ОБЛАСТЬ ТЕХНИКИ
[0001] Настоящее изобретение относится к соединениям формулы I, ингибирующим PDE2, как описано ниже, к их применению в качестве фармацевтических средств и к фармацевтическим композициям, их содержащим. Эти соединения могут использоваться, например, для лечения PDE2-опосредованных расстройств, таких как тревога, депрессия, расстройство аутистического спектра (ASD), шизофрения и когнитивное расстройство.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
[0002] PDE2 представляет собой гомодимер размером 105 кДа, который экспрессируется в широком спектре тканей и клеточных типов, включая мозг (в том числе гиппокамп, полосатое тело и префронтальную кору), сердце, тромбоциты, эндотелиальные клетки, клетки надпочечников и макрофаги. Хотя цГМФ является предпочтительным субстратом и эффекторной молекулой этого фермента, PDE2 гидролизует как циклический аденозинмонофосфат (цАМФ), так и циклический гуанозинмонофосфат (цГМФ) и, как полагают, участвует в ряде физиологических процессов. В частности, было показано, что ингибирование синтазы оксида азота (NOS), которая уменьшает передачу сигналов цГМФ, ослабляет поведенческие эффекты бензодиазепина хлордиазепоксида, анксиолитического соединения. Кроме того, было показано, что коммерчески доступные ингибиторы PDE2, такие как Bay 60-7550, увеличивают уровни циклического нуклеотида в головном мозге и оказывают значительное противотревожное и антидепрессантное действие у нормальных грызунов и грызунов в условиях стресса (Xu et al., Eur. J. Pharmacol. (2005) 518:40-46; Masood et al., J. Pharmacol. Exp. Ther. (2008) 326:369-379; Masood et al., JPET (2009) 331:690-699; Xu et al., Intl. J. Neuropsychopharmacol. (2013) 16:835-847). Было показано, что ингибирование PDE2 с помощью Bay 60-7550 повышает уровень цГМФ и цАМФ в стимулированных первичных культурах нейронов в дозозависимом режиме; усиливает LTP в срезах гиппокампа в ответ на электрическую стимуляцию; улучшает обучение на модели поведения животных с распознаванием объектов и выполнения задач социального поведения у крыс; улучшает фазы приобретения и закрепления в памяти новых объектов у крыс в возрасте; повышает показатели при выполнении задач по расположению объекта и распознавания при введении после обучения. Gomez et al., Bioorg. Med. Chem. Lett. (2013) 23:6522-6527. Было также показано, что Bay 60-7550 улучшает когнитивную функцию и функцию памяти у крыс за счет усиления активности nNOS в головном мозге. (Domek-Lopacinska et al. (2008) Brain Res. 1216:68-77). Поэтому PDE2 играет важную роль в эффективном поведении и когнитивной функции.
[0003] В дополнение к эффективному поведению и когнитивной функции было обнаружено, что в эндотелиальных клетках мРНК PDR2A и активность сильно индуцированы в ответ на стимуляцию фактором некроза опухоли in vitro. Селективное ингибирование активности PDE2 с помощью 9-(6-фенил-2-оксогекс-3-ил)-2-(3,4-диметоксибензил)пурин-6-она (PDP) значительно изменяет барьерную функцию эндотелиальных клеток, это предполагает, что PDE2, вероятно, играет важную роль в регуляции жидкости и целостности белка в системе кровообращения в патологических условиях. Поэтому PDE2 может быть хорошей фармакологической мишенью при сепсисе или более локализованных воспалительных реакциях.
[0004] В недавнем исследовании также было показано, что ингибирование PDE2 вызывает легочную дилатацию, предотвращает ремоделирование легочных сосудов и уменьшает гипертрофию правого желудочка, характерную для легочной гипертензии, что указывает на возможность применения в терапии ингибирования PDE2 при легочной гипертензии. Bubb et al., «Inhibition of Phosphodiesterase 2 Augments cGMP and cAMP Signaling to Ameliorate Pulmonary Hypertension», Circulation, August 5, 2014, p. 496-507, DOI:10.1161/CIRCULATIONAHA.114.009751.
[0005] Несмотря на многообещающие доклинические данные и идентификацию PDE2 в качестве перспективной лекарственной мишени, в настоящее время ни один ингибитор PDE2 не участвует в клинических исследованиях, отчасти из-за плохих метаболической стабильности и проникновения в головной мозг существующих соединений PDE2. Таким образом, существует потребность в соединениях, которые избирательно ингибируют активность PDE2, демонстрируя превосходные биофизические свойства.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
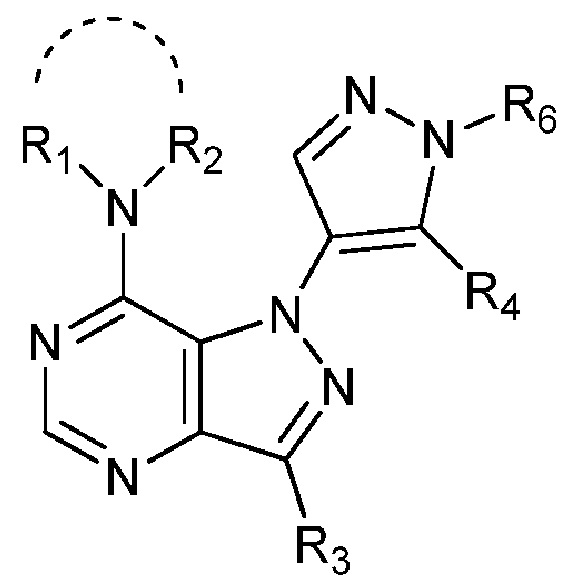
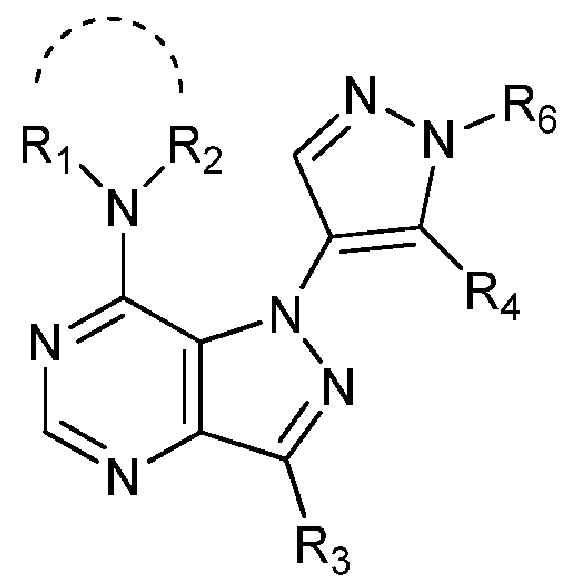
[0006] Изобретение относится к новым соединениям, обладающим сильными и селективными ингибирующими свойствами PDE2 с улучшенной пероральной доступностью и проницаемостью в мозг. Таким образом, в первом аспекте, изобретение относится к соединению формулы I:
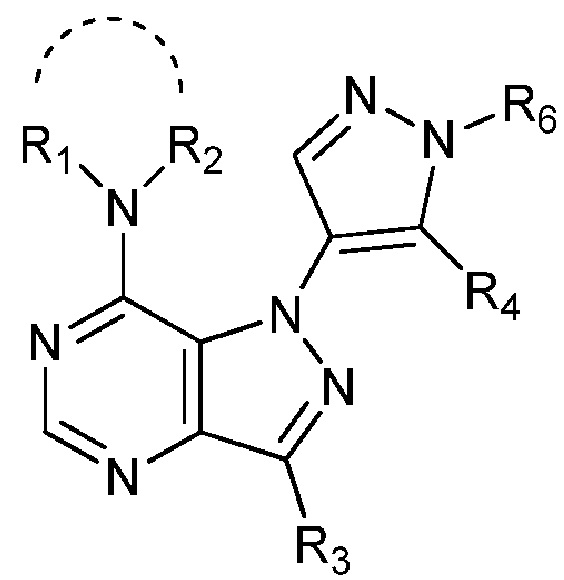
формула I,
где
(i) R1 и R2 вместе с атомом азота образуют гетероC3-7циклоалкил (например, образуют азетидин-1-ил);
(ii) R3 представляет собой H или C1-4алкил (например, метил);
(iii) R4 представляет собой гетероарил или арил (например, фенил) необязательно замещенный одной или несколькими группами, выбранными из C1-4алкила (например, этила), C3-7циклоалкила (например, циклопропила), C1-4алкокси (например, метокси) и галогенC1-4алкила (например, трифторметила);
(iv) R6 представляет собой H или C1-4алкил (например, метил);
в свободной или солевой форме.
[0007] Изобретение относится далее к соединению формула I, как изложено ниже:
1.1 формула I, где R1 и R2 вместе с атомом азота образуют гетероC3-7циклоалкил (например, азетидин-1-ил);
1.2 формула 1.1, где R1 и R2 вместе с атомом азота образуют азетидин-1-ил;
1.3 формула I или любая из 1.1-1.2, где R3 представляет собой H или C1-4алкил (например, метил);
1.4 формула I или любая из 1.1-1.2, где R3 представляет собой C1-4алкил (например, метил);
1.5 формула I или любая из 1.1-1.4, где R4 представляет собой гетероарил или арил (например, фенил) необязательно замещенный одной или несколькими группами, выбранными из C1-4алкила (например, этила), C3-7циклоалкила (например, циклопропила), C1-4алкокси (например, метокси) и галогенC1-4алкила (например, трифторметила);
1.6 формула I или любая из 1.1-1.4, где R4 представляет собой арил (например, фенил) замещенный одной или несколькими группами, выбранными из C1-4алкила (например, этила), C3-7циклоалкила (например, циклопропила), C1-4алкокси (например, метокси) и галогенC1-4алкила (например, трифторметила);
1.7 формула I или любая из 1.1-1.4, где R4 представляет собой арил (например, фенил) замещенный C1-4алкилом (например, этилом);
1.8 формула I или любая из 1.1-1.4, где R4 представляет собой арил (например, фенил) замещенный C3-7циклоалкилом (например, циклопропилом);
1.9 формула I или любая из 1.1-1.4, где R4 представляет собой арил (например, фенил) замещенный C1-4алкокси (например, метокси);
1.10 формула I или любая из 1.1-1.4, где R4 представляет собой арил (например, фенил) замещенный галогеномC1-4алкилом (например, трифторметилом);
1.11 формула I или любая из 1.1-1.10, где R6 представляет собой H или C1-4алкил (например, метил);
1.12 формула I или любая из 1.1-1.10, где R6 представляет собой C1-4алкил (например, метил);
1.13 любая из предшествующих формул, где соединение выбрано из группы, включающей:
7-(азетидин-1-ил)-3-метил-1-(1-метил-5-(4-(трифторметил)фенил)-1H-пиразол-4-ил)-1H-пиразолo[4,3-d]пиримидин;
7-(азетидин-1-ил)-1-(5-(4-метокси-2-метилфенил)-1-метил-1H-пиразол-4-ил)-3-метил-1H-пиразолo[4,3-d]пиримидин;
7-(азетидин-1-ил)-1-(5-(4-циклопропилфенил)-1-метил-1H-пиразол-4-ил)-3-метил-1H-пиразолo[4,3-d]пиримидин;
7-(азетидин-1-ил)-1-(5-(4-этилфенил)-1-метил-1H-пиразол-4-ил)-3-метил-1H-пиразолo[4,3-d]пиримидин;
1.14 любая из предшествующих формул, где соединения ингибируют опосредованный фосфодиэстеразой (например, опосредованный PDE2) гидролиз cGMP, например, с IC50 менее чем 1 мкМ, более предпочтительно, менее чем или равной 250 нМ, более предпочтительно, менее чем или равной 10 нМ в аффинном анализе PDE с иммобилизованными частицами металла, например, как описано в примере 5,
в свободной или солевой форме.
[0008] Во втором аспекте изобретение относится к фармацевтической композиции, содержащей соединение по изобретению, то есть, соединения формулы I или любой из формул 1.1-1.14, в свободном виде или в виде фармацевтически приемлемой соли, в комбинации или ассоциации с фармацевтически приемлемыми разбавителями или носителем.
[0009] Изобретение относится также к способам применения соединений по изобретению для лечения опосредованных PDE2 расстройств, например, расстройств, как изложено ниже (особенно для лечения тревоги, депрессии, расстройства аутистического спектра (ASD), шизофрении, когнитивных нарушений). Этот список не является исчерпывающим и может включать в себя другие заболевания и расстройства, как изложено ниже.
[0010] Таким образом, в третьем аспекте изобретение относится к способу лечения опосредованного PDE2 расстройства, включающего введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по изобретению, описанного в настоящем документе, то есть, соединения формулы I или любой из формул 1.1-1.14, в свободном виде или в виде фармацевтически приемлемой соли, или фармацевтической композиции, описанной в настоящем документе.
[0011] В следующем варианте осуществления третьего аспекта изобретение относится к способу лечения у субъекта, предпочтительно, млекопитающего, предпочтительно, человека, следующих нарушений:
неврологические расстройства (такие как мигрень, эпилепсия, болезнь Альцгеймера, болезнь Паркинсона, травма головного мозга; инсульт; цереброваскулярные заболевания (включая церебральный атеросклероз, церебральную амилоидную ангиопатию, наследственное внутимозговое кровоизлияние, гипоксию и ишемию мозга); спинальная мышечная атрофия; боковой амиотрофический склероз; рассеянный склероз;
когнитивные расстройства (включая амнезию, сенильную деменцию, деменцию, связанную с ВИЧ, деменцию, связанную с болезнью Альцгеймера, деменцию, связанную с болезнью Хантингтона, деменцию с тельцами Леви, сосудистую деменцию, деменцию, связанную с отравлением лекарственными средствами, делирий и умеренное когнитивное расстройство); когнитивная дисфункция, связанная с болезнью Паркинсона и депрессией;
умственная отсталость (включая синдром Дауна и синдром ломкой Х-хромосомы);
расстройства сна (включая гиперсомнию, расстройство циркадного ритма сна, бессонницу, парасомнию и депривацию сна);
психические расстройства (такие как тревожность (включая острое стрессовое расстройство, генерализированное тревожное расстройство, социальное тревожное расстройство, паническое расстройство, посттравматическое стрессовое расстройство (PTSD), обсессивно-компульсивное расстройство, специфическая фобия, социофобия, хроническое тревожное расстройство и обсессивно-компульсивное расстройство личности);
имитируемое расстройство (включая острую галлюцинаторную манию);
расстройства контроля над побуждениями (включая патологическое влечение к азартным играм, патологическое влечение к поджогам, патологическое влечение к воровству и интермиттирующее эксплозивное расстройство);
расстройства настроения (включая биполярное расстройство I типа, биполярное расстройство II типа, маниакальное состояние, смешанное аффективное состояние, большая депрессия, хроническая депрессия, сезонная депрессия, психотическая депрессия и послеродовая депрессия);
психомоторные расстройства (экстрапирамидные и двигательные расстройства, например, паркинсонизм, болезнь телец Леви, тремор, тремор, вызванный лекарственными средствами, поздняя дискинезия, вызванная лекарственными препаратами, Дискинезия, вызванная L-допа и синдром беспокойных ног);
психотические расстройства (включая шизофрению (например, непрерывное или эпизодическое, параноидальное, гебефреническое, кататоническое, недифференцированное и остаточное шизофренические расстройства), шизоаффективное расстройство, шизофреноформное и бредовое расстройство);
лекарственная зависимость (включая наркотическую зависимость, алкоголизм, зависимость от амфетамина, зависимость от кокаина, зависимость от никотина и абстинентный наркотический синдром);
расстройства пищевого поведения (включая анорексию, булимию, расстройство пищевого поведения, гиперфагию и пагофагию);
психические расстройства у детей (включая cиндром дефицита внимания, синдром дефицита внимания/гиперактивности, кондуктивное расстройство (например, тикозные расстройства, такие как временные, хронические, двигательные или голосовые тикозные расстройства), аутизм и расстройство аутического спектра (ASD));
психические и поведенческие расстройства вследствие употребления психоактивных веществ;
сердечно-сосудистое расстройство (например, легочная гипертензия и легочная артериальная гипертензия); и
боль (например, боль в костях и суставах (остеоартрит), повторяющаяся боль при движении, зубная боль, боль, связанная с раковым заболеванием, миофасциальная боль (мышечная травма, фибромиалгия), периоперационная боль (общая хирургия, гинекология), хроническая боль и невропатическая боль),
который включает введение указанному субъекту терапевтически эффективного количества соединения по изобретению, описанного в настоящем документе, то есть, соединения формулы I или любой из формул 1.1-1.14, в свободном виде или в виде фармацевтически приемлемой соли, или фармацевтической композиции, описанной в настоящем документе.
[0012] В одном варианте осуществления заболевание или расстройство выбрано из группы, включающей тревожность, депрессию, расстройство аутического спектра и шизофрению, например, тревожность и/или депрессию у аутичных и/или больных шизофренией. В другом варианте осуществления заболевание или расстройство представляет собой когнитивные нарушения, связанные с шизофренией или деменцией
[0013] В четвертом аспекте изобретение относится к соединению по изобретению, описанному в настоящем документе, то есть, соединениям формулы I или любой из формул 1.1-1.14, в свободном виде или в виде фармацевтически приемлемой соли (для использования при изготовлении лекарственного средства) для лечения расстройства, опосредованного PDE2, как описано в настоящем документе.
[0014] В пятом аспекте изобретение относится к фармацевтической композиции, содержащей соединение по изобретению, описанное в настоящем документе, то есть, соединение формулы I или любой из формул 1.1-1.14, в свободном виде или в виде фармацевтически приемлемой соли в комбинации или ассоциации с фармацевтически приемлемыми разбавителями или носителем для применения при лечении расстройства, опосредованного PDE2, как описано в настоящем документе.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0015] Если не указано иное или ясно из контекста, следующие термины здесь имеют следующие значения:
(a) «Алкил», как используется в настоящем документе, представляет собой насыщенную или ненасыщенную углеводородную группу, предпочтительно, насыщенную, предпочтительно, содержащую от одного до шести атомов углерода, предпочтительно, содержащую от одного до четырех атомов углерода, которая может быть прямой или разветвленной, и которая может необязательно моно-, ди- или три-замещенной, например, галогеном (например, хлором или фтором), гидрокси или карбокси.
(b) «Арил», как используется в настоящем документе, представляет собой моно или бициклический ароматический углеводород, предпочтительно, фенил, необязательно замещенный, например, алкилом (например, метилом), галогеном (например, хлором или фтором), галогеналкилом (например, трифторметилом) или гидрокси.
(c) «Гетероарил», как используется в настоящем документе, представляет собой ароматическую группу где один или несколько атомов, образующих ароматическое кольцо, представляет собой серу или азот вместо углерода, например, пиридил или тиадиазолил, которое необязательно может быть замещено, например, алкилом, галогеном, галогеналкилом или гидрокси.
[0016] Соединения по изобретению, например, соединения формулы I или любой из формул 1.1-1.14 могут существовать в свободной или солевой форме, например, в виде солей добавления кислот. В данном описании, если не указано иное, такую фразу как «соединения по изобретению» следует понимать как охватывающую соединения в любом виде, например, в свободном виде или в виде соли добавления кислоты, или, если соединения содержат кислотные заместители, в виде соли добавления основания. Соединения по изобретению предназначены для использования в качестве фармацевтических препаратов, поэтому предпочтительными являются фармацевтически приемлемые соли. Соли, которые непригодны для фармацевтического применения, могут быть использованы, например, для выделения или очистки соединений по изобретению в свободном виде или в виде их фармацевтически приемлемых солей, поэтому они также включены. Соединения по изобретению в некоторых случаях могут существовать в форме пролекарства. Формой пролекарства является соединение, которое в организме преобразуется в соединение по изобретению. Например, когда соединения по изобретению содержат гидрокси или карбокси заместители, эти заместители могут образовывать физиологически гидролизуемые и приемлемые сложные эфиры. Как используется в настоящем документе, «физиологически гидролизуемый и приемлемый сложный эфир» означает сложные эфиры соединений по изобретению, которые гидролизуются в физиологических условиях с образованием кислот (в случае соединений по изобретению, которые имеют гидрокси заместители) или спиртов (в случае соединений по изобретению, которые имеют карбокси заместители), которые сами по себе являются физиологически переносимыми в дозах, подлежащих введению. Таким образом, когда соединение по изобретению содержит гидрокси группу, например, соединение-OH, то ацильное сложноэфирное пролекарство такого соединения, то есть, соединение-O-C(O)-C1-4алкил, может гидролизоваться в организме с образованием физиологически гидролизуемого спирта (соединение-OH) с одной стороны и кислоты с другой (например, HOC(O)-C1-4алкил). Альтернативно, когда соединение по изобретению содержит группу карбоновой кислоты, например, соединение-C(O)OH, то сложноэфирное пролекарство такого соединения, соединение-C(O)O-C1-4алкил может гидролизоваться с образованием соединения-C(O)OH и HO-C1-4алкила. Как будет понятно, термин, таким образом, охватывает обычные фармацевтические пролекарственные формы.
[0017] Соединения по изобретению в данном документе включают их энантиомеры, диастереоизомеры и рацематы, а также их полиморфы, гидраты, сольваты и комплексы. Некоторые отдельные соединения в рамках настоящего изобретения могут иметь двойные связи. Предполагается, что характерные примеры двойных связей по данному изобретению подразумевают как E, так и Z-изомеры двойной связи. Кроме того, некоторые соединения в рамках настоящего изобретения могут содержать один или несколько асимметрических центров. Данное изобретение включает использование любого из оптически чистых стереоизомеров, а также любую комбинацию стереоизомеров.
[0018] Предполагается также, что соединения по изобретению охватывают их стабильные и нестабильные изотопы. Стабильными изотопами являются нерадиоактивные изотопы, которые содержат один дополнительный нейтрон по сравнению с распространенными нуклидами одного того же вида (то есть, элемента). Ожидается, что активность соединений, содержащих такие изотопы, будет сохранена, и такое соединение также будет полезно для измерения фармакокинетики неизотопных аналогов. Например, атом водорода в определенном положении соединения по изобретению может быть заменен на дейтерий (стабильный изотоп, который не радиоактивен). Примеры известных стабильных изотопов включают, но ими не ограничиваются, дейтерий, 13C, 15N, 18O. Альтернативно, нестабильные изотопы, представляющие собой радиоактивные изотопы, которые содержат дополнительные нейтроны по сравнению с распространенными нуклидами того же вида (то есть, элемента), например, 123I, 131I, 125I, 11C, 18F, могут заменить соответствующие распространенные е виды I, C и F. Другим примером полезного изотопа соединения по изобретению является изотоп 11С. Эти радиоизотопы могут быть использованы для радиоизображений и/или фармакокинетических исследований соединений по изобретению. Изотопно-меченные соединения формулы I обычно могут быть получены путем замены изотопно-немеченного реагента на изотопно-меченный реагент.
[0010] Фраза «соединения по изобретению» или «ингибиторы PDE 2 по изобретению» охватывает любое и все соединения, описанные в настоящем документе, например, соединения формулы I или любой из формул 1.1-1.4, как описано в настоящем документе, в свободной или солевой форме.
[0011] «лечение» и «обработка» следует понимать, соответственно, как подразумевающие лечение или улучшение симптомов заболевания, а также и лечение причины заболевания. В одном варианте осуществления изобретение относится к способу лечения заболевания или расстройства, описанного в настоящем документе. В другом варианте осуществления изобретение относится к способу профилактики заболевания или расстройства, как описано в настоящем документе.
[0012] В случае способов лечения слово «эффективное количество» предназначено для обозначения терапевтически эффективного количества для лечения конкретного заболевания или расстройства.
[0013] Термин «легочная гипертензия» обозначает легочную артериальную гипертензию.
[0014] Термин «субъект» включает человека или не человека (то есть, животное). В конкретном варианте осуществления изобретение охватывает как человека, так и не человека. В другом варианте осуществления изобретение относится к не человеку. В другом варианте осуществления термин относится к человеку.
[0015] Термин «содержащий», используемый в этом описании, предполагается как неограниченный и не исключает дополнительных, не перечисленных элементов или стадий способа.
[0016] Термин «когнитивные расстройства» относится к любому расстройству, включающему симптом когнитивного дефицита (то есть, субнормальное или субоптимальное функционирование в одном или более когнитивных аспектах, таких как память, интеллект, обучение, логика, внимание или способность к целенаправленной деятельности (рабочая память) у человека по сравнению с другими индивидуумами в группе одного и того же возраста). Таким образом, когнитивные расстройства включают, но этим не ограничиваются, амнезию, сенильную деменцию, деменцию, связанную с ВИЧ, деменцию, связанную с болезнью Альцгеймера, деменцию, связанную с болезнью Хантингтона, деменцию с тельцами Леви, сосудистую деменцию, деменцию, связанную с отравлением лекарственными средствами, делирий и умеренное когнитивное расстройство. Когнитивные расстройства также могут быть расстройствами, в первую очередь, но не исключительно, связанными с психозом (шизофрения), расстройствами настроения, биполярными расстройствами, инсультом, лобно-височной деменцией, прогрессирующим супрануклеарным параличом, травмой мозга и злоупотреблением наркотиками, синдромом Аспергера и возрастным нарушением памяти.
[0017] Соединения по изобретению, например, соединения формулы I или любой из формул 1.1-1.14, как описано в настоящем документе, в свободном виде или в виде фармацевтически приемлемой соли могут использоваться в качестве единственного терапевтического агента, но могут также использоваться в комбинации или для совместного введения с другими активными агентами.
[0018] Дозы, используемые в практике настоящего изобретения, будут, разумеется, различаться в зависимости, например, от конкретного заболевания или состояния, подлежащего лечению, конкретного соединения по изобретению, способа введения и желаемой терапии. Соединения по изобретению могут вводиться любым подходящим путем, включая пероральный, парентеральный, трансдермальный или путем ингаляции, но, предпочтительно, вводятся перорально. Как правило, удовлетворительные результаты, например, для лечения заболеваний, как указано выше, представлены для перорального введения в дозах примерно от 0,01 до 2,0 мг/кг. Для более крупных млекопитающих, например, для человека, указанная суточная доза для перорального введения будет, соответственно, находиться в диапазоне от примерно 0,75 до 150 мг, удобно вводить один раз или в разделенных дозах 2-4 раза, ежедневно или в форме пролонгированного высвобождения. Единичные дозированные формы для перорального введения, таким образом, например, могут содержать от примерно 0,2 до 75 или 150 мг, например, от примерно 0,2 или 2,0 до 50, 75 или 100 мг соединения по изобретению вместе с фармацевтически приемлемым разбавителем или носителем.
[0019] Фармацевтические композиции, содержащие соединения по изобретению, могут быть получены с использованием обычных разбавителей или вспомогательных веществ и методов, известных в области получения лекарственных средств. Фармацевтически приемлемый носитель может содержать любой обычный фармацевтический носитель или вспомогательное вещество. Подходящие фармацевтические носители включают инертные разбавители или наполнители, воду и различные органические растворители (такие как гидраты и сольваты). Фармацевтическая композиция может, при желании, содержать дополнительные ингредиенты, такие как ароматизаторы, связующие вещества, вспомогательные вещества и тому подобное. Таким образом, для перорального введения могут использоваться таблетки, содержащие различные вспомогательные вещества, такие как лимонная кислота, вместе с различными разрыхлителями, такими как крахмал, альгиновая кислота и некоторые сложные силикаты, и со связующими веществами, такими как сахароза, желатин и аравийская камедь. Кроме того, для получения таблеток часто могут быть использованы смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк. В мягких и твердых заполненных желатиновых капсулах также могут быть использованы твердые композиции аналогичного типа. Неограничивающие примеры материалов, таким образом, включают лактозу или молочный сахар и полиэтиленгликоли с высокой молекулярной массой. Когда желательны водные суспензии или эликсиры для перорального введения, активное соединение в нем может быть объединено с различными подслащивающими или ароматизирующими агентами, красителями или красителями и, при желании, эмульгаторами или суспендирующими агентами вместе с разбавителями, такими как вода, этанол, пропиленгликоль, глицерин или их комбинации. Фармацевтическая композиция может, например, быть в форме, пригодной для перорального введения, в виде таблетки, капсулы, пилюли, порошка, препарата замедленного высвобождения, раствора или суспензии для парентеральной инъекции в виде стерильного раствора, суспензии или эмульсии для местного введения в виде мази или крема, или для ректального введения в виде суппозитория.
[0020] Соединения по изобретению, описанные в настоящем документе, и их фармацевтически приемлемые соли могут быть получены с использованием способов, как описано в настоящем документе, и приведенных в настоящем документе в качестве примера, и подобных им способов, и способов, известных в области химии. Такие способы включают, но не ограничиваются ими, описанные ниже. Если они не являются коммерчески доступными, исходные вещества для этих способов могут быть получены с помощью способов, которые выбирают из известных в области химии, с использованием способов, которые подобны или аналогичны синтезу известных соединений. Все ссылки, приведенные в настоящем документе, включены посредством ссылки во всей их полноте.
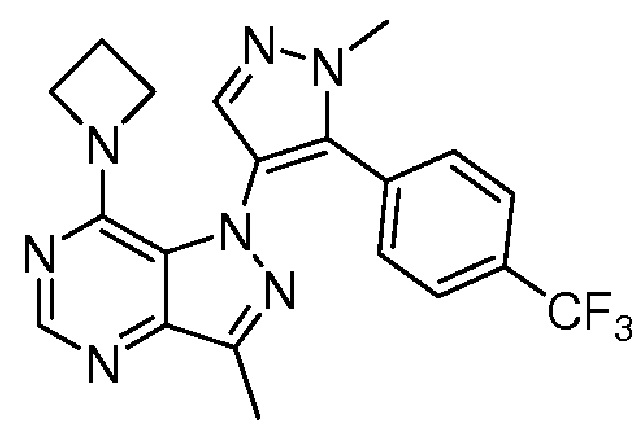
Пример 1
7-(Азетидин-1-ил)-3-метил-1-(1-метил-5-(4-(трифторметил)фенил)-1H-пиразол-4-ил)-1H-пиразолo[4,3-d]пиримидин
(a) 5-Хлор-1-метил-4-нитро-1H-пиразолe
[0021] К раствору 1-метил-4-нитро-1H-пиразола (5,50 г, 43,3 ммоль) и гексахлорэтана (10,54 г, 44,5 ммоль) в метиленхлориде (120 мл) при температуре 25°C по каплям добавляли литий бис(триметилсилил)амид (1,0M, 65 мл, 65 ммоль) в ТГФ. Реакционную смесь перемешивали при температуре 25°C в течение 60 мин, и затем гасили водой (1 мл). Смесь упаривали досуха. Остаток последовательно промывали водой (50 мл), насыщ. NaHCO3 два раза (2×30 мл) и насыщенным солевым раствором (30 мл) и затем сушили в вакууме с получением 6,50 г продукта (93%-ный выход). МС (ESI) m/z 162,0 [M+H]+. 1H ЯМР (500 МГц, CDCl3) δ 8,15 (с, 1H), 3,92 (с, 3H).
(b) Гидрохлорид 5-хлор-1-метил-1H-пиразол-4-амина
[0022] К суспензии 5-хлор-1-метил-4-нитро-1H-пиразола (6,50 г, 40,2 ммоль) в 12н HCl (15 мл) и этаноле (15 мл) добавляли хлорид олова(I) (35,5 г, 160,9 ммоль). Реакционную смесь перемешивали при температуре 90°C до полного завершения реакции. Реакционную смесь упаривали досуха. Остаток обрабатывали 12н HCl (25 мл) и затем охлаждали до температуры 5°C в течение 2 ч. После фильтрования слой на фильтре промывали 6н HCl (2×25 мл) и затем сушили в вакууме с получением 6,08 г продукта (выход: 90%). МС (ESI) 132,0 [M+H]+. 1H ЯМР (500 МГц, CD3OD) δ 7,66 (с, 1H), 3,89 (с, 3H).
(c) Гидрохлорид 5-хлор-4-гидразинил-1-метил-1H-пиразола
[0023] В перемешиваемый раствор гидрохлорида 5-хлор-1-метил-1H-пиразол-4-амина (6,08 г, 36,2 ммоль) в HCl (12н, 35 мл) при температуре 0°C добавляли водный NaNO2 (5,50 г, 80,0 ммоль). Реакционную смесь перемешивали при температуре 0°C в течение 45 мин и затем добавляли хлорид олова(II) (22,8 г, 120 ммоль). После завершения добавления реакционную смесь перемешивали при температуре 0°C в течение 30 мин и затем перемешивали при комнатной температуре в течение ночи. Полученную смесь охлаждали на ледяной бане в течение 2 ч и затем фильтровали. Слой на фильтре промывали HCl (12н, 20 мл) и затем сушили в вакууме с получением 7,77 г сырого продукта (выход: 98%), который использовали в следующей реакции без дополнительной очистки. МС (ESI) m/z 147,0 [M+H]+.
(d) 5-Бром-4-хлор-6-(1-этоксивинил)пиримидин
[0024] Суспензию 5-бром-4,6-дихлорпиримидина (5,00 г, 21,9 ммоль) и трибутил(1-этоксивинил)олова (7,92 г, 21,9 ммоль) в ДМФ (20 мл) дегазировали аргоном и затем добавляли тетракис(трифенилфосфин)палладий(0) (1,27 мг, 1,10 ммоль). Суспензию опять дегазировали и затем нагревали при температуре 110°C в атмосфере аргона в течение 8 ч. После удаления растворителей при пониженном давлении остаток очищали с помощью хроматографии на колонке с силикагелем, элюируя градиентом 0-40% этилацетата в гексане в течение 30 мин с получением 2,72 г продукта (выход: 47%). МС (ESI) m/z 263,0 [M+H]+. 1H ЯМР (500 МГц, CDCl3) δ 8,85 (с, 1H), 4,71 (д, J=3,1 Гц, 1H), 4,60 (д, J=3,1 Гц, 1H), 3,98 (кв, J=7,0 Гц, 2H), 2,67 (с, 0H), 1,42 (т, J=7,0 Гц, 3H).
(e) 5-Бром-4-хлор-6-(1-(2-(5-хлор-1-метил-1H-пиразол-4-ил)гидразоно)этил)пиримидин
[0025] Суспензию 5-бром-4-хлор-6-(1-этоксивинил)пиримидина (1,60 г, 6,07 ммоль) и гидрохлорида 5-хлор-4-гидразинил-1-метил-1H-пиразола (3,11 г, 12,1 ммоль) в уксусной кислоте (32 мл) перемешивали при температуре 60°C в течение 6 ч. После удаления растворителя при пониженном давлении остаток обрабатывали насыщ. NaHCO3 (40 мл) и затем экстрагировали этилацетатом (3×100 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (70 мл) и затем упаривали досуха. Остаток сушили в вакууме с получением 1,0 г сырого продукта (выход: 45%), который использовали на следующей стадии без дополнительной очистки. МС (ESI) m/z 362,9 [M+H]+. 1H ЯМР (500 МГц, CDCl3) δ 8,79 (с, 1H), 7,66 (с, 1H), 7,05 (с, 1H), 3,83 (с, 3H), 2,32 (с, 3H).
(f) 7-Хлор-1-(5-хлор-1-метил-1H-пиразол-4-ил)-3-метил-1H-пиразолo[4,3-d]пиримидин
[0026] Смесь 5-бром-4-хлор-6-(1-(2-(5-хлор-1-метил-1H-пиразол-4-ил)гидразоно)этил)пиримидина (334 мг, 0,92 ммоль), 1,10-фенантролина (497 мг, 2,76 ммоль) и K2CO3 (127 мг, 0,92 ммоль) в толуоле (4 мл) в герметичном сосуде нагревали при температуре 100°C в течение 1,5 ч. После охлаждения до комнатной температуры реакционную смесь разбавляли толуолом (3 мл) и затем фильтровали. Твердое вещество промывали толуолом два раза (2×3 мл). Объединенный фильтрат промывали насыщенным FeSO4·7H2O три раза (3×4 мл) и затем упаривали досуха. Объединенный остаток затем сушили в вакууме с получением 147 мг сырого продукта (выход: 57%), который использовали на следующей стадии без дополнительной очистки. МС (ESI) m/z 283,0 [M+H]+.
(g) 7-(Азетидин-1-ил)-1-(5-хлор-1-метил-1H-пиразол-4-ил)-3-метил-1H-пиразолo[4,3-d]пиримидин
[0027] Смесь 7-хлор-1-(5-хлор-1-метил-1H-пиразол-4-ил)-3-метил-1H-пиразолo[4,3-d]пиримидина (147 мг, 0,52 ммоль), гидрохлорида азетидина (64 мг, 0,68 ммоль) и Et3N (105 мг, 1,04 ммоль) в толуоле (1,5 мл) в герметичном сосуде перемешивали при комнатной температуре до полного завершения реакции. Смесь выливали в 2н NaOH (18 мл) и затем три раза экстрагировали CH2Cl2 (3×25 мл). Объединенную органическую фазу промывали насыщенным солевым раствором (20 мл) и затем упаривали досуха. Объединенный остаток затем сушили в вакууме с получением 181 г сырого продукта, который использовали на следующей стадии без дополнительной очистки. МС (ESI) 304,1 [M+H]+. 1H ЯМР (500 МГц, CDCl3) δ 8,51 (с, 1H), 7,66 (с, 1H), 3,95 (с, 3H), 3,87 (т, J=7,8 Гц, 4H), 2,61 (с, 3H), 2,33-2,25 (м, 2H).
(h) 7-(Азетидин-1-ил)-3-метил-1-(1-метил-5-(4-(трифторметил)фенил)-1H-пиразол-4-ил)-1H-пиразолo[4,3-d]пиримидин
[0028] Суспензию 7-(азетидин-1-ил)-1-(5-хлор-1-метил-1H-пиразол-4-ил)-3-метил-1H-пиразолo[4,3-d]пиримидина (58 мг, 0,19 ммоль), 4-(трифторметил)-фенилбороновой кислоты (51 мг, 0,27 ммоль) и трикалий фосфата (92 мг, 0,43 ммоль) в этаноле (0,70 мл) и воде (0,080 мл) нагревали при температуре 70°C в атмосфере аргона в течение 10 мин и затем добавляли тетракис(трифенилфосфин)палладий(0) (19 мг, 0,017 ммоль). Суспензию опять дегазировали и затем нагревали в микроволновом реакторе при температуре 130°C в течение 2 ч. Добавляли дополнительное количество тетракис(трифенилфосфин)палладия(0) (9 мг, 0,0079 ммоль). Смесь нагревали в микроволновом реакторе при температуре 140°C в течение 7 ч. После удаления растворителей остаток очищали с помощью полу-препаративной ВЭЖХ, используя градиент 0-33% ацетонитрила в воде, содержащей 0,1% муравьиной кислоты, в течение 16 мин, получая 12 мг целевого продукта в виде твердого вещества не совсем белого цвета (чистота по данным ВЭЖХ: 98%; выход: 15%). МС (ESI) m/z 414,2 [M+H]+, 1H ЯМР (500 МГц, CDCl3) δ 8,43 (с, 1H), 7,71 (с, 1H), 7,63 (д, J=8,1 Гц, 2H), 7,50 (д, J=8,1 Гц, 2H), 3,96 (с, 3H), 3,81 (т, J=7,8 Гц, 4H), 2,59 (с, 3H), 2,32-2,21 (м, 2H).
Пример 2
7-(Азетидин-1-ил)-1-(5-(4-метокси-2-метилфенил)-1-метил-1H-пиразол-4-ил)-3-метил-1H-пиразолo[4,3-d]пиримидин
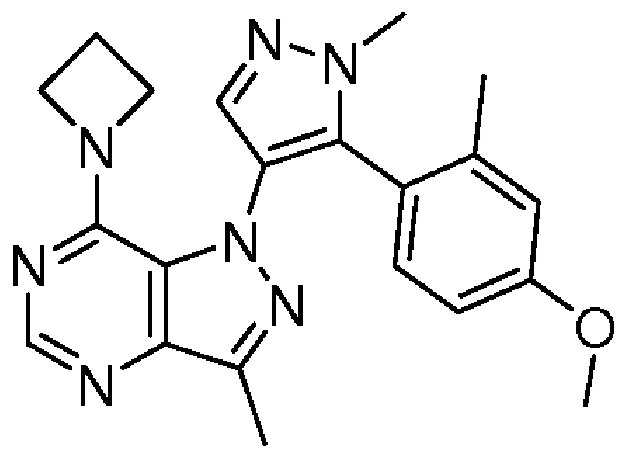
[0029] Указанное в заголовке соединение получали способом, аналогичным способу, описанному для синтеза по примеру 1, где на конечной стадии вместо 4-(трифторметил)фенилбороновой кислоты и K3PO4 добавляли 4-метокси-2-метилфенилбороновую кислоту и K2CO3, соответственно. Конечный продукт получали в виде твердого вещества не совсем белого цвета (чистота по данным ВЭЖХ: 99%; выход: 32%). МС (ESI) m/z 390,2 [M+H]+. 1H ЯМР (500 МГц, CDCl3) δ 8,37 (с, 1H), 7,77 (с, 1H), 7,06 (д, J=8,4 Гц, 1H), 6,68 (д, J=2,5 Гц, 1H), 6,64 (дд, J=8,5, 2,6 Гц, 1H), 4,04-3,82 (м, 4H), 3,74 (с, 3H), 3,71 (с, 3H), 2,55 (с, 3H), 2,35-2,26 (м, 2H), 2,08 (с, 3H).
Пример 3
7-(Азетидин-1-ил)-1-(5-(4-циклопропилфенил)-1-метил-1H-пиразол-4-ил)-3-метил-1H-пиразолo[4,3-d]пиримидин
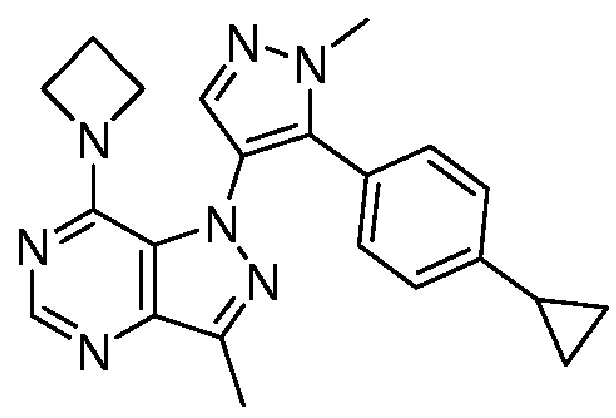
[0030] Указанное в заголовке соединение получали способом, аналогичным способу, описанному для синтеза по примеру 1, где вместо 4-(трифторметил)фенилбороновой кислоты и K3PO4 на конечной стадии добавляли циклопропилфенил-бороновую кислоту и Na2CO3, соответственно. МС (ESI) m/z 386,2 [M+H]+.
Пример 4
7-(Азетидин-1-ил)-1-(5-(4-этилфенил)-1-метил-1H-пиразол-4-ил)-3-метил-1H-пиразолo[4,3-d]пиримидин
[0031] Указанное в заголовке соединение получали способом, аналогичным способу, описанному для синтеза по примеру 1, где на конечной стадии вместо 4-(трифторметил)фенилбороновой кислоты и K3PO4 добавляли 4-этилфенил-бороновую кислоту и Cs2CO3, соответственно. МС (ESI) m/z 374,2 [M+H]+.
ПРИМЕР 5
Измерение ингибирования PDE2 in vitro
[0032] r-hPDE2A (номер доступа NM_002599, фосфодиэстераза 2A Homo sapiens, цГМФ-стимулированная, вариант транскрипции 1) Вектор экспрессии млекопитающих с копией рекомбинантной кДНК гена был приобретен у компании Origene. Белок экспрессировался посредством временной трансфекции клеток HEK293. Клетки собирали через 48 часов после трансфекции, промывали один раз буфером TBS (50 мМ Трис-HCl, pH 7,5, 150 мМ NaCl), затем лизировали ультразвуком в холодном буфере для гомогенизации (50 мМ Трис-HCl, pH 7,5, 5 мМ MgCl2, 1× смесь ингибиторов протеаз). Гомогенат центрифугировали в течение 30 мин при 15000 g при температуре 4°C с получением растворимой цитозольной фракции. Концентрацию белка в цитозоле определяли, используя набор BCA Protein Assay Kit (Pierce) вместе с бычьим сывороточным альбумином в качестве стандарта.
[0033] Анализ: PDE2A анализировали с помощью FL-цАМФ в качестве субстрата. Сначала проводили ферментативное титрование для определения рабочей концентрации PDE. Концентрация фермента, обуславливающая активность 100 ΔмП в отсутствие ингибитора, считается подходящей рабочей концентрацией для PDE.
[0034] Фермент PDE разбавляли в стандартном реакционном буфере (10 мМ Трис-HCl, pH 7,2, 10 мМ MgCl2, 0,1% БСА, 0,05% NaN3) в соответствии с кривой титрования. Для анализа PDE2 реакционный буфер дополняли 1 мкМ цГМФ для полной активации фермента. В каждую лунку добавляли 99 мкл разбавленного раствора фермента в 96-луночном полистирольном планшете с плоским дном и затем добавляли ~1 мкл испытуемого соединения, растворенного в 100% ДМСО. Соединения смешивали и предварительно инкубировали вместе с ферментом в течение 10 мин при комнатной температуре.
[0035] Реакцию конверсии FL-cNMP инициировали, добавляя субстрат (конечная 45 нМ). Смесь ферментов и ингибиторов (16 мкл) и раствор субстрата (4 мкл 0,225 мкМ) объединяли в 384-луночном планшете для микротитрования. Реакционную смесь инкубировали в темноте при комнатной температуре в течение 15 мин. Реакцию останавливали, добавляя в каждую лунку 384-луночного планшета 60 мкл связывающего реагента (гранулы IMAP с разведением 1:400 в связующем буфере с добавлением противовспенивателя с разбавлением 1:1800). Планшет инкубировали при комнатной температуре в течение 1 часа, чтобы обеспечить полное связывание IMAP, а затем помещали в многорежимный планшет-ридер Envision (PerkinElmer, Shelton, CT) для измерения поляризации флуоресценции (Δmp).
[0036] Снижение концентрации цАМФ, измеренное как уменьшение Δmp, указывало на ингибирование активности PDE. Значения IC50 определяли путем измерения активности фермента в присутствии 8-16 концентраций соединения в диапазоне от 0,00037 нМ до 80 000 нМ, а затем, рассчитывая концентрации лекарственного средства по сравнению с ΔmP. Значения тестовых лунок нормализовали с контрольными реакциями, проводимыми на том же планшете (значения, пересчитанные в % от контроля). Значения IC50 оценивали, используя программное обеспечение нелинейной регрессии, подгоняя четырех-параметровую односайтовую модель доза-реакция (XLFit; IDBS, Cambridge, MA). Нижняя часть кривой фиксировали при 0% контроля.
[0037] Контроль качества: Для того, чтобы определить IC50 ингибитора, выбирали концентрацию фермента, которая дает оптимальный диапазон сигналов 100-200 милли-поляризационных единиц. Суммарную интенсивность флуоресценции каждой лунки образца измеряли для расчета среднего и стандартного отклонения. Если общая интенсивность флуоресценции любой лунки образца не находилось в пределах среднего значения ±3SD, то значение mp этой конкретной лунки не учитывалось.
[0038] Используя описанную выше или подобную процедуру IMAP, авторы проверили проприетарную библиотеку соединений, сфокусированную на PDE, для идентификации новых соединений с наномолярной ингибиторной активностью PDE2. Представленные в описании в качестве примера соединения (например, соединения по примерам 1-4) тестировали, и они, как было показано, были активны в наномолярных концентрациях, например, следующим образом: IC50 23,1 нМ; пример 2: IC50 92 нМ; пример 3: IC50 9,5 нМ; пример 4: IC50 18,5 нМ. Соединения, кроме того, являются селективными в отношении PDE2; соединение по примеру 4 тестировали и, как было показано, оно является селективным в отношении PDE2 по сравнению с PDE1, PDE3, PDE4D, PDE5, PDE6, PDE7B, PDE8A, PDE9A, PDE10A и PDE11A более чем в 20 раз.
ПРИМЕР 6
Фармакокинетическое исследование на мышах
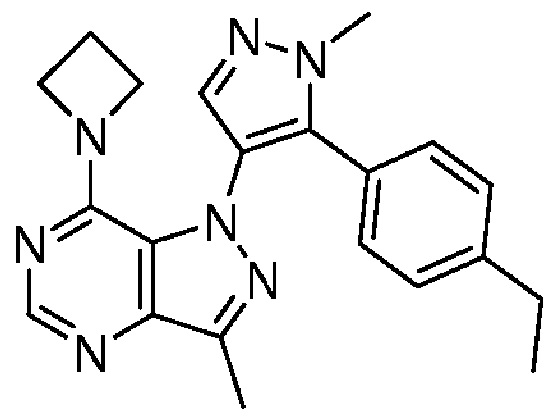
[0039] Мышам давали одну пероральную дозу тестируемого соединения (10 мг/кг, ПО) и измеряли доступность в плазме и головном мозге (0,25-4 ч), используя ВЭЖХ и ЖХ-МС, с применением способов, аналогичных способам, описанным в работах Zhao et al., J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. (2005) 819(1):73-80, и Appels, N.M., et al., Rapid Commun. Mass Spec. 2005. 19(15): p. 2187-92. Эксперимент показал, что соединение по примеру 1 имеет хорошую доступность в головной мозг, как показано на фигуре 1.
ПРИМЕР 7
Эффекты лечения ингибиторами PDE2 на работу мозга крыс Wistar в задаче распознавания объектов
[0040] Эффекты, усиливающие память, соединения по примеру 1 тестировали на когнитивной животной модели, задачей являлось распознавание объекта. смотри, Ennaceur, A., Delacour, J., 1988. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav. Brain Res. 31, 47-59. Крысы помнили объект, который они исследовали в предыдущем тесте при проверке в тесте через час. Однако при 24-часовом промежуточном интервале они не могли вспомнить, какой объект был им показан в первом тесте. Тестировали, будет ли соединение по примеру 1 ослаблять этот дефицит памяти, вызванный временем, при введении через 2 часа перед первым испытании. Соединения вводили 2-3-месячным самцам крыс Wistar.
[0010] Соединение по примеру 1 вводили перорально (2 мл/кг) за 2 ч до начала обучения в дозах 0, 0,3, 1, 3 и 10 мг/кг. Ни одна из проверенных доз не влияла на исследовательское поведение. Настоящее исследование показало, что соединение по примеру 1 способно полностью предотвращать зависящее от времени забывание в дозе 1,0 мг/кг. Улучшение промежуточной памяти обнаружено в случае 0,3 и 3,0 мг/кг.
[0011] ПРИМЕР 8
[0012] Эффекты новых ингибиторов PDE2 на передачу сигналов цГМФ в клетках гиппокампа и в мозге
[0013] Авторами было сделано предположение, что клетки НТ-22 (иммортализованные клетки-предшественники гиппокампа-нейронов мыши, субклонированные из основных клеток НТ-4) являются ценной моделью для понимания клеточных и молекулярных процессов, относящихся к зависимым от гиппокампа эмоциональным изменениям. Авторы предположили, что Aim 1 даст 10-20 новых ингибиторов PDE2 для функциональной оценки в клеточных тестах. Только соединения, которые индуцируют значительное увеличение накопления цГМФ в клеточных тестах клеток HT-22, будут улучшать поведенческую оценку. Данные клеточного анализа будут минимальными требованиями для любого ингибитора PDE2 с вероятным использованием для показаний ЦНС и будут служить руководством для выбора дозы при поведенческом тестировании.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2015 |
|
RU2702732C1 |
| КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ | 2013 |
|
RU2657540C2 |
| ТЕТРАГИДРО-ПИРАЗОЛО[1,5-a]ПИРИДО-ПИРИМИДИНЫ - АНТАГОНИСТЫ СЕРОТОНИНОВЫХ 5-HT РЕЦЕПТОРОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2008 |
|
RU2391343C1 |
| СОЕДИНЕНИЯ И СПОСОБЫ | 2015 |
|
RU2711442C2 |
| ИНГИБИТОРЫ КАТЕХОЛ-О-МЕТИЛТРАНСФЕРАЗЫ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ ПСИХИЧЕСКИХ РАССТРОЙСТВ | 2011 |
|
RU2572624C2 |
| ЗАМЕЩЕННЫЕ 2-АЛКИЛСУЛЬФАНИЛ-3-СУЛЬФОНИЛ-ПИРАЗОЛО[1,5-а]-ПИРИМИДИНЫ, АНТАГОНИСТЫ СЕРОТОНИНОВЫХ 5-HT РЕЦЕПТОРОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2008 |
|
RU2393159C1 |
| ЗАМЕЩЕННЫЕ 3-АРИЛСУЛЬФОНИЛ-ПИРАЗОЛО[1,5-а]ПИРИМИДИНЫ, АНТАГОНИСТЫ СЕРОТОНИНОВЫХ 5-HT РЕЦЕПТОРОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2008 |
|
RU2393158C1 |
| ЗАМЕЩЕННЫЕ ЦИКЛОАЛКАНО[е ИЛИ d]ПИРАЗОЛО[1,5-а]ПИРИМИДИНЫ - АНТАГОНИСТЫ СЕРОТОНИНОВЫХ 5-НТ РЕЦЕПТОРОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2008 |
|
RU2374249C1 |
| Гетероароматические соединения и их применение в качестве допаминовых D1 лигандов | 2013 |
|
RU2617842C2 |
| 2-АЛКИЛАМИНО-3-АРИЛСУЛЬФОНИЛЦИКЛОАЛКАНО[e ИЛИ d]ПИРАЗОЛО[1,5-а]ПИРИМИДИНЫ - АНТАГОНИСТЫ СЕРОТОНИНОВЫХ 5-НТ РЕЦЕПТОРОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2008 |
|
RU2377244C1 |
Изобретение относится к новому соединению формулы I, обладающему свойствами ингибитора PDE2. Соединение может найти применение при лечении заболевания, расстройства или состояния опосредованного PDE2, таких как тревожность, депрессия, расстройство аутического спектра, шизофрения, тревожность и/или депрессия у аутичных и/или больных шизофренией, и когнитивные нарушения, связанные с шизофренией или деменцией и др. В формуле I
(i) R1 и R2 вместе с атомом азота образуют азетидин-1-ил; (ii) R3 представляет собой H или C1-4алкил; (iii) R4 представляет собой фенил, необязательно замещенный одной или несколькими группами, выбранными из C1-4алкила, C1-4алкокси и галогенC1-4алкила; (iv) R6 представляет собой H или C1-4алкил. Соединение может быть в свободной форме или в виде фармацевтически приемлемой соли. Предпочтительными соединениями являются соединения, выбранные из группы, включающей 7-(азетидин-1-ил)-3-метил-1-(1-метил-5-(4-(трифторметил)фенил)-1H-пиразол-4-ил)-1H-пиразоло[4,3-d]пиримидин; 7-(азетидин-1-ил)-1-(5-(4-метокси-2-метилфенил)-1-метил-1H-пиразол-4-ил)-3-метил-1H-пиразоло[4,3-d]пиримидин; 7-(азетидин-1-ил)-1-(5-(4-циклопропилфенил)-1-метил-1H-пиразол-4-ил)-3-метил-1H-пиразоло[4,3-d]пиримидин; 7-(азетидин-1-ил)-1-(5-(4-этилфенил)-1-метил-1H-пиразол-4-ил)-3-метил-1H-пиразоло[4,3-d]пиримидин. 5 н. и 5 з.п. ф-лы, 1 ил., 8 пр.
1. Соединение формулы I
Формула I
где
(i) R1 и R2 вместе с атомом азота образуют азетидин-1-ил;
(ii) R3 представляет собой H или C1-4алкил;
(iii) R4 представляет собой фенил, необязательно замещенный одной или несколькими группами, выбранными из C1-4алкила, C1-4алкокси и галогенC1-4алкила;
(iv) R6 представляет собой H или C1-4алкил;
в свободной форме или в виде фармацевтически приемлемой соли.
2. Соединение по п. 1, где R6 представляет собой C1-4алкил, в свободной форме или в виде фармацевтически приемлемой соли.
3. Соединение по п. 1 или 2, где соединение выбрано из группы, включающей:
7-(азетидин-1-ил)-3-метил-1-(1-метил-5-(4-(трифторметил)фенил)-1H-пиразол-4-ил)-1H-пиразоло[4,3-d]пиримидин;
7-(азетидин-1-ил)-1-(5-(4-метокси-2-метилфенил)-1-метил-1H-пиразол-4-ил)-3-метил-1H-пиразоло[4,3-d]пиримидин;
7-(азетидин-1-ил)-1-(5-(4-циклопропилфенил)-1-метил-1H-пиразол-4-ил)-3-метил-1H-пиразоло[4,3-d]пиримидин;
7-(азетидин-1-ил)-1-(5-(4-этилфенил)-1-метил-1H-пиразол-4-ил)-3-метил-1H-пиразоло[4,3-d]пиримидин;
в свободной форме или в виде фармацевтически приемлемой соли.
4. Фармацевтическая композиция, обладающая свойствами ингибировать PDE2, содержащая эффективное количество соединения по любому из пп. 1-3, в комбинации или ассоциации с фармацевтически приемлемыми разбавителями или носителем.
5. Способ лечения расстройства, опосредованного PDE2, включающий введение субъекту эффективного количества соединения по любому из пп. 1-3 или фармацевтической композиции по п. 4.
6. Способ по п. 5, где расстройство выбрано из группы, включающей неврологические расстройства (такие как мигрень, эпилепсия, болезнь Альцгеймера, болезнь Паркинсона, травма головного мозга; инсульт; цереброваскулярные заболевания (включая церебральный атеросклероз, церебральную амилоидную ангиопатию, наследственное внутримозговое кровоизлияние, гипоксию и ишемию мозга); спинальная мышечная атрофия; боковой амиотрофический склероз; рассеянный склероз; когнитивные расстройства (включая амнезию, сенильную деменцию, деменцию, связанную с ВИЧ, деменцию, связанную с болезнью Альцгеймера, деменцию, связанную с болезнью Хантингтона, деменцию с тельцами Леви, сосудистую деменцию, деменцию, связанную с отравлением лекарственными средствами, делирий и умеренное когнитивное расстройство); когнитивная дисфункция, связанная с болезнью Паркинсона и депрессией; умственная отсталость (включая синдром Дауна и синдром ломкой Х-хромосомы); расстройства сна (включая гиперсомнию, расстройство циркадного ритма сна, бессонницу, парасомнию и депривацию сна); психические расстройства (такие как тревожность (включая острое стрессовое расстройство, генерализированное тревожное расстройство, социальное тревожное расстройство, паническое расстройство, посттравматическое стрессовое расстройство (PTSD), обсессивно-компульсивное расстройство, специфическая фобия, социофобия, хроническое тревожное расстройство и обсессивно-компульсивное расстройство личности); имитируемое расстройство (включая острую галлюцинаторную манию); расстройства контроля над побуждениями (включая патологическое влечение к азартным играм, патологическое влечение к поджогам, патологическое влечение к воровству и интермиттирующее эксплозивное расстройство); расстройства настроения (включая биполярное расстройство I типа, биполярное расстройство II типа, маниакальное состояние, смешанное аффективное состояние, большая депрессия, хроническая депрессия, сезонная депрессия, психотическая депрессия и послеродовая депрессия); психомоторные расстройства (экстрапирамидные и двигательные расстройства, например, паркинсонизм, болезнь телец Леви, тремор, тремор, вызванный лекарственными средствами, поздняя дискинезия, вызванная лекарственными препаратами, дискинезия, вызванная L-допа, и синдром беспокойных ног); психотические расстройства (включая шизофрению (например, непрерывное или эпизодическое, параноидальное, гебефреническое, кататоническое, недифференцированное и остаточное шизофренические расстройства), шизоаффективное расстройство, шизофреноформное и бредовое расстройство); лекарственная зависимость (включая наркотическую зависимость, алкоголизм, зависимость от амфетамина, зависимость от кокаина, зависимость от никотина и абстинентный наркотический синдром); расстройства пищевого поведения (включая анорексию, булимию, расстройство пищевого поведения, гиперфагию и пагофагию); психические расстройства у детей (включая синдром дефицита внимания, синдром дефицита внимания/гиперактивности, кондуктивное расстройство (например, тикозные расстройства, такие как временные, хронические, двигательные или голосовые тикозные расстройства), аутизм и расстройство аутического спектра (ASD)); психические и поведенческие расстройства вследствие употребления психоактивных веществ; сердечно-сосудистое расстройство (например, легочная гипертензия и легочная артериальная гипертензия); и боль (например, боль в костях и суставах (остеоартрит), повторяющаяся боль при движении, зубная боль, боль, связанная с раковым заболеванием, миофасциальная боль (мышечная травма, фибромиалгия), периоперационная боль (общая хирургия, гинекология), хроническая боль и невропатическая боль).
7. Способ по п. 6, где расстройство выбрано из следующих: тревожность, депрессия, расстройство аутического спектра, шизофрения, тревожность и/или депрессия у аутичных и/или больных шизофренией и когнитивные нарушения, связанные с шизофренией или деменцией.
8. Соединение по любому из пп. 1 или 2, в свободной форме или в виде фармацевтически приемлемой соли или фармацевтическая композиция по п. 4, для лечения или профилактического лечения заболевания, расстройства или состояния, опосредованного активностью PDE2, в соответствии с любым из пп. 5-7.
9. Применение фармацевтической композиции, содержащей соединение по любому из пп. 1-3, в свободной форме или в виде фармацевтически приемлемой соли, для изготовления лекарственного средства для лечения или профилактического лечения заболевания, расстройства или состояния в соответствии с любым из пп. 5-7.
10. Фармацевтическая композиция, содержащая соединение по любому из пп. 1-3, в свободной форме или в виде фармацевтически приемлемой соли, для применения в качестве лекарственного средства для лечения заболевания, расстройства или состояния, опосредованного активностью PDE2.
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| WO 2005083069 A1, 09.09.2005 | |||
| Приспособление к разливным машинам системы „Энцингер" для предохранения от несчастных случаев рабочих | 1927 |
|
SU13968A1 |

