Область техники, к которой относится настоящее изобретение
[0001] Настоящее изобретение относится к области фармацевтической технологии и, в частности, описывает фармацевтическую композицию, содержащую антагонист минералокортикоидных рецепторов, применение композиции в получении лекарственного средства для лечения и/или профилактики хронической болезни почек и способ лечения пациента с хронической болезнью почек с использованием композиции.
Предшествующий уровень техники настоящего изобретения
[0002] Хроническая болезнь почек (ХБП) представляет собой тип заболевания, характеризующийся (1) 3 или более месяцами поражения почек с или без сниженной скорости клубочковой фильтрации (СКФ); или (2) скоростью клубочковой фильтрации, составляющей менее 60 мл/мин/1,73 м2 в течение 3 месяцев или дольше с повреждением почек или без него; при этом повреждение почек определяют как патологические отклонения или маркеры повреждения, включая в себя отклонения в анализах крови или мочи или визуализирующих исследованиях [The National Kidney Foundation, NKF KDOQI Clinical Practice Guidelines for Chronic Kidney Disease: Evaluation, Classification, and Stratification]. Наиболее распространенные клинические симптомы включают в себя протеинурию (пенистую мочу), гематурию, отек, гипертензию, повышенное количество ночной мочи, анемию и тому подобное. Хроническая болезнь почек на поздних стадиях приводит к хронической почечной недостаточности и определенному клиническому синдрому, который развивается как системное расстройство, проявляющееся в накоплении метаболитов, дисбалансе воды/электролитов, ацидемии и т.д.
[0003] Альдостерон представляет собой стероидный гормон с минералокортикоидной активностью, который вырабатывается главным образом клубочковой зоной надпочечников [Kidney International (2012) 81, 955-968]. Альдостерон в основном функционирует для регуляции реабсорбции Na + и экскреции K + в дистальном нефроне, поддерживая электролитный баланс и объемный гомеостаз. В дополнение к задержке натрия альдостерон также может вызывать патологическое прогрессирование, приводящее к воспалению, ремоделированию и фиброзу. Альдостерон активирует минералокортикоидный рецептор (MP), воздействуя на кровеносные сосуды, вызывая сужение сосудов. Также показано, что альдостерон вызывает повреждение почечной ткани, что приводит к увеличенной протеинурии/альбуминурии. Избыточные содержания альдостерона приводят к гипертензии, сердечной недостаточности (СН) и хронической болезни почек (ХБП) [Expert Opin. Investig. Drugs (2015) 24 (8), 1-13].
[0004] Антагонист минералокортикоидных рецепторов (MRA) связывается с минералокортикоидным рецептором для блокирования взаимодействия альдостерона с минералокортикоидным рецептором [инструкция INSPRA, Pfizer, 2002]. MRA был одобрен как клинически эффективный для пациентов с сердечной недостаточностью со сниженной фракцией выброса, артериальной гипертензией и/или хронической болезнью почек [Curr Opin Nephrol Hypertens 2015, 24:417-424].
[0005] До настоящего времени для клинического применения были разработаны только два стероидных антагониста минералокортикоидных рецепторов. Спиронолактон, MRA первого поколения с относительно высокой активностью, характеризуется такими побочными эффектами, как гинекомастия, импотенция и нарушение менструального цикла из-за его структуры, сходной с прогестероном. Эплеренон, MRA второго поколения, обладает улучшенной селективностью, но сниженной активностью [Expert Opin. Investig. Drugs (2015) 24 (8), 1-13]. Как спиронолактон, так и эплеренон могут снизить вероятность госпитализации и/или смертности у пациентов с сердечной недостаточностью со сниженной фракцией выброса; уменьшить содержание альбумина в моче или отношение альбумина к креатинину в моче (UACR) и замедлить прогрессирование у пациентов с хронической болезнью почек. Тем не менее, риск развития гиперкалиемии ограничил применение этих двух MRA, особенно у пациентов с повреждениями почек [Expert Opin. Investig. Drugs (2015) 24 (8), 1-13; Kedney International 2012; 81: 955-968]. Применение спиронолактона у пациентов с тяжелым повреждением почек запрещено [инструкция Aldactone® (таблетки спиронолактона, USP)], в то время как эплеренон противопоказан пациентам с поражением почек средней и тяжелой степени тяжести с гипертензией, а также пациентам с тяжелыми повреждениями почек [инструкция INSPRA® (эплеренон), таблетки для перорального применения (USP)].
[0006] Финеренон представляет собой нестероидный MRA, разработанный Bayer, и обладает лучшей селективностью по отношению к MR по сравнению со спиронолактоном и более высокой аффинностью по отношению к MR по сравнению с эплереноном [Expert Opin. Investig. Drugs (2015) 24 (8), 1-13]. Финеренон был исследован в клинических испытаниях для лечения диабетической нефропатии (ARTS-DN), при этом UACR снижалось на 21%, 24%, 33% и 38% в группах, которым вводили финеренон в дозах, составляющих 7,5, 10, 15 и 20 мг/день, соответственно. Тем не менее, начало гиперкалиемии приводит к прекращению лечения в этих группах с частотой 2,1%, 0%, 3,2% и 1,7%, соответственно [JAMA. 2015; 314 (9): 884-894]. Проблема гиперкалиемии проявляется в группах дозировок, при которых терапевтический эффект является неудовлетворительным.
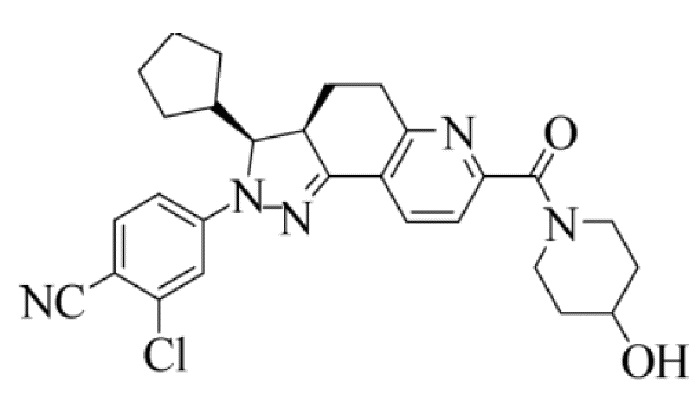
[0007] Соединение I, 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксилпиперидин-1-карбонил)-3,3а,4,5-тетрагидро-2H-пиразоло[3,4-ƒ]хинолин-2-ил]бензонитрил, характеризующееся следующей формулой, раскрыто в международных патентных публикациях WO 2012022121 A1 и WO 2014094664 A1. Это соединение также представляет собой нестероидный антагонист минералокортикоидных рецепторов (MRA), который демонстрирует относительно высокую селективность и аффинность по отношению к MR и, таким образом, является применимым в лечении хронической болезни почек. Тем не менее, поскольку лекарственные средства, нацеленно воздействующие на MR, всегда индуцируют гиперкалиемию, в предшествующем уровне техники не был раскрыт какой-либо продукт, содержащий соединение I, или какой-либо способ с использованием соединения I, который оказался безопасным и эффективным.
[0008] У здоровых субъектов приблизительно 90% калия выводится почками, при этом остальное выводится с потом и калом. У пациентов с хронической болезнью почек, с ухудшенной модулирующей способностью по отношению к ионам калия, с большой вероятностью развивается гиперкалиемия, в частности, у пациентов с заболеванием почек средней и тяжелой степени тяжести.
[0009] Повышенное содержание калия в сыворотке вызывает у пациентов повреждения, особенно повреждения сердца. Если состояние является серьезным, может возникнуть нарушение сердечного ритма, которое может привести к остановке сердца и даже смерти. В другом аспекте высокое содержание калия в сыворотке также может сделать нервно-мышечную систему менее возбужденной, что приводит к вялому параличу, или может вызывать ухудшение пищеварительной системы, что приводит к боли в животе, тошноте, рвоте или тому подобному.
[0010] Гиперкалиемия приводит к поражениям у пациентов с хронической болезнью почек. Она негативно воздействует на деполяризацию и реполяризацию кардиомиоцитов и, таким образом, вызывает замедленную проводимость электрических волн и аритмию сердца. Без лечения тяжелая гиперкалиемия может вызвать вентрикулярную фибрилляцию и остановку сердца, что приводит к внезапной сердечной смерти.
[0011] На современном этапе не существует клинически безопасного и эффективного лекарственного средства для лечения хронической болезни почек, несмотря на постоянную потребность в этом. Хотя антагонист минералокортикоидных рецепторов (MRA) продемонстрировал эффекты на лечение заболеваний почек, его клиническое применение ограничено из-за высокого содержания калия в сыворотке, вызванного механизмом, лежащим в основе его действия.
[0012] Следовательно, техническая задача состоит в том, чтобы найти безопасное и эффективное лекарственное средство или способ лечения хронической болезни почек, избегая при этом повышенных содержаний калия в сыворотке.
Краткое раскрытие настоящего изобретения
[0013] Авторы настоящего изобретения обнаружили, что соединение I не может быть использовано для безопасного и эффективного лечения хронической болезни почек, поскольку:
(1) Соединение I представляет собой антагонист минералокортикоидных рецепторов, который с высокой вероятностью вызывает повышенные содержания калия в сыворотке вследствие своего действия на мишени; и
(2) Абсорбция фармацевтической композиции, содержащей соединение I, варьируется среди пациентов, когда ее получают общепринятыми техническими средствами.
[0014] Таким образом, терапевтическое окно соединения I является достаточно узким при лечении пациентов с хронической болезнью почек. Иными словами, лекарственное средство, произведенное с помощью общепринятых технических средств, не соответствует клиническим требованиям.
[0015] С учетом вышеизложенного авторы настоящего изобретения провели обширные испытания и наконец решили проблему. В частности, авторы настоящего изобретения обнаружили корреляцию между возникновением повышенного содержания калия в сыворотке и площадью под кривой зависимости концентрации в плазме от времени (AUC), а также выяснили окно безопасности, которое может производить терапевтический эффект, не вызывая повышенные содержания калия в сыворотке. Более конкретно, безопасное и эффективное значение AUC соединения I находится в диапазоне от 188 нг*ч/мл до 3173 нг*ч/мл при введении у пациентов с хронической болезнью почек.
[0016] Для достижения окна безопасности, упомянутого выше, авторы настоящего изобретения обнаружили безопасный и эффективный диапазон доз и фармацевтическую композицию, обеспечивающую такой диапазон доз при введении субъектам. Фармацевтическая композиция согласно настоящему изобретению характеризуется биодоступностью, составляющей 50% или больше у млекопитающих. Когда фармацевтическую композицию согласно настоящему изобретению вводят пациенту с хронической болезнью почек в диапазоне доз, заявленном в настоящем изобретении, безопасное и эффективное значение AUC соединения I находится в диапазоне от 188 нг*ч/мл до 3173 нг*ч/мл.
[0017] Настоящее изобретение относится к фармацевтической композиции, содержащей соединение I и фармацевтически приемлемый носитель.
[0018] Когда пациенту с хронической болезнью почек вводят перорально фармацевтическую композицию согласно настоящему изобретению, эффективное и безопасное значение площади под кривой зависимости концентрации в плазме от времени (AUC) соединения I находится в диапазоне от 188 нг*ч/мл до 3173 нг*ч/мл. Согласно одному варианту осуществления настоящего изобретения эффективное и безопасное значение площади под кривой зависимости концентрации в плазме от I находится в диапазоне от 188 нг*ч/мл до 2893 нг*ч/мл. Согласно одному варианту осуществления настоящего изобретения эффективное и безопасное значение площади под кривой зависимости концентрации в плазме от времени (AUC) соединения I находится в диапазоне от 188 нг*ч/мл до 2613 нг*ч/мл. Согласно одному варианту осуществления настоящего изобретения эффективное и безопасное значение площади под кривой зависимости концентрации в плазме от времени (AUC) соединения I находится в диапазоне от 188 нг*ч/мл до 1117 нг*ч/мл. Согласно одному варианту осуществления настоящего изобретения эффективное и безопасное значение площади под кривой зависимости концентрации в плазме от времени (AUC) соединения I находится в диапазоне от 188 нг*ч/мл до 885 нг*ч/мл.
[0019] Учитывая, что биодоступность фармацевтической композиции согласно настоящему изобретению составляет 50% или больше у млекопитающих, для получения безопасного и эффективного значения AUC у пациентов с хронической болезнью почек, фармацевтическую композицию следует вводить в суточной дозе, составляющей 0,1 мг до 2,5 мг соединения I.
[0020] Согласно одному варианту осуществления настоящего изобретения фармацевтическую композицию согласно настоящему изобретению вводят в суточной дозе, составляющей 0,1 мг до 2,5 мг соединения I у пациента для получения безопасного и эффективного значения AUC, упомянутого выше. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,5 мг. Необязательно суточная доза соединения I составляет 0,1 мг, 0,15 мг, 0,2 мг, 0,25 мг, 0,3 мг, 0,35 мг, 0,4 мг, 0,45 мг, 0,5 мг, 0,6 мг, 0,7 мг, 0,8 мг, 0,9 мг, 1 мг, 1,5 мг, 2 мг или 2,5 мг.
[0021] Соединение I является нерастворимым в воде и характеризуется средней скоростью проникновения. Фармацевтическая композиция указанного соединения, произведенная с помощью общепринятых технических средств, обеспечивает низкую биодоступность, и при введении у субъектов наблюдается огромная индивидуальная вариабельность. Для получения клинически приемлемого эффекта, т.е. для эффективного лечения всех пациентов, общепринятым является повышение дозы. Тем не менее, из-за большой индивидуальной вариабельности у некоторых пациентов после введения соединения I наблюдается слишком высокое значение AUC, что приводит к повышенному риску повышенных содержаний калия в сыворотке. В частности, у пациентов с хронической болезнью почек способность модулировать ионы калия ухудшается, а повышенное содержание калия в сыворотке может привести к более высокому риску для здоровья.
[0022] Настоящая фармацевтическая композиция удовлетворяет требованиям клинической безопасности и эффективности за счет повышения биодоступности для снижения индивидуальной вариабельности AUC.
[0023] Для повышения биодоступности фармацевтической композиции до уровня, составляющего 50% или больше у млекопитающих, согласно одному варианту осуществления настоящего изобретения в фармацевтической композиции согласно настоящему изобретению уменьшают размер частиц соединения I. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 25 мкм или меньше. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 21,7 мкм или меньше. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 10 мкм или меньше. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 5 мкм или меньше.
[0024] Соединение I в фармацевтической композиции с различным размером частиц можно получить посредством помола, экструдирования, столкновения, разрезания, механического тонкого измельчения, вибрационного тонкого измельчения, измельчения в псевдоожиженном слое, ультразвуковой обработки, помола под высоким давлением, химического осаждения или тому подобного.
[0025] Для увеличения биодоступности фармацевтической композиции до уровня, составляющего 50% или больше у млекопитающих, поверхностно-активное вещество добавляют в фармацевтическую композицию согласно одному варианту осуществления.
[0026] Поверхностно-активное вещество представляет собой одно или несколько, выбранных из группы, состоящей из следующего: хлорид бензалкония, лаурилсульфонат натрия, додецилсульфат натрия, глицерин, холевая кислота, полоксамер, поливиниловый спирт, полисорбат 80, PVP K30 (ПВП K30) и полиэтиленгликоль. Предпочтительно поверхностно-активное вещество представляет собой одно или несколько, выбранных из группы, состоящей из хлорида бензалкония, лаурилсульфоната натрия и додецилсульфата натрия. Предпочтительно поверхностно-активное вещество представляет собой хлорид бензалкония, лаурилсульфонат натрия или додецилсульфат натрия.
[0027] В фармацевтической композиции согласно настоящему изобретению массовое отношение соединения I к поверхностно-активному веществу составляет 1:0,1-1:20, предпочтительно 1:1-1:20 и более предпочтительно 1:5-1:20.
[0028] Согласно настоящему изобретению предусмотрена фармацевтическая композиция, содержащая соединение I и фармацевтически приемлемый носитель.
[0029] Согласно одному варианту осуществления настоящего изобретения фармацевтическую композицию согласно настоящему изобретению вводят пациенту в суточной дозе, составляющей 0,1 мг до 2,5 мг соединения I. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,5 мг. Необязательно суточная доза соединения I составляет 0,1 мг, 0,15 мг, 0,2 мг, 0,25 мг, 0,3 мг, 0,35 мг, 0,4 мг, 0,45 мг, 0,5 мг, 0,6 мг, 0,7 мг, 0,8 мг, 0,9 мг, 1 мг, 1,5 мг, 2 мг или 2,5 мг.
[0030] Когда пациенту вводят фармацевтическую композицию согласно настоящему изобретению в дозе, упомянутой выше, эффективное и безопасное значение площади под кривой зависимости концентрации в плазме от времени (AUC) соединения I находится в диапазоне от 188 нг*ч/мл до 3173 нг*ч/мл. Согласно одному варианту осуществления настоящего изобретения эффективное и безопасное значение площади под кривой зависимости концентрации в плазме от времени (AUC) соединения I находится в диапазоне от 188 нг*ч/мл до 2893 нг*ч/мл. Согласно одному варианту осуществления настоящего изобретения эффективное и безопасное значение площади под кривой зависимости концентрации в плазме от времени (AUC) соединения I находится в диапазоне от 188 нг*ч/мл до 2613 нг*ч/мл. Согласно одному варианту осуществления настоящего изобретения эффективное и безопасное значение площади под кривой зависимости концентрации в плазме от времени (AUC) соединения I находится в диапазоне от 188 нг*ч/мл до 1117 нг*ч/мл. Согласно одному варианту осуществления настоящего изобретения эффективное и безопасное значение площади под кривой зависимости концентрации в плазме от времени (AUC) соединения I находится в диапазоне от 188 нг*ч/мл до 885 нг*ч/мл.
[0031] Соединение I является нерастворимым в воде и характеризуется средней скоростью проникновения. Фармацевтическая композиция указанного соединения, произведенная с помощью общепринятых технических средств, обеспечивает низкую биодоступность, и при введении у субъектов наблюдается огромная индивидуальная вариабельность. Для получения клинически приемлемого эффекта, т.е. для эффективного лечения всех пациентов, общепринятым является повышение дозы. Тем не менее, из-за значительной индивидуальной вариабельности у некоторых пациентов наблюдается слишком высокое значение AUC соединения I, что приводит к повышенному риску повышенных содержаний калия в сыворотке. В частности, у пациентов с хронической болезнью почек способность модулировать ионы калия ухудшается, а повышенное содержание калия в сыворотке приводит к более высокому риску для здоровья.
[0032] Настоящая фармацевтическая композиция удовлетворяет требованиям клинической безопасности и эффективности путем улучшения биодоступности для эффективного снижения индивидуальной вариабельности AUC.
[0033] Для повышения биодоступности фармацевтической композиции до уровня, составляющего 50% или больше у млекопитающих, согласно одному варианту осуществления настоящего изобретения в фармацевтической композиции согласно настоящему изобретению уменьшают размер частиц соединения I. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 25 мкм или меньше. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 21,7 мкм или меньше. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 10 мкм или меньше. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 5 мкм или меньше.
[0034] Соединение I в фармацевтической композиции с различным размером частиц можно получить посредством помола, экструдирования, столкновения, разрезания, механического тонкого измельчения, вибрационного тонкого измельчения, измельчения в псевдоожиженном слое, ультразвуковой обработки, помола под высоким давлением, химического осаждения или тому подобного.
[0035] Для увеличения биодоступности фармацевтической композиции до уровня, составляющего 50% или больше у млекопитающих, согласно одному варианту осуществления настоящего изобретения в фармацевтическую композицию добавляют поверхностно-активное вещество.
[0036] Поверхностно-активное вещество представляет собой одно или несколько, выбранных из группы, состоящей из следующего: хлорид бензалкония, лаурилсульфонат натрия, додецилсульфат натрия, глицерин, холевая кислота, полоксамер, поливиниловый спирт, полисорбат 80, PVP K30 и полиэтиленгликоль. Предпочтительно поверхностно-активное вещество представляет собой одно или несколько, выбранных из группы, состоящей из хлорида бензалкония, лаурилсульфоната натрия и додецилсульфата натрия. Предпочтительно поверхностно-активное вещество представляет собой хлорид бензалкония, лаурилсульфонат натрия или додецилсульфат натрия.
[0037] В фармацевтической композиции согласно настоящему изобретению массовое отношение соединения I к поверхностно-активному веществу составляет 1:0,1-1:20, предпочтительно 1:1-1:20 и более предпочтительно 1:5-1:20.
[0038] Настоящее изобретение относится к фармацевтической композиции, содержащей соединение I и фармацевтически приемлемый носитель.
[0039] Когда пациенту вводят фармацевтическую композицию согласно настоящему изобретению в суточной дозе, составляющей 0,1 мг до 2,5 мг соединения I, безопасная и эффективная площадь под кривой зависимости концентрации в плазме от времени (AUC) находится в диапазоне от 188 нг*ч/мл до 3173 нг*ч/мл.
[0040] В отношении фармацевтической композиции согласно настоящему изобретению необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,5 мг. Необязательно суточная доза соединения I составляет 0,1 мг, 0,15 мг, 0,2 мг, 0,25 мг, 0,3 мг, 0,35 мг, 0,4 мг, 0,45 мг, 0,5 мг, 0,6 мг, 0,7 мг, 0,8 мг, 0,9 мг, 1 мг, 1,5 мг, 2 мг или 2,5 мг.
[0041] Согласно одному варианту осуществления, когда пациенту вводят фармацевтическую композицию согласно настоящему изобретению, эффективное и безопасное значение площади под кривой зависимости концентрации в плазме от времени (AUC) соединения I находится в диапазоне от 188 нг*ч/мл до 2893 нг*ч/мл. Согласно одному варианту осуществления настоящего изобретения эффективное и безопасное значение площади под кривой зависимости концентрации в плазме от времени (AUC) соединения I находится в диапазоне от 188 нг*ч/мл до 2613 нг*ч/мл. Согласно одному варианту осуществления настоящего изобретения эффективное и безопасное значение площади под кривой зависимости концентрации в плазме от времени (AUC) соединения I находится в диапазоне от 188 нг*ч/мл до 1117 нг*ч/мл. Согласно одному варианту осуществления настоящего изобретения эффективное и безопасное значение площади под кривой зависимости концентрации в плазме от времени (AUC) соединения I находится в диапазоне от 188 нг*ч/мл до 885 нг*ч/мл.
[0042] Соединение I является нерастворимым в воде и характеризуется средней скоростью проникновения. Фармацевтическая композиция указанного соединения, произведенная с помощью общепринятых технических средств, обеспечивает низкую биодоступность, и при введении у субъектов наблюдается огромная индивидуальная вариабельность. Для получения клинически приемлемого эффекта, т.е. для эффективного лечения всех пациентов, общепринятым является повышение дозы. Тем не менее, из-за значительной индивидуальной вариабельности у некоторых пациентов наблюдается слишком высокое значение AUC соединения I, что приводит к повышенному риску повышенных содержаний калия в сыворотке. В частности, у пациентов с хронической болезнью почек способность модулировать ионы калия ухудшается, а повышенное содержание калия в сыворотке приводит к более высокому риску для здоровья.
[0043] Настоящая фармацевтическая композиция удовлетворяет требованиям клинической безопасности и эффективности путем улучшения биодоступности для эффективного снижения индивидуальной вариабельности AUC.
[0044] Для повышения биодоступности фармацевтической композиции до уровня, составляющего 50% или больше у млекопитающих, согласно одному варианту осуществления настоящего изобретения в фармацевтической композиции согласно настоящему изобретению уменьшают размер частиц соединения I. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 25 мкм или меньше. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 21,7 мкм или меньше. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 10 мкм или меньше. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 5 мкм или меньше.
[0045] Соединение I в фармацевтической композиции с различным размером частиц можно получить посредством помола, экструдирования, столкновения, разрезания, механического тонкого измельчения, вибрационного тонкого измельчения, измельчения в псевдоожиженном слое, ультразвуковой обработки, помола под высоким давлением, химического осаждения или тому подобного.
[0046] Для увеличения биодоступности фармацевтической композиции до уровня, составляющего 50% или больше у млекопитающих, согласно одному варианту осуществления настоящего изобретения в фармацевтическую композицию добавляют поверхностно-активное вещество.
[0047] Поверхностно-активное вещество представляет собой одно или несколько, выбранных из группы, состоящей из следующего: хлорид бензалкония, лаурилсульфонат натрия, додецилсульфат натрия, глицерин, холевая кислота, полоксамер, поливиниловый спирт, полисорбат 80, PVP K30 и полиэтиленгликоль. Предпочтительно поверхностно-активное вещество представляет собой одно или несколько, выбранных из группы, состоящей из хлорида бензалкония, лаурилсульфоната натрия и додецилсульфата натрия. Предпочтительно поверхностно-активное вещество представляет собой хлорид бензалкония, лаурилсульфонат натрия или додецилсульфат натрия.
[0048] В фармацевтической композиции согласно настоящему изобретению массовое отношение соединения I к поверхностно-активному веществу составляет 1:0,1-1:20, предпочтительно 1:1-1:20 и более предпочтительно 1:5-1:20.
[0049] Настоящее изобретение относится к применению фармацевтической композиции в получении лекарственного средства для лечения и/или профилактики хронической болезни почек.
[0050] Когда нуждающемуся в этом субъекту перорально вводят фармацевтическую композицию согласно настоящему изобретению, эффективное и безопасное значение площади под кривой зависимости концентрации в плазме от времени (AUC) соединения I находится в диапазоне от 188 нг*ч/мл до 3173 нг*ч/мл. Согласно одному варианту осуществления настоящего изобретения эффективное и безопасное значение площади под кривой зависимости концентрации в плазме от времени (AUC) соединения I находится в диапазоне от 188 нг*ч/мл до 2893 нг*ч/мл. Согласно одному варианту осуществления настоящего изобретения эффективное и безопасное значение площади под кривой зависимости концентрации в плазме от времени (AUC) соединения I находится в диапазоне от 188 нг*ч/мл до 2613 нг*ч/мл. Согласно одному варианту осуществления настоящего изобретения эффективное и безопасное значение площади под кривой зависимости концентрации в плазме от времени (AUC) соединения I находится в диапазоне от 188 нг*ч/мл до 1117 нг*ч/мл. Согласно одному варианту осуществления настоящего изобретения эффективное и безопасное значение площади под кривой зависимости концентрации в плазме от времени (AUC) соединения I находится в диапазоне от 188 нг*ч/мл до 885 нг*ч/мл.
[0051] Согласно одному варианту осуществления настоящего изобретения для обеспечения субъекта/пациента безопасным и эффективным диапазоном AUC, упомянутым выше, субъекту/пациенту вводят фармацевтическую композицию в суточной дозе, составляющей 0,1 мг до 2,5 мг соединения I. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,5 мг. Необязательно суточная доза соединения I составляет 0,1 мг, 0,15 мг, 0,2 мг, 0,25 мг, 0,3 мг, 0,35 мг, 0,4 мг, 0,45 мг, 0,5 мг, 0,6 мг, 0,7 мг, 0,8 мг, 0,9 мг, 1 мг, 1,5 мг, 2 мг или 2,5 мг.
[0052] Соединение I является нерастворимым в воде и характеризуется средней скоростью проникновения. Фармацевтическая композиция указанного соединения, произведенная с помощью общепринятых технических средств, обеспечивает низкую биодоступность, и при введении у субъектов наблюдается огромная индивидуальная вариабельность. Для получения клинически приемлемого эффекта, т.е. для эффективного лечения всех пациентов, общепринятым является повышение дозы. Тем не менее, из-за значительной индивидуальной вариабельности у некоторых пациентов наблюдается слишком высокое значение AUC соединения I, что приводит к повышенному риску повышенных содержаний калия в сыворотке. В частности, у пациентов с хронической болезнью почек способность модулировать ионы калия ухудшается, а повышенное содержание калия в сыворотке приводит к более высокому риску для здоровья.
[0053] Настоящая фармацевтическая композиция удовлетворяет требованиям клинической безопасности и эффективности путем улучшения биодоступности для эффективного снижения индивидуальной вариабельности AUC.
[0054] Для повышения биодоступности фармацевтической композиции до уровня, составляющего 50% или больше у млекопитающих, согласно одному варианту осуществления настоящего изобретения в фармацевтической композиции согласно настоящему изобретению уменьшают размер частиц соединения I. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 25 мкм или меньше. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 21,7 мкм или меньше. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 10 мкм или меньше. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 5 мкм или меньше.
[0055] Соединение I в фармацевтической композиции с различным размером частиц можно получить посредством помола, экструдирования, столкновения, разрезания, механического тонкого измельчения, вибрационного тонкого измельчения, измельчения в псевдоожиженном слое, ультразвуковой обработки, помола под высоким давлением, химического осаждения или тому подобного.
[0056] Для увеличения биодоступности фармацевтической композиции до уровня, составляющего 50% или больше у млекопитающих, согласно одному варианту осуществления настоящего изобретения в фармацевтическую композицию добавляют поверхностно-активное вещество.
[0057] Поверхностно-активное вещество представляет собой одно или несколько, выбранных из группы, состоящей из следующего: хлорид бензалкония, лаурилсульфонат натрия, додецилсульфат натрия, глицерин, холевая кислота, полоксамер, поливиниловый спирт, полисорбат 80, PVP K30 и полиэтиленгликоль. Предпочтительно поверхностно-активное вещество представляет собой одно или несколько, выбранных из группы, состоящей из хлорида бензалкония, лаурилсульфоната натрия и додецилсульфата натрия. Предпочтительно поверхностно-активное вещество представляет собой хлорид бензалкония, лаурилсульфонат натрия или додецилсульфат натрия.
[0058] В фармацевтической композиции согласно настоящему изобретению массовое отношение соединения I к поверхностно-активному веществу составляет 1:0,1-1:20, предпочтительно 1:1-1:20 и более предпочтительно 1:5-1:20.
[0059] Настоящее изобретение относится к фармацевтической композиции, содержащей соединение I и фармацевтически приемлемый носитель.
[0060] Согласно одному варианту осуществления настоящего изобретения пациенту вводят фармацевтическую композицию в суточной дозе, составляющей 0,1 мг до 2,5 мг соединения I. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,1 мг до 0,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,15 мг до 0,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,2 мг до 0,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,25 мг до 0,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 2,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 2 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 1,5 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 1 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,9 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,8 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,7 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,6 мг. Необязательно суточная доза соединения I находится в диапазоне от 0,3 мг до 0,5 мг. Необязательно суточная доза соединения I составляет 0,1 мг, 0,15 мг, 0,2 мг, 0,25 мг, 0,3 мг, 0,35 мг, 0,4 мг, 0,45 мг, 0,5 мг, 0,6 мг, 0,7 мг, 0,8 мг, 0,9 мг, 1 мг, 1,5 мг, 2 мг или 2,5 мг.
[0061] Соединение I является нерастворимым в воде и характеризуется средней скоростью проникновения. Фармацевтическая композиция указанного соединения, произведенная с помощью общепринятых технических средств, обеспечивает низкую биодоступность, и при введении у субъектов наблюдается огромная индивидуальная вариабельность. Для получения клинически приемлемого эффекта, т.е. для эффективного лечения всех пациентов, общепринятым является повышение дозы. Тем не менее, из-за значительной индивидуальной вариабельности у некоторых пациентов наблюдается слишком высокое значение AUC соединения I, что приводит к повышенному риску повышенных содержаний калия в сыворотке. В частности, у пациентов с хронической болезнью почек способность модулировать ионы калия ухудшается, а повышенное содержание калия в сыворотке приводит к более высокому риску для здоровья.
[0062] Настоящая фармацевтическая композиция удовлетворяет требованиям клинической безопасности и эффективности путем улучшения биодоступности для эффективного снижения индивидуальной вариабельности AUC.
[0063] Для повышения биодоступности фармацевтической композиции до уровня, составляющего 50% или больше у млекопитающих, согласно одному варианту осуществления настоящего изобретения в фармацевтической композиции согласно настоящему изобретению уменьшают размер частиц соединения I. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 25 мкм или меньше. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 21,7 мкм или меньше. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 5 мкм или меньше.
[0064] Соединение I в фармацевтической композиции с различным размером частиц можно получить посредством помола, экструдирования, столкновения, разрезания, механического тонкого измельчения, вибрационного тонкого измельчения, измельчения в псевдоожиженном слое, ультразвуковой обработки, помола под высоким давлением, химического осаждения или тому подобного.
[0065] Для увеличения биодоступности фармацевтической композиции до уровня, составляющего 50% или больше у млекопитающих, согласно одному варианту осуществления настоящего изобретения в фармацевтическую композицию добавляют поверхностно-активное вещество.
[0066] Поверхностно-активное вещество представляет собой одно или несколько, выбранных из группы, состоящей из следующего: хлорид бензалкония, лаурилсульфонат натрия, додецилсульфат натрия, глицерин, холевая кислота, полоксамер, поливиниловый спирт, полисорбат 80, PVP K30 и полиэтиленгликоль. Предпочтительно поверхностно-активное вещество представляет собой одно или несколько, выбранных из группы, состоящей из хлорида бензалкония, лаурилсульфоната натрия и додецилсульфата натрия. Предпочтительно поверхностно-активное вещество представляет собой хлорид бензалкония, лаурилсульфонат натрия или додецилсульфат натрия.
[0067] В фармацевтической композиции согласно настоящему изобретению массовое отношение соединения I к поверхностно-активному веществу составляет 1:0,1-1:20, предпочтительно 1:1-1:20 и более предпочтительно 1:5-1:20.
[0068] Фармацевтическую композицию согласно настоящему изобретению можно получить в виде перорального состава, более предпочтительно в виде таблеток, таблеток пролонгированного высвобождения, капсул, гранул, мягких капсул, микропилюль, микрокапсул, микросфер, липосом, самоэмульгирующейся системы лекарственной доставки, твердых дисперсий, мицелл, растворяющихся в ротовой полости таблеток, растворов, суспензий или эмульсий.
[0069] Состав фармацевтической композиции для однократного применения согласно настоящему изобретению содержит 0,01 мг до 2,5 мг соединения I, необязательно 0,01 мг до 2 мг соединения I, необязательно 0,01 мг до 1,5 мг соединения I, необязательно 0,01 мг до 1 мг соединения I, необязательно 0,01 мг до 0,9 мг соединения I, необязательно 0,01 мг до 0,8 мг соединения I, необязательно 0,01 мг до 0,7 мг соединения I, необязательно 0,01 мг до 0,6 мг соединения I, необязательно 0,01 мг до 0,5 мг соединения I, необязательно 0,025 мг до 2,5 мг соединения I, необязательно 0,025 мг до 2 мг соединения I, необязательно 0,025 мг до 1,5 мг соединения I, необязательно 0,025 мг до 1 мг соединения I, необязательно 0,025 мг до 0,9 мг соединения I, необязательно 0,025 мг до 0,8 мг соединения I, необязательно 0,025 мг до 0,7 мг соединения I, необязательно 0,025 мг до 0,6 мг соединения I, необязательно 0,025 мг до 0,5 мг соединения I, необязательно 0,05 мг до 2,5 мг соединения I, необязательно 0,05 мг до 2 мг соединения I, необязательно 0,05 мг до 1,5 мг соединения I, необязательно 0,05 мг до 1 мг соединения I, необязательно 0,05 мг до 0,9 мг соединения I, необязательно 0,05 мг до 0,8 мг соединения I, необязательно 0,05 мг до 0,7 мг соединения I, необязательно 0,05 мг до 0,6 мг соединения I, необязательно 0,05 мг до 0,5 мг соединения I, необязательно 0,1 мг до 2,5 мг соединения I, необязательно 0,1 мг до 2 мг соединения I, необязательно 0,1 мг до 1,5 мг соединения I, необязательно 0,1 мг до 1 мг соединения I, необязательно 0,1 мг до 0,9 мг соединения I, необязательно 0,1 мг до 0,8 мг соединения I, необязательно 0,1 мг до 0,7 мг соединения I, необязательно 0,1 мг до 0,6 мг соединения I, необязательно 0,1 мг до 0,5 мг соединения I, необязательно 0,15 мг до 2,5 мг соединения I, необязательно 0,15 мг до 2 мг соединения I, необязательно 0,15 мг до 1,5 мг соединения I, необязательно 0,15 мг до 1 мг соединения I, необязательно 0,15 мг до 0,9 мг соединения I, необязательно 0,15 мг до 0,8 мг соединения I, необязательно 0,15 мг до 0,7 мг соединения I, необязательно 0,15 мг до 0,6 мг соединения I, необязательно 0,15 мг до 0,5 мг соединения I, необязательно 0,2 мг до 2,5 мг соединения I, необязательно 0,2 мг до 2 мг соединения I, необязательно 0,2 мг до 1,5 мг соединения I, необязательно 0,2 мг до 1 мг соединения I, необязательно 0,2 мг до 0,9 мг соединения I, необязательно 0,2 мг до 0,8 мг соединения I, необязательно 0,2 мг до 0,7 мг соединения I, необязательно 0,2 мг до 0,6 мг соединения I, необязательно 0,2 мг до 0,5 мг соединения I, необязательно 0,25 мг до 2,5 мг соединения I, необязательно 0,25 мг до 2 мг соединения I, необязательно 0,25 мг до 1,5 мг соединения I, необязательно 0,25 мг до 1 мг соединения I, необязательно 0,25 мг до 0,9 мг соединения I, необязательно 0,25 мг до 0,8 мг соединения I, необязательно 0,25 мг до 0,7 мг соединения I, необязательно 0,25 мг до 0,6 мг соединения I, необязательно 0,25 мг до 0,5 мг соединения I, необязательно 0,3 мг до 2,5 мг соединения I, необязательно 0,3 мг до 2 мг соединения I, необязательно 0,3 мг до 1,5 мг соединения I, необязательно 0,3 мг до 1 мг соединения I, необязательно 0,3 мг до 0,9 мг соединения I, необязательно 0,3 мг до 0,8 мг соединения I, необязательно 0,3 мг до 0,7 мг соединения I, необязательно 0,3 мг до 0,6 мг соединения I, необязательно 0,3 мг до 0,5 мг соединения I, необязательно 0,01 мг, 0,025 мг, 0,05 мг, 0,1 мг, 0,15 мг, 0,2 мг, 0,25 мг, 0,3 мг, 0,35 мг, 0,4 мг, 0,45 мг, 0,5 мг, 0,6 мг, 0,7 мг, 0,8 мг, 0,9 мг, 1 мг, 1,5 мг, 2 мг или 2,5 мг соединения I.
[0070] Фармацевтическая композиция согласно настоящему изобретению может легко блокировать связывание минералокортикоидного рецептора с альдостероном. В соответствии с основным фармакологическим действием фармацевтическую композицию можно использовать специально для лечения и/или профилактики хронической болезни почек, выбранной из группы, состоящей из следующего: гипертензивная нефропатия, диабетическая нефропатия, гломерулонефрит, почечная недостаточность, альбуминурия, острое повреждение почек и киста почки; хроническая болезнь почек с гипертензией, хроническая болезнь почек с сердечной недостаточностью, хроническая болезнь почек с гипертензией и сердечной недостаточностью, хроническая болезнь почек с ожирением, хроническая болезнь почек с гиперлипидемией, хроническая болезнь почек с сахарным диабетом и кардиоренальный синдром; сердечно-сосудистых заболеваний, выбранных из группы, состоящей из следующего: гипертензия, сердечная недостаточность (т.е. застойная сердечная недостаточность, выбранная из группы, состоящей из следующего; сердечная недостаточность со сниженной фракцией выброса, сердечная недостаточность с нормальной фракцией выброса и острая сердечная недостаточность), инфаркт миокарда, стенокардия, гипертрофия сердца, миокардит, васкулярный фиброз сердца, дисфункция барорецепторов, гиперволемия и аритмия сердца, гиперлипидемия и ожирение; эндокринных заболеваний, выбранных из группы, состоящей из следующего: первичный и вторичный гиперальдостеронизм, болезнь Аддисона, синдром Иценко-Кушинга и синдром Барттера. В частности, фармацевтическую композицию согласно настоящему изобретению можно использовать для лечения и/или профилактики хронической болезни почек, включая в себя следующее: диабетическая нефропатия и гипертензивная нефропатия; хроническая болезнь почек с гипертензией и/или сердечной недостаточностью; сердечная недостаточность и/или гипертензия.
[0071] Согласно одному варианту осуществления в настоящем изобретении дополнительно раскрыто применение фармацевтической композиции в получении лекарственного средства для лечения хронической болезни почек, сердечной недостаточности и гипертензии.
[0072] Согласно другому варианту осуществления настоящее изобретение относится к способу лечения хронической болезни почек, сердечной недостаточности и/или гипертензии, предусматривающему введение нуждающемуся в этом субъекту терапевтически эффективного количества фармацевтической композиции согласно настоящему изобретению.
[0073] Субъект согласно настоящему изобретению может представлять собой млекопитающих, предпочтительно людей, в частности, тех, которые характеризуются наличием заболевания, выбранного из группы, состоящей из следующего: хроническая болезнь почек, выбранная из группы, состоящей из следующего: гипертензивная нефропатия, диабетическая нефропатия, гломерулонефрит, почечная недостаточность, альбуминурия, киста почки и гломерулосклероз; хроническая болезнь почек с гипертензией, хроническая болезнь почек с сердечной недостаточностью, хроническая болезнь почек с гипертензией и сердечной недостаточностью, хроническая болезнь почек с ожирением, хроническая болезнь почек с гиперлипидемией, хроническая болезнь почек с сахарным диабетом и кардиоренальный синдром; сердечно-сосудистые заболевания, выбранные из группы, состоящей из следующего: гипертензия, сердечная недостаточность (т.е. застойная сердечная недостаточность, выбранная из группы, состоящей из следующего: сердечная недостаточность со сниженной фракцией выброса, сердечная недостаточность с нормальной фракцией выброса и острая сердечная недостаточность), инфаркт миокарда, стенокардия, гипертрофия сердца, миокардит, васкулярный фиброз сердца, ишемия миокарда, ишемическая болезнь сердца, атеросклеротическая болезнь сердца, дисфункция барорецепторов, гиперволемия и аритмия сердца, гиперлипидемия и ожирение; эндокринные заболевания, выбранные из группы, состоящей из следующего: первичный и вторичный гиперальдостеронизм, болезнь Аддисона, синдром Иценко-Кушинга и синдром Барттера. В частности, субъект представляет собой субъекта, характеризующегося наличием заболевания, выбранного из следующего: хроническая болезнь почек, выбранная из группы, состоящей из следующего: гипертензивная нефропатия, диабетическая нефропатия, гломерулонефрит, почечная недостаточность, альбуминурия, киста почки и гломерулосклероз; хроническая болезнь почек с гипертензией, хроническая болезнь почек с сердечной недостаточностью, хроническая болезнь почек с гипертензией и сердечной недостаточностью, хроническая болезнь почек с ожирением, хроническая болезнь почек с гиперлипидемией, хроническая болезнь почек с сахарным диабетом и кардиоренальный синдром.
[0074] Согласно варианту осуществления фармацевтическую композицию согласно настоящему изобретению можно вводить один раз или несколько раз в день. Предпочтительно фармацевтическую композицию вводят один раз в день в дозе, упомянутой выше. Фармацевтическую композицию согласно настоящему изобретению можно вводить в любое время суток.
[0075] Фармацевтическую композицию согласно настоящему изобретению можно использовать для лечения нуждающегося в этом субъекта в комбинации с некоторыми другими средствами, которые могут представлять собой одно или два средства, выбранные из группы, состоящей из антигипертензивного средства, антилипемического средства и антидиабетического средства.
[0076] Антигипертензивное средство может представлять собой следующее: ингибитор ангиотензинпревращающего фермента, блокатор рецепторов ангиотензина II, ингибитор ренина, блокатор кальциевого канала, диуретик, блокатор бета-рецепторов или блокатор альфа-рецепторов. В частности, антигипертензивное средство включает в себя без ограничения ингибитор ангиотензинпревращающего фермента, выбранный из группы, состоящей из следующего: каптоприл, эналаприл, беназеприл, делаприл, лизиноприл и периндоприл; блокатор рецепторов ангиотензина II, выбранный из группы, состоящей из следующего: лозартан, валсартан, ирбесартан, кандесартан, телмисартан, эпросартан и ирбесартан; ингибитор ренина, выбранный из группы, состоящей из следующего: алискирен и алискирен; блокатор кальциевого канала, выбранный из группы, состоящей из следующего: нифедипин, амлодипин, лерканидипин, нимодипин, никардипин, нитрендипин, нисолдипин, фелодипин, бенидипин, лацидипин, дилтиазем, верапамил, флунаризин, циннаризин и лидофлазин; диуретик, выбранный из группы, состоящей из следующего: хлортиазид, хлорталидон и фуросемид; блокатор бета-рецепторов, выбранный из группы, состоящей из следующего: атенолол, метопролол, соталол гидрохлорид, пропранолол гидрохлорид и карведилол; и блокатор альфа-рецепторов, выбранный из группы, состоящей из следующего: фентоламин, толазолин, феноксибензамин и празозин.
[0077] Антилипемическое средство выбрано из группы, состоящей из следующего: аторвастатин, ловастатин, симвастатин, правастатин, флувастатин, розувастатин, ципрофибрат, безафибрат, фенофибрат и гемфиброзил.
[0078] Антидиабетическое средство выбрано из группы, состоящей из следующего: инсулинотропное средство, метформины, ингибитор альфа-глюкозидазы, сенсибилизирующее средство - производное тиазолидиндиона, инсулинотропное средство - производное меглитинида, агонист рецептора GLP-1 и ингибитор DPP-4. Антидиабетическое средство включает в себя без ограничения относящееся к сульфонилмочевинам инсулинотропное средство, выбранное из группы, состоящей из следующего: глипизид, гликлазид, глибенкламид, глиборнурид, глимепирид и гликвидон; относящееся к несульфонилмочевинам инсулинотропное средство - производное меглитинидов, выбранное из группы, состоящей из репаглинида и натеглинида; метформины, выбранные из метформин; ингибитор альфа-глюкозидазы, выбранный из группы, состоящей из Bose-100, акарбозы и воглибоза; инсулин-сенсибилизирующее средство, выбранное из росиглитазона и пиоглитазона; ингибитор дипепитилпептидазы 4 (DPP-4), выбранный из группы, состоящей из ситаглиптина, саксаглиптина и вилдаглиптина; и агонист рецептора GLP-1, выбранный из эксенатида и лираглутида.
[0079] Соединение I представляет собой антагонист минералокортикоидных рецепторов. Его клиническое применение вызывает риск повышенных содержаний калия в сыворотке из-за механизма, лежащего в основе его действия. У пациента с хронической болезнью почек, из-за ухудшения способности модулировать ионы калия, с большой вероятностью развивается гиперкалиемия при введении указанного соединения. Гиперкалиемия приводит к тяжелым повреждениям или даже представляет смертельный риск для пациента с хронической болезнью почек. Таким образом, применение лекарственных средств MRA в значительной степени ограничено, и до сих пор не было одобрено ни одно лекарственное средство MRA для лечения хронической болезни почек и связанных с ней осложнений.
[0080] Для безопасного и эффективного применения соединения I в клиническом лечении хронической болезни почек, для которой до настоящего времени не существовало клинически одобренного лекарственного средства, авторы настоящего изобретения провели обширные эксперименты на соединении I и наконец обнаружили окно безопасности и эффективности. Позже на основании окна безопасности и эффективности они обнаружили безопасный и эффективный диапазон доз соединения I и изобрели фармацевтическую композицию, характеризующуюся биодоступностью, составляющей 50% или больше у млекопитающих.
[0081] I. Фармацевтическая композиция согласно настоящему изобретению характеризуется узким окном безопасности и эффективности
[0082] С помощью клинических испытаний обнаружили, что соединение I характеризуется очень узким окном безопасности у пациентов с хронической болезнью почек, т.е. оно является безопасным и эффективным, когда AUC находится в диапазоне, составляющем 188 нг*ч/мл-3173 нг*ч/мл.
[0083] i. Широкое окно безопасности обнаружено у крыс SD
[0084] В эксперименте испытания на токсичность и фармакокинетическом исследовании, в котором самцам крыс SD внутрижелудочно вводили исследуемое соединение в течение 13 недель, а затем был период восстановления в течение 4 недель, соединение I вводили в высокой дозе, составляющей 30 мг/кг/день, равновесная AUC0-24ч составляла приблизительно 49900 нг*ч/мл. Ни один явный побочный эффект не наблюдался, и содержание калия в сыворотке не являлся повышенным.
[0085] ii. Широкое окно безопасности наблюдали у здоровых добровольцев
[0086] В одном клиническом испытании здоровым добровольцам давали однократную дозу соединения I в дозе в диапазоне, составляющем 0,5-30 мг/день. Значение AUC0-24ч находилось в диапазоне, составляющем 162,5-5016 нг*ч/мл, и повышенное содержание калия в сыворотке не наблюдалось.
[0087] В другом клиническом испытании 6 здоровым добровольцам давали многократные введения соединения I в дозе, составляющей 5 мг/день. Было доказано, что средняя равновесная AUCtau составляла 6373±1026 нг*ч/мл, и повышенные содержания калия в сыворотке наблюдали у 3 субъектов, но они являлись временными.
[0088] В дополнительном клиническом испытании соединение I давали здоровым добровольцам в дозе, составляющей 2,5 мг/день в режиме многократного введения доз. Обнаружено, что средняя равновесная AUCtau составляла 2863±822 нг*ч/мл, и повышенные содержания калия в сыворотке не наблюдались.
[0089] iii. Очень узкое окно безопасности обнаружили у пациентов с хронической болезнью почек
[0090] Пациенты с хронической болезнью почек реагировали по-разному. Если их подвергали действию многократных доз соединения I в дозе, составляющей 2,5 мг/день, безопасная равновесная AUCtau составляла 2613±280 нг*ч/мл. У одного субъекта содержание калия в сыворотке слегка повышалось.
[0091] Фармацевтические эффекты соединения I наблюдались, когда пациентам с хронической болезнью почек давали повторные дозы соединения в дозе, составляющей 0,5 мг/день. Средняя равновесная AUCtau составляла 652,5±232,2 нг*ч/мл, и повышенное содержание калия в сыворотке не наблюдалось.
[0092] С учетом вышеизложенного соединение I показало широкое окно безопасности в испытаниях с участием животных и здоровых субъектов, но очень узкое окно безопасности у пациентов с хронической болезнью почек. Окно клинической безопасности нельзя было спрогнозировать из испытаний на животных или здоровых людях. Иными словами, окно безопасности и эффективности соединения I у пациентов с хронической болезнью почек является непрогнозируемым.
[0093] В статистике 95% доверительный интервал в нормально распределенной выборке находится в диапазоне от среднего минус двукратное стандартное отклонение до среднего плюс двукратное стандартное отклонение. На основании упомянутых выше клинических испытаний на пациентах с ХБП нижняя граница значения AUC соединения I, которое демонстрирует эффекты у пациентов с ХБП, которым вводили фармацевтическую композицию, определяют как среднее значение AUC минус двукратное стандартное отклонение, измеренное при дозе, составляющей 0,5 мг, т.е. 188 нг*ч/мл; и верхняя граница значения AUC соединения I, которое является безопасным у пациентов ХБП с представляет собой среднее значение AUC плюс двукратное стандартное отклонение, измеренное в дозе, составляющей 2,5 мг, т.е. 3173 нг*ч/мл; соответственно, безопасное и эффективное значение AUC соединения I находится в диапазоне, составляющем от 188 нг*ч/мл до 3173 нг*ч/мл. Предпочтительно нижняя граница значения AUC соединения I, которое демонстрирует эффекты после абсорбции у пациентов, представляет собой среднее значение AUC минус двукратное стандартное отклонение, измеренное в дозе, составляющей 0,5 мг, т.е. 188 нг*ч/мл, и верхняя граница значения AUC соединения I, которое является безопасным у пациентов с ХБП, представляет собой среднее значение AUC плюс стандартное отклонение, измеренное в дозе, составляющей 2,5 мг, т.е. 2893 нг*ч/мл; соответственно, безопасное и эффективное значение AUC соединения I находится в диапазоне, составляющем 188 нг*ч/мл до 2893 нг*ч/мл. Предпочтительно нижняя граница значения AUC соединения I, которое демонстрирует эффекты после абсорбции у пациентов, представляет собой среднее значение AUC минус двукратное стандартное отклонение, измеренное в дозе, составляющей 0,5 мг, т.е. 188 нг*ч/мл, и верхняя граница значения AUC соединения I, которое является безопасным у пациентов с ХБП, представляет собой среднее значение AUC измеренное в дозе, составляющей 2,5 мг, т.е. 2613 нг*ч/мл; соответственно безопасное и эффективное значение AUC соединения I находится в диапазоне, составляющем 188 нг*ч/мл-2613 нг*ч/мл. Предпочтительно нижняя граница значения AUC соединения I, которое демонстрирует эффекты после абсорбции у пациентов, представляет собой среднее значение AUC минус двукратное стандартное отклонение, измеренное в дозе, составляющей 0,5 мг, т.е. 188 нг*ч/мл, и верхняя граница значения AUC соединения I, которое является безопасным у пациентов с ХБП, представляет собой среднее значение AUC плюс двукратное стандартное отклонение, измеренное в дозе, составляющей 0,5 мг, т.е. 1117 нг*ч/мл; соответственно, безопасное и эффективное значение AUC соединения I находится в диапазоне, составляющем 188 нг*ч/мл-1117 нг*ч/мл. Предпочтительно нижняя граница значения AUC соединения I, которое демонстрирует эффекты после абсорбции у пациентов, представляет собой среднее значение AUC минус двукратное стандартное отклонение, измеренное в дозе, составляющей 0,5 мг, т.е. 188 нг*ч/мл, и верхняя граница значения AUC соединения I, которое является безопасным у пациентов с ХБП, представляет собой среднее значение AUC плюс стандартное отклонение, измеренное в дозе, составляющей 0,5 мг, т.е. 885 нг*ч/мл; соответственно, безопасное и эффективное значение AUC соединения I находится в диапазоне, составляющем 188 нг*ч/мл-885 нг*ч/мл.
[0094] В эксперименте по фармакодинамике с использованием чувствительных к соли крыс с повреждением почек, индуцированном высоким содержанием солей, кривая, показывающая взаимоотношение между изменением SBP или UACR по сравнению с исходным уровнем и AUC0-24 (см. фиг. 1 и фиг. 2), выявляет точку волнообразного изменения при 100 ч*нг/мл AUC, после которой кривая имеет тенденцию быть плоской. Это указывает на то, что лекарственное средство начинает действовать, когда AUC достигает 100 ч*нг/мл.
[0095] В первый день, когда пациентам с ХБП вводили в дозу, составляющую 0,5 мг/день, обнаружили, что равновесная AUCtau составляет 105,6 ч*нг/мл или больше у этих субъектов, при этом UACR снижалось на 30,5% или больше по сравнению с исходным уровнем, указывая на то, что введение лекарственного средства оказало действие.
[0096] Фармакокинетическое исследование на группе, которой давали многократные дозы соединения I в дозе, составляющей 0,5 мг/день, использовали для прогнозирования равновесной AUCtau в других группах дозирования. Когда фармацевтическую композицию с биодоступностью, составляющей 50% у собак, давали пациентам с ХБП в дозе, составляющей 0,1 мг/день, AUCtau составляла 130,5 ч*нг/мл, что было выше нижней границы эффективной AUCtau, составляющей 100 ч*нг/мл, указывая на то, что введение лекарственного средства в дозе, составляющей 0,1 мг/день, оказало действия. Иными словами, суточная доза, составляющая 0,1 мг, оказала действие.
[0097] II. Огромная индивидуальная вариабельность обнаружена в отношении абсорбции соединения I
[0098] i. Наблюдали большую индивидуальную вариабельность абсорбции соединения
[0099] Индивидуумы по-разному реагируют на определенное лекарственное средство, что называется "индивидуальная вариабельность эффекта лекарственного средства". Например, некоторые люди не чувствительны к лекарственному средству, и стандартная доза может являться неэффективной для получения терапевтического эффекта. С другой стороны, некоторые другие люди являются особенно чувствительными к лекарственному средству, и очень низкая доза может производить очевидный эффект, тогда как стандартная доза может вызывать необычайно интенсивный эффект или даже токсичность.
[00100] Кроме того, на абсорбцию лекарственного средства также влияют условия питания. Например, на абсорбцию лекарственного средства может влиять состояние натощак или сытости или же состав пищи. В состоянии сытости абсорбция лекарственного средства будет задерживаться, а скорость выведения будет замедляться. Потребление воды, чая, алкоголя и продуктов с высоким содержанием жиров также в некоторой степени влияет на абсорбцию лекарственного средства.
[00101] Соединение I является нерастворимым в воде и характеризуется средней скоростью проникновения. Состояние пациента и рацион будут влиять на абсорбцию лекарственного средства. Фармацевтическая композиция указанного соединения, произведенная с помощью общепринятых технических средств, обеспечивает низкую биодоступность, и пациентов с ХБП наблюдается огромная индивидуальная вариабельность у. Исследования показали, что чем ниже биодоступность, тем больше будет индивидуальная вариабельность. Тем временем, соединение I характеризуется узким окном безопасности у пациентов с хронической болезнью почек, создавая большой риск для безопасности при применении этого соединения у этих пациентов. Алкоголь и рацион с высоким содержанием жиров будут особенно усиливать абсорбцию соединения I, дополнительно увеличивая индивидуальную вариабельность и создавать большую угрозу безопасности для пациентов.
[00102] Таким образом, даже если диапазон клинических доз является узким, биодоступность должна быть улучшена для снижения индивидуальной вариабельности с целью безопасного и эффективного клинического применения этого соединения.
[00103] Следовательно, безопасное и эффективное применение соединения I тесно связано с биодоступностью и дозой фармацевтической композиции согласно настоящему изобретению.
[00104] ii. Эффект соединения I с различным размером частиц на абсорбцию
[00105] а) С целью клинически безопасного и эффективного введения лекарственного средства авторы настоящего изобретения обнаружили с помощью обширных исследований, что чем меньше размер частиц соединения I, тем выше будет биодоступность при пероральном введении. Ниже приведена абсорбция соединения I, характеризующегося различным размером частиц у крыс.
Биодоступность составляет 13,2%, когда параметр распределения частиц по размерам D90 соединения I составляет 72,0 мкм;
биодоступность составляет 34,8%, когда параметр распределения частиц по размерам D90 соединения I составляет 41,5 мкм;
биодоступность составляет 54,6%, когда параметр распределения частиц по размерам D90 соединения I составляет 21,7 мкм;
биодоступность составляет 66,9%, когда параметр распределения частиц по размерам D90 соединения I составляет 3,8 мкм;
биодоступность составляет 82,3%, когда параметр распределения частиц по размерам D90 соединения I составляет 538 нм;
биодоступность составляет 76,9%, когда соединение I в составе твердой дисперсии вводят перорально;
биодоступность составляет 81,6%, когда соединение I в составе раствора вводят перорально.
[00106] Если соединение I производят общепринятыми способами, соединение I характеризуется D90, составляющим 72,2 мкм. Его биодоступность составляет только 13,2%, когда его дают перорально крысам. Биодоступность достигает 54,6%, когда D90 снижают до 21,7 мкм. Когда соединение I составляют в виде твердой дисперсии или раствора, оно распределяется в виде отдельных молекул, и его биодоступность будет составлять 76,9% и 81,6%, соответственно, когда его дают перорально крысам.
[00107] Когда соединение I характеризуется D90, составляющим 25 мкм или меньше, его доступность составляет 50% или больше, что удовлетворяет требованиям клинической безопасности и эффективности.
[00108] b) Абсорбция фармацевтической композиции согласно настоящему изобретению у собак
[00109] Капсулу 5, полученную в примере 2, испытывали в отношении ее фармакокинетических свойств у собак породы бигль. Биодоступность составляла 50% или больше, когда D90 соединения I составлял 5 мкм, что удовлетворяло требованиям клинической безопасности и эффективности.
[00110] iii. Эффект поверхностно-активного вещества на абсорбцию
[00111] В фармакокинетическом исследовании на крысах, когда не использовали никаких поверхностно-активных веществ, биодоступность составляла 34,8% с D90 соединения I, составляющим 41,5 мкм. После добавления додецилсульфата натрия (SDS) к соединению I в качестве поверхностно-активного вещества в соотношении, составляющем 10:1 (SDS: соединение I), биодоступность составляла 64,7% с D90 соединения I составляющим 52,5 мкм.
[00112] В фармакокинетическом исследовании на собаках породы бигль с использованием капсулы 5, полученной в примере 2, без какого-либо поверхностно-активного вещества, средняя биодоступность составляла 56,5%. Когда таблетку 3 получали в примере 2 с додецилсульфатом натрия (SDS), было доказано, что биодоступность оказалась равной 77,4%.
[00113] Все это указывает на то, что биодоступность можно сильно улучшить путем добавления поверхностно-активного вещества в фармацевтический состав согласно настоящему изобретению, что может эффективно снижать индивидуальную вариабельность.
[00114] vi. Абсорбция фармацевтической композиции в клинических испытаниях с участием людей
[00115] Капсулу, полученную в примере 2 в качестве предпочтительного варианта осуществления (т.е. капсулу 5, характеризующуюся D90, составляющим 5 мкм и средней биодоступностью, составляющей 56,5% у собак породы бигль), использовали в клинических испытаниях.
[00116] В клинических испытаниях капсулу соединения I вводили с помощью многократных доз в суточной дозе, составляющей 2,5 мг. В отношении индивидуальной вариабельности AUC у здоровых добровольцев AUC у субъекта с самым высоким уровнем AUC был в 2,37 раз выше этого показателя у субъекта с самым низким уровнем. У пациентов с ХБП AUC у субъекта с самым высоким уровнем AUC был в 2,27 раз выше этого показателя у субъекта с самым низким уровнем.
[00117] В клинических испытаниях капсулы соединения I также давали пациентам с ХБП в многократных дозах в суточной дозе, составляющей 0,5 мг. Тем не менее, AUC у субъекта с самым высоким уровнем AUC все еще был в 2,14 раз выше этого показателя у субъекта с самым низким уровнем.
[00118] Когда его составляли, как описано согласно предпочтительному варианту осуществления настоящего изобретения, соединение I характеризовалось биодоступностью, составляющей 56,5% у собак. Тем не менее, когда такой состав соединения давали здоровым субъектам и пациентам с ХБП, все еще наблюдалась индивидуальная вариабельность AUC (значение AUC у субъекта с самым высоким уровнем более чем в два раза превышало этот показатель у субъекта с самым низким уровнем).
[00119] Согласно вышеупомянутым вариантам осуществления, относящимся к биодоступности соединения I у млекопитающих, капсула, полученная в примере 2 в качестве предпочтительного варианта осуществления, характеризовалась биодоступностью, составляющей 50% или больше у собак. Затем эту предпочтительную капсулу вводили пациентам с ХБП в многократных дозах в суточной дозе, составляющей 0,5 мг или 2,5 мг, наблюдали индивидуальную вариабельность (значение AUC у субъекта с самым высоким уровнем более чем в два раза превышало этот показатель у субъекта с самым низким уровнем). В дополнение ко всему этому обнаружили, что соединение I индуцирует высокие содержания калия в сыворотке у пациентов с ХБП. Таким образом, определили, что биодоступность, составляющая 50% или больше у млекопитающих, являлась основным требованием, т.е. биодоступность, составляющая 50% или больше у собак породы бигль являлась обязательной для фармацевтической композиции согласно настоящему изобретению. Упомянутое требование в отношении биодоступности и диапазон доз согласно настоящему изобретению следует объединять с учетом безопасного и эффективного введения лекарственного средства.
[00120] В ходе экспериментов обнаружили, что проблемы слишком высоких уровней AUC у некоторых пациентов в связи с индивидуальной вариабельностью можно избежать, если биодоступность фармацевтической композиции согласно настоящему изобретению можно увеличить до 50% или более у млекопитающих. Таким образом, риск развития высоких содержаний калия в сыворотке будет под контролем. Для безопасного и эффективного клинического применения лекарственного средства фармацевтическая композиция согласно настоящему изобретению должна характеризоваться своей повышенной биодоступностью у пациентов с ХБП, так чтобы уменьшить индивидуальную вариабельность и обеспечить безопасность применения лекарственного средства.
[00121] Улучшение биодоступности с уменьшением размера частиц
[00122] Для увеличения биодоступности фармацевтической композиции согласно настоящему изобретению до уровня, составляющего 50% или больше у млекопитающих, согласно одному варианту осуществления параметр распределения частиц по размерам D90 соединения I, как определили, составляет 25 мкм или меньше. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 21,7 мкм или меньше. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 10 мкм или меньше. Согласно одному варианту осуществления настоящего изобретения параметр распределения частиц по размерам D90 соединения I составляет 5 мкм или меньше.
[00123] Соединение I, характеризующееся различным размером частиц можно получить посредством помола, экструдирования, столкновения, разрезания, механического тонкого измельчения, вибрационного тонкого измельчения, измельчения в псевдоожиженном слое, ультразвуковой обработки, помола под высоким давлением, химического осаждения или тому подобного.
[00124] Улучшение биодоступности с помощью добавления поверхностно-активного вещества
[00125] Для увеличения биодоступности фармацевтической композиции согласно настоящему изобретению до уровня, составляющего 50% или больше у млекопитающих, согласно одному варианту осуществления фармацевтическая композиция согласно настоящему изобретению содержит поверхностно-активное вещество.
[00126] Поверхностно-активное вещество представляет собой одно или несколько, выбранных из группы, состоящей из следующего: хлорид бензалкония, лаурилсульфонат натрия, додецилсульфат натрия, глицерин, холевая кислота, полоксамер, поливиниловый спирт, полисорбат 80, PVP K30 и полиэтиленгликоль. Предпочтительно поверхностно-активное вещество представляет собой одно или несколько, выбранных из группы, состоящей из хлорида бензалкония, лаурилсульфоната натрия и додецилсульфата натрия. Предпочтительно поверхностно-активное вещество представляет собой хлорид бензалкония, лаурилсульфонат натрия или додецилсульфат натрия.
[00127] В фармацевтической композиции согласно настоящему изобретению массовое отношение соединения I к поверхностно-активному веществу составляет 1:0,1-1:20, предпочтительно 1:1-1:20 и более предпочтительно 1:5-1:20.
[00128] Соединение I согласно настоящему изобретению может находиться в любой форме. Например, соединение I может находиться в аморфной структуре, структуре кристалла или смешанного кристалла.
[00129] III. Фармацевтическая композиция согласно настоящему изобретению характеризуется специальным диапазоном доз
[00130] После введения фармацевтической композиции согласно настоящему изобретению уровень AUC соединения I у людей является достаточно высоким, что свидетельствует о достаточно хорошей эффективности лекарственного средства. В клиническом применении очень низкая доза будет давать терапевтический эффект.
[00131] Клинические дозы таблеток спиронолактона и таблеток эплеренона, двух доступных антагонистов минералокортикоидного рецептора, составляют 40-400 мг/день и 25-50 мг/день, соответственно. Дозы являются достаточно высокими.
[00132] В испытаниях фармацевтической композиции согласно настоящему изобретению здоровым добровольцам вводили однократную дозу, составляющую 0,5-30 мг, или альтернативно вводили в многократных дозах в суточной дозе, составляющей 2,5 мг, и показали очень хорошую переносимость. Когда здоровым добровольцам введение проводили в многократных дозах в дозе, составляющей 5 мг/день, также наблюдали относительно хорошую переносимость, несмотря на однократно повышенные содержания калия в сыворотке.
[00133] Фармацевтическая композиция согласно настоящему изобретению, когда ее давали пациентам с ХБП в многократных дозах в суточной дозе, составляющей 0,5 мг, показывала хорошую терапевтическую активность, при этом повышенные содержания калия в сыворотке не наблюдались. Когда фармацевтическую композицию вводили в многократных дозах в дозе, составляющей 2,5 мг/день, обнаружили колебания содержания калия в сыворотке у одного субъекта, что указывает на то, что при такой дозе отсутствует риск безопасности для пациентов с ХБП.
[00134] Кроме того, из фармакодинамического эксперимента с использованием чувствительных к соли крыс с повреждением почек, вызванным высоким содержанием солей, а также из клинического испытания пациентов с ХБП видно, что суточная доза, составляющая 0,1 мг, показала терапевтические эффекты на пациентов с хронической болезнью почек.
[00135] Введение фармацевтической композиции согласно настоящему изобретению индуцирует очень высокое значение AUC соединения I в организме людей, указывая на относительно хорошую терапевтическую активность. С другой стороны, очень низкая доза 0,1 мг в клиническом применении дает терапевтический эффект, а это означает, что эффективная доза является достаточно низкой. Кроме того, повышенные содержания калия в сыворотке не были обнаружены при суточной дозе, составляющей 0,1 мг-0,5 мг. Таким образом, диапазон доз для безопасного и эффективного применения фармацевтической композиции согласно настоящему изобретению наконец был найден. Дозу для клинического применения фармацевтической композиции согласно настоящему изобретению нельзя спрогнозировать из лекарственных средств, доступных на рынке или клинического испытания с участием здоровых добровольцев.
[00136] Таким образом, соединение I может индуцировать повышенные содержания калия в сыворотке из-за механизма, лежащего в основе действия антагониста минералокортикоидного рецептора. Лекарственные средства MRA, доступные на рынке, принимают очень высокие клинические дозы, и соединение I также показало широкое окно безопасности в экспериментах на животных и клинических испытаниях с участием здоровых добровольцев. Таким образом, предположили, что клиническая доза соединения I будет достаточно высокой у пациентов с хронической болезнью почек. Тем не менее, в клинических испытаниях с участием пациентов с ХБП, содержания калия в сыворотке повышались при суточной дозе, составляющей 2,5 мг, указывая на то, что диапазон доз у пациентов с ХБП будет очень узким.
[00137] Авторы настоящего изобретения обнаружили окно безопасности и эффективности для соединения I с помощью серий экспериментов. Кроме того, на основании этого окна был обнаружен диапазон доз для безопасного и эффективного применения фармацевтической композиции согласно настоящему изобретению. Кроме того, авторы настоящего изобретения изобрели фармацевтическую композицию, характеризующуюся биодоступностью, составляющей 50% или больше у млекопитающих. Когда пациенту с хроническими болезнями почек вводят фармацевтическую композицию согласно настоящему изобретению с диапазоном доз, раскрытым в настоящем документе, AUC соединения I контролируется на безопасном и эффективном уровне, так что обеспечивается безопасность и эффективность клинического применения.
[00138] Биодоступность представляет собой меру скорости и степени, в которой лекарственное средство достигает системного кровообращения при введении через невнутривенные пути, и представляет собой важный параметр для оценки абсорбции лекарственного средства. Биодоступность включает в себя абсолютную биодоступность и относительную биодоступность, причем первая сравнивает абсорбцию активного лекарственного средства после невнутривенного введения с внутривенным введением, а вторая измеряет абсорбцию данного состава по сравнению с другим составом. Биодоступность в настоящем документе относится к абсолютной биодоступности.
[00139] Размер частиц в настоящем документе также может называться «крупность частиц». В фармацевтической композиции согласно настоящему изобретению соединение I характеризуется размером частиц, составляющим 100 мкм или меньше, порядка микрона или нанометра, или существует в виде молекул.
[00140] Термин D90 в настоящем документе относится к размеру, который разделяет распределение частиц по размерам на 10% выше и 90% ниже этого диаметра.
[00141] Фармацевтическая композиция согласно настоящему изобретению может эффективно блокировать связывание минералокортикоидного рецептора с альдостероном, и, таким образом, в соответствии с механизмом, лежащим в основе действия, ее можно использовать, в частности, при лечении и/или профилактике хронической болезни почек. Хроническая болезнь почек выбрана из группы, состоящей из следующего: гипертензивная нефропатия, диабетическая нефропатия, гломерулонефрит, почечная недостаточность, альбуминурия, киста почки и гломерулосклероз; хроническая болезнь почек с гипертензией, хроническая болезнь почек с сердечной недостаточностью, хроническая болезнь почек с гипертензией и сердечной недостаточностью, хроническая болезнь почек с ожирением, хроническая болезнь почек с гиперлипидемией, хроническая болезнь почек с сахарным диабетом и кардиоренальный синдром; сердечно-сосудистые заболевания, выбранные из группы, состоящей из следующего: гипертензия, сердечная недостаточность (т.е. застойная сердечная недостаточность, выбранная из группы, состоящей из сердечной недостаточности со сниженной фракцией выброса, сердечной недостаточности с нормальной фракцией выброса и острой сердечной недостаточности), острый инфаркт миокарда, стенокардия, гипертрофия сердца, миокардит, васкулярный фиброз сердца, ишемия миокарда, коронарная атеросклеротическая болезнь сердца, атеросклеротическая болезнь сердца, дисфункция барорецепторов, гиперволемия и аритмия сердца, гиперлипидемия и ожирение; эндокринные заболевания, выбранные из группы, состоящей из следующего: первичный и вторичный гиперальдостеронизм, болезнь Аддисона, синдром Иценко-Кушинга и синдром Барттера. Согласно варианту осуществления настоящее изобретение также относится к применению фармацевтической композиции в получении лекарственных средств для лечения хронической болезни почек, сердечной недостаточности и/или гипертензии.
[00142] Согласно другому варианту осуществления настоящее изобретение относится к способу лечения хронической болезни почек, сердечной недостаточности и/или гипертензии, предусматривающему введение нуждающемуся в этом субъекту терапевтически эффективного количества фармацевтической композиции согласно настоящему изобретению.
[00143] Согласно варианту осуществления настоящего изобретения фармацевтическую композицию согласно настоящему изобретению можно вводить один или несколько раз в день, предпочтительно можно вводить один раз с дозой, описанной выше. Фармацевтическую композицию согласно настоящему изобретению можно вводить в любое время суток.
[00144] Фармацевтическую композицию согласно настоящему изобретению можно вводить нуждающимся в этом пациентам в комбинации с другими лекарственными средствами, которые могут представлять собой одно или два средства, выбранные из группы, состоящей из антигипертензивного средства, антилипемического средства и антидиабетического средства.
[00145] Антигипертензивное средство выбрано из группы, состоящей из следующего: ингибитор ангиотензинпревращающего фермента, блокатор рецептора ангиотензина II, ингибитор ренина, блокатор кальциевого канала, диуретик, блокатор бета-рецепторов и блокатор альфа-рецепторов. В частности, антигипертензивное средство включает в себя без ограничения ингибитор ангиотензинпревращающего фермента, выбранный из группы, состоящей из следующего: каптоприл, эналаприл, беназеприл, делаприл, лизиноприл и периндоприл; блокатор рецептора ангиотензина II, выбранный из группы, состоящей из следующего: лозартан, валсартан, ирбесартан, кандесартан, телмисартан, эпросартан и ирбесартан; ингибитор ренина, выбранный из группы, состоящей из алискирена и алискирена; блокатор кальциевого канала, выбранный из группы, состоящей из следующего: нифедипин, амлодипин, лерканидипин, нимодипин, никардипин, нитрендипин, нисолдипин, фелодипин, бенидипин, лацидипин, дилтиазем, верапамил, флунаризин, циннаризин и лидофлазин; диуретик, выбранный из группы, состоящей из хлортиазида, хлорталидона и фуросемида; блокатор бета-рецепторов, выбранный из группы, состоящей из следующего: атенолол, метопролол, соталол гидрохлорид, пропранолол гидрохлорид и карведилол; и блокатор альфа-рецепторов, выбранный из группы, состоящей из фентоламина, толазолина, феноксибензамина и празозина; антилипемическое средство, выбранное из группы, состоящей из следующего: аторвастатин, ловастатин, симвастатин, правастатин, флувастатин, розувастатин, ципрофибрат, безафибрат, фенофибрат и гемфиброзил.
[00146] Антилипемическое средство выбрано из группы, состоящей из следующего: аторвастатин, ловастатин, симвастатин, правастатин, флувастатин, розувастатин, ципрофибрат, безафибрат, фенофибрат и гемфиброзил.
[00147] Антидиабетическое средство выбрано из группы, состоящей из следующего: инсулинотропное средство, метформины, ингибитор α-глюкозидаза, сенсибилизирующее средство - производное тиазолидиндиона, инсулинотропное средство - производное меглитинидов, агонист рецептора GLP-1 и ингибитор DPP-4. Антидиабетическое средство включает в себя без ограничения относящееся к сульфонилмочевинам инсулинотропное средство, выбранное из группы, состоящей из следующего: глипизид, гликлазид, глибенкламид, глиборнурид, глимепирид и гликвидон; относящееся к несульфонилмочевинам инсулинотропное средство - производное меглитинидов, выбранное из группы, состоящей из репаглинида и натеглинида; метформины, выбранные из метформина; ингибитор альфа-глюкозидазы, выбранный из группы, состоящей из Bose-100, акарбозы и воглибоза; инсулин-сенсибилизирующее средство, выбранное из росиглитазона и пиоглитазона; ингибитор дипепитилпептидазы 4 (DPP-4), выбранный из группы, состоящей из ситаглиптина, саксаглиптина и вилдаглиптина; и агонист рецептора GLP-1, выбранный из эксенатида и лираглутида.
[00148] Фармацевтическая композиция согласно настоящему изобретению содержит соединение I и фармацевтически приемлемый носитель, и ее можно вводить соответствующими путями, включая в себя без ограничения пероральный, парентеральный, интраперитонеальный, внутривенный, трансдермальный, подъязычный, внутримышечный, ректальный, назальный и подкожный пути введения.
[00149] Фармацевтическая композиция согласно настоящему изобретению предпочтительно представляет собой пероральный состав и более предпочтительно таблетки, таблетки пролонгированного высвобождения, капсулы или гранулы. Фармацевтическая композиция согласно настоящему изобретению также может представлять собой мягкие капсулы, микропилюли, микрокапсулы, микросферы, липосомы, самоэмульгирующуюся систему лекарственной доставки, твердые дисперсии, мицеллы, растворяющиеся в ротовой полости таблетки, растворы, суспензии или эмульсии.
[00150] Фармацевтически приемлемый носитель в фармацевтической композиции согласно настоящему изобретению представляет собой один или несколько нетоксических фармацевтических носителей. Указанные носители могут являться совместимыми с другими ингредиентами в фармацевтической композиции и не оказывают никакого вреда субъектам, принимающим композицию.
[00151] Носители
[00152] Фармацевтическая композиция согласно настоящему изобретению получена в виде перорального состава и содержит один или несколько носителей, включая в себя разбавители, наполнители, смазывающие средства, способствующие скольжению средства, связующие средства, средства для улучшения распадаемости, поверхностно-активные вещества и тому подобное. Носители, используемые в фармацевтической композиции согласно настоящему изобретению, можно выбирать и комбинировать для обеспечения лучших свойств, получая безопасные, эффективные и поддающиеся контролю фармакокинетические средства, когда композицию вводят субъекту, и удовлетворения требованиям введения лекарственного средства.
[00153] Разбавитель или наполнитель, используемый для увеличения массы состава, содержащего дозу для однократного введения, включает в себя без ограничения фосфат кальция, сульфат кальция, микрокристаллическую целлюлозу, лактозу, маннит, сорбит, крахмал и тому подобное.
[00154] Смазывающее средство, используемое для снижения трения между гранулами и стенкой формы при прессовании или выгрузке, для предотвращения прикрепления гранул к устройству для штамповки таблеток и/или для выгрузки гранул из устройства для штамповки таблеток, включает в себя без ограничения тальк, стеариновую кислоту, стеарат кальция, стеарат цинка, стеарат магния, растительные масла и тому подобное.
[00155] Способствующее скольжению средство, добавленное для улучшения подвижности гранул, включает в себя без ограничения тальк, диоксид кремния и кукурузный крахмал.
[00156] Связующее средство включает в себя без ограничения пирролидон, поливинилпирролидон, ксантановую камедь, целлюлозную камедь (такую как карбоксиметилцеллюлоза, метилцеллюлоза, гидроксиэтилцеллюлоза, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза), желатин и крахмал.
[00157] Средство для улучшения распадаемости, с помощью которого таблетки могут быстро распадаться на маленькие частицы в желудочно-кишечном соке, с тем чтобы активные ингредиенты растворялись и абсорбировались для проявления эффектов, включает в себя без ограничения крахмал, глину, целлюлозу, альгинаты, прежелатинизированный крахмал, кросполивинилпирролидон, кроскармеллозу натрия, карбоксиметилкрахмал натрия, растительный клей и тому подобное.
[00158] Поверхностно-активное вещество, способное изменять межфазное поверхностное натяжение для обеспечения увлажняющего и солюбилизирующего эффектов, включает в себя без ограничения полоксамер, додецилсульфат натрия, полисорбаты, полиэтиленгликольоктаноат, глицерилкапринат, полиэтиленгликольглицериллаурат, полиэтиленгликольглицерилстеарат и тому подобное.
[00159] Другие материалы-носители включают в себя без ограничения консерванты, антиоксиданты и другие материалы-носители, обычно используемые в фармацевтической промышленности.
[00160] Таблетки пролонгированного высвобождения могут содержать один или несколько материалов-носителей, выбранных из группы, состоящей из следующего: производные простых эфиров целлюлозы, включая в себя гидроксипропилметилцеллюлозу, этилцеллюлозу, карбоксиметилцеллюлозу натрия и гидропропилцеллюлозу; акриловые полимеры, включая в себя карбомер и акриловую смолу; хитин и производные, полимер молочной кислоты и тому подобное. Другие добавки могут быть включены дополнительно, часто используемые добавки включают в себя смачивающие средства (такие как этанол, вода и тому подобное), красители (такие как оксиды железа III), консерванты, антиоксид анты, препятствующие спадению средства (такие как глицин), поверхностно-активные вещества (такие как додецилсульфат натрия), регуляторы рН (такие как цитрат натрия или гидроксид натрия), наполнители (микрокристаллическая целлюлоза и тому подобное), и средства для улучшения распадаемости (кроскармеллоза натрия и тому подобное).
[00161] Часто используемые добавки для состава раствора, такого как пероральный состав согласно настоящему изобретению, включают в себя добавки для улучшения вкуса, улучшения прозрачности и усиления стабильности. Добавка для улучшения вкуса, как правило, включает в себя четыре класса, т.е. подсластитель, ароматические средства, слизь и шипучее средство. Подсластитель содержит встречающиеся в природе и полученные искусственно подсластители. Встречающийся в природе подсластитель может представлять собой сахарозу, сахарный сироп и ароматический сироп, тогда как искусственный подсластитель может представлять собой сахарин натрия, аспартам и тому подобное. Ароматические средства включает в себя лимонную эссенцию, масло мяты перечной, яблочную эссенцию и каучуковую эссенцию. Часто используемая слизь может представлять собой альгинат натрия, аравийскую камедь, желатин, метилцеллюлозу, карбоксиметилцеллюлозу натрия и тому подобное. Шипучее средство может представлять собой лимонную кислоту, винную кислоту и тому подобное. Добавка для усиления прозрачности представляет собой, например, хитозан, осветляющее средство на основе сока 101, ZTC1+1 встречающиеся в природе осветляющее средство, желатин, дубильную кислоту, яичные белки и тому подобное. Добавка для усиления стабильности может представлять собой сложные эфиры парагидроксибензоата, органические кислоты и их соли и другие добавки, такие как ди(ацетат) хлоргексидина, домифен и тому подобное.
[00162] Часто используемые добавки для мягких капсул могут представлять собой растительные масла, ароматические сложные эфиры, органические кислоты, глицерин, изопропанол, полиэтиленгликоль, пропандиол и поверхностно-активные вещества.
[00163] Микропилюли могут содержать один или несколько материалов-оснований, выбранных из группы, состоящей из растворимого в воде основания, не растворимого в воде основания и смешанного основания. Растворимое в воде основание, главным образом, включает в себя полиэтиленгликоль 4000, полиэтиленгликоль 6000, поливинилпирролидон (PVP), S-40 (полиоксиэтиленмоностеарат), стеарат натрия, глицерин, желатин, мочевина, полоксамер, ПЭГ вместе с поверхностно-активными веществами и простые полиэфиры. Основные не растворимые в воде основания представляют собой следующее: стеариновая кислота, глицерилмоностеарат, воск насекомых, гидрогенизированные растительные масла, стеариловый спирт, цетиловый спирт и полусинтетические жирные кислоты. Смешанное основание может представлять собой, например, полиэтиленгликоль вместе с S-40 или полоксамером. Смешанное основание используют с целью увеличения количества лекарственного средства, подлежащего распределению, регулируя предельно допустимый срок распределения или предельно допустимый срок распадаемости, что может быть полезным для состава микро пилюль.
[00164] Суспензии могут содержать один или несколько материалов-носителей, выбранных из группы, состоящей из следующего: суспендирующее средство, смачивающее средство, флоккулирующее средство и дефлоккулирующее средство.
[00165] Часто используемые суспендирующие средства включают в себя без ограничения суспендирующие средства на основе малых молекул, такие как глицерин и сиропы; и полимеры, такие как 1) встречающийся в природе полимер, который, главным образом, представляет собой растительный клей, такой как аравийская камедь, трагакантовая камедь и персиковая камедь, и растительный полисахарид, такой как альгинат натрия, агар и крахмальный клейстер; 2) синтетические или полусинтетические полимеры, включая в себя целлюлозы, такие как метилцеллюлоза, карбоксиметилцеллюлоза натрия и гидропропилцеллюлоза, и другие, такие как карбопол, повидон и глюкан; 3) диатомит; и 4) тиксотроп.
[00166] Смачивающее средство включает в себя без ограничения полисорбаты, полиоксиэтиленовое касторовое масло и полоксамер.
[00167] Флоккулирующее средство включает в себя без ограничения неорганические флоккулирующие средства, такие как сульфат алюминия, хлорид алюминия, сульфат железа (III) и хлорид железа (III); модифицированные монокатионные неорганические флоккулирующие средства, такие как полисиликат алюминия (феррит) и полифосфат алюминия (феррит); модифицированные поликатионные неорганические флоккулирующие средства, такие как сульфат полиалюминийхлорида железа (III), полисиликат железа (III), сополимер железа (III) и алюминия и тому подобное; органические полимеры, такие как полиакриламид; композитные флоккулирующие средства, такие как полиалюминийхлорид и полисульфат железа (III); и флоккулирующее средства микробиологического происхождения, такие как Phodococcus erythropolis и содержащий их NOC-1.
[00168] Дефлоккулирующее средство включает в себя без ограничения цитрат натрия, тартраты, фосфаты, карбонаты, глицинаты, сукцинат магния и дегидрохолат натрия.
[00169] Микрокапсулы могут содержать один или несколько материалов-носителей, выбранных из группы, состоящей из следующего: желатин, аравийская камедь, альбумин, крахмал, хитозан, альгинаты, метилцеллюлоза, этилцеллюлоза, карбоксиметилцеллюлоза, гидропропилметилцеллюлоза, полимер молочной кислоты, сополимер молочной и гликолевой кислот, полиалкилцианоакрилат, полиамид, поли(виниловый спирт) и полиакриловая смола.
[00170] Микросферы могут содержать один или несколько материалов-носителей, выбранных из группы, состоящей из следующего: встречающиеся в природе полимерные микросферы, такие как крахмальные микросферы, альбуминовые микросферы, желатиновые микросферы, хитозаны и тому подобное; и синтетические полимерные микросферы, такие как полимолочные микросферы и тому подобное.
[00171] Липосомы могут содержать один или несколько материалов-носителей, выбранных из группы, состоящей из фосфолипидов и холестеринов. Фосфолипиды включают в себя встречающиеся в природе и синтетические фосфолипиды. Основной встречающийся в природе фосфолипид представляет собой лецитин (фосфатидилхолин, PC), тогда как синтетический фосфолипид, главным образом, представляет собой DPPC (дипальмитоилфосфатидилхолин), DPPE (дипальмитоилфосфоэтаноламин) и DSPC (дистеароилфосфатидилхолин).
[00172] Самоэмульгирующаяся система лекарственной доставки может содержать один или несколько материалов-носителей, выбранных из группы, состоящей из масел, поверхностно-активных веществ и вспомогательных поверхностно-активных веществ.
[00173] Масла делятся на встречающиеся в природе растительные масла (такие как соевое масло, арахисовое масло и тому подобное), среднецепочечные триглицериды и полусинтетические среднецепочечные производные, такие как С8/С10 триглицериды кокосового масла, С8/С10 моноглицериды или диглицериды кокосового масла, сорбитанолеат, глицерилолеат-пропиленгликоль, глицерилолеат, пропиленгликольмонооктаноат, глицерилтрибутират и очищенные ацетилированные моноглицериды.
[00174] Поверхностно-активные вещества представляют собой, главным образом, полиоксиэтиленовое касторовое масло, полиоксиэтиленовое гидрогенизированное касторовое масло, полиоксиэтиленсорбитанолеат, полиоксиэтиленовые триглицериды, ПЭГ-8 глицериды каприловой/каприновой кислоты, С8/С10 полигликолизированные глицериды кокосового масла, ПЭГ глицерид лауриновой кислоты, Labrafil М 1944CS и Lavrafil M-22125CS.
[00175] Вспомогательные поверхностно-активные вещества включают в себя без ограничения этанол, пропиленгликоль, глицерин, изопропанол, полиэтиленгликоль и пропенилэтиленгликольлаурат.
[00176] Существует три трипа материалов-носителей для твердых дисперсий. Они представляют собой растворимые в воде материалы-носители, включая в себя полиэтиленгликоль (PEG), поливинилпирролидон (PVP), поверхностно-активные вещества, органические кислоты, сахариды, спирты и тому подобное; нерастворимые материалы-носители, включая в себя целлюлозу и полиакриловую смолу; и кишечнорастворимые материалы, включая в себя целлюлозу и полиакриловые смолы.
[00177] Мицеллы содержат поверхностно-активные вещества, включая в себя без ограничения гексадецилтриметиламмонийхлорид, гексадецилтриметиламмонийбромид, додецилтриметиламинийбромид, додецилпиридинийбромид, октансульфонат натрия, октилсульфат натрия, додецилсульфат натрия, тетрадецилсульфат натрия, гексадецилсульфат натрия, октадецилсульфат натрия, стеарат калия, олеат калия, лаурат калия, додецилсульфонат натрия, лауриловый эфир полиоксиэтилена (6), лауриловый эфир полиоксиэтилена (9), лауриловый эфир полиоксиэтилена (12), миристиловый эфир полиоксиэтилена (6), диоктилнатрийсульфосукцинат, додециламмонийхлорид, додецилбензолсульфонат натрия, лаурат сахарозы, пальмитат сахарозы, стеарат сахарозы, Tween-20, Tween-40, Tween-60, Tween-65, Tween 80 и Tween 85.
[00178] Растворяющиеся в ротовой полости таблетки могут содержать один или несколько материалов, выбранных из группы, состоящей из следующего: поверхностно-активные вещества, такие как додецилсульфат натрия, лецитин, Tween и Span; длинноцепочечные полимеры, такие как полипептиды (желатин или дегидратированный желатин); сахариды и производные, такие как декстран, глюканы, сорбит, маннит и крахмал; гели, такие как аравийская камедь, ксантановая камедь и растительный клей; целлюлозы; альгинаты; PVP; и поливиниловый спирт. Могут быть включены некоторые другие добавки, включая в себя смачивающее средство (такое как этанол), краситель (такой как оксиды трехвалентного железа), консервант, антиоксидант, препятствующее спадению средство (такое как глицин), усилитель проникновения (такой как додецилсульфат натрия), регулятор рН (такой как цитрат натрия или гидроксид натрия), ароматические средства и подсластитель.
Краткое описание графических материалов
[00179] Фигура 1 представляет собой кривую, на которой показана корреляция между вариабельностью SBP на основании исходных уровней и AUC0-24 в модели DSS с гипертензией и повреждением почек, индуцированным высоким содержанием солей, у крыс.
[00180] Фигура 2 представляет собой кривую, на которой показана корреляция между вариабельностью UACR на основании исходных уровней и AUC0-24 в модели DSS с гипертензией и повреждением почек, индуцированным высоким содержанием солей, у крыс.
Подробное раскрытие настоящего изобретения
[00181] Настоящее изобретение включает в себя без ограничения следующие примеры. Другие варианты осуществления для реализации технического решения согласно настоящему изобретению подпадают под объем, заявленный в настоящем изобретении.
[00182] Пример 1. Получение соединения I
[00183] 2-хлор-4-[(3S,3aR)-3-циклопентил-7-(4-гидроксилпиперидин-1-карбонил)-3,3а,4,5-тетрагидро-2H-пиразоло[3,4-ƒ]хинолин-2-ил]бензонитрил (в настоящем документе называемый "соединение I"), со структурой, как показано ниже.
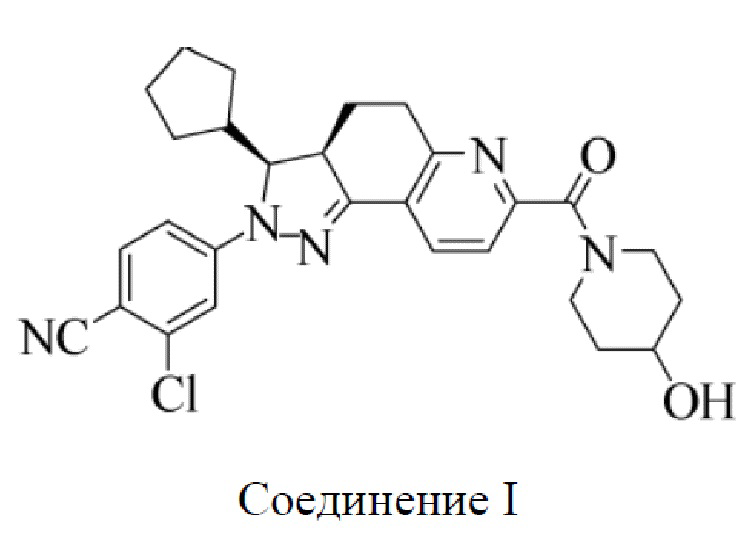
[00184] Соединение I можно получить способом, описанным в международной патентной публикации WO 2012022121 A1 или WO 2014094664 A1, или с помощью других способов синтеза.
[00185] Пример 2. Иллюстративные составы фармацевтической композиции согласно настоящему изобретению
[00186] Примеры согласно настоящему изобретению представляют часть составов фармацевтической композиции, которые являются применимыми для введения фармацевтической композиции согласно настоящему изобретению. Следует отметить, что состав в настоящем документе не ограничен представленными ниже, другие составы, характеризующиеся биодоступностью, составляющей 50% или больше, подпадают под заявленный объем настоящего изобретения.
[00187] Иллюстративные составы таблетки показаны ниже.
[00188] Состав 1. Каждая таблетка содержала 0,5 мг соединения I.

[00189] Способ изготовления:
(1) Поливинилпирролидон K30 вводили в состав водного раствора с концентрацией, составляющей 5% (масс./масс.), чтобы использовать в качестве связующего средства;
(2) Микрокристаллическую целлюлозу РН101, маннит и кроскармеллозу натрия взвешивали в количествах, как описано выше, и в достаточной мере смешивали в течение 15 минут для получения смеси 1;
(3) Суспензию, содержащую 0,5 г наноразмерного соединения I, взвешивали и затем добавляли к смеси 1 и смешивали с ней; к полученной смеси добавляли водный раствор поливинилпирролидона K30, связующего средства, для получения мягкого материала, который обрабатывали с помощью сита 24 меш для получения влажных гранул;
(4) Влажные гранулы высушивали при 55±5°С в течение 2-3 часов, контролировали, чтобы содержание воды в гранулах находилось на уровне ниже 2,5%;
(5) Высушенные гранулы снова просеивали через сито 24 меш;
(6) Стеарат магния и диоксид кремния добавляли к гранулам в количествах, описанных выше, и затем смешивали в течение 15 минут;
(7) Проводили прессование таблеток, при этом контролировали, чтобы твердость таблетки составляла 6-10 кг;
(8) Таблетки покрывали оболочкой;
(9) Таблетки упаковывали и размещали на складе.
[00190] Состав 2. Каждый таблетка содержала 0,5 мг соединения I.

[00191] Состав 3. Каждый таблетка содержала 2,5 мг соединения I.

[00192] Способ изготовления:
(1) Исходные материалы просеивали через сито 30 меш и хранили до следующего применения;
(2) Соединение I, додецилсульфат натрия, микрокристаллическую целлюлозу РН101, моногидрат лактозы, кроскармеллозу натрия и гидроксипропилцеллюлозу в достаточной мере смешивали в течение 15 минут;
(3) Воду добавляли в смесь для получения мягкого материала, который обрабатывали с помощью сита 24 меш для получения влажных гранул;
(4) Влажные гранулы высушивали при 55±5°С в течение 2-3 часов, контролировали, чтобы содержание воды в гранулах находилось на уровне ниже 2,5%;
(5) Высушенные гранулы снова просеивали через сито 24 меш;
(6) Стеарат магния и диоксид кремния добавляли к гранулам в количествах, описанных выше, и затем смешивали в течение 15 минут;
(7) Проводили прессование таблеток, при этом контролировали, чтобы твердость таблетки составляла 6-10 кг;
(8) Таблетки покрывали оболочкой;
(9) Таблетки упаковывали и размещали на складе.
[00193] Иллюстративные составы капсулы показаны ниже.
[00194] Состав 1. Каждая капсула содержала 0,1 мг соединения I.

[00195] Состав 2. Каждая капсула содержала 0,25 мг соединения I.

[00196] Способ изготовления:
(1) Поливинилпирролидон K30 вводили в состав водного раствора с концентрацией, составляющей 5% (масс./масс.), чтобы использовать в качестве связующего средства;
(2) Соединение I, микрокристаллическую целлюлозу РН101, маннит и кроскармеллозу натрия взвешивали в количествах, описанных выше, и в достаточной мере смешивали в течение 15 минут; измеряли содержание воды полученной смеси;
(3) К полученной смеси добавляли водный раствор поливинилпирролидона K30 в качестве связующего средства для получения мягкого материала, который обрабатывали с помощью сита 24 меш для получения влажных гранул;
(4) Влажные гранулы высушивали при 55±5°С в течение 2-3 часов, контролировали, чтобы содержание воды в гранулах находилось на уровне ниже 2,5%;
(5) Высушенные гранулы снова просеивали через сито 24 меш;
(6) Стеарат магния и диоксид кремния добавляли к гранулам в количествах, описанных выше, и затем смешивали в течение 15 минут;
(7) Рассчитывали количество полученной смеси, подлежащее введению в капсулу, и смесь взвешивали в этом количестве и помещали в капсулу;
(8) Капсулы помещали во флаконы из полиэтилена высокой плотности для твердых пероральных лекарственных средств и флаконы герметично закрывали;
(9) Флаконы размещали на складе.
[00197] Состав 3. Каждая капсула содержала 0,5 мг соединения I.

[00198] Состав 4. Каждая капсула содержала 2,5 мг соединения I.


[00199] Состав 5. Каждая капсула содержала 10 мг соединения I.

[00200] Способ изготовления:
(1) Получение раствора связующего средства: поливинилпирролидон K30 вводили в состав водного раствора с концентрацией, составляющей 3-5% (масс./масс.), чтобы использовать в качестве связующего средства;
(2) Смешивание: микрокристаллическую целлюлозу РН101, маннит и кроскармеллозу натрия пропускали через сито 30 меш и затем переносили с соединением I в гранулятор с большим усилием сдвига, где их смешивали в течение 2-3 минут при скорости перемешивания, составляющей 180-220 об/мин, и скорости резания, составляющей 475-525 об/мин;
(3) Грануляция: раствор связующего средства добавляли к смеси, полученной на стадии (2) не позднее 5-7 минут после завершения стадии (2), и смесь подвергали грануляции при скорости перемешивания, составляющей 180-220 об/мин, и скорости резания, составляющей 475-525 об/мин, с последующей грануляцией в течение 0,5-1,5 минут при скорости перемешивания, составляющей 180-220 об/мин, и скорости резания, составляющей 1800-2200 об/мин;
(4) Сушка: влажные гранулы высушивали при 55±5°С, контролировали, чтобы содержание воды в гранулах находилось на уровне ниже 2,5%;
(5) Дробление: высушенные гранулы помещали в мельницу Фитца и пропускали через сито №0033 при скорости вращения, составляющей 840-960 об/мин; стадию дробления можно проводить с использованием других способов или устройств, при условии, что может быть получен тот же эффект;
(6) Смешивание: стеарат магния и диоксид кремния просеивали, соответственно, с использованием сита 30 меш или некоторых других устройств; диоксид кремния затем помещали в V-образный смеситель и смешивали в течение 12 минут при 20 об/мин; добавляли стеарат магния и смешивали с диоксидом кремния в течение 3 минут при 20 об/мин;
(7) Заполнение капсул: капсулы заполняли смешанным порошком с использованием устройства для заполнения капсул (MF-30);
(8) Полировка: капсулы полировали с использованием полировальной машины для капсул;
(9) Упаковка и маркировка: капсулы помещали во флаконы из HDPE, чьи крышки плотно завинчивали и запаивали с использованием электромагнитной запаечной машины; флаконы маркировали позже.
[00201] Состав 6. Каждая капсула содержала 2,5 мг соединения I.

[00202] Способ изготовления:
(1) Исходные материалы пропускали через сито 30 меш и хранили до будущего использования;
(2) Смешивание: соединение I, микрокристаллическую целлюлозу РН101, моногидрат лактозы, кроскармеллозу натрия, стеарат магния и диоксид кремния равномерно смешивали в течение 15 минут;
(3) Рассчитывали количество, подлежащее введению в капсулу, и смесь в этом количестве помещали в капсулы;
(4) Капсулы помещали во флаконы из полиэтилена высокой плотности для твердых пероральных лекарственных средств и флаконы герметично закрывали;
(5) Флаконы размещали на складе.
[00203] Иллюстративная самоэмульгирующаяся система лекарственной доставки (SEDDS) показана ниже.
[00204] Состав SEDDS
[00205] Соединение I: этанол: Kolliphor EL : Miglyol 812N = 10 мг : 1 г : 5 г : 4 г
[00206] Способ изготовления:
[00207] Соединение I растворяли в этаноле, полученное добавляли и смешивали с Kolliphor EL и Miglyol 812N для получения эмульгированной смеси.
[00208] Иллюстративные составы мягкой капсулы показаны ниже.
[00209] Состав 1. Каждая мягкая капсула содержала 0,25 мг соединения I.
[00210] Состав 2. Каждая мягкая капсула содержала 0,5 мг соединения I.

[00211] Состав 3. Каждая мягкая капсула содержала 1,0 мг соединения I.
[00212] Способ изготовления:
(1) Соединение I смешивали с полиэтиленгликолем 400 и пропиленгликолем и растворяли в них;
(2) Смесь прессовали в мягкие капсулы;
(3) Мягкая капсулы упаковывали и размещали на складе.
[00213] Иллюстративные составы суспензии показаны ниже.
[00214] Состав 1. Суспензия содержала 0,25 мг соединения I на мл.

[00215] Способ изготовления:
(1) Метилцеллюлозу растворяли в воде, чье количество составляло приблизительно 80% от используемой всего; раствор хранили для будущего использования;
(2) Соединение I взвешивали и помещали в раствор; после достаточного смешивания соединение I суспендировали в растворе;
(3) Воду добавляли до общего объема, составляющего 1000 мл, и проводили достаточное смешивание.
[00216] Состав 2. Суспензия содержала 0,5 мг соединения I на мл.
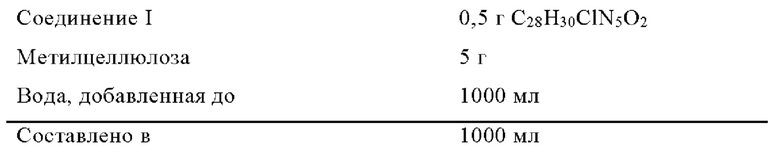
[00217] Состав 3. Суспензия содержала 1,0 мг соединения I на мл.
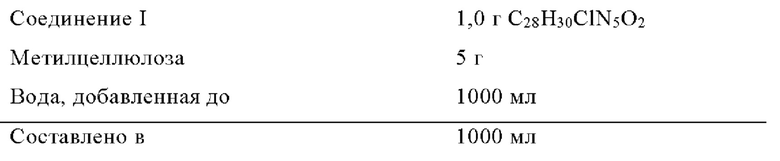
[00218] Иллюстративные составы эмульсии показаны ниже.
[00219] Состав 1. Эмульсия содержала 0,25 мг соединения I на мл.

[00220] Способ изготовления:
(1) Получение водной фазы: соединение I и полиэтиленгликоль 400 нагревали до 60°С так, чтобы соединение I растворилось; раствор хранили в тепле для будущего использования;
(2) Получение масляной фазы: глицерилмоностеарат и соевое масло нагревали до 60°С и растворяли; раствор хранили в тепле для будущего использования;
(3) Водную фазу добавляли и смешивали при 60°C с масляной фазой и полученную смесь добавляли и смешивали при 60°C с Span 80 и Tween 80; воду добавляли до общего объема, составляющего 1000 мл; полученную смесь измельчали до однородного размера с использованием коллоидной мельницы.
[00221] Состав 2. Эмульсия содержала 0,5 мг соединения I на мл.

[00222] Состав 3. Эмульсия содержала 1,0 мг соединения I на мл.
[00222] Состав 3. Эмульсия содержала 1,0 мг соединения I на мл.

[00223] Иллюстративные составы микропилюли показаны ниже.
[00224] Состав 1. Каждая пилюля содержала 0,1 мг соединения I.

[00225] Состав 2. Каждая пилюля содержала 0,25 мг соединения I.

[00226] Состав 3. Каждая пилюля содержала 0,5 мг соединения I.

[00227] Состав 4. Каждая пилюля содержала 1,0 мг соединения I.

[00228] Способ изготовления:
(1) Соединение I, полиэтиленгликоль 4000 и полиэтиленгликоль 6000 взвешивали в количествах, изложенных выше, для будущего использования;
(2) Полиэтиленгликоль 4000 и полиэтиленгликоль 6000 расплавляли на водяной бане при 80°С и затем добавляли соединение I, которое растворяли при перемешивании; смесь хранили при 80°С для будущего использования;
(3) При температуре, составляющей 80°С, смесь добавляли по каплям в конденсированный диметикон, где смесь конденсировали в твердые пилюли;
(4) Пилюли извлекали и высушивали, а затем помещали в полиэтиленовые флаконы, которые должным образом запечатывали.
[00229] Иллюстративные составы растворяющейся в ротовой полости таблетки показаны ниже.
[00230] Состав 1. Каждая растворяющаяся в ротовой полости таблетка содержала 0,25 мг соединения I.

[00231] Способ изготовления:
(1) Соединение I, микрокристаллическую целлюлозу РН101, маннит и кроскармеллозу натрия равномерно смешивали;
(2) Воду добавляли к смеси для проведения грануляции и полученные гранулы высушивали;
(3) Стеарат магния и диоксид кремния добавляли для однородного смешивания с гранулами;
(4) Проводили дражирование;
(5) Полученные таблетки покрывали оболочкой и затем упаковывали.
[00232] Состав 2. Каждая растворяющаяся в ротовой полости таблетка содержала 0,5 мг соединения I.

[00233] Состав 3. Каждая растворяющаяся в ротовой полости таблетка содержала 1,0 мг соединения I.

[00234] Иллюстративные составы таблетки длительного высвобождения показаны ниже.
[00235] Состав 1. Каждая таблетка длительного высвобождения содержала 0,25 мг соединения I.

[00236] Способ изготовления:
(1) Соединение I и добавки взвешивали;
(2) Полученное в достаточной мере смешивали в течение 15 минут;
(3) Проводили дражирование;
(4) Полученные таблетки покрывали оболочкой и затем упаковывали.
[00237] Состав 2. Каждая таблетка длительного высвобождения содержала 0,5 мг соединения I.

[00238] Состав 3. Каждая таблетка длительного высвобождения содержала 1,0 мг соединения I.


[00239] Иллюстративные составы микрокапсулы показаны ниже.
[00240] Состав 1. Каждый грамм микрокапсул содержал 0,25 мг соединения I.

[00241] Способ изготовления:
(1) Стеариновую кислоту взвешивали и расплавляли на водяной бане;
(2) Соединение I взвешивали и затем перемешивали с 10% этилцеллюлозой в этаноле и растворяли в ней;
(3) Этанол (сколько потребуется) добавляли к смеси, полученной на стадии (2), и полученную смесь добавляли к расплавленной стеариновой кислоте; смесь оставляли нагреваться на водяной бане до получения однородной жидкости;
(4) Жидкость распыляли и охлаждали, при этом микрокапсулы оседали;
(5) Микрокапсулы собирали.
[00242] Состав 2. Каждый грамм микрокапсул содержал 0,5 мг соединения I.

[00243] Состав 3. Каждый грамм микрокапсул содержал 1,0 мг соединения I.

[00244] Иллюстративный состав липосомы показан ниже.
[00245] Состав 1. Каждый мл липосом содержал 0,25 мг соединения I.


[00246] Способ изготовления:
(1) Соевый лецитин и холестерин взвешивали и растворяли в этаноле (сколько потребуется);
(2) Соединение I взвешивали и растворяли с помощью ультразвука в полиэтиленгликоле 4000; воду добавляли к раствору до общего объема, составляющего 800 мл;
(3) Раствор, полученный на стадии (1), медленно вводили в раствор, полученный на стадии (2), и полученную смесь перемешивали при 55°С; этанол полностью удаляли и воду добавляли до общего объема, составляющего 1000 мл.
[00247] Иллюстративный состав мицеллы показан ниже.
[00248] Состав 1. Каждый мл мицеллы содержал 0,25 мг соединения I.

[00249] Способ изготовления:
(1) Соединение I, соевый лецитин и полиэтиленгликоль 4000 взвешивали и растворяли в этаноле;
(2) Воду добавляли до общего объема, составляющего 1000 мл, для разведения полученной смеси.
[00250] Иллюстративные составы раствора показаны ниже.
[00251] Состав 1. Каждые 5 мл раствора содержали 0,1 мг соединения I.


[00252] Состав 2. Каждые 5 мл раствора содержали 0,25 мг соединения I.

[00253] Состав 3. Каждые 5 мл раствора содержали 0,5 мг соединения I.

[00254] Состав 4. Каждые 5 мл раствора содержали 1,0 мг соединения I.

[00255] Способ изготовления:
(1) Соединение I и добавки взвешивали;
(2) Додецилсульфат натрия растворяли в воде для инъекций (сколько потребуется) и затем добавляли соединение I, перемешивали для растворения; затем эдетат динатрия, лимонную кислоту и аспартам добавляли в полученный раствор и растворяли в нем;
(3) Полученный раствор подвергали фильтрованию и стерилизации;
(4) Ампулы заполняли раствором, по 5 мл каждая;
(5) Ампулы упаковывали.
[00256] Пример 3. Эффект размера частиц на абсорбцию
[00257] Пример 3-1. Эффект размера частиц на абсорбцию
[00258] Соединение I с различными составами и/или различными значениями D90 сравнивали в отношении показателей абсорбции у самцов крыс SD.
[00259] Образцы для испытания: соединение I получали с помощью способа, как описано в примере 1 международной патентной публикации WO 2014094664 A1, и затем тонко измельчали для получения образцов соединения I с D90, составляющим 538 нм, 3,8 мкм, 21,7 мкм, 41,5 мкм и 71,6 мкм, соответственно; непосредственно перед испытаниями указанные образцы суспендировали в 0,5% МС для получения 1 мг/мл суспензий.
Растворы: 5% DMSO + 95% (раствор, содержащий 6% HP-β-CD)
Твердые дисперсии: соединение I : поливинилпирролидон K30 = 1:8 (масс./масс.)
[00260] Крысам SD внутрижелудочно вводили однократную дозу исследуемых образцов в дозе, составляющей 1,0 мг/кг, и кровь собирали до введения и через 30 мин, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 10 ч и 24 ч после введения.
[00261] Состав для внутривенной инъекции представлял собой раствор, где соединение I растворяли в 5% DMSO + 95% (6% HP-β-CD раствор), и его вводили крысам SD в дозе, составляющей 2 мг/кг. Образцы крови крыс собирали перед введением и через 5 мин, 15 мин, 30 мин, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч и 24 ч после введения.
[00262] Забор крови: Животных фиксировали и их хвосты нагревали на водяной бане за 10 мин до забора крови; приблизительно 100 мкл крови собирали через хвостовую вену для каждого животного и помещали в пробирку с антикоагулянтом, содержащую гепарин; образцы крови подвергали центрифугированию при 8000 об/мин в течение 6 мин при 4°С для получения образцов плазмы, которые необходимо получить не позднее чем через 30 минут после забора крови; образцы плазмы хранили в морозильной камере при -80°С для будущих испытаний.
[00263] Анализ образцов: 50 мкл каждого образец плазмы из морозильной камеры переносили в центрифужную пробирку; 100 мкл воды и 400 мкл стандартного раствора МТВЕ (50 нг/мл) добавляли в пробирку и хорошо смешивали с плазмой; полученную смесь смешивали в течение 10 минут с использованием вихревого смесителя и затем подвергали центрифугированию в течение 10 минут (4000 об/мин); 300 мкл супернатанта переносили в другую центрифужную пробирку и высушивали потоком газообразного азота; полученное вещество растворяли в 200 мкл раствора, состоящего из метанола и воды в соотношении, составляющем 1:1, из чего 20 мкл использовали для испытания ЖХ-МС/МС.
[00264] Обработка данных: Концентрацию соединения выводили с помощью Analyst 1.6.1 (АВ Sciex); средние, среднеквадратические отклонения и коэффициенты вариации рассчитывали с использованием Microsoft Excel (вычисления не требовались, если указанные параметры были напрямую выведены с помощью Analyst 1.6.1) и фармакокинетические параметры определяли с использованием анализа NCA, выполненного в Pharsight Phoenix 6.3.
[00265] Результаты и обсуждение

[00266] Выводы
[00267] Из представленной выше таблицы можно увидеть, что чем ниже размер частиц соединения I, тем выше будет биодоступность. Когда соединение I характеризовалось размером частиц, составляющим 21,7 мкм, биодоступность достигала 54,6%, что удовлетворяет клиническим требованиям.
[00268] Пример 3-2. Эффект размера частиц на абсорбцию
[00269] Образец для испытания: соединение I с D90, составляющим 5 мкм, суспендировали в 0,5% МС непосредственно перед испытаниями для получения 0,25 мг/мл суспензии.
[00270] Животные: три собаки породы бигль.
[00271] Способ
[00272] Введение лекарственного средства и забор крови:
(1) Все животные голодали в течение 12 ч или дольше перед введением лекарственного средства, и корм предоставляли через 4 ч после введения лекарственного средства, с неограниченным доступом к воде в течение всего времени. Животным внутрижелудочно вводили исследуемые образцы в однократной дозе в дозе, составляющей 0,5 мг/кг, и 200 мкл крови собирали для каждого животного, через малую подкожную вену, непосредственно перед введением (t=0) и через 10 мин, 30 мин, 45 мин, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 12 ч, 24 ч, 30 ч, 48 ч и 72 ч после введения. Собранную кровь хранили в сухой пробирке с гепарином (изотонический раствор хлорида натрия, содержащий 0,1% гепарина натрия).
(2) Получение плазмы: образцы цельной крови, собранные на стадии (1), подвергали центрифугированию при низкой скорости (8000 об/мин, 6 мин, 4°С) для отделения плазмы (цельную кровь хранили в передвижной морозильной камере при приблизительно 0-4°С, и плазму было необходимо отделить не позднее чем через 30 минут после забора крови), и плазму хранили в темноте в морозильной камере при -70°С (или ниже) для будущего анализа.
[00273] Анализ образцов: 50 мкл каждого образца плазмы из морозильной камеры переносили в центрифужную пробирку; 100 мкл воды и 400 мкл стандартного раствора МТВЕ (50 нг/мл) добавляли в пробирку и хорошо смешивали с плазмой; полученную смесь смешивали в течение 10 минут с использованием вихревого смесителя и затем подвергали центрифугированию в течение 5 минут (12000 об/мин); 300 мкл супернатанта переносили в другую центрифужную пробирку и высушивали потоком газообразного азота; полученное вещество растворяли в 200 мкл раствора, состоящего из метанола и воды в соотношении, составляющем 1:1, из чего 20 мкл использовали для измерения ЖХ-МС/МС.
[00274] Обработка данных: Концентрацию соединения выводили с помощью Analyst 1.6.1 (АВ Sciex); средние, среднеквадратические отклонения и коэффициенты вариации рассчитывали с использованием Microsoft Excel (вычисления не требовались, если указанные параметры напрямую выводили с помощью Analyst 1.6.1) и фармакокинетические параметры определяли с использованием анализа NCA, выполненного в Pharsight Phoenix 6.3.
[00275] Результаты:


[00276] Пример 3-3. Добавки не оказывали никакого эффекта на абсорбцию
[00277] С помощью способов, описанных в примере 3-1, капсулу состава капсулы 6, полученную в примере 2, добавляли в воду для получения суспензии с концентрацией, составляющей 0,1 мг/мл, которую вводили внутрижелудочно крысам SD в однократной дозе в дозе, составляющей 10 мл/кг (с фактической дозой, составляющей 0,95 мг/кг).
[00278] Было доказано, что AUCINF составляла 821 нг*ч/мл (раствор при введении с помощью внутривенной инъекции в дозе, составляющей 2,0 мг/кг, давал в результате AUCINF, составляющую 3126 нг*ч/мл), с биодоступностью, составляющей 52,5%.
[00279] Пример 3-4. Эффект размера частиц на абсорбцию
[00280] Образцы для испытания:
Капсула состава капсулы 5, полученная в примере 2, 10 мг соединения I на капсулу.
Растворы: 5% DMSO + 95% (6% HP-β-CD раствор)
[00281] Животные: четыре собаки породы бигль.
[00282] Способ
[00283] Введение лекарственного средства и забор крови:
(1) Введение капсул. Все животные голодали в течение 12 ч или дольше перед введением лекарственного средства, и корм предоставляли через 4 ч после введения лекарственного средства, всегда с неограниченным доступом к воде. Собакам породы бигль перорально давали капсулу состава капсулы 5, полученную в примере 2 (две капсулы для каждой собаки породы бигль, 10 мг соединения I на капсулу) в однократной дозе, и 200 мкл крови собирали для каждого животного, через малую подкожную вену, непосредственно перед введением (0 ч) и через 0,17 ч, 0,50 ч, 0,75 ч, 1,0 ч, 2,0 ч, 4,0 ч, 6,0 ч, 8,0 ч, 12 ч и 24 ч после введения. Собранную кровь хранили в сухой пробирке с гепарином (изотонический раствор хлорида натрия, содержащий 0,1% гепарина натрия).
(2) Получение плазмы. Образцы цельной крови, собранные на стадии (1), подвергали центрифугированию при низкой скорости (8000 об/мин, 6 мин, 4°С) для отделения плазмы (цельную кровь хранили в передвижной морозильной камере при приблизительно 0-4°С, и плазму было необходимо отделить не позднее чем через 30 минут после забора крови), и плазму хранили в темноте в морозильной камере при -70°С (или ниже) для будущего анализа.
[00284] Анализ образцов: 50 мкл каждого образца плазмы из морозильной камеры переносили в центрифужную пробирку; 100 мкл воды и 400 мкл стандартного раствора МТВЕ (50 нг/мл) добавляли в пробирку и хорошо смешивали с плазмой; полученную смесь смешивали в течение 10 минут с использованием вихревого смесителя и затем подвергали центрифугированию в течение 5 минут (12000 об/мин); 300 мкл супернатанта переносили в другую центрифужную пробирку и высушивали потоком газообразного азота; полученное вещество растворяли в 200 мкл раствора, состоящего из метанола и воды в соотношении, составляющем 1:1, из чего 20 мкл использовали для измерения ЖХ-МС/МС.
[00285] Обработка данных: Концентрацию соединения выводили с помощью Analyst 1.6.1 (АВ Sciex); средние, среднеквадратические отклонения и коэффициенты вариации рассчитывали с использованием Microsoft Excel (вычисления не требовались, если указанные параметры напрямую выводили с помощью Analyst 1.6.1) и фармакокинетические параметры определяли с использованием анализа NCA, выполненного в Pharsight Phoenix 6.3.
[00286] Результаты:
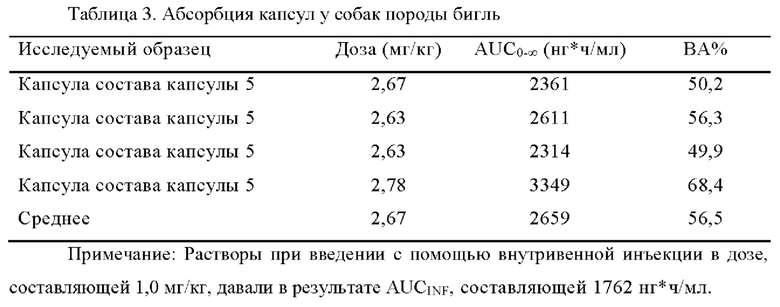
[00287] Пример 3-5. Эффект размера частиц на абсорбцию
[00288] Образец для испытания: Капсула состава капсулы 6, полученная в примере 2, 2,5 мг соединения I на капсулу.
[00289] Животные: четыре собаки породы бигль.
[00290] Собакам породы бигль перорально вводили капсулу состава капсулы 6, полученную в примере 2, по одной капсуле для каждой собаки породы бигль, 2,5 мг соединения I на капсулу. Эксперименты проводили с использованием способов, описанных в примере 3-3.

[00291] Пример 4. Эффект поверхностно-активных веществ на абсорбцию
[00292] Пример 4-1. Эффект поверхностно-активных веществ на абсорбцию
[00293] Образцы для испытания: 1 мг/мл суспензии составляли с помощью смешивания соединения I и поверхностно-активного вещества в соотношениях, составляющих 1:5, 1:10 и 1:20, соответственно.
[00294] Крысам SD внутрижелудочно вводили исследуемые образцы в однократной дозе в дозе, составляющей 1,0 мг/кг, и кровь собирали перед введением и через 30 мин, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 10 ч и 24 ч после введения.
[00295] Забор крови: Животных фиксировали и их хвосты нагревали на водяной бане за 10 мин до забора крови; приблизительно 100 мкл крови собирали через хвостовую вену для каждого животного и помещали в пробирку с антикоагулянтом, содержащую гепарин; образцы крови подвергали центрифугированию при 8000 об/мин в течение 6 мин при 4°С для получения образцов плазмы, которые необходимо получить не позднее чем через 30 минут после забора крови; образцы плазмы хранили в морозильной камере при -80°С для будущих испытаний.
[00296] Анализ образцов: 50 мкл каждого образца плазмы из морозильной камеры переносили в центрифужную пробирку; 100 мкл воды и 400 мкл стандартного раствора МТВЕ (50 нг/мл) добавляли в пробирку и хорошо смешивали с плазмой; полученную смесь смешивали в течение 10 минут с использованием вихревого смесителя и затем подвергали центрифугированию в течение 5 минут (12000 об/мин); 300 мкл супернатанта переносили в другую центрифужную пробирку и высушивали потоком газообразного азота; полученное вещество растворяли в 200 мкл раствора, состоящего из метанола и воды в соотношении, составляющем 1:1, из чего 20 мкл использовали для измерения ЖХ-МС/МС.
[00297] Обработка данных: Концентрацию соединения выводили с помощью Analyst 1.6.1 (АВ Sciex); средние, среднеквадратические отклонения и коэффициенты вариации рассчитывали с использованием Microsoft Excel (вычисления не требовались, если указанные параметры напрямую выводили с помощью Analyst 1.6.1) и фармакокинетические параметры определяли с использованием анализа NCA, выполненного в Pharsight Phoenix 6.3.
[00298] Результаты
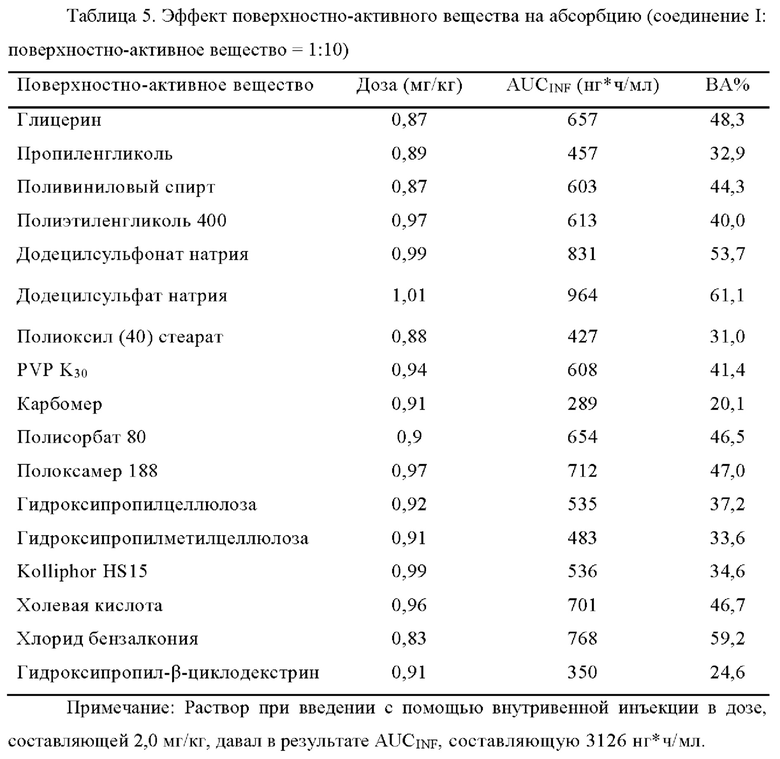
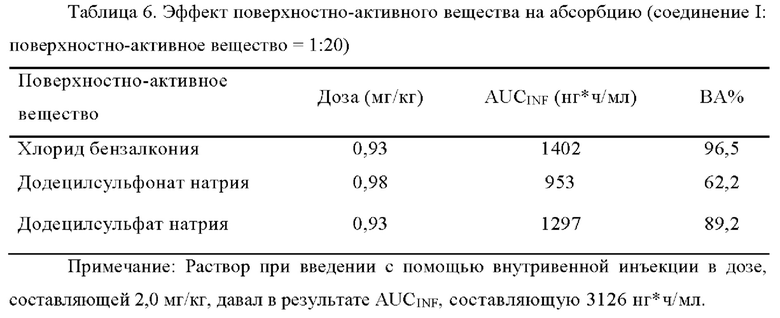


[00299] Пример 4-2. Эффект поверхностно-активных веществ на абсорбцию
[00300] Образцы для испытания: таблетка состава таблетки 3, полученная в примере 2, 2,5 мг соединения I на таблетку.
Растворы: 5% DMSO + 95% (6% HP-β-CD раствор), внутривенное (IV) введение.
[00301] Животные: восемь собак породы бигль, четыре для каждой группы.
[00302] Способ
[00303] Введение лекарственного средства и забор крови:
(1) Все животные голодали в течение 12 ч или дольше до введения лекарственного средства, и корм предоставляли через 4 ч после введения лекарственного средства, всегда с неограниченным доступом к воде. Собакам породы бигль перорально давали таблетки, полученные в примере 2, по две таблетки для каждой собаки породы бигль, 2,5 мг соединения I на таблетку, и 200 мкл крови собирали для каждого животного, через малую подкожную вену, через 30 мин, 45 мин, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 12 ч и 24 ч после введения. Собранную кровь образцы хранили в сухой пробирке с K2EDTA.
Для группы с внутривенным (IV) введением кровь собирали через 5 мин, 15 мин, 30 мин, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 12 ч и 24 ч после введения.
(2) Получение плазмы: образцы цельной крови, собранные на стадии (1), подвергали центрифугированию при низкой скорости (8000 об/мин, 6 мин, 4°С) для отделения плазмы (цельную кровь хранили в передвижной морозильной камере при приблизительно 0-4°С, и плазму было необходимо отделить не позднее чем через 30 минут после забора крови), и плазму хранили в темноте в морозильной камере при -70°С (или ниже) для будущего анализа.
[00304] Анализ образцов: 50 мкл каждого образца плазмы из морозильной камеры переносили в центрифужную пробирку; 100 мкл воды и 400 мкл стандартного раствора МТВЕ (50 нг/мл) добавляли в пробирку и хорошо смешивали с плазмой; полученную смесь смешивали в течение 10 минут с использованием вихревого смесителя и затем подвергали центрифугированию в течение 5 минут (12000 об/мин); 300 мкл супернатанта переносили в другую центрифужную пробирку и высушивали потоком газообразного азота; полученное вещество растворяли в 200 мкл раствора, состоящего из метанола и воды в соотношении, составляющем 1:1, из чего 20 мкл использовали для измерения ЖХ-МС/МС.
[00305] Обработка данных: Концентрацию соединения выводили с помощью Analyst 1.6.1 (АВ Sciex); средние, среднеквадратические отклонения и коэффициенты вариации рассчитывали с использованием Microsoft Excel (вычисления не требовались, если указанные параметры напрямую выводили с помощью Analyst 1.6.1) и фармакокинетические параметры определяли с использованием анализа NCA, выполненного в Pharsight Phoenix 6.3.
[00306] Результаты

[00307] Пример 4-3. Эффект поверхностно-активных веществ на абсорбцию
[00308] Образец для испытания:
Самоэмульгирующаяся система лекарственной доставки (SEDDS): соединение I: этанол : Kolliphor EL : Miglyol 812N = 10 мг : 1 г : 5 г : 4 г.
Растворы: 5% DMSO + 95% (6% HP-β-CD раствор)
[00309] Крысам SD внутрижелудочно вводили исследуемые образцы в однократной дозе в дозе, составляющей 1,0 мг/кг, и кровь собирали перед введением и через 30 мин, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 10 ч и 24 ч после введения.
[00310] Состав для внутривенной инъекции представлял собой раствор, где соединение I растворяли в 5% DMSO + 95% (6% HP-β-CD раствор), и его вводили крысам SD в дозе, составляющей 2 мг/кг. Образцы крови крыс собирали перед введением и через 5 мин, 15 мин, 30 мин, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч и 24 ч после введения.
[00311] Забор крови: Животных фиксировали и их хвосты нагревали на водяной бане за 10 мин до забора крови; приблизительно 100 мкл крови собирали через хвостовую вену для каждого животного и помещали в пробирку с антикоагулянтом, содержащую гепарин; образцы крови подвергали центрифугированию при 8000 об/мин в течение 6 мин при 4°С для получения образцов плазмы, которые необходимо получить не позднее чем через 30 минут после забора крови; образцы плазмы хранили в морозильной камере при -80°С для будущих испытаний.
[00312] Анализ образцов: 50 мкл каждого образца плазмы из морозильной камеры переносили в центрифужную пробирку; 100 мкл воды и 400 мкл стандартного раствора МТВЕ (50 нг/мл) добавляли в пробирку и хорошо смешивали с плазмой; полученную смесь смешивали в течение 10 минут с использованием вихревого смесителя и затем подвергали центрифугированию в течение 5 минут (12000 об/мин); 300 мкл супернатанта переносили в другую центрифужную пробирку и высушивали потоком газообразного азота; полученное вещество растворяли в 200 мкл раствора, состоящего из метанола и воды в соотношении, составляющем 1:1, из чего 20 мкл использовали для измерения ЖХ-МС/МС.
[00313] Обработка данных: Концентрацию соединения выводили с помощью Analyst 1.6.1 (АВ Sciex); средние, среднеквадратические отклонения и коэффициенты вариации рассчитывали с использованием Microsoft Excel (вычисления не требовались, если указанные параметры напрямую выводили с помощью Analyst 1.6.1) и фармакокинетические параметры определяли с использованием анализа NCA, выполненного в Pharsight Phoenix 6.3.
[00314] Результаты и выводы
[00315] Крысы, которым перорально вводили лекарственное средство SEDDS, характеризовались AUCINF, составляющей 1297 нг*ч/мл (раствор, введенный внутривенно в дозе, составляющей 2,0 мг/кг, давал в результате AUCINF, составляющую 3126 нг*ч/мл), с биодоступностью, составляющей 83,0%.
[00316] Пример 5. Токсичность и фармакокинетические исследования у крыс SD, которым внутрижелудочно вводили соединение I в течение 13 недель с восстановительным периодом, составляющим 4 недель
[00317] Образец для испытания: твердая дисперсия соединения I (соединение I : PVPK30 = 1:8 (масс./масс.))
[00318] Животные: крысы SD, уровень SPF.
[00319] Способ:
[00320] 220 крыс SD использовали в исследованиях. Трем группам, т.е. группе высокой дозы, группе промежуточной дозы и группе низкой дозы, вводили соединение I в следующих дозах, по 20 самцов и 20 самок в каждой группе. В частности, животным-самцам вводили суспензию в дозах, составляющих 3, 9 и 30 мг/кг/день, соответственно, тогда как самкам вводили в дозах, составляющих 1, 3 и 10 мг/кг/день, соответственно. Животным в контрольной группе, 20 самцов и 20 самок, давали очищенную воду. Все животные голодали в течение ночи перед заборами крови и анатомическими экспериментами.
[00321] Кроме того, для фармакокинетического исследования 9 самцов и 9 самок включили в группу введения, при этом 3 самца и 3 самки для контрольной группы.
[00322] Результаты и выводы
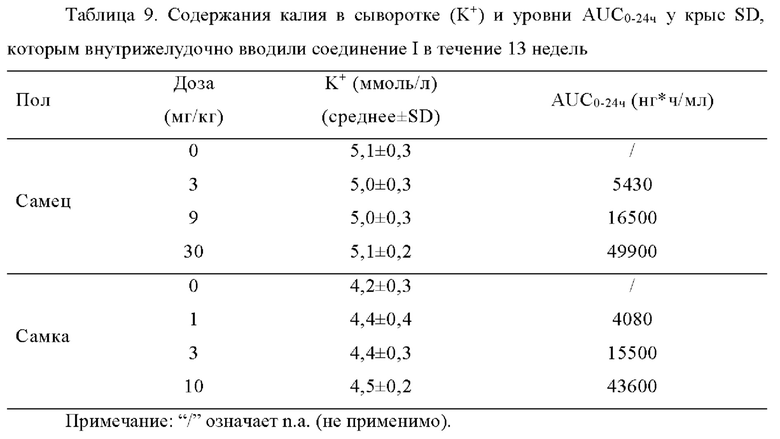
[00323] На основании приведенных выше результатов NOAEL (максимальная доза, не вызывающая наблюдаемых нежелательных эффектов) составляла 30 мг/кг/день у животных-самцов и 10 мг/кг/день у самок, с соответствующей AUC0-24ч, составляющей 49900 нг*ч/мл и 43600 нг*ч/мл, соответственно, причем ниже этого уровня AUC явное аномальное содержание калия в сыворотке не наблюдалось.
[00324] Пример 6. Испытание на эффективность у чувствительных к соли крыс с индуцированным солью повреждением почек
[00325] Животные: самцы крыс Dahl/ss
[00326] Исследуемое изделие и состав:
Твердые дисперсии соединения I (соединение I : PVPK30 = 1:8 (масс./масс.)) : Твердые дисперсии соединения I составляли с подходящими количествами стерильной воды для инъекций в суспензии с концентрациями, составляющими 0,03, 0,10, 0,30 и 1,00 мг/мл, соответственно. Суспензии получали непосредственно перед использованием.
[00327] Крыс Dahl/ss случайным образом разделили на 6 групп на основании значений артериального давления, исследованного перед введением лекарственного средства, т.е. контрольная группа 1 с нормальными животными (n=10), контрольная группа 2 с животными с индуцированным повреждением почек (4% NaCl, n=12), группы лечения с помощью доз, составляющих 0,3 мг/кг/день (n=11), 1 мг/кг/день (n=11), 3 мг/кг/день (n=11) и 10 мг/кг/день (n=11), соответственно, где n представляет собой число животных.
[00328] Способ
[00329] Крыс Dahl/ss кормили исследовательским рационом AIN-93G, содержащим 4% NaCl, чтобы индуцировать гипертензивную нефропатию, с тем чтобы провести испытания in vivo фармакодинамической активности соединения I.
[00330] За одну неделю до эксперимента значения артериального давления измеряли два раза у крыс путем измерения артериального давления с помощью хвостовой манжеты, с тем чтобы крысы могли приспособиться к операции мониторинга артериального давления. Затем, непосредственно перед испытанием, артериальное давление измеряли и использовали в качестве исходного значения. В соответствии с исходными значениями артериального давления крыс случайным образом делили на группы. На следующий день животных в контрольной группе 2 и группах лечения кормили исследовательским рационом AIN-93G, содержащим 4% NaCl и воду, ad libitum, для индукции модели повреждения почек. Построение модели заняло 42 дня. Животным в контрольной группе 1 давали нормальный рацион AIN-93G.
[00331] Животным в группах лечения внутрижелудочно вводили соединение I два раза в день в дозах, составляющих 0,3, 1, 3 и 10 мг/кг/день, соответственно, в виде суспензий соединения I, вводимых в объеме, составляющем 5 мл/кг на дозу. Двум контрольным группам давали равное количество стерильной воды для инъекций.
[00332] Измерение артериального давления (систолического артериального давления, SBP): артериальное давление измеряли в течение 6 недель, один раз в неделю и анализировали изменения артериального давления в указанных группах.
[00333] Патологическое исследование почек и сердца: в конце исследования крысы безболезненно умерщвляли, проводили билатеральный забор почек и сердца для гистопатологического анализа. Почки окрашивали с использованием гематоксилина-эозина (НЕ) для полуколичественной оценки повреждений почек. Толщину стенки левого желудочка измеряли для каждого сердца для анализа повреждения сердца.
[00334] Кроме того, образцы крови через 0 ч, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 10 ч и 24 ч после последнего введения собирали из хвостовой вены крыс до окончания исследования. Образцы крови помещали в низкотемпературную высокоскоростную центрифугу и центрифугировали в течение 6 мин при 4°С, 8000 об/мин. Образцы плазмы собирали и хранили в морозильной камере при -80°С до измерения концентрации лекарственного средства в плазме.
[00335] Результаты и выводы
[00336] В соответствии с результатами испытания строили кривую зависимости артериального давления (SBP) или соотношения альбумина к креатинину в моче (UACR) от AUC0-24 (см. фиг. 1 и фиг. 2).
[00337] Кривая зависимости SBP или UACR от AUC0-24 показала точку волнообразного изменения при 100 ч*нг/мл AUC, после которой кривая имела тенденцию быть плоской, и SBP и UACR значительно уменьшались. Это указывало на то, что соединение I характеризовалось хорошей эффективностью и оказывало действие, когда AUC достигала 100 ч*нг/мл.
[00338] Пример 7. Исследования безопасности, переносимости и фармакокинетики у здоровых субъектов с введением однократной дозы
[00339] Пяти группам здоровых субъектов давали капсулы в дозах, составляющих 0,5 мг (1 капсула, 0,5 мг соединения I на капсулу), 1,0 мг (2 капсулы, 0,5 мг соединения I на капсулу), 2,5 мг (1 капсула, 2,5 мг соединения I на капсулу), 10 мг (1 капсула, 10 мг соединения I на капсулу) и 30 мг (3 капсулы, 10 мг соединения I на капсулу), причем каждая группа содержала по 8 субъектов. Введение лекарственного средства проводили только однократно.
[00340] Капсулы, упомянутые выше, представляли собой капсулы, полученные в примере 2, т.е. капсулы состава капсулы 3 (0,5 мг соединения I на капсулу), состава капсулы 4 (2,5 мг соединения I на капсулу) и состава капсулы 5 (10 мг соединения I на капсулу).
[00341] Образцы плазмы собирали для измерения концентрации лекарственного средства в плазме и фармакокинетические параметры анализировали с использованием некомпартментной модели. Образцы сыворотки собирали для измерения содержаний калия в сыворотке.
[00342] Результаты
[00343] У здоровых субъектов, которым проводили введение в дозе, составляющей 0,5-30 мг/день, AUC0-24 соединения I составляла 162,5-5016 нг*ч/мл, и Т1/2 составляло приблизительно 60 часов, при этом повышенное содержание калия в сыворотке не наблюдалось.

[00344] Пример 8. Исследования безопасности, переносимости и фармакокинетики у здоровых субъектов с введением многократных доз
[00345] Двум группам здоровых субъектов давали капсулы один раз в день в суточных дозах, составляющих 2,5 мг (1 капсула на каждое введение, 2,5 мг соединения I на капсулу), и 5,0 мг (2 капсулы на каждое введение, 2,5 мг соединения I на капсулу), соответственно, причем каждая группа содержала по 6 субъектов. Введение проводили в течение 14 дней подряд. Капсулы, используемые в этом исследовании, представляли собой состав капсулы 4, полученный в примере 2.
[00346] Образцы плазмы собирали для измерения концентрации лекарственного средства в плазме и фармакокинетические параметры анализировали с использованием некомпартментной модели. Образцы сыворотки собирали для измерения содержаний калия в сыворотке.
[00347] Результаты
[00348] У здоровых субъектов, которым проводили введение в режиме многократных доз с суточной дозой, составляющей 2,5 мг/день, средняя равновесная AUC0-24 составляла 2865±821 нг*ч/мл, без наблюдаемого повышенного содержания калия в сыворотке.
[00349] У здоровых субъектов, которым проводили введение в режиме многократных доз с суточной дозой, составляющей 5,0 мг/день, средняя равновесная AUC0-24 составляла 6376±1028 нг*ч/мл, с временным повышенным содержанием калия в сыворотке, наблюдаемым у трех субъектов.
[00350] Пример 9. Исследования безопасности, переносимости и фармакокинетики у пациентов с хронической болезнью почек
[00351] Пациенты в настоящем примере представляли собой пациентов с хронической болезнью почек с симптомами почечной недостаточности, протеинурии, острого повреждения почек, клубочкового нефрита, почечной кисты, частого мочеиспускания, почечного камня, обструктивной уропатии и т.д. Указанные пациенты также характеризуются наличием других заболеваний или осложнений, таких как сахарный диабет, гиперлипидемия, гиперхолестеринемия, гипертензия, заболевания периферических кровеносных сосудов, атеросклеротическая болезнь сердца и т.д.
[00352] Указанные пациенты продолжали принимать лекарственные средства, которые они принимали до настоящего клинического испытания, при этом лекарственные средства представляют собой, например, ингибиторы ангиотензинпревращающего фермента, такие как лизиноприл, беназеприл, эналаприл и эналаприл малеат; блокаторы рецептора ангиотензина II, такие как валсартан и лозартан; блокаторы кальциевого канала, такие как амлодипин и нифедипин; диуретики, такие как фуросемид; блокаторы бета-рецепторов, такие как метопролол сукцинат, метопролол тартрат, карведилол и атенолол; и антилипемические лекарственные средства, такие как симвастатин, аторвастатин, фенофибрат, правастатин и розувастатин.
[00353] В настоящем примере вводимая доза относилась к количеству соединения I, хотя вводили фармацевтическую композицию. Двум группам субъектов проводили введение один раз в день в течение 56 дней подряд в суточных дозах, составляющих 0,5 мг (1 капсула для каждого введения, 0,5 мг соединения I на капсулу) и 2,5 мг (1 капсула для каждого введения, 2,5 мг соединения I на капсулу), соответственно, 6 субъектов в группе введения дозы, составляющей 0,5 мг/день, а также 6 субъектов в группе введения дозы, составляющей 2,5 мг/день. Используемые в настоящем испытании капсулы представляли собой капсулы, полученный в примере 2, т.е. капсулы состава капсулы 3 (капсула с 0,5 мг соединения I), и состава капсулы 4 (капсула, содержащая 2,5 мг соединения I).
[00354] Образцы плазмы собирали для измерения концентрации лекарственного средства в плазме и фармакокинетические параметры анализировали с использованием некомпартментной модели. Образцы сыворотки собирали для измерения содержаний калия в сыворотке.
[00355] Результаты
[00356] У пациентов с ХБП, которым введение осуществляли в многократных дозах с суточной дозой, составляющей 0,5 мг, средняя равновесная AUCtau составляла 652,5±232,2 нг*ч/мл, без наблюдаемого повышенного содержания калия в сыворотке.
[00357] У пациентов с ХБП, которым введение проводили в многократных дозах с суточной дозой, составляющей 2,5 мг, умеренное увеличение содержания калия в сыворотке наблюдали у одного субъекта. Средняя безопасная равновесная AUCtau у пациентов составляла 2613±280 нг*ч/мл.
[00358] У этих пациентов с ХБП после многократных введений в дозе, составляющей 0,5 мг/день или 2,5 мг/день, наблюдали значительное уменьшение значений артериального давления и соотношения альбумина к креатинину в моче (UACR). На 8ой неделе UACR уменьшалось на 58,9% и 50,7% в двух группах, соответственно. Фармацевтическая композиция согласно настоящему изобретению показала очевидный эффект на защиту почек.
[00359] Пациенты с ХБП характеризовались равновесной AUCtau, составляющей 105,6 нг*ч/мл или больше при введении в течение одного дня в суточной дозе, составляющей 0,5 мг, и UACR уменьшалось на 30,5% или больше по сравнению с исходным уровнем, указывая на эффективность фармацевтической композиции согласно настоящему изобретению.
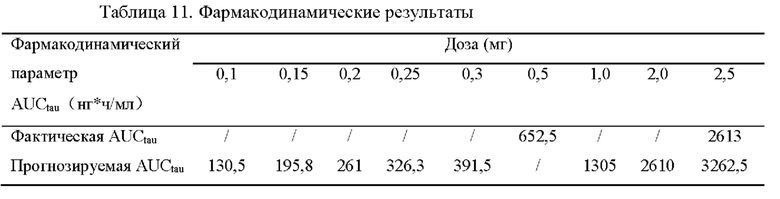
[00360] У пациентов с ХБП, принимающих фармацевтическую композицию согласно настоящему изобретению, значение AUCtau соединения I находилось в диапазоне, составляющем 188 нг*ч/мл - 3173 нг*ч/мл, указывая на превосходную эффективность. Кроме того, повышенное содержание калия в сыворотке не наблюдалось. Таким образом, фармацевтическая композиция согласно настоящему изобретению удовлетворяет требованиям клинической безопасности и эффективности.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ АНТАГОНИСТ МИНЕРАЛОКОРТИКОИДНЫХ РЕЦЕПТОРОВ, И ЕЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2750667C2 |
| СХЕМА ЛЕЧЕНИЯ ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ ФАБРИ С ИСПОЛЬЗОВАНИЕМ СТАБИЛИЗИРОВАННОЙ α-ГАЛАКТОЗИДАЗЫ | 2018 |
|
RU2793184C2 |
| ТАКРОЛИМУС ДЛЯ УЛУЧШЕННОГО ЛЕЧЕНИЯ ПАЦИЕНТОВ С ТРАНСПЛАНТАТАМИ | 2009 |
|
RU2574006C2 |
| СХЕМА ЛЕЧЕНИЯ 2-АМИНО-1-(2-(4-ФТОРФЕНИЛ)-3-(4-ФТОРФЕНИЛАМИНО)-8,8-ДИМЕТИЛ-5,6-ДИГИДРОИМИДАЗО[1,2-А]ПИРАЗИН-7(8Н)-ИЛ)ЭТАНОНОМ И ЕГО КОМБИНАЦИЯМИ | 2018 |
|
RU2791466C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ОТ HELICOBACTER PYLORI | 2014 |
|
RU2671400C2 |
| НИЗКОДОЗОВАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ИЗОТРЕТИНОИНА ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ | 2015 |
|
RU2707753C2 |
| КОМПОЗИЦИИ АКАМПРОСАТА, СПОСОБЫ ИХ ПРИМЕНЕНИЯ И СОДЕРЖАЩИЕ ИХ КОМБИНАЦИИ | 2012 |
|
RU2671399C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ 3α-ЭТИНИЛ-3β-ГИДРОКСИАНДРОСТАН-17-ОНА ОКСИМА | 2018 |
|
RU2779262C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ СОКРИСТАЛЛОВ ТРАМАДОЛА И КОКСИБОВ | 2011 |
|
RU2567843C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ БЕССОННИЦЫ | 2015 |
|
RU2703297C2 |
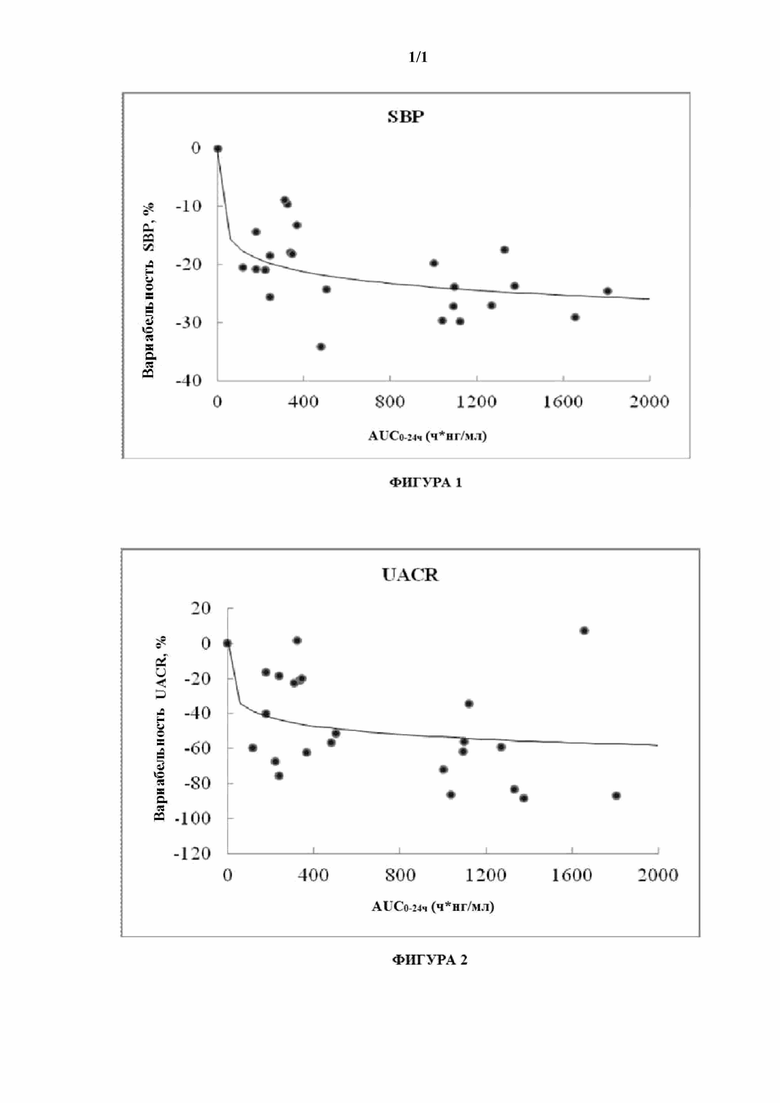
Группа изобретений относится к фармацевтической промышленности, в частности к фармацевтической композиции для лечения хронической болезни почек у человека, содержащей соединение I 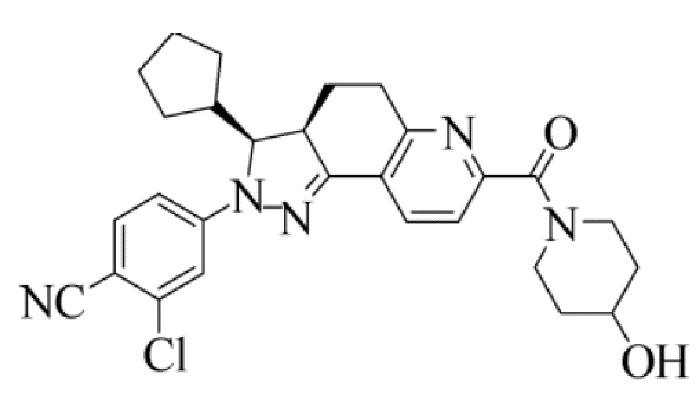 и один или несколько фармацевтически приемлемых носителей, причем композиция представляет собой пероральную лекарственную форму, содержащую от 0,1 до 1,0 мг соединения I для однократного применения. Также предложены способ лечения и/или профилактики хронической болезни почек путем введения нуждающемуся в этом пациенту-человеку указанной композиции и применение данной композиции для получения лекарственного средства для лечения и/или профилактики хронической болезни почек. Группа изобретений обеспечивает установление конкретного диапазона доз для безопасного и эффективного лечения хронической болезни почек при отсутствии побочных эффектов гиперкалиемии. 3 н. и 20 з.п. ф-лы, 2 ил., 11 табл., 9 пр.
и один или несколько фармацевтически приемлемых носителей, причем композиция представляет собой пероральную лекарственную форму, содержащую от 0,1 до 1,0 мг соединения I для однократного применения. Также предложены способ лечения и/или профилактики хронической болезни почек путем введения нуждающемуся в этом пациенту-человеку указанной композиции и применение данной композиции для получения лекарственного средства для лечения и/или профилактики хронической болезни почек. Группа изобретений обеспечивает установление конкретного диапазона доз для безопасного и эффективного лечения хронической болезни почек при отсутствии побочных эффектов гиперкалиемии. 3 н. и 20 з.п. ф-лы, 2 ил., 11 табл., 9 пр.
1. Фармацевтическая композиция для лечения хронической болезни почек у человека, содержащая соединение I:
и один или несколько фармацевтически приемлемых носителей, причем фармацевтическая композиция представляет собой пероральную лекарственную форму и от 0,1 до 1,0 мг соединения I содержится в фармацевтической композиции для однократного применения.
2. Фармацевтическая композиция по п. 1, причем от 0,1 до 0,5 мг соединения I содержится в фармацевтической композиции для однократного применения.
3. Фармацевтическая композиция по п. 1, причем от 0,2 до 0,5 мг соединения I содержится в фармацевтической композиции для однократного применения.
4. Фармацевтическая композиция по п. 1, причем введение композиции человеку обеспечивает биодоступность соединения I, составляющую 50% или больше.
5. Фармацевтическая композиция по п. 4, причем D90 частиц соединения I составляет 25 мкм или меньше.
6. Фармацевтическая композиция по п. 5, причем D90 частиц соединения I составляет 10 мкм или меньше.
7. Фармацевтическая композиция по п. 4, причем фармацевтическая композиция содержит одно или несколько поверхностно-активных веществ, выбранных из группы, состоящей из хлорида бензалкония, лаурилсульфоната натрия, додецилсульфата натрия, глицерина, холевой кислоты, полоксамера, поливинилового спирта, полисорбата 80, ПВП K30 и полиэтиленгликоля.
8. Фармацевтическая композиция по п. 7, причем фармацевтическая композиция содержит одно или несколько поверхностно-активных веществ, выбранных из группы, состоящей из хлорида бензалкония, лаурилсульфоната натрия и додецилсульфата натрия.
9. Фармацевтическая композиция по п. 8, причем фармацевтическая композиция содержит одно поверхностно-активное вещество, выбранное из группы, состоящей из хлорида бензалкония, лаурилсульфоната натрия и додецилсульфата натрия.
10. Фармацевтическая композиция по п. 7, в которой массовое отношение соединения I к поверхностно-активному веществу составляет 1:0,1-1:20.
11. Фармацевтическая композиция по п. 10, в которой массовое отношение находится между 1:1 и 1:20.
12. Фармацевтическая композиция по п. 1, причем фармацевтическая композиция представляет собой таблетку, таблетку длительного высвобождения, капсулу, гранулу, мягкую капсулу, микропилюлю, микрокапсулу, микросферу, липосому, самоэмульгирующуюся систему лекарственной доставки, твердую дисперсию, мицеллу, растворяющуюся в ротовой полости таблетку, раствор, суспензию или эмульсию.
13. Фармацевтическая композиция по п. 1, причем при пероральном введении фармацевтической композиции нуждающемуся в этом пациенту с хронической болезнью почек безопасные и эффективные диапазоны площади под кривой зависимости концентрации в плазме от времени (AUC) составляют от 188 нг⋅ч/мл до 3173 нг⋅ч/мл.
14. Фармацевтическая композиция по п. 13, причем диапазоны AUC составляют от 188 нг⋅ч/мл до 2893 нг⋅ч/мл.
15. Фармацевтическая композиция по п. 14, причем диапазоны AUC составляют от 188 нг⋅ч/мл до 2613 нг⋅ч/мл.
16. Фармацевтическая композиция по п. 15, причем диапазоны AUC составляют от 188 нг⋅ч/мл до 1117 нг⋅ч/мл.
17. Фармацевтическая композиция по п. 16, причем диапазоны AUC составляют от 188 нг⋅ч/мл до 885 нг⋅ч/мл.
18. Способ лечения и/или профилактики хронической болезни почек, предусматривающий введение нуждающемуся в этом пациенту-человеку фармацевтической композиции по любому из пп. 1-17.
19. Способ по п. 18, дополнительно предусматривающий введение пациенту-человеку одного или нескольких средств, выбранных из группы, состоящей из антигипертензивного средства, антилипемического средства и антидиабетического средства.
20. Фармацевтическая композиция по п. 1, в форме, где однократная доза составляет от 0,1 до 1,0 мг.
21. Фармацевтическая композиция по п. 1, в форме, где однократная доза составляет от 0,1 до 0,5 мг.
22. Фармацевтическая композиция по п. 1, в форме, где однократная доза составляет от 0,2 до 0,5 мг.
23. Применение фармацевтической композиции по любому из пп. 1-17 для получения лекарственного средства для лечения и/или профилактики хронической болезни почек.
| US 20130289029 A1, 31.10.2013 | |||
| US 20150336950 A1, 26.11.2015 | |||
| RU 2012140690 A, 27.03.2014. |

