Область техники, к которой относится изобретение
Настоящее изобретение относится к новой фармацевтической композиции, к ее применению в терапии, такой как лечение медицинских состояний, таких как гиперсомния и печеночная энцефалопатия.
Предшествующий уровень техники
Одна из самых больших проблем в разработке фармацевтических препаратов заключается в том, что лекарственные соединения очень часто являются нерастворимыми или плохо растворимыми в водной среде. Недостаточная растворимость лекарственного средства в свою очередь означает недостаточную биодоступность и плохой уровень лекарственного средства в плазме при введении субъектам, таким как люди и животные.
Установлено, что от 40% до 70% всех новых химических веществ, определенных в программах поиска лекарств, недостаточно растворимы в водной среде (M. Lindenberg, S et al.: European Journal of Pharmaceutics and Biopharmaceuticals, vol. 58, no.2, pp. 265-278, 2004; D.J. Hauss: Drugs and Pharmaceutical Sciences, Vol. 170, pp. 1-339, Informa Healthcare NC, 2007; Gupta et al.; Volume 2013, Article ID 848043 Review Article http://dx.doi.org/10.1155/2013/848043).
Ученые исследовали различные способы решения проблемы низкой растворимости лекарств, чтобы повысить биодоступность плохо абсорбируемых лекарств, с целью повышения их клинической эффективности при пероральном приеме. Такие технологии, как увеличение площади поверхности и, следовательно, повышение растворения, иногда могут решить проблемы растворимости. Другими способами, которые могут также решить проблемы биодоступности, являются добавление поверхностно-активных веществ и полимеров. Тем не менее, каждое химическое соединение имеет свои уникальные химические и физические свойства и, следовательно, имеет свои собственные различные проблемы при изготовлении фармацевтического препарата, который может проявлять свою клиническую эффективность.
Приготовление лекарственного средства в различных типах липидов полезно для конкретных лекарственных средств. Липидные композиции для перорального введения обычно состоят из лекарственного средства, растворенного в смеси вспомогательных веществ с широким спектром физико-химических свойств, начиная от чистых триглицеридных масел, моно- и диглицеридов и значительной части липофильных или гидрофильных поверхностно-активных веществ и сорастворителей.
Основным критерием при выборе подходящих вспомогательных веществ для любой композиции на основе липидов является определение одного или нескольких вспомогательных веществ, которые обладают способностью растворять полную дозу и в то же время обеспечивают приготовленную стандартную дозу лекарственного средства, которую можно принимать перорально, и которая имеет размер, пригодный для проглатывания пациентом. Обычно лекарственная нагрузка в сочетании с размером таблетки или капсулы является ограничивающим фактором.
Композиции на основе липидов могут содержать только один липид или смесь различных типов липидов в комбинации. Также часто при приготовлении плохо растворимого лекарственного средства требуется также включать одно или несколько дополнительных вспомогательных веществ для получения удовлетворительной растворимости, а также стабильности лекарственного средства. Фармацевтические препараты, содержащие несколько типов липидных систем в комбинациях, часто сложны в изготовлении, и, следовательно, стоимость товаров возрастает.
Самоэмульгирующиеся системы доставки лекарств (SEDDS) могут быть полезны для приготовления плохо растворимых лекарств. Однако очень немногие составы на основе липидов достигли фармацевтического рынка. Пищевые масла, которые представляют собой логический и предпочтительный выбор липидных вспомогательных веществ для разработки SEDDS, редко выбираются из-за их плохой способности к растворению больших количеств липофильных лекарств. Свойства самоэмульгирования также требуют включения в композицию относительно больших количеств поверхностно-активного вещества в дополнение к масляному носителю лекарственного средства.
Смесь моно- и диглицеридов каприловой/каприновой кислоты (Akoline) является эмульгатором природного происхождения, который является предпочтительным, поскольку он считается более безопасным, чем синтетические коммерческие поверхностно-активные вещества. Однако среди специалистов в области фармацевтики признается, что такие вспомогательные вещества имеют ограниченную эффективность самоэмульгирования (P.P. Constantinides; Pharmaceutical Research, vol. 12, no. 11. Pp. 1561-1572, 1995).
Обычно концентрация поверхностно-активного вещества составляет от 30 до 60% от общей композиции для образования SEDDS (C.W. Pouton; International Journal of Pharmaceutics, vol. 27, no. 2-3, pp. 335-348, 1985). Большие количества поверхностно-активных веществ могут вызвать раздражение желудочно-кишечного тракта. Поверхностно-активные вещества, включенные в композицию SEDDS, должны иметь относительно высокое значение ГЛБ и гидрофильность, чтобы обеспечить быстрое и легкое диспергирование в водной жидкости ЖКТ в виде очень тонкодисперсной эмульсии масло-в-воде, и, следовательно, могут быть достигнуты хорошие самоэмульгирующие свойства. Кроме того, один или несколько сорастворителей часто добавляют в композицию, чтобы способствовать растворению высоких концентраций лекарственного средства.
Соединение 3α-этинил-3β-гидроксиандростан-17-она оксим в настоящее время изучают в клинических испытаниях II фазы для лечения печеночной энцефалопатии (ПЭ) и гиперсомнии (ГС). Одна из проблем, связанных с этим соединением, состоит в том, что оно имеет плохую растворимость в водной среде, и, следовательно, существует необходимость в способе приготовления этого соединения для получения клинически и коммерчески пригодного лекарственного продукта.
Описание изобретения
Соединение 3α-этинил-3β-гидроксиандростан-17-она оксим (Соединение I)
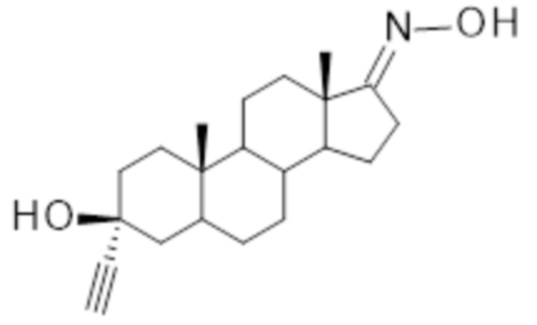 Соединение I
Соединение I
имеет плохую растворимость в водных средах, что, в свою очередь, означает, что лекарственная нагрузка является низкой, и поэтому трудно достичь клинически терапевтической дозы без назначения пациентам чрезмерного количество доз лекарственного средства. Для обеспечения возможности приготовления из этого соединения фармацевтического лекарственного продукта, обеспечивающего достаточную лекарственную нагрузку и, следовательно, достижения клинически достаточного терапевтического эффекта лекарственного продукта, была разработана новая фармацевтическая композиция.
Одним аспектом настоящего изобретения является фармацевтическая композиция, включающая:
(i) 3α-этинил-3β-гидроксиандростан-17-она оксим
; и
(ii) носитель, содержащий
(а) 45-100% сложного моноэфира;
(b) при необходимости, до 51% сложного диэфира;
(с) при необходимости, до 10% сложного триэфира;
где сложный моноэфир, сложный диэфир (если имеется) и/или сложный триэфир (если имеется) содержит жирную кислоту, выбранную из одной или нескольких из:
- каприловой кислоты (С8) в количестве 50-90%;
- каприновой кислоты (С10) в количестве 10-50%;
- капроновой кислоты (С6) в количестве до 3%;
- лауриновой кислоты (С12) в количестве до 100%;
- пальмитиновой кислоты (С16) в количестве до 12%;
- миристиновой кислоты (С14) в количестве до 3%;
- стеариновой кислоты (С18) в количестве до 6%;
- олеиновой кислоты (С18: 1) в количестве до 100%;
- линолевой кислоты (С18: 2) в количестве до 35%; и где
количество соединения 3α-этинил-3β-гидроксиандростан-17-она оксима составляет 0,1-10 масс.% от общей массы композиции.
В одном аспекте изобретения фармацевтическая композиция по настоящему изобретению включает 3α-этинил-3β-гидроксиандростан-17-она оксим в количестве от 0,1 до 10 масс.% от общей массы композиции, и наполнитель, содержащий:
(а) 45-100% сложного моноэфира;
(b) при необходимости до 51% сложного диэфира;
(с) при необходимости до 10% сложного триэфира;
где жирная кислота (кислоты), образующая сложный моноэфир (моноэфиры), и жирная кислота (кислоты), образующая необязательный сложный ди- и/или триэфир (эфиры), выбраны из одной или нескольких из каприловой кислоты (С8), каприновой кислоты (C10), капроновой кислоты (C6), лауриновой кислоты (C12), пальмитиновой кислоты (C16), миристиновой кислоты (C14), стеариновой кислоты (C18), олеиновой кислоты (C18:1) и линолевой кислоты (C18:2).
В одном аспекте общее количество вышеуказанного сложного эфира (эфиров) жирных кислот, образующего носитель, составляет 100% носителя, и носитель полностью состоит из одного или нескольких из вышеуказанных сложных эфиров жирных кислот.
В одном аспекте изобретения носитель является сложным моноэфиром.
В еще одном аспекте изобретения носитель представляет собой смесь сложного моноэфира и сложного диэфира.
В еще одном аспекте изобретения носитель представляет собой смесь сложного моноэфира и сложного триэфира.
В еще одном аспекте изобретения носитель представляет собой смесь сложного моноэфира, сложного диэфира и сложного триэфира.
Настоящее изобретение является совершенно неожиданным в том, что биодоступность и воздействие лекарственного средства 3α-этинил-3β-гидроксиандростан- 17-она оксима могут быть увеличены путем приготовления указанного соединения в соответствии с настоящим изобретением.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где включение поверхностно-активного вещества или любого другого вспомогательного вещества в композицию в дополнение к наполнителю не требуется.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, состоящая только из 3α-этинил-3β-гидроксиандростан-17-она оксима и только определенного выше носителя.
Один аспект изобретения представляет фармацевтическую композицию, как описано и заявлено в настоящем документе, состоящую из 3α-этинил-3β-гидроксиандростан-17-она оксима и только определенного выше носителя, где вышеуказанный сложный эфир (эфиры) жирной кислоты, образующий носитель, составляет 100% от носителя.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где количество 3α-этинил-3β-гидроксиандростан-17-она оксима составляет от 0,1 до 5 масс.% от общей массы композиции.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, в которой количество 3α-этинил-3β-гидроксиандростан-17-она оксима составляет 0,1-4 масс.% от общей массы композиции.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где количество носителя в указанной фармацевтической композиции составляет от 90 до 99,9 масс.% от общей массы композиции.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где количество носителя в указанной фармацевтической композиции составляет 95-99,9 масс.% от общей массы композиции.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, обеспечивающая растворимость 3α-этинил-3β-гидроксиандростан-17-она оксима в носителе до 50 мг/мл.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, обеспечивающая растворимость 3α-этинил-3β-гидроксиандростан-17-она оксима в носителе до 40 мг/мл.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, обеспечивающая растворимость 3α-этинил-3β-гидроксиандростан-17-она оксима в носителе до 60 мг/мл.
Еще одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, которая обеспечивает растворимость 3α-этинил-3β-гидроксиандростан-17-она оксима в носителе до 100 мг/мл, например, до 90 мг/мл, или до 80 мг/мл, или до 70 мг/мл, или до 60 мг/мл, или до 50 мг/мл, или до 40 мг/мл, или до 30 мг/мл, или до 20 мг/мл, или до 10 мг/мл.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где сложный моноэфир, сложный диэфир (если имеется) и/или сложный триэфир (если имеется) содержит жирную кислоту каприловую кислоту (С8) в количестве 50-90%; и жирную кислоту каприновую кислоту (С10) в количестве 10-50%.
Примерами сложных моноэфиров, которые могут быть использованы в носителе согласно изобретению, являются моноглицериды, выбранные из любого из следующих: глицерин монокапроат, глицерин монокаприлат, глицерин монодеканоат, глицерин монолаурат, глицерин мономиристат, глицерин монопальмитат, глицерин моностеарат, глицерин моноолеолат, и глицерин монолинолеат.
Одним аспектом изобретения является фармацевтическая композиция, описанная и заявленная в настоящем документе, содержащая глицериновые моноэфиры каприловой кислоты и каприновой кислоты в качестве носителя.
Еще одним примером сложных моноэфиров, которые могут быть полезны в носителе согласно изобретению, являются моноэфиры пропиленгликоля, выбранные из любого из пропиленгликоля каприлата, пропиленгликоля капроата и пропиленгликоля деканоата.
Дополнительные примеры сложных моноэфиров, которые могут быть использованы в носителе согласно изобретению, выбраны из любого из пропиленгликоля пальмитата, пропиленгликоля стеарата и пропиленгликоля линолеата.
Другими примерами сложных моноэфиров, которые могут быть использованы в носителе согласно изобретению, являются сложные эфиры пропиленгликоля и жирных кислот, выбранные из любого из пропиленгликоля изостеарата, пропиленгликоля лаурата, пропиленгликоля миристата, пропиленгликоля олеата и пропиленгликоля олеата SE.
Примерами сложных диэфиров, которые могут быть использованы в носителе согласно изобретению, являются сложные эфиры пропиленгликоля и жирных кислот, такие как любой диэфир, выбранный из пропиленгликоля дикапроата, пропиленгликоля дикаприлата, пропиленгликоля дидеканоата, пропиленгликоля дилаурата, пропиленгликоля димиристата, пропиленгликоля дипальмитата, пропиленгликоля дистеарата, пропиленгликоля диолеата и пропиленгликоля дилинолеата.
Другими примерами сложных диэфиров, которые могут быть использованы в носителе согласно изобретению, являются сложные эфиры глицерина и жирных кислот, такие как любые диэфиры, выбранные из глицерина дикапроата, глицерина дикаприлата, глицерина дидеканоата, глицерина дилаурата, глицерина димиристата, глицерина дипальмитата, глицерина дистеарата, глицерина диолеата и глицерина дилинолеата.
Аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где сложный моноэфир представляет собой моноглицерид, сложный диэфир, если присутствует, представляет собой диглицерид, а сложный триэфир, если присутствует, представляет собой триглицерид.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где носитель представляет собой смесь глицерина монокаприлата и глицерина монодеканоата, то есть смесь глицериновых моноэфиров каприлового эфира и капринового эфира, таких как, например, Imwitor 742®.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где носитель представляет собой смесь моно- и диглицеридов каприловой и каприновой кислот, таких как, например, Akoline MCM®.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где носитель представляет собой смесь моно- и диглицеридов каприловой и каприновой кислот, таких как, например, Capmul® MCM.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где носителем является глицерил моноолеат, такой как, например, Cithrol GMO®.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где носителем является пропиленгликоль монолаурат, такой как, например, Lauroglycol FCC®.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где носитель представляет собой олеоил макрогол-6 глицерид, такой как, например, Labrafil M1944 CS®.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где носитель представляет собой полиоксиэтилен-8 каприловый/каприновый глицерид, такой как, например, Acconon MC8-2®.
Одним аспектом изобретения является фармацевтическая композиция в соответствии с тем, как описано и заявлено в настоящем документе, где носителем является пропиленгликоля монокаприлат, такой как, например, Сapryol 90® или Сapryol PGMC®.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, включающая сложный моноэфир, сложный диэфир (если имеется) и/или сложный триэфир (если имеется) жирной кислоты каприловой кислоты (С8) и жирной кислоты каприновой кислоты (С10), где количество сложного эфира каприловой кислоты (С8) составляет 50-90%, а количество сложного эфира каприновой кислоты (С10) составляет 10-50% в носителе из фармацевтической композиции по изобретению.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где носителем является сложный эфир каприловой кислоты (С8) в количестве 50-90%; и сложный эфир каприновой кислоты (С10) в количестве 10-50%, причем сложный эфир каприловой кислоты (С8) и сложный эфир каприновой кислоты (С10) составляют в целом 100%.
Описание чертежей
Фиг. 1 представляет собой график, показывающий, что концентрация в плазме соединения ИНЛ (исследуемого нового лекарства) 3α-этинил-3β-гидроксиандростан-17-она оксима повышалась пропорционально увеличению дозы с константами пропорциональности β, равными 1,01 для Cмax и 1,12 для AUC0-∞ (среднее значение ± SEM), после однократной пероральной дозы (1, 3, 10, 30, 100 и 200 мг, соответственно) и для шести субъектов на дозу в исследовании SAD (исследовании действия однократных нарастающих доз).
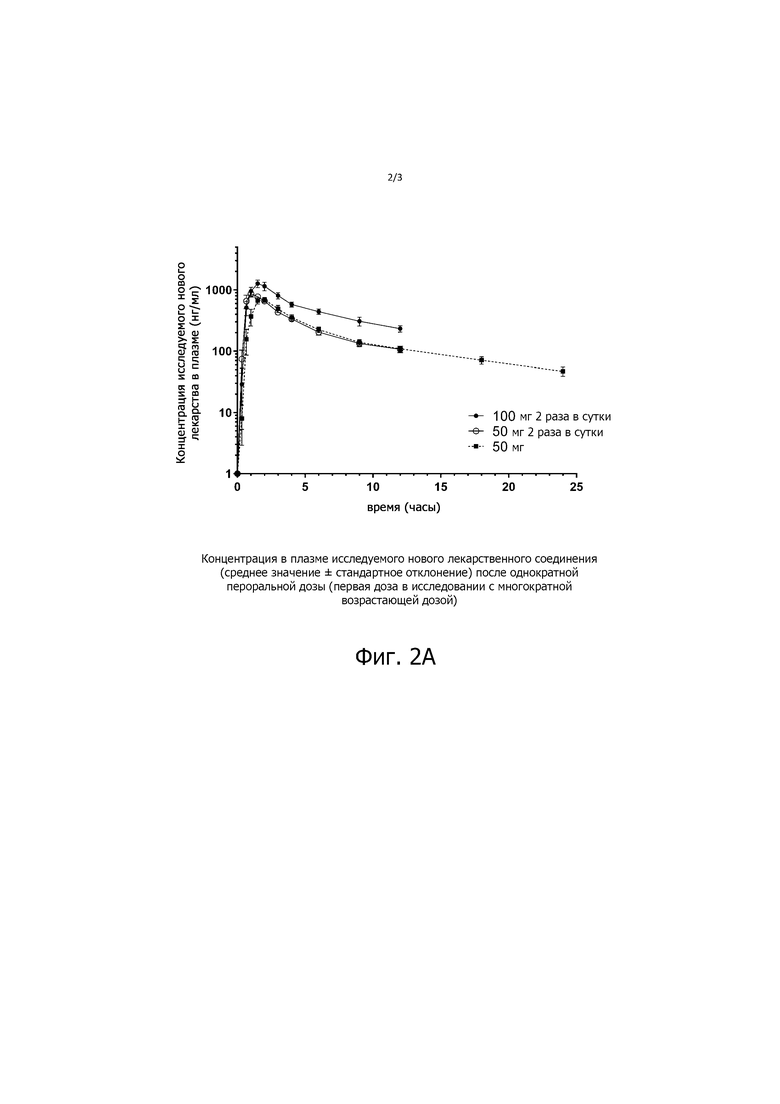
На фиг. 2А показана концентрация в плазме соединения ИНЛ 3α-этинил-3β-гидроксиандростан-17-она оксима (среднее значение ± SEM) после первой однократной пероральной дозы в исследовании MAD (исследовании многократных нарастающих доз) (исследование первой дозы MAD). Доза в исследовании MAD включала 50 мг один раз в сутки, 50 мг два раза в сутки (то есть 100 мг) и 100 мг два раза в сутки (то есть 200 мг), по шесть субъектов на дозу.
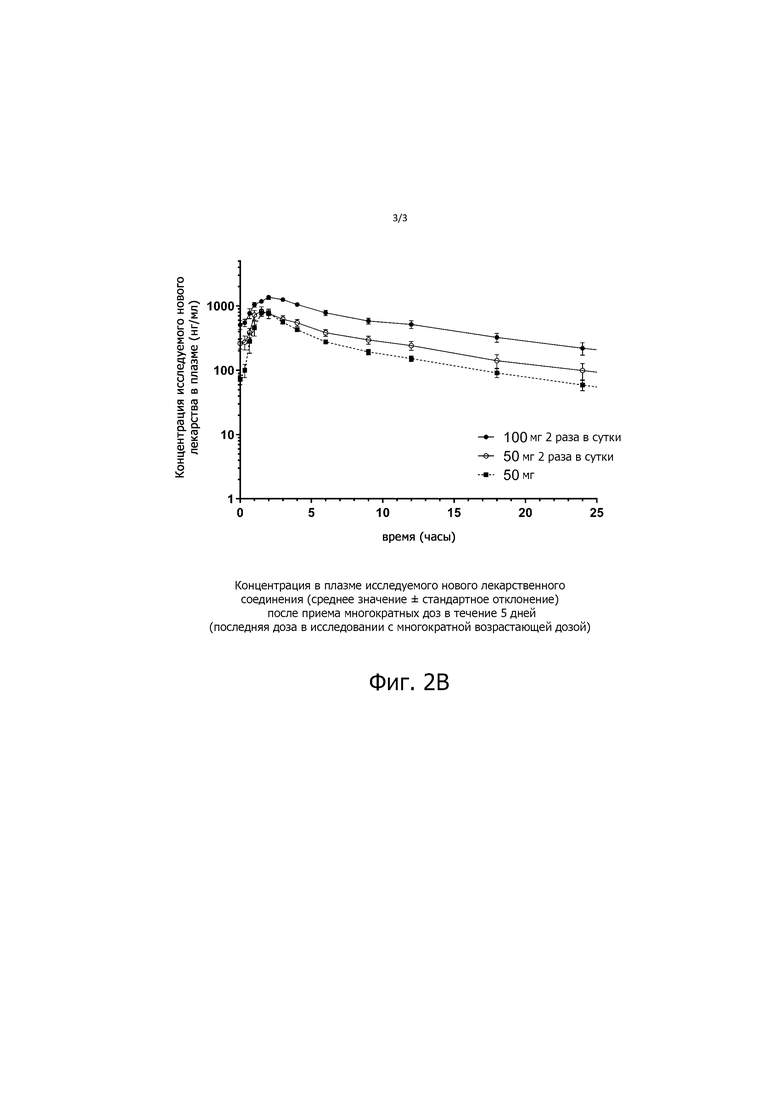
На фиг. 2В показана концентрация в плазме соединения ИНЛ 3α-этинил-3β-гидроксиандростан-17-она оксима (среднее значение ± SEM) после повторного введения в течение пяти дней (последняя доза исследования MAD). Доза составила 50 мг один раз в сутки, 50 мг два раза в сутки (т.е. 100 мг) и 100 мг два раза в сутки (то есть 200 мг), по шесть субъектов на дозу.
Определения
Выражение «плохо растворимый», используемое в данном документе при обсуждении растворимости в водной среде 3α-этинил-3β-гидроксиандростан-17-она оксима, относится к растворимости по величине мкг/мл. Показано, что растворимость 3α-этинил-3β-гидроксиандростан-17-она оксима в воде составляет всего 1,5 мкг/мл, 0,2 мкг/мл в SGF (искусственном желудочном соке), 7 мкг/мл в FaSSIF (искусственном кишечном соке в состоянии натощак) и 19 мкг/мл в FeSSIF (в искусственном кишечном соке в состоянии после приема пищи).
Выражение «соединение ИНЛ» в настоящем документе определено как 3α-этинил-3β-гидроксиандростан-17-она оксим (также упоминаемый как Соединение I в описании данной заявки).
Выражение «биоэквивалентный продукт» или «продукт, демонстрирующий биоэквивалентность» в настоящем документе определяется как продукт, который содержит 3α-этинил-3β-гидроксиандростан-17-она оксим в качестве терапевтического агента, в той же пероральной лекарственной форме и в том же количестве дозы или концентрации указанного соединения, и который имеет идентичное значение AUC±20% и/или идентичное значение Cmax±20%, и который показывает такой же или подобный терапевтический эффект.
Выражение «Cmax» в настоящем документе определяется как максимальная концентрация терапевтического соединения 3α-этинил-3β-гидроксиандростан-17-она оксима, которая достигается в конкретный момент времени от момента введения соединения человеку.
Выражение «AUC» (площадь под кривой) определяется в настоящем документе как мера абсорбции лекарственного средства. Большая величина AUC означает, что лекарственное средство имеет более высокую абсорбцию лекарства у субъекта, тогда как меньшая величина AUC означает, что лекарственное средство имеет более низкую абсорбцию лекарства.
Выражение «большое количество носителя», используемое в описании, означает 90-99,9 масс.% от общей массы композиции, например, 95-99,9 масс.% от общей массы композиции.
Выражение «состав ИНЛ», используемое во всем описании, означает фармацевтический состав, содержащий 3α-этинил-3β-гидроксиандростан-17-она оксим, и используемый в клиническом исследовании I фазы, указанном в этом описании.
Выражение «ФК», используемое в описании, означает фармакокинетические свойства исследуемого соединения.
Выражение «SAD» определяется как «однократная нарастающая доза», то есть начинающаяся с более низкой дозы и с увеличением каждой дозы 3α-этинил-3β-гидроксиандростан-17-она оксима в соответствии с протоколом клинического испытания.
Выражение «MAD» определяется как «многократная нарастающая доза», то есть начинающаяся с более низкой дозы и с увеличением каждой дозы 3α-этинил-3β-гидроксиандростан-17-она оксима в соответствии с протоколом клинического испытания.
Используемая в настоящем документе формулировка «один раз в сутки» означает, что 3α-этинил-3β-гидроксиандростан 17-она оксим применяют у субъекта только один раз в сутки.
Используемая в настоящем документе формулировка «дважды в сутки» или «BID» означает, что 3α-этинил-3β-гидроксиандростан-17-она оксим применяют в определенной дозе два раза в сутки; т.е. доза 50 мг два раза в сутки (BID) означает, что суточная доза составляет 100 мг/сутки, а формулировка 100 мг два раза в сутки (BID) означает, что суточная доза составляет 200 мг в сутки.
Используемое в настоящем документе выражение «носитель» определяется как сложный моноэфир, при необходимости в комбинации со сложным диэфиром и, при необходимости, также в комбинации со сложным триэфиром.
Используемое в настоящем документе выражение «сложный эфир (эфиры) жирной кислоты» определяется как сложный эфир, который образуется при взаимодействии жирной кислоты или карбоновой кислоты со спиртом с образованием сложного моноэфира, сложного диэфира или сложного триэфира, или их комбинации. Когда спирт представляет собой глицерин, получаемый сложный эфир (эфиры) жирных кислот может представлять собой моноглицерид, диглицерид или триглицерид, или комбинацию указанного моно-, ди- или триглицерида.
Используемый в настоящем документе процент жирных кислот означает, что общее количество конкретной жирной кислоты или общее количество двух или более конкретных жирных кислот всегда должно составлять 100%. Если в образовании сложного эфира присутствует только одна жирная кислота, то содержание жирной кислоты должно составлять 100% от общего количества жирных кислот; когда используют две или более жирные кислоты, например, если используют конкретную жирную кислоту, для которой процентные содержания, как здесь описано и заявлено, составляют менее 100%, общее количество жирных кислот должно составлять в целом 100%, и так далее.
Формы единственного числа, используемые в данном описании, также включают ссылки на множественное число, если в контексте явно не указано иное.
Выражение «печеночная энцефалопатия типа А» обычно означает печеночную энцефалопатию, ассоциированную с острой печеночной недостаточностью, обычно связанную с отеком головного мозга.
Выражение «печеночная энцефалопатия типа B» обычно означает печеночную энцефалопатию (шунтирование), вызванную портосистемным шунтированием без ассоциированного истинного заболевания печени.
Выражение «печеночная энцефалопатия типа C» обычно подразумевает печеночную энцефалопатию, возникающую у пациентов с циррозом печени - этот тип подразделяется на эпизодическую, персистентную и минимальную энцефалопатию.
Выражение «минимальная печеночная энцефалопатия» обычно означает печеночную энцефалопатию, которая не приводит к клинически выраженной когнитивной дисфункции, но может быть продемонстрирована с помощью психоневрологических исследований.
Выражение «манифестная печеночная энцефалопатия» обычно означает клинически выраженную печеночную энцефалопатию, проявляющуюся как психоневрологический синдром с большим спектром психических и моторных расстройств. Манифестная печеночная энцефалопатия может возникать эпизодически, в течение нескольких часов или дней у ранее стабильных пациентов, или у пациентов могут наблюдаться стойкие психоневрологические нарушения.
Выражение «гиперсомнолентность» означает группу расстройств, характеризующихся чрезмерной сонливостью в дневное время, которая возникает, несмотря на то, что у субъекта обычно отмечается нормальное качество и продолжительность ночного сна.
Выражение «нарколепсия» включает как нарколепсию 1 типа (с катаплексией), так и нарколепсию 2 типа (без катаплексии).
Фармацевтические композиции и дозировка
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где доза 3α-этинил-3β-гидроксиандростан-17-она оксима составляет от 1 мг до 200 мг.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где доза 3α-этинил-3β-гидроксиандростан-17-она оксима составляет от 10 мг до 100 мг.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где доза 3α-этинил-3β-гидроксиандростан-17-она оксима составляет от 3 мг до 30 мг.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где доза 3α-этинил-3β-гидроксиандростан-17-она оксима составляет от 30 мг до 60 мг.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где суточная доза 3α-этинил-3β-гидроксиандростан-17-она оксима составляет от 1 мг/сутки до 200 мг/сутки.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где суточная доза 3α-этинил-3β-гидроксиандростан-17-она оксима составляет от 50 мг/сутки до 100 мг/сутки.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где суточная доза 3α-этинил-3β-гидроксиандростан-17-она оксима составляет от 20 мг/сутки до 160 мг/сутки.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где суточная доза 3α-этинил-3β-гидроксиандростан-17-она оксима составляет от 40 мг/сутки до 160 мг/сутки.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где суточная доза 3α-этинил-3β-гидроксиандростан-17-она оксима составляет от 80 мг/сутки до 160 мг/сутки.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где указанную суточную дозу применяют один раз в сутки.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где указанную суточную дозу применяют два раза в сутки.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где указанная композиция помещена в капсулу.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где указанная капсула содержит от 5 мг до 40 мг 3α-этинил-3β-гидроксиандростан-17-она оксима.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где указанная капсула содержит от 10 мг до 20 мг 3α-этинил-3β-гидроксиандростан-17-она оксима.
Одним аспектом изобретения является фармацевтическая композиция, описанная и заявленная в настоящем документе, содержащая дополнительный терапевтический агент, используемый в качестве стандартного средства лечения печеночной энцефалопатии.
Одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, где дополнительный терапевтический агент представляет собой соединение, снижающее образование аммиака.
Одним аспектом изобретения является фармацевтическая композиция, описанная и заявленная в настоящем документе, где соединение, снижающее образование аммиака, выбрано из рифаксимина, лактулозы, орнитина фенилацетата и глицерина фенилбутирата.
Одним аспектом изобретения является фармацевтическая композиция, описанная и заявленная в настоящем документе, в которой соединением, снижающим образование аммиака, является рифаксимин.
Одним аспектом изобретения является фармацевтическая композиция, описанная и заявленная в настоящем документе, в которой соединением, снижающим образование аммиака, является лактулоза.
Еще одним аспектом изобретения является комбинированный продукт, содержащий:
(i) фармацевтическую композицию, как описано и заявлено в настоящем документе; и
(ii) соединение, снижающее образование аммиака.
В еще одном аспекте изобретения комбинированный продукт, как описано и заявлено в настоящем документе, представляет собой набор из комбинации частей.
Еще одним аспектом изобретения является фармацевтическая композиция или капсула, заполненная указанной композицией, как описано и заявлено в настоящем документе, включающая дополнительный терапевтический агент, пригодный для лечения гиперсомнии.
Еще одним аспектом изобретения является комбинированный продукт, содержащий:
(i) фармацевтическую композицию, как описано и заявлено в настоящем документе; и
(ii) терапевтический агент, пригодный для лечения гиперсомнии.
Дополнительное терапевтическое средство, пригодное для лечения гиперсомнии, может быть выбрано из любого из оксибата натрия (Xyrem®), модафинила (Provigil®), армодафинила (Nuvigil®), метилфенидата, амфетамина, (R)-2-амино-3-фенилпропилкарбамата гидрохлорида (JZP-110), JZP-507, JZP-258, оксалата, такого как эсциталопрама оксалат, и питолизанта (Wakix®).
В еще одном аспекте изобретения комбинированный продукт, как описано в настоящем документе, представляет собой набор из комбинации частей.
Еще одним аспектом изобретения является лекарственный продукт, который биоэквивалентен фармацевтическому препарату или биоэквивалентен комбинации, как описано и заявлено в настоящем документе.
Медицинское применение и медицинское лечение
Пациенты, страдающие печеночной энцефалопатией, могут проявлять симптомы, включая нарушение цикла сна-бодрствования, когнитивной функции, памяти, обучения, двигательной координации, сознания, снижение уровня энергии и изменение личности, когнитивные нарушения, дезориентацию и кому, но не ограничиваясь этим.
Одним аспектом настоящего изобретения является способ лечения печеночной энцефалопатии, где фармацевтическую композицию, как описано и заявлено в настоящем документе, применяют у субъекта, нуждающегося в таком лечении.
Еще одним аспектом изобретения является способ профилактики печеночной энцефалопатии, где фармацевтическую композицию, как описано и заявлено в настоящем документе, применяют у субъекта, нуждающегося в таком лечении.
Еще одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, для использования в способе лечения или профилактики печеночной энцефалопатии.
Еще одним аспектом изобретения является применение фармацевтической композиции, как описано и заявлено в настоящем документе, для изготовления лекарственного средства для лечения или профилактики печеночной энцефалопатии.
В одном варианте осуществления изобретения указанная печеночная энцефалопатия выбрана из любой из печеночной энцефалопатии типа A, печеночной энцефалопатии типа B, печеночной энцефалопатии типа C, минимальной печеночной энцефалопатии и манифестной печеночной энцефалопатии.
Одним аспектом настоящего изобретения является способ лечения гиперсомнолентности, где фармацевтическую композицию, как описано и заявлено в настоящем документе, применяют у субъекта, нуждающегося в таком лечении.
Еще одним аспектом изобретения является способ профилактики гипертонической болезни, где фармацевтическую композицию, описанную и заявленную в настоящем документе, применяют у субъекта, нуждающегося в таком лечении.
Еще одним аспектом изобретения является фармацевтическая композиция, как описано и заявлено в настоящем документе, для применения в способе лечения или профилактики гиперсомнолентности.
Еще одним аспектом изобретения является применение фармацевтической композиции, как описано и заявлено в настоящем документе, для приготовления лекарственного средства для лечения или профилактики гиперсомнолентности.
В одном варианте осуществления изобретения гиперсомнолентность выбрана из группы, состоящей из идиопатической гиперсомнии, рецидивирующей гиперсомнии, нарколепсии, нарушения сна во время сменной работы, чрезмерной сонливости, эндозепин-зависимого рецидивирующего ступора и амфетамин-резистентной гиперсомнии.
В некоторых других вариантах осуществления гиперсомнолентность связана с расстройством, выбранным из группы, состоящей из синдрома беспокойных ног, ночной дистонии, ночного двигательного расстройства, синдрома Клейна-Левина, болезни Паркинсона, расстройства, связанного с лекарством или веществом, психического расстройства, нарушения поведения в фазе сна с быстрыми движениями глаз (REM), ночной лобной дистонии, ночного двигательного расстройства, обструктивного апноэ во сне, цирроза печени и печеночной энцефалопатии.
Способы применения
Фармацевтическая композиция, как описано и заявлено в настоящем документе, может быть применена энтеральным способом. Примеры энтерального введения включают введение в пищевод, желудок и тонкую и толстую кишку (то есть в желудочно-кишечный тракт). Способы применения включают пероральный, сублингвальный (растворение препарата под языком) и ректальный.
В одном аспекте изобретения фармацевтическую композицию согласно настоящему изобретению применяют перорально.
Способ получения композиций
Фармацевтическую композицию в соответствии с настоящим изобретением готовят путем предварительного нагревания носителя, как определено в настоящем документе, до подходящей температуры, после чего 3α-этинил-3β-гидроксиандростан-17-она оксим, при необходимости в микронизированной форме, добавляют к указанному носителю при перемешивании при подходящей температуре до получения прозрачного раствора.
3α-этинил-3β-гидроксиандростан-17-она оксим может быть получен в соответствии с процедурой синтеза, описанной в опубликованной заявке на патент WO 2008/063128.
Одним аспектом изобретения является способ получения фармацевтической композиции, как описано и заявлено в настоящем документе, где:
(i) носитель, как здесь описано и заявлено в настоящем документе, нагревают до температуры от 15°С до 80°С; и
(ii) 3α-этинил-3β-гидроксиандростан-17-она оксим, при необходимости в микронизированной форме, добавляют в носитель при перемешивании и при температуре от 15°С до 80°С до полного растворения соединения и получения прозрачного раствора, после чего раствор при необходимости разливают в капсулы.
В одном аспекте изобретения температура для нагревания носителя на этапе (i) выше составляет от 60°C до 75°C.
В одном аспекте изобретения температуру для наполнения капсулы снижают примерно до 30°C.
Еще одним аспектом изобретения является фармацевтическая композиция, которую можно получить любым одним способом, как описано и заявлено в настоящем документе.
Примеры
Растворимость 3α-этинил-3β-гидроксиандростан-17-она оксима (Соединения I) испытывали в различных носителях.
Общее описание
Растворимость Соединения I оценивали в разных носителях. Соединение I взвешивали в маленьких флаконах и добавляли известное количество носителя. Образец нагревали, если требовалось для плавления носителя, до температуры примерно от 15°С до 80°С и перемешивали вихревой или магнитной мешалкой. Для оценки растворимости Соединения I посредством визуального контроля в систему добавляли больше Соединения I или носителя.
Пример 1
Каприновый/каприловый моноглицерид (1,02 г) [Imwitor® 742, полученный от IOI Oleo] нагревали до 40°C. Микронизированное Соединение I добавляли в аликвотах и смесь перемешивали вихревой мешалкой. Добавление продолжали до тех пор, пока получался прозрачный раствор после смешивания. Всего 28 мг Соединения I приводило к прозрачному раствору, но 31 мг не растворялось.
Пример 2
Глицерин моноолеат [Cithrol GMO от Croda Health Care] плавили при 50°C. Микронизированное Соединение I взвешивали во флаконе (3,9 мг) и добавляли глицерин моноолеат (413 мг). Образец нагревали и перемешивали вихревой мешалкой. Прозрачный раствор не был получен. Дополнительное количество глицерина моноолеата (83 мг) приводило к прозрачному раствору. По-видимому, смесь затвердевает при комнатной температуре.
Пример 3
Микронизированное Соединение I (1,3 мг) добавляли к олеоил-макрогол-6 глицериду [Labrafil M1944 CS от Gattefossé] (0,91 г). Образец нагревали примерно до 40°С и перемешивали вихревой мешалкой. Был получен прозрачный раствор. Добавляли Соединение I (3,3 мг), все еще получая прозрачный раствор после нагревания до 40°C и перемешивания вихревой мешалкой. Дальнейшее добавление Соединения I (3,0 мг) приводило к нерастворенному материалу после многократного перемешивания вихревой мешалкой и нагревания.
Пример 4
Микронизированное Соединение I взвешивали во флаконе (1,8 мг) и добавляли пропиленгликоль монолаурат [Lauroglycol FCC от Gattefossé] (100 мкл). Образец перемешивали вихревой мешалкой при комнатной температуре. Не было получено прозрачного раствора. Порциями добавляли пропиленгликоль монолаурат (2 × 100 мкл), и после перемешивания вихревой мешалкой получали прозрачный раствор.
Пример 5
Микронизированное Соединение I взвешивали во флаконе (25,9 мг) и добавляли пропиленгликоль монокаприлат типа I [Capryol PGMC от Gattefossé] (0,991 г). Образец перемешивали при комнатной температуре в течение примерно 60 минут, и появлялся нерастворенный материал. Добавляли дополнительный пропиленгликоль монокаприлат типа I (0,994 г), и после 60 минут перемешивания при комнатной температуре получали прозрачный раствор. Добавляли Соединение I (6 мг), и при перемешивании снова получали прозрачный раствор. Добавление Соединения I (3 мг) приводило к нерастворенному материалу после перемешивания при комнатной температуре в течение 60 минут.
Пример 6
Микронизированное Соединение I взвешивали во флаконе (26,2 мг) и добавляли пропиленгликоль монокаприлат типа II [Capryol 90 от Gattefossé] (0,988 г). Образец перемешивали при комнатной температуре в течение примерно 60 минут, и появлялся нерастворенный материал. Добавляли дополнительный пропиленгликоль монокаприлат типа II (0,383 г), и через 30 минут перемешивания при комнатной температуре получали прозрачный раствор. Добавляли Соединение I (4,5 мг), и при перемешивании снова получали прозрачный раствор. Добавление Соединения I (4,9 мг) приводило к нерастворенному материалу после перемешивания при комнатной температуре в течение 60 минут. Дальнейшее добавление пропиленгликоля монокаприлата типа II (0,139 г) приводило к прозрачному раствору после перемешивания в течение 60 минут.
Пример 7
Микронизированное Соединение I взвешивали во флаконе (32,5 мг) и добавляли полиоксиэтилен-8 каприловый/каприновый глицерид [Acconon MC8-2 от Abitec] (1,008 г). Перемешивание при комнатной температуре в течение 30-60 минут приводило к нерастворенному материалу. Полиоксиэтилен-8 каприловый/каприновый глицерид добавляли восемью порциями до общего количества 14,06 г. Материал перемешивали не менее 30 минут между каждым добавлением. После последнего добавления был получен прозрачный раствор.
Таблица 1. Растворимость Соединения I в разных носителях (Примеры 1-7)
Пример 8А
Липидный раствор в капсуле
Каприновый/каприловый глицерид [Imwitor® 742] нагревали до 60°С и добавляли Соединение I до концентрации 20 мг/мл. После завершения растворения температуру снижали до 30°С и раствор разливали в твердые желатиновые капсулы размером 0. Целевая масса заполнения составила 500 мг, после чего капсулы закрывали.
Пример 8В
Липидный раствор в капсуле
Каприновый/каприловый глицерид [Imwitor® 742] нагревали до 60°C и добавляли Соединение I до концентрации 2 мг/мл. После завершения растворения температуру снижали до 30°С и раствор разливали в твердые желатиновые капсулы размером 0. Целевая масса заполнения составила 500 мг, после чего капсулы закрывали.
Пример 9
Липидное твердое вещество в капсуле
Каприновый/каприловый глицерид [Imwitor® 742] нагревали до 70°C и добавляли Соединение I до концентрации 40 мг/мл. По окончании растворения температуру снижали до 30°C и раствор заливали в твердые желатиновые капсулы размером 0. Целевая масса заполнения составила 500 мг, после чего капсулы закрывали, и выдерживали при -20°C в течение 2-3 часов до хранения в холодильнике.
Повышая температуру нагревания с 60°С до 70°С, можно растворить 40 мг/мл Соединения I вместо 20 мг/мл.
Пример 10
Следующие фармацевтические составы были изготовлены и использованы в клиническом испытании I фазы, описанном ниже.
Следуя способу приготовления, описанному в Примерах 8A и 8B, следующие дозы помещали в твердую желатиновую капсулу (размер 0) для клинического применения. Для капсул по 10 мг 0,5 мл концентрации 20 мг/мл, полученной в Примере 8А, вводили в каждую капсулу. Для капсул по 1 мг 0,5 мл с концентрацией 2 мг/мл, полученной в примере 8В, вводили в каждую капсулу.
Капсулы плацебо содержали только наполнитель, который вводили в капсулы того же типа, что и для капсул, содержащих Соединение I. Целевая масса наполнения составила 500 мг.
Капсулы по 1 мг использовали как для дозы 1 мг, так и для дозы 3 мг в клиническом испытании. Капсулы по 10 мг использовали для более высоких доз в клиническом испытании.
Биологические исследования
I. Пероральная абсорбция 3α-этинил-3β-гидроксиандростан-17-она оксима
В исследовании, проведенном заявителем по настоящему изобретению, абсорбцию при пероральном приеме 3α-этинил-3β-гидроксиандростан-17-она оксима имитировали у разных видов животных и человека в разных дозах. Растворимость в разных средах показана в следующей таблице.
Модель абсорбции в ЖКТ (ACAT™) в GastroPlus™ v9.0 была использована для имитации фракции и количества абсорбированного 3α-этинил-3β-гидроксиандростан-17-она оксима после перорального применения. Абсорбцию моделировали у крысы, собаки и человека. Вводные параметры базовой линии суммированы в таблице А. Вводные параметры были выбраны из условий, использованных в предыдущих доклинических экспериментах, или из тех, которые планировались в будущих исследованиях. Размер частиц 3α-этинил-3β-гидроксиандростан-17-она оксима в клиническом материале составляет примерно 10 мкм.
Таблица A. Параметры дозирования, используемые для моделирования
* Искусственное число, отражающее, что растворимость в носителе не изменяется при прохождении через тонкую кишку.
Параметры ФК, использованные для моделирования, были взяты из сводки результатов ФК клинических испытаний I фазы у человека с однократной нарастающей дозой (SAD), описанных в пункте II ниже. Используемые данные были получены от уровней дозы 10, 100 и 200 мг. Использовали медианное значение клиренса, объема распределения, AUC, T1/2 (терминального), Cmax и Tmax. Для объема распределения использовали среднюю массу тела 70 кг для преобразования объема распределения от литров до л/кг. Для данных о концентрации в плазме по времени были использованы средние значения.
Смоделированные (прогнозируемые) профили концентрации в плазме в зависимости от времени после перорального введения в различных дозах и составе суммированы в таблице B ниже вместе с фактическими профилями концентрации в плазме в зависимости от времени, наблюдаемыми в клиническом испытании I фазы (см. таблицу 1 ниже).
Таблица B. Прогнозированные и наблюдаемые параметры ФК для 3α-этинил-3β-гидроксиандростан-17-она оксима после перорального применения у человека.
После перорального применения раствора 3α-этинил-3β-гидроксиандростан-17-она оксима у человека прогнозируемая абсорбируемая фракция завершается в интервале доз 10-200 мг. Прогнозируемая абсорбируемая фракция существенно уменьшается после введения порошка в капсуле. Фракции, абсорбированные на 78% и 14%, были смоделированы для доз 10 и 200 мг, соответственно. Для состава раствора прогнозируемые значения Cmax были приблизительно в 2 раза ниже значений Cmax, которые фактически наблюдались у человека после введения липида в капсуле. AUC и Tmax составляли от 70 до 100% наблюдаемых значений. Вводные параметры ФК, которые использовались для моделирования, не учитывают влияние, например, связывания белка, соотношения кровь/плазма и мульти-компартментной кинетики, и поэтому могут объяснять расхождения между наблюдаемыми и моделируемыми временными профилями концентрации в плазме. Другим фактором может быть использованный вводный параметр проницаемости. Более высокая проницаемость in vivo для человека, чем экспериментальное значение проницаемости in vitro, которое использовали при моделировании, может привести к более высоким пиковым концентрациям в плазме.
Тем не менее, моделирование с использованием перорального водного раствора может достаточно хорошо прогнозировать профили концентрации в плазме, которые были получены для капсул, содержащих моноглицериды со средней длиной цепи. Для моделирования пероральных водных растворов предполагалось, что вся доза 3α-этинил-3β-гидроксиандростан-17-она оксима находилась в растворе без осаждения при прохождении через тонкую кишку. Это свидетельствует, что 3α-этинил-3β-гидроксиандростан-17-она оксим в моноглицеридах со средней длиной цепи ведет себя как водный раствор без осаждения в отношении абсорбции при пероральном приеме.
Имитированная абсорбция из капсулы с порошком была намного меньше, чем абсорбция из капсулы с раствором. AUC и Cmax были уменьшены в 10 и 20 раз при пероральных дозах 100 и 200 мг. Имитация абсорбции при пероральном приеме из капсулы с порошком показывает прогнозируемую максимальную концентрацию в плазме приблизительно 100 нг/мл для пероральных доз более 100 мг.
Растворимость, а не растворение является ограничивающим параметром для пероральной абсорбции при высокой дозе (>100 мг). Микронизация 3α-этинил-3β-гидроксиандростан-17-она оксима в порошке в капсуле, приводящая к улучшенному растворению вследствие увеличения площади поверхности частиц, будет, таким образом, оказывать ограниченное влияние на общее абсорбируемое количество и максимальную концентрацию в плазме.
С другой стороны, прогнозируется, что улучшенная растворимость окажет значительное влияние на абсорбцию при пероральном приеме и максимальные концентрации в плазме, полученные после порошка в виде капсул. Улучшение растворимости в желудочно-кишечном тракте может наблюдаться в сочетании с приемом пищи. Таким образом, для порошка в капсулах прогнозируется, что условия натощак или после приема пищи приведут к значительному воздействию на концентрации 3α-этинил-3β-гидроксиандростан-17-она оксима в плазме и будут способствовать вариабельности абсорбции. Такие эффекты не ожидаются для липидной композиции из текущих моделей. Для подтверждения этого потребуются данные in vivo, так как многие другие пищевые эффекты, которые не были учтены в настоящем исследовании, могут влиять на пероральную абсорбцию.
II. Клинические испытания I фазы
Пероральную абсорбцию 3α-этинил-3β-гидроксиандростан-17-она оксима изучали в клиническом испытании I фазы на людях-добровольцах. Композиция для перорального введения в испытании I фазы представляла собой капсулу, содержащую раствор липида указанного соединения. Было обнаружено, что пероральная абсорбция у людей обеспечивает хорошее содержание в плазме с Tmax между 1 и 2 часами и линейной дозой Cmax в диапазоне доз 1-200 мг. Также было показано, что 3α-этинил-3β-гидроксиандростан-17-она оксим при пероральном введении проникает в мозг человека и противодействует эффектам нейростероидного аллопрегнанолона в отношении рецептора ГАМК-А в дозах, которые хорошо переносятся и связаны с линейной фармакокинетикой.
Дизайн исследования
Всего в исследование было включено 90 подходящих здоровых мужчин (в возрасте от 18 до 50 лет). Используемый пероральный состав представлял собой капсулу, содержащую липидный раствор 3α-этинил-3β-гидроксиандростан-17-она оксима, приготовленный в соответствии с приведенным выше Примером 10 (состав ИНЛ ниже).
Первое исследование у людей с однократной нарастающей дозой (SAD) (EudraCT 2015-004911-19) и исследование с 5-дневной многократной нарастающей дозой (MAD) (EudraCT 2016-003651-30) были проспективными, рандомизированными, двойными слепыми и плацебо-контролируемыми, и предназначались для оценки безопасности, переносимости и фармакокинетических (ФК) характеристик однократных и многократных пероральных доз 3α-этинил-3β-гидроксиандростан-17-она оксима (соединения ИНЛ) у здоровых мужчин-добровольцев.
Состав ИНЛ вводили перорально в следующих дозах:
SAD: 1, 3, 10, 30, 100 и 200 мг.
MAD: 50 мг один раз в сутки; 50 мг два раза в сутки (два раза в сутки), то есть 100 мг/сутки; или 100 мг два раза в сутки (два раза в сутки), то есть 200 мг/сутки.
Антагонистическое исследование с целью оценки биологической активности соединения ИНЛ (EudraCT 2015-004911-19) было рандомизированным и плацебо-контролируемым с двойным слепым трехкомпонентным перекрестным дизайном. Предметы исследования, принимаемые перорально: плацебо (А); 3 мг состава ИНЛ (B); или 30 мг состава ИНЛ (C); затем через 90 минут внутривенная (в/в) инъекция аллопрегнанолона (0,05 мг/кг) и повторные измерения максимальной скорости саккадированных движений глаз (SEV) (Saccadometer Plus, Ober Consulting Sp. z o.o, Польша) и седации/сонливости (визуальная аналоговая шкала, при оценке от «Отсутствия сонливости» до «Засыпания» в течение трех часов.
Анализы соединения ИНЛ и аллопрегнанолона
Соединение ИНЛ и аллопрегнанолон количественно определяли с помощью СВЭЖХ/МС/МС, Waters Acquity, колонки C18, и тройного квадрупольного масс-спектрометра Waters XEVO-TQ-S. В качестве внутренних стандартов использовали D4- [3α-этинил-3β-гидроксиандростан-17-она оксим] и D5-аллопрегнанолон, соответственно. Для анализа аллопрегнанолона использовали дериватизацию оксима для повышения чувствительности. Стандартные образцы и образцы для контроля качества готовили в пустой плазме/сыворотке человека. Предел обнаружения для 3α-этинил-3β-гидроксиандростан-17-она оксима составлял 1 нг/мл, а для аллопрегнанолона 0,05 нг/мл (0,15 нмоль/л).
Анализ данных
Фармакокинетические параметры оценивали с использованием некомпартментного анализа и программного обеспечения Phoenix WinNonlin® версии 6.3 или новее (Pharsight Corporation, США). Пропорциональность дозы для исследования SAD была основана на на AUC0-> ∞ и Cmax, и для исследования MAD на равновесном AUCτ. Коэффициент накопления между первой и последней дозой рассчитывали как (равновесное AUCτ/ первая доза AUC0->∞). Статистические расчеты проводили с использованием статистики IBM SPSS (версия 24, IBM).
Результаты
Фармакокинетика
После однократного введения препарата ИНЛ концентрация в плазме увеличивалась пропорционально увеличению дозы с константами пропорциональности β, равными 1,01 для Cмax и 1,12 для AUC0-∞ (таблица 1, фиг. 1). Кроме того, дозирование в течение 5 дней два раза в день приводило к пропорциональному увеличению концентрации в плазме соединения 3α-этинил-3β-гидроксиандростан-17-она оксима ИНЛ с дозой, с увеличением AUC0-∞ в 1,9 раза при удвоении дозы с 50 до 100 мг, и с очень небольшим накоплением, в 1,1 - 1,2 раза (таблица 1, фиг. 2).
Антагонизм ИНЛ соединения в отношении индуцированного аллопрегнанолоном снижения SEV
30 мг соединения ИНЛ значительно ингибировали индуцированное аллопрегнанолоном снижение SEV (анализ AUC, p = 0,04, таблица 2) в течение периода времени, когда субъекты реагировали на инъекцию аллопрегнанолона с уменьшением SEV (n = 11, 69%). При этом уменьшение эффекта аллопрегнанолона с применением 3 мг соединения ИНЛ было незначительным (р = 0,286).
Антагонизм ИНЛ соединения в отношении аллопрегнанолон-индуцированной седации
Для субъектов, которые ответили на инъекцию аллопрегнанолона седацией (n = 8, 50%), статистический анализ показал, что как 3 мг, так и 30 мг соединения ИНЛ значительно ингибировали седативный эффект в течение периода времени ответа (р = 0,012 и р = 0,05, соответственно, таблица 2).
Воздействие аллопрегнанолона
В трех случаях исследования кривые концентрации аллопрегнанолона в сыворотке были очень схожи с самым высоким уровнем в сыворотке крови в первый момент исследования после внутривенного введения, 74 - 82 нМ. Не было никаких существенных различий между воздействиями аллопрегнанолона в течение трех исследований (р = 0,163, сравнение AUC).
Обсуждение
Настоящие данные демонстрируют, что перорально вводимый 3α-этинил-3β-гидроксиандростан-17-она оксим, когда его готовят в сложном эфире жирной кислоты по настоящему изобретению, хорошо переносится людьми. Нежелательные явления (НЯ), как правило, были легкими, и не наблюдалось ни серьезных нежелательных явлений (СНЯ), ни токсичности, ограничивающей дозу, вплоть до самых высоких доз, вводимых здоровым взрослым мужчинам по протоколам с однократной (200 мг) или многократной (100 мг каждые 12 часов в течение 5 дней) нарастающей дозой. При всех изученных дозах 3α-этинил-3β-гидроксиандростан-17-она оксима показана линейная ФК; то есть как Cmax (SAD), так и системное воздействие, оцениваемое как AUC0-∞ (SAD) или AUCτ (равновесное состояние), были линейно связаны с пероральной дозой, и не было никаких признаков накопления лекарственного средства во время дозирования в равновесном состоянии.
Важно, что результаты контрольного исследования показывают, что 3α-этинил-3β-гидроксиандростан-17-она оксим проникает в головной мозг и противодействует эффектам аллопрегнанолона в отношении рецептора ГАМК-А. Перорально вводимый 3α-этинил-3β-гидроксиандростан-17-она оксим вызывает явное дозозависимое ингибирование эффектов аллопрегнанолона. Отмечено значительное ингибирование SEV с 30 мг 3α-этинил-3β-гидроксиандростан-17-она оксима с промежуточным, но незначительным ингибированием при 3 мг.
3α-этинил-3β-гидроксиандростан-17-она оксим также значительно подавляет седативный эффект, индуцированный аллопрегнанолоном, в дозах как 3 мг, так и 30 мг, опять же в очевидной дозозависимой форме.
Благоприятные данные о безопасности и фармакокинетике, а также результаты исследования стимуляции аллопрегнанолоном позволяют предположить, что 3α-этинил-3β-гидроксиандростан-17-она оксим, полученный в соответствии с настоящим изобретением, представляет собой многообещающее новое лечение заболеваний человека, связанных с нейростероидными эффектами в отношении ЦНС. Печеночная энцефалопатия (ПЭ), неврологическое заболевание, связанное с циррозом печени и портосистемным анастомозом, при котором сообщается о повышенных уровнях аллопрегнанолона в мозге (Ahboucha et al. 2005 и 2006), является одним из примеров.
Результаты исследования антагонистов расширяют ранее полученные данные на животных моделях, свидетельствующие о том, что 3α-этинил-3β-гидроксиандростан-17-она оксим противодействует эффектам аллопрегнанолона в концентрациях, которые могут быть патофизиологически значимыми. Концентрации аллопрегнанолона в сыворотке крови в исследовании антагонистов через 10 минут после инъекции в среднем составляли около 80 нМ, что значительно выше, чем концентрации 6–20 нмоль/кг в головном мозге пациентов с циррозом печени, умерших при печеночной коме (Ahboucha et al. 2005 & 2006). Более того, исследования на животных показывают, что зависимость концентрации от времени для 3α-этинил-3β-гидроксиандростан-17-она оксима (неопубликованные данные) и аллопрегнанолона (Johansson et al. 2002) очень схожи в мозге и плазме. Результаты показывают, что 3α-этинил-3β-гидроксиандростан-17-она оксим, вводимый в дозах до 30 мг, находящихся в диапазоне, который хорошо переносится взрослыми мужчинами, может подавлять действие нейростероидов даже в концентрациях, превышающих те, которые описаны у больных ПЭ.
Несколько недавних наблюдений также указывают на нейростероид-индуцированную аллостерическую активацию рецепторов ГАМК-A ЦНС при чрезмерной дневной сонливости (EDS), связанной с такими расстройствами, как идиопатическая гиперсомния (Billiard and Sonka, 2016). К ним относятся идентификация предполагаемого агониста ГАМК-А в СМЖ пациентов с первичной гиперсомнией (Rye 2012) и клиническое наблюдение, что антагонист ГАМК-А флумазенил может способствовать бодрствованию у пациентов с EDS (Korkmaz 1997). В самом деле, клинические проявления ПЭ и расстройств гиперсомнии проявляют сходство, и ПЭ считается расстройством сонливости (Montagnese 2015).
Таким образом, эти результаты показывают, что 3α-этинил-3β-гидроксиандростан-17-он оксим, вводимый человеку перорально в виде фармацевтической композиции, как описано и заявлено в настоящем документе, противодействует эффектам нейростероидного аллопрегнанолона в отношении рецептора ГАМК-А в головном мозге в дозах, которые хорошо переносятся и связаны с линейной фармакокинетикой. Это свидетельствует, что 3α-этинил-3β-гидроксиандростан-17-она оксим может также быть способен нормализовать ГАМК-эргическую нейротрансмиссию и представляет собой многообещающее новое терапевтическое средство для лечения расстройств, связанных с гиперсомнолентностью, или других расстройств, связанных с нейростероид-опосредованной аллостерической избыточной активацией ГАМК-А рецепторов.
Таблица 1. Концентрации в плазме 3α-этинил-3β-гидроксиандростан-17-она оксима (Соединения I) и фармакокинетические параметры, представленные в виде среднего значения (SD), шесть субъектов на дозу
(нг/мл)
CMax *
(нг⋅ч/мл)
AUC*
* кратное увеличение по сравнению со следующей более низкой дозой, aτ=24 часа, bτ =12 часов.
Таблица 2. Статистический анализ субъектов, отвечающих на аллопрегнанолон, в течение периода ответа. Изменение максимальной скорости саккадического движения глаз (ΔSEV) показано как площадь под кривой (AUC), а изменение седации показано как AUC по шкале VAS. Статистический анализ с использованием знакового рангового критерия Уилкоксона показал значительный антагонизм Соединения I по отношению к аллопрегнанолон-индуцированным изменениям SEV и седации
+
плацебо
+
Соединение I, 3 мг
+
Соединение I, 30 мг
(град/с x мин)
N.S.
p= 0,04*
(мм x мин)
p= 0,01*
p= 0,05*
* Достоверно отличается от Аллопрегнанолон + плацебо, N.S. = отличия не достоверны.

Изобретение относится к пероральной фармацевтической композиции в форме липидного раствора, включающей 3α-этинил-3β-гидроксиандростан-17-она оксим и носитель. Носитель содержит 45-100% сложного моноэфира, представляющего собой моноглицерид или моноэфир пропиленгликоля, при необходимости, до 51% сложного диэфира, представляющего собой диглицерид или диэфир пропиленгликоля и при необходимости, до 10% сложного триэфира, представляющего собой триглицерид. При этом сложный моноэфир, сложный диэфир (если имеется) и/или сложный триэфир (если имеется) содержит жирную кислоту, выбранную из каприловой кислоты (С8) в количестве 50-90%; каприновой кислоты (С10) в количестве 10-50%; капроновой кислоты (С6) в количестве до 3%; лауриновой кислоты (С12) в количестве до 100%; пальмитиновой кислоты (С16) в количестве до 12%; миристиновой кислоты (С14) в количестве до 3%; стеариновой кислоты (С18) в количестве до 6%; олеиновой кислоты (С18:1) в количестве до 100% и линолевой кислоты (С18:2) в количестве до 35%. 3α-этинил-3β-гидроксиандростан-17-она оксима составляет 0,1-10 масс.% от общей массы композиции. Изобретение также относится к способу получения указанной композиции путем нагревания носителя и добавления активного агента до полного растворения. Изобретение обеспечивает улучшенную доставку активного агента, его биодоступность и терапевтическое воздействие in vivo. 2 н. и 21 з.п. ф-лы, 3 ил., 2 табл., 10 пр.
1. Пероральная фармацевтическая композиция в форме липидного раствора, включающая:
(i) 3α-этинил-3β-гидроксиандростан-17-она оксим
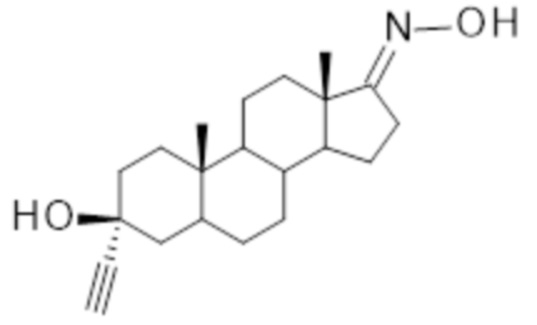 ; и
; и
(ii) носитель, содержащий
(а) 45-100% сложного моноэфира;
(b) при необходимости, до 51% сложного диэфира;
(с) при необходимости, до 10% сложного триэфира
где сложный моноэфир, сложный диэфир (если имеется) и/или сложный триэфир (если имеется) содержит жирную кислоту, выбранную из одной или нескольких из:
- каприловой кислоты (С8) в количестве 50-90%;
- каприновой кислоты (С10) в количестве 10-50%;
- капроновой кислоты (С6) в количестве до 3%;
- лауриновой кислоты (С12) в количестве до 100%;
- пальмитиновой кислоты (С16) в количестве до 12%;
- миристиновой кислоты (С14) в количестве до 3%;
- стеариновой кислоты (С18) в количестве до 6%;
- олеиновой кислоты (С18:1) в количестве до 100%;
- линолевой кислоты (С18:2) в количестве до 35%; и где
где сложный моноэфир представляет собой моноглицерид или моноэфир пропиленгликоля; сложный диэфир, если он присутствует, представляет собой диглицерид или диэфир пропиленгликоля, и сложный триэфир, если он присутствует, является триглицеридом, и
количество 3α-этинил-3β-гидроксиандростан-17-она оксима составляет 0,1-10 масс.% от общей массы композиции.
2. Фармацевтическая композиция по п.1, состоящая только из 3α-этинил-3β-гидроксиандростан-17-она оксима и носителя.
3. Фармацевтическая композиция по п.1, где сложный моноэфир представляет собой моноглицерид, сложный диэфир, если присутствует, представляет собой диглицерид, а сложный триэфир, если присутствует, представляет собой триглицерид.
4. Фармацевтическая композиция по любому из пп.1-3, где сложный моноэфир представляет собой моноглицерид, выбранный из любого из глицерина монокапроата, глицерина монокаприлата, глицерина монодеканоата, глицерина монолаурата, глицерина мономиристата, глицерина монопальмитата, глицерина моностеарата, глицерина моноолеата, и глицерина монолинолеата.
5. Фармацевтическая композиция по п.1, где сложный моноэфир представляет собой сложный моноэфир пропиленгликоля, выбранный из любого из пропиленгликоля каприлата, пропиленгликоля капроата, пропиленгликоля деканоата, пропиленгликоля пальмитата, пропиленгликоля стеарата, пропиленгликоля линолеата, пропиленгликоля изостеарата, пропиленгликоля лаурата, пропиленгликоля миристата, пропиленгликоля олеата и пропиленгликоля олеата SE.
6. Фармацевтическая композиция по п.1, где сложный диэфир представляет собой сложный диэфир пропиленгликоля, выбранный из любого из пропиленгликоля дикапроата, пропиленгликоля дикаприлата, пропиленгликоля дидеканоата, пропиленгликоля дилаурата, пропиленгликоля димиристата, пропиленгликоля дипальмитата, пропиленгликоля дистеарата, пропиленгликоля диолеата и пропиленгликоля дилинолеата.
7. Фармацевтическая композиция по любому из пп.1-3, где указанным сложным диэфиром является диглицерид, выбранный из любого из глицерина дикапроата, глицерина дикаприлата, глицерина дидеканоата, глицерина дилаурата, глицерина димиристата, глицерина дипальмитата, глицерина дистеарата, глицерина диолеата, и глицерина дилинолеата.
8. Фармацевтическая композиция по любому из пп.1-7, где количество 3α-этинил-3β-гидроксиандростан-17-она оксима составляет от 0,1 до 5 масс.% от общей массы композиции.
9. Фармацевтическая композиция по любому из пп.1-8, где количество 3α-этинил-3β-гидроксиандростан-17-она оксима составляет от 0,1 до 4 масс.% от общей массы композиции.
10. Фармацевтическая композиция по любому из пп.1-9, где количество носителя в указанной фармацевтической композиции составляет от 90 до 99,9 масс.% от общей массы композиции.
11. Фармацевтическая композиция по любому из пп.1-9, где количество носителя в указанной фармацевтической композиции составляет от 95 до 99,9 масс.% от общей массы композиции.
12. Фармацевтическая композиция по любому из предыдущих пунктов, где жирная кислота представляет собой каприловую кислоту (С8) в количестве 50-90%; и каприновую кислоту (С10) в количестве 10-50%.
13. Фармацевтическая композиция по п.1, где носитель представляет собой смесь глицерина монокаприлата и глицерина монодеканоата, смесь моно- и диглицеридов каприловой и каприновой кислот, глицерина моноолеат, пропиленгликоля монолаурат или пропиленгликоля монокаприлат.
14. Фармацевтическая композиция по любому из пп.1-13, включающая 3α-этинил-3β-гидроксиандростан-17-она оксим в количестве от 1 до 200 мг.
15. Фармацевтическая композиция по любому из пп.1-13, включающая 3α-этинил-3β-гидроксиандростан-17-она оксим в количестве от 10 до 100 мг.
16. Фармацевтическая композиция по любому из пп.1-13, включающая 3α-этинил-3β-гидроксиандростан-17-она оксим в количестве от 3 до 30 мг.
17. Фармацевтическая композиция по любому из п.п.1-13, включающая 3α-этинил-3β-гидроксиандростан-17-она оксим в количестве от 30 до 60 мг.
18. Фармацевтическая композиция по любому из пп.1-17, где композиция помещена в капсулу.
19. Фармацевтическая композиция по п.18, где капсула содержит от 5 мг до 40 мг 3α-этинил-3β-гидроксиандростан-17-она оксима.
20. Фармацевтическая композиция по п.18, где капсула содержит от 10 мг до 20 мг 3α-этинил-3β-гидроксиандростан-17-она оксима.
21. Способ получения фармацевтической композиции по любому из пп.1-20, где:
(а) носитель нагревают до температуры от 15°С до 80°С; и
(b) 3α-этинил-3β-гидроксиандростан-17-она оксим, при необходимости в микронизированной форме, добавляют в носитель при перемешивании при температуре от 15°С до 80°С до полного растворения соединения и получения прозрачного раствора, после чего раствор при необходимости разливают в капсулу.
22. Способ по п.21, где на этапе (а) носитель нагревают до температуры 60-75°С.
23. Способ по пп.21 и 22, где на этапе (b) после полного растворения 3α-этинил-3β-гидроксиандростан-17-она оксима температуру снижают примерно до 30°С.
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Токарный резец | 1924 |
|
SU2016A1 |
| COLIN CREWS et al., "Quantitation of the Main Constituents of Some Authentic Sesame Seed Oils of Different Origin", JOURNAL OF AGRICULTURAL AND FOOD CHEMISTRY, US, (20060801), vol | |||
| Видоизменение прибора для получения стереоскопических впечатлений от двух изображений различного масштаба | 1919 |
|
SU54A1 |
| Печь для сжигания твердых и жидких нечистот | 1920 |
|
SU17A1 |

