Данное изобретение относится к области медицинской химии и фармакологии и касается противоопухолевого средства для лечения рака предстательной железы в форме саше, включающего оксазолиновое производное прегн-17(20)-ена, встроенное в фосфолипидные наночастицы.
Рак предстательной железы (РПЖ) является наиболее распространенным онкологическим заболеванием мужчин. Заболеваемость РПЖ неуклонно возрастает во всем мире. Согласно статистике Министерства здравоохранения Российской Федерации каждый год количество заболевших возрастает на 8%-9% (что составляет 34 тысячи новых случаев). В настоящее время российские показатели прироста заболеваемости раком предстательной железы в 4 раза выше, чем в США и в 2,5 раза - чем у населения Европы [http://impotencija.net/rak-predstatelnoj-zhelezy]. Андрогены играют ключевую роль в развитии и прогрессии рака простаты, а снижение уровня андрогенов в опухоли лежит в основе современной стратегии терапии этого заболевания. Ключевым ферментом синтеза андрогенов является цитохром Р450 17А1 (17α-гидроксилаза-17,20-лиаза, CYP17A1). В настоящее время CYP17A1 рассматривается в качестве основной мишени для разработки лекарственных препаратов для лечения рака простаты. Известны стероидные и нестероидные соединения, обладающие способностью ингибировать активность CYP17A1, подавлять рост клеток карциномы простаты и проявлять противоопухолевую активность in vivo. В качестве стероидных соединений, являющихся ингибиторами CYP17A1, используются 3-пиридил-, 2-пиридил, 4-пиридил-, 1-пиразолил-, 1-имидазолил-, 1,2,3-триазол-1-ил-, 1,2,3-триазол-2-ил-, 1,2,4-триазол-1-ил-, 1Н-бензимидазол-1-ил-содержащие производные андрост-16-ена [1-6]. Известен ингибитор CYP17A1 - андроста-5,16-диен-17-(3-пиридил)-3β-ол (абиратерон), являющийся лекарственным препаратом против рака простаты. Ацетат абиратерона (торговое название Zytiga) в 2011 г. был рекомендован к широкому применению в качестве лекарственного препарата, а его аналог, галетерон (андроста-5,16-диен-гидрокси-17-(1H-бензимидазол-1-ил)-3β-ол, TOK-001, VN/124-1) в настоящее время проходит III фазу клинических испытаний. Ингибирование абиратероном и галетероном активности CYP17A1 вызывает подавление биосинтеза андрогенов, замедляет рост опухолевых клеток простаты и развитие опухоли у мышей, а также продлевает жизнь больных, страдающих от осложненных форм рака простаты на последних стадиях болезни [2, 3].
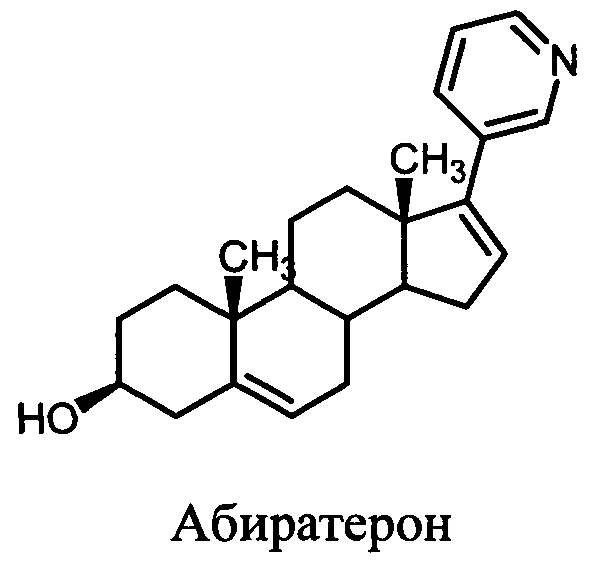
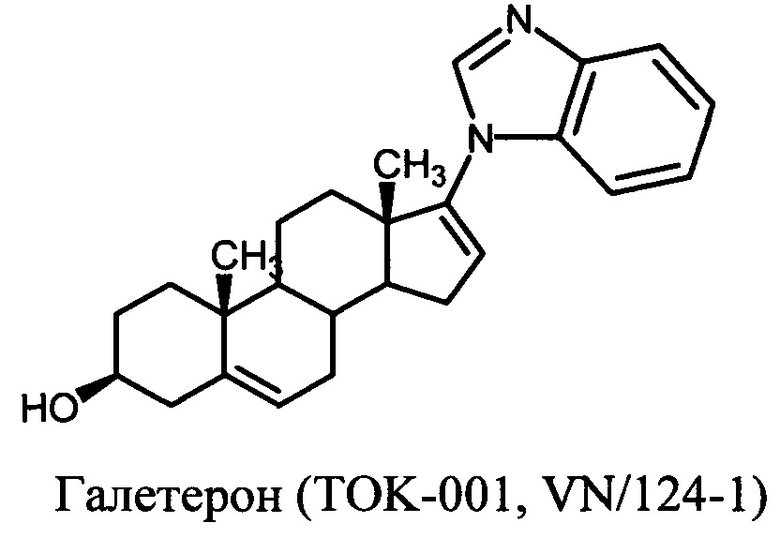
Синтез абиратерона и галетерона сложен и дорог, поскольку: 1) осуществляется в пять стадий; 2) включает использование в качестве исходного сырья гормональный препарат дегидроэпиандростерон; 3) включает использование катализатора на основе палладия. Описаны оксазолин-содержащие производные прегна-5,17(20)-диена [4, 5], однако сведений об их биологической активности ранее не приводилось.
Известна наносистема на основе растительных фосфолипидов для включения биологически активных соединений и способ ее получения (варианты) (патент РФ 2391966), включающая фосфатидилхолин растительного происхождения (78-95%), мальтозу и лекарственное средство в форме фосфолипидных наночастиц размером 10-30 нм при следующем соотношении компонентов: фосфатидилхолин - 20-43 мас. %, мальтоза - 57-80 мас. %, лекарственное средство - 2-8 мас. %.
Известна также композиция для встраивания лекарственных субстанций в липидную матрицу, композиция лекарственного средства с фосфолипидно-жирнокислотной системой и способы их получения (патент РФ 2463056), включающая фосфатидилхолин растительного происхождения (73-94%), соль жирной кислоты и мальтозу при следующем соотношений компонентов: фосфатидилхолин - 19-25 мас. %, соль жирной кислоты - 1,5-10 мас. %, мальтоза - 67-78 мас. %, лекарственная субстанция - 1-10 мас. %, в виде лиофильно высушенных наночастиц размером 10-20 нм. Недостатком композиции является введение дополнительного компонента - соли жирной кислоты.
Наиболее близким по технической сущности (прототипом) к предлагаемому противоопухолевому средству для лечения рака предстательной железы являются производные прегн-17(20)-ена, проявляющие противоопухолевую активность [1], представляющие собой производные прегн-17(20)-ена общей формулы (I), где R - 4',5'-дигидро-1',3'-оксазол-2'-ил- (Ia), либо бензо-[d]-оксазол-2'-ил- (Ib)
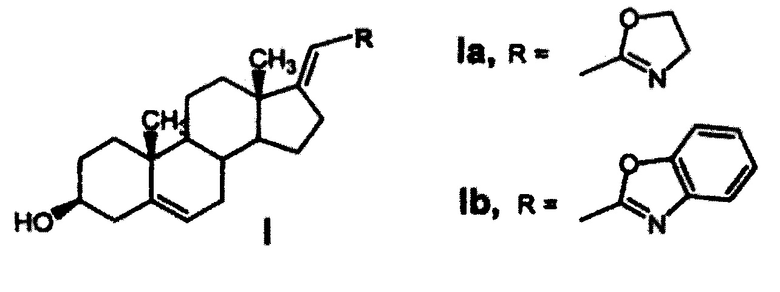
Заявляемое противоопухолевое средство отличается от объекта, раскрытого в прототипе тем, что, с целью повышения эффективности действия при уменьшении дозы и ослабления токсического воздействия на здоровые ткани, дополнительно модифицировано олеиновой кислотой с получением фармацевтической субстанции и содержит фосфолипиды и мальтозу при следующем соотношении компонентов: фармацевтическая субстанция : фосфолипиды : мальтоза 1:10:39, выполненное в форме саше.
Задачей настоящего изобретения является разработка противоопухолевого средства для лечения рака предстательной железы, включающего оксазолиновое производное прегн-17(20)-ена, встроенное в фосфолипидные наночастицы.
В Научно-исследовательском институте биомедицинской химии имени В.Н. Ореховича (ИБМХ) разработана оригинальная трехстадийная схема синтеза новых азотсодержащих производных 17(20)E]-прегна-5,17(20)-диена из доступного 3β-ацетокси-17α-бром-21-иодпрегн-5-ен-20-она, отличающаяся простотой и экономичностью по сравнению с известными схемами синтеза азотсодержащих производных андроста-5,16-диена. Структура синтезированных соединений удовлетворяет всем требованиям известного фармакофора для ингибиторов CYP17A1 (набору элементов в структуре соединения, необходимых для его связывания с молекулярной мишенью).
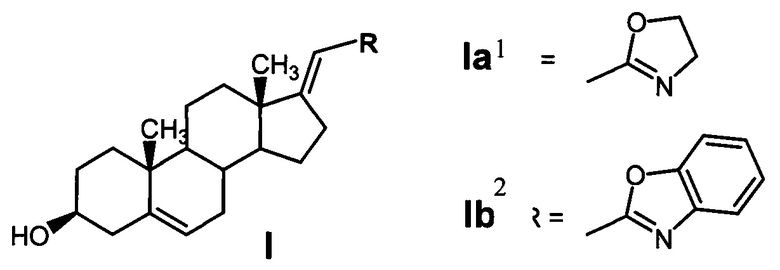
1 - 2'-{[(Е)-3β-гидроксиандрост-5-ен-17-илиден]-метил}-4',5'-дигидро-1',3'-оксазол; 2 - 2'-{[(Е)-3β-гидроксиандрост-5-ен-17-илиден]-метил}-бензо-[d]-оксазол
На основании детального сравнительного изучения физико-химических свойств соединений 1 и 2, разработки методов их синтеза, полученных результатов предварительного исследования специфической противоопухолевой активности для дальнейшей разработки лекарственного средства для лечения РПЖ было рекомендовано соединение 1 - 2'-{[(Е)-3β-гидроксиандрост-5-ен-17-илиден]-метил}-4',5'-дигидро-1',3'-оксазол, т.к. оно обладало большей ингибирующей и специфической активностью в отношении клеток карциномы простаты линии LNCaP и 17а-гидроксилазы-17,20-лиазы (CYP17A1) [1].
Исследования показали, что выбранное соединение 1 плохо растворялось в воде. С целью повышения растворимости соединение 1 модифицировали олеиновой кислотой с получением фармацевтической субстанции (ФС) -(3S,10R,13S,E)-17-((4,5-дигидрооксазол-2-ил)метилен)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[а]фе-нантрен-3-ил олеата с последующим ее включением в фосфолипидные наночастицы, как систему транспорта. Получение производного, модифицированного олеиновой кислотой, способствовало также облегчению встраивания ФС в фосфолипидные наночастицы.
Согласно изобретению, готовая лекарственная форма представляет собой порошок для приготовления раствора в пакетике саше следующего состава:
Активное вещество
Вспомогательные вещества
под условным названием Алсевирон, обладающая противоопухолевым действием.
Лекарственное средство Алсевирон не относится ни к дженерикам, ни к воспроизведенным лекарственным средствам. Активным компонентом этого лекарственного средства является новое соединение.
Основным компонентом используемого растительного соевого фосфолипида является фосфатидилхолин, содержание которого не менее 78-95% масс. В фосфолипиде содержатся также в незначительных количествах другие фосфолипидные компоненты (лизофосфатидилхолин до 4% масс., другие фосфолипиды - следовые количества).
В качестве вспомогательного вещества композиция содержит также мальтозу. Мальтоза является криопротектором и используется для получения лиофильно высушенного порошка, способного после растворения в физиологическом растворе или воде полностью восстанавливать свою структуру (в частности, размер частиц).
Изобретение характеризуется следующими примерами.
Материалы и методы
В работе использовались следующие материалы:
1. Соевый фосфолипид марки Липоид С 100
фирмы Липоид ГмбХ, Германия
2. Мальтозы моногидрат (ТУ 6-09-2108-77)
3. Вода для инъекций (по ФС №42-4587-95)
Пример 1. Способ получения ФС - (3S,10R,13S,E)-17-((4,5-дигидрооксазол-2-ил)метилен)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1H-циклопента[а]фенантрен-3-ил олеата.
Олеиновую кислоту (0,123 мл, 110 мг, 0,39 ммоль) растворяли в толуоле, добавляли дициклогексилкарбодиимид (144 мг, 0,7 ммоль), затем 2'-{[(Е)-3β-гидроксиандрост-5-ен-17-илиден]метил}-4',5'-дигидро-1',3'-оксазол* (132 мг, 0,37 ммоль) и диметиламинопиридин (226 мг, 1,85 ммоль). Раствор перемешивали при комнатной температуре 3,5 ч, затем реакционную смесь упаривали и остаток подвергали флэш-хроматографии на силикагеле в системе петролейный эфир -ацетон - уксусная кислота (75: 24: 1). Фракции, содержащие ФС, упаривали. Выход ФС 105 мг (0,17 ммоль, 46%). МСВР, рассчитанный для [C41H66NO3]+: 620,5043, найдено: 620,5032; 1Н ЯМР: 0,83 (3Н, с, Н-18); 0,87 (3Н, т, J=6,8 Гц, СН3, олеат); 1,03 (3Н, с, Н-19); 2,25 (2Н, т, J=7,4 Гц, αСН2, олеат); 2,74 (2Н, м, Н-16); 3,88 и 4,22 (каждый 2Н, м, СН2, оксазолин); 4,60 (1H, м, Н-3); 5,33 (2Н, м, СН, олеат); 5,37 (1Н, м, Н-6); 5,64 (1H, т, J=2,4 Гц, Н-20).
Пример 2. Способ получения готовой лекарственной формы (ГЛФ).
К вводному раствору мальтозы 975 мг (2,84 ммоль) прибавляли 250 мг (0,36 ммоль) соевого фосфолипида и 25,0 мг (0,04 ммоль) ФС. Смесь гомогенизировали методом роторно-статорной гомогенизации в течение 3-х мин. при температуре не выше 45 С до получения однородной первичной суспензии. Полученную суспензию переносили в приемную емкость гомогенизатора высокого давления. Гомогенизировали циклически при давлении 1000 бар ± 10%. Раствор между последующими циклами охлаждали пропусканием через холодильник с водяным охлаждением, не допуская его нагрева выше 55°С.
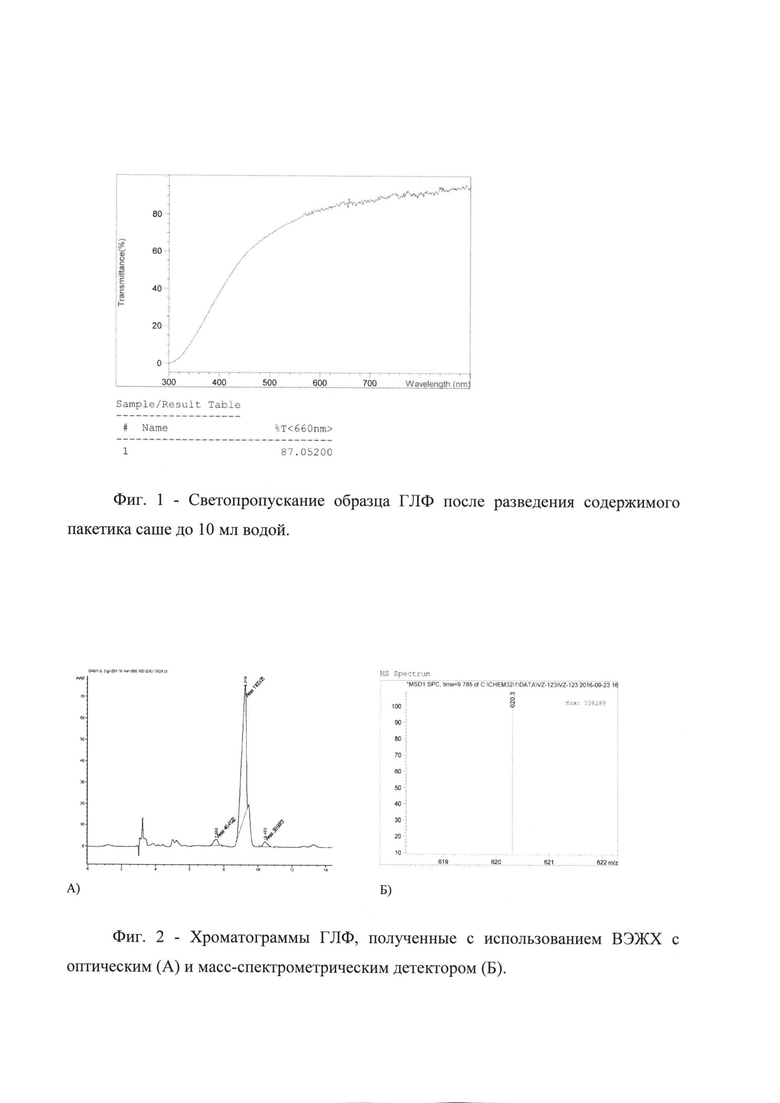
Процесс гомогенизации продолжали до достижения раствором прозрачности 50-60%. Контроль по светопропусканию проводили при 660 нм в кювете с длиной оптического пути 1 см.
Затем провели стандартизирующую фильтрацию на установке Millipore Corporation (США), последовательно пропуская раствор через фильтры из стекловолокна 1 мкм и мембранный с размером пор 0,22 мкм.
Полученную ультратонкую эмульсию разливали в поддоны из нержавеющей стали толщиной около 15 мм и лиофилизировали. Полученная лиофильно высушенная масса (порошок желтоватого цвета) представляла собой лекарственную композицию, из которой в дальнейшем после размалывания в шаровой мельнице была получена пероральная готовая форма лекарственного средства для лечения рака предстательной железы в виде порошка для приготовления раствора в пакетиках саше общим весом 2,5 г.
После регидратации содержимого пакетика (препарат Алсевирон) - определяли его физико-химические характеристики.
Основные характеристики ГЛФ Алсевирон представлены в таблице 1.

Проведена серия экспериментов при различном соотношении компонентов: ФС, фосфолипида и мальтозы. Полученные данные представлены в таблице 2.
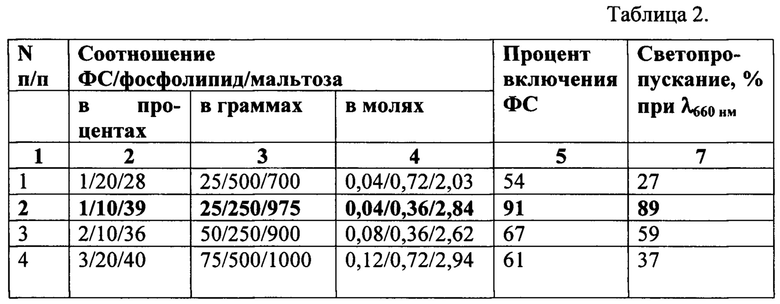
Установлено, что наиболее оптимальным для получения фосфолипидных частиц малого размера с включенной субстанцией является соотношение компонентов ФС : фосфолипид : мальтоза 1:10:39. Поэтому для дальнейших исследований и экспериментов на животных нами была выбрана композиция под номером 2 в таблице 2.
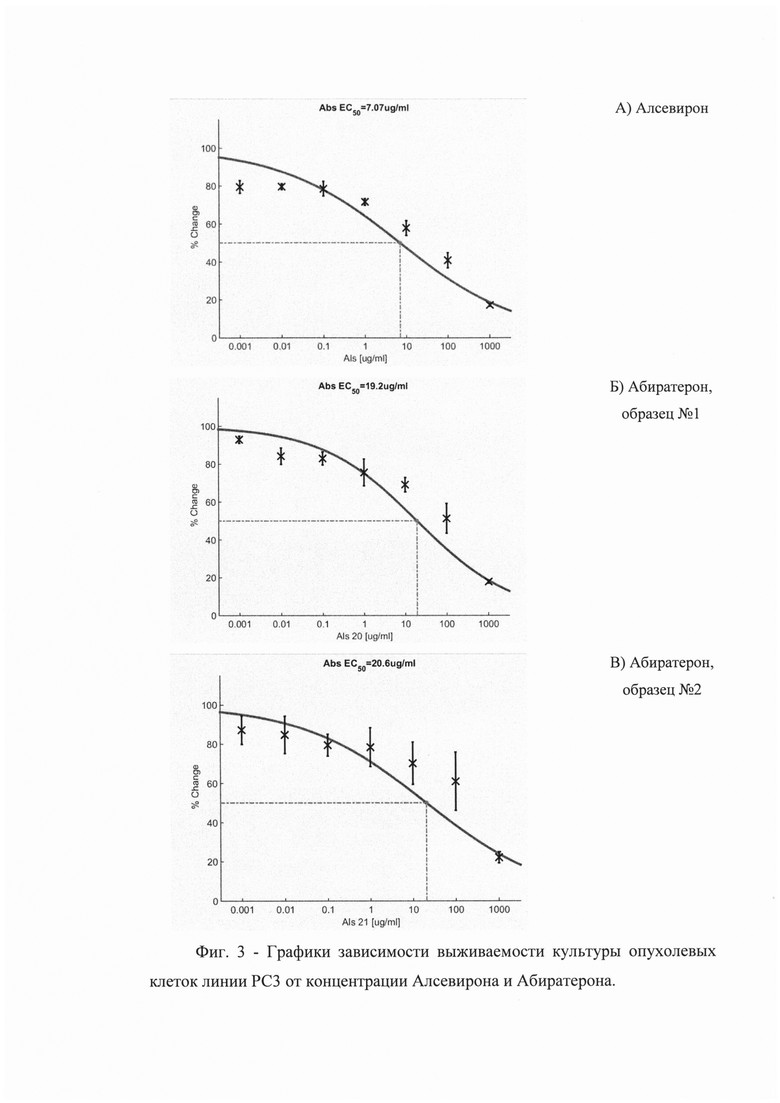
Наряду со стандартными методами контроля качества, регламентированными ГФ, для характеристики фармацевтической субстанции (ФС) были выбраны следующие методы характеристики образцов ГЛФ: метод светопропускания, метод ВЭЖХ с оптической и масс-спектрометрической детекцией на хроматографах Agilent 1100 и Agilent 1200 Series («AgilentTechnologies», США), соответственно.
Для измерения светопропускания содержимое пакетика саше разводили до 10 мл дистиллированной водой, полученный образец наливали в кварцевую кювету и определяли светопропускание при длине волны 660 нм. На Фиг. 1 приведен типичный спектр светопропускания одного из образцов ГЛФ. На Фиг. 2 приведены хроматограммы, полученные на хроматографе с оптической и масс-спетрометрической детекцией.
Пример 3. Данные специфической активности Алсевирона.
Доклинические исследования специфической активности препарата Алсевирон выполнено в параллельном сравнении с субстанцией абиратерона ацетат (Абиратерон), представляюшей собой активное вещество противоопухолевого препарата Zitiga, на сигнальных клеточных линиях рака предстательной железы (in vitro) в диапазоне концентраций 0,001-1000 μg/ml на линии клеток рака предстательной железы человека РС-3. О цитотоксичности судили по общепринятому показателю IC50 (halfmaximal inhibitory concentration) в стандартном МТТ-тесте.
Оценка цитотоксической активности. Исследуемые соединения (образцы Алсевирона и Абиратерона) растворяли в диметилсульфоксиде (ДМСО). Концентрация соединений в стоковых растворах - 1 мг/мл. Для получения рабочих растворов делали ряд последовательных десятикратных разведений в стерильном 0,9% р-ре NaCl в триплетах.
В качестве тестовой клеточной культуры использовали перевиваемую линию клеток аденокарциномы простаты человека РС-3 (из банка ФГБУ «НМИЦ онкологии имени Н.Н. Блохина» Минздрава России) в фазе логарифмического роста. Клетки в концентрации 4,2×105 кл/мл, ресуспендированные в среде RPMI-1640 с добавлением 1% сыворотки крови эмбрионов коровы, 2 мМ L-глутамина, 100 Е/мл пенициллина и 100 мкл/мл стрептомицина (все - ПанЭко, РФ), рассевали по лункам 96-луночного планшета (Nung) по 180 мкл, куда добавляли по 20 мкл рабочих растворов исследуемых субстанций в тетраплетах. В контрольные лунки вносили по 20 мкл 0,9% р-ре NaCl. Клеточную культуру с тестируемыми субстанциями инкубировали при 37°С и 5% CO2 в течение 4 суток.
Воздействие исследуемых субстанций на выживаемость опухолевых клеток определяли с использованием МТТ-теста. Оптическую плотность окрашенного раствора оценивали при длине волны 540 нм против 690 нм на планшетном фотометре Multiscan MS (ThermoLabsystems, Финляндия). Конечный результат теста рассчитывали по формулам:
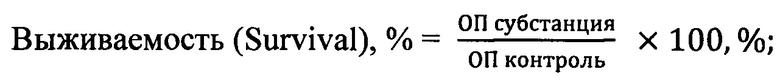
Цитотоксичность (Cytotoxicity), %=100-Survival
По результатам расчета выживаемости культуры клеток в тетраплетах в программе Combenefit software был рассчитан показатель ЕС50 - эффективная концентрация исследуемого соединения, вызвавшая гибель 50% клеток.
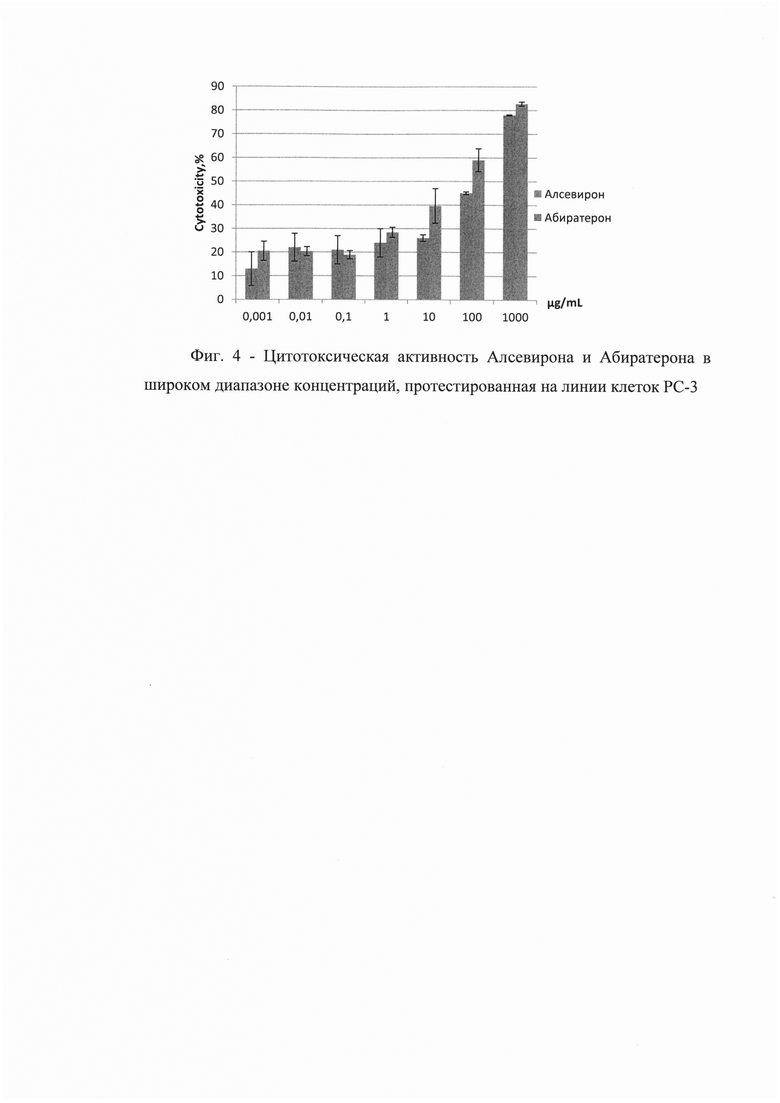
Сравнительная оценка цитотоксической активности Алсевирона и Абиратерона. На Фиг. 3 приведены результаты расчета ЕС50. Полученные результаты свидетельствуют о том, что Алсевирон обладает наибольшей активностью в сравнении с обоими образцами Абиратерона, эффективно (более чем на 50%), подавляя жизнеспособность культуры опухолевых клеток линии РС3 в концентрации более 7,1 μg/ml в сравнении 19,2 и 20,6 μg/ml Абиратерона, соответственно.
На Фиг. 4 приведены результаты расчета цитотоксичности растворов с различной концентрацией действующих веществ. Приведенные дискретные значения показывают, что из тестированных соединений наиболее широким эффективным диапазоном концентраций опосредующих значимый цитотоксический эффект, приводящий к гибели более 50% клеток в культуре, обладал Алсевирон в концентрациях 100 и 1000 мкг/мл.
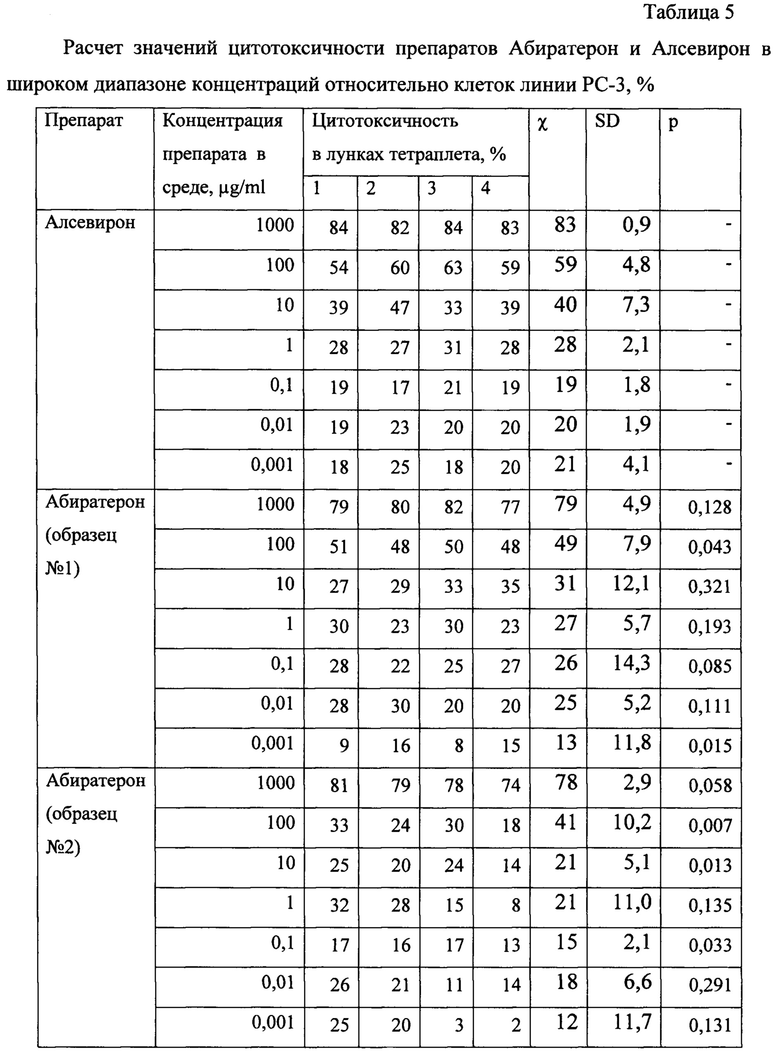
По результатам проведенных исследований было доказано, что Алсевирон обладает более высокой цитотоксической активностью в сравнении с Абиратероном. Гибель более чем 50% опухолевых клеток наблюдали в результате их коинкубации с Алсевироном в концентрации 50 μg/ml и выше, тогда как Абиратерон даже в концентрации 100 μg/ml индуцировал гибель только 41±3,4% клеток. Следует указать, что цитотоксическая активность Алсевирона в концентрации 25 μg/ml также была довольна высока - 47±2,2%, тогда как активность Абиратерона в этой концентрации соответствовала 34±2,2%. Расчет по кривым аппроксимации показателя средней эффективной концентрации препарата, вызывающей гибель 50% клеток в процессе коинкубации, показал, что ЕС50 Алсевирона в среднем соответствует 7,7 μg/ml, а Абиратерона 19-20 μg/ml, т.е. Алсевирон вызывает цитотоксический эффект клеток в концентрации в 2,5 раза меньшей, чем Абиратерон.
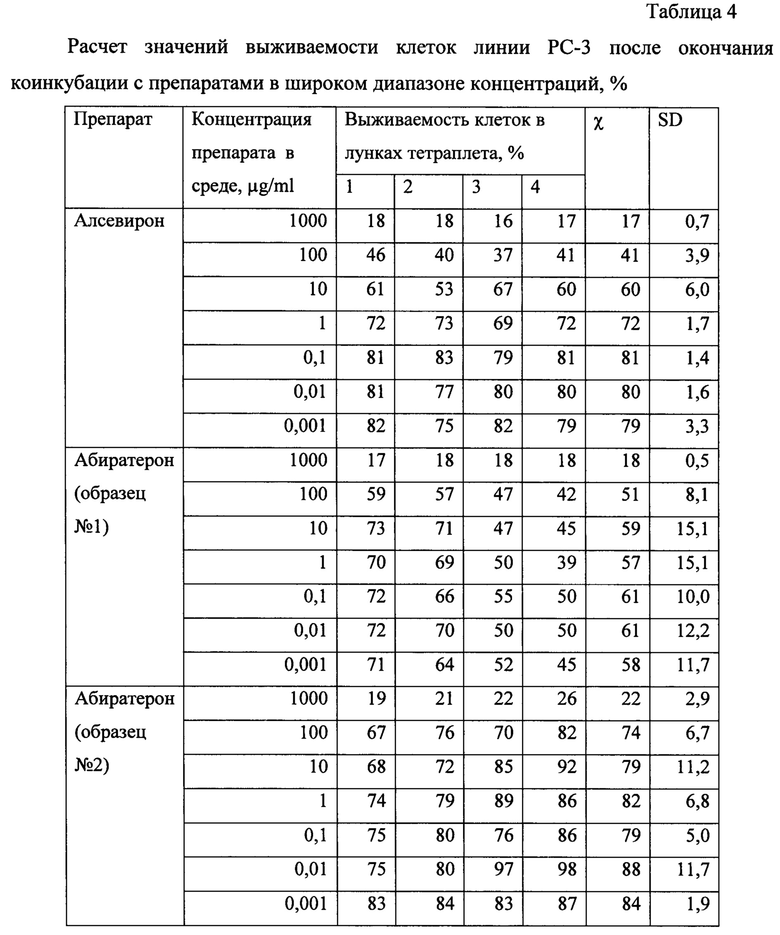
В таблицах 3-5 представлены данные для оценки цитотоксичности Алсевирона и Абиратерона в широком диапазоне концентраций.
Пример 4. Изучение острой токсичности лекарственного средства
Для экспериментов животных распределяли по группам случайным образом, используя в качестве критерия массу тела, так, чтобы в группе индивидуальная масса животных не отличалась более чем на 10% от средней массы животных одного пола. Исследуемый препарат Алсевирон вводили мышам обоего пола (n=72) и крысам обоего пола (n=72) внутрижелудочно. Животным за один прием вводили максимально возможный объем при внутрижелудочном введении: мышам 0,5 мл/20 г, крысам 4,0 мл/200 г. Для достижения нужных для введения объемов, минимальную дозу разводили водой для инъекций до соответствующего массе тела животного объема. Дозы для мышей 5,0 - 10,0 - 15,0 - 20,0 - 25,0 мл/кг по лекарственной форме; дозы для крыс: 5,0 - 10,0 - 15,0 - 20,0 - 25,0 мл/кг по лекарственной форме. Определение параметров токсичности при однократном введении проводили с использованием двухэтапного метода. На первом этапе устанавливали ориентировочная ЛД50 методом Кербера и Миллера, либо Дейхмана и Лебланка. На втором этапе определяли точные показатели ЛД16, ЛД50±m, ЛД84 методом пробит-анализа по Литчфилду и Уилкоксону. Проявления интоксикации: ни в одной из групп не отмечалось случаев летальности. После введения внутрижелудочно максимальных доз препарата Алсевирон не отмечалось видимых различий в двигательном и пищевом поведении животных, в состоянии внешних покровов и видимых слизистых, в реакциях на внешние раздражители в сравнении с контрольными группами. Общая продолжительность наблюдения за животными, после введения препаратов, составила 14 суток. В первый день наблюдение было непрерывным, т.к. этот день является наивысшей точкой интоксикации. Ежедневная регистрация клинической картины интоксикации проводилась визуально в клетке для содержания и в руках. В результате проведенных исследований на белых аутбредных мышах и крысах установлено, что при однократном внутрижелудочном введении максимальных доз препарата Алсевирон, гибели животных не наблюдалось. LD50 для мышей и крыс не была достигнута при введении максимально возможной дозы 25,0 мл/кг по лекарственной форме или 62,50 мг/кг по действующему веществу ФС (оксазолиновое производное 17(20)Е-прегна-5,17(20)диена в виде конъюгата с олеиновой кислотой), что в 88 раз больше, чем терапевтическая доза для человека 0,71 мг/кг. Не было выявлено влияния на массовые коэффициенты органов при однократном внутрижелудочном введении белым аутбредным мышам. Не выявлено разницы по приросту массы тела между группами с введением исследуемого препарата и контрольными группами при однократном внутрижелудочном введении белым аутбредным крысам в острых дозах. В экспериментах не установлено половых и межвидовых различий.
Пример 5. Изучение хронической токсичности лекарственного средства
Для проведения эксперимента по изучению хронической токсичности были использованы крысы Wistar (160 голов) и кролики породы Шиншилла (96 голов).
Исследования проводились при внутрижелудочном введении препарата в течение 180 дней ежедневно в трех дозах.
Разовая доза для человека: 50 мг/ 70 кг=0,71 мг/кг (1ТДвещ).
Поскольку препарат не обладает высокой токсичностью, (ЛД50 достигнута не была), для расчета доз крысам и кроликам были взяты ЭТД.
1 ЭТД для крыс массой 200 г=0,71×39/6,5=4,26 мг/кг (39 - коэффициент пересчета доз для человека массой 70 кг, 6,5 - коэффициент пересчета доз для крысы массой 200-250 г);
5 ЭТД (промежуточная)=4,26×5=21,3 мг/кг;
10 ЭТД (максимальная)=4,26×10=42,6 мг/кг;
1 ЭТД для кроликов массой 2 кг=0,71×39/12,8=2,16 мг/кг (39 - коэффициент пересчета доз для человека массой 70 кг, 12,8 - коэффициент пересчета доз для кроликов массой 2000 г);
5 ЭТД (промежуточная)=2,16×5=10,8 мг/кг;
10 ЭТД (максимальная)=2,16×10=21,6 мг/кг.
Были использованы следующие методы анализа:
- Общее состояние (динамика массы тела, ректальная температура). Внешний вид оценивается при ежедневном осмотре животных. Взвешивание, измерение ректальной температуры, потребления воды и корма выполняется раз в неделю;
- Поведение крыс (двигательная и исследовательская активность);
- Гематологические показатели (количество эритроцитов, лейкоцитов, тромбоцитов, уровень гемоглобина, лейкоцитарная формула);
- Миелограмма;
- Свертываемость крови;
- Биохимические показатели и активность ферментов сыворотки крови (общий белок, креатинин, мочевина, глюкоза, холестерин, общие липиды, билирубин общий и связанный, активность щелочной фосфатазы, лактатдегидрогеназы, аспартат- и аланинамино-трансфераз, кальций, калий, натрий).
- Функциональное состояние сердечно-сосудистой системы крыс (ЭКГ-исследование во II стандартном отведении, систолическое давление);
- Функциональное состояние ЦНС крыс (поведение, суммационно-подпороговый показатель);
- Функциональная активность почек крыс (по анализу мочи);
- Патоморфологическое исследование, включающее: некропсию, макроскопическое исследование, взвешивание и гистологическое исследование внутренних органов.
Местно-раздражающее действие при патоморфологическом (макроскопически и гистологически) описании места введения препарата.
Результаты. В течение 180 дней не отмечалось гибели животных при внутрижелудочном введении Алсевирон в эквитерапевтической и превышающих эквитерапевтическую в 5 и 10 раз дозах. Не выявлено местно-раздражающего действия исследуемого препарата Алсевирон при его многократном внутрижелудочном введении, не отмечено нарушений функционального состояния основных органов и систем организма.
Результаты изучения субхронической токсичности на крысах и кроликах породы Шиншилла показали, что многократное внутрижелудочное введение исследуемого препарата Алсевирон в субтоксических дозах не вызывало значительных нарушений функционального состояния основных органов и систем организма.
Пример 6. Изучение специфической токсичности лекарственного средства
Изучение специфической токсичности препарата Алсевирон в эквитерапевтических дозах и дозах, превышающих эквитерапевтическую в 10 раз, показало:
- отсутствие аллергических свойств (исследование на наличие анафилактической активности при накожном тестировании и в конъюнктивальной пробе);
- отсутствие иммунотоксической активности (препарат не вызывает активацию гуморального и клеточного звена иммунитета как у самцов, так и у самок);
- отсутствие мутагенной активности при тестировании мутаций замены пар оснований в молекуле ДНК в тесте Эймса на штаммах S. typhimurium ТА98, ТА 100, ТА1535, ТА1537 и штаммах E.coli pKM101 и uvrA, в вариантах с метаболической активацией S9 и без таковой, не имеет цитогенетической активности в тесте учета хромосомных аберраций в клетках костного мозга млекопитающих, не оказывал генотоксического действия на лимфоциты и клетки печени мышей BALB/c через 6 и 24 часа после внутрижелудочного введения.
Разработанный в ИБМХ инновационный противоопухолевый препарат для лечения рака предстательной железы на основе фосфолипидной композиции оксазолинового производного прегн-17(20)-ена получен по оригинальной технологии. Препарат планируется получать в виде лиофильно-высушенного порошка с длительным сроком хранения. При этом технология не является сложной и хорошо воспроизводима. Предварительные доклинические исследования показали, что разработанный препарат обладает неоспоримыми конкурентными преимуществами по сравнению с зарубежными аналогами как с экономической, так и с медицинской стороны.
ЛИТЕРАТУРА
1. Патент РФ 2617698
2. US 5604213 А.
3. Stulov SV, Tkachev YV, Novikov RA, Zavialova MG, Timofeev VP, Misharin AY. Synthesis of 21-nitrogen substituted pregna-5,17(20)-dienes from pregnenolone, Steroids 2012; 77: 77-84.
4. Stulov SV, Mankevich OV, Novikov RA, Tkachev YV, Timofeev VP, Dugin NO, Pozdnev VF, Fedyushkina IV, Scherbinin DS, Veselovsky AV, Misharin AY. Synthesis and molecular modeling of (4''R)- and (4''S)- 4''-substituted 2''-{[(E)-androst-5-en-17-ylidene]-methyl}-oxazolines. Steroids 2013; 78: 521-527.
5. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Meth. 1983, 65, 55-63.
Таблицы

| название | год | авторы | номер документа |
|---|---|---|---|
| Производные прегн-17(20)-ена, проявляющие противоопухолевую активность | 2015 |
|
RU2617698C2 |
| Фосфолипидная композиция доксорубицина для лечения больных раком молочной железы | 2019 |
|
RU2714137C1 |
| ФОСФОЛИПИДНАЯ КОМПОЗИЦИЯ ЭКДИСТЕНА, ОБЛАДАЮЩАЯ АДАПТОГЕННОЙ И ГЕПАТОПРОТЕКТОРНОЙ АКТИВНОСТЬЮ | 2013 |
|
RU2575561C2 |
| СПОСОБ СОЧЕТАННОЙ ТЕРАПИИ СОЕДИНИТЕЛЬНОТКАННОЙ САРКОМЫ М-1 КРЫС С ИСПОЛЬЗОВАНИЕМ КОНЬЮГАТА ДИПРОПОКСИБАКТЕРИОПУРПУРИНА С ДОКСОРУБИЦИНОМ | 2023 |
|
RU2808909C1 |
| ПРОТИВОТУБЕРКУЛЕЗНАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2011 |
|
RU2472512C1 |
| СПОСОБ ПОЛУЧЕНИЯ КОМПОЗИЦИИ ДЛЯ ФОТОДИНАМИЧЕСКОЙ ТЕРАПИИ В ФОРМЕ ФОСФОЛИПИДНЫХ НАНОЧАСТИЦ НА ОСНОВЕ ГЛЮКАМИНОВОЙ СОЛИ ХЛОРИНА Е6, МАЛЬТОЗЫ И ФОСФАТИДИЛХОЛИНА | 2014 |
|
RU2576025C1 |
| Фармацевтическая композиция на основе глюкокортикостероида будесонида и фосфатидилхолина для сухой ингаляции | 2019 |
|
RU2730488C1 |
| Липидное производное сарколизина в составе фосфолипидных наночастиц | 2017 |
|
RU2662086C1 |
| Производные 2-аминохроменов проявляющие противоопухолевую активность. Фармацевтические композиции | 2018 |
|
RU2704262C1 |
| ПРОТИВООПУХОЛЕВОЕ СРЕДСТВО ДЛЯ ПЕРОРАЛЬНОГО ПРИМЕНЕНИЯ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2004 |
|
RU2292209C2 |
Изобретение относится к области медицинской химии и фармакологии и касается противоопухолевого средства для лечения рака предстательной железы в форме саше на основе 2'-{[(Е)-3β-гидроксиандрост-5-ен-17-илиден]метил}-4',5'-дигидро-1',3'-оксазола, модифицированного олеиновой кислотой с получением фармацевтической субстанции - (3S,10R,13S,E)-17-((4,5-дигидрооксазол-2-ил)метилен)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1Н-циклопента[а]фенантрен-3-ил олеата, при этом противоопухолевое средство дополнительно содержит фосфолипид и мальтозу при следующем соотношении компонентов: фармацевтическая субстанция : фосфолипид : мальтоза 1:10:39. Использование изобретения позволяет повысить эффективность лечения при уменьшении дозы и ослаблении токсического воздействия на здоровые ткани. 4 ил., 5 табл., 6 пр.
Противоопухолевое средство для лечения рака предстательной железы в форме саше, содержащее 2'-{[(Е)-3β-гидроксиандрост-5-ен-17-илиден]метил}-4',5'-дигидро-1',3'-оксазол, отличающееся тем, что 2'-{[(Е)-3β-гидроксиандрост-5-ен-17-илиден]метил}-4',5'-дигидро-1',3'-оксазол модифицирован олеиновой кислотой с получением фармацевтической субстанции - (3S,10R,13S,E)-17-((4,5-дигидрооксазол-2-ил)метилен)-10,13-диметил-2,3,4,7,8,9,10,11,12,13,14,15,16,17-тетрадекагидро-1Н-циклопента[а]фенантрен-3-ил олеата, при этом противоопухолевое средство дополнительно содержит фосфолипид и мальтозу при следующем соотношении компонентов: фармацевтическая субстанция : фосфолипид : мальтоза 1:10:39.
| Производные прегн-17(20)-ена, проявляющие противоопухолевую активность | 2015 |
|
RU2617698C2 |
| ИЗОИНДОЛИНОВЫЕ СОЕДИНЕНИЯ ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ РАКА | 2009 |
|
RU2527952C2 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| BAMBURY R.M | |||
| et al | |||
| Novel and next-generation androgen receptor-directed therapies for prostate cancer: Beyond abiraterone and enzalutamide//Urol Oncol | |||
| Токарный резец | 1924 |
|
SU2016A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |

