ПЕРЕКРЕСТНАЯ ССЫЛКА
По настоящей заявке испрашивается приоритет предварительной заявки США № 62/188025, поданной 2 июля 2015 года, включенной в настоящий документ в качестве ссылки в полном объеме.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Метилирование ДНК представляет собой пострепликативную химическую модификацию ДНК. Различные злокачественные опухоли можно разделять по их нарушенным профилям метилирования ДНК (степень общего или специфического метилирования ДНК), и гиперметилирование конкретных генов может быть ассоциировано с прогнозом для рака желудка, легкого, пищевода, поджелудочной железы и толстого кишечника. Паттерны метилирования ДНК также можно использовать для предсказания ответа на терапию или устойчивости к терапии при глиоме и меланоме. Азацитидин и децитабин представляют собой два гипометилирующих средства, одобренных FDA, которые оказывают свой терапевтический эффект путем ингибирования уровней метилирования ДНК.
Динуклеотидные соединения, полученные из децитабина для разработки терапии по аналогичным показаниям, были описаны в патенте США №7700567 и его эквиваленте WO2007041071. Составы лекарственных средств, содержащие динуклеотидные соединения типа, описанного в WO2007041071, описаны в WO2013033176. Содержание каждого из US7700567, WO2007041071 и WO2013033176 в полном объеме включено в качестве ссылки.
Лиофилизация, часто обозначаемая как лиофильная сушка, представляет собой способ дегидратации, при котором замораживают субстрат, содержащий растворитель, а затем помещают его в вакуум, таким образом, что растворитель удаляется путем сублимации, т.е. прямого перехода из твердого замороженного состояния в газообразное состояние.
ВКЛЮЧЕНИЕ ПУТЕМ ССЫЛКИ
Каждый патент, публикация, и непатентная литература, процитированные в заявке, включены, таким образом, в качестве ссылки в полном объеме, как если бы они были включены путем ссылки по отдельности.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В некоторых вариантах осуществления изобретение относится к способу получения лиофилизированной фармацевтической композиции, способу, включающему растворение соединения формулы (1):
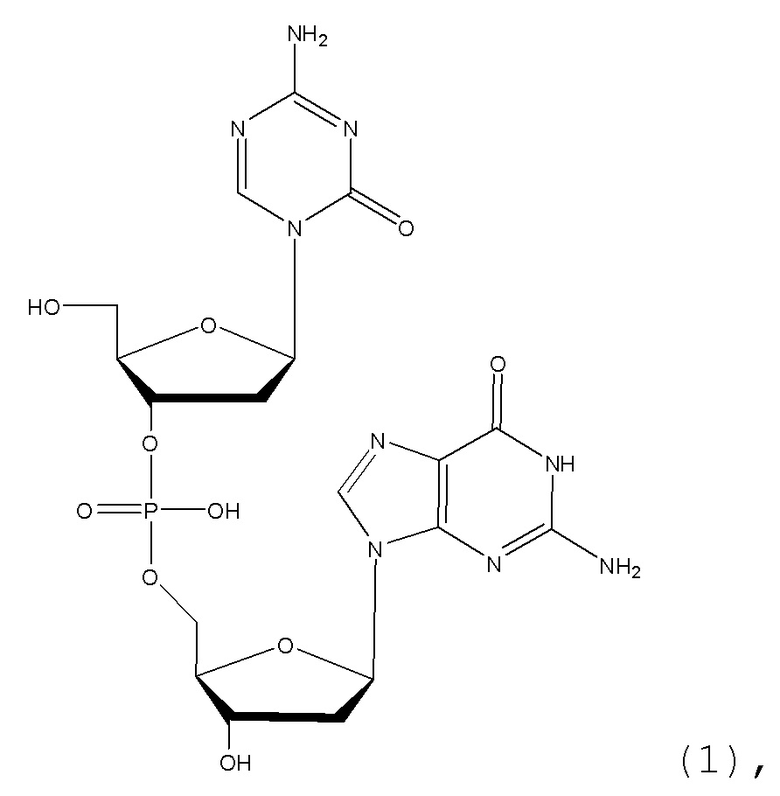
или его фармацевтически приемлемой соли в растворителе, содержащем диметилсульфоксид (ДМСО) для образования раствора, где растворитель затем удаляют способом лиофильной сушки для получения лиофилизированного продукта, где способ лиофильной сушки включает: (i) первую стадию замораживания, на которой раствор замораживают, снижая его температуру до температуры не большей, чем приблизительно -20°C; (ii) первую стадию нагревания, на которой температуру замороженного раствора индуцируют до температуры в диапазоне приблизительно от -15°C до приблизительно 5°C, где температура диапазоне приблизительно от -15°C до приблизительно 5°C сохраняет раствор замороженным; (iii) вторую стадию замораживания, на которой температуру раствора понижают до температуры не большей, чем приблизительно -20°C; (iv) стадию первичного высушивания, где стадия первичного высушивания включает этап сублимации, на котором ДМСО удаляют из раствора в замороженном состоянии путем сублимации при пониженном давлении для получения частично высушенного продукта; и (v) стадию вторичного высушивания, на которой ДМСО удаляют путем выпаривания из частично высушенного продукта в незамороженном состоянии при пониженном давлении для получения лиофилизированного продукта.
В некоторых вариантах осуществления изобретение относится к фармацевтической композиции, приготовленной способом, включающим стадии: растворения соединения формулы (1):
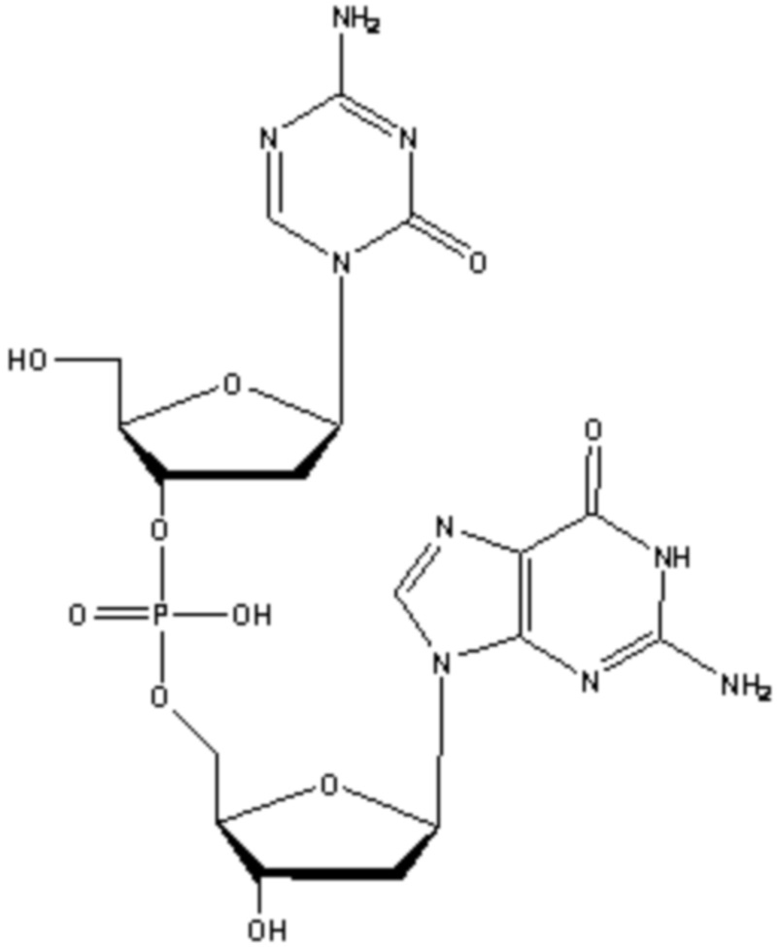 (1),
(1),
или его фармацевтически приемлемой соли в растворителе, содержащем диметилсульфоксид (ДМСО) для образования раствора, где растворитель затем удаляют способом лиофильной сушки для получения лиофилизированного продукта, где способ лиофильной сушки включает: (i) первую стадию замораживания, на которой раствор замораживают, снижая его температуру до температуры не большей, чем приблизительно -20°C; (ii) первую стадию нагревания, на которой температуру замороженного раствора индуцируют до температуры в диапазоне приблизительно от -15°C до приблизительно 5°C, где температура в диапазоне приблизительно от -15°C до приблизительно 5°C сохраняет раствор замороженным; (iii) вторую стадию замораживания, на которой температуру раствора понижают до температуры не большей, чем приблизительно -20°C; (iv) стадию первичного высушивания, где стадия первичного высушивания включает этап сублимации, на котором ДМСО удаляют из раствора в замороженном состоянии путем сублимации при пониженном давлении для получения частично высушенного продукта; и (v) стадию вторичного высушивания, на которой ДМСО удаляют путем выпаривания из частично высушенного продукта в незамороженном состоянии при пониженном давлении для получения лиофилизированного продукта.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фигура 1 представляет собой график удаления ДМСО со временем с протеканием процесса лиофилизации. Профили удаления ДМСО для четырех составов A, B, C и D с различными концентрациями показаны на Фигуре 1.
ПОДРОБНОЕ ОПИСАНИЕ
Эта заявка относится к лиофилизированным фармацевтическим композициям, содержащим динуклеотид, полученный из децитабина, и к способам получения и применения композиций с нуклеотидом, полученным из децитабина.
Настоящее изобретение относится к улучшенным лиофилизированным композициям, содержащим соединение формулы (1) или его фармацевтически приемлемую соль, и к способу получения улучшенных лиофилизированных фармацевтических композиций с использованием способа лиофильной сушки. Изобретение также относится к применению лиофилизированных фармацевтических композиций в медицине и, в частности, к их применению для лечения злокачественных опухолей.
Настоящее изобретение предлагает улучшенные способы для лиофилизации субстрата, содержащего неводный растворитель, например, ДМСО, и соединение формулы (1), или его фармацевтически приемлемую соль. В основном, способы включают две стадии замораживания с промежуточной стадией нагревания (стадия отжига) между двумя стадиями замораживания. Способы можно использовать для удаления неводного растворителя из субстрата. В некоторых конкретных вариантах осуществления, соединение в субстрате представляет собой соединение формулы (1):
или его фармацевтически приемлемую соль. Настоящее изобретение также предлагает лиофилизированные композиции, содержащие соединение формулы (1) или его фармацевтически приемлемую соль. Кроме того, настоящее изобретение предлагает применения лиофилизированных фармацевтических композиций в медицине, в частности, для лечения злокачественных опухолей.
Было обнаружено, что при использовании двух стадий замораживания и промежуточной стадии нагревания (стадии отжига) между двумя стадиями замораживания, можно удалять ДМСО значительно быстрее во время последующей стадии первичного высушивания и что, таким образом, длина стадии вторичного высушивания может быть значительно уменьшена. Без ограничения какой-либо теорией, полагают, что промежуточная стадия нагревания может обеспечить повышенную пористость, тем самым позволяя ДМСО более легко испаряться. Таким образом, значительно больше DMSO удаляется во время стадии первичного высушивания.
Исследования составов при помощи микроскопии при лиофильной сушки (FDM) показали, что даже при температурах ниже -30°C, время от времени, может присутствовать некоторое количество остаточного незамороженного растворителя или со-растворителя. Как применяют в настоящем документе, термин «замороженный», таким образом, включает состояние, в котором присутствует твердая структура, образованная из молекул растворителя и/или со-растворителя, но также может присутствовать некоторое количество растворителя и/или со-растворителя в незамороженной, или жидкой, форме.
Способ получения лиофилизированной фармацевтической композиции
Способы, предлагаемые в настоящем документе, включают способ получения лиофилизированной фармацевтической композиции, содержащей соединение, например, соединение формулы (1)) или его фармацевтически приемлемую соль, при этом способ включает растворение соединения формулы (1) или его фармацевтически приемлемой соли в неводном растворителе, содержащем диметилсульфоксид и необязательно один или несколько со-растворителей для образования раствора, а затем удаление растворителя и любых со-растворителей способом лиофильной сушки для получения лиофилизированного продукта; где способ лиофильной сушки включает одну или несколько следующих стадий: (i) первую стадию замораживания, на которой раствор замораживают, снижая его температуру до температуры не большей, чем приблизительно -20°C; (ii) первую стадию нагревания, на которой температуру замороженного раствора индуцируют до температуры в диапазоне приблизительно от -15°C до приблизительно 5°C, где температура в диапазоне приблизительно от -15°C до приблизительно 5°C сохраняет раствор замороженным; (iii) вторую стадию замораживания, которая происходит после первой стадии нагревания и на которой температуру раствора в замороженном состоянии понижают до температуры не большей, чем приблизительно -20°C; (iv) стадию первичного высушивания, включающую этап сублимации, на котором ДМСО и один или несколько со-растворителей, при наличии, удаляют из раствора в замороженном состоянии путем сублимации при пониженном давлении для получения частично высушенного продукта; и (v) стадию вторичного высушивания, на которой ДМСО и один или несколько со-растворителей, при наличии, удаляют путем выпаривания из частично высушенного продукта в незамороженном состоянии при пониженном давлении для получения лиофилизированного продукта.
Последовательность стадий замораживания и промежуточной стадии нагревания (i), (ii), и (iii) можно повторять один или несколько раз, прежде чем перейти к стадии первичного высушивания (iv). Например, после первой последовательности стадий (i), (ii), и (iii) может быть вторая последовательность стадий (i), (ii), и (iii), и, необязательно, третья и четвертая последовательности стадий (i), (ii), и (iii) до перехода к стадии первичного высушивания (iv).
Способ по изобретению может, например, уменьшать общее время для способа лиофильной сушки, по меньшей мере, на сутки, и, в некоторых вариантах осуществления изобретения, вплоть до двух суток. Способ по изобретению может дополнительно позволить более легко восстанавливать раствор, чем композиции, приготовленные при помощи способов, которые не включают промежуточную стадию нагревания. Например, в некоторых вариантах осуществления изобретения, как определено в настоящем документе, время восстановления композиций может быть снижено от времени более 30 минут до времени менее чем 20 минут и, в некоторых вариантах осуществления, до времени менее чем 10 минут.
Процедуру лиофилизации можно проводить на приборе для лиофильной сушки. Прибор для лиофильной сушки может иметь камеру, в которую можно помещать для лиофильной сушки контейнеры для лиофильной сушки (например, флаконы для лиофильной сушки), содержащие раствор. Камера может быть соединена с источником вакуума (например, вакуумной помпой), чтобы уменьшить давление внутри камеры. Прибор может также иметь компоненты для замораживания или нагревания содержимого камеры. Перед лиофилизацией, можно получать основной объем раствора соединения формулы (1) в ДМСО и необязательно в одном или нескольких со-растворителях и фильтровать через фильтр (например, фильтр для стерилизации) перед заполнением аликвотами контейнеров для лиофильной сушки (например, флаконов для лиофильной сушки) и переносом в прибор для лиофильной сушки. Перед переносом в прибор для лиофильной сушки контейнеры можно частично закрыть пробкой для предотвращения загрязнения, тем не менее, позволяющей удалять растворитель во время способа лиофильной сушки.
В следующих абзацах параметры способа лиофильной сушки изложены более подробно со ссылкой на конкретные варианты осуществления, множества, подмножества, диапазоны и индивидуальные значения для каждого параметра. Во избежание сомнений каждый вариант осуществления, множество, подмножество, диапазон и индивидуальное значение, определенное относительно одного из параметров способа лиофильной сушки, можно комбинировать с каждым вариантом осуществления, множеством, подмножеством, диапазоном и индивидуальным значением, определенным относительно любого другого параметра способа лиофильной сушки. Эта заявка, таким образом, описывает все комбинации вариантов осуществления, множества, подмножества, диапазоны и индивидуальные значения для каждого параметра способа лиофильной сушки.
Температуры, упомянутые выше и в другом месте в настоящем документе по отношению к параметрам способа лиофилизации, являются температурами полок в приборе для лиофильной сушки. Полки, как правило, охлаждаются охлаждающими жидкостями, температуры которых контролируются, и обеспечивают способ определения температуры полок. Измерения температуры, полученные для охлаждающих жидкостей, могут быть перепроверены на основании температур, полученных непосредственно в продукте лиофилизации в контейнерах для лиофильной сушки при введении температурных зондов в выбранные контейнеры для лиофильной сушки.
На первой стадии замораживания (i), раствор можно замораживать, снижая его температуру до температуры не больше, чем приблизительно -20°C, например, температуру можно снижать до значения не больше, чем приблизительно -30°C (или не больше, чем приблизительно -35°C, или не больше, чем приблизительно -40°C, или не больше, чем приблизительно -41°C, или не больше, чем приблизительно 42°C, или не больше, чем приблизительно -43°C, или не больше, чем приблизительно -44°C). Например, раствор можно замораживать, снижая температуру до значения в диапазоне приблизительно от -40°C до приблизительно -50°C, или приблизительно от -42°C до приблизительно -48°C, или приблизительно от -43°C до приблизительно -47°C, или приблизительно от -44°C до приблизительно -46°C, например, приблизительно -45°C.
Первая стадия замораживания может включать этап линейного изменения температуры, где температура снижается от начальной температуры (например, внешней среды) до целевой температуры в течение первого периода времени, например, в течение периода приблизительно до 2 часов или приблизительно до 1,5 часов или до 1,25 часа, например, приблизительно, 1 час.
После достижения целевой температуры, замороженный раствор может поддерживаться при целевой температуре в течение второго периода времени, например, приблизительно до 3 часов, или приблизительно до 2,5 часов или приблизительно до 2 часов, например, приблизительно 1,5 часа.
После первой стадии замораживания, раствор подвергают первой стадии нагревания, на которой температура замороженного раствора достигает температуры в диапазоне от -15°C до 4°C, при которой раствор остается в замороженном состоянии. Например, замороженный раствор может быть согрет до температуры в диапазоне приблизительно от -5°C до приблизительно 5°C, или приблизительно от -3°C до приблизительно 3°C, или приблизительно от -2°C до приблизительно 2°C, или приблизительно от -1°C до приблизительно 1°C, например, приблизительно 0°C.
Первая стадия нагревания может включать первый период времени, во время которого замороженный раствор нагревается до целевой температуры, и второй период времени, во время которого замороженный раствор поддерживается при целевой температуре. Например, первый период времени, при котором замороженный раствор нагревается до целевой температуры, может быть приблизительно до 2 часов, или приблизительно до 1,75 часов, или приблизительно до 1,5 часов, например, приблизительно 1,3 часа.
После первой стадии нагревания, все еще замороженный раствор можно подвергать второй стадии замораживания, на которой температуру раствора в замороженном состоянии снижают до температуры не больше, чем приблизительно -20°C. Температуру можно снижать до величины не больше, чем приблизительно -30°C (или не больше, чем приблизительно -35°C, или не больше, чем приблизительно -40°C, или не больше, чем приблизительно -41°C, или не больше, чем приблизительно 42°C, или не больше, чем приблизительно -43°C, или не больше, чем приблизительно -44°C). Например, температуру замороженного раствора можно снижать до значения в диапазоне приблизительно от -40°C до приблизительно -50°C, или приблизительно от -42°C до приблизительно -48°C, или приблизительно от -43°C до приблизительно -47°C, или приблизительно от -44°C до приблизительно -46°C, например, приблизительно -45°C.
После второй стадии замораживания, замороженный раствор можно подвергать стадии первичного высушивания, включающей этап сублимации, на котором диметилсульфоксид и один или несколько со-растворителей, при наличии, удаляются путем сублимации из раствора в замороженном состоянии при пониженном давлении для получения частично высушенного продукта. На стадии первичного высушивания замороженный раствор можно нагревать для облегчения более быстрой сублимации ДМСО, сохраняя раствор в замороженном состоянии. Например, замороженный раствор можно нагревать до температуры в диапазоне от -25°C до 0°C, или от -22°C до -2°C, например, приблизительно от -20°C до приблизительно -5°C.
На стадии первичного высушивания, замороженный раствор можно нагревать поэтапно. Например, на первом этапе нагревания, температуру можно индуцировать от температуры не больше, чем приблизительно -30°C до температуры в диапазоне приблизительно от -25°C до приблизительно -19°C (например, приблизительно -20°C), а затем поддерживать при такой температуре в течение определенного поддерживающего периода. При этой температуре остаточный незамороженный растворитель и/или со-растворитель можно удалять выпариванием.
На втором этапе нагревания температуру можно индуцировать от температуры в диапазоне приблизительно от -25°C до приблизительно -19°C (например, приблизительно -20°C), до температуры в диапазоне приблизительно от -10°C до приблизительно 0°C (например, приблизительно -5°C), а затем поддерживать при такой температуре в течение дополнительно определенного поддерживающего периода. Следует понимать, что дополнительные промежуточные стадии нагревания и поддерживающие периоды можно добавлять к первой и второй стадиям нагревания. В качестве альтернативы нагреванию замороженного раствора постадийно, нагревание можно проводить в непрерывном режиме до достижения требуемой целевой температуры.
В начале первичного периода высушивания, давление в сосуде, содержащем замороженный раствор, можно снижать (как правило, от атмосферного давления) до давления, при котором может происходить удаление ДМСО и необязательно других со-растворителей. Давление можно снижать до давления ниже, чем 1 мбар, например, ниже 500 мкбар, или менее, чем 100 мкбар, или менее, чем 50 мкбар. Например, давление можно снижать до давления менее, чем 20 мкбар, или менее, чем 10 мкбар, или от 1 до 10 мкбар, или от 4 до 8 мкбар, например, приблизительно 6 мкбар.
Стадия первичного высушивания может включать начальную стадию снижения давления, на которой поддерживается постоянная температура, а давление снижают до целевой величины, с последующим нагреванием замороженного раствора, как определено выше. Альтернативно, снижение давления и нагревание замороженного раствора можно проводить одновременно.
Стадия первичного высушивания занимает приблизительно от 20 до приблизительно 60 часов, например, приблизительно от 30 до приблизительно 50 часов.
Прогресс на стадии первичного высушивания можно контролировать при помощи одного или нескольких сенсоров или датчиков, присутствующих в камере для лиофилизации в приборе для лиофильной сушки. Сенсоры или датчики (такие как манометр Пирани) можно использовать для измерения одного или нескольких параметров внутри камеры, и, таким образом, определенные изменения по одному или нескольким параметрам могут указывать на прогресс первичного высушивания и обеспечивать способы, которые определяют, когда завершена сублимация ДМСО и необязательно любых со-растворителей. Например, сенсор или датчик могут измерять давление внутри камеры или проводимость газа в камере.
Во время процесса сублимации температура должна быть ниже критической температуры и давления продукта, таким образом, чтобы продукт оставался замороженным. Сублимация представляет собой прямой фазовый переход твердое вещество-газ для ДМСО. Если условия превышают критическую температуру и давления, продукт не заморожен и, вместо этого, является жидкостью, и ДМСО может изменяться из жидкости в газ (кипеть). Недопустимо, чтобы ДМСо кипел вместо сублимации.
Стадию первичного высушивания можно проводить под давлениями приблизительно от 5 мкбар до приблизительно 40 мкбар. Температура замерзания продукта при этих давлениях составляет приблизительно от -2°C до приблизительно -4°C. Стадию первичного высушивания можно проводить при температурах приблизительно от -3°C до приблизительно -9°C. В этом температурном диапазоне давление пара является подходящим для быстрой сублимации, которая приводит к лучшему продукту. В некоторых вариантах осуществления давление составляет приблизительно 20 мкбар. В некоторых вариантах осуществления температура составляет приблизительно -6°C.
Как только сублимация ДМСО прекратилась или упала ниже конкретного уровня, инициируют стадию вторичного высушивания. На стадии вторичного высушивания диметилсульфоксид и один или несколько со-растворителей, при наличии, удаляют путем испарения из частично высушенного продукта в незамороженном состоянии при пониженном давлении для получения лиофилизированного продукта. Таким образом, на стадии вторичного высушивания поддерживаются условия сниженного давления и частично высушенный продукт нагревают до температуры, при которой он больше не заморожен. Поскольку точка кипения ДМСО составляет приблизительно 189°C, частично высушенный продукт можно нагревать до температуры, по меньшей мере, приблизительно 40°C, дополнительно, как правило, по меньшей мере, приблизительно 45°C, например, по меньшей мере, приблизительно 50°C, или, по меньшей мере, приблизительно 55°C. В некоторых вариантах осуществления частично высушенный продукт нагревают до температуры в диапазоне приблизительно от 55°C до приблизительно 70°C, например, приблизительно 65°C.
Стадия вторичного высушивания может включать один или несколько этапов линейного изменения температуры, на которых частично высушенный продукт нагревают до целевой температуры, каждый этап линейного изменения температуры сопровождается этапом поддержания температуры. В одном из вариантов осуществления присутствует один этап линейного изменения температуры с последующим одним этапом поддержания температуры.
Во время стадии вторичного высушивания молекулы незамороженного растворителя удаляют для получения лиофилизированного продукта, который содержит только низкие уровни остаточного ДМСО.
Целесообразно, чтобы стадию вторичного высушивания проводили при температуре приблизительно 30°C до приблизительно 65°C, например, приблизительно 40°C.
В конце стадии вторичного высушивания в камеру для лиофильной сушки запускают инертный газ, такой как азот, и контейнеры (например, флаконы), содержащие лиофилизированный продукт, полностью закупоривают (например, с помощью пробки и необязательно также крышки) под инертным газом.
Процедуру лиофилизации можно проводить на растворе соединения формулы (1) или его фармацевтически приемлемой соли в неводном растворителе, содержащем диметилсульфоксид и необязательно один или несколько co-растворителей.
В некоторых вариантах осуществления загрязнения водой можно избежать на любой стадии. Не ограничиваясь теорией, полагают, что образование гидрата, в частности, нарушает структуру продукта, что не способствует легкому восстановлению.
В некоторых вариантах осуществления изобретения, по существу отсутствуют со-растворители; т.е. растворитель по существу содержит ДМСО.
В других вариантах осуществления изобретения, могут присутствовать один или несколько других неводных со-растворителей. Когда присутствует со-растворитель, общий объем co-растворителя, как правило, может составлять не более, чем приблизительно 25% (об./об.) от растворителя в целом. Дополнительно, как правило, общий объем co-растворителя, при наличии, составляет не более, чем приблизительно 20%, или не более, чем приблизительно 15%, или не более, чем приблизительно 10%, или не более, чем приблизительно 5% от общего объема растворителя. Например, общий объем co-растворителя может составлять приблизительно от 0% (об./об.) до приблизительно 5% (об./об.) от общего объема растворителя.
Раствор, предназначенный для лиофилизации, может содержать количество соединения формулы (1) или его фармацевтически приемлемой соли в диапазоне приблизительно от 5 мг/мл до приблизительно 200 мг/мл, например, в диапазоне приблизительно от 10 мг/мл до приблизительно 150 мг/мл. Например, раствор может содержать приблизительно от 20 мг/мл до приблизительно 120 мг/ мл, или приблизительно от 22 мг/мл до приблизительно 110 мг/мл, или приблизительно от 25 мг/мл до приблизительно 105 мг/мл, или приблизительно от 25 мг/мл до приблизительно 100 мг/мл соединения формулы (1) или его фармацевтически приемлемой соли.
В некоторых вариантах осуществления раствор содержит приблизительно от 40 мг/мл до приблизительно 110 мг/мл, или приблизительно от 50 мг/мл до приблизительно 105 мг/мл соединения формулы (1) или его фармацевтически приемлемой соли.
В конкретном варианте осуществления изобретения, раствор содержит или 75 мг/мл или 100 мг/мл натриевой соли соединения формулы (1).
Неограничивающие примеры давлений, которые можно использовать во время способа по изобретению включают приблизительно 1 мкбар, приблизительно 2 мкбар, приблизительно 3 мкбар, приблизительно 4 мкбар, приблизительно 5 мкбар, приблизительно 6 мкбар, приблизительно 7 мкбар, приблизительно 8 мкбар, приблизительно 9 мкбар, приблизительно 10 мкбар, приблизительно 15 мкбар, приблизительно 20 мкбар, приблизительно 25 мкбар, приблизительно 30 мкбар, приблизительно 35 мкбар, приблизительно 40 мкбар, приблизительно 45 мкбар, приблизительно 50 мкбар, приблизительно 55 мкбар, приблизительно 60 мкбар, приблизительно 65 мкбар, приблизительно 70 мкбар, приблизительно 80 мкбар, приблизительно 90 мкбар, и приблизительно 100 мкбар.
Лиофилизированные фармацевтические композиции.
Изобретение относится к лиофилизированной фармацевтической композиции, которую можно приготовить (или которая приготовлена) способом лиофильной сушки, как описано в настоящем документе.
Лиофилизированные фармацевтические композиции по изобретению характеризуются улучшеннной растворимостью по сравнению с известными лиофилизированными составами соединений формулы (1) и их солей. Таким образом, в другом варианте осуществления изобретение относится к лиофилизированной фармацевтической композиции, содержащей соединение формулы (1) или его фармацевтически приемлемую соль, которую можно получать способом лиофильной сушки, как определено в настоящем документе, и которая имеет время растворения не более, чем 20 минут при температуре окружающей среды и без помощи механического перемешивания в неводном растворителе, содержащем 65% (об./об.) пропиленгликоля, 25% (об./об.) глицерина и 10% (об./об.) этанола.
В некоторых вариантах осуществления лиофилизированная фармацевтическая композиция имеет время растворения в неводном растворителе не более чем 15 минут, или не более чем 12 минут.
В конкретных вариантах осуществления лиофилизированная фармацевтическая композиция имеет время растворения в неводном растворителе не более чем 10 минут.
Лиофилизированные фармацевтические композиции по изобретению также характеризуются сниженными уровнями остаточного растворителя ДМСО. Таким образом, в другом варианте осуществления изобретение относится к лиофилизированной фармацевтической композиции, содержащей соединение формулы (1) или его фармацевтически приемлемую соль, которую можно получать способом лиофильной сушки, как определено в настоящем документе, и где в количестве лиофилизированной композиции, полученной из 1 грамма раствора, содержание остаточного ДМСО составляет не более чем 20 мг или не более чем 19 мг. Следует понимать, что ссылка на «раствор» означает раствор его фармацевтически приемлемой соли в растворителе, содержащем диметилсульфоксид и необязательно один или несколько co-растворителей. Растворитель может быть не водным, безводным или по существу безводным.
В другом варианте осуществления предлагается лиофилизированная фармацевтическая композиция, содержащая соединение формулы (1) или его фармацевтически приемлемую соль, которую можно получать способом лиофильной сушки, как определено в настоящем документе, и где любой остаточный ДМСО присутствует в композиции в количестве, соответствующем не более чем 35 мг на 100 мг эквивалента свободного основания соединения формулы (1).
Термин «100 мг эквивалента свободного основания» относится к количеству по массе свободного основания, которое может присутствовать, или, когда соединение формулы (1) находится в форме соли, к количеству по массе свободного основания, содержащегося в соли. Например, количество остаточного ДМСО на 100 мг эквивалента свободного основания составляет не более, чем приблизительно 32 мг, или не более, чем приблизительно 31 мг, например, в диапазоне приблизительно от 15 мг до приблизительно 35 мг, или приблизительно от 20 мг до приблизительно 32 мг, или приблизительно от 25 мг до приблизительно 30 мг.
В другом варианте осуществления предлагается лиофилизированная фармацевтическая композиция, содержащая соединение формулы (1) или его фармацевтически приемлемую соль, которую можно получать способом лиофильной сушки, как определено в настоящем документе, и которая: (a) имеет время растворения при температуре окружающей среды и без помощи механического перемешивания в неводном растворителе, содержащем 65% (об./об.) пропиленгликоля, 25% (об./об.) глицерина и 10% (об./об.) этанола, не более, чем 20 минут (или не более чем 15, или 12, или 10 минут); и (b) имеет такое содержание остаточного ДМСО, что в количестве лиофилизированной композиции, полученной из 1 грамма раствора, содержание остаточного ДМСО составляет не более чем 20 мг, или не более чем 19 мг. Растворитель может быть не водным, безводным или по существу безводным.
Лиофилизированные фармацевтические композиции по изобретению, т.е. композиции, которые можно получать способом лиофильной сушки, как определено в настоящем документе, могут также быть охарактеризованы в отношении их улучшенной пористости и увеличенной удельной площади поверхности по сравнению с известными композициями. Удельную площадь поверхности можно измерять при помощи известных способов, таких как способ адсорбции Брунауэра-Эмметта-Теллера (BET).
Лиофилизированные фармацевтические композиции по изобретению можно предоставлять в запечатанных контейнерах, таких как флаконы (например, стеклянные флаконы), необязательно содержащих защитную атмосферу инертного газа, такого как азот или аргон. Запечатанные контейнеры можно открывать, когда необходимо, и восстанавливать содержимое, растворяя в востановителе, таком как не водный, безводный или по существу безводный растворитель, перед введением пациенту. Примеры растворителей, в которых можно восстанавливать лиофилизированные фармацевтические композиции по изобретению, описаны в WO2013033176.
В дополнительном аспекте, таким образом, изобретение относится к запечатанному фармацевтическому контейнеру, содержащему лиофилизированную фармацевтическую композицию, как определено в настоящем документе. Запечатанный фармацевтический контейнер может быть, например, флаконом с пробкой и необязательно дополнительными компонентами (такими как ободок) для удержания пробки на месте. Запечатанный контейнер необязательно может содержать защитную атмосферу инертного газа, такого как азот или аргон.
В конкретном варианте осуществления изобретение относится к запечатанному фармацевтическому контейнеру, содержащему лиофилизированную фармацевтическую композицию, как определено в настоящем документе, где композиция содержит соединение формулы (1) или его фармацевтически приемлемую соль в количестве соответствующем приблизительно 100 мг эквивалента свободного основания соединения формулы (1), и где в композиции присутствует не более, чем 35 мг остаточного ДМСО.
Восстановленные составы, приготовленные из лиофилизированных фармацевтических композиций
Лиофилизированные фармацевтические композиции по изобретению можно восстанавливать в растворителях, таких как не водные, безводные или по существу безводные растворители, для получения жидких инъекционных композиций для введения индивидууму. Жидкие композиции могут быть предназначены для введения путем подкожной инъекции. Таким образом, в дополнительном аспекте, изобретение относится к способу получения жидкой инъекционной композиции, включающему растворение лиофилизированной фармацевтической композиции, как определено в настоящем документе, в растворителе, в частности, в неводном растворителе.
Неограничивающие примеры подходящих растворителей включают пропиленгликоль, глицерин, этанол, и любую комбинацию вышеуказанных. Составы можно получать в виде неводных составов. Составы могут быть безводными или по существу безводными.
Смесь растворителей может содержать процентную долю пропиленгликоля на основании либо массы, либо объема. В некоторых вариантах осуществления процентная доля пропиленгликоля может составлять, по меньшей мере, приблизительно 10%, по меньшей мере, приблизительно 20%, по меньшей мере, приблизительно 30%, по меньшей мере, приблизительно 40%, по меньшей мере, приблизительно 50%, по меньшей мере, приблизительно 10%, по меньшей мере, приблизительно 20%, по меньшей мере, приблизительно 30%, по меньшей мере, приблизительно 40%, или, по меньшей мере, приблизительно 50%. В некоторых вариантах осуществления процентная доля пропиленгликоля может составлять не более 90%, не более 80%, не более 70%, не более 60%, не более приблизительно 90%, не более приблизительно 80%, не более приблизительно 70%, или не более приблизительно 60%. В некоторых вариантах осуществления процентная доля пропиленгликоля может составлять приблизительно от 30% до приблизительно 90%, приблизительно от 45% до приблизительно 85%, приблизительно от 55% до приблизительно 75%, приблизительно от 60% до приблизительно 70%, приблизительно от 30% до приблизительно 90%, приблизительно от 45% до приблизительно 85%, приблизительно от 55% до приблизительно 75%, или приблизительно от 60% до приблизительно 70%. В некоторых вариантах осуществления процентная доля пропиленгликоля может составлять 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, приблизительно 30%, приблизительно 35%, приблизительно 40%, приблизительно 45%, приблизительно 50%, приблизительно 55%, приблизительно 60%, приблизительно 65%, приблизительно 70%, приблизительно 75%, приблизительно 80%, приблизительно 85%, или приблизительно 90%.
Смесь растворителей может содержать процентную долю глицерина на основании либо массы, либо объема. В некоторых вариантах осуществления процентная доля глицерина может составлять, по меньшей мере, 5%, по меньшей мере, 10%, по меньшей мере, 15%, по меньшей мере, 25%, по меньшей мере, 30%, по меньшей мере, приблизительно 5%, по меньшей мере, приблизительно 10%, по меньшей мере, приблизительно 15%, по меньшей мере, приблизительно 25%, или, по меньшей мере, приблизительно 30%. В некоторых вариантах осуществления процентная доля глицерина может составлять не более 70%, не более 60%, не более 50%, не более 40%, не более 30%, не более приблизительно 70%, не более приблизительно 60%, не более приблизительно 50%, не более приблизительно 40%, или не более приблизительно 30%. В некоторых вариантах осуществления процентная доля глицерина может составлять от 0% до 50%, от 5% до 45%, от 15% до 35%, от 20% до 30%, от 0% до приблизительно 50%, приблизительно от 5% до приблизительно 45%, приблизительно от 15% до приблизительно 35%, или приблизительно от 20% до приблизительно 30%. В некоторых вариантах осуществления процентная доля глицерина может составлять 0%, 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, приблизительно 5%, приблизительно 10%, приблизительно 15%, приблизительно 20%, приблизительно 25%, приблизительно 30%, приблизительно 35%, приблизительно 40%, приблизительно 45%, или приблизительно 50%.
Смесь растворителей может содержать процентную долю этанола на основании либо массы, либо объема. В некоторых вариантах осуществления процентная доля этанола может составлять, по меньшей мере, 1%, по меньшей мере, 3%, по меньшей мере, 5%, по меньшей мере, 10%, по меньшей мере, 15%, по меньшей мере, приблизительно 1%, по меньшей мере, приблизительно 3%, по меньшей мере, приблизительно 5%, по меньшей мере, приблизительно 10%, или, по меньшей мере, приблизительно 15%. В некоторых вариантах осуществления процентная доля этанола может составлять не более 30%, не более 25%, не более 20%, не более 15%, не более 10%, не более приблизительно 30%, не более приблизительно 25%, не более приблизительно 20%, не более приблизительно 15%, или не более приблизительно 10%. В некоторых вариантах осуществления процентная доля этанола может составлять от 0% до 30%, от 0% до 25%, от 0% до 20%, от 5% до 15%, от 0% до приблизительно 30%, от 0% до приблизительно 25%, от 0% до приблизительно 20%, или приблизительно от 5% до приблизительно 15%. В некоторых вариантах осуществления процентная доля этанола может составлять 0%, 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, приблизительно 1%, приблизительно 2%, приблизительно 3%, приблизительно 4%, приблизительно 5%, приблизительно 6%, приблизительно 7%, приблизительно 8%, приблизительно 9%, приблизительно 10%, приблизительно 11%, приблизительно 12%, приблизительно 13%, приблизительно 14%, или приблизительно 15%.
В некоторых вариантах осуществления растворитель или смесь растворителей содержит от 45% до 85% пропиленгликоля, от 5% до 45% глицерина и от 0% до 30% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей содержит приблизительно от 45% до приблизительно 85% пропиленгликоля, приблизительно от 5% до приблизительно 45% глицерина, и от 0% до приблизительно 30% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей по существу содержит от 45% до 85% пропиленгликоля, от 5% до 45% глицерина и от 0% до 30% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей по существу содержит приблизительно от 45% до приблизительно 85% пропиленгликоля, приблизительно от 5% до приблизительно 45% глицерина и от 0% до приблизительно 30% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей представляет собой от 45% до 85% пропиленгликоля, от 5% до 45% глицерина и от 0% до 30% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей представляет собой приблизительно от 45% до приблизительно 85% пропиленгликоля, приблизительно от 5% до приблизительно 45% глицерина и от 0% до приблизительно 30% этанола.
В некоторых вариантах осуществления растворитель или смесь растворителей содержит от 55% до 75% пропиленгликоля, от 15% до 35% глицерина и от 0% до 20% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей содержит приблизительно от 55% до приблизительно 75% пропиленгликоля, приблизительно от 15% до приблизительно 35% глицерина и от 0% до приблизительно 20% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей по существу содержит от 55% до 75% пропиленгликоля, от 15% до 35% глицерина и от 0% до 20% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей включает в себя приблизительно от 55% до приблизительно 75% пропиленгликоля, приблизительно от 15% до приблизительно 35% глицерина и от 0% до приблизительно 20% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей представляет собой от 55% до 75% пропиленгликоля, от 15% до 35% глицерина и от 0% до 20% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей представляет собой приблизительно от 55% до приблизительно 75% пропиленгликоля, приблизительно от 15% до приблизительно 35% глицерина и от 0% до приблизительно 20% этанола.
В некоторых вариантах осуществления растворитель или смесь растворителей содержит от 60% до 70% пропиленгликоля, от 20% до 30% глицерина и от 5% до 15% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей содержит приблизительно от 60% до приблизительно 70% пропиленгликоля, приблизительно от 20% до приблизительно 30% глицерина и приблизительно от 5% до приблизительно 15% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей по существу содержит от 60% до 70% пропиленгликоля, от 20% до 30% глицерина и от 5% до 15% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей по существу содержит приблизительно от 60% до приблизительно 70% пропиленгликоля, приблизительно от 20% до приблизительно 30% глицерина и приблизительно от 5% до приблизительно 15% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей представляет собой 60% до 70% пропиленгликоля, от 20% до 30% глицерина и от 5% до 15% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей представляет собой приблизительно от 60% до приблизительно 70% пропиленгликоля, приблизительно от 20% до приблизительно 30% глицерина и приблизительно от 5% до приблизительно 15% этанола.
В некоторых вариантах осуществления растворитель или смесь растворителей содержит 65% пропиленгликоля, 25% глицерина и 10% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей содержит приблизительно 65% пропиленгликоля, приблизительно 25% глицерина и приблизительно 10% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей по существу содержит 65% пропиленгликоля, 25% глицерина и 10% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей по существу содержит приблизительно 65% пропиленгликоля, приблизительно 25% глицерина и приблизительно 10% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей представляет собой 65% пропиленгликоля, 25% глицерина и 10% этанола. В некоторых вариантах осуществления растворитель или смесь растворителей представляет собой приблизительно 65% пропиленгликоля, приблизительно 25% глицерина и приблизительно 10% этанола.
Эксципиенты
Фармацевтическая композиция по изобретению может быть комбинацией любых фармацевтических соединений, описываемых в настоящем документе, с другими химическими компонентами, такими как носители, стабилизаторы, разбавители, диспергирующие средства, суспендирующие средства, загущающие вещества и/или эксципиенты. Фармацевтическая композиция облегчает введение соединения в организм. Фармацевтические композиции можно вводить в терапевтически эффективных количествах в виде фармацевтических композиций при помощи различных форм и путей введения, включая, например, внутривенное, подкожное, внутримышечное, пероральное, ректальное, аэрозольное, парентеральное, офтальмологическое, легочное, трансдермальное, вагинальное, ушное, назальное и местное введение.
Фармацевтическую композицию можно вводить местным или системным способом, например, путем инъекции соединения непосредственно в орган, необязательно в виде депо-составов или составов с пролонгированным высвобождением. Фармацевтические композиции можно предоставлять в форме состава с быстрым высвобождением, в форме состава с длительным высвобождением, или в форме состава с промежуточным высвобождением. Форма с быстрым высвобождением может обеспечивать немедленное высвобождение. Состав с длительным высвобождением может обеспечивать контролируемое высвобождение или замедленное отсроченное высвобождение.
Для перорального введения, фармацевтические композиции можно легко формулировать, комбинируя активные соединения с фармацевтически приемлемыми носителями или эксципиентами. Такие носители можно использовать для формулирования таблеток, порошков, пилюль, драже, капсул, жидкостей, гелей, сиропов, эликсиров, густых суспензий и суспензий для перорального приема индивидуумом.
Фармацевтические препараты для перорального применения можно получать, смешивая один или несколько твердых эксципиентов с одним или несколькими соединениями, описываемыми в настоящем документе, необязательно перемалывая полученную смесь, и обрабатывая смесь гранул после добавления подходящих вспомогательных средств, при желании, для получения сердцевин таблеток или драже. Сердцевины можно обеспечивать подходящими покрытиями. Для этой цели можно использовать концентрированные растворы сахара, которые могут содержать эксципиент, такой как гуммиарабик, тальк, поливинилпирролидон, карбопол гель, полиэтиленгликоль, и/или диоксид титана, растворы лака и подходящие органические растворители или смеси растворителей. К покрытиям таблеток или драже можно добавлять красители или пигменты, например, для идентификации или для того чтобы охарактеризовать различные комбинации доз активного соединения.
Фармацевтические препараты, которые можно использовать перорально включают твердые капсулы, сделанные из желатина, а также мягкие запечатанные капсулы, сделанные из желатина и пластификатора, такого как глицерин или сорбит. В некоторых вариантах осуществления капсула включает твердую желатиновую капсулу, содержащую один или несколько из фармацевтического, бычьего и растительного желатинов. Желатин может подвергаться щелочной обработке. Твердые капсулы могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, связывающие средства, такие как крахмалы, и/или смазочные средства, такие как тальк или стеарат магния, и стабилизаторы. В мягких капсулах, активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как жирные масла, парафиновое масло, или жидкие полиэтиленгликоли. Можно добавлять стабилизаторы. Все составы для перорального введение предлагаются в дозировках, подходящих для такого введения.
Для буккального или сублингвального введения композиции могут представлять собой таблетки, таблетки-леденцы или гели.
Парентеральные инъекции можно формулировать для болюсной инъекции или непрерывного вливания. Фармацевтические композиции могут быть в форме, подходящей для парентеральной инъекции в виде стерильной суспензии, раствора или эмульсии в масляных или водных носителях, и могут содержать вспомогательные вещества, такие как суспендирующие, стабилизирующие и/или диспергирующие средства. Фармацевтические составы для парентерального введения включают водные растворы активных соединений в водорастворимой форме. Суспензии активных соединений можно получать в виде масляных суспензий для инъекций. Подходящие липофильные растворители или носители включают жирные масла, такие как сезамовое масло, или синтетические сложные эфиры жирных кислот, такие как этилолеат или триглицериды, или липосомы. Водные суспензии для инъекций могут содержать вещества, которые повышают вязкость суспензии, такие как карбоксиметилцеллюлоза натрия, сорбит или декстран. Суспензия может также содержать подходящие стабилизаторы или вещества, которые повышают растворимость соединений для того чтобы получать высоко концентрированные растворы. Альтернативно, активный ингредиент может быть в форме порошка для разбавления подходящим носителем перед использованием, например, стерильной апирогенной водой, 0,9% физиологическим раствором или 5% декстрозой в воде.
Активные соединения можно вводить местно и можно формулировать в ряд композиций, которые можно вводить местно, таких как растворы, суспензии, лосьоны, гели, пасты, медицинские карандаши, бальзамы, кремы и мази. Такие фармацевтические композиции могут содержать солюбилизаторы, стабилизаторы, средства, повышающие тоничность, буферы и консерванты.
Составы, подходящие для трансдермального введения активных соединений, могут использовать трансдермальные средства доставки и пластыри для трансдермальной доставки, могут представлять собой липофильные эмульсии или забуференные водные растворы, растворенные и/или диспергированные в полимере или адгезиве. Такие пластыри могут быть сконструированы для непрерывной, пульсирующей доставки фармацевтических соединений или доставки по мере необходимости. Трансдермальную доставку можно выполнять при помощи ионофоретических пластырей. Дополнительно, трансдермальные пластыри могут обеспечивать контролируемую доставку. Скорость абсорбции можно замедлять при использовании мембран, контролирующих скорость или путем захвата соединения внутри полимерной матрицы или геля. Напротив, можно использовать усилители абсорбции для повышения абсорбции. Усилитель абсорбции или носитель могут включать абсорбирующиеся фармацевтически приемлемые растворители для облегчения прохождения через кожу. Например, трансдермальные устройства могут быть в форме повязки, содержащей опорный элемент, резервуар, содержащий соединения и носители, барьер, контролирующий скорость для доставки соединений к коже индивидуума с контролируемой и предопределенной скоростью в течение длительного периода времени, и адгезивы для закрепления устройства на коже или глазу.
Для введения путем ингаляции активные соединения могут быть в форме аэрозоля, аэрозольного тумана или порошка. Фармацевтические композиции удобно доставлять в форме аэрозольного спрея из упаковок под давлением или небулайзера, с использованием подходящего пропеллента, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа. В случае аэрозоля под давлением, единицу дозирования можно определять путем обеспечения клапана для доставки дозированного количества. Капсулы и картриджи, например, желатиновые, для применения в ингаляторе или инсуфляторе можно формулировать с содержанием порошка из смеси соединений и подходящей порошковой основы, такой как лактоза или крахмал.
Соединения можно также формулировать в виде ректальных композиций, таких как клизма, ректальные гели, ректальные пены, ректальные аэрозоли, суппозитории, гелевые суппозитории, или клизма с удержанием, содержащих общепринятые основы для суппозиториев, такие как масло какао или другие глицериды, а также синтетические полимеры, такие как поливинилпирролидон и ПЭГ. В композициях в форме суппозиториев можно использовать низкоплавкий воск, такой как смесь глицеридов жирных кислот или масло какао.
В практических способах лечения или применении, предлагаемых в настоящем документе, терапевтически эффективные количества соединений, описываемых в настоящем документе, вводят в фармацевтических композициях индивидууму с заболеванием или состоянием, подлежащим лечению. В некоторых вариантах осуществления индивидуум представляет собой млекопитающего, такого как человек. Терапевтически эффективное количество может широко варьировать в зависимости от тяжести заболевания, возраста и относительного здоровья индивидуума, мощности применяемых соединений и других факторов. Соединения можно использовать по отдельности или в комбинации с одним или несколькими терапевтическими средствами в виде компонентов смесей.
Фармацевтические композиции можно формулировать с использованием одиного или нескольких физиологически приемлемых носителей, содержащих эксципиенты и вспомогательные средства, которые облегчают обработку активных соединений в препараты, которые можно использовать фармацевтически. Состав можно модифицировать в зависимости от выбранного пути введения. Фармацевтические композиции, содержащие соединения, описываемые в настоящем документе, можно производить, например, способами смешивания, растворения, гранулирования, производства драже, растирания в порошок, эмульгирования, инкапсулирования, захватывания или прессования.
Фармацевтические композиции могут включать, по меньшей мере, один фармацевтически приемлемый носитель, разбавитель, или эксципиент и соединения, описываемые в настоящем документе, в форме свободного основания или фармацевтически приемлемой соли. Способы и фармацевтические композиции, описываемые в настоящем документе, включают использование кристаллических форм (также известных, как полиморфы), и активных метаболитов этих соединений с тем же самым типом активности.
Способы получения композиций, содержащих соединения, описываемые в настоящем документе, включают формулирование соединений с одним или несколькими инертными фармацевтически приемлемыми эксципиентами или носителями для образования твердой, полутвердой, или жидкой композиции. Твердые композиции включают, например, порошки, таблетки, диспергируемые гранулы, капсулы, облатки и суппозитории. Жидкие композиции включают, например, растворы, в которых растворено соединение, эмульсии, содержащие соединение, или раствор, содержащий липосомы, мицеллы или наночастицы, содержащие соединение, как описано в настоящем документе. Полутвердые композиции включают, например, гели, суспензии и кремы. Композиции могут быть в жидких растворах или суспензиях, твердых формах, подходящих для раствора или суспензии в жидкости перед использованием, или в виде эмульсий. Эти композиции могут также содержать незначительные количества нетоксических вспомогательных веществ, таких как увлажнители или эмульгаторы, средства для буферирования pH и другие фармацевтически приемлемые добавки.
Неограничивающие примеры лекарственных форм, подходящих для использования по изобретению, включают питание, еду, пеллет, таблетку-леденец, жидкий препарат, эликсир, аэрозоль, форму для ингаляции, спрей, порошок, таблетку, пилюлю, капсулу, гель, гелевую таблетку, наносуспензию, наночастицу, микрогель, суппозиторий пастилки, водные или масляные суспензии, мазь, пластырь, лосьон, препарат для чистки зубов, эмульсию, кремы, капли, диспергируемые порошки или гранулы, эмульсию в твердых или мягких гелевых капсулах, сиропы, фитоцевтики, нутрицевтики и их любое сочетание.
Неограничивающие примеры фармацевтически приемлемых эксципиентов, подходящих для использования по изобретению, включают гранулирующие средства, связывающие средства, смазки, дезинтегрирующие средства, подсластители, способствующие скольжению средства, антиадгезивные средства, антистатические средства, поверхностно-активные вещества, антиоксиданты, камеди, покрывающие средства, красители, ароматизаторы, покрывающие средства, пластификаторы, консерванты, суспендирующие средства, эмульгаторы, противомикробные средства, целлюлозный материал растений и средства для окатывания, и их любое сочетание.
Композиция по изобретению может быть, например, формой с немедленным высвобождением или составом с контролируемым высвобождением. Состав с немедленным высвобождением можно формулировать для того чтобы позволить соединениям действовать быстро. Неограничивающие примеры составов с немедленным высвобождением включают быстрорастворимые составы. Состав с контролируемым высвобождением может быть фармацевтическим составом, который был адаптирован таким образом, что скорости высвобождения и профили высвобождения лекарственного средства могут соответствовать физиологическим и хронотерапевтическим требованиям, или, альтернативно, был сформулирован для эффективного высвобождения лекарственного средства с запрограммированной скоростью. Неограничивающие примеры составов с контролируемым высвобождением включают гранулы, гранулы с отсроченным высвобождением, гидрогели (например, синтетического или природного происхождения), другие гелеобразующие средства (например, гелеобразующие пищевые волокна), составы на основе матрицы (например, составы, содержащие полимерный материал, по меньшей мере, с одним активным ингредиентом, диспергированным в нем), гранулы внутри матрицы, полимерные смеси, и гранулярные массы.
Описанные композиции необязательно могут включать приблизительно от 0,001% до приблизительно 0,005% объемной массы фармацевтически приемлемых консервантов. Одним из неограничивающих примеров подходящего консерванта является бензиловый спирт.
В некоторых, состав с контролируемым высвобождением представляет собой форму с отсроченным высвобождением. Форму с отсроченным высвобождением можно формулировать для отсрочки действия соединения в течение длительного периода времени. Форму с отсроченным высвобождением можно формулировать для отсрочки высвобождения эффективной дозы одного или нескольких соединений, например, в течение приблизительно 4, приблизительно 8, приблизительно 12, приблизительно 16, или приблизительно 24 часов.
Состав с контролируемым высвобождением может быть в форме с пролонгированным высвобождением. Форму с пролонгированным высвобождением можно формулировать для поддержания, например, действия соединения в течение длительного периода времени. Форму с пролонгированным высвобождением можно формулировать для того, чтобы обеспечить эффективную дозу любого соединения, описываемого в настоящем документе (например, обеспечить физиологически эффективный профиль в крови) в течение приблизительно 4, приблизительно 8, приблизительно 12, приблизительно 16 или приблизительно 24 часов.
Неограничивающие примеры фармацевтически приемлемых эксципиентов можно найти, например, в Remington: The Science and Practice of Pharmacy, Nineteenth Ed (Easton, Pa.: Mack Publishing Company, 1995); Hoover, John E., Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pennsylvania 1975; Liberman, H.A. and Lachman, L., Eds., Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980; и Pharmaceutical Dosage Forms and Drug Delivery Systems, Seventh Ed. (Lippincott Williams & Wilkins1999), каждая из которых включена в качестве ссылки в полном объеме.
Описанные способы включают введение динуклеотидного производного децитабина или его фармацевтически приемлемой соли, в комбинации с фармацевтически приемлемым носителем. Носитель можно выбирать для того чтобы минимизировать любую деградацию активного ингредиента и чтобы минимизировать любые нежелательные побочные эффекты у индивидуума.
Соединение формулы (I) или его фармацевтически приемлемую соль в настоящем документе можно удобно формулировать в фармацевтические композиции, включающие один или несколько фармацевтически приемлемых носителей. См. например, Remington's Pharmaceutical Sciences, последнее издание, E.W. Martin Mack Pub. Co., Easton, PA, включенное в настоящий документ в качестве ссылки, которое описывает типичные носители и общепринятые способы получения фармацевтических композиций, которые можно использовать в сочетании с получением составов соединения, описываемого в настоящем документе. Такие фармацевтические средства могут быть стандартными носителями для введения композиций людям и являющимся людьми индивидуумам, включая растворы, такие как физиологический раствор и забуференные растворы с физиологическим pH. Другие композиции можно вводить в соответствии со стандартными процедурами. Например, фармацевтические композиции могут также включать один или несколько дополнительных активных ингредиентов, таких как противомикробные средства, противовоспалительные средства и анестетики.
Неограничивающие примеры фармацевтически приемлемых носителей в качестве неограничивающих примеров включают физиологический раствор, раствор Рингера и раствор декстрозы. pH раствора может составлять приблизительно от 5 до приблизительно 8, и может составлять приблизительно от 7 до приблизительно 7,5. Дополнительные носители включают препараты с пролонгированным высвобождением, такие как полупроницаемые матрицы твердых гидрофобных полимеров, содержащие соединение формулы (I) или его фармацевтически приемлемую соль, при этом матрицы находятся в форме изделий с определенной формой, например, пленки, липосомы, микрочастицы или микрокапсулы.
Описанные способы относятся к введению соединения формулы (I) или его фармацевтически приемлемой соли как части фармацевтической композиции. В различных вариантах осуществления композиции по изобретению могут включать жидкость, содержащую активное средство в растворе, в суспензии, или и в растворе, и в суспензии. Жидкие композиции могут включать гели. В одном из вариантов осуществления жидкая композиция является водной. Альтернативно, композиция может принимать форму мази. В другом варианте осуществления композиция представляет собой водную композицию, образующую гель in situ. В некоторых вариантах осуществления композиция представляет собой водный раствор, образующий гель in situ.
Фармацевтические составы могут включать дополнительные носители, а также загустители, разбавители, буферы, консерванты, и поверхностно-активные средства в дополнение к соединениям, описываемым в настоящем документе. Фармацевтические составы могут также включать один или несколько дополнительных активных ингредиентов, таких как противомикробные средства, противовоспалительные средства и анестетики.
Эксципиент может выполнять как простую и прямую роль, являясь инертным наполнителем, или эксципиент, как применяют в настоящем документе, может быть частью системы, стабилизирующей pH, или покрытия, чтобы обеспечить безопасную доставку ингредиентов в желудок.
Соединение формулы (I) или его фармацевтически приемлемая соль могут также присутствовать в жидкостях, эмульсиях, или суспензиях для доставки активных терапевтических средств в аэрозольной форме в полости организма, такие как нос, глотка или бронхиальные пути. Соотношение соединения формулы (I) или его фармацевтически приемлемой соли с другими составляющими в этих препаратах может варьироваться в зависимости от лекарственной формы.
В зависимости от предполагаемого способа введения, фармацевтические композиции, вводимые как часть описанных способов, могут быть в форме твердых, полутвердых или жидких лекарственных форм, таких как, например, таблетки, суппозитории, пилюли, капсулы, порошки, жидкости, суспензии, лосьоны, кремы, гели, или т.п., например, в стандартной лекарственной форме, подходящей для однократного введения точной дозы. Композиции могут содержать, как указано выше, эффективное количество соединения формулы (I) или его фармацевтически приемлемой в комбинации с фармацевтически приемлемым носителем и, кроме того, могут включать другие медицинские средства, фармацевтические средства, носители, адъюванты, разбавители, и т.д.
Для твердых композиций не токсичные твердые носители включают, например, маннит, лактозу, крахмал, стеарат магния, сахаринат натрия, тальк, целлюлозу, глюкозу, сахарозу и карбонат магния фармацевтической степени чистоты.
Фармацевтически приемлемые соли
Соединение формулы (1) и его фармацевтически приемлемые соли можно получать способами, описанными в WO2013033176, и как описано ниже в примерах.
В каждом из указанных выше аспектов и вариантов осуществления изобретения соединение формулы (1) можно использовать в форме соли или не-соли.
Фармацевтически приемлемые соли включают, например, соли присоединения кислоты и соли присоединения основания. Кислота, которую добавляют к соединению для образования соли присоединения кислоты, может быть органической кислотой или неорганической кислотой. Основание, которое добавляют к соединению для образования соли присоединения основания, может быть органическим основанием или неорганическим основанием. В некоторых вариантах осуществления фармацевтически приемлемая соль представляет собой соль металла. В некоторых вариантах осуществления фармацевтически приемлемая соль представляет собой аммонийную соль.
Соли присоединения кислот могут возникать при добавлении кислоты к соединению, описываемому в настоящем документе. В некоторых вариантах осуществления кислота является органической. В некоторых вариантах осуществления кислота является неорганической. Неограничивающие примеры подходящих кислот включают соляную кислоту, бромистоводородную кислоту, йодистоводородную кислоту, азотную кислоту, азотистую кислоту, серную кислоту, сернистую кислоту, фосфорную кислоту, никотиновую кислоту, изоникотиновую кислоту, молочную кислоту, салициловую кислоту, 4-аминосалициловую кислоту, винную кислоту, аскорбиновую кислоту, гентизиновую кислоту, глюконовую кислоту, глюкуроновую кислоту, сахаристую кислоту, муравьиную кислоту, бензойную кислоту, глутаминовую кислоту, пантотеновую кислоту, уксусную кислоту, пропионовую кислоту, масляную кислоту, фумаровую кислоту, янтарную кислоту, лимонную кислоту, щавелевую кислоту, малеиновую кислоту, гидроксималеиновую кислоту, метилмалеиновую кислоту, гликолевую кислоту, яблочную кислоту, коричную кислоту, миндальную кислоту, 2-феноксибензойную кислоту, 2-ацетоксибензойную кислоту, эмбоновую кислоту, фенилуксусную кислоту, N-циклогексилсульфаминовую кислоту, метансульфоновую кислоту, этансульфоновую кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту, 2-гидроксиэтансульфоновую кислоту, этан-1,2-дисульфоновую кислоту, 4-метилбензолсульфоновую кислоту, нафталин-2-сульфоновую кислоту, нафталин-1,5-дисульфоновую кислоту, 2-фосфоглицериновую кислоту, 3-фосфоглицериновую кислоту, глюкоза-6-фосфорную кислоту и аминокислоту.
Неограничивающие примеры подходящих солей присоединения кислот включают гидрохлоридную соль, гидробромидную соль, йодогидратную соль, нитратную соль, нитритную соль, сульфатную соль, сульфитную соль, фосфатную соль, водородфосфатную соль, диводородфосфатную соль, карбонатную соль, бикарбонатную соль, никотинатную соль, изоникотинатную соль, соль молочной кислоты, салицилатную соль, 4-аминосалицилатную соль, соль винной кислоты, аскорбатную соль, гентизинатную соль, глюконатную соль, глюкуронатную соль, сахаристую соль, формиатную соль, бензоатную соль, глутаминатную соль, пантотенатную соль, ацетатную соль, пропионатную соль, бутиратную соль, соль фумаровой кислоты, соль янтарной кислоты, соль лимонной кислоты, оксалатную соль, соль малеиновой кислоты, гидроксисоль малеиновой кислоты, метилсоль малеиновой кислоты, гликолатную соль, соль яблочной кислоты, соль коричной кислоты, соль миндальной кислоты, 2-феноксибензоатную соль, 2-ацетоксибензоатную соль, эмбонатную соль, фенилацетатную соль, N-циклогексилсульфаматную соль, метансульфонатную соль, этансульфонатную соль, бензолсульфонатную соль, п-толуолсульфонатную соль, 2-гидроксиэтансульфонатную соль, этан-1,2-дисульфонатную соль, 4-метилбензолсульфонатную соль, нафталин-2-сульфонатную соль, нафталин-1,5-дисульфонатную соль, 2-фосфоглицератную соль, 3-фосфоглицератную соль, глюкоза-6-фосфатную соль и аминокислую соль.
Соли металлов могут возникать при добавлении неорганического основания к соединению, описываемому в настоящем документе. Неорганическое основание состоит из катиона металла, спаренного с основным противоионом, таким как, например, гидроксид, карбонат, бикарбонат, или фосфат. Металл может быть щелочным металлом, щелочноземельным металлом, переходным металлом или металлом основной группы. Неограничивающие примеры подходящих металлов включают литий, натрий, калий, цезий, церий, магний, марганец, железо, кальций, стронций, кобальт, титан, алюминий, медь, кадмий и цинк.
Неограничивающие примеры подходящих солей металлов включают литиевую соль, натриевую соль, калиевую соль, соль цезия, соль церия, магниевую соль, соль марганца, соль железа, кальциевую соль, соль стронция, кобальтовую соль, титановую соль, соль алюминия, медную соль, кадмиевую соль и цинковую соль.
Аммонийные соли могут возникать при добавлении аммиака или органического амина к соединению, описываемому в настоящем документе. Неограничивающие примеры подходящих органических аминов включают триэтиламин, диизопропиламин, этаноламин, диэтаноламин, триэтаноламин, морфолин, N-метилморфолин, пиперидин, N-метилпиперидин, N-этилпиперидин, дибензиламин, пиперазин, пиридин, пиразол, пипиразол, имидазол, пиразин, пипиразин, этилендиамин, N,N'-дибензилэтилендиамин, прокаин, хлорпрокаин, холин, дициклогексиламин, и N-метилглюкамин.
Неограничивающие примеры подходящих аммонийных солей включают триэтиламиновую соль, диизопропиламиновую соль, этаноламиновую соль, диэтаноламиновую соль, триэтаноламиновую соль, морфолиновую соль, N-метилморфолиновую соль, пиперидиновую соль, N-метилпиперидиновую соль, N-этилпиперидиновую соль, дибензиламиновую соль, пиперазиновую соль, пиридиновую соль, пиразолиновую соль, пипиразолиновую соль, имидазольную соль, пиразиновую соль, пипиразиновую соль, этилендиаминовую соль, N,N'-дибензилэтилендиаминовую соль, прокаиновую соль, хлорпрокаиновую соль, холиновую соль, дициклогексиламиновую соль и N-метилглюкаминовую соль.
Одним из конкретных примеров соли соединения формулы (1) является натриевая соль.
Применение в терапевтических целях
Лиофилизированные фармацевтические композиции по настоящему изобретению можно использовать для лечения широкого спектра заболеваний, которые чувствительны к лечению децитабином, в том числе заболеваний, описываемых в настоящем документе.
Таким образом, в других аспектах, изобретение относится к (i) лиофилизированной фармацевтической композиции, как определено в настоящем документе, для применения в медицине; (ii) лиофилизированной фармацевтической композиции, как определено в настоящем документе, для применения для лечения заболевания, как определено в настоящем документе; (iii) способу лечения заболевания, как определено в настоящем документе, при этом способ включает смешивание лиофилизированной фармацевтической композиции, как определено в настоящем документе, с фармацевтически приемлемым растворителем и введение эффективного количества смеси нуждающемуся в этом индивидууму; (iv) применению лиофилизированной фармацевтической композиции, как определено в настоящем документе, для получения лекарственного средства для лечения заболевания, как определено в настоящем документе; (v) способу лечения злокачественной опухоли у нуждающегося в этом пациента, при этом способ включает восстановление лиофилизированной фармацевтической композиции, как определено в настоящем документе, в фармацевтически приемлемом растворителе для получения жидкого состава, содержащего соединение формулы (1) или его фармацевтически приемлемую соль, и введение пациенту терапевтически эффективного количества жидкого состава.
Примеры заболеваний, которые можно лечить с использованием лиофилизированных фармацевтических композиций по настоящему изобретению, включают заболевания с нежелательной или неконтролируемой клеточной пролиферацией. Такие показания включают доброкачественные опухоли, различные типы злокачественных опухолей, такие как первичные опухоли и опухолевые метастазы, рестеноз (например, коронарные, каротидные и церебральные бляшки), гематологические нарушения, нарушенную стимуляцию эндотелиальных клеток (атеросклероз), повреждения тканей организма при хирургических операциях, нарушенное заживление ран, нарушенный ангиогенез, заболевания, которые вызывают фиброз ткани, туннельные синдромы, нарушения в тканях, которые не являются высоковаскуляризированными, и пролиферативные ответы, связанные с трансплантацией органов.
В основном, клетки в доброкачественной опухоли сохраняют признаки своей дифференцировки и не делятся полностью неконтролируемым образом. Доброкачественная опухоль, как правило, является локализованной и не метастатической. Конкретные типы доброкачественных опухолей, которые можно лечить с использованием настоящего изобретения, включают гемангиомы, гепатоцеллюлярную аденому, кавернозную гемангиому, местную узловую гиперплазию, невромы слухового нерва, нейрофиброму, аденому желчного протока, цистаному желчного протока, фиброму, липомы, лейомиомы, мезотелиомы, тератомы, миксомы, узловую регенеративную гиперплазию, тархомы и пиогенные гранулемы.
В злокачественных опухолях клетки становятся недифференцированными, не отвечают на сигналы организма, контролирующие рост, и размножаются неконтролируемым образом. Злокачественная опухоль является инвазивной и способна распространяться в отдаленные участки (метастазирование). Злокачественные опухоли, в основном, делятся на две категории: первичные и вторичные. Первичные опухоли возникают непосредственно из ткани, в которой они обнаружены. Вторичная опухоль, или метастаз, представляет собой опухоль, которая зародилась в другом месте в организме, но сейчас распространилась в отдаленный орган. Общими путями для метастазирования являются прямой рост в прилежащие структуры, распространение через сосудистую или лимфатическую системы, и прокладывание путей вдоль тканевых плоскостей и пространств организма (перитонеальная жидкость, цереброспинальная жидкость, и т.д.)
Примерами злокачественных опухолей являются карциномы, например, карциномы мочевого пузыря, молочной железы, толстого кишечника, почки, эпидермиса, печени, легкого, пищевода, желчного пузыря, яичника, поджелудочной железы, желудка, шейки матки, щитовидной железы, предстательной железы, желудочно-кишечной системы, или кожи, опухоли кроветворной ткани, такие как лейкоз, B-клеточная лимфома, T-клеточная лимфома, лимфома Ходжкина, неходжкинская лимфома, волосатоклеточная лимфома, или лимфома Беркитта; опухоли кроветворной ткани миелоидного происхождения, например, острые и хронические миелогенные лейкозы, миелодиспластический синдром, или промиелоцитарный лейкоз; фолликулярный рак щитовидной железы; опухоли мезенхимального происхождения, например фибросаркома или рабдомиосаркома; опухоли центральной или периферической нервной системы, например астроцитома, нейробластома, глиома или шваннома; меланома; семинома; тератокарцинома; остеосаркома; пигментная ксеродерма; кератоксантома; фолликулярный рак щитовидной железы; или саркома Капоши.
Конкретные типы злокачественных опухолей или злокачественные опухоли, первичные или вторичные, которые можно лечить с помощью этого изобретения, включают рак мочевого пузыря, рак молочной железы, рак яичников, рак кожи, рак кости, рак предстательной железы, рак печени, рак легких, рак головного мозга, рак гортани, желчного пузыря, поджелудочной железы, прямой кишки, паращитовидной железы, щитовидной железы, надпочечника, нервной ткани, головы и шеи, толстой кишки, желудка, бронхов, почек, базально-клеточную карциному, плоскоклеточную карциному, язвенного и папиллярного типов, метастатическую карциному кожи, остеосаркому, саркому Юинга, саркому ретикулярных клеток, миелому, гигантоклеточную опухоль, мелкоклеточный рак легких, камни в желчном пузыре, опкхоль островковых клеток, первичную опухоль головного мозга, острые и хронические лимфоцитарные и гранулоцитарные опухоли, волосатоклеточную опухоль, аденому, гиперплазию, медуллярную карциному, феохромоцитому, невромы слизистой оболочки, кишечные ганглионевромы, гиперпластическую опухоль роговичного нерва, опухоль при марфаноидном внешнем виде, опухоль Вильма, семиному, опухоль яичников, лейомиому матки, дисплазию шейки матки и карциному in situ, нейробластому, ретинобластому, саркому мягких тканей, злокачественный карциноид, местное повреждение кожи, фунгоидный микоз, рабдомиосаркому, саркому Капоши, остеогенную и другую саркому, злокачественную гиперкальциемию, почечноклеточную опухоль, истинную полицитемию, аденокарциному, мультиформную глиобластому, лейкозы, лимфомы, злокачественные меланомы, эпидермоидные карциномы, и другие карциномы и саркомы.
В одном из вариантов осуществления злокачественная опухоль выбрана из миелодиспластического синдрома, острого миелогенного лейкоза, рака яичников, рака печени и колоректального рака.
Гематологические нарушения включают нарушение роста клеток крови, которое может приводить к диспластическим изменениям клеток крови и гематологическим злокачественным новообразованиям, таким как различные лейкозы. Примеры гематологических нарушений в качестве неограничивающих примеров включают острый миелолейкоз, острый промиелоцитарный лейкоз, острый лимфобластный лейкоз, хронический миелогенный лейкоз, миелодиспластические синдромы и серповидно-клеточную анемию.
Лечение нарушенной клеточной пролиферации, вызванной повреждениями ткани организма во время хирургической операции, возможно для ряда хирургических процедур, включая операцию на суставах, операцию на кишечнике и келоидный рубец. Заболевания, которые производят фиброзную ткань, включают эмфизему.
Туннельные синдромы, которые можно лечить при помощи настоящего изобретения, включают туннельный синдром запястья. Примером нарушений клеточной пролиферации, которые можно лечить при помощи изобретения, является опухоль кости.
Пролиферативные ответы, связанные с трансплантацией органа, которые можно лечить при помощи настоящего изобретения, включают пролиферативные ответы, участвующие в потенциальном отторжении органа или в связанных с ним осложнениях. Конкретно, эти пролиферативные ответы могут происходить во время пересадки сердца, легкого, печени, почки, и других органов тела или органных систем.
Нарушения ангиогенеза, которые можно лечить при помощи настоящего изобретения, включают патологический ангиогенез, сопровождающий ревматоидный артрит, ишемическую реперфузию, связанную с отеком и повреждением головного мозга, кортикальную ишемию, гиперплазию и гиперваскуляризацию яичников (синдром поликистозного яичника), эндометриоз, псориаз, диабетическую ретинопатию, и другие ангиогенные заболевания глаз, такие как ретинопатию недоношенных (ретролентальную фиброплазию), мышечную дистрофию, отторжение трансплантата роговицы, нейрососудистую глаукому и синдром Ослера-Вебера.
Заболевания, связанные с патологическим ангиогенезом, требуют роста сосудов или стимулируют рост сосудов. Например, роговичный ангиогенез включает три фазы: преваскулярный латентный период, активную неоваскуляризацию и созревание и регрессию сосудов. Природу и механизм различных ангиогенных факторов, в том числе элементов воспалительного ответа, таких как лейкоциты, тромбоциты, цитокины, и эйкозаноиды, или неустановленные составляющие плазмы, еще предстоит выявить.
В некоторых вариантах осуществления лиофилизированные фармацевтические композиции по настоящему изобретению можно использовать для лечения заболеваний, связанных с нежелательным или патологическим ангиогенезом. Способ включает введение пациенту, страдающему от нежелательного или патологического ангиогенеза, фармацевтических составов по настоящему изобретению отдельно или в комбинации с противоопухолевым средством, активность которого как противоопухолевого средства in vivo нежелательно нарушена высокими уровнями метилирования ДНК. Конкретные дозы этих средств, необходимые для ингибирования ангиогенеза и/или ангиогенных заболеваний, могут зависеть от тяжести состояния, пути введения, и сопутствующих факторов, которые может определить лечащий врач. В основном, общепризнанные и эффективные суточные дозы представляют собой количество, достаточное для эффективного ингибирования ангиогенеза и/или ангиогенных заболеваний.
Лиофилизированные фармацевтические композиции по настоящему изобретению можно использовать для лечения ряда заболеваний, связанных с нежелательным ангиогенезом, таких как ретинальная/хориоидальная неоваскуляризация неоваскуляризация роговицы. Примеры ретинальной/хориоидальной неоваскуляризации в качестве неограничивающих примеров включают дистрофию Беста, миопию, ямки диска зрительного нерва, болезнь Штаргардта, болезнь Педжета, закупорку вен, закупорку артерий, серповидно-клеточную анемию, саркоид, сифилис, эластическую псевдоксантому обструктивные заболевания сонной артерии, хронический увеит/витрит, микобактериальные инфекции, болезнь Лайма, системную красную волчанку, ретинопатию недоношенных, болезнь Илза, диабетическую ретинопатию, дегенерацию желтого пятна, болезнь Бекета, инфекции, вызванные ретинитом или хориоидитом, синдром предполагаемого гистоплазмоза глаз, парспланит, хроническую отслойку сетчатки, синдромы повышенной вязкости крови, токсоплазмоз, травму и осложнения после лазера, заболевания, связанные с рубеозом (неоваскуляризация угла) и заболевания, вызванные патологической пролиферацией фиброваскулярной или виброзной ткани, включая все формы пролиферативной витреоретинопатии. Примеры неоваскуляризации роговицы в качестве неограничивающих примеров включают эпидемический кератоконъюнктивит, недостаток витамина A, длительное ношение контактных линз, атопический кератит, верхний лимбальный кератит, сухой кератит птеригиума, синдром Шегрена, красные угри, филектенулез, диабетическую ретинопатию, ретинопатию недоношенных, отторжение трансплантата роговицы, язву Мурена, краевую дегенерацию Терриена, краевой кератолиз, полиартериит, саркоидоз Вегенера, склерит, периферическую радиальную кератотомию, неоваскулярную глаукому и ретролентальную фиброплазию, сифилис, микобактериальные инфекции, липидную дистрофию, химические ожоги, бактериальные язвы, грибковые язвы, инфекции, связанные с вирусом простого герпеса, инфекции, связанные с вирусом опоясывающего герпеса, инфекции, связанные с простейшими, и саркому Капоши.
В некоторых вариантах осуществления лиофилизированные фармацевтические композиции по настоящему изобретению можно использовать для лечения хронических воспалительных заболеваний, связанных с патологическим ангиогенезом. Способ включает введение пациенту, страдающемц от хронического воспалительного заболевания, связаннного с патологическим ангиогенезом, фармацевтических составов по настоящему изобретению отдельно или в комбинации с противоопухолевым средством, активность которого как противоопухолевого средства in vivo нежелательно нарушена высокими уровнями метилирования ДНК. Хроническое воспаление зависит от непрерывного образования капиллярных отростков для поддержания притока воспалительных клеток. Приток и присутствие воспалительных клеток продуцируют гранулемы и, таким образом, поддерживают хроническое воспалительное состояние. Ингибирование ангиогенеза при помощи фармацевтических составов по настоящему изобретению может предотвращать возникновение гранулем, таким образом, облегчая заболевание. Примеры хронического воспалительного заболевания в качестве неограничивающих примеров включают воспалительные заболевания кишечника, такие как болезнь Крона и язвенный колит, псориаз, саркоидоз и ревматоидный артрит.
Воспалительные заболевания кишечника, такие как болезнь Крона и язвенный колит, характеризуются хроническим воспалением и ангиогенезом в различных участках желудочно-кишечного тракта. Например, болезнь Крона встречается как хроническое трансмуральное воспалительное заболевание, которое наиболее часто поражает дистальный отлед подвздошной кишки и толстую кишку, но может также встречаться в любой части желудочно-кишечного тракта от рта до ануса и перианальной области. Пациенты с болезнью Крона, как правило, имеют хроническую диарею, связанную с болью в животе, лихорадку, анорексию, потерю веса и вздутие живота. Язвенный колит также представляет собой хроническое неспецифическое воспалтельное и язвенное заболевание, возникающее в слизистом слое толстой кишки, которое характеризуется кровавой диареей. Эти воспалительные заболевания кишечника, в основном, вызваны хроническим гранулематозным воспалением на всем протяжении желудочно-кишечного тракта, включающим прорастание новых капилляров, окруженных цилиндром из воспалительных клеток. Ингибирование ангиогенеза при помощи фармацевтических составов по настоящему изобретению должно препятствовать образованию прорастания и предотвращать образование гранулем. Воспалительные заболевания кишечника также демонстрируют внекишечные проявления, такие как поражения кожи. Такие поражения характеризуются воспалением и ангиогенезом и могут происходить на многих участках вне желудочно-кишечного тракта. Ингибирование ангиогенеза при помощи лиофилизированных фармацевтических композиций по настоящему изобретению должно снижать приток воспалительных клеток и предотвращать образование поражений.
Саркоидоз, еще одно хроническое воспалительное заболевание, характеризуется как мультисистемное гранулематозное нарушение. Гранулемы этого заболевания могут формироваться везде в организме, и, таким образом, симптомы зависят от расположения гранулем и от того, является ли заболевание активным. Гранулемы происходят от ангиогенного прорастания капилляров, обеспечивающих постоянное питание воспалительных клеток. При помощи лиофилизированных фармацевтических композиций по настоящему изобретению для ингибирования ангиогенеза, можно ингибировать образование таких гранулем. Псориаз, также хроническое и рецидивирующее воспалительное заболевание, is характеризуется папулами и бляшками различных размеров. Лечение с использованием фармацевтических составов по настоящему изобретению может уменьшать вероятность образования новых кровеносных сосудов, необходимых для поддержания характерных поражений, и обеспечивать пациенту облегчение симптомов.
Ревматоидный артрит (РА) также представляет собой хроническое воспалительное заболевание, которое характеризуется неспецифическим воспалением периферических суставов. Полагают, что кровеносные сосуды в синовиальной выстилке суставов подвергаются ангиогенезу. В дополнение к формированию новых сосудистых сетей, эндотелиальные клетки высвобождают факторы и активные формы кислорода, которые приводят к росту паннуса и разрушению хряща. Факторы, вовлеченные в ангиогенез, могут вносить активный вклад в состояние хронического воспаления при ревматоидном артрите и помогать поддерживать его. Лечение при помощи фармацевтических составов по настоящему изобретению по отдельности или в сочетании с другими средствами против РА может уменьшать вероятность образования новых кровеносных сосудов, необходимых для поддержания хронического воспаления и обеспечивать пациенту с РА облегчение симптомов.
В некоторых вариантах осуществления лиофилизированные фармацевтические композиции по настоящему изобретению можно использовать для лечения заболеваний, связанных с нарушенным синтезом гемоглобина. Способ включает введение фармацевтических составов по настоящему изобретению пациенту, страдающему от заболевания, связанного с нарушенным синтезом гемоглобина. Составы, содержащие децитабин, стимулируют синтез фетального гемоглобина, поскольку механизм включения в ДНК связан с гипометилированием ДНК. Примеры заболеваний, связанных с нарушенным синтезом гемоглобина, в качестве неограничивающих примеров включают серповидно-клеточную анемию и β-талассемию.
В некоторых вариантах осуществления лиофилизированные фармацевтические композиции по настоящему изобретению можно использовать для контроля внутриклеточной экспрессии гена. Способ включает введение фармацевтических составов по настоящему изобретению пациенту, страдающему заболеванием, связанным с патологическими уровнями экспрессии гена. Метилирование ДНК связано с контролем экспрессии гена. Конкретно, метилирование в промоторах или рядом с ними ингибирует транскрипции, в то время как деметилирование восстанавливает экспрессию. Примеры возможных применений описанных механизмов в качестве неограничивающих примеров включают терапевтически изменненное ингибирование роста, индукцию апоптоза и клеточную дифференцировку.
В некоторых вариантах осуществления лиофилизированные фармацевтические композиции по изобретению можно использовать для лечения пациентов с генетическими мутациями, связанными с опухолевым гиперметилированием, таких как пациенты с типами опухолей, которые содержат мутацию или недостаток сукцинат дегидрогеназы (СДГ), и включают пациентов с не-KIT мутированными желудочно-кишечными стромальными опухолями (GIST).
Облегчение активации генов при помощи лиофилизированных фармацевтических композиций по настоящему изобретению может вызывать дифференцировку клеток для терапевтических целей. Клеточная дифференцировка индуцируется через механизм гипометилирования. Примеры морфологической и функциональной дифференцировки в качестве неограничивающих примеров включают дифференцировку в направлении формирования мышечных клеток, мышечных трубочек, клеток эритроидного и лимфоидного ростка.
Миелодиспластические синдромы (МДС) представляют собой гетерогенные нарушения в клональных гематопоэтических стволовых клетках, связанные с наличием диспластических изменений в одном или нескольких гематопоэтических ростков, в том числе диспластических изменений в миелоидном, эритроидном и мегакариотическом рядах. Эти изменения приводят к цитопении в одном или нескольких из трех ростков. У индивидуумов, страдающих от МДС, как правило, развиваются осложнения, связанные с анемией, нейтропенией (инфекции) или тромбоцитопенией (кровотечение). В основном, приблизительно от 10% до приблизительно 70% индивидуумов с МДС заболевают острым лейкозом. Типичные миелодиспластические синдромы включают острый миелолейкоз, острый промиелоцитарный лейкоз, острый лимфобластный лейкоз и хронический миелогенный лейкоз.
Острый миелолейкоз (ОМЛ) представляет собой наиболее распространенный тип острого лейкоза у взрослых. Некоторые наследственные генетические нарушения и иммунодефицитные состояния ассоциированы с повышенным риском ОМЛ. Они включают нарушения с дефектами стабильности ДНК, приводящими к случайному хромосомному разрыву, такие как синдром Блума, анемия Фанкони, родословная с синдромом Ли-Фраумени, атаксия-телеангиэктазия и X-сцепленная агаммаглобулинемия.
Острый промиелоцитарный лейкоз (ОПМЛ) представляет собой отдельный подтип ОМЛ. Этот подтип характеризуется промиелоцитарными бластами, содержащими хромосомную транслокацию 15;17. Это транслокация приводит к производству слитого транскрипта, содержащего последовательность рецептора ретиноевой кислоты и последовательность промиелоцитарного лейкоза.
Острый лимфобластный лейкоз (ОЛЛ) представляет собой гетерогенное заболевание с различными подтипами, которые демонстрируют различные клинические особенности. При ОЛЛ были продемонстрированы повторяющиеся цитогенетические аномалии. Наиболее распространенной сопутствующей цитогенетической аномалией является транслокация 9;22, приводящая к развитию филадельфийской хромосомы.
В конкретном варианте осуществления лиофилизированные фармацевтические композиции по настоящему изобретению можно использовать для лечения МДС, например, МДС, выбранного из ОМЛ, ОПМЛ и ОЛЛ.
Следует понимать, что в каждом из вышеуказанных терапевтических применений, лиофилизированные фармацевтические композиции по изобретению, как правило, восстанавливают в подходящем растворителе, как определено в настоящем документе, перед введением индивидууму, например, млекопитающему индивидууму, такому как пациент-человек.
Дозировки и введение
Дозы лиофилизированных фармацевтических композиций по изобретению, восстановленных или смешанных по мере необходимости с фармацевтически приемлемым растворителем или смесью растворителей, как определено в настоящем документе, можно вводить индивидууму известными в данной области способами. Неограничивающие примеры способов введения включают подкожную инъекцию, внутривенную инъекцию и инфузию.
Доза состава содержит количество, которое является терапевтически эффективным для лечения заболевания. Терапевтически эффективное количество соединения по изобретению можно выражать в виде мг соединения на кг массы тела индивидуума. В некоторых вариантах осуществления терапевтически эффективное количество составляет 1-1000 мг/кг, 1-500 мг/кг, 1-250 мг/кг, 1-100 мг/кг, 1-50 мг/кг, 1-25 мг/кг, или 1-10 мг/кг. В некоторых вариантах осуществления терапевтически-эффективное количество составляет 5 мг/кг, 10 мг/кг, 25 мг/кг, 50 мг/кг, 75 мг/кг, 100 мг/кг, 150 мг/кг, 200 мг/кг, 250 мг/кг, 300 мг/кг, 400 мг/кг, 500 мг/кг, 600 мг/кг, 700 мг/кг, 800 мг/кг, 900 мг/кг, 1000 мг/кг, приблизительно 5 мг/кг, приблизительно 10 мг/кг, приблизительно 25 мг/кг, приблизительно 50 мг/кг, приблизительно 75 мг/кг, приблизительно 100 мг/кг, приблизительно 150 мг/кг, приблизительно 200 мг/кг, приблизительно 250 мг/кг, приблизительно 300 мг/кг, приблизительно 400 мг/кг, приблизительно 500 мг/кг, приблизительно 600 мг/кг, приблизительно 700 мг/кг, приблизительно 800 мг/кг, приблизительно 900 мг/кг, или приблизительно 1000 мг/кг.
Соединение, описываемое в настоящем документе, может присутствовать в композиции в диапазоне приблизительно от 1 мг до приблизительно 5 мг, приблизительно от 5 мг до приблизительно 10 мг, приблизительно от 10 мг до приблизительно 15 мг, приблизительно от 15 мг до приблизительно 20 мг, приблизительно от 20 мг до приблизительно 25 мг, приблизительно от 25 мг до приблизительно 30 мг, приблизительно от 30 мг до приблизительно 35 мг, приблизительно от 35 мг до приблизительно 40 мг, приблизительно от 40 мг до приблизительно 45 мг, приблизительно от 45 мг до приблизительно 50 мг, приблизительно от 50 мг до приблизительно 55 мг, приблизительно от 55 мг до приблизительно 60 мг, приблизительно от 60 мг до приблизительно 65 мг, приблизительно от 65 мг до приблизительно 70 мг, приблизительно от 70 мг до приблизительно 75 мг, приблизительно от 75 мг до приблизительно 80 мг, приблизительно от 80 мг до приблизительно 85 мг, приблизительно от 85 мг до приблизительно 90 мг, приблизительно от 90 мг до приблизительно 95 мг, приблизительно от 95 мг до приблизительно 100 мг, приблизительно от 100 мг до приблизительно 125 мг, приблизительно от 125 мг до приблизительно 150 мг, приблизительно от 150 мг до приблизительно 175 мг, приблизительно от 175 мг до приблизительно 200 мг, приблизительно от 200 мг до приблизительно 225 мг, приблизительно от 225 мг до приблизительно 250 мг или приблизительно от 250 мг до приблизительно 300 мг.
Соединение, описываемое в настоящем документе, может присутствовать в композиции в количестве приблизительно 1 мг, приблизительно 5 мг, приблизительно 10 мг, приблизительно 15 мг, приблизительно 20 мг, приблизительно 25 мг, приблизительно 30 мг, приблизительно 35 мг, приблизительно 40 мг, приблизительно 45 мг, приблизительно 50 мг, приблизительно 55 мг, приблизительно 60 мг, приблизительно 65 мг, приблизительно 70 мг, приблизительно 75 мг, приблизительно 80 мг, приблизительно 85 мг, приблизительно 90 мг, приблизительно 95 мг, приблизительно 100 мг, приблизительно 125 мг, приблизительно 150 мг, приблизительно 175 мг, приблизительно 200 мг, приблизительно 225 мг, приблизительно 250 мг или приблизительно 300 мг.
В некоторых вариантах осуществления терапевтически эффективное количество можно вводить 1-35 раз в неделю, 1-14 раз в неделю, или 1-7 раз в неделю. В некоторых вариантах осуществления терапевтически эффективное количество можно вводить 1-10 раз в сутки, 1-5 раз в сутки, 1 раз, 2 раза или 3 раза в сутки.
Предполагается, что лиофилизированные фармацевтические композиции по изобретению будут полезны по отдельности или в комбинированном лечении с другими химиотерапевтическими средствами или лучевой терапией для профилактики или лечения ряда пролиферативных заболеваний или состояний. Примеры таких заболеваний и состояний перечислены выше.
Лиофилизированные фармацевтические композиции по изобретению, вводят ли их отдельно или в комбинации с противоопухолевыми средствами и терапией, такой как лучевая терапия, как правило, вводят индивидууму, который нуждается в таком введении, например пациенту-человеку или пациенту-животному, предпочтительно человеку.
Примеры химиотерапевтических средств, которые можно совместно вводить с лиофилизированными фармацевтическими композициями по изобретению, как определено в настоящем документе, в качестве неограничивающих примеров включают ингибиторы топоизомеразы I, другие антиметаболиты, средства, нацеленные на тубулин, ДНК-связывающее средство и ингибиторы топоизомеразы II, алкилирующие средства, моноклональные антитела, противогормональные средства, ингибиторы трансдукции сигнала, ингибиторы протеасомы, ингибиторы ДНК-метилтрансферазы, цитокины, интерфероны, интерлейкины, ретиноиды, терапевтические средства, нацеленные на хроматин, например, модуляторы HDAC или HAT, T-клеточные активаторы, в том числе иммуномодулирующие антитела, вакцины против злокачественных опухолей, гормональные средства, средства растительного происхождения, биологические средства, иммуномодулирующие средства, лучевая терапия и другие терапевтические или профилактические средства, например, средства, которые уменьшают или облегчают некоторые из побочных эффектов, связанных с химиотерапией, например противорвотные средства и средства, которые предотвращают нейтропению, связанную с химиотерапией, или уменьшают ее длительность и предотвращают осложнения, которые возникают из-за сниженных уровней эритроцитов или лейкоцитов, такие как эритропоэтин (EPO), гранулоцитарно-макрофагальный колониестимулирующий фактор (GM-CSF), и гранулоцитарный колониестимулирующий фактор (G-CSF).
В одном из вариантов осуществления лиофилизированные фармацевтические композиции по изобретению применяют в комбинации с ингибиторами деацетилазы гистонов (HDAC) (или они их дополнительно содержат) для дополнительного изменения транскрипции генов синергичным образом, например, для восстановления транскрипции генов, выключенных гиперметилированием и ацетилированием гистонов.
Ингибиторы HDAC в качестве неограничивающих примеров включают следующие структурные классы: 1) гидроксамовые кислоты; 2) циклические пептиды; 3) бензамиды; и 4) короткоцепочечные жирные кислоты. Примеры гидроксамовых кислот и производных гидроксамовых кислот включают трихостатин A (TSA), субероиланилид гидроксамовой кислоты (SAHA), оксамфлатин, субериновую бисгидроксамовую кислота (SBHA), m-карбокси-коричная кислота-бисгидроксамовая кислота (CBHA) и пироксамид. TSA выделяли в качестве противогрибкового антибиотика (Tsuji et al (1976) J. Antibiot (Tokyo) 29:1-6), и было обнаружено, что он является можным ингибитором HDAC млекопитающих (Yoshida et al. (1990) J. Biol. Chem. 265:17174-17179). Открытие того, что TSA-устойчивые клеточные линии имеют измененную HDAC, свидетельствует о том, что этот фермент является важной мишенью для TSA. Другие ингибиторы HDAC на основе гидроксамовой кислоты, SAHA, SBHA, и CBHA представляют собой синтетические соединения, которые способны ингибировать HDAC в микромолярной концентрации или ниже in vitro или in vivo. Glick et al. (1999) Cancer Res. 59:4392-4399. Все эти ингибиторы HDAC на основе гидроксамовой кислоты обладают необходимым структурным признаком: полярным гидроксамовым концом, связанным через гидрофобный метиленовый спейсер (например, 6 углеродов в длину) с другим полярным участком, который присоединен к концевой гидрофобной группе (например, бензольному кольцу).
Циклические пептиды, применяемые в качестве ингибиторов HDAC, представляют собой, в основном, циклические тетрапептиды. Примеры циклических пептидов в качестве неограничивающих примеров включают трапоксин A, апицидин и FR901228. Трапоксин A представляет собой циклический тетрапептид, который содержит 2-амино-8-oxo-9,10-эпокси-деканоильную (AOE) группу. Kijima et al. (1993) J. Biol. Chem. 268:22429-22435. Апицидин представляет собой метаболит грибов, который демонстрирует мощную активность широкого спектра действия против простейших и ингибирует активность HDAC в наномолярных концентрациях. Darkin-Rattray et al. (1996) Proc. Natl. Acad. Sci. USA. 93;13143-13147. FR901228 представляет собой депсипептид, который выделен из Chromobacterium violaceum, и было показано, что он ингибирует активность HDAC в микромолярных концентрациях.
Примеры бензамидов в качестве неограничивающих примеров включают MS-27-275. Saito et al. (1990) Proc. Natl. Acad. Sci. USA. 96:4592-4597. Примеры короткоцепочечных жирных кислот в качестве неограничивающих примеров включают бутираты (например, масляная кислота, аргининбутират и фенилбутират (PB)). Newmark et al. (1994) Cancer Lett. 78:1-5; и Carducci et al. (1997) Anticancer Res. 17:3972-3973. Кроме того, депудецин, для которого было показано ингибирование HDAC в микромолярных концентрациях (Kwon et al. (1998) Proc. Natl. Acad. Sci. USA. 95:3356-3361), также включен в объем ингибитора деацетилазы гистонов по настоящему изобретению.
В одном из вариантов осуществления алкилирующее средство применяют в комбинации с лиофилизированными фармацевтическими композициями по изобретению. Примеры алкилирующих средств включают бисхлорэтиламины (азотистые иприты, например, хлорамбуцил, циклофосфамид, ифосфамид, мехлоретамин, мелфалан, урацилиприт), азиридины (например, тиотепа), алкил альконсульфонаты (например, бусульфан), нитрозомочевины (например, кармустин, ломустин, стрептозоцин), неклассические алкилирующие средства (алтретамин, дакарбазин, и прокарбазин), соединения платины (карбоплатин и цисплатин).
В другом варианте осуществления лиофилизированную фармацевтическую композицию по изобретению применяют в комбинации с соединением платины, таким как цисплатин или карбоплатин.
В другом варианте осуществления лиофилизированную фармацевтическую композицию по изобретению применяют в комбинации с членом суперсемейства ретиноидов, таким как полностью трансретинол, полностью транс-ретиноевая кислота (третиноин), 13-цис ретиноевая кислота (изотретиноин) и 9-цис ретиноевая кислота.
В дополнительном варианте осуществления лиофилизированную фармацевтическую композицию по изобретению применяют в комбинации с гормональным средством, таким как синтетический эстроген (например, диэтилстибестрол), антиэстроген (например, тамоксифен, торемифен, флуоксиместерол и ралоксифен), антиандроген (бикалутамид, нилутамид, флутамид), ингибитор ароматазы (например, аминоглутетимид, анастрозол и тетразол), кетоконазол, гозерелин ацетат, леупродид, мегестрол ацетат и мифепристон.
В еще одном варианте осуществления лиофилизированную фармацевтическую композицию по изобретению применяют в комбинации со средством растительного происхождения, таким как алкалоид барвинка (например, винкристин, винбластин, виндезин, винзолидин и винорелбин), камптотецин (20(S)-камптотецин, 9-нитро-20(S)-камптотецин, и 9-амино-20(S)-камптотецин), подофиллотоксин (например, этопозид (VP-16) и тенипозид (VM-26)), и таксан (например, паклитаксел и доцетаксел).
В конкретном варианте осуществления лиофилизированную фармацевтическую композицию по изобретению применяют в комбинации с таксаном, таким как паклитаксел и доцетаксел.
В другом варианте осуществления лиофилизированные фармацевтические композиции по изобретению можно использовать в комбинации с антрациклином, таким как даунорубицин или идарубицин.
В дополнительном варианте осуществления лиофилизированную фармацевтическую композицию по изобретению применяют в комбинации с биологическим средством, таким как иммуномодулирующий белок (например, цитокин), моноклональное антитело против опухолевого антигена, ген-опухолевый супрессор или вакцина против злокачественной опухоли.
Примеры интерлейкинов, которые можно использовать в комбинации с лиофилизированной фармацевтической композицией по изобретению, в качестве неограничивающих примеров включают интерлейкин 2 (IL-2) и интерлейкин 4 (IL-4), интерлейкин 12 (IL-12). Примеры интерферонов, которые можно использовать в комбинации с лиофилизированной фармацевтической композицией по изобретению, в качестве неограничивающих примеров включают, интерферон альфа, интерферон бета (фибробластный интерферон) и интерферон гамма (фибробластный интерферон). Примеры таких цитокинов в качестве неограничивающих примеров включают эритропоэтин (эпоэтин), гранулоцитарный CSF (филграстим) и гранулоцитарно-макрофагальный CSF (сарграмостим). Иммуномодултирующие средства, иные, чем цитокины, в качестве неограничивающих примеров включают бациллу Кальмета-Герена, левамизол и остреотид.
Примеры моноклональных антител против опухолевых антигенов, которые можно использовать в сочетании с лиофилизированной фармацевтической композицией по изобретению, в качестве неограничивающих примеров включают ГЕРЦЕПТИН(R) (трастузумаб), РИТУКСАН(R) (Ритуксимаб), МИЛОТАРГ(R) (анти-CD33), и КЭМПАС(R) (анти-CD52).
В дополнительном варианте осуществления лиофилизированную фармацевтическую композицию по изобретению можно использовать в комбинации с вакциной против злокачественной опухоли, например, с вакциной против злокачественной опухоли, выбранной из противоопухолевой вакцины на основе раково-тестикулярного антигена (CTA), такой как вакцина на основе (CTA), выбранная из NY-ESO-1, LAGE-1, MAGE-A1, -A2, -A3, -A4, -A6, -A10, -A12, CT7, CT10, GAGE1-6, GAGE 1-2, BAGE, SSX1-5, SSX 2, HAGE, PRAME, RAGE-1, XAGE-1, MUC2, MUC5B и HMW-MAA. Неограничивающие примеры вакцин на основе CTA включают вакцины на основе MAGE-A3 (например recMAGE-A3), NY-ESO-1 и PRAME.
В другом варианте осуществления лиофилизированную фармацевтическую композицию по изобретению можно использовать в комбинации с активатором T-клеток, например, с активатором T-клеток, который является антителом (необязательно МАТ), например, выбранным из (a) агониста CD137; (b) агониста CD40; (c) агониста OX40; (d) МАТ к PD-1; (e) МАТ к PD-L1; (f) МАТ к CTLA-4; и (г) комбинаций (a)-(f). В некоторых вариантах осуществления дополнительный терапевтический компонент представляет собой тремелимумаб или имилимумаб.
В другом варианте осуществления лиофилизированную фармацевтическую композицию по изобретению можно использовать в комбинации с карбоплатином для лечения резистентного к препаратам платины рецидивирующего рака яичников.
В другом варианте осуществления лиофилизированную фармацевтическую композицию по изобретению можно использовать для лечения печеночноклеточной карциномы (например, после неэффективности сорафениба).
В другом варианте осуществления лиофилизированную фармацевтическую композицию по изобретению можно использовать в комбинации с иринотеканом для лечения метастатического рака толстого кишечника.
В другом варианте осуществления лиофилизированную фармацевтическую композицию по изобретению можно использовать в комбинации с 5-фторурацилом (5-ФУ), лейковорином, оксалиплатином для лечения метастатического рака толстого кишечника.
В другом варианте осуществления лиофилизированную фармацевтическую композицию по изобретению можно использовать в комбинации с цитарабином и флударабином для лечения детского повторного/рефрактерного ОМЛ.
В другом варианте осуществления лиофилизированную фармацевтическую композицию по изобретению можно использовать в комбинации с ингибитором JAK2 для лечения миопролиферативных неоплазий.
Лиофилизированная фармацевтическая композиция по изобретению и любые другие терапевтические средства могут быть представлены раздельно или представлены вместе в фармацевтической упаковке, наборе или упаковке для пациента.
Лиофилизированную фармацевтическую композицию по изобретению и комбинации с другими терапевтическими средствами или лучевой терапией, как описано выше, можно вводить в течение длительного времени для поддержания благоприятных терапевтических эффектов или можно вводить только в течение короткого периода. Альтернативно, их можно вводить пульсирующим или непрерывным образом.
Лиофилизированную фармацевтическую композицию по изобретению можно вводить в эффективном количестве, т.е. в количестве, которое эффективно для получения желаемого терапевтического эффекта, или по отдельности (в монотерапии) или в комбинации с одним или несколькими химиотерапевтическими средствами или лучевой терапией. Например, «эффективное количество» может быть количеством соединения, которое при введении индивидууму, страдающему от злокачественной опухоли, замедляет рост опухоли, облегчает симптомы заболевания и/или повышает продолжительность жизни.
Количество лиофилизированной фармацевтической композиции по изобретению, вводимой индивидууму, может зависеть от типа и тяжести заболевания или состояния и от характеристик индивидуума, таких как общее состояние здоровья, возраст, пол, масса тела и переносимость лекарственных средств. Специалист способен определить подходящие дозы в зависимости от этих и других факторов. Эффективные дозы для широко используемых противоопухолевых лекарственных средств и лучевой терапии хорошо известны специалистам.
Чистота соединений по изобретению
Любое соединение в настоящем документе можно очищать. Соединение в настоящем документе может быть не менее 1% чистоты, по меньшей мере, 2% чистоты, по меньшей мере, 3% чистоты, по меньшей мере, 4% чистоты, по меньшей мере, 5% чистоты, по меньшей мере, 6% чистоты, по меньшей мере, 7% чистоты, по меньшей мере, 8% чистоты, по меньшей мере, 9% чистоты, по меньшей мере, 10% чистоты, по меньшей мере, 11% чистоты, по меньшей мере, 12% чистоты, по меньшей мере, 13% чистоты, по меньшей мере, 14% чистоты, по меньшей мере, 15% чистоты, по меньшей мере, 16% чистоты, по меньшей мере, 17% чистоты, по меньшей мере, 18% чистоты, по меньшей мере, 19% чистоты, по меньшей мере, 20% чистоты, по меньшей мере, 21% чистоты, по меньшей мере, 22% чистоты, по меньшей мере, 23% чистоты, по меньшей мере, 24% чистоты, по меньшей мере, 25% чистоты, по меньшей мере, 26% чистоты, по меньшей мере, 27% чистоты, по меньшей мере, 28% чистоты, по меньшей мере, 29% чистоты, по меньшей мере, 30% чистоты, по меньшей мере, 31% чистоты, по меньшей мере, 32% чистоты, по меньшей мере, 33% чистоты, по меньшей мере, 34% чистоты, по меньшей мере, 35% чистоты, по меньшей мере, 36% чистоты, по меньшей мере, 37% чистоты, по меньшей мере, 38% чистоты, по меньшей мере, 39% чистоты, по меньшей мере, 40% чистоты, по меньшей мере, 41% чистоты, по меньшей мере, 42% чистоты, по меньшей мере, 43% чистоты, по меньшей мере, 44% чистоты, по меньшей мере, 45% чистоты, по меньшей мере, 46% чистоты, по меньшей мере, 47% чистоты, по меньшей мере, 48% чистоты, по меньшей мере, 49% чистоты, по меньшей мере, 50% чистоты, по меньшей мере, 51% чистоты, по меньшей мере, 52% чистоты, по меньшей мере, 53% чистоты, по меньшей мере, 54% чистоты, по меньшей мере, 55% чистоты, по меньшей мере, 56% чистоты, по меньшей мере, 57% чистоты, по меньшей мере, 58% чистоты, по меньшей мере, 59% чистоты, по меньшей мере, 60% чистоты, по меньшей мере, 61% чистоты, по меньшей мере, 62% чистоты, по меньшей мере, 63% чистоты, по меньшей мере, 64% чистоты, по меньшей мере, 65% чистоты, по меньшей мере, 66% чистоты, по меньшей мере, 67% чистоты, по меньшей мере, 68% чистоты, по меньшей мере, 69% чистоты, по меньшей мере, 70% чистоты, по меньшей мере, 71% чистоты, по меньшей мере, 72% чистоты, по меньшей мере, 73% чистоты, по меньшей мере, 74% чистоты, по меньшей мере, 75% чистоты, по меньшей мере, 76% чистоты, по меньшей мере, 77% чистоты, по меньшей мере, 78% чистоты, по меньшей мере, 79% чистоты, по меньшей мере, 80% чистоты, по меньшей мере, 81% чистоты, по меньшей мере, 82% чистоты, по меньшей мере, 83% чистоты, по меньшей мере, 84% чистоты, по меньшей мере, 85% чистоты, по меньшей мере, 86% чистоты, по меньшей мере, 87% чистоты, по меньшей мере, 88% чистоты, по меньшей мере, 89% чистоты, по меньшей мере, 90% чистоты, по меньшей мере, 91% чистоты, по меньшей мере, 92% чистоты, по меньшей мере, 93% чистоты, по меньшей мере, 94% чистоты, по меньшей мере, 95% чистоты, по меньшей мере, 96% чистоты, по меньшей мере, 97% чистоты, по меньшей мере, 98% чистоты, по меньшей мере, 99% чистоты, по меньшей мере, 99,1% чистоты, по меньшей мере, 99,2% чистоты, по меньшей мере, 99,3% чистоты, по меньшей мере, 99,4% чистоты, по меньшей мере, 99,5% чистоты, по меньшей мере, 99,6% чистоты, по меньшей мере, 99,7% чистоты, по меньшей мере, 99,8% чистоты или по меньшей мере, 99,9% чистоты.
Примеры
ПРИМЕР 1. Получение лиофилизированного состава натриевой соли соединения формулы (1).
Натриевую соль соединения формулы (1) растворяли в ДМСО при определенной концентрации с использованием вертикальной мешалки в сосуде из нержавеющей стали (НС) соответствующего размера. После полного растворения лекарственного средства в ДМСО, образцы из основного объема раствора тестировали с использованием способа УФ или ВЭЖХ без остановки процесса для того чтобы установить, что количество натриевой соли соединения формулы 1 было в пределах 95-105% от целевой концентрации. Основной объем раствора фильтровали через серии двух заранее стерилизованных 0,2-микронных стерилизующих фильтров, которые были совместимы с ДМСО, и собирали в двухлитровый буферный резервуар из НС. Скорость фильтрации постоянно корректировали путем визуального наблюдения за количеством, доступным для заполнения буферного резервуара. Затем аликвотами по одному грамму из отфильтрованного основного объема раствора заполняли депирогенированные прозрачные стеклянные флаконы на 5 см3. Каждый флакон был автоматически частично закрыт на линии заполнения покрытой фторполимером хлорбутилкаучуковой пробкой для лиофилизации, которая была предварительно стерилизована. Флаконы с продуктом переносили в лиофилизатор в условиях асептического переноса для инициирования цикла лиофилизации. Использованный лиофилизатор представлял собой лиофилизатор полупромышленного масштаба, Lyobeta 35, IMA-Telstar, который имел объем камеры 1,02 м2, емкость для льда - 35 кг, 22 кг/24 часа - емкость конденсатора.
Флаконы, содержащие раствор, лиофилизировали с использованием параметров цикла, представленными ниже в таблице 1.
После завершения цикла лиофилизации, лиофилизатор был заполнен азотом, и флаконы были автоматически полностью закрыты. Флаконы асептически перемещали в изолятор, где каждый флакон автоматически закрывали синей алюминиевой пробкой с отрывной накладкой. Флаконы проверяли визуально, прежде чем приступить к отбору проб для контроля при выпуске продукции, и к маркировке и упаковке. Флаконы хранили при 2-8°C до готовности. Каждый флакон был помечен по его содержимому.
ПРИМЕР 2. Сравнительные испытания
I. Лиофилизированные составы, произведенные способом по изобретению:
Были приготовлены основные растворы, содержащие натриевую соль соединения формулы (1) в четырех различных концентрациях в ДМСО, и полученными растворами (обозначенными от A до D) заполнили флаконы для лиофилизации и провели лиофилизацию при помощи протокола, описанного выше в примере 1. Датчики Пирани и Баратрон применяли для определения окончания стадии первичного высушивания (сублимации). Фигура 1 показывает прогрессивное снижение содержания ДМСО с течением времени в процессе стадий первичного и вторичного высушивания.
После лиофилизации, лиофилизированные образцы анализировали на чистоту (% чистоты при помощи ВЭЖХ), остаточное содержание ДМСО и остаточную влагу. Образцы восстанавливали, растворяя их в системе неводных растворителей, описанной в ТАБЛИЦЕ 2 ниже, и анализировали время восстановления и внешний вид восстановленных составов.
Результаты анализов представлены в таблице 3 ниже.
Результаты для четырех различных концентраций, n=1
LOQ=предел количественного определения
II. Составы для сравнения:
Основные растворы натриевой соли соединения формулы (1) с концентрацией 100 мг/мл лиофилизировали с использованием прибора, описанного выше в примере 1, но с другим температурным профилем, который не включал первую стадию нагревания во время замораживания раствора, но включал замораживание состава с различной скоростью замораживания.
Характеристики составов для сравнения, приготовленных таким образом, показаны ниже в таблице 4.
>30 минут для полного растворения
>30 минут для полного растворения
>30 минут для полного растворения
III. Сравнение результатов, полученных для составов, описанных в I и II
Результаты, показанные на этапе I выше, демонстрируют, что когда промежуточная стадия нагревания («первая стадия нагревания») включена во время замораживания раствора перед первичным высушиванием в соответствие с изобретением, результатом является лиофилизированный сухой состав, который можно восстановить менее чем за 20 минут, и менее чем 15 минут в некоторых случаях.
Для сравнения, составам для сравнения FP1, FP2 и FP3, описанным в II выше и приготовленным путем процесса, в котором отсутствует промежуточная стадия нагревания, потребовалось больше времени для восстановления (более 30 минут). Не желая быть связанными какой-либо теорией, полагаем, что промежуточная стадия нагревания оказывает эффект повышения пористости лиофилизированного продукта и увеличения площади поверхности, доступной для контакта с молекулами растворителя, таким образом, повышая растворимость составов.
IV. Сравнение времени сушки с Примером 4 из WO2013/033176
Пример 4 в WO2013/033176 описывает лиофилизацию раствора натриевой соли соединения формулы (1) с использованием параметров цикла, показанных ниже в таблице 5.
Общее время лиофилизации для состава, описанного в примере 4 из WO2013/033176, составляет 109 часов и 47 минут. Для сравнения, общее время лиофилизации для состава по изобретению, как описано выше в I, составило 76,8 часов, т.е. более чем на 30 часов короче, чем общее время лиофилизации, например, для 4 в WO2013/033176. Большая часть различий объясняется значительно уменьшенной стадией вторичного высушивания в способе по настоящему изобретению по сравнению со способом, описанным в примере 4 из WO2013/033176 (21 час против 50,78 часа). В способе по настоящему изобретению, промежуточная стадия нагревания (первая) расположена между двумя стадиями замораживания, когда раствор изначально заморожен, и полагают, что это приводит к гораздо более пористой структуре, из которой ДМСО может более легко сублимироваться на стадии первичного высушивания. Таким образом, большая часть ДМСО удаляется во время стадии первичного высушивания, в результате чего можно использовать более короткую стадию вторичного высушивания.
Таким образом, подводя итоги, способ по настоящему изобретению может уменьшать время, необходимое для получения лиофилизированного продукта, что значительно улучшило характеристики растворения.
ПРИМЕР 3. Крупномасштабные исследования на составах A и B по 75 мг/мл и 100 мг/мл.
Результаты, полученные в экспериментах, описанных в примере 2, показали что наиболее низкие уровни остаточного ДМСО получали с составом B, в котором лиофилизировали основной раствор, содержащий активное соединение в концентрации 75 мг/мл Подтверждающие исследования, таким образом, проводили на растворах с концентрацией 75 мг/мл и 100 мг/мл натриевой соли соединения формулы (1) в ДМСО. Лиофилизацию проводили в формате 100 флаконов, и проводили анализ на нескольких образцах. Использовали протокол, описанный в примере 1. Свойства полученных лиофилизированных продуктов показаны ниже в таблице 6.
Анализ
* Время восстановления не включает рассеивание пузырьков, что может занять приблизительно дополнительные 10 минут. Однако восстановление проводили вручную, и оно не требовало механического перемешивающего аппарата.
** Хотя не обнаружено в этом случае, могут быть случаи, когда растворы слегка туманны и/или от слегка белого до желтого по цвету.
Результаты в таблице 6 демонстрируют, что способ по изобретению можно использовать для получения лиофилизированных составов, которые имеют время растворения менее чем десять минут (за исключением времени, необходимого для очистки от пузырьков) и что восстановление можно проводить вручную без необходимости в механических перемешивателях.
ПРИМЕР 4. Получение натриевой соли соединения формулы (1).
Натриевую соль соединения формулы (1) получали, как описано в US 7700567 (содержание которого, таким образом, включено в качестве ссылки) путем соединения 1c (где R1=карбаматная защитная группа) с фосфорамидитным элементом структуры 1d:
Твердая подложка из стекла с заданным размером пор (CPG) 1c, связанная с защищенным 2'-дезоксигуанозином (где R1=трет-бутил феноксиацетил), была соединена с 2-2,5 эквивалентов феноксиацетил децитабин фосфорамидита (1d, где R1=феноксиацетил) в присутствии 60% of 0,3 M активатора бензилтиотетразола (в ацетонитриле) в течение 10 минут. Твердую подложку из CPG, содержащую защищенный DpG динуклеотид обрабатывали 20 мл 50 мМ K2CO3 в метаноле в течение 1 часа и 20 минут. Связанный продукт окисляли, удаляли защитную группу, и отмывали полученное соединение, фильтровали и очищали при помощи ВЭЖХ ÄKTA Explorer 100 с препаративной колонкой Gemini C18 (Phenomenex), 250×21,2 мм, 10 мкм с защитной колонкой (Phenomenex), 50×21,2 мм, 10 мкм, с 50 мМ триэтиламмоний ацетатом (pH 7) в воде MilliQ (подвижная фаза A) и 80% ацетонитрилом в воде MilliQ (подвижная фаза B), от 2% до 20/25% подвижной фазы B в объемах колонки.
Масс-спектрометрия с отрицательной ионизацией распылением (ESI-MS (-ve) DpG динуклеотида 2b:
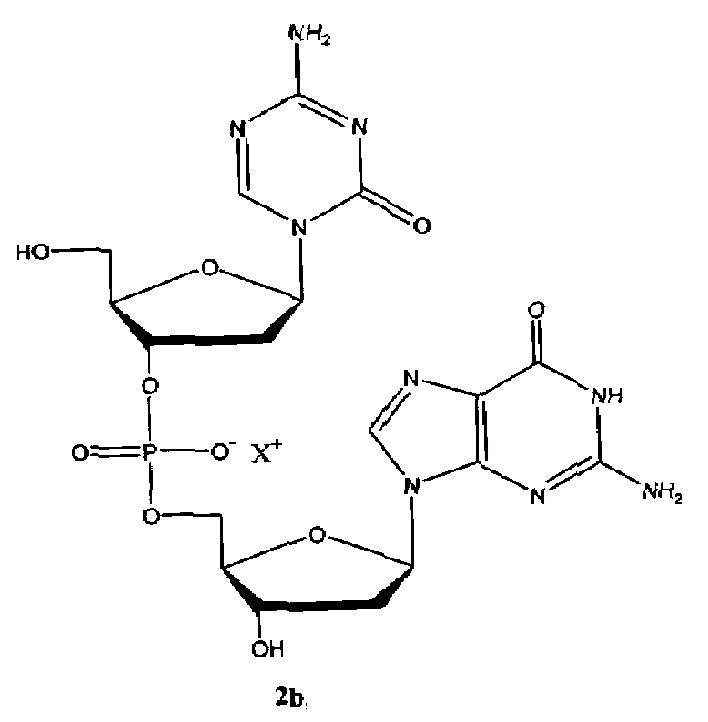
где X+=триэтиламмоний (рассчитанная точная масса для нейтрального соединения C18H24N9O10P составляет 557,14), показал масса/заряд 556,1 [M-H]- и 1113,1 for [2M-H] - (см. масс-спектр на фигура 31 в US 7700567).
Натриевую соль соединения формулы (1), т.е. DpG динуклеотид 2b, где X+=натрий, получали путем повторного растворения триэтиламмонийной соли в 4 мл воды, 0,2 мл 2M раствора NaClO4. Когда добавляли 36 мл ацетона, динуклеотид выпадал в осадок. Раствор хранили при -20°C в течение нескольких часов и центрифугировали при 4000 об./мин. в течение 20 минут. Супернатант удаляли и твердый осадок отмывали 30 мл ацетона с последующим дополнительным центрифугированием при 4000 об./мин. в течение 20 минут. Осадок, который растворяли в воде и лиофилизировали, показал масса/заряд 556,0 [M-H]- (см. масс-спектр на фигуре 36 из US 7700567).
ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ
Следующие неограничивающие варианты осуществления предлагают иллюстративные примеры по изобретению, но не ограничивают объем изобретения.
Вариант осуществления 1. Способ получения лиофилизированной фармацевтической композиции, включающий растворение соединения формулы (1):
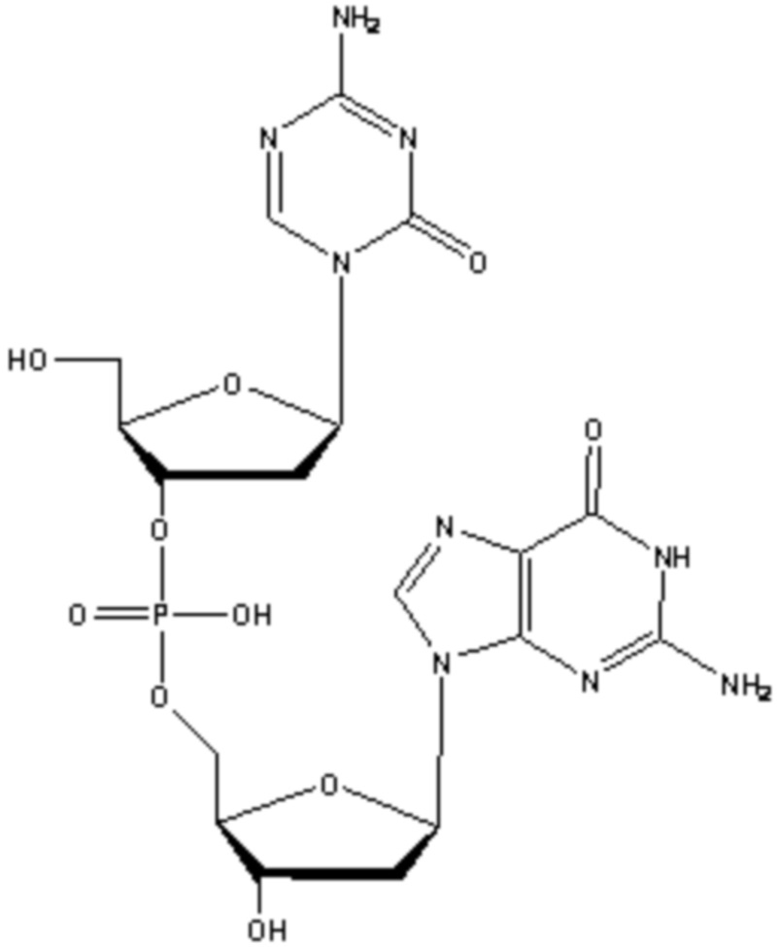 (1),
(1),
или его фармацевтически приемлемой соли в растворителе, содержащем диметилсульфоксид (ДМСО) для образования раствора, где растворитель затем удаляют способом лиофильной сушки для получения лиофилизированного продукта, где способ лиофильной сушки включает: (i) первую стадию замораживания, на которой раствор замораживают, снижая его температуру до температуры не большей, чем приблизительно -20°C; (ii) первую стадию нагревания, на которой температуру замороженного раствора индуцируют до температуры в диапазоне приблизительно от -15°C до приблизительно 5°C, где температура в диапазоне приблизительно от -15°C до приблизительно 5°C сохраняет раствор замороженным; (iii) вторую стадию замораживания, на которой температуру раствора понижают до температуры не большей, чем приблизительно -20°C; (iv) стадию первичного высушивания, где стадия первичного высушивания включает этап сублимации, на котором ДМСО удаляют из раствора в замороженном состоянии путем сублимации при пониженном давлении для получения частично высушенного продукта; и (v) стадию вторичного высушивания, на которой ДМСО удаляют путем выпаривания из частично высушенного продукта в незамороженном состоянии при пониженном давлении для получения лиофилизированного продукта.
Вариант осуществления 2. Способ по варианту осуществления 1, где соединение формулы (1) находится в форме натриевой соли.
Вариант осуществления 3. Способ по любому из вариантов осуществления 1-2, где растворитель является неводным.
Вариант осуществления 4. Способ по любому из вариантов осуществления 1-3, где лиофилизированная фармацевтическая композиция имеет время растворения при температуре окружающей среды и без помощи механического перемешивания в неводном растворителе, содержащем 65% (об./об.) пропиленгликоля, 25% (об./об.) глицерина и 10% (об./об.) этанола, не более чем приблизительно 20 минут.
Вариант осуществления 5. Способ по любому из вариантов осуществления 1-4, где в количестве лиофилизированной фармацевтической композиции, полученной из 1 грамма раствора, содержание остаточного ДМСО составляет не более чем приблизительно 20 мг.
Вариант осуществления 6. Способ по любому из вариантов осуществления 1-5, где любой остаточный ДМСО присутствует в лиофилизированной фармацевтической композиции в количестве, соответствующем не более чем 35 мг на 100 мг эквивалента свободного основания соединения формулы (1).
Вариант осуществления 7. Способ по любому из вариантов осуществления 1-6, дополнительно включающий упаковку лиофилизированного фармацевтического средства в запечатанный фармацевтический контейнер.
Вариант осуществления 8. Способ по любому из вариантов осуществления 1-7, дополнительно включающий растворение лиофилизированной фармацевтической композиции в растворителе для получения жидкой композиции для инъекций.
Вариант осуществления 9. Способ по варианту осуществления 8, где растворитель представляет собой неводный растворитель.
Вариант осуществления 10. Способ по любому из вариантов осуществления 1-9, где раствор дополнительно содержит co-растворитель.
Вариант осуществления 11. Способ по варианту осуществления 1, дополнительно включающий восстановление лиофилизированной фармацевтической композиции в фармацевтически приемлемом растворителе для получения жидкого состава, содержащего соединение формулы (1) или его фармацевтически приемлемую соль.
Вариант осуществления 12. Способ по любому из вариантов осуществления 1-11, где сниженное давление на стадии первичного высушивания составляет приблизительно от 5 мкбар до приблизительно 40 мкбар.
Вариант осуществления 13. Способ по любому из вариантов осуществления 1-12, где температура на стадии первичного высушивания составляет приблизительно от -3°C до приблизительно -9°C.
Вариант осуществления 14. Способ по любому из вариантов осуществления 1-13, где температура на стадии вторичного высушивания составляет приблизительно от 30°C до приблизительно 65°C.
Вариант осуществления 15. Фармацевтическая композиция, полученная способом, включающим стадии: растворения соединения формулы (1):
(1),
или его фармацевтически приемлемой соли в растворителе, содержащем диметилсульфоксид (ДМСО) для образования раствора, где растворитель затем удаляют способом лиофильной сушки для получения лиофилизированного продукта, где способ лиофильной сушки включает: (i) первую стадию замораживания, на которой раствор замораживают, снижая его температуру до температуры не большей, чем приблизительно -20°C; (ii) первую стадию нагревания, на которой температуру замороженного раствора индуцируют до температуры в диапазоне приблизительно от -15°C до приблизительно 5°C, где температура в диапазоне приблизительно от -15°C до приблизительно 5°C сохраняет раствор замороженным; (iii) вторую стадию замораживания, на которой температуру раствора понижают до температуры не большей, чем приблизительно -20°C; (iv) стадию первичного высушивания, где стадия первичного высушивания включает этап сублимации, на котором ДМСО удаляют из раствора в замороженном состоянии путем сублимации при пониженном давлении для получения частично высушенного продукта; и (v) стадию вторичного высушивания, на которой ДМСО удаляют путем выпаривания из частично высушенного продукта в незамороженном состоянии при пониженном давлении для получения лиофилизированного продукта.
Вариант осуществления 16. Фармацевтическая композиция по варианту осуществления 15, где соединение формулы (1) находится в форме натриевой соли.
Вариант осуществления 17. Фармацевтическая композиция по любому из вариантов осуществления 15-16, где растворитель является неводным.
Вариант осуществления 18. Фармацевтическая композиция по любому из вариантов осуществления 15-17, где лиофилизированная фармацевтическая композиция имеет время растворения при температуре окружающей среды и без помощи механического перемешивания в неводном растворителе, содержащем 65% (об./об.) пропиленгликоля, 25% (об./об.) глицерина и 10% (об./об.) этанола, не более чем приблизительно 20 минут.
Вариант осуществления 19. Фармацевтическая композиция по любому из вариантов осуществления 15-18, где в количестве лиофилизированной фармацевтической композиции, полученной из 1 грамма раствора, содержание остаточного ДМСО составляет не более чем приблизительно 20 мг.
Вариант осуществления 20. Фармацевтическая композиция по любому из вариантов осуществления 15-19, где любой остаточный ДМСО присутствует в лиофилизированной фармацевтической композиции в количестве, соответствующем не более чем 35 мг на 100 мг эквивалента свободного основания соединения формулы (1).
Вариант осуществления 21. Фармацевтическая композиция по любому из вариантов осуществления 15-20, где способ дополнительно включает упаковку лиофилизированного фармацевтического средства в запечатанный фармацевтический контейнер.
Вариант осуществления 22. Фармацевтическая композиция по любому из вариантов осуществления 15-21, где способ дополнительно включает растворение лиофилизированной фармацевтической композиции в растворителе для получения жидкой композиции для инъекций.
Вариант осуществления 23. Фармацевтическая композиция по варианту осуществления 22, где растворитель представляет собой неводный растворитель.
Вариант осуществления 24. Фармацевтическая композиция по любому из вариантов осуществления 15-23, где раствор дополнительно содержит co-растворитель.
Вариант осуществления 25. Фармацевтическая композиция по варианту осуществления 15, где способ дополнительно включает восстановление лиофилизированной фармацевтической композиции в фармацевтически приемлемом растворителе для получения жидкого состава, содержащего соединение формулы (1) или его фармацевтически приемлемую соль.
Вариант осуществления 26. Фармацевтическая композиция по любому из вариантов осуществления 15-25, где сниженное давление на стадии первичного высушивания составляет приблизительно от 5 мкбар до приблизительно 40 мкбар.
Вариант осуществления 27. Фармацевтическая композиция по любому из вариантов осуществления 15-26, где температура на стадии первичного высушивания составляет приблизительно от -3°C до приблизительно -9°C.
Вариант осуществления 28. Фармацевтическая композиция по любому из вариантов осуществления 15-27, где температура на стадии вторичного высушивания составляет приблизительно от 30°C до приблизительно 65°C.
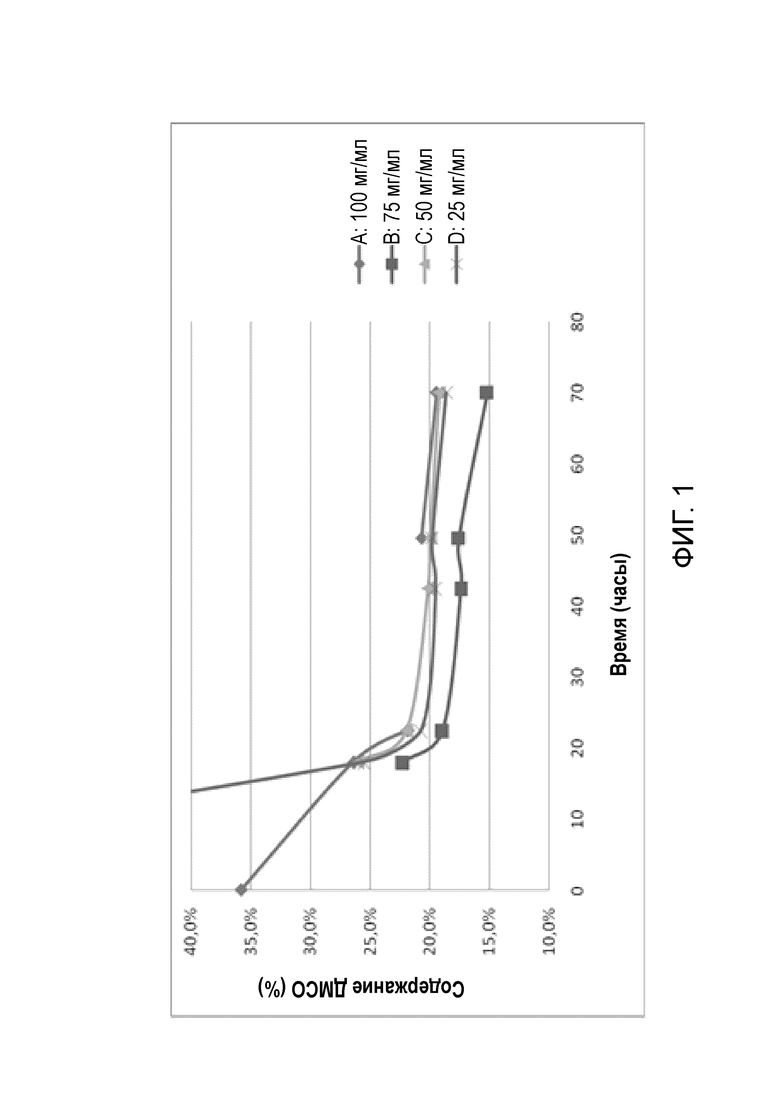
Изобретение относится к медицине. Способ получения лиофилизированной фармацевтической композиции, содержащей соединение формулы (I):
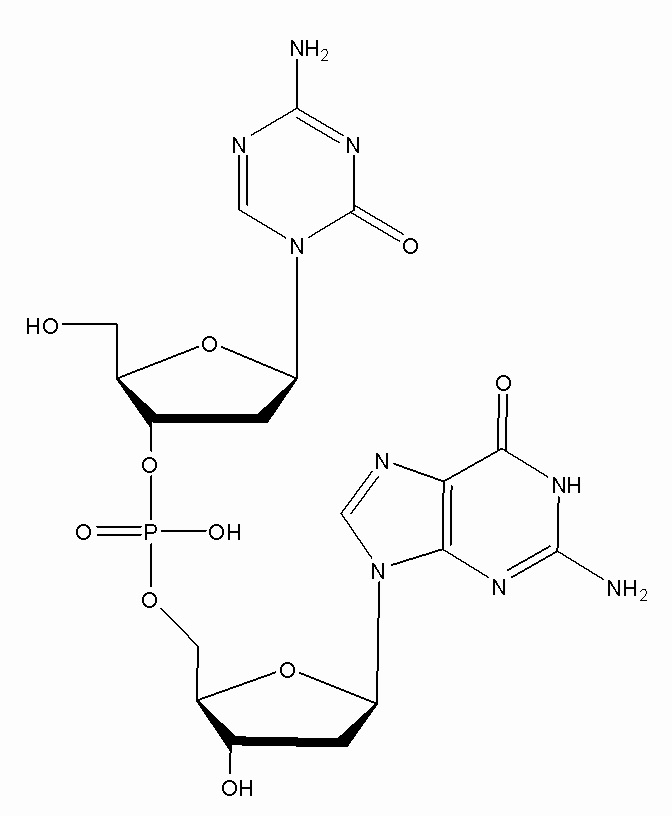
или его фармацевтически приемлемую соль, включает растворение соединения в растворителе, содержащем диметилсульфоксид и необязательно один или несколько со-растворителей для образования раствора, а затем удаление растворителя и любых со-растворителей способом лиофильной сушки, при этом осуществляют первую стадию замораживания, на которой раствор замораживают, снижая его температуру до температуры -20°C; первую стадию нагревания, на которой температуру замороженного раствора индуцируют до температуры в диапазоне от -15°C до 5°C, где раствор сохраняют замороженным; вторую стадию замораживания, на которой температуру раствора понижают до температуры, не большей -20°C; стадию первичного высушивания, включающую этап сублимации, на котором ДМСО удаляют из раствора в замороженном состоянии путем сублимации при пониженном давлении для получения частично высушенного продукта; и стадию вторичного высушивания, на которой ДМСО удаляют путем выпаривания из частично высушенного продукта в незамороженном состоянии при пониженном давлении для получения лиофилизированного продукта. Также изобретение относится к лиофилизированным фармацевтическим композициям и их применению в медицине, в частности, для лечения злокачественной опухоли. Предлагаемый способ получения лиофилизированной фармацевтической композиции, содержащей соединение формулы (I), позволяет более эффективно очистить получаемый лиофилизированный продукт от растворителя, сократить время сушки, а также уменьшить время, необходимое для получения раствора лиофилизированного продукта. 2 н. и 13 з.п. ф-лы, 1 ил., 6 табл., 4 пр.
1. Способ получения лиофилизированной фармацевтической композиции, включающий растворение соединения формулы:
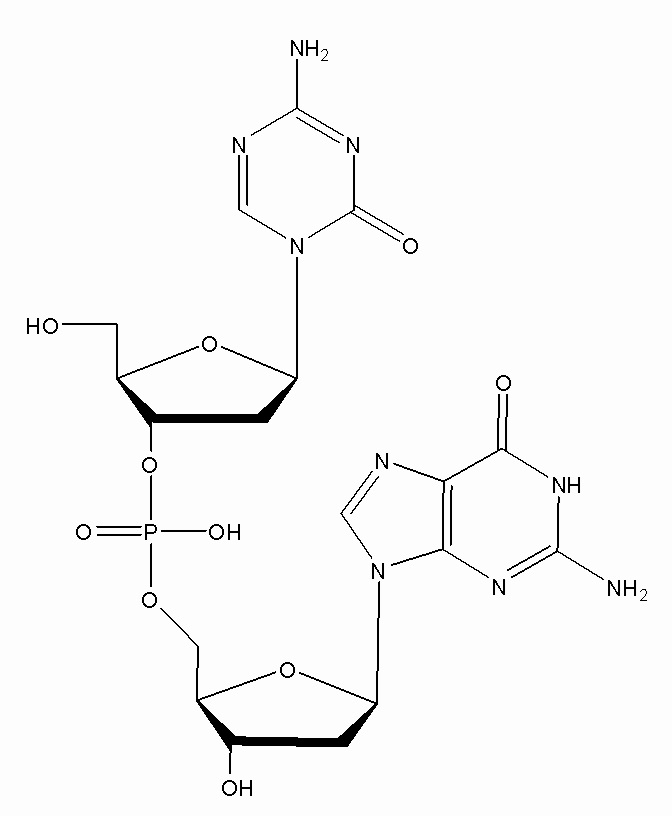
далее указанного как соединение формулы (1),
или его фармацевтически приемлемой соли в растворителе, содержащем диметилсульфоксид (ДМСО) для образования раствора, где растворитель затем удаляют способом лиофильной сушки для получения лиофилизированного продукта, где способ лиофильной сушки включает:
(i) первую стадию замораживания, на которой раствор замораживают, снижая его температуру до температуры, не большей чем -20°C;
(ii) первую стадию нагревания, на которой температуру замороженного раствора индуцируют до температуры в диапазоне от -15°C до 5°C, где температура диапазоне от -15°C до 5°C сохраняет раствор замороженным;
(iii) вторую стадию замораживания, на которой температуру раствора понижают до температуры, не большей чем -20°C;
(iv) стадию первичного высушивания, где стадия первичного высушивания включает этап сублимации, на котором ДМСО удаляют из раствора в замороженном состоянии путем сублимации при пониженном давлении для получения частично высушенного продукта; и
(v) стадию вторичного высушивания, на которой ДМСО удаляют путем выпаривания из частично высушенного продукта в незамороженном состоянии при пониженном давлении для получения лиофилизированного продукта.
2. Способ по п. 1, где соединение формулы (1) находится в форме натриевой соли.
3. Способ по п. 1, где растворитель является неводным.
4. Способ по п. 1, где лиофилизированная фармацевтическая композиция имеет время растворения при температуре окружающей среды и без помощи механического перемешивания в неводном растворителе, содержащем 65% (об./об.) пропиленгликоля, 25% (об./об.) глицерина и 10% (об./об.) этанола, не более чем 20 минут, где лиофилизированная фармацевтическая композиция содержит 100 мг эквивалента свободного основания соединения формулы (1) и где в композиции присутствует не более 35 мг остаточного ДМСО.
5. Способ по п. 1, где в количестве лиофилизированной фармацевтической композиции, полученной из 1 грамма раствора, содержание остаточного ДМСО составляет не более чем 20 мг.
6. Способ по п. 1, где любой остаточный ДМСО присутствует в лиофилизированной фармацевтической композиции в количестве, соответствующем не более чем 35 мг на 100 мг эквивалента свободного основания соединения формулы (1).
7. Способ по п. 1, дополнительно включающий упаковку лиофилизированной фармацевтической композиции в запечатанный фармацевтический контейнер.
8. Способ по п. 1, дополнительно включающий растворение лиофилизированной фармацевтической композиции в растворителе для получения жидкой композиции для инъекций.
9. Способ по п. 8, где растворитель представляет собой неводный растворитель.
10. Способ по п. 1, где раствор дополнительно содержит co-растворитель.
11. Способ по п. 1, дополнительно включающий восстановление лиофилизированной фармацевтической композиции в фармацевтически приемлемом растворителе для получения жидкого состава, содержащего соединение формулы (1) или его фармацевтически приемлемую соль.
12. Способ по п. 1, где сниженное давление на стадии первичного высушивания составляет от 0,5 Па (5 мкбар) до 4 Па (40 мкбар).
13. Способ по п. 1, где температура на стадии первичного высушивания составляет от -3°C до -9°C.
14. Способ по п. 1, где температура на стадии вторичного высушивания составляет от 30°C до 65°C.
15. Лиофилизированная фармацевтическая композиция, содержащая соединения формулы (1):
(1)
или его фармацевтически приемлемой соли, полученное способом по любому из пп. 1-14.
| WO 2014134355 A1, 04.09.2014 | |||
| WO 2013033176 A1, 07.03.2013 | |||
| US 20140193456 A1, 10.07.2014 | |||
| US 7700567 B2, 20.04.2010. |

