Область техники
Данное изобретение относится к химии органических соединений, фармакологии и медицине и касается новых химических соединений, характеризующихся высокой эффективностью для профилактики и терапии инфекционных заболеваний, в частности, заболеваний стафилококковой этиологии, в том числе заболеваний, вызванных резистентными формами стафилококковых инфекций.
Уровень техники
Антибиотикорезистентность - явление устойчивости штаммов возбудителей инфекций к действию одного или нескольких антибактериальных препаратов, снижение чувствительности культуры микроорганизмов к действию антибактериального вещества, которая может носить нативный или приобретенный характер. Устойчивость бактерий делает многие антибиотики клинически неэффективными.
Устойчивость микроорганизмов к антибиотическим препаратам является приоритетной проблемой для системы здравоохранения. Ежегодно инфекции, вызванные резистентными бактериями, уносят более 50000 жизней только в США и Европе, в то время как в развивающихся странах и Китае эти цифры значительно выше. Ущерб мировой экономике, связанный с антибиотикорезистентностью, составляет около 30 млрд. долларов в год. При сохранении текущей тенденции развития и распространения лекарственной устойчивости, к 2050 году суммарные потери мировой экономики превысят 100 трлн. долларов, а суммарные человеческие потери составят 300 млн. смертей. Существование проблем мульти- и кросс-резистентности делает необходимым поиск антимикробных препаратов с принципиально новыми механизмами действия. Однако в течение последних 45 лет в клиническую практику были введены всего два новых класса антибиотиков. Из находящихся сейчас в разработке лекарств новые антибиотики составляют менее 5%. Стоит также отметить и тот факт, что большинство подходов при разработке новых антибиотических препаратов связано с получением модификаций уже существующих молекул, что позволяет лишь временно снизить, но не решить проблему резистентности патогенных микроорганизмов. Для ее решения необходимо искать принципиально новые классы антимикробных препаратов.
Метициллин-резистентный золотистый стафилококк (MRSA) является самым распространенным антибиотикорезистентным патогеном. В 15 странах Евросоюза более 10% всех септических инфекций вызвано MRSA, распространенность резистентности в некоторых из этих стран достигает 50% всех штаммов. В России доля устойчивых к антибиотикам штаммов золотистого стафилококка на протяжении последних 10 лет сохраняется в среднем на уровне 40%. MRSA занимает первое место в структуре возбудителей послеоперационных инфекционных осложнений. Расходы лечебных учреждений на приобретение лекарственных средств в среднем составляют 15-20% от бюджета, из них на долю антиинфекционных препаратов приходится 50-60%, что заставляет искать прежде всего наиболее эффективные препараты, в частности, позволяющие преодолеть резистентность возбудителей инфекций. Возрастающая частота циркуляции нозокоминальной формы MRSA приводит к увеличению числа полирезистентных штаммов CA-MRSA, что вовлекает в группы риска людей, не находящихся в лечебно-профилактических учреждениях, прежде всего, детей, спортсменов, военнослужащих. Частота распространения MRSA в некоторых отделениях реанимации, онкологии и гематологии превышает 50-60%, что создает крайне серьезные проблемы для терапии. При этом, независимо от результатов оценки in vitro при инфекциях, вызываемых MRSA, все антибиотика, относящиеся к классу β-лактамов, следует считать клинически неэффективными и не использовать в терапии MRSA.
Таким образом, поиск и разработка новых эффективных антибактериальных препаратов для применения в клинической практике для лечения инфекционных заболеваний и преодоления антибиотикорезистентности является крайне актуальной задачей.
Раскрытие изобретения
Задачей настоящего изобретения является разработка новых эффективных соединений для терапии инфекционных заболеваний, в том числе вызванных антибиотикорезистентными патогенами, в частности патогенами стафилококковой этиологии.
Техническим результатом данного изобретения является разработка и получение новых эффективных соединений, обладающих высокой антибактериальной активностью и низкой токсичностью, для лечения инфекционных заболеваний, в том числе вызванных антибиотикорезистентными патогенами (включая мультирезистентные штаммы), в частности заболеваний стафилококковой этиологии, например, инфекционных заболеваний кожи и/или мягких тканей, пневмонии, эндокардита или остеомиелита. Применение соединений по изобретению в терапии инфекционных заболеваний обеспечивает снижение скорости возникновения резистентности к новой терапии у патогенов, вызывающих инфекционные заболевания, в том числе стафилококков.
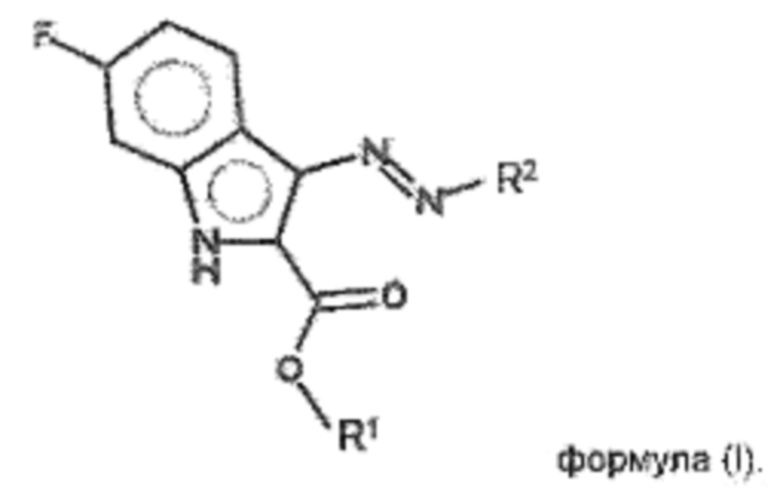
Указанные технические результаты достигаются посредством разработки и создания соединений общей формулы (I):
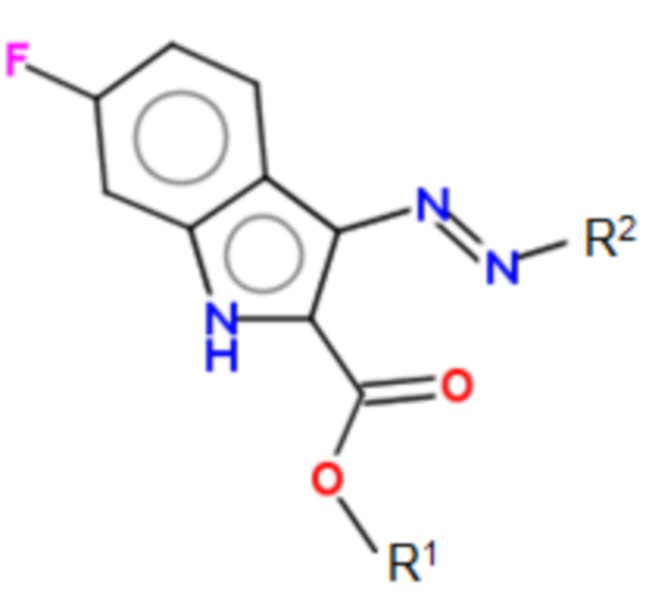 формула (I),
формула (I),
или их фармацевтически приемлемых солей, таутомеров, сольватов или гидратов, где:
R1 выбирается независимо и представляет собой -C1-10-алкил;
R2 выбирается независимо и представляет собой C5-10-гетероциклил, содержащий 1-2 атома N и необязательно содержащий 1-2 атома О, причем R2 присоединяется через атом азота в цикле к диазеновому фрагменту; или -NR3R4,
причем заместитель R2 может быть необязательно замещен 1-3 заместителями R5;
R3 выбирается независимо и представляет собой H, -C1-6-алкил, -C1-6-алкил-NH-C1-6-алкил, -C1-6-алкил-C5-6-гетероарил, содержащий 1-2 атома N;
R4 выбирается независимо и представляет собой H, -C1-6-алкил, -C1-6-алкил-NH-C1-6-алкил, -C1-6-алкил-C5-6-гетероарил, содержащий 1-2 атома N;
R5 выбирается независимо и представляет собой -C1-6-алкил.
При этом заместители R5 независимо могут быть как одинаковыми, так и различающимися.
Отдельный подкласс соединений, представляющих интерес, включает соединения формулы (I), в которых:
R1 выбирается независимо и представляет собой -C1-6-алкил;
R2 выбирается независимо и представляет собой морфолин, пиперидин, пиперазин, пирролидин, пергидроазепин, пиридин, метилпиперидин, метилпиперазин или -NR3R4,
R3 выбирается независимо и представляет собой -C1-6-алкил, -C1-6-алкил-NH-C1-6-алкил, -C1-6-алкил-C5-6-гетероарил,
R4 выбирается независимо и представляет собой -C1-6-алкил, -C1-6-алкил-NH-C1-6-алкил, -C1-6-алкил-C5-6-гетероарил;
R5 выбирается независимо и представляет собой -C1-6-алкил.
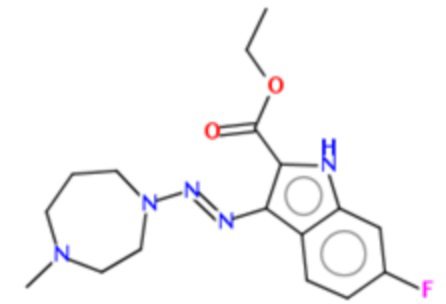
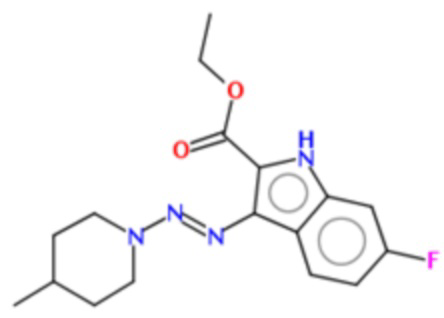
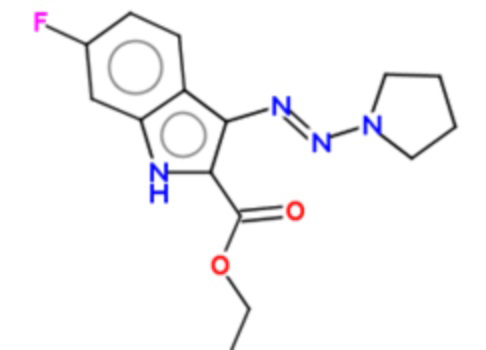
В частных вариантах воплощения изобретения соединения, представляющие интерес, могут быть выбраны из следующих соединений общей формулы (I):
этил-6-фтор-3-[4-метил-пиперазин-1-ил-азо]-1Н-индол-2-карбоксилат (BX-SI035):
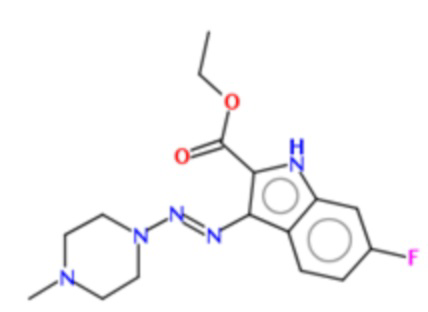 ;
;
этил-6-фтор-3-[4-метил-[1,4]диазепан-1-ил-азо]-1Н-индол-2-карбоксилат (BX-SI039):
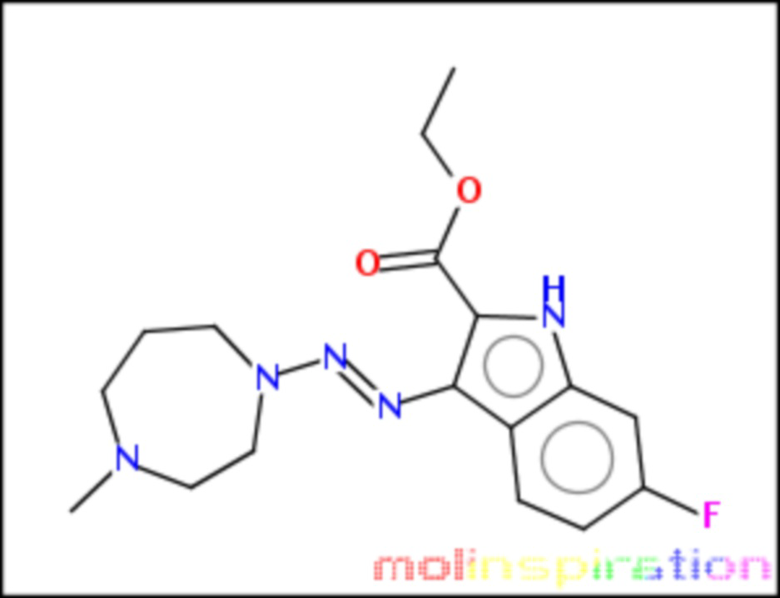 ;
;
этил-6-фтор-3-[пирролидин-1-ил-азо]-1Н-индол-2-карбоксилат (BX-SI043):
 ;
;
этил-6-фтор-3-[N,N’-диметилэтан-1,2-диамин-1-ил-азо]-1Н-индол-2-карбоксилат (BX-SI044):
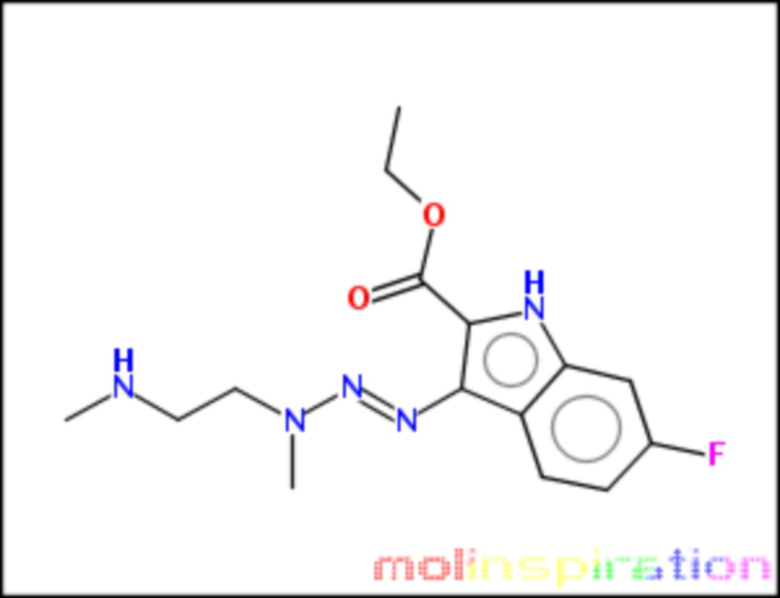 ;
;
этил-6-фтор-3-(4-метил-пиперидин-1-ил-азо)-1Н-индол-2-карбоксилат (BX-SI045):
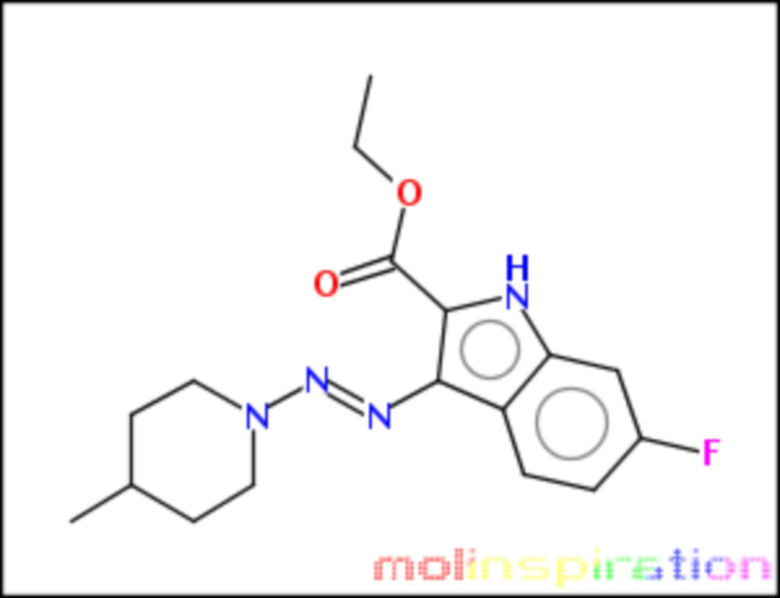 ;
;
этил-6-фтор-3[(1E)-3-метил-3-(2-пиридин-2-ил-этил)-триаз-1-ен-1-ил]-1H-индол-2-карбоксилат (BX-SI048):
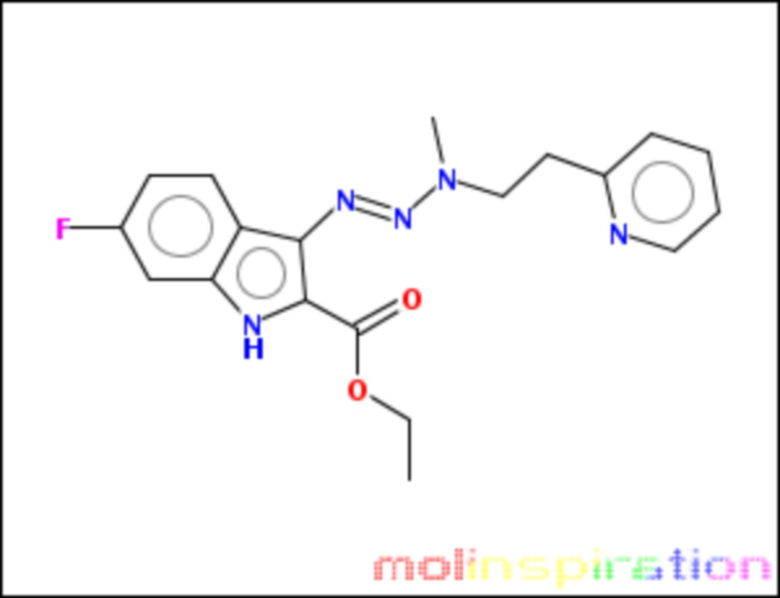 .
.
Настоящее изобретение относится к применению соединений, являющихся предметом изобретения, в качестве антибактериального лекарственного средства для лечения и/или профилактики инфекционного заболевания у субъекта, вызванного бактериальной инфекцией.
В частных вариантах воплощения изобретения инфекционное заболевание вызвано антибиотикорезистентным патогеном.
В частных вариантах воплощения изобретения инфекционное заболевание вызвано патогеном стафилококковой этиологии.
В частных вариантах воплощения изобретения инфекционное заболевание вызвано Staphylococcus aureus, Methicillin-resistant Staphylococcus aureus.
В частных вариантах воплощения изобретения субъект представляет собой млекопитающее. В предпочтительных вариантах воплощения изобретения субъект представляет собой человека.
Настоящее изобретение также включает применение соединений, являющихся предметом изобретения, для получения фармацевтической композиции для лечения и/или профилактики инфекционного заболевания, вызванного бактериальной инфекцией.
Кроме того, изобретением предусматриваются фармацевтические композиции для лечения и/или профилактики инфекционного заболевания, вызванного бактериальной инфекцией, включающие терапевтически эффективное количество соединения, являющегося предметом изобретения, и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество.
В некоторых вариантах воплощения изобретения вспомогательное вещество может представлять собой носитель, наполнитель и/или растворитель.
В некоторых вариантах воплощения изобретения инфекционное заболевание вызвано антибиотикорезистентным патогеном.
В некоторых вариантах воплощения изобретения инфекционное заболевание вызвано патогеном стафилококковой этиологии, в том числе патогеном с множественной лекарственной устойчивостью. В частных вариантах воплощения изобретения инфекционное заболевание вызвано Staphylococcus aureus, в том числе Methicillin-resistant Staphylococcus aureus.
В частных вариантах воплощения изобретения фармацевтическая композиция предназначена для лечения и/или предотвращения заболевания, которое представляет собой инфекционное заболевание кожи и/или мягких тканей, пневмонию, эндокардит или остеомиелит.
Настоящее изобретение также относится к способу лечения и/или профилактики инфекционных заболеваний, включающему введение (в качестве монотерапии или в комбинации с одним или несколькими агентами) терапевтически эффективного количества соединения, являющегося предметом изобретения, в организм человека или животного, нуждающегося в лечении и/или профилактики таких заболеваний. Термин «введение» в организм соединения настоящего изобретения включает доставку к реципиенту соединения, описанного в настоящем изобретении, пролекарства, или другого фармакологически приемлемого производного такого соединения, используя любые допустимые препараты или пути введения в организм, хорошо известные специалистам.
Изобретение также включает получение соединений общей формулы (I).
Краткое описание чертежей
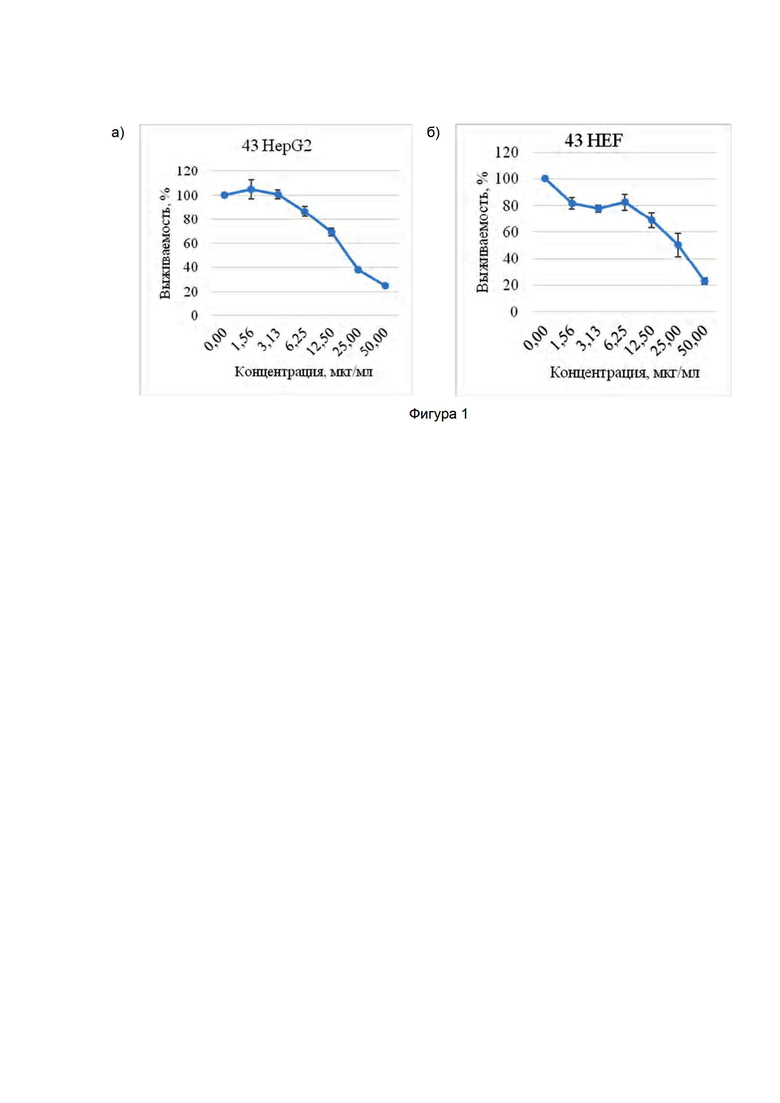
Фигура 1. Кривые цитотоксичности соединения по изобретению этил-6-фтор-3-[ пирролидин-1-ил-азо]-1H-индол-2-карбоксилат :
а) исследования на клетках гепатокарциномы человека HepG2;
б) исследования на эмбриональных фибробластах человека HEF.
Подробное раскрытие изобретения
Определения (термины)
Для лучшего понимания настоящего изобретения ниже приведены некоторые термины, использованные в настоящем описании изобретения. Кроме того, если не указано иное, все вхождения функциональных групп выбираются независимо, два вхождения могут быть как одинаковыми, так и разными.
В описании данного изобретения термины «включает» и «включающий» интерпретируются как означающие «включает, помимо всего прочего». Указанные термины не предназначены для того, чтобы их истолковывали как «состоит только из».
Термин «алкил» сам по себе или как часть другого заместителя, относится к насыщенным углеводородным группам с прямой или разветвленной цепью, включая углеводородные группы, имеющие указанное число атомов углерода (то есть С1-6 подразумевает от одного до шести атомов углерода). Примеры алкилов включают метил, этил, н-пропил, изо-пропил, но не ограничиваются ими.
Термин «гетероцикл» или «гетероциклил» означает в настоящем документе неароматические циклические системы (насыщенные или частично ненасыщенные), имеющие от пяти до семи атомов в цикле, содержащие 1 или 2 гетероатома N. Примеры гетероциклических колец, включают, но не ограничиваются, пирролидин, пиперидин и другие.
Термин «гетероарил» в настоящем документе означает группы, содержащие ароматический цикл, имеющий от пяти до шести атомов в цикле, содержащий 1 или 2 гетероатома N.
Данное изобретение содержит только такие комбинации заместителей и производных, которые образуют стабильное или химически возможное соединение. Стабильным или химически возможным соединением называется такое соединение, стабильности которого достаточно для его синтеза и аналитического детектирования. Предпочтительные соединения данного изобретения являются достаточно стабильными и не разлагаются при температуре до 40°C в отсутствие химически активных условий, в течение, по крайней мере, одной недели.
Если не указано иначе, приведенные в материалах заявки структуры соединений также подразумевают и все стереоизомеры, то есть R- и S- изомеры для каждого ассиметричного центра. Кроме того, отдельные стереохимические изомеры, равно как и энантиомеры и диастереомерные смеси настоящих соединений, также являются предметом данного изобретения. Таким образом, данное изобретение охватывает каждый диастереомер или энантиомер, свободный в значительной степени от других изомеров (>90%; более предпочтительно, >95% мольной чистоты), так же как и смесь таких изомеров.
Конкретный оптический изомер может быть получен разделением рацемической смеси в соответствии со стандартной процедурой, например, путем получения диастереоизомерных солей путем обработки оптически активной кислотой или основанием с последующим разделением смеси диастереомеров кристаллизацией с последующим выделением оптически активных оснований из этих солей. Примерами соответствующих кислот являются винная, диацетилвинная, дибензоилвинная, дитолуолвинная и камфорсульфоновая кислота. Другая методика разделения оптических изомеров заключается в использовании хиральной хроматографической колонки. Кроме того, другой метод разделения включает синтез ковалентных диастереомерных молекул путем реакции соединений изобретения с оптически чистой кислотой в активированной форме или оптически чистым изоцианатом. Полученные диастереомеры можно разделить обычными способами, например, хроматографией, дистилляцией, кристаллизаций или сублимацией, а затем гидролизовать для получения энантиомерно чистого соединения.
Оптически активные соединения данного изобретения могут быть получены с использованием оптически активных исходных материалов. Такие изомеры могут находиться в форме свободной кислоты, свободного основания, эфира или соли.
Термин «сольват» относится к ассоциации или комплексу из одной или нескольких молекул растворителя и соединения по изобретению. Примеры растворителей, образующих сольваты, включают, но ими не ограничиваются, воду, изопропанол, этанол, метанол, ДМСО, этилацетат, уксусную кислоту и этаноламин.
Термин «гидрат» относится к комплексу, где молекулами растворителя является вода.
Соединения настоящего изобретения могут существовать в свободной форме или, если требуется, в виде фармацевтически приемлемой соли или другого производного. Используемый здесь термин «фармацевтически приемлемая соль» относится к таким солям, которые, в рамках проведенного медицинского заключения, пригодны для использования в контакте с тканями человека и животных без излишней токсичности, раздражения, аллергической реакции и т.д., и отвечают разумному соотношению пользы и риска. Фармацевтически приемлемые соли аминов, карбоновых кислот, фосфонатов и другие типы соединений хорошо известны в медицине. Соли могут быть получены in situ в процессе выделения или очистки соединений изобретения, а также могут быть получены отдельно, путем взаимодействия свободной кислоты или свободного основания соединения изобретения с подходящим основанием или кислотой, соответственно. Примером фармацевтически приемлемых, нетоксичных солей кислот могут служить соли аминогруппы, образованные неорганическими кислотами, такими как соляная, бромоводородная, фосфорная, серная и хлорная кислоты, или органическими кислотами, такими как уксусная, щавелевая, малеиновая, винная, янтарная или малоновая кислоты, или полученные другими методами, используемыми в данной области, например, с помощью ионного обмена. К другим фармацевтически приемлемым солям относятся адипинат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, гептанат, гексанат, гидройодид, 2-гидрокси-этансульфонат, лактобионат, лактат, лаурат, лаурил сульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат, ундеканат, валериат и подобные. Типичные соли щелочных и щелочноземельных металлов содержат натрий, литий, калий, кальций, магний и другие. Кроме того, фармацевтически приемлемые соли могут содержать, если требуется, нетоксичные катионы аммония, четвертичного аммония и амина, полученные с использованием таких противоионов, как галогениды, гидроксиды, карбоксилаты, сульфаты, фосфаты, нитраты, низшие алкил сульфонаты и арил сульфонаты.
Настоящее изобретение включает все фармацевтически приемлемые изотопно меченые соединения по настоящему изобретению, в которых один или несколько атомов замещен атомами, имеющими такой же атомный номер, но атомную массу или массовое число, отличные от атомной массы или массового числа, обычно встречающихся в природе.
Примеры изотопов, подходящих для включения в соединения по изобретению, включают изотопы водорода, такие как 2H и 3H, углерода, такие как 11C, 13C и 14C, хлора, такие как 36CI, фтора, такие как 18F, йода, такие как 123I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O, фософора, такие как 32P, и серы, такие как 35S.
Некоторые изотопно меченые соединения формулы (I), например, те, которые включают радиоактивный изотоп, используют в исследованиях распределения лекарственного препарата и/или субстрата в тканях. В частности, с этой целью используют радиоактивные изотопы, такие как тритий, то есть 3H, и углерод-14, то есть 14C, ввиду легкости их введения и доступности средств их обнаружения.
Замещение более тяжелыми изотопами, такими как дейтерий, то есть 2H, может обеспечить определенные терапевтические эффекты, обусловленные метаболической стабильностью, например, увеличением периода полувыведения in vivo или снижением норм дозирования, и, следовательно, может быть предпочтительным в некоторых случаях.
Изотопно меченые соединения по изобретению могут быть получены обычными способами, известными специалисту в данной области или способами, аналогичными описанным в прилагаемых примерах способов синтеза, при использовании соответствующих изотопно меченых реагентов вместо немеченого ранее применяемого реагента.
Фармацевтически приемлемые сольваты в соответствии с изобретением включают сольваты, где растворитель кристаллизации может быть изотопно замещен, например, D2O, d6-ацетон, d6-ДМСО.
Термин «субъект» охватывает все виды млекопитающих, предпочтительно человека.
Осуществление изобретения
Обзор методов получения соединений изобретения
Соединения, являющиеся предметом настоящего изобретения, могут быть получены с использованием описанных ниже синтетических методов. Перечисленные методы не являются исчерпывающими и допускают введение разумных модификаций. Указанные реакции должны проводиться с использованием подходящих растворителей и материалов. При реализации данных общих методик для синтеза конкретных веществ необходимо учитывать присутствующие в веществах функциональные группы и их влияние на протекание реакции. Для получения некоторых веществ необходимо изменить порядок стадий либо отдать предпочтение одной из нескольких альтернативных схем синтеза. Следует понимать, что эти и все приведенные в материалах заявки примеры не являются ограничивающими и приведены только для иллюстрации настоящего изобретения.
Синтез соединений по изобретению
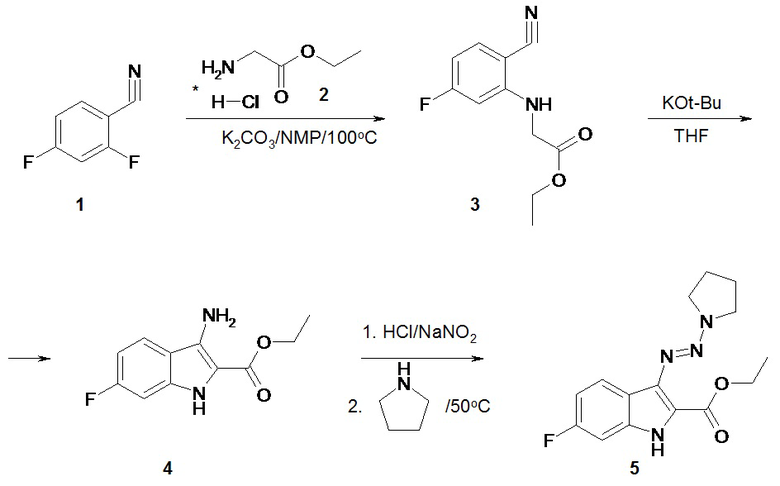
Схема 1. Синтез соединения по изобретению этил-6-фтор-3-[ пирролидин-1-ил-азо]-1H-индол-2-карбоксилата (BX-SI043).
В круглодонную колбу объемом 500 мл помещали 13.9 г (0.1 моль) соединения 1 (см. схему 1), 27.8 г (0.2 моль) гидрохлорида этилового эфира глицина 2, 41.1 г (0.3 моль) K2CO3 и 250 мл N-метил-2-пирролидона. Реакционную смесь перемешивали 6-8 часов при 100°C. После охлаждения обрабатывали 1000 мл воды и 200 мл этилацетата. Органический слой отделяли, сушили безводным K2CO3 и упаривали до объема ~ 50 мл. Остаток пропускали через слой силикагеля 5*10 см в системе этилацетат/гексан - 1/2 (для отделения смолообразных примесей). Фракции, содержащие соединение 3, упаривали, растворяли в 150 мл абсолютного тетрагидрофурана и обрабатывали 11.2 г (0.1 моль) трет-бутилата калия. Реакционную смесь перемешивали при комнатной температуре 16 часов, упаривали, обрабатывали 200 мл насыщенного раствора NaHCO3 в воде и 200 мл этилацетата. Органический слой сушили безводным K2CO3 и упаривали. Остаток очищали на силикагеле 5*20 см в системе этилацетат/гексан - 1/2. Фракции, содержащие соединение 4 (контроль по ЖХ/МС) упаривали, остаток кристаллизовали из 20 мл хлористого метилена. Выход на две стадии составил 1.5 г (7%).
Методика синтеза соединения по изобретению BX-SI043 (этил-6-фтор-3-[пирролидин-1-ил-азо]-1H-индол-2-карбоксилата)
В круглодонную колбу объемом 50 мл помещали 0.22 г (0.001 моль) соединения 4 (см. схему 1), 5 мл воды и 5 мл диметилформамида. При помощи бани со льдом охлаждали реакционную смесь до 5°С. Далее прибавляли 0.3 мл концентрированной соляной кислоты. Через 15 минут обрабатывали реакционную смесь 0.086 г (0.00125 моль) нитрита натрия и оставляли перемешиваться при этой температуре на 30 минут. После этого в реакционную смесь вносили 0.71 г (0.01 моль) пирролидина, грели 50°С 1 час. После охлаждения реакционную смесь разбавляли 25 мл воды. Выпавший осадок фильтровали и сушили. Выход соединения 5 BX-SI043 составил 0.13 г (45%).
1H NMR (300 MHz, DMSO-d6) δ 11.53 (s, 1H), 8.07 (dd, J = 8.8, 5.7 Hz, 1H), 7.16 - 7.04 (m, 1H), 6.91 (dd, J = 10.7, 8.3 Hz, 1H), 4.29 (q, J = 7.0 Hz, 2H), 4.01-3.52 (m, 4H), 2.00 (s, 4H), 1.33 (t, J = 7.0 Hz, 3H).
Получение этил-6-фтор-3-[4-метил-пиперазин-1-ил-азо]-1Н-индол-2-карбоксилата (BX-SI035)
Получено согласно вышеописанной методике из соединения 4 (см. схему 1), путем присоединения соответствующего вторичного амина.
1H NMR (300 MHz, DMSO-d6) δ 11.72 (s, 1H), 8.11 (dd, J = 8.9, 5.8 Hz, 1H), 7.11 (dd, J = 9.9, 2.1 Hz, 1H), 6.94 (td, J = 9.1, 2.1 Hz, 1H), 4.31 (q, J = 7.1 Hz, 2H), 3.77 (m, 4H), 2.54 (m, 4H), 2.26 (s, 3H), 1.33 (t, J = 7.1 Hz, 3H).
Получение этил-6-фтор-3-[4-метил-[1,4]диазепан-1-ил-азо]-1Н-индол-2-карбоксилата (BX-SI039)
Получено согласно вышеописанной методике из соединения 4 (см. схему 1), путем присоединения соответствующего вторичного амина.
1H NMR (300 MHz, DMSO-d6) δ 11.54 (s, 1H), 8.11 (dd, J = 9.0, 5.8 Hz, 1H), 7.08 (dd, J = 10.0, 2.4 Hz, 1H), 6.98 - 6.84 (m, 1H), 4.29 (q, J = 7.1 Hz, 2H), 4.16-4.03 (m, 2H), 3.87-3.73 (m, 2H), 2.81-2.61 (m, 2H), 2.59-2.51 (m, 2H), 2.29 (s, 3H), 2.06-1.82 (m, 2H), 1.33 (t, J = 7.1 Hz, 3H).
Получение этил-6-фтор-3-[N,N’-диметилэтан-1,2-диамин-1-ил-азо]-1Н-индол-2-карбоксилата (BX-SI044)
Получено согласно вышеописанной методике из соединения 4 (см. схему 1), путем присоединения соответствующего вторичного амина.
1H NMR (300 MHz, DMSO-d6) δ 11.55 (s, 1H), 8.12 (dd, J = 9.1, 5.8 Hz, 1H), 7.08 (dd, J = 9.7, 2.0 Hz, 1H), 6.92 (t, J = 9.0 Hz, 1H), 4.30 (q, J = 7.1 Hz, 2H), 3.91 (t, J = 6.4 Hz, 2H), 3.20 (s, 3H), 2.56 (t, J = 5.9 Hz, 2H), 2.20 (s, 6H), 1.33 (t, J = 7.1 Hz, 3H).
Получение этил-6-фтор-3-(4-метил-пиперидин-1-ил-азо)-1Н-индол-2-карбоксилата (BX-SI045)
Получено согласно вышеописанной методике из соединения 4 (см. схему 1), путем присоединения соответствующего вторичного амина.
1H NMR (300 MHz, DMSO-d6) δ 11.64 (s, 1H), 8.11 (ddd, J = 8.6, 5.9, 2.0 Hz, 1H), 7.10 (dt, J = 10.0, 2.3 Hz, 1H), 6.93 (tt, J = 9.4, 2.3 Hz, 1H), 4.6-4.3 (s, 2H), 4.31 (q, J = 7.2, 2H), 3.21-3.03 (s, 2H), 1.82-1.68 (m, 3H), 1.33 (t, J = 7.1, 3H), 1.29-1.16 (m, 2H), 1.00 0 - 0.92 (m, 3H).
Получение этил-6-фтор-3[(1E)-3-метил-3-(2-пиридин-2-ил-этил)-триаз-1-ен-1-ил]-1H-индол-2-карбоксилата (BX-SI048)
Получено согласно вышеописанной методике из соединения 4 (см. схему 1), путем присоединения соответствующего вторичного амина.
1H NMR (300 MHz, DMSO-d6) δ 11.53 (s, 1H), 8.52 (d, J = 4.2 Hz, 1H), 7.75-7.6 (m, 2H), 7.36-7.29 (m, 1H), 7.24-7.16 (m, 1H), 7.06 (d, J = 10.0 Hz, 1H), 6.92-6.82 (m, 1H), 4.29 (q, J = 7.0 Hz, 2H), 4.23-4.15 (m, 2H), 3.25-3.13 (m, 3H), 2.65-2.55 (m, 2H), 1.31 (t, J = 7.0 Hz, 3H).
Применение химических соединений по изобретению
Применение соединений по медицинским показаниям
Соединения, описанные в данном изобретении, могут применяться для лечения и/или профилактики инфекционных заболеваний.
Способ терапевтического применения соединений
Предмет данного изобретения также включает введение субъекту, нуждающемуся в соответствующем лечении, терапевтически эффективного количества соединения по изобретению. Под терапевтически эффективным количеством подразумевается такое количество соединения, вводимого или доставляемого пациенту, при котором у пациента с наибольшей вероятностью проявится желаемая реакция на лечение (профилактику). Точное требуемое количество может меняться от субъекта к субъекту в зависимости от возраста, массы тела и общего состояния пациента, тяжести заболевания, методики введения препарата, комбинированного лечения с другими препаратами и т.п.
Соединение по изобретению или фармацевтическая композиция, содержащая соединение, может быть введено в организм пациента в любом количестве и любым путем введения, эффективным для лечения или профилактики заболевания.
После смешения лекарственного препарата с конкретным подходящим фармацевтически допустимым носителем в желаемой дозировке, композиции, составляющие суть изобретения, могут быть введены в организм человека или других животных перорально, парентерально, местно и т.п.
В том случае, когда соединение по изобретению используется как часть режима комбинированной терапии, доза каждого из компонентов комбинированной терапии вводится в течение требуемого периода лечения. Соединения, составляющие комбинированную терапию, могут вводиться в организм пациента как единовременно, в виде дозировки, содержащей все компоненты, так и в виде индивидуальных дозировок компонентов.
Фармацевтические композиции
Изобретение также относится с фармацевтическим композициям, которые содержат соединение общей формулы (I) (или пролекарственную форму, фармацевтически приемлемую соль или другое фармацевтически приемлемое производное) и один или несколько фармацевтически приемлемых носителей, адъювантов, растворителей и/или наполнителей, таких, которые могут быть введены в организм пациента совместно с соединением, составляющем суть данного изобретения, и которые не разрушают фармакологической активности этого соединения, и являются нетоксичными при введении в дозах, достаточных для доставки терапевтического количества соединения.
Фармацевтические композиции, заявляемые в данном изобретении, содержат соединения данного изобретения совместно с фармацевтически приемлемыми носителями, которые могут включать в себя любые растворители, разбавители, дисперсии или суспензии, поверхностно-активные вещества, изотонические агенты, загустители и эмульгаторы, консерванты, вяжущие вещества, смазочные материалы и т.д., подходящие для конкретной формы дозирования. Материалы, которые могут служить фармацевтически приемлемыми носителями, включают, но не ограничиваются, моно- и олигосахариды, а также их производные; желатин; тальк; эксципиенты, такие как какао-масло и воск для суппозиториев; масла, такие как арахисовое, хлопковое, сафроловое, кунжутное, оливковое, кукурузное и соевое масло; гликоли, такие как пропиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные вещества, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический раствор, раствор Рингера; этиловый спирт и фосфатные буферные растворы. Также в составе композиции могут быть другие нетоксичные совместимые смазочные вещества, такие как лаурилсульфат натрия и стеарат магния, а также красители, разделительные жидкости, пленкообразователи, подсластители, вкусовые добавки и ароматизаторы, консерванты и антиоксиданты.
Предметом данного изобретения являются также лекарственные формы - класс фармацевтических композиций, состав которых оптимизирован для определенного пути введения в организм в терапевтически эффективной дозе, например, для введения в организм орально, местно, внутриглазным способом, пульмональным, например, в виде ингаляционного спрея, или внутрисосудистым способом, интраназально, подкожно, внутримышечно, а также инфузионным способом, в рекомендованных дозировках.
Лекарственные формы согласно изобретению могут содержать составы, полученные методами использования липосом, методами микрокапсулирования, методами приготовления наноформ препарата, или другими методами, известными в фармацевтике.
При получении композиции, например, в форме таблетки, активное начало смешивают с одним или несколькими фармацевтическими эксципиентами, такими как желатин, крахмал, лактоза, стеарат магния, тальк, кремнезем, аравийская камедь, маннит, микрокристаллическая целлюлоза, гипромеллоза или аналогичные соединения.
Таблетки можно покрыть сахарозой, целлюлозным производным или другими веществами, подходящими для нанесения оболочки. Таблетки могут быть получены различными способами, такими как непосредственное сжатие, сухое или влажное гранулирование или горячее сплавление в горячем состоянии.
Фармацевтическую композицию в форме желатиновой капсулы можно получить, смешивая активное начало с растворителем и заполняя полученной смесью мягкие или твердые капсулы.
Для введения парентеральным путем используются водные суспензии, изотонические солевые растворы или стерильные растворы для инъекций, которые содержат фармакологически совместимые агенты, например, пропиленгликоль или бутиленгликоль.
Характеристика биологической активности соединений
Исследование антибактериальной активности соединений по изобретению в условиях in vitro
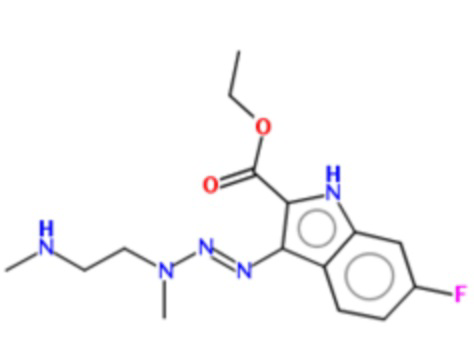
В данном эксперименте были проанализированы соединения по изобретению, в частности, структуры которых представлены в Таблице 1.
Таблица 1. Примеры соединений по изобретению, которые исследовались в настоящем эксперименте.
Клинические изоляты S. aureus с множественной лекарственной устойчивостью были получены из коллекции НИИ Антимикробной Химиотерапии Смоленской Государственной Медицинской Академии (Табл.2). Сток бактерий хранили при -80°С на среде LB c 25% содержанием глицерина. Для получения тестовой культуры свежеразмороженную культуру культивировали на неселективной питательной агаровой среде TSA при 37°С. Профиль устойчивости штаммов к различным антибактериальным препаратам приведен ниже.
Таблица 2. Профиль устойчивости тестовых штаммам к антибактериальным препаратам. R - устойчив; S - чувствителен.
Обозначения в таблице: Ц - Цефотаксим, К - Клиндамицин, Э - Эритромицин, Г - Гентамицин, Тг - Тигециклин, Тт - Тетрациклин.
Исследования антибактериальной активности веществ проводили согласно методикам, изложенным в действующем стандарте ГОСТ Р ИСО 20776-1-2010 «Клинические лабораторные исследования и диагностические тест-системы in vitro. Исследование чувствительности инфекционных агентов и оценка функциональных характеристик изделий для исследования чувствительности к антимикробным средствам».
Референтный метод лабораторного исследования активности антимикробных агентов против быстрорастущих аэробных бактерий, вызывающих инфекционные болезни.
Испытания выполняли в планшетах для микроразведения. Метод основан на подготовке рабочих растворов антибактериального агента объемом 50 мкл на лунку (с добавлением прививочного материала также в объеме 50 мкл). В качестве среды использовали бульон Mueller-Hinton. Рабочие растворы готовили методом двойных серийных разведений в бульоне Mueller-Hinton согласно процедуре, представленной в Таблице 3.
Исследуемые 6-фтор-3-триазеноиндолы по изобретению растворяли в ДМСО, а затем доводили дистиллированой водой до нужной концентрации. Рабочие растворы использовали в день приготовления. Бульон, использованный для разведения, затем использовали для теста чувствительности.
Таблица 3. Схема разведения рабочих растворов.
Рабочие растворы распределяли в планшеты для микроразведения по 50 мкл на лунку с удвоенной желательной окончательной концентрацией антибактериального агента. Для каждой концентрации антибактериального агента использовалось 4 тестовые лунки на один тестовый штамм.
В каждом ряду планшета одна лунка, содержащая 50 мкл или 100 мкл антибактериальной среды без агента, была использована как контроль роста для каждого проверяемого штамма. Аналогично, лунка, содержащая 100 мкл среды без антибактериального агента, была использована как неинокулированная лунка отрицательного контроля для каждого проверяемого штамма.
Инокулюм готовили методом суспензии колоний. Три колонии из неселективной питательной агаровой средой (инкубированной при 37°С в течение 24 ч) брали петлей и перемещали в стерильный солевой раствор. Суспензию регулировали с помощью солевого раствора для получения мутности, эквивалентной 0,5 стандарта McFarland. Стандарт мутности готовили, добавляя аликвоту 0.5 мл 0.048 моль/л BaCl2 (11.72 г/л BaCl2*2H2O) к 99.5 мл из 0.18 моль/л H2SO4 при постоянном встряхивании для сохранения суспензии. Отрегулированный инокулюм разводили в бульоне, чтобы получить окончательную концентрацию числа клеток 5х105 КОЕ/мл.
Инокулирование планшетов проводили следующим образом: к каждой лунке, содержащей 50 мкл антибактериального агента, разведенного в бульоне, добавляли 50 мкл бактериальной суспензии S. aureus. Планшеты инкубировали в термостате при 37°С в течение 20 ч.
Результаты считывали при наличии достаточного роста испытуемого организма (то есть при явном пятне или определенном помутнении в положительном контроле роста), когда не было никакого роста в неинокулированном или отрицательном контроле роста (если присутствует) и когда были установлены чистота и соответствующая концентрация числа клеток инокулюма. Размер роста в каждой лунке сравнивали с размером роста в положительном контроле роста, а наиболее низкую концентрацию агента, которая полностью тормозит видимый рост, регистрировали как минимальную подавляющую концентрацию (МПК).
Определение цитотоксичности и индекса селективности.
Влияние веществ на жизнеспособность эукариотических клеток исследовали с помощью МТТ теста, позволяющего оценивать суммарную активность дыхательных ферментов митохондрий. Этот тест основан на способности дегидрогеназ живых клеток восстанавливать неокрашенные формы 3-4,5-диметилтиазол-2-ил-2,5-дифенилтетразола бромида - МТТ-реагента до голубого кристаллического формазана. Данный метод широко распространен для изучения токсического действия различных соединений на клетки, в том числе и на этапе скрининга веществ-прототипов лекарственных препаратов.
Цитотоксичность исследовали на двух адгезионных клеточных линиях: клетках гепатокарциномы человека HepG2 и эмбриональных фибробластах человека HEF. Время инкубации с веществом составляло 24 часа для всех клеточных линий (см. Фиг. 1).
Предварительно, за 16 часов до добавления вещества, проводили открепление клеток трипсином, центрифугирование, подсчет в камере Горяева. Клетки помещали в лунки 96-луночного планшета. Количество клеток подбиралось в зависимости от свойств клеточной линии и конечной оптической плотности в эксперименте. Фибробласты и гепатоциты использовали в количестве 10 тыс. клеток на лунку в объеме 100 мкл среды DMEM/F12 c добавлением 10% FBS.
Исследуемые 6-фтор-3-триазеноиндолы по изобретению добавляли в 100 мкл среды в лунки таким образом, что конечные концентрации составили 50, 25, 12.5, 6.25, 3.125, 1.6, 0.8, 0.4 и 0.2 мкг/мл. Итоговый объем лунки составлял 200 мкл. В качестве отрицательного контроля добавляли среду c сывороткой.
После 24 часов излишнее количество среды отбрасывали. В каждую лунку добавляли по 30 мкл раствора MTT (5 мг/мл в PBS). После выпадения кристаллов формазана (2-4 ч, в зависимости от клеточной линии) его растворяли добавлением 100 мкл DMSO. Оптическое поглощение измеряли на планшетном спектрофотометре при длине волны 570 нм.
Выживаемость клеток рассчитывалась по формуле: (OD обработанных клеток - OD blank)/(OD контрольных клеток - OD blank) × 100%, где OD blank - значения OD в лунках без клеток, но с добавлением раствора МТТ и ДМСО. Конечный результат представляли в виде значений концентраций полумаксимального ингибирования (IC50).
Эксперимент с каждой клеточной линией проводился минимум трижды. В каждом эксперименте каждая концентрация была представлена минимум тремя лунками-повторностями.
Вещество растворяли в ДМСО (стоковый раствор 10 мг/мл), раствор вещества в ДМСО хранился при температуре +4°С. Содержание ДСМСО в среде с клетками не превышало 0,5%.
Индекс селективности рассчитывали путем деления значения концентрации полумаксимального ингибирования (IC50) на значение минимальной подавляющей концентрации (МПК).
Результаты определения антибактериальной активности и индекса селективности 3-триозеноиндолов по изобретению приведены ниже.
Таблица 4. Антимикробная активность (МПК) исследуемых соединений по изобретению.
Как следует из приведенных примеров, соединения по изобретению, в частности соединения BX-SI043 и BX-SI045, эффективнее подавляют рост резистентных штаммов золотистого стафилококка по сравнению с применяемыми в клинике препаратами ванкамцином и линезолидом.
Таблица 5. Цитотоксичность и индекс селективности исследуемых соединений по изобретению.
В результате проведенных исследований установлено, что соединения по изобретению, в частности соединения, представленные в Таблице 1, продемонстрировали высокую антибактериальную активность in vitro по отношению к панели клинических изолятов MRSA с множественной устойчивостью. Минимальные подавляющие концентрации соединений по изобретению значительно превосходят аналоги в клинике (ванкомицин, линезолид), как и высокие значения индекса селективности, которые свидетельствуют о достаточно низкой цитотоксичности исследуемых веществ. Приведенные данные свидетельствуют о том, что соединения по изобретению являются эффективными агентами, обладающими высокой антибактериальной активностью и низкой токсичностью, и могут быть использованы для лечения и профилактики инфекционных заболеваний, вызванных бактериальной инфекцией, в том числе вызванных антибиотикорезистентными патогенами, такими Staphylococcus aureus, включая мультирезистентные штаммы, в том числе Methicillin-resistant Staphylococcus aureus. Приведенные данные также свидетельствуют о том, что применение соединений по изобретению в терапии инфекционных заболеваний способно обеспечить снижение скорости возникновения резистентности к новой терапии у патогенов, вызывающих инфекционные заболевания, в том числе стафилококков, поскольку соединения по изобретению способы преодолевать устойчивость патогенов к разным группам препаратов.
Несмотря на то, что изобретение описано со ссылкой на раскрываемые варианты воплощения, для специалистов в данной области должно быть очевидно, что конкретные подробно описанные эксперименты приведены лишь в целях иллюстрирования настоящего изобретения, и их не следует рассматривать как каким-либо образом ограничивающие объем изобретения. Должно быть понятно, что возможно осуществление различных модификаций без отступления от сути настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТИАЗОЛИНО-2-ПИРИДОНЫ С КОНДЕНСИРОВАННЫМИ ЦИКЛАМИ В КОМБИНАЦИИ С ЛЕКАРСТВЕННЫМ СРЕДСТВОМ ПРОТИВ ТУБЕРКУЛЕЗА | 2018 |
|
RU2791467C2 |
| ЗАМЕЩЕННЫЕ 4-{ [4-(3,3-ДИОКСИДО-1,3-БЕНЗОКСАТИОЛ-6-ИЛ)АРИЛОКСИ]МЕТИЛ} ПИПЕРИДИНЫ КАК АГОНИСТЫ РЕЦЕПТОРОВ GPR119, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2014 |
|
RU2576037C1 |
| НОВЫЕ ПЕПТИДНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2699572C2 |
| ПЕПТИДНЫЕ МАКРОЦИКЛЫ ПРОТИВ ACINETOBACTER BAUMANNII | 2016 |
|
RU2729609C2 |
| ПИПЕРАЗИНО[1,2-А]ИНДОЛ-1-ОНЫ И [1,4]ДИАЗЕПИНО[1,2-А]ИНДОЛ-1-ОН | 2014 |
|
RU2628126C2 |
| Способ ингибирования бактериальной цистатионин-γ-лиазы | 2023 |
|
RU2841071C1 |
| НОВЫЕ ПРОТИВОМИКРОБНЫЕ СРЕДСТВА | 2009 |
|
RU2522582C2 |
| ЗАМЕЩЕННЫЕ 2-ТИОКСО-ИМИДАЗОЛИДИН-4-ОНЫ И ИХ СПИРОАНАЛОГИ, ПРОТИВОРАКОВЫЙ АКТИВНЫЙ КОМПОНЕНТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ, СПОСОБ ЛЕЧЕНИЯ РАКА ПРОСТАТЫ | 2014 |
|
RU2557235C1 |
| ПРОП-2-ИН-1-ИЛ (2Z)-2-[(3β,4α,8α,11α,14β,16β)-16-(АЦЕТИЛОКСИ)-3-(БУТИЛАМИНО)-11-ГИДРОКСИ-4,8,10,14-ТЕТРАМЕТИЛГОНАН-17-ИЛИДЕН]-6-МЕТИЛГЕПТ-5-ЕНОАТ, ПРОЯВЛЯЮЩИЙ АНТИБАКТЕРИАЛЬНУЮ И ФУНГИЦИДНУЮ АКТИВНОСТЬ | 2024 |
|
RU2841387C1 |
| НОВЫЕ ДИСПИРО-ИНДОЛИНОНЫ, ИНГИБИТОРЫ MDM2/p53 ВЗАИМОДЕЙСТВИЯ, СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2015 |
|
RU2629750C2 |
Изобретение относится к соединениям, характеризующимся высокой антибактериальной активностью, общей формулы (I), где R1-R2 имеют значения, указанные в описании. Изобретение также относится к фармацевтическим композициям, содержащим указанные соединения, и к применению вышеуказанных соединений для лечения и/или профилактики инфекционных заболеваний, в том числе вызванных антибиотикорезистентными патогенами (включая мультирезистентные штаммы), в частности заболеваний стафилококковой этиологии, например, инфекционных заболеваний кожи и/или мягких тканей, пневмонии, эндокардита или остеомиелита. 4 н. и 10 з.п. ф-лы, 1 ил., 5 табл., 1 пр.
1. Соединение формулы (I):
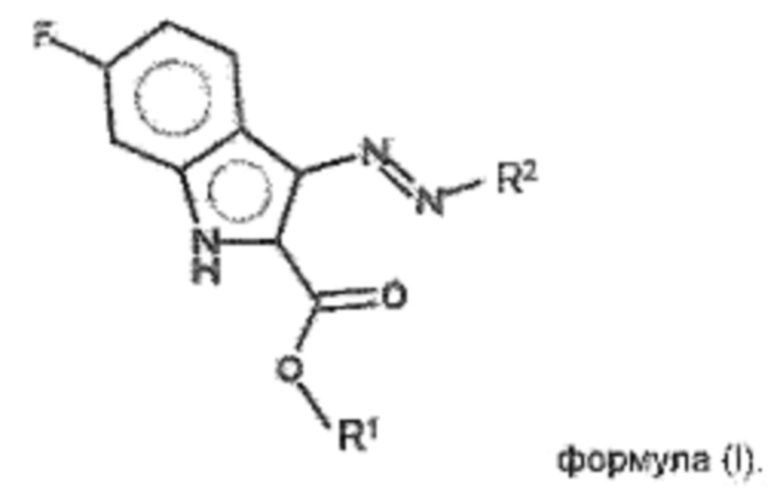
или его фармацевтически приемлемая соль, таутомер, где;
R1 выбирается независимо и представляет собой -C1-10-алкил;
R2 выбирается независимо и представляет собой С5-10-гетероциклил, содержащий 1-2-атома N и необязательно содержащий 1-2 атома О, причем R2 присоединяется через атом азота в цикле к диазеновому фрагменту, или -NR3R4;
причем заместитель R2 может быть необязательно замещен 1-3 заместителями R5;
R3 выбирается независимо и представляет собой Н, -С1-6алкил, -С1-6-алкил-NH-C1-6-алкил, -С1-6-алкил-С5-6-гетероарил, содержащий 1-2 атома N;
R4 выбирается независимо и представляет собой Н, -С1-6-алкил, -С1-6-алкил-NH-С1-6-алкил, С1-6-алкил-С5-6-гетероарил, содержащий 1-2 атома N;
R5 выбирается независимо и представляет собой -С1-6-алкил.
2. Соединение по п. 1, в котором:
R1 выбирается независимо и представляет собой -C1-6-алкил;
R2 выбирается независимо и представляет собой морфолин, пиперидин, пиперазин, пирролидин, пергидроазепин, пиридин, метилпиперидин, метилпиперазин или -NR3R4,
R3 выбирается независимо и представляет собой -С1-6-алкил, -С1-6-алкил-NH-C1-6-алкил, -C1-6-алкил-С5-6-гетероарил;
R4 выбирается независимо и представляет собой -С1-6-алкил, -С1-6-алкил-NH-C1-6-алкил, -C1-6-алкил-С5-6-гетероарил;
R5 выбирается независимо и представляет собой -C1-6-алкил.
3. Соединение по п. 1, выбранное из группы:
Этил-6-фтор-3-[4-метил-пиперазин-1-ил-азо]-1Н-индол-2-карбоксилат;
Этил-6-фтор-3-[4-метил-[1,4]-диазепан-1-ил-азо]-1Н-индол-2-карбоксилат;
Этил-6-фтор-3-[пирролидин-1-ил-азо]-1Н-индол-2-карбоксилат;
Этил-6-фтор-3-[N,N'-диметилэтан-1,2-диамин-1-ил-азо]-1Н-индол-2-карбоксилат;
Этил-6-фтор-3-[4-метил-пиперадин-1-ил-азо]-1Н-индол-2-карбоксилат;
Этил-6-фтор-3-[(1Е)-3-метил-3-(2-пиридин-2-ил-этил)-триаз-1-ен-1-ил]-1Н-индол-2-карбоксилат.
4. Применение соединения по п. 1 в качестве антибактериального лекарственного средства для лечения и/или профилактики инфекционного заболевания у субъекта, вызванного бактериями рода Staphylococcus.
5. Применения по п. 4, в котором инфекционное заболевание вызвано антибиотикорезистентным патогеном.
6. Применение по п. 4, в котором инфекционное заболевание вызвано Staphylococcus aureus, Methicillin-resestant Staphylococcus aureus.
7. Применение по п. 4, в котором субъект представляет собой млекопитающее.
8. Применение по п. 7, в котором млекопитающее представляет собой человека.
9. Применение соединения по п. 1 для получения фармацевтической композиции для лечения и/или профилактики инфекционного заболевания, вызванного бактериями рода Staphylococcus.
10. Фармацевтическая композиция для лечения и/или профилактики инфекционного заболевания, вызванного бактериями рода Staphylococcus, включающая терапевтически эффективное количество соединения по п. 1 и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
11. Фармацевтическая композиция по п. 10, в которой вспомогательное вещество представляет совой носитель, растворитель и/или наполнитель.
12. Фармацевтическая композиция по п. 10, в которой инфекционное заболевание вызвано антибиотикорезистентным патогеном.
13. Фармацевтическая композиция по п. 10, характеризующаяся тем, что инфекционное заболевание вызвано Staphylococcus aureus, Methicillin-resistant Staphylococcus aureus.
14. Фармацевтическая композиция по п. 10, характеризующаяся тем, что заболевание представляет собой инфекционное заболевание кожи и/или мягких тканей, пневмонию, эндокардит или остеомиелит.
| WO 2018088920 A1, 05.17.2018 | |||
| Нестерова И.Н | |||
| и др | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Симаков С.В | |||
| и др | |||
| "Синтез и противоопухолевая активность | |||

