Изобретение относится к технологии получения стабильных изотопов бора (10В и 11В).
Известны способы разделения изотопов бора методом ректификации соединений бора общего состава ВХ3, где X - атом галогена: Cl (Севрюгова Н.Н., Уваров О.В., Жаворонков Н.М. Разделение стабильных изотопов бора // Атомная энергия, 1960, т. 9, №2, с. 110-125; Андриец С.П., Гущин А.А., Калашников А.Л., Козырев А.С. и др. Создание и испытание пилотной установки для разделения изотопов бора ректификацией BCl3 // Перспективные материалы. - Спец. выпуск (8). - февр. 2010, с. 193-198) или F (Nettley Р.Т., Cartwright D.K., Kronberger Н. The production of 10Boron by low-temperature distillation of boron trifluoride // In: Proc. of the Intern. Symp. on isotope separation. Amsterdam: North Holl. Pub. Co., 1958, p. 385-407; Амирханова И.Б., Асатиани П.Я., Борисов A.B., Гвердцители И.Г. и др. Влияние давления на разделение изотопов бора // Атомная энергия, 1967, т. 23, №4, с. 336-339).
Известен способ разделения изотопов бора методом химобменной ректификации комплексного соединения галогенида бора (трифторид бора) с диметиловым эфиром (CH3)2O⋅BF3 (Каминский В.А., Карамян А.Т., Гиоргадзе И.А. и др. Концентрирование изотопа В10 методом химобменной дистилляции (CH3)2O⋅BF3 при атмосферном давлении // В кн.: Производство изотопов. Сб. статей, М.: Атомиздат, 1973, с. 466-468).
Известны также способы разделения изотопов бора методом химического изотопного обмена между газообразным трифторидом бора и его комплексными соединениями с метилфениловым (анизол) или этилфениловым (фенетол) эфирами, то есть, в двухфазных системах «BF3, газ - BF3⋅CH3OC6H5, жидк.» или «BF3, газ - BF3⋅C2H5OC6H5, жидк.» соответственно (Волощук A.M., Джиджийшвили Ш.И., Катальников С.Г. и др. Разделение изотопов бора на опытной установке с использованием комплексных соединений BF3 с анизолом и фенетолом // В кн: Производство изотопов. Сб. статей, М.: Атомиздат, 1973, с. 443-450; Katalnikov S.G. Physico-chemical and Engineering Principles of Boron Isotopes Separation by Using BF3-Anisole⋅BF3 System // Separation Science and Technology, 2001, Vol. 36, №8&9, p. 1737-1768) или нитрометаном в системе «BF3, газ - BF3⋅CH3NO2, жидк.» (Patent US №5419887 А, опубл. 30.05.1995; Khoroshilov A.V., Lizunov A.V., Stepanov A.V., Cherednichenko S.A. Thermal dissociation of the complex BF3⋅D and boron isotope separation in the system BF3-BF3⋅CH3NO2 // Radiochemistry, 2009, Vol. 51, №4, p. 400-402; Хорошилов A.B., Степанов A.B., Лизунов A.B., Зернова Е.В. Первое разделение изотопов бора методом химического обмена при пониженной температуре в системе трифторид бора - его комплексное соединение с нитрометаном // Перспективные материалы. - Спец. выпуск (8). - февр. 2010, с. 258-262).
Недостатком вышеуказанных способов, основанных на методе ректификации, химобменной ректификации и химического изотопного обмена в системе газ-жидкость, является необходимость использования токсичных галогенидов бора, как в виде жидкости (BCl3,жидк., BF3,жидк., BF3⋅СН3ОС6Н5,жидк., BF3⋅C2H5OC6H5,жидк., BF3⋅CH3NO2,жидк.), так и в виде газа (BCl3,газ, BF3,газ), когда предельно допустимая концентрация в воздухе рабочей зоны (ПДКр.з.), например, для BCl3 и BF3 равна 1 мг/м3, класс опасности 2, а ориентировочные безопасные уровни воздействия загрязняющего атмосферу вещества (ОБУВа.в.) составляют 0,03 мг/м3 и 0,005 мг/м3 соответственно (Бора трихлорид [Электронный ресурс]. - Режим доступа: http:/toxi.dyndns.org/base/nonorganic/Borum4/Trihloridboran.htm (дата обращения: 01.12.2017); Бора трифторид [Электронный ресурс]. - Режим доступа: http://toxi.dyndns.org/base/nonorganic/Borum4/Bora_triftorid.htm (дата обращения: 01.12.2017) и Вредные вещества в промышленности. Справочник для инженеров, химиков и врачей. В 3-х т. / Под ред. Н.В. Лазарева и Э.Н. Левиной / - Л., Химия, 1977. Т. 3. Неорганические и элементоорганические соединения, с. 312-313).
Для уменьшения токсичности рабочих систем, применяемых в способах разделения изотопов бора и основанных на использовании его галогенидов общего состава ВХ3, где X - атом галогена (Cl, F), предложен химический обмен в экстракционной жидкость-жидкостной системе «водный раствор борной кислоты - Н3ВО3 в изоамиловом спирте» или в системе «Н3ВО3/H2O//Н3ВО3/(СН3)2СНСН2СН2ОН» с однократным коэффициентом разделения изотопов бора α=1,0027 и, соответственно, коэффициентом обогащения ε=α-1=2,7⋅10-3 (Кузнецова Е.М., Панченков Г.М., Филиппова Р.С., Малахов В.Ф. О новом способе разделения изотопов бора // ЖФХ, 1960, т. 34, №10, с. 2370-2371).
Основным недостатком этого способа является относительно малое значение однократного коэффициента разделения (обогащения).
Наиболее близким по технической сущности к заявляемому изобретению (прототип) можно считать способ разделения изотопов бора химическим обменом в экстракционной двухфазной жидкость-жидкостной системе «раствор борной кислоты H3BO3 в воде - комплексное соединение бора с азотсодержащим фенолформальдегидным резольным олигомером (марка «Яррезин Б»), полученным поликонденсацией п-трет-бутилфенола с формальдегидом в присутствии аммиака, имеющим среднюю молекулярную массу 450-500 атомных единиц и растворенным в смеси органических растворителей - октанола и тридекана» или, что более приемлемо, «водный раствор H3BO3 - Н3ВО3 с Яррезином Б в растворе октанола и тридекана» или еще более кратко «H3BO3/H2O//H3BO3⋅Яррезин Б/С8Н17ОН+C13H28». При этом обращение потоков фаз по изотопу 10В, как указано в прототипе, может быть осуществлено реэкстракцией борной кислоты водой с последующим упариванием полученного раствора (Патент РФ №2311949 от 10.12.2007, Бюл. №34) - прототип.
Значения однократного коэффициента разделения изотопов бора α в прототипе (табл. 2) обозначены как Ri (изменение отношения концентрации изотопов 11В к 10В, то есть, [11В]/[10В]) и найдены как

где R0 и R - [11B]/[10B] в водной фазе до и после контактов соответственно; n - общее число контактов.
Численные значения α при соотношении объемов фаз O:В=1:1 при n=3 (пример 1) и n=4 (пример 2), как указано в табл. 2, составили 1,00274 и 1,00316 соответственно, а при O:В=2:1 и n=2 (пример 3) - 1,00856. По этим данным среднее значение однократного изотопного эффекта для серии из трех опытов равно α=1,005±0,003, a ε=(5±3)⋅10-3 и это значение почти в два раза выше ε в системе с изоамиловым спиртом: (5/2,7)⋅10-3≈1,9.
Необходимо отметить, что реэкстракция борной кислоты из органической фазы в воду производится при уравновешивании органической фазы с существенно большим объемом воды с последующим упариванием водного раствора борной кислоты до значения ее равновесной концентрации в водной фазе.
Недостатками прототипа, как способа разделения изотопов бора, являются:
- многокомпонентность органической фазы (собственно Яррезин Б, октанол и тридекан), требующая, как минимум, четырех операций для ее приготовления: измерение массы твердого Яррезина Б; измерение объема каждого из растворителей (октанол, тридекан); образование общего раствора;
- невысокие значения однократного коэффициента разделения (обогащения) изотопов бора и, соответственно, большое число теоретических ступеней разделения, необходимое для достижения заданной концентрации изотопа 10В (или 11В);
- малая производительность процесса экстракции борной кислоты;
- большой расход воды и, соответственно, высокие энергетические затраты при обращении потоков фаз по изотопу 10В за счет реэкстракции борной кислоты водой.
Задача настоящего изобретения - уменьшение числа компонентов органической фазы и, соответственно, снижение затрат на ее приготовление, увеличение однократного коэффициента разделения (обогащения) изотопов бора и, как следствие, снижение числа теоретических ступеней разделения, необходимых для достижения требуемой концентрации изотопа 10В (или 11В), увеличение производительности процесса экстракции борной кислоты, уменьшение расхода воды и энергетических затрат на обращение потоков фаз по изотопу 10В.
Поставленная задача решается способом разделения изотопов бора химическим обменом в двухфазной жидкость-жидкостной системе с использованием водного раствора борной кислоты в качестве водной фазы и органической фазы, состоящей из комплексного соединения борной кислоты с органическим веществом, и с обращением потоков реэкстракцией борной кислоты водой с последующим упариванием водного раствора при этом в качестве органического вещества используют трибутиловый эфир фосфорной кислоты, а реэкстракцию проводят в условиях перекрестного тока органической фазы и воды.
Пример 1.
Двухфазную жидкость-жидкостную систему готовили при комнатной температуре (295±2) К растворением борной кислоты в воде с приготовлением 0,80 М раствора и контактированием полученного раствора с трибутиловым эфиром фосфорной кислоты (трибутилфосфатом) в качестве органической фазы при соотношении объемов фаз 1:1. Сравнительные данные по составу фаз для прототипа и предлагаемого изобретения, когда рабочая система «водный раствор Н3ВО3 - комплексное соединение Н3ВО3 с ТБФ) имеет вид «Н3ВО3/Н2О//Н3ВО3⋅ТБФ/ТБФ» приведены в табл. 1.

Из табл. 1 следует, что решение задачи приготовления двухфазной системы существенно отличается от прототипа числом компонентов, используемых для ее образования: вместо трех компонентов по прототипу предлагается использование единственного вещества, что приводит к значительному сокращению числа технологических операций на стадии ее приготовления: вместо четырех операций по прототипу требуется лишь одна - измерение объема ТБФ, причем это сопоставление дано без учета контроля качества каждого из компонентов.
Пример 2.
После уравновешивания водного раствора борной кислоты с исходной концентрацией С0=0,80 М с трибутилфосфатом (ТБФ) при температуре (295±2) К в течение 24 ч отобраны пробы водной фазы и методом масс-спектрометрии индуктивно связанной аргоновой плазмы выполнен их изотопный анализ, а также изотопный анализ исходного раствора Н3ВО3, с определением значений изотопного отношения по уравнению:

Полученные значения R использованы для определения однократного коэффициента разделения изотопов бора α в системе «Н3ВО3/Н2О//Н3ВО3/ТБФ»:

где R0 и R* - значения изотопного отношения для бора борной кислоты в водной фазе до (R0) и после (R*) уравновешивания с ТБФ.
Результаты троекратных измерений в виде средних значений приведены в табл. 2, откуда следует, что однократный коэффициент разделения изотопов бора в экстракционной системе «Н3ВО3/H2O//Н3ВО3⋅ТБФ/ТБФ» при комнатной температуре составляет α=1,0067±0,0034 или α=1,007±0,003. Сравнение измеренного значения коэффициента разделения со средним α по прототипу (α=1,005±0,003) показывает, что однократный изотопный эффект, выраженный коэффициентом обогащения ε, в заявляемом способе в 1,4 раза выше по сравнению с прототипом (7⋅10-3/5⋅10-3=1,4).

Это означает, что, например, минимальное число теоретических ступеней разделения для изменения концентрации изотопа 10В (или 11В) от природного уровня до заданного значения будет в 1,4 раза меньше. Дополнительно можно отметить, что по сравнению с системой на основе изоамилового спирта значение коэффициента обогащения более, чем в два с половиной раза больше: 7⋅10-3/2,7⋅10-3≈2,6 со всеми вытекающими из этого последствиями.
Пример 3.
В условиях, аналогичных указанным в прототипе (исходная концентрация H3BO3, два значения соотношения объемов фаз O:В = 1 и 2; разное число контактов фаз или уравновешиваний n) выполнены опыты по экстракции борной кислоты из ее водного раствора трибутилфосфатом. При этом время однократного контакта в экспериментах с ТБФ принято равным τ=5 мин, так как равновесие в системе «H3BO3/H2O//H3BO3⋅ТБФ/ТБФ» достигается, как показали исследования, менее чем за 5 мин. По окончании n уравновешиваний определяли равновесную концентрацию борной кислоты в водной фазе С*, рассчитывали количество экстрагированной трибутилфосфатом H3BO3

где Vв - объем водной фазы,
и производительность процесса экстракции борной кислоты, как изменение концентрации H3BO3 в единицу времени, то есть, как

Полученные результаты в сравнении с данными прототипа приведены в табл. 3.
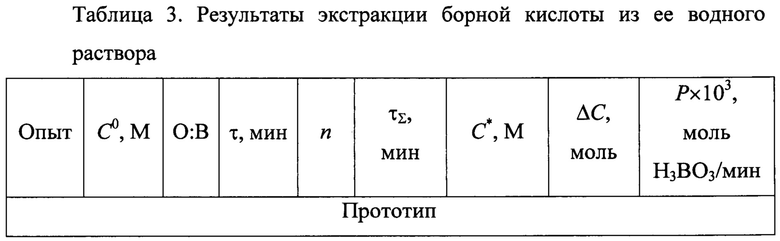
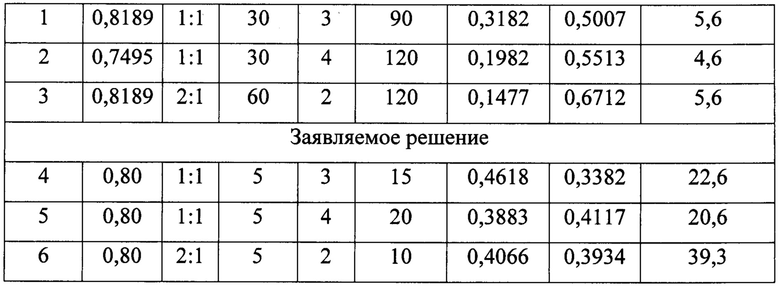
Примечания к таблице:
С0 - концентрация Н3ВО3 в исходном водном растворе;
O:В - соотношение объемов фаз в органической (О) и водной фазе (В);
τ - время одного контакта фаз (время однократного уравновешивания);
n - число контактов (уравновешиваний);
τΣ - общее время n контактов (уравновешиваний);
С* - концентрация H3BO3 в водной фазе после n контактов (уравновешиваний);
ΔС - количество экстрагированной Н3ВО3, причем ΔС=(С0-С*)×Vв, где Vв - объем водной фазы;
Р - производительность процесса экстракции борной кислоты (бора), причем P=ΔС/τΣ.
По данным табл. 3 при одинаковых условиях экстракции H3BO3 (серия опытов 4-6 в сравнении с опытами 1-3 по прототипу) использование ТБФ в качестве экстрагента позволяет увеличить производительность процесса экстракции борной кислоты в (22,6/5,6)×10-3=4,0; (20,6/4,6)×10-3=4,5 и (39,3/5,6)×10-3=7,0 раз соответственно, то есть, в 4-7 раз.
Пример 4.
Из находящейся в состоянии равновесия при комнатной температуре двухфазной системы «водный раствор H3BO3 - комплексное соединение H3BO3 с ТБФ или «Н3ВО3/H2O//H3BO3⋅ТБФ/ТБФ» отбирали 20 мл органической фазы, то есть, содержащего борную кислоту трибутилфосфата. В первой серии опытов («1» в табл. 4) этот объем уравновешивали в течение примерно 10 мин с разным объемом дистиллированной воды (прямая реэкстрация), варьируя соотношение O:В от 1:1 до 1:3 и определяя значения концентраций H3BO3 в каждой из фаз (С*вод и С*орг), а также рассчитывая степень извлечения (Гр) борной кислоты:
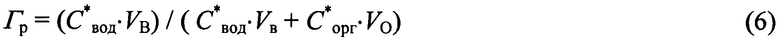
Во второй серии («2» в табл. 4) тот же объем органической фазы первоначально так же в течение 10 мин уравновешивали с таким же объемом воды (O:В=1;1), но после расслоения фаз не только определяли концентрации С*вод, С*орг и Гр, а и декантировали органическую фазу и вновь уравновешивали ее с исходной водой, проводя в целом операцию уравновешивания трижды и расходуя суммарно тот же объем воды (перекрестная реэкстракция).
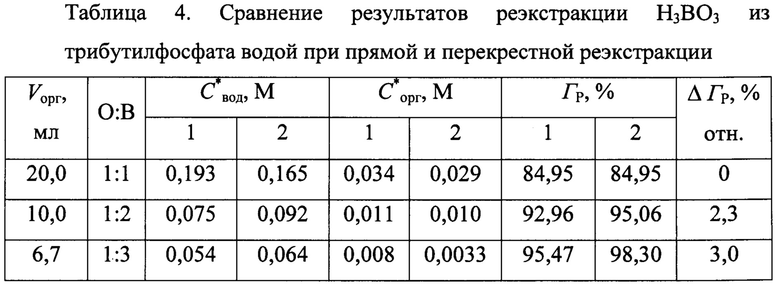
Сопоставление полученных результатов, представленное в табл. 4, показывает, что при равном объеме воды, расходуемой на реэкстракцию борной кислоты из трибутилфосфата, когда отношение объемов O:В=1:3, в случае перекрестной реэкстракции степень извлечения борной кислоты составила 98,30% против 95,47% для прямой реэкстракции, что при заданных значениях остаточной концентрации борной кислоты в ТБФ (С*орг) и степени извлечения Гр приведет к снижению расхода воды, как минимум, на 3% отн.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ КОНЦЕНТРИРОВАНИЯ СТАБИЛЬНОГО ИЗОТОПА БОРА В | 2023 |
|
RU2815423C1 |
| СПОСОБ РАЗДЕЛЕНИЯ ИЗОТОПОВ БОРА | 2006 |
|
RU2311949C1 |
| Способ разделения изотопов бора | 2021 |
|
RU2777556C1 |
| МАССООБМЕННАЯ КОЛОННА | 2019 |
|
RU2729797C1 |
| СПОСОБ ЭКСТРАКЦИОННОГО РАЗДЕЛЕНИЯ СКАНДИЯ И ТОРИЯ | 2016 |
|
RU2611001C1 |
| СПОСОБ РАЗДЕЛЬНОГО ИЗВЛЕЧЕНИЯ ЗОЛОТА И СЕРЕБРА ИЗ ТИОЦИАНАТНЫХ РАСТВОРОВ | 2008 |
|
RU2385958C1 |
| СПОСОБ ОЧИСТКИ ОКСИДОВ УРАНА ОТ ПРИМЕСЕЙ | 2009 |
|
RU2384902C1 |
| СПОСОБ РАЗДЕЛЕНИЯ ИЗОТОПОВ КРЕМНИЯ | 2021 |
|
RU2778866C1 |
| СПОСОБ ЭКСТРАКЦИОННОГО РАЗДЕЛЕНИЯ И КОНЦЕНТРИРОВАНИЯ ЦИРКОНИЯ И ГАФНИЯ | 2000 |
|
RU2190677C2 |
| СПОСОБ ВЫДЕЛЕНИЯ РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ ИЗ РАСТВОРА СОЛЯНОКИСЛОТНОГО РАЗЛОЖЕНИЯ ЭВДИАЛИТОВОГО КОНЦЕНТРАТА | 2005 |
|
RU2288171C1 |
Изобретение относится к технологии получения стабильных изотопов бора. Способ разделения изотопов бора осуществляется путем химического обмена в двухфазной жидкость-жидкостной системе с использованием водного раствора борной кислоты в качестве водной фазы и органической фазы, состоящей из комплексного соединения борной кислоты с органическим веществом, в качестве которого используют трибутиловый эфир фосфорной кислоты, при этом осуществляют реэкстракцию борной кислоты водой с последующим упариванием водного раствора в условиях перекрестного тока органической фазы и воды. Изобретение обеспечивает уменьшение числа компонентов органической фазы, увеличение однократного коэффициента разделения, снижение числа теоретических ступеней разделения, необходимых для достижения требуемой концентрации изотопа бора, увеличение производительности процесса экстракции борной кислоты, уменьшение расхода воды и снижение энергетических затрат. 4 табл., 4 пр.
Способ разделения изотопов бора химическим обменом в двухфазной жидкость-жидкостной системе с использованием водного раствора борной кислоты в качестве водной фазы и органической фазы, состоящей из комплексного соединения борной кислоты с органическим веществом, и с обращением потоков реэкстракцией борной кислоты водой с последующим упариванием водного раствора, отличающийся тем, что в качестве органического вещества используют трибутиловый эфир фосфорной кислоты, а реэкстракцию проводят в условиях перекрестного тока органической фазы и воды.
| СПОСОБ РАЗДЕЛЕНИЯ ИЗОТОПОВ БОРА | 2006 |
|
RU2311949C1 |
| 1й:СНСиЮЗНАЯ | 0 |
|
SU375088A1 |
| СЕВРЮГОВА Н.Н., УВАРОВ О.В., ЖАВОРОНКОВ О.М., журнал Атомная энергния, 1960 г., том 9, вып | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Прялка для изготовления крученой нити | 1920 |
|
SU112A1 |
| US 5419887 A1, 30.05.1995. | |||

