ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая международная заявка испрашивает приоритет по ранее поданным предварительным заявкам США 62/051735 от 17 сентября 2014 и 62/052283 от 18 сентября 2014, каждая из которых включена в настоящий документ в виде ссылки во всей своей полноте.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001] Область изобретения в основном касается соединений и способов лечения и/или профилактики заболеваний, расстройств и/или повреждений центральной нервной системы (ЦНС). В одном аспекте область касается ингибиторов фосфодиэстеразы 1 (PDE1) как нейропротекторных агентов и/или нейрорегенеративных агентов. Еще в одном аспекте область изобретения касается предотвращения развития заболевания или расстройства ЦНС у индивидуума с риском развития заболевания или расстройства ЦНС.
УРОВЕНЬ ТЕХНИКИ
[0002] Фосфодиэстеразы циклических нуклеотидов (PDE) понижающе регулируют внутриклеточную cAMP- и cGMP-передачу сигналов посредством гидролиза этих циклических нуклеотидов до их соответствующих 5ʹ-монофосфатов (5ʹAMP и 5ʹGMP). Идентифицировано одиннадцать семейств фосфодиэстераз, но только PDE в семействе I, Ca2+/кальмодулин-зависимые фосфодиэстеразы (CaM-PDE), которые активируются Ca2+-кальмодулином, как показано, опосредуют пути передачи сигналов через кальций и циклический нуклеотид (например, cAMP и cGMP). Три известных CaM-PDE гена, PDE1A, PDE1B и PDE1C, все экспрессируются в ткани центральной нервной системы. PDE1A экспрессируется повсюду в головном мозге, с высокими уровнями экспрессии в слоях с CA1 по CA3 гиппокампа и мозжечка и с низким уровнем в полосатом теле. PDE1A также экспрессируется в легких и сердце. PDE1B преимущественно экспрессируется в полосатом теле, зубчатой извилине, обонятельном тракте и мозжечке, и его экспрессия коррелирует с областями головного мозга, имеющими высокие уровни дофаминергической иннервации. Хотя PDE1B экспрессируется главным образом в центральной нервной системе, его также детектируют в обонятельном эпителии, церебеллярных клетках-зернах, полосатом теле, сердце и сосудистых гладких мышцах.
[0003] CaM-PDE играют решающую роль в опосредовании трансдукции сигнала в клетках головного мозга, особенно в области головного мозга, известной как базальные ганглии или полосатое тело. Например, активация глутаматного рецептора NMDA-типа и/или активация дофаминового рецептора D2 дает в результате повышенные внутриклеточные концентрации кальция, приводя к активации эффекторов, таких как кальмодулин-зависимая киназа II (CaMKII) и кальциневрин, и к активации CaM-PDE, результатом чего является пониженное содержание cAMP и cGMP. С другой стороны, активация дофаминового рецептора D1 ведет к активации аденилатциклаз, резултатом которой является повышенное содержание cAMP. Этот циклический нуклеотид, в свою очередь, активирует протеинкиназу A (PKA; cAMP-зависимую протеинкиназу). Известно, что производство cGMP происходит в тканях, вовлеченных в когнитивную функцию посредством различных стимуляций, например, производство оксида азота, индуцированное высокими внутриклеточными уровнями кальция, и впоследствии активирует протеинкиназу G (PKG; cGMP-зависимую протеинкиназу). PKG и PKA фосфорилируют далее по ходу элементы пути трансдукции сигнала, такие как DARPP-32 (дофамин и cAMP-регулируемый фосфопротеин) и белок, связывающий cAMP реактивный элемент (CREB). Фосфорилированный DARPP-32, в свою очередь, ингибирует активность протеинфосфатов-1 (PP-1), увеличивая тем самым состояние фосфорилирования субстратных белков, таких как прогестероновый рецептор (PR), приводя к индукции физиологических реакций. При шизофрении нарушается передача сигналов рецепторов D1, что способствует когнитивному расстройству при заболевании. Роль cAMP и cGMP в когнитивной функции основательно установлена в исследованиях на животных. Исследования на грызунах навели на мысль, что индуцируя cAMP и cGMP синтез посредством активации дофаминового рецептора D1 или прогестеронового рецептора усиливает прогестероновую передачу сигналов, связанную с различными физиологическими реакциями, включая лордозную реакцию, связанную с восприимчивостью к спариванию у некоторых грызунов. Смотри работу Mani и др., Science (2000) 287: 1053, содержание которой включено здесь посредством ссылки.
[0004] Следовательно, CaM-PDE могут воздействовать на дофамин-регулируемый и другие внутриклеточные пути передачи сигналов в базальных ганглиях (полосатом теле), включая, но не ограничиваясь этим, внутриклеточные пути передачи сигналов через оксид азота, норадренергический, нейротензиновый, CCK, VIP, серотониновый, глютаматный (например, через NMDA рецептор, AMPA рецептор), GABA, ацетилхолиновый, аденозиновый (например, через A2A рецептор) путь, через каннабиноидный рецептор, через натрийуретический пептид (например, ANP, BNP, CNP), DARPP-32 и эндорфиновый внутриклеточный путь передачи сигналов.
[0005] Фосфодиэстеразная (PDE) активность, в частности активность фосфодиэстеразы 1 (PDE1), действует в ткани головного мозга как регулятор локомоторной активности, обучаемости и памяти. PDE1 представляет собой терапевтическую мишень для регуляции внутриклеточного пути передачи сигналов, предпочтительно в нервной системе, включая, но не ограничиваясь этим, путь внутриклеточной передачи сигналов через дофаминовый рецептор D1, дофаминовый рецептор D2, оксид азота, норадренергический, нейротензиновый, CCK, VIP, серотониновый, глютаматный (например, через NMDA рецептор, AMPA рецептор), GABA, ацетилхолиновый, аденозиновый (например, через A2A рецептор) путь передачи сигналов, путь через каннабиноидный рецептор, натрийуретический пептид (например, ANP, BNP, CNP), эндорфиновый путь внутриклеточной передачи сигналов и прогестероновый путь передачи сигналов. Например, ингибирование PDE1B должно действовать, усиливая эффект агониста дофаминового рецептора D1 посредством защиты cGMP и cAMP от деградации, и ингибировать пути передачи сигналов через дофаминовый рецептор D2 посредством ингибирования активности PDE1, что является следствием опосредованного рецептором D2 повышения внутриклеточного кальция. Хроническое повышение уровней внутриклеточного кальция связано с гибелью клеток при многих расстройствах, особенно при нейродегенеративных заболеваниях, таких как болезни Альцгеймера, Паркинсона и Гентингтона, и при расстройствах системы кровообращения, приводя к инсульту и инфаркту миокарда. Следовательно, ингибиторы PDE1 потенциально пригодны при заболеваниях, характеризуемых пониженной активностью передачи сигналов дофаминовым рецептором D1, таких как болезнь Паркинсона, синдром беспокойных ног, депрессия, нарколепсия и когнитивное расстройство, например, когнитивное расстройство, связанное с шизофренией. Ингибиторы PDE1 пригодны также при заболеваниях, которые можно облегчить, усиливая прогестероновую передачу сигналов, таких как сексуальная дисфункция у женщин.
[0006] Кроме того, нейрогенез является жизненно важным процессом в головном мозге животных и людей, посредством которого постоянно генерируются новые нервные клетки на протяжении всей жизни организма. Вновь образовавшиеся клетки способны дифференцироваться в функциональные клетки центральной нервной системы и интегрироваться в существующие нейронные контуры в головном мозге. Нейрогенез, как известно, продолжается на протяжении всего периода полового созревания в двух областях головного мозга млекопитающих: cубвентрикулярной зоне (SVZ) боковых желудочков и зубчатой извилине гиппокампа. В этих областях полипотентные клетки-предшественники нейронов (NPC) продолжают делиться и дают новые функциональные нейроны и глиальные клетки. Показано, что различные факторы могут стимулировать гиппокампальный нейрогенез у взрослого человека, например, адреналэктомия, добровольное применение, обогащенная среда, гиппокамп-зависимое обучение и антидепрессанты. На нейрогенез негативно влияют другие факторы, такие как гормоны коры надпочечников, стресс, возраст и наркотики.
[0007] Хотя важность нейрогенеза не может быть преувеличена, недостаток аксонов для восстановления после повреждения спинного мозга все еще остается одной из главнейших проблем, стоящих перед медициной и нейробиологией. В отличие от миелинизированных аксонов периферической нервной системы миелинизированные аксоны центральной нервной системы не регенерируются после разрыва. Важной разработкой однако является идентификация ингибиторных белков в миелиновых оболочках, которые окружают аксоны ЦНС. Некоторые биоактивные молекулы, по-видимому, ингибируют разрастание нейритов, приводя к недостаточной регенерации нейронов ЦНС. Миелин содержит ряд белков, которые, как показано, ингибируют процесс разрастания нейритов. Член семейства ретикулонов NogoA представляет собой первый белок, идентифицированный как ингибитор разрастания нейритов. Он экспрессируется олигодендроцитами и некоторыми нейронами и может быть обнаружен внутри клеток и на поверхности клеток (в частности на миелиновых оболочках аксонов). Другие белки, которые вносят вклад в ингибирование регенерации аксонов, включают миелин-связанный гликопротеин (MAG), олигодендроцит-миелиновый гликопротеин (OMgp) и протеогликан верзикан.
[0008] Таким образом, по-видимому, ЦНС среда ограничивает аксональную регенерацию после повреждения. Действительно, миелин ЦНС идентифицирован как главный фактор, приводящий к регенеративной неудаче. Имеются доказательства, которые показывают, что белки ЦНС, присутствующие в миелиновой оболочке, ингибируют аксональный рост и регенерацию.
[0009] Предложены различные стратегии преодоления ингибирования аксональной регенерации. Один подход, который являлся эффективным, состоит в повышении уровней внутриклеточного cAMP. Это можно выполнить несколькими способами, такими как нарушение периферического кондиционирования, введение аналогов cAMP, примирование нейротрофинами или обработка ингибитором фосфодиэстеразы ролипрамом (ингибитором PDE4). Эффекты cAMP могут быть транскрипционно зависимы, и cAMP-опосредованная активация CREB может давать повышающую регуляцию и экспрессию генов, например, аргиназы I и интерлейкина-6. Полагают, что продукты этих генов промотируют аксональную регенерацию, которая дает возможность другим cAMP-регулируемым генам производить дополнительные агенты, которые будут полезны при лечении повреждения спинного мозга. Однако, что касается повышения экспрессии IL-6, один существенный недостаток этого механизма действия может состоять в том, что IL-6 является потенциально опасным провоспалительным цитокином, это означает возможность того, что высокие уровни IL-6 могут фактически обострять воспаление, которое возникает после повреждения спинного мозга, что далее может вести к увеличению гибели клеток. Действительно, фактором, поддерживающим эту проблему, является то, что по наблюдениям IL-6 трансгенные мыши имеют обширный астроглиоз, нейродегенерацию и разрушение гемоэнцефалического барьера.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0010] Изобретение касается соединения формулы V:
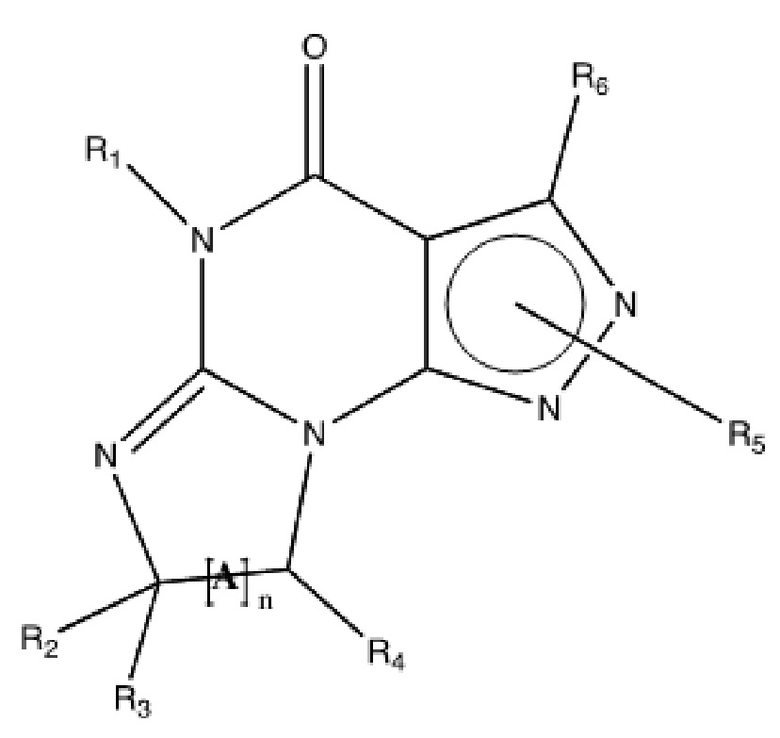
Формула V
где
R1 обозначает C1-4 алкил (например, метил);
R4 обозначает H, и R2 и R3 независимо обозначают H или C1-4 алкил (например, R2 и R3 оба обозначают метил, или R2 обозначает H, и R3 обозначает изопропил);
(iii) R5 присоединен по одному из атомов азота на пиразольной части формулы V и представляет собой фрагмент формулы A
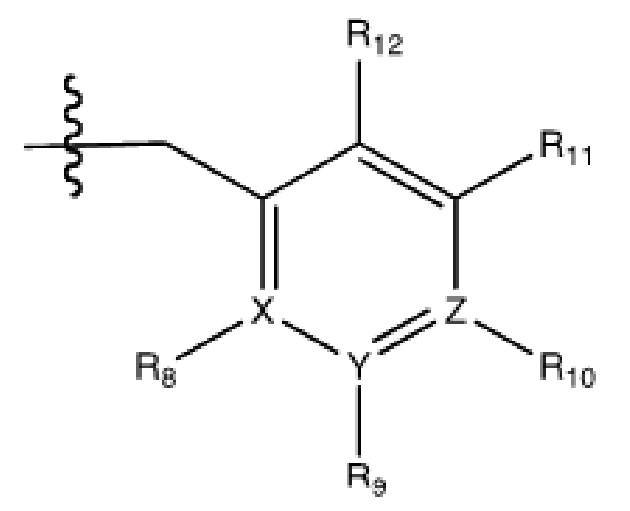
Формула A
где X, Y и Z обозначают C, и R8, R9, R11 и R12 обозначают H, и R10 обозначает атом галогена (например, хлора) или гетероарил, необязательно замещенный атомом галогена, алкилом, галогеналкилом, гидрокси или карбокси (например, пиридил или 2-галогенпиридил, (например, пирид-2-ил, 5-фторпирид-2-ил или 6-фторпирид-2-ил)); и
(iv) R6 обозначает H, C1-4алкил (например, метил, этил или пропил), ариламино необязательно замещенный C1-4алкилом или атомом галогена (например, фениламино или 4-фторфениламино), или тиоC1-4алкил (например, тиоэтил); и
(v) n=0;
в свободном виде, в виде соли или пролекарства, включая его энантиомеры, диастереомеры и рацематы.
[0011] Еще в одном аспекте изобретение предполагает, что ингибиторы PDE1 (например, формулы V) представляют собой соединения формулы V, соответствующие любой из следующих формул:
1.1 соединение формулы V, где R1 обозначает метил;
1.2 соединение формулы V или 1.1, где R2 и R3 обозначают C1-4 алкил;
1.3 соединение формулы V или любой из 1.1-1,2, где R2 и R3 оба обозначают метил;
1.4 соединение формулы V или любой из 1.1-1.3, где R10 обозначает гетероарил, необязательно замещенный атомом галогена;
1.5 соединение формулы V или любой из 1.1-1,4, где R10 обозначает пирид-2-ил;
1.6 соединение формулы V или любой из 1.1-1,4, где R10 обозначает 5-фтор-пирид-2-ил;
1.7 соединение формулы V или любой из 1.1-1,4, где R10 обозначает 6-фтор-пирид-2-ил;
1.8 соединение формулы V или любой из 1.1-1,7, где R6 обозначает C1-4алкил;
1.9 соединение формулы V или любой из 1.1-1.8, где R6 обозначает этил;
1.10 соединение формулы V или любой из 1.1-1.8, где R6 обозначает пропил;
1.11 соединение формулы V или любой из 1.1-1,7, где R6 обозначает ариламино, необязательно замещенный C1-4алкилом или атомом галогена;
1.12 соединение формулы V или любой из 1.1-1,7, где R6 обозначает 4-фторфениламино;
1.13 любая из предыдущих формул, где соединение выбрано из группы, включающей
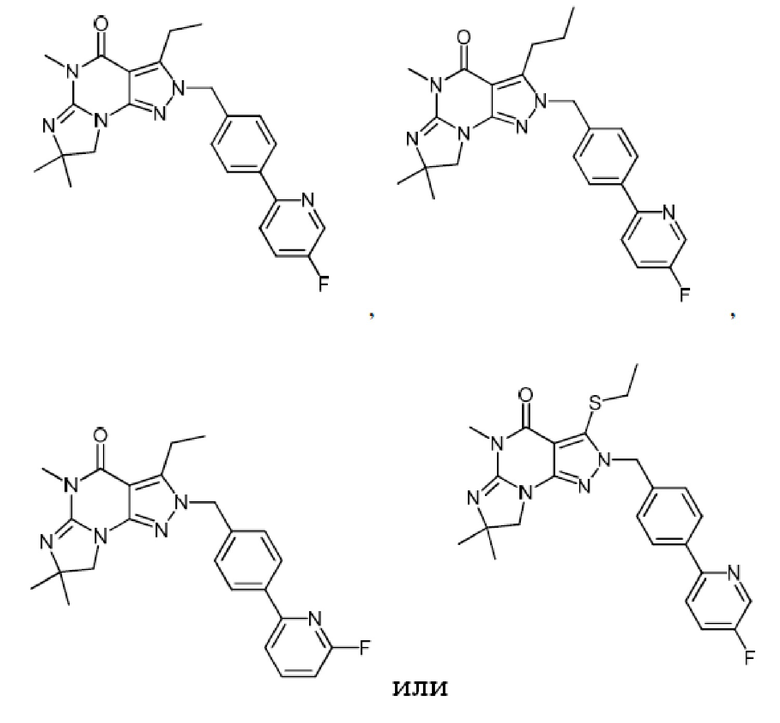

или
в свободном виде, в виде соли или пролекарства, включая его энантиомеры, диастереомеры и рацематы;
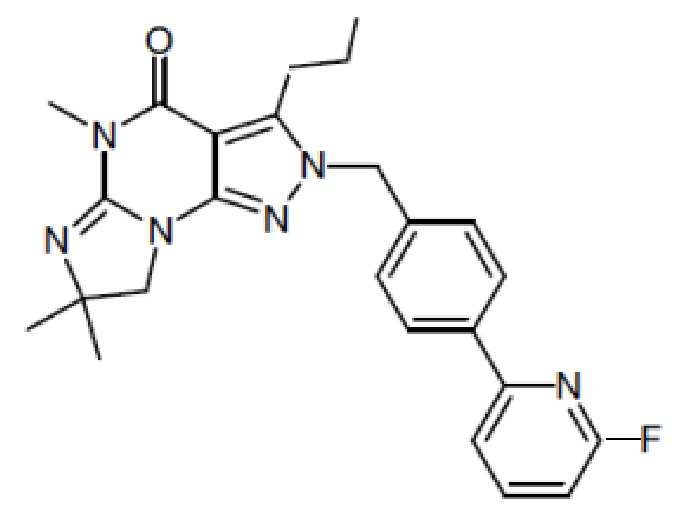
1.14 любая из предыдущих формул, где соединение выбрано из группы, включающей:
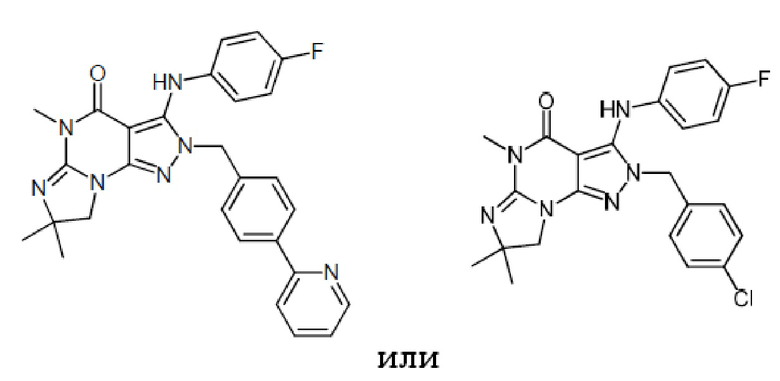
в свободном виде, в виде соли или пролекарства, включая его энантиомеры, диастереомеры и рацематы.
[0012] В одном аспекте селективные ингибиторы PDE1 любой из предыдущих формул (например, формулы V или 1.1-1.14) представляют собой соединения, которые ингибируют фосфодиэстераза-опосредуемый (например, PDE1-опосредуемый, главным образом PDE1A- или PDE1C-опосредуемый) гидролиз cGMP, например, предпочтительные соединения имеют IC50 меньше 1М, предпочтительно меньше 500 нМ и более предпочтительно меньше 50 нМ в PDE исследовании с использованием реагента с иммобилизованными металл аффинными частицами, в свободном виде или в виде соли.
[0013] Одним преимуществом настоящего изобретения является то, что ингибитор PDE1 (например, соединение, соответствующее любой из формул V или 1.1-1.14) может действовать как нейропротекторный агент и/или нейрорегенеративный агент. В случае повреждения ЦНС (например, повреждения спинного мозга), заболевания или расстройства ЦНС раскрытые здесь соединения и способы можно применять для содействия или усиления разрастания нейритов и аксональной регенерации даже в присутствии ингибиторов аксональной регенерации.
[0014] Не связываясь никакой конкретной теорией, полагают, что, по меньшей мере, одним преимуществом настоящего изобретения является то, что введение ингибитора PDE1 (например, любого соединения формулы V или 1.1-1.14) может влиять на увеличение уровней внутриклеточного cAMP и инициировать транскрипцию генов, что необходимо для преодоления ингибирования аксональной регенерации и промотирования разрастания нейритов и/или аксональной регенерации в случае заболевания, расстройства или повреждения ЦНС. Например, повышенный уровень внутриклеточного cAMP, такой как уровень, являющийся результатом ингибирования PDE1, будет приводить к повышенной активности cAMP-зависимых белков, таких как протеинкиназа C (PKC).
[0015] Кроме того, полагают, что введение ингибитора PDE1 (например, соединения, соответствующего любой из формул V или 1.1-1.14) может повышать внутриклеточные уровни обоих фосфатов cAMP и cGMP. Без связи с теорией, это повышение уровней обоих cAMP и cGMP может служить для уравновешивания потенциально вредных влияний, которые могут быть связаны с хронически повышенными уровнями внутриклеточного кальция. Наблюдают, что повышенные уровни внутриклеточного кальция могут быть связаны с развитием различных дегенеративных заболеваний. Например, одним возможным объяснением является то, что повышенные уровни внутриклеточного кальция (например, хронически повышенные уровни внутриклеточного кальция) ведут к активации PDE1, которая далее стимулирует гидролиз cAMP. Затем пониженная концентрация cAMP дезактивирует cAMP-зависимые белки, такие как протеинкиназа C (PKC).
[0016] Однако не связываясь никакой теорией, полагают, что другим потенциальным преимуществом введения ингибитора PDE1 (например, соединения, соответствующего любой из формул V или 1.1-1.14) является повышение уровня внутриклеточного cGMP. Это повышение внутриклеточного cGMP может вести к повышению активности PKG, предотвращению дальнейшего роста уровней внутриклеточного кальция. Таким образом, без связи с какой-либо теорией, введение ингибитора PDE1 (например, соединения, соответствующего любой из формул V или 1.1-1.14) может иметь двойное преимущество, например, играя благотворную роль в аксональной регенерации (и/или нейропротекции) и одновременно с этим ослабляя вредные воздействия, которые могут быть связаны с повышенными уровнями внутриклеточного кальция.
[0017] В одном варианте осуществления изобретение включает композиции и способы лечения и предупреждения заболевания, расстройства или повреждения ЦНС (например, повреждения спинного мозга, например, спинальной мышечной атрофии, например, повреждения двигательных нейронов), где способ включает введение эффективного количества ингибитора PDE1 (например, соединения, соответствующего любой из формул V или 1.1-1.14) с целью модуляции внутриклеточных уровней cAMP и/или cGMP. В одном варианте это повышение внутриклеточного cAMP оказывает нейропротекторное действие и/или способствует повышению или стимуляции нейрогенеза (например, ингибитор PDE1 увеличивает разрастание нейритов и/или аксональной регенерации).
[0018] Еще в одном варианте осуществления изобретение включает композиции и способы лечения и предупреждения повреждений периферической нервной системы (ПНС), где способ включает введение ингибитора PDE1 с целью повышения внутриклеточных уровней cAMP и/или cGMP, что прямо или опосредовано повышает регенерацию нервов и/или оказывает защитное действие против дальнейшего повреждения нерва.
[0019] В одном варианте осуществления изобретение включает композиции и способы предупреждения заболевания или расстройства ЦНС у субъекта, который находится в группе риска развития указанного заболевания или расстройства, где способ включает:
1) получение ЦНС-образца от субъекта;
2) измерение уровней внутриклеточного кальция в образце;
3) сравнение уровней внутриклеточного кальция в биологическом образце с эталоном;
4) определение, находится ли пациент в группе риска развития заболевания или расстройства ЦНС, на основании уровня внутриклеточного кальция по сравнению с эталоном;
5) введение субъекту ингибитора PDE1 (например, соединения, соответствующего любой из формул V или 1.1-1.14) на основании уровня внутриклеточного кальция у субъекта (например, введение субъекту ингибитора PDE1, так как он имеет повышенный уровень внутриклеточного кальция по сравнению с эталоном).
[0020] Если не определено по-другому или не ясно из контекста, указанные здесь далее термины имеют следующие значения:
(a) Используемый здесь термин «алкил» обозначает насыщенный или ненасыщенный углеводородный фрагмент, предпочтительно насыщенный фрагмент, предпочтительно имеющий от одного до шести атомов углерода, который может быть линейным или разветвленным и необязательно может быть моно-, ди- или тризамещенным, например, атомами галогена (например, хлора или фтора), гидрокси или карбокси.
(b) Используемый здесь термин «циклоалкил» обозначает насыщенный или ненасыщенный неароматический углеводородный фрагмент, предпочтительно насыщенный фрагмент, предпочтительно содержащий от трех до девяти атомов углерода, по меньшей мере, некоторые из которых образуют неароматическую моно- или бициклическую или мостиковую циклическую структуру, и который необязательно может быть замещенным, например, атомом галогена (например, хлора или фтора), гидрокси или карбокси. Если циклоалкил необязательно содержит один или более атомов, выбранных из N и O и/или S, то указанный циклоалкил также может представлять собой гетероциклоалкил.
(c) Если не указано иначе, термин «гетероциклоалкил» обозначает насыщенный или ненасыщенный неароматический углеводородный фрагмент, предпочтительно насыщенный фрагмент, предпочтительно содержащий от трех до девяти атомов углерода, по меньшей мере, некоторые из которых образуют неароматическую моно- или бициклическую или мостиковую циклическую структуру, где, по меньшей мере, один атом углерода заменен на N, O или S, причем гетероциклоалкил может быть необязательно замещен, например, атомом галогена (например, хлора или фтора), гидрокси или карбокси.
(d) Используемый здесь термин «арил» обозначает моно- или бициклический ароматический углеводород, предпочтительно фенил, необязательно замещенный, например, алкилом (например, метилом), атомом галогена (например, хлора или фтора), галогеналкилом (например, трифторметилом), гидрокси, карбокси или дополнительным арилом или гетероарилом (например, бифенилом или пиридилфенилом).
(e) Используемый здесь термин «гетероарил» обозначает ароматический фрагмент, где один или более атомов, составляющих ароматическое кольцо, являются атомами серы или азота вместо атоммов углерода, например, пиридил или тиадиазолил, который необязательно может быть замещенным, например, алкилом, атомом галогена, галогеналкилом, гидрокси или карбокси.
(f) Подразумевается, что если название заместителей оканчивается на «ен», например, алкилен, фенилен или арилалкилен, указанные заместители являются мостиковыми или соединены с двумя другими заместителями. Следовательно, подразумевается, что метилен представляет собой -CH2-, фенилен представляет собой -C6H4-, и арилалкилен представляет собой, например, -C6H4-CH2- или-CH2-C6H4-.
[0021] В данной спецификации, если не указано иначе, такое выражение как «соединения по изобретению» следует понимать как охватывающее соединения в любом виде, например, в свободном виде или в виде аддитивной соли кислоты, или, если соединения содержат кислотные заместители, в виде аддитивной соли основания. Соединения по изобретению предназначены для применения в качестве лекарственных препаратов, следовательно, предпочтительными являются фармацевтически приемлемые соли. Соли, которые не подходят для фармацевтических применений, могут быть пригодны, например, для выделения и очистки свободных соединений по изобретению или их фармацевтически приемлемых солей, следовательно, также включены в область изобретения.
[0022] Соединения по изобретению, охватывающие любые из раскрытых здесь соединений, могут существовать в свободном виде или в виде солей, например, аддитивных солей кислот. В данной спецификации, если не указано иначе, такое выражение как «соединения по изобретению» следует понимать как охватывающее соединения в любом виде, например, в свободном виде или в виде аддитивной соли кислоты, или, если соединения содержат кислотные заместители, в виде аддитивной соли основания. Соединения по изобретению предназначены для применения в качестве лекарственных препаратов, следовательно, предпочтительными являются фармацевтически приемлемые соли. Соли, которые не подходят для фармацевтических применений, могут быть пригодны, например, для выделения и очистки свободных соединений по изобретению или их фармацевтически приемлемых солей, следовательно, также включены в область изобретения.
[0023] В некоторых случаях соединения по изобретению могут также существовать в виде пролекарства. Форма пролекарства представляет собой соединение, которое в организме преобразуется в соединение по изобретению. Например, если соединения по изобретению содержат гидрокси или карбоксизаместители, то эти заместители могут образовывать физиологически гидролизуемые и приемлемые сложные эфиры. Используемое здесь выражение «физиологически гидролизуемый и приемлемый сложный эфир» обозначает сложные эфиры соединений по изобретению, которые являются гидролизуемыми в физиологических условиях с образованием кислот (в случае соединений по изобретению, которые имеют гидроксизаместители) или спиртов (в случае соединений по изобретению, которые имеют карбоксизаместители), которые сами являются физиологически допустимыми при дозах, подлежащих введению. Следовательно, если соединение по изобретению содержит гидроксигруппу, например, соединение-OH, то пролекарственный сложный эфир ацила такого соединения, то есть соединение-O-C(O)-C1-4алкил, может гидролизоваться в организме с образованием физиологически гидролизуемого спирта (соединение-OH), с одной стороны, и карбоновой кислоты, с другой стороны (например, HOC(O)-C1-4алкил). По-другому, если соединение по изобретению содержит карбоновую кислоту, например, соединение-C(O)OH, то пролекарственный сложный эфир кислоты такого соединения, соединение-C(O)O-C1-4алкил может гидролизоваться с образованием соединение-C(O)OH и спирта HO-C1-4алкил. Как будет понятно, данный термин охватывает, таким образом, общеизвестные фармацевтические пролекарственные формы.
[0024] В другом варианте осуществления изобретение также касается фармацевтической композиции, содержащей соединение по изобретению в свободном виде или в виде фармацевтически приемлемой соли в смеси с фармацевтически приемлемым носителем.
Способы получения соединений по изобретению.
[0025] Соединения по изобретению и их фармацевтически приемлемые соли можно получить, применяя способы, которые здесь описаны и проиллюстрированы, аналогичные способы и способы, известные в области химии. Такие способы включают, но не ограничены этим, способы, описанные ниже. Если исходные материалы для этих процессов не доступны коммерчески, их можно получить способами, которые выбраны из химической практики, применяя методики, которые подобны или аналогичны синтезу известных соединений.
[0026] Разнообразные исходные материалы и/или соединения по изобретению можно получить, применяя способы, описанные в US 2008-0188492 A1, US 2010-0173878 A1, US 2010-0273754 A1, US 2010-0273753 A1, WO 2010/065153, WO 2010/065151, WO 2010/065151, WO 2010/065149, WO 2010/065147, WO 2010/065152, WO 2011/153129, WO 2011/133224, WO 2011/153135, WO 2011/153136, WO 2011/153138, US 2014/0194396, PCT/US14/30412, и каждая работа включена здесь в виде ссылки во всей своей полноте.
[0027] Соединения по изобретению включают свои энантиомеры, диастереомеры и рацематы, а также их полиморфы, гидраты, сольваты и комплексы. Некоторые индивидуальные соединения, входящие в область этого изобретения, могут содержать двойные связи. Имеется в виду, что изображения двойных связей в этом изобретении включают оба изомера двойной связи, E и Z. Кроме того, некоторые соединения, входящие в область этого изобретения, могут содержать один или более асимметрических центра. Это изобретение включает применение любого из оптически чистых стереомеров, а также любой комбинации стереомеров.
[0028] Подразумевается также, что соединения по изобретению охватывают их стабильные и нестабильные изотопы. То есть соединения по изобретению включают замещение или обогащение каких-либо атомов или нескольких атомов структуры стабильными или нестабильными изотопными вариантами этих атомов. Изотопы представляют собой атомы одного и того же элемента, которые содержат разное количество нейтронов. Изотопный вариант представляет собой любой изотоп любого элемента, отличный от его наиболее широко распространенного в природе изотопа. Изотопный вариант содержит один или более дополнительных нейтронов, или на один или на несколько нейтронов меньше по сравнению с наиболее широко распространенным в природе нуклидом одного элемента. Изотопы могут быть либо стабильными (нерадиоактивными) или нестабильными (радиоактивными). Например, наиболее широко распространенным в природе нуклидом углерода является 12C, и одним известным стабильным изотопом углерода является 13C. Изотопы элемента в основном имеют одинаковые характеристические электронные и химические свойства. Ожидается, что активность соединений, содержащих такие изотопы, будет сохраняться, и такое соединение также будет полезно для определения фармакокинетики неизотопных аналогов. Например, атом водорода в одном или нескольких положениях соединений по изобретению может быть замещен (или обогащен) дейтерием. Примеры известных стабильных изотопов включают, но не ограничены этим, дейтерий (2H), 13C, 15N и 18O. Примеры известных нестабильных изотопов включают 3H, 123I, 131I, 125I, 11C, 18F. Нестабильные изотопы могут быть пригодны для исследования соединений по изобретению с получением радиоизображения и/или исследования фармакокинетики. Атомы в одном или нескольких положениях в соединении по изобретению могут быть замещены или обогащены любым известным изотопным вариантом. Природные источники химических веществ и реагенты в основном не являются изотопно чистыми, так что соединения по изобретению, полученные традиционными химическими способами, в основном имеют некоторую обычные, природные вариации изотопного содержания. Например, природный состав элемента углерода имеет примерно 98,93% 12C и 1,07% 13C. Следовательно, соединения по изобретению, полученные традиционными химическими способами, обычно содержат около 98,93% 12C и 1,07% 13C для каждого атома углерода структуры. Термин «обогащение» касается присутствия в химической структуре меньшего изотопа, превышающее его природное содержание. Таким образом, например, соединение по изобретению можно обогатить присутствием 13C по одному или нескольким положениям атома углерода. Используемый здесь термин «замещение» касается обогащения изотопным вариантом более чем примерно на 95%.
[0029] Температуры плавления не скорректированы, и «разл.» обозначает разложение. Температуры приведены в градусах Цельсия (°C); пока не указано по-другому, операции выполняют при комнатной температуре или температуре окружающей среды, то есть при температуре в диапазоне 18-25°C. Термин «хроматография» обозначает флэш-хроматографию на силикагеле; тонкослойную хроматографию (ТСХ) выполняют на пластинах с силикагелем. ЯМР-данные представлены с использованием дельта-значений основных диагностических протонов, выраженных в миллионных долях (м.д.) относительно тетраметилсилана (ТМС) в качестве внутреннего стандарта. Используют общеизвестные сокращения для вида сигнала. Константы взаимодействия (J) даны в Гц. Для масс-спектров (МС) приводят основной ион с наименьшей массой для молекул, где изотопное расщепление дает в результате несколько масс-спектральных пиков. Составы смесей растворителей даны в объемных процентах или посредством объемных соотношений. В случаях, где ЯМР-спектры обозначают как «сложные», приводят только диагностические сигналы.
[0030] Термины и сокращения:
BOC=трет-бутоксикарбонил
BOP=бензотриазол-1-ил-окси-трис(диметиламино)фосфоний гексафторфосфат
BuLi=н-бутиллитий
ButOH=трет-бутиловый спирт,
CAN=нитрат церия(IV)аммония,
DBU=1,8-диазабицикло[5,4,0]ундец-7-ен,
ДИПЭА=диизопропилэтиламин,
ДМФА=N,N-диметилформамид,
ДМСО=диметилсульфоксид,
Et2O=диэтиловый эфир,
EtOAc=этилацетат,
экв.=эквивалент(ы),
час.=час(ы),
ВЭЖХ=высокоэффективная жидкостная хроматография,
LDA=литийдиизопропиламид,
MeOH=метанол,
NBS=N-бромсукцинимид,
NCS=N-хорсукцинимид,
NaHCO3=бикарбонат натрия,
NH4OH=гидроксид аммония,
Pd2(dba)3=трис[дибензилиденацетон]дипалладий(0)
PMB=п-метоксибензил,
POCl3=оксихлорид фосфора,
SOCl2=тионилхлорид,
ТФУ=трифторуксусная кислота,
TFMSA=трифторметансульфоновая кислота,
ТГФ=тетрогидрофуран.
[0031] Синтетические способы, применимые в этом изобретении, проиллюстрированы ниже. Определения для групп R соответсвуют сформулированным выше для любой из формул V или 1.1-1.14, если не указано иначе.
[0032] Промежуточные соединения формулы IIb можно получить взаимодействием соединения формулы IIa с малоновой кислотой и уксусным ангидридом в уксусной кислоте, необязательно при нагревании (например, примерно до 90°C в течение примерно 3 час.):
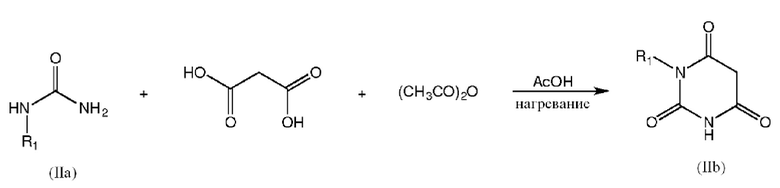
где R1 обозначает C1-4 алкил, например, метил.
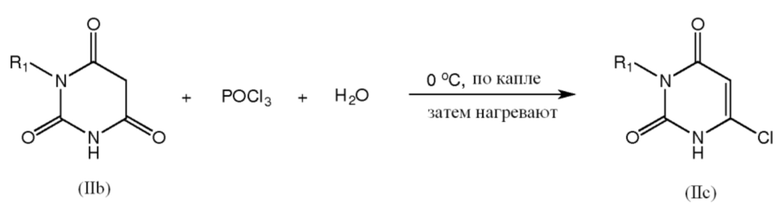
[0033] Промежуточные соединения формул IIc можно получить взаимодействием соединения формулы IIb с хлорирующим соединением, таким как POCl3, необязательно при небольших количествах воды и/или нагревании (например, нагревании примерно до 80°C в течение примерно 4 час.):
[0034] Промежуточные соединения формулы IId можно получить взаимодействием соединения формулы IIc, например, с реагентом P1-L в растворителе, таком как ДМФА, в присутствии основания, такого как карбонат калия, бикарбонат натрия, карбонат цезия, гидроксид натрия, триэтиламин, диизопропилэтиламин или подобные, при комнатной температуре или при нагревании:
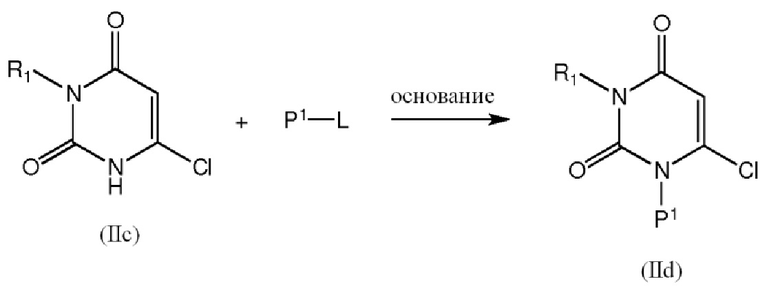
где P1 обозначает защитную группу (например, PMB или BOC); и L обозначает уходящую группу, такую как атом галогена, мезилат или тозилат. Предпочтительно P1 представляет собой PMB, и основание представляет собой карбонат калия.
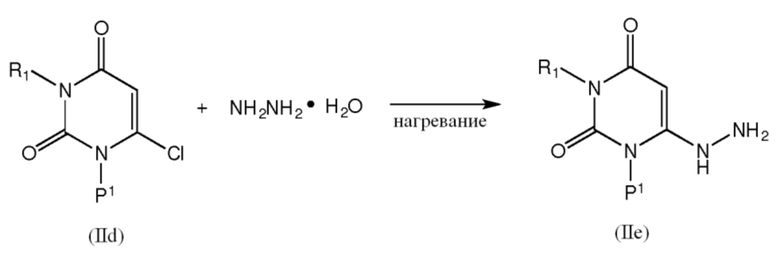
[0035] Промежуточные соединения формулы IIe можно получить взаимодействием соединения формулы IId с гидразином или гидразингидратом в растворителе, таком как метанол, предпочтительно при нагревании (например, кипячении с обратным холодильником в течение примерно 4 час.):
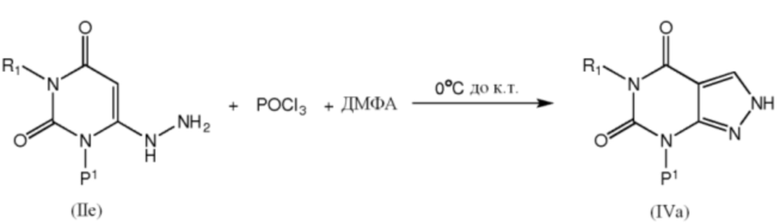
[0036] Промежуточные соединения формулы IVa можно получить взаимодействием соединения формулы IIe с POCl3 и ДМФА:
[0037] Промежуточные соединения формулы IVb можно получить взаимодействием соединения формулы IVс реагентом формулы F1-X в растворителе, таком как ДМФА, в присутствии основания, такого как карбонат калия при комнатной температуре:
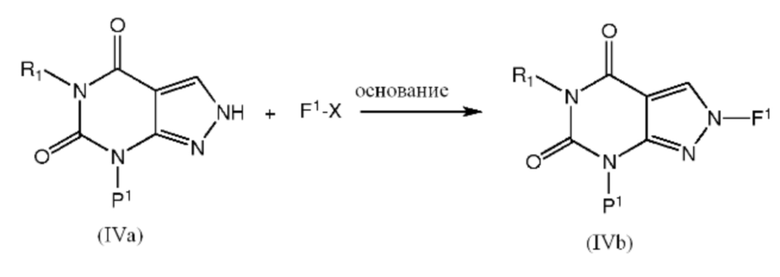
где F1 обозначает защитную группу (например, замещенную бензильную группу, такую как 4-бромбензил), и X обозначает атом галогена (например, Br).
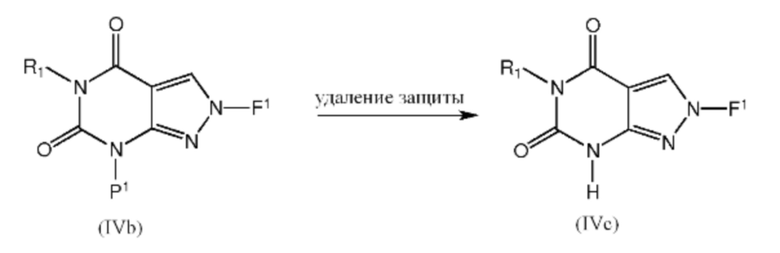
[0038] Промежуточные соединения формулы IVc можно получить из соединений формулы IVb посредством удаления защитной группы P1, применяя подходящий способ. Например, если P1 представляет собой группу PMB, то ее можно удалить при помощи ТФУ/TFMSA при температуре окружающей среды или повышенной температуре, при этом, если P1 представляет собой BOC, то его можно удалить, используя кислоту, такую как ТФУ, или водный раствор соляной кислоты:
[0039] Промежуточные соединения формулы IVd можно получить взаимодействием соединения формулы IVc с хлорирующим соединением, таким как POCl3, необязательно при нагревании (например, при кипячении с обратным холодильником в течение 2 дней или более или микроволновом облучении при 150-200°C в течение 5-10 мин. в закупоренной пробирке):
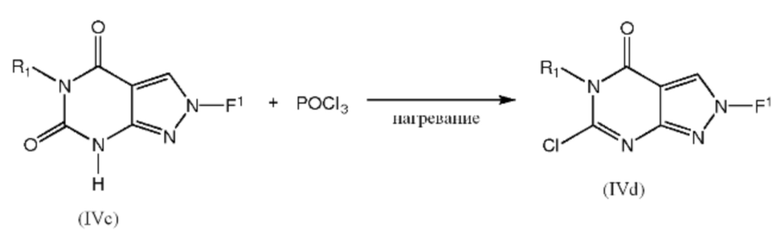
[0040] Промежуточные соединения формулы IVe можно получить взаимодействием соединения формулы IVd с аминоспиртом в основных условиях, в растворителе, таком как ДМФА, необязательно при нагревании:
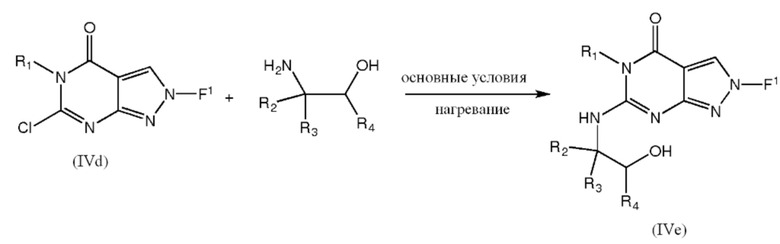
где R1, R2, R3 и R4 такие, как определено ранее для любой из формул V или 1.1-1.14.
[0041] По-другому, промежуточные соединения IVe можно получить непосредственно из соединений формулы IVc взаимодействием с аминоспиртом и связующим агентом, таким как BOP, в присутствии основания, такого как DBU:
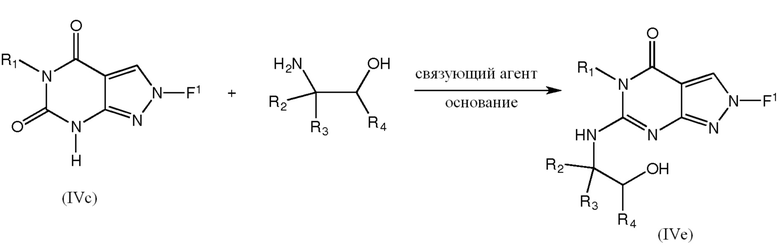
где R1, R2, R3 и R4 такие, как определено ранее для любой из формул V или 1.1-1.14.
[0042] Промежуточные соединения формулы IVf можно получить взаимодействием соединения формулы IVe с дегидратирующим/галогенирующим агентом, таким как SOCl2, в растворителе, таком как дихлорметан, при комнатной температуре или нагревании при 35°C:

[0043] Промежуточные соединения формулы IVg можно получить взаимодействием соединения формулы IVf с катализаторами, такими как соли меди и 2,2,6,6-тетраметилгептан-3,5-дион, и основанием, таким как карбонат цезия, в растворителе, таком как NMP, при нагревании:

где, F2 представляет собой диариловый эфир.
[0044] Промежуточные соединения формулы IVh можно получить взаимодействием соединения формулы IVg с кислотной системой, такой как ТФУ и TFMSA, в растворителе, таком как дихлорметан, при комнатной температуре:
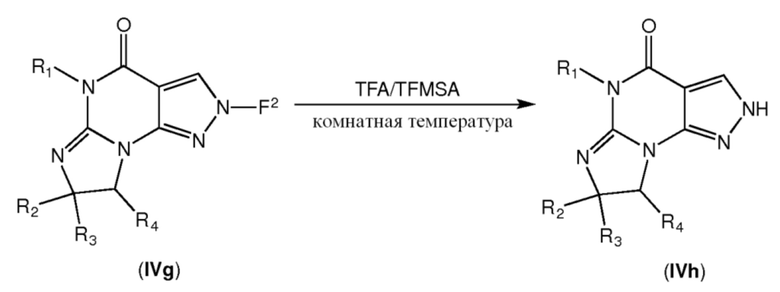
[0045] Промежуточные соединения формулы IVi можно получить взаимодействием соединения формулы IVh с реагентом формулы R5-(CH2)n-L в присутствии основания, такого как карбонат калия, в растворителе, таком как ДМФА, при комнатной температуре:
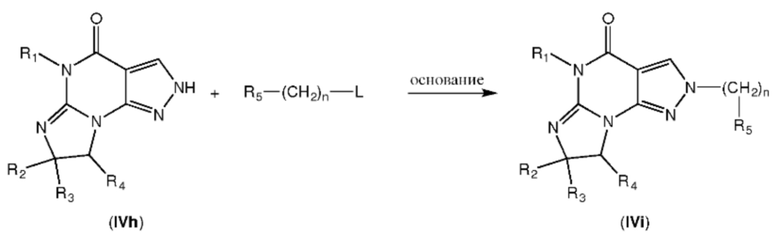
где n равно 0, и R5 представляет собой фрагмент формулы A, который определен ранее для любой из формул V или 1.1-1.14, и L обозначает уходящую группу, такую как атом галогена (например, Br).
[0046] Промежуточные соединения формулы IVj, где X обозначает атом галогена (например, Cl), можно получить взаимодействием соединения формулы IVi с галогенирующим агентом (например, NCS или NBS) и основанием, таким как LiHMDS, в растворителе, таком как ТГФ, при низкой температуре:
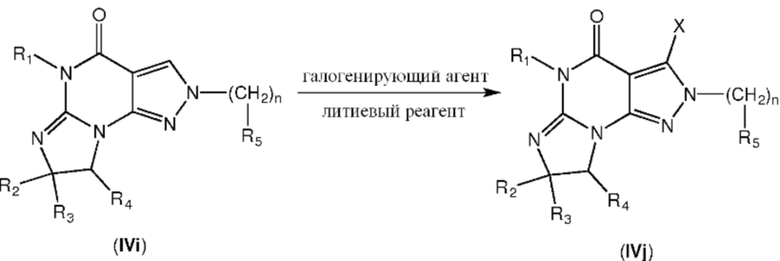
[0047] Затем из соединений формулы IVj можно получить соединения по изобретению, способами, известными специалистам в данной области. Например, замещением атома галогена X ариламином или алкилмеркаптаном.
Способы применения соединений по изобретению
[0048] Изобретение также касается способа I, где способ I включает профилактику и/или лечение заболеваний, расстройств и повреждений центральной нервной системы, где способ включает введение эффективного количества ингибитора PDE1 (например, любого соединения формулы V или 1.1-1.14) с целью модуляции уровня внутриклеточного cAMP.
[0049] Например, способ I также включает:
1.1. Способ I, где введение ингибитора PDE1 усиливает аксональный рост или регенерацию и/или замедляет или отменяет утрату таких клеток в нейродегенеративных условиях.
1.2. Любой способ из предыдущего способа I и всех последующих, где заболевание, расстройство или повреждение ЦНС касается повреждения, которое прямо или опосредовано воздействует на нормальное функционирование ЦНС.
1.3. Любой способ из предыдущего способа I и всех последующих, где заболевание, расстройство или повреждение ЦНС может быть структурным, физическим или механическим повреждением и может быть вызвано физическим импульсом, например, раздавливанием, сжатием или растяжением нервных волокон.
1.4. Любой способ из предыдущего способа I и всех последующих, где заболевание, расстройство или повреждение ЦНС представляет собой повреждение спинного мозга.
1.5. Способ по пункту 1.4, где ингибитор PDE1 замедляет или тормозит прогрессирование повреждения спинного мозга.
1.6. Любой способ из предыдущего способа I и всех последующих, где ингибитор PDE1 замедляет или тормозит деградацию аксональных волокон.
1.7. Любой способ из предыдущего способа I и всех последующих, где заболевание, расстройство или повреждение ЦНС касается повреждения двигательных нейронов.
1.8. Любой способ из предыдущего способа I и всех последующих, где заболевание, расстройство или повреждение выбрано из группы, включающей: неврологические травмы и повреждения, травму и/или повреждение, связанное с хирургическим вмешательством, ретинальное повреждение и травму, повреждение, связанное с эпилепсией, повреждение спинного мозга, повреждение головного мозга, хирургическую операцию на головном мозге, связанное с травмой повреждение головного мозга, связанное с травмой повреждение спинного мозга, повреждение головного мозга, связанное с лечением рака, повреждение спинного мозга, связанное с лечением рака, повреждение головного мозга, связанное с инфекцией, повреждение головного мозга, связанное с воспалением, повреждение спинного мозга, связанное с инфекцией, повреждение спинного, мозга связанное с воспалением, повреждение головного мозга, связанное с токсинами в окружающей среде, и повреждение спинного мозга, связанное с токсинами в окружающей среде.
1.9. Любой способ из предыдущего способа I и всех последующих, где заболевание, расстройство или повреждение ЦНС включает разрушенные или деградированные в результате болезни нейроны или нервные волокна (например, вследствие болезни Паркинсона), химический дисбаланс или физиологическую дисфункцию, такую как аноксия (например, инсульт), аневризму или реперфузионное повреждение.
1.10. Любой способ из предыдущего способа I и всех последующих, где заболевание, расстройство или повреждение ЦНС является нейродегенеративным расстройством.
1.11. Способ по пункту 1.10, где нейродегенеративное заболевание, расстройство или повреждение выбрано из группы, включающей: болезнь Альцгеймера, множественный склероз, спинальную мышечную атрофию, глаукому, лобно-височную деменцию, деменцию с тельцами Леви, кортикобазальную дегенерацию, прогрессирующий супрануклеарный паралич, прионовые болезни, болезнь Гентингтона, множественную системную атрофию, болезнь Паркинсона, боковой амиотрофический склероз, наследственную спастическую параплегию, спиноцеребеллярные атрофии, атаксию Фридрейха, амилоидоз, метаболические (связанные с диабетом) расстройства, расстройства, связанные с токсинами, хроническое воспаление ЦНС, болезнь Шарко-Мари-Тута, диабетическую невропатию, повреждение в результате химиотерапии рака (например, при действии алкалоидов Винка и доксорубицина), повреждение головного мозга, связанное с инсультом, ишемию, связанную с инсультом, и неврологические расстройства, включающие, но не ограниченные этим, разнообразные периферические невропатические и неврологические расстройства, связанные с нейродегенерацией, в том числе, но не ограничиваясь этим: тригеминальную невралгию, глоссофарингеальную невралгию, паралич Белла, злокачественную миастению, мышечную дистрофию, боковой амиотрофический склероз, прогрессирующую мышечную атрофию, прогрессирующую бульбарную наследственную мышечную атрофию, синдромы ущемления грыжи, перелома или выпадения позвоночного диска, шейный спондилез, заболевания нервных сплетений, синдром деструкции торокального прохода, периферические невропатии, такие как невропатии, вызванные, например, свинцом, акриламидами, гамма-дикетонами, дисульфидом углерода, дапсоном, клещами, порфирией, и синдром Гийена-Барре.
1.12. Любой способ из предыдущего способа I и всех последующих, где заболевание, расстройство или повреждение ЦНС представляет собой поражение ЦНС, судорогу или повреждение вследствие острых приступов (например, эпилептических припадков), радиационного поражения, повреждения в результате химиотерапии, и/или инсульта, или другого ишемического повреждения.
1.13. Любой способ из предыдущего способа I и всех последующих, где введение ингибитора PDE1 применяют для обновления, замещения и/или пополнения нейронов и/или глиальных клеток.
1.14. Любой способ из предыдущего способа I и всех последующих, где ингибитор PDE1 вводят нуждающемуся в этом субъекту или пациенту.
1.15. Любой способ из предыдущего способа I и всех последующих, где ингибитор PDE1 повышает уровень или экспрессию внутриклеточного cAMP.
1.16. Любой способ из предыдущего способа I и всех последующих, где ингибитор PDE1 снижает уровень или экспрессию внутриклеточного cAMP.
1.17. Любой способ из предыдущего способа I и всех последующих, где PDE1 модулирует активность PKA или PKG.
1.18. Любой способ из предыдущего способа I и всех последующих, где ингибитор PDE1 повышает активность PKA или PKG.
1.19. Любой способ из предыдущего способа I и всех последующих, где введение ингибитора PDE1 повышает уровень cAMP и cGMP.
1.20. Любой способ из предыдущего способа I и всех последующих, где введение ингибитора PDE1 повышает уровень внутриклеточного cAMP, и где этот повышенный уровень внутриклеточного cAMP оказывает нейропротекторные и/или нейрорегенеративные эффекты.
1.21. Любой способ из предыдущего способа I и всех последующих, включающий введение эффективного количества ингибитора PDE1 пациенту, который страдает от заболевания или расстройства, связанного с повышенными (например, хронически повышенными) уровнями внутриклеточного кальция, и где ингибитор PDE1 предупреждает дальнейший рост указанных уровней кальция.
1.22. Любой способ из предыдущего способа I и всех последующих, где ингибитор PDE1 вводят сам по себе или в комбинации с другим активным агентом.
1.23. Любой способ из предыдущего способа I и всех последующих, где заболевание, расстройство или повреждение связано с двигательными нейронами, и где заболевание, расстройство или повреждение двигательных нейронов представляет собой множественный склероз.
1.24. Любой из предыдущего способа II и всех последующих, где ингибитор PDE1 вводят в комбинации с другим активным агентом с целью лечения множественного склероза.
1.25. Способ по пункту 2.11, где активный агент выбран из группы, включающей: интерферон, глатирамер ацетат, натализумаб, Гилениа® (финголимод), Фампира®, иммунодепрессанты и кортикоиды.
[0050] В другом варианте осуществления изобретение касается способа II, где способ II включает композиции и способы лечения или профилактики заболевания, расстройства или повреждения периферической нервной системы (ПНС), где способ включает введение эффективного количества ингибитора PDE1 (например, любого соединения формулы V или 1.1-1.14) для повышения внутриклеточных уровней cAMP.
Например, способ II также включает:
2.1. Способ II, где заболевание, расстройство или повреждение ПНС касается повреждения, которое прямо или опосредовано воздействовует на нормальное функционирование ЦНС.
2.2. Любой из предыдущего способа II и всех последующих, где ингибитор PDE1 вводят нуждающемуся в этом субъекту или пациенту.
2.3. Любой из предыдущего способа II и всех последующих, где ингибитор PDE1 повышает уровень или экспрессию внутриклеточного cAMP.
2.4. Любой из предыдущего способа II и всех последующих, где ингибитор PDE1 (например, прямо или опосредовано) модулирует активность PKA и/или PKG.
2.5. Любой из предыдущего способа II и всех последующих, где ингибитор PDE1 (например, прямо или опосредовано) повышает активность PKA и/или PKG.
2.6. Любой из предыдущего способа II и всех последующих, где введение ингибитора PDE1 повышает уровень cAMP и/или cGMP.
2.7. Любой из предыдущего способа II и всех последующих, где введение ингибитора PDE1 повышает уровень внутриклеточного cAMP, и где этот повышенный уровень внутриклеточного cAMP защищает нервные волокна, регенерирует нервные волокна или промотирует рост нервных волокон (например, аксональной регенерации).
2.8. Любой из предыдущего способа II и всех последующих, включающий введение эффективного количества ингибитора PDE1 пациенту, который страдает от заболевания или расстройства, связанного с повышенными (например, хронически повышенными) уровнями внутриклеточного кальция.
2.9. Любой из предыдущего способа II и всех последующих, где ингибитор PDE1 вводят сам по себе или в комбинации с другим активным агентом.
2.10. Способ по пункту 2.9, где активный агент выбран из IGF (например, IGF-1) или стероида.
2.11. Любой из предыдущего способа II и всех последующих, где заболевание, расстройство или повреждение ПНС выбрано из группы, включающей: невропатию (например, периферическую невропатию, автономную невропатию и мононевропатию), ишиас, кистевой туннельный синдром, полиневропатию, диабетическую невропатию, постгерпетичекую невралгию и синдром торокального прохода.
[0051] В другом варианте осуществления изобретение касается способа III, где способ III включает композиции и способы предупреждения заболевания или расстройства ЦНС у субъекта, который находится в группе риска развития указанного заболевания или расстройства, где способ включает:
1) получение образца от субъекта;
2) измерение уровней внутриклеточного кальция в образце;
3) сравнение уровней внутриклеточного кальция в биологическом образце с эталоном;
4) определение, находится ли пациент в группе риска развития заболевания или расстройства ЦНС на основании уровня внутриклеточного кальция по сравнению с эталоном;
5) введение ингибитора PDE1 (например, соединения, соответствующего любой из формул V или 1.1-1.14) субъекту на основании уровня внутриклеточного кальция у субъекта (например, введение ингибитора PDE1 субъекту, так как он имеет повышенные уровни внутриклеточного кальция по сравнению с эталоном).
Например, способ III также включает:
3.1. Способ III, где образец является биологическим образцом.
3.2. Любой из предыдущего способа III и всех последующих, где уровни внутриклеточного кальция пациента определяют, применяя химический флуоресцентный зонд.
3.3. Любой из предыдущего способа III и всех последующих, где уровни внутриклеточного кальция пациента повышены по сравнению с контролем (например, эталон).
3.4. Любой из предыдущего способа III и всех последующих, где ингибитор PDE1 вводят пациенту, который, как показано, имеет повышенные уровни внутриклеточного кальция по сравнению с контролем (например, эталоном).
3.5. Любой из предыдущего способа III и всех последующих, где введение ингибитора PDE1 замедляет или предупреждает развитие заболевания или расстройства ЦНС и/или ПНС, где заболевание или расстройство ЦНС представляет собой заболевание или расстройство, которое коррелирует с повышенными (например, хронически повышенными) уровнями внутриклеточного кальция.
3.6. Любой из предыдущего способа III и всех последующих, где введение ингибитора PDE1 снижает вероятность развития у индивидуума заболевания или расстройства ЦНС и/или ПНС, где заболевание или расстройство ЦНС и/или ПНС представляет собой заболевание или расстройство, которое коррелирует с повышенными (например, хронически повышенными) уровнями внутриклеточного кальция (например, любое из заболеваний, расстройств или повреждений, перечисленных в способе I и последующих и способе II и последующих).
3.7. Любой из предыдущего способа III и всех последующих, где способ необязательно включает определение внутриклеточных уровней cAMP или cGMP пациента.
3.8. Любой из предыдущего способа III и всех последующих, где ингибитор PDE1 вводят сам по себе или в комбинации с другим активным агентом.
3.9. Любой из предыдущего способа III и всех последующих, где вводят ингибитор PDE1, так как пациент имеет низкие уровни cAMP и/или cGMP по сравнению с контрольным субъектом.
[0052] Соединения по изобретению пригодны для лечения заболеваний, характеризуемых нарушением или повреждением cAMP- и cGMP-опосредуемых путей, например, вследствие повышенной экспрессии PDE1 или пониженной экспрессии cAMP и cGMP в результате ингибирования или пониженных уровней индукторов синтеза циклических нуклеотидов, таких как дофамин и оксид азота (NO). Предотвращая деградацию cAMP и cGMP посредством PDE1, повышая тем самым внутриклеточные уровни cAMP и cGMP, соединения по изобретению усиливают активность индукторов синтеза циклических нуклеотидов.
[0053] В другом варианте осуществления изобретение также касается способов лечения, где способ включает введение эффективного количества ингибитора PDE1 (например, любого соединения формулы V или 1.1-1.14) для лечения любого одного или нескольких из следующих состояний:
(i) нейродегенеративные заболевания, в том числе болезнь Паркинсона, синдром беспокойных ног, треморы, дискинезия, болезнь Гентингтона, болезнь Альцгеймера и обусловленные действием лекарства нарушения движений;
(ii) ментальные расстройства, в том числе депрессия, синдром дефицита внимания, синдром дефицита внимания и гиперактивности, биполярное расстройство, состояние тревоги, нарушения сна, например, нарколепсия, когнитивное расстройство, например, когнитивное расстройство при шизофрении, деменции, синдроме Туретта, аутизме, синдроме ломкой Х-хромосомы, отмене психостимуляторов и наркомании;
(iii) расстройства кровообращения и сердечно-сосудистые расстройства, в том числе цереброваскулярное заболевание, инсульт, застойная сердечная недостаточность, гипертензия, легочная гипертензия, например, легочная артериальная гипертензия и сексуальная дисфункция, в том числе сердечно-сосудистые заболевания и родственные расстройства, которые описаны в международной заявке PCT/US2014/16741, содержание которой включено здесь посредством ссылки;
(iv) респираторные и воспалительные расстройства, в том числе астма, хроническая обструктивная болезнь легких и аллергический ринит, а также аутоиммунные и воспалительные заболевания;
(v) заболевания, которые можно облегчить, усиливая прогестероновую передачу сигналов, например, сексуальная дисфункция у женщин;
(vi) заболевание или расстройство, такое как психоз, глаукома или повышенное внутриглазное давление;
(vii) травматическое повреждение головного мозга;
(viii) любое заболевание или состояние, характеризуемое низкими уровнями cAMP и/или cGMP (или ингибированием cAMP- и/или cGMP-путей передачи сигналов) в клетках, экспрессирующих PDE1; и/или
(ix) любое заболевание или состояние, характеризуемое пониженной активностью передачи сигналов дофаминовым рецептором D1,
включающих введение эффективного количества соединения по изобретению, например, любого соединения формулы V или 1.1-1.14 в свободном виде, или в виде фармацевтически приемлемой соли, или в виде пролекарства, нуждающемуся в этом пациенту, человеку или животному.
[0054] В одном аспекте изобретение касается способов лечения или профилактики нарколепсии. В этом варианте осуществления можно использовать ингибиторы PDE1 (например, любое из соединений формул V или 1.1-1.14) в качестве единственного терапевтического агента, но также можно использовать в комбинации или для совместного введения с другими активными агентами. Таким образом, изобретение также включает способ лечения нарколепсии, включающий введение вместе, последовательно или одновременно терапевтически эффективных количеств
(i) ингибитора PDE1, например, соединения, соответствующего любой из формул V или 1.1-1.14) и
(ii) соединения для содействия бодрствованию или регулирования сна, например, выбранного из (a) стимуляторов центральной нервной системы амфетаминов и амфетамин-подобных соединений, например, метилфенидата, декстроамфетамина, метамфетамина и пемолина; (b) модафенила, (c) антидепрессантов, например, трициклических соединений (в том числе имипрамина, дезипрамина, кломипрамина и протриптилина) и селективных ингибиторов обратного захвата серотонина (в том числе флуоксетина и сертралина); и/или (d) гамма-гидроксибутирата (GHB),
в свободном виде, или в виде фармацевтически приемлемой соли, или в виде пролекарства, нуждающемуся в этом пациенту, человеку или животному.
[0055] В другом аспекте изобретение также касается способов лечения или профилактики состояния, которое можно облегчить, усиливая прогестероновую передачу сигналов, включающих введение нуждающемуся в этом пациенту, человеку или животному, эффективного количества соединения по изобретению, например, соединения, соответствующего любой из формул V или 1.1-1.14, в свободном виде, или в виде фармацевтически приемлемой соли, или в виде пролекарства. Заболевания или состояния, которые можно облегчить посредством усиления прогестероновой передачи сигналов, включают, но не ограничены этим, сексуальную дисфункцию у женщин, вторичную аменорею (например, аменорею при физических нагрузках, ановуляцию, менопаузу, симптомы менопаузы, гипотиреоз), предменструальный синдром, преждевременные роды, бесплодие, например, бесплодие вследствие повторных выкидышей, нерегулярные менструальные циклы, аномальное маточное кровотечение, остеопороз, аутоиммунное заболевание, множественный склероз, увеличение простаты, рак простаты и гипотиреоз. Например, при усилении прогестероновой передачи сигналов можно использовать ингибиторы PDE1 для содействия имплантации яйцеклетки посредством воздействия на выстилку матки, и в помощь сохранения беременности у женщин, которые подвержены выкидышам из-за иммунной реакции на беременность или низкой функции прогестерона. Новые ингибиторы PDE1, например, которые описаны здесь, также могут быть пригодны для усиления эффективности гормонозаместительной терапии, например, будучи введенными в комбинации с эстрогеном/эстрадиолом/эстриолом и/или прогестероном/прогестинами женщине в постклимактерическом периоде, и в случае эстроген-индуцированной эндометриальной гиперплазии и карциномы. Способы по изобретению пригодны также для животноводства, например, индуцируя сексуалную восприимчивость и/или эструс у млекопитающих женского пола, отличных от человека, подлежащих размножению.
[0056] В этом аспекте ингибиторы PDE1 можно использовать в предыдущих способах лечения или профилактики в качестве единственного терапевтического агента, а также можно использовать в комбинации или для совместного введения с другими активными агентами, например в сопряжении с гормонозаместительной терапией. Таким образом, изобретение также включает способ лечения расстройств, которые можно облегчить посредством усиления прогестероновой передачи сигналов, включающий введение вместе, последовательно или одновременно терапевтически эффективных количеств
(i) ингибитора PDE1, например, соединения, соответствующего любой из формул V или 1.1-1.14, и
(ii) гормона, например, выбранного из эстрогена и аналогов эстрогена (например, эстрадиола, эстриола, сложных эфиров эстрадиола), прогестерона и аналогов прогестерона (например, прогестинов),
в свободном виде, или в виде фармацевтически приемлемой соли, или в виде пролекарства нуждающемуся в этом пациенту, человеку или животному.
[0057] Изобретение также касается способа повышения или усиления активности внутриклеточной передачи сигналов дофаминовым рецептором D1 в клетке или ткани, включающего взаимодействие указанной клетки или ткани с количеством соединения по изобретению, например, соединения, соответствующего любой из формул V или 1.1-1.14, в свободном виде, или в виде фармацевтически приемлемой соли, или в виде пролекарства, достаточным для ингибирования активности PDE1.
[0058] Изобретение также касается способа лечения PDE1-связанного расстройства, нарушения пути внутриклеточной передачи сигналов дофаминовым рецептором D1 или расстройств, которые можно облегчить, усиливая прогестероновый путь передачи сигналов у нуждающегося в этом пациента, включающего введение пациенту эффективного количества соединения по изобретению, например, соединения, соответствующего любой из формул V или 1.1-1.14, в свободном виде, или в виде фармацевтически приемлемой соли, или в виде пролекарства, которое ингибирует PDE1, где активность PDE1 модулирует фосфорилирование DARPP-32 и/или GluR1 AMPA рецептора.
[0059] В другом аспекте изобретение также касается способа лечения глаукомы или повышенного внутриглазного давления, включающий локальное введение терапевтически эффективного количества ингибитора PDE1 по изобретению, например, соединения, соответствующего любой из формул V или 1.1-1.14, в свободном виде или в виде фармацевтически приемлемой соли в офтальмологически приемлемом носителе в глаз нуждающегося в этом пациента. Однако, по-другому, лечение может включать системную терапию. Системная терапия включает, например, лечение, при котором лекарство может попадать непосредственно в кровоток, или пероральные способы введения.
[0060] Изобретение также касается фармацевтической композиции для локального офтальмологического применения, содержащей ингибитор PDE1; например, офтальмологического раствора, суспензии, крема или мази, содержащей ингибитор PDE1 по изобретению, например, соединение, соответствующее любой из формул V или 1.1-1.14, в свободном виде или в виде офтальмологически приемлемой соли в комбинации или ассоциации с офтальмологически приемлемым разбавителем или носителем.
[0061] Необязательно ингибитор PDE1 (например, соединение, соответствующее любой из формул V или 1.1-1.14) можно вводить последовательно или одновременно со вторым лекарством, пригодным для лечения глаукомы или повышенного внутриглазного давления. Если вводят два активных агента, то терапевтически эффективные количества каждого агента могут быть ниже количества, необходимого для активности в качестве монотерапевтического агента. Таким образом, подпороговое количество (то есть количество ниже уровня, необходимого для эффективности в качестве монотерапевтического агента) может считаться терапевтически эффективным и также может альтернативно обозначаться как эффективное количество. Действительно, преимуществом введения различных агентов с разными механизмами действия и разными профилями побочных эффектов может быть снижение дозировки и побочных эффектов одгого или обоих агентов, а также повышение или усиление их активности в качестве монотерапевтических агентов.
[0062] Таким образом, изобретение касается способа лечения состояния, выбранного из глаукомы и повышенного внутриглазного давления, включающего введение нуждающемуся в этом пациенту эффективного количества, например, подпорогового количества агента, снижающего, как известно, внутриглазное давление, вместе, одновременно или последовательно с эффективным количеством, например, подпороговым количеством ингибитора PDE1 по изобретению, например, соединением, соответствующим любой из формул V или 1.1-1.14, в свободном виде или в виде фармацевтически приемлемой соли, так чтобы количество агента, снижающего, как известно, внутриглазное давление, и количество ингибитора PDE1 в комбинации было эффективным для лечения состояния.
[0063] В одном аспекте один или оба агента вводят локально в глаз. Таким образом, изобретение касается способа снижения побочных эффектов лечения глаукомы или повышенного внутриглазного давления посредством введения уменьшенной дозы агента, как известно, снижающего внутриглазное давление, вместе, одновременно или последовательно с эффективным количеством ингибитора PDE1. Однако также можно применять способы, отличные от локального введения, например системное терапевтическое введение.
[0064] Необязательный дополнительный агент или агенты для использования в комбинации с ингибитором PDE1 могут быть выбраны, например, из существующих лекарств и обычно включают вводимый капельным способом простагландин, пилокарпин, эпинефрин или бета-блокатор для локального лечения, например, тимол, а также вводимые системно ингибиторы карбоангидразы, например, ацетазоламид. Ингибиторы холинэстеразы, такие как физостигмин и эхотиопат, также могут быть использованы и оказывают действие, подобное действию пилокарпина. Таким образом, лекарства, применяемые в настоящее время для лечения глаукомы, включают, например:
1. Аналоги простагландина, такие как латанопрост (ксалатан), биматопрост (лумиган) и травопрост (траватан), которые увеличивают увеосклеральный отток внутриглазной жидкости. Биматопрост также повышает трабекулярный отток.
2. Локальные антагонисты бета-адренергических рецепторов, такие как тимол, левобунолол (бетаган) и бетаксолол, которые снижают производство внутриглазной жидкости цилиарным телом.
3. Альфа2-адренергические агонисты, такие как бримонидин (альфаган), которые действуют по двойственному механизму, снижая производство воды и увеличивая увеосклеральный отток.
4. Менее селективные симпатомиметики подобно эпинефрину и дипивефрину (пропин) увеличивают отток внутриглазной жидкости через трабекулярную сеть и, возможно, через увеосклеральный путь оттока, вероятно, посредством действия бета2-агониста.
5. Миотические агенты (пара-симпатомиметики), подобные пилокарпину, действуют посредством противодействия цилиарной мышце, сжимая трабекулярную сеть и допуская увеличенный отток водянистой влаги.
6. Ингибиторы карбоангидразы, подобные дорзоламиду (трусопту), бринзоламиду (азопту), ацетазоламиду (диамоксу), снижают секрецию внутриглазной жидкости посредством ингибирования карбоангидразы в цилиарном теле.
7. Физостигмин также используют для лечения глаукомы и замедленной эвакуации содержимого желудка.
[0065] Например, изобретение касается фармацевтических композиций, содержащих ингибитор PDE1 по изобретению, например, соединение, соответствующее любой из формул V или 1.1-1.14, в свободном виде или в виде фармацевтически приемлемой соли, и агент, выбранный из (i) простаноидов, унопростона, латанопроста, травопроста или биматопроста; (ii) альфа адренергического агониста, такого как бримонидин, апраклонидин или дипивефрин, и (iii) мускаринового агониста, такого как пилокарпин, в комбинации или ассоциации с фармацевтически приемлемым разбавителем или носителем. Например, изобретение касается глазных препаратов, содержащих ингибитор PDE1 по изобретению, например, соединение, соответствующее любой из формул V или 1.1-1.14, вместе с биматопростом, aбримонидином, бримонидином, тимолом или их комбинацией, в свободном виде или в виде офтальмологически приемлемой соли, в комбинации или ассоциации с офтальмологически приемлемым разбавителем или носителем. Однако кроме выбора комбинации рядовой специалист в данной области может выбрать подходящий селективный агонист или антагонист подтипа рецептора. Например, для альфа-адренергического агониста, можно выбрать агонист, селективный относительно альфа1-адренергического рецептора, или агонист, селективный относительно альфа2-адренергического рецептора, такой как бримонидин. Для антагониста бета-адренергических рецепторов можно выбрать антагонист, селективный относительно β1, или β2, или β3 в зависимости от подходящего терапевтического применения. Можно также выбрать мускариновый агонист, селективный относительно конкретного подтипа рецептора, например, M1-M5.
[0066] Ингибитор PDE1 можно вводить в виде офтальмологической композиции, которая включает офтальмологический раствор, крем или мазь. Офтальмологическая композиция может дополнительно включать агент, снижающий внутриглазное давление.
[0067] Еще в одном примере раскрытые ингибиторы PDE1 могут быть объединены с подпороговым количеством агента, снижающего внутриглазное давление, который может представлять собой офтальмологический раствор биматопроста, офтальмологический раствор бримонидин тартрата или офтальмологический раствор бримонидин тартрата/тимол малеата.
[0068] Кроме указанных выше способов также было неожиданно обнаружено, что ингибиторы PDE1 (например, соединения, соответствующие любой из формул V или 1.1-1.14) пригодны для лечения психоза, например, любых состояний, характеризуемых психотическими симптомами, такими как галлюцинации, параноидальные или аномальные идеи или дезорганизованная речь и мышление, например, шизофрении, шизоаффективного расстройства, шизофреноформного расстройства, психотического расстройства, бредового расстройства и мании, например, при острых маниакальных эпизодах и биполярном расстройстве. Не предполагая связываться какой-либо теорией, считают, что типичные и нетипичные антипсихотические лекарства, такие как клозапин, обладают главным образом антагонистической активностью относительно дофаминового рецептора D2. Однако ингибиторы PDE1 действуют главным образом, усиливая передачу сигналов дофаминовым рецептором D1. Усиливая передачу сигналов рецептором D1, ингибиторы PDE1 могут повышать функцию рецептора NMDA в различных областях головного мозга, например, в нейронах прилегающего ядра и в префронтальной коре. Это усиление функции можно видеть, например, в рецепторах NMDA, содержащих субъединицу NR2B, и оно может происходить, например, через активацию Src- и протеинкиназа A-семейства киназ.
[0069] Следовательно, изобретение касается нового способа лечения психоза, например, шизофрении, шизоаффективного расстройства, шизофреноформного расстройства, психотического расстройства, бредового расстройства и мании, например, при острых маниакальных эпизодах и биполярном расстройстве, включающего введение нуждающемуся в этом пациенту терапевтически эффективного количества ингибитора фосфодиэстеразы-1 (PDE1) по изобретению, например, соединения, соответствующего любой из формул V или 1.1-1.14, в свободном виде или в виде фармацевтически приемлемой соли.
[0070] Ингибиторы PDE1 можно использовать в предыдущих способах лечения и профилактики в качестве единственного терапевтического агента, а также можно использовать в комбинации или для совместного введения с другими активными агентами. Таким образом, изобретение также включает способ лечения психоза, например, шизофрении, шизоаффективного расстройства, шизофреноформного расстройства, психотического расстройства, бредового расстройства или мании, включающий введение вместе, последовательно или одновременно терапевтически эффективных количеств:
(i) ингибитора PDE1 по изобретению в свободном виде или в виде фармацевтически приемлемой соли и
(ii) антипсихотических средств, например, типичных антипсихотических средств, таких как
бутирофеноны, например, галоперидол (Haldol, Serenace), дроперидол (Droleptan);
фенотиазины, например, хлорпромазин (Thorazine, Largactil), флуфеназин (Prolixin), перфеназин (Trilafon), прохлорперазин (Compazine), тиоридазин (Mellaril, Melleril), трифторперазин (Stelazine), мезоридазин, перициазин, промазин, трифлупромазин (Vesprin), левомепромазин (Nozinan), прометазин (Phenergan), пимозид (Orap);
тиоксантены, например, хлорпротиксен, флупентиксол (Depixol, Fluanxol), Тиотиксен (Navane), зуклопентиксол (Clopixol, Acuphase);
неатипичные антипсихотические средства, например, клозапин (Clozaril), оланзапин (Zyprexa), рисперидон (Risperdal), кветиапин (Seroquel), зипрасидон (Geodon), амисулприд (Solian), палиперидон (Invega), арипипразол (Abilify), буфепринокс; норклозапин,
в свободном виде или в виде фармацевтически приемлемой соли нуждающемуся в этом пациенту.
[0071] В конкретном варианте осуществления соединения по изобретению особенно подходят для лечения или профилактики шизофрении.
[0072] Соединения по изобретению в свободном виде или в виде фармацевтически приемлемой соли особенно подходят для лечения болезни Паркинсона, шизофрении, нарколепсии, глаукомы и сексуальной дисфункции у женщин.
[0073] Еще в одном аспекте изобретение касается способа удлинения или усиления роста ресниц посредством введения эффективного количества аналога простагландина, например, биматопроста, вместе, одновременно или последовательно с эффективным количеством ингибитора PDE1 по изобретению в свободном виде или в виде фармацевтически приемлемой соли в глаз нуждающегося в этом пациента.
[0074] Еще в одном аспекте изобретение касается способа лечения или профилактики травматического повреждения головного мозга, включающего введение терапевтически эффективного количества ингибитора PDE1 по изобретению, например, соединения, соответствующего любой из формул V или 1.1-1.14, в свободном виде или в виде фармацевтически приемлемой соли нуждающемуся в этом пациенту. Травматическое повреждение головного мозга (TBI) охватывает первичное повреждение, а также вторичное повреждение, в том числе фокальные и диффузные повреждения головного мозга. Вторичные повреждения представляют собой множественные, параллельные, взаимодействующие и взаимозависимые каскады биологических реакций происходящих в результате дискретных субклеточных процессов (например, токсичность, обусловленная реактивными видами кислорода, чрезмерной стимуляцией глутаматных рецепторов, избыточным притоком кальция и воспалительной повышающей регуляцией) которые являются результатом или обостряются вследствие воспалительной реакции и прогрессируют после начального (первичного) повреждения.
[0075] Настоящее изобретение также касается
(i) соединения по изобретению, например, соединения, соответствующего любой из формул V или 1.1-1.14, которые описаны здесь ранее, в свободном виде или в виде фармацевтически приемлемой соли, например, для использования в любом способе или для лечения любого заболевания или состояния, которое указано здесь ранее;
(ii) применения соединения по изобретению, например, соединения, соответствующего любой из формул V или 1.1-1.14, которое описано здесь ранее, в свободном виде или в виде фармацевтически приемлемой соли, (для производства медикамента) для лечения любого заболевания или состояния, которое указано здесь ранее;
(iii) фармацевтической композиции, содержащей соединение по изобретению, например, соединение, соответствующее любой из формул V или 1.1-1.14, которое описано здесь ранее, в свободном виде или в виде фармацевтически приемлемой соли в комбинации или ассоциации с фармацевтически приемлемым разбавителем или носителем; и
(iv) фармацевтической композиции, содержащей соединение по изобретению, например, соединение, соответствующее любой из формул V или 1.1-1.14, которое описано здесь ранее, в свободном виде или в виде фармацевтически приемлемой соли в комбинации или ассоциации с фармацевтически приемлемым разбавителем или носителем для использования при лечении любого заболевания или состояния, которое указано здесь ранее.
[0076] Следовательно, изобретение касается применения соединения по изобретению, например, соединения, соответствующего любой из формул V или 1.1-1.14, которое описано здесь ранее, в свободном виде или в виде фармацевтически приемлемой соли, или соединения по изобретению в виде фармацевтической композиции (для производства медикамента) для лечения или профилактики любого одного или нескольких из следующих заболеваний: болезнь Паркинсона, синдром беспокойных ног, треморы, дискинезии, болезнь Гентингтона, болезнь Альцгеймера и/или вызванные лекарственными средствами двигательные расстройства; депрессия, расстройство, характеризующееся дефицитом внимания, синдром дефицита внимания и гиперактивности, биполярное заболевание, состояние тревоги, расстройство сна, нарколепсия, когнитивное расстройство, например, когнитивное расстройство при шизофрении, деменция, синдром Туретта, аутизм, синдром ломкой Х-хромосомы, отмена психостимуляторов и/или наркомания; цереброваскулярное заболевание, инсульт, застойная сердечная недостаточность, гипертензия, легочная гипертензия, например, легочная артериальная гипертензия и/или сексуальная дисфункци; астма, хроническая обструктивная болезнь легких и/или аллергический ринит, а также аутоиммунные и воспалительные заболевания; и/или сексуальная дисфункция у женщин, аменорея при физических нагрузках, ановуляция, менопауза, симптомы менопаузы, гипотиреоз, предменструальный синдром, преждевременные роды, бесплодие, нерегулярные менструальные циклы, аномальное маточное кровотечение, остеопороз, множественный склероз, увеличение простаты, рак простаты, гипотиреоз и/или эстроген-индуцированная эндометриальная гиперплазия и/или карцинома; и/или любое заболевание или состояние, характеризуемое низкими уровнями cAMP и/или cGMP (или ингибирование cAMP и/или cGMP пути передачи сигналов) в клетках, экспрессирующих PDE1, и/или пониженной активностью передачи сигналов дофаминовым рецептором D1; и/или любое заболевание или состояние, которое можно облегчить, усиливая прогестероновую передачу сигналов.
[0077] Изобретение также касается применения соединения по изобретению в свободном виде или в виде фармацевтически приемлемой соли (для производства лекарственного средства) для лечения или профилактики любого одного или нескольких из следующих заболеваний:
a) глаукома, повышенное внутриглазное давление,
b) психоз, например, любые состояния, характеризуемые психотическими симптомами, такими как галлюцинации, параноидальные или аномальные идеи, или дезорганизованная речь и мышление, например, шизофрения, шизоаффективное расстройство, шизофреноформное расстройство, психотическое расстройство, бредовое расстройство и мания, например, при острых маниакальных эпизодах и биполярное расстройство;
c) травматическое повреждение головного мозга и/или
d) центральные и периферические дегенеративные расстройства, особенно расстройства с воспалительными компонентами.
[0078] Выражение «соединение по изобретению» или «ингибиторы PDE1 по изобретению» охватывает любое и все раскрытые здесь соединения, например, соединение формулы V или 1.1-1.14.
[0079] Термины «лечение» и «обработка» следует понимать как охватывающие профилактику, лечение или облегчение симптомов заболевания, а так же лечение причины заболевания.
[0080] Для способов лечения используемое здесь выражение «терапевтически эффективное количество» обозначает количество лекарственного средства (например, ингибитора PDE1), достаточное для лечения или облегчения патологических эффектов заболевания, расстройства или повреждения ЦНС или ПНС. Например, терапевтически эффективное количество ингибитора PDE1 может составлять количество, достаточное, например, для повышения внутриклеточных уровней cAMP или cGMP, снижения внутриклеточных уровней кальция и/или повышения нейрорегенерации. Где уместно, терапевтически эффективное количество может также составлять количество ингибитора PDE1, необходимое для снижения или предупреждения развития заболевания или расстройства ЦНС или ПНС.
[0081] Термин «пациент» или «субъект» касается человека или пациента, отличного от человека (то есть животного). В конкретном варианте осуществления изобретение охватывает пациентов людей и пациентов, отличных от людей. В другом варианте осуществления изобретение охватывает пациентов, отличных от людей. В другом варианте осуществления термин охватывает пациентов людей.
[0082] Используемый здесь термин «контрольный субъект» обозначает любой организм, человеческий или отличный от человеческого, в котором не имеется и/или не подозревается о наличии расстройства, синдрома, заболевания, состояния и/или симптома заболевания ЦНС или ПНС. Используемый здесь термин «эталон» относится к предшествующему измерению и получению результатов для контрольного субъекта или популяции контрольных субъектов. В другом аспекте термин «эталон» относится к предшествующему измерению и получению результатов для пациента до развития у него или нее расстройства, синдрома, заболевания, состояния и/или симптома заболевания ЦНС или ПНС.
[0083] Используемый здесь термин «биологический образец» может включать любой образец, содержащий биологический материал, полученный, например, из организма, биологической жидкости, продуктов выделения, клетки или части клетки, клеточной линии, биопсии, культуры ткани или другого источника, содержащего внутриклеточный кальций, cAMP или cGMP.
[0084] «Нейрогенный агент» определяют как химический агент или реагент, который промотирует, стимулирует или другим образом повышает количество, или степень, или природу нейрогенеза in vivo, или ex vivo, или in vitro, относительно количества, степени или природы нейрогенеза в отсутствие агента или реагента.
[0085] Используемый здесь термин «повреждение ЦНС» может включать, например, повреждение клеток ретинального ганглия, травматическое повреждение головного мозга, повреждение, связанное с инсультом, повреждение, связанное с церебральной аневризмой, повреждение спинного мозга или повреждение, включающее моноплегию, диплегию, параплегию, гемиплегию и квадраплегию, нейропролиферативное расстройство или невропатический болевой синдром. Используемый здесь термин «повреждение ПНС» может включать, например, повреждение спинальных или черепно-мозговых нервов, где повреждение может включать поражение или некоторое острое или хроническое повреждение.
[0086] Соединения по изобретению (например, любое соединение формулы V или 1.1-1.14), которые описаны здесь ранее, в свободном виде или в виде фармацевтически приемлемой соли можно использовать в качестве единственного терапевтического агента, а также можно использовать в комбинации или для совместного введения с другими активными агентами.
[0087] Конечно, используемые в практике настоящего изобретения дозы очень зависят, например, от подлежащего лечению конкретного заболевания или состояния, конкретного используемого соединения по изобретению, способа введения и желательной терапии. Соединения по изобретению можно вводить подходящим способом, в том числе перорально, парентерально, трансдермально или посредством ингаляции, но предпочтительно вводят перорально. Вообще, удовлетворительные результаты, например, лечения перечисленных здесь ранее заболеваний, как указано, получены при пероральном введении доз примерно от 0,01 до 2,0 мг/кг. Для более крупных млекопитающих, например людей, указанная дневная доза при пероральном приеме, соответственно, находится в диапазоне примерно от 0,75 до 150 мг, и ее удобно вводить один раз в день, или поделенными дозами от 2 до 4 раз в день, или в виде формы с длительным высвобождением. Таким образом, стандартные дозированные формы для перорального введения могут содержать, например, от примерно 0,2 до 75 или 150 мг, например, от примерно 0,2 или 2,0 до 50, 75 или 100 мг соединения по изобретению вместе с фармацевтически приемлемым разбавителем или носителем.
[0088] Фармацевтические композиции, содержащие соединения по изобретению, можно получить, используя общеизвестные разбавители или наполнители и методики, известные в области медицины. Таким образом, дозированные формы для перорального введения могут включать таблетки, капсулы, растворы, суспензии и подобное.
ПРИМЕРЫ
Пример 1
7,8-Дигидро-2-(4-(пиридин-2-ил)бензил)-3-(4-фторфениламино)-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он
(a) 7-(4-Метоксибензил)-5-метил-2-(4-(пиридин-2-ил)бензил)-2H-пиразоло[3,4-d]пиримидин-4,6(5H,7H)-дион
[0089] Суспензию 7-(4-метоксибензил)-5-метил-2H-пиразоло[3,4-d]пиримидин-4,6(5H,7H)-диона (8,43 г, 29,4 ммоль), 2-(4-(хлорметил)фенил)пиридина (6,0 г, 29,4 ммоль) и K2CO3 (4,07 г, 29,4 ммоль) в ДМФА (100 мл) перемешивают при комнатной температуре в течение ночи. Растворитель удаляют при пониженном давлении. Полученный остаток обрабатывают водой (150 мл) и гексаном (25 мл). Смесь перемешивают при комнатной температуре в течение часа и затем фильтруют. Осадок на фильтре промывают водой три раза (3×50 мл) и затем сушат в вакууме, получая 13 г сырого продукта (выход: 97%), который используют на следующей стадии без дополнительной очистки. МС (ESI) m/z 454,2 [M+H]+.
(b) 5-Метил-2-(4-(пиридин-2-ил)бензил)-2H-пиразоло[3,4-d]пиримидин-4,6(5H,7H)-дион
[0090] Добавляют ТФУ (50 мл) в суспензию 7-(4-метоксибензил)-5-метил-2-(4-(пиридин-2-ил)бензил)-2H-пиразоло[3,4-d]пиримидин-4,6(5H,7H)-диона (13 г, 28,7 ммоль) в метиленхлориде (80 мл), получая желто-коричневый раствор, и затем добавляют TFMSA (4 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворители удаляют при пониженном давлении. Полученный остаток обрабатывают водой (150 мл), охлаждают до 0°C и затем доводят pH до 8-9 при помощи 28% гидроксида аммония (примерно 35 мл). После фильтрования полученные твердые вещества промывают водой три раза (3×50 мл) и затем сушат в вакууме, получая 12,8 г сырого продукта (выход сырого продукта: 134%), который используют на следующей стадии без дополнительной очистки. МС (ESI) m/z 334,1 [M+H]+.
(c) 6-Хлор-5-метил-2-(4-(пиридин-2-ил)бензил)-2H-пиразоло[3,4-d]пиримидин-4(5H)-он
[0091] 5-Метил-2-(4-(пиридин-2-ил)бензил)-2H-пиразоло[3,4-d]пиримидин-4,6(5H,7H)-дион (8,5 г, 25,5 ммоль) суспендируют в POCl3 (300 мл) и затем медленно нагревают до кипения с обратным холодильником. После кипячения смеси с обратным холодильником в течение 30 час. удаляют POCl3 при пониженном давлении. Полученный остаток обрабатывают водой (300 мл), охлаждают до 0°C и затем доводят pH до 8-9 при помощи 28% гидроксида аммония (примерно 30 мл). После фильтрования полученные твердые вещества промывают водой пять раз (5×50 мл) и затем сушат в вакууме, получая 8,6 г сырого продукта (выход сырого продукта: 96%), который используют на следующей стадии без дополнительной очистки. МС (ESI) m/z 352,1 [M+H]+.
(d) 6-(1-Гидрокси-2-метилпропан-2-иламино)-5-метил-2-(4-(пиридин-2-ил)бензил)-2H-пиразоло[3,4-d]пиримидин-4(5H)-он
[0092] Смесь 6-хлор-5-метил-2-(4-(пиридин-2-ил)бензил)-2H-пиразоло[3,4-d]пиримидин-4(5H)-она (4,0 г, 11 ммоль), 2-амино-2-метилпропан-1-ола (6,5 мл, 71 ммоль) и ДИПЭА (3,4 мл, 20 ммоль) в DMA (20 мл) нагревают при 130°C в течение часа. Растворитель удаляют при пониженном давлении. Полученный остаток обрабатывают водой (200 мл). После фильтрования осадок на фильтре промывают водой дважды (2×50 мл) и затем сушат в вакууме, получая 3,7 г сырого продукта (выход сырого продукта: 80%), который используют на следующей стадии без дополнительной очистки. МС (ESI) m/z 405,2 [M+H]+.
(e) 7,8-Дигидро-2-(4-(пиридин-2-ил)бензил)-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он
[0093] Добавляют тионилхлорид (756 мкл, 10,4 ммоль) по капле к раствору сырого 6-(1-гидрокси-2-метилпропан-2-иламино)-5-метил-2-(4-(пиридин-2-ил)бензил)-2H-пиразоло[3,4-d]пиримидин-4(5H)-она (4,2 г, 10,4 ммоль) в ДМФА (84 мл). Реакционную смесь перемешивают при комнатной температуре в течение 20 мин. Добавляют воду (5 мл), чтобы погасить реакцию. Растворители удаляют при пониженном давлении. Полученный остаток обрабатывают метиленхлоридом и затем промывают 5% водным раствором NaHCO3 три раза. Органическую фазу выпаривают досуха, получая 6,1 г сырого продукта (выход сырого продукта: 152%), который используют на следующей стадии без дополнительной очистки. МС (ESI) m/z 387,2 [M+H]+.
(f) 7,8-Дигидро-2-(4-(пиридин2-ил)бензил)-3-хлор-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он
[0094] Добавляют 1,0M LiHMDS (55,4 мл, 55,4 ммоль) в ТГФ по капле к раствору сырого 7,8-дигидро-2-(4-(пиридин2-ил)бензил)-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-она (4,6 г, 11,9 ммоль) и гексахлорэтана (2,58 г, 10,9 ммоль) в метиленхлориде (130 мл) при 0°C. Реакционную смесь перемешивают при 0°C в течение 30 мин. и затем гасят водой (100 мл) и метиленхлоридом (150 мл). Органическую фазу промывают водой три раза (3×70 мл) и затем выпаривают досуха. Полученный сырой продукт очищают на колонке с нейтральным оксидом алюминия, получая 1,5 г чистого продукта (ВЭЖХ чистота: 96%; выход: 30%). МС (ESI) m/z 421,1 [M+H]+.
(g) 7,8-Дигидро-2-(4-(пиридин2-ил)бензил)-3-(4-фторфениламино)-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он
[0095] 7,8-Дигидро-2-(4-(пиридин2-ил)бензил)-3-хлор-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он (550 мг, 1,31 ммоль), 4-фторбензоламин (125 мкл, 1,31 ммоль) и карбонат калия (361 мг, 2,61 ммоль) в трет-амиловом спирте (3 мл) дегазируют аргоном и затем добавляют ксантфос (15 мг, 0,026 ммоль) и Pd2(dba)3 (12 мг, 0,013 ммоль). Суспензию снова дегазируют и затем медленно нагревают до 110°C. Реакционную смесь перемешивают при 110°C в атмосфере аргона в течение ночи. Добавляют еще одну порцию Pd2(dba)3 (12 мг) и ксантфоса (15 мг). Реакционную смесь нагревают при 110°C еще 24 час. для полной конверсии. После рутинной обработки сырой продукт очищают методом колоночной хроматографии на силикагеле, получая 352 мг конечного продукта в виде бежевого твердого вещества (ВЭЖХ-чистота: 97,4%; выход: 54%). 1H ЯМР (500 MГц, хлороформ-d) δ: 8,68 (дт, J=4,7, 1,3 Гц, 1H), 7,88 (д, J=8,3 Гц, 2H), 7,74 (тд, J=7,7, 1,8 Гц, 1H), 7,68 (д, J=8,0 Гц, 2H), 7,23 (ддд, J=7,4, 4,8, 1,2 Гц, 1H), 7,06 (д, J=8,3 Гц, 2H), 7,00-6,93 (м, 2H), 6,94-6,87 (м, 2H), 6,79 (с, 1H), 4,90 (с, 2H), 3,71 (с, 2H), 3,35 (с, 3H), 1,40 (с, 6H). МС (ESI) m/z 496,2 [M+H]+.
[0096] Соединение из примера 1 демонстрирует хорошую селективность относительно PDE1 и ингибирует активность PDE при значении IC50, равном или меньшем 5 нМ.
Пример 2
7,8-Дигидро-2-(4-(6-фторпиридин-2-ил)бензил)-3-(4-фторфениламино)-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он
[0097] Способ синтеза аналогичен способу примера 1, где на стадии (a) добавляют 2-(4-(хлорметил)фенил)-6-фторпиридин вместо 2-(4-(хлорметил)фенил)пиридина. Конечный продукт получают в виде не совсем белого твердого вещества (ВЭЖХ-чистота: 99%). 1H ЯМР (500 MГц, хлороформ-d) δ: 7,89 (д, J=8,4 Гц, 2H), 7,83 (кв., J=8,0 Гц, 1H), 7,58 (дд, J=7,5, 2,3 Гц, 1H), 7,05 (д, J=8,3 Гц, 2H), 7,00-6,84 (м, 6H), 4,91 (с, 2H), 3,76 (с, 2H), 3,39 (с, 3H), 1,47 (с, 6H). МС (ESI) m/z 514,3 [M+H]+
Пример 3
7,8-Дигидро-2-(4-(пиридин-2-ил)бензил)-3-(3,4-дифторфениламино)-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он
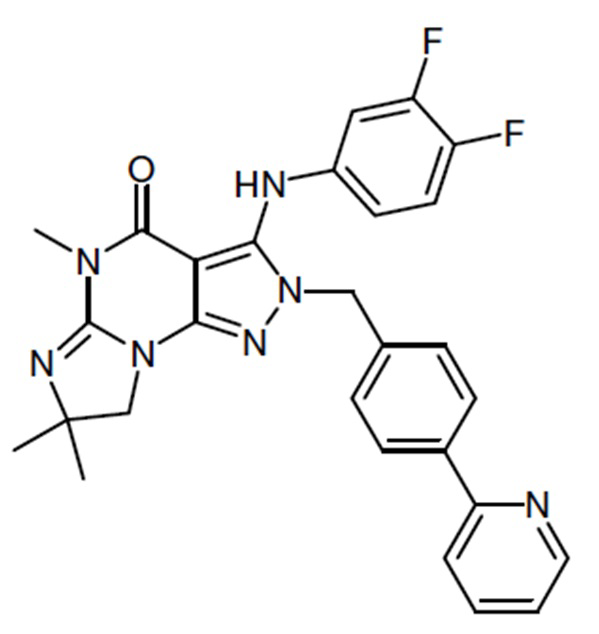
(a) 2-(4-Бромбензил)-7,8-дигидро-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он
[0098] Указанное в заголовке соединение синтезируют применяя методику, аналогичную методике, описанной для стадий (a)-(e) примера 1, где на стадии (a) добавляют 1-бром-4-(бромметил)бензол вместо 2-(4-(хлорметил)фенил)пиридина. МС (ESI) m/z 388,1 [M+H]+.
(b) 2-(4-Феноксибензил)-7,8-дигидро-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он
[0099] 2-(4-Бромбензил)-7,8-дигидро-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он (118 г, 304 ммоль) добавляют к суспензии фенола (57 г, 606 ммоль) и карбоната цезия (200 г, 614 ммоль) в NMP (900 мл) с последующим добавлением 2,2,6,6-тетраметилгептан-3,5-диона (7 мл, 33,5 ммоль) и CuCl (15 г, 152 ммоль). Реакционную смесь нагревают при 120°C в атмосфере азота в течение 10 час. По завершении взаимодействия смесь разбавляют водой (4 л) и затем экстрагируют этилацетатом. Объединенную органическую фазу выпаривают досуха. Полученный сырой продукт очищают методом колоночной хроматографии на силикагеле, получая 103 г продукта (выход: 84%). МС (ESI) m/z 402,2 [M+H]+.
(c) 7,8-Дигидро-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он
[0100] Добавляют ТФУ (600 мл) к суспензии 2-(4-феноксибензил)-7,8-дигидро-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-она (103 г, 257 ммоль) в метиленхлориде (210 мл), получая желто-коричневый раствор, и затем добавляют TFMSA (168 мл). Реакционную смесь перемешивают при комнатной температуре до исчезновения исходного материала. Реакционную смесь выливают в холодную воду (3 л). После фильтрования осадок на фильтре дважды промывают водой и затем подщелачивают водным раствором гидроксида аммония, а затем добавляют этилацетат при перемешивании. Осажденные твердые вещества отфильтровывают, промывают последовательно три раза водой, два раза этилацетатом и один раз метанолом и затем сушат в вакууме, получая 45 г продукта (выход: 80%). МС (ESI) m/z 220,2 [M+H]+.
(d) 7,8-Дигидро-2-(4-(пиридин-2-ил)бензил)-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он
[0101] Суспензию 7,8-дигидро-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-она (1,5 г, 6,84 ммоль), 2-(4-(бромметил)фенил)пиридина (1,7 г, 6,84 ммоль) и K2CO3 (2,83 г, 20,5 ммоль) в ДМФА (60 мл) перемешивают при комнатной температуре в течение 2-3 дней. Растворитель удаляют при пониженном давлении. Полученный остаток обрабатывают водой (100 мл), воздействуют ультразвуком и затем фильтруют. Осадок на фильтре сушат в вакууме, получая 2,19 г сырого продукта (выход: 83%), который используют на следующей стадии без дополнительной очистки. МС (ESI) m/z 387,1 [M+H]+.
(e) 7,8-Дигидро-2-(4-(пиридин-2-ил)бензил)-3-хлор-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он
[0102] 1,0M LiHMDS (3,0 мл, 3,0 ммоль) в ТГФ добавляют по капле к раствору сырого 7,8-дигидро-2-(4-(пиридин2-ил)бензил)-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-она (1,16 г, 3,0 ммоль) и гексахлорэтана (2,13 г, 9,0 ммоль) в метиленхлориде (30 мл). Реакционную смесь перемешивают при комнатной температуре в течение 90 мин. и затем гасят холодной водой (200 мл). Смесь экстрагируют метиленхлоридом три раза (50 мл×3) и объединенную органическую фазу промывают насыщенным солевым раствором (30 мл), а затем выпаривают досуха при пониженном давлении. Полученный остаток очищают на колонке с нейтральным оксидом алюминия, получая 960 мг чистого продукта в виде не совсем белого твердого вещества (ВЭЖХ чистота: 96,8%; выход: 76%). МС (ESI) m/z 421,2 [M+H]+.
(f) 7,8-Дигидро-2-(4-(пиридин-2-ил)бензил)-3-(3,4-дифторфениламино)-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он
[0103] 7,8-Дигидро-2-(4-(пиридин2-ил)бензил)-3-хлор-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он (230 мг, 0,546 ммоль), 3,4-дифторбензоламин (106 мг, 0,821 ммоль) и карбонат калия (300 мг, 2,17 ммоль) в трет-амиловом спирте (2,8 мл) дегазируют аргоном и затем добавляют ксантфос (26 мг, 0,045 ммоль) и Pd2(dba)3 (20 мг, 0,022 ммоль). Суспензию снова дегазируют и затем нагревают до 110°C. Реакционную смесь перемешивают при 110°C в атмосфере аргона в течение ночи. После рутинной обработки сырой продукт очищают на колонке с основным оксидом алюминия, получая 194 мг конечного продукта в виде бежевого твердого вещества (ВЭЖХ-чистота: 99%; выход: 69%). 1H ЯМР (500 MГц, хлороформ-d) δ: 8,69 (д, J=4,5 Гц, 1H), 7,88 (д, J=8,2 Гц, 2H), 7,76 (тд, J=7,8, 1,6 Гц, 1H), 7,67 (д, J=7,9 Гц, 1H), 7,26-7,17 (м, 2H), 7,15 (д, J=8,2 Гц, 2H), 7,03 (м, 1H), 6,69 (м, 1H), 6,60 (м, 1H), 5,05 (с, 2H), 3,79 (с, 2H), 3,29 (с, 3H), 1,47 (с, 6H). МС (ESI) m/z 514,2 [M+H]+.
Пример 4
7,8-Дигидро-2-(4-(пиридин2-ил)бензил)-3-(4-фтор-3-метилфениламино)-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он
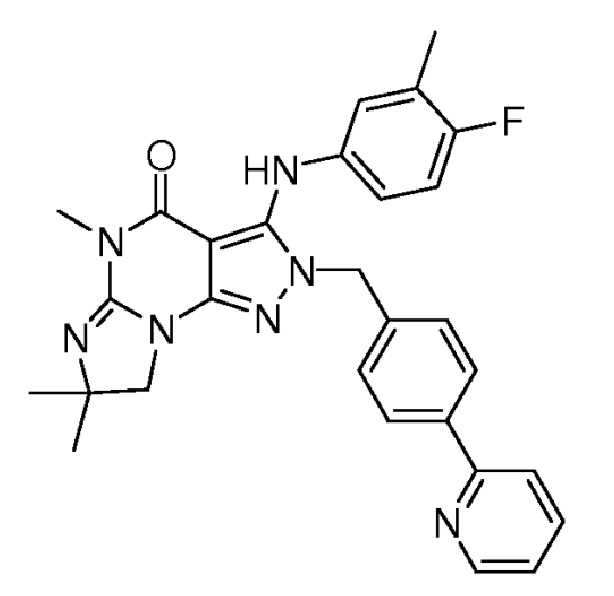
[0104] Способ синтеза аналогичен способу примера 3, где на стадии (f) добавляют 4-фтор-3-метилбензоламин вместо 3,4-дифторбензоламина. Конечный продукт получают в виде не совсем белого твердого вещества (ВЭЖХ-чистота: 97%). 1H ЯМР (500 MГц, хлороформ-d) δ: 8,70 (ддд, J=4,8, 1,9, 1,0 Гц, 1H), 7,86 (д, J=8,3 Гц, 2H), 7,77 (тд, J=7,7, 1,9 Гц, 1H), 7,68 (д, J=8,0 Гц, 1H), 7,26 (м, 1H), 7,06 (д, J=8,3 Гц, 2H), 6,97-6,86 (м, 2H), 6,81-6,69 (м, 2H), 4,91 (с, 2H), 3,81 (с, 2H), 3,40 (с, 3H), 2,13 (д, J=1,4 Гц, 3H), 1,49 (с, 6H). МС (ESI) m/z 510,2 [M+H]+
Пример 5
7,8-Дигидро-2-(4-(5-фторпиридин-2-ил)бензил)-3-этил-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он
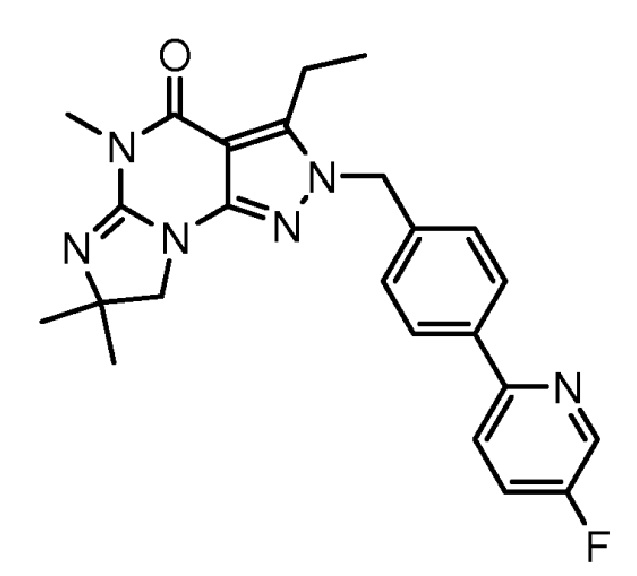
(a) 7,8-Дигидро-2-(4-(5-фторпиридин-2-ил)бензил)-3-хлор-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он
[0105] Указанное в заголовке соединение получают, применяя методику, аналогичную методике, описанной на стадиях (a)-(f) примера 1, где на стадии (a) добавляют 2-(4-(хлорметил)фенил)-5-фторпиридин вместо 2-(4-(хлорметил)фенил)пиридина. МС (ESI) m/z 439,2 [M+H]+.
(b) 7,8-Дигидро-2-(4-(5-фторпиридин-2-ил)бензил)-3-этил-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он
[0106] Добавляют по капле этилмагнийбромид (3,0 M в эфире, 3 мл) в реакционную пробирку, содержащую ZnCl2 (1,2 г, 8,8 ммоль), при 0°C в атмосфере аргона. Смесь перемешивают при комнатной температуре в течение 20 мин. и затем охлаждают до -78°C. Добавляют по капле 9-метокси-9-борабицикло[3,3,1]нонан (1,0M в гексане, 8 мл). По завершении добавления смесь перемешивают при комнатной температуре в течение 40 мин. Медленно добавляют к смеси 7,8-дигидро-2-(4-(5-фторпиридин-2-ил)бензил)-3-хлор-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он (352 мг, 0,8 ммоль) в безводном ДМФА (15 мл), а затем 2-дициклогексилфосфино-2′,6′-диметоксибифенил (S-Phos, 38 мг) и ацетат палладия (13 мг). Реакционную пробирку закупоривают и перемешивают при комнатной температуре в течение 30 мин., а затем нагревают при 100°C в течение 4 дней. Смесь разбавляют водой (150 мл) и затем экстрагируют дихлорметаном (60 мл×3). Объединенную органическую фазу выпаривают досуха при пониженном давлении. Остаток очищают при помощи системы полупрепаративной ВЭЖХ, снабженной колонкой С18 с обращенной фазой, применяя градиент 0-26% ацетонитрила в воде, содержащей 0,1% муравьиной кислоты, за 16 мин. и получая 177 мг продукта в виде светло-желтого твердого вещества (ВЭЖХ-чистота: 99,5%; выход: 51%). 1H ЯМР (500 MГц, хлороформ-d) δ: 8,53 (д, J=2,9 Гц, 1H), 7,92 (д, J=8,3 Гц, 2H), 7,69 (дд, J=8,8, 4,2 Гц, 1H), 7,47 (тд, J=8,4, 2,9 Гц, 1H), 7,25 (д, J=9,0 Гц, 2H), 5,29 (с, 2H), 3,73 (с, 2H), 3,41 (с, 3H), 2,95 (кв., J=7,6 Гц, 2H), 1,42 (с, 6H), 1,18 (т, J=7,5 Гц, 3H). МС (ESI) m/z 433,3 [M+H] +.
Пример 6
7,8-Дигидро-2-(4-(6-фторпиридин2-ил)бензил)-3-этил-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он

[0107] Указанное в заголовке соединение получают применяя методику, аналогичную методике, описанной в примере 5, где на стадии (a) добавляют 2-(4-(хлорметил)фенил)-6-фторпиридин вместо 2-(4-(хлорметил)фенил)-5-фторпиридина. 1H ЯМР (400 MГц, хлороформ-d) δ: 7,98 (д, J=8,4 Гц, 2H), 7,84 (м, 1H), 7,59 (дд, J=7,5, 2,4 Гц, 2H), 7,25 (д, J=8,4 Гц, 3H), 6,87 (дд, J=8,1, 3,0 Гц, 1H), 5,28 (с, 2H), 3,71 (с, 2H), 3,38 (с, 3H), 2,94 (кв., J=7,5 Гц, 2H), 1,40 (с, 6H), 1,17 J=7,5 Гц, 3H). МС (ESI) m/z 433,2 [M+H]+.
[0108] Соединение из примера 5 демонстрирует хорошую селективность относительно PDE1 и ингибирует активность PDE при значении IC50, равном или меньшем 30 нМ.
Пример 7
7,8-Дигидро-2-(4-(5-фторпиридин-2-ил)бензил)-3-пропил-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он
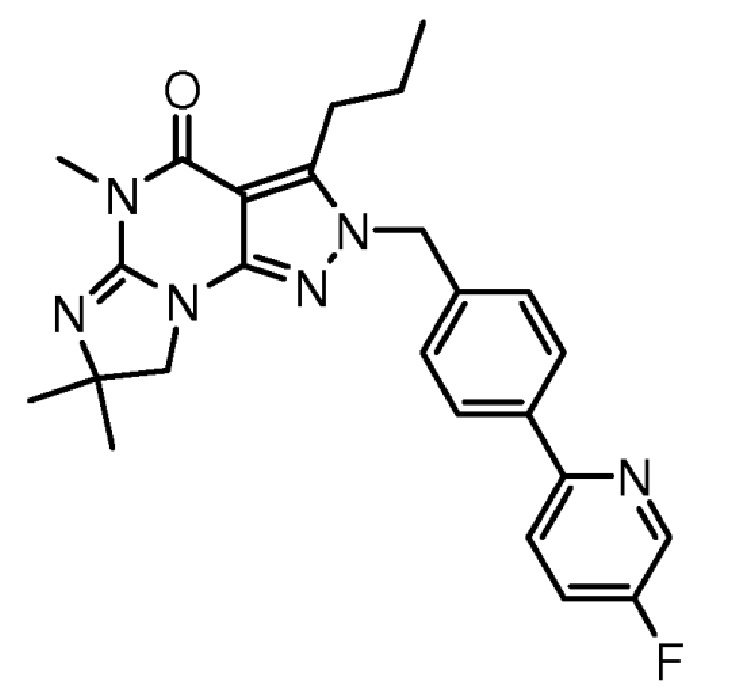
[0109] Указанное в заголовке соединение получают, применяя методику, аналогичную методике, описанной в примере 5, где на стадии (b) добавляют пропилмагнийбромид вместо этилмагнийбромида. МС (ESI) m/z 447,2 [M+H]+.
[0110] Соединение из примера 7 демонстрирует хорошую селективность относительно PDE1 и ингибирует активность PDE при значении IC50, равном или меньшем 15 нМ.
Пример 8
7,8-Дигидро-2-(4-(6-фторпиридин2-ил)бензил)-3-пропил-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он
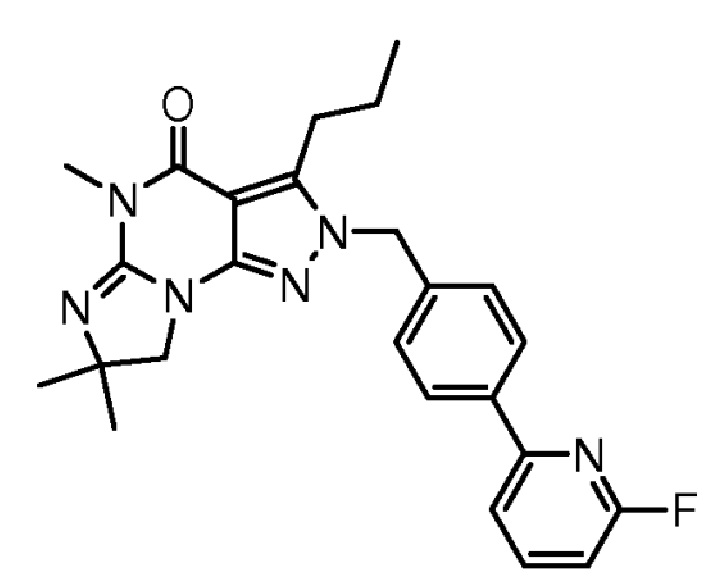
[0111] Указанное в заголовке соединение получают применяя методику, аналогичную методике, описанной в примере 5, где на стадии (b) добавляют пропилмагнийбромид вместо этилмагнийбромида и на стадии (a) добавляют 2-(4-(хлорметил)фенил)-6-фторпиридин вместо 2-(4-(хлорметил)фенил)-5-фторпиридина. МС (ESI) m/z 447,2 [M+H]+.
Пример 9
7,8-Дигидро-2-(4-хлорбензил)-3-(4-фторфениламино)-5,7,7-триметил-[2H]-имидазо-[1,2-a]пиразоло[4,3-e]пиримидин-4(5H)-он
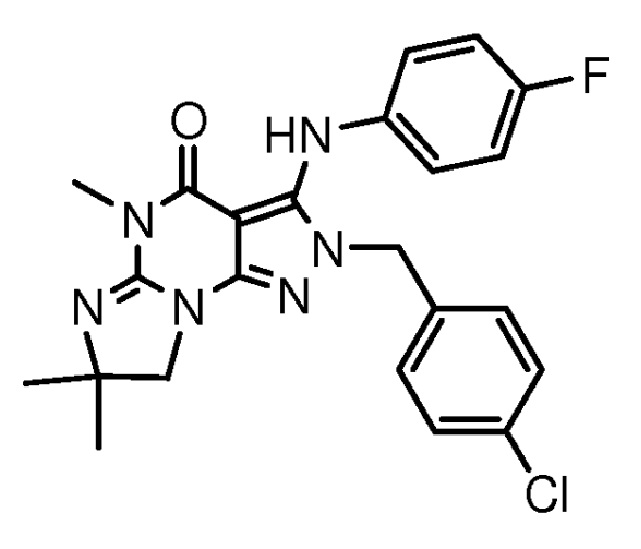
[0112] Указанное в заголовке соединение получают, применяя методику, аналогичную методике, описанной в примере 1, где на стадии (a) добавляют 1-хлор-4-(хлорметил)бензол вместо 2-(4-(хлорметил)фенил)пиридина. МС (ESI) m/z 453,2 [M+H]+.
[0113] Соединение из примера 9 демонстрирует хорошую селективность относительно PDE1 и ингибирует активность PDE при значении IC50, равном или меньшем 5 нМ.
Пример 10: определение ингибирования PDEIB in vitro, применяя IMAP набор для исследования фосфодиэстераз.
[0114] Фосфодиэстераза IB (PDEIB) представляет собой кальций/кальмодулин-зависимый фермент фосфодиэстеразу, которая конвертирует циклический гуанозинмонофосфат (cGMP) в 5'-гуанозинмонофосфат (5'-GMP). PDEIB также может конвертировать модифицированный cGMP субстрат, например, флуоресцентный cGMP-флуоресцеин в соответствующий GMP-флуоресцеин. Образование GMP-флуоресцеина из cGMP-флуоресцеина можно определить количественно, используя, например, IMAP (Molecular Devices, Sunnyvale, CA) реагент с иммобилизованными металл аффинными частицами.
[0115] Короче говоря, IMAP реагент связывается с высоким сродством со свободным 5'-фосфатом, то есть обнаруживается в GMP-флуоресцеине и не обнаруживается в cGMP-флуоресцеине. Результирующий комплекс GMP-флуоресцеин-IMAP имеет больший размер относительно cGMP-5 флуоресцеина. Небольшие флуорофоры, которые связываются в более крупный, медленно переворачивающийся комплекс, можно отличить от несвязанных флуорофоров, так как испускаемые фотоны, когда они флуоресцируют, сохраняют такую же полярность, как фотоны, используемые для возбуждения флуоресценции.
[0116] При исследовании фосфодиэстераз cGMP-флуоресцеин, который не может связываться с IMAP, и следовательно, сохраняет небольшую поляризацию флуоресценции, конвертируется в GMP-флуоресцеин, который, если связан с IMAP, дает большое повышение поляризации флуоресценции (Δmp). Следовательно, ингибирование фосфодиэстеразы детектируют как уменьшение Δmp.
Исследование ферментов
[0117] Материалы: все химикаты доступны от Sigma-Aldrich (St. Louis, MO) за исключением IMAP-реагентов (реакционный буфер, буфер для связывания, FL-GMP и IMAP гранулы), которые доступны от Molecular Devices (Sunnyvale, CA).
[0118] Исследование: можно использовать следующие ферменты фосфодиэстеразы: 3',5'-циклический нуклеотид-специфическая фосфодиэстераза головного мозга коров (Sigma, St. Louis, MO) (преимущественно PDEIB) и рекомбинантные полноразмерные человеческие PDE1A и PDE1B (r-hPDE1A и r-hPDE1B, соответственно) которые могут производиться, например, в клетках HEK или SF9 специалистом в данной области. Фермент PDE1 восстанавливают 50% глцерином до 2,5 ед./мл. Одна единица фермента гидролизует 1,0 мкмоль 3',5'-cAMP до 5'-AMP в мин. при pH 7,5 и 30°C. Одну часть фермента добавляют к 1999 частям реакционного буфера (30 мкМ CaCl2, 10 ед./мл кальмодулина (Sigma P2277), 10 мМ Трис-HCl pH 7,2, 10 мМ MgCl2, 0,1% BSA, 0,05% NaN3), получая конечную концентрацию 1,25 мед./мл. Добавляют 99 мкл разбавленного раствора фермента в каждую ячейку плоскодонного 96-ячеечного полистирольного планшета, к которой добавлен 1 мкл тестируемого соединения, растворенного в 100% ДМСО. Соединения перемешивают и предварительно инкубируют с ферментом в течение 10 мин. при комнатной температуре.
[0119] Реакцию преобразования FL-GMP инициируют, объединяя 4 части смеси фермента и ингибитора с 1 частью раствора субстрата (0,225 мкл) на 384-ячеечном микротитровальном планшете. Реакционную смесь инкубируют в темноте при комнатной температуре в течение 15 мин. Взаимодействие останавливают, добавляя 60 мкл связующего реагента (разведение 1:400 гранул IMAP в буфере для связывания, дополненном 1:1800 разведением антивспенивателя) в каждую ячейку 384-ячеечного планшета. Планшет инкубируют при комнатной температуре в течение 1 час., позволяя связыванию IMAP продолжаться до завершения, и затем помещают в многофункциональный анализатор для микропланшетов Envision (PerkinElmer, Shelton, CT) для определения поляризации флуоресценции (Δmp).
[0120] Снижение концентрации GMP, определяемое как уменьшенное Δmp, является показателем ингибирования активности PDE. Значения IC50 определяют, измеряя активность фермента в присутствии 8-16 концентраций соединения в диапазоне от 0,0037 нМ до 80000 нМ и затем отмечая на графике концентрацию лекарства относительно AmP, что позволяет определять значения IC50, применяя нелинейный регрессионный анализ с использованием программного обеспечения (XLFit; IDBS, Cambridge, MA).
[0121] В настоящем исследовании различные соединения из примеров 1-9 демонстрируют хорошую селективность относительно PDE1 и могут ингибировать PDE1 при значениях IC50, равных или меньших 50 нМ.
ПРИМЕР 11
[0122] Селективный ингибитор PDE1 по настоящему изобретению демонстрирует микросомальную стабильность в исследованиях микросомальной стабильности у человека. Упоминаемый выше селективный ингибитор PDE1 демонстрирует значение K меньше 0,01 и демонстрирует период полураспада T1/2 примерно 100-1800 мин.
ПРИМЕР 12
[0123] Селективный ингибитор PDE1 по настоящему изобретению демонстрирует способность проходить гематоэнцефалический барьер. После инъекции 10 мг/кг на подходящей мышиной модели упоминаемый выше селективный ингибитор PDE1 может быть обнаружен примерно при 3 мкМ меньше чем приблизительно через 0,5 час. после инъекции.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРИМЕНЕНИЯ | 2019 |
|
RU2836384C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2014 |
|
RU2679142C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2015 |
|
RU2709786C2 |
| СПОСОБЫ ЛЕЧЕНИЯ РАКОВЫХ ЗАБОЛЕВАНИЙ И ОПУХОЛЕЙ С ИСПОЛЬЗОВАНИЕМ ИНГИБИТОРОВ PDE1 | 2019 |
|
RU2811918C2 |
| ТЕТРАГИДРО-ПИРАЗОЛО[1,5-a]ПИРИДО-ПИРИМИДИНЫ - АНТАГОНИСТЫ СЕРОТОНИНОВЫХ 5-HT РЕЦЕПТОРОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2008 |
|
RU2391343C1 |
| СПОСОБЫ ЛЕЧЕНИЯ | 2020 |
|
RU2822394C1 |
| ЗАМЕЩЕННЫЕ 2-АМИНО-3-СУЛЬФОНИЛ-ТЕТРАГИДРО-ПИРАЗОЛО[1,5-a]ПИРИДО-ПИРИМИДИНЫ - АНТАГОНИСТЫ СЕРОТОНИНОВЫХ 5-HT РЕЦЕПТОРОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2008 |
|
RU2384581C2 |
| КРИСТАЛЛЫ СОЛИ | 2013 |
|
RU2652116C2 |
| ЗАМЕЩЕННЫЕ 2-АЛКИЛСУЛЬФАНИЛ-3-СУЛЬФОНИЛ-ПИРАЗОЛО[1,5-а]-ПИРИМИДИНЫ, АНТАГОНИСТЫ СЕРОТОНИНОВЫХ 5-HT РЕЦЕПТОРОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2008 |
|
RU2393159C1 |
| КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE 2, ТАКИЕ КАК 1-АРИЛ-4-МЕТИЛ-[1,2,4]ТРИАЗОЛО[4,3-А]ХИНОКСАЛИНОВЫЕ СОЕДИНЕНИЯ, И ИНГИБИТОРЫ PDE 10, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ НЕВРОЛОГИЧЕСКИХ ИЛИ МЕТАБОЛИЧЕСКИХ РАССТРОЙСТВ | 2013 |
|
RU2657540C2 |
Изобретение относится к применимому в медицине ингибитору PDE1, характеризующемуся структурой, выбранной из следующих:
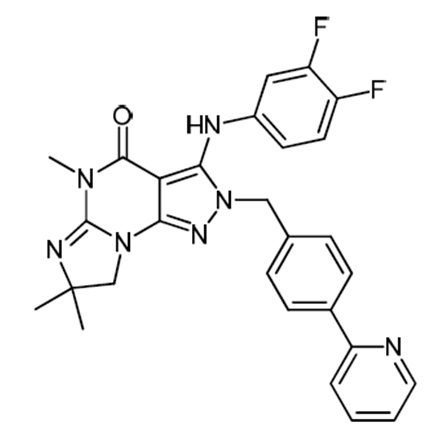
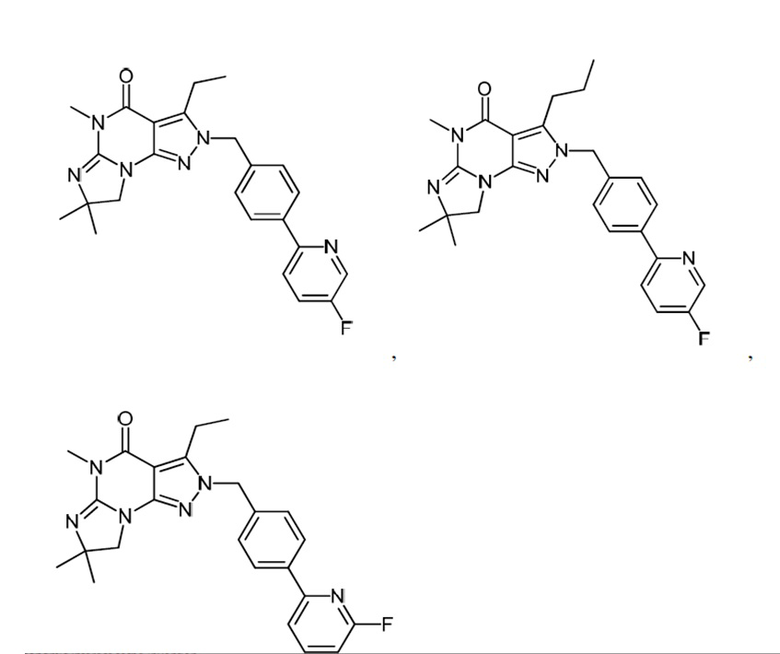 ,
, 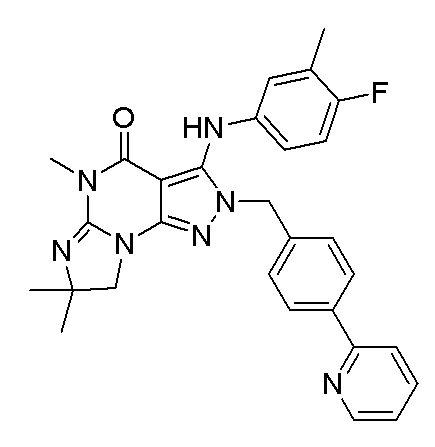 ,
,
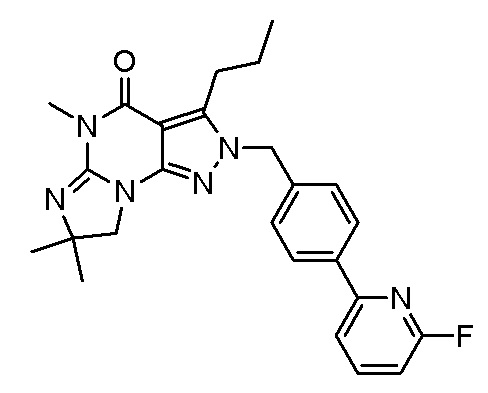 ,
,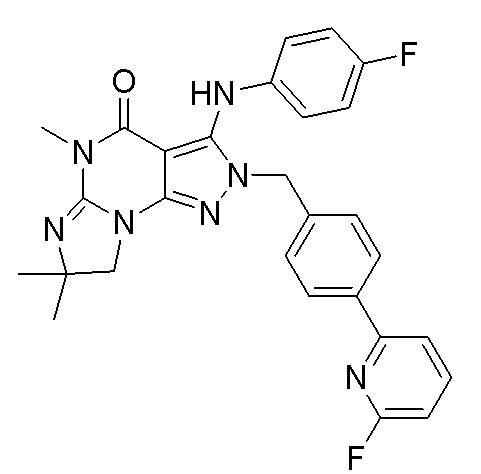 ,
,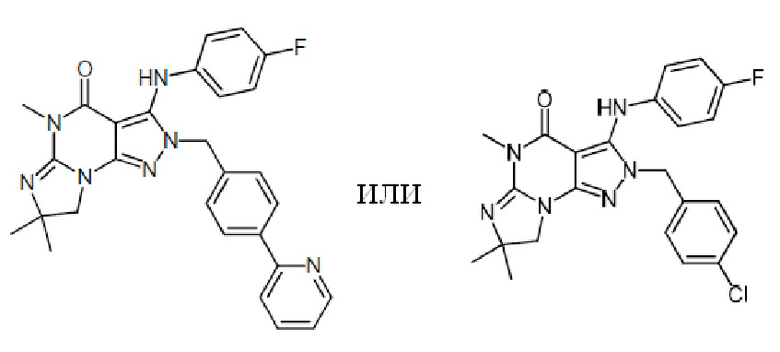 , фармацевтической композиции на его основе для лечения и профилактики заболевания, расстройства или повреждения ЦНС. Предложены новые эффективные ингибиторы PDE1 с удлиненным временем полураспада in vivo, для лечения заболеваний, расстройств и повреждений головного и спинного мозга. 5 н. и 6 з.п. ф-лы, 12 пр.
, фармацевтической композиции на его основе для лечения и профилактики заболевания, расстройства или повреждения ЦНС. Предложены новые эффективные ингибиторы PDE1 с удлиненным временем полураспада in vivo, для лечения заболеваний, расстройств и повреждений головного и спинного мозга. 5 н. и 6 з.п. ф-лы, 12 пр.
1. Соединение, выбранное из следующих:
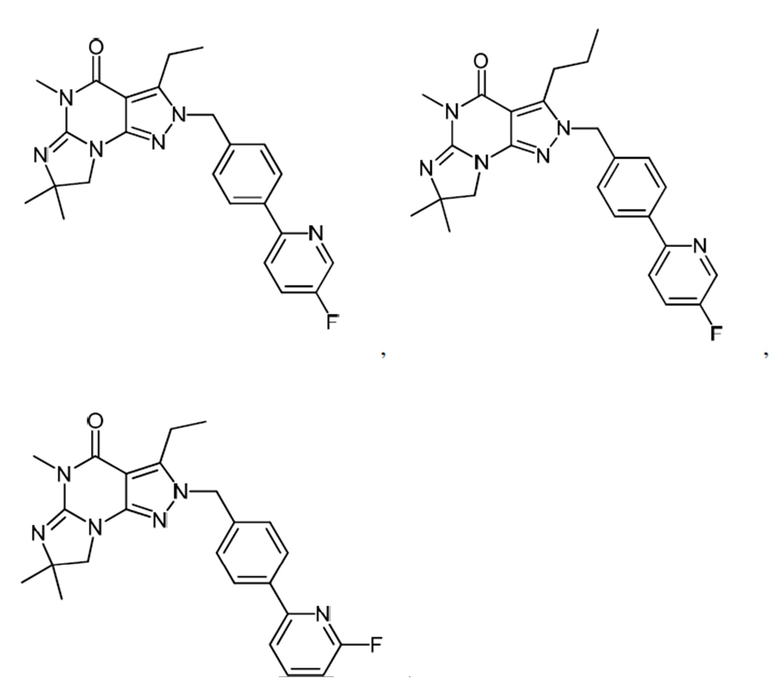
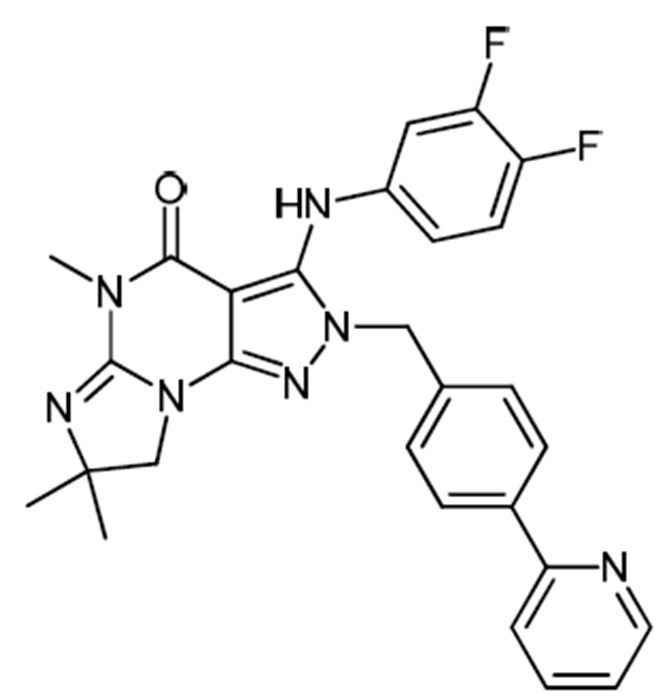 ,
, 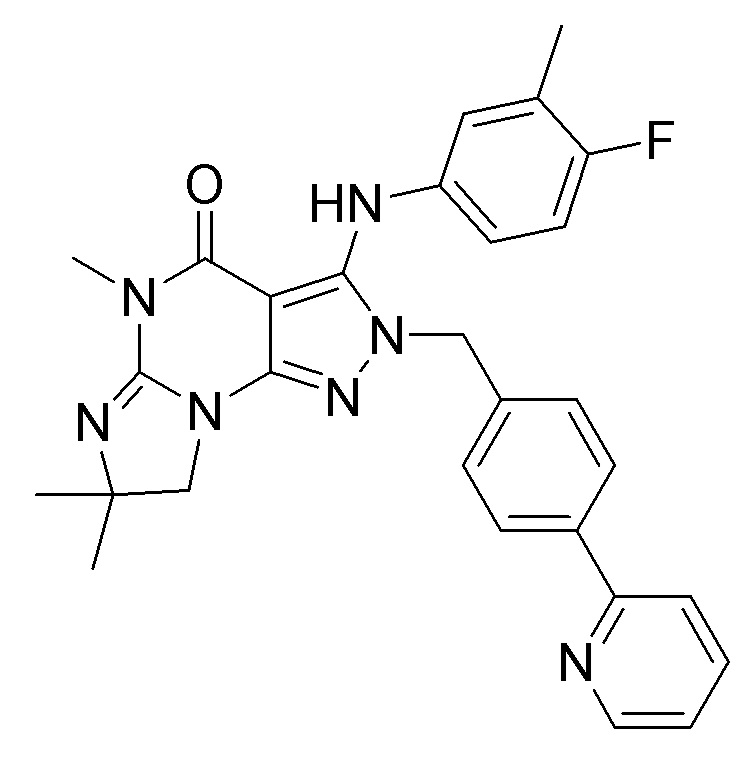 ,
,
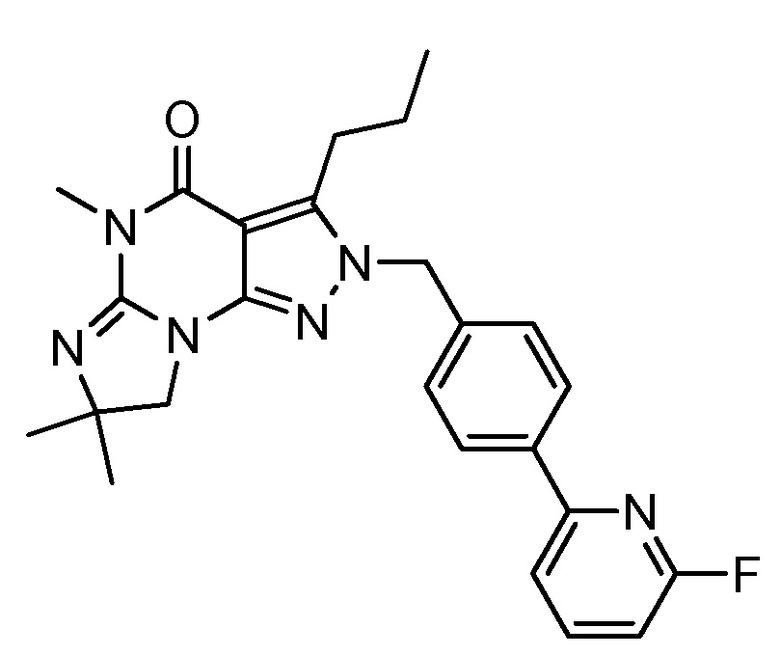 ,
,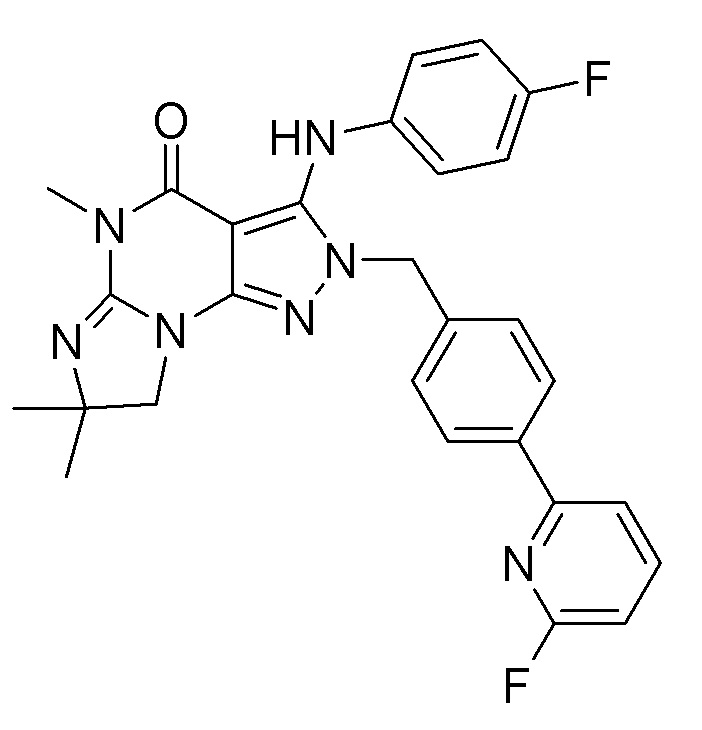
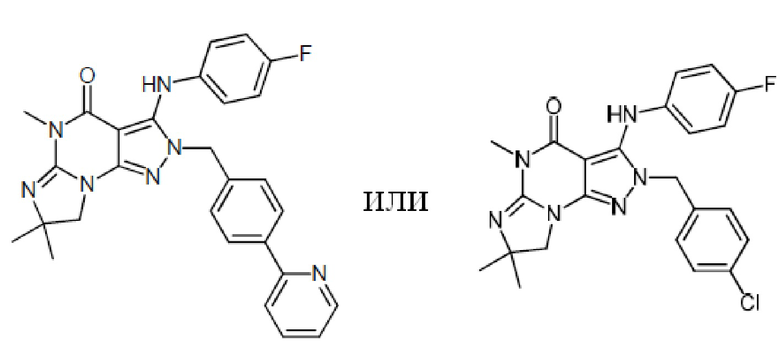 .
.
2. Фармацевтическая композиция, обладающая ингибирующей PDE1 активностью, содержащая терапевтически эффективное количество соединения по п. 1 в смеси, по меньшей мере, с одним фармацевтически приемлемым носителем или наполнителем.
3. Способ профилактики и/или лечения заболевания, расстройства и/или повреждения ЦНС, где способ включает введение субъекту терапевтически эффективного количества ингибитора PDE1, где введение ингибитора PDE1 модулирует уровень внутриклеточного cAMP у субъекта, где ингибитор PDE1 представляет собой соединение по п. 1 или фармацевтическую композицию по п. 2.
4. Способ по п. 3, где заболевание, расстройство или повреждение ЦНС представляет собой повреждение спинного мозга.
5. Способ по п. 3, где заболевание, расстройство или повреждение ЦНС касается повреждения двигательных нейронов.
6. Способ по любому из пп. 3-5, где заболевание, расстройство или повреждение ЦНС выбрано из группы, включающей: неврологические травмы и повреждения, травмы и/или повреждения, связанные с хирургическим вмешательством, ретинальные травмы и повреждения, повреждение, связанное с эпилепсией, повреждение спинного мозга, повреждение головного мозга, хирургическую операцию на головном мозге, повреждение, связанное с травмой головного мозга, повреждение, связанное с травмой спинного мозга, повреждение головного мозга, связанное с лечением рака, повреждение спинного мозга, связанное с лечением рака, повреждение головного мозга, связанное с инфекцией, повреждение головного мозга, связанное с воспалением, повреждение спинного мозга, связанное с инфекцией, повреждение спинного мозга, связанное с воспалением, повреждение головного мозга, связанное с токсинами в окружающей среде, и повреждение спинного мозга, связанное с токсинами в окружающей среде.
7. Способ по любому из пп. 3-6, где заболевание, расстройство или повреждение ЦНС представляет собой нейродегенеративное расстройство.
8. Способ по п. 7, где нейродегенеративное заболевание, расстройство или повреждение выбрано из группы, включающей: болезнь Альцгеймера, множественный склероз, глаукому, лобно-височную деменцию, деменцию с тельцами Леви, кортикобазальную дегенерацию, прогрессирующий супрануклеарный паралич, прионовые болезни, болезнь Гентингтона, множественную системную атрофию, болезнь Паркинсона, боковой амиотрофический склероз, наследственную спастическую параплегию, спиноцеребеллярные атрофии, атаксию Фридрейха, амилоидоз, метаболические (связанные с диабетом) расстройства, расстройства, связанные с токсинами, хроническое воспаление ЦНС и болезнь Шарко-Мари-Тута.
9. Способ лечения или профилактики заболевания, расстройства или повреждения ПНС, где способ включает введение субъекту терапевтически эффективного количества ингибитора PDE1 с целью повышения внутриклеточных уровней cAMP у субъекта, где ингибитор PDE1 представляет собой соединение по п. 1 или фармацевтическую композицию по п. 2.
10. Способ по любому из пп. 3-9, где ингибитор PDE1 вводят пациенту, который, как показано, имеет повышенные уровни внутриклеточного кальция по сравнению с контрольным субъектом.
11. Способ профилактики развития заболевания или расстройства ЦНС у субъекта, который находится в группе риска развития заболевания или расстройства ЦНС, где способ включает:
1) получение ЦНС-образца от субъекта;
2) измерение уровней внутриклеточного кальция в образце;
3) сравнение уровней внутриклеточного кальция в биологическом образце с эталоном;
4) определение, находится ли пациент в группе риска развития заболевания или расстройства ЦНС, на основании уровня внутриклеточного кальция по сравнению с эталоном;
5) введение субъекту терапевтически эффективного количества ингибитора PDE1 на основании уровня внутриклеточного кальция у субъекта, поставившего его под риск развития заболевания или расстройства ЦНС, где ингибитор PDE1 представляет собой соединение по п. 1 или фармацевтическую композицию по п. 2.
| WO2006133261 A2, 14.12.2006 | |||
| WO2008063505 A1, 29.05.2008 | |||
| WO2010098839 A, 02.09.2010 | |||
| US 20120094966 A1, 19.04.2012 | |||
| US 20130239234 A1, 12.09.2013 | |||
| Фотографический аппарат | 1928 |
|
SU16510A1 |

