ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к кристаллическим материалам, имеющим каркас типа каркаса MFI и/или MEL, содержащим кристаллы небольшого размера и имеющим большую площадь поверхности, обозначаемым ЕММ-30, к их синтезу и их применению в качестве адсорбентов и катализаторов в реакциях органического синтеза.
УРОВЕНЬ ТЕХНИКИ
Классификацию кристаллических материалов производит Структурная Комиссия Международной ассоциации по цеолитам (англ. Structure Commission of the International Zeolite Association) в соответствии с правилами, установленными Комиссией IUPAC по номенклатуре цеолитов. Согласно этой классификации, цеолитам каркасного типа и другим микропористым кристаллическим материалам, для которых была установлена структура, присваивается трехбуквенный код, и они внесены в Атлас цеолитов каркасного типа, шестое дополненное издание, Elsevier (2007).
Одним из известных кристаллических материалов, структура которого была установлена, является материал, отнесенный каркасному типу MFI, который, как хорошо известно, включает ZSM-5. Кристаллический ZSM-5 и его традиционный синтез с применением в качестве структурообразующего агента катионов тетрапропиламмония рассмотрен в патентах US 3702886 и RE 29948, содержания которых полностью включены в настоящее описание посредством ссылки Традиционный ZSM-5 имеет характерную дифрактограмму, которая позволяет отличать его от других известных кристаллических материалов, и представляет собой чрезвычайно универсальный катализатор, применяемый в ряде различных реакций органического синтеза.
Другой известной структурой кристаллического материала является структура MEL, также называемого ZSM-11, которая подробно рассмотрена в патенте US 3709979, содержание которого полностью включено в настоящее описание посредством ссылки. Кристаллические материалы каркасного типа MFI и MEL имеют схожие структуры, и их часто совместно получают в способах синтеза цеолита в виде взаимно проросших или разупорядоченных материалов.
Для некоторых проводимых на цеолитах реакциях, катализируемых кислотой, было бы полезно снизить длину диффузии реагентов и/или получаемых молекул за счет применения цеолита с уменьшенными размерами кристаллов и, следовательно, с повышенной площадью наружной поверхности. Это может позволить снизить селективное влияние цеолита на частицы определенной формы, хотя это может быть неважным для тех реакций, в которых требуется лишь высокая активность. Например, увеличенная площадь наружной поверхности позволяет проводить реакции с более крупными молекулами, которые не могут проникать в поры цеолита. Кроме того, при проведении некоторых способов наблюдали, что при увеличении площади наружной поверхности цеолита ZSM-5 скорость его деактивации снижается. См., например, М. Choi с соавт., Nature 461 (2009) 246-249 J. Kim, М. Choi, R. Ryoo, J. Catal. 269 (2010) 219-228). Также сообщали, что увеличение площади наружной поверхности приводит к увеличению выходов пропилена при конверсии метанола. См., например, М. Firoozi с соавт.. Catal. Commun. 10 (2009) 1582-1585.
Один из примеров мелкокристаллического ZSM-5 рассмотрен в патенте US 5240892, в котором описан ZSM-5 в виде пластинок, имеющих первый и второй основные размеры, составляющие по меньшей мере приблизительно 0,05 микрона, предпочтительно по меньшей мере приблизительно 0,1 микрона, и меньший третий размер, составляющий менее приблизительно 0,02 микрона, предпочтительно менее приблизительно 0,01 микрона. Сорбционная способность ZSM-5 в отношении мезитилена составляет по меньшей мере 3,0% масс, его получают из такого источника оксида кремния, как осажденный оксид кремния, в отсутствие органического структурообразующего агента или в присутствии такого структурообразующего агента, как н-пропиламин.
Кроме того, в публикации Chem. Mater. 21 (2009) 641-654, D. Serrano с соавт., представлен синтез ZSM-5 кристаллов размером от 5 до 10 нм с применением двойной матрицы из ионов тетрапропиламмония (ТПА) и фениламинопропилтриметоксисилана. Согласно этому способу, силанизирующий агент вводят после кратковременного предварительного подогрева синтез-геля перед началом кристаллизации цеолита. На Фиг. 1 публикации Serrano с соавт. схематично представлены продукты кристаллизации, в то время как на Фиг. 2 представлены полученные с помощью ТЭМ (трансмиссионного электронного микроскопа) изображения продукта. Несмотря на то, что на полученных с помощью ТЭМ изображениях показаны очень мелкие частицы, пики на порошковых рефрактограммах продукта, полученного в этой работе, указывают на размеры кристаллов, превышающие диапазон от 5 до 10 нм.
В работе Ryoo с соавт., опубликованной в виде статьи "Stable single-unit-cell nanosheets of zeolite MFI as active and long-lived catalysts (Стабильные нанолисты цеолита MFI толщиной в одну элементарную ячейку в качестве активных катализаторов с длительным сроком службы)", Nature, 461, 246-249 (10 сентября 2009 г.), рассмотрен синтез варианта ZSM-5 толщиной в одну элементарную ячейку с помощью одного направляющего агента, состоящего из алкильной цепочки, содержащей 22 атома углерода, и двух четвертичных аммонийных групп, разделенных метиленовой цепочкой, состоящей из 6 атомов углерода. В этом материале четвертичные аммонийные группы расположены внутри нанолистов толщиной в одну элементарную ячейку, которые отделены друг от друга длинными алкильными цепочками. На Фиг. 3 в работе Ryoo с соавторами схематично представлен вариант однопластинчатых и многопластинчатых кристаллов ZSM-5 толщиной в одну элементарную ячейку.
В патентной заявке US 2015/0298981 рассмотрен кристаллический материал, обозначенный ЕММ-20 и имеющий каркасную структуру ZSM-5, включающий кристаллы, площадь наружной поверхности которых превышает 100 м2/г (определяемую способом физической адсорбции азота с построением t-графика), и имеющий уникальную дифрактограмму. Кристаллический материал может быть синтезирован в присутствии органического структурообразующего агента (Q), выбранного из одного или более следующих дикатионов: дикатионов 1,4-бис(N-пентилпирролидиний)бутана, дикатионов 1,5-бис(N-пентилпирролидиний)пентана или дикатионов 1,6-бис(N-пентилпирролидиний)гексана.
В работе 2012 года Zhang с соавторами представлен синтез "самособирающегося в стержни" материала типа MFI или MFI/MEL с применением гидроксида тетрабутилфосфония (ТВРОН) и/или гидроксида тетрабутиламмония (ТВАОН) в качестве структурообразующего агента (сокращенно "СОА"). См. "Synthesis of Self-Pillared Zeolite Nanosheets by Repetitive Branching (Синтез самособирающихся в стержни цеолитовых нанолистов многократным ветвлением)", Science, 336 (2012), 1684-1687. Полученные с помощью ТЭМ изображения описанных продуктов показывают, что морфологически эти продукты состоят из тонких листов цеолита шириной приблизительно 1 элементарную ячейку. Тонкие листы взаимно проникают друг в друга под прямыми углами, образуя составленный из расположенных крест-накрест слоев каркас кристаллов, внутри которого имеются мезопоры размером 2-7 нм. Полученная структура "карточного домика" из нанолистов образует постоянную сетку из мезопор размерами от 2 до 7 нанометров, которая, наряду с большой площадью наружной поверхности и сниженной длиной диффузии в микропорах, обеспечивает более высокие скорости реакций объемных молекул по сравнению со скоростями, достигаемыми при использовании других мезопористых и традиционных цеолитов MFI. В этой публикации рассмотрены только материалы с Si/Al геля ≥ 75.
Несмотря на описанные выше успехи, имеется необходимость создания новых форм кристаллических материалов каркасного типа MFI и MEL, содержащих ультрамелкие кристаллы, имеющих большие площади наружной поверхности, а также необходимость расширения диапазона Si/Al, в котором эти материалы могут быть синтезированы.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Согласно изобретению, в настоящее время было обнаружено, что несмотря на эффект самосборки в виде стержней, который наблюдали Zhang с соавторами, который сложно воспроизвести, особенно при соотношениях геля Si/Al<75, кристаллические материалы каркасного типа MFI и/или MEL с большой площадью поверхности также можно получить при небольших соотношениях Si/Al, применяя в качестве СОА ТВРОН и ТВАОН при пониженном содержании воды и/или пониженном содержании катиона Группы 1 в геле. Кроме того, было обнаружено, что кристаллические материалы каркасного типа MFI и/или MEL с большой площадью поверхности также могут быть получены при использовании новых СОА, а именно дикатионов 5-бис(N-трибутиламмоний)пентана и/или дикатионов 1,6-бис(N-трибутиламмоний)гексана. В самом деле, в некоторых случаях можно было получить кристаллические материалы каркасного типа MFI и/или MEL с наружной и/или общей площадью поверхности, превышающей опубликованные ранее соответствующие значения площадей поверхности.
Таким образом, первый аспект изобретения относится к способу получения кристаллического материала, имеющего тип каркаса MFI и/или MEL, где способ включает: (i) приготовление смеси для синтеза, подходящей для образования кристаллического материала, где смесь включает источник оксида четырехвалентного элемента Y, необязательно источник трехвалентного элемента X, необязательно источник щелочного или щелочноземельного металла (М), воду и структурообразующий агент (Q1), включающий катионы тетрабутиламмония и/или катионы тетрабутилфосфония, причем смесь для синтеза имеет по меньшей мере одно из следующих молярных отношений: (а) молярное отношение H2O/YO2, составляющее менее 10, и (b) молярное отношение YO2/X2O3, составляющее менее 150; (ii) нагревание смеси в условиях кристаллизации, которые включают температуру, составляющую от 80°С до 220°С, и время, составляющее от 1 суток до 28 суток, до образования кристаллов кристаллического материала; и (iii) извлечение кристаллического материала, полученного в этапе (ii).
Второй аспект изобретения относится к способу получения кристаллического материала, имеющего тип каркаса MFI и/или MEL, где способ включает: (i) приготовление смеси для синтеза, подходящей для образования кристаллического материала, где указанная смесь включает источник оксида четырехвалентного элемента Y, необязательно источник трехвалентного элемента X, необязательно источник щелочного или щелочноземельного металла (М), воду и структурообразующий агент (Q2), включающий дикатионы 1,5-бис(N-трибутиламмоний)пентана и/или дикатионы 1,6-бис(N-трибутиламмоний)гексана; (ii) нагревание смеси в условиях кристаллизации, которые включают температуру, составляющую от 80°С до 220°С, и время, составляющее от 1 суток до 28 суток, до образования кристаллов кристаллического материала; и (iii) извлечение кристаллического материала, полученного в этапе (ii).
Третий аспект изобретения относится к кристаллическому материалу, содержащему в своей пористой структуре дикатионы 1,5-бис(N-трибутиламмоний)пентана и/или дикатионы 1,6-бис(N-трибутиламмоний)гексана.
Четвертый аспект изобретения относится к кристаллическому материалу, имеющему каркасный тип MFI и/или MEL и химический состав, включающий молярное соотношение (n) YO2:Х2О3, где n представляет собой число, составляющее по меньшей мере 20, но менее 2000, например менее 150, X представляет собой трехвалентный элемент, и Y представляет собой четырехвалентный элемент, и при этом кристаллический материал включает кристаллы, общая площадь поверхности которых (определяемая способом физической адсорбции азота с построением t-графика) выше 770 м2/г, и/или площадь наружной поверхности (определяемая способом физической адсорбции азота с построением t-графика) которых выше 400 м2/г.
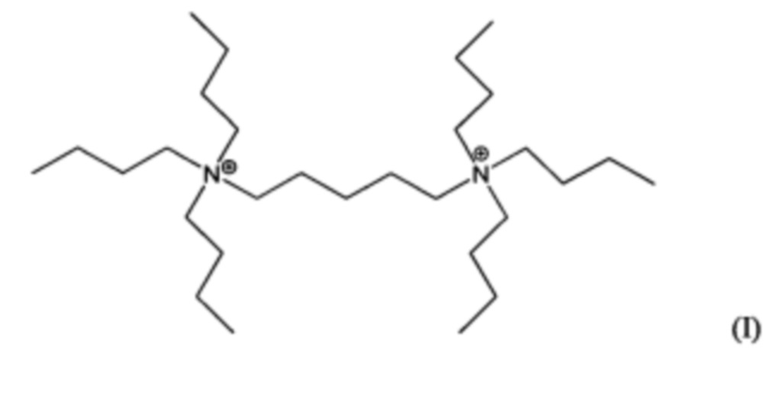
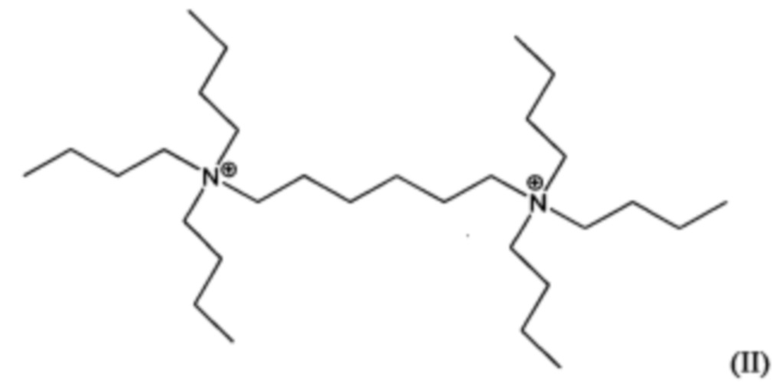
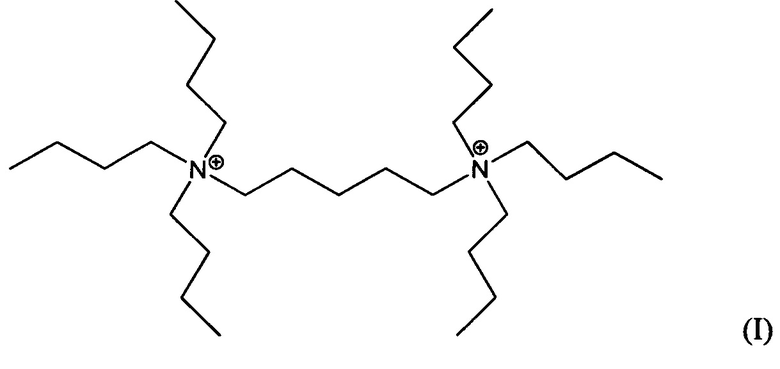
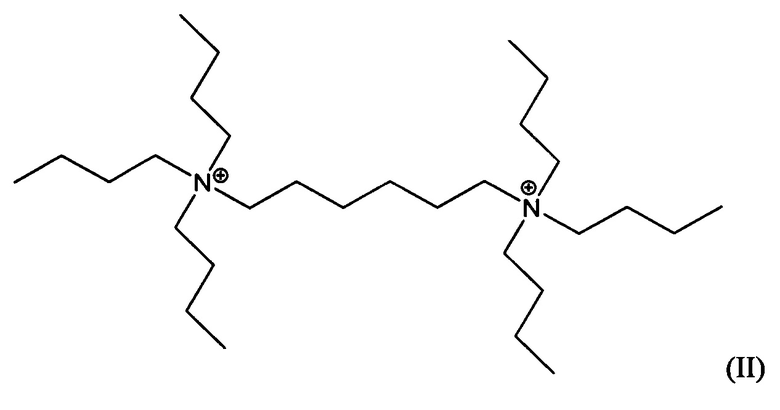
Пятый аспект изобретения относится к органическому азотсодержащему соединению, включающему дикатион, имеющий формулу (I) или (II):
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
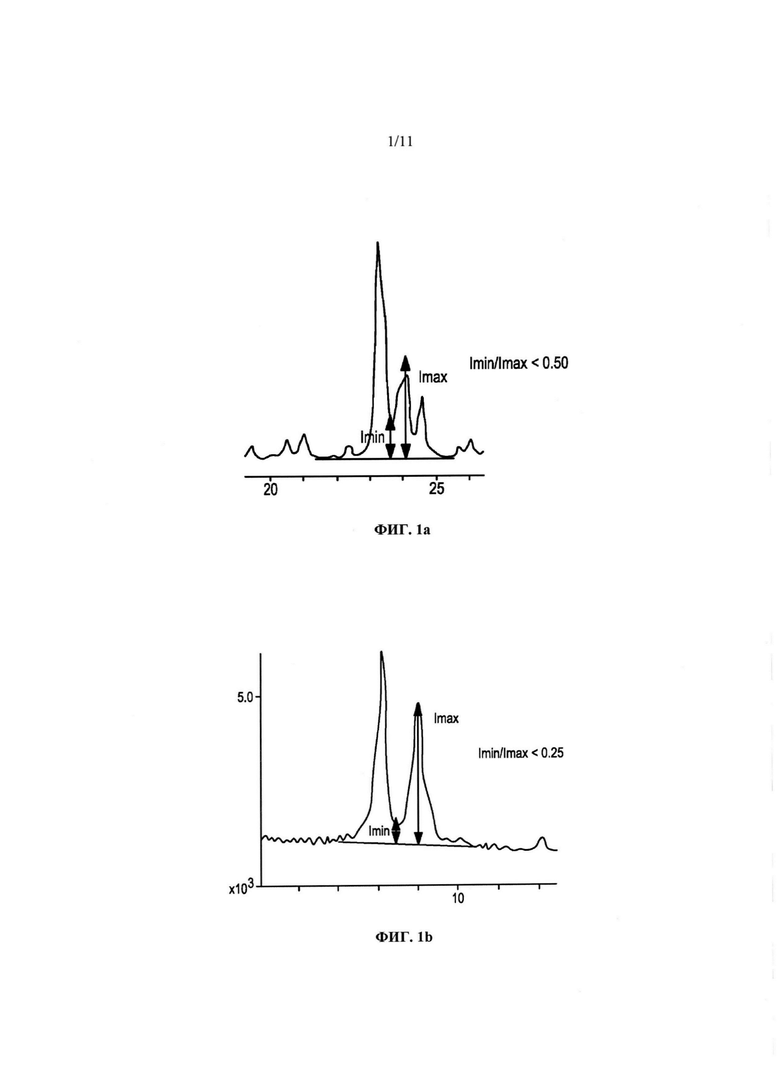
На Фиг. 1 (а) и (b) представлены части дифрактограммы традиционного продукта ZSM-5, полученного как указано в патенте US 5240892, в области от 20 до 28 градусов два-тета (2θ) и от 5 до 10 градусов два-тета, соответственно.
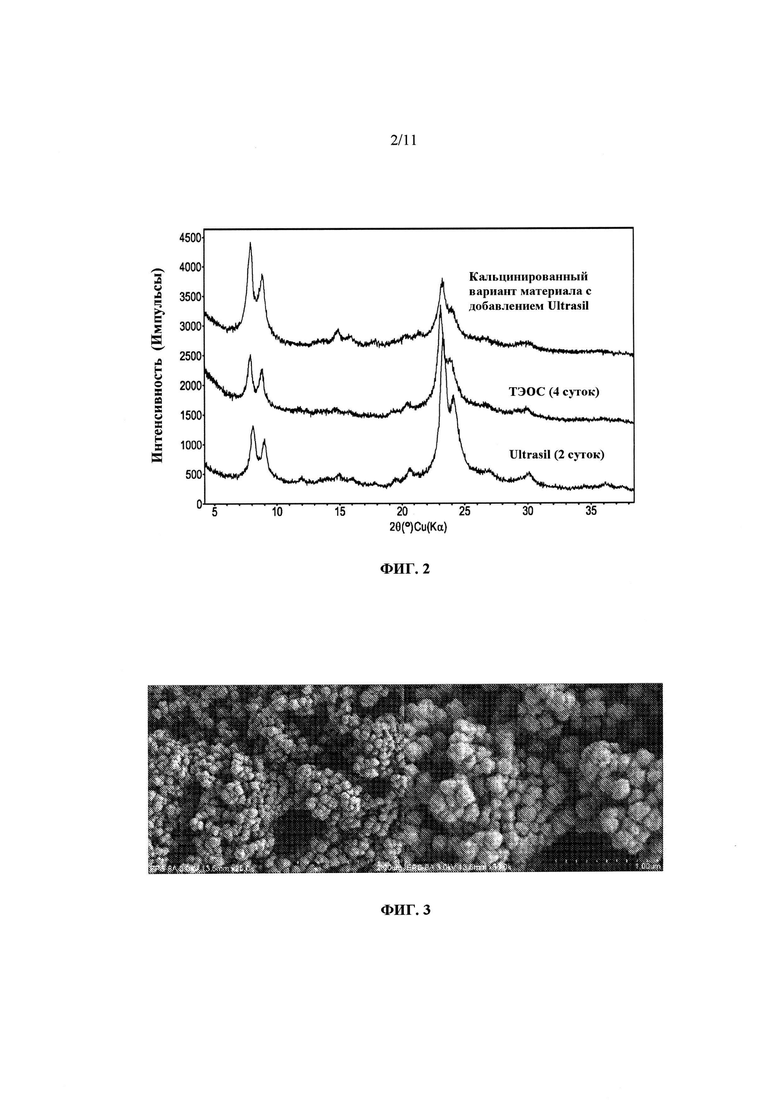
На Фиг. 2 представлено сравнение дифрактограммы материала Примера 1 в том виде, в котором он синтезирован, с дифрактограммами продуктов Примера 2 в том виде, в котором они синтезированы, а также в кальцинированном виде.
На Фиг. 3 представлены полученные с помощью сканирующего электронного микроскопа (СЭМ) изображения продукта Примера 1.
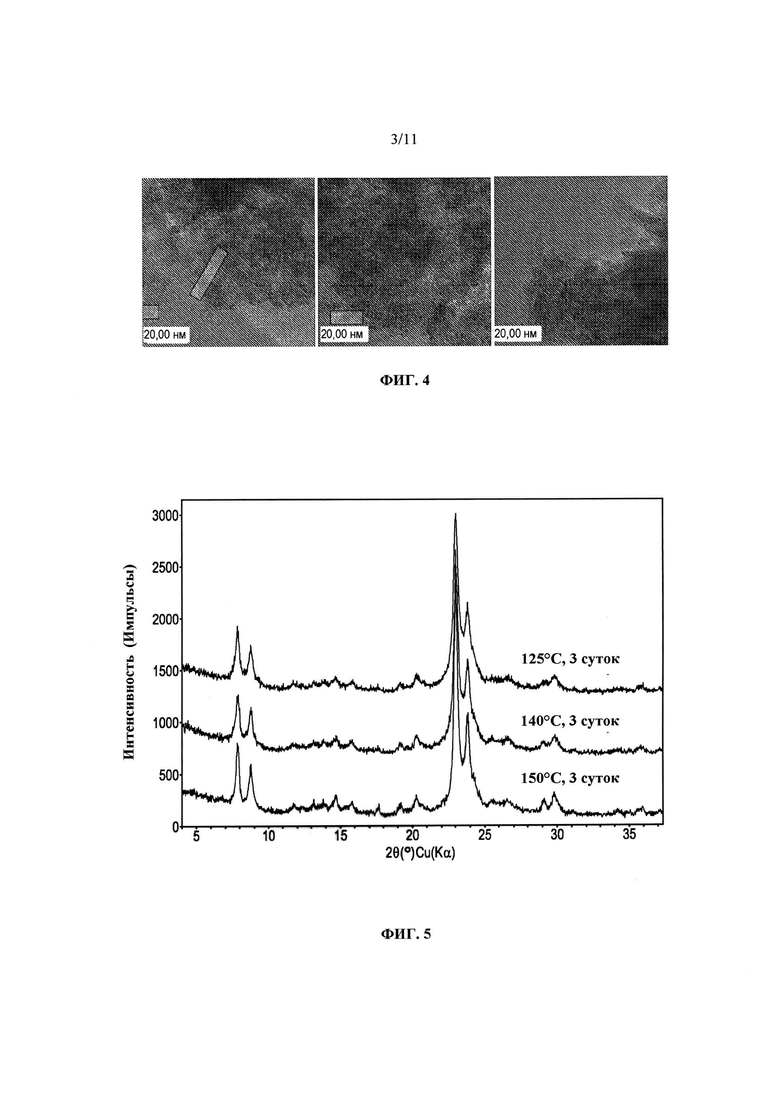
На Фиг. 4 представлены полученные с помощью трансмиссионного электронного микроскопа высокого разрешения (ТЭМВР) изображения продукта Примера 1.
На Фиг. 5 представлено сравнение дифрактограмм, полученных при повторении синтеза Примера 7 при температурах, изменяемых в диапазоне от ~125°С до ~150°С.
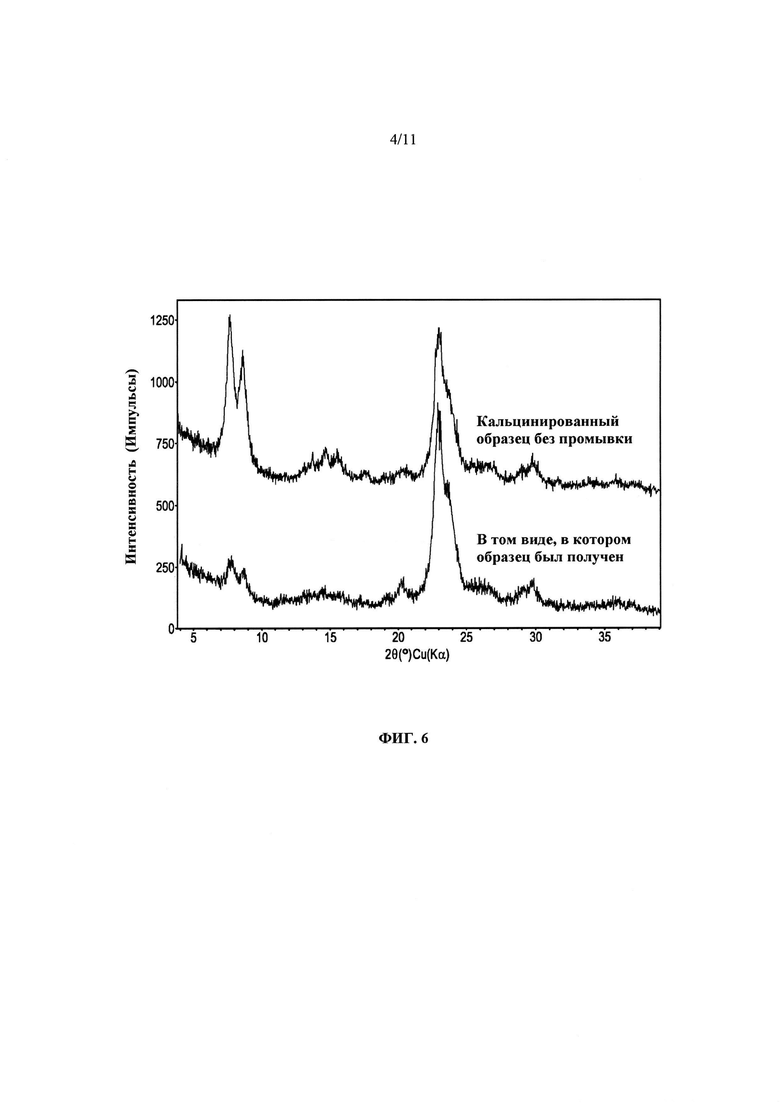
На Фиг. 6 представлено сравнение дифрактограмм продукта Примера 14(а) после синтеза и без промывки и (b) после кальцинации.
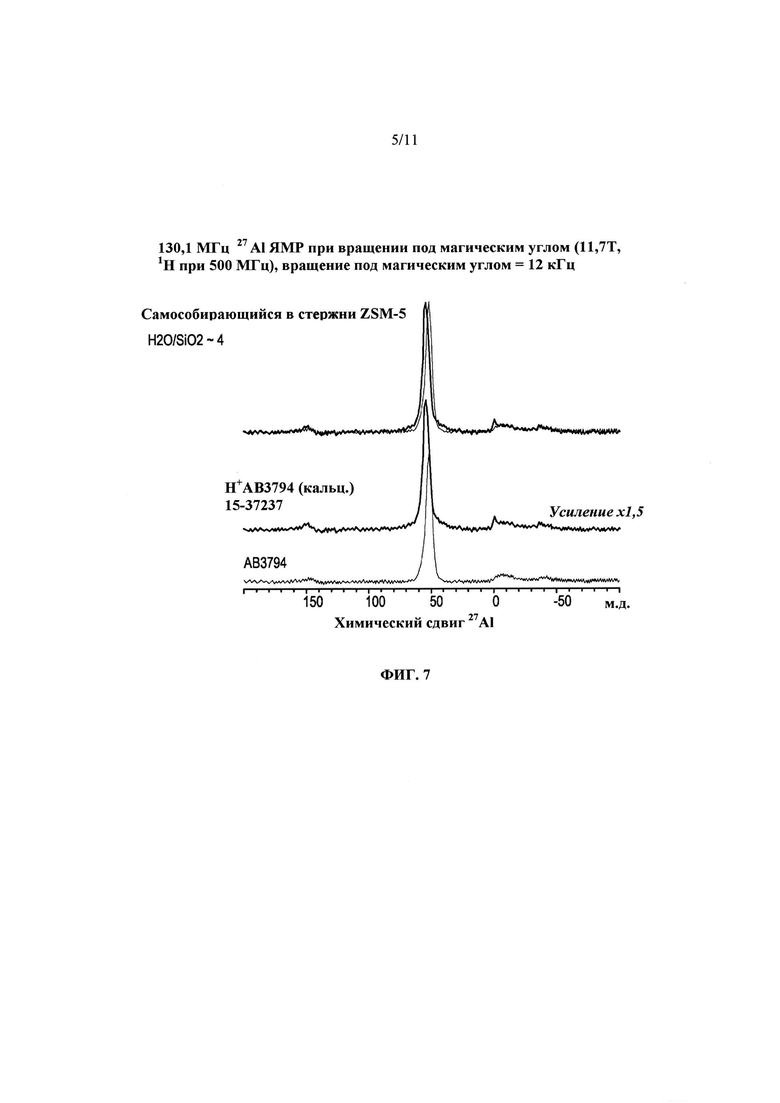
На Фиг. 7 представлены 27Al ЯМР спектры продукта Примера 14 в том виде, в котором он был получен, выделенного без этапа промывки (внизу), кальцинированного варианта этого продукта (в середине) и два спектра в наложении (вверху).
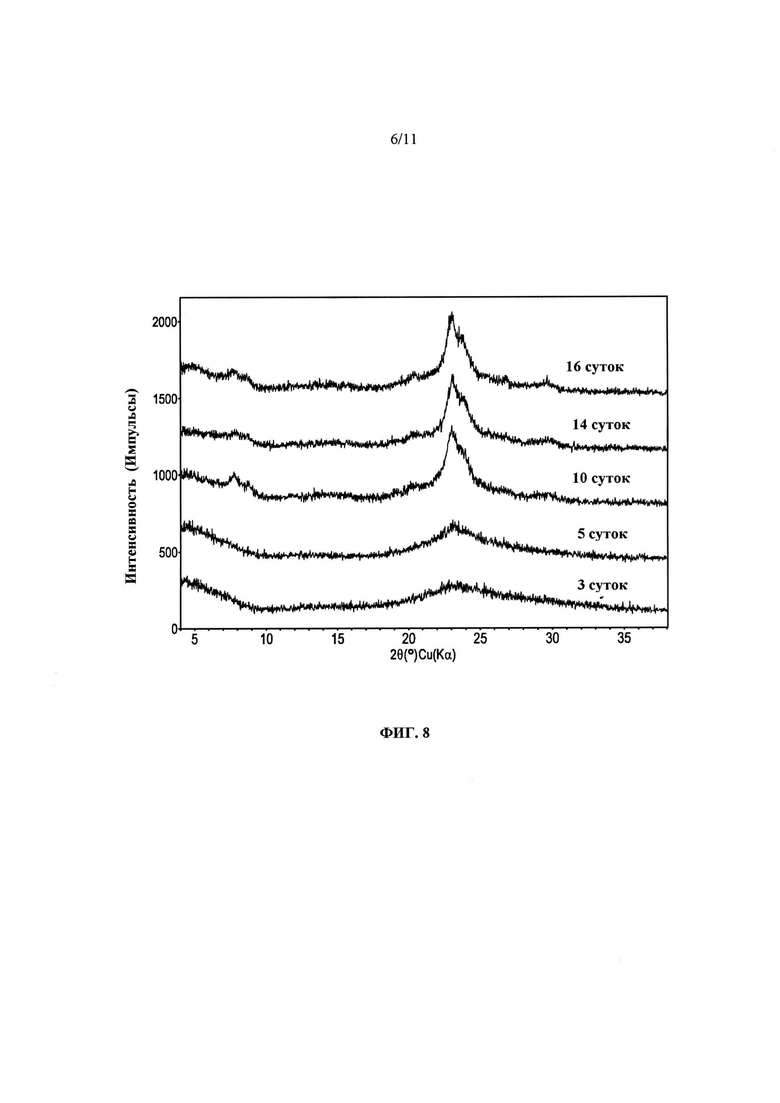
На Фиг. 8 представлены дифрактограммы продуктов Примера 16, полученные после нагревания при ~100°С в течение различных периодов времени.
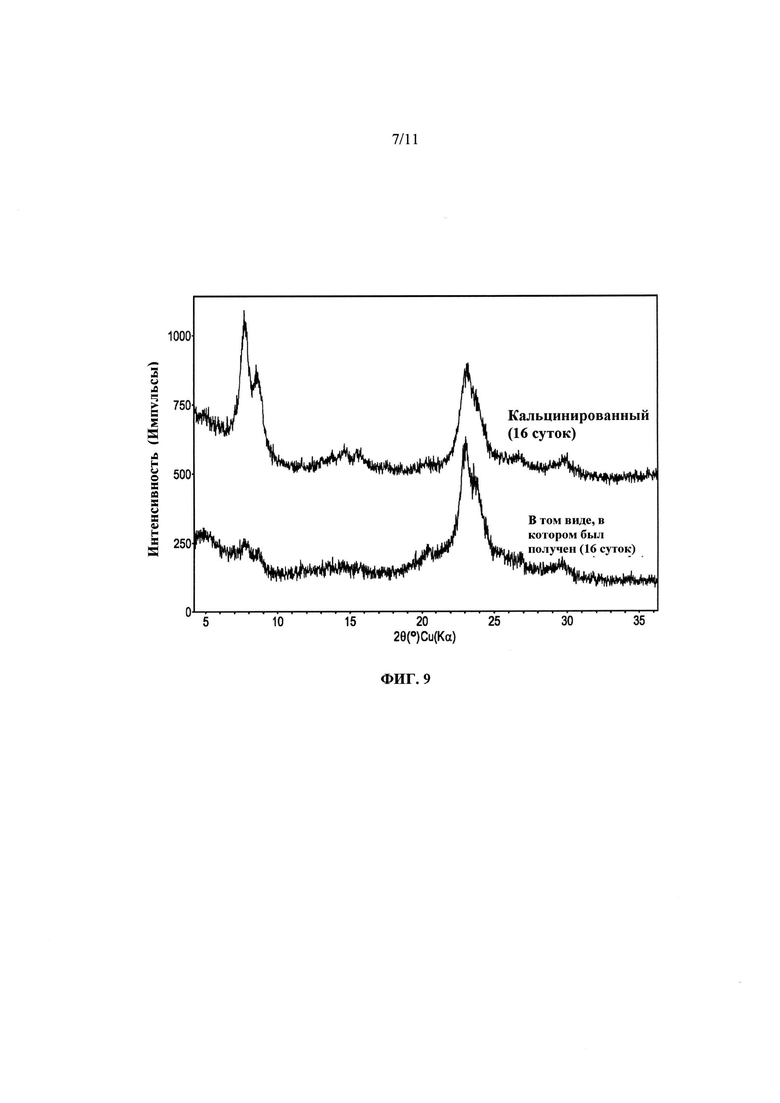
На Фиг. 9 представлено сравнение дифрактограмм, зарегистрированных сразу после получения, и дифрактограмм кальцинированных продуктов Примера 16, полученных а результате ~16 суток нагревания при ~100°С.
На Фиг. 10 представлены полученные с помощью ТЭМ изображения кальцинированного продукта Примера 16, полученного после ~16 суток нагревания при ~100°С.
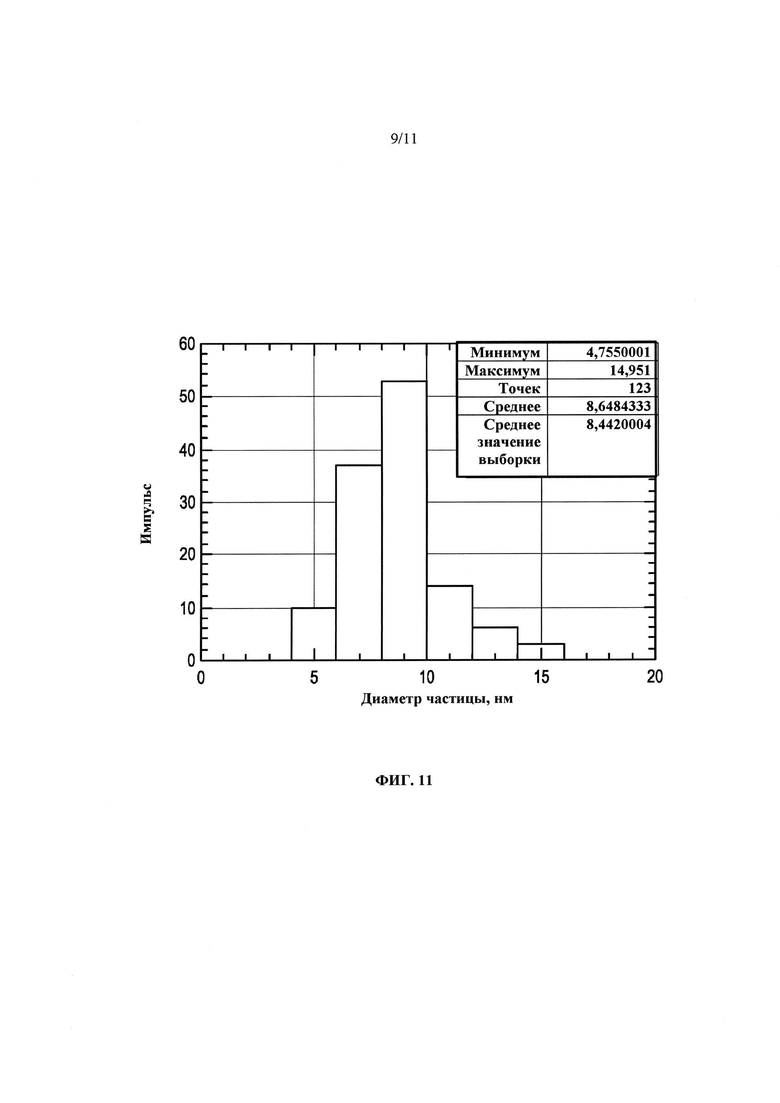
На Фиг. 11 представлена гистограмма распределения размера кристаллов кальцинированного продукта Примера 16, полученного после ~16 суток нагревания при 100°С.
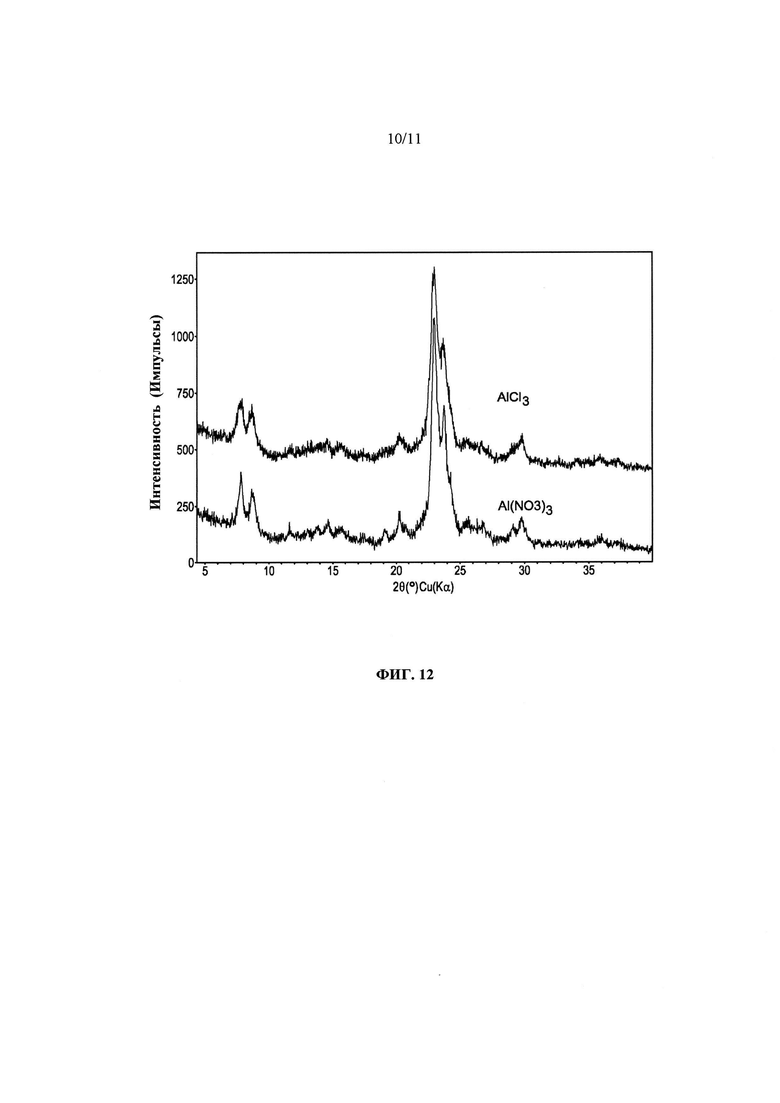
На Фиг. 12 представлено сравнение дифрактограмм продуктов Примера 18 (с использованием хлорида алюминия в качестве источника алюминия) и Примера 19 (с использованием нитрата алюминия в качестве источника алюминия).
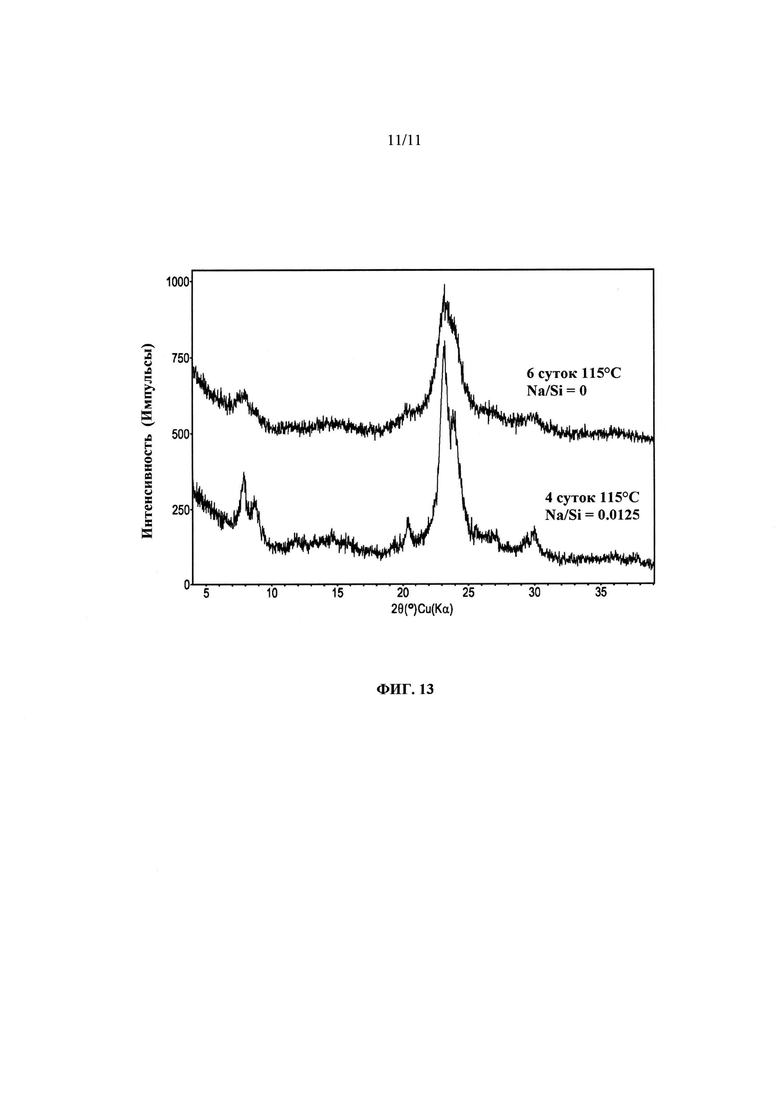
На Фиг. 13 представлено сравнение дифрактограмм продуктов Примеров 22 и 23.
СВЕДЕНИЯ, ПОДТВЕРЖДАЮЩИЕ ВОЗМОЖНОСТЬ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
В настоящей работе рассмотрены способы получения кристаллических материалов, имеющих тип каркаса MFI и/или MEL, и мелкокристаллических форм таких кристаллических материалов, имеющих чрезвычайно большую наружную и/или общую площадь поверхности.
В некоторых воплощениях кристаллическая фаза кристаллического материала, полученного способом согласно настоящему изобретению, в основном включает материал, имеющий каркас типа каркаса MFI, в частности, ZSM-5. В некоторых воплощениях кристаллическая фаза кристаллического материала может содержать нетривиальные количества материала, имеющего каркас типа каркаса MEL, в частности, ZSM-11, то есть по меньшей мере 5% масс. или по меньшей мере 10% масс. или по меньшей мере 20% масс., или по меньшей мере 30% масс., или по меньшей мере 40% масс., или по меньшей мере 50% масс., или по меньшей мере 60% масс., или по меньшей мере 70% масс., или по меньшей мере 80% масс., или по меньшей мере 90% масс. материала, имеющего каркас типа каркаса MEL. В некоторых воплощениях кристаллическая фаза кристаллического материала, полученного способом согласно настоящему изобретению, может включать по существу только материал, имеющий каркас типа каркаса MEL, в частности, ZSM-11.
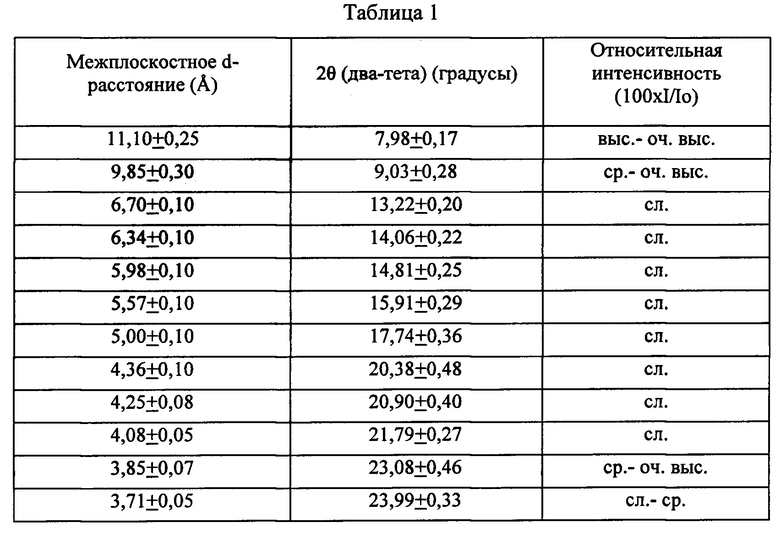
Синтезированный традиционным образом, например, в присутствии катионов тетрапропиламмония, как описано в патентах US 3702886 и RE 29948, типичный препарат ZSM-5 имеет дифрактограмму, включающую характеристические полосы, перечисленные ниже в Таблице 1:

Примечание:
оч. выс. - очень высокая интенсивность;
выс. - высокая интенсивность;
ср. - средняя интенсивность;
сл. - слабая интенсивность.
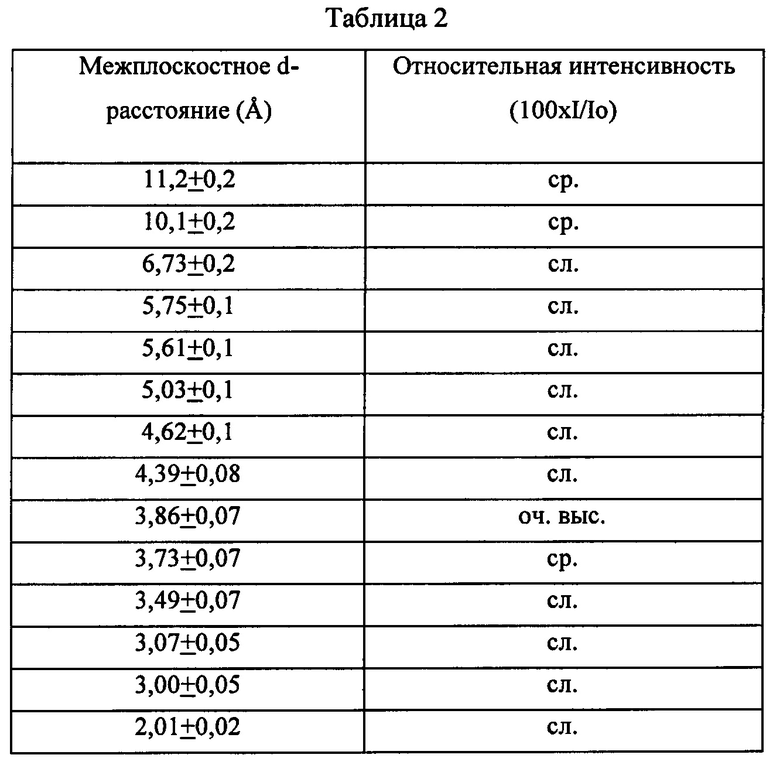
Синтезированный традиционным образом, например, в присутствии катионов тетрабутиламмония, как описано в патенте US 3709979, типичный препарат ZSM-11 имеет дифрактограмму, включающую характеристические полосы, перечисленные ниже в Таблице 2:
Кристаллический материал, полученный способом согласно изобретению, может иметь пики рентгеновской дифракции, связанные только с материалами ZSM-5 (MFI) и/или с ZSM-11 (MEL). В частности, кристаллический материал, полученный способом согласно изобретению, может, в том виде, в котором он синтезирован, иметь дифрактограмму, включающую максимумы характеристических пиков, перечисленные ниже в Таблице 3:
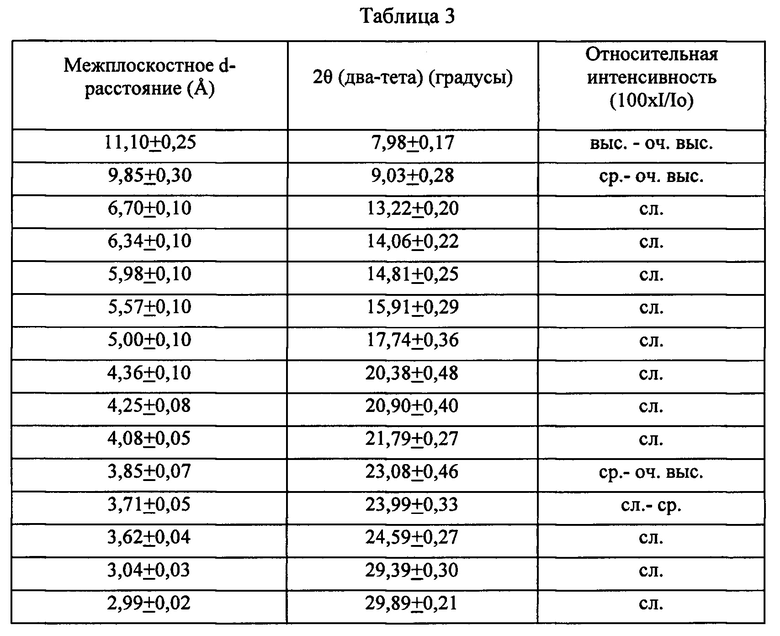
Приведенные данные рентгеновской дифракции были получены с помощью дифракционной системы Panalytical X'Pert Pro™ с многоканальным детектором Xcelerator™, снабженным детектором из твердотельного германия, с применением излучения K-альфа меди. Данные рентгеновской дифракции были записаны при ступенчатом сканировании с шагом ~0,02 градуса два-тета, где тета - угол Брэгга, с эффективным временем счета, составляющим ~2 секунды для каждого шага. Межплоскостные расстояния, т.е. d-расстояния, были рассчитаны в Ангстремах, и относительные интенсивности полос (линий), I/Io, представляют собой отношение интенсивности пика к интенсивности самой сильно выраженной полосы, возвышающейся над фоном. Коррекцию интенсивностей с учетом эффекта Лоренца и поляризационного эффекта не проводили. Относительные интенсивности имеют следующие обозначения: оч. выс. = очень высокая (75-100), выс. = высокая (50-74), ср. = средняя (25-49) и сл. = слабая (0-24). В некоторых воплощениях одна, несколько или все относительные интенсивности, которые, как указано, включают сл. (слабую), могут иметь отличные от нуля величины.
Известно, что некоторые полосы в дифрактограммах цеолитов могут иметь тенденцию к уширению по мере снижения соответствующего размера кристалла цеолита, в результате чего соседние полосы могут перекрываться и проявляться в виде лишь частично разрешенных пиков или неразрешенных уширенных пиков. В некоторых воплощениях кристаллических материалов, рассмотренных в настоящем описании, в особенности, кристаллических материалов с ультрамелкими кристаллами, описанных ниже, такое уширение полос может приводить к образованию в дифрактограмме только одного диффузного (размытого) составного элемента в области двух-тета, находящейся от приблизительно 21,5° до приблизительно 26° (диапазон d-расстояния от ~4,13 до ~3,42). В таких случаях максимум составного пика вблизи 24,0±0,30 градусов два-тета и максимум составного пика вблизи 24,6±0,30 градусов два-тета может проявляться либо в виде "плеча", либо может образовывать часть крупного размытого составного пика с максимумом вблизи 23,1 (±0,20) градуса двух-тета.
до ~3,42). В таких случаях максимум составного пика вблизи 24,0±0,30 градусов два-тета и максимум составного пика вблизи 24,6±0,30 градусов два-тета может проявляться либо в виде "плеча", либо может образовывать часть крупного размытого составного пика с максимумом вблизи 23,1 (±0,20) градуса двух-тета.
В порошковой дифрактограмме типичного (более крупные кристаллиты) образца ZSM-5 составной пик с максимумом вблизи 23,1 градуса два-тета и составной пик вблизи 24,0 градусов два-тета могут пересекаться, образуя ясно видимый локальный минимум [см. Фиг. 1(a)]. В таких типичных препаратах ZSM-5 отношение относительной интенсивности этого локального минимума (Imin) за вычетом фона к относительной интенсивности составного пика вблизи 24,0 градусов два-тета (Imax) за вычетом фона может составлять менее 0,40 как для полученного без дальнейшей обработки, так и для кальцинированного цеолитного продукта. В некоторых воплощениях кристаллического материала согласно изобретению, содержащего ультрамелкие кристаллы, локальный минимум может все же явно отличаться от составного пика вблизи 24,0 градусов двух-тета, даже если отношение Imin/Imax составляет по меньшей мере приблизительно 0,70. В некоторых воплощениях кристаллы могут быть настолько мелкими и пики настолько уширенными, что максимум пика вблизи 24,0 градусов двух-тета может проявляться либо как точка перегиба крупного размытого составного пика с максимумом вблизи 23,1 (±0,20) градуса двух-тета, либо локальный максимум или точка перегиба у составного пика вблизи 24,0 (±0,30) градусов двух-тета может отсутствовать. В этих крайних случаях отношение Imin/Imax может приближаться к 1,0.
Аналогично, в типичных препаратах ZSM-5 составной пик с максимумом вблизи 7,96 (±0,30) градусов двух-тета и составной пик с максимумом вблизи 8,90 (±0,30) градусов двух-тета могут пересекаться, образуя ясно видимый локальный минимум [см. Фиг. 1(b)], в котором отношение относительной интенсивности этого локального минимума (Imin) за вычетом фона к относительной интенсивности составного пика вблизи 8,90 градусов два-тета (Imax) за вычетом фона может составлять менее 0,20 как для полученного без дальнейшей обработки, так и для кальцинированной формы цеолита. Напротив, в кальцинированном варианте кристаллического материала согласно изобретению, содержащего ультрамелкие кристаллы, отношение Imin/Imax может составлять по меньшей мере 0,20 или более 0,20. В некоторых ситуациях (при наличии ультрамелких кристаллов согласно изобретению), отношение Imin/Imax может составлять по меньшей мере 0,40. Следует учитывать, что в тех случаях, когда имеется эффект предпочтительной ориентации, следует принять меры для снижения/минимизации влияния таких эффектов на дифрактограммы.
Большинство рассмотренных в настоящем описании синтезов приводит к получению кристаллических материалов каркасного типа MFI и/или MEL, содержащих мелкие кристаллы, такие, что общая площадь поверхности кристаллической фазы может составлять по меньшей мере приблизительно 500 м2/г, и площадь наружной поверхности может составлять по меньшей мере приблизительно 150 м2/г. Однако, в некоторых воплощениях ультрамелких кристаллов согласно настоящему изобретению кристаллические материалы, рассмотренные в настоящем описании, могут включать кристаллы с общей площадью поверхности, составляющей по меньшей мере приблизительно 750 м2/г, например по меньшей мере приблизительно 770 м2/г или по меньшей мере приблизительно 800 м2/г, например, от приблизительно 800 м2/г до приблизительно 1500 м2/г. Альтернативно или дополнительно содержащие ультрамелкие кристаллы кристаллические материалы, рассмотренные в настоящем описании, могут включать кристаллы, площадь наружной поверхности которых составляет по меньшей мере 350 м2/г, например по меньшей мере приблизительно 400 м2/г или по меньшей мере приблизительно 450 м2/г, например, от приблизительно 450 м2/г до приблизительно 1200 м2/г или от приблизительно 500 м2/г до приблизительно 1000 м2/г. Предположительно, эти значения превышают любые опубликованные ранее для кристаллических материалов каркасного типа MFI и MEL величины. Все приведенные в настоящем описании величины площади поверхности определены по данным физической адсорбции азота способом с построением t-графика. Подробное описание этого способа имеется в публикации Lippens, B.C., deBoer, J.H., "Studies on pore systems in catalysts (Исследования систем пор в катализаторах: V. Способ t)", J. Catal., 4, 319 (1965), содержание которой полностью включено в настоящее описание посредством ссылки.
Поскольку с помощью рассмотренных в настоящем описании синтезов могут быть получены кристаллы ультрамалых размеров (например, менее 50 микрон), любое измерение размер кристаллов/определение распределения размера кристаллов должно быть проведено (и проводилось, согласно изобретению) с применением трансмиссионного электронного микроскопа (ТЭМ) в трансмиссионном режиме. Измерения среднего размера кристаллов (диаметра) представляет собой усредненный размер (диаметр) по меньшей мере 100 различных кристаллов. Таким образом, в некоторых воплощениях ультрамелких кристаллов согласно настоящему изобретению кристаллические материалы, рассмотренные в настоящем описании, могут включать кристаллы, средние размеры (диаметры) которых составляют приблизительно 35 нм или менее (например, приблизительно 30 нм или менее, приблизительно 25 нм или менее, приблизительно 20 нм или менее, приблизительно 15 нм или менее, или приблизительно 10 нм или менее) и необязательно по меньшей мере приблизительно 3 нм (например, по меньшей мере приблизительно 4 нм, по меньшей мере приблизительно 5 нм, по меньшей мере приблизительно 6 нм, по меньшей мере приблизительно 7 нм, по меньшей мере приблизительно 8 нм, по меньшей мере приблизительно 9 нм, или по меньшей мере приблизительно 10 нм). Дополнительно или альтернативно кристаллические материалы, рассмотренные в настоящем описании, могут включать кристаллы, имеющие распределение средних размеров (диаметров), в котором по существу не имеется размеров (диаметров), превышающих приблизительно 50 нм (например, превышающих приблизительно 40 нм, превышающих приблизительно 35 нм, превышающих приблизительно 30 нм, превышающих приблизительно 25 нм, превышающих приблизительно 20 нм или превышающих приблизительно 15 нм). В этом контексте распределения размеров фраза "по существу не имеется" означает 3% или менее, например, 2% или менее, 1% или менее, 0,5% или менее или полное отсутствие.
Следует учитывать, что при анализе таких кристаллических материалов, содержащих ультрамелкие кристаллы, рентгеновской дифракции может быть недостаточно для установления того, имеет ли материал структуру MFI или MEL; в этом случае для подтверждения наличия структуры каркасного типа MFI и/или MEL в материале может возникнуть необходимость применения других аналитических способов, таких как трансмиссионная электронная микроскопия высокого разрешения и дифракция электронов.
Рассмотренные в настоящем описании кристаллические материалы каркасного типа MFI и/или MEL могут иметь состав, включающий молярное соотношение:
X2O3:(n)YO2,
где X представляет собой необязательный трехвалентный элемент, такой как бор, алюминий, железо и/или галлий, желательно по меньшей мере включающий алюминий, Y представляет собой четырехвалентный элемент, такой как кремний, германий, олово, титан и/или цирконий, желательно по меньшей мере включающий кремний, и n составляет по меньшей мере приблизительно 20, например по меньшей мере приблизительно 30 или по меньшей мере приблизительно 40. В тех воплощениях, в которых присутствует трехвалентный элемент, n может составлять приблизительно 2000 или менее, например, приблизительно 1000 или менее или приблизительно 500 или менее. В особенно предпочтительном воплощении n может составлять менее приблизительно 150, необязательно вместе с одним или более минимальными значениями n, рассмотренными выше.
В некоторых воплощениях способа согласно настоящему изобретению кристаллические материалы каркасного типа MFI и/или MEL, включающие формы кристаллов материала ультрамалого размера, могут быть получены в присутствии катионов тетрабутиламмония и/или катионов тетрабутилфосфония, применяемых в качестве структурообразующего агента (Q1). В таких воплощениях может быть получена смесь для синтеза, содержащая источник Q1 катионов, такой как гидроксид и/или галогенид (например, фторид, хлорид, бромид и/или йодид, в некоторых воплощениях, необязательно исключая фторид), наряду с источником оксида четырехвалентного элемента Y, необязательно источником трехвалентного элемента X, необязательно источником щелочного и/или щелочноземельного металла (М) и водой; таким образом, смесь для синтеза имеет молярный состав, включающий по меньшей мере одно и в некоторых воплощениях оба следующих соотношения: (а) молярное отношение H2O/YO2, составляющее менее приблизительно 10, например по меньшей мере приблизительно 2, такое как от приблизительно 2 до приблизительно 9; и (b) молярное отношение YO2/X2O3, составляющее менее приблизительно 150, например по меньшей мере приблизительно 20, такое как от приблизительно 30 до приблизительно 140.
В некоторых воплощениях смесь для синтеза может иметь молярное отношение M/YO2, составляющее от 0 до менее приблизительно 0,04, например от 0 до приблизительно 0,0125, от более 0 до менее приблизительно 0,04 или от более 0 до приблизительно 0,0125.
В частности, как показано ниже в Примерах, было обнаружено, что получению мелкокристаллических материалов с большой общей площадью и большой площадью наружной поверхности способствуют пониженное молярное отношение H2O/YO2 и/или пониженное молярное отношение M/YO2.
Кроме того, смесь для синтеза может иметь молярный состав, включающий следующие соотношения:

В других воплощениях способа согласно настоящему изобретению материалы каркасного типа MFI и/или MEL, включающие кристаллы ультрамалого размер таких материалов, могут быть получены в присутствии дикатионов 1,5-бис(N-трибутиламмоний)пентана, и/или дикатионов 1,6-бис(N-трибутиламмоний)гексана, применяемых в качестве структурообразующего агента (Q2). Предположительно, такие дикатионы являются новыми и имеют формулы (I) и (II), соответственно:
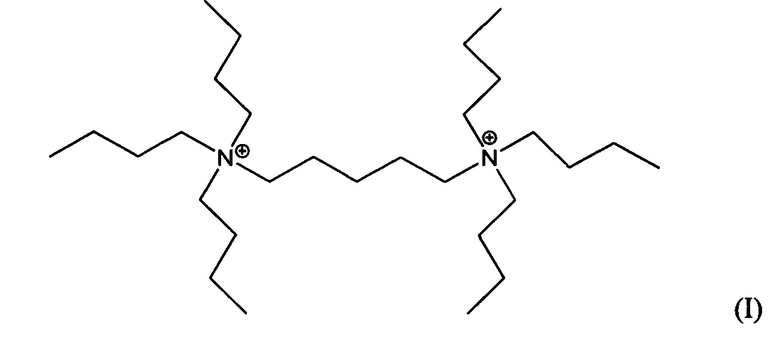
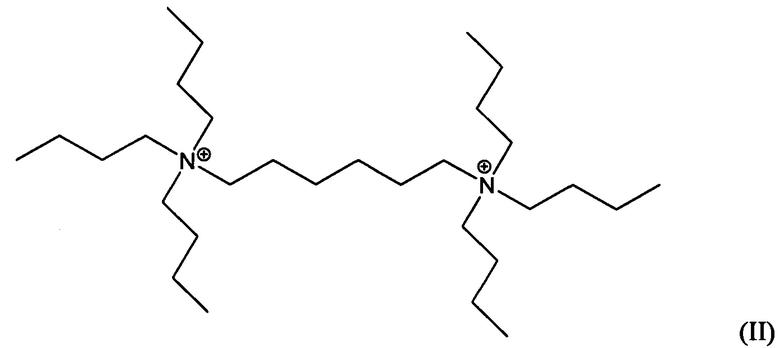
Материалы, имеющие формулы (I) и (II), могут быть легко получены по реакции трибутиламина с 1,5-дибромпентаном или 1,6-дибромгексаном.
В этих других воплощениях способа, в которых применяют Q2 в качестве структурообразующего агента, получают смесь для синтеза, содержащую источник катионов Q2, такой как гидроксид и/или галогенид (например, фторид, хлорид, бромид и/или йодид, в некоторых воплощениях необязательно за исключением фторида), наряду с источником оксида четырехвалентного элемента Y, необязательно источником трехвалентного элемента X, необязательно источником щелочного или щелочноземельного металла (М) и водой, в результате чего смесь для синтеза имеет молярный состав, включающий следующие соотношения:

Также неожиданно было обнаружено, что получению мелкокристаллических материалов с большой общей площадью и большой площадью наружной поверхности способствует пониженное молярное отношение H2O/YO2 и/или пониженное молярное отношение M/YO2. Таким образом, в некоторых воплощениях молярное отношение H2O/YO2 может составлять приблизительно 10 или менее, например, приблизительно 5 или менее. Дополнительно или в альтернативном варианте молярное отношение M/YO2 в смеси для синтеза может составлять приблизительно 0,02 или менее, например, приблизительно 0,015 или менее или приблизительно 0,0125 или менее.
Подходящие источники четырехвалентного элемента Y в смеси для синтеза, рассмотренной выше, могут зависеть от типа выбранного элемента Y, но в некоторых предпочтительных воплощениях, в которых Y включает/представляет собой кремний и/или германий, они могут включать коллоидные суспензии оксида кремния, пирогенные оксиды кремния, осажденные оксиды кремния, силикаты щелочных металлов, тетраалкилортосиликаты, оксид германия или подобные материалы или их комбинацию. Если трехвалентный элемент X присутствует в смеси для синтеза, то он обычно может включать алюминий, и неограничивающие примеры подходящих источников алюминия могут включать гидратированный оксид алюминия и/или водорастворимые соли алюминия, такие как нитрат алюминия. Комбинированные источники алюминия и кремния могут включать глины и/или обработанные глины, такие как метакаолин. Дополнительно или в альтернативном варианте могут быть применены другие комбинированные источники X и Y, включающие алюмосиликаты, такие как цеолит Y.
В некоторых воплощениях смесь для синтеза может включать затравочные кристаллы кристаллического материала, такого как ZSM-5, полученные в предыдущем синтезе, желательно в количестве, составляющем от приблизительно 0,01 масс. частей на миллион до приблизительно 10000 масс. частей на миллион, например, от приблизительно 100 масс. частей на миллион до приблизительно 5000 масс. частей на миллион, в пересчете на массу смеси для синтеза.
Кристаллизация кристаллических материалов каркасного типа MFI и/или MEL из рассмотренных выше смесей для синтеза может быть проведена в статических условиях, при встряхивании или при перемешивании в подходящей реакционной емкости, например, в полипропиленовых бочках или в автоклавах футерованных Тефлоном™ или автоклавах из нержавеющей стали, при температуре, составляющей от приблизительно 80°С до приблизительно 220°С, например, от приблизительно 100°С до приблизительно 150°С, в течение времени, достаточного для протекания кристаллизации при установленной температуре, например, от приблизительно 4 часов до приблизительно 28 суток, от приблизительно 12 часов до приблизительно 21 суток или от приблизительно 1 суток до приблизительно 14 суток. Затем кристаллы могут быть отделены от жидкости и извлечены.
В материале, в том виде, в котором он был синтезирован, любые катионы щелочных и/или щелочноземельных металлов могут быть заменены до требуемой степени и в зависимости от молярного отношения X2O3/YO2 в соответствии с методиками, хорошо известными в данной области техники, например, ионным обменом с другими катионами. Примеры заменяющих катионов могут включать ионы металлов, ионы водорода, предшественники ионов водорода (например, ионы аммония) или их смеси. В некоторых воплощениях особенно предпочтительные катионы могут включать катионы, обладающие каталитической активностью, которая может быть специально отрегулирована для проведения определенных реакций конверсии углеводородов. Они могут включать, без ограничений, водород, редкоземельные металлы, металлы Групп 2-15 Периодической Таблицы элементов и их комбинации. В настоящем изобретении применяют схему нумерации Групп Периодической Таблицы, приведенную в публикации Chemical and Engineering News, 63(5), 27 (1985).
В том виде, в котором он синтезирован, кристаллический материал дополнительно может быть подвергнут обработке для удаления всего или части органического структурообразующего агента (агентов) Q1/Q2, применяемого в его синтезе. Предпочтительно, это может быть выполнено/предпринято, например, с помощью термической обработки, в которой материал, в том виде, в котором он синтезирован, может быть нагрет до температуры, составляющей по меньшей мере приблизительно 370°С, в течение по меньшей мере 1 минуты и обычно не более чем в течение 20 часов. Несмотря на то, что для термической обработки может быть применено давление ниже атмосферного, из соображений удобства обычно желательно применение атмосферного давления и/или давления выше атмосферного. Термическая обработка может быть выполнена при температуре, составляющей до приблизительно 925°С. Альтернативно, органический структурообразующий агент (агенты) Q1/Q2 может быть удален обработкой озоном (см., например, публикацию Parikh с соавторами, Microporous and Mesoporous Materials (Микропористые и мезопористые материалы) 76 (2004) 17-22). Органический-разложенный/-свободный продукт, в особенности, в металлической, водородной и/или аммонийной формах, может быть особенно подходящим для катализа некоторых реакций органического синтеза (например, конверсии углеводородов).
Если требуется наличие функции гидрирования-дегидрирования, то кристаллический материал согласно изобретению может быть тщательно перемешан с гидрирующим компонентом, например, включающим молибден, рений, никель, кобальт, хром, марганец и/или благородный металл (такой как платина и/или палладий). Такой компонент может быть включен в композицию посредством сокристаллизации, введен в структуру композиции ионным обменом с элементом Группы IIIA (например, алюминием), внесен пропитыванием или тщательно смешан с композицией, образуя физическую смесь. Такой компонент может быть внесен пропитыванием в/на композицию, как, например, в случае платины, обработкой силиката раствором, содержащим ион, содержащий платину. Таким образом, подходящие для этой цели соединения платины могут включать хлорплатиновую кислоту, хлорид платины (II) и/или различные соединения, содержащие комплекс платины с амином.
При применении кристаллического материала согласно изобретению либо в качестве адсорбента, либо в качестве катализатора, он должен быть по меньшей мере частично дегидратирован. Это может быть осуществлено нагреванием до температуры, например, составляющей от 200°С до приблизительно 370°С, в такой атмосфере, так воздух, азот и т.д., при атмосферном давлении, давлении ниже атмосферного или давлении выше атмосферного, в течение достаточного времени, например, составляющего от 30 минут до 48 часов. Дополнительно или альтернативно дегидратация может быть проведена при комнатной температуре простым приложением вакуума к кристаллическому материалу, но для требуемой степени дегидратации может потребоваться более длительная обработка.
Кристаллические материалы, рассмотренные в настоящем описании, могут быть применены в качестве адсорбента или, в особенности, в алюмосиликатной форме, в качестве катализатора, ускоряющего один или более из разнообразных способов конверсии органических соединений, многие из которых в настоящее время имеют большое коммерческое/промышленное значение. Примеры способов конверсии химических веществ, которые могут эффективно катализироваться кристаллическими материалами согласно изобретению, предпочтительно могут включать способы, в которых важно наличие относительно высокой кислотной активности и большой площади поверхности.
Как и в случаях многих других катализаторов, может быть желательно включать в кристаллические материалы согласно изобретению другой материал, устойчивый к температурному воздействию и другим условиям, создаваемым в способах органического синтеза. Такие материалы могут включать активные и неактивные материалы и синтетические или натуральные цеолиты, а также неорганические материалы, такие как глины, оксид кремния и/или оксиды других металлов, такие как оксид алюминия. Последний может быть природным или в виде гелеобразных осадков или гелей, включающих смеси оксида кремния и оксиды других металлов. Применение материала в сочетании с кристаллическими материалами согласно изобретению (т.е. в смешанном с ними виде или присутствующими во время синтеза нового кристалла), которые находятся в активной форме (формах), может изменять степень конверсии и/или селективность катализатора в некоторых способах органического синтеза. Неактивные материалы могут подходящим образом служить разбавителями, которые позволяют регулировать степень конверсии в заданном способе, чтобы продукты могли быть получены экономически эффективным и упорядоченным образом без применения других средств регулирования скорости реакции. Эти материалы могут быть включены в природные глины, например, бентонит и каолин, для повышения прочности катализатора на раздавливание в условиях коммерческого функционирования. Указанные материалы (т.е. глины, оксиды и т.д.) могут функционировать как связующие вещества для катализатора. Может быть желательным получение катализатора, имеющего высокую прочность на раздавливание, поскольку при коммерческом применении может быть особенно желательным предотвращение истирания катализатора с образованием порошкообразных материалов. Таким образом, связующие вещества на основе глины и/или оксида обычно применяют с целью повышения прочности катализатора на раздавливание.
Природные глины, которые могут образовывать композитные материалы с кристаллическими материалами согласно изобретению, могут включать семейства монтмориллонита и каолина, и эти семейства включают суббентониты и каолины, общеизвестные под наименованиями глин Dixie, McNamee, Georgia и Florida, или другие материалы, в которых основным минеральным составляющим является галлуазит, каолинит, диккит, накрит или анауксит. Такие глины могут быть использованы в необработанном состоянии, в котором они быть изначально добыты, или сначала подвергнуты кальцинации, кислотной обработке или химической модификации. Другие подходящие связующие вещества могут включать неорганические оксиды, такие как оксид кремния, оксид циркония, оксид титана, оксид магния, оксид бериллия, оксид алюминия и их смеси.
Кроме перечисленного выше, кристаллические материалы согласно изобретению могут быть соединены в виде композитного материала с материалом, имеющим пористую матрицу, таким как диоксид кремния-оксид алюминия, диоксид кремния-оксид магния, диоксид кремния-оксид циркония, диоксид кремния-оксид тория, диоксид кремния-оксид бериллия, диоксид кремния-оксид титана, или с тройными композициями, такими как диоксид кремния-оксид алюминия-оксид тория, диоксид кремния-оксид алюминия-оксид циркония диоксид кремния-оксид алюминия-оксид магния и диоксид кремния-оксид магния-оксид циркония.
Относительные пропорции кристаллического материала каркасного типа MFI и/или MEL и матрицы из неорганического оксида могут изменяться в широких пределах, с содержанием сит (англ. sieve content), составляющим от приблизительно 1% масс. до приблизительно 90% масс., и обычно, в особенности, если композитный материал получают в виде гранул, составляющим от приблизительно 2% масс. до приблизительно 80% масс. от массы композитного материала.
Дополнительно или альтернативно изобретение может включать одно или более из приведенных ниже воплощений.
Воплощение 1. Способ получения кристаллического материала, имеющего тип каркаса MFI и/или MEL, где способ включает: (i) приготовление смеси для синтеза, подходящей для образования кристаллического материала, где смесь включает источник оксида четырехвалентного элемента Y, необязательно источник трехвалентного элемента X, необязательно источник щелочного и/или щелочноземельного металла (М), воду и структурообразующий агент (Q1), включающий катионы тетрабутиламмония и/или катионы тетрабутилфосфония, причем смесь для синтеза имеет по меньшей мере одно из следующих молярных отношений: (а) молярное отношение H2O/YO2, составляющее приблизительно 10 или менее (например, приблизительно 5 или менее); и (b) молярное отношение YO2/X2O3, составляющее менее приблизительно 150; (ii) нагревание смеси в условиях кристаллизации, включающих температуру, составляющую от приблизительно 80°С до приблизительно 220°С, и время, составляющее от приблизительно 4 часов до приблизительно 28 суток, до образования кристаллов кристаллического материала; и (iii) извлечение кристаллического материала, полученного в этапе (ii).
Воплощение 2. Способ получения кристаллического материала, имеющего тип каркаса MFI и/или MEL, где способ включает: (i) приготовление смеси для синтеза, подходящей для образования указанного кристаллического материала, где указанная смесь включает источник оксида четырехвалентного элемента Y, необязательно источник трехвалентного элемента X, необязательно источник щелочного и/или щелочноземельного металла (М), воду и структурообразующий агент (Q2), включающий дикатионы 1,5-бис(N-трибутиламмоний)пентана и/или дикатионы 1,6-бис(N-трибутиламмоний)гексана; (ii) нагревание смеси в условиях кристаллизации, которые включают температуру, составляющую от приблизительно 80°С до приблизительно 220°С, и время, составляющее от приблизительно 4 часов до приблизительно 28 суток, до образования кристаллов кристаллического материала; и (iii) извлечение кристаллического материала, полученного в этапе (ii).
Воплощение 3. Способ воплощения 1 или воплощения 2, в котором X включает один или более из В, Al, Fe, и Ga, и Y включает один или более из Si, Ge, Sn, Ti, и Zr.
Воплощение 4. Способ любого из предшествующих воплощений, в котором X включает алюминий, и Y включает кремний.
Воплощение 5. Способ любого из предшествующих воплощений, в котором смесь для синтеза имеет молярное отношение YO2/X2O3, составляющее по меньшей мере приблизительно 20.
Воплощение 6. Способ любого из предшествующих воплощений, в котором молярное отношение H2O/YO2 в смеси для синтеза составляет по меньшей мере приблизительно 2, например, от приблизительно 2 до приблизительно 9.
Воплощение 7. Способ любого из предшествующих воплощений, в котором молярное отношение M/YO2 в смеси для синтеза составляет приблизительно 0,04 или менее, например, приблизительно 0,02 или менее или приблизительно 0,0125 или менее.
Воплощение 8. Способ любого из предшествующих воплощений, в котором условия кристаллизации включают температуру, составляющую от приблизительно 100°С до 150°С.
Воплощение 9. Способ любого из предшествующих воплощений, в котором кристаллический материал, извлеченный в этапе (iii) имеет общую площадь поверхности (определяемую способом физической адсорбции азота с построением t-графика), составляющую по меньшей мере приблизительно 750 м2/г, и/или площадь наружной поверхности (определяемую способом физической адсорбции азота с построением t-графика), составляющую по меньшей мере приблизительно 350 м2/г.
Воплощение 10. Способ любого из воплощений 2-9, в котором смесь для синтеза имеет состав, в котором молярные отношения находятся в следующих диапазонах: YO2/X2O3 составляет по меньшей мере приблизительно 20; H2O/YO2 составляет от приблизительно 2 до приблизительно 60 (например, приблизительно 10 или менее или приблизительно 5 или менее); OH-/YO2 составляет от приблизительно 0,1 до приблизительно 0,8; M/YO2 составляет от 0 до приблизительно 0,4; и Q2/YO2 составляет от приблизительно 0,05 до приблизительно 0,4.
Воплощение 11. Способ любого из предшествующих воплощений, в котором кристаллический материал, извлеченный в этапе (iii) имеет: (а) средний размер (диаметр), составляющий приблизительно 35 нм или менее (например, приблизительно 30 нм или менее, приблизительно 25 нм или менее, приблизительно 20 нм или менее, приблизительно 15 нм или менее, или приблизительно 10 нм или менее) и необязательно по меньшей мере приблизительно 3 нм (например, по меньшей мере приблизительно 4 нм, по меньшей мере приблизительно 5 нм, по меньшей мере приблизительно 6 нм, по меньшей мере приблизительно 7 нм, по меньшей мере приблизительно 8 нм, по меньшей мере приблизительно 9 нм, или по меньшей мере приблизительно 10 нм); и/или (b) распределение среднего размера (диаметра), в котором по существу отсутствуют размеры (диаметры), превышающие приблизительно 50 нм (например, превышающие приблизительно 40 нм, превышающие приблизительно 35 нм, превышающие приблизительно 30 нм, превышающие приблизительно 25 нм, превышающие приблизительно 20 нм или превышающие приблизительно 15 нм).
Воплощение 12. Кристаллический материал, имеющий тип каркаса MFI и/или MEL, полученный способом одного или более предшествующих воплощений.
Воплощение 13. Кристаллический материал, такой как материал, имеющий тип каркаса MFI и/или MEL, имеющий внутреннюю пористую структуру дикатионов 1,5-бис(N-трибутиламмония)пентана и/или дикатионов 1,6-бис(N-трибутиламмоний)гексана.
Воплощение 14. Кристаллический материал, имеющий тип каркаса MFI и/или MEL и химический состав, отличающийся молярным соотношением (n)YO2:X2O3, в котором n представляет собой число, составляющее по меньшей мере приблизительно 20 и менее приблизительно 2000, X представляет собой трехвалентный элемент, и Y представляет собой четырехвалентный элемент, и при этом кристаллический материал включает кристаллы, общая площадь поверхности которых (определяемая способом физической адсорбции азота с построением t-графика) превышает приблизительно 770 м2/г, и/или площадь наружной поверхности (определяемая способом физической адсорбции азота с построением t-графика) превышает приблизительно 400 м2/г.
Воплощение 15. Органическое азотсодержащее соединение, включающее дикатион, имеющий формулу (I) или (II):
ПРИМЕРЫ
Ниже изобретение более подробно рассмотрено с помощью приведенных Примеров и сопроводительных фигур.
Пример 1
Проводили синтез MFI, аналогичный синтезу, описанному Zhang с соавт. (Science, 336 (2012), 1684-1687, обсуждаемый выше), с применением в качестве структурообразующего агента (СОА) гидроксида тетрабутилфосфония (ТВРОН) при следующих молярных соотношениях состава геля: ~0,30 ТВРОН : ~0,0125 NaOH : ~0,005 Al2O3 : ~1 SiO2 : ~10 Н2О : ~4 EtOH. Присутствие этанола в этом синтезе обусловлено применением в качестве источника оксида кремния тетраэтилортосиликата (сокращенно "ТЭОС") - после гидролиза в воде каждая молекула ТЭОС может создавать приблизительно четыре молекулы этанола. Источником алюминия был сульфат алюминия. Спустя ~4 суток нагревания до ~115°С при встряхивании, в результате этого синтеза получили продукт с очень большой общей площадью поверхности, составляющей ~620 м2/г, и площадью наружной поверхности, составляющей ~317 м2/г. Полученные большие значения площадей поверхности согласуются со значениями, ожидаемыми для материалов, содержащих очень мелкие кристаллы.
Порошковая дифрактограмма продукта Примера 1 в том виде, в котором он синтезирован, представлена на Фиг. 2, а полученные с помощью СЭМ изображения продукта представлены на Фиг. 3. При увеличении, обеспечиваемом СЭМ, фасетчатые области не просматриваются. На Фиг. 4 представлены полученные с помощью ТЭМВР изображения того же материала. Несколько изображений были получены в различных участках продукта, и на них также не наблюдается аморфных областей. В отличие от продуктов, описанных Zhang с соавт., продукты согласно изобретению, по-видимому, не имеют упорядоченной "самособирающейся в стержни" структуры. Однако, продукты включают множество мелких кристаллов, которые перекрываются и расположены в различных ориентациях относительно друг друга. Как видно, толщины различных кристаллов неодинаковы. В меньшинстве случаев можно различить кристаллы, которые имеют толщину приблизительно ~1-2 элементарных ячейки - они отмечены прозрачными рамками. Бахромки внутри этих кристаллов, по-видимому, имеют характеристики материалов MFI и/или MEL. В середине изображения видны поры каркасного типа MFI - расстояние между порами соответствует расстояниям в других материалах MFI и/или MEL.
Пример 2
Повторяли синтез Примера 1, применяя вместо ТЭОС осажденный диоксид кремния Ultrasil™ и гидроксид тетрабутиламмония (ТВАОН) в качестве структурообразующего агента (СОА). Молярный состав геля был следующим ~0,30 ТВАОН : ~0,010 NaOH : ~0,005 A12O3 : ~1 SiO2 : ~10 H2O. В этом синтезе этанол не добавляли. В этом случае оказалось, что синтез завершился после нагревания до ~115°С в течение ~2 суток. Не прибегая к какой-либо теории, можно предположить, что, несмотря на то, что в этом синтезе применяли другой СОА и другой источник диоксида кремния, более короткая продолжительность кристаллизации может объясняться отсутствием этанола, а не другой природой источников диоксида кремния. Общая площадь поверхности этого материала составила ~760 м2/г, и площадь наружной поверхности составила ~370 м2/г. Общая площадь поверхности заметно превышала величину 710 м2/г, полученную для однослойного препарата Ryoo с соавт. (Nature 461, 246-249, рассмотрено в настоящем описании), и полагают, что это одно из самых больших значений площади поверхности, опубликованных для материала типа MFI.
На Фиг. 2 представлено сравнение порошковых дифрактограмм продуктов, полученных в Примерах 1 и 2, каждая из которых, как видно, содержит уширенные полосы, характерные для материалов, содержащих очень мелкие кристаллы. Как показано на Фиг. 2, после кальцинации продукта Примера 2 происходит повышение относительных интенсивностей малоугловых элементов, часто наблюдаемых в дифрактограммах микропористых материалов. Одним из очевидных последствий малых размеров кристаллов может быть то, что многие отчетливые пики "нормального" ZSM-5 проявляются в виде "плеч" или образуют широкие выступы с соседними пиками. Следует отметить, что впадина между первыми малоугловыми пиками имеет интенсивность, составляющую более половины интенсивности пика вблизи 8,9° два-тета.
Пример 3
Повторяли синтез Примера 1 во встряхиваемых автоклавах емкостью ~23 мл и ~45 мл, применяя в качестве источника диоксида кремния ТЭОС, а в качестве источника алюминия - изопропилат алюминия. Молярный состав геля был следующим ~0,30 ТВРОН : ~0,0125 NaOH : ~0,0125 A12O3 : ~1 SiO2: ~10 H2O : ~4 EtOH. Поскольку для препаратов ZSM-5, содержащих большее количество алюминия, обычно требуется большее время, чем для препаратов с более высокими отношениями Si/Al, способ выполняли при более высокой температуре, т.е. при ~160°С. После нагревания в течение ~8 суток и последующей кальцинации при ~600°С, был получен продукт с общей площадью поверхности, определенной способом BET, составляющей ~640 м2/г, и площадью наружной поверхности, составляющей ~381 м2/г. При анализе некальцинированного продукта было получено то же значение площади наружной поверхности, но объем микропор измерению не поддавался (по-видимому, из-за того, что внутри микропор оставался СОА).
Пример 4
Синтез Примера 3 повторяли в более крупном масштабе в автоклаве с верхним перемешиванием емкостью ~300 мл. После нагревания в течение ~8 суток, был получен продукт с общей площадью по BET, составляющей всего ~419 м2/г, и площадью наружной поверхности, составляющей ~44 м2/г. Такие величины больше подходят для типичных образцов ZSM-5. Элементы в порошковой дифрактограмме этого материала имели более заостренные формы, чем элементы в дифрактограмме материала, полученного во встряхиваемых реакторах. Не прибегая к какой-либо теории, следует указать на то, что, несмотря на отсутствие в настоящий момент внятного объяснения изменения размера кристаллов, площади поверхностей продуктов, получаемых в системах с верхним перемешиванием, часто оказываются меньше, чем площади поверхностей продуктов, получаемых в системах со встряхиванием.
Примеры 5 и 6
Синтезы Примеров 3 и 4 повторяли, применяя сульфат алюминия и Ultrasil™ в качестве источников алюминия и диоксида кремния, соответственно, при тех же остальных условиях. После нагревания в течение ~6 суток при встряхивании, был получен продукт с общей площадью, составляющей ~568 м2/г, и площадью наружной поверхности, составляющей ~191 м2/г. При повторении синтеза в автоклаве с верхним перемешиванием емкостью ~300 мл, общая площадь и площадь наружной поверхности уменьшались до ~510 м2/г и ~129 м2/г (510/129), соответственно. В этом случае оказалось, что в системе с верхним перемешиванием получают продукт с меньшей площадью поверхности, но различия не настолько существенны, как в системах Примеров 3 и 4, в которых применяли ТЭОС и изопропилат алюминия.
Пример 7
Была выполнена серия экспериментов при различных температурах и молярном отношении SiO2/Al2O3, составляющем ~100, в системе сульфата алюминия/Ultrasil™. Каждый синтез проводили в автоклаве с верхним перемешиванием емкостью ~300 мл, применяя ТВРОН в качестве СОА, при молярном отношении Н2О/SiO2, составляющем ~10. После нагревания в течение ~4 суток при ~160°С, порошковая дифрактограмма продукта включала относительно заостренные элементы. При повторении синтеза при ~150°С в течение ~3 суток, продукт имел общую площадь/площадь наружной поверхности, составляющие ~503/~103 м2/г. При ~140°С в течение ~3 суток, общая площадь/площадь наружной поверхности повышалась до ~538/~169 м2/г. При ~125°С в течение ~3 суток, соответствующие величины составляли ~534/~196 м2/г. Таким образом, оказалось, что снижение температуры с ~140°С до ~125°С не приводит к большим изменениям общих площадей поверхности, определяемых способом BET, но вклад площади наружной поверхности увеличивается. На Фиг. 5 представлено уширение элементов порошковых дифрактограмм продуктов, получаемых по мере понижения температуры синтеза.
Пример 8
Была проведена серия экспериментов, аналогичных Примеру 7, в которых композиция геля имела молярное отношение SiO2/Al2O3, составляющее ~50. В результате первого эксперимента, проводимого в автоклаве емкостью ~300 мл при ~160°С в течение ~6 суток, был получен продукт с общей площадью/площадью наружной поверхности, составляющими ~514/~155 м2/г. При повторении эксперимента с нагреванием лишь в течение ~4 суток, величины площадей поверхности повышались до ~551/~192 м2/г. При нагревании до ~150°С во встряхиваемом автоклаве емкостью ~23 мл в течение ~5 суток эти величины дополнительно повышались до ~592/~228 м2/г. Другой синтез был выполнен в автоклаве с верхним перемешиванием емкостью ~300 мл при поэтапном нагревании, согласно которому гель нагревали до ~140°С в течение ~2 суток и затем до ~150°С в течение еще ~3 суток. В этом случае общая площадь/площадь наружной поверхности составляли ~548/~205 м2/г, что приблизительно совпадало с этими параметрами продукта, полученного в результате нагревания в течение ~4 суток при ~160°С.
Пример 9
При повторении экспериментов Примера 8 во встряхиваемых реакторах емкостью ~125 мл наблюдали выраженную тенденцию изменения наблюдаемых значений общей площади/площади наружной поверхности: при ~140°С в течение ~4 суток полученные площади поверхности составляли ~496/~130 м2/г, при ~130°С в течение ~4 суток полученные площади поверхности составляли ~538/~206 м2/г, и при ~120°С в течение ~6 суток полученные площади поверхности составляли ~559/~298 м2/г.
Пример 10
Вновь повторяли эксперименты Примера 8, но при пониженных молярных отношениях SiO2/Al2O3, составляющих ~40 и ~30. В эксперименте с молярным отношением SiO2/Al2O3, составляющем ~40, применяли ТЭОС и изопропилат алюминия. В эксперименте с молярным отношением SiO2/Al2O3, составляющем ~30, применяли Ultrasil™ и сульфат алюминия. После нагревания в течение по меньшей мере ~40 суток при ~170°С и ~160°С, соответственно, в обоих экспериментах получили только аморфные фазы. Было отмечено, что при более низких отношениях Si/Al в геле и в любом получаемом продукте для улучшения кристаллизации обычно возникает необходимость применения более высокой концентрации натрия. Молярное отношение Na/SiO2 в гелях в этих экспериментах оставалось низким и составляло ~0,0125.
При повторении экспериментов с гелем, в котором молярное отношение SiO2/Al2O3 составляло ~32, и с повышенным молярным отношением Na/SiO2, составляющим ~0,05, были получены относительно выраженные, полностью кристаллические продукты ZSM-5 после нагревания до ~160°С и ~140°С лишь в течение ~3 суток. Даже при ~125°С был получен относительно выраженный продукт ZSM-5. Введение в систему дополнительного количества натрия может привести к образованию продукта с более высоким содержанием алюминия, но повышенное содержание натрия также может способствовать образованию более крупных кристаллитов. Соответственно, если в системе желательно получение очень мелких кристаллов, то для предотвращения образования более крупных кристаллов может быть полезным снижение и/или минимизация концентрации натрия.
Пример 11
Была проведена серия экспериментов, в масштабе ~23 мл и в условиях повышенной концентрации (отношение Н2О/Si ≈ 3), в которой в качестве СОА применяли ТВАОН. Для достижения малых отношений Н2О/Si, из ~40%-ного раствора ТВАОН с помощью роторного испарителя при ~70°С отгоняли воду, получая ~70%-ный раствор ТВАОН (~2,7 ммоль/г). Обычно концентрирование раствора СОА при повышенных температурах нежелательно, поскольку это может приводить к отщеплению по Гофману. В этом случае, несмотря на изменение цвета геля после удаления воды, по данным ЯМР разложения геля не происходило. Синтез цеолита при молярном отношении SiO2/Al2O3 в геле, составляющем ~50, при ~150°С в течение ~5 суток привел к получению продукта с общей площадью/площадью наружной поверхности, составляющими ~523/~237 м2/г. Аналогичный синтез при молярном отношении SiO2/Al2O3, составляющем ~64, при ~140°С в течение ~4 суток к получению продукта с общей площадью/площадью наружной поверхности, составляющими ~533/~219 м2/г.
Пример 12
Была проведена серия экспериментов, в которой в качестве СОА применяли гидроксид 1,5-бис(N-трибутил-аммония)пентана. Это С5-дичетвертичное соединение ("дикват") было получено добавлением ~17,74 г трибутиламина к ~60 мл ацетонитрила с последующим добавлением ~10,0 г 1,5-дибромпентана. Затем смесь нагревали в течение ~4 суток при ~80°С в запаянном контейнере из Тефлона™. Затем продукт извлекали испарением растворителя на роторном испарителе, промывкой маслом с простым эфиром, декантацией простого эфира и последующим испарением на роторном испарителе в вакууме (при пониженном давлении) при ~80°С до появления твердых веществ. Затем твердые вещества суспендировали в смеси ацетон/эфир, извлекали фильтрованием и оставляли сушиться. Чистоту продукта определяли с помощью анализов 1H и 13С ЯМР. Затем твердое вещество превращали в гидроксидную форму ионным обменом с ионообменной смолой Dowex™.
Полученный раствор СОА концентрировали испарением на роторном испарителе до приблизительно 60% масс. и затем использовали для приготовления геля, имеющего следующий молярный состав: ~0,30 С5-Дикват : ~0,0125 NaOH : ~0,02 Al2O3 : ~1 SiO2 : ~3,5 Н2О. После нагревания в течение ~9 суток при ~160°С гель превращался в продукт с общей площадью/площадью наружной поверхности, составляющими ~548/~203 м2/г. При повторении эксперимента при отношении Н2О/SiO2, повышенном до ~10, продукт, полученный после нагревания в течение ~9 суток при ~160°С, имел общую площадь/площадь наружной поверхности, составляющие ~589/~222 м2/г. В другом эксперименте другую часть геля, в которой отношение H2O/SiO2 составляло ~10, нагревали при ~150°С в течение ~21 суток, получая продукт с общей площадью/площадью наружной поверхности, составляющими ~539/~205 м2/г.
В некоторых экспериментах с применением СОА на основе С5-диквата требовалось больше времени и/или более высокие температуры для получения аналогичных по структуре продуктов, чем в экспериментах с применением СОА на основе ТВАОН.
Пример 13
Была проведена серия экспериментов аналогичных экспериментам Примера 12, но в качестве СОА применяли гидроксид 1,6-бис(N-трибутиламмоний)пентана. Этот С6-дикват был получен аналогично С5-диквату, но с использованием 1,6-дибромгексана вместо 1,5-дибромпентана. В этом случае полученный раствор СОА перед применением в реакциях синтеза, описанных ниже, также концентрировали испарением на роторном испарителе до приблизительно 60% масс.
Первые синтезы с применением С6-диквата проводили при ~150°С в течение ~7 суток. При отношении Н2О/SiO2, составляющем ~10, продукт имел общую площадь/площадь наружной поверхности, составляющие ~512/~241 м2/г, а при отношении Н2О/SiO2, составляющем ~3,5, продукт имел общую площадь/площадь наружной поверхности, составляющие ~577/~269 м2/г. при повторении синтеза с отношением Н2О/SiO2, составляющем ~3,5, при ~135°С в течение ~8 суток, общая площадь/площадь наружной поверхности составили ~548/~269 м2/г. Затем синтезы повторяли с отношением H2O/SiO2, составляющем ~3,5, при ~125°С и ~115°С, применяя затравочные кристаллы. После нагревания в течение ~4 суток при ~125°С измеренные общая площадь/площадь наружной поверхности продукта составили ~575/~289 м2/г, и после нагревания в течение ~4 суток при ~115°С измеренные общая площадь/площадь наружной поверхности продукта составили ~594/~346 м2/г.
Не прибегая к какой-либо теории, можно предположить, что значительное снижение продолжительности кристаллизации при использовании С6-диквата по сравнению с С5-дикватом говорит о том, что расстояния между N-центрами в С6-диквате могут удовлетворительно соответствовать расстояниям между пересечениями структуры каркасного типа в продукте. Кроме того, как видно, несмотря на большую скорость кристаллизации, сохраняется способность образовывать материалы, содержащие мелкие кристаллы.
Пример 14
Продукты, получаемые в высококонцентрированных средах, обычно извлекают центрифугированием с последующей промывкой, поскольку частицы обычно фильтруют через стеклянные фильтры. Таким образом, был проведен эксперимент для того, чтобы установить возможность устранения этапов центрифугирования/промывки, выполняя простую сушку кристаллизованного геля и последующую кальцинацию всего продукта (для удаления СОА). Такой упрощенный способ конечной обработки может быть предпочтительным для крупномасштабного получения. Одной из потенциальных проблем такого рационализированного способа является то, что избыток щелочных катионов может разрушать цеолит во время кальцинации, если они функционируют как частицы основного характера. Однако, маловероятно, что такое может произойти в синтезах согласно изобретению, которые проводят при относительно низких отношениях натрий/Si. Другой потенциальных проблем такого рационализированного способа является то, что в большинстве систем существенное количество диоксида кремния обычно остается растворенным в растворе - кальцинация такого высушенного продукта с большой вероятностью приводит к образованию аморфного материала или к образованию других нежелательных фаз с высоким содержанием диоксида кремния/разбавителей. Однако, весьма маловероятно, что такое может произойти в синтезах согласно изобретению, поскольку в системах с относительно низким отношением вода/оксид кремния имеется небольшой избыток воды, и большая часть этого избытка воды обычно связана с поверхностями очень мелких продуктов-кристаллитов, в результате чего выходы в таких концентрированных системах относительно высоки (близки к 100% или по меньшей мере >90%).
Таким образом, часть продукта Примера 13, полученного при ~135°С в течение ~8 суток, использовали в том виде, в котором он синтезирован, без промывки продукта. Рентгенодифракционный спектр этого продукта представлен в нижней части Фиг. 6. Непромытый продукт затем кальцинировали при ~600°С и снова анализировали рентгенодифракционным способом. Полученная дифрактограмма представлена в верхней части Фиг. 5, и на ней не отмечено значительного перехода продукта в аморфное состояние. На Фиг. 7 представлен 27Al ЯМР спектр до и после кальцинации продукта при ~600°С для удаления СОА. До кальцинации весь алюминий может находиться в тетраэдральном окружении. Неожиданно было обнаружено, что после кальцинации, ионного обмера и последующей повторной кальцинации лишь приблизительно 3-4% алюминия перешли в октаэдральное состояние. Напротив, в типичных ZSM-5 материалах по меньшей мере 10% алюминия после кальцинации переходит в октаэдральное состояние.
Пример 15
Повторяли способ Примера 13 с применением С6-диквата при молярном отношении SiO2/Al2O3 в геле, составляющем ~60 и молярном отношении Н2О/SiO2, составляющем ~3,5, без добавления натрия и в присутствии ~5% масс. затравочных кристаллов ZSM-5. После нагревания в течение приблизительно пяти суток при ~135°С был получен кристаллический продукт с общей площадью/площадью наружной поверхности, составляющими ~623/~337 м2/г. Поскольку в этом синтезе не применяли натрий, продукт можно кальцинировать непосредственно, и при этом не требуется предварительного или последующего ионного обмена для превращения цеолита в полностью кислотную форму.
Примеры 16 и 17
Повторяли способ Примера 15 с молярными отношениями SiO2/Al2O3, составляющими ~80 (Пример 16) и ~150 (Пример 17), при температуре, составляющей ~100°С. После нагревания в течение ~16 и ~12 суток, соответственно, были получены продукты с общей площадью/площадью наружной поверхности, составляющими ~861/~550 м2/г и ~757/~409 м2/г. Как оказалось, снижение температуры приводило к сильному увеличению площади поверхности продукта, но для завершения кристаллизации требовалось больше времени. На Фиг. 8 представлены порошковые дифрактограммы продуктов Примера 16, полученные после различных периодов нагревания. При нагревании от ~10 до ~16 суток появляется лишь небольшое заострение среднеугольных элементов, указывающее на то, что синтез может завершаться за ~10 суток. На Фиг. 9 представлено сравнение дифрактограмм продуктов в том виде, в котором они были получены, и кальцинированных продуктов Примера 16, получаемых после нагревания в течение ~16 суток при ~100°С. На Фиг. 10 представлены зарегистрированные с помощью ТЭМ изображения кальцинированного продукта Примера 16, полученного после нагревания в течение ~16 суток при ~100°С, и на Фиг. 11 представлена гистограмма распределения размеров кристаллов. Поскольку предполагается, что кристаллы находятся в виде тонких пластинок, представленные распределения могут показывать только кристаллы больших размеров (диаметров). В распределении, показанном на Фиг. 11, представлены измерения размеров кристаллов, сделанные с помощью ТЭМ в трансмиссионном режиме. Измерения проводили в случайных областях каждой сетки/держателя ТЭМ, содержащего образец. Количество случайных областей может быть различным в зависимости от количества различаемых частиц в любой заданной области, например, по меньшей мере 100 частиц могут быть определены во всех областях каждого образца. Во избежание пристрастности, лучше всего исследовать по меньшей мере 3 случайные области. Для определения среднего размера (диаметра) каждой различимой частицы, для каждой визуально наблюдаемой частицы устанавливали подходящее увеличение. Для оценки "среднего" размера (диаметра) кристалла брали среднее распределение размеров всех исследованных частиц.
Примеры 18 и 19
Результаты Примеров 15-17 показывают, что даже относительно небольшие концентрации натрия могут оказывать сильное влияние на продолжительность кристаллизации и на морфологию конечного продукта. Для исследования возможного влияния аниона, связанного с источником алюминия, были проведены два одинаковых синтеза, за исключением того, что в одном из них в качестве источника алюминия применяли хлорид алюминия (Пример 18), а в другом применяли нитрат алюминия (Пример 19). В каждом случае в гель для синтеза вводили С6-дикват Примера 13, и гель имел следующий молярный состав: ~1 SiO2 : ~0,04 Al+3 : ~3,5 H2O : ~0,15 СОА(ОН)2 : ~0,0125 NaOH. Каждый синтез проводили с новой футеровкой из Тефлона™ во избежание возможного влияния различных концентраций натрия, имеющихся в использованных футеровках (которые обычно после каждого использования очищают в растворе гидроксида натрия). Материалы для обоих синтезов помещали в одну и ту же печь и обрабатывать одинаковое время при встряхивании, и затем извлекали после нагревания в течение одинакового периода времени (~4 суток при ~150°С). На Фиг. 12 представлено сравнение порошковых рентгенодифрактограмм продуктов. Спектр продукта, полученного в синтезе с хлоридом алюминия, содержал несколько более уширенные пики, чем спектр продукта, полученного с нитратом алюминия. Общая площадь поверхности первого продукта была чуть больше (~530 в сравнении с ~512 м2/г), но площадь наружной поверхности была больше почти на 50 м2/г. Полученные результаты показывают, что природа и концентрация анионов в синтезе также оказывает влияние на морфологию соответствующих получаемых продуктов.
Пример 20
Повторяли способ Примера 13 при молярном отношении С6-дикват/SiO2, сниженном с ~0,3 до ~0,14, и молярном отношении H2O/SiO2, повышенном до ~5,5-6. Кроме того, в качестве источника алюминия применяли алюминий Alcoa С-31™ (тригидрат алюминия). После нагревания в течение ~8 суток при ~150°С был получен продукт с общей площадью/площадью наружной поверхности, составляющими ~540/~240 м2/г.
Пример 21
Была проведена серия экспериментов, аналогичных экспериментам Примера 12, но с применением в качестве СОА гидроксида 1,4-бис(N-трибутиламмония)бутана. Этот С4-дикват был получен аналогично С5-диквату, с заменой 1,5-дибромпентана 1,4-дибромбутаном.
Оказалось, что препараты С4-диквата не позволяют получить какие-либо кристаллические фазы после нагревания в течение нескольких недель при ~160°С и отношениях H2O/Si, составляющих ~3,5 и ~10.
Примеры 22 и 23
Был получен ряд боросиликатных аналогов материалов, полученных в предшествующих Примерах; для этого в качестве источника бора применяли борную кислоту, и в качестве СОА применяли С6-дикват Примера 13. В первом синтезе (Пример 22) использовали отношение Na/SiO2, составляющее ~0,0125, и синтез проводили при ~115°С в течение ~4 суток. После кальцинации продукта, он имел общую площадь/площадь наружной поверхности, составляющие ~319/~175 м2/г. Не прибегая к какой-либо теории, можно предположить, что величина площади поверхности, которая оказалась меньше ожидаемой, может быть обусловлена присутствием натрия во время синтеза и образующемся продукте. При повторении синтеза без щелочных катионов (Пример 23) при нагревании до ~115°С в течение ~6 суток, площади поверхности увеличивались до ~665/~483 м2/г. На Фиг. 13 представлены порошковые дифрактограммы продуктов каждого из этих синтезов в том виде, в котором эти продукты были получены. Как было указано выше для алюмосиликатных систем, отсутствие натрия приводит к увеличению продолжительности кристаллизации, но его отсутствие также приводит к получению продуктов, в порошковых дифрактограммах которых пики уширены (что указывает на наличие более мелких кристаллов).
Примеры 24 и 25
Повторяли синтезы Примеров 16 и 17, применяя ТВАОН вместо С6-диквата в качестве СОА. После нагревания в течение ~13 суток при ~100°С из смеси для синтеза с молярным отношением SiO2/Al2O3, составляющим ~80 (Пример 24), получали продукт с общей площадью/площадью наружной поверхности, составляющими ~846/~518 м2/г, в то время как после нагревания в течение ~11 суток при ~100°С из смеси для синтеза с молярным отношением SiO2/Al2O3, составляющим ~150 (Пример 25), получали продукт с общей площадью/площадью наружной поверхности, составляющими ~821/~489 м2/г.
Несмотря на то, что настоящее изобретение было рассмотрено и проиллюстрировано с помощью конкретных воплощений, специалистам в данной области техники должно быть понятно, что изобретение может включать варианты, не обязательно проиллюстрированные в настоящем описании. По этой причине, для определения истинного объема настоящего изобретения следует учитывать только прилагаемые пункты формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| Новый синтетический кристаллический материал EMM-26, его получение и применение | 2015 |
|
RU2688542C2 |
| EMM-28 - новый синтетический кристаллический материал, его получение и применение | 2016 |
|
RU2721569C2 |
| СИНТЕТИЧЕСКИЙ ПОРИСТЫЙ КРИСТАЛЛИЧЕСКИЙ МАТЕРИАЛ ITQ-13, ЕГО СИНТЕЗ И ПРИМЕНЕНИЕ | 2002 |
|
RU2293058C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЦЕОЛИТА ТИПА MEL | 2018 |
|
RU2712549C1 |
| МОЛЕКУЛЯРНОЕ СИТО EMM-23, ЕГО СИНТЕЗ И ПРИМЕНЕНИЕ | 2012 |
|
RU2599745C2 |
| ЦЕОЛИТНЫЕ МАТЕРИАЛЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ С ПРИМЕНЕНИЕМ АЛКЕНИЛТРИАММОНИЕВЫХ СОЕДИНЕНИЙ | 2013 |
|
RU2622300C2 |
| КАТАЛИЗАТОР ДЛЯ АРОМАТИЗАЦИИ АЛКАНОВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ АРОМАТИЗАЦИИ УГЛЕВОДОРОДОВ С ПРИМЕНЕНИЕМ КАТАЛИЗАТОРА | 2003 |
|
RU2307117C2 |
| МОЛЕКУЛЯРНОЕ СИТО ЕММ-22, ЕГО СИНТЕЗ И ПРИМЕНЕНИЕ | 2012 |
|
RU2601462C2 |
| ВЫСОКОАКТИВНЫЕ МОЛЕКУЛЯРНЫЕ СИТА СО СТРУКТУРОЙ ТИПА МТТ | 2009 |
|
RU2501735C2 |
| МЕЛКОКРИСТАЛЛИЧЕСКИЙ ZSM-5, ЕГО СИНТЕЗ И ПРИМЕНЕНИЕ | 2013 |
|
RU2640759C2 |

Изобретение относится к синтезу цеолитов. Предложен способ получения мелкокристаллических материалов, имеющих тип каркаса MFI и/или MEL, большую площадь поверхности и обозначаемых ЕММ-30. В способе в качестве структурообразующего агента используют катионы тетрабутиламмония и/или катионы тетрабутилфосфония или дикатионы 1,5-бис(N-трибутиламмоний)пентана и/или дикатионы 1,6-бис(N-трибутиламмоний)гексана. Изобретение обеспечивает возможность получения цеолитов с каркасом типа MFI и/или MEL, имеющих ультрамелкие кристаллы, из реакционной смеси, имеющей широкий диапазон Si/Al-соотношения. 2 н. и 10 з.п. ф-лы, 13 ил., 25 пр.
1. Способ получения кристаллического материала, имеющего тип каркаса MFI и/или MEL, где способ включает:
(i) приготовление смеси для синтеза, подходящей для образования кристаллического материала, где смесь включает источник оксида четырехвалентного элемента Y, источник трехвалентного элемента X, источник щелочного или щелочноземельного металла (М), воду и структурообразующий агент (Q1), включающий катионы тетрабутиламмония и/или катионы тетрабутилфосфония, где смесь для синтеза имеет состав, содержащий:
(a) молярное отношение H2O/YO2, составляющее приблизительно 10 или менее;
(b) молярное отношение YO2/X2O3, составляющее менее приблизительно 150; и
(c) молярное отношение M/YO2, составляющее приблизительно 0,04 или менее;
(ii) нагревание смеси в условиях кристаллизации, включающих температуру, составляющую от приблизительно 80°С до приблизительно 220°С, и время, составляющее от приблизительно 4 часов до приблизительно 28 суток, до образования кристаллов кристаллического материала; и
(iii) извлечение кристаллического материала, полученного на этапе (ii).
2. Способ по п. 1, в котором X включает один или более из В, Al, Fe и Ga и Y включает один или более из Si, Ge, Sn, Ti и Zr.
3. Способ по п. 1, в котором X включает алюминий и Y включает кремний.
4. Способ по п. 1, в котором смесь для синтеза имеет молярное отношение YO2/X2O3, составляющее по меньшей мере приблизительно 20.
5. Способ по п. 1, в котором молярное отношение H2O/YO2 в смеси для синтеза составляет по меньшей мере приблизительно 2.
6. Способ по п. 1, в котором условия кристаллизации включают температуру, составляющую от приблизительно 100°С до приблизительно 150°С.
7. Способ по п. 1, в котором кристаллический материал, извлеченный на этапе (iii), имеет общую площадь поверхности, определяемую способом физической адсорбции азота с построением t-графика и составляющую по меньшей мере приблизительно 750 м2/г, и/или площадь наружной поверхности, определяемую способом физической адсорбции азота с построением t-графика и составляющую по меньшей мере приблизительно 350 м2/г.
8. Способ получения кристаллического материала, имеющего тип каркаса MFI и/или MEL, где способ включает:
(i) приготовление смеси для синтеза, подходящей для образования указанного кристаллического материала, где указанная смесь включает источник оксида четырехвалентного элемента Y, необязательно источник трехвалентного элемента X, необязательно источник щелочного или щелочноземельного металла (М), воду и структурообразующий агент (Q2), включающий дикатионы 1,5-бис(N-трибутиламмоний)пентана и/или дикатионы 1,6-бис(N-трибутиламмоний)гексана;
(ii) нагревание смеси в условиях кристаллизации, которые включают температуру, составляющую от 80°С до 220°С, и время, составляющее от приблизительно 4 часов до приблизительно 28 суток, до образования кристаллов кристаллического материала; и
(iii) извлечение кристаллического материала, полученного на этапе (ii).
9. Способ по п. 8, в котором смесь для синтеза имеет состав, в котором молярные отношения находятся в следующих диапазонах:
YO2/X2O3 составляет по меньшей мере приблизительно 20;
H2O/YO2 составляет от приблизительно 2 до приблизительно 60;
OH-/YO2 составляет от приблизительно 0,1 до приблизительно 0,8;
M/YO2 составляет от 0 до приблизительно 0,4; и
Q2/YO2 составляет от приблизительно 0,05 до приблизительно 0,4.
10. Способ по п. 9, в котором молярное отношение H2O/YO2 в смеси для синтеза составляет приблизительно 10 или менее.
11. Способ по п. 9, в котором молярное отношение M/YO2 в смеси для синтеза составляет приблизительно 0,02 или менее.
12. Способ по п. 8, в котором условия кристаллизации включают температуру, составляющую от приблизительно 100°С до приблизительно 150°С.
| US 20130059722 A1, 07.03.2013 | |||
| US 7767192 B1, 03.08.2010 | |||
| WO 2015091078 A1, 25.06.2015 | |||
| СПОСОБ ПОЛУЧЕНИЯ ЦЕОЛИТОВЫХ КАТАЛИЗАТОРОВ, СПОСОБ ПОЛУЧЕНИЯ СВЯЗАННЫХ ЦЕОЛИТОВ, ЦЕОЛИТОВЫЕ КАТАЛИЗАТОРЫ | 1998 |
|
RU2240866C2 |
| КАТАЛИЗАТОР АРОМАТИЗАЦИИ МЕТАНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ КОНВЕРСИИ МЕТАНА С ПОЛУЧЕНИЕМ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 2015 |
|
RU2585289C1 |

