Настоящее изобретение относится к фармацевтической лекарственной форме с контролируемым (регулируемым) высвобождением 3-(2-диметиламинометилциклогексил)фенола, предпочтительно его (1R,2R)-стереоизомера, или одной из его фармацевтически приемлемых солей.
3-(2-Диметиламинометилциклогексил)фенол известен из уровня техники. Речь при этом идет о принимаемом внутрь лекарственном веществе, обладающем анальгетическим действием (см., например, DE 19525137, WO 02/43712 и WO 02/67916).
3-(2-Диметиламинометилциклогексил)фенол имеет два центра хиральности и поэтому встречается в виде четырех стереоизомеров (двух пар энантиомеров), а именно: в виде (1R,2R-), (1S,2S)-, (1R,2S)- и (1S,2R)-3-(2-диметиламинометилциклогексил)фенола. Эти четыре стереоизомера имеют следующие химические структуры:
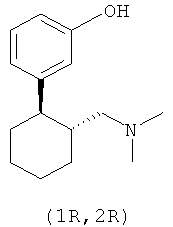
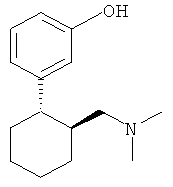
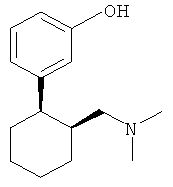
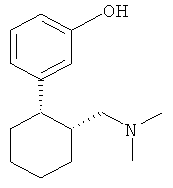
Традиционные лекарственные формы для приема внутрь, содержащие 3-(2-диметиламинометилциклогексил)фенол, исключительно быстро высвобождают все количество этого действующего вещества в желудочно-кишечном тракте, из-за чего анальгетическое действие препарата быстро проходит. Поэтому в настоящее время при лечении сильных хронических болей 3-(2-диметиламинометилциклогексил)фенолом содержащее его лекарственное средство требуется принимать через сравнительно короткие интервалы времени, например от четырех до шести раз в сутки, для поддержания таким путем достаточной концентрации действующего вещества в плазме крови пациента на протяжении всех 24 ч.
Однако необходимость частого приема лекарственного средства легко может привести к ошибкам в его применении, а также к нежелательным колебаниям концентрации действующего вещества в плазме крови, что вредит соблюдению пациентом режима и схемы лечения и снижает эффективность обезболивающего лечения, прежде всего при лечении хронических болей. Еще один известный недостаток состоит в том, что прием внутрь 3-(2-диметиламинометилциклогексил)фенола в составе традиционных, содержащих его лекарственных форм может сопровождаться побочными действиями, прежде всего тошнотой и рвотой.
Известно, что для увеличения продолжительности непрерывного высвобождения действующего вещества можно использовать лекарственные формы с замедленным высвобождением содержащихся в них действующих веществ (так называемые дюрантные лекарственные формы).
Из уровня техники известны дюрантные лекарственные формы для большого числа действующих веществ. Обычно для замедления высвобождения действующего вещества используют покрытие из пригодных для этой цели материалов и/или действующее вещество внедряют в регулирующую его высвобождение матрицу.
При использовании покрытий для замедления высвобождения действующего вещества содержащую его сердцевину снабжают замедляющим высвобождение действующего вещества покрытием из гидрофильных и/или гидрофобных полимеров. При использовании матриц для замедления высвобождения действующего вещества его внедряют в полимерную матрицу, регулирующую высвобождение действующего вещества.
Однако для обеспечения определенного профиля высвобождения действующего вещества невозможно просто лишь заменить одно действующее вещество, содержащееся в известной из уровня техники композиции с требуемым профилем его высвобождения, на другое действующее вещество. Более того, в каждом отдельном случае необходимо учитывать индивидуальные физические и химические свойства конкретного действующего вещества. Так, в частности, на профиль высвобождения действующего вещества значительное влияние могут оказывать его многочисленные индивидуальные свойства. За такие специфические характеристики высвобождения действующего вещества могут быть ответственны, например, доза, в которой его требуется вводить в организм, размер и форма его частиц, твердость, гигроскопичность, растворимость, ее зависимость от значения pH, гидрофильность/липофильность, кислотность/основность и иные параметры.
В основу изобретения была положена задача предложить фармацевтическую композицию (лекарственную форму) с 3-(2-диметиламинометилциклогексил)фенолом, предпочтительно его (1R,2R)-стереоизомером, соответственно с одной из его фармацевтически приемлемых солей, которая (фармацевтическая композиция) обладала бы преимуществами перед известными из уровня техники фармацевтическими композициями.
Такая лекарственная форма должна, в частности, обеспечивать поддержание концентрации действующего вещества - 3-(2-диметиламинометилциклогексил)фенола - в плазме на фармакологически эффективном уровне в течение продолжительного периода времени, предпочтительно в течение по меньшей мере 12 ч (за счет его контролируемого или регулируемого высвобождения), и при этом должна обладать лишь минимально возможным спектром побочных действий, к которым относятся прежде всего тошнота и/или рвота. Помимо этого фармакокинетические характеристики подобной лекарственной формы должны по многим аспектам отличаться от фармакокинетических характеристик сравнительной лекарственной формы без контролируемого высвобождения действующего вещества (раствор действующего вещества, жидкая лекарственная форма, немедленное высвобождение (англ. "immediate release")).
Указанная задача решается с помощью объектов, заявленных в формуле изобретения.
При создании изобретения неожиданно было установлено, что возможно приготовление лекарственной формы с 3-(2-диметиламинометилциклогексил)фенолом или одной из его фармацевтически приемлемых солей в качестве действующего вещества, которая контролируемо высвобождает его и при этом обладает преимуществами перед известными из уровня техники лекарственными формами.
В соответствии с этим в изобретении предлагается лекарственная форма для контролируемого высвобождения 3-(2-диметиламинометилциклогексил)фенола, предпочтительно (1R,2R)-3-(2-диметиламинометилциклогексил)фенола, или одной из его фармацевтически приемлемых солей в качестве действующего вещества, которая
(I) in vivo обеспечивает достижение максимального уровня действующего вещества в плазме через 2-10 ч и/или
(II) in vitro при измерении в соответствии с Европейской фармакопеей с использованием аппарата с плосколопастной мешалкой в буфере со значением рН 6,8 (предпочтительно 900 мл) при температуре 37°С и при скорости вращения мешалки 75 об/мин
- через 0,5 ч высвобождает от 3,0 до 37 мас.%,
- через 1 ч высвобождает от 5,0 до 56 мас.%,
- через 2 ч высвобождает от 10 до 77 мас.%,
- через 3 ч высвобождает от 15 до 88 мас.%,
- через 6 ч высвобождает по меньшей мере 30 мас.%,
- через 12 ч высвобождает по меньшей мере 50 мас.%,
- через 18 ч высвобождает по меньшей мере 70 мас.% и
- через 24 ч высвобождает по меньшей мере 80 мас.%
первоначально содержащегося в ней действующего вещества.
В приведенной ниже таблице представлены наиболее предпочтительные профили высвобождения действующего вещества из предлагаемых в изобретении лекарственных форм (№1-8):
Предлагаемая в изобретении лекарственная форма, предпочтительно после ее приема внутрь, замедленно высвобождает действующее вещество - 3-(2-диметиламинометилциклогексил)фенол - и поэтому пригодна для приема с по меньшей мере 12-часовыми интервалами. В соответствии с этим предлагаемая в изобретении лекарственная форма может использоваться для обезболивающего лечения, при котором действующее вещество - 3-(2-диметиламинометилциклогексил)фенол для поддержания его концентрации в плазме на достаточном уровне требуется вводить в организм один раз в сутки, например с 24-часовыми интервалами, или два раза в сутки, предпочтительно с 12-часовыми интервалами.
При создании изобретения при этом неожиданно было установлено, что по сравнению с традиционными лекарственными формами, предназначенными для перорального введения 3-(2-диметиламинометилциклогексил)фенола в их составе, удается существенно уменьшить побочные действия, прежде всего тошноту и/или рвоту. Связанное с этим преимущество состоит в увеличении широты терапевтического действия 3-(2-диметиламинометилциклогексил)-фенола (соотношение между терапевтической и токсической дозами действующего вещества), что позволяет помимо прочего увеличить дозировку действующего вещества, а тем самым и повысить его терапевтическую эффективность.
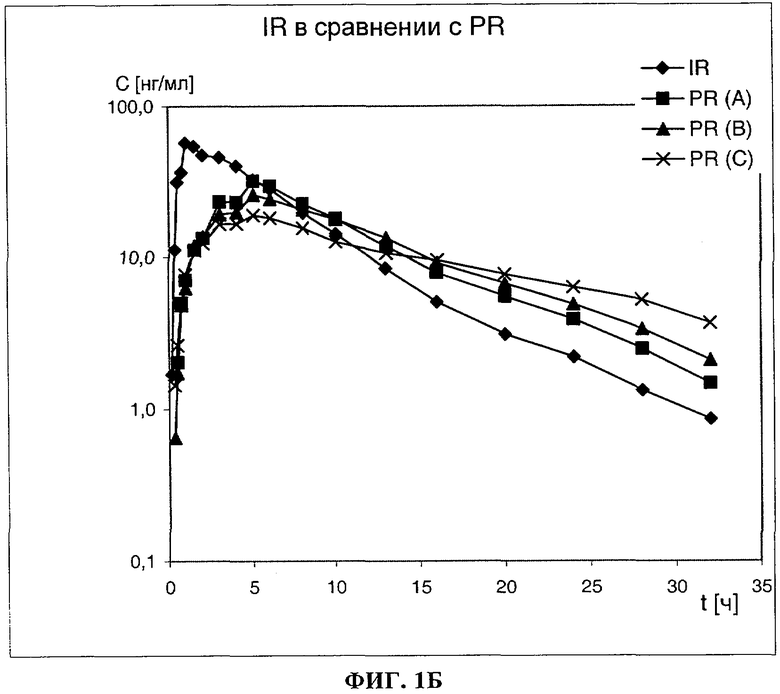
На фиг.1 показаны графики изменения средней концентрации 3-(2-диметиламинометилциклогексил)фенола в плазме после приема внутрь содержащих его предлагаемых в изобретении лекарственных форм PR (A), PR (В) и PR (С) с контролируемым высвобождением действующего вещества в сравнении с традиционной лекарственной формой (сравнительная лекарственная форма без контролируемого высвобождения действующего вещества, раствор действующего вещества, жидкая лекарственная форма, IR от англ. "immediate release", немедленное высвобождение). На фиг.1А значения по ординате нанесены в линейном, а на фиг.1Б - в логарифмическом масштабе.
При применении традиционной лекарственной формы без контролируемого высвобождения действующего вещества (немедленное высвобождение IR, раствор действующего вещества, жидкая лекарственная форма) обычно следует исходить из того, что характер изменения концентрации действующего вещества в плазме во времени достаточно быстро после ее приема в основном определяется только кинетикой превращения действующего вещества в ходе обмена веществ и выведения действующего вещества из организма. Эта кинетика различна у разных действующих веществ (определяется постоянной действующего вещества), однако практически не зависит от технологии лекарственной формы.
В отличие от этого при применении традиционной лекарственной формы с контролируемым высвобождением действующего вещества обычно следует исходить из того, что технология лекарственной формы влияет на характер изменения концентрации действующего вещества в плазме во времени до тех пор, пока действующее вещество все еще продолжает высвобождаться из лекарственной формы. В течение этого промежутка времени кинетика до поступления действующего вещества из лекарственной формы и кинетика превращения уже высвободившегося действующего вещества в ходе обмена веществ и его выведения из организма накладываются друг на друга. Однако в некоторый момент времени высвобождение действующего вещества из лекарственной формы достигает фазы, в которой поступление действующего вещества в организм практически полностью прекращается. По достижении этого момента времени кинетика до поступления действующего вещества из лекарственной формы в крайнем случае оказывает еще лишь незначительное влияние на характер изменения концентрации действующего вещества во времени. Более того, в этой фазе характер изменения концентрации действующего вещества во времени в основном все еще определяется кинетикой превращения действующего вещества в ходе обмена веществ и его выведения из организма, в соответствии с чем в течение этого интервала времени кривая, отражающая изменение концентрации действующего вещества в плазме во времени, по своей форме приближается к такой же кривой, характерной для лекарственных форм без контролируемого высвобождения действующего вещества.
Однако из приведенных на фиг.1 графиков следует, что иногда даже по истечении многих часов после приема предлагаемых в изобретении лекарственных форм (PR (A), PR (В) и PR (С)) соответствующие им кривые изменения концентрации действующего вещества в плазме во времени не приближаются к кривой, отражающей изменение концентрации действующего вещества в плазме во времени после приема сравнительной лекарственной формы без контролируемого высвобождения действующего вещества (раствор действующего вещества, жидкая лекарственная форма, IR). Как следует прежде всего из приведенных на фиг.1Б диаграмм, у предлагаемых в изобретении лекарственных форм с контролируемым высвобождением действующего вещества (PR (A), PR (В) и PR (С)) наклон кривых, отражающих изменение концентрации действующего вещества в плазме во времени, в их конечной части даже по истечении многих часов после приема лекарственных форм явно отличается от наклона аналогичной кривой, характеризующей сравнительную лекарственную форму без контролируемого высвобождения действующего вещества (конечные участки кривых проходят не параллельно, а в виде семейства кривых).
В конечной фазе выведения (элиминации) на кривой изменения концентрации действующего вещества в плазме во времени при логарифмической регрессии угловой коэффициент нисходящих прямых регрессии численно равен постоянной скорости выведения (элиминации) λz. Поэтому наличие у предлагаемой в изобретении лекарственной формы с контролируемым высвобождением действующего вещества неожиданных свойств в сравнении с обычной лекарственной формой без контролируемого высвобождения действующего вещества можно обнаружить по показателю λz, соответственно по пропорциональному ему времени полувыведения.
Преимущество, связанное с наличием у предлагаемой в изобретении лекарственной формы подобных неожиданных свойств, состоит в том, что она in vivo все еще сохраняет эффект замедленного высвобождения действующего вещества, обеспечивая поддержание его концентрации в плазме на определенном уровне, даже в те моменты времени, в которые на основании данных о высвобождении действующего вещества in vitro уже невозможно было бы ожидать значительного замедления высвобождения действующего вещества. В соответствии с этим эффект замедленного высвобождения действующего вещества in vivo (по данным измерения концентрации действующего вещества в плазме) гораздо более выражен по сравнению с замедлением высвобождения действующего вещества in vitro (по данным измерения показателей высвобождения действующего вещества) и носит прогрессивный характер. Тем самым возрастает ожидаемая продолжительность действия действующего вещества, и дополнительно улучшается его переносимость пациентом по сравнению с ожидаемой ситуацией без этого эффекта in vivo.
Согласно изобретению под "контролируемым" высвобождением действующего вещества в контексте настоящего описания может подразумеваться отсроченное или задержанное высвобождение (англ. "extended release", "delayed release"), ступенчатое или поэтапное высвобождение (англ. "repeat action release"), пролонгированное высвобождение (англ. "prolonged release") или равномерно пролонгированное высвобождение (англ. "sustained release"). Немедленное же высвобождение действующего вещества (англ. "immediate release", IR), происходящее, например, при применении раствора действующего вещества, не должно трактоваться как контролируемое высвобождение действующего вещества в указанном выше смысле.
В одном из предпочтительных вариантов предлагаемая в изобретении лекарственная форма содержит полимерную матрицу, которая обеспечивает замедленное высвобождение по меньшей мере части от всего количества содержащегося в лекарственной форме действующего вещества (замедление высвобождения действующего вещества с помощью матрицы). Для этого по меньшей мере эта часть от всего количества действующего вещества, предпочтительно (1R,2R)-3-(2-диметиламинометилциклогексил)фенола или одной из его фармацевтически приемлемых солей, должна присутствовать во внедренном в подобную полимерную матрицу виде.
В другом предпочтительном варианте предлагаемая в изобретении лекарственная форма имеет пленочное покрытие, которое обеспечивает замедленное высвобождение по меньшей мере части от всего количества содержащегося в лекарственной форме действующего вещества (замедление высвобождения действующего вещества с помощью покрытия).
Помимо этого можно также комбинировать между собой замедление высвобождения действующего вещества с помощью матрицы и с помощью покрытия. Согласно изобретению более предпочтительно, однако, обеспечивать замедленное высвобождение действующего вещества в основном только с помощью полимерной матрицы.
В одном из предпочтительных вариантов предлагаемая в изобретении лекарственная форма содержит полимерную матрицу, которая в предпочтительном варианте образована одним или несколькими гидрофильными или гидрофобными фармацевтически приемлемыми полимерами, такими, например, как простые эфиры целлюлозы, сложные эфиры целлюлозы, полиэтиленгликоли (ПЭГ), камеди, (мет-)акрилаты, производные белков, жиры, воски, жирные спирты и/или эфиры жирных кислот. При использовании гидрофильных полимеров в качестве образующих матрицу материалов массовая доля полимерной матрицы предпочтительно должна составлять от 5,0 до 85 мас.%, более предпочтительно от 20 до 60 мас.%, в пересчете на общую массу предлагаемой в изобретении лекарственной формы.
Предлагаемая в изобретении лекарственная форма в предпочтительном варианте содержит полимерную матрицу, в которую внедрена по меньшей мере часть от всего предусмотренного количества действующего вещества, предпочтительно (1R,2R)-3-(2-диметиламинометилциклогексил)фенола или одной из его фармацевтически приемлемых солей, и которая представляет собой полимерную матрицу на основе простого и/или сложного эфира целлюлозы, который в концентрации 2,0 мас.% в водном растворе при 20°С имеет вязкость, предпочтительно определяемую путем капиллярной вискозиметрии в соответствии с Европейской фармакопеей, в пределах от 3000 до 150000 мПа·с, предпочтительно от 5000 до 145000 мПа·с, более предпочтительно от 10000 до 140000 мПа·с, особенно предпочтительно от 25000 до 135000 мПа·с, наиболее предпочтительно от 50000 до 130000 мПа·с, прежде всего от 80000 до 120000 мПа·с.
В одном из предпочтительных вариантов полимерная матрица в предлагаемой в изобретении лекарственной форме содержит по меньшей мере один простой и/или сложный эфир целлюлозы, выбранный из группы, включающей метилцеллюлозу (МЦ), этилцеллюлозу (ЭЦ), гидроксиэтилцеллюлозу (ГЭЦ), гидроксипропилцеллюлозу (ГПЦ), карбоксиметилцеллюлозу (КМЦ) и гидроксипропилметилцеллюлозу (ГПМЦ). Особенно предпочтительны ГПМЦ с вязкостью примерно 100000 мПа·с, измеренной в 2%-ном по массе водном растворе при 20°С. Вместо простых и/или сложных эфиров целлюлозы или в дополнение к ним в полимерной матрице могут также содержаться полиэтиленгликоли (ПЭГ) в количестве от 0 до 60 мас.%.
Массовая доля полимерной матрицы предпочтительно должна составлять от 5,0 до 85 мас.%, более предпочтительно от 10 до 50 мас.%, особенно предпочтительно от 25 до 45 мас.%, в пересчете на общую массу лекарственной формы. При определении массовой доли полимеров в предлагаемой в изобретении лекарственной форме предпочтительно при этом учитывать только те из них, которые образуют матрицу, в которую внедрена по меньшей мере часть от всего предусмотренного количества действующего вещества. Обычные же полимерные вспомогательные вещества, такие, например, как микрокристаллическая целлюлоза, не включаются в массовую долю полимеров в предлагаемой в изобретении лекарственной форме, если они практически не участвуют в образовании матрицы.
В одном из предпочтительных вариантов массовая доля действующего вещества, предпочтительно (1R,2R)-3-(2-диметиламинометилциклогексил)фенола или одной из его фармацевтически приемлемых солей, в предлагаемой в изобретении лекарственной форме составляет от 0,5 до 85 мас.%, более предпочтительно от 5,0 до 50 мас.%, особенно предпочтительно от 15 до 35 мас.%, в пересчете на общую массу лекарственной формы.
Содержание действующего вещества в предлагаемых в изобретении лекарственных формах предпочтительно должно составлять от 0,5 до 85 мас.%, а содержание полимерной матрицы - от 8,0 до 40 мас.%. Особенно предпочтительны лекарственные формы с содержанием действующего вещества от 3,0 до 70 мас.%, прежде всего от 8,0 до 66 мас.%, и с содержанием полимерной матрицы от 10 до 35 мас.%, прежде всего от 10 до 30 мас.%, в пересчете на общую массу фармацевтической композиции.
Относительное массовое соотношение между полимерной матрицей и действующим веществом, предпочтительно (1R,2R)-3-(2-диметиламинометилциклогексил)фенолом или одной из его фармацевтически приемлемых солей, предпочтительно должно составлять от 3:1 до 1:10, более предпочтительно от 2,5:1 до 1:8, особенно предпочтительно от 2,2:1 до 1:5, наиболее предпочтительно от 2:1 до 1:2.
В одном из особенно предпочтительных вариантов полимерная матрица в предлагаемой в изобретении лекарственной форме содержит гидроксипропилметилцеллюлозу, которая в концентрации 2,0 мас.% в водном растворе при 20°С имеет вязкость, предпочтительно определяемую путем капиллярной вискозиметрии в соответствии с Европейской фармакопеей, в пределах от 50000 до 130.000 мПа·с и массовая доля которой одновременно составляет от 15 до 35 мас.% в пересчете на общую массу лекарственной формы.
В предлагаемых в изобретении лекарственных формах могут далее в качестве других их компонентов содержаться также обычно используемые в фармацевтике вспомогательные вещества, к которым относятся, например, наполнители, в частности лактоза, микрокристаллическая целлюлоза (МЦС) или гидрофосфат кальция, и/или скользящие вещества, смазывающие вещества и регуляторы текучести, в частности тальк, стеарат магния, стеариновая кислота и/или высокодисперсный диоксид кремния, и общее содержание которых в таблетке предпочтительно должно составлять от 0 до 80 мас.%, более предпочтительно от 5,0 до 65 мас.%.
Подобные вспомогательные вещества известны специалистам в данной области. Дополнительную информацию о вспомогательных веществах можно найти, например, в публикации Н.Р.Fiedler, Lexikon der Hilfsstoffe für Pharmazie, Kosmetik und angrenzende technische Gebiete, изд-во Editio Cantor Aulendorff, 2002, которая полностью включена в настоящее описание в качестве ссылки.
В предпочтительном варианте предлагаемая в изобретении лекарственная форма содержит по меньшей мере один наполнитель при относительном массовом соотношении между ним или суммарно всеми наполнителями и полимерной матрицей менее 6:1, более предпочтительно от 5:1 до 1:2, особенно предпочтительно от 4:1 до 1:1,5, наиболее предпочтительно от 3:1 до 1:1.
В одном из предпочтительных вариантов предлагаемая в изобретении лекарственная формы содержит наполнитель, выбранный из группы, включающей растворимые в водной среде наполнители, например лактозу, нерастворимые в водной среде ненабухающие наполнители, например гидрофосфат кальция, и нерастворимые в водной среде набухающие наполнители, например микрокристаллическую целлюлозу.
Предпочтительно наполнитель выбирать из группы, включающей микрокристаллическую целлюлозу, гидрофосфат кальция и лактозу.
Профиль высвобождения действующего вещества, предпочтительно (1R,2R)-3-(2-диметиламинометилциклогексил)фенола или одной из его фармацевтически приемлемых солей, из предлагаемой в изобретении лекарственной формы предпочтительно не зависит от значения pH, которое может встречаться в физиологических условиях при прохождении лекарственной формы через желудочно-кишечный тракт. Характеристики высвобождения действующего вещества из предлагаемой в изобретении лекарственной формы предпочтительно должны быть в основном идентичны между собой не только при значениях рН окружающей среды, равных 1,2 и 6,8, но и при высвобождении действующего вещества на протяжении всего времени изменения значения рН с 1,2 вплоть до 7,2 через промежуточные значения 2,3 и 6,8.
Предлагаемая в изобретении лекарственная форма содержит в качестве действующего вещества 3-(2-диметиламинометилциклогексил)фенол или одну из его фармацевтически приемлемых солей. Такое действующее вещество может быть при этом представлено в виде смеси двух или более его стереоизомеров (энантиомеров и/или диастереомеров). В предлагаемой в изобретении лекарственной форме 3-(2-диметиламинометилциклогексил)фенол может присутствовать в виде смеси всех четырех его диастереомеров в любом их соотношении между собой, но также может присутствовать в виде смеси двух или трех из четырех его стереоизомеров либо в виде одного из его чистых стереоизомеров. В одном из предпочтительных вариантов действующее вещество представлено в виде рацемической смеси из пары (1R,2R)/(1S,2S)энантиомеров и предпочтительно не содержит ни (1R,2S)-, ни (1S,2R)диастереомер либо содержит их в относительном массовом количестве менее 2,0 мас.% в пересчете на общую массу действующего вещества.
К фармацевтически приемлемым солям действующего вещества согласно настоящему изобретению относятся те его соли, которые физиологически совместимы при их фармацевтическом применении, прежде всего при их введении в организм млекопитающих и/или человека. Подобные фармацевтически приемлемые соли могут быть образованы, например, с неорганическими или органическими кислотами. В качестве примера солей с неорганическими кислотами можно назвать гидрохлориды, гидробромиды, сульфаты, фосфаты, гидрофосфаты и дигидрофосфаты. В качестве примера солей с органическими кислотами можно назвать формиаты, ацетаты, пропионаты, фумараты, глутараты, пируваты, малаты, тартраты, бензоаты, цитраты, аскорбаты, малеаты и другие.
В одном из особенно предпочтительных вариантов предлагаемая в изобретении лекарственная форма содержит стереоизомер (1R,2R)-3-(2-диметиламинометилциклогексил)фенол или одну из его фармацевтически приемлемых солей в энантиомерном избытке (англ. "enantiomeric excess") предпочтительно по меньшей мере 90% ее, более предпочтительно по меньшей мере 95% ее, особенно предпочтительно по меньшей мере 97% ее, прежде всего по меньшей мере 98% ее.
Действующее вещество 3-(2-диметиламинометилциклогексил)фенол может содержаться в предлагаемой в изобретении лекарственной форме как таковое, т.е. в виде основания, но также в виде одной из его фармацевтически приемлемых солей, например, в виде гидрохлорида. Получение гидрохлоридов этого соединения известно, например, из DE 19525137. Из уровня техники известны способы, позволяющие переводить гидрохлорид в свободное основание или в иную фармацевтически приемлемую соль. Из уровня техники достаточно хорошо известны также методы разделения энантиомеров, соответственно диастереомеров. Так, например, диастереомеры можно разделять посредством ЖХВД, а энантиомеры можно разделять посредством ЖХВД на хиральных неподвижных фазах.
Предлагаемая в изобретении лекарственная форма наряду с 3-(2-диметиламинометилциклогексил)фенолом, соответственно одной из его фармацевтически приемлемых солей может также содержать другие фармацевтически активные вещества. В предпочтительном же варианте предлагаемая в изобретении лекарственная форма содержит только 3-(2-диметиламинометилциклогексил)фенол, предпочтительно (1R,2R)-3-(2-диметиламинометилциклогексил)фенол или одну из его фармацевтически приемлемых солей, а в остальном не содержит никаких иных фармацевтически активных веществ.
Предпочтительные предлагаемые в изобретении лекарственные формы (№1-5) содержат следующие компоненты в указанных ниже количествах (данные о содержании каждого из компонентов в процентах указаны в пересчете на общую массу лекарственной формы):
В качестве других компонентов предлагаемая в изобретении лекарственная форма может дополнительно содержать необязательно перевариваемые незамещенные или замещенные углеводороды с длинной цепью (т.е. с 8-50 атомами углерода, предпочтительно с 12-40 атомами углерода), такие, например, как жирные спирты, глицериловые эфиры жирных кислот, минеральные и растительные масла, а также воски, среди которых предпочтительны углеводороды с температурой плавления от 25 до 90°С.Особенно предпочтительны жирные спирты, а наиболее предпочтительны лауриловый спирт, миристиловый спирт, стеариловый спирт, цетиловый спирт и цетилстеариловый спирт. Их содержание в лекарственной форме предпочтительно должно составлять от 0 до 20 мас.%.
Предлагаемая в изобретении лекарственная форма характеризуется предпочтительными фармакокинетическими параметрами.
В настоящем описании используются следующие фармакокинетические параметры, которые можно определить исходя из концентрации 3-(2-диметиламинометилциклогексил)фенола в плазме крови:
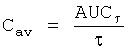
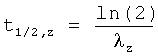
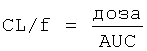
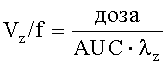
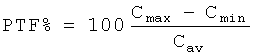
Значение каждого из указанных выше фармакокинетических параметров выражают в виде среднего значения, рассчитываемого путем усреднения по отдельным значениям, полученным для всех исследуемых пациентов/испытуемых.
Специалистам в данной области хорошо известно, каким образом можно рассчитать фармакокинетические параметры для 3-(2-диметиламинометил-циклогексил)фенола исходя из измеренной концентрации этого действующего вещества в плазме крови. В этом отношении можно сослаться, например, на публикацию "Parameters for Compartment-free Pharmacokinetics", под ред. Cawello Willi, изд-во Shaker Verlag, Aachen, 1999.
В предпочтительном варианте усредненный максимальный уровень действующего вещества в плазме (Cmax) in vivo, предпочтительно после приема внутрь предлагаемой в изобретении лекарственной формы, достигается в среднем по истечении времени tmax, составляющего от 2 до 10 ч, более предпочтительно от 3 до 8 ч, особенно предпочтительно от 3,5 до 6 ч, наиболее предпочтительно от 4,0 до 5,5 ч, прежде всего от 4,2 до 5,2 ч.
В предпочтительном варианте среднее значение параметра MRT in vivo, предпочтительно после приема внутрь предлагаемой в изобретении лекарственной формы, превышает 7,5 ч, более предпочтительно превышает 8,0 ч, особенно предпочтительно превышает 9,0 ч, наиболее предпочтительно составляет от 10,0 до 25,0 ч, прежде всего от 11,0 до 20,0 ч.
В предпочтительном варианте среднее значение параметра HVD in vivo, предпочтительно после приема внутрь предлагаемой в изобретении лекарственной формы, превышает 5,0 ч, более предпочтительно превышает 6,0 ч, особенно предпочтительно превышает 7,0 ч, наиболее предпочтительно составляет от 8,0 до 20,0 ч, прежде всего от 9,0 до 18,0 ч.
В предпочтительном варианте среднее значение параметра λz меньше, чем у сравнительной лекарственной формы без контролируемого высвобождения действующего вещества, при одной и той же его дозе. В предпочтительном варианте среднее значение параметра λz in vivo, предпочтительно после приема внутрь предлагаемой в изобретении лекарственной формы, составляет менее 0,125 ч-1, более предпочтительно менее 0,122 ч-1, особенно предпочтительно менее 0,118 ч-1, наиболее предпочтительно составляет от 0,050 до 0,115 ч-1, прежде всего от 0,060 до 0,112 ч-1.
В предпочтительном варианте среднее значение параметра t1/2,z больше, чем у сравнительной лекарственной формы без контролируемого высвобождения действующего вещества, при одной и той же его дозе. В предпочтительном варианте среднее значение параметра t1/2,z in vivo, предпочтительно после приема внутрь предлагаемой в изобретении лекарственной формы, превышает 5,7 ч, более предпочтительно превышает 6,0 ч, особенно предпочтительно превышает 6,2 ч, наиболее предпочтительно составляет от 6,4 до 20,0 ч, прежде всего от 6,6 до 15,0 ч.
Под "сравнительной лекарственной формой без контролируемого высвобождения действующего вещества" в контексте настоящего описания подразумевается лекарственная форма с немедленным высвобождением действующего вещества, например, жидкая лекарственная форма, в частности раствор действующего вещества или дисперсия действующего вещества, содержащая его в такой же дозировке. В одном из предпочтительных вариантов под сравнительной лекарственной формой подразумевается раствор действующего вещества, который содержит следующие компоненты из расчета на 1 мл его объема:
В другом предпочтительном варианте под "сравнительной лекарственной формой без контролируемого высвобождения действующего вещества" подразумевается лекарственная композиция в капсулах, содержащая в качестве вспомогательных веществ микрокристаллическую целлюлозу, гидроксипропилцеллюлозу с низкой степенью замещения, стеарат магния и диоксид кремния, предпочтительно лекарственная композиция следующего состава:
Общая масса содержимого капсулы, предпочтительно твердожелатиновой капсулы размера №0el, составляет 360 мг.
В предпочтительном варианте при применении действующего вещества в дозе D среднее значение отношения Cmax/D in vivo, предпочтительно после приема внутрь предлагаемой в изобретении лекарственной формы, лежит в пределах
7,0·10-5 л-1 ≤Cmax/D≤1,05·10-3 л-1,
более предпочтительно в пределах
8,0·10-5 л-1 ≤Cmax/D≤1,0·10-3 л-1,
особенно предпочтительно в пределах
9,0·10-5 л-1 ≤Cmax/D≤9,0·10-4 л-1,
наиболее предпочтительно в пределах
1,0·10-4 л-1 ≤Cmax/D≤8,0·10-4 л-1,
прежде всего в пределах
2,0·10-4 л-1 ≤Cmax/D≤7,0·10-4 l-1.
В предпочтительном варианте среднее значение отношения Cmax/AUC in vivo, предпочтительно после приема внутрь предлагаемой в изобретении лекарственной формы, составляет от 0,150 до 0,010 ч-1, более предпочтительно от 0,125 до 0,020 ч-1, особенно предпочтительно от 0,100 до 0,030 ч-1, наиболее предпочтительно от 0,095 до 0,040 ч-1, прежде всего от 0,090 до 0,050 ч-1.
Отношение Cmax/AUC можно трактовать как параметр, используемый вместо скорости всасывания.
В предпочтительном варианте среднее значение параметра PTF при двукратном в сутки применении лекарственной формы, предпочтительно с интервалом в 12 ч, составляет менее 80%, более предпочтительно не более 75%, особенно предпочтительно не более 70%, наиболее предпочтительно не более 65%, прежде всего не более 60%.
Предлагаемая в изобретении лекарственная форма содержит в качестве действующего вещества 3-(2-диметиламинометилциклогексил)фенол, предпочтительно (1R,2R)-3-(2-диметиламинометилциклогексил)фенол, как таковой и/или в виде его фармацевтически приемлемой соли в количестве, которое из расчета на одну дозированную единицу обычно составляет от 2,5 до 800 мг, прежде всего от 5 до 400 мг, особенно предпочтительно от 10 до 250 мг (количество в миллиграммах указано в пересчете на 3-(2-диметиламинометилциклогексил)фенол в виде гидрохлорида), при этом то или иное конкретное количество действующего вещества, при условии, что его содержание в лекарственной форме остается в указанных выше пределах, практически не влияет на характеристики высвобождения действующего вещества из предлагаемой в изобретении лекарственной формы.
В одном из предпочтительных вариантов предлагаемая в изобретении лекарственная форма предназначена для перорального или ректального применения, предпочтительно один или два раза в сутки. При применении пациентом предлагаемой в изобретении лекарственной формы один или два раза в сутки надежно достигается высокая терапевтическая эффективность при длительных сильных болях.
Предлагаемые в изобретении лекарственные формы могут быть представлены в виде простых таблеток, а также в виде таблеток с покрытием, например в виде таблеток с пленочным покрытием (филм-таблеток) или драже. Обычно таблетки имеют круглую (шарообразную) или двояковыпуклую форму, но могут также иметь продолговатую форму, которая позволяет при необходимости делить таблетку на две или более частей. Возможны также лекарственные формы в виде гранулятов, сфероидов, крупинок либо пеллетов или микрокапсул, расфасовываемых в пакеты-саше или в капсулы или спрессовываемых в распадающиеся таблетки.
Дополнительно к замедляющей высвобождение действующего вещества матрице или вместо нее в предлагаемой в изобретении лекарственной форме можно также использовать замедляющее высвобождение действующего вещества покрытие. При этом содержащиеся в лекарственной форме действующее вещество, которое предпочтительно, но не обязательно внедрять в полимерную матрицу, и возможно используемые другие фармацевтические вспомогательные вещества, такие, например, как связующие, наполнители, скользящие вещества, смазывающие вещества и регуляторы текучести, покрывают материалом, который регулирует и/или модулирует замедленное высвобождение действующего вещества в водной среде. В качестве примера пригодных для нанесения таких покрытий материалов можно назвать нерастворимые в воде воски и полимеры, такие как полиметакрилаты (эудрагит или иные аналогичные полимеры) либо нерастворимые в воде целлюлозы, прежде всего этилцеллюлозу. При необходимости в материале покрытия могут также содержаться водорастворимые полимеры, такие как поливинилпирролидон, водорастворимые целлюлозы, такие как гидроксипропилметилцеллюлоза или гидроксипропилцеллюлоза, иные водорастворимые вещества, такие как полисорбат 80, или гидрофильные порообразователи, такие как полиэтиленгликоль, лактоза или маннит.
В лекарственных формах, предпочтительно таблетках, с покрытием оно может быть одно- или многослойным. Для применения в качестве материалов таких покрытий пригодны известные гидроксипропилметилцеллюлозы с низкой вязкостью от примерно 1,0 до 100 мПа·с и низкой молекулярной массой менее 10000 г/моль (например, материал Pharmacoat 606 с вязкостью 6,0 мПа·с, измеренной в 2,0%-ном по массе водном растворе при 20°С), которые практически не влияют или лишь незначительно влияют на профиль высвобождения действующего вещества из предлагаемых в изобретении лекарственных средств (лекарственных форм).
Известные специалистам в данной области диффузионные покрытия, например, на основе набухающих, но нерастворимых в воде поли(мет)акрилатов, приводят к модуляции замедления высвобождения действующего вещества из предлагаемых в изобретении лекарственных форм. Содержащую действующее вещество, предпочтительно замедленно высвобождающую его сердцевину, содержание в которой действующего вещества предпочтительно составляет от 0,5 до 85 мас.%, особенно предпочтительно от 3,0 до 70 мас.%, наиболее предпочтительно от 8,0 до 66 мас.%, можно различными, известными специалистам в данной области методами, например дражированием, распылением из растворов или суспензий либо нанесением порошковых покрытий, покрывать оболочкой из дополнительного действующего вещества, которое высвобождается не замедленно, а быстро в начальной дозе, хотя в принципе для замедленного высвобождения действующего вещества при одновременном быстром его поступлении в кровь в целях быстрого утоления боли при первом применении предлагаемой в изобретении лекарственной формы нанесение такого покрытия и не является строго обязательным условием.
В других вариантах предлагаемая в изобретении лекарственная форма может представлять собой многослойную или заключенную в оболочку таблетку, при этом в случае многослойной таблетки с содержанием действующего вещества предпочтительно от 0,5 до 85 мас.%, особенно предпочтительно от 3,0 до 70 мас.%, наиболее предпочтительно от 8,0 до 66 мас.%, действующее вещество замедленно высвобождается полимерной матрицей из одного или нескольких слоев многослойной таблетки, соответственно в случае таблетки в оболочке с содержанием действующего вещества предпочтительно от 0,5 до 85 мас.%, особенно предпочтительно от 3,0 до 70 мас.%, наиболее предпочтительно от 8,0 до 66 мас.%, действующее вещество замедленно высвобождается полимерной матрицей из сердцевины таблетки в оболочке, тогда как высвобождение действующего вещества из одного или нескольких других слоев многослойной таблетки, соответственно из наружного слоя оболочки заключенной в нее таблетки происходит без замедления. Многослойные и заключенные в оболочку таблетки могут также иметь одно или несколько покрытий без действующего вещества.
Предлагаемые в изобретении лекарственные формы можно получать, например, следующим общим способом.
После отмеривания взвешиванием необходимых количеств компонентов лекарственной формы (действующего вещества, полимеров для образования полимерной матрицы [образующих матрицу материалов] и других возможно используемых компонентов) их просеивают на обычной просеивающей машине. Для этого можно использовать, например, просеивающую машину Quadro Comil U10, обычно с размером отверстий сита примерно 0,813 мм. Затем просеянные материалы смешивают между собой в контейнерном смесителе, например в контейнерном смесителе Bohle, при следующих типичных рабочих условиях: продолжительность смешения примерно 15 мин ±45 с при скорости вращения 20±1 об/мин. После этого из полученной порошковой смеси в таблеточном прессе прессуют таблетки. Для этого можно использовать, например, таблеточный пресс Korsch EKO, оснащенный круглым пуансоном с вогнутой по форме драже рабочей поверхностью и с диаметром 10 мм. В другом варианте порошковую смесь можно также сначала уплотнять, а затем просеивать полученные таким путем спрессованные изделия (сначала через протирочное сито Comil с размером отверстий 3 мм, а затем через сито с круглыми отверстиями размером 1,2 мм), после чего из полученного в результате гранулята можно с добавлением смазывающего вещества (например, стеарата магния) прессовать описанным выше путем таблетки, например, на таблеточном прессе EK0, оснащенном круглыми пуансонами диаметром 10 мм. Гранулят можно также получать путем мокрой грануляции, используя водные или органические растворители, предпочтительно водный растворитель с добавкой приемлемого связующего или без него. Подобный способ получения предлагаемых в изобретении лекарственных форм можно без особого труда хорошо известным из уровня техники путем согласовывать с конкретными требованиями и с необходимым типом лекарственной формы.
Способ получения предлагаемых в изобретении лекарственных форм, содержащих 3-(2-диметиламинометилциклогексил)фенол или одну из его фармацевтически приемлемых солей, позволяет получать такие лекарственные формы с высокой воспроизводимостью характеристик высвобождения из них действующего вещества. Профиль высвобождения действующего вещества из предлагаемых в изобретении лекарственных форм остается неизменным после их хранения в течение по меньшей мере одного года в обычных условиях согласно рекомендациям ICH Q1AR Stability Testing Guideline.
Следующим объектом изобретения является фармацевтическая композиция, включающая в качестве действующего вещества 3-(2-диметиламинометилцикло-гексил)фенол или одну из его фармацевтически приемлемых солей, предпочтительно (1R,2R)-3-(2-диметиламинометилциклогексил)фенол как таковой и/или в виде его фармацевтически приемлемой соли, и простой или сложный эфир целлюлозы, который в концентрации 2,0 мас.% в водном растворе при 20°С имеет вязкость в пределах от 3000 до 150000 мПа·с.
Предлагаемая в изобретении композиция пригодна для получения из нее предлагаемой в изобретении лекарственной формы.
В одном из предпочтительных вариантов простой, соответственно сложный эфир целлюлозы в предлагаемой в изобретении композиции выбран из группы, включающей метилцеллюлозу, этилцеллюлозу, гидроксиэтилцеллюлозу, гидроксипропилцеллюлозу, карбоксиметилцеллюлозу и гидроксипропилметилцеллюлозу.
Другие предпочтительные простые, соответственно сложные эфиры целлюлозы рассмотрены выше в описании предлагаемой в изобретении лекарственной формы и соответственно могут также использоваться в предлагаемой в изобретении композиции.
Следующим объектом изобретения является применение 3-(2-диметиламинометилциклогексил)фенола или одной из его фармацевтически приемлемых солей для получения описанной выше лекарственной формы или приготовления описанной выше композиции, предназначенной для борьбы с болью. В предпочтительном варианте боль, которая может быть слабой, умеренной, сильной или особо сильной, выбрана из группы, включающей острую боль и хроническую боль, прежде всего боль при воспалении или невропатическую боль.
В предпочтительном варианте борьба с болью сопровождается при этом значительным по сравнению с лекарственной формой без контролируемого высвобождения действующего вещества уменьшением побочных действий, к которым относятся тошнота и/или рвота. Действующее вещество предпочтительно принимать внутрь.
Следующим объектом изобретения является применение 3-(2-диметиламинометилциклогексил)фенола или одной из его фармацевтически приемлемых солей в качестве действующего вещества для приготовления лекарственной формы с контролируемым его высвобождением, предназначенной для борьбы с болью при значительном по сравнению с лекарственной формой без контролируемого высвобождения действующего вещества уменьшении побочных действий, к которым относятся тошнота и/или рвота. Действующее вещество предпочтительно принимать внутрь. В предпочтительном варианте боль выбрана из группы, включающей острую боль и хроническую боль.
Следующим объектом изобретения является способ борьбы с болью, заключающийся в том, что в организм пациента в фармацевтически эффективном количестве вводят 3-(2-диметиламинометилциклогексил)фенол или одну из его фармацевтически приемлемых солей в качестве действующего вещества, максимальный уровень которого в плазме достигается по истечении 2-10 ч. Действующее вещество предпочтительно принимать внутрь. В предпочтительном варианте боль выбрана из группы, включающей острую боль и хроническую боль.
Следующим объектом изобретения является способ борьбы с болью, заключающийся в том, что в организм пациента вводят лекарственную форму с контролируемым высвобождением действующего вещества, в качестве которого она в фармацевтически эффективном количестве содержит 3-(2-диметиламинометилциклогексил)фенол или одну из его фармацевтически приемлемых солей, при значительном по сравнению с лекарственной формой без контролируемого высвобождения действующего вещества уменьшении побочных действий, к которым относятся тошнота и/или рвота. В предпочтительном варианте лекарственная форма с контролируемым высвобождением действующего вещества предназначена для приема внутрь. В предпочтительном варианте боль выбрана из группы, включающей острую боль и хроническую боль.
Ниже настоящее изобретение и предпочтительные варианты его осуществления проиллюстрированы на примерах, которые, однако, не ограничивают объем изобретения.
Пример 1
Описанным ниже способом изготавливали партию из 1000 матричных таблеток указанного в приведенной ниже таблице состава из расчета на одну таблетку.
После отмеривания взвешиванием необходимых количеств всех компонентов их просеивали на просеивающей машине Quadro Comil U10, используя сито с размером отверстий 0,813 мм, после чего просеянные материалы смешивали между собой в контейнерном смесителе (Bohle LM 40) в течение 15 мин ±45 с при скорости вращения 20±1 об/мин и затем из полученной порошковой смеси в эксцентриковом таблеточном прессе Korsch EK0 прессовали выпуклые по форме драже таблетки с диаметром 10 мм, радиусом кривизны выпуклых поверхностей 8 мм и средней массой 240 мг.
Характеристики высвобождения действующего вещества из полученных таким путем таблеток in vitro определяли с использованием аппарата с лопастной мешалкой в соответствии с методом, описанным в Европейской фармакопее, при 75 об/мин в 900 мл буфера с pH 6,8 согласно Европейской фармакопее и при 37°С с обнаружением путем ультрафиолетовой спектрометрии, получив приведенные ниже в таблице результаты.
Пример 2
Аналогично описанному в примере 1 способу изготавливали матричные таблетки указанного в приведенной ниже таблице состава из расчета на одну таблетку.
Характеристики высвобождения действующего вещества из таких таблеток in vitro определяли аналогично примеру 1, получив следующие результаты.
Пример 3
Аналогично описанному в примере 1 способу изготавливали партию из 75 матричных таблеток указанного в приведенной ниже таблице состава из расчета на одну таблетку.
Характеристики высвобождения действующего вещества из таких таблеток in vitro определяли аналогично примеру 1, получив следующие результаты.
Пример 4
Аналогично описанному в примере 1 способу изготавливали партию из 100 матричных таблеток указанного в приведенной ниже таблице состава из расчета на одну таблетку.
Характеристики высвобождения действующего вещества из таких таблеток in vitro определяли при следующих условиях:
(A): аналогично примеру 1;
(Б): с использованием аппарата с лопастной мешалкой в соответствии с методом, описанным в Европейской фармакопее, при 75 об/мин в 900 мл буфера с рН 1,2 согласно фармакопее США 22 и при 37°С с обнаружением путем ультрафиолетовой спектрометрии;
(B): с использованием аппарата с лопастной мешалкой в соответствии с методом, описанным в Европейской фармакопее, при 75 об/мин, при этом с 0-й по 30-ю мин значение рН составляло 1,2, с 30-й по 120-ю мин значение рН составляло 2,3, с 120-й по 180-ю мин значение pH составляло 6,5, а в течение оставшегося времени эксперимента значение pH составляло 7,2.
Результаты, полученные при соответствующих экспериментальных условиях, приведены ниже в таблице.
Пример 5
Аналогично описанному в примере 1 способу изготавливали партию из 100 матричных таблеток указанного в приведенной ниже таблице состава из расчета на одну таблетку.
Таблетки прессовали с разными усилиями прессования, получая таблетки со следующими показателями сопротивления разрушению: (А) 60 Н, (Б) 80 Н, (В) 100 Н, (Г) 150 Н.
Характеристики высвобождения действующего вещества из таких таблеток in vitro определяли аналогично примеру 1, получив следующие результаты.
Пример 6
Аналогично описанному в примере 1 способу изготавливали партию из 200 матричных таблеток указанного в приведенной ниже таблице состава из расчета на одну таблетку.
Характеристики высвобождения действующего вещества из таких таблеток in vitro определяли аналогично примеру 1, получив следующие результаты.
Пример 7
Описанным ниже способом изготавливали партию из 1000 матричных таблеток указанного в приведенной ниже таблице состава из расчета на одну таблетку.
Для этого после отмеривания взвешиванием необходимых количеств всех компонентов таблетки их просеивали через сито с размером отверстий 0,315 мм, смешивали, полученную смесь гранулировали в присутствии воды в смесителе Kenwood, гранулят протирали через сито с размером отверстий 1 мм, сушили в полочном сушильном шкафу при 50°С и высушенный материал прессовали на эксцентриковом таблеточном прессе ЕК0, оснащенном пуансонами с продолговатой формой рабочих поверхностей размером 7×17 мм, получая таблетки массой по 550 мг.
При определении характеристик высвобождения действующего вещества из таких таблеток in vitro получили следующие результаты.
Пример 8
а) Описанным ниже способом изготавливали партию из 1000 таблеток с пленочным покрытием указанного в приведенной ниже таблице состава из расчета на одну таблетку.
Для этого после отмеривания взвешиванием необходимых количеств всех компонентов сердцевины таблетки их просеивали через сито с размером отверстий 0,315 мм, смешивали и из полученной смеси на ротационном таблеточном прессе Fette P1200, оснащенном пуансонами с диаметром вогнутых рабочих поверхностей 10 мм и с радиусом их кривизны 8 мм, прессовали сердцевины таблеток. После этого на сердцевины таблеток (композиции А и В) наносили пленочное покрытие из водной лаковой суспензии (содержание твердой фазы около 29%), состоявшей из гипромеллозы с вязкостью 6 мПа·с, макрогола 6000, пропиленгликоля, талька, диоксида титана и очищенной воды, до увеличения массы каждой таблетки на 12 мг.
При определении характеристик высвобождения действующего вещества из таких таблеток с пленочным покрытием in vitro получили следующие результаты.
Из приведенных выше в таблице данных следует, что в экспериментах in vitro действующее вещество высвобождалось из лекарственных форм PR (A), PR (В) и PR (С) на 80% по истечении примерно 360 мин, 480 мин, соответственно 720 мин.
б) Фармакокинетические параметры трех этих предлагаемых в изобретении лекарственных форм с разными характеристиками высвобождения из них действующего вещества [лекарственные формы с замедленным высвобождением действующего вещества PR (A), PR (В) и PR (С), где сокращение "PR" означает "пролонгированное высвобождение" от англ. "prolonged release"] определяли in vivo проведением открытого рандомизированного четырехэтапного перекрестного исследования в фазе 1 и сравнивали с фармакокинетическими параметрами раствора действующего вещества [лекарственная форма с немедленным высвобождением действующего вещества, англ. "immediate release", сокращенно IR] в качестве сравнительной лекарственной формы. Подобный раствор действующего вещества с немедленным его высвобождением содержал следующие компоненты из расчета на 1 мл его объема:
Раствор приготавливали стандартным методом приготовления инъекционных растворов и разливали в ампулы порциями по 1 мл.
В исследовании принимали участие 8 добровольных испытуемых женского пола, каждой из которых действующее вещество (1R,2R)-3-(2-диметиламинометилциклогексил)фенол вводили в дозе D, равной 60 мг, и концентрацию действующего вещества в плазме измеряли на протяжении 32 ч.
Концентрацию (1R,2R)-3-(2-диметиламинометилциклогексил)фенола в плазме крови количественно оценивали с использованием О-десметилтрамадола (Ml) в качестве внутреннего стандарта. Действующие вещества экстрагировали из проб крови путем жидкостной экстракции трет-бутилметиловым эфиром. Экстракты анализировали жидкостной хроматографией высокого давления (ЖХВД) путем флуорометрического обнаружения. На калибровочных кривых сигналы при концентрации действующего вещества в плазме в интервале от 0,23 до 92 нг/мл имели линейный характер.
Полученные результаты в графическом виде представлены на фиг.1А и 1Б. На фиг.1А графики изменения концентрации действующего вещества в плазме (ордината) показаны в линейном масштабе, а на фиг.1Б - в логарифмическом масштабе.
Фармакокинетические параметры вычисляли путем некомпартментального (бескамерного) анализа с использованием утвержденного программного обеспечения MODUNA, разработанного фирмой Grünenthal GmbH.
Вычисления проводили со всеми имеющимися знаками после запятой без округления.
Для вычисления площади первой части поверхности под кривой на участке от момента введения действующего вещества до момента, в который впервые удается обнаружить присутствие действующего вещества в плазме, значение концентрации Ctdose в зависимости от метода введения действующего вещества в организм экстраполировали до момента введения действующего вещества в организм tdose. При значении tlag больше 0 ч площадь первой части поверхности под кривой вычисляли, начиная с этого значения tlag.
Значение AUC вычисляли путем суммирования площади части поверхности под кривой AUC0-t (площадь под кривой изменения концентрации действующего вещества в плазме во времени, построенной по измеренным значениям концентрации действующего вещества, находящимися выше предела обнаружения) и площади остальной поверхности под кривой 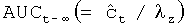 ; где
; где 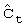 представляет собой определенную на основании логарифмической регрессии концентрацию действующего вещества в плазме в момент, в который в последний раз удается измерить концентрацию действующего вещества в плазме, все еще остающуюся выше предела обнаружения.
представляет собой определенную на основании логарифмической регрессии концентрацию действующего вещества в плазме в момент, в который в последний раз удается измерить концентрацию действующего вещества в плазме, все еще остающуюся выше предела обнаружения.
Значение AUC0-t вычисляли путем численного интегрирования с использованием формулы трапеций (Gibaldi и Perrier, 1982). До достижения значения Cmax использовали формулу трапеций в линейных величинах, а затем - формулу трапеций в логарифмических величинах. При наличии нескольких максимумов концентрации действующего вещества в плазме значение Cmax определяли для первого из них.
Постоянную скорости выведения в конечной фазе выведения (элиминации) λz определяли путем логарифмического регрессионного анализа конечного участка кривых изменения концентрации действующего вещества в плазме во времени (Cawello, 1999). Временные интервалы λz [ч] для регрессионного анализа задавали в соответствии со следующей таблицей:
При определении продолжительности полувыведения HVD путем линейной интерполяции определяли момент, в который концентрация действующего вещества в плазме снижалась на 50% от максимальной Cmax.
Фармакокинетические параметры, вычисленные на основании измеренных значений концентрации действующего вещества в плазме, приведены ниже в таблице (сокращение "KB" означает "коэффициент вариации").
Лекарственные формы с замедленным высвобождением действующего вещества (PR (A), PR (В) и PR (С)), которые в условиях in vitro высвобождают его на 80% по истечении примерно 360 мин, 480 мин, соответственно 720 мин, в исследованиях in vivo на человеке также проявляют возрастающее замедляющее высвобождение действующего вещества действие.
В то время как среднее значение Cmax при применении раствора действующего вещества составило 64,2 нг/мл (сравнительная лекарственная форма с немедленным высвобождением действующего вещества, IR), при применении предлагаемых в изобретении лекарственных форм это значение снижалось до 32,2 нг/мл (лекарственная форма PR(A)), до 26,8 нг/мл (лекарственная форма PR(B)), соответственно до 19,7 нг/мл (лекарственная форма PR(C)).
Среднее значение tmax у всех трех предлагаемых в изобретении лекарственных форм было схожим и составляло от 4,5 до 5,0 ч, однако однозначно превышало тот же показатель, полученный для раствора действующего вещества (1,25 ч).
Средняя продолжительность полувыведения HVD возрастала с 4,78 ч при применении раствора действующего вещества до 9,40 ч (лекарственная форма PR (А)), до 12,05 ч (лекарственная форма PR (В)), соответственно до 15,07 ч (лекарственная форма PR (С)). Подобная тенденция подтверждается результатами определения средней продолжительности пребывания действующего вещества в организме, которая возрастала с 7,28 ч при применении раствора действующего вещества до 19,59 ч (лекарственная форма PR (С)).
Среднее время полувыведения в конечной фазе выведения (элиминации) возрастало с 5,69 ч при применении раствора действующего вещества до 6,68 ч (лекарственная форма PR (А)), до 7,44 ч (лекарственная форма PR (В)), соответственно до 12,10 ч (лекарственная форма PR (С)). Достаточно выраженный эффект замедления высвобождения действующего вещества из предлагаемых в изобретении лекарственных форм в сравнении с лекарственной формой с немедленным высвобождением действующего вещества (раствор действующего вещества, жидкая лекарственная форма, IR) проявляется в уменьшении вдвое концентрации Cmax или в удвоении продолжительности HVD при одновременном увеличении времени t1/2,z, в идеальном случае - без изменения при этом значения AUC.
В одном из исследований отдельные значения PTF% после двукратного в сутки приема предлагаемых в изобретении лекарственных форм лежали в интервале от 49 до 88% при суточной дозе действующего вещества в пределах от 160 до 400 мг. Средние значения этого параметра по отдельным группам, различавшимся между собой дозой действующего вещества, в которой его получали испытуемые, варьировались от 57 до 64%.
в) По результатам клинических исследований было установлено, что по сравнению с традиционными лекарственными формами, предназначенными для перорального введения 3-(2-диметиламинометилциклогексил)фенола в их составе, применение предлагаемых в изобретении лекарственных форм позволяет значительно уменьшить побочные действия, прежде всего тошноту и/или рвоту.
Связанное с этим преимущество состоит помимо прочего в увеличении широты терапевтического действия 3-(2-диметиламинометилциклогексил)фенола (соотношение между терапевтической и токсической дозами действующего вещества).
Лекарственная форма для борьбы с болью и контролируемого высвобождения 3-(2-диметиламинометилциклогексил)фенола или одной из его фармацевтически приемлемых солей включает указанное действующее вещество и полимерную матрицу на основе простого эфира целлюлозы, который в концентрации 2,0 мас.% в водном растворе при 20°С имеет вязкость в пределах от 3000 до 150000 мПа·с. Простой эфир целлюлозы выбран из группы, включающей метилцеллюлозу, этилцеллюлозу, гидроксиэтилцеллюлозу, гидроксипропилцеллюлозу, карбоксиметилцеллюлозу и гидроксипропилметилцеллюлозу. Лекарственная форма in vivo обеспечивает достижение максимального уровня действующего вещества в плазме через 2-10 ч. Лекарственная форма по изобретению обеспечивает поддержание концентрации действующего вещества 3-(2-диметиламинометилциклогексил)фенола в плазме на фармакологически эффективном уровне в течение, по меньшей мере, 12 ч и обладает минимально возможным спектром побочных действий, к которым относятся тошнота и/или рвота. 6 н. и 29 з.п. ф-лы, 1 ил., 8 пр.
1. Лекарственная форма для борьбы с болью и контролируемого высвобождения 3-(2-диметиламинометилциклогексил)фенола или одной из его фармацевтически приемлемых солей в качестве действующего вещества с полимерной матрицей на основе простого эфира целлюлозы, который в концентрации 2,0 мас.% в водном растворе при 20°С имеет вязкость в пределах от 3000 до 150000 мПа·с, причем эта форма in vivo обеспечивает достижение максимального уровня действующего вещества в плазме через 2-10 ч.
2. Лекарственная форма по п.1, которая in vitro при измерении в соответствии с Европейской фармакопеей с использованием аппарата с плосколопастной мешалкой в буфере со значением рН 6,8 при температуре 37°С и при скорости вращения мешалки 75 об/мин
- через 0,5 ч высвобождает от 3,0 до 37 мас.%,
- через 1 ч высвобождает от 5,0 до 56 мас.%,
- через 2 ч высвобождает от 10 до 77 мас.%,
- через 3 ч высвобождает от 15 до 88 мас.%,
- через 6 ч высвобождает по меньшей мере 30 мас.%,
- через 12 ч высвобождает по меньшей мере 50 мас.%,
- через 18 ч высвобождает по меньшей мере 70 мас.% и
- через 24 ч высвобождает по меньшей мере 80 мас.% первоначально содержащегося в ней действующего вещества.
3. Лекарственная форма по п.1 или 2, отличающаяся тем, что отношение Cmax/AUC составляет не менее 0,010 ч-1 и не более 0,150 ч-1.
4. Лекарственная форма по п.1 или 2, отличающаяся тем, что у нее значение параметра t1/2,z больше, чем у сравнительной лекарственной формы без контролируемого высвобождения действующего вещества при одной и той же его дозе.
5. Лекарственная форма по п.1 или 2, отличающаяся тем, что значение параметра t1/2,z составляет более 5,7 ч.
6. Лекарственная форма по п.1 или 2, отличающаяся тем, что значение параметра MRT составляет более 7,5 ч.
7. Лекарственная форма по п.1 или 2, отличающаяся тем, что значение параметра HVD составляет более 5,0 ч.
8. Лекарственная форма по п.1 или 2, отличающаяся тем, что при применении действующего вещества в дозе D значение отношения Cmax/D составляет не менее 7,0·10-5 л-1 и не более 1,05·10-3 л-1.
9. Лекарственная форма по п.1 или 2, отличающаяся тем, что при ее двукратном в сутки применении значение параметра PTF составляет менее 80%.
10. Лекарственная форма по п.1 или 2, отличающаяся тем, что полимерная матрица обеспечивает замедленное высвобождение по меньшей мере части от всего количества содержащегося в лекарственной форме действующего вещества.
11. Лекарственная форма по п.1 или 2, отличающаяся тем, что она имеет пленочное покрытие, которое обеспечивает замедленное высвобождение по меньшей мере части от всего количества содержащегося в лекарственной форме действующего вещества.
12. Лекарственная форма по п.1, отличающаяся тем, что в полимерную матрицу внедрена по меньшей мере часть от всего предусмотренного количества действующего вещества.
13. Лекарственная форма по п.12, отличающаяся тем, что простой эфир целлюлозы выбран из группы, включающей метилцеллюлозу, этилцеллюлозу, гидроксиэтилцеллюлозу, гидроксипропилцеллюлозу, карбоксиметилцеллюлозу и гидроксипропилметилцеллюлозу.
14. Лекарственная форма по п.12, отличающаяся тем, что массовая доля полимерной матрицы составляет от 5,0 до 85 мас.% в пересчете на общую массу лекарственной формы.
15. Лекарственная форма по п.12, отличающаяся тем, что относительное массовое соотношение между полимерной матрицей и действующим веществом составляет от 3:1 до 1:10.
16. Лекарственная форма по п.12, отличающаяся тем, что полимерная матрица содержит гидроксипропилметилцеллюлозу, которая в концентрации 2,0 мас.% в водном растворе при 20°С имеет вязкость в пределах от 50000 до 130000 мПа·с и массовая доля которой составляет от 15 до 35 мас.% в пересчете на общую массу лекарственной формы.
17. Лекарственная форма по п.12, отличающаяся тем, что она содержит наполнитель при относительном массовом соотношении между ним и полимерной матрицей менее 6:1.
18. Лекарственная форма по п.17, отличающаяся тем, что наполнитель выбран из группы, включающей растворимые в водной среде наполнители, не растворимые в водной среде не набухающие наполнители и не растворимые в водной среде набухающие наполнители.
19. Лекарственная форма по п.1 или 2, отличающаяся тем, что действующее вещество представляет собой (1R,2R)-3-(2-диметиламинометилциклогексил)фенол или одну из его фармацевтически приемлемых солей.
20. Лекарственная форма по п.1 или 2, отличающаяся тем, что массовая доля действующего вещества составляет от 0,5 до 85 мас.% в пересчете на общую массу лекарственной формы.
21. Лекарственная форма по п.1 или 2, отличающаяся тем, что максимальный уровень действующего вещества в плазме in vivo достигается по истечении 3-8 ч.
22. Лекарственная форма по п.1 или 2, отличающаяся тем, что она рассчитана на однократное или двукратное в сутки применение.
23. Лекарственная форма по п.1 или 2, отличающаяся тем, что она предназначена для перорального или ректального применения.
24. Лекарственная форма по п.1 или 2, отличающаяся тем, что она представлена в виде таблетки.
25. Фармацевтическая композиция контролируемого высвобождения для борьбы с болью, включающая в качестве действующего вещества 3-(2-диметиламинометилциклогексил)фенол или одну из его фармацевтически приемлемых солей и простой эфир целлюлозы, который в концентрации 2,0 мас.% в водном растворе при 20°С имеет вязкость в пределах от 3000 до 150000 мПа·с.
26. Композиция по п.25, отличающаяся тем, что простой эфир целлюлозы выбран из группы, включающей метилцеллюлозу, этилцеллюлозу, гидроксиэтилцеллюлозу, гидроксипропилцеллюлозу, карбоксиметилцеллюлозу и гидроксипропилметилцеллюлозу.
27. Применение 3-(2-диметиламинометилциклогексил)фенола или одной из его фармацевтически приемлемых солей в качестве действующего вещества для приготовления лекарственной формы по одному из пп.1-24 или композиции по п.25, предназначенной для борьбы с болью.
28. Применение по п.27, отличающееся тем, что борьба с болью сопровождается значительным по сравнению с лекарственной формой без контролируемого высвобождения действующего вещества уменьшением побочных действий, к которым относятся тошнота и/или рвота.
29. Применение по п.27, отличающееся тем, что лекарственную форму принимают внутрь.
30. Применение по п.27, отличающееся тем, что боль выбрана из острой боли и хронической боли.
31. Применение 3-(2-диметиламинометилциклогексил)фенола или одной из его фармацевтически приемлемых солей в качестве действующего вещества для приготовления лекарственной формы с контролируемым его высвобождением по любому из пп.1-24, предназначенной для борьбы с болью при значительном по сравнению с лекарственной формой без контролируемого высвобождения действующего вещества уменьшении побочных действий, к которым относятся тошнота и/или рвота.
32. Способ борьбы с болью, заключающийся в том, что в организм пациента вводят лекарственную форму с контролируемым высвобождением, которая содержит фармацевтически эффективное количество 3-(2-диметиламинометилциклогексил)фенола или одной из его фармацевтически приемлемых солей в качестве действующего вещества, с полимерной матрицей на основе простого эфира целлюлозы, который в концентрации 2,0 мас.% в водном растворе при 20°С имеет вязкость в пределах от 3000 до 150000 мПа·с, при этом максимальный уровень действующего вещества в плазме достигается по истечении 2-10 ч.
33. Способ по п.32, отличающийся тем, что лекарственную форму принимают внутрь.
34. Способ по п.32, отличающийся тем, что боль выбрана из острой боли и хронической боли.
35. Способ борьбы с болью, заключающийся в том, что в организм пациента вводят лекарственную форму с контролируемым высвобождением действующего вещества, в качестве которого она в фармацевтически эффективном количестве содержит 3-(2-диметиламинометилциклогексил)фенол или одну из его фармацевтически приемлемых солей, с полимерной матрицей на основе простого эфира целлюлозы, который в концентрации 2,0 мас.% в водном растворе при 20°С имеет вязкость в пределах от 3000 до 150000 мПа·с, причем эта форма по сравнению с лекарственной формой без контролируемого высвобождения действующего вещества значительно уменьшает побочные действия, к которым относятся тошнота и/или рвота.
| RU 2003127396 А, 20.03.2005 | |||
| US 5601842 А, 11.02.1997 | |||
| DE 4329794 A1, 09.03.1995 | |||
| ЛЕКАРСТВЕННОЕ СРЕДСТВО С ЗАМЕДЛЕННЫМ ВЫСВОБОЖДЕНИЕМ АКТИВНОДЕЙСТВУЮЩЕГО ВЕЩЕСТВА | 1994 |
|
RU2138253C1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| DE 19525137 A1, 16.01.1997 | |||
| 6-ДИМЕТИЛАМИНОМЕТИЛ-1-ФЕНИЛЦИКЛОГЕКСАНОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИ ДЕЙСТВУЮЩИХ ВЕЩЕСТВ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1996 |
|
RU2178409C2 |

