Уровень техники
Настоящее изобретение относится к новым соединениям, к фармацевтическим композициям, содержащим данные соединения, к способам и промежуточным соединениям для получения данных соединений, а также к применению данных соединений в терапии. Более конкретно, оно относится к 1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-ил)мочевине или ее фармацевтически приемлемой соли, проявляющей ингибирование TrkA киназы и применимой в лечении боли, рака, воспаления/воспалительных заболеваний, нейродегенеративных заболеваний, некоторых инфекционных заболеваний, синдрома Шегрена, эндометриоза, диабетической периферической нейропатии, простатита, синдрома тазовой боли, заболеваний, связанных с дисбалансом регуляции ремоделирования костной ткани, а также заболеваний, обусловленных аберрантной передачей сигналов фактора роста соединительной ткани.
Существующие схемы лечения болевых состояний используют несколько классов соединений. Опиоиды (такие как морфин) имеют несколько недостатков, в том числе вызывают рвоту, запор и побочные эффекты со стороны дыхательной системы, а также обладают потенциалом для наркотической зависимости. Нестероидные противовоспалительные болеутоляющие средства (НПВС, такие как СОХ-1 или СОХ-2) также имеют недостатки, в том числе обладают недостаточной эффективностью в лечении сильной боли. Кроме того, ингибиторы СОХ-1 могут привести к образованию язв слизистой оболочки. Соответственно, существует постоянная необходимость в новых и более эффективных методах для облегчения боли, особенно хронической боли.
Trk представляют собой высокоаффинные рецепторные тирозинкиназы, активированные группой растворимых факторов роста, называемых нейротрофинами (NT). Семейство рецепторов Trk состоит из трех представителей: TrkA, TrkB и TrkC. К числу нейротрофинов относятся (i) фактор роста нервов (NGF), который активирует TrkA, (ii) нейротрофический фактор головного мозга (BDNF) и NT-4/5, которые активируют TrkB, и (iii) NT3, который активирует TrkC. Trk широко экспрессируются в нервной ткани и участвуют в поддержании, передаче сигналов и выживании нейрональных клеток (Patapoutian, A. et al., Current Opinion in Neurobiology, 2001,11, 272-280).
Ингибиторы пути Trk/нейротрофины демонстрировали эффективность в многочисленных доклинических моделях боли на животных. Например, антагонистические антитела NGF и TrkA, такие как RN-624, продемонстрировали эффективность в моделях воспалительной и нейропатической боли на животных (Woolf, С.J. et al. (1994) Neuroscience 62, 327-331; Zahn, P.K. et al. (2004) J. Pain 5, 157-163; McMahon, S.B. et al., (1995) Nat. Med. 1, 774-780; Ma, Q.P. и Woolf, C.J. (1997) NeuroReport 8, 807-810; Shelton, D.L. et al. (2005) Pain 116, 8-16; Delafoy, L. et al. (2003) Pain 105, 489-497; Lamb, K. et al. (2003) Neurogastroenterol. Motil. 15, 355-361; Jaggar, S. I. et al. (1999) Br. J. Anaesth. 83, 442-448) и моделях нейропатической боли на животных (Ramer, М.S. и Bisby, М.А. (1999) Eur. J. Neurosci. 11, 837-846; Ro, L.S. et al. (1999) Pain 79,265-274; Herzberg, U. et al., (1997) Neuroreport 8, 1613-1618; Theodosiou, M. et al. (1999) Pain 81, 245-255; Li, L. et al. (2003) Mol. Cell. Neurosci. 23, 232-250; Gwak, Y.S. et al. (2003) Neurosci. Lett. 336, 117-120).
Также было показано, что NGF, секретируемый опухолевыми клетками и опухолевыми инвазивными макрофагами, непосредственно стимулирует TrkA, расположенную на периферических болевых волокнах. С помощью различных опухолевых моделей на мышах и крысах было показано, что нейтрализация NGF моноклональными антителами ингибирует связанную с раковым заболеванием боль до степени, подобной или превосходящей самую высокую переносимую дозу морфина. Так как TrkA киназа может служить медиатором обусловленных NGF биологических ответов, ингибиторы TrkA и/или других Trk киназ могут обеспечивать эффективное лечение состояний хронической боли и обусловленной раковым заболеванием боли.
Последние публикации также показали, что сверхэкспрессия, активация, амплификация и/или мутация Trk киназ обусловлены многими раковыми заболеваниями, в том числе нейробластомой (Brodeur, G.М., Nat. Rev. Cancer 2003, 3, 203-216), раком яичников (Davidson. В., et al., Clin. Cancer Res. 2003, 9, 2248-2259), раком ободочной и прямой кишки (Bardelli, A., Science 2003, 300, 949), меланомой (Truzzi, F., et al., Dermato-Endocrinology 2011, 3(1), pp. 32-36), раком головы и шеи (Yilmaz, Т., et al., Cancer Biology and Therapy 2010, 10(6), pp. 644-653), карциномой желудка (Du, J. et al., World Journal of Gastroenterology 2003, 9(7), pp. 1431-1434), карциномой легких (Ricci A., et al., American Journal of Respiratory Cell and Molecular Biology 25 (4), pp. 439-446), раком молочной железы (Jin, W., et al., Carcinogenesis 2010, 31(11), pp. 1939-1947), секреторным раком молочной железы (Euthus, D.M., et al., Cancer Cell 2002, 2(5), pp. 347-348), глиобластомой (Wadhwa, S., et al., Journal of Biosciences 2003, 28(2), pp. 181-188), медуллобластомой (Gruber-Olipitz, M., et al., Journal of Proteome Research 2008, 7(5), pp. 1932-1944), раком слюнных желез (Li, Y.-G., et al., Chinese Journal of Cancer Prevention and Treatment 2009, 16 (6), pp. 428-430), папиллярной карциномой щитовидной железы (Greco, A., et al., Molecular and Cellular Endocrinology 2010, 321(1), pp. 44-49) и миелоидным лейкозом у взрослых (Eguchi, М., et al., Blood 1999, 93(4), pp. 1355-1363). В доклинических моделях рака неселективные низкомолекулярные ингибиторы TrkA, В и С проявляли эффективность как в ингибировании развития опухоли, так и в остановке опухолевых метастазов (Nakagawara, А. (2001) Cancer Letters 169:107-114; Meyer, J., et al. (2007) Leukemia, 21(10): 2171-2180; Pierottia, M.A. и Greco A., (2006) Cancer Letters 232:90-98; Eric Adriaenssens, E., et al. Cancer Res (2008) 68:(2) 346-351). Эти данные подтверждают обоснованность применения ингибиторов Trk в лечении рака.
Кроме того, ингибирование пути нейротрофин/Trk продемонстрировало эффективность в лечении доклинических моделей воспалительных заболеваний антителами NGF или неселективными низкомолекулярными ингибиторами TrkA. Например, ингибирование пути нейротрофин/Trk было задействовано в доклинических моделях воспалительных заболеваний легкого, в том числе астмы (Freund-Michel, V; Frossard, N., Pharmacology & Therapeutics (2008) 117(1), 52-76), интерстициального цистита (Hu, Vivian Y; et. al. The Journal of Urology (2005), 173(3), 1016-21), синдрома болезненного мочевого пузыря (Liu, Н.-Т., et al., (2010) BJU International, 106(11), pp. 1681-1685), воспалительных заболеваний кишечника, в том числе язвенного колита и болезни Крона (Di Mola, F.F, et. al., Gut (2000) 46(5), 670-678), и воспалительных кожных заболеваний, таких как атопический дерматит (Dou, Y.-C, et. al. Archives of Dermatological Research (2006) 298(1), 31-37), экзема и псориаз (Raychaudhuri, S.P., et al., J. Investigative Dermatology (2004) 122(3), 812-819). Эти данные подтверждают обоснованность применения ингибиторов Trk в лечении воспалительных заболеваний.
Считается, что рецептор TrkA также имеет решающее значение в патогенезе заболевания паразитарной инфекцией Trypanosoma cruzi (болезнь Шагаса) у хозяев-людей (de Melo-Jorge, М. et al., Cell Host & Microbe (2007) 1(4), 251-261).
Ингибиторы Trk также могут находить применение в лечении заболеваний, связанных с дисбалансом регуляции ремоделирования костной ткани, таких как остеопороз, ревматоидный артрит и метастазы в кости. Метастазы в кости являются частым осложнением рака, встречающимся вплоть у 70 процентов пациентов с раком молочной железы или предстательной железы на поздней стадии и у около 15-30 процентов пациентов с карциномой легких, толстой кишки, желудка, мочевого пузыря, матки, прямой кишки, щитовидной железы или почек. Остеолитические метастазы могут вызывать сильную боль, патологические переломы, угрожающую жизни гиперкальциемию, сдавление спинного мозга и другие синдромы сдавления нервов. По этим причинам костный метастаз является серьезным и дорогостоящим осложнением рака. В связи с этим вещества, способные индуцировать апоптоз пролиферирующих остеобластов, были бы очень полезными. Экспрессия рецепторов TrkA наблюдалась в области образования костной ткани в моделях перелома костей на мышах (K. Asaumi, et al., Bone (2000) 26(6) 625-633). Кроме того, локализация NGF наблюдалась практически во всех костеобразующих клетках (K Asaumi, et al., Bone (2000) 26(6) 625-633). Недавно было продемонстрировано, что ингибитор Trk ингибирует передачу сигналов, активированную нейротрофинами, связывающимися со всеми тремя рецепторами Trk в остеобластах hFOB человека (J. Pinski, et al., Cancer Research (2002) 62, 986-989). Эти данные подтверждают обоснованность применения ингибиторов Trk в лечении заболеваний, связанных с ремоделированием костной ткани, таких как метастазы в кости у больных раком.
Ингибиторы Trk также могут находить применение в лечении заболеваний и расстройств, таких как синдром Шегрена (Fauchais, A.L., et al., (2009) Scandinavian Journal of Rheumatology, 38(1), pp. 50-57), эндометриоз (Barcena De Arellano, M.L., et al., (2011) Reproductive Sciences, 18(12), pp. 1202-1210; Barcena De Arellano, et al., (2011) Fertility and Sterility, 95(3), pp. 1123-1126; Cattaneo, A., (2010) Current Opinion in Molecular Therapeutics, 12(1), pp. 94-106), диабетическая периферическая нейропатия (Kim, H.C., et al., (2009) Diabetic Medicine, 26 (12), pp. 1228-1234; Siniscalco, D., et al., (2011) Current Neuropharmacology, 9(4), pp. 523-529; Ossipov, M.H., (2011) Current Pain and Headache Reports, 15(3), pp. 185-192), а также простатит и синдром тазовой боли (Watanabe, Т., et al., (2011) BJU International, 108(2), pp. 248-251; и Miller, L. J., et al., (2002) Urology, 59(4), pp. 603-608).
Ингибиторы TrkA могут также служить для лечения заболеваний, обусловленных аберрантной передачей сигналов фактора роста соединительной ткани (ФРСТ, также называемого CCN2), например, заболеваний, включающих ремоделирование тканей и фиброз. ФРСТ является ключевым медиатором ремоделирования ткани и фиброза (Lipson, K.Е., et al., (2012), Fibrogenesis & Tissue Repair 2012, 5(Suppl 1):S24), а методы лечения, которые снижают передачу сигналов ФРСТ, доказали свою эффективность в лечении фиброза (Li, G., et al., J. Gene Med., 2006, 8:889-900). ФРСТ взаимодействует с TrkA и активирует ее (Wahab, N.А., (2005) J. Am. Soc. Nephrol. 16:340-351), а ингибирование этого пути ингибиторами TrkA может оказаться полезным в лечении различных фиброзных заболеваний, таких как синдром Рейно, идиопатический фиброз легких, рубцевание (гипертрофическое, келоидное и другое), цирроз, эндомиокардиальный фиброз, предсердный фиброз, миелофиброз, прогрессирующий массивный фиброз (легких), нефрогенный системный фиброз, склеродермия, системный склероз, артрофиброз и фиброз глаза.
Известно несколько классов низкомолекулярных ингибиторов Trk киназ, которые, как известно, служат для лечения боли или рака (Wang, Т et al., Expert Opin. Ther. Patents (2009) 19(3), 305-319; McCarthy С. и Walker E., Expert Opin. Ther. Patents 2014, 24(7):731-744).
В публикации международной заявки № WO 2010/032856 описаны соединения, представленные формулой
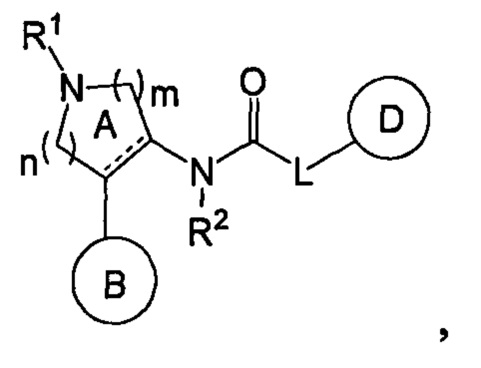
где кольцо В представляет собой ароматическое кольцо, кольцо D представляет собой ароматическое кольцо, a L представляет собой NR3, NR3C(R4aR4b), О или OC(R4aR4b), которые, как утверждается, являются антагонистами рецепторов тахикининов.
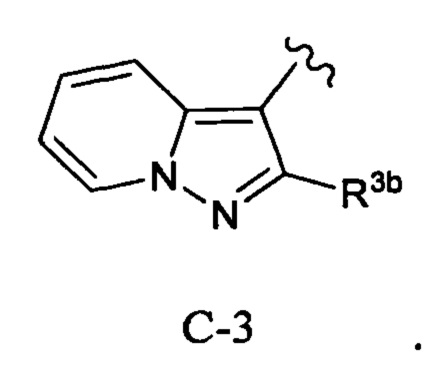
В публикации международной заявки № WO 2012/158413 раскрывается подгруппа соединений пирролидинилмочевины в качестве ингибиторов TrkA, имеющих общую формулу:
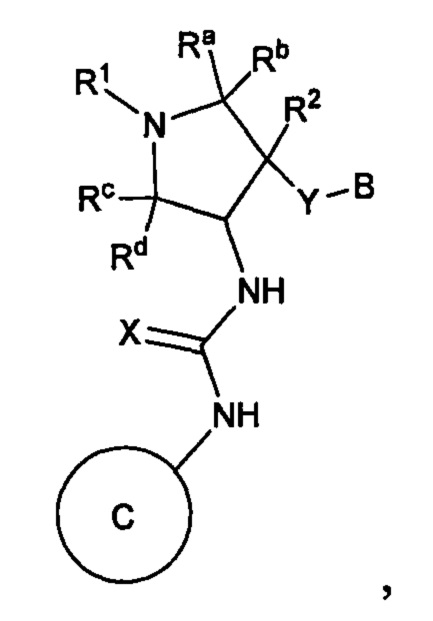
где
Y представляет собой связь, -О- или -ОСН2-;
X представляет собой О, S или NH;
R1 представляет собой (1-3С алкокси)(1-6С)алкил, (трифторметокси)(1-6С)алкил, (1-3С сульфанил)(1-6С) алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, циано(1-6С)алкил, аминокарбонил(1-6С)алкил, гидрокси(1-6С)алкил, дигидрокси(2-6С)алкил, (1-6С)алкил, (1-3С алкиламино)(1-3С)алкил, (1-4С алкоксикарбонил)(1-6С)алкил, амино(1-6С)алкил, гидрокси(1-3С алкокси)(1-6С)алкил, ди(1-3С алкокси)(1-6С)алкил, (1-3С алкокси)трифтор(1-6С)алкил, гидрокситрифтор(1-6С)алкил, (1-4С алкоксикарбонил)(1-3С алкокси)(1-6С) алкил, гидроксикарбонил(1-3С алкокси)(1-6С)алкил, гетAr5(СН2)0-1 или Ar5(CH2)0-1;
гетAr5 представляет собой 5-6-членное гетероарильное кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, О или S, при этом данное кольцо необязательно замещено одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила, (1-6С)алкокси и CF3;
Ar5 представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из галогена, (1-6С)алкила, (1-6С)алкокси, CF3O-, (1-4С)алкоксикарбонила и аминокарбонила;
В представляет собой фенил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена, CF3, CF3O-, (1-4С)алкокси, гидрокси(1-4С)алкила, (1-6С)алкила и CN; 5-6-членный гетероарил, имеющий 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенный 1-2 группами, независимо выбранными из (1-6С)алкила, галогена, ОН, CF3, NH2 и гидрокси(1-2С)алкила; 1-6С-алкил; или (1-6С)алкокси; и
Кольцо С представляет собой формулу С-1, С-2 или С-3:
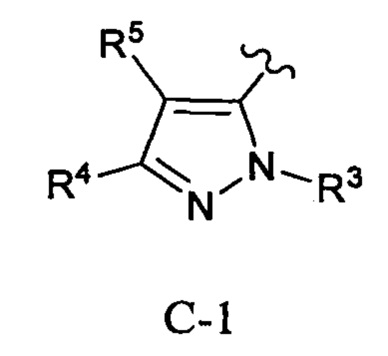
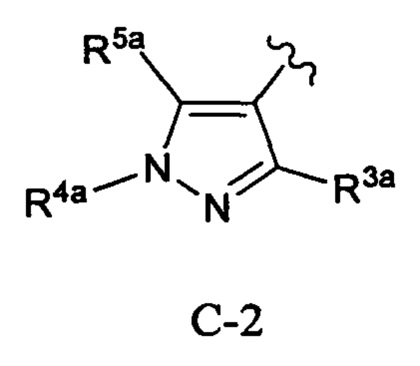
Примеры таких соединений в WO 2012/158413 включают соединение примера 511, имеющее структуру:
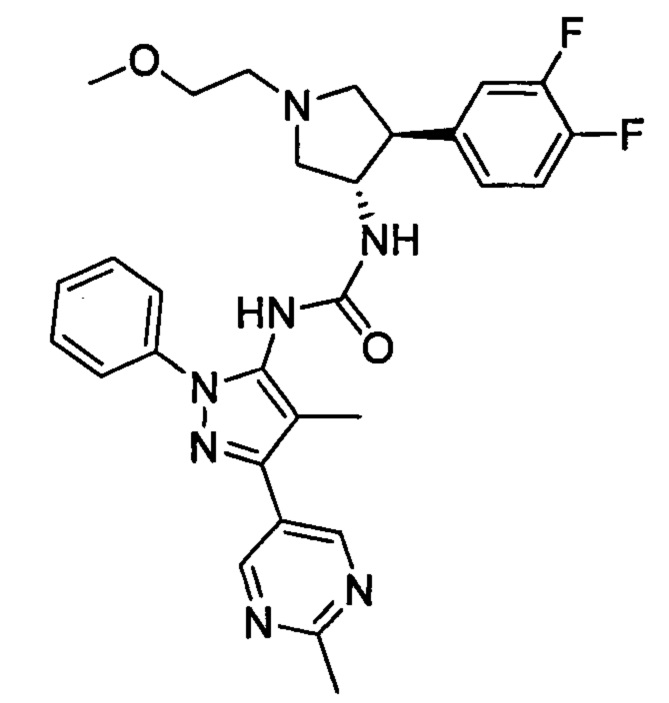
которое также известно как 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-ил)мочевина (далее по тексту в данном документе "соединение 2"), и соединение примера 441, имеющее структуру:
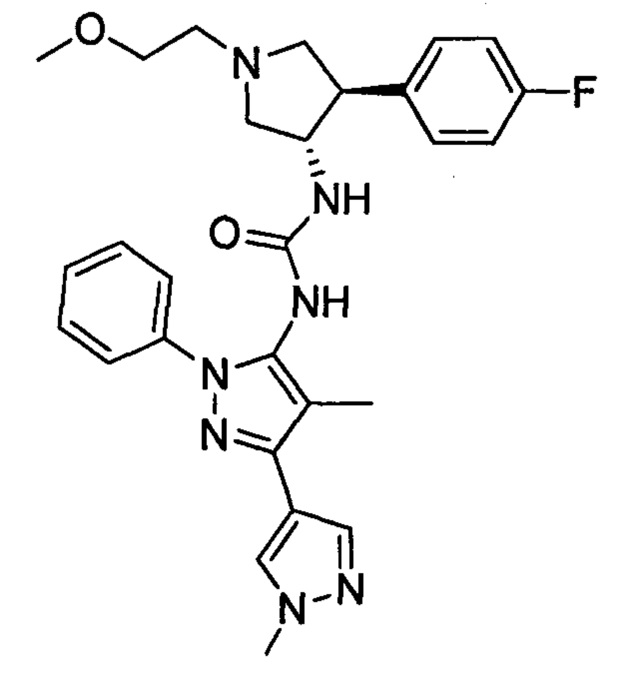
которое также известно как 1-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина (далее по тексту в данном документе "соединение 3").
На данный момент обнаружено, что соединение, обладающее неожиданными и особенно желательными свойствами, может быть получено посредством выбора 2-метилпиримидин-5-ила в качестве группы R4 и 3-фторфенила в качестве группы В.
Сущность изобретения
В данном документе предлагается соединение 1:
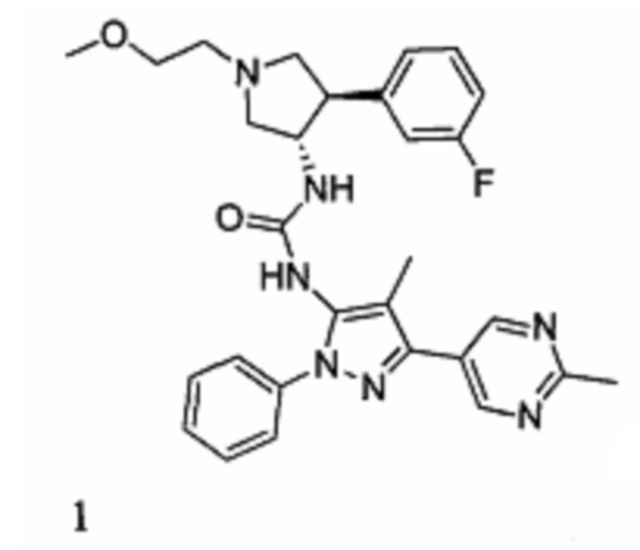
или его фармацевтически приемлемая соль, которая является ингибитором TrkA. Данное соединение также можно описать химическим названием 1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-ил)мочевина.
Неожиданно было обнаружено, что соединение 1 имеет значительно более низкий расчетный свойственный человеку в/в (внутривенный) клиренс у крыс по сравнению с соединениями 2 и 3. Также неожиданно было обнаружено, что соединение 1 обеспечивает более высокую AUC при пероральном введении (площадь под кривой зависимости концентрации лекарственного средства в плазме крови от времени) после приема пероральной дозы, составляющей 10 мг/кг, у крыс по сравнению с соединениями 2 и 3. Также неожиданно было обнаружено, что соединение 1 обеспечивает более высокую Cmax (максимальную концентрацию, достигнутую в кривой зависимости концентрации лекарственного средства в плазме крови от времени) после приема пероральной дозы, составляющей 10 мг/кг, у крыс по сравнению с соединениями 2 и 3. Также неожиданно было обнаружено, что соединение 1 обеспечивает более высокую остаточную концентрацию (минимальную концентрацию, достигнутую в кривой зависимости концентрации лекарственного средства в плазме крови от времени) после приема пероральной дозы, составляющей 10 мг/кг, у крыс по сравнению с Соединениями 2 и 3. Также неожиданно было обнаружено, что соединение 1 имеет более высокое оценочное ингибирование TrkA по сравнению с соединениями 2 и 3. соединение 1 также имеет неожиданно улучшенное распределение от периферической к центральной нервной системе по сравнению с соединением 2, о чем свидетельствует более высокое отношение концентрации в головном мозге к концентрации в плазме крови после перорального введения у крыс, соединение 1 также имеет неожиданное снижение ингибирующей активности канала hERG (ген специфических калиевых каналов сердца человека) по сравнению с соединением 2.
Также в данном документе предлагаются способы лечения заболевания или расстройства, модулируемого TrkA, включающие введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения 1 или его фармацевтически приемлемой соли.
Также в данном документе предлагаются способы лечения хронической и острой боли, в том числе, без ограничения ими, воспалительной боли, нейропатической боли и боли, обусловленной раком, хирургическим вмешательством или переломом кости, включающие введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения 1 или его фармацевтически приемлемой соли.
Также в данном документе предлагаются способы лечения рака, включающие введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения 1 или его фармацевтически приемлемой соли.
Также в данном документе предлагаются способы лечения воспаления и воспалительных заболеваний, включающие введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения 1 или его фармацевтически приемлемой соли.
Также в данном документе предлагаются способы лечения нейродегенеративных заболеваний, включающие введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения 1 или его фармацевтически приемлемой соли.
Также в данном документе предлагаются способы лечения простатита или синдрома тазовой боли, включающие введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения 1 или его фармацевтически приемлемой соли.
Также в данном документе предлагаются способы лечения заболеваний, связанных с дисбалансом регуляции ремоделирования костной ткани, таких как остеопороз, ревматоидный артрит и метастазы в кости, включающие введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения 1 или его фармацевтически приемлемой соли.
Также в данном документе предлагаются способы лечения заболеваний и расстройств, выбранных из некоторых инфекционных заболеваний, синдрома Шегрена, эндометриоза и диабетической периферической нейропатии, включающие введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения 1 или его фармацевтически приемлемой соли.
Также в данном документе предлагаются способы лечения заболеваний, обусловленных аберрантной передачей сигналов фактора роста соединительной ткани, включающие введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения 1 или его фармацевтически приемлемой соли.
В одном варианте реализации изобретения любой из приведенных выше способов лечения включает введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения 1 или его фармацевтически приемлемой соли в комбинации с дополнительным терапевтическим средством.
Также в данном документе предлагается фармацевтическая композиция, содержащая соединение 1 или его фармацевтически приемлемую соль.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в терапии.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении хронической и острой боли, в том числе, без ограничения ими, воспалительной боли, нейропатической боли и боли, обусловленной раком, хирургическим вмешательством или переломом кости.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении рака.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении воспаления или воспалительных заболеваний.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении нейродегенеративных заболеваний.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении простатита или синдрома тазовой боли.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении заболеваний, связанных с дисбалансом регуляции ремоделирования костной ткани, таких как остеопороз, ревматоидный артрит и метастазы в кости.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении некоторых инфекционных заболеваний, синдрома Шегрена, эндометриоза и диабетической периферической нейропатии.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении заболеваний, обусловленных аберрантной передачей сигналов фактора роста соединительной ткани.
Также в данном документе предлагается применение соединения 1 или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения заболеваний и расстройств, таких как хроническая и острая боль, в том числе, без ограничения ими, воспалительная боль, нейропатическая боль и боль, обусловленная раком, хирургическим вмешательством или переломом кости.
Также в данном документе предлагается применение соединения 1 или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения заболеваний и расстройств, выбранных из рака, воспаления или воспалительных заболеваний, нейродегенеративных заболеваний, некоторых инфекционных заболеваний, синдрома Шегрена, эндометриоза, диабетической периферической нейропатии, простатита, синдрома тазовой боли и заболеваний, связанных с дисбалансом регуляции ремоделирования костной ткани, таких как остеопороз, ревматоидный артрит и метастазы в кости.
Также в данном документе предлагается применение соединения 1 или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения заболеваний, обусловленных аберрантной передачей сигналов фактора роста соединительной ткани.
Также в данном документе предлагаются промежуточные соединения, применяемые в получении соединений, ингибирующих TrkA, таких как соединение 1, а также способы получения таких промежуточных соединений.
Также в данном документе предлагаются способы получения, способы разделения и способы очистки соединения 1 или его фармацевтически приемлемой соли.
Краткое описание фигур
На фигуре 1 представлен график сравнения концентрации лекарственного средства в плазме крови (мкг/мл) в зависимости от времени (в часах) в случае соединения 1 (треугольники) и соединения 2 (квадраты) после приема пероральной дозы, составляющей 10 мг/кг, у крыс.
На фигуре 2 представлен график сравнения концентрации лекарственного средства в плазме крови (мкг/мл) в зависимости от времени (в часах) в случае соединения 1 (треугольники) и соединения 3 (кружки) после приема пероральной дозы, составляющей 10 мг/кг, у крыс.
На фигуре 3 представлено сравнение оценочного процента ингибирования TrkA в зависимости от времени (в часах) в случае соединения 1 (треугольники) и соединения 2 (квадраты) после приема пероральной дозы, составляющей 10 мг/кг, у крыс.
На фигуре 4 представлено сравнение оценочного процента ингибирования TrkA в зависимости от времени (в часах) в случае соединения 1 (треугольники) и соединения 3 (кружки) после приема пероральной дозы, составляющей 10 мг/кг, у крыс.
Подробное описание сущности изобретения
В данном документе предлагается соединение 1
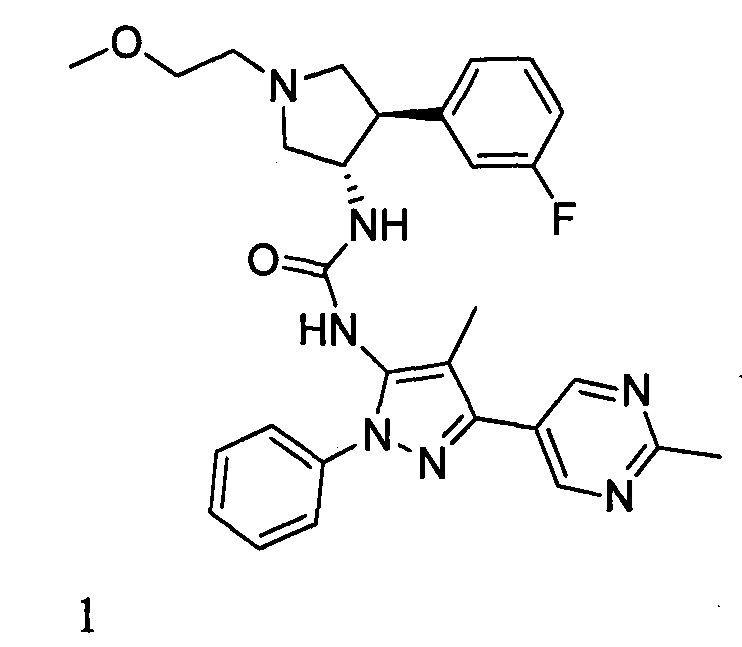
или его фармацевтически приемлемая соль, которая является ингибитором TrkA. Данное соединение также можно описать химическим названием 1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-ил)мочевина.
Было обнаружено, что соединение 1 обладает определенными неожиданными и желательными свойствами. Одним из особенно полезных свойств соединения 1 является его расчетный в/в клиренс у человека, который значительно ниже по сравнению с расчетным в/в клиренсом у человека как в случае соединения 2, так и соединения 3, как показано в таблице 1.

* Нет данных
** Наблюдаемую у доклинических видов среднюю степень поправки использовали для расчета в/в клиренса у человека.
*** Доклинический средний поправочный коэффициент для всех испытуемых видов
В таблице 1 значения расчетного в/в клиренса у человека рассчитывали из данных концентрации соединения в плазме крови, собранной у доклинических видов (мыши, крысы, собаки и обезьяны), в зависимости от времени после введения внутривенных (в/в) доз, составляющих 1 мг/кг. Концентрации соединения в плазме крови определяли с помощью ЖХ-МС (жидкостной хроматографии-масс-спектрометрии). Значения AUCinf (площадь под кривой зависимости концентрации в плазме крови от времени для интервала дозирования от 0 времени, экстраполированная до бесконечности) для отдельных кривых Cp/t (концентрация лекарственного средства в плазме крови в любой момент времени t) определяли с помощью интегрирования линейным методом трапеций и экстраполяции первого порядка от конечных точек кривых Cp/t, а клиренс (CL) рассчитывали с помощью отношения доза/AUCinf.
Значение расчетного печеночного клиренса (CLh) рассчитывали посредством поправки периода полувыведения in vitro (t1/2) на стабильность соединения в микросомах печени (1 мг/мл; 20 минут) с помощью физических и физиологических поправочных коэффициентов, применяемых для расчета собственного клиренса (CLint) и печеночного клиренса (CLhep), приведенных в таблице 2 и применяемых в следующих уравнениях:
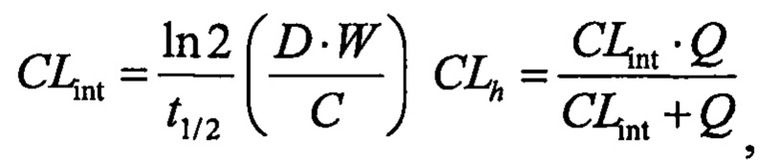
где D представляет собой отношение количества цитохром Р450-подобного белка к массе печени, W представляет собой отношение средней массы рассматриваемой печени к массе животного, С представляет собой концентрацию в микросомах печени и Q представляет собой зависящий от вида печеночный кровоток.
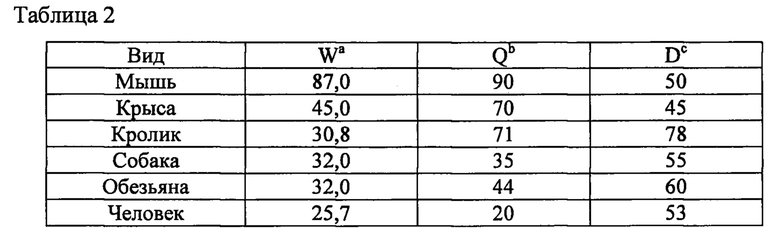
a) W = отношение средней массы печени (г) к массе животного (кг).
b) Q = средний печеночный кровоток (мл/мин/кг).
c) D (мик.) = отношение количества цитохром Р450-подобного белка (мг) к массе печени (г).
Затем определяли поправочный коэффициент in vivo/in vitro посредством деления значения микросомального CLh на значение в/в CL. Для каждого соединения применяли среднее арифметическое доклинических поправочных коэффициентов, чтобы рассчитать в/в клиренс у человека, исходя из измеренного микросомального CLh у человека (CLh/поправочный коэффициент).
Кроме того, соединение 1 неожиданно обеспечивает более высокую AUC при пероральном введении (площадь под кривой зависимости концентрации лекарственного средства в плазме крови от времени), более высокую Сmax (максимальную концентрацию, достигнутую в кривой зависимости концентрации лекарственного средства в плазме крови от времени) и более высокую остаточную концентрацию (минимальную концентрацию, достигнутую в кривой зависимости концентрации лекарственного средства в плазме крови от времени) после приема пероральной дозы (10 мг/кг), как продемонстрировано на примере их фармакокинетических кривых у крыс, по сравнению с соединениями 2 и 3, как показано на фигурах 1 и 2 и обобщено в таблице 3.
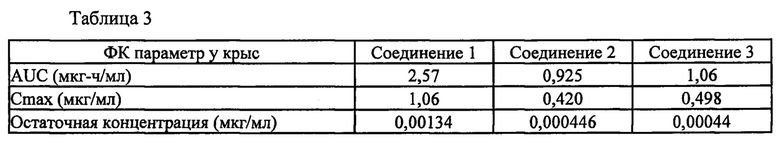
На фигурах 3 и 4 проиллюстрировано оценочное ингибирование TrkA в случае соединения 1 после приема пероральной дозы, составляющей 10 мг/кг, у крыс по сравнению с соединением 2 и в случае соединения 1 по сравнению с соединением 3, соответственно. Кривые на фигурах 3 и 4 рассчитывали с помощью следующего уравнения:
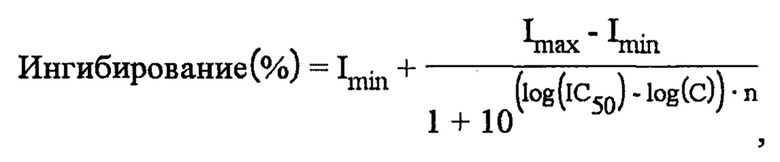
где Imin и Imax представляют собой минимальное и максимальное возможное ингибирование мишени, соответственно, IC50 представляет собой концентрацию, при которой мишень ингибируется на 50%, С представляет собой концентрацию ингибитора, а n представляет собой угловой коэффициент Хилла. Как показано на фигурах 3 и 4, оценочное ингибирование TrkA в случае соединения 1 выше, чем в случае соединения 2 и соединения 3.
Более низкий клиренс, более высокая AUC, более высокая Сmax и более высокая остаточная концентрация соединения 1 приводят к большему ингибированию рецептора TrkA в течение дня после введения соединения 1 пациенту по сравнению с соединениями 2 и 3. Это обеспечивает более высокую терапевтическую эффективность в случае соединения 1 по сравнению с соединениями 2 и 3. Возможность достижения улучшенной терапевтической эффективности имеет важное значение в лечении пациентов, страдающих, например, от болевого синдрома средней и высокой интенсивности, некоторых воспалительных состояний или рака (в том числе раковых заболеваний, обусловленных аберрантной передачей сигналов TrkA).
Кроме того, более низкий клиренс соединения 1 приводит к более длительному периоду полувыведения соединения после введения по сравнению с соединениями 2 и 3, приводя к более устойчивому ингибированию рецептора TrkA после перорального, внутривенного или подкожного введения соединения. Увеличенный период полувыведения имеет преимущества с точки зрения обеспечения более устойчивой эффективности и/или уменьшенной частоты приема лекарственного средства. Кроме того, более низкий клиренс у человека и более высокая AUC при пероральном введении соединения 1 имеют преимущество в том аспекте, что требуется более низкая доза соединения 1 для эффективного ингибирования рецептора TrkA и обеспечения заданной терапевтической эффективности по сравнению с соединениями 2 и 3. Снижение дозы соединения, необходимого для эффективности, приводит к улучшению переносимости лекарственного средства, стоимости лечения, а также соблюдению пациентами режима и схемы лечения.
Соединение 1 также имеет неожиданно улучшенное распределение от периферической к центральной нервной системе по сравнению с соединением 2, о чем свидетельствует более высокое отношение концентрации в головном мозге к концентрации в плазме крови после перорального введения у крыс, как показано в таблице 4. Поддержание низкой концентрации в головном мозге имеет преимущество снижения риска со стороны центральной нервной системы, связанного с побочными эффектами, такими как побочные эффекты, связанные с когнитивной или двигательной недостаточностью. Сниженная концентрация в центральной нервной системе является благоприятной в том аспекте, что у пациента возникает меньше побочных эффектов или они менее выражены для заданного терапевтического эффекта и/или достигается больший терапевтический эффект, прежде чем начнут наблюдаться какие-либо дозолимитирующие побочные эффекты.
Соединение 1 также имеет неожиданно сниженную ингибирующую активность в отношении канала hERG (ген специфических калиевых каналов сердца человека) по сравнению с соединением 2, как показано в таблице 4. Ингибирование функции канала hERG может привести к серьезному побочному медикаментозному (приобретенному) синдрому удлиненного интервала QT, который может создавать сопутствующий риск внезапной смерти. Из-за серьезного характера этого побочного эффекта снижение ингибирующей активности в отношении hERG является весьма желательным.

Описанные выше неожиданные свойства соединения 1 по сравнению с соединением 2 нельзя объективно возлагать на незначительное структурное различие между этими соединениями. Единственным структурным различием между соединением 1 и соединением 2 является удаление одного атома фтора. Тем не менее, в данной области техники демонстрировалось, что удаление одного атома фтора из соединения, как правило, не приводит к получению соединения, имеющего улучшенное изменение расчетного клиренса у человека, как наблюдалось в случае соединения 1 по сравнению с соединением 2. По сути, в данной области техники существует ряд примеров с другими фрагментами лекарственных средств, демонстрирующих что аналоги, имеющие больше атомов фтора, как правило, имеют более низкий клиренс in vivo по сравнению с их аналогами, имеющими меньше атомов фтора (смотри, например, Barker, A.J., et al., Bioorg. Med. Chem. Lett. 11 (2001) 1911-1914; Rosenblum, S.B. et al., J. Med. Chem., 41, (1998) 973-980; Brown, M.F. et al., Bioorg. Med. Chem. Lett. 14 (2004) 2175-2179). В противоположность этому, монофтористое соединение 1 имеет более низкий расчетный клиренс у человека по сравнению с дифтористым соединением 2. Аналогичным образом, фармакокинетические (ФК) преимущества более высокой Сmах, более высокой AUC, более высокой остаточной концентрации и более высокого нокдауна мишени TrkA в случае соединения 1 по сравнению с соединением 2, как показано на доклинических ФК данных крысы, являются весьма значительными и неожиданными для такого небольшого структурного изменения.
Кроме того, сравнение ФК параметров соединения 1 и соединения 3 демонстрирует, что улучшенная фармакокинетика соединения 1 не является общим явлением для всех монофтористых аналогов. По сути, соединение 3 продемонстрировало худший прогнозный клиренс у человека и худшие ФК значения в отношении Сmах, AUC, остаточной концентрации и нокдауна мишени TrkA по сравнению с соединением 1, несмотря на наличие монофтор-замещенной фенильной группы. Уникальная комбинация метилпиримидинила и монофтор-замещенных фенильных фрагментов в соединении 1 неожиданно обеспечивает превосходные свойства, описанные выше.
Также в данном документе предлагаются фармацевтически приемлемые соли соединения 1. Конкретные соли включают гидрохлоридные соли. В одном варианте реализации изобретения в данном документе предлагается моногидрохлоридная соль соединения 1. В одном варианте реализации изобретения в данном документе предлагается дигидрохлоридная соль соединения 1.
В одном варианте реализации изобретения в данном документе предлагается щелочная форма соединения 1.
В одном варианте реализации изобретения соединение 1 или его соли можно выделить в форме сольватов и, следовательно, любой такой сольват входит в объем настоящего изобретения. Например, соединение 1 или его соли могут существовать как в несольватированной, так и в сольватированной формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и тому подобное.
Термин "фармацевтически приемлемый" означает, что вещество или композиция являются химически и/или токсикологически совместимыми с другими содержащими данный препарат компонентами и/или млекопитающим, подлежащим лечению ими.
Также в данном документе предлагается способ получения соединения 1 или его фармацевтически приемлемой соли, который включает:
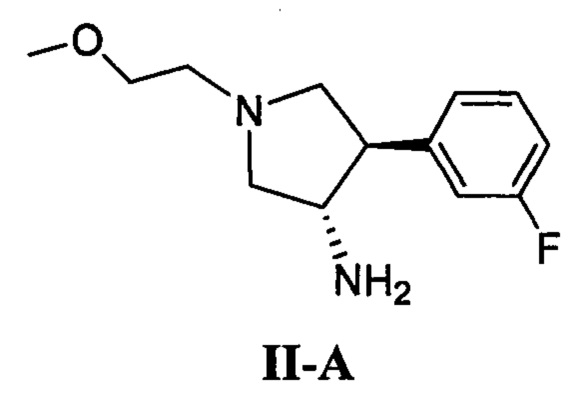
(а) приведение в контакт соединения, имеющего формулу II-А
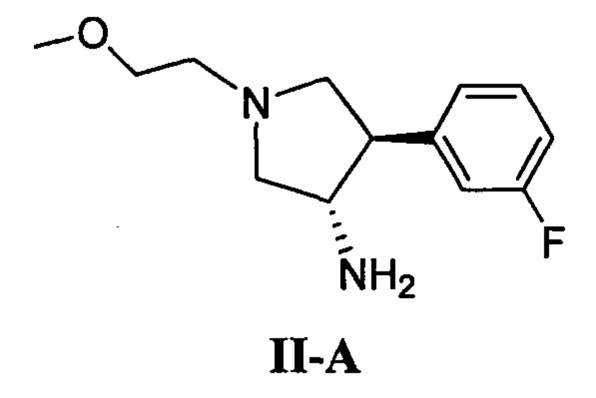
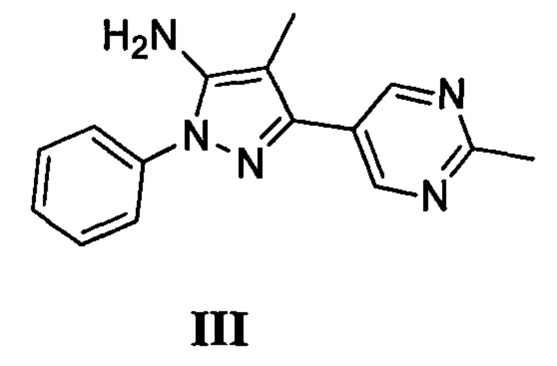
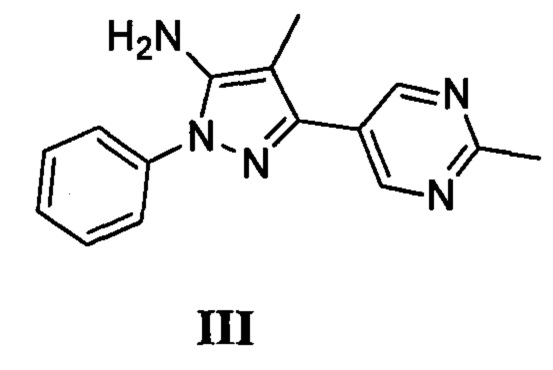
с соединением, имеющим формулу III
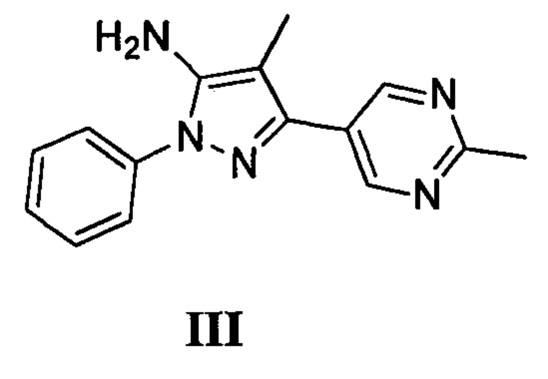
в присутствии карбонилдиимидазола или трифосгена и основания; или
(б) приведение в контакт соединения, имеющего формулу II-А
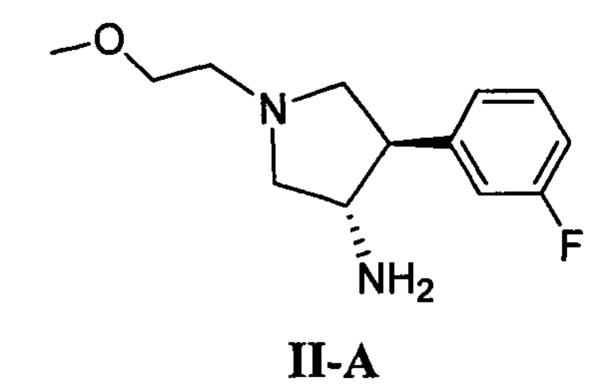
с соединением, имеющим формулу IV
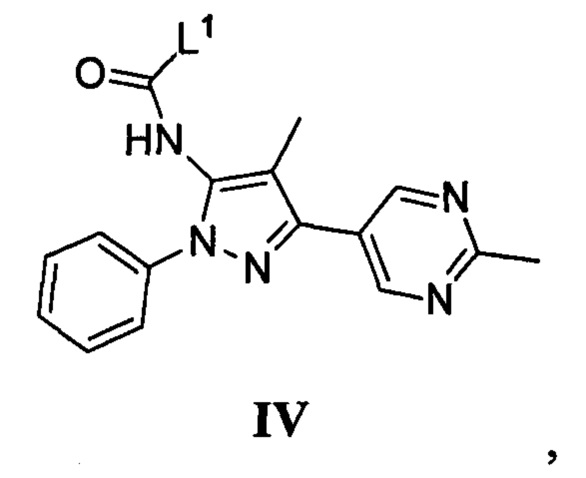
где L1 представляет собой уходящую группу, в присутствии основания; или
(в) приведение в контакт соединения, имеющего формулу V
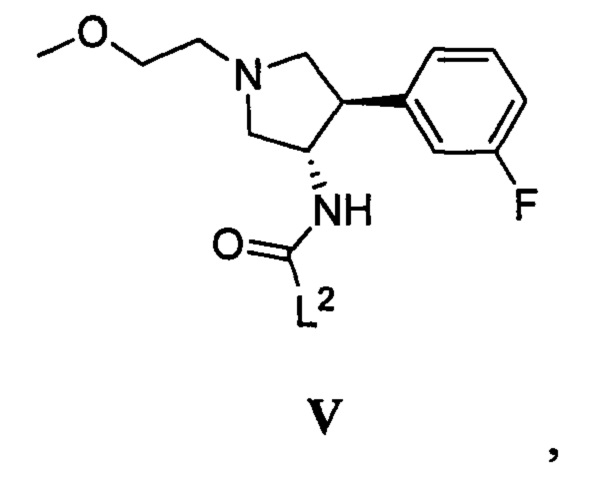
где L2 представляет собой уходящую группу, с соединением, имеющим формулу III
в присутствии основания; или
(г) приведение в контакт соединения, имеющего формулу VI
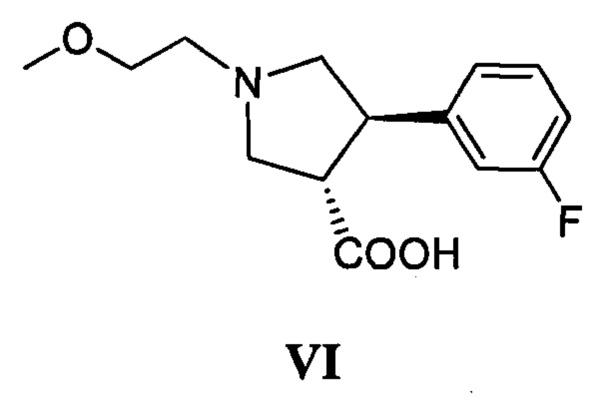
с дифенилфосфорилазидом с образованием промежуточного соединения, с последующим приведением в контакт данного промежуточного соединения с соединением, имеющим формулу III
в присутствии основания; или
(д) приведение в контакт соединения, имеющего формулу II-А
с соединением, имеющим формулу VII
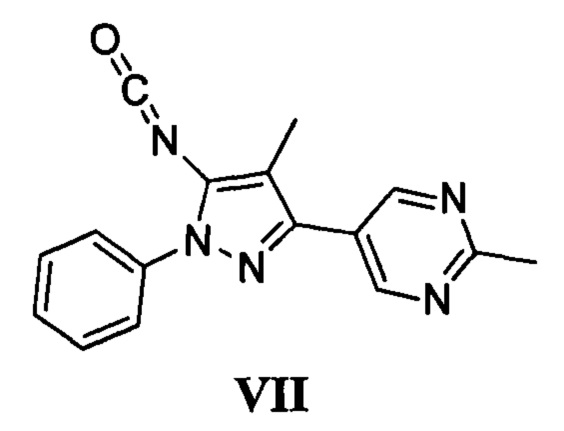
в присутствии основания; и
удаление защитных групп, в случае присутствия, и необязательно получение его фармацевтически приемлемой соли.
Со ссылкой на способ (а), основание может представлять собой аминное основание, такое как триэтиламин или диизопропиламин. Подходящие растворители включают дихлорметан, дихлорэтан, ТГФ, ДМА и ДМФА. Реакцию обычно проводят при температуре окружающей среды.
Со ссылкой на способ (б), замещаемая группа может представлять собой, например, фенокси или 4-нитрофенокси. Основание может представлять собой аминное основание, такое как триэтиламин или диизопропиламин. Подходящие растворители включают ДМА, ДМФА и ДХЭ. Реакцию обычно проводят при температуре окружающей среды.
Со ссылкой на способ (в), замещаемая группа может представлять собой, например, фенокси или 4-нитрофенокси. Основание может представлять собой аминное основание, такое как триэтиламин или диизопропиламин. Подходящие растворители включают ДХЭ, ДМА и ДМФА. Реакцию обычно проводят при температуре окружающей среды.
Со ссылкой на способ (г), основание может представлять собой аминное основание, такое как триэтиламин или диизопропиламин. Подходящие растворители включают толуол и ДМФА. Реакцию обычно проводят при повышенных температурах, например, при температуре кипения растворителя.
Со ссылкой на способ (д), основание может представлять собой аминное основание, такое как триэтиламин или диизопропиламин. Подходящие растворители включают ДХМ, ДХЭ, ДМФА и ТГФ. Реакцию обычно проводят при температуре от около 0°С до температуры окружающей среды.
Также в данном документе предлагается способ получения рацемической смеси соединения формулы II
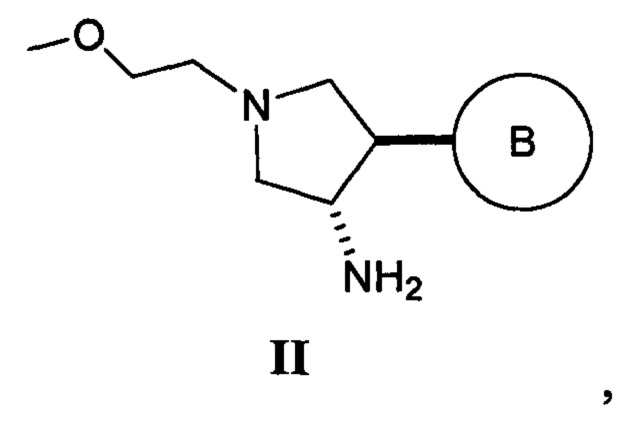
где кольцо В и фрагмент NH2 находятся в транс-конфигурации. Соединения формулы II (такие как соединения формулы II-А) можно применять для получения соединений, таких как соединения, ингибирующие TrkA, таких как соединение 1.
Соответственно, в данном документе предлагается способ получения рацемической смеси соединения формулы II
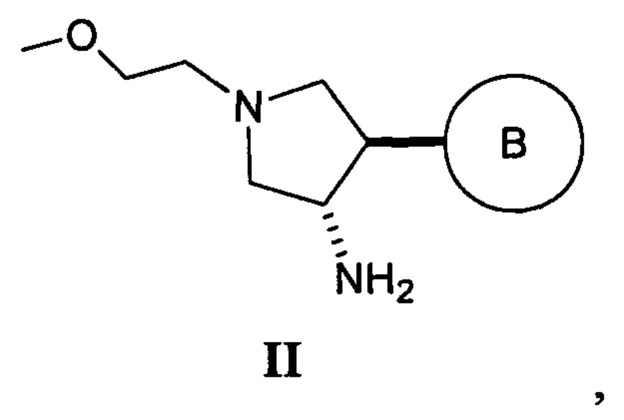
где
кольцо В представляет собой Ar1 или гетAr1;
Ar1 представляет собой фенил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена, CF3, CF3O-, (1-4С)алкокси, гидрокси(1-4С)алкила, (1-6С)алкила и CN; и
гетAr1 представляет собой 5-6-членный гетероарил, имеющий 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенный 1-2 группами, независимо выбранными из (1-6С)алкила, галогена, ОН, CF3, NH2 и гидрокси(1-2С)алкила, при этом указанный способ включает:
(а) приведение в контакт соединения формулы (а)
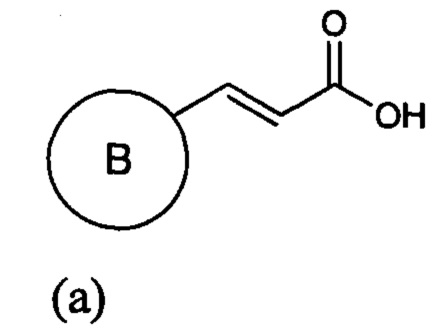
где кольцо В является таким, как определено для формулы II, с 2-метокси-N-(метоксиметил)-N((триметилсилил)метил)этанамином, имеющим формулу
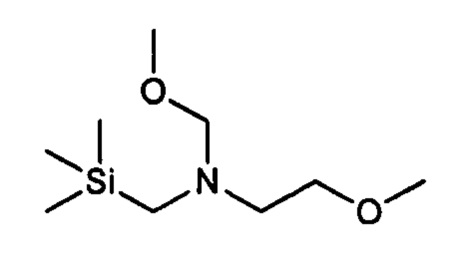
в присутствии каталитического количества кислоты, с получением соединения формулы (б)
где кольцо В является таким, как определено для формулы II;
(б) приведение в контакт указанного соединения формулы (б) с карбонилдиимидазолом в присутствии каталитического количества гидрохлорида имидазола, с последующей обработкой аммиаком, с получением соединения, имеющего формулу (в)
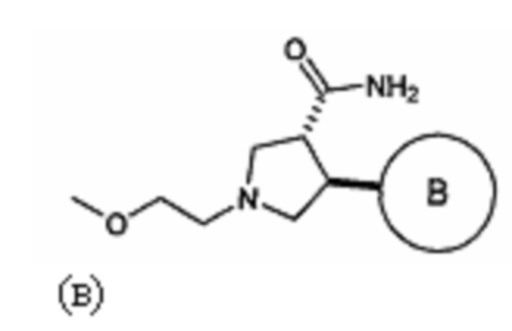
где кольцо В является таким, как определено для формулы II; и
(в) приведение в контакт указанного соединения формулы (в) с гипохлоритом натрия, с последующей обработкой KOH, с последующей обработкой НСl, с получением указанного соединения формулы II в виде рацемической смеси транс формулы II.
Со ссылкой на стадию (а), кислота может представлять собой органическую кислоту, такую как трифторуксусная кислота.
В одном варианте реализации указанного выше способа получения рацемической смеси соединения формулы II кольцо В представляет собой фенил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена, CF3, CF3O-, (1-4С)алкокси, гидрокси(1-4С)алкила, (1-6С)алкила и CN.
В одном варианте реализации указанного выше способа получения рацемической смеси соединения формулы II кольцо В представляет собой фенил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена.
В одном варианте реализации указанного выше способа получения рацемической смеси соединения формулы II кольцо В представляет собой фенил, необязательно замещенный одним или более атомами фтора.
В одном варианте реализации указанного выше способа получения рацемической смеси соединения формулы II кольцо В представляет собой 3-фторфенил.
В одном варианте реализации указанного выше способа получения рацемической смеси соединения формулы II кольцо В представляет собой 3,4-дифторфенил.
В одном варианте реализации указанного выше способа получения рацемической смеси соединения формулы II кольцо В представляет собой 5-6-членный гетероарил, имеющий 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенный 1-2 группами, независимо выбранными из (1-6С)алкила, галогена, ОН, CF3, NH2 и гидрокси(1-2С)алкила.
В одном варианте реализации указанного выше способа кольцо В представляет собой пиридильное, тиофенильное, тиазолильное, оксазолильное или изоксазолильное кольцо, необязательно замещенное 1-2 группами, независимо выбранными из (1-6С)алкила, галогена, ОН, CF3, NH2 и гидрокси(1-2С)алкила.
В одном варианте реализации указанного выше способа получения рацемической смеси соединения формулы II кольцо В представляет собой пиридильное, тиофенильное, тиазолилыюе, оксазолильное или изоксазолильное кольцо, необязательно замещенное 1-2 группами, независимо выбранными из галогена и (1-6С)алкила.
В одном варианте реализации указанного выше способа получения рацемической смеси соединения формулы II 2-метокси-N-(метоксиметил)-N-((триметилсилил)метил)этанамин, имеющий формулу
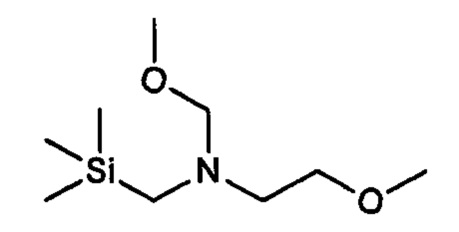
может быть получен способом, включающим:
(i) приведение в контакт (хлорметил)триметилсилана с 2-метоксиэтанамином с получением (2-метокси-N-((триметилсилил)метил)этанамина, имеющего структуру
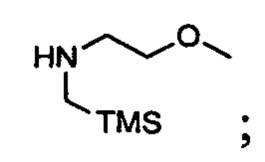 и
и
(ii) обработку указанного (2-метокси-N-((триметилсилил)метил)этанамина формальдегидом в метаноле с получением указанного 2-метокси-N-(метоксиметил)-N-((триметилсилил)метил)этанамина.
В данном документе дополнительно предлагается способ выделения энантиомера 1 транс формулы II, либо в виде свободного основания, либо в виде соли ди-п-толуоил-D-винной кислоты, включающий:
обработку рацемической транс формулы II ди-п-толуоил-D-винной кислотой с получением соли ди-п-толуоил-D-винной кислоты рацемической транс II;
перекристаллизацию соли ди-п-толуоил-D-винной кислоты транс II с получением соли ди-п-толуоил-D-винной кислоты энантиомера 1 транс II; и
необязательную обработку соли ди-п-толуоил-D-винной кислоты энантиомера 1 транс II неорганическим основанием с получением свободного основания энантиомера 1 транс формулы II, имеющего абсолютную конфигурацию, изображенную ниже:
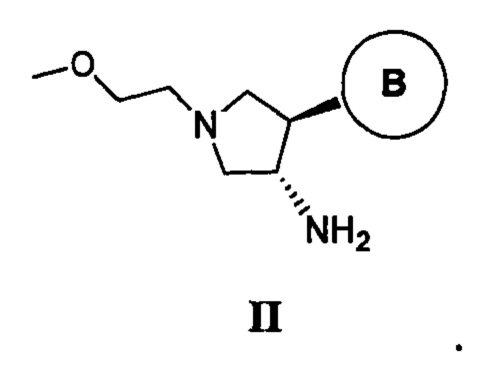
В одном варианте реализации изобретения соль ди-п-толуоил-D-винной кислоты энантиомера 1 транс II применяют для получения соединений, таких как соединения, ингибирующие TrkA, таких как соединение 1. Например, в одном варианте реализации изобретения соль ди-п-толуоил-D-винной кислоты энантиомера 1 транс формулы II применяют для получения соединений, таких как соединения, ингибирующие TrkA, таких как Соединение 1, в условиях Шоттен-Баумана, при этом свободное основание соли ди-п-толуоил-D-винной кислоты энантиомера 1 транс формулы II получают с применением двухфазной системы растворителей, состоящей из воды и органического растворителя (такого как дихлорметан), в присутствии основания, такого как гидроксид натрия, и подвергают реакции с соединением, имеющим формулу IV.
В одном варианте реализации изобретения свободное основание энантиомера 1 транс формулы II применяют для получения соединений, таких как соединения, ингибирующие TrkA, таких как соединение 1.
В одном варианте реализации указанного выше способа выделения энантиомера 1 транс формулы II кольцо В представляет собой фенил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена, CF3, CF3O-, (1-4С)алкокси, гидрокси(1-4С)алкила, (1-6С)алкила и CN.
В одном варианте реализации указанного выше способа выделения энантиомера 1 транс формулы II кольцо В представляет собой фенил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена.
В одном варианте реализации указанного выше способа выделения энантиомера 1 транс формулы II кольцо В представляет собой фенил, необязательно замещенный одним или более атомами фтора.
В одном варианте реализации указанного выше способа выделения энантиомера 1 транс формулы II кольцо В представляет собой 3-фторфенил.
В одном варианте реализации указанного выше способа выделения энантиомера 1 транс формулы II кольцо В представляет собой 3,4-дифторфенил.
В одном варианте реализации изобретения указанный выше способ выделения энантиомера 1 транс формулы II относится к способу получения (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина.
В одном варианте реализации указанного выше способа выделения энантиомера 1 транс формулы II кольцо В представляет собой 5-6-членный гетероарил, имеющий 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенный 1-2 группами, независимо выбранными из (1-6С)алкила, галогена, ОН, CF3, NH2 и гидрокси(1-2С)алкила.
В одном варианте реализации указанного выше способа выделения энантиомера 1 транс формулы II кольцо В представляет собой пиридильное, тиофенильное, тиазолильное, оксазолильное или изоксазолильное кольцо, необязательно замещенное 1-2 группами, независимо выбранными из (1-6С)алкила, галогена, ОН, CF3, NH2 и гидрокси(1-2С)алкила.
В одном варианте реализации указанного выше способа выделения энантиомера 1 транс формулы II кольцо В представляет собой пиридильное, тиофенильное, тиазолильное, оксазолильное или изоксазолильное кольцо, необязательно замещенное 1-2 группами, независимо выбранными из галогена и (1-6С)алкила.
Указанный выше способ получения рацемической смеси соединения формулы II имеет ряд преимуществ по сравнению со способом, описанным в публикации международной заявки № WO 2012/158413. Например, способ, описанный в WO 2012/158413, включает нитроальдольную реакцию между реагентом бензальдегида и нитрометаном. Затем полученный в результате промежуточный 2-нитровинилбензол восстанавливают гидрированием при высоком давлении. Обе эти стадии реакции не пригодны для синтеза в больших масштабах, так как нитроальдольные реакции и полученные с их помощью промежуточные нитросоединения являются неустойчивыми и могут быть опасны. В противоположность этому, в описанном выше способе получения рацемической смеси соединения формулы II не применяются особо опасные химические соединения, и он позволяет избежать применения нитросодержащих промежуточных соединений и гидрирования. Кроме того, все условия, используемые в указанном выше способе получения рацемической смеси соединения формулы II, находятся в пределах стандартных рабочих параметров, типичных для опытных установок крупномасштабного синтеза, например, в данном способе нет экстремальных температур, и не требуются сосуды высокого давления.
Способность соединения 1 или его фармацевтически приемлемой соли выступать в качестве ингибитора TrkA может быть продемонстрирована с помощью анализов, описанных в примерах А и Б.
Соединение 1 или его фармацевтически приемлемую соль применяют в лечении заболеваний и расстройств, включающих, без ограничения ими, боль, рак, воспаление/воспалительные заболевания, нейродегенеративные заболевания, некоторые инфекционные заболевания, синдром Шегрена, эндометриоз, диабетическую периферическую нейропатию, простатит, синдром тазовой боли, заболевания, связанные с дисбалансом регуляции ремоделирования костной ткани, а также заболевания, обусловленные аберрантной передачей сигналов фактора роста соединительной ткани.
При использовании по тексту данного документа, термины "лечить" или "лечение" относятся к терапевтическим или паллиативным мерам. Благоприятные или желаемые клинические результаты включают, без ограничения ими, ослабление, полностью или частично, симптомов, связанных с расстройством или состоянием, снижение степени заболевания, стабилизацию (то есть без ухудшения) болезненного состояния, задержку или замедление развития заболевания, уменьшение интенсивности или временное облегчение болезненного состояния и ремиссию (частичную или полную), обнаруживаемую или необнаруживаемую. Термин "лечение" может также означать продление времени выживания по сравнению с ожидаемым выживанием при отсутствии лечения.
В некоторых вариантах реализации изобретения соединение 1 или его фармацевтически приемлемую соль применяют для профилактики заболеваний и расстройств, указанных в данном документе. Термин "профилактика", при использовании по тексту данного документа, означает предотвращение появления, рецидива или распространения, полностью или частично, заболевания или состояния, описанного в данном документе, или его симптомов, и включает введение соединения 1 или его фармацевтически приемлемой соли до появления симптомов.
Соединение 1 или его фармацевтически приемлемая соль могут применяться в комбинации с одним или более дополнительными терапевтическими средствами, которые имеют такой же или другой механизм действия.
Термин "фармацевтическая комбинация", при использовании по тексту данного документа, относится к фармацевтическому препарату, полученному в результате смешивания или комбинирования более чем одного действующего вещества, и включает как фиксированные, так и нефиксированные комбинации действующих веществ. Термин "фиксированная комбинация" означает, что соединение 1 или его фармацевтически приемлемую соль и по меньшей мере одно дополнительное терапевтическое средство вводят пациенту одновременно в виде одного препарата или дозированной формы. Термин "нефиксированная комбинация" означает, что соединение 1 или его фармацевтически приемлемую соль и по меньшей мере одно дополнительное терапевтическое средство вводят пациенту в виде отдельных препаратов либо одновременно, либо последовательно без наложения специальных ограничений по времени, при этом такое введение обеспечивает эффективные уровни двух или более соединений в организме пациента. Последнее также относится к смешанным препаратам, например, введение 3 или более действующих веществ.
При использовании по тексту данного документа, термин "совместное введение" означает введение выбранных терапевтических средств одному пациенту, и предназначен для включения схем лечения, при которых средства вводят одним или различными способами введения или одновременно, или в разное время. Этот термин охватывает введение млекопитающему двух или более средств таким образом, чтобы средства и/или их метаболиты попадали в организм млекопитающего одновременно. Он включает одновременное введение отдельных композиций, введение в разное время отдельных композиций и/или введение композиции, в которой присутствуют оба средства. В одном варианте реализации соединение (соединения) по данному изобретению и другое терапевтическое средство (средства) вводят в одной композиции. В одном варианте реализации изобретения соединение 1 или его фармацевтически приемлемую соль и другое средство (средства) смешивают в композиции.
В одном варианте реализации изобретения соединение 1 или его фармацевтически приемлемую соль применяют в лечении боли, в том числе хронической и острой боли. Например, Соединение 1 или его фармацевтически приемлемую соль применяют в лечении нескольких типов боли, в том числе воспалительной боли, нейропатической боли, позвоночной боли и боли, обусловленной раком, хирургическим вмешательством или переломом кости.
В одном варианте реализации изобретения соединение 1 или его фармацевтически приемлемую соль применяют в лечении острой боли. Острая боль, по определению Международной ассоциации по изучению боли, является следствием заболевания, воспаления или повреждения тканей. Этот тип боли, как правило, возникает внезапно, например, после травмы или хирургического вмешательства, и может сопровождаться тревожным расстройством или стрессом, и ограничивается определенным периодом времени и выраженностью. В некоторых случаях она может стать хронической.
В одном варианте реализации изобретения соединение 1 или его фармацевтически приемлемую соль применяют в лечении хронической боли. Существует широко распространенное мнение, что хроническая боль, по определению Международной ассоциации по изучению боли, представляет собой заболевание как таковое. Она может усугубляться экологическими и психологическими факторами. Хроническая боль сохраняется в течение более длительного периода, чем острая боль и не поддается лечению большинством терапевтических способов, как правило, в течение 3 месяцев или более. Она может и зачастую вызывает серьезные проблемы у пациентов.
Соответственно, в данном документе предлагается способ лечения боли у млекопитающего, включающий введение указанному млекопитающему, нуждающемуся в этом, Соединения 1 или его фармацевтически приемлемой соли в количестве, эффективном для лечения указанной боли. В одном варианте реализации изобретения боль представляет собой острую боль. В одном варианте реализации изобретения боль представляет собой хроническую боль. В одном варианте реализации изобретения боль представляет собой хроническую позвоночную боль. В одном варианте реализации изобретения боль представляет собой нейропатическую боль, такую как боль, обусловленная диабетической периферической нейропатией или недиабетической (например, обусловленная химиотерапией) периферической нейропатией. В одном варианте реализации изобретения боль представляет собой воспалительную боль, такую как боль, обусловленная остеоартритом. В одном варианте реализации изобретения боль представляет собой боль, обусловленную раком. В одном варианте реализации изобретения боль представляет собой боль, обусловленную хирургическим вмешательством. В одном варианте реализации изобретения боль представляет собой боль, обусловленную переломом кости.
Также в данном документе предлагается способ профилактики боли у млекопитающего, включающий введение указанному млекопитающему, нуждающемуся в этом, соединения 1 или его фармацевтически приемлемой соли в количестве, эффективном для предотвращения указанной боли. В одном варианте реализации изобретения боль представляет собой острую боль. В одном варианте реализации изобретения боль представляет собой хроническую боль. В одном варианте реализации изобретения боль представляет собой хроническую позвоночную боль. В одном варианте реализации изобретения боль представляет собой нейропатическую боль, такую как боль, обусловленная диабетической периферической нейропатией или недиабетической (например, обусловленная химиотерапией) периферической нейропатией. В одном варианте реализации изобретения боль представляет собой воспалительную боль, такую как боль, обусловленная остеоартритом. В одном варианте реализации изобретения боль представляет собой боль, обусловленную раком. В одном варианте реализации изобретения боль представляет собой боль, обусловленную хирургическим вмешательством. В одном варианте реализации изобретения боль представляет собой боль, обусловленную переломом кости.
Также в данном документе предлагается способ лечения боли у млекопитающего, включающий совместное введение млекопитающему, нуждающемуся в этом, эффективного количества: (а) соединения 1 или его фармацевтически приемлемой соли; и (б) по меньшей мере одного дополнительного терапевтического средства, выбранного из противовоспалительных соединений, стероидов (например, дексаметазон, кортизон и флутиказон), болеутоляющих средств, таких как НПВС (например, аспирин, ибупрофен, индометацин и кетопрофен), опиоидов (таких как морфин), антагонистов рецепторов кальцитонин ген-родственного пептида, подтип-селективных модуляторов ионных каналов, противосудорожных средств (например, прегабалин и габапентин), двойных ингибиторов обратного захвата серотонина-норадреналина (например, дулоксетин, венлафаксин и милнаципран), ингибиторов киназ семейства JAK (например, руксолитиниб или тофацитиниб) и трициклических антидепрессантов (например, амитриптилин, нортриптилин и дезипрамин). Эти дополнительные терапевтические средства можно вводить с соединением 1 или его фармацевтически приемлемой солью в качестве части одной или разных лекарственных форм, одним или различными способами введения и с одним или разными режимами применения в соответствии со стандартной фармацевтической практикой, известной специалистам в данной области техники.
Соответственно, также в данном документе предлагается фармацевтическая комбинация, содержащая эффективное количество: (а) соединения 1 или его фармацевтически приемлемой соли и (б) по меньшей мере одного дополнительного терапевтического средства, выбранного из противовоспалительных соединений, стероидов (например, дексаметазон, кортизон и флутиказон), болеутоляющих средств, таких как НПВС (например, аспирин, ибупрофен, индометацин и кетопрофен) и опиоидов (таких как морфин), для применения в лечении боли у млекопитающего, при этом (а) и (б) могут находиться в разных лекарственных формах или в одной лекарственной форме. В одном варианте реализации изобретения в данном документе предлагается фармацевтическая комбинация, содержащая (а) эффективное количество соединения 1 или его фармацевтически приемлемой соли и (б) эффективное количество болеутоляющего средства, такого как НПВС (например, аспирин, ибупрофен, индометацин и кетопрофен).
Соединение 1 или его фармацевтически приемлемую соль также применяют в лечении рака. Конкретные примеры включают нейробластому, рак яичников, поджелудочной железы, ободочной и прямой кишки, и предстательной железы.
Соответственно, в данном документе также предлагается способ лечения рака у млекопитающего, включающий введение указанному млекопитающему, нуждающемуся в этом, соединения 1 или его фармацевтически приемлемой соли в количестве, эффективном для лечения указанного рака.
Соединение 1 или его фармацевтически приемлемую соль также применяют в лечении рака, связанного с дисрегуляцией TrkA.
Соответственно, также в данном документе предлагается способ лечения пациента, у которого диагностирован рак, связанный с дисрегуляцией TrkA, включающий введение пациенту терапевтически эффективного количества соединения 1 или его фармацевтически приемлемой соли.
В одном варианте реализации изобретения дисрегуляция TrkA включает сверхэкспрессию TrkA дикого типа (аутокринная/паракринная активация).
В одном варианте реализации изобретения дисрегуляция TrkA включает амплификацию гена или одну или более хромосомных транслокаций или инверсий, приводящих к слиянию генов TrkA. В одном варианте реализации изобретения дисрегуляция является результатом генетических транслокаций, при этом экспрессируемый белок представляет собой слитый белок, содержащий остатки не-TrkA и TrkA белков, и как минимум домен киназы TrkA. В одном варианте реализации изобретения слитый белок TrkA представляет собой LMNA-TrkA, TFG-TrkA, ТРМ3-TrkA, CD74-TrkA, NFASC-TrkA, MPRIP-TrkA, BCAN-TrkA или TPR-TrkA. В одном варианте реализации изобретения слитый белок TrkA представляет собой LMNA-TrkA, TFG-TrkA, ТРМ3-TrkA, CD74-TrkA, NFASC-TrkA, MPRIP-TrkA, BCAN-TrkA, TP53-TrkA, RNF213-TrkA, RABGAP1L-TrkA, IRF2BP2-TrkA, SQSTM1-TrkA, SSBP2-TrkA или TPR-TrkA, где
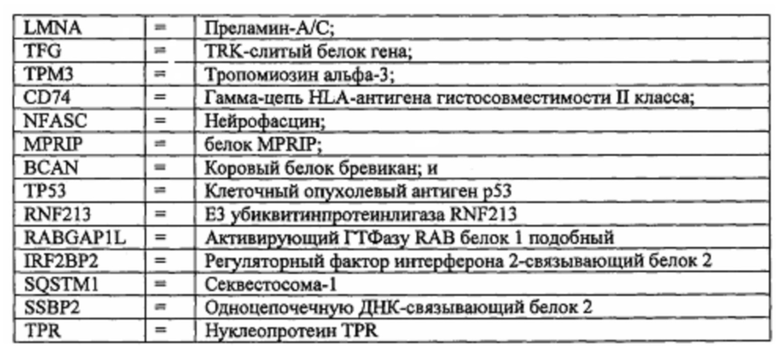
В одном варианте реализации изобретения дисрегуляция TrkA включает одну или более делеций, инсерций или мутаций в белке TrkA. В одном варианте реализации изобретения дисрегуляция включает делецию одного или более остатков белка TrkA, приводящую к конститутивной активности TrkA киназы. В одном варианте реализации изобретения делеция включает делецию остатков 303-377 в изоформе 2 TrkA.
В одном варианте реализации изобретения дисрегуляция TrkA включает сплайс-вариант, при этом экспрессируемый белок представляет собой альтернативно сплайсированный вариант TrkA с делецией одного или более остатков, приводящей к конститутивной активности TrkA киназы. В одном варианте реализации изобретения альтернативно сплайсированная форма TrkA с конститутивной активностью имеет делеций экзонов 8, 9 и 11, приводящие к потере остатков 192-284 и 393-398 в экспрессируемом белке по сравнению с изоформой 2 TrkA.
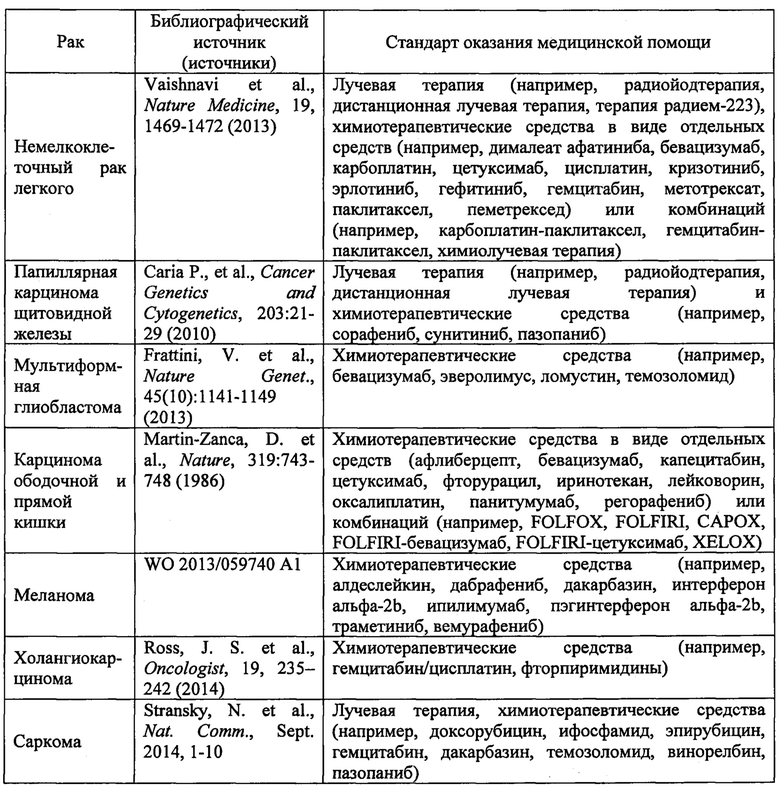
Раковые заболевания, которые установлены в качестве связанных с дисрегуляцией TrkA (смотри приведенные ниже библиографические источники; смотри также www.cancer.gov и www.nccn.org), включают:
(А) Раковые заболевания, при которых дисрегуляция TrkA включает амплификацию гена или одну или более хромосомных транслокаций или инверсий, приводящих к слиянию генов TrkA, включающие:
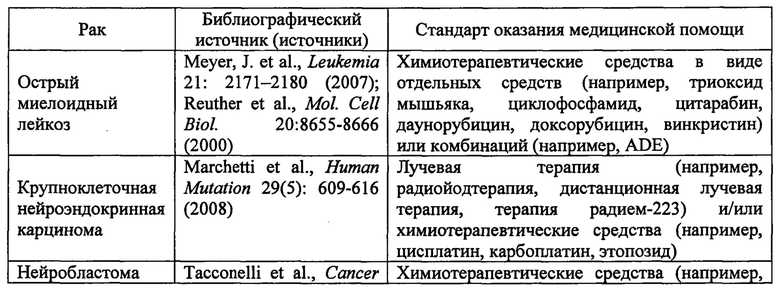
(Б) Раковые заболевания, при которых дисрегуляция TrkA включает одну или более делеций, инсерций или мутаций в белке TrkA, включающие

(В) Раковые заболевания, обусловленные сверхэкспрессией TrkA дикого типа (аутокринная активация), включающие:
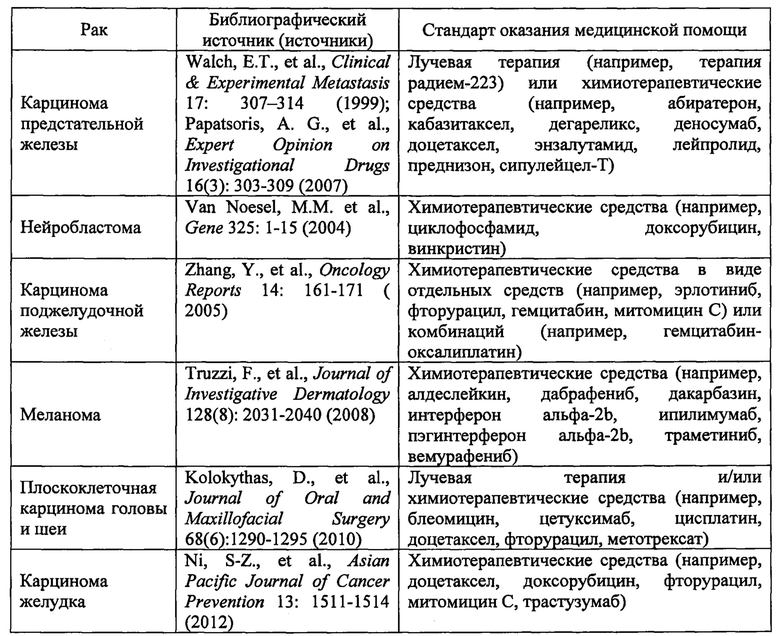
В одном варианте реализации изобретения в данном документе предлагается способ лечения пациента, у которого диагностирован рак, связанный с дисрегуляцией TrkA, включающий введение пациенту соединения 1 или его фармацевтически приемлемой соли в количестве, эффективном для лечения указанного рака, при этом рак выбран из немелкоклеточного рака легкого, папиллярной карциномы щитовидной железы, мультиформной глиобластомы, острого миелоидного лейкоза, карциномы ободочной и прямой кишки, крупноклеточной нейроэндокринной карциномы, рака предстательной железы, нейробластомы, карциномы поджелудочной железы, меланомы, плоскоклеточной карциномы головы и шеи и карциномы желудка. В одном варианте реализации изобретения в данном документе предлагается способ лечения пациента, у которого диагностирован рак, связанный с дисрегуляцией TrkA, включающий введение пациенту соединения 1 или его фармацевтически приемлемой соли в количестве, эффективном для лечения указанного рака, при этом рак выбран из немелкоклеточного рака легкого, папиллярной карциномы щитовидной железы, мультиформной глиобластомы, острого миелоидного лейкоза, карциномы ободочной и прямой кишки, крупноклеточной нейроэндокринной карциномы, рака предстательной железы, нейробластомы, карциномы поджелудочной железы, меланомы, плоскоклеточной карциномы головы и шеи, карциномы желудка, холангиокарциномы и саркомы.
Также предлагается способ лечения рака у млекопитающего, нуждающегося в этом, включающий: (а) определение того, обусловлен ли рак дисрегуляцией TrkA киназы; и (б) в случае, если было установлено, что рак обусловлен дисрегуляцией TrkA киназы, введение млекопитающему терапевтически эффективного количества соединения 1 или его фармацевтически приемлемой соли. В одном варианте реализации изобретения соединение 1 представляет собой дигидрохлоридную соль.
В одном варианте реализации изобретения дисрегуляция TrkA включает одну или более хромосомных транслокаций или инверсий, приводящих к слиянию генов TrkA. В одном варианте реализации изобретения слияние генов TrkA представляет собой LMNA-TrkA, TFG-TrkA, ТРМ3-TrkA, CD74-TrkA, NFASC-TrkA, MPRIP-TrkA, BCAN-TrkA, TP53-TrkA, RNF213-TrkA, RABGAP1L-TrkA, IRF2BP2-TrkA, SQSTM1-TrkA, SSBP2-TrkA или TPR-TrkA. В одном варианте реализации изобретения рак представляет собой немелкоклеточный рак легкого, папиллярную карциному щитовидной железы, мультиформную глиобластому, карциному ободочной и прямой кишки, меланому, холангиокарциному или саркому.
В одном варианте реализации изобретения дисрегуляция TrkA включает одну или более делеций, инсерций или мутаций в белке TrkA. В одном варианте реализации изобретения рак представляет собой острый миелоидный лейкоз, крупноклеточную нейроэндокринную карциному или нейробластому.
В одном варианте реализации изобретения дисрегуляция TrkA представляет собой сверхэкспрессию TrkA дикого типа (аутокринная активация). В одном варианте реализации изобретения рак представляет собой карциному предстательной железы, нейробластому, карциному поджелудочной железы, меланому, плоскоклеточную карциному головы и шеи или карциному желудка.
В одном варианте реализации изобретения любой из указанных выше способов лечения рака у млекопитающего, нуждающегося в этом, дополнительно включает введение эффективного количества соединения 1 или его фармацевтически приемлемой соли в комбинации с эффективным количеством по меньшей мере одного дополнительного терапевтического средства, выбранного из одной или более дополнительных терапий или химиотерапевтических средств.
В одном варианте реализации изобретения любой из указанных выше способов лечения рака у млекопитающего, нуждающегося в этом, включает введение указанному млекопитающему, нуждающемуся в этом, эффективного количества соединения 1 или его фармацевтически приемлемой соли в комбинации с одним или более дополнительными химиотерапевтическими средствами.
В одном варианте реализации изобретения дополнительное химиотерапевтическое средство (средства) выбирают из терапевтических средств, направленных на рецепторную тирозинкиназу, включающих кабозантиниб, кризотиниб, эрлотиниб, гефитиниб, иматиниб, лапатиниб, нилотиниб, пазопаниб, пертузумаб, регорафениб, сунитиниб и трастузумаб.
В одном варианте реализации изобретения дополнительное химиотерапевтическое средство (средства) выбирают из ингибиторов пути сигнальной трансдукции, в том числе ингибиторов пути Ras-Raf-MEK-ERK (например, биниметиниб, селуметиниб, энкорафениб, сорафениб, траметиниб, вемурафениб), ингибиторов пути PI3K-Akt-mTOR-S6K (например, эверолимус, рапамицин, перифозин, темсиролимус) и модуляторов пути апоптоза (например, обатоклакс).
В одном варианте реализации изобретения дополнительное химиотерапевтическое средство (средства) выбирают из цитотоксических химиотерапевтических средств, включающих триоксид мышьяка, блеомицин, кабазитаксел, капецитабин, карбоплатин, цисплатин, циклофосфамид, цитарабин, дакарбазин, даунорубицин, доцетаксел, доксорубицин, этопозид, фторурацил, гемцитабин, иринотекан, ломустин, метотрексат, митомицин С, оксалиплатин, паклитаксел, пеметрексед, темозоломид и винкристин.
В одном варианте реализации изобретения дополнительное химиотерапевтическое средство (средства) выбирают из направленных на ангиогенез препаратов, включающих афлиберцепт и бевацизумаб.
В одном варианте реализации изобретения дополнительное химиотерапевтическое средство (средства) выбирают из иммунонаправленных средств, включающих алдеслейкин, ипилимумаб, ламбролизумаб, ниволумаб, сипулейцел-Т.
В одном варианте реализации изобретения дополнительное химиотерапевтическое средство (средства) выбирают из средств, активных по отношению к пути TrkA, в том числе биофармацевтических препаратов, направленных на NGF, таких как антитела к NGF, а также неселективных ингибиторов Trk.
В одном варианте реализации изобретения дополнительное химиотерапевтическое средство или терапия представляет собой лучевую терапию, в том числе радиойодтерапию, дистанционную лучевую терапию и терапию радием-223.
В одном варианте реализации изобретения дополнительное химиотерапевтическое средство (средства) включает любую из приведенных выше терапий или терапевтических средств, которые являются стандартами оказания медицинской помощи при раковых заболеваниях, отличающихся тем, что рак связан с дисрегуляцией TrkA.
В одном варианте реализации изобретения в данном документе предлагается способ лечения рака у пациента, нуждающегося в этом, включающий введение указанному пациенту соединения 1 или его фармацевтически приемлемой соли в комбинации по меньшей мере с одной дополнительной терапией или химиотерапевтическим средством, выбранным из лучевой терапии (например, радиойодтерапия, дистанционная лучевая терапия, терапия радием-223), цитотоксических химиотерапевтических средств (например, триоксид мышьяка, блеомицин, кабазитаксел, капецитабин, карбоплатин, цисплатин, циклофосфамид, цитарабин, дакарбазин, даунорубицин, доцетаксел, доксорубицин, этопозид, фторурацил, гемцитабин, иринотекан, ломустин, метотрексат, митомицин С, оксалиплатин, паклитаксел, пеметрексед, темозоломид, винкристин), терапевтических средств, направленных на тирозинкиназу (например, афатиниб, кабозантиниб, цетуксимаб, кризотиниб, дабрафениб, эрлотиниб, гефитиниб, иматиниб, лапатиниб, нилотиниб, пазопаниб, панитумумаб, пертузумаб, регорафениб, сунитиниб, трастузумаб), модуляторов апоптоза и ингибиторов сигнальной трансдукции (например, эверолимус, перифозин, рапамицин, биниметиниб, селуметиниб, энкорафениб, сорафениб, темсиролимус, траметиниб, вемурафениб), иммунонаправленных препаратов (например, алдеслейкин, интерферон альфа-2b, ипилимумаб, ламбролизумаб, ниволумаб, преднизон, сипулейцел-Т) и направленных на ангиогенез препаратов (например, афлиберцепт, бевацизумаб), при этом количество соединения 1 или его фармацевтически приемлемой соли, в комбинации с дополнительной терапией или терапевтическим средством, является эффективным для лечения указанного рака. Эти дополнительные терапевтические средства можно вводить с соединением 1 или его фармацевтически приемлемой солью в качестве части одной или разных лекарственных форм, одним или различными способами введения и с одним или разными режимами применения в соответствии со стандартной фармацевтической практикой, известной специалистам в данной области техники.
Также в данном документе предлагается (i) фармацевтическая комбинация для лечения рака у пациента, нуждающегося в этом, содержащая (а) соединение 1 или его фармацевтически приемлемую соль, (б) дополнительное терапевтическое средство и (в) необязательно по меньшей мере один фармацевтически приемлемый носитель для одновременного, раздельного или последовательного применения в лечении рака, при этом совокупное количество соединения 1 или его фармацевтически приемлемой соли и дополнительного терапевтического средства является эффективным для лечения указанного рака; (ii) фармацевтическая композиция, содержащая такую комбинацию; (iii) применение такой комбинации для получения лекарственного средства, предназначенного для лечения рака; и (iv) серийная упаковка или продукт, содержащий такую комбинацию в виде комбинированного препарата для одновременного, раздельного или последовательного применения; а также способ лечения рака у пациента, нуждающегося в этом.
В одном варианте реализации изобретения комплексная терапия служит для лечения рака, выбранного из немелкоклеточного рака легкого, папиллярной карциномы щитовидной железы, мультиформной глиобластомы, острого миелоидного лейкоза, карциномы ободочной и прямой кишки, крупноклеточной нейроэндокринной карциномы, рака предстательной железы, нейробластомы, карциномы поджелудочной железы, меланомы, плоскоклеточной карциномы головы и шеи и карциномы желудка. В одном варианте реализации изобретения комплексная терапия служит для лечения рака, выбранного из немелкоклеточного рака легкого, папиллярной карциномы щитовидной железы, мультиформной глиобластомы, острого миелоидного лейкоза, карциномы ободочной и прямой кишки, крупноклеточной нейроэндокринной карциномы, рака предстательной железы, нейробластомы, карциномы поджелудочной железы, меланомы, плоскоклеточной карциномы головы и шеи, карциномы желудка, холангиокарциномы и саркомы.
Соединение 1 или его фармацевтически приемлемую соль также применяют в лечении воспаления или воспалительного заболевания или расстройства.
Соответственно, в данном документе предлагается способ лечения воспаления или воспалительного заболевания или расстройства у млекопитающего, включающий введение указанному млекопитающему, нуждающемуся в этом, соединения 1 или его фармацевтически приемлемой соли в количестве, эффективном для лечения указанного воспаления. В одном варианте реализации изобретения воспалительное заболевание выбрано из воспалительных заболеваний легкого (таких как астма), интерстициального цистита (ИЦ), синдрома болезненного мочевого пузыря (СБМП), воспалительных заболеваний кишечника (в том числе неспецифического язвенного колита и болезни Крона), а также воспалительных кожных заболеваний, таких как атопический дерматит и псориаз.
В одном варианте реализации изобретения способ лечения воспаления или воспалительного заболевания или расстройства включает введение млекопитающему, нуждающемуся в этом, соединения 1 или его фармацевтически приемлемой соли в комбинации с одним или более дополнительными агентами. Примеры дополнительных агентов включают анти-ФНО агенты (например, моноклональное антитело, такое как инфликсимаб (Ремикейд), адалимумаб (Хумира), цертолизумаба пэгол (Симзия) и голимумаб (Симпони), или слитый белок циркулирующих рецепторов, такой как этанерцепт (Энбрел)), антиметаболические и антифолатные лекарственные средства (например, Метотрексат) или специфические ингибиторы киназ (например, ингибиторы семейства JAK, такие как руксолитиниб, тофацитиниб, CYT387, лестауртиниб, пакритиниб и TG101348). Эти дополнительные терапевтические средства можно вводить с соединением 1 или его фармацевтически приемлемой солью в качестве части одной или разных лекарственных форм, одним или различными способами введения и с одним или разными режимами применения в соответствии со стандартной фармацевтической практикой, известной специалистам в данной области техники.
Соединение 1 или его фармацевтически приемлемую соль также применяют в лечении нейродегенеративного заболевания у млекопитающего. В одном варианте реализации изобретения соединение 1 или его фармацевтически приемлемую соль также можно применять в лечении демиелинизации и дисмиелинизации посредством стимуляции миелинизации, нейронального выживания и дифференцировки олигодендроцитов посредством блокировки взаимодействия Sp35-TrkA.
Соответственно, в данном документе также предлагается способ лечения нейродегенеративного заболевания у млекопитающего, включающий введение указанному млекопитающему, нуждающемуся в этом, соединения 1 или его фармацевтически приемлемой соли в количестве, эффективном для лечения указанного нейродегенеративного заболевания. В одном варианте реализации изобретения нейродегенеративное заболевание представляет собой рассеянный склероз. В одном варианте реализации изобретения нейродегенеративное заболевание представляет собой болезнь Паркинсона. В одном варианте реализации изобретения нейродегенеративное заболевание представляет собой болезнь Альцгеймера.
Также в данном документе предлагается способ лечения некоторых инфекционных заболеваний, таких как инфекция Trypanosoma cruzi у млекопитающего, включающий введение указанному млекопитающему, нуждающемуся в этом, соединения 1 или его фармацевтически приемлемой соли в количестве, эффективном для лечения указанной инфекции Trypanosoma cruzi.
Также в данном документе предлагается способ лечения синдрома Шегрена у млекопитающего, включающий введение указанному млекопитающему, нуждающемуся в этом, соединения 1 или его фармацевтически приемлемой соли в количестве, эффективном для лечения указанного синдрома.
Также в данном документе предлагается способ лечения эндометриоза у млекопитающего, включающий введение указанному млекопитающему, нуждающемуся в этом, соединения 1 или его фармацевтически приемлемой соли в количестве, эффективном для лечения указанного эндометриоза.
Также в данном документе предлагается способ лечения диабетической периферической нейропатии у млекопитающего, включающий введение указанному млекопитающему, нуждающемуся в этом, соединения 1 или его фармацевтически приемлемой соли в количестве, эффективном для лечения указанной диабетической периферической нейропатии.
Также в данном документе предлагается способ лечения простатита у млекопитающего, включающий введение указанному млекопитающему, нуждающемуся в этом, соединения 1 или его фармацевтически приемлемой соли в количестве, эффективном для лечения указанного простатита.
Также в данном документе предлагается способ лечения синдрома тазовой боли у млекопитающего, включающий введение указанному млекопитающему, нуждающемуся в этом, соединения 1 или его фармацевтически приемлемой соли в количестве, эффективном для лечения указанного синдрома тазовой боли.
Также в данном документе предлагается способ лечения заболеваний, связанных с дисбалансом регуляции ремоделирования костной ткани у млекопитающего, включающий введение указанному млекопитающему, нуждающемуся в этом, соединения 1 или его фармацевтически приемлемой соли в количестве, эффективном для лечения указанного заболевания. В одном варианте реализации изобретения данное заболевание представляет собой остеопороз, ревматоидный артрит или метастазы в кости.
В одном варианте реализации изобретения способ лечения заболеваний, связанных с дисбалансом регуляции ремоделирования костной ткани у млекопитающего, нуждающегося в этом, включает введение соединения 1 или его фармацевтически приемлемой соли в комбинации с одним или более дополнительными терапевтическими средствами или препаратами. Примеры дополнительных терапевтических средств или препаратов включают анти-ФНО средства (например, моноклональное антитело, такое как инфликсимаб (Ремикейд), адалимумаб (Хумира), цертолизумаба пэгол (Симзия) и голимумаб (Симпони), или слитый белок циркулирующих рецепторов, такой как этанерцепт (Энбрел)), антиметаболические и антифолатные лекарственные средства (например, Метотрексат) или специфические ингибиторы киназ (например, ингибиторы семейства JAK, такие как руксолитиниб, тофацитиниб, CYT387, лестауртиниб, пакритиниб и TG101348). Эти дополнительные терапевтические средства можно вводить с соединением 1 или его фармацевтически приемлемой солью в качестве части одной или разных лекарственных форм, одним или различными способами введения и с одним или разными режимами применения в соответствии со стандартной фармацевтической практикой, известной специалистам в данной области техники.
Также в данном документе предлагается способ лечения заболеваний, обусловленных аберрантной передачей сигналов фактора роста соединительной ткани у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему соединения 1 или его фармацевтически приемлемой соли в количестве, эффективном для лечения указанного заболевания. В одном варианте реализации изобретения заболевание, обусловленное аберрантной передачей сигналов фактора роста соединительной ткани представляет собой синдром Рейно, идиопатический фиброз легких, рубцевание (гипертрофическое, келоидное и другое), цирроз, эндомиокардиальный фиброз, предсердный фиброз, миелофиброз, прогрессирующий массивный фиброз (легких), нефрогенный системный фиброз, склеродермию, системный склероз, артрофиброз и фиброз глаза.
При использовании по тексту данного документа, термин "эффективное количество" означает количество соединения, которое при введении млекопитающему, нуждающемуся в таком лечении, является достаточным для (i) лечения конкретного заболевания, состояния или расстройства, подающегося лечению соединением 1 или его фармацевтически приемлемой солью, или (ii) подавления, уменьшения интенсивности или устранения одного или более симптомов конкретного заболевания, состояния или расстройства, описанного в данном документе. Количество соединения 1, которое будет соответствовать такому количеству, будет варьироваться в зависимости от таких факторов, как болезненное состояние и его выраженность, а также идентификационные характеристики (например, вес) млекопитающего, нуждающегося в лечении, но тем не менее может быть рутинно определено специалистом в данной области техники.
При использовании по тексту данного документа, термин "млекопитающее" относится к теплокровному животному, которое болеет или подвержено риску развития заболевания, описанного в данном документе, и включает, без ограничения ими, морских свинок, собак, кошек, крыс, мышей, хомяков и приматов, в том числе людей.
Соединение 1 или его фармацевтически приемлемую соль можно вводить любым удобным способом, например, в желудочно-кишечный тракт (например, ректально или перорально), нос, легкие, мышечную или сосудистую систему, промыванием раны, чрескожно или посредством нанесения на кожу. Соединение 1 или его фармацевтически приемлемую соль можно вводить в любой удобной для введения форме, например, таблетки, порошки, капсулы, растворы, дисперсии, суспензии, сиропы, спреи, суппозитории, гели, эмульсии, пластыри и т.д. Такие композиции могут содержать компоненты, обычные для фармацевтических препаратов, например, разбавители, носители, модификаторы рН, подсластители, объемообразующие препараты и другие активные вещества. Если необходимо парентеральное введение, композиции должны быть стерильными и в форме раствора или суспензии, подходящей для инъекции или инфузии. Такие композиции образуют дополнительный аспект данного изобретения.
Другой препарат может быть получен посредством смешивания соединения 1 или его фармацевтически приемлемой соли с носителем или вспомогательным веществом. Подходящие носители и вспомогательные вещества хорошо известны специалистам в данной области техники. Данные препараты могут также включать один или более буферных растворов, стабилизирующих веществ, поверхностно-активных веществ, смачивающих веществ, смазывающих веществ, эмульгаторов, суспендирующих веществ, консервантов, антиоксидантов, опакеров, способствующих скольжению веществ, вспомогательных технологических добавок, красителей, подсластителей, отдушек, ароматизаторов, разбавителей и других известных добавок для обеспечения красивой формы выпуска лекарственного средства (то есть соединения, описанного в данном документе, или его фармацевтической композиции) или способствования производству данного фармацевтического препарата (то есть лекарственного средства).
Соответственно, также в данном документе предлагается фармацевтическая композиция, которая содержит соединение 1 или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым разбавителем или носителем. В одном варианте реализации изобретения фармацевтическую композицию, содержащую соединение 1 или его фармацевтически приемлемую соль, готовят для перорального введения. В одном варианте реализации изобретения фармацевтическая композиция, содержащая соединение 1 или его фармацевтически приемлемую соль, приготовленная для перорального введения, имеет форму таблетки или капсулы.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении боли у млекопитающего. В одном варианте реализации изобретения боль представляет собой хроническую боль. В одном варианте реализации изобретения боль представляет собой острую боль. В одном варианте реализации изобретения боль представляет собой хроническую позвоночную боль, воспалительную боль, нейропатическую боль или боль, обусловленную раком, хирургическим вмешательством или переломом кости.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении рака у млекопитающего. В одном варианте реализации изобретения рак выбран из немелкоклеточного рака легкого, папиллярной карциномы щитовидной железы, мультиформной глиобластомы, острого миелоидного лейкоза, карциномы ободочной и прямой кишки, крупноклеточной нейроэндокринной карциномы, рака предстательной железы, нейробластомы, карциномы поджелудочной железы, меланомы, плоскоклеточной карциномы головы и шеи, карциномы желудка, холангиокарциномы и саркомы.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении воспаления или воспалительного заболевания или расстройства у млекопитающего. В одном варианте реализации изобретения воспалительное заболевание представляет собой воспалительные заболевания легких (такие как астма), интерстициальный цистит, синдром болезненного мочевого пузыря, воспалительные заболевания кишечника (в том числе неспецифический язвенный колит и болезнь Крона), а также воспалительные кожные заболевания, такие как атопический дерматит.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении инфекционных заболеваний, например, инфекции Trypanosoma cruzi, у млекопитающего.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении синдрома Шегрена у млекопитающего.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении эндометриоза у млекопитающего.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении диабетической периферической нейропатии у млекопитающего.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении простатита у млекопитающего.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении синдрома тазовой боли у млекопитающего.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении нейродегенеративного заболевания у млекопитающего.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении заболевания, связанного с дисбалансом регуляции ремоделирования костной ткани.
Также в данном документе предлагается соединение 1 или его фармацевтически приемлемая соль для применения в лечении заболеваний, обусловленных аберрантной передачей сигналов фактора роста соединительной ткани.
Также в данном документе предлагается применение соединения 1 или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения состояния, выбранного из боли, рака, воспаления, нейродегенеративного заболевания, инфекции Trypanosoma cruzi, а также заболеваний, обусловленных аберрантной передачей сигналов фактора роста соединительной ткани. В одном варианте реализации изобретения состояние представляет собой хроническую боль. В одном варианте реализации изобретения состояние представляет собой острую боль. В одном варианте реализации изобретения боль представляет собой хроническую позвоночную боль. В одном варианте реализации изобретения боль представляет собой воспалительную боль, нейропатическую боль или боль, обусловленную раком, хирургическим вмешательством или переломом кости. В одном варианте реализации изобретения состояние представляет собой рак. В одном варианте реализации изобретения состояние представляет собой воспаление. В одном варианте реализации изобретения состояние представляет собой нейродегенеративное заболевание. В одном варианте реализации изобретения состояние представляет собой инфекцию Trypanosoma cruzi. В одном варианте реализации изобретения состояние представляет собой синдром Шегрена. В одном варианте реализации изобретения состояние представляет собой эндометриоз. В одном варианте реализации изобретения состояние представляет собой диабетическую периферическую нейропатию. В одном варианте реализации изобретения состояние представляет собой простатит. В одном варианте реализации изобретения состояние представляет собой синдром тазовой боли. В одном варианте реализации изобретения заболевание представляет собой заболевание, связанное с дисбалансом регуляции ремоделирования костной ткани. В одном варианте реализации изобретения заболевание представляет собой заболевание, обусловленное аберрантной передачей сигналов фактора роста соединительной ткани.
Также в данном документе предлагается лекарственное средство, содержащее соединение 1 или его фармацевтически приемлемую соль, для лечения боли у млекопитающего в комбинации с дополнительным терапевтическим средством, выбранным из противовоспалительных соединений, стероидов (например, дексаметазон, кортизон и флутиказон), болеутоляющих средств, таких как НПВС (например, аспирин, ибупрофен, индометацин и кетопрофен), опиоидов (таких как морфин), антагонистов рецепторов кальцитонин ген-родственного пептида, подтип-селективных модуляторов ионных каналов, противосудорожных средств (например, прегабалин и габапентин), двойных ингибиторов обратного захвата серотонина-норадреналина (например, дулоксетин, венлафаксин и милнаципран), ингибиторов киназ семейства JAK (например, руксолитиниб или тофацитиниб) и трициклических антидепрессантов (например, амитриптилин, нортриптилин и дезипрамин). В одном варианте реализации изобретения в данном документе предлагается лекарственное средство, содержащее соединение 1 или его фармацевтически приемлемую соль в комбинации с НПВС, для лечения боли у млекопитающего.
Также в данном документе предлагается лекарственное средство, содержащее терапевтическое средство, выбранное из противовоспалительных соединений, стероидов (например, дексаметазон, кортизон и флутиказон), болеутоляющих средств, таких как НПВС (например, аспирин, ибупрофен, индометацин и кетопрофен) и опиоидов (таких как морфин), в комбинации с соединением 1 или его фармацевтически приемлемой солью для лечения боли у млекопитающего.
Примеры
Получение синтетических промежуточных соединений
Препарат А
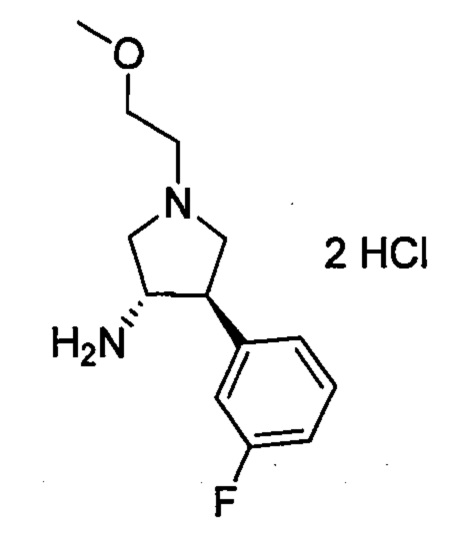
(3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид
Стадия А: Получение трет-бутил (3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-илкарбамата: Раствор трет-бутил (3S,4R)-4-(3-фторфенил)пирролидин-3-илкарбамата (250 мг, 0,89 ммоль, коммерчески доступный), DIEA (0,48 мл, 2,68 ммоль) и 1-бром-2-метоксиэтана (149 мг, 1,07 ммоль) в ДМФА (3 мл) перемешивали при температуре окружающей среды в течение 2 часов, затем нагревали до 60°С в течение 4 часов, затем охлаждали до температуры окружающей среды в течение 16 часов. После распределения между ЕtOАс и насыщенным раствором NaHCO3 (10 мл каждого), органический слой промывали водой и насыщенным раствором хлорида натрия (2 х 10 мл каждого), сушили над Na2SO4, фильтровали и выпаривали с образованием трет-бутил (3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-илкарбамата (250 мг, выход 83%) в виде вязкого масла оранжевого цвета. ЖХМС (APCI) m/z = 339,1 (М+Н).
Стадия Б: Получение (3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорида: Раствор трет-бутил (3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-илкарбамата (250 мг, 0,74 ммоль) в 5-6 N НСl в изопропиловом спирте (14,8 мл, 73,9 ммоль) перемешивали при температуре окружающей среды в течение 1 часа. Смесь выпаривали в вакууме и растирали с Et2O с получением указанного в заголовке соединения (230 мг, выход 100%) в виде бежевого твердого вещества. ЖХМС (APCI) m/z = 239,1 (М+Н).
Препарат Б
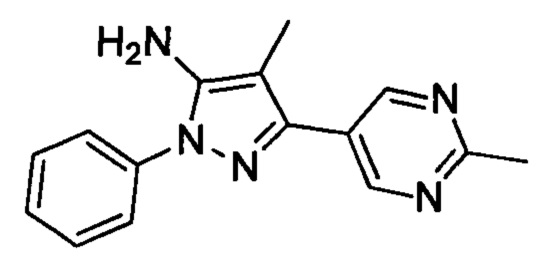
4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-амин
Стадия А: Получение 5-амино-4-метил-1-фенил-1Н-пиразол-3(2Н)-она: Смесь этил 2-цианопропаноата (50,5 г, 397,2 ммоль) и фенилгидразина (39 мл, 397,2 ммоль) в диоксане (100 мл) нагревали при 110°С в течение 5 дней. Охлажденную смесь выпаривали до 1/2 объема, а затем охлаждали на льду и растирали в порошок с холодным Et2O. Полученные твердые вещества фильтровали, тщательно промывали Et2O и сушили в вакууме с получением 5-амино-4-метил-1-фенил-1Н-пиразол-3(2Н)-она (34,69 г, выход 46%) в виде рассыпчатого белого порошка. МС (APCI) m/z =190,1 (М+Н).
Стадия Б: Получение 5-амино-4-метил-1-фенил-1Н-пиразол-3-ил трифторметансульфоната: Суспензию 5-амино-4-метил-1-фенил-1Н-пиразол-3(2Н)-она (13,72 г, 72,5 ммоль) и N-фенилбис(трифторметилсульфонамид) (27,2 г, 76,1 ммоль) в ДМФА (100 мл) обрабатывали DIEA (37,9 мл, 217,5 ммоль), и полученную смесь перемешивали при температуре окружающей среды в течение 16 часов. Смесь распределяли между насыщенным раствором NaHCO3 (400 мл) и EtOAc (200 мл), и водный слой экстрагировали EtOAc (2 × 200 мл). Объединенные органические фазы промывали водой (5 х 50 мл) и насыщенным раствором хлорида натрия (50 мл), затем сушили над Na2SO4, фильтровали и выпаривали в вакууме. Остаток очищали с помощью хроматографии на колонке с силикагелем, элюируемой смесью 4:1 гексаны/EtOAc, с получением 5-амино-4-метил-1-фенил-1Н-пиразол-3-ил трифторметансульфоната (23,1 г, выход 99%) в виде бледно-желтого твердого вещества. МС (APCI) m/z = 322,0 (М+Н).
Стадия В: Получение 4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-амина: 5-Амино-4-метил-1-фенил-1Н-пиразол-3-ил трифторметансульфонат (900 мг, 2,8 ммоль), 2-метил-5-(4,4,5,5-тетраметил-1,3, 2-диоксаборолан-2-ил)пиримидин (925 мг, 4,2 ммоль), K2СО3 (1,55 г, 11,2 ммоль) и Pd(PPh3)4 (324 мг, 0,28 ммоль) объединяли в смеси толуола (10 мл), воды (5 мл) и EtOH (2,5 мл) и нагревали до 95°С в герметично закрытой пробирке в течение 16 часов. Охлажденную смесь фильтровали, и фильтрат распределяли между водой (50 мл) и EtOAc (50 мл). Водный слой экстрагировали EtOAc (2 х 30 мл), и объединенные органические фазы промывали насыщенным раствором хлорида натрия (30 мл), сушили над Na2SO4, фильтровали и выпаривали в вакууме. Остаток очищали с помощью хроматографии на колонке с силикагелем, элюируемой смесью 2% МеОН/ДХМ, с получением указанного в заголовке соединения (533 мг, выход 72%) в виде твердого вещества розового цвета. МС (APCI) m/z = 266,1 (М+Н).
Препарат В
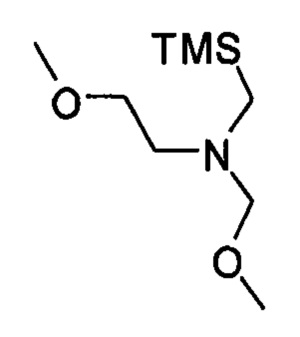
2-метокси-N-(метоксиметил)-N-((триметилсилил)метил)этанамин
Стадия А: Получение 2-метокси-N-((триметилсилил)метил)этанамина: К ДМСО-раствору (15 мл) 2-метоксиэтанамина (14,2 мл, 163 ммоль) при 90°С добавляли ДМСО-раствор (10 мл) (хлорметил)триметилсилана (11,4 мл, 81,5 ммоль) с помощью капельной воронки в течение 40 минут. Смесь нагревали при 90°С в течение 3,5 часов, затем охлаждали до температуры окружающей среды. Затем смесь разбавляли Н2О (150 мл) и экстрагировали EtOAc (2 × 150 мл). Объединенные органические экстракты промывали насыщенным раствором хлорида натрия (150 мл), сушили с помощью MgSO4, фильтровали и выпаривали с получением 2-метокси-N-((триметилсилил)метил)этанамина (8,14 г, выход 62%) в виде желтого масла. МС (APCI) m/z = 162,0 (М+Н).
Стадия Б: Получение 2-метокси-N-(метоксиметил)-N-((триметилсилил)метил)этанамина: МеОН-раствор (2,45 мл) формальдегида (37%-ный водный раствор, 4,91 г, 60,6 ммоль) охлаждали до 0°С и обрабатывали добавлением по каплям 2-метокси-N-((триметилсилил)метил)этанамина (8,14 г, 50,5 ммоль). Двухфазную смесь перемешивали при 0°С в течение 3 часов, затем добавляли K2СО3 (6,97 г, 50,5 ммоль), и полученную смесь перемешивали при 0°С в течение 1 часа. Желтое масло декантировали в K2СО3 (2,00 г, 14,4 ммоль), и полученную смесь перемешивали при температуре окружающей среды в течение 2 часов. После декантирования желтого масла твердый K2СО3 промывали Et2O (2 × 10 мл), и промытый Et2O объединяли с декантируемым желтым маслом и выпаривали на ротационном испарителе с образованием указанного в заголовке соединения (9,92 г, выход 96%) в виде желтого масла. 1Н ЯМР (CDCl3) δ 4,00 (с, 2Н), 3,37-3,43 (м, 2Н), 3,29 (с, 3Н), 3,19 (с, 3Н), 2,77-2,82 (м, 2Н), 2,18 (с, 2Н), 0,00 (с, 9Н) м.д.
Препарат Г
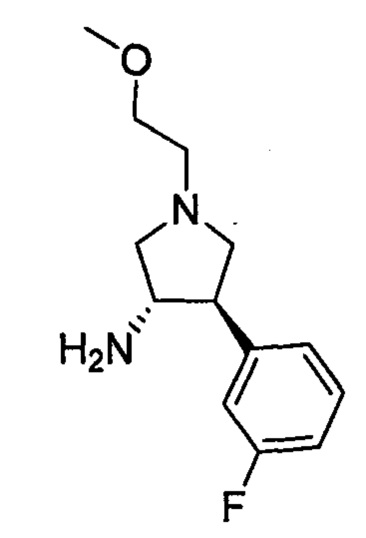
(3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амин
Стадия А: Получение (транс)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-карбоновой кислоты: (Е)-3-(3-фторфенил)акриловую кислоту (25,20 г, 151,7 ммоль) последовательно обрабатывали EtOAc (75 мл) и гептаном (75 мл), а затем ТФУК (1,17 мл, 15.17 ммоль). Смесь помещали в баню с холодной водой (внутренняя температура 15°С) и по каплям добавляли 2-метокси-N-(метоксиметил)-N-((триметилсилил)метил)этанамин [Препарат В] (69,82 г, 197,2 ммоль) из капельной воронки в течение 20 минут, добавляя в водяную баню лед для поддержания внутренней температуры в диапазоне 13-19°С во время добавления. Баню со льдом убирали и перемешивали смесь при температуре окружающей среды в течение 18 часов, затем охлаждали (внутренняя температура 13°С) и по каплям добавляли дополнительный объем 2-метокси-N-метоксиметил)-N-((триметилсилил)метил)этанамина (Препарат В; 34 г) из капельной воронки в течение 6 минут. Баню удаляли и после перемешивания в течение еще 4 часов выпаривали смесь при температуре окружающей среды до половины объема. Добавляли гептан (100 мл) и смесь частично выпаривали с удалением около 150 мл растворителя. Эту процедуру повторяли дважды с удалением 100 мл растворителя каждый раз. Остатки суспензии смывали на дно колбы гептаном (50 мл), затем герметично закрывали и перемешивали при температуре окружающей среды в течение 64 часов. Твердые вещества фильтровали, промывали гептаном (2 х 50 мл) и сушили в вакууме с получением (транс)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-карбоновой кислоты (44,99 г, выход 111%) в виде бледно-желтого твердого вещества, которое непосредственно использовали на следующей стадии, предполагая 100% выход. МС (APCI) m/z = 268,1 (М+Н).
Стадия Б: Получение (транс)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-карбоксамида: К суспензии (транс)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-карбоновой кислоты (40,54 г, 151,7 ммоль) в ТГФ (440 мл) добавляли КДИ (31,97 г, 197,2 ммоль) с последующим добавлением гидрохлорида имидазола (3,171 г, 30,33 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 16 часов, а затем добавляли аммиак (135,0 мл, 2М в изоPrOH, 270,0 ммоль) с помощью капельной воронки в течение 30 минут. После перемешивания в течение еще 3 часов полученные твердые вещества фильтровали, промывали EtOAc и фильтрат выпаривали. Остаток растворяли в EtOAc (200 мл) и промывали 25/75 соотношением вода: насыщенный раствор хлорида натрия (2 х 200 мл). Органическую фазу сушили над MgSO4, фильтровали, выпаривали и сушили в вакууме с получением (транс)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-карбоксамида (54,97 г, выход 136%) в виде желтого твердого вещества, которое использовали, предполагая 100% выход. МС (APCI) m/z = 267,2 (М+Н).
Стадия В: Получение (транс)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина: К охлажденному на бане со льдом раствору (транс)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-карбоксамида (29,33 г, 85,90 ммоль) в МеОН (345 мл) в 3-горлой круглодонной колбе емкостью 1 литр, снабженной температурным датчиком, добавляли NaOCl (10-15% активного Cl, водный раствор) (116,3 мл, 189,0 ммоль) из капельной воронки в течение 20 минут [внутренняя температура в пределах 7-13°С]. Баню со льдом убирали и гетерогенную смесь перемешивали при температуре окружающей среды в течение 90 минут, затем при 55°С в течение 3 часов. Добавляли раствор KOH (67,48 г, 1203 ммоль) в Н2О (225,9 мл, 12542 ммоль) и раствор перемешивали при 75°С в течение 19 часов. Смесь охлаждали до 0°С и медленно добавляли концентрированную НСl (147,1 мл, 4802 ммоль) в течение 30 минут. Значение рН доводили до 6 добавлением водного раствора K3РО4 (30% масс, 155 мл), и смесь частично выпаривали для удаления органических растворителей. Добавляли водный раствор K3РО4 (30% масс, 280 мл) до тех пор, пока значение рН не достигло 10, и смесь экстрагировали EtOAc (2 х 450 мл). Объединенные органические экстракты выпаривали и сушили в вакууме с получением (транс)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина (28,08 г, выход 91%) в виде темно-янтарного сиропа. МС (APCI) m/z = 239,2 (М+Н).
Стадия Г: Получение (3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амин(2S,3S)-2,3-бис((4-метилбензоил)окси)сукцината: (транс)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (125 мг, 0,52 ммоль) и (2S,3S)-2,3-бис((4-метилбензоил)окси)янтарную кислоту (222,9 мг, 0,58 ммоль) засыпали в 4 мл виалу, затем обрабатывали МеОН (1,57 мл) с последующим добавлением воды (0,175 мл). Виалу закрывали крышкой и реакционную смесь перемешивали при 50°С в течение 5 минут, затем давали возможность медленно остыть до температуры окружающей среды в течение 17 часов. Полученные твердые вещества фильтровали, промывали Et2O (4 х 0,2 мл) и сушили в вакууме с получением (3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амин(2S,3S)-2,3-бис((4-метилбензоил)окси)сукцината (131,6 мг, выход 40%) в виде белого твердого вещества. Хиральный СФХ анализ свободного основания пробы выявил > 95% э.и. Это вещество использовали в качестве затравочных кристаллов на следующей стадии:
(Транс)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (7,39 г, 18,61 ммоль) растворяли в МеОН (52,7 мл) и обрабатывали (2S,3S)-2,3-бис((4-метилбензоил)окси)янтарной кислоты (10,78 г, 27,91 ммоль), а затем Н2О (9,3 мл). Смесь перемешивали при 50°С в течение 5 минут. Затравочные кристаллы (90 мг) добавляли к раствору, и давали возможность смеси медленно нагреться до температуры окружающей среды в течение 16 часов. Полученные твердые вещества фильтровали, промывали Et2O (4 х 5 мл) и сушили в вакууме. Остаток суспендировали в МеОН (15 мл) и Н2О (2,7 мл), перемешивали при 50°С в течение 15 минут, затем давали возможность медленно остыть до температуры окружающей среды в течение 19 часов. Твердые вещества фильтровали, промывали Et2O (3 x 5 мл) и сушили в вакууме с получением (3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амин(2S,3S)-2,3-бис((4-метилбензоил)окси)сукцината (3,12 г, выход 27%) в виде белого твердого вещества. Хиральный анализ свободного основания пробы выявил > 99,7% э.и.
Стадия Д: Получение (3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина: Суспензию (3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амин(2S,3S)-2,3-бис((4-метилбензоил)окси)сукцината (2,89 г, 4,63 ммоль) в ДХМ (25 мл) промывали 1 М водным раствором NaOH (2 x 15 мл). Объединенные водные фазы экстрагировали ДХМ (25 мл), а объединенные органические экстракты промывали насыщенным раствором хлорида натрия (30 мл), сушили над Na2SO4, фильтровали, выпаривали и сушили в вакууме с получением (3S,4R)-4-(3-фторофенил)-1-(2-метоксиэтил)пирролидин-3-амина (998 мг, выход 90%) в виде светло-бежевого масла. МС (APCI) m/z = 239,2 (М+Н).
Примеры синтеза
Пример 1
1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-ил)мочевины дигидрохлорид
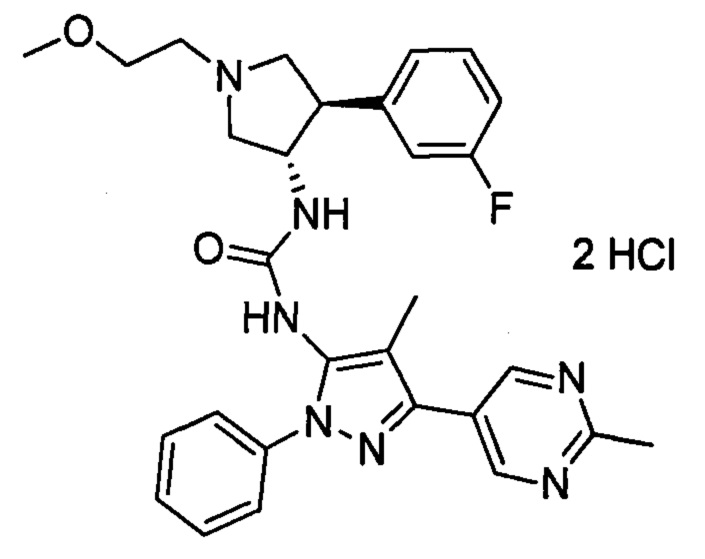
Стадия А: Получение 1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-ил)мочевины: К раствору 4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-амина (Препарат Б; 61 мг, 0,16 ммоль) в ДХМ (2 мл) добавляли трифосген (24 мг, 0,08 ммоль) с последующим добавлением DIEA (84 мкл, 0,48 ммоль). Смесь перемешивали при температуре окружающей среды в течение 15 минут и затем обрабатывали (3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амином (Препарат А; 50 мг, 0,16 ммоль) и DIE А (84 мкл, 0,48 ммоль). Реакционную смесь перемешивали в течение 16 часов. Смесь распределяли между водой (10 мл) и ДХМ (10 мл), и водный слой экстрагировали ДХМ (2 х 10 мл). Объединенные органические фазы промывали насыщенным раствором хлорида натрия (10 мл), сушили над Na2SO4, фильтровали и выпаривали. Остаток очищали с помощью хроматографии на колонке с силикагелем, элюируемой 2,5-5% МеОН/ДХМ. Фракции, содержащие продукт, дополнительно очищали с помощью обращенно-фазовой ВЭЖХ (5-95% НАК/вода/0,1% ТФУК) с получением после основной водной обработки указанного в заголовке соединения (28 мг, выход 33%) в виде белого твердого вещества. МС (APCI) m/z = 530,3 (М+Н).
Стадия Б: Получение 1-((3S,4R)-4-(3-фтopфeнил)-1-(2-мeтoкcиэтил)пиppoлидин-3-ил)-3-(4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-ил)мочевины дигидрохлорида: К охлажденному до 0°C раствору 4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-амина (Препарат Б; 2,56 г, 9,64 ммоль) в ДХМ (96 мл) добавляли трифосген (1,72 г, 5,78 ммоль) в виде одной аликвоты с последующим добавлением по каплям DIEA (8,40 мл, 48,20 ммоль). Смесь перемешивали при 0°С в течение 1 часа. (3S,4R)-4-(3-фтopфeнил)-1-(2-мeтoкcиэтил)пиppoлидин-3-aминa дигидрохлорид (Препарат А, 3,0 г, 9,64 ммоль) добавляли к смеси небольшими аликвотами в течение 10 минут. Реакционную смесь перемешивали при температуре окружающей среды в течение 40 минут и затем выливали в воду (100 мл). После разделения фаз водный слой экстрагировали ДХМ (2×100 мл). Объединенные органические фазы промывали насыщенным раствором хлорида натрия (100 мл), фильтровали и выпаривали. Остаток очищали с помощью обращенно-фазовой хроматографии (SNAP 120 г С18, 5-75% МеОН/вода с 0,1% НСl) и сушили в вакууме с получением указанного в заголовке соединения (3,33 г, выход 57%) в виде пены кремового цвета. МС (APCI) m/z = 530,3 (М+Н).
Пример 2
1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-ил)мочевина
К суспензии 4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-амина (Препарат Б; 800 мг, 3,02 ммоль) в ДХМ (15 мл) добавляли триэтиламин (2,1 мл, 15,1 ммоль) с последующим добавлением КДИ (587 мг, 3,62 ммоль). Смесь герметично закрывали в атмосфере N2 и перемешивали при температуре окружающей среды в течение 22 часов, затем обрабатывали (3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амином (Препарат Г; 826 мг, 3,47 ммоль) в ДХМ (4 мл). Перемешивание продолжали в течение 100 минут. Смесь распределяли между ДХМ (60 мл) и водой (100 мл). После разделения фаз слой ДХМ экстрагировали 0,5 N НСl (50 мл) и затем 0,2 N НСl (2×25 мл). Кислотные вытяжки объединяли, декантировали в чистую колбу, помещали в баню с водой комнатной температуры и подщелачивали 6 н NaOH (водн.) и затем 1 н NaOH (водн.) с доведением значения рН до 9-10 при перемешивании. Суспензию перемешивали в бане со льдом в течение 5 минут. Полученные твердые вещества фильтровали, промывали водой и простым эфиром (3×20 мл каждого) и сушили в вакууме с получением указанного в заголовке соединения (1,44 г, выход 90%) в виде белого твердого вещества. МС (APCI) m/z = 530,3 (М+Н).
Пример 3
Получение (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин(2S,3S)-2,3-бис((4-метилбензоил)окси)сукцината
Стадия А: Получение (2-метокси-N-((триметилсилил)метил)этанамина: В колбу загружали (хлорметил)триметилсилан (3300,0 г, 1,0 экв.), ацетонитрил (5,28 л) и 2-метоксиэтанамин (4041,0 г, 2,0 экв.). Реакционную смесь перемешивали при 74°С в течение 16 часов, а затем давали возможность реакционной смеси остыть до 25°С. Добавляли пентан (16 л), и реакционную смесь перемешивали в течение 1 минуты. Слои разделяли. Нижний слой (слой ацетонитрила) добавляли обратно в делительную воронку и добавляли в делительную воронку пентан (16 л). После перемешивания слои разделяли. Повторяли экстракцию пентаном (16 л). Объединенные слои пентана добавляли в делительную воронку и добавляли воду (3,3 л). После перемешивания слои разделяли. МеОН (3,0 л) добавляли в 22 л реактор, оснащенный паром, механической мешалкой, конденсатором, температурным датчиком J-Kem и капельной воронкой. Атмосферную перегонку растворителя осуществляли при 25°-45°С. Во время перегонки раствор пентана добавляли в перегонную колбу с помощью капельной воронки со скоростью, необходимой для поддержания объема на уровне около 6,6 л, пока весь раствор пентана не оказывался в перегонной колбе. Перегонку продолжали до тех пор, пока общий объем в колбе не доходил до около 6,6 л. Реакционную смесь охлаждали до 25°С и перемешивали при температуре окружающей среды в атмосфере азота с образованием (2-метокси-N-((триметилсилил)метил)этанамина (выход 72%).
Стадия Б: Получение 2-метокси-N-(метоксиметил)-1-((триметилсилил)метил)этанамина: В колбу загружали (2-метокси-N-((триметилсилил)метил)этанамин (380,0 г, 1,0 экв.), метанол (332 мл) и пентан (193 мл). Реакционную смесь перемешивали в бане с льдо-водяной смесью температуры ниже 5°С. 37%-ный водный раствор формальдегида (97,9 мл, 1,2 экв.) добавляли к реакционной смеси со скоростью, необходимой для поддержания внутренней температуры ниже 10°С, и реакционную смесь перемешивали при температуре ниже 10°С в течение 2 часов. Трет-бутилметиловый эфир (700 мл, 5,2 экв.) и воду (300,0 мл) добавляли к реакционной смеси, и полученную смесь перемешивали при температуре окружающей среды в течение около 1 минуты. Слои разделяли, и органический слой добавляли обратно в делительную воронку. Добавляли в делительную воронку насыщенный раствор хлорида натрия (300 мл). Смесь перемешивали и слои разделяли. Карбонат калия K2СО3 (10,0 г) добавляли к органическому слою, и после смешивания фильтровали органический слой через фильтровальную бумагу. Осадок на фильтре промывали трет-бутилметиловым эфиром. Объединенные органические слои выпаривали с получением 2-метокси-N-(метоксиметил)-N-((триметилсилил)метил)этанамина в виде масла (выход 75%).
Стадия В: Получение транс-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-карбоновой кислоты: В колбу загружали (Е)-3-(3,4-дифторфенил)акриловую кислоту (57,0 г, 1,0 экв.), этилацетат (143 мл) и гептан (143 мл), и полученную смесь перемешивали. Добавляли трифторуксусную кислоту (2,4 мл, 0,1 экв.) и реакционную смесь охлаждали до температуры ниже 20°С. 2-Метокси-N-(метоксиметил)-N-((триметилсилил)метил)этанамин добавляли с помощью капельной воронки со скоростью, необходимой для поддержания внутренней температуры в пределах 20°С - 25°С, и реакционную смесь перемешивали при 20°С в течение 2 часов. Реакционную смесь выпаривали при пониженном давлении при температуре от 10°С до 27°С. Добавляли этилацетат (200 мл) к остатку, и полученную смесь выпаривали при пониженном давлении. Остаток перемешивали в течение ночи при температуре 20°С. Азеотропное удаление примесей осуществляли с помощью вакуумной перегонки посредством добавления гептана аликвотами (общий объем: 1200 мл). Полученной суспензии давали возможность осесть в течение ночи при 20°С, а затем фильтровали через фильтровальную бумагу. Осадок на фильтре промывали гептаном и сушили в вакуумной печи при 45°С в течение около 4 часов с получением транс-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-карбоновой кислоты (выход 90%).
Стадия Г: Получение транс-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-карбоксамида: В колбу загружали транс-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-карбоновую кислоту (98,3 г, 1,0 экв.), карбонилдиимидазол (69,8 г, 1,3 экв.), имидазол-НСl (7,2 г, 0,2 экв.) и тетрагидрофуран (1000 мл). Реакционную смесь перемешивали в течение 1 часа при 25°С с последующим перемешиванием в течение ночи при температуре окружающей среды (около 20°С) в атмосфере азота. В течение 15 минут к реакционной смеси добавляли аммиак (2,0 М в изопропиловом спирте) (307 мл, 1,8 экв.). Реакционную смесь перемешивали при 25°С в течение около 30 минут. Реакционную смесь фильтровали через фильтровальную бумагу и промывали отфильтрованные твердые вещества с помощью EtOAc (200 мл). Объединенные органические слои выпаривали при пониженном давлении при 37°С. К полученному неочищенному маслу добавляли этилацетат (200 мл) и полученную смесь выпаривали при пониженном давлении при 37°С. К остатку добавляли этилацетат (200 мл), воду (500 мл) и NaCl (82,0 г, 4,1 экв.) и перемешивали смесь в течение около 2 минут при температуре окружающей среды. Слои разделяли и выпаривали объединенные органические слои при пониженном давлении при 37°С с получением транс-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-карбоксамида (выход 99%).
Стадия Д: Получение транс-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина: В колбу загружали транс-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-карбоксамид (96,8 г, 1,0 экв.) и метанол (1450 мл) и охлаждали реакционную смесь до 5°С. 10%-ый раствор гипохлорита натрия (462,2 мл, 2,2 экв.) добавляли с помощью капельной воронки со скоростью, необходимой для поддержания внутренней температуры ниже 20°С. Реакционную смесь перемешивали в течение 1 часа при 20°С, а затем при 55°С в течение 1 часа. В течение 5 минут к реакционной смеси добавляли гидроксид калия (314,6 г, 14,0 экв.) в виде раствора в воде (100 мл) и перемешивали реакционную смесь при 72°С - 75°С в течение 12 часов. Реакционную смесь охлаждали до 55°С и в течение 10 минут добавляли 37% масс. НСl (583,8 мл, 55,9 экв.) для доведения значения рН до ниже 2. Добавляли трехосновный K3РО4 (30% масс, 100,0 мл) для доведения значения рН реакционной смеси до 6-7. Реакционную смесь выпаривали при пониженном давлении при 37°С. Полученную водную смесь переносили в делительную воронку и добавляли 30% масс, трехосновного K3РО4 (540,0 мл) и 37% масс, трехосновного K3РО4 (50,0 мл) для доведения значения рН до ≥10. Добавляли EtOAc (1000 мл) и перемешивали полученную смесь в течение около 2 минут при температуре окружающей среды. Слои разделяли, и водный слой добавляли обратно в делительную воронку. Добавляли EtOAc (500 мл) в делительную воронку и смесь перемешивали. Слои разделяли и выпаривали объединенные органические слои при пониженном давлении с получением транс-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина (выход 99%).
Стадия Д: Получение (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин(2S,3S)-2,3-бис((4-метилбензоил)окси)сукцината: В колбу загружали транс-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (123,3 г, 1,0 экв.) и метанол (1603 мл). Полученную смесь перемешивали при температуре окружающей среды. Добавляли (2S,3S)-2,3-бис((4-метилбензоил)окси)янтарную кислоту (145,0 г, 1,5 экв.) и воду (295 мл), и нагревали реакционную смесь до 65°С. Реакционную смесь затравливали (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин(2S,3S)-2,3-бис((4-метилбензоил)окси)сукцинатом (0,5 г, 0,7 экв.) (затравку готовили осуществлением реакции в небольшом масштабе, которая проходила с получением продукта в виде твердого вещества), и реакционную смесь медленно охлаждали до 25°С в течение 16 часов. Суспензию фильтровали через фильтровальную бумагу. Осадок на фильтре промывали трет-бутилметиловым эфиром (700 мл), а затем сушили в вакуумной печи при 45°С в течение около 4 часов с получением (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин(2S,3S)-2,3-бис((4-метилбензоил)окси)сукцината (выход 35% за 2 стадии).
Биологические анализы
В биологических анализах соединение 1 относится к 1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-ил)мочевине; соединение 2 относится к 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-ил)мочевине (описанной в WO 2012/158413); и соединение 3 относится к 1-(1',4-диметил-1-фенил-1Н,1'H-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевине (описанной в WO 2012/158413).
Пример А
Анализ связывания TrkA киназы
TrkA связывающую активность определяли с помощью анализа связывания TrkA киназы LanthaScreen™ Eu. 5 нМ His-меченой рекомбинантной TrkA человека (6His меченый цитоплазматический домен от Invitrogen, каталожный № PV 3144) инкубировали с 4 нМ Alexa-Fluor® Tracer 236 (Invitrogen, каталожный № PV 5592), 2 нМ биотинилированного антитела к His-метке (Invitrogen, каталожный № PV 6090) и 2 нМ меченого европием стрептавидина (Invitrogen, каталожный № PV 5899) в буфере (25 мМ MOPS, рН 7,5, 5 мМ MgCl2, 0,005% Triton Х-100). Трехкратные серийные разведения соединения 1, соединения 2 или соединения 3 в ДМСО добавляли до доведения конечного процентного содержания ДМСО до 2%. После 60-минутной инкубации при 22°С реакционную смесь измеряли с применением многорежимного сканирующего спектрофотометра для прочтения планшетов EnVision (PerkinElmer) с помощью двухволнового детектирования TR-FRET при 615 нм и 665 нм. Процент контроля рассчитывали с помощью логометрического коэффициента эмиссии. Значение IC50 определяли посредством согласования четырех-параметрической модели с процентом данных контроля.
У соединения 1 было усредненное значение IC50, составляющее 1,1 нМ при испытании в анализе примера А. У соединения 2 было усредненное значение IC50, составляющее 1,5 нМ при испытании в анализе примера А. У соединения 3 было усредненное значение IC50, составляющее 1,7 нМ при испытании в анализе примера А.
пример Б
Клеточный анализ TrkA
Клетки СНО-K1, трансфицированные TrkA дикого типа человека, высевали в 96-луночный плоскодонный планшет при концентрации 3×105 клеток/лунка в полной среде DMEM, содержащей 10% FBS и давали возможность прикрепиться в течение 24 часов при 37°С, 5% СО2. Затем среду заменяли полной бессывороточной средой для анализа и содержали клетки в бессывороточной среде в течение 1 часа при 37°С, 5% СО2. Клетки обрабатывали соединением 1, 2 или 3 при серийных разведениях 1:3, при этом конечные концентрации находились в диапазоне от 1 мкМ до 152 пМ. Соединение инкубировали на клетках в течение 1 часа при 37°С, 5% СО2. Затем клетки стимулировали конечной концентрацией из 30 нг/мл NGF человека в течение 7 минут при 37°С, 5% СО2. Среду удаляли и лизировали клетки буфером для лизиса, содержащим ингибиторы фосфатазы и протеазы. Фосфо-TrkA измеряли с помощью ELISA (R&D, каталожный № DYC 2578). ELISA охватывал всю TrkA и детектировал весь фосфо-тирозин. Оптическую плотность измеряли для каждой лунки с помощью ридера Versamax при длине волны, составляющей 450 нм. Значения IC50 определяли посредством согласования четырех-параметрической модели с процентом данных контроля.
У соединения 1 было усредненное значение IC50, составляющее 1,9 нМ при испытании в анализе примера Б. У соединения 2 было усредненное значение IC50, составляющее 1,3 нМ при испытании в анализе примера Б. У соединения 3 было усредненное значение IC50, составляющее 6,1 нМ при испытании в анализе примера Б.
Пример В
Клиренс микросом человека и гепатоцитов человека у соединений 1, 2 и 3
Микросомальная инкубация печени
100 мМ раствора калий-фосфатного аналитического буфера (КРВ) получали следующим образом. KН2РО4 и K2НРО4 по отдельности растворяли в воде чистой для анализа с получением в результате конечной концентрации, составляющей 100 мМ. Готовили 75:25 смесь об./об. из K2НРO4:KН2РO4 и доводили значение рН раствора до 7,4 с помощью разбавленной НСl или разбавленных растворов NaOH. Исходные растворы соединения 1, соединения 2 и соединения 3 готовили в концентрации 10 мМ (активного соединения) в ДМСО. Исходный раствор разбавляли до 2,5 мкМ непосредственно перед применением с помощью раствора КРВ для создания рабочего стандартного образца. При визуальном осмотре все исследуемые соединения были полностью растворимы в ДМСО при комнатной температуре. НАДФ-восстанавливающий раствор (НВР) получали в день анализа разбавлением одного объема 17 мг/мл НАДФ+ одним объемом 78 мг/мл глюкозо-6-фосфата (оба получены в KРВ, рН 7,4) и 7,9 объемами 20 мМ MgCl2. Конечные концентрации НАДФ+ и глюкозо-6-фосфата составляли 1,7 мг/мл и 7,8 мг/мл, соответственно. Непосредственно перед применением, НВР активировали посредством добавления 10 мкл глюкозо-6-фосфатдегидрогеназы (150 ед/мл в KРВ, рН 7,4) на мл исходного раствора НВР. Микросомы печени разбавляли с помощью КРВ до 2,5 мг белка/мл.
В случае соединения 1, соединения 2 и соединения 3, а также каждого положительного контроля (то есть декстрометорфана, диазепама, дилтиазема, фенацетина, толбутамида и верапамила), 20 мкл 2,5 мкМ рабочего стандартного образца раствора исследуемого соединения и 20 мкл микросом (2,5 мг белка/мл) добавляли в каждую лунку полипропиленового 96-луночного планшета (Costar, VWR, Вест Честер, штат Пенсильвания) в двух повторностях. Планшеты помещали в инкубатор при 37°С в течение 5 минут перед добавлением стартового раствора. 10-мкл аликвоту раствора НВР добавляли в каждую исходную лунку для инициации метаболизма. Концентрация исследуемого соединения в начале инкубации составляла 1 мкМ. Один инкубационный планшет готовили для каждого момента времени (то есть 0 и 20 минут). Инкубацию проводили при температуре 37°С и 100% относительной влажности. В каждый момент времени соответствующий инкубационный планшет вынимали из инкубатора и добавляли в каждую лунку раствор, содержащий внутренний стандарт (150 мкл, 0,25 мкМ лабеталола в 60% ацетонитриле). Планшет немедленно центрифугировали при 2,095 × g в течение 7 минут при комнатной температуре с применением настольной центрифуги Allegra (Beckman Coulter, Фуллертон, штат Калифорния). 200-мкл аликвоту надосадочной жидкости переносили из каждой лунки в 96-луночный планшет с неглубокими лунками (Costar). Планшеты герметично закрывали с помощью мата для закрывания планшетов многократного применения.
Инкубация гепатоцитов
Исходные растворы соединения 1, соединения 2 и соединения 3 готовили в концентрации 10 мМ (активного соединения) в ДМСО. Стабильность соединения 1, соединения 2 и соединения 3 (1 мкМ) in vitro оценивали в присутствии гепатоцитов следующим образом. Криоконсервированные гепатоциты размораживали, выделяли из транспортной среды и разбавляли до плотности, составляющей 1×106 жизнеспособных клеток/мл, в соответствии с рекомендациями поставщика, с применением модифицированной по способу Дульбекко среды Игла, IX, с высоким содержанием глюкозы (DMEM, Invitrogen, Карлсбад, штат Калифорния). Жизнеспособность определяли с помощью трипанового синего с применением гемоцитометра (3500 Hausser, VWR, Вест Честер, штат Пенсильвания). 10 мМ исходные растворы соединения 1, соединения 2 и соединения 3 разбавляли до 2 мкМ с применением DMEM с добавками для создания рабочего стандартного образца. 20-мкл аликвоту исследуемого соединения или контроля (то есть антипирина, диазепама, дилтиазема, лоразепама, пропранолола, верапамила и 7-этил-10-гидроксикамптотецина [СН-38]) добавляли в каждую исследуемую лунку 96-луночного полипропиленового планшета (Costar, VWR, Вест Честер, штат Пенсильвания) с немедленным добавлением 20 мкл суспензии гепатоцитов. Для каждого момента времени (то есть 0, 60 и 120 минут) получали один инкубационный планшет с пробами, полученными в двух повторностях. Инкубацию проводили при температуре 37°С и 100% относительной влажности. В каждый момент времени соответствующий инкубационный планшет вынимали из инкубатора и добавляли в каждую лунку раствор, содержащий ВС (200 мкл, 0,25 мкМ лабеталола в 60% ацетонитриле). Содержимое планшета смешивали при 700 об/мин в течение 1 минуты на планшетном шейкере (ПСА MTS 2/4 Digital Microliter Shaker, VWR) и немедленно центрифугировали при 2095 × g в течение 10 минут при комнатной температуре с применением настольной центрифуги Allegra (Beckman Coulter, Фуллертон, штат Калифорния). 200-мкл аликвоту надосадочной жидкости переносили из каждой лунки в 96-луночный планшет с неглубокими лунками (Costar). Планшеты герметично закрывали с помощью матов для закрывания планшетов многократного применения.
Расчеты
Все расчеты осуществляли с применением BioAssay Enterprise. Средние коэффициенты площади пика рассчитывали посредством усреднения коэффициентов площади пика (n = 2) соединения 1, соединения 2, соединения 3 и внутреннего стандарта для каждой пробы. Оставшийся процент рассчитывали посредством определения отношения коэффициента площади пика в каждый момент времени к коэффициенту площади пика проб в нулевой момент времени. Коэффициент потерь исследуемого соединения (km) определяли с помощью линейной регрессии -ln(f(t)) в зависимости от времени. В регрессии использовалась формула "y = mx", поэтому модель принудительно задавала свободный член, составляющий 100% остатка, и предполагала, что метаболизм происходил согласно кинетике первого порядка. t1/2 определяли делением ln(2) на km. Расчетный собственный клиренс (CLint) рассчитывали посредством поправки периода полувыведения in vitro на стабильность каждого из соединения 1, соединения 2 и соединения 3 с помощью физических и физиологических поправочных коэффициентов, приведенных в таблице 5 и применяемых в следующем уравнении:
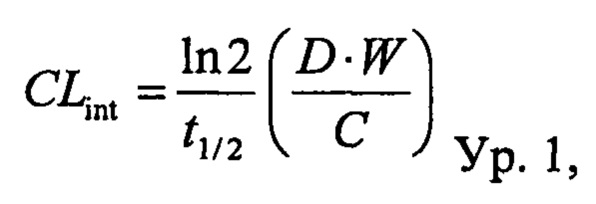
где D представляет собой отношение количества гепатоцитов к массе печени для конкретного вида. W представляет собой отношение средней массы рассматриваемой печени к массе животного, а С представляет собой отношение количества гепатоцитов, присутствующих во время инкубации, к единице объема. Расчетный печеночный клиренс (CLh) рассчитывали с помощью физических и физиологических поправочных коэффициентов, приведенных в таблице 5 и применяемых в следующем уравнении:
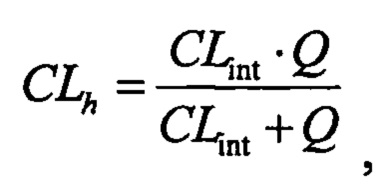
где Q представляет собой зависящий от вида печеночный кровоток. Поправку на несвязанную фракцию исследуемого соединения (fu) не делали. Расчетный коэффициент экстракции печенью (КЭП) определяли с помощью расчета отношения расчетного CLh к печеночному кровотоку.
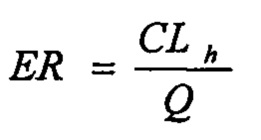
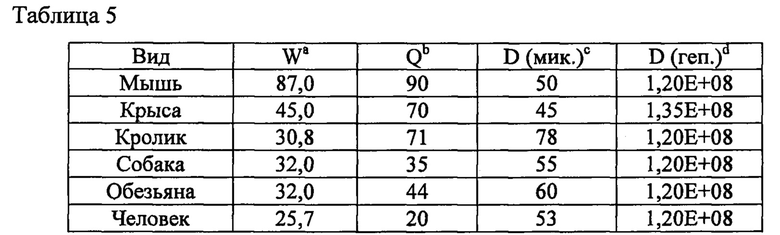
a) W = отношение средней массы печени (г) к массе животного (кг).
b) Q = средний печеночный кровоток (мл/мин/кг).
c) D (мик.) = отношение количества цитохром Р450-подобного белка (мг) к массе печени (г).
d) D (геп.) = отношение количества гепатоцитов к массе печени (г).
Пример Г
Фармакокинетика у самцов крыс линии Спрег-Доули
Каждое из соединения 1, соединения 2 и соединения 3 готовили посредством добавления соединения (сухого) к 20% Trappsol® (гидроксипропил-бета-циклодекстрин, CDT, Inc.) (водн.) с рН 5,0 и перемешивания на вортексе до получения однородной суспензии (п/о) или раствора (в/в).
В случае внутривенного введения дозы животных анестезировали 3% изофлураном, сбалансированным О2. 1 мг/кг соединения 1, соединения 2 и соединения 3 вводили в объеме дозы, составляющем 1 мл/кг, в латеральную хвостовую вену. Пока животное находилось под наркозом (3% изофлуран, сбалансированный О2,0,2 мл/момент времени) кровь забирали из противоположной латеральной хвостовой вены в пробирки с NaЭДТА (1,5% об./об.) в следующие моменты времени: 1, 5, 15, 30 минут и 1, 2, 4, 8 и 24 часа, и тщательно смешивали.
В случае перорального введения дозы крысы получали одну пероральную дозу после ночного голодания. Пока животное находилось под наркозом 3% изофлураном, сбалансированным О2 (0,2 мл/момент времени) кровь забирали из латеральной хвостовой вены в пробирки с NaЭДТА (1,5% об./об.) в следующие моменты времени: 5, 15 и 30 минут, а также 1, 2, 4, 8, 24 и 26 часов, и тщательно смешивали. Спустя 24 часа после забора крови животным вводили повторную пероральную дозу и через 2 часа после введения дозы производили забор плазмы и головного мозга для анализа соединения. Перед удалением головной мозг перфузировали солевым раствором, взвешивали и подвергали сверхбыстрому замораживанию в жидком азоте до проведения анализа с помощью ЖХМС/МС. Плазму отделяли центрифугированием при 14000 об/мин в течение 10 минут и переносили пробы плазмы в пробирки с резиновыми крышками 96-луночного планшета и хранили при -20±5°С до проведения анализа.
Анализ плазмы
Единую 12-точечную калибровочную кривую получали в первую очередь посредством серийного разведения (3-кратного) 40 мкг/мл исходного раствора соединения 1, соединения 2 и соединения 3 в ДМСО с получением стандартных растворов. Затем добавляли плазму (20 мкл) в планшет для экстракции и добавляли 2,5 мкл каждого стандартного раствора к ранее не подверженной экспериментам плазме. Исходный раствор внутреннего стандарта (10,0 мкл 2,5 мкг/мл в ацетонитриле) впоследствии добавляли к каждому стандартному раствору и раствору пробы. Белковый материал осаждали из 20 мкл плазмы посредством добавления 317,5 мкл ацетонитрила при общем объеме, составляющем 350 мкл. Пробы перемешивали на вортекс-миксере в течение 5 минут и центрифугировали в центрифуге Allegra X-12R (Beckman Coulter, Фуллертон, штат Калифорния) в течение 15 минут при около 1500 × g при температуре 4°С. 100-мкл аликвоту каждой надосадочной жидкости переносили с помощью 550 мкл пипеточного дозатора Personal Pipettor (Apricot Designs, Монровия, штат Калифорния) в 96-луночные планшеты и разбавляли 1:1 водой для ВЭЖХ. Полученные планшеты герметично закрывали матами для закрывания планшетов и анализировали количество соединения в плазме с помощью ЖХМС/МС.
Система ЖХМС/МС состояла из автоматического дозатора HTC-PAL (Leap Technologies, Inc., Каррборо, штат Северная Каролина), ВЭЖХ HP 1100 (Agilent Technologies Inc., Санта-Клара, штат Калифорния) и тройного квадрупольного масс-спектрометра API4000 (Applied Biosystems, Фостер Сити, штат Калифорния). Хроматографическое удерживание анализируемого вещества и внутреннего стандарта достигали с помощью колонки Phenomenex® Synergi 4μ Hydro-RP 80А (2,1×30 мм, размер частиц 4 мкм; Phenomenex, Торранс, штат Калифорния) в сочетании с градиентными условиями с применением подвижных фаз А (10 мМ ацетата аммония в водном растворе 0,1% муравьиной кислоты и 1% изопропилового спирта) и Б (10 мМ ацетата аммония в 89,9% ацетонитриле, 10% метаноле, 0,1% муравьиной кислоте). Общее время хроматографирования, включая в том числе уравновешивания, для одной инъекции составляло 3,5 минуты, а скорость потока составляла 0,8 мл/минута. Количество растворителя Б увеличивали с 5 до 95% за 1,0 минуту, выдерживали на уровне 95% в течение 1,0 минуты, а затем снижали до 5% за 0,1 минуты.
Анализ головного мозга
Единую 12-точечную калибровочную кривую получали в первую очередь посредством серийного разведения (3-кратного) 40 мкг/мл исходного раствора соединения 1, соединения 2 и соединения 3 в ДМСО. Гомогенат головного мозга получали посредством добавления 5,0 мл воды в конические 50 мл пробирки, содержащие головной мозг. Содержимое пробирки гомогенизировали с помощью многоместного гомогенизатора Omni-Prep (Кеннесо, штат Джорджия) при 22000 об/мин в течение 180 секунд. Гомогенат (100 мкл) добавляли в планшет для экстракции и добавляли 2,5 мкл каждого стандартного раствора к гомогенату головного мозга, ранее не подверженному экспериментам. Исходный раствор внутреннего стандарта (10,0 мкл 2,5 мкг/мл в ацетонитриле) впоследствии добавляли к каждому стандартному раствору и раствору пробы. Белковый материал осаждали из 100 мкл гомогената опухоли мыши посредством добавления 237,5 мкл ацетонитрила при общем объеме, составляющем 350 мкл. Пробы перемешивали на вортекс-миксере в течение 5 минут и центрифугировали в центрифуге Allegra X-12R (Beckman Coulter, Фуллертон, штат Калифорния) в течение 15 минут при около 1500 × g при температуре 4°С. 100-мкл аликвоту каждой надосадочной жидкости переносили с помощью 550 мкл пипеточного дозатора Personal Pipettor (Apricot Designs, Монровия, штат Калифорния) в 96-луночные планшеты и разбавляли 1:1 водой для ВЭЖХ. Полученные планшеты герметично закрывали матами для закрывания планшетов и анализировали количество соединения в головном мозге с помощью ЖХМС/МС.
Система ЖХМС/МС состояла из автоматического дозатора HTC-PAL (Leap Technologies, Inc., Каррборо, штат Северная Каролина), ВЭЖХ HP 1100 (Agilent Technologies Inc., Санта-Клара, штат Калифорния) и тройного квадрупольного масс-спектрометра API4000 (Applied Biosystems, Фостер Сити, штат Калифорния). Хроматографическое удерживание анализируемого вещества и внутреннего стандарта достигали с помощью колонки Phenomenex® Synergi 4μ Hydro-RP 80А (2,1×30 мм, размер частиц 4 мкм; Phenomenex, Торранс, штат Калифорния) в сочетании с градиентными условиями с применением подвижных фаз А (10 мМ ацетата аммония в водном растворе 0,1% муравьиной кислоты и 1% изопропилового спирта) и Б (10 мМ ацетата аммония в 89,9% ацетонитриле, 10% метаноле, 0,1% муравьиной кислоте).
Общее время хроматографирования, в том числе время уравновешивания, для одной инъекции составляло 3,5 минуты, а скорость потока составляла 0,8 мл/мин. Количество растворителя Б увеличивали с 5 до 95% за 1,0 минуту, выдерживали на уровне 95% в течение 1,0 минуты, а затем снижали до 5% за 0,1 минуты. Масс-спектрометрическое детектирование анализируемых веществ осуществляли с применением режима ионизации ESI+. Во время инфузии исходного раствора соединения 1, соединения 2 и соединения 3 оптимизировали ионный ток. Реакцию анализируемых веществ измеряли посредством мониторинга множественных реакций (MRM) переходов, уникальных для каждого соединения. Фармакокинетические параметры рассчитывали посредством стандартного некомпартментного моделирования с применением проприетарного программного обеспечения для внутреннего пользования (Sherlock версия 2.1). AUC рассчитывали с помощью интегрирования линейным методом трапеций.
Пример Д
Фармакокинетика у собак
Препарат в/в введения для доставки 1 мг/кг в объем дозы 1 мл/кг готовили следующим образом. Несущей средой для препарата являлся 20% Trappsol® (гидроксипропил-бета-циклодекстрин, CDT, Inc.) (водн.) в воде. К каждому из соединения 1, соединения 2 и соединения 3 (в виде сухих соединений) добавляли конечный объем несущей среды и перемешивали полученную смесь до получения однородного раствора. Значение рН конечного раствора составляло 7. Перед доставкой животному раствор фильтровали с применением 0,2 мкМ фильтра.
Самцы собак породы бигль получали однократную внутривенную дозу после ночного голодания. Кровь/плазму собирали (1,5 мл/момент времени) в пробирки для забора крови с K2ЭДТА в следующие моменты времени после введения дозы: 0, 1,5, 15 и 30 минут, а также 1, 2, 4, 8, 12, 24 и 48 часов, и тщательно смешивали. Производили забор проб крови из яремной, головной или подкожной вен находящихся в сознании животных. Пробы крови держали на льду до проведения анализа плазмы. Пробы крови центрифугировали при 3200 об/мин в течение 10 минут при температуре около 5°С. Пробы плазмы переносили непосредственно в пробирки 96-луночного планшета (MatrixTech, 0,75 мл). На пробирках 96-луночного планшета размещали резиновые инъекционные колпачки. Пробы плазмы хранили при -20±5°С до проведения анализа.
Анализ плазмы
Единую 12-точечную калибровочную кривую получали в первую очередь посредством серийного разведения (3-кратного) 40 мкг/мл исходного раствора соединения 1, соединения 2 и соединения 3 в ДМСО с получением стандартных растворов. Затем добавляли плазму (20 мкл) в планшет для экстракции и добавляли 2,5 мкл каждого стандартного раствора к ранее не подверженной экспериментам плазме. Исходный раствор внутреннего стандарта (10,0 мкл 2,5 мкг/мл в ацетонитриле) впоследствии добавляли к каждому стандартному раствору и раствору пробы. Белковый материал осаждали из 20 мкл плазмы посредством добавления 317,5 мкл ацетонитрила при общем объеме, составляющем 350 мкл. Пробы перемешивали на вортекс-миксере в течение 5 минут и центрифугировали в центрифуге Allegra X-12R (Beckman Coulter, Фуллертон, штат Калифорния) в течение 15 минут при около 1500 × g при температуре 4°С. 100-мкл аликвоту каждой надосадочной жидкости переносили с помощью 550 мкл пипеточного дозатора Personal Pipettor (Apricot Designs, Монровия, штат Калифорния) в 96-луночные планшеты и разбавляли 1:1 водой для ВЭЖХ. Полученные планшеты герметично закрывали матами для закрывания планшетов и анализировали количество соединения в плазме с помощью ЖХМС/МС.
Система ЖХМС/МС состояла из автоматического дозатора HTC-PAL (Leap Technologies, Inc., Каррборо, штат Северная Каролина), ВЭЖХ HP 1100 (Agilent Technologies Inc., Санта-Клара, штат Калифорния) и тройного квадрупольного масс-спектрометра API4000 (Applied Biosystems, Фостер Сити, штат Калифорния). Хроматографическое удерживание анализируемого вещества и внутреннего стандарта достигали с помощью колонки Phenomenex® Synergi 4μ Hydro-RP 80А (2,1×30 мм, размер частиц 4 мкм; Phenomenex, Торранс, штат Калифорния) в сочетании с градиентными условиями с применением подвижных фаз А (10 мМ ацетата аммония в водном растворе 0,1% муравьиной кислоты и 1% изопропилового спирта) и Б (10 мМ ацетата аммония в 89,9% ацетонитриле, 10% метаноле, 0,1% муравьиной кислоте). Общее время хроматографирования, в том числе время уравновешивания, для одной инъекции составляло 3,5 минуты, а скорость потока составляла 0,8 мл/минута. Количество растворителя Б увеличивали с 5 до 95% за 1,0 минуту, выдерживали на уровне 95% в течение 1,0 минуты, а затем снижали до 5% за 0,1 минуты.
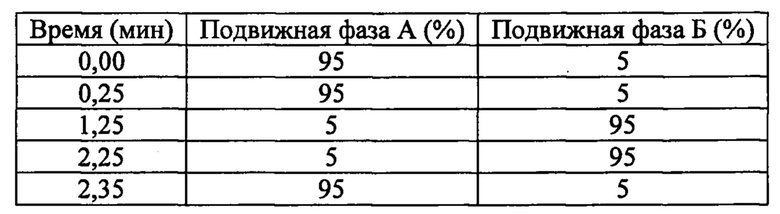

Масс-спектрометрическое детектирование анализируемых веществ осуществляли с применением режима ионизации ESI+. Во время инфузии исходного раствора соединения 1, соединения 2 и соединения 3 оптимизировали ионный ток. Реакцию анализируемых веществ измеряли посредством мониторинга множественных реакций (MRM) переходов, уникальных для каждого соединения. Фармакокинетические параметры рассчитывали посредством стандартного некомпартментного моделирования с применением проприетарного программного обеспечения для внутреннего пользования (Sherlock версия 2.1). AUC рассчитывали с помощью интегрирования линейным методом трапеций.
Пример Е
Анализ hERG FASTPatch®
Этот анализ использовали для измерения воздействия соединения 1, соединения 2 и соединения 3 на клонированные калиевые каналы hERG, экспрессируемые в клетках эмбриональной почки человека (НЕК293). Воздействие соединения 1, соединения 2 и соединения 3 in vitro на ток калиевых каналов (суррогат IKr, кардиальный калиевый ток задержанного выпрямления) hERG (ген специфических калиевых каналов сердца человека), экспрессируемых в клетках млекопитающих, оценивали при комнатной температуре с помощью QPatch НТ® (Sophion Bioscience A/S, Дания), системы автоматической параллельной фиксации потенциала. Исследуемый препарат оценивали при 0,3, 1, 3, 10, 30 и 100 мкМ, при этом каждую концентрацию испытывали на трех или более клетках (n≥3). Продолжительность воздействия каждой концентрации исследуемого препарата составляла 3 минуты. Положительный контроль подтвердил чувствительность тест-системы к ингибированию hERG.
Пример Ё
Анализ связывания р38 киназы
р38α связывающую активность соединения 1 определяли с помощью анализа связывания р38α киназы LanthaScreen™ Eu. 5 нМ неактивной, GST-меченой рекомбинантной р38α человека (GST-меченый цитоплазматический домен от Invitrogen, каталожный № PV 3305) инкубировали с 5 нМ Alexa-Fluor® Tracer 199 (Invitrogen, каталожный № PV 5830) и 2 нМ меченого европием антитела к GST (Invitrogen, каталожный № PV 5594) в буфере (25 мМ [Na+] ГЭПЭС рН 7,3, 10 мМ MgCl2, 100 мкМ NaVO4). Трехкратные серийные разведения соединения 1 в ДМСО добавляли до доведения конечного процентного содержания ДМСО до 2%. После 60-минутной инкубации при 22°С реакционную смесь измеряли с применением многорежимного сканирующего спектрофотометра для прочтения планшетов EnVision (PerkinElmer) с помощью двухволнового детектирования TR-FRET при 615 нМ и 665 нМ. Процент контроля рассчитывали с помощью логометрического коэффициента эмиссии. Значение IC50 определяли посредством согласования четырех-параметрической модели с процентом данных контроля. Было установлено, что соединение 1 является в 1000 раз более сильным в отношении TrkA, чем р38α.
Пример Ж
Нецелевой анализ профиля киназ
Соединение 1 испытывали на нецелевую активность киназ при концентрации, составляющей 10 мкМ, по методу Millipore, Inc. в рамках их услуги KinaseProfiler™ в отношении всех киназ, имеющихся в их единой панели киназ. Соединение 1 хроматографировали в двух повторностях при концентрации АТФ, близкой к Км (константе Михаэлиса) для каждой отдельной киназы в соответствии со спецификациями Millipore. Результаты показаны в приведенной ниже таблице. Данные представлены в виде процента от контроля (ПОК) и представляют собой среднее значение двух повторностей.
В KinaseProfiler™ соединение 1 продемонстрировало удивительную и неожиданную селективность в отношении ингибирования TrkA по сравнению с другими киназами панели. По сути, соединение 1 было преимущественно неактивным в отношении нецелевых киназ при концентрации, составляющей 10 мкМ, и поэтому не ожидалось, что оно станет ингибировать нецелевые киназы при терапевтических дозах у млекопитающих. Способность соединения 1 селективно ингибировать путь Trk без ингибирования других нецелевых киназ могла бы обеспечить профиль лекарственного средства, по сути, без побочных эффектов, связанных с ингибированием нецелевых киназ. Такой профиль лекарственного средства будет означать более безопасный подход к лечению боли, воспаления, рака и некоторых кожных заболеваний, чем сообщалось ранее.
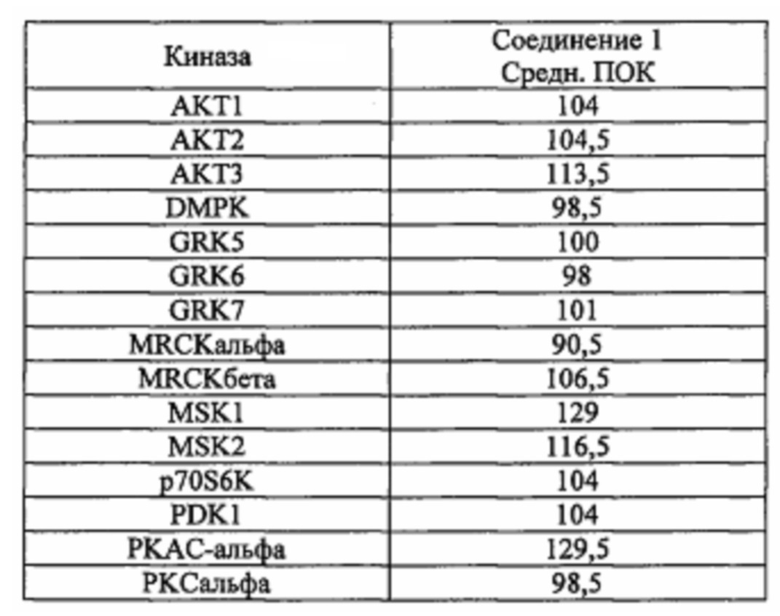
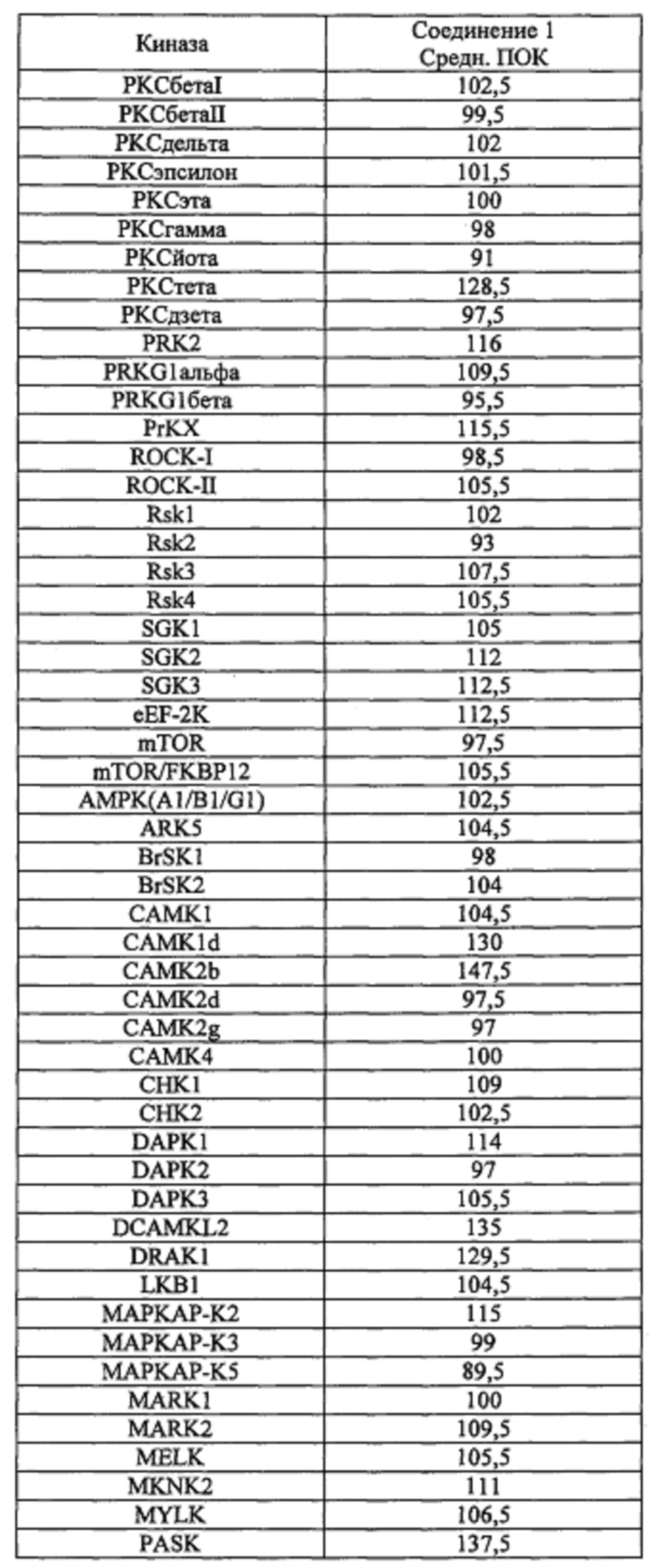
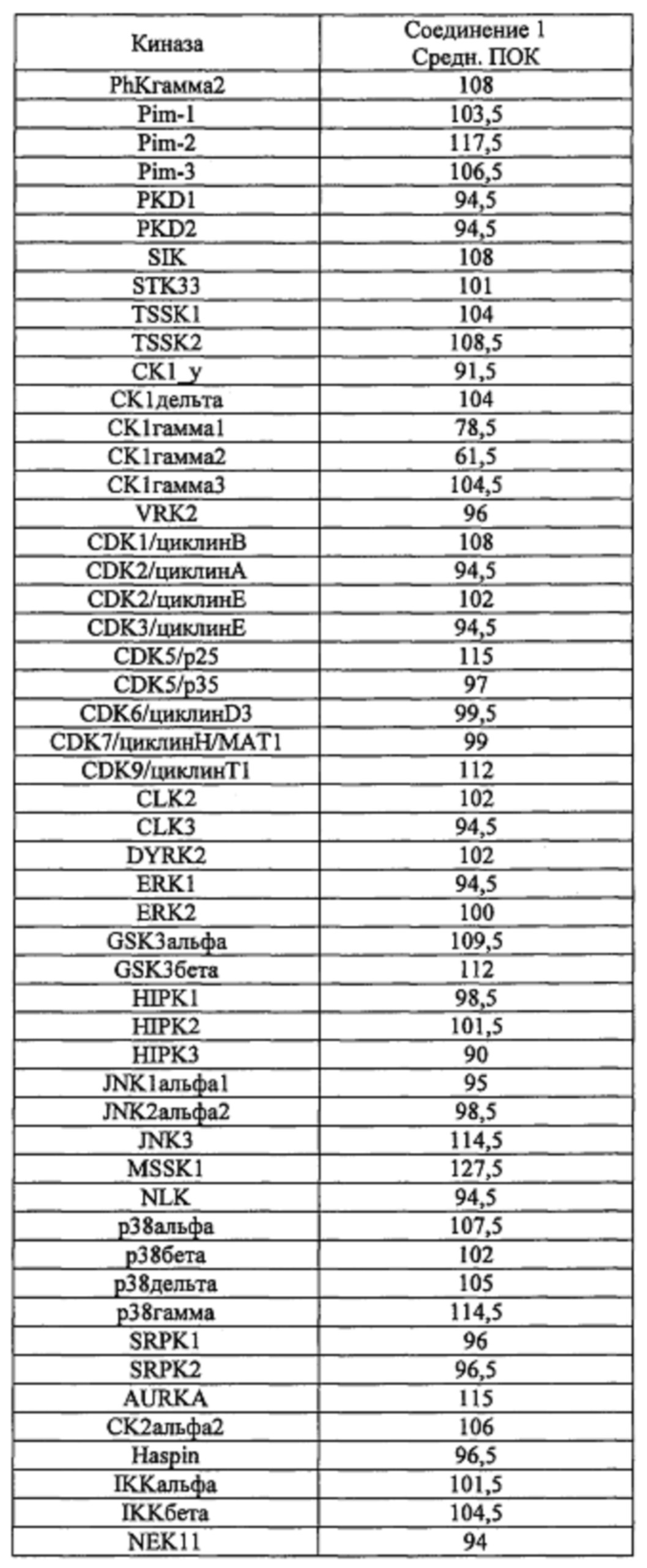
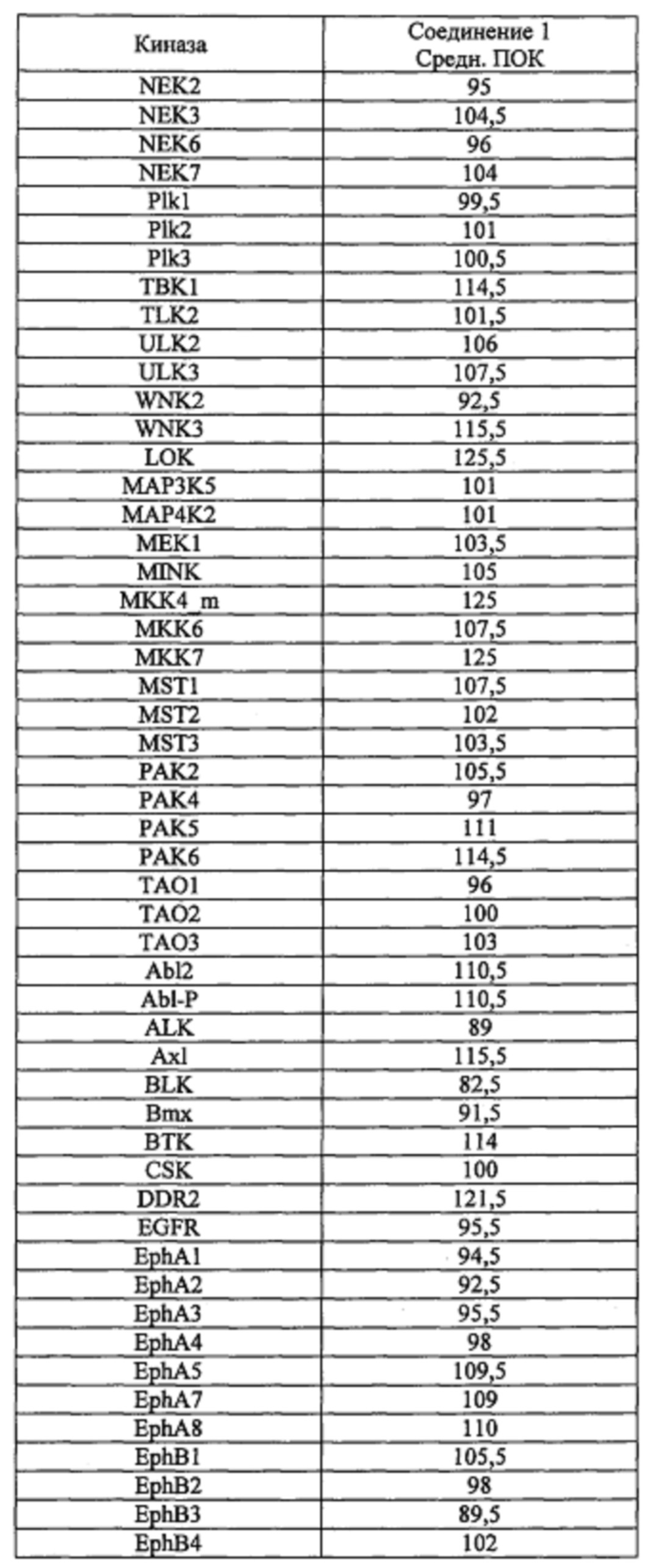
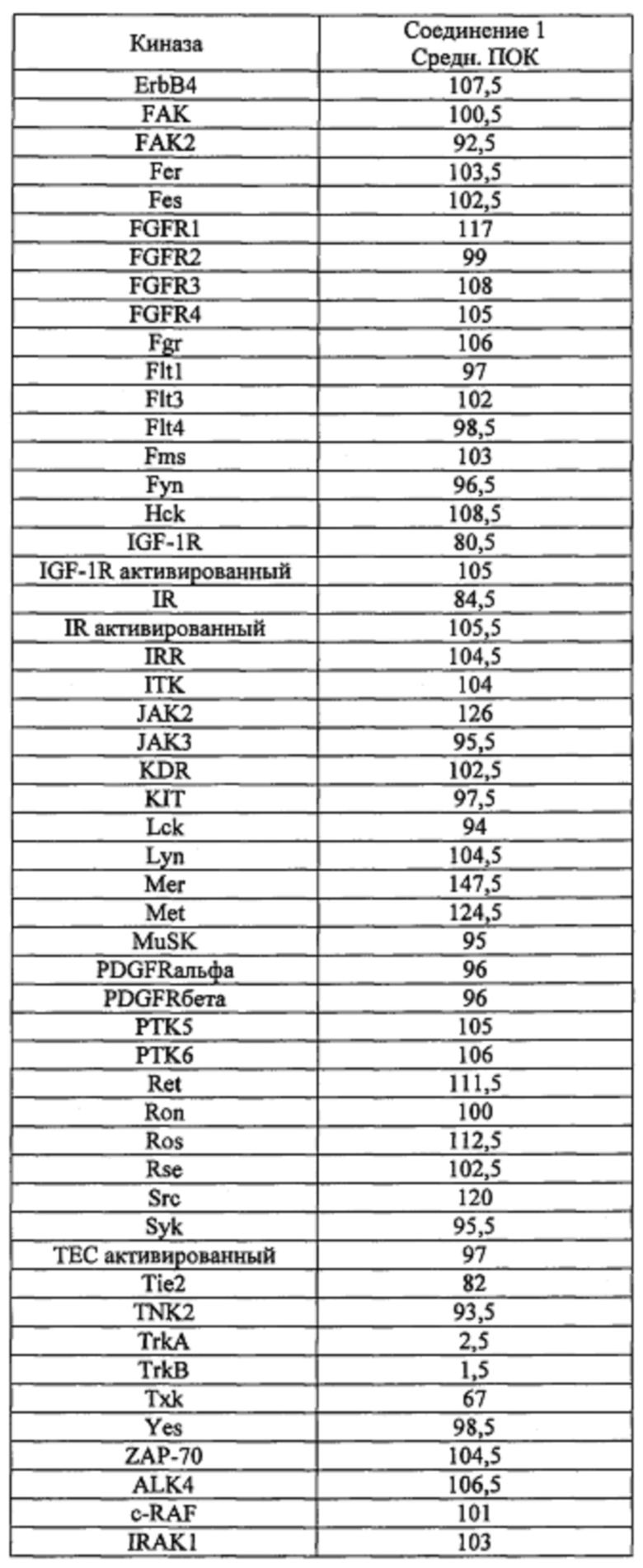
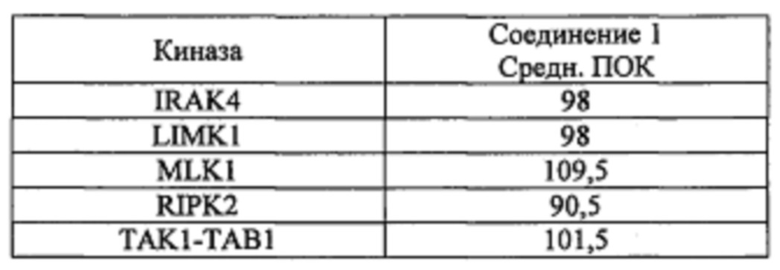
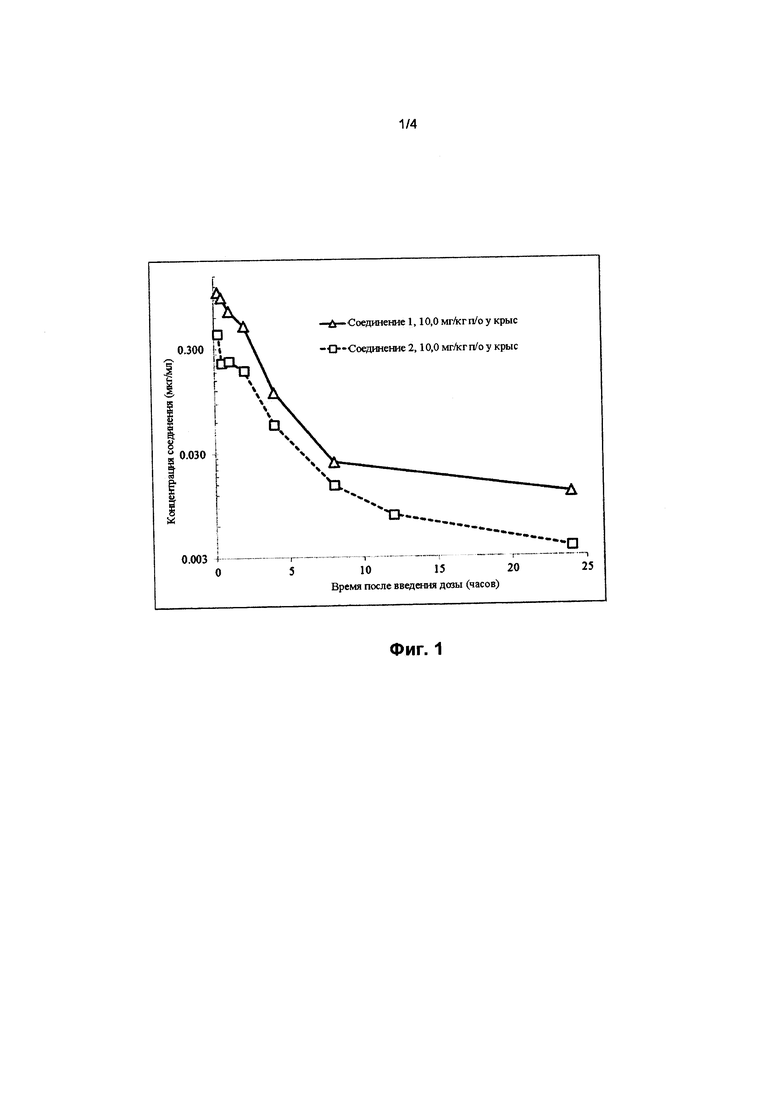
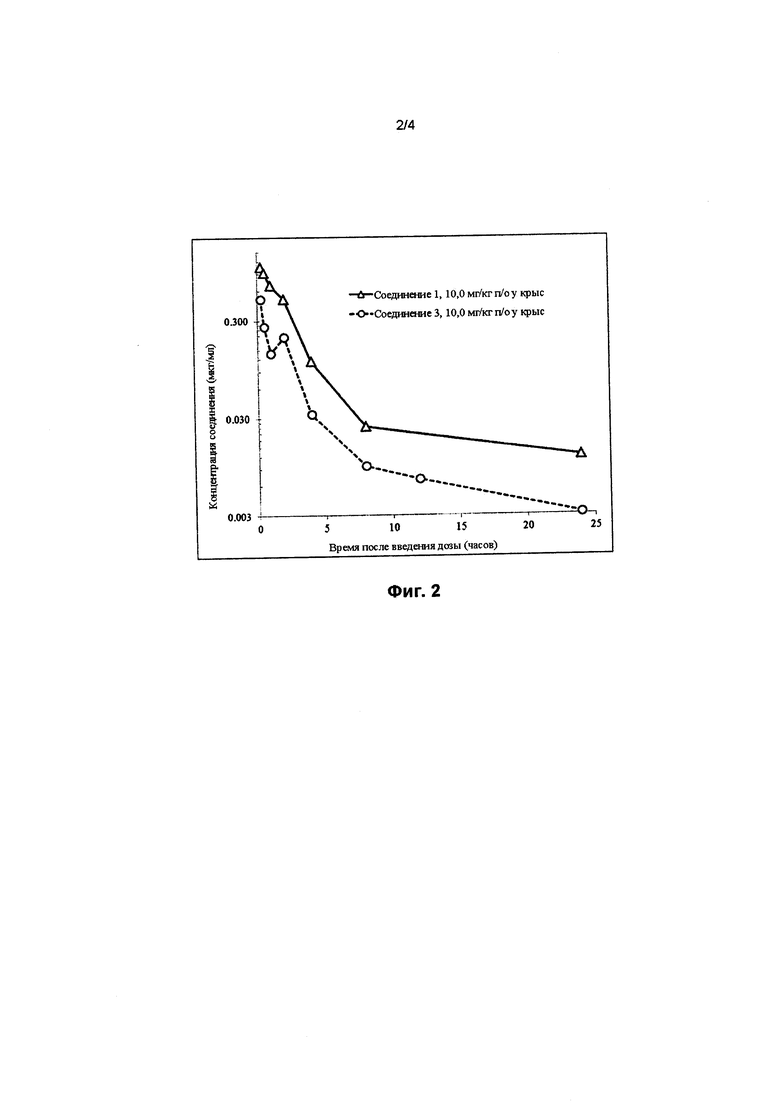
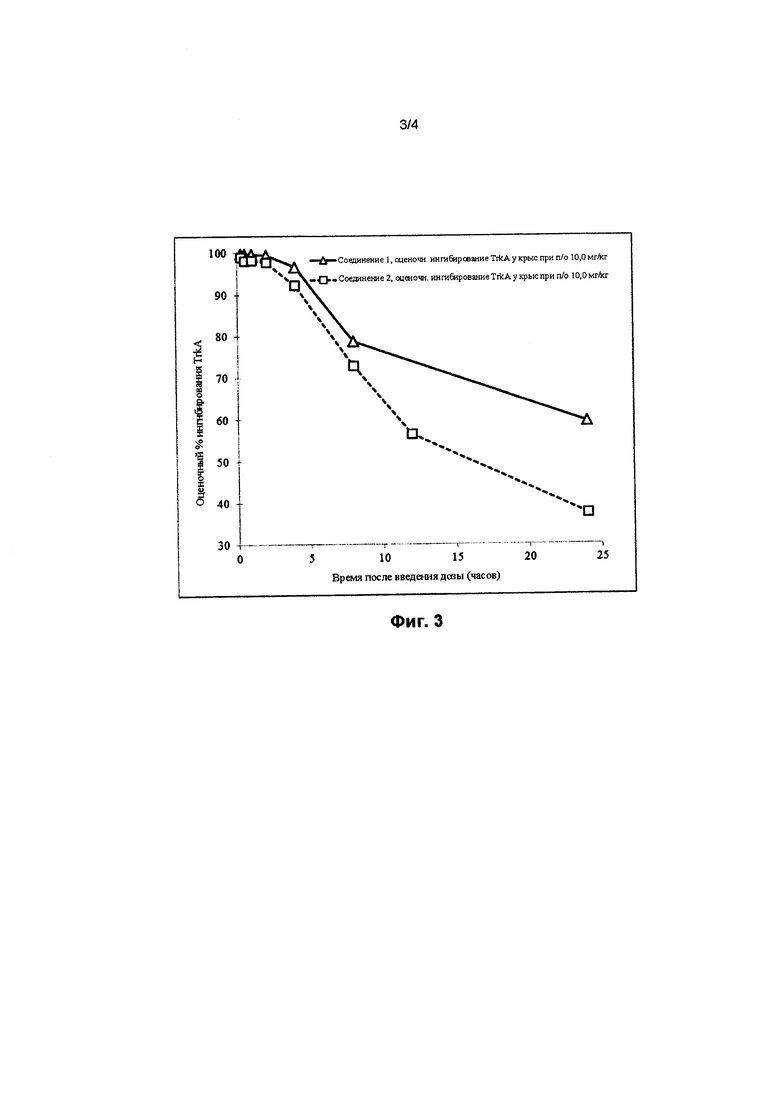
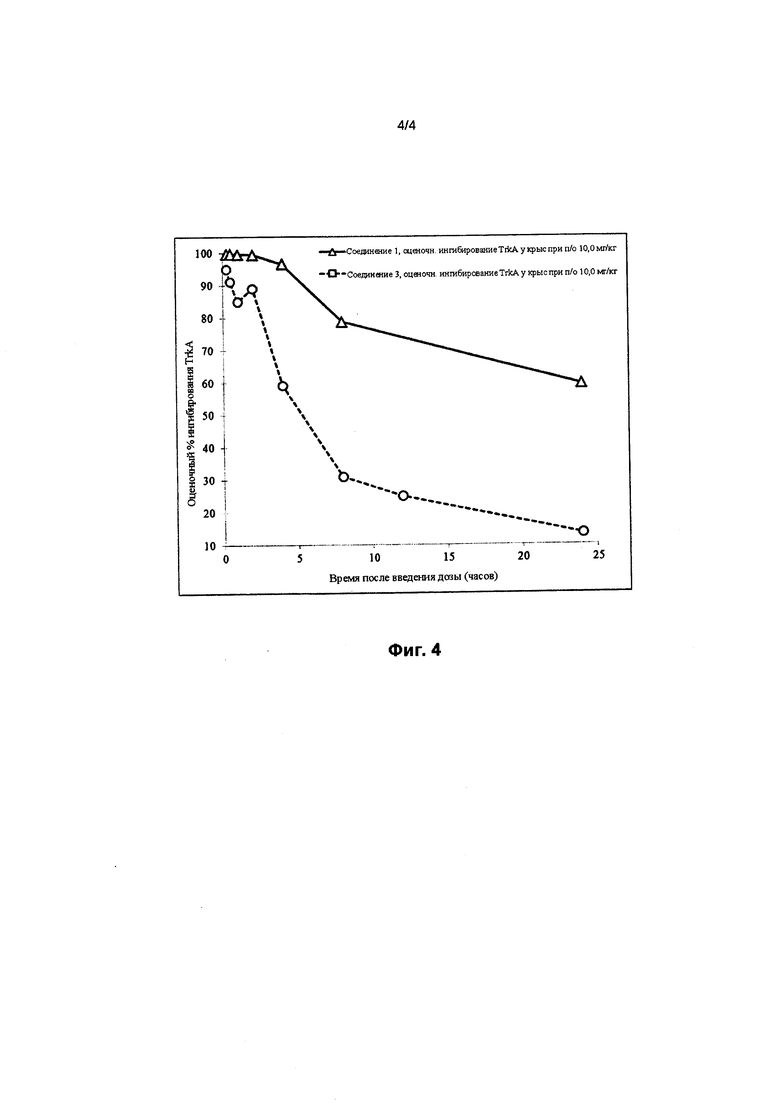
Изобретение относится к новому соединению - 1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-ил)мочевине формулы I или ее дигидрохлоридной соли. Соединения обладают свойствами селективного ингибитора TrkA киназы и могут быть использованы для лечения заболевания или расстройств выбранных из группы, состоящей из боли, рака, воспаления или воспалительных заболеваний, эндометриоза, диабетической периферической нейропатии, простатита, синдрома тазовой боли, заболеваний, связанных с дисбалансом регуляции ремоделирования костной ткани, а также заболеваний, обусловленных аберрантной передачей сигналов фактора роста соединительной ткани. Новое соединение соответствует формуле I
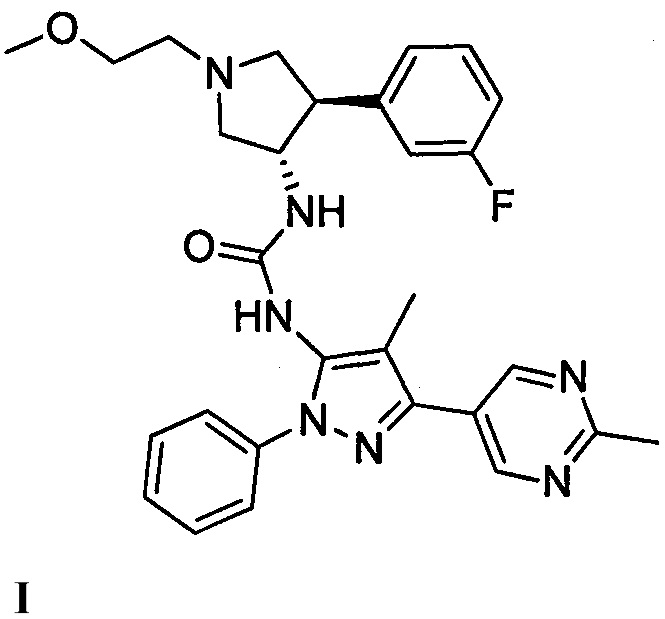
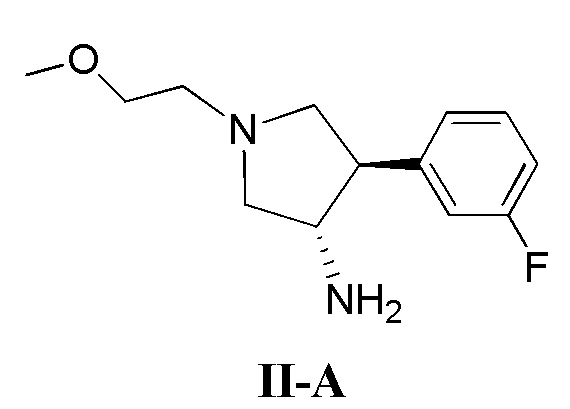
и может быть получено реакцией соединения формулы II-A с соединением III, или IV, или VII
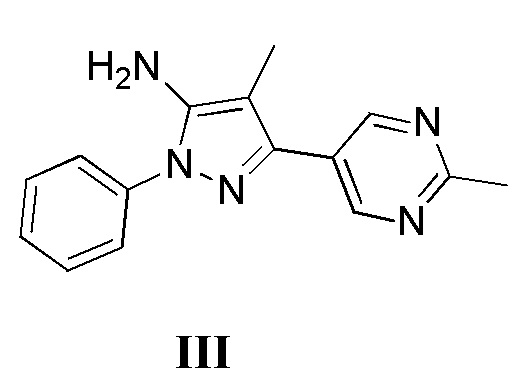
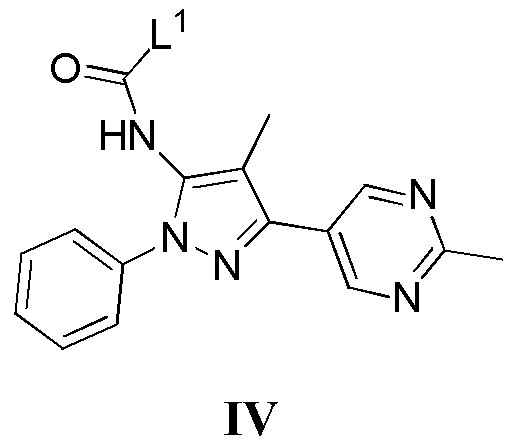
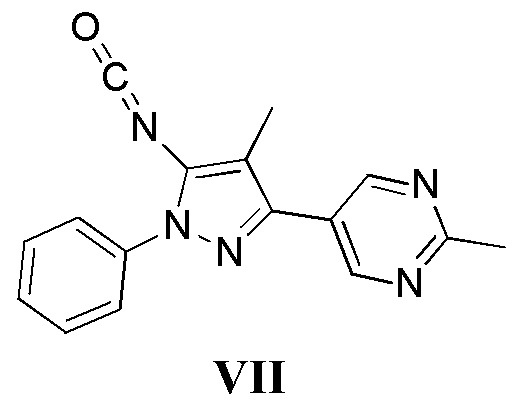
с удалением защитных групп, в случае их присутствия, и необязательным получением дигидрохлоридной соли соединения I. Либо соединение формулы I может быть получено реакцией соединения V или VI
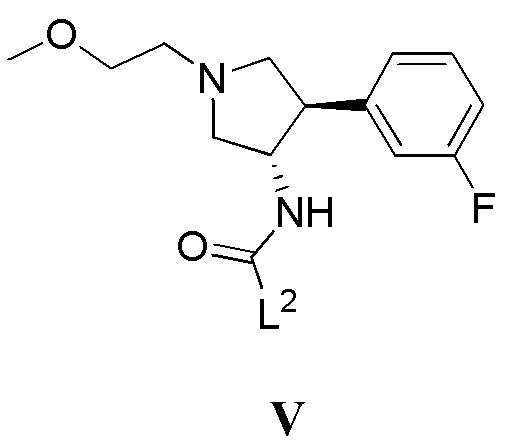
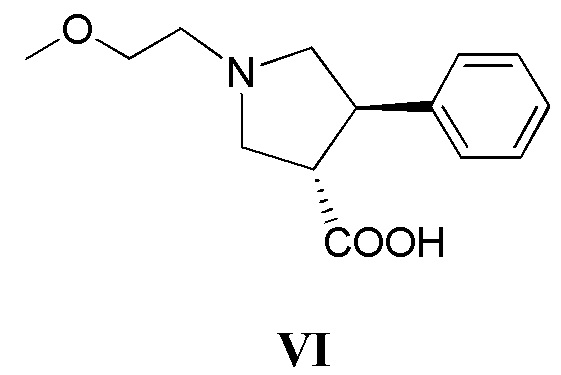
с соединением III, и удалением защитных групп, в случае их присутствия, и необязательным получением дигидрохлоридной соли соединения I. Условия реакций и значения радикалов указаны в формуле изобретения. Соединение формулы I и его дигидрохлоридная соль обладают значительно более высокими показателями активности при их использовании по сравнению с ближайшими аналогами и предназначены для перорального применения. 8 н. и 19 з.п. ф-лы, 4 ил., 13 табл., 9 пр.
1. Соединение
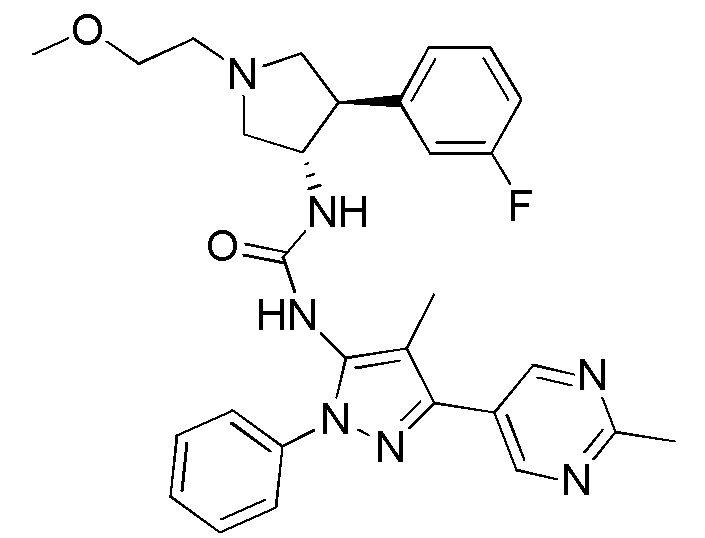
1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-ил)мочевина или его дигидрохлоридная соль.
2. Соединение по п. 1, которое представляет собой дигидрохлоридную соль 1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-ил)мочевины.
3. Фармацевтическая композиция, обладающая селективной Trk ингибиторной активностью, которая содержит соединение по п. 1 или 2 и фармацевтически приемлемый разбавитель или носитель.
4. Фармацевтическая композиция по п. 3, отличающаяся тем, что указанная фармацевтическая композиция предназначена для перорального применения.
5. Фармацевтическая композиция по п. 4, отличающаяся тем, что указанная фармацевтическая композиция имеет форму таблетки или капсулы.
6. Способ лечения заболевания или расстройства у млекопитающего, где заболевание или расстройство выбрано из группы, состоящей из боли, рака, воспаления или воспалительных заболеваний, эндометриоза, диабетической периферической нейропатии, простатита, синдрома тазовой боли, заболеваний, связанных с дисбалансом регуляции ремоделирования костной ткани, а также заболеваний, обусловленных аберрантной передачей сигналов фактора роста соединительной ткани, включающий введение указанному млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения по п. 1 или 2, где указанное соединение является селективным ингибитором Trk.
7. Способ по п. 6, отличающийся тем, что предназначен для лечения боли.
8. Способ по п. 7, отличающийся тем, что указанная боль представляет собой хроническую боль.
9. Способ по п. 7, отличающийся тем, что указанная боль выбрана из группы, состоящей из нейропатической боли, воспалительной боли, боли, обусловленной раком, боли, обусловленной переломом кости.
10. Способ по п. 9, отличающийся тем, что указанная боль представляет собой нейропатическую боль.
11. Способ по п. 9, отличающийся тем, что указанная боль представляет собой воспалительную боль.
12. Способ по п. 9, отличающийся тем, что указанная боль представляет собой боль, обусловленную раком.
13. Способ по п. 9, отличающийся тем, что указанная боль представляет собой боль, обусловленную переломом кости.
14. Способ по п. 6, отличающийся тем, что указанное заболевание представляет собой воспалительное заболевание.
15. Способ по п. 14, отличающийся тем, что воспалительное заболевание выбрано из группы, состоящей из воспалительных заболеваний легкого, интерстициального цистита, синдрома болезненного мочевого пузыря, воспалительных заболеваний кишечника и воспалительных кожных заболеваний.
16. Способ по п. 15, отличающийся тем, что воспалительное заболевание представляет собой интерстициальный цистит.
17. Способ по п. 15, отличающийся тем, что воспалительное заболевание представляет собой синдром болезненного мочевого пузыря.
18. Способ по п. 6, отличающийся тем, что заболевание представляет собой заболевание, обусловленное аберрантной передачей сигналов фактора роста соединительной ткани.
19. Способ по п. 18, отличающийся тем, что заболевание выбрано из группы, состоящей из синдрома Рейно, идиопатического фиброза легких, рубцевания (гипертрофического, келоидного и другого), цирроза, эндомиокардиального фиброза, предсердного фиброза, миелофиброза, прогрессирующего массивного фиброза (легких), нефрогенного системного фиброза, склеродермии, системного склероза, артрофиброза и фиброза глаза.
20. Способ по п. 6, отличающийся тем, что указанное заболевание представляет собой рак.
21. Соединение по п. 1 или 2 или его дигидрохлоридная соль, где указанное соединение является селективным ингибитором Trk, для применения в лечении боли, рака, воспаления/воспалительных заболеваний, эндометриоза, диабетической периферической нейропатии, простатита, синдрома тазовой боли, заболеваний, связанных с дисбалансом регуляции ремоделирования костной ткани, или заболеваний, обусловленных аберрантной передачей сигналов фактора роста соединительной ткани.
22. Применение соединения по п. 1 или 2 или его дигидрохлоридной соли, где указанное соединение является селективным ингибитором Trk, для изготовления лекарственного средства для лечения боли, рака, воспаления/воспалительных заболеваний, эндометриоза, диабетической периферической нейропатии, простатита, синдрома тазовой боли, заболеваний, связанных с дисбалансом регуляции ремоделирования костной ткани, или заболеваний, обусловленных аберрантной передачей сигналов фактора роста соединительной ткани.
23. Способ лечения рака у млекопитающего, нуждающегося в этом, включающий:
(а) определение того, обусловлен ли рак дисрегуляцией TrkA киназы; и
(б) в случае если было установлено, что рак обусловлен дисрегуляцией TrkA киназы, введение млекопитающему терапевтически эффективного количества соединения
1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-ил)мочевины или его дигидрохлоридной соли.
24. Способ по п. 23, отличающийся тем, что соединение представляет собой дигидрохлоридную соль 1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-ил)мочевины.
25. Способ получения соединения по п. 1 или 2, включающий:
(а) приведение в контакт соединения, имеющего формулу II-A
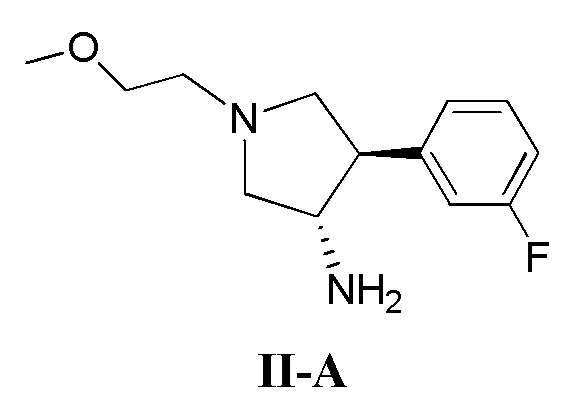 ,
,
с соединением, имеющим формулу III
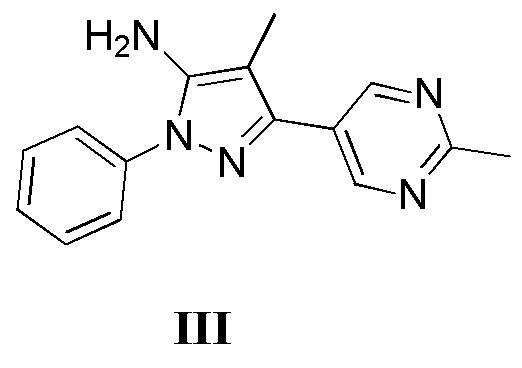 ,
,
в присутствии карбонилдиимидазола или трифосгена и основания; или
(б) приведение в контакт соединения, имеющего формулу II-A
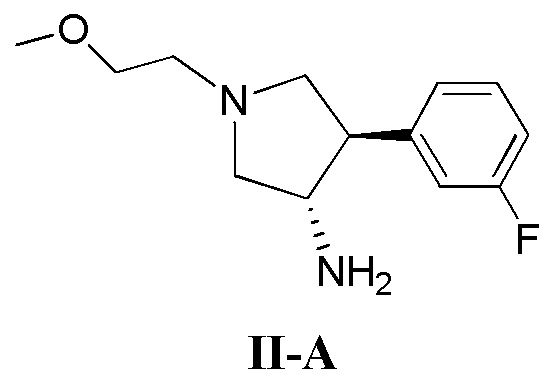 ,
,
с соединением, имеющим формулу IV
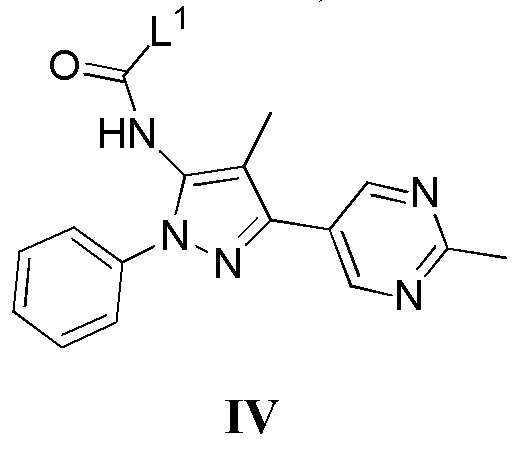 ,
,
где L1 представляет собой уходящую группу, в присутствии основания; или
(в) приведение в контакт соединения, имеющего формулу II-A
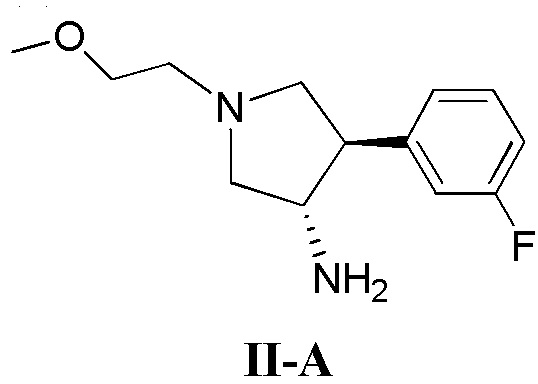 ,
,
с соединением, имеющим формулу VII
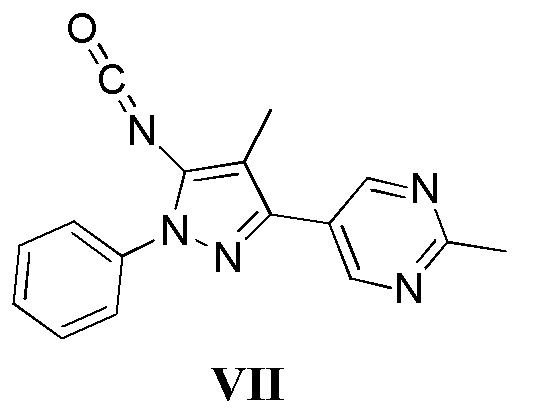 ,
,
в присутствии основания; и
удаление защитных групп, в случае присутствия, и необязательно получение его дигидрохлоридной соли.
26. Способ получения соединения по п. 1 или 2, включающий:
приведение в контакт соединения, имеющего формулу V
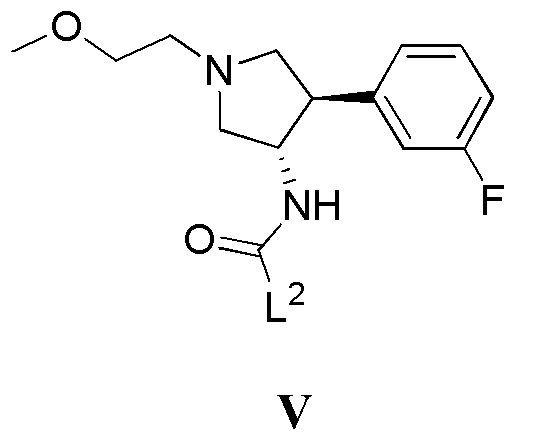 ,
,
где L2 представляет собой уходящую группу, с соединением, имеющим формулу III
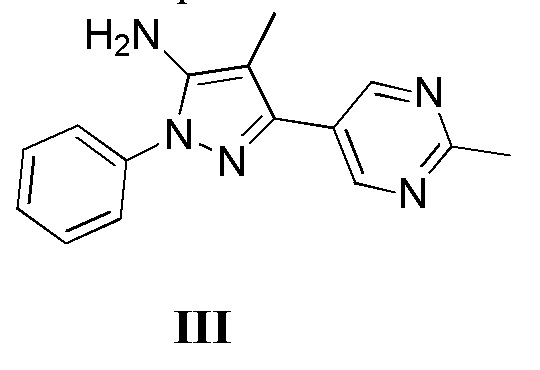 ,
,
в присутствии основания; и
удаление защитных групп, в случае присутствия, и необязательно получение его дигидрохлоридной соли.
27. Способ получения соединения по п. 1 или 2, включающий:
приведение в контакт соединения, имеющего формулу VI
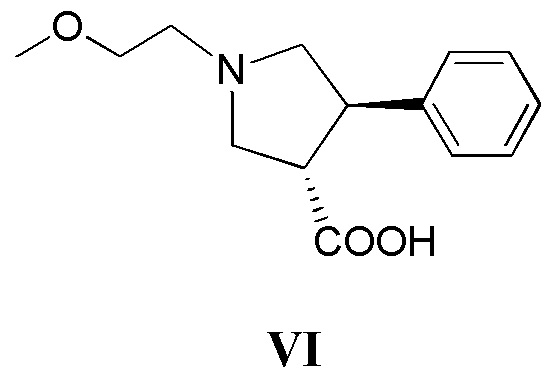 ,
,
с дифенилфосфорилазидом с образованием промежуточного соединения, с последующим приведением в контакт данного промежуточного соединения с соединением, имеющим формулу III
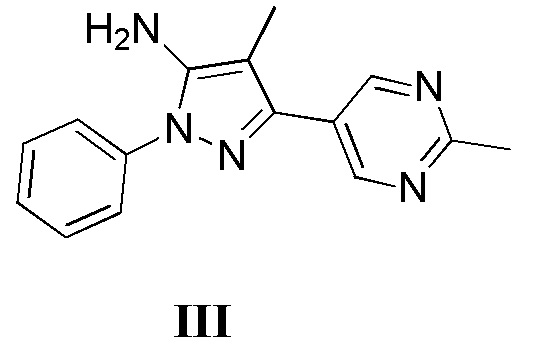 ,
,
в присутствии основания; и
удаление защитных групп, в случае присутствия, и необязательно получение его дигидрохлоридной соли.
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Jason C | |||
| Wong et al | |||
| Pharmacokinetic Optimization of Class-Selective Histone Deacetylase Inhibitors and Identification of Associated Candidate Predictive Biomarkers of Hepatocellular Carcinoma Tumor Response, Journal of Medicinal | |||

