РОДСТВЕННАЯ ЗАЯВКА
[0001] По настоящей заявке испрашивается приоритет по предварительной заявке U.S. № 61/947748, поданной 4 марта 2014 г., полное содержание указанной выше заявки включено в настоящее изобретение в качестве ссылки.
УРОВЕНЬ ТЕХНИКИ
[0002] Грелин является содержащим 28 аминокислот пептидным гормоном, который продуцируется в кишечнике и который играет главную роль в регуляции питания, всасывании питательных веществ, моторике ЖК (желудочно-кишечный тракт) и энергетическом гомеостазе. Секреция грелина усиливается при патологических состояниях с отрицательным энергетическим балансом - при голодании, кахексии и нервной анорексии - в то время как его экспрессия уменьшается при патологических состояниях с положительным энергетическим балансом - при питании, гипергликемии и ожирении. Он является эндогенным лигандом для усиливающего секрецию рецептора гормона роста (GHSR) и GHSR-1a, который обусловливает по меньшей мере некоторую часть его функции путем активации GHSR-1a, включающей стимулирование секреции гормона роста при выбранных физиологических состояниях.
[0003] Аналоги грелина применяются в терапии в многочисленных различных случаях (см., например, патенты U.S. №№ 7456253 и 7932231, полные содержания которых включены в настоящее изобретение в качестве ссылки.
[0004] Особенно перспективным с терапевтической точки зрения аналогом грелина является H-Inp-D-Bal-D-Trp-Phe-Apc-NH2 (формула (I), SEQ ID NO: 1). До настоящего времени этот аналог получали только с помощью твердофазного синтеза. Необходимы жидкофазные методики синтеза, которые позволяют в промышленном масштабе получить аналог грелина, H-Inp-D-Bal-D-Trp-Phe-Apc-NH2 (SEQ ID NO: 1), и его фармацевтически приемлемые соли. Например, необходимы жидкофазные методики, обеспечивающие желательный выход, высокую чистоту (например, стереохимическую чистоту), экономичность или их комбинацию.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0005] Настоящее изобретение относится к новым способам синтеза аналога грелина H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2 (SEQ ID NO: 1) и его фармацевтически приемлемых солей, которые можно с успехом использовать для проводимого в промышленном масштабе синтеза аналога грелина H-Inp-D-Bal-D-Trp-Phe-Apc-NH2 (SEQ ID NO: 1).
[0006] Одним вариантом осуществления настоящего изобретения является способ синтеза пептида формулы (I)
H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2, (I)
или его фармацевтически приемлемой соли. Способ включает по меньшей мере одну стадию сочетания двух аминокислот пептида формулы (I) в жидкой фазе.
[0007] Другим вариантом осуществления настоящего изобретения является пептидный фрагмент структурной формулы (II)
Boc-Inp-(D)Bal-(D)Trp-Phe-Apc(Boc)-NH2, (II)
или его соль.
[0008] Другим вариантом осуществления настоящего изобретения является пептидный фрагмент структурной формулы (III)
Boc-Inp-DBal-DTrp-OH, (III)
или его соль.
[0009] Другим вариантом осуществления настоящего изобретения является пептидный фрагмент структурной формулы (IV)
H-Phe-Apc(Boc)-NH2, (IV)
или его соль.
[0010] Другим вариантом осуществления настоящего изобретения является пептидный фрагмент структурной формулы (V)
H-DBal-DTrp-OH, (V)
или его соль.
[0011] Другим вариантом осуществления настоящего изобретения является пептидный фрагмент структурной формулы (VI)
Z-Phe-Apc(Boc)-NH2, (VI)
или его соль.
[0012] Способы жидкофазного синтеза пептида, раскрытые в настоящем изобретении, характеризуются целым рядом преимуществ. Например, жидкофазный способ синтеза, раскрытый в настоящем изобретении, обеспечивает конвергентную, а не ступенчатую схему синтеза и тем самым увеличение полного выхода. Кроме того, использование силилирующих реагентов позволяет использовать апротонные органические растворители, что устраняет недостатки водных растворителей, такие как образование примесей, которые следует удалять. Использование силилированных промежуточных продуктов дополнительно позволяет использовать незащищенных по главной цепи аминокислотных остатков в качестве промежуточных продуктов, что приводит к уменьшению количества стадий синтеза и увеличению выхода. Дополнительным преимуществом раскрытого способа проявляется в проведении амидирования N-концевого аминокислотного остатка (Apc) на стадии использования дипептида, а не на стадии использования одиночного аминокислотного остатка. Такое амидирование приводит к уменьшению загрязнения аммиаком и также к исключению преждевременного обрыва пептидной цепи вследствие аминолиза активированной карбоциклической группы растворенным аммиаком.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0013] Приведенное ниже должно быть очевидно из последующего более конкретного описания приведенных в качестве примеров вариантов осуществления настоящего изобретения, что проиллюстрировано на прилагаемых чертежах, на которых для одних и тех же компонентов используются одинаковые. Чертежи необязательно приведены в масштабе, основное внимание направлено на иллюстрацию вариантов осуществления настоящего изобретения.
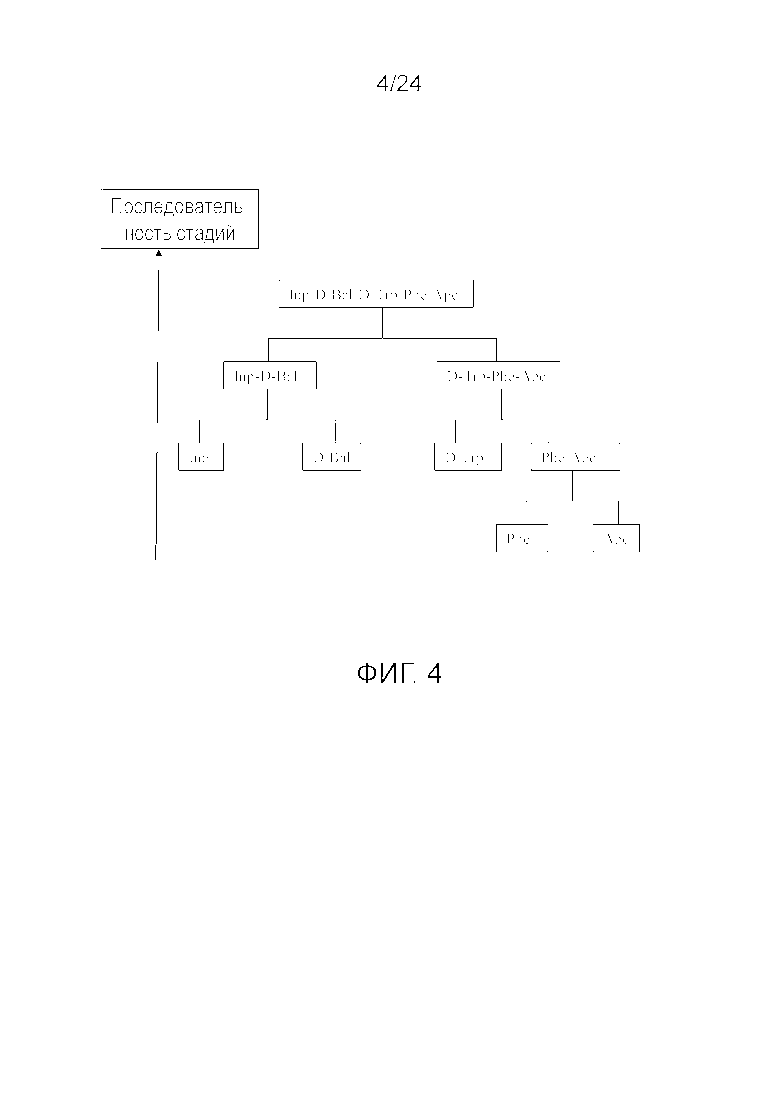
[0014] На фиг. 1 представлена блок-схема, иллюстрирующая последовательность стадий, использующуюся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении.
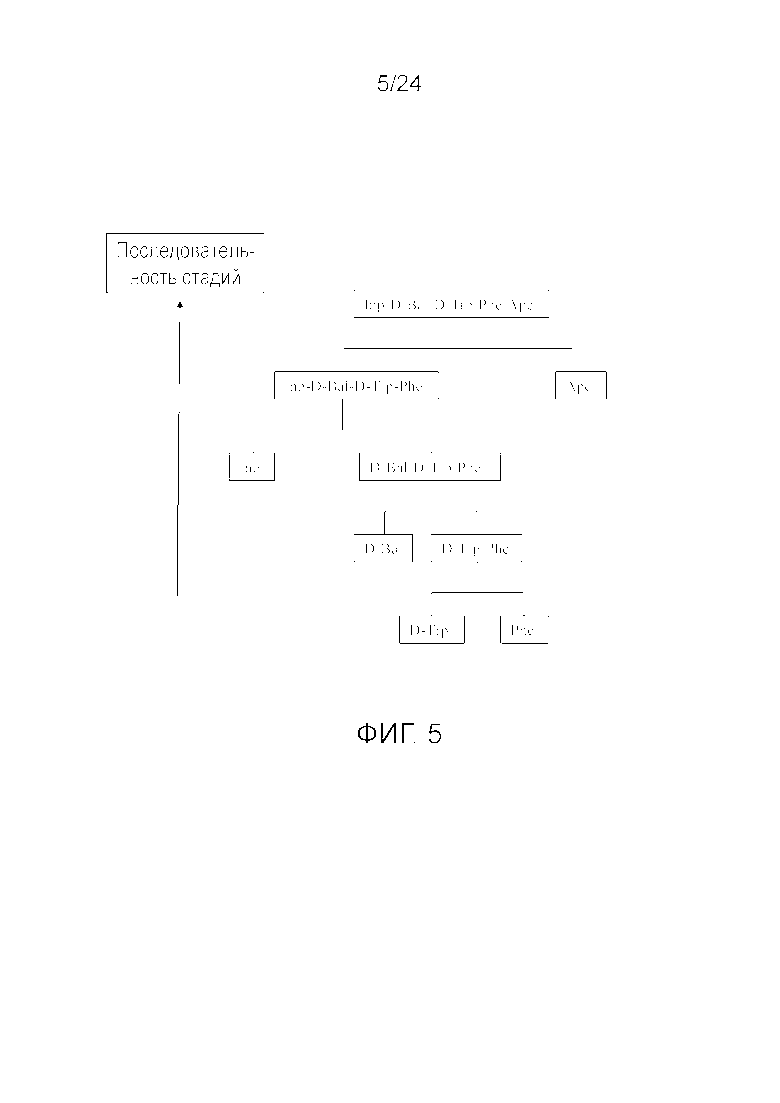
[0015] На фиг. 2 представлена блок-схема, иллюстрирующая последовательность стадий, использующуюся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении.
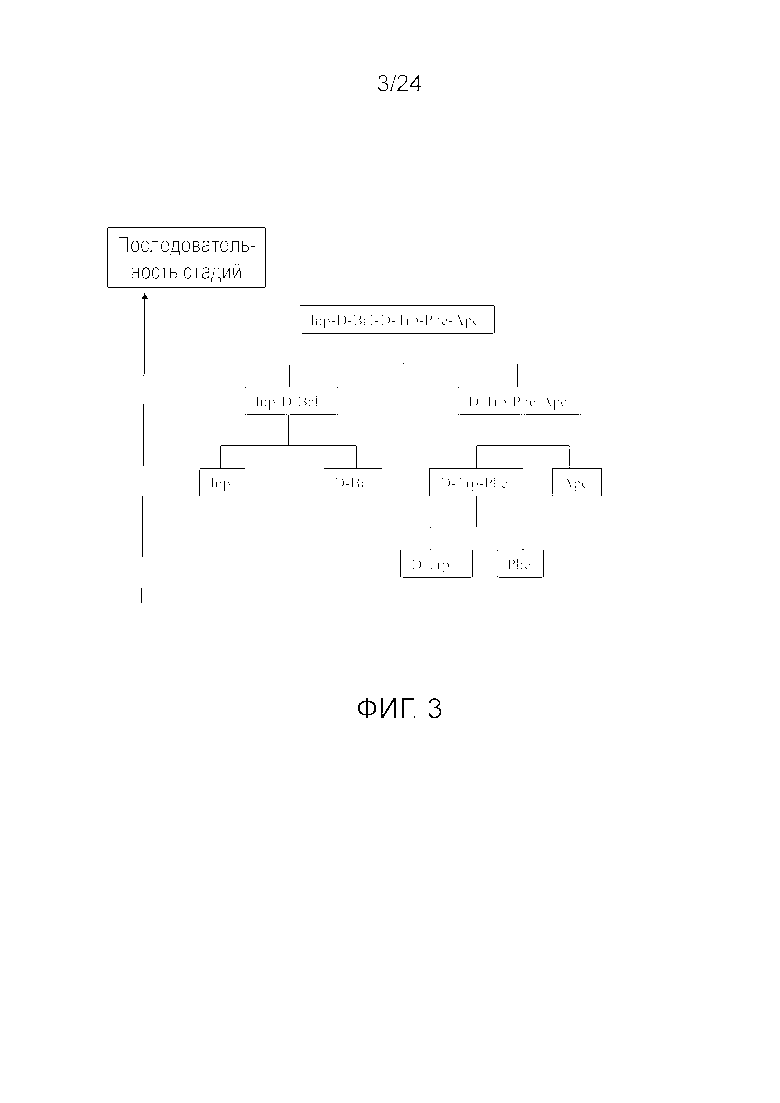
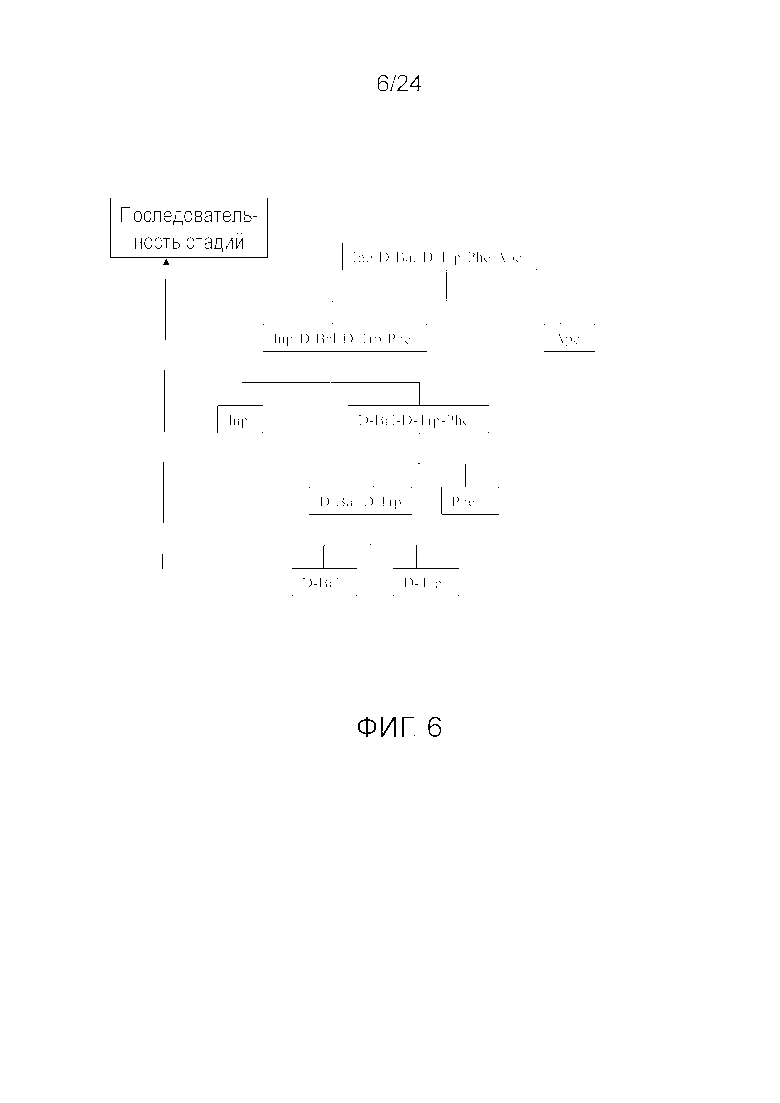
[0016] На фиг. 3 представлена блок-схема, иллюстрирующая последовательность стадий, использующуюся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении.
[0017] На фиг. 4 представлена блок-схема, иллюстрирующая последовательность стадий, использующуюся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении.
[0018] На фиг. 5 представлена блок-схема, иллюстрирующая последовательность стадий, использующуюся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении.
[0019] На фиг. 6 представлена блок-схема, иллюстрирующая последовательность стадий, использующуюся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении.
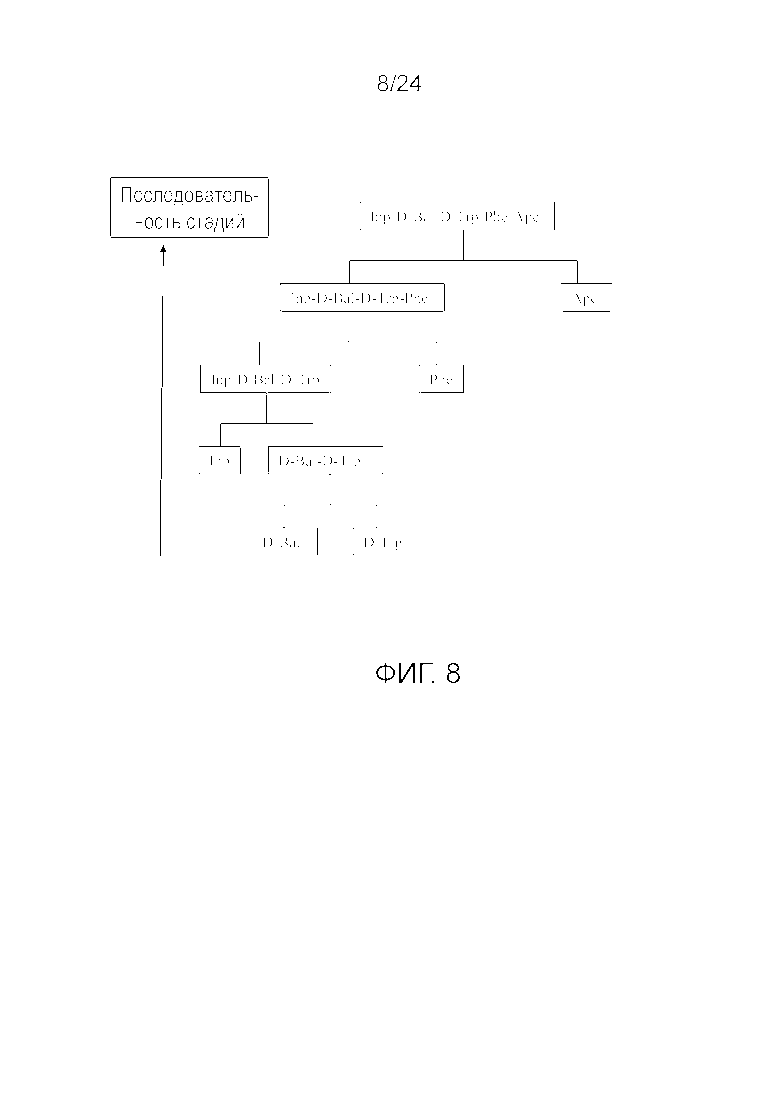
[0020] На фиг. 7 представлена блок-схема, иллюстрирующая последовательность стадий, использующуюся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении.
[0021] На фиг. 8 представлена блок-схема, иллюстрирующая последовательность стадий, использующуюся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении.
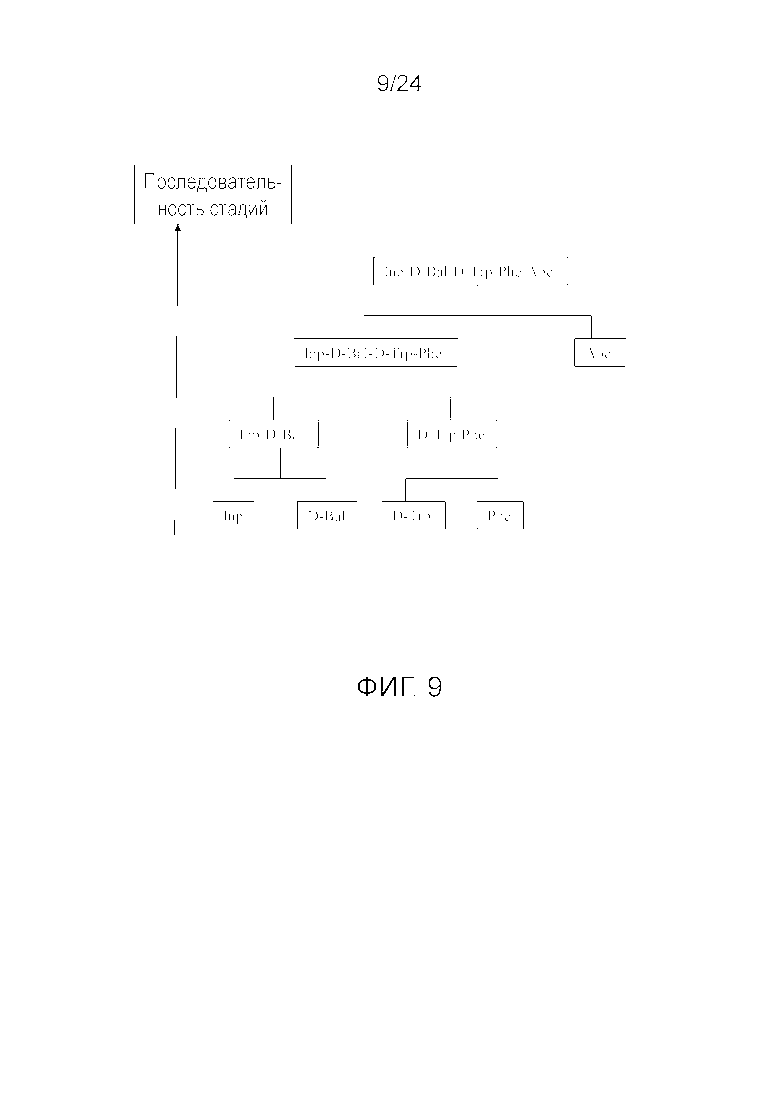
[0022] На фиг. 9 представлена блок-схема, иллюстрирующая последовательность стадий, использующуюся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении.
[0023] На фиг. 10 представлена блок-схема, иллюстрирующая последовательность стадий, использующуюся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении.
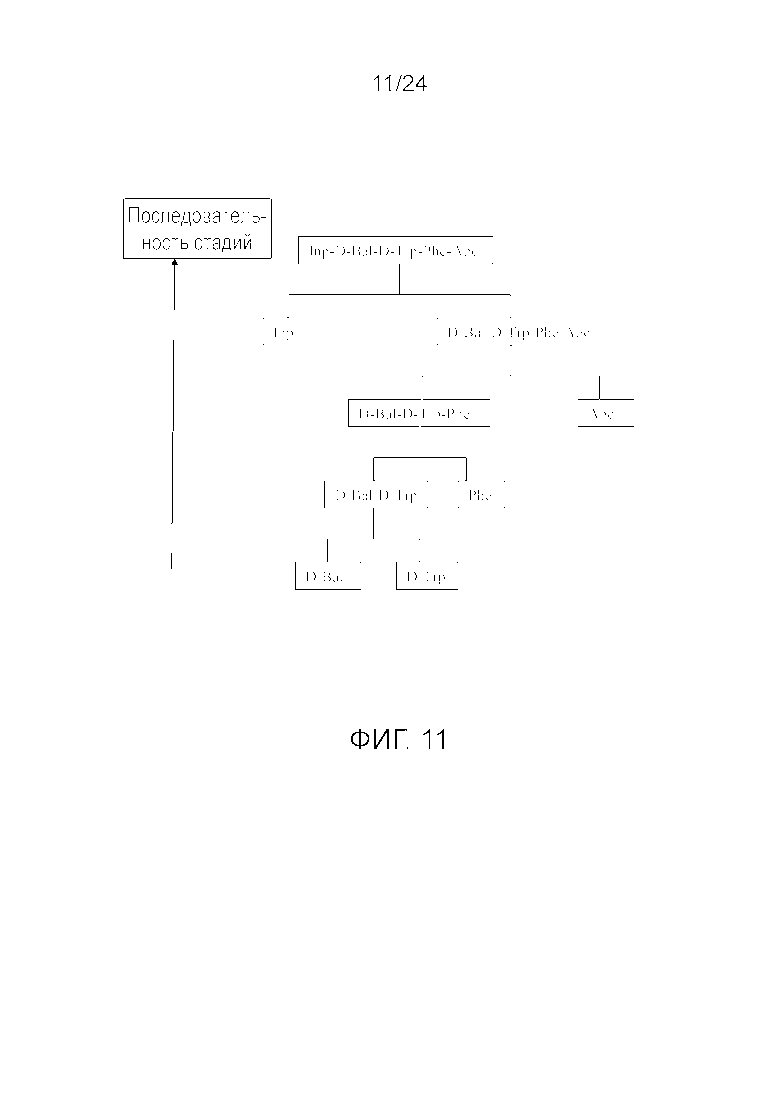
[0024] На фиг. 11 представлена блок-схема, иллюстрирующая последовательность стадий, использующуюся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении.
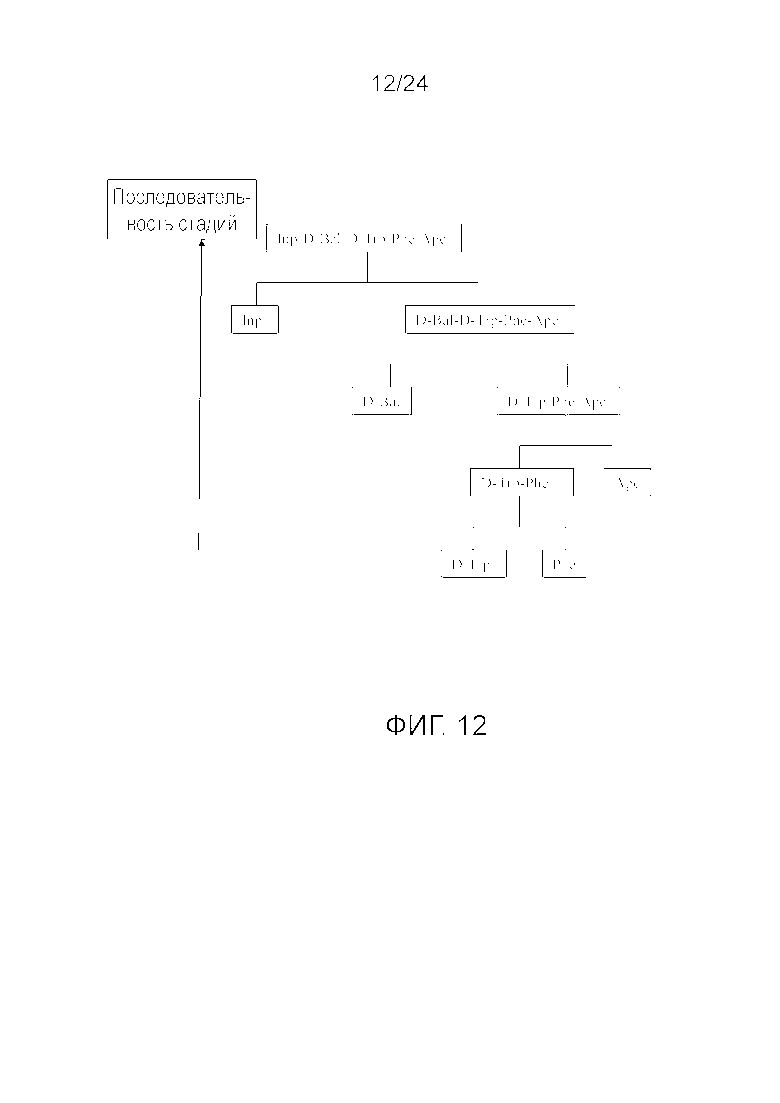
[0025] На фиг. 12 представлена блок-схема, иллюстрирующая последовательность стадий, использующуюся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении.
[0026] На фиг. 13 представлена блок-схема, иллюстрирующая последовательность стадий, использующуюся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении.
[0027] На фиг. 14 представлена блок-схема, иллюстрирующая последовательность стадий, использующуюся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении.
[0028] На фиг. 15 представлена блок-схема, иллюстрирующая последовательность стадий, использующуюся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении.
[0029] Фиг. 16 является иллюстрацией схемы синтеза, использующейся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении, для получения промежуточного продукта, применяющегося при практическом осуществлении настоящего изобретения.
[0030] Фиг. 17 является иллюстрацией схемы синтеза, использующейся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении, для получения промежуточного продукта, применяющегося при практическом осуществлении настоящего изобретения.
[0031] Фиг. 18 является иллюстрацией схемы синтеза, использующейся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении, для получения промежуточного продукта, применяющегося при практическом осуществлении настоящего изобретения.
[0032] Фиг. 19 является иллюстрацией схемы синтеза, использующейся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении, для получения промежуточного продукта, применяющегося при практическом осуществлении настоящего изобретения.
[0033] Фиг. 20 является иллюстрацией схемы синтеза, использующейся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении, для получения промежуточного продукта, применяющегося при практическом осуществлении настоящего изобретения.
[0034] Фиг. 21 является иллюстрацией схемы синтеза, использующейся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении, для получения промежуточного продукта, применяющегося при практическом осуществлении настоящего изобретения.
[0035] Фиг. 22 является иллюстрацией схемы синтеза, использующейся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении, для получения промежуточного продукта, применяющегося при практическом осуществлении настоящего изобретения.
[0036] Фиг. 23 является иллюстрацией схемы синтеза, использующейся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении, для получения промежуточного продукта, применяющегося при практическом осуществлении настоящего изобретения.
[0037] Фиг. 24 является иллюстрацией схемы синтеза, использующейся в приведенном в качестве примера варианте осуществления способа, раскрытого в настоящем изобретении, для получения соединения
формулы (I).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0038] Обозначения, использующиеся для определения пептидов, являются такими, которые обычно применяются в данной области техники, и аминогруппа на N-конце указывается слева и карбоксигруппа на C-конце указывается справа.
[0039] При использовании в настоящем изобретении термин "аминокислота" включает и природную аминокислоту, и неприродную аминокислоту. Термин "аминокислота", если не указано иное, включает отдельные молекулы аминокислоты (т. е. молекулы, которые содержат и присоединенный к аминогруппе водород, и присоединенную к карбонильному атому углерода гидроксигруппу) и остатки аминокислот (т. е. молекулы, в которых удалены присоединенный к аминогруппе водород или присоединенная к карбонильному атому углерода гидроксигруппа, или они оба). Аминогруппа может представлять собой альфа-аминогруппу, бета-аминогруппу и т. п. Например, термин "аминокислота аланин" может означать отдельный аланин H-Ala-OH или любой из аланиновых остатков H-Ala-, -Ala-OH или -Ala-. Если не указано иное, все аминокислоты, содержащиеся в соединениях, описанных в настоящем изобретении, могут находиться в D- или L-конфигурации. Термин "аминокислота" включает ее соли, включая ее фармацевтически приемлемые соли. Любая аминокислота может быть защищенной или незащищенной. Защитные группы могут быть присоединены к аминогруппе (например, альфа-аминогруппе), карбоксигруппе главной цепи или к любой функциональной группе боковой цепи. В качестве примера фенилаланин, защищенный бензилоксикарбонильной группой (Z) по альфа-аминогруппе, представляется в виде Z-Phe-OH.
[0040] При использовании в настоящем изобретении термин "пептидный фрагмент" означает две или большее количество аминокислот, ковалентно связанных по меньшей мере одной амидной связью (т. е. связью между аминогруппой одной аминокислоты и карбоксигруппой другой аминокислоты, выбранной из числа аминокислот пептидного фрагмента). Термины "полипептид" и "пептидные фрагменты" используются взаимозаменяемым образом. Термин "пептидный фрагмент" включает его соли, включая его фармацевтически приемлемые соли.
[0041] При использовании в настоящем изобретении термин "сочетание" означает стадию взаимодействия двух химических фрагментов с образованием ковалентной связи. При указании на сочетание аминокислот термин "сочетание" означает стадию взаимодействия двух аминокислот и тем самым образование ковалентной амидной связи между аминогруппой одного аминокислотного остатка и карбоксигруппой (например, карбоксигруппой главной цепи) другой аминокислоты.
[0042] При использовании в настоящем изобретении термин "группа, активирующая карбоксигруппу" означает группу, которая модифицирует карбоксигруппу аминокислоты или карбоксильный конец пептидного фрагмента, восприимчивый по отношению к аминолизу. Обычно группа, активирующая карбоксигруппу, является электроноакцепторным фрагментом, который замещает гидроксильный фрагмент карбоксигруппы. Такой электроноакцепторный фрагмент увеличивает поляризацию и тем самым электрофильность карбонильного атома углерода. При использовании в настоящем изобретении термин "активированная карбоксигруппа" означает карбоксигруппу, в которой гидроксигруппа заменена группой, активирующей карбоксигруппу.
[0043] При использовании в настоящем изобретении термин "нуклеофильная добавка" означает химическое соединение или звено, которое используется в органическом синтезе для регулирования его стереохимического результата.
[0044] При использовании в настоящем изобретении термин "силилированная аминокислота" означает аминокислоту, которая модифицирована силилсодержащим фрагментом по меньшей мере в одном способном к модификации положении. Примеры способных к модификации положений включают функциональные группы -NH и -OH. Такие модификации являются результатом взаимодействия аминокислоты с силилирующим реагентом, описанного ниже. В приведенном в качестве примера варианте осуществления силилированная аминокислота является персилилированной, т. е. модифицированной силилсодержащим фрагментом по всем способным к модификации положениям.
[0045] Для облегчения синтеза аналога грелина H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2 (SEQ ID NO: 1) в промышленном масштабе в настоящем изобретении разработаны новые способы его синтеза. Обычно весь способ проводят в растворе, т. е. без проведения твердофазных реакций, таких как сочетание аминокислоты со связанной со смолой аминокислотой.
[0046] Имеется все больше данных в пользу существования отдельного пути грелина, который в некоторой степени перекрывается с GHSR-1a и который приводит к увеличению массы тела и усилению моторики ЖК без высвобождения GH (гормон роста). Наиболее очевидные данные получены для пептидных аналогов грелина, которые являются полными антагонистами GHSR-1a и не стимулируют высвобождение GH, но влияют на моторику ЖК и приводят к увеличению массы тела.
[0047] Фармакологические исследования аналога грелина H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2 (SEQ ID NO: 1), небольшого пептидного агониста грелина и, проведенные с использованием полноразмерного грелина человека на людях клинические исследования для рака, сердечных и COPD кахексий, продемонстрировали увеличение аппетита, массы тела и минутного сердечного выброса без явной токсичности. С учетом активных прокинетических эффектов грелина объектами клинического воздействия агониста грелина также являются нарушения моторики ЖК, в частности, послеоперационная кишечная непроходимость, вызванная опиоидом констипация, гастропарез, синдром раздраженной толстой кишки и хроническая констипация. Грелин и аналог грелина H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2 (SEQ ID NO: 1) также оказывают противовоспалительное воздействие, подавляя целый ряд воспалительных цитокинов, так что дополнительными возможными объектами клинического воздействия являются воспалительные патологические состояния ЖК, такие как воспалительная болезнь кишечника.
[0048] Ниже представлено описание приведенных в качестве примеров вариантов осуществления настоящего изобретения.
[0049] Первым вариантом осуществления настоящего изобретения является способ синтеза H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2 или его фармацевтически приемлемой соли, включающий стадию взаимодействия двух аминокислот в жидкой фазе.
[0050] Вторым вариантом осуществления настоящего изобретения является способ синтеза H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2 или его фармацевтически приемлемой соли, включающий получение силилированной аминокислоты путем силилирования незащищенной или защищенной аминокислоты или незащищенного или защищенного пептидного фрагмента по реакции с силилирующим реагентом в полярном апротонном органическом растворителе.
[0051] Защищенной аминокислотой является аминокислота, в которой одна или большее количество функциональных групп защищены защитной группой. Защищенным пептидным фрагментом является дипептид, трипептид или тетрапептид, в котором одна или большее количество функциональных групп аминокислоты пептидного фрагмента защищены защитной группой. Предпочтительно, если защищенная аминокислота и/или защищенный пептидный фрагмент, предлагаемый в настоящем изобретении, содержит защищенную аминогруппу. Термин "защитная группа аминогруппы" означает защитные группы, которые можно использовать для замены кислого протона аминогруппы для уменьшения ее нуклеофильности.
[0052] Примеры защитных групп аминогруппы (например, X1, X2, X3, X4 и т. п.) включают, но не ограничиваются только ими, замещенные или незамещенные группы ацильного типа, такие как формил, акрилил (Acr), бензоил (Bz), ацетил (Ac), трифторацетил, замещенные или незамещенные группы арилалкилоксикарбонильного типа, такие как бензилоксикарбонильная (Z), п-хлорбензилоксикарбонильная, п-бромбензилоксикарбонильная, п-нитробензилоксикарбонильная, п-метоксибензилоксикарбонильная, бензгидрилоксикарбонильная, 2(п-бифенилил)изопропилоксикарбонильная, 2-(3,5-диметоксифенил)изопропилоксикарбонильная, п-фенилазобензилоксикарбонильная, трифенилфосфоноэтилоксикарбонильная или 9-флуоренилметилоксикарбонильная группа (Fmoc), замещенные или незамещенные группы алкилоксикарбонильного типа, такие как трет-бутилоксикарбонильная (BOC), трет-амилоксикарбонильная, диизопропилметилоксикарбонильная, изопропилоксикарбонильная, этоксикарбонильная, аллилоксикарбонильная, 2-метилсульфонилэтилоксикарбонильная или 2,2,2-трихлорэтилоксикарбонильная группа, группы циклоалкилоксикарбонильного типа, такие как циклопентилоксикарбонильная, циклогексилоксикарбонильная, адамантилоксикарбонильная или изоборнилоксикарбонильная группа, и группы, содержащие гетероатом, такие как бензосульфонильная, п-толуолсульфонильная, мезитиленсульфонильная, метокситриметилфенилсульфонильная, 2-нитробензолсульфонильная, 2-нитробензолсульфенильная, 4- нитробензолсульфонильная или 4-нитробензолсульфенильная группа. Из этих групп X предпочтительными являются содержащие карбонильную, сульфенильную или сульфонильную группу. Защитные группы аминогруппы X1, X2, X3, X4 и т. п. предпочтительно выбраны из группы, включающей аллилоксикарбонильные группы, трет-бутилоксикарбонил (BOC), бензилоксикарбонил (Z), 9-флуоренилметилоксикарбонил (Fmoc), 4-нитробензолсульфонил (Nosyl), 2-нитробензолсульфенил (Nps) и замещенные производные.
[0053] Предпочтительными защитными группами аминогруппы X1, X2, X3, X4 и т. п. для способа, предлагаемого в настоящем изобретении, являются трет-бутилоксикарбонил (Boc), 9-флуоренилметилоксикарбонил (Fmoc) и бензилоксикарбонил (Z). Еще более предпочтительными защитными группами аминогруппы для способа, предлагаемого в настоящем изобретении, являются трет-бутилоксикарбонил (Boc) и бензилоксикарбонил (Z).
[0054] Защитные группы аминогруппы X1, X2, X3, X4 и т. п. можно ввести по различным методикам, известным в данной области техники. Например, по реакции с подходящими галогенангидридами кислот или ангидридами кислот. С другой стороны, защитные группы аминогруппы X1, X2, X3, X4 и т. п. можно удалить (т. е. провести стадию удаления защитной группы), например, с помощью ацидолиза, гидрогенолиза (например, в присутствии водорода (например, пропускаемого через жидкую реакционную среду) и катализатора, такого как палладиевый катализатор), путем обработки разбавленным раствором гидроксида аммония, путем обработки гидразином, путем обработки натрием и путем обработки амидом натрия.
[0055] В предпочтительном варианте осуществления способ, соответствующий любому из вариантов осуществления, описанных в настоящем изобретении, проводят без защиты карбоксигрупп аминокислот. Каждая стадия сочетания при синтезе аминокислоты включает сочетание аминокислоты, содержащей защищенную аминогруппу и необязательно активированную карбоксигруппу, с аминокислотой, содержащей незащищенную аминогруппу и незащищенную карбоксигруппу.
[0056] Предпочтительно, если силилирование незащищенной или защищенной аминокислоты или незащищенного или защищенного пептидного фрагмента включает силилирование незащищенной аминогруппы незащищенной или защищенной аминокислоты, или незащищенного или защищенного пептидного фрагмента.
[0057] Силилированный фрагмент, полученный способом, предлагаемым в настоящем изобретении (например, способом, соответствующим второму варианту осуществления), при желании можно выделить и очистить; однако предпочтительно использовать силилированный фрагмент in situ.
[0058] Типичные силилирующие реагенты включают N,O-бис(триметилсилил)ацетамид, N,O-бис(триметилсилил)трифторацетамид, гексаметилдисилазан, N-метил-N-триметилсилилацетамид, N,-метил-N-триметилсилилтрифторацетамид, N-(триметилсилил)ацетамид, N-(триметилсилил)диэтиламин, N-(триметилсилил)диметиламин, 1-(триметилсилил)имидазол, 3-(триметилсилил)-2-оксазолидон и (триметилсилил)-N-диметилацетамид. Предпочтительным силилирующим реагентом является (триметилсилил)-N-диметилацетамид.
[0059] Реакции силилирования, предлагаемые в настоящем изобретении, обычно проводят при температуре, равной от 0°C до 100°C и предпочтительно от 25°C до 50°C.
[0060] Обычно используют от 0,5 до 5, предпочтительно от 0,7 до 3, более предпочтительно от 1 до 2,5 и еще более предпочтительно примерно 2 или от 1,8 до 2,2 экв. силилирующего реагента в пересчете на количество молей силилируемых аминогрупп.
[0061] Обычно силилирование, предлагаемое в настоящем изобретении, проводят в присутствии полярного апротонного органического растворителя. Чаще растворителем является апротонный органический растворитель, обладающий статической относительной диэлектрической проницаемостью, равной от 5 до 10. Предпочтительно, если растворителем является этилацетат.
[0062] В третьем варианте осуществления настоящего изобретения способ, соответствующий любому из описанных вариантов осуществления, включает взаимодействие силилированного (например, силилированного фрагмента, соответствующего второму варианту осуществления) с (1) защищенной и активированной аминокислотой или (2) защищенным и активированным пептидным фрагментом, содержащим защитную группу аминогруппы и активированную карбоксигруппу.
[0063] Обычно взаимодействие силилированного фрагмента (например, силилированного фрагмента, соответствующего второму или третьему варианту осуществления) с (1) защищенной и активированной аминокислотой или (2) защищенным и активированным пептидным фрагментом, содержащим защитную группу аминогруппы и активированную карбоксигруппу, проводят в присутствии полярного апротонного органического растворителя. Чаще растворителем является апротонный органический растворитель, обладающий статической относительной диэлектрической проницаемостью, равной от 5 до 10. Предпочтительно, если растворителем является этилацетат.
[0064] Обычно раствор реакционной смеси, использующийся для силилирования и/или использующийся в последующей реакции сочетания аминокислоты или пептида с силилированным фрагментом, содержит от 10 мас.% до 90 мас.% полярного апротонного растворителя в пересчете на полную массу раствора.
[0065] Обычно взаимодействие силилированного фрагмента, предлагаемую в настоящем изобретении, с (1) защищенной и активированной аминокислотой или (2) защищенным и активированным пептидным фрагментом, аминокислотой или пептидным фрагментом, содержащим защитную группу аминогруппы и активированную карбоксигруппу, проводят при температуре от -50°C до 50°C.
[0066] Подходящие активирующие карбоксигруппу реагенты (также называющиеся в настоящем изобретении "активаторами") включают, но не ограничиваются только ими, N-гидроксисукцинимид (HOSu), N-гидроксифталимид, пентафторфенол (PfpOH) и ди-(п-хлортетрафторфенил)карбонат. В данной области техники также известно, что эти активаторы образуют активные сложные эфиры. Предпочтительно, если активатором является N-гидроксисукцинимид (HOSu).
[0067] В предпочтительном варианте осуществления настоящего изобретения в способе синтеза аналога грелина используют X1-(D)Bal-OSu, X4-Inp-OSu и X3-Phe-OSu, где каждый X1, X3 и X4 независимо означает защитную группу аминогруппы.
[0068] В четвертом варианте осуществления настоящего изобретения способ, соответствующий любому из вариантов осуществления, описанных в настоящем изобретении, дополнительно включает силилирование аминокислоты H-(D)Trp-OH с получением силилированного остатка аминокислоты H-(D)Trp-OH и взаимодействие силилированного остатка аминокислоты H-(D)Trp-OH с аминокислотой X1-(D)Bal-Y1, где X1 означает защитную группу аминогруппы и Y1 означает активированную карбоксигруппу. В предпочтительном варианте осуществления X1 означает Boc и Y1 означает -OSu. В более предпочтительном варианте осуществления, X1 означает Boc и Y1 означает -OSu и обе реакции силилирования и сочетание проводят в этилацетате.
[0069] В пятом варианте осуществления настоящего изобретения способ, соответствующий любому из вариантов осуществления, описанных в настоящем изобретении, дополнительно включает силилирование аминокислоты H-Apc(X2)-OH с получением силилированного остатка аминокислоты H-Apc(X2)-OH и взаимодействие силилированного остатка аминокислоты H-Apc(X2)-OH с аминокислотой X3-Phe-Y2, где X2 означает защитную группу аминогруппы и Y2 означает активированную карбоксигруппу. X3 является таким, как определено выше. В предпочтительном варианте осуществления H-Apc(X2)-OH представляет собой H-Apc(Boc)-OH и X3-Phe-Y2 представляет собой Z-Phe-OSu. В более предпочтительном варианте осуществления, H-Apc(X1)-OH представляет собой H-Apc(Boc)-OH и X2-Phe-Y3 представляет собой Z-Phe-OSu, и обе реакции силилирования и сочетания проводят в этилацетате.
[0070] Шестым вариантом осуществления настоящего изобретения является способ, соответствующий любому из вариантов осуществления, описанных в настоящем изобретении, где фрагмент X3-Phe-Apc(X2)-NH2 получают путем сочетания силилированного остатка аминокислоты H-Apc(X2)-OH и аминокислоты X3-Phe-Y2 в органическом растворителе с последующим амидированием карбоксигруппы. В предпочтительном варианте осуществления аминокислоту H-Apc(X2)-OH силилируют в этилацетате по ее реакции с (триметилсилил)-N-диметилацетамидом. В более предпочтительном варианте осуществления готовят суспензию H-Apc(X2)-OH, этилацетата и (триметилсилил)-N-диметилацетамида, суспензию нагревают (до температуры, равной от 35°C до 50°C; предпочтительно равной примерно 45°C) и после того, как силилирование в основном завершается, добавляют X3-Phe-Y2. Предпочтительно, если X3-Phe-Y-2 представляет собой Z-Phe-OSu и H-Apc(X2)-OH представляет собой H-Apc(Boc)-OH. Также предпочтительно, если амидирование карбоксигруппы проводят в присутствии аммиака и DCC. Кроме того, более предпочтительно, если X3-Phe-Y2 представляет собой Z-Phe-OSu и H-Apc(X2)-OH представляет собой H-Apc(Boc)-OH, и амидирование карбоксигруппы проводят в присутствии аммиака и DCC.
[0071] Седьмым вариантом осуществления настоящего изобретения является способ, соответствующий любому из вариантов осуществления, описанных в настоящем изобретении, где пептидный фрагмент X4-Inp-(D)Bal-(D)Trp-OH получают из пептидного фрагмента H-(D)Bal-(D)Trp-OH и X4-Inp-Y3 в присутствии основания, где Y3 означает активированную карбоксигруппу. В предпочтительном варианте осуществления основанием является диизопропилэтиламин, X4 означает Boc и Y3 означает -OSu. В другом предпочтительном варианте осуществления, HCl.H-(D)Bal-(D)Trp-OH солюбилизируют при температуре, равной от 10°C до 70°C (предпочтительно, примерно 40°C) в органическом растворителе (например, DMA) в присутствии основания с образованием раствора, затем раствор охлаждают (например, до температуры, равной 0°C) и к раствору добавляют Boc-Inp-OSu при температуре, равной от 10°C до 30°C.
[0072] Восьмым вариантом осуществления настоящего изобретения является способ, соответствующий любому из вариантов осуществления, описанных в настоящем изобретении, дополнительно включающий получение X4-Inp-(D)Bal-(D)Trp-Phe-Apc(X2)-NH2 из X4-Inp-(D)Bal-(D)Trp-OH и H-Phe-Apc(X2)-NH2 в присутствии нуклеофильной добавки и реагента сочетания. В предпочтительном варианте осуществления нуклеофильной добавкой является HOPO. В другом предпочтительном варианте осуществления нуклеофильной добавкой является HOPO и реагентом сочетания является EDC. В еще одном предпочтительном варианте осуществления H-Phe-Apc(X2)-NH2, X4-Inp-(D)Bal-(D)Trp-OH и нуклеофильную добавку солюбилизируют в органическом растворителе и затем добавляют реагент сочетания. В еще одном предпочтительном варианте осуществления H-Phe-Apc(X2)-NH2, X4-Inp-(D)Bal-(D)Trp-OH и HOPO солюбилизируют в органическом растворителе при температуре, равной от 10°C до 30°C (предпочтительно равной примерно 25°C), с образованием раствора, раствор охлаждают (например, до температуры, равной от 2°C до 10°C) и затем добавляют EDC. Предпочтительно, если H-Phe-Apc(X2)-NH2 представляет собой H-Phe-Apc(Boc)-NH2 и X4-Inp-(D)Bal-(D)Trp-OH представляет собой Boc-Inp-(D)Bal-(D)Trp-OH. Кроме того, предпочтительно, если органическим растворителем является диметилацетамид. В другом предпочтительном варианте осуществления способ дополнительно включает синтез Boc-Inp-(D)Bal-(D)Trp-Phe-Apc(Boc)-NH2 по реакции Boc-Inp-(D)Bal-(D)Trp-OH и H-Phe-Apc(Boc)-NH2 в органическом растворителе и в присутствии 2-гидроксипиридин-N-оксида и 1-(3-диметиламинопропил)-3-этилкарбодиимида.
[0073] Девятым вариантом осуществления настоящего изобретения является способ, соответствующий любому из вариантов осуществления, описанных в настоящем изобретении, дополнительно включающий удаление защитной группы Z-Phe-Apc(Boc)-NH2 путем гидрогенолиза с образованием H-Phe-Apc(Boc)-NH2. В предпочтительном варианте осуществления Z-Phe-Apc(Boc)-NH2 солюбилизируют в органическом растворителе (например, метаноле) и удаление защитной группы включает добавление катализатора (например, палладиевого катализатора) к органическому растворителю и пропускание или выработку водорода в органическом растворителе. Предпочтительно, если органическим растворителем является метанол.
[0074] Девятым вариантом осуществления настоящего изобретения является способ, соответствующий любому из вариантов осуществления, описанных в настоящем изобретении, дополнительно включающий удаление защитной группы Boc-Inp-(D)Bal-(D)Trp-Phe-Apc(Boc)-NH2 путем ацидолиза с образованием H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2.2HCl. В предпочтительном варианте осуществления ацидолиз проводят в присутствии 4-метилтиофенила и HCl в изопропаноле.
[0075] Десятым вариантом осуществления настоящего изобретения является способ жидкофазного синтеза аналога грелина H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2 или его фармацевтически приемлемой соли, включающий (a) синтез фрагмента H-(D)Bal-(D)Trp-OH из силилированных H-(D)Trp-OH и X1-(D)Bal-Y1 в органическом растворителе, (b) синтез фрагмента X3-Phe-Apc(X2)-NH2 из силилированных H-Apc(X2)-OH и X3-Phe-Y4 в органическом растворителе, (c) синтез фрагмента X4-Inp-(D)Bal-(D)Trp-OH из H-(D)Bal-(D)Trp-OH и X4-Inp-Y3 в органическом растворителе и в присутствии основания, и (d) синтез X-Inp-(D)Bal-(D)Trp-Phe-Apc(X2)-NH2 из X4-Inp-(D)Bal-(D)Trp-OH и H-Phe-Apc(X2)-NH2 в присутствии нуклеофильной добавки и реагента сочетания. В предпочтительных вариантах осуществления одну или большее количество стадий (a), (b), (c) и (d), соответствующих десятому варианту осуществления, можно провести независимо, как описано выше для первого - девятого вариантов осуществления, включая то, как описано в соответствующих конкретных и предпочтительных вариантах осуществления. В других конкретных вариантах осуществления одну или большее количество стадий (a), (b), (c) и (d) можно провести, как описано в соответствующих приведенных ниже примерах. Предпочтительно, если каждый реагент сочетания независимо представляет собой карбодиимидный реагент.
[0076] Другие варианты осуществления жидкофазного синтеза аналога грелина H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2 (SEQ ID NO: 1) или его фармацевтически приемлемой соли (например, ацетата H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2) схематично представлены на фиг. 1-14 и включают линейный синтез, представленный на фиг. 7 и 12, и конвергентный синтез, представленный на фиг. 1-6, 8-11, 13 и 14. Конвергентные синтезы являются предпочтительными и особенно предпочтительным является синтез, схематично представленный на фиг. 1. Аминогруппы аминокислот и пептидных фрагментов, приведенные на фиг. 1-14, можно защитить, как описано в настоящем изобретении, предпочтительно защитными группами аминогруппы Boc и Z, карбоксигруппы можно активировать, как описано в настоящем изобретении (например, с помощью HOSu), и эти аминокислоты и пептидные фрагменты можно последовательно ввести в реакции сочетания, как показано на всех фиг. 1-14, с реагентами сочетания и по реакциям сочетания, описанным в настоящем изобретении.
[0077] Аналог грелина H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2 (SEQ ID NO: 1) или его фармацевтически приемлемую соль (например, гидрохлорид H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2) можно дополнительно очистить и лиофилизировать и получить лиофилизированный аналог грелина. Соответственно, другим вариантом осуществления настоящего изобретения является способ получения лиофилизированного аналога грелина, способ включает получение неочищенного продукта, включающего H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2, или его фармацевтически приемлемой соли, соответствующей любому из вариантов осуществления, описанных в настоящем изобретении, и дополнительно включает очистку неочищенного продукта с помощью высокоэффективной жидкостной хроматографии и получение очищенного продукта, и лиофилизацию очищенного продукта и получение лиофилизированного аналога грелина. В предпочтительном варианте осуществления способ включает элюирование неочищенного продукта из колонки (предпочтительно, содержащей привитой с помощью C18 диоксид кремния) с помощью буфера ацетонитрил/ацетат аммония в градиентном режиме с получением элюата, фракционирование элюата, объединение фракций, обладающих необходимой чистотой (например, >95%) и получение объединенной фракции, разбавление объединенной фракции водой и получение разбавленной объединенной фракции, элюирование разбавленной объединенной фракции обогащенной ацетонитрилом смесью в градиентном режиме и получение второго элюата, фракционирование второго элюата, объединение вторых фракций, обладающих необходимой чистотой, и получение объединенной фракции высокой чистоты, выпаривание ацетонитрила в вакууме из объединенной фракции высокой чистоты и получение водного раствора, и сушку вымораживанием водного раствора, и получение лиофилизированного аналога грелина.
[0078] На фиг. 15 приведена схематичная диаграмма получения лиофилизированного аналога грелина, описывающегося последовательностью H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2 (SEQ ID NO: 1), включающей синтетические последовательности синтеза аналога грелина H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2 (SEQ ID NO: 1).
[0079] Реагенты сочетания, предлагаемые в настоящем изобретении, обычно являются карбодиимидными реагентами. Примеры карбодиимидных реагентов включают, но не ограничиваются только ими, N,Nʹ-дициклогексилкарбодиимид (DCC), 1-(3-диметиламинопропил)-3-этилкарбодиимид (EDC), N-циклогексил-Nʹ-изопропилкарбодиимид (CIC), N,Nʹ-диизопропилкарбодиимид (DIC), N-трет-бутил-Nʹ-метилкарбодиимид (BMC), N-трет-бутил-Nʹ-этилкарбодиимид (BEC), бис[[4-(2,2-диметил-1,3-диоксолил)]-метил]карбодиимид (BDDC) и N,N-дициклопентилкарбодиимид. DCC является предпочтительным реагентом сочетания.
[0080] Нуклеофильные добавки, предлагаемые в настоящем изобретении, обычно выбраны из группы, включающей 2-гидроксипиридин-N-оксид (HOPO), 1-гидрокси-7-азабензотриазол (HOAt), 1-гидроксибензотриазол (HOBt), 3,4-дигидро-3-гидрокси-4-оксо-1,2,3-бензотриазин (HODhbt) и этил-1-гидрокси-1H-1,2,3-триазол-4-карбоксилат (HOCt).
[0081] Обычно силилирующий реагент, предлагаемый в настоящем изобретении, выбран из группы, включающей N,O-бис(триметилсилил)ацетамид, N,O-бис(триметилсилил)трифторацетамид, гексаметилдисилазан, N-метил-N-триметилсилилацетамид, N-метил-N-триметилсилилтрифторацетамид, триметилхлорсилан+основание, N-(триметилсилил)ацетамид, триметилсилилцианид, N-(триметилсилил)диэтиламин, N-(триметилсилилдиметиламин, 1-(триметилсилил)имидазол и 3-триметилсилил-2-оксазолидинон. В приведенном в качестве примера варианте осуществления силилирующим реагентом является (триметилсилил)-N-диметилацетамид.
[0082] Способы, описанные в настоящем изобретении, обычно могут дополнительно включать стадии остановки реакции (например, путем добавления 3-(диметиламино)пропиламина), стадии промывки (например, органическим растворителем (например, ацетонитрилом, диизопропиловым эфиром, изопропанолом или циклогексаном), раствором KHSO4 (например, 4 (мас./об.)% раствором KHSO4), раствором NaCl (например, 2 (мас./об.)% раствором NaCl), деминерализованной водой, раствором NaHCO3 (например, 4 (мас./об.)% раствором NaHCO3)), стадии концентрирования (например, концентрирования в вакууме, кристаллизации, фильтрования, осаждения) и стадии сушки (например, сушки в вакууме или азеотропной перегонки).
[0083] Обозначения, использующиеся для определения пептидов, являются такими, которые обычно применяются в данной области техники, и аминогруппа на N-конце указывается слева и карбоксигруппа на C-конце указывается справа.
[0084] При использовании в настоящем изобретении термин "аминокислота" включает и природную аминокислоту, и неприродную аминокислоту.
[0085] Некоторые аминокислоты, содержащиеся в соединениях, предлагаемых в настоящем изобретении, можно представить и представлены в настоящем изобретении следующим образом:
[0086] Apc означает следующую структуру, описывающую 4-аминопиперидин-4-карбоновую кислоту:
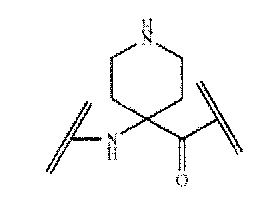 ,
,
[0087] Bal означает следующую структурную формулу, описывающую 3-бензотиенилаланин:
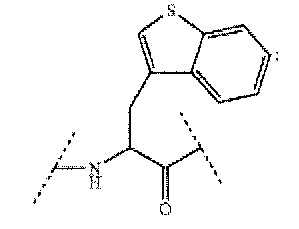 ,
,
[0088] Inp означает следующую структурную формулу, описывающую изонипекотиновую кислоту:
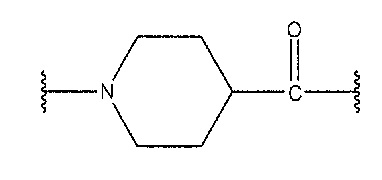 ,
,
[0089] Phe означает следующую структурную формулу, описывающую фенилаланин:
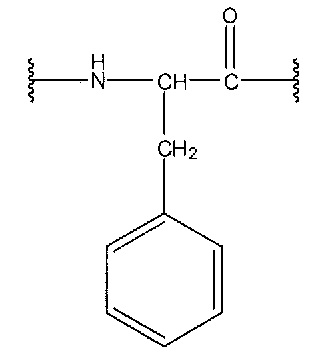 , и
, и
[0090] Trp означает следующую структурную формулу, описывающую триптофан:
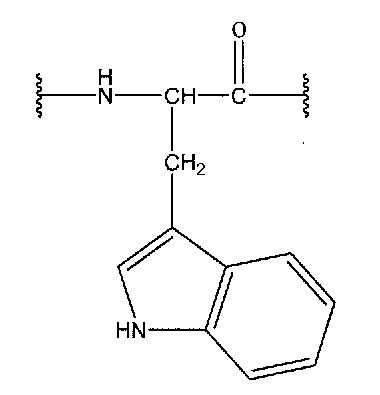
[0091] Некоторые другие аббревиатуры, использующиеся в настоящем изобретении, определяются следующим образом:
BDDC означает бис[[4-(2,2-диметил-1,3-диоксолил)]-метил]карбодиимид,
BEC означает N-трет-бутил-Nʹ-этилкарбодиимид,
BMC означает N-трет-бутил-Nʹ-метилкарбодиимид,
Boc означает трет-бутилоксикарбонил,
CIC означает N-циклогексил-Nʹ-изопропилкарбодиимид;
DMA означает диметиламин,
DCC означает N,Nʹ-дициклогексилкарбодиимид
DCU означает N,Nʹ-дициклогексилмочевину
DIC означает N,Nʹ-диизопропилкарбодиимид,
DIEA или DIPEA означает диизопропилэтиламин,
EDC означает 1-(3-диметиламинопропил)-3-этилкарбодиимид,
Fmoc означает флуоренилметилоксикарбонил,
HOAt означает 1-гидрокси-7-азабензотриазол,
HOBt означает 1-гидроксибензотриазол,
HOCt означает этил-1-гидрокси-1H-1,2,3-триазол-4-карбоксилат,
HODhbt означает 3,4-дигидро-3-гидрокси-4-оксо-1,2,3-бензотриазин,
HOPO означает 2-гидроксипиридин-N-оксид,
HOSu или SucOH означает N-гидроксисукцинимид,
PfpOH означает пентафторфенол, и
Z означает бензилоксикарбонил.
[0092] За исключением N-концевой аминокислоты все аббревиатуры аминокислот (например, Phe) в настоящем изобретении означают структуру -NH-C(R)(R')-CO-, где R и R' все независимо означают водород или боковую цепь аминокислоты (например, R= бензил и R'=H для Phe), или R и R' могут быть соединены друг с другом с образованием кольцевой системы, как в случае Apc и Inp. Соответственно, 4-аминопиперидин-4-карбоновой кислотой является H-Apc-OH, 3-бензотиенилаланином является H-Bal-OH, изонипекотиновой кислотой является H-Inp-OH, фенилаланином является H-Phe-OH и триптофаном является H-Trp-OH. Обозначение "OH" для этих аминокислот или для пептидов (например, Boc-Inp-(D)Bal-(D)Trp-OH) показывает, что C-конец является свободной кислотой. Обозначение "NH2", например, для промежуточного продукта, защищенного дипептида Z-Phe-Apc(Boc)-NH2 или для пептида H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2 показывает, что амидируют C-конец защищенного пептидного фрагмента. Кроме того, некоторые R и Rʹ по отдельности или в комбинации в виде кольцевой структуры могут включать функциональные группы для которых во время жидкофазного синтеза необходима защита, например, группу R и Rʹ в Apc, можно защитить другой группой, например, группой Boc: Apc(Boc). Кроме того, N-конец аминокислот можно защитить защитной группой аминогруппы X, такой как Boc, что приводит к следующему обозначению: X-Inp-OH, X-Bal-OH и т. п. (например, Boc-Inp-OH, Boc-Bal-OH и т. п.). Карбоксигруппу аминокислот можно активировать, например, активатором Y, таким как N-гидроксисукцинимид (HOSu), что приводит к следующему обозначению H-Inp-Y (например, H-Inp-OSu).
[0093] Если аминокислота обладает изомерными формами, то представлена L-форма аминокислоты, если явно не указана D-форма, например, (D)Bal или D-Bal.
[0094] Аналог грелина H-Inp-DBal-D-Trp-Phe-Apc-NH2 (SEQ ID NO: 1) можно получить в виде солей с кислотами или основаниями. Фармацевтически приемлемые соли (в виде растворимых в воде или растворимых или диспергирующихся в масле продуктов) включают обычные нетоксичные соли или четвертичные аммониевые соли, которые образуются, например, из неорганических или органических кислот или оснований. Примеры таких солей включают соли присоединения с кислотами, такие как ацетат, адипат, альгинат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, цитрат, камфорат, камфорсульфонат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, фумарат, глюкогептаноат, глицерофосфат, гемисульфат, гептаноат, гексаноат, гидрофторид, гидробромид, гидройодид, 2-гидроксиэтансульфонат, лактат, малеат, метансульфонат, 2-нафталинсульфонат, никотинат, оксалат, памоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, тиоцианат, тозилат и ундеканоат; и соли присоединения с основаниями, такие как соли аммония, соли щелочных металлов, такие как соли натрия и калия, соли щелочноземельных металлов, такие как соли кальция и магния, соли с органическими основаниями, такие как соли с дихлоргексиламином, N-метил-D-глюкамином, и соли с аминокислотами, такие как соли с аргинином и лизином. Предпочтительно, если аналог грелина H-Inp-DBal-DTrp-Phe-Apc-NH2 (SEQ ID NO: 1) получают в виде ацетата.
Примеры
Синтез H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2 по схеме, приведенной на фиг. 15
[0095] В описанном ниже синтезе используют (1) защищенную аминокислоту в качестве исходного вещества, а именно, Boc-Inp-OH, Boc-(D)Bal-OH, Z-Phe-OH и H-Apc(Boc)-OH, и (2) незащищенную аминокислоту H-(D)Trp-OH. Эти аминокислоты имеются в продаже или можно синтезировать по методикам, известным в данной области техники.
1. Синтез Boc-Inp-OSu
[0096] Boc-Inp-OSu синтезировали по схеме синтеза, приведенной на фиг. 16.
[0097] А именно, Boc-Inp-OH (1,15 г, 5 ммоля) и N-гидроксисукцинимид (SucOH) (0,69 г, 6 ммоля) солюбилизировали в 12,3 мл ацетонитрила при комнатной температуре. После растворения твердых веществ раствор охлаждали до 0°C и по каплям добавляли DCC (1,08 г 5,25 ммоля), растворенный в 1,4 мл ацетонитрила. Температуру поддерживали равной 0°C в течение 1 ч и затем постепенно повышали до комнатной температуры в течение 4 ч. После проведения реакции в течение ночи двумя порциями добавляли DCC (0,10 г, 0,5 ммоля), растворенный в 0,15 мл ацетонитрила. После завершения реакции образовавшийся DCC удаляли фильтрованием и дважды промывали с помощью 3,8 мл ацетонитрила. Маточные растворы объединяли и концентрировали в вакууме с получением раствора объемом 5 мл. Затем этот концентрированный раствор добавляли к 10,4 мл изопропанола, что приводило к осаждению Boc-Inp-OSu. Суспензию концентрировали в вакууме до объема, равного 8 мл, и затем разбавляли с помощью 12,5 мл изопропанола. Твердое вещество отфильтровывали, дважды промывали с помощью 3,8 мл изопропанола и сушили в вакууме при 45°C и получали 1,51 г белого порошкообразного вещества (выход 90%).
2. Синтез Boc-(D)Bal-OSu
[0098] Boc-DBal-OH синтезировали по схеме синтеза, приведенной на фиг. 17.
[0099] А именно, Boc-(D)Bal-OH (1,61 г, 5 ммоля) и N-гидроксисукцинимид (SucOH) (0,69 г, 6 ммоля) солюбилизировали в 17,6 мл ацетонитрила при комнатной температуре. После растворения твердых веществ раствор охлаждали до 0°C и по каплям добавляли DCC (1,03 г 5 ммоля), растворенный в 1,3 мл ацетонитрила. Температуру поддерживали равной 0°C в течение 1 ч и затем постепенно повышали до комнатной температуры в течение 4 ч. После проведения реакции в течение ночи двумя порциями добавляли DCC (0,10 г, 0,5 ммоля), растворенный в 0,15 мл ацетонитрила. После завершения реакции образовавшийся DCC удаляли фильтрованием и дважды промывали с помощью 12 мл ацетонитрила. Маточные растворы объединяли и концентрировали в вакууме с получением раствора объемом 13 мл. Затем этот концентрированный раствор добавляли к 27 мл изопропанола. Во время дополнительного концентрирования в вакууме Boc-(D)Bal-OSu кристаллизовался. Ацетонитрил дополнительно удаляли с помощью еще 43 мл изопропанола. Конечный объем суспензии составлял 53 мл. Твердое вещество отфильтровывали, дважды промывали с помощью 9 мл изопропанола, затем с помощью 9 мл диизопропилового эфира и сушили в вакууме при 45°C, и получали 1,83 г белого порошкообразного вещества (выход 85%).
3. Синтез H-(D)Bal-(D)Trp-OH
[00100] H-(D)Bal-(D)Trp-OH синтезировали по схеме синтеза, приведенной на фиг. 19.
[00101] А именно, H-(D)Trp-OH (0,91 г, 4,34 ммоля) добавляли к (триметилсилил)-N-диметилацетамиду (1,27 г, 8,67 ммоля) и 4,1 мл этилацетата. Реакционную среду нагревали при 45°C до образования раствора (в течение примерно 2 ч). Раствор охлаждали до 0°C и добавляли к холодному раствору Boc-(D)Bal-OSu (1,83 г 4,25 ммоля) в 17,6 мл этилацетата. Через 15 мин после добавления температуру реакционной среды доводили до комнатной температуры. После достижения необходимой степени превращения (примерно через 5 ч) реакцию останавливали с помощью 3-(диметиламино)пропиламина (0,11 г 1,06 ммоля), затем дважды промывали с помощью 14,5 мл 4 (мас./об.)% раствора KHSO4 и один раз промывали с помощью 17 мл 2 (мас./об.)% раствора NaCl и в заключение один раз промывали с помощью 14 мл деминерализованной воды. Полученную органическую фазу концентрировали в вакууме, добавляли 13,4 мл ледяной уксусной кислоты и раствор дополнительно концентрировали до конечного объема, равного 9,7 мл. Добавляли 4-метилтиофенол (1,82 г 12,75 ммоля) и 4 н. раствор HCl в диоксане (2,23 г 8,5 ммоля). Через 2 ч реакцию останавливали и реакционную среду осаждали в 106 мл диизопропилового эфира. Твердое вещество отфильтровывали и дважды промывали с помощью 20 мл диизопропилового эфира. После сушки в течение ночи в вакууме при 45°C получали 1,98 г HCl H-(D)Bal-(D)Trp-OH (выход 90%).
4. Синтез Boc-Inp-(D)Bal-(D)Trp-OH
[00102] Boc-Inp-(D)Bal-(D)Trp-OH синтезировали по схеме синтеза, приведенной на фиг. 20.
[00103] А именно, HCl H-(D)Bal-(D)Trp-OH (1,74 г 3,83 ммоля) солюбилизировали при 40°C в 13,8 мл DMA в присутствии DIPEA (1,03 г, 7,86 ммоля). После образования раствора смесь охлаждали до 0°C и к этому раствору при комнатной температуре добавляли Boc-Inp-OSu (1,31 г 4,02 ммоля) в виде твердого вещества. Через 1 ч после добавления температуру реакционной среды доводили до комнатной температуры. После проведения реакции в течение ночи превращение становилось полным и реакцию останавливали путем добавления 3-(диметиламино)пропиламина (0,08 г 0,8 ммоля). Затем смесь разбавляли с помощью 56 мл этилацетата и трижды промывали с помощью 28 мл 4 (мас./об.)% раствора KHSO4, затем один раз промывали с помощью 25 мл деминерализованной воды. Полученную органическую фазу концентрировали в вакууме и сушили с помощью азеотропной перегонки. Всего дополнительно добавляли 68 мл этилацетата. Раствор концентрировали до конечного объема, равного 14 мл, и осаждали в 128 мл диизопропилового эфира. Твердое вещество отфильтровывали, дважды промывали с помощью 24 мл диизопропилового эфира и сушили в вакууме при 45°C и получали 1,6 г твердого вещества (выход 81%).
5. Синтез Z-Phe-OSu
[00104] Z-Phe-OSu синтезировали по схеме синтеза, приведенной на фиг. 18.
[00105] А именно, Z-Phe-OH (1,53 г, 5 ммоля) и N-гидроксисукцинимид (SucOH) (0,69 г, 6 ммоля) солюбилизировали в 16,3 мл ацетонитрила при комнатной температуре. После растворения твердых веществ раствор охлаждали до 0°C и по каплям добавляли DCC (1,08 г 5,25 ммоля), растворенный в 1,3 мл ацетонитрила. Температуру поддерживали равной 0°C в течение 1 ч и затем постепенно повышали до комнатной температуры в течение 4 ч. После проведения реакции в течение ночи двумя порциями добавляли DCC (0,10 г, 0,5 ммоля), растворенный в 0,15 мл ацетонитрила. После завершения реакции образовавшийся DCC удаляли фильтрованием и дважды промывали с помощью 4 мл ацетонитрила. Маточные растворы объединяли и концентрировали в вакууме с получением раствора объемом 6 мл. Затем этот концентрированный раствор добавляли к 12 мл изопропанола. Во время дополнительного концентрирования в вакууме Z-Phe-OSu кристаллизовался. Ацетонитрил дополнительно удаляли с помощью еще 14,5 мл изопропанола. Конечный объем суспензии составлял 24 мл. Твердое вещество отфильтровывали, дважды промывали с помощью 4 мл изопропанола и сушили в вакууме при 45°C и получали 1,7 г белого порошкообразного вещества (выход 87%).
6. Синтез Z-Phe-Apc(Boc)-NH2
[00106] Z-Phe-Apc(Boc)-NH2 синтезировали по схеме синтеза, приведенной на фиг. 21.
[00107] А именно, H-Apc(Boc)-OH (1,06 г 4,2 ммоля) добавляли к 8,8 мл этилацетата, содержащего (триметилсилил)-N-диметилацетамид (1,23 г 8,4 ммоля). Суспензию нагревали при 45°C. После образования раствора добавляли раствор Z-Phe-OSu (1,7 г 4,28 ммоля) в 16,1 мл этилацетата. Температуру поддерживали равной 45°C и после проведения реакции в течение ночи реакцию останавливали с помощью 3-(диметиламино)пропиламина (0,11 г 1,07 ммоля). Затем смесь дважды промывали с помощью 11 мл 4 (мас./об.)% раствора KHSO4, затем с помощью 11 мл 2 (мас./об.)% раствора NaCl и в заключение с помощью 11 мл деминерализованной воды. Промытую органическую фазу сушили с помощью азеотропной перегонки путем добавления 28 мл этилацетата. Раствор концентрировали до конечного объема, равного 28,2 мл, и охлаждали до 0°C. Добавляли DCC (0,78 г 4,63 ммоля), предварительно солюбилизированный в 1 мл этилацетата, затем по каплям добавляли 9,261 мл 0,5M раствора аммиака (4,63 ммоля) в диоксане. После завершения добавления температуру смеси доводили до комнатной температуры и через 1 ч превращение становилось полным. Реакцию останавливали путем добавления 0,83 мл воды и нагревали в течение 30 мин при 35°C. DCU удаляли фильтрованием и полученный раствор дважды промывали с помощью 33 мл 4 (мас./об.)% раствора KHSO4, 33 мл 4 (мас./об.)% раствора NaHCO3 и в заключение с помощью 33 мл деминерализованной воды. Промытую органическую фазу сушили с помощью азеотропной перегонки. Затем добавляли 29 мл этилацетата. Конечный объем составлял 7,8 мл. К этому раствору добавляли 7,7 мл горячего циклогексана. Z-Phe-Apc(Boc)-NH2 кристаллизовался в течение ночи при 5°C. После отфильтровывания кристаллов дважды промывали с помощью 15 мл циклогексана, твердое вещество сушили в вакууме при 45°C. Получали 1,98 г белых кристаллов (выход 87%).
7. Синтез H-Phe-Apc(Boc)-NH2
[00108] H-Phe-Apc(Boc)-NH2 синтезировали по схеме синтеза, приведенной на фиг. 22.
[00109] А именно, Z-Phe-Apc(Boc)-NH2 (1,97 г 3,65 ммоля) солюбилизировали в 6,15 мл метанола. После добавления 0,194 г палладиевого катализатора, нанесенного на древесный уголь (0,18 ммоля) реакцию останавливали путем пропускания N2 в течение 30 мин и затем через раствор при 35°C пропускали водород. Через 2 ч реакция завершалась и катализатор отфильтровывали. Полученный раствор концентрировали в вакууме и добавляли 9 мл ацетонитрила. Раствор дополнительно концентрировали и H-Phe-Apc(Boc)-NH2 кристаллизовался. После установления объема, равного 4,1 мл, суспензию фильтровали и твердое вещество дважды промывали с помощью 10 мл диизопропилового эфира. Твердое вещество сушили в вакууме при 45°C и получали 1,3 г твердого вещества (выход 87%).
8. Синтез Boc-Inp-(D)Bal-(D)Trp-Phe-Apc(Boc)-NH2
[00110] Boc-Inp-(D)Bal-(D)Trp-Phe-Apc(Boc)-NH2 синтезировали по схеме синтеза, приведенной на фиг. 23.
[00111] А именно, H-Phe-Apc(Boc)-NH2 (0,95 г 2,35 ммоля), Boc-Inp-(D)Bal-(D)Trp-OH (1,6 г 2,47 ммоля) и 2-гидроксипиридин-N-оксид (0,32 г 2,84 ммоля) солюбилизировали в 11,3 мл диметилацетамида при комнатной температуре. После образования раствора смесь охлаждали до 5°C и добавляли этил-Nʹʹ-диметилпропиламинкарбодиимид (0,55 г 2,84 ммоля). Через 1 ч температуру устанавливали равной 10°C и через 5 ч смесь нагревали до комнатной температуры. После проведения реакции в течение ночи обеспечивалась удовлетворительная степень превращения и смесь разбавляли с помощью 40 мл этилацетата. Полученный раствор промывали с помощью 19 мл 4 (мас./об.)% раствора KHSO4, трижды с помощью 14 мл 4 (мас./об.)% раствора NaHCO3 и в заключение с помощью 15 мл деминерализованной воды. Промытую органическую фазу сушили с помощью азеотропной перегонки. Затем добавляли 29 мл этилацетата. Конечный объем составлял 16,3 мл. К этому раствору добавляли 19 мл горячего циклогексана. Boc-Inp-(D)Bal-(D)Trp-Phe-Apc(Boc)-NH2 кристаллизовали в течение ночи при 5°C. После отфильтровывания кристаллов дважды промывали с помощью 15 мл циклогексана, твердое вещество сушили в вакууме при 45°C. Получали 2 г белых кристаллов (выход 86%).
9. Синтез H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2 (неочищенного)
[00112] H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2 синтезировали по схеме синтеза, приведенной на фиг. 24.
[00113] А именно, Boc-Inp-(D)Bal-(D)Trp-Phe-Apc(Boc)-NH2 (2 г 2,02 ммоля) и 4-метилтиофенол солюбилизировали в 9 мл изопропанола. Добавляли 5 н. раствор HCl в изопропаноле (3,3 мл 20,2 ммоля) и смесь нагревали при 40°C. После проведения реакции в течение ночи реакция завершалась и образовывалась суспензия. Суспензию разбавляли с помощью 83 мл диизопропилового эфира и фильтровали. Твердое вещество трижды промывали с помощью 10 мл диизопропилового эфира. После сушки в вакууме при 45°C получали 1,7 г твердого вещества (выход 70%).
10/11. Очистка/лиофилизация неочищенного H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2
[00114] Неочищенный продукт элюировали из колонки, содержащей привитой с помощью C18 диоксид кремния при элюировании с помощью буфера ацетонитрил/ацетат аммония в градиентном режиме. Элюат фракционировали и фракции, обладающие чистотой более 95%, объединяли. Фракции разбавляли водой, повторно вводили в колонку и элюировали обогащенной ацетонитрилом смесью в градиентном режиме. Ацетонитрил выпаривали в вакууме и полученный водный раствор сушили вымораживанием и получали конечный продукт, который являлся ацетатом искомого полипептида.
[00115] Положения всех патентов, опубликованных заявок и литературы, цитированных в настоящем изобретении, во всей своей полноте включены в настоящее изобретение в качестве ссылки.
[00116] Хотя настоящее изобретение подробно представлено и описано с помощью его приведенных в качестве примеров вариантов осуществления, специалисты в данной области техники должны понимать, что без отклонения от объема настоящего изобретения, определяющегося прилагаемой формулой изобретения, в него можно внести различные по форме и подробностям изменения.

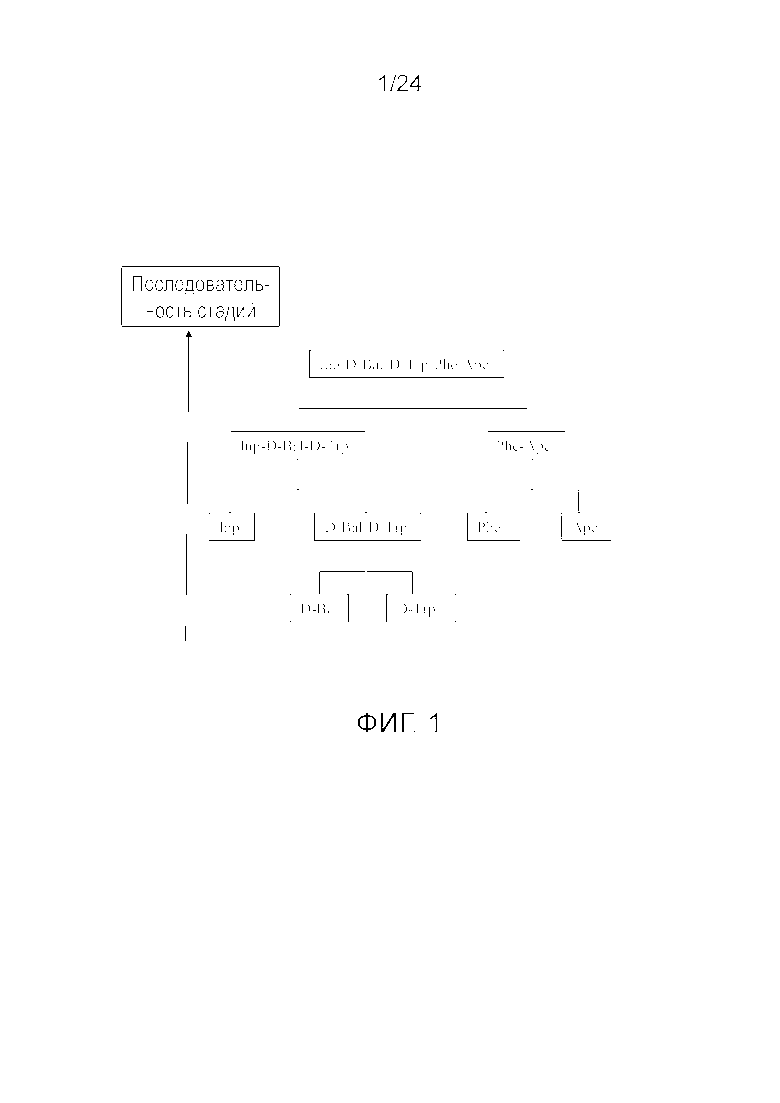
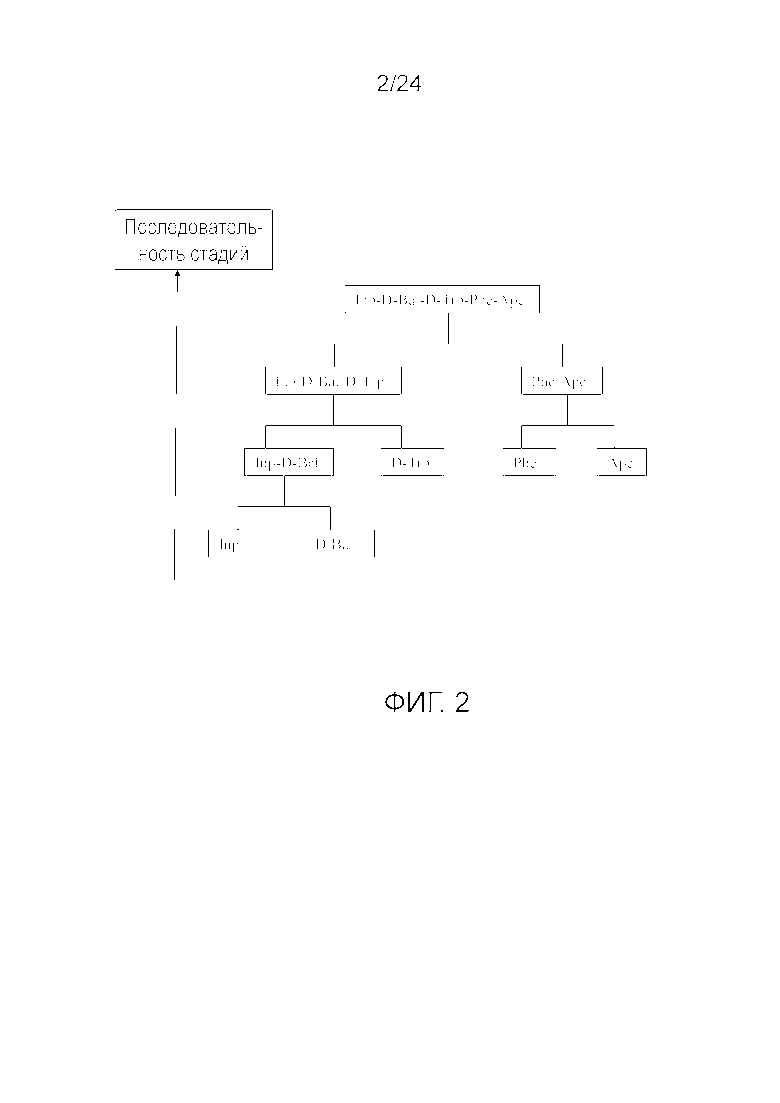

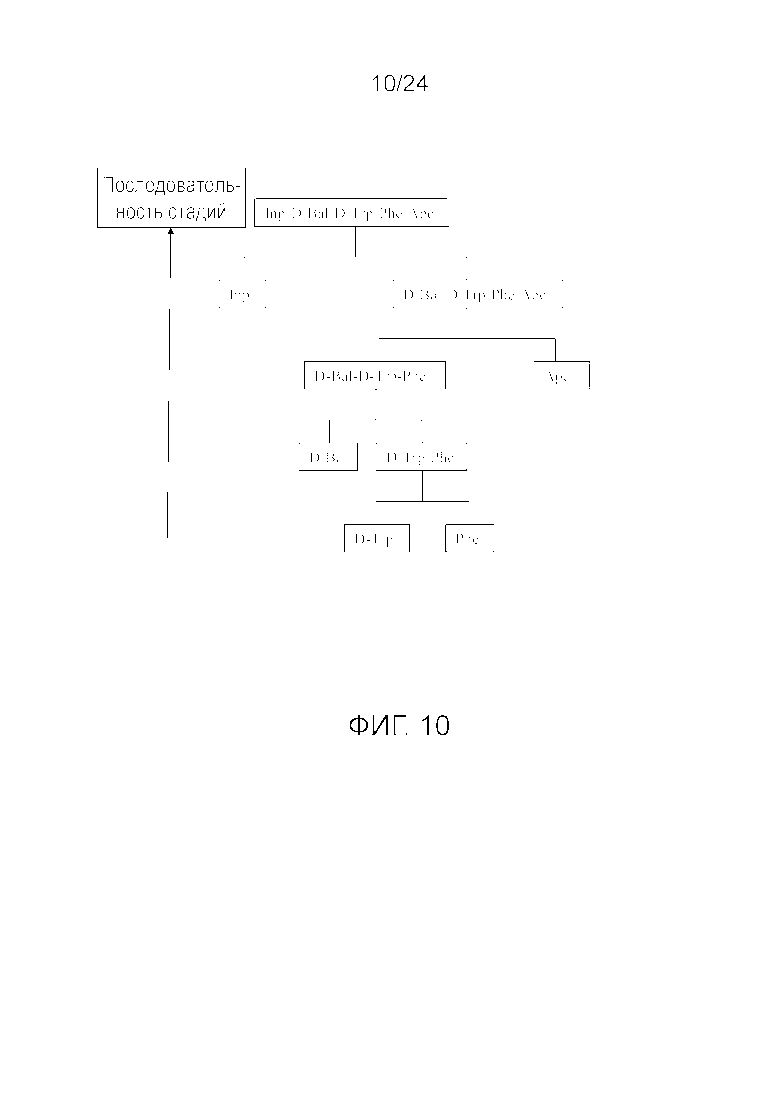

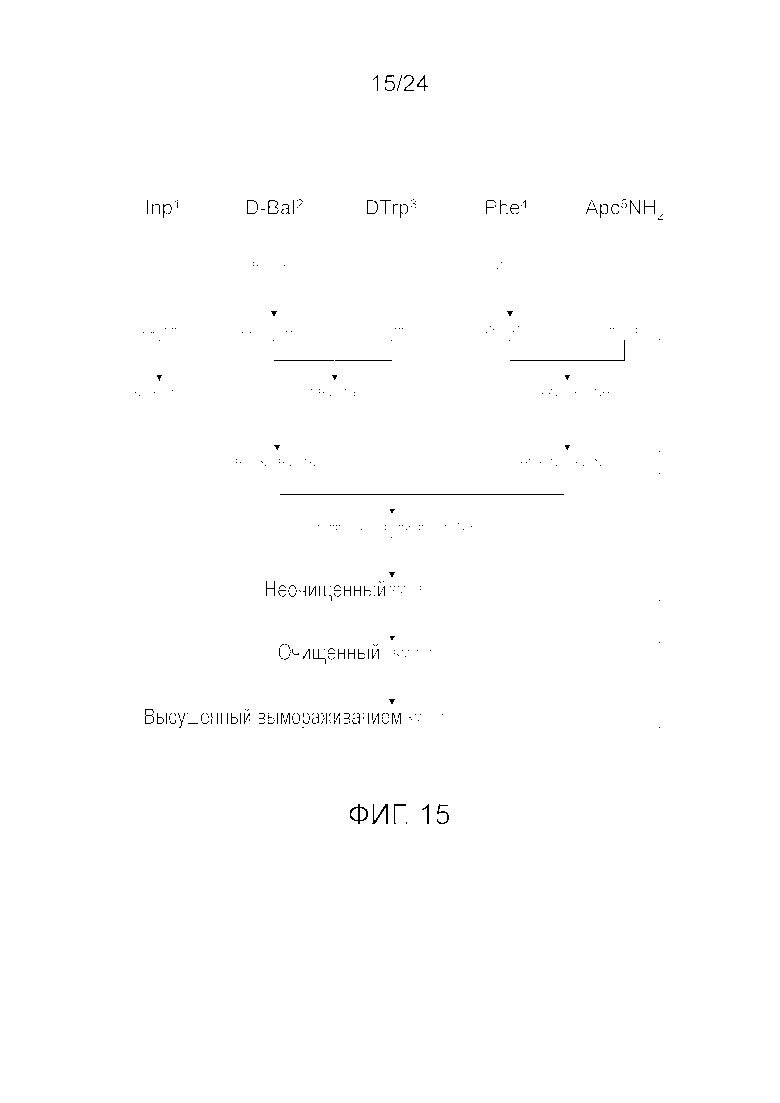









Изобретение относится к способу жидкофазного синтеза аналога грелина H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2 (SEQ ID NO: 1, формула (I)) и его фармацевтически приемлемых солей. Способ позволяет в промышленном масштабе получить аналог грелина SEQ ID NO: 1 и обеспечивает высокую чистоту. 6 н. и 12 з.п. ф-лы, 24 ил., 1 пр.
1. Способ синтеза пептида формулы (I):
H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2,
или его фармацевтически приемлемой соли, включающий стадию взаимодействия пептидного фрагмента следующей формулы:
X4-Inp-(D)Bal-(D)Trp-OH,
с пептидным фрагментом следующей формулы:
H-Phe-Apc(X2)-NH2,
в присутствии нуклеофильной добавки
и тем самым получение пептидного фрагмента следующей формулы:
X4-Inp-(D)Bal-(D)Trp-Phe-Apc(X2)-NH2,
или его соли,
где каждый из X2 и X4 независимо означает защитную группу аминогруппы.
2. Способ по п. 1, дополнительно включающий стадию удаления защитной группы пептидного следующей формулы:
X4-Inp-(D)Bal-(D)Trp-Phe-Apc(X2)-NH2,
и тем самым получение пептида формулы (I)
H-Inp-(D)Bal-(D)Trp-Phe-Apc-NH2 (I)
или его соли.
3. Способ по п. 1 или 2, дополнительно включающий стадии:
(i) взаимодействия первого силилирующего реагента с аминокислотой H-(D)Trp-OH в первом жидком растворителе и тем самым получение силилированного аминокислотного остатка аминокислоты H-(D)Trp-OH или его соли;
(ii) взаимодействия силилированного аминокислотного остатка аминокислоты H-(D)Trp-OH с аминокислотой X1-(D)Bal-Y1 во втором жидком растворителе и тем самым получение пептидного фрагмента следующей формулы:
X1-(D)Bal-(D)Trp-OH,
или его соли,
где X1 означает защитную группу аминогруппы и Y1 означает группу, активирующую карбоксигруппу;
(iii) взаимодействия второго силилирующего реагента с аминокислотой H-Apc(X2)-OH в третьем жидком растворителе и тем самым получение силилированного аминокислотного остатка аминокислоты H-Apc(X2)-OH, где X2 означает защитную группу аминогруппы;
(iv) взаимодействия силилированного аминокислотного остатка аминокислоты H-Apc(X2)-OH с аминокислотой X3-Phe-Y2 в четвертом жидком растворителе и тем самым получение пептидного фрагмента следующей формулы:
X3-Phe-Apc(X2)-OH,
или его соли,
где X3 означает защитную группу аминогруппы и Y2 означает группу, активирующую карбоксигруппу;
(v) взаимодействия пептидного фрагмента следующей формулы:
X3-Phe-Apc(X2)-OH,
с амидирующим реагентом в пятом жидком растворителе и тем самым получение пептидного фрагмента следующей формулы
X3-Phe-Apc(X2)-NH2,
или его соли;
(vi) удаления защитной группы пептидного фрагмента следующей формулы:
X3-Phe-Apc(X2)-NH2,
и тем самым получение пептидного фрагмента следующей формулы:
H-Phe-Apc(X2)-NH2,
или его соли;
(vii) удаления защитной группы пептидного фрагмента следующей формулы:
X1-(D)Bal-(D)Trp-OH,
и тем самым получение пептидного фрагмента следующей формулы:
H-(D)Bal-(D)Trp-OH,
или его соли; и
взаимодействия аминокислоты X4-Inp-Y3 с пептидным фрагментом следующей структурной формулы:
H-(D)Bal-(D)Trp-OH,
в шестом жидком растворителе и тем самым получение пептидного фрагмента следующей формулы:
X4-Inp-(D)Bal-(D)Trp-OH,
или его соли,
где X4 означает защитную группу аминогруппы и Y3 означает группу, активирующую карбоксигруппу.
4. Способ по п. 3, в котором первый-шестой жидкий растворитель каждый независимо является органическим растворителем.
5. Способ по п. 3, в котором первый и второй силилирующие реагенты оба представляют собой (триметилсилил)-N-диметилацетамид.
6. Способ по п. 1 или 3, в котором нуклеофильная добавка выбрана из группы, включающей 2-гидроксипиридин-N-оксид (HOPO), 1-гидрокси-7-азабензотриазол (HOAt), 1-гидроксибензотриазол (HOBt), 3,4-дигидро-3-гидрокси-4-оксо-1,2,3-бензотриазин (HODhbt) и этил-1-гидрокси-1H-1,2,3-триазол-4-карбоксилат (HOCt).
7. Способ по п. 6, в котором нуклеофильной добавкой является 2-гидроксипиридин-N-оксид (HOPO).
8. Способ по п. 1, в котором группы, активирующие карбоксигруппу Y1, Y2 и Y3, все независимо выбраны из группы, включающей N-гидроксисукцинимид (HOSu), N-гидроксифталимид, пентафторфенол (PfpOH) и ди-(п-хлортетрафторфенил)карбонат.
9. Пептидный фрагмент структурной формулы (II)
Boc-Inp-(D)Bal-(D)Trp-Phe-Apc(Boc)-NH2 (II)
или его соль, полученный способом по п. 3.
10. Пептидный фрагмент структурной формулы (III)
Boc-Inp-DBal-DTrp-OH (III)
или его соль, полученный способом по п. 3.
11. Пептидный фрагмент структурной формулы (IV)
H-Phe-Apc(Boc)-NH2 (IV)
или его соль, полученный способом по п. 3.
12. Пептидный фрагмент структурной формулы (V)
H-DBal-DTrp-OH (V)
или его соль, полученный способом по п. 3.
13. Пептидный фрагмент структурной формулы (VI)
Z-Phe-Apc(Boc)-NH2 (VI)
или его соль, полученный способом по п. 3.
14. Способ по п.1, дополнительно включающий взаимодействие аминокислоты X1-(D)Bal-Y1 с аминокислотой H-(D)Trp-OH, тем самым получая пептидный фрагмент следующей формулы X1-(D)Bal-(D)Trp-OH или его соль, где X1 представляет собой защитную группу аминогруппы и Y1 представляет собой активирующую карбоксигруппу.
15. Способ по п.14, дополнительно включающий удаление защитной группы пептидного фрагмента следующей формулы X1-(D)Bal-(D)Trp-OH, тем самым получая пептидный фрагмент следующей формулы H-(D)Bal-(D)Trp-OH или его соль.
16. Способ по п.15, дополнительно включающий взаимодействие аминокислоты X4-Inp-Y3 с пептидным фрагментом H-(D)Bal-(D)Trp-OH, тем самым получая пептидный фрагмент следующей формулы X4-Inp-(D) Bal-(D)Trp-OH или его соль, где X4 представляет собой защитную группу аминогруппы и Y3 представляет собой активирующую карбоксигруппу.
17. Способ по п.1, дополнительно включающий взаимодействие аминокислоты X4-Inp-Y3 с H-(D)Bal-OH с образованием пептидного фрагмента X4-Inp-(D)Bal-OH или его соли где X4 представляет собой защитную группу аминогруппы и Y3 представляет собой активирующую карбоксигруппу.
18. Способ по п.17, дополнительно включающий взаимодействие пептидного фрагмента X4-Inp-(D)Bal-OH с аминокислотой H-(D)Trp-OH с образованием пептидного фрагмента X4-Inp-(D)Bal-(D)Trp-OH или его соли.
| РИЛИЗИНГ-ПЕПТИДЫ РОСТОВОГО ГОРМОНА | 2003 |
|
RU2323941C2 |
| WO 2013093639 A1, 19.07.2017 | |||
| WO 2009065836 A1, 28.05.2009 | |||
| WO 2013113916 A2, 08.08.2013. | |||

