Область техники
Настоящее изобретение относится к фармацевтическим композициям, которые содержат лобеглитазон, для перорального введения с усовершенствованной растворимостью и скоростью растворения.
Уровень техники
Лобеглитазон представляет собой лекарственное средство ряда глитазона, которое разработано в Chong Kun Dang Pharmaceutical Corp. и одобрено в качестве 20-го нового лекарственного средства в Корее. Лобеглитазон используют в качестве антидиабетического средства для перорального введения, которое обладает активностью усовершенствования резистентности к инсулину. В частности, лобеглитазон используют в качестве средства для диабета 2-го типа. Дополнительно, лобеглитазон может быть очень эффективен для тучных пациентов с диабетом, поскольку лобеглитазон не имеет сердечнососудистых побочных эффектов, которые вызывали полемику вокруг существующих лекарственных средств ряда глитазона, в частности, вокруг розиглитазона из GSK.
Лобеглитазон имеет фрагмент 2,4-тиазолидиндиона, и N-метиламиноэтоксибензиловую группу конъюгируют со фрагментом 2,4-тиазолидиндиона в качестве линкера, как проиллюстрировано далее в формуле I. Лобеглитазон представляет собой порошок белого или бледно-желтоватого цвета и почти не имеет запаха.
[Формула I]
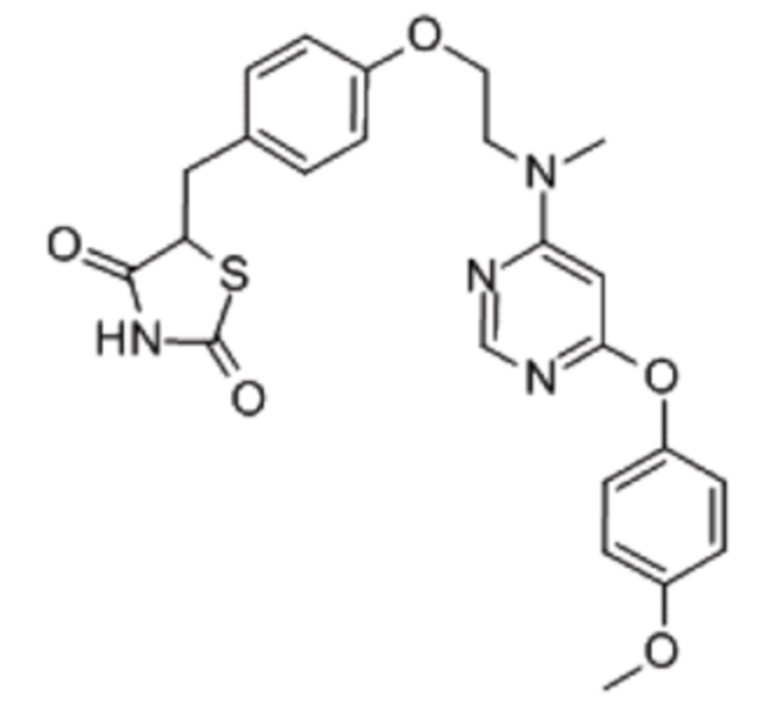
Между тем, лобеглитазон изначально разрабатывали в виде сульфатной формы, но сульфат лобеглитазона нерастворим в воде и обнаружен феномен межмолекулярной кластеризации, а также имели место проблемы со снижением растворимости, когда сульфат лобеглитазона растворяли и перекристаллизовывали. В последствии проблема снижения растворимости сульфата лобеглитазона возникла при формулировании сульфата лобеглитазона в качестве активного ингредиента.
Скорость растворения лекарственного средства тесно связана с растворимостью самого лекарственного средства, и в целом известно, что если растворимость лекарственного средства низка, скорость растворения лекарственного средства также низка.
Как изложено выше, когда растворение лекарственного средства в фармацевтической дозированной форме происходит слишком медленно, концентрация лекарственного средства в крови не может достигать эффективного уровня и ожидаемый эффект не может быть достигнут. Следовательно, определенные уровни скорости растворения и паттерн растворения, которые могут максимизировать эффекты лекарственного средства, являются очень важными вопросами для составов.
Следовательно, для разработки дозированной формы для перорального введения необходимо усовершенствование скорости растворения для быстрого проявления эффектов. Следующие способы можно использовать для усовершенствования скорости растворения препаратов для перорального введения: 1) уменьшение размера частицы посредством помола активных ингредиентов; 2) добавление поверхностно-активного средства или солюбилизирующего средства; 3) наноразмеры; и 4) использование твердой дисперсии. Первый способ среди приведенных выше четырех способов является самым простым путем усовершенствования скорости растворения и используется наиболее широко.
При усовершенствовании скорости растворения лекарственного средства для перорального введения посредством уменьшения размера частицы активного ингредиента, характерно в целом то, что усовершенствование скорости растворения происходит, когда размер частицы активных ингредиентов меньше. Тем не менее, проблема снижения стабильности лекарственного средства для перорального введения возникает из-за увеличения возможностей вступать в реакцию с другими добавками, включенными в состав, по причине увеличения площади поверхности посредством снижения размера частицы активного ингредиента.
В корейской публикации № 10-2005-0084316 раскрыто, что усовершенствование скорости растворения происходит при использовании водорастворимых добавок, таких как d-маннит, взамен использования лактозы, а в корейской публикации № 10-2007-0093259 раскрыто, что скорость растворения можно усовершенствовать при использовании конкретного полимера, такого как сополимеры метакрилата и сложного эфира метакриловой кислоты. Однако получение твердой дисперсии с использованием чрезмерного полимера или с использованием чрезмерного поверхностно-активного средства для того, чтобы усовершенствовать скорость растворения оказывается бессмысленным, поскольку его эффект заметно снижается из-за сниженной доли растворенного количества плохо растворимого лекарственного средства по сравнению с дозой поверхностно-активного средства.
Дополнительно, стабильность можно обеспечивать с использованием контролирующего pH средства, создавая лекарственное средство, которое разлагается при определенном уровне pH, чтобы существовать в стабильном диапазоне pH, в котором лекарственное средство не может разлагаться, но его использование чрезвычайно ограничено при рассмотрении различных уровней pH организма человека и его применение главным образом ограничено жидкими составами.
Авторы настоящего изобретения подтвердили, что растворимость сохраняется и происходит заметное усовершенствование скорости растворения при влажном гранулировании терапевтически эффективного количества лобеглитазона или его фармацевтически приемлемой соли с использованием воды в качестве связывающего растворителя и с использованием производных целлюлозы в качестве эксципиента, при этом исследуя способы сохранения растворимости и целевой скорости растворения лобеглитазона, и, соответственно, выполнили настоящее изобретение.
Описание изобретения
Техническая проблема
Настоящее изобретение предусматривает фармацевтические композиции, которые содержат лобеглитазон или его фармацевтически приемлемые соли для перорального введения с улучшенной растворимостью и скоростью растворения. В частности, настоящее изобретение предусматривает фармацевтические композиции для перорального введения, которые проявляют быстрые эффекты через сохранение растворимости и усовершенствование скорости растворения посредством влажного гранулирования плохо растворимого лобеглитазона или его фармацевтически приемлемых солей с очищенной водой в качестве связывающего растворителя и с использованием производного целлюлозы в качестве эксципиента.
Решение проблемы
Настоящее изобретение предусматривает фармацевтические композиции для перорального введения лобеглитазона или его фармацевтически приемлемых солей. Фармацевтические композиции по настоящему изобретению сохраняют растворимость активных ингредиентов, т. е., лобеглитазона или его фармацевтически приемлемых солей, и проявляют усовершенствованную скорость растворения.
Фармацевтические композиции по настоящему изобретению могут содержать от 0,1 до 10,0 массовой части лобеглитазона или его фармацевтически приемлемых солей и от 10,0 до 40,0 массовой части производных целлюлозы.
Что касается фармацевтических композиций по настоящему изобретению, основу без лобеглитазона или какие-либо фармацевтически приемлемые соли можно использовать в качестве указанного лобеглитазона или его фармацевтически приемлемых солей. Предпочтительно, сульфат лобеглитазона можно использовать в качестве указанного лобеглитазона или его фармацевтически приемлемых солей. Дополнительно, лобеглитазон или его фармацевтически приемлемые соли можно получать, как раскрыто в корейском патенте № 10-0450700.
Также можно вводить фармакологически эффективное количество лобеглитазона или его фармацевтически приемлемых солей. В частности, может содержаться от 0,1 до 1000 мг, предпочтительно от 0,1 до 500 мг и наиболее предпочтительно от 0,1 до 250 мг лобеглитазона или его фармацевтически приемлемых солей.
Кроме того, производные целлюлозы обозначают производные, сформированные посредством превращения одной или нескольких гидроксильных групп, существующих в одном или нескольких повторяющихся звеньях безводной глюкозы полимера целлюлозы, чтобы обеспечивать одну или несколько конъюгированных групп на полимере целлюлозы, таким как ацетат целлюлозы, простой эфиры целлюлозы и сложный эфиры целлюлозы, среди прочих.
В одном из вариантов осуществления настоящего изобретения простой эфир целлюлозы представляет собой «подходящую конъюгированную с простым эфиром группу, которая может присутствовать на повторяющихся звеньях безводной глюкозы полимера целлюлозы», и он включает (C1-C4)алкил, который необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из гидрокси, карбокси, (C1-C4)алкокси и гидрокси(C1-C4)алкокси. Например, «простой эфир целлюлозы» включает (C1-C4)алкил, такой как метил или этил, среди прочих; гидрокси(C1-C4)алкил, такой как 2-гидроксиэтил, 2-гидроксипропил или 3-гидроксипропил, среди прочих; (C1-C4)алкокси(C1-C4)алкил, такой как 2-метоксиэтил, 3-метоксипропил, 2-метоксипропил или 2-этоксиэтил, среди прочих; гидрокси(C1-C4)алкокси(C1-C4)алкил, такой как 2-(2-гидроксиэтокси)этил или 2-(2-гидроксипропокси)пропил, среди прочих; карбокси(C1-C4)алкил такой как карбоксиметил, среди прочих; или группу формулы H-[O-(C1-C4)алкил-]m (m представляет собой целое число в пределах от 1 до 5, например, 1, 2 или 3), такую как H-[O-CH(CH3)CH2-]m или H-[O-CH2CH2-]m, среди прочих. Для прояснения, термин «конъюгированная с простым эфиром группа» обозначает одну или несколько групп среди указанных функциональных групп, которые конъюгированы с полимером целлюлозы посредством атома кислорода. Например, одна или несколько гидроксигрупп повторяющихся звеньев безводной глюкозы превращаются в метоксигруппу, когда конъюгированной с простым эфиром группой является метил.
Также простой эфир целлюлозы может включать идентичные конъюгированные с простым эфиром группы, например, метилцеллюлоза может содержать метильные группы. Альтернативно, простой эфир целлюлозы может содержать множество неодинаковых конъюгированных с простым эфиром групп. Например, гидроксипропилметилцеллюлоза обозначает целлюлозу, которая содержит как метильную группу, так и гидроксипропильную (например, 2-гидроксипропильную) конъюгированную с простым эфиром группу.
Что касается фармацевтических композиций по настоящему изобретению, предпочтительно использовать водорастворимый простой эфир целлюлозы или сложный эфир водорастворимого простого эфира целлюлозы для указанного производного целлюлозы. Водорастворимый простой эфир целлюлозы или сложный эфир водорастворимого простого эфира целлюлозы может присутствовать в композициях в количестве, достаточном для того, чтобы ингибировать скорость преципитации лекарственного средства in vivo из раствора в случае, когда лекарственное средство проходит из кислой среды желудка в желудочно-кишечный тракт (например, область высокого pH в верхних отделах кишечника), где предположительно происходит абсорбция лекарственного средства. Подробно, указанный водорастворимый простой эфир целлюлозы или сложный эфир водорастворимого простого эфира целлюлозы может составлять от 0,1 до 90 массовых частей, предпочтительно от 1 до 80 массовых частей, более предпочтительно от 5 до 75 массовых частей и наиболее предпочтительно от 10 до 40 массовых частей в композициях на основании от 0,1 до 10 массовых частей лобеглитазона или его фармацевтически приемлемых солей (т. е. активного ингредиента).
Что касается фармацевтических композиций по настоящему изобретению, предпочтительный водорастворимый простой эфир целлюлозы можно выбирать из группы, состоящей из метилцеллюлозы, гидроксиэтилцеллюлозы, гидроксипропилцеллюлозы, гидроксипропилметилцеллюлозы, гидроксибутилметилцеллюлозы, гидроксиэтилэтилцеллюлозы, солей карбоксиметилцеллюлозы (например, карбоксиметилцеллюлозы натрия) и солей карбоксиметилгидроксиэтилцеллюлозы (например, карбоксиметилгидроксиэтилцеллюлозы натрия), среди прочих. Более предпочтительно, указанный водорастворимый простой эфир целлюлозы можно выбирать из группы, состоящей из гидроксиэтилцеллюлозы, гидроксипропилцеллюлозы, гидроксипропилметилцеллюлозы и солей карбоксиметилцеллюлозы (например, карбоксиметилцеллюлозы натрия), среди прочих.
Дополнительно, в одном из вариантов осуществления настоящего изобретения указанный водорастворимый простой эфир целлюлозы можно выбирать из группы, состоящей из гидроксиэтилцеллюлозы и гидроксипропилцеллюлозы. Когда водорастворимым простым эфиром целлюлозы является гидроксипропилцеллюлоза, содержание гидроксипропокси может составлять 16,0% масс. или больше, предпочтительно от 20 до 40% масс. Также предпочтительно кинематическая вязкость указанной гидроксипропилцеллюлозы должна составлять от 100 до 600 спз (например, от 150 до 450 спз), и в этот момент вязкость измеряют с использованием 2% масс./об. раствора гидроксипропилцеллюлозы при 25°C. Альтернативно, гидроксипропилцеллюлоза с низкой степенью замещения, в которой содержание гидроксипропокси составляет приблизительно от 5 приблизительно до 16% масс., можно использовать в качестве гидроксипропилцеллюлозы. Гидроксипропилцеллюлоза с низкой степенью замещения обычно по-видимому является водонерастворимой, но обнаружено, что указанная гидроксипропилцеллюлоза с низкой степенью замещения является достаточно гидрофильной в случае композиций по настоящему изобретению, чтобы предотвращать преципитацию лекарственных средств из раствора, и что указанная гидроксипропилцеллюлоза с низкой степенью замещения представляет собой одно из водорастворимых производных целлюлозы по настоящему изобретению. Предпочтительно степень замещения указанной гидроксипропилцеллюлозы с низкой степенью замещения составляет приблизительно от 0,1 приблизительно до 1,0, например, от 0,2 до 0,4 (например приблизительно 0,2), и термин «степень замещения» обозначает усредненное число гидроксипропильных групп на цикл безводной глюкозы целлюлозы. Кроме того, в соответствии с указанной гидроксипропилцеллюлозой с низкой степенью замещения, содержание гидроксипропокси может составлять приблизительно от 5 приблизительно до 16% масс. и предпочтительно от 9 до 13% масс. Также предпочтительно усредненный размер частицы указанной гидроксипропилцеллюлозы с низкой степенью замещения составляет от 10 до 75 мкм и более предпочтительно от 50 до 60 мкм.
Кроме того, фармацевтические композиции по настоящему изобретению дополнительно могут содержать один или несколько фармацевтически приемлемых эксципиентов. Разбавители, связывающие средства, разрыхлители и/или смазывающие средства можно использовать в качестве добавок.
Что касается настоящего изобретения, в качестве разбавителей предпочтительно использовать водорастворимые разбавители. Водорастворимые разбавители могут представлять собой сахарид (например, лактозу, сахарозу и декстрозу), полисахарид (например, декстран и мальтодекстрин), полиол (например, маннит, ксилит и сорбит) и циклодекстрин, модифицированный крахмал (например, гидролизат или прежелатинизированный крахмал, который можно модифицировать с помощью тепла, механического воздействия или химической реакции), микрокристаллический крахмал, маннит, сорбит, трегалозу, мальтозу, минеральную соль (например, карбонат кальция, карбонат магния, двухосновный фосфат кальция (безводный/дигидрат), трехосновный фосфат кальция), целлюлозу, микрокристаллическую целлюлозу, сульфат кальция, ксилит и лактит, среди прочих, без ограничения. В отношении настоящего изобретения для водорастворимых разбавителей предусмотрен достаточный объем препаратов, и они могут существовать в препаратах в количестве, достаточном для того, чтобы приступать к получению таблеток. Водорастворимые разбавители по настоящему изобретению могут составлять от 1 до 25 массовых частей, предпочтительно от 1 до 20 массовых частей и более предпочтительно от 2 до 15 массовых частей в композициях на основании от 0,1 до 10 массовых частей лобеглитазона или его фармацевтически приемлемых солей.
В настоящем изобретении в качестве связывающих средств предпочтительно использовать гидрофильные связывающие средства. Гидрофильные связывающие средства предоставляют в количестве, достаточном для того, чтобы действовать в качестве поддержки лобеглитазона или его фармацевтически приемлемых солей (т. е. активного ингредиента), наряду с другими эксципиентами в композициях. Гидрофильные связывающие средства облегчают получение и обработку гранул, а также усовершенствуют стабильность активных ингредиентов. Надлежащие гидрофильные связывающие средства для настоящего изобретения можно выбирать из группы, состоящей из поливинилпирролидона (например, повидон K25-32, в частности K29-32, значение «k» представляет собой диапазон усредненной молекулярной массы, получаемой с помощью уравнения Фикенчера), лактозы (может существовать в форме дегидрата или гидрата, например, моногидрата лактозы), крахмала, модифицированного крахмала, сахара, сенегальской камеди, трагакантовой камеди, гуаровой камеди, пектина, воскового связывающего средства, микрокристаллической целлюлозы, кополивидона, желатина и альгината (например, альгината натрия), среди прочих. Предпочтительно, гидрофильные связывающие средства по настоящему изобретению можно выбирать из группы, состоящей из поливинилпирролидона, крахмала, модифицированного крахмала и микрокристаллической целлюлозы. Гидрофильные связывающие средства по настоящему изобретению могут составлять от 1 до 20 массовых частей, предпочтительно от 1 до 10 массовых частей и более предпочтительно от 2 до 8 массовых частей в композициях на основании от 0,1 до 10 массовых частей лобеглитазона или его фармацевтически приемлемых солей.
Разрыхлители по настоящему изобретению можно выбирать из группы, состоящей из кроскармелозы натрия, кросповидона, поливинилпирролидона, крахмалгликолята натрия, крахмала и альгиновой кислоты, среди прочих, и предпочтительно использовать кроскармелозу натрия или кросповидон. Разрыхлители по настоящему изобретению могут составлять от 0,1 до 20 массовых частей, предпочтительно от 0,1 до 10 массовых частей и более предпочтительно от 1 до 10 массовых частей в композициях на основании от 0,1 до 10 массовых частей лобеглитазона или его фармацевтически приемлемых солей.
Надлежащие смазывающие средства по настоящему изобретению обозначают фармацевтически приемлемые добавки, которые в целом используют для получения твердых составов в данной области в качестве смазывающего средства или модификатора скольжения. Смазывающие средства можно выбирать из группы, состоящей из талька, стеарата магния, стеарата кальция, коллоидного диоксида, силиката кальция, крахмала, минерального масла, воска, глицерилбегената, полиэтиленгликоля, бензоата натрия, ацетата натрия, стеарилфумарата натрия и гидрогенизированного растительного масла, без ограничения. Предпочтительно, смазывающие средства выбирают из группы, состоящей из стеарата магния, стеарилфумарата натрия и стеариновой кислоты, и наиболее предпочтительно смазывающим средством является стеарат магния. Предпочтительно предоставлять достаточное количество смазывающих средств, поскольку смазывающие средства снижают трение о стенки формы при прессовании препаратов в таблетки. Кроме того, смазывающие средства облегчают обработку сухого порошка препарата. Предпочтительно смазывающие средства по настоящему изобретению составляют от 0,1 до 2 массовых частей в композициях на основании от 0,1 до 10 массовых частей лобеглитазона или его фармацевтически приемлемых солей.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения, фармацевтические композиции содержат:
(a) от 0,1 до 10 массовых частей лобеглитазона или его сульфата;
(b) от 10 до 40 массовых частей производных целлюлозы, выбранных из группы, состоящей из гидроксиэтилцеллюлозы, гидроксипропилцеллюлозы, гидроксипропилметилцеллюлозы и солей карбоксиметилцеллюлозы;
(c) от 2 до 15 массовых частей одного или нескольких водорастворимых разбавителей, выбранных из группы, состоящей из лактозы (в частности, моногидрата лактозы), маннита и микрокристаллической целлюлозы;
(d) от 2 до 8 массовых частей одного или нескольких гидрофильных связывающих средств, выбранных из группы, состоящей из поливинилпирролидона (например, повидона K25-32, в частности K29-32), крахмала, модифицированного крахмала, сахара и гидроксипропилметилцеллюлозы;
(e) от 1 до 10 массовых частей одного или нескольких разрыхлителей, выбранных из группы, состоящей из кроскармелозы натрия или кросповидона; и
(f) от 0,1 до 2,0 массовой части одного или нескольких смазывающих средств (например, стеарата магния), и
сумма ингредиентов (a)+(b)+(c)+(d)+(e)+(f) составляет 100 массовых частей.
Дополнительные эксципиенты, которые можно добавлять в фармацевтические композиции по настоящему изобретению, представляют собой консервант, стабилизатор, антиоксидант, модификатор текучести диоксида кремния и подавитель прилипания, среди прочих, и в препарате можно смешивать quantum satis дополнительных случайных ингредиентов (например, красителя или ароматизатора), которые не оказывают нежелательных эффектов на таблетки или порошки, полученные с использованием препарата, а также для того, чтобы осуществлять ожидаемые функции.
Фармацевтические композиции по настоящему изобретению можно вводить через различные надлежащие пути, включая пероральное введение, офтальмическое введение, трансдермальное введение, подкожное введение, внутривенное введение или внутримышечное введение, среди прочих. Фармацевтические композиции по настоящему изобретению можно формулировать для перорального или парентерального введения в соответствии с выбранным путем введения, но пероральное введение является наиболее предпочтительным.
В качестве препарата для перорального введения фармацевтические композиции по настоящему изобретению можно формулировать в форме порошков, гранул, таблеток, пилюль, таблеток, покрытых сахаром, капсул, жидких составов, гелевых составов, составов со взвесями и суспензиями, среди прочих, с помощью способов, известных в данной области. Например, в соответствии с препаратами для перорального введения, таблетки или покрытые таблетки можно получать посредством смешивания активных ингредиентов и твердых эксципиентов с их последующим перемалыванием и перемешиванием и добавления достаточных добавок с последующей переработкой в гранулярную смесь.
Препарат для перорального введения по настоящему изобретению можно получать с помощью различных способов, известных в данной области. Примерами этих способов являются: сушка после влажного гранулирования, помол и прессование таблеток с мембранным покрытием или без него; помол после сухого гранулирования и прессование таблеток с использованием мембранного покрытия или без него; прессование таблеток с использованием мембранного покрытия или без него после получения сухой смеси; формирование таблеток; влажное гранулирование, сушка и заполнение гранулами желатиновой капсулы; заполнение желатиновой капсулы сухой смесью; или заполнение желатиновой капсулы суспензией или раствором, среди прочих. В целом, чтобы различать, препарат маркируют посредством выдавливания или печати на его поверхности.
Предпочтительно, фармацевтические композиции по настоящему изобретению можно получать с помощью способа получения, который включает: (S1) смешивание лобеглитазона или его фармацевтически приемлемых солей, разбавителя, связывающего средства и разрыхлителя и обработку через влажное гранулирование и (S2) смешивание продуктов влажного гранулирования, производных целлюлозы, связывающего средства, разрыхлителя и смазывающего средства.
Кроме того, наиболее предпочтительно использовать очищенную воду в качестве растворителя для влажного гранулирования на стадии (S1).
Фармацевтические композиции по настоящему изобретению, полученные способом по настоящему изобретению, обладают превосходящей физической стабильностью (твердостью или ломкостью) и демонстрируют заметно превосходящую скорость растворения.
Полезные эффекты изобретения
Фармацевтические композиции по настоящему изобретению могут проявлять незамедлительный фармакологический эффект посредством усовершенствования скорости растворения, при этом сохраняя растворимость лобеглитазона или его фармацевтически приемлемых солей.
Краткое описание фигур
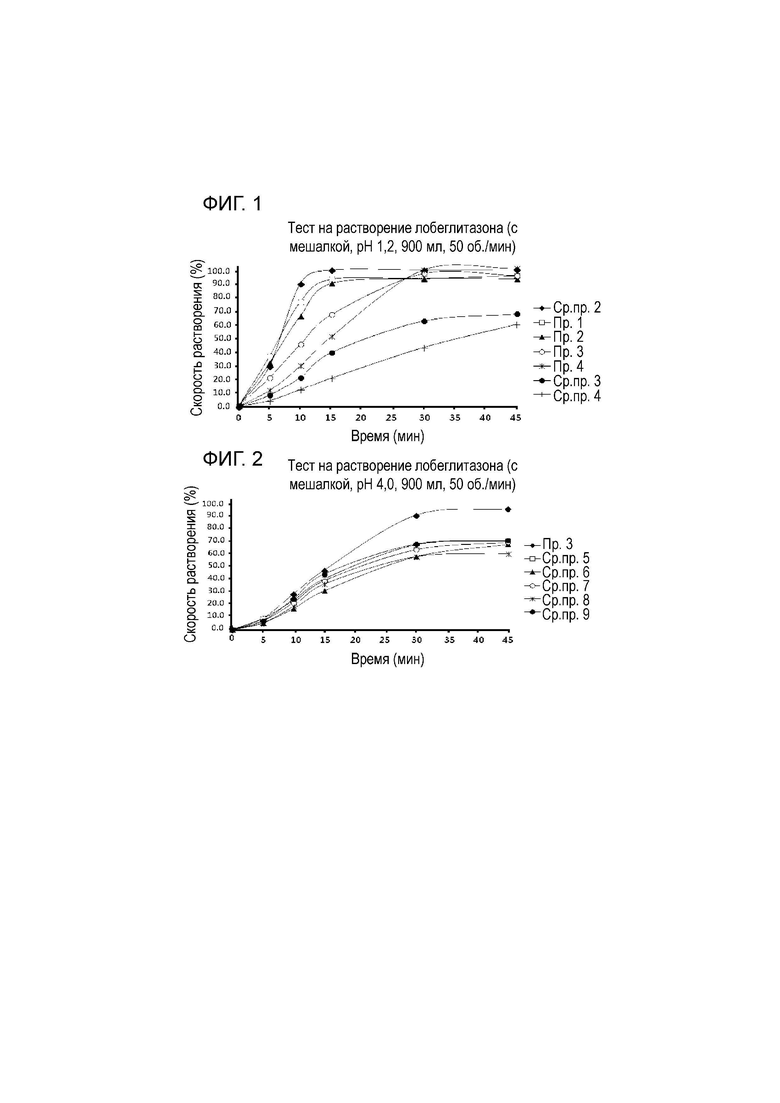
На фиг. 1 представлен график, показывающий результат сравнения скоростей растворения среди таблеток, полученных с использованием композиций с содержанием производного целлюлозы (гидроксипропилцеллюлозы с низкой степенью замещения) 5% масс. (сравнительный пример 2), 10% масс. (пример 1), 15% масс. (пример 2), 32% масс. (пример 3), 40% масс. (пример 4), 50% масс. (сравнительный пример 3) и 63% масс. (сравнительный пример 4), соответственно.
На фиг. 2 представлен график, показывающий результат сравнения скоростей растворения среди таблеток, полученных с использованием композиций, в которых растворителем для связывающего раствора, используемого в процессе влажного гранулирования, соответственно является очищенная вода (пример 3), смешанный раствор очищенная вода:безводный этанол 1:1 (сравнительный пример 5), смешанный раствор очищенная вода:дихлорметан 4:1 (сравнительный пример 6), смешанный раствор очищенная вода:дихлорметан 1:4 (сравнительный пример 7), смешанный раствор очищенная вода:изопропиловый спирт 4:1 (сравнительный пример 8) и смешанный раствор очищенная вода:изопропиловый спирт 1:4 (сравнительный пример 9).
Вариант осуществления изобретения
Настоящее изобретение описано более полно далее в настоящем документе со ссылкой на сопроводительные примеры, сравнительные примеры и экспериментальные примеры. Однако настоящее изобретение можно осуществлять во многих различных формах, и его не следует толковать в качестве ограниченного вариантами осуществления, изложенными в настоящем документе.
Примеры с 1 до 4 и сравнительные примеры с 1 до 4: получение таблеток, которые содержат сульфат лобеглитазона по настоящему изобретению
Фармацевтические композиции, которые содержат сульфат лобеглитазона, получали с использованием композиций из следующей таблицы 1.
1) Растирание активного ингредиента
Сульфат лобеглитазона растирали в порошок с использованием гидрата лактозы (распылительная сушка) и смешивали с кроскармелозой натрия (Ac-di-sol, DMV-Fonterra Excipients GmBH, Germany).
2) Получение связывающего раствора
Краситель синий № 2 добавляли в очищенную воду и его измельчали и перемешивали посредством гомогенизатора (HG-150, Daihan Scientific). Здесь добавляли повидон K-30 (DaebongLS, Korea) и смесь перемешивали и растворяли. Затем связывающий раствор получали посредством просеивания через сито 350 меш.
3) Получение гранул
Смесь из приведенного выше 1) и связывающий раствор из приведенного выше 2) помещали в высокоскоростной смешиватель (LFS-GS-2J, Fukae Powtec, Japan) и затем объединяли и гранулировали с использованием высокоскоростного смешивателя. Затем гранулы сушили с использованием сушилки с псевдоожиженным слоем (FBCG-1, Cheongjin Technology Industry, Korea). После сушки гранулы деагломерировали с помощью Co-mil (U3, Quadro, Canada).
4) Финальное смешивание и прессование таблеток
Высушенные гранулы из 3) помещали в смешиватель и осуществляли первое смешивание с добавлением кроскармелозы натрия, микрокристаллической целлюлозы (Avicel PH 102, FMC BioPolymer, U.S.A.) и гидроксипропилцеллюлозы с низкой степенью замещения (LH-11, Shin-Etsu, Japan). После завершения первого смешивания, осуществляли второе смешивание, помещая стеарат магния, просеянный через сито 30 меш, и получали конечную смесь. Таблетки получали через прессование таблеток с использованием автоматической машины для прессования таблеток (XP1, Korsch, Germany).
[Таблица 1]
(количество ингредиентов: мг/таблетка;
Пр.: пример; Ср. пр.: сравнительный пример;)
Экспериментальный пример 1: измерение твердости и ломкости получаемых таблеток
Твердость, ломкость и угол естественного откоса таблеток, полученных в примерах 1-4 и сравнительных примерах 1-4 по настоящему изобретению измеряют следующими способами, и результат измерения представлен далее в таблице 2.
[Измерение твердости]
- Измеритель твердости: BH-225 (Pharmatest Apparatebau, GmbH, Germany)
- Способ измерения: вычисленное среднее значение для 10 раз циклического измерения
[Измерение ломкости]
- Прибор для тестирования ломкости: SVM-102 (Pharmatest Apparatebau, GmbH, Germany)
- Способ измерения: 20 таблеток, измеренная ломкость после вращения 100 раз
[Таблица 2]
Как показано в таблице 2, полученные таблетки имели высокую твердость и низкую ломкость, когда содержали 10% масс. или больше водорастворимых производных целлюлозы. Подтверждено, что сравнительный пример 1 был недостаточным для композиций для перорального введения, поскольку таблетки не формировали из-за недостаточной прочности связывания внутри гранул, и, соответственно, измерение не было доступно. Также сравнительный пример 2 демонстрировал низкую твердость 5 килофунтов и высокую ломкость 0,7%. Однако примеры 1-4 или сравнительные примеры 3 и 4, содержащие 10% масс. или больше водорастворимых производных целлюлозы, демонстрировали заметно усовершенствованные твердость и ломкость, которые составляли 10 килофунтов или больше и 0,1% или меньше, соответственно.
Экспериментальный пример 2: тест на растворимость
Растворение полученных препаратов из примеров 1-4 и сравнительных примеров 2-4 тестировали в соответствии со вторым способом из теста на растворимость (с мешалкой), приведенного в Корейской фармакопее (10-е издание), и анализировали с помощью следующих способов. Результат теста на растворимость представлен в таблице 3 и на фиг. 1.
[Способ из теста на растворимость]
- Аппарат: Hanson SR 8 (Hanson Research, U.S.A.)
- Среда для растворения: среда с pH 1,2, 900 мл
- Температура среды для растворения: 37±0,5°C
- Скорость вращения: 50 об./мин
- Количество образца: одна таблетка на каждый сосуд
[Способ анализа: условия ВЭЖХ жидкостной хроматографии]
- Детектор: Спектрометр ультрафиолетового и видимого спектра (длина волны измерения: 250 нм)
- Колонка: X-Terra RP 18 (4,6×250 мм, 5 мкм) или эквивалентные
- Подвижная фаза: буферный раствор нитрата аммония 50 мМ (pH 5,0):ацетонитрил=55:45
- Вводимый объем: 100 мкл
- Скорость потока: 1,5 мл/мин
- Температура колонки: 40°C
[Таблица 3]
Как показано в таблице 3 и на фиг. 1, в отношении примеров 1-4 и сравнительных примеров 3 и 4, в которых обеспечивали достаточную твердость и ломкость для формирования таблеток, примеры 1-4 демонстрировали скорость растворения 95% только за 30 минут, тогда как сравнительные примеры 3 и 4 демонстрировали заметно более низкую скорость растворения 70% или меньше даже за 45 минут. Соответственно, в настоящем изобретении подтверждали, что композиции, содержащие лобеглитазон, демонстрировали надлежащие характеристики растворения для фармакокинетики сульфата лобеглитазона, который имеет время поступления один час или меньше до достижения максимальной концентрации в крови, когда композиция, содержащая лобеглитазон, содержала от 10 до 40% масс. производных целлюлозы.
Пример 3 и сравнительные примеры 5-9: получение таблеток, которые содержат сульфат лобеглитазона по настоящему изобретению
Получали пример 3 и сравнительные примеры 5-9 с использованием композиций, приведенных в следующей таблице 4, посредством изменения связывающих растворов на основании примера 3, который демонстрирует характеристики растворения, достаточные для фармакокинетики.
[Таблица 4]
(количество ингредиентов: мг/таблетка; Пр.: пример; Ср.пр.: сравнительный пример;)
Экспериментальный пример 3: тест на растворимость
Растворение полученных препаратов из примера 3 и сравнительных примеров 5-9 тестировали в соответствии со вторым способом из теста на растворимость (с мешалкой), приведенного в Корейской фармакопее (10-е издание), и анализировали с помощью следующих способов. Результат теста на растворимость представлен в таблице 5 и на фиг. 2.
[Способ из теста на растворимость]
- Аппарат: Hanson SR 8 (Hanson Research, U.S.A.)
- Среда для растворения: среда с pH 4,0, 900 мл
- Температура среды для растворения: 37±0,5°C
- Скорость вращения: 50 об./мин
- Количество образца: одна таблетка на каждый сосуд
[Способ анализа: условия ВЭЖХ жидкостной хроматографии]
- Детектор: Спектрометр ультрафиолетового и видимого спектра (длина волны измерения: 250 нм)
- Колонка: X-Terra RP 18 (4,6×250 мм, 5 мкм) или эквивалентные
- Подвижная фаза: буферный раствор нитрата аммония 50 мМ (pH 5,0):ацетонитрил=55:45
- Вводимый объем: 100 мкл
- Скорость потока: 1,5 мл/мин
- Температура колонки: 40°C
[Таблица 5]
Скорости растворения для различных связывающих растворителей не различались значительно в ходе влажного гранулирования при тестировании растворения в среде с pH 1,2, но в тесте на растворимость в среде с pH 4,0 пример 3, в котором в качестве связывающего растворителя использовали только очищенную воду, демонстрировал 97% скорости растворения за 45 минут, как показано в таблице 5 и на фиг. 2. Однако сравнительные примеры 5-9, в которых органический растворитель или смесь очищенной воды и органического растворителя использовали в качестве связывающего растворителя, демонстрировали от 60 до 70% заметно сниженной скорости растворения. Следовательно, подтверждено, что скорость растворения удивительно снижалась при использовании смеси очищенной воды и органического растворителя в качестве связывающего растворителя по сравнению с влажным гранулированием с использованием только очищенной воды. В настоящем изобретении подтверждали с помощью приведенных выше экспериментальных примеров, что растворимость и скорость растворения из-за плохой растворимости лобеглитазона удивительно усовершенствована, когда влажное гранулирование осуществляют с использованием только очищенной воды в качестве связывающего растворителя.
Промышленная применимость
Фармацевтические композиции по настоящему изобретению могут проявлять быстрый фармакологический эффект посредством усовершенствования скорости растворения при сохранении растворимости лобеглитазона или его фармацевтически приемлемых солей. Следовательно, фармацевтические композиции по настоящему изобретению можно использовать в качестве фармацевтического препарата для перорального введения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЙ КОМБИНИРОВАННЫЙ СОСТАВ | 2016 |
|
RU2736942C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИПРОЛИФЕРАТИВНЫМ ДЕЙСТВИЕМ (ВАРИАНТЫ), СПОСОБ ЕЕ ПОЛУЧЕНИЯ И СПОСОБЫ С ЕЕ ИСПОЛЬЗОВАНИЕМ | 2003 |
|
RU2318518C2 |
| КОМПОЗИЦИЯ ТАБЛЕТКИ ЛЕНАЛИДОМИДА ДЛЯ ПЕРОРАЛЬНОГО ПРИЕМА | 2017 |
|
RU2725074C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2011 |
|
RU2589842C2 |
| ДЮРАНТНАЯ ЛЕКАРСТВЕННАЯ ФОРМА, СОДЕРЖАЩАЯ 3-(2-ДИМЕТИЛАМИНОМЕТИЛЦИКЛОГЕКСИЛ)ФЕНОЛ | 2006 |
|
RU2445081C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИБУПРОФЕН И ДОМПЕРИДОН ДЛЯ ЛЕЧЕНИЯ МИГРЕНИ | 1998 |
|
RU2195935C2 |
| ТВЕРДЫЕ ПРЕПАРАТЫ, СОДЕРЖАЩИЕ ТОФОГЛИФЛОЗИН, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2700164C2 |
| ТВЕРДАЯ ДИСПЕРСИЯ С УЛУЧШЕННОЙ РАСТВОРИМОСТЬЮ, СОДЕРЖАЩАЯ ПРОИЗВОДНОЕ ТЕТРАЗОЛА В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2013 |
|
RU2662819C2 |
| АНТИРЕТРОВИРУСНАЯ КОМБИНАЦИЯ | 2008 |
|
RU2531089C2 |
| ПЕРОРАЛЬНЫЙ КОМБИНИРОВАННЫЙ ПРЕПАРАТ, ВКЛЮЧАЮЩИЙ ГЕМИГЛИПТИН И ДАПАГЛИФЛОЗИН, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2021 |
|
RU2827705C1 |
Группа изобретений относится к фармацевтической промышленности, в частности к фармацевтической композиции для перорального введения, включающей 0,1-10 мас.ч. лобеглитазона или его фармацевтически приемлемой соли и 10-40 мас.ч. гидроксипропилцеллюлозы с низкой степенью замещения, в которой содержание гидроксипропокси составляет от 5,0 до 16,0 мас.%. Также предложен способ получения указанной фармацевтической композиции, в соответствии с которым лобеглитазон или его фармацевтически приемлемую соль смешивают с водорастворимым разбавителем, гидрофильным связывающим средством и разрыхлителем; далее посредством влажного гранулирования получают гранулы, которые затем смешивают с гидроксипропилцеллюлозой, гидрофильным связывающим средством, разрыхлителем и смазывающим средством. Группа изобретений обеспечивает создание фармацевтических композиций, проявляющих быстрый фармакологический эффект за счет улучшения скорости растворения при сохранении растворимости лобеглитазона или его фармацевтически приемлемых солей. 2 н. и 13 з.п. ф-лы, 3 пр., 5 табл., 2 ил.
1. Фармацевтическая композиция для перорального введения, которая содержит от 0,1 до 10 массовых частей лобеглитазона или его фармацевтически приемлемой соли и от 10 до 40 массовых частей производного целлюлозы, где производное целлюлозы представляет собой гидроксипропилцеллюлозу с низкой степенью замещения, в которой содержание гидроксипропокси составляет от 5,0 до 16,0% мас.
2. Фармацевтическая композиция для перорального введения по п.1, в которой фармацевтически приемлемой солью лобеглитазона является сульфат лобеглитазона.
3. Фармацевтическая композиция для перорального введения по п.1, в которой содержание гидроксипропокси в гидроксипропилцеллюлозе с низкой степенью замещения составляет от 9,0 до 13,0% мас.
4. Фармацевтическая композиция для перорального введения по п.1, в которой усредненный размер частицы гидроксипропилцеллюлозы с низкой степенью замещения составляет от 10 до 75 мкм.
5. Фармацевтическая композиция для перорального введения по п.4, в которой усредненный размер частицы гидроксипропилцеллюлозы с низкой степенью замещения составляет от 50 до 60 мкм.
6. Фармацевтическая композиция для перорального введения по п.1, которая дополнительно содержит от 2 до 15 массовых частей водорастворимых разбавителей, от 2 до 8 массовых частей гидрофильных связывающих средств, от 1 до 10 массовых частей разрыхлителей и от 0,1 до 2 массовых частей смазывающих средств.
7. Фармацевтическая композиция для перорального введения по п.6, в которой водорастворимые разбавители выбраны из группы, состоящей из сахарида, полисахарида, полиола, циклодекстрина и их смесей.
8. Фармацевтическая композиция для перорального введения по п.6, в которой гидрофильные связывающие средства выбраны из группы, состоящей из поливинилпирролидона, крахмала, модифицированного крахмала, сахарида, микрокристаллической целлюлозы и их смесей.
9. Фармацевтическая композиция для перорального введения по п.6, в которой разрыхлители выбраны из группы, состоящей из кроскармеллозы натрия, кросповидона и их смесей.
10. Фармацевтическая композиция для перорального введения по п.6, в которой смазывающие средства выбраны из группы, состоящей из талька, стеарата магния, стеарата кальция, стеариновой кислоты, коллоидного диоксида, силиката кальция, минерального масла, воска, глицерилбегената, полиэтиленгликоля, бензоата натрия, ацетата натрия, стеарилфумарата натрия и их смесей.
11. Фармацевтическая композиция для перорального введения по п.1, где композиция имеет форму таблетки.
12. Фармацевтическая композиция для перорального введения по п.11, где фармацевтическая композиция содержит
а) гранулу, содержащую лобеглитазон или его фармацевтически приемлемую соль; и
б) производное целлюлозы,
при этом производное целлюлозы представляет собой гидроксипропилцеллюлозу с низкой степенью замещения, в которой содержание гидроксипропокси составляет от 5,0 до 16,0% мас.
13. Фармацевтическая композиция для перорального введения по п.12, в которой гранула дополнительно содержит водорастворимые разбавители, гидрофильные связывающие средства или разрыхлители.
14. Способ получения фармацевтической композиции для перорального введения, включающий:
смешивание лобеглитазона или его фармацевтически приемлемой соли, водорастворимого разбавителя, гидрофильного связывающего средства и разрыхлителя и получение гранулы посредством влажного гранулирования; и
смешивание гранулы, производного целлюлозы, гидрофильного связывающего средства, разрыхлителя и смазывающего средства,
где фармацевтическая композиция содержит от 0,1 до 10 массовых частей лобеглитазона или его фармацевтически приемлемой соли и от 10 до 40 массовых частей производного целлюлозы,
где производное целлюлозы представляет собой гидроксипропилцеллюлозу с низкой степенью замещения, в которой содержание гидроксипропокси составляет от 5,0 до 16,0% мас.
15. Способ получения фармацевтической композиции для перорального введения по п.14, где растворителем для влажного гранулирования является очищенная вода.
| Sin Gon Kim et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| СПОСОБ ЛЕЧЕНИЯ ДИАБЕТА | 2007 |
|
RU2442585C2 |
| RU 2012131509 A, 27.01.2014. | |||

