Изобретение относится к области органической химии, медицинской химии, способу ингибирования бактериальной цистатионин-g-лиазы с использованием соединений формулы (I), включая соединения Ia – Ik. Соединения, входящие в формулу (I), представляют собой малые молекулы-потенциаторы, ингибиторы бактериальной цистатионин-γ-лиазы (цистатионин-гамма-лиазы, сystathionine gamma-lyase, или bCSE), применяют самостоятельно или в качестве потенциатора совместно с антибиотиками для подавления роста антибиотикорезистентных бактерий.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
В настоящее время актуальной является проблема разработки малых молекул, обладающих способностью выступать потенциаторами известных, апробированных в клинической практике, антибактериальных препаратов. Это объясняется возникновением значительного количества клинически значимых штаммов патогенных микроорганизмов, обладающих устойчивостью к антибиотикам, что является следствием того, что происходит повсеместное нерациональное применение антибиотиков наряду с естественной способностью микроорганизмов мутировать и образовывать резистентные к антибиотикам штаммы.
Инфекции, вызываемые грамотрицательными бактериями Pseudomonas aeruginosa и Acinetobacter baumannii, несут большую угрозу для госпитализированных пациентов, особенно это касается штаммов с множественной лекарственной устойчивостью. Множественная лекарственная устойчивость P. aeruginosa и A. baumannii все чаще вызывает внутрибольничные инфекции; штаммы с множественной лекарственной устойчивостью распространяются в новые географические районы, и восприимчивые штаммы приобретают гены устойчивости. Появляются новые бета-лактамазы и карбапенемазы расширенного спектра действия, что приводит к появлению панрезистентных штаммов. Текущие исследования сосредоточены на влиянии антибиотиков на экспрессию генов в биопленках P. aeruginosa и их вкладе в устойчивость к терапии. Варианты лечения инфекций P. aeruginosa и A. baumannii с множественной лекарственной устойчивостью в большинстве случаев ограничены карбапенемами. Сульбактам является вариантом лечения панрезистентного A. baumannii, и/или рассматривается возможность возобновления применения старого препарата колистин для панрезистентного A. baumannii и P. aeruginosa. Иммунотерапия является многообещающим новым методом, который в настоящее время изучается. Изучаются профилактика возникновения резистентности с помощью комбинированной терапии и фармакокинетические стратегии.
Появление и распространение P. aeruginosa и A. baumannii с множественной лекарственной устойчивостью и их генетический потенциал по переносу различных детерминант устойчивости к антибиотикам представляют серьезную угрозу в больницах. Сложное взаимодействие клонального распространения, персистенции, передачи элементов резистентности и межклеточного взаимодействия усугубляют трудности в лечении инфекций, вызванных этими штаммами с множественной лекарственной устойчивостью (Navon-Venezia et al., 2005) [1].
Инфекции, вызываемые грамположительными бактериями Staphylococcus аureus, также несут большую угрозу для госпитализированных пациентов, особенно штаммы с множественной лекарственной устойчивостью, в том числе MRSA (метициллинрезистентный золотистый стафилококк).
В отсутствие новых антибиотиков существует необходимость разработки новых методов лечения.
Одной из перспективных молекулярных мишеней в жизненном цикле бактерий, воздействие на которую повышает их восприимчивость к антибиотикотерапии, является фермент – бактериальная цистатионин-γ-лиаза. Бактериальная цистатионин-γ-лиаза (bCSE) является основным продуцентом H2S у патогенных бактерий, таких как золотистый стафилококк, синегнойная палочка и др. Подавление активности bCSE значительно повышает чувствительность бактерий к антибиотикам. Одна из функций этого фермента заключается в его участии в продукции бактериями сероводорода, представляющего фактор естественной защиты от окислительного стресса, (WO2019232083(А1)) [2]. В то же время цистатионин-γ-лиаза играет важную роль в жизнедеятельности организмов млекопитающих, в том числе – человека. Таким образом, задача создания потенциаторов антибиотиков на основе блокаторов этого фермента несколько осложняется тем, что целевые соединения должны обладать высокой селективностью в отношении фермента бактериального происхождения, при минимальном (или отсутствующем) ингибирующем воздействии на соответствующий фермент человеческого организма.
УРОВЕНЬ ТЕХНИКИ
Роль бактерий-персистеров в распространении резистентности к антибиотикам становится все более очевидной, вследствие чего использование потенцирующих препаратов, повышающих чувствительность бактерий-персистеров к антибиотикам, является актуальной стратегией решения вышеуказанной проблемы.
В этой связи было исследовано несколько подходов, проведен поиск и анализ источников информации из общедоступного уровня техники до даты испрашиваемого приоритета.
Многообещающий, но малоизученный подход к разработке новых потенциаторов включают в себя нарушение системы общей защиты патогенов от различных антибиотиков. Одна из таких систем производит сероводород (H2S), который защищает бактерии от окислительного стресса (L. Luhachack, E. Nudler, 2014) [3].
Практически все бактерии генерируют H2S с помощью ферментов CSE, CBS или 3-меркаптопируватсертрансферазы (K. Shatalin, et al., 2011) [4]. Нарушение биогенеза H2S сенсибилизирует широкий спектр патогенов, включая S. aureus и P. aeruginosa, к различным классам бактерицидных препаратов (K. Shatalin, et al., 2021) [5].
Один из новых подходов заключается в ингибировании источника бактериальной инфекции с целью потенцирования бактерицидных антибиотиков.
В 2013 году после тестирования (Asimakopoulou, A., et al. 2013) [6] различных часто используемых ингибиторов CSE и CBS было обнаружено, что L-аминоэтоксивинилглицин (АВГ) является в достаточной степени мощным и селективным ингибитором CSE. Также было выявлено, что пропаргилглицин (ПАГ) аналогично проявляет избирательность по отношению к CSE, но его эффективность ниже, чем у АВГ. Тем не менее данные вещества не обладают селективностью именно к бактериальной цистатионин-γ-лиазе, в сравнении с человеческой.
В 2021 году было обнаружено, что индолсодержащие соединения являются селективными ингибиторами bCSE (K. Shatalin, et al., 2021) [5]. Были получены результаты, подтверждающие эффективность концепции потенцирования действия антибактериальных средств. Однако несмотря на то что применение этих соединений в сочетании с бактерицидными антибиотиками позволяет снизить дозировку антибиотиков, путь их синтеза длинный (семь стадий) и включает такие сложные для исполнения этапы, как катализируемая родием циклизация и кросс-сочетание Сузуки.
Из уровня техники выявлены нижеследующие аналоги предлагаемого изобретения.
Известен источник информации RU 2640418 C2 от 09.01.2018 «Ингибиторы цистатионин-гамма-лиазы (CSE)» [8].
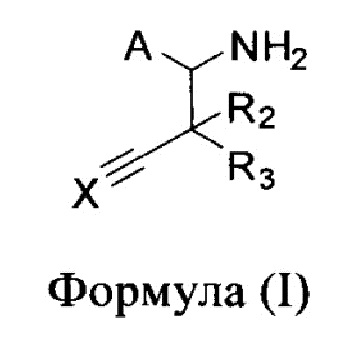
Изобретение относится к соединениям Формулы (I) или их фармацевтически приемлемым солям, ингибирующим активность цистатионин-гамма-лиазы (CSE). В Формуле (I) А представляет собой 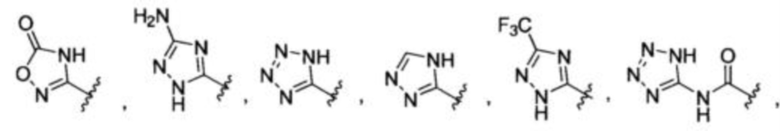
или -CONHSO2R4, где R4 представляет собой независимо незамещенный алкил или незамещенный арил; X представляет собой CR1 или N; R1 представляет собой Н; каждый R2 и R3 представляет собой Н. Изобретение относится также к фармацевтической композиции, содержащей указанные соединения, и способу лечения или предупреждения различных заболеваний, связанных с активностью CSE.
Недостатками аналога по сравнению с предлагаемым изобретением являются отсутствие тестирования активности веществ по отношению к мишени – бактериальной цистатионин-гамма-лиазе, таким образом, подтверждение действия именно на эту мишень отсутствует. В чистом виде не был выделен и фермент грызунов, на котором проводились испытания. Целью данных испытаний было исследование ингибирования фермента CSE для лечения заболеваний небактериальной природы. Действие данных веществ на бактерии не изучалось.
Известно техническое решение US9725426B2 от 08.08.2017 г. «Cystathionine-γ-lyase (CSE) inhibitors» [7], в котором описаны соединения и фармацевтические композиции, содержащие такие соединения, которые ингибируют цистатионин-γ-лиазу (CSE). Также описаны способы применения таких ингибиторов CSE, отдельно или в сочетании с другими соединениями, для лечения заболеваний или состояний, при которых ингибирование CSE может принести пользу. Изобретение относится к ингибиторам CSE, представленных следующей формулой:
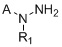
или их фармацевтически приемлемым солям, которые ингибируют цистатионин- γ-лиазу. Соединения могут быть использованы для лечения или предупреждения, или снижения числа случаев состояния, выбранного из ноцицептивной боли, острой послеоперационной боли, нейропатической боли, невралгии тройничного нерва (тригеминальной невралгии), диабетической периферической нейропатии, герпетической невралгии, постгерпетической невралгии, воспалительной боли, смешанной боли нейропатического и воспалительного характера, ревматоидного артрита, воспалительного заболевания кишечника, синдрома раздраженного кишечника, остеоартрита, острого панкреатита, хронического панкреатита, боли, ассоциируемой с острым панкреатитом, боли, ассоциируемой с хроническим панкреатитом, мигрени, подагры, анкилозирующего спондилоартрита, системной красной волчанки (SLE), синдрома системного воспалительного ответа (SIRS), синдрома полиорганной дисфункции (MODS), астмы, хронической обструктивной легочной болезни (COPD), чувствительной кожи, акне, розацеа (красных угрей), контактного дерматита и боли, ассоциируемой с раком.
Недостатками являются сложность многостадийного синтеза и отсутствие изучения ингибирования бактериальной цистатионин-гамма-лиазы.
Известен источник информации K. Shatalin, et al., 2021 ««Inhibitors of bacterial H2S biogenesis targeting antibiotic resistance and tolerance» [5], в котором описан новый подход, заключающийся в ингибировании источника бактериальной инфекции с целью потенцировать бактерицидные антибиотики. Был установлен основной ферментативный источник H2S в двух распространенных у человека патогенах, грамположительных бактерий Staphylococcus aureus и грамотрицательных бактерий Pseudomonas aeruginosa, которые относятся к ведущим причинам госпитальных инфекций и характеризуются резистентностью к антибиотикам.
Недостатками являются сложность синтеза целевых соединений, сложность их очистки, отсутствие исследования активности на Acinetobacter baumannii, отсутствие изучения потенцирования действия цефепима, меропенема, смеси пиперициллина и тазобактама.
Известно техническое решение, описанное в заявке на патент на изобретение WO2019232083(А1), 05.12.2019 «Compounds and methods for treating bacterial infections» [2], который представляет собой наиболее близкий аналог предлагаемого изобретения и выбран за прототип. Изобретение относится к ингибитору bCSE, представленному формулой (I):
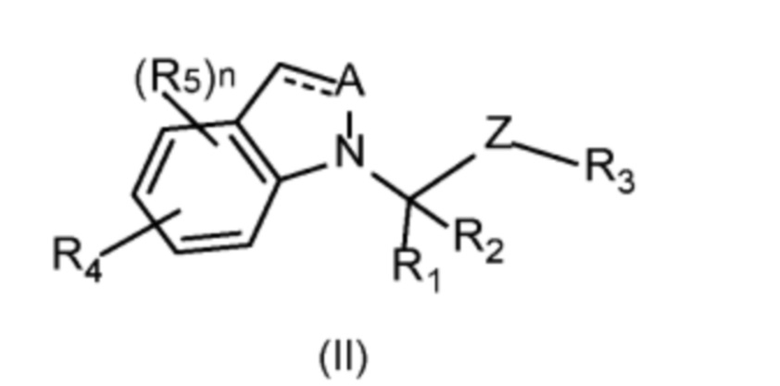
или его фармацевтически приемлемой соли или изомеру, способу его получения и применению ингибитора bCSE для лечения заболеваний, вызванных антибиотикорезистентностью. Соединения по этому изобретению получают посредством серии реакций, таких как сочетание по Сузуки, замещение, гидролиз. Лекарственное средство на основе фармацевтической композиции используют для лечения заболеваний, связанных с активностью bCSE: пневмония, кожные инфекции, некротический фасциит, пиомиозит, некротические пневмонии, инфекционный эндокардит.
Недостатками прототипа по сравнению с предлагаемым изобретением являются сложность синтеза целевых соединений, сложность их очистки, отсутствие исследования активности на Acinetobacter baumannii, отсутствие изучения потенцирования действия цефепима, меропенема, смеси пиперициллина и тазобактама.
Целью настоящего изобретения является разработка:
- нового способа ингибирования бактериальной цистатионин-g-лиазы с использованием нафтилсодержащих карбоновых кислот, представляющих собой соединения формулы (I), для ингибирования бактериальной цистатионин-g-лиазы, которые применяют самостоятельно или в качестве потенциаторов совместно с антибиотиками для лечения заболеваний, вызываемых антибиотикорезистентными бактериями.
В результате проведенного анализа уровня техники можно сделать вывод о том, что предлагаемое изобретение может быть признано соответствующим критериям патентоспособности «новизна» и «изобретательский уровень» до даты испрашиваемого приоритета.
Технический результат заключается:
- в увеличении эффективности ингибирования бактериальной цистатионин- γ -лиазы (bCSE) за счёт использования нафтилсодержащих карбоновых кислот, представляющих собой соединение формулы (I) - молекулы-потенциаторы Ic-Ih, для подавления роста антибиотикорезистентных бактерий;
- в увеличении эффективности применения синтезированных соединений формулы (I) – веществ Ic-Ih, в качестве потенциаторов антибиотиков - селективных ингибиторов бактериальной цистатионин- γ -лиазы веществ, более доступных синтетически, чем опубликованные ранее аналоги;
- в расширении арсенала способов ингибирования белка (bCSE) за счёт применения потенциаторов антибиотиков для подавления роста антибиотикорезистентных штаммов микроорганизмов.
РАСКРЫТИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Для достижения технического результата заявителем выполнена разработка способа синтеза и применения структурного типа ингибиторов белка цистатионин-γ-лиазы – потенциаторов антибиотиков, охарактеризованных общей формулой (I). В предлагаемой группе изобретений объединены структурные, биохимические, микробиологические подходы к скринингу и подтверждению активности низкомолекулярных соединений. Показан способ синтеза предлагаемых соединений, проведены ферментативные, микробиологические исследования новых структурных аналогов, описанных ранее в литературе блокаторов цистатионин-γ-лиазы – вероятных потенциаторов антибиотиков. В ходе исследования биологических свойств этих соединений было установлено, что в структуре соединений необходимо наличие двух составляющих: 1) индольного или пиррольного фрагмента с карбоксильной группой при гетероциклическом ядре, ответственного за образование водородных связей и прочих сильных взаимодействий лиганда с ферментом; 2) 1- или 2-нафтильного фрагмента, образующего необходимые контакты в гидрофобном кармане в активном центре.
Чтобы устранить недостатки имеющихся аналогов и прототипа [2-8], авторы настоящего изобретения разработали новый способ:
Способ ингибирования бактериальной цистатионин-g-лиазы с использованием нафтилсодержащих карбоновых кислот, представляющих собой соединение формулы (I):
 I,
I,
в котором кольцо, имеющее связи - - -, отсутствует или представляет собой бензольное кольцо, непосредственно присоединённое к ароматической системе пиррола посредством двух соседних атомов углерода, характеризующийся тем, что соединение формулы (I) применяют самостоятельно или в качестве потенциатора совместно с антибиотиками для подавления роста антибиотикорезистентных бактерий, при этом для подавления роста грамотрицательных бактерий Acinetobacter baumannii или Pseudomonas aeruginosa антибиотики выбирают из группы: цефепим, меропенем, смесь пиперациллина и тазобактама, а для подавления роста грамположительных бактерий вида Staphylococcus aureus, в том числе MRSA, антибиотики выбирают из группы: канамицин, ампициллин, норфлоксацин.
Выбор этих соединений связан с наличием в их составе фрагментов, ответственных за эффективное связывание с целевым белком: карбоксильной группы при гетероцикле, формирующей прочные полярные взаимодействия; нафтильного фрагмента, эффективно связывающегося с липофильной частью активного сайта.
КРАТКОЕ ОПИСАНИЕ ФИГУР, ТАБЛИЦ, ИНЫХ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фигуре 1 представлена ингибирующая активность соединений Ia-Ik по отношению к bCSE и hCSE.
На фигуре 2 представлены кривые зависимости интенсивности флуоресценции WSP5 от концентрации ингибитора для ферментов бактериального (А) и человеческого (B) CSE для ряда соединений PAG, NL2, Ic.
На фигуре 3 изображено увеличение активности антибиотиков в присутствии веществ Ia-Ik против: (A) S. aureus ATCC 25923; (B) A. baumannii GIMC5509:ABT-52Ts19; (C) P. aeruginosa GIMC5016:PA1840/36/2015. Результаты представлены в виде соотношения минимальной ингибирующей концентрации антибиотика без добавления потенциатора к минимальной ингибирующей концентрации антибиотика в присутствии потенциатора (показывает во сколько раз усилилась активность антибиотика).
На фигуре 4 представлено исследование цитотоксичности на клеточной линии почки эмбриона человека HEK293. Приведены значения 50% клеточной цитотоксичности (CC50) для соединений Ia-Ik.
В таблице 1 приведены структура и свойства соединений формулы (I).
В таблице 2 приведены оптические настройки прибора для детектирования сигнала флуоресценции.
В таблице 3 приведены результаты ингибирующей активности соединений в ферментативных тестах на bCSE и hCSE.
В таблице 4 приведены результаты микробиологических тестов потенцирующей активности в комбинации с антибиотиками против S. aureus ATCC 25923 и S. aureus INA00761 (MRSA).
В таблице 5 приведены результаты микробиологических тестов потенцирующей активности в комбинации с антибиотиками против клинических изолятов A. baumannii и P. aeruginosa.
В таблице 6 приведено исследование цитотоксичности соединений на клеточной линии почки эмбриона человека HEK293.
В таблице 7 показаны результаты определения растворимости лидерного соединения Ic.
В таблице 8 показаны результаты определения стабильности в микросомах печени крыс лидерного соединения Ic.
В таблице 9 показаны результаты определения проницаемости на клетках линии Caco-2 лидерного соединения Ic.
В таблице 10 показаны результаты определения стабильности в плазме крови человека лидерного соединения Ic.
В таблице 11 показаны результаты определения стабильности в искусственном кишечном соке (SIF) и искусственном желудочном соке (SGF) лидерного соединения Ic.
В таблице 12 показаны результаты определения доли лидерного соединения Ic, связавшегося с белками плазмы крови человека.
В таблице 13 показаны результаты определения коэффициента распределения logD7.4 лидерного соединения Ic, между 1-октанолом и 0.1 М фосфатным буферным раствором.
ОБЪЕКТ ИЗОБРЕТЕНИЯ
Указанный технический результат достигается совокупностью существенных признаков, представленных в формуле изобретения, где заявлен способ ингибирования бактериальной цистатионин-g-лиазы с использованием соединения формулы (I), как указано ниже.
В одной неограничивающей реализации изобретения предложен способ ингибирования бактериальной цистатионин-g-лиазы с использованием нафтилсодержащих карбоновых кислот, представляющих собой соединение формулы (I), а именно:
I,
в котором кольцо, имеющее связи - - -, отсутствует или представляет собой бензольное кольцо, непосредственно присоединённое к ароматической системе пиррола посредством двух соседних атомов углерода, характеризующийся тем, что соединение формулы (I) применяют самостоятельно или в качестве потенциатора совместно с антибиотиками для подавления роста антибиотикорезистентных бактерий, при этом для подавления роста грамотрицательных бактерий Acinetobacter baumannii или Pseudomonas aeruginosa антибиотики выбирают из группы: цефепим, меропенем, смесь пиперациллина и тазобактама, а для подавления роста грамположительных бактерий вида Staphylococcus aureus, в том числе MRSA, антибиотики выбирают из группы: канамицин, ампициллин, норфлоксацин.
Ещё в одном примере реализации представлен способ ингибирования бактериальной цистатионин-g-лиазы, в котором нафтилсодержащая карбоновая кислота представляет собой соединение, имеющее формулу:
 Ic.
Ic.
Ещё в одном примере реализации представлен способ ингибирования бактериальной цистатионин-g-лиазы, в котором нафтилсодержащая карбоновая кислота представляет собой соединение, имеющее формулу:
 Id.
Id.
Ещё в одном примере реализации представлен способ ингибирования бактериальной цистатионин-g-лиазы, в котором нафтилсодержащая карбоновая кислота представляет собой соединение, имеющее формулу:
 Ie.
Ie.
Ещё в одном примере реализации представлен способ ингибирования бактериальной цистатионин-g-лиазы, в котором нафтилсодержащая карбоновая кислота представляет собой соединение, имеющее формулу:
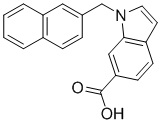 If.
If.
Ещё в одном примере реализации представлен способ ингибирования бактериальной цистатионин-g-лиазы, в котором нафтилсодержащая карбоновая кислота представляет собой соединение, имеющее формулу:
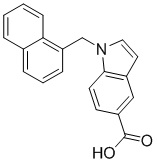 Ig.
Ig.
Ещё в одном примере реализации представлен способ ингибирования бактериальной цистатионин-g-лиазы, в котором нафтилсодержащая карбоновая кислота представляет собой соединение, имеющее формулу:
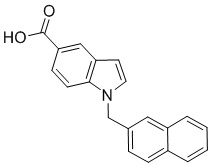 Ih.
Ih.
Множество соединений формулы (I) по примерам в соответствии с настоящим изобретением, их структура и свойства представлены в таблице 1.
Таблица 1. Структура и свойства соединений формулы (I).
ATCC 25923, мкг/мл
HEK293,
мкМ
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения и общая терминология.
Далее будут приведены ссылки на определенные варианты осуществления изобретения, примеры которых проиллюстрированы прилагаемыми структурами и формулами. Подразумевается, что изобретение охватывает все альтернативы, модификации и эквиваленты, которые могут быть включены в объем настоящего изобретения, как определено в формуле изобретения. Специалисту в данной области известны многие способы и материалы, аналогичные или эквивалентные описанным в данном документе, которые могут быть использованы при практическом осуществлении настоящего изобретения. Настоящее изобретение не ограничивается изложенными в данном документе способами и материалами. В случае, если один или несколько из включенных литературных ссылок, патентов и аналогичных материалов отличается от или противоречит данной заявке, включая, в частности, определенные термины, использование терминов, описанные приемы и т.п., преимущественную силу имеет настоящая заявка.
Далее следует принять во внимание, что определенные признаки изобретения, которые для ясности описаны в контексте отдельных вариантов осуществления изобретения, могут быть представлены также совместно в одном варианте. И наоборот, разнообразные признаки изобретения, которые для краткости описаны в контексте одного варианта осуществления изобретения, могут быть представлены отдельно или в любой подходящей подкомбинации.
Для описания настоящего изобретения используются следующие термины.
Все технические и научные термины, использованные в данном документе, имеют такое значение, какое обычно понимается специалистами в области, к которой относится изобретение, если не указано иное. Все патенты и публикации, на которые имеются ссылки в данном документе, полностью включены в изобретение посредством ссылки.
В контексте данного документа будут применяться следующие определения, если не указано иное. В целях предложенного изобретения химические элементы обозначены в соответствии с Периодической таблицей элементов, версия CAS, и Справочником по химии и физике, 75-й выпуск, 1994 и т.д.
В тех случаях, когда термин конкретно не определен, этот термин имеет принятое специалистами в данной области значение и означает, что этот термин в данном контексте используется для описания настоящего изобретения.
Если представлен диапазон значений, то следует понимать, что каждое промежуточное значение, до десятых долей единицы нижнего предела, если из контекста явно не следует иное (так как в случае группы, содержащей некоторое число атомов углерода, в этом случае предусмотрено каждое число атомов углерода, попадающее в указанный интервал), между верхней и нижней границами этого диапазона и любые другие указанные или промежуточные значения в этом установленном интервале охватываются настоящим изобретением. Верхние и нижние пределы этих меньших диапазонов могут быть независимо включены в меньшие диапазоны, что также входит в объем изобретения, с учетом любого конкретно исключенного предела в указанном диапазоне. Когда указано, что диапазон включает один или оба предела, диапазоны, исключающие любое из обоих включенных пределов, также входят в объем изобретения.
Термин «соединение», как он используется в настоящем документе, если не указано иное, относится к любому конкретному химическому соединению, раскрытому в данном документе, и включает таутомеры, изомеры положения, геометрические изомеры и, где это применимо, стереоизомеры, включая оптические изомеры (энантиомеры) и другие стереоизомеры (диастереомеры), а также их фармацевтически приемлемые соли и производные (включая формы пролекарств), когда это применимо в контексте. Согласно его использованию в контексте, термин «соединение» обычно относится к единственному соединению, но также может включать другие соединения, такие как стереоизомеры, изомеры положения и/или оптические изомеры (включая рацемические смеси), а также конкретные энантиомеры или энантиомерно обогащенные смеси раскрытых соединений. Термин относится также, в данном контексте, к пролекарственным формам соединений, которые должны быть модифицированы для облегчения введения и доставки соединений к месту проявления их активности.
Отмечается, что при описании настоящих соединений, среди прочих, описано множество заместителей и связанных с ними вариантов. Специалисту в данной области понятно, что молекулы, которые описаны в настоящем документе, представляют собой стабильные соединения, как это обычно предусматривается далее.
Термин «эффективный» используется для описания количества соединения, композиции или компонента, которое, при использовании в контексте его предназначения, приводит в предполагаемому результату. Термин «эффективный» охватывает все другие термины, относящееся к эффективному количеству или эффективным концентрациям, которые иным образом описаны или использованы в настоящей заявке.
Ссылки, которые цитируются, включены в данный документ в качестве ссылок во всей их полноте.
Термин «заболевание или состояние» используется для описания какой-либо группы патологий, возбудителями которых являются патогенные и условно-патогенные микроорганизмы. Ингибирование роста данных патогенных микроорганизмов в организме пациента может обеспечить успешное лечение или облегчение симптомов пациенту, нуждающемуся в этом. В некоторых случаях заболевание или состояние может быть вылечено.
Термин «фармацевтически приемлемые соли» относится к органическим или неорганическим солям предложенного в настоящем изобретении соединения. Термин «фармацевтически приемлемая соль» используется в описании для обозначения, где это применимо, солевой формы одного или нескольких соединений, описанных в настоящем документе, которые предложены для увеличения растворимости соединения в желудочных соках желудочно-кишечного тракта пациента для способствования растворения и биодоступности соединений. Фармацевтически приемлемые соли включают полученные с фармацевтически приемлемыми неорганическими или органическими основаниями и кислотами, где это применимо. Подходящие соли включают соли, полученные со щелочными металлами, такими как калий и натрий, щелочноземельными металлами, такими как кальций, магний, и соли аммония, среди многочисленных других кислот и оснований, хорошо известных в фармацевтической области. Соли натрия и калия являются особенно предпочтительными в качестве солей нейтрализации фосфатов в соответствии с настоящим изобретением. Фармацевтически приемлемые соли хорошо известны специалистам. Например, S.М. Berge et al., подробно описывает фармацевтически приемлемые соли в публикации в журнале J. Pharmaceutical Sciences, 1977, 66: 1-19.
Термин «фармацевтически приемлемое производное» используется в описании для обозначения любой фармацевтически приемлемой пролекарственной формы (такой как сложноэфирная, амидная, другая пролекарственная группа), которая при введении пациенту обеспечивает непосредственно или косвенно настоящее соединение или его активный метаболит по настоящему соединению.
Термин «пролекарство» относится к соединению, которое превращается in vivo в соединение по формуле (I). Такое превращение может осуществляться в результате, например, гидролиза пролекарства в крови или ферментативного превращения исходной формы в крови или тканях. Пролекарствами соединений, предложенных в настоящем изобретении, могут быть, например, сложные эфиры. Некоторыми распространенными сложными эфирами, которые были использованы в качестве пролекарств, являются фениловые эфиры, сложные эфиры алифатических кислот (С1-24), ацилоксиметиловые эфиры, карбонаты, карбаматы и эфиры аминокислот. Другие пролекарственные формы включают фосфаты. Всестороннее обсуждение пролекарств представлено в публикации Т. Хигучи и В. Стелла Пролекарства, как инновационные системы доставки, Том 14 серии Симпозиум А.К.С., под редакцией Эдварда Б. Рош, Биообратимые носители в разработке лекарственных средств, Американская фармацевтическая ассоциация и Пергамон Пресс, 1987, Дж. Рауцио с сотр. Пролекарства: разработка и клиническое применение, Nature Review Drug , 2008, 7, 255-270, и С.Дж. Хекер с сотр., Пролекарства, представляющие собой фосфаты и фосфонаты, Journal of Chemistry, 2008, 51, 2328-2345 (Т. Higuchi и V. Stella, Pro-drugs as Novel Delivery Systems, Vol. 14 of the A.C.S. Symposium Series, Edward B. Roche, ed., Carriersin Drug Design, American Pharmaceutical Association и Pergamon Press, 1987, J. Rautio et al., Prodrugs: Design и Clinical Applications, Nature Review Drug , 2008, 7, 255-270, и S.J. Hecker et al., Prodrugs of Phosphates и Phosphonates, Journal of Chemistry, 2008, 51, 2328-2345), все эти публикации полностью включены в настоящее изобретение в форме ссылки.
Термин «сольват» относится к ассоциации или комплексу одной или нескольких сольватных молекул и соединению, предложенному в изобретении. Не имеющие ограничительного характера примеры растворителей, образующих сольваты, включают воду, изопропанол, этанол, метанол, диметилсульфоксид, этилацетат, уксусную кислоту и этаноламин.
Термин «гидрат» относится к комплексу, в котором молекулой растворителя является вода.
Термин «амиды» относится к аналогам представленных веществ, в которых карбоксильная группа замещена на амидную.
Термин «независимо» используется в настоящем документе, чтобы показать, что переменная, которая применяется независимо, меняется независимо от применения к применению.
Термин «алкил» будет обозначать в своем контексте линейный, разветвленный или циклический полностью насыщенный углеводородный радикал или алкильную группу, предпочтительно C1-C10, более предпочтительно C1-C6, альтернативно C1-C3 алкильную группу, которая может быть необязательно замещена. Примерами алкильных групп являются, среди прочих, метил, этил, н-бутил, втор-бутил, н-гексил, н-гептил, н-октил, н-нонил, н-децил, изопропил, 2-метилпропил, циклопропил, циклопропилметил, циклобутил, циклопентил, циклопентилэтил, циклогексилэтил и циклогексил.
Термин «замещенный» (каждый заместитель является независимым от любого другого заместителя) также будет обозначать в своем контексте использование C1-C6 алкила, C1-C6 алкокси, галогена, фенила, нафтила, гетероциклов и др.
Термин «гетероцикл», «гетероциклическое ядро» или «гетероциклическое соединение», используемые взаимозаменяемо в настоящем документе, относятся к ароматическому, моноциклическому или бициклическому кольцу, содержащему 5-9 кольцевых атомов, среди которых, по меньшей мере, один кольцевой атом является азотом, и среди которых может быть одно или несколько мест соединения с остальной молекулой.
Термины «лечение», «лечить» или «подвергать лечению» и т.д., как используется в настоящем документе, относятся к какому-либо действию, обеспечивающему благоприятное воздействие на пациента, которому могут быть введены настоящие соединения.
Фармацевтические композиции, содержащие комбинации эффективного количества, по меньшей мере, одного соединения, в соответствии с настоящим изобретением, и одного или нескольких соединений, иным образом описанных в настоящем документе, все в эффективном количестве, в комбинации с фармацевтически эффективным количеством носителя, добавки или инертного наполнителя, представляют следующий аспект настоящего изобретения. Настоящее изобретение включает, где это применимо, композиции, содержащие фармацевтически приемлемые соли, в частности, соли добавления кислот или оснований соединений по настоящему изобретению. Кислоты, которые используются для получения фармацевтически приемлемых кислотно-аддитивных солей вышеуказанного основного соединения, используемые в данном изобретении, представляют собой такие, которые образуют нетоксичные кислотно-аддитивные соли, то есть соли, содержащие фармакологически приемлемые анионы, такие как соли, среди многочисленных других, гидрохлорид, гидробромид, гидройодид, нитрат, сульфат, бисульфат, фосфат, кислый фосфат, ацетат, лактат, цитрат, кислый цитрат, тартрат, битартрат, сукцинат, малеат, фумарат, глюконат, сахарат, бензоат,метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат и памоат [то есть, 1,1′-метилен-бис-(2-гидрокси-3-нафтоат)]. Фармацевтически приемлемые соли добавления оснований также могут использоваться для получения фармацевтически приемлемых солевых форм соединений или производных в соответствии с настоящим изобретением. Химические основания, которые могут быть использованы в качестве реагентов для получения солей фармацевтически приемлемых оснований с настоящими соединениями, являющимися кислотными по природе, представляют собой такие, которые образуют нетоксичные соли оснований с такими соединениями. Такие нетоксичные соли оснований включают, но этим не ограничиваются, производные таких фармакологически приемлемых катионов, как катионы щелочных металлов (например, калия и натрия) и катионы щелочноземельных металлов (например, кальция, цинка и магния), аммония, или водорастворимые соли добавления аминов, таких как N-метилглюкамин-(меглумин), и соли низшего алканоламмония и другого основания фармацевтически приемлемых органических аминов, среди прочих.
Для описания настоящего изобретения используются следующие обозначения:
Антибиотикорезистентные грамотрицательные бактерии, использованные в заявленном способе, - это штаммы клинических изолятов бактерий: Acinetobacter baumannii GIMC5509:ABT-52Ts19 и Pseudomonas aeruginosa GIMC5016:PA1840/36/2015.
Антибиотикорезистентные грамположительные бактерии, использованные в заявленном способе, - это Staphylococcus aureus, в том числе MRSA.
ВЭЖХ-УФ/МС – высокоэффективная жидкостная хроматография с ультрафиолетовым детектированием и масс-спектрометрией.
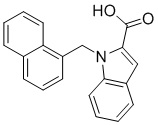
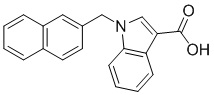
Лидерное соединение - соединение, которое имеет оптимальные показатели ингибирования, токсичности по результатам ферментативного и микробиологического тестирования и в дальнейшем может быть использовано в исследовании in vivo. В настоящем изобретение это соединение Iс.
Культура клеток HEK293T - производная клеточной линии человека, экспрессирующая мутантную версию большого Т-антигена SV40, часто используются в биологических исследованиях для производства белков и получения рекомбинантных ретровирусов.
Тациллин – смесь антибиотиков пиперациллина и тазобактама.
ЯМР – ядерный магнитный резонанс - резонансное поглощение или излучение электромагнитной энергии веществом, содержащим ядра с ненулевым спином во внешнем магнитном поле, на частоте ν (называемой частотой ЯМР), обусловленное переориентацией магнитных моментов ядер.
ADME – «Absorption, Distribution, Мetabolism, and Excretion» (Абсорбция, Распределение, Метаболизм и Выведение).
bCSE – бактериальная цистатионин-γ-лиаза (Cystathionine gamma-lyase).
hCSE – человеческая цистатионин-γ-лиаза (Cystathionine gamma-lyase).
HRMS – масс-спектрометрия высокого разрешения.
IC50 - половинная максимальная ингибирующая концентрация (IC50) является мерой эффективности вещества в ингибировании определенной биологической или биохимической функции. IC50 — это количественная мера, которая показывает, сколько конкретного ингибирующего вещества (например, лекарственного препарата) необходимо для ингибирования in vitro данного биологического процесса или биологического компонента на 50%. Биологическим компонентом может быть фермент, клетка, клеточный рецептор или микроорганизм. Значения IC50 обычно выражаются в молярной концентрации (М) (https://www.vniim.ru/files/SI-2019.pdf). В настоящем документе может быть использовано обозначение мкМ – микромоль.
MIC – минимальная ингибирующая концентрация антибиотика, при которой не наблюдается роста бактерий
CC50 - цитотоксичная доза – концентрация препарата, вызывающая гибель 50% клеток в культуре, мкг/мл. Чем выше значение СС50, тем меньше токсичность вещества для человека.
MRSA - антибиотикорезистентные грамположительные бактерии, представляющие собой бактериальный штамм метициллинрезистентного золотистого стафилококка Staphylococcus aureus. В предлагаемом способе использованы штаммы стафилококка: S. aureus ATCC 25923, S. aureus INA00761.
DL – пропаргилглицин (PAG) - рацемический 2-пропаргилглицин, являющийся необратимым и неселективным ингибитором цистатионин-γ-лиазы, использованный в качестве контрольного ингибитора.
NL-1 – (2-(6-бром-1H-индол-1-ил)aцетил)глицин.
NL-2 – (5-((6-бром-1H-индол-1-ил)метил)-2-метилфуран-3-карбоновая кислота).
PBS-буфер - фосфатно-солевой буфер — буферный раствор, являющийся водным раствором солей, содержащим хлорид натрия, гидрофосфат натрия, хлорид калия и дигидрофосфат калия.
ОБЩИЕ СИНТЕТИЧЕСКИЕ ПОДХОДЫ
Общая методика 1: N-алкилирование индолкарбоновых кислот
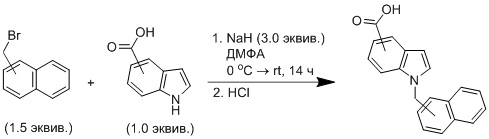
В сухую круглодонную колбу вносили соответствующее азотсодержащее гетероциклическое соединение (1.0 экв.) в инертной атмосфере. Далее растворяли в сухом ДМФА. Добавляли гидрид натрия (3.0 экв., 60%-ная суспензия в минеральном масле) при 0 °C в атмосфере азота. Полученный раствор непрерывно перемешивали при этой температуре в течение 30 мин. Затем к смеси при 0 °C добавляли 1-(бромметил)нафталин или 2-(бромметил)нафталин (1.5 экв.). Реакционную смесь перемешивали в течение 1 ч при этой температуре, далее полученному раствору давали нагреться до комнатной температуры и перемешивали до завершения реакции (около 14 ч). Натриевая соль индолкарбоновой кислоты обильно выпадает в осадок во время экстракции, препятствуя тем самым эффективному разделению фаз. Проблема была решена добавлением концентрированного раствора хлорида натрия. Тем не менее мутность сохранялась до тех пор, пока дихлорметан присутствовал в водной фазе. Водную фазу прокипятили для удаления остатка дихлорметана: был получен прозрачный раствор. После охлаждения до комнатной температуры водную фазу подкисляли соляной кислотой до рН 3-4 – обильно выпадал осадок. Осадок отфильтровывали, промыли четыре раза 5 мл дистиллированной воды и сушили при пониженном давлении в течение ночи для получения конечного соединения.
Общая методика 2: N-алкилирование пиррол- и индолкарбоксилатов
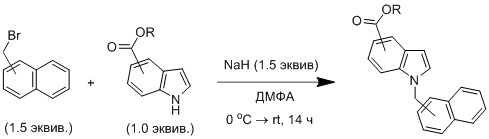
В сухую круглодонную колбу вносили соответствующее азотсодержащее гетероциклическое соединение (1.0 экв.) в инертной атмосфере. Далее растворяли в сухом ДМФА. Добавляли гидрид натрия (1.5 экв., 60%-ная суспензия в минеральном масле) при 0 °C в атмосфере азота. Полученный раствор непрерывно перемешивали при этой температуре в течение 30 мин. Затем при 0 °C добавляли 1-(бромметил)нафталин или 2-(бромметил)нафталин (1.5 экв.). Реакционную смесь перемешивали в течение 1 ч при этой температуре, далее полученному раствору давали нагреться до комнатной температуры и перемешивали до завершения реакции (около 14 ч). После этого реакционную смесь разбавляли 2,5%-ным раствором соляной кислоты (30 мл), а водный слой экстрагировали этилацетатом (3 × 15 мл). Объединенные органические слои промывали водой, насыщенным раствором гидрокарбоната натрия и соляным раствором и высушивали над сульфатом натрия, фильтровали и концентрировали в вакууме.
Общая методика 3: Омыление N-алкилированных пиррол- и индолкарбоксилатов
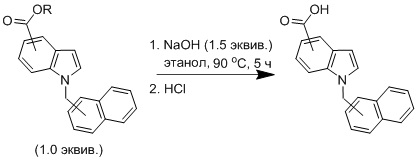
В круглодонную колбу вносили соответствующий сложный эфир (1 экв.), добавляли этанол, 1.5 М раствор гидроксида натрия в воде (1.5 экв.). Полученный раствор перемешивали и нагревали с обратным холодильником на масляной бане (90 °C) в течение 5 ч. Смесь охлаждали до комнатной температуры и концентрировали в вакууме. Полученный сухой остаток растворяли в 20 мл воды и экстрагировали с помощью дихлорметана (3 × 15 мл). Водный слой подкисляли соляной кислотой до рН 3-4, что приводило к обильному образованию белого осадка. Осадок отфильтровывали, четыре раза промывали 5 мл дистиллированной воды и сушили при пониженном давлении в течение ночи для получения конечного соединения.
Целевые вещества были получены путём N-алкилирования 1-(бромметил)нафталином / 2-(бромметил)нафталином либо эфиров соответствующих гетероциклических карбоновых кислот с последующим гидролизом и подкислением, либо самих гетероциклических кислот с последующим подкислением. Идентификация соединений была проведена методами ЯМР 1H, 13C-спектроскопии, HRMS, чистота подтверждена методом ВЭЖХ-УФ/МС.
На основании вышеприведенного следует отметить, что авторами предложен более короткий способ синтеза малых молекул - ингибиторов bCSE, выгодно отличающийся от описанных в литературе способов синтеза ингибиторов bCSE.
Пример 1: Синтез 1-(нафталин-1-илметил)-1Н-индол-3-карбоновой кислоты (Ia)
Получена из 1Н-индол-3-карбоновой кислоты (150.0 мг, 0.93 ммоль), 1-(бромметил)нафталина (308.7 мг, 1.40 ммоль) и NaH (111.7 мг, 2.79 ммоль) в ДМФА (5 мл) по общей методике 1. Осадок отфильтровывали, промывали дистиллированной водой и сушили при пониженном давлении в течение ночи, получая Ia в виде белого порошка (142.1 мг, 51%): Тпл 238 °C.
1H ЯМР (300 МГц, ДМСО-d6) δ 12.04 (с, 1H), 8.18–8.12 (м, 1H), 8.08 (дд, J = 6.3, 2.9 Гц, 1H), 8.04–7.96 (м, 2H), 7.89 (д, J = 8.3 Гц, 1H), 7.61–7.52 (м, 3H), 7.45–7.38 (м, 1H), 7.25–7.18 (м, 2H), 6.97 (д, J = 7.1 Гц, 1H), 6.01 (с, 2H).
13С ЯМР (75 МГц, ДМСО-d6) δ 165.49, 136.72, 135.30, 133.31, 132.47, 130.42, 128.67, 128.24, 126.62, 126.51, 126.12, 125.50, 124.95, 123.07, 122.48, 121.48, 120.96, 111.04, 107.13, 47.39.
Расчет HRMS (ESI+) для C20H16NO2 [M+H]+: 302.1181; найдено: 302.1174. Расчет HRMS (ESI-) для C20H14NO2 [M-H]-: 300.1025; найдено: 300.0995.
Пример 2: Синтез 1-(нафталин-2-илметил)-1Н-индол-3-карбоновой кислоты (Ib)
Получена из 1Н-индол-3-карбоновой кислоты (72.90 мг, 0.45 ммоль), 2-(бромметил)нафталина (150.00 мг, 0.68 ммоль) и NaH (42.20 мг, 1.13 ммоль) в ДМФА (5 мл) по общей методике 1. Осадок отфильтровывали, промывали дистиллированной водой и сушили при пониженном давлении в течение ночи, получая Ib в виде белого порошка (78.4 мг, 58%): Тпл 216 °C.
1H ЯМР (300 МГц, ДМСО-d6) δ 12.08 (с, 1H), 8.29 (с, 1H), 8.07–8.01 (м, 1H), 7.86 (дд, J = 10.2, 6.5 Гц, 4H), 7.60–7.54 (м, 1H), 7.52-7.46 (м, 2H), 7.41 (дд, J = 8.5, 1.7 Гц, 1H), 7.21–7.14 (м, 2H), 5.67 (с, 2H).
13С ЯМР (75 МГц, ДМСО-d6) δ 165.56, 136.34, 135.58, 134.69, 132.75, 132.32, 128.38, 127.67, 127.55, 126.68, 126.42, 126.13, 125.96, 125.34, 122.34, 121.37, 120.90, 111.10, 106.94, 49.75.
Расчет HRMS (ESI+) для C20H16NO2 [M+H]+: 302.1181; найдено: 302.1179. Расчет HRMS (ESI-) для C20H14NO2 [M-H]-: 300.1025; найдено: 300.0995.
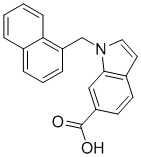
Пример 3: Синтез 1-(нафталин-1-илметил)-1Н-индол-6-карбоновой кислоты (Ie)
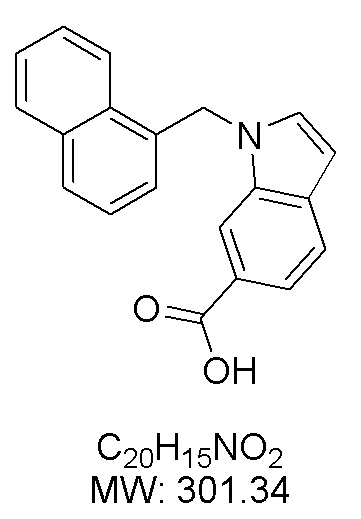
Получена из 1Н-индол-6-карбоновой кислоты (150.00 мг, 0.93 ммоль), 1-(бромметил)нафталина (308.70 мг, 1.40 ммоль) и NaH (111.70 мг, 2.79 ммоль) в ДМФА (5 мл) по общей методике 1. Осадок отфильтровывали, промывали дистиллированной водой и сушили при пониженном давлении в течение ночи, получая Ie в виде коричневого порошка (274.0 мг, 98%): Тпл 228 °C.
1H ЯМР (400 МГц, ДМСО-d6) δ 12.57 (с, 1H), 8.18 (д, J = 7.8 Гц, 1H), 8.07 (с, 1H), 8.01–7.96 (м, 1H), 7.86 (д, J = 8.2 Гц, 1H), 7.67 (с, 2H), 7.64 (д, J = 3.1 Гц, 1H), 7.62–7.56 (м, 2H), 7.37 (т, J = 7.7 Гц, 1H), 6.72 (д, J = 7.0 Гц, 1H), 6.63 (д, J = 2.9 Гц, 1H), 6.05 (с, 2H).
13С ЯМР (101 МГц, ДМСО-d6) δ 168.19, 135.47, 133.44, 133.26, 132.79, 131.70, 130.32, 128.61, 127.90, 126.49, 126.10, 125.48, 123.88, 123.64, 123.15, 120.26, 120.20, 112.19, 101.61, 47.19.
Расчет HRMS (ESI+) для C20H16NO2 [M+H]+: 302.1181; найдено: 302.1176. Расчет HRMS (ESI-) для C20H14NO2 [M-H]-: 300.1025; найдено: 300.0990.
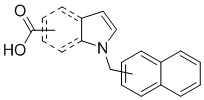
Пример 4: Синтез 1-(нафталин-1-илметил)-1Н-индол-5-карбоновой кислоты (Ig)
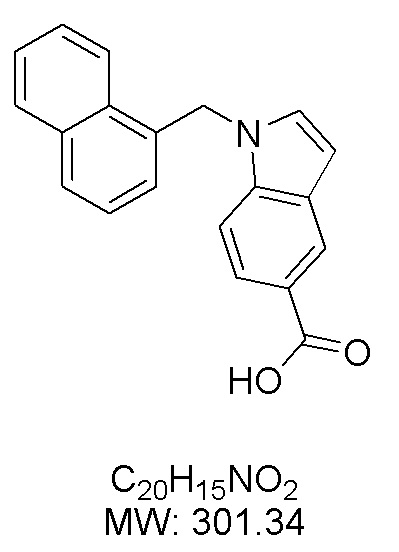
Получена из 1Н-индол-5-карбоновой кислоты (150.00 мг, 0.93 ммоль), 1-(бромметил)нафталина (308.70 мг, 1.40 ммоль) и NaH (111.70 мг, 2.79 ммоль) в ДМФА (5 мл) по общей методике 1. Осадок отфильтровывали, промывали дистиллированной водой и сушили при пониженном давлении в течение ночи, получая Ig в виде коричневого порошка (256.4 мг, 91%): Тпл 225 °C.
1H ЯМР (400 МГц, ДМСО-d6) δ 12.47 (с, 1H), 8.29 (д, J = 1,2 Гц, 1H), 8.18–8.15 (м, 1H), 7.97 (дд, J = 7.0, 2.4 Гц, 1H), 7.86 (д, J = 8.3 Гц, 1H), 7.73 (дд, J = 8.7, 1.6 Гц, 1H), 7.58 (ддд, J = 7.1, 5.0, 1.6 Гц, 2H), 7.55–7.51 (м, 2H), 7.41–7.35 (м, 1H), 6.81 (д, J = 6.7 Гц, 1H), 6.68 (д, J = 2.7 Гц, 1H), 5.98 (с, 2H).
13С ЯМР (101 МГц, ДМСО-d6) δ 168.23, 138.47, 133.29, 133.25, 130.82, 130.39, 128.61, 127.97, 127.76, 126.50, 126.07, 125.47, 124.30, 123.25, 123.14, 122.52, 121.87, 109.92, 102.78, 47.23.
Расчет HRMS (ESI+) для C20H16NO2 [M+H]+: 302.1181; найдено: 302.1174. Расчет HRMS (ESI-). для C20H14NO2 [M-H]-: 300.1025; найдено: 300.0993.
Пример 5: Синтез 1-(нафталин-2-илметил)-1Н-индол-5-карбоновой кислоты (Ih)
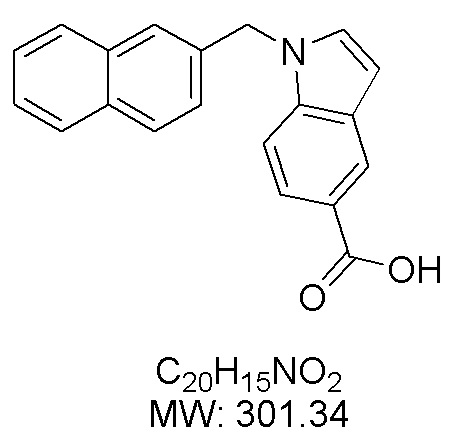
Получена из 1Н-индол-5-карбоновой кислоты (72.90 мг, 0.45 ммоль), 2-(бромметил)нафталина (150.00 мг, 0.68 ммоль) и NaH (42.20 мг, 1.13 ммоль) в ДМФА (5 мл) по общей методике 1. Осадок отфильтровывали, промывали дистиллированной водой и сушили при пониженном давлении в течение ночи, получая Ih в виде коричневого порошка (110.70 мг, 81%): Тпл 212 °C.
1H ЯМР (300 МГц, ДМСО-d6) δ 12.46 (с, 1Н), 8.26 (д, J = 1.1 Гц, 1Н), 7.88–7.80 (м, 3Н), 7.75–7.66 (м, 3H), 7.58 (д, J = 8.7 Гц, 1H), 7.51 – 7.45 (м, 2H), 7.35 (дд, J = 8.5, 1.8 Гц, 1H), 6.67 (дд, J = 3.2, 0.6 Гц, 1H), 5.63 (с, 2H).
13С ЯМР (75 МГц, ДМСО-d6) δ 168.20, 138.13, 135.42, 132.77, 132.27, 130.82, 128.29, 127.88, 127.62, 127.54, 126.38, 126.01, 125.56, 125.25, 123.17, 122.40, 121.80, 109.90, 102.66, 49.46.
Расчет HRMS (ESI+) для C20H16NO2 [M+H]+: 302.1181; найдено: 302.1173. Расчет HRMS (ESI-) для C20H14NO2 [M-H]-: 300.1025; найдено: 300.0984.
Пример 6: Синтез этил 1-(нафталин-1-илметил)-1Н-индол-2-карбоксилата
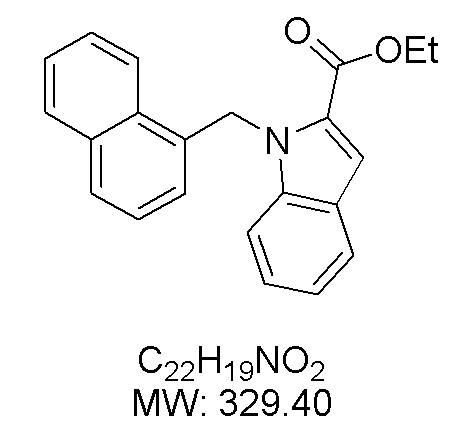
Получен из этил-1Н-индол-2-карбоксилата (150.00 мг, 0.79 ммоль), 1-(бромметил)нафталина (262.90 мг, 1.19 ммоль) и NaH (47.60 мг, 1.19 ммоль) в ДМФА (5 мл) по общей методике 2. Полученную смесь очищали методом колоночной хроматографии на силикагеле (с градиентом гексан/этилацетат 20:1→ 15:1), получая продукт в виде желтого масла (104.5 мг, 40%).
1H ЯМР (300 МГц, ДМСО-d6) δ 8.28 (д, J = 8.4 Гц, 1Н), 8.01–7.95 (м, 1Н), 7.83–7.75 (м, 2Н), 7.64 (ддд, J = 11.9, 8.0, 1.2 Гц, 2Н), 7.49 (д, J = 0.8 Гц, 1Н), 7.46–7.41 (м, 1Н), 7.30–7.15 (м, 3Н), 6.36 (с, 2Н), 6.11–6.04 (м, 1Н), 4.17 (к, J = 7.2 Гц, 2Н), 1.16 (т, J = 7.1 Гц, 3Н).
13С ЯМР (75 МГц, ДМСО-d6) δ 160.99, 139.39, 134.38, 133.09, 129.90, 128.53, 127.56, 127.02, 126.33, 126.02, 125.53, 125.43, 125.37, 122.90, 122.58, 120.96, 120.92, 111.10, 110.74, 60.31, 45.39, 13.92.
Расчет HRMS (ESI+). для C22H20NO2 [M+H]+: 330.1494; найдено: 330.1481.
Пример 7: Синтез этил 1-(нафталин-2-илметил)-1Н-индол-2-карбоксилата
Получен из этил-1Н-индол-2-карбоксилата (300.00 мг, 1.58 ммоль), 2-(бромметил)нафталина (526.00 мг, 2.38 ммоль) и NaH (95.20 мг, 2.38 ммоль) в ДМФА (10 мл по общей методике 2. Полученную смесь очищали методом колоночной хроматографии на силикагеле (с градиентом гексан/этилацетат 20:1→15:1), получая продукт в виде желтого масла (501.10 мг, 96%).
1H ЯМР (300 МГц, ДМСО-d6) δ 7.83 (т, J = 7.2 Гц, 2Н), 7.75 (д, J = 7.8 Гц, 2Н), 7.61 (д, J = 8.5 Гц, 1Н), 7.50 (с, 1Н), 7.47 – 7.41 (м, 3Н), 7.30 (т, J = 7.7 Гц, 1Н), 7.22 (д, J = 8.5 Гц, 1Н), 7.15 (т, J = 7.5 Гц, 1Н), 6.02 (с, 2Н), 4.28 (к, J = 7.1 Гц, 2Н), 1.28 (т, J = 7.1 Гц, 3Н).
13С ЯМР (75 МГц, ДМСО-d6) δ 161.28, 139.12, 136.14, 132.76, 132.11, 128.16, 127.55, 127.50, 127.32, 126.29, 125.82, 125.59, 125.29, 124.64, 124.52, 122.54, 120.86, 111.32, 110.74, 60.47, 47.36, 14.09.
Расчет HRMS (ESI+). для C22H20NO2 [M+H]+: 330.1494; найдено: 330.1483.
Пример 8: Синтез метил 1-(нафталин-2-илметил)-1Н-индол-6-карбоксилата
Получен из метил-1Н-индол-6-карбоксилата (200.00 мг, 1.14 ммоль), 2-(бромметил)нафталина (378.80 мг, 1.71 ммоль) и NaH (68.50 мг, 1.71 ммоль) в ДМФА (5 мл) по общей методике 2. Полученную смесь очищали методом колоночной хроматографии на силикагеле (гексан/этилацетат в соотношении 4:1), получая продукт в виде желтого порошка (129.9 мг, 65%): Тпл 123 °C.
1H ЯМР (300 МГц, ДМСО-d6) δ 8.13 (к, J = 1.0 Гц, 1Н), 7.89–7.85 (м, 2Н), 7.81 (дд, J = 5.5, 3.2 Гц, 2Н), 7.66 (дд, J = 3.3, 1.0 Гц, 3Н), 7.50–7.46 (м, 2Н), 7.32 (дд, J = 8.5, 1.8 Гц, 1Н), 6.64 (дд, J = 3.1, 0.8 Гц, 1Н), 5.71 (с, 2Н), 3.80 (с, 3Н).
13С ЯМР (75 МГц, ДМСО-d6) δ 167.04, 135.55, 135.09, 133.13, 132.77, 132.25, 132.06, 128.32, 127.57, 127.54, 126.40, 125.99, 125.15, 124.98, 122.36, 120.41, 119.83, 112.01, 101.60, 51.74, 49.33.
Расчет HRMS (ESI+). для C21H18NO2 [M+H]+: 316.1338; найдено: 316.1328.
Пример 9: Синтез этил 1-(нафталин-1-илметил)-1Н-пиррол-2-карбоксилата
Получен из этил-1Н-пиррол-2-карбоксилата (100.00 мг, 0.72 ммоль), 1-(бромметил)нафталина (190.90 мг, 1.08 ммоль) и NaH (43.20 мг, 1.08 ммоль) в ДМФА (5 мл) по общей методике 2. Полученную смесь очищали методом колоночной хроматографии на силикагеле (гексан/этилацетат в соотношении 94:6) для выделения продукта в виде бесцветного масла (129.9 мг, 65%).
1H ЯМР (300 МГц, ДМСО-d6) δ 8.10 (д, J = 7.4 Гц, 1Н), 8.00–7.94 (м, 1Н), 7.83 (д, J = 8.2 Гц, 1Н), 7.59 (ддд, J = 6.8, 4.5, 1.6 Гц, 2Н), 7.41–7.34 (м, 1Н), 7.20 (т, J = 2.1 Гц, 1Н), 7.00 (дд, J = 3.9, 1.8 Гц, 1Н), 6.49 (д, J = 7.1 Гц, 1Н), 6.24 (дд, J = 3.9, 2.6 Гц, 1Н), 6.06 (s, 2Н), 4.07 (к, J = 7.1 Гц, 2Н), 1.11 (т, J = 7.1 Гц, 3Н).
13С ЯМР (75 МГц, ДМСО-d6) δ 160.20, 135.11, 133.01, 130.33, 129.91, 128.53, 127.39, 126.43, 126.01, 125.58, 122.78, 122.40, 121.79, 118.03, 108.44, 59.35, 49.34, 14.09.
Расчет HRMS (ESI+) для C18H18NO2 [M+H]+: 280.1338; найдено: 280.1331.
Пример 10: Синтез этил 1-(нафталин-2-илметил)-1Н-пиррол-2-карбоксилата
Получен из этил-1Н-пиррол-2-карбоксилата (150.00 мг, 1.09 ммоль), 2-(бромметил)нафталина (357.50 мг, 1.62 ммоль) и NaH (64.70 мг, 1.62 ммоль) в ДМФА (5 мл) по общей методике 2. Полученную смесь очищали методом колоночной хроматографии на силикагеле (гексан/этилацетат в соотношении 94:6) для получения продукта в виде бесцветного масла (192.3 мг, 64%).
1H ЯМР (300 МГц, хлороформ-d) δ 7.82–7.75 (м, 3Н), 7.51 (с, 1Н), 7.48–7.43 (м, 2Н), 7.28 (дд, J = 8.5, 1.7 Гц, 1Н), 7.06 (дд, J = 3.9, 1.8 Гц, 1Н), 6.96–6.89 (м, 1Н), 6.22 (дд, J = 3.9, 2.6 Гц, 1Н), 5.73 (с, 2Н), 4.24 (к, J = 7.1 Гц, 2Н), 1.30 (т, J = 7.1 Гц, 3Н).
13С ЯМР (75 МГц, хлороформ-d) δ 161.31, 136.00, 133.56, 132.94, 129.09, 128.55, 128.04, 127.80, 126.30, 126.01, 125.75, 125.18, 122.66, 118.47, 108.65, 59.96, 52.35, 14.50.
Расчет HRMS (ESI+) для C18H18NO2 [M+H]+: 280.1338; найдено: 280.1331.
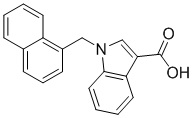
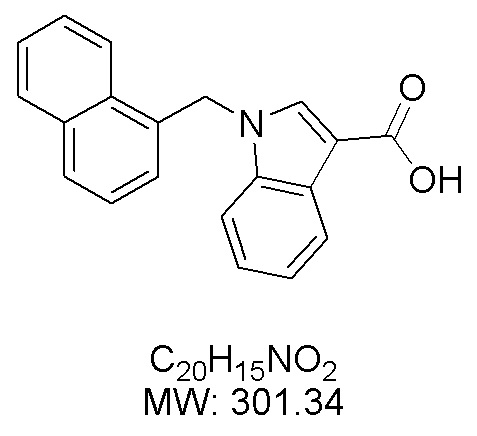
Пример 11: Синтез 1-(нафталин-1-илметил)-1Н-индол-2-карбоновой кислоты (Ic)
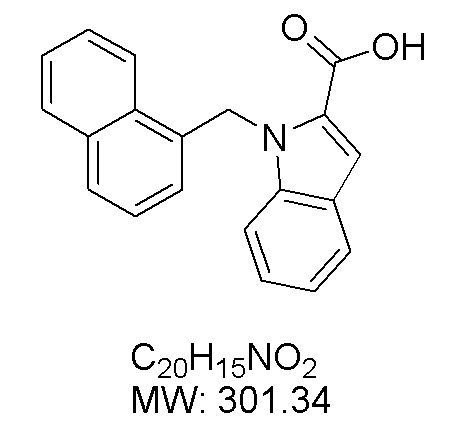
Получена из этил 1-(нафталин-1-илметил)-1Н-индол-2-карбоксилата (95.00 мг, 0.29 ммоль) и NaOH (1.5 М в воде, 0.38 мл, 0.57 ммоль) в этаноле (5 мл) по общей методике 3. Осадок отфильтровывали, промывали дистиллированной водой и сушили при пониженном давлении в течение ночи, получая Ic в виде белого порошка (256.4 мг, 91%): Тпл 228 °C.
1H ЯМР (400 МГц, ДМСО-d6) δ 12.89 (с, 1Н), 8.29 (д, J = 8.3 Гц, 1Н), 7.98 (д, J = 7.6 Гц, 1Н), 7.77 (дд, J = 8.0, 5.2 Гц, 2Н), 7.69–7.65 (м, 1Н), 7.63–7.59 (м, 1Н), 7.45–7.43 (м, 1Н), 7.38 (д, J = 8.4 Гц, 1Н), 7.26–7.21 (м, 2Н), 7.18–7.13 (м, 1Н), 6.39 (с, 2Н), 6.08 (д, J = 7.1 Гц, 1Н).
13С ЯМР (101 МГц, ДМСО-d6) δ 162.66, 139.27, 134.54, 133.10, 129.96, 128.55, 126.99, 126.35, 126.02, 125.62, 125.47, 125.03, 122.98, 122.47, 121.09, 120.75, 111.08, 110.55, 45.22.
Расчет HRMS (ESI+) для C20H16NO2 [M+H]+: 302.1181; найдено: 302.1176. Расчет HRMS (ESI-) для C20H14NO2 [M-H]-: 300.1025; найдено: 300.0997.
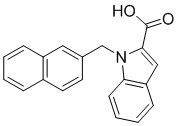
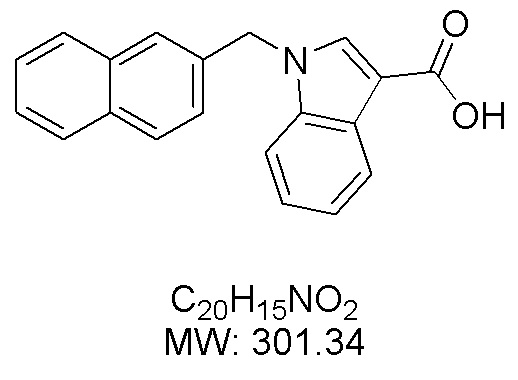
Пример 12: Синтез 1-(нафталин-2-илметил)-1Н-индол-2-карбоновой кислоты (Id)
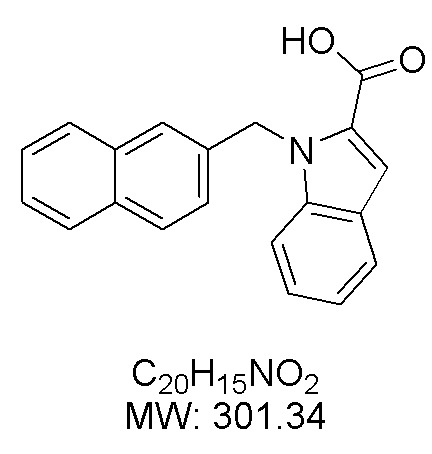
Получена из этил-1-(нафталин-2-илметил)-1Н-индол-2-карбоксилата (35.7 мг, 0.11 ммоль) и NaOH (1.5 М в воде, 0.10 мл, 0.14 ммоль) в этаноле (2 мл) по общей методике 3. Осадок отфильтровывали, промывали дистиллированной водой и сушили при пониженном давлении в течение ночи, получая Id в виде белого порошка (20.6 мг, 63%): Тпл 219 °C.
1H ЯМР (300 МГц, ДМСО-d6) δ 12.98 (с, 1Н), 7.85–7.80 (м, 2Н), 7.77–7.71 (м, 2Н), 7.59–7.55 (м, 1Н), 7.51 (с, 1Н), 7.45 (дд, J = 6.2, 3.3 Гц, 2Н), 7.37 (д, J = 0.8 Гц, 1Н), 7.29–7.20 (м, 2Н), 7.16–7.10 (м, 1Н), 6.05 (с, 2Н).
13С ЯМР (75 МГц, ДМСО-d6) δ 162.91, 138.99, 136.29, 132.75, 132.08, 128.21, 128.10, 127.50, 127.48, 126.25, 125.77, 125.65, 124.91, 124.69, 124.54, 122.39, 120.64, 111.24, 110.49, 47.15.
Расчет HRMS (ESI+) для C20H16NO2 [M+H]+: 302.1181; найдено: 302.1175. Расчет HRMS (ESI-) для C20H14NO2 [M-H]-: 300.1025; найдено: 300.1022.
Пример 13: Синтез 1-(нафталин-2-илметил)-1Н-индол-6-карбоновой кислоты (If)
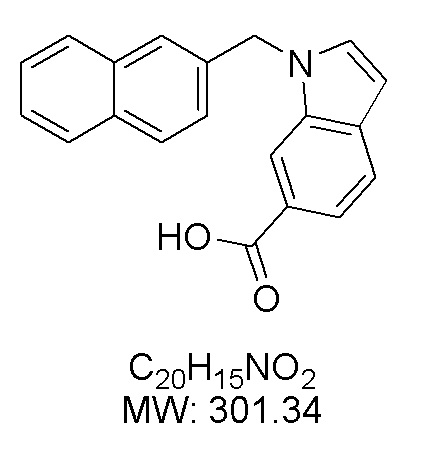
Получена из метил-1-(нафталин-1-илметил)-1Н-индол-6-карбоксилата (300.00 мг, 0.95 ммоль) и NaOH (1.5 М в воде, 1.27 мл, 1.90 ммоль) в этаноле (7 мл) по общей методике 3. Осадок отфильтровывали, промывали дистиллированной водой и сушили при пониженном давлении в течение ночи, получая If в виде белого порошка (234.5 мг, 82%): Тпл 208 °C.
1H ЯМР (300 МГц, ДМСО-d6) δ 12.52 (с, 1Н), 8.10 (к, J = 1.1 Гц, 1Н), 7.89–7.84 (м, 2Н), 7.83–7.77 (м, 2Н), 7.69 (с, 1Н), 7.65 (д, J = 0.9 Гц, 2Н), 7.50–7.46 (м, 2Н), 7.32 (дд, J = 8.5, 1.7 Гц, 1Н), 6.63 (дд, J = 3.1, 0.8 Гц, 1Н), 5.70 (с, 2Н).
13С ЯМР (75 МГц, ДМСО-d6) δ 168.14, 135.62, 135.13, 132.81, 132.26, 131.83, 128.31, 127.58, 127.56, 126.40, 125.99, 125.18, 125.01, 123.51, 120.17, 120.13, 112.13, 101.48, 49.37.
Расчет HRMS (ESI+) для C20H16NO2 [M+H]+: 302.1181; найдено: 302.1171. Расчет HRMS (ESI-) для C20H14NO2 [M-H]-: 300.1025; найдено: 300.1013.
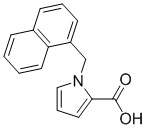
Пример 14: Синтез 1-(нафталин-1-илметил)-1Н-пиррол-2-карбоновой кислоты (Ij)
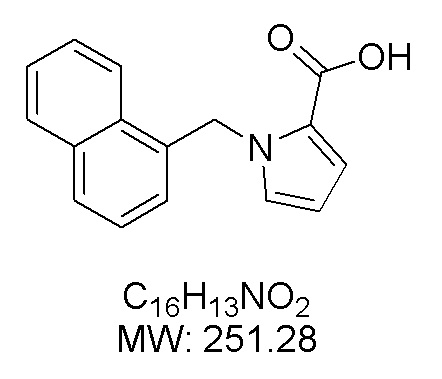
Получена из этил-1-(нафталин-1-илметил)-1Н-пиррол-2-карбоксилата (108.90 мг, 0.39 ммоль) и NaOH (1.5 М в воде, 0.34 мл, 0.51 ммоль) в этаноле (5 мл) по общей методике 3. Осадок отфильтровывали, промывали дистиллированной водой и сушили при пониженном давлении в течение ночи, получая Ij в виде белого порошка (61.2 мг, 61%): Тпл 160 °C.
1H ЯМР (300 МГц, ДМСО-d6) δ 12.14 (с, 1Н), 8.10 (д, J = 7.5 Гц, 1Н), 7.99–7.94 (м, 1Н), 7.83 (д, J = 8.2 Гц, 1Н), 7.62–7.54 (м, 2Н), 7.39 (т, J = 7.7 Гц, 1Н), 7.12 (с, 1Н), 6.96 (с, 1Н), 6.54 (д, J = 7.0 Гц, 1Н), 6.20 (с, 1Н), 6.08 (с, 2Н).
13С ЯМР (75 МГц, ДМСО-d6) δ 161.84, 135.28, 133.02, 130.01, 129.74, 128.53, 127.38, 126.41, 125.98, 125.61, 122.88, 122.69, 117.88, 108.19, 49.09.
Расчет HRMS (ESI+) для C16H14NO2 [M+H]+: 252.1025; найдено: 252.1018. Расчет HRMS (ESI-) для C16H12NO2 [M-H]-: 250.0868; найдено: 250.0830.
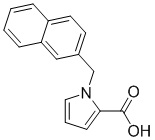
Пример 15: Синтез 1-(нафталин-2-илметил)-1Н-пиррол-2-карбоновой кислоты (Ik)
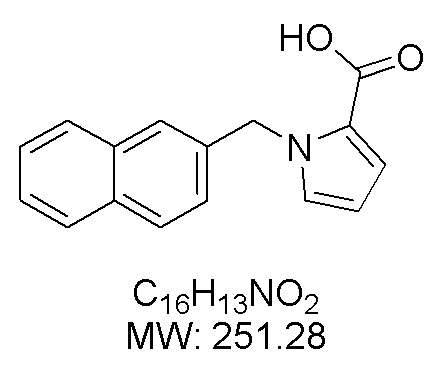
Получена из этил-1-(нафталин-2-илметил)-1Н-пиррол-2-карбоксилата (170.00 мг, 0.61 ммоль) и NaOH (1.5 М в воде, 0.65 мл, 0.97 ммоль) в этаноле (5 мл) по общей методике 3. Осадок отфильтровывали, промывали дистиллированной водой и сушили при пониженном давлении в течение ночи, получая Ik в виде белого порошка (128.00 мг, 82%): Тпл 160 °C.
1H ЯМР (300 МГц, ДМСО-d6) δ 12.15 (с, 1Н), 7.86 (дд, J = 8.9, 4.1 Гц, 2Н), 7.82–7.78 (м, 1Н), 7.52 (с, 1Н), 7.50–7.46 (м, 2Н), 7.31-7.26 (м, 2Н), 6.90 (дд, J = 3.9, 1.8 Гц, 1Н), 6.18 (дд, J = 3.9, 2.6 Гц, 1Н), 5.73 (с, 2Н).
13С ЯМР (75 МГц, ДМСО-d6) δ 161.83, 136.79, 132.79, 132.15, 129.61, 127.98, 127.56, 127.50, 126.26, 125.83, 125.04, 124.92, 122.10, 117.96, 108.11, 51.02.
Расчет HRMS (ESI+) для C16H14NO2 [M+H]+: 252.1025; найдено: 252.1019. Расчет HRMS (ESI-) для C16H12NO2 [M-H]-: 250.0868; найдено: 250.0836.
Пример 16: Ферментативная активность, определение IC50
Значения IC50 ингибирующей активности веществ по отношению к ферментативной активности бактериального и человеческого фермента CSE (цистатионин-γ-лиазы) определяли в ферментативном тесте в неклеточной системе. В качестве контрольного ингибитора использовали DL-пропаргилглицин (PAG), являющийся необратимым ингибитором цистатионин-γ-лиазы.
Фермент в концентрации 0,01 мкМ и исследуемые малые молекулы согласно изобретению в диапазоне концентраций от 0,05 до 500 мкМ прединкубировали в 96-луночном, черном планшете (Greiner, Австрия) с низкой сорбцией в термошейкере PST-60HL-4 (Biosan, Латвия) в течении 40 минут при температуре 30 °С на 300 оборотах в минуту, в реакционном 50 мM Na-фосфатном буфере с рН = 8, содержащем 10 мкМ пиридоксаль фосфата (Sigma-aldrich, США), 1,2 мкМ флуоресцентного зонда Washington State Probe-5 (Cayman Chemical, США), 0,1% Твин 20 (Sigma-aldrich, США). По окончании прединкубации в систему вносили 100 мкМ L-цистеина (TCI, Индия) в 50 мM Na-фосфатном буфере с рН= 8. Планшет закрывали пленкой (SOVTECH, Россия), чтобы исключить испарения смеси и инкубировали планшет при комнатной температуре в темном, защищенном от света месте в течение 18 часов. Детектировали уровень получаемого флуоресцентного сигнала на спектрофотометре CLARIOstar Plus (BMG Labtech, Германия). Настройка оптики для детектирования сигнала флуоресценции приведена в таблице 2.
Таблица 2. Оптические настройки прибора для детектирования сигнала флуоресценции.
Обработка данных, полученных при детектировании интенсивности флуоресцентного сигнала, производилась с помощью программы MARS Data Analysis. Для этого осуществлялось построение 4-х параметрической кривой (4-parameter fit) зависимости интенсивность флуоресцентного сигнала (linear, Y value) WSP5 от логарифма концентрации ингибитора (logarithmic, X values). С помощью функции «fit results» проводился программный расчет значения концентрации полумаксимального ингибирования (IC50) для каждого соединения.
В результате проведенного исследования на бактериальном и человеческом ферментах CSE были получены значения IC50 для ряда малых молекул (таблица 3, фигура 1).
Таблица 3. Результаты ингибирующей активности соединений в ферментативных тестах на bCSE и hCSE
В качестве веществ-сравнения в тестах использовались следующие вещества: NL2 как молекула с доказанным ингибирующим эффектов относительно bCSE (положительный контроль), PAG как молекула, лучше ингибирующая hCSE (отрицательный контроль). Эффективность ингибирования наших веществ в тестах устанавливали в сравнении с данными, получаемыми для этих молекул в каждом конкретном эксперименте. Соединения Ig, Ih продемонстрировали сопоставимую с NL2 активность. Соединения, содержащие моноциклическую ароматическую систему пиррола (Ij, Ik), проявили пониженную активность, соединения, содержащие индол с карбоксильной группой либо во втором, либо в пятом положении – повышенную. Среди них соединение Ic оказалось самым эффективным ингибитором (фигура 2).
Пример 17: Микробиологические исследования, определение MIC
Бактериальную ночную культуру (НК) специфического штамма выращивали на жидкой питательной среде LB (BD Difco) в течение 18 часов при 37 °С при перемешивании 180 об/мин на орбитальном шейкере Innova 44R (Eppendorf). По истечении указанного времени, 50 мкл НК помещали в 5 мл LB и выращивали 3 часа при 37 °C с перемешиванием. Бактериальный рост в жидкой среде оценивался и стандартизировался денситометрически с помощью Densi-La-Meter II (Erba Lachema, Чехия). Бактериальную суспензию развели до плотности 106 кл/мл стерильной средой LB и использовали в качестве «посевной культуры».
Для определения минимальной ингибирующей концентрации (MIC) использовались 96-луночные планшеты. Каждая лунка была заполнена 100 мкл посевной культуры.
Затем по 10 мкл раствора каждого исследуемого соединения (потенциатора) было добавлено в экспериментальные лунки. Затем в ряды планшета было добавлено по 90 мкл серийных двукратных разведений соответствующего антибиотика в стерильной LB. Итоговая концентрация потенциаторов была 50 мкг/мл. Итоговый объем в лунках составлял 200 мкл. В качестве контролей мы использовали лунки с 200 мкл бактериальной культуры, без добавления антибиотика или потенциатора, и лунки со стерильной LB.
В качестве референсного потенциатора мы использовали NL2, с показанной согласно литературным данным активностью. Планшеты инкубировали в термостате 37 °C в течение 20 часов и затем бактериальный рост регистрировался по OD 600 нм на микропланшетном сканере ClarioStar (BMG LABTECH).
Полученные данные микробиологического тестирования на грамположительных бактериях приведены в таблице 4.
Таблица 4. Результаты микробиологических тестов потенцирующей активности в комбинации с антибиотиками против S. aureus ATCC 25923 и S. aureus INA00761 (MRSA)
*резистентность во всех концентрациях ниже максимальной тестируемой.
В случае S. aureus ATCC 25923, все потенциаторы были взяты в концентрации 50 мкг/мл, в которой они сами по себе (без добавления антибиотика) не обладали ингибирующей активностью по отношению к бактериальной культуре. Неожиданно S. aureus INA00761 проявил большую чувствительность к этой концентрации потенциаторов, и более низкие концентрации веществ были взяты для оценки их потенцирующего действия: 50 мкг/мл для Ij, Ia и NL2; 25 мкг/мл для Ik и If; 12.5 мкг/мл для Ig, Ic, Ib, Ih, и Id; 6 мкг/мл для Ie (фигура 3).
Как видно из полученных данных, все протестированные вещества проявили активность по отношению к штамму S. aureus ATCC 25923, но только вещество Ic потенцировало действие ампициллина против метициллин-резистентного золотистого стафилококка S. aureus INA00761, что является важным результатом, т. к. ампициллин является антибиотиком первой линии терапии, не обладающим серьёзными побочными эффектами. Данное соединение было выбрано в качестве лидерного и дальнейшие исследования на клинических изолятах грамотрицательных бактерий проводили именно для этого соединения. Результаты данного исследования приведены в таблице 5.
Таблица 5. Результаты микробиологических тестов потенцирующей активности в комбинации с антибиотиками против клинических изолятов A. baumannii и P. aeruginosa.
В случае A. baumannii GIMC5509:ABT-52Ts19 и P. aeruginosa GIMC5016:PA1840/36/2015, потенциаторы были взяты в концентрации 50 мкг/мл, в которой они сами по себе (без добавления антибиотика) не обладали ингибирующей активностью по отношению к бактериальной культуре. Показано, что лидерное соединение значительно потенцирует действие цефепима и менее сильно - смеси пиперациллина и тазобактама против P. aeruginosa GIMC5016:PA1840/36/2015. В случае A. baumannii GIMC5509:ABT-52Ts19 потенцирование наблюдалось при совместном использовании лидерного соединения с тремя антибиотиками: цефепимом, смесью пиперациллина и тазобактама (тациллин), меропенемом.
Пример 18: Исследование цитотоксичности, определение СС50
Общую цитотоксичность определяли в тесте на клеточной линии почки эмбриона человека HEK293 (1-1-609, Институт Цитологии Российской Академии Наук, Россия).
Клетки культивировали в среде ДМЕМ (ПанЭко, Россия) с добавлением 10% фетальной бычьей сыворотки (HyClone, Cytiva, Австрия), 2 мМ L-глутамина и 1% пенициллин/стрептомицин (100X, Capricorn Scientific, Германия) не менее 1-2 пассажей после разморозки. Клетки трипсинизировали 0.05% раствором Тринсин-ЭДТА (ПанЭко, Россия) и повторно высевали в 96-луночные планшеты (Corning, США) в концентрации 1*10^4 клеток/100 мкл на лунку. Инкубировали во влажной атмосфере в СО2 инкубаторе при 5% СО2 и 37 °С в течение 24 часов.
Исследуемые соединения титровали последовательно в ДМСО и в клеточной среде, переносили полученные после титрования растворы в 96-луночные планшеты с клетками и оставляли на инкубацию в течение 72 часов в СО2 инкубаторе при 5% CО2 и 37 °С.
После инкубации проводили детекцию при помощи витального красителя АlamarBlue (Invitrogen, США), в лунки клеточного планшета вносили по 10 мкл красителя, и оставляли планшет на 3 часа инкубации в СО2 инкубаторе при 5% CО2 и 37 °С. Флуоресцентный сигнал детектировали на спектрофотометре (BMG Labtech, Германия) при длине волны возбуждения 545 нм и испускания 600 нм. Для определения соответствующих значений 50% клеточной цитотоксичности (CC50) к полученным данным использовали четырехпараметрическую модель кривой при помощи программного обеспечения GraphPad Prism 8.
В качестве контрольного соединения с высокой токсичностью использовали Доксорубицин (Sigma-Aldrich, США).
Данные исследования цитотоксичности представлены в таблице 6.
Таблица 6. Исследование цитотоксичности соединений на клеточной линии почки эмбриона человека HEK293.
Данные показаны как средние ± СО (n = 3).
Анализ цитотоксичности показал, что СС50 полученных соединений относительно клеточной линии HEK293 от 2 до 6 раз выше, чем IC50 соответствующих соединений (фигура 4). Лидерное соединение обладает удовлетворительным терапевтическим индексом.
Пример 19: Исследование ADME-свойств лидерного соединения Ic
Кинетическая растворимость
Исходные растворы анализируемых соединений готовили в ДМСО с концентрацией 10 мМ. В полипропиленовую пробирку объемом 1.5 мл вносят 990 мкл 0.1 М фосфатного буферного раствора (pH = 7.4) и добавляют 10 мкл исходного раствора анализируемого соединения. Эксперимент проводили в трех повторах. Полученные растворы помещали в термошейкер и перемешивали в течение 2 часов при 300 об/мин при 25 °С, далее фильтровали с использованием шприцевых фильтров (PVDF, 0.45 мкм). В виалу вместимостью 2 мл переносили 320 мкл фильтрата и добавляли 80 мкл ацетонитрила, перемешивали и проводили ВЭЖХ-УФ анализ на жидкостном хроматографе Vanquish Flex (Thermo Scientific, США) с диодно-матричным детектором DAD FG (Thermo Scientific, США). Разделение определяемых соединений проводили на колонке Agilent Poroshell 120 EC-C18 (100 мм × 2.0 мм, размер частиц 1.9 мкм) в следующих хроматографических условиях: с 0 по 3.5 мин – элюирование от 5% до 95% подвижной фазы Б, с 3.5 по 4.5 мин – элюирование в 95% подвижной фазы Б, с 4.5 по 8 минуту – уравновешивание хроматографической колонки в 95% подвижной фазы А. Подвижная фаза А – 0.1% муравьиная кислота в воде, подвижная фаза Б – 0.1% муравьиная кислота в ацетонитриле. Температура колонки – 40°С. Объем вводимой пробы – 5 мкл, скорость потока – 0.4 мл/мин. Концентрацию анализируемого соединения в фильтратах (растворимость) определяли по калибровочной кривой, построенной в диапазоне от 1 мкМ до 100 мкМ. В качестве контрольных соединений использовали диклофенак натрия (высокая растворимость) и никардипин (низкая растворимость).
На основе полученных данных оценивали растворимость лидерного соединения Ic. Лидерное соединение обладает высокой растворимостью (>100 мкМ). Результаты приведены в таблице 7.
Таблица 7. Результаты определения растворимости лидерного соединения Ic. Данные представлены в виде M ± σ, где M – среднеарифметическое значение результатов 3 параллельных определений, σ – стандартное отклонение.
Стабильность в микросомах печени
1 мкM анализируемого соединения инкубировали с микросомальной фракцией печени крыс (Gibco, США) при 37 °C в присутствии NADPH (кофактор ферментов фазы I метаболизма) или UDPGA (кофактор ферментов фазы II метаболизма). В качестве буферного раствора использовали 0.1 М фосфатный буферный раствор (pH = 7,4). Объем инкубационной смеси 600 мкл. Во временные точки 0, 5, 15, 30, 45 мин отбирали в полипропиленовую пробирку объемом 0.5 мл 50 мкл инкубационной смеси и останавливали реакцию путем добавления 100 мкл ацетонитрила. Центрифугировали пробирки при 10000 x g в течение 10 мин при температуре 4 °С. 100 мкл супернатанта переносили в виалу вместимостью 2 мл со стеклянной вставкой и проводили ВЭЖХ-МС/МС анализ на жидкостном хроматографе UltiMate 3000 UHPLC (Thermo, США) c масс-спектрометрическим детектором EVOQ Elite (Bruker, США) с ионизацией электрораспылением. По кинетике убыли тестируемого соединения в процессе инкубации рассчитывали период полувыведения (t1/2) и клиренс (CLint). C помощью программы MS Workstation software (version 8.2.1; Bruker, Германия) рассчитывали площадь пика анализируемого соединения. Далее строили график в координатах «натуральный логарифм площади пика» – «время инкубации».
Период полувыведения рассчитывали по формуле:
t1/2 = -0.693/slope, где
t1/2 – период полувыведения, мин
slope – угол наклона графика зависимости натурального логарифма площади пика от времени инкубации.
Клиренс рассчитывали по формуле:
CLint = (0.693/t1/2) × (incubation volume/microsomal protein), где
CLint – клиренс, мкл/мин/мг
t1/2 – период полувыведения, мин
incubation volume – объем инкубационной смеси, мкл
microsomal protein – содержание микросомального белка в инкубационной смеси, мг
Эксперимент проводили в двух повторах. В качестве контрольных соединений использовали верапамил (субстрат цитохрома P450) и 7-гидроксикумарин (субстрат УДФ-глюкуронилтрансферазы).
Результаты экспериментов показали, что лидерное соединение Ic обладает высокой стабильностью к действию ферментов I фазы метаболизма (изоферменты цитохрома Р450) и ферментов II фазы метаболизма (УДФ-глюкуронилтрансферазы). Результаты экспериментов приведены в таблице 8.
Таблица 8. Результаты определения стабильности в микросомах печени крыс лидерного соединения Ic. Данные представлены в виде M ± σ, где M – среднеарифметическое значение результатов 2 параллельных определений, σ – стандартное отклонение.
УДФ-ГТ
УДФ-ГТ
Проницаемость через монослой клеток Caco-2
На полупроницаемую мембрану (ПЭТ-мембрана, размер пор: 0,4 мкм, площадь поверхности: 0.33 см2, Corning, США) планшета, состоящего из двух камер, высеивали клетки колоректальной аденокарциномы Caco-2 и культивировали их в течение 21 дня до образования монослоя. Целостность монослоя оценивали с использованием красителя Lucifer yellow. Для оценки транспорта в прямом направлении A-B (от апикальной к базолатеральной мембране) в апикальную камеру (камера-донор) добавляли 300 мкл раствора анализируемого соединения (10 мкМ в растворе Хенкса). В базолатеральную камеру (камера-реципиент) добавляли 800 мкл раствора Хенкса и инкубировали планшет при 37 °C в атмосфере CO2 (5%) в течение 2 часов. Для оценки транспорта в направлении B-A (от базолатеральной к апикальной мембране) в апикальную камеру (камера-реципиент) добавляли 300 мкл раствора Хенкса. В базолатеральную камеру (камера-донор) добавляли 800 мкл раствора анализируемого соединения (10 мкМ в растворе Хенкса) и инкубировали планшет при 37 °C в атмосфере CO2 (5%) в течение 2 часов. В полипропиленовые пробирки объемом 0.5 мл отбирали аликвоты (100 мкл) из базолатеральной и апикальной камер, добавляли 300 мкл ацетонитрила и перемешивали. Центрифугировали пробирки при 10000 x g в течение 10 мин при температуре 4 °С. 100 мкл супернатанта переносили в виалу вместимостью 2 мл со стеклянной вставкой и проводили ВЭЖХ-МС/МС анализ на жидкостном хроматографе UltiMate 3000 UHPLC (Thermo, США) c масс-спектрометрическим детектором EVOQ Elite (Bruker, США) с ионизацией электрораспылением.
Коэффициент кажущейся проницаемости рассчитывали по формуле:
Papp = (V × ∆C)/(∆t × A × C0), где
Рарр – коэффициент кажущейся проницаемости, см/сек
V – объем раствора Хенкса в камере-реципиенте (в тесте А-В – 0.8 см3; в тесте В-А – 0.3 см3), см3
∆t – время инкубации (7200 сек), сек
A – площадь поверхности мембраны, на которой культивируются клетки (0.33 см2), см2
C0 – начальная концентрация анализируемого соединения в камере-доноре (10 мкМ), мкМ
∆C – изменение концентрации анализируемого соединения после инкубации (в тесте А-В – концентрация в образце из базолатеральной камеры; в тесте В-А – концентрация в образце из апикальной камеры), мкМ.
Эксперимент проводили в двух повторах. В качестве контрольных соединений использовали атенолол (соединение с низкой проницаемостью) и пропранолол (соединение с высокой проницаемостью).
На основе полученных данных оценивали проницаемость лидерного соединения Ic. Лидерное соединение обладает высокой проницаемостью. Отношение коэффициентов кажущейся проницаемости Papp(B-A) к Papp(A-B) меньше 2, что свидетельствует о том, что лидерное соединение не является субстратом транспортных белков гликопротеина-P и белка резистентности рака молочной железы (BCRP). Результаты приведены в таблице 9.
Таблица 9. Результаты определения проницаемости на клетках линии Caco-2 лидерного соединения Ic. Данные представлены в виде M ± σ, где M – среднеарифметическое значение результатов 2 параллельных определений, σ – стандартное отклонение.
(10–6 см/сек)
Стабильность в плазме крови человека
В полипропиленовую пробирку объемом 1.5 мл вносили 98 мкл пулированной плазмы крови человека, добавляли 2 мкл раствора анализируемого соединения с концентрацией 500 мкМ. Помещали пробирки в термошейкер на 4 часа при 37 °C, 400 rpm. Вынимали пробирки из термошейкера, вносили 400 мкл ацетонитрила и перемешивали. Центрифугировали при 10000 x g в течение 10 мин при температуре 4 °С. 100 мкл супернатанта переносили в виалу вместимостью 2 мл со стеклянной вставкой и проводили ВЭЖХ-МС/МС анализ на жидкостном хроматографе UltiMate 3000 UHPLC (Thermo, США) c масс-спектрометрическим детектором EVOQ Elite (Bruker, США) с ионизацией электрораспылением.
Процент оставшегося после инкубации соединения рассчитывали по следующей формуле:
% remaining plasma = (Sпосле инкубации, плазма /S0, плазма) × 100
% remaining plasma – процент оставшегося соединения после инкубации с плазмой крови в течение 4 часов при 37 °C, %
где Sпосле инкубации, плазма – площадь хроматографического пика, измеренная в образце после инкубации с плазмой крови в течение 4 часов при 37˚C
S0, плазма – площадь хроматографического пика, измеренная в образце плазмы крови без проведения инкубации (нулевая точка).
Эксперимент проводили в трех повторах. В качестве контрольных соединений использовали верапамил (стабильное в плазме крови соединение) и пропантелин бромид (нестабильное в плазме крови соединение).
Лидерное соединение Ic стабильно в плазме крови человека. Результаты приведены в таблице 10.
Таблица 10. Результаты определения стабильности в плазме крови человека лидерного соединения Ic. Данные представлены в виде M ± σ, где M – среднеарифметическое значение результатов 3 параллельных определений, σ – стандартное отклонение.
Стабильность в искусственном кишечном соке и искусственном желудочном соке
Искусственный кишечный сок (SIF) готовили путем добавления панкреатина к искусственному кишечному соку без фермента (Ricca, США, кат. номер 7109.75-16). Концентрация панкреатина в искусственном кишечном соке 10 мг/мл. Искусственный желудочный сок (SGF) готовили путем добавления пепсина к искусственному желудочному соку без фермента (Ricca, США, кат. номер 7108-16). Концентрация пепсина в искусственном желудочном соке 1.6 мг/мл. В полипропиленовую пробирку объемом 1.5 мл вносили 98 мкл искусственного кишечного сока или искусственного желудочного сока, добавляли 2 мкл раствора анализируемого соединения с концентрацией 500 мкМ. Помещали пробирки в термошейкер на 2 часа при 37 °C, 400 rpm. Вынимали пробирки из термошейкера, вносили 400 мкл ацетонитрила и перемешивали. Центрифугировали при 10000 x g в течение 10 мин при температуре 4 °С. 100 мкл супернатанта переносили в виалу вместимостью 2 мл со стеклянной вставкой и проводили ВЭЖХ-МС/МС анализ на жидкостном хроматографе UltiMate 3000 UHPLC (Thermo, США) c масс-спектрометрическим детектором EVOQ Elite (Bruker, США) с ионизацией электрораспылением.
Процент оставшегося после инкубации соединения рассчитывали по следующей формуле:
% remaining SIF/SGF = (Sпосле инкубации, SIF/SGF / S0, SIF/SGF) × 100
% remaining SGF/SIF – процент оставшегося соединения после инкубации в искусственном кишечном соке или искусственном желудочном соке в течение 2 часов при 37 °C, %
где Sпосле инкубации, SIF/SGF – площадь хроматографического пика, измеренная в образце после инкубации в искусственном кишечном соке или искусственном желудочном соке в течение 2 часов при 37 °C
S0,SIF/SGF – площадь хроматографического пика, измеренная в образце искусственного кишечного сока или искусственного желудочного сока без проведения инкубации (нулевая точка).
Эксперимент проводили в трех повторах. В качестве контрольных соединений использовали верапамил (стабильное соединение) и омепразол (нестабильное в кислой среде соединение).
Лидерное соединение Ic стабильно в искусственном кишечном соке и искусственном желудочном соке. Результаты приведены в таблице 11.
Таблица 11. Результаты определения стабильности в искусственном кишечном соке (SIF) и искусственном желудочном соке (SGF) лидерного соединения Ic. Данные представлены в виде M ± σ, где M – среднеарифметическое значение результатов 3 параллельных определений, σ – стандартное отклонение.
Определение свободной и связанной фракции соединения в плазме крови методом равновесного диализа
В плашку для проведения равновесного диализа RED Device Reusable Base Plate (Thermo, США) помещали вставки RED Device Inserts (Thermo, США). В правую камеру вставки приливали 500 мкл фосфатного буферного солевого раствора. В левую камеру (красного цвета) вставки приливали 300 мкл пулированной плазмы крови человека с концентрацией исследуемого соединения 5 мкМ. Плашку инкубировали при 37 °C в атмосфере CO2 (5%) в течение 6 часов. Вынимали плашку из инкубатора, отбирали в новую полипропиленовую пробирку объемом 1.5 мл 100 мкл плазмы из левой камеры, приливали 100 мкл фосфатного буферного солевого раствора и 600 мкл ацетонитрила, перемешивали. Из правой камеры отбирали в новую полипропиленовую пробирку объемом 1.5 мл 100 мкл фосфатного буферного солевого раствора, приливали 100 мкл пулированной плазмы крови человека и 600 мкл ацетонитрила, перемешивали. Центрифугировали пробирки при 10000 x g в течение 10 мин при температуре 4 °С. 100 мкл супернатанта переносили в виалу вместимостью 2 мл со стеклянной вставкой и проводили ВЭЖХ-МС/МС анализ на жидкостном хроматографе UltiMate 3000 UHPLC (Thermo, США) c масс-спектрометрическим детектором EVOQ Elite (Bruker, США) с ионизацией электрораспылением.
Долю соединения, связавшегося с белками плазмы рассчитывали по следующей формуле:
fb (%) = [1 − (Cb/Cp)] × 100, где
fb (%) – доля соединения, связавшегося с белками плазмы
Cb – концентрация соединения в камере с буферным раствором (правая камера)
Cp – концентрация соединения в камере с плазмой (левая камера).
Эксперимент проводили в двух повторах. В качестве контрольных соединений использовали пропранолол (высокая степень связывания с белками плазмы крови) и атенолол (низкая степень связывания с белками плазмы крови).
Лидерное соединение Ic обладает (таблица 12) очень высокой степенью связывания с белками плазмы крови человека (>99%), что вероятно, связано с наличием в структуре молекулы Ic карбоксильной группы, которая взаимодействует с альбумином, содержащимся в плазме крови (Gardiner et al., 2019) [9]. Многие зарегистрированные лекарственные средства обладают очень высокой степенью связывания с белками плазмы крови (Smith, Dennis A. Et al., 2010) [10], (Zhang, Fengling et al., 2012) [11]. В дальнейшем будут проведены фармакокинетические исследования in vivo с целью подтвердить, что лидерное соединение не обладает неудовлетворительными фармакокинетическим параметрами из-за высокой степени связывания с белками плазмы крови.
Таблица 12. Результаты определения доли лидерного соединения Ic, связавшегося с белками плазмы крови человека. Данные представлены в виде M ± σ, где M – среднеарифметическое значение результатов 2 параллельных определений, σ – стандартное отклонение.
Определение коэффициента распределения logD7.4
В полипропиленовую пробирку объемом 1.5 мл вносили 245 мкл 1-октанола, насыщенного 0.1 М фосфатным буферным раствором. Далее добавляли 5 мкл раствора анализируемого соединения (10 мМ в ДМСО) и 500 мкл 0.1 М фосфатного буферного раствора, насыщенного 1-октанолом. Полученную смесь интенсивно перемешивали на вортексе в течении 3 часов при комнатной температуре. Центрифугировали пробирки при 1500 x g в течение 5 мин при комнатной температуре и оставляли их на 3 часа при комнатной температуре для полного разделения слоев. 50 мкл верхней фазы (1-октанол) переносили в полипропиленовую пробирку объемом 0.5 мл, добавляли 100 мкл 1-октанола, насыщенного 0.1 М фосфатным буферным раствором и перемешивали. 50 мкл нижней фазы (0.1 М фосфатный буферный раствор) переносили в полипропиленовую пробирку объемом 0.5 мл, добавляли 100 мкл 0.1 М фосфатного буферного раствора, насыщенного 1-октанолом и перемешивали. Далее 100 мкл каждого из растворов переносили в виалы вместимостью 2 мл со стеклянной вставкой и проводили ВЭЖХ-МС/МС анализ на жидкостном хроматографе UltiMate 3000 UHPLC (Thermo, США) c масс-спектрометрическим детектором EVOQ Elite (Bruker, США) с ионизацией электрораспылением.
Коэффициент распределения между 1-октанолом и 0.1 М фосфатным буферным раствором рассчитывали по следующей формуле:
logD7.4 = log10 (Soctanol/Sbuffer), где
logD7.4 – коэффициент распределения анализируемого соединения между 1-октанолом и 0.1 М фосфатным буферным раствором
Soctanol – площадь хроматографического пика, измеренная в образце 1-октанола (верхний слой)
Sbuffer – площадь хроматографического пика, измеренная в образце 0.1 М фосфатного буферного раствора (нижний слой)
Эксперимент проводили в трех повторах. В качестве контрольных соединений использовали парацетамол (logD7.4 <1) и кетоконазол (logD7.4 >3).
С помощью программы ACD/Labs Percepta Platform (ACD/Labs Release 2021.2.2, Advanced Chemistry Development, Канада, https://www.acdlabs.com) определяли расчетные значения logD7.4 и сравнивали их с экспериментальными.
Измеренное значение logD7.4 лидерного соединения Ic находится в пределах оптимального диапазона от 1 до 3 (Waring, Michael J. et al., 2010) [12]. Предсказанное с помощью программы ACD/Labs Percepta Platform значение logD7.4 близко к измеренному. Результаты представлены в таблице 13.
Таблица 13. Результаты определения коэффициента распределения logD7.4 лидерного соединения Ic, между 1-октанолом и 0.1 М фосфатным буферным раствором. Данные представлены в виде M ± σ, где M – среднеарифметическое значение результатов 3 параллельных определений, σ – стандартное отклонение.
Биологические исследования полученных соединений проходили в два параллельных этапа. С одной стороны, в энзиматических тестах была оценена способность полученных соединений селективно угнетать цистатионин-g-лиазу бактериального происхождения по сравнению с человеческой цистатионин-g-лиазой. С другой стороны, в микробиологических тестах была исследована способность целевых веществ усиливать действие антибиотиков в отношении болезнетворных микроорганизмов, содержащих указанный фермент. В качестве внешнего контроля использовались вещества NL-1 и NL-2, синтезированные согласно известным прописям [4].
В итоге выполненных исследований авторами были получены неожиданные результаты:
1) Наличие карбоксильной группы во втором положении индольного кольца и самой бициклической системы индола необходимо для эффективного ингибирования bCSE и потенцирования действия антибиотиков.
2) Соединения формулы (I) особенно сильно потенцируют действие канамицина против грамположительных бактерий, смеси пиперациллина и тазобактама против Acinetobacter Baumannii, цефепима против Pseudomonas aeruginosa.
3) Лидерное соединение проявляет ADME-свойства, которые позволяют использовать его в дальнейших in vivo исследованиях
Каждый способ представлен со ссылкой на конкретное соединение или группу соединений, подробные детали синтеза которого представлены выше. Все пронумерованные соединения в рамках настоящего изобретения (см. таблицу 1), входящие в предлагаемую структуру Маркуша, обозначенную формулой (I), могут быть синтезированы относительно легко, используя напрямую способы, которые были изложены. В некоторых случаях для некоторых предпочтительных вариантов осуществления представлено больше подробностей синтеза, чтобы предоставить такие сведения, которые могут служить в качестве модели для синтеза ряда других соединений, описанных в данном документе.
Притязания по формуле изобретения подтверждаются приведенными примерами реализации заявляемого изобретения, но не ограничиваются ими.
Заявителем предприняты меры, направленные на повышение достоверности данных исследования, минимизацию/исключение субъективности оценки. Молекулярная структура и чистота синтезированных веществ подтверждены с помощью комплекса физико-химических методов анализа, что сводит вероятность ошибки и необъективной интерпретации результатов к минимуму. Все конечные результаты настоящего исследования являются объективными, поскольку получены с использованием инструментальных методов анализа, в которых возможность влияния на результаты измерений сведена к минимуму. Меры, направленные на повышение достоверности данных исследования, включают в себя использование подходящих вспомогательных и расходных материалов высокого качества, однозначное установление структуры полупродуктов и конечных веществ ЯМР 1H, 13C-спектроскопией, HRMS, подтверждение чистоты методом ВЭЖХ-УФ/МС, использование подходящих методов во время выделения и очистки веществ.
Соединения, которые показывают оптимальную активность в тест-системах, будут затем образовывать основу для следующего этапа разработки лекарственных средств. На следующем этапе эти выбранные соединения подвергают общепризнанному пути разработки лекарственных средств. Путь разработки лекарственных средств определяет потенциальную ценность соединения путем оценки ряда факторов, включая биодоступность, токсикологию, фармакологию и эффективность на животных моделях, перед тем как соединения будут признаны возможными для тестирования на человеке.
Предлагаемый способ ингибирования бактериальной цистатионин-гамма-лиазы с использованием нафтилсодержащих карбоновых кислот, представляющих собой соединение формулы (I), может быть востребован фармацевтическими компаниями для создания на их основе терапевтического препарата для лечения заболеваний, вызываемых антибиотикорезистентными бактериями, который может применяться самостоятельно или в качестве потенциатора совместно с антибиотиками.
Эквиваленты.
Специалистам в данной области будут очевидны, или они смогут достоверно установить при помощи обычных экспериментальных методик, многочисленные эквиваленты конкретных вариантов реализации, описанные в настоящем документе. Предполагается, что такие эквиваленты входят в объем охраны следующей формулы изобретения.
Резюмируя вышесказанное, предлагаемое изобретение может быть признано соответствующей критерию патентоспособности «Промышленная применимость» во всех частных формах воплощения признаков формулы изобретения до даты испрашиваемого приоритета.
Источники информации
1. Navon-Venezia, et al. «Update on Pseudomonas aeruginosa and Acinetobacter baumannii infections in the healthcare setting». Curr. Opin. Infect. Dis. 2005, 18, 306-313, DOI: 10.1097/01.qco.0000171920.44809.f0.
2. Заявка на патент на изобретение WO2019232083(А1) (UNIV NEW YORK [US]), 05.12.2019 «Compounds and methods for treating bacterial infections» (прототип).
3. L. Luhachack, E. Nudler «Bacterial gasotransmitters: An innate defense against antibiotics». Curr. Opin. Microbiol. 2014, 21, 13–17, DOI: 10.1016/j.mib.2014.06.017.
4. K. Shatalin, et al. «H2S: a universal defense against antibiotics in bacteria». Science 2011, 334, 986–990, DOI: 10.1126/science.1209855.
5. K. Shatalin, et al., 2021 «Inhibitors of bacterial H2S biogenesis targeting antibiotic resistance and tolerance». Science 2021, 372, 1169-1175, DOI:10.1126/science.abd8377.
6. A. Asimakopoulou, et al. «Selectivity of Commonly Used Pharmacological Inhibitors for Cystathionine β-Synthase (CBS) and Cystathionine γ-Lyase (CSE)». Br. J. Pharmacol. 2013, 169, 922–932, DOI:10.1111/bph.12171.
7. US9725426B2 (SOVA PHARMACEUTICALS INC [US]), 08.08.2017 г. «Cystathionine-γ-lyase (CSE) inhibitors».
8. Патент на изобретение RU2640418 C2 (SOVA FARMASYUTIKALS INK), 09.01.2018 «Ингибиторы цистатионин-гамма-лиазы (CSE)».
9. P. Gardiner; R. Cox; K. Grime «Plasma Protein Binding as an Optimizable Parameter for Acidic Drugs». Drug Metab. Dispos. 2019, 47, 865–873, DOI:10.1124/dmd.119.087163.
10. D. Smith; L. Di; E. Kerns «The Effect of Plasma Protein Binding on in Vivo Efficacy: Misconceptions in Drug Discovery». Nat. Rev. Drug Discov. 2010, 9, 929–939, DOI:10.1038/nrd3287.
11. F. Zhang, et al. «Compilation of 222 drugs’ plasma protein binding data and guidance for study designs». Drug Discov. Today 2012, 17, 475-485, DOI: 10.1016/j.drudis.2011.12.018.
12. Waring, Michael J. «Lipophilicity in drug discovery». Expert Opin. Drug Discov. 2010, 5, 235–248, DOI:10.1517/17460441003605098.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГИДРОКСАМОВЫЕ КИСЛОТЫ, СОДЕРЖАЩИЕ ЯДРА 1,2,4- И 1,3,4-ОКСАДИАЗОЛА В КАЧЕСТВЕ ПОТЕНЦИАТОРОВ ДЕЙСТВИЯ АНТИБИОТИКОВ | 2023 |
|
RU2830361C1 |
| 2,4,5-ТРИ(МЕТОКСИФЕНИЛ) ЦИС-ИМИДАЗОЛИН И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2019 |
|
RU2730497C1 |
| ТИЛОЗИНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2714482C2 |
| Производные 4-((арил)(метил)амино)хиназолин-7-карбоновой кислоты с противоопухолевым действием и способ их получения | 2024 |
|
RU2834688C1 |
| ПРИМЕНЕНИЕ ТРИЦИКЛИЧЕСКОГО СЕРУСОДЕРЖАЩЕГО ПРОИЗВОДНОГО 1,2-ДИГИДРОХИНОЛИНА ДЛЯ ИНГИБИРОВАНИЯ РЕПЛИКАЦИИ БЕТА-КОРОНАВИРУСОВ, ВКЛЮЧАЯ SARS-CoV-2 | 2021 |
|
RU2780247C1 |
| АНТИБАКТЕРИАЛЬНЫЕ СРЕДСТВА НА ОСНОВЕ ЧЕТВЕРТИЧНЫХ АММОНИЕВЫХ СОЛЕЙ | 2014 |
|
RU2561281C1 |
| Соединения фторхинолонового ряда на основе производных пиридоксина, обладающие антибактериальными свойствами | 2019 |
|
RU2713932C1 |
| Фторхинолоны на основе 4-дезоксипиридоксина | 2016 |
|
RU2634122C1 |
| ЗАМЕЩЕННЫЕ ПИРИМИДИНИЛПИРРОЛЫ, АКТИВНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2012 |
|
RU2621732C2 |
| ИНГИБИТОРЫ ЦИСТАТИОНИН-γ-ЛИАЗЫ (CSE) | 2013 |
|
RU2661879C2 |
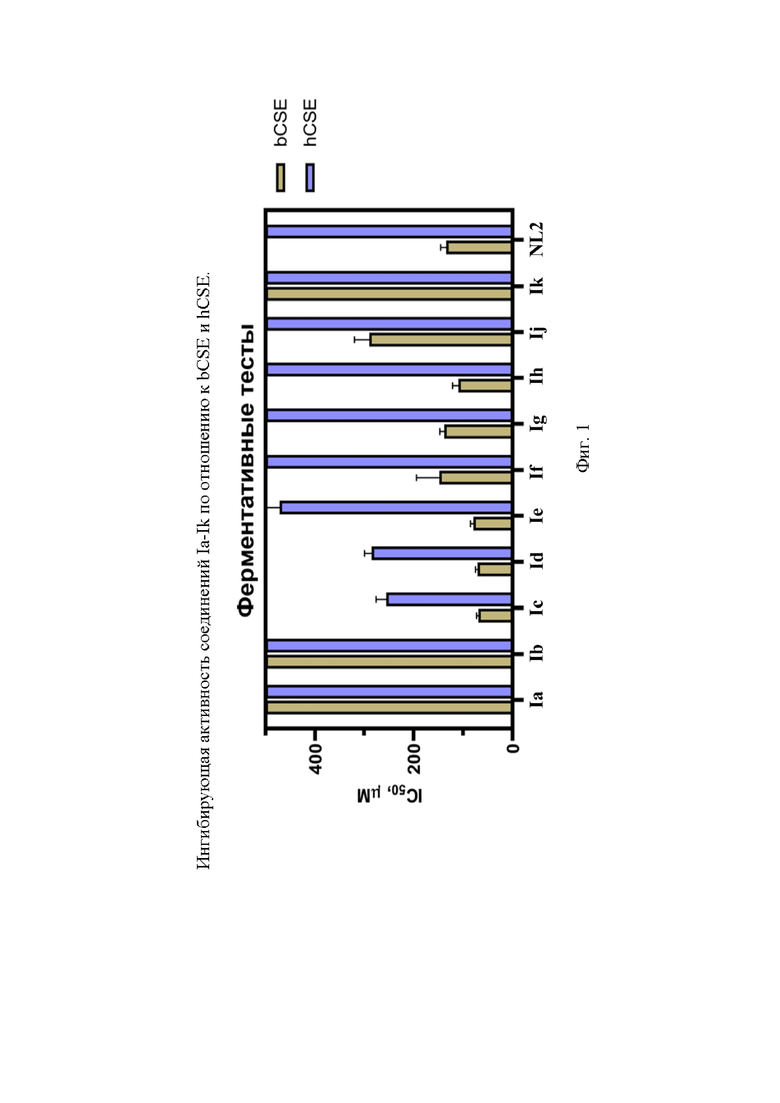
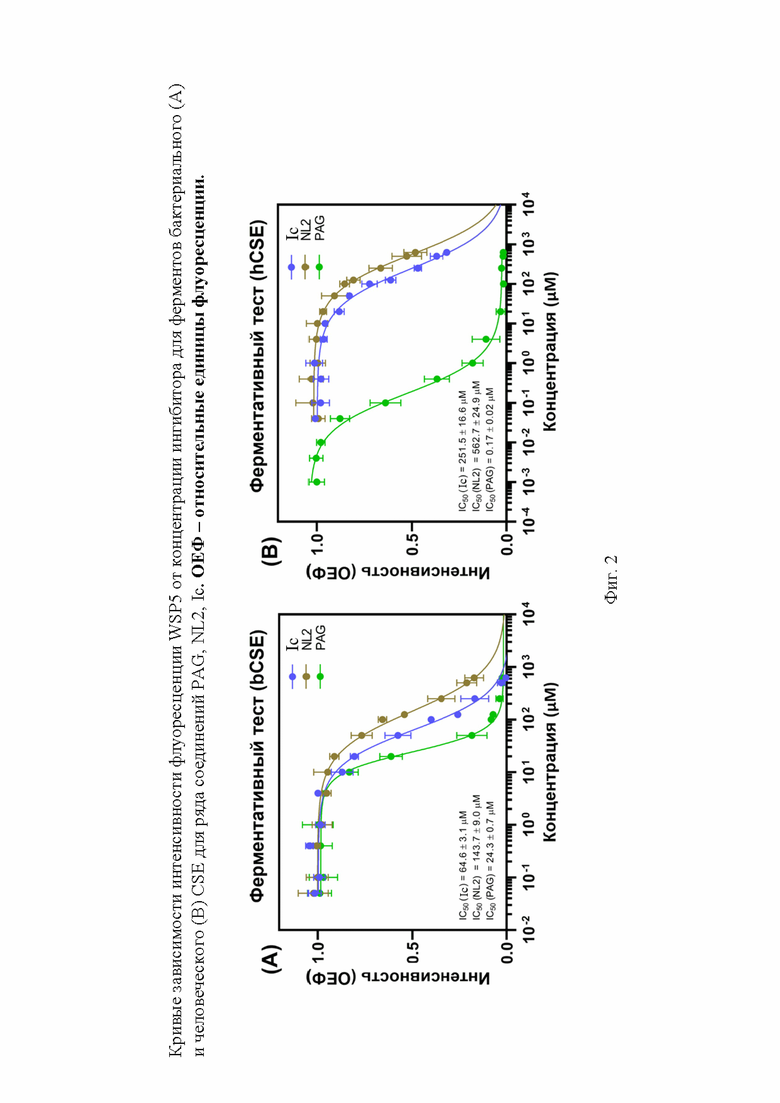
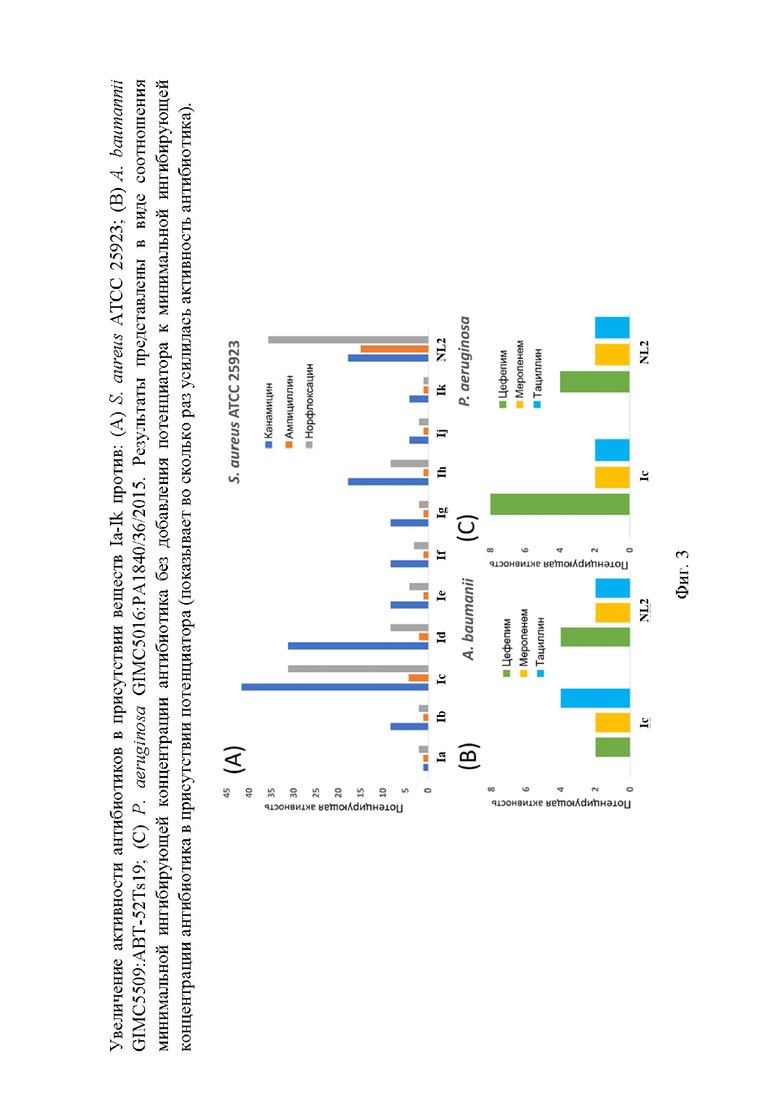
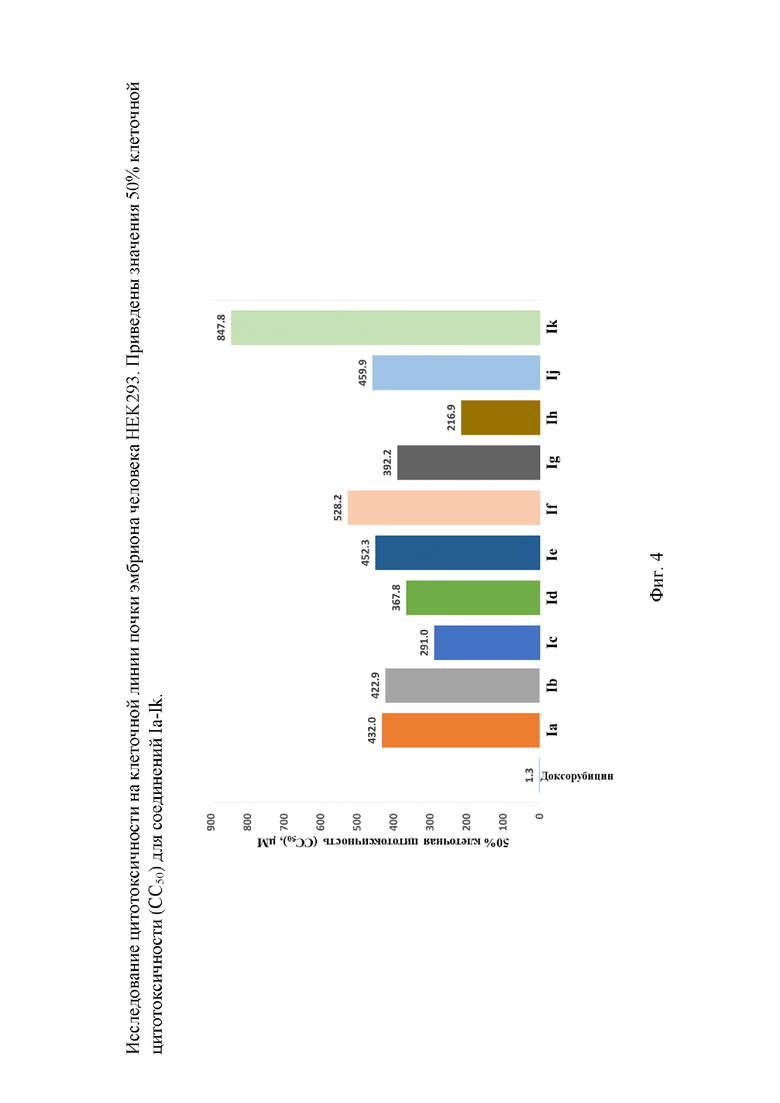
Изобретение относится к области органической и медицинской химии и представляет собой способ ингибирования бактериальной цистатионин-γ-лиазы (цистатионин-гамма-лиазы, сystathionine gamma-lyase, или bCSE) с использованием соединений формулы (I), где кольцо, имеющее связи «- - -», отсутствует или представляет собой бензольное кольцо, непосредственно присоединённое к ароматической системе пиррола посредством двух соседних атомов углерода. Способ характеризуется тем, что соединение формулы (I) применяют самостоятельно или в качестве потенциатора совместно с антибиотиками для подавления роста антибиотикорезистентных бактерий. Для подавления роста грамотрицательных бактерий Acinetobacter baumannii или Pseudomonas aeruginosa антибиотики выбирают из группы: цефепим, меропенем, смесь пиперациллина и тазобактама. Для подавления роста грамположительных бактерий вида Staphylococcus aureus, в том числе MRSA, антибиотики выбирают из группы: канамицин, ампициллин, норфлоксацин. Технический результат: увеличение эффективности ингибирования бактериальной цистатионин-γ-лиазы (bCSE); увеличение эффективности применения более доступных синтетически соединений формулы (I) в качестве потенциаторов антибиотиков; расширение арсенала способов ингибирования белка (bCSE) за счёт применения потенциаторов антибиотиков для подавления роста антибиотикорезистентных штаммов микроорганизмов. 6 з.п. ф-лы, 4 ил., 13 табл., 19 пр.
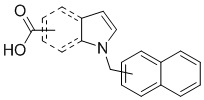 (I)
(I)
1. Способ ингибирования бактериальной цистатионин-γ-лиазы с использованием нафтилсодержащих карбоновых кислот, представляющих собой соединение формулы (I):
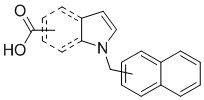 I,
I,
в котором кольцо, имеющее связи «- - -», отсутствует или представляет собой бензольное кольцо, непосредственно присоединённое к ароматической системе пиррола посредством двух соседних атомов углерода, характеризующийся тем, что соединение формулы (I) применяют самостоятельно или в качестве потенциатора совместно с антибиотиками для подавления роста антибиотикорезистентных бактерий, при этом для подавления роста грамотрицательных бактерий Acinetobacter baumannii или Pseudomonas aeruginosa антибиотики выбирают из группы: цефепим, меропенем, смесь пиперациллина и тазобактама, а для подавления роста грамположительных бактерий вида Staphylococcus aureus, в том числе MRSA, антибиотики выбирают из группы: канамицин, ампициллин, норфлоксацин.
2. Способ ингибирования бактериальной цистатионин-γ-лиазы по п.1, в котором нафтилсодержащая карбоновая кислота представляет собой соединение, имеющее формулу:
 Ic.
Ic.
3. Способ ингибирования бактериальной цистатионин-γ-лиазы по п.1, в котором нафтилсодержащая карбоновая кислота представляет собой соединение, имеющее формулу:
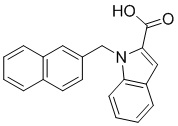 Id.
Id.
4. Способ ингибирования бактериальной цистатионин-γ-лиазы по п.1, в котором нафтилсодержащая карбоновая кислота представляет собой соединение, имеющее формулу:
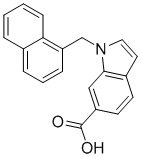 Ie.
Ie.
5. Способ ингибирования бактериальной цистатионин-γ-лиазы по п.1, в котором нафтилсодержащая карбоновая кислота представляет собой соединение, имеющее формулу:
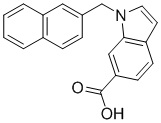 If.
If.
6. Способ ингибирования бактериальной цистатионин-γ-лиазы по п.1, в котором нафтилсодержащая карбоновая кислота представляет собой соединение, имеющее формулу:
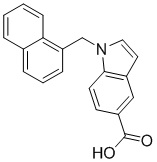 Ig.
Ig.
7. Способ ингибирования бактериальной цистатионин-γ-лиазы по п.1, в котором нафтилсодержащая карбоновая кислота представляет собой соединение, имеющее формулу:
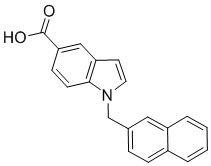 Ih.
Ih.
| WO 2019232083 A1, 05.12.2019 | |||
| ИНГИБИТОРЫ ЦИСТАТИОНИН-γ-ЛИАЗЫ (CSE) | 2013 |
|
RU2661879C2 |
| ИНГИБИТОРЫ ЦИСТАТИОНИН-Г-ЛИАЗЫ (CSE) | 2013 |
|
RU2640418C2 |
| ПРЕОБРАЗОВАТЕЛЬ ПОСТОЯННОГО НАПРЯЖЕНИЯ В ПЕРЕМЕННОЕ | 0 |
|
SU381141A1 |

