Настоящее изобретение относится к препаратам. В частности, оно относится к композициям и препаратам соединений, родственных джорумицину, рениерамицину, сафрацину и сафрамицину, таких как соединения PM00104 и PM00121.
Предпосылки к созданию изобретения
Джорумицин представляет собой природное соединение, выделенное из кожи и из слизи тихоокеанского голожаберного моллюска Jorunna funebris (Fontana A. et al., Tetrahedron (2000), 56, 7305-8). Кроме того, известно, что семейство рениерамицинов выделено из губок и оболочников (James M.F. et al. J. Am. Chem. Soc. (1982), 104, 265-269; Oku N. et al. Journal Natural Products (2003), 66, 1136-9). Соединения сафрацина и сафрамицина раскрыты в публикации: Manzanares I. et al. Curr. Med. Chem. Anti-Cancer Agents (2001), 1, 257-276, а также в WO 00/18233 и WO 01/87894.
Вследствие наличия подробного описания в данных публикациях и приведенных в них ссылках структурные характеристики данных соединений в настоящей заявке детально не приведены; любой квалифицированный специалист в данной области способен получить такую информацию непосредственно из процитированных и родственных источников. По крайней мере два из данных соединений, PM00104 и PM00121, будут специально упомянуты здесь для иллюстрации отличительных характеристик данного изобретения.
PM00104 и PM00121 представляют собой синтетические алкалоиды, родственные джорумицину и рениерамицинам, а также соединениям сафрацина и сафрамицина. Они имеют следующие химические структуры:
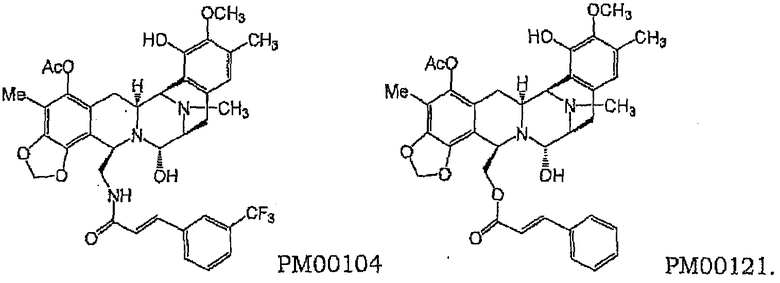
Фармацевтическая композиция, включающая PM00104 или PM00121 вместе с фармацевтически приемлемым носителем, заявлена в WO 01/87894.
Соединение PM00104 продемонстрировало значительную активность in vitro против клеточных линий солидных и несолидных опухолей, а также значительную активность in vivo в некоторых ксенотрансплантированных мышам клеточных линиях человека, таких как линии грудной железы и простаты. Предварительные представления о механизме действия PM00104 предполагали изменения в клеточном цикле, ДНК-связывающие свойства и ингибирование транскрипции. Кроме того, в настоящее время для соединения PM00104 проводятся клинические испытания I этапа. Дополнительные подробности, касающиеся данных по активности PM00104 и PM00121, см. в WO 01/87894.
PM00104 и PM00121, а также родственные соединения представляют собой сложные химические объекты, что следует из их структурных характеристик. Кроме того, они проявляют ограниченную растворимость в воде, и их стабильность, особенно в биосовместимых формах и препаратах, является труднопредсказуемой и труднодостижимой. Данные характеристики создают затруднения для специалистов и для стандартных методологий в данной области, в частности, когда дело касается получения препаратов данных соединений, которые должны без труда использоваться в медицинских целях. Такие применения предпочтительно основываются на препаратах, характеристики которых включают одно или более из следующего: биосовместимость, стабильность в условиях окружающей среды или в условиях, которые, насколько это возможно, близки к условиям окружающей среды, с максимально возможным сроком хранения и способностью к легкому восстановлению с образованием восстановленных растворов, которые стабильны в условиях окружающей среды или близких к ним условиях, в течение максимально возможного времени.
Ввиду потенциальной возможности применения данных соединений как противоопухолевых агентов существует необходимость предоставить препарат, который может решить проблемы, которые стандартные препараты и производственные методологии не решают или решают не полностью. Данные проблемы включают проблему стабильности данных соединений. Варианты осуществления препаратов PM00104, PM00121 и родственных соединений предпочтительно должны показывать благоприятные лиофилизационные свойства, предпочтительно должны поддаваться легкому восстановлению, и они предпочтительно должны обладать свойствами подвергаться разведению, например разведению инфузионной жидкостью, проявляя в то же время в максимально возможной степени желаемые характеристики препаратов для медицинского применения. Как указано выше, варианты осуществления данных препаратов должны быть стабильными в течение длительного хранения. Кроме того, препарат и технология его получения должны удовлетворять стандартам биосовместимости и должны, таким образом, допускать возможность эффективного применения наполнителя препарата, который нетоксичен, по крайней мере, в концентрациях, используемых для инфузии.
Общий обзор взаимодействий эксципиент-лекарственное средство в исходных препаратах дан в работе: Akers M.J., Journal of Pharmaceutical Sciences, 91, 2002, 2283-2300. Данная публикация содержит среди прочего раздел о наполнителях и лиопротекторах, включая указанный объект в контексте лиофилизации.
Предполагается, что методологии и препараты, разработанные в контексте данного изобретения, применимы к другим родственным соединениям помимо PM00104 и PM00121.
Объекты изобретения
Изобретение относится к композициям и препаратам соединений общей формулы (I):
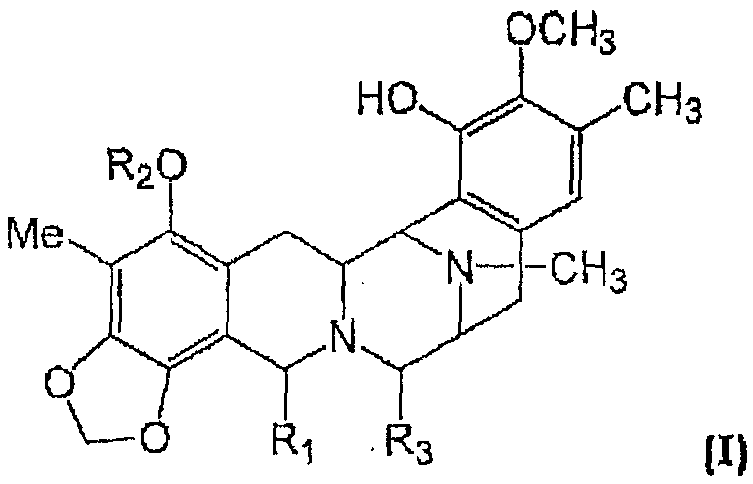
где R1 выбран из группы, состоящей из -CH2-N(Ra)2 и -CH2-ORa, где каждый Ra независимо выбран из группы, состоящей из H, алкил-CO-, галогеналкил-CO-, циклоалкилалкил-CO-, галогеналкил-O-CO-, арилалкил-CO-, арилалкенил-CO-, гетероарил-CO-, алкенил-CO-, алкила, алкенила и аминокислотного ацила, или же две Ra-группы вместе с N-атомом фрагмента -CH2-N(Ra)2 образуют гетероциклическую группу;
R2 выбран из алкил-CO-, циклоалкил-CO- и галогеналкил-CO- и
R3 представляет собой OH или CN, или
к их фармацевтически приемлемой соли, производному, пролекарству или стереоизомеру. Различные группы могут быть незамещенными или замещенными.
Таким образом, настоящее изобретение предоставляет стабильные препараты соединений общей формулы (I) и способы получения таких препаратов.
Цель настоящего изобретения заключается в том, чтобы предоставить новый стабильный препарат соединений общей формулы (I). В частности, необходим препарат со значительным сроком хранения. Кроме того, особенно важно избежать образования примесей.
Сущность изобретения
Согласно настоящему изобретению предоставлены композиции, которые включают соединение общей формулы (I) и дисахарид, и способы получения таких композиций. Предпочтительные варианты таких композиций обладают фармацевтической чистотой.
Некоторые варианты таких композиций представлены лиофилизированными препаратами, которые включают соединение общей формулы (I) и дисахарид. Даны способы получения таких препаратов.
Подробное описание изобретения
В контексте данного изобретения было обнаружено, что дисахариды стабилизируют препараты соединений общей формулы (I), определенной выше.
В данных соединениях заместители могут быть выбраны в соответствии со следующим руководством:
Алкильные группы предпочтительно содержат от 1 до 12 атомов углерода. Один более предпочтительный класс алкильных групп содержит от 1 до примерно 6 атомов углерода и, наиболее предпочтительно, 1, 2, 3 или 4 атома углерода. Метил, этил и пропил, включая изопропил, представляют собой особенно предпочтительные алкильные группы в соединениях настоящего изобретения. Термин алкил, если не указано особо, относится как к циклическим, так и к нециклическим группам, хотя циклические группы будут включать, по крайней мере, три углеродных циклических члена.
Предпочтительные алкенильные группы в соединениях настоящего изобретения содержат один или несколько ненасыщенных связей и от 2 до примерно 12 атомов углерода. Один более предпочтительный класс алкенильных групп содержит от 2 до примерно 6 атомов углерода и, наиболее предпочтительно, 2, 3 или 4 атома углерода. Термин алкенил относится как к циклическим, так и к нециклическим группам.
Подходящие арильные группы в соединениях настоящего изобретения включают моно- и полициклические соединения, включающие полициклические соединения, которые содержат изолированные и/или конденсированные арильные группы. Типичные арильные группы содержат от 1 до 3 изолированных или сконденсированных колец и от 6 до примерно 18 циклических атомов углерода. Особенно предпочтительные арильные группы включают замещенные или незамещенные фенил, нафтил, бифенил, фенантрил или антрацил.
Подходящие гетероциклические группы включают гетероароматические и гетероалициклические группы. Подходящие гетероароматические группы в соединениях настоящего изобретения содержат один, два или три гетероатома, выбранных из атомов N, O или S, и включают, например, кумаринильную, включающую 8-кумаринильную, хинолинильную, включающую 8-хинолинильную, пиридильную, пиразинильную, пиримидильную, фурильную, пирролильную, тиенильную, тиазолильную, оксазолильную, имидазолильную, индолильную, бензофуранильную и бензотиазольную группы. Подходящие гетероалициклические группы в соединениях настоящего изобретения содержат один, два или три гетероатома, выбранных из атомов N, O или S, и включают, например, тетрагидрофуранильную, тетрагидропиранильную, пиперидинильную, морфолиновую и пирролиндинильную группы. Фталимидо представляет собой другую возможную гетероциклическую группу.
Подходящие аминокислотные ацильные группы включают аланил, аргинил, аспартил, цистил, глутамил, глутаминил, глицил, гистидил, гидроксипролил, изолейцил, лейцил, лизил, метионил, фенилаланил, пролил, серил, треонил, тиронил, триптофил, тирозил, валил, а также другие аминокислотные группы, которые могут представлять собой L- или D-.
Упомянутые здесь группы могут быть замещенными в одном или более доступных положениях одной или более подходящими группами, такими как R', OR', =O, SR', SOR', SO2R', NO2, NHR', N(R')2, =N-R', NHCOR', N(COR')2, NHSO2R', CN, галоген, C(=O)R', CO2R', OC(=O)R', где каждая из групп R' независимо выбрана из группы, состоящей из водорода, OH, NO2, NH2, SH, CN, галогена, =O, C(=O)H, C(=O)алкила, CO2H, замещенного или незамещенного C1-C12-алкила, замещенного или незамещенного C2-C12-алкенила, замещенного или незамещенного C2-C12-алкинила и замещенного или незамещенного арила. Подходящие галогеновые заместители в соединениях настоящего изобретения включают F, Cl, Br и I. Если такие группы сами являются замещенными, заместители могут быть выбраны из вышеприведенного списка.
Термин “фармацевтически приемлемые соли, производные, пролекарства” относится к любой фармацевтически приемлемой соли, сложному эфиру, сольвату, гидрату или любому другому соединению, которое при введению реципиенту способно давать (прямо или опосредованно) соединение, как описано здесь. Однако будет принято во внимание, что соли, не являющиеся фармацевтически приемлемыми, также входит в объем изобретения, поскольку они могут быть пригодны для получения фармацевтически приемлемых солей. Получение солей, пролекарств и производных может быть проведено способами, известными в данной области.
Например, фармацевтически приемлемые соли предоставленных здесь соединений синтезируют из исходного соединения, которое содержит основный или кислотный фрагмент, стандартными химическими способами. В общем, такие соли получают, например, проводя реакцию свободных кислых или основных форм данных соединений со стехиометрическим количеством подходящих основания или кислоты в воде, или в органическом растворителе, или в смеси их обоих. В общем, неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил, являются предпочтительными. Примеры кислых аддитивных солей включают аддитивные соли минеральных кислот, такие как, например, гидрохлорид, гидробромид, гидроиодид, сульфат, нитрат, фосфат, и аддитивные соли органических кислот, такие как, например, ацетат, малеат, фумарат, цитрат, оксалат, сукцинат, тартрат, малат, манделат, метансульфонат и п-толуолсульфонат. Примеры щелочных аддитивных солей включают неорганические соли, такие как, например, натриевые, калиевые, кальциевые и аммонийные соли, и органические щелочные соли, такие как, например, этилендиаминовые, этаноламиновые, N,N-диалкиленэтаноламиновые, триэтаноламиновые и основные аминокислотные соли.
Соединения изобретения могут находиться в кристаллической форме либо как свободные соединения, либо как сольваты (например, гидраты), и предполагается, что обе формы входят в объем настоящего изобретения. Способы сольватации, в общем, известны в данной области.
Любое соединение, которое представляет собой пролекарство соединения формулы (I), входит в объем и сущность изобретения. Термин “пролекарство” использован в своем наиболее широком смысле и охватывает такие производные, которые in vivo превращаются в соединения изобретения. Такие производные легко представили бы себе специалисты в данной области, и они включают, например, соединения, где свободная гидроксильная группа превращена в сложноэфирное производное.
Соединения настоящего изобретения, представленные вышеописанной формулой (I), могут включать энантиомеры в зависимости от их асимметрии или диастереоизомеры. Стереоизомерия за счет двойной связи также возможна, следовательно, в некоторых случаях молекула могла бы существовать как (E)-изомер или как (Z)-изомер. Единственные изомеры и смеси изомеров подпадают под объем настоящего изобретения.
Примеры соединений настоящего изобретения включают соединения, раскрытые, например, в WO 00/18233 и WO 01/87894. Авторы настоящего изобретения включают посредством специальной ссылки каждое из соединений, определенных в соответствующих примерах данных PCT-заявок. В более общем смысле авторы настоящего изобретения включают посредством специальной ссылки содержание данных двух PCT-заявок, поскольку они раскрывают соединения настоящей формулы (I). Авторы признают упоминание предпочтительных групп, данных в указанных текстах, в частности, так как они относятся к настоящим группам R1 и R2, особенно R2.
R3 обычно представляет собой OH.
Предпочтительные соединения данного изобретения представляют собой соединения следующей химической структуры:
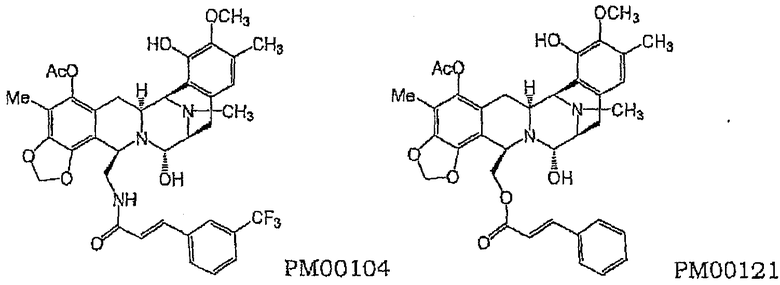
Соединения общей формулы (I), включая соединения PM00104 и PM00121, представляют собой сложные химические объекты, чье поведение в препаратах непредсказуемо в терминах поведения других неродственных химических веществ. Такое поведение еще сложнее предсказать, когда, по крайней мере, одно соединение общей формулы (I) включено в качестве активного вещества в препарат, который должен удовлетворять стандартам биосовместимости, включая медицинские стандарты. Авторы, далее, нашли в данном отношении, что применение дисахаридов в качестве наполнителей может радикально уменьшить образование примесей в ходе лиофилизации и хранения композиций соединений PM00104 и PM00121.
Кроме того, применение дисахаридов также улучшает условия хранения, делая возможным долговременное хранение лиофилизированного препарата в широком температурном диапазоне, включая условия охлаждения и комнатную температуру. Термин “стабильный”, использованный здесь, например, в выражении “стабильный препарат соединений PM00104 и PM00121”, относится к препарату, который удовлетворяет характеристикам стабильности, приведенным здесь, и их эквивалентам, которыми не обладают стандартные препараты и которые не достижимы, когда препарат получен стандартными производственными методологиями.
Примеры осуществлений настоящего изобретения представлены новыми фармацевтически приемлемыми композициями, включающими соединение общей формулы (I) и дисахарид. Примеры подходящих дисахаридов включают лактозу, трегалозу, сахарозу и их комбинации. Дополнительные примеры дисахаридов, которые могут быть использованы в некоторых осуществлениях данного изобретения, включают, по крайней мере, одну из: мальтозу, изомальтозу, целлобиозу, изосахарозу, изотрегалозу, сорбозу, туранозу, мелибиозу, гентиобиозу и их смеси. На данный момент предпочтительна сахароза. В других осуществлениях изобретения композиция включает соединение общей формулы (I) и свободный дисахарид лактозу. В других осуществлениях изобретения композиция включает соединение общей формулы (I) и свободный дисахарид трегалозу. В других осуществлениях изобретения композиция включает соединение общей формулы (I) и свободный дисахарид сахарозу. В других осуществлениях изобретения композиция включает соединение общей формулы (I) и свободный дисахарид мальтозу. В других осуществлениях изобретения композиция включает соединение общей формулы (I) и свободный дисахарид изомальтозу. В других осуществлениях изобретения композиция включает соединение общей формулы (I) и свободный дисахарид целлобиозу. В других осуществлениях изобретения композиция включает соединение общей формулы (I) и свободный дисахарид изосахарозу. В других осуществлениях изобретения композиция включает соединение общей формулы (I) и свободный дисахарид изотрегалозу. В других осуществлениях изобретения композиция включает соединение общей формулы (I) и свободный дисахарид сорбозу. В других осуществлениях изобретения композиция включает соединение общей формулы (I) и свободный дисахарид туранозу. В других осуществлениях изобретения композиция включает соединение общей формулы (I) и свободный дисахарид мелибиозу. В других осуществлениях изобретения композиция включает соединение общей формулы (I) и свободный дисахарид гентиобиозу. Таким образом, в некоторых осуществлениях композиция данного изобретения содержит, по крайней мере, один из дисахаридов, предпочтительно каждый из: лактозы, трегалозы, сахарозы, мальтозы, изомальтозы, целлобиозы, изосахарозы, изотрегалозы, сорбозы, туранозы, мелибиозы и гентиобиозы в количестве менее чем 2%, или менее чем 1%, или менее чем 0,5%, или менее чем 0,2%, или менее чем 0,1% по массе.
Термины “их смеси” и “их комбинации”, использованные здесь, относятся к, по крайней мере, двум объектам, которые обеспечивают антецедентный базис для терминов “их смеси” или “их комбинации”. В качестве иллюстрации, но не в качестве ограничения термины “продукт, включающий, по крайней мере, один из: A, B, C и их смеси” относится к осуществлениям продукта, для которых выполнено любое из следующего: A находится в продукте; B находится в продукте; C находится в продукте; A и B находятся в продукте; A и C находятся в продукте; B и C находятся в продукте и A, B и C находятся в продукте.
Более того, понятно, что термины, такие как “реагирование”, “образование” и родственные термины, примененные в настоящей заявке к химическому объекту, относятся к любому из: (a) к химическому объекту самому по себе и (b) к химическому объекту в форме, в которой такой объекту присутствует в реакционной среде. Аналогично, чтобы назвать химический объект или дать его формулу в контексте операции или реакционной стадии или для того, чтобы назвать его или дать его формулу, когда он находится в среде, твердой или жидкой, включающей продукты, препараты и комбинации, необходимо учесть, что в настоящей заявке название относится к любому из: (a) к химическому объекту самому по себе и (b) к химическому объекту в форме, в которой такой объект присутствует в среде. Например, наименование кислого химического объекта относится в настоящей заявке к любой форме или к любым формам, в которых такой объект присутствует в контексте, в котором он назван. В качестве иллюстрации, но не в качестве ограничения наименование химического объекта “хлорид натрия” или указание его химической формулы относится в настоящей заявке к объекту NaCl как такой двухатомной молекуле, если это представляет собой форму, в которой хлорид натрия присутствует в релевантной среде; наименование также относится к совокупности недиссоциированных и/или диссоциированных химических частиц, если хлорид натрия в релевантной среде полностью или частично диссоциирован, включая частицы в такой среде, которые сольватированы, являются частью каркасов, ассоциированы с другими частицами и так далее.
Чтобы дать более краткое описание, некоторые количественные выражения, данные здесь, не квалифицируются термином “примерно”. Понятно, что независимо от того, использован термин “примерно” явно или нет, подразумевается, что каждое количество, данное здесь, относится к фактически данной величине, а также подразумевается, что оно относится к приближенному значению такой данной величины, которое было бы обоснованно включено на основе стандартных знаний в данной области, включая эквиваленты и приближенные значения, обусловленные условиями экспериментов и/или измерений для такой данной величины.
Активное вещество или активные вещества в контексте данного изобретения могут иметь природное, полусинтетическое или синтетическое происхождение, включая комбинации происхождений. В осуществлениях, где активное вещество представляет собой соединение, такое как PM00104 или PM00121, данные соединения имеют синтетическое или полусинтетическое происхождение и могут быть получены, следуя раскрытию публикации WO 01/87894, которая полностью включена посредством ссылки.
Отношение активного вещества к наполнителю в осуществлениях данного изобретения определяется в соответствии с растворимостью наполнителя и, когда препарат лиофилизирован, также в соответствии со способностью наполнителя к лиофилизации. Предполагается, что данное отношение (мас./мас.) может составлять примерно 1:1 в некоторых осуществлениях, примерно 1:5 в других осуществлениях, примерно 1:10 в еще одних других осуществлениях, тогда как другие осуществления иллюстрируют отношения в диапазоне от примерно 1:10 до примерно 1:1. Предполагается, что другие осуществления имеют такие отношения в диапазоне от примерно 1:10 до примерно 1:80, и другие дополнительные осуществления имеют такие отношения в диапазоне от примерно 1:80 до примерно 1:1500. Когда активное соединение представляет собой PM00104 или PM00121, отношение (мас./мас.) активного ингредиента к наполнителю типично равно от примерно 1:80 до примерно 1:1500, предпочтительно от примерно 1:100 до примерно 1:800, более предпочтительно от примерно 1:100 до примерно 1:400 и наиболее предпочтительно примерно 1:200.
Лиофилизированный материал обычно присутствует во флаконе, который содержит определенное количество активного соединения. Когда активное соединение представляет собой PM00104, активные количества иллюстрируются 2,5 мг/флакон. Когда активное соединение представляет собой PM00121, активные количества иллюстрируются 1 мг/флакон.
Настоящее изобретение не ограничено определенными формами контейнера или дизайнами контейнера, если контейнер подходит для применения по назначению и удовлетворяет предусмотренным стандартам. Осуществления данного изобретения представлены препаратом, содержащимся во флаконах.
Лиофилизированные препараты данного изобретения могут быть восстановлены и разбавлены, давая композицию данного изобретения в форме раствора, готового для внутривенной инъекции. Фактические количества восстановленной жидкости не являются ограничивающими характеристиками осуществлений данного изобретения. В качестве иллюстрации, но не в качестве ограничения осуществления лиофилизированных препаратов по данному изобретению восстановлены с использованием некоторого объема воды. Большинство данных объемов не превышают примерно 20 мл, причем предпочтительные объемы находятся в диапазоне от примерно 1 мл до примерно 15 мл, более предпочтительно в диапазоне от примерно 1 мл до примерно 10 мл и даже более предпочтительно в диапазоне от примерно 3 мл до примерно 8 мл и наиболее предпочтительно объемы равны примерно 5 мл. Когда активное соединение представлено соединением PM00104, восстановленный раствор в таких осуществлениях содержит концентрацию PM00104 вплоть до 5 мг/мл, причем концентрации примерно 2,5 мг/мл, примерно 1 мг/мл и примерно 0,5 мг/мл являются предпочтительными.
Восстановленные осуществления настоящего изобретения могут быть далее разбавлены по желанию, причем данное дальнейшее разбавление не является ограничением настоящего изобретения. Данное дальнейшее разбавление предпочтительно осуществляют водной системой, которая обычно представляет собой 0,9% хлорид натрия или 5% глюкозу. Восстановленный раствор будет разбавлен в зависимости от концентрации в восстановленном растворе и желаемой концентрации в разбавленном растворе.
Осуществления препаратов соединений формулы (I) по данному изобретению могут быть использованы для лечения различных раковых заболеваний. Понятно, что “лечение” в данном контексте относится к действию, которое ведет к уменьшению симптомов ракового(ых) состояния(ий). Более того, осуществления препаратов по данному изобретению могут быть использованы в исследованиях с использованием лабораторных тканей, включая названные, но не ограничиваясь ими: клинические исследования, аналитические исследования и моделирующие анализы.
Осуществления данного изобретения, которые включают соединения формулы (I), предпочтительно вводят инфузией. Инфузионную стадию типично повторяют по циклическому базису, который по необходимости может быть повторен, например, в 1 до 20 циклов. Цикл включает фазу инфузии препарата соединения формулы (I) и обычно также фазу, не включающую инфузию активного вещества. Типично, цикл разрабатывают в течение нескольких недель, и, таким образом, цикл обычно включает одну или несколько недель фазы инфузии активного вещества и одну или несколько недель для завершения цикла. Авторы предпочитают, чтобы были использованы времена инфузии вплоть до 24 часов, более предпочтительно 1-12 часов, причем 1-6 часов являются наиболее предпочтительными временами. Короткие времена инфузии, которые позволяют проводить лечение без необходимости ночного пребывания в больнице, являются особенно желательными. Однако инфузия может занимать от 12 до 24 часов или даже быть более продолжительной, если это необходимо.
Осуществления препаратов данного изобретения, которые содержат соединение формулы (I), могут быть получены лиофилизацией композиции данного изобретения в форме основного раствора, включающего соединение формулы (I) и дисахарид. Обычно основной раствор будет забуферен, например, до pH примерно 4. Подходящие буферные агенты включают фосфатный буфер, цитратный буфер, фосфатный/цитратный буфер (смесь фосфатного буфера и цитратного буфера), лактатный буфер, аскорбатный буфер, тартарный/цитратный буфер, бикарбонатный/солянокислый буфер, ацетатный буфер, сукцинатный буфер и глициновый/солянокислый буфер. Могут быть использованы смеси буферов. Биосовместимые буферы, которые позволяют контролировать pH при желаемом значении, представляют дополнительные осуществления данного изобретения.
Другие компоненты могут быть включены в основной раствор, например поверхностно-активные вещества, такие как полиоксиэтиленсорбитанмоноолеат (также известный как полисорбат) или полиоксил-40-стеарат. Другие возможные поверхностно-активные вещества включают фосфолипиды, такие как лецитин; сополимеры полиоксиэтилена-полиоксипропилена, такие как поверхностно-активное вещество Pluronic; полиоксиэтиленовые сложные эфиры 12-гидроксистеариновой кислоты, такие как поверхностно-активное вещество Solutol, этоксилаты холестерина, такие как диацилглицерин, диалкилглицерин; соли желчных кислот, такие как холат натрия, деоксихолат натрия; сложные эфиры сахарозы, такие как монолаурат сахарозы, моноолеат сахарозы; поливинилпирролидон (PVP) или поливиниловый спирт (PVA).
Препарат обычно поставляют в виде флакона, содержащего лиофилизированный продукт. Данная форма для поставки, однако, не является ограничением настоящего изобретения. Чтобы предоставить флакон, содержащий лиофилизированный продукт, основной раствор добавляют во флакон и проводят его лиофилизацию.
Лиофилизацию проводят в некоторых осуществлениях данного изобретения, используя сокращенные времена вторичной сушки. Предпочтительный протокол включает охлаждение до температуры от примерно -40°C до примерно -50°C, первичное высушивание при 80-85 мкбар в течение 25-50 часов и вторичное высушивание при более низком давлении и при температуре выше 0°C в течение 3-20 часов.
Осуществления данного изобретения включают лиофилизацию охлаждением продукта ниже -40°C. Первичную сушку проводят при температуре от примерно -20°C до примерно -27°C и давлении, равном примерно 85 мкбар, в течение приблизительно 35-46 часов. Вторичную сушку выполняют при температуре от примерно 20°C до примерно 25°C в течение приблизительно 30-45 часов.
Осуществления препаратов данного изобретения подходят для хранения при температурах, значительно больших, чем температуры хранения стандартных препаратов. Примеры температур хранения препаратов по данному изобретению составляют около +5°C. Данные температуры легко обеспечиваются обычными холодильниками.
Описание чертежа изобретения
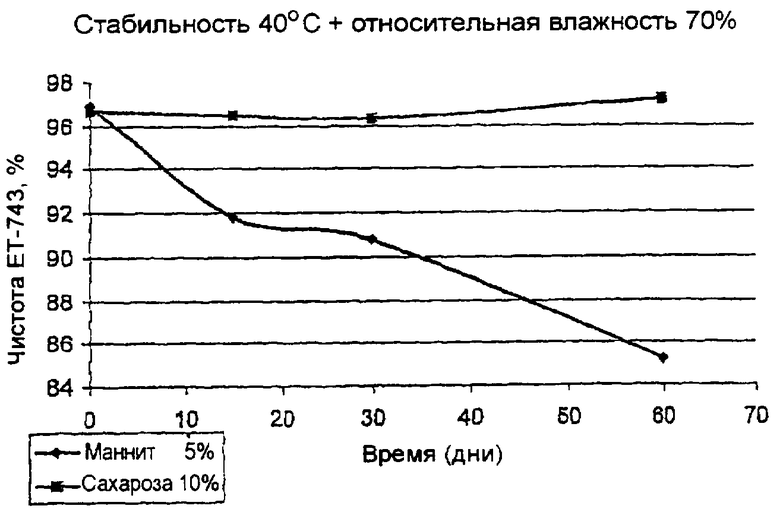
Чертеж - Сравнительная эволюция чистоты PM00104 в % двух препаратов PM00104, причем один включает сахарозу, а другой - маннит, хранившихся при 40°C/относительной влажности 70% в течение 3 месяцев.
Примеры
Пример 1
Данный пример иллюстрирует настоящее изобретение и раскрывает сравнительное исследование стабильности двух препаратов PM00104, где в одном препарате в качестве наполнителя использован маннит, а в другом препарате использована сахароза, которая представляет собой дисахарид.
Композиция основного раствора каждого из препаратов имела следующий состав (Таблица I):
Основные растворы получали и лиофилизировали стандартизованным способом.
Препарат с маннитом
Получали препарат с маннитом в объеме 50 мл:
40 мл 0,05 М (pH 4) раствора дигидрофосфата калия добавляли к 5,493 мг соединения PM00104 и поддерживали перемешивание смеси в течение 1 часа.
Затем добавляли 2,5 г маннита, промывая чашку 5 мл раствора фосфатного буфера (pH 4). Смесь перемешивали в течение еще одного часа.
Вслед за этим pH раствора доводили до pH 4, используя 1 N фосфорную кислоту, и раствор доводили до конечной массы 52 г, используя 0,05 M (pH 4) фосфатный буфер.
Раствор фильтровали через PVDF-фильтр, и профильтрованный раствор помещали в стеклянные флаконы объемом 10 мл в количестве 2 мл/флакон, и флаконы лиофилизировали согласно следующей методике (Таблица II):
После лиофилизации флаконы герметизировали и переносили в охлаждаемое место (-20°C).
Препарат с сахарозой
Получали препарат с сахарозой в объеме 300 мл:
32,615 мг соединения PM00104 добавляли к 100 мл раствора 0,05 М (pH 4) дигидрофосфата калия, промывая чашку дополнительными 110 мл раствора 0,05 М (pH 4) дигидрофосфата калия. Затем поддерживали перемешивание смеси в течение 1 часа.
Добавляли 30 г сахарозы, промывая чашку 30 мл раствора фосфатного буфера (pH 4). Поддерживали перемешивание смеси в течение еще одного часа.
Вслед за этим pH раствора доводили до pH 4, используя 1 M фосфорную кислоту, и раствор доводили до конечной массы 300 г, используя воду для инъекций.
Раствор фильтровали через фильтр Millipore-Optiscale, и профильтрованный раствор помещали в стеклянные флаконы объемом 10 мл в количестве 2 мл/флакон, и флаконы лиофилизировали.
Испытание на стабильность проводили при температуре 5°C, 25°C/относительная влажность 60% и 40°C/относительная влажность 75% в случае препарата с сахарозой и при 40°C/относительная влажность 75% в случае препарата с маннитом.
В Таблице III и на чертеже показана эволюция хроматографической чистоты по соединению PM00104 исследуемых препаратов:
Данные Таблицы III и чертеж свидетельствуют, что препарат, содержащий сахарозу, показывал лучшую стабильность при 40°C и относительной влажности 75% при незначительном понижении чистоты. Данное понижение значительно меньше, чем понижение, замеченное для препарата с маннитом.
Пример 2
Получали препарат соединения PM00121, включающий сахарозу в качестве наполнителя, и оценивали его стабильность при температуре 5°C, 25°C/относительная влажность 60% и 40°C/относительная влажность 75%.
В каждом флаконе композиция основного раствора имела следующий состав (Таблица IV):
Препарат соединения PM00121 получали следующим образом:
100 мл 0,1% (pH 2,5) раствора полисорбата 80 добавляли к 161,05 мг соединения PM00121 и впоследствии также добавляли дополнительные 110 мл 0,1% (pH 2,5) раствора полисорбата 80. Поддерживали перемешивание смеси в течение 1 часа.
Затем добавляли 2,04 г дигидрофосфата калия, промывая чашку 15 мл раствора 0,1% (pH 2,5) раствора полисорбата 80.
Вслед за этим 30 г сахарозы взвешивали и добавляли к раствору, промывая чашку 15 мл 0,1% (pH 2,5) раствора полисорбата 80. Затем поддерживали перемешивание смеси более чем 1 час.
Вслед за этим pH раствора доводили до pH 4, используя 1 M фосфорную кислоту, и раствор доводили до конечной массы 300 г, используя воду для инъекций.
Раствор фильтровали, используя фильтр Millipore-Optiscale. Профильтрованный раствор помещали в стеклянные флаконы объемом 10 мл в количестве 2 мл/флакон и флаконы выдерживали при -20°C до проведения лиофилизации.
Лиофилизацию проводили согласно нижеследующей Таблице V:
После лиофилизации флаконы герметизировали и переносили в охлаждаемое место (-20°C).
Испытание на стабильность проводили при температуре 5°C, 25°C/относительная влажность 60% и 40°C/относительная влажность 75%.
В Таблице VI приведена хроматографическая чистота по соединению PM00121 исследуемого препарата:
Было отмечено, что препарат, включающий дисахарид, стабилен при 5°C и 25°C/относительной влажности 60%.
Пример 3
Получали два препарата, 104-FA и 104-FB, соединения PM00104, включающие сахарозу в качестве наполнителя, и оценивали их стабильность при температуре -20°C, 5°C, 25°C/относительная влажность 60% и 45°C/относительная влажность 75%.
Для каждого препарата композиция основного раствора в каждом флаконе имела следующий состав (Таблица VII):
Основные растворы получали и лиофилизировали, используя следующие протоколы:
Препарат 104-FA
1,750 л основного раствора получали следующим образом:
153,125 мл 0,05 N фосфорной кислоты добавляли к 905,61 мг соединения PM00104. Смесь перемешивали в течение 15 минут. Затем добавляли 1400 мл воды для инъекций, за чем следовало добавление 11,9 г дигидрофосфата калия и 175 г сахарозы. Смесь вновь перемешивали в течение 1 ч 15 мин.
Не было необходимости доводить pH раствора до 3,8≤pH≤4, поскольку значение его pH составляло 3,91. Раствор доводили до конечной массы 1820 г, используя воду для инъекций.
Затем раствор фильтровали через фильтр Millipack®-20 0,22 мкм. Профильтрованный раствор помещали в стеклянные флаконы объемом 25 мл в количестве 5,4 мл основного раствора/флакон и флаконы выдерживали при -20°C до проведения лиофилизации.
Лиофилизацию проводили согласно нижеследующей Таблице VIII:
Флаконы герметизировали и переносили в охлаждаемое место (-20°C).
Препарат 104-FB
2,271 г соединения PM00104 добавляли к 100 мл 0,05 N фосфорной кислоты, промывая чашку 265 мл 0,05 N фосфорной кислоты. Смесь перемешивали в течение 15 минут. Затем добавляли 3360 мл воды для инъекций, за чем следовало добавление 28,56 г дигидрофосфата калия. Смесь перемешивали в течение 3 минут и добавляли 420 г сахарозы. Смесь вновь перемешивали в течение 1 ч 15 мин.
Не было необходимости доводить pH раствора до 3,8≤pH≤4, поскольку значение его pH составляло 3,84. Раствор доводили до конечной массы 4369 г, используя воду для инъекций.
Затем раствор фильтровали через фильтр 0,22 мкм. Профильтрованный раствор помещали в стеклянные флаконы объемом 25 мл в количестве 5 мл основного раствора/флакон и флаконы выдерживали при -20°C до проведения лиофилизации.
Лиофилизацию проводили согласно нижеследующей Таблице IX:
После лиофилизации флаконы герметизировали и переносили в охлаждаемое место (-20°C).
Испытание на стабильность проводили на обоих препаратах при температуре -20°C±5°C, 5°C±3°C, 25°C±2°C/относительная влажность 60%±5% и 40°C±2°C/относительная влажность 70%±5%.
В Таблице X приведена эволюция хроматографической чистоты по соединению PM00104 препарата 104-FA во время хранения при -20°C, 5°C, 25°C/относительная влажность 60% и 40°C/относительная влажность 75%.
относительная влажность 60%
относительная влажность 75%
В Таблице XI приведена эволюция хроматографической чистоты по соединению PM00104 препарата 104-FB во время хранения при -20°C, 5°C, 25°C/относительная влажность 60% и 40°C/относительная влажность 70%.
относительная влажность 60%
относительная влажность 75%
Данные Таблиц X и XI показывают, что эволюция чистоты препаратов, хранившихся при 5°C и 25°C/относительная влажность 60%, сопоставима с эволюциями препарата, хранившегося при -20°C. Следовательно, существенное разложение не обнаруживалось при 5°C и 25°C/относительная влажность 60%, что указывает на то, что препараты, включающие дисахарид, можно хранить, по крайней мере, при +5°C в течение продолжительного периода времени.
Все процитированные здесь ссылки включены посредством ссылки во всей полноте. Отличительные признаки и преимущества данного изобретения очевидны в свете данного здесь раскрытия. На основе данного раскрытия могут быть произведены модификации и изменения, подходящие для разных условий и применений, образующие, таким образом, осуществления в пределах объема данного изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЭКТИНЭСАЙДИН И ДИСАХАРИД | 2005 |
|
RU2382647C2 |
| Фармацевтическая композиция нейропротекторного действия для парентерального применения на основе гексаметилендиамида бис-(N-моносукцинил-L-глутамил-L-лизина) в лиофилизированной лекарственной форме | 2017 |
|
RU2678203C2 |
| ПРЕПАРАТ ДЛЯ ИНЪЕКЦИЙ НА ОСНОВЕ САПОНИНА B4 PULSATILLA | 2017 |
|
RU2759382C2 |
| СТАБИЛЬНЫЙ ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ НА ОСНОВЕ АНТИТЕЛА К PD-1 И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2016 |
|
RU2731418C2 |
| УПАКОВКА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2019 |
|
RU2798169C2 |
| ЛИОФИЛИЗИРОВАННАЯ ИНЪЕЦИРУЕМАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ПОЛУСИНТЕТИЧЕСКИХ АЛКАЛОИДОВ VINCA И УГЛЕВОДА, СТАБИЛЬНАЯ ПРИ КОМНАТНОЙ ТЕМПЕРАТУРЕ | 2007 |
|
RU2449791C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ АНТИТЕЛА К PCSK-9, И ЕЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2782792C2 |
| ЛИОФИЛИЗИРОВАННЫЙ ПРЕПАРАТ ЦИТОТОКСИЧЕСКИХ ДИПЕПТИДОВ | 2012 |
|
RU2597154C2 |
| ЛИОФИЛИЗИРОВАННЫЕ ЛИПОСОМЫ | 2012 |
|
RU2780489C2 |
| ЛИОФИЛИЗИРОВАННЫЕ ЛИПОСОМЫ | 2012 |
|
RU2648753C2 |
Изобретение относится к области фармацевтики и медицины и касается фармацевтической композиции, обладающей противоопухолевой активностью, включающей дисахарид и соединение формулы (1), способа получения данной композиции, изделия, включающего данную композицию, и способа лечения пролиферативных заболеваний с помощью данной композиции. 5 н. и 17 з.п. ф-лы, 11 табл., 1 ил.
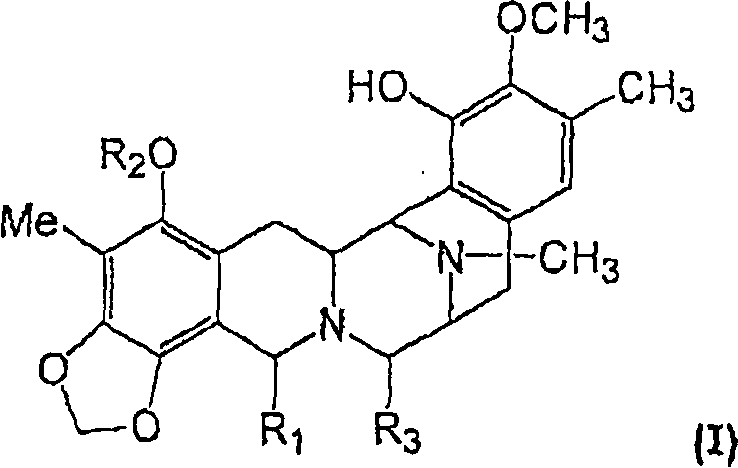
1. Фармацевтическая композиция, обладающая противоопухолевой активностью, которая включает дисахарид и соединение общей формулы (I)
где R1 выбран из группы, состоящей из -CH2-N(Ra)2 и -CH2-ORa, где каждый Ra независимо выбран из группы, состоящей из Н, алкил-СО-, галогеналкил-СО-, циклоалкилалкил-СО-, галогеналкил-O-СО-, арилалкил-СО-, арилалкенил-СО-, гетероарил-СО-, алкенил-СО-, алкила, алкенила и аминокислотного ацила, или две Ra-группы вместе с N-атомом фрагмента -CH2-N(Ra)2 образуют гетероциклическую группу;
R2 выбран из алкил-СО-, циклоалкил-СО- и галогеналкил-СО- и
R3 представляет собой ОН или СN; или
его фармацевтически приемлемая соль, производное, пролекарство или стереоизомер, и где различные группы могут быть незамещенными или замещенными.
2. Композиция по п.1, где указанное соединение выбрано из РМ00104 и РМ00121.
3. Композиция по любому предшествующему пункту, где указанный дисахарид выбран из группы, состоящей из лактозы, трегалозы, сахарозы и их смесей.
4. Композиция по п.3, где указанный дисахарид представляет собой сахарозу.
5. Композиция по п.1, где отношение (мас./мас.) соединения к дисахариду равно от примерно 1:80 до примерно 1:1500.
6. Композиция по п.5, где отношение (мас./мас.) соединения к дисахариду составляет от примерно 1:100 до примерно 1:400.
7. Композиция по п.6, где отношение (мас./мас.) соединения к дисахариду составляет примерно 1:200.
8. Композиция по п.1, которая дополнительно включает буферный агент.
9. Композиция по п.8, где указанный буферный агент представляет собой фосфатный буфер.
10. Композиция по п.1, которая дополнительно включает поверхностно-активное вещество.
11. Композиция по п.10, где поверхностно-активное вещество представляет собой полиоксиэтиленсорбитанмоноолеат.
12. Композиция по п.1, которая находится в форме лиофилизированной препаративной формы.
13. Композиция по п.12, где лиофилизированная препаративная форма находится во флаконе и включает количество соединения РМ00104 или РМ00121.
14. Композиция по п.13, где указанное количество соединения РМ00104 равно примерно 2,5 мг.
15. Композиция по п.14, где указанный флакон содержит препаративную форму, включающую: примерно 2,5 мг РМ00104, примерно 500 мг сахарозы и примерно 34 мг фосфата, где указанные 34 мг фосфата рассчитаны как дигидрофосфат калия.
16. Способ изготовления флакона, содержащего лиофилизированную композицию по п.12, включающий добавление раствора, включающего композицию по любому из пп.1-11 во флакон и лиофилизацию указанного раствора.
17. Способ по п.16, где соединение представляет собой РМ00104.
18. Способ уменьшения образования примесей в лиофилизированной препаративной форме по п.12, включающий лиофилизацию основного раствора, который включает указанное соединение и дисахарид.
19. Способ по п.18, где соединение представляет собой РМ00104.
20. Способ получения раствора для внутривенной инфузии, включающий добавление воды к лиофилизированной препаративной форме по п.12 для образования восстановленного раствора и разбавление указанного восстановленного раствора водной системой.
21. Способ по п.20, где соединение представляет собой РМ00104.
22. Способ лечения рака, который включает внутривенную инфузию раствора, полученного способом по любому из пп.20 и 21.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| WO 00/18233 A1, 06.04.2000 | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| СПОСОБЫ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ЭСТЕИНАСЦИДИНА, СОЕДИНЕНИЯ, ПРОМЕЖУТОЧНЫЙ ПРОДУКТ | 1997 |
|
RU2194709C2 |

