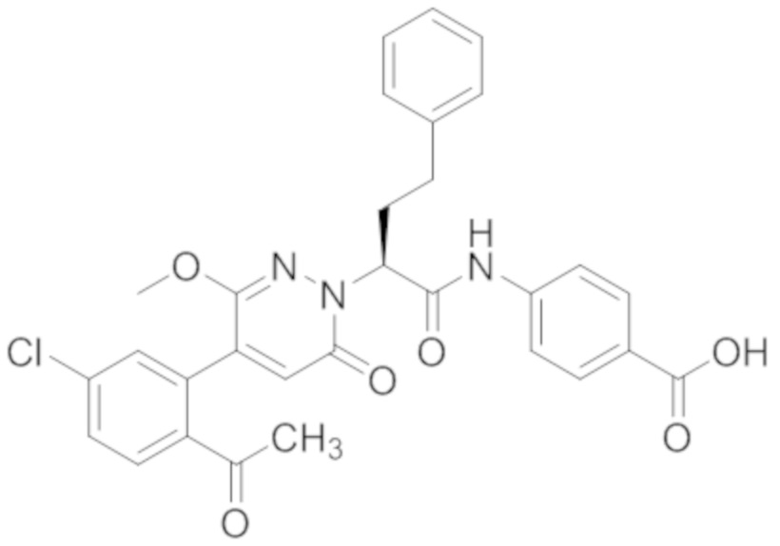
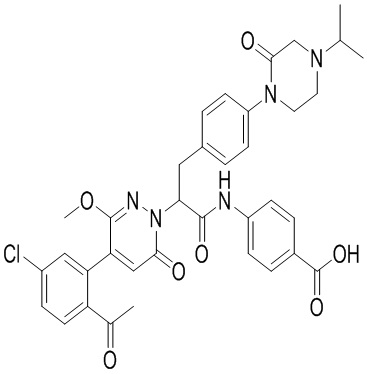
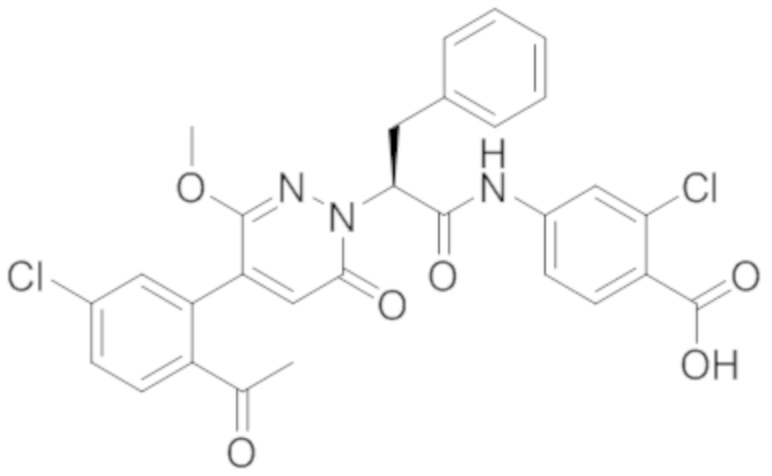
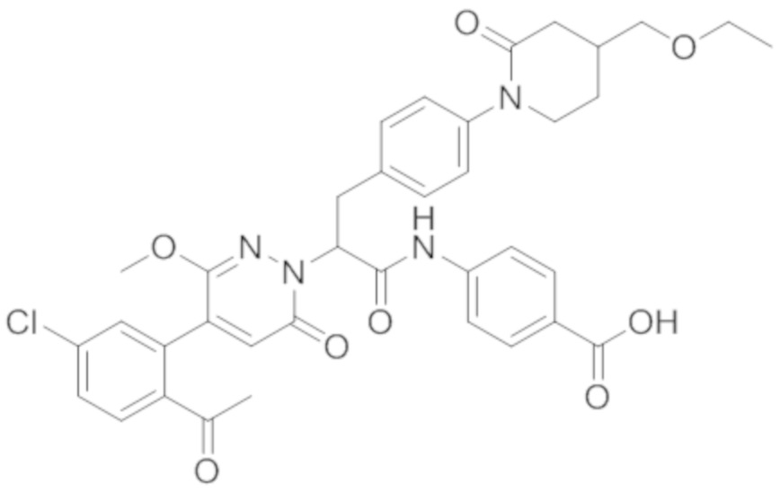
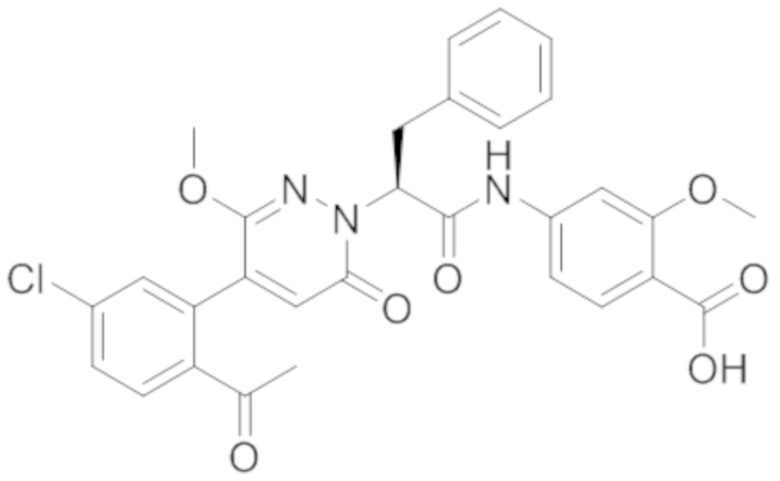
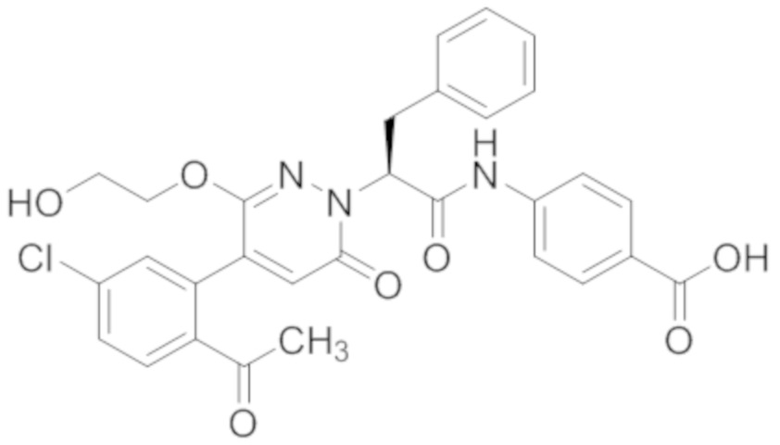
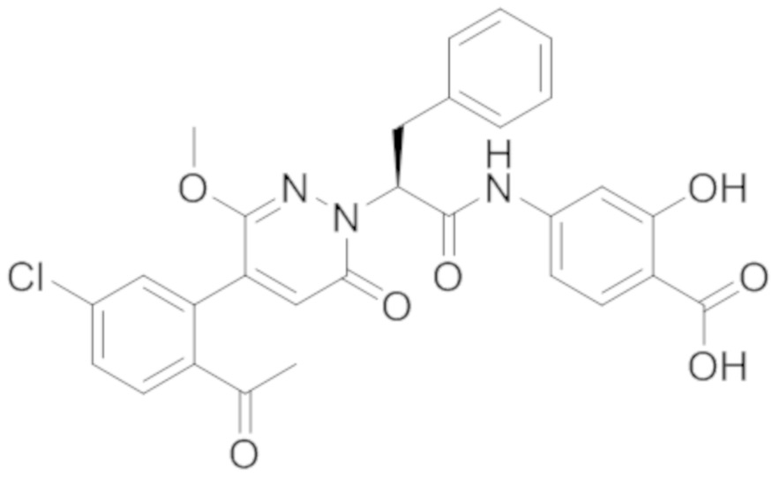
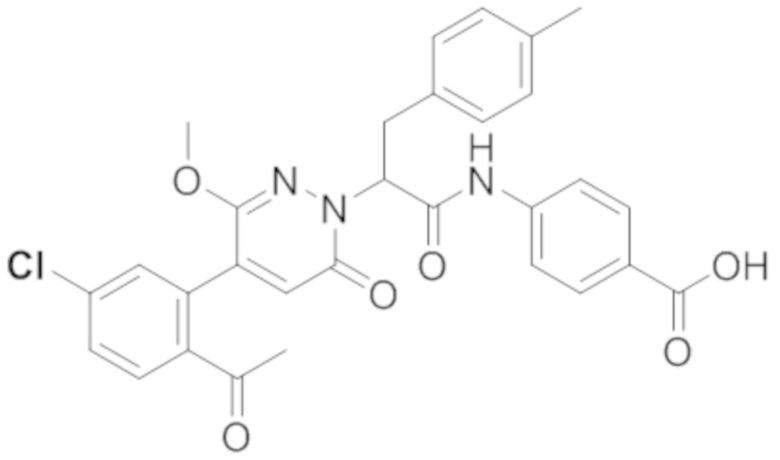
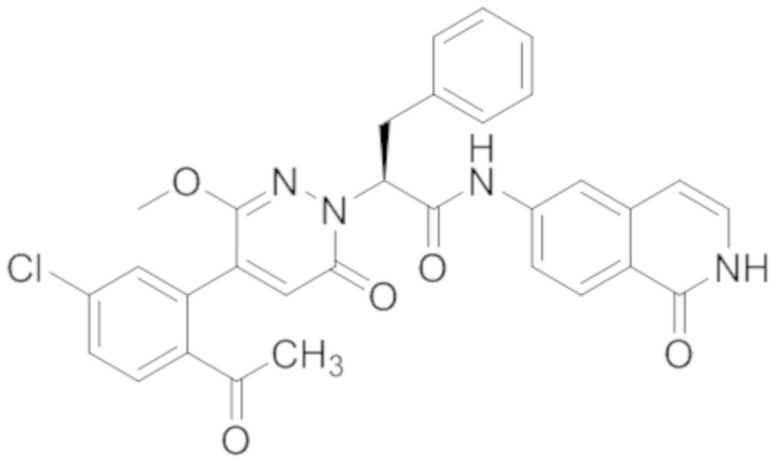
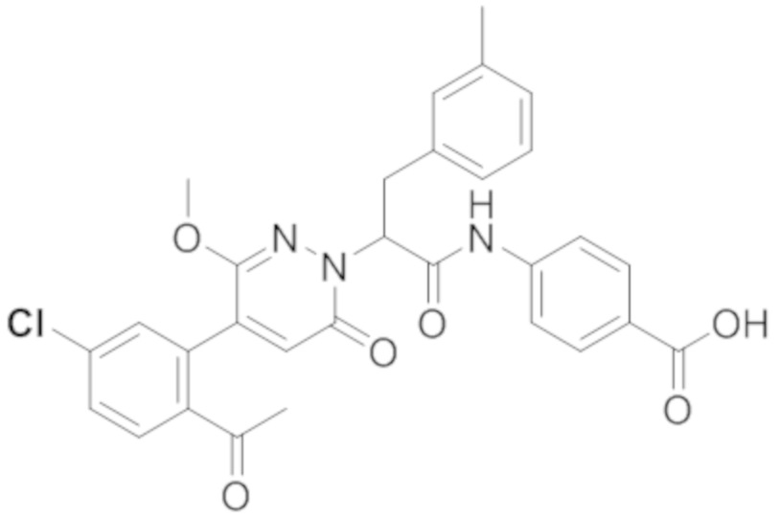
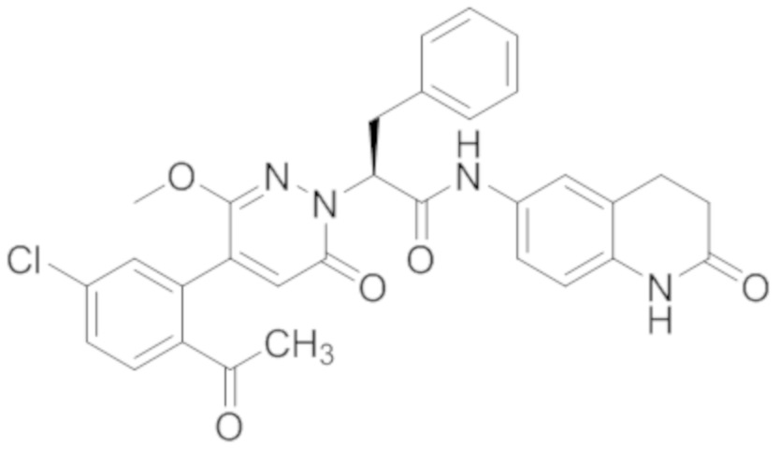
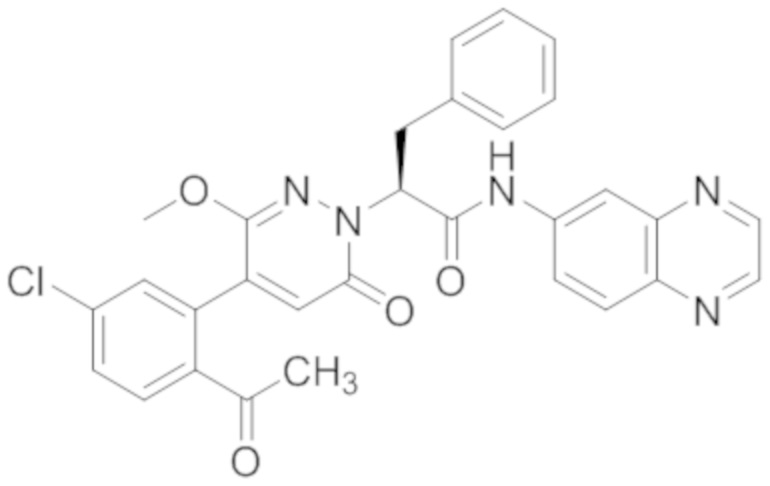
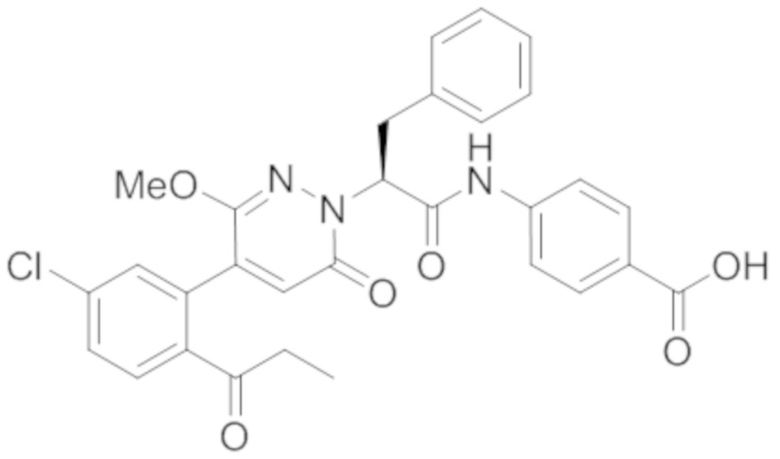
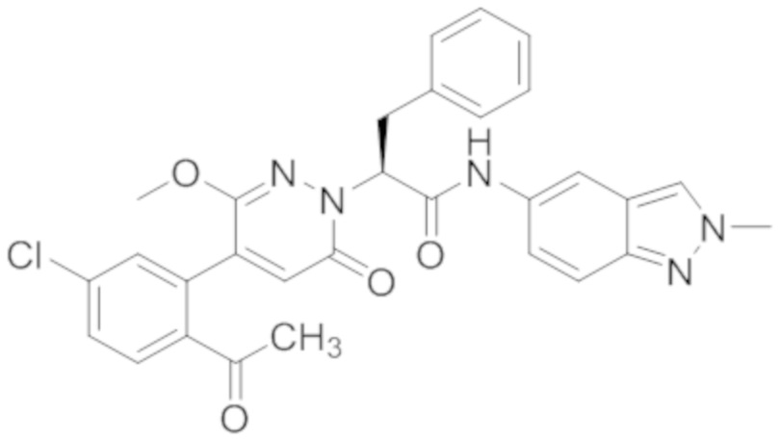
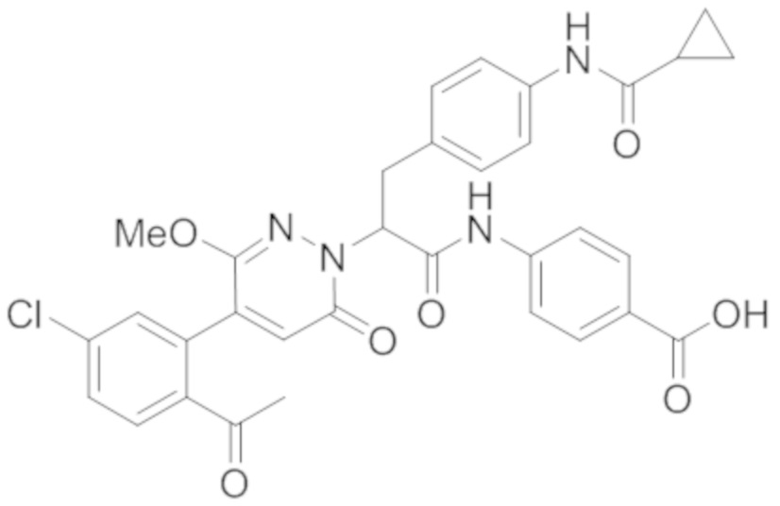
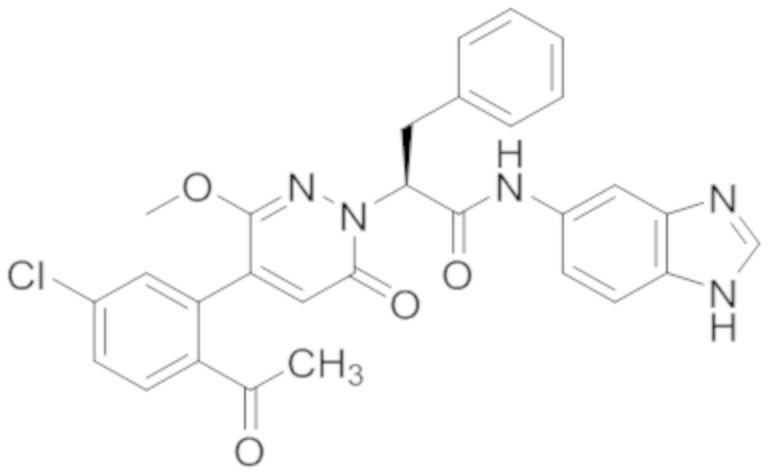
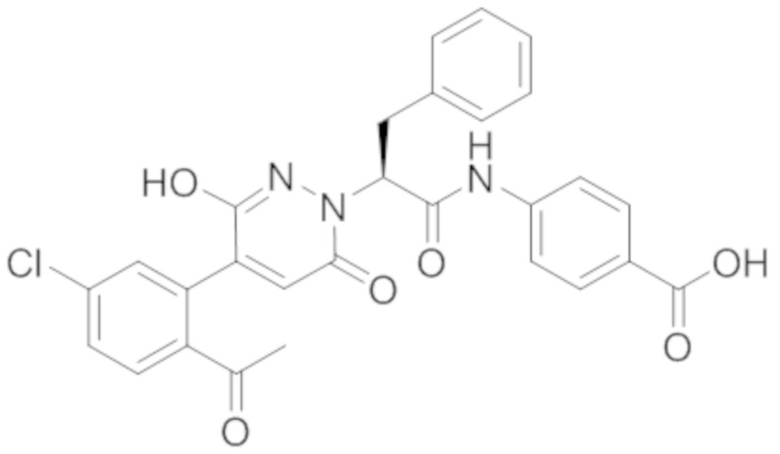
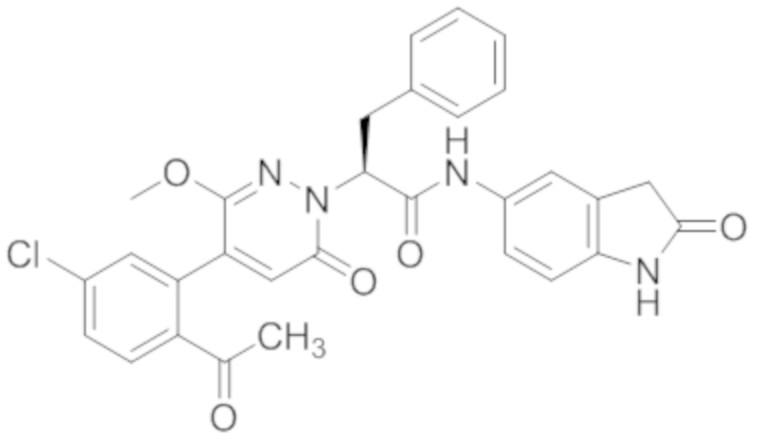
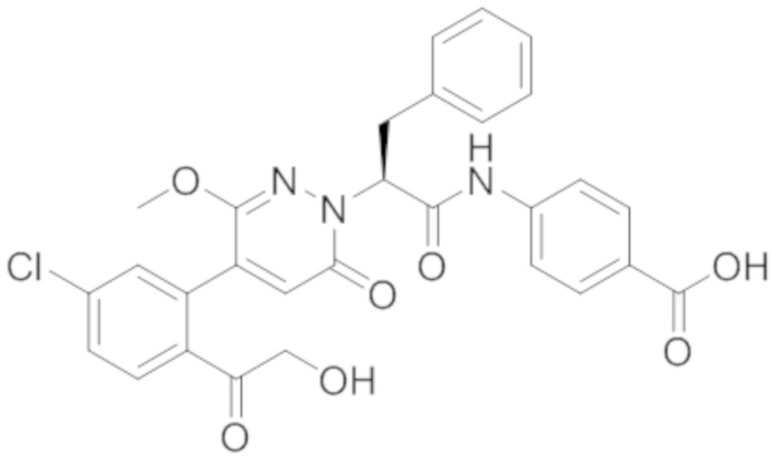

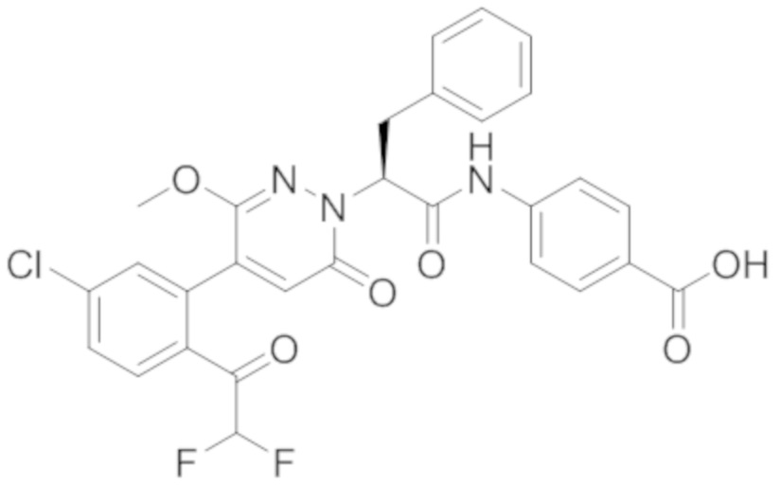
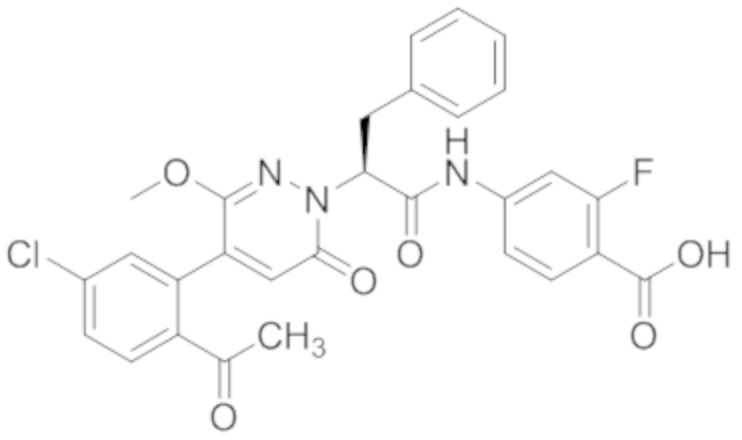
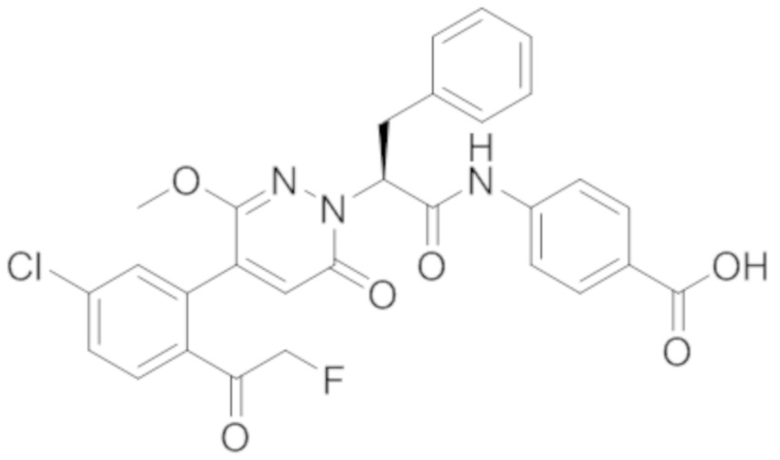
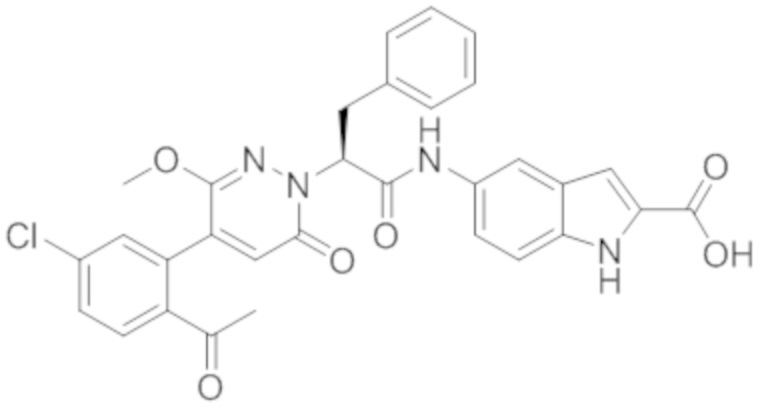
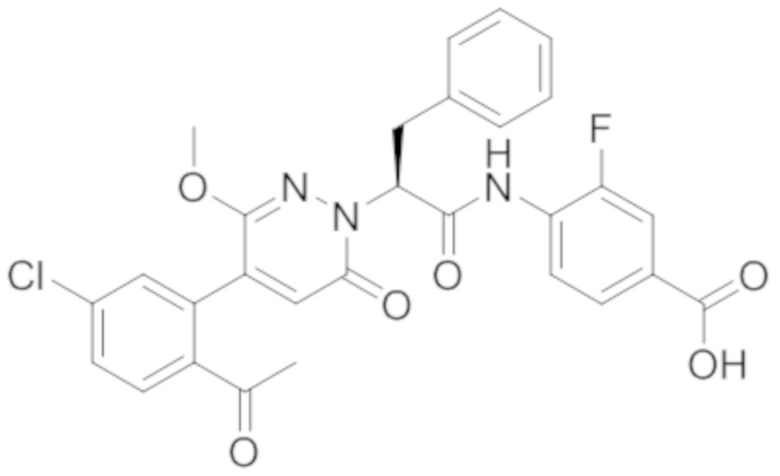
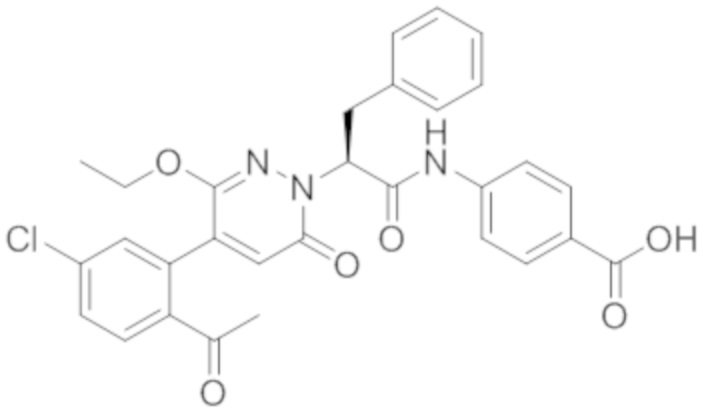
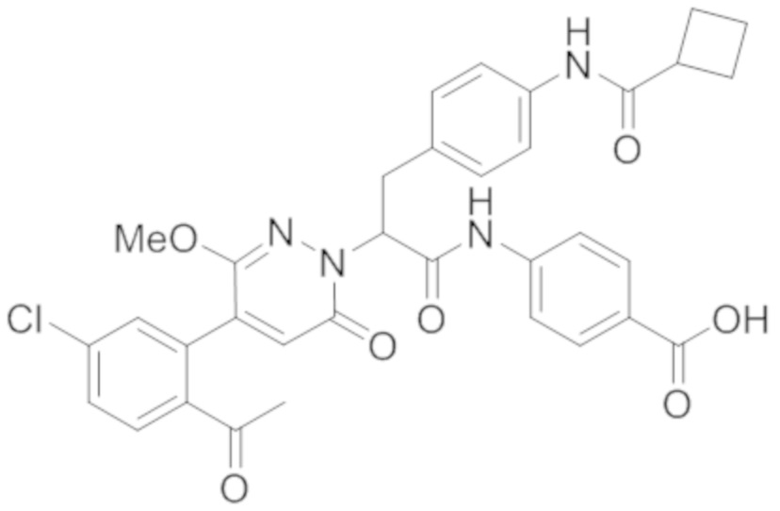
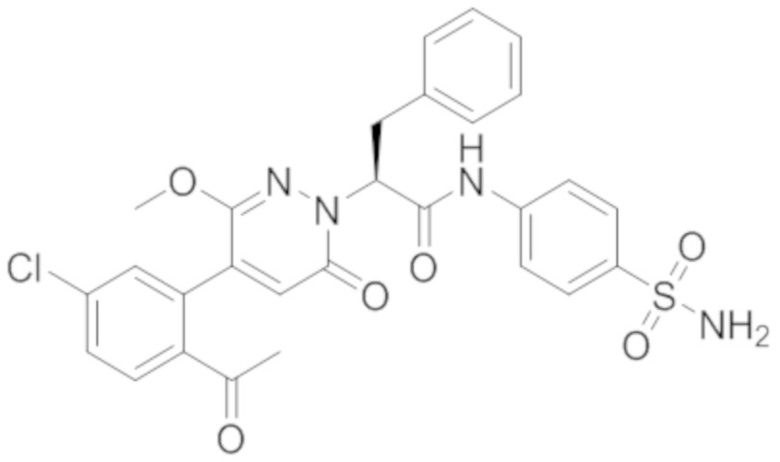
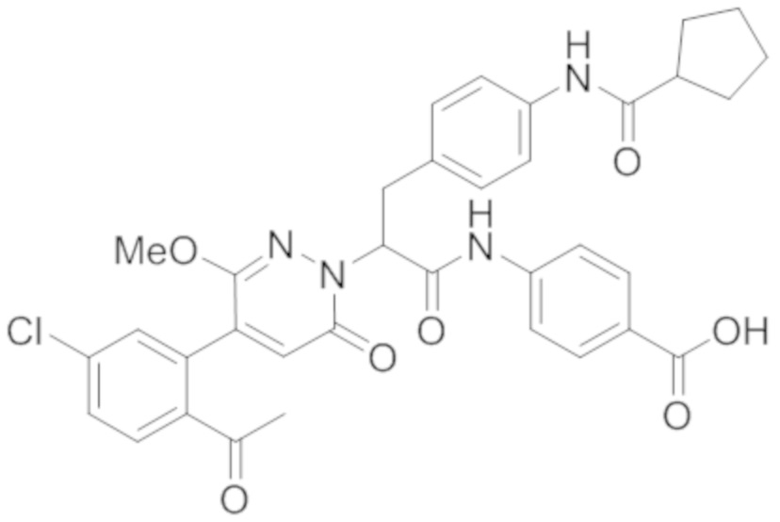
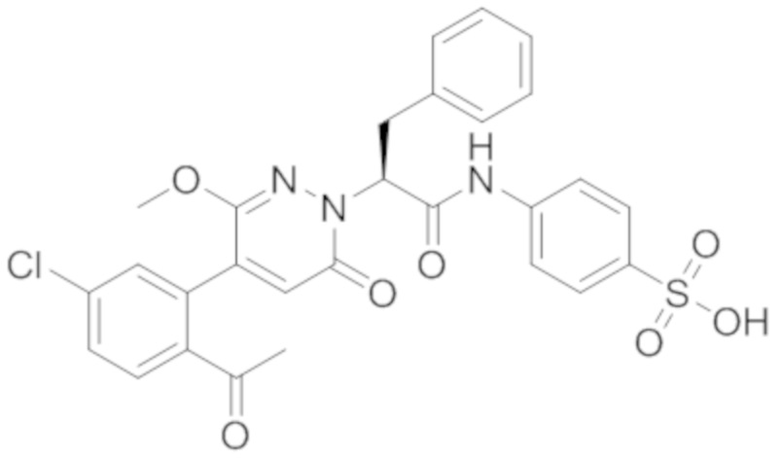
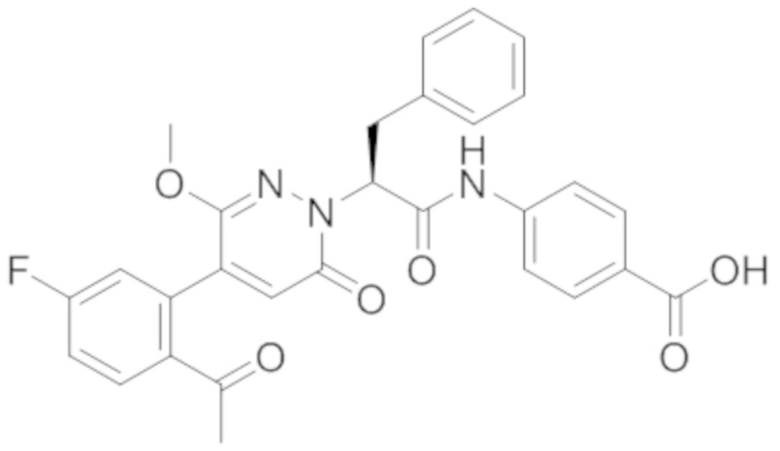
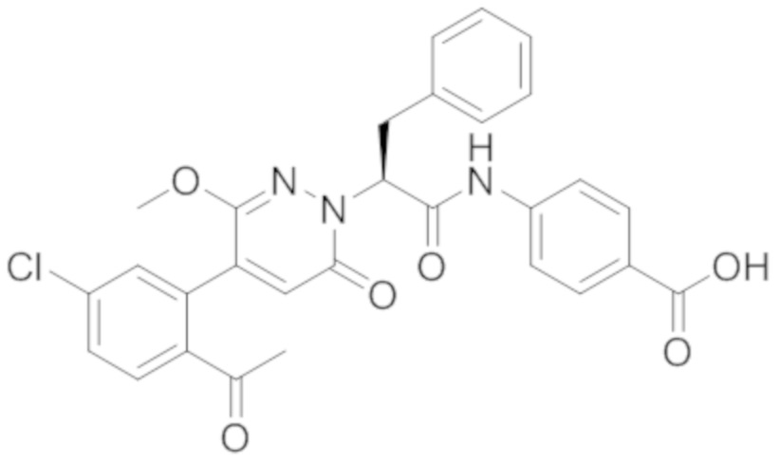
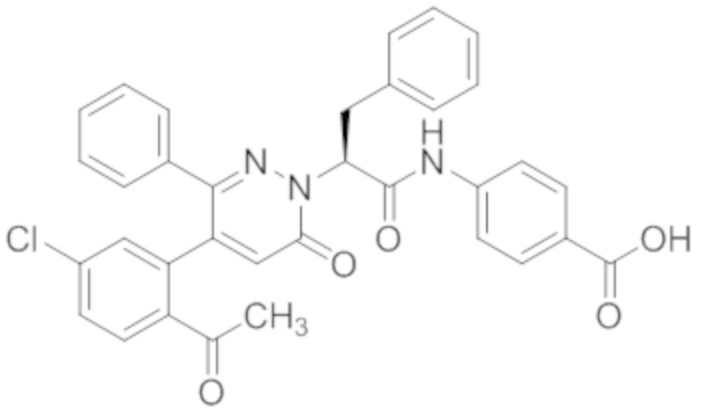

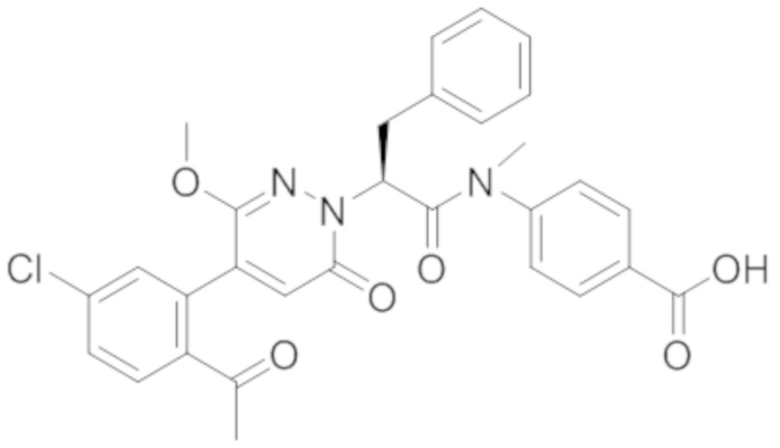
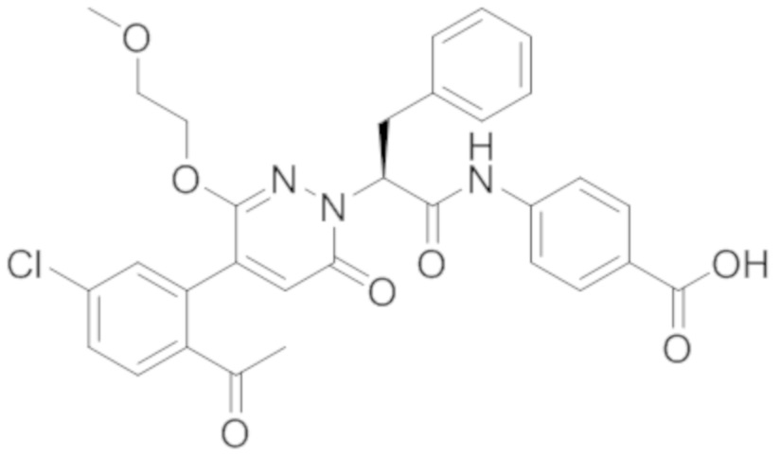
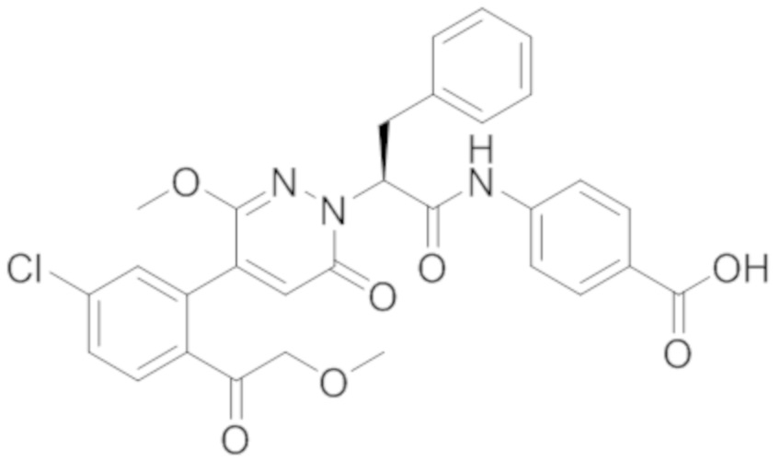
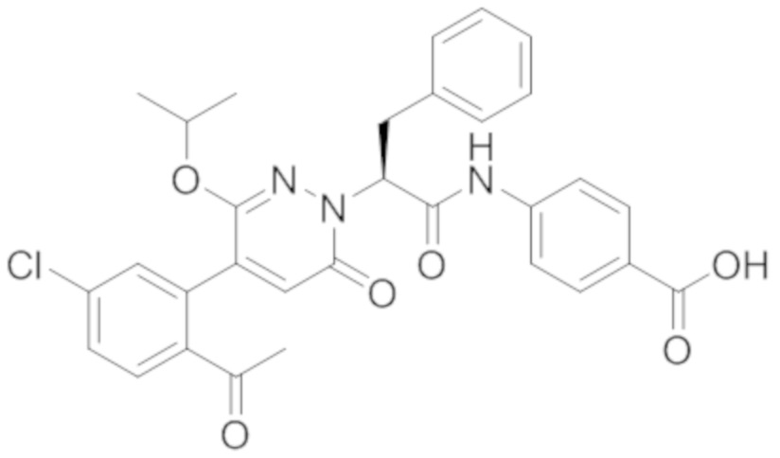

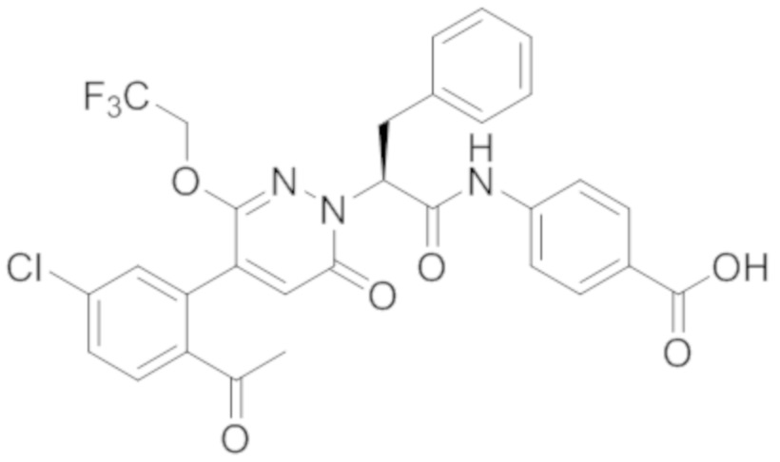
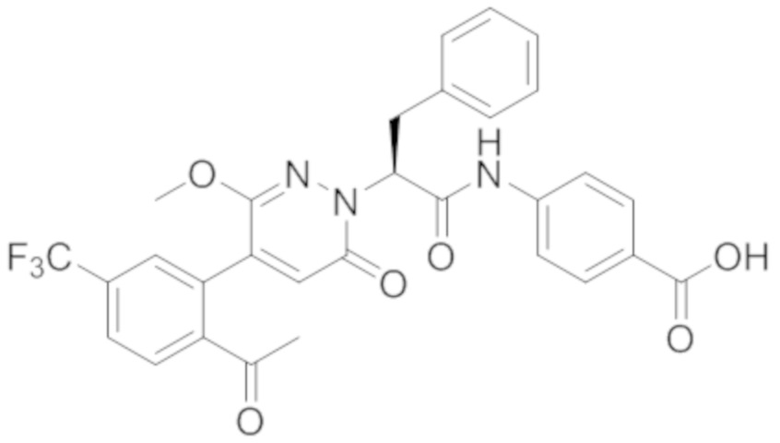
В предпочтительном варианте осуществления настоящего изобретения, фармацевтически приемлемая соль означает соль, полученную из соединения и фармацевтически приемлемой кислоты или основания.
В предпочтительном варианте осуществления настоящего изобретения, больше одного атома водорода в соединении заменены на изотоп дейтерий.
Другой целью настоящего изобретения является разработка фармацевтической композиции, содержащей описанное выше соединение формулы (I) или его стереоизомер, таутомер, фармацевтически приемлемую соль и один или больше фармацевтически приемлемых носителей.
Другой целью настоящего изобретения является разработка применения описанного выше соединения формулы (I) или его стереоизомера, таутомера, фармацевтически приемлемой соли в производстве лекарственного средства для лечения заболеваний, связанных с FXIa, в частности настоящее изобретение касается применения лекарственного средства для лечения заболеваний, связанных с тромбозом.
Если не указано иное, перечисленные далее термины и выражения, применяющиеся в настоящем тексте, имеют указанные ниже значения. Термины или выражения, для которых не указано конкретное значение, не должны пониматься как неопределенные или неясные, а должны пониматься в своем обычном общепринятом значении. Когда в настоящем тексте указана торговая марка, то подразумевается, что она обозначает соответствующий коммерческий продукт или его действующее вещество. При использовании в настоящем тексте, термин «фармацевтически приемлемый» означает такие соединения, материалы, композиции и/или дозированные формы, которые, согласно общепринятым медицинским представлениям, подходят для применения в контакте с тканями человека и животных, не вызывая избыточной токсичности, раздражения, аллергических реакций или других проблем или осложнений, и имеют приемлемое соотношение польза/риск.
Термин “фармацевтически приемлемая соль” означает соль соединения по настоящему изобретению, полученную из соединения по настоящему изобретению и фармацевтически приемлемой кислоты или основания.
Помимо солевых форм, соединения по настоящему изобретению существуют также в форме пролекарств. Пролекарства соединений по настоящему изобретению в физиологических условиях легко трансформируются в соединения по настоящему изобретению. Кроме того, пролекарства могут превращаться в соединения по настоящему изобретению под воздействием химических или биохимических механизмов в in vivo окружении.
Некоторые соединения по настоящему изобретению могут существовать в несольватированной и сольватированной форме, включая форму гидратов. В целом, несольватированные и сольватированные формы эквивалентны и все входят в объем настоящего изобретения.
Соединения по настоящему изобретению могут существовать в определенных геометрических или стереоизомерных формах. Настоящее изобретение охватывает все такие соединения, включая цис- и транс-изомеры, (-)- и (+)-энантиомеры, (R)- и (S)-энантиомеры, диастереомерные изомеры, (D)-изомеры, (L)-изомеры, и их рацемические и другие смеси, такие как энантиомерно или диастереомерно обогащенные смеси, и все такие смеси входят в объем настоящего изобретения. Дополнительные асимметричные атомы углерода могут присутствовать в заместителях, таких как алкил. Все такие изомеры, а также их смеси, входят в объем настоящего изобретения.
Оптически активные (R)- и (S)-изомеры, а также D и L изомеры, могут быть получены хиральным синтезом или с использованием хиральных реагентов или другими общеизвестными методами. Если целевым является один энантиомер соединения по настоящему изобретению, его можно получить асимметрическим синтезом или с помощью модификации вспомогательным хиральным реагентом, где полученную смесь диастереомеров разделяют, и вспомогательную хиральную группу отщепляют, получая чистый целевой эннатимоер. Альтернативно, когда молекула содержит основную функциональную группу (такую как амино-группа) или кислотную функциональную группу (такую как карбоксильная группа), можно получить диастереомерную соль с подходящей оптически активной кислотой или основанием, затем диастереомеры разделяют известными в данной области методами, и выделяют чистые энантиомеры. Кроме того, разделение энантиомеров и диастереомеров обычно можно осуществить с помощью хроматографии на хиральной неподвижной фазе, опционально в комбинации с химической дериватизацией (например, из амина в карбамат).
Атомы в молекулах соединений по настоящему изобретению состоят из изотопов, и изменение изотопного состава может увеличить время полураспада, снизить скорость выведения, стабилизировать метаболизм и улучшить активность in vivo. Также в настоящее изобретение входят варианты осуществления, в которых по меньшей мере один атом заменен на атом, имеющий то же атомное число (число протонов), но отличающееся массовое число (сумма протонов и нейтронов). Примеры изотопов для соединений по настоящему изобретению включают изотопы атома водорода, атома углерода, атома азота, атома кислорода, атома фосфора, атома серы, атома фтора, атома хлора, которые соответственно включают 2H, 3H, 13C, 14C, 15N, 17O, 18O, 31P, 32P, 35S, 18F, 36Cl. В частности, радиоактивные изотопы, которые испускают радиацию во время своего распада, такие как 3H или 14C, могут применяться в топологических исследованиях фармацевтических композиций или соединений in vivo. Стабильные изотопы не распадаются и не изменяют своего количества, и они не являются радиоактивными, поэтому безопасны для применения. Когда атомы в составе молекул соединений по настоящему изобретению представляют собой изотопы, замену на изотопы можно осуществить известными в данной области методами, заменяя использующиеся в синтезе реагенты на реагенты, содержащие соответствующие изотопы.
Соединения по настоящему изобретению могут содержать неприродный процент изотопов атомов по одному или больше атомам в составе их молекул. Например, соединения могут быть помечены радиоактивными изотопами, такими как дейтерий (2H), иод-125 (125I) или C-14 (14C). Все вариации изотопного состава в соединениях по настоящему изобретению, вне зависимости от того радиоактивные они или нет, включены в объем настоящего изобретения.
Кроме того, один или больше атомов водорода в соединениях по настоящему изобретению могут быть заменены на изотоп дейтерий (2H). После дейтерирования соединения по настоящему изобретению обладают увеличенным временем полураспада, сниженной скоростью выведения, у них стабилизирован метаболизм и улучшенная активность in vivo.
Способ получения изотопных производных обычно включает применение метода межфазного катализа. Например, предпочтительный метод дейтерирования использует межфазный катализатор (например, соли тетраалкиламмония, NBu4HSO4). Метиленовые протоны в дифенилметановых соединениях подвергаются обмену под воздействием межфазного катализатора, что приводит к более высокому проценту дейтерирования, чем восстановление дейтерированными силанами (например, триэтилдейтеросиланом) в присутствии кислоты (например, метансульфокислоты), или применение кислоты Льюиса, такой как трихлорид алюминия, с дейтероборатом натрия.
Термин “фармацевтически приемлемый носитель” означает любой носитель или среду в препарате, способный доставлять эффективное количество действующего вещества по настоящему изобретению, не нарушая биологическую активность этого действующего вещества и не оказывая токсичного побочного действия на хозяина или пациента. Репрезентативные примеры носителя включают воду, масло, вещества растительного происхождения, минеральные вещества, кремовую основу, матрикс лосьона, матрикс мази и т.п. Такие матриксы включают суспендирующие агенты, реагенты, придающие липкость, усилители проникновения и т.п. Их препараты хорошо известны квалифицированным специалистам в области косметики или фармацевтических средств для наружного применения. Дополнительную информацию о носителях можно найти в работе Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott, Williams & Wilkins (2005), содержание которой включено в настоящий текст посредством ссылки.
Термин “вспомогательное вещество” обычно означает носитель, разбавитель и/или среду, необходимые для приготовления эффективной фармацевтической композиции.
Термин “эффективное количество” или “терапевтически эффективное количество” в отношении лекарственного средства или фармакологически активного вещества означает нетоксичное, но достаточное для достижения желаемого эффекта количество лекарственного средства или фармакологически активного вещества. Для пероральных дозированных форм по настоящему изобретению, “эффективное количество” одного действующего вещества в композиции означает количество, необходимое для достижения целевого эффекта при использовании в комбинации с другим действующим веществом в композиции. Конкретное эффективное количество варьируется от пациента к пациенту и зависит от возраста и общего состояния здоровья пациента, а также от конкретного действующего вещества. Подходящее эффективное количество в каждом конкретном случае может определить квалифицированный специалист, основываясь на стандартных экспериментах.
Термины “действующее вещество”, “терапевтическое средство”, “активное вещество” или “активная субстанция” означают химическое вещество, эффективное для лечения целевого нарушения, заболевания или состояния.
Термины “опционально” или “необязательно” означают, что описанное далее событие или обстоятельство может иметь место или не иметь место, и такое описание включает случаи, в которых указанное событие или обстоятельство имеет место, и случаи, в которых указанное событие или обстоятельство не имеет место.
“ ” означает связь.
” означает связь.
Соединения по настоящему изобретению можно получить различными методами синтеза, известными квалифицированным специалистам в данной области, включая описанные ниже частные варианты осуществления, а также варианты осуществления в комбинации с другими методами химического синтеза и эквивалентные альтернативы, хорошо известные квалифицированным специалистам в данной области, при этом предпочтительные варианты осуществления включают (но не ограничиваются только ими) примеры по настоящему изобретению.
Подробное описание изобретения
Настоящее изобретение далее описано более подробно с привлечением примеров, но варианты осуществления настоящего изобретения не ограничиваются приведенными примерами.
Структуру соединений определяли методами ядерного магнитного резонанса (ЯМР) или масс-спектрометрии (МС). Химические сдвиги в спектрах ЯМР (δ) даны в единицах 10-6 (м.д.). Спектры ЯМР записывали на спектрометре Bruker AVANCE-III, и растворителями служили дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный хлороформ (CDCl3), а в качестве внутреннего стандарта применялся тетраметилсилан (ТМС).
Масс-спектры регистрировали на масс-спектрометре ISQ EC (производитель: Thermo, модель: ISQ EC).
Анализ методом высокоэффективной жидкостной хроматографии (ВЭЖХ) проводили с помощью ВЭЖХ-хроматографа Thermo U3000 HPLC DAD.
Использовали систему CombiFlash Rf+ LUMEN (TELEDYNE ISCO).
Пластинами для тонкослойной хроматографии на силикагеле служили силикагелевые пластины Yantai Yinlong HSGF254 или GF254, пластины для аналитической тонкослойной хроматографии (ТСХ) имели толщину слоя 0.17 мм - 0.23 мм, а препаративные пластины ТСХ для разделения и очистки продуктов имели толщину слоя 0.4 мм - 0.5 мм.
Для колоночной хроматографии на силикагеле применяли Rushan Shangbang силикагель 100 - 200 меш.
rt означает комнатную температуру.
Пример 1
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксопирролидин-1(6H)-ил)-3- фенилпропанамидо)бензойной кислоты
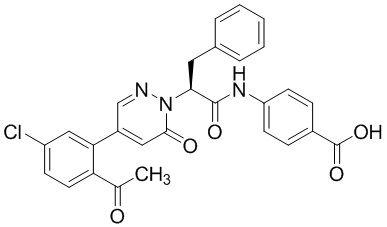
Использовался следующий путь синтеза:
Стадия A: Синтез 1-бром-4-хлор-2-винилбензола
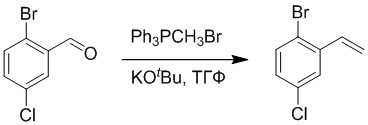
2-Бром-5-хлорбензальдегид (3.00 г, 13.6 ммоль) растворяли в тетрагидрофуране (40.0 мл). После этого добавляли в полученный раствор бромметил трифенилфосфин (5.86 г, 16.0 ммоль) и трет-бутоксид калия (3.00 г, 27.0 ммоль), и атмосферу в реакционной колбе три раза заменяли на азот. Реакционную смесь перемешивали при 60°C в течение 4 часов.
Реакционный раствор разбавляли, медленно добавляя по каплям 50 мл воды. Полученный смешанный раствор экстрагировали этилацетатом (100 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия, водой (100 мл), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/15). Получали 2.00 г маслянистого продукта 1-бром-4-хлор-2-винилбензола (выход: 66.0%). LCMS: RT = 4.56 мин, [M+H]+ = 217.14.
Стадия B: Синтез 2-(2-бром-5-хлорфенил)ацетальдегида
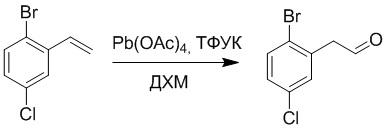
1-Бром-4-хлор-2-винилбензол (1.20 г, 5.5 ммоль) и ацетат свинца (9.70 г, 22.0 ммоль) растворяли в дихлорметане (30.0 мл). После этого добавляли в полученный раствор трифторуксусную кислоту (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов.
Объединяли небольшие образцы (500 мг сырого продукта) и добавляли в реакционный раствор насыщенный водный раствор бикарбоната натрия (100 мл), чтобы погасить реакцию. Полученный смешанный раствор экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (50 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/10). Получали 1.50 г твердого 2-(2-бром-5-хлорфенил)ацетальдегида (выход: 82.0%). LCMS: RT = 4.33 мин, [M+H]+ = 233.26.
Стадия C: Синтез 4-(2-бром-5-хлорфенил)-5-гидроксифуран-2(5H)-она
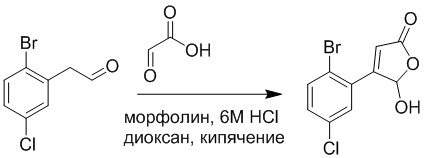
2-(2-Бром-5-хлорфенил)ацетальдегид (1.50 г, 6.4 ммоль) и 2-оксоуксусную кислоту (713 мг, 9.60 ммоль) растворяли в 1,4-диоксане (20.0 мл). После этого добавляли в полученный раствор морфолин (547 мг, 6.4 ммоль) и соляную кислоту (6 моль/л, 4.0 мл). Реакционную смесь перемешивали при 110°C в течение 14 часов.
Добавляли в реакционный раствор насыщенный водный раствор бикарбоната натрия (50 мл), чтобы погасить реакцию. Полученный смешанный раствор экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (50 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/5). Получали 900 мг твердого 4-(2-бром-5-хлорфенил)-5-гидроксифуран-2(5H)-она (выход: 51.0%). LCMS: RT = 3.88 мин, [M+H]+ = 289.16.
Стадия D: Синтез 5-(2-бром-5-хлорфенил)пиридазин-3(2H)-она
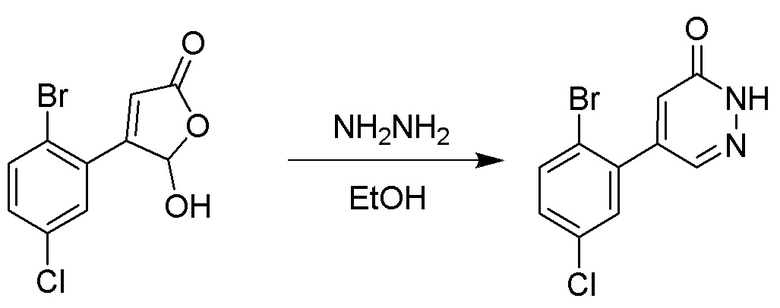
4-(2-бром-5-хлорфенил)-5-гидроксифуран-2(5H)-он (600 мг, 2.0 ммоль) и гидразин (132 мг, 4.1 ммоль) растворяли в этаноле (10.0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов.
Для разбавления реакционного раствора добавляли воду (50 мл). Полученный смешанный раствор экстрагировали этилацетатом (40 мл × 3 раза). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (30 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/5). Получали 380 мг белого твердого 5-(2-бром-5-хлорфенил)пиридазин-3(2H)-она (выход: 64.2%). LCMS: RT = 2.87 мин, [M+H]+ = 285.20.
Стадия E: Синтез трет-бутил (R)- 4-(2-гидрокси-3-фенилпропанамидо)бензоата
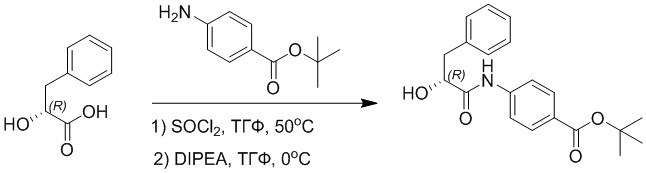
D-фенилмолочную кислоту (23.0 г, 138 ммоль) растворяли в сухом тетрагидрофуране (400 мл), помещали в сухую трехгорлую колбу, перемешивали на ледяной бане в течение 15 минут в инертной атмосфере азота. Тионилхлорид (20 мл, 207 ммоль) медленно добавляли по каплям в реакционный раствор, прикапывание заканчивалось примерно за 30 минут. Реакционный раствор нагревали до 50°C и перемешивали при постоянной температуре в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры, упаривали на роторном испарителе и сушили на масляном насосе 15 минут, затем растворяли в ТГФ, получая раствор A. Трет-бутил 4-аминобензоат (20 г, 110 ммоль) и диизопропилэтиламин (68 мл, 414 ммоль) растворяли в сухом тетрагидрофуране (200 мл), помещали в сухую трехгорлую колбу. Полученный смешанный раствор перемешивали на ледяной бане в течение 15 минут в инертной атмосфере азота. Раствор A медленно добавляли по каплям в смешанный раствор на ледяной бане в течение 1 часа.
Добавляли в реакционный раствор воду, чтобы погасить реакцию, смешанный раствор экстрагировали этилацетатом (200 мл × 3 раза), органические фазы объединяли, Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (100 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/4). Получали 19 г желтого твердого трет-бутил (R)-4-(2-гидрокси-3-фенилпропанамидо)бензоата (выход: 53%). LCMS: RT = 4.16 мин, [M-H]- = 340.09.
Стадия F: Синтез трет-бутил (R)-4-(2-(((4-нитрофенил)сульфонил)окси)-3- фенилпропанамидо)бензоата
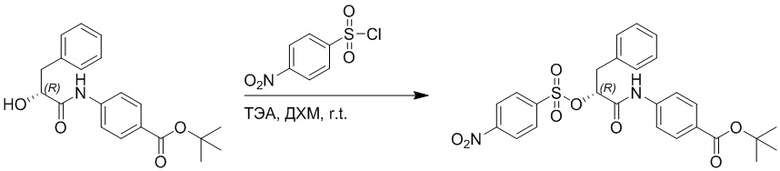
Трет-бутил (R)-4-(2-гидрокси-3-фенилпропанамидо)бензоат (19 г, 55.7 ммоль) и триэтиламин (21.6 мл, 167.1 ммоль) растворяли в дихлорметане (100.0 мл). Добавляли в реакционный раствор 4-нитробензолсульфонил хлорид (18.5 г, 165.6 ммоль) на ледяной бане. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов.
Насыщенный водный раствор бикарбоната натрия (100 мл) добавляли в реакционный раствор, чтобы погасить реакцию. Полученный смешанный раствор экстрагировали этилацетатом (200 мл × 3 раза). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (100 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток растворяли в дихлорметане (40 мл), и добавляли по каплям в н-гексан (400 мл) при перемешивании. В осадок выпадало большое количество белого твердого вещества, его отфильтровывали, и осадок на фильтре собирали, получая 11.2 г белого твердого трет-бутил (R)-4-(2-(((4-нитрофенил)сульфонил)окси)-3-фенилпропанамидо)бензоата (выход: 38%). LCMS: RT = 4.39 мин.
Стадия G: Синтез трет-бутил (S)-4-(2-(4-(2-бром-5-хлорфенил)-6-оксопиридазин- 1(6H)-ил)-3-фенилпропанамидо)бензоата
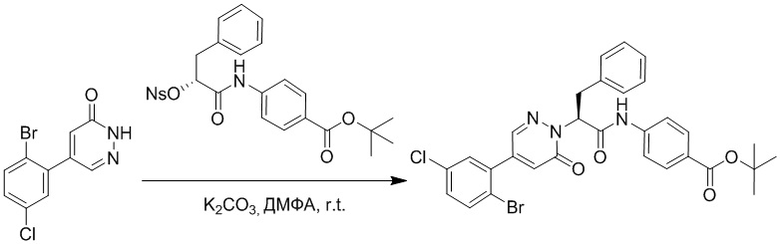
5-(2-бром-5-хлорфенил)пиридазин-3(2H)-он (380 мг, 1.33 ммоль) и трет-бутил (R)-4-(2-((((4-нитрофенил)сульфонил)окси)-3-фенилпропанамидо)бензоат (840 мг, 1.60 ммоль) растворяли в этаноле (10.0 мл). После этого добавляли в полученный раствор карбонат калия (367 мг, 2.66 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов.
Добавляли воду (50 мл), чтобы разбавить реакционный раствор. Полученный смешанный раствор экстрагировали этилацетатом (40 мл × 3 раза). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (30 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/5). Получали 460 мг белого твердого трет-бутил (S)-4-(2-(4-(2-бром-5-хлорфенил)-6-оксопиридазин- 1(6H)-ил)-3-фенилпропанамидо)бензоата (выход: 56.0%). LCMS: RT = 3.95 мин, [M+H]+ = 608.06.
Стадия H: Синтез трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата
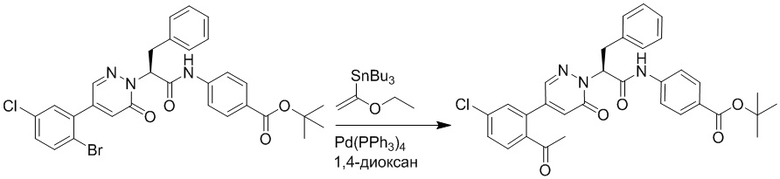
Трет-бутил (S)-4-(2-(4-(2-бром-5-хлорфенил)-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)бензоат (300 мг, 0.49 ммоль) и трибутил(1-этоксивинил)станнан (213 мг, 0.59 ммоль) растворяли в 1,4-диоксане (15.0 мл). После этого добавляли в полученный раствор тетракис(трифенилфосфин)палладий (56 мг, 0.049 ммоль). Реакционную смесь перемешивали при 100°C в течение 18 часов.
В реакционный раствор добавляли соляную кислоту (1 моль/л, 10 мл), затем перемешивали в течение 1 часа. Полученный смешанный раствор экстрагировали этилацетатом (40 мл × 3 раза). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (30 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/5). Получали 250 мг белого твердого трет-бутил (S)-4- (2-(4-(2-ацетил-5-хлорфенил)-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата (выход: 70.0%). LCMS: RT = 4.17 мин, [M+H]+ = 572.03.
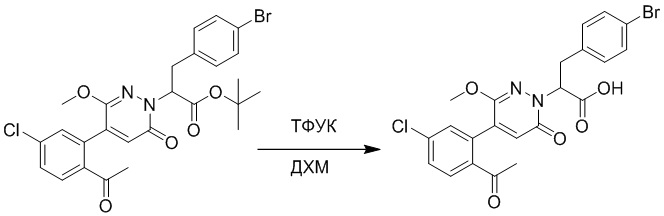
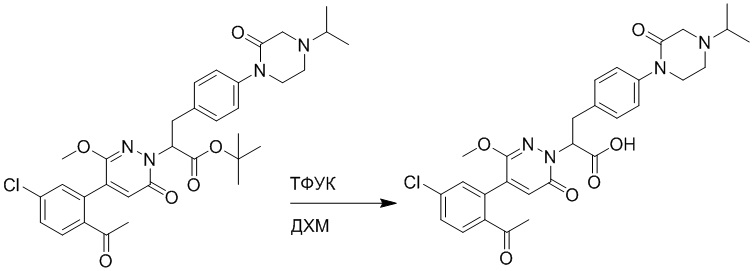
Стадия I: Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксопирролидин-1(6H)- ил)-3-фенилпропанамидо)бензойной кислоты
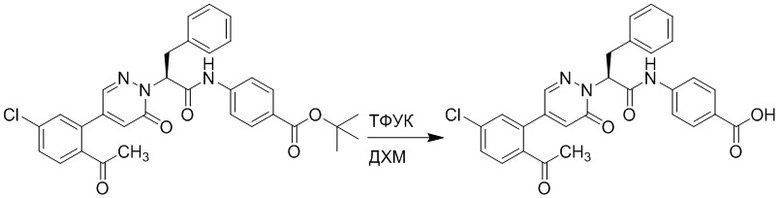
Трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)бензоат (240 мг, 350 ммоль) растворяли в дихлорметане (4 мл). После этого добавляли в полученный раствор трифторуксусную кислоту (1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов.
Реакционный раствор упаривали при пониженном давлении и очищали методом препаративной высокоэффективной жидкостной хроматографии. Получали 207 мг белой твердой (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксопирролидин-1(6H)-ил)-3- фенилпропанамидо)бензойной кислоты (выход: 95.0%). LCMS: RT = 3.94 мин, [M+H]+ = 516.10. 1H ЯМР (400 МГц, ДМСО) δ 12.72 (с, 1H), 10.59 (с, 1H), 8.03 (д, J = 8.4 Гц, 1H) , 7.93 – 7.86 (м, 3H), 7.74 – 7.66 (м, 3H), 7.55 (д, J = 2.1 Гц, 1H), 7.31 – 7.22 (м, 4H), 7.20 – 7.14 (м, 1H), 6.87 (д, J = 2.2 Гц, 1H), 5.82 (дд, J = 9.6, 5.5 Гц, 1H), 3.46 (ддд, J = 19.7, 14.1, 7.6 Гц, 2H), 2.55 (с, 3H).
Пример 2
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксо-3-фенилпиридазин-1(6H)-ил)- 3-фенилпропанамидо)бензойной кислоты
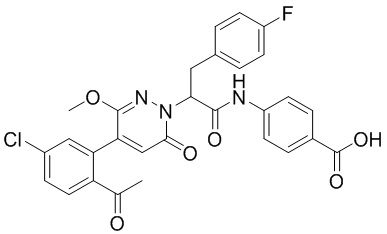
Использовался следующий путь синтеза:
Стадия A: Синтез трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)ацетата
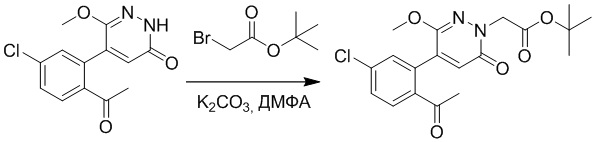
5-(2-Ацетил-5-хлорфенил)-6-метилпиридин-3(2H)-он (800 мг, 2.8 ммоль) и трет-бутил 2-бромацетат (671 мг, 3.4 ммоль) растворяли в N,N-диметилформамиде (20.0 мл). После этого добавляли в полученный раствор карбонат калия (793 мг, 5.7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов.
Добавляли воду (50 мл), чтобы разбавить реакционный раствор. Полученный смешанный раствор экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (30 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток суспендировали в смеси н-гексан/этилацетат. Получали 780 мг желтого твердого трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)ацетата (выход: 69.0%). LCMS: RT = 3.35 мин, [M+H]+ = 393.06.
Стадия B: трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-(4-фторфенил)пропаноат
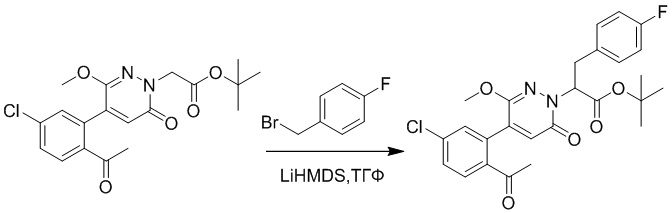
Трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)- ил)ацетат (70 мг, 0.17 ммоль ) и 1-(бромметил)-4-фторбензол (40 мг, 0.20 ммоль) растворяли в тетрагидрофуране (5.0 мл). После этого добавляли в полученный раствор бис(триметилсилил)амид лития (0.3 мл, 0.30 ммоль). Реакционную смесь перемешивали при -50°C в течение 2 часов.
Добавляли воду (10 мл), чтобы разбавить реакционный раствор. Полученный смешанный раствор экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (30 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/5). Получали 50 мг желтого твердого трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3- метокси-6-оксопиридазин-1(6H)-ил)-3-(4-фторфенил)пропаноата (выход: 56.0%). LCMS: RT = 3.87 мин, [M+H]+ = 501.02.
Стадия C: 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)- 3-(4-фторфенил)пропановая кислота
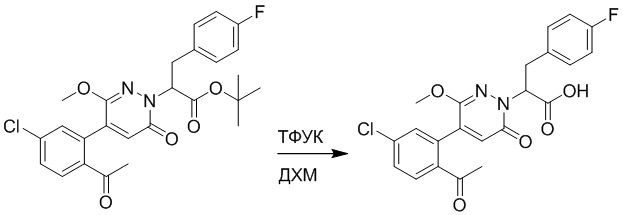
Трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(4- фторфенил)пропаноат (50 мг, 0.1 ммоль) растворяли в дихлорметане (4.0 мл). После этого добавляли в полученный раствор трифторуксусную кислоту (1.0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов.
Реакционный раствор упаривали при пониженном давлении. Получали 40 мг желтой твердой 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(4- фторфенил)пропановой кислоты (выход: 90.0%). LCMS: RT = 3.29 мин, [M+H]+ = 445.01.
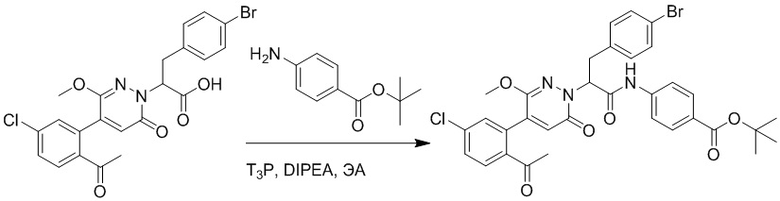
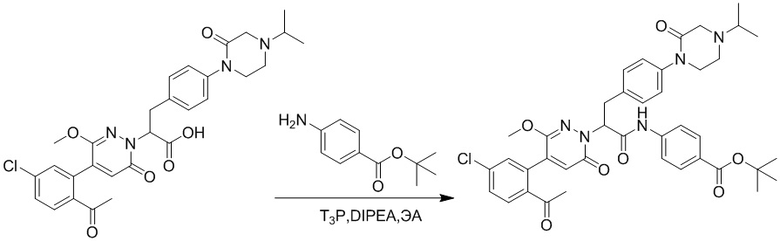
Стадия D: Синтез трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-4-фторфенил)пропанамидо)бензоата
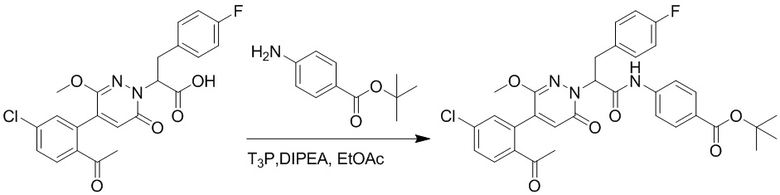
2-(4-(2-Ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(4- фторфенил)пропановую кислоту (40 мг, 0.10 ммоль) и трет-бутил 4-аминобензоат (22 мг, 0.12 ммоль) растворяли в этилацетате (10.0 мл). После этого добавляли в полученный раствор 1-пропилфосфорный ангидрид (151 мг, 0.50 ммоль) и N,N-диизопропилэтиламин (37 мг, 0.30 ммоль). Реакционную смесь перемешивали при 50°C в течение 3 часов.
Добавляли воду (30 мл), чтобы разбавить реакционный раствор. Полученный смешанный раствор экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (30 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1). Получали 35 мг желтого твердого трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)- 3-метокси-6-оксопиридазин-1(6H)-ил)-3-4-фторфенил)пропанамидо)бензоата (выход: 58.0%). LCMS: RT = 4.14 мин, [M+H]+ = 620.14.
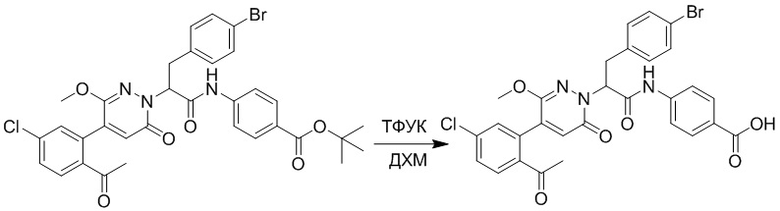
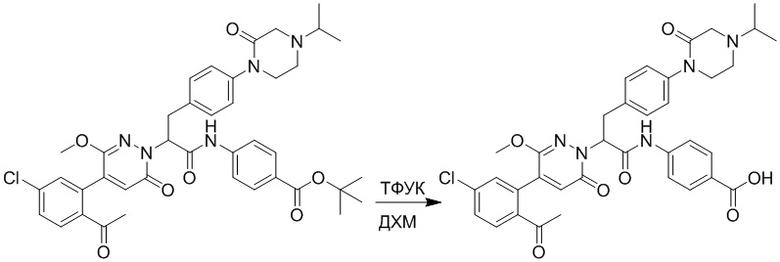
Стадия E: Синтез 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-4- фторфенил)пропанамидо)бензойной кислоты
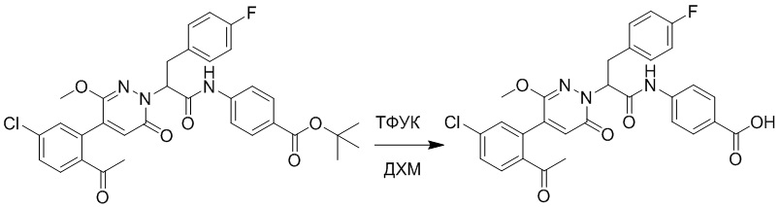
Трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- 4-фторфенил)пропанамидо)бензоат (40 мг, 0.060 ммоль) растворяли в дихлорметане (4.0 мл). После этого добавляли в полученный раствор трифторуксусную кислоту (1.0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов.
Реакционный раствор упаривали при пониженном давлении, и остаток от упаривания очищали методом препаративной высокоэффективной жидкостной хроматографии. Получали 18 мг белой твердой 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-4-фторфенил) пропанамидо)бензойной кислоты (выход: 50.0%). LCMS: RT = 3.98 мин, [M-H]- = 562.08. 1H ЯМР (500 МГц, ДМСО) δ 10.51 (с, 1H), 8.02 (д, J = 8.4 Гц, 1H), 7.92 (д, J = 8.8 Гц , 2H), 7.73 (д, J = 8.7 Гц, 2H), 7.71 (дд, J = 8.4, 2.2 Гц, 1H), 7.52 (д, J = 2.2 Гц, 1H), 7.35 (дд, J = 8.6, 5.6 Гц, 2H), 7.11 (дд, J = 12.3, 5.5 Гц, 2H), 6.92 (с, 1H), 5.72 (дд, J = 10.2, 5.0 Гц, 1H), 3.68 (с, 3H), 3.51 (дд, J = 14.1, 10.2 Гц, 1H), 3.42 (дд, J = 14.0, 4.7 Гц, 1H), 2.55 (с, 3H).
Пример 3
Синтез 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- (4-бромфенил)пропанамидо)бензойной кислоты
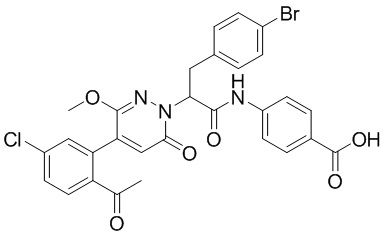
Использовался следующий путь синтеза:
Стадия A: Синтез трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-(4-бромфенил)пропаноата
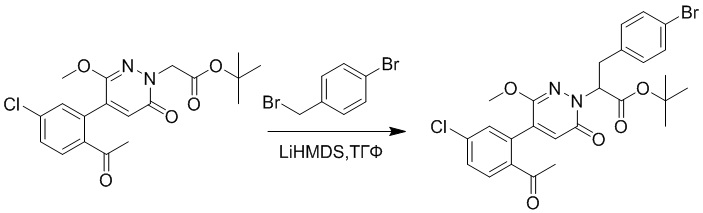
Трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)- ил)ацетат (150 мг, 0.38 ммоль) и 1-бром-4-(бромметил)бензол (114.7 мг, 0.46 ммоль) растворяли в тетрагидрофуране (5.0 мл). После этого добавляли в полученный раствор бис(триметилсилил)амид лития (0.5 мл, 0.50 ммоль). Реакционную смесь перемешивали при 50°C в течение 2 часов.
Добавляли воду (10 мл), чтобы разбавить реакционный раствор. Полученный смешанный раствор экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (30 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/5). Получали 120 мг желтого твердого трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-(4-бромфенил)пропаноата (выход: 56.0%). LCMS: RT = 3.89 мин, [M+H]+ = 561.14.
Стадия B: Синтез 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-(4-бромфенил)пропановой кислоты
Трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(4- бромфенил)пропаноат (30 мг, 0.050 ммоль) растворяли в дихлорметане (4.0 мл). После этого добавляли в полученный раствор трифторуксусную кислоту (1.0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов.
Реакционный раствор упаривали при пониженном давлении. Получали 24 мг желтой твердой 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(4- бромфенил)пропановой кислоты (выход: 88.8%). LCMS: RT = 3.35 мин, [M+H]+ = 505.02.
Стадия C: Синтез трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-(4-бромфенил)пропанамидо)бензоата
2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(4- бромфенил)пропановую кислоту (24 мг, 0.047 ммоль) и трет-бутил 4-аминобензоат (11 мг, 0.057 ммоль) растворяли в этилацетате (5.0 мл). После этого добавляли в полученный раствор 1-пропилфосфорный ангидрид (59 мг, 0.19 ммоль) и N,N-диизопропилэтиламин (18.4 мг, 0.14 ммоль). Реакционную смесь перемешивали при 50°C в течение 3 часов.
Добавляли воду (30 мл), чтобы разбавить реакционный раствор. Полученный смешанный раствор экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (30 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1). Получали 28 мг желтого твердого трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси- 6-оксопиридазин-1(6H)-ил)-3-(4-бромфенил)пропанамидо)бензоата (выход: 87.5%). LCMS: RT = 4.27 мин, [M+H]+ = 680.10.
Стадия D: Синтез 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-(4 -бромфенил)пропанамидо)бензойной кислоты
Трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- (4-бромфенил))пропанамидо)бензоат (28 мг, 0.040 ммоль) растворяли в дихлорметане (4.0 мл). После этого добавляли в полученный раствор трифторуксусную кислоту (1.0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов.
Реакционный раствор упаривали при пониженном давлении, и остаток от упаривания очищали методом препаративной высокоэффективной жидкостной хроматографии. Получали 12 мг белой твердой 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(4-бромфенил) пропанамидо)бензойной кислоты (выход: 48%). LCMS: RT = 4.15 мин, [M-H]- = 623.94. 1H ЯМР (400 МГц, ДМСО) δ 12.76 (с, 1H), 10.51 (с, 1H), 7.96 (дд, J = 46.8, 7.5 Гц, 3H), 7.73 (д, J = 7.7 Гц, 3H), 7.55 – 7.42 (м, 3H), 7.28 (с, 2H), 6.92 (с, 1H), 5.73 (с, 1H), 3.66 (с, 3H), 3.48 (д, J = 10.9 Гц, 2H), 2.55 (с, 3H).
Пример 4
Синтез 4-((2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- (4-(4-изопропил-2-оксопиперазин-1-ил)фенил)пропанил)окси)бензойной кислоты
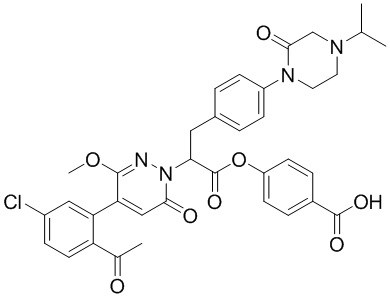
Использовался следующий путь синтеза:
Стадия A: Синтез (4-бромфенил)метанола
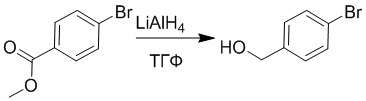
Метил 4-бромбензоат (2.0 г, 9.3 ммоль) растворяли в тетрагидрофуране (100.0 мл). После этого добавляли в полученный раствор литийалюминий гидрид (700 мг, 18.6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов.
Добавляли в реакционный раствор метанол (100 мл), чтобы погасить реакцию, и затем смесь упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1). Получали 1.3 г (4-бромфенил)метанола в виде белого масла (выход: 87.5%). LCMS: RT=4.16 мин, [M+H]+=187.02.
Стадия B: Синтез 4-изопропилпиперазин-2-она
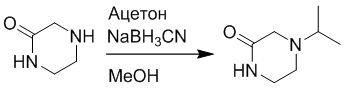
Пиперазин-2-он (2.0 г, 20 ммоль) растворяли в метаноле (50.0 мл). После этого добавляли в полученный раствор ацетон (5.8 мг, 150 ммоль) и цианоборгидрид натрия (2.6 г, 40 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов.
Добавляли в реакционный раствор метанол (50 мл), чтобы погасить реакцию, и затем смесь упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1). Получали 2.8 г 4-изопропилпиперазин-2-она в виде белого масла (выход: 100%). LCMS: RT = 0.82 мин, [M+H]+ = 143.04.
Стадия C: Синтез 1-(4-(гидроксиметил)фенил)-4-изопропил-2-она
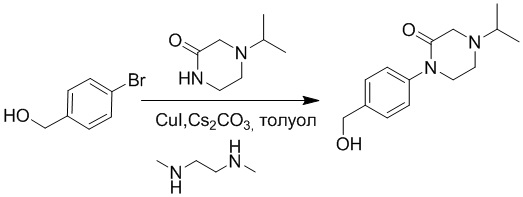
(4-Бромфенил)метанол (700 мг, 3.7 ммоль) и 4-изопропилпиперазин-2-он (1 г, 7.4 ммоль) растворяли в толуоле (20.0 мл). После этого добавляли в полученный раствор карбонат цезия (2.4 г, 7.4 ммоль), N,N-диметилэтан-1,2-диамин (659 мг, 7.4 ммоль) и иодид меди (712 мг, 3.7 ммоль). Атмосферу в системе три раза заменяли на азот, и смесь перемешивали при 100°C в течение 18 часов.
Добавляли воду (100 мл), чтобы разбавить реакционный раствор. Полученный смешанный раствор экстрагировали этилацетатом (20 мл × 3 раза). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (20 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1). Получали 750 мг белого твердого 1-(4-(гидроксиметил)фенил)-4-изопропил-2-она (выход: 80.8%). LCMS: RT = 3.16 мин, [M+H]+ = 249.16.
Стадия D: Синтез 1-(4-(бромметил)фенил)-4-изопропил-2-она
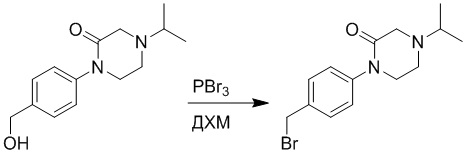
1-(4-(Гидроксиметил)фенил)-4-изопропил-2-он (150 мг, 0.6 ммоль) растворяли в дихлорметане (10.0 мл). После этого добавляли в полученный раствор трибромид фосфора (324 мг, 1.2 ммоль). Реакционную смесь перемешивали при комнатной температуре 1 час.
Добавляли воду (10 мл), чтобы разбавить реакционный раствор. Полученный смешанный раствор экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (20 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Получали 90 мг 1-(4-(бромметил)фенил)-4-изопропил-2-она в виде белого масла (выход: 80.8%). LCMS: RT = 3.46 мин, [M+H]+ = 311.12.
Стадия E: Синтез трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-(4-(4-изопропил-2-оксопиперазин-1-)трет-бутил)фенил) пропаноата

Трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)- ил)ацетат (50 мг, 0.12 ммоль) и 1-(4-(бромметил)фенил)-4-изопропил-2-он (47 мг, 0.15 ммоль) растворяли в тетрагидрофуране (5.0 мл). После этого добавляли в полученный раствор бис(триметилсилил)амид лития (0.19 мл, 0.19 ммоль). Реакционную смесь перемешивали при -50°C в течение 2 часов.
Добавляли воду (10 мл), чтобы разбавить реакционный раствор. Полученный смешанный раствор экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (30 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1). Получали 20 мг трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(4-(4- изопропил-2-оксопиперазин-1-)трет-бутил)фенил)пропаноата в виде желтого масла (выход: 25.0%). LCMS: RT = 3.26 мин, [M+H]+ = 623.10.
Стадия F: Синтез 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-(4-(4-изопропил-2-оксопиперазин-1-)трет-бутил)фенил)пропановой кислоты
Трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(4- (4-изопропил)-2-оксопиперазин-1-)трет-бутил)фенил)пропаноат (20 мг, 0.030 ммоль) растворяли в дихлорметане (4.0 мл). После этого добавляли в полученный раствор трифторуксусную кислоту (1.0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов.
Реакционный раствор упаривали при пониженном давлении, и остаток от упаривания очищали методом препаративной высокоэффективной жидкостной хроматографии. Получали 17 мг белой твердой 2-(4-(2-ацетил-5-хлорфенил)- 3-метокси-6-оксопиридазин-1(6H)-ил)-3-(4-(4-изопропил-2-оксопиперазин-1-)трет-бутил)фенил)пропановой кислоты (выход: 93.0%) . LCMS: RT = 2.56 мин, [M+H]+ = 568.25.
Стадия G: Синтез трет-бутил 4-((2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-(4-(4-изопропил-2-оксопиперазин)трет-бутил-1-ил)фенил) пропанамидо)бензоата
2-(4-(2-Ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(4-(4- изопропил)-2-оксопиперазин-1-)трет-бутил)фенил)пропановую кислоту (17 мг, 0.029 ммоль) и трет-бутил 4-аминобензоат (7 мг, 0.035 ммоль) растворяли в этилацетате (5.0 мл). После этого добавляли в полученный раствор 1-пропилфосфорный ангидрид (45 мг, 0.14 ммоль) и N,N-диизопропилэтиламин (11 мг, 0.087 ммоль). Реакционную смесь перемешивали при 50°C в течение 3 часов.
Добавляли воду (30 мл), чтобы разбавить реакционный раствор. Полученный смешанный раствор экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (30 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток от упаривания очищали методом колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1). Получали 8 мг желтого твердого трет-бутил 4-((2-(4-(2-ацетил-5-хлорфенил)-3-метокси- 6-оксопиридазин-1(6H)-ил)-3-(4-(4-изопропил-2-оксопиперазин)трет-бутил-1-ил)фенил) пропанамидо)бензоата (выход: 87.5%). LCMS: RT = 3.42 мин, [M+H]+ = 742.16.
Стадия H: Синтез 4-((2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-(4-(4-изопропил-2-оксопиперазин)трет-бутил-1-ил)фенил)пропанамидо) бензойной кислоты
Трет-бутил 4-((2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)- 3-(4-(4-изопропил-2-оксопиперазин)трет-бутил-1-ил)фенил)пропанамидо)бензоат (8 мг, 0.01 ммоль) растворяли в дихлорметане (4.0 мл). После этого добавляли в полученный раствор трифторуксусную кислоту (1.0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов.
Реакционный раствор упаривали при пониженном давлении, и остаток от упаривания очищали методом препаративной высокоэффективной жидкостной хроматографии. Получали 1.7 мг белой твердой 4-((2-(4-(2-ацетил-5-хлорфенил)-3- метокси-6-оксопиридазин-1(6H)-ил)-3-(4-(4-изопропил-2-оксопиперазин)трет-бутил-1-ил) фенил)пропанамидо)бензойной кислоты (выход: 23.0%). LCMS: RT = 2.88 мин, [M-H]-= 684.21.
Пример 5
Синтез 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопирролидин-1(6H)-ил)- 3-(4-(4-(этоксиэтил)-2-оксопиридин-1-ил)фенил)пропанамидо)бензойной кислоты
Использовали следующий путь синтеза:
Стадия A: Синтез 4-(гидроксиметил)пиперидин-2-она
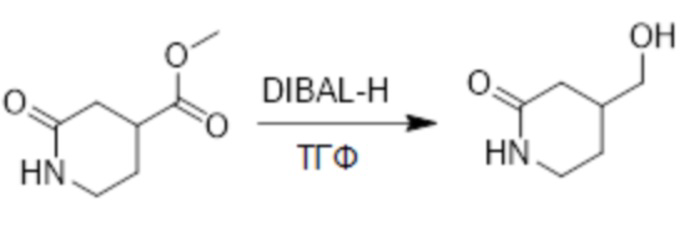
Метил 2-оксопиперидин-4-карбоксилат (1.0 г, 6.3 ммоль) растворяли в смеси тетрагидрофуран/метанол = 1:1 (100.0 мл). После этого добавляли в полученный раствор диизобутилалюминий гидрид (550 мг, 25.0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов.
Добавляли в реакционный раствор метанол (100 мл), чтобы погасить реакцию, и затем смесь упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1), получая 500 мг 4-(гидроксиметил)пиперидин-2-она в виде белого масла (выход: 61.0%). LCMS: RT = 1.53 мин, [M+H]+ = 130.10.
Стадия B: Синтез (2-оксопиридин-4-ил)метил 4-метилбензолсульфоната
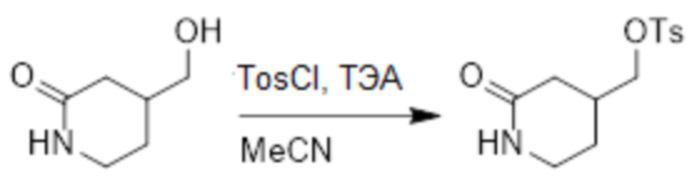
4-(Гидроксиметил)пиперидин-2-он (500 мг, 3.8 ммоль) растворяли в ацетонитриле (20.0 мл). После этого добавляли в полученный раствор 4-метилбензолсульфонил хлорид (1.46 г, 7.7 ммоль) и триэтиламин (959 мг, 9.5 ммоль). Реакционную смесь перемешивали при 50°C в течение 3 часов.
Реакционный раствор упаривали при пониженном давлении, и остаток очищали методом колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1), получая 530 мг белого твердого (2-оксопиридин-4-ил)метил 4-метилбензолсульфоната (выход:48.0%). LCMS: RT = 3.04 мин, [M+H]+ = 284.15.
Стадия C: Синтез 4-(этоксиметил)пиперидин-2-она
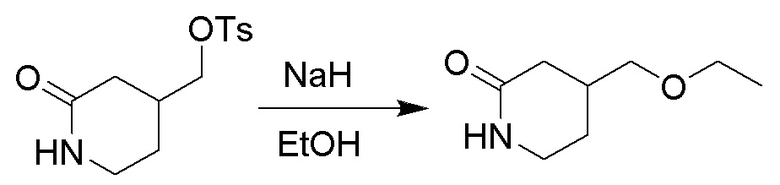
(2-Оксопиридин-4-ил)метил 4-метилбензолсульфонат (380 мг, 1.3 ммоль) растворяли в этаноле (10.0 мл). После этого добавляли в полученный раствор гидрид натрия (107 мг, 2.6 ммоль). Реакционную смесь перемешивали при 50°C в течение 3 часов.
Реакционный раствор упаривали при пониженном давлении, и остаток очищали методом колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1), получая 180 мг белого твердого 4-(этоксиметил)пиперидин-2-она (выход: 85.0%). LCMS: RT = 2.95 мин, [M+H]+ = 158.06.
Стадия D: Синтез 3-(4-бромфенил)-2-((трет-бутоксикарбонил)амино) пропановой кислоты
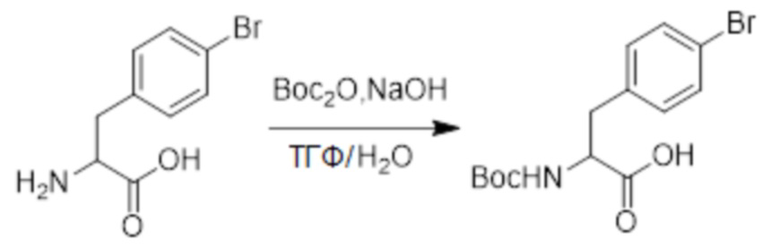
2-Амино-3-(4-бромфенил)пропановую кислоту (4.0 г, 16 ммоль) растворяли в смеси тетрагидрофуран/вода = 2:1 (60.0 мл). После этого добавляли в полученный раствор гидрид натрия (1.3 г, 32 ммоль) и ди-трет-бутил дикарбонат (5.36 г, 24.5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов.
Разбавленный раствор соляной кислоты (1.0 моль/л) медленно добавляли по каплям в реакционный раствор, доводя значение pH до 3-4. Выпадал белый твердый осадок. Полученный смешанный раствор экстрагировали этилацетатом (30 мл x 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (30 мл x 3), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 20/1), получая 6.3 г белого твердого 3-(4-бромфенил)-2-((трет-бутоксикарбонил)амино)пропановой кислоты (выход: 112.0%). LCMS: RT = 3.73 мин, [M+H]+ = 345.04.
Стадия E: Синтез трет-бутил 4-(3-(4-бромфенил)-2-((трет-бутоксикарбонил) амино)пропанамидо)бензоата
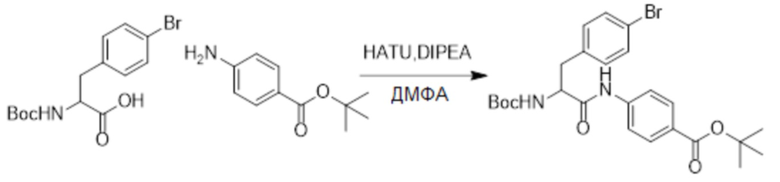
3-(4-Бромфенил)-2-((трет-бутоксикарбонил)амино)пропановую кислоту (6.3 г, 18.3 ммоль) растворяли в N,N-диметилформамиде (60.0 мл). После этого добавляли в полученный раствор трет-бутил 4-аминобензоат (3.9 г, 20.1 ммоль), N,N-диизопропилэтиламин (4.7 г, 36.6 ммоль) и 2-(7-оксибензотриазол)-N,N,N',N'- тетраметилмочевины гексафторфосфат (10.4 г, 27.4 ммоль). Атмосферу в реакционной колбе три раза заменяли на азот, и смесь перемешивали при комнатной температуре в течение 4 часов.
Добавляли насыщенный раствор хлорида аммония, чтобы погасить реакцию. Полученный смешанный раствор экстрагировали этилацетатом (100 мл x 3). Органические фазы объединяли. Органическую фазу промывали насыщенным водным раствором хлорида натрия (100 мл), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/10). Получали 7.8 г белого твердого трет-бутил 4-(3-(4-бромфенил)-2-((трет-бутоксикарбонил)амино) пропанамидо)бензоата (выход: 82.0%). LCMS: RT = 4.26 мин, [M+H]+ = 520.23.
Стадия F: Синтез трет-бутил 4-(2-((трет-бутоксикарбонил)амино)-3-(4-(4- (этоксиметил)-2-оксопиридин-1-ил)фенил)пропанамидо)бензоата
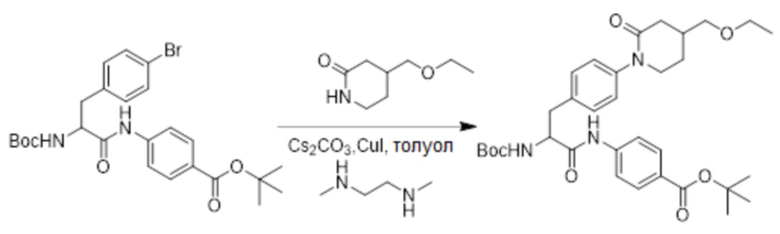
Трет-бутил 4-(3-(4-бромфенил)-2-((трет-бутоксикарбонил)амино)пропанамидо) бензоат (2 г, 3.8 ммоль) растворяли в толуоле (40.0 мл). После этого добавляли в полученный раствор 4-(этоксиметил)пиперидин-2-он (302 мг, 1.9 ммоль), карбонат цезия (1.25 г, 3.8 ммоль), N,N-диметилэтил-1,2-диамин (339 мг, 3.8 ммоль), иодид меди (366 мг, 1.9 ммоль), и атмосферу в реакционной колбе три раза заменяли на азот. Смесь перемешивали при 100°C в течение 18 часов.
Реакционный раствор разбавляли, добавляя воду (100 мл). Полученный смешанный раствор экстрагировали этилацетатом (20 мл x 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (20 мл x 3), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1), получая 330 мг белого твердого трет-бутил 4-(2-((трет-бутоксикарбонил)амино)-3-(4-(4-(этоксиметил)-2-оксопиридин-1- ил)фенил)пропанамидо)бензоата (выход: 15.0%). LCMS: RT = 3.66 мин, [M+H]+ = 596.11.
Стадия G: Синтез 4-(2-амино-3-(4-(4-(этоксиметил)-2-оксопиридин-1-ил)фенил) пропанамидо)бензойной кислоты
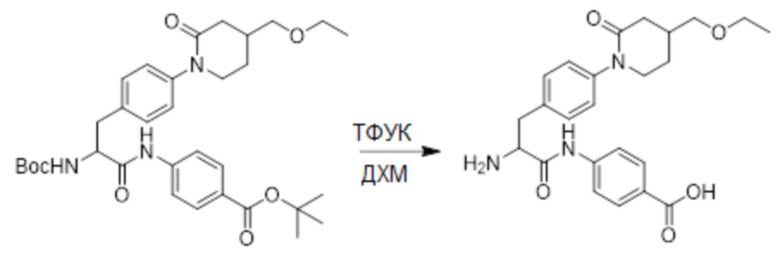
Трет-бутил 4-(2-((трет-бутоксикарбонил)амино)-3-(4-(4-(этоксиметил)-2- оксопиридин-1-ил)фенил)пропанамидо)бензоат (330 мг, 0.55 ммоль) растворяли в дихлорметане (4.0 мл). После этого добавляли в полученный раствор трифторуксусную кислоту (1.0 мл) и перемешивали при комнатной температуре в течение 2 часов.
Реакционный раствор упаривали при пониженном давлении, получая 230 мг белой твердой 4-(2-амино-3-(4-(4-(этоксиметил)-2-оксопиридин-1-ил)фенил)пропанамидо) бензойной кислоты (выход: 96.0%). LCMS: RT = 2.79 мин, [M+H]+ = 440.26.
Стадия H: Синтез 4-(2-хлор-3-(4-(4-(этоксиметил)-2-оксопиридин-1-ил)фенил) пропанамидо)бензойной кислоты
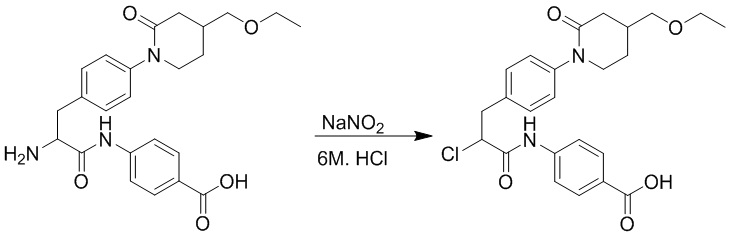
2-Амино-3-(4-(4-(этоксиметил)-2-оксопиридин-1-ил)фенил)пропанамидо)бензойную кислоту (230 мг, 0.52 ммоль) растворяли в соляной кислоте (6 моль/л, 10 мл). После этого добавляли в полученный раствор нитрит натрия (80 мг, 1.10 ммоль) при 0°C и перемешивали при 0°C в течение 4 часов.
Реакционный раствор экстрагировали дихлорметаном (20 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (20 мл), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: дихлорметан/метанол = 10/1), получая 150 мг белой твердой 4-(2-хлор-3-(4-(4-(этоксиметил)-2-оксопиридин-1-ил)фенил) пропанамидо) бензойной кислоты (выход: 62.0%). LCMS: RT = 3.66 мин, [M+H]+ = 459.15.
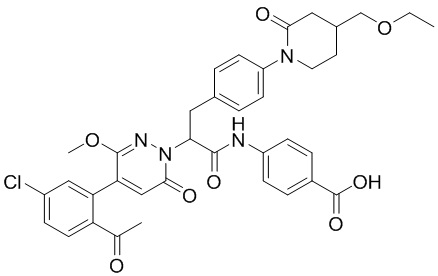
Стадия I: Синтез 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопирролидин- 1(6H)-ил)-3-(4-(4-(этоксиэтил)-2-оксопиридин-1-ил)фенил)пропанамидо)бензойной кислоты

4-(2-Хлор-3-(4-(4-(этоксиметил)-2-оксопиридин-1-ил)фенил)пропанамидо)бензойную кислоту (150 мг, 0.33 ммоль) растворяли в N,N-диметилформамиде (10 мл). После этого добавляли в полученный раствор 5-(2-ацетил-5-хлорфенил)-6-метилпиридин-3(2H)-он (139 мг, 0.50 ммоль), карбонат калия (92 мг, 0.66 ммоль) и иодид калия (5 мг, 0.030 ммоль) и перемешивали при 70°C в течение 2 часов.
Реакционный раствор разбавляли добавлением насыщенного водного раствора хлорида аммония (20 мл). Полученный смешанный раствор экстрагировали этилацетатом (20 мл x 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (20 мл), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток от упаривания очищали методом ВЭЖХ, получая 5 мг 4-(2-(4-(2-ацетил-5-хлорфенил)-3- метокси-6-оксопирролидин-1(6H)-ил)-3-(4-(4-(этоксиэтил)-2-оксопиридин-1-ил)фенил) пропанамидо)бензойной кислоты (выход: 2.0%). LCMS: RT = 3.78 мин, [M+H]+ = 701.37.
Пример 6
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(2-гидроксиэтокси)-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
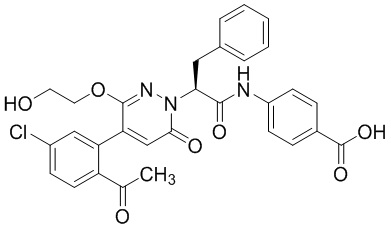
Использовали следующий путь синтеза:
Стадия A: Синтез 5-бром-6-(2-((трет-бутилдиметилсилил)окси)этокси)-2- (4-метоксибензил)пиридазин-3(2H)-она
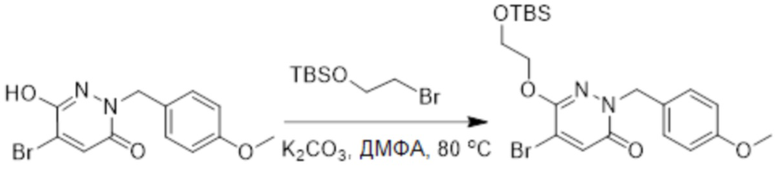
5-Бром-6-гидрокси-2-(4-метоксибензил)пиридазин-3(2H)-он (402 мг, 1.29 ммоль), (2-бромэтокси)(трет-бутил)диметилсилан (1.24 г, 5.17 ммоль) и карбонат калия (714 мг, 5.17 ммоль) растворяли в N,N-диметилформамиде (5 мл) в инертной атмосфере азота и перемешивали при 80°C в течение 3 часов.
Реакционный раствор охлаждали, разбавляли этилацетатом (100 мл) и промывали водой (20 мл × 2) и насыщенным водным раствором хлорида натрия (20 мл), соответственно. Затем органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали, и сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир: этилацетат = 5/1). Получали 200 мг белого твердого 5-бром-6-(2-((трет-бутилдиметилсилил)окси)этокси)-2-(4-метоксибензил)пиридазин- 3(2H)-она (выход: 33.0%). LC-MS: RT = 5.13 мин, [M+H]+ = 469.10.
Стадия B: Синтез 5-(2-ацетил-5-хлорфенил)-6-(2-((трет-бутилдиметилсилил) окси)этокси)-2-(4-метоксибензил)пиридазин-3(2H)-она
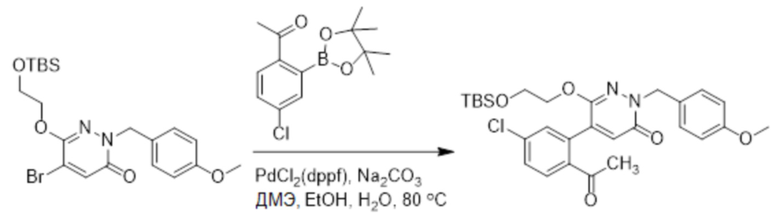
1-(4-Хлор-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этан-1-он (156 мг, 0.55 ммоль), 5-бром-6-(2-((трет-бутилдиметилсилил)окси)этокси)-2-(4-метоксибензил) пиридазин-3(2H)-он (200 мг, 0.43 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (35 мг, 0.043 ммоль) и карбонат натрия (92 мг, 0.86 ммоль) растворяли в смеси диметиловый эфир этиленгликоля (3 мл)/этанол (0.4 мл)/вода (0.4 мл) в инертной атмосфере азота, и проводили реакцию при 90°C в течение 2.5 часов.
Реакционный раствор охлаждали, разбавляли этилацетатом (100 мл) и промывали водой (20 мл × 2) и насыщенным водным раствором хлорида натрия (20 мл), соответственно. Затем органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали, и сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир: этилацетат = 5/2). Получали 85 мг белого твердого 5-(2-ацетил-5-хлорфенил)-6-(2-((трет-бутилдиметилсилил)окси)этокси)-2-(4- метоксибензил)пиридазин-3(2H)-она (выход: 36.0%). LC-MS: RT = 4.93 мин, [M+H]+ = 543.20.
Стадия C: Синтез 5-(2-ацетил-5-хлорфенил)-6-(2-гидроксиэтокси)пиридазин-3 (2H)-она
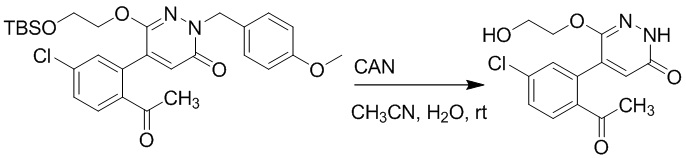
5-(2-Ацетил-5-хлорфенил)-6-(2-((трет-бутилдиметилсилил)окси)этокси)-2-(4- метоксибензил)пиридазин-3(2H)-он (85 мг, 0.156 ммоль), церийаммоний нитрат (258 мг, 0.470 ммоль) растворяли в смеси ацетонитрил (3.0 мл)/вода (1.0 мл), и через 3 часа добавляли дополнительное количество церийаммоний нитрата (602 мг, 1.10 ммоль), реакцию продолжали при перемешивании при комнатной температуре в течение 2.5 часов.
Реакцию гасили водой, разбавляли этилацетатом (100 мл) и промывали водой (10 мл × 2) и насыщенным водным раствором хлорида натрия (20 мл), соответственно. Затем органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали, и сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир: этилацетат = 2/1). Получали 30 мг белого твердого 5-(2-ацетил-5-хлорфенил)-6-(2-гидроксиэтокси)пиридазин-3(2H)-она (выход: 63.0%). LC-MS: RT = 2.80 мин, [M+H]+ = 309.10.
Стадия D: Синтез трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(2- гидроксиэтокси)-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата
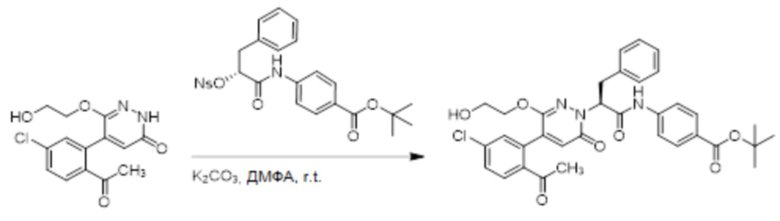
5-(2-Ацетил-5-хлорфенил)-6-(2-гидроксиэтокси)пиридазин-3(2H)-он (44 мг, 0.143 ммоль), трет-бутил (R)-4-(2-((4-нитрофенил)сульфонил)окси)-3-фенилпропанамидо) бензоат (83 мг, 0.157 ммоль) и карбонат калия (40 мг, 0.286 ммоль) растворяли в N,N-диметилформамиде (3 мл) и перемешивали при 45°C в течение 6.5 часов.
Реакционный раствор охлаждали, разбавляли этилацетатом (50 мл) и промывали водой (10 мл × 2) и насыщенным водным раствором хлорида натрия (10 мл), соответственно. Затем органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали, и сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир: этилацетат = 1/1). Получали 31 мг белого твердого трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(2-гидроксиэтокси)-6-оксопиридазин-1 (6H)-ил)-3-фенилпропанамидо)бензоата (выход: 34.0%). LC-MS: RT = 4.19 мин, [M+H]+ = 632.16.
Стадия E: Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(2-гидроксиэтокси)-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
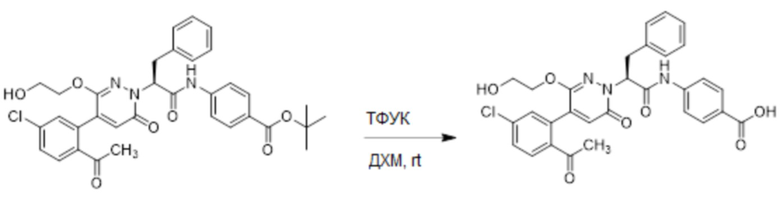
Трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(2-гидроксиэтокси)-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоат (31 мг, 0.049 ммоль) растворяли в дихлорметане (5.0 мл), добавляли по каплям трифторуксусную кислоту (1.0 мл) при комнатной температуре, и реакцию продолжали при комнатной температуре 2.5 часа.
Реакционный раствор упаривали на роторном испарителе, вакуумировали на масляном насосе, и растворяли в метаноле. Затем в раствор по каплям добавляли н-гексан, выпадало большое количество твердого осадка, перемешивали при комнатной температуре в течение 1 часа и фильтровали, получая 10 мг белой твердой (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(2-гидроксиэтокси)-6-оксипиридазин-1(6H)-ил)-3- фенилпропанамидо)бензойной кислоты (выход: 35.0%). LC-MS: RT = 3.64 мин, [M-H]- = 574.10. 1H ЯМР (500 МГц, ДМСО) δ 12.73 (с, 1H), 10.51 (с, 1H), 8.01 (д, J = 8.4 Гц, 1H), 7.91 (д, J = 8.6 Гц, 2H), 7.72 (д, J = 8.6 Гц, 2H), 7.69 (дд, J = 8.4, 1.9 Гц, 1H), 7.50 (д, J = 1.8 Гц, 1H), 7.34 – 7.25 (м, 5H), 7.19 (т, J = 6.8 Гц, 1H), 6.89 (с, 1H), 5.75 (дд, J = 10.0, 5.0 Гц, 1H), 4.69 (т, J = 5.3 Гц, 1H), 4.08 (с, 1H), 4.06 – 3.94 (м, 2H), 3.52 (ддд, J = 24.4, 12.4, 7.8 Гц, 3H), 3.40 (дд, J = 14.1, 4.8 Гц, 1H), 2.55 (с, 3H).
Пример 7
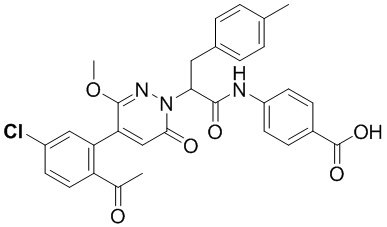
Синтез 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- (п-толил)пропанамидо)бензойной кислоты
Использовали следующий путь синтеза:
Стадия A: Синтез трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)ацетата
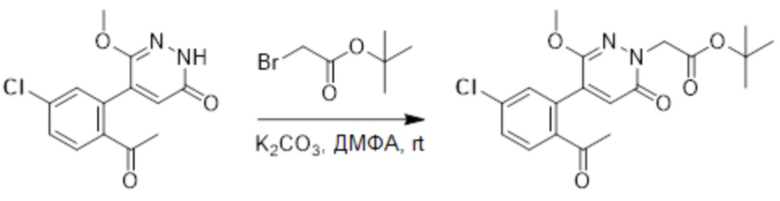
5-(2-Ацетил-5-хлорфенил)-6-метоксипиридазин-3(2H)-он (1.1 г, 3.9 ммоль) и карбонат калия (1.1 г, 7.9 ммоль) растворяли в N,N-диметилформамиде (10 мл) в инертной атмосфере азота, добавляли трет-бутил 2-бромацетат (922 мг, 4.7 ммоль) при комнатной температуре и перемешивали 4 часа.
Реакцию гасили водой и экстрагировали этилацетатом (50 мл × 3), органическую фазу промывали насыщенным водным раствором хлорида натрия (20 мл × 2), сушили над безводным сульфатом натрия, фильтровали и упаривали. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир: этилацетат = 2/1). Получали 1.3 г трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)ацетата (выход: 85.0%). MS (ESI) M/Z: 393.3 [M+H]+
Стадия B: Синтез трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-(п-толил)пропаноата
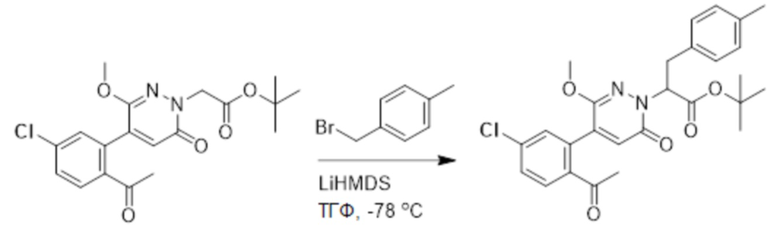
Трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)- ил)ацетат (70 мг, 0.18 ммоль) и 1-(бромметил)-4-метилбензол (66 мг, 0.35 ммоль) растворяли в безводном тетрагидрофуране (4.0 мл), охлаждали до -78°C, перемешивали 10 минут, добавляли по каплям бис(триметилсилил)аминолитий (700 мкл, 1.0 моль/л, раствор в тетрагидрофуране) и перемешивали при -78°C в течение 2 часов.
Реакцию гасили водой и экстрагировали этилацетатом (20 мл × 3), органическую фазу промывали насыщенным водным раствором хлорида натрия (10 мл × 2), сушили над безводным сульфатом натрия, фильтровали и упаривали. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир: этилацетат = 2/1). Получали 80 г трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-(п-толил)пропаноата (выход: 89.0%).
Стадия C: Синтез 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-(п-толил)пропановой кислоты

Трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- (п-толил)пропаноат (80 мг, 0.16 ммоль) растворяли в дихлорметане (1.5 мл), добавляли по каплям трифторуксусную кислоту (0.5 мл) при комнатной температуре, и реакцию продолжали при комнатной температуре 4 часа.
Реакционный раствор упаривали на роторном испарителе и сушили на масляном насосе, получая 70 мг 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1 (6H)-ил)-3-(п-толил)пропановой кислоты (выход: 99.0%).
Стадия D: Синтез трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)- 3-метокси-6- оксопиридазин-1(6H)-ил)-3-(п-толил)пропанамидо)бензоата
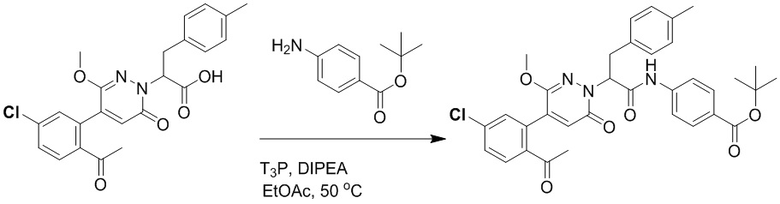
2-(4-(2-Ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(п-толил) пропановую кислоту (25 мг, 0.057 ммоль), трет-бутил 4-аминобензоат (11 мг, 0.057 ммоль), 1-пропилфосфорный ангидрид (91 мг, 0.285 ммоль) и N,N-диизопропилэтиламин (29 мкл, 0.171 ммоль) растворяли в этилацетате (2.0 мл), перемешивали при 50°C в течение 3 часов.
Реакционный раствор охлаждали, разбавляли этилацетатом (50 мл) и промывали водой (10 мл × 2) и насыщенным водным раствором хлорида натрия (10 мл), соответственно. Затем органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали, и сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир: этилацетат = 3/1). Получали 11 мг трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксипиридазин-1(6H)-ил)-3-(п-толил) пропанамидо)бензоата (выход: 32.0%). LC-MS: RT = 4.67 мин, [M+H]+ = 616.19.
Стадия E: Синтез 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-(п-толил)пропанамидо)бензойной кислоты
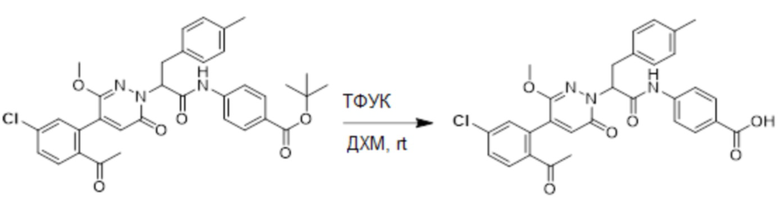
Трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- (п-толил)пропанамидо)бензоат (11 мг) растворяли в дихлорметане (2.5 мл), добавляли по каплям трифторуксусную кислоту (0.5 мл) при комнатной температуре, и реакционную смесь перемешивали при комнатной температуре в течение 50 минут.
Реакционный раствор упаривали на роторном испарителе, вакуумировали на масляном насосе, и растворяли остаток в метаноле. Затем в раствор добавляли по каплям н-гексан, выпадало большое количество твердого осадка, смесь перемешивали при комнатной температуре в течение 1 часа и фильтровали, получая 7.6 мг белой твердой 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(п-толил) пропанамидо)бензойной кислоты (выход: 76.0%). LC-MS: RT = 4.08 мин, [M+H]+ = 560.10. 1H ЯМР (500 МГц, ДМСО-d6, м.д.) δ 12.72 (с, 1H), 10.52 (с, 1H), 7.99 (д, J = 8.4 Гц, 1H), 7.91 (д, J = 8.7 Гц, 2H), 7.73 (д, J = 8.8 Гц, 2H), 7.69 (дд, J = 8.3, 2.1 Гц, 1H), 7.51 (д, J = 2.1 Гц, 1H), 7.20 (д, J = 8.0 Гц, 2H), 7.09 (д, J = 7.9 Гц, 2H), 6.91 (с, 1H), 5.71 (дд, J = 10.3, 4.7 Гц, 1H), 3.67 (с, 3H), 3.48 (дд, J = 14.1, 10.4 Гц, 1H), 3.37-3.32 (м, 1H), 2.53 (с, 3H), 2.24 (с, 3H).
Пример 8
Синтез 4 (2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H) -ил)-3- (м-толил)пропанамидо)бензойной кислоты
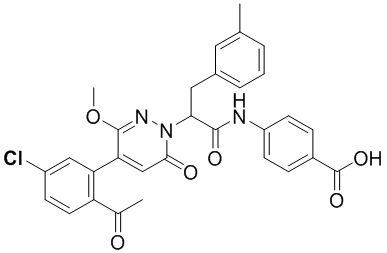
Использовали следующий путь синтеза:
Стадия A: Синтез трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-(м-толил)пропаноата

Трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)- ил)ацетат (70 мг, 0.18 ммоль) и 1-(бромметил)-3-метилбензол (66 мг, 0.35 ммоль) растворяли в безводном тетрагидрофуран (4 мл), охлаждали до -78°C, перемешивали 10 минут, добавляли по каплям бис(триметилсилил)аминолитий (700 мкл, 1.0 моль/л, раствор в тетрагидрофуране) и перемешивали при -78°C в течение 2 часов.
Реакцию гасили водой и экстрагировали этилацетатом (20 мл × 3), органическую фазу промывали насыщенным водным раствором хлорида натрия (10 мл × 2), сушили над безводным сульфатом натрия, фильтровали и упаривали. Сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир: этилацетат = 2/1). Получали 80 г трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-(м-толил)пропаноата (выход: 89.0%).
Стадия B: Синтез 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-(м-толил)пропановой кислоты
Трет-бутил 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- (м-толил)пропаноат (80 мг, 0.16 ммоль) растворяли в дихлорметане (1.5 мл), добавляли по каплям трифторуксусную кислоту (0.5 мл) при комнатной температуре, и реакцию продолжали при комнатной температуре 4 часа.
Реакционный раствор упаривали на роторном испарителе и сушили на масляном насосе, получая 70 мг 2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-(м-толил)пропановой кислоты (выход: 99.0%).
Стадия C: Синтез трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-(м-толил)пропанамидо)бензоата
2-(4-(2-Ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(м-толил) пропановую кислоту (38 мг, 0.086 ммоль), трет-бутил 4-аминобензоат (17 мг, 0.086 ммоль), 1-пропилфосфорный ангидрид (137 мг, 0.430 ммоль) и N,N-диизопропилэтиламин (43 мкл, 0.258 ммоль) растворяли в этилацетате (2.0 мл), перемешивали при 50°C в течение 3 часов.
Реакционный раствор охлаждали, разбавляли этилацетатом (50 мл) и промывали водой (10 мл × 2) и насыщенным водным раствором хлорида натрия (10 мл), соответственно. Затем органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали, и сырой продукт очищали методом колоночной хроматографии на силикагеле (петролейный эфир: этилацетат = 3/1). Получали 19 мг трет-бутил 4-(2-(4- (2-ацетил-5-хлорфенил)-3-метокси-6-оксипиридазин-1(6H)-ил)-3-(м-толил)пропанамидо) бензоата (выход: 36.0%). LC-MS: RT = 4.67 мин, [M+H]+ = 616.19.
Стадия D: Синтез 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-(м-толил)пропанамидо)бензойной кислоты
Трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- (м-толил)пропанамидо)бензоат (19 мг) растворяли в дихлорметане (2.5 мл), добавляли по каплям трифторуксусную кислоту (0.5 мл) при комнатной температуре, и реакционную смесь перемешивали при комнатной температуре в течение 2 часов.
Реакционный раствор упаривали на роторном испарителе, вакуумировали на масляном насосе, и остаток растворяли в метаноле. Затем н-гексан добавляли по каплям в раствор, выпадало большое количество твердого осадка, смесь перемешивали при комнатной температуре в течение 1 часа и фильтровали, получая 9.5 мг белой твердой 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(м-толил) пропанамидо)бензойной кислоты (выход: 55.0%). LC-MS: RT = 4.08 мин, [M+H]+ = 560.12. 1H ЯМР (400 МГц, ДМСО-d6, м.д.) δ 12.73 (с, 1H), 10.53 (с, 1H), 7.99 (д, J = 8.3 Гц, 1H), 7.91 (д, J = 8.6 Гц, 2H), 7.71 (дд, J = 12.2, 8.7 Гц, 3H), 7.51 (с, 1H), 7.21 – 7.09 (м, 3H), 7.00 (д, J = 7.2 Гц, 1H), 6.91 (с, 1H), 5.71 (дд, J = 10.3, 4.6 Гц, 1H), 3.69 (с, 3H), 3.49 (дд, J = 13.9, 10.4, 1H), 3.38-3.32 (м, 1H), 2.53 (с, 3H), 2.26 (с, 3H).
Пример 9
Синтез (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1 (6H)-ил)- 3-фенил-N-(хиноксалин-6-ил)пропанамида
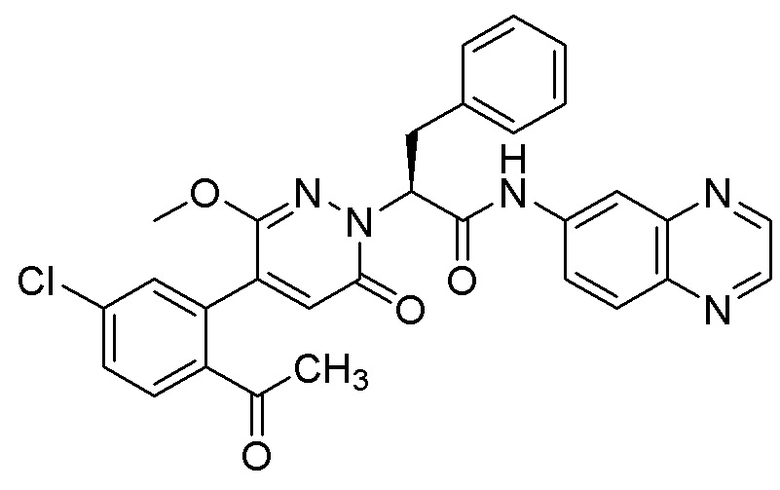
Использовали следующий путь синтеза:
Стадия A: Синтез метил (R)-3-фенил-2-((трифторметил)сульфонил)окси) пропаноата
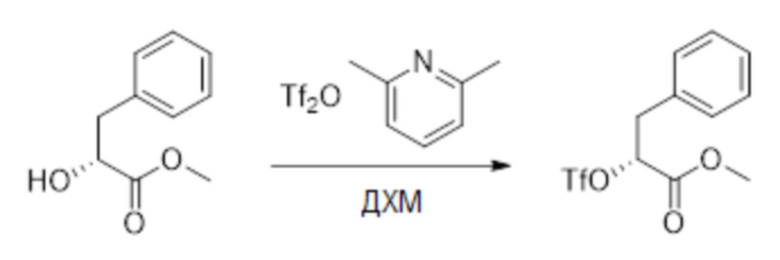
Метил (R)-2-гидрокси-3-фенилпропаноат (5.00 г, 27.8 ммоль) растворяли в дихлорметане (30.0 мл), добавляли 2,6-диметилпиридин (3.47 г, 33.3 ммоль), затем медленно добавляли ангидрид трифторметилсульфокислоты (5.4 мл, 33.3 ммоль) при -10°C и перемешивали 30 минут.
Добавляли воду (20 мл) в реакционный раствор, чтобы погасить реакцию, добавляли в реакционный раствор этилацетат (100 мл), и полученную смесь промывали насыщенным водным раствором хлорида натрия (20 мл × 3), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 100/5). Получали 5.1 г метил (R)-3-фенил-2-((трифторметил)сульфонил)окси) пропаноата в виде бесцветного масла (выход: 59.0%). LCMS: RT = 3.24 мин, [M+H]+ = 313.28.
Стадия B: Синтез метил (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропаноата
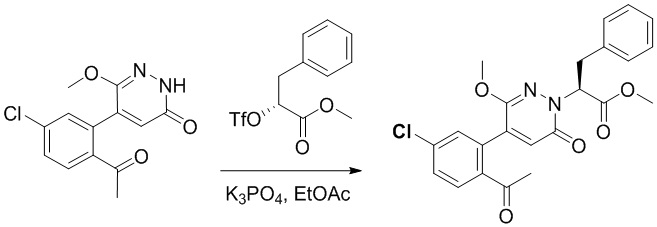
5-(2-Ацетил-5-хлорфенил)-6-метоксипиридин-3(2H)-он (1.2 г, 4.3 ммоль) и фосфат калия (5.4 г, 25.8 ммоль) растворяли в этилацетате (20.0 мл). После этого добавляли в полученный раствор метил (R)-3-фенил-2-((трифторметил)сульфонил)окси)пропаноат (6.7 г, 21.6 ммоль) и перемешивали при комнатной температуре в течение 3 часов.
Добавляли в реакционный раствор воду (5 мл), чтобы погасить реакцию. Добавляли в раствор этилацетат (100 мл), и смесь промывали насыщенным водным раствором хлорида натрия (20 мл × 3), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/1). Получали 1.2 г желтого твердого метил (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропаноата (выход: 63.0%). LCMS: RT = 4.08 мин, [M+H]+ = 441.11.
Стадия C: Синтез метил (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропановой кислоты

Метил (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропаноат (1.20 г, 2.72 ммоль) растворяли в метаноле (15.0 мл) и воде (6.0 мл). После этого добавляли в полученный раствор моногидрат гидроксида лития (217 мг, 5.44 ммоль), перемешивали при комнатной температуре в течение 2 часов.
Разбавленный раствор соляной кислоты (1.0 моль/л) медленно добавляли по каплям в реакционный раствор, доводя значение pH до 4-5. Добавляли в раствор этилацетат (100 мл), и смесь промывали насыщенным водным раствором хлорида натрия (20 мл × 3), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/10). Получали 460 г желтой твердой (S)-2-(4-(2- ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-фенилпропановой кислоты (выход: 39.6%). LCMS: RT = 3.88 мин, [M+H]+ = 427.09.
Стадия D: Синтез (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-фенил-N-(хиноксалин-6-ил)пропанамида
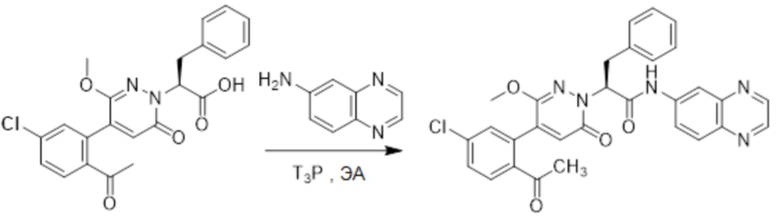
(S)-2-(4-(2-Ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропановую кислоту (59 мг, 0.138 ммоль) и хиноксалин-6-амин (24 мг, 0.166 ммоль) растворяли в этилацетате (5.0 мл), добавляли N,N-диизопропилэтиламин (89 мг, 0.690 ммоль). После этого добавляли в полученный раствор 1-пропилфосфорный ангидрид (175 мг, 0.552 ммоль). Реакционный раствор нагревали до 60°C и перемешивали 8 часов.
Реакционный раствор охлаждали до комнатной температуры и упаривали при пониженном давлении. Остаток очищали на пластинке ТСХ (метанол / дихлорметан = 1:16), получая 80 мг сырого продукта, который очищали методом препаративной высокоэффективной жидкостной хроматографии. Использовали следующие условия разделения: хроматографическая колонка: X select C18 19 мм * 150 м; Подвижная фаза: вода (содержит 0.05% трифторуксусной кислоты) и ацетонитрил; Скорость потока: 25 мл/мин; Градиент: содержание ацетонитрила повышали с 5% до 100% за 7 минут; Длина волны детектора: 254 нм. После очистки получали 9 мг желтого твердого (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксипиридазин-1(6H)-ил)-3-фенил-N- (хиноксалин-6-ил)пропанамида (выход: 11.8%). LCMS: RT = 3.96 мин, [M+H]+ = 554.15. 1H ЯМР (500 МГц, ДМСО) δ 10.71 (с, 1H), 8.88 (д, J = 1.8 Гц, 1H), 8.82 (д, J = 1.8 Гц, 1H), 8.49 (д, J = 2.2 Гц, 1H), 7.98 (ддд, J = 18.2, 11.4, 5.7 Гц, 3H), 7.68 (дд, J = 8.3, 2.1 Гц, 1H), 7.50 (д, J = 2.1 Гц, 1H), 7.31 (т, J = 6.9 Гц, 2H), 7.23 (дт, J = 36.4, 7.1 Гц, 3H), 6.93 (д, J = 7.2 Гц, 1H), 5.77 (дд, J = 9.8, 5.1 Гц, 1H), 3.66 (с, 3H), 3.51 (дд, J = 14.0, 10.2 Гц, 1H), 3.41 (дд, J = 14.1, 4.8 Гц, 1H) 2.52 (с, 3H).
Пример 10
Синтез (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)- N-(2-метил-2H-индазол-5-ил)-3-фенилпропанамида
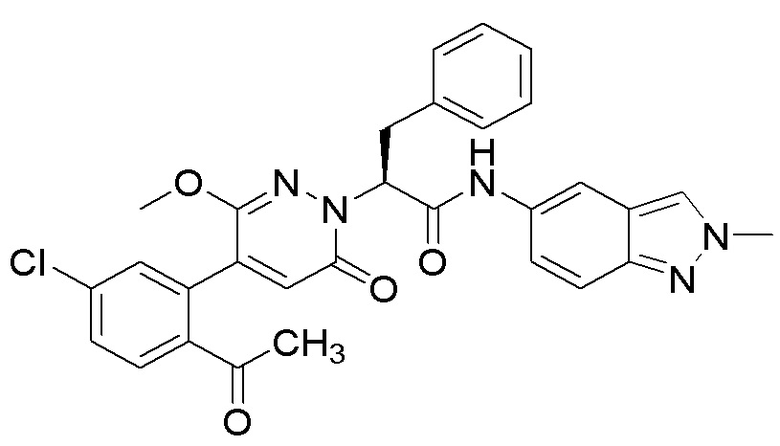
Стадия A: Синтез (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-N-(2-метил-2H-индазол-5-ил)-3-фенилпропанамида
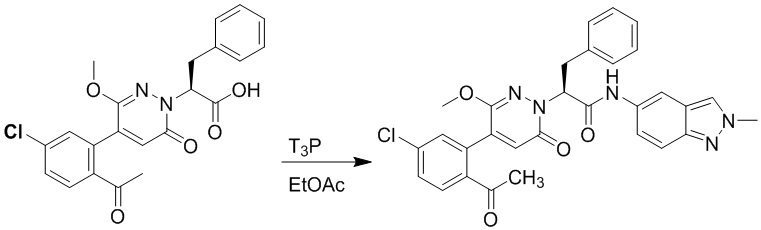
(S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропановую кислоту (83 мг, 0.195 ммоль) и хиноксалин-6-амин (28.6 мг, 0.195 ммоль) растворяли в этилацетате (3.0 мл), добавляли N,N-диизопропилэтиламин (8251 мг, 1.95 ммоль). После этого добавляли в полученный раствор 1-пропилфосфорный ангидрид (248 мг, 0.78 ммоль). Реакционный раствор нагревали до 60°C и перемешивали 18 часов.
Реакционный раствор охлаждали до комнатной температуры и упаривали при пониженном давлении. Остаток очищали на пластинке ТСХ (метанол / дихлорметан = 1:15), получая 80 мг сырого продукта, который очищали методом препаративной высокоэффективной жидкостной хроматографии. Использовали следующие условия разделения: хроматографическая колонка: X select C18 19 мм * 150 мм; Подвижная фаза: вода (содержит 0.05% трифторуксусной кислоты) и ацетонитрил; Скорость потока: 25 мл/мин; Градиент: содержание ацетонитрила повышали с 5% до 100% за 7 минут; Длина волны детектора: 254 нм. После очистки получали 50 мг желтого твердого (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-N-(2-метил-2h- индазол-5-ил)-3-фенилпропанамида (выход: 46.3%). LCMS: RT = 3.87 мин, [M+H]+ = 556.16 . 1H ЯМР (400 МГц, ДМСО) δ 10.08 (с, 1H), 8.24 (с, 1H), 8.07 (с, 1H), 7.98 (д, J = 8.4 Гц, 1H), 7.67 (дд, J = 8.3, 2.2 Гц, 1H), 7.51 (дд, J = 18.5, 5.6 Гц, 2H), 7.24 (ддт, J = 32.0, 30.0, 6.9 Гц, 6H), 6.89 (с, 1H), 5.73 (дд, J = 10.2, 4.8 Гц, 1H), 4.11 (с, 3H), 3.67 (с, 3H), 3.51 (дд, J = 14.0, 10.2 Гц, 1H), 3.41 (дд, J = 14.1, 4.8 Гц, 1H), 2.53 – 2.50 (м, 3H).
Пример 11
Синтез (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)- N-(1H-бензо[d]имидазол-5-ил)-3-фенилпропанамида
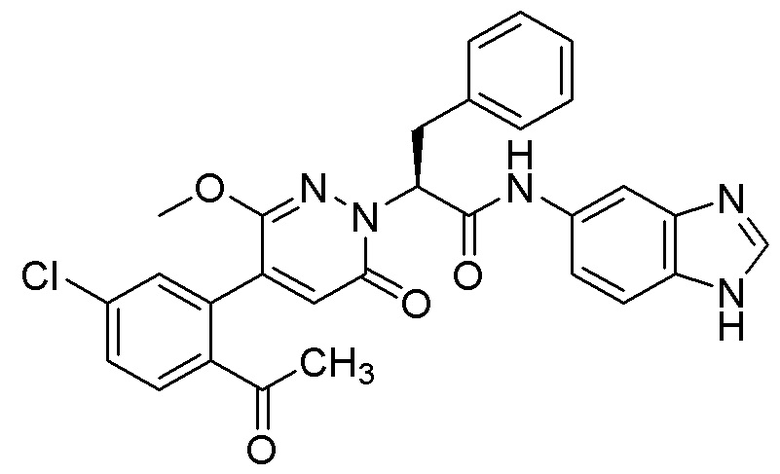
Использовался следующий путь синтеза.
Стадия A: Синтез 1H-бензо[d]имидазол-5-амина
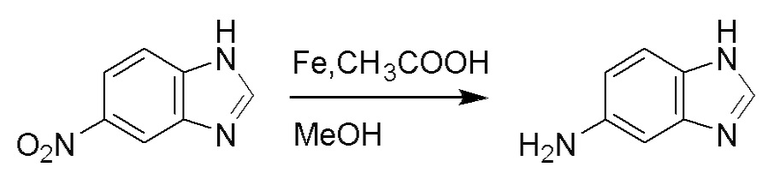
5-Нитро-1H-бензо[d]имидазол (200 мг, 1.23 ммоль) растворяли в метаноле (10 мл), добавляли уксусную кислоту (1 мл) и порошок железа (687 мг, 12.3 ммоль), нагревали до 80°C и вели реакцию в течение 4 часов.
Насыщенный раствор бикарбоната натрия добавляли в реакционный раствор до pH = 7-8. Полученный смешанный раствор экстрагировали этилацетатом (100 мл × 3) и упаривали. Получали 150 мг желтого твердого 1H-бензо[d]имидазол-5-амина (выход: 92.0%). LCMS: RT = 0.60 мин, [M+H]+ = 134.06.
Стадия B: Синтез (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-N-(1H-бензо[d]имидазол-5-ил)-3-фенилпропанамида
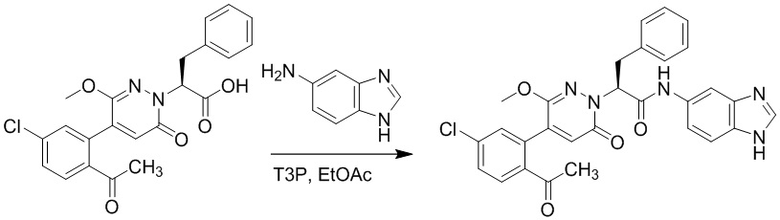
(S)-2-(4-(2-Ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропановую кислоту (65 мг, 0.150 ммоль) и 1H-бензо[d]имидазол-5-амин (22 мг, 0.167ммоль) растворяли в этилацетате (3.0 мл) и добавляли N,N-диизопропилэтиламин (193 мг, 1.5 ммоль). После этого добавляли в полученный раствор 1-пропилфосфоновый ангидрид (190 мг, 0.6 ммоль). Реакционный раствор нагревали до 60°C и перемешивали 4 часа.
Реакционную смесь охлаждали до комнатной температуры и упаривали при пониженном давлении. Сырой продукт очищали методом препаративной высокоэффективной жидкостной хроматографии. Использовали следующие условия разделения: хроматографическая колонка: X select C18 19 мм * 150 мм; подвижная фаза: вода (содержит 0.05% трифторуксусной кислоты) и ацетонитрил; скорость потока: 25 мл/мин; градиент: повышение содержания ацетонитрила с 5% до 100% за 7 минут; длина волны детектора: 254 нм. После очистки получали 6.05 мг желтого твердого (S)-2-(4- (2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-N-(1H-бензо[d]имидазол- 5-ил)-3-фенилпропанамида (выход: 7.0%). LCMS: RT = 3.07 мин, [M+H]+ = 542.15.
Пример 12
Синтез (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)- N-(2-оксодигидроиндол-5-ил)-3-фенилпропанамида

Использовался следующий путь синтеза.
Стадия A: Синтез 5-амино-2,3-дигидро-1H-индол-2-она
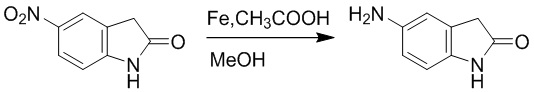
5-Нитроиндол-2-он (200 мг, 1.12 ммоль) растворяли в метаноле (10 мл), добавляли уксусную кислоту (1 мл) и порошок железа (629 мг, 11.2 ммоль), нагревали до 80°C и вели реакцию в течение 4 часов.
Насыщенный раствор бикарбоната натрия добавляли в реакционный раствор до pH = 7-8. Полученный смешанный раствор экстрагировали этилацетатом (100 мл × 3) и упаривали. Получали 150 мг желтого твердого 5-амино-2,3-дигидро-1H-индол-2-она (выход: 88.0%). LCMS: RT = 0.61 мин, [M+H]+ = 165.09.
Стадия B: (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)- ил)-N-(2-оксодигидроиндол-5-ил)-3-фенилпропанамид
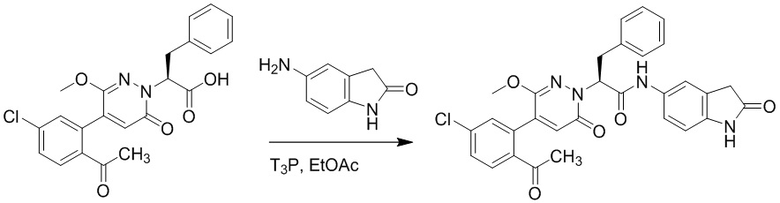
(S)-2-(4-(2-Ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропановую кислоту (65 мг, 0.150 ммоль) и 5-амино-2,3-дигидро-1H-индол-2-он (25 мг, 0.167 ммоль) растворяли в этилацетате (3.0 мл) и добавляли N,N-диизопропилэтиламин (193 мг, 1.5 ммоль). После этого добавляли в полученный раствор 1-пропилфосфоновый ангидрид (190 мг, 0.6 ммоль). Реакционный раствор нагревали до 60°C и перемешивали 3 часа.
Реакционную смесь охлаждали до комнатной температуры и упаривали при пониженном давлении. Сырой продукт очищали методом препаративной высокоэффективной жидкостной хроматографии. Использовали следующие условия разделения: хроматографическая колонка: X select C18 19 мм * 150 мм; подвижная фаза: вода (содержит 0.05% трифторуксусной кислоты) и ацетонитрил; скорость потока: 25 мл/мин; градиент: повышение содержания ацетонитрила с 5% до 100% за 7 минут; длина волны детектора: 254 нм. После очистки получали 2.42 мг желтого твердого (S)-2-(4-(2- ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-N-(2-оксодигидроиндол-5- ил)-3-фенилпропанамида (выход: 2.8%). LCMS: RT = 3.72 мин, [M+H]+ = 557.15.
Пример 13
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)- ил)-3-фенилпропанамидо)-2-фторбензамида
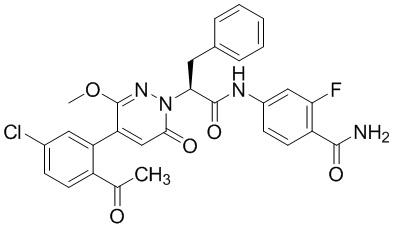
Использовался следующий путь синтеза.
Стадия A: Синтез 2-фтор-4-нитробензамида
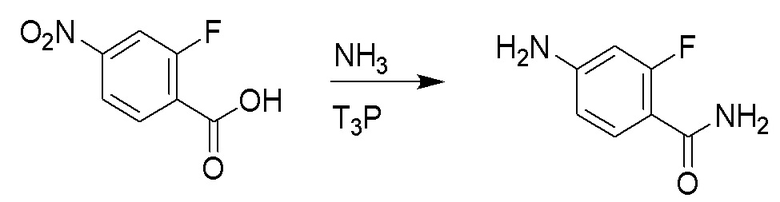
2-Фтор-4-нитробензойную кислоту (200 мг, 1.08 ммоль) и аммиак (4.3 мл, 0.5М раствор в тетрагидрофуране) растворяли в этилацетате (5.0 мл). Добавляли в полученный раствор 1-пропилфосфоновый ангидрид (1.37 г, 4.32 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов.
Реакционный раствор охлаждали до комнатной температуры и упаривали при пониженном давлении. Получали 180 мг 2-фтор-4-нитробензамида в виде не совсем белого твердого вещества (выход: 90.0%) после очистки методом колоночной хроматографии на силикагеле. LCMS: RT = 3.45 мин, [M+H]+ = 185.02.
Стадия B: Синтез 4-амино-2-фторбензамида
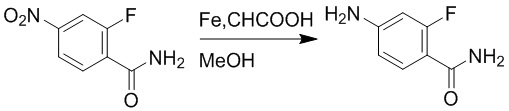
2-Фтор-4-нитробензамид (180 мг, 0.97 ммоль) растворяли в метаноле (10 мл), добавляли уксусную кислоту (1 мл) и порошок железа (669 мг, 9.7 ммоль), нагревали до 80°C и вели реакцию в течение 4 часов.
Насыщенный раствор бикарбоната натрия добавляли в реакционный раствор до pH = 7-8. Полученный смешанный раствор экстрагировали этилацетатом (100 мл × 3) и упаривали. Получали 140 мг желтого твердого 4-амино-2-фторбензамида (выход: 93.0%). LCMS: RT = 1.15 мин, [M+H]+ = 155.05.
Стадия C: Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)-2-фторбензамида
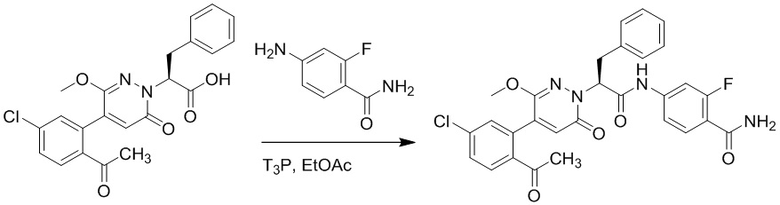
(S)-2-(4-(2-Ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропановую кислоту (40 мг, 0.09 ммоль) и 4-амино-2-фторбензамид (15.9 мг, 0.1 ммоль) растворяли в этилацетате (3.0 мл) и добавляли N,N-диизопропилэтиламин (116 мг, 0.9 ммоль). После этого добавляли в полученный раствор 1-пропилфосфоновый ангидрид (114 мг, 0.36 ммоль). Реакционный раствор нагревали до 60°C и перемешивали 3 часа.
Реакционную смесь охлаждали до комнатной температуры и упаривали при пониженном давлении. Сырой продукт очищали методом препаративной высокоэффективной жидкостной хроматографии. Использовали следующие условия разделения: хроматографическая колонка: X select C18 19 мм * 150 мм; подвижная фаза: вода (содержит 0.05% трифторуксусной кислоты) и ацетонитрил; скорость потока: 25 мл/мин; градиент: повышение содержания ацетонитрила с 5% до 100% за 7 минут; длина волны детектора: 254 нм. После очистки получали 2.47 мг желтого твердого (S)-4-(2-(4- (2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)-2- фторбензамида (выход: 4.8%). LCMS: RT = 3.83 мин, [M+H]+ = 563.14.
Пример 14
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)- ил)-3-фенилпропанамидо)-2-фторбензойной кислоты

Использовался следующий путь синтеза.
Стадия A: Синтез 4-амино-2-фторбензойной кислоты
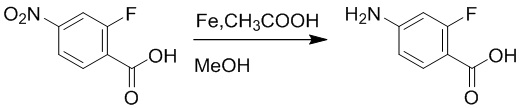
2-Фтор-4-нитробензойную кислоту (200 мг, 1.12 ммоль) растворяли в метаноле (10 мл), добавляли уксусную кислоту (1 мл) и порошок железа (629 мг, 11.2 ммоль), нагревали до 80°C и вели реакцию в течение 4 часов.
Насыщенный раствор бикарбоната натрия добавляли в реакционный раствор до pH = 7-8. Полученный смешанный раствор экстрагировали этилацетатом (100 мл × 3) и упаривали. Получали 150 мг желтой твердой 4-амино-2-фторбензойной кислоты (выход: 89.0%). LCMS: RT = 1.66 мин, [M+H]+ = 156.03.
Стадия B: Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)-2-фторбензойной кислоты

(S)-2-(4-(2-Ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропановую кислоту (40 мг, 0.09 ммоль) и 4-амино-2-фторбензойную кислоту (16 мг, 0.10 ммоль) растворяли в этилацетате (3.0 мл) и добавляли N,N-диизопропилэтиламин (116 мг, 0.9 ммоль). После этого добавляли в полученный раствор 1-пропилфосфоновый ангидрид (114 мг, 0.36 ммоль). Реакционный раствор нагревали до 60°C и перемешивали 3 часа.
Реакционную смесь охлаждали до комнатной температуры и упаривали при пониженном давлении. Сырой продукт очищали методом препаративной высокоэффективной жидкостной хроматографии. Использовали следующие условия разделения: хроматографическая колонка: X select C18 19 мм * 150 мм; подвижная фаза: вода (содержит 0.05% трифторуксусной кислоты) и ацетонитрил; скорость потока: 25 мл/мин; градиент: повышение содержания ацетонитрила с 5% до 100% за 7 минут; длина волны детектора: 254 нм. После очистки получали 1.28 мг (S)-4-(2-(4-(2-ацетил-5- хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)-2- фторбензойной кислоты в виде желтого твердого вещества (выход: 2.4%). LCMS: RT = 3.99 мин, [M+H]+ = 564.12.
Пример 15
Синтез (S)-5-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)- ил)-3-фенилпропанамидо)-1H-индол-2-муравьиной кислоты
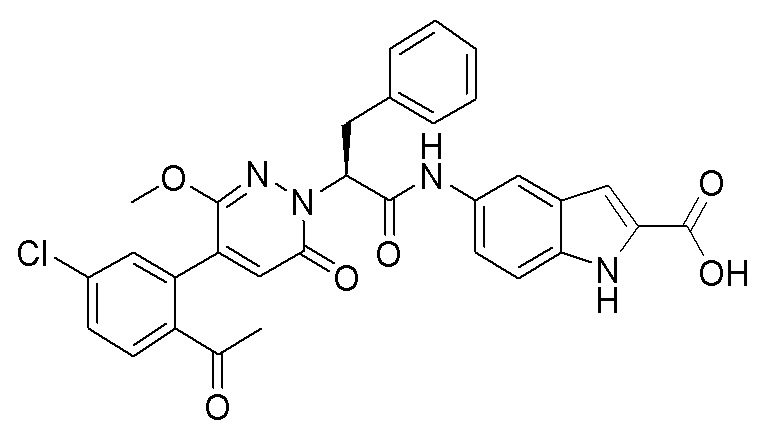
Использовался следующий путь синтеза.
Стадия A: Синтез ди-трет-бутил (R)-5-(2-гидрокси-3-фенилпропанамидо)-1H- индол-1,2-диформиата
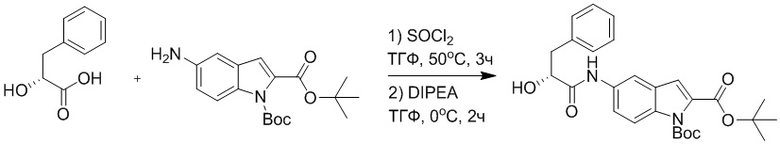
D-Фенилмолочную кислоту (1.00 г, 6.0 ммоль) растворяли в сухом тетрагидрофуране (40.0 мл) в сухой трехгорлой колбе и перемешивали на ледяной бане 15 минут в инертной атмосфере азота. Тионилхлорид (0.7 мл, 9.0 ммоль) медленно добавляли в реакционный раствор по каплям в течение 30 минут. Реакционный раствор нагревали до 50°C, перемешивали 3 часа при постоянной температуре, охлаждали до комнатной температуры и упаривали на роторном испарителе. Остаток вакуумировали на масляном насосе 15 минут и растворяли в ТГФ, получая раствор A. Ди-трет-бутил 5-амино-1H-индол-1,2-дикарбоксилат (1.6 г, 4.8 ммоль) и диизопропилэтиламин (3.0 мл, 18 ммоль) растворяли в сухом тетрагидрофуране (20.0 мл) в сухой трехгорлой колбе и перемешивали на ледяной бане 15 минут в инертной атмосфере азота. Раствор A медленно добавляли в полученный смешанный раствор по каплям и перемешивали в течение 1 часа на ледяной бане. Мониторинг полноты прохождения реакции осуществляли методом LCMS.
Реакцию гасили добавлением воды в реакционный раствор, и полученный смешанный раствор экстрагировали этилацетатом (200 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (100 мл × 3), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/4). Получали 1.0 г желтого твердого ди-трет-бутил (R)-5-(2-гидрокси-3-фенилпропанамидо)- 1H-индол-1,2-диформиата (выход: 34.7 %). LCMS: RT = 4.53 мин, [M+H]+ = 481.38.
Стадия B: Синтез ди-трет-бутил (R)-5-(2-(((4-нитрофенил)сульфонил)оксо)- 3-фенилпропанамидо)-1H-индол-1,2-диформиата
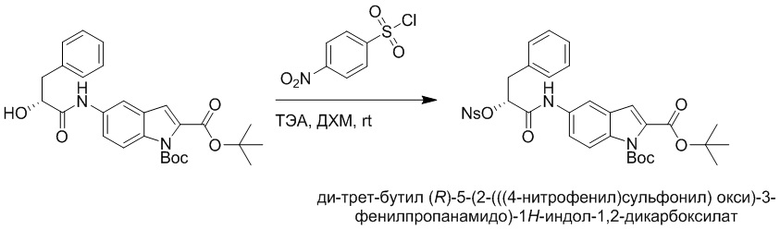
Ди-трет-бутил (R)-5-(2-гидрокси-3-фенилпропанамидо)-1H-индол-1,2-диформиат (330 мг, 0.7 ммоль) и триэтиламин (0.3 мл, 2.1 ммоль) растворяли в дихлорметане (10.0 мл). Добавляли в реакционный раствор 4-нитробензол сульфонилхлорид (221 мг, 1.0 ммоль) на ледяной бане и перемешивали при комнатной температуре в течение 2 часов.
Реакцию гасили добавлением насыщенного водного раствора бикарбоната натрия (10 мл) в реакционный раствор. Полученный смешанный раствор экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (10 мл × 3), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток растворяли в дихлорметане (4 мл), который затем добавляли в н-гексан (60 мл) по каплям при перемешивании. В осадок выпадало большое количество белого твердого вещества, его отфильтровывали. Осадок на фильтре собирали, получая 440 мг белого твердого ди-трет-бутил (R)-5-(2-(((4-нитрофенил)сульфонил)оксо)-3- фенилпропанамидо)-1H-индол-1,2-диформиата (выход: 96.7%). LCMS: RT = 4.75 мин, [M+H]+ = 688.09.
Стадия C: Синтез ди-трет-бутил (S)-5-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси- 6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)-1H-индол-1,2-диформиата
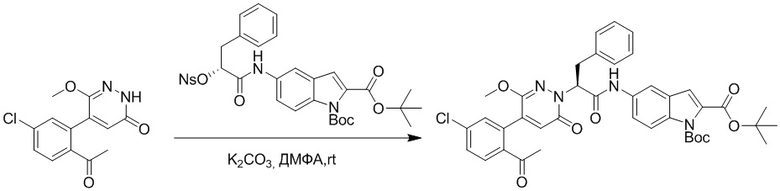
5-(2-Ацетил-5-хлорфенил)-6-метоксипиридазин-3(2H)-он (100 мг, 0.36 ммоль) растворяли в N,N-диметилформамиде (2.0 мл). После этого добавляли в полученный раствор карбонат калия (100 мг, 0.72 ммоль) и ди-трет-бутил (R)-5-(2-(((4-нитрофенил) сульфонил)оксо)-3-фенилпропанамидо)-1H-индол-1,2-диформиат (240.0 мг, 0.36 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов.
Реакцию гасили добавлением воды в реакционный раствор. Полученный смешанный раствор экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, и объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (10 мл × 3), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/1). Получали 65 мг желтого твердого ди-трет-бутил (S)-5-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-фенилпропанамидо)-1H-индол-1,2-диформиата (выход: 24.4%). LCMS: RT = 4.92 мин, [M+H]+ = 741.20.
Стадия D: Синтез (S)-5-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)-1H-индол-2-муравьиной кислоты
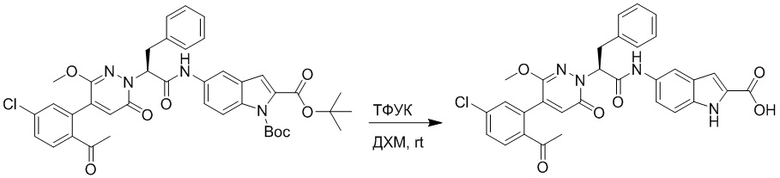
Ди-трет-бутил (S)-5-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-фенилпропанамидо)-1H-индол-1,2-диформиат (65 мг, 0.09 ммоль) растворяли в дихлорметане (2.0 мл). После этого добавляли в полученный раствор трифторуксусную кислоту (1.0 мл) и перемешивали при комнатной температуре в течение 1 часа.
Реакционный раствор упаривали при пониженном давлении на воздушной бане. Остаток очищали методом препаративной высокоэффективной жидкостной хроматографии и получали 7 мг желтой твердой (S)-5-(2-(4-(2-ацетил-5-хлорфенил)-3- метокси-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)-1H-индол-2-муравьиной кислоты (выход: 11.0%). LCMS: RT = 3.88 мин, [M+H]+ = 585.10. 1H ЯМР (500 МГц, ДМСО) δ 12.85 (с, 1H), 11.70 (с, 1H), 10.03 (с, 1H), 7.98 (д, J = 8.3 Гц, 2H), 7.68 (дд, J = 8.3, 2.1 Гц, 1H), 7.49 (д, J = 2.1 Гц, 1H), 7.40 – 7.24 (м, 6H), 7.19 (дд, J = 13.7, 6.6 Гц, 1H), 7.04 (д, J = 1.8 Гц, 1H), 6.89 (с, 1H), 5.74 (дд, J = 10.1, 4.9 Гц, 1H), 3.68 (с, 3H), 3.52 (дд, J = 14.1, 10.4 Гц, 1H), 3.42 (дд, J = 14.0, 4.8 Гц, 1H), 2.52 (с, 3H).
Пример 16
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-этокси-6-оксопиридазин-1(6H)-ил)- 3-фенилпропанамидо)бензойной кислоты
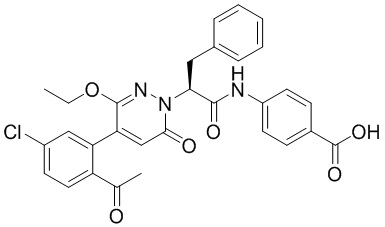
Использовался следующий путь синтеза.
Стадия A: Синтез 5-бром-6-этокси-2-(4-метоксибензил)пиридазин-3(2H)-она
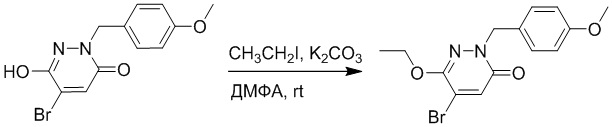
5-Бром-6-гидрокси-2-(4-метоксибензил)пиридазин-3(2H)-он (0.5 г, 1.6 ммоль) растворяли в N,N-диметилформамиде (2.0 мл) при комнатной температуре. После этого добавляли в полученный раствор карбонат калия (442 мг, 3.2 ммоль) и иодэтан (380 мг, 2.4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов.
Реакцию гасили добавлением воды в реакционный раствор, и полученный смешанный раствор экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (20 мл × 3), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/4), и получали 280 мг белого твердого 5-бром-6-этокси-2-(4-метоксибензил)пиридазин- 3(2H)-она (выход: 51.3%). LCMS: RT = 4.06 мин, [M+H]+ = 341/339.
Стадия B: Синтез 5-(2-ацетил-5-хлорфенил)-6-этокси-2-(4- метоксибензил)пиридазин-3(2H)-она
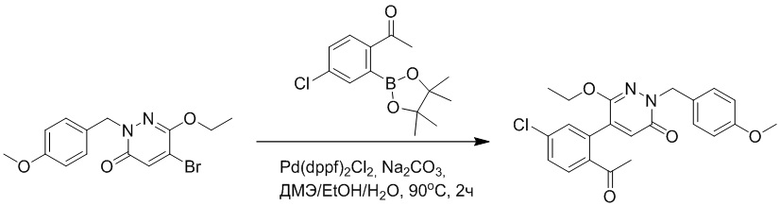
5-Бром-6-этокси-2-(4-метоксибензил)пиридазин-3(2H)-он (280 мг, 0.82 ммоль), 1-(4-хлор-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этан-1-он (280 мг, 1.0 ммоль) и карбонат натрия (180 мг, 1.7 ммоль) помешали в трехгорлую колбу при комнатной температуре и заменяли атмосферу в колбе на азот. Добавляли смесь растворителей (10 мл, ДМЭ: EtOH: H2O = 8:1:1) и продували колбу азотом. Добавляли комплекс [1,1'-бис(дифенилфосфино)ферроцен]палладий дихлорида с дихлорметаном (59 мг, 0.07 ммоль) и продували колбу азотом. Смесь нагревали до 90°C и вели реакцию в течение 2 часов.
Реакционный раствор охлаждали до комнатной температуры и фильтровали через диатомит. Осадок на фильтре промывали этилацетатом (30 мл × 2), фильтрат и промывной раствор объединяли и упаривали при пониженном давлении. В полученный остаток добавляли воду (50 мл), и смешанный раствор экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (20 мл × 3), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/2). Получали 120 мг желтого твердого 5-(2-ацетил-5-хлорфенил)-6-этокси-2-(4- метоксибензил)пиридазин-3(2H)-она, (выход: 50.0%). LCMS: RT = 4.12 мин, [M+H]+ = 413.12.
Стадия C: Синтез 5-(2-ацетил-5-хлорфенил)-6-этоксипиридазин-3(2H)-она
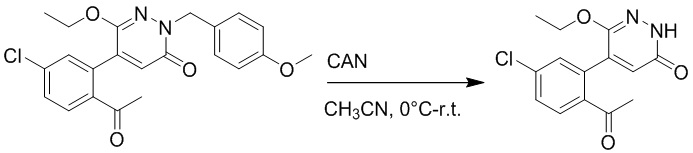
5-(2-Ацетил-5-хлорфенил)-6-этокси-2-(4-метоксибензил)пиридазин-3(2H)-он (120 мг, 0.29 ммоль) добавляли в смесь растворителей (4 мл, ацетонитрил: вода = 3:1) при 0°C, и затем медленно добавляли церийаммоний нитрат (1.5 г, 2.9 ммоль), после этого вели реакцию 30 минут при комнатной температуре.
После окончания реакции, реакционную смесь гасили добавлением воды, и смешанный раствор экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (30 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/1). Получали 64 мг желтого твердого 5-(2-ацетил-5-хлорфенил)-6-этоксипиридазин-3(2H)-она (выход: 75.6%). LCMS: RT = 3.41 мин, [M+H]+ = 293.09.
Стадия D: Синтез трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-этокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата
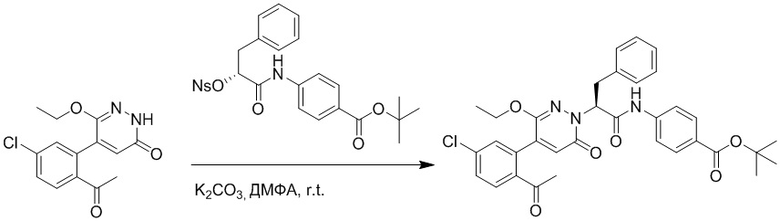
5-(2-Ацетил-5-хлорфенил)-6-этоксипиридазин-3(2H)-он (64 мг, 0.22 ммоль), трет-бутил (R)-4-(2-(((4-нитрофенил)сульфонил)оксо)-3-фенилпропанамидо)бензоат (170 мг, 0.33 ммоль) и карбонат калия (61 мг, 0.44 ммоль) добавляли в N,N-диметилформамид (2.0 мл) при комнатной температуре и оставляли для прохождения реакции на ночь при комнатной температуре.
После окончания реакции, реакционную смесь гасили добавлением воды, и смешанный раствор экстрагировали этилацетатом (10 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (10 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 58 мг бледно-желтого твердого трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-этокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата (выход: 43.0%). LCMS: RT = 4.67 мин, [M+H]+ = 616.18.
Стадия G: Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-этокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты

Трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-этокси-6-оксопиридазин-1(6H)-ил)- 3-фенилпропанамидо)бензоат (58 мг, 0.09 ммоль) растворяли в дихлорметане (2.0 мл). После этого добавляли в полученный раствор трифторуксусную кислоту (0.5 мл) и перемешивали при комнатной температуре в течение 1 часа.
Реакционный раствор упаривали при пониженном давлении на воздушной бане. Полученный остаток очищали, суспендируя его в дихлорметане и н-гексане, и получали 19 мг желтой твердой (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-этокси-6-оксопиридазин- 1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты (выход: 36.0%). LCMS: RT = 4.08 мин, [M+H]+ = 560.08. 1H ЯМР (500 МГц, ДМСО) δ 12.76 (с, 1H), 10.52 (с, 1H), 8.02 (д, J = 8.4 Гц, 1H), 7.92 (д, J = 8.7 Гц, 2H), 7.79 – 7.67 (м, 3H), 7.51 (д, J = 2.1 Гц, 1H), 7.38 – 7.25 (м, 4H), 7.21 (д, J = 6.4 Гц, 1H), 6.91 (с, 1H), 5.77 (дд, J = 9.8, 5.1 Гц, 1H), 4.10 (дд, J = 17.0, 7.0 Гц, 2H), 3.44 (ддд, J = 22.3, 18.9, 11.2 Гц, 2H), 2.55 (с, 3H), 1.15 (т, J = 7.0 Гц, 3H).
Пример 17
Синтез (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенил-N-(4-аминосульфонилфенил)пропанамида
Использовался следующий путь синтеза.
Стадия A: Синтез метил (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропаноата
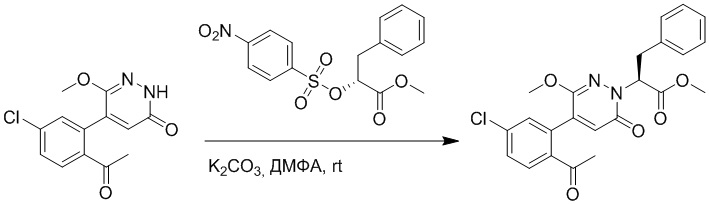
5-(2-Ацетил-5-хлорфенил)-6-этоксипиридазин-3(2H)-он (230 мг, 0.83 ммоль), метил (R)-2-(2-(((4-нитрофенил)сульфонил)оксо)-3-фенилпропаноат (450 мг, 1.2 ммоль) и карбонат калия (230 мг, 1.67 ммоль) добавляли в N,N-диметилформамид (4.0 мл) при комнатной температуре и оставляли для прохождения реакции при комнатной температуре на 6 часов.
После окончания реакции, реакционную смесь гасили добавлением воды, и смешанный раствор экстрагировали этилацетатом (40 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (20 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 270 мг бледно-желтого твердого метил (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропаноата (выход: 74.0%). LCMS: RT = 4.08 мин, [M+H]+ = 441.09.
Стадия B: Синтез (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-фенилпропановой кислоты
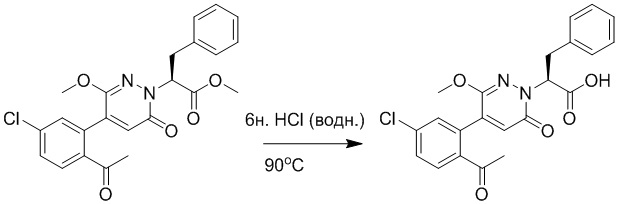
Метил (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропаноат (270 мг, 0.61 ммоль) растворяли в 6н. растворе соляной кислоты (10.0 мл). Смесь нагревали до 50°C и перемешивали 12 часов при постоянной температуре.
Реакцию гасили добавлением насыщенного водного раствора бикарбоната натрия, и значение pH доводили до слабокислого. Полученный смешанный раствор экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (20 мл × 3), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Получали 240 мг желтой твердой (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропановой кислоты (выход: 92.0%). LCMS: RT = 3.87 мин, [M+H]+ = 427.05.
Стадия C: Синтез (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенил-N-(4-аминосульфонилфенил)пропанамида
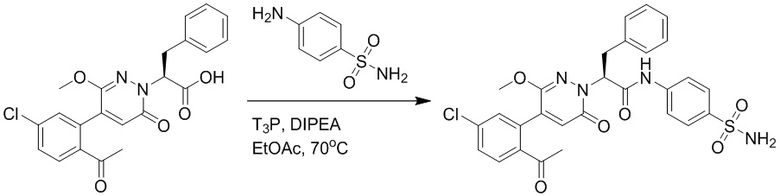
(S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропановую кислоту (33 мг, 0.12 ммоль), сульфаниламид (52 мг, 0.35 ммоль) и N,N-диизопропилэтиламин (0.2 мл) растворяли в этилацетате (2.0 мл). После этого добавляли в полученный раствор 1-пропилфосфоновый ангидрид (0.2 мл, 50%-ный раствор в этилацетате). Смесь нагревали до 70°C и перемешивали 15 часов при постоянной температуре.
Реакцию гасили добавлением воды (10 мл) в реакционный раствор. Полученный смешанный раствор экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (10 мл × 3), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом препаративной высокоэффективной жидкостной хроматографии и получали 6 мг белого твердого (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-фенил-N-(4- аминосульфонилфенил)пропанамида (выход: 15.0%). LCMS: RT = 3.81 мин, [M+H]+ = 581.06.
Пример 18
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)- ил)-3-фенилпропанамидо)фенилсульфоновой кислоты
Использовался следующий путь синтеза.
Стадия A: Синтез (2R,2'S)-N,N'-(дитио(-4,1-фенилен))бис(2-хлор-3- фенилпропанамида)
D-Фенилмолочную кислоту (0.6 г, 3.6 ммоль) растворяли в сухом тетрагидрофуране (40.0 мл) в сухой трехгорлой колбе и перемешивали на ледяной бане 15 минут в инертной атмосфере азота. Тионилхлорид (0.8 мл, 10.8 ммоль) медленно добавляли в реакционный раствор по каплям в течение 30 минут. Реакционный раствор нагревали до 70°C, перемешивали 5 часов при постоянной температуре, охлаждали до комнатной температуры и упаривали на роторном испарителе. Остаток вакуумировали на масляном насосе 15 минут и растворяли в ТГФ, получая раствор A. 4,4-Дитиоиоданилин (890 мг, 3.6 ммоль) и диизопропилэтиламин (2 мл, 10.8 ммоль) растворяли в сухом тетрагидрофуране (20.0 мл) в сухой трехгорлой колбе и перемешивали на ледяной бане 15 минут в инертной атмосфере азота. Раствор A медленно добавляли в смешанный раствор по каплям и перемешивали в течение 1 часа на ледяной бане. Мониторинг полноты прохождения реакции осуществляли методом LCMS.
Реакцию гасили добавлением воды, и полученный смешанный раствор экстрагировали этилацетатом (40 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (20 мл × 3), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/4). Получали 1.2 г желтого твердого (2R, 2'S)-N,N'-(дитио(-4,1-фенилен))бис(2-хлор-3-фенилпропанамида) (выход: 38.0%). LCMS: RT = 4.72 мин, [M+H]+ = 581.08.
Стадия B: Синтез (R)-4-(2-хлор-3-фенилпропанамидо)фенилсульфоновой кислоты
(2R,2'S)-N,N'-(дитио(-4,1-фенилен))бис(2-хлор-3-фенилпропанамид) (300 мг, 0.52 ммоль) растворяли в ацетонитриле (10.0 мл). После этого добавляли в полученный раствор 30%-ный раствор пероксида водорода (2.0 мл). Смесь нагревали до 50°C и перемешивали 5 часов при постоянной температуре.
Реакцию гасили добавлением насыщенного водного раствора бикарбоната натрия в реакционный раствор и доводили значение pH раствора до слабокислого. Полученный смешанный раствор экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (10 мл × 3), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Получали 300 мг желтой твердой (R)-4-(2-хлор-3- фенилпропанамидо)фенилсульфоновой кислоты (выход: 86.0%). LCMS: RT = 6.83 мин, [M-H]- = 337.95.
Стадия C: Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)фенилсульфоновой кислоты
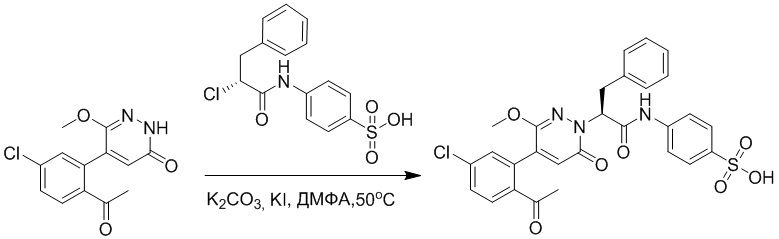
H)-он (33 мг, 0.12 ммоль) и карбонат калия (33 мг, 0.24 ммоль) растворяли в N,N-диметилформамиде (2.0 мл). После этого добавляли в полученный раствор иодид калия (40 мг, 0.24 ммоль) и (R)-4-(2-хлор-3-фенилпропанамидо)фенилсульфоновую кислоту (40 мг, 0.18 ммоль). Смесь нагревали до 50°C и перемешивали 40 часов при постоянной температуре.
Реакцию гасили добавлением воды (10 мл). Полученный смешанный раствор экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (10 мл × 3), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом препаративной высокоэффективной жидкостной хроматографии и получали 16 мг желтой твердой (S)-4-(2-(4- (2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо) фенилсульфоновой кислоты (выход: 23.0%). LCMS: RT = 7.96 мин, [M+H]+ = 582.12.
Пример 19
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)- ил)-3-фенилпропанамидо)бензойной кислоты
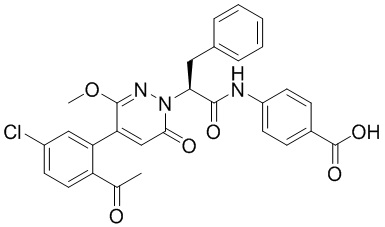
Использовался следующий путь синтеза.
Стадия A: Синтез 5-бром-6-гидрокси-2-(4-метоксибензил)пиридазин-3(2H)-она
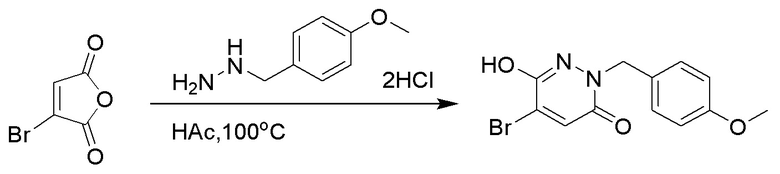
Броммалеиновый ангидрид (2.00 г, 11.3 ммоль) и (4-метоксибензил)гидразин гидрохлорид (2.13 г, 11.3 ммоль) добавляли в ледяную уксусную кислоту (50.0 мл) и вели реакцию при 100°C в течение 3 часов.
Реакционный раствор охлаждали до комнатной температуры после окончания реакции и выливали в воду. Выпадало большое количество твердого осадка, который некоторое время перемешивали и отфильтровали. Осадок на фильтре промывали водой и сушили. Получали 1.50 г бледно-желтого твердого 5-бром-6-гидрокси-2-(4- метоксибензил)пиридазин-3(2H)-она, который напрямую использовали в следующей стадии без очистки. LCMS: RT = 3.44 мин, [M+H]+ = 311.03.
Стадия B: Синтез 5-бром-6-метокси-2-(4-метоксибензил)пиридазин-3(2H)-она
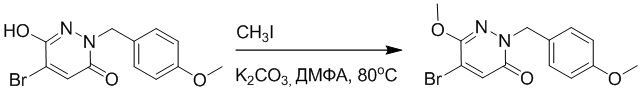
5-Бром-6-гидрокси-2-(4-метоксибензил)пиридазин-3(2H)-он (1.50 г, 4.82 ммоль) и карбонат калия (2.66 г, 19.29 ммоль) добавляли в N,N-диметилформамид (15.0 мл) при комнатной температуре и перемешивали при 80°C в течение 15 минут. Иодметан (1.2 мл) добавляли в раствор при этой же температуре и вели реакцию еще 30 минут.
После окончания реакции, реакционную смесь гасили добавлением воды, и смешанный раствор экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (50 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/3). Получали 1.10 г белого твердого 5-бром-6-метокси-2-(4-метоксибензил)пиридазин-3(2H)-она (выход: 70.3%). LCMS: RT = 3.87 мин, [M+H]+ = 325.01.
Стадия C: Синтез пинаколового эфира 6-ацетил-3-хлорфенилбороновой кислоты
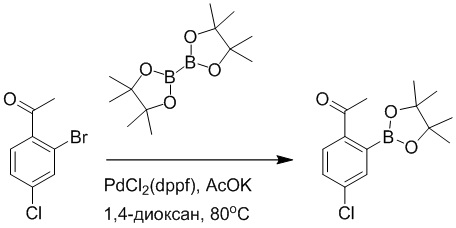
2-Бром-4-хлорацетофенон (5.00 г, 21.41 ммоль), бис(пинаколато)диборон (8.16 г, 32.12 ммоль) и ацетат калия (4.20 г, 42.82 ммоль) помещали в трехгорлую колбу при комнатной температуре и продували колбу азотом. Добавляли 1,4-диоксан (60.0 мл) и продували колбу азотом. Добавляли [1,1'-бис(дифенилфосфино)ферроцен]палладий хлорид (1.75 г, 2.14 ммоль) и продували колбу азотом. Смесь нагревали до 80°C и вели реакцию в течение 3 часов.
После окончания реакции, реакционную смесь гасили добавлением воды и фильтровали через диатомит. Осадок на фильтре промывали этилацетатом, и фильтрат экстрагировали этилацетатом (80 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (50 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/50). Получали 2.1 г желтого твердого пинаколового эфира 6-ацетил-3-хлорфенилбороновой кислоты (выход: 35.0%). LCMS: RT = 4.26 мин, [M-H]- = 279.08.
Стадия D: Синтез 5-(2-ацетил-5-хлорфенил)-6-метокси-2-(4- метоксибензил)пиридазин-3(2H)-она
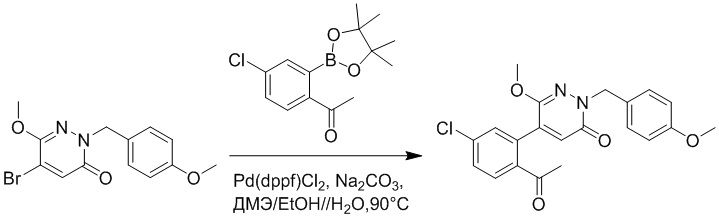
5-Бром-6-метокси-2-(4-метоксибензил)пиридазин-3(2H)-он (1.10 г, 3.39 ммоль), пинаколовый эфир 6-ацетил-3-хлорфенилбороновой кислоты (949 мг, 3.39 ммоль) и карбонат натрия (718 мг, 6.78 ммоль) добавляли в трехгорлую колбу при комнатной температуре и продували колбу азотом. Добавляли смесь растворителей (10 мл, 1, 2-диметоксиэтан:EtOH:H2O = 8:1:1) и продували колбу азотом. Добавляли [1,1'-бис (дифенилфосфино)ферроцен]палладий хлорид (249 мг, 0.34 ммоль) и продували колбу азотом. Смесь нагревали до 90°C и вели реакцию в течение 1 часа.
После окончания реакции, реакционную смесь гасили добавлением воды, и смешанный раствор экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (50 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 676 мг желтого твердого 5-(2-ацетил-5-хлорфенил)-6-метокси-2-(4-метоксибензил)пиридазин- 3(2H)-она (выход: 50.2%). LCMS: RT = 3.99 мин, [M+H]+ = 399.07.
Стадия E: Синтез 5-(2-ацетил-5-хлорфенил)-6-метоксипиридазин-3(2H)-она
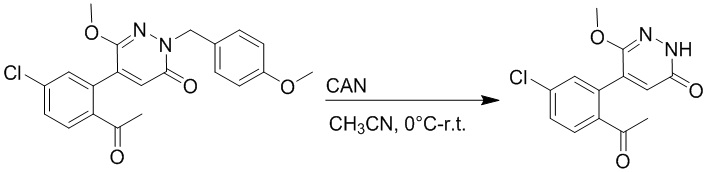
5-(2-Ацетил-5-хлорфенил)-6-метокси-2-(4-метоксибензил)пиридазин-3(2H)-он (676 мг, 1.70 ммоль) добавляли в смесь растворителей (4 мл, ацетонитрил: вода = 3:1) при 0°C, затем медленно добавляли церийаммоний нитрат (7.46 г, 13.60 ммоль), после этого вели реакцию 30 минут при комнатной температуре.
После окончания реакции, реакционную смесь гасили добавлением воды, и смешанный раствор экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (30 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/1). Получали 238 мг желтого твердого 5-(2-ацетил-5-хлорфенил)-6-метоксипиридазин-3(2H)-она (выход: 50.0%). LCMS: RT = 3.23 мин, [M+H]+ = 279.08.
Стадия E: Синтез трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата
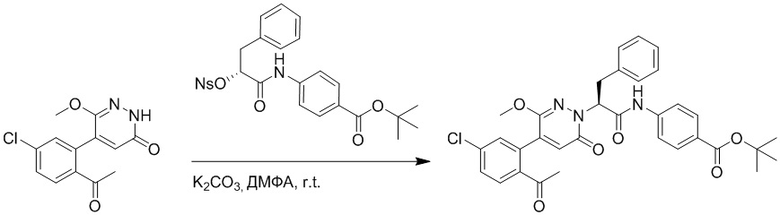
5-(2-Ацетил-5-хлорфенил)-6-метоксипиридазин-3(2H)-он (50 мг, 0.18 ммоль), трет-бутил (R)-4-(2-(((4-нитрофенил)сульфонил)оксо)-3-фенилпропанамидо)бензоат (113 мг, 0.22 ммоль) и карбонат калия (50 мг, 0.36 ммоль) добавляли в N,N-диметилформамид (2.0 мл) при комнатной температуре и оставляли для прохождения реакции на ночь при комнатной температуре.
После окончания реакции, реакционную смесь гасили добавлением воды, и смешанный раствор экстрагировали этилацетатом (10 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (10 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 75 мг бледно-желтого твердого трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата (выход: 66.7%). LCMS: RT = 4.53 мин, [M+H]+ = 602.13.
Стадия G: Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
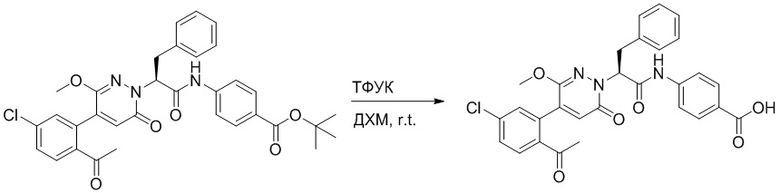
Трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)- ил)-3-фенилпропанамидо)бензоат (75 мг, 0.12 ммоль) растворяли в дихлорметане (2.0 мл) при комнатной температуре. Трифторуксусную кислоту (0.25 мл) добавляли по каплям и оставляли для прохождения реакции при комнатной температуре на 3 часов.
После окончания реакции упаривали дихлорметан и отгоняли на масляном насосе трифторуксусную кислоту. Остаток растворяли в дихлорметане (1.0 мл) и добавляли в н-гексан (10.0 мл) по каплям. Выпадал белый твердый осадок, его отфильтровали. Осадок на фильтре промывали н-гексаном и сушили. Получали 50 мг белой твердой (S)-4-(2- (4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо) бензойной кислоты (выход: 76.5%). LCMS: RT = 3.98 мин, [M-H]- = 544.10. 1H ЯМР (500 МГц, ДМСО) δ 12.79 (с, 1H), 10.52 (с, 1H), 7.99 (д, J = 8.4 Гц, 1H), 7.91 (д, J = 8.7 Гц, 2H), 7.72 (д, J = 8.7 Гц, 2H), 7.69 (дд, J = 8.3, 2.1 Гц, 1H), 7.50 (д, J = 2.1 Гц, 1H), 7.37–7.23 (м, 4H), 7.19 (т, J = 7.1 Гц, 1H), 6.91 (с, 1H), 5.74 (дд, J = 10.2, 4.9 Гц, 1H), 3.67 (с, 3H), 3.52 (дд, J = 14.1, 10.3 Гц, 1H), 3.41 (дд, J = 14.1, 4.7 Гц, 1H), 2.53 (с, 3H).
Пример 20
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(циклопропилметокси)-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
Использовался следующий путь синтеза.
Стадия A: Синтез 5-бром-6-(циклопропилметокси)-2-(4-метоксибензил) пиридазин-3(2H)-она
5-Бром-6-гидрокси-2-(4-метоксибензил)пиридазин-3(2H)-он (1.50 г, 4.82 ммоль) и карбонат калия (2.66 г, 19.29 ммоль) добавляли в N,N-диметилформамид (15.0 мл) при комнатной температуре и перемешивали при 80°C в течение 15 минут. Циклопропилметилбромид (1.8 мл) добавляли в раствор при этой же температуре и вели реакцию в течение еще 30 минут.
После окончания реакции, реакционную смесь гасили добавлением воды, и смешанный раствор экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (50 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/3). Получали 1.32 г белого твердого 5-бром-6-(циклопропилметокси)-2-(4-метоксибензил)пиридазин- 3(2H)-она (выход: 74.9%). LCMS: RT = 4.20 мин, [M+H]+ = 365.39.
Стадия B: Синтез 5-(2-ацетил-5-хлорфенил)-6-(циклопропилметокси)-2-(4- метоксибензил)пиридазин-3(2H)-она
5-Бром-6-(циклопропилметокси)-2-(4-метоксибензил)пиридазин-3(2H)-он (1.32 г, 3.61 ммоль), пинаколовый эфир 6-ацетил-3-хлорфенилбороновой кислоты (1.01 г, 3.61 ммоль) и карбонат натрия (765 мг, 7.22 ммоль) добавляли в трехгорлую колбу при комнатной температуре и продували колбу азотом. Добавляли смесь растворителей (10 мл, 1,2-диметоксиэтан:EtOH:H2O = 8:1:1) и продували колбу азотом. Добавляли [1,1'-бис (дифенилфосфино)ферроцен]палладий хлорид (263.2 мг, 0.36 ммоль) и продували колбу азотом. Смесь нагревали до 90°C и вели реакцию в течение 1 часа.
После окончания реакции, реакционную смесь гасили добавлением воды, и смешанный раствор экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (50 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 685 мг желтого твердого 5-(2-ацетил-5-хлорфенил)-6-(циклопропилметокси)-2-(4-метоксибензил) пиридазин-3(2H)-она (выход: 43.2%). LCMS: RT = 4.26 мин, [M+H]+ = 439.13.
Стадия C: Синтез 5-(2-ацетил-5-хлорфенил)-6-(циклопропилметокси) пиридазин-3(2H)-она

5-(2-Ацетил-5-хлорфенил)-6-(циклопропилметокси)-2-(4-метоксибензил)пиридазин- 3(2H)-он (685 мг, 1.56 ммоль) добавляли в смесь растворителей (4 мл, ацетонитрил: вода = 3:1) при 0°C и затем медленно добавляли церийаммоний нитрат (6.85 г, 12.59 ммоль), после этого вели реакцию 30 минут при комнатной температуре.
После окончания реакции, реакционную смесь гасили добавлением воды, и смешанный раствор экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (30 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/1). Получали 223 мг желтого твердого 5-(2-ацетил-5-хлорфенил)-6-(циклопропилметокси)пиридазин- 3(2H)-она (выход: 44.8%). LCMS: RT = 3.59 мин, [M+H]+ = 319.04.
Стадия D: Синтез трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3- (циклопропилметокси)-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата
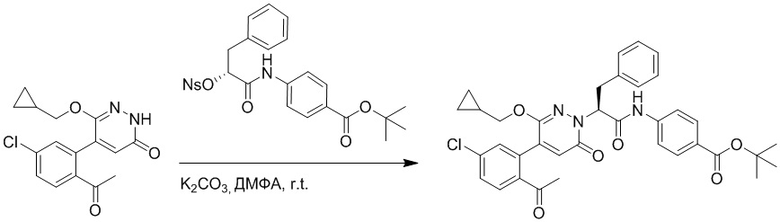
5-(2-Ацетил-5-хлорфенил)-6-(циклопропилметокси)пиридазин-3(2H)-он (50 мг, 0.16 ммоль), трет-бутил (R)-4-(2-(((4-нитрофенил)сульфонил)оксо)-3-фенилпропанамидо) бензоат (100 мг, 0.19 ммоль) и карбонат калия (44 мг, 0.32 ммоль) добавляли в N,N-диметилформамид (2.0 мл) при комнатной температуре и оставляли для прохождения реакции на ночь при комнатной температуре.
После окончания реакции, реакционную смесь гасили добавлением воды, и смешанный раствор экстрагировали этилацетатом (10 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (10 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 63 мг трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(циклопропилметокси)-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата в виде светло-желтого твердого вещества, (выход: 62.5%). LCMS: RT= 4.77 мин, [M+H]+ = 642.19.
Стадия E: Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(циклопропилметокси)- 6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
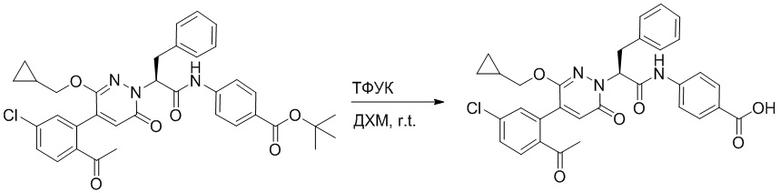
Трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(циклопропилметокси)-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоат (63 мг, 0.10 ммоль) растворяли в дихлорметане (2.0 мл) при комнатной температуре. Трифторуксусную кислоту (0.25 мл) добавляли по каплям и оставляли для прохождения реакции при комнатной температуре на 3 часов.
После окончания реакции дихлорметан упаривали и трифторуксусную кислоту отгоняли на масляном насосе. Остаток растворяли в дихлорметане (1.0 мл) и добавляли в н-гексан (10.0 мл) по каплям. Выпадал белый твердый осадок, его отфильтровывали. Осадок на фильтре промывали н-гексаном и сушили. Получали 50 мг (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(циклопропилметокси)-6-оксопиридазин-1(6H)-ил)- 3-фенилпропанамидо)бензойной кислоты в виде белого твердого вещества (выход: 85.3%). LCMS:RT = 4.20 мин, [M-H]- = 584.14.
Пример 21
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(2-метоксиэтокси)-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
Использовался следующий путь синтеза.
Стадия A: Синтез 5-бром-2-(4-метоксибензил)-6-(2-метоксиэтокси)пиридазин- 3(2H)-она
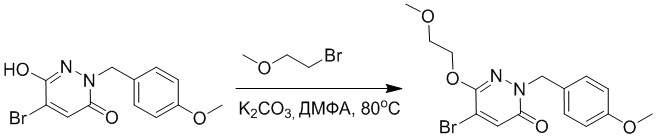
5-Бром-6-гидрокси-2-(4-метоксибензил)пиридазин-3(2H)-он (500 мг, 1.61 ммоль) и карбонат калия (900 мг, 6.52 ммоль) добавляли в N,N-диметилформамид (5.0 мл) при комнатной температуре и перемешивали при 80°C в течение 15 минут. Добавляли в раствор 1-бром-2-метоксиэтан (0.6 мл) при этой же температуре и вели реакцию в течение еще 30 минут.
После окончания реакции, реакционную смесь гасили добавлением воды, и смешанный раствор экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (50 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/3). Получали 333 мг белого твердого 5-бром-2-(4-метоксибензил)-6-(2-метоксиэтокси)пиридазин-3(2H)-она (выход: 55.9%). LCMS: RT = 3.81 мин, [M+H]+ = 369.03.
Стадия B: Синтез 5-(2-ацетил-5-хлорфенил)-2-(4-метоксибензил)-6-(2- метоксиэтокси)пиридазин-3(2H)-она
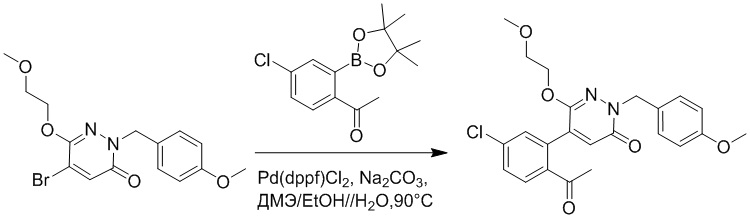
5-Бром-2-(4-метоксибензил)-6-(2-метоксиэтокси)пиридазин-3(2H)-он (333 мг, 0.90 ммоль), пинаколовый эфир 6-ацетил-3-хлорфенилбороновой кислоты (253 мг, 0.90 ммоль) и карбонат натрия (192 мг, 1.81 ммоль) добавляли в трехгорлую колбу при комнатной температуре и продували колбу азотом. Добавляли смесь растворителей (10 мл, 1, 2-диметоксиэтан:EtOH:H2O = 8:1:1) и продували колбу азотом. Добавляли [1,1'-бис (дифенилфосфино)ферроцен]дихлорпалладий (74 мг, 0.09 ммоль) и продували колбу азотом. Смесь нагревали до 90°C и вели реакцию в течение 1 часа.
После окончания реакции, реакционную смесь гасили добавлением воды, и смешанный раствор экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (50 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 186 мг желтого твердого 5-(2-ацетил-5-хлорфенил)-2-(4-метоксибензил)-6-(2-метоксиэтокси) пиридазин-3(2H)-она, (выход: 46.7%). LCMS: RT = 3.97 мин, [M+H]+ = 443.15.
Стадия C: Синтез 5-(2-ацетил-5-хлорфенил)-6-(2-метоксиэтокси) пиридазин-3(2H)-она
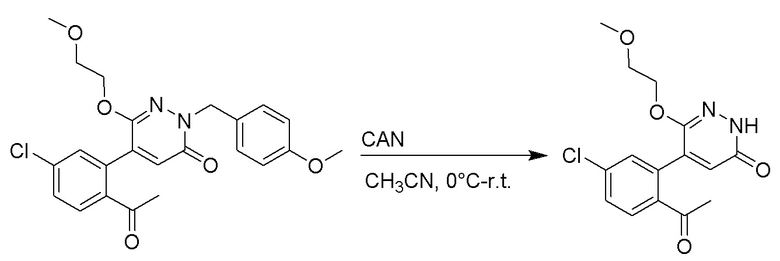
5-(2-Ацетил-5-хлорфенил)-2-(4-метоксибензил)-6-(2-метоксиэтокси)пиридазин- 3(2H)-он (186 мг, 0.42 ммоль) добавляли в смесь растворителей (4 мл, ацетонитрил: вода = 3:1) при 0°C, затем медленно добавляли церийаммоний нитрат (2.30 г, 4.20 ммоль), после этого вели реакцию 30 минут при комнатной температуре.
После окончания реакции, реакционную смесь гасили добавлением воды, и смешанный раствор экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (30 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/1). Получали 89 мг желтого твердого 5-(2-ацетил-5-хлорфенил)-6-(2-метоксиэтокси)пиридазин-3(2H)-она (выход: 66.7%). LCMS: RT = 3.25 мин, [M+H]+ = 323.10.
Стадия D: Синтез трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(2- метоксиэтокси)-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата
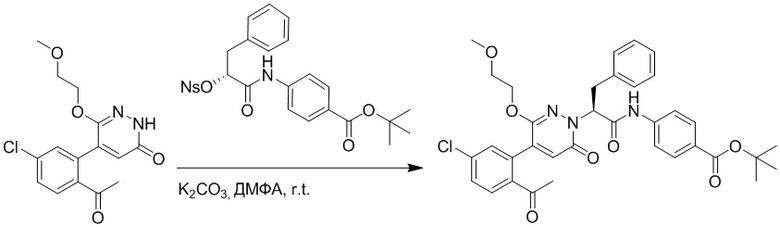
5-(2-Ацетил-5-хлорфенил)-6-(2-метоксиэтокси)пиридазин-3(2H)-он (89 мг, 0.28 ммоль), трет-бутил (R)-4-(2-(((4-нитрофенил)сульфонил)оксо)-3-фенилпропанамидо) бензоат (160 мг, 0.30 ммоль) и карбонат калия (77 мг, 0.56 ммоль) добавляли в N,N-диметилформамид (2.0 мл) при комнатной температуре и оставляли для прохождения реакции на ночь при комнатной температуре.
После окончания реакции, реакционную смесь гасили добавлением воды, и смешанный раствор экстрагировали этилацетатом (10 мл × 3). Органические фазы объединяли. Объединенные органические фазы промывали насыщенным водным раствором хлорида натрия (10 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 140 мг бледно-желтого твердого трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(2- метоксиэтокси)-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата (выход: 78.6%). LCMS: RT = 4.52 мин, [M+H]+ = 646.20.
Стадия E: Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(2-метоксиэтокси)-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
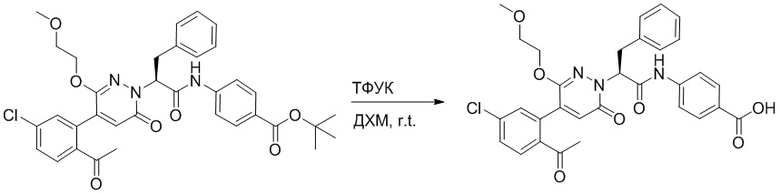
Трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(2-метоксиэтокси)-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоат (140 мг, 0.22 ммоль) растворяли в дихлорметане (2.0 мл) при комнатной температуре. Трифторуксусную кислоту (0.25 мл) добавляли по каплям и оставляли для прохождения реакции при комнатной температуре на 3 часа.
После окончания реакции дихлорметан упаривали и трифторуксусную кислоту отгоняли на масляном насосе. Остаток растворяли в дихлорметане (1.0 мл) и добавляли в н-гексан (10.0 мл) по каплям. Выпадал белый твердый осадок, его отфильтровывали. Осадок на фильтре промывали н-гексаном и сушили. Получали 68 мг белой твердой (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(2-метоксиэтокси)-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)бензойной кислоты (выход: 52.4%). LCMS: RT = 3.96 мин, [M-H]- = 588.10. 1H ЯМР (500 МГц, ДМСО) δ 12.71 (с, 1H), 10.51 (с, 1H), 8.02 (д, J = 8.4 Гц, 1H), 7.90 (д, J = 7.0 Гц, 2H), 7.72 (д, J = 8.8 Гц, 2H), 7.69 (дд, J = 8.4, 2.1 Гц, 1H), 7.49 (д, J = 2.2 Гц, 1H), 7.32 – 7.25 (м, 4H), 7.23 – 7.16 (м, 1H), 6.89 (с, 1H), 5.75 (дд, J = 9.9, 5.1 Гц, 1H), 4.19 – 4.07 (м, 2H), 3.53 – 3.42 (м, 4H), 3.12 (с, 3H), 2.54 (с, 3H).
Пример 22
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-изопропокси-6-оксопиридазин- 1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
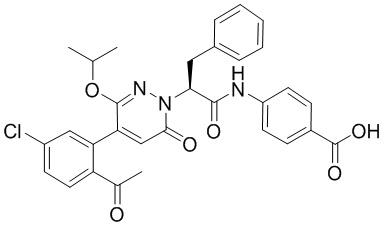
Использовался следующий путь синтеза.
Стадия A: Синтез 5-бром-6-изопропил-2-(4-метоксибензил)пиридазин-3(2H)-она
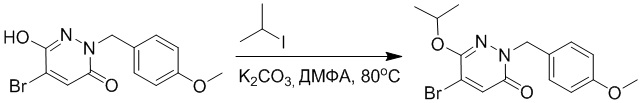
5-Бром-6-гидрокси-2-(4-метоксибензил)пиридазин-3(2H)-он (500 мг, 1.61 ммоль) и карбонат калия (900 мг, 6.52 ммоль) добавляли в N,N-диметилформамид (5.0 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°C в течение 15 минут. При этой же температуре добавляли 2-иодметан (0.6 мл) и реакцию продолжали еще 30 минут.
После завершения реакции, реакционную смесь гасили добавлением воды и экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (50 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/3). Получали 212 мг белого твердого 5-бром-6-изопропил-2-(4-метоксибензил)пиридазин-3(2H)-она (выход: 37.3%). LCMS: RT = 4.21 мин, [M+H]+ = 353.02.
Стадия B: Синтез 5-(2-ацетил-5-хлорфенил)-6-изопропил-2-(4-метоксибензил) пиридазин-3(2H)-она
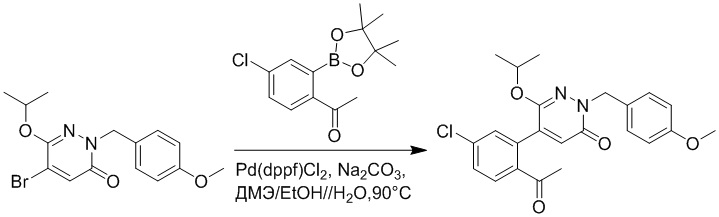
5-Бром-6-изопропил-2-(4-метоксибензил)пиридазин-3(2H)-он (212 мг, 0.60 ммоль), пинаколовый эфир 6-ацетил-3-хлорфенилбороновой кислоты (169 мг, 0.60 ммоль) и карбонат натрия (128 мг, 1.20 ммоль) добавляли в трехгорлую колбу при комнатной температуре и продували колбу азотом. Добавляли смесь растворителей (10 мл, 1,2-диметоксиэтан:этанол:вода = 8:1:1) и продували колбу азотом. Добавляли 1,1'-бисдифенилфосфиноферроцен палладий дихлорид (49 мг, 0.06 ммоль) и продували колбу азотом. Смесь нагревали до 90°C и вели реакцию в течение 1 часа.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (50 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 127 мг 5-(2-ацетил-5-хлорфенил)-6-изопропил-2-(4-метоксибензил)пиридазин-3(2H)-она в виде желтого твердого вещества (выход: 50.0%). LCMS: RT = 4.24 мин.
Стадия C: Синтез 5-(2-ацетил-5-хлорфенил)-6-изопропилпиридазин-3(2H)-она
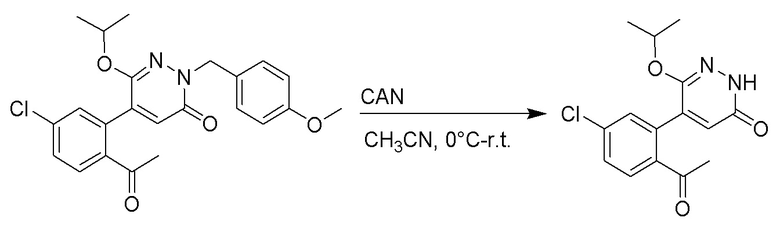
5-(2-Ацетил-5-хлорфенил)-6-изопропил-2-(4-метоксибензил)пиридазин-3(2H)-он (127 мг, 0.30 ммоль) добавляли в смесь растворителей (4 мл, ацетонитрил:вода = 3:1) при 0°C, и затем медленно добавляли церийаммоний нитрат (1.64 г, 2.99 ммоль). После окончания добавления вели реакцию при комнатной температуре 30 минут.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (30 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/1). Получали 47 мг 5-(2-ацетил-5-хлорфенил)-6-изопропилпиридазин-3(2H)-она в виде желтого твердого вещества (выход: 50.0%). LCMS: RT = 3.55 мин, [M+H]+ = 307.08.
Стадия D: Синтез трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-изопропокси- 6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата
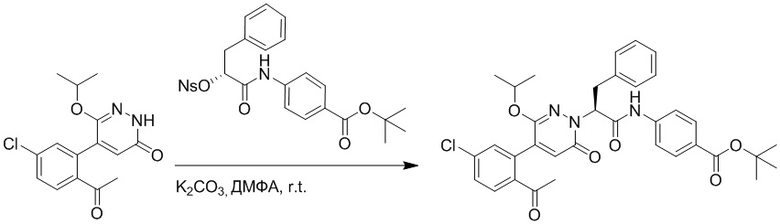
5-(2-Ацетил-5-хлорфенил)-6-изопропилпиридазин-3(2H)-он (47 мг, 0.15 ммоль), трет-бутил (R)-4-(2-(((4-нитрофенил)сульфонил)окси)-3-фенилпропанамидо)бензоат (89 мг, 0.17 ммоль) и карбонат калия (43 мг, 0.31 ммоль) добавляли в N,N-диметилформамид (2.0 мл) при комнатной температуре, и реакцию вели при комнатной температуре в течение ночи.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 80 мг трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-изопропокси-6-оксопиридазин-1(6H)-ил)- 3-фенилпропанамидо)бензоата в виде бледно-желтого твердого вещества (выход: 86.7%). LCMS: RT = 4.85 мин, [M+H]+ = 630.15.
Стадия E: Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-изопропокси- 6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
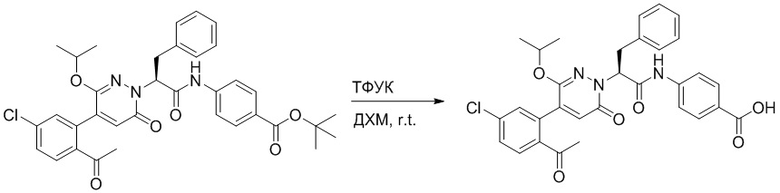
Трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-изопропокси-6-оксопиридазин- 1(6H)-ил)-3-фенилпропанамидо)бензоат (80 мг, 0.13 ммоль) добавляли в дихлорметан (2.0 мл) при комнатной температуре и добавляли по каплям трифторуксусную кислоту (0.25 мл). Реакцию вели при комнатной температуре 3 часа.
После окончания реакции, дихлорметан упаривали досуха и отгоняли трифторуксусную кислоту на масляном насосе. Полученный остаток растворяли в дихлорметане (1.0 мл), и раствор добавляли по каплям в н-гексан (10.0 мл), при этом выпадало в осадок белое твердое вещество, которое отфильтровывали при отсасывании. Осадок на фильтре промывали н-гексаном и сушили, получая 40 мг белой твердой (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-изопропокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)бензойной кислоты (выход: 53.6%). LCMS: RT = 4.16 мин, [M-H]- = 572.07. 1H ЯМР (400 МГц, ДМСО) δ 12.72 (с, 1H), 10.50 (с, 1H), 8.01 (д, J = 8.4 Гц, 1H), 7.90 (д, J = 8.7 Гц, 2H), 7.72 (д, J = 8.8 Гц, 2H), 7.69 (дд, J = 8.4, 2.2 Гц, 1H), 7.47 (д, J = 2.1 Гц, 1H), 7.28 (д, J = 4.4 Гц, 4H), 7.19 (дт, J = 8.7, 4.4 Гц, 1H), 6.88 (с, 1H), 5.77 (дд, J = 8.7, 6.2 Гц, 1H), 4.85 (дт, J = 12.4, 6.1 Гц, 1H), 3.43 (дд, J = 7.4, 3.2 Гц, 2H), 2.55 (с, 3H), 1.10 (дд, J = 9.9, 6.2 Гц, 6H).
Пример 23
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксо-3-(2,2,2-трифторэтокси) пиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
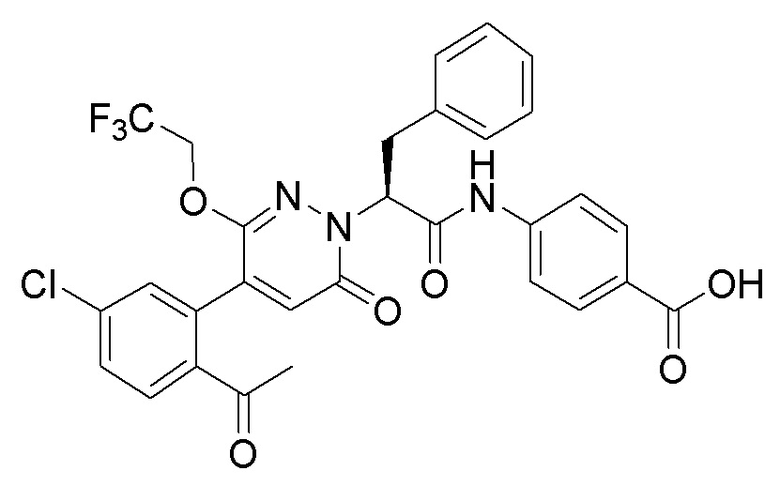
Использовался следующий путь синтеза.
Стадия A: Синтез 5-бром-2-(4-метоксибензил)-6-(2,2,2-трифторэтокси) пиридазин-3(2H)-она
5-Бром-6-гидрокси-2-(4-метоксибензил)пиридазин-3(2H)-он (350 мг, 1.13 ммоль) и карбонат калия (535 мг, 3.88 ммоль) добавляли в N,N-диметилформамид (5.0 мл) при комнатной температуре. Реакционную смесь перемешивали при 80°C в течение 15 минут. При этой же температуре добавляли 2,2,2-трифторэтил трифторметансульфонат (0.6 мл), и реакцию продолжали 30 минут.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (50 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 2/5). Получали 123 мг белого твердого 5-бром-2-(4-метоксибензил)-6-(2,2,2-трифторэтокси)пиридазин-3(2H)-она (выход: 27.4%). LCMS: RT = 4.04 мин, [M+H]+ = 392.97.
Стадия B: Синтез 5-(2-ацетил-5-хлорфенил)-2-(4-метоксибензил)-6-(2,2,2- трифторэтокси)пиридазин-3(2H)-она

5-Бром-2-(4-метоксибензил)-6-(2,2,2-трифторэтокси)пиридазин-3(2H)-он (123 мг, 0.31 ммоль), пинаколовый эфир 6-ацетил-3-хлорфенилбороновой кислоты (88 мг, 0.32 ммоль) и карбонат натрия (66 мг, 0.62 ммоль) добавляли в трехгорлую колбу при комнатной температуре и продували колбу азотом. Добавляли смесь растворителей (10 мл, 1,2-диметоксиэтан:этанол:вода = 8:1:1) и продували колбу азотом. Добавляли 1,1'-бисдифенилфосфиноферроцен палладий дихлорид (22 мг, 0.03 ммоль) и продували колбу азотом. Смесь нагревали до 90°C и вели реакцию в течение 1 часа.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (50 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/3). Получали 64 мг желтого твердого 5-(2-ацетил-5-хлорфенил)-2-(4-метоксибензил)-6-(2,2,2-трифторэтокси) пиридазин-3(2H)-она (выход: 45.2%). LCMS: RT = 4.15 мин, [M+H]+ = 467.09.
Стадия C: Синтез 5-(2-ацетил-5-хлорфенил)-6-(2,2,2-трифторэтокси)пиридазин- 3(2H)-она
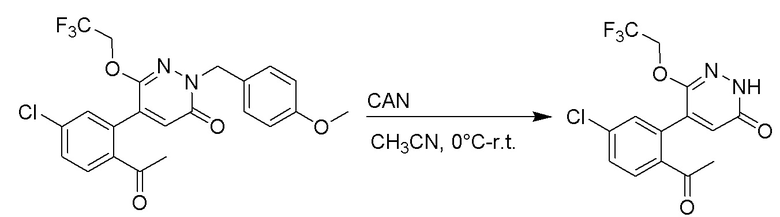
5-(2-Ацетил-5-хлорфенил)-2-(4-метоксибензил)-6-(2,2,2-трифторэтокси)пиридазин- 3(2H)-он (64 мг, 0.14 ммоль) добавляли в смесь растворителей (4 мл, ацетонитрил: вода = 3:1) при 0°C, и затем медленно добавляли церийаммоний нитрат (751 мг, 1.37 ммоль). После окончания добавления вели реакцию при комнатной температуре 30 минут.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (30 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/1). Получали 30 мг 5-(2-ацетил-5-хлорфенил)-6-(2,2,2-трифторэтокси)пиридазин-3(2H)-она в виде желтого твердого вещества (выход: 64.3%). LCMS: RT = 3.57 мин, [M+H]+ = 347.04.
Стадия D: Синтез трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксо-3-(2,2,2- трифторэтокси)пиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата
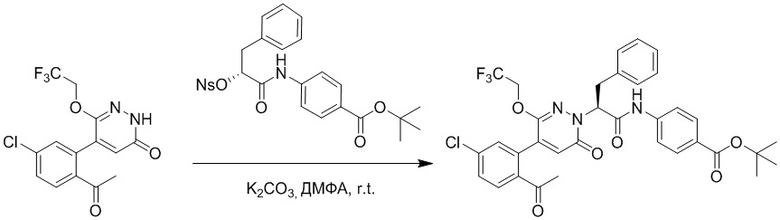
5-(2-Ацетил-5-хлорфенил)-6-(2,2,2-трифторэтокси)пиридазин-3(2H)-он (30 мг, 0.09 ммоль), трет-бутил (R)-4-(2-(((4-нитрофенил)сульфонил)окси)-3-фенилпропанамидо) бензоат (69 мг, 0.13 ммоль) и карбонат калия (24 мг, 0.18 ммоль) добавляли в N,N-диметилформамид (2.0 мл) при комнатной температуре, и реакцию вели при комнатной температуре в течение ночи.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 80 мг трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксо-3-(2,2,2-трифторэтокси)пиридазин- 1(6H)-ил)-3-фенилпропанамидо)бензоата в виде бледно-желтого твердого вещества (выход: 55.6%). LCMS: RT = 4.60 мин, [M+H]+ = 670.18.
Стадия E: Синтез (S)-4-2-4-(2-ацетил-5-хлорфенил)-6-оксо-3-(2,2,2- трифторэтокси)пиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
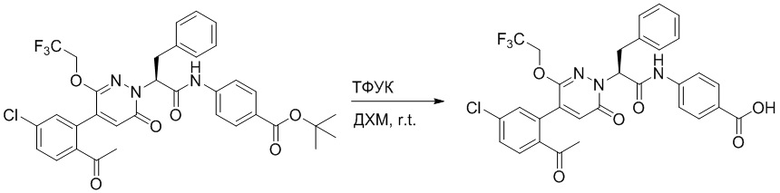
Трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксо-3-(2,2,2-трифторэтокси) пиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоат (30 мг, 0.05 ммоль) добавляли в дихлорметан (2.0 мл) при комнатной температуре, и добавляли по каплям трифторуксусную кислоту (0.25 мл). Реакцию вели при комнатной температуре 3 часа.
После окончания реакции, дихлорметан упаривали досуха и трифторуксусную кислоту отгоняли на масляном насосе. Полученный остаток растворяли в дихлорметане (1.0 мл), и раствор добавляли по каплям в н-гексан (10.0 мл), при этом выпадало в осадок белое твердое вещество, которое отфильтровывали при отсасывании. Осадок на фильтре промывали н-гексаном и сушили, получая 14 мг белой твердой (S)-4-2-4-(2-ацетил-5-хлорфенил)-6-оксо-3-(2,2,2-трифторэтокси)пиридазин-1(6H)-ил)-3- фенилпропанамидо)бензойной кислоты (выход: 45.7%). LCMS: RT = 4.08 мин, [M-H]- = 612.06. 1H ЯМР (400 МГц, ДМСО) δ 12.71 (с, 1H), 10.53 (с, 1H), 8.10 (д, J = 8.4 Гц, 1H), 7.91 (д, J = 8.8 Гц, 2H), 7.78 – 7.67 (м, 3H), 7.52 (д, J = 2.1 Гц, 1H), 7.37 – 7.24 (м, 4H), 7.19 (т, J = 6.3 Гц, 1H), 6.95 (с, 1H), 5.75 (дд, J = 9.8, 5.3 Гц, 1H), 4.70 (кв, J = 8.8 Гц, 2H), 3.55 (дд, J = 14.2, 10.2 Гц, 1H), 3.42 (дд, J = 14.2, 4.9 Гц, 1H), 2.53 (с, 3H).
Пример 24
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксо-3-(оксетан-3-илокси)- пиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
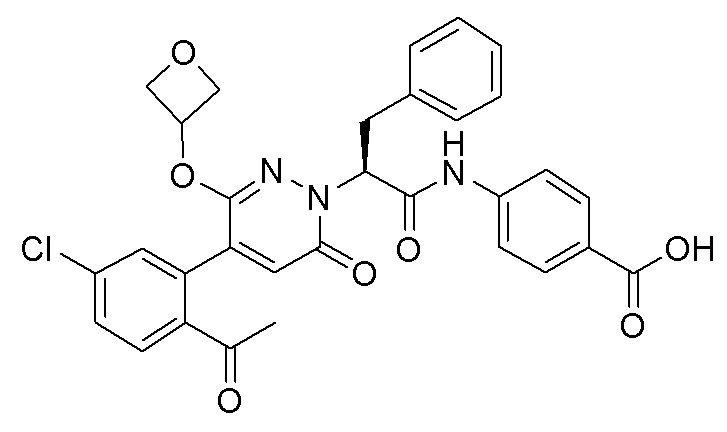
Использовался следующий путь синтеза.
Стадия A: Синтез 5-бром-2-(4-метоксибензил)-6-(оксетан-3-илокси)пиридазин- 3(2H)-она
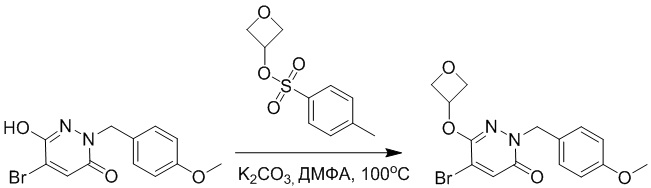
5-Бром-6-гидрокси-2-(4-метоксибензил)пиридазин-3(2H)-он (500 мг, 1.61 ммоль) и карбонат калия (890 мг, 6.45 ммоль) добавляли в N,N-диметилформамид (5.0 мл) при комнатной температуре. Реакционную смесь перемешивали при 100°C в течение 15 минут. При этой же температуре добавляли 3-п-тозилоксиоксетан (1.47 г, 6.44 ммоль), и реакцию продолжали в течение ночи.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (50 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 330 мг белого твердого 5-бром-2-(4-метоксибензил)-6-(оксетан-3-илокси)пиридазин-3(2H)-она (выход: 55.9%). LCMS: RT = 3.62 мин, [M+H]+ = 367.03.
Стадия B: Синтез 5-(2-ацетил-5-хлорфенил)-2-(4-метоксибензил)-6-(оксетан-3- илокси)пиридазин-3(2H)-она
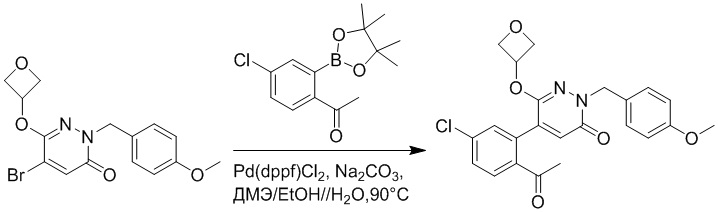
5-Бром-2-(4-метоксибензил)-6-(оксетан-3-илокси)пиридазин-3(2H)-он (330 мг, 0.90 ммоль), пинаколовый эфир 6-ацетил-3-хлорфенилбороновой кислоты (252 мг, 0.90 ммоль) и карбонат натрия (191 мг, 1.80 ммоль) помещали в трехгорлую колбу при комнатной температуре и продували колбу азотом. Добавляли смесь растворителей (10 мл, 1,2-диметоксиэтан:этанол:вода = 8:1:1) и продували колбу азотом. Добавляли 1,1'-бисдифенилфосфиноферроцен палладий дихлорид (66 мг, 0.09 ммоль) и продували колбу азотом. Смесь нагревали до 90°C и вели реакцию в течение 1 часа.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (50 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/3). Получали 80 мг 5-(2-ацетил-5-хлорфенил)-2-(4-метоксибензил)-6-(оксетан-3-илокси)пиридазин-3(2H)-она в виде желтого твердого вещества (выход: 20.0%). LCMS: RT = 3.82 мин, [M+H]+ = 441.08.
Стадия C: Синтез 5-(2-ацетил-5-хлорфенил)-6-(оксетан-3-илокси)пиридазин- 3(2H)-она

5-(2-Ацетил-5-хлорфенил)-2-(4-метоксибензил)-6-(оксетан-3-илокси)пиридазин- 3(2H)-он (80 мг, 0.18 ммоль) добавляли в смесь растворителей (4 мл, ацетонитрил: вода = 3:1) при 0°C, и затем медленно добавляли церийаммоний нитрат (790 мг, 1.44 ммоль). После окончания добавления вели реакцию при комнатной температуре 30 минут.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (30 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/1). Получали 30 мг 5-(2-ацетил-5-хлорфенил)-6-(оксетан-3-илокси)пиридазин-3(2H)-она в виде желтого твердого вещества (выход: 50.0%). LCMS: RT = 3.09 мин, [M+H]+ = 321.07.
Стадия D: Синтез трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксо-3- (оксетан-3-илокси)-пиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата
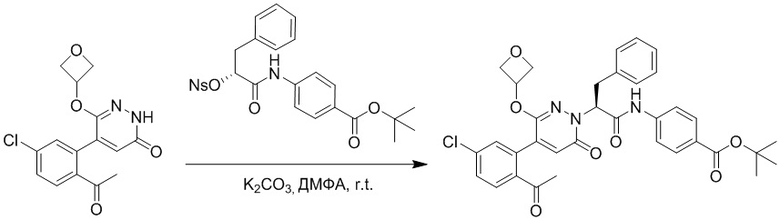
5-(2-Ацетил-5-хлорфенил)-6-(оксетан-3-илокси)пиридазин-3(2H)-он (30 мг, 0.09 ммоль), трет-бутил (R)-4-(2-(((4-нитрофенил)сульфонил)окси)-3-фенилпропанамидо) бензоат (69 мг, 0.13 ммоль) и карбонат калия (24 мг, 0.18 ммоль) добавляли в N,N-диметилформамид (2.0 мл) при комнатной температуре, и реакцию вели при комнатной температуре в течение ночи.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 80 мг трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксо-3-(оксетан-3-илокси)-пиридазин- 1(6H)-ил)-3-фенилпропанамидо)бензоата в виде бледно-желтого твердого вещества (выход: 77.8%). LCMS: RT = 4.38 мин, [M+H]+ = 644.16.
Стадия E: Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксо-3-(оксетан-3- илокси)-пиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
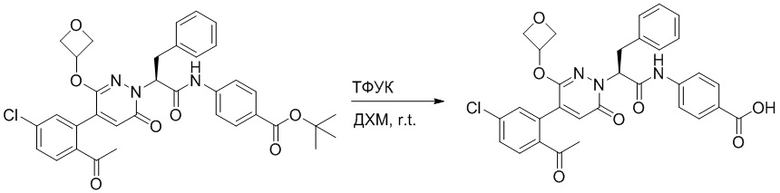
Трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксо-3-(оксетан-3-илокси)- пиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоат (45 мг, 0.07 ммоль) добавляли в дихлорметан (2.0 мл) при комнатной температуре, и добавляли по каплям трифторуксусную кислоту (0.25 мл). Реакцию вели при комнатной температуре 3 часа.
После окончания реакции, дихлорметан упаривали досуха и трифторуксусную кислоту отгоняли на масляном насосе. Полученный остаток растворяли в дихлорметане (1.0 мл), и раствор добавляли по каплям в н-гексан (10.0 мл), при этом выпадало в осадок белое твердое вещество, которое отфильтровывали при отсасывании. Осадок на фильтре промывали н-гексаном и сушили, получая 11 мг белой твердой (S)-4-(2-(4-(2-ацетил-5- хлорфенил)-6-оксо-3-(оксетан-3-илокси)-пиридазин-1(6H)-ил)-3-фенилпропанамидо) бензойной кислоты (выход: 26.7%). LCMS: RT = 3.82 мин, [M-H]- = 586.07. 1H ЯМР (400 МГц, ДМСО) δ 12.70 (с, 1H), 10.48 (с, 1H), 8.05 (д, J = 8.4 Гц, 1H), 7.90 (д, J = 8.7 Гц, 2H), 7.76 – 7.70 (м, 2H), 7.55 (д, J = 2.0 Гц, 1H), 7.31 – 7.23 (м, 4H), 7.23 – 7.16 (м, 1H), 6.98 (с, 1H), 5.73 (дд, J = 8.9, 6.3 Гц, 1H), 5.25 – 5.21 (м, 1H), 4.72 (т, J = 6.6 Гц, 2H), 4.30 (дд, J = 7.4, 5.2 Гц, 2H), 3.41 – 3.35 (м, 2H), 2.60 (с, 3H).
Пример 25
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(дифторметокси)-6-оксопиридазин- 1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
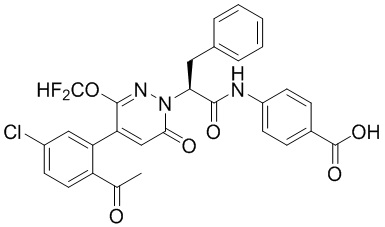
Использовался следующий путь синтеза.
Стадия A: Синтез 5-бром-6-(дифторметокси)-2-(4-метоксибензил)пиридазин- 3(2H)-она
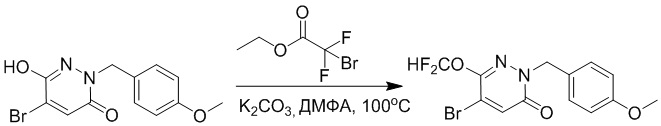
5-Бром-6-гидрокси-2-(4-метоксибензил)пиридазин-3(2H)-он (600 мг, 1.93 ммоль) и карбонат калия (1.07 г, 7.72 ммоль) добавляли в N,N-диметилформамид (5.0 мл) при комнатной температуре. Реакционную смесь перемешивали при 100°C в течение 15 минут. При этой же температуре добавляли этил дифторбромацетат (1.0 мл), и реакцию продолжали в течение 1 часа.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (50 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 270 мг белого твердого, 5-бром-6-(дифторметокси)-2-(4-метоксибензил)пиридазин-3(2H)-она (выход: 38.9%). LCMS: RT = 3.92 мин, [M+H]+ = 360.97.
Стадия B: Синтез 5-(2-ацетил-5-хлорфенил)-6-(дифторметокси)-2-(4- метоксибензил)пиридазин-3(2H)-она
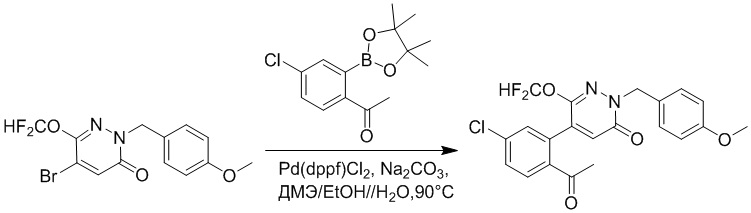
5-Бром-6-(дифторметокси)-2-(4-метоксибензил)пиридазин-3(2H)-он (270 мг, 0.75 ммоль), пинаколовый эфир 6-ацетил-3-хлорфенилбороновой кислоты (210 мг, 0.75 ммоль) и карбонат натрия (160 мг, 1.50 ммоль) добавляли в трехгорлую колбу при комнатной температуре и продували колбу азотом. Добавляли смесь растворителей (10 мл, 1,2-диметоксиэтан:этанол:вода = 8:1:1) и продували колбу азотом. Добавляли 1,1’-бисдифенилфосфиноферроцен палладий дихлорид (59 мг, 0.08 ммоль) и продували колбу азотом. Смесь нагревали до 90°C и вели реакцию в течение 1 часа.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (50 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/3). Получали 110 мг 5-(2-ацетил-5-хлорфенил)-6-(дифторметокси)-2-(4-метоксибензил)пиридазин-3(2H)-она в виде желтого твердого вещества (выход: 33.3%). LCMS: RT = 4.08 мин, [M+H]+ = 435.07.
Стадия C: Синтез 5-(2-ацетил-5-хлорфенил)-6-(дифторметокси)-пиридазин- 3(2H)-она
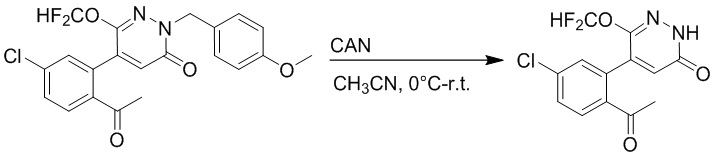
5-(2-Ацетил-5-хлорфенил)-6-(дифторметокси)-2-(4-метоксибензил)пиридазин-3(2H)-он (110 мг, 0.25 ммоль) добавляли в смесь растворителей (4 мл, ацетонитрил: вода = 3:1) при 0°C, и затем медленно добавляли церийаммоний нитрат (1.10 г, 2.00 ммоль). После окончания добавления вели реакцию при комнатной температуре 30 минут.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (30 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/1). Получали 50 мг 5-(2-ацетил-5-хлорфенил)-6-(дифторметокси)-пиридазин-3(2H)-она в виде желтого твердого вещества (выход: 64.0%). LCMS: RT = 3.47 мин, [M+H]+ = 315.02.
Стадия D: Синтез трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3- (дифторметокси)-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата
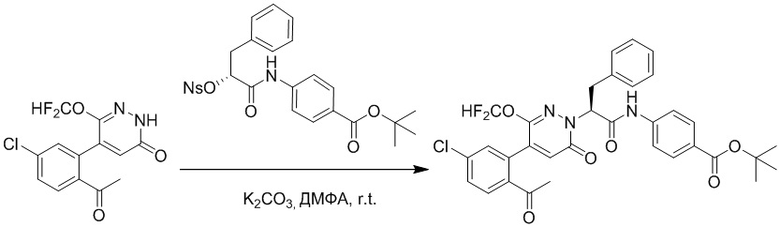
Твердый 5-(2-ацетил-5-хлорфенил)-6-(дифторметокси)пиридазин-3(2H)-он (50 мг, 0.16 ммоль), трет-бутил (R)-4-(2-(((4-нитрофенил)сульфонил)окси)-3-фенилпропанамидо) бензоат (92 мг, 0.18 ммоль) и карбонат калия (44 мг, 0.32 ммоль) добавляли в N,N-диметилформамид (2.0 мл) при комнатной температуре, и реакцию вели при комнатной температуре в течение ночи.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 55 мг трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(дифторметокси)-6-оксопиридазин- 1(6H)-ил)-3-фенилпропанамидо)бензоата в виде бледно-желтого твердого вещества (выход: 56.3%). LCMS: RT = 4.54 мин, [M+H]+ = 638.14.
Стадия E: Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксо-3-(оксетан-3- илокси)-пиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
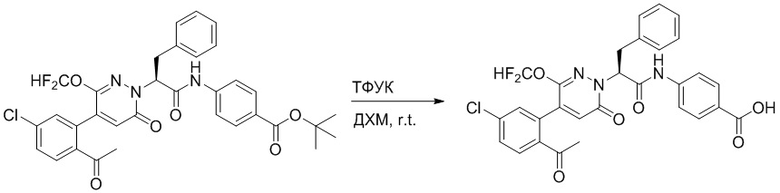
Трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(дифторметокси)-6-оксопиридазин- 1(6H)-ил)-3-фенилпропанамидо)бензоат (55 мг, 0.09 ммоль) добавляли в дихлорметан (2.0 мл) при комнатной температуре, и добавляли трифторуксусную кислоту (0.25 мл) по каплям. Реакцию вели при комнатной температуре 3 часа.
После окончания реакции, дихлорметан упаривали досуха и трифторуксусную кислоту отгоняли на масляном насосе. Полученный остаток растворяли в дихлорметане (1.0 мл), и раствор добавляли по каплям в н-гексан (10.0 мл), при этом выпадало в осадок белое твердое вещество, которое отфильтровывали при отсасывании. Осадок на фильтре промывали н-гексаном и сушили, получая 20 мг белой твердой (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксо-3-(оксетан-3-илокси)-пиридазин-1(6H)-ил)-3- фенилпропанамидо)бензойной кислоты (выход: 38.3%). LCMS: RT = 4.04 мин, [M-H]- = 580.04. 1H ЯМР (400 МГц, ДМСО) δ 12.74 (с, 1H), 10.59 (с, 1H), 8.12 (д, J = 8.4 Гц, 1H), 7.91 (д, J = 8.7 Гц, 2H), 7.76 (дд, J = 8.4, 2.2 Гц, 1H), 7.72 (д, J = 8.7 Гц, 2H), 7.57 (д, J = 1.9 Гц, 1H), 7.36 (т, J = 62.5 Гц, 1H), 7.35 – 7.25 (м, 4H), 7.19 (дд, J = 12.2, 5.1 Гц, 1H), 7.03 (с, 1H), 5.75 (дд, J = 10.2, 5.0 Гц, 1H), 3.52 (дд, J = 14.1, 10.5 Гц, 1H), 3.41 (дд, J = 14.3, 4.8 Гц, 1H), 2.57 (с, 3H).
Пример 26
Синтез (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенил-N-(хиноксалин-6-ил)пропанамида

Использовался следующий путь синтеза.
Стадия A: Метил (R)-3-фенил-2-(((трифторметил)сульфонил)окси)бутират
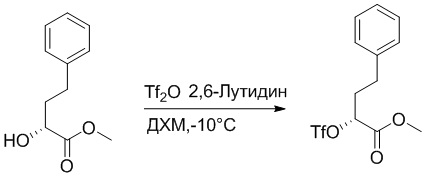
Метил (R)-2-гидрокси-3-фенилбутират (3.00 г, 14.41 ммоль) растворяли в дихлорметане (25.0 мл), добавляли 2,6-диметилпиридин (2.0 мл) и затем медленно добавляли ангидрид трифторметансульфокислоты (3.0 мл) при -10°C. Реакционную смесь перемешивали 30 минут.
В реакционный раствор добавляли воду (30 мл), чтобы погасить реакцию. Этилацетат (100 мл) добавляли в реакционный раствор. Полученный смешанный раствор промывали насыщенным водным раствором хлорида натрия (30 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 20/1). Получали 2.0 г метил (R)-3-фенил-2- (((трифторметил)сульфонил)окси)бутирата в виде бесцветного масла (выход: 42.4%). LCMS: RT = 4.43 мин.
Стадия B: Синтез метил (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилбутирата
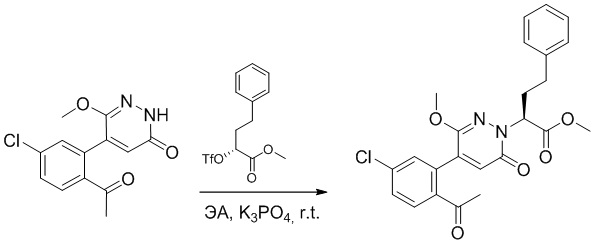
5-(2-Ацетил-5-хлорфенил)-6-метоксипиридазин-3(2H)-он (200 мг, 0.50 ммоль) и фосфат калия (425 мг, 2.00 ммоль) растворяли в этилацетате (6.0 мл). После этого добавляли в полученный раствор метил (R)-3-фенил-2-(((трифторметил)сульфонил)окси) бутират (204 мг, 0.60 ммоль), и раствор перемешивали при комнатной температуре в течение 3 часов.
Воду (1 мл) добавляли в реакционный раствор, чтобы погасить реакцию. Добавляли этилацетат (50 мл), и смешанный раствор промывали насыщенным водным раствором хлорида натрия (10 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/1). Получали 110 мг метил (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилбутирата в виде желтого твердого вещества (выход: 46.0%). LCMS: RT = 4.29 мин, [M+H]+ = 469.06.
Стадия C: Синтез (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилмасляной кислоты
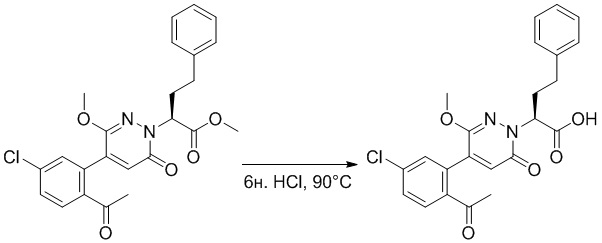
Метил (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)- 3-фенилбутират (110 мг, 0.23 ммоль) растворяли в 6н. HCl (4.0 мл). Реакцию вели при 90°C в течение ночи.
После окончания реакции раствор охлаждали до -10°C, и значение pH доводили до 3-4 добавлением 6н. раствора NaOH. Добавляли этилацетат (30 мл), и смешанный раствор промывали насыщенным водным раствором хлорида натрия (10 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Получали 100 мг (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)- ил)-3-фенилмасляной кислоты в виде желтого твердого вещества. Полученный продукт напрямую использовали в следующей стадии без дополнительной очистки. LCMS: RT = 3.97 мин, [M+H]+ = 441.10.
Стадия D: Синтез трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-4-фенилбутанамидо)бензоата
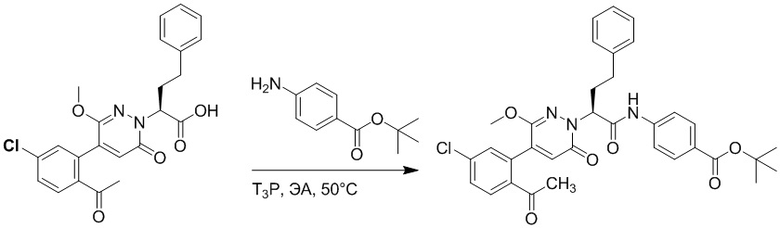
(S)-2-(4-(2-Ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилмасляную кислоту (100 мг, 0.23 ммоль) и трет-бутил 4-амино-бензоат (53 мг, 0.27 ммоль) растворяли в этилацетате (3.0 мл) при комнатной температуре и добавляли N,N-диизопропилэтиламин (0.1 мл). После этого добавляли в полученный раствор 1-пропилфосфорный ангидрид (0.5 мл). Реакционный раствор нагревали до 50°C и перемешивали 4 часа.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/3). Получали 100 мг трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-4- фенилбутанамидо)бензоата в виде бледно-желтого твердого вещества (выход: 69.6%). LCMS: RT = 4.64 мин, [M+H]+ = 616.20.
Стадия E: Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-4-фенилбутанамидо)бензойной кислоты
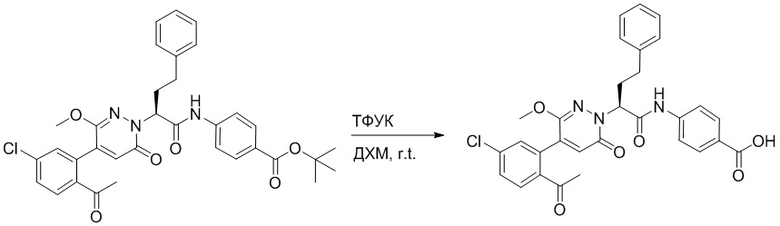
трет-Бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)- 4-фенилбутанамидо)бензоат (100 мг, 0.16 ммоль) добавляли в дихлорметан (2.0 мл) при комнатной температуре, и добавляли по каплям трифторуксусную кислоту (0.25 мл). Реакцию вели при комнатной температуре 3 часа.
После окончания реакции, дихлорметан упаривали досуха и трифторуксусную кислоту отгоняли на масляном насосе. Полученный остаток растворяли в дихлорметане (1.0 мл), и раствор добавляли по каплям в н-гексан (10.0 мл), при этом выпадало в осадок белое твердое вещество, которое отфильтровывали при отсасывании. Осадок на фильтре промывали н-гексаном и сушили, получая 70 мг белой твердой (S)-4-(2-(4-(2-ацетил- 5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-4-фенилбутанамидо)бензойной кислоты (выход: 78.3%). LCMS: RT = 4.05 мин, [M-H]- = 558.06. 1H ЯМР (500 МГц, ДМСО) δ 12.72 (с, 1H), 10.49 (с, 1H), 8.03 (д, J = 8.4 Гц, 1H), 7.90 (д, J = 8.8 Гц, 2H), 7.76 – 7.68 (м, 3H), 7.58 (д, J = 2.1 Гц, 1H), 7.33 – 7.21 (м, 4H), 7.19 (т, J = 7.2 Гц, 1H), 7.00 (с, 1H), 5.38 (дд, J = 10.2, 4.4 Гц, 1H), 3.66 (с, 3H), 2.67 (т, J = 7.8 Гц, 2H), 2.57 (с, 3H), 2.45 – 2.14 (м, 2H).
Пример 27
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-фенил пропанамидо)-2-хлорбензойной кислоты
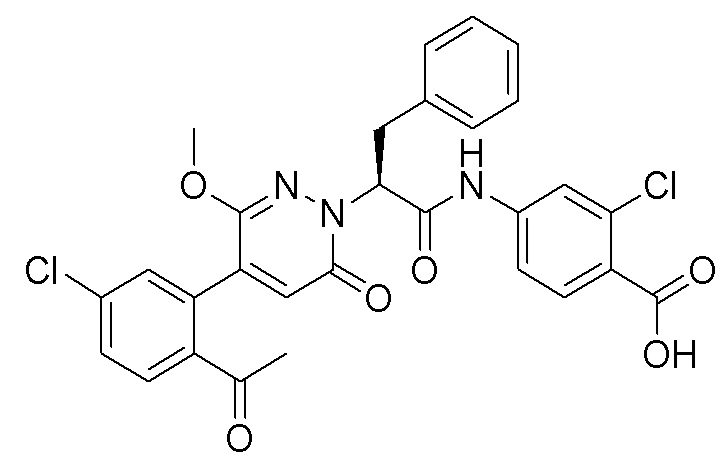
Использовался следующий путь синтеза.
Стадия A: Синтез метил (S)-4-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)-2-хлорбензоата
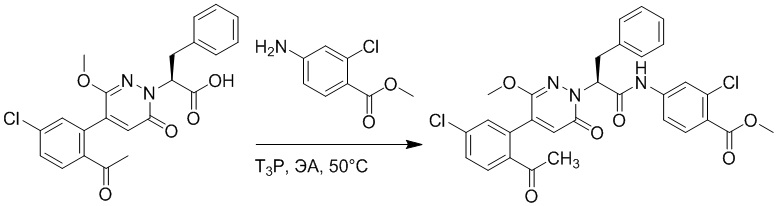
(S)-2-(4-(2-Ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропановую кислоту (40 мг, 0.09 ммоль), метил 2-хлор-4-амино-бензоат (21 мг, 0.11 ммоль) растворяли в этилацетате (3.0 мл) при комнатной температуре и добавляли N,N-диизопропилэтиламин (0.05 мл). После этого добавляли в полученный раствор 1-пропилфосфорный ангидрид (0.2 мл). Реакционный раствор нагревали до 50°C и перемешивали 4 часа.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 35 мг метил (S)-4-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)-2-хлорбензоата в виде бледно-желтого твердого вещества (выход: 66.7%). LCMS: RT = 4.31 мин, [M+H]+ = 594.05.
Стадия B: Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенил пропанамидо)-2-хлорбензойной кислоты
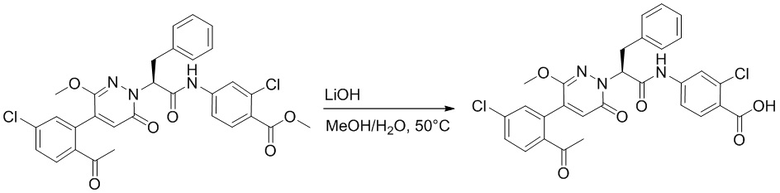
Метил (S)-4-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)-2-хлорбензоат (35 мг, 0.06 ммоль) растворяли в метаноле (2.0 мл). После этого добавляли в полученный раствор водный раствор (1.0 мл) моногидрата гидроксида лития (5 мг, 0.11 ммоль). Реакционную смесь перемешивали при 50°C в течение ночи.
Разбавленный раствор соляной кислоты (1.0 моль/л) медленно добавляли по каплям в реакционный раствор, доводя значение pH до 3-4. Добавляли этилацетат (50 мл), и смешанный раствор промывали насыщенным водным раствором хлорида натрия (10 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 2/1). Получали 6 мг белой твердой (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)-2-хлорбензойной кислоты (выход: 17.3%). LCMS: RT = 4.08 мин, [M-H]- = 578.04.
Пример 28
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)- 3-фенилпропанамидо)-2-метоксибензойной кислоты
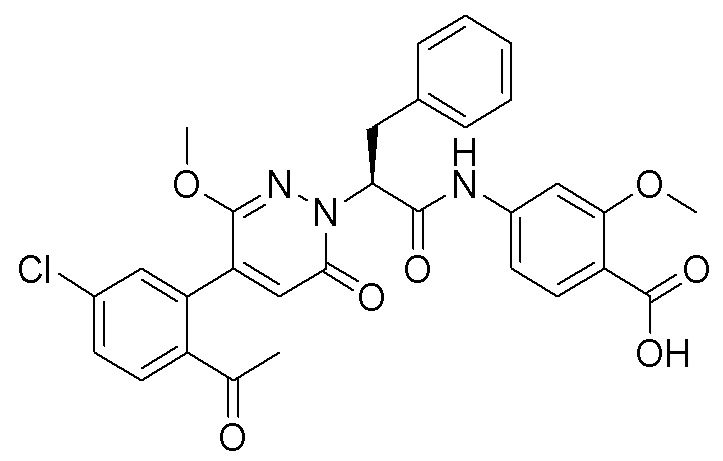
Использовался следующий путь синтеза.
Стадия A: Синтез метил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)-2-метоксибензоата
(S)-2-(4-(2-Ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропановую кислоту (40 мг, 0.09 ммоль), метил 2-метокси-4-амино-бензоат (22 мг, 0.12 ммоль) растворяли в этилацетате (3.0 мл) при комнатной температуре и добавляли N,N-диизопропилэтиламин (0.05 мл). После этого добавляли в полученный раствор 1-пропилфосфорный ангидрид (0.2 мл). Реакционный раствор нагревали до 50°C и перемешивали 4 часа.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 50 мг метил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)-2-метоксибензоата в виде бледно-желтого твердого вещества (выход: 94.2%). LCMS: RT = 4.09 мин, [M+H]+ = 590.11.
Стадия B: Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)-2-метоксибензойной кислоты

Метил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)-2-метоксибензоат (50 мг, 0.08 ммоль) растворяли в метаноле (2.0 мл). После этого в полученный раствор добавляли водный раствор (1.0 мл) моногидрата гидроксида лития (8.4 мг, 0.16 ммоль). Реакционную смесь перемешивали при 50°C в течение 4 часов.
Разбавленный раствор соляной кислоты (1.0 моль/л) медленно добавляли по каплям в реакционный раствор, доводя значение pH до 3-4. Добавляли этилацетат (50 мл), и смешанный раствор промывали насыщенным водным раствором хлорида натрия (10 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 2/1). Получали 6 мг белой твердой (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)-2-метоксибензойной кислоты (выход: 13.0%). LCMS: RT = 3.93 мин, [M-H]- = 574.09.
Пример 29
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)- 3-фенилпропанамидо)-2-гидроксибензойной кислоты
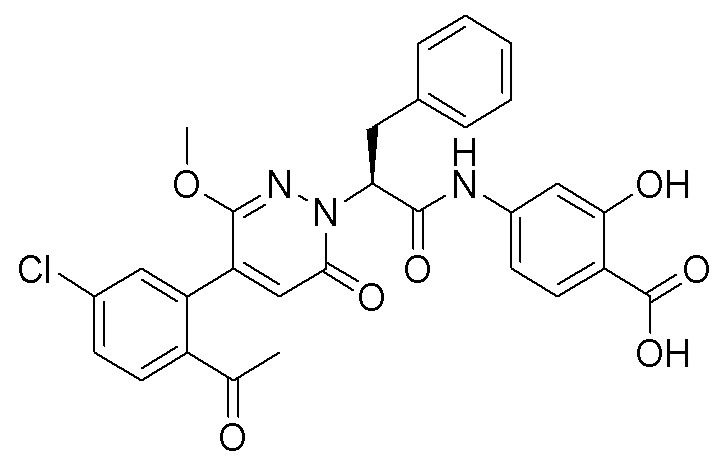
Использовался следующий путь синтеза.
Стадия A: Синтез метил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)-2-гидроксибензоата
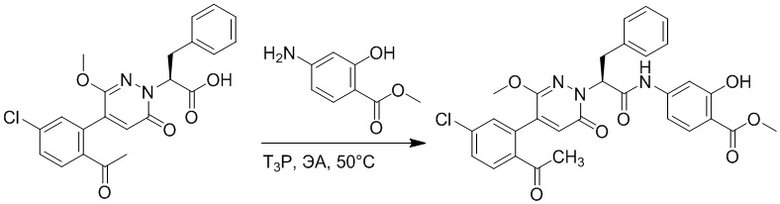
(S)-2-(4-(2-Ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропановую кислоту (40 мг, 0.09 ммоль), метил 2-гидрокси-4-амино-бензоат (19 мг, 0.12 ммоль) растворяли в этилацетате (3.0 мл) при комнатной температуре и добавляли N,N-диизопропилэтиламин (0.05 мл). После этого добавляли в полученный раствор 1-пропилфосфорный ангидрид (0.2 мл). Реакционный раствор нагревали до 50°C и перемешивали 4 часа.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 6 мг метил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)-2-гидроксибензоата в виде бледно-желтого твердого вещества (выход: 55.6%). LCMS: RT = 4.27 мин, [M-H]- = 576.23.
Стадия B: Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)-2-гидроксибензойной кислоты
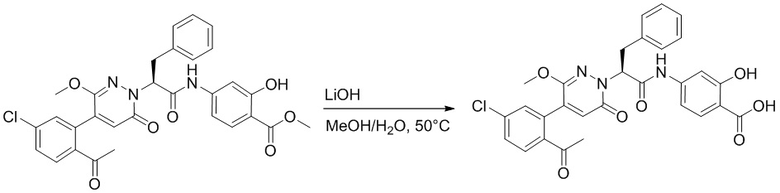
Метил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)-2-гидроксибензоат (30 мг, 0.05 ммоль) растворяли в метаноле (2.0 мл). После этого добавляли в полученный раствор водный раствор (1.0 мл) моногидрата гидроксида лития (5 мг, 0.11 ммоль). Реакционную смесь перемешивали при 50°C в течение 4 часов.
Разбавленный раствор соляной кислоты (1.0 моль/л) медленно добавляли по каплям в реакционный раствор, доводя значение pH до 3-4. Добавляли этилацетат (50 мл), и смешанный раствор промывали насыщенным водным раствором хлорида натрия (10 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 2/1). Получали 5 мг белой твердой (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)-2-гидроксибензойной кислоты (выход: 17.9%). LCMS: RT = 4.36 мин, [M-H]- = 560.07.
Пример 30
Синтез (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-N- (1-оксо-1,2-дигидроизохинолин-6-ил)-3-фенилпропанамида
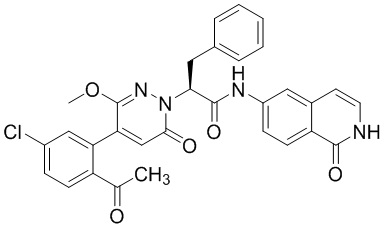
Использовался следующий путь синтеза.
Стадия A: Синтез (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-N-(1-оксо-1,2-дигидроизохинолин-6-ил)-3-фенилпропанамида
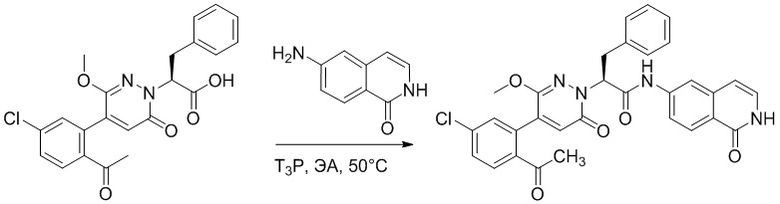
(S)-2-(4-(2-Ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропановую кислоту (40 мг, 0.09 ммоль), 6-аминоизохинолин-1(2H)-он (18 мг, 0.12 ммоль) растворяли в этилацетате (3.0 мл) при комнатной температуре и добавляли N,N-диизопропилэтиламин (0.05 мл). После этого добавляли в полученный раствор 1-пропилфосфорный ангидрид (0.2 мл). Реакционный раствор нагревали до 50°C и перемешивали 4 часа.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 10 мг (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-N-(1-оксо-1,2- дигидроизохинолин-6-ил)-3-фенилпропанамида в виде бледно-желтого твердого вещества (выход: 19.6%). LCMS: RT = 3.80 мин, [M-H]- = 567.07. 1H ЯМР (400 МГц, ДМСО) δ 13.15 (с, 1H), 10.59 (с, 1H), 8.00 (д, J = 8.4 Гц, 1H), 7.89 (д, J = 1.9 Гц, 1H), 7.84 (д, J = 8.6 Гц, 1H), 7.69 (дд, J = 8.3, 2.1 Гц, 1H), 7.57 (дд, J = 8.6, 2.0 Гц, 1H), 7.50 (д, J = 2.1 Гц, 1H), 7.34 – 7.25 (м, 4H), 7.19 (т, J = 7.5 Гц, 1H), 6.91 (с, 1H), 5.70 (дд, J = 9.8, 5.1 Гц, 1H), 3.66 (с, 3H), 3.52 – 3.38 (м, 2H), 2.53 (с, 3H).
Пример 31
Синтез (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)- N-(2-оксо-1,2,3,4-тетрагидрохинолин-6-ил)-3-фенилпропанамида
Использовался следующий путь синтеза.
Стадия A: Синтез (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-N-(2-оксо-1,2,3,4-тетрагидрохинолин-6-ил)-3-фенилпропанамида
(S)-2-(4-(2-Ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропановую кислоту (40 мг, 0.09 ммоль), 6-амино-3,4-дигидро-2(1H)-хинолинон (18 мг, 0.12 ммоль) растворяли в этилацетате (3.0 мл) при комнатной температуре и добавляли N,N-диизопропилэтиламин (0.05 мл). После этого добавляли в полученный раствор 1-пропилфосфорный ангидрид (0.2 мл). Реакционный раствор нагревали до 50°C и перемешивали 4 часа.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: pure этилацетат). Получали 15 мг (S)-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-N-(2-оксо-1,2,3,4- тетрагидрохинолин-6-ил)-3-фенилпропанамида в виде бледно-желтого твердого вещества (выход: 29.2%). LCMS: RT = 3.79 мин, [M-H]- = 570.17. 1H ЯМР (400 МГц, ДМСО) δ 10.03 (с, 1H), 7.99 (д, J = 8.4 Гц, 1H), 7.69 (дд, J = 8.3, 2.2 Гц, 1H), 7.49 (д, J = 2.1 Гц, 1H), 7.45 (с, 1H), 7.33 – 7.25 (м, 5H), 7.18 (т, J = 6.7 Гц, 1H), 6.89 (с, 1H), 6.78 (д, J = 8.5 Гц, 1H), 5.69 (дд, J = 10.3, 4.8 Гц, 1H), 3.68 (с, 3H), 3.49 (дд, J = 13.9, 10.6 Гц, 1H), 3.38 (дд, J = 14.2, 4.7 Гц, 1H), 2.84 (т, J = 7.5 Гц, 2H), 2.53 (с, 3H), 2.45 – 2.39 (м, 2H).
Пример 32
Синтез (S)-4-(2-(4-(5-хлор-2-пропанилфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
Использовался следующий путь синтеза.
Стадия A: Синтез 1-(2-бром-4-хлорфенил)пропан-1-она
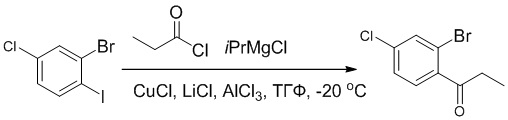
Согласно хорошо известной методике (Angewandte Chemie, International Edition, 2010, 49(46), 8729-8732), 2-бром-4-хлор-1-иодбензол (2.00 г, 6.30 ммоль) растворяли в 2 мл тетрагидрофурана и охлаждали до -20°C. Добавляли по каплям раствор изопропилмагний хлорида в н-гексане (2M концентрация) (4.1 мл, 8.2 ммоль), и смесь перемешивали при этой же температуре в течение 1 часа.
Пропанилхлорид (716 мкл, 8.20 ммоль), хлорид лития (23 мг, 378 мкмоль), хлорид меди (19 мг, 189 мкмоль) и трихлорид алюминия (25 мг, 189 мкмоль) добавляли в 2 мл тетрагидрофурана. Реакционную смесь перемешивали при комнатной температуре, и охлаждали на ледяной бане. Полученную реакционную смесь после прохождения реакции в течение 1 часа медленно добавляли по каплям в описанный выше раствор. Реакцию вели при комнатной температуре 2 часа. Реакцию гасили добавлением 40 мл насыщенного раствора хлорида аммония, экстрагировали дихлорметаном (40 мл × 3 раза), и органические фазы объединяли. Органическую фазу промывали насыщенным водным раствором хлорида натрия (50 мл), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 20/1). Получали 1.37 г 1-(2-бром-4-хлорфенил)пропан-1-она в виде бесцветной прозрачной жидкости (выход: 87.8%). LCMS: RT = 4.30 мин, нет пика молекулярного иона.
Стадия B: Синтез 1-(4-хлор-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2- ил)фенил)пропан-1-она
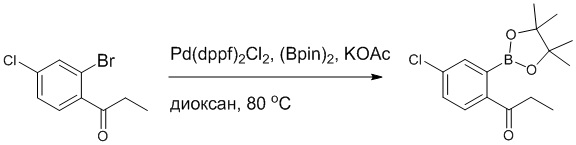
В раствор 1-(2-бром-4-хлорфенил)пропан-1-она (1.37 г, 5.53 ммоль), бис(пинаколато)диборона (2.83 г, 11.1 ммоль), ацетата калия (1.09 г, 11.1 ммоль) в 1,4-диоксане (21 мл) добавляли Pd(dppf)2Cl2·2CH2Cl2 (227 мг, 0.28 ммоль) в инертной атмосфере азота. Полученный смешанный раствор перемешивали при 80°C в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры и экстрагировали этилацетатом (50 мл × 2 раза). Органические фазы объединяли и последовательно промывали водой (50 мл) и насыщенным водным раствором хлорида натрия (50 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 10/1). Получали 1.0 г 1-(4-хлор-2-(4,4,5,5- тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропан-1-она в виде бледно-желтого твердого вещества (выход: 61.3%). LCMS: RT = 4.46 мин, [M+H]+ = 293.13.
Стадия C: Синтез 5-(5-хлор-2-пропанилфенил)-6-метокси-2-(4- метоксибензил)пиридазин-3(2H)-она
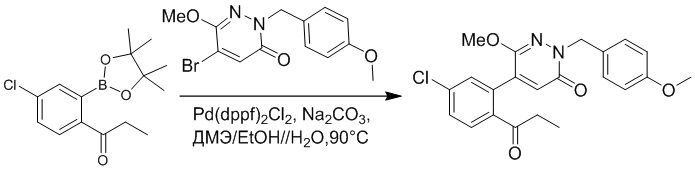
В раствор 1-(4-хлор-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропан- 1-она (268 мг, 0.91 ммоль), 5-бром-6-метокси-2-(4-метоксибензил)пиридазин-3(2H)-она (269 мг, 0.83 ммоль), карбоната натрия (176 мг, 1.66 ммоль) в диметиловом эфире этиленгликоля (5.6 мл) добавляли этанол (0.7 мл), воду (0.7 мл) и Pd(dppf)2Cl2 (30 мг, 0.040 ммоль) в инертной атмосфере азота. Полученный смешанный раствор перемешивали при 90°C в течение 1 часа. Реакционный раствор охлаждали до комнатной температуры и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли и последовательно промывали водой (40 мл) и насыщенным водным раствором хлорида натрия (40 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 10/1-2/1). Получали 321 мг 5-(5-хлор-2-пропанилфенил)-6-метокси-2-(4-метоксибензил)пиридазин- 3(2H)-она в виде светло-желтого маслянистого вещества (выход: 94.0%). LCMS: RT = 4.11 мин, [M+H]+ = 413.11.
Стадия D: Синтез 5-(5-хлор-2-пропанилфенил)-6-метоксипиридазин-3(2H)-она
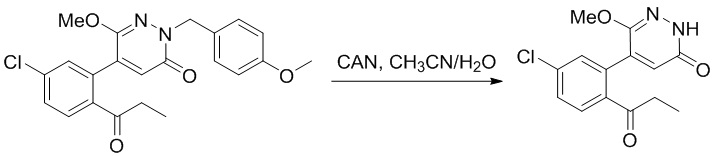
Раствор 5-(5-хлор-2-пропанилфенил)-6-метокси-2-(4-метоксибензил)пиридазин- 3(2H)-она (290 мг, 0.70 ммоль) в смеси ацетонитрил/вода (4.7 мл/1.6 мл) добавляли церийаммоний нитрат (3.09 г, 5.63 ммоль) при охлаждении на ледяной бане. После окончания добавления охлаждающую баню убирали, и реакцию вели при комнатной температуре в течение 0.5 часа. Реакционный раствор экстрагировали этилацетатом (50 мл × 2 раза). Органические фазы объединяли и последовательно промывали водой (50 мл × 2 раза) и насыщенным водным раствором хлорида натрия (50 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1.5/1-1/1.5). Получали 65 мг белого твердого 5-(5-хлор-2- пропанилфенил)-6-метоксипиридазин-3(2H)-она (выход: 31.8%). LCMS: RT = 3.42 мин, [M+H]+ = 293.01.
Стадия E: Синтез трет-бутил (S)-4-(2-(4-(5-хлор-2-пропанилфенил)-3-метокси- 6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата
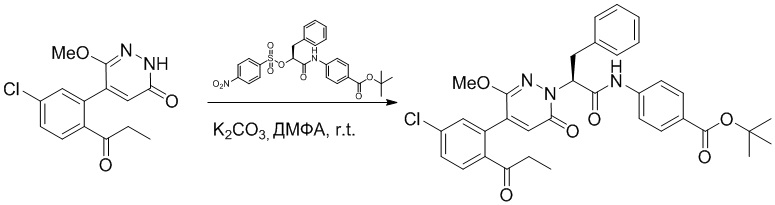
В раствор 5-(5-хлор-2-пропанилфенил)-6-метоксипиридазин-3(2H)-она (65 мг, 0.22 ммоль) в N,N-диметилметанамиде (2.2 мл) добавляли карбонат калия (61 мг, 0.44 ммоль) и трет-бутил (R)-4-(2-(((4-нитрофенил)сульфонил)окси)-3-фенилпропанамидо)бензоат (128 мг, 0.24 ммоль) при комнатной температуре. После окончания добавления смесь нагревали при 40°C в течение ночи.
Реакционный раствор экстрагировали этилацетатом (50 мл × 2 раза). Органические фазы объединяли и последовательно промывали водой (50 мл × 2 раза) и насыщенным водным раствором хлорида натрия (50 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 3/1). Получали 127 мг трет-бутил (S)-4-(2-(4-(5-хлор-2-пропанилфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата в виде бесцветного масла (выход: 94.0%). LCMS: RT = 4.68 мин, [M-H]- = 614.14.
Стадия F: Синтез (S)-4-(2-(4-(5-хлор-2-пропанилфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
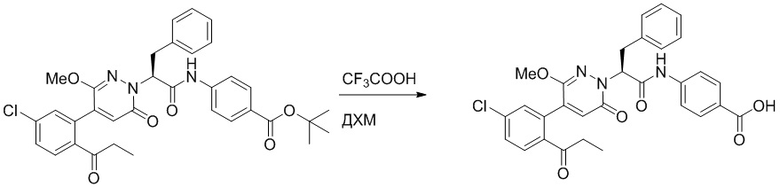
В раствор трет-бутил (S)-4-(2-(4-(5-хлор-2-пропанилфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата (170 мг, 0.28 ммоль) в дихлорметане (3.0 мл) добавляли трифторуксусную кислоту (0.5 мл) при комнатной температуре. После окончания добавления вели реакцию при комнатной температуре в течение 0.5 часа, и растворитель упаривали при пониженном давлении. Остаток растворяли в абсолютном этаноле (2.0 мл), раствор медленно добавляли по каплям в 40 мл н-гексана, при этом выпадало большое количество твердого осадка. Его отфильтровывали при пониженном давлении, получая 110 мг белой твердой (S)-4-(2-(4-(5-хлор-2- пропанилфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты (выход: 70.0%). LCMS: RT = 4.09 мин, [M-H]- = 559.15. 1H ЯМР (500 МГц, ДМСО) 1H ЯМР (500 МГц, ДМСО) δ 12.75 (ушир.с, 1H), 10.53 (с, 1H), 7.97 (д, J = 8.4 Гц, 1H), 7.91 (д, J = 8.7 Гц, 2H), 7.73 (д, J = 8.7 Гц, 2H), 7.67 (дд, J = 8.3, 2.1 Гц, 1H), 7.51 (д, J = 2.0 Гц, 1H), 7.36–7.26 (м, 4H), 7.21 (т, J = 7.1 Гц, 1H), 6.91 (с, 1H), 5.72 (дд, J = 10.3, 4.8 Гц, 1H), 3.64 (с, 3H), 3.55 (дд, J = 14.0, 10.3 Гц, 1H), 3.40 (дд, J = 14.0, 4.6 Гц, 1H), 2.99 (ушир.с, 2H), 1.03 (т, J = 7.1 Гц, 3H).
Пример 33
Синтез 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- (4-(циклопропанформамидо)фенил)пропанамидо)бензойной кислоты
Применяли описанный ниже путь синтеза.
Стадия A: Синтез 2-гидрокси-3-(4-нитрофенил)пропановой кислоты
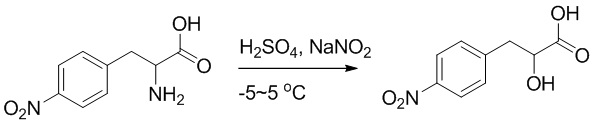
2-Амино-3-(4-нитрофенил)пропановую кислоту (4.2 г, 20.0 ммоль) растворяли в 80 мл 0.5 M серной кислоты, охлаждали на ледяной бане и медленно по каплям добавляли раствор нитрита натрия (5.52 г растворяли в 18 мл воды, 80.0 ммоль). Поддерживая температуру не выше 5°C, перемешивали реакционную смесь 3.5 часа. Реакционный раствор экстрагировали этилацетатом (100 мл × 2), органические фазы объединяли и промывали насыщенным водным раствором хлорида натрия (80 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученную сырую 2-гидрокси-3-(4-нитрофенил)пропановую кислоту использовали напрямую в следующей реакции (выход: 61.0%). LCMS: RT = 2.99 мин, [M-H]- = 209.98.
Стадия B: Синтез трет-бутил 4-(2-гидрокси-3-(4-нитрофенил)пропанамидо) бензоата
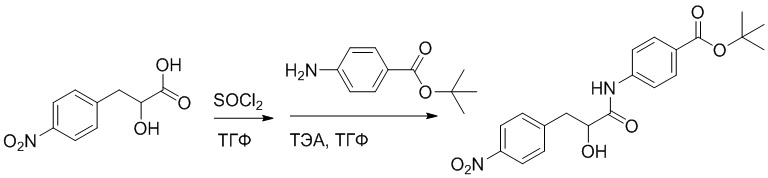
В инертной атмосфере азота медленно добавляли тионилхлорид (1.8 мл, 24.5 ммоль) по каплям в раствор 2-гидрокси-3-(4-нитрофенил)пропановой кислоты (2.59 г, 12.3 ммоль) в тетрагидрофуране (61.5 мл). После окончания добавления смесь нагревали до 45°C и перемешивали в течение ночи. Растворитель упаривали досуха при пониженном давлении, и полученный остаток напрямую использовали в следующей стадии.
Описанный выше сырой продукт растворяли в 30 мл тетрагидрофурана, добавляли триэтиламин (4.1 мл, 29.5 ммоль) и затем медленно добавляли по каплям раствор трет-бутил 4-аминобензоата (1.9 г в 19 мл ТГФ, 9.84 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 0.5 часа. Реакцию гасили водой (50 мл), экстрагировали этилацетатом (100 мл × 2 раза), органические фазы объединяли и промывали насыщенным водным раствором хлорида натрия (50 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 3/1-2/1). Получали 1.1 г трет-бутил 4-(2-гидрокси-3-(4- нитрофенил)пропанамидо)бензоата в виде желтого твердого вещества (выход: 23.0%). LCMS: RT = 4.11 мин, нет пика молекулярного иона.
Стадия C: Синтез трет-бутил 4-(3-(4-нитрофенил)-2-((4-нитрофенил)сульфонил) окси)пропанамидо)бензоата
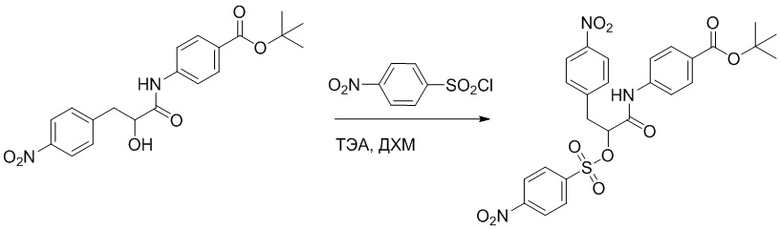
Триэтиламин (1.2 мл, 8.6 ммоль) и 4-нитробензолсульфонил хлорид (947 мг, 4.3 ммоль) добавляли последовательно в раствор трет-бутил 4-(2-гидрокси-3-(4-нитрофенил) пропанамидо)бензоата (1.1 г, 2.85 ммоль) в дихлорметане (28.5 мл) при комнатной температуре. После окончания добавления реакционную смесь перемешивали при комнатной температуре 3 часа.
Реакционный раствор разбавляли дихлорметаном (100 мл), последовательно промывали водой (50 мл) и насыщенным водным раствором хлорида натрия (50 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 4/1-2/1). Получали 1.02 г трет-бутил 4-(3-(4-нитрофенил)-2-((4-нитрофенил)сульфонил)окси)пропанамидо)бензоата в виде желтого твердого вещества (выход: 63.0%). LCMS: RT = 4.29 мин, нет пика молекулярного иона.
Стадия D: Синтез трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-4-нитрофенил)пропанамидо)бензоата
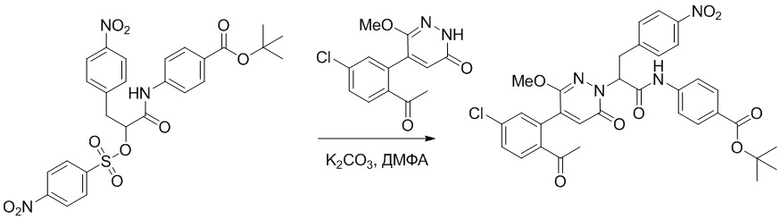
5-(2-Ацетил-5-хлорфенил)-6-метоксипиридазин-3(2H)-он (219 мг, 0.79 ммоль) и карбонат калия (218 мг, 1.58 ммоль) добавляли в раствор трет-бутил 4-(3-(4-нитрофенил)-2-((4-нитрофенил)сульфонил)окси)пропанамидо)бензоата (585 мг, 1.02 моль) в N,N-диметилформамиде (4.0 мл) при комнатной температуре, и реакционную смесь перемешивали при комнатной температуре в течение ночи.
Реакционный раствор экстрагировали этилацетатом (50 мл × 2), и органические фазы объединяли и последовательно промывали водой (40 мл × 2) и насыщенным водным раствором хлорида натрия (40 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 4/1-2/1). Получали 500 мг трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)- 3-4-нитрофенил)пропанамидо)бензоата в виде желтого твердого вещества (выход: 63.0%). LCMS: RT = 4.46 мин, [M-H]- = 645.05.
Стадия E: Синтез трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-(4-аминофенил)пропанамидо)бензоата
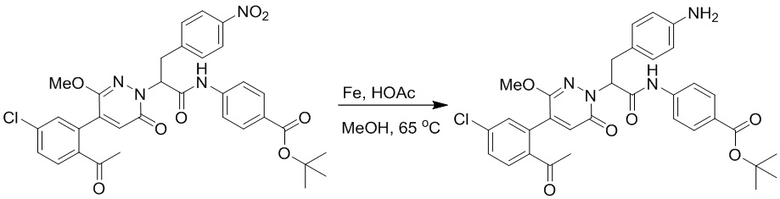
В инертной атмосфере азота последовательно добавляли уксусную кислоту (0.78 мл) и порошок восстановленного железа (438 мг, 7.8 ммоль) в раствор трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-4-нитрофенил) пропанамидо)бензоата (505 мг, 0.78 ммоль) в метаноле (7.8 мл), и реакционную смесь нагревали до 65°C и перемешивали в течение 1 часа.
Реакционный раствор доводили до pH = 8 насыщенным раствором бикарбоната натрия, разбавляли этилацетатом (100 мл), фильтровали через диатомит, органическую фазу последовательно промывали водой (40 мл) и насыщенным водным раствором хлорида натрия (40 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток представлял собой сырой трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(4-аминофенил) пропанамидо)бензоат, который напрямую использовали в следующей стадии. LCMS: RT = 4.00 мин, [M-H]-= 615.16.
Стадия F: Синтез 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-(4-(циклопропанформамидо)фенил)пропанамидо)бензойной кислоты
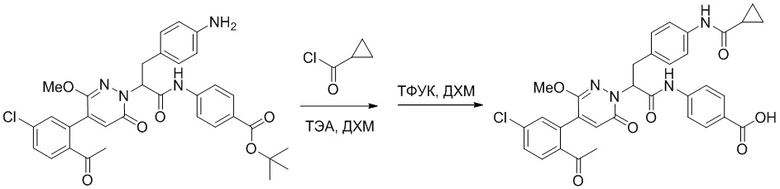
В инертной атмосфере азота добавляли триэтиламин (46 мкл, 0.33 ммоль) и циклопропанкарбонил хлорид (20 мкл, 0.22 ммоль) в раствор трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(4-аминофенил) пропанамидо)бензоата (65 мг, 0.11 ммоль) в дихлорметане (1.1 мл), и реакционную смесь перемешивали при комнатной температуре в течение 0.5 часа. Реакцию гасили водой (1 мл), разбавляли дихлорметаном (50 мл) и последовательно промывали водой (30 мл) и насыщенным водным раствором хлорида натрия (30 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный сырой продукт использовали напрямую в следующей реакции. LCMS: RT = 4.28 мин, [M+H]+= 685.23.
Описанный выше сырой продукт растворяли в дихлорметане (1.0 мл) и добавляли трифторуксусную кислоту (0.2 мл). Реакционную смесь перемешивали при комнатной температуре 45 минут. Растворитель упаривали при пониженном давлении, и полученный остаток очищали методом препаративной ВЭЖХ, получая 20 мг 4-(2-(4-(2-ацетил-5- хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(4-(циклопропанформамидо)фенил) пропанамидо)бензойной кислоты в виде бледно-желтого твердого вещества (выход: 29.0%). LCMS: RT = 3.72 мин, [M+H]+= 629.18. 1H ЯМР (500 МГц, ДМСО) δ 12.71 (ушир.с, 1H), 10.48 (с, 1H), 10.10 (с, 1H), 8.00 (д, J = 8.4 Гц, 1H), 7.90 (д, J = 8.7 Гц, 2H), 7.72 (д, J = 8.7 Гц, 2H), 7.69 (дд, J = 8.4, 2.1 Гц, 1H), 7.51 (д, J = 2.0 Гц, 1H), 7.49 (д, J = 8.4 Гц, 2H), 7.21 (д, J = 8.4 Гц, 2H), 6.90 (с, 1H), 5.69 (дд, J = 10.2, 4.8 Гц , 1H), 3.68 (с, 3H), 3.46 (дд, J = 14.0, 10.4 Гц, 1H), 3.38 – 3.34 (м, 1H), 2.54 (с, 3H), 1.77 – 1.71 (м, 1H), 0.88 – 0.82 (м, 2H), 0.79 – 0.72 (м, 2H).
Пример 34
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-гидрокси-6-оксопиридазин-1(6H)- ил)-3-фенилпропанамидо)бензойной кислоты

Применяли описанный ниже путь синтеза.
Стадия A: Синтез 5-(2-ацетил-5-хлорфенил)-6-гидрокси-2-(4- метоксибензил)пиридазин-3(2H)-она
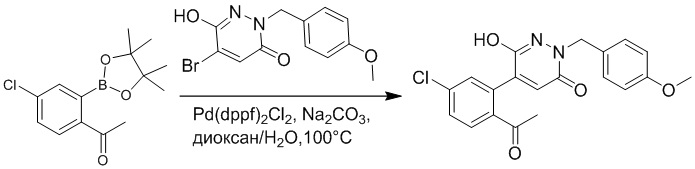
В инертной атмосфере азота добавляли воду (5 мл) и Pd(dppf)2Cl2 (370 мг, 0.48 ммоль) в раствор 1-(4-хлор-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этан- 1-она (1.6 г, 5.7 ммоль), 5-бром-6-гидрокси-2-(4-метоксибензил)пиридазин-3(2H)-она (1.5 г, 4.8 ммоль), карбоната калия (1.3 г, 9.6 ммоль) в 1,4-диоксане (20 мл), и смесь перемешивали при 100°C в течение 3 часов.
Реакционный раствор охлаждали до комнатной температуры и экстрагировали этилацетатом (100 мл × 2). Органические фазы объединяли и последовательно промывали водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 2/1). Получали 500 мг 5-(2-ацетил-5-хлорфенил)-6- гидрокси-2-(4-метоксибензил)пиридазин-3(2H)-она в виде желтого твердого вещества (выход: 27.0%). LCMS: RT = 3.72 мин, [M+H]+ = 383.03.
Стадия B: Синтез 5-(2-ацетил-5-хлорфенил)-6-(аллилокси)-2-(4- метоксибензил)пиридазин-3(2H)-она
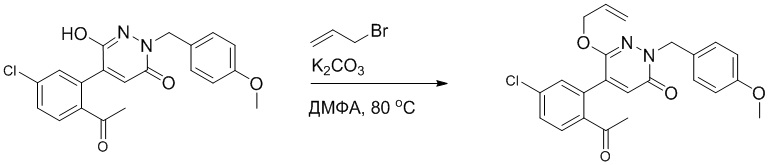
Карбонат калия (205 мг, 1.49 ммоль) добавляли в раствор 5-(2-ацетил-5-хлорфенил)- 6-гидрокси-2-(4-метоксибензил)пиридазин-3(2H)-она (200 мг, 0.52 ммоль) в N,N-диметилформамиде (2.5 мл) при комнатной температуре. Смесь нагревали до 80°C и перемешивали 15 минут, затем добавляли аллибромид (180 мкл, 2.1 ммоль), и реакционную смесь перемешивали при этой же температуре 0.5 часа.
После охлаждения до комнатной температуры, реакционный раствор экстрагировали этилацетатом (50 мл × 2 раза), органические фазы объединяли и последовательно промывали водой (40 мл × 2 раза) и насыщенным водным раствором хлорида натрия (40 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 3/1). Получали 150 мг 5-(2-ацетил-5-хлорфенил)-6-(аллилокси)-2-(4-метоксибензил)пиридазин-3(2H)-она в виде желтого маслянистого вещества (выход: 68.0%). LCMS: RT = 4.17 мин, [M+H]+= 425.10.
Стадия C: Синтез 5-(2-ацетил-5-хлорфенил)-6-(аллилокси)пиридазин-3(2H)-она
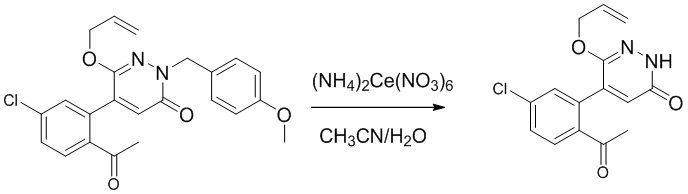
При охлаждении на ледяной бане добавляли церийаммоний нитрат (903 мг, 1.65 ммоль) в раствор 5-(2-ацетил-5-хлорфенил)-6-(аллилокси)-2-(4-метоксибензил)пиридазин- 3(2H)-она (140 мг, 0.33 ммоль) в смеси ацетонитрил/вода (2.4 мл/0.8 мл). После окончания добавления охлаждающую баню убирали, и реакционную смесь перемешивали при комнатной температуре в течение 0.5 часа. Реакционный раствор экстрагировали этилацетатом (50 мл × 2 раза), органические фазы объединяли и последовательно промывали водой (50 мл × 2 раза) и насыщенным водным раствором хлорида натрия (50 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 1/1-1/1.5). Получали 85 мг 5-(2-ацетил-5-хлорфенил)-6-(аллилокси)пиридазин-3(2H)-она в виде желтого маслянистого вещества (выход: 85.0%). LCMS: RT = 3.48 мин, [M+H]+ = 305.10.
Стадия D: Синтез трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(аллилокси)-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата
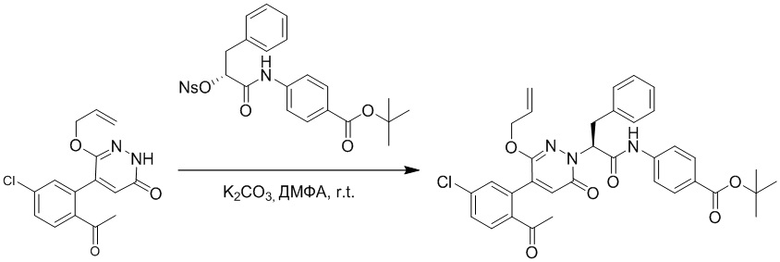
Карбонат калия (77 мг, 0.56 ммоль) и трет-бутил (R)-4-(2-(((4-нитрофенил) сульфонил)окси)-3-фенилпропанамидо)бензоат (161 мг, 0.31 ммоль) добавляли в раствор 5-(2-ацетил-5-хлорфенил)-6-(аллилокси)пиридазин-3(2H)-она (85 мг, 0.28 ммоль) в N,N-диметилформамиде (3.0 мл) при комнатной температуре, и реакционную смесь перемешивали при комнатной температуре в течение ночи.
Реакционный раствор экстрагировали этилацетатом (50 мл × 2 раза), органические фазы объединяли и последовательно промывали водой (30 мл × 2 раза) и насыщенным водным раствором хлорида натрия (30 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 4/1-2/1), получая 104 мг трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(аллилокси)-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата в виде светло-желтого маслянистого вещества (выход: 59.0%). LCMS: RT = 4.69 мин, [M-H]- = 626.13.
Стадия E: Синтез трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-гидрокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата
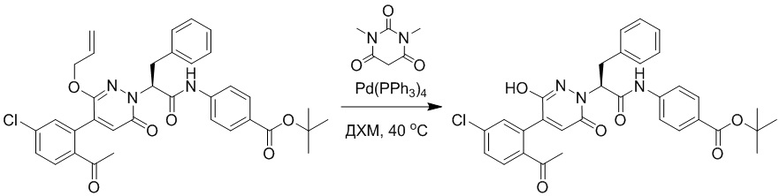
В инертной атмосфере азота добавляли 1,3-диметилбарбитуровую кислоту (149 мг, 0.95 ммоль) и тетракис(трифенилфосфин)палладий (7 мг, 0.006 ммоль) в раствор трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-(аллилокси)-6-оксопиридазин-1(6H)-ил)- 3-фенилпропанамидо)бензоата (100 мг, 0.16 ммоль) в дихлорметане (4.0 мл), и реакционный раствор нагревали при 40°C в течение 1.5 часов.
Реакционный раствор разбавляли дихлорметаном (100 мл) и последовательно промывали насыщенным водным раствором бикарбоната натрия (40 мл), водой (40 мл) и насыщенным водным раствором хлорида натрия (30 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 4/1 - 2/1), получая 52 мг трет-бутил (S)-4-(2-(4-(2-ацетил-5- хлорфенил)-3-гидрокси-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата в виде бесцветного масла (выход: 55.0% ). LCMS: RT = 4.31 мин, [M+H]+ = 588.16.
Стадия F: Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-гидрокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
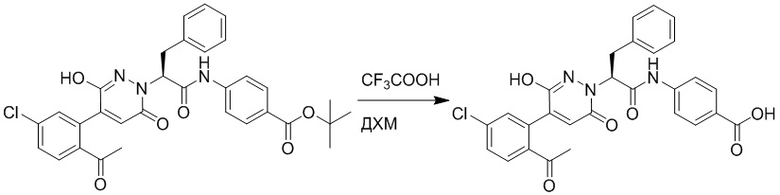
При комнатной температуре добавляли трифторуксусную кислоту (0.5 мл) в раствор трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-гидрокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамид)бензоата (52 мг, 0.088 ммоль) в дихлорметане (2.0 мл). После окончания добавления реакционную смесь перемешивали при комнатной температуре 0.5 часа, и растворитель упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 2/1-1/1), получая 30 мг (S)-4-(2-(4-(2- ацетил-5-хлорфенил)-3-гидрокси- 6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты в виде белого твердого вещества (выход: 64.0%). LCMS: RT = 3.78 мин, [M+H]+= 532.09.
Пример 35
Синтез (S)-4-(2-(4-(5-хлор-2-(2-гидроксиацетил)фенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
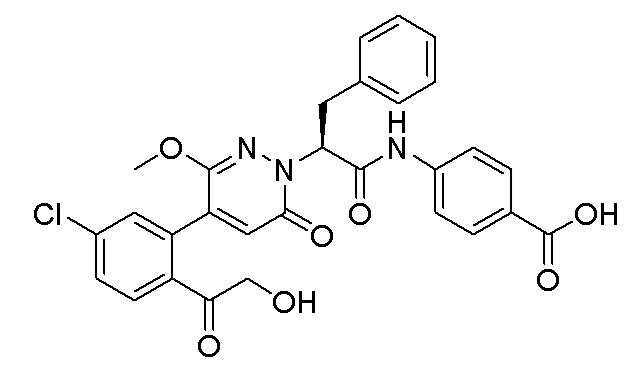
Применяли описанный ниже путь синтеза.
Стадия A: Синтез (S)-4-(2-(4-(5-хлор-2-(2-гидроксиацетил)фенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты

(S)-4-(2-(4-(5-хлор-2-(2-метоксиацетил)фенил)-3-метокси-6-оксопиридазин-1(6H)-ил)- 3-фенилпропанамидо)бензойную кислоту (17 мг, 0.03 ммоль) растворяли в метаноле (4.0 мл) и добавляли метанольный раствор хлороводорода (0.5 мл). Смесь перемешивали при комнатной температуре в течение 1 часа.
Реакционный раствор упаривали при пониженном давлении, и полученный остаток очищали методом препаративной высокоэффективной жидкостной хроматографии, получая 8 мг (S)-4-(2-(4-(5-хлор-2-(2-гидроксиацетил))фенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты в виде белого твердого вещества (выход: 47.0%). LCMS: RT = 3.69 мин, [MH]- = 560.13. 1H ЯМР (400 МГц, ДМСО) δ 12.87 – 12.61 (м, 1H), 10.49 (с, 1H), 7.91 (дд, J = 13.3, 8.6 Гц, 2H), 7.70 (д, J = 8.7 Гц, 2H), 7.65 (дд, J = 8.3, 2.1 Гц, 1H), 7.52 (д, J = 2.1 Гц, 1H), 7.34 – 7.25 (м, 3H), 7.18 (т, J = 6.7 Гц, 1H), 6.91 (с, 1H), 5.71 (дд, J = 9.7, 5.2 Гц, 1H), 4.65 (с, 2H), 3.66 (с, 3H), 3.55 – 3.45 (м, 1H), 3.40 (дд, J = 14.0, 5.3 Гц, 1H) , 1.22 (с, 2H).
Пример 36
Синтез (S)-4-(2-(4-(5-хлор-2-(2,2-дифторацетил)фенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
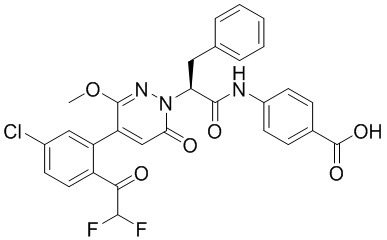
Применяли описанный ниже путь синтеза.
Стадия A: Синтез (Z)-5-(2-(1-(бутилимино)этил)-5-хлорфенил)-6-метокси-2- (4-метоксибензил)пиридазин-3(2H)-она
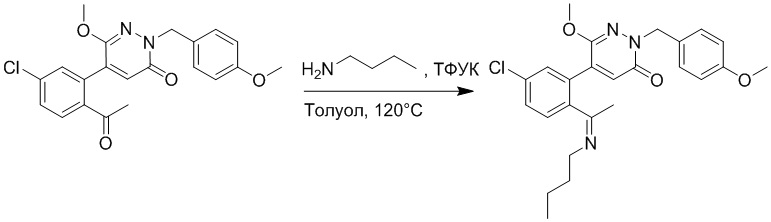
5-(2-Ацетил-5-хлорфенил)-6-метокси-2-(4-метоксибензил)пиридазин-3(2H)-он (200 мг, 0.50 ммоль), н-бутиламин (2.0 мл) добавляли в толуол (2.0 мл) и добавляли по каплям трифторуксусную кислоту (8.0 мкл). Устанавливали насадку Дина-Старка, и реакционную смесь перемешивали при 120°C в течение 8 часов.
После окончания реакции толуол упаривали досуха, полученный остаток растворяли в метил-трет-бутиловом эфире (30 мл), промывали насыщенным водным раствором хлорида натрия (10 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Получали 220 мг (Z)-5-(2-(1- (бутилимино)этил)-5-хлорфенил)-6-метокси-2-(4-метоксибензил)пиридазин-3(2H)-она в виде желтого масла, который напрямую использовали в следующей стадии без дополнительной очистки. LCMS: RT = 3.10 мин, [M+H]+ = 454.17.
Стадия B: Синтез 5-(5-хлор-2-(2,2-дифторацетил)фенил)-6-метокси-2-(4- метоксибензил)пиридазин-3(2H)-она
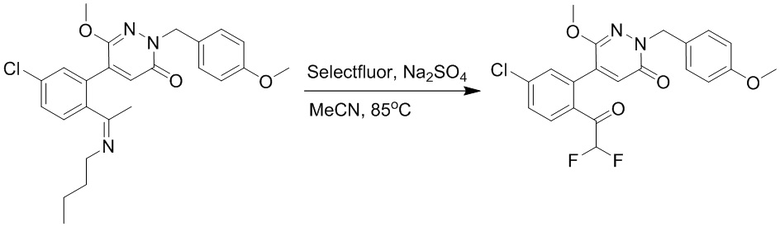
(Z)-5-(2-(1-(Бутилимино)этил)-5-хлорфенил)-6-метокси-2-(4-метоксибензил) пиридазин-3(2H)-он (220 мг, 0.49 ммоль), Selectfluor (206 мг, 0.58 ммоль) и небольшое количество безводного сульфата натрия добавляли в безводный ацетонитрил (2.0 мл) при комнатной температуре, и смесь перемешивали при 85°C в течение ночи .
После окончания реакции, реакционную смесь гасили добавлением воды, и значение pH доводили до 3-4 добавлением 1M разбавленной соляной кислоты. Смесь экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (20 мл × 3), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 2/5). Получали 60 мг 5-(5-хлор-2- (2,2-дифторацетил)фенил)-6-метокси-2-(4-метоксибензил)пиридазин-3(2H)-она в виде желтого твердого вещества (выход: 28.6%). LCMS: RT = 4.06 мин, [M+H]+ = 435.04.
Стадия C: Синтез 5-(5-хлор-2-(2,2-дифторацетил)фенил)-6-метоксипиридазин- 3(2H)-она

5-(5-Хлор-2-(2,2-дифторацетил)фенил)-6-метокси-2-(4-метоксибензил)пиридазин- 3(2H)-он (60 мг, 0.14 ммоль) добавляли в смесь растворителей (4 мл, ацетонитрил: вода = 3:1) при 0°C, затем медленно добавляли церийаммоний нитрат (605 мг, 1.10 ммоль). После окончания добавления реакционную смесь перемешивали при комнатной температуре 30 минут.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/1). Получали 25 мг 5-(5-хлор-2-(2,2- дифторацетил)фенил)-6-метоксипиридазин-3(2H)-она в виде желтого твердого вещества (выход: 57.2%). LCMS: RT = 3.42 мин, [M+H]+ = 315.03.
Стадия D: Синтез трет-бутил (S)-4-(2-(4-(5-хлор-2-(2,2-дифторацетил)фенил)-3- метокси-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата
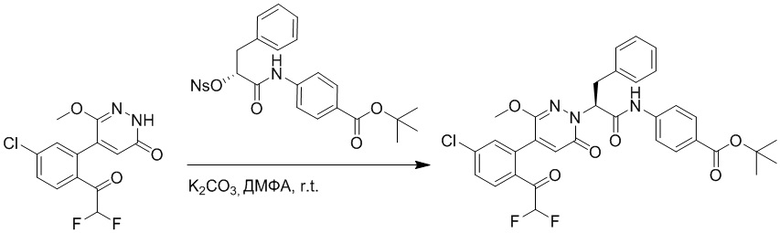
5-(5-Хлор-2-(2,2-дифторацетил)фенил)-6-метоксипиридазин-3(2H)-он (25 мг, 0.08 ммоль), трет-бутил (R)-4-(2-(((4-нитрофенил)сульфонил)окси)-3-фенилпропанамидо) бензоат (46 мг, 0.08 ммоль) и карбонат калия (22 мг, 0.16 ммоль) добавляли в N,N-диметилформамид (2.0 мл) при комнатной температуре, и реакционную смесь перемешивали при комнатной температуре в течение ночи.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (10 мл × 3). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 32 мг трет-бутил (S)-4- (2-(4-(5-хлор-2-(2,2-дифторацетил)фенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)бензоата в виде бледно-желтого твердого вещества (выход: 62.5%). LCMS: RT = 4.13 мин, [M+H]+ = 638.15.
Стадия E: Синтез (S)-4-(2-(4-(5-хлор-2-(2,2-дифторацетил(фенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
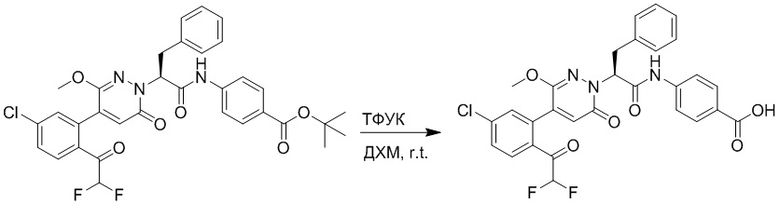
трет-Бутил (S)-4-(2-(4-(5-хлор-2-(2,2-дифторацетил)фенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоат (32 мг, 0.05 ммоль) добавляли в дихлорметан (2.0 мл) при комнатной температуре и добавляли по каплям трифторуксусную кислоту (0.5 мл). Реакционную смесь перемешивали при комнатной температуре 3 часа.
После окончания реакции дихлорметан упаривали досуха и трифторуксусную кислоту отгоняли досуха на масляном насосе. Полученный остаток растворяли в дихлорметане (1.0 мл), и раствор добавляли по каплям в н-гексан (10.0 мл), при этом выпадало в осадок белое твердое вещество, которое отфильтровывали при отсасывании. Осадок на фильтре промывали н-гексаном и сушили, получая 18 мг (S)-4-(2-(4-(5-хлор-2-(2,2-дифторацетил(фенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)бензойной кислоты в виде бледно-желтого твердого вещества (выход: 62.0%). LCMS: RT = 4.00 мин, [MH]- = 580.07. 1H ЯМР (500 МГц, ДМСО) δ 12.71 (с, 1H), 10.53 (с, 1H), 8.03 (д, J = 8.4 Гц, 1H), 7.91 (д, J = 8.7 Гц, 2H), 7.80 (дд, J = 8.4, 2.1 Гц, 1H), 7.73 (д, J = 8.7 Гц, 2H), 7.66 (д, J = 2.0 Гц, 1H), 7.37 –7.25 (м, 4H), 7.19 (т, J = 7.2 Гц, 1H), 7.12 (т, J = 52.4 Гц, 1H), 7.01 (с, 1H), 5.78–5.71 (м, 1H), 3.63 ( s, 3H), 3.53 (дд, J = 14.1, 10.3 Гц, 1H), 3.42 (дд, J = 14.2, 4.6 Гц, 1H).
Пример 37
Синтез (S)-4-(2-(4-(5-хлор-2-(2-фторацетил)фенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
Применяли описанный ниже путь синтеза.
Стадия A: Синтез 5-(5-хлор-2-(2-метоксиацетил)фенил)-6-метокси-2-(4- метоксибензил)пиридазин-3(2H)-она
5-(2-Ацетил-5-хлорфенил)-6-метокси-2-(4-метоксибензил)пиридазин-3(2H)-он (300 мг, 0.75 ммоль) и гидроксид калия (210 мг, 3.75 ммоль) добавляли в метанол (5.0 мл) при комнатной температуре, перемешивали при 0°C в течение 5 мин и добавляли по каплям иодбензол диацетат (364 мг, 1.13 ммоль). Реакционную смесь перемешивали при этой же температуре 15 минут.
После окончания реакции добавляли раствор сульфита натрия и раствор хлорида аммония на ледяной бане, и смесь экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (30 мл × 3), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 230 мг 5-(5-хлор-2-(2-метоксиацетил)фенил)-6-метокси-2-(4-метоксибензил)пиридазин-3(2H)-она в виде желтого твердого вещества (выход: 72.0%). LCMS: RT = 3.99 мин, [M+H]+ = 429.34.
Стадия B: Синтез 5-(5-хлор-2-(2-гидроксиацетил)фенил)-6-метокси-2-(4- метоксибензил)пиридазин-3(2H)-она
5-(5-Хлор-2-(2-метоксиацетил)фенил)-6-метокси-2-(4-метоксибензил)пиридазин- 3(2H)-он (230 мг, 0.54 ммоль) добавляли в метанол (8.0 мл) при комнатной температуре, затем добавляли по каплям метанольный раствор хлороводорода (0.8 мл, 3.20 ммоль, 4.0 моль/л).
После окончания реакции, реакционную смесь гасили добавлением воды, и значение pH доводили до нейтрального насыщенным раствором бикарбоната натрия. Смесь экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (20 мл × 3), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Получали 200 мг 5-(5-хлор-2-(2-гидроксиацетил)фенил)-6-метокси-2-(4- метоксибензил)пиридазин-3(2H)-она в виде желтого твердого вещества, которое напрямую использовали в следующей стадии без дополнительной очистки. LCMS: RT = 3.69 мин, [M+H]+ = 415.07.
Стадия C: Синтез 5-(5-хлор-2-(2-гидроксиацетил)фенил)-6-метокси-2-(4- метоксибензил)пиридазин-3(2H)-она
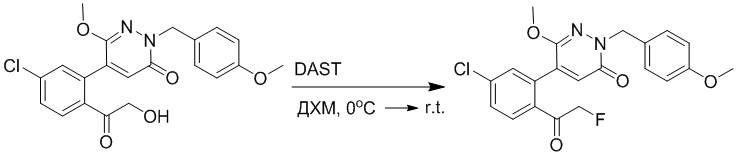
5-(5-Хлор-2-(2-гидроксиацетил)фенил)-6-метокси-2-(4-метоксибензил)пиридазин- 3(2H)-он (200 мг, 0.48 ммоль) добавляли в дихлорметан (10.0 мл) при 0°C, и в полученный раствор медленно добавляли по каплям трифторид диэтиламиносеры (90 мкл, 0.72 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи.
После окончания реакции, реакционную смесь гасили добавлением воды, и значение pH доводили до нейтрального насыщенным раствором бикарбоната натрия. Смесь экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 70 мг 5-(5-хлор-2-(2-фторацетил)фенил)-6-метокси-2-(4-метоксибензил)пиридазин-3(2H)-она в виде желтого твердого вещества (выход: 35.4%). LCMS: RT = 3.94 мин, [M+H]+ = 417.11.
Стадия D: Синтез 5-(5-хлор-2-(2-фторацетил)фенил)-6- метоксипиридазин-3(2H)-она
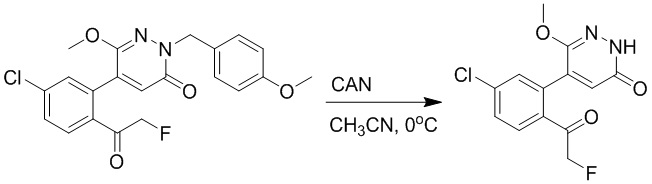
5-(5-Хлор-2-(2-фторацетил)фенил)-6-метокси-2-(4-метоксибензил)пиридазин-3(2H)-он (70 мг, 0.17 ммоль) добавляли в смесь растворителей (4 мл, ацетонитрил: вода = 3:1) при 0°C, и затем медленно добавляли церийаммоний нитрат (595 мг, 1.09 ммоль). После окончания добавления реакционную смесь перемешивали при комнатной температуре 30 минут.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/1). Получали 30 мг 5-(5-хлор-2-(2- фторацетил)фенил)-6-метоксипиридазин-3(2H)-она в виде желтого твердого вещества (выход: 47.1%). LCMS: RT = 3.19 мин, [M+H]+ = 297.02.
Стадия E: Синтез трет-бутил (S)-4-(2-(4-(5-хлор-2-(2-фторацетил)фенил)-3- метокси-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата
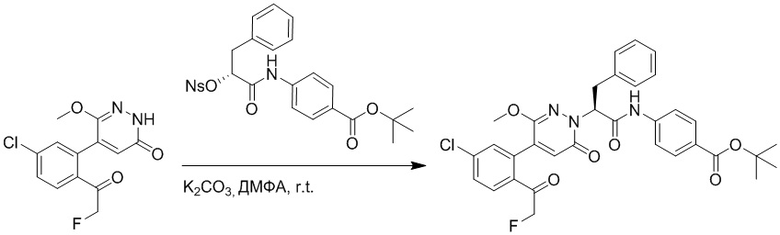
5-(5-Хлор-2-(2-фторацетил)фенил)-6-метоксипиридазин-3(2H)-он (30 мг, 0.08 ммоль), трет-бутил (R)-4-(2-(((4-нитрофенил)сульфонил)окси)-3-фенилпропанамидо)бензоат (46 мг, 0.08 ммоль) и карбонат калия (20 мг, 0.16 ммоль) добавляли в N,N-диметилформамид (2.0 мл) при комнатной температуре, и реакционную смесь перемешивали в течение ночи при комнатной температуре.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (10 мл × 3). Органические фазы объединяли, органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/1). Получали 22 мг трет-бутил (S)-4-(2-(4-(5-хлор- 2-(2-фторацетил)фенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо) бензоата в виде бледно-желтого твердого вещества (выход: 50.0%). LCMS: RT = 4.53 мин, [M-H]- = 618.13.
Стадия F: Синтез (S)-4-(2-(4-(5-хлор-2-(2-фторацетил)фенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
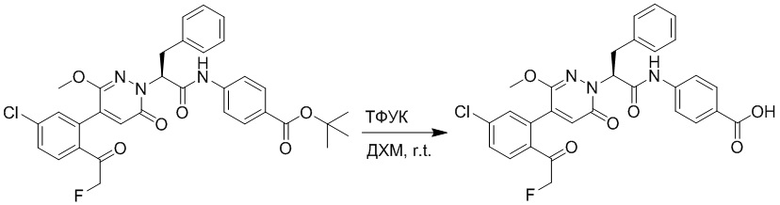
трет-Бутил (S)-4-(2-(4-(5-хлор-2-(2-дифторацетил)фенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоат (22 мг, 0.04 ммоль) добавляли в дихлорметан (2.0 мл) при комнатной температуре и добавляли по каплям трифторуксусную кислоту (0.5 мл). Реакционную смесь перемешивали при комнатной температуре 3 часа.
После окончания реакции, дихлорметан упаривали досуха и трифторуксусную кислоту отгоняли досуха на масляном насосе. Полученный остаток растворяли в дихлорметане (1.0 мл), и раствор добавляли по каплям в н-гексан (10.0 мл), при этом выпадало в осадок белое твердое вещество, которое отфильтровывали при отсасывании. Осадок на фильтре промывали н-гексаном и сушили, получая 5 мг (S)-4-(2-(4-(5-хлор-2- (2-фторацетил)фенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо) бензойной кислоты в виде бледно-желтого твердого вещества (выход: 22.2%). LCMS: RT = 3.96 мин, [M-H]- = 562.06.
Пример 38
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-фенилпропанамидо)-3-фторбензойной кислоты
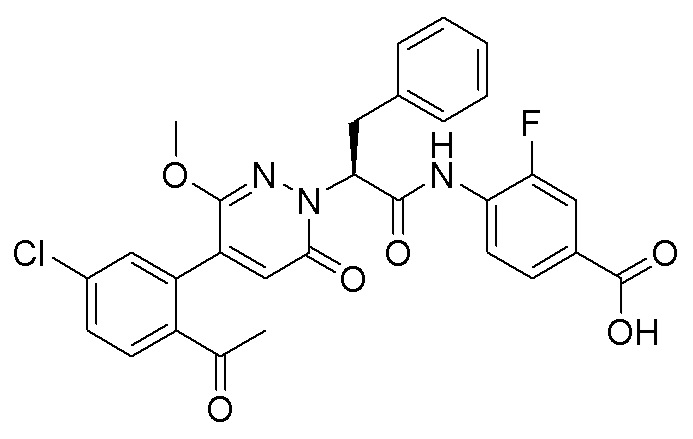
Применяли описанный ниже путь синтеза.
Стадия A: Синтез метил (S)-4-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)-3-фторбензоата
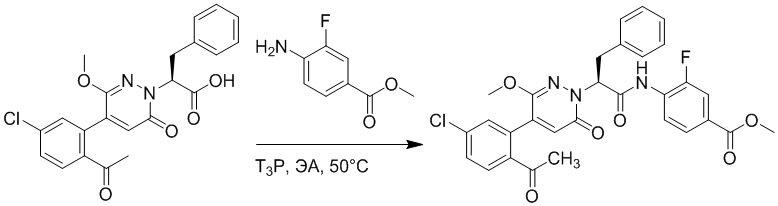
(S)-2-(4-(2-Ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропановую кислоту (80 мг, 0.19 ммоль), метил 3-фтор-4-амино-бензоат (38 мг, 0.22 ммоль) растворяли в этилацетате (2.0 мл) при комнатной температуре и добавляли N,N-диизопропилэтиламин (0.09 мл). После этого добавляли в полученный раствор 1-пропилфосфорный ангидрид (0.4 мл). Реакционный раствор нагревали до 50°C и перемешивали 3 часа.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 2), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 80 мг метил (S)-4-2-(4-(2- ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)-3- фторбензоата в виде бледно-желтого твердого вещества (выход: 73.7%). LCMS: RT = 4.28 мин, [M+H]+ = 578.06.
Стадия B: Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)-3-фторбензойной кислоты
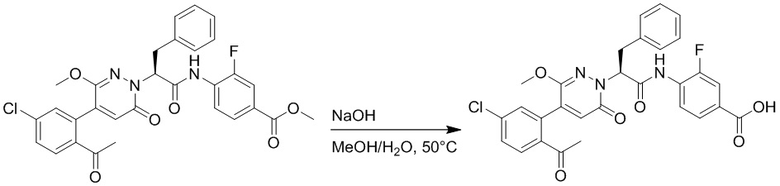
Метил (S)-4-2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)-3-фторбензоат (80 мг, 0.14 ммоль) растворяли в метаноле (2.0 мл). После этого добавляли в полученный раствор водный раствор (1.0 мл) гидроксида натрия (11 мг, 0.28 ммоль). Реакционный раствор перемешивали при 50°C в течение 3 часов.
Разбавленный раствор соляной кислоты (1.0 моль/л) медленно добавляли по каплям в реакционный раствор, доводя значение pH до 3-4. Добавляли этилацетат (30 мл), и смесь промывали насыщенным водным раствором хлорида натрия (10 мл × 3), сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток растворяли в дихлорметане (1.0 мл), и раствор добавляли по каплям в н-гексан (10.0 мл), при этом выпадало в осадок белое твердое вещество, которое отфильтровывали при отсасывании. Осадок на фильтре промывали н-гексаном и сушили, получая 38.42 мг (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)-3-фторбензойной кислоты в виде белого твердого вещества (выход: 48.7%). LCMS: RT = 4.09 мин, [MH]- = 562.07. 1H ЯМР (500 МГц, ДМСО) δ 13.13 (с, 1H), 10.28 (с, 1H), 7.99 (д, J = 8.4 Гц, 1H), 7.79 –7.73 (м, 2H), 7.72 (д, J = 1.8 Гц, 1H), 7.68 (дд, J = 8.3, 2.2 Гц, 1H), 7.49 (д, J = 2.1 Гц, 1H), 7.31 (дт, J = 15.2, 6.7 Гц, 5H), 7.19 (т, J = 7.3 Гц, 1H), 6.91 (с, 1H), 5.94 (дд, J = 10.6, 4.8 Гц, 1H), 3.68 (с, 3H), 3.42 (дд, J = 14.6, 4.7 Гц, 1H), 3.37–3.34 (м, 1H), 2.52 (с, 3H).
Пример 39
Синтез 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)- 3-(4-(циклобутанформамидо)фенил)пропанамидо)бензойной кислоты
Стадия A: Синтез 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-(4-(циклобутанформамидо)фенил)пропанамидо)бензойной кислоты
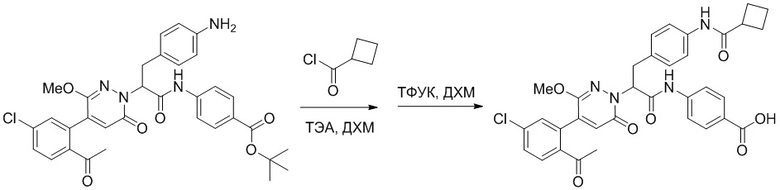
В инертной атмосфере азота добавляли триэтиламин (27 мкл, 0.17 ммоль) и циклопропанкарбонилхлорид (15 мкл, 0.12 ммоль) в раствор трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(4-аминофенил) пропанамидо)бензоата (40 мг, 0.065 ммоль) в дихлорметане (1.0 мл), и реакционную смесь перемешивали при комнатной температуре в течение 0.5 часа. Реакцию гасили водой (1 мл), разбавляли дихлорметаном (50 мл) и последовательно промывали водой (30 мл) и насыщенным водным раствором хлорида натрия (30 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный сырой продукт использовали напрямую в следующей реакции. LCMS: RT = 4.36 мин, [M+H]+ = 699.24.
Описанный выше сырой продукт растворяли в дихлорметане (1.0 мл) и добавляли трифторуксусную кислоту (0.2 мл). Реакционную смесь перемешивали при комнатной температуре 45 минут. Растворитель упаривали при пониженном давлении, и полученный остаток очищали методом препаративной ВЭЖХ, получая 20 мг 4-(2-(4-(2- ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(4-(циклобутанформамидо) фенил)пропанамидо)бензойной кислоты в виде бледно-желтого твердого вещества (выход: 48.0%). LCMS: RT = 3.82 мин, [M+H]- = 641.15. 1H ЯМР (500 МГц, ДМСО) δ 12.73 (с, 1H), 10.60 – 10.36 (м, 1H), 9.62 (с, 1H), 8.00 (д, J =8.4 Гц, 1H), 7.90 (д, J =8.7 Гц, 2H), 7.72 (д, J =8.8 Гц, 2H), 7.69 (дд, J =8.3, 2.1 Гц, 1H), 7.54 – 7.47 (м, 3H), 7.21 (д, J =8.5 Гц, 2H), 6.89 (с, 1H), 5.70 (дд, J =10.3, 4.8 Гц, 1H), 3.68 (с, 3H), 3.48 – 3.43 (м, 1H), 3.34 (дд, J =14.1, 4.7 Гц, 1H), 3.22 – 3.14 (м, 1H), 2.54 (с, 3H), 2.24 – 2.15 (м, 2H), 2.12 – 2.03 (м, 2H), 1.96 – 1.87 (м, 1H), 1.83 – 1.73 (м, 1H).
Пример 40
Синтез 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- (4-(циклопентанформамидо)фенил)пропанамидо)бензойной кислоты
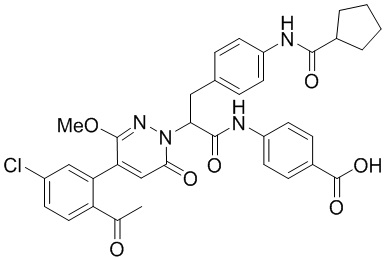
Стадия A: Синтез 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-(4-(циклопентанформамидо)фенил)пропанамидо)бензойной кислоты
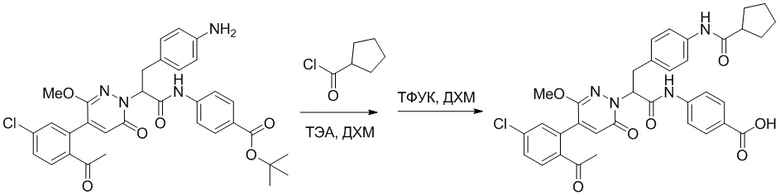
В инертной атмосфере азота добавляли триэтиламин (27 мкл, 0.17 ммоль) и циклопентанкарбонилхлорид (15 мкл, 0.12 ммоль) в раствор трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3-(4-аминофенил) пропанамидо)бензоата (40 мг, 0.065 ммоль) в дихлорметане (1.0 мл), и реакционную смесь перемешивали при комнатной температуре в течение 0.5 часа. Реакцию гасили водой (1 мл), разбавляли дихлорметаном (50 мл) и последовательно промывали водой (30 мл) и насыщенным водным раствором хлорида натрия (30 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный сырой продукт использовали напрямую в следующей реакции. LCMS: RT = 4.44 мин, [M+H]+ = 711.12.
Описанный выше сырой продукт растворяли в дихлорметане (1.0 мл) и добавляли трифторацетат (0.2 мл). Реакционную смесь перемешивали при комнатной температуре 45 минут. Растворитель упаривали при пониженном давлении, и полученный остаток очищали методом препаративной ВЭЖХ, получая 10 мг 4-(2-(4-(2-ацетил-5-хлорфенил)-3- метокси-6-оксопиридазин-1(6H)-ил)-3-(4-(циклопентанформамидо)фенил)пропанамидо) бензойной кислоты в виде бледно-желтого твердого вещества (выход: 23.0%). LCMS: RT = 3.93 мин, [M+H]-= 655.12. 1H ЯМР (500 МГц, ДМСО) δ 12.72 (с, 1H), 10.49 (с, 1H), 9.77 (с, 1H), 8.00 (д, J =8.4 Гц, 1H), 7.90 (д, J =8.8 Гц, 2H), 7.73 (д, J =8.8 Гц, 2H), 7.69 (дд, J = 8.4, 2.2 Гц, 1H), 7.54 – 7.48 (м, 3H), 7.21 (д, J =8.5 Гц, 2H), 6.90 (с, 1H), 5.70 (дд, J =10.3, 4.8 Гц, 1H), 3.68 (с, 3H), 3.50 – 3.44 (м, 2H), 2.76 – 2.69 (м, 1H), 2.54 (с, 3H), 1.86 – 1.76 (м, 2H), 1.73 – 1.62 (м, 4H), 1.57 – 1.49 (м, 2H).
Пример 41
Синтез (S)-4-(2-(4-(2-ацетил-5-фторфенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
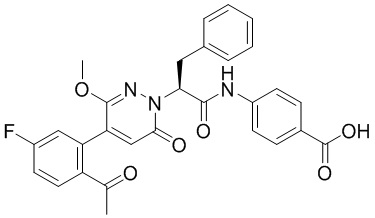
Применяли описанный ниже путь синтеза.
Стадия A: Синтез 1-(4-фтор-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2- ил)фенил)этан-1-она
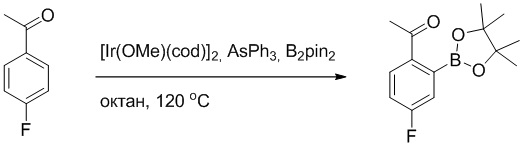
В инертной атмосфере азота добавляли трифенилмышьяк (80 мг, 0.26 ммоль) и [Ir(OMe)(cod)]2 (44 мг, 0.066 ммоль) в раствор 4-фторацетофенона (3.0 г, 21.7 ммоль), бис(пинаколато)диборона (1.15 г, 4.34 ммоль) в н-октане (21.8 мл), и смесь перемешивали при 120°C в течение 18 часов. Реакционный раствор охлаждали до комнатной температуры, разбавляли этилацетатом (100 мл), последовательно промывали водой (80 мл) и насыщенным водным раствором хлорида натрия (80 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 50/1-20/1). Получали 615 мг 1-(4-фтор-2-(4,4,5,5- тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этан-1-она в виде бледно-желтого твердого вещества (выход: 54.0%). LCMS: RT = 4.01 мин, [M-H]- = 263.04.
Стадия B: Синтез 5-(2-ацетил-5-фторфенил)-6-метокси-2-(4- метоксибензил)пиридазин-3(2H)-она
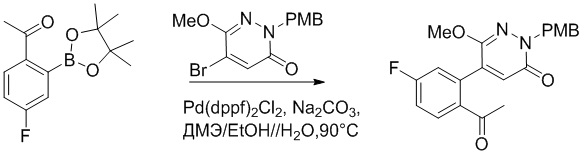
В инертной атмосфере азота добавляли этанол (0.38 мл), воду (0.38 мл) и Pd(dppf)2Cl2 (24 мг, 0.034 ммоль) в раствор 1-(4-фтор-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан- 2-ил)фенил)этан-1-она (205 мг, 0.67 ммоль), 5-бром-6-метокси-2-(4-метоксибензил) пиридазин-3(2H)-она (146 мг, 0.45 ммоль), карбоната натрия (142 мг, 1.34 ммоль) в диметиловом эфире этиленгликоля (3.0 мл), и полученный раствор перемешивали при 90°C в течение 1 часа. Реакционный раствор охлаждали до комнатной температуры, экстрагировали этилацетатом (30 мл × 2), органические фазы объединяли и последовательно промывали водой (40 мл) и насыщенным водным раствором хлорида натрия (40 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 2/1). Получали 120 мг 5-(2-ацетил-5-фторфенил)-6-метокси-2-(4-метоксибензил)пиридазин-3(2H)-она в виде светло-желтого маслянистого вещества (выход: 70.0%). LCMS: RT = 3.39 мин, [M+H]+ = 383.16.
Стадия C: Синтез 5-(2-ацетил-5-фторфенил)-6-метоксипиридазин-3(2H)-она
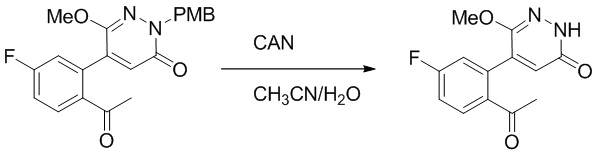
Церийаммоний нитрат (1.38 г, 2.51 ммоль) добавляли в раствор 5-(2-ацетил-5-фторфенил)-6-метокси-2-(4-метоксибензил)пиридазин-3(2H)-она (120 мг, 0.31 ммоль) в смеси ацетонитрил/вода (2.4 мл/0.8 мл) при охлаждении на ледяной бане. После окончания добавления охлаждающую баню убирали, и реакционную смесь перемешивали при комнатной температуре в течение 0.5 часа. Реакционный раствор экстрагировали этилацетатом (50 мл × 2), органические фазы объединяли и последовательно промывали водой (50 мл × 2) и насыщенным водным раствором хлорида натрия (50 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный сырой продукт использовали напрямую в следующей реакции. LCMS: RT = 3.00 мин, [M+H]+ = 263.11.
Стадия D: Синтез трет-бутил (S)-4-(2-(4-(2-ацетил-5-фторфенил)-3-метокси- 6-оксопиридазин-1(6H)-ил)3-фенилпропанамидо)бензоата
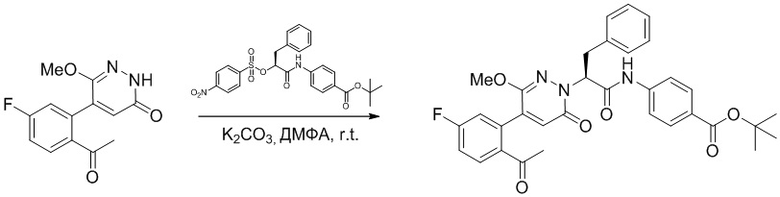
Карбонат калия (87 мг, 0.63 ммоль) и трет-бутил (R)-4-(2-(((4-нитрофенил) сульфонил)окси)-3-фенилпропанамидо)бензоат (182 мг, 0.35 ммоль) добавляли в раствор описанного выше сырого продукта в N,N-диметилформамиде (3.1 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение ночи.
Реакционный раствор экстрагировали этилацетатом (50 мл × 2), органические фазы объединяли и последовательно промывали водой (50 мл × 2) и насыщенным водным раствором хлорида натрия (50 мл). Затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 3/1 - 2/1). Получали 153 мг трет-бутил (S)-4-(2-(4-(2-ацетил-5-фторфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)3-фенилпропанамидо)бензоата в виде бесцветного масла (выход: 83.0%). LCMS: RT = 4.40 мин, [M+H]+ = 586.21.
Стадия E: Синтез (S)-4-(2-(4-(2-ацетил-5-фторфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
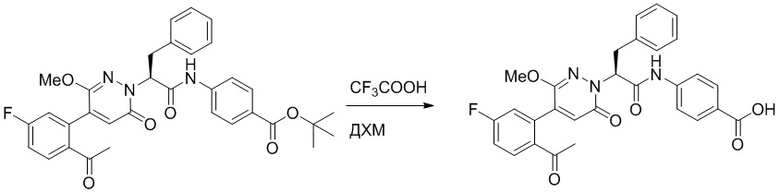
Трифторацетат (0.3 мл) добавляли в раствор трет-бутил (S)-4-(2-(4-(2-ацетил- 5-фторфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)3-фенилпропанамидо)бензойной кислоты (153 мг, 0.26 ммоль) в дихлорметане (2.0 мл) при комнатной температуре. После окончания добавления реакционную смесь перемешивали при комнатной температуре в течение 0.5 часа, и растворитель упаривали при пониженном давлении. Остаток растворяли в дихлорметане (1.0 мл), и раствор медленно добавляли по каплям в 30 мл н-гексана, при этом выпадало большое количество твердого осадка. Его отфильтровывали при пониженном давлении и получали 85 мг (S)-4-(2-(4-(2-ацетил-5-фторфенил)-3- метокси-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты в виде бледно-желтого твердого вещества (выход: 62.0%). LCMS: RT = 3.84 мин, [M-H]- = 528.07.
Пример 42
Синтез (S)-4-(2-(4-(3-хлор-2,6-дифторфенил)-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)бензойной кислоты
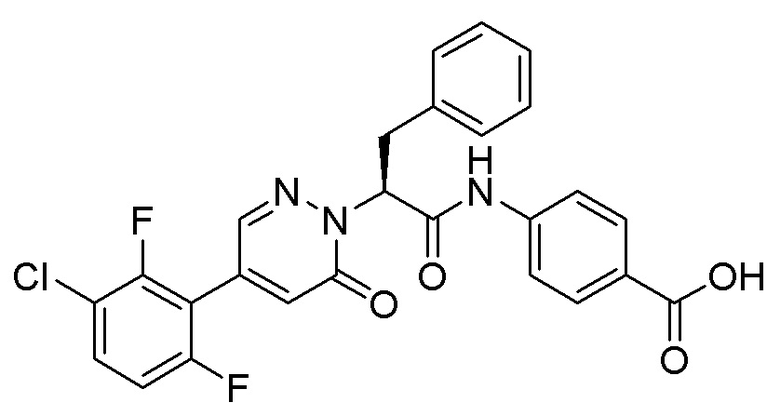
Применяли описанный ниже путь синтеза.
Стадия A: Синтез 1-хлор-2,4-дифтор-3-винилбензола
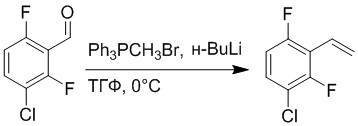
Тетрагидрофурановый раствор н-бутиллития (21.3 мл, 33.60 ммоль, 1.6 моль/л) медленно добавляли по каплям в тетрагидрофуран (50.0 мл), содержащий метилтрифенилфосфин бромид (12.18 г, 34.00 ммоль) поддерживая температуру равной 0°C, и смесь перемешивали при той же температуре в течение 1 часа. После этого добавляли в полученный раствор 5-хлор-2,6-дифторбензальдегид (5.00 г, 28.32 ммоль) в тетрагидрофуране (5.0 мл). Продолжали перемешивание при этой же температуре еще 30 минут, нагревали до комнатной температуры и вели реакцию в течение 2 часов.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (100 мл × 3 раза). Органические фазы объединяли и сначала промывали их насыщенным водным раствором хлорида натрия (50 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: pure н-гексан). Получали 1.50 г 1-хлор-2,4-дифтор-3-винилбензола в виде бесцветной прозрачной жидкости (выход: 25.2%). LCMS: RT = 4.48 мин.
Стадия B: синтез 2-(3-хлор-2,6-дифторфенил)ацетальдегида
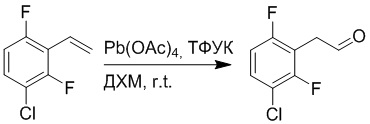
Тетраацетат свинца и трифторуксусную кислоту помещали в реакционную колбу при 0°C. После этого добавляли по каплям раствор 1-хлор-2,4-дифтор-3-винилбензола (1.50 мг, 8.57 ммоль) в дихлорметане (15.0 мл). Через 2 минуты убирали охлаждающую баню, и реакционную смесь перемешивали при комнатной температуре 2 часа.
После окончания реакции, реакционную смесь гасили добавлением воды, затем добавляли насыщенный раствор хлорида натрия до прекращения выпадения белого осадка. После фильтрования при отсасывании, осадок на фильтре промывали дихлорметаном, фильтрат доводили до нейтрального pH насыщенным раствором бикарбоната натрия, и слои разделяли. Органическую фазу промывали насыщенным водным раствором хлорида натрия (50 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Получали 1.50 г, 2-(3-хлор-2,6-дифторфенил)ацетальдегида в виде желтого масла, которое использовали напрямую в следующей реакции без очистки.
Стадия C: синтез 4-(3-хлор-2,6-дифторфенил)-5-гидроксифуран-2(5H)-она
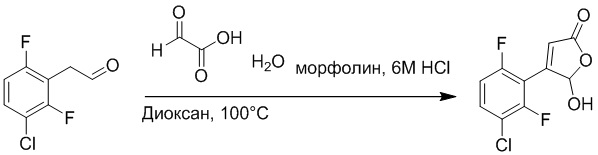
При комнатной температуре, морфолин (719 мг, 8.26 ммоль), соляную кислоту (1.4 мл, 8.6 ммоль, 6.0 моль/л), гидрат глиоксиловой кислоты (688 мг, 7.48 ммоль) и 2-(3-хлор-2,6-дифторфенил)ацетальдегид (1.5 г, 0.48 ммоль) последовательно добавляли в 1,4-диоксан (10.0 мл), и реакционную смесь кипятили 2 часов.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (30 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/10). Получали 1.00 г 4-(3-хлор-2,6- дифторфенил)-5-гидроксифуран-2(5H)-она в виде желтого твердого вещества (выход: 47.3%). LCMS: RT = 3.45 мин, [M-H]- = 244.93.
Стадия D: синтез 5-(3-хлор-2,6-дифторфенил)пиридазин-3(2H)-она
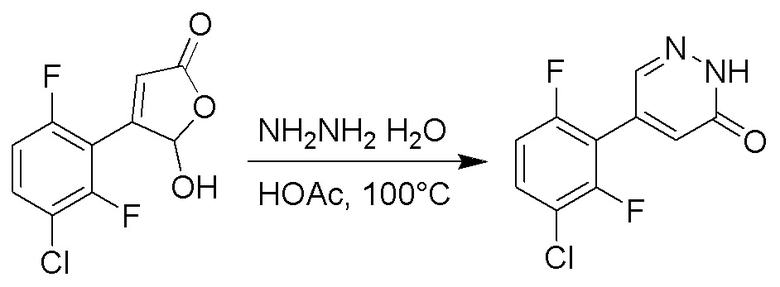
При комнатной температуре добавляли 4-(3-хлор-2,6-дифторфенил)-5- гидроксифуран-2(5H)-он (500 мг, 2.03 ммоль) в ледяную уксусную кислоту (3 мл), и затем медленно добавляли 80%-ный гидразин гидрат (246 мкл, 4.06 ммоль). Реакционную смесь перемешивали при 100°C в течение 1 часа.
После окончания реакции смесь охлаждали до комнатной температуры, при этом выпадало большое количество твердого осадка, смесь разбавляли небольшим количеством этилацетата, чтобы диспергировать твердый осадок. После фильтрования при отсасывании, осадок на фильтре промывали небольшим количеством этилацетата и сушили, получая 330 мг белого твердого 5-(3-хлор-2,6-дифторфенил)пиридазин-3(2H)-она (выход: 67.0%). LCMS: RT = 3.28 мин, [M+H]+ = 242.98.
Стадия E: синтез (S)-трет-бутил 4-(2-(4-(3-хлор-2,6-дифторфенил)-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата
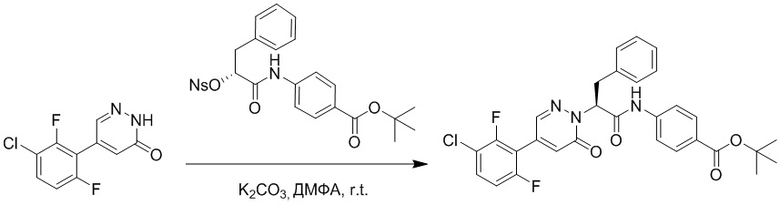
При комнатной температуре 5-(3-хлор-2,6-дифторфенил)пиридазин-3(2H)-он (100 мг, 0.42 ммоль), трет-бутил (R)-4-(2-(((4-нитрофенил)сульфонил)окси)-3- фенилпропанамидо)бензоат (239 мг, 0.45 ммоль) и карбонат калия (285 мг, 2.10 ммоль) добавляли в N,N-диметилформамид (5.0 мл) и оставляли для прохождения реакции при комнатной температуре на 3 часа.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (20 мл × 3 раза). Органические фазы объединяли и сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 230 мг трет-бутил (S)-4-(2-(4-(3-хлор-2,6- дифторфенил)-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата в виде бледно- желтого твердого вещества (выход: 96.8%). LCMS: RT = 4.60 мин, [M+H]+ = 566.13.
Стадия F: синтез (S)-4-(2-(4-(3-хлор-2,6-дифторфенил)-6-оксопиридазин- 1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
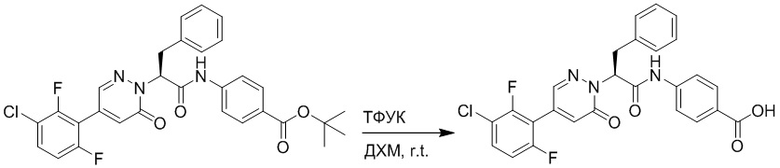
При комнатной температуре добавляли трет-бутил (S)-4-(2-(4-(3-хлор-2,6- дифторфенил)-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоат (230 мг, 0.40 ммоль) в дихлорметан (4.0 мл), затем добавляли по каплям трифторуксусную кислоту (1.0 мл). Смесь оставляли для прохождения реакции при комнатной температуре на 3 часа.
После окончания реакции, дихлорметан упаривали досуха и трифторуксусную кислоту досуха отгоняли на масляном насосе. Остаток от упаривания растворяли в дихлорметане (2.0 мл) и добавляли по каплям в н-гексан (30.0 мл), при этом выпадало в осадок белое твердое вещество. После фильтрования при отсасывании, осадок на фильтре промывали н-гексаном и сушили, получая 5 мг (S)-4-(2-(4-(3-хлор-2,6-дифторфенил)-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты в виде светло-желтого твердого вещества (выход: 75.8%). LCMS: RT = 4.02 мин, [M-H]- = 508.06. 1H ЯМР (500 МГц, ДМСО) δ 12.76 (с, 1H), 10.66 (с, 1H), 8.19 (д, J = 1.4 Гц, 1H), 7.92 (д, J = 8.8 Гц, 2H), 7.88–7.81 (м, 1H), 7.72 (д, J = 8.8 Гц, 2H), 7.40 (тд, J = 9.0, 1.4 Гц, 1H), 7.33–7.25 (м, 4H), 7.20 (т, J = 7.6 Гц, 2H), 5.90 (дд, J = 9.3, 6.0 Гц, 1H), 3.57–3.46 (м, 2H).
Пример 43
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксо-3-фенилпиридазин-1(6H)-ил)- 3-фенилпропанамидо)бензойной кислоты
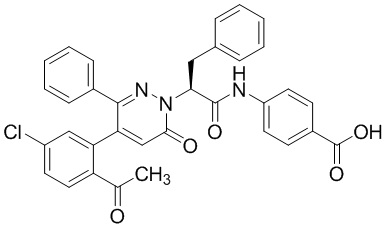
Применяли описанный ниже путь синтеза.
Стадия A: синтез 5-(2-ацетил-5-хлорфенил)-6-фенилпиридазин-3(2H)-она
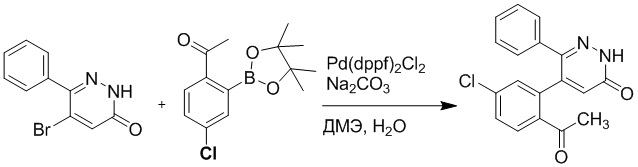
5-Бром-6-фенилпиридазин-3(2H)-он (150 мг, 0.60 ммоль) и 1-(4-хлор-2-(4,4,5,5- тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этан-1-он (181 мг, 0.64 ммоль) растворяли в диметиловом эфире этиленгликоля (10.0 мл) и воде (2.0 мл). После этого добавляли в полученный раствор карбонат натрия (126 мг, 1.10 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]палладий дихлорид (69 мг, 0.090 ммоль). Реакционную смесь перемешивали при 120°C в течение 4 часов.
Добавляли воду (50 мл) для разбавления реакционного раствора. Смесь экстрагировали этилацетатом (20 мл × 3 раза). Органические фазы объединяли. Органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (20 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/2). Получали 30 мг твердого 5-(2-ацетил-5-хлорфенил)-6-фенилпиридазин-3(2H)-она (выход: 16.0%). LCMS: RT = 3.55 мин, [M+H]+ = 299.20.
Стадия B: синтез трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксо-3- фенилпиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата
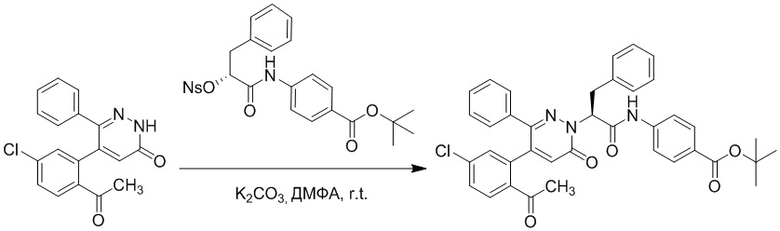
5-(2-Ацетил-5-хлорфенил)-6-фенилпиридазин-3(2H)-он (30 мг, 0.092 ммоль) и трет-бутил (R)-4-(2-(((4-нитрофенил)сульфонил)окси)-3-фенилпропанамидо)бензоат (58 мг, 0.11 ммоль) растворяли в N,N-диметилформамиде (10.0 мл). После этого добавляли в полученный раствор карбонат калия (25 мг, 0.18 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов.
Добавляли воду (20 мл) для разбавления реакционного раствора. Смесь экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли. Органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Получали 18 мг сырого трет-бутил (S)-4-(2-(4-(2-ацетил-5- хлорфенил)-6-оксо-3-фенилпиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата (выход: 30.0%). LCMS: RT = 4.41 мин, [M+H]+ = 647.13.
Стадия C: синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксо-3- фенилпиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
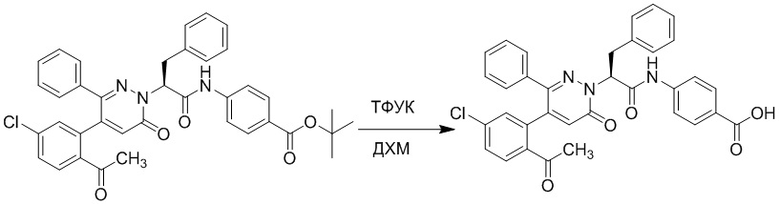
трет-Бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксо-3-фенилпиридазин-1(6H)-ил)- 3-фенилпропанамидо)бензоат (18 мг, 0.027 ммоль) растворяли в дихлорметане (4.0 мл). После этого добавляли в полученный раствор трифторуксусную кислоту (1.0 мл), и смесь перемешивали при комнатной температуре в течение 2 часов.
Реакционный раствор упаривали при пониженном давлении и очищали методом препаративной высокоэффективной жидкостной хроматографии, получая 13 мг белой твердой (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-6-оксо-3-фенилпиридазин-1(6H)-ил)-3- фенилпропанамидо)бензойной кислоты (выход: 81.0%). LCMS: RT = 4.20 мин, [M+H]+ = 592.14.
Пример 44
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)- N-метил-3-фенилпропанамидо)бензойной кислоты
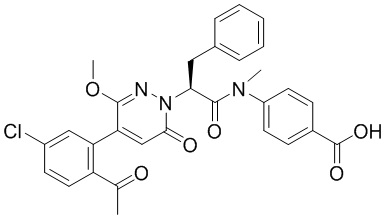
Применяли описанный ниже путь синтеза.
Стадия A: синтез метил (R)-4-(2-хлор-N-метил-3-фенилпропанамидо)бензоата
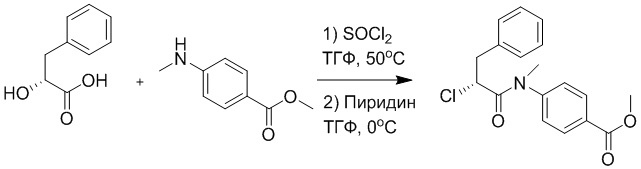
D-фенилмолочную кислоту (0.5 г, 3.0 ммоль) растворяли в сухом тетрагидрофуране (40.0 мл) и помещали в сухую трехгорлую колбу. В инертной атмосфере азота перемешивали на ледяной бане 15 минут и медленно добавляли по каплям в реакционный раствор тионилхлорид (0.7 мл, 9.0 ммоль). Через 30 минут прикапывание было завершено. Смесь нагревали до 70°C и перемешивали при постоянной температуре в течение 5 часов. Реакционный раствор охлаждали до комнатной температуры, упаривали на роторном испарителе, упаривали на масляном насосе 15 минут и затем растворяли в ТГФ, получая раствор A. Метил 4-(метиламино)бензоат (500 мг, 3.0 ммоль) и диизопропилэтиламин (1.5 мл, 9.0 ммоль) растворяли в сухом тетрагидрофуран (20.0 мл) и помещали в сухую трехгорлую колбу. В инертной атмосфере азота перемешивали на ледяной бане 15 минут и медленно добавляли по каплям раствор A. Реакционную смесь перемешивали на ледяной бане в течение 1 часа и отслеживали полноту протекания реакции методом LCMS.
Добавляли в реакционный раствор воду, чтобы погасить реакцию. Смесь экстрагировали этилацетатом (40 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (20 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/4), получали 600 мг метил (R)-4-(2-хлор-N-метил-3-фенилпропанамидо)бензоата в виде желтого твердого вещества (выход: 67.0%). LCMS: RT = 4.10 мин, [M+H]+ = 332.11.
Стадия B: синтез метил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-N-метил-3-фенилпропанамидо)бензоата
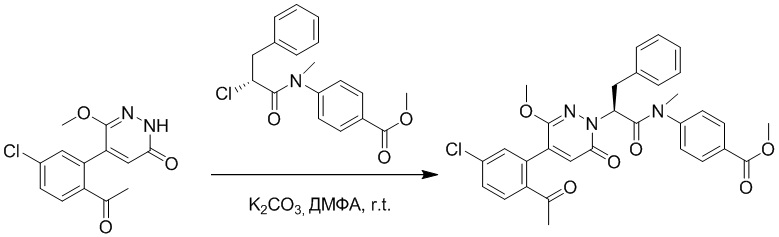
5-(2-Ацетил-5-хлорфенил)-6-этоксипиридазин-3(2H)-он (60 мг, 0.22 ммоль) растворяли в N,N-диметилформамиде (2.0 мл). После этого добавляли в полученный раствор карбонат калия (60 мг, 0.44 ммоль) и метил (R)-4-(2-хлор-N-метил-3- фенилпропанамидо)бензоат (100 мг, 0.32 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов.
Добавляли в реакционный раствор воду, чтобы погасить реакцию. Смесь экстрагировали этилацетатом (20 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/1). Получали 40 мг метил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-N- метил-3-фенилпропанамидо)бензоата в виде желтого твердого вещества (выход: 32.0%). LCMS: RT = 4.11 мин, [M+H]+ = 572.08.
Стадия C: синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-N-метил-3-фенилпропанамидо)бензойной кислоты
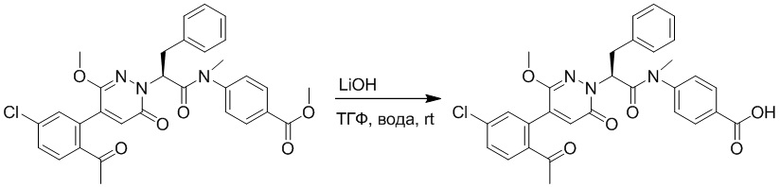
Метил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)- N-метил-3-фенилпропанамидо)бензоат (40 мг, 0.07 ммоль) растворяли в тетрагидрофуране (4.0 мл) и воде (1.0 мл). После этого добавляли гидроксид лития (6 мг, 0.14 ммоль) в полученный раствор, затем перемешивали при комнатной температуре в течение 6 часов.
Добавляли в реакционный раствор воду (10 мл), и смесь экстрагировали этилацетатом (20 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом препаративной высокоэффективной жидкостной хроматографии, получая 16 мг (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-N-метил-3-фенилпропанамидо)бензойной кислоты в виде желтого твердого вещества (выход: 41.0%). LCMS: RT = 3.87 мин, [M+H] + = 560.14. 1H ЯМР (500 МГц, ДМСО) δ 12.97 (с, 1H), 7.99 (д, J = 8.4 Гц, 1H), 7.88 (д, J = 8.4 Гц, 2H), 7.68 (дд, J = 8.4, 2.2 Гц, 1H), 7.34 (м, 3H), 7.15 (м, 3H), 6.99 (с, 2H), 6.55 (с, 1H), 5.57 (с, 1H), 3.58 (с, 3H), 3.29 (д, J = 14.1, 5.6 Гц, 1H), 3.19 (с, 3H), 3.16 (с, 1H), 2.54 (с, 3H).
Пример 45
Синтез (S)-4-(2-(4-(5-хлор-2-(2-метоксиацетил)фенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
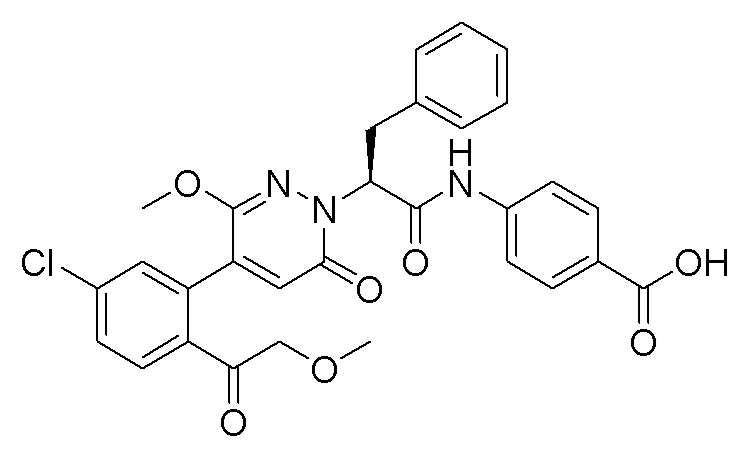
Применяли описанный ниже путь синтеза.
Стадия A: синтез (S)-4-(2-(4-(5-хлор-2-(2-метоксиацетил)фенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
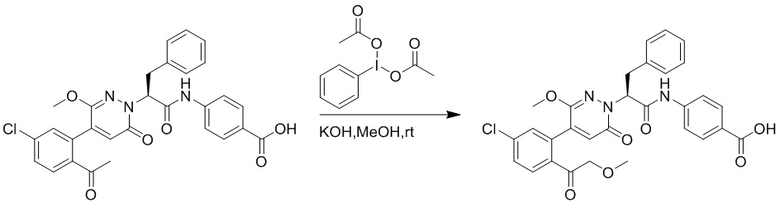
(S)-4-(2-(4-(2-Ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)бензойную кислоту (80 мг, 0.15 ммоль) растворяли в метаноле (4.0 мл). После этого добавляли в полученный раствор гидроксид калия (33 мг, 0.60 ммоль) и диацетилиодбензол (60 мг, 0.18 ммоль), и смесь перемешивали при комнатной температуре в течение 1 часа.
Добавляли воду (10 мл) в реакционный раствор, добавляли разбавленную соляную кислоту (1.0 мл), и смесь экстрагировали этилацетатом (20 мл × 3 раза). Органические фазы объединяли, и органическую фазу промывали насыщенным водным раствором хлорида натрия (10 мл × 3 раза), сушили над безводным сульфатом натрия, фильтровали и упаривали при пониженном давлении. Остаток от упаривания очищали методом препаративной высокоэффективной жидкостной хроматографии, получая 47 мг (S)-4-(2-(4-(5-хлор-2-(2-метоксиацетил)фенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)бензойной кислоты в виде желтого твердого вещества (выход: 55.0%). LCMS: RT = 3.97 мин, [M+H]+ = 576.15.
Пример 46
Синтез 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)- 4-метилпентанамидо)бензойной кислоты
Применяли описанный ниже путь синтеза.
Стадия A: синтез 2-гидрокси-4-метилпентановой кислоты
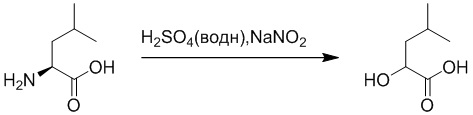
L-лейцин (2.0 г, 15.2 ммоль) растворяли в 1 M серной кислоте (40.0 мл), медленно добавляли по каплям водный раствор нитрита натрия (8.0 g) при охлаждении на бане из соли со льдом. Температуре давали естественным образом подняться до комнатной, и смесь перемешивали в течение ночи.
Реакционный раствор упаривали при пониженном давлении, и полученный остаток очищали методом препаративной высокоэффективной жидкостной хроматографии, получая 2.0 г жидкой 2-гидрокси-4-метилпентановой кислоты (выход: 100.0%). LCMS: RT = 3.69 мин.
Стадия B: синтез трет-бутил 4-(2-гидрокси-4-метилпентанамидо)бензоата
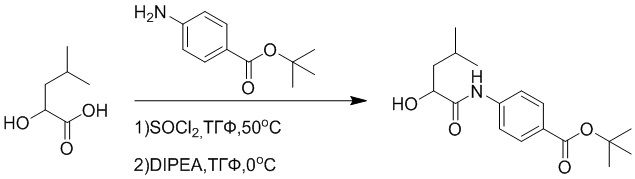
2-Гидрокси-4-метилпентановую кислоту (2.0 г, 15.0 ммоль) растворяли в сухом тетрагидрофуране (40.0 мл) и помещали в сухую трехгорлую колбу. В инертной атмосфере азота смесь перемешивали на ледяной бане 15 минут и медленно добавляли по каплям тионилхлорид (3.6 г, 30.0 ммоль) в реакционный раствор. Прикапывание заканчивалось примерно за 30 минут. Нагревали реакционную смесь до 50°C и перемешивали при постоянной температуре 3 часа, затем раствор охлаждали до комнатной температуры, упаривали на роторном испарителе, откачивали на масляном насосе 15 минут, и остаток растворяли в ТГФ, получая раствор A. Растворяли трет-бутил 4-аминобензоат (2.5 г, 12.8 ммоль) и диизопропилэтиламин (5.3 мл, 45.0 ммоль) в сухом тетрагидрофуране (20.0 мл) и помещали в сухую трехгорлую колбу. В инертной атмосфере азота реакционную смесь перемешивали на ледяной бане 15 минут и медленно добавляли по каплям раствор A. Перемешивали на ледяной бане в течение 1 часа, затем протекание реакции мониторили методом LCMS до полного завершения.
Добавляли в реакционный раствор воду, чтобы погасить реакцию. Смесь экстрагировали этилацетатом (40 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (30 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/4), получая 224 мг трет-бутил 4-(2-гидрокси-4-метилпентанамидо)бензоата в виде желтого твердого вещества (выход: 6.0%). LCMS: RT = 4.16 мин, [M-H]- = 306.06.
Стадия C: синтез трет-бутил 4-(4-метил-2-(((4-нитрофенил)сульфонил)окси) пентанамидо)бензоата
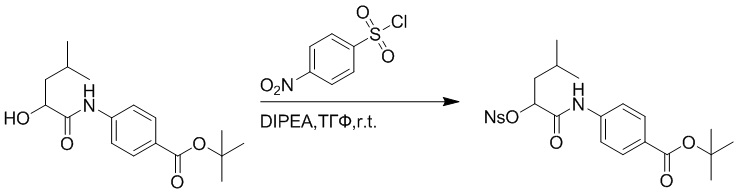
трет-Бутил 4-(2-гидрокси-4-метилпентанамидо)бензоат (224 мг, 0.73 ммоль) и триэтиламин (0.3 мл, 2.1 ммоль) растворяли в дихлорметане (10.0 мл). Добавляли в реакционный раствор 4-нитробензолсульфонилхлорид (221 мг, 1.0 ммоль) при охлаждении на ледяной бане, и смесь перемешивали при комнатной температуре в течение 2 часов.
Насыщенный водный раствор бикарбоната натрия (10 мл) добавляли в реакционный раствор, чтобы погасить реакцию. Смесь экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли. Органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток растворяли в дихлорметане (4 мл), и раствор добавляли по каплям в н-гексан (60 мл) при перемешивании. В осадок выпадало большое количество белого твердого вещества, его отфильтровывали, и осадок на фильтре собирали, получая 300 мг белого твердого трет-бутил 4-(4-метил-2-(((4-нитрофенил)сульфонил)окси)пентанамидо)бензоата (выход: 83.0%). LCMS: RT = 4.44 мин.
Стадия D: синтез трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-4-метилпентанамидо)бензоата
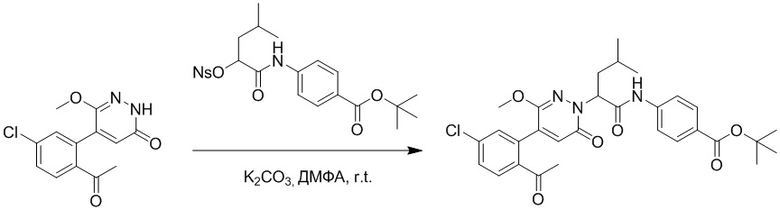
5-(2-Ацетил-5-хлорфенил)-6-метоксипиридазин-3(2H)-он (58 мг, 0.20 ммоль) растворяли в N,N-диметилформамиде (2.0 мл). После этого добавляли в полученный раствор карбонат калия (83 мг, 0.60 ммоль) и трет-бутил 4-(4-метил-2-(((4- нитрофенил)сульфонил)окси)пентаноиламино)бензоат (150.0 мг, 0.30 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов.
Добавляли в реакционный раствор воду, чтобы погасить реакцию. Смесь экстрагировали этилацетатом (20 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/1). Получали 66 мг трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-4- метилпентанамидо)бензоата в виде желтого твердого вещества (выход: 24.0%). LCMS: RT = 4.60 min , [M+H]+ = 568.18.
Стадия E: синтез 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-4-метилпентанамидо)бензойной кислоты
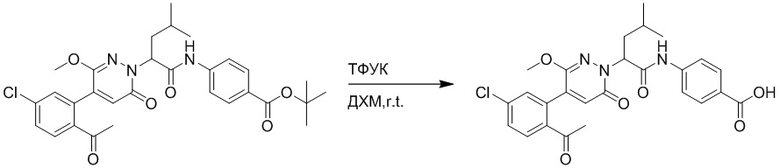
трет-Бутил 4-(2-(4-(5-хлор-2-(2-гидроксиацетил)фенил)-3-метокси-6-оксопиридазин- 1(6H)-ил)-4-метилпентанамидо)бензоат (66 мг, 0.12 ммоль) растворяли в дихлорметане (2.0 мл). После этого добавляли в полученный раствор трифторуксусную кислоту (1.0 мл), и смесь перемешивали при комнатной температуре в течение 1 часа.
Реакционный раствор упаривали при пониженном давлении на воздушной бане. Полученный остаток очищали, суспендируя его в дихлорметане и н-гексане, получая 19 мг трет-бутил 4-(2-(4-(2-ацетил-5-хлорфенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-4- метилпентанамидо)бензоата в виде желтого твердого вещества (выход: 32.0%). LCMS: RT = 3.98 мин, [M-H]- = 510.10. 1H ЯМР (400 МГц, ДМСО) δ 10.45 (с, 1H), 8.01 (д, J = 8.4 Гц, 1H), 7.88 (д, J = 8.8 Гц, 2H), 7.74 – 7.66 (м, 3H), 7.56 (д, J = 2.2 Гц, 1H), 6.95 (с, 1H), 5.45 (дд, J = 10.9, 4.3 Гц, 1H), 3.63 (с, 3H), 2.55 (с, 3H), 2.24 (дд, J = 17.7, 6.8 Гц, 1H), 1.78 (дд, J = 11.6, 6.7 Гц, 1H), 1.58 (с, 1H), 0.94 (д, J = 6.6 Гц, 6H).
Пример 47
Синтез (S)-4-(2-(4-(2-ацетил-5-(трифторметил)фенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-6-фенилпропанамидо)бензойной кислоты
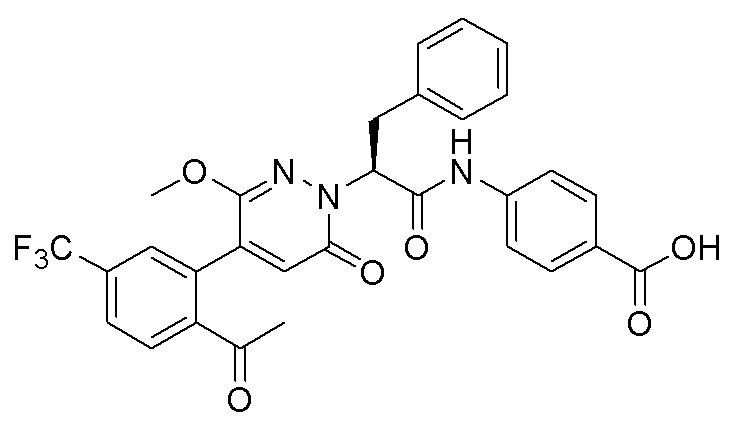
Применяли описанный ниже путь синтеза.
Стадия A: синтез 1-(2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-4- (трифторметил)фенил)этан-1-она
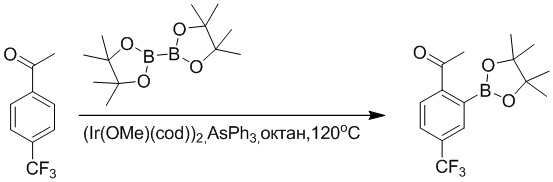
При комнатной температуре растворяли 1-(4-(трифторметил)фенил)этан-1-он (8 мл, 39.6 ммоль) и 4,4,4',4',5,5,5',5'-октаметил-2,2'-бис(1,3,2-диоксаборолан) (2.0 г, 7.9 ммоль) в н-октане (50.0 мл). После этого добавляли в полученный раствор метокси(циклооктадиен)иридий(I) димер (159 мг, 0.24 ммоль) и трифенилмышьяк (146 мг, 0.48 ммоль). Реакционную смесь перемешивали при 120°C в течение 18 часов.
Реакцию гасили добавлением воды в реакционный раствор. Смесь экстрагировали этилацетатом (100 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (50 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/4), получая 1.2 г 1-(2-(4,4,5,5- тетраметил-1,3,2-диоксаборолан-2-ил)-4-(трифторметил)фенил)этан-1-она в виде желтого твердого вещества (выход: 49.0%). LCMS: RT = 3.91 мин.
Стадия B: синтез 5-(2-ацетил-5-(трифторметил)фенил)-6-метокси-2- (4-метоксибензил)пиридазин-3(2H)-она
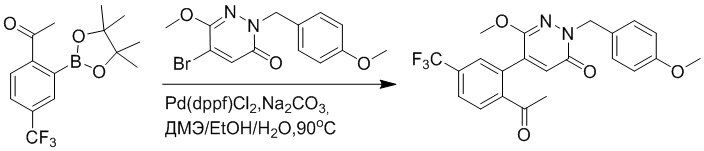
При комнатной температуре помещали в трехгорлую колбу 5-бром-6-метокси- 2-(4-метоксибензил)пиридазин-3(2H)-он (310 мг, 1.0 ммоль), 1-(2-(4,4,5,5-тетраметил- 1,3,2-диоксаборолан-2-ил)-4-(трифторметил)фенил)этан-1-он (470 мг, 1.5 ммоль) и карбонат натрия (210 мг, 2.0 ммоль). После заполнения колбы азотом добавляли диметиловый эфир этиленгликоля (8 мл), этанол (1 мл) и воду (1 мл). Продували колбу азотом и добавляли комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия с дихлорметаном (59 мг, 0.07 ммоль). Продували колбу азотом, повышали температуру до 90°C и продолжали реакцию 2 часа.
Реакционный раствор охлаждали до комнатной температуры, фильтровали через слой диатомита, осадок на фильтре промывали этилацетатом (30 мл × 2 раза). Фильтрат и промывной раствор объединяли и упаривали при пониженном давлении. В полученный остаток добавляли воду (50 мл). Смесь экстрагировали этилацетатом (50 мл × 3 раза), и органические фазы объединяли. Органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (20 мл × 3 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/петролейный эфир = 1/2). Получали 180 мг 5-(2-ацетил-5-(трифторметил)фенил)-6-метокси-2-(4- метоксибензил)пиридазин-3(2H)-она в виде желтого твердого вещества (выход: 42.0%). LCMS: RT = 4.04 мин, [M+H]+ = 433.10.
Стадия C: синтез 5-(2-ацетил-5-(трифторметил)фенил)-6-метоксипиридазин- 3(2H)-она
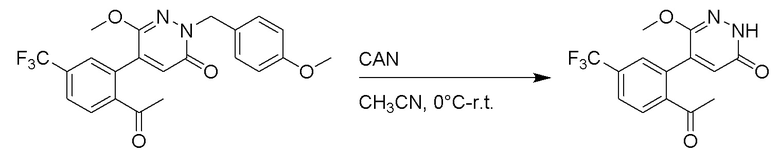
При 0°C добавляли 5-(2-ацетил-5-(трифторметил)фенил)-6-метокси-2-(4- метоксибензил)пиридазин-3(2H)-он (180 мг, 0.41 ммоль) в ацетонитрил (6 мл) и воду (2 мл), затем медленно добавляли церийаммоний нитрат (2.1 г, 4.1 ммоль). После окончания добавления реакционную смесь перемешивали при комнатной температуре 30 минут.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (30 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/1). Получали 80 мг 5-(2-ацетил-5- (трифторметил)фенил)-6-метоксипиридазин-3(2H)-она в виде желтого твердого вещества (выход: 63.0%). LCMS: RT = 3.33 мин, [M+H] + = 313.07.
Стадия D: синтез трет-бутил (S)-4-(2-(4-(2-ацетил-5-(трифторметил)фенил)-3- метокси-6-оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоата
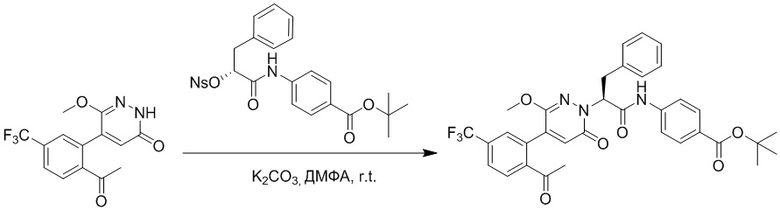
При комнатной температуре добавляли 5-(2-ацетил-5-(трифторметил)фенил)-6- метоксипиридазин-3(2H)-он (45 мг, 0.14 ммоль), трет-бутил (R)-4-(2-(((4-нитрофенил) сульфонил)окси)-3-фенилпропанамидо)бензоат (114 мг, 0.22 ммоль) и карбонат калия (38 мг, 0.28 ммоль) в N,N-диметилформамид (2.0 мл) и оставляли для прохождения реакции на ночь при комнатной температуре.
После окончания реакции, реакционную смесь гасили добавлением воды, и смесь экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (10 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/1). Получали 58 мг трет-бутил (S)-4-(2-(4-(2-ацетил-5-(трифторметил)фенил)-3-метокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)бензоата в виде светло-желтого твердого вещества (выход: 43.0%). LCMS: RT = 4.52 мин, [M-H]- = 634.15.
Стадия E: синтез (S)-4-(2-(4-(2-ацетил-5-(трифторметил)фенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
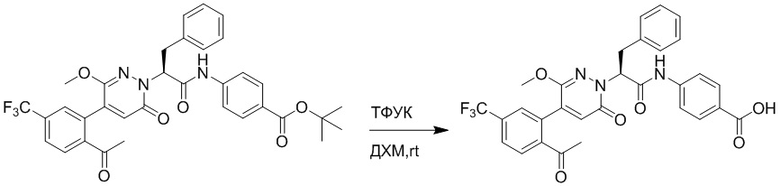
трет-Бутил (S)-4-(2-(4-(2-ацетил-5-(трифторметил)фенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензоат (80 мг, 0.12 ммоль) растворяли в дихлорметане (2.0 мл). После этого добавляли в полученный раствор трифторуксусную кислоту (0.5 мл), и смесь перемешивали при комнатной температуре в течение 1 часа.
Реакционный раствор упаривали при пониженном давлении на воздушной бане. Остаток от упаривания очищали, суспендируя его в дихлорметане и н-гексане, получая 33 мг желтой твердой (S)-4-(2-(4-(2-ацетил-5-(трифторметил)фенил)-3-метокси-6- оксопиридазин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты (выход: 45.0%). LCMS: RT = 3.99 мин, [M+H]+ = 580.09. 1H ЯМР (400 МГц, ДМСО) δ 10.48 (с, 1H), 8.14 (д, J = 8.0 Гц, 1H), 7.98 (д, J = 8.0 Гц, 1H), 7.89 (д, J = 8.8 Гц, 2H), 7.77 (с, 1H), 7.71 (д, J = 8.8 Гц, 2H), 7.34 – 7.23 (м, 5H), 7.18 (т, J = 6.8 Гц, 1H), 6.99 (с, 1H), 5.74 (дд, J = 10.1, 4.9 Гц, 1H), 3.67 (с, 3H), 3.56 – 3.46 (м, 1H), 3.41 (дд, J = 14.1, 5.1 Гц, 1H), 2.57 (с, 3H).
Пример 48. Соединение A
Синтез (S)-4-(2-(4-(5-хлор-2-(4-хлор-1H-1,2,3-триазол-1-ил)фенил)-6- оксопиримидин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
Соединение A
Применяли описанный ниже путь синтеза.
Стадия A: синтез 4-хлор-2-(тетраметил-1,3,2-диоксаборолан-2-ил)анилина
2-Бром-4-хлоранилин (3.0 г, 14.5 ммоль), 4,4,5,5-тетраметил-2-(тетраметил-1,3,2- диоксаборолан-2-ил)-1,3,2-диоксаборолан (38 г, 150.0 ммоль), ацетат калия (2.9 г, 30.0 ммоль) и комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия с дихлорметаном (1.1 г, 1.5 ммоль) растворяли в диметилсульфоксиде (75 мл). В инертной атмосфере азота реакционную смесь нагревали при 80°C в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры. Добавляли воду для растворения соли, и реакционный раствор фильтровали. Твердый осадок суспендировали в дихлорметане, и нерастворенный осадок отфильтровывали. Фильтрат упаривали и затем очищали методом колоночной хроматографии на силикагеле, получая 5.2 г белого твердого 4-хлор-2-(тетраметил-1,3,2-диоксаборолан-2-ил)анилина (выход: 100%). LCMS: RT = 4.40 мин, [M+H]+ = 254.10.
Стадия B: синтез 4-хлор-2-(6-метоксипиримидин-4-ил)анилина
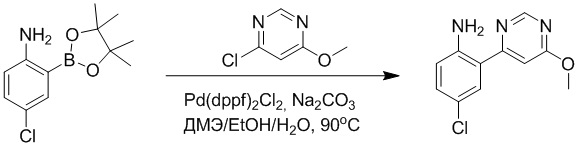
4-Хлор-6-метоксипиримидин (3.9 г, 15.4 ммоль), карбонат натрия (3.2 г, 30.8 ммоль), диметиловый эфир этиленгликоля (16 мл), этанол (2 мл) и воду (2 мл) помещали в трехгорлую колбу. В инертной атмосфере азота добавляли комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия с дихлорметаном (1.3 г, 1.5 ммоль). Добавляли 4-хлор-2-(тетраметил-1,3,2-диоксаборолан-2-ил)анилин (3.31 г, 23.1 ммоль) в диметиловом эфире этиленгликоля (8 мл), и реакционный раствор нагревали при 90°C в течение 2 часов. После окончания реакции согласно данным LCMS мониторинга, смесь охлаждали до комнатной температуры, фильтровали через слой диатомита, и осадок на фильтре промывали три раза этилацетатом (30 мл). Фильтрат и промывной раствор объединяли, промывали один раз водой и два раза насыщенным раствором хлорида аммония. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и упаривали на роторном испарителе. Остаток очищали методом колоночной хроматографии на силикагеле, получая 1.0 г 4-хлор-2-(6-метоксипиримидин-4-ил)анилина в виде желтого твердого вещества (выход: 28%). LCMS: RT = 3.95 мин, [M+H]+ = 236.04.
Стадия C: синтез 4-{5-хлор-2-[4-(триметилсилил)-1H-1,2,3-триазол-1-ил]-фенил}- 6-метокси-пиримидина
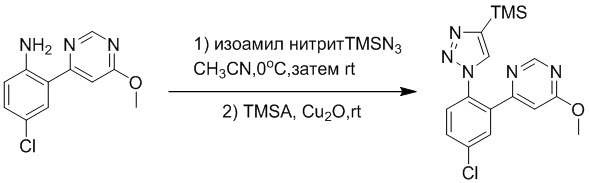
4-Хлор-2-(6-метоксипиримидин-4-ил)анилин (0.9 г, 3.8 ммоль) растворяли в ацетонитриле (60 мл). Добавляли 3-метилбутилнитрит (0.6 мл, 5.8 ммоль) при 0°C, затем добавляли по каплям азидотриметилсилан (0.6 мл, 5.8 ммоль). Наблюдалось выделение газа. Через 10 минут охлаждающую баню убирали, и реакционную смесь нагревали до комнатной температуры. Через 1 час добавляли этинилтриметилсилан (1.8 мл, 11.4 ммоль) и оксид меди (0.06 г, 0.36 ммоль), и реакционную смесь перемешивали еще 1 час. Добавляли в реакционный раствор этилацетат и насыщенный водный раствор хлорида аммония для разделения слоев. Органическую фазу промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и упаривали. Дальнейшую очистку проводили методом колоночной хроматографии на силикагеле, получая 730 мг 4-{5-хлор-2-[4-(триметилсилил)-1H-1,2,3-триазол-1-ил]-фенил}-6-метокси- пиримидина в виде желтого твердого вещества (выход: 45%). LCMS: RT = 2.04 мин, [M+H]+ = 360.10.
Стадия D: синтез 4-[5-хлор-2-(4-хлор-1H-1,2,3-триазол-1-ил)фенил]-6- метоксипиримидина
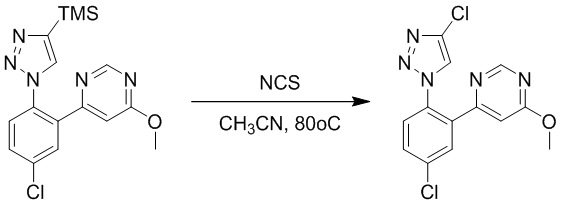
4-{5-Хлор-2-[4-(триметилсилил)-1H-1,2,3-триазол-1-ил]фенил}-6-метоксипиримидин (700 мг, 1.94 ммоль) растворяли в ацетонитриле (20 мл) и добавляли в раствор N-хлорсукцинимид (0.9 г, 7.2 ммоль) и силикагель (2.9 г, 50.44 ммоль). Реакционный раствор перемешивали при 80°C в течение 1 час. Затем раствор фильтровали, и отделенный силикагель промывали этилацетатом. Фильтрат промывали водой и насыщенным водным раствором хлорида натрия и упаривали. Остаток далее очищали методом колоночной хроматографии на силикагеле, получая 450 мг 4-[5-хлор-2-(4-хлор-1H-1,2,3-триазол-1-ил)фенил]-6-метоксипиримидина в виде желтого твердого вещества (выход 72%). LCMS: RT = 2.00 мин, [M+H]+ = 322.05.
Стадия E: синтез 6-[5-хлор-2-(4-хлор-1H-1,2,3-триазол-1-ил)фенил]пиримидин- 4-ола
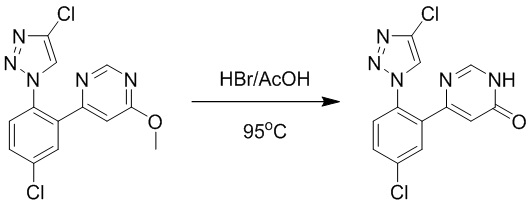
48%-ный водный раствор бромистоводородной кислоты (1.5 мл, 13.3 ммоль) добавляли в раствор 4-[5-хлор-2-(4-хлор-1H-1,2,3-триазол-1-ил)фенил]-6- метоксипиримидина (450 мг, 1.4 ммоль) в уксусной кислоте (3 мл). Смесь перемешивали при 95°C в течение 1 часа. Реакционный раствор упаривали досуха, затем разделяли между этилацетатом и насыщенным водным раствором бикарбоната натрия. Органическую фазу упаривали, и остаток очищали методом колоночной хроматографии на силикагеле, получая 190 мг 6-[5-хлор-2-(4-хлор-1H-1,2,3-триазол-1- ил)фенил]пиримидин-4-ола в виде желтого твердого вещества (выход: 44%). LCMS: RT = 1.74 мин, [M-H]- = 305.97.
Стадия F: синтез трет-бутил (S)-4-(2-(4-(5-хлор-2-(4-хлор-1H-1,2,3-триазол-1-ил) фенил)-6-оксопиримидин-1(6H)-ил)-3-фенилпропанамидо)бензоата
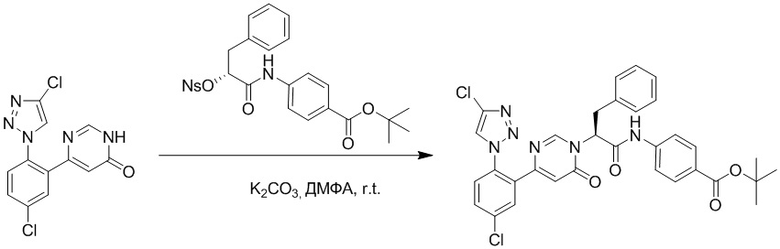
При комнатной температуре добавляли 6-[5-хлор-2-(4-хлор-1H-1,2,3-триазол- 1-ил)фенил]пиримидин-4-ол (45 мг, 0.15 ммоль), трет-бутил (R)-4-(2-(((4-нитрофенил) сульфонил)окси)-3-фенилпропанамидо)бензоат (93 мг, 0.18 ммоль) и карбонат калия (40 мг, 0.3 ммоль) в N,N-диметилформамид (3.0 мл), и смесь перемешивали при комнатной температуре в течение ночи. Добавляли в реакционный раствор воду, чтобы погасить реакцию. Смесь экстрагировали этилацетатом (40 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (30 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле, получая 150 мг желтый жидкий трет-бутил (S)-4-(2-(4-(5-хлор-2-(4-хлор-1H-1,2,3-триазол-1-ил)фенил)-6-оксопиримидин-1(6H)-ил)-3- фенилпропанамидо)бензоат (выход: 59%). LCMS: RT =2.00 мин, [M+H]+ =631.18.
Стадия F: синтез (S)-4-(2-(4-(5-хлор-2-(4-хлор-1H-1,2,3-триазол-1-ил)фенил)-6- оксопиримидин-1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты
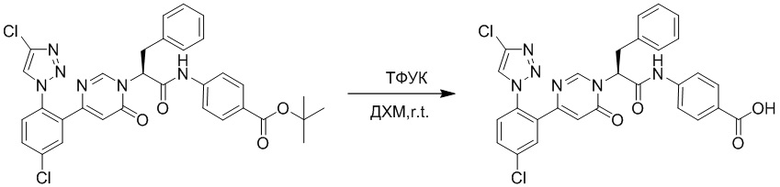
трет-Бутил (S)-4-(2-(4-(5-хлор-2-(4-хлор-1H-1,2,3-триазол-1-ил)фенил)-6- оксопиримидин-1(6H)-ил)-3-фенилпропанамидо)бензоат (150 мг, 0.25 ммоль) растворяли в дихлорметане (2.0 мл). После этого добавляли в полученный раствор трифторуксусную кислоту (0.5 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционный раствор упаривали при пониженном давлении на воздушной бане. Остаток от упаривания очищали методом препаративной хроматографии, получая 70 мг белой твердой (S)-4-(2-(4-(5-хлор-2-(4-хлор-1H-1,2,3-триазолe-1-ил)фенил)-6-оксопиримидин- 1(6H)-ил)-3-фенилпропанамидо)бензойной кислоты (выход: 59%). LCMS: RT = 2.00 мин, [M+H]+ = 573.16. 1H ЯМР (400 МГц, CD3OD) δ 10.36 (с, 1H), 8.36 (с, 1H), 8.18 (с, 1H), 7.87 (дд, J = 12.0, 5.1 Гц, 2H), 7.72 (д, J = 2.3 Гц, 1H), 7.66–7.47 (м, 4H), 7.28–7.07 (м, 5H), 6.22 (д, J = 0.8 Гц, 1H), 5.74 (дд, J = 10.5, 6.2 Гц, 1H), 3.49 (дд, J = 14.1, 6.3 Гц, 1H), 3.34–3.24 (м, 1H).
Пример 49. Соединение B
Синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-5-метокси-2-оксопиридиний-1(2H)- ил)-3-фенилпропанамидо)бензойной кислоты
Соединение B
Применяли описанный ниже путь синтеза.
Стадия A: синтез (2,5-диметоксипиридин-4-ил)бороновой кислоты
2,5-Диметоксипиридин (10.0 г, 71.9 ммоль) растворяли в сухом тетрагидрофуране (40 мл) и помещали в сухую трехгорлую колбу. В инертной атмосфере азота смесь перемешивали при охлаждении в бане сухой лед/этанол в течение 15 минут и медленно добавляли по каплям в реакционный раствор диизопропиламид лития (20 мл, 2.0M раствор в ТГФ). Прикапывание заканчивалось примерно за 30 минут. После 3 часов перемешивания при охлаждении в бане сухой лед/этанол, добавляли тиизопропилборат (33.0 мл, 143.8 ммоль), реакционной смеси позволяли нагреться естественным образом до комнатной температуры и перемешивали при постоянной температуре 18 часов. После окончания реакции согласно данным LCMS мониторинга, добавляли в реакционный раствор разбавленную соляную кислоту, доводя значение pH до 3-4. После 15 минут перемешивания отгоняли растворитель на роторном испарителе, и остаток суспендировали в ацетонитриле, получая 10.6 г (2,5-диметоксипиридин-4-ил)бороновой кислоты в виде белого твердого вещества (выход: 80%). LCMS: RT = 1.73 мин, [M+H]+ = 184.08.
Стадия B: синтез 1-(4-хлор-2-(2,5-диметоксипиридин-4-ил)фенил)этан-1-она
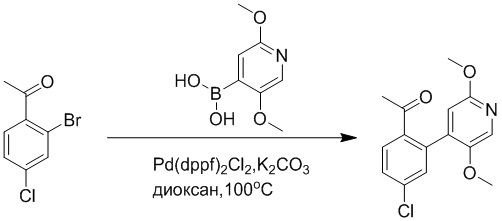
2-Бром-4-хлорацетофенон (14.8 г, 63.6 ммоль) и (2,5-диметоксипиридин-4- ил)бороновую кислоту (9.7 г, 53.0 ммоль) растворяли в 1,4-диоксане (40 мл), добавляли раствор карбоната калия (14.6 г, 106 моль) в воде (10 мл) и помещали смесь в сухую трехгорлую колбу. В инертной атмосфере азота добавляли в реакционный раствор комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия с дихлорметаном (3.87 г, 5.3 ммоль). В инертной атмосфере азота смесь нагревали до 100°C и перемешивали при постоянной температуре 18 часов. После окончания реакции согласно данным LCMS мониторинга, смесь охлаждали до комнатной температуры и фильтровали через слой диатомита. Осадок на фильтре промывали три раза этилацетатом (30 мл), фильтрат и промывной раствор объединяли, промывали один раз водой и два раза насыщенным раствором хлорида аммония. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и упаривали на роторном испарителе досуха. Остаток очищали методом колоночной хроматографии на силикагеле, получая 8.2 г 1-(4-хлор-2-(2,5-диметоксипиридин-4-ил)фенил)этан-1-она в виде желтого твердого вещества (выход: 53%). LCMS: RT = 4.03 мин, [M+H]+ = 292.03.
Стадия C: синтез 4-(2-ацетил-5-хлорфенил)-5-метоксипиридин-2(1H)-она
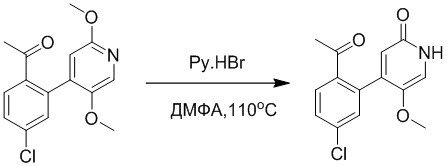
1-(4-Хлор-2-(2,5-диметоксипиридин-4-ил)фенил)этан-1-он (8.2 г, 28 ммоль) и пиридин гидробромид (22 г, 140 ммоль) растворяли в N,N-диметилформамиде (20 мл) и помещали в сухую колбу. В инертной атмосфере азота смесь нагревали до 110°C и перемешивали при постоянной температуре 4 часа. После окончания реакции согласно данным LCMS мониторинга, смесь охлаждали до комнатной температуры. Реакционный раствор добавляли по каплям в 100 мл воды и добавляли 5%-ный раствор карбоната натрия, доводя значение pH до 10-11. Смесь экстрагировали четыре раза дихлорметаном (40 мл × 4). Органические фазы объединяли, и органическую фазу сушили над безводным сульфатом натрия, фильтровали и упаривали на роторном испарителе. Остаток растворяли в ДХМ (10 мл), затем раствор добавляли по каплям в н-гексан (120 мл), при этом выпадало большое количество твердого осадка, который отфильтровывали. Осадок на фильтре, представляющий собой сырой продукт, собирали и далее очищали методом колоночной хроматографии на силикагеле, получая 6.4 г 4-(2-ацетил-5-хлорфенил)-5- метоксипиридин-2(1H)-она в виде желтого твердого вещества (выход: 82%). LCMS: RT = 3.81 мин, [M-H]- = 277.04.
Стадия D: синтез трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-5-метокси-2- оксопиридиний-1(2H)-ил)-3-фенилпропанамидо)бензоата
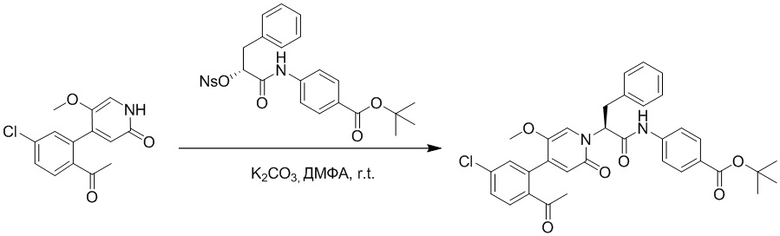
При комнатной температуре добавляли 4-(2-ацетил-5-хлорфенил)-5- метоксипиридин-2(1H)-он (1.5 г, 5.4 ммоль), трет-бутил (R)-4-(2-(((4-нитрофенил) сульфонил)окси)-3-фенилпропанамидо)бензоат (4.0 г, 7.6 ммоль) и карбонат калия (1.5 г, 10.8 ммоль) в N,N-диметилформамид (20.0 мл) и оставляли для прохождения реакции на ночь при комнатной температуре. Добавляли в реакционный раствор воду, чтобы погасить реакцию. Смесь экстрагировали этилацетатом (40 мл × 3 раза). Органические фазы объединяли, и органическую фазу сначала промывали насыщенным водным раствором хлорида натрия (30 мл × 2 раза), затем сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии на силикагеле (элюент: этилацетат/н-гексан = 1/2). Получали 1.9 г трет-бутил (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-5-метокси-2-оксопиридиний-1(2H)-ил)-3- фенилпропанамидо)бензоата в виде желтого твердого вещества (выход: 59%). LCMS: RT = 4.42 мин, [M+H]+ = 601.18.
Стадия E: синтез (S)-4-(2-(4-(2-ацетил-5-хлорфенил)-5-метокси-2- оксопиридиний-1(2H)-ил)-3-фенилпропанамидо)бензойной кислоты
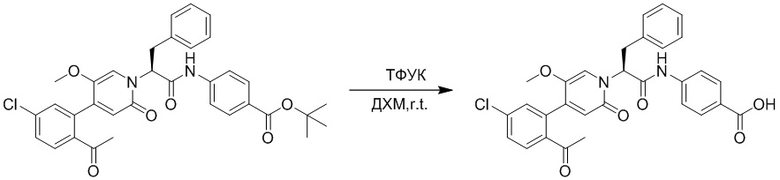
(Трет-бутил S)-4-(2-(4-(2-ацетил-5-хлорфенил)-3-этокси-6-оксопиридазин-1(6H)-ил)-3- фенилпропанамидо)бензоат (1.9 г, 3.2 ммоль) растворяли в дихлорметане (12.0 мл). После этого добавляли в полученный раствор трифторуксусную кислоту (0.5 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционный раствор упаривали при пониженном давлении на воздушной бане. Полученный остаток суспендировали в метаноле и очищали, получая 1.0 г (S)-4-(2-(4-(2-ацетил-5- хлорфенил)-5-метокси-2-оксопиридиний-1(2H)-ил)-3-фенилпропанамидо)бензойной кислоты в виде желтого твердого вещества (выход: 59%). LCMS: RT = 3.88 мин, [M-H]- =543.06. 1H ЯМР (400 МГц, ДМСО) δ 10.82 (с, 1H), 7.92 (д, J = 8.8 Гц, 2H), 7.82 (д, J = 8.3 Гц, 1H), 7.76 (д, J = 8.8 Гц, 2H), 7.61 (дд, J = 8.4, 2.3 Гц, 2H), 7.42 (с, 1H), 7.38 (с, 1H), 7.33–7.23 (м, 4H), 7.22–7.14 (м, 1H), 6.30 (с, 1H), 6.02 (дд, J = 9.5, 6.6 Гц, 1H), 3.53 (с, 3H), 3.49-3.44 (м, 2H), 2.36 (с, 3H).
Пример 50. Определение биологической активности соединений по настоящему изобретению в ингибировании человеческого фактора свертывания крови XIa методом абсорбциометрии
1. Материалы для проведения эксперимента
Фермент: Человеческий Фактор XIa (ENZYME RESEARCH, Cat. No. HFXIa 1111a)
Субстрат: S-2366TM: (CHROMOGENIX, Cat. No. 82109039)
Буфер: 145 мM NaCl, 5 мМ KCl, 1 мг/мл ПЭГ 8000, 30 мМ HEPES, pH 7.4
2. Методика проведения эксперимента
10 мM раствор тестируемого соединения в 100%-ном ДМСО разбавляли 100%-ным ДМСО до концентрации 1000, 200, 40, 8, 1.6, 0.32, 0.064, 0.0128, 0.00256, 0.00128 мкM; добавляли в каждую лунку 96-луночного микропланшета 98 мкл раствора (77.7 нг/мл) фермента FXIa, и в бланковые лунки добавляли 98 мкл буфера. Добавляли 2 мкл соединений в разных концентрациях, а в бланковые и контрольные лунки вместо них добавляли ДМСО. Перемешивали на шейкере и инкубировали при 37°C в течение 20 минут.
Наконец, добавляли в каждую лунку по 100 мкл раствора (800 мкМ) субстрата и измеряли поглощение при 405 нм.
3. Обработка результатов
Построение кривой осуществляли с помощью программы GraphPad Prism, и вычисляли значения IC50, представленные в таблице 1.
Таблица 1. Значения IC50 для соединений по настоящему изобретению в ингибировании человеческого FXIa
Вывод: Соединения по настоящему изобретению обладают очевидной ингибирующей активностью в отношении человеческого FXIa.
Пример 51. Определение антикоагулянтного действия у соединений по настоящему изобретению в плазме крови человека in vitro
1. Материалы для проведения эксперимента
Плазма: Кровь человека собирали в вакуумную пробирку для забора крови, содержащую 3.2% цитрат натрия (объемное соотношение 1:9), центрифугировали при 3000 об/мин в течение 10 минут при комнатной температуре, затем плазму отделяли, распределяли по EP пробиркам и хранили при -80°C.
Реагенты: набор для анализа APTT (набор для определения активированного частичного тромбопластинового времени, mindray), раствор хлорида кальция.
Прибор: анализатор коагуляции крови (mindray, C2000-A)
2. Методика проведения эксперимента
Аликвоты замороженной плазмы крови человека размораживали при комнатной температуре и хорошо перемешивали. 10 мM раствор тестируемого соединения в 100%-ном ДМСО разбавляли 100%-ным ДМСО до концентрации 1500, 750, 375, 187.5, 93.75, 46.88, 23.44, 11.72 мкМ. 98 мкл человеческой плазмы крови помещали в EP пробирку объемом 1.5 мл, затем добавляли 2 мкл соединений в разных концентрациях, а в бланковой группе добавляли 2 мкл 100%-ного ДМСО. Инкубировали на водяной бане при 37°C в течение 10 минут, и образцы помещали в соответствующие положения анализатора коагуляции крови для определения APTT для каждого соединения.
3. Обработка результатов
Построение кривой осуществляли с помощью программы GraphPad Prism, и вычисляли значения EC1.5× и EC2× соответственно, т.е. концентрацию соединений, соответствующую 1.5× и 2× APTT в бланковой контрольной группе. Полученные результаты представлены в таблице 2.
Таблица 2. Антикоагулянтное действие соединений по настоящему изобретению в плазме крови человека in vitro
Вывод: Из результатов, представленных в таблице 2, видно, что соединения по настоящему изобретению обладают очевидным антикоагулянтным действием в плазме крови человека.
Пример 52. Исследование селективности соединений по настоящему изобретению к факторам свертывания крови
1. Материалы для проведения эксперимента
Фермент: hFXa: Человеческий Фактор Xa: 71nkat. hFIIa: HT5146L. hFVIIa: Человеческий Фактор VIIa: hFVIIa 4591L. калликреин: LOT180223.
Субстрат: S-2222TM: CHROMOGENIX, NO864682. S-2238TM: CHROMOGENIX, NO770996. S-2288TM: CHROMOGENIX,NO378902. ADG302.
Буфер:
Буфер для hFXa: 100 мМ NaCl, 5 мМ CaCl2, 33% этиленгликоль, 50 мМ Tris (pH 7.5).
Буфер для hFIIa: 0.145 M NaCl, 0.005 M KCl, 1 мг/мл ПЭГ-8000, 0.030 M HEPES (pH 7.4).
Буфер для hFVIIa: 0.145 M NaCl, 0.005 M KCl, 1 мг/мл ПЭГ-8000, 0.030 M HEPES (pH 7.4).
Буфер для калликреина: 50 мМ Tris, 50 мМ имидазол и 150 мМ NaCl (pH 8.2).
2. Методика проведения эксперимента
10 мM раствор тестируемого соединения в 100%-ном ДМСО разбавляли 100%-ным ДМСО до концентрации 1000, 200, 40, 8, 1.6 мкМ. 98 мкл раствора фермента добавляли в каждую лунку 96-луночного микропланшета, а в бланковые лунки добавляли 98 мкл буфера. Добавляли 2 мкл соединений в разных концентрациях, а в бланковые и контрольные лунки вместо них добавляли ДМСО. Перемешивали на шейкере и инкубировали при 37°C в течение 20 минут.
Концентрации hFXa и S-2222TM составляли: FXa (1:28) и 800 мкмоль/л, соответственно. Концентрации hFIIa и S-2238TM составляли: hFIIa (0.06 Ед/мл) и 500 мкмоль/л, соответственно. Концентрации hFVIIa и S-2288TM составляли: hFVIIa (80 нM) и 1600 мкмоль/л, соответственно. Концентрации калликреина и субстрата составляли: калликреин (20 нM) и 1600 мкмоль/л, соответственно.
Наконец, добавляли в каждую лунку 100 мкл субстрата и измеряли их поглощение при 405 нм.
3. Обработка результатов
Построение кривой осуществляли с помощью программы GraphPad Prism, и вычисляли значения IC50, которые показаны в Таблице 3.
Таблица 3. Исследование селективности соединений по настоящему изобретению к факторам свертывания крови
IC50 (мкM)
IC50 (мкM)
IC50 (мкM)
IC50 (нM)
Вывод: Соединения по настоящему изобретению обладают хорошей селективностью к другим факторам свертывания крови.
Пример 53. Исследование фармакокинетических параметров соединений по настоящему изобретению
1. Материалы для проведения эксперимента
SD крысы: самцы, 180-250 г, куплены в Guangdong Medical Laboratory Animal Center. Яванские макаки: самцы, 4-6 кг, куплены в Guangzhou Chunsheng Biological Research Institute Co., Ltd. Собаки породы бигль: самцы, 8-12 кг, от Kanglong Chemical (Ningbo) New Drug Technology Co., Ltd.
Реагенты: ДМСО (диметилсульфоксид), ПЭГ-400 (полиэтиленгликоль 400), нормальный физраствор, гепарин, ацетонитрил, муравьиная кислота и пропранолол (внутренний стандарт) приобретались у коммерческих поставщиков.
Прибор: Thermo Fisher Scientific LC-MS (U300 UPLC, TSQ QUANTUMN ULTRA тройной квадрупольный масс-спектрометр).
2. Методика проведения эксперимента
Соединение отвешивали и растворяли в системе ДМСО-ПЭГ-400-физраствор (5:60:35, об/об/об). После введения крысам/обезьянам внутривенно или через желудочный зонд, брали 200 мкл венозной крови через 5 минут (не брали при введении через зонд), 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов и 24 часа в гепаринизированные EP пробирки, центрифугировали при 12000 об/мин в течение 2 минут, и полученную плазму замораживали при -80°C до проведения тестирования. Определенное количество тестируемого соединения точно отвешивали и растворяли в ДМСО до концентрации 1 мг/мл, далее этот раствор использовали как стоковый. Необходимое количество стокового раствора соединения аккуратно отбирали пипеткой и разбавляли ацетонитрилом, готовя серию стандартных растворов. Аккуратно отбирали пипеткой 20 мкл каждого из описанных выше растворов из серии стандартов, добавляли 180 мкл бланковой плазмы, перемешивали на вихревой мешалке и перемешивали, получая образцы плазмы, эквивалентные концентрации в плазме 1, 3, 10, 30, 100, 300, 1000, 3000 и 5000 нг/мл. Анализ для каждой концентрации проводили в двух повторностях образцов, строя стандартную кривую. Отбирали 20 мкл плазмы, добавляли 200 мкл внутреннего стандарта пропранолола (5 нг/мл) в ацетонитриле, перемешивали на вихревой мешалке. Затем центрифугировали при 4000 об/мин в течение 5 минут и собирали надосадочный раствор для проведения анализа методом LC-MS. Условия LC-MS детектирования описаны ниже:
Хроматографическая колонка: Thermo Scientific HYPERSIL GOLD C-18 UPLC column, 100*2.1 мм, 1.9 мкм.
Подвижная фаза: система вода (0.1% муравьиной кислоты) / ацетонитрил, элюирование в градиенте согласно приведенной ниже таблице.
3. Обработка результатов
После определения концентрации лекарственного средства в крови методом LC-MS, использовали программу WinNonlin 6.1 для вычисления фармакокинетических параметров некомпартментным модельным методом. Полученные результаты приведены в таблицах 3 и 4.
Таблица 4. Фармакокинетические параметры соединений по настоящему изобретению (крысы)
(ч)
(нг/мл)
(ч*нг/мл)
(ч)
(мл/мин/кг)
(л/кг)
(%)
(CN201680058331)
Таблица 5. Фармакокинетические параметры соединений по настоящему изобретению (яванские макаки)
(ч)
(нг/мл)
(ч*нг/мл)
(ч)
(мл/мин/кг)
(л/кг)
(%)
нение B
Таблица 6. Фармакокинетические параметры соединений по настоящему изобретению (собаки породы бигль)
Вывод: Соединения по настоящему изобретению демонстрируют определенную пероральную абсорбцию у крыс и обезьян, демонстрируют хорошую пероральную абсорбцию у собак, и скорость выведения in vivo сравнительно низкая. Большинство соединений имеют продолжительное время полужизни при пероральном введении и хорошие фармакокинетические параметры.
Пример 54. Исследование соединений по настоящему изобретению на клетках Caco-2
Материалы для проведения эксперимента:
Среда: Минимальная эссенциальная среда Игла, модифицированная по способу Дульбекко (DMEM) (Corning), фетальная бычья сыворотка (FBS) (Corning), двойное антитело (Solarbio), 96-луночный трансвел-микропланшет для высокопроизводительного скрининга (Corning), клетки Caco-2.
Методика проведения эксперимента: клетки Caco-2 выращивали в 96-луночном трансвел-микропланшете для высокопроизводительного скрининга 14-18 дней, и определяли трансэпителиальное электрическое сопротивление (TEER) для каждой лунки, чтобы убедиться, что в каждой лунке сформировался сплошной монослой. Добавляли лекарственное средство и инкубировали 2 часа, после чего определяли концентрацию лекарственного средства A-B и B-A.
Обработка результатов: вычисляли значения PappA-B и PappB-A: Papp = (VA×[лекарство]акцептор)/(Площадь×Время×[лекарство]изначальное,донор); Вычисляли коэффициент эффлюкса: Коэффициент эффлюкса = Papp(B-A)/Papp(A-B).
Таблица 7: Полученные на Caco-2 результаты для соединений по настоящему изобретению
Вывод: Соединение по настоящему изобретению обладает хорошей проницаемостью через мембрану.
Пример 55. Анализ ингибирования CYP фермента соединениями по настоящему изобретению
Материалы для проведения эксперимента:
Микросомы печени (150-donor, Corning, Cat. 452117; Lot. 38292), NADPH.
Методика проведения эксперимента:
Готовили 0.2 мг/мл систему микросом, добавляли каждое тестируемое вещество и субстрат, при этом финальная концентрация тестируемого вещества составляла 50 мкМ. После 8-минутного предварительного инкубирования добавляли 10 мМ НАДФН для начала реакции, при этом финальная концентрация НАДФН составляла 1 мМ. После периода инкубации добавляли внутренний стандарт, такой как метанол, останавливая реакцию. Определяли количество метаболитов субстрата, образовавшихся в каждой реакции.
Обработка результатов: Принимая образование метаболитов в бланковой лунке за 100%, вычисляли уменьшение образования метаболитов в каждой лунке с тестируемым веществом и вычисляли коэффициент ингибирования.
Таблица 8. Ингибирование CYP фермента соединениями по настоящему изобретению
Вывод: Соединения по настоящему изобретению не оказывают ингибирующего действия на основные CYP ферменты, и риск межлекарственного взаимодействия (DDI) небольшой.
Пример 56. Анализ действия соединений по настоящему изобретению на hERG
Материалы для проведения эксперимента:
Стабильно трансфицированная линия клеток HEK293-hERG (invitrogen). Среда DMEM (Gibco), HEPES (invitrogen), бластицидин (invitrogen)
Методика проведения эксперимента:
Использовали для экспериментов стабильно трансфицированные клетки HEK293-hERG, выращенные до степени полимеризации 40%-80%. Сначала к клеткам добавляли чистый растворитель для определения базового уровня. Соединения тестировали после того как ток калиевых каналов hERG был стабильным в течение 5 минут. Регистрировали ток калиевых каналов hERG в присутствии тестируемых соединений в течение примерно 5 минут до достижения устойчивого состояния, и затем записывали 5 полос частот свипа. Для контроля правильности работы клеток и техники эксперимента, эту же порцию клеток тестировали также с положительным контролем – дофетилидом.
Обработка результатов:
Подавление пикового тока = (1 – пиковый следовой ток с соединением/ пиковый следовой ток с чистым растворителем)*100
Таблица 9. Результаты экспериментального исследования действия соединений по настоящему изобретению на hERG
Вывод: Соединения по настоящему изобретению имеют высокие значения IC50 по влиянию на ток калиевых каналов hERG и обладают высокой безопасностью для сердца.
Описанные выше варианты осуществления представляют собой предпочтительные варианты осуществления настоящего изобретения. Однако, варианты осуществления настоящего изобретения не ограничиваются только описанными выше примерами, и любые другие изменения, модификации, замены, комбинации и упрощения, не выходящие за рамки сути и объема настоящего изобретения, должны рассматриваться как эквивалентные способы замены, и все они включены в объем притязаний настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА | 2016 |
|
RU2740019C1 |
| ЛЕЧЕНИЕ МИОДИСТРОФИИ ДЮШЕНА | 2007 |
|
RU2462458C2 |
| АЗАБИЦИКЛИЧЕСКИЕ И ДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ ОФТАЛЬМОЛОГИЧЕСКИХ НАРУШЕНИЙ | 2018 |
|
RU2795521C2 |
| ПРОЛЕКАРСТВЕННЫЕ СРЕДСТВА ИНГИБИТОРОВ ОБРАТНОЙ ТРАНСКРИПТАЗЫ ВИЧ | 2015 |
|
RU2693622C2 |
| ИНГИБИРУЮЩИЕ JAK СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИМИДИНА И СПОСОБЫ | 2010 |
|
RU2567238C2 |
| ИНГИБИРУЮЩИЕ JAK СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИМИДИНА И СПОСОБЫ | 2010 |
|
RU2675857C2 |
| БИЦИКЛИЧЕСКИЕ КЕТОНЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2018 |
|
RU2797922C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛОПИРИДАЗИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ФОСФОДИЭСТЕРАЗЫ IV, И/ИЛИ ПРОДУЦИРОВАНИЯ TNF | 2003 |
|
RU2328499C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ МЕК | 2009 |
|
RU2509078C2 |
| СОЕДИНЕНИЯ, МОДУЛИРУЮЩИЕ ЭСТРОГЕНОВЫЕ РЕЦЕПТОРЫ | 2019 |
|
RU2826635C2 |
Изобретение относится к соединению формулы (I), 35, 36, 37, 44 или 45 или его стереоизомеру, фармацевтически приемлемой соли, которые обладают способностью ингибировать фактор FXIa. В формуле (I) R1 представляет собой C1-4 алкил; X представляет собой галоген; R3 представляет собой атом водорода; Y представляет собой атом кислорода; R2 выбран из группы, состоящей из атома водорода, метила, этила, фенила, гидроксиэтила, циклопропилметила, метоксиэтила, изопропила, дифторметила,  и CF3CH2-; R4 выбран из группы, состоящей из фенила, 4-фторфенила, 4-бромфенила, 3-метилфенила, 4-метилфенила, бензила, изопропила,
и CF3CH2-; R4 выбран из группы, состоящей из фенила, 4-фторфенила, 4-бромфенила, 3-метилфенила, 4-метилфенила, бензила, изопропила,  ,
,  и т.д.; Ar выбран из группы, состоящей из
и т.д.; Ar выбран из группы, состоящей из  ,
, 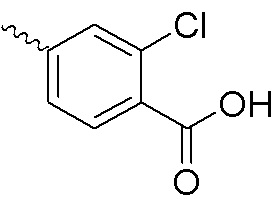 ,
, 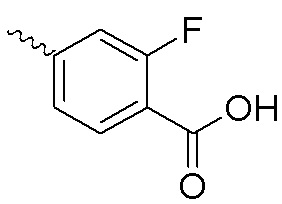 ,
, 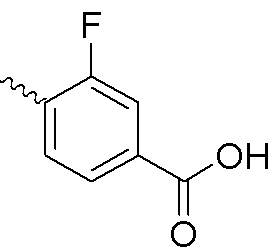 ,
, 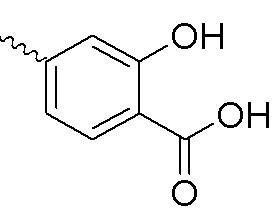 и т.д.; R7 представляет собой атом водорода. Изобретение относится также к фармацевтической композиции для ингибирования фактора FXIa, содержащей указанные соединения, и их применению в производстве лекарственного средства для лечения заболеваний, связанных с FXIa, предпочтительно заболеваний, связанных с тромбозом. 3 н. и 6 з.п. ф-лы, 9 табл., 56 пр.
и т.д.; R7 представляет собой атом водорода. Изобретение относится также к фармацевтической композиции для ингибирования фактора FXIa, содержащей указанные соединения, и их применению в производстве лекарственного средства для лечения заболеваний, связанных с FXIa, предпочтительно заболеваний, связанных с тромбозом. 3 н. и 6 з.п. ф-лы, 9 табл., 56 пр.
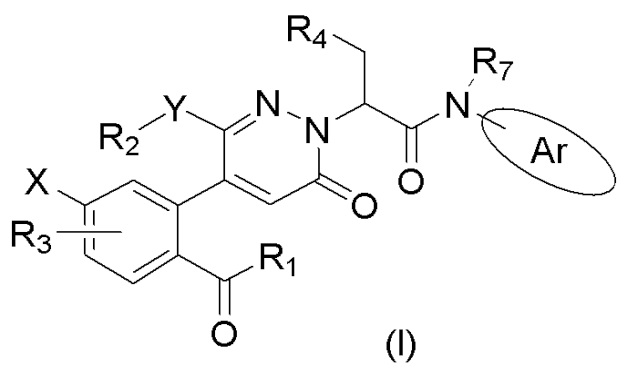
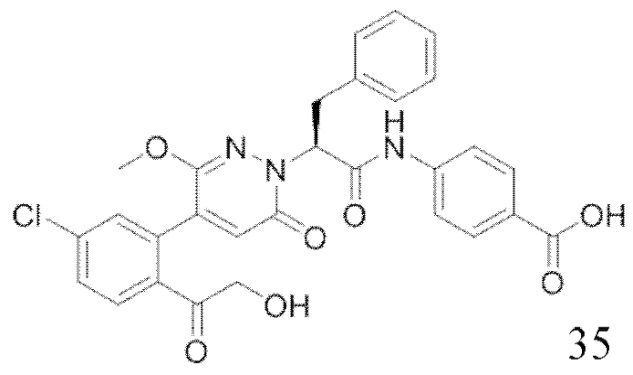
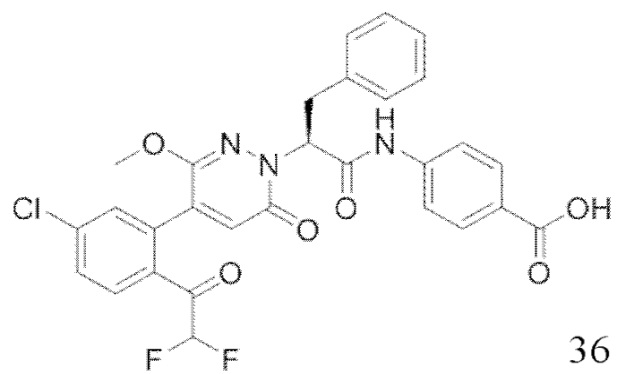
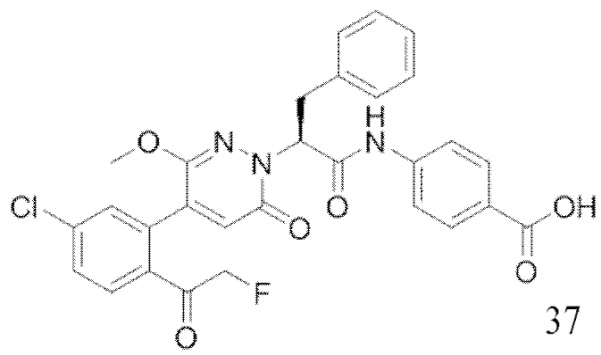
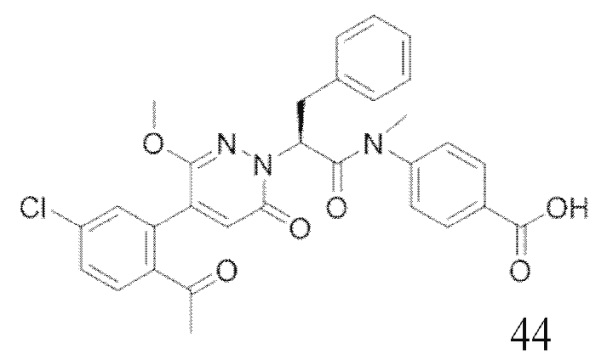
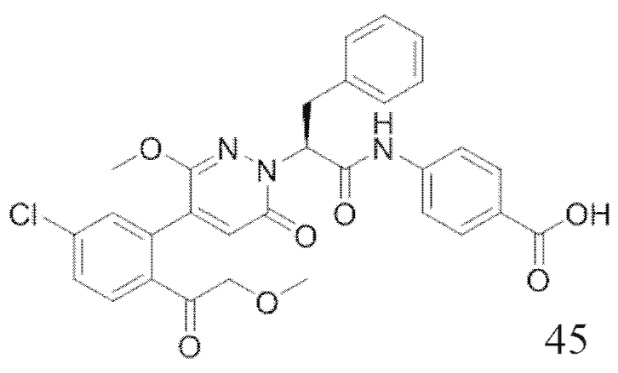
1. Соединение формулы (I), 35, 36, 37, 44 или 45 или его стереоизомер, фармацевтически приемлемая соль,
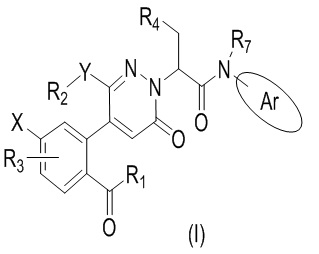
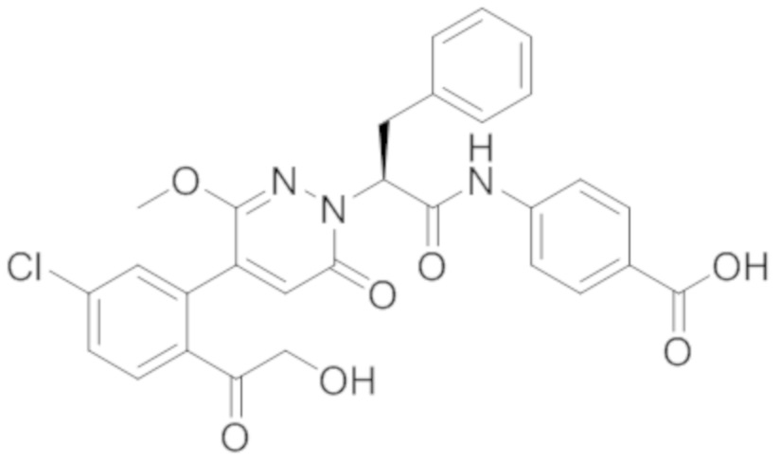 35
35
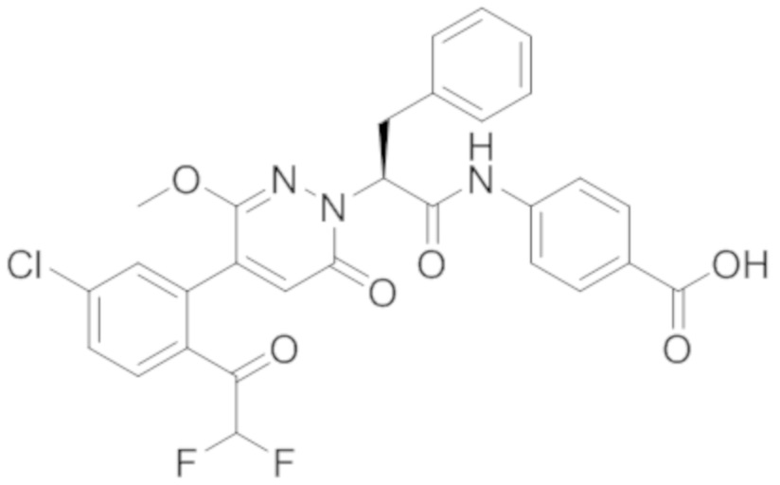 36
36  37
37
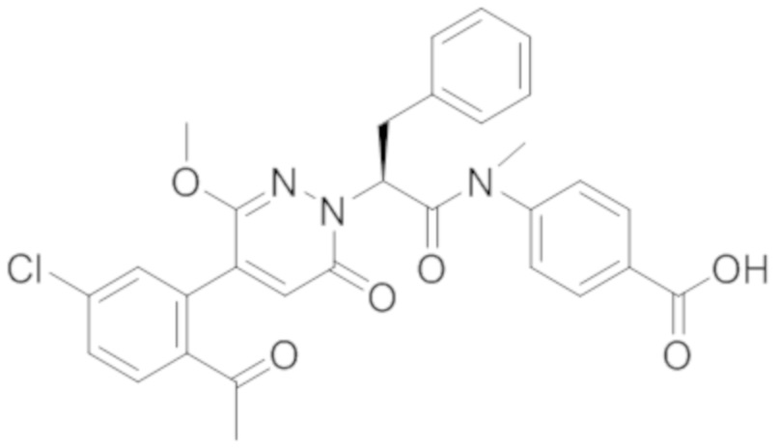 44
44  45
45
где R1 представляет собой C1-4 алкил;
X представляет собой галоген;
R3 представляет собой атом водорода;
Y представляет собой атом кислорода;
R2 выбран из группы, состоящей из атома водорода, метила, этила, фенила, гидроксиэтила, циклопропилметила, метоксиэтила, изопропила, дифторметила,  и CF3CH2-;
и CF3CH2-;
R4 выбран из группы, состоящей из фенила, 4-фторфенила, 4-бромфенила, 3-метилфенила, 4-метилфенила, бензила, изопропила,  ,
,  ,
,  ,
,  и
и  ;
;
Ar выбран из группы, состоящей из  ,
,  ,
,  ,
, 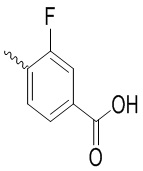 ,
, 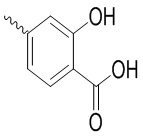 ,
, 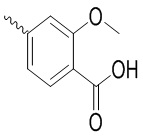 ,
, 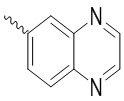 ,
, 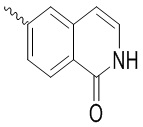 ,
, 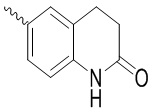 ,
, 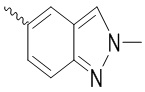 ,
, 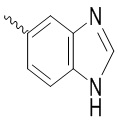 ,
, 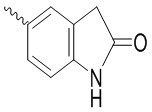 ,
, 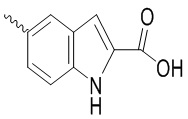 ,
, 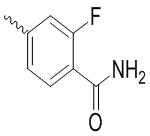 ,
, 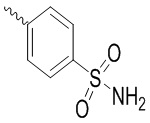 и
и 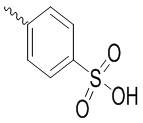 ; и
; и
R7 представляет собой атом водорода.
2. Соединение или его стереоизомер, фармацевтически приемлемая соль по п. 1, где C1-4 алкил выбран из группы, состоящей из метила, этила, пропила, изопропила, н-бутила, изо-бутила, втор-бутила и трет-бутила.
3. Соединение или его стереоизомер, фармацевтически приемлемая соль по п. 1, где галоген выбран из группы, состоящей из фтора, хлора, брома и иода.
4. Соединение или его стереоизомер, фармацевтически приемлемая соль по п. 1, где соединение или его фармацевтически приемлемая соль выбраны из следующих соединений:
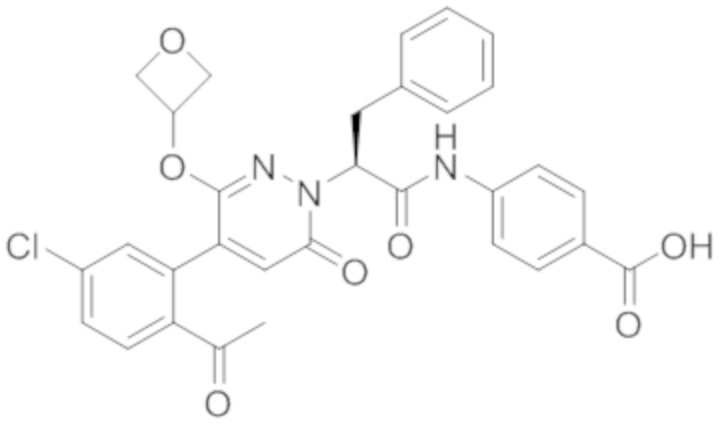
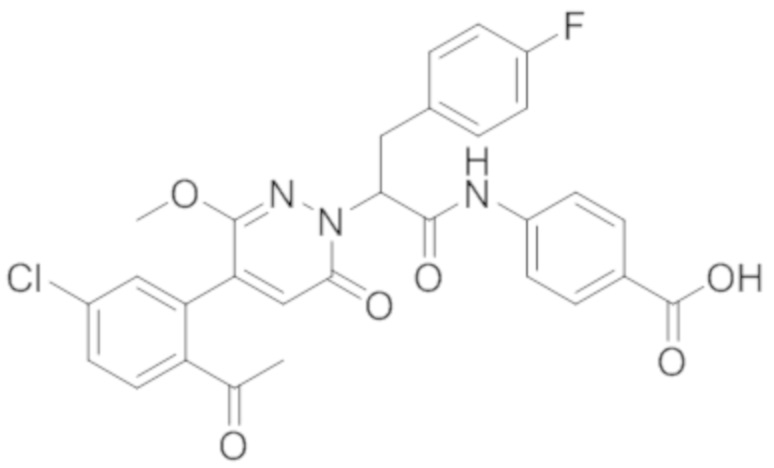
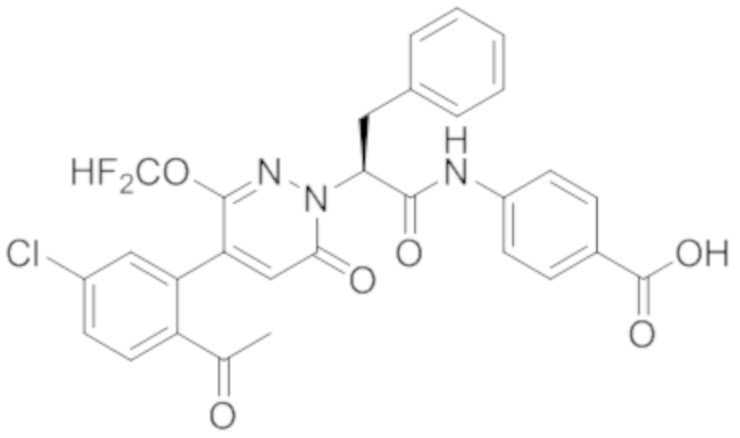
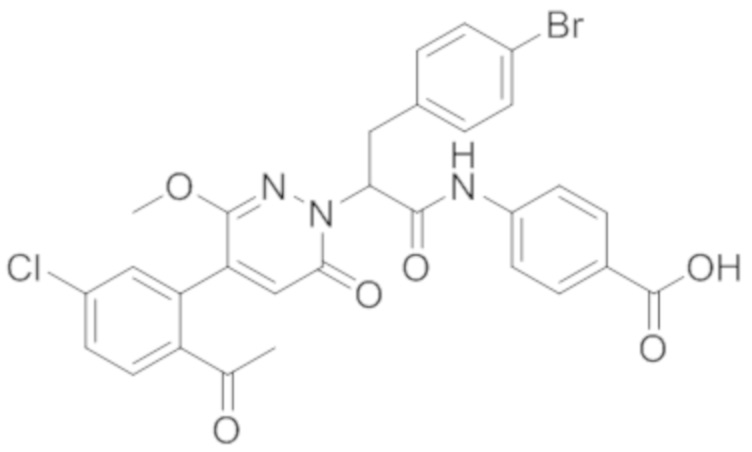
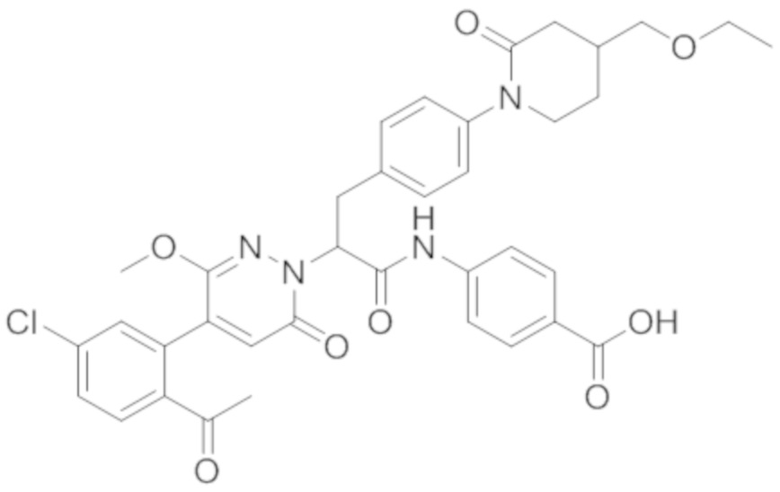
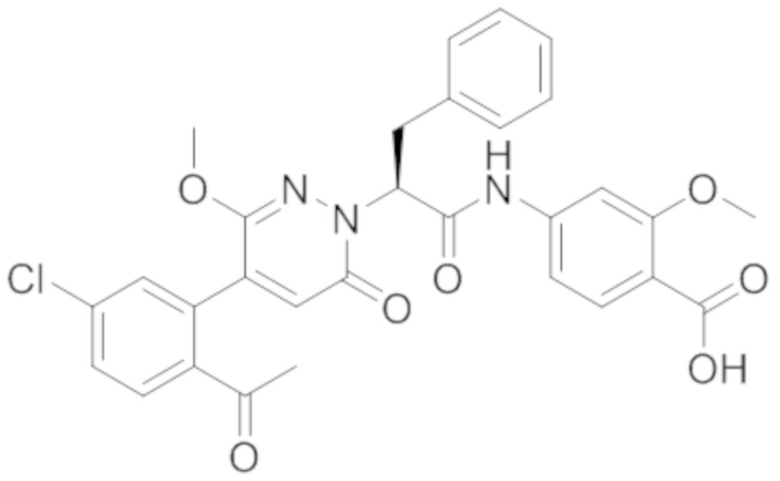
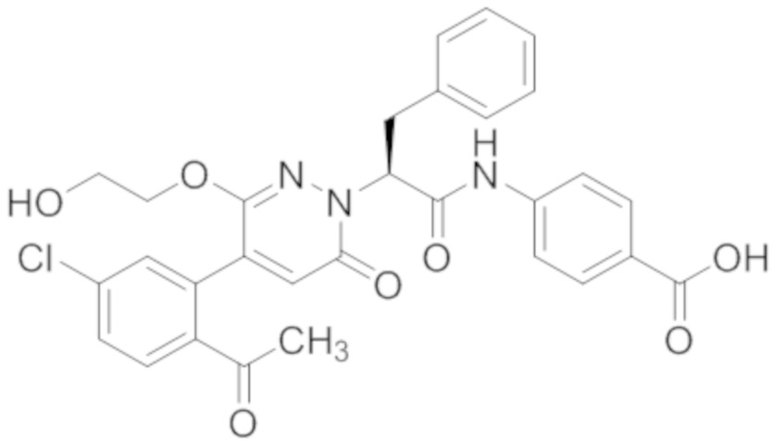
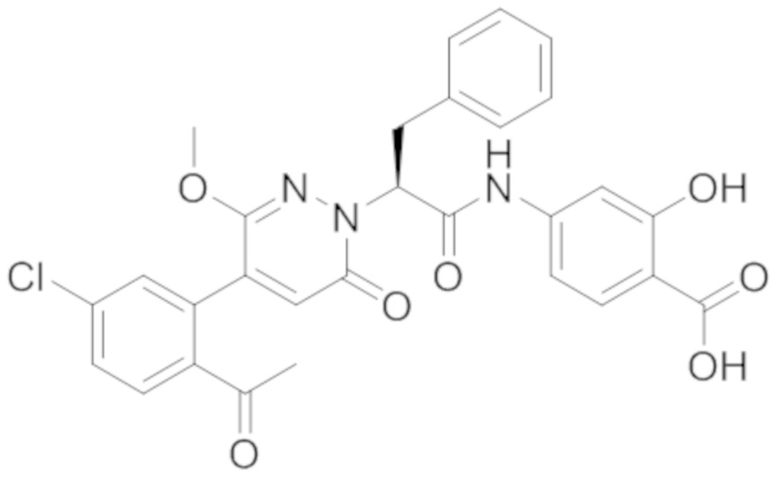
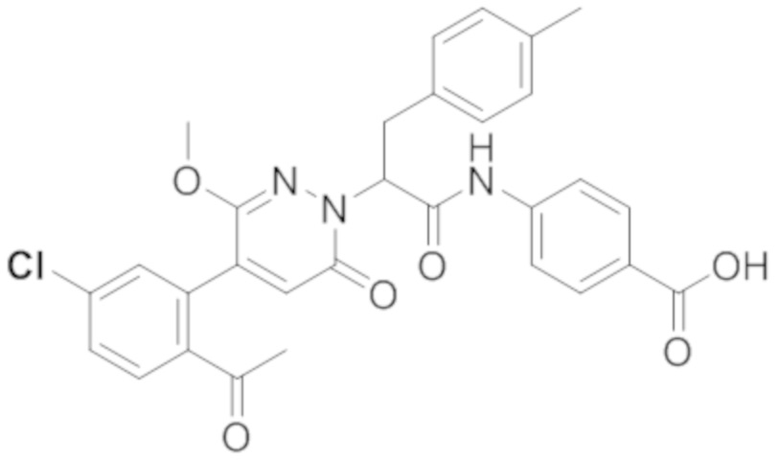
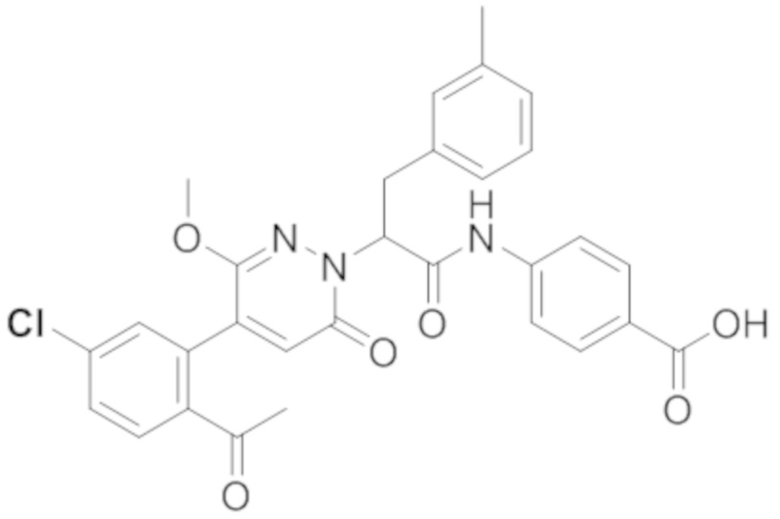
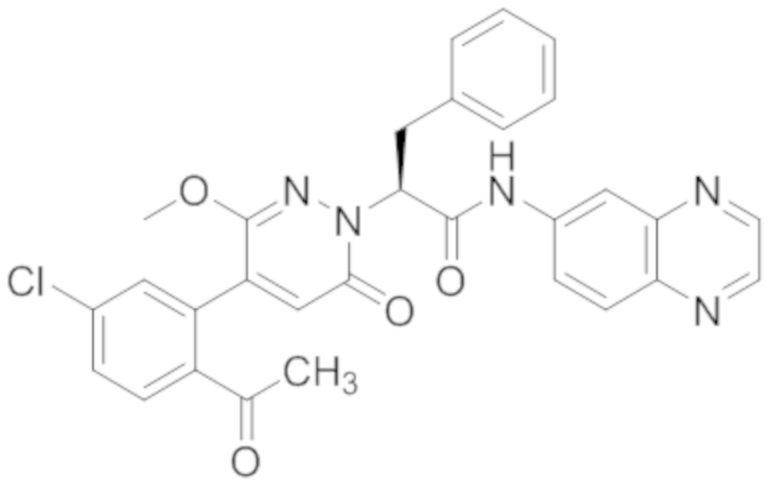
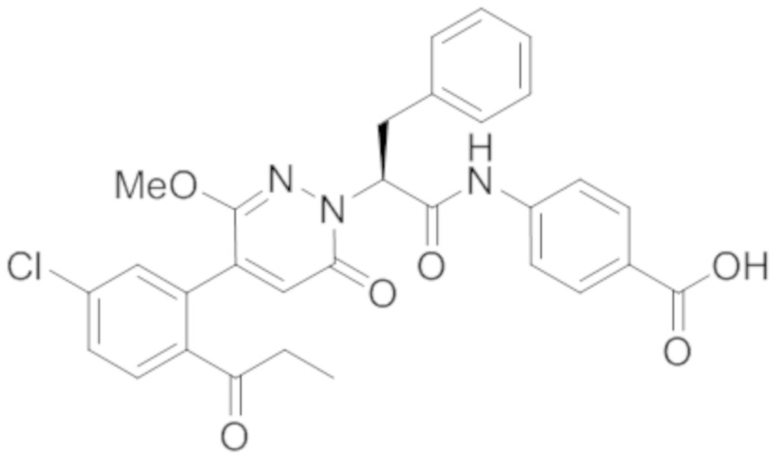
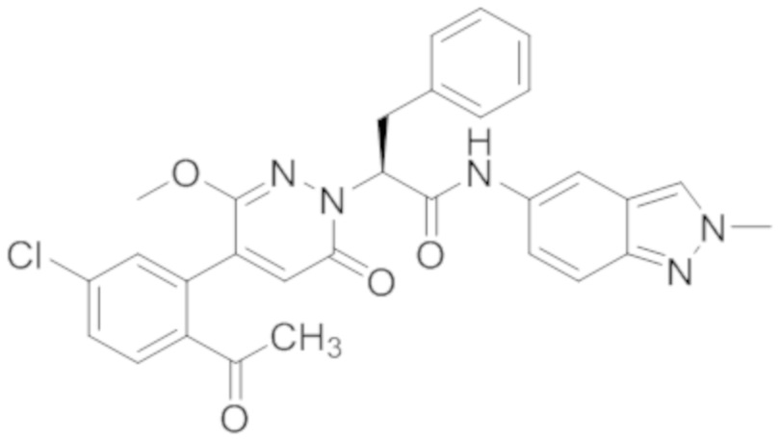
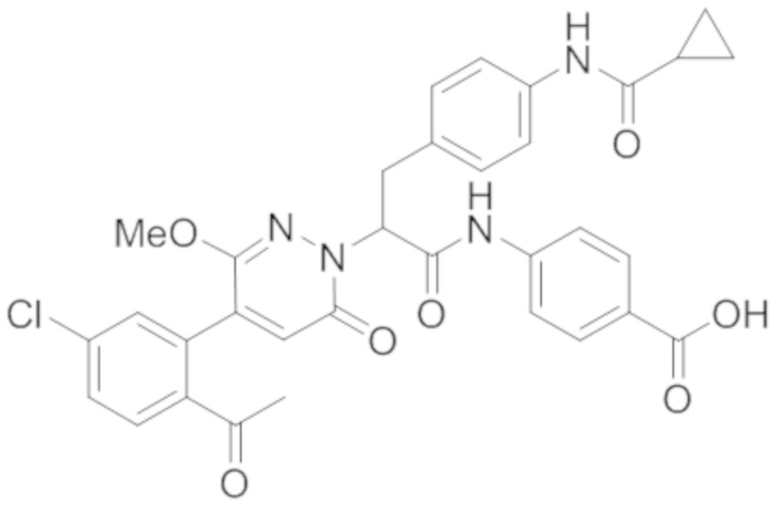
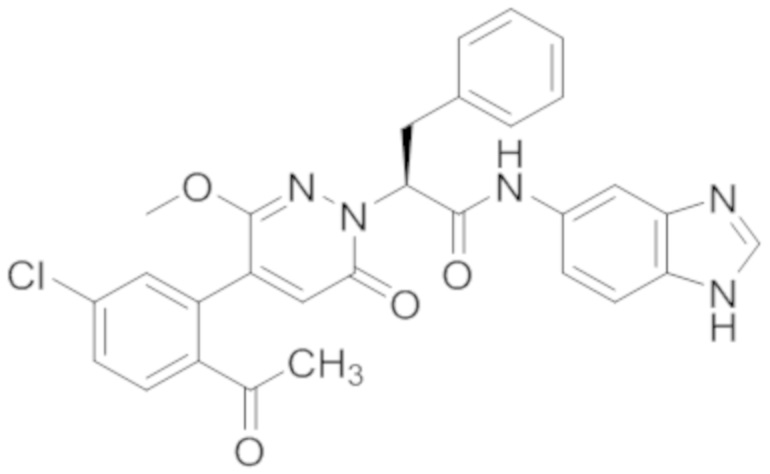
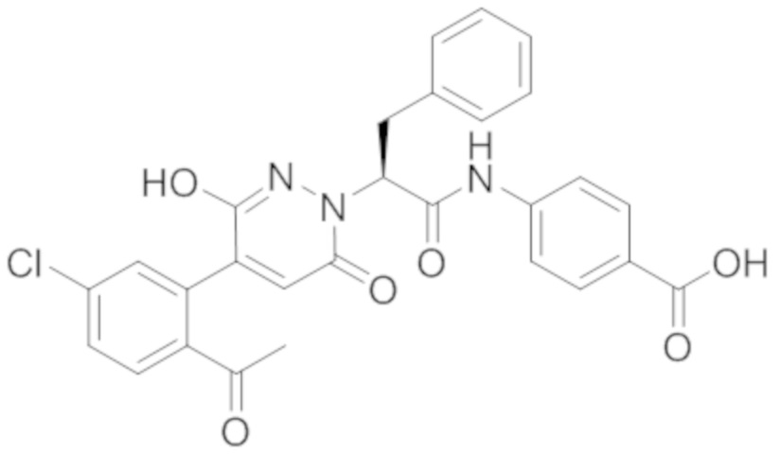
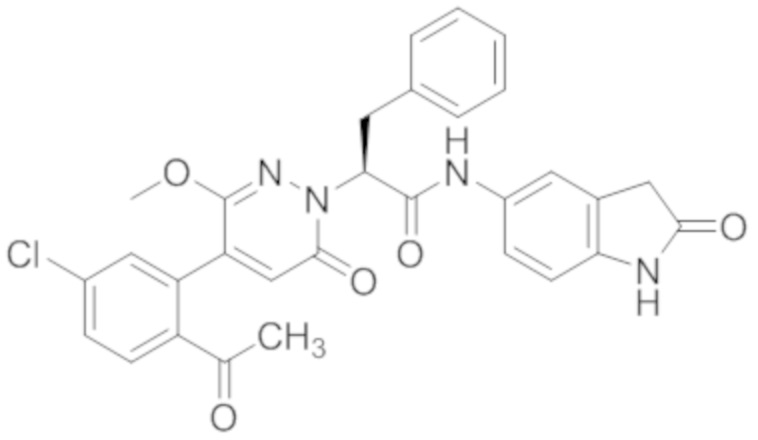
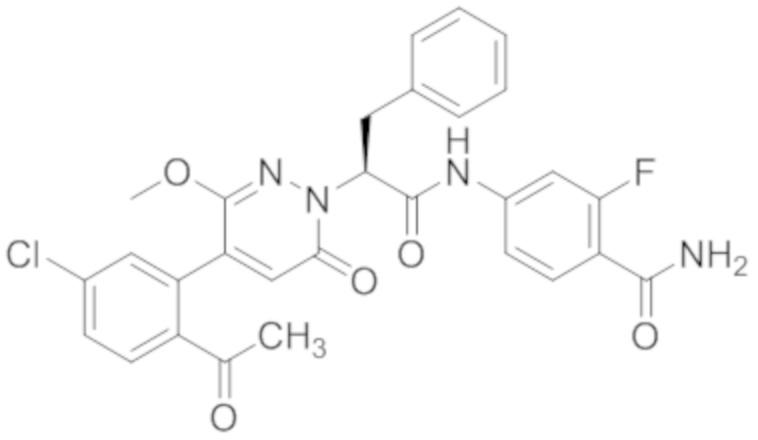
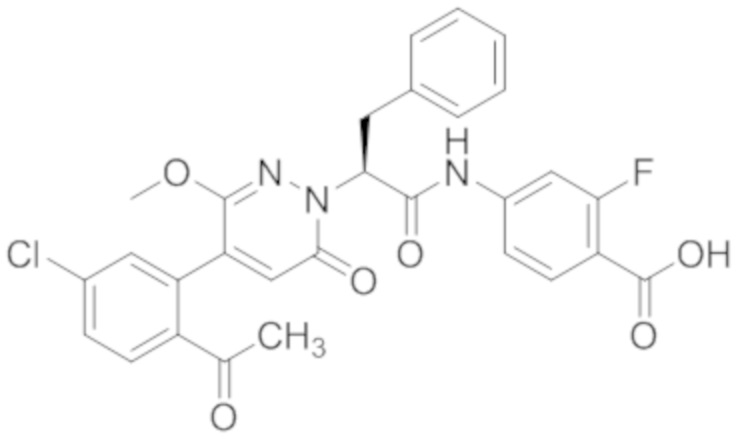

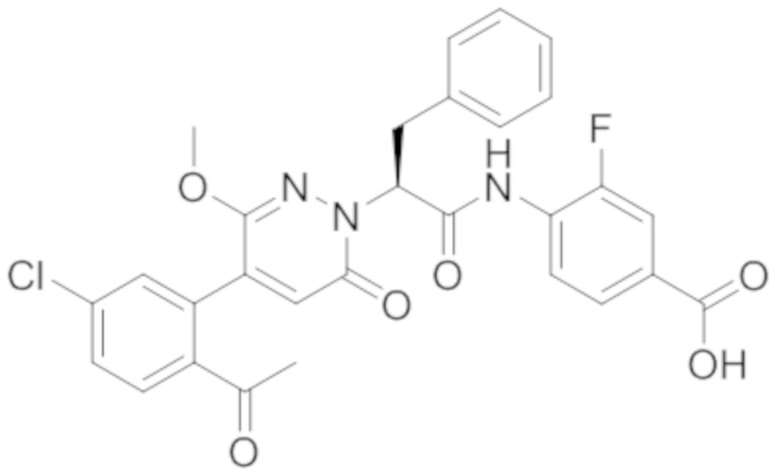
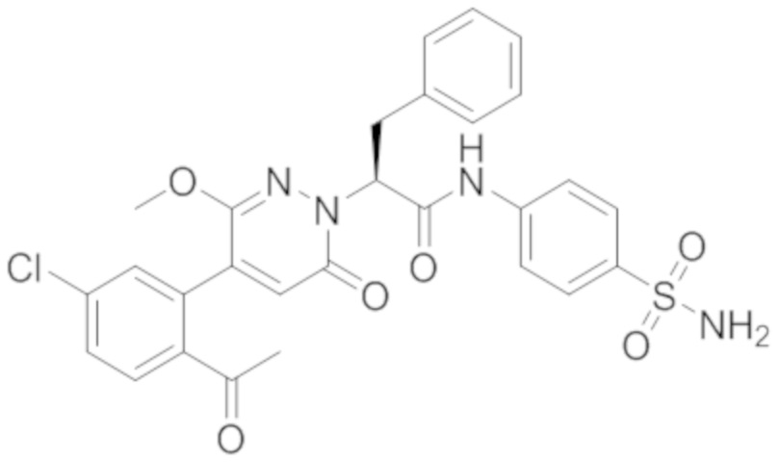
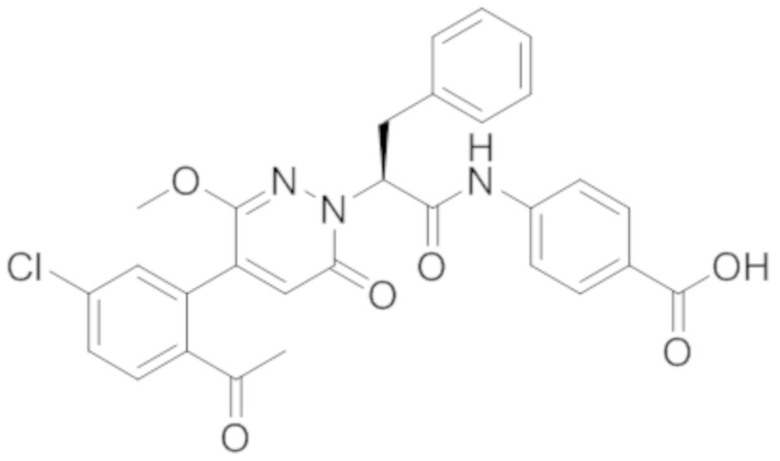
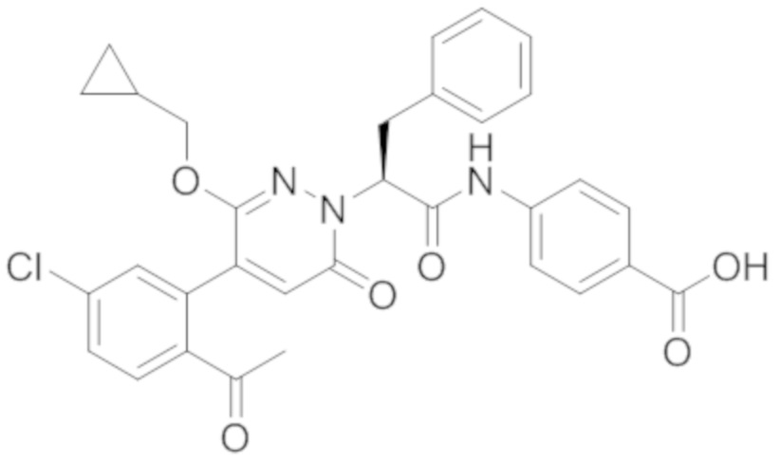
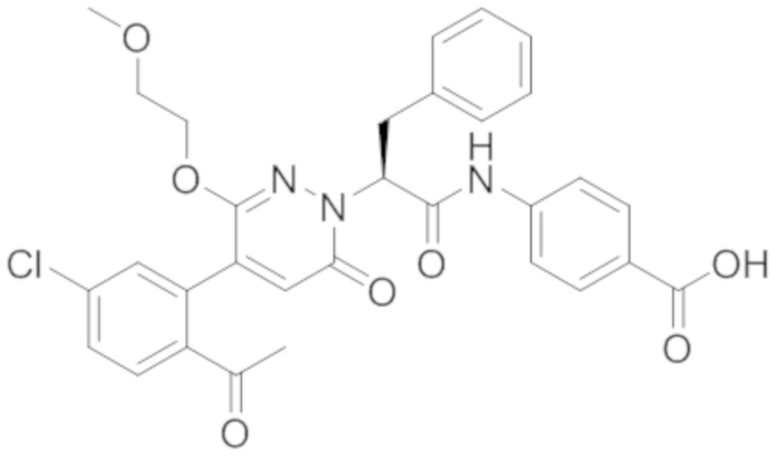
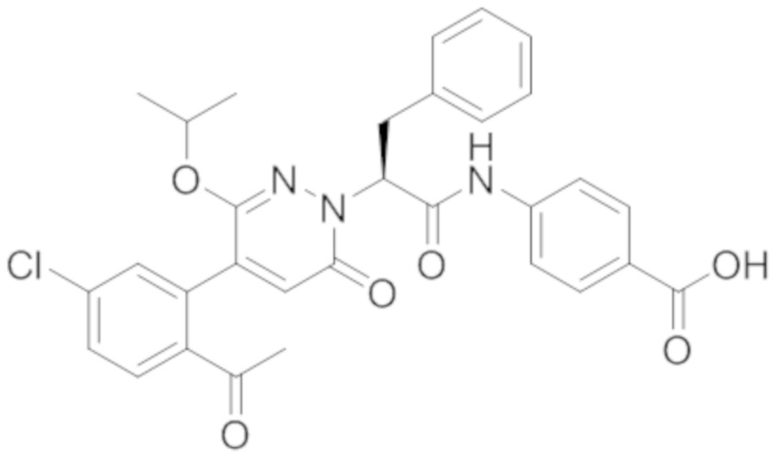
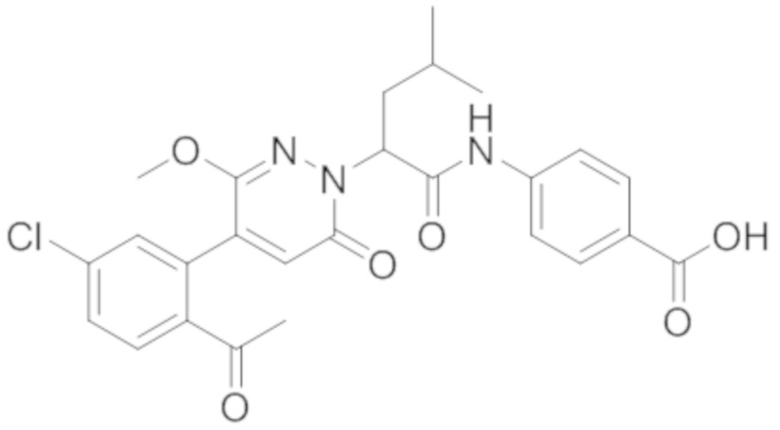
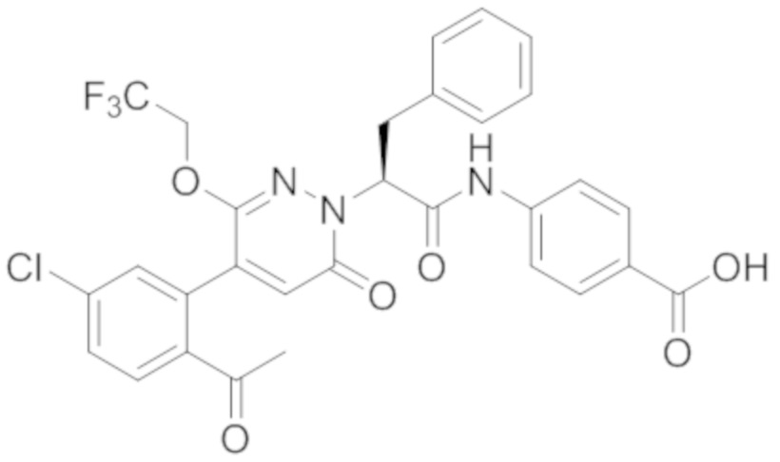
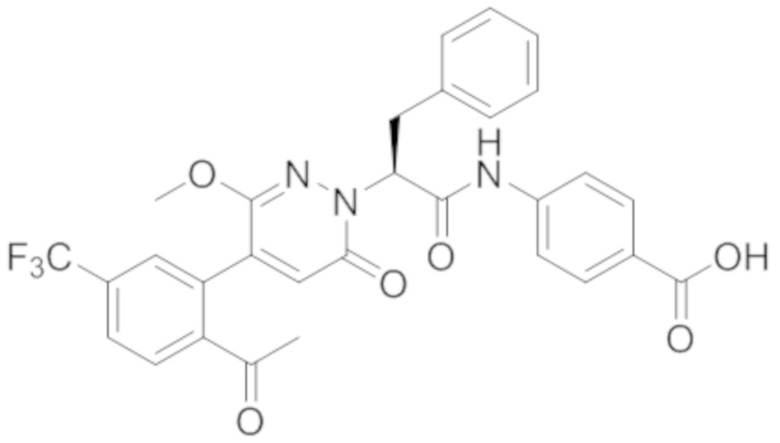
5. Соединение или его стереоизомер, фармацевтически приемлемая соль по п. 1, где фармацевтически приемлемая соль получена из соединения и фармацевтически приемлемой кислоты или основания.
6. Соединение или его стереоизомер, фармацевтически приемлемая соль по любому из пп. 1-5, где более одного атома водорода в соединении заменены на изотоп дейтерий.
7. Фармацевтическая композиция для ингибирования фактора FXIa, содержащая эффективное количество соединения или его стереоизомера, или его фармацевтически приемлемой соли по любому из пп. 1-6 и один или больше фармацевтически приемлемых носителей.
8. Применение соединения или его стереоизомера, фармацевтически приемлемой соли по любому из пп. 1-6 в производстве лекарственного средства для лечения заболеваний, связанных с FXIa, предпочтительно заболеваний, связанных с тромбозом.
9. Применение соединения или его стереоизомера, фармацевтически приемлемой соли по п. 8, где заболевания, связанные с FXIa, представляют собой заболевания, связанные с тромбозом.
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| Способ получения цианистых соединений | 1924 |
|
SU2018A1 |
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОКАРБАЗОЛА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ, СПОСОБ ЛЕЧЕНИЯ И/ИЛИ ПРОФИЛАКТИКИ ФИЗИОЛОГИЧЕСКИХ И/ИЛИ ПАТОЛОГИЧЕСКИХ СОСТОЯНИЙ, ОПОСРЕДОВАННЫХ LHRH РЕЦЕПТОРОМ, ПОСРЕДСТВОМ ВЫШЕНАЗВАННЫХ ПРОИЗВОДНЫХ | 2008 |
|
RU2497806C9 |
| СОЕДИНЕНИЯ | 2012 |
|
RU2630677C2 |
