Уровень техники
Настоящее изобретение относится к новым соединениям, к фармацевтическим композициям, содержащим эти соединения, к способам получения этих соединений и к применению этих соединений в терапии. Более конкретно, оно относится к соединениям пирролидинилмочевины и пирролидинилтиомочевины, которые демонстрируют ингибирование киназы TrkA, и которые пригодны для лечения боли, рака, воспаления, нейродегенеративных заболеваний и некоторых инфекционных заболеваний.
В существующих схемах лечения болевых состояний используют различные классы соединений. Опиоиды (такие как морфин) имеют несколько недостатков, включая рвоту, запор и негативные дыхательные эффекты, а также могут вызывать зависимость. Нестероидные противовоспалительные анальгетики (NSAID, такие как анальгетики типа СОХ-1 или СОХ-2), также имеют недостатки, включая недостаточную эффективность при лечении сильной боли. Кроме того, ингибиторы СОХ-1 могут вызывать язву слизистой оболочки. Соответственно, существует необходимость в новых и более эффективных средствах лечения для облегчения боли, особенно хронической боли.
Trk представляют собой тирозинкиназы с высокой аффинностью к рецептору, которые активируются группой растворимых факторов роста, называемых нейротрофинами (NT). В семейство рецепторов Trk входят три члена: TrkA, TrkB и TrkC. Среди нейротрофинов существует (i) фактор роста нервов (NGF), который активирует TrkA, (ii) нейротрофный фактор головного мозга (BDNF) и NT-4/5, которые активируют TrkB и (iii) NT3, который активирует TrkC. Trk широко экспрессируются в нейронной ткани и участвуют в сохранении, передаче сигналов и выживании нейронных клеток (Patapoutian, A. et al., Current Opinion in Neurobiology, 2001, 11, 272-280).
Ингибиторы пути Trk/нейротрофина демонстрируют эффективность во многих доклинических моделях боли на животных. Например, антагонистические антитела NGF и TrkA, такие как RN-624, демонстрируют эффективность в моделях воспалительной и невропатической боли на животных (Woolf, C.J. et al. (1994) Neuroscience 62, 327-331; Zahn, P.K. et al. (2004) J. Pain 5, 157-163; McMahon, SB. et al., (1995) Nat. Med. 1, 774-780; Ma, Q. P. and Woolf, C. J. (1997) NeuroReport 8, 807-810; Shelton, D.L. et al. (2005) Pain 116, 8-16; Delafoy, L. et al. (2003) Pain 105, 489-497; Lamb, K. et al. (2003) Neurogastroenterol. Motil. 15, 355-361; Jaggar, S.I. et al. (1999) Br. J Anaesth. 83, 442-448) и моделях невропатической боли на животных (Ramer, М.S. and Bisby, М.А. (1999) Eur. J. Neurosci. 11, 837-846; Ro, L. S. et al. (1999); Herzberg, U. et al., Pain 79, 265-274 (1997) Neuroreport 8, 1613-1618; Theodosiou, M. et al. (1999) Pain 81, 245-255; Li, L. et al. (2003) Mol. Cell. Neurosci. 23, 232-250; Gwak, Y.S. et al. (2003) Neurosci. Lett. 336, 117-120).
Показано также, что NGF, секретируемый клетками опухоли и макрофагами в опухоли, напрямую стимулирует TrkA, расположенную на периферийных болевых волокнах. Используя различные модели опухоли на мышах и крысах, было показано, что нейтрализация NGF моноклональными антителами ингибирует боль, связанную с раком, до уровня, аналогичного или более низкого по сравнению с максимальной допустимой дозой морфина. Поскольку киназа TrkA может служить как медиатор NGF-обусловленных биологических реакций, то ингибиторы TrkA и/или других киназ Trk могут обеспечивать эффективное лечение хронических болевых состояний.
Недавно в литературе было также показано, что сверхэкспрессия, активация, амплификация и/или мутация киназ Trk связаны со многими видами рака, включая нейробластому (Brodeur, G.М., Nat. Rev. Cancer 2003, 3, 203-216), рак яичников (Davidson. В., et al., Clin. Cancer Res. 2003, 9, 2248-2259), рак толстой и прямой кишок (Bardelli, А., Science 2003, 300, 949), меланому (Truzzi, F., et al., Dermato-Endocrinology 2008, 3 (1), pp.32-36), рак головы и шеи (Yilmaz, Т., et al., Cancer Biology and Therapy 2010, 10 (6), cc.644-653), карциному желудка (Du, J. et al., World Journal of Gastroenterology 2003, 9 (7), cc.1431-1434), карциному легких (RicciA., et al., American Journal of Respiratory Cell and Molecular Biology 25 (4), cc.439-446), рак молочной железы (Jin, W., et al., Carcinogenesis 2010, 31 (11), cc.1939-1947), глиобластому (Wadhwa, S., et al., Journal of Biosciences 2003, 28 (2), cc.181-188), медуллобластому (Gruber-Olipitz, M., et al., Journal of Proteome Research 2008, 7 (5), cc.1932-1944), секреторный рак молочной железы (Euthus, D.M., et al., Cancer Cell 2002, 2 (5), cc.347-348), рак слюнной железы (Li, Y.-G., et al., Chinese Journal of Cancer Prevention and Treatment 2009, 16 (6), cc.428-430), папиллярную карциному щитовидной железы (Greco, A., et al., Molecular and Cellular Endocrinology 2010, 321 (1), cc.44-49) и миелоидный лейкоз взрослых (Eguchi, М., et al., Blood 1999, 93 (4), cc.1355-1363). В доклинических моделях рака неселективные низкомолекулярные ингибиторы Trk А, В и С были эффективными для ингибирования роста опухоли и остановки метастазирования опухоли (Nakagawara, А. (2001) Cancer Letters 169: 107-114; Meyer, J. et al. (2007) Leukemia, 1-10; Pierottia, M.A. и Greco A., (2006) Cancer Letters 232: 90-98; Eric Adriaenssens, E., et al. Cancer Res (2008) 68: (2) 346-351).
Кроме того, показано, что ингибирование пути нейротрофин/Trk является эффективным для лечения доклинических моделей воспалительных заболеваний с антителами NGF или неселективными низкомолекулярными ингибиторами Trk A. Например, ингибирование пути нейротрофин/Trk изучали в доклинических моделях воспалительных заболеваний легких, включая астму (Freund-Michel, V; Frossard, N.; Pharmacology & Therapeutics (2008), 117 (1), 52-76), интерстициального цистита (Hu Vivian Y; et. al. The Journal of Urology (2005), 173 (3), 1016-21), воспалительных заболеваний кишечника, включая неспецифический язвенный колит и болезнь Крона (Di Mola, F. F, et. al., Gut (2000), 46 (5), 670-678), и воспалительных кожных заболеваний, таких как атопический дерматит (Dou, Y.-C; et. al. Archives of Dermatological Research (2006), 298 (1), 31-37), экзема и псориаз (Raychaudhuri, S. P., et al., J. Investigative Dermatology (2004), 122 (3), 812-819).
Предполагается также, что рецептор TrkA отвечает за развитие заболевания в инфекционном заболевании паразитарной инфекции Trypanosoma cruzi (Chagas disease) в организме человека-хозяина (de Melo-Jorge, М. et al., Cell Host & Microbe (2007) 1 (4), 251-261).
Ингибиторы Trk также могут найти применение для лечения заболеваний, связанных с дисбалансом регуляции ремоделирования костей, такого как остеопороз, ревматоидный артрит и костные метастазы. Костные метастазы являются частым осложнением рака, возникающим у 70% пациентов с распространенным раком молочной железы или простаты и примерно у 15-30% пациентов с карциномой легких, ободочной кишки, желудка, мочевого пузыря, матки, прямой кишки, щитовидной железы или почек. Остеолитические метастазы могут вызывать острую боль, патологические трещины, угрожающую жизни гиперкальцемию, компрессию спинного мозга и другие синдромы сдавливания нервов. По этим причинам костные метастазы являются серьезным и дорогостоящим осложнением рака. Поэтому агенты, которые могут вызывать апоптоз разрастающихся остеобластов, очень полезны. Экспрессию рецепторов TrkA наблюдали в областях формирования костей в моделях перелома костей на мышах (К. Asaumi, et al., Bone (2000) 26 (6) 625-633). Кроме того, наблюдали локализацию NGF практически во всех клетках, формирующих костную ткань (К. Asaumi, et al.). Недавно было показано, что ингибитор Trk ингибирует сигнальные пути тирозина, активированные нейротрофинами, связанными со всеми тремя Trk рецепторами в остеобластах hFOB человека (J. Pinski, et al., (2002) 62, 986-989). Эти данные дают логическое обоснование применения ингибиторов Trk для лечения заболеваний ремоделирования костей, таких как костные метастазы у пациентов с раком.
Известно, что для лечения боли или рака пригодны несколько классов низкомолекулярных ингибиторов Trk киназ (Expert Opin. Ther. Patents (2009) 19 (3), 305-319).
Краткое описание изобретения
Было обнаружено, что соединения пирролидинилмочевины, пирролидинилтиомочевины и пирролидинилгуанидина представляют собой ингибиторы TrkA, и они могут быть пригодны для лечения расстройств и заболеваний, таких как боль, включая хроническую и острую боль. Соединения настоящего изобретения могут быть полезны для лечения множества типов боли, включая воспалительную боль, невропатическую боль и боль, связанную с раком, хирургией и переломами костей. Кроме того, соединения настоящего изобретения могут быть полезны для лечения рака, воспалений, нейродегенеративных заболеваний и некоторых инфекционных заболеваний.
Более конкретно, в настоящем документе представлены соединения Формулы I:
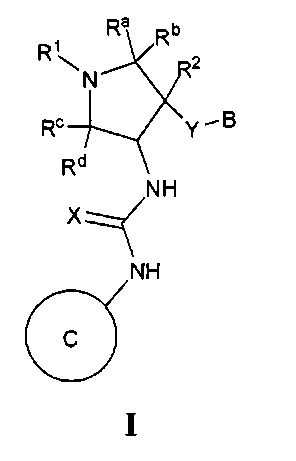
или их стереоизомеры, таутомеры или фармацевтически приемлемые соли, сольваты или пролекарства, причем фрагмент Y-B и фрагмент NH-C(=X)-NH находятся в транс-конфигурации, a R1, R2, Ra, Rb, Rc, Rd, X, Y, В и кольцо C являются такими, как описано в настоящем документе.
В одном варианте реализации, в настоящем документе представлены соединения Формулы I':
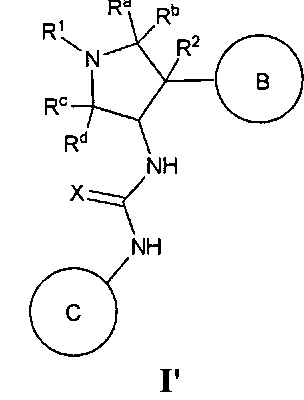
или их стереоизомеры, таутомеры или фармацевтически приемлемые соли, сольваты или пролекарства, причем кольцо B и фрагмент NH-C(=X)-NH находятся в транс-конфигурации, a R1, R2, Ra, Rb, Rc, Rd, X, кольцо B и кольцо C являются такими, как описано в настоящем документе.
В другом аспекте настоящего изобретения представлены способы предупреждения или лечения заболевания или расстройства, которое модулируется TrkA, включающие введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения настоящего изобретения или его стереоизомера, сольвата или фармацевтически приемлемой соли. В одном варианте реализации, заболевание и расстройства включают хроническую и острую боль, включая, но, не ограничиваясь этим, воспалительную боль, невропатическую боль и боль, связанную с раком, хирургией и переломами костей. В другом варианте реализации, заболевание и расстройства включают, но не ограничиваясь этим, рак, воспаление, нейродегенеративные заболевания и некоторые инфекционные заболевания.
В другом аспекте настоящего изобретения представлена фармацевтическая композиция, содержащая соединение настоящего изобретения или его фармацевтически приемлемую соль.
В другом аспекте настоящего изобретения представлены соединения настоящего изобретения для применения в терапии.
В другом аспекте настоящего изобретения представлены соединения настоящего изобретения для применения при лечении заболевания и расстройств хронической и острой боли, включая, но, не ограничиваясь этим, воспалительную боль, невропатическую боль и боль, связанную с раком, хирургией и переломами костей. В другом аспекте настоящего изобретения представлены соединения настоящего изобретения для применения при лечении заболевания и расстройств, выбранных из рака, воспаления, нейродегенеративных заболеваний и некоторых инфекционных заболеваний.
В другом аспекте настоящего изобретения представлено применение соединения настоящего изобретения в производстве лекарственных средств для лечения заболевания и расстройств, таких как хроническая и острая боли, включая, но не ограничиваясь этим, воспалительную боль, невропатическую боль и боль, связанную с раком, хирургией и переломами костей.
В другом аспекте настоящего изобретения представлено применение соединения настоящего изобретения в производстве лекарственных средств для лечения заболевания и расстройств, выбранных из рака, воспаления, нейродегенеративных заболеваний и некоторых инфекционных заболеваний.
В другом аспекте настоящего изобретения представлены промежуточные соединения для получения соединений Формулы I.
Другой аспект настоящего изобретения включает способы получения, способы разделения и способы очистки соединений настоящего изобретения.
Подробное описание изобретения
B настоящем документе представлены соединения и их фармацевтические композиции, которые потенциально пригодны для лечения заболеваний, состояний и/или расстройств, модулируемых TrkA.
В одном варианте реализации представлено соединение Формулы I:
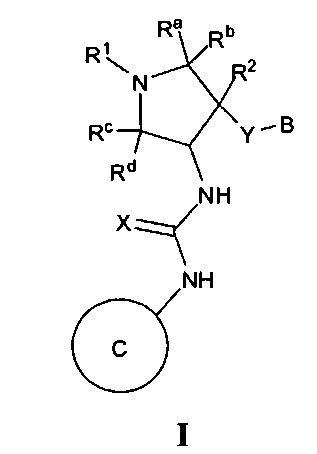
или его стереоизомеры, таутомеры или фармацевтически приемлемые соли, сольваты или пролекарства, где:
фрагмент Y-B и фрагмент NH-C(=X)-NH находятся в транс-конфигурации;
Ra, Rb, Rc и Rd независимо выбраны из Н и (1-3С)алкила;
X представляет собой O, S или NH;
R1 представляет собой (1-3С алкокси)(1-6С)алкил, (трифторметокси)(1-6С)алкил, (1-3С сульфанил)(1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, циано(1-6С)алкил, аминокарбонил(1-6С)алкил, гидрокси(1-6С)алкил, дигидрокси(2-6С)алкил, (1-6С)алкил, (1-3С алкиламино)(1-3С)алкил, (1-4Салкоксикарбонил)(1-6С)алкил, амино(1-6С)алкил, гидрокси(1-3Салкокси)(1-6С)алкил, ди(1-3С алкокси)(1-6С)алкил, (1-3С алкокси)трифтор(1-6С)алкил, гидрокситрифтор(1-6С)алкил, (1-4С алкоксикарбонил)(1-3С алкокси)(1-6С)алкил, гидроксикарбонил(1-3С алкокси)(1-6С)алкил, гетAr5(CH2)0-1 или Ar5(CH2)0-1;
R2 представляет собой Н, F или ОН;
Y представляет собой связь, -O- или -OCH2-;
В представляет собой Ar1, гетAr1, 1-6С алкил или (1-6С)алкокси;
Ar1 представляет собой фенил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена, CF3, CF3O-, (1-4С)алкокси, гидрокси(1-4С)алкила, (1-6С)алкила и CN;
гетAr1 представляет собой 5-6-членный гетероарил, имеющий 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенный 1-2 группами, независимо выбранными из (1-6С)алкила, галогена, OH, CF3, NH2 и гидрокси(1-2С)алкила;
кольцо C представляет собой формулу С-1, С-2 или С-3
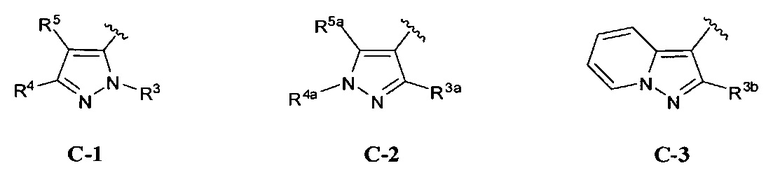
R3 представляет собой Н, (1-6С)алкил, гидрокси(1-6С)алкил, Ar2, гетСус1, (3-7С)циклоалкил или гетAr2;
Ar2 представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из галогена, (1-6С)алкила и гидроксиметила;
гетСус1 представляет собой 5-6-членное насыщенное или частично ненасыщенное гетероциклическое кольцо, имеющее 1-2 кольцевых гетероатома, независимо выбранных из N и O;
гетAr2 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, O и S, и необязательно замещенное одной или более группами, независимо выбранными из (1-6С)алкила и галогена;
R4 представляет собой Н, ОН, (1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, циано(1-6С)алкил, гидрокси(1-6С)алкил, дигидрокси(2-6С)алкил, (1-3С алкокси)(1-6С)алкил, амино(1-6С)алкил, аминокарбонил(1-6С)алкил, (1-3С)алкилсульфонамидо(1-6С)алкил, сульфамидо(1-6С)алкил, гидроксикарбонил(1-6С)алкил, гетAr3(1-6С)алкил, Ar3(1-6С)алкил, (1-6С)алкокси, монофтор(1-6С)алкокси, дифтор(1-6С)алкокси, трифтор(1-6С)алкокси, тетрафтор(2-6С)алкокси, пентафтор(2-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, дигидрокси(2-6С)алкокси, амино(2-6С)алкокси, аминокарбонил(1-6С)алкокси, гидроксил-карбонил(1-6С)алкокси, гетСус2(1-6С)алкокси, гетAr3(1-6С)алкокси, Ar3(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (1-3С алкилсульфонил)(1-6С)алкокси, (3-6С)циклоалкил [необязательно замещенный F, ОН, (1-6С алкил), (1-6С) алкокси или (1-3С алкокси)(1-6С)алкилом], гетAr4, Ar4, гетСус2(O)СН2-, (1-4С алкоксикарбонил)(1-6С)алкокси, гидроксикарбонил(1-6С)алкокси, аминокарбонил(1-6С)алкокси, гетСус2С(=O)(1-6С)алкокси, гидрокси(1-3С алкокси)(1-6С)алкокси, гидрокситрифтор(1-6С)алкокси, (1-3С)алкилсульфонамидо(1-6С)алкокси, (1-3С)алкиламидо(1-6С)алкокси, ди(1-3С алкил)амино-карбокси, гетСус2С(=O)O-, гидроксидифтор(1-6С)алкил, (1-4С алкилкарбокси)(1-6С)алкил, (1-6С)алкоксикарбонил, гидрокси/карбонил, аминокарбонил, (1-3С алкокси)аминокарбонил, гетСус3, галоген, CN, трифторметилсульфонил, N-(1-3С алкил)пиридинонил, N-(1-3С трифторалкил)пиридинонил, (1-4С алкилсилокси)(1-6С)алкокси, изоиндолин-1,3-дионил(1-6С)алкокси или N-(1-3С алкил)оксадиазолонил;
гетСус2 представляет собой 4-6-членное гетероциклическое кольцо, имеющее 1-2 кольцевых гетероатома, независимо выбранных из N и О, и необязательно замещенное 1-2 группами, независимо выбранными из (1-6С)алкила, (1-4С алкилкарбокси)(1-6С)алкила и (1-6С)ацила;
гетСус3 представляет собой 4-7-членный гетероцикл, имеющий 1-2 кольцевых гетероатома, независимо выбранных из N и О, и необязательно замещенный одним или более заместителями, независимо выбранными из F, CN, CF3, (1-6С)алкила, гидрокси(1-6С)алкила, (1-3С алкокси)(1-6С)алкила, (1-6С)ацил-, (1-6С)алкилсульфонила, трифторметилсульфонила и (1-4С алкокси)карбонила;
гетAr3 представляет собой 5-членное гетероариловое кольцо, имеющее 1-3 кольцевых атома, независимо выбранных из N, О и S, и необязательно замещенное (1-6С)алкилом;
Ar3 представляет собой фенил, необязательно замещенный (1-4С)алкокси;
гетAr4 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенное 1-2 заместителями, независимо выбранными из (1-6С)алкила, галогена, CN, гидрокси(1-6С)алкила, трифтор(1-6С)алкила, (3-6С)циклоалкила, (3-6С циклоалкил)СН2- (3-6С циклоалкил)С(=O)-, (1-3С алкокси)(1-6С)алкила, (1-6С)алкокси, (1-6С)алкилсульфонила, NH2, (1-6С алкил)амино, ди(1-6С алкил)амино, (1-3С трифторалкокси), (1-3С)трифторалкила и метоксибензила; или 9-10-членный бициклический гетероарил, имеющий 1-3 кольцевых атома азота;
Ar4 представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из (1-6С)алкила, галогена, CN, CF3, CF3O-, (1-6С)алкокси, (1-6Салкил)ОС(=O)-, аминокарбонила, (1-6С)алкилтио, гидрокси(1-6С)алкила, (1-6С алкил)SO2-, НОС(=O)- и (1-3С алкокси)(1-3С алкил)ОС(=O)-;
R5 представляет собой Н, (1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, галоген, CN, (1-4С)алкокси, гидрокси(1-4С)алкил, (1-3С алкокси)(1-4С)алкил, (1-4С алкил)ОС(=O)-, (1-6С)алкилтио, фенил [необязательно замещенный одной или более группами, независимо выбранными из галогена, (1-6С)алкила и (1-6С)алкокси], (3-4С)циклоалкил, амино, аминокарбонил или трифтор(1-3С алкил)амидо; или
R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное, частично ненасыщенное или ненасыщенное карбоциклическое кольцо, необязательно замещенное одним или более заместителями, независимо выбранными из (1-6С)алкила, или
R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное, частично ненасыщенное или ненасыщенное гетероциклическое кольцо, имеющее кольцевой гетероатом, выбранный из N, О или S, причем указанное гетероциклическое кольцо необязательно замещено одним или двумя заместителями, независимо выбранными из (1-6С алкил)С(=O)O-, (1-6)ацила, (1-6С)алкила и оксо, и указанный кольцевой атом серы необязательно окислен до S(=O) или SO2;
гетAr5 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, O или S, причем указанное кольцо необязательно замещено одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила, (1-6С)алкокси и CF3;
Ar5 представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из галогена, (1-6С)алкила, (1-6С)алкокси, CF3O-, (1-4С)алкоксикарбонила и аминокарбонила;
R3a представляет собой водород, галоген, (1-6С)алкил, трифтор(1-6С)алкил, (3-6С)циклоалкил, фенил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила и гидроксиметила, или 5-6-членное гетероариловое кольцо, имеющее 1-3-кольцевых гетероатома, независимо выбранных из N, О и S, и необязательно замещенное одной или более группами, независимо выбранными из (1-6С)алкила и галогена;
R3b представляет собой водород, (1-6С)алкил, трифтор(1-6С)алкил, (3-6С)циклоалкил, фенил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила и гидроксиметила, или 5-6-членное гетероариловое кольцо, имеющее 1-3- кольцевых гетероатома, независимо выбранных из N, О и S, и необязательно замещенное одной или более группами, независимо выбранными из (1-6С)алкила и галогена;
R4a представляет собой водород, (1-6С)алкил, трифтор(1-6С)алкил, фенил [необязательно замещенный одной или более группами, независимо выбранными из (1-6С)алкила, галогена, CN, CF3, CF3O-, (1-6С)алкокси, (1-6Салкил)ОС(=O)-, аминокарбонила, (1-6С)алкилтио, гидрокси(1-6С)алкила, (1-6С алкил)SO2-, НОС(=O)- и (1-3С алкокси)(1-3С алкил)ОС(=O)-], или 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенное 1-2 заместителями, независимо выбранными из (1-6С)алкила, гидрокси(1-6С)алкила, трифтор(1-6С)алкила, (3-6С)циклоалкила, (3-6С циклоалкил)СН2-(3-6С циклоалкил)С(=O)-, (1-3С алкокси)(1-6С)алкила, (1-6С)алкокси, (1-6С)алкилсульфонила, NH2, (1-6С алкил)амино, ди(1-6С алкил)амино, (1-3С трифторалкокси)(1-3С)трифторалкила и метоксибензила; и
R5a представляет собой водород, галоген, (1-6С)алкил, трифтор(1-6С)алкил, (3-6С)циклоалкил, фенил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила и гидроксиметила, или 5-6-членное гетероариловое кольцо, имеющее 1-3-кольцевых гетероатома, независимо выбранных из N, О и S, и необязательно замещенное одной или более группами, независимо выбранными из (1-6С)алкила и галогена.
В одном варианте реализации, соединения Формулы I имеют структуру I':
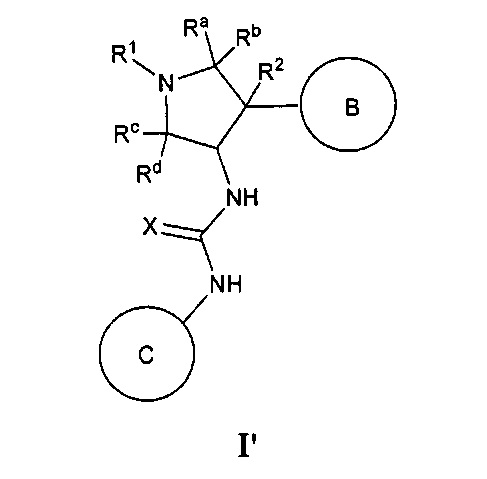
или их стереоизомеры, таутомеры или фармацевтически приемлемые соли, сольваты или пролекарства, где:
кольцо B и фрагмент NH-C(=X)-NH находятся в транс-конфигурации;
Ra, Rb, Rc и Rd независимо выбраны из Н и (1-3С)алкила;
X представляет собой O или S;
R1 представляет собой (1-3С алкокси)(1-6С)алкил, (трифторметокси)(1-6С)алкил, (1-3С сульфанил)(1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, циано(1-6С)алкил, аминокарбонил(1-6С)алкил, гидрокси(1-6С)алкил, дигидрокси(2-6С)алкил, (1-6С)алкил или (1-3С алкиламино)(1-3С)алкил;
R2 представляет собой Н, F или OH;
кольцо B представляет собой Ar1 или гетAr1;
Ar1 представляет собой фенил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена, CF3, CF3O-, (1-4С)алкокси, гидрокси(1-4С)алкила и (1-6С)алкила;
гетAr1 представляет собой 5-6-членный гетероарил, имеющий 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенный 1-2 группами, независимо выбранными из (1-6С)алкила, галогена, ОН, CF3, NH2 и гидрокси(1-2С)алкила;
кольцо C представляет собой
 ;
;
R3 представляет собой Н, (1-6С)алкил, гидрокси(1-6С)алкил, Ar2, гетСус1, (3-7С)циклоалкил или гетAr2;
Ar2 представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из галогена, (1-6С)алкила и гидроксиметила;
гетСус1 представляет собой 5-6-членное насыщенное или частично ненасыщенное гетероциклическое кольцо, имеющее 1-2 кольцевых гетероатома, независимо выбранных из N и O;
гетAr2 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, О и S, и необязательно замещенное (1-6С)алкилом;
R4 представляет собой Н, ОН, (1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, циано(1-6С)алкил, гидрокси(1-6С)алкил, дигидрокси(2-6С)алкил, (1-3С алкокси)(1-6С)алкил, амино(1 -6С)алкил, аминокарбонил(1-6С)алкил, (1-3С)алкилсульфонамидо(1-6С)алкил, сульфамидо(1-6С)алкил, гидроксикарбонил(1-6С)алкил, гетAr3(1-6С)алкил, Ar3(1-6С)алкил, (1-6С)алкокси, монофтор(1-6С)алкокси, дифтор(1-6С)алкокси, трифтор(1-6С)алкокси, тетрафтор(2-6С)алкокси, пентафтор(2-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, дигидрокси(2-6С)алкокси, амино(2-6С)алкокси, аминокарбонил(1-6С)алкокси, гидроксикарбонил(1-6С)алкокси, гетСус2(1-6С)алкокси, гетAr3(1-6С)алкокси, Ar3(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (1-3С алкилсульфонил)(1-6С)алкокси, (3-6С)циклоалкил, гетAr4 или Ar4;
гетСус2 представляет собой 4-6-членное гетероциклическое кольцо, имеющее 1-2 кольцевых гетероатома, независимо выбранных из N и О, и необязательно замещенное 1-2 группами, независимо выбранными из (1-6С)алкила;
гетAr3 представляет собой 5-членное гетероариловое кольцо, имеющее 1-3 кольцевых атома, независимо выбранных из N, О и S, и необязательно замещенное (1-6С)алкилом;
Ar3 представляет собой фенил, необязательно замещенный (1-4С)алкокси;
гетAr4 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенное 1-2 заместителями, независимо выбранными из (1-6С)алкила, или 9-10-членный бициклический гетероарил, имеющий 1-3 кольцевых атома азота;
Ar4 представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из (1-6С)алкила, галогена, CN, CF3, CF3O-, (1-6С)алкокси, (1-6Салкил)ОС(-O)-, аминокарбонила, (1-6С)алкилтио, гидрокси(1-6С)алкила, (1-6С алкил)SO2-, НОС(=O)- и (1-3С алкокси)(1-3С алкил)ОС(=O)-; и
R5 представляет собой Н, (1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, галоген, CN, (1-4С)алкокси, гидрокси(1-4С)алкил, (1-3С алкокси)(1-4С)алкил, (1-4С алкил)ОС(=O)-, (1-6С)алкилтио или фенил, необязательно замещенный одной или более группами, независимо выбранными из галогена, (1-6С)алкила и (1-6С)алкокси; или
R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное карбоциклическое кольцо, необязательно замещенное одним или более заместителями, независимо выбранными из (1-6С)алкила, или
R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное гетероциклическое кольцо, имеющее кольцевой гетероатом, выбранный из N, О или S, причем указанный кольцевой атом азота необязательно замещен (1-6С алкил)С(=O)O- или (1-6)ацилом, и указанный кольцевой атом серы необязательно окислен до S(=O) или SO2.
Следует понимать, что при определении последовательности присоединения заместителя к структуре, в случаях использования двух или более радикалов, первый названный радикал считается концевым, а последний названный радикал считается присоединенным к той структуре, о которой идет речь. Так, например, радикал «алкоксиалкил» присоединен к рассматриваемой структуре алкиловой группой.
Термины «(1-6С)алкил», «(1-4С)алкил» и «(1-3С)алкил», используемые в настоящем документе, относятся к насыщенным линейным одновалентным углеводородным радикалам, содержащим от одного до шести углеродных атомов, от одного до четырех углеродных атомов и от одного до трех углеродных атомов, соответственно, или к разветвленным насыщенным одновалентным углеводородным радикалам, содержащим от трех до шести углеродных атомов, от трех до четырех углеродных атомов или три углеродных атома, соответственно. Примеры включают, но не ограничиваясь этим, метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-метил-1 -пропил, 2-бутил, 2-метил-2-пропил, 2,2-диметилпропил, 1-пентил, 2-пентил, 3-пентил, 2-метил-2-бутил, 3-метил-2-бутил, 3-метил-1-бутил, 2-метил-1-бутил, 1-гексил, 2-гексил, 3-гексил, 2-метил-2-пентил, 3-метил-2-пентил, 4-метил-2-пентил, 3-метил-3-пентил, 2-метил-3-пентил, 2,3-диметил-2-бутил и 3,3-диметил-2-бутил.
«(1-4С)Алкокси», «(1-3С)алкокси», «(1-6С)алкокси» и «(2-6С)алкокси» относятся к радикалу -OR, в котором R представляет собой (1-4С)алкил, (1-3С)алкил, (1-6С)алкил или (2-6С)алкил, соответственно, как описано выше. Примеры включают метокси, этокси и тому подобные.
«(1-6С)Ацил» обозначает радикал RC(=O)-, в котором R представляет собой линейный насыщенный одновалентный углеводородный радикал, содержащий от одного до пяти углеродных атомов, или разветвленный насыщенный одновалентный углеводородный радикал, содержащий от трех до пяти углеродных атомов, например, метилкарбонил и тому подобные.
«(1-3С Алкокси)(1-6С)алкил» и «(1-3С алкокси)(1-4С)алкил» обозначает линейный насыщенный углеводородный радикал, содержащий от одного до шести углеродных атомов или от одного до четырех углеродных атомов, или разветвленный насыщенный одновалентный углеродный радикал, содержащий от трех до шести углеродных атомов или от трех до четырех углеродных атомов, соответственно, причем один из углеродных атомов замещен одной (1-3С)алкокси-группой, как описано в настоящем документе.
«(1-3С Алкокси)(1-6С)алкокси» обозначает (1-6С)алкокси-группу, как описано в настоящем документе, в которой один из углеродных атомов замещен (1-3С)алкокси-группой, как описано в настоящем документе. Примеры включают метоксиметокси, метоксиэтокси и тому подобные.
«(1-3С Алкокси)аминокарбонил» обозначает (1-3С алкил)-O-NH-С(=O)-группу.
«(1-6С)Алкоксикарбонил» и «(1-4С)алкоксикарбонил» обозначает (1-6С)-O-С(=O)-и (1-4С)-O-С(=O)- группу, соответственно.
(1-4С Алкоксикарбонил)(1-6С)алкокси обозначает (1-6С)алкиловую группу, как описано в настоящем документе, в которой один из углеродных атомов замещен (1-4С алкокси)карбонильной группой, как описано в настоящем документе.
«(1-3С Алкокси)трифтор(1-6С)алкил» обозначает (1-6С)алкиловую группу, как описано в настоящем документе, в которой один из углеродных атомов замещен тремя атомами фтора, а другой углеродный атом замещен (1-3С)алкокси-группой, как описано в настоящем документе.
«(1-4С Алкоксикарбонил)(1-6С алкокси)» обозначает (1-6С) алкокси-группу, как описано в настоящем документе, в которой один из углеродных атомов замещен (1-4С) алкоксикарбонильной группой, то есть алкил-O-С(=O)- группой.
«(1-4С Алкоксикарбонил)(1-3С алкокси)(1-6С)алкил» обозначает (1-3С алкокси)(1-6С)алкиловую группу, как описано в настоящем документе, в которой один из углеродных атомов замещен (1-4С) алкоксикарбонильной группой, то есть алкил-O-С(=O)- группой.
«(1-3 Алкокси)гидроксикарбонилалкил» обозначает гидроксикарбонилалкиловую группу, как описано в настоящем документе, в которой один из углеродных атомов замещен одной (1-3С алкокси) группой.
«Амино» обозначает группу -NRR', в которой R и R' независимо выбраны из водорода или (1-3С)алкила, как описано в настоящем документе. Примеры включают H2N-, CH3NH-, (CH3)2N и тому подобные.
«Амино(1-6С)алкил» обозначает линейный насыщенный одновалентный углеводородный радикал, содержащий от одного до шести углеродных атомов, или разветвленный насыщенный одновалентный углеводородный радикал, содержащий от трех до шести углеродных атомов, в котором один из углеродных атомов замещен одной группой -NRR', причем R и R' независимо выбраны из водорода или (1-3С)алкила, как описано в настоящем документе. Примеры включают аминометил, метиламиноэтил, 2-этиламино-2-метилэтил и тому подобные.
«Амино(2-6С)алкокси» обозначает (2-6С)алкокси-группу, как описано в настоящем документе, причем один из углеродных атомов замещен одной группой -NRR', при этом R и R' независимо выбраны из водорода или (1-3С)алкила, как описано в настоящем документе.
«Аминокарбонил» обозначает радикал RR'NCO-, в котором R и R' независимо выбраны из водорода или (1-6С)алкила, как описано в настоящем документе. Примеры включают H2NCO-, диметиламинокарбонил и тому подобные.
«Аминокарбонил(1-6С)алкил» обозначает линейный насыщенный углеводородный радикал, содержащий от одного до шести углеродных атомов, или разветвленный насыщенный одновалентный углеводородный радикал, содержащий от трех до шести углеродных атомов, причем один из углеродных атомов замещен одной аминокарбонильной группой, как описано в настоящем документе, например, 2-аминокарбонилэтил, 1-, 2- или 3-диметиламинокарбонилпропил, и тому подобные.
«Аминокарбонил(1-6С)алкокси» обозначает (1-6С)алкокси, как описано в настоящем документе, причем один из углеродных атомов замещен одной аминокарбонильной группой, как описано в настоящем документе.
«(1-3С)Алкиламидо(1-6С)алкокси» обозначает (1-6С)алкокси, как описано в настоящем документе, причем один из углеродных атомов замещен одной алкиламино-группой, то есть замещен (1-3C)C(=O)NH- группой.
«(1-4С алкил)карбокси» обозначает группу R'-C(=O)O-, причем R' представляет собой (1-4С)алкил.
«(1-4С алкилсилокси)(1-6С)алкокси» обозначает (1-6С)алкокси-группу, как описано в настоящем документе, в которой один из углеродных атомов замещен (1-4С алкил)силокси-группой, например, (1-4С алкил)Si-O- группой, такой как трет-бутилсилокси-группой.
«(1-3С)Алкилсульфонамидо» обозначает радикал (1-3C)алкилSO2NH-, причем (1-3С)алкил является таким, как описано в настоящем документе.
«(1-3С Алкилсульфонамидо)(1-6С)алкил» обозначает линейный насыщенный углеводородный радикал, содержащий от одного до шести углеродных атомов, или разветвленный насыщенный одновалентный углеводородный радикал, содержащий от трех до шести углеродных атомов, замещенный одной (1-3С)алкилсульфонамидо-группой, как описано в настоящем документе.
«(1-3С)Алкилсульфонамидо(1-6С)алкокси» обозначает (1-6С)алкокси-группу, как описано в настоящем документе, в которой один из углеродных атомов замещен одной (1-3С)алкилсульфонамидо-группой, как описано в настоящем документе.
«(1-3С)Алкилсульфонил» обозначает радикал -SO2R, в котором R представляет собой (1-3С)алкил, как описано выше, например, метилсульфонил и тому подобные.
«(1-3С Алкилсульфонил)(1-6С)алкокси» обозначает (1-6С)алкокси-группу, как описано в настоящем документе, в которой один из углеродных атомов замещен (1-3С)алкилсульфониловой группой.
«Гидроксикарбонил» обозначает НОС(=O)-.
«(1-4С алкил)карбокси(1-6С)алкил» обозначает (1-6С)алкиловую группу, как описано в настоящем документе, в которой один из углеродных атомов замещен (1-4С алкил)карбокси-группой, как описано в настоящем документе.
«Циано(1-6С)алкил» обозначает линейный насыщенный углеводородный радикал, содержащий от одного до шести углеродных атомов, или разветвленный насыщенный одновалентный углеводородный радикал, содержащий от трех до шести углеродных атомов, замещенный циано (CN) группой.
«(3-6С)Циклоалкил» обозначает циклический насыщенный одновалентный углеводородный радикал, содержащий углеродные атомы, например, циклопропил, циклобутил, циклопентил или циклогексил.
«Ди(1-3С алкокси)(1-6С)алкил» обозначает (1-6С)алкиловую группу, как описано в настоящем документе, в которой два углеродных атома, каждый замещен (1-3С)алкокси-группой, как описано в настоящем документе.
«Дигидрокси(2-6)алкил» обозначает линейный насыщенный углеводородный радикал, содержащий от двух до шести углеродных атомов, или разветвленный насыщенный одновалентный углеводородный радикал, содержащий от трех до шести углеродных атомов, замещенный двумя гидрокси (ОН) группами, при условии, что две гидрокси-группы не находятся у одного атома углерода.
«Дигидрокси(2-6С)алкокси» обозначает (2-6С)алкокси-группу, как описано в настоящем документе, в которой два углеродных атома замещены гидрокси-группой.
«Галоген», используемый в настоящем документе, обозначает F, Cl, Br или I.
«Гетероцикл» относится к насыщенной или частично ненасыщенной кольцевой системе, имеющей один или более кольцевых гетероатомов, упомянутых для определенной гетероциклической группы, причем указанный гетероцикл необязательно замещен заместителями, определенными для этой конкретной гетероциклической группы.
«Гетероарил» относится к 5-6-членной ненасыщенной кольцевой системе, имеющей один или более кольцевых гетероатомов, упомянутых для определенной гетероариловой группы, причем указанный гетероарил необязательно замещен заместителями, определенными для этой конкретной гетероариловой группы.
«гетСус2С(=O)(1-6С)алкокси» обозначает (1-6С)алкокси, как описано в настоящем документе, причем один из углеродных атомов замещен гетСус2C(=O) группой, и при этом гетСус2 является таким, как описано в настоящем документе.
«Гидрокси(1-6)алкил» и «гидрокси(1-4С)алкил» обозначает линейный насыщенный углеводородный радикал, содержащий от одного до шести углеродных атомов или от одного до четырех углеродных атомов, соответственно, или разветвленный насыщенный одновалентный углеводородный радикал, содержащий от трех до шести углеродных атомов или от трех до четырех углеродных атомов, соответственно, причем один из углеродных атомов замещен гидрокси (ОН) группой.
«Гидрокси(1-6С)алкокси» обозначает (1-6С)алкокси-группу, как описано в настоящем документе, в которой один из углеродных атомов замещен гидрокси-группой.
«Гидрокси(1-3Салкокси)(1-6С)алкил» обозначает (1-3С алкокси)(1-6С)алкиловую группу, как описано в настоящем документе, в которой один из углеродных атомов замещен гидрокси-группой.
«Гидрокси(1-3С алкокси)(1-6С)алкокси» обозначает (1-3С алкокси)(1-6С)алкокси, как описано в настоящем документе, причем один из углеродных атомов замещен гидрокси-группой.
«Гидроксидифтор(1-6С)алкил» обозначает дифтор(1-6С)алкиловую группу, как описано в настоящем документе, в которой один из углеродных атомов замещен гидрокси-группой.
«Гидрокситрифтор(1-6С)алкокси» обозначает трифтор(1-6С)алкокси-группу, как описано в настоящем документе, в которой один из углеродных атомов замещен гидрокси-группой.
«Гидроксикарбонилалкил» обозначает линейный насыщенный углеводородный радикал, содержащий от одного до шести углеродных атомов, или разветвленный насыщенный одновалентный углеводородный радикал, содержащий от трех до шести углеродных атомов, замещенный одной -COOH группой. Примеры включают 2-гидроксикарбонилэтил, 1-, 2- или 3-гидроксикарбонилпропил и тому подобные.
«Изоиндолин-1,3-дионил(1-6С)алкокси» обозначает (1-6С)алкокси-группу, как описано в настоящем документе, в которой один из углеродных атомов замещен изоиндолин-1,3-диониловой группой.
«Монофтор(1-6С)алкил», «дифтор(1-6С)алкил» и «трифтор(1-6С)алкил» относится к (1-6С)алкиловой группе, как описано в настоящем документе, в которой от одного до трех атомов водорода, соответственно, замещены группами фтора.
«Тетрафтор(2-6С)алкил» и «пентафтор(2-6С)алкил» относится к линейному насыщенному одновалентному углеводородному радикалу, содержащему от двух до шести углеродных атомов, или разветвленному насыщенному одновалентному углеводородному радикалу, содержащему от трех до шести углеродных атомов, причем от четырех до пяти водородных атомов, соответственно, замещены группами фтора.
«(Трифторметокси)(1-6С)алкил» обозначает линейный насыщенный углеводородный радикал, содержащий от одного до шести углеродных атомов, или разветвленный насыщенный одновалентный углеводородный радикал, содержащий от трех до шести углеродных атомов, замещенный одной CF3O-группой.
«Трифтор(1-3С алкил)амидо» обозначает (1-3С алкил)С(=O)NH-группу, в которой один из атомов углерода замещен тремя атомами фтора.
«Трифтор(1-6С)алкокси» обозначает (1-6С)алкокси-группу, как описано в настоящем документе, в которой один из углеродных атомов замещен тремя атомами фтора.
«Сульфамидо(1-6С)алкил» обозначает линейный насыщенный углеводородный радикал, содержащий от одного до шести углеродных атомов, или разветвленный насыщенный одновалентный углеводородный радикал, содержащий от трех до шести углеродных атомов, замещенный одной сульфамидо (H2NSO2NH-) группой.
Следует отметить, что соединения настоящего изобретения могут содержать группы, которые могут существовать в таутомерных формах, такие как гетероатом-замещенные гетероариловые или гетероциклические группы и тому подобные, которые иллюстрируются следующими общими и частными примерами:
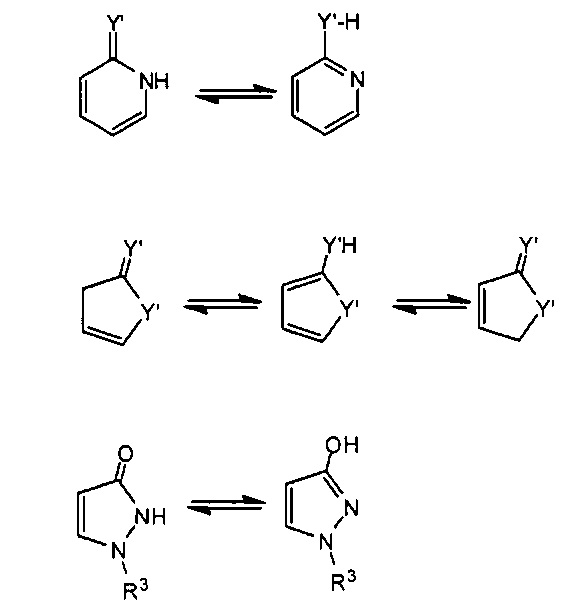
где Y'=О, S или NR, и хотя в настоящем документе названа, описана, изображена и/или заявлена одна форма, предполагается, что все таутомерные формы, по сути, включены в это название, описание, изображение и/или заявление.
В одном варианте реализации формулы I, Ra, Rb, Rc и Rd независимо выбраны из Н и метила. В одном варианте реализации, Ra, Rb, Rc и Rd представляют собой водород. В одном варианте реализации, Ra представляет собой метил, a Rb, Rc и Rd представляют собой водород. В одном варианте реализации, Ra и Rb представляют собой метил, a Rc и Rd представляют собой водород. В одном варианте реализации, Ra, Rb, Rc представляют собой водород, a Rd представляет собой метил. В одном варианте реализации, Ra и Rb представляют собой водород, a Rc и Rd представляют собой метил.
В одном варианте реализации, X представляет собой О.
В одном варианте реализации, X представляет собой S.
В одном варианте реализации, X представляет собой NH.
В одном варианте реализации, R1 представляет собой (1-3С алкокси)(1-6С)алкил, например, метоксиэтил, метоксипропил, этоксиэтил и 2-метоксипропил. Конкретные примеры включают метоксиэтил и 2-метоксипропил, имеющие структуры:

В одном варианте реализации, R1 представляет собой (трифторметокси)(1-6С)алкил, например, трифторметоксиэтил, трифторметоксипропил и тому подобные. Конкретный пример представляет собой трифторметоксиэтил.
В одном варианте реализации, R1 представляет собой (1-3С сульфанил)(1-6С)алкил, например, метилсульфанилэтил, этилсульфанилэтил и тому подобные. Конкретный пример представляет собой метилсульфанилэтил.
В одном варианте реализации, R1 представляет собой монофтор(1-6С)алкил, дифтор(1-6С)алкил или трифтор(1-6С)алкил. Конкретные примеры включают 1,3-дифторпроп-2-ил, 2,2-дифторэтил, 2,2,2-трифторэтил и 2,2,2-трифторпропил, имеющие структуры:
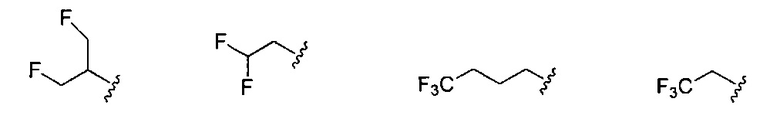 .
.
В одном варианте реализации, R1 представляет собой тетрафтор(2-6С)алкил или пентафтор(2-6С)алкил. Конкретный пример представляет собой 3,3,4,4,4-пентафторбутил.
В одном варианте реализации, R1 представляет собой циано(1-6С)алкил. Конкретный пример представляет собой 2-цианоэтил.
В одном варианте реализации, R1 представляет собой аминокарбонил(1-6С)алкил. Конкретный пример представляет собой аминокарбонилметил. Другой пример представляет собой метиламинокарбонилметил, имеющий формулу MeNHC(=O)CH2-.
В одном варианте реализации, R1 представляет собой гидрокси(1-6С)алкил. Конкретный пример представляет собой 2-гидроксиэтил. Другой пример представляет собой 2-гидроксипропил.
В одном варианте реализации, R1 представляет собой дигидрокси(2-6С)алкил. Конкретный пример представляет собой структуру:
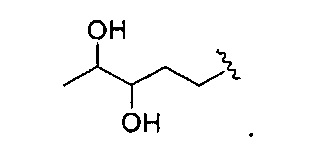
В одном варианте реализации, R1 представляет собой (1-6С)алкил. Примеры включают метил, этил и пропил.
В одном варианте реализации, R1 представляет собой (1-3Салкиламино)(1-3С)алкил, то есть (1-3С)алкиловую группу, которая замещена (1-3С алкил)аминогруппой, например, (1-3Салкил)NH-группой, такой как метиламино. Конкретный пример представляет собой (2-метиламино)этил.
В одном варианте реализации, R1 представляет собой (1-4С алкоксикарбонил)(1-6С)алкил. Конкретный пример представляет собой метоксикарбонилметил, имеющий структуру:
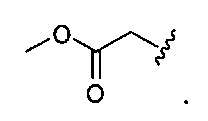
В одном варианте реализации, R1 представляет собой амино(1-6С)алкил, такой как метиламино(1-6С)алкил. Конкретный пример представляет собой 2-метиламиноэтил.
В одном варианте реализации, R1 представляет собой гидрокси(1-3С алкокси)(1-6С)алкил. Примеры включают гидроксиметокси(1-6С)алкил. Конкретные примеры включают структуры:
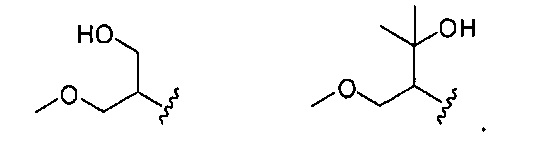
В одном варианте реализации, R1 представляет собой ди(1-3С алкокси)(1-6С)алкил. Примеры включают диметокси(1-6С)алкил. Конкретный пример включает 1,3-диметоксипроп-2-ил, имеющий структуру:
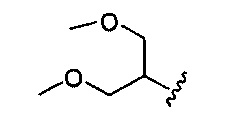 .
.
В одном варианте реализации, R1 представляет собой (1-3С алкокси)трифтор(1-6С)алкил. Примеры включают метокситрифтор(1-6С)алкил. Конкретный пример включает 3,3,3 -трифтор-2-метоксипропил.
В одном варианте реализации, R1 представляет собой гидрокситрифтор(1-6С)алкил. Конкретный пример включает 3,3,3-трифтор-2-гидроксипропил.
В одном варианте реализации, R1 представляет собой (1-4С алкоксикарбонил)(1-3С алкокси)(1-6С)алкил. Примеры включают (метоксикарбонил)метокси(1-6С)алкил. Конкретный пример включает структуру:
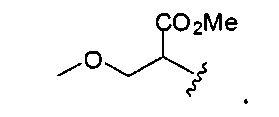
В одном варианте реализации, R1 представляет собой гидроксикарбонил(1-3С алкокси)(1-6С)алкил. Примеры включают (метоксикарбонил)гидрокси(1-6С)алкил. Конкретный пример включает структуру:
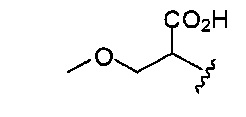 .
.
В одном варианте реализации, R1 представляет собой гетAr5(СН2)0-1.
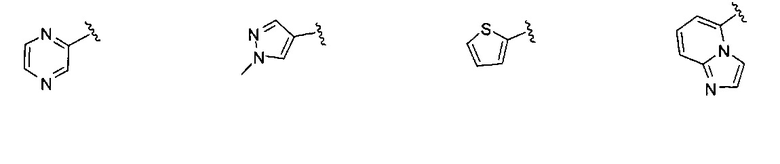
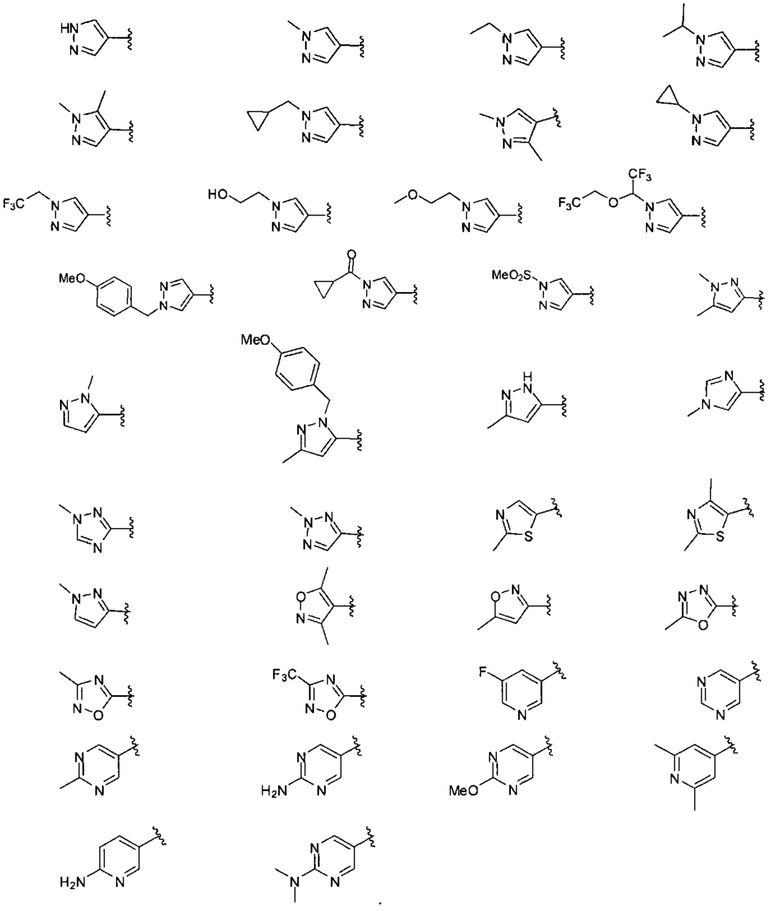
В одном варианте реализации, R1 представляет собой гетAr5, причем гетAr5 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, О или S, при этом указанное кольцо необязательно замещено одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила, (1-6С)алкокси и CF3. Примеры включают пирролиловые, пиразолиловые, имидазолиловые, фураниловые, изоксазолиловые, оксазолиловые, тиофениловые, тиазолиловые, тиадиазолиловые, триазолиловые, тиадиазолиловые, пиридиловые, пиримидиловые и пиразиниловые кольца, причем указанное кольцо необязательно замещено одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила, (1-6С)алкокси и CF3.
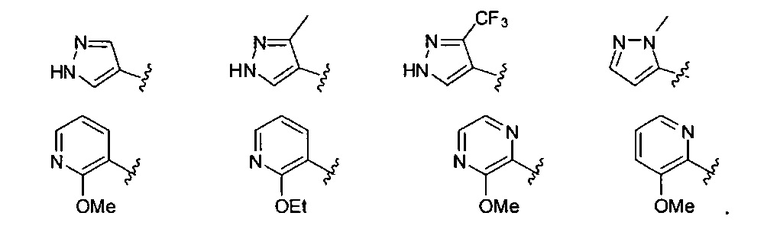
В одном варианте реализации, R1 представляет собой гетAr5, причем гетAr5 представляет собой пиразолил, пиридил или пиразинил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила, (1-6С)алкокси и CF3. В одном варианте реализации, гетAr5 замещен одним из указанных заместителей. В одном варианте реализации, R1 представляет собой пиразолил, пиридил или пиразинил, необязательно замещенный метилом, трифторметилом, метокси или этокси.
Конкретные примеры R1, представленного гетAr5, включают структуры:
В одном варианте реализации, R1 представляет собой гетAr5(СН2)-, причем гетAr5 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, О или S, при этом указанное кольцо необязательно замещено одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила, (1-6С)алкокси и CF3. Примеры включают пирролиловые, пиразолиловые, имидазолиловые, фураниловые, изоксазолиловые, оксазолиловые, тиофениловые, тиазолиловые, тиадиазолиловые, триазолиловые, тиадиазолиловые, пиридиловые, пиримидиловые и пиразиниловые кольца, причем указанное кольцо необязательно замещено одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила, (1-6С)алкокси и CF3.
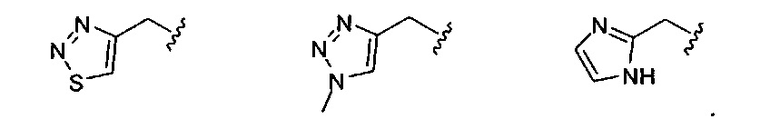
В одном варианте реализации, R1 представляет собой гетAr5(СН2)-, причем гетAr5 представляет собой имидазолил, тиадиазолил или триазолил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила, (1-6С)алкокси и CF3. В одном варианте реализации, гетAr5 замещен одним или более заместителями, независимо выбранными из метила, метокси, этокси и трифторметила. В одном варианте реализации, гетAr5 замещен одним из указанных заместителей.
Конкретные примеры R1, представленного гетAr5(СН2)-, включают структуры:
В одном варианте реализации, R1 представляет собой Ar5(CH2)0-1.
В одном варианте реализации, R1 представляет собой Ar5, причем Ar5 представляет собой фенил, необязательно замещенный одной или более заместителями, независимо выбранными из галогена, (1-6С)алкила, (1-6С)алкокси, CF3O-, (1-4С)алкоксикарбонила и аминокарбонила. В одном варианте реализации, Ar5 представляет собой фенил, необязательно замещенный одним или более заместителями, независимо выбранными из F, О, метила, метокси, трифторметокси и СН3С(=O)O-.
Примеры R1, представленного Ar5, включают фенил, 2-метоксифенил, 2-фторфенил, 2-метилфенил, 2-хлорфенил, 2-трифторметоксифенил, 2-(метоксикарбонил)фенил, 4-фторфенил и 2,6-дифторфенил.
В одном варианте реализации, R1 выбран из (1-3С алкокси)(1-6С)алкила, дифтор(1-6С)алкила и трифтор(1-6С)алкила.
В одном варианте реализации, R2 представляет собой Н.
В одном варианте реализации, R2 представляет собой F.
В одном варианте реализации, R2 представляет собой ОН.
В одном варианте реализации, группа Y Формулы I, соединяющая пирролидиниловое кольцо и группу B, представляет собой связь.
В одном варианте реализации, группа Y Формулы I, соединяющая пирролидиниловое кольцо и группу B, представляет собой -O-.
В одном варианте реализации, группа Y Формулы I, соединяющая пирролидиниловое кольцо и группу B, представляет собой -OCH2-, причем кислород группы Y связан с пирролидиниловым кольцом.
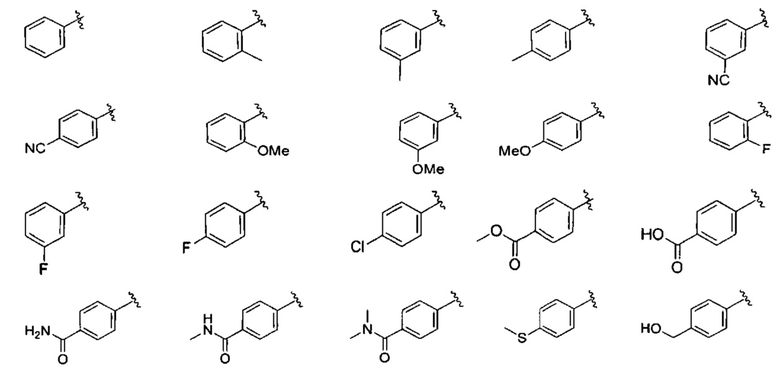
В одном варианте реализации, В представлен кольцом В, причем кольцо В представляет собой Ar1, а Ar1 представляет собой фенил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена, CF3, CF3O-, (1-4С)алкокси, гидрокси(1-4С)алкила, (1-6С)алкила и CN. Примеры включают фенил, фторфенил, дифторфенил, трифторфенил, хлорфенил, трифторметилфенил, метоксифенил, цианофенил, хлорфторфенил, цианофторфенил и хлорцианофенил.
Конкретные примеры кольца B, представленного Ar1, включают фенил, 2-фторфенил, 3-фторфенил, 4-фторфенил, 3,4-дифторфенил, 3,5-дифторфенил, 2,4-дифторфенил, 2,5-дифторфенил, 3,4,5-трифторфенил, 2-хлорфенил, 3-хлорфенил, 4-хлорфенил, 3-трифторметилфенил, 3-метоксифенил, 3-хлор-4-фторфенил, 4-хлор-3-фторфенил, 3-хлор-5-фторфенил, 3-циано-5-фторфенил, 2-цианофенил, 4-цианофенил и 3-циано-4-фторфенил.
В одном варианте реализации, В представлен кольцом В, причем кольцо В представляет собой Ar1, а Ar1 представляет собой фенил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена, CF3, CF3O-, (1-4С)алкокси, гидрокси(1-4С)алкила и (1-6С)алкила. Примеры включают фенил, фторфенил, дифторфенил, трифторфенил, хлорфенил, трифторметилфенил и метоксифенил. Конкретные примеры Кольца B, представленного Ar1, включают фенил, 2-фторфенил, 3-фторфенил, 4-фторфенил, 3,4-дифторфенил, 3,5-дифторфенил, 2,4-дифторфенил, 2,5-дифторфенил, 3,4,5-трифторфенил, 2-хлорфенил, 3-хлорфенил, 4-хлорфенил, 3-трифторметилфенил и 3-метоксифенил.
В одном варианте реализации, В представлен кольцом B, причем кольцо B представляет собой Ar1, а Ar1 представляет собой фенил, необязательно замещенный одним или более галогенами.
В одном варианте реализации, В представлен кольцом B, причем кольцо B представляет собой Ar1, как описано для Формулы I, a Y представляет собой связь.
В одном варианте реализации, В представлен кольцом B, причем кольцо B представляет собой Ar1, как описано для Формулы I, a Y представляет собой -O-. Конкретные примеры включают группы -Y-B, имеющие структуры:
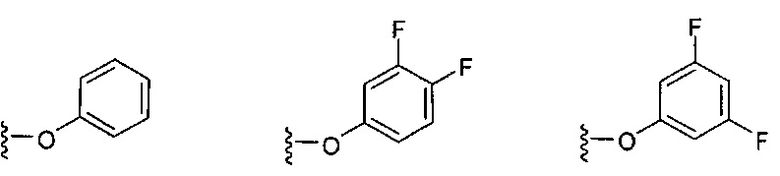 .
.
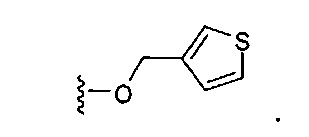
В одном варианте реализации, В представлен кольцом B, причем кольцо B представляет собой Ar1, как описано для Формулы I, a Y представляет собой -OCH2-. Конкретный пример включает группу -Y-B, имеющую структуру:
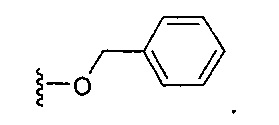
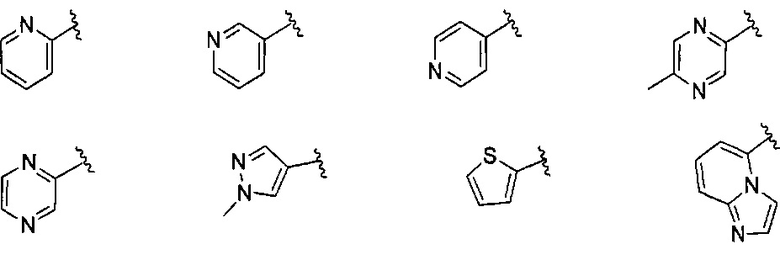
В одном варианте реализации, В представлен кольцом В, причем кольцо В представляет собой гетAr1, а гетAr1 представляет собой 5-6-членный гетероарил, имеющий 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенный 1-2 группами, независимо выбранными из (1-6С)алкила, галогена, ОН, CF3, NH2 и гидрокси(1-2С)алкила. В одном варианте реализации, кольцо B представляет собой гетAr1, причем гетAr1 представляет собой 5-6-членный гетероарил, имеющий 1-2 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенный 1-2 группами, независимо выбранными из (1-6С)алкила, галогена, ОН, CF3, NH2 и гидрокси(1-2С)алкила. Примеры Кольца В включают пиридиловое, тиофениловое, тиазолиловое, оксазолиловое и изоксазолиловое кольцо, необязательно замещенное 1-2 группами, независимо выбранными из (1-6С)алкила, галогена, ОН, CF3, NH2 и гидрокси(1-2С)алкила. В одном варианте реализации, кольцо В представляет собой пиридиловое, тиофениловое, тиазолиловое, оксазолиловое или изоксазолиловое кольцо, необязательно замещенное 1-2 группами, независимо выбранными из галогена и (1-6С)алкила.
Примеры кольца B, представленного гетAr1, включают пирид-4-ил, пиридн-3-ил, пирид-2-ил, 5-фторпирид-3-ил, тиен-2-ил, тиазол-2-ил, 2,4-диметилтиазол-5-ил, оксазол-5-ил, изоксазол-5-ил, тиен-2-ил, 5-хлорпирид-3-ил, 5-фторпирид-2-ил, 3-фторпирид-4-ил, 1-метилпиразол-4-ил, имеющие структуры:
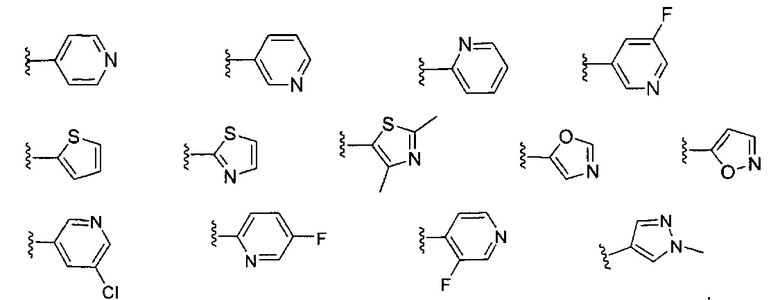
В некоторых вариантах реализации, примеры кольца B, представленного гетAr1, включают пирид-4-ил, пирид-3-ил, пирид-2-ил, 5-фторпирид-3-ил, тиен-2-ил, тиазол-2-ил, 2,4-диметилтиазол-5-ил, оксазол-5-ил и изоксазол-5-ил, имеющие структуры:
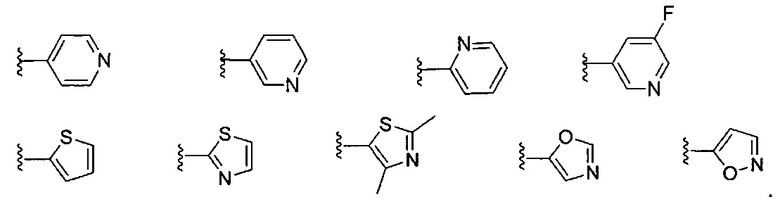
В одном варианте реализации, кольцо В представляет собой пиридиловое кольцо, необязательно замещенное 1-2 группами, независимо выбранными из (1-6С)алкила и галогена.
В одном варианте реализации, В представлен кольцом B, причем кольцо B представляет собой гетAr1, как описано для Формулы I, a Y представляет собой связь.
В одном варианте реализации, В представлен кольцом B, причем кольцо B представляет собой гетAr1, как описано для Формулы I, a Y представляет собой -O-. Конкретный пример группы -Y-B представляет собой структуру:
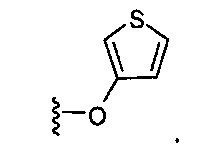
В одном варианте реализации, В представлен кольцом B, причем кольцо B представляет собой гетAr1, как описано для Формулы I, a Y представляет собой -OCH2-. Конкретный пример группы -Y-B представляет собой структуру:
В одном варианте реализации, В представляет собой (1-6С)алкил. Примеры включают метил и этил, изопропил.
В одном варианте реализации, В представляет собой (1-6С)алкокси. Пример представляет собой изопропокси.
Далее будет сделана ссылка на кольцо С.
В одном варианте реализации, кольцо C представляет собой формулу С-1:
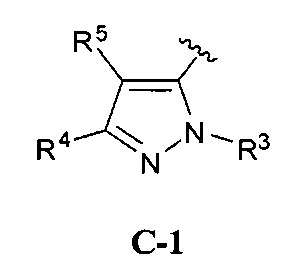
где R3, R4 и R5 являются такими, как описано для Формулы I.
В одном варианте реализации, R3 представляет собой Н.
В одном варианте реализации, R представляет собой (1-6С)алкил. Примеры R3 включают метил или этил.
В одном варианте реализации, R представляет собой гидрокси(1-6С)алкил. Пример R3 представляет собой 2-гидроксиэтил.
В одном варианте реализации, R3 представляет собой Ar2, причем Ar2 представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из галогена, (1-6С)алкила и гидроксиметила. Примеры включают фенил, фторфенил, метилфенил и гидроксиметилфенил.
Примеры R3, представленного Ar2, включают фенил, 2-фторфенил, 3-фторфенил, 4-фторфенил, 2-метилфенил, 3-метилфенил, 4-метилфенил, 3-(гидроксиметил)фенил, 3-хлорфенил, 3-хлор-4-фторфенили 3-хлор-3-фторфенил. Конкретные примеры R3, представленного Ar2, включают фенил, 2-фторфенил, 3-фторфенил, 4-фторфенил, 2-метилфенил, 3-метилфенил, 4-метилфенил и 3-(гидроксиметил)фенил.
В одном варианте реализации, R3 представляет собой гетСус1, причем гетСус1 представляет собой 5-6-членное насыщенное или частично ненасыщенное гетероциклическое кольцо, имеющее 1-2 кольцевых гетероатома, независимо выбранных из N и О. В одном варианте реализации, R3 представляет собой пирролидиниловое, тетрагидрофураниловое, имидазолидиниловое, пиперидиниловое, пиперазиниловое, тетрагидропираниловое или морфолиниловое кольцо. Пример R3 представляет собой тетрагидро-2Н-пиран-4-ил.
В одном варианте реализации, R3 представляет собой (3-7С)циклоалкил. В одном варианте реализации, R3 представляет собой циклогексил.
В одном варианте реализации, R3 представляет собой гетAr2, причем гетAr2 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, О и S, и необязательно замещенное одним или более заместителями, независимо выбранными из (1-6С)алкила и галогена. В одном варианте реализации, R3 представляет собой тиенил, фурил, имидазолил, пиразолил, тиазолил, изотиазолил, оксазолил, изоксазолил, триазолил, тиадиазолил, оксадиазолил, пиридил, пиримидил, пиразинил или пиридазинил, необязательно замещенный одним или более заместителями, независимо выбранными из (1-6С)алкила и галогена. В одном варианте реализации, R3 представляет собой пиразолил, пиридил или пиридазинил, необязательно замещенный одной или более группами, независимо выбранными из (1-6С)алкила и галогена. В одном варианте реализации, R3 представляет собой пиразолил, пиридил или пиридазинил, необязательно замещенный (1-6С)алкилом или галогеном. Примеры R3, представленного гетAr2, включают 1-метил-1Н-пиразол-4-ил, пирид-2-ил, пирид-3-ил, пирид-4-ил, пиридазинил и 3-хлорпирид-5-ил.
В одном варианте реализации, R3 представляет собой гетAr2, причем гетAr2 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, О и S, и необязательно замещенное (1-6С)алкилом. В одном варианте реализации, R3 представляет собой тиенил, фурил, имидазолил, пиразолил, тиазолил, изотиазолил, оксазолил, изоксазолил, триазолил, тиадиазолил, оксадиазолил, пиридил, пиримидил, пиразинил или пиридазинил, необязательно замещенный (1-6С)алкилом. В одном варианте реализации, R3 представляет собой пиразолил, пиридил или пиридазинил, необязательно замещенный (1-6С)алкилом. Примеры R3 включают 1-метил-1Н-пиразол-4-ил, пирид-2-ил, пирид-3-ил, пирид-4-ил и пиридазинил.
В одном варианте реализации, R3 выбран из Н, Ar2, гетAr2 и (1-6С)алкила.
В одном варианте реализации, R3 выбран из Н, Ar2 и (1-6С)алкила.
В одном варианте реализации, R3 выбран из Ar2 и (1-6С)алкила.
В одном варианте реализации, R3 выбран из Ar2, гетAr2 и (1-6С)алкила.
В одном варианте реализации, R4 представляет собой Н.
В одном варианте реализации, R4 представляет собой ОН.
В одном варианте реализации, R4 представляет собой (1-6С)алкил. Примеры R4 включают метил, этил, изопропил и трет-бутил.
В одном варианте реализации, R4 представляет собой монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил или пентафтор(2-6С)алкил. Примеры R4 включают фторметил, 2-фторэтил, дифторметил и 2,2-дифторэтил, трифторметил, 2,2,2-трифторэтил, 3,3,3-трифторпропил, 2,2,3,3-тетрафторпропил и 2,2,3,3,3-пентафторпропил.
В одном варианте реализации, R4 представляет собой трифтор(1-6С)алкил. Пример R4 включает CF3.
В одном варианте реализации, R4 представляет собой циано(1-6С)алкил. Пример R4 включает цианометил и 2-цианопропан-2-ил.
В одном варианте реализации, R4 представляет собой гидрокси(1-6С)алкил. Примеры R4 включают гидроксиметил, 2-гидроксиэтил, 2-гидроксипропил, 2-гидрокси-2-метилпропил и 1-гидрокси-2-метилпропан-2-ил.
В одном варианте реализации, R4 представляет собой дигидрокси(2-6С)алкил. Пример R4 включает 2,3-дигидроксипропил.
В одном варианте реализации, R4 представляет собой (1-3С алкокси)(1-6С)алкил. Примеры R4 включают метоксиметил, 2-метоксиэтил и 3-метоксипропил.
В одном варианте реализации, R4 представляет собой амино(1-6С)алкил. Примеры R4 включают аминометил, 2-аминоэтил и 3-аминопропил.
В одном варианте реализации, R4 представляет собой аминокарбонил(1-6С)алкил. Примеры R4 включают аминокарбонилметил и 2-(аминокарбонил)этил.
В одном варианте реализации, R4 представляет собой (1-3С)алкилсульфонамидо(1-6С)алкил. Примеры включают CH3SO2NHCH2- и CH3SO2NHCH2CH2-.
В одном варианте реализации, R4 представляет собой гидроксикарбонил(1-6С)алкил. Примеры включают НОС(=O)СН2- и НОС(=O)СН2СН2-.
В одном варианте реализации, R4 представляет собой гетAr3(1-6С)алкил, причем гетAr3 представляет собой 5-членное гетероариловое кольцо, имеющее 1-3 кольцевых атома, независимо выбранных из N, О и S, и необязательно замещенное (1-6С)алкилом. В одном варианте реализации, гетAr3 представляет собой тиениловое, фуриловое, имидазолиловое, пиразолиловое, тиазолиловое, изотиазолиловое, оксазолиловое, изоксазолиловое, триазолиловое, тиадиазолиловое или оксадиазолиловое кольцо, необязательно замещенное (1-6С)алкилом. Примеры R4, представленного гетAr3(1-6С)алкилом, включают (1-метил-1Н-1,2,4-триазол-3-ил)метил и (5-метил-1,3,4-оксадиазол-2-ил)метил.
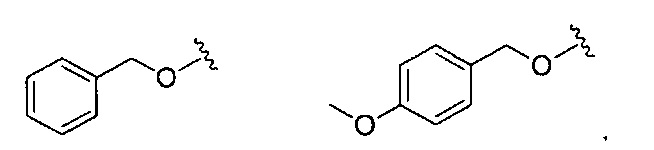
В одном варианте реализации, R4 представляет собой Ar3(1-6С)алкил, причем фенил необязательно замещен (1-4С)алкокси или гидрокси(1-4С)алкилом. В одном варианте реализации, Ar3 представляет собой фенил или 4-метоксифенил. Примеры R4, представленного Ar3(1-6С)алкилом, включают бензил и 4-метоксибензил.
В одном варианте реализации, R4 представляет собой (1-6С)алкокси. Примеры включают метокси и этокси.
В одном варианте реализации, R4 представляет собой монофтор(1-6С)алкокси, дифтор(1-6С)алкокси, трифтор(1-6С)алкокси, тетрафтор(2-6С)алкокси или пентафтор(2-6С)алкокси. Примеры R4 включают фторметокси, 2-фторэтокси, 2,2-дифторметокси, трифторметокси, 2,2,2-трифторэтокси и 2,2-дифторэтокси. В одном варианте реализации, R4 представляет собой 2-фторэтокси.
В одном варианте реализации, R4 представляет собой циано(1-6С)алкокси. Пример R4 включает цианометокси и 2-цианоэтокси.
В одном варианте реализации, R4 представляет собой гидрокси(1-6С)алкокси. Примеры R4 включают 2-гидрокси-2-метилпропокси, 2-гидроксиэтокси, 2-гидроксипропокси, 2-гидрокси-2-метилпропокси и 2-гидроксибутокси.
В одном варианте реализации, R4 представляет собой дигидрокси(2-6С)алкокси. Примеры R4 включают 2,3-дигидроксипропокси и 3-гидрокси-2-(гидроксиметил)пропокси.
В одном варианте реализации, R4 представляет собой амино(2-6С)алкокси. Пример представляет собой H2NCH2CH2O-.
В одном варианте реализации, R4 представляет собой аминокарбонил(1-6С)алкокси. Примеры включают H2NC(=O)CH2O- и H2NC(=O)CH2CH2O-.
В одном варианте реализации, R4 представляет собой гетСус2(1-6С)алкокси, причем гетСус2 представляет собой 4-6-членное гетероциклическое кольцо, имеющее 1-2 кольцевых гетероатома, независимо выбранных из N и О, и при этом гетСус2 необязательно замещен 1-2 группами, независимо выбранными из (1-6С)алкила, (1-4С алкокси)карбонила и (1-6С)ацила. Примеры гетСус2 включают оксетаниловое, тетрагидрофураниловое, тетрагидропираниловое, азетидиниловое, пирролидиниловое, пиперидиниловое, пиперазиниловое, морфолиниловое и 1,3-диоксоланиловое кольцо, необязательно замещенное 1-2 группами, независимо выбранными из (1-6С)алкила, (1-4С алкокси)карбонила и (1-6С)ацила. Примеры R4, представленного гетСус2(1-6С)алкокси, включают оксетан-2-илметокси, 2-(оксетан-2-ил)пропокси, (2,2-диметил-1,3-диоксолан-4-ил)метокси, (1,3-диоксолан-4-ил)метокси, 2-морфолиноэтокси, пиперазинилэтокси и пиперидинилэтокси группы, необязательно замещенные 1-2 группами, независимо выбранными из (1-6С)алкила, (1-4С алкокси)карбонила и (1-6С)ацила. Конкретные примеры включают структуры:
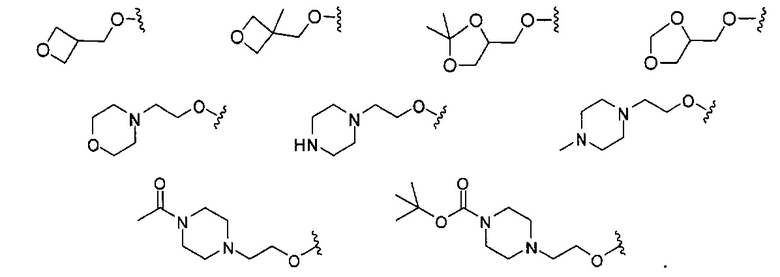
В одном варианте реализации, R4 представляет собой гетСус2(1-6С)алкокси, причем гетСус2 представляет собой 4-6-членное гетероциклическое кольцо, имеющее 1-2 кольцевых гетероатома, независимо выбранных из N и О, и при этом гетСус2 необязательно замещен 1-2 группами, независимо выбранными из (1-6С)алкила. Примеры гетероциклических колец включают оксетаниловое, тетрагидрофураниловое, тетрагидропираниловое, азетидиниловое, пирролидиниловое, пиперидиниловое, пиперазиниловое, морфолиниловое и 1,3-диоксоланиловое кольцо, необязательно замещенное 1-2 группами, независимо выбранными из (1-6С)алкила. Примеры R4, представленного гетСус2(1-6С)алкокси, включают оксетан-2-илметокси, 2-(оксетан-2-ил)пропокси, (2,2-диметил-1,3-диоксолан-4-ил)метокси, (1,3-диоксолан-4-ил)метокси и 2-морфолиноэтокси, пиперазинилэтокси-кольца, необязательно замещенные (1-6С)алкилом, такие как:
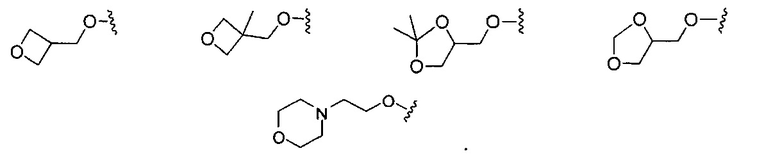
В одном варианте реализации, R4, представленный гетСус2(1-6С)алкокси, выбран из оксетан-2-илметокси, 2-(оксетан-2-ил)пропокси, (2,2-диметил-1,3-диоксолан-4-ил)метокси, (1,3-диоксолан-4-ил)метокси и 2-морфолиноэтокси.
В одном варианте реализации, R4 представляет собой гетAr3(1-6С)алкокси, причем гетAr3 представляет собой 5-членное гетероариловое кольцо, имеющее 1-3 кольцевых атома, независимо выбранных из N, О и S, и необязательно замещенное (1-6С)алкилом. В одном варианте реализации, гетAr3 представляет собой тиениловое, фуриловое, имидазолиловое, пиразолиловое, тиазолиловое, изотиазолиловое, оксазолиловое, изоксазолиловое, триазолиловое, тиадиазолиловое или оксадиазолиловое кольцо, необязательно замещенное (1-6С)алкилом. В одном варианте реализации, гетAr3 представляет собой триазолиловое или оксадиазолиловое кольцо, необязательно замещенное (1-6С)алкиловой группой, такой как метиловая группа. Примеры R4, представленного гетAr3(1-6С)алкокси, включают (1-метил-1Н-1,2,4-триазол-3-ил)метокси и (5-метил-1,3,4-оксадиазол-2-ил)метокси, которые могут быть представлены структурами:
 .
.
В одном варианте реализации, R4 представляет собой Ar3(1-6С)алкокси, причем Ar3 представляет собой фенил, необязательно замещенный (1-4С)алкокси. Примеры включают фенилметокси и (4-метоксифенил)метокси, имеющие структуры:
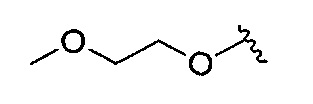
В одном варианте реализации, R4 представляет собой (1-4С алкокси)(1-6С)алкокси. Примеры включают (2-метокси)этокси, имеющий структуру:
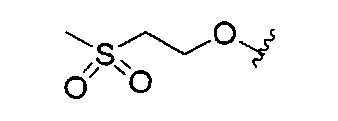
В одном варианте реализации, R4 представляет собой (1-3Салкилсульфонил)(1-6С)алкокси. Примеры включают (2-метилсульфонил)этокси, имеющий структуру:
В одном варианте реализации, R4 представляет собой (3-6С)циклоалкил, необязательно замещенный F, ОН, (1-6С алкил), (1-6С)алкокси или (1-3С алкокси)(1-6С)алкилом. Примеры включают циклопропил, циклобутил, циклопентил, циклогексил, 2-гидроксициклобутил. В одном варианте реализации, R4 представляет собой 2-гидроксициклобутил. В одном варианте реализации, R4 представляет собой циклопропил.
В одном варианте реализации, R4 представляет собой (3-6С)циклоалкил. Примеры включают циклопропил, циклобутил, циклопентил, циклогексил, 2-гидроксициклобутил. В одном варианте реализации, R4 представляет собой циклопропил.
В одном варианте реализации, R4 представляет собой гетAr4, причем гетAr4 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенное 1-2 заместителями, независимо выбранными из (1-6С)алкила, галогена, CN, гидрокси(1-6С)алкила, трифтор(1-6С)алкила, (3-6С)циклоалкила, (3-6С циклоалкил)СН2- (3-6С циклоалкил)С(=O)-, (1-3С алкокси)(1-6С)алкила, (1-6С)алкокси, (1-6С)алкилсульфонила, NH2, (1-6С алкил)амино, ди(1-6С алкил)амино, (1-3С трифторалкокси)(1-3С)трифторалкила и метоксибензила; или 9-10-членный бициклический гетероарил, имеющий 1-3 кольцевых атома азота.
Примеры включают пиридиловое, пиримидиниловое, пиридазиниловое, пиразолиловое, имидазолиловое, тиениловое, 1,2,4-триазолиловое, 1,2,3-триазолиловое, тиазолиловое, оксазолиловое, 1,3,4-оксадиазолиловое, 1,2,4-осадиазолиловое и имидазо[1,2-а]пиридиниловое кольцо, необязательно замещенное 1-2 заместителями, независимо выбранными из (1-6С)алкила, гидрокси(1-6С)алкила, трифтор(1-6С)алкила, (3-6С)циклоалкила, (3-6С циклоалкил)CH2-, (3-6С циклоалкил)С(=O)-, (1-3С алкокси)(1-6С)алкила, (1-6С)алкокси, (1-6С)алкилсульфонила, NH2, (1-6С алкил)амино, ди(1-6С алкил)амино, (1-3С трифторалкокси)(1-3С)трифторалкила и метоксибензила.
В одном варианте реализации, R4 представляет собой гетAr4, причем гетAr4 представляет собой пиридиловое, пиримидиниловое, пиридазиниловое, пиразолиловое, имидазолиловое, тиониловое, 1,2,4-триазолиловое, 1,2,3-триазолиловое, тиазолиловое, оксазолиловое, 1,3,4-оксадиазолиловое, 1,2,4-оксадиазолиловое или имидазо[1,2-а]пиридиниловое кольцо, необязательно замещено 1-2 заместителями, независимо выбранными из фтора, метила, этила, изопропила, циклопропилметила, циклопропила, трифторметила, 2,2,2-трифторэтила, метокси, H2N-, (CH3)2N-, 2-гидроксиэтила, 2-метоксиэтила, 1-(2,2,2-трифторэтокси)-2,2,2-трифторэтила, циклопропилкарбонила, метилсульфонила и 4-метоксибензила.
В одном варианте реализации, примеры R4, представленного гетAr4, включают структуры:

В одном варианте реализации, R4 представляет собой гетAr4, причем гетAr4 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенное 1-2 заместителями, независимо выбранными из (1-6С)алкила, или 9-10-членный бициклический гетероарил, имеющий 1-3 кольцевых атома азота. В одном варианте реализации, гетAr4 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-2 кольцевых гетероатома, независимо выбранных из N и S, и необязательно замещенное 1-2 заместителями, независимо выбранными из (1-6С)алкила, или 9-членный бициклический гетероарил, имеющий 1-2 кольцевых атома азота. Примеры включают пиридиловое, пиридазиниловое, пиразолиловое, тиениловое и имидазо[1,2-а]пиридиниловое кольцо, необязательно замещенное 1-2 заместителями, независимо выбранными из (1-6С)алкила.
В одном варианте реализации, примеры R4, представленного гетAr4, включают пиридин-2-ил, пиридин-3-ил, пиридин-4-ил, 5-метилпиридазин-2-ил, пиридазин-2-ил, 1-метилпиразол-4-ил, тиен-2-ил и имидазо[1,2-а]пиридин-5-ил, имеющие структуры:
 .
.
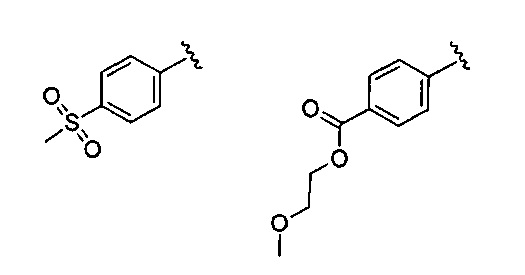
В одном варианте реализации R4 представляет собой Ar4, причем Ar4 представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из (1-6С)алкила, галогена, CN, CF3, CF3O-, (1-6С)алкокси, (1-6С алкил)ОС(=O)-, аминокарбонила, (1-6С)алкилтио, гидрокси(1-6С)алкила, (1-6С алкил)SO2-, НОС(=O)- и (1-3С алкокси)(1-3С алкил)ОС(=O)-. Примеры включают фенил, необязательно замещенный одной или более группами, независимо выбранными из метила, F, Cl, CN, метокси, CH3OC(=O)-, аминокарбонила, метиламинокарбонила, диметиламинокарбонила, метилтио, гидроксиметила, CH3SO2-, НОС(=O)- и CH3OCH2CH2OC(=O)-. В некоторых вариантах реализации, R4 представляет собой фенил, необязательно замещенный одним или двумя из указанных заместителей. Конкретные примеры включают структуры:
 .
.
В одном варианте реализации, R4 представляет собой гетСус2(O)CH2, причем гетСус2 представляет собой 4-6-членное гетероциклическое кольцо, имеющее 1-2 кольцевых гетероатома, независимо выбранных из N и О, и при этом гетСус2 необязательно замещен 1-2 группами, независимо выбранными из (1-6С)алкила, (1-4С алкокси)карбонила и (1-6С)ацила. Примеры гетСус2 включают оксетаниловое, тетрагидрофураниловое, тетрагидропираниловое, азетидиниловое, пирролидиниловое, пиперидиниловое, пиперазиниловое, морфолиниловое и 1,3-диоксоланиловое кольцо, необязательно замещенное 1-2 группами, независимо выбранными из (1-6С)алкила, (1-4С алкокси)карбонила и (1-6С)ацила. Примеры R4, представленного гетСус2(1-6С)алкокси, включают пиперазинилэтокси и пиперидинилэтокси группы, необязательно замещенные 1-2 группами, независимо выбранными из (1-6С)алкила, (1-4С алкокси)карбонила и (1-6С)ацила. Конкретные примеры включают структуры:
В одном варианте реализации, R4 представляет собой (1-4С алкоксикарбонил)(1-6С)алкокси. Примеры включают метоксикарбонил(1-6С)алкокси и этилкарбонил(1-6С)алкокси. Конкретный пример представляет собой этоксикарбонилметокси.
В одном варианте реализации, R4 представляет собой гидроксикарбонил(1-6С)алкокси. Конкретный пример представляет собой гидроксикарбонилметокси.
В одном варианте реализации, R4 представляет собой аминокарбонил(1-6С)алкокси. Примеры включают Н2NC(=O)(1-6С)алкокси, (1-6С алкил)NHC(=O)(1-6С)алкокси и ди(1-6С алкил)NC(=O)(1-6С)алкокси. Конкретный пример представляет собой CH3CH2NC(=O)CH2O-.
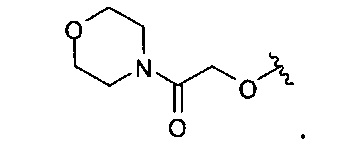
В одном варианте реализации, R4 представляет собой гетСус2С(=O)(1-6С)алкокси, причем гетСус2 представляет собой 4-6-членное гетероциклическое кольцо, имеющее 1-2 кольцевых гетероатома, независимо выбранных из N и О, и необязательно замещенное 1-2 группами, независимо выбранными из (1-6С)алкила, (1-4С алкокси)карбонила и (1-6С)ацила. Примеры гетСус2 включают оксетаниловое, тетрагидрофураниловое, тетрагидропираниловое, азетидиниловое, пирролидиниловое, пиперидиниловое, пиперазиниловое, морфолиниловое и 1,3-диоксоланиловое кольцо, необязательно замещенное 1-2 группами, независимо выбранными из (1-6С)алкила, (1-4С алкокси)карбонила и (1-6С)ацила. В одном варианте реализации, гетСус2 представляет собой морфолинил. Конкретный пример R4, представленного гетСус2С(=O)(1-6С)алкокси, представляет собой структуру:
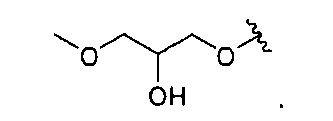
В одном варианте реализации, R4 представляет собой гидрокси(1-3С алкокси)(1-6С)алкокси. Конкретный пример представляет собой 2-гидрокси-3-метоксипропокси, имеющий структуру:
В одном варианте реализации, R4 представляет собой гидрокситрифтор(1-6С)алкокси. Конкретный пример представляет собой 3,3,3-дифтор-2-гидроксипропокси, имеющий структуру:
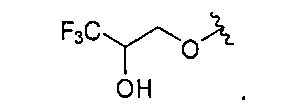
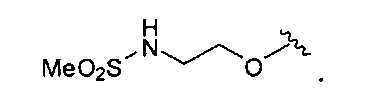
В одном варианте реализации, R4 представляет собой (1-3С)алкилсульфонамидо(1-6С)алкокси. Примеры включают метансульфонамидо(1-6С)алкокси. Конкретный пример представляет собой 2-метансульфонамидоэтокси, имеющий структуру:
В одном варианте реализации, R4 представляет собой (1-3С)алкиламидо(1-6С)алкокси. Конкретный пример представляет собой 2-(метиламидо)этокси, имеющий структуру:
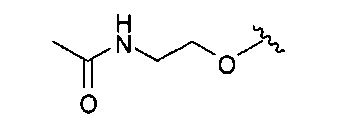 .
.
В одном варианте реализации, R4 представляет собой ди(1-3С алкил)аминокарбокси. Конкретный пример представляет собой диметиламинокарбокси, имеющий структуру:
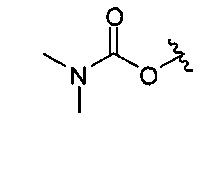 .
.
В одном варианте реализации, R4 представляет собой гетСус2С(=O)O-, причем гетСус2 представляет собой 4-6-членное гетероциклическое кольцо, имеющее 1-2 кольцевых гетероатома, независимо выбранных из N и О, и необязательно замещенное 1-2 группами, независимо выбранными из (1-6С)алкила, (1-4С алкокси)карбонила и (1-6С)ацила. Примеры гетСус2 включают оксетаниловое, тетрагидрофураниловое, тетрагидропираниловое, азетидиниловое, пирролидиниловое, пиперидиниловое, пиперазиниловое, морфолиниловое и 1,3-диоксоланиловое кольцо, необязательно замещенное 1-2 группами, независимо выбранными из (1-6С)алкила, (1-4С алкокси)карбонила и (1-6С)ацила. В одном варианте реализации, гетСус2 представляет собой морфолинил. Конкретный пример R4, представленного гетСус2С(=O)O-, представляет собой структуру:
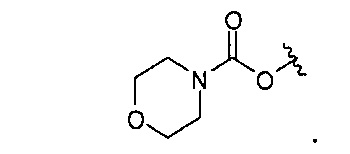
В одном варианте реализации, R4 представляет собой гидроксидифтор(1-6С)алкил. Пример включают 2,2-дифтор-2-гидроксиэтил.
В одном варианте реализации, R4 представляет собой (1-4С алкилкарбонил)(1-6С)алкил. Примеры включают метилкарбокси(1-6С)алкил. Конкретный пример представляет собой (2-метилкарбокси)этил.
В одном варианте реализации, R4 представляет собой (1-6С)алкоксикарбонил. Примеры включают метоксикарбонил и этоксикарбонил.
В одном варианте реализации, R4 представляет собой гидроксикарбонил.
В одном варианте реализации, R4 представляет собой аминокарбонил, то есть радикал RR'NCO-, в котором R и R' независимо представляют собой водород или (1-6С)алкил, как описано в настоящем документе. Примеры включают аминокарбонил, метиламинокарбонил, диметиламинокарбонил, этилкарбонил и изопропиламинокарбонил.
В одном варианте реализации, R4 представляет собой (1-3С алкокси)аминокарбонил. Пример включает метоксиаминокарбонил.
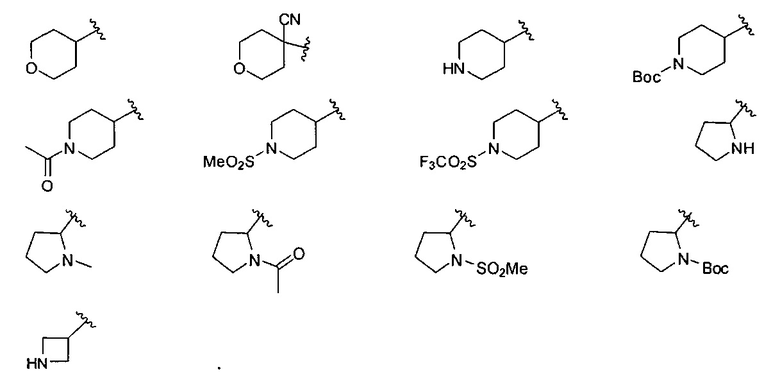
В одном варианте реализации, R4 представляет собой гетСус3, который представляет собой 4-7-членный гетероцикл, имеющий 1-2 кольцевых гетероатома, независимо выбранных из N и О, и необязательно замещенный одним или более заместителями, независимо выбранными из F, CN, CF3, (1-6С)алкила, гидрокси(1-6С)алкила, (1-3С алкокси)(1-6С)алкила, (1-6С)ацил-, (1-6С)алкилсульфонил, трифторметилсульфонила и (1-4С алкокси)карбонила. В одном варианте реализации, гетСус3 представляет собой тетрагидропиранил, пиперидинил, пирролидинил или азетидинил, необязательно замещенный одним или более заместителями, независимо выбранными из F, CN, CF3, (1-6С)алкила, гидрокси(1-6С)алкила, (1-3С алкокси)(1-6С)алкила, (1-6С)ацил-, (1-6С)алкилсульфонила, трифторметилсульфонила и (1-4С алкокси)карбонила. В одном варианте реализации, гетСус3 необязательно замещен одним или двумя из указанных заместителей. В одном варианте реализации, гетСус3 представляет собой тетрагидропиранил, пиперидинил, пирролидинил или азетидинил, необязательно замещенный CN, Me, CH3C(=O)-, MeSO2-, CF3SO2- или (СН3)3СОС(=O)-. Конкретные примеры R4, представленного гетСус3, включают структуры:
В одном варианте реализации, R4 представляет собой галоген. В одном варианте реализации, R4 представляет собой Br.
В одном варианте реализации, R4 представляет собой CN.
В одном варианте реализации, R4 представляет собой трифторметилсульфонил.
В одном варианте реализации, R4 представляет собой N-(1-3C алкил)пиридинонил. Примеры включают N-(1-3 С алкил) замещенный пиридин-2(1Н)-он-4-иловые и N-(1-3С алкил) замещенный пиридин-2(1Н)-он-5-иловые группы. Конкретные примеры включают структуры:
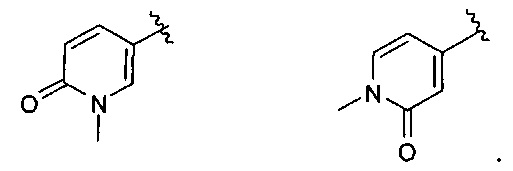
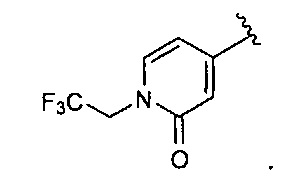
В одном варианте реализации, R4 представляет собой N-(1-3С трифторалкил)пиридинонил. Примеры включают N-(1-3C трифторалкил)-замещенный пиридин-2(1Н)-он-4-иловые и N-(1-3C трифторалкил)-замещенный пиридин-2(1Н)-он-5-иловые группы. Конкретный пример включает структуру:
В одном варианте реализации, R4 представляет собой (1-4С алкилсилокси)(1-6С)алкокси. Примеры включают трет-бутилсилокси(1-6С)алкокси-группы. Конкретный пример представляет собой (2-трет-бутилсилокси)пропокси.
В одном варианте реализации, R4 представляет собой изоиндолин-1,3-дионил(1-6С)алкокси. Конкретный пример включает структуру:
В одном варианте реализации, R4 представляет собой N-(1-3C алкил)оксадиазолонил. Конкретные примеры включают структуры:

В одном варианте реализации, R4 выбран из Н, (1-6С)алкила, трифтор(1-6С)алкила, гидрокси(1-6С)алкила, циано(1-6С)алкила, (1-3С алкокси)(1-6С)алкила, (1-6С)алкокси, монофтор(1-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, дигидрокси(2-6С)алкокси, гетСус2(1-6С)алкокси, Ar3(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (1-3С алкилсульфонил)(1-6С)алкокси, (3-6С)циклоалкила [необязательно замещенного F, (1-6С алкил), (1-6С) алкокси или (1-3С алкокси)(1-6С)алкилом], гетAr4, Ar4, (1-4С алкоксикарбонил)(1-6С)алкокси, гидроксикарбонил(1-6С)алкокси, аминокарбонил(1-6С)алкокси, гетСус2С(=O)(1-6С)алкокси, гидрокси(1-3С алкокси)(1-6С)алкокси, гидрокситрифтор(1-6С)алкокси, (1-3С)алкилсульфонамидо(1-6С)алкокси, (1-3С)алкиламидо(1-6С)алкокси, ди(1-3С алкил)аминокарбокси, гетСус2С(=O)O-, гидроксидифтор(1-6С)алкила, (1-4С алкилкарбокси)(1-6С)алкила, (1-6С)алкокси-карбонила, гидроксикарбонила, аминокарбонила, (1-3С алкокси)аминокарбонила, гетСус2, галогена, CN, трифторметилсульфонила, N-(1-3С алкил)пиридинонила, N-(1-3Стрифторалкил)пиридинонила, (1-4С алкилсилокси)(1-6С)алкокси, изоиндолин-1,3-дионил(1-6С)алкокси и N-(1-3С алкил)оксадиазолонила.
В одном варианте реализации, R4 выбран из Н, (1-6С)алкила, трифтор(1-6С)алкила, гидрокси(1-6С)алкила, циано(1-6С)алкила, (1-3С алкокси)(1-6С)алкила, (1-6С)алкокси, монофтор(1-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, дигидрокси(2-6С)алкокси, гетСус2(1-6С)алкокси, Ar3(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (1-3С алкилсульфонил)(1-6С)алкокси, (3-6С)циклоалкила, гетAr4 и Ar4.
В одном варианте реализации, R4 выбран из Н, (1-6С)алкила, трифтор(1-6С)алкила, гидрокси(1-6С)алкила, циано(1-6С)алкила, (1-3С алкокси)(1-6С)алкила, (1-6С)алкокси, монофтор(1-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, дигидрокси(2-6С)алкокси, гетСус2(1-6С)алкокси, Ar3(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (1-3С алкилсульфонил)(1-6С)алкокси, (3-6С)циклоалкила, гетAr4 и Ar4.
В одном варианте реализации, R4 выбран из Н, (1-6С)алкила, трифтор(1-6С)алкила, циано(1-6С)алкила, (1-3С алкокси)(1-6С)алкила, (1-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (3-6С)циклоалкила, гетAr4 и Ar4.
В одном варианте реализации, R4 выбран из (1-6С)алкила, трифтор(1-6С)алкила, циано(1-6С)алкила, (1-3С алкокси)(1-6С)алкила и (3-6С)циклоалкила.
В одном варианте реализации, R4 выбран из (1-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси и (1-4С алкокси)(1-6С)алкокси.
В одном варианте реализации, R4 выбран из гетAr4 и Ar4.
В одном варианте реализации, R5 представляет собой Н.
В одном варианте реализации, R5 представляет собой (1-6С)алкил. Примеры включают метил, этил, пропил, изопропил и бутил.
В одном варианте реализации, R5 представляет собой монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил или пентафтор(2-6С)алкил. Примеры включают фторметил, 2-фторэтил, дифторметил, 2,2-дифторэтил, 1,3-дифторпроп-2-ил, трифторметил, 2,2,2-трифторэтил, 3,3,3-трифторпропил, 1,1,2,2-тетрафторпропан и 2,2,3,3,3-пентафторпропил.
В одном варианте реализации, R5 представляет собой галоген. В одном варианте реализации, R5 представляет собой F. В одном варианте реализации, R5 представляет собой О. В одном варианте реализации, R5 представляет собой Br.
В одном варианте реализации, R5 представляет собой CN.
В одном варианте реализации, R5 представляет собой (1-4С)алкокси. Примеры включают метокси и этокси.
В одном варианте реализации, R5 представляет собой гидрокси(1-4С)алкил. Примеры включают гидроксиметил и 3-гидроксипропил.
В одном варианте реализации, R5 представляет собой (1-4С алкил)ОС(=O)-. Примеры включают СН3СН2OC(=O)-.
В одном варианте реализации, R5 представляет собой (1-6С)алкилтио. Пример представляет собой метилтио (MeS-).
В одном варианте реализации, R5 представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из галогена, (1-6С)алкила и (1-6С)алкокси. В одном варианте реализации, R5 представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из F, Cl, метила, этила, метокси и этокси. В одном варианте реализации, R5 представляет собой фенил.
В одном варианте реализации, R5 представляет собой (3-4С)циклоалкил. В одном варианте реализации, R5 представляет собой циклопропил. В одном варианте реализации, R5 представляет собой циклобутил.
В одном варианте реализации, R5 представляет собой амино. В одном варианте реализации, R5 представляет собой NH2.
В одном варианте реализации, R5 представляет собой аминокарбонил. В одном варианте реализации, R5 представляет собой H2NC(=O)-.
В одном варианте реализации, R5 представляет собой трифтор(1-3С алкил)амидо. В одном варианте реализации, R5 представляет собой CF3C(=O)NH-.
В одном варианте реализации, R5 выбран из Н, галогена, CN, (1-6С)алкила, (1-4С)алкокси, гидрокси(1-4С)алкила или фенила, необязательно замещенного одной или более группами, независимо выбранными из галогена, (1-6С)алкила и (1-6С)алкокси.
В одном варианте реализации, R5 выбран из Н, галогена или (1-6С)алкила.
В одном варианте реализации, R5 выбран из Н, метила, Cl или Br.
В одном варианте реализации Формулы I, R4 представляет собой Н, ОН, (1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, циано(1-6С)алкил, гидрокси(1-6С)алкил, дигидрокси(2-6С)алкил, (1-3С алкокси)(1-6С)алкил, амино(1-6С)алкил, аминокарбонил(1-6С)алкил, (1-3С)алкилсульфонамидо(1-6С)алкил, сульфамидо(-6С)алкил, гидроксил-карбонил(1-6С)алкил, гетAr3(1-6С)алкил, Ar3(1-6С)алкил, (1-6С)алкокси, монофтор(1-6С)алкокси, дифтор(1-6С)алкокси, трифтор(1-6С)алкокси, тетрафтор(2-6С)алкокси, пентафтор(2-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, дигидрокси(2-6С)алкокси, амино(2-6С)алкокси, аминокарбонил(1-6С)алкокси, гидроксикарбонил(1-6С)алкокси, гетСус2(1-6С)алкокси, гетAr3(1-6С)алкокси, Ar3(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (1-3С алкилсульфонил)(1-6С)алкокси, (3-6С)циклоалкил [необязательно замещенный F, (1-6С алкил), (1-6С) алкокси или (1-3С алкокси)(1-6С)алкилом], гетAr4, Ar4, (1-4С алкоксикарбонил)(1-6С)алкокси, гидроксикарбонил(1-6С)алкокси, аминокарбонил(1-6С)алкокси, гетСус2С(=O)(1-6С)алкокси, гидрокси(1-3С алкокси)(1-6С)алкокси, гидрокситрифтор(1-6С)алкокси, (1-3С)алкилсульфонамидо(1-6С)алкокси, (1-3С)алкиламидо(1-6С)алкокси, ди(1-3С алкил)аминокарбокси, гетСус2С(=O)O-, гидроксидифтор(1-6С)алкил, (1-4С алкилкарбокси)(1-6С)алкил, (1-6С)алкоксикарбонил, гидроксикарбонил, аминокарбонил, (1-3С алкокси)аминокарбонил, гетСус3, галоген, CN, трифторметилсульфонил, N-(1-3С алкил)пиридинонил, N-(1-3С трифторалкил)пиридинонил, (1-4С алкилсилокси)(1-6С)алкокси, изоиндолин-1,3-дионил(1-6С)алкокси или N-(1-3С алкил)оксадиазолонил; и R5 представляет собой Н, (1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, галоген, CN, (1-4С)алкокси, гидрокси(1-4С)алкил, (1-3С алкокси)(1-4С)алкил, (1-4С алкил)ОС(=O)-, (1-6С)алкилтио, фенил [необязательно замещенный одной или более группами, независимо выбранным из галогена, (1-6С)алкила и (1-6С)алкокси], (3-4С)циклоалкил, амино, аминокарбонил или трифтор(1-3С алкил)амидо.
В одном варианте реализации Формулы I, R4 представляет собой Н, ОН, (1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, циано(1-6С)алкил, гидрокси(1-6С)алкил, дигидрокси(2-6С)алкил, (1-3С алкокси)(1-6С)алкил, амино(1-6С)алкил, аминокарбонил(1-6С)алкил, (1-3 С)алкилсульфонамидо(1-6С)алкил, сульфамидо(1-6С)алкил, гидроксил-карбонил(1-6С)алкил, гетAr3(1-6С)алкил, Ar3(1-6С)алкил, (1-6С)алкокси, монофтор(1-6С)алкокси, дифтор(1-6С)алкокси, трифтор(1-6С)алкокси, тетрафтор(2-6С)алкокси, пентафтор(2-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, дигидрокси(2-6С)алкокси, амино(2-6С)алкокси, аминокарбонил(1-6С)алкокси, гидроксикарбонил(1-6С)алкокси, гетСус2(1-6С)алкокси, гетAr3(1-6С)алкокси, Ar3(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (1-3С алкилсульфонил)(1-6С)алкокси, (3-6С)циклоалкил, гетAr4 или Ar4; и R5 представляет собой Н, (1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, галоген, CN, (1-4С)алкокси, гидрокси(1-4С)алкил, (1-3С алкокси)(1-4С)алкил, (1-4С алкил)ОС(=O)-, (1-6С)алкилтио или фенил, необязательно замещенный одной или более группами, независимо выбранным из галогена, (1-6С)алкила и (1-6С)алкокси.
В одном варианте реализации Формулы I, R4 выбран из Н, (1-6С)алкила, трифтор(1-6С)алкила, циано(1-6С)алкила, (1-3С алкокси)(1-6С)алкила, (1-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (3-6С)циклоалкила, гетAr4 и Ar4, и R5 выбран из Н, галогена, CN, (1-6С)алкила, (1-4С)алкокси, гидрокси(1-4С)алкила, (1-6С)алкилтио или фенила, необязательно замещенного одной или более группами, независимо выбранными из галогена, (1-6С)алкила и (1-6С)алкокси.
В одном варианте реализации, R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное, частично ненасыщенное или ненасыщенное карбоциклическое кольцо, необязательно замещенное одним или более заместителями, независимо выбранными из (1-6С)алкила, или R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное, частично ненасыщенное или ненасыщенное гетероциклическое кольцо, имеющее кольцевой гетероатом, выбранный из N, О или S, причем указанное гетероциклическое кольцо необязательно замещено одним или двумя заместителями, независимо выбранными из (1-6С алкил)С(=O)O-, (1-6)ацила, (1-6С)алкила и оксо, и указанный кольцевой атом серы необязательно окислен до S(=O) или SO2.
В одном варианте реализации, R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное, частично ненасыщенное или ненасыщенное карбоциклическое кольцо, необязательно замещенное одним или более заместителями, независимо выбранными из (1-6С)алкила. Примеры кольца C, если R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное или ненасыщенное карбоциклическое кольцо, включают структуры:
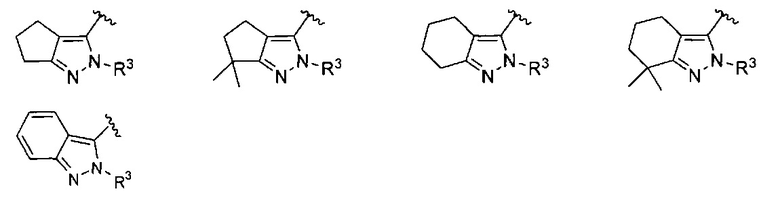
где R3 является таким, как описано для Формулы I.
В одном варианте реализации, R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное карбоциклическое кольцо, необязательно замещенное одним или более заместителями, независимо выбранными из (1-6С)алкила, или R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное гетероциклическое кольцо, имеющее кольцевой гетероатом, выбранный из N, О или S, причем указанный кольцевой атом азота необязательно замещен (1-6С алкил)С(=O)O- или (1-6)ацилом, а указанный кольцевой атом серы необязательно окислен до S(=O) или SO2.
В одном варианте реализации, R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное карбоциклическое кольцо, необязательно замещенное одним или более заместителями, независимо выбранными из (1-6С)алкила. Примеры кольца C, если R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное карбоциклическое кольцо, включают структуры:
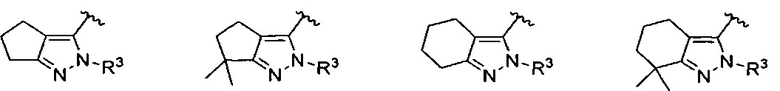
где R3 является таким, как описано для Формулы I.
В одном варианте реализации, R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное, частично ненасыщенное или ненасыщенное гетероциклическое кольцо, имеющее кольцевой гетероатом, выбранный из N, О или S, причем указанный кольцевой атом N необязательно замещен (1-6С алкил)C(=O)O-, (1-6С алкил)C(=O)-, (1-6)алкилом или оксо, и указанный кольцевой атом S серы необязательно окислен до S(=O) или SO2. Примеры кольца C, если R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное гетероциклическое кольцо, включают структуры:
где R3 является таким, как описано для Формулы I.
В одном варианте реализации, R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное гетероциклическое кольцо, имеющее кольцевой гетероатом, выбранный из N, О или S, причем указанный кольцевой атом N необязательно замещен (1-6С алкил)C(=O)O- или (1-6С алкил)С(=O)-, и указанный кольцевой атом S необязательно окислен до S(=O) или SO2. Примеры кольца C, если R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное гетероциклическое кольцо, включают структуры:
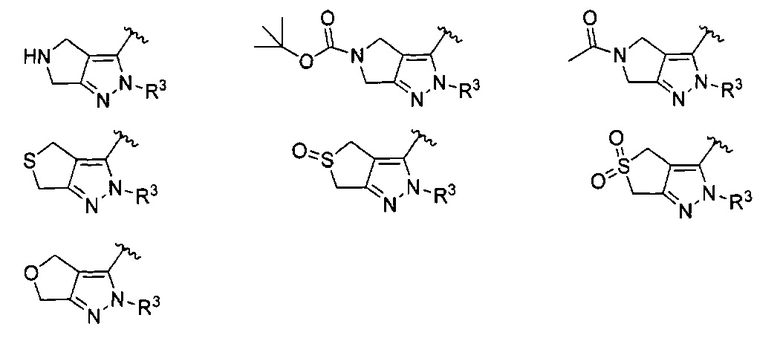
где R3 является таким, как описано для Формулы I.
В одном варианте реализации, кольцо C представляет собой формулу С-2
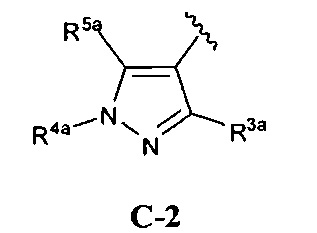
где R3a, R4a и R5a являются такими, как описано для Формулы I.
В одном варианте реализации, R3a представляет собой водород.
В одном варианте реализации, R3a представляет собой галоген.
В одном варианте реализации, R3a представляет собой (1-6С)алкил. В одном варианте реализации, R3a представляет собой метил.
В одном варианте реализации, R3a представляет собой трифтор(1-6С)алкил. В одном варианте реализации, R3a представляет собой CF3.
В одном варианте реализации, R3a представляет собой (3-6С)циклоалкил. В одном варианте реализации, R3a представляет собой циклопропил.
В одном варианте реализации, R3a представляет собой фенил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила и гидроксиметила. Примеры включают фенил, фторфенил, метилфенил и гидроксиметилфенил, например, включают фенил, 2-фторфенил, 3-фторфенил, 4-фторфенил, 2-метилфенил, 3-метилфенил, 4-метилфенил, 3-(гидроксиметил)фенил, 3-хлорфенил, 3-хлор-4-фторфенил и 3-хлор-2-фторфенил. В одном варианте реализации R3a представляет собой фенил.
В одном варианте реализации, R3a представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, О и S, и необязательно замещенное одной или более группами, независимо выбранными из (1-6С)алкила и галогена. В одном варианте реализации, R3a представляет собой тиениловое, фуриловое, имидазолиловое, пиразолиловое, тиазолиловое, изотиазолиловое, оксазолиловое, изоксазолиловое, триазолиловое, тиадиазолиловое, оксадиазолиловое, пиридиловое, пиримидиловое, пиразиниловое или пиридазиниловое кольцо, необязательно замещенное (1-6С)алкилом или галогеном. В одном варианте реализации, R3a представляет собой пиразолил, пиридил или пиридазинил, необязательно замещенный одной или более группами, независимо выбранными из (1-6С)алкила и галогена. В одном варианте реализации, R3a представляет собой пиразолил, пиридил или пиридазинил, необязательно замещенный (1-6С)алкилом или галогеном.
В одном варианте реализации, R4a представляет собой водород.
В одном варианте реализации, R4a представляет собой (1-6С)алкил. В одном варианте реализации, R4a представляет собой метил, этил или изопропил.
В одном варианте реализации, R4a представляет собой трифтор(1-6С)алкил. В одном варианте реализации, R4a представляет собой 2,2,2-трифторэтил.
В одном варианте реализации, R4a представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из (1-6С)алкила, галогена, CN, CF3, CF3O-, (1-6С)алкокси, (1-6С алкил)ОС(=O)-, аминокарбонила, (1-6С)алкилтио, гидрокси(1-6С)алкила, (1-6С алкил)SO2-, НОС(=O)- и (1-3С алкокси)(1-3С алкил)ОС(=O)-. Примеры включают фенил, необязательно замещенный одной или более группами, независимо выбранными из метила, F, Cl, CN, метокси, СН3OC(=O)-, аминокарбонила, метиламинокарбонила, диметиламинокарбонила, метилтио, гидроксиметила, CH3SO2-, НОС(=O)- и СН3ОСН2СН2OC(=О)-. В некоторых вариантах реализации, R4a представляет собой фенил, необязательно замещенный одним или двумя из указанных заместителей. В одном варианте реализации, R4a представляет собой фенил.
В одном варианте реализации, R4a представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенное 1-2 заместителями, независимо выбранными из (1-6С)алкила, гидрокси(1-6С)алкила, трифтор(1-6С)алкила, (3-6С)циклоалкила, (3-6С циклоалкил)СН2-(3-6С циклоалкил)С(=O)-, (1-3С алкокси)(1-6С)алкила, (1-6С)алкокси, (1-6С)алкилсульфонила, NH2, (1-6С алкил)амино, ди(1-6С алкил)амино, (1-3С трифторалкокси)(1-3С)трифторалкила и метоксибензила. Примеры включают пиридиловое, пиримидиниловое, пиридазиниловое, пиразолиловое, имидазолиловое, тиониловое, 1,2,4-триазолиловое, 1,2,3-триазолиловое, тиазолиловое, оксазолиловое, 1,3,4-оксадиазолиловое, 1,2,4-осадиазолиловое и имидазо[1,2-а]пиридиниловое кольцо, необязательно замещенное 1-2 заместителями, независимо выбранными из (1-6С)алкила, гидрокси(1-6С)алкила, трифтор(1-6С)алкила, (3-6С)циклоалкила, (3-6С циклоалкил)СН2-, (3-6С циклоалкил)С(=O)-, (1-3С алкокси)(1-6С)алкила, (1-6С)алкокси, (1-6С)алкилсульфонила, NH2, (1-6С алкил)амино, ди(1-6С алкил)амино, (1-3С трифторалкокси)(1-3С)трифторалкила и метоксибензила. В одном варианте реализации, R4a представляет собой пиразинил.
В одном варианте реализации, R5a является таким, как описано для Формулы I.
В одном варианте реализации, R5a выбран из водорода, галогена, (1-6С)алкила и фенила.
В одном варианте реализации, R5a представляет собой водород.
В одном варианте реализации, R5a представляет собой галоген.
В одном варианте реализации, R5a представляет собой (1-6С)алкил. В одном варианте реализации, R5a представляет собой метил.
В одном варианте реализации, R5a представляет собой фенил.
В одном варианте реализации, кольцо С представляет собой формулу С-2, в которой R3a представляет собой (1-6С)алкил, трифтор(1-6С)алкил или фенил; R4a представляет собой (1-6С)алкил, трифтор(1-6С)алкил, фенил или пиразинил; и R5a представляет собой водород, (1-6С)алкил или фенил.
В одном варианте реализации, кольцо C представляет собой формулу С-3
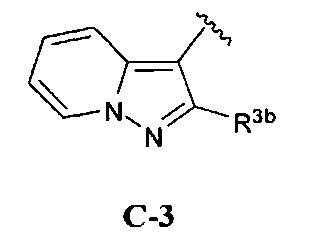
где R3b является таким, как описано для Формулы I.
В одном варианте реализации, R3b представляет собой водород.
В одном варианте реализации, R3b представляет собой (1-6С)алкил. В одном варианте реализации, R3b представляет собой метил.
В одном варианте реализации, R3b представляет собой трифтор(1-6С)алкил. В одном варианте реализации, R3b представляет собой CF3.
В одном варианте реализации, R3b представляет собой (3-6С)циклоалкил. В одном варианте реализации, R3b представляет собой циклопропил.
В одном варианте реализации, R3b представляет собой фенил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила и гидроксиметила. В одном варианте реализации R3b представляет собой фенил.
В одном варианте реализации, R3b представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, О и S, и необязательно замещенное одной или более группами, независимо выбранными из (1-6С)алкила и галогена. В одном варианте реализации, R представляет собой тиенил, фурил, имидазолил, пиразолил, тиазолил, изотиазолил, оксазолил, изоксазолил, триазолил, тиадиазолил, оксадиазолил, пиридил, пиримидил, пиразинил или пиридазинил, необязательно замещенный (1-6С)алкилом или галогеном. В одном варианте реализации, R3b представляет собой пиразолил, пиридил или пиридазинил, необязательно замещенный одной или более группами, независимо выбранными из (1-6С)алкила и галогена. В одном варианте реализации, R3b представляет собой пиразолил, пиридил или пиридазинил, необязательно замещенный (1-6С)алкилом или галогеном.
В одном варианте реализации, кольцо С представляет собой формулу С-3, в которой R3b представляет собой водород или фенил.
В другом варианте реализации настоящего изобретения представлено соединение по Формуле I, которое обозначено как Формула I-а, в которой:
X представляет собой O;
B представляет собой Ar1;
Y представляет собой связь;
кольцо C представляет собой
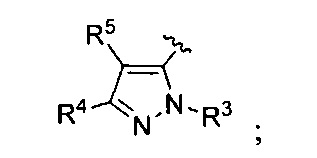
R4 представляет собой Н, ОН, (1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, циано(1-6С)алкил, гидрокси(1-6С)алкил, дигидрокси(2-6С)алкил, (1-3С алкокси)(1-6С)алкил, амино(1-6С)алкил, аминокарбонил(1-6С)алкил, (1-3С)алкилсульфонамидо(-6С)алкил, сульфамидо(1-6С)алкил, гидроксикарбонил(1-6С)алкил, гетAr3(1-6С)алкил, Ar3(1-6С)алкил, (1-6С)алкокси, монофтор(1-6С)алкокси, дифтор(1-6С)алкокси, трифтор(1-6С)алкокси, тетрафтор(2-6С)алкокси, пентафтор(2-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, дигидрокси(2-6С)алкокси, амино(2-6С)алкокси, аминокарбонил(1-6С)алкокси, гидроксикарбонил(1-6С)алкокси, гетСус2(1-6С)алкокси, гетAr3(1-6С)алкокси, Ar3(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (1-3С алкилсульфонил)(1-6С)алкокси, (3-6С)циклоалкил, гетAr4 или Ar4;
R5 представляет собой Н, (1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, галоген, CN, (1-4С)алкокси, гидрокси(1-4С)алкил, (1-3С алкокси)(1-4С)алкил, (1-4С алкил)ОС(=O)- или фенил, необязательно замещенный одной или более группами, независимо выбранными из галогена, (1-6С)алкила и (1-6С)алкокси; и Ra, Rb, Rc, Rd, R1, R2, Ar1, R3, гетСус2, гетAr3, Ar3, гетAr4 и Ar4 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-a, Ra, Rb, Rc и Rd представляют собой водород; R2 представляет собой водород; и R1, Ar1, R3, гетСус2, гетAr3, Ar3, гетAr4 и Ar4 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-a, Ra, Rb, Rc и Rd представляют собой водород; R2 представляет собой водород; R1 представляет собой (1-3С алкокси)(1-6С)алкил, дифтор(1-6С)алкил или трифтор(1-6С)алкил; и Ar1, R3, гетСус2, гетAr3, Ar3, гетAr4 и Ar4 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-a, Ra, Rb, Rc и Rd представляют собой водород; R2 представляет собой водород; R1 представляет собой (1-3С алкокси)(1-6С)алкил, дифтор(1-6С)алкил или трифтор(1-6С)алкил; R3 представляет собой Ar2, гетAr2 или (1-6С)алкил; и Ar1, Ar2, гетAr2, гетСус2, гетAr3, Ar3, гетAr4 и Ar4 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-a, Ra, Rb, Rc и Rd представляют собой водород; R2 представляет собой водород; R1 представляет собой (1-3С алкокси)(1-6С)алкил, дифтор(1-6С)алкил или трифтор(1-6С)алкил; R3 представляет собой Ar2, гетAr2 или (1-6С)алкил; R4 представляет собой Н, (1-6С)алкил, трифтор(1-6С)алкил, циано(1-6С)алкил, (1-3С алкокси)(1-6С)алкил, (1-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (3-6С)циклоалкил, гетAr4 или Ar4; и Ar1, Ar2, гетAr2, гетСус2, гетAr3, Ar3, гетAr4 и Ar4 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-a, Ra, Rb, Rc и Rd представляют собой водород; R2 представляет собой водород; R1 представляет собой (1-3С алкокси)(1-6С)алкил, дифтор(1-6С)алкил или трифтор(1-6С)алкил; R3 представляет собой Ar2, гетAr2 или (1-6С)алкил; R4 представляет собой гетAr4 или Ar4; и Ar1, Ar2, гетAr2, гетСус2, гетAr3, Ar3, гетAr4 и Ar4 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-a, Ra, Rb, Rc и Rd представляют собой водород; R2 представляет собой водород; R1 представляет собой (1-3С алкокси)(1-6С)алкил, дифтор(1-6С)алкил или трифтор(1-6С)алкил; R3 представляет собой Ar2, гетAr2 или (1-6С)алкил; R4 представляет собой Н, (1-6С)алкил, трифтор(1-6С)алкил, циано(1-6С)алкил, (1-3С алкокси)(1-6С)алкил, (1-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (3-6С)циклоалкил, гетAr4 или Ar4; R5 представляет собой Н, галоген, CN, (1-6С)алкил, (1-4С)алкокси, гидрокси(1-4С)алкил или фенил, необязательно замещенный одной или более группами, независимо выбранными из галогена, (1-6С)алкила и (1-6С)алкокси; и Ar1, Ar2, гетAr2, гетСус2, гетAr3, Ar3, гетAr4 и Ar4 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-a, Ra, Rb, Rc и Rd представляют собой водород; R2 представляет собой водород; R1 представляет собой (1-3С алкокси)(1-6С)алкил, дифтор(1-6С)алкил или трифтор(1-6С)алкил; R3 представляет собой Ar2, гетAr2 или (1-6С)алкил; R4 представляет собой Н, (1-6С)алкил, трифтор(1-6С)алкил, циано(1-6С)алкил, (1-3С алкокси)(1-6С)алкил, (1-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (3-6С)циклоалкил, гетAr4 или Ar4; R5 представляет собой Н, галоген или (1-6С)алкил; и Ar1, Ar2, гетAr2, гетAr4 и Ar4 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-a, Ra, Rb, Rc и Rd представляют собой водород; R2 представляет собой водород; R1 представляет собой (1-3С алкокси)(1-6С)алкил, дифтор(1-6С)алкил или трифтор(1-6С)алкил; R3 представляет собой Ar2, гетAr2 или (1-6С)алкил; R4 представляет собой гетAr4 или Ar4; R5 представляет собой Н, галоген или (1-6С)алкил; и Ar1, Ar2, гетAr2, гетAr4 и Ar4 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-a, R4 представляет собой (1-6С)алкил, трифтор(1-6С)алкил, циано(1-6С)алкил, (1-3С алкокси)(1-6С)алкил, (1-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (3-6С)циклоалкил, гетAr4 или Ar4; и Ra, Rb, Rc, Rd, R1, R2, R3, гетAr4 и Ar4 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-a, R4 представляет собой (1-6С)алкил, трифтор(1-6С)алкил, циано(1-6С)алкил, (1-3С алкокси)(1-6С)алкил, (1-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (3-6С)циклоалкил, гетAr4 или Ar4; R3 представляет собой Н, (1-6С)алкил или Ar2; и Ra, Rb, Rc, Rd, R1, R2, гетAr4, Ar4 и Ar2 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-a, R4 представляет собой (1-6С)алкил, трифтор(1-6С)алкил, циано(1-6С)алкил, (1-3С алкокси)(1-6С)алкил, (1-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (3-6С)циклоалкил, гетAr4 или Ar4; R3 представляет собой Н, (1-6С)алкил или Ar2; R5 представляет собой Н, (1-6С)алкил или галоген; и Ra, Rb, Rc, Rd, R1, R2, гетAr4, Ar4 и Ar2 являются такими, как описано для Формулы I.
В другом варианте реализации настоящего изобретения представлено соединение по Формуле I, которое обозначено как Формула I-b, в которой:
X представляет собой O;
В представляет собой гетAr1;
Y представляет собой связь;
кольцо C представляет собой формулу С-1:
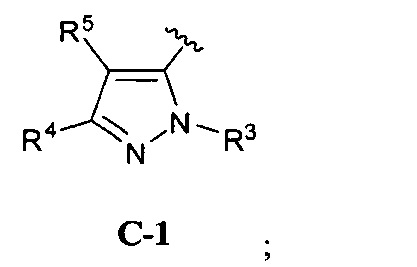
R4 представляет собой Н, ОН, (1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, циано(1-6С)алкил, гидрокси(1-6С)алкил, дигидрокси(2-6С)алкил, (1-3С алкокси)(1-6С)алкил, амино(1-6С)алкил, аминокарбонил(1-6С)алкил, (1-3С)алкилсульфонамидо(1-6С)алкил, сульфамидо(1-6С)алкил, гидроксикарбонил(1-6С)алкил, гетAr3(1-6С)алкил, Ar3(1-6С)алкил, (1-6С)алкокси, монофтор(1-6С)алкокси, дифтор(1-6С)алкокси, трифтор(1-6С)алкокси, тетрафтор(2-6С)алкокси, пентафтор(2-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, дигидрокси(2-6С)алкокси, амино(2-6С)алкокси, аминокарбонил(1-6С)алкокси, гидроксикарбонил(1-6С)алкокси, гетСус2(1-6С)алкокси, гетAr3(1-6С)алкокси, Ar3(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (1-3С алкилсульфонил)(1-6С)алкокси, (3-6С)циклоалкил, гетAr4 или Ar4;
R5 представляет собой Н, (1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, галоген, CN, (1-4С)алкокси, гидрокси(1-4С)алкил, (1-3С алкокси)(1-4С)алкил, (1-4Салкил)ОС(=O)- или фенил, необязательно замещенный одной или более группами, независимо выбранными из галогена, (1-6С)алкила и (1-6С)алкокси; и Ra, Rb, Rc, Rd, R1, R2, гетAr1, R3, гетСус2, гетAr3, Ar3, гетAr4 и Ar4 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-b, Ra, Rb, Rc и Rd представляют собой водород; R2 представляет собой водород; и R1, гетAr1, R3, гетСус2, гетAr3, Ar3, гетAr4 и Ar4 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-b, Ra, Rb, Rc и Rd представляют собой водород; R2 представляет собой водород; R1 представляет собой (1-3С алкокси)(1-6С)алкил, дифтор(1-6С)алкил или трифтор(1-6С)алкил; и гетAr1, R3, гетСус2, гетAr3, Ar3, гетAr4 и Ar4 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-b, Ra, Rb, Rc и Rd представляют собой водород; R2 представляет собой водород; R1 представляет собой (1-3С алкокси)(1-6С)алкил, дифтор(1-6С)алкил или трифтор(1-6С)алкил; R3 представляет собой Ar2, гетAr2 или (1-6С)алкил; и гетAr1, Ar2, гетAr2, гетСус2, гетAr3, Ar3, гетAr4 и Ar4 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-b, Ra, Rb, Rc и Rd представляют собой водород; R2 представляет собой водород; R1 представляет собой (1-3С алкокси)(1-6С)алкил, дифтор(1-6С)алкил или трифтор(1-6С)алкил; R3 представляет собой Ar2, гетAr2 или (1-6С)алкил; R4 представляет собой Н, (1-6С)алкил, трифтор(1-6С)алкил, циано(1-6С)алкил, (1-3С алкокси)(1-6С)алкил, (1-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (3-6С)циклоалкил, гетAr4 или Ar4; и гетAr1, Ar2, гетAr2, гетСус2, гетAr3, гетAr4 и Ar4 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-b, Ra, Rb, Rc и Rd представляют собой водород; R2 представляет собой водород; R1 представляет собой (1-3С алкокси)(1-6С)алкил, дифтор(1-6С)алкил или трифтор(1-6С)алкил; R3 представляет собой Ar2, гетAr2 или (1-6С)алкил; R4 представляет собой Н, (1-6С)алкил, трифтор(1-6С)алкил, циано(1-6С)алкил, (1-3С алкокси)(1-6С)алкил, (1-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (3-6С)циклоалкил, гетAr4 или Ar4; R5 представляет собой Н, галоген, CN, (1-6С)алкил, (1-4С)алкокси, гидрокси(1-4С)алкил или фенил, необязательно замещенный одной или более группами, независимо выбранными из галогена, (1-6С)алкила и (1-6С)алкокси; и гетAr1, Ar2, гетAr2, гетСус2, гетAr3, гетAr4 и Ar4 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-b, Ra, Rb, Rc и Rd представляют собой водород; R2 представляет собой водород; R1 представляет собой (1-3С алкокси)(1-6С)алкил, дифтор(1-6С)алкил или трифтор(1-6С)алкил; R3 представляет собой Ar2, гетAr2 или (1-6С)алкил; R4 представляет собой Н, (1-6С)алкил, трифтор(1-6С)алкил, циано(1-6С)алкил, (1-3С алкокси)(1-6С)алкил, (1-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (3-6С)циклоалкил, гетAr4 или Ar4; R5 представляет собой Н, галоген или (1-6С)алкил; и гетAr1, Ar2, гетAr2, гетAr3, гетAr4 и Ar4 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-b, R4 представляет собой (1-6С)алкил, трифтор(1-6С)алкил, циано(1-6С)алкил, (1-3С алкокси)(1-6С)алкил, (1-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (3-6С)циклоалкил, гетAr4 или Ar4; и Ra, Rb, Rc, Rd, R1, R2, R3, гетAr4 и Ar4 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-b, R4 представляет собой (1-6С)алкил, трифтор(1-6С)алкил, циано(1-6С)алкил, (1-3С алкокси)(1-6С)алкил, (1-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (3-6С)циклоалкил, гетAr4 или Ar4; R3 представляет собой Н, (1-6С)алкил или Ar2; и Ra, Rb, Rc, Rd, R1, R2, гетAr4, Ar4 и Ar2 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-b, R4 представляет собой гетAr4 или Ar4; R3 представляет собой Н, (1-6С)алкил или Ar2; R5 представляет собой Н, (1-6С)алкил или галоген; и Ra, Rb, Rc, Rd, R1, R2, гетAr4, Ar4 и Ar2 являются такими, как описано для Формулы I.
В другом варианте реализации настоящего изобретения представлено соединение по Формуле I, которое обозначено как Формула I-c, в которой:
X представляет собой О;
В представляет собой Ar1;
Y представляет собой связь;
кольцо C представляет собой формулу С-1:
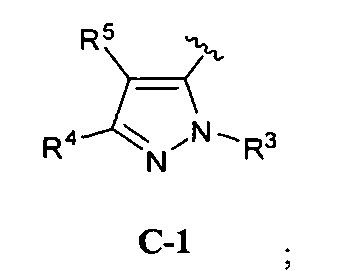
R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное, частично ненасыщенное или насыщенное карбоциклическое кольцо, необязательно замещенное одним или более заместителями, независимо выбранными из (1-6С)алкила, или R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное, частично ненасыщенное или насыщенное гетероциклическое кольцо, имеющее кольцевой гетероатом, выбранный из N, О или S, причем указанное гетероциклическое кольцо необязательно замещено (1-6С алкил)С(=O)O- или (1-6)ацилом, а указанный кольцевой атом серы необязательно окислен до S(=O) или SO2; причем Ra, Rb, Rc, Rd, R1, R2, Ar1 и R3 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-c, R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное карбоциклическое кольцо, необязательно замещенное одним или более заместителями, независимо выбранными из (1-6С)алкила, или R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное гетероциклическое кольцо, имеющее кольцевой гетероатом, выбранный из N, О или S, причем указанный кольцевой атом азота необязательно замещен (1-6С алкил)С(=O)O- или (1-6)ацилом, а указанный кольцевой атом серы необязательно окислен до S(=O) или SO2; причем Ra, Rb, Rc, Rd, R1, R2, Ar1 и R3 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-c, Ra, Rb, Rc, Rd и R2 представляют собой водород; и R1, Ar1 и R3 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-c, Ra, Rb, Rc, Rd и R2 представляют собой водород; R1 представляет собой (1-3С алкокси)(1-6С)алкил, (трифторметокси)(1-6С)алкил, (1-3С сульфанил)(1-6С)алкил, трифтор(1-6С)алкил, циано(1-6С)алкил или (1-3Салкиламино)(1-3С)алкил; и Ar1 и R3 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-c, Ra, Rb, Rc, Rd и R2 представляют собой водород; R1 представляет собой (1-3С алкокси)(1-6С)алкил, (трифторметокси)(1-6С)алкил, (1-3С сульфанил)(1-6С)алкил, трифтор(1-6С)алкил, циано(1-6С)алкил или (1-3Салкиламино)(1-3С)алкил; R3 представляет собой Н, (1-6С)алкил или фенил, необязательно замещенный одной или более группами, независимо выбранными из галогена, (1-6С)алкила и гидроксиметила; и Ar1 является таким, как описано для Формулы I.
В одном варианте реализации Формулы I-c, Ra, Rb, Rc, Rd и R2 представляют собой водород; R1 представляет собой (1-3С алкокси)(1-6С)алкил, (трифторметокси)(1-6С)алкил, (1-3С сульфанил)(1-6С)алкил, трифтор(1-6С)алкил, циано(1-6С)алкил или (1-3Салкиламино)(1-3С)алкил; R3 представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из галогена, (1-6С)алкила и гидроксиметила; и Ar1 является таким, как описано для Формулы I.
В другом варианте реализации настоящего изобретения представлено соединение по Формуле I, которое обозначено как Формула I-d, в которой:
X представляет собой O;
B представляет собой гетAr1;
Y представляет собой связь;
кольцо C представляет собой формулу С-1:
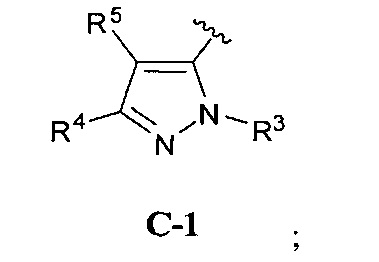
R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное, частично ненасыщенное или насыщенное карбоциклическое кольцо, необязательно замещенное одним или более заместителями, независимо выбранными из (1-6С)алкила, или R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное, частично ненасыщенное или насыщенное гетероциклическое кольцо, имеющее кольцевой гетероатом, выбранный из N, О или S, причем указанное гетероциклическое кольцо необязательно замещено (1-6С алкил)С(=O)O- или (1-6)ацилом, а указанный кольцевой атом серы необязательно окислен до S(=O) или SO2; причем Ra, Rb, Rc, Rd, R1, R2, Ar1 и R3 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-d, R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное карбоциклическое кольцо, необязательно замещенное одним или более заместителями, независимо выбранными из (1-6С)алкила, или R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное гетероциклическое кольцо, имеющее кольцевой гетероатом, выбранный из N, О или S, причем указанный кольцевой атом азота необязательно замещен (1-6С алкил)С(=O)O- или (1-6)ацилом, а указанный кольцевой атом серы необязательно окислен до S(=O) или SO2; причем Ra, Rb, Rc, Rd, R1, R2, гетAr1 и R3 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-d, Ra, Rb, Rc, Rd и R2 представляют собой водород; и R1, гетAr1 и R3 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-d, Ra, Rb, Rc, Rd и R2 представляют собой водород; R1 представляет собой (1-3С алкокси)(1-6С)алкил, (трифторметокси)(1-6С)алкил, (1-3С сульфанил)(1-6С)алкил, трифтор(1-6С)алкил, циано(1-6С)алкил или (1-3Салкиламино)(1-3С)алкил; и гетAr1 и R3 являются такими, как описано для Формулы I.
В одном варианте реализации Формулы I-d, Ra, Rb, Rc, Rd и R2 представляют собой водород; R1 представляет собой (1-3С алкокси)(1-6С)алкил, (трифторметокси)(1-6С)алкил, (1-3С сульфанил)(1-6С)алкил, трифтор(1-6С)алкил, циано(1-6С)алкил или (1-3Салкиламино)(1-3С)алкил; R представляет собой Н, (1-6С)алкил или фенил, необязательно замещенный одной или более группами, независимо выбранными из галогена, (1-6С)алкила и гидроксиметила; и гетAr1 является таким, как описано для Формулы I.
В одном варианте реализации Формулы I-d, Ra, Rb, Rc, Rd и R2 представляют собой водород; R1 представляет собой (1-3С алкокси)(1-6С)алкил, (трифторметокси)(1-6С)алкил, (1-3С сульфанил)(1-6С)алкил, трифтор(1-6С)алкил, циано(1-6С)алкил или (1-3С алкиламино)(1-3С)алкил; R3 представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из галогена, (1-6С)алкила и гидроксиметила; и гетAr1 является таким, как описано для Формулы I.
Как было отмечено, фрагмент Y-B и фрагмент -NH-C(=X)-NH- в Формуле I находятся в транс-конфигурации в пирролидинового кольца, относительная стереохимия которого может быть представлена формулами A или B:
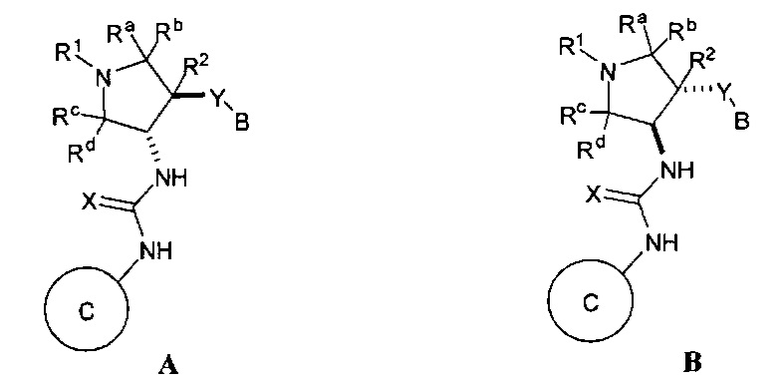
в которых прямые закрашенные прямоугольники  и прямые пунктирные прямоугольники
и прямые пунктирные прямоугольники  указывают относительную стереохимию. В одном варианте реализации представленных выше Формул А и В, Y представляет собой связь, а В представляет собой кольцо В, причем кольцо В представляет собой Ar1 или гетAr1.
указывают относительную стереохимию. В одном варианте реализации представленных выше Формул А и В, Y представляет собой связь, а В представляет собой кольцо В, причем кольцо В представляет собой Ar1 или гетAr1.
В одном варианте реализации Формулы I, фрагменты Y-B и -NH-C(=X)-NH- находятся в абсолютной транс-конфигурации, что может быть представлено Формулами C и D:
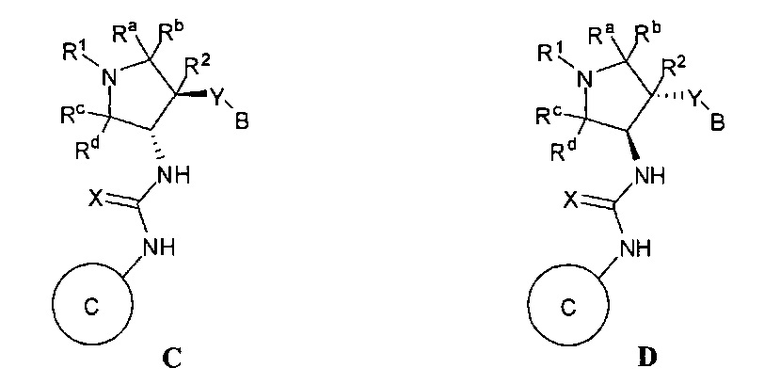
в которых сплошные клинья  и пунктирные клинья
и пунктирные клинья  указывают абсолютную стереохимию. В одном варианте реализации представленных выше Формул C и D, Y представляет собой связь, а B представляет собой кольцо B, причем кольцо B представляет собой Ar1 или гетAr1.
указывают абсолютную стереохимию. В одном варианте реализации представленных выше Формул C и D, Y представляет собой связь, а B представляет собой кольцо B, причем кольцо B представляет собой Ar1 или гетAr1.
Следует понимать, что некоторые соединения по настоящему изобретению могут содержать один или несколько центров асимметрии, и поэтому могут быть получены и выделены в виде смеси изомеров, такой как рацемическая смесь, или в виде энантиомерно чистой формы.
Дополнительно следует понимать, что соединения Формулы I или их соли могут быть выделены в форме сольватов, и, соответственно, что любой такой сольват включен в рамки настоящего изобретения. Например, соединения Формулы I могут существовать в несольватированных или сольватированных формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и тому подобными.
Соединения формулы I включают их фармацевтически приемлемые соли. Кроме того, соединения формулы I включают также другие соли таких соединений, которые не обязательно являются фармацевтически приемлемыми солями, и которые могут быть пригодными в качестве промежуточных соединений для получения и/или очистки соединений формулы I и/или для разделения энантиомеров соединений формулы I. Конкретные примеры солей включают гидрохлоридные соли.
Термин «фармацевтически приемлемый» показывает, что вещество или композиция совместима химически и/или токсикологически с другими ингредиентами, входящими в состав композиции, и/или с организмом млекопитающего, подлежащим лечению.
В настоящем изобретении представлен также способ получения соединения формулы I или его соли, как описано в настоящем документе, который включает:
(а) для соединения Формулы I, где X представляет собой O, связывание соответствующего соединения, имеющего формулу II
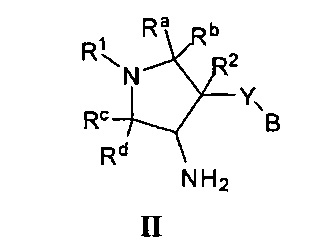
с соответствующим соединением, имеющим формулу III
в присутствии карбонилдиимидазола или трифосгена и основания; или
(б) для соединения Формулы I, где X представляет собой S, связывание соответствующего соединения, имеющего формулу II
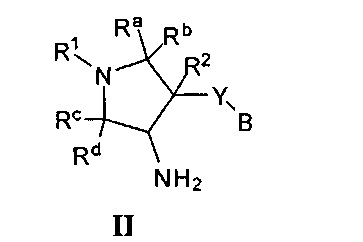
с соответствующим соединением, имеющим формулу III
в присутствии ди(1Н-имидазол-2-ил)метантиона и основания; или
(в) для соединения Формулы I, где X представляет собой O, связывание соответствующего соединения, имеющего формулу II
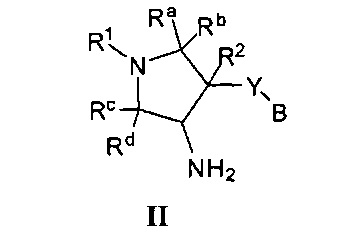
с соответствующим соединением, имеющим формулу IV
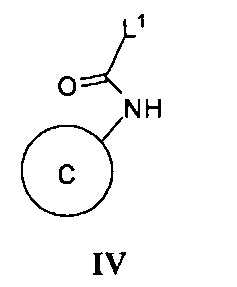
где L1 представляет собой уходящую группу, в присутствии основания; или
(г) для соединения Формулы I, где X представляет собой O, связывание соответствующего соединения, имеющего формулу V
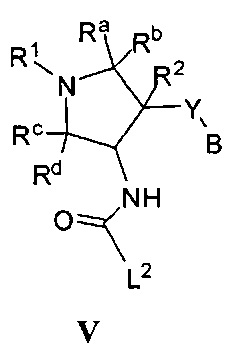
где L2 представляет собой уходящую группу, с соответствующим соединением, имеющим формулу III
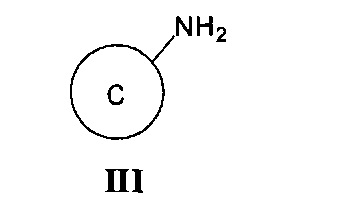
в присутствии основания; или
(д) для соединения Формулы I, где X представляет собой O, активацию соответствующего соединения, имеющего формулу VI
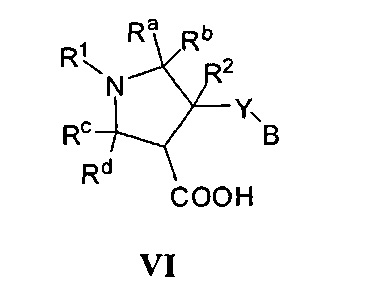
дифенилфосфорил азидом с последующим связыванием активированного промежуточного соединения с соответствующим соединением, имеющим формулу III
в присутствии основания; или
(е) для соединения Формулы I, где X представляет собой O, связывание соответствующего соединения, имеющего формулу II
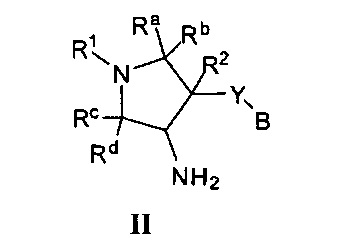
с соответствующим соединением, имеющим формулу VII
в присутствии основания; или
(ж) для соединения Формулы I, где R1 представляет собой (трифторметокси)(1-6С)алкил, (1-3С сульфанил)(1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил или пентафтор(2-6С)алкил, взаимодействие соответствующего соединения, имеющего формулу VIII
с соответствующим соединением, имеющие (трифторметокси)(1-6С)алкил-L3, (1-3С сульфанил)(1-6С)алкил-L3, монофтор(1-6С)алкил-L3, дифтор(1-6С)алкил-L3, трифтор(1-6С)алкил-L3, тетрафтор(2-6С)алкил-L3 или пентафтор(2-6С)алкил-L3, где L3 представляет собой уходящий атом или уходящую группу, в присутствии основания; или
(з) для соединения Формулы I, где X представляет собой O, R4 представляет собой CH3OCH2-, и R5 представляет собой OHCH2-, обработку соответствующего соединения, имеющего общую формулу IX
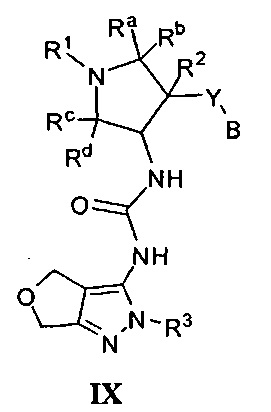
неорганической кислотой; и
необязательно удаление защитных групп, и необязательно получение его фармацевтически приемлемой соли.
В представленных выше способах, термин «соответствующий» обозначает, что определения «соответствующего соединения» являются такими, как описаны для Формулы I, если не указано иное.
В одном варианте реализации любого из представленных выше способов, Y представляет собой связь, а B представляет собой кольцо B, причем кольцо B представляет собой Ar1 или гетAr1, как описано для Формулы I.
В отношении способа (а), основание может быть аминным основанием, таким как триэтиламин или диизопропиламин. Соответствующие растворители включают дихлорметан, дихлорэтан, ТГФ, ДМА и ДМФ. Реакцию легко выполняют при комнатной температуре.
В отношении способа (б), основание может быть аминным основанием, таким как триэтиламин или диизопропиламин. Соответствующие растворители включают дихлорметан, дихлорэтан, ТГФ, ДМА и ДМФ. Реакцию легко выполняют при комнатной температуре.
В отношении способа (в), уходящая группа может быть, например, фенокси или 4-нитрофенокси. Основанием может быть аминное основание, такое как триэтиламин или диизопропиламин. Соответствующие растворители включают ДМА, ДМФ и ДХЭ. Реакцию легко выполняют при комнатной температуре.
В отношении способа (г), уходящая группа может быть, например, фенокси или 4-нитрофенокси. Основанием может быть аминное основание, такое как триэтиламин или диизопропиламин. Соответствующие растворители включают ДХЭ, ДМА и ДМФ. Реакцию легко выполняют при комнатной температуре.
В отношении способа (д), основание может быть аминным основанием, таким как триэтиламин или диизопропиламин. Соответствующие растворители включают толуол и ДМФ. Реакцию легко выполняют при повышенных температурах, например, при температуре дефлегмации растворителя.
В отношении способа (е), основание может быть аминным основанием, таким как триэтиламин или диизопропиламин. Соответствующие растворители включают ДХМ, ДХЭ, ДМФ и ТГФ. Реакцию легко выполняют при температурах приблизительно от 0°C до комнатной температуры.
Соединение Формулы VII может быть получено взаимодействием соединения Формулы III с бис(трихлорметил)карбонатом в присутствии основания, такого как аминное основание.
В отношении способа (ж), основание может быть аминным основанием, таким как триэтиламин или диизопропиламин. Соответствующие растворители включают ДМФ, ДМА и ТГФ. Реакцию легко выполняют при температурах от комнатной температуры до 60°C.
В отношении способа (з), кислота может быть, например, хлороводородной кислотой. Соответствующие растворители включают ДХМ. Реакцию легко выполняют при комнатной температуре.
Аминные группы в соединениях, описанных в любом из представленных выше способов, могут быть защищены любой удобной аминозащитной группой, например, как описано в книге Greene & Wuts, ред., "Protecting Groups in Organic Synthesis", 2oe изд. Нью-Йорк; John Wiley & Sons, Inc., 1991. Примеры аминозащитных групп включают ацил и алкоксикарбонильные группы, такие как трет-бутоксикарбонил (ВОС), феноксикарбонил и [2-(триметилсилил)этокси]метил (SEM). Точно так же, карбоксильные группы могут быть защищены любой удобной карбоксил-защитной группой, например, как описано в книге Greene & Wuts, eds., "Protecting Groups in Organic Synthesis", 2ое изд., Нью-Йорк; John Wiley & Sons, Inc., 1991. Примеры карбоксил-защитных групп включают (1-6С)алкиловые группы, такие как метил, этил и трет-бутил. Спиртовые группы могут быть защищены любой удобной защитной группой для спирта, например, как описано в книге Greene & Wuts, eds., "Protecting Groups in Organic Synthesis", 2ое изд., Нью-Йорк; John Wiley & Sons, Inc., 1991. Примеры защитных групп для спирта включают бензил, тритил, силиловые эфиры и тому подобные.
Соединения формул II, III, III, IV, V, VI, VII, VIII, и IX предположительно, также являются новыми и представлены в качестве дополнительных аспектов настоящего изобретения.
В одном варианте реализации описанных выше способов (а), (б), (в) и (е), где B представляет собой Ar1, a Ra, Rb, Rc, Rd и R2 представляют собой водород, один энантиомер промежуточного соединения II, а именно энантиомер II-A, получают хиральной кристаллизацией до его применения. Соответственно, в одном варианте реализации, способ получения энантиомера 1 II-А включает:
получение рацемического транс-II-А
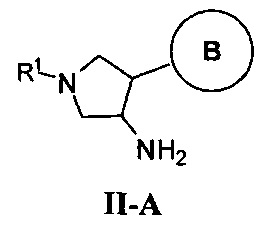
где кольцо В и группа NH2 находятся в транс-конфигурации; кольцо В представляет кольцо В представляет собой Ar1 или гетAr1; Ar1 представляет собой фенил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена, CF3, CF3O-, (1-4С)алкокси, гидрокси(1-4С)алкила, (1-6С)алкила и CN; и гетAr1 представляет собой 5-6-членный гетероарил, имеющий 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенный 1-2 группами, независимо выбранными из (1-6С)алкила, галогена, ОН, CF3, NH2 и гидрокси(1-2С)алкила; указанный способ включает:
обработку рацемического транс-II-А ди-п-толуоил-D-винной кислотой для получения ди-п-толуоил-D-виннокислой соли рацемического II-А;
перекристаллизацию ди-п-толуоил-D-виннокислой соли транс-II-А для получения ди-п-толуолил-D-виннокислой соли энантиомера 1 транс-II-А; и
обработку ди-п-толуоил-D-виннокислой соли энантиомера 1 транс-II-А неорганическим основанием для получения свободного основания энантиомера 1 транс-II-А, имеющего абсолютную конфигурацию, изображенную ниже:
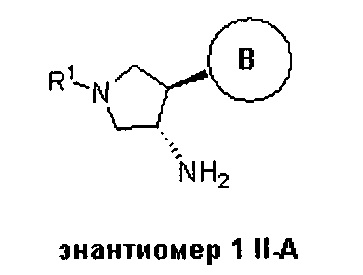
В одном варианте рацемический транс-II-А, R1 представляет собой 2-метоксиэтокси, и кольцо B представляет собой 4-фторфенил, и рацемический транс-II-А получают по способу, который включает:
взаимодействие 4-фторбензальдегида с нитрометаном в присутствии уксусной кислоты и ацетата аммония для получения (Е)-1-фтор-4-(2-нитровинил)бензола
взаимодействие (Е)-1-фтор-4-(2-нитровинил)бензола с 2-метокси-N-(метоксиметил)-N-((триметилсилил)метил)этанамином в присутствии каталитического количества кислоты (такой как ТФК) для получения транс-3-(4-фторфенил)-1-(2-метоксиэтил)-4-нитропирролидина
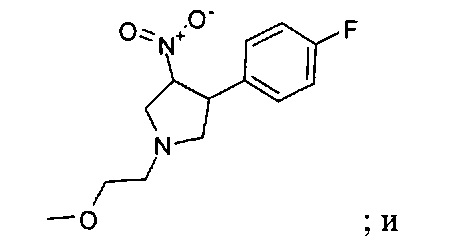
обработку транс-3-(4-фторфенил)-1-(2-метоксиэтил)-4-нитропирролидина оксидом платины (IV) или никелем Ренея в атмосфере водорода для получения транс-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина
где 4-фторфениловая и аминогруппа находятся в транс-конфигурации.
В одном варианте реализации, неорганическое основание представляет собой гидроксид щелочного металла, такой как гидроксид натрия.
Такой же процесс, как описан выше, может быть использован с применением ди-п-толуолил-D-винной кислоты для получения энантиомера 2 II-A:
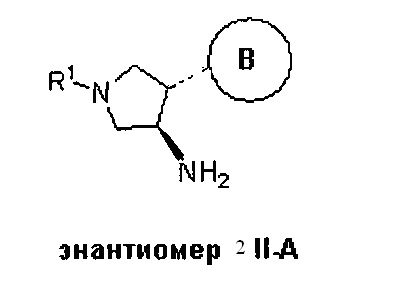
Способность соединений настоящего изобретения действовать в качестве ингибиторов TrkA может быть продемонстрирована анализом, описанным в Примере А.
Соединения формулы I пригодны для лечения боли, включая хроническую и острую боль. Например, соединения Формулы I могут быть применимы для лечения множества типов боли, включая воспалительную боль, невропатическую боль и боль, связанную с раком, хирургией и переломами костей.
В одном варианте реализации, соединения Формулы I пригодны для лечения острой боли. Острая боль, как определено Международной Ассоциацией по изучению боли, является результатом болезни, воспаления или повреждения тканей. Этот тип боли обычно наступает внезапно, например, после травмы или хирургической операции, и может сопровождаться тревогой или стрессом. Причина обычно может быть диагностирована и вылечена, и боль ограничивается данным периодом времени и тяжестью. В некоторых случаях она может стать хронической.
В одном варианте реализации, соединения Формулы I пригодны для лечения хронической боли. Хроническая боль, по определению Международной Ассоциации по изучению боли, сама представляет собой заболевание. Она может стать гораздо сильнее под действием внешних и психологических факторов. Хроническая боль существует в течение более длительного периода, чем острая боль, и является устойчивой к большинству медицинских способов лечения, обычно в течение более 3 месяцев или более. Она может вызывать и зачастую вызывает существенные проблемы у пациентов.
Соединения формулы I пригодны также для лечения рака. Конкретные примеры включают нейробластому, рак яичников, поджелудочной железы, толстой и прямой кишок, рак простаты.
Соединения Формулы I также пригодны для лечения воспаления и некоторых инфекционных заболеваний.
Кроме того, соединения Формулы I могут также быть использованы для лечения интерстициального цистита (IC), синдрома раздраженного мочевого пузыря (PBS), недержания мочи, астмы, анорексии, атопического дерматита и псориаза.
Соединения Формулы I также пригодны для лечения нейродегенеративных заболеваний у млекопитающих, включая введение указанному млекопитающему одного или более соединений Формулы I или их фармацевтически приемлемых солей в количестве, эффективном для лечения или предупреждения указанного нейродегенеративного заболевания. В одном варианте реализации, соединения Формулы I могут также быть использованы для лечения демиелинизации и дисмиелинизации путем промотирования миелинизации, нейронного выживания и дифференцировки олигодендроцитов посредством блокирования взаимодействия Sp35-TrkA. В одном варианте реализации, нейродегенеративное заболевание представляет собой рассеянный склероз. В одном варианте реализации, нейродегенеративное заболевание представляет собой болезнь Паркинсона. В одном варианте реализации, нейродегенеративное заболевание представляет собой болезнь Альцгеймера.
При использовании в настоящем документе, термины «лечить» или «лечение» относятся к терапевтическим или паллиативным мерам. Преимущественные или желательные клинические результаты включают, но не ограничиваясь этим, полное или частичное облегчение симптомов, связанных с расстройством или состоянием, уменьшение распространения заболевания, стабилизацию (то есть не ухудшение) состояния заболевания, отсрочку или замедление прогрессирования заболевания, улучшение или ослабление болезненного состояния и ремиссию (частичную или полную), явные или неявные. «Лечение» также означает продление продолжительности жизни по сравнению с ожидаемой продолжительностью жизни без лечения.
В некоторых вариантах реализации, соединения Формулы I пригодны для предупреждения заболеваний или расстройств, описанных в настоящем документе. Термин «предупреждение», используемый в настоящем документе, обозначает полное или частичное предупреждение возникновения, рецидива или распространения заболевания или состояния, описанного в настоящем документе, или его симптома.
Соответственно, в одном варианте реализации настоящего изобретения представлен способ лечения боли у млекопитающего, включающий введение указанному млекопитающему одного или более соединений Формулы I или их фармацевтически приемлемых солей в количестве, эффективном для лечения или предупреждения указанной боли. В одном варианте реализации, боль представляет собой хроническую боль. В одном варианте реализации, боль представляет собой острую боль. В одном варианте реализации, боль представляет собой воспалительную боль, невропатическую боль и боль, связанную с раком, хирургией и переломами костей.
В другом варианте настоящего изобретения представлен способ лечения воспаления у млекопитающего, включающий введение указанному млекопитающему одного или более соединений Формулы I или их фармацевтически приемлемых солей в количестве, эффективном для лечения или предупреждения указанного воспаления.
В другом варианте настоящего изобретения представлен способ лечения нейродегенеративного заболевания у млекопитающего, включающий введение указанному млекопитающему одного или более соединений Формулы I или их фармацевтически приемлемых солей в количестве, эффективном для лечения или предупреждения указанного нейродегенеративного заболевания.
В другом варианте настоящего изобретения представлен способ лечения инфекции Trypanosoma cruzi у млекопитающего, включающий введение указанному млекопитающему одного или более соединений Формулы I или их фармацевтически приемлемых солей в количестве, эффективном для лечения или предупреждения указанной инфекции Trypanosoma cruzi.
Выражение «эффективное количество» означает такое количество соединения, которое при введении млекопитающему, нуждающемуся в таком лечении, является достаточным для (i) лечения или предупреждения конкретного заболевания, состояния или расстройства, которое может лечиться соединением Формулы I, (ii) облегчения, улучшения или исключения одного или более симптомов конкретного заболевания, состояния или расстройства, или (iii) предупреждения или отсрочки возникновения одного или более симптомов конкретного заболевания, состояния или расстройства, описанных в настоящем документе.
Количество соединения Формулы I, которое соответствует такому количеству, варьируется в зависимости от факторов, таких как конкретное соединение, состояние заболевания и его степень, свойства (например, вес) млекопитающего, нуждающегося в лечении, но, тем не менее, может быть определено обычным способом специалистом в данной области.
Используемый в настоящем документе термин «млекопитающее» относится к теплокровному животному, страдающему или имеющему риск развития заболевания, описанного в настоящем документе, и включает, но не ограничиваясь этим, морских свинок, собак, кошек, крыс, мышей, хомяков и приматов, включая людей.
Соединения настоящего изобретения могут быть использованы в комбинации с одним или более дополнительными лекарствами, которые работают по такому же или по другому механизму действия. Примеры включают противовоспалительные соединения, стероиды (например, дексаметазон, кортизон и флутиказон), анальгетики, такие как нестероидные противовоспалительные препараты (NSAID) (например, аспирин, ибупрофен, индометацин и кетопрофен) и опиоиды (такие как морфин) и химиотерапевтические агенты.
Соединения настоящего изобретения могут быть введены любым удобным путем, например, в желудочно-кишечный тракт (например, ректально или перорально), в нос, легкие, мышцы или сосудистую сеть, или трансдермально, или дермально. Соединения могут быть введены в любой удобной форме введения, например, в таблетках, порошках, капсулах, растворах, дисперсиях, суспензиях, сиропах, спреях, суппозиториях, гелях, эмульсиях, пластырях и тому подобных. Такие композиции могут содержать компоненты, обычные в фармацевтических препаратах, например, разбавители, носители, модификаторы pH, подсластители, наполнители и дополнительные активные агенты. При необходимости парентерального введения соединения должны быть стерильными и в форме раствора или суспензии, пригодной для инъекции или инфузии. Такие композиции составляют дополнительный аспект настоящего изобретения. Примером подходящей пероральной лекарственной формы является таблетка, содержащая около 25 мг, 50 мг, 100 мг, 250 мг или 500 мг соединения настоящего изобретения, смешанного с 90-30 мг безводной лактозы, около 5-40 мг кроскармеллозы натрия, около 5-30 мг поливинилпирролидона («ПВП») К30 и около 1-10 мг стеарата магния. Порошковые компоненты сначала смешивают вместе, а затем смешивают с раствором ПВП. Полученная композиция может быть высушена, гранулирована, смешана со стеаратом магния и спрессована в форму таблетки при помощи стандартного оборудования. Аэрозольная композиция может быть получена растворением соединения по настоящему изобретению, например, 5-400 мг, в соответствующем буферном растворе, например, фосфатном буфере, с добавлением, при необходимости, регулятора тоничности, например, соли, такой как хлорид натрия. Раствор обычно фильтруют, например, с использованием 0,2-микронного фильтра, для удаления примесей и загрязнителей.
Другую композицию можно получить смешиванием соединения, описанного в настоящем документе, и носителя или формообразующего средства. Соответствующие носители и формообразующие средства хорошо известны специалистам в данной области и подробно описаны, например, в книге Ansel, Howard С, et al., Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems. Philadelphia: Lippincott, Williams & Wilkins, 2004; Gennaro, Alfonso R., et al. Remington: The Science and Practice of Pharmacy. Philadelphia: Lippincott, Williams & Wilkins, 2000; и Rowe, Raymond C. Handbook of Pharmaceutical Excipients. Chicago, Pharmaceutical Press, 2005. Композиции могут также включать один или более буферов, стабилизаторов, поверхностно-активных веществ, увлажняющих агентов, смазывающих агентов, эмульгаторов, суспендирующих средств, консервантов, антиоксидантов, матирующих средств, глидантов, технологических добавок, красителей, подсластителей, отдушек, ароматизаторов, разбавителей и других известных добавок для получения простых форм лекарства (то есть соединения, описанного в настоящем документе, или его фармацевтической композиции) или добавки в производстве фармацевтического продукта (то есть лекарственного средства).
Соответственно, в другом аспекте настоящего изобретения представлена фармацевтическая композиция, которая содержит соединение Формулы I или его фармацевтически приемлемую соль, как описано выше, вместе с фармацевтически приемлемым разбавителем или носителем.
В соответствии с другим аспектом, в настоящем изобретении представлено соединение Формулы I или его фармацевтически приемлемая соль, для использования при лечении боли у млекопитающих. В одном варианте реализации, боль представляет собой хроническую боль. В одном варианте реализации, боль представляет собой острую боль. В одном варианте реализации, боль представляет собой воспалительную боль, невропатическую боль и боль, связанную с раком, хирургией и переломами костей.
В другом варианте реализации, в настоящем изобретении представлено соединение Формулы I или его фармацевтически приемлемая соль, для использования при лечении воспаления у млекопитающих.
В другом варианте реализации, в настоящем изобретении представлено соединение Формулы I или его фармацевтически приемлемая соль, для использования при лечении инфекционных заболеваний, например, инфекции Trypanosoma cruzi, у млекопитающих.
В другом варианте реализации, в настоящем изобретении представлено соединение Формулы I или его фармацевтически приемлемая соль, для использования при лечении нейродегенеративных заболеваний у млекопитающих.
В соответствии со следующим аспектом, в настоящем изобретении представлено применение соединения Формулы I или его фармацевтически приемлемой соли в производстве лекарственного средства для лечения состояния, выбранного из боли, рака, воспаления, нейродегенеративного заболевания или инфекции Trypanosoma cruzi. В одном варианте реализации, состояние представляет собой хроническую боль. В одном варианте реализации, состояние представляет собой острую боль. В одном варианте реализации, боль представляет собой воспалительную боль, невропатическую боль и боль, связанную с раком, хирургией и переломами костей. В одном варианте реализации, состояние представляет собой рак. В одном варианте реализации, состояние представляет собой воспаление. В одном варианте реализации, состояние представляет собой нейродегенеративное заболевание. В одном варианте реализации, состояние представляет собой инфекцию Trypanosoma cruzi.
Сокращения, используемые в настоящем документе, имеют следующие значения:

Примеры
Следующие примеры иллюстрируют настоящее изобретение. В примерах, описанных ниже, если не указано иное, все температуры представлены далее в градусах Цельсия. Реагенты были закуплены у коммерческих поставщиков, таких как Aldrich Chemical Company, Lancaster, TCI или Maybridge, и были использованы без дополнительной очистки, если не указано иное. ТГФ, ДХМ, толуол, ДМФ и диоксан приобрели у компании Aldrich в бутылях Sure/Seal™ и использовали такими, как при получении.
Реакции, указанные далее ниже, были выполнены, в основном, при положительном давлении азота или аргона, или с использованием сушильной трубки (если не указано иное), в безводных растворителях, и реакционные колбы обычно снабжали резиновой мембраной для ввода веществ и реагентов через шприц. Стеклянные изделия были высушены в печи и/или при нагревании.
Колоночную хроматографию выполнили на системе Biotage (производитель: Dyax Corporation), имеющей силикагелевую или обращенно-фазовую колонку С-18, или на картридже из диоксида кремния SepPak (Waters).
Биологический анализ
Пример А
Анализ TrkA Omnia
Ферментативную селективность оценили при помощи аналитических реагентов киназы Omnia™ производства Invitrogen Corp. Фермент (TrkA производства Invitrogen Corp.) и исследуемое соединение (различные концентрации) инкубировали в течение 10 минут при комнатной температуре в 384-луночном белом полипропиленовом планшете (Nunc, № по каталогу 267462). Затем на планшет добавили пептид Omnia Tyr №4, а также АТФ. Конечные концентрации составили: 20 нМ фермента, 500 мкМ АТФ, 10 мкМ пептидного субстрата. Аналитический буфер содержал 25 мМ MOPS рН 7,5, 0,005% (об./об.) Triton Х-100 и 5 мМ MgCl2. Выработку фосфорилированного пептида контролировали непрерывно в течение 70 минут, используя микропланшет-ридер FlexStation II384 компании Molecular Devices (возбуждение=360 нм; испускание=485 нм). Исходные значения рассчитали по кривым прогресса. Значения IC50 рассчитали по этим величинам, используя 4 или 5-параметрическую логистическую кривую.
При испытании в этом анализе, соединения настоящего изобретения имели среднее значение IC50 ниже 1000 нМ. При испытании в этом анализе, некоторые соединения имели среднее значение IC50 ниже 100 нМ.
В Таблице А представлены средние значения IC50 для соединений настоящего изобретения, испытанных в анализе Примера А, причем А представляет собой усредненное значение IC50<100 нМ; В представляет собой усредненное значение IC50 от 100 до 1000 нМ; и С представляет собой усредненное значение IC50 от 1000 до 10000 нМ.
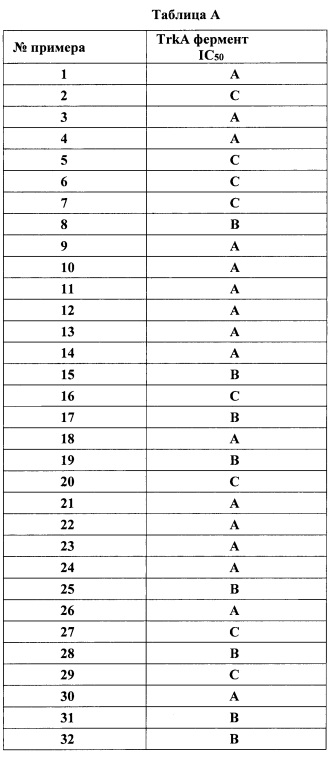
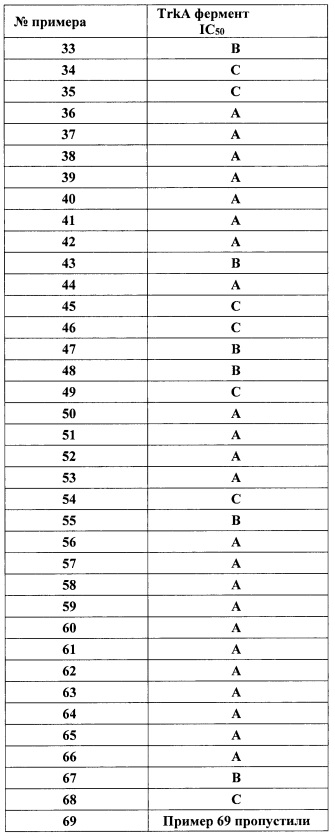
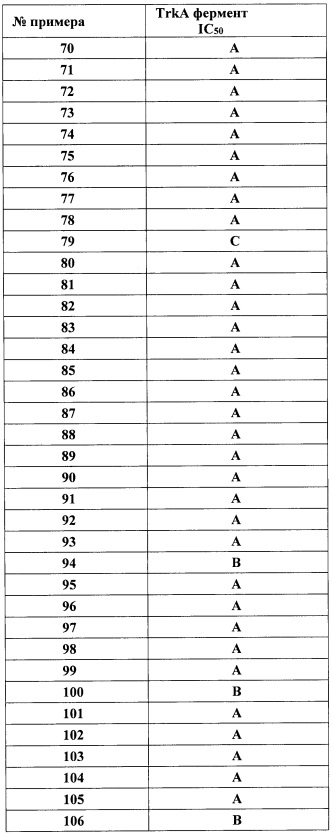
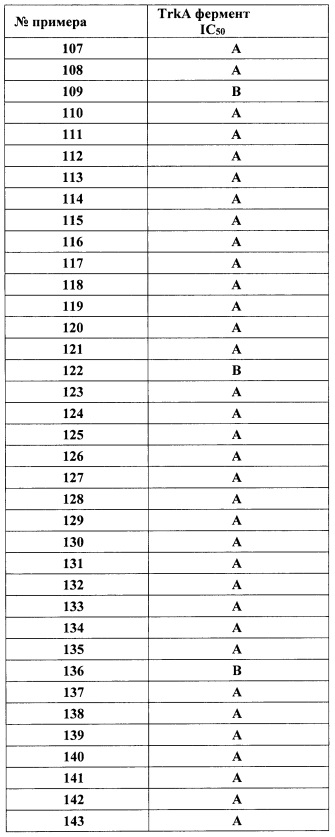
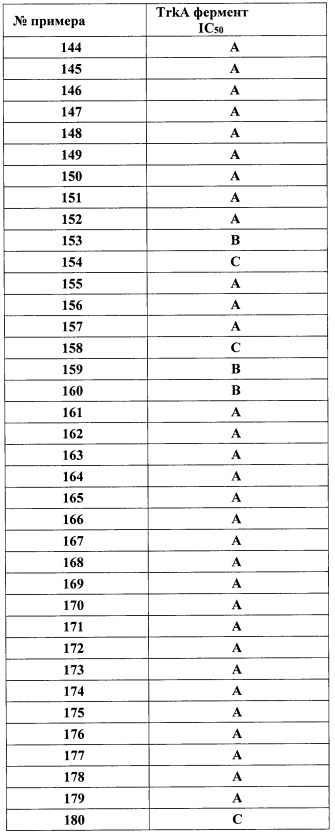
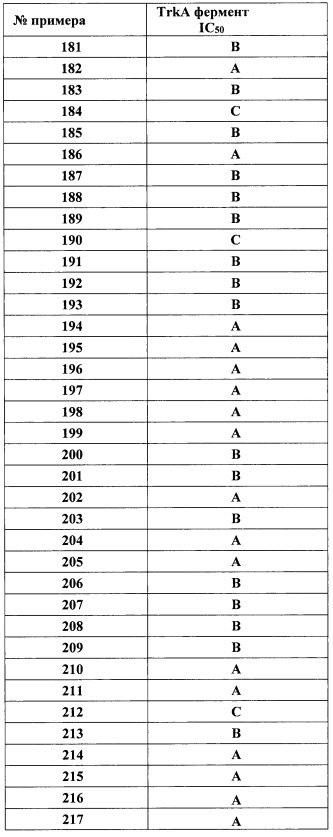
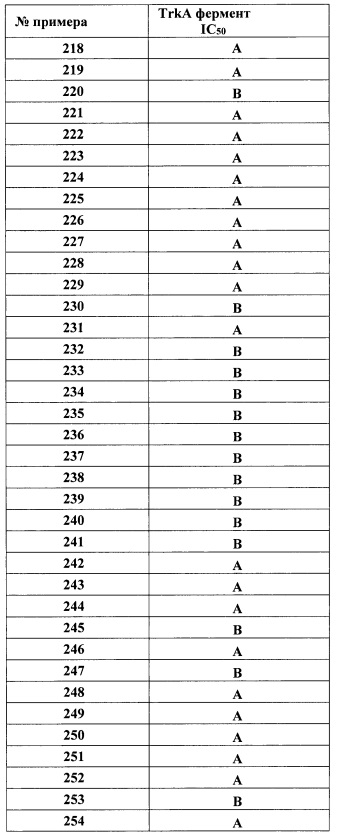
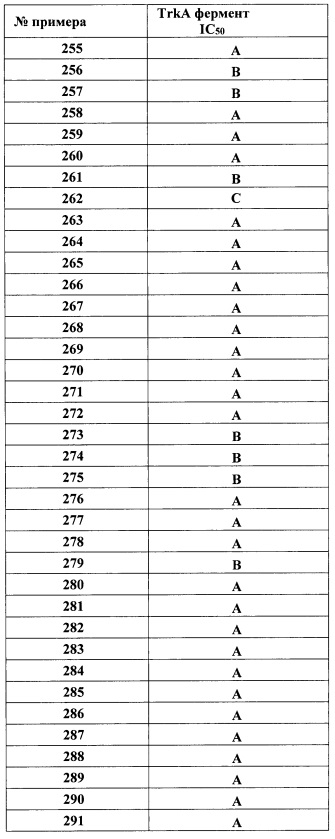
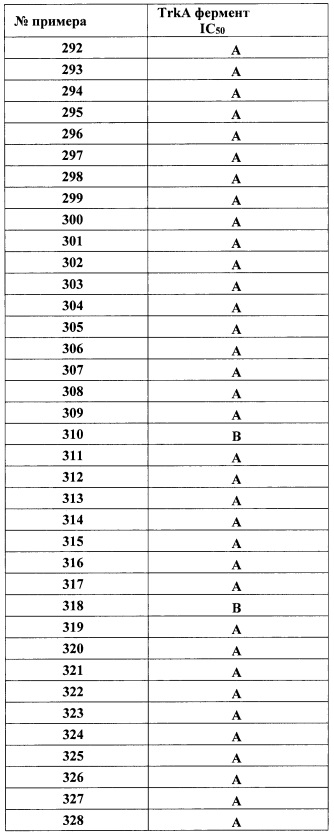
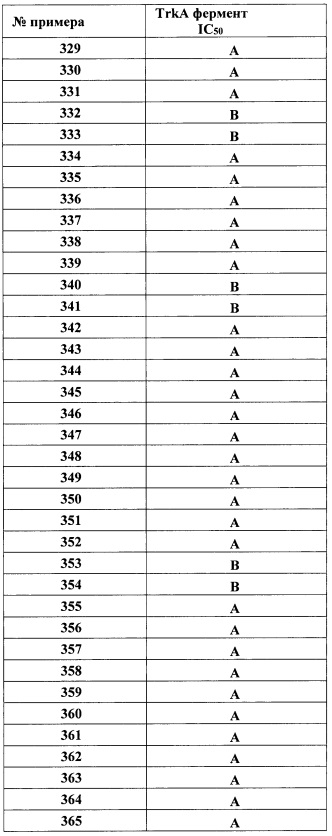
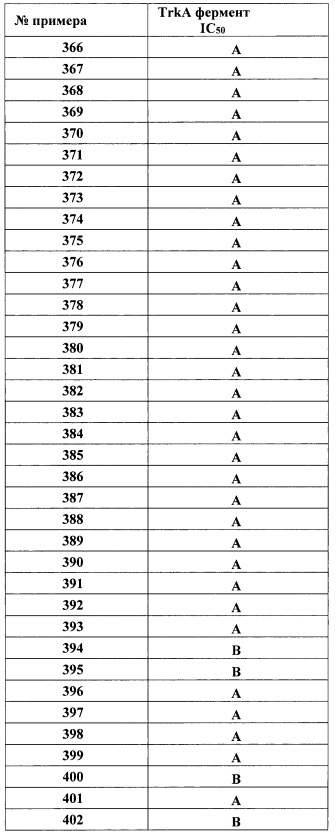

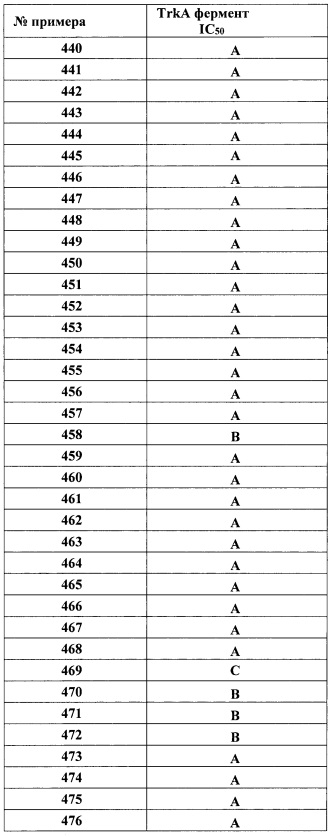
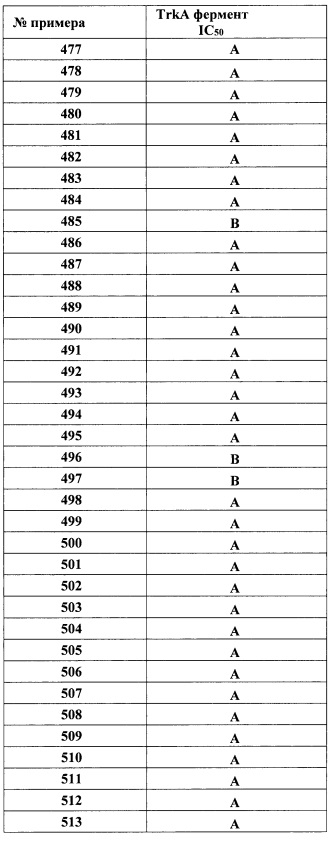
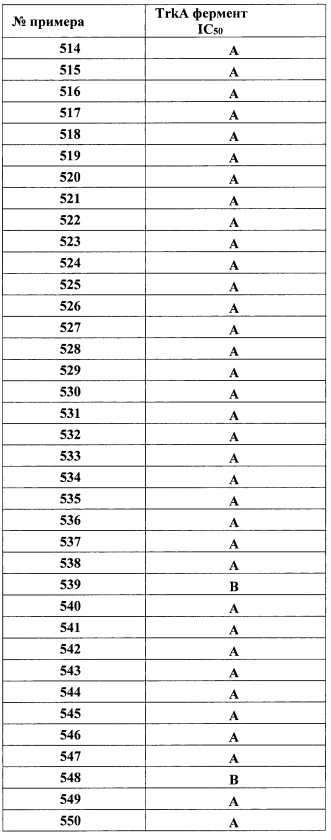
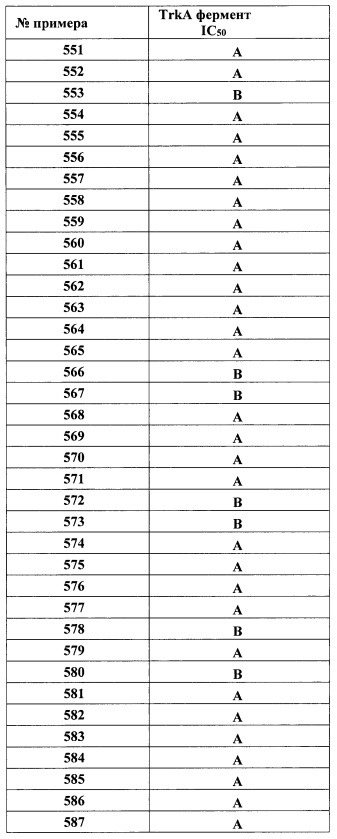
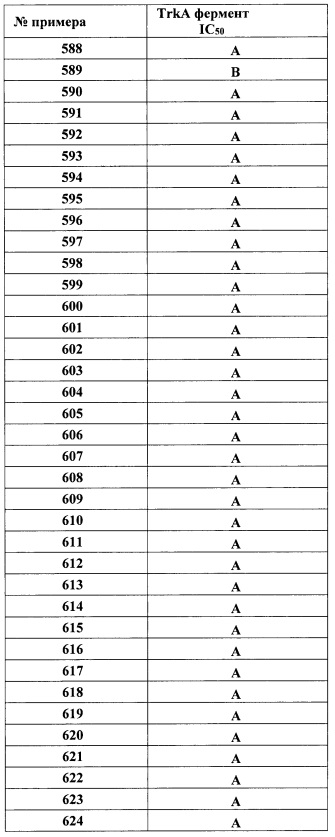
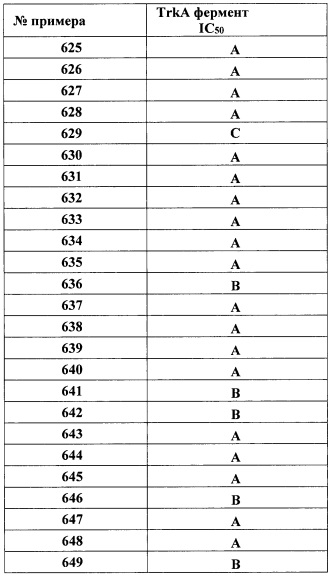
Получение синтетических Промежуточных соединений
Получение А
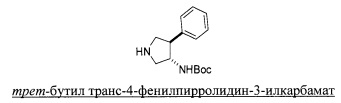
Стадия А: Получение транс-1-бензил-3-нитро-4-фенилпирролидина: К ДХМ (2 л) раствору (Е)-(2-нитровинил)бензола (149 г, 1,00 моль) добавили ТФК (19,5 мл, 0,250 поддерживая температуру реакции между -15 и -10°C. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 18 часов, затем промыли 2 н. NaOH (500 мл) и обработали 2 н. HCl (1 л). Полученную белую суспензию перемешивали в течение 1 часа, затем отфильтровали и промыли ДХМ. Затем к собранному белому твердому веществу добавили ДХМ (1 л) и 2 н. NaOH (750 мл), и перемешивали до полного растворения твердого вещества. После разделения фаз водный слой экстрагировали ДХМ (2×1 л). Объединенные органические слои высушили при помощи MgSO4, отфильтровали и концентрировали для получения указанного в заголовке соединения в виде грязновато-белого твердого вещества (205 г, выход 73%). МС (apci) m/z=283,1 (М+Н).
Стадия B: Получение транс-1-бензил-4-фенилпирролидин-3-амина: К суспензии транс-1-бензил-3-нитро-4-фенил-пирролидина (93,9 г, 333 ммоль) в EtOH (1,20 л) добавили концентрированную HCl (450 мл), затем небольшими частями добавили цинковую пыль (173 г, 2,66 моль) за 1,5 часа, поддерживая температуру в диапазоне 55-60°C. Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, затем охладили на бане изо льда и воды, затем добавили концентрированный раствор NH4OH (900 мл). Смесь (pH=10-11) отфильтровали, а собранный цинк промыли CHCl3. Затем выполнили разделение фаз фильтрата, а водный слой экстрагировали CHCl3 (2×400 мл). Объединенные органические слои промыли Н2O, насыщенным солевым раствором, высушили при помощи MgSO4, отфильтровали и концентрировали для получения указанного в заголовке соединения в виде маслянистого вещества янтарного цвета (85,0 г, выход 100%). МС (apci) m/z=253,2 (М+Н).
Стадия С: Получение транс-(1-бензил-4-фенил-пирролидин-3-ил)карбаминовой кислоты трет-бутилового эфира: К смеси транс-1-бензил-4-фенилпирролидин-3-амина (85,0 г, 333 ммоль), ТГФ (750 мл) и триэтиламина (69,6 мл, 500 ммоль) частями медленно добавили (Вос)2O (72,7 г, 333 ммоль) за 30 минут. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, и концентрировали in vacuo. Остаток растворили в CHCl3 и промыли водным раствором Na2CO3 и насыщенным солевым раствором. Органический слой высушили MgSO4, отфильтровали и концентрировали для получения указанного в заголовке соединения в виде бледно-желтого твердого вещества (116 г, выход 99%). МС (apci) m/z=353,0 (М+Н).
Стадия D: Получение трет-бутил транс-4-фенилпирролидин-3-илкарбамата: В реактор Парра объемом 2 галлона (8,8 л) загрузили транс-(1-бензил-4-фенил-пирролидин-3-ил)-карбаминовой кислоты трет-бутиловый эфир (114 г, 323 ммоль), EtOH (2 л) и 10% Pd/C (влажность 50%, 11,0 г). Реактор несколько раз продули N2, наполнили H2 до давления 56-57 psi и встряхивали при 80°C. Когда реакция завершилась, по данным анализа ВЭЖХ, реакционную смесь отфильтровали, а фильтрат концентрировали для получения неочищенного продукта в виде желтого твердого вещества. Неочищенный материал растерли с толуолом для получения указанного в заголовке продукта в виде белого твердого вещества (68,4 г, выход 78%). МС (apci) m/z=262,9 (М+Н).
Получение А2
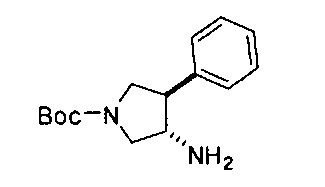
транс-трет-бутил 3-амино-4-фенилпирролидин-1-карбоксилат
Стадия А: Получение транс-Н-(1-бензил-4-фенилпирролидин-3-ил)-2,2,2-трифторацетамида: К раствору транс-1-бензил-4-фенилпирролидин-3-амина (Получение А, стадия В, 61,9 г, 245 ммоль) в ДХМ (400 мл) добавили ДИЭА (64,1 мл, 368 ммоль) и охладили смесь на ледяной бане. По каплям добавили трифторуксусный ангидрид (38,1 мл, 270 ммоль) за 30 минут, под атмосферой N2. После добавления смесь перемешивали в течение 30 минут, а затем концентрировали in vacuo. Остаток растворили в ДХМ и промыли насыщенным водным NaHCO3 и насыщенным солевым раствором. Раствор высушили при помощи MgSO4, отфильтровали и концентрировали in vacuo. Неочищенный материал обработали гексанами, а полученную желтую суспензию перемешивали при комнатной температуре в течение 1 часа. Твердое вещество собрали фильтрацией, промыли гексанами и высушили под вакуумом для получения указанного в заголовке соединения (78,7 г, выход 92%) в виде желтого твердого вещества. МС (apci) m/z=349,1 (М+Н).
Стадия В: Получение транс-трет-бутил-3-фенил-4-(2,2,2-трифторацетамидо)пирролидин-1-карбоксилата: Раствор транс-N-(1-бензил-4-фенилпирролидин-3-ил)-2,2,2-трифторацетамида (78,7 г, 226 ммоль) в EtOH (400 мл) продули N2 и обработали 20% Pd(OH)2 на активированном углероде (31,7 г, 45,2 ммоль). Смесь встряхивали при комнатной температуре под давлением H2 30 psi в реакторе Парра в течение 7 часов, а затем отфильтровали через стекловолоконную фильтровальную бумагу (GF/F) и концентрировали in vacuo. Остаток растворили в ДХМ (250 мл), затем добавили ТЭА (49,4 мл, 355 ммоль) и охладили на ледяной бане. Медленно добавили Boc2O (56,8 г, 260 ммоль) за 15 минут, и нагревали реакционную смесь до комнатной температуры, и перемешивали в течение 1 часа. Смесь промыли насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, затем высушили при помощи MgSO4. Раствор отфильтровали, концентрировали, а остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 40% EtOAc в гексанах для получения указанного в заголовке соединения в виде белого твердого вещества (63,2 г, выход 75%). 1Н ЯМР (CDCl3) δ 7,23-7,39 (m, 5Н), 6,36 (br s, 1Н), 4,47-4,55 (m, 1Н), 3,92-4,00 (m, 1Н), 3,78-4,00 (m, 1H), 3,50-3,59 (m, 1Н), 3,22-3,45 (m, 2Н), 1,49 (s, 9Н).
Стадия C: Получение транс-трет-бутил 3-амино-4-фенилпирролидин-1-карбоксилата: Раствор транс-трет-бутил 3-фенил-4-(2,2,2-трифторацетамидо)пирролидин-1-карбоксилата (63,2 г, 176 ммоль) в МеОН (200 мл) охладили на ледяной бане и добавили 2 н. NaOH (220 мл, 440 ммоль). Реакционную смесь оставили нагреваться до комнатной температуры в течение ночи, затем концентрировали приблизительно до 200 мл и разбавили Н2О (200 мл). Водную смесь экстрагировали ДХМ, а объединенные экстракты промыли насыщенным солевым раствором и высушили над Na2SO4. Раствор отфильтровали и концентрировали для получения указанного в заголовке соединения в виде светло-желтого маслянистого вещества (46,2 г, выход 99%). МС (apci) m/z=163,0 (М+Н-Вос).
Получение B
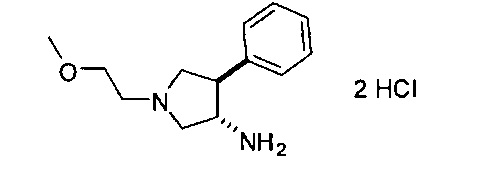
транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-амина дигидрохлорид
Стадия А: Получение трет-бутил транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-илкарбамата. К раствору трет-бутил транс-4-фенилпирролидин-3-илкарбамата (Получение А, 4,82 г, 17,5 ммоль) в сухом ДМФ (50 мл) последовательно добавили ДИЭА (9,12 мл, 52,4 ммоль) и 1-бром-2-метоксиэтан (1,97 мл, 20,9 ммоль). Смесь перемешивали при комнатной температуре в течение 46 часов, а затем вылили в Н2О (300 мл). Смесь экстрагировали ЕtOАс (3×150 мл), а объединенные экстракты промыли насыщенным солевым раствором, высушили над MgSO4/активированным углем, отфильтровали через слой SiO2, закрытый упакованным MgSO4, и элюировали EtOAc. Раствор концентрировали и высушили in vacuo, получив продукт в виде белого твердого вещества (5,15 г, выход 92%).МС (apci) m/z=321,1 (М+Н).
Стадия B: Получение транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-амина дигидрохлорида: К раствору трет-бутил транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-илкарбамата (5,10 г, 15,9 ммоль) в 2:1 ЕtOАс-МеОН (150 мл) добавили 4 н. HCl в диоксане (59,7 мл, 239 ммоль). Смесь перемешивали при комнатной температуре в течение 90 минут, а затем концентрировали in vacuo. Полученную пену обработали EtOAc (200 мл), обрабатывали ультразвуком в течение 5 минут и энергично перемешивали до образования тонкодисперсной белой суспензии. Суспензию отфильтровали, промыли EtOAc и высушили под вакуумом для получения указанного в заголовке соединения в виде белого порошка (5,10 г, выход 100%). МС (apci) m/z=221,1 (М+Н).
Получение C
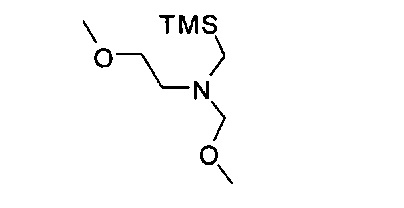
2-метокси-N-(метоксиметил)-N-((триметилсилил)метил)этанамин
Стадия А: Получение 2-метокси-N-((триметилсилил)метил)этанамина: К ДМСО (15 мл) раствору 2-метоксиэтанамина (14,2 мл, 163 ммоль) при 90°C через капельную воронку добавили ДМСО (10 мл) раствор (хлорметил)триметилсилана (11,4 мл, 81,5 ммоль) за 40 минут. Смесь нагревали при 90°C в течение 3,5 часов, затем охладили до комнатной температуры. Затем ее разбавили Н2O (150 мл) и экстрагировали EtOAc (2×150 мл). Объединенные органические экстракты промыли насыщенным солевым раствором (150 мл), высушили MgSO4, отфильтровали и концентрировали для получения продукта в виде желтого маслянистого вещества (8,14 г, выход 62%). МС (apci) m/z=162,0 (М+Н).
Стадия В: Получение 2-метокси-N-(метоксиметил)-N-((триметилсилил)метил)этанамина: МеОН (2,45 мл) раствор формальдегида (37% водный, 4,91 г, 60,6 ммоль) охладили до 0°C и обработали добавлением по каплям 2-метокси-N-((триметилсилил)метил)этанамина (8,14 г, 50,5 ммоль). Двухфазную смесь перемешивали при 0°C в течение 3 часов, затем добавили K2CO3 (6,97 г, 50,5 ммоль) и перемешивали смесь при 0°C в течение 1 часа. Желтое маслянистое вещество декантировали на K2CO3 (2,00 г, 14,4 ммоль) и перемешивали смесь при комнатной температуре в течение 2 часов. После декантации желтого маслянистого вещества, твердый K2CO3 промыли Et2O (2×10 мл), а промывочные растворы Et2O объединили с декантированным желтым маслянистым веществом и концентрировали на ротационном испарителе для получения продукта в виде желтого маслянистого вещества (9,92 г, выход 96%). 1Н ЯМР (CDCl3) δ 4,00 (s, 2Н), 3,37-3,43 (m, 2Н), 3,29 (s, 3Н), 3,19 (s, 3Н), 2,77-2,82 (m, 2Н), 2,18 (s, 2Н), 0,00 (s, 9Н).
Получение D
(3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-амина дигидрохлорид
Стадия A: Получение (R)-3-циннамоил-4-фенилоксазолидин-2-она: ТГФ (50 мл) раствор (R)-4-фенилоксазолидин-2-она (5,90 г, 36,2 ммоль) охладили до -78°C и по каплям обработали бис(триметилсилил)амидом лития (36,9 мл, 36,9 ммоль, 1,0 М в ТГФ) за 15 минут. После 15-минутного перемешивания при 78°C, затем ввели ТГФ (10 мл) раствор циннамоилхлорида (6,33 г, 38,0 ммоль). Смесь перемешивали в течение 1 часа при -78°C и 2 часа при комнатной температуре, затем погасили насыщенным раствором NaHCO3 (50 мл) и перемешивали в течение 1 часа. Смесь разбавили EtOAc (200 мл), промыли водой и насыщенным солевым раствором, высушили над MgSO4, отфильтровали и концентрировали для получения продукта в виде бледно-желтого твердого вещества (10,6 г, выход 99,9%). МС (apci) m/z=293,9 (М+Н).
Стадия B: Получение (R)-3-((3S,4R)-(2-метоксиэтил)-фенилпирролидин-3-карбонил)-4-фенилоксазолидин-2-она: Толуольный (500 мл) раствор (11)-3-циннамоил-4-фенилоксазолидин-2-она (8,00 г, 27,3 ммоль) и ТФК (0,210 мл, 2,73 ммоль) сначала охладили до 5-10°C, затем по каплям добавили толуольный (30 мл) раствор 2-метокси-N-(метоксиметил)-N-((триметилсилил)метил)этанамина (Получение C, 8,40 г, 40,9 ммоль). Полученную смесь нагревали до комнатной температуры и перемешивали в течение 3 часов, затем промыли насыщенным раствором NaHCO3 и водой, высушили MgSO4, отфильтровали и концентрировали in vacuo. Неочищенный материал очистили колоночной хроматографией на диоксиде кремния, элюируя 16-20% EtOAc в гексанах, для получения продукта (6,5 г, выход 60%). МС (apci) m/z=395,2 (М+Н).
Стадия C: Получение (3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-карбоновой кислоты: К 1 М водному раствору LiOH (41,2 мл, 41,2 ммоль) при 0°C добавили H2O2 (3,37 мл, 33,0 ммоль, 30 вес.%). Затем смесь добавили к раствору (R)-3-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-карбонил)-4-фенилоксазолидин-2-она (6,50 г, 16,5 ммоль) в ТГФ (100 мл) за 10 минут при 0°C. После перемешивания в течение 1 часа, ввели 2,0 М водный раствор Na2SO3 (33,0 мл, 65,9 ммоль) при 0°C и нагрели реакционную смесь до комнатной температуры. После перемешивания в течение 10 минут, смесь промыли EtOAc (50 мл). Водный слой подкислили 1 н. HCl до pH 3-5, затем обработали NaCl (10 г), затем экстрагировали 10% iPrOH/ДХМ. Органический слой высушили при помощи MgSO4, отфильтровали и концентрировали для получения продукта (4,11 г, выход 100%). МС (apci) m/z=250,1 (М+Н).
Стадия D: Получение бензил (3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-илкарбамата: К раствору (3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-карбоновой кислоты (4,11 г, 16,5 ммоль) в толуоле (70 мл) добавили ТЭА (5,74 мл, 41,2 ммоль), затем дифенилфосфорилазид (4,99 мл, 23,1 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, а затем нагревали с дефлегматором в течение 1 часа. Затем добавили бензиловый спирт (3,42 мл, 33,0 ммоль) и нагревали реакционную смесь с дефлегматором в течение 15 часов. Реакционную смесь обработали EtOAc, промыли водой, высушили над MgSO4, отфильтровали и концентрировали in vacuo. Неочищенный материал очистили колоночной хроматографией на диоксиде кремния, элюируя 1% МеОН/ДХМ для получения продукта (2,5 г, выход 43%). МС (apci) m/z=355,2 (М+Н).
Стадия Е: Получение (3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-амина дигидрохлорида: Бензил (3S,4R)-1-(2-метоксиэтил-4-фенилпирролидин-3-илкарбамат (0,257 г, 0,725 ммоль) и ТФК (3,91 мл, 50,8 ммоль) нагревали при 60°C в течение 17 часов. Реакционную смесь концентрировали in vacuo, используя толуол для азеотропной перегонки, затем обработали 2 н. HCl в Et2O и снова концентрировали для получения указанного в заголовке соединения (0,21 г, выход 100%) в виде грязновато-белого твердого вещества. МС (apci) m/z=221,2 (М+Н).
Получение Е
(3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина трифторацетат
Стадия А: Получение (R,Е)-3-(3-(3,5-дифторфенил)акрилоил)-4-фенилоксазолидин-2-она: К раствору (E)-3-(3,5-дифторфенил)акриловой кислоты (10,0 г, 54,3 ммоль) в Еt2О (150 мл) при 0°C добавили ДИЭА (9,48 мл, 54,3 ммоль), затем пивалоилхлорид (6,69 мл, 54,3 ммоль). Смесь перемешивали при 0°C в течение 1 часа и охладили до -78°C. В это же время (R)-4-фенилоксазолидин-2-он (8,86 г, 54,3 ммоль) в ТГФ (200 мл) охладили до -78°C и медленно добавили бутиллитий (21,7 мл, 2,5 М, 54,3 ммоль). Смесь перемешивали в течение 20 минут при -78°C и перенесли через канюлю в раствор смешанного ангидрида. Объединенную смесь перемешивали при -78°C в течение 15 минут, оставили нагреваться до 0°C и перемешивали еще 30 минут. Реакционную смесь погасили насыщенным раствором NH4Cl (25 мл), разбавили EtOAc (600 мл), промыли водой, NaHCO3 и насыщенным солевым раствором, высушили над MgSO4 и концентрировали in vacuo. Неочищенный материал очистили колоночной хроматографией на диоксиде кремния, элюируя 10-20% этилацетата в гексанах, для получения продукта (11,0 г, выход 61,5%).
Стадия В: Получение (3S,4R)-4-(3,5-дифторфенил)- -(2-метоксиэтил)пирролидин-3-амина трифторуксуснокислой соли: Получили по способам, описанным в Получении D, стадии В-Е, заменяя (R)-3-циннамоил-4-фенилоксазолидин-2-он на (R,E)-3-(3-(3,5-дифторфенил)акрилоил)-4-фенилоксазолидин-2-он, для получения указанного в заголовке соединения (1,70 г, выход 102%). МС (apci) m/z=257,2 (М+Н).
Получение F
(3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин
Получили по способу, описанному в Получении D, заменяя циннамоилхлорид на (Е)-3-(3,4-дифторфенил)акрилоилхлорид. МС (apci) m/z=257,1 (М+Н).
Получение G
(3S,4R)-1-(2-метоксиэтил)-4-(3-(трифторметил)фенил)пирролидин-3-амина дигидрохлорид
Стадия А: Получение трет-бутил (3S,4R)-1-(2-метоксиэтил)-4-(3-(трифторметил)фенил)пирролидин-3-илкарбамата: Раствор трет-бутил (3S,4R)-4-(3-(трифторметил)фенил)-пирролидин-3-илкарбамата (100 мг, 0,303 ммоль, имеется в продаже), N,N-диэтилпропан-2-амина (0,145 мл, 0,908 ммоль) и 1-бром-2-метоксиэтана (0,0361 мл, 0,363 ммоль) в ДМФ (1 мл) перемешивали при комнатной температуре в течение 2 часов, затем нагревали до 60°C в течение 4 часов, затем охлаждали до комнатной температуры в течение ночи. После разделения между EtOAc и насыщенным раствором NaHCO3 (по 10 мл каждого), органический слой промыли водой и насыщенным солевым раствором (2×10 мл каждого), высушили над Na2SO4, отфильтровали и концентрировали для получения неочищенного продукта в виде белого твердого вещества (80 мг, выход 68%). ЖХМС (apci) m/z=389,1 (М+Н).
Стадия В: Получение (3S,4R)-1-(2-метоксиэтил)-4-(3-(трифторметил)фенил)пирролидин-3-амина дигидрохлорида: Раствор трет-бутил (3S,4R)-1-(2-метоксиэтил)-4-(3-(трифторметил)фенил)пирролидин-3-илкарбамата (80,0 мг, 0,206 ммоль) в 5-6 н. HCl в IPA (4,12 мл, 20,6 ммоль) перемешивали при комнатной температуре в течение 1 часа, затем концентрировали in vacuo и растерли с Еt2О для получения продукта в виде бежевого твердого вещества (74 мг, выход 99,5%). ЖХМС (apci) m/z=289,1 (М+Н).
Получение Н
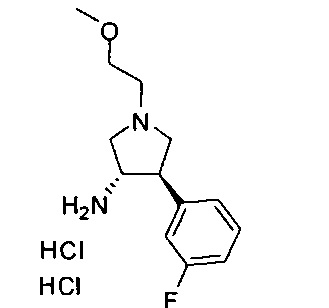
(3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид
Получили по способу Получения G, заменяя трет-бутил (3S,4R)-4-(3-(трифторметил)фенил)пирролидин-3-илкарбамат на трет-бутил (3S,4R)-4-(3-фторфенил)пирролидин-3-илкарбамат, для получения указанного в заголовке соединения. ЖХМС (apci) m/z=239,1 (М+Н).
Получение I
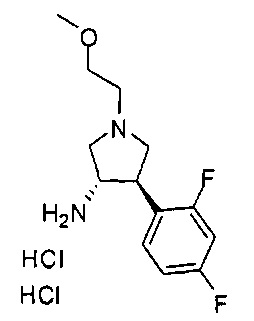
(3S,4R)-4-(2,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид
Получили по способу Получения G, заменяя трет-бутил (3S,4R)-4-(3-(трифторметил)фенил)пирролидин-3-илкарбамат на трет-бутил (3S,4R)-4-(2,4-дифторфенил)пирролидин-3-илкарбамат, для получения указанного в заголовке соединения. ЖХМС (apci) m/z=257,1 (М+Н).
Получение J
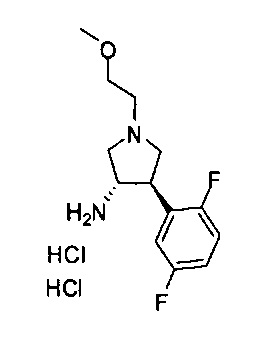
(3S,4R)-4-(2,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид
Получили по способу Получения G, заменяя трет-бутил (3S,4R)-4-(3-(трифторметил)фенил)пирролидин-3-илкарбамат на трет-бутил (3S,4R)-4-(2,5-дифторфенил)пирролидин-3-илкарбамат, для получения указанного в заголовке соединения. ЖХМС (apci) m/z=257,1 (М+Н).
Получение K
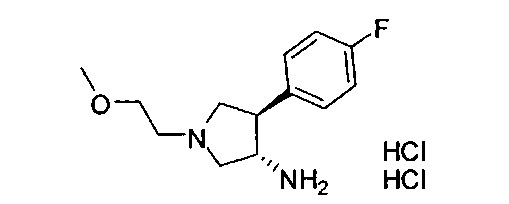
(3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид
Получили по способу, описанному в Получении D, заменяя циннамоилхлорид на (Е)-3-(4-фторфенил)акрилоилхлорид. МС (apci) m/z=239,1 (М+Н).
Получение L1
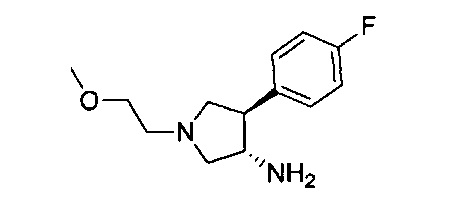
(3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амин
Стадия А: Получение (Е)-1-фтор-4-(2-нитровинил)бензола: Уксусную кислоту (2,0 л, 35,5 моль) и ацетат аммония (310,5 г, 4,03 моль) перемешивали при комнатной температуре в течение 1 часа, затем добавили нитрометан (611 мл, 11,3 моль) и 4-фторбензальдегид (200 г, 1,61 ммоль), и нагревали реакционную смесь до 90°C в течение 3 часов. Реакционную смесь оставили остывать до комнатной температуры, затем за 2 часа добавили Н2О (4 л) при механическом перемешивании. Суспензию перемешивали в течение 1 часа, затем отфильтровали и промыли 2:1 водой/уксусной кислотой (500 мл). Твердые вещества высушили в вакуумной печи (50°C) для получения указанного в заголовке продукта в виде бледно-желтого твердого вещества (238 г, 1,42 моль, выход 88%). 1Н ЯМР (CDCl3) δ 7,98 (1Н), 7,55 (3Н), 7,16 (2Н).
Стадия В: Получение транс-3-(4-фторфенил)-1-(2-метоксиэтил)-4-нитро-пирролидина: К суспензии (Е)-1-фтор-4-(2-нитровинил)бензола (201 г, 1,20 моль) в ДХМ (1,09 л) и ТФК (9,3 мл, 120 ммоль) за 30 минут по каплям добавили 2-метокси-N-(метоксиметил)-N-((триметилсилил)метил)этанамин (Получение C; 383 г, 1,86 моль), а внутреннюю температуру реакционной смеси поддерживали в диапазоне 23-36°C, охлаждая на ледяной бане. Реакционную смесь вылили в водный фосфатный буферный раствор (pH 7, 500 мл) и разбавили ДХМ (300 мл). Фазы разделили, а водную фазу экстрагировали ДХМ (400 мл). Органические фазы объединили, промыли насыщенным солевым раствором (300 мл), высушили (MgSO4), отфильтровали и концентрировали под пониженным давлением. Неочищенное маслянистое вещество очистили колоночной хроматографией на диоксиде кремния, элюируя 40% EtOAc в гептане, для получения указанного в заголовке соединения в виде желтого маслянистого вещества (245 г, выход 76%). МС (apci) m/z=269,1 (М+Н).
Стадия С: Получение транс-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина: К раствору транс-3-(4-фторфенил)-1-(2-метоксиэтил)-4-нитропирролидина (289 г, 1,08 моль) в EtOH (1 л) добавили оксид платины (IV) (24,5 г, 108 ммоль) в сосуде Парра и установили на встряхиватель Парра. Сосуд вакуумировали и наполнили азотом (3х), затем вакуумировали и наполнили водородом (60 psi). Сосуд при необходимости пополняли водородом до завершения реакции. Реакционную смесь отфильтровали через Celite® и промыли МеОН (50 мл), затем концентрировали под пониженным давлением для получения указанного в заголовке соединения в виде желтого маслянистого вещества (243 г, выход 95%). МС (apci) m/z=239,1 (М+Н).
Стадия D: Получение (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина (2S,3S)-2,3-бис(4-метилбензоилокси)сукцината: К раствору (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина (120 г, 504 ммоль) в ТГФ (3,0 л) и Н2О (333 мл) добавили ди-п-толуоил-D-винную кислоту (195 г, 504 ммоль). Перемешивали при комнатной температуре в течение 1 часа, затем поместили в морозильную камеру (-11°C) на 18 часов. Смесь перемешивали для получения суспензии, отфильтровали и промыли Et2O (4×100 мл). Твердое вещество высушивали в вакуумной печи (40°C) в течение 4 часов, затем дважды перекристаллизовали по следующему способу: твердое вещество растворили в ТГФ (1,06 мл) и H2O (118 мл) при нагревании до 45°C, затем оставили остывать до комнатной температуры на 2 часа, затем поместили в морозильную камеру (-1°C) на 18 часов; смесь перемешивали для получения суспензии, отфильтровали и промыли Et2O (4×100 мл). После двух перекристаллизации твердое отфильтровали и промыли Et2O (4×100 мл). После двух перекристаллизации твердое вещество высушивали в вакуумной печи (40°С) в течение 18 часов для получения указанного в заголовке соединения в виде белого кристаллического вещества (96 г, выход 31%). МС (apci) m/z=239,2 (М+Н).
Стадия Е: Получение (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина: (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина (2S,3S)-2,3-бис(4-метилбензоилокси)сукцинат (20 г, 32,0 ммоль) растворили в ДХМ (300 мл) и промыли 1 М NaOH (2×200 мл). Объединенные водные фазы экстрагировали ДХМ (200 мл). Объединенные органические экстракты промыли насыщенным солевым раствором (200 мл), высушили (MgSO4), отфильтровали и концентрировали, затем высушили под вакуумом для получения указанного в заголовке соединения в виде желтого маслянистого вещества (6,17 г, 81%, э.и.>99%). МС (apci) m/z=239,1 (М+Н).
Следующие пирролидиновые промежуточные соединения получили по способу Получения L1, используя соответствующий бензальдегид на Стадии А и заменяя EtOH и оксид платины (IV) на МеОН и никель Ренея, соответственно, на Стадии С. Для получения L3, 90% ТГФ/Н2О на Стадии D заменили на 85% МеОН/Н2О.
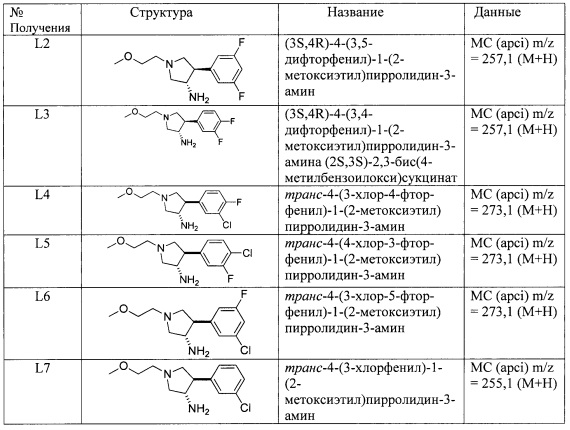
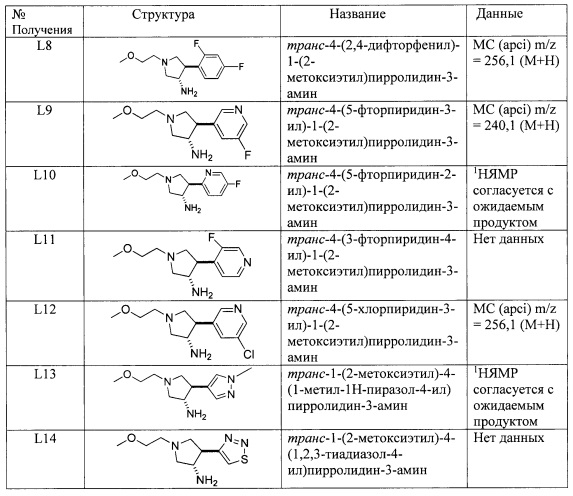
Получение L15
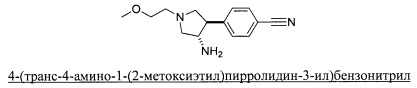
Получили по способу, описанному в Получении L1, Стадии А-С, заменяя 4-фторбензальдегид на 4-формилбензонитрил на Стадии А, и заменяя EtOH и оксид платины (IV) на МеОН, Zn (пыль) и насыщенный раствор NH4Cl, соответственно, на Стадии С. МС (apci) m/z=246,1 (М+Н).
Получение L16
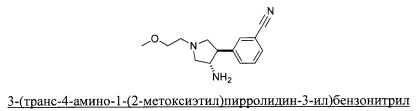
Получили по способу, описанному в Получении L1, Стадии A-С, заменяя 4-фторбензальдегид на 3-формилбензонитрил на Стадии A, и заменяя EtOH и оксид платины (IV) на MeOH, Zn (пыль) и насыщенный раствор NH4Cl, соответственно, на Стадии С. МС (apci) m/z=246,2 (М+Н).
Получение М
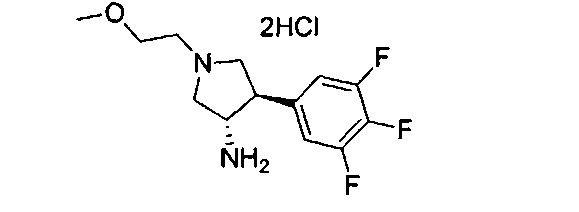
(3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-амина дигидрохлорид
Получили по способу, описанному в Получении D, заменяя циннамоилхлорид на (Е)-3-(3,4,5-трифторфенил)акрилоилхлорид. 1Н ЯМР (D2O) δ 7,06-7,10 (m, 2Н), 4,13-4,20 (m, 1Н), 3,92-3,99 (m, 2H), 3,71-3,74 (m, 1H), 3,57-3,63 (m, 3H), 3,41-3,49 (m, 3H), 3,25 (s, 3H).
Получение N
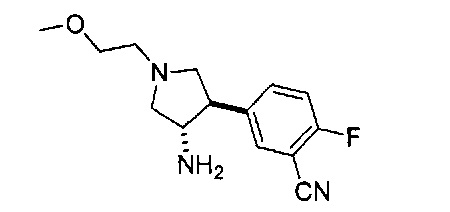
Транс-5-(4-амино-1-(2-метоксиэтил)пирролидин-3-ил)-2-фторбензонитрил
Стадия А: (Е)-2-фтор-5-(2-нитровинил)бензонитрил. К раствору 2-фтор-5-формилбензонитрила (3,84 г, 25,0 ммоль) в 3:1 CH3NO2/CH3CN (25 мл) добавили DMAP (0,305 г, 2,50 ммоль) и перемешивали смесь при комнатной температуре в течение 23 часов. Смесь охладили на ледяной бане и добавили Ac2O (3,54 мл, 37,5 ммоль). Смесь перемешивали в течение 5 минут, оставили нагреваться до комнатной температуры и перемешивали в течение 1 часа. Смесь концентрировали до желтого твердого вещества. Твердое вещество суспендировали в iPrOH (70 мл) и перемешивали в течение 10 минут. Суспензию собрали вакуумной фильтрацией, осадок на фильтре промыли iPrOH и высушили в вакууме для получения указанного в заголовке соединения в виде светлого желто-коричневого порошка (3,36 г, 70%). 1Н ЯМР (CDCl3) δ 7,96 (d, 1H), 7,79-7,88 (m, 2Н), 7,57 (d, 1H), 7,36 (t, 1H).
Стадия B: Транс-2-фтор-5-(1-(2-метоксиэтил)-4-нитропирролидин-3-ил)бензонитрил: Используя (Е)-2-фтор-5-(2-нитровинил)бензонитрил на Стадии B способа, описанного в Получении L1, получили указанное в заголовке соединение в виде светло-золотистого сиропообразного вещества (1,56 г, 53%). МС (apci) m/z=294,1 (М+Н).
Стадия С: Транс-5-(4-амино-1-(2-метоксиэтил)пирролидин-3-ил)-2-фторбензонитрил: Раствор транс-2-фтор-5-(1-(2-метоксиэтил)-4-нитропирролидин-3-ил)бензонитрила (450 мг, 1,53 ммоль) в MeOH (6,0 мл) охладили до 0°C. Последовательно добавили Zn пыль (1,00 мг, 15,3 ммоль) и насыщенный водный раствор NH4Cl (1,0 мл) и перемешивали смесь в течение 5 минут. Смесь оставили достигать комнатной температуры и перемешивали до завершения реакции, по данным анализа ЖХМС. Смесь отфильтровали через упакованный Celite®, используя MeOH для промывания и элюирования, а фильтрат концентрировали до бесцветного сиропообразного вещества. Сиропообразное вещество обработали 1 М K2CO3 (15 мл), смешали и экстрагировали CH2Cl2 (3Х). Объединенные CH2Cl2 экстракты высушили над Na2SO4, отфильтровали и концентрировали для получения указанного в заголовке соединения в виде бесцветного сиропообразного вещества (412 мг, 100%). МС (apci) m/z=264,1 (М+Н).
Получение O
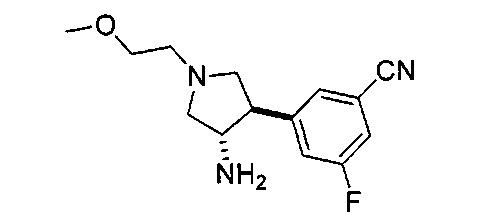
Транс-3-(4-амино-1-(2-метоксиэтил)пирролидин-3-ил)-5-фторбензонитрил
Стадия А: 3-фтор-5-формилбензонитрил: Раствор 3-бром-5-фторбензонитрила (5,00 г, 25,0 ммоль) в сухом ТГФ (25 мл) охладили до 0°C и по каплям добавили 2 М iPrMgCl (15,0 мл, 30,0 ммоль) в ТГФ за 5 минут. Смесь перемешивали при 0°C в течение 15 минут, затем при комнатной температуре в течение 1 часа. Смесь охладили до 0°C и добавили сухой ДМФ (5,81 мл, 75,0 ммоль). Смесь перемешивали в течение 17 часов, за это время температура достигла комнатной температуры через 2 часа. Смесь добавили к ледяной воде (150 мл) и Et2O (100 мл). Двухфазную смесь перемешали и обработали 6 М HCl до водного pH=3. Органический слой удалили, а водный слой экстрагировали Et2O (2Х). Объединенные Et2O фракции промыли насыщенным раствором NaCl и высушили над MgSO4/активированным углем. Высушенный раствор отфильтровали через слой SiO2, элюируя Et2O. Фильтрат концентрировали для получения указанного в заголовке соединения в виде твердого желтого вещества, которое высушили в вакууме (3,68 г, 99%). 1Н ЯМР (CDCl3) δ 10,0 (s, 1Н), 8,00 (s, 1Н), 7,81-7,86 (m, 1Н), 7,62-7,67 (m, 1Н).
Стадия В: Транс-3-(4-амино-1-(2-метоксиэтил)пирролидин-3-ил)-5-фторбензонитрил: Указанное в заголовке соединение получили, используя 3-фтор-5-формилбензонитрил в способе, описанном для получения транс-5-(4-амино-1-(2-метоксиэтил)пирролидин-3-ил)-2-фторбензонитрила (Получение N). Соединение выделили в виде бесцветного сиропообразного вещества (542 мг, 93%). МС (apci) m/z=264,1 (М+Н).
Получение Р
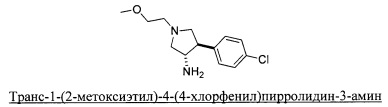
Стадия А: Транс-3-(4-хлорфенил)-1-(2-метоксиэтил)-4-нитропирролидин: Используя (Е)-1-хлор-4-(2-нитровинил)бензол на стадии В способа, описанного в Получении L1, получили указанное в заголовке соединение в виде вязкого бесцветного маслянистого вещества (5,10 г, 64%). МС (apci) m/z=285,0 (М+Н).
Стадия В: Транс-1-(2-метоксиэтил)-4-(4-хлорфенил)пирролидин-3-амин: К суспензии никеля Ренея 2800 (50 вес. % в Н2О, 0,873 г, 5,10 ммоль) в МеОН (25 мл) добавили транс-3-(4-хлорфенил)-1-(2-метоксиэтил)-4-нитропирролидин (2,90 г, 10,2 ммоль) в МеОН (25 мл). Смесь продули газообразным Н2 и перемешивали под атмосферой Н2 из баллона в течение 16 часов. Смесь продули газообразным N2 и отфильтровали через упакованный Celite®, используя МеОН для промывания и элюирования. Фильтрат концентрировали до мутного маслянистого вещества. Маслянистое вещество растворили в CH2Cl2 и высушили над NaSO4/активированным углем. Раствор отфильтровали и концентрировали для получения указанного в заголовке соединения в виде светлого золотистого маслянистого вещества, которое высушили в вакууме (2,46 г, 95%). МС (apci) m/z=255,1 (М+Н).
В Таблице 1 представлен список имеющихся в продаже пиразоловых промежуточных соединений, которые были использованы в синтезе соединений, описанных в Примерах.

Промежуточное соединение P1
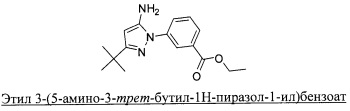
К суспензии этил 3-гидразинилбензоата гидрохлорида (500 мг, 2,31 ммоль) в EtOH (20 мл) добавили 4,4-диметил-3-оксопентаннитрил (318 мг, 2,54 ммоль). Реакционную смесь нагревали с дефлегматором в течение 18 часов, затем охладили до комнатной температуры и концентрировали in vacuo. Неочищенный продукт очистили колоночной хроматографией на диоксиде кремния, элюируя 0-5% МеОН в ДХМ, для получения продукта в виде желтого маслянистого вещества (154 мг, выход 23%). МС (apci) m/z=288,2 (М+Н).
Соединения в Таблице 2 были получены по способу, описанному для Промежуточного соединения Р1, с заменой 4,4-диметил-3-оксопентаннитрила на соответствующий цианокетон, и этил 3-гидразинилбензоата гидрохлорида на соответствующий гидразин.
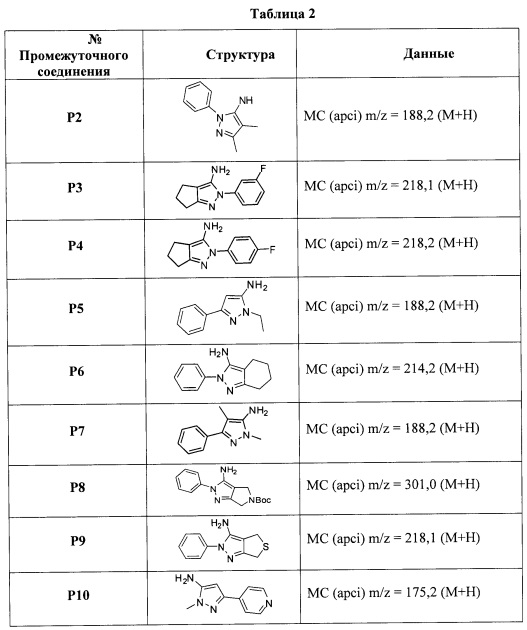
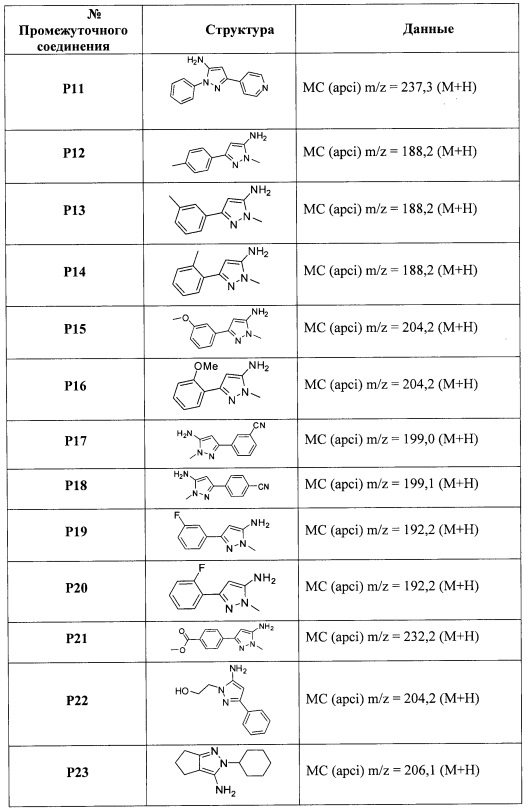
Промежуточное соединение P101
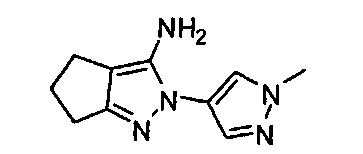
2-(1-метил-1Н-пиразол-4-ил)-2,4,5,6-тетрагидроциклопента-[с]пиразол-3-амин
Стадия А: Получение ди-трет-бутил 1-(1-метил-1Н-пиразол-4-ил)гидразин-1,2-дикарбоксилата: К раствору 4-бром-1-метил-1Н-пиразола (1,93 мл, 18,6 ммоль) в эфире (37,3 мл), охлажденному до -78°C, добавили nBuLi (23,3 мл, 37,3 ммоль). После перемешивания при -78°C в течение 30 минут, по каплям добавили раствор ди-трет-бутил азодикарбоксилата (4,29 г, 18,6 ммоль) в Et2O (37,3 мл, 18,6 ммоль). Через 1 час реакционную смесь нагрели до -20°C и погасили льдом. После нагревания до комнатной температуры, смесь отфильтровали и промыли Et2O. Полученное твердое вещество растворили в смеси ДХМ и воды, и эту смесь разделили на фазы. Органический слой высушили при помощи MgSO4, отфильтровали и концентрировали in vacuo для получения первой партии продукта в виде белого твердого вещества (1,64 г, выход 28%). Вторую партию продукта выделили из фильтрата колоночной хроматографией на диоксиде кремния, элюируя 40-60% гексанов в EtOAc (0,51 г, выход 8,8%). МС (apci) m/z=313,0 (М+Н).
Стадия В: Получение 2-(1-метил-1Н-пиразол-4-ил)-2,4,5,6-тетрагидроциклопента-[с]пиразол-3-амина: К раствору ди-трет-бутил 1-(1-метил-1Н-пиразол-4-ил)гидразин-1,2-дикарбоксилата (103 мг, 0,330 ммоль) в EtOH (1,65 мл, 0,330 ммоль) добавили концентрированную HCl (137 мкл, 1,65 ммоль). Смесь перемешивали при комнатной температуре в течение 5 минут, затем охладили на ледяной бане, затем добавили 2-оксоциклопентанкарбонитрил (36,0 мг, 0,330 ммоль). После перемешивания в течение 5 минут, реакционную смесь нагревали до комнатной температуры в течение ночи. Реакционную смесь концентрировали и разделили в воде и ДХМ. После разделения фаз водный слой подщелочили (pH 10), а затем экстрагировали ДХМ (3×10 мл). Объединенные органические экстракты высушили при помощи MgSO4, отфильтровали и концентрировали in vacuo. Неочищенный материал очистили обращенно-фазовой колоночной хроматографией, элюируя 0-100% ацетонитрила в воде, для получения продукта в виде твердого желтого вещества (4,5 мг, выход 6,7%). МС (apci) m/z=204,1 (М+Н).
Промежуточное соединение P102
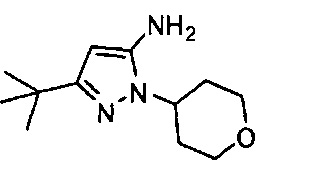
3-трет-бутил-1-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразол-5-амин
Стадия А: Получение (тетрагидро-2Н-пиран-4-ил)гидразина гидрохлорида: Суспензию дигидро-2Н-пиран-4(3Н)-она (2,00 г, 20,0 ммоль) и трет-бутил гидразинкарбоксилата (2,64 г, 20,0 ммоль) в гексанах (20,0 мл) нагревали с дефлегматором в течение 2 часов. После охлаждения добавили комплекс ВН3-ТГФ (20,0 мл, 20,0 ммоль) и перемешивали реакционную смесь в течение 1 часа. Затем смесь обработали 4 н. HCl в диоксане (20,0 мл, 79,9 ммоль), затем 3 каплями воды. После перемешивания при комнатной температуре в течение 1 часа, реакционную смесь отфильтровали и промыли EtOAc для получения продукта в виде твердого вещества (2,39 г, выход 78,4%). МС (apci) m/z=117,0 (М+Н).
Стадия B: Получение 3-трет-бутил-1-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразол-5-амина: Получили по способу, описанному для получения Промежуточного соединения Р1, заменяя (тетрагидро-2Н-пиран-4-ил)гидразин дигидрохлоридом этил 3-гидразинбензоата гидрохлорид, для получения продукта в виде желтого маслянистого вещества (0,472 г, выход 99,9%). МС (apci) m/z=224,1 (М+Н).
Промежуточное соединение Р103
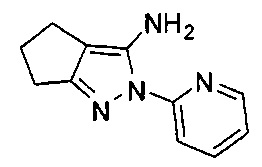
2-(пиридин-2-ил)-2,5,6-тетрагидроциклопента[с]пиразол-3-амин
Стадия А: Получение 2-(2-(пиридин-2-ил)гидразоно)циклопентан-карбонитрила: Раствор 2-гидразинилпиридина (0,200 г, 1,83 ммоль) и 2-оксоциклопентанкарбонитрила (0,200 г, 1,83 ммоль) в MeOH (9,16 мл) обработали концентрированной HCl (0,764 мл, 9,16 ммоль) и нагревали с дефлегматором в течение 16 часов. Реакционную смесь концентрировали in vacuo, а затем разделили в воде и ДХМ. После разделения фаз, водный слой промыли ДХМ, подщелочили (насыщенным раствором NaHCO3, pH 10) и экстрагировали ДХМ. Объединенные органические экстракты высушили над MgSO4, отфильтровали и концентрировали. Неочищенный материал очистили колоночной хроматографией на диоксиде кремния, элюируя 100% EtOAc, для получения продукта (0,289 г, выход 78,6%). МС (apci) m/z=201,2 (М+Н).
Стадия В: Получение 2-(пиридин-2-ил)-2,4,5,6-тетрагидроциклопента[с]пиразол-3-амина: Раствор 2-(2-(пиридин-2-ил)гидразоно)циклопентанкарбонитрила (0,243 г, 1,21 ммоль) в EtOH (6,06 мл, 1,21 ммоль) обработали 6 М HCl (0,202 мл, 1,21 ммоль) и нагревали с дефлегматором в течение 3 дней. После удаления растворителя, неочищенный остаток разбавили в воде, подщелочили (насыщенным NaHCO3, pH 10) и экстрагировали ДХМ. Объединенные органические экстракты высушили над MgSO4, отфильтровали и концентрировали. Неочищенный материал очистили колоночной хроматографией на диоксиде кремния, элюируя 50% EtOAc в гексанах, для получения продукта (0,198 г, выход 81,6%). МС (apci) m/z=201,2 (М+Н).
Промежуточное соединение Р104
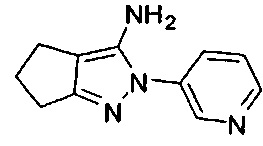
2-(пиридин-3-ил)-2,5,6-тетрагидроциклопента[с]пиразол-3-амин
Получили по способу, описанному выше для Промежуточного соединения Р103, заменяя 3-гидразинилпиридином 2-гидразинилпиридин, для получения указанного в заголовке продукта. МС (apci) m/z=201,1 (М+Н).
Промежуточное соединение Р105
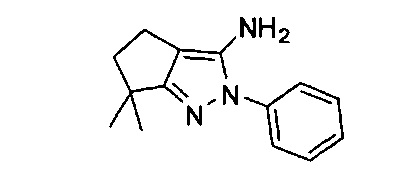
6,6-диметил-2-фенил-2,5,6-тетрагидроциклопента[с]пиразол-3-амин
Стадия A: Получение 5-хлор-2,2-диметилпентаннитрила: Изобутиронитрил (1,38 г, 20,0 ммоль) и 1-бром-3-хлорпропан (3,46 г, 22,0 ммоль) последовательно добавили к 1 М раствору бис(триметилсилил)амида лития (20,0 мл, 20,0 ммоль) при перемешивании. После перемешивания при 70°C в течение 16 часов, реакционную смесь погасили водой, затем экстрагировали ДХМ. Объединенные органические слои высушили при помощи MgSO4, отфильтровали и концентрировали in vacuo для получения 5-хлор-2,2-диметилпентаннитрила (2,91 г, выход 100%).1Н ЯМР (CDCl3) δ 3,57-3,61 (m, 2Н), 1,94-2,02 (m, 2Н), 1,67-1,72 (m, 2Н), 1,37 (s, 6Н).
Стадия B: Получение 2,2-диметилгександинитрила: Суспензию 5-хлор-2,2-диметилпентаннитрила (2,91 г, 20,0 ммоль) и NaCN (1,57 г, 32,0 ммоль) в ДМФ (20,0 мл) и воде (1 мл) нагревали при 100°C в течение 16 часов. После охлаждения реакционную смесь разбавили водой и нагревали с дефлегматором в течение 30 минут, затем охладили, вылили в воду и перемешивали в течение 3 часов. Затем раствор экстрагировали Et2O. Объединенные Et2O экстракты промыли H2O, высушили при помощи MgSO4, отфильтровали и концентрировали in vacuo для получения продукта (2,20 г, выход 80,7%). 1Н ЯМР (CDCl3) δ 2,42-2,47 (m, 2Н), 1,83-1,92 (m, 2Н), 1,67-1,72 (m, 2Н), 1,39 (s, 6Н).
Стадия C: Получение 3,3-диметил-2-оксоциклопентанкарбонитрила: Суспензию KOtBu (0,511 г, 4,55 ммоль) в толуоле (18,4 мл) обработали толуольным (2,0 мл) раствором 2,2-диметилгександинитрила (1,00 г, 7,34 ммоль) и нагревали при 80°C в течение 2 часов. Затем реакционную смесь охладили до комнатной температуры и погасили водой. Смесь разделили, а органический слой перемешивали в 2 н. HCl (20 мл) в течение 16 часов. Смесь разделили, а органический слой высушили при помощи MgSO4, отфильтровали и концентрировали in vacuo для получения желто-белого твердого вещества. Неочищенное твердое вещество очистили колоночной хроматографией на диоксиде кремния, элюируя 10-40% EtOAc в гексанах, для получения продукта (0,250 г, выход 24,8%). 1Н ЯМР (CDCl3) δ 3,20-3,26 (m, 1Н), 2,38-2,47 (m, 1Н), 2,14-2,25 (m, 1Н), 1,97-2,05 (m, 1Н), 1,74-1,83 (m, 1Н), 1,14 (s, 6Н).
Стадия D: Получение 6,6-диметил-2-фенил-2,4,5,6-тетрагидроциклопента[с] пиразол-3-амина: Получили по способу, описанному для Промежуточного соединения P1, заменяя фенилгидразином этил 3-гидразинилбензоата гидрохлорид, и 3,3-диметил-2-оксоциклопентанкарбонитрилом 4,4-диметил-3-оксопентаннитрил, для получения продукта (0,192 г, выход 46,2%) в виде желтого твердого вещества. МС (apci) m/z=228,2 (М+Н).
Промежуточное соединение Р106
7,7-диметил-2-фенил-4,5,6,7-тетрагидро-2Н-индазол-3-амин
Стадия A: Получение 2,2-диметилгептандинитрила: Получили по способу, описанному для Промежуточного соединения Р105, Стадии A и B, заменяя 1-бром-4-хлорбутаном 1-бром-3-хлорпропан, для получения продукта (2,21 г, выход 73,7%). 1Н ЯМР (CDCl3) δ 2,37-2,42 (m, 2Н), 1,53-1,77 (m, 6Н), 1,36 (s, 6Н).
Стадия B: Получение 3,3-диметил-2-оксоциклогексанкарбонитрила: Суспензию KOtBu (0,463 г, 4,13 ммоль) в толуоле (16,6 мл) обработали раствором 2,2-диметилгептандинитрила (1,00 г, 6,66 ммоль) в толуоле (2,0 мл) и нагревали при 80°C в течение 48 часов. После охлаждения до комнатной температуры, реакционную смесь погасили водой и разделили фазы, а органический слой перемешивали с 2 н. HCl (20 мл) в течение 16 часов. После разделения фаз органический слой высушили при помощи MgSO4, отфильтровали и концентрировали in vacuo. Неочищенный материал очистили колоночной хроматографией на диоксиде кремния, элюируя 10-20% EtOAc в гексанах, для получения продукта (0,374 г, выход 37,2%). 1Н ЯМР (CDCl3) δ 3,72-3,78 (m, 1Н), 2,42-2,50 (m, 1H), 1,78-2,04 (m, 4H), 1,60-1,70 (m, 1H), 1,21 (s, 3H), 1,16 (s, 3H).
Стадия C: Получение 7,7-диметил-2-фенил-4,5,6,7-тетрагидро-2Н-индазол-3-амина: Получили по способу, описанному для Промежуточного соединения P1, заменяя фенилгидразином этил 3-гидразинилбензоата гидрохлорид, и 3,3-диметил-2-оксоциклогексанкарбонитрилом 4,4-диметил-3-оксопентаннитрил, для получения продукта в виде грязновато-белого твердого вещества (0,490 г, выход 54,2%, чистота 66%). МС (apci) m/z=242,2 (М+Н).
Промежуточное соединение Р107
3-изопропил-4-метил-1 -фенил-1Н-пиразол-5-амин
Стадия А: Получение 2,4-диметил-3-оксопентаннитрила: К раствору пропиононитрила (518 мг, 9,40 ммоль) в ТГФ (50 мл, 7,83 ммоль) при -78°C под N2 медленно добавили бис(триметилсилил)амид лития (1 M в ТГФ) (7,83 мл, 7,83 ммоль). Через 30 минут по каплям добавили метилизобутират (0,898 мл, 7,83 ммоль), и нагрели реакционную смесь до 0°C. Образовался желтый осадок, реакционную смесь перемешивали в течение 1 часа, затем разбавили Н2О (50 мл) для растворения твердого вещества. Смесь экстрагировали Et2O (25 мл), а щелочную водную фазу подкислили при помощи 2 М HCl (5 мл) и экстрагировали Et2O (2×50 мл). Объединенные органические фазы промыли насыщенным солевым раствором (50 мл), высушили при помощи MgSO4, отфильтровали и концентрировали для получения продукта (421 мг, выход 42,9%).
Стадия B: Получение 3-изопропил-4-метил-1-фенил-1Н-пиразол-5-амина: Получили по способу, описанному для Промежуточного соединения P1, заменяя фенилгидразином этил 3-гидразинилбензоата гидрохлорид, и 4,4-диметил-3-оксопентаннитрил на 2,4-диметил-3-оксопентаннитрил, для получения продукта в виде желтого сиропообразного вещества (0,587 г, выход 81,1%). МС (apci) m/z=216,2 (М+Н).
Промежуточное соединение Р108
2-фенил-4,6-дигидро-2Н-фуро[3,4-с]пиразол-3-амин
Стадия A: Получение 4-оксотетрагидрофуран-3-карбонитрила: К суспензии KOtBu (996,6 мг, 8,881 ммоль) в ТГФ (640,4 мг, 8,881 ммоль), охлажденной до 0°C, по каплям добавили метил 2-гидроксиацетат (675,5 мкл, 8,881 ммоль) и перемешивали в течение 10 минут. Затем добавили акрилонитрил (589,1 мкл, 8,881 ммоль) и перемешивали реакционную смесь при комнатной температуре. Через 2 часа реакционную смесь разбавили H2O (50 мл), затем экстрагировали Et2O (25 мл) для удаления исходного эфира. Щелочную водную фазу подкислили 2М раствором HCl (5 мл), затем экстрагировали Et2O (2×50 мл). Объединенные органические фазы высушили при помощи MgSO4, отфильтровали и концентрировали для получения светло-коричневого маслянистого вещества (446 мг, выход 45,2%). 1Н ЯМР (CDCl3) δ 4,63 (t, 1Н), 4,24 (t, 1Н), 4,14 (d, 1Н), 4,02 (d, 1Н), 3,57 (t, 1Н).
Стадия В: Получение 2-фенил-4,6-дигидро-2Н-фуро[3,4-с]пиразол-3-амина: Получили по способу, описанному для Промежуточного соединения P1, заменяя фенилгидразином этил 3-гидразинилбензоата гидрохлорид, и 4,4-диметил-3-оксопентаннитрил на 4-оксатетрагидрофуран-3-карбонитрил, для получения продукта в виде красновато-коричневого сиропообразного вещества (182 мг, выход 22,5%). МС (apci) m/z=202,1 (М+Н).
Промежуточное соединение Р109
3-(метоксиметил)-1-фенил-1Н-пиразол-5-амин
Стадия A: Получение 4-метокси-3-оксобутаннитрила: К раствору метил 2-метоксиацетата (0,4753 мл, 4,803 ммоль) в ТГФ (20 мл, 4,803 ммоль) при -78°C под N2 добавили ацетонитрил (0,3033 мл, 5,763 ммоль), затем бис(триметилсилил)амид лития (1 М в ТГФ) (4,803 мл, 4,803 ммоль). После перемешивания в течение 1 часа, реакционную смесь нагрели до 0°C и перемешивали в течение 1 часа. Затем реакционную смесь разбавили H2O (25 мл), промыли Et2O (25 мл), затем нейтрализовали при помощи 2 М HCl (1,5 мл). Смесь экстрагировали Et2O (2×25 мл), а объединенные органические фазы промыли насыщенным солевым раствором (25 мл), высушили помощи MgSO4, отфильтровали и концентрировали для получения продукта (169 мг, выход 31,1%). 1Н ЯМР (CDCl3) δ 4,09 (s, 2Н), 3,66 (s, 2Н), 3,46 (s, 3Н)
Стадия B: Получение 3-(метоксиметил)-1-фенил-1Н-пиразол-5-амина: Получили по способу, описанному для Промежуточного соединения P1, заменяя фенилгидразином этил 3-гидразинилбензоата гидрохлорид, и 4,4-диметил-3-оксопентаннитрил на 4-метокси-3-оксобутаннитрил, для получения продукта в виде бледно-желтого остатка (6,0 мг, выход 2,0%). МС (apci) m/z=204,0 (М+Н).
Промежуточное соединение Р110
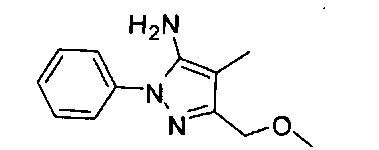
3-(метоксиметил)-4-метил-1-фенил-1Н-пиразол-5-амин
Получили по способу, описанному для Промежуточного соединения Р109, заменяя ацетонитрил на пропионитрил, для получения продукта в виде оранжевого остатка. МС (apci) m/z=218,0 (М+Н).
Промежуточное соединение Р111
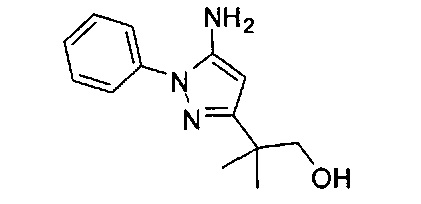
2-(5-амино-1-фенил-1Н-пиразол-3-ил)-2-метилпропан-1-ол
Стадия A: Получение метил 3-(трет-бутилдиметилсилилокси)-2,2-диметил-пропаноата: Метил 3-гидрокси-2,2-диметилпропаноат (1,000 г, 7,567 ммоль), TBDMS-C1 (1,140 г, 7,567 ммоль) и имидазол (0,5666 г, 8,323 ммоль) растворили в ДМФ (5 мл, 7,567 ммоль) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавили H2O (25 мл) и экстрагировали с EtOAc (2×25 мл). Объединенные органические фазы промыли насыщенным солевым раствором (25 мл), высушили помощи MgSO4, отфильтровали и концентрировали для получения продукта (1,92 г, выход 103%). 1Н ЯМР (CDCl3) δ 3,66 (s, 3Н), 3,57 (s, 2Н), 1,15 (s, 6Н), 0,87 (s, 9Н), 0,02 (s, 6Н).
Стадия B: Получение 5-(трет-бутилдиметилсилилокси)-4,4-диметил-3-оксопентаннитрила: Получили по способу, описанному для Промежуточного соединения Р109, заменяя метил-2-метоксиацетат на метил 3-(трет-бутилдиметилсилилокси)-2,2-диметилпропаноат, для получения продукта в виде бледно-желтого остатка. 1Н ЯМР (CDCl3) δ 3,70 (s, 2Н), 3,55 (s, 2Н), 1,15 (s, 6Н), 0,89 (s, 9Н), 0,06 (s, 6Н).
Стадия C: Получение 2-(5-амино-1-фенил-1Н-пиразол-3-ил)-2-метилпропан-1-ола: Получили по способу, описанному для Промежуточного соединения P1, заменяя фенилгидразином этил 3-гидразинилбензоата гидрохлорид, и 4,4-диметил-3-оксопентаннитрил на метил 3-(трет-бутилдиметилсилилокси)-2,2-диметилпропаноат, для получения продукта в виде желтого сиропообразного вещества (74 мг, выход 66%). МС (apci) m/z=232,2 (М+Н).
Промежуточное соединение Р112
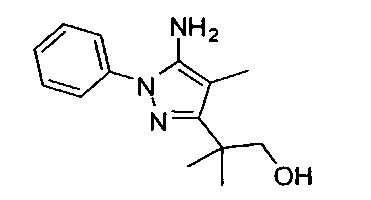
2-(5-амино-4-метил-1-фенил-1Н-пиразол-3-ил)-2-метилпропан-1-ол
Получили по способу, описанному для Промежуточного соединения P111, заменяя ацетонитрил на пропионитрил, для получения продукта в виде желтого остатка. МС (apci) m/z=246,2 (М+Н).
Промежуточное соединение Р113
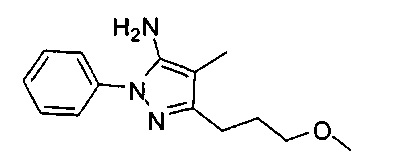
3-(3-метоксипропил)-4-метил-1-фенил-1Н-пиразол-5-амин
Получили по способу, описанному для Промежуточного соединения Р109, заменяя метил 2-метоксиацетат на метил 4-метоксибутаноат, и заменяя ацетонитрил на пропионитрил на Стадии А, для получения продукта в виде оранжево-коричневого сиропообразного вещества. МС (apci) m/z=246,1 (М+Н).
Промежуточное соединение Р114
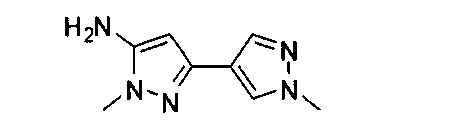
1,1'-диметил-1H,1'H-3,4'-бипиразол-5-амин
Стадия A: Получение 3-(1-метил-1Н-пиразол-4-ил)-3-оксопропаннитрила: Раствор этил 1-метил-1Н-пиразол-4-карбоксилата (500 мг, 3,24 ммоль), толуола (7,50 мл, 70,4 ммоль) и ацетонитрила (346 мкл, 6,49 ммоль) обработали одной порцией KOtBu (1092 мг, 9,73 ммоль), получив мутный раствор. Реакционную смесь оставили перемешиваться при комнатной температуре на один час и по анализу ВЭЖХ определили, что реакция завершена. Смесь обработали водой (7,5 мл) и перемешивали в течение 1 минут, затем подкислили при помощи 3 М HCl (3027 мкл, 9,08 ммоль) до pH 5,5-6. Водный слой экстрагировали этилацетатом (3×5 мл), а объединенные органические экстракты концентрировали in vacuo для получения желтого вязкого маслянистого вещества, которое полностью затвердело при помещении под высокий вакуум, с образованием продукта (102 мг, выход 21,1%). 1Н ЯМР (CDCl3) δ 8,02 (s, 1H), 7,94 (s, 1H), 3,98 (s, 3H), 3,82 (s, 2H)
Стадия B: Получение 1,1'-диметил-1H,1'H-3,4'-бипиразол-5-амина: Получили по способу, описанному для Промежуточного соединения P1, заменяя метилгидразином этил 3-гидразинилбензоата гидрохлорид, и заменяя 4,4-диметил-3-оксопентаннитрил на 3-(1-метил-1Н-пиразол-4-ил)-3-оксопропаннитрил, для получения продукта в виде твердого вещества цвета слоновой кости (45 мг, выход 44,6%). МС (apci) m/z=178,1 (М+Н).
Промежуточное соединение Р115
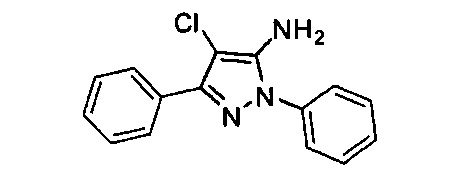
4-хлор-1,3-дифенил-1Н-пиразол-5-амин
К раствору 1,3-дифенил-1Н-пиразол-5-амина (Таблица 1; 0,100 г, 0,425 ммоль) в ацетонитриле (2 мл) добавили N-хлорсукцинимид (0,0568 г, 0,425 ммоль). Бледно-желтый раствор перемешивали при комнатной температуре в течение 3 часов, затем концентрировали in vacuo и очистили колоночной хроматографией на диоксиде кремния, элюируя 20% EtOAc в гексанах, для получения продукта в виде светло-коричневого маслянистого вещества (0,10 г, выход 87%). МС (apci) m/z=270,0 (М+Н).
Промежуточное соединение Р116
4-бром-1,3-дифенил-1Н-пиразол-5-амин
Получили по способу, описанному для Промежуточного соединения Р115, заменяя N-хлор-сукцинимид на N-бром-сукцинимид. МС (apci) m/z=313,9 (М+Н).
Промежуточное соединение Р117
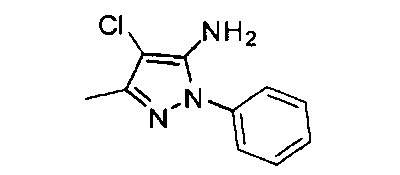
4-хлор-3-метил-1-фенил-1Н-пиразол-5-амин
Получили по способу, описанному для Промежуточного соединения Р115, заменяя 1,3-дифенил-1Н-пиразол-5-амин на 3-метил-1-фенил-1Н-пиразол-5-амин. МС (apci) m/z=207,9 (М+Н).
Промежуточное соединение Р118
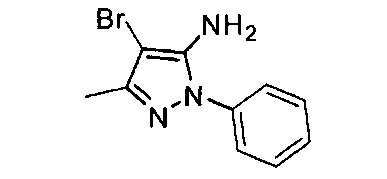
4-бром-3-метил-1-фенил-1Н-пиразол-5-амин
Получили по способу, описанному для Промежуточного соединения Р117, заменяя N-хлор-сукцинимид на N-бром-сукцинимид. МС (apci) m/z=251,9 (М+Н).
Промежуточное соединение Р119
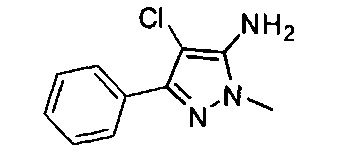
4-хлор-1-метил-3-фенил-1Н-пиразол-5-амин
Получили по способу, описанному для Промежуточного соединения Р115, заменяя 1,3-дифенил-1Н-пиразол-5-амин на 1-метил-3-фенил-1Н-пиразол-5-амин (Таблица 1). МС (apci) m/z=208,0 (М+Н).
Промежуточное соединение Р120
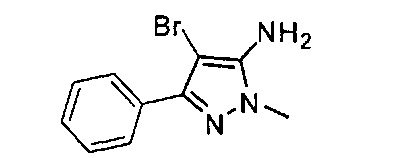
4-бром-1-метил-3-фенил-1Н-пиразол-5-амин
Получили по способу, описанному для Промежуточного соединения Р119, заменяя N-хлор-сукцинимид на N-бром-сукцинимид. МС (apci) m/z=251,9 (М+Н).
Промежуточное соединение Р121
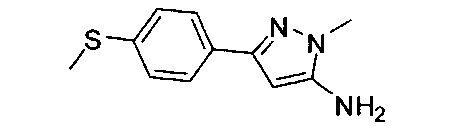
1-метил-3-(4-(метилтио)фенил)-1Н-пиразол-5-амин
Стадия А: Получение 3-(4-(метилтио)фенил)-3-оксопропаннитрила: К суспензии NaH (60% в минеральном масле) (154 мг, 3,84 ммоль) в диоксане (25,0 мл, 2,74 ммоль) добавили ацетонитрил (0,217 мл, 4,12 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, затем обработали метил 4-(метилтио)бензоатом (500 мг, 2,74 ммоль) и нагревали с дефлегматором в течение 15 часов. Суспензию охладили, затем разбавили водой (25 мл) и промыли Et2O (25 мл). Водный слой нейтрализовали 2 М раствором HCl (1,8 мл) и экстрагировали Et2O (2×25 мл). Объединенные органические фазы промыли насыщенным солевым раствором (25 мл), высушили помощи MgSO4, отфильтровали и концентрировали in vacuo. Полученный остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 0-5% MeOH/ДХМ, для получения продукта (317 мг, выход 60,4%). 1Н ЯМР (CDCl3) δ 7,82 (d, 2Н), 7,30 (d, 2Н), 4,02 (s, 2Н), 2,54 (s, 3Н).
Стадия В: Получение 1-метил-3-(4-(метилтио)фенил)-1Н-пиразол-5-амина: Получили по способу, описанному для Промежуточного соединения Р1, заменяя метилгидразином этил 3-гидразинилбензоата гидрохлорид, и заменяя 3-(4-(метилтио)фенил)-3-оксопропаннитрилом 4,4-диметил-3-оксопентаннитрил, для получения продукта в виде желтого твердого вещества (0,307 г, выход 96,7%). МС (apci) m/z=220,0 (М+Н).
Промежуточное соединение Р122
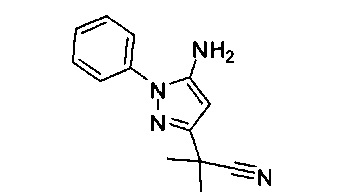
2-(5-амино-1-фенил-1Н-пиразол-3-ил)-2-метилпропаннитрил
Получили по способу, описанному для Промежуточного соединения Р121, заменяя метил 4-(метилтио)бензоат на этил 2-циано-2-метилпропаноат на Стадии A, и фенилгидразина гидрохлоридом метилгидразин на Стадии В. МС (apci) m/z=227,1 (М+Н).
Промежуточное соединение Р123
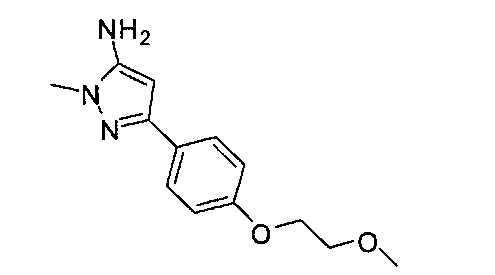
3-(4-(2-метоксиэтокси)фенил)-1-метил-1Н-пиразол-5-амин
Стадия А: Получение 3-(4-(бензилокси)фенил)-3-оксопропаннитрила: Получили по способу, описанному для Промежуточного соединения 121, заменяя метил 4-(метилтио)бензоат на метил 4-(бензилокси)бензоат на Стадии А. 1Н ЯМР (CDCl3) δ 7,90 (d, 2Н), 7,42 (m, 4Н), 7,37 (m, 1Н), 7,05 (d, 2Н), 5,16 (s, 2Н), 4,00 (s, 2Н).
Стадия В: Получение 3-(4-(бензилокси)фенил)-1-метил-1Н-пиразол-5-амина: Получили по способу, описанному для Промежуточного соединения Р1, заменяя метилгидразином этил 3-гидразинилбензоата гидрохлорид, и 3-(4-(бензилокси)фенил)-3-оксопропаннитрилом 4,4-диметил-3-оксопентаннитрил, для получения продукта в виде желтого твердого вещества. МС (apci) m/z=280,1 (М+Н).
Стадия С: Получение 4-(5-амино-1-метил-1Н-пиразол-3-ил)фенола: К раствору 3-(4-(бензилокси)фенил)-1-метил-1Н-пиразол-5-амина (47 мг, 0,17 ммоль) в EtOH (5,0 мл) добавили 5% Pd/C (9,0 мг, 0,0084 ммоль) и перемешивали под Н2 из баллона в течение 17 часов. Реакционную смесь отфильтровали через Celite®, промыли EtOH и концентрировали in vacuo для получения продукта (28 мг, выход 88%). МС (apci) m/z=190,1 (М+Н).
Стадия D: Получение 3-(4-(2-метоксиэтокси)фенил)-1-метил-1Н-пиразол-5-амина: К раствору 4-(5-амино-1-метил-1Н-иразол-3-ил)фенола (14 мг, 0,074 ммоль) в ДМСО (0,50 мл, 7,0 ммоль) добавили Cs2CO3 (48 мг, 0,15 ммоль) и 1-бром-2-метоксиэтан (9,7 мкл, 0,10 ммоль). Реакционную смесь перемешивали в течение 16 часов, затем разбавили водой (10 мл) и экстрагировали ДХМ (3×10 мл). Объединенные органические экстракты промыли насыщенным солевым раствором (10 мл), высушили помощи MgSO4, отфильтровали и концентрировали для получения неочищенного продукта (22 мг, выход 120%). Неочищенный продукт использовали без очистки на следующих стадиях. МС (apci) m/z=248,0 (М+Н).
Промежуточное соединение Р124
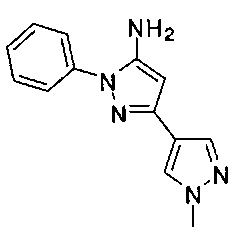
1'-метил-1-фенил-1Н,1'H-3,4'-бипиразол-5-амин
Получили по способу, описанному для Промежуточного соединения Р114, заменяя метилгидразин на фенилгидразин на Стадии В. МС (apci) m/z=240,0 (М+Н).
Промежуточное соединение Р125
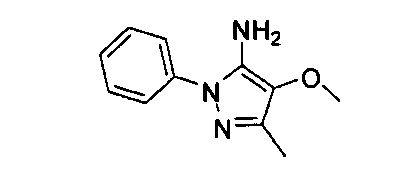
4-метокси-3-метил-1-фенил-1Н-пиразол-5-амин
Получили по способу, описанному для Промежуточного соединения Р121, заменяя метил 4-(метилтио)бензоат на этилацетат, и заменяя ацетонитрил на 2-метоксиацетонитрил на Стадии A, и фенилгидразина гидрохлоридом метилгидразин на стадии В. МС (apci) m/z=204,0 (М+Н).
Промежуточное соединение Р126
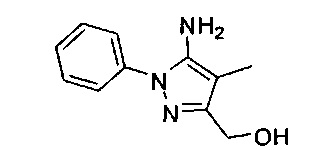
(5-амино-4-метил-1-фенил-1Н-пиразол-3-ил)метанол
Получили по способу, описанному для Промежуточного соединения Р112, заменяя метил 3-гидрокси-2,2-диметилпропаноат на этил 2-гидроксиацетат на Стадии А. МС (apci) m/z=204,1 (М+Н).
Промежуточное соединение Р127
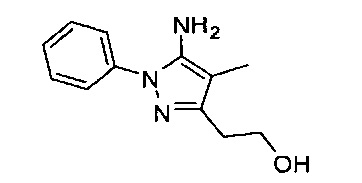
2-(5-амино-4-метил-1-фенил-1Н-пиразол-3-ил)этанол
Получили по способу, описанному для Промежуточного соединения Р112, заменяя метил 3-гидрокси-2,2-диметилпропаноат на метил 3-гидроксипропаноат на Стадии А. МС (apci) m/z=218,0 (М+Н).
Промежуточное соединение Р128
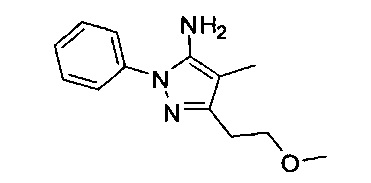
3-(2-метоксиэтил)-4-метил-1-фенил-1Н-пиразол-5-амин
Стадия A: Получение 5-метокси-2-метил-3-оксопентаннитрила: К суспензии NaNH2 (50 вес.% суспензия в толуоле) (330 мг, 4,23 ммоль) в ТГФ (25 мл, 4,23 ммоль) под N2 при -78°C добавили пропиононитрил (0,448 мл, 6,35 ммоль) и перемешивали реакционную смесь в течение 30 минут. Добавили метил 3-метоксипропаноат (0,495 мл, 4,23 ммоль) и перемешивали реакционную смесь при -78°C в течение 1 часа, затем при 0°C в течение 2,5 часов. Реакционную смесь разбавили Н2О (25 мл) и промыли Et2O (25 мл). Щелочную водную фазу нейтрализовали 2 М раствором HCl (1,6 мл), затем экстрагировали Et2O (3×25 мл). Объединенные органические фазы промыли насыщенным солевым раствором (25 мл), высушили помощи MgSO4, отфильтровали и концентрировали для получения неочищенного продукта в виде бледного зеленоватого маслянистого вещества (171 мг). Неочищенную смесь использовали непосредственно на следующей стадии.
Стадия B: Получение 3-(2-метоксиэтил)-4-метил-1-фенил-1Н-пиразол-5-амина: Получили по способу, описанному для Промежуточного соединения P1, заменяя 5-метокси-2-метил-3-оксопентаннитрилом 4,4-диметил-3-оксопентаннитрил, и заменяя фенилгидразина гидрохлоридом этил 3-гидразинилбензоата гидрохлорид, для получения продукта в виде желтого твердого вещества (56 мг, выход 20%). МС (apci) m/z=232,0 (М+Н).
Промежуточное соединение Р129
Фенил (5-оксидо-2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-ил)карбамат
ТГФ (4 мл) раствор фенил 2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-илкарбамата (Промежуточное соединение Р130, Стадия B; 50 мг, 0,15 ммоль) охладили до -50°C при помощи внешней бани из сухого льда и MeCN, и обработали ТГФ (2 мл) раствором 3-хлорпероксибензойной кислоты (33 мг, 0,13 ммоль). После перемешивания в течение 1 часа, смесь погасили Na2S2O3 и водой, экстрагировали EtOAc, промыли NaHCO3 и насыщенным солевым раствором, высушили помощи MgSO4, отфильтровали и концентрировали для получения продукта, который использовали напрямую на следующей стадии, без дополнительной очистки. МС (apci) m/z=354,1 (М+Н).
Промежуточное соединение P130
Фенил (5,5-диоксидо-2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-ил)карбамат
Стадия A: Получение 2-фенил-4,6-дигидро-2Н-тиено[3,4-с] пиразол-3-амина: Суспензию 4-оксотетрагидротиофен-3-карбонитрила (1,00 г, 7,86 ммоль) и фенилгидразина гидрохлорида (1,25 г, 8,65 ммоль) в абсолютном EtOH (40 мл) нагревали с дефлегматором в течение 2 часов. После удаления растворителя под пониженным давлением, белый твердый остаток растерли с 1 н. NaOH (40 мл). Твердое вещество собрали фильтрацией, промыли 0,1 н. NaOH, водой и гексанами (приблизительно по 10 мл каждого), затем высушили под высоким вакуумом для получения продукта в виде белого твердого вещества (1,6 г, выход 95%). МС (apci полож.) m/z=218,1 (М+Н).
Стадия B: Получение фенил 2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-илкарбамата. К суспензии 2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-амина (500 мг, 2,30 ммоль) в EtOAc (10 мл) добавили NaOH (2 М, водный, 2,3 мл, 4,60 ммоль), затем по каплям добавили фенилкарбонохлоридат (0,400 мл, 3,22 ммоль). После перемешивания при комнатной температуре в течение 2 часов, по каплям добавили еще одну порцию фенилкарбонохлоридата (0,16 мл, 1,3 ммоль), и перемешивали реакционную смесь при комнатной температуре в течение 15 часов. Реакционную смесь разбавили EtOAc (20 мл) и разделили фазы. Органический слой промыли H2O, насыщенным солевым раствором (по 25 мл каждого), затем высушили при помощи Na2SO4, отфильтровали и концентрировали. Неочищенный материал очистили обращенно-фазовой колоночной хроматографией, элюируя 5-70% ацетонитрила в воде, для получения продукта в виде белого твердого вещества (0,5 г, выход 64%). МС (apci полож.) m/z=338,1 (М+Н).
Стадия С: Получение фенил (5,5-диоксидо-2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-ил)карбамата. К мутному раствору фенил 2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-илкарбамата (50 мг, 0,15 ммоль) в ДХМ (1,5 мл) при 0°C добавили МСРВА (91 мг, 0,37 ммоль, 70-75% комплекс с водой) и перемешивали смесь при комнатной температуре в течение 10 минут. Затем смесь разбавили ДХМ (3 мл) и промыли насыщенным водным раствором NaHCO3 (3×2 мл) и насыщенным водным раствором Na2S2O3 (3×2 мл). Органический слой высушили помощи MgSO4, отфильтровали и концентрировали под пониженным давлением для получения указанного в заголовке продукта в виде светло-желтоватого пенистого вещества (31 мг, выход 57%, чистота 95%). МС (apci полож.) m/z=371,0 (М+Н).
Промежуточное соединение Р131
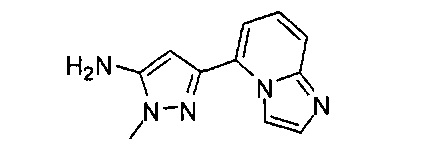
3-(имидазо[1,2-а]пиридин-5-ил)-1-метил-1Н-пиразол-5-амин
Стадия A: Метил имидазо[1,2-а]пиридин-5-карбоксилат: К суспензии метил 6-аминопиколината (1,52 г, 10,0 ммоль) в iPrOH (10 мл) добавили 2-хлорацетальдегид (2,57 мл, 20,0 ммоль) и перемешивали смесь при комнатной температуре в течение 30 минут. Полученный раствор нагревали при 70°C в течение 16 часов, затем охладили до комнатной температуры и концентрировали in vacuo. Остаток разбавили H2O (40 мл) и обработали 1 М K2CO3 до pH=10. Смесь экстрагировали EtOAc (3x), а объединенные экстракты промыли насыщенным раствором NaCl и высушили над MgSO4/активированным углем. Высушенный раствор элюировали через слой SiO2, покрытый слоем MgSO4, используя для элюирования EtOAc. Раствор концентрировали для получения указанного в заголовке соединения в виде кремово-белого твердого вещества (1,73 г, выход 98,2%). МС (apci) m/z=177,0 (М+Н).
Стадия В: 3-(имидазо[1,2-а]пиридин-5-ил)3-оксопропаннитрил: 1 М раствор LiHMDS (3,15 мл, 3,15 ммоль) в сухом ТГФ охладили до -78°C и по каплям добавили ацетонитрил (0,172 мл, 3,30 ммоль) за 1 минуту. Смесь перемешивали при -78°C в течение 1 часа и добавили раствор метил имидазо[1,2-а]пиридин-5-карбоксилата (0,529 г, 3,00 ммоль) в сухом ТГФ (2,0 мл). Смесь оставили достигать комнатной температуры и перемешивали в течение 2,5 часов. Смесь вылили в охлажденную H2O (30 мл), а полученный водный раствор экстрагировали Et2O (3x). Водную часть охладили до 0°C и медленно добавили 6 М HCl до pH=6. Полученную желтую суспензию отфильтровали, а собранное твердое вещество промыли H2O и EtOAc. Твердое вещество высушили в вакууме для получения указанного в заголовке соединения в виде желтого твердого вещества (317 мг, выход 57,1%). МС (apci) m/z=186,0 (М+Н).
Стадия С: 3-(имидазо[1,2-а]пиридин-5-ил)-1 -метил-1Н-пиразол-5-амин: К тонкодисперсной суспензии 3-(имидазо[1,2-а]пиридин-5-ил)-3-оксопропаннитрила (229 мг, 1,24 ммоль) в абсолютном EtOH (4 мл) добавили метилгидразин (78,1 мкл, 1,45 ммоль), и полученный раствор перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь нагревали с дефлегматором в течение 5 часов и добавили дополнительное количество метилгидразина (200 мкл). Смесь нагревали с дефлегматором в течение 15 часов, охладили до комнатной температуры и концентрировали. Остаточное желто-коричневое твердое вещество растворили в 5% MeOH/CH2Cl2 и элюировали через слой SiO2, элюируя 5% MeOH/CH2Cl2. Элюент концентрировали, а остаточное желтое твердое вещество промыли МТБЭ и высушили в вакууме для получения указанного в заголовке соединения в виде светло-желтого порошка (150 мг, выход 56,9%). МС (apci) m/z=214,0 (М+Н).
Промежуточное соединение Р132
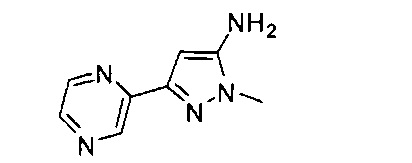
1-метил-3-(пиразин-2-ил)-1Н-пиразол-5-амин
Стадия А: Получение 3-оксо-3-(пиразин-2-ил)пропаннитрила: К суспензии NaH (60% в минеральном масле, 81,1 мг, 2,03 ммоль) в диоксане (15 мл) добавили ацетонитрил (0,114 мл, 2,17 ммоль), затем метилпиразин-2-карбоксилат (200 мг, 1,45 ммоль) и нагревали реакционную смесь с дефлегматором в течение 2,5 часов. Реакционную смесь охладили до комнатной температуры и разбавили H2O (25 мл) и экстрагировали Et2O (25 мл). Водную фазу нейтрализовали 2 М водным раствором HCl (0,7 мл), затем экстрагировали 10% MeOH/ДХМ (3×25 мл). Объединенные органические фазы промыли насыщенным солевым раствором (25 мл), высушили помощи MgSO4, отфильтровали и концентрировали для получения неочищенного продукта в виде оранжевого сиропообразного вещества (134 мг, выход 62,9%). 1Н ЯМР (CDCl3) δ 9,32 (d, 1Н), 8,87 (d, 1Н), 8,68 (dd, 1H), 4,34 (s, 2H).
Стадия B: Получение 1-метил-3-(пиразин-2-ил)-1Н-пиразол-5-амина: К суспензии 3-оксо-3-(пиразин-2-ил)пропаннитрила (67,0 мг, 0,455 ммоль) в EtOH (5 мл) добавили метилгидразин (0,024 мл, 0,455 ммоль). Реакционную смесь нагревали с дефлегматором в течение 15 часов, затем концентрировали in vacuo. Неочищенный продукт очистили колоночной хроматографией на диоксиде кремния, элюируя 0-5% MeOH/ДХМ, для получения продукта в виде коричневого остатка (33 мг, выход 41%). МС (apci) m/z=176,2 (М+Н).
Промежуточное соединение Р133
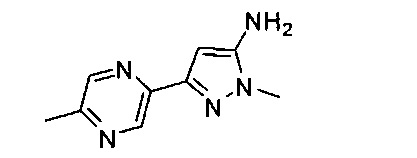
1-метил-3-(5-метилпиразин-2-ил)-1Н-пиразол-5-амин
Получили по способу, описанному для Промежуточного соединения Р107, заменяя метилизобутират на Стадии A на метил 5-метилпиразин-2-карбоксилат, и пропионитрил на ацетонитрил, для получения 3-(5-метилпиразин-2-ил)-3-оксопропаннитрила. На Стадии B фенилгидразин заменили метилгидразином для получения указанного в заголовке пиразола. МС (apci) m/z=190,2 (М+Н).
Промежуточное соединение Р134
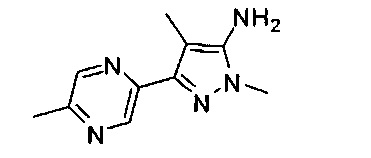
1,4-диметил-3-(5-метилпиразин-2-ил)-1Н-пиразол-5-амин
Получили по способу, описанному для Промежуточного соединения Р107, заменяя метилизобутират на Стадии А на метил 5-метилпиразин-2-карбоксилат, для получения 2-метил-3-(5-метилпиразин-2-ил)-3-оксопропаннитрила. На Стадии B фенилгидразин заменили метилгидразином для получения указанного в заголовке соединения. МС (apci) m/z=204,1 (М+Н).
Промежуточное соединение Р135
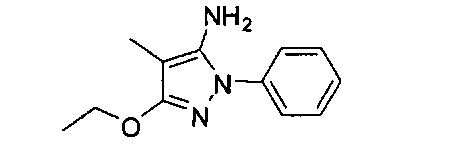
3-этокси-4-метил-1-фенил-1Н-пиразол-5-амин
Стадия А: Получение 5-амино-4-метил-1-фенил-1Н-пиразол-3(2Н)-она: Смесь этил 2-цианопропаноата (5,0 г, 46 ммоль) и фенилгидразина (5,9 г, 46 ммоль) в диоксане (10 мл) нагревали при 110°С в течение 17 часов. Неочищенный материал охладили до комнатной температуры, концентрировали и растерли с холодным EtOH и Et2O. Полученное твердое вещество отфильтровали, промыли Et2O и высушили под вакуумом для получения продукта в виде белого твердого вещества (3,4 г, выход 39%). МС (apci) m/z=190,0 (М-Н).
Стадия В: Получение 3-этокси-4-метил-1-фенил-1Н-пиразол-5-амина: К суспензии 5-амино-4-метил-1-фенил-1Н-пиразол-3(2Н)-она (10,0 г, 52,9 ммоль) в ДМФ (100 мл) добавили K2CO3 (14,6 г, 106 ммоль) и бромэтан (4,34 мл, 58,1) при комнатной температуре. После перемешивания в течение 17 часов, реакционную смесь обработали EtOAc и промыли водой (3х, для получения продукта N-алкилирования) и насыщенным солевым раствором, высушили при помощи MgSO4, отфильтровали и концентрировали для получения продукта (5,35 г, выход 47%). МС (apci) m/z=218,1 (М+Н).
Соединения в Таблице 3 получили по способу, описанному для Промежуточного соединения Р135, заменяя бромэтан на соответствующий алкилгалогенид или алкилметансульфонат.
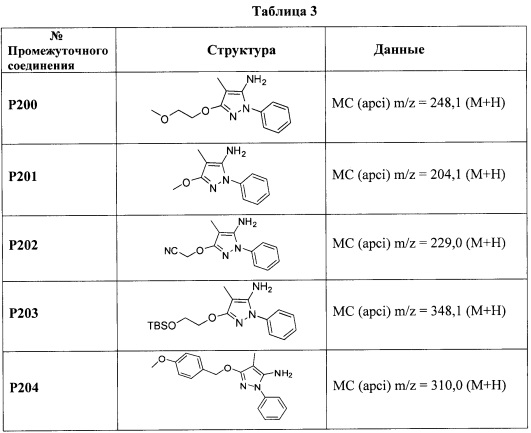
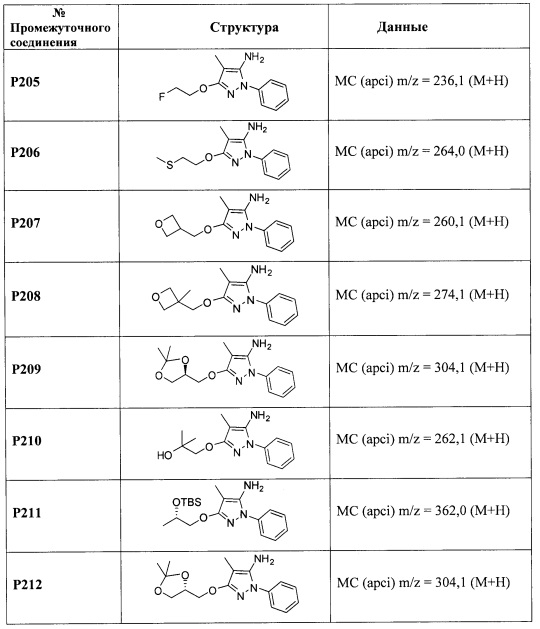
Промежуточное соединение Р136
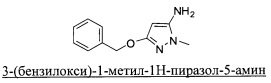
Стадия А: Получение 5-амино-1-метил-4-фенил-1Н-пиразол-3(2H)-она: К суспензии этил 2-циано-2-фенилацетата (2,56 г, 13,3 ммоль) в EtOH (10 мл) по каплям добавили метилгидразин (1,09 мл, 19,9 ммоль). Реакционную смесь нагревали при 85°С в течение 15 часов. Реакционную смесь охладили до 0°С и отфильтровали. Полученное твердое вещество промыли холодным EtOH (20 мл) и Et2O (20 мл) для получения заданного продукта (2,10 г, выход 83,7%). МС (apci) m/z=190,2 (М+Н)
Стадия В: Получение 3-(бензилокси)-1-метил-1Н-пиразол-5-амина: Суспензию 5-амино-1-метил-1Н-пиразол-3(2Н)-она (0,35 г, 3,1 ммоль), бензилхлорида (0,43 г, 3,4 ммоль) и K2CO3 (1,3 г, 9,3 ммоль) в ДМФ (4 мл) нагревали при 70°С в течение 17 часов. После охлаждения реакционную смесь обработали EtOAc, промыли водой и насыщенным солевым раствором, высушили при помощи MgSO4 и концентрировали. Неочищенный продукт очистили колоночной хроматографией на диоксиде кремния, элюируя 2-6% МеОН в ДХМ, для получения указанного в заголовке соединения (0,16 г, выход 25%). МС (apci) m/z=204,0 (М+Н).
Промежуточное соединение Р137
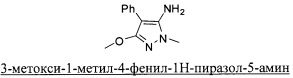
К суспензии 5-амино-1-метил-4-фенил-1Н-пиразол-3(2Н)-она (Стадия А получения Промежуточного соединения Р136; 208 мг, 1,10 ммоль) и K2CO3 (456 мг, 3,30 ммоль) в ДМФ (5 мл) по каплям добавили йодметан (172 мг, 1,21 ммоль). Реакционную смесь перемешивали в течение 15 часов. Растворитель удалили под пониженным давлением, а остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 33% EtOAc в гексанах, для получения указанного в заголовке пиразола (66,0 мг, выход 30,4%). МС (apci) m/z=204,1 (М+Н).
Промежуточное соединение Р138
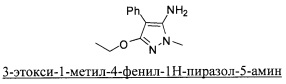
Получили так, как описано в Промежуточном соединении Р137, заменяя йодметан на йодэтан на Стадии В, для получения указанного в заголовке соединения. МС (apci) m/z=218,2 (М+Н).
Промежуточное соединение Р139
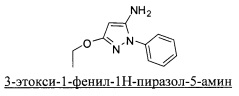
Получили по способу, описанному для Промежуточного соединения 135, заменяя этил-2-цианопропаноат на этил-2-цианоацетат на Стадии А. МС (apci) m/z=204,0 (М+Н).
Соединения в следующей Таблице получили по способу, описанному для Промежуточного соединения Р135, заменяя бромэтан на соответствующий алкилгалогенид, алкилметансульфонат или эпоксид.
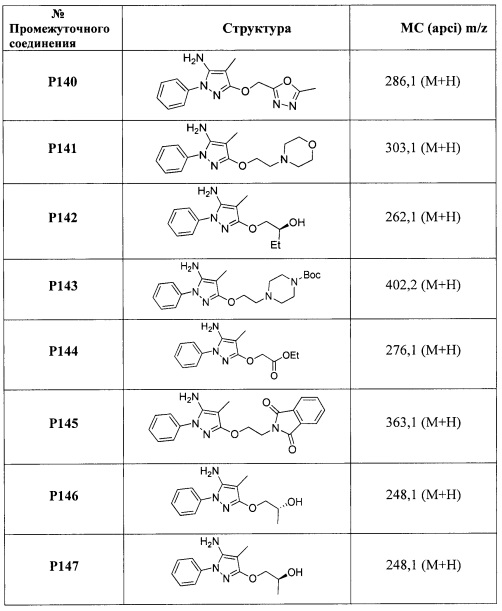
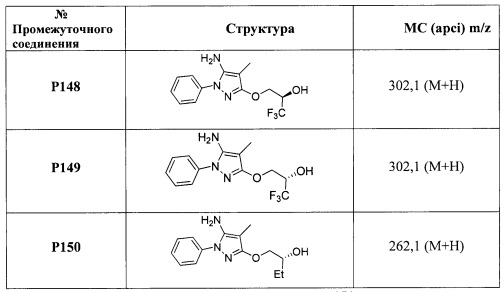
Промежуточное соединение 151
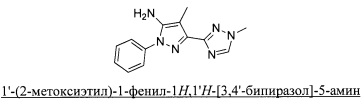
Стадия А: Получение метил 1-етил-1Н-1,2,4-триазол-3-карбоксилата: К перемешанной суспензии NaH (60% дисперсия в масле, 0,346 г, 8,66 ммоль) в ДМФ (20 мл) по каплям добавили раствор метил 1Н-1,2,4-триазол-3-карбоксилата (1,00 г, 7,87 ммоль) в ДМФ (20 мл) при 0°С под азотом. Реакционную смесь перемешивали при 0°С в течение 1 часа. По каплям добавили MeI (0,982 мл, 15,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь вылили в холодную воду и экстрагировали EtOAc. Объединенные органические слои промыли насыщенным солевым раствором, высушили и концентрировали. Остаток очистили колоночной хроматографией (3:1 гексаны/EtOAc) для получения указанного в заголовке соединения (0,380 г, выход 34%) в виде белого твердого вещества. МС (apci) m/z=142,1 (М+Н).
Стадия В: Получение 1'-(2-метоксиэтил)-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-амина: Получили по способу, описанному для Промежуточного соединения Р109, используя метил 1-метил-1Н-1,2,4-триазол-3-карбоксилат вместо метил 2-метоксиацетата, и заменяя пропионитрилом ацетонитрил на Стадии А. МС (apci) m/z=255,1 (М+Н).
Промежуточное соединение 152
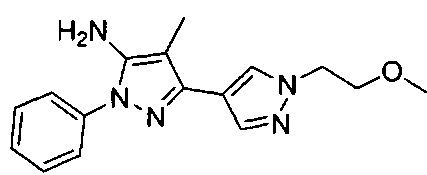
1'(2-метоксиэтил)-4-метил-1-фенил-1H,1'H-[3,4'-бипиразол]-5-амин
Получили по способу, описанному для Промежуточного соединения Р109, используя этил 1-(2-метоксиэтил)-1Н-пиразол-4-карбоксилат вместо метил 2-метоксиацетата, и заменяя пропионитрилом ацетонитрил на Стадии А.
Промежуточное соединение 153
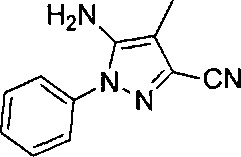
5-амино-4-метил-1-фенил-1Н-пиразол-3-карбонитрил
К перемешанному раствору анилина (2,02 г, 21,7 ммоль) в 6 н. HCl (22 мл) по каплям добавили раствор NaNO4 (1,50 г, 21,7 ммоль) в воде (20 мл) при 0-5°C. Реакционную смесь перемешивали при 0°C в течение 15 минут. Добавили уксусную кислоту (10 мл). Этот раствор по каплям добавили к перемешанному раствору этил 2,3-дицианобутаноата (полученного по способу, описанному в публикации Bioorganic & Medicinal Chemistry, 2004, 12, 3345-3356, 3,60 г, 21,7 ммоль) в уксусной кислоте (12 мл) и воде (18 мл) при 0°C. После перемешивания в течение 1 часа, по каплям добавили концентрированный гидроксид аммония (50 мл), затем ТГФ (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Отделили органический слой. Водный слой экстрагировали EtOAc. Объединенные органические слои промыли насыщенным солевым раствором, высушили и концентрировали. Остаток очистили флэш-хроматографией на силикагеле (3:1 гексаны/EtOAc) для получения указанного в заголовке соединения (2,95 г, выход 69%). МС (apci) m/z=198,9 (М+Н).
Промежуточное соединение 154
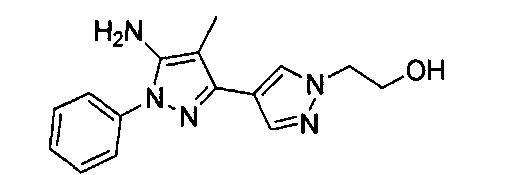
2-(5-амино-4-метил-1-фенил-1H,1'H-3,4'-бипиразол-1'-ил)этанол
Стадия А: Получение этил 1-(2-((трет-бутилдиметилсилил)окси)этил)-1Н-пиразол-4-карбоксилата: Получили по способу, описанному для Примера 556, заменяя 1-бром-2-метоксиэтан на (2-бромэтокси)(трет-бутил)диметилсилан на Стадии А. МС (apci) m/z=298,9 (М+Н).
Стадия В: Получение 2-(5-амино-4-метил-1-фенил-1Н,1'H-3,4'-бипиразол-1'-ил)этанола: Получили по способу, описанному для Промежуточного соединения Р109, используя этил 1-(2-((трет-бутилдиметилсилил)окси)этил)-1Н-пиразол-4-карбоксилат вместо метил 2-метоксиацетата, и заменяя пропионитрилом ацетонитрил на Стадии А. МС (apci) m/z=283,9 (М+Н).
Промежуточное соединение 155
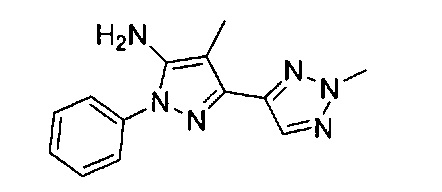
4-метил-3-(2-метил-2Н-1,2,3-триазол-4-ил)-1-фенил-1Н-пиразол-5-амин
Стадия A: Получение этил 2-метил-2Н-1,2,3-триазол-4-карбоксилата: Смесь этил 2Н-1,2,3-триазол-4-карбоксилата (2,00 г, 14,2 ммоль), K2CO3 (3,53 г, 25,5 ммоль) и метилйодида (3,54 мл, 56,7 ммоль) в ацетонитриле (40 мл) перемешивали при 50°C под азотом в течение ночи. После охлаждения до комнатной температуры, смесь отфильтровали через Celite®. Фильтрат концентрировали in vacuo. Остаток очистили флэш-хроматографией на силикагеле (4:1 гексан/EtOAc) для получения указанного в заголовке соединения (0,780 г, выход 35%). МС (apci) m/z=156,0 (М+Н).
Стадия B: Получение 4-метил-3-(2-метил-2Н-1,2,3-триазол-4-ил)-1-фенил-1Н-пиразол-5-амина: Получили по способу, описанному для Промежуточного соединения Р109, используя этил 2-метил-2Н-1,2,3-триазол-4-карбоксилат вместо метил 2-метоксиацетата, и заменяя пропионитрилом ацетонитрил на Стадии А. МС (apci) m/z=254,9 (М+Н).
Промежуточное соединение 156
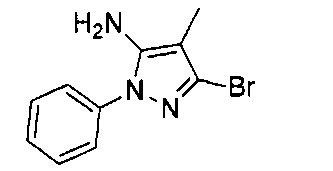
3-бром-4-метил-1-фенил-1Н-пиразол-5-амин:
К перемешанному раствору 5-амино-4-метил-1-фенил-1Н-пиразол-3(2Н)-она (Промежуточное соединение Р135, Стадия А, 1,00 г, 5,29 ммоль) в MeCN (20 мл) добавили POBr3 (2,27 г, 7,93 ммоль). Реакционную смесь нагревали с дефлегматором в течение 3 часов. Реакционную смесь концентрировали in vacuo. Остаток растворили в ДХМ. Осторожно добавили насыщенный водный раствор NaHCO3. Водный слой экстрагировали ДХМ. Объединенные органические слои промыли насыщенным солевым раствором, высушили и концентрировали. Остаток очистили флэш-хроматографией на силикагеле (1:2 гексан/EtOAc) для получения указанного в заголовке соединения (0,23 г, выход 17%). МС (apci) m/z=251,8 (М+Н).
Промежуточное соединение 157
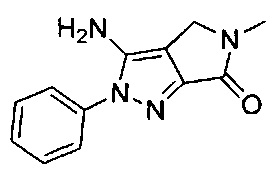
3-амино-5-метил-2-фенил-4,5-дигидропирроло[3,4-с]пиразол-6(2Н)-он
Стадия А: Получение этил 5-амино-4-((метиламино)метил)-1-фенил-1Н-пиразол-3-карбоксилата: К перемешанному раствору этил 5-амино-4-формил-1-фенил-1Н-пиразол-3-карбоксилата (полученного по способу, описанному в публикации J. Heterocyclic Chemistry, 2010, 47, с. 287-291, 142 мг, 0,548 ммоль) в ДХМ (3 мл) добавили 2,0 М MeNH2 в ТГФ (0,822 мл, 1,64 ммоль). Добавили две капли уксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавили MeOH (0,4 мл), затем частями добавили NaBH2 (31 мг, 0,82 ммоль). Реакцию погасили медленным добавлением воды. Смесь экстрагировали ДХМ. Объединенные органические слои промыли насыщенным солевым раствором, высушили и концентрировали. Неочищенный продукт использовали на следующей стадии без дополнительной очистки. МС (apci) m/z=275,0 (М+Н).
Стадия В: Получение 3-амино-5-метил-2-фенил-4,5-дигидропирроло [3,4-с]пиразол-6(2Н)-она: К перемешанному раствору этил 5-амино-4-((метиламино)метил)-1-фенил-1Н-пиразол-3-карбоксилата (неочищенный, 65 мг, 0,24 ммоль) в MeOH (0,5 мл) и ТГФ (0,5 мл) добавили 2 н. NaOH (0,24 мл, 0,47 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, а затем концентрировали in vacuo. К остатку добавили воду. pH довели до 4-5 при помощи 1 н. раствора HCl. Воду выпарили под пониженным давлением. Неочищенную кислоту (58 мг) растворили в ДМФ (3 мл). Добавили Et3N (66 мкл, 0,47 ммоль), затем EDCI (90 мг, 0,47 ммоль) и HOBt (32 мг, 0,24 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, а затем разделили между EtOAc и водой. Водный слой экстрагировали EtOAc. Объединенные органические слои промыли водой и насыщенным солевым раствором, высушили и концентрировали. Остаток очистили флэш-хроматографией на силикагеле (2% MeOH в ДХМ) для получения указанного в заголовке соединения (15 мг, 28%) в виде белого твердого вещества. МС (apci) m/z=228,9 (М+Н)
Промежуточное соединение 158
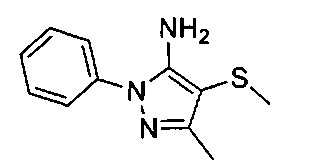
3-метил-4-(метилтио)-1-фенил-1Н-пиразол-5-амин
Получили по способу, описанному для Промежуточного соединения Р109, заменяя метил 2-метоксиацетат на этилацетат, и заменяя ацетонитрил на 2-(метилтио)ацетонитрил на стадии A, для получения продукта в виде коричневого маслянистого вещества. МС (apci) m/z=220,1 (М+Н).
Промежуточное соединение 159
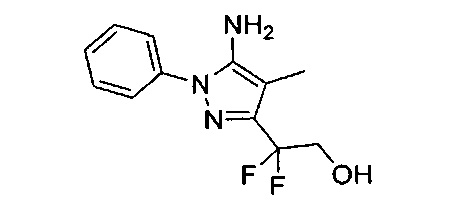
2-(5-амино-4-метил-1-фенил-1Н-пиразол-3-ил)-2,2-дифторэтанол
Получили по способу, описанному для Промежуточного соединения Р111, заменяя ацетонитрил на пропионитрил, и заменяя метил 3-гидрокси-2,2-диметилпропаноат на этил 2,2-дифтор-3-гидроксипропаноат, для получения продукта в виде бледно-желтого твердого вещества. МС (apci) m/z=254,1 (М+Н).
Промежуточное соединение 160
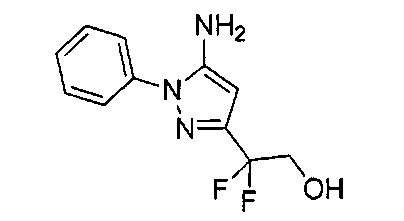
2-(5-амино-1-фенил-1Н-пиразол-3-ил)-2,2-дифторэтанол
Получили по способу, описанному для Промежуточного соединения P111, заменяя метил 3-гидрокси-2,2-диметилпропаноат на этил 2,2-дифтор-3-гидроксипропаноат, для получения продукта в виде бледно-желтого твердого вещества. МС (apci) m/z=240,0 (М+Н).
Промежуточное соединение 161
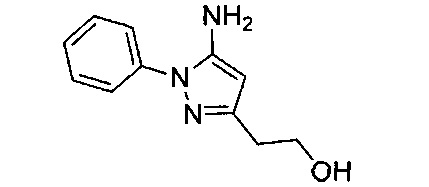
2-(5-амино-1-фенил-1Н-пиразол-3-ил)этанол
Получили по способу, описанному для Промежуточного соединения P111, заменяя метил 3-гидрокси-2,2-диметилпропаноат на метил 3-гидроксипропаноат на Стадии А. МС (apci) m/z=204,1 (М+Н).
Промежуточное соединение 162
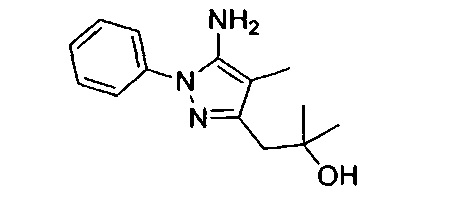
1-(5-амино-4-метил-1-фенил-1Н-пиразол-3-ил)-2-метилпропан-2-ол
Стадия А: Получение этил 3-гидрокси-3-метилбутаноата: К раствору бис(триметилсилил)амида лития (1 M в ТГФ) (100 мл, 100 ммоль) в ТГФ (100 мл) под N2, и охлажденному до -78°C, добавили этилацетат (9,74 мл, 100 ммоль). Реакционную смесь перемешивали в течение 30 минут, а затем добавили ацетон (8,81 мл, 120 ммоль). Реакционную смесь перемешивали в течение 10 минут, а затем погасили при помощи HCl (2 М водный, 70 мл, 140 ммоль) и оставили нагреваться до комнатной температуры. Реакционную смесь экстрагировали EtOAc (2×150 мл). Органические фазы объединили и промыли насыщенным водным раствором NaHCO3 (2×50 мл), высушили (MgSO4), отфильтровали и концентрировали для получения продукта в виде желтого маслянистого вещества (12,8 г, выход 88%). 1Н ЯМР (CDCl3) δ 4,18 (q, 3Н), 2,49 (s, 2Н), 1,29 (m, 9Н).
Стадия В: Получение 5-гидрокси-5-метил-3-оксогексаннитрила: К раствору пропиононитрила (1,77 мл, 30,5 ммоль) в ТГФ (100 мл) под N2 при -78°C добавили бис(триметилсилил)амид лития (1 M в ТГФ) (27,9 мл, 27,9 ммоль). Перемешивали в течение 1 часа, затем добавили этил 3-гидрокси-3-метилбутаноат (1,86 г, 12,7 ммоль). Реакционную смесь перемешивали при -78°C в течение 1 часа, затем перемешивали при 0°C в течение 1,5 часов, затем разбавили Н2О (100 мл) и экстрагировали Et2O (50 мл). Фазы разделили, а щелочную водную фазу нейтрализовали раствором HCl (6 М водный, 4,5 мл), затем экстрагировали Et2O (3×75 мл). Объединенные органические фазы промыли насыщенным солевым раствором (75 мл), высушили (MgSO4), отфильтровали и концентрировали для получения продукта в виде бледно-желтого маслянистого вещества (1,24 г, выход 63%). 1Н ЯМР (CDCl3) δ 3,54 (m, 1Н), 2,89 (s, 2Н), 1,50 (d, 3Н), 1,32 (s, 3Н), 1,31 (s, 3Н).
Стадия C: Получение 1-(5-амино-4-метил-1-фенил-1Н-пиразол-3-ил)-2-метилпропан-2-ола: К суспензии фенилгидразина (0,793 мл, 7,99 ммоль) и HCl (5-6 MeiPrOH, 1,60 мл, 7,99 ммоль) в EtOH (25 мл) добавили раствор 5-гидрокси-2,5-диметил-3-оксогексаннитрила (1,24 г, 7,99 ммоль) в EtOH (25 мл). Реакционную смесь нагревали с дефлегматором в течение 17 часов, затем охладили до комнатной температуры, разбавили насыщенным водным раствором NaHCO3 (10 мл), экстрагировали 10:90 МеОН/ДХМ (3×25 мл), а объединенные органические фазы высушили (MgSO4), отфильтровали и концентрировали. Очистили колоночной хроматографией на диоксиде кремния, элюируя 0-75% ацетона в гексанах, для получения указанного в заголовке соединения в виде оранжевого маслянистого вещества (1,13 г, выход 58%). МС (apci) m/z=246,1 (М+Н).
Следующие пиразоловые промежуточные соединения были получены по способу, использованному для получения Промежуточного соединения 162, Стадии В и С, с использованием соответствующего исходного материала. Для получения Промежуточных соединений 168 и 169 исходный материал (приобретенный у Oakwood) представлял собой смесь цис- и транс-диастереомеров.
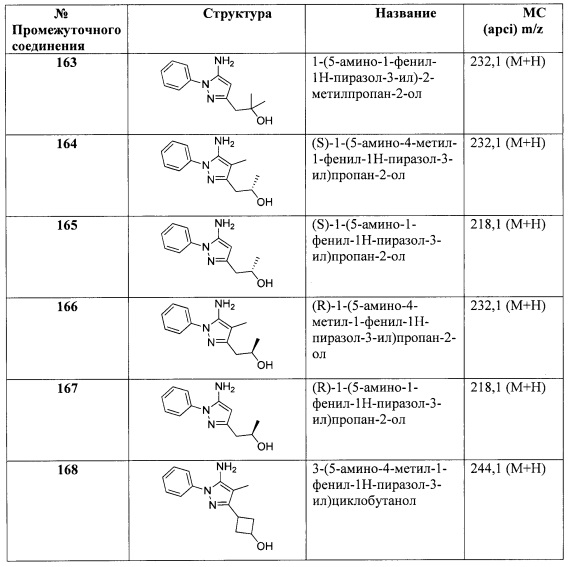
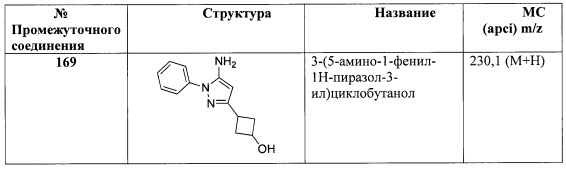
Промежуточное соединение 170
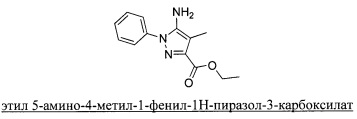
Получили по способу, описанному для Промежуточного соединения Р109, заменяя метил 2-метоксиацетат на диэтилоксалат, и заменяя ацетонитрил на пропионитрил на Стадии А, для получения продукта в виде желтого твердого вещества. МС (apci) m/z=246,1 (М+Н).
Промежуточное соединение 171
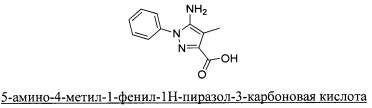
К раствору этил 5-амино-4-метил-1-фенил-1Н-пиразол-3-карбоксилата (Промежуточное соединение 170, 1,52 мг, 6,21 ммоль) в ТГФ (12 мл) и МеОН (6 мл) добавили LiOH (2 М, водный, 9,31 мл, 18,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 19 часов, затем частично концентрировали под пониженным давлением, затем нейтрализовали при помощи 6 М HCl (3,2 мл), экстрагировали 10:90 МеОН/ДХМ (3×25 мл), а объединенные органические экстракты промыли насыщенным солевым раствором (50 мл), высушили (MgSO4), отфильтровали и концентрировали для получения указанного в заголовке соединения в виде желтого твердого вещества (1,3 г, выход 96%). МС (apci) m/z=218,1 (М+Н).
Промежуточное соединение 172
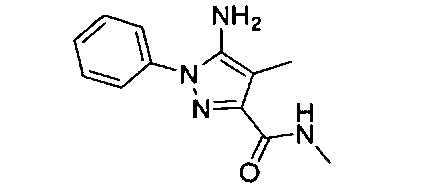
5-амино-N,4-диметил-1-фенил-1Н-пиразол-3-карбоксамид
К раствору 5-амино-4-метил-1-фенил-1Н-пиразол-3-карбоновой кислоты (Промежуточное соединение 171, 223 мг, 1,02 ммоль) в ацетонитриле (10 мл) добавили ДИЭА (0,71 мл, 4,10 ммоль), метанамина гидрохлорид (138 мг, 2,05 ммоль), ДМФ (2 мл), а затем НАТУ (428 мг, 1,13 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 19 часов, а затем частично концентрировали под пониженным давлением. Смесь очистили обращенно-фазовой колоночной хроматографией, элюируя 5-60% ацетонитрила в воде, для получения указанного в заголовке соединения в виде бледно-желтого твердого вещества (182 мг, выход 77%). МС (apci) m/z=231,1 (М+Н).
Промежуточное соединение 173
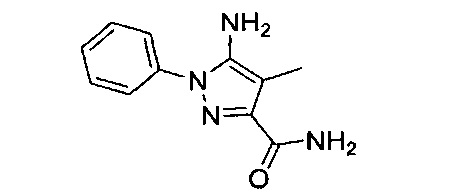
5-амино-4-метил-1-фенил-1Н-пиразол-3-карбоксамид
Раствор 5-амино-4-метил-1-фенил-1Н-пиразол-3-карбонитрил а (150 мг, 0,757 ммоль) в концентрированной H2SO4 (0,5 мл) перемешивали при комнатной температуре в течение 17 часов. Реакционную смесь охладили и нейтрализовали добавлением водного раствора NaOH (2 М, 11 мл), затем экстрагировали 10% MeOH/ДХМ (5×10 мл), а объединенные органические экстракты промыли насыщенным солевым раствором, высушили (MgSO4), отфильтровали и концентрировали под пониженным давлением для получения указанного в заголовке соединения в виде белого твердого вещества (151 мг, выход 95%). МС (apci) m/z=239,1 (M+Na).
Промежуточное соединение 174
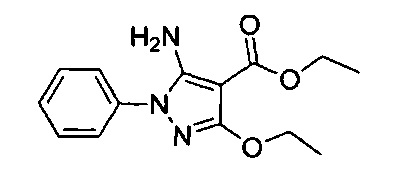
этил 5-амино-3-этокси-1-фенил-1Н-пиразол-4-карбоксилат
Стадия А: Получение диэтил 2-цианомалоната: К суспензии NaH (60 вес.% i минеральном масле, 499 мг, 12,49 ммоль) в ТГФ (100 мл) под N2 при 0°C добавили диэтилмалонат (1,90 мл, 12,49 ммоль). Ледяную баню убрали, а реакционную смесь перемешивали при комнатной температуре в течение 30 минут, затем охладили до 0°C и добавили бромциан (5 М в MeCN, 2,5 мл, 12,49 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 19 часов, затем разбавили H2O (50 мл), экстрагировали Et2O (50 мл). Водную фазу нейтрализовали HCl (2 М, водный, 3 мл), затем экстрагировали ДХМ (2×50 мл). Объединенные ДХМ экстракты высушили (MgSO4), отфильтровали и концентрировали для получения продукта в виде желтого маслянистого вещества (837 мг, выход 36%). 1Н ЯМР (CDCl3) δ 4,46 (s, 1Н), 4,35 (q, 4H), 1,35 (t, 6H).
Стадия B: Получение этил 5-амино-3-этокси-1-фенил-1Н-пиразол-4-карбоксилата: Получили по способу, описанному для Промежуточного соединения Р135, заменяя этил 2-цианопропаноат на диэтил 2-цианомалонат на Стадии A, для получения продукта в виде коричневого сиропообразного вещества (400 мг, выход 32%). МС (apci) m/z=276,1 (М+Н).
Промежуточное соединение 175
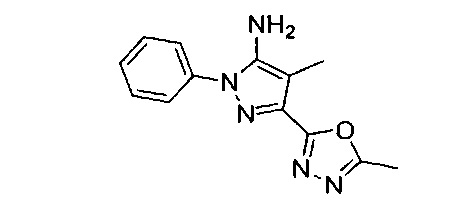
4-метил-3-(5-метил-1,3,4-оксадиазол-2-ил)-1-фенил-1Н-пиразол-5-амин
Стадия A: Получение N'-ацетил-5-амино-4-метил-1-фенил-1Н-пиразол-3-карбогидразида: К раствору 5-амино-4-метил-1-фенил-1Н-пиразол-3-карбоновой кислоты (Промежуточное соединение 171, 93 мг, 0,428 ммоль) в ДХМ (5 мл) и ДИЭА (0,149 мл, 0,856 ммоль) добавили изобутил карбонохлоридат (0,061 мл, 0,471 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем добавили ацетогидразид (48 мг, 0,642 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, затем разбавили H2O (10 мл), экстрагировали ДХМ (2×10 мл), высушили (MgSO4), отфильтровали и концентрировали под пониженным давлением для получения продукта в виде бледно-желтого твердого вещества (119 мг, выход 101%). МС (apci) m/z=274,1 (М+Н).
Стадия B: Получение 4-метил-3-(5-метил-1,3,4-оксадиазол-2-ил)-1-фенил-1Н-пиразол-5-амина: Смесь N'-ацетил-5-амино-4-метил-1-фенил-1Н-пиразол-3-карбогидразида (117 мг, 0,428 ммоль) и POCl3 (0,5 мл) нагревали в пробирке для работы под давлением до 90°C в течение 1 часа. Реакционную смесь перенесли в делительную воронку с EtOAc (5 мл), затем разбавили насыщенным водным раствором NaHCO3 (20 мл), экстрагировали EtOAc (2×15 мл), высушили (MgSO4), отфильтровали и концентрировали. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 0-75% ацетона в гексанах, для получения указанного в заголовке соединения в виде желтого твердого вещества (19,6 мг, выход 18%). МС (apci) m/z=256,1 (М+Н).
Промежуточное соединение 176
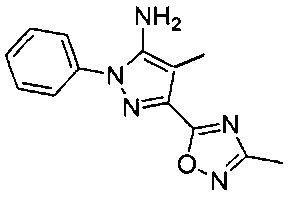
4-метил-3-(3-метил-1,2,4-оксадиазол-5-ил)-1-фенил-1Н-пиразол-5-амин
К суспензии NaH (60% в минеральном масле, 36 мг, 0,897 ммоль) в ТГФ (5 мл) под N2 добавили N-гидроксиацетимидамид (66 мг, 0,897 ммоль). Реакционную смесь нагревали с дефлегматором в течение 1 часа, затем охладили до комнатной температуры и добавили этил 5-амино-4-метил-1-фенил-1Н-пиразол-3-карбоксилат (Промежуточное соединение 170, 200 мг, 0,815 ммоль). Реакционную смесь нагревали с дефлегматором в течение 18 часов, затем охладили до комнатной температуры и добавили дополнительное количество NaH (60% в минеральном масле, 18 мг, 0,449 ммоль). Реакционную смесь нагревали с дефлегматором в течение 4 часов, затем разбавили H2O (10 мл), экстрагировали ДХМ (2×15 мл), а объединенные органические экстракты высушили (MgSO4), отфильтровали и концентрировали под пониженным давлением. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 0-50% ацетона в гексанах, для получения указанного в заголовке соединения в виде оранжевого твердого вещества (84 мг, выход 40%). МС (apci) m/z=256,1 (М+Н).
Промежуточное соединение 177
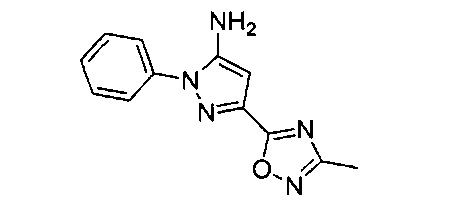
3-(3-метил-1,2,4-оксадиазол-5-ил)-1-фенил-1Н-пиразол-5-амин
Получили по способу, описанному для Промежуточного соединения 176, заменяя этил 5-амино-4-метил-1-фенил-1Н-пиразол-3-карбоксилат на этил 5-амино-1-фенил-1Н-пиразол-3-карбоксилат (Nanjing Chemlin Chemical Co.), для получения продукта в виде желто-коричневого твердого вещества (83 мг, выход 53%). МС (apci) m/z=242,1 (М+Н).
Промежуточное соединение 178
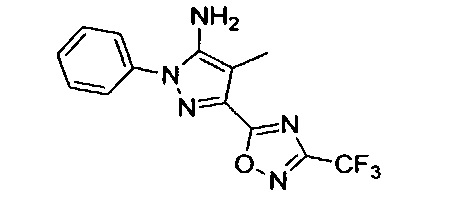
4-метил-1-фенил-3-(3-(трифторметил)-1,2,4-оксадиазол-5-ил)-1Н-пиразол-5-амин
Стадия A: Получение 2,2,2-трифтор-N'-гидроксиацетимидамида: К суспензии гидроксиламина гидрохлорида (5,45 г, 78,4 ммоль) в MeOH (100 мл) добавили NaOMe (25 вес.% раствор в MeOH, 17,9 мл, 78,4 ммоль) и перемешивали смесь при комнатной температуре в течение 10 минут, затем отфильтровали, а твердое вещество промыли MeOH. Фильтрат охладили до 0°C, а затем через раствор продували газообразный 2,2,2-трифторацетонитрил (7,45 г, 78,4 ммоль) в течение 30 минут. Затем реакционную смесь оставили нагреваться до комнатной температуры в течение 19 часов. Раствор концентрировали под пониженным давлением до 50 мл, а твердые вещества отфильтровали. Фильтрат концентрировали, повторно суспендировали в холодном MeOH и отфильтровали. Фильтрат концентрировали, снова повторно суспендировали в холодном MeOH и отфильтровали. Фильтрат концентрировали для получения продукта в виде воскообразного белого вещества (6,7 г, выход 67%). 1Н ЯМР (CD3CN) δ 8,32 (s, 1Н), 5,25 (br s, 2H), 19F ЯМР (CD3CN) δ - 71,8 (s).
Стадия В: Получение 4-метил-1-фенил-3-(3-трифторметил)-1,2,4-оксадиазол-5-ил)-1Н-пиразол-5-амина: К суспензии NaH (60% в минеральном масле, 356 мг, 0,897 ммоль) в ТГФ (5 мл, 0,815 ммоль) под N2 добавили 2,2,2-трифтор-N'-гидроксиацетимидамид (115 мг, 0,897 ммоль). Реакционную смесь нагревали с дефлегматором в течение 1 часа, затем охладили до комнатной температуры и добавили порошкообразные молекулярные сита 4А (200 мг) и этил 5-амино-4-метил-1-фенил-1Н-пиразол-3-карбоксилат (Промежуточное соединение 170; 200 мг, 0,815 ммоль), и нагревали с дефлегматором. Реакционную смесь нагревали с дефлегматором в течение 18 часов, затем отфильтровали, разбавили Н2О (15 мл), экстрагировали ДХМ (2×25 мл), а объединенные органические экстракты промыли насыщенным солевым раствором (25 мл), высушили (MgSO4), отфильтровали и концентрировали под пониженным давлением. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 0-50% ацетона в гексанах, для получения указанного в заголовке соединения в виде белого твердого вещества (44 мг, выход 17%). МС (apci) m/z=310,1 (М+Н).
Промежуточное соединение 179
2-фенил-2Н-индазол-3-амин
Стадия А: Получение 1-(2-йодфенил)-2-фенилдиазена: К раствору 2-йоданилина (1,00 г, 4,57 ммоль) в уксусной кислоте (46 мл) добавили нитрозобензол (0,880 г, 8,22 ммоль) и нагревали смесь при 85°C в течение 16 часов. Смесь охладили до комнатной температуры, вылили в воду и медленно обработали насыщенным раствором NaHCO3 до щелочной среды. Смесь экстрагировали EtOAc (3х), а объединенные экстракты промыли водой, насыщенным раствором NaCl и высушили над MgSO4. Раствор отфильтровали, концентрировали, а остаток очистили обращенно-фазовой хроматографией для получения указанного в заголовке соединения в виде красного твердого вещества (0,880 г, выход 63%). 1H ЯМР (CDCl3) δ 7,23-7,39 (m, 3Н), 7,64 (d, 1Н), 7,56-7,51 (m, 3Н), 7,45 (t, 1H), 7,1 (t, 1H).
Стадия В: 2-(фенилдиазенил)бензонитрил; К раствору 1-(2-йодфенил)-2-фенилдиазена (0,44 г, 1,4 ммоль) в 1-пропаноле (14 мл) добавили CuCN (0,900 г, 10,0 ммоль) и нагревали реакционную смесь с дефлегматором в течение 16 часов. Смесь охладили до комнатной температуры, отфильтровали, а собранное твердое вещество промыли CH2Cl2. Объединенный фильтрат и промывочные растворы концентрировали для получения указанного в заголовке соединения в виде красновато-оранжевого твердого вещества, которое высушили в вакууме (0,280 г, выход 95%). 1Н ЯМР (CDCl3) δ 8,03-8,06 (m, 2Н), 7,88 (dd, 2Н), 7,71 (t, 1Н), 7,54-7,58 (m, 4Н).
Стадия C: 2-фенил-2Н-индазол-3-амин: Смесь 2-(фенилдиазенил)бензонитрила (0,28 г, 1,35 ммоль) и дигидрата SnCl2 (0,562 мл, 6,76 ммоль) в EtOH (14 мл) нагревали с дефлегматором в течение 16 часов. Смесь охладили до комнатной температуры и концентрировали. Остаток разбавили EtOAc и водой, и отфильтровали. Водный слой удалили, а слой EtOAc промыли водой. Объединенные водные фракции подщелочили насыщенным раствором NaHCO3 и экстрагировали CH2Cl2 (2Х). Объединенные органические слои высушили над MgSO4, отфильтровали и концентрировали для получения указанного в заголовке соединения в виде светло-пурпурного твердого вещества, которое высушили в вакууме (0,241 г, выход 85%). 1Н ЯМР (CDCl3) δ 7,69 (d, 2Н), 7,52-7,58 (m, 3Н), 7,47 (d, 2Н), 7,26 (t, 1Н), 6,90 (t, 1Н), 4,28 (br s, 2Н).
Промежуточное соединение 180
3-этокси-4-метил-1-(пиразин-2-ил)-1Н-пиразол-5-амин
Стадия А: 5-амино-4-метил-1-(пиразин-2-ил)-1Н-пиразол-3(2H)-он: К смеси 2-гидразинилпиразина (0,551 г, 5,00 ммоль) и этил 2-цианопропаноата (0,669 г, 5,00 ммоль) в абсолютном EtOH (10 мл) добавили 3 М NaOEt в EtOH (0,167 мл, 0,501 ммоль) и нагревали смесь с дефлегматором в течение 64 часов. Смесь концентрировали, а остаточное желто-коричневое твердое вещество обработали EtOAc (30 мл) и обработали ультразвуком. Полученную желто-коричневую суспензию энергично перемешивали в течение 8 часов. Твердое вещество собрали вакуумной фильтрацией, промыли EtOAc и высушили в вакууме для получения указанного в заголовке соединения в виде светлого желто-коричневого порошка (682 мг, 71%). 1Н ЯМР (ДМСО d6) δ 10,3 (br s, 1Н), 8,82 (s, 1H), 8,30 (d, 2H), 6,55 (s, 2H), 1,71 (s, 3H).
Стадия B: 3-этокси-4-метил-1-(пиразин-2-ил)-1Н-пиразол-5-амин: Смесь 5-амино-4-метил-1-(пиразин-2-ил)-1Н-пиразол-3(2Н)-она (382 мг, 2,00 ммоль) и порошкообразного K2CO3 (552 мг, 4,00 ммоль) в сухом ДМФ (3,0 мл) перемешивали при комнатной температуре в течение 10 минут. Смесь охладили до 0°C и добавили бромэтан (229 мг, 2,10 ммоль). Смесь оставили достигать до комнатной температуры и перемешивали в течение 24 часов. Реакционную смесь вылили в холодную H2O (12 мл), оставили достигать комнатной температуры и экстрагировали EtOAc (3Х). Объединенные экстракты промыли насыщенным раствором NaCl (2Х), высушили над MgSO4 и активированным углем. Высушенный раствор разбавили равным объемом гексанов и отфильтровали через слой SiO2, покрытый слоем MgSO4, элюируя 50% EtOAc в гексанах. Фильтрат концентрировали, а остаточное желтое твердое вещество промыли гексанами (3Х) и высушили в вакууме для получения указанного в заголовке соединения в виде светло-желтого кристаллического вещества (195 мг, 45%). 1Н ЯМР (CDCl3) δ 9,10 (s, 1Н), 8,23 (s, 1H), 8,14 (s, 1Н), 5,50 (br s, 2Н), 4,33 (q, 2Н), 1,80 (s, 3Н), 1,42 (t, 3Н).
Промежуточное соединение 181
2-(пиридазин-4-ил)-2,4,5,6-тетрагидроциклопента[с]пиразол-3-амин
Суспензию 4-гидразинилпиридазина гидробромида (0,368 г, 1,93 ммоль) в абсолютном EtOH (5 мл) обработали 2-оксоциклопентанкарбонитрилом (0,191 г, 1,75 ммоль) и нагревали смесь с дефлегматором в течение 22 часов. Смесь охладили до комнатной температуры и концентрировали до оранжевого твердого вещества. Твердое вещество суспендировали в 1 М NaOH и перемешивали в течение 10 минут. Твердое вещество собрали, тщательно промыли H2O и Et2O и высушили в вакууме для получения указанного в заголовке соединения в виде желто-коричневого порошка (,323 г, 92%). МС (apci) m/z=202,1 (М+Н).
Промежуточное соединение 182
(5-амино-1-фенил-1Н-пиразол-3-ил)метанол
Стадия А: Этил 2-(трет-бутилдиметилсилилокси)ацетат: Смесь этил 2-гидроксиацетата (3,00 г, 28,8 ммоль), TBDMS-Cl (5,21 г, 34,6 ммоль) и имидазола (2,55 г, 37,5 ммоль) перемешивали при комнатной температуре в течение 60 часов. Смесь концентрировали, а остаток очистили хроматографией на SiO2, элюируя 10% EtOAc в гексанах, для получения указанного в заголовке соединения в виде бесцветного маслянистого вещества (4,12 г, 65%). 1Н ЯМР (CDCl3) δ 4,12 (s, 2Н), 4,09 (q, 2Н), 1,17 (t, 3Н), 0,18(s, 9Н), 0,00 (s, 6Н).
Стадия В: (5-амино-1-фенил-1Н-пиразол-3-ил)метанол: Раствор ацетонитрила (0,526 мл, 10,1 ммоль) в сухом ТГФ (20,4 мл, 9,16 ммоль) охладили до -78°C и по каплям добавили 2,5 М nBuLi в гексанах (4,21 мл, 10,5 ммоль). Реакционную смесь перемешивали в течение 15 минут и добавили этил 2-(трет-бутилдиметилсилилокси)ацетат (2,00 г, 9,16 ммоль). Реакционную смесь оставили нагреваться до комнатной температуры и перемешивали в течение 2 часов. Реакционную смесь разбавили ледяной водой и концентрировали. Остаточную водную смесь подкислили до pH=5 и экстрагировали EtOAc (3Х). Объединенные органические экстракты промыли насыщенным солевым раствором, высушили над MgSO4, отфильтровали и концентрировали. Остаточное коричневое маслянистое вещество растворили в MeOH (23 мл) и добавили фенилгидразин (0,907 мл, 9,14 ммоль). Смесь обработали концентрированной HCl (3,81 мл, 45,7 ммоль) и нагревали с дефлегматором в течение 18 часов. После охлаждения смесь концентрировали, а остаток разделили между H2O и CH2Cl2. Смесь отфильтровали, а органический слой удалили из фильтрата. Водную часть промыли CH2Cl2 и обработали насыщенным раствором NaHCO3 до щелочной среды. Водную смесь экстрагировали CH2Cl2 (3Х), а объединенные органические фракции высушили над MgSO4, отфильтровали и концентрировали. Остаток очистили колоночной хроматографией на диоксиде кремния, используя элюирование градиентом 70-100% EtOAc в гексанах, а затем 0-5% MeOH/EtOAc. Растворы, содержащие продукт, объединили и концентрировали для получения указанного в заголовке соединения в виде желтого пенистого вещества (0,760 г, выход 44%). МС (apci) m/z=190,1 (М+Н).
Промежуточное соединение 183
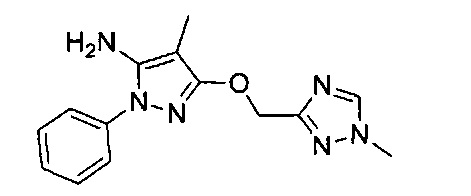
4-метил-3-((1-метил-1Н-1,2,4-триазол-3-ил)метокси)-1-фенил-1Н-пиразол-5-амин
Указанное в заголовке соединение получили по способу, описанному для Промежуточного соединения Р135, заменяя бромэтан на 3-(хлорметил)-1-метил-1Н-1,2,4-триазола гидрохлорид. Продукт выделили в виде золотистого сиропообразного вещества (110 мг, 27%).МС (apci) m/z=285,1 (М+Н).
Промежуточное соединение 184
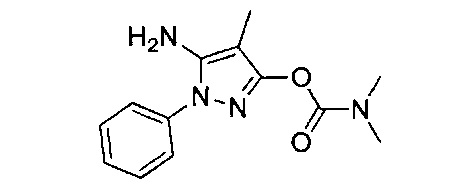
5-амино-4-метил-1-фенил-1Н-пиразол-3-ил диметилкарбамат
Смесь 5-амино-4-метил-1-фенил-1Н-пиразол-3(2Н)-она (Промежуточное соединение Р135, Стадия A, 0,378 г, 2,00 ммоль) и порошкообразного K2CO3 (0,553 г, 4,00 ммоль) в сухом ДМФ (4 мл) перемешивали при комнатной температуре в течение 5 минут. Добавили диметилкарбамоил хлорид (0,206 мл, 2,20 ммоль) и перемешивали смесь в течение 6 часов. Смесь вылили в ледяную H2O (40 мл) и экстрагировали EtOAc (3Х). Объединенные экстракты промыли насыщенным раствором NaCl (2Х), высушили над MgSO4 и отфильтровали через слой SiO2, покрытый слоем MgSO4 (элюирование с EtOAc). Фильтрат концентрировали, а остаток высушили в вакууме для получения указанного в заголовке соединения в виде светло-золотистого сиропообразного вещества (,507 г, 97%). МС (apci) m/z=261,1 (М+Н).
Промежуточное соединение 185
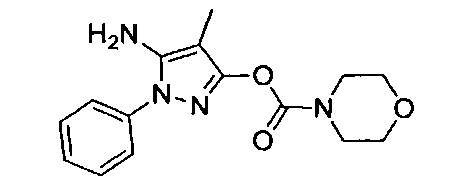
5-амино-4-метил-1-фенил-1Н-пиразол-3-ил морфолин-4-карбоксилат
Указанное в заголовке соединение получили, используя морфолин-4-карбонилхлорид в способе, описанном для 5-амино-4-метил-1-фенил-1Н-пиразол-3-ил диметилкарбамата (Промежуточное соединение 184). Соединение выделили в виде светло-желтого воскообразного вещества (0,285 г, 47%). 1Н ЯМР (CDCl3) δ 7,54 (d, 2Н), 7,43 (t, 2Н), 7,31 (t, 1H), 3,66-3,78 (m, 8H), 3,57 (brs, 2H), 1,85 (s, 3Н).
Промежуточное соединение 186
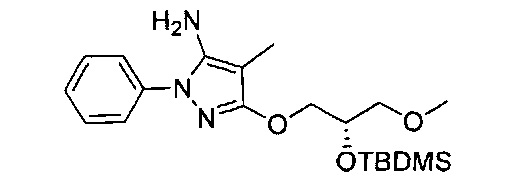
(S)-3-(2-((трет-бутилдиметилсилил)окси)-3-метоксипропил)-4-метил-1-фенил-1Н-пиразол-5-амин
Стадия A: (S)-1-(5-амино-4-метил-1-фенил-1Н-пиразол-3-илокси)-3-метоксипропан-2-ол: Смесь 5-амино-4-метил-1-фенил-1Н-пиразол-3(2Н)-она (Р135, Стадия А, 1,21 г, 6,40 ммоль) и порошкообразного K2CO3 (1,77 г, 12,8 ммоль) в сухом ДМФ (12 мл) перемешивали при комнатной температуре в течение 10 минут. Добавили (S)-2-(метоксиметил)оксиран (0,622 мл, 6,72 ммоль) и перемешивали смесь при 80°C в течение 6 часов. Смесь охладили до комнатной температуры, вылили в ледяную H2O (25 мл) и экстрагировали EtOAc (3Х). Объединенные экстракты промыли насыщенным раствором NaCl (2Х), высушили над и отфильтровали через слой SiO2, покрытый слоем MgSO4, элюируя EtOAc. Фильтрат концентрировали для получения указанного в заголовке соединения в виде бесцветного, вязкого маслянистого вещества (701 мг, 40%). МС (apci) m/z=278,1 (М+Н).
Стадия B: (S)-3-(2-((трет-бутилдиметилсилил)окси)-3-метоксипропил)-4-метил-1-фенил-1Н-пиразол-5 -амин: К раствору TBDMS-C1 (725 мг, 4,81 ммоль) и имидазола (390 мг, 5,72 ммоль) в сухом ДМФ (7,0 мл) добавили (S)-1-(5-амино-4-метил-1-фенил-1Н-пиразол-3-илокси)-3-метоксипропан-2-ол (635 мг, 2,29 ммоль) в сухом ДМФ (2 мл). Смесь перемешивали при комнатной температуре в течение 2,5 часов. Смесь добавили к H2O (70 мл), перемешивали в течение 5 минут и экстрагировали Et2O (3Х). Объединенные экстракты промыли насыщенным раствором NaCl (2Х) и высушили над MgSO4. Высушенный раствор отфильтровали через слой SiO2, покрытый слоем MgSO4 (элюирование с Et2O). Фильтрат концентрировали для получения указанного в заголовке соединения в виде бесцветного маслянистого вещества, которое высушили в вакууме (940 мг, 105%). МС (apci) m/z=392,2 (М+Н). 1Н ЯМР (CDCl3) δ 7,50 (d, 2Н), 7,40 (t, 2Н), 7,23 (t, 1Н), 4,09-4,30 (m, 3Н), 3,57 (br s, 2H), 3,38-3,44 (m, 2H), 3,32 (s, 3H), 1,83 (s, 3H), 0,88 (s, 9H), 0,11 (s, 6H).
Промежуточное соединение 187
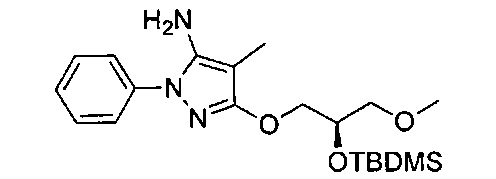
(R)-3-(2-((трет-бутилдиметилсилил)окси)-3-метоксипропил)-4-метил-1-фенил-1Н-пиразол-5-амин
Указанное в заголовке соединение получили, используя способ, описанный для (S)-3-(2-((трет-бутилдиметилсилил)окси)-3-метоксипропокси)-4-метил-1-фенил-1Н-пиразол-5-амина (Промежуточное соединение 186), заменяя (S)-2-(метоксиметил)оксиран на (R)-2-(метоксиметил)оксиран на Стадии А. Продукт получили в виде бесцветного сиропообразного вещества (921 мг, 38% за 2 стадии). МС (apci) m/z=392,2 (М+Н). 1Н ЯМР (CDCl3) δ 7,50 (d, 2Н), 7,40 (t, 2Н), 7,23 (t, 1Н), 4,09-4,30 (m, 3Н), 3,57 (br s, 2H), 3,38-3,44 (m, 2H), 3,32 (s, 3H), 1,83 (s, 3H), 0,88 (s, 9H), 0,11 (s, 6H).
Промежуточное соединение 188
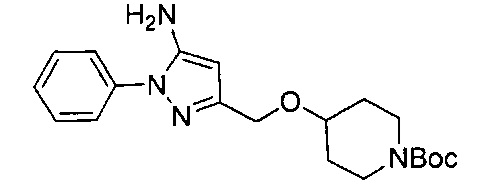
трет-бутил 4-((5-амино-1-фенил-1Н-пиразол-3-ил)метокси)пиперидин-1-карбоксилат
Стадия А: трет-бутил 4-(2-этокси-2-оксоэтокси)пиперидин-1-карбоксилат: Раствор трет-бутил 4-гидроксипиперидин-1-карбоксилата (2,00 г, 9,94 ммоль) в сухом ТГФ (25 мл) охладили до 0°C и добавили KOtBu (1,12 г, 9,94 ммоль). Смесь оставили достигать до комнатной температуры и перемешивали в течение 10 минут. Смесь охладили до 0°C и по каплям добавили этил 2-бромацетат (1,65 мл, 14,9 ммоль). Реакционную смесь оставили достигать до комнатной температуры и перемешивали в течение 17 часов. Смесь разделили в H2O и EtOAc, перемешали и удалили органический слой. Органический слой высушили над MgSO4, отфильтровали и концентрировали. Остаточное густое желтое маслянистое вещество очистили хроматографией на диоксиде кремния, используя градиентное элюирование 10-25% EtOAc в гексанах, для получения указанного в заголовке соединения в виде бесцветного маслянистого вещества (0,967 г, выход 34%). 1Н ЯМР (CDCl3) δ 4,22 (q, 2Н), 4,12 (s, 2Н), 3,67-3,84 (m, 2Н), 3,52-3,63 (m, 1Н), 3,05-3,11 (m, 2H), 1,81-1,90 (m, 2H), 1,53-1,62 (m, 2H), 1,45 (s, 9H), 1,29 (t, 3Н).
Стадия В: трет-бутил 4-((5-амино-1-фенил-1Н-пиразол-3-ил)метокси)пиперидин-1-карбоксилат: Раствор диизопропиламина (1,08 мл, 7,74 ммоль) в сухом ТГФ (5 мл) охладили до 0°C и медленно добавили 2,5 М nBuLi в гексанах (2,96 мл, 7,41 ммоль). Смесь перемешивали при 0°C в течение 10 минут и охладили до -78°C. Добавили ацетонитрил (0,404 мл, 7,74 ммоль) и перемешивали смесь в течение 15 минут. Добавили раствор трет-бутил 4-(2-этокси-2-оксоэтокси)пиперидин-1-карбоксилата (0,967 г, 3,37 ммоль) в ТГФ (2,5 мл) и перемешивали смесь при -78°C в течение 1 часа. Смесь оставили достигать комнатной температуры, погасили ледяной водой и концентрировали. Остаточную водную смесь нейтрализовали при помощи 2 М HCl и экстрагировали CH2Cl2 (3Х). Объединенные органические фракции высушили над MgSO4, отфильтровали и концентрировали для получения неочищенного циано-кетона в виде желтого маслянистого вещества, которое сразу использовали на следующей стадии.
Стадия С: трет-бутил 4-((5-амино-1-фенил-1Н-пиразол-3-ил)метокси)пиперидин-1-карбоксилат: Неочищенное маслянистое вещество, полученное на Стадии B, растворили в EtOH (17 мл) и добавили фенилгидразин (0,396 мл, 3,99 ммоль). Смесь нагревали при 60°C в течение 60 часов, охладили до комнатной температуры и концентрировали. Остаток разделили в EtOAc и воде, перемешали и удалили органический слой. Водный слой экстрагировали EtOAc (2Х), а объединенные EtOAc части высушили над MgSO4, отфильтровали и концентрировали. Остаточное оранжевое маслянистое вещество очистили хроматографией на диоксиде кремния, используя градиентное элюирование 10-100% EtOAс в гексанах. Объединенные фракции, содержащие продукт, концентрировали, а остаточное желто-оранжевое маслянистое вещество повторно очистили обращенно-фазовой ВЭЖХ, используя градиент 0-100% ацетонитрила в воде, для получения указанного в заголовке соединения в виде оранжевого пенистого вещества (0,264 г, выход 21%). МС (apci) m/z=373,2 (М+Н).
Промежуточное соединение 189
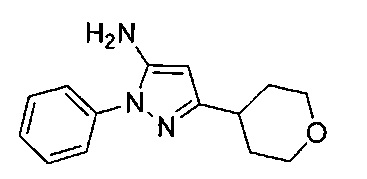
1-фенил-3-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразол-5-амин
Стадия А: 3-оксо-3-(тетрагидро-2Н-пиран-4-ил)пропаннитрил: 1 М раствор LHMDS в сухом ТГФ (26,3 мл, 26,3 ммоль) охладили до -78°C и по каплям добавили ацетонитрил (1,43 мл, 27,5 ммоль) за 2 минуты. Смесь перемешивали при -78°C в течение 1 часа и добавили раствор метил тетрагидро-2Н-пиран-4-карбоксилата (3,41 мл, 25,0 ммоль) в сухом ТГФ (12 мл). Смесь перемешивали в течение 1 часа, убрали баню из сухого льда и оставили смесь достигать комнатной температуры. Смесь вылили в ледяную H2O (250 мл) и экстрагировали Et2O (3Х). Водную часть охладили до 0°C и по каплям добавили 6 М HCl до pH=3 (первоначальный pH=12). Смесь экстрагировали EtOAc (3Х), а объединенные экстракты высушили над MgSO4. Раствор элюировали через слой SiO2, элюируя EtOAc. Фильтрат концентрировали для получения указанного в заголовке соединения в виде бесцветного маслянистого вещества (2,52 г, 66%). 1Н ЯМР (CDCl3) δ 3,99-4,06 (m, 2Н), 3,54 (s, 2Н), 3,46 (t, 2Н), 2,76-2,86 (m, 1Н), 1,70-1,86 (m, 4Н).
Стадия B: 1-фенил-3-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразол-5-амин: К раствору 3-оксо-3-(тетрагидро-2Н-пиран-4-ил)пропаннитрила (2,30 г, 12,8 ммоль) в абсолютном EtOH (35 мл) добавили фенилгидразина гидрохлорид (2,21 г, 15,3 ммоль) и нагревали смесь с дефлегматором до завершения реакции, по данным ТСХ (5 часов). Смесь охладили до комнатной температуры и концентрировали. Остаток разделили в H2O (75 мл) и EtOAc (40 мл). Добавили 2 М NaOH до pH=5 при энергичном перемешивании, органический слой удалили, а водный слой экстрагировали EtOAc (2Х). Объединенные EtOAc фракции промыли H2O и насыщенным раствором NaCl. Раствор разбавили равным объемом гексанов, высушили над MgSO4/активированным углем и элюировали через слой SiO2, элюируя 50% EtOAc в гексанах. Фильтрат концентрировали для получения золотистого сиропообразного вещества. Сиропообразное вещество обработали Et2O и перемешивали до образования тонкодисперсной, зернистой суспензии. Твердое вещество собрали, промыли Et2O и высушили в вакууме для получения указанного в заголовке соединения в виде белого твердого вещества (2,01 г, 65%). 1Н ЯМР (CDCl3) δ 7,55 (d, 2Н), 7,46 (t, 2Н), 7,32 (t, 1Н), 5,49 (s, 1H), 4,00-4,08 (m, 2Н), 3,97 (br s, 2Н), 3,52 (dt, 2Н), 2,86 (m, 1Н) 1,73-1,93 (m, 4Н).
Следующие соединения были получены по способу, использованному для получения 1-фенил-3-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразол-5-амина (Промежуточное соединение 189), с использованием ацетонитрила или пропионитрила на Стадии А, вместе с соответствующим сложным эфиром.
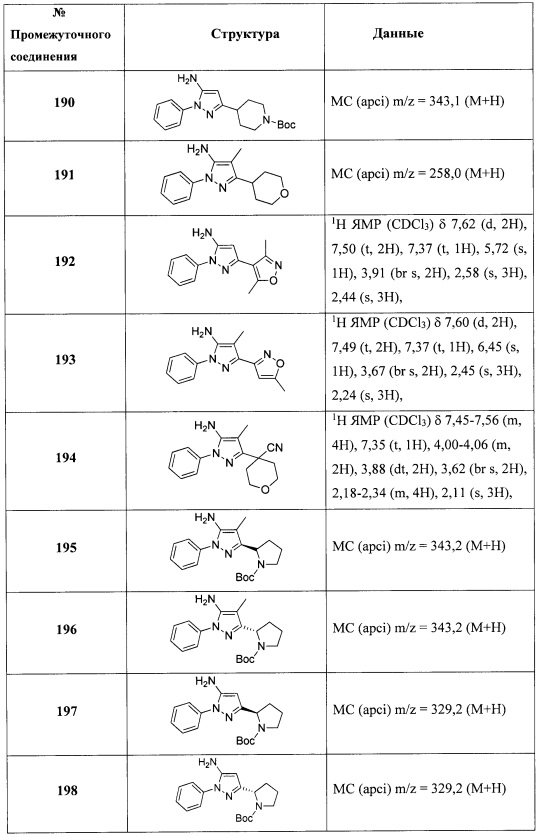
Промежуточное соединение 199
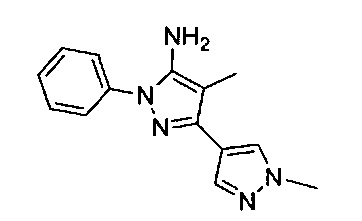
Фенил 1',4-диметил-1-фенил-1Н, 1'Н-3,4'-бипиразол-5-илкарбамат
Стадия А: этил 1-метил-1Н-пиразол-4-карбоксилат: В 3000-мл трехгорлую колбу добавили этил 2-формил-3-оксопропаноат (100 г, 694 ммоль), затем безводный EtOH 200 марки (694 мл) для получения прозрачного желтоватого раствора. Реакционную смесь охладили на ледяной бане до 5°C, затем по каплям добавили метилгидразин (35,8 мл, 680 ммоль). При добавлении гидразина наблюдали бурную экзотерму, а температуру поддерживали ниже 12°C контролированием скорости добавления. После завершения добавления гидразина, ледяную баню убрали, а реакционную смесь оставили перемешиваться при комнатной температуре в течение ночи. Реакционную смесь концентрировали на ротационном испарителе до неочищенного оранжевого маслянистого вещества. Неочищенное вещество растворили в ДХМ и повторно концентрировали, затем под высоким вакуумом в течение 2 дней для получения коричнево-оранжевого маслянистого вещества. ЖХ/МС и 1Н ЯМР показали практически чистый этил 1-метил-1Н-пиразол-4-карбоксилат (106 г, 99,1%).
Стадия B: 2-метил-3-(1-метил-1Н-пиразол-4-ил)-3-оксопропаннитрил: В четырехгорлую 5-литровую кругл о донную колбу, оснащенную верхней мешалкой и капельной воронкой, загрузили LHMDS (1444 мл, 1444 ммоль) (1,0 М в ТГФ). Раствор сначала охладили на бане из ацетона и сухого льда (внутренняя температура -79°C) под азотом, затем через капельную воронку медленно добавили пропиононитрил (103 мл, 1444 ммоль). Смесь перемешивали при -80°C в течение 90 минут. Затем по каплям, через капельную воронку ввели этил 1-метил-1Н-пиразол-4-карбоксилат (106 г, 688 ммоль) в безводном ТГФ (500 мл) (время добавления: около 45 минут; внутренняя температура при добавлении оставалась ниже -76°C). После завершения добавления, реакционную смесь оставили медленно нагреваться до комнатной температуры и перемешивали в течение ночи. На дне колбы в осадок выпало оранжевое стекловидное вещество. Органические вещества декантировали, а стекловидное вещество растворили в теплой воде. Смесь промыли эфиром (3×1000 мл). Затем pH водной фазы довели до 5 (pH-индикаторная бумага), используя концентрированную HCl и насыщенный раствор бикарбоната. Водный слой экстрагировали ДХМ (3×1000 мл). Объединенные органические экстракты высушили над MgSO4, отфильтровали и концентрировали для получения 2-метил-3-(1-метил-1Н-пиразол-4-ил)-3-оксопропаннитрила в виде маслянистого вещества янтарного цвета (92 г, 82%). МС (apci) m/z=162,1 (М-Н).
Стадия C: 1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-амин: В 3 л, 3-горлую кругло донную колбу загрузили 2-метил-3-(1-метил-1Н-пиразол-4-ил)-3-оксопропаннитрил (60 г, 368 ммоль), абсолютный безводный этанол (1000 мл) и фенилгидразина гидрохлорид (58 г, 404 ммоль) при комнатной температуре, для получения желтоватой суспензии. Реакционный сосуд оснастили водяным холодильником и нагревали с дефлегматором (используя колбонагреватель) в течение ночи. Реакционную смесь концентрировали и добавили 1 М NaOH (1 л), а твердое вещество раздробили и собрали. Твердое вещество промыли водой и гексанами. Из фильтрата собрали вторую партию продукта, выпавшего в осадок. Объединенные твердые вещества раздробили и растерли с эфиром (500 мл). Твердое вещество собрали фильтрацией, промыли гексанами и высушили на воздухе под вакуумом для получения 1',4-диметил-1-фенил-1Н,1'H-3,4-бипиразол-5-амина (93 г, 100%).
Стадия D: фенил 1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-илкарбамат: В 3 л круглодонную колбу загрузили диметил-1-фенил-1Н,1'Н-3,4-бипиразол-5-амин (50 г, 197,4 ммоль) и EtOAc(1000 мл) для получения прозрачного коричневатого раствора. К нему одной порцией добавили NaOH (2 М, водный) (500 мл) для получения мутной смеси (водный и органический слои были прозрачными, но между слоями наблюдали осадок). Через 3 минуты медленно добавили фенил карбонохлоридат (74,29 мл, 592,2 ммоль) при комнатной температуре с экзотермой до 33°C. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавили дополнительное количество фенил карбонохлоридата (10 мл). Через 30 минут органические вещества отделили, промыли насыщенным солевым раствором и концентрировали in vacuo. Продукт очистили силикагелевой хроматографией (элюируя 75% этилацетата в гексанах) для получения фенил диметил-1-фенил-1Н,1Н-3,4-бипиразол-5-илкарбамата (60 г, 81,4%).
Промежуточное соединение 200
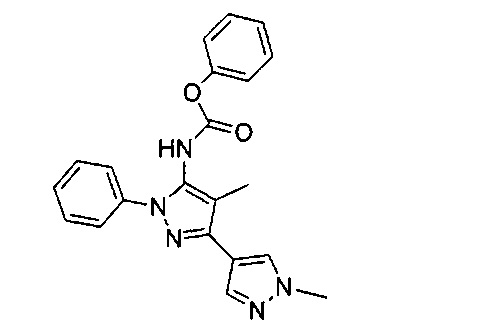
фенил 1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-илкарбамат
В 3 л круглодонную колбу загрузили диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-амин (50 г, 197,4 ммоль) и EtOAc (1000 мл) для получения прозрачного коричневатого раствора. К нему одной порцией добавили NaOH (2 М, водный) (500 мл) для получения мутной смеси (водный и органический слои были прозрачными, но между слоями наблюдали осадок). Через 3 минуты медленно добавили фенилкарбонохлоридат (74,29 мл, 592,2 ммоль) при комнатной температуре (температура реакционной смеси при добавлении повысилась до 33°C). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавили дополнительное количество фенил карбонохлоридата (10 мл). Через 30 минут органические слои отделили, промыли насыщенным солевым раствором и концентрировали in vacuo. Остаток очистили силикагелевой хроматографией (элюируя 75% этилацетата в гексанах) для получения фенил 1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-илкарбамата (60 г, 81,4%).
Промежуточное соединение 201
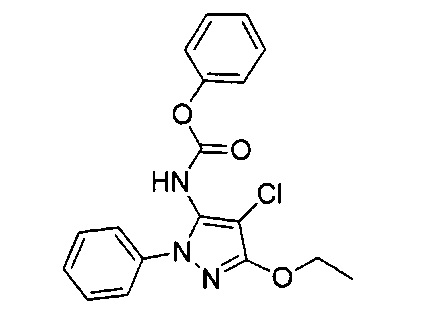
фенил (4-хлор-3-этокси-1-фенил-1Н-пиразол-5-ил)карбамат
Стадия А: Получение фенил (3-этокси-1-фенил-1Н-пиразол-5-ил)карбамата: К суспензии 3-этокси-1-фенил-1Н-пиразол-5-амина (Промежуточное соединение Р139, 169 мг, 0,832 ммоль) в EtOAc(5 мл) при 0°C добавили 2,0 М водный раствор NaOH (1,25 мл, 2,50 ммоль), затем по каплям добавили фенилкарбонохлоридат (0,178 мл, 1,41 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов. Реакционную смесь разбавили EtOAc и разделили фазы. Органический слой промыли водой и насыщенным солевым раствором, высушили над MgSO4 и концентрировали. Остаток очистили флэш-хроматографией на силикагеле (6:1 гексаны/EtOAc) для получения указанного в заголовке соединения (219 мг, выход 81%). МС (apci) m/z=324,1 (М+Н).
Стадия В: Получение фенил (4-хлор-3-этокси-1-фенил-1Н-пиразол-5-ил)карбамата: К раствору фенил 3-этокси-1-фенил-1Н-пиразол-5-илкарбамата (92 мг, 0,28 ммоль) и пиридиния 4-метилбензолсульфоната (7,2 мг, 0,028 ммоль) в ДХМ (2 мл) добавили N-хлорсукцинимид (42 мг, 0,31 ммоль) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 2 дней, а затем концентрировали под пониженным давлением. Остаток очистили флэш-хроматографией на силикагеле (9:1, гексаны/EtOAc) для получения указанного в заголовке соединения (76 мг, выход 75%). МС (apci) m/z=358,1 (М+Н).
Промежуточное соединение 202
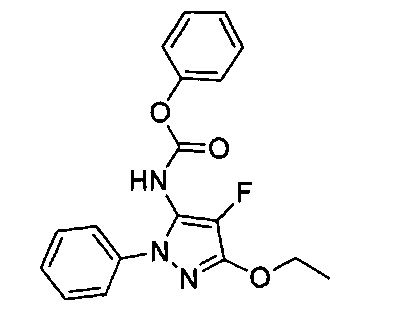
Фенил (3-этокси-4-фтор-1-фенил-1Н-пиразол-5-ил)карбамат
Получили по способу, описанному в Примере 167, стадия B, используя фенил 3-этокси-1-фенил-1Н-пиразол-5-илкарбамат вместо фенил 3-метил-1-фенил-1Н-пиразол-5-илкарбамата.
Промежуточное соединение 203
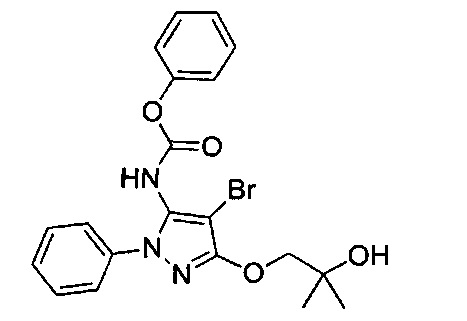
Фенил (4-бром-3-(2-гидрокси-2-метилпропокси)-1-фенил-1Н-пиразол-5-ил)карбамат
Стадия А: Получение 5-амино-1-фенил-1Н-пиразол-3(2Н)-она: Получили по способу, описанному для Промежуточного соединения Р1, заменяя 4,4-диметил-3-оксопентаннитрил на этил 2-цианоацетат, и заменяя фенилгидразином этил 3-гидразинилбензоата гидрохлорид. МС (apci) m/z=176,0 (М+Н).
Стадия В: Получение 1-((5-амино-1-фенил-1Н-пиразол-3-ил)окси)-2-метилпропан-2-ола: Смесь 5-амино-1-фенил-1Н-пиразол-3(2Н)-она (0,330 г, 1,88 ммоль), 2,2-диметилоксирана (0,143 г, 1,98 ммоль) и K2CO3 (0,521 г, 3,77 ммоль) в ДМА (5 мл) нагревали при 80°C в течение 3 дней. После охлаждения реакционную смесь разбавили EtOAc, промыли водой и насыщенным солевым раствором и высушили над MgSO4. Смесь отфильтровали через слой Sid, элюируя EtOAc, для получения указанного в заголовке соединения. МС (apci) m/z=248,1 (М+Н).
Стадия С: Получение фенил (3-(2-гидрокси-2-метилпропокси)-1-фенил-1Н-пиразол-5-ил)карбамата: Получили по способу, описанному для Промежуточного соединения 201, Стадия А, используя 1-((5-амино-1-фенил-1Н-пиразол-3-ил)окси)-2-метилпропан-2-ол вместо 3-этокси-1-фенил-1Н-пиразол-5-амина. МС (apci) m/z=368,1 (М+Н).
Стадия D: Получение фенил (4-бром-3-(2-гидрокси-2-метилпропокси)-1-фенил-1Н-пиразол-5-ил)карбамата: Получили по способу, описанному для Промежуточного соединения 201, Стадия В, используя N-бромсукцинимид вместо N-хлорсукцинимида, и заменяя фенил (3-(2-гидрокси-2-метилпропокси)-1-фенил-1Н-пиразол-5-ил)карбаматом фенил 3-этокси-1-фенил-1H-пиразол-5-илкарбамат. МС (apci) m/z=446,1 (М+Н).
Следующие соединение были получены по способу, описанному для получения Промежуточного соединения 200, с использованием соответствующего аминопиразолового промежуточного соединения:
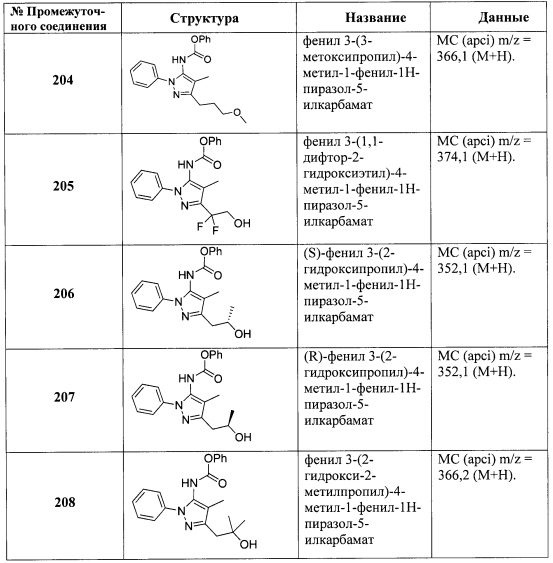
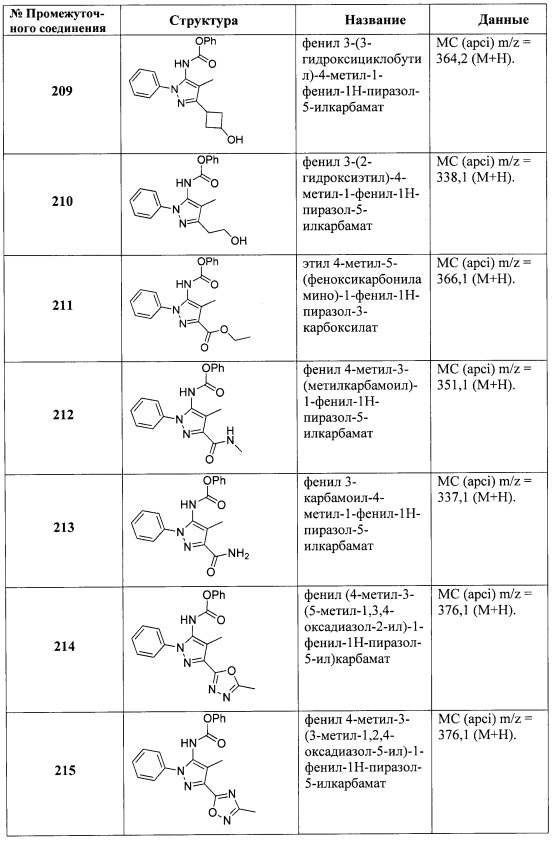
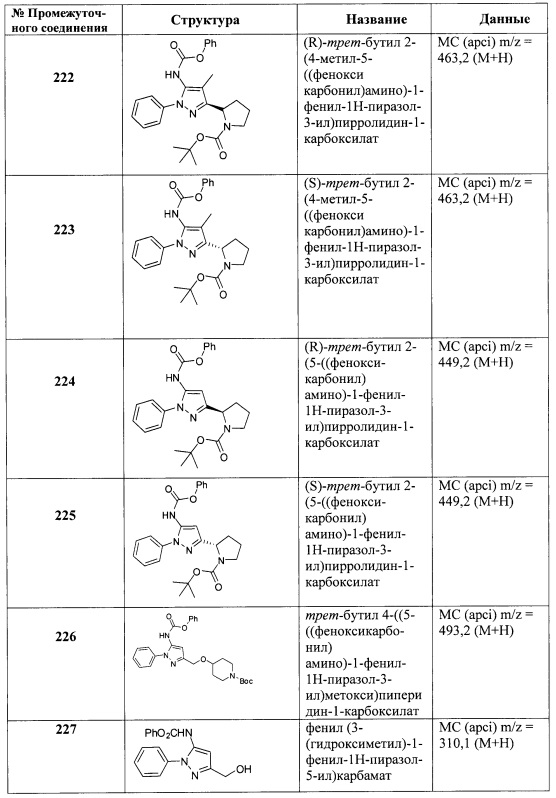
Промежуточное соединение 228
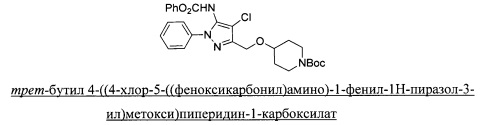
К суспензии трет-бутил 4-((5-(феноксикарбониламино)-1-фенил-1Н-пиразол-3-ил)метокси)пиперидин-1-карбоксилата (Промежуточное соединение 226, 98,5 мг, 0,200 ммоль) в ДХМ (2,0 мл) добавили пиридиния 4-метилбензолсульфонат (PPTS) (5,03 мг, 0,020 ммоль) и N-хлорсукцинимид (40,1 мг, 0,300 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 8 дней. Смесь разбавили водой и CH2Cl2, органический слой отделили, а водный слой экстрагировали CH2Cl2 (2Х). Объединенные органические фракции высушили над MgSO4, отфильтровали и концентрировали. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя градиентом 30-40% EtOAc в гексанах, для получения указанного в заголовке соединения в виде оранжевого маслянистого вещества (73,5 мг, выход 70%). МС (apci) m/z=527,2 (М+Н).
Промежуточное соединение 229
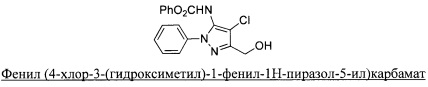
Получили из фенил 3-(гидроксиметил)-1-фенил-1Н-пиразол-5-илкарбамата (Промежуточное соединение 227), используя способ, описанный для получения трет-бутил 4-((4-хлор-5-((феноксикарбонил)амино)-1-фенил-1Н-пиразол-3-ил)метокси)-пиперидин-1-карбоксилата (Промежуточное соединение 228). В этом случае соединение выделили в виде белого твердого вещества (108 мг, 28%). МС (apci) m/z=344,0 (М+Н).
Промежуточное соединение 230
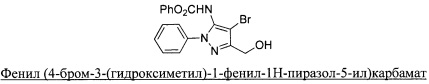
К суспензии фенил 3-(гидроксиметил)-1-фенил-1Н-пиразол-5-илкарбамата (Промежуточное соединение 227, 100 мг, 0,323 ммоль) в CH2Cl2 (1,6 мл) добавили пиридиния 4-метилбензолсульфонат (PPTS) (8,12 мг, 0,0323 ммоль) и N-бромсукцинимид (86,3 мг, 0,485 ммоль). Реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Полученную суспензию отфильтровали, а собранное твердое вещество быстро промыли CH2Cl2 и высушили в вакууме для получения указанного в заголовке соединения в виде белого твердого вещества (48,5 мг, выход 39%). МС (apci) m/z=388,0 (М+Н).
Следующие пиразоловые промежуточные соединения были получены по способам, описанным для получения Промежуточного соединения 228, 229 или 230.
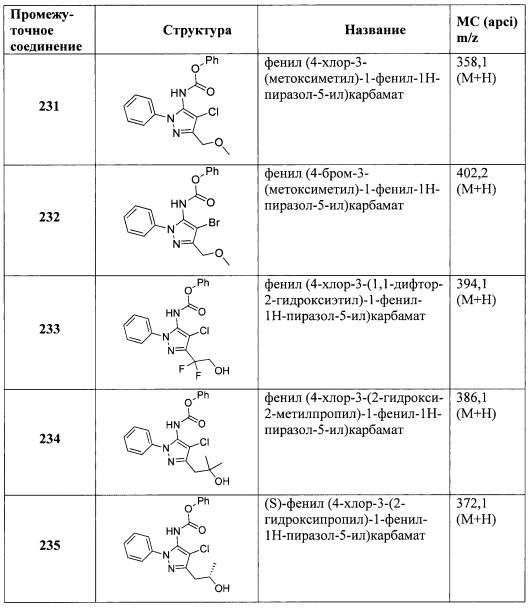
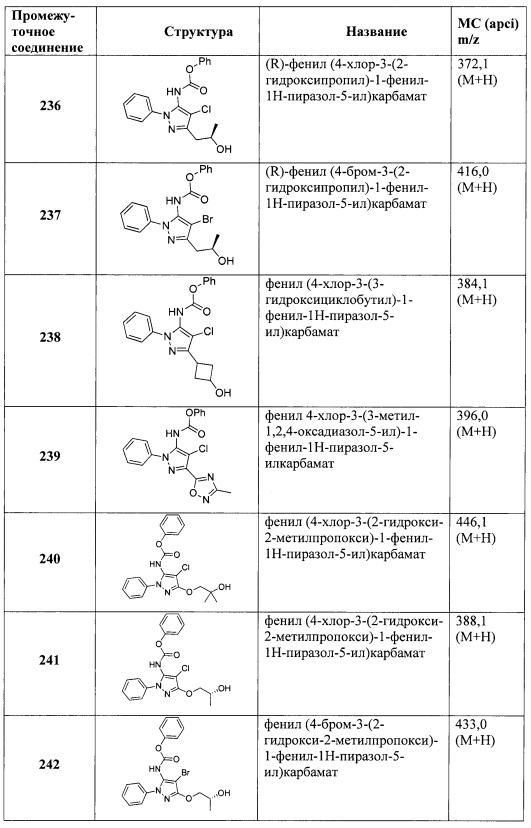
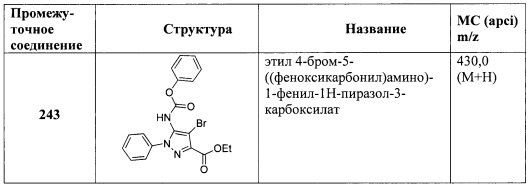
Промежуточное соединение 244
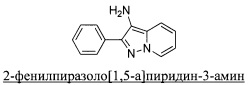
Стадия А: Этил 2-фенилпиразоло[1,5-а]пиридин-3-карбоксилат: К раствору 1-аминопиридиния йодида (2,22 г, 10,0 ммоль) в ДМФ (20 мл) добавили K2CO3 (1,93 г, 14,0 ммоль) и этил 3-фенилпропиолат (3,30 мл, 20,0 ммоль). Смесь перемешивали при комнатной температуре в течение 18 часов и вылили в холодную воду (100 мл). Смесь перемешивали в течение 30 минут и отфильтровали через упакованный Celite®, элюируя EtOAc и Н2О. Органический слой удалили и промыли H2O (4Х), высушили над MgSO4, отфильтровали и концентрировали. Остаточное темное красно-оранжевое маслянистое вещество очистили хроматографией на диоксиде кремния, используя градиентное элюирование 10-50% EtOAc в гексанах, для получения указанного в заголовке соединения в виде желтого твердого вещества (1,75 г, выход 65,7%). МС (apci) m/z=267,0 (М+Н).
Стадия В: 2-фенилпиразоло[1,5-а]пиридин-3-карбоновой кислоты гидрохлорид: К раствору этил 2-фенилпиразоло[1,5-а]пиридин-3-карбоксилата (1,72 г, 6,46 ммоль) в EtOH (10 мл) добавили 2 М NaOH (16,1 мл, 32,3 ммоль) и нагревали реакционную смесь с дефлегматором в течение 7 часов. Реакционную смесь охладили до комнатной температуры и охладили на ледяной бане. Смесь подкислили концентрированной HCl, а полученную суспензию собрали вакуумной фильтрацией. Осадок на фильтре промыли водой и высушили в вакууме для получения указанного в заголовке соединения в виде золотистого твердого вещества (1,69 г, выход 95,2%). МС (apci) m/z=195,2 ((М+Н)-СО2).
Стадия С: 2-фенилпиразоло[1,5-а]пиридин: Смесь 2-фенилпиразоло[1,5-а]пиридин-3-карбоновой кислоты гидрохлорида (1,00 г, 3,64 ммоль) в полифосфорной кислоте (41,6 мл, 364 ммоль) нагревали при 80°С в течение 17 часов. Полученную студенистую смесь охладили до комнатной температуры и добавили ледяную воду (150мл). Смесь перемешивали до начала образования зернистой суспензии. Суспензию перенесли в воду (350 мл) и перемешивали смесь до образования тонко дисперсной зернистой суспензии. Твердое вещество собрали вакуумной фильтрацией, промыли водой и высушили в вакууме для получения указанного в заголовке соединения в виде оранжевого твердого вещества (0,84 г, 118%), которое использовали напрямую на следующей стадии. МС (apci) m/z=195,2 (М+Н).
Стадия D: 3 -нитрозо-2-фенилпиразоло [1,5-а]пиридин: К раствору 2-фенилпиразоло[1,5-а]пиридина (0,84 г, 2,85 ммоль) в уксусной кислоте (2,85 мл, 51,4 ммоль) по каплям добавили NaNO2 (0,295 г, 4,28 ммоль) в воде (1,43 мл) за 10 минут (наблюдали бурное выделение пузырьков газа). Смесь перемешивали в течение 20 минут и погасили льдом. Полученную суспензию нагрели до комнатной температуры, а твердое вещество собрали вакуумной фильтрацией. Собранное твердое вещество промыли водой и высушили в вакууме для получения указанного в заголовке соединения в виде темно-зеленого твердого вещества (0,380 г, 59,6%). МС (apci) m/z=224,1 (М+Н).
Стадия Е: 2-фенилпиразоло[1,5-а]пиридин-3-амин: К смеси 3-нитрозо-2-фенилпиразоло[1,5-а]пиридина (0,380 г, 1,70 ммоль) и Zn пыли (0,557 г, 8,51 ммоль) в EtOH (3,81 мл) добавили концентрированную HCl (0,156 мл, 1,87 ммоль). Смесь нагревали с дефлегматором в течение 3 часов и охладили до комнатной температуры. Смесь разбавили MeOH, отфильтровали, a Zn осадок на фильтре промыли MeOH. Объединенный фильтрат и промывочные растворы концентрировали, а остаток разделили в воде и ДХМ. Органический слой удалили, а водную часть обработали насыщенным раствором NaHCO3 для достижения рН=10. Смесь экстрагировали ДХМ (3Х), а объединенные экстракты промыли насыщенным раствором NaCl, высушили над MgSO4, отфильтровали и концентрировали. Остаток высушили в вакууме для получения указанного в заголовке соединения в виде коричневого твердого вещества (0,304 г, выход 85,5%). МС (apci) m/z=210,1 (М+Н). 1Н ЯМР (CDCl3) δ 8,31 (d, 1H), 7,91 (d, 2H), 7,49 (t, 2H), 7,38 (t, 2H), 6,96 (dd, 1H), 6,64 (t, 1H), 3,21 (br s, 2H).
Промежуточное соединение 245
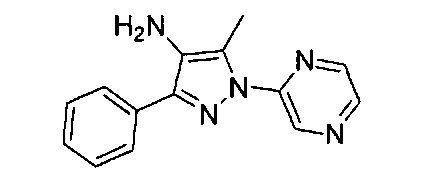
5-метил-3-фенил-1-(пиразин-2-ил)-1 Н-пиразол-4-амин
Стадия А: 2-(5-метил-4-нитрозо-3-фенил-1Н-пиразол-1-ил)пиразин. К раствору 2-гидразинилпиразина (0,485 г, 4,40 ммоль) в НОАс (6 мл) небольшими частями за 2 минуты добавили (2-(гидроксиимино)-1-фенилбутан-1,3-дион (0,765 г, 4,00 ммоль). Смесь перемешивали в течение 5 минут, а полученную светло-оранжевую суспензию перемешивали при 60°C в течение 6 часов. Добавили EtOH (1 мл) и нагревали смесь при 60°C еще 6 часов. Полученную темно-зеленую суспензию охладили до комнатной температуры и разбавили смесь H2O (30 мл). Зеленую суспензию перемешивали в течение 1 часа, а твердое вещество собрали вакуумной фильтрацией. Собранное твердое вещество промыли H2O и высушили в вакууме. Твердое вещество суспендировали в EtOH (25 мл) и добавили концентрированную HCl (500 мкл). Смесь нагревали с дефлегматором в течение 20 часов, охладили до комнатной температуры и разбавили холодной H2O (75 мл). Смесь обработали 1 М NaOH до рН=7 и экстрагировали Et2O (3Х). Объединенные экстракты промыли насыщенным раствором NaCl и высушили над MgSO4. Высушенный раствор отфильтровали через упакованный Celite® и концентрировали. Остаточное зеленовато-желтое твердое вещество очистил на SiO2 колонке, используя поэтапное градиентное элюирование (25% CH2Cl2, 50% EtOAc/гексаны), для получения указанного в заголовке соединения в виде бирюзового твердого вещества (325 мг, 31%). МС (apci) m/z=266,1 (М+Н).
Стадия B: 5-метил-3-фенил-1-(пиразин-2-ил)-1Н-пиразол-4-амин. К смеси 2-(5-метил-4-нитрозо-3-фенил-1Н-пиразол-1-ил)пиразина (325 мг, 1,04 ммоль) и Zn пыли (340 мг, 5,21 ммоль) в EtOH (10 мл) добавили концентрированную HCl (95,5 мкл, 1,15 ммоль). Смесь перемешивали при комнатной температуре в течение 17 часов, затем при 65°C в течение 3 часов. Смесь охладили до комнатной температуры и отфильтровали через упакованный Celite®, элюируя MeOH. Элюент концентрировали, а остаток обработали H2O и перемешали. Полученную оранжевую суспензию обработали 2 М HCl до рН=1, и экстрагировали смесь Et2O (3Х). Водную часть обработали 2 М NaOH до рН=8 и экстрагировали EtOAc (3Х). Объединенные EtOAc экстракты промыли насыщенным раствором NaCl и высушили над MgSO4/активированным углем. Раствор элюировали через слой SiO2, элюируя EtOAc. Элюент концентрировали для получения указанного в заголовке соединения в виде светло-желтого воскообразного вещества (33 мг, 13%). МС (esi) m/z=252,2 (М+Н).
Промежуточное соединение 246
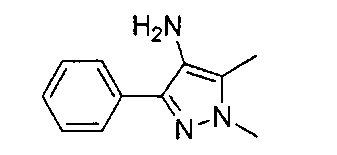
1,5-диметил-3-фенил-1Н-пиразол-4-амин
Стадия A: 1,5-диметил-4-нитрозо-3-фенил-1Н-пиразол: К раствору метилгидразина (0,484 г, 10,5 ммоль) в НОАс (10 мл) небольшими частями за 5 минут добавили 2-(гидроксиимино)-1-фенилбутан-1,3-дион (2,01 г, 10,5 ммоль). Реакционную смесь нагревали при 60°C в течение 1 часа и охладили до комнатной температуры. К смеси добавили Et2O (50 мл) и H2O (10 мл), затем медленно добавили насыщенный раствор Na2CO3 для получения рН=8. Органический слой удалили, а водный слой экстрагировали Et2O (2Х). Объединенные органические фракции высушили над Na2SO4, отфильтровали и концентрировали. Остаток очистили силикагелевой хроматографией (1:5 EtOAc/гексаны) для получения указанного в заголовке соединения в виде зеленого твердого вещества (1,32 г, 63%). МС (apci) m/z=202,1 (М+Н).
Стадия B: 1,5-диметил-3-фенил-1 Н-пиразол-4-амин: К раствору 1,5-диметил-4-нитрозо-3-фенил-1Н-пиразола (1,32 г, 6,60 ммоль) в MeOH (50 мл) добавили Pd(OH)2 на углероде (200 мг, 20 вес.%, 0,286 ммоль) и встряхивали реакционную смесь под давлением 50 psi Н2 в течение 3 часов при комнатной температуре. Реакционную смесь вакуумировали, продули N2, отфильтровали через слой Celite® с элюированием MeOH. Элюент концентрировали, а остаток высушили в вакууме для получения указанного в заголовке соединения в виде желто-коричневого твердого вещества (1,23 г, 100%). МС (apci) m/z - 188,1 (М+Н).
Промежуточное соединение 247
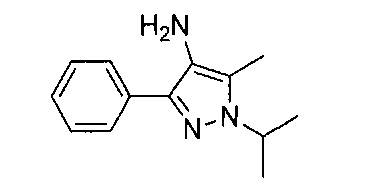
1-изопропил-5-метил-3-фенил-1Н-пиразол-4-амин
Указанное в заголовке соединение получили по способу, описанному для Промежуточного соединения 246, используя изопропилгидразина гидрохлорид вместо метилгидразина на Стадии A, для получения 620 мг (57%) указанного в заголовке соединения за 2 стадии. МС (apci) m/z=216,1 (М+Н).
Промежуточное соединение 248
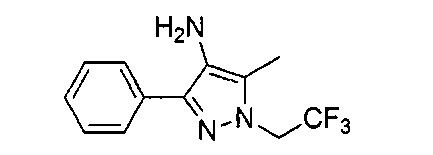
5-метил-3-фенил-1-(2,2,2-трифторэтил)-1Н-пиразол-4-амин
Стадия A: 5-метил-4-нитрозо-3-фенил-1-(2,2,2-трифторэтил)-1Н-пиразол: Указанное в заголовке соединение получили, используя (2,2,2-трифторэтил)гидразин вместо метилгидразина на Стадии А способа, описанного для получения 1,5-диметил-3-фенил-1Н-пиразол-4-амина (Промежуточное соединение 246). Соединение выделили в виде зеленого твердого вещества (999 мг, 71%). 1Н ЯМР (CDCl3) δ 7,60-7,73 (m, 5Н), 4,70 (q, 2H), 2,27 (t, 3Н).
Стадия B: 5-метил-3-фенил-1-(2,2,2-трифторэтил)-1Н-пиразол-4-амин: К смеси 5-метил-4-нитрозо-3-фенил-1-(2,2,2-трифторэтил)-1Н-пиразола (50 мг, 0,186 ммоль) и Zn пыли (60,7 мг, 0,929 ммоль) в EtOH (0,4 мл) добавили концентрированную HCl (17,0 мкл, 0,204 ммоль) и нагревали смесь с дефлегматором в течение 3 часов. Смесь охладили до комнатной температуры и разбавили MeOH, и отфильтровали. Фильтрат концентрировали, а остаток разбавили в воде. Водную смесь обрабатывали насыщенным раствором NaHCO3 до достижения рН=10. Смесь экстрагировали ДХМ (3Х), а объединенные экстракты высушили над Na2SO4, отфильтровали и концентрировали для получения указанного в заголовке соединения в виде желтого маслянистого вещества (47,1 мг, выход 99,4%). МС (apci) m/z=256,1 (М+Н).
Промежуточное соединение 249
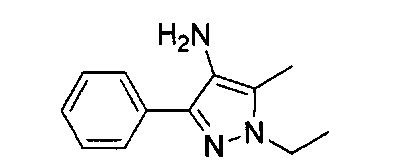
1-этил-5-метил-3-фенил-1Н-пиразол-4-амин
Стадия A: 1 -этил-5-метил-4-нитрозо-3-фенил-1 Н-пиразол: Указанное в заголовке соединение получили по способу, описанному для получения Промежуточного соединения 246, используя этилгидразина оксалат вместо метилгидразина на Стадии А. 1-Этил-5-метил-4-нитрозо-3-фенил-1Н-пиразол выделили в виде зеленого маслянистого вещества (288 мг, 26%). 1Н ЯМР (CDCl3) δ 8,19 (d, 2Н), 7,46-7,50 (m, 3Н), 4,15 (q, 2Н), 2,43 (s, 3Н), 1,50 (t, 3Н). Получили также неосновной региоизомер, 1-этил-3-метил-4-нитрозо-5-фенил-1Н-пиразол, в виде голубовато-зеленого твердого вещества (165 мг, 15%). 1Н ЯМР (CDCl3) δ 7,71 (dd, 2Н), 7,59 (m, 3Н), 4,17 (q, 2Н), 2,28 (s, 3Н), 1,51 (t, 3Н).
Стадия B: 1-этил-5-метил-3-фенил-1Н-пиразол-4-амин: Получили по способу, описанному для получения Промежуточного соединения 248, используя 1-этил-5-метил-4-нитрозо-3-фенил-1Н-пиразол на Стадии В. Указанное в заголовке соединение выделили в виде светло-пурпурного твердого вещества (281 мг, 104%). МС (apci) m/z=202,1 (М+Н).
Промежуточное соединение 250
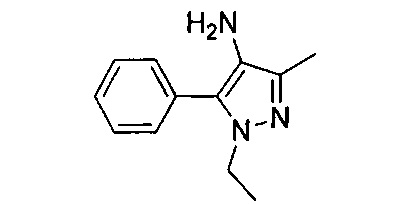
1-этил-3-метил-5-фенил-1Н-пиразол-4-амин
Получили по способу, описанному для получения Промежуточного соединения 249, используя 1-этил-3-метил-4-нитрозо-5-фенил-1Н-пиразол на Стадии A. Указанное в заголовке соединение получили по Стадии B. Это соединение выделили в виде бесцветного маслянистого вещества (82,4 мг, 52,5%) после очистки обращенно-фазовой хроматографией. МС (apci) m/z=202,1 (М+Н).
Промежуточное соединение 251
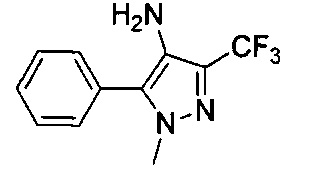
1-метил-5-фенил-3-(трифторметил)-1Н-пиразол-4-амин
Стадия А: 4,4,4-трифтор-2-(гидроксиимино)-1-фенилбутан-1,3-дион: Раствор 4,4,4-трифтор-1-фенилбутан-1,3-диона (5,00 г, 23,1 ммоль) в НОАс (46,3 мл) охладили до 10°C и добавили нитрит натрия (1,84 г, 26,6 ммоль) в воде (6,0 мл). Смесь перемешивали при комнатной температуре в течение 90 минут и разбавили H2O (150 мл). Смесь экстрагировали Et2O 3), а объединенные органические фракции осторожно промыли насыщенным раствором NaHCO3 до рН=9. Et2O раствор промыли H2O и насыщенным раствором NaCl и высушили над MgSO4. Высушенный раствор отфильтровали и концентрировали для получения указанного в заголовке соединения в виде желтого пенистого вещества (4,21 г, выход 74,2%). МС (apci) m/z=244,1 (М-Н).
Стадия B: 4-нитрозо-3-фенил-5-(трифторметил)-1Н-пиразол: Раствор гидразина моногидрата (0,204 г, 4,08 ммоль) в EtOH (5 мл) охладили до 0°C и добавили 4,4,4-трифтор-2-(гидроксиимино)-1-фенилбутан-1,3-дион (1,00 г, 4,08 ммоль) в EtOH (15 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, добавили избыток порошкообразного MgSO4 и нагревали смесь при 60°C в течение 16 часов. Смесь охладили до комнатной температуры, отфильтровали и концентрировали для получения неочищенного указанного в заголовке соединения в виде зеленого твердого вещества (78,7 мг, 8,0%), которое использовали напрямую на следующей стадии. МС (apci) m/z=240,0 (М-Н).
Стадия C: 1-метил-5-фенил-3-(трифторметил)-1Н-пиразол-4-амин: К раствору 4-нитрозо-3-фенил-5-(трифторметил)-1Н-пиразола (78,7 мг, 0,326 ммоль) в ДМФ (1,6 мл) добавили NaH (14,4 мг, 0,359 ммоль) и перемешивали смесь при комнатной температуре в течение 30 минут. Смесь обработали метилйодидом (40,6 мкл, 0,653 ммоль) и перемешивали в течение 17 часов. Реакционную смесь напрямую очистили обращенно-фазовой ВЭЖХ, используя градиентное элюирование 20-100% ацетонитрила в воде, для получения светло-голубого твердого вещества (40,2 мг). Твердое вещество растворили в EtOH (0,35 мл) и подвергли восстановлению, описанному на Стадии B получения 5-метил-3-фенил-1-(2,2,2-трифторэтил)-1Н-пиразол-4-амина (Промежуточное соединение 248). Указанное в заголовке соединение получили в виде белого твердого вещества (25,1 мг, 66,1%).
Промежуточное соединение 252
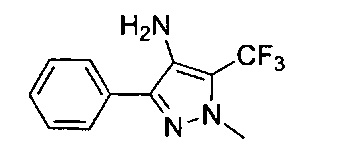
1-метил-3-фенил-5-(трифторметил)-1Н-пиразол-4-амин
Стадия А: 1-метил-4-нитрозо-3-фенил-5-(трифторметил)-1Н-пиразол. К раствору метилгидразина (0,214 мл, 4,08 ммоль) в EtOH (20 мл) добавили 4,4,4-трифтор-2-(гидроксиимино)-1-фенилбутан-1,3-дион (Промежуточное соединение 251, Стадия A; 1,00 г, 4,079 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и добавили избыток MgSO4. Смесь перемешивали при 60°C в течение 48 часов и охладили при комнатной температуре. Смесь отфильтровали, а фильтрат концентрировали до зеленого остатка. Остаток очистили силикагелевой хроматографией, используя для элюирования градиент 10-30% EtOAc в гексанах, для получения указанного в заголовке соединения в виде зеленого твердого вещества (482 мг, 46%). 1Н ЯМР (CDCl3) δ 7,89 (d, 2Н), 7,45-7,52 (m, 3Н), 4,15 (s, 3Н).
Стадия B: 1-метил-3-фенил-5-(трифторметил)-1Н-пиразол-4-амин. Получили из 1-метил-4-нитрозо-3-фенил-5-(трифторметил)-1Н-пиразола по способу, описанному для получения Промежуточного соединения 248, Стадия В. Указанное в заголовке соединение получили в виде белого твердого вещества (309 мг, 68%). 1Н ЯМР (CDCl3) δ 7,65 (d, 2Н), 7,45 (t, 2Н), 7,35 (t, 1Н), 3,93 (s, 3Н), 3,52 (br s, 2Н).
Получение U-1
1-(3-(2-(трет-бутилдиметилсилилокси)этокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу Примера 1, заменяя транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-амина дигидрохлорид (Получение В) на (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (Получение F) и используя Промежуточное соединение Р203. МС (apci) m/z - 630,1 (М+Н).
Получение U-2
1-(3-((S)-2-(трет-бутилдиметилсилилокси)пропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу Примера 1, заменяя транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-амина дигидрохлорид (Получение В) на (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (Получение F) и используя Промежуточное соединение Р211. MC (apci) m/z=644,4 (М+Н).
Синтетические Примеры
Пример 1
1-(3-трет-бутил-1-фенил-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина
Стадия A: Получение фенил 3-трет-бутил-1-фенил-1Н-пиразол-5-илкарбамата: К раствору 3-трет-бутил-1-фенил-1Н-пиразол-5-амина (Таблица 1; 200,0 мг, 0,9290 ммоль) в EtOAc (10 мл) добавили 2 М водный раствор NaOH (0,9290 мл, 1,858 ммоль), затем фенилхлорформиат (0,1632 мл, 1,301 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, а затем разделили фазы. Органическую фазу промыли H2O (10 мл), насыщенным солевым раствором (10 мл), высушили при помощи MgSO4, отфильтровали и концентрировали для получения продукта в виде коричневого кристаллического вещества (320 мг, выход 103%). МС (apci) m/z=336,1 (М+Н).
Стадия В: Получение 1-(3-трет-бутил-1-фенил-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевины: К раствору транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-амина дигидрохлорида (Получение В, 30,0 мг, 0,102 ммоль) и ДИЭА (0,0535 мл, 0,307 ммоль) в ДМА (1 мл) добавили фенил 3-трет-бутил-1-фенил-1Н-пиразол-5-илкарбамат (34,3 мг, 0,102 ммоль), и перемешивали реакционную смесь при комнатной температуре в течение 1 часа. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-60% ацетонитрила в воде, для получения продукта в виде желто-коричневого твердого вещества (38 мг, выход 81%). МС (apci) m/z=462,2 (М+Н).
Соединения в Таблице 4 были получены по способу Примера 1, с использованием соответствующих исходных материалов.
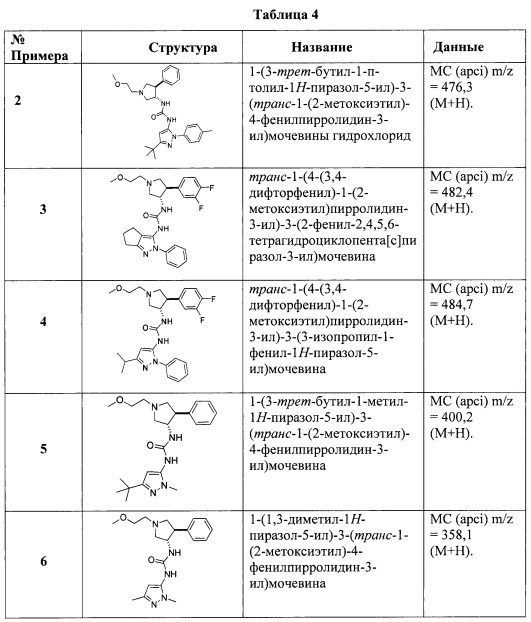
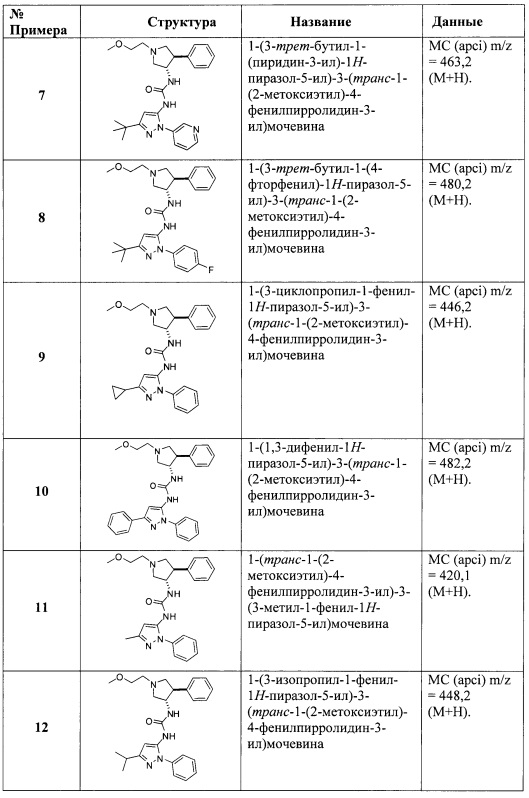
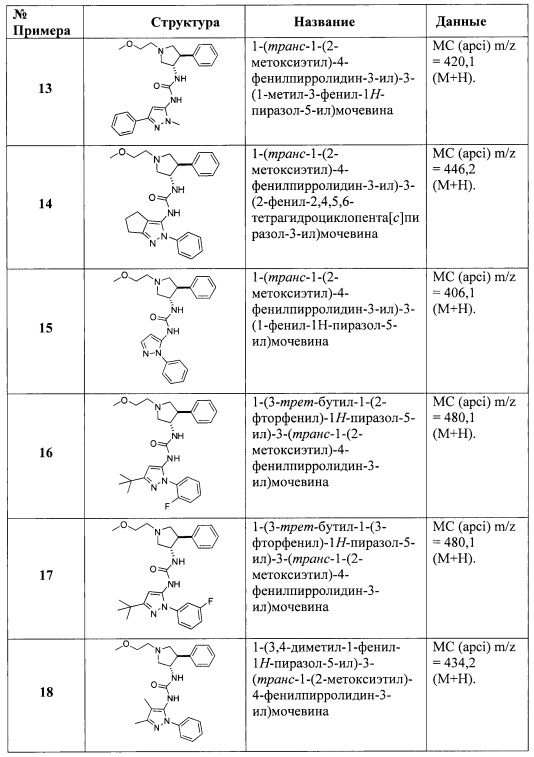
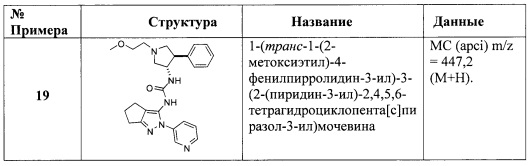
Пример 20
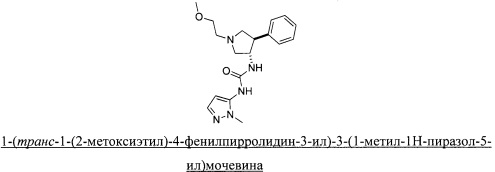
Стадия А: Получение 4-нитрофенил транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-илкарбамата: К суспензии транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-амина дигидрохлорида (Получение В; 0,180 г, 0,614 ммоль) в ДХМ (3 мл) при 0°С добавили триэтиламин (0,428 мл, 3,07 ммоль), затем по каплям добавили раствор 4-нитрофенилкарбонохлоридата (0,136 г, 0,675 ммоль) в ДХМ (0,5 мл). Ледяную баню убрали, а реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Смесь разбавили насыщенным раствором NaHCO3 (30 мл) и экстрагировали ДХМ (3×20 мл). Объединенные органические слои промыли насыщенным солевым раствором (20 мл), высушили при помощи MgSO4, отфильтровали и концентрировали in vacuo для получения неочищенного промежуточного соединения, которое использовали на следующей стадии без очистки. МС (apci) m/z=386,2 (М+Н).
Стадия В: Получение 1-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(1-метил-1Н-пиразол-5-ил)мочевины: К раствору 4-нитрофенил транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-илкарбамата (80 мг, 0,21 ммоль) в ДХЭ (1 мл) добавили 1-метил-1H-пиразол-5-амин (Таблица 1; 24 мг, 0,25 ммоль), затем ДИЭА (0,15 мл, 0,83 ммоль). Смесь нагревали при 55°С в течение 18 часов, затем разбавили 1 мл ДХМ, промыли насыщенным раствором бикарбоната (2×1 мл) и концентрировали in vacuo. Неочищенный остаток очистили обращенно-фазовой колоночной хроматографией, элюируя 5-45% ацетонитрила в воде, для получения указанного в заголовке продукта (10 мг, выход 14%). МС (apci) m/z=344,2 (М+Н).
Пример 21
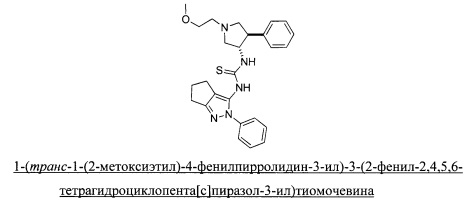
К CHCl3 (0,5 мл) суспензии 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-амина (Таблица 1; 45,2 мг, 0,227 ммоль) добавили ди(1Н-имидазол-1-ил)метантион (40,4 мг, 0,227 ммоль), затем ДИЭА (0,158 мл, 0,908 ммоль). После перемешивания при комнатной температуре в течение 16 часов, к реакционной смеси одной порцией добавили транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-амин (Получение В, 50,0 мг, 0,227 ммоль). Через 2 часа реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-68% ацетонитрила в воде, для получения указанного в заголовке продукта в виде белого твердого вещества (70 мг, выход 67%). МС (apci) m/z=462,1 (М+Н).
Пример 22
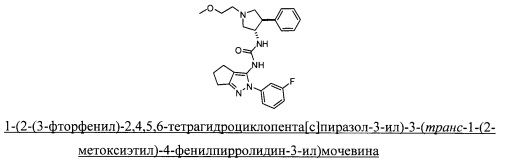
Раствор 2-(3-фторфенил)-2,4,5,6-тетрагидроциклопента[с]пиразол-3-амина (Промежуточное соединение Р3; 25,0 мг, 0,115 ммоль) в ДХМ (575 мкл, 0,115 ммоль) обработали 1,1'-карбонилдиимидазолом (18,7 мг, 0,115 ммоль) и ТЭА (32,1 мкл, 0,230 ммоль). После перемешивания при комнатной температуре в течение 3 часов, добавили транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-амина дигидрохлорид (Получение В, 33,7 мг, 0,115 ммоль), затем ТЭА (64,2 мкл, 0,460 ммоль). После перемешивания при комнатной температуре в течение 30 минут, неочищенную реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 0-100% ацетонитрила в воде, для получения указанного в заголовке соединения в виде прозрачного маслянистого вещества (17,6 мг, выход 33,0%). МС (apci) m/z=464,1 (М+Н).
Соединения в Таблице 5 были получены по способу Примера 22, с использованием соответствующих исходных материалов. Время превращения для активированного промежуточного соединения с 1,1'-карбонилдиимидазолом варьировалось, и его контролировали отбором аликвоты и погашением в МеОН. Для контролирования полного превращения в метилкарбамат использовали анализ ЖХМС (от 30 минут до 16 часов).
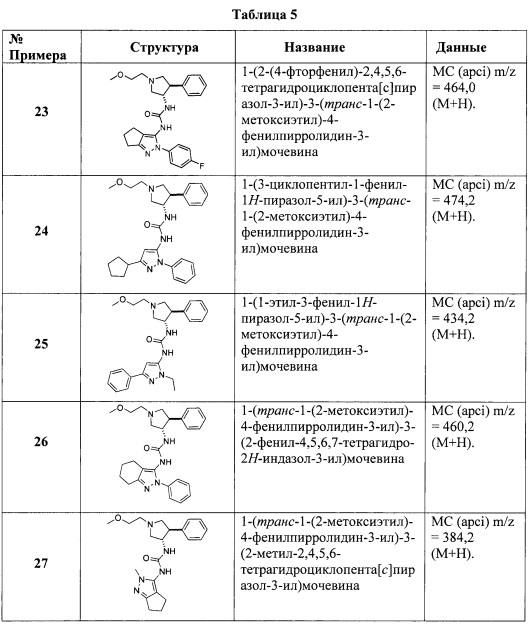
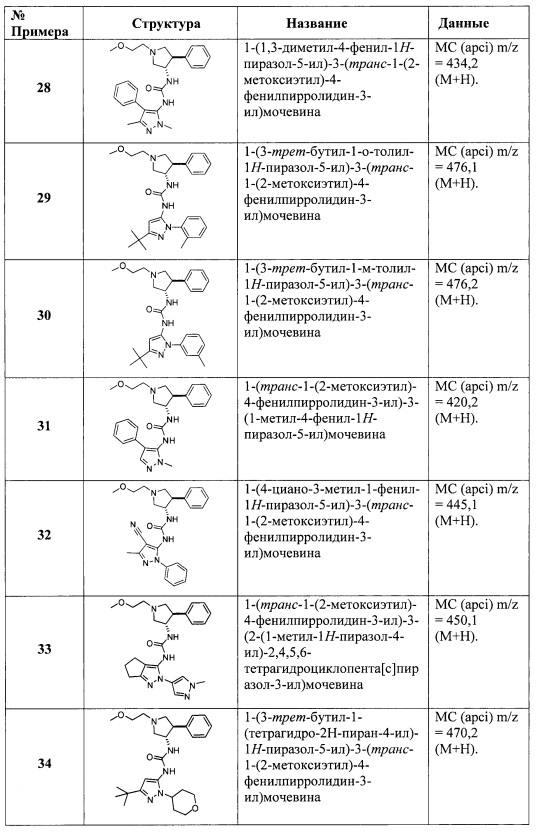
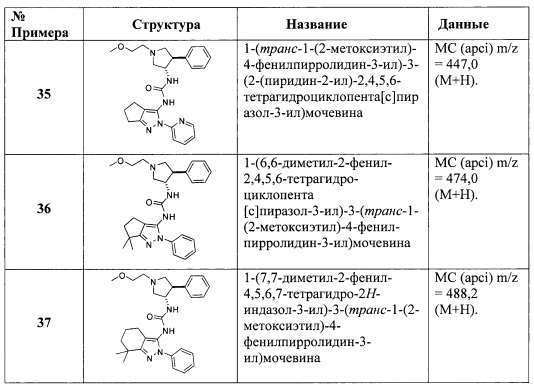
Пример 38
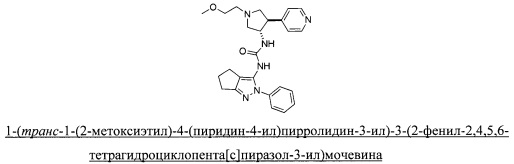
Стадия А: Получение транс-этил 1-(2-метоксиэтил)-4-(пиридин-4-ил)пирролидин-3-карбоксилата: К раствору (Е)-этил 3-(пиридин-4-ил)акрилата (200 мг, 1,13 ммоль) в ДХМ (10 мл), охлажденному до 0°С, последовательно одной частью добавили ТФК (0,017 мл, 0,226 ммоль), затем по каплям добавили 2-метокси-N-(метоксиметил)-N-((триметилсилил)метил)этанамин (Получение С, 278 мг, 1,35 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Смесь погасили насыщенным раствором NaHCO3 (10 мл), фазы разделили, а водный слой экстрагировали ДХМ (15 мл). Объединенные органические фазы высушили при помощи MgSO4, отфильтровали и концентрировали in vacuo. Неочищенный продукт очистили колоночной хроматографией на диоксиде кремния, элюируя от 0 до 5% МеОН в ДХМ, для получения продукта в виде бесцветного маслянистого вещества (276 мг, выход 88%). МС (apci) m/z=279,2 (М+Н).
Стадия В: Получение транс-1-(2-метоксиэтил)-4-(пиридин-4-ил)пирролидин-3-карбоксилата лития: К раствору транс-этил 1-(2-метоксиэтил)-4-(пиридин-4-ил)пирролидин-3-карбоксилата (276 мг, 0,992 ммоль) в ТГФ (6 мл) и MeOH (3 мл) добавили 2 М водный раствор LiOH (0,496 мл, 0,992 ммоль).Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, а полученный белый осадок собрали вакуумной фильтрацией, промыли Et2O и высушили на воздухе. Фильтрат концентрировали до желтого твердого вещества, растерли с MeOH (1 мл), отфильтровали, а белое твердое вещество промыли Et2O и высушили на воздухе. Две порции твердого вещества объединили для получения продукта в виде белого твердого вещества (197 мг, выход 88%). 1H ЯМР (d6-ДМСО) δ 8,37-8,39 (m, 2Н), 7,26-7,28 (m, 2Н), 3,36-3,43 (m, 3Н), 3,24 (s, 3Н), 2,75-2,86 (m, 1H), 2,41-2,68 (m, 6Н).
Стадия С: Получение бензил транс-1 -(2-метоксиэтил)-4-(пиридин-4-ил)пирролидин-3-илкарбамата: К суспензии транс-1-(2-метоксиэтил)-4-пиридин-4-ил)пирролидин-3-карбоксилата лития (25 мг, 0,098 ммоль) в толуоле (1,8 мл) и ДМФ (0,2 мл) последовательно добавили ДИЭА (0,034 мл, 0,20 ммоль) и дифенилфосфорилазид (0,029 мл, 0,14 ммоль).Реакционную смесь перемешивали при комнатной температуре в течение 20 минут, затем нагревали с дефлегматором в течение 10 минут. Ввели бензиловый спирт (100 мг, 0,98 ммоль) и нагревали реакционную смесь с дефлегматором в течение 17 часов. Реакционную смесь напрямую очистили колоночной хроматографией на диоксиде кремния, элюируя 0-10% MeOH в ДХМ, для получения продукта в виде желтого маслянистого вещества (11 мг, выход 32%). МС (apci) m/z=356,1 (М+Н).
Стадия D: Получение транс-1-(2-метоксиэтил)-4-(пиридин-4-ил)пирролидин-3-амина бис(2,2,2-трифторацетата): Раствор бензил транс-1-(2-метоксиэтил)-4-(пиридин-4-ил)пирролидин-3-илкарбамата (11 мг, 0,031 ммоль) в ТФК (1 мл) нагревали в закрытой пробирке при 60°C в течение 18 часов. Реакционную смесь перенесли в EtOH (5 мл) и концентрировали для получения неочищенного продукта в виде коричневого маслянистого вещества, которое использовали напрямую на Стадии F, предположив количественный выход. МС (apci) m/z=222,1 (М+Н).
Стадия Е: Получение фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамата: Суспензию 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3 -амина (Таблица 1; 100 мг, 0,50 ммоль) в EtOAc (2,0 мл) и 2М водном NaOH (0,50 мл, 1,0 ммоль) по каплям обработали фенилкарбонохлоридатом (0,088 мл, 0,70 ммоль) и перемешивали в течение 16 часов при комнатной температуре. Затем реакционную смесь разделили на фазы, а органическую фазу промыли водой (5 мл) и насыщенным солевым раствором (5 мл), высушили над Na2SO4, отфильтровали и концентрировали in vacuo для получения продукта (160 мг, выход 100%). МС (apci) m/z=320,1 (М+Н).
Стадия F: Получение 1-(транс-1-(2-метоксиэтил)-4-(пиридин-4-ил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-етрагидроциклопента[с]пиразол-3-ил)мочевины: Получили по способу, описанному в Примере 1, заменяя фенил 3-трет-бутил-фенил-1Н-пиразол-5-илкарбамат на Стадии В на фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат, и заменяя отранс-1-(2-метоксиэтил)-4-фенилпирролидин-3-амина дигидрохлорид на Стадии В на транс-1-(2-метоксиэтил)-4-(пиридин-4-ил)пирролидин-3-амина бис(2,2,2-трифторацетат), для получения конечного продукта в виде белого твердого вещества (6,0 мг, выход 42%). МС (apci) m/z=447,2 (М+Н).
Пример 39
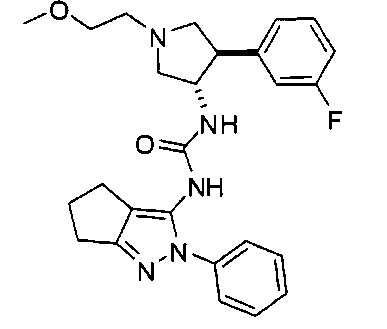
транс-1-(4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Стадия А: Получение транс-трет-бутил 3-(3-фторфенил)-4-(3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)уреидо)пирролидин-1-карбоксилата: К суспензии транс-1-(трет-бутоксикарбонил)-4-(3-фторфенил)пирролидин-3-карбоновой кислоты (приобрели у компании ChemImpex; 100 мг, 0,32 ммоль) в безводном толуоле (2 мл) добавили триэтиламин (180 мкл, 1,29 ммоль), затем дифенилфосфорилазид (98 мкл, 0,45 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 1 часа, а затем нагревали с дефлегматором в течение 1 часа. После охлаждения реакционную смесь обработали 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-амином (Таблица 1; 193 мг, 0,97 ммоль) и перемешивали при нагревании с дефлегматором в течение 15 часов. Охлажденную смесь разделили между насыщенным раствором NaHCO3 (20 мл) и EtOAc (20 мл), а водный слой экстрагировали EtOAc (2×10 мл). Объединенные органические фазы промыли насыщенным солевым раствором (10 мл), высушили над Na2SO4 и концентрировали in vacuo. Неочищенный материал очистили колоночной хроматографией на диоксиде кремния, элюируя 1% MeOH в ДХМ, затем 5% MeOH в ДХМ, для получения продукта в виде коричневого смолянистого вещества. Анализ ЖХМС смолянистого вещества показал, что оно представляет собой приблизительно 2:1 смесь заданного продукта и исходного материала, симметричной мочевины 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-амина (по ЖХМС). Смесь использовали непосредственно на следующей стадии. МС (apci) m/z=506,2 (М+Н).
Стадия В: Получение 1-(транс-4-(3-фторфенил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины: К смеси, выделенной на Стадии А (90 мг, 0,18 ммоль), в CH2Cl2 (8 мл) добавили ТФК (2 мл). Раствор перемешивали при комнатной температуре в течение 2 часов, а затем концентрировали, а остаток разделили между 1 н. NaOH (20 мл) и CH2Cl2 (20 мл). Водный слой экстрагировали CH2Cl2 (2×10 мл), а объединенные органические фазы промыли насыщенным солевым раствором (10 мл), высушили над Na2SO4 и концентрировали для получения коричневого смолянистого вещества. В результате очистки колоночной хроматографией на диоксиде кремния, элюируя от 2% MeOH в ДХМ до 10% MeOH/CH2Cl2/0,1% 7 н. NH3/MeOH, получили продукт в виде бледно-желтого твердого вещества (31 мг, выход 43%). МС (apci) m/z=406,1 (М+Н).
Стадия С: Получение транс-1-(4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины: К раствору 1-(транс-4-(3-фторфенил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]-пиразол-3-ил)мочевины (31 мг, 0,076 ммоль) в безводном ДМФ (1 мл) добавили 1-бром-2-метоксиэтан (9,1 мкл, 0,09 ммоль), затем ДИЭА (40 мкл, 0,23 ммоль). Смесь нагрели до 60°C и перемешивали в течение 15 часов. Охлажденную смесь разделили между насыщенным раствором NaHCO3 (20 мл) и EtOAc(10 мл), а водный слой экстрагировали EtOAc (2×10 мл). Объединенные органические фазы промыли водой (5×10 мл) и насыщенным солевым раствором (10 мл), затем высушили над Na2SO4 и концентрировали. Неочищенный материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2,5% MeOH/CH2Cl2, для получения продукта в виде бесцветного стекловидного вещества (14 мг, выход 40%). МС (apci) m/z=464,1 (М+Н).
Пример 40
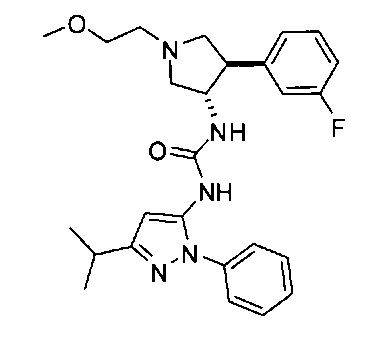
транс-1-(4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-изопропил-1-фенил-1Н-пиразол-5-ил)мочевина
Получили по способу, описанному для Примера 39, заменяя 3-изопропил-1-фенил-1Н-пиразол-5-амином (Таблица 1) 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-амин на Стадии А. МС (apci) m/z=466,2 (М+Н).
Пример 41
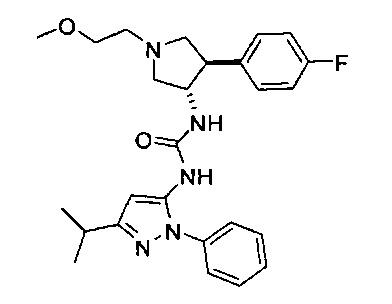
транс-1-(4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-изопропил-1-фенил-1Н-пиразол-5-ил)мочевина
Получили по способам, описанным для Примера 39, заменяя транс-1-(трет-бутоксикарбонил)-4-(4-фторфенил)пирролидин-3-карбоновой кислотой транс-1-(трет-бутоксикарбонил)-4-(3-фторфенил)пирролидин-3-карбоновую кислоту на Стадии А. МС (apci) m/z=466,2 (М+Н).
Пример 42
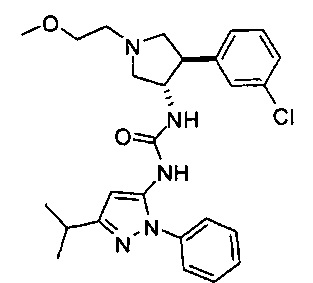
транс-1-(4-(3-хлорфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-изопропил-1-фенил-1Н-пиразол-5-ил)мочевина
Получили по способам, описанным для Примера 40, заменяя транс-1-(трет-бутоксикарбонил)-4-(3-хлорфенил)пирролидин-3-карбоновой кислотой транс-1-(трет-бутоксикарбонил)-4-(3-фторфенил)пирролидин-3-карбоновую кислоту на Стадии А. МС (apci) m/z=482,1 (М+Н).
Пример 43
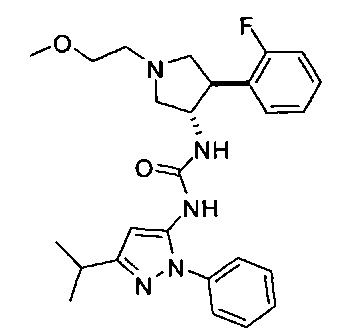
транс-1-(4-(2-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3 -изопропил-1-фенил-1Н-пиразол-5-ил)мочевина
Получили по способам, описанным для Примера 40, заменяя транс-1-(трет-бутоксикарбонил)-4-(2-фторфенил)пирролидин-3-карбоновой кислотой транс-1-(трет-бутоксикарбонил)-4-(3-фторфенил)пирролидин-3-карбоновую кислоту на Стадии А. МС (apci) m/z=466,2 (М+Н).
Пример 44
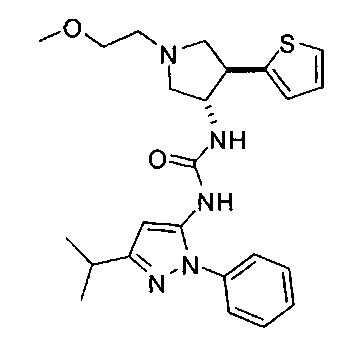
транс-1-(3-изопропил-1-фенил-1Н-пиразол-5-ил)-3-(1-(2-метоксиэтил)-4-(тиофен-2-ил)пирролидин-3-ил)мочевина
Получили по способам, описанным для Примера 40, заменяя транс-1-(трет-бутоксикарбонил)-4-(тиофен-2-ил)пирролидин-3-карбоновой кислотой транс-1-(трет-бутоксикарбонил)-4-(3-фторфенил)пирролидин-3-карбоновую кислоту на Стадии А. МС (apci) m/z=454,1 (М+Н).
Пример 45
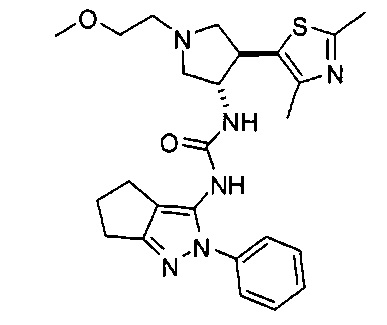
1-((3,4-транс)-4-(2,4-диметилтиазол-5-ил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Стадия А: Получение (Е)-метил 3-(2,4-диметилтиазол-5-ил)акрилата: Раствор метил (трифенил-фосфоранилиден)ацетата (2,61 г, 7,80 ммоль) в CH2Cl2 (10 мл) охладили до 0°С, а затем по каплям добавили раствор 2,4-диметилтиазол-5-карбоксальдегида (приобретенного у компании Maybridge, 1,00 г, 7,08 ммоль) в сухом CH2Cl2 (10 мл) за период 15 минут. Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 24 часов. После удаления растворителя под пониженным давлением, полученный желтоватый твердый остаток растворили в CH2Cl2 и очистили силикагелевой флэш-хроматографией, элюируя 15% EtOAc в гексанах, для получения продукта в виде белого порошка (1,32 г, выход 94,5%). 1Н-ЯМР (400 МГц, ДМСО-d6) δ 7,73 (d, J=15,62 Гц, 1Н), 6,07 (d, J=15,62 Гц, 1Н), 3,71 (s, 3Н, ОСН3), 2,63 (s, 3Н, СН3), 2,42 (s, 3Н, СН3). МС (apci) m/z=198 (М+Н).
Стадия В: Получение (3,4-транс)-метил 4-(2,4-диметилтиазол-5-ил)-1-(2-метоксиэтил)пирролидин-3-карбоксилата: Раствор (Е)-метил 3-(2,4-диметилтиазол-5-ил)акрилата (200 мг, 1,01 ммоль) и ТФК (7,81 мкл, 0,101 ммоль) в толуоле (15 мл) охладили на ледяной бане, затем по каплям добавили раствор 2-метокси-N-(метоксиметил)-N-((триметилсилил)метил)этанамина (312 мг, 1,52 ммоль) в толуоле (15 мл). Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 3 дней. Реакционную смесь промыли насыщенным раствором NaHCO3 (25 мл) и водой (2×25 мл), высушили при помощи Na2SO4, отфильтровали и концентрировали in vacuo. Неочищенный материал очистили обращенно-фазовой колоночной хроматографией, элюируя 5-50% ацетонитрила в воде, для получения продукта в виде прозрачного маслянистого вещества (43 мг, выход 14%). МС (apci) m/z=299,1 (М+Н).
Стадия С: Получение (3,4-транс)-4-(2,4-диметилтиазол-5-ил)-1-(2-метоксиэтил)пирролидин-3-карбоновой кислоты: К раствору (3,4-транс)-метил 4-(2,4-диметилтиазол-5-ил)-1-(2-метоксиэтил)пирролидин-3-карбоксилата (41 мг, 0,14 ммоль) в смесевой системе растворителей 2:2:1 ТГФ/МеОН/вода (0,7 мл) добавили LiOH-H2O (17 мг, 0,41 ммоль) и перемешивали смесь при комнатной температуре в течение ночи. После удаления растворителя, твердый остаток растворили в воде (0,2 мл) и подкислили 1 н. раствором HCl до рН 4-5. Смесь напрямую очистили обращенно-фазовой хроматографией, элюируя от 5 до 33% ацетонитрила в воде, для получения продукта в виде прозрачного маслянистого вещества (30 мг, выход 77%). МС (apci) m/z=285,1 (М+Н).
Стадия D: Получение бензил (3,4-транс)-4-(2,4-диметилтиазол-5-ил)-1-(2-метоксиэтил)пирролидин-3-илкарбамата: К мутному раствору (3,4-транс)-4-(2,4-диметилтиазол-5-ил)-1-(2-метоксиэтил)пирролидин-3-карбоновой кислоты (25,6 мг, 0,09 ммоль) в толуоле (0,9 мл) добавили ТЭА (31 мкл, 0,22 ммоль), затем дифенилфосфоразидат (27 мкл, 0,13 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, а затем при 100°C в течение 1 часа. Добавили бензиловый спирт (19 мкл, 0,18 ммоль) и нагревали смесь при 100°C в течение 18 часов. После охлаждения смесь разбавили EtOAc (2 мл), промыли водой (2×1 мл), высушили при помощи MgSO4, отфильтровали и концентрировали. Неочищенный остаток очистили обращенно-фазовой колоночной хроматографией, элюируя от 5 до 60% ацетонитрила в воде, для получения продукта в виде желтоватого маслянистого вещества (18 мг, выход 51%). МС (apci) m/z=390,1 (М+Н).
Стадия Е: Получение (3,4-транс)-4-(2,4-диметилтиазол-5-ил)-1-(2-метоксиэтил)пирролидин-3-амина бис(2,2,2-трифторацетата): Раствор бензил (3,4-транс)-4-(2,4-диметилтиазол-5-ил)-1-(2-метоксиэтил)пирролидин-3-илкарбамата (18,0 мг, 0,0462 ммоль) в ТФК (178 мкл, 2,31 ммоль) нагревали при 60°C в течение 18 часов. Реакционную смесь разбавили толуолом/EtOH и концентрировали. Неочищенный материал очистили обращенно-фазовой колоночной хроматографией, элюируя 5-38% ацетонитрила в воде, для получения продукта в виде бесцветного стекловидного вещества (14 мг, выход 63%). МС (apci) m/z=256,1 (М+Н).
Стадия F: Получение 1-((3,4-транс)-4-(2,4-диметилтиазол-5-ил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины: К прозрачному раствору (3,4-транс)-4-(2,4-диметилтиазол-5-ил-1-(2-метоксиэтил)пирролидин-3-аминабис(2,2,2-трифторацетата) (14 мг, 0,029 ммоль) в ДМА (0,3 мл) добавили фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат (11 мг, 0,035 ммоль). Смесь охладили на ледяной бане и добавили ДИЭА (0,020 мл, 0,12 ммоль). Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 5 минут, затем напрямую очистили обращенно-фазовой хроматографией, элюируя от 5 до 50% ацетонитрила в воде, для получения указанного в заголовке продукта в виде белого твердого вещества (10 мг, выход 72%).МС (apci) m/z=481,2 (М+Н).
Пример 46
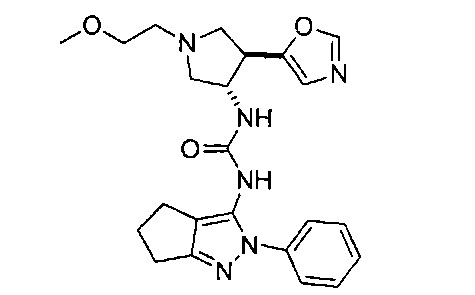
1-(транс-1-(2-метоксиэтил)-4-(оксазол-5-ил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Стадия А: Получение (Е)-этил 3-(оксазол-5-ил)акрилата: К суспензии NaH (60 вес.% в минеральном масле, 227 мг, 5,67 ммоль) в ТГФ (40 мл) под N2 при 0°C по каплям добавили этил 2-(диэтоксифосфорил)ацетат (1,12 мл, 5,67 ммоль). Смесь перемешивали при 0°C в течение 30 минут, а затем добавили раствор оксазол-5-карбальдегида (500 мг, 5,15 ммоль) в ТГФ (5 мл). Ледяную баню убрали, а реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь разбавили H2O (30 мл) и экстрагировали EtOAc (50 мл). Органическую фазу промыли насыщенным солевым раствором (50 мл), высушили при помощи MgSO4, отфильтровали и концентрировали in vacuo. Неочищенный продукт очистили колоночной хроматографией на диоксиде кремния, элюируя от 0 до 1% MeOH в ДХМ, для получения продукта в виде бледно-желтого маслянистого вещества (354 мг, выход 41,1%). 1Н ЯМР (CDCl3) δ 7,92 (s, 1Н), 7,48 (d, 1Н), 7,29 (s, 1Н), 6,40(d, 1H), 4,27 (q, 2Н), 1,29 (t, 3Н).
Стадия В: Получение транс-этил 1-(2-метоксиэтил)-4-(оксазол-5-ил)пирролидин-3-карбоксилата: К раствору (Е)-этил 3-(оксазол-5-ил)акрилата (354 мг, 2,12 ммоль) в ДХМ (20 мл) при 0°C добавили ТФК (0,033 мл, 0,42 ммоль), затем по каплям добавили 2-метокси-N-(метоксиметил)-N-((триметилсилил)метил)этанамин (Получение С, 522 мг, 2,54 ммоль). Ледяную баню убрали, а реакционную смесь перемешивали при комнатной температуре в течение 18 часов, затем реакционную смесь разбавили насыщенным водным раствором NaHCO3 (20 мл), разделили, а водную фазу экстрагировали ДХМ (20 мл). Объединенные органические фазы высушили при помощи MgSO4, отфильтровали и концентрировали in vacuo. Неочищенный продукт очистили колоночной хроматографией на диоксиде кремния, элюируя 0-3% MeOH в ДХМ, для получения продукта в виде бледно-желтого маслянистого вещества (321 мг, выход 56,5%). МС (apci) m/z=269,2 (М+Н).
Стадия С: Получение транс-1-(2-метоксиэтил)-4-(оксазол-5-ил)пирролидин-3-карбоксилата лития: К раствору транс-этил 1-(2-метоксиэтил)-4-(оксазол-5-ил)пирролидин-3-карбоксилата (321 мг, 1,20 ммоль) в ТГФ (6 мл) и MeOH (3 мл) добавили 2 М водный раствор LiOH (0,837 мл, 1,67 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов, а затем концентрировали до вязкого желтого вещества. Неочищенный продукт растворили в MeOH (10 мл) и концентрировали для получения продукта в виде желтого пенистого вещества (234 мг, выход 79,4%). МС (apci отр.) m/z=239,2 (M-Li).
Стадия D: Получение бензил транс-1-(2-метоксиэтил)-4-(оксазол-5-ил)пирролидин-3-илкарбамата: К раствору транс-этил 1-(2-метоксиэтил)-4-(оксазол-5-ил)пирролидин-3-карбоксилата (234 мг, 0,872 ммоль) в ДМФ (0,8 мл) добавили ДИЭА (0,304 мл, 1,74 ммоль), затем толуол (10 мл). Добавили дифенилфосфорилазид (0,263 мл, 1,22 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 1 часа, затем с дефлегматором в течение 1 часа. Добавили бензиловый спирт (0,903 мл, 8,72 ммоль) и нагревали реакционную смесь с дефлегматором в течение 17 часов. Смесь охладили и разбавили H2O (25 мл), и экстрагировали ДХМ (3×20 мл), а объединенные органические фазы промыли насыщенным солевым раствором (25 мл), высушили при помощи MgSO4, отфильтровали, концентрировали. Неочищенный продукт очистили обращенно-фазовой колоночной хроматографией, элюируя 5-70% ацетонитрила в воде, для получения продукта в виде бледно-желтого сиропообразного вещества (55 мг, выход 18%). МС (apci) m/z=346,1 (М+Н).
Стадия Е: Получение транс-1-(2-метоксиэтил)-4-(оксазол-5-ил)пирролидин-3-амина бис(2,2,2-трифторацетата): Раствор бензил транс-1-(2-метоксиэтил)-4-(оксазол-5-ил)пирролидин-3-илкарбамата (55 мг, 0,16 ммоль) в ТФК (2 мл) нагревали в закрытой пробирке при 60°C в течение 16 часов. Реакционную смесь перенесли в колбу, содержащую EtOH (10 мл) и концентрировали in vacuo. Неочищенный продукт растворили в толуоле (15 мл) и три раза перегнали азеотропной перегонкой для получения неочищенного продукта в виде коричневого сиропообразного вещества (130 мг, выход 186%). МС (apci) m/z=212,1 (М+Н).
Стадия F: Получение 1-(транс-1-(2-метоксиэтил)-4-(оксазол-5-ил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины: Получили по способу, описанному в Примере 45, Стадия F, используя транс-1-(2-метоксиэтил)-4-(оксзаол-5-ил)пирролидин-3-амина бис(2,2,2-трифторацетат) для получения продукта в виде бесцветного остатка (5,0 мг, выход 18%). МС (apci) m/z=437,3 (М+Н).
Пример 47
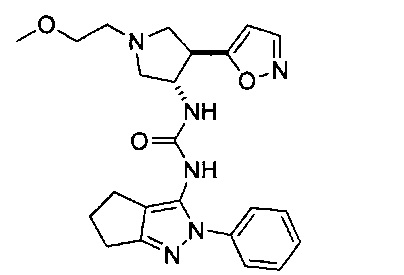
1-(транс-4-(изоксазол-5-ил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Получили по способу, описанному в примере 46, заменяя оксазол-5-карбальдегид на Стадии А на изоксазол-5-карбальдегид. МС (apci) m/z=437,0 (М+Н).
Пример 48
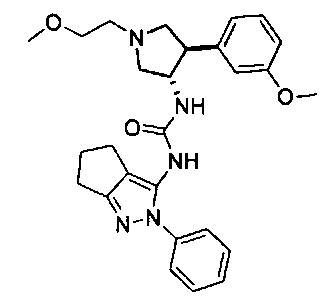
1-((3,4-транс)-1-(2-метоксиэтил)-4-(3-метоксифенил)пирролидин-3-ил)-3-(2-(фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Стадия А: Получение (3,4-транс)-трет-бутил 3-(3-метоксифенил)-4-(3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)уреидо)пирролидин-1-карбоксилата: К смеси (3,4-транс)-трет-бутил 3-амино-4-(3-метоксифенил)пирролидин-1-карбоксилата (30 мг, 0,095 ммоль, приобрели у компании BroadPharm) и фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразоли-3-илкарбамата (37 мг, 0,11 ммоль) добавили ДМА (0,5 мл), охладили на ледяной бане, затем добавили ДИЭА (0,050 мл, 0,29 ммоль). Ледяную баню убрали, а реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем реакционную смесь напрямую очистили обращенно-фазовой хроматографией, элюируя 5-75% ацетонитрила в воде, от 5 до 75%, для получения продукта в виде белого твердого вещества (15 мг, выход 30%).МС (apci) m/z=518,0 (М+Н).
Стадия В: Получение 1-((3,4-транс)-4-(3-метоксифенил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины гидрохлорида: Раствор (3,4-транс)-трет-бутил 3-(3-метоксифенил)-4-(3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)уреидо)пирролидин-1-карбоксилата (15 мг, 0,029 ммоль) в 5-6 н. HCl в IPA (58 мкл, 0,29 ммоль) перемешивали при комнатной температуре в течение 3 часов, затем концентрировали in vacuo, обработали эфиром и высушили под высоким вакуумом, получив неочищенный продукт в виде грязновато-белого твердого вещества. Твердое вещество напрямую использовали на следующей стадии без дополнительной очистки. МС (apci) m/z=418,1 (М+Н).
Стадия C: Получение 1-((3,4-транс)-1-(2-метоксиэтил)-4-(3-метоксифенил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины: К ДМФ (0,3 мл) раствору 1-((3,4-транс)-4-(3-метоксифенил)пирролидин-3-ил-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины гидрохлорида (13 мг, 0,029 ммоль) добавили N-этил-N-изопропилпропан-2-амин (16 мкл, 0,086 ммоль) и 1-бром-2-метоксиэтан (4,8 мг, 0,034 ммоль), и перемешивали реакционную смесь при комнатной температуре в течение 3 дней. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-55% ацетонитрила в воде, получив указанный в заголовке продукт в виде белого твердого вещества (10 мг, выход 73%). МС (apci) m/z=476,2 (М+Н).
Пример 49
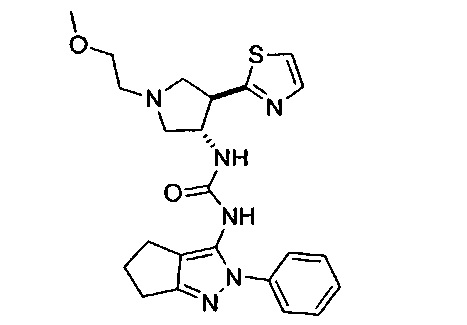
1-(1-(2-метоксиэтил)-4-(тиазол-2-ил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Стадия А: Получение транс-0-(2-метоксиэтил)-4-нитропирролидин-3-ил)тиазола: ПС-ДМАП (3,52 г, 5,00 ммоль) небольшими частями добавили к раствору тиазол-2-карбальдегида (2,83 г, 25,0 ммоль) в ацетонитриле (5 мл) и нитрометане (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов и добавили ацетонитрил (50 мл), затем уксусный ангидрид (2,59 мл, 27,5 ммоль). Реакционную смесь перемешивали в течение 1 часа, отфильтровали и концентрировали in vacuo для получения (Е)-2-(2-нитровинил)тиазола (3,99 г). Часть (Е)-2-(2-нитровинил)тиазола (1,00 г, 6,40 ммоль) растворили в ДХМ (5 мл), охладили до 0°C и обработали ТФК (0,0987 мл, 1,28 ммоль), затем по каплям добавили 2-метокси-N-(метоксиметил)-М-((триметилсилил)метил)этанамин (1,32 г, 6,40 ммоль). Реакционную смесь оставили нагреваться до комнатной температуры на ночь. Добавили 1 н. раствор NaOH (5 мл) и экстрагировали реакционную смесь несколькими порциями ДХМ на фазоразделительном стеклянном фильтре. Объединенные ДХМ экстракты концентрировали для получения неочищенного указанного в заголовке соединения (1,61 г, выход 94,1%). МС (apci) m/z=258,0 (М+Н).
Стадия В: Получение транс-1-(2-метоксиэтил)-4-(тиазол-2-ил)пирролидин-3-амина: транс- 1-(2-Метоксиэтил)-4-нитропирролидин-3-ил)тиазол (80 мг, 0,31 ммоль) растворили в 1 мл MeOH и обработали 10% Pd/C (33 мг, 0,031 ммоль). Реакционную смесь перемешивали под водородом из баллона в течение ночи, отфильтровали через Celite® и концентрировали для получения неочищенного указанного в заголовке соединения (68 мг, выход 96%). МС (apci) m/z=228,1 (М+Н).
Стадия С: Получение 1-(транс-1-(2-метоксиэтил)-4-(тиазол-2-ил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины: транс-1-(2-Метокси-этил)-4-(тиазол-2-ил)пирролидин-3-амин (15,0 мг, 0,0660 ммоль) растворили в 1 мл ДХМ и обработали ДИЭА (23,0 мкл, 0,132 ммоль), затем фенил (2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)карбаматом (25,4 мг, 0,0792 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, концентрировали и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-50% ацетонитрила в воде, для получения указанного в заголовке соединения (3,2 мг, выход 10,7%). МС (apci) m/z=453,1 (М+Н).
Пример 50
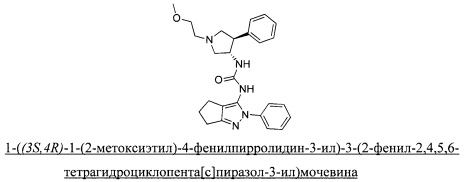
К ДХМ (5 мл) раствору (3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-амина дигидрохлорида (Получение D; 0,10 г, 0,34 ммоль) в ДХМ (5 мл) при 0°С последовательно добавили фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат (Пример 38, Стадия С; 0,13 г, 0,41 ммоль) и ТЭА (0,14 мл, 1,0 ммоль). Полученную смесь оставили нагреваться до комнатной температуры и перемешивали в течение 2 часов. Затем ее обработали EtOAc, промыли насыщенным раствором NH4Cl, насыщенным раствором NaHCO3 и насыщенным солевым раствором. Объединенные органические слои высушили при помощи MgSO4, отфильтровали и концентрировали. Неочищенный материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2-2,5% МеОН в ДХМ, для получения продукта (0,11 г, выход 72%). МС (apci) m/z=446,2 (М+Н).
Пример 51
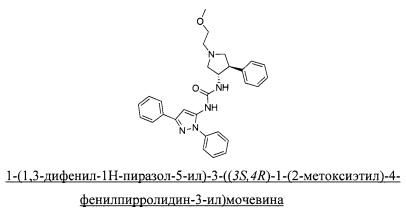
1,3-Дифенил-1Н-пиразол-5-амин (Таблица 1; 32,0 мг, 0,136 ммоль), ДИЭА (35,6 мкл, 0,204 ммоль) и 1,1'-карбонилдиимидазол (19,3 мг, 0,119 ммоль) смешали в CHCl3 (0,5 мл) в закрытой пробирке и нагревали при 60°С в течение 4 часов. Смесь охладили до комнатной температуры и добавили (3S,4R)-1-(2-метоксиэтил)-4-фенипирролидин-3-амина дигидрохлорид (Получение D; 15,0 мг, 0,0681 ммоль). После нагревания при 100°С в течение 15 часов, реакционную смесь концентрировали и напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 0-70% ацетонитрила в воде, для получения указанного в заголовке соединения (5,6 мг, выход 17%). МС (apci) m/z=482,2 (М+Н).
Пример 52
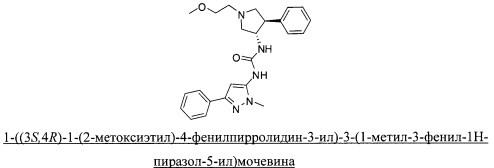
К раствору 1-метил-3-фенил-1Н-пиразол-5-амина (Таблица 1; 49 мг, 0,28 ммоль) в ДХМ (2 мл) добавили КДИ (46 мг, 0,28 ммоль), затем ДИЭА (200 мкл, 1,1 ммоль). Через 2 часа при комнатной температуре, добавили раствор (3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-амина дигидрохлорида (Получение D, 83 мг, 0,28 ммоль) в ДХМ (0,6 мл). Реакционную смесь перемешивали в течение 15 минут, затем напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-50% ацетонитрила в воде, для получения указанного в заголовке продукта в виде белого твердого вещества (45 мг, выход 38%). МС (apci) m/z=420,1 (М+Н).
Соединения в Таблице 6 были получены по способу Примера 52, с использованием соответствующих исходных материалов. Время превращения для активированного промежуточного соединения с КДИ варьировалось, и его контролировали отбором аликвоты и погашением в МеОН. Для контролирования полного превращения в метилкарбамат использовали анализ ЖХМС (от 30 минут до 16 часов).
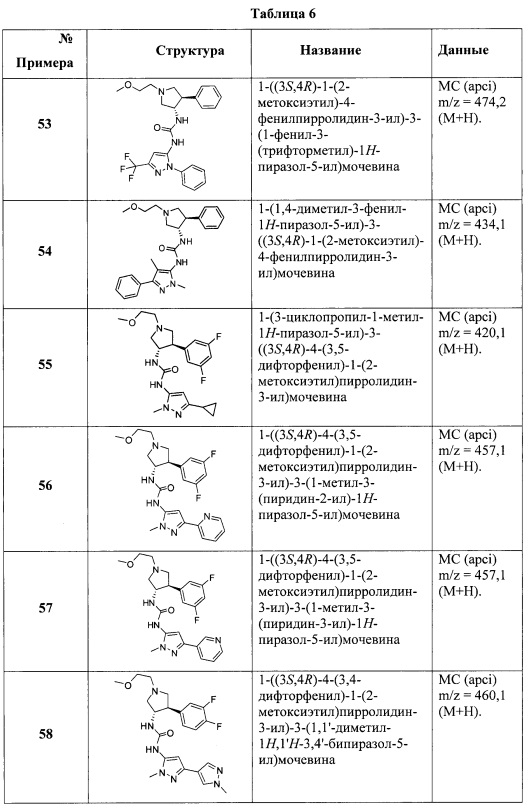
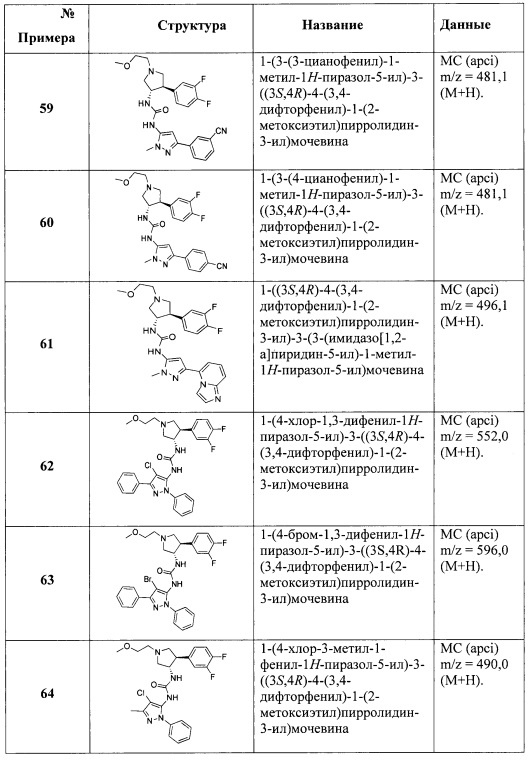
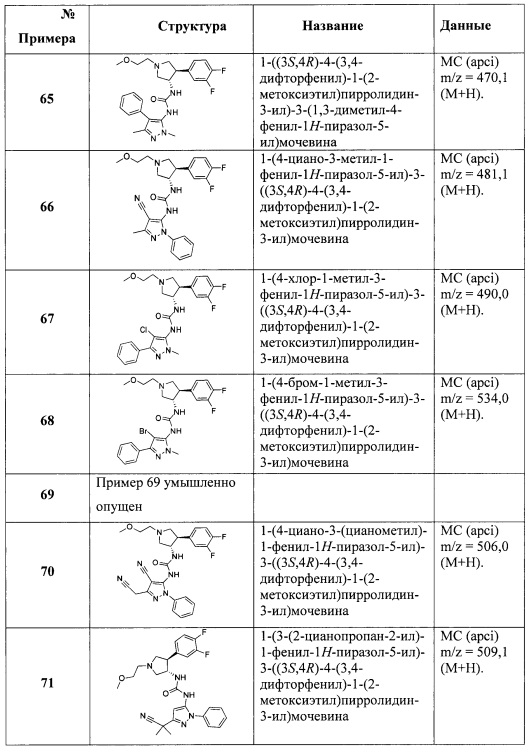
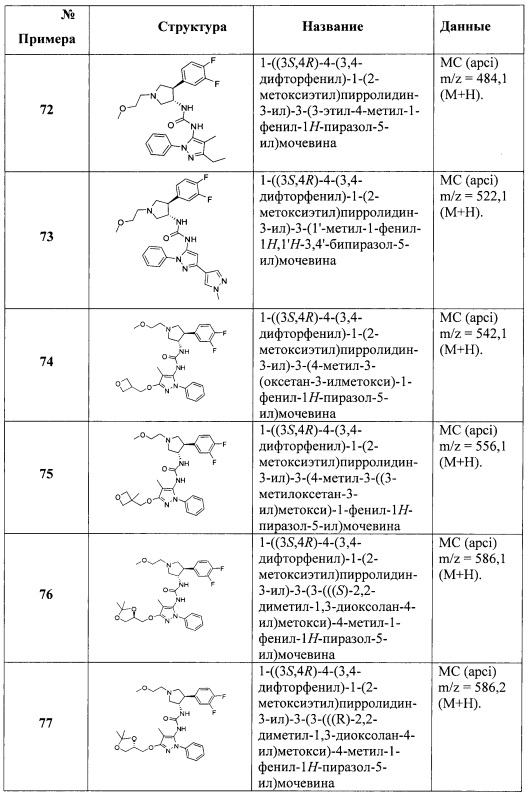
Соединения Таблицы 7 были получены по способу Примера 1 с заменой соединения Получения В на соединение Получения D, Е, F, G, Н, J или K и с использованием соответствующего пиразолового промежуточного соединения.
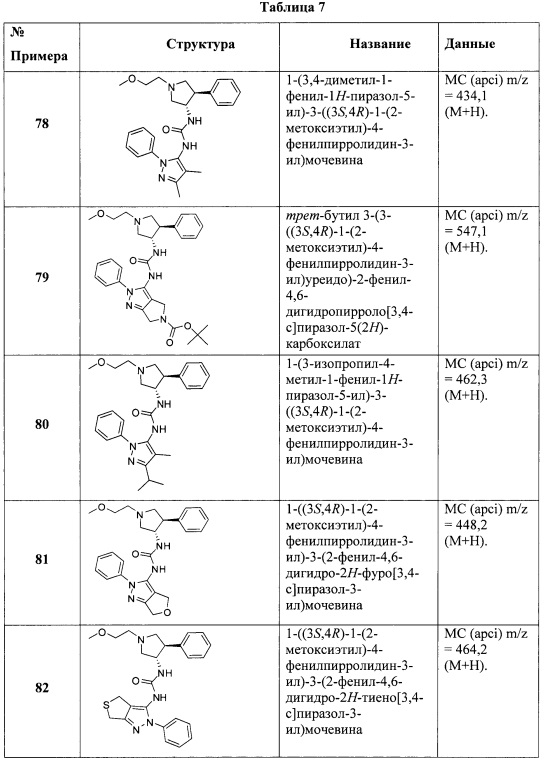
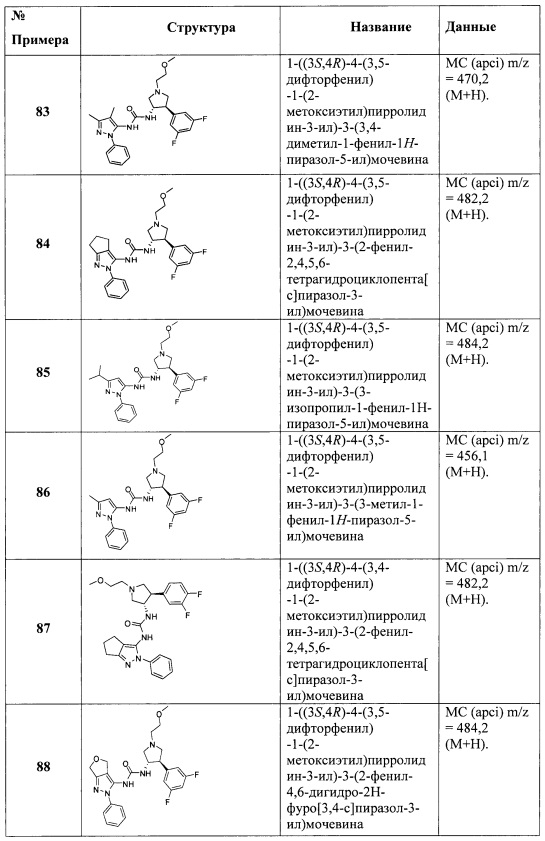
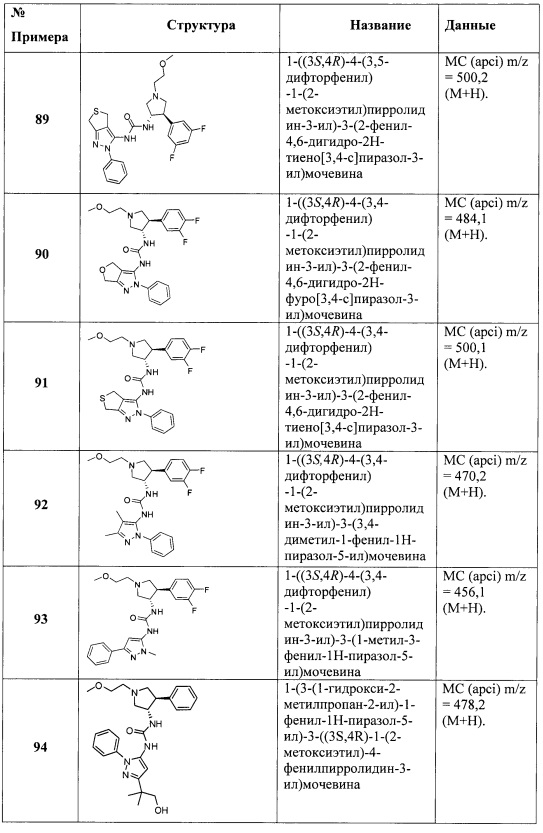
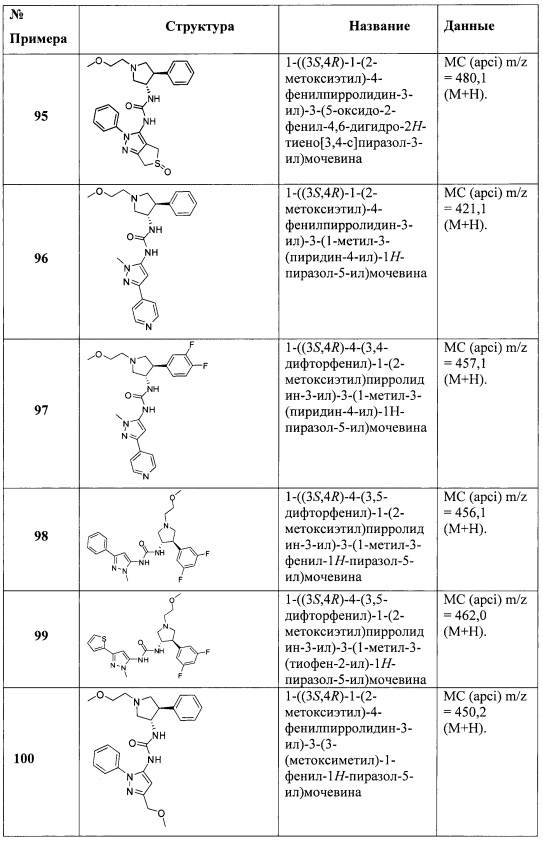
Соединения Таблицы 8 были получены по способу Примера 1 с заменой соединения Получения В на соединение Получения F или K и с использованием соответствующего пиразолового промежуточного соединения.
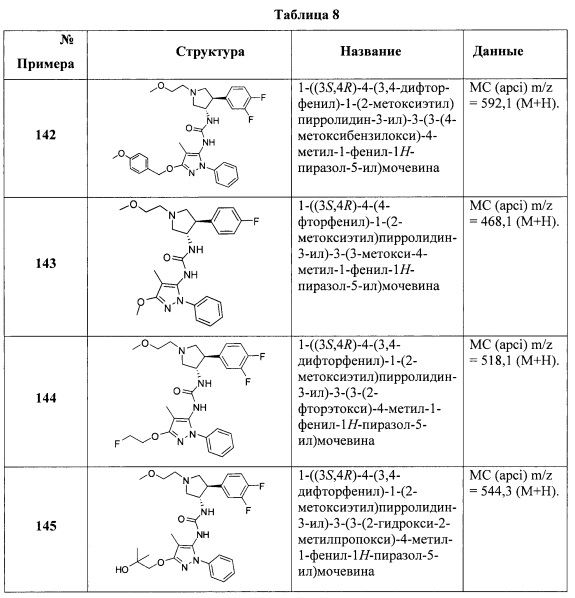
Соединения Таблицы 9 были получены по способу Примера 1 с заменой соединения Получения В на соединение Получения Е, F, Н или K и с использованием соответствующего пиразолового промежуточного соединения.

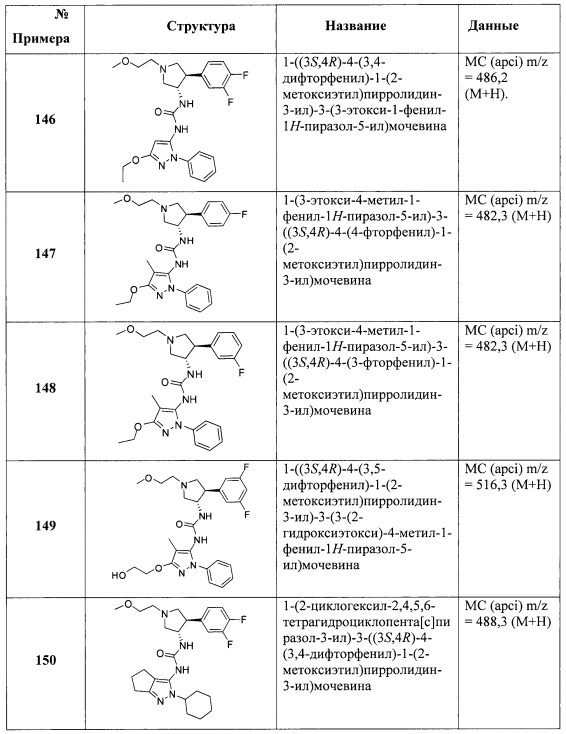
Пример 151
Стадия А: Получение 2-(пиридин-4-ил)-2,4,5,6-тетрагидроциклопента[с]пиразол-3-амина: Раствор 2-оксоциклопентанкарбонитрила (0,4 г, 3,7 ммоль, приобрели у компании ААТ Pharmaceutical) и 4-гидразинилпиридина гидрохлорида (0,53 г, 3,7 ммоль) в метаноле (35 мл) закрыли в сосуде для работы под давлением и нагревали при 80°С в течение ночи. После удаления растворителя in vacuo, остаток растерли с 1 н. раствором NaOH (20 мл) и экстрагировали ДХМ (3×25 мл). Объединенные органические слои промыли насыщенным солевым раствором, высушили при помощи MgSO4, отфильтровали и концентрировали для получения неочищенного продукта в виде коричневатого твердого вещества, которое напрямую использовали на следующей стадии. МС (apci) m/z=201,2 (М+Н).
Стадия В: Получение 1-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-(пиридин-4-ил)-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины: К смеси 2-(пиридин-4-ил)-2,4,5,6-тетрагидроциклопента[с]пиразол-3-амина (43 мг, 0,21 ммоль) в ДХМ (2 мл) при 0°С добавили ДИЭА (0,075 мл, 0,43 ммоль), затем одной порцией добавили трифосген (25 мг, 0,086 ммоль). Реакционную смесь нагрели до комнатной температуры и перемешивали в течение 2 часов. Взяли аликвоту (0,5 мл) реакционной смеси (содержащей 3-изоцианато-2-(пиридин-4-ил)-2,4,5,6-тетрагидроциклопента[с]пиразол (12,1 мг, 0,0535 ммоль)) и последовательно обработали (3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-амина бис(2,2,2-трифторацетатом) (Получение D, 20 мг, 0,0446 ммоль) и N-этил-N-изопропилпропан-2-амином (38,8 мкл, 0,223 ммоль). После перемешивания в течение 30 минут реакционную смесь напрямую очистили обращенно-фазовой хроматографией, элюируя 5-50% ацетонитрила в воде, для получения конечного продукта в виде грязновато-белого твердого вещества (5 мг, выход 25%). МС (apci полож.) m/z=447,2 (М+Н).
Соединения Таблицы 10 были получены по способу Примера 151, с заменой пиразола на соответствующий аналог и с заменой соединения Получения D на соединение Получения F.
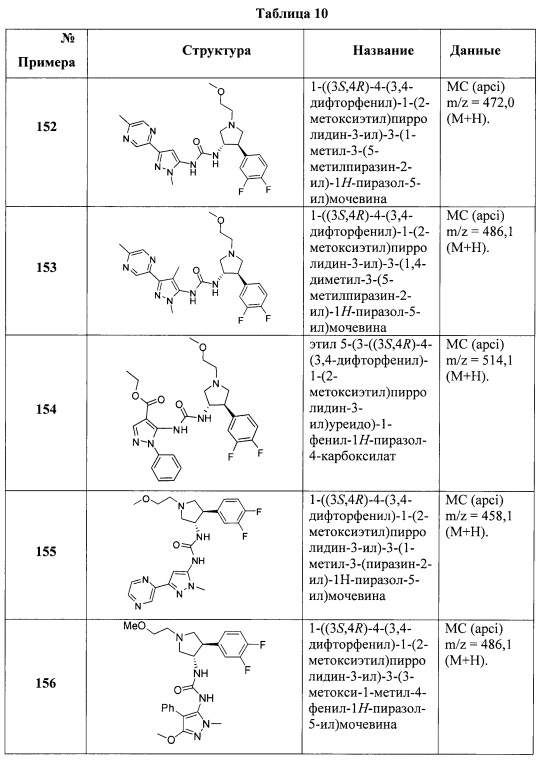
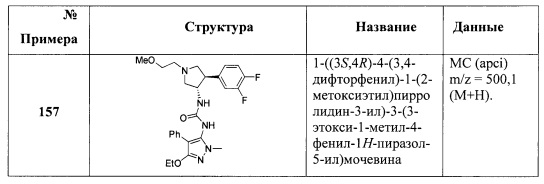
Пример 158
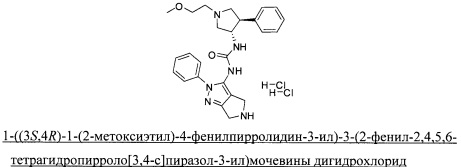
трет-Бутил 3-(3-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)уреидо)-2-фенил-4,6-дигидропирроло[3,4-с]пиразол-5(2Н)-карбоксилат (Пример 79) обработали 4 н. HCl в диоксане (2,0 мл, 0,017 ммоль) и перемешивали при комнатной температуре в течение 30 минут. Полученную бежевую суспензию отфильтровали, а твердые вещества промыли Et2O для получения продукта в виде желто-коричневого твердого вещества (6,4 мг, выход 74%). МС (apci) m/z=447,1 (М+Н).
Пример 159
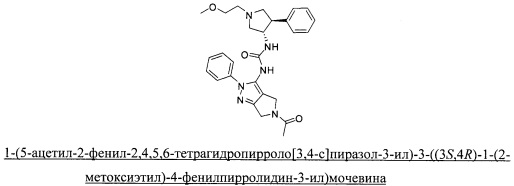
К раствору 1-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-ил)мочевины дигидрохлорида (Пример 158, 2,0 мг, 0,0039 ммоль) и ДИЭА (0,0067 мл, 0,039 ммоль) в ацетонитриле (1,0 мл, 19 ммоль) добавили уксусную кислоту (0,0011 мл, 0,019 ммоль), затем НАТУ (2,9 мг, 0,0077 ммоль). После перемешивания в течение 1 часа при комнатной температуре, реакционную смесь напрямую очистили колоночной хроматографией на диоксиде кремния, элюируя 0-10% МеОН в ДХМ, для получения продукта (0,7 мг, выход 37%). МС (apci) m/z=489,2 (М+Н).
Пример 160
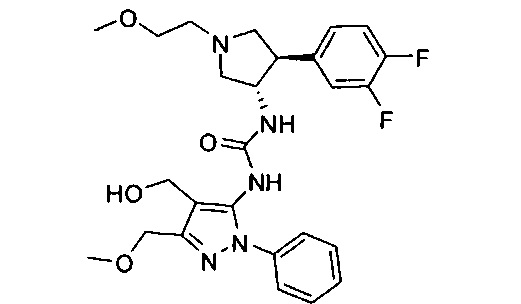
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-(гидроксиметил)-3-(метоксиметил)-1-фенил-1Н-пиразол-5-ил)мочевина
Раствор 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-4,6-дигидро-2Н-фуро[3,4-с]пиразол-3-ил)мочевины (Пример 90, 250 мг, 0,517 ммоль) в ДХМ (20 мл) обработали 4 н. HCl в диоксане (5 мл). После концентрирования до сухости, остаток преобразовали в свободное основание разделением с 1 н. NaOH и ДХМ, затем очистили колоночной хроматографией на диоксиде кремния, элюируя 2-4% МеОН в ДХМ, для получения продукта (29 мг, выход 11%). МС (apci) m/z=516,2 (М+Н).
Пример 161
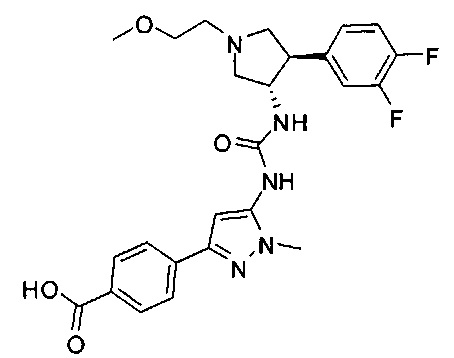
4-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-метил-1Н-пиразол-3-ил)бензойная кислота
К раствору метил 4-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-метил-1Н-пиразол-3-ил)бензоата (Пример 121, 79 мг, 0,15 ммоль) в ТГФ (4,0 мл, 0,15 ммоль) и МеОН (2,0 мл, 49 ммоль) при 0°C добавили LiOH (2 М водный) (0,15 мл, 0,31 ммоль). Эту реакционную смесь перемешивали при комнатной температуре и добавляли дополнительное количество LiOH до завершения превращения, наблюдаемого по анализу ВЭЖХ (приблизительно 2 дня). Подкислив 2 М раствором HCl (1 мл), смесь разбавили водой (10 мл), экстрагировали ДХМ (20 мл), затем экстрагировали 10% МеОН в ДХМ (3×10 мл). Объединенные органические фазы промыли насыщенным солевым раствором (25 мл), высушили при помощи MgSO4, отфильтровали и концентрировали для получения продукта в виде белого твердого вещества (70 мг, выход 91%). МС (apci) m/z=500,1 (М+Н).
Пример 162
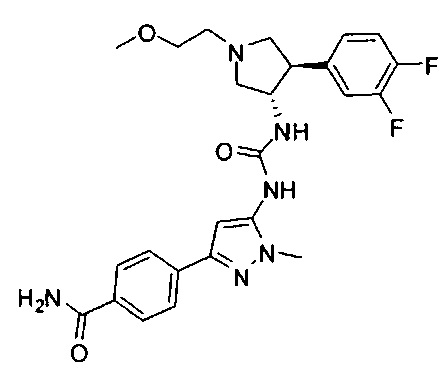
4-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-метил-1Н-пиразол-3-ил)бензамид
К раствору 4-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-метил-1Н-пиразол-3-ил)бензойной кислоты (Пример 161, 12 мг, 0,024 ммоль) в ДМФ (0,5 мл, 0,024 ммоль) последовательно добавили N-метилморфолин (0,0079 мл, 0,072 ммоль), NH3 (0,5 М в диоксане) (0,096 мл, 0,048 ммоль) и НАТУ (10 мг, 0,026 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре и очистили обращенно-фазовой колоночной хроматографией, элюируя 5-50% ацетонитрила в воде, для получения указанного в заголовке соединения (5,1 мг, выход 43%).МС (apci) m/z=499,1 (М+Н).
Пример 163
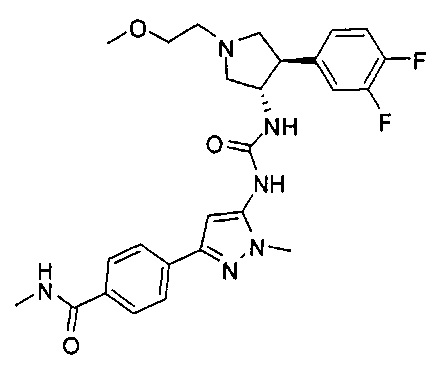
4-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-метил-1Н-пиразол-3-ил)-N-метилбензамид
Получили по способу, описанному в Примере 162, заменяя NH3 на метиламин (2 М в ТГФ). МС (apci) m/z=513,1 (М+Н).
Пример 164
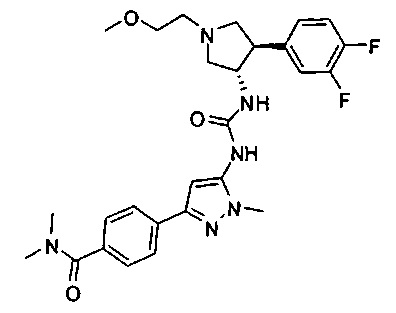
4-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-метил-1Н-пиразол-3-ил)-N,N-диметилбензамид
Получили по способу, описанному в Примере 162, заменяя NH3 на диметиламин (2 М в ТГФ). МС (apci) m/z=5527,1 (М+Н).
Пример 165
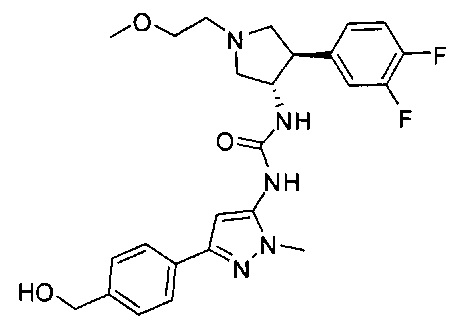
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(4-(гидроксиметил)фенил)-1-метил-1Н-пиразол-5-ил)мочевина
К суспензии метил 4-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-метил-1Н-пиразол-3-ил)бензоата (Пример 121; 20 мг, 0,039 ммоль) в ТГФ (2 мл, 0,081 ммоль) при 0°C под N2 добавили алюмогидрид лития (1 М бис-ТГФ раствор в толуоле) (0,078 мл, 0,078 ммоль). Реакционную смесь перемешивали при 0°C в течение 1 часа, затем погасили добавлением 3 мкл Н2О и 3 мкл 1 М водного раствора NaOH, затем 9 мкл H2O. Смесь перемешивали при комнатной температуре в течение 4 часов, затем отфильтровали через шприцевой фильтр, промыли ТГФ (2 мл) и концентрировали до белого твердого вещества. Твердое вещество очистили обращенно-фазовой хроматографией, элюируя 5-50% ацетонитрила в воде, для получения указанного в заголовке соединения (10 мг, выход 53%).МС (apci) m/z=486,1 (М+Н).
Пример 166
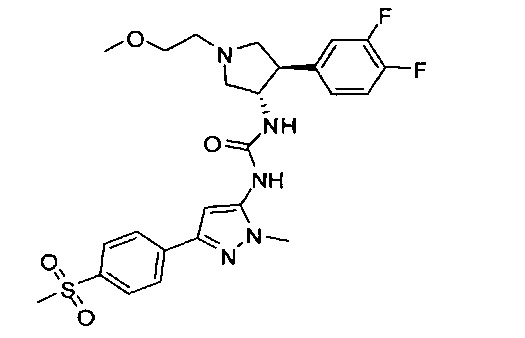
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-метил-3-(4-(метилсульфонил)фенил)-1Н-пиразол-5-ил)мочевина
Стадия А: Получение фенил 1-метил-3-(4-(метилтио)фенил)-1Н-пиразол-5-илкарбамата: Получили по Стадии А Примера 1, заменяя 3-трет-бутил-1-фенил-1Н-пиразол-5-амин на 1-метил-3-(4-(метилтио)фенил)-1Н-пиразол-5-амин (Промежуточное соединение Р121), для получения продукта. МС (apci) m/z=340,0 (М+Н).
Стадия В: Получение фенил 1-метил-3-(4-(метилсульфонил)фенил)-1Н-пиразол-5-илкарбамата: К раствору фенил 1-метил-3-(4-(метилтио)фенил)-1Н-пиразол-5-илкарбамата (110 мг, 0,324 ммоль) в ДХМ (5 мл, 0,295 ммоль) при 0°C одной порцией добавили МСРВА (70-75% в H2O) (72,6 мг, 0,295 ммоль). Реакционную смесь оставили нагреваться до комнатной температуры и перемешивали в течение 2 часов, а затем добавили еще одну порцию МСРВА (72,6 мг, 0,295 ммоль). После перемешивания в течение 5 часов при комнатной температуре, смесь разбавили ДХМ (25 мл) и промыли насыщенным водным раствором NaHCO3 (2×10 мл) и насыщенным водным раствором Na2S2O3 (3×10 мл). Органический слой высушили при помощи MgSO4, отфильтровали и концентрировали in vacuo для получения неочищенного продукта (101 мг, выход 92,3%). МС (apci) m/z=372,0 (М+Н).
Стадия С: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-метил-3-(4-(метилсульфонил)фенил)-1Н-пиразол-5-ил)мочевины: Получили по Стадии В Примера 1, заменяя фенил 1-метил-3-(4-(метилсульфонил)фенил)-1Н-пиразол-5-илкарбаматом фенил 3-трет-бутил-1-фенил-1Н-пиразол-5-илкарбамат, и заменяя соединением Получения F соединение Получения В. МС (apci) m/z=534,1 (М+Н).
Пример 167
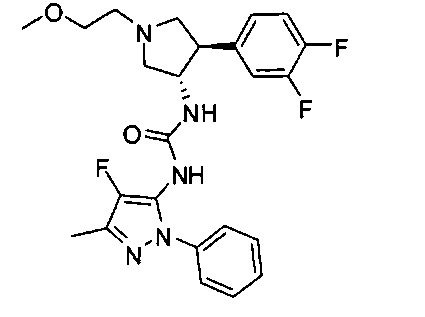
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-фтор-3-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Стадия A: Получение фенил 3-метил-1-фенил-1Н-пиразол-5-илкарбамата: Получили по способу Примера 1, Стадия А, заменяя 3-отрет-бутил-1-фенил-1Н-пиразол-5-амин на 3-метил-1-фенил-1Н-пиразол-5-амин, для получения продукта. МС (apci) m/z=294,1 (М+Н).
Стадия B: Получение фенил 4-фтор-3-метил-1-фенил-1Н-пиразол-5-илкарбамата: К раствору фенил 3-метил-1-фенил-1Н-пиразол-5-илкарбамата (20 мг, 0,0682 ммоль) в ацетонитриле (0,5 мл) небольшими частями добавили Selectfluor (26,6 мг, 0,0750 ммоль) при комнатной температуре, и перемешивали реакционную смесь в течение ночи. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-65% ацетонитрила в воде, для получения продукта в виде белого пенистого вещества (12,4 мг, выход 58,4%).МС (apci) m/z=312,0 (М+Н).
Стадия С: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-фтор-3-метил-1-фенил-1Н-пиразол-5-ил)мочевины: Получили по способу Примера 1, Стадия B, заменяя фенил 4-фтор-3-метил-1-фенил-1Н-пиразол-5-илкарбаматом фенил 3-трет-бутил-1-фенил-1Н-пиразол-5-илкарбамат, и заменяя соединением Получения F соединение Получения В. МС (apci) m/z=474,1 (М+Н).
Пример 168
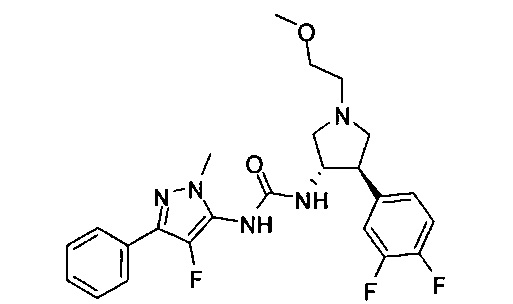
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-фтор-1-метил-3-фенил-1Н-пиразол-5-ил)мочевина
Получили по такому же способу, как Пример 167, заменяя 1-метил-3-фенил-1Н-пиразол-5-амином 3-метил-1-фенил-1Н-пиразол-5-амин. МС (apci) m/z=474,1 (М+Н).
Пример 169
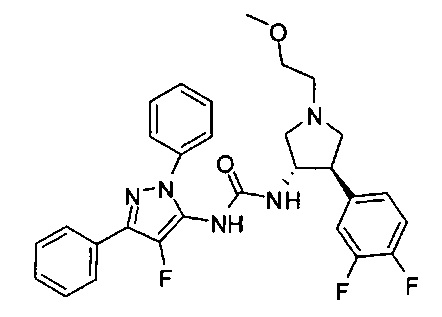
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-фтор-1,3-дифенил-1Н-пиразол-5-ил)мочевина
Получили по такому же способу, как Пример 167, заменяя 1,3-ди-фенил-1Н-пиразол-5-амином 3-метил-1-фенил-1Н-пиразол-5-амин. МС (apci) m/z=536,1 (М+Н).
Пример 170
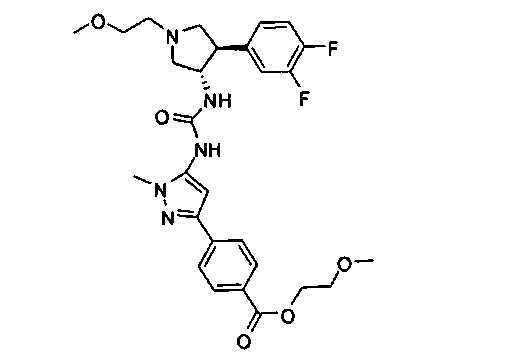
2-метоксиэтил 4-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-метил-1Н-пиразол-3-ил)бензоат
Стадия А: Получение 4-(5-(3-((3S,4R)A-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-метил-1Н-пиразол-3-ил)бензоата лития: К раствору метил 4-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-метил-1Н-пиразол-3-ил)бензоата (Пример 121; 158 мг, 0,308 ммоль) в ТГФ (4 мл, 0,308 ммоль) и МеОН (2,00 мл, 49,4 ммоль) при 0°C добавили LiOH (2 М водный) (0,308 мл, 0,615 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 48 часов. Добавили еще одну порцию LiOH (70 мкл, 0,4 экв.) и перемешивали реакционную смесь еще 4 дня. Реакционную смесь концентрировали до сухости и использовали напрямую на следующей стадии, предположив количественный выход. МС (apci) m/z=500,1 (М+Н).
Стадия В: Получение 2-метоксиэтил 4-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-метил-1Н-пиразол-3-ил)бензоата: К раствору 4-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-метил-1Н-пиразол-3-ил)бензоата лития (15 мг, 0,030 ммоль) в ДМФ (0,5 мл, 0,030 ммоль) добавили ДИЭА (0,016 мл, 0,089 ммоль) и 2-метоксиэтанол (9,0 мг, 0,12 ммоль), затем НАТУ (17 мг, 0,045 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-65% ацетонитрила в воде, для получения продукта в виде белого пенистого вещества (1,8 мг, выход 11%). МС (apci) m/z=558,0 (М+Н).
Пример 171
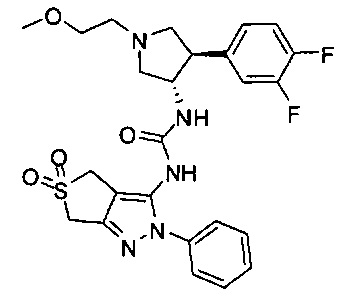
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(5,5-диоксидо-2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-ил)мочевина
Стадия А: Получение 2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-амина: Суспензию 4-оксотетрагидротиофен-3-карбонитрила (1,00 г, 7,86 ммоль) и фенилгидразина гидрохлорида (1,25 г, 8,65 ммоль) в абсолютном EtOAc (40 мл, 7,86 ммоль) нагревали с дефлегматором в течение 2 часов. Смесь концентрировали, а остаток растерли с 1 н. водным раствором NaOH (40 мл). Твердое вещество собрали фильтрацией, последовательно промыли 0,1 н. водным раствором NaOH, водой и гексанами, затем высушили in vacuo для получения продукта (1,62 г, выход 95%) в виде белого твердого вещества. МС (apci) m/z=218,1.
Стадия В: Получение фенил 2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-илкарбамата: К суспензии 2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-амина (500 мг, 2,30 ммоль) в EtOAc (10,0 мл, 2,30 ммоль) добавили NaOH (2,30 мл, 2 М, водный, 4,60 ммоль), затем по каплям добавили фенилхлорформиат (0,40 мл, 3,22 ммоль) при комнатной температуре. После перемешивания в течение 2 часов, добавили дополнительное количество фенилхлорформиата (0,14 мл). Перемешивание продолжали в течение 5 минут, а затем добавили еще одну порцию фенилхлорформиата (0,081 мл) и перемешивали смесь еще 16 часов. Реакционную смесь разбавили EtOAc и разделили фазы. Органическую фазу промыли водой и насыщенным солевым раствором (каждого по 25 мл), высушили над Na2SO4, отфильтровали и концентрировали. Остаток очистили обращенно-фазовой колоночной хроматографией, элюируя 5-70% ацетонитрила в воде, для получения продукта (0,50 г, выход 64%) в виде белого твердого вещества (чистота 83%). МС (apci) m/z=338,1.
Стадия С: Получение фенил (5,5-диоксидо-2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-ил)карбамата: К молочно-белому раствору фенил 2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-илкарбамата (100 мг, 0,29 ммоль) в ДХМ (5 мл) при 0°C добавили МСРВА (170 мг, 70-75% водный комплекс, 0,74 ммоль). Смесь сняли с бани и перемешивали при комнатной температуре в течение 10 минут, затем разбавили ДХМ (20 мл) и последовательно промыли насыщенным раствором NaHCO3 (3×10 мл), насыщенным раствором Na2S2O3 (2×10 мл) и насыщенным солевым раствором (10 мл). Органический слой высушили над Na2SO4 и концентрировали для получения продукта (107 мг, выход 98%) в виде бледно-оранжевого пенистого вещества, которое использовали без очистки. МС (apci) m/z=371,4.
Стадия D: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(5,5-диоксидо-2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-ил)мочевины: К раствору (3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина трифторацетата (Получение Е; 60 мг, 0,12 ммоль) и фенил (5,5-диоксидо-2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-ил)карбамата (50,3 мг, 0,14 ммоль) в безводном ДМА (2 мл) добавили ДИЭА (97 мкл, 0,56 ммоль). Смесь перемешивали при комнатной температуре в течение 15 часов. Затем реакционную смесь разделили между насыщенным раствором NH4Cl (20 мл) и EtOAc (10 мл). Водный слой экстрагировали EtOAc (2×10 мл), а объединенные органические фазы промыли водой (5×10 мл) и насыщенным солевым раствором (10 мл), затем высушили над Na2SO4 и концентрировали. В результате очистки неочищенного продукта колоночной хроматографией на диоксиде кремния, элюируя 2% МеОН в ДХМ, получили продукт в виде бледно-желтого пенистого вещества (33 мг, выход 50%). МС (apci) m/z=532,1.
Пример 172
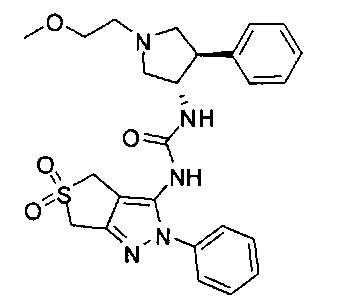
1-(5,5-диоксидо-2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина
Получили по способу, использованному для Примера 171, заменяя (3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина трифторацетат (Получение Е) на (3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-амина дигидрохлорид (Получение D) на Стадии D. МС (apci) m/z=496,0 (М+Н).
Пример 173
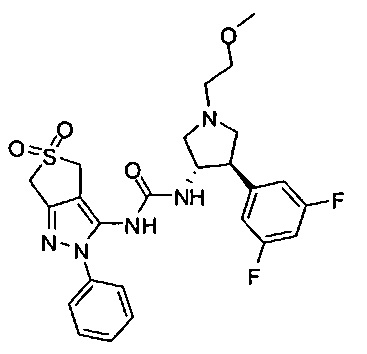
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(5,5-диоксидо-2-фенил-4,6-дигидро-2Н-тиено [3,4-с] пиразол-3-ил)мочевина
Получили по такому же способу, как Пример 171, заменяя (3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина трифторацетатом (Получение Е) (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (Получение F). МС (apci) m/z=532,0 (М+Н).
Пример 174
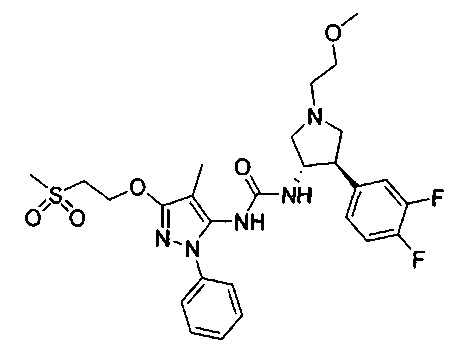
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-(метилсульфонил)этокси)-1-фенил-1Н-пиразол-5-ил)мочевина
Стадия А: Получение фенил 4-метил-3-(2-(метилтио)этокси)-1-фенил-1Н-пиразол-5-илкарбамата: Получили по способу, описанному для Примера 171, Стадия В, заменяя 2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-амин на 4-метил-3-(2-(метилтио)этокси)-1-фенил-1Н-пиразол-5-амин (Промежуточное соединение Р206). МС (apci) m/z=384,0 (М+Н).
Стадия В: Получение фенил 4-метил-3-(2-(метилсульфонил)этокси)-1-фенил-1Н-пиразол-5-илкарбамата: Фенил 4-метил-3-(2-(метилтио)этокси)-1-фенил-1Н-пиразол-5-илкарбамат (0,217 г, 0,566 ммоль) обработали ТГФ (10 мл) и охладили до 0°C. К реакционной смеси добавили раствор 3-хлорпероксибензойной кислоты (МСРВА) с ТГФ (4 мл). После перемешивания в течение 1 часа, смесь нагрели до комнатной температуры и перемешивали в течение 2 часов. Реакционную смесь обработали EtOAc, погасили Na2S2O3 и водой, экстрагировали EtOAc, промыли NaHCO3 и насыщенным солевым раствором, высушили над MgSO4, отфильтровали и концентрировали in vacuo. Полученный неочищенный продукт очистили колоночной хроматографией на диоксиде кремния для получения продукта (0,207 г, выход 88,0%). МС (apci) m/z=416,0 (М+Н).
Стадия C: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-(метилсульфонил)этокси)-1-фенил-1Н-пиразол-5-ил)мочевины: К (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амину (Получение F; 50 мг, 0,15 ммоль) в ДХМ (5 мл) при 0°C добавили фенил 4-метил-3-(2-(метилсульфонил)этокси)-1-фенил-1H-пиразол-5-илкарбамат (63 мг, 0,15 ммоль), затем добавили ТЭА (0,064 мл, 0,46 ммоль). Полученную смесь оставили нагреваться до комнатной температуры и перемешивали в течение 17 часов. Затем реакционную смесь обработали EtOAc, промыли насыщенным раствором NH4Cl, насыщенным раствором NaHCO3 и насыщенным солевым раствором, высушили при помощи MgSO4, отфильтровали, концентрировали и очистили колоночной хроматографией на диоксиде кремния, элюируя 3% МеОН в ДХМ, для получения указанного в заголовке продукта в виде белого твердого вещества (47 мг, выход 53%). МС (apci) m/z=578,0 (М+Н).
Пример 175
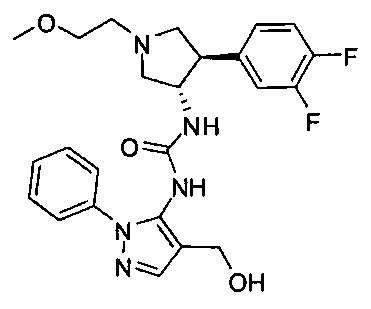
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-(гидроксиметил)-1-фенил-1Н-пиразол-5-ил)мочевина
К раствору этил 5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-4-карбоксилата (Пример 154; 55 мг, 0,11 ммоль) в ТГФ (2 мл) при 0°C под N2 по каплям добавили алюмогидрид лития (1 М бис-ТГФ раствор в толуоле, 0,21 мл, 0,21 ммоль). Реакционную смесь перемешивали при 0°C в течение 1,5 часов, затем при комнатной температуре в течение 3 часов Реакцию погасили последовательным добавлением H2O (0,008 мл), 1 М водного раствора NaOH (0,008 мл) и H2O (0,024 мл). После перемешивания при комнатной температуре в течение 2 часов, реакционную смесь отфильтровали, промыли ТГФ (2 мл) и концентрировали in vacuo. Неочищенный продукт очистили препаративной ТСХ (пластина 0,5 мм), элюируя 10% МеОН в ДХМ, для получения продукта в виде белого твердого вещества (6 мг, выход 11%). МС (apci) m/z=472,0 (М+Н).
Пример 176
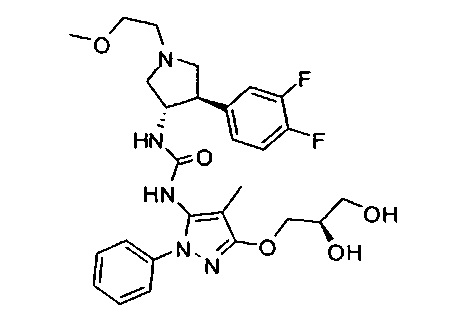
1-((3S,4R)-4-(3,4-Дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((S)-2,3-дигидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
К раствору 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(((S)-2,2-диметил-1,3-диоксолан-4-ил)метокси)-4-метил-1 -фенил-1 Н-пиразол-5 -ил)мочевины (Пример 76; 65,0 мг, 0,111 ммоль) в ТГФ (2 мл) добавили 1 М водный раствор HCl (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 75 минут, а затем концентрировали для удаления ТГФ. Оставшийся водный раствор разбавили H2O (2 мл) и обработали 2 М NaOH до рН=10. Полученную молочно-белую смесь обработали NaCl до насыщения и экстрагировали EtOAc (2Х). Объединенные органические экстракты высушили над MgSO4 и отфильтровали через упакованный Celite®. Элюент концентрировали до бесцветного гелеобразного вещества, которое промыли Et2O и высушили в вакууме для получения указанного в заголовке соединения в виде белого твердого вещества (53 мг, выход 88%). МС (apci) m/z=546,1 (М+Н).
Пример 177
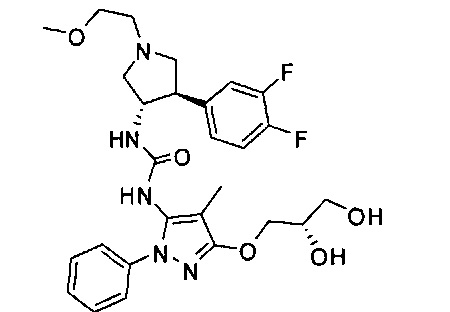
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((S)-2,3-дигидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Получили из 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(((R)-2,2-диметил-1,3-диоксолан-4-ил)метокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины (Пример 77; 50,0 мг, 0,0854 ммоль) по способу, описанному для Примера 176, для получения указанного в заголовке соединения в виде белого твердого вещества (38 мг, выход 82%). МС (apci) m/z=546,2 (М+Н).
Пример 178
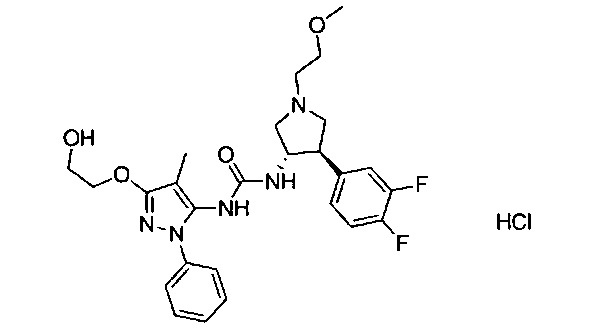
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-гидроксиэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины гидрохлорид
К 1-(3-(2-(трет-бутилдиметилсилилокси)этокси)-4-метил-1-фенил-1-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевине (Получение U-1; 46 мг, 0,073 ммоль) в ДХМ (2 мл) при комнатной температуре добавили 2 н. HCl (22 мл, 0,44 ммоль). После перемешивания в течение 1 часа, реакционную смесь концентрировали in vacuo и промыли Et2O для получения продукта в виде соли HCl (45 мг, выход 100%). МС (apci) m/z=516,1 (М+Н).
Пример 179
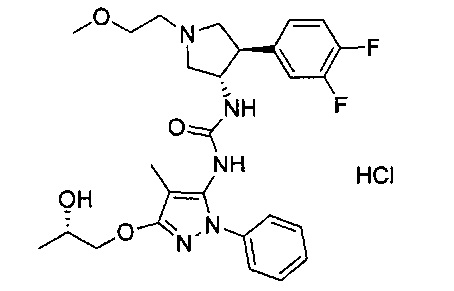
1-((3R,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((S)-2-гидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины гидрохлорид
К 1-(3-((S)-(2-(трет-бутилдиметилсилилокси)пропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевине (Получение U-2; 33 мг, 0,051 ммоль) в ДХМ (2 мл) при комнатной температуре добавили 2 н. HCl (0,15 мл, 0,31 ммоль). После перемешивания в течение 1 часа, реакционную смесь концентрировали in vacuo и промыли Et2O для получения HCl соли продукта (29 мг, выход 100%). МС (apci) m/z=530,3 (М+Н).
Пример 180
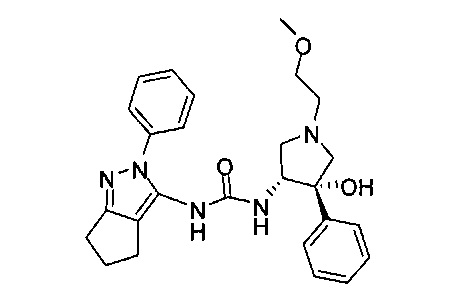
1-((3S,4R)-4-гидрокси-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Стадия А: Получение (3S,4R)-трет-бутил 3-азидо-4-гидроксипирролидин-1-карбоксилата: трет-Бутил 6-окса-3-азабицикло[3.1.0]гексан-3-карбоксилат (15,42 г, 83,25 ммоль), (1S,2S)-(-)-[1,2-циклогександиамино-N,N'-бис(3,5-ди-трет-бутилсалицилиден)]хрома (III) хлорид (1,181 г, 1,665 ммоль) и азидотриметилсилан (12,79 мл, 91,58 ммоль) перемешивали при комнатной температуре под атмосферой азота в течение 18 часов. Полученную темную красно-коричневую смесь обработали МеОН (100 мл) и K2CO3 (13,81 г, 99,90 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 5 часов. Раствор отфильтровали через слой Celite®, концентрировали и растворили в EtOAc (100 мл) и воде (50 мл). Слои разделили, и водный слой экстрагировали EtOAc. Объединенные органические экстракты промыли насыщенным водным раствором NaHCO3, водой и насыщенным солевым раствором, высушили при помощи MgSO4 и концентрировали для получения коричневого маслянистого вещества. Маслянистое вещество очистили колоночной хроматографией на диоксиде кремния, элюируя 20% EtOAc в гексанах, для получения указанного в заголовке соединения (э.и.=93%, 3,99 г, выход 102%). МС (apci) m/z=129,0 (М+Н-Вос).
Стадия B: Получение (3R,4R)-4-азидопирролидин-3-ола гидрохлорида: (3R,4R)-трет-бутил 3-азидо-4-гидроксипирролидин-1-карбоксилат (9,0 г, 39 ммоль) и 4 н. HCl в диоксане (15 мл, 59 ммоль) смешали в ДХМ (30 мл) и перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь концентрировали in vacuo для получения указанного в заголовке соединения (6,5 г, выход 100%) в виде желтого маслянистого вещества. МС (apci) m/z=129,0 (М+Н).
Стадия С: Получение (3R,4R)-4-азидо-1-(2-метоксиэтил)пирролидин-3-ола: (3R,4R)-4-азидопирролидин-3-ола гидрохлорид (6,5 г, 39,5 ммоль), 1-бром-2-метоксиэтан (6,59 г, 47,4 ммоль) и ДИЭА (13,8 мл, 79,0 ммоль) смешали в 10 мл ДМФ и перемешивали при комнатной температуре в течение 18 часов. Добавили МР-ТсОН (39,5 г, 158 ммоль) и встряхивали реакционную смесь в течение 1 часа, отфильтровали, а смолу промыли ДХМ. Амин выделили из смолы встряхиванием с 7 н. раствором NH3 в МеОН (113 мл, 790 ммоль) и ДХМ (113 мл) в течение 1 часа. Реакционную смесь отфильтровали, а смолу промыли ДХМ. Объединенные фильтраты концентрировали для получения неочищенного указанного в заголовке соединения (7,09 г, выход 96,4%). МС (apci) m/z=187,0 (М+Н).
Стадия D: Получение (3R,4R)-4-амино-1-(2-метоксиэтил)пирролидин-3-ола: (3R,4R)-4-азидо-1-(2-метоксиэтил)пирролидин-3-ол (3,0 г, 16,1 ммоль) и 1% Pd/C (1,71 г, 1,61 ммоль) смешали в 40 мл МеОН и встряхивали реакционную смесь под давлением H2 40 psi во встряхивателе Парра в течение трех дней. Реакционную смесь отфильтровали через Celite® и концентрировали для получения указанного в заголовке соединения (2,53 мг, выход 98,0%) в виде коричневого маслянистого вещества. МС (apci) m/z=161,1 (М+Н).
Стадия E: Получение трет-бутил (3R,4R)-4-гидрокси-1-(2-метоксиэтил)пирролидин-3-илкарбамата: (3R,4R)-4-Амино-1-(2-метоксиэтил)пирролидин-3-ол (2,50 г, 15,6 ммоль), Boc2O (4,09 г, 18,7 ммоль) и ПС-ДМАП (0,191 г, 1,56 ммоль) смешали в 50 мл ДХМ и встряхивали при комнатной температуре в течение 18 часов. Реакционную смесь отфильтровали, концентрировали и очистили колоночной хроматографией на диоксиде кремния, элюируя 5-20% EtOAc в гексанах, для получения указанного в заголовке соединения (3,17 г, выход 78,0%). МС (apci) m/z=261,0 (М+Н).
Стадия F: Получение (R)-трет-бутил 1-(2-метоксиэтил)-4-оксопирролидин-3-илкарбамата: Раствор оксалилхлорида (33,51 мкл, 0,3841 ммоль) в 5 мл ДХМ охладили до -78°C и по каплям добавили ДМСО (54,52 мкл, 0,7683 ммоль). Реакционную смесь перемешивали в течение 15 минут и по каплям добавили раствор трет-бутил (3R,4R)-4-гидрокси-1-(2-метоксиэтил)пирролидин-3-илкарбамата (50 мг, 0,1921 ммоль) в 2 мл ДХМ. Реакционную смесь оставили нагреваться до -40°C в течение 1 часа, а затем охладили до -78°C. По каплям добавили триэтиламин (267,7 мкл, 1,921 ммоль) и оставили реакционную смесь нагреваться до 0°C в течение 1 часа, затем погасили водой и экстрагировали эфиром (40 мл). Органический слой высушили над MgSO4, отфильтровали и концентрировали для получения неочищенного указанного в заголовке соединения (32 мг, выход 65%). МС (apci) m/z=259,0 (М+Н).
Стадия G: Получение трет-бутил (3R,4S)-4-гидрокси-1-(2-метоксиэтил)-4-фенилпирролидин-3-илкарбамата: (R)трет-бутил 1-(2-метоксиэтил)-4-оксопирролидин-3-илкарбамат (17,0 мг, 0,0658 ммоль) растворили в ТГФ (2 мл) и охладили раствор до -78°C. По каплям добавили раствор фениллития в дибутиловом эфире (395 мкл, 0,197 ммоль) и перемешивали реакционную смесь при -78°C в течение 1 часа, а затем оставили нагреваться до комнатной температуры в течение 1 часа. Реакционную смесь вылили в насыщенный солевой раствор (10 мл) и экстрагировали несколькими порциями эфира. Объединенные органические экстракты высушили, концентрировали и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-50% ацетонитрила в воде, для получения указанного в заголовке соединения в виде примерно 4:1 смеси с трет-бутил (3R,4R)-4-гидрокси-1-(2-метоксиэтил)-4-фенилпирролидин-3-илкарбаматом (8,5 мг, выход 38%). МС (apci) m/z=337,1 (М+Н).
Стадия Н: Получение (3S,4R)-4-амино-1-(2-метоксиэтил)-3-фенилпирролидин-3-ола дигидрохлорида: К раствору продукта из Стадии G (7,0 мг, 0,0208 ммоль) в 0,1 мл изопропанола добавили раствор HCl в изопропаноле (29,7 мкл, 0,208 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем концентрировали для получения указанного в заголовке соединения в виде примерно 4:1 смеси с (3R,4R)-4-амино-1-(2-метоксиэтил)-3-фенилпирролидин-3-ола дигидрохлоридом (6,5 мг, выход 101%). МС (apci) m/z=237,1 (М+Н).
Стадия I: Получение 1-((3R,4S)-4-гидрокси-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины: Продукт из Стадии Н (6,5 мг, 0,021 ммоль) и ДИЭА (11 мкл, 0,063 ммоль) смешали в 0,5 мл ДХМ и охладили до 0°C. Добавили фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат (7,4 мг, 0,023 ммоль) и оставили реакционную смесь нагреваться до комнатной температуры в течение 1 часа. Реакционную смесь концентрировали и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-60% ацетонитрила в воде, для получения указанного в заголовке соединения (3,8 мг, выход 39%). МС (apci) m/z=462,2 (М+Н).
Пример 181
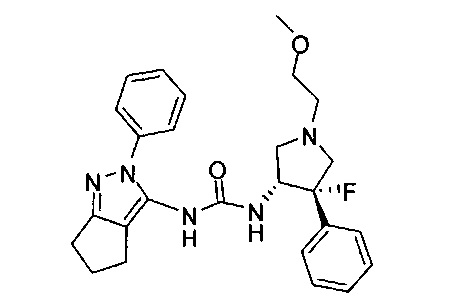
1-((3R,4S)-4-фтор-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Приблизительно 2:1 смесь 1-((3R,4S)-4-гидрокси-1-(2-метоксиэтил)-4-фенил-пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины и 1-((3R,4R)-4-гидрокси-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины (5,0 мг, 0,011 ммоль) (полученную так, как описано в Примере 177, Стадия I) растворили в ДХМ (2 мл) и охладили до -78°C. Добавили DAST (1,7 мг, 0,011 ммоль) и оставили реакционную смесь медленно нагреваться до комнатной температуры в течение ночи. Реакцию погасили МеОН, концентрировали и очистили обращенно-фазовой ЖХСД для получения указанного в заголовке соединения в виде примерно 1: 3 смеси с 1-((3R,4R)-4-фтор-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевиной (2,6 мг, выход 40%). Изомеры не разделяли. МС (apci) m/z=464,1 (М+Н).
Пример 182
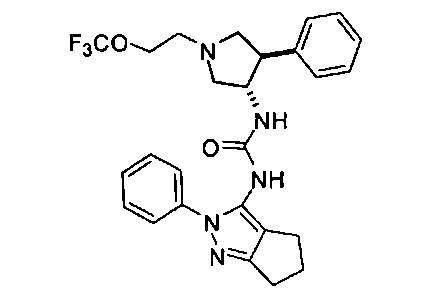
1-(транс-4-фенил-1-(2-(трифторметокси)этил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Стадия А: Получение транс-трет-бутил 3-фенил-4-(3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)уреидо)пирролидин-1-карбоксилата: К раствору транс-трет-бутил 3-амино-4-фенилпирролидин-1-карбоксилата (Получение А2; 40 мг, 0,15 ммоль) в ДМА (0,5 мл, 0,15 ммоль) добавили фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат (Пример 38, Стадия С; 58 мг, 0,18 ммоль), затем охладили на ледяной бане. К реакционной смеси добавили ДИЭА (0,080 мл, 0,46 ммоль), а затем смесь оставили нагреваться до комнатной температуры в течение ночи. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-75% ацетонитрила в воде, для получения продукта в виде белого твердого вещества (30 мг, выход 41%). МС (apci) m/z=488,0 (М+Н).
Стадия B: Получение 1-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)-3-(транс-4-фенилпирролидин-3-ил)мочевины гидрохлоридной соли: транс-трет-Бутил 3-фенил-4-(3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)уреидо)пирролидин-1-карбоксилат (30 мг, 0,062 ммоль) обработали 4 н. HCl в диоксане и перемешивали при комнатной температуре в течение 15 часов, затем концентрировали in vacuo и растерли в Et2O для получения продукта (20 мг, выход 83%). МС (apci) m/z=388,1 (М+Н).
Стадия С: Получение 1-(транс-4-фенил-1-(2-(трифторметокси)этил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины: К ДМФ (0,5 мл) раствору 1-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)-3-(транс-4-фенилпирролидин-3-ил)мочевины гидрохлоридной соли (16 мг, 0,038 ммоль) добавили N-этил-N-изопропилпропан-2-амин (21 мкл, 0,11 ммоль) и 1-бром-2-(трифторметокси)этан (8,7 мг, 0,045 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 3 часов, затем нагревали до 40°C в течение 15 часов. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-75% ацетонитрила в воде, для получения указанного в заголовке соединения (10 мг, выход 50%). МС (apci) m/z=499,9 (М+Н).
Пример 183
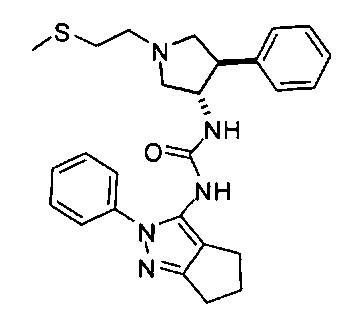
1-(транс-1-(2-(метилтио)этил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Получили так, как описано в Примере 182, заменяя 1-бром-2-(трифторметокси)этан на (2-хлорэтил)(метил)сульфан, для получения продукта (2,6 мг, выход 24%) в виде бежевого твердого вещества. МС (apci) m/z=462,1 (М+Н).
Пример 184
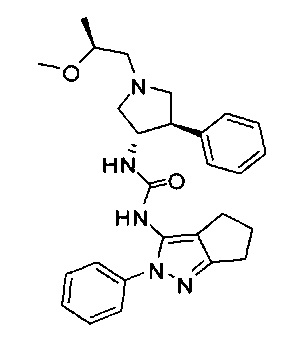
1-((3S,4R)-1-((S)-2-метоксипропил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента|с] пиразол-3-ил)мочевина
Стадия A: (S)-2-метоксипропил метансульфонат: Раствор (S)-2-метоксипропан-1-ола (451 мг, 5,00 ммоль) и ДИЭА (1,74 мл, 10,0 ммоль) в сухом CH2Cl2 (4 мл) охладили до 0°C и за 2 минуты добавили MsCl (0,406 мл, 5,25 ммоль). Смесь перемешивали в течение 3 часов, и за это время смесь достигла комнатной температуры. Смесь промыли холодной H2O, насыщенным раствором NaHCO3 и высушили над Na2SO4. Высушенный раствор отфильтровали через упакованный Celite® и концентрировали для получения указанного в заголовке продукта в виде светло-золотистого маслянистого вещества (821 мг, выход 98%). 1Н ЯМР (CDCl3) δ 4,22 (m, 1H), 4,13 (m, 1Н), 3,63 (m, 1Н), 3,40 (s, 3Н), 3,06 (s, 3Н), 1,20 (d, J=6,4 Гц, 3Н).
Стадия B: Трет-бутил (3S,4R)-1-((S)-2-метоксипропил)-4-фенилпирролидин-3-илкарбамат: К раствору трет-бутил (3S,4R)-4-фенилпирролидин-3-илкарбамата (имеется в продаже, 262 мг, 1,00 ммоль) и ДИЭА (348 мкл, 2,00 ммоль) в ДМФ (2,0 мл) добавили (S)-2-метоксипропилметансульфонат (252 мг, 1,50 ммоль). Реакционную смесь нагревали при 60°C в течение 21 часа и добавили дополнительное количество (S)-2-метоксипропил метансульфоната (84,0 мг). Реакционную смесь нагревали при 60°C в течение 2 часов, охладили до комнатной температуры и добавили к H2O (8 мл). Смесь экстрагировали EtOAc (3Х), а объединенные экстракты промыли насыщенным раствором NaCl (2Х) и высушили при помощи MgSO4. Высушенный раствор отфильтровали через слой SiO2, элюируя EtOAc. Раствор концентрировали для получения неочищенного указанного в заголовке соединения в виде светло-золотистого сиропообразного вещества (462 мг, выход 138%), которое напрямую использовали на следующей стадии. МС (apci) m/z=335,1 (М+Н).
Стадия С: (3S,4R)-1-((S)-2-метоксипропил)-4-фенилпирролидин-3-аминадигидрохлорид: К раствору неочищенного трет-бутил (3S,4R)-1-((S)-2-метоксипропил)-4-фенилпирролидин-3-илкарбамата в EtOAc (10 мл) добавили 4 М HCl в диоксане (10,0 мл, 40,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, а затем разбавили МТБЭ (50 мл). Полученный осадок собрали, промыли МТБЭ и высушили в вакууме для получения указанного в заголовке соединения в виде липкого белого вещества (276 мг, выход 90%). МС (apci) m/z=235,1 (М+Н).
Стадия D: 1-((3S,4R)-1-((S)-2-метоксипропил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина: К раствору (3S,4R)-1-((S)-2-метоксипропил)-4-фенилпирролидин-3-аминадигидрохлорида (56,2 мг, 0,240 ммоль) в сухом ДМФ (0,8 мл) добавили ДИЭА (139 мкл, 0,796 ммоль) и перемешивали смесь при комнатной температуре в течение 5 минут. Добавили фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат (Пример 38, Стадия E; 75,1 мг, 0,200 ммоль) и перемешивали смесь при комнатной температуре в течение 4 часов. Смесь добавили к H2O (5 мл) и экстрагировали EtOAc (3Х). Объединенные экстракты промыли 1 М раствором NaOH (2Х), H2O и насыщенным раствором NaCl. Раствор высушили при помощи MgSO4, отфильтровали и концентрировали in vacuo. Остаток очистили хроматографией на диоксиде кремния, элюируя EtOAc для получения указанного в заголовке соединения в виде воскообразного белого вещества (31 мг, выход 34%). МС (apci) m/z=460,1 (М+Н).
Пример 185
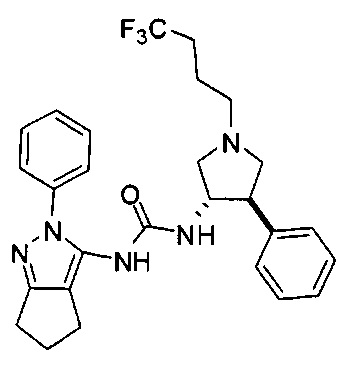
1-((3,4-транс)-4-фенил-1-(4,4,4-трифторбутил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина Получили так, как описано в Примере 182, заменяя 1-бром-2-(трифторметокси)этан на 4-бром-1,1,1-трифторбутан, для получения продукта (10 мг, выход 57%) в виде белого твердого вещества. МС (apci) m/z=498,2 (М+Н).
Пример 186
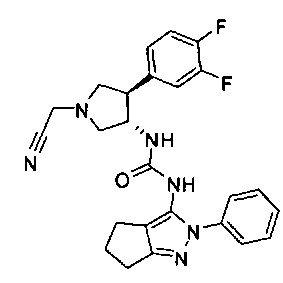
1-((3S,4R)-1-(цианометил)-4-(3,4-дифторфенил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Стадия А: Получение трет-бутил (3S,4R)-1-(цианометил)-4-(3,4-дифторфенил)пирролидин-3-илкарбамата: К раствору трет-бутил (3S,4R)-4-(3,4-дифторфенил)пирролидин-3-илкарбамата (100,0 мг, 0,3352 ммоль, приобрели у компании ACS Scientific) и ТЭА (51,39 мкл, 0,3687 ммоль) в ТГФ (1,5 мл) по каплям добавили 2-бромацетонитрил (25,68 мкл, 0,3687 ммоль). После перемешивания в течение двух часов при комнатной температуре, реакционную смесь отфильтровали и концентрировали. Неочищенный материал очистили колоночной хроматографией на диоксиде кремния, элюируя 33% EtOAc в гексанах, для получения продукта в виде белого твердого вещества (98 мг, выход 87%). МС (apci полож.) m/z=338,0 (М+Н).
Стадия B: Получение 2-((3S,4R)-3-амино-4-(3,4-дифторфенил)пирролидин-1-ил)ацетонитрила гидрохлорида: Смесь трет-бутил (3S,4R)-1-(цианометил)-4-(3,4-дифторфенил)пирролидин-3-илкарбамата (20 мг, 0,059 ммоль) в HCl (150 мкл, 0,59 ммоль, 4 н. в диоксане) перемешивали при комнатной температуре в течение 15 минут, затем концентрировали in vacuo, растерли с эфиром и высушили под высоким вакуумом для получения продукта в виде белого твердого вещества (16 мг, выход 99%). МС (apci полож.) m/z=238,0 (М+Н).
Стадия C: Получение 1-((3S,4R)-1-(цианометил)-4-(3,4-дифторфенил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины: К прозрачному раствору 2-((3S,4R)-3-амино-4-(3,4-дифторфенил)пирролидин-1-ил)ацетонитрила гидрохлорида (16 мг, 0,058 ммоль) и фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамата (19 мг, 0,058 ммоль) в ДМА (0,5 мл) по каплям добавили ДИЭА (0,041 мл, 0,23 ммоль) при комнатной температуре. После перемешивания в течение 1 часа, реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-55% ацетонитрила в воде, для получения продукта в виде белого твердого вещества (18 мг, выход 65%). МС (apci полож.) m/z=463,0 (М+Н).
Пример 187
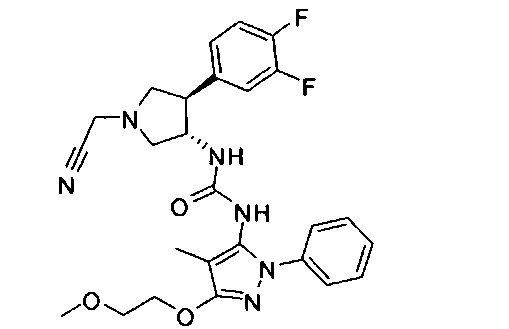
1-((3S,4R)-1-(цианометил)-4-(3,4-дифторфенил)пирролидин-3-ил)-3-(3-(2-метоксиэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
К прозрачному раствору 2-((3S,4R)-3-амино-4-(3,4-дифторфенил)пирролидин-1-ил)ацетонитрила гидрохлорида (Пример 186, Стадия B, 10 мг, 0,037 ммоль) и фенил 3-(2-метоксиэтокси)-4-метил-1-фенил-1Н-пиразол-5-илкарбамата (13 мг, 0,037 ммоль) в ДМА (180 мкл, 0,037 ммоль) по каплям добавили ДИЭА (32 мкл, 0,18 ммоль) при комнатной температуре. Реакционную смесь быстро нагрели до 60°C (примерно за 1 минуту), затем охладили до комнатной температуры и перемешивали в течение 20 минут. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-65% ацетонитрила в воде, для получения продукта в виде белого твердого вещества (12 мг, выход 64%). МС (apci полож.) m/z=511,1 (М+Н).
Пример 188
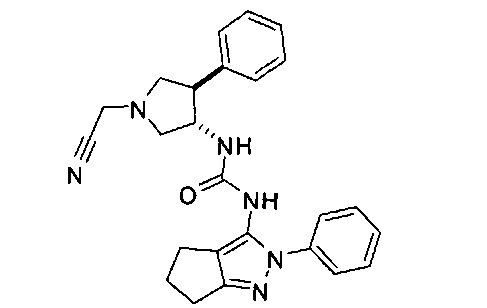
1-((3,4-транс)-1-(цианометил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с] пиразол-3-ил)мочевина
Стадия А: Получение трет-бутил (3,4-транс)-1-(цианометил)-4-фенилпирролидин-3-илкарбамата: К раствору трет-бутил (3,4-транс)-4-фенилпирролидин-3-илкарбамата (206,0 мг, 0,7852 ммоль, Получение А) и ТЭА (120,4 мкл, 0,8637 ммоль) в ТГФ (3 мл) по каплям добавили 2-бромацетонитрил (60,16 мкл, 0,8637 ммоль). После перемешивания при комнатной температуре в течение ночи, реакционную смесь отфильтровали и концентрировали для получения продукта в виде белого твердого вещества (230 мг, выход 97%). МС (apci полож.) m/z=302,1 (М+Н).
Стадия В: Получение 2-((3,4-транс)-3-амино-4-фенилпирролидин-1-ил)ацетонитрила гидрохлорида: Смесь трет-бутил (3,4-транс)-1-(цианометил)-4-фенилпирролидин-3-илкарбамата (230 мг, 0,763 ммоль) и HCl (4770 мкл, 19,1 ммоль, 4 н. в диоксане) перемешивали при комнатной температуре в течение 2 часов, затем концентрировали in vacuo, обработали эфиром и высушили под высоким вакуумом для получения продукта в виде бледно-желтоватого твердого вещества (180 мг, выход 99%). МС (apci полож.) m/z=202,1 (М+Н).
Стадия С: Получение 1-((3,4-транс)-1-(цианометил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины: К прозрачному раствору 2-(3,4-транс)-3-амино-4-фенилпирролидин-1-ил)ацетонитрила гидрохлорида (33 мг, 0,14 ммоль) и фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамата (40 мг, 0,13 ммоль) в ДМА (630 мкл) по каплям добавили ДИЭА (110 мкл, 0,63 ммоль) при комнатной температуре. После перемешивания при комнатной температуре в течение ночи, реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-55% ацетонитрила в воде, для получения продукта в виде белого твердого вещества (45 мг, выход 84%). МС (apci полож.) m/z=427,1 (М+Н).
Пример 189
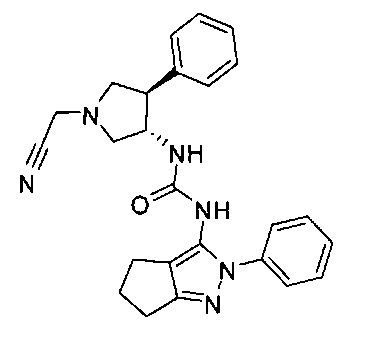
1-((3S,4R)-1-(цианометил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Стадия А: Получение трет-бутил (3S,4R)-1-(цианометил)-4-фенилпирролидин-3-илкарбамата: К раствору трет-бутил (3S,4R)-4-фенилпирролидин-3-илкарбамата (535 мг, 2,04 ммоль, приобрели у компании ACS Scientific) и ТЭА (313 мкл, 2,24 ммоль) в ТГФ (8 мл) по каплям добавили 2-бромацетонитрил (156 мкл, 2,24 ммоль). После перемешивания при комнатной температуре в течение ночи, реакционную смесь отфильтровали и концентрировали in vacuo. Неочищенный материал очистили колоночной хроматографией на диоксиде кремния, элюируя 50% гексанов в EtOAc, для получения продукта в виде белого твердого вещества (510 мг, выход 83%). МС (apci полож.) m/z=302,0 (М+Н).
Стадия В: Получение 2-((3S,4R)-3-амино-4-фенилпирролидин-1-ил)ацетонитрила гидрохлорида: Смесь трет-бутил (3S,4R)-1-(цианометил)-4-фенилпирролидин-3-илкарбамата (490 мг, 1,63 ммоль) и HCl (20 мл, 80 ммоль, 4 н. в диоксане) перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали in vacuo, растерли с эфиром и высушили под высоким вакуумом для получения бледно-желтоватого твердого вещества. Данные ЖХМС показали, что это смесь двух продуктов: 2-((3S,4R)-3-амино-4-фенилпирролидин-1-ил)ацетонитрила гидрохлорида и 2-((3S,4R)-3-амино-4-фенилпирролидин-1-ил)ацетамида гидрохлорида с соотношением приблизительно 1:2; МС (apci полож.) m/z=202,1 и 220,1 (М+Н), соответственно. Эту смесь двух продуктов использовали напрямую на следующей стадии без дополнительной очистки.
Стадия С: Получение 1-((3S,4R)-(цианометил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,45,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины: К прозрачному раствору неочищенного продукта Стадии В (39 мг, 0,16 ммоль) в ДМА (420 мкл, 0,13 ммоль) добавили фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат (40 мг, 0,13 ммоль), затем ДИЭА (110 мкл, 0,63 ммоль) при комнатной температуре, и перемешивали реакционную смесь в течение 18 часов. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-54% ацетонитрила в воде, для получения указанного в заголовке продукта в виде белого твердого вещества (11 мг, выход 21%). МС (apci полож.) m/z=427,1 (М+Н).
Пример 190
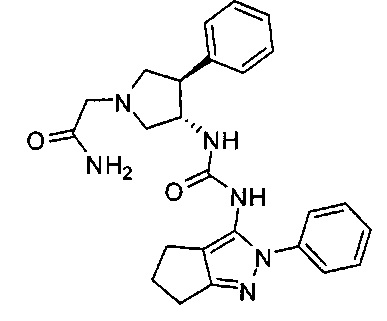
2-((3R,4S)-3-фенил-4-(3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)уреидо)пирролидин-1-ил)ацетамид
К прозрачному раствору неочищенного продукта Примера 189, Стадия В, содержащему 2-((3S,4R)-3-амино-4-фенилпирролидин-1-ил)ацетамида гидрохлорид (39 мг, 0,16 ммоль), в ДМА (420 мкл, 0,13 ммоль) добавили фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат (40 мг, 0,13 ммоль), затем ДИЭА (110 мкл, 0,63 ммоль) при комнатной температуре, и перемешивали реакционную смесь в течение 18 часов. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-54% ацетонитрила в воде, для получения указанного в заголовке продукта в виде белого твердого вещества (20 мг, выход 36%). МС (apci полож.) m/z=445,1 (М+Н).
Пример 191
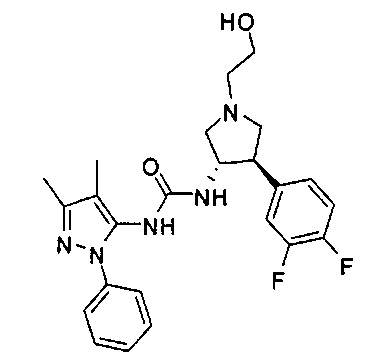
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-гидроксиэтил)пирролидин-3-ил)-3-(3,4-диметил-1-фенил-1Н-пиразол-5-ил)мочевина
Стадия А: Получение трет-бутил (3S,4R)-4-(3,4-дифторфенил)-1-(2-гидроксиэтил)пирролидин-3-илкарбамата: Трет-бутил (3S,4R)-4-(3,4-дифторфенил)пирролидин-3-илкарбамат (ACS Scientific, 410 мг, 1,37 ммоль), 2-бромэтанол (180 мг, 1,44 ммоль) и ДИЭА (533 мг, 4,12 ммоль) смешали в 1 мл ДМФ и перемешивали при комнатной температуре в течение 72 часов. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 0-50% ацетонитрила в воде, для получения указанного в заголовке соединения (290 мг, выход 61,6%). МС (apci) m/z=343,0 (М+Н).
Стадия В: Получение 2-((3S,4R)-3-амино-4-(3,4-дифторфенил)пирролидин-1-ил)этанола гидрохлорида: Трет-бутил (3S,4R)-4-(3,4-дифторфенил)-1-(2-гидроксиэтил)пирролидин-3-илкарбамат (230 мг, 0,672 ммоль) и хлороводород в изопропаноле (480 мкл, 3,36 ммоль) смешали и перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь концентрировали для получения указанного в заголовке соединения (166 мг, выход 102%). МС (apci) m/z=243,0 (М+Н).
Стадия C: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-гидроксиэтил)пирролидин-3-ил)-3-(3,4-диметил-1-фенил-1Н-пиразол-5-ил)мочевины: 2-((3S,4R)-3-Амино-4-(3,4-дифторфенил)пирролидин-1-ил)этанола гидрохлорид (160 мг, 0,574 ммоль), фенил 3,4-диметил-1-фенил-1Н-пиразол-5-илкарбамат (168 мг, 0,547 ммоль) и ДИЭА (286 мкл, 1,64 ммоль) смешали в 0,5 мл ДМФ и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 0-50% ацетонитрила в воде, для получения указанного в заголовке соединения (127 мг, выход 51,0%). МС (apci) m/z=456,0 (М+Н).
Пример 192
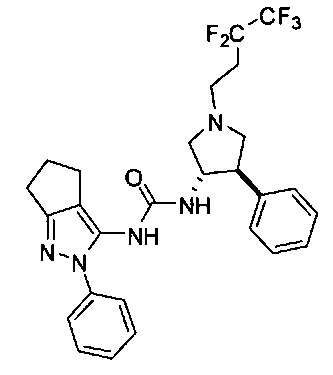
1-((транс)-1-(3,3,4,4,4-пентафторбутил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
1-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)-3-((транс)-4-фенилпирролидин-3-ил)мочевину (Пример 182, Стадия В, 8,0 мг, 0,021 ммоль), 1,1,1,2,2-пентафтор-4-йодбутан (5,7 мг, 0,021 ммоль) и ДИЭА (3,6 мкл, 0,021 ммоль) смешали в 0,1 мл ДМФ и перемешивали при 60°C в течение 2 часов. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 0-65% ацетонитрила в воде, для получения указанного в заголовке соединения (3,2 мг, выход 29%). МС (apci) m/z=534,1 (М+Н).
Пример 193
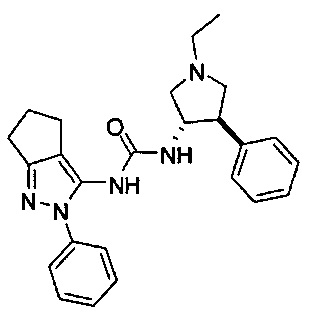
1-((транс)-1-этил-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
1-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)-3-((транс)-4-фенилпирролидин-3-ил)мочевину (Пример 182, Стадия B, 5,0 мг, 0,012 ммоль), бромэтан (1,3 мг, 0,012 ммоль) и ДИЭА (4,1 мкл, 0,024 ммоль) смешали в 0,1 мл ДМФ и перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 0-65% ацетонитрила в воде, для получения указанного в заголовке соединения (4,3 мг, выход 88%). МС (apci) m/z=416,1 (М+Н).
Пример 194
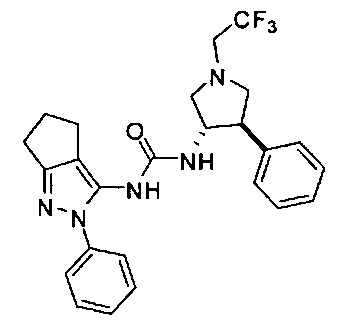
1-((транс)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с] пиразол-3-ил)мочевина
Стадия А: Получение трет-бутил (транс)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-илкарбамата: Транс-трет-бутил-4-фенилпирролидин-3-илкарбамат (1,00 г, 3,81 ммоль), 2,2,2-трифторэтил трифторметансульфонат (1,06 г, 4,57 ммоль) и ДИЭА (1,48 г, 11,4 ммоль) смешали в 2 мл ДМФ и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 0-75% ацетонитрила в воде, для получения указанного в заголовке соединения (1,19 мг, выход 90,7%). МС (apci) m/z=345,0 (М+Н).
Стадия В: Получение (транс)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-амина гидрохлорида: Трет-бутил (транс)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-илкарбамат (1,19 г, 3,46 ммоль) и HCl (5 н. в изопропаноле, 1,48 мл, 10,4 ммоль) смешали и перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь концентрировали для получения указанного в заголовке соединения (0,85 г, выход 101%). МС (apci) m/z=245,0 (М+Н).
Стадия С: Получение 1-((транс)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-етрагидроциклопента[c]пиразол-3-ил)мочевины: (транс)-4-Фенил-1-(2,2,2-трифтор-этил)пирролидин-3-амина гидрохлорид (10,0 мг, 0,0356 ммоль), фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат (9,48 мг, 0,0297 ммоль) и ДИЭА (15,5 мкл, 0,0891 ммоль) смешали в 0,2 мл ДМФ и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 0-75% ацетонитрила в воде, для получения указанного в заголовке соединения (11,5 мг, выход 82,5%). МС (apci) m/z=470,0 (М+Н).
Пример 195
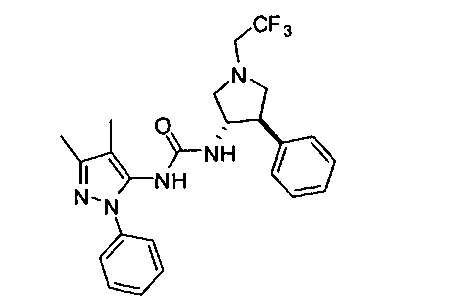
1-(3,4-диметил-1-фенил-1Н-пиразол-5-ил)-3-((транс)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному в Примере 194, заменяя фенил 3,4-диметил-1-фенил-1Н-пиразол-5-илкарбаматом фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат на Стадии С. Материал очистили обращенно-фазовой колоночной хроматографией, элюируя 0-70% ацетонитрила в H2O, для получения указанного в заголовке соединения (6,5 мг, выход 48%). МС (apci) m/z=458,1 (М+Н).
Пример 196
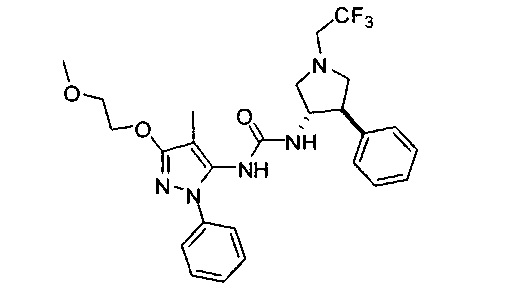
1-(3-(2-метоксиэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((транс)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному в Примере 194, заменяя фенил 3-(2-метоксиэтокси)-4-метил-1-фенил-1Н-пиразол-5-илкарбаматом фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат на Стадии С. Материал очистили обращенно-фазовой колоночной хроматографией, элюируя 0-70% ацетонитрила в H2O, для получения указанного в заголовке соединения (9,3 мг, выход 61%). МС (apci) m/z=518,1 (М+Н).
Пример 197
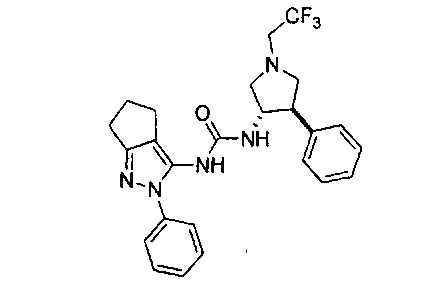
1-((транс)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Получили по способу, описанному в Примере 194, Стадии А-С, используя трет-бутил (3S,4R)-4-фенилпирролидин-3-илкарбамат (приобретенный у компании ACS Scientific, № по каталогу 3-1005) вместо транс-трет-бутил-4-фенилпирролидин-3-илкарбамата на Стадии А. Конечный продукт очистили обращенно-фазовой колоночной хроматографией, элюируя 0-60% ацетонитрила в H2O, для получения указанного в заголовке соединения (7,2 мг, выход 52%). МС (apci) m/z=470,0 (М+Н).
Пример 198
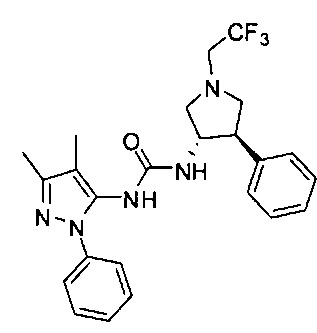
1-(3,4-диметил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному в Примере 197, заменяя фенил 3,4-диметил-1-фенил-1Н-пиразол-5-илкарбаматом фенил 2-фенил-2,4,5,6-тетрагидро-циклопента[с]пиразол-3-илкарбамат на Стадии С. Материал очистили обращенно-фазовой колоночной хроматографией, элюируя 0-60% ацетонитрила в Н2О, для получения указанного в заголовке соединения (10,0 мг, выход для образования мочевины 67,5%). МС (apci) m/z=458,0 (М+Н).
Пример 199
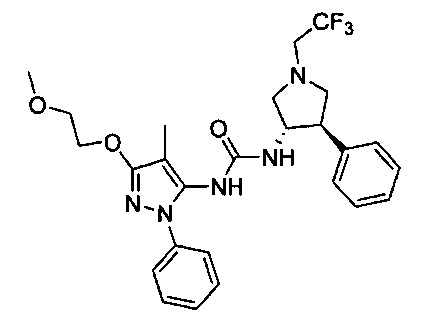
1-(3-(2-метоксиэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному в Примере 197, заменяя фенил 3-(2-метоксиэтокси)-4-метил-1-фенил-1Н-пиразол-5-илкарбаматом фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат. Материал очистили обращенно-фазовой колоночной хроматографией, элюируя 0-70% ацетонитрила в H2O, для получения указанного в заголовке соединения (8,9 мг, выход 53%). МС (apci) m/z=518,1 (М+Н).
Пример 200
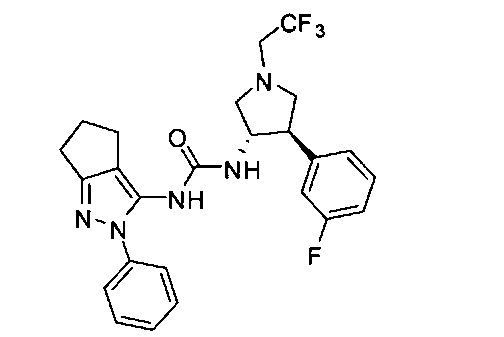
1-((3S,4R)-4-(3-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Получили по способу, описанному в Примере 194, Стадии А-С, используя трет-бутил (3S,4R)-4-(3-фторфенил)пирролидин-3-илкарбамат (приобретенный у компании ACS Scientific, № по каталогу 3-1029) вместо транс-трет-бутил-4-фенилпирролидин-3-илкарбамата на Стадии А. Конечный продукт очистили обращенно-фазовой колоночной хроматографией, элюируя 0-75% ацетонитрила в Н2О, для получения указанного в заголовке соединения (12 мг, выход 92%). МС (apci) m/z=488,1 (М+Н).
Пример 201
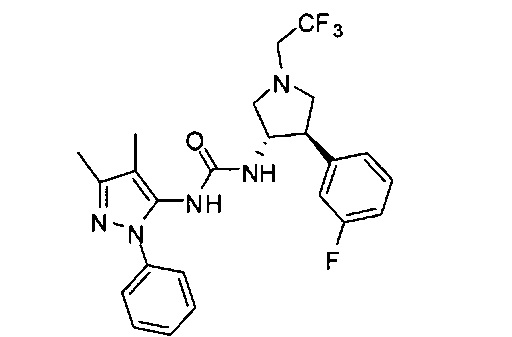
1-(3,4-диметил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному в Примере 200, заменяя фенил 3,4-диметил-1-фенил-1Н-пиразол-5-илкарбаматом фенил 2-фенил-2,4,5,6-тетрагидро-циклопента[с]пиразол-3-илкарбамат на Стадии С. Материал очистили обращенно-фазовой колоночной хроматографией, элюируя 5-80% ацетонитрила в Н2О, для получения указанного в заголовке соединения (5,3 мг, выход 48%). МС (apci) m/z=476,0 (М+Н).
Пример 202
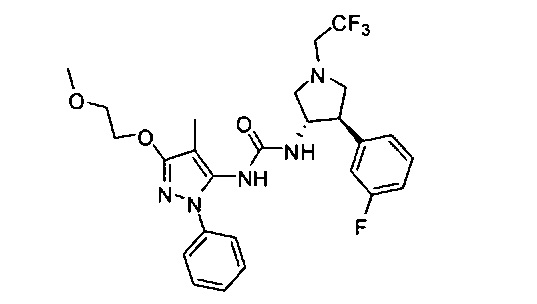
1-((3S,4R)-4-(3-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(3-(2-метоксиэтоксиУ4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Получили по способу, описанному в Примере 200, заменяя фенил 3-(2-метоксиэтокси)-4-метил-1-фенил-1Н-пиразол-5-илкарбаматом фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат. Материал очистили обращенно-фазовой колоночной хроматографией, элюируя 5-80% ацетонитрила в H2O, для получения указанного в заголовке соединения (6,6 мг, выход 58%). МС (apci) m/z=536,1 (М+Н).
Пример 203
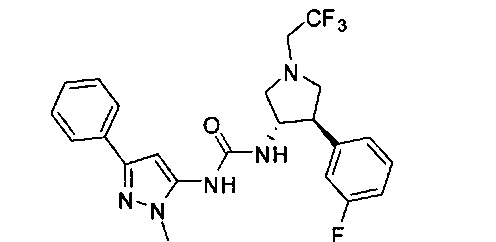
1-((3S,4R)-4-(3-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(1-метил-3-фенил-1Н-пиразол-5-ил)мочевина
Получили по способу, описанному в Примере 200, заменяя фенил 1-метил-3-фенил-1Н-пиразол-5-илкарбаматом фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат. Материал очистили обращенно-фазовой колоночной хроматографией, элюируя 0-75% ацетонитрила в H2O, для получения указанного в заголовке соединения (6,3 мг, выход 64%). МС (apci) m/z=462,0 (М+Н).
Пример 204
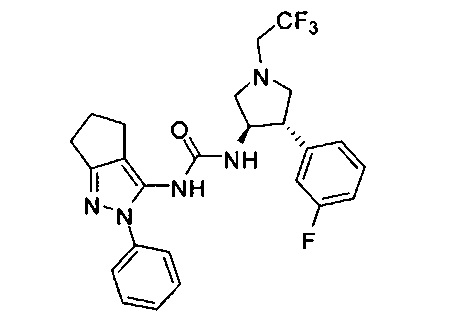
1-((3R,4S)-4-(3-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Стадия А: Получение (Е)-3-(4-хлор-3-фторфенил)акрилоилхлорида: (Е)-3-(4-хлор-3-фторфенил)акриловую кислоту (28,1 г, 140 ммоль) суспендировали в хлороформе (250 мл) и добавили ДМФ (0,1 мл), затем оксалилхлорид (20,0 мл, 229 ммоль). Реакционную смесь перемешивали в течение 18 часов, а затем выпарили до сухости. Добавили гептан и концентрировали смесь для получения продукта (30,6 г, выход 99,7%) в виде бледно-желтого твердого вещества.
Стадия B: Получение (R)-4-бензил-3-((3R,4S)-1-бензил-4-(4-хлор-3-фторфенил)пирролидин-3-карбонил)оксазолидин-2-она: (R)-4-бензилоксазолидин-2-он (21,8 г, 123 ммоль) растворили в ТГФ (600 мл) и охладили до -78°C. По каплям добавили бис(триметилсилил)амид лития в ТГФ (127 мл, 127 ммоль) за 15 минут, и перемешивали смесь еще 15 минут при -78°C. Добавили раствор (Е)-3-(4-хлор-3-фторфенил)акрилоилхлорида (28,3 г, 129 ммоль) в ТГФ (100 мл) и перемешивали смесь в течение 1 часа при -78°C, затем оставили нагреваться до комнатной температуры и перемешивали еще один час. Добавили насыщенный водный раствор бикарбоната натрия (50 мл) и перемешивали реакционную смесь в течение 1 часа. ТГФ удалили in vacuo, добавили этилацетат (1 л) и промыли реакционную смесь водой (2х) и насыщенным солевым раствором, высушили при помощи MgSO4, отфильтровали и выпарили для получения (R,E)-4-бензил-3-(3-(4-хлор-3-фторфенил)акрилоил)оксазолидин-2-она (44,3 г, выход 100%) в виде желто-коричневого твердого вещества. Это твердое вещество растворили в толуоле (500 мл) и добавили 2,2,2-трифторуксусную кислоту (0,9486 мл, 12,31 ммоль). Температуру повысили до 35°C и за 20 минут добавили N-бензил-1-метокси-N-((триметилсилил)метил)метанамин (52,50 мл, 184,7 ммоль), поддерживая температуру при 25-30°C при помощи внешней водяной бани. Смесь промыли насыщенным водным раствором бикарбоната натрия и концентрировали in vacuo для получения маслянистого остатка, который растерли с гексанами, отфильтровали и промыли гексанами для получения белого твердого вещества (55,7 г). Это твердое вещество (55,7 г) суспендировали в гексанах (200 мл) и нагревали с дефлегматором. Добавляли бензол (220 мл) до растворения твердого вещества при дефлегмации. Раствор оставили медленно охлаждаться до комнатной температуры, а затем поместили в морозильную камеру на 4 часа. Полученное твердое вещество собрали фильтрацией и промыли 100 мл холодной смеси 1:1 гексана/бензола. Полученное твердое вещество (7,6 г) очистили колоночной хроматографией на диоксиде кремния, элюируя 20-40%) EtOAc в гексанах. Первым элюировали (R)-4-бензил-3-((3S,4R)-1-бензил-4-(4-хлор-3-фторфенил)пирролидин-3-карбонил)оксазолидин-2-он (3,2 г, выход 42%), затем (R)-4-бензил-3-((3R,4S)-1-бензил-4-(4-хлор-3-фторфенил)пирролидин-3-карбонил)оксазолидин-2-он (3,0 г, выход 39%). МС (apci) m/z=493,0 (М+Н).
Стадия С: Получение (3R,4S)-1-бензил-4-(4-хлор-3-фторфенил)пирролидин-3-карбоновой кислоты: Пероксид водорода (30%, водный, 2,38 мл, 23,1 ммоль) по каплям добавили к смеси гидроксида лития моногидрата (0,638 г, 15,2 ммоль) и ледяной воды (50 мг). Смесь перемешивали в течение 30 минут, а полученный раствор добавили к раствору (R)-4-бензил-3-((3R,4S)-1-бензил-4-(4-хлор-3-фторфенил)пирролидин-3-карбонил)оксазолидин-2-она (3,00 г, 6,09 ммоль) в ТГФ (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 5 часов, погасили добавлением 2 М водного раствора Na2SO3 (20 мл) и перемешивали в течение ночи. pH довели до 6 добавлением твердого KHSO4 и экстрагировали смесь EtOAc (2×100 мл). Объединенные органические экстракты промыли насыщенным солевым раствором, высушили при помощи Mg2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 10% МеОН в ДХМ, для получения продукта (1,80 г, выход 88,6%) в виде белого твердого вещества. МС (apci) m/z=334,0 (М+Н).
Стадия D: Получение трет-бутил (3R,4S)-1-бензил-4-(4-хлор-3-фторфенил)пирролидин-3-илкарбамата: (3R,4S)-1-бензил-4-(4-хлор-3-фторфенил)пирролидин-3-карбоновую кислоту (120 мг, 0,360 ммоль), NEt3 (150 мкл, 1,08 ммоль) и дифенилфосфорилазид (116 мкл, 0,539 ммоль) смешали в 2 мл толуола в закрытой емкости и перемешивали при 100°C в течение 30 минут. Реакционную смесь оставили остывать до комнатной температуры и добавили 2-метилпропан-2-олат лития в ТГФ (1,44 мл, 0,719 ммоль). Реакционную смесь перемешивали при 100°C в течение 5 часов, охладили, концентрировали и очистили обращенно-фазовой колоночной хроматографией, элюируя 20-90%) ацетонитрила в Н2О, для получения продукта (61,0 мг, выход 41,9%). МС (apci) m/z=405,0 (М+Н).
Стадия Е: Получение трет-бутил (3R,4S)-4-(3-фторфенил)пирролидин-3-илкарбамата: трет-бутил(3R,4S)-1-бензил-4-(4-хлор-3-фторфенил)пирролидин-3-илкарбамат (60 мг, 0,15 ммоль) растворили в 5 мл МеОН и добавили 10%) Pd/C (32 мг, 0,030 ммоль). Реакционную смесь перемешивали под водородом из баллона в течение 18 часов, отфильтровали через Celite®, а затем концентрировали для получения указанного в заголовке соединения (36 мг, выход 87%). МС (apci) m/z=281,1 (М+Н).
Стадия F: Получение трет-бутил (3R,4S)-4-(3-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-илкарбамата: Трет-бутил (3R,4S)-4-(3-фторфенил)пирролидин-3-илкарбамат (35 мг, 0,12 ммоль), 2,2,2-трифторэтилтрифторметансульфонат (38 мг, 0,16 ммоль) и ДИЭА (65 мкл, 0,37 ммоль) смешали в 0,5 мл ДМФ и перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 0-80% ацетонитрила в H2O, для получения продукта (30 мг, выход 66%). МС (apci) m/z=363,0 (М+Н).
Стадия G: Получение (3R,4S)-4-(3-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-амина гидрохлорида: Трет-бутил (3R,4S)-4-(3-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-илкарбамат (30 мг, 0,083 ммоль) и хлороводород в изопропаноле (50 мкл, 0,25 ммоль) смешали в 1 мл изопропанола и перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь концентрировали in vacuo для получения продукта (21 мг, выход 97%). МС (apci) m/z=263,0 (М+Н).
Стадия Н: Получение 1-((3R,4S)-4-(3-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины: (3R,4S)-4-(3-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-амин (10,0 мг, 0,0381 ммоль), фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат (10,1 мг, 0,0318 ммоль) и ДИЭА (16,6 мкл, 0,0953 ммоль) смешали в 0,2 мл ДМФ и перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь очистили обращенно-фазовой колоночной хроматографией, элюируя 0-70% ацетонитрила в H2O, для получения указанного в заголовке соединения (12,5 мг, выход 80,7%). МС (apci) m/z=488,1 (М+Н).
Пример 205
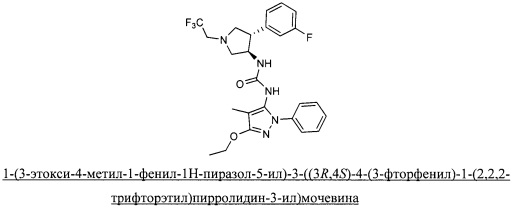
Получили по способу, описанному в Примере 204, Стадия Н, заменяя фенил 3-этокси-4-метил-1-фенил-1Н-пиразол-5-илкарбаматом фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат. Материал очистили обращенно-фазовой колоночной хроматографией, элюируя 0-80% ацетонитрила в Н2О, для получения указанного в заголовке соединения (9,3 мг, выход 58%). МС (apci) m/z=476,0 (М+Н).
Пример 206
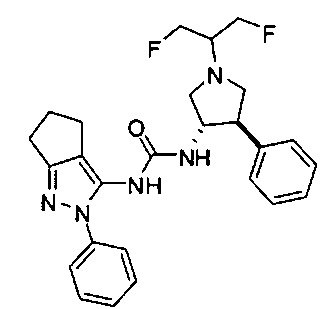
1-((транс)-1-(1,3-дифторпропан-2-ил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента [с]пиразол-3-ил)мочевина
Стадия A: Получение трет-бутил (транс)-1-(1,3-дифторпропан-2-ил)-4-фенил-пирролидин-3-илкарбамата; 1,3-Дифторпропан-2-ол (150,0 мг, 1,561 ммоль) растворили в ДХМ (5 мл) и охладили раствор до 0°C. Добавили ДИЭА (339,9 мкл, 1,952 ммоль), затем трифторметансульфоновый ангидрид (197,0 мкл, 1,171 ммоль). Реакционную смесь перемешивали при 0°C в течение 1 часа и добавили трет-бутил 4-фенилпирролидин-3-илкарбамат (102,4 мг, 0,3903 ммоль). Реакционную смесь оставили нагреваться до комнатной температуры в течение 2 часов, концентрировали, загрузили на картридж Samplet при помощи МеОН и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-70% ацетонитрила в H2O, для получения продукта (119,0 мг, выход 89,56%). МС (apci) m/z=341,1 (М+Н).
Стадия В: Получение (транс)-1-(1,3-дифторпропан-2-ил)-4-фенилпирролидин-3-амина гидрохлорида: Трет-бутил (транс)-1-(1,3-дифторпропан-2-ил)-4-фенилпирролидин-3-илкарбамат (119 мг, 0,350 ммоль) и хлороводород (5 н. в изопропаноле, 210 мкл, 1,05 ммоль) смешали в 1 мл изопропанола и перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь концентрировали in vacuo для получения продукта (85,0 мг, выход 101%). МС (apci) m/z=241,1 (М+Н).
Стадия С: Получение 1-((транс)-1-(1,3-дифторпропан-2-ил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-етрагидроциклопента[с]пиразол-3-ил)мочевины:(транс)-1-(1,3-Дифторпропан-2-ил)-4-фенилпирролидин-3-амина гидрохлорид (10,0 мг, 0,0361 ммоль), фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат (9,62 мг, 0,0301 ммоль) и ДИЭА (15,7 мкл, 0,0903 ммоль) смешали в 0,2 мл ДМФ и перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь очистили обращенно-фазовой колоночной хроматографией, элюируя 0-70% ацетонитрила в H2O, для получения продукта (11,3 мг, выход 80,6%). МС (apci) m/z=466,1 (М+Н).
Пример 207
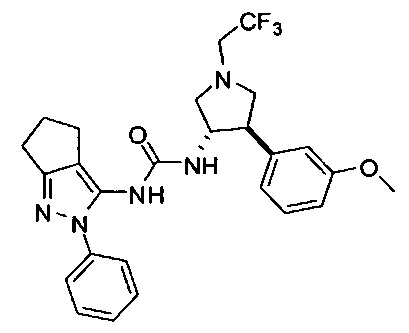
(транс)-трет-бутил 3-(3-метоксифенил)-4-(3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)уреидо)пирролидин-1-карбоксилат
Стадия А: Получение трет-бутил (транс)-1-(1,3-дифторпропан-2-ил)-4-фенилпирролидин-3-илкарбамата: Транс-трет-бутил 3-амино-4-(3-метоксифенил)пирролидин-1-карбоксилат (100,0 мг, 0,3420 ммоль), фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат (99,30 мг, 0,3109 ммоль) и ДИЭА (162,5 мкл, 0,9328 ммоль) смешали в 0,2 мл ДМФ и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь очистили обращенно-фазовой колоночной хроматографией, элюируя 0-80% ацетонитрила в H2O, для получения продукта (102,0 мг, выход 63,38%). МС (apci) m/z=518,1 (М+Н).
Стадия В: Получение 1-((транс)-4-(3-метоксифенил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины гидрохлорида: (Транс)-трет-бутил 3-(3-метокси-фенил)-4-(3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)уреидо)пиррлидин-1-карбоксилат (102 мг, 0,197 ммоль) и хлороводород в изопропаноле (118 мкл, 0,591 ммоль) смешали в 1 мл изопропанола и перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь концентрировали in vacuo для получения продукта (80,0 мг, выход 97,2%). МС (apci) m/z=418,1 (М+Н).
Стадия С: Получение 1-((транс)-4-(3-метоксифенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины: 1-((транс)-4-(3-метоксифенил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины гидрохлорид (8,0 мг, 0,018 ммоль), 2,2,2-трифторэтил трифторметансульфонат (6,1 мг, 0,026 ммоль) и ДИЭА (9,2 мкл, 0,053 ммоль) смешали в 0,2 мл ДМФ и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 0-80% ацетонитрила в H2O, для получения указанного в заголовке соединения (6,7 мг, выход 76%). МС (apci) m/z=500,1 (М+Н).
Пример 208
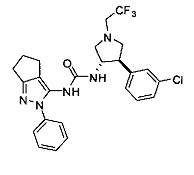
1-((транс)-4-(3-хлорфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Стадия А: Получение (транс)-трет-бутил 3-(3-хлорфенил)-4-(3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)уреидо)пирролидин-1-карбоксилата: (транс)-1-(трет-Бутокси-карбонил)-4-(3-хлорфенил)пирролидин-3-карбоновую кислоту (150 мг, 0,460 ммоль), NEt3 (193 мкл, 1,38 ммоль) и дифенилфосфорилазид (149 мкл, 0,691 ммоль) смешали в 2 мл толуола в закрытой емкости и перемешивали при 100°C в течение 30 минут. Реакционную смесь охладили и добавили 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-амин (Таблица 1; 183 мг, 0,921 ммоль). Реакционную смесь перемешивали при 100°C в течение 16 часов, охладили, концентрировали и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-60% ацетонитрила в Н2О, для получения продукта (42 мг, выход 18%)). МС (apci) m/z=522,1 (М+Н).
Стадия В: Получение 1-((транс)-4-(3-хлорфенил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента|с]пиразол-3-ил)мочевины гидрохлорида: (транс)-трет-бутил 3-(3-хлор-фенил)-4-(3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)уреидо)пирролидин-1-карбоксилат (40 мг, 0,077 ммоль) и хлороводород в изопропаноле (46 мкл, 0,23 ммоль) смешали в 10,1 мл IPA и перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали in vacuo для получения продукта (32 мг, выход 99%). МС (apci) m/z=422,0 (М+Н).
Стадия С: Получение 1-((транс)-4-(3-хлорфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины: 1-((транс)-4-(3-Хлорфенил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины гидрохлорид (15 мг, 0,033 ммоль), 2,2,2-трифторэтил трифторметансульфонат (11 мг, 0,049 ммоль) и ДИЭА (17 мкл, 0,098 ммоль) смешали в 0,2 мл ДМФ и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 0-80% ацетонитрила в Н2О, для получения указанного в заголовке соединения (8,2 мг, выход 50%). МС (apci) m/z=504,0 (М+Н).
Пример 209
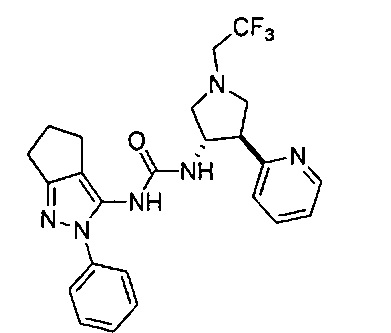
1-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)-3-((транс)-4-(пиридин-2-ил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному в Примере 208, заменяя (транс)-1-(трет-бутоксикарбонил)-4-(2-пиридил)пирролидин-3-карбоновой кислотой (транс)-1-(трет-бутоксикарбонил)-4-(3-хлорфенил)пирролидин-3-карбоновую кислоту на Стадии А. Неочищенный конечный продукт очистили обращенно-фазовой колоночной хроматографией, элюируя 0-60% ацетонитрила в Н2О, для получения указанного в заголовке соединения (4,2 мг, выход 23% за 3 стадии). МС (apci) m/z=471,1 (М+Н).
Пример 210
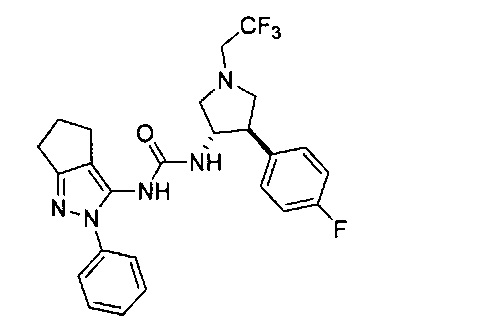
1-((транс)-4-(4-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Получили по способу, описанному в Примере 208, заменяя (транс)-1-(трет-бутоксикарбонил)-4-(4-фторфенил)пирролидин-3-карбоновой кислотой (транс)-1-(трет-бутоксикарбонил)-4-(3-хлорфенил)пирролидин-3-карбоновую кислоту на Стадии А. Конечный продукт очистили обращенно-фазовой колоночной хроматографией, элюируя 0-70% ацетонитрила в H2O, для получения указанного в заголовке соединения (10,0 мг, выход 65% за 3 стадии). МС (apci) m/z=488,2 (М+Н).
Пример 211
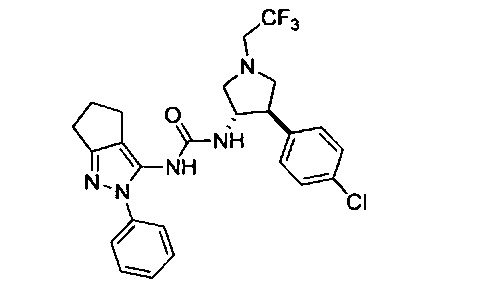
1-((транс)-4-(4-хлорфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Получили по способу, описанному в Примере 208, используя (транс)-1-(трет-бутоксикарбонил)-4-(4-хлорфенил)пирролидин-3-карбоновую кислоту вместо (транс)-1-(трет-бутоксикарбонил)-4-(3-хлорфенил)пирролидин-3-карбоновой кислоты на Стадии А. Конечный продукт очистили обращенно-фазовой колоночной хроматографией, элюируя 0-70% ацетонитрила в H2O, для получения указанного в заголовке соединения (7,3 мг, выход 14% за 3 стадии). МС (apci) m/z=504,2 (М+Н).
Пример 212
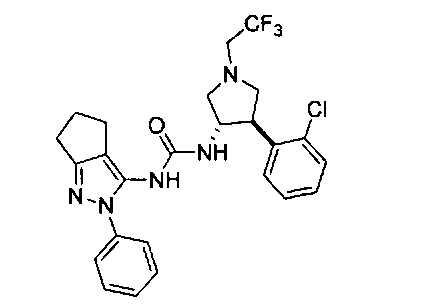
1-((транс)-4-(2-хлорфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Получили по способу, описанному в Примере 208, заменяя (транс)-1-(трет-бутоксикарбонил)-4-(2-хлорфенил)пирролидин-3-карбоновой кислотой (транс)-1-(трет-бутоксикарбонил)-4-(3-хлорфенил)пирролидин-3-карбоновую кислоту на Стадии А. Конечный продукт очистили обращенно-фазовой колоночной хроматографией, элюируя 0-70%) ацетонитрила в H2O, для получения указанного в заголовке соединения (5,2 мг, выход 34% за 3 стадии). МС (apci) m/z=504,2 (М+Н).
Пример 213
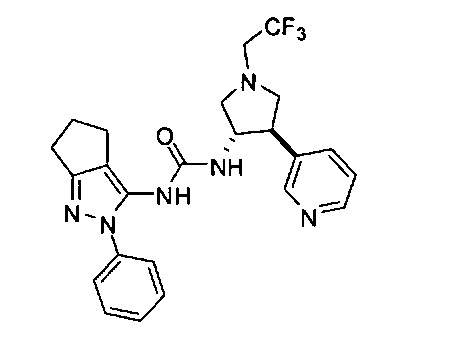
1-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)-3-((транс)-4-(пиридин-3-ил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному в Примере 208, заменяя (транс)-1-(трет-бутоксикарбонил)-4-(3-пиридил)пирролидин-3-карбоновой кислотой (транс)-1-(трет-бутоксикарбонил)-4-(3-хлорфенил)пирролидин-3-карбоновую кислоту на Стадии А. Конечный продукт очистили обращенно-фазовой колоночной хроматографией, элюируя 0-60% ацетонитрила в H2O, для получения указанного в заголовке соединения (1,2 мг, выход 8% за 3 стадии). МС (apci) m/z=471,2 (М+Н).
Пример 214
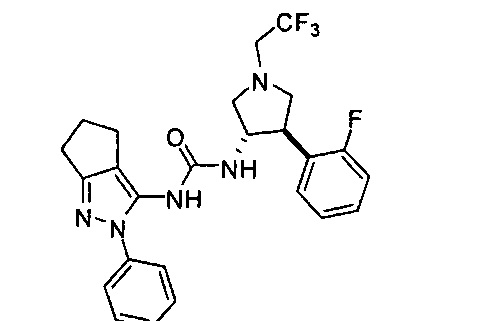
1-((транс)-4-(2-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Получили по способу, описанному в Примере 208, заменяя (транс)-1-(трет-бутоксикарбонил)-4-(2-фторфенил)пирролидин-3-карбоновой кислотой (транс)-1-(трет-бутоксикарбонил)-4-(3-хлорфенил)пирролидин-3-карбоновую кислоту на Стадии А. Конечный продукт очистили обращенно-фазовой колоночной хроматографией, элюируя 0-70% ацетонитрила в Н2О, для получения указанного в заголовке соединения (7,7 мг, выход 57% за 3 стадии). МС (apci) m/z=488,2 (М+Н).
Пример 215
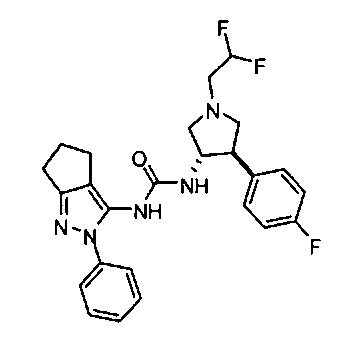
1-((транс)-4-(4-фторфенил)-1-(2,2-дифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Получили по способу, описанному в Примере 210, заменяя 2,2-дифторэтил трифторметансульфонатом 2,2,2-трифторэтил трифторметансульфонат. Конечный продукт очистили обращенно-фазовой колоночной хроматографией, элюируя 0-60% ацетонитрила в H2O, для получения указанного в заголовке соединения (4,9 мг, выход для алкилирования 46%). МС (apci) m/z=471,2 (М+Н).
Пример 216
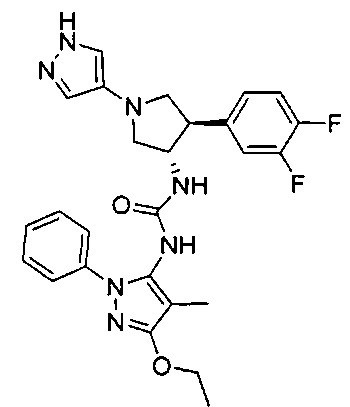
1-((3S,4R)-4-(3,4-дифторфенил)-1-(1Н-пиразол-4-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Стадия А: Получение 4-йод-1-(4-метоксибензил)-1H-пиразола: К раствору 4-йод-1Н-пиразола (5,54 г, 28,6 ммоль) и K2CO3 (4,74 г, 34,3 ммоль) в ДМФ (20 мл) добавили 1-(хлорметил)-4-метоксибензол (4,67 мл, 34,3 ммоль) и перемешивали при комнатной температуре в течение 2 часов. Добавили эфир (80 мл) и воду (30 мл). Органическую фазу отделили, промыли насыщенным солевым раствором и высушили над сульфатом натрия. После удаления растворителя, остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 25% EtOAc в гексанах, для получения указанного в заголовке соединения в виде белого твердого вещества (8,0 г, выход 89%). 1Н ЯМР (d6-ДМСО) δ 7,97 (s, 1Н), 7,52 (s, 1Н), 7,21 (d, 2Н), 6,89 (d, 2Н), 5,24 (s, 2Н), 3,73 (s, 3Н).
Стадия В: Получение трет-бутил (3S,4R)-4-(3,4-дифторфенил)-1-(1-(4-метоксибензилУ 1H-пиразол-4-ил)пирролидин-3-илкарбамата: Смесь трет-бутил(3S,4R)-4-(3,4-дифторфенил)пирролидин-3-илкарбамата (ACS Scientific, 522 мг, 1,75 ммоль), 4-йод-1-(4-метоксибензил)-1Н-пиразола (500 мг, 1,59 ммоль), K2CO3 (660 мг, 4,78 ммоль), (S)-пирролидин-2-карбоновой кислоты (73,3 мг, 0,637 ммоль) и Cu(I)I (60,6 мг, 0,318 ммоль) смешали в ДМСО (4 мл) в закрытой емкости и нагревали до 100°C в течение 18 часов. Реакционную смесь разбавили ДХМ (40 мл), промыли H2O (2×20 мл), высушили (MgSO4), отфильтровали и концентрировали. Очистили колоночной хроматографией на диоксиде кремния, элюируя 1% МеОН в ДХМ, для получения указанного в заголовке соединения в виде коричневого маслянистого вещества (376 мг, выход 49%). МС (apci) m/z=485,2 (М+Н).
Стадия С: Получение (3S,4R)-4-(3,4-дифторфенил)-1-(1-(4-метоксибензил)-1Н-пиразол-4-ил)пирролидин-3-амина дигидрохлорида: Раствор трет-бутил (3S,4R)-4-(3,4-дифторфенил)-1-(1-(4-метоксибензил)-1H-пиразол-4-ил)пирролидин-3-илкарбамата (376 мг, 0,776 ммоль) в EtOH (2 мл) и HCl (5-6 М в iPrOH) (3,10 мл, 15,5 ммоль) перемешивали при комнатной температуре в течение 19 часов. Концентрировали под вакуумом, разбавили Et2O (3×20 мл) и концентрировали для получения продукта в виде зеленовато-коричневого твердого вещества (401 мг, 113%). МС (apci) m/z=385,2 (М+Н).
Стадия D: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(1-(4-метоксибензил)-1Н-пиразол-4-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)мочевины: К раствору (3S,4R)-4-(3,4-дифторфенил)-1-(1-(4-метоксибензил)-1H-пиразол-4-ил)пирролидин-3-амина дигидрохлорида (40 мг, 0,088 ммоль) в ДИЭА (0,061 мл, 0,35 ммоль) и ДМА (1 мл) добавили фенил 3-этокси-4-метил-1-фенил-1Н-пиразол-5-илкарбамат (полученный так, как в Примере 1, Стадия А, исходя из Промежуточного соединения Р135; 29,5 мг, 0,088 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 1 часа. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-70% ацетонитрила в воде, для получения продукта в виде белого твердого вещества (24 мг, выход 44%). МС (apci) m/z=628,3 (М+Н).
Стадия Е: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(1H-пиразол-4-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)мочевины: Раствор 1-((3S,4R)-4-(3,4-дифторфенил)-1-(1-(4-метоксибензил)-1H-пиразол-4-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины (22 мг, 0,035 ммоль) в ТФК (2 мл) нагревали в пробирке для работы под давлением до 60°C в течение 18 часов. Реакционную смесь перенесли в круглодонную колбу, концентрировали и перегнали азеотропной перегонкой с толуолом (2×10 мл). Неочищенный продукт растворили в МеОН (5 мл), а остаточную ТФК удалили пропусканием через смолу на полимерной подложке (StratoSpheres PL-HCO3 MP). Неочищенный продукт очистили препаративной ТСХ (пластина 0,5 мм, элюировали 10% МеОН в ДХМ) для получения указанного в заголовке соединения в виде грязновато-белого твердого вещества (13 мг, выход 73%). МС (apci) m/z=508,2 (М+Н).
Пример 217
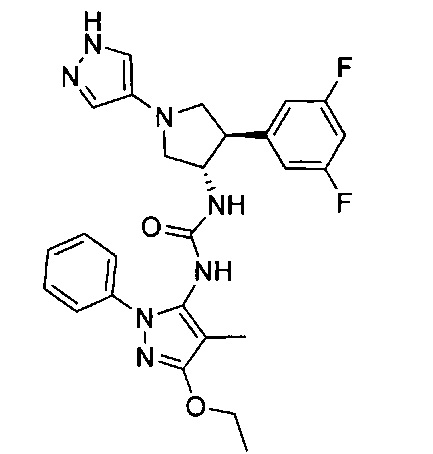
1-((3S,4R)-4-(3,5-дифторфенил)-1-(1Н-пиразол-4-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)мочевина
Получили по способу, описанному в Примере 216, заменяя трет-бутил (3S,4R)-4-(3,4-дифторфенил)пирролидин-3-илкарбамат на трет-бутил (3S,4R)-4-(3,5-дифторфенил)пирролидин-3-илкарбамат на Стадии В. МС (apci) m/z=508,2 (М+Н).
Пример 218
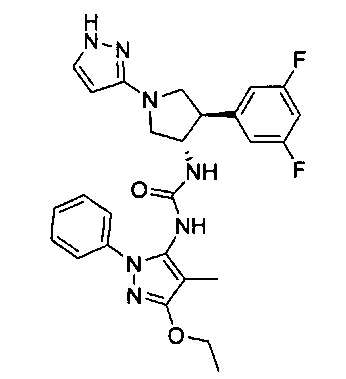
1-((3S,4R)-4-(3,5-дифторфенил)-1-(1H-пиразол-3-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)мочевина
Получили по способу, описанному в Примере 216, заменяя 4-йод-1Н-пиразол на 3-йод-1Н-пиразол на Стадии А, и заменяя трет-бутил (3S,4R)-4-(3,4-дифторфенил)пирролидин-3-илкарбамат на трет-бутил (3S,4R)-4-(3,5-дифторфенил)пирролидин-3-илкарбамат на Стадии В. МС (apci) m/z=508,2 (М+Н).
Пример 219
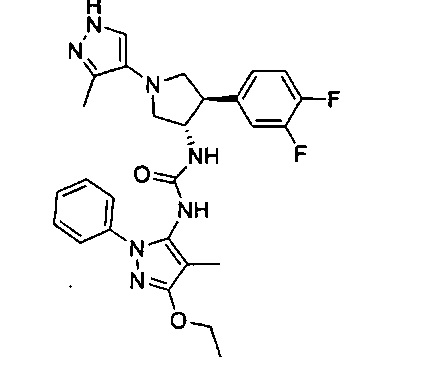
1-((3S,4R)-4-(3,4-дифторфенил)-1-(3-метил-1H-пиразол-4-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)мочевина
Получили по способу, описанному в Примере 216, заменяя 4-йод-1Н-пиразол на 4-йод-3-метил-1Н-пиразол на Стадии А. МС (apci) m/z=522,2 (М+Н).
Пример 220
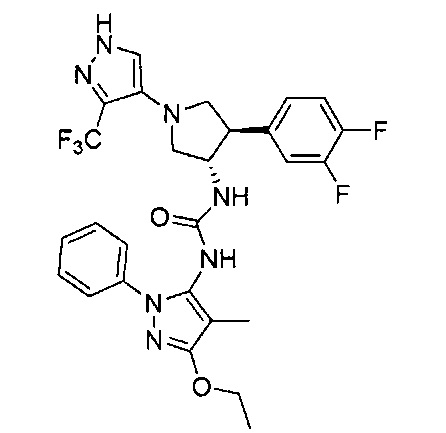
1-((3S,4R)-4-(3,4-дифторфенил)-1-(3-(трифторметил)-1H-пиразол-4-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)мочевина
Получили по способу, описанному в Примере 216, заменяя 4-йод-1Н-пиразол на 4-йод-3-метил-1Н-пиразол на Стадии А. МС (apci) m/z=576,2 (М+Н).
Пример 221
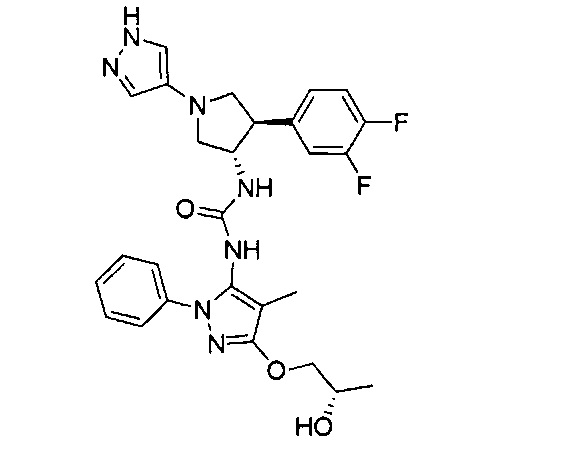
1-((3S,4R)-4-(3,4-дифторфенил)-1-(1H-пиразол-4-ил)пирролидин-3-илУ3-(3-((S)-2-гидроксипропокси)-4-метил-1-фенил-1H-пиразол-5-ил)мочевина
Стадия А: Получение 1-(3-((S)-2-(трет-бутилдиметилсилилокси)пропокси)-4-метил-1-фенил-1H-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(1-(4-метоксибензил)-1Н-пиразол-4-ил)пирролидин-3-ил)мочевины: К раствору (3S,4R)-4-(3,4-дифторфенил)-1-(1-(4-метоксибензил)-1Н-пиразол-4-ил)пирролидин-3-амина дигидрохлорида (Пример 216, Стадия С, 35 мг, 0,077 ммоль) в ДИЭА (0,053 мл, 0,31 ммоль) и ДМА (1 мл) добавили (S)-фенил 3-(2-(трет-бутилдиметилсилилокси)пропокси)-4-метил-1-фенил-1Н-пиразол-5-илкарбамат (полученный так, как в Примере 1, Стадия А, исходя из Промежуточного соединения Р211; 37 мг, 0,077 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 1 часа. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-95% ацетонитрила в воде, для получения продукта в виде белого твердого вещества (40 мг, выход 68%). МС (apci) m/z=772,4 (М+Н).
Стадия В: Получение (S)-1-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(1H-пиразол-4-ил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-илокси)пропан-2-ил 2,2,2-трифторацетата: Раствор 1-(3-((S)-2-(трет-бутилдиметилсилилокси)пропокси)-4-метил-1-фенил-1H-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(1-(4-метоксибензил)-1Н-пиразол-4-ил)пирролидин-3-ил)мочевины (39 мг, 0,051 ммоль) в ТФК (2 мл) нагревали в пробирке для работы под давлением до 60°C в течение 16 часов. Реакционную смесь перенесли в круглодонную колбу, концентрировали и перегнали азеотропной перегонкой с толуолом (2×10 мл) для получения продукта в виде желто-коричневого твердого вещества (32 мг, 99%). МС (apci) m/z=634,2 (М+Н).
Стадия С: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(1H-пиразол-4-ил)пирролидин-3-ил)-3-(3-((S)-2-гидроксипропоксиУ 4-метил-1-фенил-1H-пиразол-5-ил)мочевины: К раствору (S)-1-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(1H-пиразол-4-ил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-илокси)пропан-2-ил 2,2,2-трифторацетата (32 мг, 0,051 ммоль) в МеОН (1 мл) и ТГФ (2 мл) добавили 2 М водный раствор LiOH (0,5 мл, 1,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, разбавили H2O (20 мл), экстрагировали 10: 90 МеОН/ДХМ (2×20 мл), а объединенные органические фазы высушили (MgSO4), отфильтровали и концентрировали под пониженным давлением. Очистили колоночной хроматографией на диоксиде кремния, элюируя 10% МеОН в ДХМ, для получения указанного в заголовке соединения в виде белого твердого вещества (19 мг, выход 72%). МС (apci) m/z=538,2 (М+Н).
Пример 222
Получили по способу, описанному в Примере 221, заменяя (S)-фенил 3-(2-(трет-бутилдиметилсилилокси)пропокси)-4-метил-1-фенил-1Н-пиразол-5-илкарбамат на (S)-фенил 3-((2,2-диметил-1,3-диоксолан-4-ил)метокси)-4-метил-1-фенил-1Н-пиразол-5-илкарбамат (полученный так, как в Примере 1, Стадия А, исходя из Промежуточного соединения Р209) на Стадии А. МС (apci) m/z=554,2 (М+Н).
Соединения в следующей таблице были получены по способу Примера 216, с заменой соответствующего исходного материала на Стадиях А и В и с заменой соответствующего пиразолового промежуточного соединения на Стадии D.
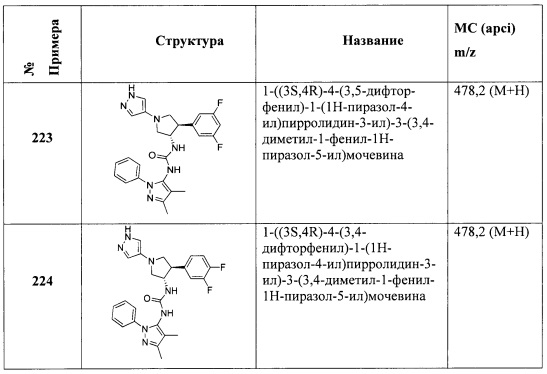
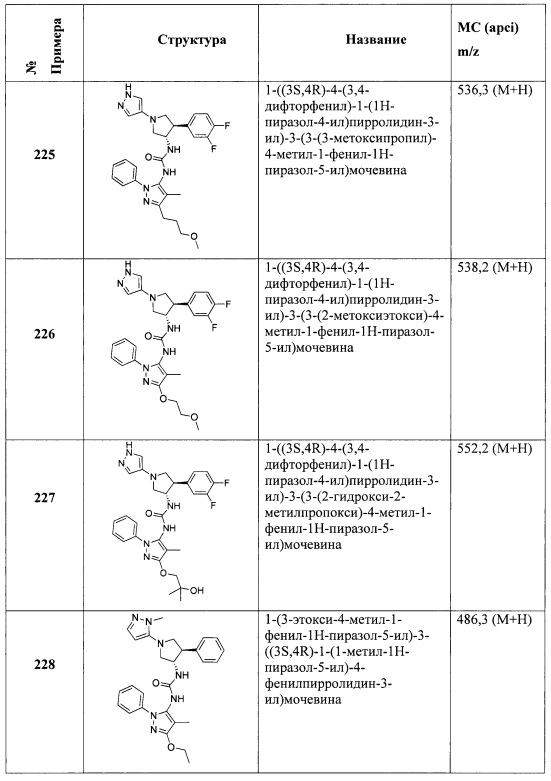
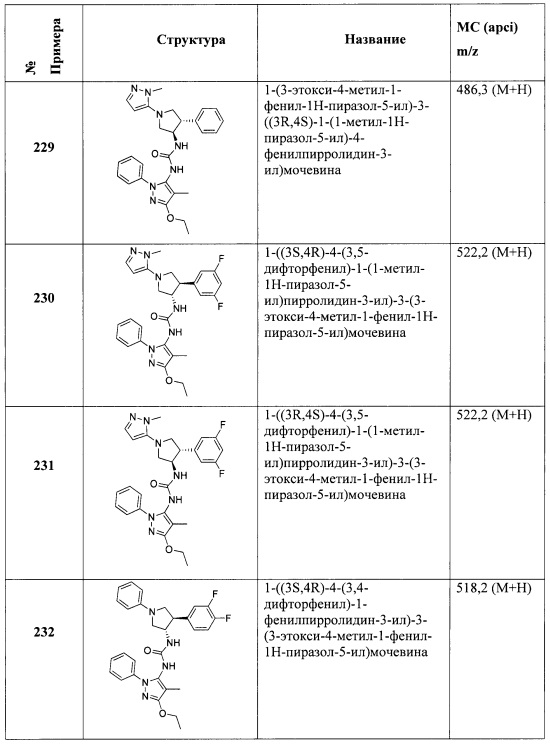
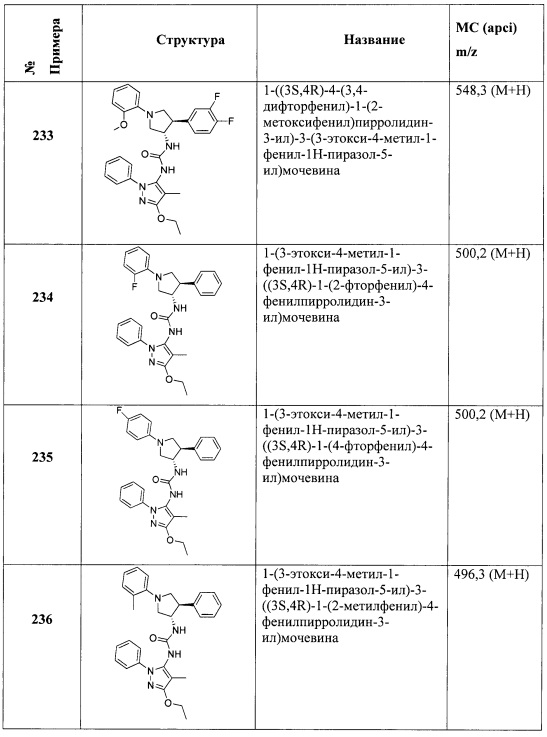
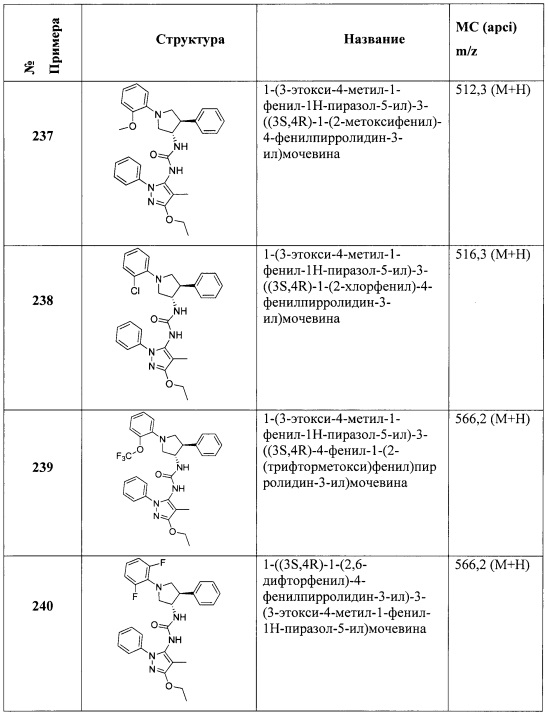
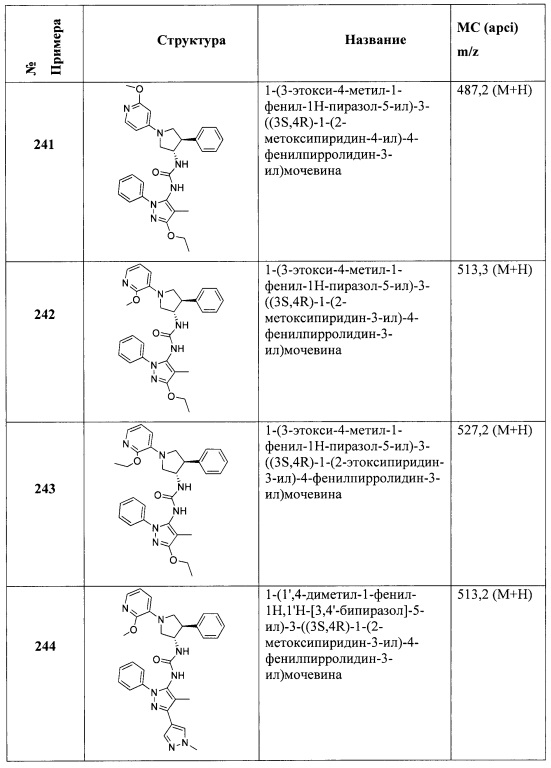
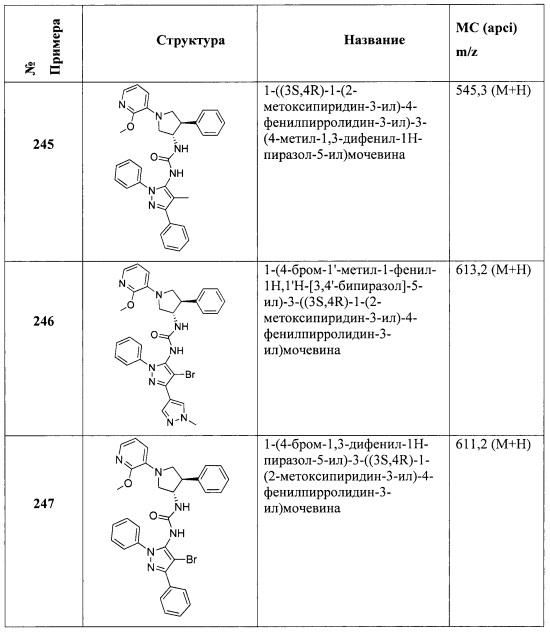
Пример 248
1-((3S,4R)-1-((1,2,3-тиадиазол-4-ил)метил)-4-(3,4-дифторфенил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Стадия А: Получение трет-бутил ((3S,4R)-1-((1,2,3-тиадиазол-4-ил)метил)-4-(3,4-дифторфенил)пирролидин-3-ил)карбамата. Трет-бутил (3S,4R)-4-(3,4-дифторфенил)пирролидин-3-илкарбамат (150 мг, 0,503 ммоль), 1,2,3-тиадиазол-4-карбальдегид (68,9 мг, 0,603 ммоль) и ДИЭА (186 мкл, 1,01 ммоль) смешали в 1 мл ДХМ и перемешивали при комнатной температуре в течение 30 минут. Добавили триацетоксиборгидрид натрия (213 мг, 1,01 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение ночи, и концентрировали. Неочищенный материал очистили обращенно-фазовой колоночной хроматографией, элюируя 0-50% ацетонитрила в воде, для получения продукта (168 мг, 0,424 ммоль, выход 84,3%). МС (apci) m/z=397,1 (М+Н).
Стадия В: Получение (3S,4R)-1-((1,2,3-тиадиазол-4-ил)метил)-4-(3,4-дифторфенил)пирролидин-3-амина дигидрохлорида. Трет-бутил (3S,4R)-1-((1,2,3-тиадиазол-4-ил)метил)-4-(3,4-дифторфенил)пирролидин-3-илкарбамат (150 мг, 0,378 ммоль) и 5 н. HCl в IPA (378 мкл, 1,89 ммоль) смешали в 1 мл IPA и оставили стоять при комнатной температуре в течение ночи. Реакционную смесь концентрировали для получения продукта (140 мг, 0,379 ммоль, выход 100%). МС (apci) m/z=297,0 (М+Н).
Стадия С: Получение 1-((3S,4R)-1-((1,2,3-тиадиазол-4-ил)метил)-4-(3,4-дифторфенил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины: (3S,4R)-1-((1,2,3-тиадиазол-4-ил)метил)-4-(3,4-дифторфенил)пирролидин-3-амина дигидрохлорид (20 мг, 0,054 ммоль), фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат (16 мг, 0,049 ммоль, полученный так, как описано для Примера 38, Стадия Е) и ДИЭА (26 мкл, 0,15 ммоль) смешали в 0,2 мл ДМФ и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь загрузили на картридж Samplet и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-80% ацетонитрила в воде, для получения указанного в заголовке соединения (24 мг, 0,046 ммоль, выход 93%). МС (apci) m/z=522,2 (М+Н).
Пример 249
1-((3S,4R)-1-((1,2,3-тиадиазол-4-ил)метил)-4-(3,4-дифторфенил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)мочевина
Получили по способу, описанному в Примере 216, Стадия С, используя фенил 3-этокси-4-метил-1-фенил-1Н-пиразол-5-илкарбамат (который получили по способу Примера 1, Стадия А, исходя из Промежуточного соединения Р135) вместо фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамата. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-80% ацетонитрила в Н2О, для получения указанного в заголовке соединения (23 мг, 0,043 ммоль, выход 87%). МС (apci) m/z=540,2 (М+Н).
Пример 250
1-((3S,4R)-1-((1,2,3-тиадиазол-4-ил)метил)-4-(3,4-дифторфенил)пирролидин-3-ил)-3-(3-(цианометокси)-4-метил-1-фенил-1H-пиразол-5-ил)мочевина
Получили по способу, описанному в Примере 216, Стадия С, используя фенил 3-(цианометокси)-4-метил-1-фенил-1Н-пиразол-5-илкарбамат вместо фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамата. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-80% ацетонитрила в H2O, для получения указанного в заголовке соединения (3,4 мг, 0,0062 ммоль, выход 50%). МС (apci) m/z=551,2 (М+Н).
Пример 251
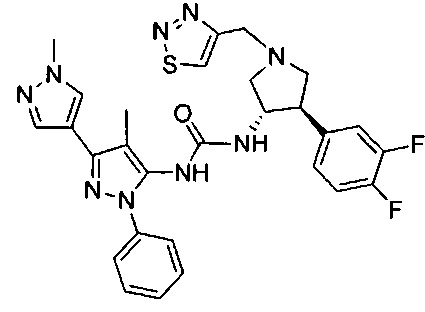
1-((35,4R)-1-((1,2,3-тиадиазол-4-ил)метил)-4-(3,4-дифторфенил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)мочевина
Получили по способу, описанному в Примере 216, Стадия С, используя фенил 1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-илкарбамат (9,2 мг, 0,025 ммоль) вместо фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамата. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-70% ацетонитрила в H2O, для получения указанного в заголовке соединения (4,2 мг, 0,0073 ммоль, выход 30%). МС (apci) m/z=576,2 (М+Н).
Пример 252
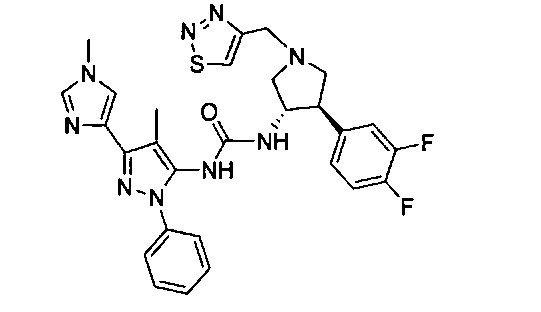
1-((3S,4R)-1-((1,2,3-тиадиазол-4-ил)метил)-4-(3,4-дифторфенил)пирролидин-3-ил)-3-(4-метил-3-(1-метил-1H-имидазол-4-ил)-1-фенил-1H-пиразол-5-ил)мочевина
Получили по способу, описанному в Примере 216, Стадия С, используя 1-метил-4-(4-метил-5-(феноксикарбониламино)-1-фенил-1Н-пиразол-3-ил)-3-(феноксикарбонил)-1Н-имидазол-3-ия хлорид вместо фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамата и добавляя дополнительный эквивалент (3S,4R)-1-((1,2,3-тиадиазол-4-ил)метил)-4-(3,4-дифторфенил)пирролидин-3-амина дигидрохлорида. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-80% ацетонитрила в Н2О, для получения указанного в заголовке соединения (6,2 мг, 0,011 ммоль, выход 42%). МС (apci) m/z=576,2 (М+Н).
Пример 253
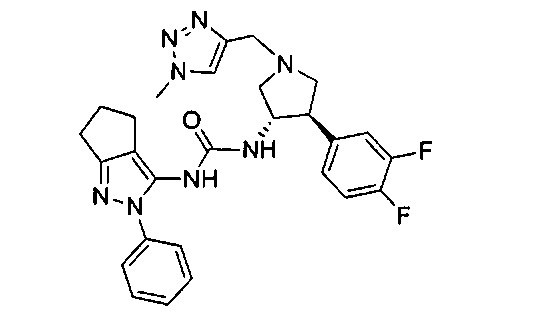
1-((3S,4R)-4-(3,4-дифторфенил)-1-((1-метил-1Н-1,2,3-триазол-4-ил)метил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Получили по способу, описанному в Примере 216, используя 1-метил-1Н-1,2,3-триазол-4-карбальдегид вместо 1,2,3-тиадиазол-4-карбальдегида на Стадии А. Конечный продукт очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-80% ацетонитрила в H2O, для получения указанного в заголовке соединения (17 мг, 0,033 ммоль, выход 76% за 3 стадии). МС (apci) m/z=519,2 (М+Н).
Пример 254
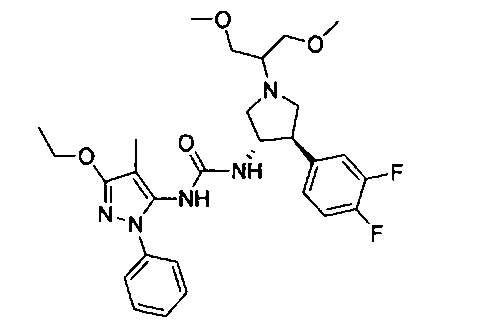
1-((3S,4R)-4-(3,4-дифторфенил)-1-(1,3-диметоксипропан-2-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)мочевина
Получили по способу, описанному в Примере 249, используя 1,3-диметоксипропан-2-он вместо 1,2,3-тиадиазол-4-карбальдегида на Стадии А. Конечный продукт очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 5-80% ацетонитрила в H2O, для получения указанного в заголовке соединения (17,3 мг, 0,0318 ммоль, выход 57,4%) за 3 стадии). МС (apci) m/z=544,3 (М+Н).
Пример 255
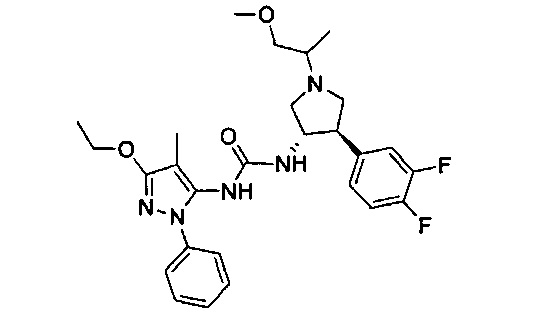
1-((3S,4R)-4-(3,4-дифторфенил)-1-(1-метоксипропан-2-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)мочевина
Получили по способу, описанному в Примере 249, используя 1-метоксипропан-2-он вместо 1,2,3-тиадиазол-4-карбальдегида на Стадии А. Конечный продукт очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-80% ацетонитрила в H2O, для получения указанного в заголовке соединения (8,2 мг, 0,016 ммоль, выход 70,1%) за 3 стадии). МС (apci) m/z=514,3 (М+Н).
Пример 256
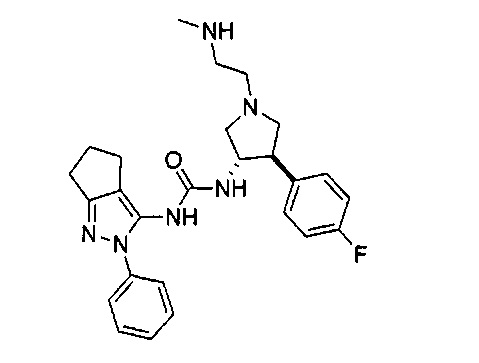
1-(транс)-4-(4-фторфенил)-1-(2-(метиламино)этил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
1-((3S,4R)-4-(4-фторфенил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидро-циклопента[с]пиразол-3-ил)мочевины гидрохлорид (11 мг, 0,0249 ммоль, полученный так, как в Примере 210), 2-йод-N-метил-N-тритилэтанамин (19,1 мг, 0,0448 ммоль) и ДИЭА (9,65 мг, 0,0747 ммоль) смешали в 0,2 мл ДМФ и перемешивали при комнатной температуре в течение ночи. Добавили HCl (7 н. в IPA, 35,6 мкл, 0,249 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 4 часов. Добавили 1 н. раствор NaOH (2 мл) и экстрагировали реакционную смесь несколькими порциями ДХМ на фазоразделительном стеклянном фильтре. Объединенные органические экстракты концентрировали и очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-60% ацетонитрила в H2O, для получения указанного в заголовке соединения (2,2 мг, 0,00476 ммоль, выход 19,1%). МС (apci) m/z=463,3 (М+Н).
Пример 257
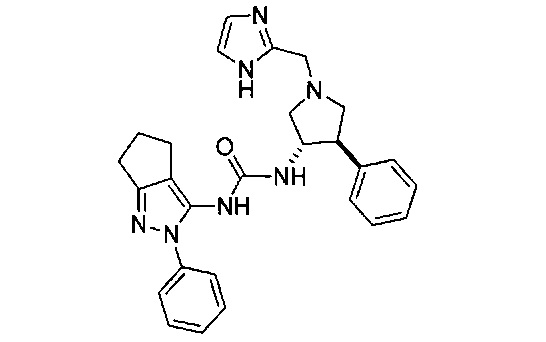
1-((транс)-1-((1H-имидазол-2-ил)метил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
1-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)-3-((транс)-4-фенилпирролидин-3-ил)мочевину (15 мг, 0,039 ммоль, полученную так, как описано в Примере 182, Стадия В), 2-(хлорметил)-1Н-имидазола гидрохлорид (5,9 мг, 0,039 ммоль) и ДИЭА (14 мкл, 0,077 ммоль) смешали в 0,2 мл ДМФ и перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь загрузили на картридж Samplet и очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-70%) ацетонитрила в H2O, для получения указанного в заголовке соединения (7,9 мг, 0,017 ммоль, выход 44%). МС (apci) m/z=468,2 (М+Н).
Пример 258
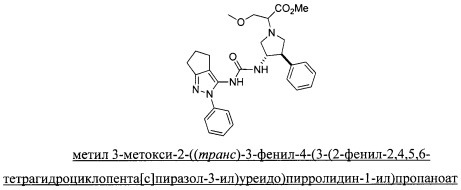
1-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)-3-((транс)-4-фенилпирролидин-3-ил)мочевину (30 мг, 0,077 ммоль, полученную так, как описано в Примере 182, Стадия В), метил 2-бром-3-метоксипропаноат (15 мг, 0,077 ммоль) и ДИЭА (29 мкл, 0,15 ммоль) смешали в 0,2 мл ДХМ и перемешивали при комнатной температуре в течение 3 дней. Реакционную смесь загрузили на картридж Samplet и очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 5-80% ацетонитрила в Н2О, для получения указанного в заголовке соединения (24 мг, 0,048 ммоль, выход 62%) в виде смеси диастереомеров. МС (apci) m/z=504,3 (М+Н).
Пример 259
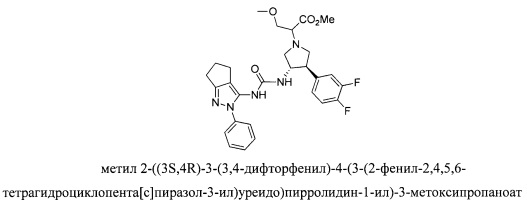
Получили по способу, описанному в Примере 194, используя метил 2-бром-3-метоксипропаноат вместо 2,2,2-трифторэтил трифторметансульфоната и перемешивая в течение 3 дней вместо 1 часа на Стадии А. Конечный продукт очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 5-80% ацетонитрил в Н2О, для получения указанного в заголовке соединения (260 мг, 0,482 ммоль, выход 83,5% за 3 стадии) в виде смеси диастереомеров. МС (apci) m/z=540,3 (М+Н).
Пример 260
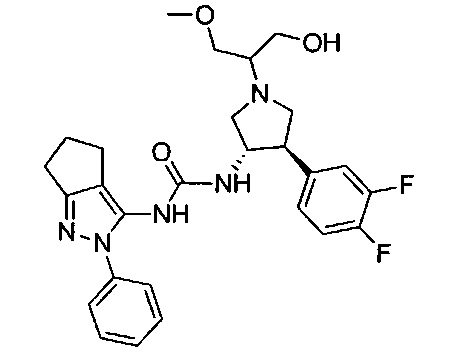
1-((3S,4R)-4-(3,4-дифторфенил)-1-(1-гидрокси-3-метоксипропан-2-ил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Метил 2-((3R,4S)-3-(3,4-дифторфенил)-4-(3-(2-фенил-2,4,5,6-тетрагидро-циклопента[с]пиразол-3-ил)уреидо)пирролидин-1-ил)-3-метоксипропаноат (200 мг, 0,371 ммоль, полученный так, как описано в Примере 259) растворили в 5 мл ТГФ и охладили раствор до 0°C. Добавили LiAlH4 (28,1 мг, 0,741 ммоль) и оставили реакционную смесь нагреваться до комнатной температуры в течение 2 часов. Добавили декагидрат сульфата натрия (1194 мг, 3,71 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 2 часов, отфильтровали и концентрировали. Неочищенный продукт очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-70% ацетонитрила в Н2О, для получения указанного в заголовке соединения (20 мг, 0,0391 ммоль, выход 10,5%) в виде смеси диастереомеров. МС (apci) m/z=512,3 (М+Н).
Пример 261
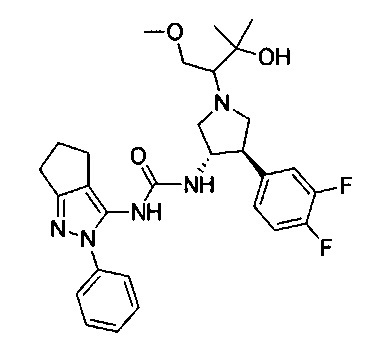
1-((3S,4R)-4-(3,4-дифторфенил)-1-(3-гидрокси-1-метокси-3-метилбутан-2-ил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Метил 2-((3R,4S)-3-(3,4-дифторфенил)-4-(3-(2-фенил-2,4,5,6-тетрагидро-циклопента[с]пиразол-3-ил)уреидо)пирролидин-1-ил)-3-метоксипропаноат (20 мг, 0,037 ммоль, полученный так, как описано в Примере 12) растворили в 1 мл ТГФ и охладили до 0°C. Добавили метилмагния бромид (2 M в ТГФ, 93 мкл, 0,19 ммоль) и оставили реакционную смесь нагреваться до комнатной температуры в течение ночи. Добавили H2O (2 мл) и экстрагировали реакционную смесь ДХМ (2×5 мл) на фазоразделительном стеклянном фильтре. Объединенные органические экстракты концентрировали и очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-70% ацетонитрила в Н2О, для получения указанного в заголовке соединения (7,9 мг, 0,015 ммоль, выход 39%) в виде смеси диастереомеров. МС (apci) m/z=539,6 (М+Н).
Пример 262
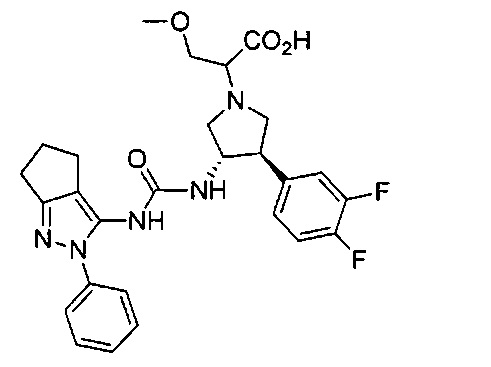
2-((3R,4S)-3-(3,4-дифторфенил)-4-(3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)уреидо)пирролидин-1-ил)-3-метоксипропановой кислоты гидрохлорид
Метил 2-((3R,4S)-3-(3,4-дифторфенил)-4-(3-(2-фенил-2,4,5,6-тетрагидро-циклопента[с]пиразол-3-ил)уреидо)пирролидин-1-ил)-3-метоксипропаноат (300 мг, 0,556 ммоль, полученный так, как описано в Примере 259) и NaOH (1 н., водный, 834 мкл, 0,834 ммоль) смешали в 1 мл МеОН и перемешивали при комнатной температуре в течение ночи. Реакционную смесь загрузили на картридж Samplet и очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-50% ацетонитрила в 0,01 М водном растворе HCl, для получения указанного в заголовке соединения (298 мг, 0,530 ммоль, выход 95,4%) в виде смеси диастереомеров. МС (apci) m/z=526,2 (М+Н).
Пример 263
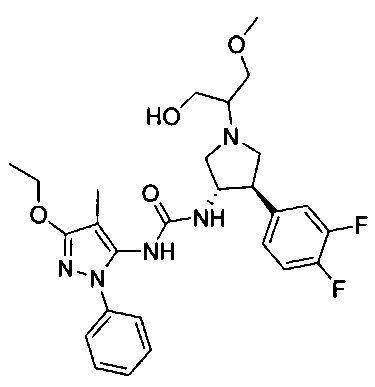
1-((3S,4R)-4-(3,4-дифторфенил)-1-(1-гидрокси-3-метоксипропан-2-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)мочевина
Стадия А: Получение трет-бутил (3S,4R)-4-(3,4-дифторфенил)-1-(1-гидрокси-3-метоксипропан-2-ил)пирролидин-3-илкарбамата. Метил 2-((3S,4R)-3-(трет-бутоксикарбониламино)-4-(3,4-дифторфенил)пирролидин-1-ил)-3-метоксипропаноат (220 мг, 0,531 ммоль, полученный так, как описано в Примере 259, Стадия А) растворили в 5 мл ТГФ и небольшими частями добавили NaBH4 (100 мг, 2,65 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, при 40°C в течение 3 часов, а затем при 50°C в течение ночи. Реакционную смесь отфильтровали, концентрировали и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-70% ацетонитрила в воде, для получения указанного в заголовке соединения (154 мг, 0,399 ммоль, выход 75,1%). МС (apci) m/z=387,2 (М+Н).
Стадия В: Получение 2-((3S,4R)-3-амино-4-(3,4-дифторфенил)пирролидин-1-ил)-3-метоксипропан-1-ола дигидрохлорида. Трет-бутил (3S,4R)-4-(3,4-дифторфенил)-1-(1-гидрокси-3-метоксипропан-2-ил)пирролидин-3-илкарбамат (120 мг, 0,311 ммоль) и HCl (5 н. в IPA, 186 мкл, 0,932 ммоль) смешали в 5 мл ДХМ и. перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь концентрировали для получения указанного в заголовке соединения (87 мг, 0,304 ммоль, выход 97,9%).
Стадия С: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(1-гидрокси-3-метоксипропан-2-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)мочевины. 2-((3S,4R)-3-амино-4-(3,4-дифторфенил)пирролидин-1-ил)-3-метоксипропан-1-ола дигидрохлорид (13,0 мг, 0,0362 ммоль), фенил 3-этокси-4-метил-1-фенил-1Н-пиразол-5-илкарбамат (полученный по способу Примера 1, Стадия А, исходя из Промежуточного соединения Р135; 10,2 мг, 0,0302 ммоль) и ДИЭА (11,7 мг, 0,0905 ммоль) смешали в 0,2 мл ДМФ и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь загрузили на картридж Samplet и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-60% ацетонитрила в воде, для получения указанного в заголовке соединения (10,9 мг, 0,0206 ммоль, выход 68,3%). МС (apci) m/z=530,2 (М+Н).
Пример 264
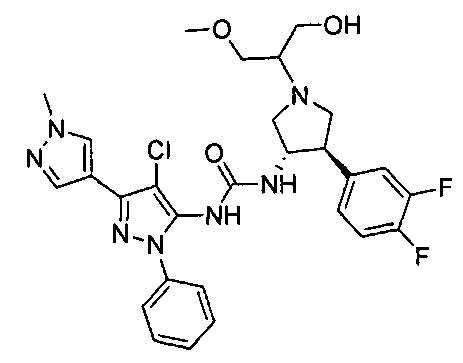
1-(4-хлор-1'-метил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(1-гидрокси-3-метоксипропан-2-ил)пирролидн-3-ил)мочевина
Получили по способу, описанному в Примере 263, используя фенил 4-хлор-1'-метил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-илкарбамат вместо фенил 3-этокси-4-метил-1-фенил-1H-пиразол-5-илкарбамата. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-80% ацетонитрила в Н2О, для получения указанного в заголовке соединения (17,2 мг, 0,0293 ммоль, выход 63,3%). МС (apci) m/z=586,2 (М+Н).
Пример 265
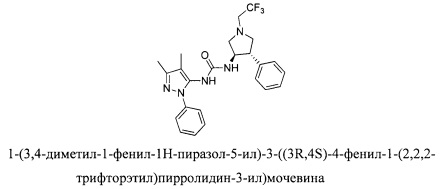
Стадия А: Разделение рацемического (транс)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-амина гидрохлорида. (транс)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-амина гидрохлорид (3,80 г, 11,0 ммоль, полученный по способу, описанному для Примера 194, Стадия В) разделили сверхкритической жидкостной хроматографией (СЖХ), используя колонку 20 мм × 250 мм Chiral Tech Chiralcell OD-H, деталь №14345. В качестве элюента использовали 9:1 сверхкритический СО2:МеОН с 0,1% NH4OH в качестве модификатора. Выделили 1 пик для получения (3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-амина (1,07 г, 3,81 ммоль, выход 34,6%, э.и. 98%). МС (apci) m/z=245,1 (М+Н).
Стадия В: Получение 1-(3,4-диметил-1-фенил-1Н-пиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевины. (3R,4S)-4-фенил-1-(2,2,2-трифтор-этил)пирролидин-3-амин (15,0 мг, 0,0614 ммоль), фенил 3,4-диметил-1-фенил-1Н-пиразол-5-илкарбамат (17,2 мг, 0,0558 ммоль) и ДИЭА (21,6 мг, 0,167 ммоль) смешали в 0,2 мл ДМФ и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь загрузили на картридж Samplet и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-80% ацетонитрила в воде, для получения указанного в заголовке соединения (7,2 мг, 0,0157 ммоль, выход 28,2%). МС (apci) m/z=458,2 (М+Н).
Пример 266
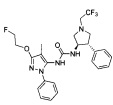
1-(3-(2-фторэтокси)-4-метил-1-фенил-1H-пиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному в Примере 1, Стадия B, используя фенил 3-(2-фторэтокси)-4-метил-1-фенил-1Н-пиразол-5-илкарбамат вместо фенил 3,4-диметил-1-фенил-1Н-пиразол-5-илкарбамата. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-80% ацетонитрила в H2O, для получения указанного в заголовке соединения (2,2 мг, 0,0044 ммоль, выход 26%). МС (apci) m/z=506,2 (М+Н).
Пример 267
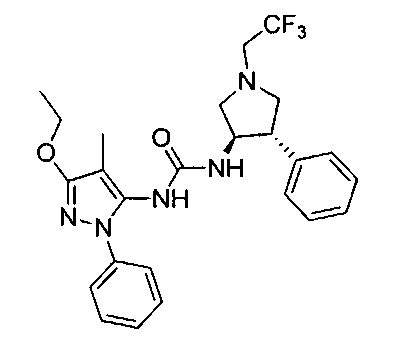
1-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному в Примере 1, Стадия B, используя фенил 3-этокси-4-метил-1-фенил-1Н-пиразол-5-илкарбамат (который получили по способу Примера 1, Стадия А, исходя из Промежуточного соединения Р135) вместо фенил 3,4-диметил-1-фенил-1Н-пиразол-5-илкарбамата. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-80% ацетонитрила в Н2О, для получения указанного в заголовке соединения (7,1 мг, 0,0146 ммоль, выход 42,7%). МС (apci) m/z=488,2 (М+Н).
Пример 268
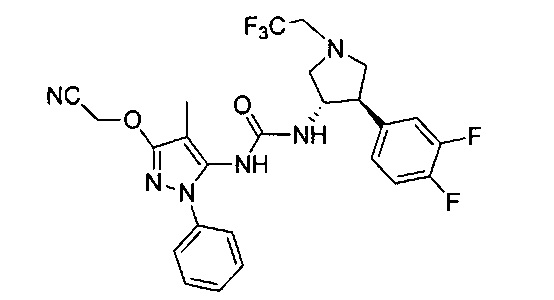
1-(3-(цианометокси)-4-метил-1-фенил-1H-пиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному в Примере 1, Стадия В, используя фенил 3-(цианометокси)-4-метил-1-фенил-1Н-пиразол-5-илкарбамат вместо фенил 3,4-диметил-1-фенил-1Н-пиразол-5-илкарбамата. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-80% ацетонитрила в H2O, для получения указанного в заголовке соединения (11,4 мг, 0,0229 ммоль, выход 67,0%). МС (apci) m/z=499,2 (М+Н).
Пример 269
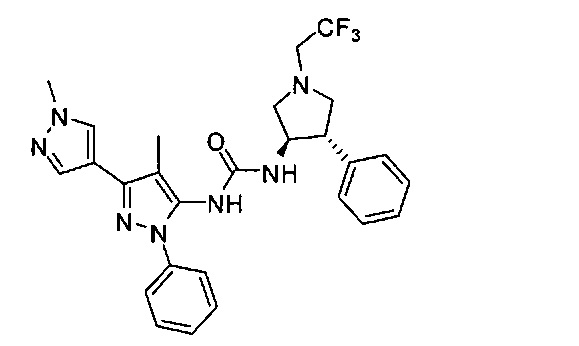
1-(1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному в Примере 1, Стадия В, используя фенил 1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-илкарбамат вместо фенил 3,4-диметил-1-фенил-1Н-пиразол-5-илкарбамата. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-70% ацетонитрила в H2O, для получения указанного в заголовке соединения (6,1 мг, 0,012 ммоль, выход 68%). МС (apci) m/z=524,2 (М+Н).
Пример 270
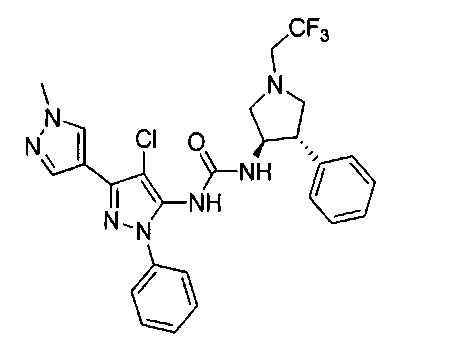
1-(4-хлор-1'-метил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному в Примере 1, Стадия В, используя фенил 4-хлор-1'-метил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-илкарбамат вместо фенил 3,4-диметил-1-фенил-1Н-пиразол-5-илкарбамата. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-70% ацетонитрила в Н2О, для получения указанного в заголовке соединения (11,3 мг, 0,0208 ммоль, выход 60,9%). МС (apci) m/z=488,2 (М+Н).
Пример 271
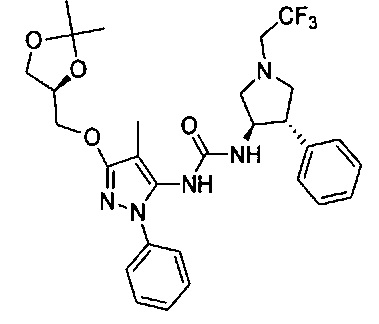
1-(3-(((S)-2,2-диметил-1,3-диоксолан-4-ил)метокси)-4-метил-1-фенил-1H-пиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному в Примере 1, Стадия В, используя (S)-фенил 3-((2,2-диметил-1,3-диоксолан-4-ил)метокси)-4-метил-1-фенил-1Н-пиразол-5-илкарбамат вместо фенил 3,4-диметил-1-фенил-1Н-пиразол-5-илкарбамата. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-80% ацетонитрила в Н2О, для получения указанного в заголовке соединения (5,6 мг, 0,00976 ммоль, выход 28,6%). МС (apci) m/z=574,3 (М+Н).
Пример 272
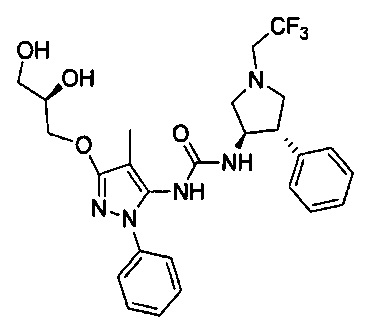
1-(3-((R)-2,3-дигидроксипропокси)-4-метил-1-фенил-1H-пиразол-5 -ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина
1-(3-(((S)-2,2-диметил-1,3-диоксолан-4-ил)метокси)-4-метил-1-фенил-1H-пиразол-5-ил)-3-((3R,4S)-4-фенил-l-(2,2,2-трифторэтил)пирролидин-3-ил)мочевину (4,4 мг, 0,0077 ммоль, полученную так, как описано в Примере 7) и HCl (7 н. в IPA, 3,3 мкл, 0,023 ммоль) смешали в 0,2 мл IPA и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали и добавили ДИЭА (1,3 мкл, 0,0077 ммоль). Неочищенный материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-60% ацетонитрила в Н2О, для получения указанного в заголовке соединения (3,7 мг, 0,0069 ммоль, выход 90%). МС (apci) m/z=534,2 (М+Н).
Пример 273
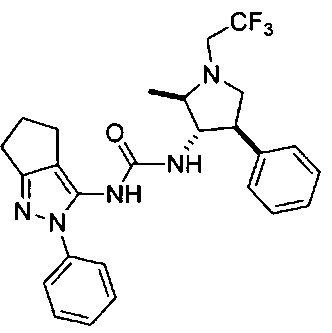
(R,S)-1-((2α,3β,4α)-2-метил-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Стадия А: Получение Н-бензил-1-(триметилсилил)этанамина. (1-хлорэтил)триметилсилан (6,3 г, 46,1 ммоль) и бензиламин (14,8 г, 138 ммоль) смешали в сосуде для работы под давлением и нагревали при 180°C в течение ночи. Реакционную смесь растворили в EtOAc (200 мл), промыли 1 н. раствором NaOH (100 мл), высушили (MgSO4) и концентрировали. Неочищенный продукт очистили силикагелевой колоночной хроматографией, элюируя 10% МеОН в CH2Cl2, для получения указанного в заголовке соединения (8,10 г, 39,1 ммоль, выход 84,7%). МС (apci) m/z=208,1 (М+Н).
Стадия В: Получение N-бензил-N-(метоксиметил)-1-(триметилсилил)этанамина. Смесь формальдегида (37% водный раствор, 3,45 мл, 46,3 ммоль) и метанола (1,88 мл, 46,3 ммоль) при 0°C небольшими порциями, за 10 минут обработали N-бензил-1 (триметилсилил)этанамином (8,00 г, 38,6 ммоль). Для промывания остаточного амина использовали дополнительное количество МеОН (2 мл). Смесь перемешивали при 0°C в течение 3 часов, добавили K2CO3 (8,00 г, 57,9 ммоль) и оставили смесь нагреваться до комнатной температуры в течение ночи. Смесь декантировали в новую колбу с добавлением небольшого количества Et2O и добавили дополнительное количество K2CO3 (50 г). Смесь перемешивали в течение 30 минут и отфильтровали, промыли небольшим количеством Et2O и осторожно концентрировали для получения указанного в заголовке соединения (9,60 г, 38,2 ммоль, выход 99,0%) в виде бледно-желтой жидкости. МС (apci) m/z=208,1 (М+2Н-CH2OCH3).
Стадия С: Получение (R,S)(2α,3β,4α)-1-бензил-2-метил-3-нитро-4-фенилпирролидина и (R,S)(3β,4α,5α)-1-бензил-2-метил-4-нитро-3-фенилпирролидина Раствор (Е)-(2-нитровинил)бензола (0,500 г, 3,35 ммоль) и ТФК (0,0258 мл, 0,335 ммоль) в 10 мл CH2Cl2 охладили до 0°C и по каплям добавили N-бензил-N-(метоксиметил)-1-(триметилсилил)этанамин (1,01 г, 4,02 ммоль). Реакционную смесь перемешивали при 0°C в течение 2 часов, а затем оставили нагреваться до комнатной температуры в течение ночи. Реакционную смесь концентрировали, загрузили на картридж Samplet, используя МеОН (2 мл) и ДИЭА (0,584 мл, 3,35 ммоль), и очистили обращенно-фазовой колоночной хроматографией, элюируя 5-95% ацетонитрила в воде с 0,1% буферного ацетата аммония, для получения указанных в заголовке соединений (287 мг, 0,925 ммоль, выход 56,5%) в виде смеси 5:3. МС (apci) m/z=297,1 (М+Н).
Стадия D: Получение (R,S)-1-((2α,3β,4α)-1-бензил-2-метил-4-фенил-пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины и (R,S)-1-((3β,4α,5α)-1-бензил-5-метил-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины. ((2α,3β,4α)-1-бензил-2-метил-3-нитро-4-фенилпирролидини (3β,4α,5α)-1-бензил-2-метил-4-нитро-3-фенилпирролидин (110 мг смеси 5: 3, 0,371 ммоль) растворили в 10 мл ТГФ и добавили никель Ренея (3,18 мг, 0,0371 ммоль). Реакционную смесь перемешивали под Н2 из баллона в течение 3 дней, отфильтровали через Celite®, концентрировали и растворили в 0,2 мл ДМФ. Добавили фенил 2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-илкарбамат (149 мг, 0,467 ммоль) и ДИЭА (222 мкл, 1,27 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 1 часа. Реакционную смесь загрузили на картридж Samplet и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-80% ацетонитрила в воде, для получения указанных в заголовке соединений (172 мг, 0,350 ммоль, выход 82%) в виде смеси ~7:3. МС (apci) m/z=492,3 (М+Н).
Стадия Е: Получение (R,S)-1-((2α,3β,4α)-2-метил-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины и 1-((3β,4α,5α)-5-метил-4-фенил-пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины. 1-((2α,3β,4α)-1-бензил-2-метил-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевину и 1-((3β,4α,5α)-1-бензил-5-метил-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевину (170 мг смеси 7: 3, 0,346 ммоль) растворили в 10 мл ТГФ и добавили 10% Pd/C (36,8 мг, 0,0346 ммоль). Реакционную смесь перемешивали под Н2 из баллона в течение 16 часов, отфильтровали через Celite и концентрировали для получения указанных в заголовке соединений (133 мг, 0,335 ммоль, выход 96%) в виде смеси ~7: 3. МС (apci) m/z=402,2 (М+Н).
Стадия F: Получение (R,S)-1-((2α,3β,4α)-2-метил-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевины. 1-((2α,3β,4α)-2-метил-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с] пиразол-3-ил)мочевину и 1-((3β,4α,5α)-5-метил-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевину (20 мг смеси 7: 3, 0,050 ммоль) и ДИЭА (8,7 мкл, 0,050 ммоль) смешали в 0,2 мл ДМФ и добавили 2,2,2-трифторэтилтрифторметансульфонат (35 мг, 0,15 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, загрузили на картридж Samplet и разделили обращенно-фазовой колоночной хроматографией, элюируя 0-80% ацетонитрила в воде. Выделили пик 1 для получения указанного в заголовке соединения (3,0 мг, 0,0062 ммоль, выход 12%). МС (apci) m/z=484,2 (М+Н).
Пример 274
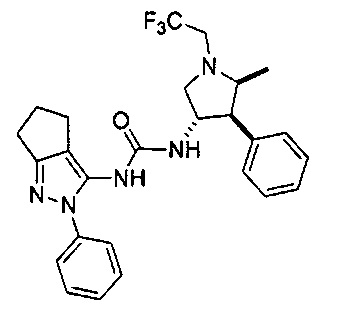
(R,S)-1-((3β,4α,5α)-5-метил-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина
Получили по способу, описанному в Примере 9, выделив пик 2 вместо пика 1 на стадии F, для получения указанного в заголовке соединения (1,9 мг, 0,0039 ммоль, выход 7,9%). МС (apci) m/z=484,2 (М+Н).
Пример 275
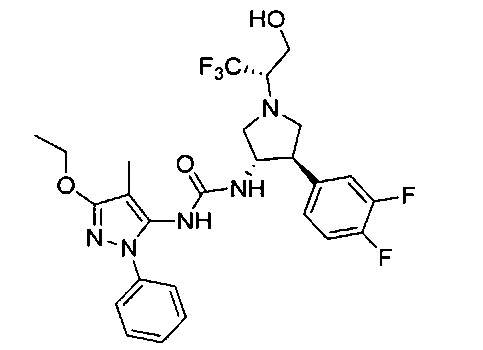
1-((3S,4R)-4-(3,4-дифторфенил)-1-((S)-1,1,1-трифтор-3-гидроксипропан-2-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)мочевина
Стадия А: Получение трет-бутил (3S,4R)-4-(3,4-дифторфенил)-1-((R)-3,3,3-трифтор-2-гидроксипропил)пирролидин-3-илкарбамата. Трет-бутил (3S,4R)-4-(3,4-дифторфенил)пирролидин-3-илкарбамат (900 мг, 3,017 ммоль), (R)-2-(трифторметил)оксиран (338,0 мг, 3,017 ммоль) и ДИЭА (779,8 мг, 6,034 ммоль) смешали в 2 мл ДМФ и перемешивали при комнатной температуре в течение ночи. Реакционную смесь загрузили на картридж Samplet и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-80% ацетонитрила в воде, для получения указанного в заголовке соединения (1144 мг, 2,788 ммоль, выход 92,40%). МС (apci) m/z=411,2 (М+Н).
Стадия В: Получение (R)-3-((3S,4R)-3-(трет-бутоксикарбониламино)-4-(3,4-дифторфенил)пирролидин-1-ил)-1,1,1-трифторпропан-2-ил метансульфоната. Трет-бутил (3S,4R)-4-(3,4-дифторфенил)-1-((R)-3,3,3-трифтор-2-гидроксипропил)пирролидин-3-илкарбамат (900 мг, 2,19 ммоль) растворили в 5 мл CH2Cl2 и охладили до 0°C. Добавили ДИЭА (1146 мкл, 6,58 ммоль), затем MsCl (204 мкл, 2,63 ммоль). Реакционную смесь оставили нагреваться до комнатной температуры в течение 1 часа, концентрировали и очистили обращенно-фазовой колоночной хроматографией, элюируя 5-90% ацетонитрила в воде, для получения указанного в заголовке соединения (805 мг, 1,65 ммоль, выход 75,1%). МС (apci) m/z=489,1 (М+Н).
Стадия С: Получение (S)-2-((3S,4R)-3-амино-4-(3,4-дифторфенил)пирролидин-1-ил)-3,3,3-трифторпропан-1-ола. N,N,N-триметилгексадекан-1-аминия хлорид (25% раствор в H2O, 3144 мг, 2,46 ммоль) и (R)-3-((3S,4R)-3-(трет-бутоксикарбониламино)-4-(3,4-дифторфенил)пирролидин-1-ил)-1,1,1-трифторпропан-2-ил метансульфонат (800 мг, 1,64 ммоль) смешали в 0,5 мл ТГФ и нагревали при 150°C в течение 3 дней. Реакционную смесь загрузили на картридж Samplet и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-50% ацетонитрила в воде, для получения указанного в заголовке соединения (287 мг, 0,925 ммоль, выход 56,5%). МС (apci) m/z=311,1 (М+Н).
Стадия D: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-((S)-1,1,1-трифтор-3-гидроксипропан-2-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)мочевины. (S)-2-((3S,4R)-3-амино-4-(3,4-дифторфенил)пирролидин-1-ил)-3,3,3-трифторпропан-1-ол (10 мг, 0,032 ммоль), фенил-3-этокси-4-метил-1-фенил-1Н-пиразол-5-илкарбамат (полученный по способу Примера 1, Стадия А, исходя из Промежуточного соединения Р135; 9,1 мг, 0,027 ммоль) и ДИЭА (10 мг, 0,081 ммоль) смешали в 0,2 мл ДМФ и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь загрузили на картридж Samplet и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-60% ацетонитрила в воде, для получения указанного в заголовке соединения (8,1 мг, 0,015 ммоль, выход 54%). МС (apci) m/z=554,2 (М+Н).
Пример 276
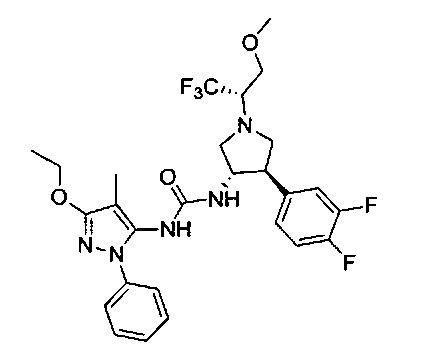
1-((3S,4R)-4-(3,4-дифторфенил)-1-((S)-1,1,1-трифтор-3-метоксипропан-2-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Стадия А: Получение трет-бутил (3S,4R)-4-(3,4-дифторфенил)-1-((S)-1,1,1-трифтор-3-гидроксипропан-2-ил)пирролидин-3-илкарбамата.(S)-2-((3S,4R)-3-амино-4-(3,4-дифторфенил)пирролидин-1-ил)-3,3,3-трифторпропан-1-ол (120 мг, 0,387 ммоль, полученный так, как описано в Примере 11, Стадия С), Boc2O (92,9 мг, 0,425 ммоль) и ПС-ДМАП (27,2 мг, 0,0387 ммоль) смешали в 5 мл CH2Cl2 и оставили стоять при комнатной температуре в течение ночи. Добавили дополнительное количество Boc2O (30 мг, 0,14 ммоль), затем дополнительное количество ПС-ДМАП (27,2 мг, 0,0387 ммоль). Реакционную смесь оставили стоять в течение 2 часов, отфильтровали, концентрировали и очистили обращенно-фазовой колоночной хроматографией, элюируя 5-95% ацетонитрила в воде, для получения указанного в заголовке соединения (63 мг, 0,154 ммоль, выход 39,7%). МС (apci) m/z=411,2 (М+Н).
Стадия В: Получение трет-бутил (3S,4R)-4-(3,4-дифторфенил)-1-((S)-1,1,1-трифтор-3-метоксипропан-2-ил)пирролидин-3-илкарбамата. Трет-бутил (3S,4R)-4-(3,4-дифторфенил)-1-((S)-1,1,1-трифтор-3-гидроксипропан-2-ил)пирролидин-3-илкарбамат (25 мг, 0,061 ммоль) и Ag2O (28 мг, 0,12 ммоль) смешали в 1 мл толуола и перемешивали при 0°C. Добавили йодметан (10 мг, 0,073 ммоль) и оставили реакционную смесь нагреваться до комнатной температуры, и перемешивали в течение 5 часов. Добавили ацетонитрил (0,5 мл) и дополнительное количество йодметана (10 мг, 0,073 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение ночи, отфильтровали через Celite®, концентрировали и очистили обращенно-фазовой колоночной хроматографией, элюируя 5-95% ацетонитрила в воде, для получения указанного в заголовке соединения (22 мг, 0,052 ммоль, выход 85%). МС (apci) m/z=425,2 (М+Н).
Стадия С: Получение (3S,4R)-4-(3,4-дифторфенил)-1-((S)-1,1,1-трифтор-3-метоксипропан-2-ил)пирролидин-3-амина дигидрохлорида. Трет-бутил (3S,4R)-4-(3,4-дифторфенил)-1-((S)-1,1,1-трифтор-3-метоксипропан-2-ил)-3-илкарбамат (20 мг, 0,0471 ммоль) и HCl (5 н. в IPA, 37,7 мкл, 0,188 ммоль) смешали в 5 мл CH2Cl2 и перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь концентрировали для получения указанного в заголовке соединения (15 мг, 0,0463 ммоль, выход 98,2%). МС (apci) m/z=325,1 (М+Н).
Стадия D: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-((S)-1,1,1-трифтор-3-метоксипропан-2-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)мочевины.
(3S,4R)-4-(3,4-дифторфенил)-1-((S)-1,1,1-трифтор-3-метоксипропан-2-ил)пирролидин-3-амина дигидрохлорид (7,5 мг, 0,019 ммоль), фенил 3-этокси-4-метил-1-фенил-1Н-пиразол-5-илкарбамат (полученный по способу Примера 1, Стадия A, исходя из Промежуточного соединения Р135; 5,8 мг, 0,017 ммоль) и ДИЭА (6,7 мг, 0,051 ммоль) смешали в 0,2 мл ДМФ и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь загрузили на картридж Samplet и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-90% ацетонитрила в воде, для получения указанного в заголовке соединения (6,8 мг, 0,012 ммоль, выход 70%). МС (apci) m/z=568,2 (М+Н).
Пример 277
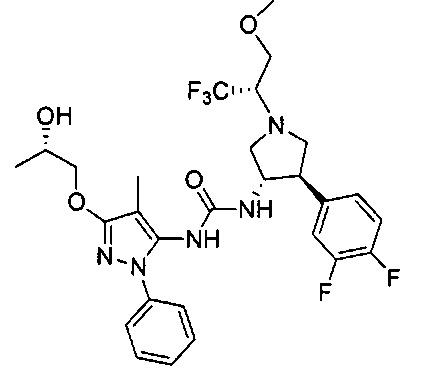
1-((3S,4R)-4-(3,4-дифторфенил)-1-((S)-1,1,1-трифтор-3-метоксипропан-2-ил)пирролидин-3-ил)-3-(3-((S)-2-гидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Получили по способу, описанному в Примере 12, Стадия D, используя (S)-фенил 3-(2-гидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-илкарбамат вместо фенил 3-этокси-4-метил-1-фенил-1Н-пиразол-5-илкарбамата. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-70% ацетонитрила в H2O, для получения указанного в заголовке соединения (7,7 мг, 0,013 ммоль, выход 75%). МС (apci) m/z=598,3 (М+Н).
Пример 278
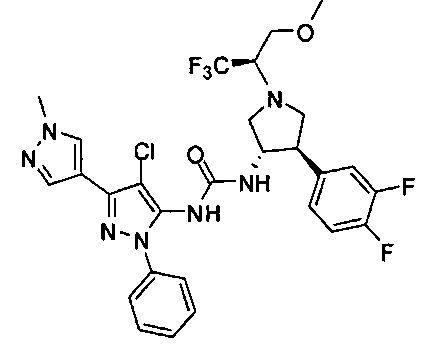
1-(4-хлор-1'-метил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-((R)-1,1,1-трифтор-3-метоксипропан-2-ил)пирролидн-3-ил)мочевина
Стадия А: Получение (S)-3-((3S,4R)-3-(трет-бутоксикарбониламино)-4-(3,4-дифторфенил)пирролидин-1-ил)-1,1,1-трифторпропан-2-ил метансульфоната. (трет-бутил (3S,4R)-4-(3,4-дифторфенил)пирролидин-3-илкарбамат (1000 мг, 3,352 ммоль), (S)-2-(трифторметил)оксиран (413,2 мг, 3,687 ммоль) и ДИЭА (866,4 мг, 6,704 ммоль) смешали в 2 мл ДМФ и перемешивали при комнатной температуре в течение ночи. Добавили метансульфонилхлорид (285,4 мкл, 3,687 ммоль) и перемешивали реакционную смесь в течение 1 часа. Еще два раза добавили дополнительное количество метансульфонилхлорида (285,4 мкл, 3,687 ммоль), каждый раз перемешивая в течение 20 минут. Реакционную смесь загрузили на картридж Samplet и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-80% ацетонитрила в воде, для получения указанного в заголовке соединения (1194 мг, 2,444 ммоль, выход 72,92%). МС (apci) m/z=411.1 (М+Н).
Стадия В: Получение (3S,4R)-4-(3,4-дифторфенил)-1-((R)-1,1,1-трифтор-3-метоксипропан-2-ил)пирролидин-3-амина. N,N,N-триметилгексадекан-1-аминия гидросульфат (281 мг, 0,737 ммоль) и (S)-3-((3S,4R)-3-(трет-бутоксикарбониламино)-4-(3,4-дифторфенил)пирролидин-1-ил)-1,1,1-трифторпропан-2-ил метансульфонат (180 мг, 0,368 ммоль) смешали в 10 мл МеОН в сосуде для работы под давлением и нагревали при 170°C в течение 20 часов. Реакционную смесь загрузили на картридж Samplet и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-95% ацетонитрила в воде, для получения указанного в заголовке соединения (67 мг, 0,207 ммоль, выход 56,1%). МС (apci) m/z=425,1 (М+Н).
Стадия С: 1-(4-хлор-1'-метил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-((R)-1,1,1-трифтор-3-метоксипропан-2-ил)пирролидн-3-ил)мочевина. Фенил 4-хлор-1'-метил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-илкарбамат (25 мг, 0,0635 ммоль), (3S,4R)-4-(3,4-дифторфенил)-1-((R)-1,1,1-трифтор-3-метоксипропан-2-ил)пирролидин-3-аминадигидрохлорид (27,7 мг, 0,069 ммоль) и ДИЭА (24,6 мг, 0,190 ммоль) смешали в 0,2 мл ДМФ и перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь загрузили на картридж Samplet и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-80% ацетонитрила в воде, для получения указанного в заголовке соединения (17,2 мг, 0,0276 ммоль, выход 43,4%). МС (apci) m/z=624.2 (М+Н).
Пример 279
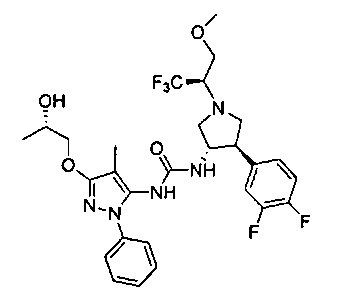
1-((3S,4R)-4-(3,4-дифторфенил)-1-((R)-1,1,1-трифтор-3-метоксипропан-2-ил)пирролидин-3-ил)-3-(3-((S)-2-гидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Получили по способу, описанному в Примере 14, Стадия С, используя (S)-фенил 3-(2-гидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-илкарбамат вместо фенил 4-хлор-1'-метил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-илкарбамата. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-75% ацетонитрила в Н2О, для получения указанного в заголовке соединения (13,0 мг, 0,0218 ммоль, выход 95,1%). МС (apci) m/z=598,3 (М+Н).
Следующее соединение было получено по способу Примера 52, с использованием соответствующих исходных материалов.
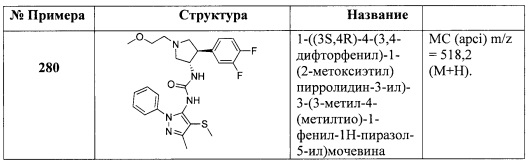
Следующие соединения были получены по способу Примера 1, Стадия В, с использованием соответствующих исходных материалов.
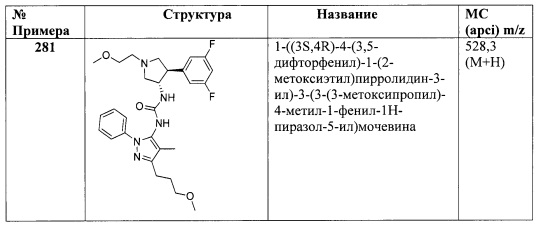
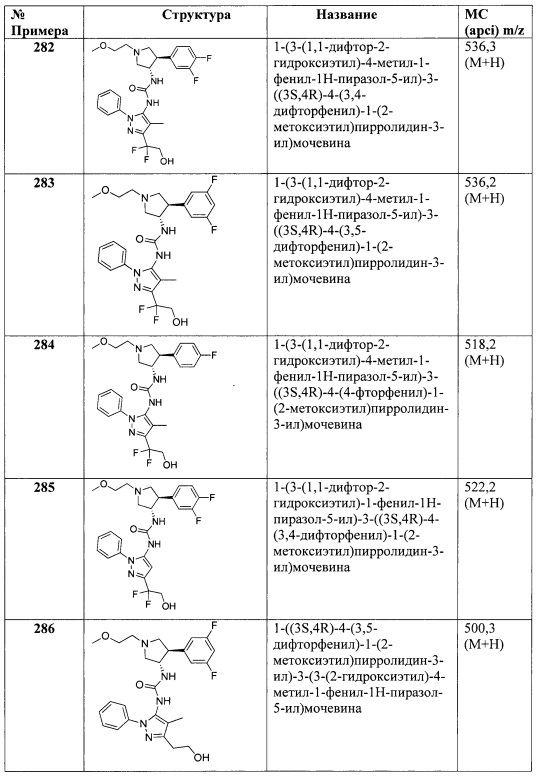
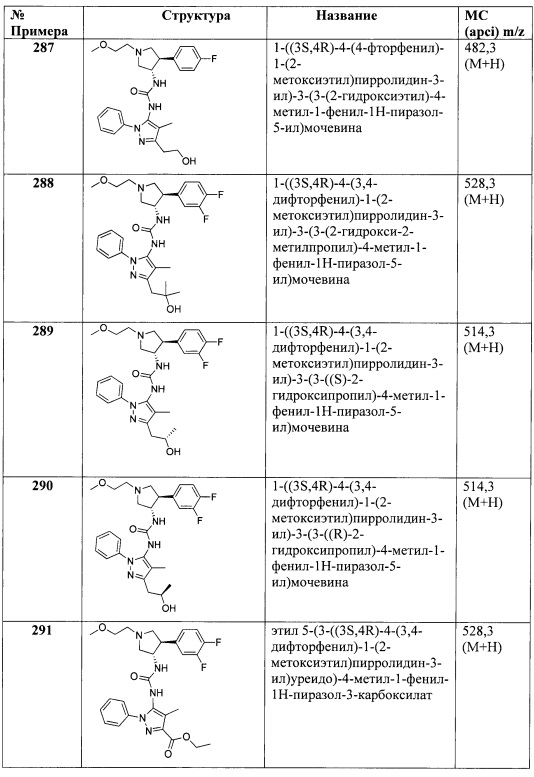
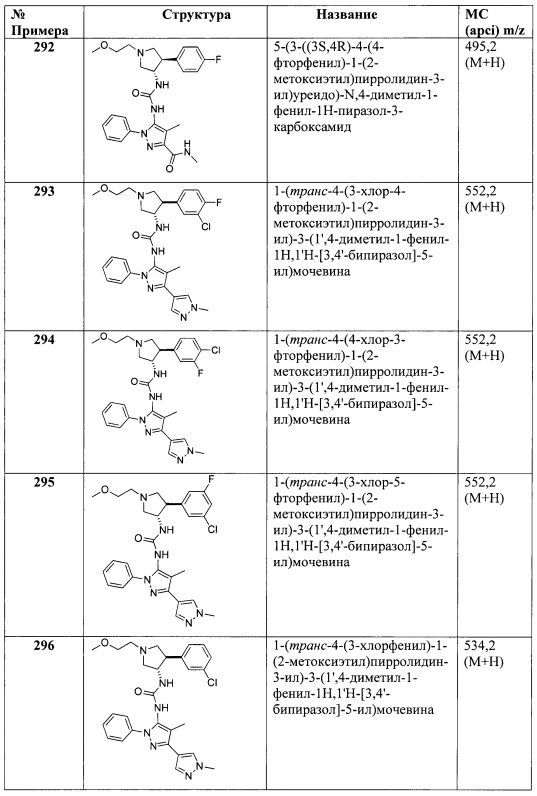
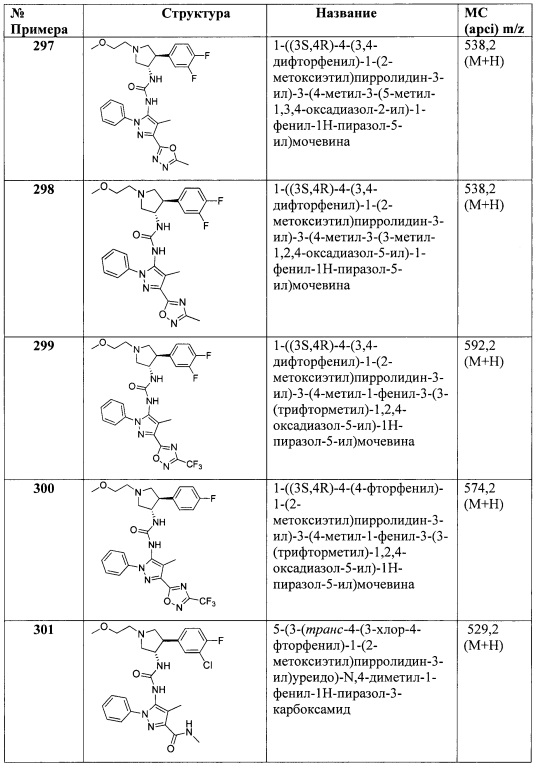
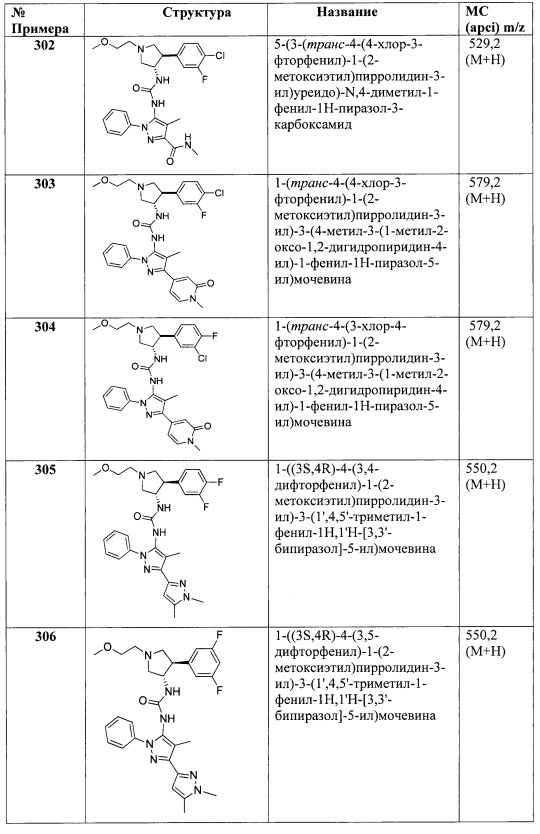
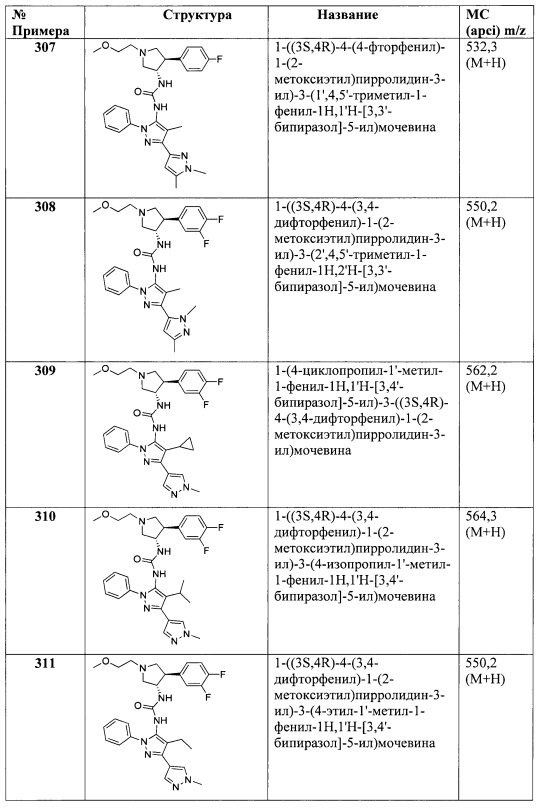
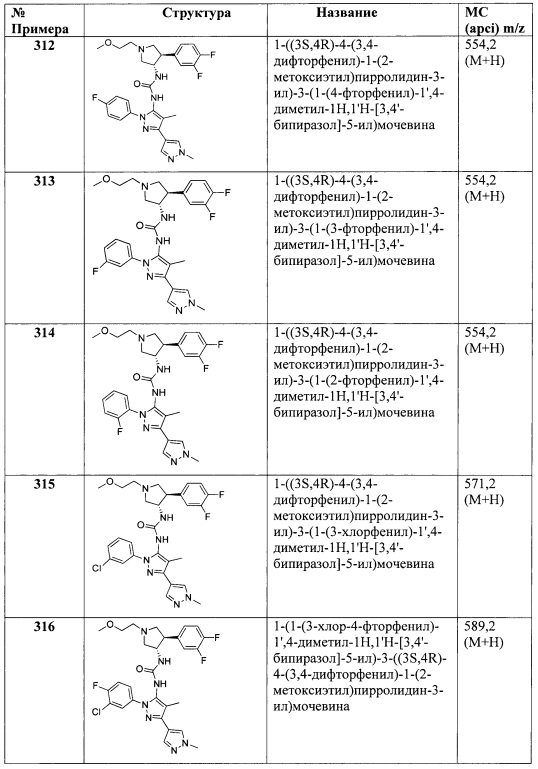
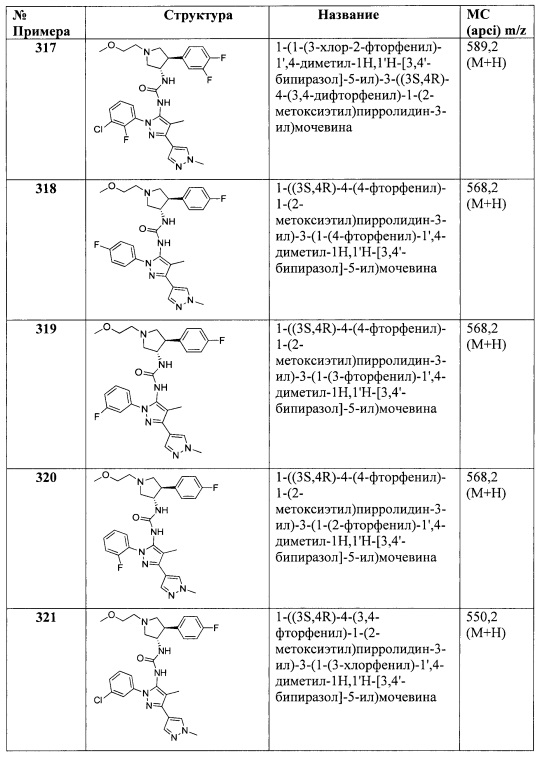
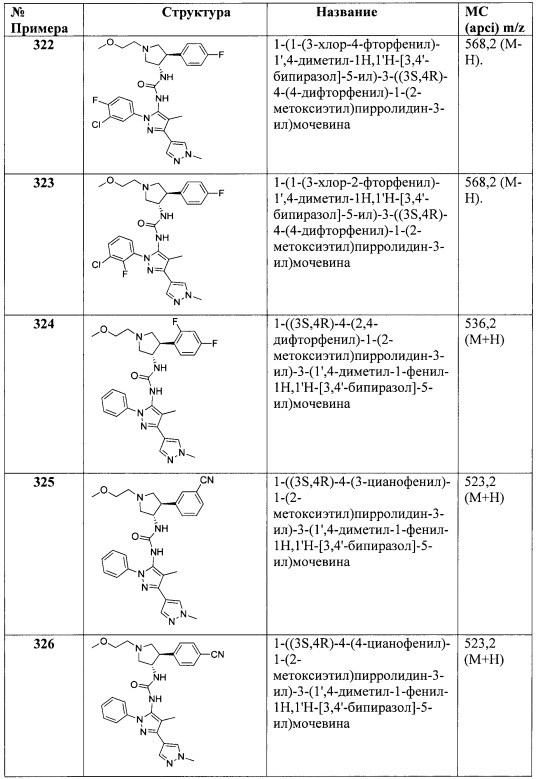
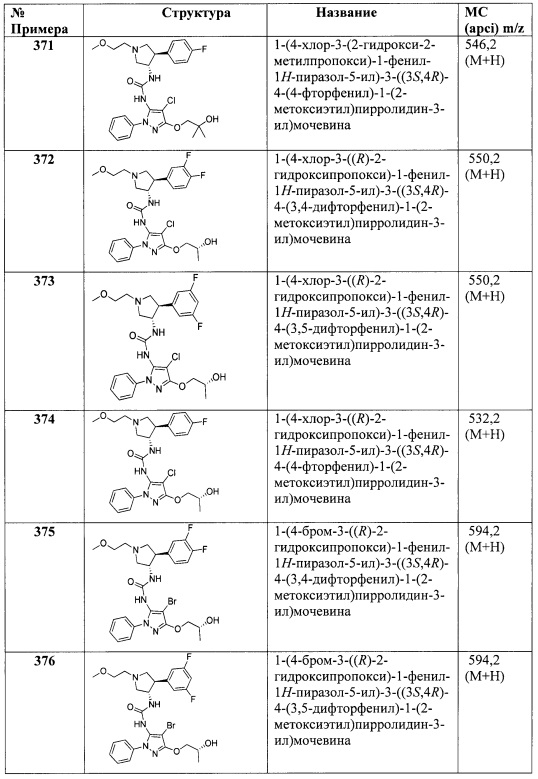
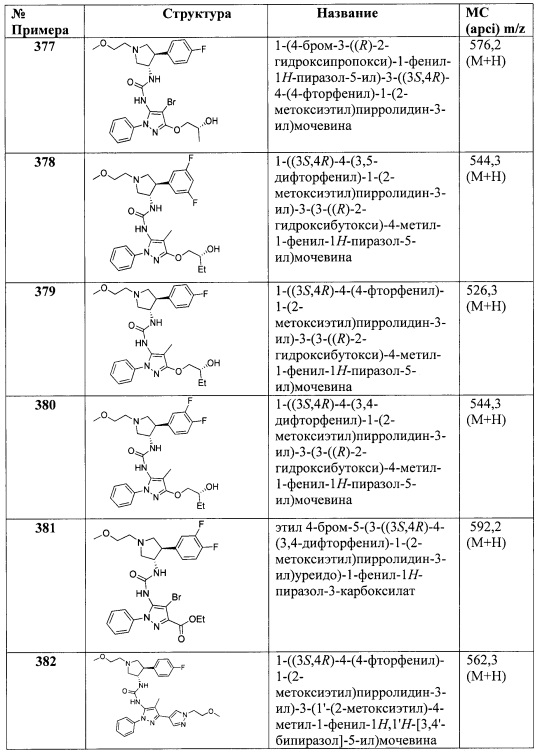
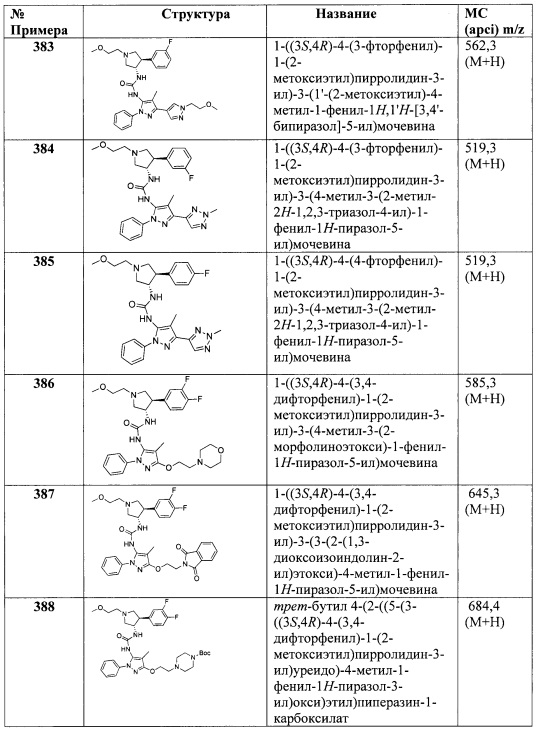
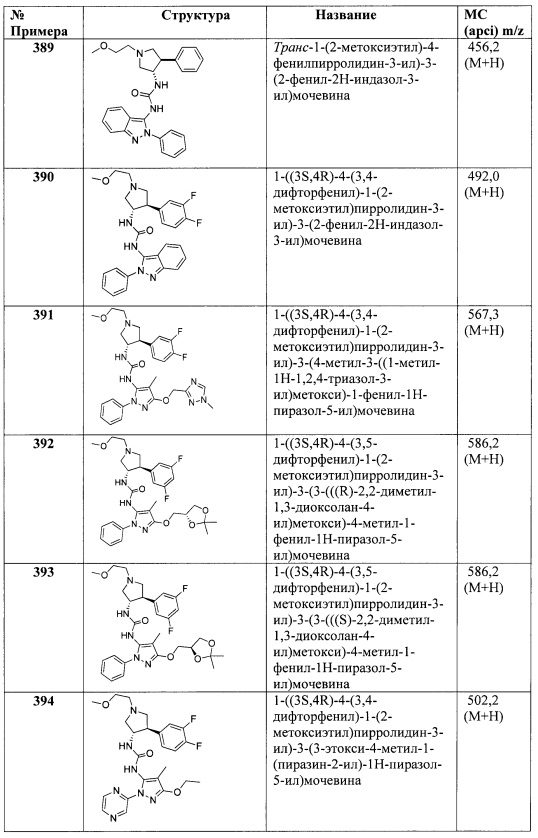
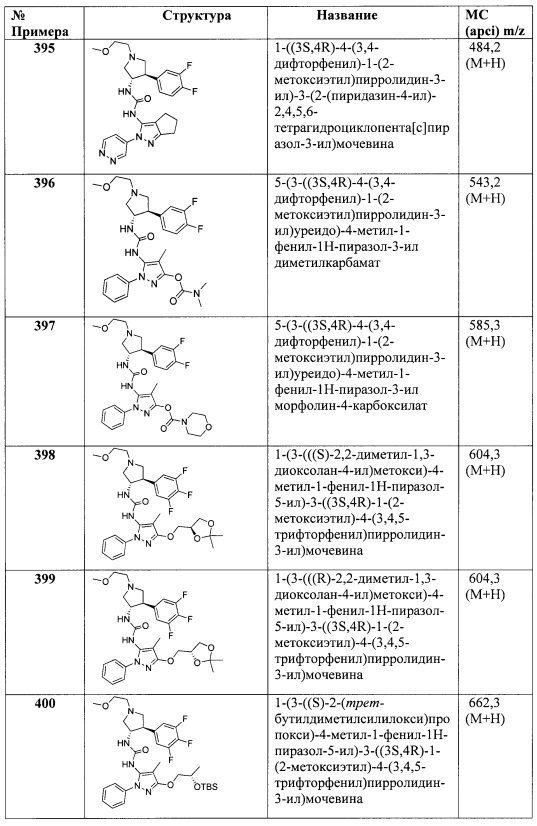
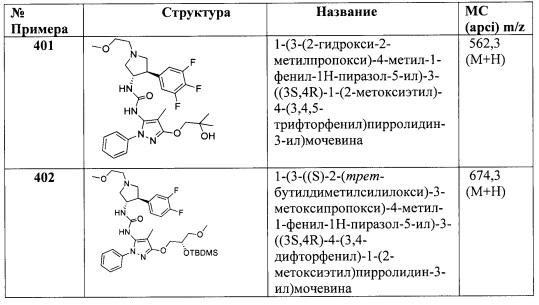
Следующие соединения были получены по способу Примера 1, Стадия В, с использованием соответствующих галогенированных пиразол-карбаматных исходных материалов.
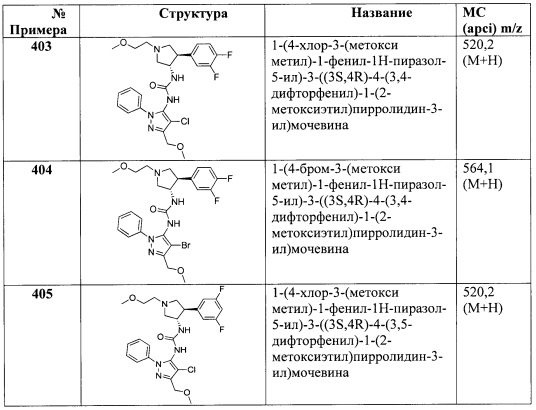
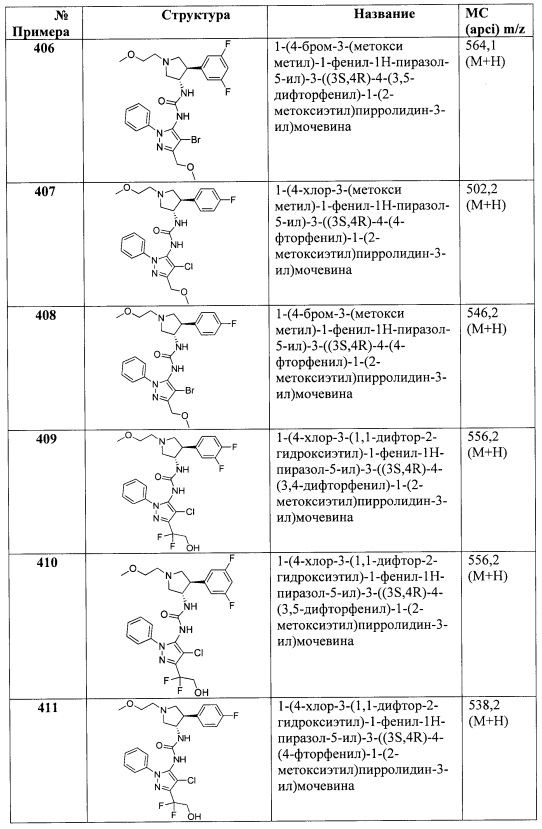
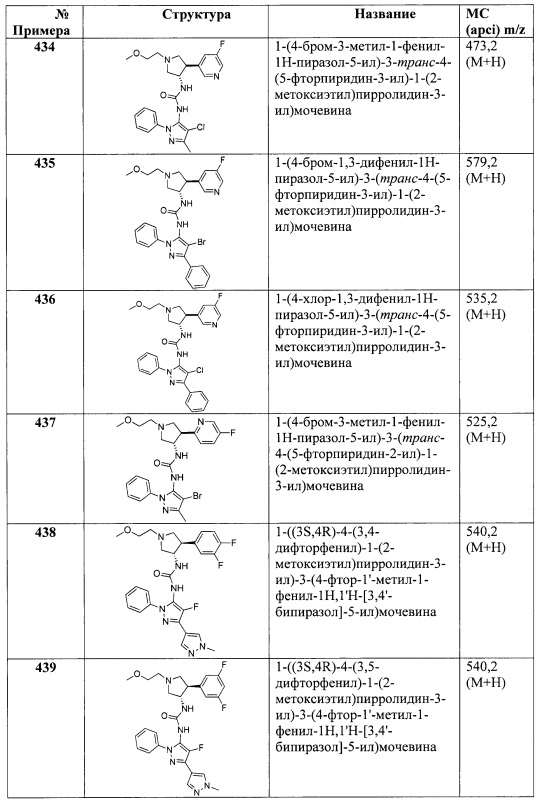
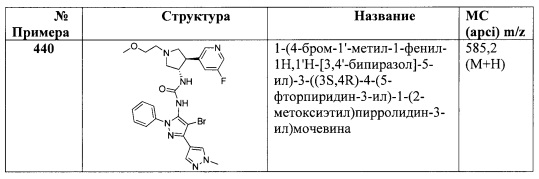
Пример 441
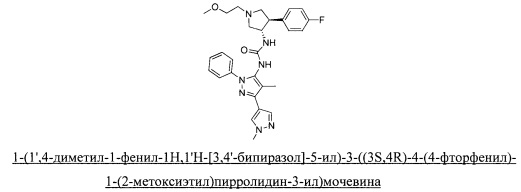
К суспензии (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорида (Получение L1; 40 мг, 0,13 ммоль) в ДМА (428 мкл) добавили ДИЭА (112 мкл, 0,64 ммоль) для получения прозрачного раствора. К этому раствору добавили фенил 1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-илкарбамат (Промежуточное соединение 199; 53 мг, 0,14 ммоль). После перемешивания в течение ночи, реакционную смесь напрямую очистили обращенно-фазовой хроматографией (С18, от 5 до 42% ацетонитрила в воде) для получения 1-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины в виде белого твердого вещества (42 мг, 63%). МС (apci) m/z=518,3 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 7,85 (s, 1Н), 7,74 (s, 1H), 7,48-7,5 (m, 2H), 7,34-7,38 (m, 2H), 7,27-7,31 (m, 1H), 7,05-7,1 (m, 2H), 6,81-6,85 (m, 2H), 5,37 (br s, 1H), 4,27 (br s, 1H), 3,96 (s, 3H), 3,33-3,43 (m, 2H), 3,25 (br s, 3H), 3,16-3,19 (m, 1H), 2,98 (br s, 1H), 2,70-2,83 (m, 2H), 2,49-2,65 (m, 2H), 2,27 (t, 1H), 2,09 (s, 3H).
Пример 442
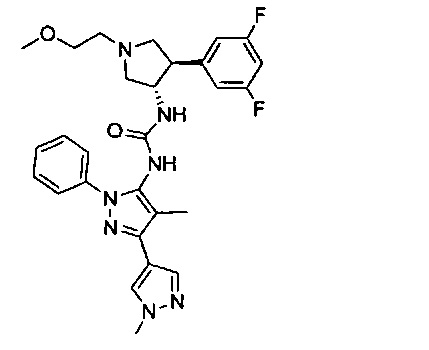
1-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному в Примере 441, заменяя (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид на (3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид на стадии F. МС (apci) m/z=536,3 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 7,86 (s, 1Н), 7,74 (s, 1H), 7,49-7,52 (m, 2H), 7,36-7,41 (m, 2H), 7,28-7,33 (m, 1H), 6,62-6,70 (m, 3H), 5,33 (br s, 1H), 4,27 (br s, 1H), 3,96 (s, 3H), 3,36-3,45 (m, 2H), 3,27 (br s, 3H), 3,15-3,20 (m, 1H), 2,96 (br s, 1H), 2,72-2,81 (m, 2H), 2,54-2,67 (m, 2H), 2,31 (t, 1H), 2,11 (s, 3H).
Пример 443
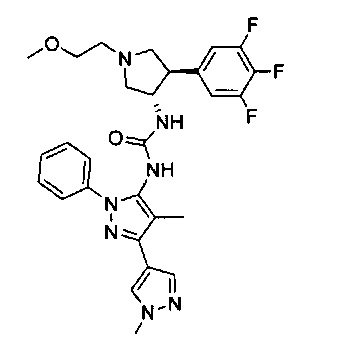
1-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4,5-трифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному для Примера 441, заменяя (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид на (3S,4R)-4-(3,4,5-трифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид на Стадии F. МС (apci) m/z=554,3 (М+Н).
Пример 444
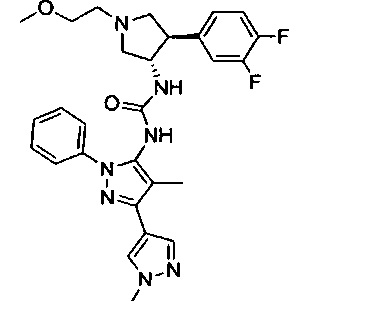
1-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному в Примере 441, заменяя(3S,4R)-4-(2,4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид на (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид на стадии F. МС (apci) m/z - 554,3 (М+Н). 1Н ЯМР (400 МГц, CDCl3) δ 7,86 (s, 1Н), 7,74 (s, 1H), 7,48-7,52 (m, 2H), 7,34-7,39 (m, 2H), 7,28-7,32 (m, 1H), 6,92-7,04 (m, 2H), 6,81-6,85 (m, 1H), 5,32 (br s, 1H), 4,24 (br s, 1H), 3,96 (s, 3H), 3,38-3,40 (m, 2H), 3,26 (br s, 3H), 3,12-3,17 (m, 1H), 2,91 (br s, 1H), 2,72-2,77 (m, 2H), 2,52-2,66 (m, 2H), 2,27 (t, 1H), 2,1 (s, 3H).
Пример 445
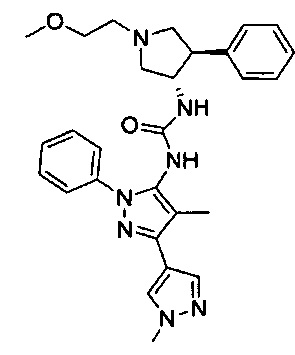
1-(1',4-диметил-1-фенил-1H,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному в Примере 441, заменяя (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид на (3S,4R)-4-фенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид на Стадии F. МС (apci) m/z=500,3 (М+Н).
Пример 446
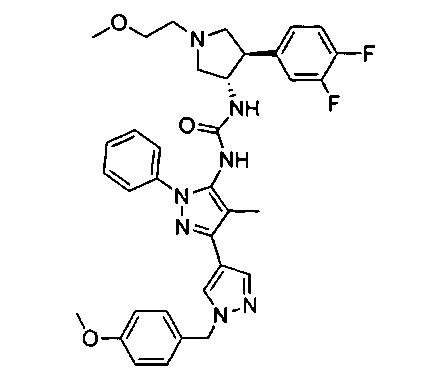
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-[1'(4-метоксибензил)-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил')мочевина
Получили по способу, описанному в Примере 441, заменяя (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид на (3S,4R)-4-(3,4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид и фенил 1,4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-илкарбамат на фенил (1'-(4-метоксибензил)-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)карбамат на Стадии F. МС (apci) m/z=642,3 (М+Н).
Пример 447
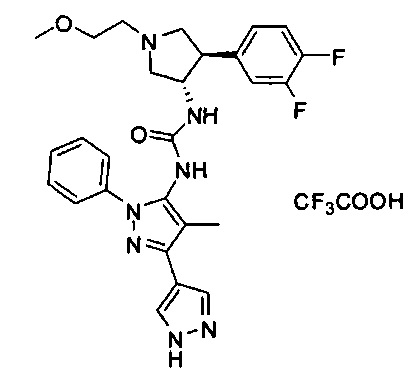
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-4-метоксибензил)-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол1-5-ил)мочевины трифторацетат
Смесь 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-(4-метоксибензил)-4-метил-1-фенил-1Н,1'Н-[-3,4'-бипиразол]-5-ил)мочевины (300 мг, 0,468 ммоль) и ТФК (720 мкл, 9,35 ммоль) в сосуде для работы под давлением закрыли и нагревали при 60°С. Реакционную смесь нагревали в течение 18 часов, затем охладили и добавили эфир (30 мл), а полученную смесь обработали ультразвуком для получения неочищенного продукта в виде бежевого твердого вещества. Твердое вещество растворили в минимальном количестве МеОН и очистили обращенно-фазовой хроматографией (С18, от 5 до 35%, до 50% ацетонитрила в воде) для получения 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-(4-метоксибензил)-4-метил-1-фенил-1-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевины в виде трифлатной соли (135 мг, 38%).
Пример 448
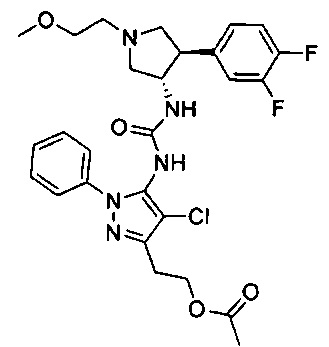
2-(4-хлор-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-3-ил)этилацетат
Стадия А: Получение 2-(4-хлор-5-((феноксикарбонил)амино)-1-фенил-1Н-пиразол-3-ил)этилацетата и фенил (3-(2-((феноксикарбонил)окси)этил)-1-фенил-1Н-пиразол-5-ил)карбамата: К раствору 2-(5-амино-1-фенил-1Н-пиразол-3-ил)этанола (Промежуточное соединение 161, 242 мг, 1,191 ммоль) в EtOAc (10 мл) добавили водный раствор гидроксида натрия (2 М, 1,19 мл, 2,38 ммоль), затем фенилкарбонохлоридат (0,179 мл, 1,43 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, а затем разделили фазы. Органическую фазу промыли H2O (5 мл), насыщенным солевым раствором (5 мл), высушили при помощи MgSO4, отфильтровали и концентрировали для получения продукта в виде желто-коричневого твердого вещества (250 мг), смеси двух компонентов: 2-(4-хлор-5-((феноксикарбонил)амино)-1-фенил-1Н-пиразол-3-ил)этилацетата и фенил (3-(2-((феноксикарбонил)окси)этил)-1-фенил-1Н-пиразол-5-ил)карбамата, которую использовали без очистки на следующей стадии.
Стадия В: Получение 2-(4-хлор-5-((феноксикарбонил)амино)-1-фенил-1Н-пиразол-3-ил)этилацетата и фенил (4-хлор-3-(2-((феноксикарбонил)окси)этил)-1-фенил-1Н-пиразол-5-ил)карбамата: К раствору 2-(4-хлор-5-((феноксикарбонил)амино)-1-фенил-1 Н-пиразол-3-ил)этилацетата и фенил (3-(2-((феноксикарбонил)окси)этил)-1-фенил-1Н-пиразол-5-ил)карбамата (250 мг, 0,773 ммоль) в ДХМ (10 мл) добавили пиридиния 4-метилбензолсульфонат (PPTS) (19,4 мг, 0,077 ммоль) и н-хлорсукцинимид (155 мг, 1,16 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 дней, затем разбавили H2O (10 мл), разделили фазы, а водную фазу экстрагировали ДХМ (2×20 мл). Объединенные органические фазы высушили (MgSO4), отфильтровали и концентрировали. Неочищенное маслянистое вещество очистили колоночной хроматографией на диоксиде кремния, элюируя 40% ацетона в гексанах, для получения продукта в виде оранжевого маслянистого вещества (63 мг), смеси двух компонентов: 2-(4-хлор-5-((феноксикарбонил)амино)-1-фенил-1Н-пиразол-3-ил)этилацетата, МС (apci) m/z=400,1 (М+Н), и фенил (4-хлор-3-(2-((феноксикарбонил)окси)этил)-1-фенил-1Н-пиразол-5-ил)карбамата, МС (apci) m/z=478,1 (М+Н).
Стадия С: Получение 2-(4-хлор-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-3-ил)этилацетата: К раствору (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорида (Получение F, 52 мг, 0,158 ммоль) в ДМА (0,6 мл) добавили ДИЭА (0,110 мл, 0,630 ммоль), добавили смесь 2-(4-хлор-5-((феноксикарбонил)амино)-1-фенил-1Н-пиразол-3-ил)этилацетата и фенил (4-хлор-3-(2-((феноксикарбонил)окси)этил)-1-фенил-1Н-пиразол-5-ил)карбамата (63 мг). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-80% ацетонитрила в воде, и собирая Пик 1 для получения указанного в заголовке соединения в виде оранжевого твердого вещества (21,8 мг). МС (apci) m/z=562,2 (М+Н).
Пример 449
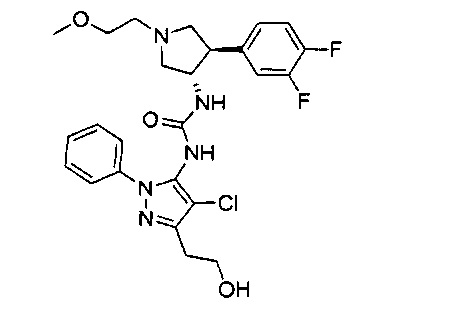
1-(4-хлор-3-(2-гидроксиэтил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Стадия А: Получение 2-(4-хлор-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-3-ил)этилфенилкарбоната: Реакционную смесь из Примера 448, Стадия С, напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-80% ацетонитрила в воде, и собирая Пик 2 для получения указанного в заголовке соединения в виде бледно-желтого смолянистого вещества (28 мг). МС (apci) m/z=640,2 (М+Н).
Стадия В: Получение 1-(4-хлор-3-(2-гидроксиэтил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины: К раствору 2-(4-хлор-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-3-ил)этилфенилкарбоната (28 мг, 0,044 ммоль) в ТГФ (2 мл) и МеОН (1 мл) добавили водный раствор LiOH (2 М, 0,066 мл, 0,132 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем разбавили водным раствором HCl (2 М, 0,06 мл) и Н2О (5 мл), и экстрагировали ДХМ (10 мл), затем 10: 90 МеОН/ДХМ (2×10 мл). Объединенные органические экстракты высушили (MgSO4), отфильтровали и концентрировали. Неочищенный продукт очистили колоночной хроматографией на диоксиде кремния, элюируя 0-10% МеОН в ДХМ, для получения продукта в виде грязновато-белого твердого вещества (14 мг, выход 61%). МС (apci) m/z=520,2 (М+Н).
Пример 450
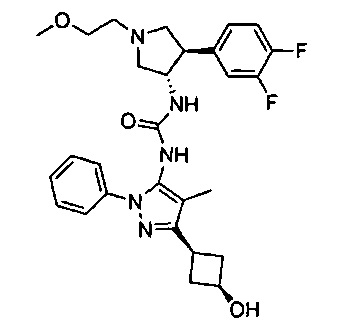
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(цис-3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Стадия А: Получение цис- и транс-1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины: К раствору (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорида (Получение F, 67 мг, 0,204 ммоль) в ДМА (1 мл) и ДИЭА (0,142 мл, 0,815 ммоль) добавили фенил 3-(3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-илкарбамат (Промежуточное соединение 209; 74 мг, 0,204 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 1 часа. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-70% ацетонитрила в воде, для получения продукта в виде белого твердого вещества (59 мг, выход 55%). МС (apci) m/z=526,3 (М+Н).
Стадия B: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(цис-3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины: Смесь цис- и транс-1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины (55 мг, 0,105 ммоль) разделили препаративной ВЭЖХ на бензамидной колонке (Princeton Analytical, 4,6 мм×250 мм, 5 мкм), элюируя 10% EtOH в гексанах. Собрали Пик 1 для получения указанного в заголовке соединения в виде белого твердого вещества (21,1 мг, выход 38%). МС (apci) m/z=526,3 (М+Н). 1Н ЯМР (d6-ДМСО) δ 7,79 (br s, 1Н), 7,42 (m, 4Н), 7,30 (m, 3Н), 7,06 (m, 1Н), 6,70 (d, 1H), 5,06 (d, 1Н), 4,04 (m, 2Н), 3,43 (t, 2Н), 3,24 (s, 3H), 3,05 (m, 2Н), 2,85 (m, 2Н), 2,47-2,66 (m, 6Н), 2,08 (m, 2Н), 1,77 (s, 3H).
Пример 451
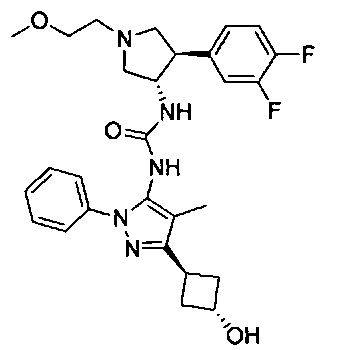
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(транс-3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Смесь цис- транс-гидроксициклобутиловых диастереомеров 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины (Пример 450, Стадия А, 55 мг, 0,105 ммоль) разделили препаративной ВЭЖХ на бензамидной колонке (Princeton Analytical, 4,6 мм×250 мм, 5 мкм), элюируя 10% EtOH в гексанах. Собрали Пик 2 для получения указанного в заголовке соединения в виде белого твердого вещества (27,5 мг, выход 50%). МС (apci) m/z=526,3 (М+Н). 1Н ЯМР (d6-ДМСО) δ 7.80 (br s, 1Н), 7,42 (m, 4H), 7,31 (m, 3H), 7,06 (m, 1H), 6,70 (d, 1H), 5,02 (d, 1H), 4,33 (m, 1H), 4,04 (m, 1H), 3,43 (t, 2H), 3,34 (m, 1H), 3,24 (s, 3H), 3,05 (m, 2H), 2,88 (t, 1H), 2,43-2,67 (m, 6H), 2,19 (m, 2H), 1,74 (s, 3H).
Пример 452
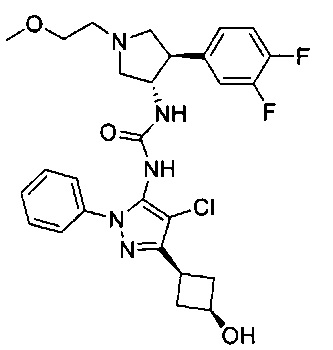
1-(4-хлор-3-(цис-3-гидроксициклобутил)-1-фенил-1H-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Стадия А: цис- и транс-1-(4-хлор-3-(3-гидроксициклобутил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина: Получили по способу, описанному в Примере 450, Стадия А, заменяя фенил 3-(3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-илкарбамат на фенил (4-хлор-3-(3-гидроксициклобутил)-1-фенил-1Н-пиразол-5-ил)карбамат (Промежуточное соединение 238) на Стадии А, для получения продукта в виде смеси цис- и транс-гидроксициклобутиловых диастереомеров. МС (apci) m/z=546,2 (М+Н).
Стадия В: Получение 1-(4-хлор-3-(цис-3-гидроксициклобутил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины: Смесь цис- и транс-гидроксициклобутиловых диастереомеров 1-(4-хлор-3-(3-гидроксициклобутил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины (41 мг, 0,075 ммоль) разделили препаративной ВЭЖХ на колонке DEAP (Princeton Analytical, 4,6 мм×250 мм, 5 мкм), элюируя 10% EtOH в гексанах. Собрали Пик 1 для получения указанного в заголовке соединения в виде белого твердого вещества (10,2 мг, выход 25%). МС (apci) m/z=546,2 (М+Н). 1Н ЯМР (CDCl3) 7,50 (d, 2Н), 7,41 (t, 2Н), 7,30 (m, 1Н), 7,05 (m, 1Н), 6,97 (m, 1H), 6,86 (m, 1H), 5,48 (m, 1Н), 4,30 (m, 1Н), 3,43 (br m, 2Н), 3,33 (m, 1Н), 3,25 (br m, 2Н), 3,10 (m, 2Н), 2,96 (br m, 1Н), 2,60-2,83 (m, 5Н), 2,33 (m, 3H).
Пример 453
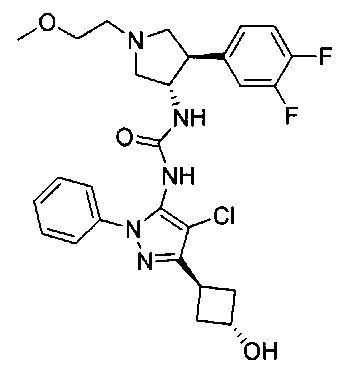
1-(4-хлор-3-((1r,3S)-3-гидроксициклобутил)-1-фенил-1Н-пиразол-5-ил)-3-(транс-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Смесь цис- и транс-1-(4-хлор-3-(3-гидроксициклобутил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины (Пример 452, Стадия А, 41 мг, 0,075 ммоль) разделили препаративной ВЭЖХ на колонке DEAP (Princeton Analytical, 4,6 мм×250 мм, 5 мкм), элюируя 10% EtOH в гексанах. Собрали Пик 1 для получения указанного в заголовке соединения в виде белого твердого вещества (16,4 мг, выход 40%). МС (apci) m/z=546,2 (M+H). 1H ЯМР (CDCl3) 7,51 (d, 2H), 7,41 (t, 2H), 7,33 (m, 1H), 7,04 (m, 1H), 6,96 (m, 1H), 6,85 (m, 1H), 5,53 (br d, 1H), 4,65 (m, 1H), 3,60 (m, 1H), 3,41 (br m, 2H), 3,25 (br m, 5H), 3,08 (br m, 1H), 2,89 (br m, 1H), 2,57-2,77 (m, 7H), 2,37 (m, 3H).
Пример 454
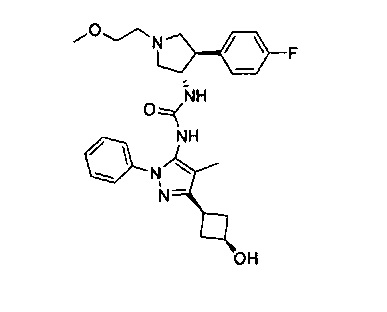
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(цис-3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Стадия А: Получение цис- и транс-1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины: Получили по способу, описанному в Примере 450, Стадия А, заменяя (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-аминадигидрохлорид на (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (Получение К) на Стадии А, для получения продукта в виде смеси цис- и транс-гидроксициклобутиловых диастереомеров. МС (apci) m/z=508,3 (М+Н).
Стадия В: Получение 1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(цис-3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины: смесь цис- и транс-1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины (49 мг, 0,096 ммоль) разделили сверхкритической жидкостной хроматографией (СЖХ) на цианоколонке (YMC-PackCN, 250×20 мм, 10 мкм), подвижная фаза 95% CO2 и 5% 80/20/0,1 MeOH/IPA/диэтиламина. Собрали Пик 1 для получения указанного в заголовке соединения в виде белого твердого вещества (16,4 мг, выход 34%). МС (apci) m/z=508,2 (М+Н). 1Н ЯМР (d6-ДМСО) δ 7,85 (br s, 1H), 7,42 (m, 4H), 7,28 (m, 3H), 7,10 (t, 2H), 6,75 (br d, 1H), 5,06 (br s, 1H), 4,04 (m, 2H), 3,42 (t, 2H), 3,24 (s, 3H), 3,06 (m, 2H), 2,84 (m, 2H), 2,46-2,64 (m, 6H), 2,08 (m, 2H), 1,76 (s, 3H).
Пример 455
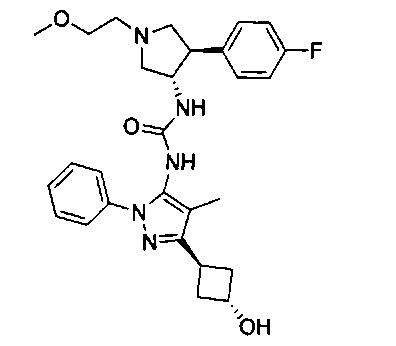
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(транс-3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Смесь цис- и транс-1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины (Пример 454, Стадия А, 49 мг, 0,096 ммоль) разделили сверхкритической жидкостной хроматографией (СЖХ) на цианоколонке (YMC-Pack CN, 250×20 мм, 10 мкм), подвижная фаза 95% CO2 и 5% 80/20/0,1 MeOH/IPA/диэтиламина. Собрали Пик 2 для получения указанного в заголовке соединения в виде белого твердого вещества (19,0 мг, выход 39%). МС (apci) m/z=508,2 (М+Н). 1Н ЯМР (d6-ДМСО) δ 7,86 (br s, 1Н), 7,42 (m, 4Н), 7,28 (m, 3H), 7,10 (t, 2Н), 6,75 (br d, 1Н), 5,03 (br s, 1H), 4,33 (m, 1H), 4,03 (m, 1H), 3,42 (t, 2H), 3,34 (m, 1H), 3,24 (s, 3H), 3,07 (m, 2H), 2,84 (t, 1H), 2,43-2,64 (m, 6H), 2,19 (m, 2H), 1,73 (s, 3H).
Пример 456
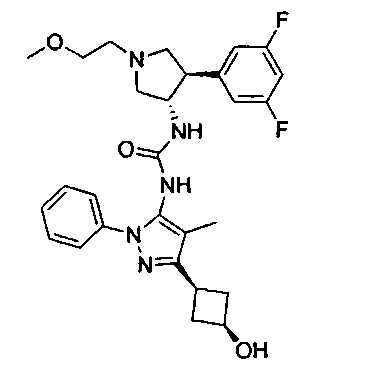
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(цис-3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Стадия А: Получение цис- и транс-1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины: Получили по способу, описанному в Примере 450, Стадия А, заменяя (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид на (3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (Получение Е) на Стадии А, для получения продукта в виде смеси цис- и транс-гидроксициклобутиловых диастереомеров. МС (apci) m/z=526,3 (М+Н).
Стадия B: Получение 1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(цис-3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины: Смесь цис- и транс-гидроксициклобутиловых диастереомеров 1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины (53 мг, 0,10 ммоль) разделили сверхкритической жидкостной хроматографией (СЖХ) на цианоколонке (YMC-Pack CN, 250×20 мм, 10 мкм), подвижная фаза 95% CO2 и 5% 80/20/0,1 MeOH/IPA/диэтиламина. Собрали Пик 1 для получения указанного в заголовке соединения в виде белого твердого вещества (17,6 мг, выход 33%). МС (apci) m/z=526,2 (М+Н). 1Н ЯМР (d6-ДМСО) 5 7,95 (br s, 1H), 7,45 (d, 2H), 7,39 (t, 2H), 7,28 (t, 1H), 7,05 (tt, 1H), 6,97 (m, 2H), 6,85 (m, 1H), 5,08 (br s, 1H), 4,05 (m, 2H), 3,43 (t, 2H), 3,24 (s, 3H), 3,08 (m, 1H), 3,02 (m, 1H), 2,89 (m, 1H), 2,84 (m, 1H), 2,45-2,65 (m, 6H), 2,08 (m, 2H), 1,77 (s, 3H).
Пример 457
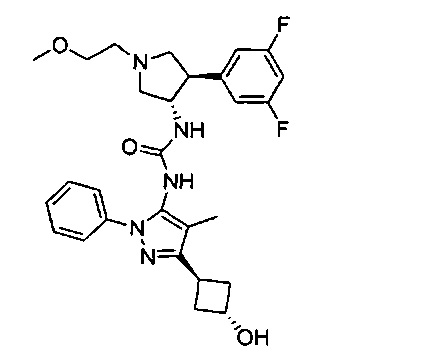
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(транс-3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Смесь цис- и транс-гидроксициклобутиловых диастереомеров 1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины (Пример 456, Стадия А, 53 мг, 0,10 ммоль) разделили сверхкритической жидкостной хроматографией (СЖХ) на цианоколонке (YMC-Pack CN, 250×20 мм, 10 мкм), подвижная фаза 95% CO2 и 5% 80/20/0,1 МеОН/IPA/диэтиламина. Собрали Пик 2 для получения указанного в заголовке соединения в виде белого твердого вещества (20,7 мг, выход 39%). МС (apci) m/z=526,2 (М+Н). 1Н ЯМР (d6-ДМСО) δ 7,96 (br s, 1H), 7,46 (d, 2Н), 7,40 (t, 2Н), 7,28 (t, 1Н), 7,05 (tt, 1Н), 6,97 (m, 2Н), 6,85 (m, 1H), 5,04 (br s, 1Н), 4,34 (m, 1Н), 4,06 (m, 1Н), 3,43 (t, 2Н), 3,35 (m, 1Н), 3,24 (s, 3H), 3,09 (q, 1Н), 3,02 (t, 1H), 2,90 (t, 1Н), 2,44-2,66 (m, 6Н), 2,19 (m, 2Н), 1,74 (s, 3H).
Пример 458
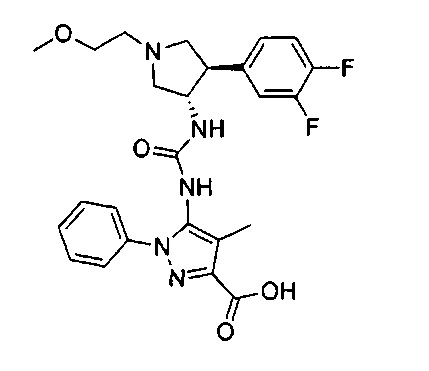
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-карбоновая кислота
К раствору этил 5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-карбоксилата (Пример 291; 333 мг, 6,21 ммоль) в ТГФ (4 мл) и МеОН (2 мл) добавили водный раствор LiOH (2 М, 0,95 мл, 1,89 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, затем частично концентрировали под пониженным давлением, затем нейтрализовали водным раствором HCl (1 М, 1 мл). Суспензию отфильтровали, а белый осадок собрали для получения указанного в заголовке соединения в виде белого твердого вещества (247 мг, выход 78%). МС (apci) m/z=500,2 (М+Н).
Пример 459
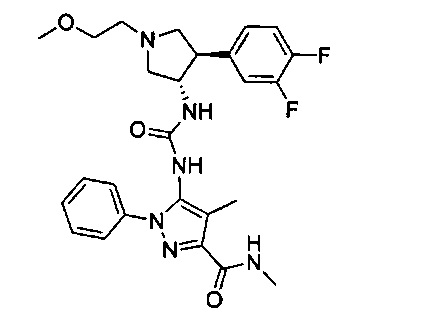
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-N,4-диметил-1-фенил-1Н-пиразол-3-карбоксамид
К раствору 5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-карбоновой кислоты (Пример 458, 25 мг, 0,050 ммоль) в ДМФ (0,5 мл) добавили ДИЭА (0,026 мл, 0,150 ммоль), метанамина гидрохлорид (6,8 мг, 0,100 ммоль), затем НАТУ (20,9 мг, 0,055 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 19 часов. Реакционную смесь напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-60% ацетонитрила в воде, для получения продукта в виде белого твердого вещества (13,5 мг, выход 53%). МС (apci) m/z=513,3 (М+Н).
Пример 460
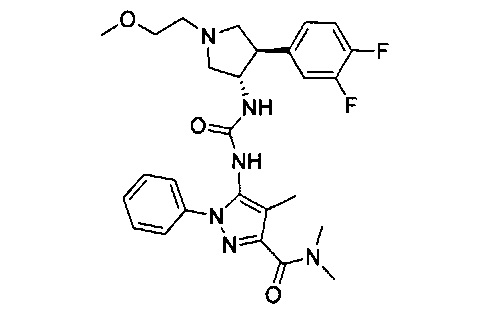
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-N,N,4-триметил-1-фенил-1Н-пиразол-3-карбоксамид
Получили по способу, описанному в Примере 459, заменяя метанамина гидрохлорид на диметиламин (2M в ТГФ), для получения продукта в виде белого твердого вещества (13,0 мг, выход 49%). МС (apci) m/z=527,2 (М+Н).
Пример 461
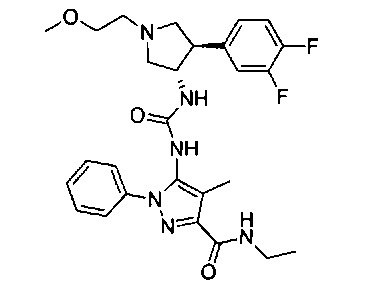
5-(3-((3S,4R-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-N-этил-4-метил-1-фенил-1Н-пиразол-3-карбоксамид
Получили по способу, описанному в Примере 459, заменяя метанамина гидрохлорид на этанамин (2 M в ТГФ), для получения продукта в виде белого твердого вещества (11,2 мг, выход 48%). МС (apci) m/z=527,2 (М+Н).
Пример 462
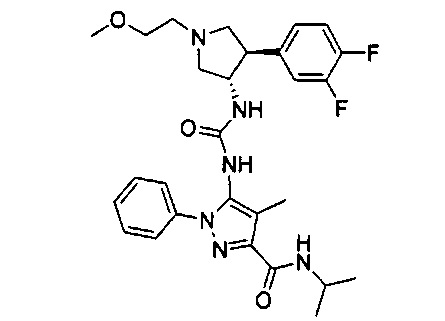
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-уреидо)N-изопропил-4-метил-1-фенил-1Н-пиразол-3-карбоксамид
Получили по способу, описанному в Примере 459, заменяя метанамина гидрохлорид на пропан-2-амин, для получения продукта в виде белого твердого вещества (5,6 мг, выход 24%). МС (apci) m/z=541,3 (М+Н).
Пример 463
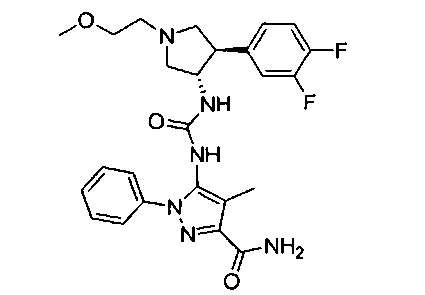
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-карбоксамид
Раствор 1-(3-циано-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины (Пример 349, 25 мг, 0,52 ммоль) в концентрированной H2SO4 (0,2 мл) перемешивали при комнатной температуре в течение 20 часов. Реакционную смесь охладили до 0°C и нейтрализовали добавлением водного раствора NaOH (15 вес.%, 4 мл), затем экстрагировали 10% МеОН/ДХМ (3×10 мл), а объединенные органические экстракты промыли насыщенным солевым раствором, высушили (MgSO4), отфильтровали и концентрировали под пониженным давлением. Неочищенный продукт очистили обращенно-фазовой колоночной хроматографией, элюируя 5-60% ацетонитрила в воде, для получения продукта в виде белого твердого вещества (1,4 г, выход 5%). МС (apci) m/z=499,2 (М+Н).
Пример 464
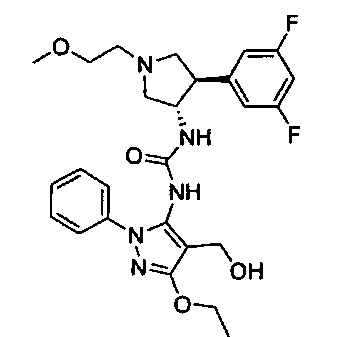
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-этокси-4-(гидроксиметил)-1-фенил-1Н-пиразол-5-ил)мочевина
Стадия А: Получение этил 5-(3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-3-этокси-1-фенил-1Н-пиразол-4-карбоксилата: К раствору этил 5-амино-3-этокси-1-фенил-1Н-пиразол-4-карбоксилата (Промежуточное соединение 174, 68 мг, 0,25 ммоль) в ДХМ (1 мл) добавили ДИЭА (0,086 мл, 0,49 ммоль), затем трифосген (26 мг, 0,086 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем добавили дополнительное количество трифосгена (26 мг, 0,086 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 22 часов, а затем добавили дополнительное количество трифосгена (26 мг, 0,086 ммоль). Реакционную смесь перемешивали при комнатной температуре еще 5 часов, затем добавили раствор (3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорида (Получение E, 81 мг, 0,25 ммоль) и ДИЭА (0,13 мл, 0,74 ммоль) в ДХМ (0,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем концентрировали под пониженным давлением и очистили обращенно-фазовой колоночной хроматографией, элюируя 5-70% ацетонитрила в воде, для получения продукта в виде белого твердого вещества (12 мг, выход 9%). МС (apci) m/z=558,3 (М+Н).
Стадия B: Получение 1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-этокси-4-(гидроксиметил)-1-фенил-1Н-пиразол-5-ил)мочевины: К раствору этил 5-(3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-3-этокси-1-фенил-1Н-пиразол-4-карбоксилата (18 мг, 0,032 ммоль) в ТГФ (2 мл) под N2, охлажденному до 0°C, добавили LiAlH4 (1 Мбис-ТГФ в толуоле, 0,032 мл, 0,032 ммоль). Реакционную смесь перемешивали при 0°C в течение 2 часов, затем при комнатной температуре в течение 90 минут, затем охладили до 0°C и погасили добавлением Н2О (0,005 мл), 5 мкл водного раствора NaOH (1 М, 0,005 мл), затем H2O (0,015 мл), перемешивали в течение 10 минут, затем отфильтровали, промыли ТГФ (2×5 мл) и концентрировали. Неочищенный продукт очистили препаративной ТСХ (пластина 0,5 мм, элюировали 5% MeOH в ДХМ) для получения продукта в виде бесцветного остатка (1,6 мг, выход 10%). МС (apci) m/z=516,3 (М+Н).
Пример 465
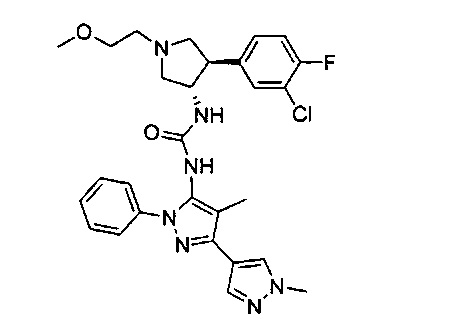
1-((3S,4R)-4-(3-хлор-4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1H'-3,4'-бипиразол-5-ил)-мочевина
Рацемическую смесь 1-(транс-(4-(3-хлор-4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)мочевины (Пример 293, 30 мг, 0,054 ммоль) разделили хиральной ВЭЖХ (колонка Chiralcel OD), элюируя 10% EtOH в гексанах. Собрали Пик 1 для получения указанного в заголовке соединения в виде белого твердого вещества (9,8 мг, выход 33%). МС (apci) m/z=552,2 (М+Н).
Пример 466
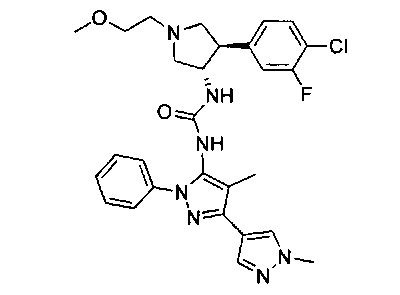
1-((3S,4R)-4-(4-хлор-3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)мочевина
Рацемическую смесь 1-(транс-4-(4-хлор-3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)мочевины (Пример 294, 42 мг, 0,076 ммоль) разделили хиральной ВЭЖХ (колонка Chiralcel OD), элюируя 10% EtOH в гексанах. Собрали Пик 1 для получения указанного в заголовке соединения в виде белого твердого вещества (17,1 мг, выход 41%). МС (apci) m/z=552,2 (М+Н).
Пример 467
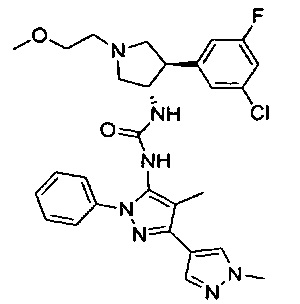
1-((3S,4R)-4-(3-хлор-5-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)мочевина
Рацемическую смесь 1-(транс-4-(3-хлор-5-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)мочевины (Пример 295, 32 мг, 0,058 ммоль) разделили хиральной ВЭЖХ (колонка Chiralpak IA), элюируя 15% EtOH в гексанах. Собрали Пик 2 для получения указанного в заголовке соединения в виде белого твердого вещества (12,5 мг, выход 39%). МС (apci) m/z=552,2 (М+Н).
Пример 468
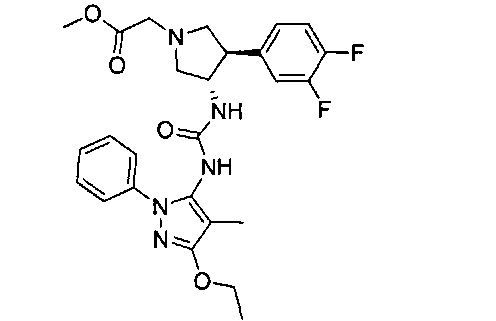
метил 2-((3R,4S)-3-(3,4-дифторфенил)-4-(3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)уреидо)пирролидин-1-ил)ацетат
Получили по способу, описанному в Примере 191, заменяя 2-бромэтанол на метил 2-бромацетат на Стадии A, и заменяя фенил 3,4-диметил-1-фенил-1Н-пиразол-5-илкарбамат на фенил 3-этокси-4-метил-1-фенил-1Н-пиразол-5-илкарбмат (полученный по способу Примера 1, Стадия А, исходя из Промежуточного соединения Р135) на Стадии C, для получения продукта в виде белого твердого вещества (23 мг, выход 50%). МС (apci) m/z=514,2 (М+Н).
Пример 469
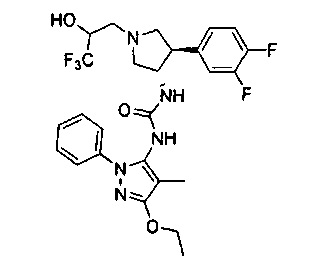
1-((3S,4R)-4-(3,4-дифторфенил)-1-(3,3,3-трифтор-2-гидроксипропил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Получили по способу, описанному в Примере 191, заменяя 2-бромэтанол на метил 3-бром-1,1,1-трифторпропан-2-ол на Стадии A, и заменяя фенил 3,4-диметил-1-фенил-1Н-пиразол-5-илкарбамат на фенил 3-этокси-4-метил-1-фенил-1Н-пиразол-5-илкарбмат (полученный по способу Примера 1, Стадия A, исходя из Промежуточного соединения Р135) на Стадии C, для получения продукта в виде белого твердого вещества (38 мг, выход 78%). МС (apci) m/z=554,2 (М+Н).
Пример 470
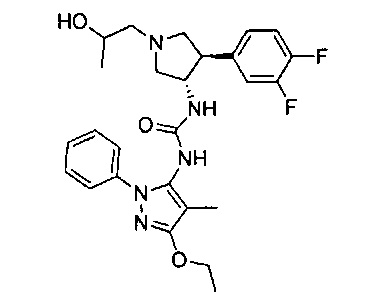
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-гидроксипропил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Получили по способу, описанному в Примере 191, заменяя 2-бромэтанол на метил 1-хлорпропан-2-ол (Aldrich, чистота 70% с <25% 2-хлорпропан-1-ола) на Стадии А, и заменяя фенил 3,4-диметил-1-фенил-1Н-пиразол-5-илкарбамат на фенил 3-этокси-4-метил-1-фенил-1Н-пиразол-5-илкарбмат (полученный по способу Примера 1, Стадия А, исходя из Промежуточного соединения Р135) на Стадии С, для получения продукта в виде белого твердого вещества (10 мг, выход 53%). МС (apci) m/z=500,2 (М+Н).
Пример 471
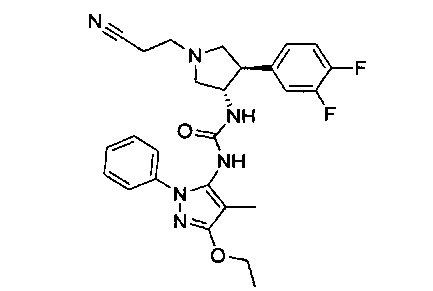
1-((3S,4R)-1-(2-пианоэтил)-4-(3,4-дифторфенил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Получили по способу, описанному в Примере 191, заменяя акрилонитрил на 1-хлорпропан-2-ол на Стадии А, и заменяя фенил 3,4-диметил-1-фенил-1Н-пиразол-5-илкарбамат на фенил 3-этокси-4-метил-1-фенил-1Н-пиразол-5-илкарбмат (полученный по способу Примера 1, Стадия А, исходя из Промежуточного соединения Р135) на стадии С, для получения продукта в виде белого твердого вещества (16 мг, выход 59%). МС (apci) m/z=459,3 (М+Н).
Пример 472
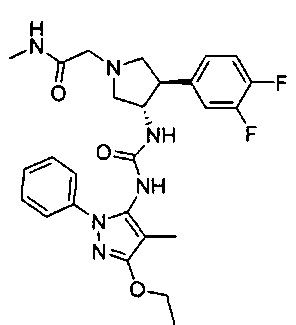
2-((3R,4S)-3-(3,4-дифторфенил)-4-(3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)уреидо)пирролидин-1-ил)-N-метилацетамид
Стадия А: Получение 2-((3R,4S)-3-(3,4-дифторфенил)-4-(3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)уреидо)пирролидин-1-ил)уксусной кислоты: К раствору метил 2-((3R,4S)-3-(3,4-дифторфенил)-4-(3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)уреидо)пирролидин-1-ил)ацетата (Пример 468; 15 мг, 0,029 ммоль) в ТГФ (0,8 мл) и MeOH (0,4 мл) добавили водный раствор LiOH (2 М, 0,044 мл, 0,088 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем разбавили водным раствором HCl (1 М, 1 мл) и насыщенным солевым раствором (2 мл) и экстрагировали ДХМ (2×5 мл). Объединенные органические экстракты высушили (MgSO4), отфильтровали и концентрировали для получения продукта в виде грязновато-белого твердого вещества (13,0 мг, выход 89%). МС (apci) m/z=500,2 (М+Н).
Стадия В: Получение 2-((3R,4S)-3-(3,4-дифторфенил)-4-(3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)уреидо)пирролидин-1-ил)-N-метилацетамида: К суспензии 2-((3R,4S)-3-(3,4-дифторфенил)-4-(3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)уреидо)пирролидин-1-ил)уксусной кислоты (7,4 мг, 0,015 ммоль) в ДМФ (0,5 мл) добавили N-метилморфолин (0,005 мл, 0,044 ммоль), метиламин (2 M в ТГФ, 0,009 мл, 0,018 ммоль), затем НАТУ (6,8 мг, 0,018 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 19 часов, затем напрямую очистили обращенно-фазовой колоночной хроматографией, элюируя 5-70% ацетонитрила в воде, для получения указанного в заголовке соединения в виде бледно-белого твердого вещества (4,2 мг, выход 55%). МС (apci) m/z=513,3 (М+Н).
Пример 473
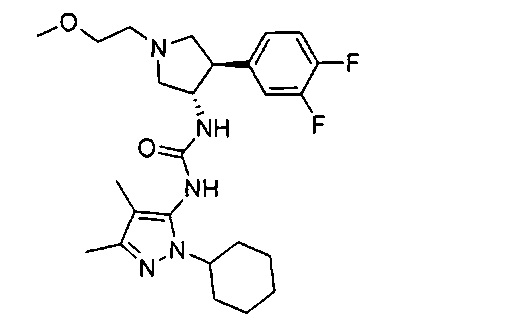
1-(1-циклогексил)-3,4-диметил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Стадия А: Получение 1-циклогексил-3,4-диметил-1Н-пиразол-5-амина: К суспензии циклогексилгидразина гидрохлорида (0,465 г, 3,09 ммоль) в этаноле (30 мл) добавили 2-оксоциклопентанкарбонитрил (0,30 г, 3,09 ммоль). Смесь нагревали с дефлегматором в течение 18 часов, затем охладили до комнатной температуры и концентрировали in vacuo. Остаток разделили между насыщенным раствором NaHCO3 (30 мл) и EtOAc (30 мл). Водный слой экстрагировали EtOAc (2×20 мл), а объединенные органические фазы промыли насыщенным солевым раствором (20 мл), высушили над Na2SO4, отфильтровали и концентрировали для получения 1-циклогексил-3,4-диметил-1Н-пиразол-5-амина (0,523 г, выход 88%) в виде кремового твердого вещества. 1Н ЯМР (CDCl3) δ 3,78-3,93 (m, 1Н), 3,13 (br s, 2Н), 2,12 (s, 3Н), 1,85-1,95 (m, 6Н), 1,83 (s, 3Н), 1,63-1,73 (m, 1Н), 1,18-1,44 (m, 3Н) ppm.
Стадия В: Получение фенил (1-циклогексил-3,4-диметил-1Н-пиразол-5-ил)карбамата: К раствору 1-циклогексил-3,4-диметил-1Н-пиразол-5-амина (200 мг, 1,04 ммоль) в EtOAc(5 мл) добавили 2 н. NaOH (1,04 мл, 2,1 ммоль), затем фенилхлорформиат (182 мкл, 1,45 ммоль). Смесь перемешивали при комнатной температуре в течение 5 часов, затем разбавили водой (30 мл) и экстрагировали EtOAc (3×20 мл). Объединенные органические фазы промыли насыщенным раствором NaHCO3 (20 мл) и насыщенным солевым раствором (20 мл), затем высушили над Na2SO4 и концентрировали in vacuo для получения фенил (1-циклогексил-3,4-диметил-1Н-пиразол-5-ил)карбамата в виде бледно-пурпурного пенистого вещества, которое использовали без очистки, предположив количественный выход.
Стадия С: Получение 1-(1-циклогексил)-3,4-диметил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины: К раствору (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина (Получение F) (50 мг, 0,15 ммоль) и фенил (1-циклогексил-3,4-диметил-1Н-пиразол-5-ил)карбамата (52 мг, 0,17 ммоль) в ДМА (2 мл) добавили ДИЭА (93 мкл, 0,53 ммоль). Смесь перемешивали при комнатной температуре в течение 18 часов, затем разделили между насыщенным раствором NH4Cl (20 мл) и EtOAc (20 мл), а водный слой экстрагировали EtOAc (2×10 мл). Объединенные органические фазы промыли водой (5×10 мл) и насыщенным солевым раствором (10 мл), затем высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 2% MeOH в ДХМ, для получения указанного в заголовке соединения (42 мг, выход 58%) в виде бесцветного стекловидного вещества. МС (apci) m/z=476,3 (М+Н).
Пример 474
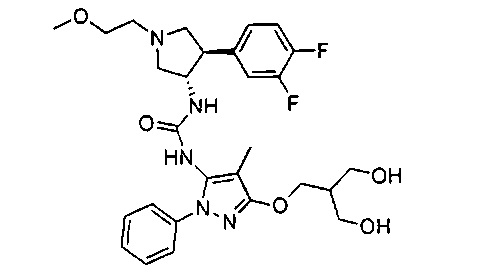
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(3-гидрокси-2-(гидроксиметил)пропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Стадия А: Получение (2,2-диметил-1,3-диоксан-5-ил)метанола: К суспензии 2-(гидроксиметил)пропан-1,3-диола (5,0 г, 47,1 ммоль) в ТГФ (100 мл) добавили п-толуолсульфоновой кислоты моногидрат (269 мг, 1,41 ммоль), затем 2,2-диметоксипропан (6,72 мл, 54,7 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов, затем добавили еще 200 мг п-толуолсульфоновой кислоты моногидрата и продолжали перемешивание еще 60 часов. Раствор обработали триэтиламином (3 мл), затем концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 5% MeOH в ДХМ, для получения (2,2-диметил-1,3-диоксан-5-ил)метанола (5,04 г, выход 73%) в виде бесцветной жидкости. 1Н ЯМР (CDCl3) δ 4,02 (dd, J=12,0, 4,1 Гц, 2Н), 3,74-3,80 (m, 4Н), 1,90 (t, J=5,1 Гц, 1H), 1,80-1,88 (m, 1Н), 1,45 (s, 3Н), 1,40 (s, 3Н) ppm.
Стадия В: Получение (2,2-диметил-1,3-диоксан-5-ил)метилметансульфоната: К раствору (2,2-диметил-1,3-диоксан-5-ил)метанола (1,0 г, 6,84 ммоль) в ДХМ (30 мл) при 0°C добавили триэтиламин (1,43 мл, 10,3 ммоль), затем мезилхлорид (0,58 мл, 7,52 ммоль). Смесь оставили медленно нагреваться до комнатной температуры и перемешивали в течение 18 часов. Смесь разделили между 0,5 М HCl (40 мл) и ДХМ (20 мл), а водный слой экстрагировали ДХМ (2×20 мл). Объединенные органические фазы промыли насыщенным солевым раствором (20 мл), высушили над Na2SO4, отфильтровали и концентрировали для получения (2,2-диметил-1,3-диоксан-5-ил)метилметансульфоната (1,29 г, выход 84%) в виде бесцветного маслянистого вещества. 1Н ЯМР (CDCl3) δ 4,42 (d, J=7,3 Гц, 2Н), 4,08 (dd, J=12,5, 3,5 Гц, 2Н), 3,77 (dd, J=12,5, 3,9 Гц, 2Н), 3,04 (s, 3Н), 1,98-2,03 (m, 1Н), 1,46 (s, 3Н), 1,39 (s, 3Н) ppm.
Стадия С: Получение 3-((2,2-диметил-1,3-диоксан-5-ил)метокси)-4-метил-1-фенил-1Н-пиразол-5-амина: К раствору 5-амино-4-метил-1-фенил-1Н-пиразол-3(2Н)-она (Промежуточное соединение Р135, Стадия А; 500 мг, 2,64 ммоль) в ДМФ (5 мл) добавили K2CO3 (1,10 г, 7,93 ммоль), затем раствор (2,2-диметил-1,3-диоксан-5-ил)метилметансульфоната (711 мг, 3,17 ммоль) в ДМФ (2 мл). Смесь перемешивали при 50°C в течение 18 часов, затем охладили, обработали водой (30 мл) и экстрагировали EtOAc (3×20 мл). Объединенные органические фазы промыли водой (5×10 мл) и насыщенным солевым раствором (10 мл), затем высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя от 4:1 до 2:1 смесью гексанов/EtOAc, для получения 3-((2,2-диметил-1,3-диоксан-5-ил)метокси)-4-метил-1-фенил-1Н-пиразол-5-амина (198 мг, 24%) в виде желтого смолянистого вещества. МС (apci) m/z=318,1 (М+Н).
Стадия D: Получение фенил (3-(3-гидрокси-2-(гидроксиметил)пропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)карбамата: К раствору 3-((2,2-диметил-1,3-диоксан-5-ил)метокси)-4-метил-1-фенил-1Н-пиразол-5-амина (198 мг, 0,62 ммоль) в EtOAc (5 мл) добавили 2 М раствор NaOH (780 мкл, 1,56 ммоль), затем фенилхлорформиат (117 мкл, 0,94 ммоль). Смесь перемешивали при комнатной температуре в течение 18 часов, затем разделили между водой (20 мл) и EtOAc (20 мл), а водный слой экстрагировали EtOAc (2×20 мл). Объединенные органические фазы промыли насыщенным раствором (20 мл) и насыщенным солевым раствором (20 мл), затем высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 2-4% MeOH в ДХМ, для получения (3-(3-гидрокси-2-(гидроксиметил)пропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)карбамата (75 мг, выход 30%)) в виде кремового пенистого вещества. МС (apci) m/z=398,2 (М+Н).
Стадия Е: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(3-гидрокси-2-(гидроксиметил)пропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины: Получили по способу Примера 473, Стадия С, заменяя фенил (1-циклогексил-3,4-диметил-1Н-пиразол-5-ил)карбамат на (3-(3-гидрокси-2-(гидроксиметил)пропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)карбамат. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2-5% MeOH в ДХМ, для получения указанного в заголовке соединения (26 мг, выход 55%) в виде бесцветного стекловидного вещества. МС (apci) m/z=560,3 (М+Н).
Пример 475
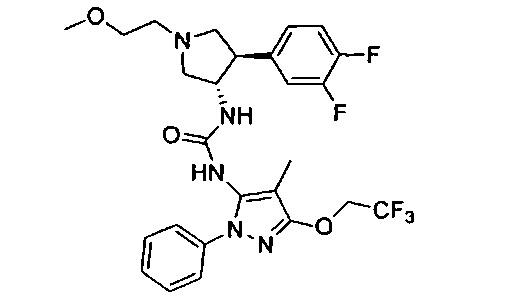
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-ил)мочевина
Стадия A: Получение 4-метил-1-фенил-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-амина: Получили по способу Примера 474, Стадия С, заменяя (2,2-диметил-1,3-диоксан-5-ил)метилметансульфонат на 1,1,1-трифтор-2-йодэтан. МС (apci) m/z=272,1 (М+Н).
Стадия В: Получение фенил (4-метил-1-фенил-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-ил)карбамата: Получили по способу Примера 474, Стадия D, заменяя 3-((2,2-диметил-1,3-диоксан-5-ил)метокси)-4-метил-1-фенил-1Н-пиразол-5-амин на 4-метил-1-фенил-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-амин. МС (apci) m/z=392,1 (М+Н).
Стадия C: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-ил)мочевины: Получили по способу Примера 473, Стадия С, заменяя фенил (1-циклогексил-3,4-диметил-1Н-пиразол-5-ил)карбамат на фенил (4-метил-1-фенил-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-ил)карбамат. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 5% MeOH в ДХМ, затем препаративной ВЭЖХ (5-95% ACN/H2O/0,1% ТФК, за 20 минут), для получения указанного в заголовке соединения (16 мг, выход 38%) после экстрактивного выделения продукта (ДХМ/1 н. NaOH) в виде белого твердого вещества. МС (apci) m/z=554,2 (М+Н).
Пример 476
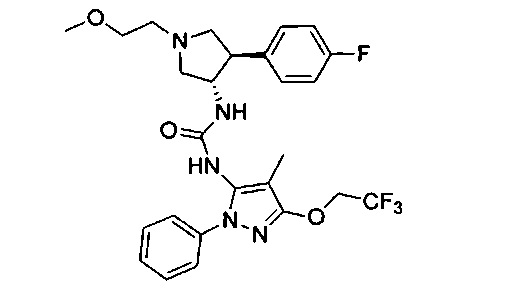
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-ил)мочевина
Получили по способу Примера 475, заменяя (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (Получение F) на (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (Получение К). Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2% MeOH в ДХМ, для получения указанного в заголовке соединения (56 мг, выход 65%) в виде белого твердого вещества. МС (apci) m/z=536,2 (М+Н).
Пример 477
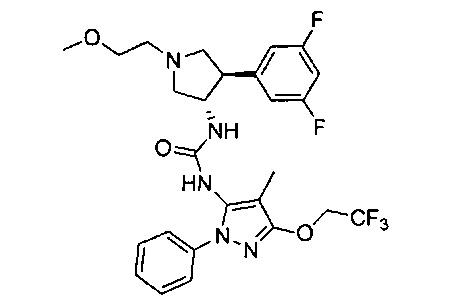
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-ил)мочевина
Получили по способу Примера 475, заменяя (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (Получение F) на (3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина трифторацетат (Получение Е). Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2% MeOH в ДХМ, для получения указанного в заголовке соединения (53 мг, выход 63%) в виде бесцветного стекловидного вещества. МС (apci) m/z=554,2 (М+Н).
Пример 478
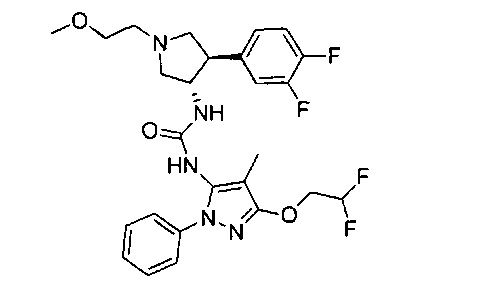
1-(3-(2,2-дифторэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу Примера 475, заменяя 1,1,1-трифтор-2-йодэтан на 1,1-дифтор-2-йодэтан на Стадии А. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2% MeOH в ДХМ, для получения указанного в заголовке соединения (55 мг, выход 68%) в виде белого твердого вещества. МС (apci) m/z=536,2 (М+Н).
Пример 479
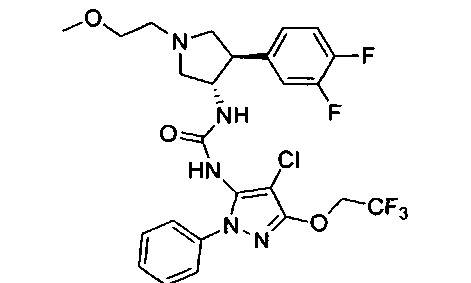
1-(4-хлор-1-фенил-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Стадия А: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-фенил-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-ил)мочевины: Получили по способу Примера 475, заменяя 5-амино-4-метил-1-фенил-1Н-пиразол-3(2Н)-он (Промежуточное соединение Р135, Стадия А) на 5-амино-1-фенил-1Н-пиразол-3(2Н)-он (Промежуточное соединение Р136, Стадия А).
Стадия В: Получение 1-(4-хлор-1-фенил-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-мочевины: К раствору 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-фенил-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-ил)мочевины (50 мг, 0,09 ммоль) в ДХМ (1 мл) добавили N-хлорсукцинимид (15 мг, 0,11 ммоль), затем пиридин-1-ия 4-метилбензолсульфонат (2 мг, 0,009 ммоль). Смесь перемешивали при комнатной температуре в течение 18 часов, затем обработали добавлением еще 5 мг N-хлорсукцинимида и перемешивали в течение 2,5 часов. Смесь разделили между насыщенным раствором NaHCO3 (20 мл) и ДХМ (20 мл), а водный слой экстрагировали ДХМ (2×10 мл). Объединенные органические фазы промыли насыщенным солевым раствором (10 мл), высушили над Na2SO4 и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 2,5% MeOH в ДХМ, для получения указанного в заголовке соединения (28 мг, выход 53%) в виде бледно-желтого пенистого вещества. МС (apci) m/z=574,2 (М+Н).
Пример 480
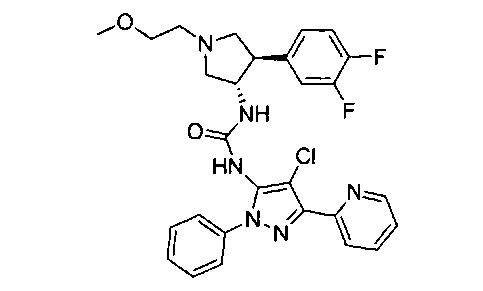
1-(4-хлор-1-фенил-3-(пиридин-2-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Стадия А: Получение 1-фенил-3-(пиридин-2-ил)-1Н-пиразол-5-амина: Получили по способу Примера 473, Стадия А, заменяя циклогексилгидразина гидрохлорид на фенилгидразина гидрохлорид, и 2-оксоциклопентанкарбонитрил на 3-оксо-3-(пиридин-2-ил)пропаннитрил. МС (apci) m/z=237,1 (М+Н).
Стадия B: Получение 4-хлор-1-фенил-3-(пиридин-2-ил)-1Н-пиразол-5-амина: Раствор 1-фенил-3-(пиридин-2-ил)-1Н-пиразол-5-амина (300 мг, 1,27 ммоль) растворили в ДХМ (20 мл) и обработали N-хлорсукцинимидом (187 мг, 1,40 ммоль), затем пиридин-1-ия 4-метилбензолсульфонатом (32 мг, 0,13 ммоль). Раствор перемешивали при комнатной температуре в течение 3 часов, затем разделили между ДХМ (20 мл) и насыщенным раствором NaHCO3 (20 мл), а водный слой экстрагировали ДХМ (2×20 мл). Объединенные органические фазы промыли насыщенным солевым раствором (20 мл), высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 2:1 смесью гексанов/EtOAc, для получения 4-хлор-1-фенил-3-(пиридин-2-ил)-1Н-пиразол-5-амина (211 мг, 61%) в виде розового пенистого вещества. МС (apci) m/z=271,0 (М+Н).
Стадия С: Получение 1-(4-хлор-1-фенил-3-(пирпидин-2-ил)-1Н-пиразол-5-ил)-3-((3S,4R-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины: К раствору 4-хлор-1-фенил-3-(пиридин-2-ил)-1H-пиразол-5-амина (37 мг, 0,14 ммоль) в ДХМ (2 мл) добавили трифосген (21 мг, 0,07 ммоль), затем ДИЭА (72 мкл, 0,41 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, затем обработали (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амином (Получение F) (50 мг, 0,15 ммоль), затем ДИЭА (72 мкл, 0,41 ммоль). После перемешивания в течение еще 18 часов, смесь разделили между насыщенным раствором NH4Cl (20 мл) и ДХМ (20 мл), и экстрагировали водный слой ДХМ (2×10 мл). Объединенные органические фазы промыли насыщенным солевым раствором (20 мл), высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 2,5% MeOH в ДХМ, затем обращенно-фазовой ВЭЖХ очисткой (5-95% АСN/вода/0,5% ТФК за 20 минут). Указанное в заголовке соединение (10 мг, выход 13%) получили после выделения продукта из воды (1 н. NaOH/ДХМ), в виде белого твердого вещества. МС (apci) m/z=553,2 (М+).
Пример 481
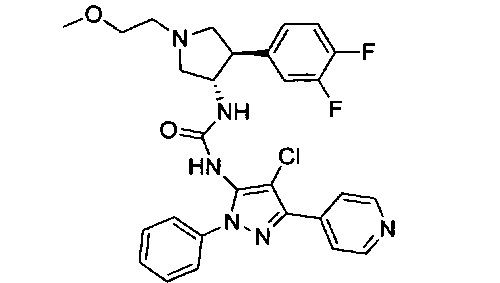
1-(4-хлор-1-фенил-3-(пиридин-4-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу Примера 480, заменяя 3-оксо-3-(пиридин-2-ил)пропаннитрил на 3-оксо-3-(пиридин-4-ил)пропаннитрил на Стадии А. Материал очистили колоночной хроматографией, элюируя 2% MeOH в ДХМ, затем обращенно-фазовой ВЭЖХ очисткой (5-95% ACN/вода/0,5% ТФК за 20 минут). Указанное в заголовке соединение (3 мг, выход 4%) получили после выделения продукта из воды (1 н. NaOH/ДХМ), в виде белого твердого вещества. МС (apci) m/z=553,2 (М+).
Пример 482
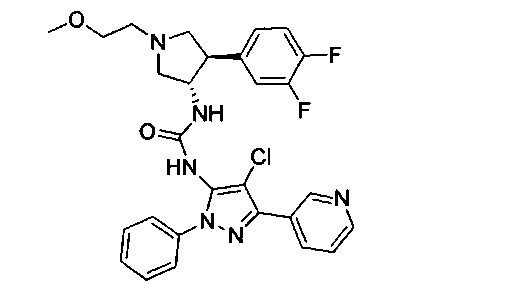
1-(4-хлор-1-фенил-3-(пиридин-3-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Стадия А: Получение 1-фенил-3-(пиридин-3-ил)-1Н-пиразол-5-амина: Получили по способу Примера 480, Стадия А, заменяя 3-оксо-3-(пиридин-2-ил)пропаннитрил на 3-оксо-3-(пиридин-3-ил)пропаннитрил. МС (apci) m/z=237,1 (М+Н).
Стадия В: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-фенил-3-(пиридин-3-ил)-1Н-пиразол-5-ил)мочевины: К раствору 1-фенил-3-(пиридин-3-ил)-1Н-пиразол-5-амина (50 мг, 0,21 ммоль) и КДИ (72 мг, 0,44 ммоль) в ДМФ (2 мл) добавили ДИЭА (147 мкл, 0,85 ммоль) и перемешивали смесь при 50°C в течение 4 часов. К охлажденной смеси добавили (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (Получение F) (146 мг, 0,44 ммоль) и ДИЭА (147 мкл, 0,85 ммоль) и продолжали перемешивание при комнатной температуре в течение 18 часов. Смесь разделили между насыщенным раствором NH4Cl (20 мл) и EtOAc (20 мл), а водный слой экстрагировали EtOAc (2×10 мл). Объединенные органические фазы промыли водой (5×10 мл) и насыщенным солевым раствором (10 мл), затем высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 5% MeOH в ДХМ, для получения 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-фенил-3-(пиридин-3-ил)-1Н-пиразол-5-ил)мочевины (80 мг, 73%) в виде белого твердого вещества. МС (apci) m/z=519,3 (М+Н).
Стадия С: Получение 1-(4-хлор-1-фенил-3-(пиридин-3-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины: К раствору 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-фенил-3-(пиридин-3-ил)-1Н-пиразол-5-ил)мочевины (40 мг, 0,08 ммоль) в ДХМ (1 мл) добавили N-хлорсукцинимид (12 мг, 0,09 ммоль), затем пиридин-1-ия 4-метилбензолсульфонат (2 мг, 0,008 ммоль). Смесь перемешивали при комнатной температуре в течение 18 часов, затем разделили между насыщенным раствором (20 мл) и ДХМ (20 мл), а водный слой экстрагировали ДХМ (2×10 мл). Объединенные органические фазы промыли насыщенным солевым раствором (10 мл), высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 2,5-5% MeOH в ДХМ, для получения указанного в заголовке соединения (16 мг, выход 38%) в виде бледно-желтого твердого вещества. МС (apci) m/z=553,2 (М+).
Пример 483
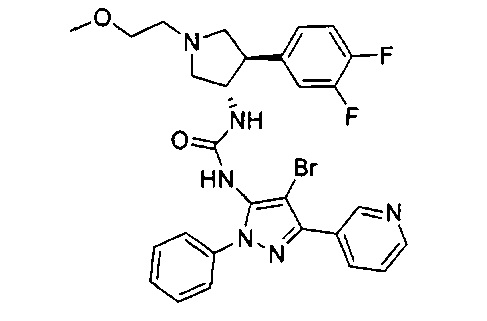
1-(4-бром-1-фенил-3-(пиридин-3-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу Примера 482, заменяя N-хлорсукцинимид на N-бромсукцинимид на Стадии С. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 3% MeOH в ДХМ, для получения указанного в заголовке соединения (31 мг, выход 67%) в виде желтого твердого вещества. МС (apci) m/z=597,2 (М+Н).
Пример 484
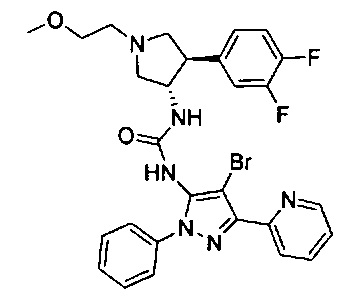
1-(4-бром-1-фенил-3-(пиридин-2-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Стадия А: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-фенил-3-(пиридин-2-ил)-1Н-пиразол-5-ил)мочевины: Получили по способу Примера 480, заменяя 4-хлор-1-фенил-3-(пиридин-2-ил)-1Н-пиразол-5-амин на 1-фенил-3-(пиридин-2-ил)-1Н-пиразол-5-амин на Стадии С. МС (apci) m/z=519,2 (М+Н).
Стадия В: Получение 1-(4-бром-1-фенил-3-(пиридин-2-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины: Получили по способу Примера 483, заменяя 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-фенил-3-(пиридин-3-ил)-1Н-пиразол-5-ил)мочевину на 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-фенил-3-(пиридин-2-ил)-1Н-пиразол-5-ил)мочевину на Стадии С. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2,5-5% MeOH в ДХМ, для получения указанного в заголовке соединения (14 мг, выход 40%) в виде бесцветного стекловидного вещества. МС (apci) m/z=597,2 (М+).
Пример 485
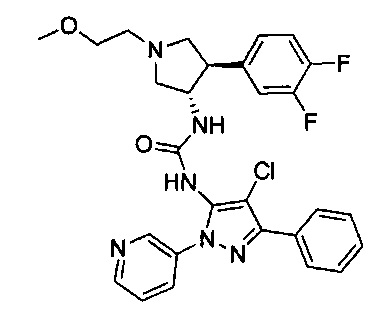
1-(4-хлор-3-фенил-1-(пиридин-3-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу Примера 482, заменяя 3-оксо-3-(пиридин-3-ил)пропаннитрил на 3-оксо-3-фенилпропаннитрил, и фенилгидразина гидрохлорид на 3-гидразинилпиридина гидрохлорид на Стадии А. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2,5% MeOH в ДХМ, для получения указанного в заголовке соединения (22 мг, выход 49%) в виде бежевого твердого вещества. МС (apci) m/z=553,2 (М+).
Пример 486
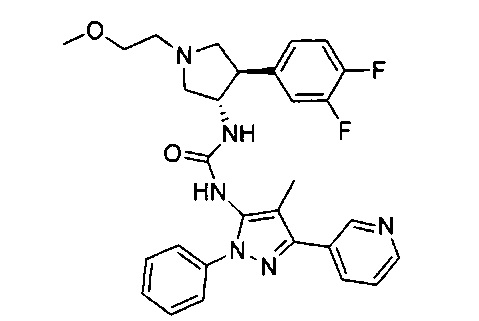
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(пиридин-3-ил)-1Н-пиразол-5-ил)мочевина
Стадия А: Получение 2-метил-3-оксо-3-(пиридин-3-ил)пропаннитрила: Раствор LiHMDS (13,6 мл, 1,0 М в ТГФ, 13,6 ммоль) охладили до -78°C под атмосферой N2 и по каплям обработали пропионитрилом (991 мкл, 13,9 ммоль). Полученную желтую взвесь перемешивали при этой температуре в течение 2 часов, затем по каплям обработали раствором этилникотината (1,0 г, 6,62 ммоль) в ТГФ (5 мл) за 10 минут. Смесь оставили медленно нагреваться до комнатной температуры в течение 18 часов, затем вылили в ледяную воду (100 мл) и экстрагировали Et2O (2×30 мл). Водную фазу охладили на льду, подкислили до pH 5 при помощи 1 н. HCl и экстрагировали ДХМ (3×30 мл). Объединенные ДХМ экстракты и промыли насыщенным солевым раствором (30 мл), высушили над Na2SO4, отфильтровали и концентрировали для получения 2-метил-3-оксо-3-(пиридин-3-ил)пропаннитрила (1,06 г, выход 100%) в виде желтого маслянистого вещества. МС (apci) m/z=161,1 (М+Н).
Стадия B: Получение 4-метил-1-фенил-3-(пиридин-3-ил)-1Н-пиразол-5-амина: Суспензию 2-метил-3-оксо-3-(пиридин-3-ил)пропаннитрила (1,06 г, 6,62 ммоль) и фенилгидразина гидрохлорида (1,05 г, 7,28 ммоль) в EtOH (30 мл) перемешивали при нагревании с дефлегматором в течение 18 часов, затем охладили до комнатной температуры. Смесь концентрировали, затем обработали насыщенным раствором (50 мл) и экстрагировали ДХМ (3×30 мл). Объединенные органические фазы промыли насыщенным солевым раствором (30 мл), высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя от 1:1 до 1:2 смесью гексанов/EtOAc, для получения 4-метил-1-фенил-3-(пиридин-3-ил)-1Н-пиразол-5-амина (984 мг, выход 59%) в виде бледно-желтого пенистого вещества. МС (apci) m/z=251,1 (М+Н).
Стадия С: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(пиридин-3-ил)-1Н-пиразол-5-ил)мочевины: К раствору 4-метил-1-фенил-3-(пиридин-3-ил)-1Н-пиразол-5-амина (100 мг, 0,40 ммоль) в ДХМ (2 мл) добавили трифосген (59 мг, 0,20 ммоль), затем ДИЭА (209 мкл, 1,20 ммоль). Смесь перемешивали в течение 1 часа при комнатной температуре, затем обработали (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амином (Получение F; 145 мг, 0,44 ммоль) и ДИЭА (209 мкл, 1,20 ммоль). После перемешивания при комнатной температуре в течение еще 18 часов, смесь разделили между насыщенным раствором NH4Cl (20 мл) и ДХМ (20 мл), и экстрагировали водный слой ДХМ (2×10 мл). Объединенные органические фазы промыли насыщенным солевым раствором (10 мл), высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 3-4% МеОН в ДХМ, для получения указанного в заголовке соединения (99 мг, выход 47%) в виде белого твердого вещества. МС (apci) m/z=533,2 (М+Н).
Следующие соединения были получены по способу Примера 486, с заменой этилникотината на соответствующий реагент на Стадии А, а для Примера 490 также с заменой фенилгидразина гидрохлорида на 3-гидразинилпиридина гидрохлорид на Стадии В.
Пример 492
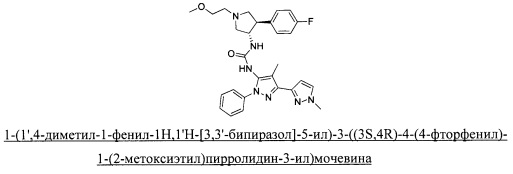
Получили по способу Примера 491, заменяя (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (Получение F) на (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (Получение К) на Стадии С. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 3% МеОН в ДХМ, для получения указанного в заголовке соединения (52 мг, выход 51%) в виде бледно-желтого пенистого вещества. МС (apci) m/z=518,2 (М+Н).
Пример 493
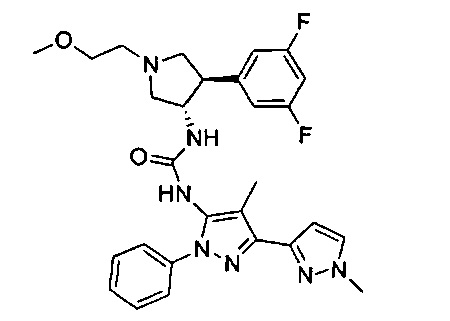
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-[3,3'-бипиразол1-5-ил)мочевина
Получили по способу Примера 491, заменяя (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (Получение F) на (3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина трифторацетат (Получение Е) на Стадии С. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 3% MeOH в ДХМ, для получения указанного в заголовке соединения (33 мг, выход 31%) в виде белого твердого вещества. МС (apci) m/z=536,2 (М+Н).
Пример 494
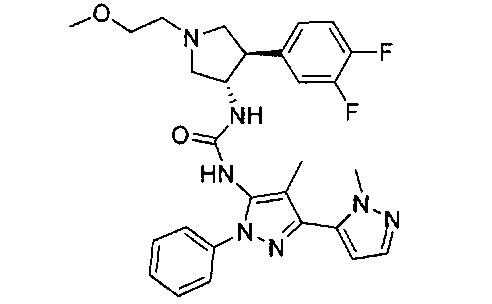
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2',4-диметил-1-фенил-1Н,2'Н-[3,3'-бипиразол -5-ил)мочевина
Стадия А: Получение 2',4-диметил-1-фенил-1H,2'H-[3,3'-бипиразол]-5-амина: Получили по способу Примера 486, Стадия А, заменяя этилникотинат на этил 1-метил-1Н-пиразол-5-карбоксилат. 1Н ЯМР (CDCl3) δ 7,55 (d,J=2,2 Гц, 1H, 7,01 (d, J=2,2 Гц, 1H, 4,19 (s, 3Н), 4,12 (q, J=7,2 Гц, 1Н), 1,65 (d, J=7,2 Гц, 3Н) ppm.
Стадия В: Получение фенил (2',4-диметил-1-фенил-1H,2'H-[3,3'-бипиразол]-5-ил)карбамата: К раствору 2',4-диметил-1-фенил-1Н,2'Н-[3,3'-бипиразол]-5-амина (100 мг, 0,39 ммоль) в EtOAc (2 мл) добавили 2 н. NaOH (395 мкл, 0,79 ммоль), затем фенилхлорформиат (75 мкл, 0,59 ммоль). Смесь перемешивали при комнатной температуре в течение 4 часов, затем обработали дополнительной аликвотой фенилхлорформиата (50 мкл) и перемешивали в течение 18 часов. Смесь разделили между водой (20 мл) и EtOAc (10 мл), а водный слой экстрагировали EtOAc (2×10 мл). Объединенные органические фазы промыли насыщенным раствором NaHCO3 (10 мл) и насыщенным солевым раствором (10 мл), затем высушили над Na2SO4, отфильтровали и концентрировали in vacuo для получения фенил (2',4-диметил-1-фенил-1Н,2'Н-[3,3'-бипиразол]-5-ил)карбамата (140 мг, выход 95%) в виде бледно-желтого смолянистого вещества. МС (apci) m/z=374,2 (М+Н).
Стадия C: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2',4-диметил-1-фенил-1Н,2'Н-[3,3'-бипиразол]-5-ил)мочевины: К раствору (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина (Получение F; 68 мг, 0,21 ммоль) и фенил (2',4-диметил-1-фенил-1Н,2'Н-[3,3'-бипиразол]-5-ил)карбамата (70 мг, 0,19 ммоль) в ДХМ (2 мл) добавили ДИЭА (114 мкл, 0,66 ммоль). После перемешивания при комнатной температуре в течение еще 3 часов, смесь разделили между насыщенным раствором NH4Cl (20 мл) и ДХМ (20 мл), и экстрагировали водный слой ДХМ (2×10 мл). Объединенные органические фазы промыли насыщенным солевым раствором (10 мл), затем высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 1-3%) MeOH в ДХМ, для получения указанного в заголовке соединения (68 мг, выход 68%) в виде белого твердого вещества. МС (apci) m/z=536,2 (М+Н).
Пример 495
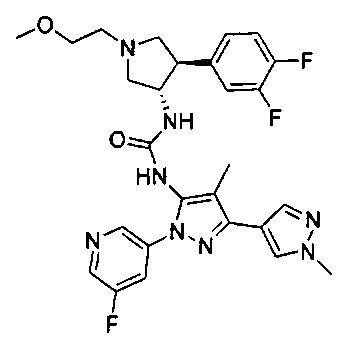
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-(5-фторпиридин-3-ил)-1',4-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина Стадия А: Получение 3-(2-(дифенилметилен)гидразинил)-5-фторпиридина: Раствор 3-бром-5-фторпиридина (5,0 г, 28,4 ммоль), бензофенонгидразона (6,13 г, 31,3 ммоль) и Xantphos (164 мг, 0,28 ммоль) дегазировали N2 в течение 10 минут, затем обработали трет-бутоксидом натрия (3,82 г, 39,8 ммоль) и ацетатом палладия (II) (64 мг, 0,28 ммоль). Гетерогенную смесь перемешивали при 85°C в закрытой емкости в течение 18 часов. Охлажденную смесь разделили между водой (100 мл) и EtOAc(100 мл), а водный слой экстрагировали EtOAc (2×50 мл). Объединенные органические фазы промыли насыщенным солевым раствором (50 мл), высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток растерли с Et2O, отфильтровали и высушили in vacuo для получения 3-(2-(дифенилметилен)гидразинил)-5-фторпиридина (6,3 г, выход 72%) в виде бежевого порошка. МС (apci) m/z=292,1 (М+Н).
Стадия В: Получение 1-(5-фторпиридин-3-ил)-1',4-диметил-1H,1'Н-[3,4'-бипиразол]-5-амина: Раствор 2-метил-3-(1-метил-1Н-пиразол-4-ил)-3-оксопропаннитрила (Пример 491, Стадия А; 100 мг, 0,61 ммоль), 3-(2-(дифенилметилен)гидразинил)-5-фторпиридина (162 мг, 0,56 ммоль) и п-толуолсульфоновой кислоты моногидрата (530 мг, 2,79 ммоль) в ЕtOН (3 мл) перемешивали при 80°C в закрытой пробирке в течение 18 часов. Охлажденную смесь обработали насыщенным раствором (30 мл) и экстрагировали ДХМ (3×10 мл). Объединенные органические фазы промыли насыщенным солевым раствором (10 мл), высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 1-3% MeOH в ДХМ, для получения 1-(5-фторпиридин-3-ил)-1',4-диметил-1Н,1'Н-[3,4'-бипиразол]-5-амина (71 мг, 47%) в виде бледно-желтого твердого вещества. МС (apci) m/z=273,1 (М+Н).
Стадия C: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-(5-фторпиридин-3-ил)-1',4-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевины: К раствору 1-(5-фторпиридин-3-ил)-1',4-диметил-1Н,1'Н-[3,4'-бипиразол]-5-амина (35 мг, 0,13 ммоль) в ДХМ (2 мл) добавили трифосген (19 мг, 0,06 ммоль), затем ДИЭА (67 мкл, 0,39 ммоль). Смесь перемешивали в течение 1 часа при комнатной температуре, затем обработали (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амином (Получение F; 42 мг, 0,13 ммоль) и ДИЭА (67 мкл, 0,39 ммоль), и продолжали перемешивание в течение 18 часов. Смесь разделили между насыщенным раствором NH4Cl (20 мл) и ДХМ (20 мл), а водный слой экстрагировали ДХМ (2×10 мл). Объединенные органические фазы промыли насыщенным солевым раствором (10 мл), высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 2,5-4% MeOH в ДХМ, для получения указанного в заголовке соединения (33 мг, 46%) в виде белого твердого вещества. МС (apci) m/z=555,2 (М+Н).
Пример 496
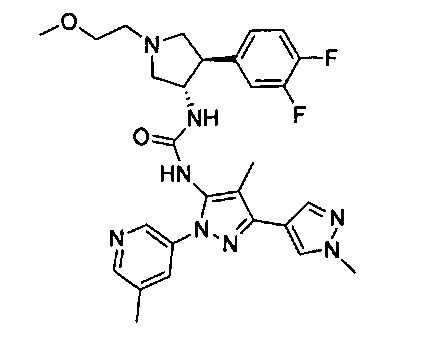
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-(5-метилпиридин-3-ил)-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина
Получили по способу Примера 495, заменяя 3-бром-5-фторпиридин на 3-бром-5-метилпиридин на Стадии А. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 5-10% MeOH в ДХМ, для получения указанного в заголовке соединения (41 мг, выход 56%) в виде кремового твердого вещества. МС (apci) m/z=551,2 (М+Н).
Пример 497
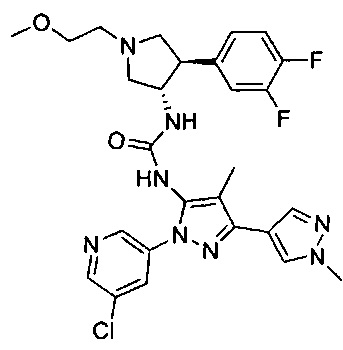
1-(1-(5-хлорпиридин-3-ил)-1',4-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3 S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу Примера 495, заменяя 3-бром-5-фторпиридин на 3-бром-5-хлорпиридин на Стадии А. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2-5% MeOH в ДХМ, для получения указанного в заголовке соединения (85 мг, выход 86%) в виде бледно-розового твердого вещества. МС (apci) m/z=571,2 (М+).
Пример 498
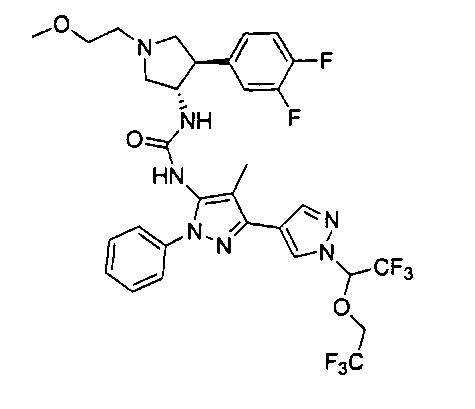
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-1'-(2,2,2-трифтор-1-(2,2,2-трифторэтокси)этил)-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина
Стадия А: Получение этил 1-(4-метоксибензил)-1Н-пиразол-4-карбоксилата: К смеси этил 1Н-пиразол-4-карбоксилата (3 г, 21,4 ммоль) и K2CO3 (3,55 г, 25,7 ммоль) в ДМФ (10 мл) добавили 1-(хлорметил)-4-метоксибензол (3,50 мл, 25,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, затем добавили эфир (30 мл) и воду (10 мл). Органический слой отделили, промыли насыщенным солевым раствором, высушили над Na2SO4 и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 3:1 смесью гексанов/EtOAc, для получения этил 1-(4-метоксибензил)-1Н-пиразол-4-карбоксилата (5,7 г, 102%) в виде бесцветного маслянистого вещества. 1Н ЯМР (CDCl3) δ 7,92 (m, 1Н), 7,80 (m, 1Н), 7,21 (m, 2Н), 6,89 (m, 2Н), 5,23 (s, 2Н), 4,27 (m, 2Н), 3,80 (s, 3Н), 1,32 (m, 3Н) ppm.
Стадия В: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-(4-метоксибензил)-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевины: Получили по способу Примера 494, заменяя этил 1-метил-1Н-пиразол-5-карбоксилат на этил 1-(4-метоксибензил)-1Н-пиразол-4-карбоксилат на Стадии А. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя от 1:1 до 1:1,2 смесью гексанов/ацетона плюс 0,5% NH4OH, для получения 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-(4-метоксибензил)-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевины (800 мг, выход 60%) в виде белого твердого вещества. МС (apci) m/z=642,3 (М+Н).
Стадия С: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевины: 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-(4-метоксибензил)-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевину (241 мг, 0,38 ммоль) смешали с ТФК (2 мл) в закрытой пробирке и перемешивали при 70°C в течение 18 часов. Охлажденную смесь концентрировали in vacuo, а остаток разделили между 1 н. раствором NaOH (20 мл) и ДХМ (10 мл). Водный слой экстрагировали ДХМ (2×10 мл), а объединенные органические фазы промыли насыщенным раствором NaHCO3 (10 мл) и насыщенным солевым раствором (10 мл), затем высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 2-5% MeOH в ДХМ, для получения 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевины (144 мг, выход 74%) в виде белого твердого вещества. МС (apci) m/z=522,2 (М+).
Стадия D: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-1'-(2,2,2-трифтор-1-(2,2,2-трифторэтокси)этил)-1Н',1'Н-[3,4'-бипиразол]-5-ил)мочевины: К раствору 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевины (50 мг, 0,10 ммоль) в ДМФ (2,5 мл) при -78°C добавили трет-бутоксид калия (264 мкл, 1 М в ТГФ, 0,264 ммоль). Смесь перемешивали в течение 10 минут, затем обработали 2,2,2-трифторэтил трифторметансульфонатом (13,1 мкл, 0,09 ммоль). После перемешивания при комнатной температуре в течение еще 2 часов, смесь разделили между насыщенным раствором NH4Cl (20 мл) и EtOAc (10 мл), и экстрагировали водный слой EtOAc (2×10 мл). Объединенные органические фазы промыли водой (2×10 мл) и насыщенным солевым раствором (10 мл), затем высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 2,5% MeOH в ДХМ, для получения указанного в заголовке соединения (29 мг, выход 43%) в виде бесцветного стекловидного вещества. МС (apci) m/z=702,2 (М+Н).
Пример 499
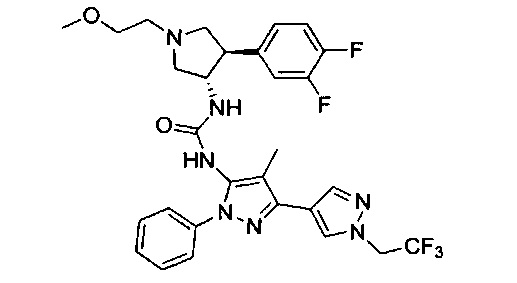
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-1'-(2,2,2-трифторэтил)-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина
К раствору 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевины (Пример 498, Стадия C; 20 мг, 0,04 ммоль) в ДМФ (0,5 мл) добавили K2CO3 (16 мг, 0,12 ммоль), затем трифторэтилтрифлат (6 мкл, 0,04 ммоль). Смесь закрыли и перемешивали при комнатной температуре в течение 5 часов. Добавили дополнительную аликвоту трифторэтилтрифлата (30 мкл) и продолжали перемешивание в течение 18 часов. Смесь разделили между водой (10 мл) и EtOAc (10 мл), а водный слой экстрагировали EtOAc (2×10 мл). Объединенные органические фазы промыли водой (4×10 мл) и насыщенным солевым раствором (10 мл), затем высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 2-5% MeOH в ДХМ, для получения указанного в заголовке соединения (8 мг, выход 35%) в виде бесцветного стекловидного вещества. МС (apci) m/z=604,2 (М+Н).
Пример 500
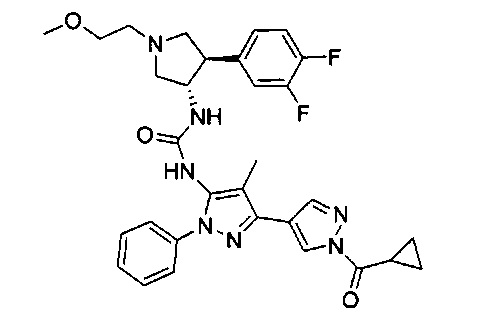
1-(1'-(циклопропилметил)-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу Примера 499, заменяя трифторэтилтрифлат на (бромметил)циклопропан. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2,5-5% MeOH в ДХМ, для получения указанного в заголовке соединения (16 мг, выход 31%) в виде белого твердого вещества. МС (apci) m/z=576,3 (М+Н).
Пример 501
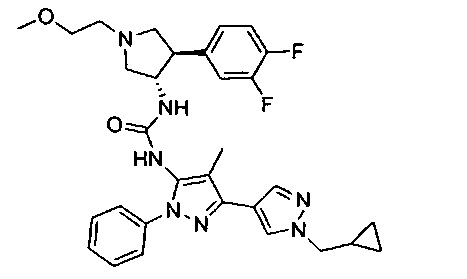
1-(1'-(циклопропанкарбонитрил)-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
К раствору 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевины (Пример 498, Стадия C; 50 мг, 0,09 ммоль) в ДХМ (2 мл) при 0°C добавили циклопропилкарбонилхлорид (13 мкл, 0,14 ммоль), затем ДИЭА (67 мкл, 0,38 ммоль). Смесь оставили медленно нагреваться до комнатной температуры в течение 18 часов, затем разделили между насыщенным раствором NaHCO3 (20 мл) и ДХМ (10 мл), а водный слой экстрагировали ДХМ (2×10 мл). Объединенные органические фазы промыли насыщенным солевым раствором (10 мл), высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 2,5% MeOH в ДХМ, для получения указанного в заголовке соединения (26 мг, выход 46%) в виде белого твердого вещества. МС (apci) m/z=590,2 (М+Н).
Пример 502
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1'-(метилсульфонил)-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина
Получили по способу Примера 501, заменяя циклопропилкарбонилхлорид на мезилхлорид. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2-5% MeOH в ДХМ, для получения указанного в заголовке соединения (39 мг, выход 68%) в виде бесцветного стекловидного вещества. МС (apci) m/z=600,2 (М+Н).
Пример 503
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-изопропил-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина
Стадия А: Получение 3-бром-4-метил-1-фенил-1Н-пиразол-5-амина: К суспензии 5-амино-4-метил-1-фенил-1Н-пиразол-3(2Н)-она (Промежуточное соединение Р135, Стадия А; 1,60 г, 8,46 ммоль) в ацетонитриле (30 мл) одной порцией добавили оксибромид фосфора (3,64 г, 12,7 ммоль). Смесь перемешивали при нагревании с дефлегматором в течение 3 часов, затем охладили и концентрировали in vacuo. Остаток обработали ДХМ (50 мл), затем медленно добавили насыщенный раствор (50 мл). Смесь перемешивали в течение 30 минут, затем разделили слои, а водный слой экстрагировали ДХМ (2×50 мл). Объединенные органические фазы промыли насыщенным солевым раствором (20 мл), высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 2:1 смесью гексанов/EtOAc, для получения 3-бром-4-метил-1-фенил-1Н-пиразол-5-амина (273 мг, выход 13%о) в виде белого твердого вещества. МС (apci) m/z=254,0 (М+Н).
Стадия В: Получение фенил (3-бром-4-метил-1-фенил-1Н-пиразол-5-ил)карбамата: К раствору 3-бром-4-метил-1-фенил-1Н-пиразол-5-амина (339 мг, 1,34 ммоль) в EtOAc (10 мл) добавили 2 н. NaOH (2 мл, 4,0 ммоль), затем фенилхлорформиат (337 мкл, 2,69 ммоль). Смесь перемешивали при комнатной температуре в течение 5 часов, затем разделили между водой (30 мл) и EtOAc (30 мл), а водный слой экстрагировали EtOAc (2×20 мл). Объединенные органические фазы промыли насыщенным раствором (30 мл) и насыщенным солевым раствором (30 мл), затем высушили над Na2SO4, отфильтровали и концентрировали in vacuo для получения фенил (3-бром-4-метил-1-фенил-1Н-пиразол-5-ил)карбамата, который использовали напрямую, предположив количественный выход. МС (apci) m/z=374,0 (М+Н).
Стадия С: Получение 1-(3-бром-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины: К раствору (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина (Получение F; 464 мг, 1,41 ммоль) и фенил (3-бром-4-метил-1-фенил-1Н-пиразол-5-ил)карбамата (500 мг, 1,34 ммоль) в ДХМ (10 мл) добавили ДИЭА (819 мкл, 4,7 ммоль). Раствор перемешивали при комнатной температуре в течение 18 часов, затем разделили между насыщенным раствором NH4Cl (30 мл) и ДХМ (30 мл), а водный слой экстрагировали ДХМ (2×20 мл). Объединенные органические фазы промыли насыщенным солевым раствором (20 мл), высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 2% MeOH в ДХМ, для получения 1-(3-бром-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины (483 мг, выход 67%) в виде белого твердого вещества. МС (apci) m/z=534,1 (М+).
Стадия D: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-изопропил-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевины: 1-(3-Бром-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевину (30 мг, 0,06 ммоль), 1-изопропил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (20 мг, 0,08 ммоль), трициклогексилфосфин (3 мг, 0,01 ммоль) и Pd2(dba)3 (5 мг, 0,006 ммоль) смешали в закрытой пробирке и добавили 1,4-диоксан (561 мкл). Раствор продували N2 в течение 30 секунд, затем обработали K3PO4 (130 мкл, 1,3 М, 0,17 ммоль), закрыли и перемешивали при 100°C в течение 1 часа. Охлажденную смесь концентрировали in vacuo, а остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 2,5-10% MeOH в ДХМ, для получения указанного в заголовке соединения (12 мг, выход 38%) в виде бесцветного стекловидного вещества. МС (apci) m/z=564,2 (М+Н).
Следующие соединения были получены по способу Примера 503, с заменой 1-изопропил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразола на соответствующий реагент на Стадии D.
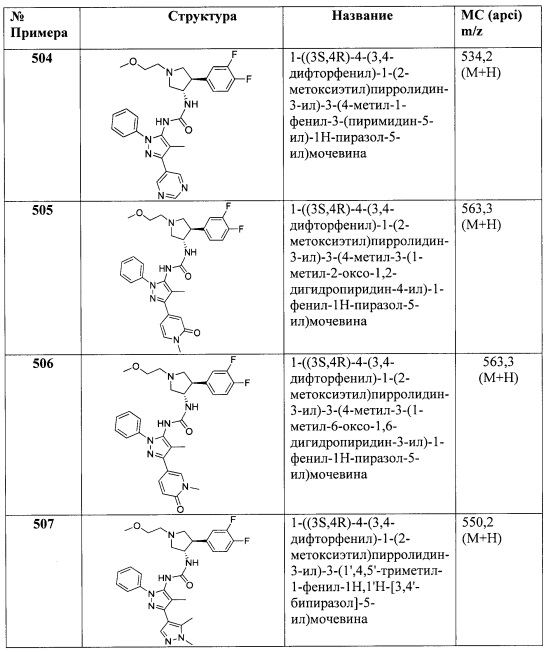
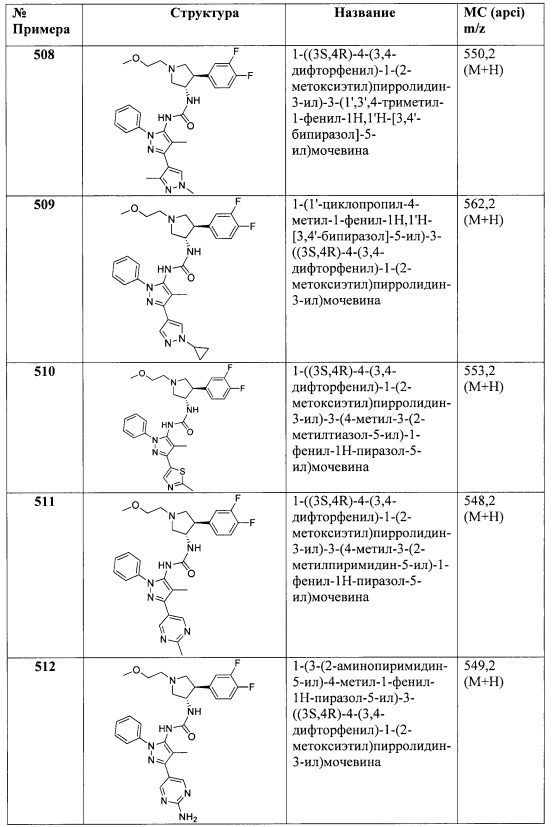
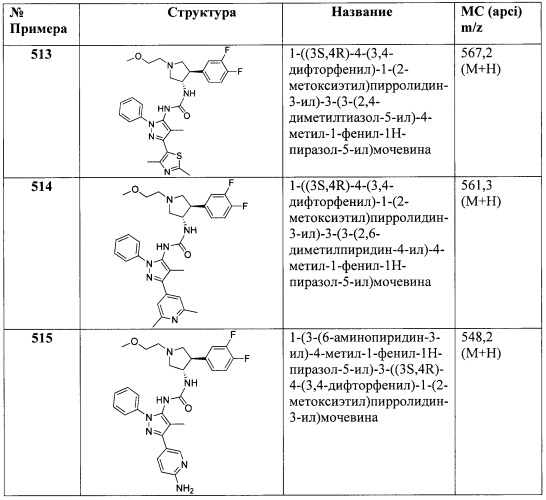
Пример 516
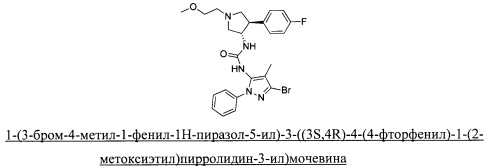
Получили по способу Примера 503, Стадия С, заменяя (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (Получение F) на (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (Получение К). Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2-3% МеОН в ДХМ, для получения указанного в заголовке соединения (526 мг, 63%) в виде кремового твердого вещества. МС (apci) m/z=518,1 (М+Н).
Пример 517
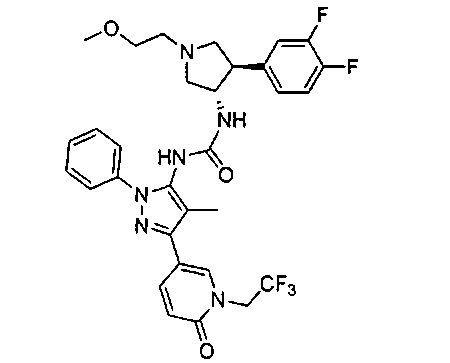
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(6-оксо-1-(2,2,2-трифторэтил)-1,6-дигидропиридин-3-ил)-1-фенил-1Н-пиразол-5-ил)мочевина
Получили по способу Примера 503, заменяя 1-изопропил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол на 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1-(2,2,2-трифторэтил)пиридин-2(1Н)он на Стадии D. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2% MeOH в ДХМ, затем обращенно-фазовой ВЭЖХ очисткой (5-95% ACN/вода/0,5% ТФК за 20 минут). Указанное в заголовке соединение (5,5 мг, выход 9%) получили после выделения продукта из воды (1 н. NaOH/ДХМ), в виде белого твердого вещества. МС (apci) m/z=631,2 (М+Н).
Пример 518
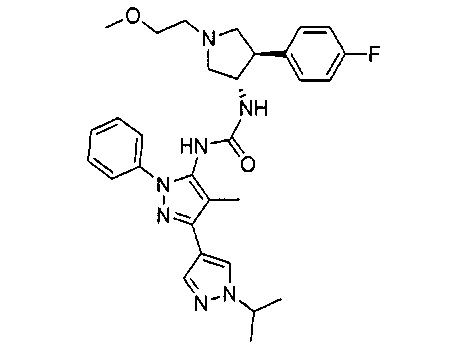
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-изопропил-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина
Получили по способу Примера 503, заменяя (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (Получение F) на (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (Получение К) на Стадии С. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2-3% MeOH в ДХМ, затем обращенно-фазовой ВЭЖХ очисткой (5-95% ACN/вода/0,5% ТФК за 20 минут). Указанное в заголовке соединение (37 мг, выход 18%) получили после выделения продукта из воды (1 н. NaOH/ДХМ), в виде бесцветного смолянистого вещества. МС (apci) m/z=546,3 (М+Н).
Пример 519
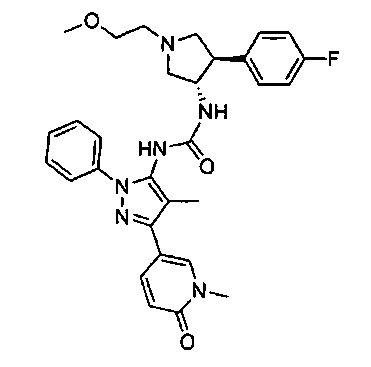
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)-1-фенил-1Н-пиразол-5-ил)мочевина
Получили по способу Примера 518, заменяя 1-изопропил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол на 1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он на Стадии D. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2-5% MeOH в ДХМ, для получения указанного в заголовке продукта (29 мг, выход 27%) в виде белого твердого вещества. МС (apci) m/z=545,2 (М+Н).
Пример 520
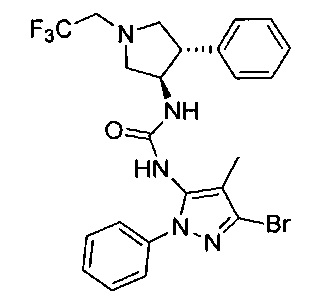
1-(3-бром-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина
Получили по способу Примера 516, заменяя (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (Получение К) на (3S,4R)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-амина гидрохлорид (Пример 265, Стадия А). Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2:1 смесью гексанов/EtOAc, для получения указанного в заголовке соединения (173 мг, выход 62%) в виде белого твердого вещества. МС (apci) m/z=522,1 (М+).
Пример 521
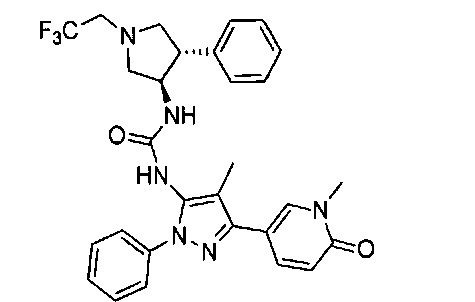
1-(4-метил-3-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)-1-фенил-1Н-пиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина
Получили по способу Примера 519, заменяя (3R,4S)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (Получение К) на (3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-амина гидрохлорид (Пример 265, Стадия А) на Стадии С. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2% MeOH в ДХМ, для получения указанного в заголовке продукта (28 мг, выход 33%) в виде бледно-желтого твердого вещества. МС (apci) m/z=550,2 (М+).
Пример 522
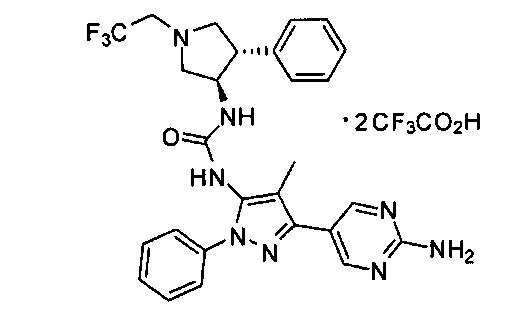
1-(3-(2-аминопиримидин-5-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевины бис(2,2,2-трифторацетат)
Получили по способу Примера 512, заменяя (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (Получение К) на (3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-амина гидрохлорид (Пример 265, Стадия А) на Стадии С. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2-5% MeOH в ДХМ, с последующей очисткой обращенно-фазовой ВЭЖХ (5-95% ACN/вода/0,5% ТФК за 20 минут). Указанное в заголовке соединение (3 мг, выход 3%) получили в виде белого твердого вещества как ди-ТФК соль. МС (apci) m/z=537,2 (М+Н).
Пример 523
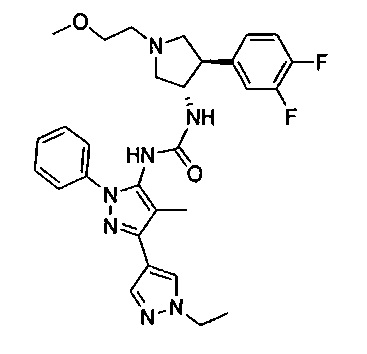
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-этил-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина
Стадия А: Получение 1'-этил-4-метил-1-фенил-1H,1'H-[3,4'-бипиразол]-5-амина: 3-Бром-4-метил-1-фенил-1Н-пиразол-5-амин (Пример 503, Стадия А; 100 мг, 0,39 ммоль), 1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (176 мг, 0,79 ммоль), K2CO3 (219 мг, 1,59 ммоль) и Pd(PPh3)4 (46 мг, 0,039 ммоль) смешали в толуоле (2 мл), воде (1 мл) и EtOH (0,5 мл) и перемешивали при 95°C в закрытой пробирке в течение 18 часов. Охлажденную смесь отфильтровали через стекловолоконную бумагу, а фильтрат разделили между водой (10 мл) и EtOAc (10 мл). Водный слой экстрагировали EtOAc (2×10 мл) и объединенные органические фазы промыли насыщенным солевым раствором (10 мл), высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 1,5% МеОН в ДХМ, для получения 1'-этил-4-метил-1-фенил-1H,1'H-[3,4'-бипиразол]-5-амина (78 мг, выход 74%) в виде бесцветного смолянистого вещества. МС (apci) m/z=268,1 (М+Н).
Стадия В: Получение фенил (1'-этил-4-метил-1-фенил-1Н,1'H-[3,4'-бипиразол]-5-ил)карбамата: К раствору 1'-этил-4-метил-1-фенил-1H,1'H-[3,4'-бипиразол]-5-амина (78 мг, 0,29 ммоль) в EtOAc (5 мл) добавили 2 н. NaOH (0,44 мл, 0,87 ммоль), затем фенилхлорформиат (73 мкл, 0,58 ммоль). Смесь перемешивали при комнатной температуре в течение 18 часов, затем разделили между водой (20 мл) и EtOAc (20 мл), а водный слой экстрагировали EtOAc (2×10 мл). Объединенные органические фазы промыли насыщенным раствором NаНСО3 (10 мл) и насыщенным солевым раствором (10 мл), затем высушили над Na2SO4, отфильтровали и концентрировали in vacuo для получения фенил (1'-этил-4-метил-1-фенил-1H,1'H-[3,4'-бипиразол]-5-ил)карбамата в виде бледно-желтого маслянистого вещества. Использовали напрямую, предположив количественный выход. МС (apci) m/z=388,2 (М+Н).
Стадия С: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-этил-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевины: К раствору (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина (Получение F; 56 мг, 0,17 ммоль) и фенил (1'-этил-4-метил-1-фенил-1H,1'H-[3,4'-бипиразол]-5-ил)карбамата (66 мг, 0,17 ммоль) в ДХМ (2 мл) добавили ДИЭА (150 мкл, 0,85 ммоль). После перемешивания при комнатной температуре в течение еще 18 часов, смесь разделили между насыщенным раствором NH4Cl (10 мл) и ДХМ (10 мл), и экстрагировали водный слой ДХМ (2×10 мл). Объединенные органические фазы промыли насыщенным солевым раствором (10 мл), затем высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 2,5-3,5% МеОН в ДХМ, для получения указанного в заголовке соединения (35 мг, выход 37%) в виде бесцветного стекловидного вещества. МС (apci) m/z=550,2 (М+Н).
Пример 524
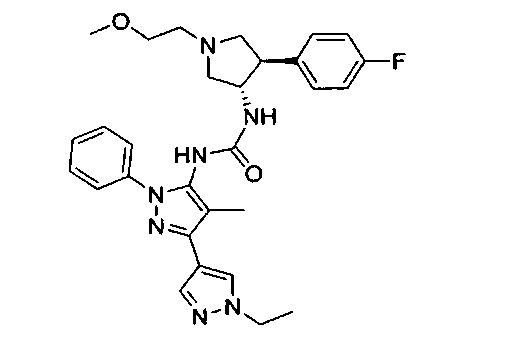
1-(1'-этил-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил1пирролидин-3-ил)мочевина
Получили по способу Примера 523, заменяя (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (Получение F) на (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (Получение К) на Стадии С. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 3% МеОН в ДХМ, для получения указанного в заголовке продукта (29 мг, выход 30%) в виде белого твердого вещества. МС (apci) m/z=532,3 (М+Н).
Пример 525
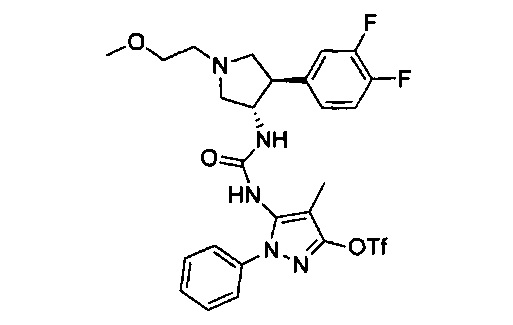
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-ил трифторметансульфонат
Стадия А: Получение 5-амино-4-метил-1-фенил-1Н-пиразол-3-ил трифторметансульфоната: Суспензию 5-амино-4-метил-1-фенил-1Н-пиразол-3(2Н)-она (Промежуточное соединение Р135, Стадия А; 0,50 г, 2,64 ммоль) и N-фенилбис(трифторметилсульфонамида) (0,99 г, 2,77 ммоль) в ДМФ (5 мл) обработали ДИЭА (1,38 мл, 7,93 ммоль) и перемешивали смесь при комнатной температуре в течение 64 часов. Смесь разделили между насыщенным раствором NaHCO3 (30 мл) и EtOAc (30 мл), а водный слой экстрагировали EtOAc (2×20 мл). Объединенные органические фазы промыли водой (5×10 мл) и насыщенным солевым раствором (10 мл), затем высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 2:1 смесью гексанов/EtOAc, для получения 5-амино-4-метил-1-фенил-1Н-пиразол-3-ил трифторметансульфоната (817 мг, выход 92%) в виде бледно-желтого маслянистого вещества. МС (apci) m/z=322,0 (М+Н).
Стадия В: Получение 4-метил-5-((феноксикарбонил)амино)-1-фенил-1Н-пиразол-3-ил трифторметансульфоната: Получили по способу Примера 503, Стадия B, заменяя 3-бром-4-метил-1-фенил-1Н-пиразол-5-амин на 5-амино-4-метил-1-фенил-1Н-пиразол-3-ил трифторметансульфонат. МС (apci) m/z=442,0 (М+Н).
Стадия С: Получение 5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-ил трифторметансульфоната: Получили по способу Примера 503, Стадия С, заменяя фенил (3-бром-4-метил-1-фенил-1Н-пиразол-5-ил)карбамат на 4-метил-5-((феноксикарбонил)амино)-1-фенил-1Н-пиразол-3-ил трифторметансульфонат. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 1,5-4% МеОН в ДХМ, для получения указанного в заголовке соединения (191 мг, выход 62%) в виде белого твердого вещества. МС (apci) m/z=604,2 (М+Н).
Пример 526
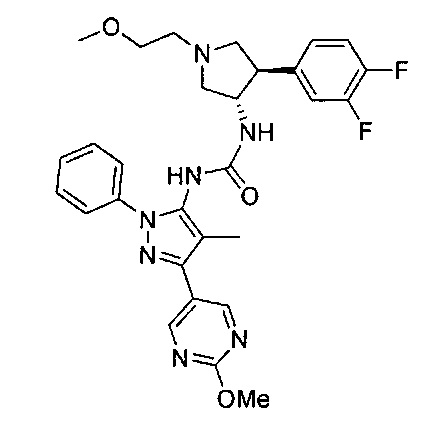
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-метоксипиримидин-5-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Стадия А: Получение 3-(2-метоксипиримидин-5-ил)-4-метил-1-фенил-1Н-пиразол-5-амина: 5-Амино-4-метил-1-фенил-1Н-пиразол-3-ил трифторметансульфонат (Пример 525, Стадия А; 200 мг, 0,62 ммоль), 2-метоксипиримидин-5-илбороновую кислоту (192 мг, 1,25 ммоль), K2CO3 (344 мг, 2,49 ммоль) и Pd(PPh3)4 (72 мг, 0,06 ммоль) смешали в толуоле (2 мл), воде (1 мл) и EtOH (0,5 мл) и перемешивали при 95°C в закрытой пробирке в течение 18 часов. Охлажденную смесь отфильтровали через стекловолоконную бумагу, а фильтрат разделили между водой (20 мл) и EtOAc (20 мл). Водный слой экстрагировали EtOAc (2×20 мл) и объединенные органические фазы промыли насыщенным солевым раствором (20 мл), высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 1% МеОН в ДХМ, для получения 3-(2-метоксипиримидин-5-ил)-4-метил-1-фенил-1Н-пиразол-5-амина (138 мг, выход 79%) в виде кремового пенистого вещества. МС (apci) m/z=282,1 (М+Н).
Стадия В: Получение фенил (3-(2-метоксипиримидин-5-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)карбамата: Получили по способу Примера 503, Стадия В, заменяя 3-бром-4-метил-1-фенил-1Н-пиразол-5-амин на 3-(2-метоксипиримидин-5-ил)-4-метил-1-фенил-1Н-пиразол-5-амин. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 1% МеОН в ДХМ, для получения фенил (3-(2-метоксипиримидин-5-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)карбамата (92 мг, выход 47%) в виде кремового пенистого вещества. МС (apci) m/z=402,1 (М+Н).
Стадия С: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-метоксипиримидин-5-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины: Получили по способу Примера 503, Стадия С, заменяя фенил (3-бром-4-метил-1-фенил-1Н-пиразол-5-ил)карбамат на фенил (3-(2-метоксипиримидин-5-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)карбамат. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2,5% МеОН в ДХМ, для получения указанного в заголовке соединения (31 мг, выход 48%) в виде белого твердого вещества. МС (apci) m/z=564,2 (М+Н).
Пример 527
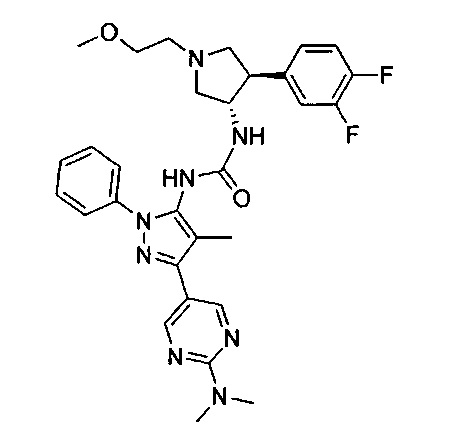
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-(диметиламино)пиримидин-5-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Получили по способу Примера 526, заменяя 2-метоксипиримидин-5-илбороновую кислоту на N,N-диметил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин-2-амин на Стадии А. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 3% МеОН в ДХМ, для получения указанного в заголовке соединения (22 мг, выход 32%) в виде белого твердого вещества. МС (apci) m/z=577,3 (М+Н).
Пример 528
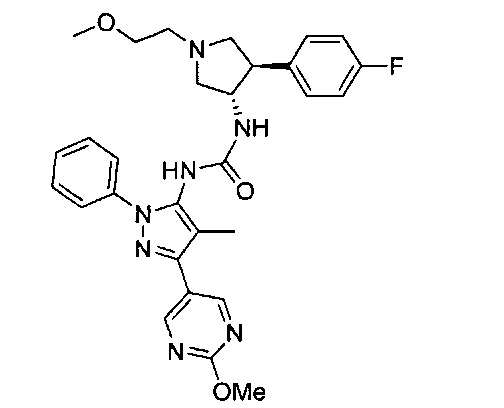
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-метоксипиримидин-5-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Получили по способу Примера 526, заменяя (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (Получение F) на (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина (2S,3S)-2,3-бис((4-метилбензоил)окси)сукцинат (Получение L1, Стадии A-D) на Стадии С.Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 3% МеОН в ДХМ, для получения указанного в заголовке соединения (32 мг, выход 51%) в виде белого твердого вещества. МС (apci) m/z=546,2 (М+Н).
Пример 529
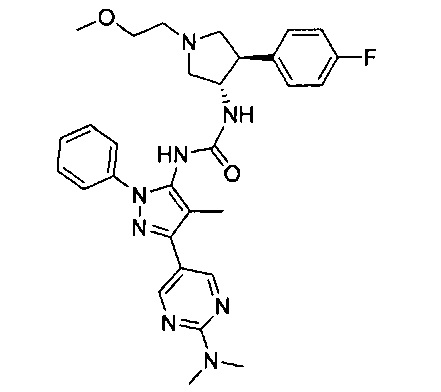
1-(3-(2-(диметиламино)пиримидин-5-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу Примера 527, заменяя (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (Получение F) на (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (Получение К) на Стадии С. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 3% МеОН в ДХМ, для получения указанного в заголовке соединения (27 мг, выход 40%) в виде белого твердого вещества. МС (apci) m/z=559,3 (М+Н).
Пример 530
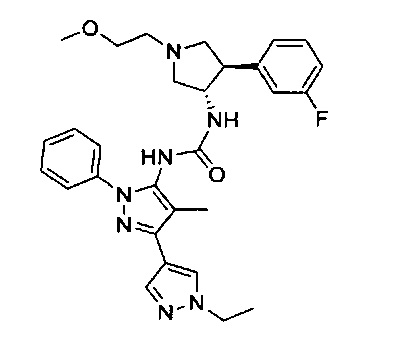
1-(1'-этил-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол)-5-ил)-3-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу Примера 526, заменяя 2-метоксипиримидин-5-илбороновую кислоту на 1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол на Стадии А, и (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (Получение F) на (3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (Получение L11) на Стадии С. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 3% МеОН в ДХМ, для получения указанного в заголовке соединения (39 мг, выход 57%) в виде белого твердого вещества. МС (apci) m/z=532,3 (М+Н).
Пример 531
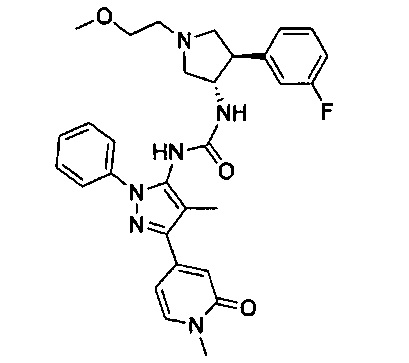
1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)-1-фенил-1Н-пиразол-5-ил)мочевина
Получили по способу Примера 526, заменяя 2-метоксипиримидин-5-илбороновую кислоту на 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он на Стадии А, и (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (Получение F) на (3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (Получение L1) на Стадии С. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 3-8% МеОН в ДХМ, для получения указанного в заголовке соединения (38 мг, выход 54%) в виде белого твердого вещества. МС (apci) m/z=545,2 (М+Н).
Пример 532
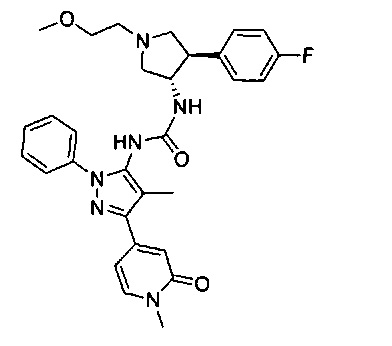
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)-1-фенил-1Н-пиразол-5-ил)мочевина
Получили по способу Примера 526, заменяя 2-метоксипиримидин-5-илбороновую кислоту на 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он на Стадии А, и (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (Получение F) на (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина (2S,3S)-2,3-бис((4-метилбензоил)окси)сукцинат (Получение L1, Стадии A-D) на Стадии С. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 3-8% МеОН в ДХМ, для получения указанного в заголовке соединения (34 мг, выход 49%) в виде белого твердого вещества. МС (apci) m/z=545,3 (М+Н).
Пример 533
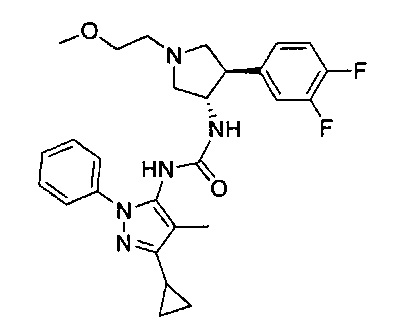
1-(3-циклопропил-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Стадия А: Получение 3-циклопропил-4-метил-1-фенил-1Н-пиразол-5-амина: Суспензию 5-амино-4-метил-1-фенил-1H-пиразол-3-ил трифторметансульфоната (Пример 525, Стадия А; 200 мг, 0,62 ммоль) в толуоле:воде, 10:1 (5,5 мл), в закрытой пробирке дегазировали аргоном в течение 5 минут. Затем добавили циклопропилтрифторборат калия (368 мг, 2,49 ммоль), Pd(OAc)2 (21 мг, 0,09 ммоль) и K3PO4 (396 мг, 1,87 ммоль), затем дициклогексил(2',6'-диизопропилбифенил-2-ил)фосфин (87 мг, 0,19 ммоль). Смесь дегазировали аргоном еще 5 минут, затем закрыли и перемешивали при 110°C в течение 18 часов. Охлажденную смесь разбавили водой (30 мл) и экстрагировали с EtOAc (3×20 мл). Объединенные органические фазы промыли насыщенным солевым раствором (20 мл), высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 1% МеОН в ДХМ, для получения 3-циклопропил-4-метил-1-фенил-1Н-пиразол-5-амина (100 мг, выход 75%) в виде желтого маслянистого вещества. МС (apci) m/z=214,1 (М+Н).
Стадия В: Получение 1-(3-циклопропил-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-Дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины: Получили по способу Примера 526, Стадии В и С, заменяя 3-(2-метоксипиримидин-5-ил)-4-метил-1-фенил-1Н-пиразол-5-амин на 3-циклопропил-4-метил-1-фенил-1Н-пиразол-5-амин на Стадии В. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 1-3% МеОН в ДХМ, для получения указанного в заголовке продукта (29 мг, выход 39%) в виде бесцветного стекловидного вещества. МС (apci) m/z=496,3 (М+Н).
Пример 534
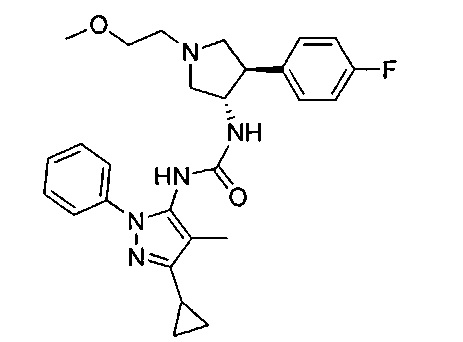
1-(3-циклопропил-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу Примера 533, заменяя (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (Получение F) на (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина (2S,3S)-2,3-бис((4-метилбензоил)окси)сукцинат (Получение L1, Стадии A-D) на Стадии С. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2-4% МеОН в ДХМ, для получения указанного в заголовке соединения (11 мг, выход 15%) в виде бесцветного стекловидного вещества. МС (apci) m/z=478,3 (М+Н).
Пример 535
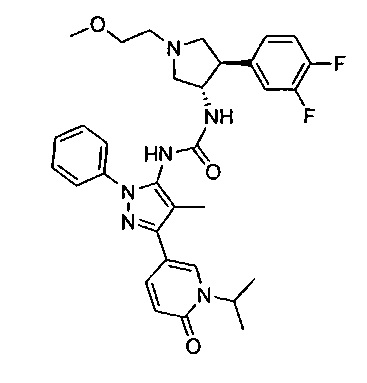
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(1-изопропил-6-оксо-1,6-дигидропиридин-3-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Стадия А: Получение 1-изопропил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-она: 5-Бром-1-изопропилпиридин-2(1Н)-он (500 мг, 2,31 ммоль), бис(пинаколато)дибор (881 мг, 3,47 ммоль) и ацетат калия (681 мг, 6,94 ммоль) смешали в закрытой емкости в 1,4-диоксане (5 мл) и продували аргоном в течение 5 минут. Затем добавили PdCl2(dppf)dcm (189 мг, 0,23 ммоль), продолжали продувание в течение 1 минуты, затем сосуд закрыли и нагревали при 100°C в течение 18 часов. Охлажденную смесь отфильтровали через стекловолоконную бумагу и промыли EtOAc и ДХМ. Фильтрат концентрировали in vacuo, а остаток очистили колоночной хроматографией на диоксиде кремния, элюируя 1% МеОН в ДХМ, затем на второй колонке, элюируя 1:1 смесью гексанов/EtOAc. Полученное твердое вещество растерли с Et2O, отфильтровали, а фильтрат концентрировали in vacuo для получения 1-изопропил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-она (308 мг, выход 51%) в виде твердого вещества персикового цвета. МС (apci) m/z=264,2 (М+Н).
Стадия В: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(1-изопропил-6-оксо-1,6-дигидропиридин-3-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины: Получили по способу Примера 526, заменяя 2-метоксипиримидин-5-илбороновую кислоту на 1-изопропил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2(1Н)-он. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 2,5-4% МеОН в ДХМ, для получения указанного в заголовке соединения (37 мг, выход 53%) в виде бесцветного стекловидного вещества. МС (apci) m/z=591,3 (М+Н).
Пример 536
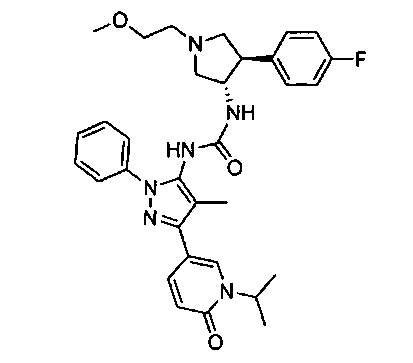
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(1-изопропил-6-оксо-1,6-дигидропиридин-3-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Получили по способу Примера 535, заменяя (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (Получение F) на (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина (2S,3S)-2,3-бис((4-метилбензоил)окси)сукцинат (Получение L1, Стадии A-D) на последней стадии. Материал очистили колоночной хроматографией на диоксиде кремния, элюируя 3-5% МеОН в ДХМ, для получения указанного в заголовке соединения (34 мг, выход 50%) в виде бесцветного стекловидного вещества. МС (apci) m/z=573,3 (М+Н).
Пример 537
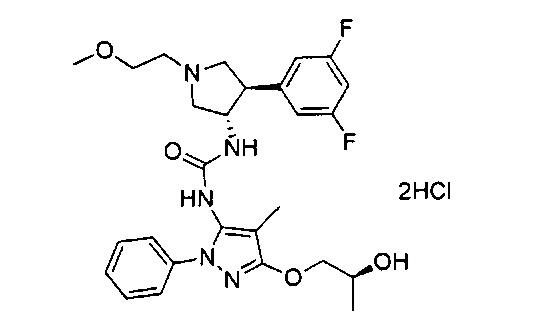
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((S)-2-гидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины дигидрохлорид
Стадия А: Получение 1-(3-((S)-2-((трет-бутилдиметилсилил)окси)пропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины: Получили по способу, описанному для Примера 1, используя Промежуточное соединение Р211 вместо 3-трет-бутил-1-фенил-1Н-пиразол-5-амина на Стадии А, и заменяя (3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлоридом транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-амина дигидрохлорид (Получение В) на Стадии В. МС (apci) m/z=644,4 (М+Н).
Стадия В: Получение 1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((S)-2-гидроксипролокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины дигидрохлорида: Получили по способу, описанному для Примера 179, используя 1-(3-((S)-2-((трет-бутилдиметилсилил)окси)пропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевину вместо 1-(3-((S)-2-((трет-бутилдиметилсилил)окси)пропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины (Получение U-2). МС (apci) m/z=530,3 (М+Н).
Пример 538
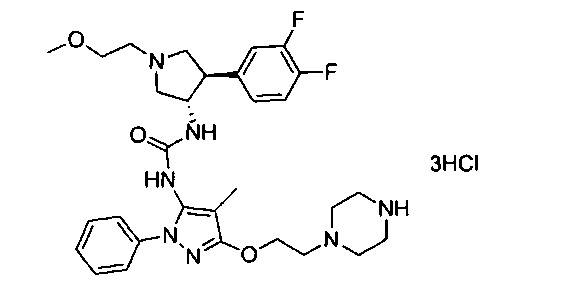
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(2-(пиперазин-1-ил)этокси)-1Н-пиразол-5-ил)мочевины тригидрохлорид
К перемешанному раствору трет-бутил 4-(2-((5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-ил)окси)этил)пиперазин-1-карбоксилата (Пример 388, 52 мг, 0,076 ммоль) в ДХМ (3 мл) добавили 2 н. раствор HCl в эфире (0,15 мл). После перемешивания при комнатной температуре в течение 1 часа, растворители выпарили под пониженным давлением для получения указанного в заголовке соединения (50 мг, выход 110%). МС (apci) m/z=584,3 (М+Н).
Пример 539
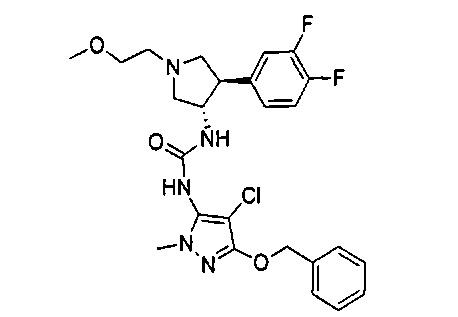
1-(3-(бензилокси)-4-хлор-1-метил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному для Промежуточного соединения 201, Стадия В, используя 1-(3-(бензилокси)-1-метил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевину (Пример 136) вместо фенил 3-этокси-1-фенил-1Н-пиразол-5-илкарбамата. МС (apci) m/z=520,2 (М+Н).
Пример 540
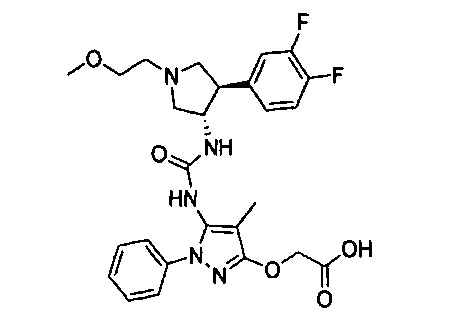
2-((5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-ил)окси)уксусная кислота
Смесь этил 2-((5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-ил)окси)ацетата (Пример 361, 190 мг, 0,341 ммоль) и 1,0 н. водного раствора LiOH (0,682 мл, 0,682 ммоль) в ТГФ (4 мл) и МеОН (2 мл) перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь разбавили водой. По каплям добавили 1,0 н. водный раствор HCl (0,8 мл), чтобы довести pH до 4. Смесь экстрагировали EtOAc. Объединенные экстракты промыли насыщенным солевым раствором, высушили над MgSO4, отфильтровали и концентрировали для получения указанного в заголовке соединения в виде грязновато-белого твердого вещества (150 мг, выход 83%). МС (apci) m/z=530,2 (М+Н).
Пример 541
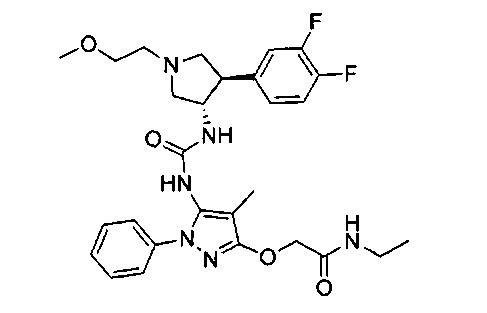
2-((5-(3-((3S,4R))-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-ил)окси)-1-N-этилацетамид
К перемешанному раствору 2-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-илокси)уксусной кислоты (Пример 540, 22 мг, 0,042 ммоль) в ДМФ (2 мл) добавили EDCI (24 мг, 0,12 ммоль) и HOBt (17 мг, 0,12 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 10 минут. Добавили этиламин (2,0 М в ТГФ, 0,062 мл, 0,12 ммоль), затем ТЭА (0,017 мл, 0,12 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 дней. Смесь разбавили EtOAc, последовательно промыли насыщенным водным раствором NH4Cl, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, высушили над MgSO4 и концентрировали. Остаток очистили флэш-хроматографией на силикагеле (5% МеОН в ДХМ) для получения указанного в заголовке соединения (16 мг, выход 69%). МС (apci) m/z=557,3 (М+Н).
Пример 542
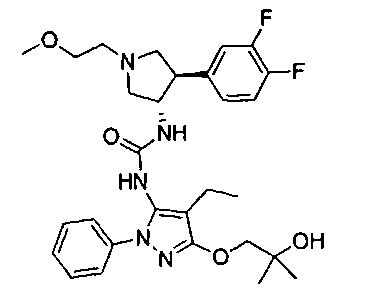
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-этил-3-(2-гидрокси-2-метилпропокси)-1-фенил-1Н-пиразол-5-ил)мочевина
Стадия А: Получение 5-амино-4-этил-1-фенил-1Н-пиразол-3(2H)-она: Смесь этил 2-цианобутаноата (10,0 г, 70,8 ммоль), фенилгидразина (7,66 г, 70,8 ммоль), диоксана (20 мл), EtOH (50 мл) и NaOEt (3,0 М в EtOH, 2,36 мл, 7,08 ммоль) нагревали при 90°C в течение 7 дней. После охлаждения реакционную смесь концентрировали. Остаток обработали Et2O.Твердое вещество собрали фильтрацией, промыли Et2O и высушили в вакууме для получения указанного в заголовке соединения (7,50 г, выход 52%). МС (apci) m/z=204,1 (М+Н).
Стадия В: Получение 1-((5-амино-4-этил-1-фенил-1Н-пиразол-3-ил)окси)-2-метилпропан-2-ола: Получили по способу, описанному для Промежуточного соединения 203, используя 5-амино-4-этил-1-фенил-1Н-пиразол-3(2Н)-он вместо 5-амино-1-фенил-1Н-пиразол-3(2Н)-она. МС (apci) m/z=276,2 (М+Н).
Стадия С: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-этил-3-(2-гидрокси-2-метилопропокси)-1-фенил-1Н-пиразол-5-ил)мочевины: Получили по способу, описанному для Примера 1, используя 1-((5-амино-4-этил-1-фенил-1Н-пиразол-3-ил)окси)-2-метилпропан-2-ол вместо 3-трет-бутил-1-фенил-1Н-пиразол-5-амина на Стадии А, и заменяя (3S,4R))-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлоридом транс-1-(2-метокиэтил)-4-фенилпирролидин-3-амина дигидрохлорид (Получение В) на Стадии В. МС (apci) m/z=558,3 (М+Н).
Пример 543
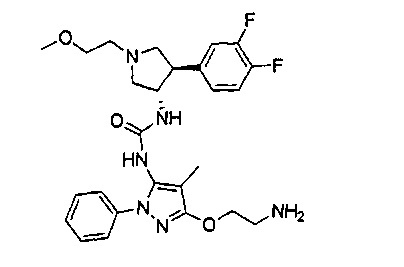
1-(3-(2-аминоэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
К раствору 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-(1,3-диоксоизоиндолин-2-ил)этокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины (Пример 387, 170 мг, 0,264 ммоль) в 1:1 МеОН: ТГФ (10 мл) при комнатной температуре добавили гидразина моногидрат (132 мг, 2,64 ммоль). Реакционную смесь нагревали при 50°C в течение 17 часов. После охлаждения смесь концентрировали. Остаток растерли с ДХМ, а твердое вещество удалили фильтрацией. Фильтрат концентрировали для получения указанного в заголовке соединения (106 мг, выход 78%). МС (apci) m/z=515,3 (М+Н).
Пример 544
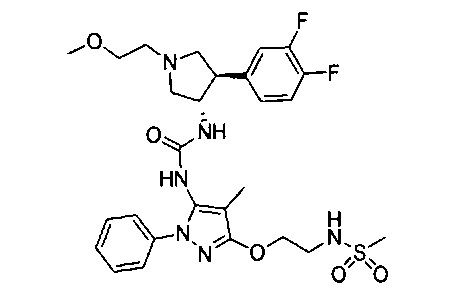
N-(2-((5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-ил)окси)этил)метансульфонамид
Смесь 1-(3-(2-аминоэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины (Пример 543, 30 мг, 0,058 ммоль), метансульфонилхлорида (7,3 мг, 0,064 ммоль) и ТЭА (0,016 мл, 0,12 ммоль) в ДХМ (3 мл) перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь разбавили EtOAc, промыли водой и насыщенным солевым раствором, высушили над MgSO4 и концентрировали. Остаток очистили флэш-хроматографией на силикагеле (3% МеОН в ДХМ) для получения указанного в заголовке соединения (25 мг, выход 72%). МС (apci) m/z=593,2 (М+Н).
Пример 545
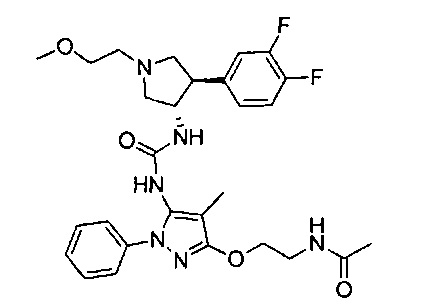
N-(2-((5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-ил~)окси)этил)ацетамид
Получили по способу, описанному для Примера 544, используя уксусный ангидрид вместо метансульфонилхлорида. МС (apci) m/z=557,3 (М+Н).
Пример 546
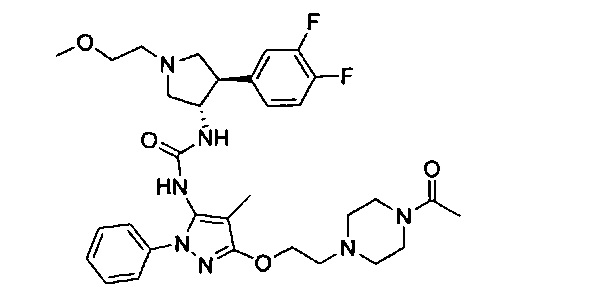
1-(3-(2-(4-ацетилпиперазин-1-ил)этокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному для Примера 544, используя уксусный ангидрид вместо метансульфонилхлорида, и заменяя 1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(2-(пиперазин-1-ил)этокси)-1Н-пиразол-5-ил)мочевины тригидрохлоридом (Пример 538) 1-(3-(2-аминоэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевину (Пример 543). МС (apci) m/z=626,4 (М+Н).
Пример 547
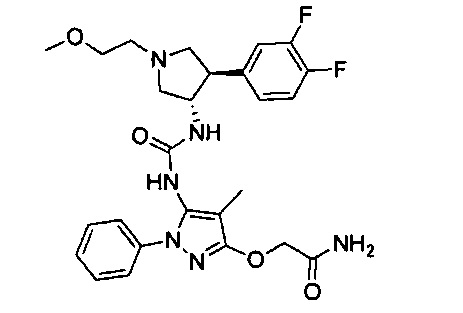
2-((5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-ил)окси)ацетамид
Получили по способу, описанному для Примера 544, используя хлорид аммония вместо этиламина. МС (apci) m/z=529,2 (М+Н).
Пример 548
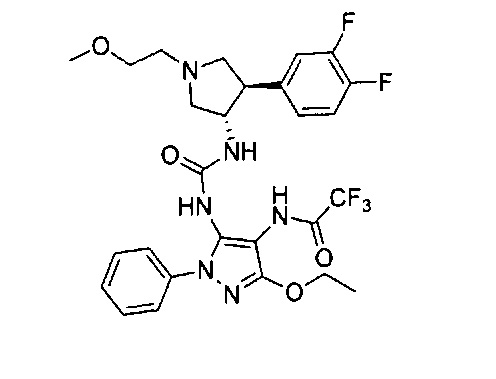
N-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-3-этокси-1-фенил-1Н-пиразол-4-ил)-2,2,2-трифторацетамид
Стадия А: Получение бензил (5-амино-3-оксо-1-фенил-2,3-дигидро-1Н-пиразол-4-ил)карбамата: Получили по способу, описанному для Примера 542, Стадия А, используя этил 2-(бензилоксикарбониламино)-2-цианоацетат вместо этил 2-цианобутаноата. МС (apci) m/z=325,1 (М+Н).
Стадия В: Получение бензил (5-амино-3-этокси-1-фенил-1Н-пиразол-4-ил)карбамата: Получили по способу, описанному для Промежуточного соединения Р135, Стадия В, используя бензил (5-амино-3-оксо-1-фенил-2,3-дигидро-1Н-пиразол-4-ил)карбамат вместо 5-амино-4-метил-1-фенил-1Н-пиразол-3(2Н)-она. МС (apci) m/z=353,1 (М+Н).
Стадия С: Получение бензил (5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-3-этокси-1-фенил-1Н-пиразол-4-ил)карбамата: Получили по способу, описанному для Примера 151, Стадия В, используя бензил (5-амино-3-этокси-1-фенил-1Н-пиразол-4-ил)карбамат вместо 2-(пиридин-4-ил)-2,4,5,6-тетрагидроциклопента[с]пиразол-3-амина, и заменяя (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлоридом (3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-амина бис(2,2,2-трифторацетат) (Получение D). МС (apci) m/z=635,3 (М+Н).
Стадия D: Получение N-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-3-этокси-1-фенил-1Н-пиразол-4-ил)-2,2,2-трифторацетамида: Раствор бензил (5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-3-этокси-1-фенил-1Н-пиразол-4-ил)карбамата (75 мг, 0,12 ммоль) в ТФК (1 мл) нагревали при 60°C в течение 17 часов. Реакционную смесь концентрировали под пониженным давлением. К остатку добавили 5% EtOH в толуоле и снова концентрировали смесь для получения неочищенного продукта в виде соли ТФК. Неочищенный материал растворили в EtOAc, промыли насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, высушили над MgSO4 и концентрировали под пониженным давлением. Остаток очистили колоночной хроматографией на силикагеле (2% МеОН в ДХМ) для получения указанного в заголовке соединения (9 мг, 13%) в качестве неосновного продукта. МС (apci) m/z=597,2 (М+Н).
Пример 549
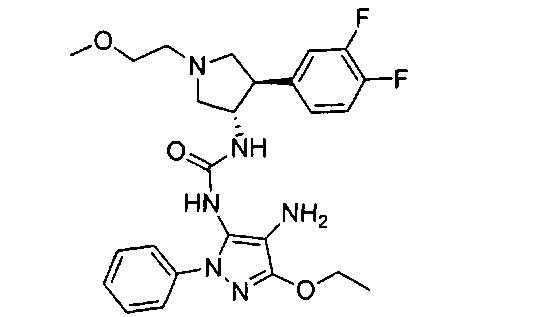
1-(4-амино-3-этокси-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному для Примера 548, Стадия С.Указанное в заголовке соединение получили в качестве основного продукта колоночной хроматографией на силикагеле (5% МеОН в ДХМ). МС (apci) m/z=501,2 (М+Н).
Пример 550
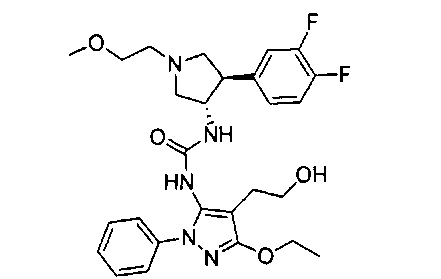
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-этокси-4-(2-гидроксиэтил)-1-фенил-1Н-пиразол-5-ил)мочевина
Стадия А: Получение 5-амино-4-(2,2-диэтоксиэтил)-1-фенил-1Н-пиразол-3(2Н)-она: Получили по способу, описанному для Примера 542, Стадия А, используя этил 2-циано-4,4-диэтоксибутаноат вместо этил 2-цианобутаноата. МС (apci) m/z=292,1 (М+Н).
Стадия В: Получение 4-(2,2-диэтоксиэтил)-3-этокси-1-фенил-1Н-пиразол-5 -амина: Получили по способу, описанному для Промежуточного соединения Р135, Стадия В, используя 5-амино-4-(2,2-диэтоксиэтил)-1-фенил-1Н-пиразол-3(2Н)-он вместо 5-амино-4-метил-1-фенил-1Н-пиразол-3(2Н)-она. МС (apci) m/z=320,2 (М+Н).
Стадия С: Получение 1-(4-(2,2-диэтоксиэтил)-3-этокси-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины: Получили по способу, описанному для Примера 1, используя 4-(2,2-диэтоксиэтил)-3-этокси-1-фенил-1Н-пиразол-5-амин вместо 3-трет-бутил-1-фенил-1Н-пиразол-5-амина на Стадии А, и заменяя (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлоридом транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-амина дигидрохлорид (Получение В) на Стадии В. МС (apci) m/z=602,3 (М+Н).
Стадия D: Получение 1-((3,5,4R))-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-этокси-4-(2-оксоэтил)-1-фенил-1Н-пиразол-5-ил)мочевины: Смесь 1-(4-(2,2-диэтоксиэтил)-3-этокси-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины (0,13 г, 0,22 ммоль), уксусной кислоты (1 мл) и воды (0,2 мл) перемешивали при комнатной температуре в течение 17 часов. По данным ВЭЖХ, реакция не была завершена. Добавили две капли 30 вес.% раствора HBr в АсОН. Реакционную смесь перемешивали еще в течение 17 часов. Реакцию погасили добавлением насыщенного водного раствора NaHCO3, экстрагировали EtOAc, промыли насыщенным водным раствором NaHCO3 (2х) и насыщенным солевым раствором, высушили над MgSO4 и концентрировали для получения указанного в заголовке соединения, которое использовали на следующей стадии без дополнительной очистки. МС (apci) m/z=528,2 (М+Н).
Стадия Е: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-этокси-4-(2-гидроксиэтил)-1-фенил-1Н-пиразол-5-ил)мочевины: К перемешанному раствору 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-этокси-4-(2-оксоэтил)-1-фенил-1Н-пиразол-5 -ил)мочевины (40 мг, 0,076 ммоль) в ТГФ (1 мл) по каплям добавили 2,0 М раствор LiBH4 в ТГФ (0,038 мл, 0,076 ммоль) при 0°C. Реакционную смесь оставили нагреваться до комнатной температуры и перемешивали в течение 3 часов. Реакционную смесь разбавили EtOAc, промыли 0,1 н. раствором HCl, насыщенным водным раствором NaHCO3 и насыщенным солевым раствором, высушили над MgSO4 и концентрировали. Остаток очистили флэш-хроматографией на силикагеле (4% МеОН в ДХМ) для получения указанного в заголовке соединения (4 мг, выход 10%). МС (apci) m/z=530,3 (М+Н).
Пример 551
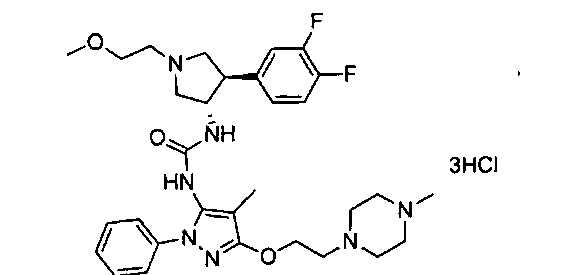
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-(4-метилпиперазин-1-ил)этокси-1-фенил-1Н-пиразол-5-ил)мочевины тригидрохлорид
К смеси 1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(2-(пиперазин-1-ил)этокси)-1Н-пиразол-5-ил)мочевины тригидрохлорида (Пример 538, 50 мг, 0,086 ммоль), NaBH(OAc)3 (73 мг, 0,34 ммоль) и ТГФ (2 мл) добавили формальдегид (37% водный раствор, 14 мг, 0,17 ммоль) при 0°C. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 17 часов. Смесь разбавили H2O (20 мл) и экстрагировали ДХМ. Объединенные органические слои высушили над MgSO4 и концентрировали. Остаток очистили флэш-хроматографией на силикагеле (3% 7 н. аммиака-МеОН в ДХМ) для получения свободного основания, которое обработали 2 н. HCl в эфире (3 капли). Смесь концентрировали и растерли с Et2O для получения указанного в заголовке соединения (22 мг, выход 43%). МС (apci) m/z=598,3 (М+Н).
Пример 552
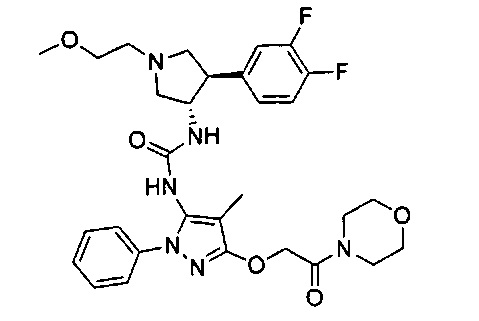
1-((3S,4R))-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-морфолино-2-оксоэтокси)-1-фенил-1Н-пиразол-5-ил)мочевина
Получили по способу, описанному для Примера 541, используя морфолин вместо этиламина. МС (apci) m/z=599,3 (М+Н).
Пример 553
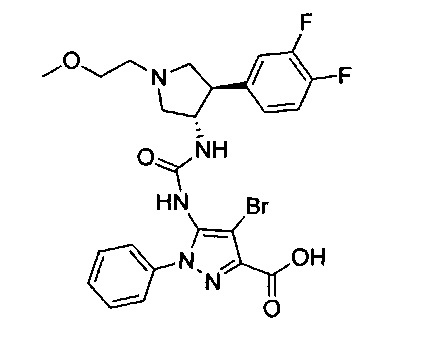
4-бром-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-3-карбоновая кислота
Получили по способу, описанному для Примера 540, используя этил 4-бром-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-3-карбоксилат (Пример 381) вместо этил 2-((5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-ил)окси)ацетата (Пример 361). МС (apci) m/z=564,2 (М+Н).
Пример 554
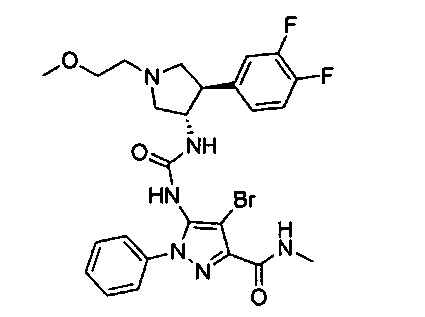
4-бром-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-N-метил-1-фенил-1Н-пиразол-3-карбоксамид
Получили по способу, описанному для Примера 541, используя этил 4-бром-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-3-карбоновую кислоту (Пример 553) вместо 2-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-илокси)уксусной кислоты (Пример 540) и заменяя метиламином этиламин. МС (apci) m/z=577,1 (М+Н).
Пример 555
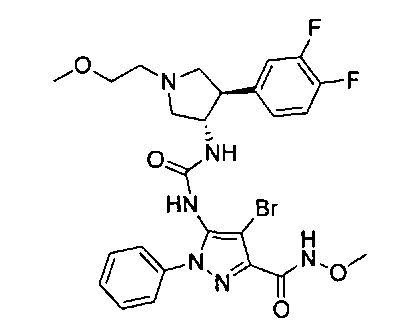
4-бром-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-N-метокси-1-фенил-1Н-пиразол-3-карбоксамид
Получили по способу, описанному для Примера 541, используя этил 4-бром-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-3-карбоновую кислоту (Пример 553) вместо 2-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-илокси)уксусной кислоты (Пример 540) и заменяя этиламин О-метилгидроксиламина гидрохлоридом. МС (apci) m/z=593,1 (М+Н).
Пример 556
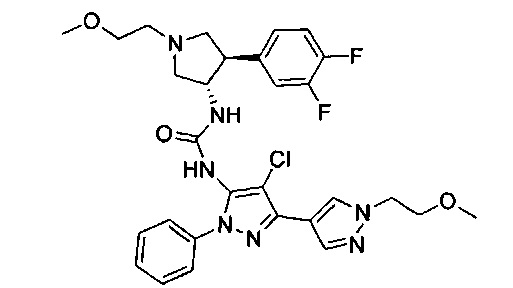
1-(4-хлор-1'-(2-метоксиэтил)-1-фенил-1Н,1'H-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Стадия А: Получение этил 1-(2-метоксиэтил)-1Н-пиразол-4-карбоксилата: Смесь этил 1Н-пиразол-4-карбоксилата (5,00 г, 35,7 ммоль), ДМФ (120 мл), K2CO3 (19,7 г, 143 ммоль) и 1-бром-2-метоксиэтана (9,92 г, 71,4 ммоль) перемешивали при 80°C в течение 4 часов. После охлаждения, реакционную смесь вылили в воду и экстрагировали EtOAc. Объединенные экстракты промыли водой и насыщенным солевым раствором, высушили и концентрировали. Остаток очистили колоночной хроматографией (4:1 гексаны/EtOAc) для получения указанного в заголовке соединения (5,57 г, выход 79%) в виде бесцветного маслянистого вещества. МС (apci) m/z=199,1 (М+Н).
Стадия В: Получение 1'-(2-метоксиэтил)-1-фенил-1Н,1'H-(3,4'-бипиразол)-5-амина: Получили по способу, описанному для Промежуточного соединения Р109, заменяя метил 2-метоксиацетат на этил 1-(2-метоксиэтил)-1Н-пиразол-4-карбоксилат на Стадии А. МС (apci) m/z=284,1 (М+Н).
Стадия С: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пиррол идин-3-ил)-3-(1'-(2-метоксиэтил)-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)мочевины: Получили по способу, описанному для Примера 1, используя 1'-(2-метоксиэтил)-1-фенил-1Н,1'H-[3,4'-бипиразол]-5-амин вместо 3-трет-бутил-1-фенил-1Н-пиразол-5-амина на Стадии А, и заменяя (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлоридом транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-амина дигидрохлорид (Получение В) на Стадии В. МС (apci) m/z=566,2 (М+Н).
Стадия D: Получение 1-(4-хлор-1'-(2-метоксиэтил)-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины: К раствору 1-((3S,4R))-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-(2-метоксиэтил)-1-фенил-1H,1'H-3,4'-бипиразол-5-ил)мочевины (40 мг, 0,071 ммоль) в ДХМ (1 мл) добавили N-хлорсукцинимид (11 мг, 0,085 ммоль), затем каталитическое количество пиридиния 4-метилбензолсульфоната (PPTS). Смесь перемешивали при комнатной температуре в течение ночи. Добавили дополнительное количество N-хлорсукцинимида (4 мг). Реакционную смесь перемешивали при комнатной температуре еще в течение 4 часов. Реакционную смесь разделили между ДХМ и насыщенным водным раствором NaHCO3. Водный слой экстрагировали ДХМ. Объединенные органические слои промыли насыщенным солевым раствором, высушили и концентрировали. Остаток очистили обращенно-фазовой препаративной ВЭЖХ (5-95% ацетонитрила в воде) для получения указанного в заголовке соединения (15 мг, выход 35%) в виде белого твердого вещества. МС (apci) m/z=600,2 (М+Н).
Пример 557
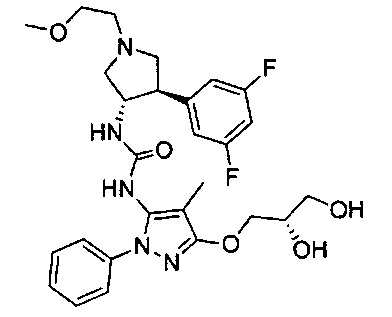
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((S)-2,3-дигидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Получили из 1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(((R)-2,2-диметил-1,3-диоксолан-4-ил)метокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины (Пример 392; 38,0 мг, 0,065 ммоль) по способу, описанному для Примера 176, для получения указанного в заголовке соединения в виде белого твердого вещества (32 мг, выход 90%). МС (apci) m/z=546,2 (М+Н).
Пример 558
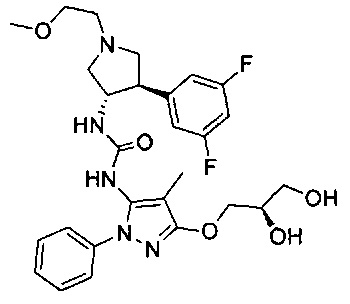
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((R)-2,3-дигидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Получили из 1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(((S)-2,2-диметил-1,3-диоксолан-4-ил)метокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины (Пример 393; 82,0 мг, 0,130 ммоль) по способу, описанному для Примера 176, для получения указанного в заголовке соединения в виде белого твердого вещества (67 мг, выход 97%). МС (apci) m/z=546,2 (М+Н).
Пример 559
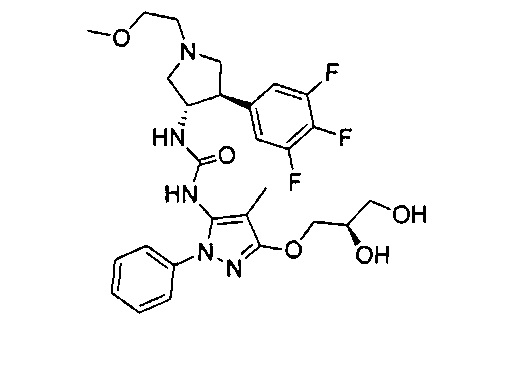
1-(3-((R)-2,3-дигидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевина
Получили из 1-(3-(((S)-2,2-диметил-1,3-диоксолан-4-ил)метокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевины (Пример 398; 90,0 мг, 0,149 ммоль) по способу, описанному для Примера 176, для получения указанного в заголовке соединения в виде белого твердого вещества (83 мг, выход 99%). МС (apci) m/z=564,2 (М+Н).
Пример 560
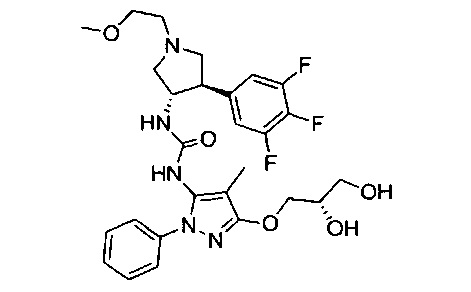
1-(3-((S)-2,3-дигидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевина
Получили из 1-(3-(((R)-2,2-диметил-1,3-диоксолан-4-ил)метокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевины (Пример 399; 64,0 мг, 0,096 ммоль) по способу, описанному для Примера 176, для получения указанного в заголовке соединения в виде белого твердого вещества (44 мг, выход 81%). МС (apci) m/z=564,2 (М+Н).
Пример 561
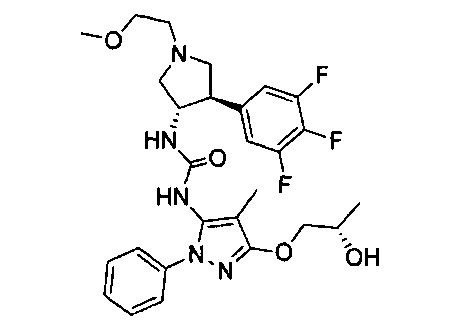
1-(3-((S)-2-гидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевина
Получили из 1-(3-((S)-2-(трет-бутилдиметилсилилокси)пропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевины (Пример 400; 64,0 мг, 0,097 ммоль) по способу, описанному для Примера 176, для получения указанного в заголовке соединения в виде белого твердого вещества (44 мг, выход 83%). МС (apci) m/z=548,3 (М+Н).
Пример 562
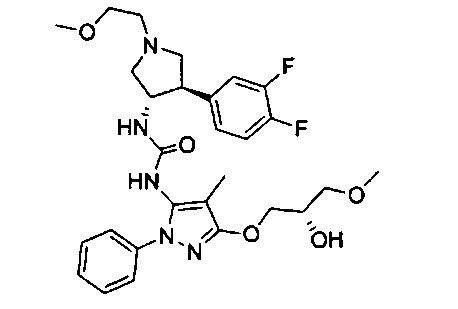
1-(3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((S)-2-гидрокси-3-метоксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
К раствору 1-(3-((S)-2-(трет-бутилдиметилсилилокси)-3-метоксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины (Пример 402, 110 мг, 0,163 ммоль) в ТГФ (5 мл) добавили 1 М HCl (5 мл) и перемешивали смесь при комнатной температуре в течение 90 минут. Смесь концентрировали до 3 мл и разбавили 1 М HCl (3 мл). Смесь промыли Et2O (2Х), а водный раствор обработали 50% раствором NaOH до рН=7. Добавили NaCl до насыщения и экстрагировали смесь EtOAc (3Х). Объединенные экстракты высушили над MgSO4, отфильтровали через упакованный Celite® (элюирование с EtOAc) и концентрировали для получения белого твердого вещества. Твердое вещество измельчили в порошок и высушили в вакууме для получения указанного в заголовке соединения в виде белого порошка (64 мг, 70%). МС (apci) m/z=560,3 (М+Н).
Пример 563
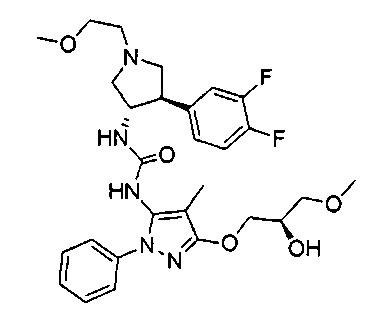
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((R)-2-гидрокси-3-метоксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Получили из 1-(3-((R)-2-(трет-бутилдиметилсилилокси)-3-метоксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины (полученной по способу Примера 402, 104 мг, 0,154 ммоль) по способу, описанному для Примера 564. Указанное в заголовке соединение получили в виде белого твердого вещества (76 мг, выход 88%). МС (apci) m/z=560,3 (М+Н).
Пример 564
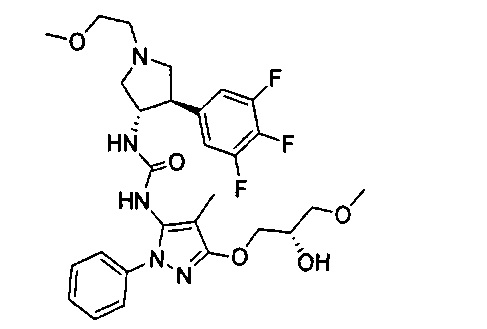
1-(3-((S)-2-гидрокси-3-метоксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевина
Стадия А: 1-(3-((S)-2-(трет-бутилдиметилсилилокси)-3-метоксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевина: Получили по способу, описанному для Примера 1, используя соответствующие исходные материалы. Указанное в заголовке соединение получили в виде бесцветного воскообразного вещества (122 мг, выход 88%). МС (apci) m/z=692,3 (М+Н).
Стадия В: 1-(3-((S)-2-гидрокси-3-метоксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевина: К раствору 1-(3-((S)-2-(трет-бутилдиметилсилилокси)-3-метоксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевины (120 мг, 0,173 ммоль) в ТГФ (5 мл) добавили 1 М HCl (5 мл) и перемешивали смесь при комнатной температуре в течение 3,5 часов. Смесь концентрировали до 5 мл и промыли Et2O (3Х). Водный раствор обработали 50% раствором NaOH до рН=7. Добавили NaCl до насыщения и экстрагировали смесь EtOAc (3Х). Объединенные органические экстракты высушили над MgSO4 и отфильтровали через упакованный Celite®, элюируя EtOAc. Фильтрат концентрировали, а остаточное белое твердое вещество промыли Et2O (3Х) и высушили в вакууме для получения указанного в заголовке соединения (76 мг, 76%). МС (apci) m/z=578,3 (М+Н).
Пример 565
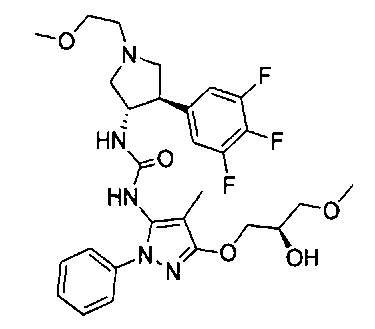
1-(3-((R)-2-гидрокси-3-метоксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-(3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевина
Стадия А: 1-(3-((R)-2-(трет-бутилдиметилсилилокси)-3-метоксипропокс)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевина: Получили по способу, описанному для Примера 1, используя соответствующие исходные материалы. Соединение получили в виде бесцветного воскообразного вещества (131 мг, выход 95%). МС (apci) m/z=692,3 (М+Н).
Стадия В: 1-(3-((R)-2-гидрокси-3-метоксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевина: Используя 1-(3-((R)-2-гидрокси-3-метоксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевину в способе, описанном для Примера 563, Стадия В, получили указанное в заголовке соединение в виде белого твердого вещества (77 мг, 73%). МС (apci) m/z=578,3 (М+Н).
Пример 566
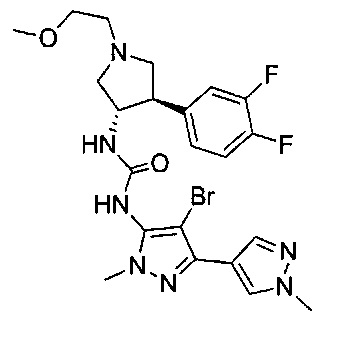
1-(4-бром-1,1'-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Стадия А: фенил 1,1'-диметил-1H,1'H-3,4'-бипиразол-5-илкарбамат: Тонкодисперсную суспензию 1,1'-диметил-1Н,1'Н-3,4'-бипиразол-5-амина (Промежуточное соединение Р114, 159 мг, 0,897 ммоль) в EtOAc (4 мл) охладили до 0°C. Последовательно добавили NaOH (987 мкл, 1,97 ммоль) и фенилхлорформиат (135 мкл, 1,08 ммоль) и перемешивали смесь в течение 5 минут. Смесь оставили достигать до комнатной температуры и перемешивали в течение 24 часов. Смесь промыли H2O (2Х) и насыщенным раствором NaCl. Раствор высушили над MgSO4/активированным углем и элюировали через слой SiO2, элюируя EtOAc. Фильтрат концентрировали, а остаток промыли гексанами (3Х) и высушили в вакууме для получения указанного в заголовке соединения в виде бесцветного воскообразного вещества (232 мг, 87%). МС (apci) m/z=298,1 (М+Н).
Стадия В: 1-(4-бром-1,1'-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина: Раствор фенил 1,1'-диметил-1Н,1'Н-3,4'-бипиразол-5-илкарбамата (44,6 мг, 0,150 ммоль) в сухом СН2Cl2 (1,0 мл) охладили до 0°C и одной порцией добавили N-бромсукцинимид (28,0 мг, 0,157 ммоль). Смесь перемешивали при 0°C в течение 5 минут, оставили достигать комнатной температуры и перемешивали до наблюдения полного расходования исходного материалов в анализе ТСХ (16 часов). К смеси добавили (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (Получение F, 59,3 мг, 0,180 ммоль), затем ДИЭА (78,4 мкл, 0,450 ммоль) и перемешивали смесь при комнатной температуре в течение 3 часов. Смесь разбавили CH2Cl2 (3 мл) и промыли Н2О (3Х). CH2Cl2 раствор высушили над Na2SO4, а высушенный раствор элюировали через короткую колонку SiO2, элюируя CH2Cl2, EtOAc и 10% (9:1 MeOH/NH4OH)/EtOAc. Растворы, содержащие продукт, объединили и концентрировали для получения бесцветного стекловидного вещества. Стекловидное вещество растворили в EtOAc, а мутный раствор отфильтровали через упакованный Celite®. Фильтрат концентрировали, а остаточное белое твердое вещество промыли Et2O и высушили в вакууме для получения указанного в заголовке соединения (57 мг, 71%). МС (apci) m/z=538,2 (М+Н).
Пример 567
1-(4-хлор-1,1'-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Указанное в заголовке соединение получили по способу, описанному для Примера 566, заменяя N-бромсукцинимид на N-хлорсукцинимид на Стадии В. Соединение выделили в виде белого твердого вещества (51 мг, 69%). МС (apci) m/z=494,1 (М+Н).
Пример 568
1-(4-хлор-1-фенил-3-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Раствор фенил 1-фенил-3-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразол-5-илкарбамата (90,9 мг, 0,250 ммоль) в сухом CH2Cl2 (1,0 мл) обработали одной порцией N-хлорсукцинимида (37,5 мг, 0,275 ммоль) и перемешивали смесь при комнатной температуре до завершения реакции, по данным анализа ТСХ (72 часа). К смеси добавили (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (Получение F, 98,8 мг, 0,300 ммоль), затем ДИЭА (131 мкл, 0,750 ммоль) и перемешивали смесь при комнатной температуре в течение 5 часов. Смесь разбавили CH2Cl2 (3 мл) и промыли Н2О (2Х), 1 М NaOH (2Х) и Н2О. CH2Cl2 раствор высушили над Na2SO4/активированным углем, а высушенный раствор отфильтровали через упакованный Celite® и концентрировали. Остаток очистили на SiO2 колонке, используя пошаговое градиентное элюирование: 50% EtOAc в гексанах, EtOAc, а затем 5% MeOH/EtOAc. Объединенные растворы, содержащие продукт, концентрировали для получения бесцветного сиропообразного вещества. Сиропообразное вещество обработали 50% Et2O в гексанах и обрабатывали ультразвуком до образования зернистой белой суспензии. Растворитель декантировали, твердое вещество промыли 50% Et2O в гексанах (2Х) и высушили в вакууме для получения указанного в заголовке соединения в виде белого твердого вещества (106 мг, выход 76%). МС (apci) m/z=560,3 (М+Н).
Соединения в следующей таблице были получены по способу Примера 568, с использованием соответствующих фенилкарбаматных и аминопирролидиновых промежуточных соединений.
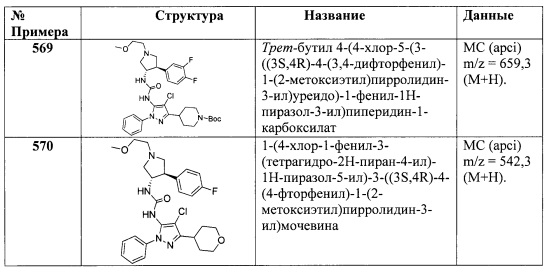
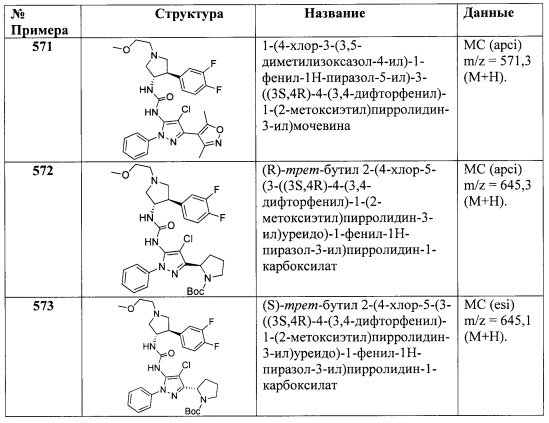
Пример 574
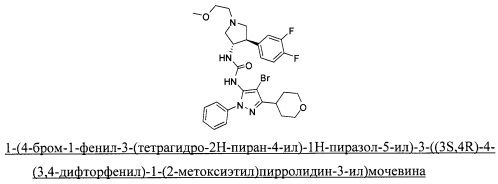
Раствор фенил 1-фенил-3-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразол-5-илкарбамата (90,9 мг, 0,250 ммоль) в сухом CH2Cl2 (1,0 мл) обработали одной порцией N-бромсукцинимида (53,4 мг, 0,300 ммоль) и перемешивали смесь при комнатной температуре до завершения реакции, по данным анализа ТСХ (4 часа). К смеси добавили (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (Получение F, 98,8 мг, 0,300 ммоль), затем ДИЭА (131 мкл, 0,750 ммоль) и перемешивали смесь при комнатной температуре в течение 3 часов. Смесь разбавили CH2Cl2 (3 мл) и промыли Н2О (2Х), 1 М NaOH (2Х) и H2O. CH2Cl2 раствор высушили над Na2SO4/активированным углем, а высушенный раствор отфильтровали через упакованный Celite® и концентрировали. Остаток очистили на SiO2 колонке, элюируя пошаговым градиентом: 50% EtOAc в гексанах, EtOAc, затем 5% МеОН/EtOAc. Объединенные растворы, содержащие продукт, концентрировали для получения бесцветного стекловидного вещества. Стекловидное вещество растворили в Et2O и обрабатывали гексанами до образования суспензии. Суспензию концентрировали для получения указанного в заголовке соединения в виде твердого белого вещества, которое высушили в вакууме (142 мг, 94%). МС (apci) m/z=604,2 (М+Н).
Соединения в следующей таблице были получены по способу Примера 574, с использованием соответствующих фенилкарбаматных и аминопирролидиновых промежуточных соединений.
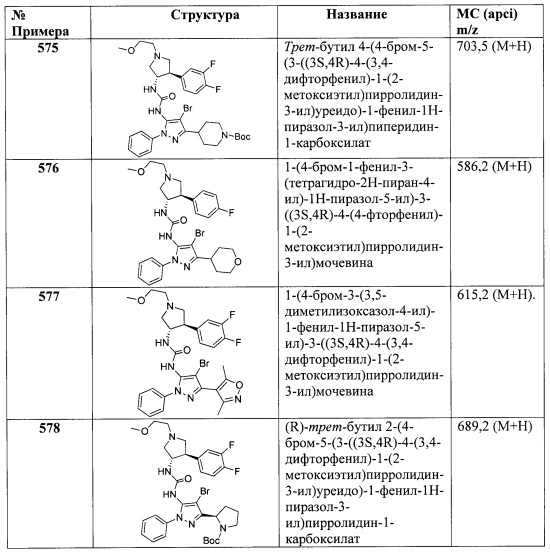
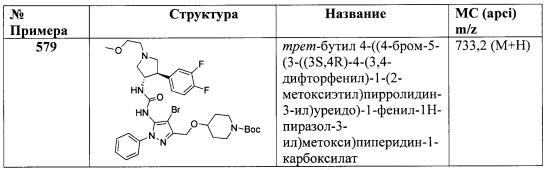
Пример 580
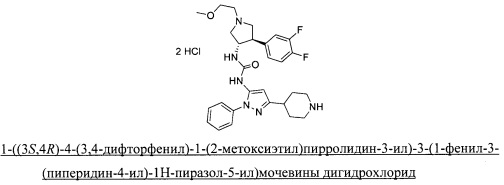
К раствору трет-бутил 4-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-3-ил)пиперидин-1-карбоксилата (полученного по способу Примера 569; 73 мг, 0,12 ммоль) в 2:1 EtOAc/MeOH (4 мл) добавили 4 М HCl в диоксане (3 мл) и перемешивали реакционную смесь при комнатной температуре до завершения реакции, по данным анализа ВЭЖХ (2 часа). Смесь концентрировали, а остаток обработали 50% EtOAc в гексанах. Смесь обрабатывали ультразвуком до образования тонкодисперсной зернистой суспензии. Твердое вещество собрали, промыли 50% EtOAc в гексанах и высушили в вакууме для получения указанного в заголовке соединения в виде белого твердого вещества (70 мг, 100%). МС (apci) m/z=525,8 (М+Н).
Соединения в следующей таблице были получены по способу Примера 580, с использованием соответствующих N-Boc промежуточных соединений.
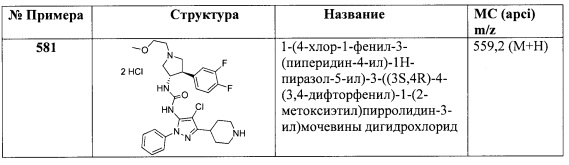
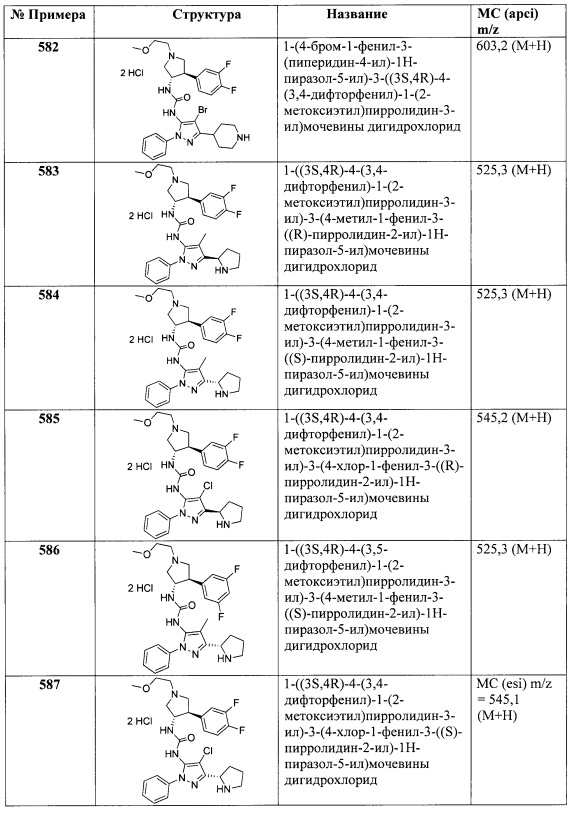
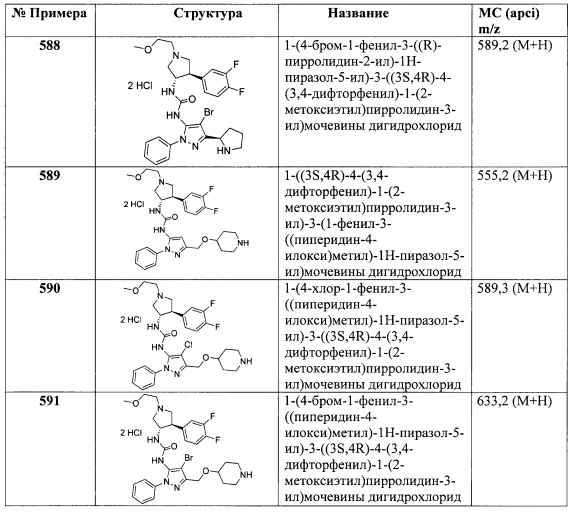
Пример 592
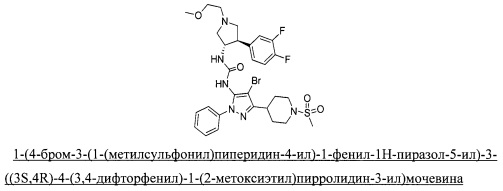
Суспензию 1-(4-бром-1-фенил-3-(пиперидин-4-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины дигидрохлорида (Пример 582, 70 мг, 0,103 ммоль) в сухом CH2Cl2 (1 мл) обработали ДИЭА (72,1 мкл, 0,414 ммоль) и перемешивали смесь при комнатной температуре в течение 5 минут. Полученный гомогенный раствор охладили до 0°C и добавили McCl (8,41 мкл, 0,109 ммоль). Смесь перемешивали в течение 2 часов, и за это время температура постепенно повысилась до комнатной температуры. Смесь разбавили CH2Cl2 (3 мл) и промыли Н2О (2Х). CH2Cl2 раствор высушили над Na2SO4, отфильтровали и концентрировали. Остаток очистили на SiO2, элюируя 50% EtOAc в гексанах, затем 5% MeOH/EtOAc. Объединенные растворы, содержащие продукт, концентрировали, а остаточное бесцветное стекловидное вещество обрабатывали ультразвуком под 50% Et2O в гексанах до образования зернистой суспензии. Растворитель декантировали, а твердое вещество высушили в вакууме для получения указанного в заголовке соединения в виде бледно-розового твердого вещества (54 мг, 77%). МС (apci) m/z=681,2 (М+Н).
Пример 593
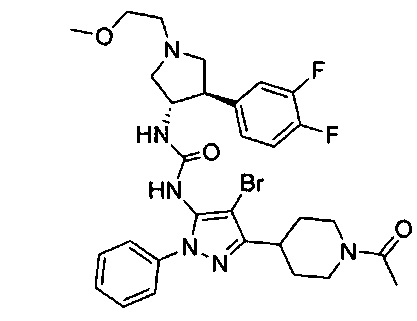
1-(3-(1-ацетилпиперидин-4-ил)-4-бром-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Суспензию 1-(4-бром-1-фенил-3-(пиперидин-4-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины дигидрохлорида (Пример 582, 71 мг, 0,105 ммоль) в сухом CH2Cl2 (1 мл) обработали ДИЭА (73,3 мкл, 0,420 ммоль) и перемешивали смесь при комнатной температуре в течение 5 минут. Полученный гомогенный раствор охладили до 0°C и добавили Ас2О (10,4 мкл, 0,110 ммоль). Смесь перемешивали в течение 1 часа, и за это время температура постепенно достигла 10°C. Смесь разбавили CH2Cl2 (3 мл) и промыли Н2О (2Х). Раствор высушили над Na2SO4, отфильтровали и концентрировали. Остаток очистили на SiO2, элюируя пошаговым градиентом: EtOAc, 5%, затем 10% MeOH/EtOAc. Объединенные растворы, содержащие продукт, концентрировали, а остаточное бесцветное стекловидное вещество растворили в 50% CH2Cl2/гексанах. Раствор концентрировали для получения указанного в заголовке в виде твердого вещества цвета слоновой кости, высушили в вакууме (50 мг, 74%). МС (apci) m/z=645,2 (М+Н).
Пример 594
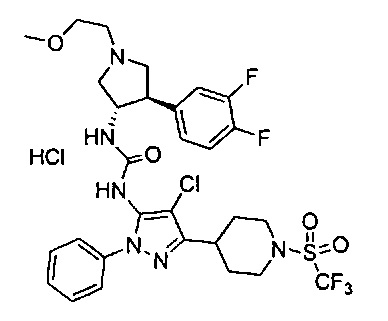
1-(4-хлор-1-фенил-3-(1-(трифторметилсульфонил)пиперидин-4-ил)-1Н-пиразол-5-ил)-3-(3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины гидрохлорид
Суспензию 1-(4-хлор-1-фенил-3-(пиперидин-4-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины дигидрохлорида (Пример 581, 60 мг, 0,0949 ммоль) в сухом CH2Cl2 (1,5 мл) обработали ДИЭА (66,3 мкл, 0,380 ммоль) и перемешивали смесь при комнатной температуре в течение 5 минут. Полученный гомогенный раствор охладили до -40°C (баня из сухого льда, CH3CN) и добавили трифторметансульфоновый ангидрид (17,3 мкл, 0,103 ммоль). Смесь перемешивали в течение 1 часа, и за это время температура достигла -5°C. Реакционную смесь концентрировали, а остаток промыли Н2О (3Х с обработкой ультразвуком) и обработали 50% EtOAc в гексанах. Смесь обработали ультразвуком, обработали MgSO4 и перемешивали в течение 30 минут. Смесь отфильтровали через упакованный Celite®, покрытый слоем MgSO4, используя 50% EtOAc в гексанах для промывания и элюирования. Фильтрат концентрировали для получения бесцветного пенистого вещества. Пенистое вещество растворили в EtOAc (3 мл) и обработали 2 М HCl в Et2O (300 мкл). Полученную мутную белую смесь перемешивали в течение 5 минут и отфильтровали через упакованный Celite® (промывание EtOAc). Фильтрат концентрировали для получения указанного в заголовке в виде твердого вещества цвета слоновой кости, высушили в вакууме (32 мг, 46%). МС (apci) m/z=691,2 (М+Н).
Пример 595
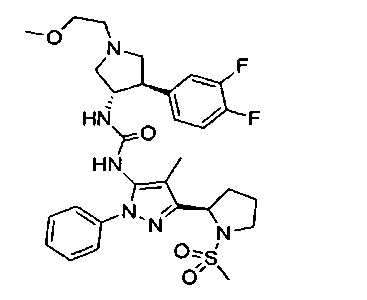
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-((R)-1-(метилсульфонил)пирролидин-2-ил)-1-фенил-1Н-пиразол-5-ил)мочевина
Получили по способу, описанному для Примера 592, используя 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-((R)-пирролидин-2-ил)-1Н-пиразол-5-ил)мочевины дигидрохлорид (Пример 583), для получения указанного в заголовке соединения в виде белого твердого вещества (84 мг, 83%). МС (apci) m/z=603,3 (М+Н).
Пример 596
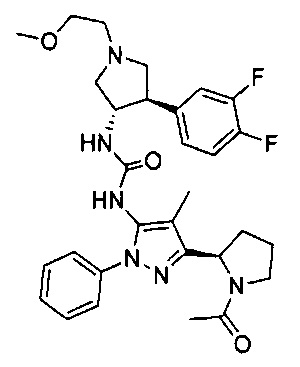
1-(3-((R)-1-ацетилпирролидин-2-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-[3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному для Примера 593, используя 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-((R)-пирролидин-2-ил)-1Н-пиразол-5-ил)мочевины дигидрохлорид (Пример 583), для получения указанного в заголовке соединения в виде твердого вещества цвета слоновой кости (89 мг, 94%). МС (apci) m/z=567,3 (М+Н).
Пример 597
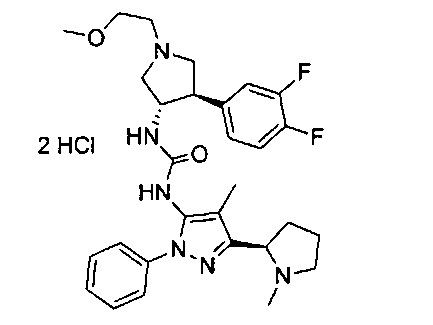
1-((3S,4R))-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-((R)-1-метилпирролидин-2-ил)-1-фенил-1Н-пиразол-5-ил)мочевины дигидрохлорид
Раствор 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-(метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-((R)-пирролидин-2-ил)-1Н-пиразол-5-ил)мочевины дигидрохлорида (Пример 583, 80 мг, 0,134 ммоль) и ДИЭА (93,5 мкл, 0,536 ммоль) в сухом CH2Cl2 (2 мл) охладили до 0°C и добавили йодметан (9,26 мкл, 0,147 ммоль). Смесь перемешивали в течение 3 часов, и за это время температура постепенно достигла комнатной температуры. Смесь разбавили CH2Cl2 (2 мл) и H2O (3 мл), и добавили 1 М NaOH до рН=11. Водный слой удалили, a CH2Cl2 фракцию промыли Н2О (2Х), высушили над Na2SO4, отфильтровали и концентрировали. Остаток очистили SiO2 хроматографией, используя пошаговое градиентное элюирование (EtOAc, 10% MeOH/EtOAc и 10% (9:1 MeOH/NH4OH/EtOAc). Объединенные растворы, содержащие продукт, концентрировали для получения свободного основания продукта в виде бесцветного воскообразного вещества, которое высушили в вакууме. Свободное основание продукта растворили в EtOAc (3 мл) и обработали 2 М HCl в Et2O (0,4 мл). Полученную суспензию перемешивали в течение 10 минут и концентрировали для получения указанного в заголовке соединения в виде светлого желто-коричневого твердого вещества, которое высушили в вакууме (25 мг, 31%). МС (apci) m/z=539,3 (М+Н).
Пример 598
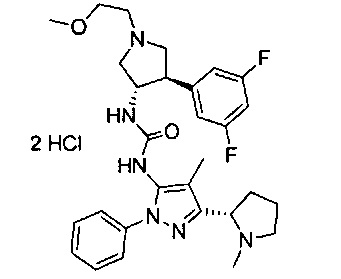
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-((S)-1-метилпирролидин-2-ил)-1-фенил-1Н-пиразол-5-ил)мочевины дигидрохлорид
Получили по способу, описанному для Примера 597, используя 1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-((S)-пирролидин-2-ил)-1Н-пиразол-5-ил)мочевины дигидрохлорид (Пример 586), для получения указанного в заголовке соединения в виде твердого вещества цвета слоновой кости (31 мг, 39%). МС (apci) m/z=539,3 (М+Н).
Пример 599
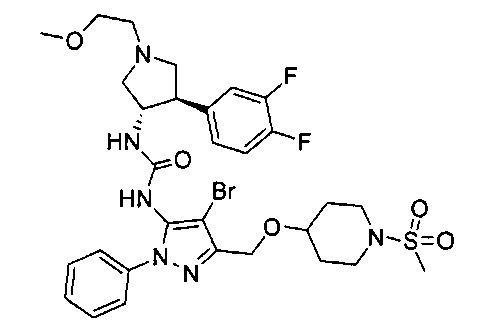
1-(4-бром-3-((1-(метилсульфонил)пиперидин-4-илокси)метил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Используя 1-(4-бром-1-фенил-3-((пиперидин-4-илокси)метил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил-1-(2-метоксиэтил)пирролидин-3-ил)мочевины дигидрохлорид (Пример 591) в способе, описанном для синтеза Примера 592, получили указанное в заголовке соединение в виде белого твердого вещества (23 мг, 88%). МС (apci) m/z=711,2 (М+Н).
Пример 600
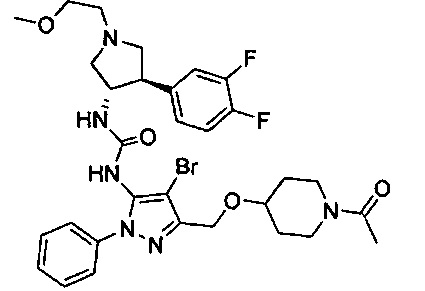
1-(3-((1-ацетилпиперидин-4-илокси)метил)-4-бром-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина
Получили по способу, описанному для Примера 593, используя 1-(4-бром-1-фенил-3-((пиперидин-4-илокси)метил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины дигидрохлорид (Пример 591), для получения указанного в заголовке соединения в виде белого твердого вещества (24 мг, 96%). МС (apci) m/z=675,2 (М+Н).
Пример 601
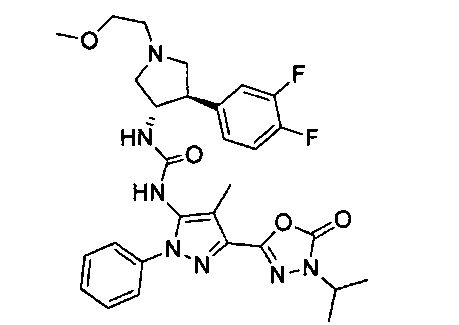
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(4-изопропил-5-оксо-4,5-дигидро-1,3,4-оксадиазол-2-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина
Стадия А: 5-амино-N'-изопропил-4-метил-1-фенил-1Н-пиразол-3-карбогидразид: К раствору 5-амино-4-метил-1-фенил-1Н-пиразол-3-карбоновой кислоты (Промежуточное соединение 171, 146 мг, 0,672 ммоль) и ДИЭА (468 мкл, 2,69 ммоль) в сухом CH2Cl2 (3,0 мл) добавили изобутилхлорформиат (98,1 мкл, 0,739 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа и охладили до 0°C. Одной порцией добавили изопропилгидразина гидрохлорид (149 мг, 1,34 ммоль) и перемешивали смесь при комнатной температуре в течение 48 часов. Смесь промыли H2O (2Х) и высушили над Na2SO4. Высушенный раствор элюировали через слой SiO2, элюируя 50% EtOAc в гексанах. Элюент концентрировали до сиропообразного вещества. Сиропообразное вещество промыли гексанами и растворили в Et2O. Раствор концентрировали для получения неочищенного указанного в заголовке соединения в виде маслянистого белого твердого вещества (148 мг, 54%). МС (apci) m/z=274,1 (М+Н). Неочищенный продукт содержал 30% побочного региоизомера (5-амино-N-изопропил-4-метил-1-фенил-1Н-пиразол-3-карбогидразид), и его использовали напрямую на следующей стадии.
Стадия В: 5-(5-амино-4-метил-1-фенил-1Н-пиразол-3-ил)-3-изопропил-1,3,4-оксадиазол-2(3H)-он: К раствору неочищенного 5-амино-N'-изопропил-4-метил-1-фенил-1Н-пиразол-3-карбогидразида (148 мг, 0,379 ммоль) в сухом CH2Cl2 (3 мл) одной порцией добавили трифосген (57,4 мг, 0,190 ммоль), а полученную белую суспензию перемешивали в течение 17 часов при комнатной температуре. Медленно добавили ДИЭА (264 мкл, 1,52 ммоль), а полученный гомогенный раствор перемешивали при комнатной температуре в течение 4 часов. Смесь промыли Н2О (3Х), высушили над Na2SO4, отфильтровали и концентрировали. Остаток очистили на SiO2 колонке, используя для элюирования 25% EtOAc в гексанах. Продукт получили в виде белого пенистого вещества, которое высушили в вакууме (35 мг, 31%). МС (apci) m/z=300,1 (М+Н).
Стадия С: Фенил 3-(4-изопропил-5-оксо-4,5-дигидро-1,3,4-оксадиазол-2-ил)-4-метил-1-фенил-1Н-пиразол-5-илкарбамат: Раствор 5-(5-амино-4-метил-1-фенил-1Н-пиразол-3-ил)-3-изопропил-1,3,4-оксадиазол-2(3Н)-она (30 мг, 0,100 ммоль) в EtOAc (1 мл) охладили до 0°C. Последовательно добавили NaOH (251 мкл, 0,501 ммоль) и фенилхлорформиат (37,7 мкл, 0,301 ммоль) и перемешивали смесь в течение 5 минут. Ледяную баню убрали, а смесь перемешивали при комнатной температуре в течение 16 часов. Смесь промыли Н2О (2Х), 1 М HCl, Н2О и насыщенным раствором NaCl. Органический слой разбавили 1 равным объемом гексанов и высушили над MgSO4. Раствор элюировали через слой SiO2, элюируя 50% EtOAc в гексанах. Элюент концентрировали, а остаточное белое твердое вещество промыли гексанами (3Х с обработкой ультразвуком) и высушили в вакууме для получения указанного в заголовке соединения (28 мг, 67%). МС (apci) m/z=420,1 (М+Н).
Стадия D: 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(4-изопропил-5-оксо-4,5-дигидро-1,3,4-оксадиазол-2-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина: К смеси фенил 3-(4-изопропил-5-оксо-4,5-дигидро-1,3,4-оксадиазол-2-ил)-4-метил-1-фенил-1Н-пиразол-5-илкарбамата (26 мг, 0,0620 ммоль) и (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-аминадигидрохлорида (Получение F, 26,5 мг, 0,0806 ммоль) в CH2Cl2 (1 мл) добавили ДИЭА (54,1 мкл, 0,310 ммоль). Полученный гомогенный раствор перемешивали при комнатной температуре в течение 3 часов. Смесь разбавили CH2Cl2 (2 мл) и промыли H2O (3Х), 1 М NaOH (2Х) и H2O. СН2С2 раствор высушили над Na2SO4, отфильтровали и концентрировали. Остаток очистили на SiO2 колонке, с пошаговым градиентным элюированием: 50% EtOAc в гексанах, EtOAc, затем 5% MeOH/EtOAc. Объединенные растворы, содержащие продукт, концентрировали для получения бесцветного стекловидного вещества. Стекловидное вещество растворили в 1:1 CH2Cl2/гексанах и концентрировали для получения указанного в заголовке соединения в виде белого твердого вещества, высушили в вакууме (20 мг, 56%). МС (apci) m/z=582,2 (М+Н).
Пример 602
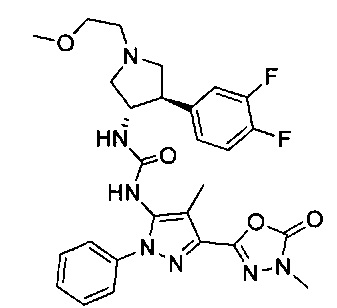
1-((3S,4R-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(4-метил-5-оксо-4,5-дигидро-1,3,4-оксадиазол-2-ил)-1-фенил-1Н-пиразол-5-ил)мочевина
Стадия А: трет-бутил 2-(5-амино-4-метил-1-фенил-1Н-пиразол-3-карбонил)-1-метилгидразинкарбоксилат: К тонко дисперсной суспензии 5-амино-4-метил-1-фенил-1Н-пиразол-3-карбоновой кислоты (Промежуточное соединение 171, 0,434 г, 2,00 ммоль) в сухом CH2Cl2 (5 мл) добавили ДИЭА (0,766 мл, 4,40 ммоль) и перемешивали смесь при комнатной температуре в течение 5 минут. Полученный раствор обработали изобутилхлорформиатом (0,292 мл, 2,20 ммоль) и перемешивали смесь в течение 2 часов. Добавили трет-бутил 1-метилгидразинкарбоксилат (0,308 мл, 2,00 ммоль) и перемешивали смесь при комнатной температуре в течение 23 часов. Смесь промыли Н2О (2Х), высушили над Na2SO4, а высушенный CH2Cl2 раствор элюировали через слой SiO2 (50% EtOAc в гексанах для элюирования). Элюент концентрировали до бесцветного воскообразного вещества. Твердое вещество обработали 50% Et2O в гексанах и перемешивали до образования зернистой белой суспензии. Растворитель декантировали, а остаточное твердое вещество промыли 50% Et2O в гексанах и высушили в вакууме, получили указанное в заголовке соединение в виде белого порошка (550 мг, 80%). МС (apci) m/z=346,2 (М+Н).
Стадия В: 5-амино-N',4-диметил-1-фенил-1Н-пиразол-3-карбогидразида дигидрохлорид: К раствору трет-бутил 2-(5-амино-4-метил-1-фенил-1Н-пиразол-3-карбонил)-1-метилгидразинкарбоксилата (540 мг, 1,56 ммоль) в EtOAc (10 мл) добавили 4 М HCl (7,82 мл, 31,3 ммоль) в диоксане и перемешивали смесь при комнатной температуре в течение 17 часов. Полученную белую суспензию концентрировали, а остаточное белое твердое вещество высушили в вакууме для получения указанного в заголовке соединения (490 мг, 99%). МС (apci) m/z=246,1 (М+Н). 1Н ЯМР (ДМСО d6) δ 11,6 (br s, 2Н), 11,3 (s, 1Н), 7,63 (d, 2H), 7,54 (t, 2H), 7,44 (t, 1H), 2,82 (s, 3H), 2,13 (s, 3H).
Стадия С: 5-(5-амино-4-метил-1-фенил-1Н-пиразол-3-ил)-3-метил-1,3,4-оксадиазол-2(3H)-он: Раствор 5-амино-N',4-диметил-1-фенил-1Н-пиразол-3-карбогидразида дигидрохлорида (484 мг, 1,52 ммоль) и ДИЭА (1,32 мл, 7,61 ммоль) в сухом CH2Cl2 (10 мл) охладили до 0°C и одной порцией добавили трифосген (230 мг, 0,761 ммоль). Смесь перемешивали в течение 17 часов, за это время температура достигла комнатной температуры через 2 часа. Добавили дополнительное количество ДИЭА (0,40 мл) и перемешивали смесь в течение 5 часов. Смесь промыли H2O (3Х), высушили над Na2SO4, а высушенный раствор элюировали через короткую колонку SiO2, элюируя 25% EtOAc в гексанах. Элюент концентрировали, а остаточное воскообразное вещество обработали 50% Et2O в гексанах и перемешивали до образования тонко дисперсной зернистой суспензии. Растворитель декантировали, а остаточное твердое вещество промыли 50% Et2O в гексанах (2Х) и высушили в вакууме для получения указанного в заголовке соединения в виде твердого вещества цвета слоновой кости (197 мг, 48%). МС (apci) m/z=272,1 (М+Н).
Стадия D: Фенил 4-метил-3-(4-метил-5-оксо-4,5-дигидро-1,3,4-оксадиазол-2-ил)-1-фенил-1Н-пиразол-5-илкарбамат: Используя 5-(5-амино-4-метил-1-фенил-1Н-пиразол-3-ил)-3-метил-1,3,4-оксадиазол-2(3Н)-он в способе, описанном для получения Примера 601, Стадия С, получили указанное в заголовке соединение в виде белого твердого вещества (80 мг, 59%). МС (apci) m/z=272,1 (М+Н) (аминопиразольный фрагмент М+Н).
Стадия Е: 1-((3S,4R)-4-(3,4-Дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(4-метил-5-оксо-4,5-дигидро-1,3,4-оксадиазол-2-ил)-1-фенил-1Н-пиразол-5-ил)мочевина: Получили по способу Примера 601, Стадия D, используя фенил 4-метил-3-(4-метил-5-оксо-4,5-дигидро-1,3,4-оксадиазол-2-ил)-1-фенил-1Н-пиразол-5-илкарбамат.Указанное в заголовке соединение выделили в виде белого твердого вещества (50 мг, 60%). МС (apci) m/z=554,2 (М+Н).
Пример 603
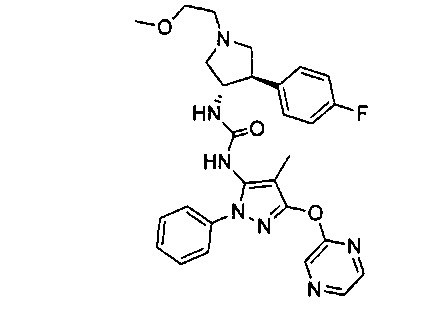
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(пиразин-2-илокси)-1Н-пиразол-5-ил)мочевина
Стадия А: 4-метил-1-фенил-3-(пиразин-2-илокси)-1Н-пиразол-5-амин: В закрытой пробирке смешали 5-амино-4-метил-1-фенил-1Н-пиразол-3(2Н)он (Получение Р135, Стадия А, 300 мг, 1,59 ммоль), 2-хлорпиразин (185 мг, 1,59 ммоль), CuCN (14,2 мг, 0,159 ммоль), карбонат цезия (620 мг, 1,90 ммоль) и сухой ДМФ (3,2 мл). Добавили этилендиамин (23,3 мкл, 0,349 ммоль) и продули пробирку N2, и закрыли. Смесь перемешивали при 110°C в течение 18 часов и охладили до комнатной температуры. Смесь добавили к ледяной H2O (30 мл) и перемешивали в течение 10 минут. Смесь экстрагировали EtOAc (3Х), а объединенные органические фракции промыли насыщенным раствором NaCl (2Х), высушили над MgSO4, отфильтровали через упакованный Celite®. Фильтрат концентрировали, а остаточное сиропообразное вещество очистили на SiO2 колонке, элюируя 40% EtOAc в гексанах, для получения указанного в заголовке соединения в виде белого твердого вещества (310 мг, выход 73%). МС (apci) m/z=268,0 (М+Н). 1Н ЯМР (CDCl3) δ 8,54 (s, 1Н), 8,28 (s, 1Н), 8,16 (s, 1Н), 7,58 (d, 2Н), 7,44 (t, 2Н), 7,31 (t, 1Н), 3,74 (br s, 2Н), 1,81 (s, 3Н).
Стадия В: 1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(пиразин-2-илокси)-1Н-пиразол-5-ил)мочевина: К раствору 4-метил-1-фенил-3-(пиразин-2-илокси)-1Н-пиразол-5-амина (50,0 мг, 0,187 ммоль) в сухом ДМФ (1,0 мл) добавили КДИ (36,4 мг, 0,224 ммоль) и ДИЭА (49,0 мкл, 0,281 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов. Добавили дополнительное количество КДИ (31 мг) и перемешивали смесь в течение 24 часов. К смеси добавили (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амин (Получение L1, 98,1 мг, 0,412 ммоль) и перемешивали смесь при комнатной температуре в течение 5 часов. Смесь разбавили охлажденной H2O (4 мл), а полученную молочно-белую суспензию обработали 2 М HCl до pH=7 (исходный pH=11). Смесь экстрагировали EtOAc (3Х), а объединенные EtOAc фракции промыли насыщенным раствором NaCl (3Х). EtOAc раствор высушили над MgSO4, отфильтровали через упакованный Celite® и концентрировали. Остаточное бесцветное стекловидное вещество очистили на SiO2 колонке с пошаговым градиентным элюированием (EtOAc, 5% МеОН/EtOAc, 10% (9:1 CH3OH/NH4OH)/EtOAc). Полученное белое пенистое вещество перекристаллизовали из 50% EtOAc в гексанах для получения указанного в заголовке соединения в виде белых сфер, которые разрушили и высушили в вакууме (44 мг, 44%). МС (apci) m/z=532,2 (М+Н).
Пример 604
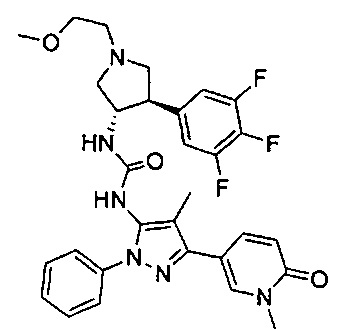
1-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)-3-(4-метил-3-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)-1-фенил-1Н-пиразол-5-ил)мочевина Раствор трифосгена (23,1 мг, 0,074 ммоль) в сухом CH3CN (1 мл) охладили до 0°C и за 45 минут добавили раствор (3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидик-3-амина дигидрохлорида (Получение М, 76,4 мг, 0,220 ммоль) и ДИЭА (115 мкл, 0,660 ммоль) в сухом CH3CN (0,5 мл). Смесь перемешивали в течение 1 часа, и за это время температура достигла 15°C. Одной порцией добавили 5-(5-амино-4-метил-1-фенил-1Н-пиразол-3-ил)-1-метилпиридин-2(1Н)-он (56,1 мг, 0,200 ммоль) и перемешивали смесь при комнатной температуре в течение 7 часов, затем перемешивали при 40°C в течение 17 часов. Смесь охладили до комнатной температуры и разбавили охлажденной H2O (4 мл). Холодную смесь (pH=5) обработали 2 М NaOH до pH=10. Смесь экстрагировали EtOAc (3Х), а объединенные экстракты промыли H2O и насыщенным раствором NaCl (2Х). EtOAc раствор высушили над MgSO4 и элюировали через короткую SiO2 колонку, элюируя EtOAc, 10% МеОН/EtOAc, затем 10% (9:1/CH3OH-NH4OH)/EtOAc. Объединенные растворы, содержащие продукт, концентрировали. Остаток обработали Et2O и встряхивали до образования белой суспензии. Растворитель декантировали, а оставшееся твердое вещество промыли Et2O (2Х) и высушили в вакууме для получения указанного в заголовке соединения в виде белого твердого вещества (34 мг, 29%). МС (apci)m/z=581,2 (М+Н).
Пример 605
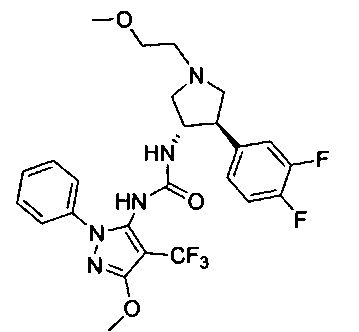
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-метокси-1-фенил-4-(трифторметил)-1Н-пиразол-5-ил)мочевина
Стадия А: Получение 5-фтор-3-метокси-1-фенил-4-(трифторметил)-1Н-пиразола. Смесь NEt3 (0,657 мл, 4,72 ммоль) и фенилгидразина (0,561 г, 5,19 ммоль) в EtOH (2 мл) по каплям добавили к раствору 1,3,3,3-тетрафтор-1-метокси-2-(трифторметил)проп-1-ена (1,00 г, 4,72 ммоль) в EtOH (3 мл). После завершения добавления, реакционную смесь перемешивали при комнатной температуре в течение ночи, концентрировали и очистили силикагелевой колоночной хроматографией, элюируя 0-10% EtOAc в гексанах, для получения указанного в заголовке соединения (372 мг, 1,43 ммоль, выход 30,3%). МС (apci)m/z=261,1 (М+Н).
Стадия В: Получение 3-метокси-1-фенил-4-(трифторметил)-1Н-пиразол-5-амина 5-фтор-3-метокси-1-фенил-4-(трифторметил)-1Н-пиразол (400 мг, 1,54 ммоль), гидразин (148 мг, 4,61 ммоль) и NEt3 (643 мкл, 4,61 ммоль) смешали в ДМЭ (3 мл) в закрытой емкости и нагревали на песочной бане, нагретой до 90°C, в течение 3 часов. Реакционную смесь охладили и добавили никель Ренея (132 мг, 1,54 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, отфильтровали через Celite®, концентрировали и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-80% ацетонитрила в воде, для получения указанного в заголовке соединения (322 мг, 1,25 ммоль, выход 81,4%). МС (apci) m/z=258,1 (М+Н).
Стадия С: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-метокси-1-фенил-4-(трифторметил)-1Н-пиразол-5-ил)мочевины. 3-метокси-1-фенил-4-(трифторметил)-1Н-пиразол-5-амин (77 мг, 0,2994 ммоль), КДИ (50,97 мг, 0,3143 ммоль) и ДИЭА (521,4 мкл, 2,994 ммоль) смешали в 0,8 мл ДМФ и перемешивали при комнатной температуре в течение ночи. Затем 0,6 мл полученного раствора добавили к (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлориду (60 мг, 0,18 ммоль) и перемешивали смесь при комнатной температуре в течение 2 часов, загрузили на картридж Samplet и очистили обращенно-фазовой колоночной хроматографией, элюируя 5-80% ацетонитрила в воде, для получения указанного в заголовке соединения (40 мг, 0,074 ммоль, выход 52%). МС (apci) m/z=540,2 (М+Н).
Пример 606
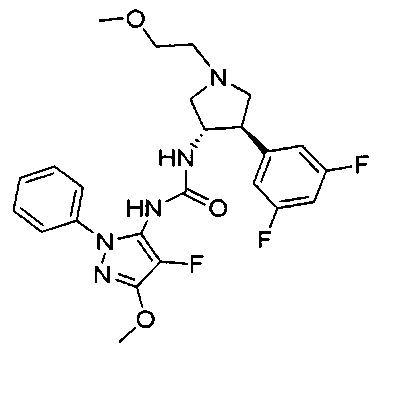
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-метокси-1-фенил-4-(трифторметил)-1Н-пиразол-5-ил)мочевина
Получили по такому же способу, как описан в Примере 605, Стадия С, используя (3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид вместо (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорида. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 5-80% ацетонитрила в Н2О, для получения указанного в заголовке соединения (11 мг, 0,021 ммоль, выход 30%). МС (apci) m/z=540,2 (М+Н).
Пример 607
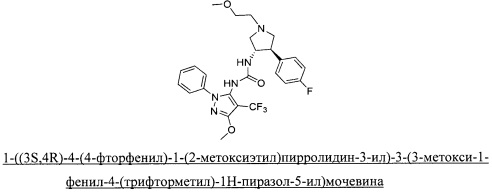
Получили по такому же способу, как описан в Примере 605, Стадия С, используя (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид вместо (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорида. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 5-80% ацетонитрила в H2O, для получения указанного в заголовке соединения (22 мг, 0,042 ммоль, выход 60%). МС (apci) m/z=522,2 (М+Н).
Пример 608
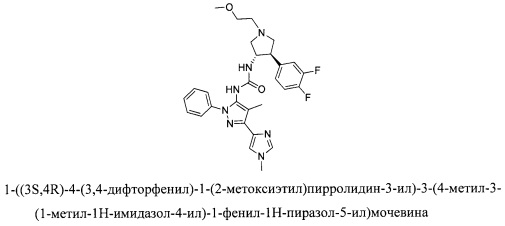
Стадия А: Получение 2-метил-3-(1-метил-1Н-имидазол-4-ил)-3-оксопропаннитрила. Пропионитрил (0,893 г, 16,2 ммоль) по каплям добавили к 1 М раствору LHMDS (13,0 мл, 13,0 ммоль) в ТГФ при -78°С. Смесь перемешивали в течение 30 минут и по каплям добавили раствор этил 1-метил-1Н-имидазол-4-карбоксилата (1,00 г, 6,49 ммоль) в ТГФ (20 мл, нагретый для растворения исходного материала). Реакционную смесь оставили нагреваться до комнатной температуры, перемешивали в течение ночи, вылили в ледяную воду (50 мл) и экстрагировали EtOAc (100 мл). рН довели до 6,5, используя 2 н. HCl, и экстрагировали смесь EtOAc (100 мл). Затем рН довели до 6, используя 2 н. HCl, и экстрагировали смесь EtOAc (2×100 мл).Объединенные экстракты из экстракций с рН 6,5 и рН 6,0 высушили (MgSO4), отфильтровали и концентрировали для получения указанного в заголовке соединения (1,02 г, 6,25 ммоль, выход 96,4%). МС (apci) m/z=164,2 (М+Н).
Стадия В: Получение 4-метил-3-(1-метил-1Н-имидазол-4-ил)-1-фенил-1Н-пиразол-5-амина гидрохлорида. В сосуд для работы под давлением загрузили 2-метил-3-(1-метил-1Н-имидазол-4-ил)-3-оксопропаннитрил (1,00 г, 6,13 ммоль), абсолютный EtOH (12,3 мл, 6,13 ммоль) и фенилгидразина гидрохлорид (0,975 г, 6,74 ммоль). Реакционную смесь закрыли, нагревали при 80°С в течение ночи и концентрировали для получения указанного в заголовке соединения (1,70 г, 5,87 ммоль, выход 95,7%). МС (apci) m/z=254,1 (М+Н).
Стадия С: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(1-метил-1Н-имидазол-4-ил)-1-фенил-1Н-пиразол-5-ил)мочевины. 4-метил-3-(1-метил-1Н-имидазол-4-ил)-1-фенил-1Н-пиразол-5-амин (20 мг, 0,07896 ммоль) растворили в 2 мл EtOAc и добавили NaOH (789,6 мкл, 0,7896 ммоль), затем фенилхлорформиат (29,72 мкл, 0,2369 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, добавили 10 мл EtOAc, а органический слой промыли насыщенным солевым раствором, высушили (MgSO4), концентрировали и растворили в CH2Cl2 (1 мл). Добавили (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (20 мг, 0,061 ммоль) и ДИЭА (88 мкл, 0,51 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение ночи, концентрировали и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-70% ацетонитрила в воде, для получения указанного в заголовке соединения (2,4 мг, 0,0045 ммоль, выход 8,9%). МС (apci) m/z=536,2 (М+Н).
Пример 609
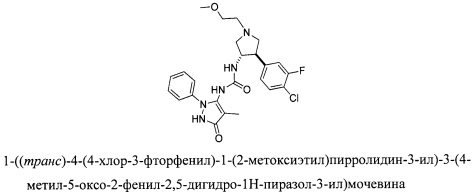
КДИ (360 мг, 2,22 ммоль), 5-амино-4-метил-1-фенил-1Н-пиразол-3(2Н)-он (350 мг, 1,85 ммоль) и ДИЭА (805 мкл, 4,62 ммоль) смешали в 3 мл ДМФ и перемешивали при комнатной температуре в течение ночи. Добавили дополнительное количество КДИ (360 мг, 2,22 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 24 часов. 0,2 мл полученного раствора добавили к раствору (транс)-4-(4-хлор-3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорида (67,5 мг, 0,195 ммоль) и ДИЭА (80,9 мкл, 0,465 ммоль) в ДМФ (2 мл) и перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь загрузили на картридж Samplet и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-70% ацетонитрила в воде, для получения указанного в заголовке соединения (39 мг, 0,0799 ммоль, выход 86,0%). МС (apci) m/z=488,1 (М+Н).
Следующие соединения были получены по способу Примера 609, с заменой (3S,4R)-4-(4-хлор-3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорида на соответствующее пирролидиновое промежуточное соединение.
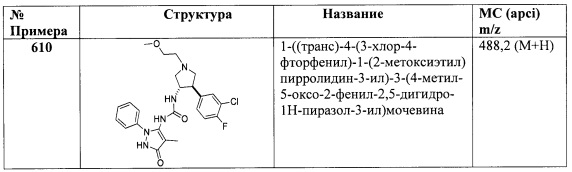
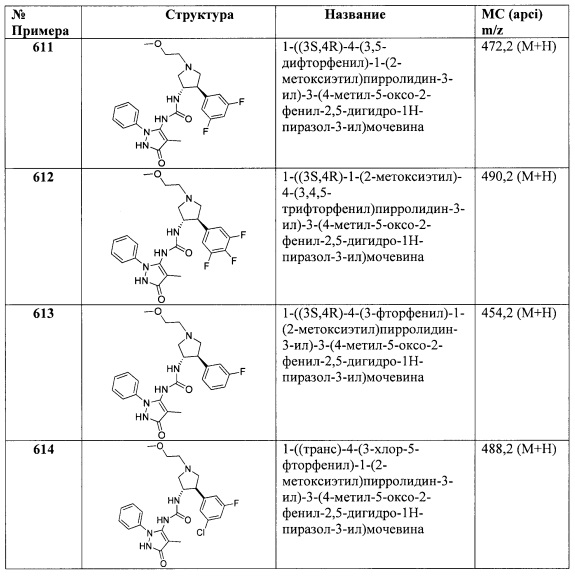
Пример 615
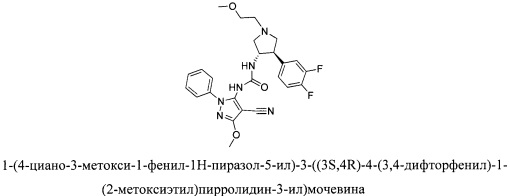
Стадия А: Получение 5-амино-3-метокси-1-фенил-1Н-пиразол-4-карбонитрила. К раствору фенилгидразина (0,783 г, 7,24 ммоль) в этаноле (5 мл) в пробирке для работы под давлением добавили 2-(диметоксиметилен)малононитрил (1,0 г, 7,24 ммоль). Смесь нагревали до 100°С в течение 18 часов. Растворитель выпарили, а неочищенный продукт очистили силикагелевой колоночной хроматографией, элюируя 5-35% ацетона в гексанах, для получения указанного в заголовке соединения (708 мг, выход 45,6%). МС (apci) m/z=215,1 (М+Н).
Стадия В: Получение 1-(4-циано-3-метокси-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины. 5-амино-3-метокси-1-фенил-1Н-пиразол-4-карбонитил (160 мг, 0,747 ммоль), КДИ (133 мг, 0,822 ммоль) и ДИЭА (650 мкл, 3,73 ммоль) смешали в 5 мл ДМФ и перемешивали при комнатной температуре в течение 3 дней. Добавили (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (295 мг, 0,896 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 1 часа, загрузили на картридж Samplet и очистили обращенно-фазовой колоночной хроматографией, элюируя 0-70% ацетонитрила в воде, для получения указанного в заголовке соединения (328 мг, 0,661 ммоль, выход 88,4%). МС (apci) m/z=497,2 (М+Н).
Пример 616
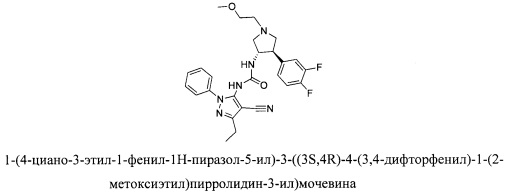
Получили так, как описано в Примере 615, Стадия В, заменяя 5-амино-3-этил-1-фенил-1Н-пиразол-4-карбонитрилом 5-амино-3-метокси-1-фенил-1Н-пиразол-4-карбонитрил. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-70% ацетонитрила в Н2О, для получения указанного в заголовке соединения (890 мг, 1,80 ммоль, выход 76,4%). МС (apci) m/z=495,2 (М+Н).
Пример 617
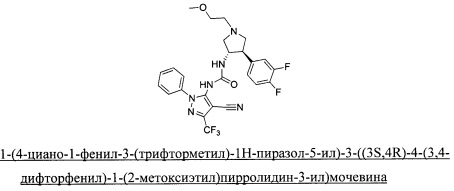
Получили так, как описано в Примере 615, Стадия В, заменяя 5-амино-1-фенил-3-(трифторметил)-1Н-пиразол-4-карбонитрилом 5-амино-3-метокси-1-фенил-1Н-пиразол-4-карбонитрил. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-70% ацетонитрила в Н2О, для получения указанного в заголовке соединения (0,81 г, 1,52 ммоль, выход 76,4%). МС (apci) m/z=535,2 (М+Н).
Пример 618
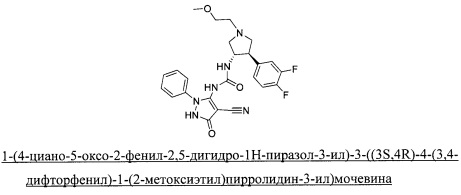
1-(4-циано-3-метокси-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин3-ил)мочевину (30 мг, 0,06042 ммоль, полученную так, как описано в Примере 615) растворили в HCl (61,20 мг, 0,6042 ммоль) и перемешивали при комнатной температуре в течение двух дней. Реакционную смесь вылили в 2 н. раствор NaOH (5 мл) и экстрагировали EtOAc (2×25 мл). Объединенные органические экстракты высушили (MgSO4), концентрировали и очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-70% ацетонитрила в H2O. Выделили пик 1 для получения указанного в заголовке соединения (11 мг, 0,02280 ммоль, выход 37,73%). МС (apci) m/z=483,2 (М+Н).
Пример 619
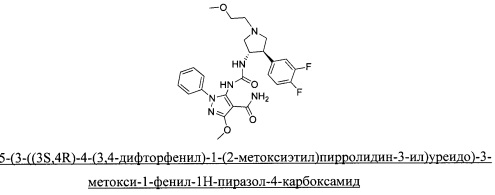
Получили так, как описано в Примере 618, выделив пик 2 вместо пика 1, для получения указанного в заголовке соединения (1,9 мг, 0,0037 ммоль, выход 6,1%). МС (apci) m/z=515,2 (М+Н).
Следующие соединения были получены по способу Примера 618, заменяя 5-амино-3-метокси-1-фенил-1Н-пиразол-4-карбонитрил на соответствующий карбонитрил, а для Примеров 623-626 заменяя также (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид на соответствующее пирролидиновое промежуточное соединение.
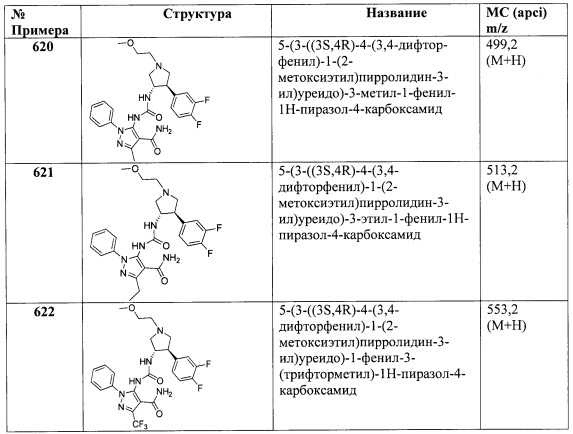
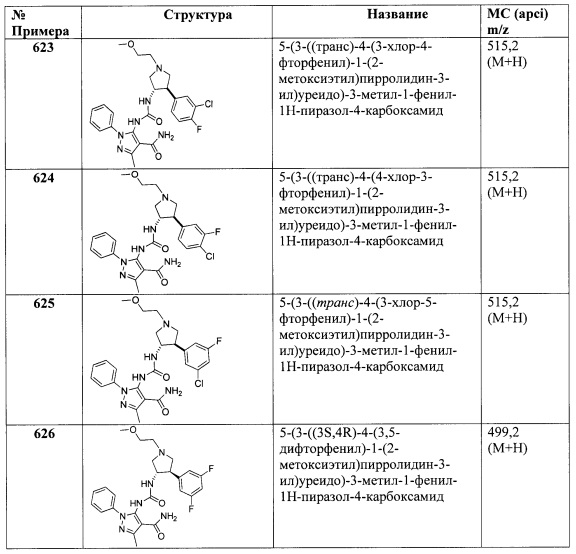
Пример 627
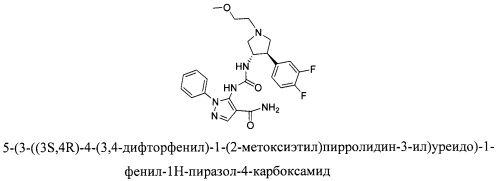
Стадия А: Активация 5-амино-1-фенил-1Н-пиразол-4-карбоксамида. 5-амино-1-фенил-1Н-пиразол-4-карбоксамид (500 мг, 2,47 ммоль) растворили в 2 мл CHCl3 и добавили пиридин (600 мкл, 7,42 ммоль), затем фенилхлорформиат (682 мкл, 5,44 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и погасили 2 мл 2 н. NaOH. Реакционную смесь экстрагировали несколькими порциями CH2Cl2 в фазоразделительном стеклянном фильтре, а объединенные органические экстракты концентрировали. Неочищенный материал очистили силикагелевой колоночной хроматографией, элюируя 5-40% ацетона в гексанах, для получения бис-фенилкарбаматного аддукта (191 мг, 0,432 ммоль, выход 17,5%). МС (apci) m/z=443,1 (М+Н).
Стадия В: Получение 5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-4-карбоксамида. (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (33 мг, 0,099 ммоль), продукт стадии А (20 мг, 0,045 ммоль) и ДИЭА (39 мкл, 0,23 ммоль) смешали в 0,2 мл ДМФ и перемешивали при комнатной температуре в течение ночи. Смесь загрузили на картридж Samplet и очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-70% ацетонитрила в H2O, для получения указанного в заголовке соединения (17 мг, 0,035 ммоль, выход 78%). МС (apci) m/z=485,2 (М+Н).
Пример 628
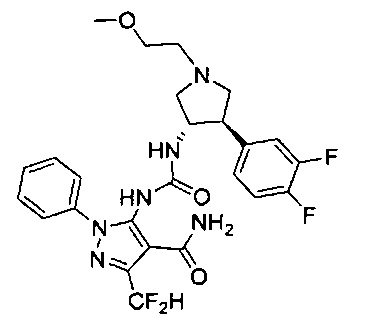
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-4-карбоксамид
Стадия А: Получение 2-(2,2-дифтор-1-гидроксиэтилиден)малононитрила. Раствор малононитрила (4 г, 61 ммоль) в метаноле наполнили метоксидом натрия (14 мг, 67 ммоль), затем метил 2,2-дифторацетатом (8,0 г, 73 ммоль). Реакционную смесь нагревали до 60°C в течение 4 часов и концентрировали in vacuo для получения указанного в заголовке соединения (8,7 г, 60 ммоль, выход 100%). 1Н ЯМР (400 МГц, CDCl3) δ ppm 6,13 (t, J=54 Гц, 1Н).
Стадия В: Получение 2-(1-хлор-2,2-дифторэтилиден)малононитрила. К суспензии 2-(2,2-дифтор-1-гидроксиэтилиден)малононитрила (1,9 г, 13 ммоль) в CH2Cl2 добавили PCl5 (2,7 г, 13 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 16 часов. Реакционную смесь разбавили CH2Cl2, промыли водой и насыщенным солевым раствором, высушили над MgSO4 и концентрировали для получения указанного в заголовке соединения (2,3 г, 14 ммоль, выход 107%).
Стадия С: Получение 5-амино-3-(дифторметил)-1-фенил-1Н-пиразол-4-карбонитрила. К раствору 2-(1-хлор-2,2-дифторэтилиден)малононитрила в этаноле добавили фенилгидразина гидрохлорид (2,3 г, 16 ммоль) и нагревали реакционную смесь до 70°C в течение 4 часов. Реакционную смесь концентрировали in vacuo и разделили материал между EtOAc и водой. Слои разделили, а органический слой промыли насыщенным солевым раствором, высушили (MgSO4) и концентрировали in vacuo. Неочищенный материал очистили силикагелевой колоночной хроматографией, используя в качестве элюента 5% EtOAc/CH2Cl2, для получения указанного в заголовке соединения (0,25 г, 1,1 ммоль, выход 7,5%). МС (apci) m/z=233,1 (М-Н).
Стадия D: Получение 3-(дифторметил)-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-4-карбоксамида. 5-амино-3-(дифторметил)-1-фенил-1Н-пиразол-4-карбонитрил получили так, как описано для 5-амино-3-метокси-1-фенил-1Н-пиразол-4-карбонитрила в Примере 618. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 5-80% ацетонитрила в H2O, затем препаративной ВЭЖХ Гильсона с использованием колонки Chiral Technologies OD-H и гексанов/EtOH, 9:1, в качестве элюента, для получения указанного в заголовке соединения (1,7 мг, 0,0032 ммоль, выход за две стадии 0,30%). МС (apci) m/z=533,2 (М-Н).
Пример 629
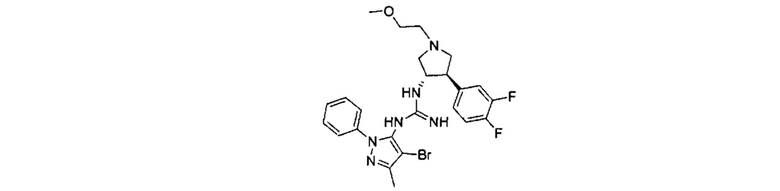
1-(4-бром-3-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)гуанидина гидрохлорид
1-(4-бром-3-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)тиомочевину (40 мг, 0,07267 ммоль, полученную по способу, описанному в Примере 630) и AgOTf (46,68 мг, 0,1817 ммоль) смешали в 5 мл CH2Cl2 и охладили на бане из ледяного МеОН. Через раствор пропускали газообразный аммиак в течение 1 минуты и оставили реакционную смесь нагреваться до комнатной температуры. Добавили HCl (5 н. в IPA, 60,7 мкл, 0,303 ммоль) и МеОН (2 мл) и отфильтровали реакционную смесь через Celite®. Твердые вещества промыли несколькими порциями МеОН, а объединенный фильтрат концентрировали и очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 0-70% ацетонитрила в 0,1 н. водном растворе HCl, для получения указанного в заголовке соединения (11 мг, 0,01814 ммоль, выход 24,97%»). МС (apci) m/z=535,1 (М+Н).
Пример 630
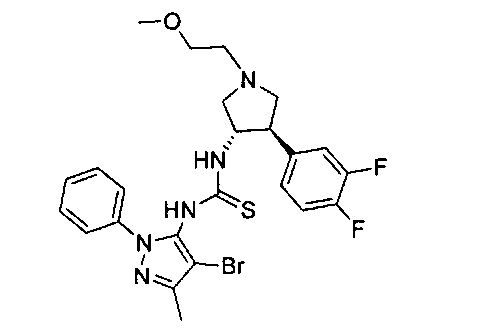
1-(4-бром-3-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)тиомочевина
4-Бром-3-метил-1-фенил-1Н-пиразол-5-амин (250 мг, 0,992 ммоль), ДИЭА (864 мкл, 4,96 ммоль) и ди(1Н-имидазол-1-ил)метантион (177 мг, 0,992 ммоль) смешали в 1 мл ДМФ и перемешивали при комнатной температуре в течение 3 дней, а затем при 70°C в течение ночи. Добавили (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (326 мг, 0,992 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 24 часов. Смесь загрузили на картридж Samplet и очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 5-80%) ацетонитрила в H2O, для получения указанного в заголовке соединения (364 мг, 0,661 ммоль, выход 66,7%). МС (apci) m/z=550,1 (М+Н).
Пример 631
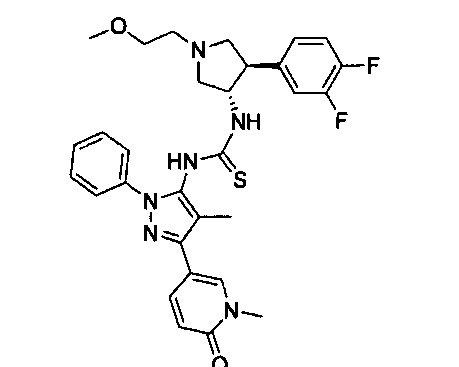
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)-1-фенил-1Н-пиразол-5-ил)тиомочевина
Получили так, как описано для Примера 630, заменяя 5-(5-амино-4-метил-1-фенил-1Н-пиразол-3-ил)-1-метилпиридин-2(1Н)-оном 4-бром-3-метил-1-фенил-1Н-пиразол-5-амин. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 5-80% ацетонитрила в H2O, для получения указанного в заголовке соединения (4,5 мг, 0,00778 ммоль, выход 12,5%). МС (apci) m/z=579,2 (М+Н).
Пример 632
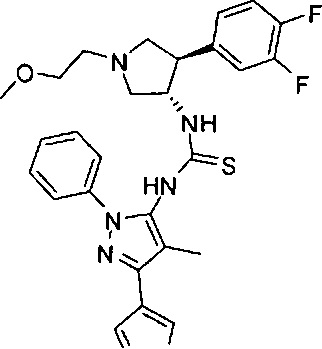
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)-тиомочевина
Получили так, как описано для Примера 630, заменяя 1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-амином 4-бром-3-метил-1-фенил-1Н-пиразол-5-амин. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 5-80% ацетонитрила в H2O, для получения указанного в заголовке соединения (71 мг, 0,129 ммоль, выход 75,8%). МС (apci) m/z=552,2 (М+Н).
Пример 633
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)тиомочевина
Получили так, как описано для Примера 630, заменяя 1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-амином 4-бром-3-метил-1-фенил-1Н-пиразол-5-амин, и (3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлоридом (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид. Материал очистили обращенно-фазовой колоночной хроматографией, используя в качестве элюента 5-80% ацетонитрила в H2O, для получения указанного в заголовке соединения (50 мг, 0,0937 ммоль, выход 69,0%). МС (apci) m/z=534,2 (М+Н).
Соединения были получены по способу Примера 52, с использованием соответствующих исходных материалов в подходящем растворителе, таком как CH2Cl2, ДМФ, ДМА или CH3CN.
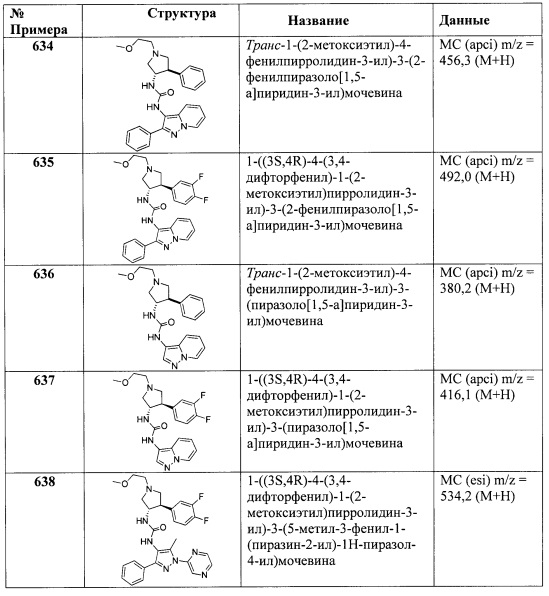
Следующие соединения были получены по способу Примера 1, с использованием соответствующих исходных материалов в подходящем растворителе, таком как CH2Cl2, ДМФ, ДМА или CH3CN.
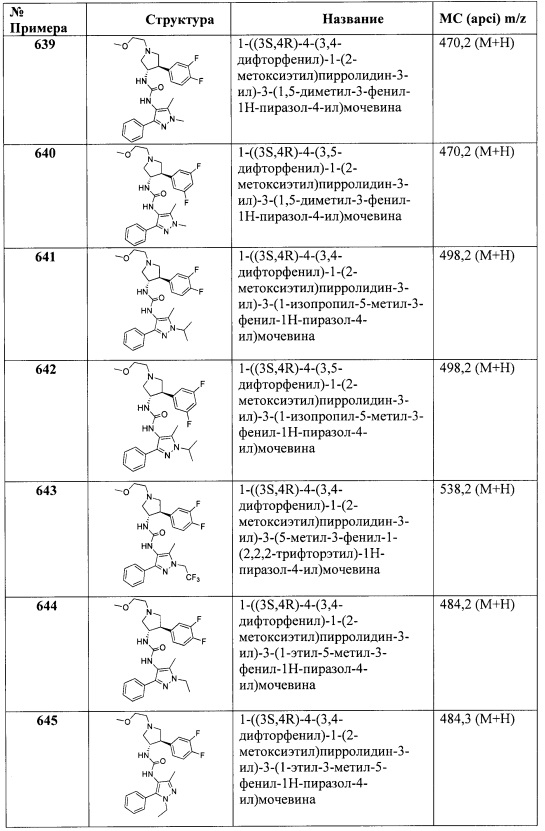
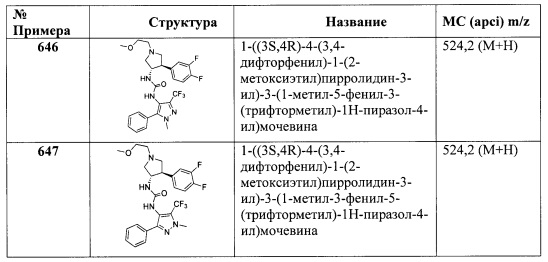
Пример 648
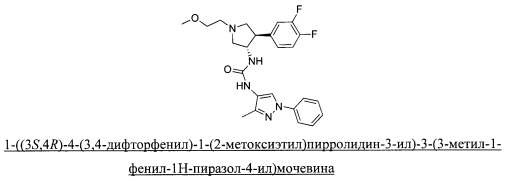
Стадия А: Получение этил 3-метил-1-фенил-1Н-пиразол-4-карбоксилата: Смесь этил 3-метил-1Н-пиразол-4-карбоксилата (0,600 г, 3,89 ммоль), фенилбороновой кислоты (0,498 г, 4,09 ммоль), Cu(ОАс)2 (0,530 г, 2,92 ммоль), пиридина (0,630 мл, 7,78 ммоль) в сухом ДМФ (39 мл) перемешивали при комнатной температуре в течение 3 дней. Реакционную смесь разделили между EtOAc и водой, а органический слой удалили. Водный слой экстрагировали EtOAc (2Х), а объединенные органические слои промыли водой и насыщенным раствором NaCl. EtOAc раствор высушили над MgSO4, отфильтровали и концентрировали. Остаток очистили флэш-хроматографией на силикагеле (4:1 гексаны/EtOAc) для получения указанного в заголовке соединения (0,428 г, 48%). МС (apci) m/z=231,1 (М+Н).
Стадия В: Получение 3-метил-1-фенил-1Н-пиразол-4-карбоновой кислоты: К раствору этил 3-метил-1-фенил-1Н-пиразол-4-карбоксилата (0,428 г, 1,86 ммоль) в 1:1 МеОН/ТГФ (8,0 мл) добавили 1 М LiOH (3,72 мл, 3,72 ммоль) и перемешивали смесь при комнатной температуре в течение 16 часов. Растворители удалили в вакууме, остаток разбавили водой и экстрагировали Et2O (2Х). Водный слой обработали 1 М HCl до рН 4-5и экстрагировали EtOAc (3Х). Объединенные органические фракции промыли водой и насыщенным раствором NaCl. EtOAc раствор высушили над MgSO4, отфильтровали и концентрировали для получения неочищенного продукта (0,280 г, 75%), который использовали на следующей стадии без дополнительной очистки. МС (apci) m/z=203,1 (М+Н).
Стадия С: Получение 1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-метил-1-фенил-1Н-пиразол-4-ил)мочевины: К раствору 3-метил-1-фенил-1Н-пиразол-4-карбоновой кислоты (50 мг, 0,25 ммоль) и Et3N (0,039 мл, 0,30 ммоль) в толуоле (2 мл) добавили дифенилфосфоразидат (0,064 мл, 0,30 ммоль). Раствор нагревали с дефлегматором в течение 1 часа и охладили до комнатной температуры. Смесь разбавили ТГФ (1 мл) и добавили (3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-амина дигидрохлорид (98 мг, 0,30 ммоль), затем Et3N (0,108 мл, 0,90 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Смесь разделили между EtOAc и насыщенным водным раствором NaHCO3. Органический слой удалили, а водный слой экстрагировали EtOAc (2Х). Объединенные органические слои промыли насыщенным раствором NaCl, высушили над MgSO4, отфильтровали и концентрировали. Остаток очистили флэш-хроматографией на силикагеле (5% МеОН в ДХМ) для получения указанного в заголовке соединения (66 мг, 59%). МС (apci) m/z=456,2 (М+Н).
Пример 649
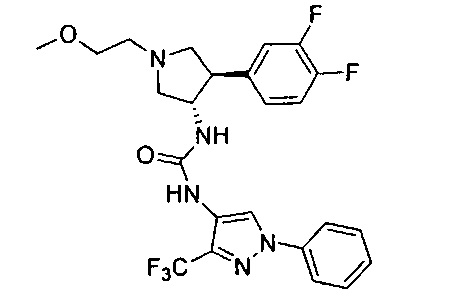
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-фенил-3-(трифторметил)-1Н-пиразол-4-ил)мочевина
Получили по способу, описанному для Примера 648, Стадия C, используя 1-фенил-3-(трифторметил)-1Н-пиразол-4-карбоновую кислоту вместо 3-метил-1-фенил-1Н-пиразол-4-карбоновой. МС (apci) m/z=510,2 (М+Н).
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ N-ПИРРОЛИДИНИЛМОЧЕВИНЫ, N'-ПИРАЗОЛИЛМОЧЕВИНЫ, ТИОМОЧЕВИНЫ, ГУАНИДИНА И ЦИАНОГУАНИДИНА КАК ИНГИБИТОРЫ КИНАЗЫ TRKA | 2013 |
|
RU2677667C2 |
| 1-((3S,4R)-4-(3-ФТОРФЕНИЛ)-1-(2-МЕТОКСИЭТИЛ)ПИРРОЛИДИН-3-ИЛ)-3-(4-МЕТИЛ-3-(2-МЕТИЛПИРИМИДИН-5-ИЛ)-1-ФЕНИЛ-1Н-ПИРАЗОЛ-5-ИЛ)МОЧЕВИНА В КАЧЕСТВЕ ИНГИБИТОРА TrkA КИНАЗЫ | 2015 |
|
RU2719489C2 |
| ЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ ПИРАЗОЛО[1,5-a]ПИРИМИДИНА КАК ИНГИБИТОРЫ КИНАЗЫ TRK | 2010 |
|
RU2584157C2 |
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ МОЧЕВИНЫ, ТИОМОЧЕВИНЫ, ГУАНИДИНА И ЦИАНОГУАНИДИНА, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ БОЛИ | 2013 |
|
RU2664541C2 |
| ТОЧЕЧНЫЕ МУТАЦИИ В УСТОЙЧИВЫХ К ИНГИБИТОРУ TRK ЗЛОКАЧЕСТВЕННЫХ ОПУХОЛЯХ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ | 2016 |
|
RU2744852C2 |
| ТРИАЗОЛОПИРИДИНОВЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ КИНАЗЫ PIM | 2012 |
|
RU2598846C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛО[1,5-a]ПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ ТРК КИНАЗЫ | 2009 |
|
RU2523544C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛО[1,5-а]ПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ ТРК КИНАЗЫ | 2014 |
|
RU2666367C2 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИНОВЫХ КИНАЗ | 2013 |
|
RU2650501C2 |
| АГОНИСТЫ РЕЦЕПТОРА МЕЛАНОКОРТИНА | 2007 |
|
RU2411240C2 |
Изобретение относится к соединениям формулы I, приведенной ниже, или к их стереомерам, таутомерам или фармацевтически приемлемым солям. R1, R2, Ra, Rb, Rc, Rd, X, Y, B и кольцо C являются такими, как определено в формуле изобретения. При этом фрагмент Y-B и фрагмент NH-C(=X)-NH находятся в транс-конфигурации. Соединения формулы I являются ингибиторами киназы TrkA и пригодны при лечении заболеваний, которые могут лечиться ингибитором киназы TrkA, таких как боль, рак, воспаление, нейродегенеративные заболевания и некоторые инфекционные заболевания. 9 н. и 47 з.п. ф-лы, 31 табл., 649 пр.
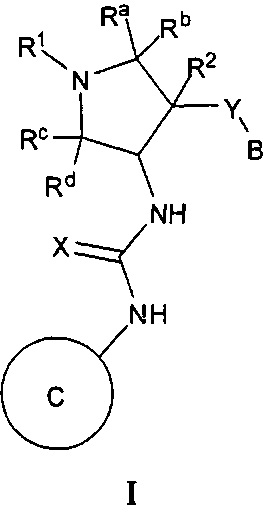
1. Соединение Формулы I:
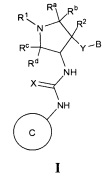
или его стереоизомеры, таутомеры или фармацевтически приемлемые соли или сольваты, где:
фрагмент Y-B и фрагмент NH-C(=X)-NH находятся в транс-конфигурации;
Ra, Rb, Rc и Rd независимо выбраны из Н и (1-3С)алкила;
X представляет собой О, S или NH;
R1 представляет собой (1-3С алкокси)(1-6С)алкил, (трифторметокси)(1-6С)алкил, (1-3С сульфанил)(1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, циано(1-6С)алкил, аминокарбонил(1-6С)алкил, гидрокси(1-6С)алкил, дигидрокси(2-6С)алкил, (1-6С)алкил, (1-3С алкиламино)(1-3С)алкил, (1-4С алкоксикарбонил)(1-6С)алкил, амино(1-6С)алкил, гидрокси(1-3С алкокси)(1-6С)алкил, ди(1-3С алкокси)(1-6С)алкил, (1-3С алкокси)трифтор(1-6С)алкил, гидрокситрифтор(1-6С)алкил, (1-4С алкоксикарбонил)(1-3С алкокси)(1-6С)алкил, гидроксикарбонил(1-3С алкокси)(1-6С)алкил, гетAr5(СН2)0-1 или Ar5(CH2)0-1;
R2 представляет собой Н, F или ОН;
Y представляет собой связь;
В представляет собой Ar1 или гетAr1;
Ar1 представляет собой фенил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена, CF3, CF3O-, (1-4С)алкокси, (1-6С)алкила и CN;
гетAr1 представляет собой 5-6-членный гетероарил, имеющий 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенный 1-2 группами, независимо выбранными из (1-6С)алкила или галогена;
кольцо С представляет собой формулу С-1, С-2 или С-3
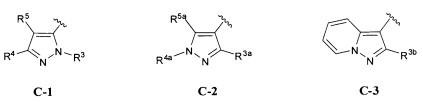
R3 представляет собой Н, (1-6С)алкил, гидрокси(1-6С)алкил, Ar2, гетСус1, (3-7С)циклоалкил или гетAr2;
Ar2 представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из галогена и (1-6С)алкила;
гетСус1 представляет собой 5-6-членное насыщенное или частично ненасыщенное гетероциклическое кольцо, имеющее 1-2 кольцевых гетероатома, независимо выбранных из N и О;
гетAr2 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, О и S, и необязательно замещенное одной или более группами, независимо выбранными из (1-6С)алкила и галогена;
R4 представляет собой Н, ОН, (1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, циано(1-6С)алкил, гидрокси(1-6С)алкил, дигидрокси(2-6С)алкил, (1-3С алкокси)(1-6С)алкил, (1-6С)алкокси, монофтор(1-6С)алкокси, дифтор(1-6С)алкокси, трифтор(1-6С)алкокси, тетрафтор(2-6С)алкокси, пентафтор(2-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, дигидрокси(2-6С)алкокси, амино(2-6С)алкокси, аминокарбонил(1-6С)алкокси, где амино в указанных группах означает -NRR', где R и R' независимо выбраны из Н и (1-3С)алкила, гетСус2(1-6С)алкокси, гетAr3(1-6С)алкокси, Ar3(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (1-3С алкилсульфонил)(1-6С)алкокси, (3-6С)циклоалкил [необязательно замещенный ОН], гетAr4, Ar4, гетСус2(О)СН2-, (1-4С алкоксикарбонил)(1-6С)алкокси, гидроксикарбонил(1-6С)алкокси, гетСус2С(=О)(1-6С)алкокси, гидрокси(1-3С алкокси)(1-6С)алкокси, гидрокситрифтор(1-6С)алкокси, (1-3С)алкилсульфонамидо(1-6С)алкокси, (1-3С)алкиламидо(1-6С)алкокси, ди(1-3С алкил)амино-карбокси, гетСус2С(=О)О-, гидроксидифтор(1-6С)алкил, (1-4С алкилкарбокси)(1-6С)алкил, (1-6С)алкоксикарбонил, гидроксикарбонил, аминокарбонил, (1-3С алкокси)аминокарбонил, где амино в указанной группе означает -NRR', где R и R' независимо выбраны из Н и (1-3С)алкила, гетСус3, галоген, CN, N-(1-3C алкил)пиридинонил, N-(1-3С трифторалкил)пиридинонил, (1-4С алкилсилокси)(1-6С)алкокси, изоиндолин-1,3-дионил(1-6С)алкокси или N-(1-3C алкил)оксадиазолонил;
гетСус2 представляет собой 4-6-членное гетероциклическое кольцо, имеющее 1-2 кольцевых гетероатома, независимо выбранных из N и О, и необязательно замещенное 1-2 группами, независимо выбранными из (1-6С)алкила, (1-4С алкилкарбокси)(1-6С)алкила и (1-6С)ацила;
гетСус3 представляет собой 4-7-членный гетероцикл, имеющий 1-2 кольцевых гетероатома, независимо выбранных из N и О, и необязательно замещенный одним или более заместителями, независимо выбранными из (1-6С)алкила, (1-3С алкокси)(1-6С)алкила, (1-6С)ацила-, (1-6С)алкилсульфонила, трифторметилсульфонила и (1-4С алкокси)карбонила;
гетAr3 представляет собой 5-членное гетероариловое кольцо, имеющее 1-3 кольцевых атома, независимо выбранных из N, О и S, и необязательно замещенное (1-6С)алкилом;
Ar3 представляет собой фенил, необязательно замещенный (1-4С)алкокси;
гетAr4 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенное 1-2 заместителями, независимо выбранными из (1-6С)алкила, галогена, CN, гидрокси(1-6С)алкила, трифтор(1-6С)алкила, (3-6С)циклоалкила, (3-6С циклоалкил)СН2- (3-6С циклоалкил)С(=О)-, (1-3С алкокси)(1-6С)алкила, (1-6С)алкокси, (1-6С)алкилсульфонила, NH2, (1-6С алкил)амино, ди(1-6С алкил)амино, (1-3С трифторалкокси), (1-3С)трифторалкила и метоксибензила; или 9-10-членный бициклический гетероарил, имеющий 1-3 кольцевых атома азота;
Ar4 представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из (1-6С)алкила, галогена, CN, CF3, CF3O-, (1-6С)алкокси, (1-6С алкил)ОС(=О)-, аминокарбонила, (1-6С)алкилтио, гидрокси(1-6С)алкила, (1-6С алкил)SO2-, НОС(=О)- и (1-3С алкокси)(1-3С алкил)ОС(=O)-;
R5 представляет собой Н, (1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, галоген, CN, (1-4С)алкокси, гидрокси(1-4С)алкил, (1-3С алкокси)(1-4С)алкил, (1-4С алкил)ОС(=O)-, (1-6С)алкилтио, фенил, (3-4С)циклоалкил, амино, аминокарбонил или трифтор(1-3С алкил)амидо; или
R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное, частично ненасыщенное или ненасыщенное карбоциклическое кольцо, необязательно замещенное одним или более заместителями, независимо выбранными из (1-6С)алкила, или
R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное, частично ненасыщенное или ненасыщенное гетероциклическое кольцо, имеющее кольцевой гетероатом, выбранный из N, О или S, причем указанное гетероциклическое кольцо необязательно замещено одним или двумя заместителями, независимо выбранными из (1-6С алкил)С(=О)О-, (1-6)ацила, (1-6С)алкила и оксо, и указанный кольцевой атом серы необязательно окислен до S(=О) или SO2;
гетAr5 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, О или S, причем указанное кольцо необязательно замещено одним или более заместителями, независимо выбранными из галогена, (1-6С)алкила, (1-6С)алкокси и CF3;
Ar5 представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из галогена, (1-6С)алкила, (1-6С)алкокси и CF3O-;
R3a представляет собой водород, галоген, (1-6С)алкил, трифтор(1-6С)алкил или фенил;
R3b представляет собой водород или фенил;
R4a представляет собой водород, (1-6С)алкил, трифтор(1-6С)алкил или фенил; и
R5a представляет собой водород, галоген, (1-6С)алкил, трифтор(1-6С)алкил или фенил.
2. Соединение по п. 1, отличающееся тем, что:
В и фрагмент NH-C(=X)-NH находятся в транс-конфигурации;
Ra, Rb, Rc и Rd независимо выбраны из Н и (1-3С)алкила;
X представляет собой О или S;
R1 представляет собой (1-3С алкокси)(1-6С)алкил, (трифторметокси)(1-6С)алкил, (1-3С сульфанил)(1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, циано(1-6С)алкил, аминокарбонил(1-6С)алкил, гидрокси(1-6С)алкил, дигидрокси(2-6С)алкил, (1-6С)алкил или (1-3С алкиламино)(1-3С)алкил;
R2 представляет собой Н, F или ОН;
В представляет собой Ar1 или гетAr1;
Ar1 представляет собой фенил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена, CF3, CF3O-, (1-4С)алкокси и (1-6С)алкила;
гетAr1 представляет собой 5-6-членный гетероарил, имеющий 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенный 1-2 группами, независимо выбранными из (1-6С)алкила и галогена;
кольцо С представляет собой
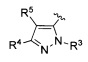 ;
;
R3 представляет собой Н, (1-6С)алкил, гидрокси(1-6С)алкил, Ar2, гетСус1, (3-7С)циклоалкил или гетAr2;
Ar2 представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из галогена и (1-6С)алкила;
гетСус1 представляет собой 5-6-членное насыщенное или частично ненасыщенное гетероциклическое кольцо, имеющее 1-2 кольцевых гетероатома, независимо выбранных из N и О;
гетAr2 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, О и S, и необязательно замещенное (1-6С)алкилом;
R4 представляет собой Н, ОН, (1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, циано(1-6С)алкил, гидрокси(1-6С)алкил, дигидрокси(2-6С)алкил, (1-3С алкокси)(1-6С)алкил, (1-6С)алкокси, монофтор(1-6С)алкокси, дифтор(1-6С)алкокси, трифтор(1-6С)алкокси, тетрафтор(2-6С)алкокси, пентафтор(2-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, дигидрокси(2-6С)алкокси, амино(2-6С)алкокси, аминокарбонил(1-6С)алкокси, гидроксикарбонил(1-6С)алкокси, гетСус2(1-6С)алкокси, гетAr3(1-6С)алкокси, Ar3(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (1-3С алкилсульфонил)(1-6С)алкокси, (3-6С)циклоалкил, гетAr4 или Ar4;
гетСус2 представляет собой 4-6-членное гетероциклическое кольцо, имеющее 1-2 кольцевых гетероатома, независимо выбранных из N и О, и необязательно замещенное 1-2 группами, независимо выбранными из (1-6С)алкила;
гетAr3 представляет собой 5-членное гетероариловое кольцо, имеющее 1-3 кольцевых атома, независимо выбранных из N, О и S, и необязательно замещенное (1-6С)алкилом;
Ar3 представляет собой фенил, необязательно замещенный (1-4С)алкокси;
гетAr4 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенное 1-2 заместителями, независимо выбранными из (1-6С)алкила, или 9-10-членный бициклический гетероарил, имеющий 1-3 кольцевых атома азота;
Ar4 представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из (1-6С)алкила, галогена, CN, CF3, CF3O-, (1-6С)алкокси, (1-6С алкил)ОС(=О)-, аминокарбонила, (1-6С)алкилтио, гидрокси(1-6С)алкила, (1-6С алкил)SO2-, НОС(=О)- и (1-3С алкокси)(1-3С алкил)ОС(=О)-;
R5 представляет собой Н, (1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, галоген, CN, (1-4С)алкокси, гидрокси(1-4С)алкил, (1-3С алкокси)(1-4С)алкил, (1-4С алкил)ОС(=О)-, (1-6С)алкилтио или фенил; или
R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное карбоциклическое кольцо, необязательно замещенное одним или более заместителями, независимо выбранными из (1-6С)алкила, или
R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное гетероциклическое кольцо, имеющее кольцевой гетероатом, выбранный из N, О или S, причем указанный кольцевой атом азота необязательно замещен (1-6С алкил)С(=О)О- или (1-6)ацилом и указанный кольцевой атом серы необязательно окислен до S(=О) или SO2.
3. Соединение по п. 1 или 2, отличающееся тем, что X представляет собой О.
4. Соединение по п. 1 или 2, отличающееся тем, что X представляет собой S.
5. Соединение по п. 1 или 2, отличающееся тем, что R1 выбран из (1-3С алкокси)(1-6С)алкила, дифтор(1-6С)алкила и трифтор(1-6С)алкила.
6. Соединение по п. 5, отличающееся тем, что R1 представляет собой (1-3С алкокси)(1-6С)алкил.
7. Соединение по п. 1 или 2, отличающееся тем, что В представляет собой Ar1.
8. Соединение по п. 7, отличающееся тем, что Ar1 представляет собой фенил, необязательно замещенный одним или более галогенами.
9. Соединение по п. 1 или 2, отличающееся тем, что В представляет собой гетAr1.
10. Соединение по п. 9, отличающееся тем, что В представляет собой пиридил, необязательно замещенный 1-2 группами, независимо выбранными из (1-6С)алкила или галогена.
11. Соединение по п. 1 или 2, отличающееся тем, что кольцо С представляет собой формулу С-1.
12. Соединение по п. 11, отличающееся тем, что:
R4 представляет собой Н, ОН, (1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, циано(1-6С)алкил, гидрокси(1-6С)алкил, дигидрокси(2-6С)алкил, (1-3С алкокси)(1-6С)алкил, (1-6С)алкокси, монофтор(1-6С)алкокси, дифтор(1-6С)алкокси, трифтор(1-6С)алкокси, тетрафтор(2-6С)алкокси, пентафтор(2-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, дигидрокси(2-6С)алкокси, амино(2-6С)алкокси, аминокарбонил(1-6С)алкокси, где амино в указанных группах означает -NRR', где R и R' независимо выбраны из Н и (1-3С)алкила, гидроксикарбонил(1-6С)алкокси, гетСус2(1-6С)алкокси, гетAr3(1-6С)алкокси, Ar3(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (1-3С алкилсульфонил)(1-6С)алкокси, (3-6С)циклоалкил, гетAr4 или Ar4; и
R5 представляет собой Н, (1-6С)алкил, монофтор(1-6С)алкил, дифтор(1-6С)алкил, трифтор(1-6С)алкил, тетрафтор(2-6С)алкил, пентафтор(2-6С)алкил, галоген, CN, (1-4С)алкокси, гидрокси(1-4С)алкил, (1-3С алкокси)(1-4С)алкил, (1-4С алкил)ОС(=О)-, (1-6С)алкилтио или фенил.
13. Соединение по п. 12, отличающееся тем, что R4 выбран из Н, (1-6С)алкила, трифтор(1-6С)алкила, циано(1-6С)алкила, (1-3С алкокси)(1-6С)алкила, (1-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси, (1-4С алкокси)(1-6С)алкокси, (3-6С)циклоалкила, гетAr4 и Ar4.
14. Соединение по п. 13, отличающееся тем, что R4 выбран из (1-6С)алкила, трифтор(1-6С)алкила, циано(1-6С)алкила, (1-3С алкокси)(1-6С)алкила и (3-6С)циклоалкила.
15. Соединение по п. 13, отличающееся тем, что R4 выбран из (1-6С)алкокси, циано(1-6С)алкокси, гидрокси(1-6С)алкокси и (1-4С алкокси)(1-6С)алкокси.
16. Соединение по п. 13, отличающееся тем, что R4 выбран из гетAr4 и Ar4.
17. Соединение по п. 16, отличающееся тем, что R4 представляет собой Ar4.
18. Соединение по п. 1 или 2, отличающееся тем, что R5 выбран из Н, галогена, CN, (1-6С)алкила, (1-4С)алкокси, гидрокси(1-4С)алкила, (1-6С)алкилтио или фенила.
19. Соединение по п. 18, отличающееся тем, что R5 выбран из Н, галогена и (1-6С)алкила.
20. Соединение по п. 19, отличающееся тем, что R5 представляет собой (1-6С)алкил.
21. Соединение по п. 1 или 2, отличающееся тем, что:
R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное карбоциклическое кольцо, необязательно замещенное одним или более заместителями, независимо выбранными из (1-6С)алкила, или
R4 и R5 вместе с атомами, к которым они присоединены, образуют 5-6-членное насыщенное гетероциклическое кольцо, имеющее кольцевой гетероатом, выбранный из N, О или S, причем указанный кольцевой атом азота необязательно замещен (1-6С алкил)С(=О)О- или (1-6)ацилом и указанный кольцевой атом серы необязательно окислен до S(=О) или SO2.
22. Соединение по п. 1 или 2, отличающееся тем, что R3 выбран из Н, Ar2, гетAr2 и (1-6С)алкила.
23. Соединение по п. 1 или 2, отличающееся тем, что R3 выбран из Ar2 и (1-6С)алкила.
24. Соединение по п. 23, отличающееся тем, что R3 представляет собой Ar2.
25. Соединение по п. 1 или 2, отличающееся тем, что R2 представляет собой Н.
26. Соединение по п. 1 или 2, отличающееся тем, что Ra, Rb, Rc и Rd представляют собой Н.
27. Соединение по п. 1 или 2, отличающееся тем, что фрагмент Y-B и фрагмент -NH-C(=X)-NH- Формулы I находятся в абсолютной транс-конфигурации, показанной в формуле С:
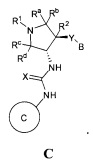
28. Соединение по п. 1 или 2, отличающееся тем, что В и фрагмент -NH-C(=X)-NH- Формулы I находятся в абсолютной транс-конфигурации, показанной в формуле D:
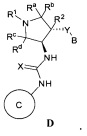
29. Соединение по п. 1 или 2, отличающееся тем, что Y представляет собой связь.
30. Соединение по п. 1, выбранное из следующих:
1-(3-трет-бутил-1-фенил-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевины гидрохлорид,
транс-1-(4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
транс-1-(4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-изопропил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-трет-бутил-1-метил-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(1,3-диметил-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(3-трет-бутил-1-(пиридин-3-ил)-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(3-трет-бутил-1-(4-фторфенил)-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(3-циклопропил-1-фенил-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(3-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-изопропил-1-фенил-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(1-метил-3-фенил-1Н-пиразол-5-ил)мочевина,
1-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-трет-бутил-1-(2-фторфенил)-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(3-трет-бутил-1-(3-фторфенил)-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-(пиридин-3-ил)-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(1-метил-1Н-пиразол-5-ил)мочевина,
1-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)тиомочевина,
1-(2-(3-фторфенил)-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(2-(4-фторфенил)-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(3-циклопентил-1-фенил-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(1-этил-3-фенил-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-фенил-4,5,6,7-тетрагидро-2Н-индазол-3-ил)мочевина,
1-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-метил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(1,3-диметил-4-фенил-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(3-трет-бутил-1-о-толил-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(3-трет-бутил-1-м-толил-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(1-метил-4-фенил-1Н-пиразол-5-ил)мочевина,
1-(4-циано-3-метил-1-фенил-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-(1-метил-1Н-пиразол-4-ил)-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(3-трет-бутил-1-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-(пиридин-2-ил)-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(6,6-диметил-2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(7,7-диметил-2-фенил-4,5,6,7-тетрагидро-2Н-индазол-3-ил)-3-(транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(транс-1-(2-метоксиэтил)-4-(пиридин-4-ил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
транс-1-(4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-изопропил-1-фенил-1Н-пиразол-5-ил)мочевина,
транс-1-(4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-изопропил-1-фенил-1Н-пиразол-5-ил)мочевина,
транс-1-(4-(3-хлорфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-изопропил-1-фенил-1Н-пиразол-5-ил)мочевина,
транс-1-(4-(2-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-изопропил-1-фенил-1Н-пиразол-5-ил)мочевина,
транс-1-(3-изопропил-1-фенил-1Н-пиразол-5-ил)-3-(1-(2-метоксиэтил)-4-(тиофен-2-ил)пирролидин-3-ил)мочевина,
1-((3,4-транс)-4-(2,4-диметилтиазол-5-ил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(транс-1-(2-метоксиэтил)-4-(оксазол-5-ил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(транс-4-(изоксазол-5-ил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((3,4-транс)-1-(2-метоксиэтил)-4-(3-метоксифенил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(1-(2-метоксиэтил)-4-(тиазол-2-ил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(1,3-дифенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(1-метил-3-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(1-фенил-3-(трифторметил)-1Н-пиразол-5-ил)мочевина,
1-(1,4-диметил-3-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-(3-циклопропил-1-метил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-метил-3-(пиридин-2-ил)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-метил-3-(пиридин-3-ил)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1,1'-диметил-1Н,1'Н-3,4'-бипиразол-5-ил)мочевина,
1-(3-(3-цианофенил)-1-метил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(3-(4-цианофенил)-1-метил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(имидазо[1,2-а]пиридин-5-ил)-1-метил-1Н-пиразол-5-ил)мочевина,
1-(4-хлор-1,3-дифенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-1,3-дифенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1,3-диметил-4-фенил-1Н-пиразол-5-ил)мочевина,
1-(4-циано-3-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-1-метил-3-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-1-метил-3-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-циано-3-(цианометил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(3-(2-цианопропан-2-ил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-этил-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-метил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(оксетан-3-илметокси)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-((3-метилоксетан-3-ил)метокси)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(((S)-2,2-диметил-1,3-диоксолан-4-ил)метокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(((R)-2,2-диметил-1,3-диоксолан-4-ил)метокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3,4-диметил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
трет-бутил 3-(3-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)уреидо)-2-фенил-4,6-дигидропирроло[3,4-с]пиразол-5(2Н)-карбоксилат,
1-(3-изопропил-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-фенил-4,6-дигидро-2Н-фуро[3,4-с]пиразол-3-ил)мочевина,
1-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3,4-диметил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-изопропил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-4,6-дигидро-2Н-фуро[3,4-с]пиразол-3-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-4,6-дигидро-2Н-фуро[3,4-с]пиразол-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3,4-диметил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-метил-3-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-(1-гидрокси-2-метилпропан-2-ил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(5-оксидо-2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-ил)мочевина,
1-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(1-метил-3-(пиридин-4-ил)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-метил-3-(пиридин-4-ил)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-метил-3-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-метил-3-(тиофен-2-ил)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(3-(метоксиметил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(3-(метоксиметил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-метил-3-п-толил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-метил-3-м-толил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-метил-3-о-толил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(3-метоксифенил)-1-метил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-метоксифенил)-1-метил-1Н-пиразол-5-ил)мочевина,
1-(3-(4-фторфенил)-1-метил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(3-(4-метоксифенил)-1-метил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-1-(2-метоксиэтил)-4-(3-(трифторметил)фенил)пирролидин-3-ил)-3-(1-метил-3-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-метил-3-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(2,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-метил-3-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(4-фторфенил)-1-метил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(4-фторфенил)-1-метил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(3-фторфенил)-1-метил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-фторфенил)-1-метил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(3-(1-гидрокси-2-метилпропан-2-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(1-гидрокси-2-метилпропан-2-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-(4-хлорфенил)-1-метил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(2,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(4-фторфенил)-1-метил-1Н-пиразол-5-ил)мочевина,
метил 4-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-метил-1Н-пиразол-3-ил)бензоат,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-(2-гидроксиэтил)-3-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(метоксиметил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(метоксиметил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-метил-3-(4-(метилтио)фенил)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1,3-дифенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(3-метоксипропил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(3,4-диметил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(4-(2-метоксиэтокси)фенил)-1-метил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метокси-3-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(гидроксиметил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-гидроксиэтил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-метоксиэтил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-(бензилокси)-1-метил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-метоксиэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
транс-1-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)-3-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-метокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-(цианометокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(4-метоксибензилокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-метокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-фторэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-гидрокси-2-метилпропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-этокси-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-гидроксиэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(2-циклогексил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-(пиридин-4-ил)-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-метил-3-(5-метилпиразин-2-ил)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1,4-диметил-3-(5-метилпиразин-2-ил)-1Н-пиразол-5-ил)мочевина,
этил 5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-4-карбоксилат,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-метил-3-(пиразин-2-ил)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-метокси-1-метил-4-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-этокси-1-метил-4-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-ил)мочевины дигидрохлорид,
1-(5-ацетил-2-фенил-2,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-(гидроксиметил)-3-(метоксиметил)-1-фенил-1Н-пиразол-5-ил)мочевина,
4-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-метил-1Н-пиразол-3-ил)бензойная кислота,
4-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-метил-1Н-пиразол-3-ил)бензамид,
4-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-метил-1Н-пиразол-3-ил)-N-метилбензамид,
4-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-метил-1Н-пиразол-3-ил)-N,N-диметилбензамид,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(4-(гидроксиметил)фенил)-1-метил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-метил-3-(4-(метилсульфонил)фенил)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-фтор-3-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-фтор-1-метил-3-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-фтор-1,3-дифенил-1Н-пиразол-5-ил)мочевина,
2-метоксиэтил 4-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-метил-1Н-пиразол-3-ил)бензоат,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(5,5-диоксидо-2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-ил)мочевина,
1-(5,5-диоксидо-2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(5,5-диоксидо-2-фенил-4,6-дигидро-2Н-тиено[3,4-с]пиразол-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-(метилсульфонил)этокси)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-(гидроксиметил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((R)-2,3-дигидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((S)-2,3-дигидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-гидроксиэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины гидрохлорид,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((S)-2-гидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины гидрохлорид,
1-((3R,4S)-4-гидрокси-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((3R,4S)-4-фтор-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(транс-4-фенил-1-(2-(трифторметокси)этил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(транс-1-(2-(метилтио)этил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((3S,4R)-1-((S)-2-метоксипропил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((3,4-транс)-4-фенил-1-(4,4,4-трифторбутил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((3S,4R)-1-(цианометил)-4-(3,4-дифторфенил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((3S,4R)-1-(цианометил)-4-(3,4-дифторфенил)пирролидин-3-ил)-3-(3-(2-метоксиэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-1-(цианометил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
2-((3R,4S)-3-фенил-4-(3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)уреидо)пирролидин-1-ил)ацетамид,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-гидроксиэтил)пирролидин-3-ил)-3-(3,4-диметил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((транс)-1-(3,3,4,4,4-пентафторбутил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((транс)-1-этил-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((транс)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(3,4-диметил-1-фенил-1Н-пиразол-5-ил)-3-((транс)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина,
1-(3-(2-метоксиэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((транс)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина,
1-((транс)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(3,4-диметил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина,
1-(3-(2-метоксиэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(3,4-диметил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(3-(2-метоксиэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(1-метил-3-фенил-1Н-пиразол-5-ил)мочевина,
1-((3R,4S)-4-(3-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3R,4S)-4-(3-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина,
1-((транс)-1-(1,3-дифторпропан-2-ил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
(транс)-трет-6утил 3-(3-метоксифенил)-4-(3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)уреидо)пирролидин-1-карбоксилат,
1-((транс)-4-(3-хлорфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)-3-((транс)-4-(пиридин-2-ил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина,
1-((транс)-4-(4-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((транс)-4-(4-хлорфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((транс)-4-(2-хлорфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)-3-((транс)-4-(пиридин-3-ил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина,
1-((транс)-4-(2-фторфенил)-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((транс)-4-(4-фторфенил)-1-(2,2-дифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(1Н-пиразол-4-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(1Н-пиразол-4-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(1Н-пиразол-3-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(3-метил-1Н-пиразол-4-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(3-(трифторметил)-1Н-пиразол-4-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(1Н-пиразол-4-ил)пирролидин-3-ил)-3-(3-((S)-2-гидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(1Н-пиразол-4-ил)пирролидин-3-ил)-3-(3-((R)-2,3-дигидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(1Н-пиразол-4-ил)пирролидин-3-ил)-3-(3,4-диметил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(1Н-пиразол-4-ил)пирролидин-3-ил)-3-(3,4-диметил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(1Н-пиразол-4-ил)пирролидин-3-ил)-3-(3-(3-метоксипропил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(1Н-пиразол-4-ил)пирролидин-3-ил)-3-(3-(2-метоксиэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(1Н-пиразол-4-ил)пирролидин-3-ил)-3-(3-(2-гидрокси-2-метилпропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(1-метил-1Н-пиразол-5-ил)-4-фенилпирролидин-3-ил)мочевина,
1-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)-3-((3R,4S)-1-(1-метил-1Н-пиразол-5-ил)-4-фенилпирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(1-метил-1Н-пиразол-5-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3R,4S)-4-(3,5-дифторфенил)-1-(1-метил-1Н-пиразол-5-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-фенилпирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксифенил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-фторфенил)-4-фенилпирролидин-3-ил)мочевина,
1-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(4-фторфенил)-4-фенилпирролидин-3-ил)мочевина,
1-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метилфенил)-4-фенилпирролидин-3-ил)мочевина,
1-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксифенил)-4-фенилпирролидин-3-ил)мочевина,
1-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-хлорфенил)-4-фенилпирролидин-3-ил)мочевина,
1-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)-3-((3S,4R)-4-фенил-1-(2-(трифторметокси)фенил)пирролидин-3-ил)мочевина,
1-((3S,4R)-1-(2,6-дифторфенил)-4-фенилпирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксипиридин-4-ил)-4-фенилпирролидин-3-ил)мочевина,
1-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксипиридин-3-ил)-4-фенилпирролидин-3-ил)мочевина,
1-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)-3-((3S,4R)-1-(2-этоксипиридин-3-ил)-4-фенилпирролидин-3-ил)мочевина,
1-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-1-(2-метоксипиридин-3-ил)-4-фенилпирролидин-3-ил)мочевина,
1-((3S,4R)-1-(2-метоксипиридин-3-ил)-4-фенилпирролидин-3-ил)-3-(4-метил-1,3-дифенил-1Н-пиразол-5-ил)мочевина,
1-(4-бром-1'-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-1-(2-метоксипиридин-3-ил)-4-фенилпирролидин-3-ил)мочевина,
1-(4-бром-1,3-дифенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксипиридин-3-ил)-4-фенилпирролидин-3-ил)мочевина,
1-((3S,4R)-1-((1,2,3-тиадиазол-4-ил)метил)-4-(3,4-дифторфенил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((3S,4R)-1-((1,2,3-тиадиазол-4-ил)метил)-4-(3,4-дифторфенил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-1-((1,2,3-тиадиазол-4-ил)метил)-4-(3,4-дифторфенил)пирролидин-3-ил)-3-(3-(цианометокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-1-((1,2,3-тиадиазол-4-ил)метил)-4-(3,4-дифторфенил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)мочевина,
1-((3S,4R)-1-((1,2,3-тиадиазол-4-ил)метил)-4-(3,4-дифторфенил)пирролидин-3-ил)-3-(4-метил-3-(1-метил-1Н-имидазол-4-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-((1-метил-1Н-1,2,3-триазол-4-ил)метил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(1,3-диметоксипропан-2-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(1-метоксипропан-2-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((транс)-4-(4-фторфенил)-1-(2-(метиламино)этил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((транс)-1-((1Н-имидазол-2-ил)метил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
метил 3-метокси-2-((транс)-3-фенил-4-(3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)уреидо)пирролидин-1-ил)пропаноат,
метил 2-((3S,4R)-3-(3,4-дифторфенил)-4-(3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)уреидо)пирролидин-1-ил)-3-метоксипропаноат,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(1-гидрокси-3-метоксипропан-2-ил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(3-гидрокси-1-метокси-3-метилбутан-2-ил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
2-((3R,4S)-3-(3,4-дифторфенил)-4-(3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)уреидо)пирролидин-1-ил)-3-метоксипропановой кислоты гидрохлорид,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(1-гидрокси-3-метоксипропан-2-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(4-хлор-1'-метил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(1-гидрокси-3-метоксипропан-2-ил)пирролидин-3-ил)мочевина,
1-(3,4-диметил-1-фенил-1H-пиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина,
1-(3-(2-фторэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина,
1-(3-этокси-4-метил-1-фенил-1H-пиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина,
1-(3-(цианометокси)-4-метил-1-фенил-1H-пиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина,
1-(1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-1'-метил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина,
1-(3-(((S)-2,2-диметил-1,3-диоксолан-4-ил)метокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина,
1-(3-((R)-2,3-дигидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина,
(R,S)1-((2α,3β,4α)-2-метил-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
(R,S)-1-((3β,4α,5α)-5-метил-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-((S)-1,1,1-трифтор-3-гидроксипропан-2-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-((S)-1,1,1-трифтор-3-метоксипропан-2-ил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-((S)-1,1,1-трифтор-3-метоксипропан-2-ил)пирролидин-3-ил)-3-(3-((S)-2-гидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(4-хлор-1'-метил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-((R)-1,1,1-трифтор-3-метоксипропан-2-ил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-((R)-1,1,1-трифтор-3-метоксипропан-2-ил)пирролидин-3-ил)-3-(3-((S)-2-гидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-метил-4-(метилтио)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(3-метоксипропил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-(1,1-дифтор-2-гидроксиэтил)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(3-(1,1-дифтор-2-гидроксиэтил)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(3-(1,1-дифтор-2-гидроксиэтил)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(3-(1,1-дифтор-2-гидроксиэтил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-гидроксиэтил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-гидроксиэтил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-гидрокси-2-метилпропил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((S)-2-гидроксипропил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((R)-2-гидроксипропил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
этил 5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-карбоксилат,
5-(3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-N,4-диметил-1-фенил-1Н-пиразол-3-карбоксамид,
1-(транс-4-(3-хлор-4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-(транс-4-(4-хлор-3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-(транс-4-(3-хлор-5-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-(транс-4-(3-хлорфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(5-метил-1,3,4-оксадиазол-2-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(3-метил-1,2,4-оксадиазол-5-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(3-(трифторметил)-1,2,4-оксадиазол-5-ил)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(3-(трифторметил)-1,2,4-оксадиазол-5-ил)-1Н-пиразол-5-ил)мочевина,
5-(3-(транс-4-(3-хлор-4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-N,4-диметил-1-фенил-1Н-пиразол-3-карбоксамид,
5-(3-(транс-4-(4-хлор-3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-N,4-диметил-1-фенил-1Н-пиразол-3-карбоксамид,
1-(транс-4-(4-хлор-3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(транс-4-(3-хлор-4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4,5'-триметил-1-фенил-1Н,1'Н-[3,3'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4,5'-триметил-1-фенил-1Н,1'Н-[3,3'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4,5'-триметил-1-фенил-1Н,1'Н-[3,3'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2',4,5'-триметил-1-фенил-1Н,2'Н-[3,3'-бипиразол]-5-ил)мочевина,
1-(4-циклопропил-1'-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-изопропил-1'-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-этил-1'-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-(4-фторфенил)-1',4-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-(3-фторфенил)-1',4-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-(2-фторфенил)-1',4-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-(3-хлорфенил)-1',4-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-(1-(3-хлор-4-фторфенил)-1',4-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(1-(3-хлор-2-фторфенил)-1',4-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-(4-фторфенил)-1',4-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-(3-фторфенил)-1',4-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-(2-фторфенил)-1',4-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3,4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-(3-хлорфенил)-1',4-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-(1-(3-хлор-4-фторфенил)-1',4-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(1-(3-хлор-2-фторфенил)-1',4-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(2,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3-цианофенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(4-цианофенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(п-толил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1,3-дифенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1,3-дифенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4,5-трифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1,3-дифенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1,3-дифенил-1Н-пиразол-5-ил)мочевина,
1-(4-бром-3-метил-1-фенил-1Н-пиразол-5-ил)-3-транс-1-(2-метоксиэтил)-4-(1-метил-1Н-пиразол-4-ил)пирролидин-3-ил)мочевина,
1-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-(транс-1-(2-метоксиэтил)-4-(1-метил-1Н-пиразол-4-ил)пирролидин-3-ил)мочевина,
1-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((транс-1-(2-метоксиэтил)-4-(1,2,3-тиадиазол-4-ил)пирролидин-3-ил)мочевина,
1-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)-3-(транс-1-(2-метоксиэтил)-4-(3-(трифторметил)фенил)пирролидин-3-ил)мочевина,
1-(3,4-диметил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3-(трифторметил)фенил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(5-фторпиридин-3-ил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-((3R,4S)-4-(5-фторпиридин-3-ил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(3-(2-фторэтокси)-4-метил-1-фенил-1H-пиразол-5-ил)-3-((3S,4R)-4-(5-фторпиридин-3-ил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(3-(2-фторэтокси)-4-метил-1-фенил-1H-пиразол-5-ил)-3-((3R,4S)-4-(5-фторпиридин-3-ил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(транс-4-(5-фторпиридин-3-ил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
1-(транс-4-(5-хлорпиридин-3-ил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(транс-4-(5-хлорпиридин-3-ил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3,4-диметил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(транс-4-(5-фторпиридин-3-ил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1,3-дифенил-1Н-пиразол-5-ил)мочевина,
1-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-(транс-4-(5-фторпиридин-2-ил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3-фторпиридин-4-ил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(1-метил-1Н-1,2,4-триазол-3-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-(2-метоксиэтил)-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-(3-циано-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-(2-гидроксиэтил)-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-метил-2Н-1,2,3-триазол-4-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-бром-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(5-метил-6-оксо-2-фенил-2,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(5-метил-6-оксо-2-фенил-2,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-((5-метил-1,3,4-оксадиазол-2-ил)метокси)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(4-хлор-3-этокси-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-этокси-4-фтор-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(4-бром-3-(2-гидрокси-2-метилпропокси)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-(2-гидрокси-2-метилпропокси)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((S)-2-гидроксибутокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
этил 2-((5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-ил)окси)ацетат,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-гидрокси-2-метилпропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-гидрокси-2-метилпропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((R)-2-гидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((R)-2-гидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((R)-2-гидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-((R)-3,3,3-трифтор-2-гидроксипропокси)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((S)-2-гидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-((S)-3,3,3-трифтор-2-гидроксипропокси)-1Н-пиразол-5-ил)мочевина,
1-(4-хлор-3-(2-гидрокси-2-метилпропокси)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-(2-гидрокси-2-метилпропокси)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-((R)-2-гидроксипропокси)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-((R)-2-гидроксипропокси)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-((R)-2-гидроксипропокси)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-3-((R)-2-гидроксипропокси)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-3-((R)-2-гидроксипропокси)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-3-((R)-2-гидроксипропокси)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((R)-2-гидроксибутокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((R)-2-гидроксибутокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((R)-2-гидроксибутокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
этил 4-бром-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-3-карбоксилат,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-(2-метоксиэтил)-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-(2-метоксиэтил)-4-метил-1-фенил-1Н,1'H-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-метил-2Н-1,2,3-триазол-4-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-метил-2Н-1,2,3-триазол-4-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-морфолиноэтокси)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-(1,3-диоксоизоиндолин-2-ил)этокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
трет-бутил 4-(2-((5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-ил)окси)этил)пиперазин-1-карбоксилат,
транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-фенил-2Н-индазол-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенил-2Н-индазол-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-((1-метил-1Н-1,2,4-триазол-3-ил)метокси)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(((R)-2,2-диметил-1,3-диоксолан-4-ил)метокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(((S)-2,2-диметил-1,3-диоксолан-4-ил)метокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-(пиразин-2-ил)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-(пиридазин-4-ил)-2,4,5,6-тетрагидроциклопента[с]пиразол-3-ил)мочевина,
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-ил диметилкарбамат,
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-ил морфолин-4-карбоксилат,
1-(3-(((S)-2,2-диметил-1,3-диоксолан-4-ил)метокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевина,
1-(3-(((R)-2,2-диметил-1,3-диоксолан-4-ил)метокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевина,
1-(3-((S)-2-(трет-бутилдиметилсилилокси)пропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевина,
1-(3-(2-гидрокси-2-метилпропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевина,
1-(3-((S)-2-(трет-бутилдиметилсилилокси)-3-метоксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-(метоксиметил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-3-(метоксиметил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-(метоксиметил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-3-(метоксиметил)-1-фенил-1H-пиразол-5-ил)-3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-(метоксиметил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-3-(метоксиметил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-(1,1-дифтор-2-гидроксиэтил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-(1,1-дифтор-2-гидроксиэтил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-(1,1-дифтор-2-гидроксиэтил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-((S)-2-гидроксипропил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-((R)-2-гидроксипропил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-3-((R)-2-гидроксипропил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-(2-гидрокси-2-метилпропил)-1-фенил-1H-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-(3-метил-1,2,4-оксадиазол-5-ил)-1-фенил-1H-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-3-(2-цианопропан-2-ил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-(2-цианопропан-2-ил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-1'-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-1'-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-1'-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-1'-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-1'-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-1'-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-1'-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-фенил-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-1,3-дифенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-1,3-дифенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-1,3-дифенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-3-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-1'-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(5-фторпиридин-3-ил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-фтор-1'-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(5-фторпиридин-3-ил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-3-метил-1-фенил-1Н-пиразол-5-ил)-3-транс-4-(5-фторпиридин-3-ил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-3-метил-1-фенил-1Н-пиразол-5-ил)-3-транс-4-(5-фторпиридин-3-ил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-1,3-дифенил-1Н-пиразол-5-ил)-3-(транс-4-(5-фторпиридин-3-ил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-1,3-дифенил-1Н-пиразол-5-ил)-3-(транс-4-(5-фторпиридин-3-ил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-3-метил-1-фенил-1Н-пиразол-5-ил)-3-(транс-4-(5-фторпиридин-2-ил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-фтор-1'-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-фтор-1'-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-(4-бром-1'-метил-1-фенил-1H,1'H-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(5-фторпиридин-3-ил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4,5-трифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(1',4-диметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-(4-метоксибензил)-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-(4-метоксибензил)-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевины трифторацетат,
2-(4-хлор-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-3-ил)этилацетат,
1-(4-хлор-3-(2-гидроксиэтил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(цис-3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(транс-3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(4-хлор-3-(цис-3-гидроксициклобутил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-((1r,3S)-3-гидроксициклобутил)-1-фенил-1Н-пиразол-5-ил)-3-(транс-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(цис-3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(транс-3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(цис-3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(транс-3-гидроксициклобутил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-карбоновая кислота,
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-N,4-диметил-1-фенил-1Н-пиразол-3-карбоксамид,
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-N,N,4-триметил-1-фенил-1Н-пиразол-3-карбоксамид,
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-N-этил-4-метил-1-фенил-1Н-пиразол-3-карбоксамид,
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-N-изопропил-4-метил-1-фенил-1Н-пиразол-3-карбоксамид,
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-карбоксамид,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-этокси-4-(гидроксиметил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3-хлор-4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)мочевина,
1-((3S,4R)-4-(4-хлор-3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)мочевина,
1-((3S,4R)-4-(3-хлор-5-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)мочевина,
метил 2-((3R,4S)-3-(3,4-дифторфенил)-4-(3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)уреидо)пирролидин-1-ил)ацетат,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(3,3,3-трифтор-2-гидроксипропил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-гидроксипропил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-1-(2-цианоэтил)-4-(3,4-дифторфенил)пирролидин-3-ил)-3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
2-((3R,4S)-3-(3,4-дифторфенил)-4-(3-(3-этокси-4-метил-1-фенил-1Н-пиразол-5-ил)уреидо)пирролидин-1-ил)-N-метилацетамид,
1-(1-циклогексил)-3,4-диметил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(3-гидрокси-2-(гидроксиметил)пропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(2,2,2-трифторэтокси)-1Н-пиразол-5-ил)мочевина,
1-(3-(2,2-дифторэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-1-фенил-3-(2,2,2-трифторэтокси)-1H-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-1-фенил-3-(пиридин-2-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-1-фенил-3-(пиридин-4-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-1-фенил-3-(пиридин-3-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-1-фенил-3-(пиридин-3-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-1-фенил-3-(пиридин-2-ил)-1H-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-фенил-1-(пиридин-3-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(пиридин-3-ил)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(пиридин-4-ил)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(пиридин-2-ил)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(5-фторпиридин-3-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(5-фторпиридин-3-ил)-4-метил-1-(пиридин-3-ил)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-[3,3'-бипиразол]-5-ил)мочевина,
1-(1',4-диметил-1-фенил-1H,1'H-[3,3'-бипиразол]-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-[3,3'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2',4-диметил-1-фенил-1Н,2'Н-[3,3'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-(5-фторпиридин-3-ил)-1',4-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-(5-метилпиридин-3-ил)-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-(1-(5-хлорпиридин-3-ил)-1',4-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-1'-(2,2,2-трифторэтил)-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-(1'-(циклопропилметил)-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(1'-(циклопропанкарбонитрил)-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1'-(метилсульфонил)-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-изопропил-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(пиримидин-5-ил)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4,5'-триметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',3',4-триметил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-(1'-циклопропил-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-метилтиазол-5-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-метилпиримидин-5-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-(2-аминопиримидин-5-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2,4-диметилтиазол-5-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2,6-диметилпиридин-4-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-(6-аминопиридин-3-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(3-бром-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(6-оксо-1-(2,2,2-трифторэтил)-1,6-дигидропиридин-3-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-изопропил-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-бром-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина,
1-(4-метил-3-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)-1-фенил-1Н-пиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевина,
1-(3-(2-аминопиримидин-5-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3R,4S)-4-фенил-1-(2,2,2-трифторэтил)пирролидин-3-ил)мочевины бис(2,2,2-трифторацетат),
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1'-этил-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)мочевина,
1-(1'-этил-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-метоксипиримидин-5-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-(диметиламино)пиримидин-5-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(2-метоксипиримидин-5-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-(2-(диметиламино)пиримидин-5-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(1'-этил-4-метил-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-циклопропил-4-метил-1-фенил-1H-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(3-циклопропил-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(1-изопропил-6-оксо-1,6-дигидропиридин-3-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(1-изопропил-6-оксо-1,6-дигидропиридин-3-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((S)-2-гидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевины дигидрохлорид,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-(2-(пиперазин-1-ил)этокси)-1Н-пиразол-5-ил)мочевины тригидрохлорид,
1-(3-(бензилокси)-4-хлор-1-метил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
2-((5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-ил)окси)уксусная кислота,
2-((5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-ил)окси)-N-этилацетамид,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-этил-3-(2-гидрокси-2-метилпропокси)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-(2-аминоэтокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
N-(2-((5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-ил)окси)этил)метансульфонамид,
N-(2-((5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-ил)окси)этил)ацетамид,
1-(3-(2-(4-ацетилпиперазин-1-ил)этокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
2-((5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-4-метил-1-фенил-1Н-пиразол-3-ил)окси)ацетамид,
N-(5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-3-этокси-1-фенил-1Н-пиразол-4-ил)-2,2,2-трифторацетамид,
1-(4-амино-3-этокси-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-этокси-4-(2-гидроксиэтил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-(4-метилпиперазин-1-ил)этокси-1-фенил-1Н-пиразол-5-ил)мочевины тригидрохлорид,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(2-морфолино-2-оксоэтокси)-1-фенил-1Н-пиразол-5-ил)мочевина,
4-бром-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-3-карбоновая кислота,
4-бром-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-N-метил-1-фенил-1Н-пиразол-3-карбоксамид,
4-бром-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-N-метокси-1-фенил-1Н-пиразол-3-карбоксамид,
1-(4-хлор-1'-(2-метоксиэтил)-1-фенил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((S)-2,3-дигидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((R)-2,3-дигидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-((R)-2,3-дигидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевина,
1-(3-((S)-2,3-дигидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевина,
1-(3-((S)-2-гидроксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((S)-2-гидрокси-3-метоксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-((R)-2-гидрокси-3-метоксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-((S)-2-гидрокси-3-метоксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевина,
1-(3-((R)-2-гидрокси-3-метоксипропокси)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)мочевина,
1-(4-бром-1,1'-диметил-1Н,1'Н-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-1,1'-диметил-1H,1'H-[3,4'-бипиразол]-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-1-фенил-3-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
трет-бутил 4-(4-хлор-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-3-ил)пиперидин-1-карбоксилат,
1-(4-хлор-1-фенил-3-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-3-(3,5-диметилизоксазол-4-ил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
(R)-трет-бутил 2-(4-хлор-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-3-ил)пирролидин-1-карбоксилат,
(S)-трет-бутил 2-(4-хлор-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-3-ил)пирролидин-1-карбоксилат,
1-(4-бром-1-фенил-3-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
трет-бутил 4-(4-бром-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-3-ил)пиперидин-1-карбоксилат,
1-(4-бром-1-фенил-3-(тетрагидро-2Н-пиран-4-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-бром-3-(3,5-диметилизоксазол-4-ил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
(R)-трет-бутил 2-(4-бром-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-3-ил)пирролидин-1-карбоксилат,
трет-бутил 4-((4-бром-5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-3-ил)метокси)пиперидин-1-карбоксилат,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-фенил-3-(пиперидин-4-ил)-1Н-пиразол-5-ил)мочевины дигидрохлорид,
1-(4-хлор-1-фенил-3-(пиперидин-4-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины дигидрохлорид,
1-(4-бром-1-фенил-3-(пиперидин-4-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины дигидрохлорид,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-((R)-пирролидин-2-ил)-1Н-пиразол-5-ил)мочевины дигидрохлорид,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-((S)-пирролидин-2-ил)-1Н-пиразол-5-ил)мочевины дигидрохлорид,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-хлор-1-фенил-3-((R)-пирролидин-2-ил)-1Н-пиразол-5-ил)мочевины дигидрохлорид,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-1-фенил-3-((S)-пирролидин-2-ил)-1Н-пиразол-5-ил)мочевины дигидрохлорид,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-хлор-1-фенил-3-((S)-пирролидин-2-ил)-1Н-пиразол-5-ил)мочевины дигидрохлорид,
1-(4-бром-1-фенил-3-((R)-пирролидин-2-ил)-1H-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины дигидрохлорид,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-фенил-3-((пиперидин-4-илокси)метил)-1Н-пиразол-5-ил)мочевины дигидрохлорид,
1-(4-хлор-1-фенил-3-((пиперидин-4-илокси)метил)-1H-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины дигидрохлорид,
1-(4-бром-1-фенил-3-((пиперидин-4-илокси)метил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины дигидрохлорид,
1-(4-бром-3-(1-(метилсульфонил)пиперидин-4-ил)-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(3-(1-ацетилпиперидин-4-ил)-4-бром-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-хлор-1-фенил-3-(1-(трифторметилсульфонил)пиперидин-4-ил)-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевины гидрохлорид,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-((R)-1-(метилсульфонил)пирролидин-2-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-(3-((R)-1-ацетилпирролидин-2-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-((R)-1-метилпирролидин-2-ил)-1-фенил-1Н-пиразол-5-ил)мочевины дигидрохлорид,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-((S)-1-метилпирролидин-2-ил)-1-фенил-1Н-пиразол-5-ил)мочевины дигидрохлорид,
1-(3-((1-ацетилпиперидин-4-илокси)метил)-4-бром-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-(4-изопропил-5-оксо-4,5-дигидро-1,3,4-оксадиазол-2-ил)-4-метил-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(4-метил-5-оксо-4,5-дигидро-1,3,4-оксадиазол-2-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-метокси-1-фенил-4-(трифторметил)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-метокси-1-фенил-4-(трифторметил)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-метокси-1-фенил-4-(трифторметил)-1Н-пиразол-5-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(1-метил-1Н-имидазол-4-ил)-1-фенил-1Н-пиразол-5-ил)мочевина,
1-((транс)-4-(4-хлор-3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-5-оксо-2-фенил-2,5-дигидро-1Н-пиразол-3-ил)мочевина,
1-((транс)-4-(3-хлор-4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-5-оксо-2-фенил-2,5-дигидро-1Н-пиразол-3-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-5-оксо-2-фенил-2,5-дигидро-1Н-пиразол-3-ил)мочевина,
1-((3S,4R)-1-(2-метоксиэтил)-4-(3,4,5-трифторфенил)пирролидин-3-ил)-3-(4-метил-5-оксо-2-фенил-2,5-дигидро-1Н-пиразол-3-ил)мочевина,
1-((3S,4R)-4-(3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-5-оксо-2-фенил-2,5-дигидро-1Н-пиразол-3-ил)мочевина,
1-((транс)-4-(3-хлор-5-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-5-оксо-2-фенил-2,5-дигидро-1Н-пиразол-3-ил)мочевина,
1-(4-циано-3-метокси-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-циано-3-этил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-циано-1-фенил-3-(трифторметил)-1H-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
1-(4-циано-5-оксо-2-фенил-2,5-дигидро-1H-пиразол-3-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)мочевина,
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-3-метокси-1-фенил-1Н-пиразол-4-карбоксамид,
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-3-метил-1-фенил-1Н-пиразол-4-карбоксамид,
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-3-этил-1-фенил-1Н-пиразол-4-карбоксамид,
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-3-(трифторметил)-1Н-пиразол-4-карбоксамид,
5-(3-((транс)-4-(3-хлор-4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-3-метил-1-фенил-1Н-пиразол-4-карбоксамид,
5-(3-((транс)-4-(4-хлор-3-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-3-метил-1-фенил-1Н-пиразол-4-карбоксамид,
5-(3-((транс)-4-(3-хлор-5-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-3-метил-1-фенил-1Н-пиразол-4-карбоксамид,
5-(3-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-3-метил-1-фенил-1Н-пиразол-4-карбоксамид,
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-4-карбоксамид,
5-(3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)уреидо)-1-фенил-1Н-пиразол-4-карбоксамид,
1-(4-бром-3-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)гуанидина гидрохлорид,
1-(4-бром-3-метил-1-фенил-1Н-пиразол-5-ил)-3-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)тиомочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(4-метил-3-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)-1-фенил-1Н-пиразол-5-ил)тиомочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)тиомочевина,
1-((3S,4R)-4-(4-фторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1',4-диметил-1-фенил-1Н,1'Н-3,4'-бипиразол-5-ил)тиомочевина,
транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(2-фенилпиразоло[1,5-а]пиридин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(2-фенилпиразоло[1,5-а]пиридин-3-ил)мочевина,
транс-1-(2-метоксиэтил)-4-фенилпирролидин-3-ил)-3-(пиразоло[1,5-а]пиридин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(пиразоло[1,5-а]пиридин-3-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(5-метил-3-фенил-1-(пиразин-2-ил)-1Н-пиразол-4-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1,5-диметил-3-фенил-1Н-пиразол-4-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1,5-диметил-3-фенил-1Н-пиразол-4-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-изопропил-5-метил-3-фенил-1Н-пиразол-4-ил)мочевина,
1-((3S,4R)-4-(3,5-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-изопропил-5-метил-3-фенил-1Н-пиразол-4-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(5-метил-3-фенил-1-(2,2,2-трифторэтил)-1Н-пиразол-4-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-этил-5-метил-3-фенил-1Н-пиразол-4-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-этил-3-метил-5-фенил-1Н-пиразол-4-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-метил-5-фенил-3-(трифторметил)-1Н-пиразол-4-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-метил-3-фенил-5-(трифторметил)-1Н-пиразол-4-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(3-метил-1-фенил-1Н-пиразол-4-ил)мочевина,
1-((3S,4R)-4-(3,4-дифторфенил)-1-(2-метоксиэтил)пирролидин-3-ил)-3-(1-фенил-3-(трифторметил)-1Н-пиразол-4-ил)мочевина.
31. Фармацевтическая композиция, способная ингибировать киназу TrkA, которая содержит соединение Формулы I, описанное в любом из пп. 1-30, или его фармацевтически приемлемую соль и фармацевтически приемлемый разбавитель или носитель.
32. Способ лечения заболевания или расстройства, выбранного из боли, рака, воспаления, нейродегенеративного заболевания или инфекции Trypanosoma cruzi у млекопитающего, который включает введение указанному млекопитающему терапевтически эффективного количества соединения Формулы I, описанного в любом из пп. 1-30, или его фармацевтически приемлемой соли.
33. Способ по п. 32, отличающийся тем, что заболевание или расстройство представляет собой боль.
34. Соединение Формулы I, описанное в любом из пп. 1-30, или его фармацевтически приемлемая соль для применения при лечении боли, рака, воспаления, нейродегенеративного заболевания или инфекции Trypanosoma cruzi.
35. Применение соединения Формулы I, описанного в любом из пп. 1-30, или его фармацевтически приемлемой соли для получения лекарственного средства для лечения боли, рака, воспаления, нейродегенеративного заболевания или инфекции Trypanosoma cruzi.
36. Способ получения соединения по п. 1, где X представляет собой О, который включает:
(а) связывание соответствующего соединения, имеющего формулу II
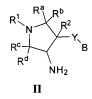
с соответствующим соединением, имеющим формулу III
в присутствии карбонилдиимидазола и основания; или
(в) связывание соответствующего соединения, имеющего формулу II
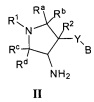
с соответствующим соединением, имеющим формулу IV
где L1 представляет собой уходящую группу, в присутствии основания; или
(е) связывание соответствующего соединения, имеющего формулу II
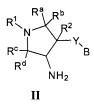
с соответствующим соединением, имеющим формулу VII
в присутствии основания; и
необязательно удаление защитных групп, и необязательно получение его фармацевтически приемлемой соли.
37. Соединение по п. 1 или его фармацевтически приемлемая соль, причем:
фрагмент Y-B и фрагмент NH-C(=X)-NH находятся в транс-конфигурации;
X представляет собой О;
В представляет собой Ar1;
Y представляет собой связь;
кольцо С представляет собой
Ra, Rb, Rc и Rd представляют собой атомы водорода;
R1 представляет собой (1-3С алкокси)(1-6С)алкил, дифтор(1-6С)алкил или трифтор(1-6С)алкил;
R2 представляет собой водород;
R3 представляет собой Ar2, гетAr2 или (1-6С)алкил;
R4 представляет собой гетAr4 или Ar4;
R5 представляет собой Н, галоген или (1-6С)алкил;
Ar1 представляет собой фенил, необязательно замещенный одним или более заместителями, независимо выбранными из галогена, CF3, CF3O-, (1-4С)алкокси, (1-6С)алкила и CN;
Ar2 представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из галогена и (1-6С)алкила;
гетAr2 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, О и S, и необязательно замещенное одной или более группами, независимо выбранными из (1-6С)алкила и галогена;
гетAr4 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенное 1-2 заместителями, независимо выбранными из (1-6С)алкила, галогена, CN, гидрокси(1-6С)алкила, трифтор(1-6С)алкила, (3-6С)циклоалкила, (3-6С циклоалкил)СН2- (3-6С циклоалкил)С(=О)-, (1-3С алкокси)(1-6С)алкила, (1-6С)алкокси, (1-6С)алкилсульфонила, NH2, (1-6С алкил)амино, ди(1-6С алкил)амино, (1-3С трифторалкокси), (1-3С)трифторалкила и метоксибензила; или 9-10-членный бициклический гетероарил, имеющий 1-3 кольцевых атома азота; и
Ar4 представляет собой фенил, необязательно замещенный одной или более группами, независимо выбранными из (1-6С)алкила, галогена, CN, CF3, CF3O-, (1-6С)алкокси, (1-6С алкил)ОС(=О)-, аминокарбонила, (1-6С)алкилтио, гидрокси(1-6С)алкила, (1-6С алкил)SO2-, НОС(=О)- и (1-3С алкокси)(1-3С алкил)ОС(=О)-.
38. Соединение по п. 37, в котором:
R1 представляет собой (1-3С алкокси)(1-6С)алкил;
R3 представляет собой Ar2;
R4 представляет собой гетAr4; и
R5 представляет собой (1-6С)алкил.
39. Соединение по п. 38, в котором Ar1 представляет собой фенил, необязательно замещенный одним или более атомами галогена.
40. Соединение по п. 39, в котором гетAr4 представляет собой 5-6-членное гетероариловое кольцо, имеющее 1-3 кольцевых гетероатома, независимо выбранных из N, S и О, и необязательно замещенное 1-2 заместителями, независимо выбранными из (1-6С)алкила.
41. Соединение по п. 37, выбранное из
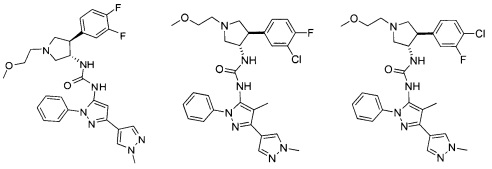
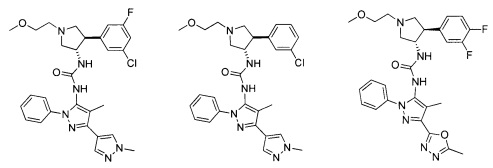
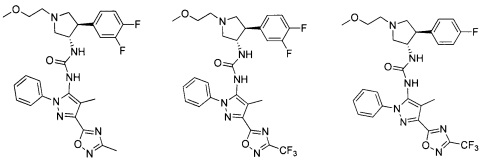
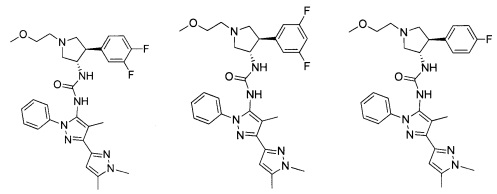
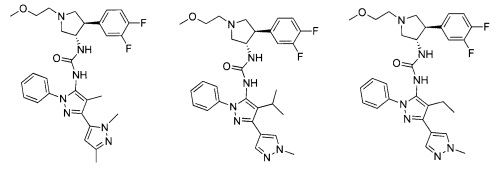
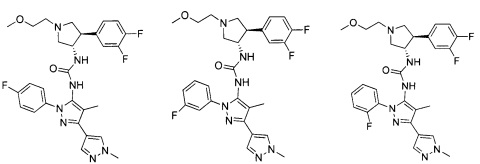
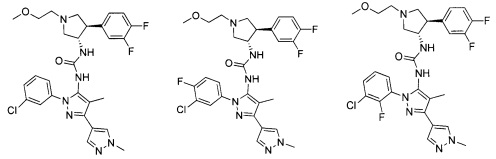
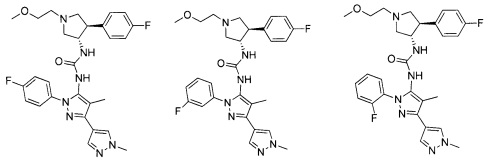
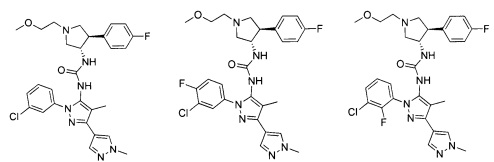
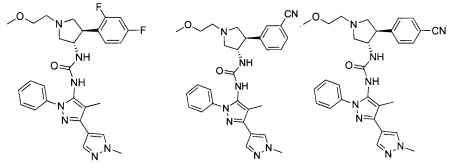
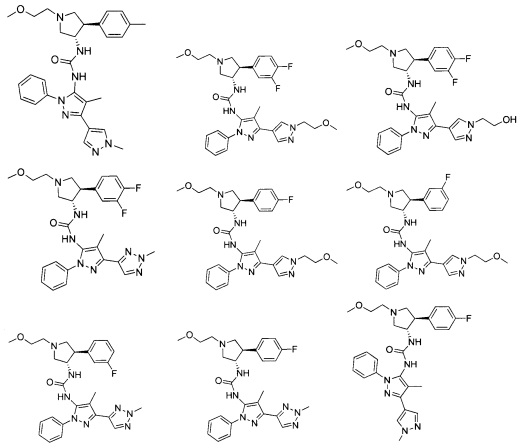
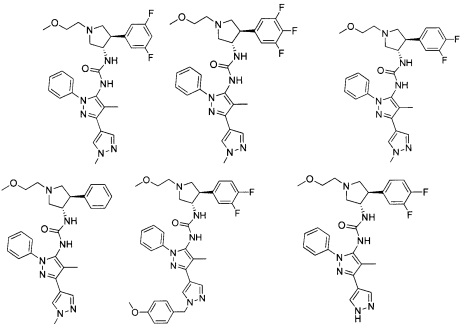
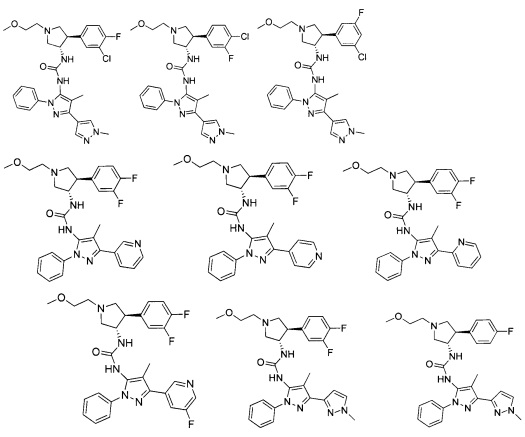
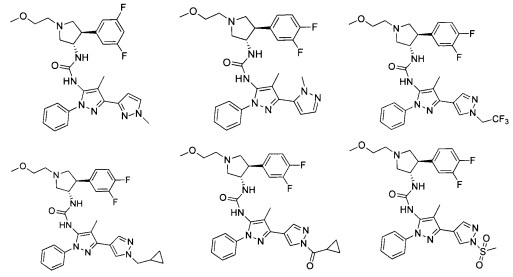
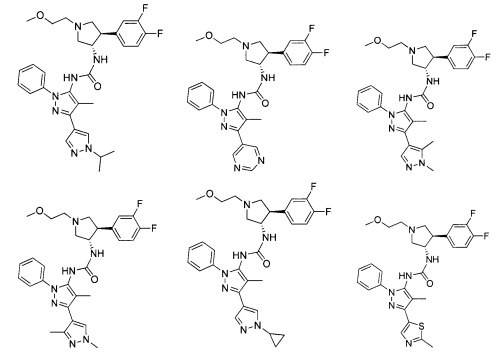
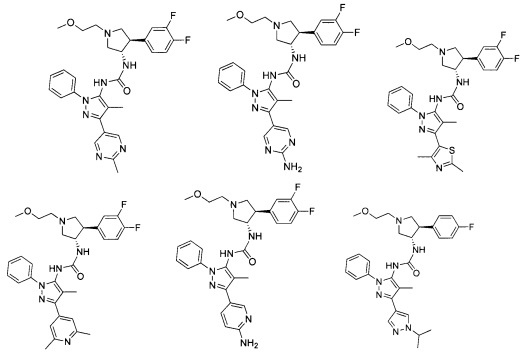
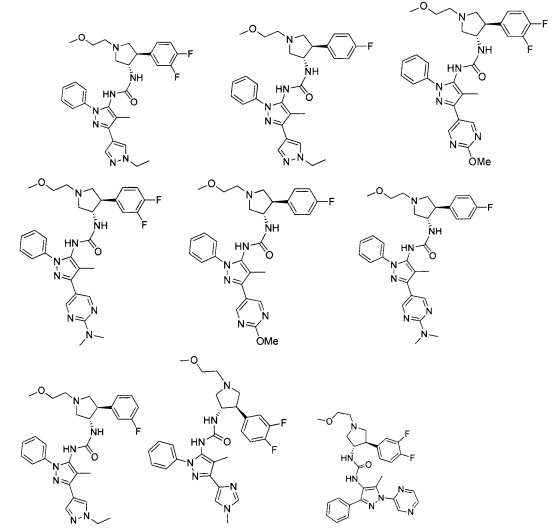
или его фармацевтически приемлемая соль.
42. Соединение по п. 41, выбранное из:
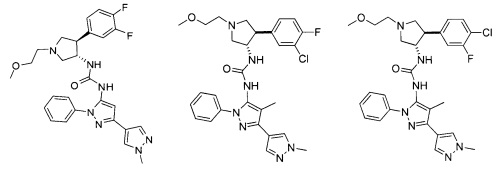
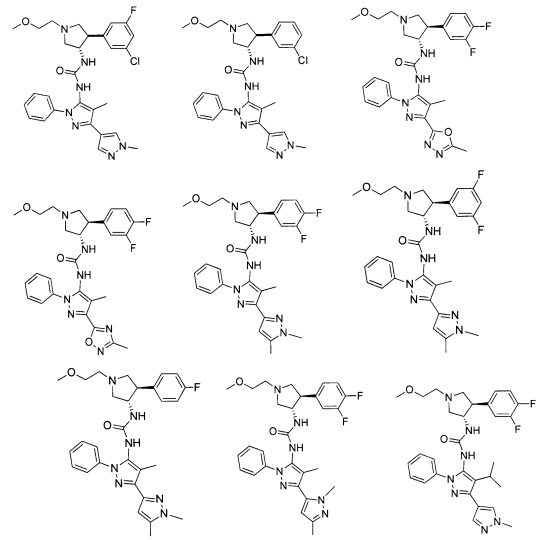
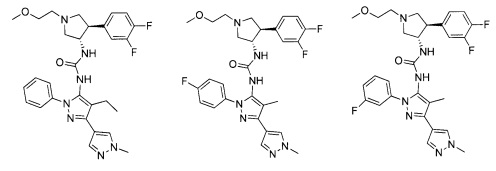
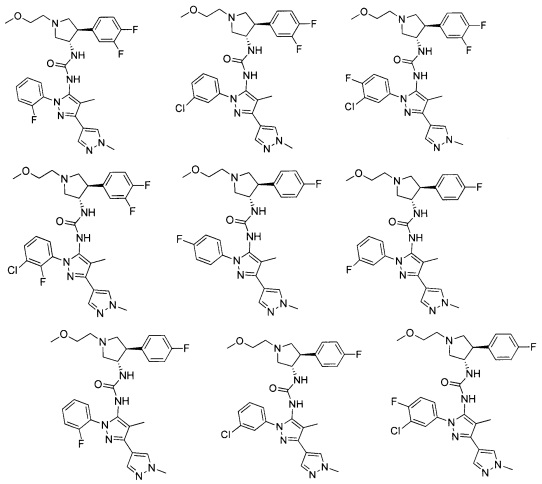
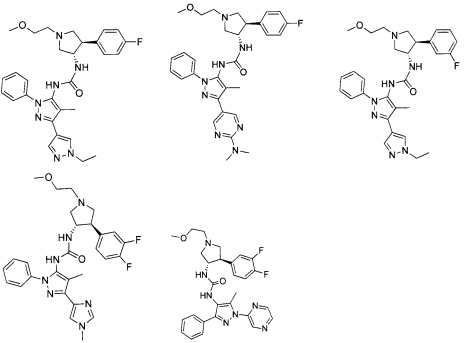
или его фармацевтически приемлемая соль.
43. Фармацевтическая композиция, способная ингибировать киназу TrkA, которая содержит соединение, описанное в любом из пп. 37-42, или его фармацевтически приемлемую соль и фармацевтически приемлемый разбавитель или носитель.
44. Способ лечения заболевания или расстройства, выбранного из боли, рака, воспаления, нейродегенеративного заболевания или инфекции Trypanosoma cruzi у млекопитающего, который включает введение указанному млекопитающему терапевтически эффективного количества соединения, описанного в любом из пп. 37-42, или его фармацевтически приемлемой соли.
45. Способ по п. 44, отличающийся тем, что заболевание или расстройство представляет собой боль.
46. Способ по п. 45, отличающийся тем, что боль представляет собой хроническую боль.
47. Способ по п. 45, отличающийся тем, что боль представляет собой острую боль.
48. Способ по п. 45, отличающийся тем, что боль является воспалительной болью.
49. Способ по п. 45, отличающийся тем, что боль является невропатической болью.
50. Способ по п. 45, отличающийся тем, что боль является болью, связанной с раком.
51. Способ по п. 45, отличающийся тем, что боль является болью, связанной с хирургией.
52. Способ по п. 45, отличающийся тем, что боль является болью, связанной с переломом кости.
53. Способ по п. 44, в котором указанным заболеванием является рак.
54. Способ по п. 53, в котором раком является нейробластома, рак яичников, рак поджелудочной железы, рак толстой и прямой кишок, рак простаты.
55. Способ получения соединения по п. 1, где X представляет собой О, включающий:
связывание соответствующего соединения, имеющего формулу V
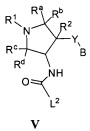
где L2 представляет собой уходящую группу, с соответствующим соединением, имеющим формулу III
в присутствии основания; и
необязательно удаление защитных групп, и необязательно получение его фармацевтически приемлемой соли.
56. Способ получения соединения по п. 1, где X представляет собой О, включающий:
активацию соответствующего соединения, имеющего формулу VI
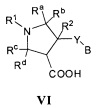
дифенилфосфорил азидом с последующим связыванием активированного промежуточного соединения с соответствующим соединением, имеющим формулу III
в присутствии основания; и
необязательно удаление защитных групп, и необязательно получение его фармацевтически приемлемой соли.
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЦЕПТОРНЫХ ТИРОЗИНКИНАЗ | 2004 |
|
RU2413727C2 |

