Область изобретения
Настоящее изобретение относится к новому синтетическому кристаллическому материалу, ЕММ-28, и к способу его получения. Настоящее изобретение относится также к применению пористых форм ЕММ-28 в реакциях превращения органических соединений и процессах сорбции.
Уровень техники
Известно, что цеолитные материалы используются в качестве сорбентов и обладают каталитическими свойствами в отношении различных типов реакций превращения углеводородов. Некоторые цеолитные материалы являются упорядоченными, пористыми кристаллическими материалами, имеющими определенную кристаллическую структуру по данным рентгеновской дифракции, внутри которой существует большое количество полостей меньшего размера, которые могут быть соединены между собой рядом каналов или пор еще меньшего размера. Эти полости и поры являются однородными по размеру внутри специфического цеолитного материала. Поскольку размеры таких пор такие, что сорбируют молекулы определенного размера и не принимают молекулы большего размера, такие материалы стали известны как "молекулярные сита" и их применяют различными способами, позволяющими использовать преимущество таких свойств.
Такие молекулярные сита, как природные, так и синтетические, включают широкий ряд содержащих положительно заряженные ионы кристаллических силикатов и замещенных силикатов, в которых кремний частично или полностью замещен другими четырехвалентными элементами. Такие силикаты можно описать как жесткие трехмерные каркасы из SiO4 тетраэдров и возможно тетраэдров оксида трехвалентного элемента, например, АlO4 и/или ВО4, в которых тетраэдры перекрестно связаны с помощью общих атомов кислорода, причем локальное соотношение общего числа атомов трехвалентного элемента и кремния к атомам кислорода составляет 1:2. Электровалентность тетраэдров, содержащих трехвалентный элемент, уравновешивается путем включения в кристалл катиона, например, катиона щелочного металла или щелочноземельного металла. Это можно выразить, когда соотношение трехвалентного элемента, например, алюминия, к ряду различных катионов, таких как Са/2, Sr/2, Na, K или Li, равно единице. Один тип катиона может обмениваться полностью или частично с другим типом катиона посредством ионного обмена обычным способом. С помощью такого катионного обмена было возможно изменять свойства выбранного силиката путем подходящего выбора катиона.
Молекулярные сита, которые находят применение в катализаторах, включают любые встречающиеся в природе или синтетические кристаллические молекулярные сита. Примеры таких молекулярных сит включают цеолиты с порами большого размера, цеолиты с промежуточным размером пор и цеолиты с порами небольшого размера. Такие цеолиты и их изотипы описаны в "Atlas of Zeolite Framework Types", eds. Ch. Baerlocher, L.B. McCusker, D.H. Olson, Elsevier, Sixth Revised Edition, 2007, который включен в данную заявку посредством ссылки. Цеолит с порами большого размера обычно имеет размер пор по меньшей мере 0,7 нм (7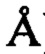 ) и включает цеолиты структурного типа LTL, VFI, MAZ, FAU, OFF, *ВЕА и MOR (комиссия IUPAC по номенклатуре цеолитов). Примеры цеолитов с порами большого размера включают мазит, оффретит, цеолит L, VPI-5, цеолит Y, цеолит X, омега и бета. Цеолит с промежуточным размером пор обычно имеет размер пор от примерно 0,5 нм до менее 0,7 нм (от примерно 5
) и включает цеолиты структурного типа LTL, VFI, MAZ, FAU, OFF, *ВЕА и MOR (комиссия IUPAC по номенклатуре цеолитов). Примеры цеолитов с порами большого размера включают мазит, оффретит, цеолит L, VPI-5, цеолит Y, цеолит X, омега и бета. Цеолит с промежуточным размером пор обычно имеет размер пор от примерно 0,5 нм до менее 0,7 нм (от примерно 5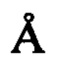 до менее 7
до менее 7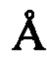 ) и включает, например, цеолиты структурного типа MFI, MEL, EUO, МТТ, MFS, AEL, AFO, HEU, FER, MWW и TON (Комиссия IUPAC по номенклатуре цеолитов). Примеры цеолитов с промежуточным размером пор включают ZSM-5, ZSM-11, ZSM-22, МСМ-22, силикалит-1 и силикалит-2. Цеолит с порами небольшого размера имеет размер пор от примерно 0,3 нм до менее 0,5 нм (от примерно 3
) и включает, например, цеолиты структурного типа MFI, MEL, EUO, МТТ, MFS, AEL, AFO, HEU, FER, MWW и TON (Комиссия IUPAC по номенклатуре цеолитов). Примеры цеолитов с промежуточным размером пор включают ZSM-5, ZSM-11, ZSM-22, МСМ-22, силикалит-1 и силикалит-2. Цеолит с порами небольшого размера имеет размер пор от примерно 0,3 нм до менее 0,5 нм (от примерно 3 до менее 5
до менее 5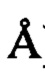 ) и включает, например, цеолиты структурного типа СНА, ERI, KFI, LEV, SOD и LTA (Комиссия IUPAC по номенклатуре цеолитов). Примеры цеолитов с порами небольшого размера включают ZK-4, SAPO-34, SAPO-35, ZK-14, SAPO-42, ZK-21, ZK-22, ZK-5, ZK-20, цеолит А, шабазит, цеолит Т и ALPO-17.
) и включает, например, цеолиты структурного типа СНА, ERI, KFI, LEV, SOD и LTA (Комиссия IUPAC по номенклатуре цеолитов). Примеры цеолитов с порами небольшого размера включают ZK-4, SAPO-34, SAPO-35, ZK-14, SAPO-42, ZK-21, ZK-22, ZK-5, ZK-20, цеолит А, шабазит, цеолит Т и ALPO-17.
Многие цеолиты синтезируют в присутствии органического направляющего агента для формирования структуры, такого как органическое азотсодержащее соединение. Например, ZSM-5 можно синтезировать в присутствии катионов тетрапропиламмония, а цеолит МСМ-22 можно синтезировать в присутствии гексаметиленимина. Известно также, что цеолиты и соответствующие молекулярные сита можно синтезировать в присутствии дичетвертичных направляющих агентов. Например, в опубликованной международной заявке США No. 2010/0178241 описан синтез EU-1 в присутствии катионов гексаметония.
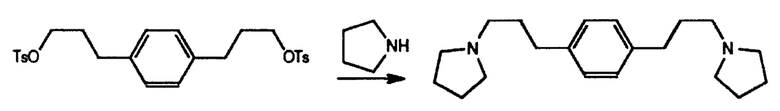
Согласно настоящему изобретению, новая структура цеолита, обозначенная EMM-28 и имеющая отличительный профиль рентгенодифрактограммы, была синтезирована с помощью органического направляющего агента для формирования структуры, нового диквата с N-метилпирролидиниевыми группами, соединенными с фенильным кольцом линейной полиметиленовой группой формулы (СН2)3 в 1,3-положениях (мета) или в 1,4-положениях (пара).
Сущность изобретения
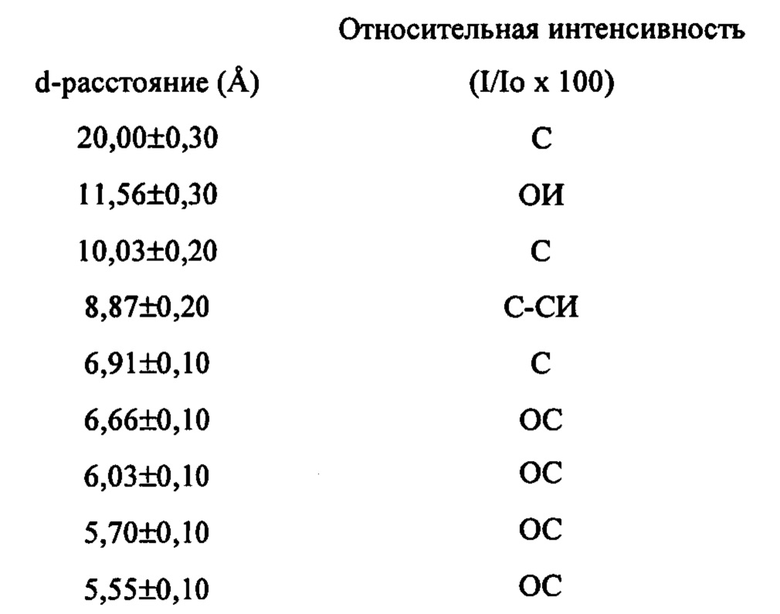
В первом аспекте, изобретение заключается в синтетическом кристаллическом материале, имеющем в только что прокаленном состоянии профиль рентгенодифрактограммы, включающий пики, указанные в Таблице 1:


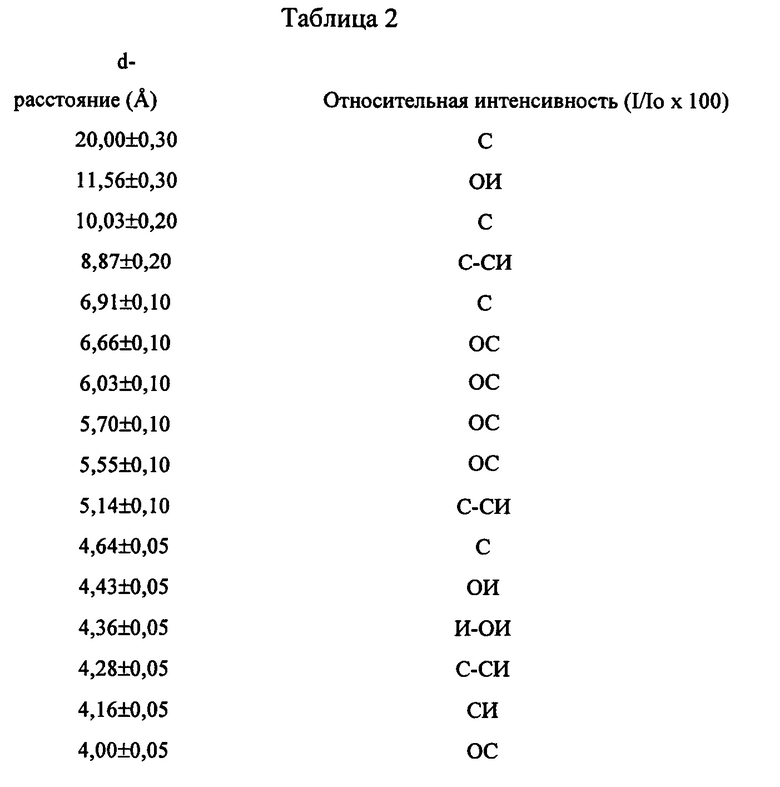
Во втором аспекте, изобретение заключается в синтетическом кристаллическом материале, имеющем сразу после синтеза профиль рентгенодифрактограммы, включающий пики, указанные в Таблице 2:

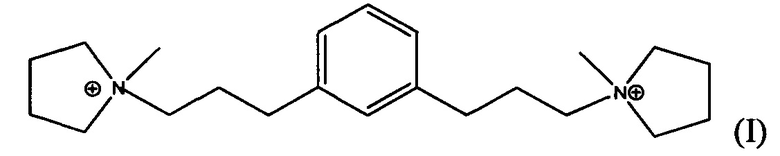
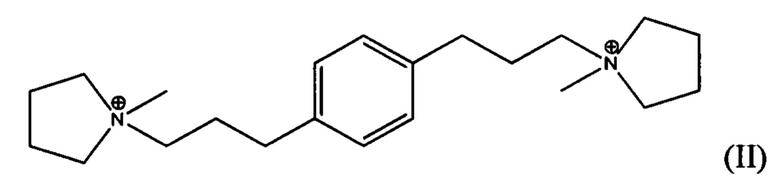
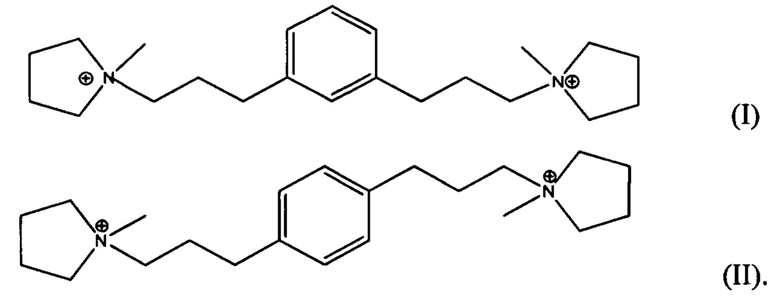
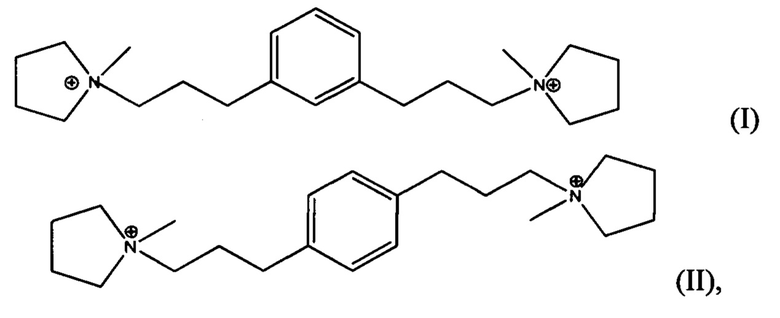
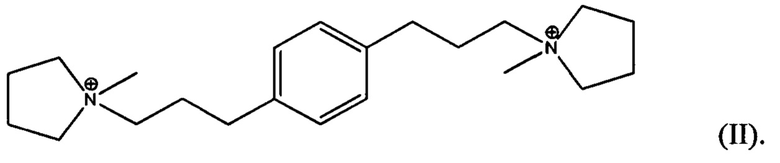
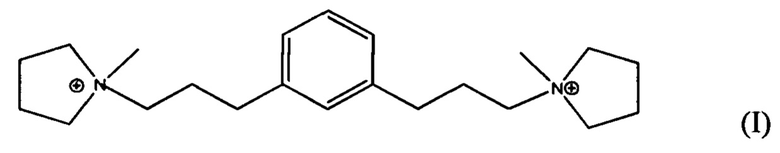
В третьем аспекте, изобретение заключается в синтетическом пористом кристаллическом материале, содержащем в структуре его пор дикатион формулы (I) или (II):
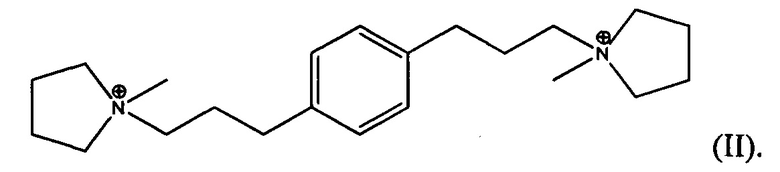
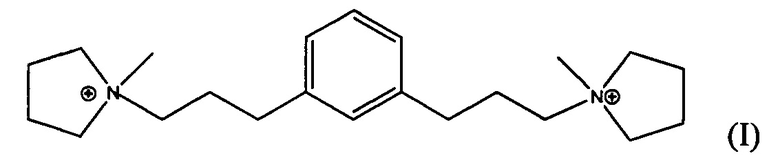
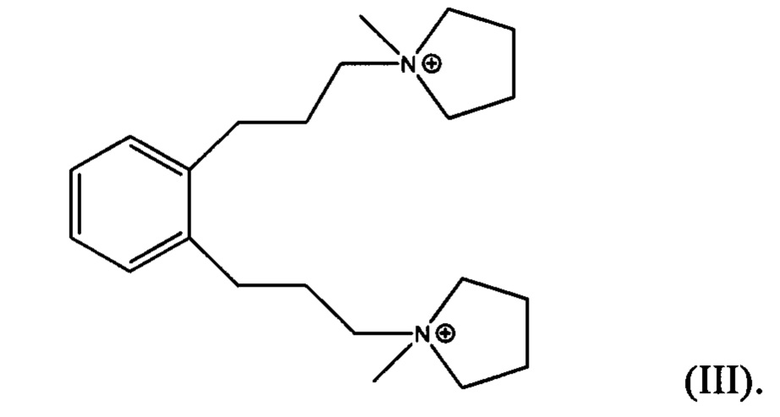
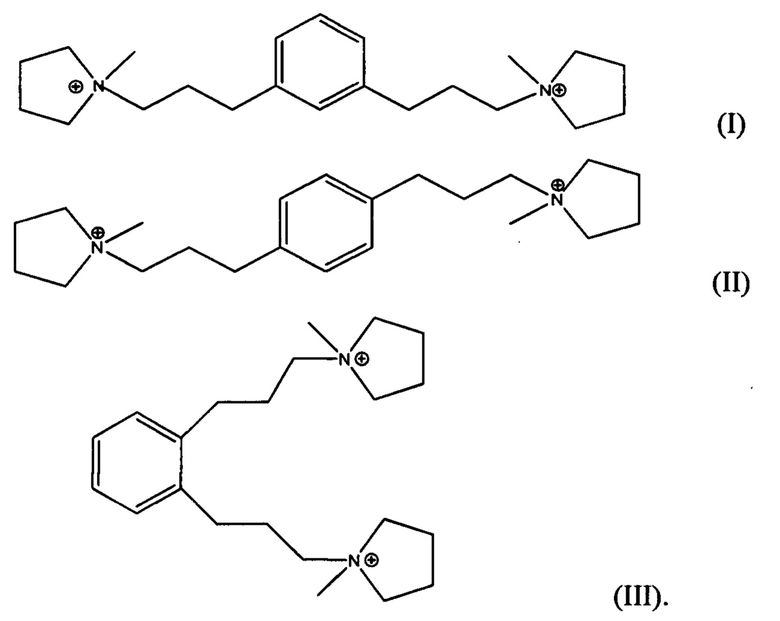
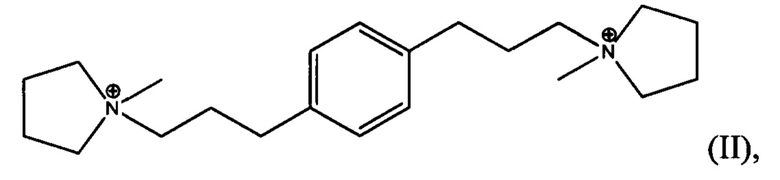
В четвертом аспекте, изобретение заключается в органическом азотсодержащем соединении, содержащем дикатион формулы (I), (II) или (III):
В пятом аспекте, изобретение состоит в способе получения соединения 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1), где х равно 2, 3 или 4, где способ включает:
взаимодействие 1,х-bis(галогенметил)бензола с диалкилмалонатом с получением тетраалкил 2,2'-(1,х-фениленbis(метилен))дималоната;
превращение по меньшей мере части тетраалкил-2,2'-(1,х-фениленbis(метилен))дималоната в 3,3'-(1,х-фенилен)дипропановую кислоту;
восстановление по меньшей мере части 3,3'-(1,х-фенилен)дипропановой кислоты или ее сложного эфира в 3,3'-(1,х-фенилен)bis(пропан-1-ол));
взаимодействие по меньшей мере части 3,3'-(1,х-фенилен)bis(пропан-1-ола)) с алкил- или арилсульфонилгалогенидом с получением соответствующего сульфонатного сложного диэфира;
взаимодействие по меньшей мере части сульфонатного сложного диэфира с пирролидином с получением 1,х-bis(3-(пирролидин-1-ил)пропил)бензола; и
взаимодействие по меньшей мере части 1,х-bis(3-(пирролидин-1-ил)пропил)бензола с метилгалогенидом с получением соединения 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1).
В другом воплощении пятого аспекта изобретение заключается в способе получения соединения 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1), где х представляет собой 2, 3 или 4, где способ включает:
взаимодействие по меньшей мере части сульфонатного сложного диэфира 3,3'-(1,х-фенилен)bis(пропан-1-ола)) с 1-метилпирролидином с получением соединения 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1).
В шестом аспекте изобретение заключается в способе получения соединения 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1), где х представляет собой 2, 3 или 4, где способ включает:
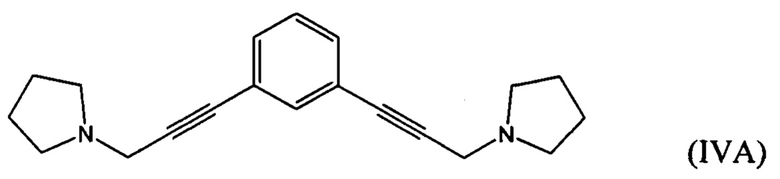
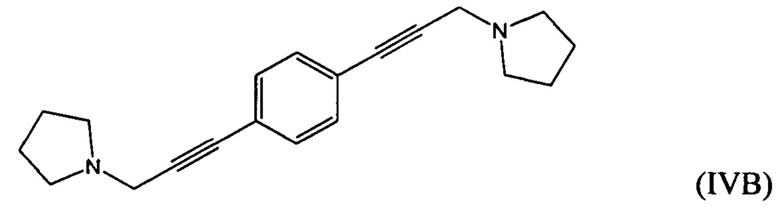
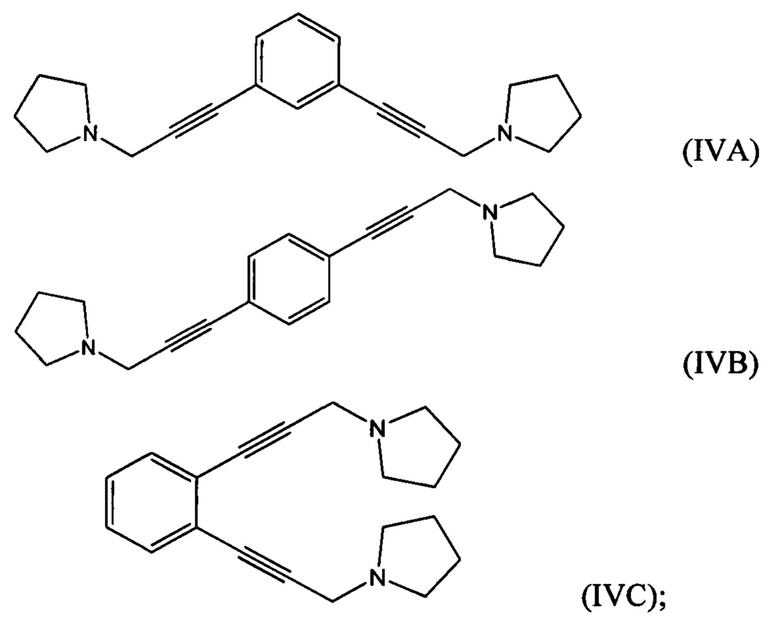
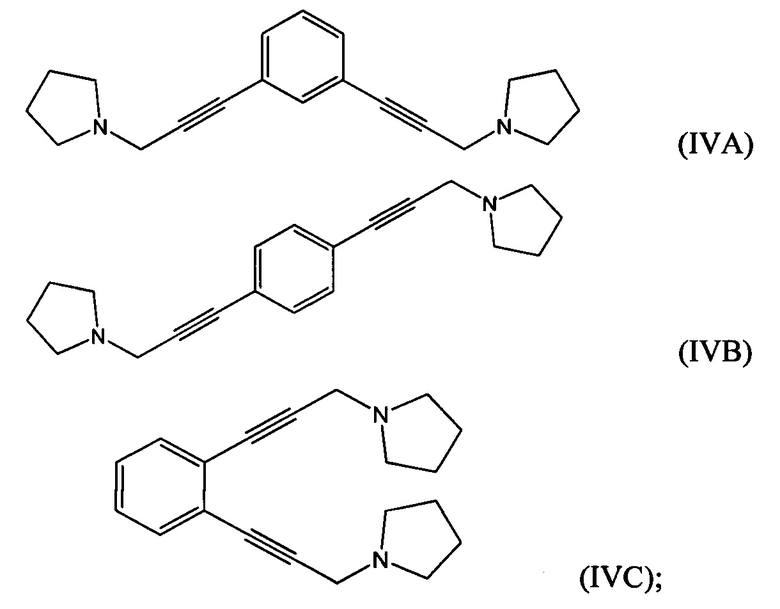
взаимодействие 1-(проп-2-ин-1-ил)пирролидина с 1,х-дигалогензамещенным бензолом с получением соединения формулы (IVA), (IVB) или (IVC):
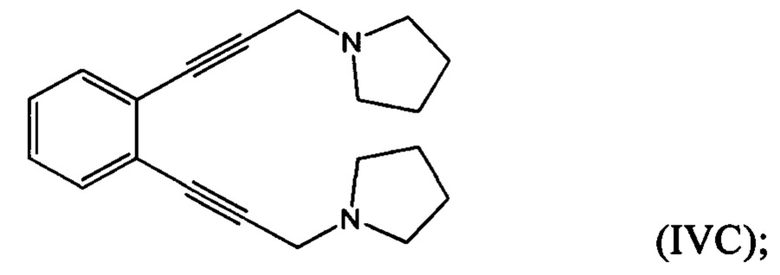
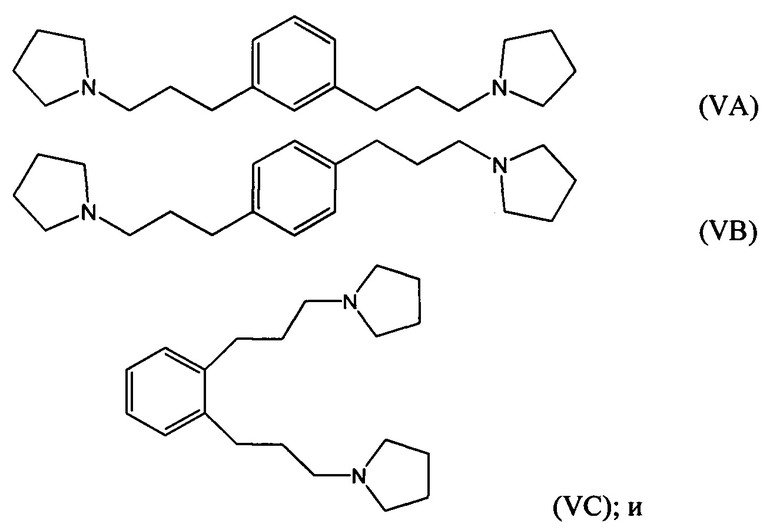
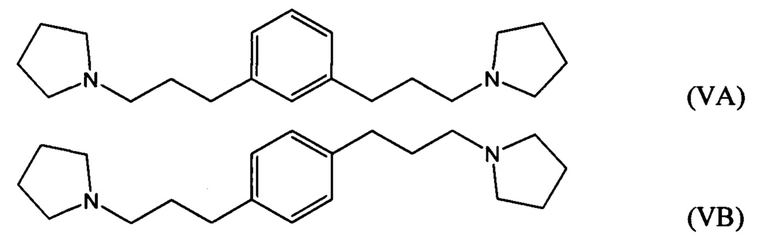
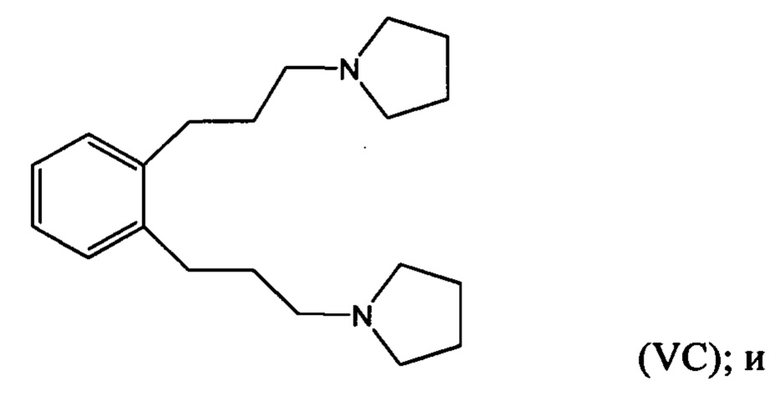
гидрирование по меньшей мере части соединение формулы (IVA), (IVB) или (IVC) с получением соединения формулы (VA), (VB) или (VC), соответственно:
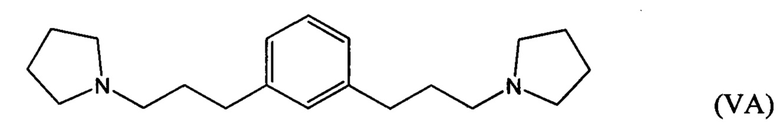
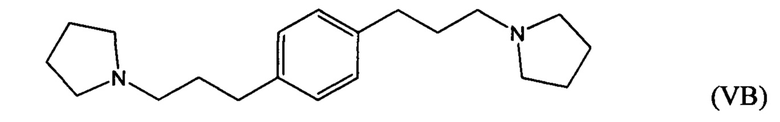
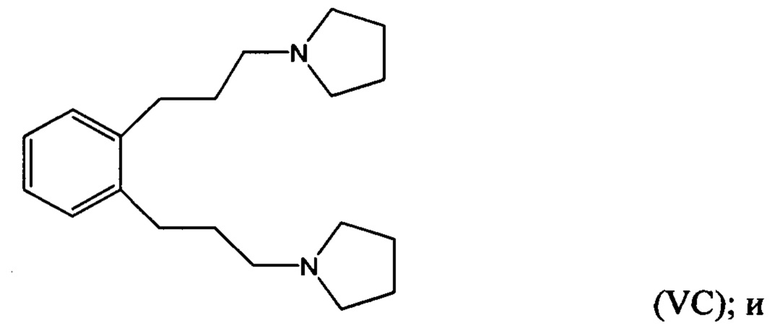
взаимодействие по меньшей мере части соединения формулы (VA), (VB) или (VC) с метилгалогенидом с получением соединения 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1).
В седьмом аспекте изобретение заключается в способе получения соединения 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1), где х представляет собой 2, 3 или 4, где способ включает:
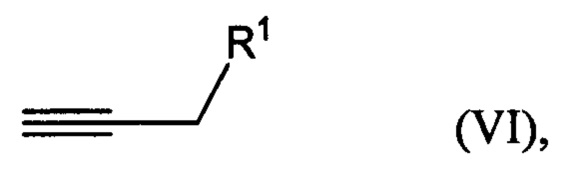
взаимодействие соединения формулы (VI)
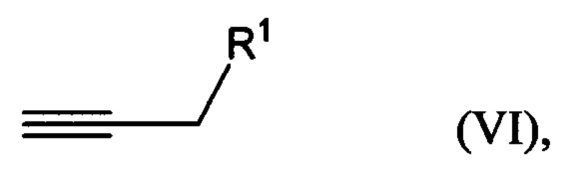
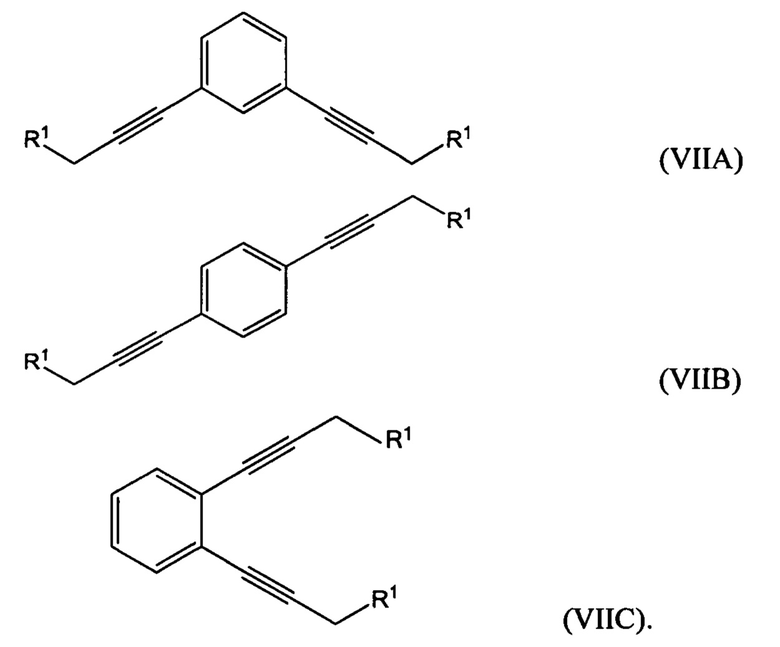
где R1 представляет собой гидроксильную группу или алкил- или арилсульфонатную группу, с 1,х-дигалогензамещенным бензолом с получением соединения формулы (VIIA), (VIIB) или (VIIC):
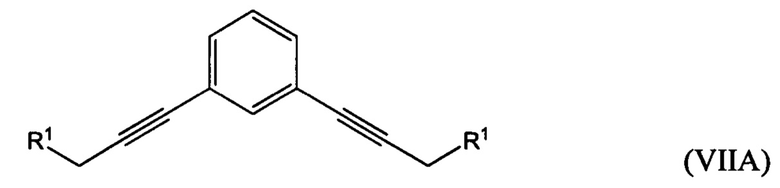
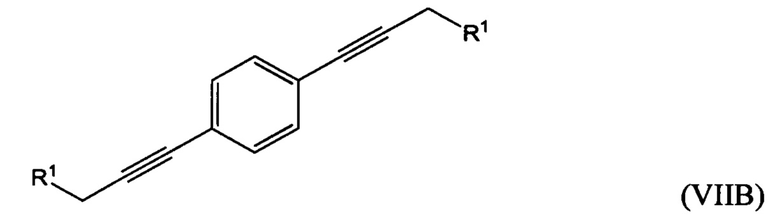
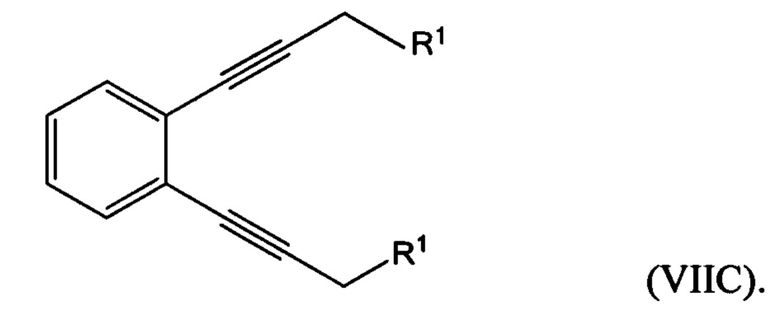
В одном из воплощений седьмого аспекта R1 представляет собой алкил- или арилсульфонатную группу, при этом способ дополнительно включает:
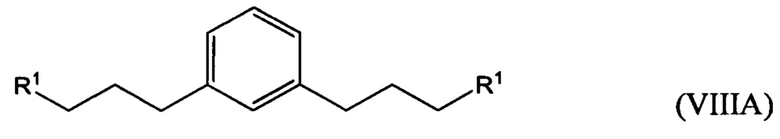
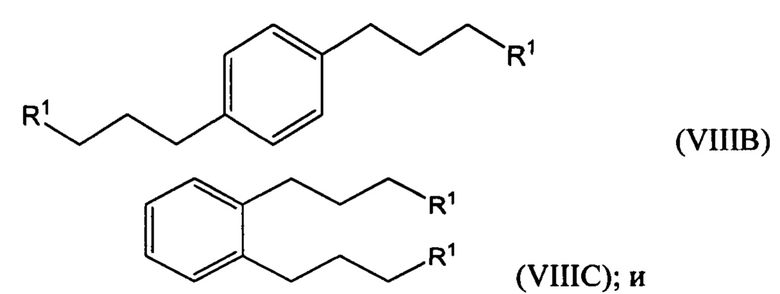
гидрирование по меньшей мере части соединения формулы (VILA), (VIIB) или (VIIC) с получением соединения формулы (VIIIA), (VIIIB) или (VIIIC), соответственно:
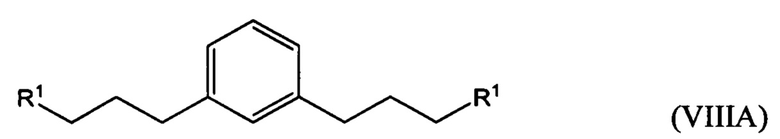
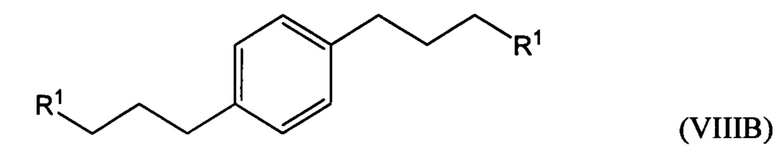
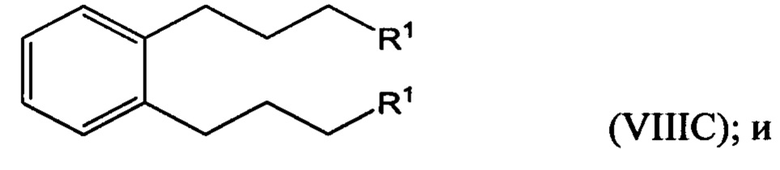
превращение по меньшей мере части соединения формулы (VIIIA), (VIIIB) или (VIIIC) в соединение 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1) (i) по реакции с 1-метилпирролидином или (ii) по реакции с пирролидином и затем с метил галогенидом.
В другом воплощении седьмого аспекта R1 представляет собой алкил- или арилсульфонатную группу, при этом способ дополнительно включает:
взаимодействие по меньшей мере части соединения формулы (VILA), (VIIB) или (VIIC) с пирролидином с получением соединения формулы (IVA), (IVB) или (LVC), соответственно:
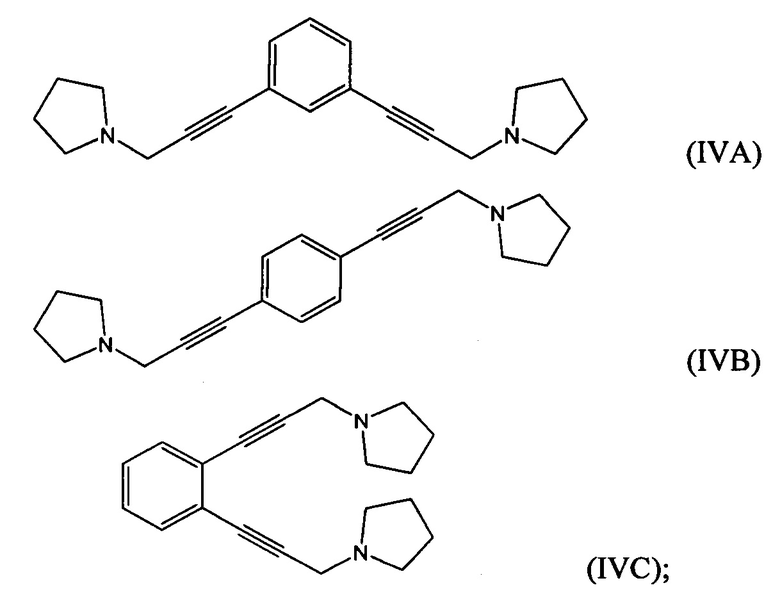
гидрирование по меньшей мере части соединения формулы (IVA), (IVB) или (IVC) с получением соединения формулы (VA), (VB) или (VC), соответственно:
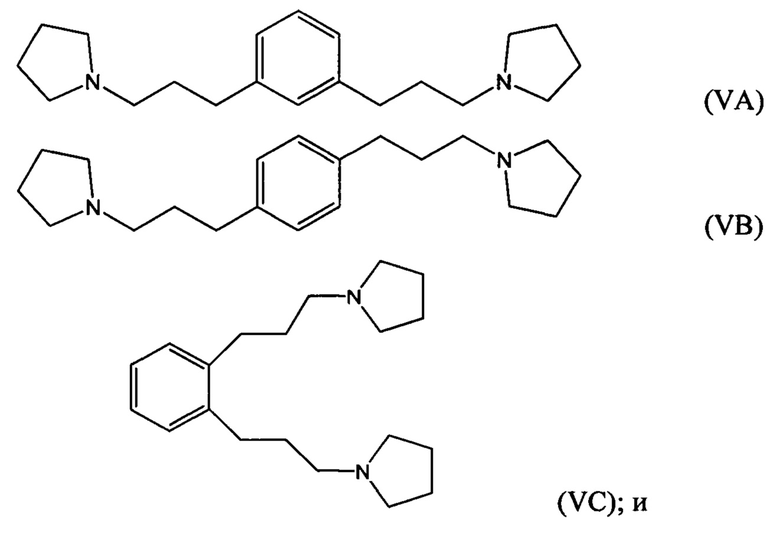
взаимодействие по меньшей мере части соединения формулы (VA), (VB) или (VC) с метилгалогенидом с получением соединения 1,1'-(1,х-фениленbis, (пропан-3,1-диил))bis(1-метилпирролидиния-1).
В еще одном воплощении седьмого аспекта R1 представляет собой гидроксильную группу, при этом способ дополнительно включает:
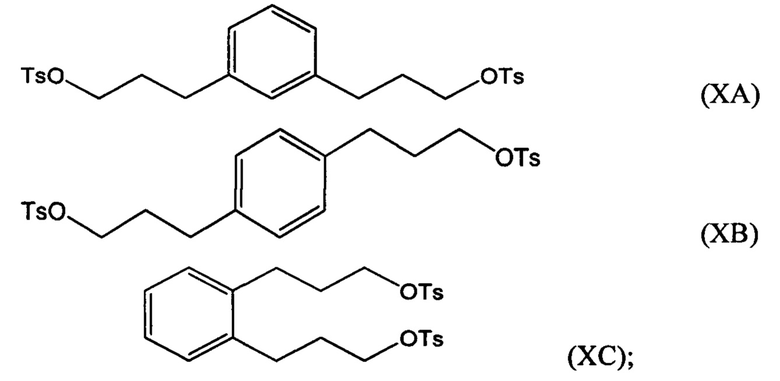
взаимодействие по меньшей мере части соединения формулы (VIIA), (VIIB) или (VIIC) с алкил- или арилсульфонилгалогенидом, таким как n-толуолсульфонилхлорид, с получением соединения формулы (IXA), (IXB) или (IXC), соответственно:
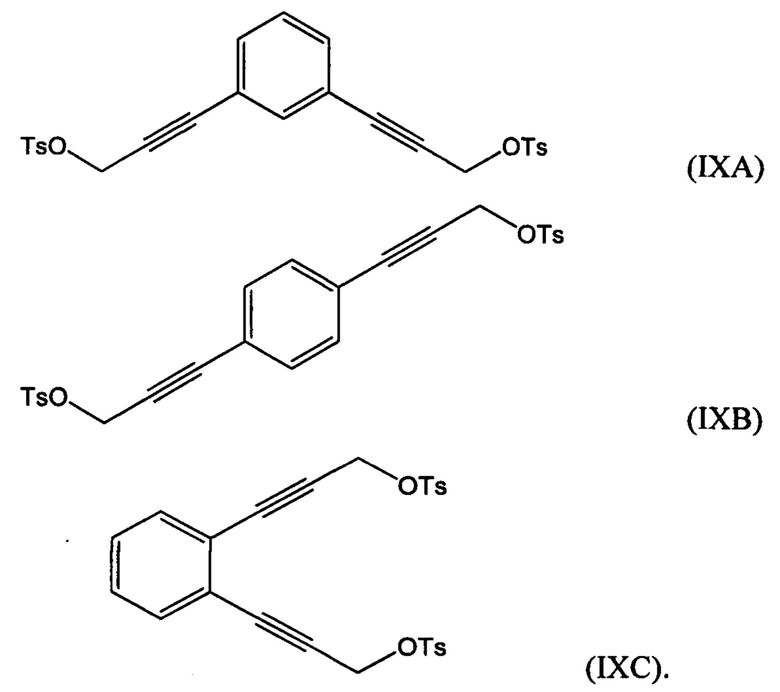
Соединение формулы (IXA), (IXB) или (IXC) можно затем гидрировать с получением соединения формулы (ХА), (ХВ) или ХС, соответственно:
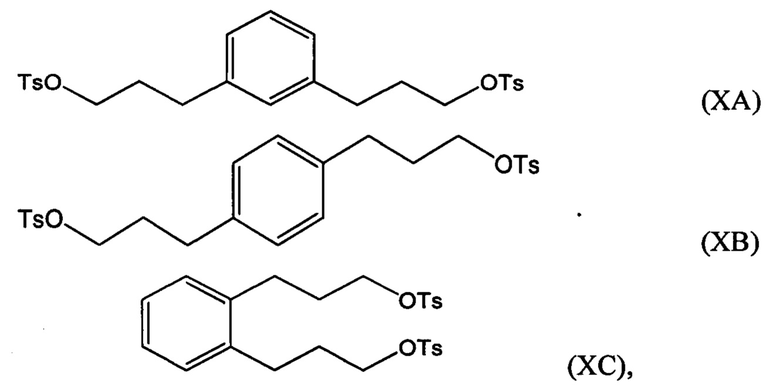
которое затем можно превратить в соединение 3,3-(1,х-фенилен)bis(пропан-3,1-диил)bis(1-метилпирролидиния) (i) по реакции с 1-метилпирролидином или (ii) по реакции с пирролидином и затем с метилгалогенидом.
В восьмом аспекте изобретение заключается в способе получения соединения 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1), где х представляет собой 2, 3 или 4, при этом способ включает:
взаимодействие соединения формулы (VI)
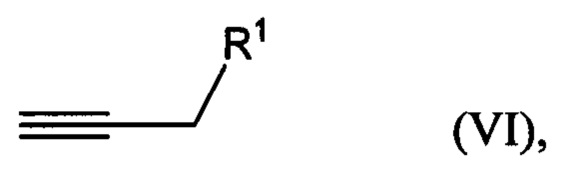
где R1 представляет собой гидроксильную группу, с 1,х-дигалогензамещенным бензолом с получением соединения формулы (VIIA), (VIIB) или (VIIC):
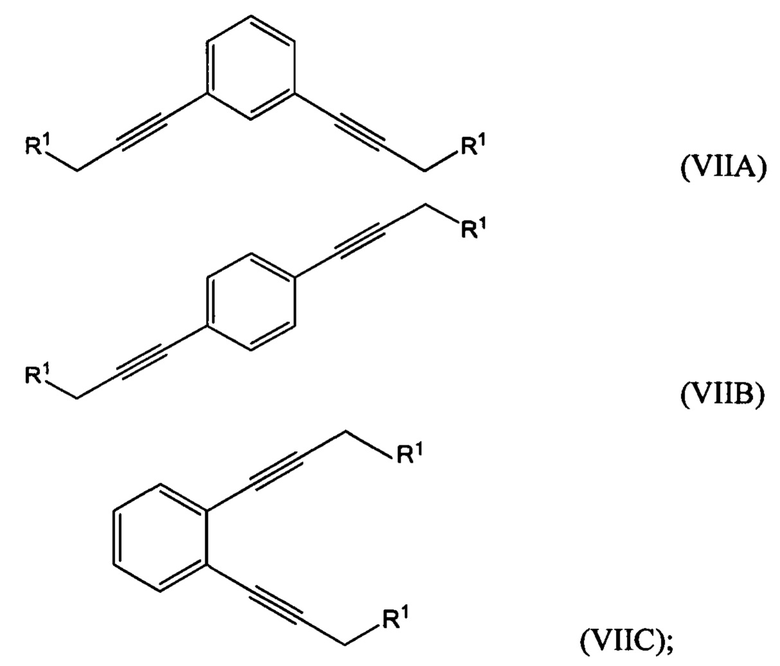
гидрирование по меньшей мере части соединения формулы (VILA), (VIIB) или (VIIC) с получением соединения формулы (VIIIA), (VIIIB) или (VIIIC), соответственно:
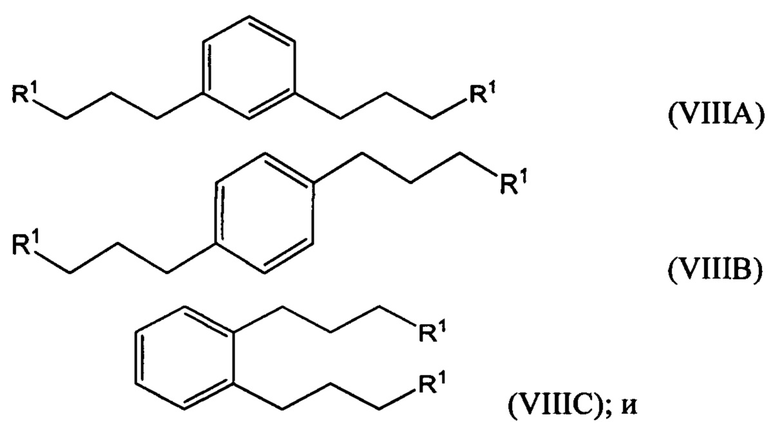
взаимодействие по меньшей мере части соединения формулы (VIIIA), (VIIIB) или (VIIIC) с алкил- или арилсульфонилгалогенидом, таким как n-толуолсульфонилхлорид, с получением соединения формулы (ХА), (ХВ) или ХС, соответственно:
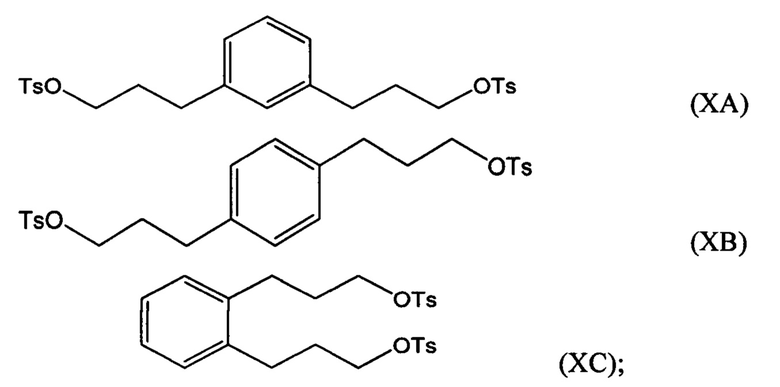
превращение по меньшей мере части соединения формулы (ХА), (ХВ) или (ХС) в соединение 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1) (i) по реакции с 1-метилпирролидином или (ii) по реакции с пирролидином и затем с метилгалогенидом.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
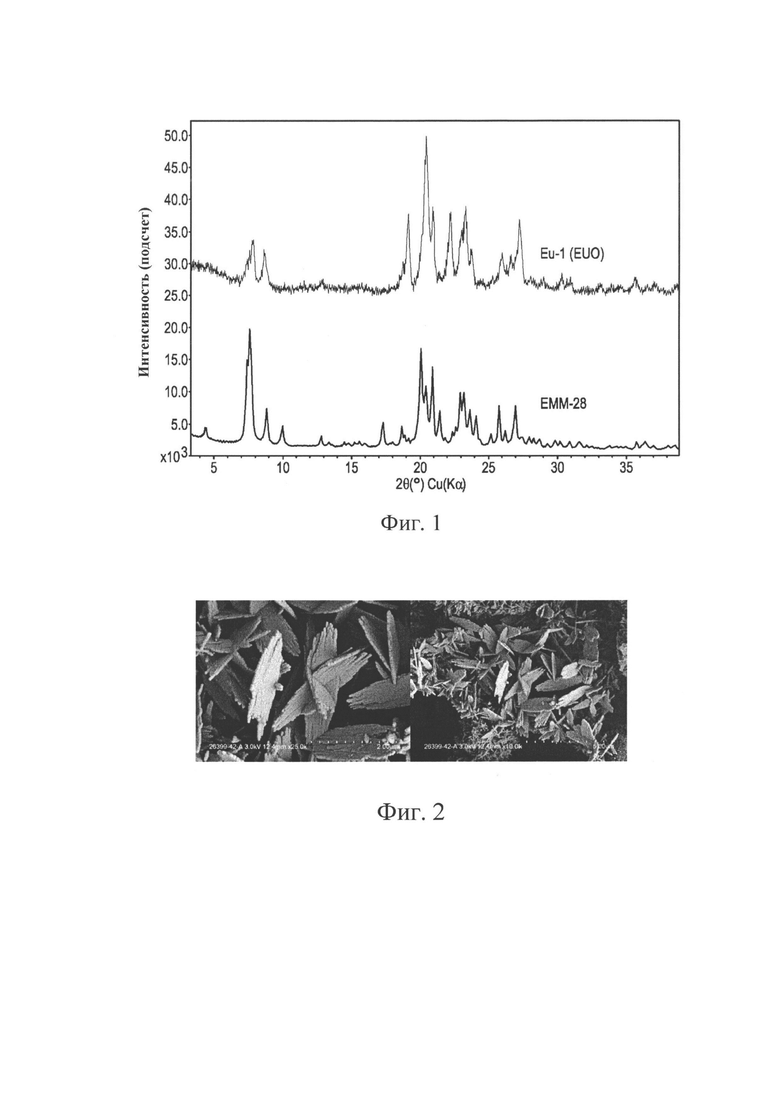
На Фиг. 1 приведено сравнение профилей рентгенодифрактограмм только что синтезированного ЕММ-28, полученного в Примере 8, и только что синтезированного материала EU-1, полученного в Примере 9.
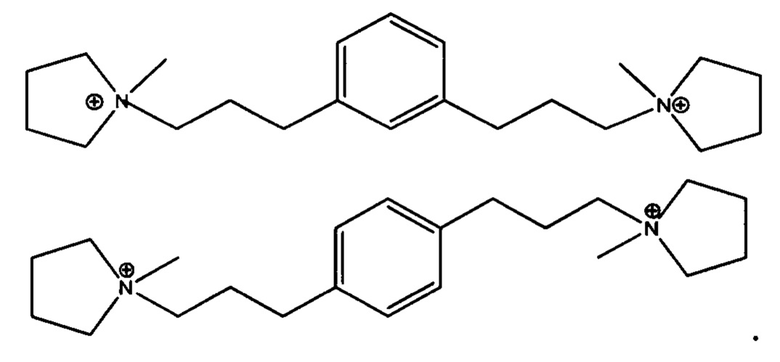
На Фиг. 2 приведены изображения, полученные с помощью сканирующего электронного микроскопа (СЭМ) ЕММ-28, полученного в Примере 15 при различном увеличении.
ПОДРОБНОЕ ОПИСАНИЕ ВОПЛОЩЕНИЙ ИЗОБРЕТЕНИЯ
В данной заявке описан новый материал молекулярного сита, который обозначен ЕММ-28, его синтез в присутствии органического направляющего агента для формирования структуры, содержащего одно или более новых дичетвертичных аммониевых соединений, и его применение в качестве сорбента и катализатора для реакций превращения органических соединений. Кроме того, описаны способы получения новых дичетвертичных аммониевых соединений.
Материал молекулярного сита ЕММ-28
Новый материал молекулярного сита ЕММ-28 отличается профилем рентгенодифрактограммы, которая для прокаленной формы молекулярного сита включает по меньшей мере пики, указанные в Таблице 1 выше, и которая для формы молекулярного сита, полученной сразу после синтеза, включает по меньшей мере пики, указанные в Таблице 2 выше.
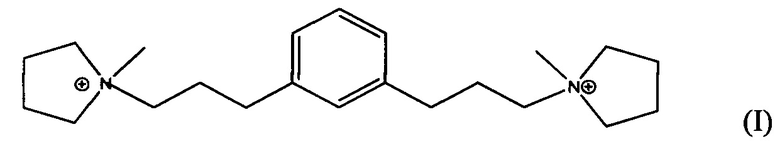
Данные рентгеноструктурной дифракции, приведенные в данной заявке, получали с помощью дифракционной системы PANalytical X-Pert Pro, снабженной детектором X'Celerator, при использовании медного источника K-alpha излучения. Данные дифракции записывали с помощью пошагового сканирования при 0,017 градусах двух тета, где тета является брэгговским углом, а время подсчета составляет 21 секунду для каждого шага. Межплоскостные расстояния, d-расстояния, вычисляли в ангстремах, причем относительные интенсивности площади пика линий, I/I(о), составляли одну сотую интенсивности самой интенсивной линии относительно базовой линии, и определяли их с помощью алгоритма аппроксимации профиля пика MDI Jade. Интенсивности не корректировали на эффекты Лоренца и поляризационные эффекты. Относительные интенсивности имеют следующие обозначения: ОИ = очень интенсивная (интенсивнее 60 до 100), И = интенсивная (интенсивнее 40 до 60), СИ = средней интенсивности (интенсивнее 20 до 40), С = слабой интенсивности (от 10 до 20) и ОС = очень слабая интенсивности (менее 10). В некоторых воплощениях, некоторые или все линии, обозначенные как "очень слабой интенсивности (англ. - very weak)", могут иметь относительную интенсивность больше нуля.
Следует понимать, что данные дифракции, приведенные для данного образца в виде отдельных линий, могут состоять из множества перекрывающихся линий, которые в определенных условиях, таких как различия по кристаллографии, могут проявляться в виде разрешенных или частично разрешенных линий. Как правило, кристаллографические изменения могут включать небольшие изменения параметров элементарной ячейки и/или изменение в симметрии кристалла без изменения структуры. Эти небольшие эффекты, включающие изменения относительных интенсивностей, также могут происходить в результате различий, связанных с наличием катиона, составом каркасной структуры, природой и степенью заполнения пор, размером и формой кристалла, предпочтительной ориентацией и предшествующими термическими и гидротермическими операциями.
Профили рентгенодифрактограмм ЕММ-28 имеют качественное подобие с рентгенограммами материалов EUO-типа (например, EU-1 и/или ZSM-50). Сравнение рентгенограмм, полученных на порошках, приведено ниже (Фиг. 1). По-видимому, уширенность экспериментального профиля ЕММ-28 обусловлена тонкой кристаллической морфологией (см. Фиг. 2). Схожесть рентгенограмм на порошке предполагает, что каркас ЕММ-28 может быть родственным, но отличным от каркаса EUO.
В прокаленной форме молекулярное сито ЕММ-28 имеет химический состав, характеризующийся следующим молярным отношением:
(n)X2O3:YO2,
где n является числом меньше 0,05, например, меньше 0,025, например, меньше 0,01, как, например, меньше 0,005, например, меньше 0,001, как, например, меньше 0,0005, например, меньше 0,00025, X является трехвалентным элементом, например, одним или более из В, Al, Fe и Ga, особенно Al, a Y является четырехвалентным элементом, таким как Si, Ge, Sn, Ti и Zr, особенно Ge и/или Si. Из допустимых значений n понятно, что ЕММ-28 можно синтезировать в полностью кремниевой форме, в которой трехвалентный элемент X отсутствует или по существу отсутствует.
Молекулярное сито ЕММ-28 сразу после синтеза может иметь химический состав, характеризующийся следующим молярным отношением:
(m)M:(b)Q:(n)X2O3:YO2:(z)H2O,
где m является числом со значением от равного или больше 0 до меньше или равного 0,1, b является числом со значением от больше 0 до меньше или равного 0,05, n является числом меньше 0,025, например, меньше 0,001, таким как, меньше 0,0005, например, меньше 0,00025, и z является числом со значением от больше или равного 0 до меньше или равного 0,2, и где М является катионом щелочного металла, X является трехвалентным элементом, таким как один или более из В, Al, Fe и Ga, особенно Al, Y является четырехвалентным элементом, таким как Si, Ge, Sn, Ti и Zr или их смесями, особенно Ge и/или Si, и Q является органическим направляющим агентом для формирования структуры.
В другом воплощении ЕММ-28 сразу после синтеза может иметь химический состав, характеризующийся следующим молярным отношением:
(k)F:(b)Q:(n)X2O3:YO2:(z)H2O,
где k является числом со значением от равного или больше 0 до меньше или равного 0,01, b является числом со значением от больше 0 до меньше или равного 0,05, n является числом меньше 0,025, например, меньше 0,001, таким как меньше 0,0005, например, меньше 0,00025, и z является числом со значением от больше или равного 0 до меньше или равного 0,2, и где F является фторид-ионом, X является трехвалентным элементом, таким как один или более из В, Al, Fe и Ga, особенно Al, Y является четырехвалентным элементом, таким как Si, Ge, Sn, Ti и Zr или их смеси, особенно Ge и/или Si, и Q является органическим направляющим агентом для формирования структуры.
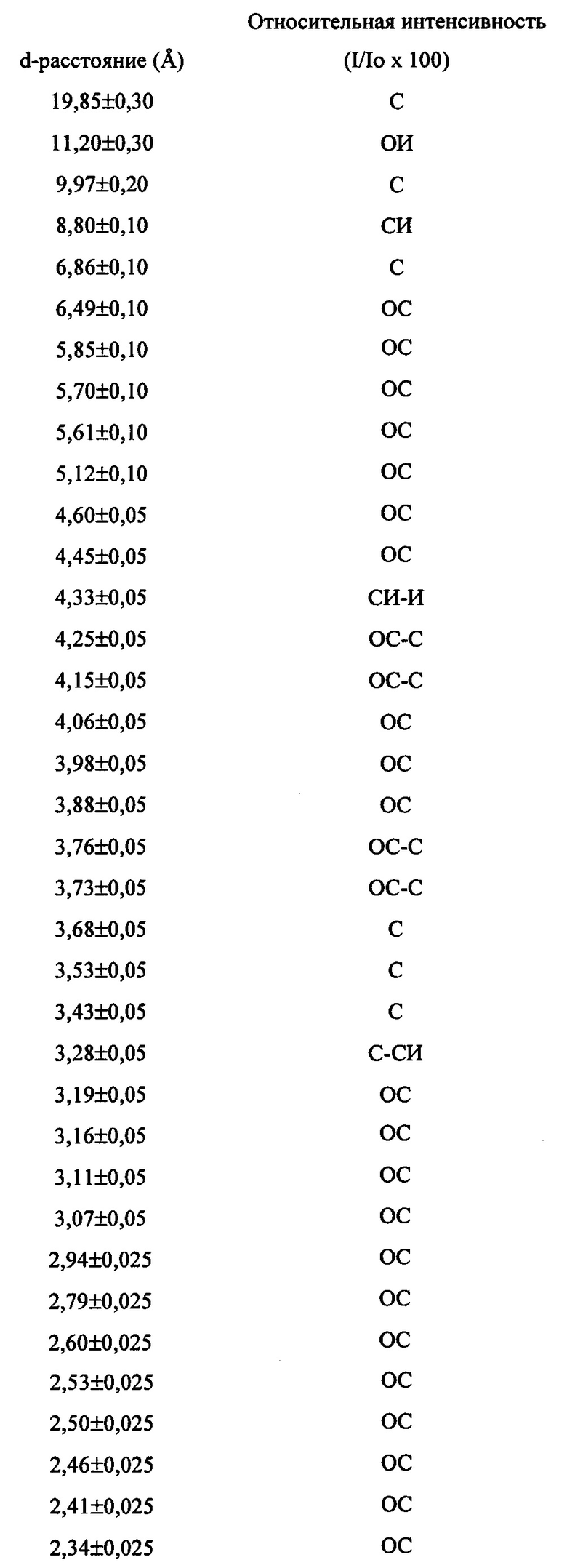
В одном из воплощений, Q выбирают из одного или более из дикатиона 1,1'-(1,3-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1) и дикатиона 1,1'-(1,4-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1), имеющих формулу I (мета) и II (пара), соответственно:
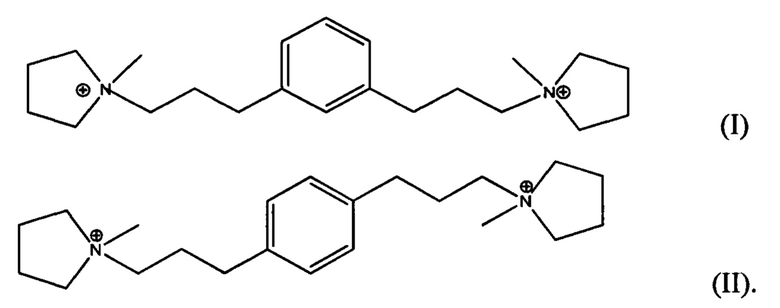
Компонент Q, который связан с материалом сразу после синтеза из-за присутствия во время кристаллизации, можно легко удалить с помощью обычных способов перекристаллизации.
Молекулярное сито ЕММ-28 является термически стабильным и в прокаленной форме обладает высокой площадью поверхности и значительной сорбционной емкостью в отношении углеводородов. Примеры сорбционной емкости и относительных скоростей сорбции перечислены ниже:

ЕММ-28 можно получить из смеси для синтеза, включающей источники воды, гидроксил-ионов, оксида четырехвалентного элемента Y, возможно трехвалентного элемента X, возможно источник фторид-ионов (F-) и органический направляющий агент (Q) для формирования структуры, описанный выше, при этом смесь имеет состав, выраженный в виде молярных отношений оксидов, находящихся в следующих диапазонах:


Подходящие источники четырехвалентного элемента Y зависят от выбранного элемента Y; но в предпочтительных воплощениях, в которых Y является кремнием и/или германием, включают коллоидные суспензии диоксида кремния, осажденный диоксид кремния, пирогенный диоксид кремния, силикаты щелочных металлов, тетраалкилортосиликаты и оксид германия. Если присутствует, трехвалентный элемент X, как правило, является алюминием, а подходящие источники алюминия включают гидратированный оксид алюминия, гидроксид алюминия, алюминаты щелочных металлов, алкоксиды алюминия и водорастворимые соли алюминия, такие как нитрат алюминия. Если присутствуют, подходящие источники фторид-ионов включают один или более из HF, NH4F и NH4HF2.
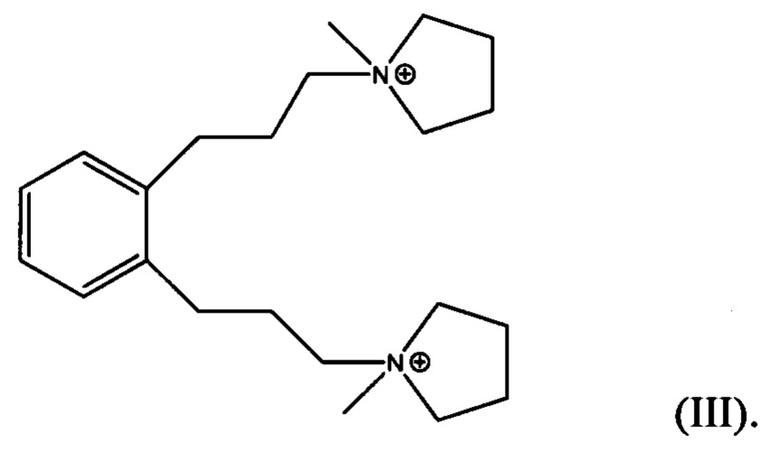
Подходящими источниками Q являются гидроксиды и/или соли соответствующих дичетвертичных аммониевых соединений. Считают, что сами соединения являются новыми и могут быть синтезированы из 1,3-bis(галогенметил)бензолов (для соединений формулы I) и из 1,4-bis, (галогенметил)бензолов (для соединений формулы II) согласно методике, описанной ниже. Кроме того, соединения формулы I и II можно получить по реакции 1,3- и 1,4-дигалогенбензолов с определенными производными пропина по реакции сочетания Соногаширы (Sonogashira coupling reaction). Более подробно указанный режим синтеза тоже обсуждается ниже. Аналогичные реакции с 1,2-bis(галогенметил)бензолами и 1,2-дигалогенбензолами в качестве исходных можно использовать для получения орто-изомера соединений формулы I и II, а именно дикатиона 1,1'-(1,2-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1), имеющего формулу III, приведенную ниже:
Реакционная смесь для получения молекулярного сита ЕММ-28 может также содержать затравку кристаллического материала, такого как ЕММ-28, из предшествующего синтеза, желательно в количестве от 0,01 ppm по массе до 500000 ppm по массе, например, от 100 ppm по массе до 5000 ppm по массе реакционной смеси.
Реагенты, как правило, обычно смешивают механическим путем, например, перемешиванием или смешиванием с высокой скоростью сдвига, для обеспечения подходящей гомогенизации смеси для синтеза. В зависимости от природы реагентов может оказаться необходимым уменьшить количество воды в смеси перед кристаллизацией для получения предпочтительного молярного отношения H2O/YO2. Подходящие способы снижения количества воды представляют собой упаривание в статичной или проточной среде, такой как воздух окружающей среды, сухой азот, сухой воздух, или с помощью сушки распылением или лиофильной сушки.
Кристаллизацию ЕММ-28 можно проводит в статических условиях или при перемешивании в подходящем сосуде для проведения реакции, таком, например, как полипропиленовые сосуды или автоклавы с тефлоновым вкладышем или из нержавеющей стали, при температуре от примерно 100°С до примерно 200°С в течение периода времени, достаточного для протекания кристаллизации при применяемой температуре, например, от примерно 1 дня до примерно 14 дней. Затем кристаллы отделяют от жидкости и извлекают.
В степени, достаточной и зависящей от молярного отношения X2O3/YO2 в материале, любые катионы в ЕММ-28 сразу после синтеза можно замещать в соответствии с методиками, хорошо известными в уровне техники, путем ионного обмена на другие катионы. Предпочтительные замещающие катионы включают ионы металлов, ионы водорода, предшественник водорода, например, ионы аммония и их смеси. Особенно предпочтительными являются катионы, которые обеспечивают каталитическую активность в некоторых реакций превращения углеводородов. Они включают водород, редкоземельные металлы и металлы групп 2-15 Периодической системы химических элементов. Используемая в данной заявке система нумерации групп Периодической системы раскрыта в Chemical and Engineering News, 63(5), 27 (1985).
Молекулярное сито, описанное в данной заявке, может быть подвергнуто обработке для удаления части или всего органического направляющего агента Q для формирования структуры, используемого в процессе синтеза молекулярного сита. Это удобно осуществлять путем термической обработки, при которой материал сразу после синтеза нагревают при температуре по меньшей мере примерно 370°С в течение по меньшей мере 1 минуты и обычно не дольше 20 часов. Хотя при термообработке можно применять пониженное атмосферное давление, атмосферное давление является желательным по причинам удобства. Термообработку можно осуществлять при температуре до примерно 925°С. Термически обработанный продукт, особенно в соответствующей металлической, водородной и аммониевой формах, особенно подходит для применения при катализе некоторых реакций превращения органических соединений, например, углеводородов.
Молекулярное сито по настоящему изобретению можно тщательно объединить с гидрирующим компонентом, таким как молибден, вольфрам, рений, никель, кобальт, хром, марганец или благородный металл, такой как платина или палладий, в тех случаях, когда должна быть осуществлена функция гидрирования-дегидрирования. Такой компонент может присутствовать в составе композиции при совместной кристаллизации, быть введен путем обмена в композицию до степени элемента группы IIIА, например, алюминия, присутствующего в структуре, быть пропитан в ней или тщательно смешан с ней физически. Такой компонент может быть пропитан в ней или на ней, так чтобы, например, в случае платины, при обработке силиката раствором, содержащим ион платинового металла. Таким образом, подходящие соединения плтины для этой цели включают хлорплатиновую кислоту, хлористую платину и различные соединения, содержащие аминные комплексные соединения платины.
Молекулярное сито по настоящему изобретению при применении либо в качестве сорбента, либо катализатора, нужно по меньшей мере частично дегидратировать. Это можно осуществить путем нагревания до температуры от 200°С до примерно 370°С в такой атмосфере, как воздух, азот и т.д., и при атмосферном, пониженном атмосферном или повышенном атмосферном давлении в течение от 30 минут до 48 часов. Дегидратацию можно еще осуществлять при комнатной температуре, в основном путем размещения ЕММ-28 под вакуумом, но требуется больше времени для получения достаточной степени дегидратации.
Молекулярное сито по настоящему изобретению можно использовать в качестве сорбента или, в частности в его алюмосиликатной форме, в качестве катализатора для катализа широкого спектра процессов превращения органических соединений, включая многие из тех, что имеют актуальную коммерческую/промышленную значимость. Примеры процессов химических превращений, которые эффективно катализируются с помощью кристаллического материала по настоящему изобретению, взятого отдельно или в сочетании с одним или более другими каталитически активными веществами, включая другие кристаллические катализаторы, включают процессы, в которых требуется катализатор с кислотной активностью. Примеры процессов превращения органических соединений, которые можно катализировать с помощью ЕММ-28, включают крекинг, гидрокрекинг, диспропорционирование, алкилирование, олигомеризацию и изомеризацию.
Как в случае многих катализаторов, может быть желательным включать ЕММ-28 с другим материалом, устойчивым к температурам и другим условиям, применяемым в процессах превращения органических соединений. Такие материалы включают активные и неактивные материалы, синтетические или природные цеолиты, а также неорганические материалы, такие как глины, диоксид кремния и/или оксиды металлов, такие как оксид алюминия. Последний может быть как природным, так и в форме желатиновых осадков или гелей, включая смеси диоксида кремния и оксидов металлов. Применение материала в сочетании с ЕММ-28, то есть соединенного с ним или присутствующего во время синтеза нового кристалла, являющегося активным, имеет склонность к изменению направления превращения и/или селективности катализатора в некоторых процессах превращения органических соединений. Неактивные материалы подходящим образом служат в качестве разбавителей для контроля степени превращения в заданном процессе, так что продукты можно получить экономичным и упорядоченным способом, без применения других средств для регулирования скорости реакции. Такие материалы можно включать в природные глины, например, бентонит и каолин, для улучшения устойчивости к дроблению катализатора в коммерческих условиях работы. Указанные материалы, то есть глины, оксиды и т.д., функционируют в качестве связующих для катализатора. Желательно обеспечить катализатор, имеющий хорошую устойчивость к дроблению, так как при коммерческом применении желательно предотвратить разрушение катализатора до порошкообразных материалов. Такие связующие на основе глины и/или оксида обычно использовали только в целях улучшения устойчивости к дроблению катализатора.
Природные глины, которые могут находиться в составе с ЕММ-28, включают виды монтмориллонита и каолина, виды которых включают суббентониты, при этом каолины широко известны как глины месторождений Dixie, McNamee, Georgia и Florida или другие, в которых основным компонентом минерал является галлуазит, каолинит, диккит, накрит или аноксит. Такие глины можно использовать как в необработанном состоянии в виде оригинально добытых или сначала подвергнутых прокаливанию, кислотной обработке или химической модификации. Связующие, используемые для смешивания с ЕММ-28, включают также неорганические оксиды, такие как диоксид кремния, диоксид циркония, диоксид титана, оксид магния, оксид бериллия, оксид алюминия и их смеси.
Помимо указанных выше материалов, ЕММ-28 может быть смешан с пористым матричным материалом, таким как диоксид кремния-оксид алюминия, диоксид кремния-оксид магния, диоксид кремния-диоксид циркония, диоксид кремния-оксид тория, диоксид кремния-оксид бериллия, диоксид кремния-диоксид титана, а также в виде третичных композиций, таких как диоксид кремния-оксид алюминия-оксид тория, диоксид кремния-оксид алюминия-диоксид циркония, диоксид кремния-оксид алюминия-оксид магния и диоксид кремния-оксид магния-диоксид циркония.
Относительные доли ЕММ-28 и матрицы в виде неорганических оксидов могут широко варьироваться, причем содержание ЕММ-28, находящееся в диапазоне от примерно 1 до примерно 90 процентов по массе и более предпочтительно, когда смесь получают в форме стеклянных шариков, в диапазоне от примерно от 2 до примерно 80 массовых процентов смеси.
Примеры
Настоящее изобретение будет теперь более подробно описано со ссылкой на следующие неограничивающие примеры и прилагаемые чертежи.
Синтез органических направляющих агентов для формирования структуры (Формулы I, II и III)
Как обсуждалось выше, органические направляющие агенты для формирования структуры, Q, используемые в синтезе ЕММ-28, можно получить из 1,3-bis(галогенметил)бензолов (для соединений формулы I) и из 1,4-bis(галогенметил)бензолов (для соединений формулы II).
Подходящий режим синтеза соединения формулы I из 1,3-bis(хлорметил)бензола описан ниже.
Получение 3,3'-(1,3-фенилен)дипропионовой кислоты
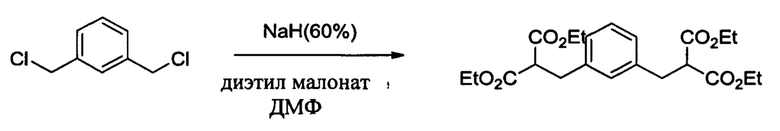
Высушенную в термостате 2 литровую 3-горлую колбу с кожухом, снабженную механической мешалкой, собирали в горячем виде и охлаждали под током N2, затем загружали 67,2 г (1680 ммоль) 60%-ного гидрида натрия в минеральном масле. Содержимое охлаждали до 0°С с помощью циркулирующей смеси гликоль-вода и 670 мл безводного ДМФ, добавляемого через мензурку. 360 мл (2,37 моль) диэтилмалоната по каплям добавляли в колбу в течение 40 минут. Примерно через половину времени процесса добавления сливали охладитель и обеспечивали рост температуры до 35°С. Весь NaH растворяли и раствор становился прозрачным. К нему добавляли 102,1 г (582 ммоль) 1,3-bis(хлорметил)бензола весь за одну порцию. Температура росла до 65°С и образовывалось твердое вещество. После нагревания паром в течение 1 часа колбу охлаждали до 0°С и добавляли раствор 37 мл концентрированной НСl в 1000 мл Н2O. Содержимое колбы затем перемещали в разделительную воронку, где вязкий нижний слой сливали в качестве продукта. После удаления летучих веществ на роторном испарителе продукт перегоняли при 125°С и 250 мТорр (примерно 33 кПа) с получением 228 г (86%) белого жидкого продукта.
Получение тетраэтил-2,2'-(1,3-феншен-bis(метилен))дималоната
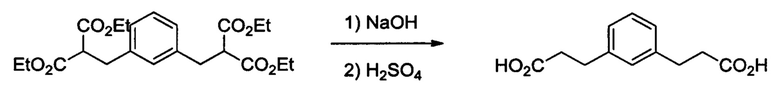
3,3'-(1,3-фенилен)дипропионовой кислоты выливали в 121 г (3,02 моль) NaOH в 640 мл Н2O и промывали 25 мл этанола. Смесь нагревали при кипении с обратным холодильником в течение 45 мин (гомогенной по достижении кипения), затем перегоняли 325 мл через 6'' дистилляционную колонку Вигро (Vigreux). Температура кипения последних 100 мл составляла 100-101°С. Раствор охлаждали и 152 г концентрированной H2SO4 добавляли со скоростью, поддерживающей кипение. Добавляли кипелки и смесь нагревали при кипении с обратным холодильником до тех пор, пока не переставал выделяться СО2 (в течение ночи). Получали желтое масло, плавающее в колбе, и некоторое количество твердого вещества. Смесь выливали в 2 л Н2O, экстрагировали 2×200 мл диэтилового эфира, затем экстракт промывали 1×200 мл насыщенного NaCl и фильтровали через 0,4 нм (4) молекулярное сито. Растворитель удаляли на роторном испарителе и отгоняли оставшиеся летучие вещества при температуре 120°С и 260 мТорр (примерно 34,5 кПа) с получением 110,5 г (100%) бледно-коричневого воска. Продукт имел ожидаемые спектры ЯМР 13С и 1Н. 13С ЯМР(СОСl3): δ, м.д.: 174,2 (Cq), 141,3 (Cq), 128,7 (СН), 128,6 (СН), 126,3 (СН), 35,7 (СН2), 30,8 (СН). 1Н ЯМР (СDСl3): δ, м.д.: 12,12 (s, 1,9Н), 7,19 (m, 1,1Н), 7,05 (m, 3,2H), 2,80 (t, 3,9H), 2,53 (t, 3,8Н).
Получение 3,3'-(1,3-фенилен)bis(пропан-1-ола)
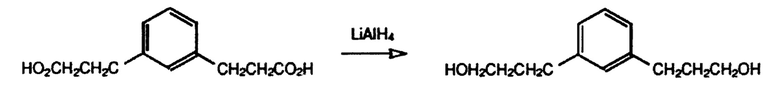
Высушенную в термостате 3-горлую колбу на 3 л с кожухом, снабженную уравновешивающей капельной воронкой, обратным холодильником и механической мешалкой, собирали в горячем виде и охлаждали под током N2, затем загружали в нее 920 г безводного ТГФ и 30,55 г (805 ммоль) пластинок LiAlH4. Смесь перемешивали в течение 30 мин, затем по каплям в течение 1 час добавляли 110 г (498 ммоль) тетраэтил-2,2'-(1,3-фениленbis(метилен))дималоната в 250 г безводного ТГФ. Через три четверти периода добавления твердое вещество стало очень трудно перемешивать. Добавление 300 мл безводного ТГФ снова сделало суспензию перемешиваемой. Реакция в процессе добавления была экзотермической и наблюдалось образование газа (Н2). Смесь кипятили с обратным холодильником в течение 20 мин, охлаждали до 0°С и выливали в 150 мл 1:1 (об/об) Н2O:ТГФ, затем 42,7 г NaOH в 427 г Н2O. Продукт отфильтровывали через воронку Бюхнера и твердый остаток промывали 500 мл диэтилового эфира. После удаления растворителя на роторном испарителе, оставшиеся летучие вещества удаляли путем вакуумной перегонки при 100°С и 2 мТорр (266 Па) с получением 70,2 г (74%) белого полутвердого вещества. Продукт имел ожидаемые спектры 13С ЯМР и 1Н ЯМР. 13С ЯМР (СDСl3): δ, м.д.: 142,0 (Cq), 128,7 (СН), 128,4 (СН), 125,9 (СН), 61,9 (СН2), 34,2 (СН2), 32,0 (СН2). 1Н ЯМР (СDСl3): δ, м.д.: 7,20 (m, 1,2Н), 7,04 (m, 3,3Н), 3,64 (m, 3,8Н), 3,05 (b, 1,8Н), 2,68 (m, 4,1Н), 1,89 (m, 3,8Н).
Получение 1,3-фениленbis(пропан-3,1-диил)bis(4-метилбензолсульфоната)
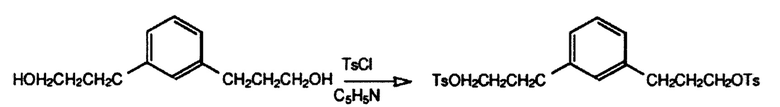
1 литровую колбу, содержащую 70,2 г (362 ммоль) 3,3'-(1,3-фенилен)bis(пропан-1-ола), 260 мл пиридина и 480 мл СНСl3 (стабилизированого амиленом) охлаждали до -5°С с помощью циркулирующей смеси гликоль-вода и в одну порцию добавляли 138 г (723 ммоль) n-толуолсульфонилхлорида. Температура росла до 25°С, охладление прекращали и после перемешивания в течение 45 мин смесь выливали в 1000 мл Н2О + 212 мл конц. НСl. Нижний слой отделяли и промывали 100 мл насыщенного раствора NaCl. Оставшийся растворитель удаляли на роторном испарителе и оставшиеся летучие вещества удаляли путем вакуумной перегонки при 60°С и 650 мТорр (86 кПа) с получением 171 г (94%) коричневой смолы. Продукт имел ожидаемые спектры 13С ЯМР и 1Н ЯМР. 13С ЯМР (СDСl3): δ, м.д.: 144,8 (Cq), 140,6 (СН), 133,0 (СН), 129,0 (СН), 127,8 (СН), 126,3 (СН), 69,8 (СН2), 31,4 (СН2), 30,3 (СН2), 21,6 (СН3). 1Н ЯМР (СDСl3): δ, м.д.: 7,81 (d, 4,1Н), 7,36 (d, 3,8Н), 7,13 (m, 1,4Н), 6,90 (m, 3,3Н), 4,04 (t, 4,1H), 2,61 (t, 4,7H), 2,46 (s, 4,7Н), 1,94 (m, 4,1Н).
Получение 1,3-bis(3-(пирролидин-1-ил)пропил)бензола
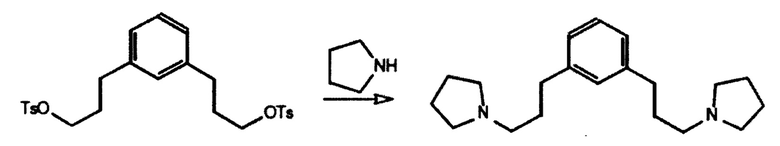
116 г (231 ммоль) 1,3-фениленbis(пропан-3,1-диил)bis(4-метилбензолсульфоната) обрабатывали 160 мл (1,92 моль) пирролидина. Смесь темнела и экзотерма достигала температуры кипения. Смесь выливали горячей в 800 мл Н2O, содержащей 80 г NaOH. Слои разделяли и водный слой экстрагировали 1х350 мл диэтилового эфира. Органические слои объединяли, промывали 1×200 мл Н2O и удаляли летучие вещества на роторном испарителе перед перегонкой продукта при 220°С и 180 мТорр (24 кПа) с получением 54,9 г желтого масла и твердого вещества. ГХ-МС (газовая хроматография с масс-спектроскопией) показывает ожидаемый продукт с m/z=300 (интенсивный М-1 пик), который, однако, в большой степени загрязнен 1-тозилпирролидином. По оценкам методом 1Н ЯМР, выход продукта составляет 77% и 81%, по оценке методом 13С ЯМР, получают, что общий выход составляет примерно 140 ммоль. 13С ЯМР соответствует ожидаемому продукту вместе с 1-тозилпирролидином.
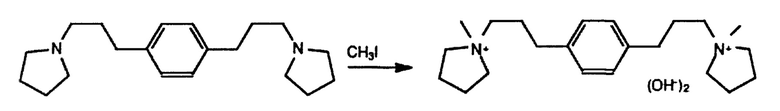
Получение гидроксида 1,1'-(1,3-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1)
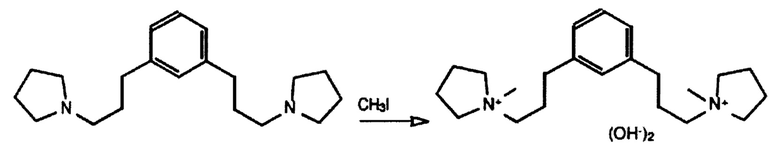
1,3-bis(3-(пирролидин-1-ил)пропил)бензол растворяли в 150 мл ацетона в колбе Эрленмейера и 40 мл (680 ммоль) йодометана постепенно добавляли в течение примерно 15 мин. Раствор доводили до спокойного кипения с обратным холодильником и осаждалось много твердого вещества. Колбу фиксировали, оборачивали в Аl фольгу, оставляли при стоянии в течение двух дней при комнатной температуре, отфильтровывали, промывали диэтиловым эфиром и сушили до постоянной массы при 65°С с получением 77,3 г розового твердого вещества (94% на основе оцененной чистоты диамина). Его подвергали ионному обмену в периодическом режиме с получением 371 г светло-желтого раствора. На титрование 2,62 мл указанного раствора, разбавленного до 25 мл, потребовалось 6,19 мл для титрования 91,1 мг фталата калия. Вычисления показывают, что получено 12,5% в виде дигидроксида. Интегрирование протонных сигналов органического вещества в спектре 1Н ЯМР относительно сигнала воды дало 12,7% в виде дигидроксида. Продукт имел ожидаемые спектры 13С ЯМР, 1Н ЯМР и 14N ЯМР. 13С ЯМР (d6-ОМSO): δ, м.д.: 141,1 (Сq), 129,0 (СН), 128,9 (СН), 126,6 (СН), 64,0 (СН2), 63,2 (СН2), 48,3 (СН3), 32,2 (СН2), 25,3 (СН2), 21,6 (СН2). 1Н ЯМР (D2O): δ, м.д.: 7,25 (m, 2,1Н), 7,14 (m, 2,1Н), 3,48 (m, 12,2Н), 3,04 (s, 5,9Н), 2,62 (t, 3,9H), 2,07 (m, 11,8Н) 14N ЯМР (D2O): δ, м.д.: 52,6 (⊗v1/2=8 Гц, вычисленный сдвиг 54,0 ppm).
В модификации указанного выше режима синтеза дикатиона формулы I, 1,3-фениленbis(пропан-3,1-диил)bis(4-метилбензолсульфонат), взаимодействует с 1-метилпирролидином в хлороформе или ацетонитриле с непосредственным получением дикатиона без промежуточного образования диамина.
Дополнительно и альтернативно, другой подходящий режим синтеза соединения формулы I из 1,3-дийодобензола описан ниже.
Получение 3,3'-(1,3-феншен)bis(про-2-ин-1-ола)
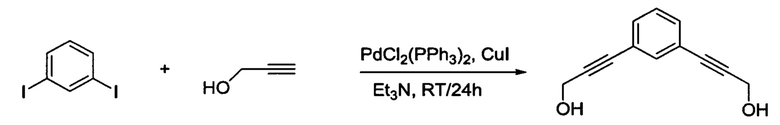
В высушенную в термостате 3-горлую круглодонную колбу на 2 л, снабженную механической мешалкой, добавляли 1,3-дийодобензол (36,0 г; 109,1 ммоль) в 225 мл сухого триэтиламина под током азота. В светло-коричневый раствор после иодида меди(I) (0,33 г; 1,74 ммоль, 0,015 мол. %) добавляли дихлорид bis(трифенилфосфин)палладия(II) (4,2g; 6,0 ммоль, 0,05 мол. %). Темно-зеленую смесь перемешивали в течение 5 минут. Пропаргиловый спирт (21,5 мл; 371,2 ммоль) по каплям добавляли через капельную воронку. Наблюдали небольшую экзотерму после добавления. Темно-коричневую смесь перемешивали в течение 3 часов при комнатной температуре. ТСХ (5% этилацета / гексана при УФ-детектировании) показала, что не осталось исходного материала. Реакционную смесь перемешивали в течение ночи, добавляли 1500 мл этилацетата и продолжали перемешивание в течение дополнительных 24 часов. Реакционную смесь отфильтровывали и фильтрат концентрировали in vacuo с извлечением 30,4 г коричневого масла. Неочищенный продукт очищали на силикагеле при использовании постоянного градиента 40-100% этилацетат/гексан с извлечением 15,0 г (74%) целевого продукта. 1Н ЯМР 400 МГц (CDCl3) δ, м.д.: 7,51 (1Н), 7,40, (1Н), 7,38 (1Н), 7,29 (1Н), 4,50-4,49 (d, 4Н), 1,71-1,68 (2Н). 13С ЯМР 100 МГц (CDCl3) δ, м.д.: 134,6, 131,6, 128,4, 122,8, 87,9, 84,7 и 51,4.
Получение 3,3'-(1,3-феншен)bis(пропан-1-ола)
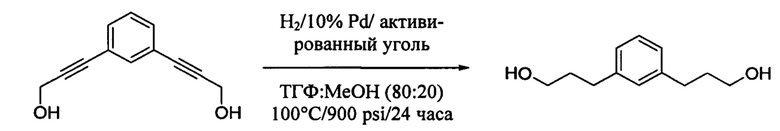
3,3'-(1,3-фенилен)bis(проп-2-ин-1-ол) (2,0 г; 10,7 ммоль) растворяли в 11 мл безводного метанола и помещали в автоклав с тефлоновым вкладышем. Суспензию палладия на активированном угле (0,4 г; 10%-ный палладий на активированном угле) в 40 мл сухого ТГФ добавляли во вкладыш под шапкой азота. Автоклав закрывали и под давлением заполняли Н2. Через 24 часа реакционный раствор отфильтровывали через слой целита. Фильтрат концентрировали in vacuo с извлечением 2,1 г (100%) неочищенного целевого продукта. Данный продукт использовали дальше без очистки. 1Н ЯМР 400 МГц (CDCl3) δ, м.д.: 7,22-7,18 (1Н), 7,04 (1Н), 7,02 (2Н), 3,69-3,64 (4Н), 2,70-2,63 (4Н), 1,92-1,85 (4Н) 1,51-1,49 (2Н). 13С ЯМР 100 МГц (CDCl3) δ, м.д.: 141,9, 128,6, 128,4, 125,9, 62,3, 34,2 и 32,0.
Получение 1,3-фениленbis(пропан-3,1-диил)bis(4-метшбензолсулъфоната)
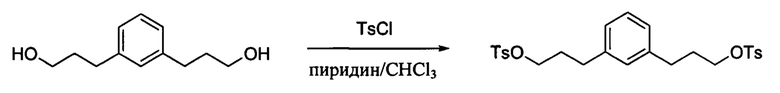
В сухой флакон объемом 25 мл с резиновой прокладкой добавляли при комнатной температуре под током азота 3,3'-(1,3-фенилен)bis(пропан-1-ола) (0,2 г; 1,0 ммоль), растворенного в 2,0 мл безводного хлороформа. Добавляли пиридин (0,17 мл; 2,1 ммоль) и охлаждали раствор до 0°С (ледяная баня). Добавляли n-толуолсульфонилхлорид (0,43 г; 2,2 ммоль) и оставляли светло-оранжевый раствор нагреваться до комнатной температуры. Через 24 часа реакционную смесь разбавляли 10 мл 5% НСl и разделяли слои. Органический слой промывали 10 мл солевого раствора и предварительно адсорбировали на силикагеле. Неочищенный продукт очищали на силикагеле при постоянном градиенте от 0 до 100% этилацетата/гексана с извлечением 0,28 г (55%) целевого соединения. 1Н ЯМР 400 МГц (CDCl3) δ, м.д.: 7,79-7,77 (4Н), 7,35-7,33 (4Н), 7,11 (1Н), 6,90-6,85 (3Н), 4,03-4,00 (4H), 2,61-2,58 (4Н), 2,45 (6Н), 1,94-1,91 (4Н). 13С ЯМР 100 МГц (CDCl3) δ, м.д.: 144,7, 140,6, 133,1, 129,8, 128,6, 127,9, 126,2, 69,6, 31,4, 30,4 и 21,6.
Получение 4-метшбензолсулъфоната 1,1'-(1,3-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1)

В сухой флакон на 20 мл с предохранительной крышкой и магнитным элементом добавляли 1,0 г (1,9 ммоль) 1,3-фениленbis(пропан-3,1-диил)bis(4-метилбензолсульфоната) под током азота. Добавляли 2 мл сухого ацетонитрила и слабо-желтый раствор перемешивали в течение 5 минут. По каплям добавляли метилпирролидин (0,64 мл; 6,0 ммоль) и перемешивали раствор при комнатной температуре в течение одного часа. ТСХ (2:1 гексан: этилацетат, УФ-детектирование) показала, что остался исходный материал. Раствор нагревали до 80°С. Через один час ТСХ показала, что исходный материал израсходован. Реакционную смесь охлаждали до комнатной температуры и выдерживали в течение ночи. Реакционный раствор концентрировали in vacuo при 45°С с извлечением 1,2 г (96%) целевого продукта. 1Н ЯМР 400 МГц (CD3CN) δ, м.д.: 7,64-7,62 (4Н), 7,26-7,23 (2Н), 7,18 (4Н), 7,11-7,09 (2Н), 3,45-3,34 (8Н), 3,33-3,31 (4Н), 2,95 (6Н), 2,64-2,58 (4Н), 2,34 (6Н), 2,08-2,04 (12Н). 13С ЯМР 100 МГц (CD3CN) δ, м.д.: 145,4, 140,4, 138,3, 128,3, 128,2, 128,0, 126,0, 125,3, 117,0, 63,7, 63,0, 47,5, 31,4, 24,7, 23,3, 20,8 и 19,9.
Получение гидроксида 1,1'-(1,3-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1)
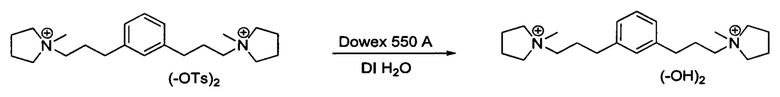
545 г смолы Dowex Monosphere 550А помещали в бутыль Nalgene с завинчивающейся крышкой на 500 мл. Смолу промывали 3×500 мл деионизированной водой для удаления любых мелких частиц. 27,3 г (40,5 ммоль) 4-метилбензолсульфоната 1,1'-(1,3-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1) растворяли в 100 мл деионизированной воды и добавляли в контейнер Nalgene. Деионизированную воду добавляли в контейнер до заполнения 80%. Верх контейнера закрывали и запечатывали.
Контейнер размещали на механическом вращающемся устройстве в течение ночи для ускорения анионного обмена. Суспензию отфильтровывали через воронку Бюхнера и промывали деионизированной водой до рН 9. Водный раствор концентрировали in vacuo при 40°С до желательной концентрации для получения дигидроксида. 1Н ЯМР 400 МГц (D2O) δ, м.д.: 7,22-7,18 (1Н), 7,04-7,03 (3Н), 3,34-3,18 (7Н), 3,17-3,14 (4Н), 2,84 (6Н), 2,57-2,54 (4Н), 2,18-1,92 (12Н). 13С ЯМР 100 МГц (D2O) δ, м.д.: 140,9, 129,0, 128,5, 126,5, 64,2, 63,4, 47,9,31,5, 24,7 и 21,1.
Подходящий режим синтеза соединения формулы II из 1,4-bis, (хлорметил)бензола описан ниже.
Получение тетраэтил 2,2'-(1,4-фениленbis(метилен))дималоната
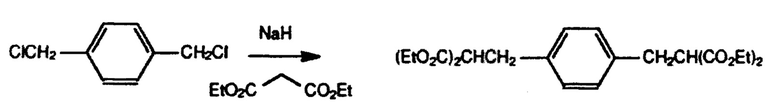
Высушенную в термостате 3-горлую колбу на 1 л с кожухом, снабженную механической мешалкой, собирали в горячем состоянии и охлаждали под током N2, затем загружали 29,6 г 60%-ного гидрида натрия в минеральном масле. Содержимое охлаждали до -5°С циркулирующей смесью гликоль-вода и с помощью мензурки добавляли 400 мл безводного ДМФ. 150 мл (1 моль) диэтилмалоната по каплям добавляли в колбу в течение 40 минут и затем нагревали содержимое до 35°С. Через один час весь NaH был растворен и полученный раствор был прозрачным. К нему добавляли 40 г (228 ммоль) 1,4-bis(хлорметил)бензола за одну порцию. Температура возрастала до 68°С и образовывалось твердое вещество. После нагревания паром в течение 30 мин колбу охлаждали до 15°С и добавляли раствор 20 мл концентрированной НСl в 400 мл Н2O. Содержимое колбы переносили в делительную воронку, где в качестве продукта сливали вязкий нижний слой. После удаления летучих веществ на роторном испарителе продукт перегоняли при 80°С и 190 мТорр (примерно 25 кПа) с получением 94,3 г (98%) целевого жидкого продукта. ЯМР показывает наличие следовых количеств диэтилмалоната. Продукт имел ожидаемые спектры 13С ЯМР и 1Н ЯМР. 13С ЯМР (СDСl3): δ, м.д.: 168,7 (Cq), 128,8 (СН), 136,4 (Cq), 61,3 (СН2), 53,7 (СН), 34,1 (СН2), 13,9 (СН3) 1Н ЯМР (СDСl3): δ, м.д.: 7,07 (s, 3,9H), 3,56 (t, 2,1H), 3,12 (d, 3,9Н).
Получение 3,3'-(1,4-фенилен)дипропановой кислоты

94,3 г тетраэтил-2,2'-(1,4-фениленbis(метилен))дималоната нагревали при кипении с обратным холодильником в течение 90 мин с 300 мл Н2O, 50 мл этанола и 50,3 г (1250 ммоль) NaOH. Этанол удаляли на роторном испарителе с получением 320 г остатка. Остаток экстрагировали 100 мл пентана для удаления минерального масла, оставшегося от гидрида натрия (2,1 г масла, полученного при упаривании пентана). Водный раствор подкисляли до рН 1,5 при использовании 63 г (642 ммоль) H2SO4, которую добавляли по каплям в течение 20 минут. Температура возрастала до 62°С. Барботер присоединяли для наблюдения за выделением СО2 и смесь нагревали при кипении с обратным холодильником до полного исчезновения выделения газа (кипятили с обратным холодильником в течение 48 часов, но выделение газа наблюдали только в течение первых 6 часов). Продукт отфильтровывали и сушили до постоянной массы при 80°С с получением 43,8 г (101%) продукта. Продукт имел ожидаемые спектры 13С ЯМР и 1Н ЯМР. 13С ЯМР (D2O+NaOH): δ, м.д.: 182,5 (Cq), 139,7 (Cq), 128,4 (СН), 39,4 (СН2), 31,5(СН2). 1Н ЯМР (D2O+NaOH): δ, м.д.: 7,12 (s, 3,9Н), 2,78 (t, 4,1Н), 2,40 (t, 3,9Н).
Получение диэтил 3,3'-(1,4-фенилен)дипропаноата

К 153 г 3,3'-(1,4-фенилен)дипропановой кислоты добавляли 800 мл USP этанола и 5 мл концентрированной H2SO4 и нагревали смесь при кипении с обратным холодильником в течение 2 часов, охлаждали, сливали с 14 г K2СО3 в 30 мл Н2O и помещали в холодильник при 0°С. После кристаллизации в течение ночи отфильтровывали твердое вещество и сушили до постоянной массы при 55°С в течение 2 недель. Образцы, взятые в промежуточные моменты времени, показывали наличие остаточной Н2O. Полученное твердое вещество 134,8 г (70%) было свободно от воды по данным метода ЯМР. ГХ-МС показал, что продукт имеет ожидаемый m/z=278, а содержащийся в следовых количествах компонент с более длительным временем удерживания и m/z=277, как полагают, является сложным триэфиром из не полностью декарбоксилированной тетракислоты. Продукт имел ожидаемые спектры 13С ЯМР и 1Н ЯМР. 13С ЯМР (СDСl3): δ, м.д.: 172,0 (Cq), 138,5 (Сq), 128,4 (СН), массы (3 часа) при 60°С и 1 мТорр (примерно 133 Па) с получением 219 г (91%) материала приемлемой чистоты по данным метода 13С ЯМР.
Получение 1,4-bis(3-(пирролидин-1-ил)пропил)бензола
146 г (291 ммоль) 1,4-фениленbis(пропан-3,1-Диил)bis(4-метилбензолсульфоната) растворяли в 440 мл теплого СНСl3 (стабилизированного амиленом) и добавляли 141 г (1,99 моль) пирролидина за одну порцию. При нагревании наблюдали небольшую экзотерму. Смесь оставляли при кипении с обратным холодильником в течение ночи, во время чего она становилась черной. Ее охлаждали до 0°С и выливали в 80 г NaOH в 800 мл Н2O, добавляли 400 мл Н2O и 200 мл диэтилового эфира и сливали нижний, очень черный слой. К оставшейся смеси добавляли 300 мл Н2O и снова сливали нижний слой. Объединенные нижние слои упаривали на роторном испарителе при 95°С и 50 мбар (5 кПа) и перегоняли черное масло при 200°С и 140 мТорр (примерно 19 кПа) с получением 82,8 г (95%) желтого масла, которое подвергали дробной кристаллизации при стоянии. ГХ-МС продукта показал целевое соотношение m/z=300 и примерно 3% продукта (m/z=225) идентифицировали как 1-тозилпирролидин на основании базы данных ГХ-МС с достоверностью 97%. Продукт имел ожидаемые спектры 13С ЯМР и 1Н ЯМР. 13С ЯМР (СDСl3): δ, м.д.: 139,5 (Сq), 128,2 (СН), 56,0 (СН2), 54,1 (СН2), 33,5 (СН2), 30,7 (СН2), 23,4 (СН2). 1Н ЯМР (СDСl3): δ, м.д.: 7,04 (s, 4,2Н), 2,57 (t, 4,3H), 2,43 (m, 11H), 1,80-1,71 (m, 12,5Н).
Получение гидроксида 1,1'-(1,4-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиний-1)
К 41,4 г (138 ммоль) 1,4-bis(3-(пирролидин-1-ил)пропил)бензола в 150 мл ацетона в колбе Эрленмейера на 500 мл порциями добавляли 41 мл (99 g, 699 ммоль) йодометана в течение 15 мин. Раствор нагревали почти до кипения и осаждалось твердое вещество. Колбу фиксировали, оборачивали А1 фольгой и оставляли при стоянии при комнатной температуре в течение 48 часов, отфильтровывали, промывали 100 мл диэтилового эфира и сушили до постоянной массы при 60°С с получением 65,2 60,4 (СН2), 35,9 (СН2), 30,5 (СН2), 14,2 (СН3). 1Н ЯМР (СDСl3): δ, м.д.: 7,13 (s, 4,5Н), 4,13 (q, 3,8Н), 2,92 (t, 4,2Н), 2,60 (t, 3,8Н), 1,25 (t, 5,6Н). Получение 3,3'-(1,4-фенилен)bis(пропан-1-ола)
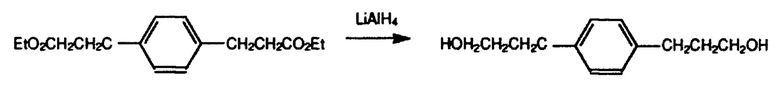
В 3-гордую высушенную в термостате колбу на 2 л с рубашкой, снабженную механической мешалкой, капельной воронкой и обратным холодильником, собранную в горячем виде и охлажденную под током N2, добавляли 300 мл безводного ТГФ и 8,4 г (221 ммоль) гранул LiАlН4. После быстрого перемешивания добавляли по каплям 49,7 г (179 ммоль) диэтил-3,3'-(1,4-фенилен)дипропаноата в 100 мл ТГФ в течение 30 мин и после этого 50 мл безводного ТГФ для промывания воронки. Смесь нагревалась и образовывалось значительное количество твердого вещества. Смесь нагревали при кипении с обратным холодильником в течение 20 минут, охлаждали и оставляли при стоянии в течение ночи, когда ее охлаждали путем добавления по каплям 42 мл Н2O + 42 мл ТГФ, а затем 12 г NaOH в 120 г Н2O. Надосадочную жидкость декантировали через сложенную воронкой фильтровальную бумагу, а твердое вещество промывали 100 мл Et2O и удаляли летучие вещества на роторном испарителе при поддержании температуры 85°С с помощью бани. После этого перегоняли остаток при 110°С и 250 мТорр (примерно 33 кПа) с получением 33,9 г (97%) продукта. Продукт имел ожидаемые спектры 13С ЯМР и 1Н ЯМР. 13С ЯМР (СDСl3): δ, м.д.: 139,3 (Сq), 128,4 (СН), 62,0 (СН2), 34,2 (СН2), 31,6 (СН2). 1Н ЯМР (СDСl3): δ, м.д.: 7,12 (s, 3,9Н), 3,65 (q, 3,9Н), 2,97 (t, 2,3Н), 1,88 (m, 4,0Н).
Получение 1,4-фениленbis(пропан-3,1-диил)bis(4-метилбензолсульфоната)

92,8 г (478 ммоль) 3,3'-(1,4-фенилен)bis(пропан-1-ола), 275 мл СНСl3 (стабилизированного амиленом) и 63 мл пиридина помещали в колбу с кожухом и охлаждали до 0°С при перемешивании магнитной мешалкой. К этому небольшими порциями добавляли 190 г (1000 ммоль) n-толуолсульфонилхлорида в течение 10 мин. Температура возрастала до 34°С. Смесь перемешивали 1 час, затем выливали в 655 мл Н2O + 140 мл концентрированной НСl, получая в результате рН<1. Нижний вязкий слой сливали и промывали 1×250 мл насыщенного раствора NaCl. Полученное масло высаживали в виде твердого вещества, которое размалывали и сушили до постоянной г (80%). Этот материал подвергали ионному обмену с помощью периодического способа, описанного ранее, с получением 163 г раствора. Для титрования 2,13 г этого раствора, разбавленного до 25 мл, понадобилось 7,41 мл, чтобы оттитровать 83,0 мг гидрофталата калия. Это соответствует вычислениям 11,7% дигидроксида. Интегрирование подвергнутого обмену раствора путем сравнения органического водорода с сигналом воды дало 12,0% дигидроксида. Продукт имел ожидаемые спектры 13С ЯМР, 1Н ЯМР и 14N ЯМР. 13С ЯМР (D2O): δ, м.д.: 139,6 (Cq), 128,9 (СН), 64,4 (СН2), 63,6 (СН2), 48,3 (СН3), 31,3 (СН2), 24,9 (СН2), 21,4 (СН2). 1Н ЯМР (D2O): δ, м.д.: 7,25 (s, 4,0Н), 3,43 (m, 8,3Н), 3,29 (m, 4,0Н), 2,97 (s, 5,9Н), 2,68 (t, 3,9H), 2,17 (m, 12,0Н). 14N ЯМР (D2O): δ, м.д.: 57,8 (⊗v1/2=14 Гц при вычисленном сдвиге 54,0 ppm).
При модификации указанного выше режима синтеза дикатиона формулы II 1,4-фениленbis(пропан-3,1-диил)bis(4-метилбензолсульфонат) реагировал с 1-метилпирролидином в хлороформе или ацетонитриле с непосредственным получением дикатиона без промежуточного получения диамина.
Специалисту в данной области техники будет понятно, что аналогичные реакции можно использовать для получения дикатиона 1,1'-(1,2-фениленbis(пропан-3,1-диил)bis(1-метилпирролидиния-1) формулы III из 1,2-bis(галогенметил)бензолов.
Аналогично, специалисту в данной области техники будет понятно, что аналогичные реакции можно использовать для получения дикатиона 1,1'-(1,4-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1) формулы II из 1,4-дигалогенбензолов.
Аналогично, специалисту в данной области техники будет понятно, что реакции, аналогичные описанным выше, можно использовать для получения дикатиона 1,1'-(1,2-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1) формулы III из 1,2-дигалогенбензолов.
Синтез ЕММ-28
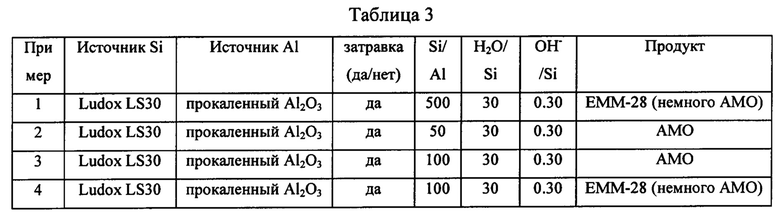
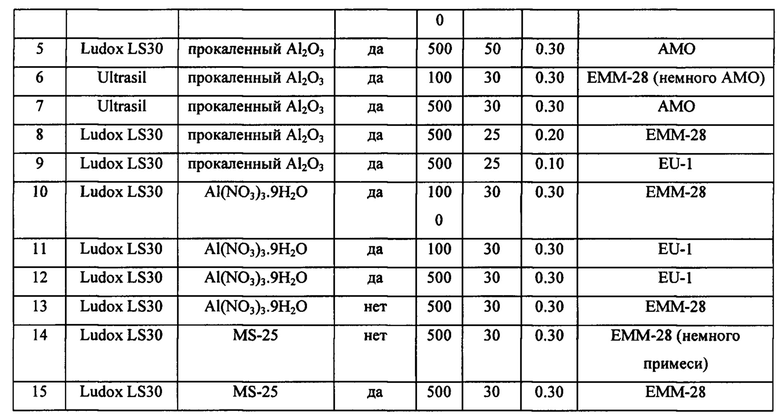
Пример 1
Синтетический гель получали при использовании Ludox LS30 в качестве источника кремния, прокаленного оксида алюминия в качестве источника алюминия и дикатиона формулы I (мета) в качестве органического направляющего агента Q для формирования структуры. Синтетический гель имел молярные отношения H2O/SiO2=30, OH-/SiO2=0,30, Q/SiO2=0,15 и Si/Al (атомное отношение) = 500.
Получение и кристаллизацию синтетического геля проводили в соответствии со следующей методикой. 9,6 г органического направляющего агента для формирования структуры смешивали с 0,5 г дистиллированной воды и затем в смесь добавляли 2,26 мг прокаленного оксида алюминия, а после этого 4,4 г Ludox LS30 и затравку 13,35 мг EMM-28. Смесь непрерывно перемешивали во время всей процедуры добавления и после этого перемешивали в течение еще одного часа, после чего смесь выливали в реактор Парра на 23 мл с тефлоновым вкладышем. После этого реактор нагревали в опрокидывающейся печи при 160°С в течение 14 дней.
Полученный продукт анализировали методом рентгеноструктурного анализа порошка, и, как показано в Таблице 3, обнаружили ЕММ-28 с небольшим количеством аморфной (АМО) примеси.
Примеры 2-4
Способ из Примера 1 повторяли при различных значениях Si/Al от 50 до 1000 и результаты показаны в Таблице 3.
Пример 5
Повторяли способ из Примера 1, но с молярным отношением H2O/SiO2=50. Полученный продукт анализировали методом рентгеноструктурного анализа порошка и обнаружили, что он по существу является аморфным (Таблица 3).
Примеры 6 и 7
Способ из Примера 1 повторяли при использовании Ultrasil в качестве источника кремния при значениях Si/Al от 100 до 500. Как показано в таблице 3, при значении Si/Al 100, при рентгеноструктурном анализе порошка выявляют, что продукт представляет собой ЕММ-28 с небольшим количеством аморфной (АМО) примеси, а при значении Si/Al 500 обнаружили, что продукт является по существу аморфным.
Пример 8
Способ из Примера 1 повторяли, снова используя Ludox LS30 в качестве источника кремния, но при Q/SiO2=0,1, H2O/SiO2=25, OH-/SiO2=0,20 и с 1% (масс.) затравки EMM-28 (в пересчете на диоксид кремния). Полученный продукт анализировали методом рентгеноструктурного анализа порошка, и, как показано в Таблице 3, он представляет собой ЕММ-28. Спектр порошковой дифрактограммы только что синтезированного материала из Примера 8 показан на Фиг. 1 (верхний спектр).
Пример 9
Способ из Примера 1 повторяли, снова используя Ludox LS30 в качестве источника кремния, но при Q/SiO2=0,05, H2O/SiO2=25, OH-/SiO2=0,10 и с 1% (масс.) затравки EMM-28 (в пересчете на диоксид кремния). Полученный продукт анализировали методом рентгеноструктурного анализа порошка, и, как показано в Таблице 3, он представляет собой EU-1. Спектр порошковой дифрактограммы только что синтезированного материала из Примера 9 показан на Фиг. 1 (нижний спектр).
Примеры 10-13
Повторяли способ из Примера 1, но при использовании Аl(NО3)3.9Н2O в качестве источника алюминия и при значениях Si/Al 1000 (Пример 10), 100 (Пример 11) и 500 (Примеры 12 и 13). В Примерах 10-12 реакционная смесь включала добавление той же затравки, которую использовали в Примере 1, а в Примере 13 затравку не использовали. Как показано в таблице 3, рентгеноструктурный анализ порошка показал, что продукты из Примеров 10 и 13 являются ЕММ-28, а продукты из Примеров 11 и 12 являются EU-1.
Пример 14
Повторяли способ получения без затравки из Примера 13, но с заменой А1(NО3)3.9Н2О на MS-25 диоксид кремния-оксид алюминия, поставляемый W.R. Grace & Со. Рентгеноструктурный анализ порошка показал, что продукт является ЕММ-28 с небольшим количеством примесной фазы.
Пример 15
Синтетический гель состава, приведенного в Таблице 3, получали при исходном смешивании 49,76 г дикатиона формулы I (мета) в качестве органического направляющего агента Q для формирования структуры с 2,838 г воды. В полученную смесь добавляли 0,054 г MS-25 диоксида кремния-оксида алюминия, 19,041 г Ludox LS30 диоксида кремния и 0,132 г затравки ЕММ-28. Смесь непрерывно перемешивали в процессе всей процедуры добавления и затем перемешивали в течение еще одного часа, после чего смесь выливали во вкладыш автоклава на 60 мл с перемешиванием. Вкладыш нагревали в опрокидывающейся печи при 160°С в течение 7 дней. Рентгеноструктурный анализ порошка показал, что продукт является ЕММ-28 (Таблица 3). Изображения, полученные с помощью сканирующей электронной микроскопии (СЭМ) продукта, показаны на Фиг. 2.
Пример 16
Способ из Примера 1 повторяли, но при использовании Аl(NО3)3.9Н2O в качестве источника алюминия и дикатиона формулы II (пара) в качестве органического направляющего агента Q для формирования структуры. Полученный продукт анализировали методом рентгеноструктурного анализа порошка и обнаружили, что он представляет собой МСМ-22.
Пример 17
Способ из Примера 16 повторяли, но при использовании смеси дикатиона формулы I (мета) и дикатиона формулы II (пара) в массовом отношении от 60 до 40 в качестве органического направляющего агента Q для формирования структуры. В этом случае, методом рентгеноструктурного анализа порошка обнаружили, что продукт является ЕММ-28 с небольшим количеством примесной фазы.
Пример 18
Повторяли способ из Примера 17 (смешанный мета/пара направляющий агент), но без затравки. Методом рентгеноструктурного анализа порошка обнаружили, что продукт является неизвестной кристаллической фазой.
Пример 19
Способ из Примера 1 повторяли, но при использовании Аl(NО3)3.9Н2О в качестве источника алюминия и дикатион формулы III (орто) в качестве органического направляющего агента Q для формирования структуры. Полученный продукт анализировали методом рентгеноструктурного анализа порошка и обнаружили, что он представляет собой RUB-35. Тот же результат получили при использовании Ultrasil, а не Ludox LS30, в качестве источника диоксида кремния.
Пример 20
Способ из Примера 19 повторяли, но при использовании органического направляющего агента Q для формирования структуры, представляющего собой смесь дикатиона формулы III (орто) и дикатиона формулы II (пара) в массовом отношении 50 на 50. Метод рентгеноструктурного анализа порошка показал, что продукт представляет собой смесь EU-1 и ZSM-12. Если не использовать затравку, обнаружили, что продукт представляет собой только ZSM-12.
Пример 21
Синтетический гель без добавления алюминия и с молярными отношениями H2O/SiO2=32, OH-/SiO2=0,4 и Q/SiO2=0,2 получали при использовании тетраметилортосиликата (TMOS) в качестве первого источника кремния, триметоксифенилсилана (MePhSi) в качестве второго источника кремния и дикатиона формулы I (мета) в качестве органического направляющего агента Q для формирования структуры.
Получение и кристаллизацию синтетического геля проводили в соответствии со следующей методикой. 0,61 мл MePhSi смешивали с 2,42 мл TMOS и в смесь добавляли 11,62 мл органического направляющего агента Q для формирования структуры и 0,012 г затравки ЕММ-28. Смесь непрерывно перемешивали во время всей процедуры добавления и затем перемешивали в течение еще 10 минут, после чего смесь помещали в лиофилизатор для удаления всей воды. После удаления смеси из лиофилизатора обратно добавляли 11,7 мл дистиллированной воды и смесь перемешивали. Затем смесь переносили в реактор Парра на 23 мл и нагревали в опрокидывающейся печи при 160°С в течение 5 дней. Полученный продукт анализировали методом рентгеноструктурного анализа порошка и обнаружили, что он представляет собой ЕММ-28.
Пример 22
Повторяли способ из Примера 21, но без использования затравки. Методом рентгеноструктурного анализа порошка обнаружили, что продукт представляет собой ЕММ-28 вместе с неизвестной кристаллической фазой.
Примеры 23 и 24
Повторяли способ из Примера 21, но при температуре кристаллизации 180°С как с затравкой в синтез-смеси (Пример 23), так и без использования затравки (Пример 24). Полученные продукты анализировали методом рентгеноструктурного анализа порошка и обнаружили, что они представляют собой ЕММ-28 в случае синтеза с затравкой и ZSM-12 в случае синтеза без затравки.
Пример 25
Способ из Примера 22 (без затравки) повторяли, но при температуре кристаллизации 170°С. Полученный продукт анализировали методом рентгеноструктурного анализа порошка и обнаружили, что он является аморфным.
Пример 26
Повторяли способ из Примера 21 (без затравки), но при температуре кристаллизации 200°С. Полученный продукт анализировали методом РСА на порошке и обнаружили, что он представляет собой кристобаллит.
Дополнительно или альтернативно, изобретение может включать одно или более из следующих воплощений.
Воплощение 1. Синтетический кристаллический материал, имеющий в только что прокаленном состоянии профиль рентгенодифрактограммы, включающий следующие пики:


где материал возможно имеет состав, характеризующийся следующим молярным отношением (n)Х2O3:YO2, где n является числом меньше 0,05, X является трехвалентным элементом и Y является четырехвалентным элементом; где X возможно включает один или более из В, Al, Fe и Ga (например, включает по меньшей мере Аl); где Y возможно включает один или более из Si, Ge, Sn, Ti и Zr (например, включает по меньшей мере Si и/или Ge).
Воплощение 2. Синтетический кристаллический материал, имеющий сразу после синтеза профиль рентгенодифрактограммы, включающий следующие пики:

где материал возможно имеет состав, характеризующийся следующим молярным отношением (m)M:(b)Q:(n)X2O3:YO2:(z)H2O, где m является числом, имеющим значение от равного или больше 0 до меньше или равного 0,1, b является числом, имеющим значение от больше 0 до меньше или равного 0.05, n является числом меньше 0,025, z является числом, имеющим значение от больше или равного 0 до меньше или равного 0,2, М является катионом щелочного металла, X является трехвалентным элементом, возможно включающим один или более из В, Al, Fe и Ga (например, включающим по меньшей мере Al); Y является четырехвалентным элементом, возможно включающим один или более из Si, Ge, Sn, Ti и Zr (например, включающим по меньшей мере Si и/или Ge) и Q является органическим направляющим агентом для формирования структуры, выбранным из одного или более из дикатионов формулы I и II:
Воплощение 3. Материал по воплощению 2, который имеет состав, характеризующийся следующим молярным отношением (k)F:(b)Q:(n)X2O3:YO2:(z)H2O, где к является числом, имеющим значение от равного или больше 0 до меньше или равного 0,01, b является числом, имеющим значение от больше 0 до меньше или равного 0,05, n является числом меньше 0,025, z является числом, имеющим значение от больше или равного 0 до меньше или равного 0,2, F является фторид-ионом, X является трехвалентным элементом, возможно включающим один или более из В, Al, Fe и Ga (например, включающим по меньшей мере Al); Y является четырехвалентным элементом, возможно включающим один или более из Si, Ge, Sn, Ti и Zr (например, включающим по меньшей мере Si и/или Ge), и Q является органическим направляющим агентом для формирования структуры, например, выбранным из одного или более из дикатионов формулы I и II.
Воплощение 4. Способ получения синтетического кристаллического материала по любому из предшествующих воплощений, включающий стадии:
(i) получения синтетической смеси, способной к образованию указанного материала, причем указанная смесь содержит воду, источник гидроксил ионов, источник оксида четырехвалентного элемента Y, источник трехвалентного элемента X и органический направляющий агент (Q) для формирования структуры, выбранный из одного или более из дикатионов формулы I и II:
где смесь возможно имеет состав, выраженный в виде молярных отношений, находящихся в пределах следующих диапазонов: X2O3/YO2 от 0 до примерно 0,05; H2O/YO2 от примерно 2 до примерно 100; ОН-/YО2 от примерно 0,05 до примерно 0,6; и Q/YO2 от примерно 0,04 до примерно 0,30, причем указанная смесь возможно дополнительно включает источник затравки;
(ii) нагревания смеси в условиях кристаллизации, включающих температуру от примерно 100°С до примерно 200°С до образования кристаллов указанного материала;
(iii) извлечения кристаллического материала, полученного на стадии (ii); и
(iv) возможно обработки кристаллов, извлеченных на стадии (iii), для удаления по меньшей мере части органического направляющего агента (Q) для формирования структуры.
Воплощение 5. Синтетический пористый кристаллический материал, полученный способом по воплощению 4.
Воплощение 6. Способ превращения исходного материала, содержащего органическое соединение, в продукт превращения, включающий стадии:
(i) приведения указанного исходного материала в контакт с катализатором в условиях превращения органического соединения с получением потока продукта, содержащего продукт превращения, причем указанный катализатор содержит активную форму синтетического пористого кристаллического материала по воплощению 1 или воплощению 5; и
(ii) извлечения продукта превращения из потока продукта.
Воплощение 7. Способ селективного выделения одного или более целевых компонентов исходного материала из оставшихся компонентов исходного материала, включающий стадии:
(i) приведения указанного исходного материала в контакт с сорбентом в условиях эффективной сорбции, причем указанный сорбент содержит активную форму синтетического пористого кристаллического материала по воплощению 1 или воплощению 5, с образованием при этом сорбированного продукта и выделяемого продукта; и
(ii) извлечения одного или более целевых компонентов (например, включающих, по существу состоящих из или представляющих собой СО2, H2S, NH3, SO3, алифатический углеводород, такой как СH4, ароматический углеводород, такой как одноядерный ароматический углеводород и/или олефин, такой как этилен, пропилен, бутен, пентен, гексен или т.п., или любую их комбинацию), из сорбированного продукта или из выделяемого продукта.
Воплощение 8. Синтетический пористый кристаллический материал, имеющий внутри пор структуру дикатиона формулы I или II:
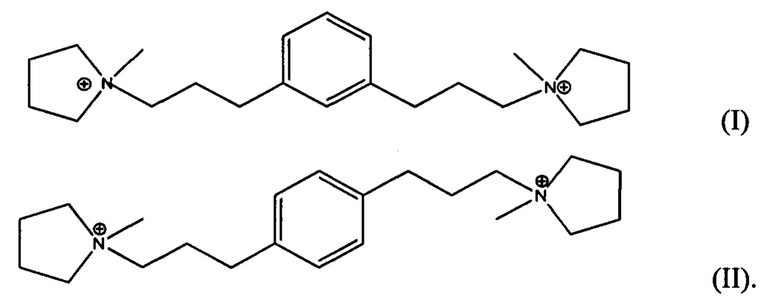
Воплощение 9. Органическое азотистое соединение, содержащее дикатион формулы I, II и III:
Воплощение 10. Способ получения соединения 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1), где х составляет 2, 3 или 4, включающий:
взаимодействие 1,х-bis(галогенметил)бензола с диалкилмалонатом с получением тетраалкил 2,2'-(1,х-фениленbis(метилен))дималоната;
превращение по меньшей мере части тетраалкил-2,2'-(1,х-фениленbis(метилен))дималоната в 3,3'-(1,х-фенилен)дипропановую кислоту;
восстановление по меньшей мере части 3,3'-(1,х-фенилен)дипропановой кислоты или ее сложного эфира в 3,3'-(1,х-фенилен)bis(пропан-1-ол);
взаимодействие по меньшей мере части 3,3'-(1,х-фенилен)bis(пропан-1-ола) с алкил- или арилсульфонилгалогенидом (например, n-толуолсульфонилхлорид) с получением соответствующего сложного сульфонатного диэфира;
взаимодействие по меньшей мере части сложного сульфонатного диэфира с пирролидином с получением 1,х-bis(3-(пирролидиний-1-ил)пропил)бензола; и
взаимодействие по меньшей мере части 1,х-bis(3-(пирролидин-1-ил)пропил)бензола с метилгалогенидом с получением галогенида 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1).
Воплощение 11. Способ получения соединения 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1), где х составляет 2, 3 или 4, включающий:
взаимодействие по меньшей мере части сложного сульфонатного диэфира 3,3'-(1,х-фенилен)bis(пропан-1-ола)) по воплощению 10 с 1-метилпирролидином с получением соединения 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1).
Воплощение 12. Способ получения соединения 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1), где х составляет 2, 3 или 4, включающий:
взаимодействие 1-(проп-2-ин-1-ил)пирролидина (который возможно получен путем взаимодействия 3-галогенпроп-1-ина с пирролидином) с 1,х-дигалогензамещенным бензолом с получением соединения формулы (IVA), (IVB) или (IVC):
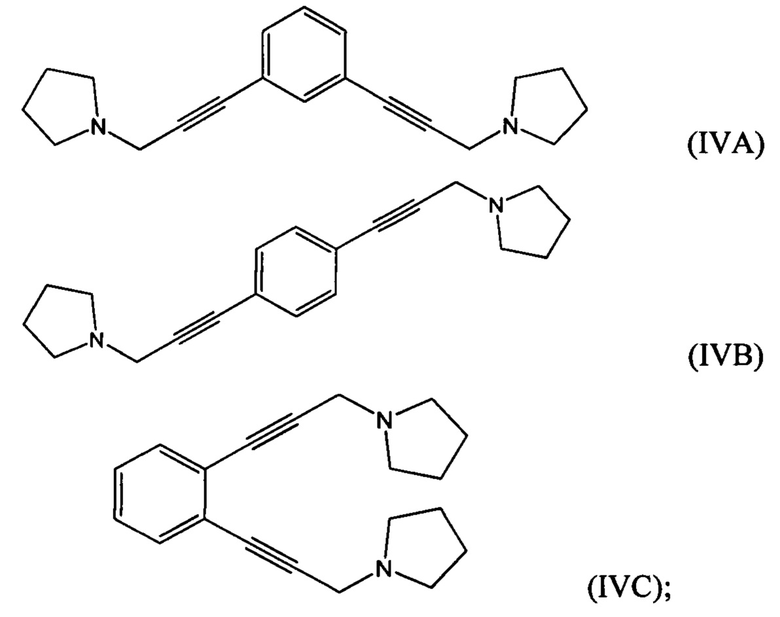
гидрирование по меньшей мере части соединения формулы (IVA), (IVB) или (IVC) с получением соединения формулы (VA), (VB) или (VC):
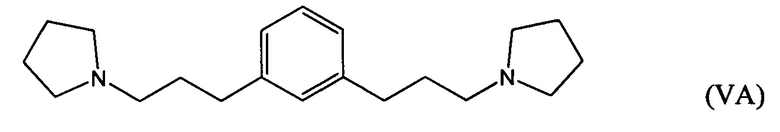
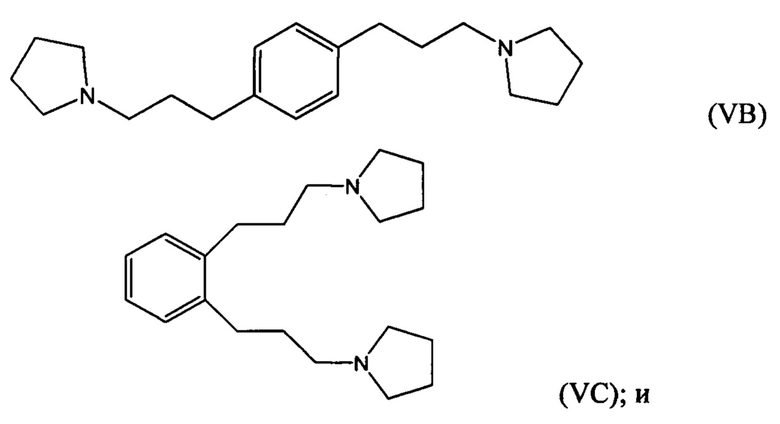
взаимодействие по меньшей мере части соединения формулы (VA), (VB) или (VC) с метилгалогенидом с получением галогенида 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1).
Воплощение 13. Способ получения соединения 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1), где х составляет 2, 3 или 4, включающий:
взаимодействие соединения формулы (VI)
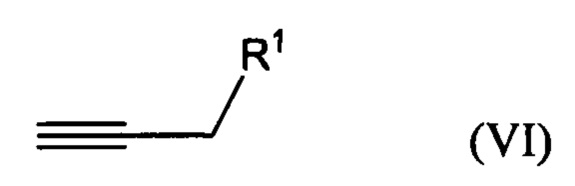
где R1 представляет собой гидроксильную группу или алкил- или арилсульфонатную группу, с 1,х-дигалогензамещенным бензолом с получением соединения формулы (VIIA), (VIIB) или (VIIC):
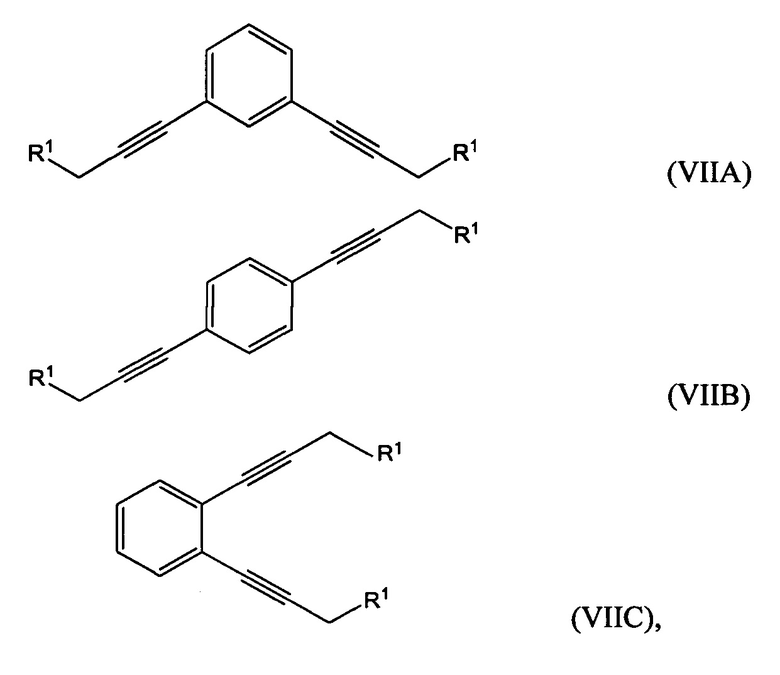
где, когда R1 представляет собой алкил- или арилсульфонатную группу (например, 4-метилбензолсульфонат), способ дополнительно включает:
гидрирование по меньшей мере части соединения формулы (VIIA), (VIIB) или (VIIC) с получением соединения формулы (VIIIA), (VIIIB) или (VIIIC):
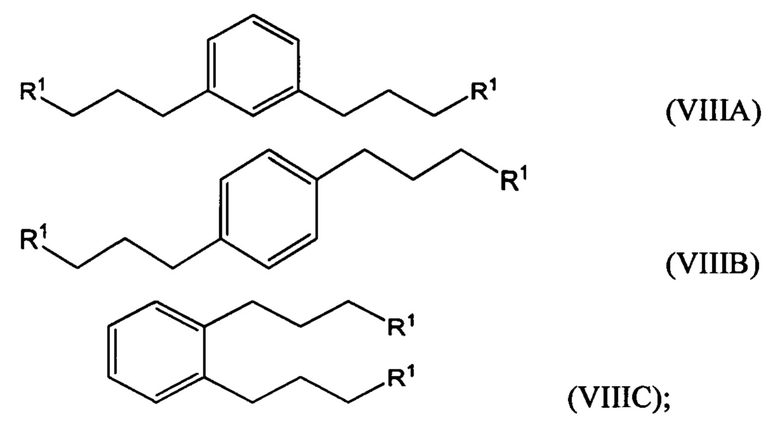
превращение по меньшей мере части соединения формулы (VIIIA), (VIIIB) или (VIIIC) в соединение 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1) (i) по реакции с 1-метилпирролидином или (ii) по реакции с пирролидином и затем с метилгалогенидом;
возможно взаимодействие по меньшей мере части соединения формулы (VIIA), (VIIB) или (VIIC), когда R1 представляет собой алкил- или арилсульфонатную группу, с пирролидином с получением соединения формулы (IVA), (IVB) или (IVC):
возможно гидрирование по меньшей мере части соединения формулы (IVA), (IVB) или (IVC) с получением соединения формулы (VA), (VB) или (VC):
возможно взаимодействие по меньшей мере части соединения формулы (VA), (VB) или (VC) с метилгалогенидом с получением галогенида 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1).
Воплощение 14. Способ по воплощению 13, в котором R1 является гидроксильной группой, причем способ дополнительно включает:
взаимодействие по меньшей мере части соединения формулы (VIIA), (VIIB) или (VIIC) с алкил- или арилсульфонилгалогенидом, таким как n-толуолсульфонилхлорид, с получением соединения формулы (IXA), (IXB) или (LXC):
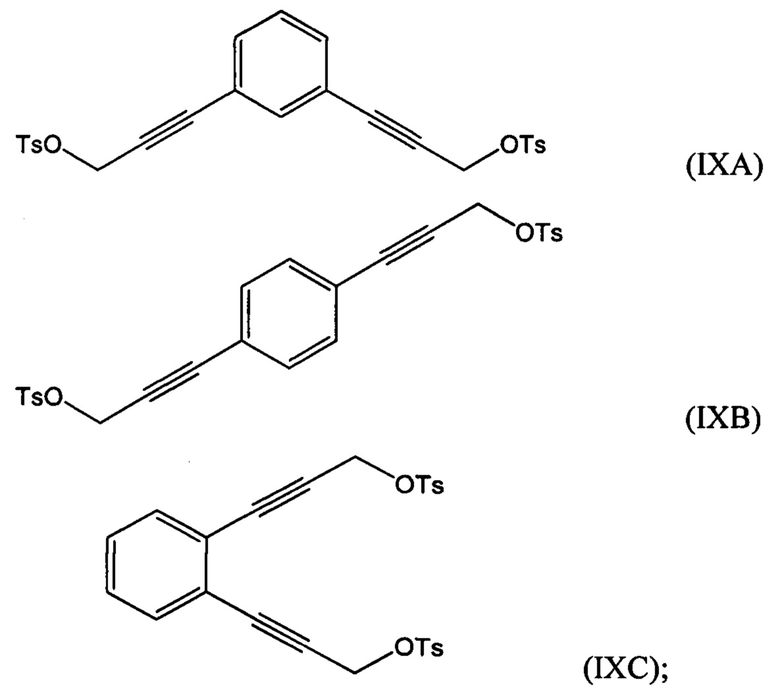
возможно гидрирование по меньшей мере части соединения формулы (IXA), (IXB) или (IXC) с получением соединения (ХА), (ХВ) или (ХС):
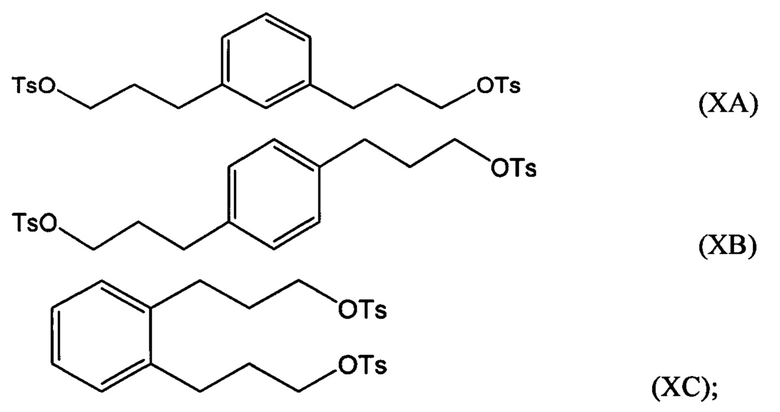
возможно превращение по меньшей мере части соединения формулы (ХА), (ХВ) или (ХС) в соединение 3,3-(1,х-фенилен)bis(пропан-3,1-диил)bis(1-метилпирролидиния) (i) по реакции с 1-метилпирролидином или (ii) по реакции с пирролидином и после этого с метилгалогенидом;
возможно взаимодействие по меньшей мере части соединения формулы (IXA), (IXB) или (IXC) с пирролидином с получением соединения формулы (IVA), (IVB) или (IVC):
возможно гидрирование по меньшей мере части соединения формулы (IVA), (IVB) или (IVC) с получением соединения формулы (VA), (VB) или (VC):
возможно взаимодействие по меньшей мере части соединения формулы (VA), (VB) или (VC) с метилгалогенидом с получением галогенида 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1).
Воплощение 15. Способ получения соединения 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1), где х составляет 2, 3 или 4, включающий:
взаимодействие соединения формулы (VI)
где R1 представляет собой гидроксильную группу, с 1,х-дигалогензамещенным бензолом с получением соединения формулы (VIIA), (VIIB) или (VIIC):
гидрирование по меньшей мере части соединения формулы (VIIA), (VIIB) или (VIIC) с получением соединения формулы (VIIIA), (VIIIB) или (VIIIC), соответственно:
взаимодействие по меньшей мере части соединения формулы (VIIIA), (VIIIB) или (VIIIC) с алкил- или арилсульфонилгалогенидом, таким как n-толуолсульфонилхлорид, с получением соединения формулы (ХА), (ХВ) или ХС, соответственно:
превращение по меньшей мере части соединения формулы (ХА), (ХВ) или (ХС) в соединение 1,1'-(1,х-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1) (i) по реакции с 1-метилпирролидином или (ii) по реакции с пирролидином и после этого с метилгалогенидом.
Хотя настоящее изобретение описано и проиллюстрировано со ссылкой на конкретные воплощения, специалистам в данной области техники понятно, что изобретение обеспечивает модификации изобретения, которые не обязательно проиллюстрированы в данной заявке. Следовательно, для определения запрашиваемого объема притязаний по настоящему изобретению, необходимо обращаться только к прилагаемой формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| Новый синтетический кристаллический материал EMM-26, его получение и применение | 2015 |
|
RU2688542C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕРФТОРАЛКИЛЦИАНО- ИЛИ ПЕРФТОРАЛКИЛЦИАНОФТОРБОРАТОВ | 2011 |
|
RU2575352C2 |
| Дикатионные ионные жидкости с полисилоксановым фрагментом в составе катиона в качестве теплоносителей | 2016 |
|
RU2627658C1 |
| МОЛЕКУЛЯРНОЕ СИТО ЕММ-22, ЕГО СИНТЕЗ И ПРИМЕНЕНИЕ | 2012 |
|
RU2601462C2 |
| ЭНАНТИОМЕРНО ЧИСТЫЕ ОСНОВНЫЕ ЭФИРЫ АРИЛ-ЦИКЛОАЛКИЛГИДРОКСИКАРБОНОВЫХ КИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В ЛЕКАРСТВЕННЫХ СРЕДСТВАХ | 1997 |
|
RU2238936C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛИДИН-2,4-ДИОНА, ОБЛАДАЮЩИЕ АНТИПРОЛИФЕРАТИВНОЙ АКТИВНОСТЬЮ | 2010 |
|
RU2555999C2 |
| Материалы EMM-23, способы их получения и их применение | 2018 |
|
RU2763758C2 |
| КОМПОЗИЦИЯ ДЛЯ ОКРАШИВАНИЯ КЕРАТИНОВЫХ ВОЛОКОН, СОДЕРЖАЩАЯ ПО МЕНЬШЕЙ МЕРЕ ОДНО ПРОИЗВОДНОЕ ДИАМИНО-N,N-ДИГИДРОПИРАЗОЛОНА | 2004 |
|
RU2330909C2 |
| ИОННЫЕ ЖИДКОСТИ С СИЛОКСАНОВЫМ ФРАГМЕНТОМ В СОСТАВЕ КАТИОНА В КАЧЕСТВЕ ТЕПЛОНОСИТЕЛЕЙ | 2015 |
|
RU2600932C1 |
| Четвертичные аммониевые соединения на основе производных пентаэритрита и пиридоксина, обладающие антибактериальной активностью | 2023 |
|
RU2811203C1 |
Изобретение относится к новому синтетическому кристаллическому материалу ЕММ-28, который синтезирован в присутствии органического направляющего агента (Q) для формирования структуры, выбранного из одного или более из следующих дикатионов:
ЕММ-28 можно использовать в реакциях превращения органических соединений и сорбционных процессах. 14 н. и 4 з.п. ф-лы, 26 пр., 3 табл., 2 ил.
1. Синтетический кристаллический материал, имеющий в только что прокаленном состоянии профиль рентгенодифрактограммы, включающий следующие пики:
где материал имеет состав, характеризующийся следующим молярным отношением (n)X2O3:YO2, где n является числом меньше 0,05, X является трехвалентным элементом и Y является четырехвалентным элементом, где X включает один или более из B, Al, Fe и Ga, и Y включает один или более из Si, Ge, Sn, Ti и Zr.
2. Синтетический кристаллический материал, имеющий сразу после синтеза профиль рентгенодифрактограммы, включающий следующие пики:


где материал имеет состав, характеризующийся следующим молярным отношением (m)M:(b)Q:(n)X2O3:YO2:(z)H2O, где m является числом, имеющим значение от равного или больше 0 до меньше или равного 0,1, b является числом, имеющим значение от больше 0 до меньше или равного 0,05, n является числом меньше 0,025, z является числом, имеющим значение от больше или равного 0 до меньше или равного 0,2, M является катионом щелочного металла, X является трехвалентным элементом, включающим один или более из B, Al, Fe и Ga; Y является четырехвалентным элементом, включающим один или более из Si, Ge, Sn, Ti и Zr, и Q является органическим направляющим агентом для формирования структуры, выбранным из одного или более из дикатионов формулы I и II:
3. Материал по п. 2, который имеет состав, характеризующийся следующим молярным отношением (k)F:(b)Q:(n)X2O3:YO2:(z)H2O, где k является числом, имеющим значение от равного или больше 0 до меньше или равного 0,01, b является числом, имеющим значение от больше 0 до меньше или равного 0,05, n является числом меньше 0,025, z является числом, имеющим значение от больше или равного 0 до меньше или равного 0,2, F является фторид-ионом, X является трехвалентным элементом, включающим один или более из B, Al, Fe и Ga (например, включающим по меньшей мере Al); Y является четырехвалентным элементом, включающим один или более из Si, Ge, Sn, Ti и Zr (например, включающим по меньшей мере Si и/или Ge), и Q является органическим направляющим агентом для формирования структуры, выбранным из одного или более из дикатионов формулы I и II.
4. Способ получения синтетического кристаллического материала по п. 2 или 3, включающий стадии:
(i) получения синтетической смеси, способной к образованию указанного материала, причем указанная смесь содержит воду, источник гидроксил ионов, источник оксида четырехвалентного элемента Y, источник трехвалентного элемента X и органический направляющий агент (Q) для формирования структуры, выбранный из одного или более из дикатионов формулы I и II:
где смесь имеет состав, выраженный в виде молярных отношений, находящихся в пределах следующих диапазонов: X2O3/YO2 от 0 до примерно 0,05; H2O/YO2 от примерно 2 до примерно 100; OH-/YO2 от примерно 0,05 до примерно 0,6; и Q/YO2 от примерно 0,04 до примерно 0,30;
(ii) нагревания смеси в условиях кристаллизации, включающих температуру от примерно 100°C до примерно 200°C до образования кристаллов указанного материала;
(iii) извлечения кристаллического материала, полученного на стадии (ii).
5. Способ по п. 4, в котором смесь на стадии (i) дополнительно включает источник затравки.
6. Способ получения синтетического кристаллического материала по п. 1, включающий получение материала по п. 2 или 3 способом по любому из пп. 4 и 5 и обработку полученного материала для удаления по меньшей мере части органического направляющего агента (Q) для формирования структуры.
7. Синтетический пористый кристаллический материал, полученный способом по любому из пп. 4 и 5.
8. Способ превращения исходного материала, содержащего органическое соединение, в продукт превращения, включающий стадии:
(i) приведения указанного исходного материала в контакт с катализатором в условиях превращения органического соединения с получением потока продукта, содержащего продукт превращения, причем указанный катализатор содержит активную форму синтетического кристаллического материала по п. 1 или 7; и
(ii) извлечения продукта превращения из потока продукта.
9. Способ селективного выделения одного или более целевых компонентов исходного материала из оставшихся компонентов исходного материала, включающий стадии:
(i) приведения указанного исходного материала в контакт с сорбентом в условиях эффективной сорбции, причем указанный сорбент содержит активную форму синтетического кристаллического материала по п. 1 или 7, с образованием при этом сорбированного продукта и выделяемого продукта; и
(ii) извлечения одного или более целевых компонентов из сорбированного продукта или из выделяемого продукта.
10. Способ по п. 9, в котором целевые компоненты выбраны из включающих, по существу состоящих из или представляющих собой CO2, H2S, NH3, SO3, алифатический углеводород, такой как CH4, ароматический углеводород, такой как одноядерный ароматический углеводород, и/или олефин, такой как этилен, пропилен, бутен, пентен, гексен, или любую их комбинацию.
11. Синтетический пористый кристаллический материал, имеющий внутри своей структуры пор дикатион формулы I или II:
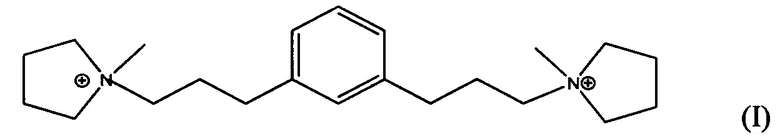
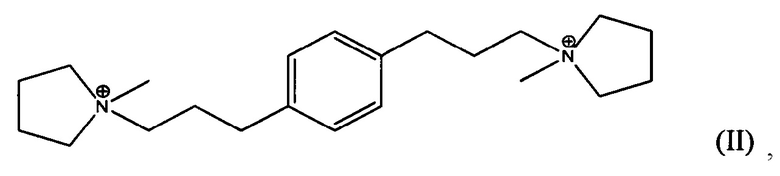
причем этот материал представляет собой молекулярное сито, выбранное из EMM-28, EU-1, MCM-22, ZSM-12 и кристобаллита.
12. Органическое азотистое соединение, содержащее дикатион формулы I и II:
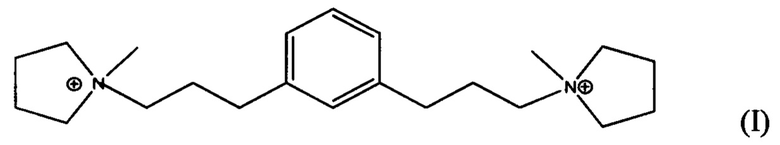
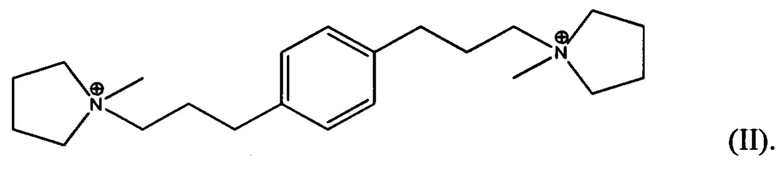
13. Способ получения соединения 1,1’-(1,x-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1), где x составляет 3 или 4, включающий:
взаимодействие 1,x-bis(галогенметил)бензола с диалкилмалонатом с получением тетраалкил-2,2'-(1,x-фениленbis(метилен))дималоната;
превращение тетраалкил-2,2'-(1,x-фениленbis(метилен))дималоната в 3,3'-(1,x-фенилен)дипропановую кислоту;
восстановление 3,3'-(1,x-фенилен)дипропановой кислоты или ее сложного эфира в 3,3'-(1,x-фенилен)bis(пропан-1-ол);
взаимодействие 3,3'-(1,x-фенилен)bis(пропан-1-ола) с алкил- или арилсульфонилгалогенидом с получением соответствующего сложного сульфонатного диэфира;
взаимодействие сложного сульфонатного диэфира с пирролидином с получением 1,x-bis(3-(пирролидиний-1-ил)пропил)бензола; и
взаимодействие 1,x-bis(3-(пирролидин-1-ил)пропил)бензола с метилгалогенидом с получением галогенида 1,1’-(1,x-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1).
14. Способ получения соединения 1,1’-(1,x-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1), где x составляет 3 или 4, включающий
взаимодействие сложного сульфонатного диэфира 3,3'-(1,x-фенилен)bis(пропан-1-ола)) по п. 13 с 1-метилпирролидином с получением соединения 1,1’-(1,x-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1).
15. Способ получения соединения 1,1’-(1,x-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1), где x составляет 3 или 4, включающий:
взаимодействие 1-(проп-2-ин-1-ил)пирролидина, который получен путем взаимодействия 3-галогенпроп-1-ина с пирролидином, с 1,x-дигалогензамещенным бензолом с получением соединения формулы (IVA) или (IVB):
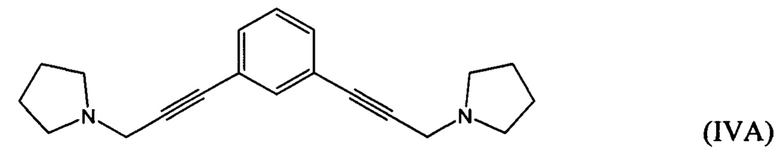
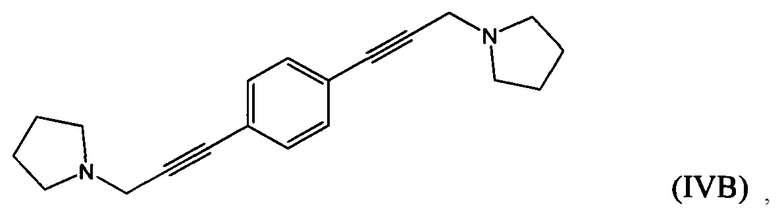
гидрирование соединения формулы (IVA) или (IVB) с получением соединения формулы (VA) или (VB):
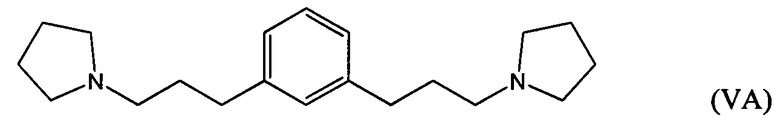
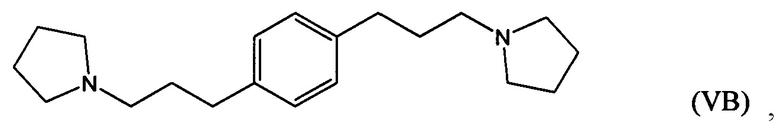
взаимодействие соединения формулы (VA) или (VB) с метилгалогенидом с получением галогенида 1,1’-(1,x-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1).
16. Способ получения соединения 1,1’-(1,x-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1), где x составляет 3 или 4, включающий:
взаимодействие соединения формулы (VI)
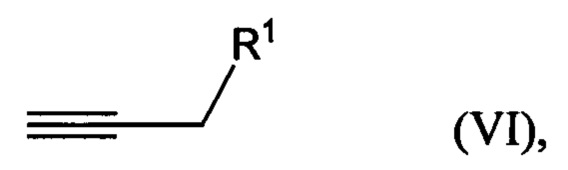
где R1 представляет собой гидроксильную группу или алкил- или арилсульфонатную группу, с 1,x-дигалогензамещенным бензолом с получением соединения формулы (VIIA) или (VIIB):
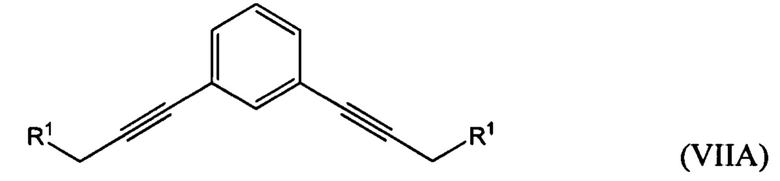
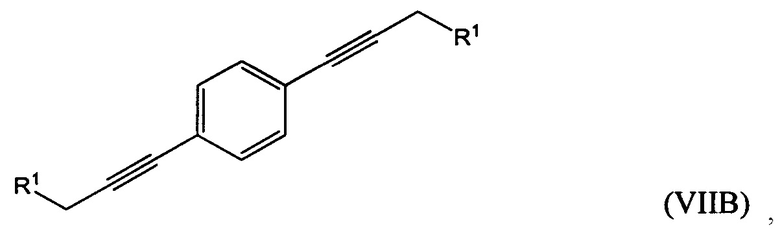
где, когда R1 представляет собой алкил- или арилсульфонатную группу, способ дополнительно включает:
гидрирование соединения формулы (VIIA) или (VIIB) с получением соединения формулы (VIIIA) или (VIIIB):
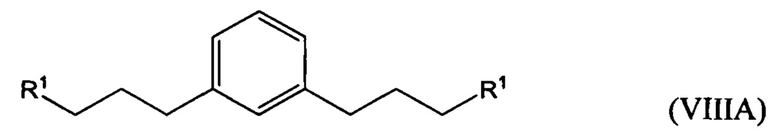
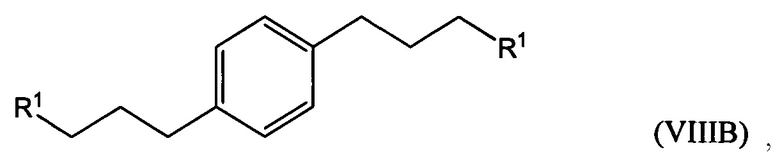
превращение соединения формулы (VIIIA) или (VIIIB) в соединение 1,1’-(1,x-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1) по реакции (i) с 1-метилпирролидином или по реакции (ii) с пирролидином и затем с метилгалогенидом;
взаимодействие соединения формулы (VIIA) или (VIIB), когда R1 представляет собой алкил- или арилсульфонатную группу, с пирролидином с получением соединения формулы (IVA) или (IVB)
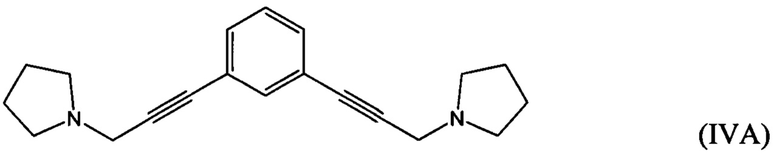
гидрирование соединения формулы (IVA) или (IVB) с получением соединения формулы (VA) или (VB):
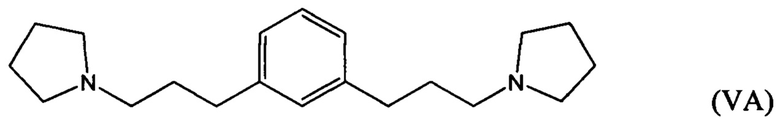
взаимодействие соединения формулы (VA) или (VB) с метилгалогенидом с получением галогенида 1,1’-(1,x-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1).
17. Способ по п. 16, в котором R1 является гидроксильной группой, причем способ
дополнительно включает:
взаимодействие соединения формулы (VIIA) или (VIIB) с алкил- или арилсульфонилгалогенидом, таким как п-толуолсульфонилхлорид, с получением соединения формулы (IXA) или (IXB):
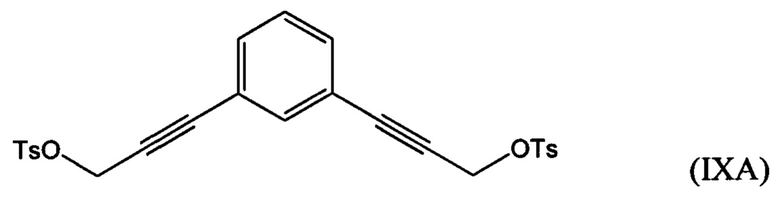
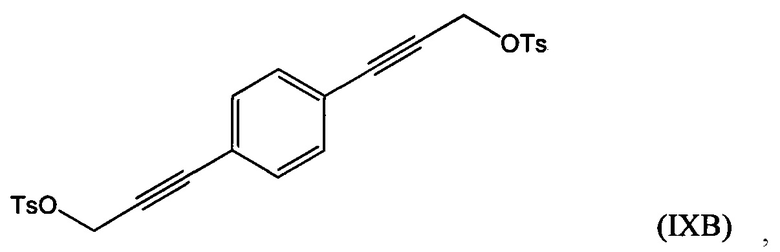
гидрирование соединения формулы (IXA) или (IXB) с получением соединения (XA) или (XB):
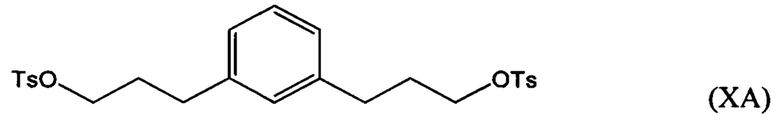
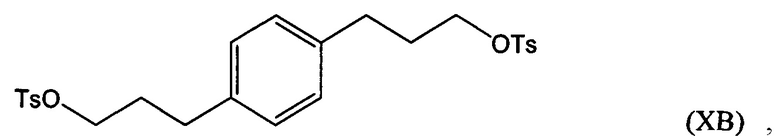
превращение соединения формулы (XA) или (XB) в соединение 3,3-(1,x-фенилен)bis(пропан-3,1-диил)bis(1-метилпирролидиния) по реакции (i) с 1-метилпирролидином или по реакции (ii) с пирролидином и после этого с метилгалогенидом;
взаимодействие соединения формулы (IXA) или (IXB) с пирролидином с получением соединения формулы (IVA) или (IVB):
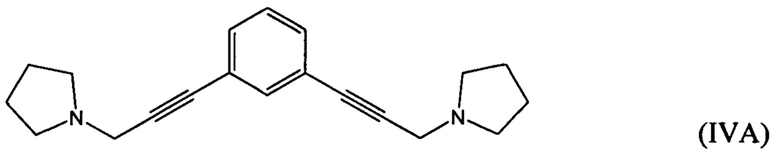
гидрирование соединения формулы (IVA) или (IVB) с получением соединения формулы (VA) или (VB):
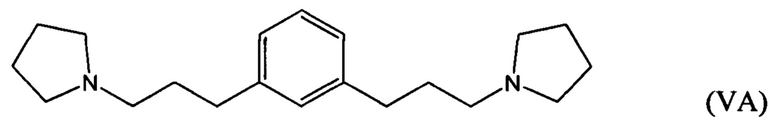
взаимодействие соединения формулы (VA) или (VB) с метилгалогенидом с получением галогенида 1,1’-(1,x-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1).
18. Способ получения соединения 1,1’-(1,x-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1), где x составляет 3 или 4, включающий:
взаимодействие соединения формулы (VI)
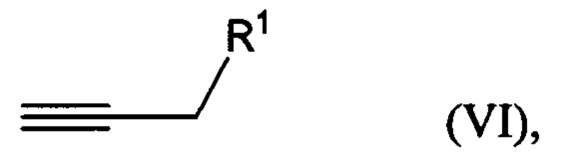
где R1 представляет собой гидроксильную группу, с 1,x-дигалогензамещенным бензолом с получением соединения формулы (VIIA) или (VIIB):
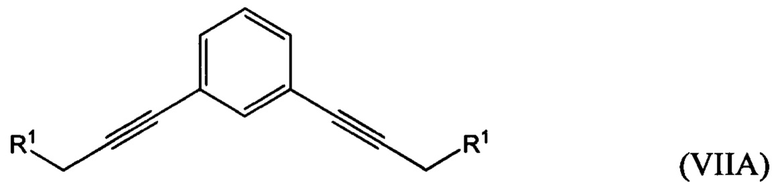
гидрирование соединения формулы (VIIA) или (VIIB) с получением соединения формулы (VIIIA) или (VIIIB) соответственно:
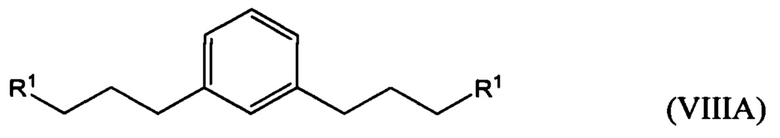
взаимодействие соединения формулы (VIIIA) или (VIIIB) с алкил- или арилсульфонилгалогенидом с получением соединения формулы (XA) или (XB) соответственно:
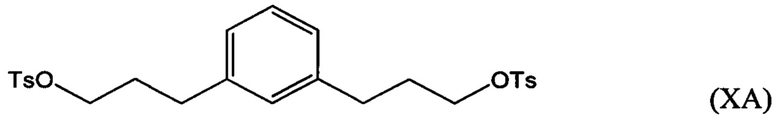
превращение соединения формулы (XA) или (XB) в соединение 1,1’-(1,x-фениленbis(пропан-3,1-диил))bis(1-метилпирролидиния-1) по реакции (i) с 1-метилпирролидином или по реакции (ii) с пирролидином и после этого с метилгалогенидом.
| WO 2014100218 A1, 26.06.2014 | |||
| US 2011166177 A1, 07.07.2011 | |||
| US 7651677 B1, 26.01.2010 | |||
| WO 2013019462 A1, 07.02.2013 | |||
| US 2010178241 A1, 15.07.2010 | |||
| СИНТЕТИЧЕСКИЙ ПОРИСТЫЙ КРИСТАЛЛИЧЕСКИЙ МАТЕРИАЛ ITQ-13, ЕГО СИНТЕЗ И ПРИМЕНЕНИЕ | 2002 |
|
RU2293058C2 |

