Настоящее изобретение относится к новому способу получения тиено-индольных производных соответствующих формуле (Ia) или (Ib), используемых в асимметрическом синтезе для приготовления ключевых промежуточных соединений (8S) или (8R) 8-(галометил)-1-алкил-7,8-дигидро-6Н-тиено[3,2-e]индол-4-ола, и необходимых промежуточных соединений данного способа.
Тиено-индольные производные описаны и заявлены в GB2344818, WO2013/149948 и WO2013/149946, где также описаны способы их получения, фармацевтической композиции, которая их содержит и ее применение в лечении некоторых опухолей млекопитающих.
Такие соединения описаны как алкилирующие соединения с цитотоксической активностью, поэтому пригодны в лечение различных видов рака и клеточных пролиферативных нарушениях. Кроме того, подобные соединения также подходят для конъюгации с различными типами нуклеофилов и являются, таким образом, пригодными в приготовлении, например, производных конъюгатов антитела с лекарственным средством.
Эти тиено-индольные производные и их аналоги, могут быть приготовлены в соответствии с известными химическими процессами, таким как, например, приготовление, описанное в J. Am. Chem. Soc. 2007, 129, 14092-14099. В частности, синтез энантиомерно чистых тиено-индольных производных, описанный в вышеуказанных документах известного уровня техники, выполнен через способ, в котором энантиомерно чистые ключевые промежуточные соединения 8-(галометил)-1-алкил-7,8-дигидро-6Н-тиено[3,2-e]индол-4-олы получены хиральным разделением из рацемической смеси, с применением хиральной хроматографии с обращенной фазой, с привлечением затратных по времени и дорогостоящих стадий.
В связи с этим, мы неожиданно обнаружили, что вышеупомянутые ключевые промежуточные соединения 8-(галометил)-1-алкил-7,8-дигидро-6Н-тиено[3,2-e]индол-4-олы, могут быть эффективно приготовлены асимметрическим синтезом, который исключает стадию хиральной очистки, обеспечивая преимущества с точки зрения уменьшения времени и стоимости всего процесса для получения конечных тиено-индольных производных, поскольку это предотвращает потерю материала, когда образуется только один из двух энантиомеров конечного соединения. Кроме того, новый способ показывает высокий общий выход по сравнению с ранее известными способами.
Примеры асимметрического синтеза, но используемого для получения фенилиндольных аналогов дуокармициновых алкилирующих ДНК субъединиц, описаны в литературе, см., например, Mol. Pharmaceutics 2015, 12, 1813-1835, J. Org. Chem. 2011, 76, 583-587 и J. Org. Chem. 2014, 79, 9699-9703.
Однако, синтетический подход, описанный в Mol. Pharmaceutics приводит в некоторых случаях к образованию нежелательных побочных продуктов, которые требуют добавления дополнительных стадий очистки и разделения. Этот недостаток минимизирован настоящим изобретением, вероятно из-за применения менее основных условий реакции.
Кроме того, в соответствии со статьями в журнале J. Org. Chem., наличие алкилтиофенового кольца в настоящем изобретении, которое менее реакционноспособное и более обогащенное электронами чем фенильное кольцо, однако позволяет применять более мягкие условия реакции для стадии циклизации, что обусловлено введением реагента Гриньяра в связь С-I.
Таким образом, первым объектом настоящего изобретения является приготовление тиено-индольных производных соответствующих формуле (Ia) или (Ib):
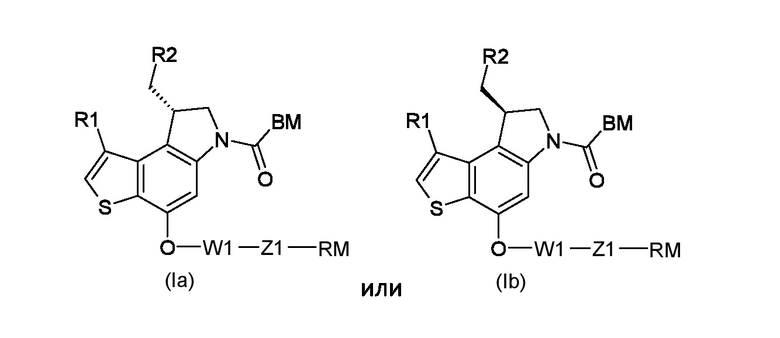
Где:
R1 представляет собой водород или линейный или разветвленный С1-С6 алкил;
R2 представляет собой уходящую группу, выбранную из галогеновых и сульфонатных групп;
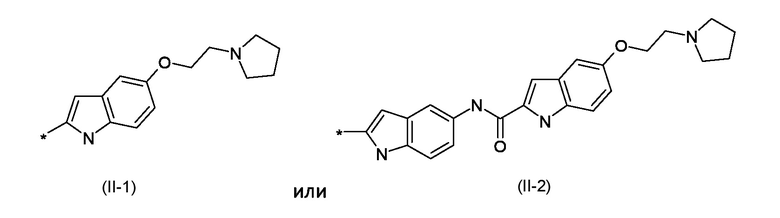
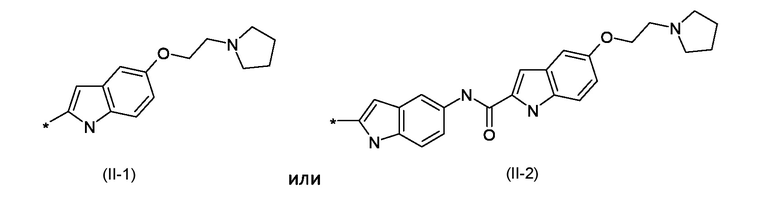
BM представляет собой ДНК-связывающий фрагмент соответствующий формуле (II-1) или (II-2):
W1 представляет собой саморасщепляющую систему, соответствующую формуле (III):
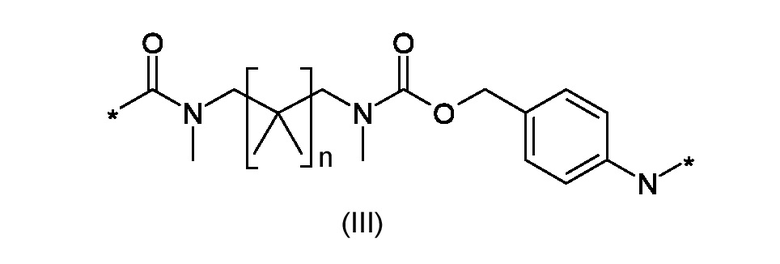
Где n представляет собой 0 или 1;
Z1 представляет собой линкер соответствующий формуле (IV-1) или (IV-2):
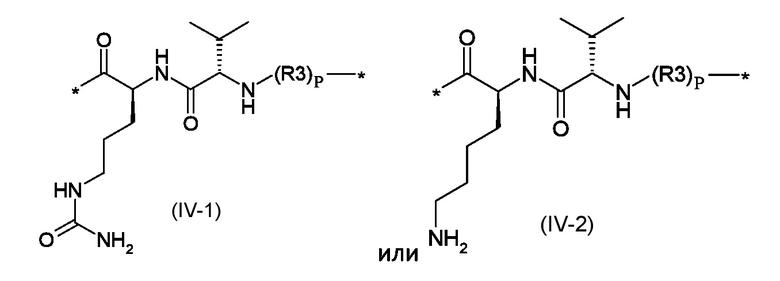
Где p представляет собой целое число от 0 до 1, а R3 - полиоксиэтиленовую цепь соответствующая формуле (V):
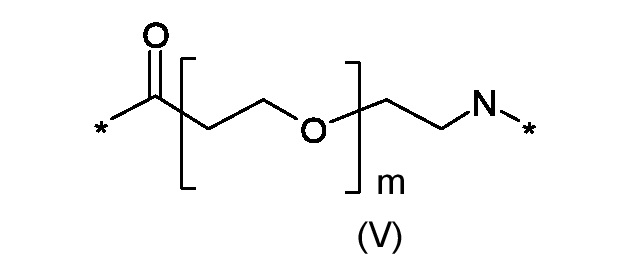
Где m представляет собой целое число от 0 до 5; и
RM представляет собой реакционноспособную частицу соответствующую формуле (VI):
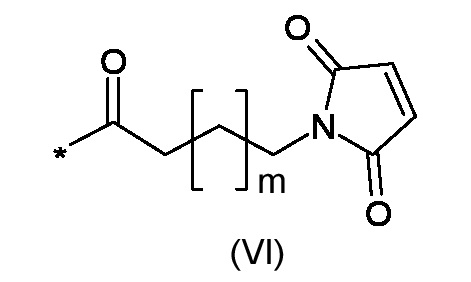
Где m является таким, как указано выше;
которое включает следующие стадии:
Стадия а) алкилирование соединения соответствующего формуле (VII):
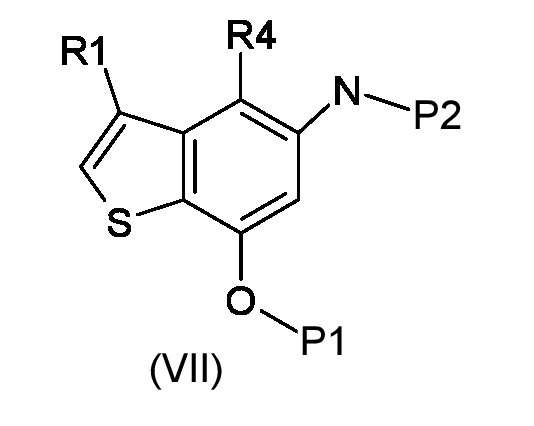
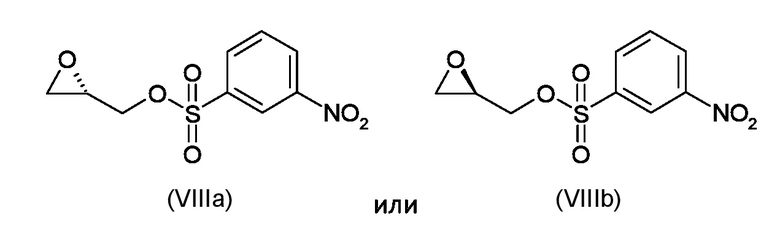
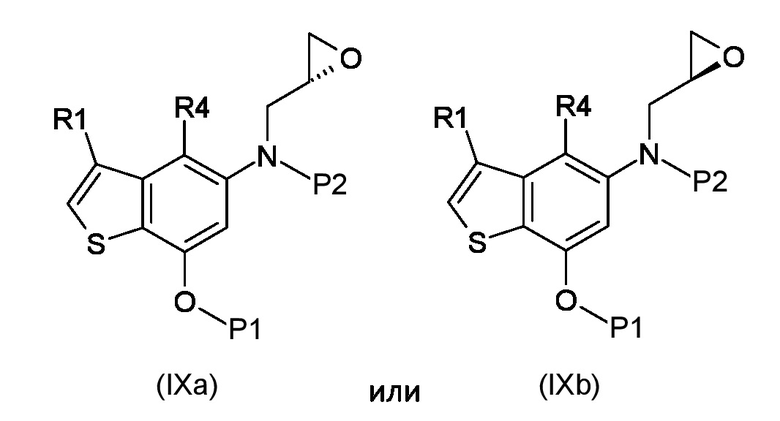
Где R4 представляет собой галоген, P1 представляет собой гидроксизащитную группу, P2 представляет собой азотзащитную группу и R1 является таким, как указано выше, (S)-глицидил 3-нозилатом (VIIIa) или (R)-глицидил 3-нозилатом (VIIIb):
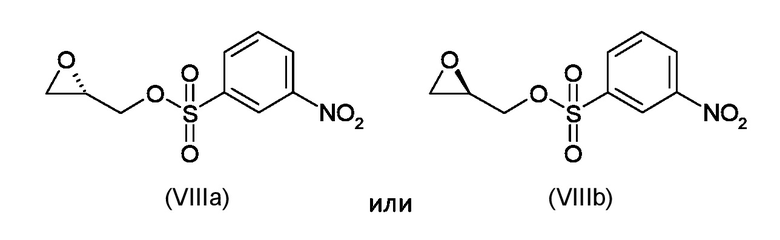
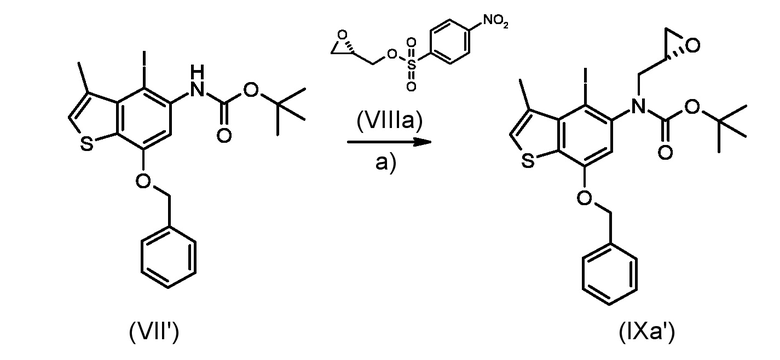
Стадия b) реакция полученного соединения, соответствующего формуле (IXa) или (IXb):
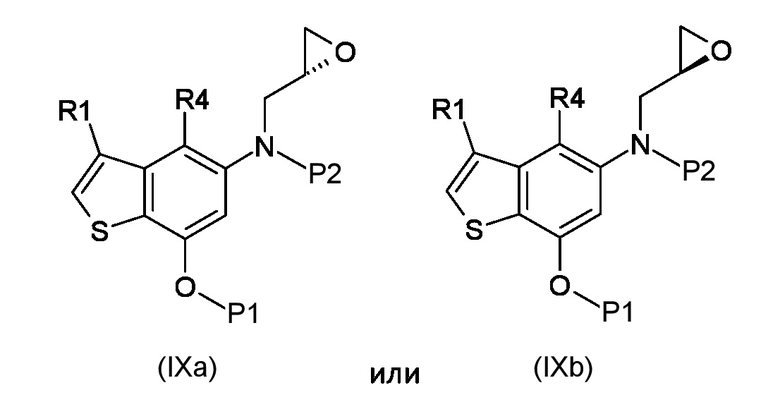
Где R1, R4, P1 и P2 являются такими, как указано выше, с металлоорганическим реагентом;
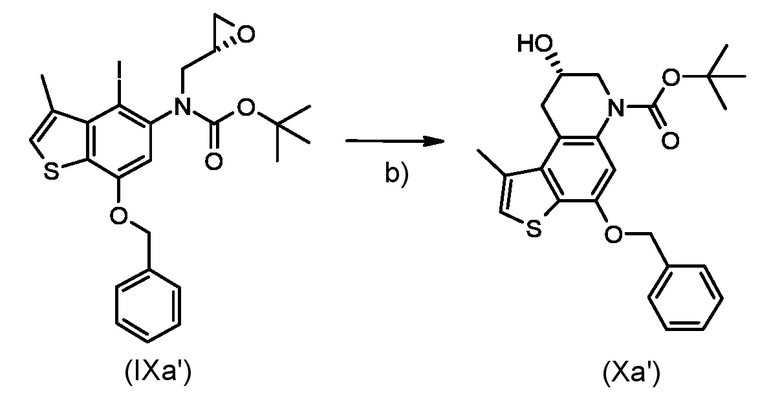
Стадия с) удаление гидроксизащитной группы P1 из полученного соединения, соответствующего формуле (Xa) или (Xb):
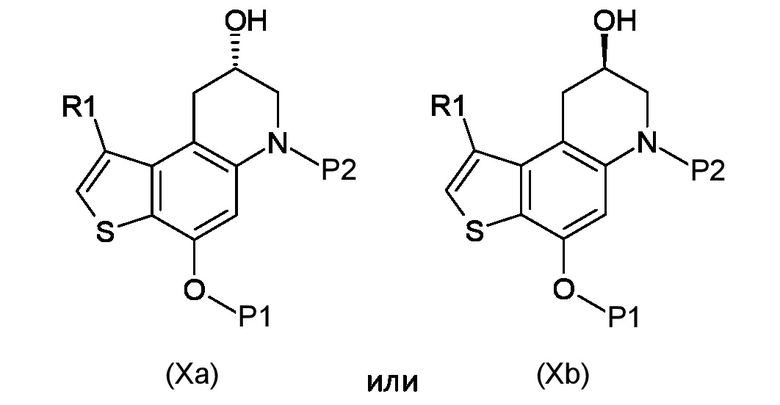
Где R1, P1 и P2 являются такими, как указано выше;
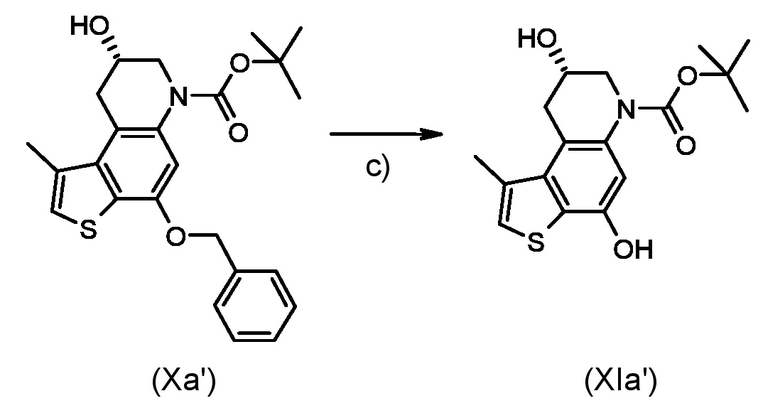
Стадия d) осуществление внутримолекулярной спироциклизации полученного соединения, соответствующего формуле (XIa) или (XIb):
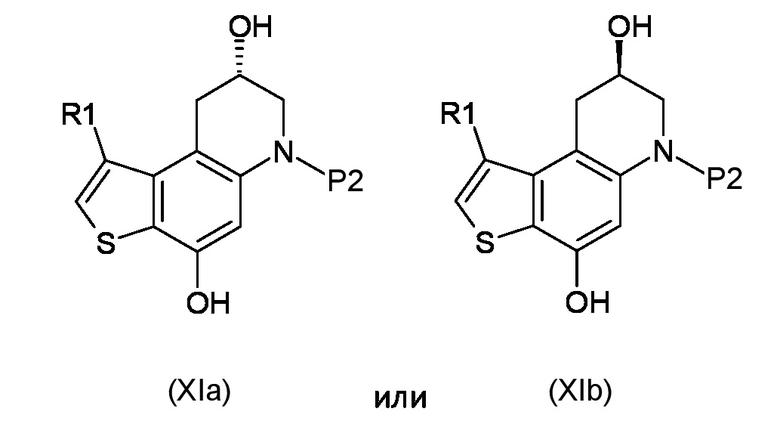
Где R1 и P2, являются такими, как указано выше;
Стадия e) достижение стереоконтролируемого региоселективного циклопропанового раскрытия полученного соединения, соответствующего формуле (XIIa) или (XIIb)
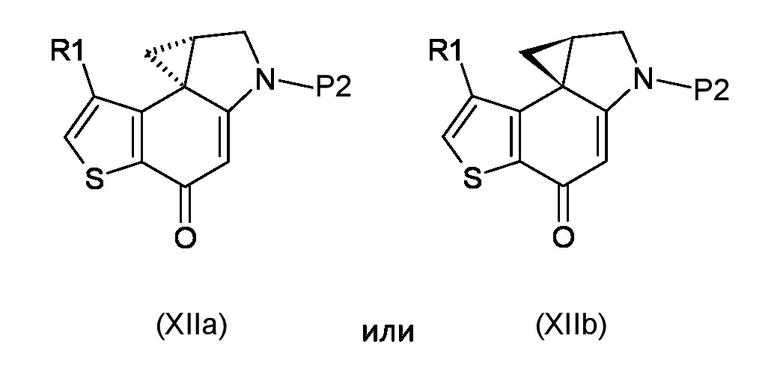
Где R1 и P2 являются такими, как указано выше, реакцией с кислотой, которая имеет формулу HR2, где R2 является таким, как указано выше;
Стадия f) удаление азотзащитной группы P2 из полученного соединения, соответствующего формуле (XIIIa) или (XIIIb)
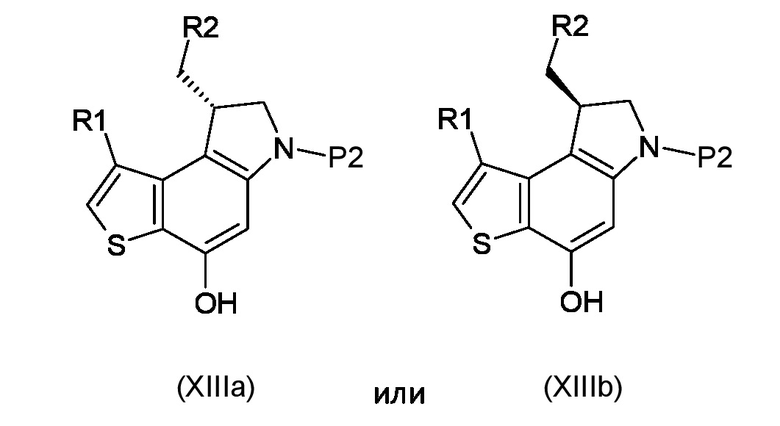
Где R1, R2 и P2 являются такими, как указано выше, таким образом, чтобы получить ключевое энантиомерно чистое промежуточное соединение 8-(галометил)-1-алкил-7,8-дигидро-6H-тиено[3,2-e]индол-4-ол, соответствующий формуле (XIVa) или (XIVb), соответственно
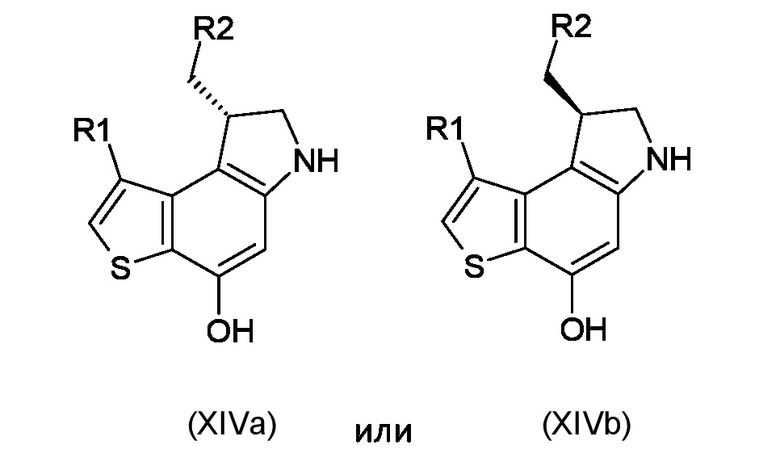
где R1 и R2 являются такими, как указано выше;
Стадия g) сочетание интермедиата, соответствующего формуле (XIVa) или (XIVb), с остатком кислоты BM-COOH, таким образом, чтобы получить интермедиат, соответствующий формуле (XVa) или (XVb), соответственно
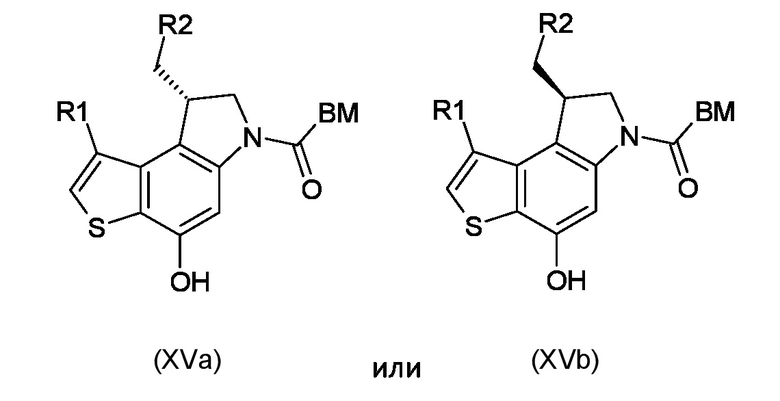
где BM, R1 и R2 являются такими, как указано выше.
Также объектом настоящего изобретения является вышеупомянутый асимметрический синтез, который, кроме того, включает получение производных интермедиата соответствующих формуле (XVa) или (XVb) по фенольному фрагменту в соответствии с приготовлениями и способами, описанными в ссылках известного уровня техники, таких, как те, что описаны в WO2013/149948 на странице 63 (стадия e), чтобы получить конечные тиено-индольные производные, соответствующие формуле (Ia) или (Ib).
Более подробно, объектом настоящего изобретения также является вышеупомянутый асимметрический синтез, который кроме того содержит следующие стадии:
Стадия h) преобразование интермедиата, соответствующего формуле (XVa) или (XVb), в карбонатные производные, соответствующие формуле (XVIa) или (XVIb), соответственно
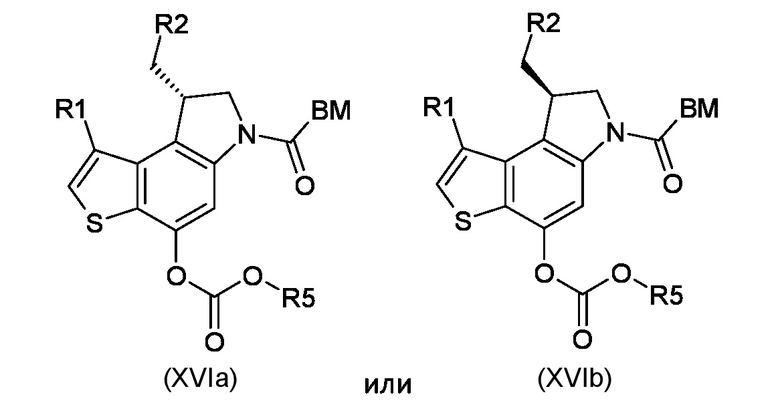
где BM, R1 и R2 являются такими, как указано выше, а R5 представляет собой сукцинимидильный или 4-нитро-фенильный остаток;
Стадия i) взаимодействие полученного интермедиата, соответствующего формуле (XVIa) или (XVIb), с амином, соответствующим формуле (XVII)
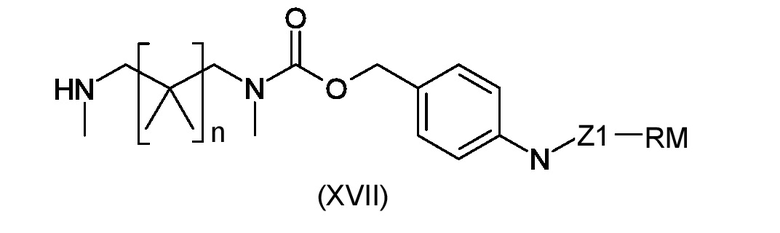
где n, Z1 и RM являются такими, как указано выше, таким образом, чтобы получить конечные соединения, соответствующие формуле (Ia) или (Ib), соответственно
где R1, R2, BM, W1 Z1 и RM являются такими, как указано выше.
При желании, полученные соединения, соответствующие формуле (Ia) или (Ib), которые указаны выше, могут быть переведены в фармацевтически приемлемые соли.
Соединения, соответствующие формуле (Ia) или (Ib), которые указаны выше, или их фармацевтически приемлемые соли, могут быть в последующем объединены с фармацевтически приемлемым носителем или растворителем для получения фармацевтической композиции.
Новый процесс синтеза объекта настоящего изобретения показан на следующей схеме 1, связанной с синтезом энантиомера, соответствующего формуле (Ia):
Схема 1
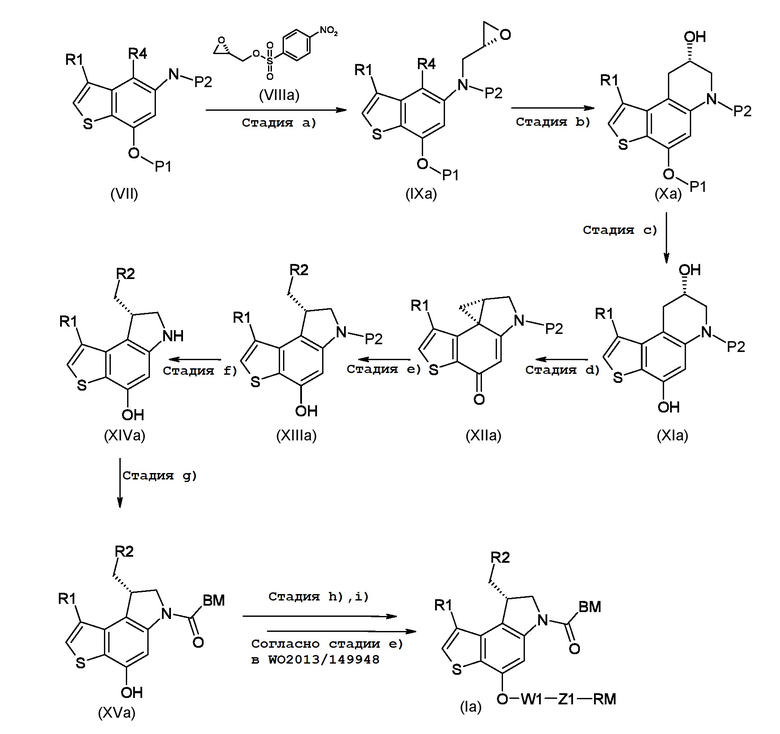
где R1, R2, R4, P1, P2, BM, W1, Z1 и RM являются такими, как указано выше.
Синтез начинается с N-алкилирования 5-амино-4-гало-3-алкил-1-бензотиофен-7-ол производного энантимерно чистым коммерческим глицидил 3-нозилатом (VIIIa, стадия a), с последующей внутримолекулярной 6-эндо-тет циклизацией с применением алкильных реагентов Гриньяра и дает энантиомерно чистое 6,7,8,9-тетрагидротиено[3,2-f]хинолин-8-ол производное (Xa, стадия b); удаление гидроксизащитной группы (стадия с) с последующей активацией Мицунобу вторичного спирта (стадия d), способствовало внутримолекулярной спироциклизации, с получением 4,4a,5,6-тетрагидро-8H-циклопропа[с]тиено[3,2-e]индол-8-он производного (XIIa, стадия d); затем, стереоэлектронный контроль региоселективного циклопропанового раскрытия (стадия е), с последующим удалением аминозащитной группы, дал ключевой энантиомерно чистый интермедиат 8-(галометил)-1-алкил-7,8-дигидро-6H-тиено[3,2-e]индол-4-ол (XIVa), стадия f); в заключение, сочетание с остатком подходящей кислоты BM-COOH, дает интермедиат (XVa)(Стадия g).
Промежуточные соединения, приготовленные в соответствии со способом настоящего изобретения, могут в дальнейшем быть применены для создания производных по фенольному кольцу, следуя предшествующим идеям известного уровня техники, таким, которые описаны в WO2013/149948, для приготовления конечных тиено-индольных производных, соответствующих формуле (Ia) или (Ib)(Стадия h, i).
Специалисту в данной области ясно, что в способе, где производное энантиомера глицидола, соответствующее формуле (VIIIb), применяется в алкилировании начального материала (VII), получают соединение, соответствующее формуле (IXb) и, следовательно, другой соответствующий тиено-индольный энантиомер, соответствующий формуле (Ib).
Также специалисту в данной области ясно, что когда P2 защитная группа представляет собой кислотно-лабильный фрагмент, такой как, например, трет-бутоксикарбонильный фрагмент, ключевые промежуточные соединения, соответствующие формуле (XIVa) или (XIVb), могут быть напрямую получены из соединения (XIIa) или (XIIb) в соответствии с условиями стадии е).
Следует отметить, что гидроксильный, W1, Z1 и RM1 фрагменты, связаны между собой через карбаматную или амидную связи, и что фрагменты, соответствующие формуле (III),(Z1) и (VI), всегда ориентированы таким образом, что, соответственно, образуется карбаматная связь между гидроксильной группой и W1, амидная связь между W1 и Z1 (используя терминальную анилиновую функцию W1)и амидная связь между Z1 и RM1.
Любые интермедиаты и/или конечные соединения могут быть выделены и очищены с использованием стандартных процедур, например хроматографии и/или кристаллизации и солеобразования.
Кроме того, промежуточные соединения, соответствующие формуле (XIa) или (XIb), пригодны для асимметрического синтеза тиено-индольных производных, соответствующих формуле (Ia) или (Ib), как указано выше, а также способ их приготовления представляет собой еще один объект настоящего изобретения.
Если не указано другого, следующие термины и фразы, которые используются здесь, имеют следующие значения.
Термин «уходящая группа» обозначает группу, которая может быть замещена другой группой в реакции замещения. Такие уходящие группы хорошо известны в данной области техники и примеры включают галоген (фтор, хлор, бром и иод) и сульфонат (например, в некоторых случаях замещенные С1-С6 алкансульфонаты, такие как метансульфонат и трифторметансульфонат, или в некоторых случаях замещенные С7-С12 алкилбензолсульфонаты, такие как п-толуолсульфонат), но не ограничиваются ими. Термин «уходящая группа» также обозначает группу, которая элиминируется как следствие реакции элиминирования, например, электронная каскадная реакция или реакция спироциклизации. В этом случае, галогеновая или сульфонатная группа могут, например, быть применены в качестве уходящей группы. Более предпочтительными уходящими группами являются хлор или бром.
Термин «галоген» обозначает бром, хлор, иод или фтор, более предпочтительно хлор или иод.
Термин «С1-С6 акил» обозначает линейные или разветвленные насыщенные алифатические углеводородные группы, имеющие от 1 до 6 атомов углерода; примером для этого термина являются такие группы как: метил, этил, н-пропил, изо-пропил, н-бутил, изобутил, сек-бутил, трет-бутил и подобные.
Термин «защитная группа» обозначает группу применяемую для защиты реакционных центров в химических синтезах, например, гидроксильной группы (-ОН), амино группы (-NH2), тиольной группы (-SH), карбонильной группы (-С=O), карбоксильной группы (-COOH). Примеры защитных групп приведены в литературе (смотреть, например, Green, Theodora W. And Wuts, Peter G.M. - Protective Groups in Organic Synthesis, Third Edition, John Wiley & Sons Inc., New York (NY), 1999).
Термин «азотзащитная группа» обозначает группу, которая вместе с атомом азота, образует карбаматы, амиды, циклические имиды, N-алкил и N-арил амины. Такие защитные группы хорошо известны в данной области техники (смотреть в тех же источниках). Неограничивающими примерами карбаматных защитных групп являются, например, метил и этил карбамат, 9-флуоренилметил карбамат (Fmoc), 2,2,2-трихлорэтилкарбамат (Troc), трет-бутил карбамат (BOC), винил карбамат (Voc), аллил карбамат (Alloc), бензил карбамат (Cbz), п-нитробензил карбамат и подобные. Неограничивающими примерами амидов являются, например, N-трихлорацетамид, N-трифторацетамид (TFA) и подобные. Неограничивающими примерами защитных групп на основе циклических имидов являются, например, N-фталимид, N-дитиасукциноилимид (Dts) и подобные. Неограничивающими примерами N-алкил и N-арил защитными группами являются, например, N-аллиламин, N-бензиламин и подобные.
Термин «гидроксизащитная группа» обозначает группу, которая вместе с атомом кислорода, образует эфиры, сложные эфиры, циклические ацетали или кетали. Такие защитные группы хорошо известны в данной области техники (смотреть в тех же источниках). Неограничивающими примерами эфирных защитных групп являются, например, алкиловые эфиры и бензиловые эфиры, такие как метоксиметиловый эфир (MOM-OR), тетрагидропираниловый эфир (THP-OR), аллиловый эфир (Allyl-OR), бензиловый эфир (Bn-OR), трифенилметиловый эфир (Tr-OR) и подобные, или силиловые эфиры, такие как триметилсилиловый эфир (TMS-OR), трет-бутилдиметилсилиловый эфир (TBS-OR или TBDMS-OR), трет-бутилдифенилсилиловый эфир (TBDPS-OR), дифенилметилсилиловый эфир (DPMS-OR) и подобные. Неограничивающими примерами сложноэфирных защитных групп являются, например, трифторацетат, бензоат (Bz-OR) и карбонаты, такие как этилкарбонат и подобные. Неограничивающими примерами защитных групп на основе циклических ацеталей или кеталей являются, например, метиленацеталь, этилиденацеталь, метоксиметиленацеталь и подобные.
Термин «связывающая частица» обозначает частицу, которая связывает или ассоциирует соединение, соответствующее формуле (Ia) или (Ib) с двухцепочечной ДНК. Связывающая частица может повысить сродство производных к ДНК или увеличить алкилирующую активность алкилирующих агентов или выбрать различные последовательности ДНК для того, чтобы изменить выбранную специфичность соединения.
Термин «саморасщепляющая группа» обозначает химическую группу, способную образовывать связь с атомом кислорода тиено-индольного остова, которая становится неустойчивой при активации, что приводит к быстрому распаду тиено-индольных производных. Саморасщепляющие системы известны специалистам в данной области техники, смотреть, например, описанные в WO2002/083180 и WO2004/043493, или описанные в Anticancer Agents in Medicinal Chemistry, 2008, 8, 618-637 или в Polym. Chem. 2011, 2, 773-790.
Термин «реакционноспособный фрагмент» обозначает химическую группу, способную реагировать с другой группой в относительно мягких условиях и без необходимости предварительной функционализации; Вышеупомянутая реакция может потребовать только применения некоторого количества тепла, давления, катализатора, кислоты и/или основания. Наиболее предпочтительный реакционноспособный фрагмент представляет собой группу с электрофильной функцией, которая реагирует с нуклеофилами, т.е. молекулами, которые содержат нуклеофильную группу.
Термин «нуклеофильная группа» относится к группам, которые отдают электронную пару электрофильной группе с образованием химической связи в ходе химической реакции. Примеры таких нуклеофильных групп включают галогены, амины, нитриты, азиды, спирты, алкоксид анионы, карбоксилат анионы, тиолы, тиоляты и др., но не ограничиваются ими.
Специалисту в данной области техники известно, что превращение одной химической функциональной группы в другую, может требовать защиты одного или более реакционных центров в соединении для того, чтобы избежать нежелательных побочных реакций. Защита таких реакционноспособных центров, и последующее снятие защиты в конце синтетических превращений, может быть выполнено с применением стандартных процедур описанных в литературе (смотреть, например, Green, Theodora W. and Wuts, Peter G.M. - Protective Groups in Organic Synthesis, Third Edition, John Wiley & Sons Inc., New York (NY), 1999).
Предпочтительными соединениями, приготовленными способом настоящего изобретения, представляют собой соединения, соответствующие формуле (Ia), как описано выше.
Более предпочтительными, являются соединения, соответствующие формуле (Ia) где R1 представляет собой метил, R2 представляет собой хлор, а BM, W1, Z1 и RM являются такими, как указано выше.
Более предпочтительными, являются соединения, соответствующие формуле (Ia) где BM представляет собой ДНК-связывающий фрагмент, соответствующий формуле (II-1), как описано выше.
Наиболее предпочтительными конкретными соединениями, являются соединения, соответствующие формуле (Ia) представленные ниже:
N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[{3-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)амино]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]-2,2-диметилпропил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 1a);
N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[{3-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]-2,2-диметилпропил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 2a);
N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[{2-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)амино]-1Н-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил) фенил]-L-орнитинамид (соединение 3a);
N-[19-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-17-оксо-4,7,10,13-тетраокса-16-азанонадекан-1-оил]-L-валил-N5-карбамоил-N-[4-({[{2-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)амино]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 4a);
N-[19-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-17-оксо-4,7,10,13-тетраоксо-16-азанонадекан-1-иол]-L-валил-N5-карбамоил-N-[4-({[{2-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено [3,2-e]индол-4-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 5a) и
N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[{2-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 6a).
Как указано выше, настоящее изобретение также обеспечивает получение промежуточного соединения, соответствующего формуле (XIa) или (XIb):
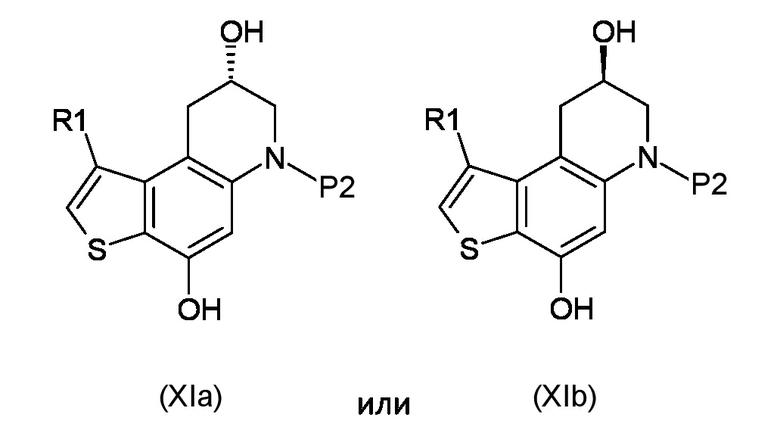
где R1 и P2 являются такими, как указано выше.
Предпочтительными промежуточными соединениями, полученными способом настоящего изобретения, являются соединения, соответствующие формуле (XIa) или (XIb), где R1 представляет собой метил, а P2 представляет собой трет-бутокси карбонил.
Еще одной задачей настоящего изобретения является способ получения промежуточного соединения, соответствующего формуле (XIa) или (XIb), как указано выше, причем этот способ включает стадии
а) N-алкилирование соединения, соответствующего формуле (VII), энантиочистым соединением, соответствующим формуле (VIIIa) или (VIIIb), для получения соединений, соответствующих формуле (IXa) или (IXb), с применением сильно основных условий.
b) внутримолекулярная 6-эндо-тет циклизация полученных соединений, соответствующих формуле (IXa) или (IXb)
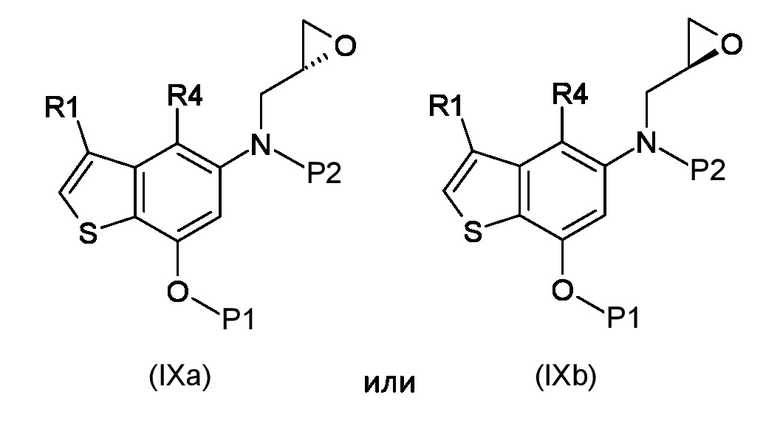
где R1, R4, P1 и P2 являются такими, как указано выше, с металлоорганическими реагентами и
c)селективное удаление гидроксизащитной группы P1, как указано выше.
Согласно стадии а), сочетание 5-амино-4-гало-3-алкил-1-бензотиофен-7-ола, соответствующего формуле (VII), с (S)-глицидил 3-нозилатом (VIIIa) или (R) глицидил 3-нозилатом (VIIIb), для получения соединения, соответствующего формуле (IXa) или (IXb), соответственно, выполняется с применением сильно основных условий, таких как, например, н-бутиллитий, t-BuOK или предпочтительно NaH. Предпочтительно, реакция выполняется при температуре в диапазоне от -10°С до 50°С в органическом растворителе, таком как, например, THF, Et2O, DMA, DMF или их смеси.
Согласно стадии b), превращение соединения, соответствующего формуле (IXa) или (IXb) в соединение, соответствующее формуле (Xa) или (Xb), соответственно, осуществляется с применением реагента Гриньяра, таким как, например, изо-PrMgCl·LiCl, сек-Bu2MgLi·Cl, MeMgBr, изо-PrMgBr или предпочтительно EtMgBr. Предпочтительно, реакция выполняется при температуре в диапазоне от -5°С до 50°С в органическом растворителе, таком как, например, THF, Et2O, DCM или их смеси.
Согласно стадии с), удаление гидроксизащитной группы из соединения, соответствующего формуле (Xa) или (Xb), для получения соединения, соответствующего формуле (XIa) или (XIb), соответственно, осуществляется известными способами, например, такими, которые описаны в Protective Groups in Organic Synthesis; Theodora W. Green, Peter G. M. Wuts. В частности, когда необходимо удалить бензильную защитную группу, реакцию проводят в условиях каталитического гидрирования с гидрирующим катализатором, предпочтительно 10% Pd/C и источником водорода, предпочтительно HCO2NH4. Предпочтительно, реакцию проводят при температуре в диапазоне от 0°С до температуры кипения, в органическом рстворителе, таком как, например, THF, Et2O, DCM, MeOH, EtOH или их смеси.
Согласно стадии d), внутримолекулярная спироциклизация преобразовывает соединение, соответствующее формуле (XIa) или (XIb) в соединение, соответствующее формуле (XIIa) или (XIIb), соответственно, ускоряя химическую реакцию Мицунобу активацией вторичного спирта органофосфорными(III) соединениями, предпочтительно фосфинами, такими как, например Ph3P, Bu3P и в присутствии азопроизводных соединений, предпочтительно диизопропил азодикарбоксилата (DAED) или 1,1'-(азодикарбонил)-дипиперидина (ADDP). Предпочтительно, реакцию проводят при температуре в диапазоне от -10°С до 50°С в органическом растворителе, таком как, например, THF, Et2O, DCM или их смеси.
Согласно стадии е), стереоконтролируемое региоселективное раскрытие циклопропанового кольца соединения, соответствующего формуле (XIIa) или (XIIb), для получения соединения, соответствующего формуле (XIIIa) или (XIIIb), соответственно, проводят в контролируемых кислых условиях, предпочтительно с HCl. Предпочтительно, реакцию проводят при температуре в диапазоне от -80°С до 25°С в органическом растворителе, таком как, например, EtOAc, Et2O, DCM или их смеси.
Согласно стадии f), удаление аминозащитной группы из соединения, соответствующего формуле (XIVa) или (XIVb), соответственно, выполняется известными способами, например, такими, которые описаны в Protective Groups in Organic Synthesis; Theodora W. Green, Peter G. M. Wuts. Предпочтительно, когда необходимо удалить трет-бутоксикарбонильную защитную группу, реакцию проводят в кислых условиях с TFA или предпочтительно с HCl. Предпочтительно, реакцию проводят при температуре в диапазоне от 0°С до температуры кипения в органическом растворителе, таком как, например, EtOAc, DCM, MeOH или их смеси.
Согласно стадии g) реакцию сочетания промежуточного соединения, соответствующего формуле (XIVa) или (XIVb), с остатком BM-COOH, для получения соединения, соответствующего формуле (XVa) или (XVb), проводят в присутствии конденсирующего агента, такого как, например,DCC, EDC или предпочтительно EDCl. Реакцию проводят при температуре от 0°С до 100°С в органическом растворителе, таком как, например, DMF.
Согласно стадии h) и i) создание производных ключевого энантиочистого интермедиата, соответствующего формуле (XVa) или (XVb), для получения соединения, соответствующего формуле (Ia) или (Ib), осуществляется в соответствии с методами, описанными в ссылках известного уровня техники, таких как те, что описаны в стадии e) на странице 63 WO2013/149948.
Экспериментальная часть
В примерах ниже, а также во всем документе, следующие сокращения имеют следующие значения. Если они не определены, термины имеют общепринятые значения.
1Н-ЯМР спектры были зарегистрированы при постоянной температуре 25°С на спектрометре Varian INOVA 500, работающем на частоте 499.7 МГц и оборудованный 5 мм датчиком тройного резонанса с непрямым детектированием (1Н{13C,15N}). Химические сдвиги были соотнесены в соответствии с сигналом остаточного растворителя, ДМСО-d6 при 2,50 мд, для 1Н. Данные изложены следующим образом: химический сдвиг, мультиплетность (с=синглет, д=дублет, к=квартет, ус=уширенный синглет, дд=дублет дублетов, тд=триплет дублетов, м=мультиплет), константа спин-спинового взаимодействия и число протонов. ВЭЖХ-МС/УФ анализы были выполнены на LCQ DecaXP (Thermo, San Jose, US) ионной ловушке, оснащенной источником электрораспыления ионов (ESI). Масс спектрометр подключен к Surveor ВЭЖХ системе (Thermo, San Jose, US) с диодно-матричным детектором (УФ-детектирование при 215-400 нм). Была использована колонка Waters XSelect CSH C18 (50×4,6 мм) с размером части 3,5 мкм. Подвижная фаза А представляла собой 5 мМ буффер (рН 4.5 с уксусной кислотой):ацетонитрил 95:5, а подвижная фаза Б представляла собой 5мМ буфер (рН 4,5 с уксусной кислотой): ацетонитрил 5:95. Градиент от 0 до 100% Б в течение 7 минут, с задержкой на 100% Б в течение 2 минут. Скорость потока 1 мл/мин. Объем вкола 10 мкл. Время удерживания (Rt) указано в минутах. Диапазон детектируемых масс от 100 до 1200 а.е.м. Температура капилляра была 275°С, а значение напряжения распыления было установлено на уровне 4 кВ. Масса задана как отношение массы к заряду.
Управление прибором, накопление и обработка данных выполнялись с применением программного обеспечения Xcalibur 1.2 (Thermo). Масс спектры высокого разрешения (HRMS) были получены на TOF Waters LCT Premier XE масс детекторе с ESI интерфейсом. Анализ основывался на стандартной градиентной хроматографии с обращенной фазой, выполненной на Waters Alliance liquid chromatograph mod. 2795. Элюент из колонки ВЭЖХ разделяли и 25 мкл/мин смешивали с потоком 100 мкл/мин смеси MeOH/iPrOH/H2O (30/10/60 (по объему)) содержащей 0,01% w/v муравьиной кислоты и 80 нМ триметоприма исходящего из насоса Waters Reagent Manager перед тем как в МС источник. ESI источник был использован при 100°С, капиллярном напряжении 2,5 кВ, конусный скиммер 60 В, потоке азота 600 л/час для десольвации при 350°С и потоке азота из конуса 10 л/час. Триметоприм был выбран как стабильное, растворимое и подходящее стандартное соединение для одноточечной масс-коррекции в реальном времени. ES(+) усредненное накопление данных в пределах всей области от 80 до 1200 а.е.м. было выполнено с частотой регистрации 2 Гц с применением «W» мода. LCT встроенное в ПК, обеспечило как центрирование данных в реальном времени, так и корректировку массы в реальном времени, на основе Trimethoprim.H+ с контрольной массой 291.1452 Да. Необходимые интенсивности МС спектров (от 40 до 2000 аналитов) были усреднены для получения конечного результата.
Энантиомерная чистота (%ее) соединений была определена хиральной ВЭЖХ, а разделение смеси энантиомеров осуществлялось применением следующих условий: температура: 25°C; скорость потока: 0,7 мл/мин; колонка: CHIRACEL OD 4.6×250 мм, 20 мкм; объем вкола: 20 мкл; подвижная фаза А: гексан; подвижная фаза Б: EtOH; изократическое элюирование при 55% Б.
Пример 1
Синтез N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[{2-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамида (соединение 6a)
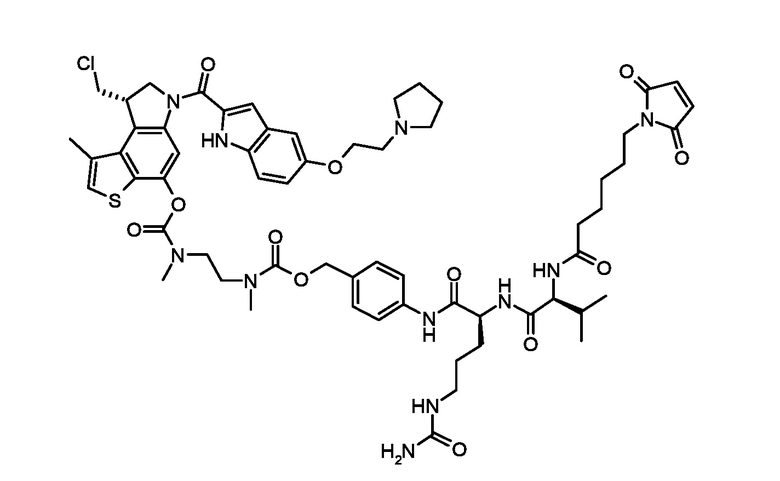
Стадия а)
Приготовление промежуточного соединения трет-бутил [7-(бензилокси)-4-иод-3-метил-1-бензотиофен-5-ил][(2S)-оксиран-2-илметил]карбамата (IXa')
Раствор трет-бутил [7-(бензилокси)-4-иод-3-метил-1-бензотиофен-5-ил]карбамата (VII') приготовленного в соответствии с GB2344818 (515 мг, 1,04 ммоль) и коммерчески доступного (S)-глицидил 3-носилата (337 мг, 1,3 ммоль) в сухом DMF (12 мл) был охлажден до 0°C и обработан NaH (60% дисперсией в минеральном масле, 61 мг, 1,56 ммоль). Полученный раствор перемешивали при 0°C в течение 5 часов, затем вылили в ледяную воду и экстрагировали этилацетатом (200 мл). Органический слой промывали водой (100 мл), насыщенным водным раствором NaCl (100 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (градиентным элюированием 6-12% EtOAc/н-гексан) для получения необходимого промежуточного соединения (500 мг, 87%) в виде белого твердого вещества.
1H ЯМР (ДМСО-d6,500МГц): δ=7,59 (уш. с., 1 H), 7,44-7,52 (м, 2 H), 7,30-7,43 (м, 3 H), 7,00-7,17 (м, 1 H), 5,25-5,39 (м, 2 H), 3,87-3,97 (м, 1 H), 3,06-3,27 (м, 2 H), 2,68 (с, 3 H), 2,57-2,67 (м, 1 H), 2,25-2,47 (м, 1 H), 1,16-1,57 (м, 9 H)
ВЭЖХ-МС(ESI)/УФ(215-400 нм): [M+H]+ 552; Rt 8,43 мин.
Стадия b)
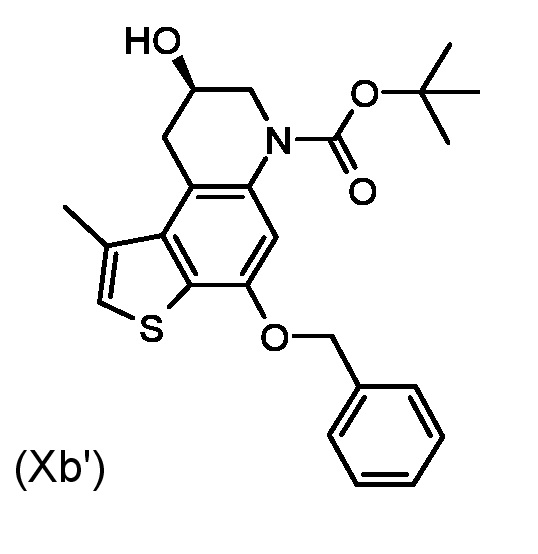
Приготовление промежуточного соединения трет-бутил (8S)-4-(бензилокси)-8-гидрокси-1-метил-8,9-дигидротиено [3,2-f]хинолин-6(7H)-карбоксилата (Xa')
Раствор промежуточного соединения (IXa') из стадии а) (360 мг, 0,661 ммоль) в сухом ТГФ (4 мл), смешивали с EtMgBr (1,3 мл, 1,0 М в ТГФ) при 0°C в атмосфере азота. Реакционная смесь перемешивали при комнатной температуре в течение 1 часа, реакцию останавливали добавлением насыщенного водного раствора NH4Cl и дважды экстрагировали EtOAc (2×40 vk). Водный раствор моногидрата п-толуолсульфокислоты (2 г в 4 мл воды) добавили к объединенным органическим слоям и перемешивали реакционную смесь в течение 15 минут. Реакцию останавливали добавлением 1 М водного раствора Na2CO3. Слои были разделены и органический слой промывали насыщенным водным раствором NaCl и сушили над Na2SO4. Растворитель удаляли при пониженном давлении, а остаток очищали флеш-хроматографией (градиентным элюированием 20-40% диэтиловый эфир/н-гексан) для получения необходимого промежуточного соединения (Xa') в виде белого твердого вещества (185 мг, 65%).
1H ЯМР (ДМСО-d6,500МГц): δ=7,45-7,52 (м, 2 H), 7,41 (т, J=7,4 Гц, 2 H), 7,33-7,37 (м, J=7,3 Гц, 1 H), 7,31 (с, 1 H), 7,06 (с, 1 H), 5,19-5,27 (м, 2 H), 5,18 (д, J=4.0 Гц, 1 H), 3,98 (м, 1 H), 3,82 (дд, J=12,4, 3,2 Гц, 1 H), 3,54 (дд, J=16,7, 6,1 Гц, 1 H), 3,29 (дд, J=12,4, 7,8 Гц, 1 H), 3,03 (дд, J=16,7, 6,4 Гц, 1 H), 2,58 (с, 3 H), 1,43 (с, 9 H)
ВЭЖХ-МС(ESI)/УФ(215-400 нм): [M+H]+ 426; Rt 7.50 мин
Стадия с)
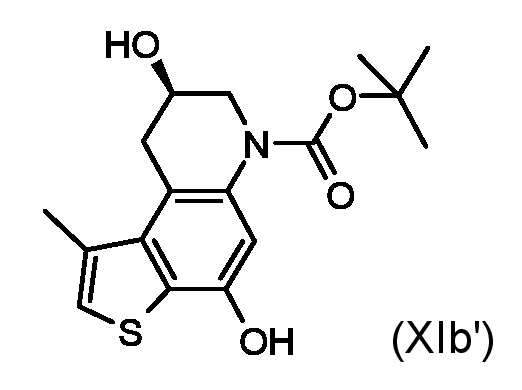
Приготовление промежуточного соединения трет-бутил (8S)-4,8-дигидрокси-1-метил-8,9-дигидротиено[3,2-f]хинолин-6(7H)-карбоксилата (XIa')
Раствор промежуточного соединения (Xa') из стадии б) (148 мг, 0,347 ммоль) в ТГФ (30 мл) смешивали с 10% Pd/C (70 мг) и 25% водным раствором HCO2NH4 (2 мл) и перемешивали в течение 2 часов. Смесь отфильтровали через слой целита, растворитель удаляли при пониженном давлении и остаток очищали флэш-хроматографией (элюированием 30% диэтиловый эфир/толуол) для получения необходимого промежуточного соединения (XIa') в виде белого твердого вещества (90 мг, 80%).
1H ЯМР (ДМСО-d6,500МГц): δ=10,07 (с, 1 H), 7,24 (с, 1 H), 6,92 (с, 1 H), 5,14 (д, J=4,0 Гц, 1 H), 3,95 (м, 1 H), 3,80 (дд, J=12,4, 3,4 Гц, 1 H), 3,51 (дд, J=16,5, 6,1 Гц, 1 H), 3,27 (дд, J=12,4, 7,8 Гц, 1 H), 2,99 (дд, J=16,5, 6,6 Гц, 1 H), 2,56 (с, 3 H), 1,46 (с, 9 H)
ВЭЖХ-МС(ESI)/УФ(215-400 нм): [M+H]+ 336; Rt 5,59 мин
Стадия d)
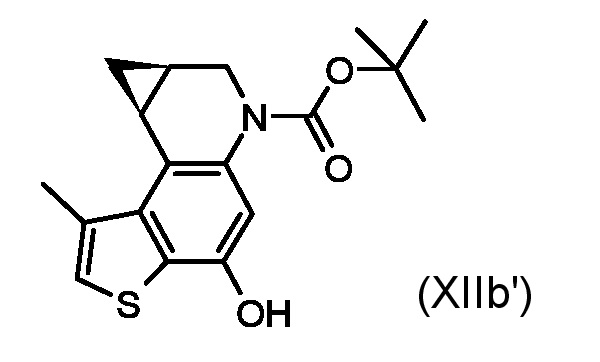
Приготовление промежуточного соединения трет-бутил (7aS,8aR)-4-гидрокси-1-метил-7,7a,8,8a-тетрагидро-6H-циклопропа[c]тиено[3,2-f]хинолин-6-карбоксилата (XIIa')
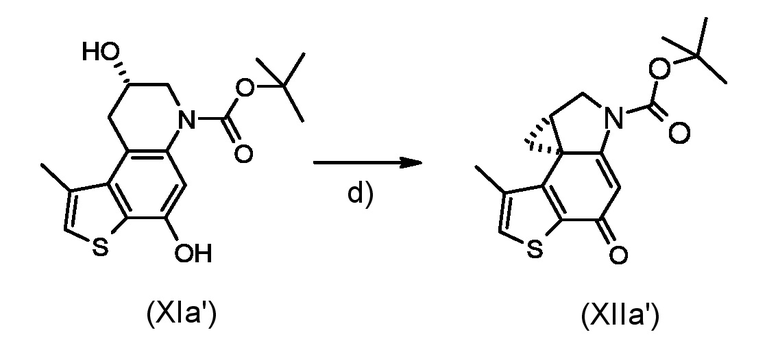
Раствор промежуточного соединения (XIa') из стидии с) (60 мг, 0,178 ммоль) в сухом ТГФ (15 мл) смешали с трибутилфосфином (0,258 мл, 0,89 ммоль) и 1,1'-(азодикарбонил)-дипиперидином (ADDP, 225 мг, 0,89 ммоль) в атмосфере азота. Реакционную смесь перемешивали в течение 1 часа при комнатной температуре, останавливали реакцию добавлением воды и экстрагировали диэтиловым эфиром. Органический слой промыли водой и насыщенным водным раствором NaCl, сушили над Na2SO4 и концентрировали. Остаток очищали флеш-хроматографией (элюированием 50% диэтиловый эфир/н-гексан) для получения необходимого промежуточного соединения (XIIa') в виде белого твердого вещества (48 мг, 85%).
1H ЯМР (ДМСО-d6, 500МГц): δ=7,54 (д, J=0,9 Гц, 2 H), 6,57 (уш. с., 1 H), 3,90-4,00 (м, 2 H), 3,37 (м, 1 H), 2,16 (д, J=0,9 Гц, 3 H), 2,13 (дд, J=7,7, 4,5 Гц, 1 H), 1,50 (с, 9 H), 1,37 (т, J=4,9 Гц, 2 H)
ВЭЖХ-МС(ESI)/УФ(215-400 нм): [M+H]+ 318; Rt 6,06 мин
Стадии e-f)
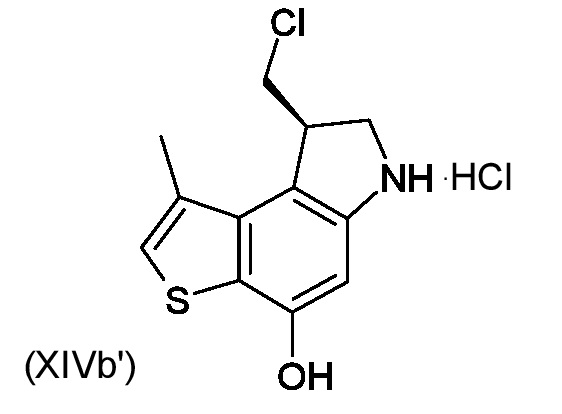
Приготовление ключевого промежуточного соединения (8S)-8-(хлорметил)-1-метил-7,8-дигидро-6H-тиено[3,2-e]индол-4-ола (XIVa')
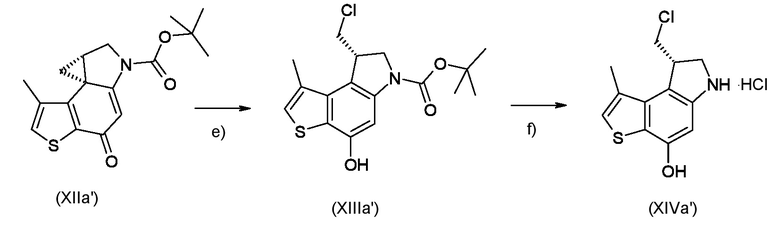
Раствор промежуточного соединения (XIIa') из стадии d) (22 мг, 0,0693 ммоль) в EtOAc (1 мл) при -78°C смешивали с 3,5 Н HCl в EtOAc (2,0 мл) и перемешивали при -78°C в течение 45 минут. Раствор нагревали до комнатной температуры и перемешивали в течение 2 часов. Растворитель и газоообразный HCl удаляли потоком азота и остаток сушили под вакуумом для получения необходимого промежуточного соединения (XIVa')(19 мг, 90%), которое вводили в следующую стадию без какой либо очистки.
Статдия g)
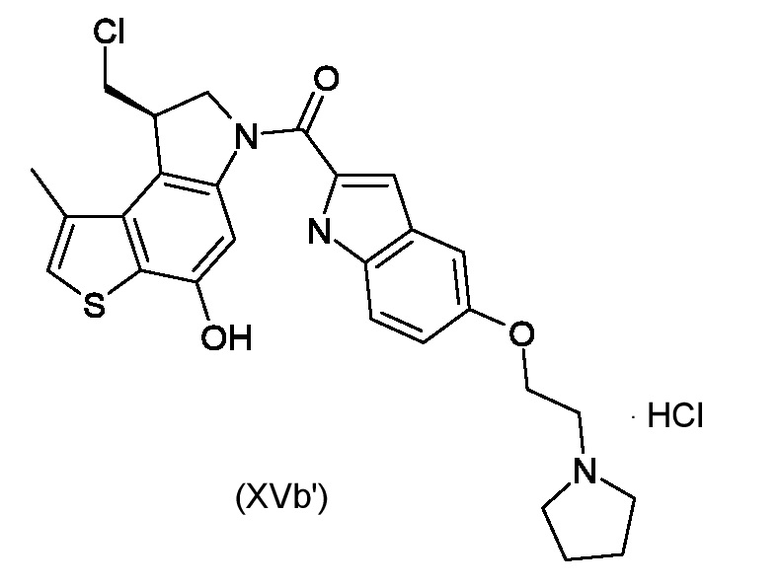
Приготовление ключевого промежуточного соединения [(8S)-8-(хлорметил)-4-гидрокси-1-метил-7,8-дигидро-6H-тиено[3,2-e]индол-6-ил]{5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}метанона гидрохлорида (XVa')
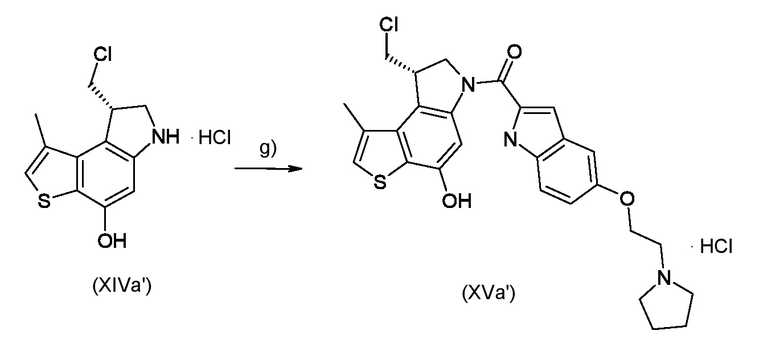
Не обработанное промежуточное соединение (XIVa') из стадии е) растворили в сухом DMF (3,5 мл) и EDCI (53 мг, 0,277 ммоль) и добавили 5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-карбоновую кислоту (1,7 экв.). Полученную реакционную смесь перемешивали всю ночь в атмосфере азота. После доваления 300 мг силикагеля, растворитель удаляли при пониженном давлении и остаток очищали флэш-хроматографией (элюированием системой DCM/MeOH/HCl (100/8/0,2) в диоксане) для получения промежуточного соединения, соответствующего формуле (XVa')(32 мг, 84%) в виде белого твердого вещества.
ВЭЖХ-МС(ESI)/УФ(215-400 нм): [M+H]+ 510; Rt 5,98 мин
Хиральная ВЭЖХ, rt=10,68; ee>99%.
1H ЯМР (ДМСО-d6, 500МГц): δ=11,69 (с, 1 H), 10,54 (с, 1 H), 10,03 (уш. с., 1 H), 7,85 (уш. с., 1 H), 7,41-7,46 (м, 2 H), 7,26 (с, 1 H), 7,08 (д, J=1,5 Гц, 1 H), 7,00 (дд, J=8,9, 2,2 Гц, 1 H), 4,67 (дд, J=10,7, 8,1 Гц 1 H), 4,56 (д, J=10,7 Гц, 1 H), 4,31 (уш. с., 2 H), 4,17 (тд, J=8,4, 2,3 Гц, 1 H), 3,89 (дд, J=11,1, 2,7 Гц, 1 H), 3,59 (м, 4 H), 3,14 (м, 2 H), 2,54 (д, J=0,9 Гц, 3 H), 1,92 (уш.с, 4 H)
Стадии h), i)
Синтез требуемого конечного тиено-индольного соединения (соединение 6а)
Начиная с ключевого энантиомерно чистого промежуточного соединения (XVa') полученного, как описано в приготовлениях выше от стадии а) до стадии g) и в соответствии с указаниями, описанными в WO2013/149948 на странице 63, было получено необходимое ключевое соединение N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[{2-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 6a).
ESI МС: m/z 1222 (MH+)
1H ЯМР (400 МГц, DMF-d7) δ м.д. 0,95 (т, J=7,8 Гц, 6 H) 2,16 (м, 1 H) 2,66 (уш. с., 3 H) 2,99-3,09 (м, 3 H) 3,09-3,29 (м, 3 H) 3,59 (уш. с., 2 H) 3,69 (уш. с., 2 H) 3,81 (уш. с., 2 H) 4,04 (д, J=10,7 Гц, 1 H) 4,31-4,47 (м, 2 H) 4,61 (уш. с., 1 H) 4,83 (уш. с., 2 H) 5,11 (д, J=15,7 Гц, 2 H) 5,60 (с, 2 H) 6,29 (уш. с., 1 H) 7,00 (м, 3 H) 7,30 (м, 2 H) 7,53 (д, J=8,5 Гц, 2 H) 7,88 (д, J=8,2 Гц, 1 H) 8,13 (д, J=7,8 Гц, 1 H) 8,27 (с, 1 H) 10,1 (м, 1 H) 11,61 (уш.с., 1 H)
Пример 2
Синтез N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[{2-[({[(8R)-8-(хлорметил)-1-метил-6-({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 6b).
По методике, аналогичной Примеру 1 и начиная с коммерчески доступного (R)-глицидил 3-нозилата, были приготовлены следующие промежуточные соединения:
трет-бутил [7-(бензилокси)-4-иод-3-метил-1-бензотиофен-5-ил][(2R)-оксиран-2-илметил]карбамат (IXb', стадия a)
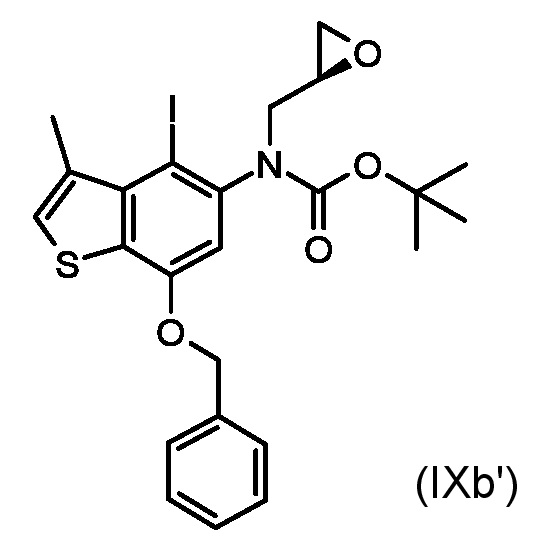
1H ЯМР (ДМСО-d6, 500МГц): δ=7,59 (уш. с., 1 H), 7,44-7,52 (м, 2 H), 7,30-7,43 (м, 3 H), 7,00-7,17 (м, 1 H), 5,25-5,39 (м, 2 H), 3,87-3,97 (м, 1 H), 3,06-3,27 (м, 2 H), 2,68 (с, 3 H), 2,57-2,67 (м, 1 H), 2,25-2,47 (м, 1 H), 1,16-1,57 (м, 9 H)
ВЭЖХ-МС(ESI)/УФ(215-400 нм): [M+H]+ 552; Rt 8,43 мин.
трет-бутил (8R)-4-(бензилокси)-8-гидрокси-1-метил-8,9-дигидротиено [3,2-f]хинолин-6(7H)-карбоксилат (Xb', стадия b)
1H ЯМР (ДМСО-d6, 500МГц): δ=7,45-7,52 (м, 2 H), 7,41 (т, J=7,4 Гц, 2 H), 7,33-7,37 (м, J=7,3 Гц, 1 H), 7,31 (с, 1 H), 7,06 (с, 1 H), 5,19-5,27 (м, 2 H), 5,18 (д, J=4,0 Гц, 1 H), 3,98 (м, 1 H), 3,82 (дд, J=12,4, 3,2 Гц, 1 H), 3,54 (дд, J=16,7, 6,1 Гц, 1 H), 3,29 (дд, J=12,4, 7,8 Гц, 1 H), 3,03 (дд, J=16,7, 6,4 Гц, 1 H), 2,58 (с, 3 H), 1,43 (с, 9 H)
ВЭЖХ-МС(ESI)/УФ(215-400 нм): [M+H]+ 426; Rt 7,50 мин
трет-бутил (8R)-4,8-дигидрокси-1-метил-8,9-дигидротиено [3,2-f]хинолин-6(7H)-карбоксилат (XIb', стадия c)
1H ЯМР (ДМСО-d6, 500МГц): δ=10,07 (с, 1 H), 7,24 (с, 1 H), 6,92 (с, 1 H), 5,14 (д, J=4,0 Гц, 1 H), 3,95 (м, 1 H), 3,80 (дд, J=12,4, 3,4 Гц, 1 H), 3,51 (дд, J=16,5, 6,1 Гц, 1 H), 3,27 (дд, J=12,4, 7,8 Гц, 1 H), 2,99 (дд, J=16,5, 6,6 Гц, 1 H), 2,56 (с, 3 H), 1,46 (с, 9 H)
ВЭЖХ-МС(ESI)/УФ(215-400 нм): [M+H]+ 336; Rt 5,59 мин
трет-бутил (7aR,8aS)-4-гидрокси-1-метил-7,7a,8,8a-тетрагидро-6H-циклопропа[c]тиено[3,2-f]хинолин-6-карбоксилат (XIIb', стадия d)
1H ЯМР (ДМСО-d6, 500МГц): δ=7,54 (д, J=0,9 Гц, 2 H), 6,57 (уш. с., 1 H), 3,90-4,00 (м, 2 H), 3,37 (м, 1 H), 2,16 (д, J=0,9 Гц, 3 H), 2,13 (дд, J=7,7, 4,5 Гц, 1 H), 1,50 (с, 9 H), 1,37 (т, J=4,9 Гц, 2 H)
ВЭЖХ-МС(ESI)/УФ(215-400 нм): [M+H]+ 318; Rt 6,06 мин
(8R)-8-(хлорметил)-1-метил-7,8-дигидро-6H-тиено[3,2-e]индол-4-ол (XIVb', стадии e-f)
[(8R)-8-(хлорметил)-4-гидрокси-1-метил-7,8-дигидро-6H-тиено[3,2-e]индол-6-ил]{5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}метанон гидрохлорид (XVb', стадия g)
Хиральная ВЭЖХ, rt=13,20; ee>99%.
1H ЯМР (ДМСО-d6, 500МГц): δ=11,69 (с, 1 H), 10,54 (с, 1 H), 10,03 (уш. с., 1 H), 7,85 (уш. с., 1 H), 7,41-7,46 (м, 2 H), 7,26 (с, 1 H), 7,08 (д, J=1,5 Гц, 1 H), 7,00 (дд, J=8,9, 2,2 Гц, 1 H), 4,67 (дд, J=10,7, 8,1 Гц 1 H), 4,56 (д, J=10,7 Гц, 1 H), 4,31 (уш. с., 2 H), 4,17 (тд, J=8,4, 2,3 Гц, 1 H), 3,89 (дд, J=11,1, 2,7 Гц, 1 H), 3,59 (м, 4 H), 3,14 (м, 2 H), 2,54 (д, J=0,9 Гц, 3 H), 1,92 (уш.с, 4 H)
Синтез требуемого конечного тиено-индольного соединения: N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[{2-[({[(8R)-8-(хлорметил)-1-метил-6-({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамида (соединение 6b) (стадии h, i)
Начиная с ключевого энантимерно чистого промежуточного соединения (XVIb'), полученного как описано в приготовлениях выше, и в WO2013/149948 на странице 63Б, был получен соответствующий энантимерно чистый N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[{2-[({[(8R)-8-(хлорметил)-1-метил-6-({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 6b)
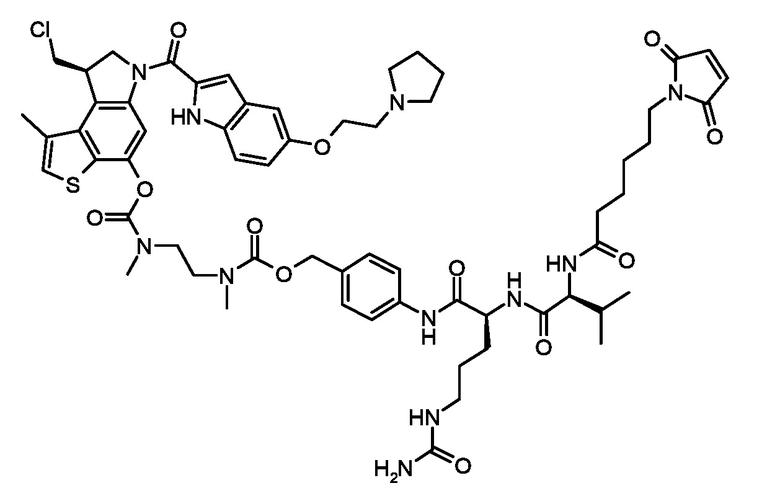
ESI МС: m/z 1222 (MH+)
1H ЯМР (400 МГц, DMF-d7) δ м.д. 0,95 (т, J=7,8 Гц, 6 H) 2,16 (м, 1 H) 2,66 (уш. с., 3 H) 2,99-3,09 (м, 3 H) 3,09-3,29 (м, 3 H) 3,59 (уш. с., 2 H) 3,69 (уш. с., 2 H) 3,81 (уш. с., 2 H) 4,04 (д, J=10,7 Гц, 1 H) 4,31-4,47 (м, 2 H) 4,61 (уш. с., 1 H) 4,83 (уш. с., 2 H) 5,11 (д, J=15,7 Гц, 2 H) 5,60 (с, 2 H) 6,29 (уш. с., 1 H) 7,00 (м, 3 H) 7,30 (м, 2 H) 7,53 (д, J=8,5 Гц, 2 H) 7,88 (д, J=8,2 Гц, 1 H) 8,13 (д, J=7,8 Гц, 1 H) 8,27 (с, 1 H) 10,1 (м, 1 H) 11,61 (уш.с., 1 H)
Пример 3
Начиная с различных ключевых промежуточных соединений, соответствующих формуле (XIVa) или (XIVb) и в соответствии с методиками, описанными выше в примерах 1 и 2 и в WO2013/149948 со страницы 43 до страницы 79, были приготовлены следующие соединения:
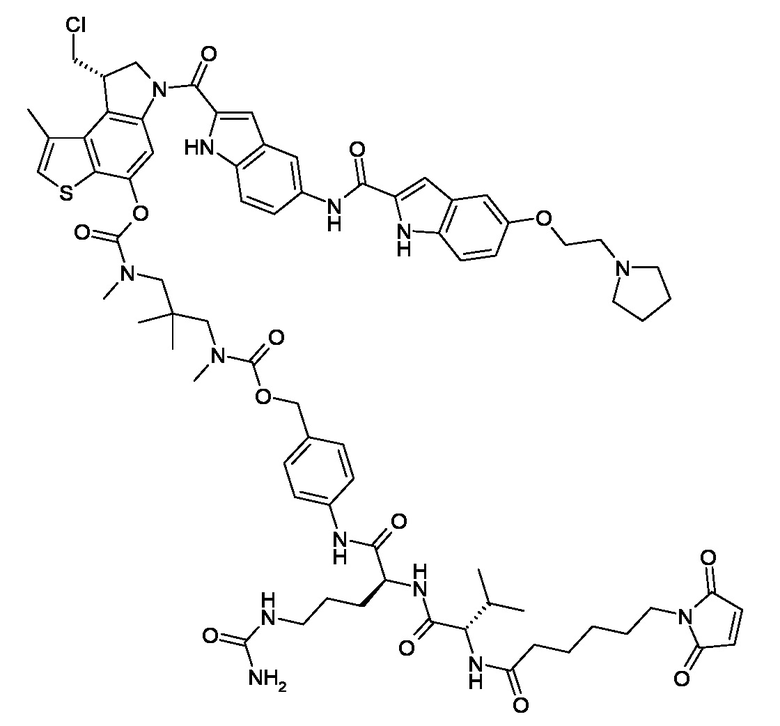
N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[{3-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)амино]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]-2,2-диметилпропил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 1a).
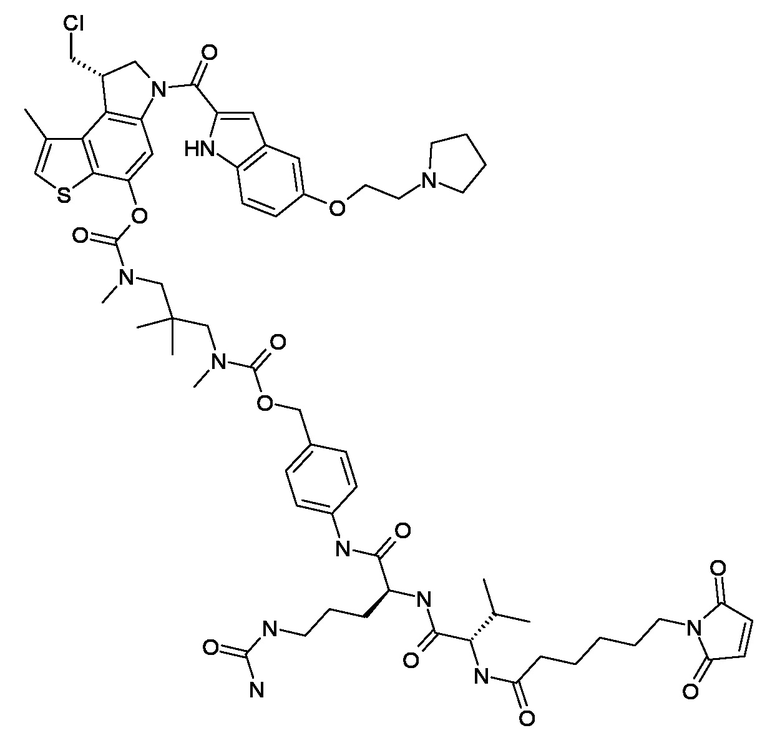
N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[{3-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]-2,2-диметилпропил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 2a).
N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[{2-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)амино]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 3a).
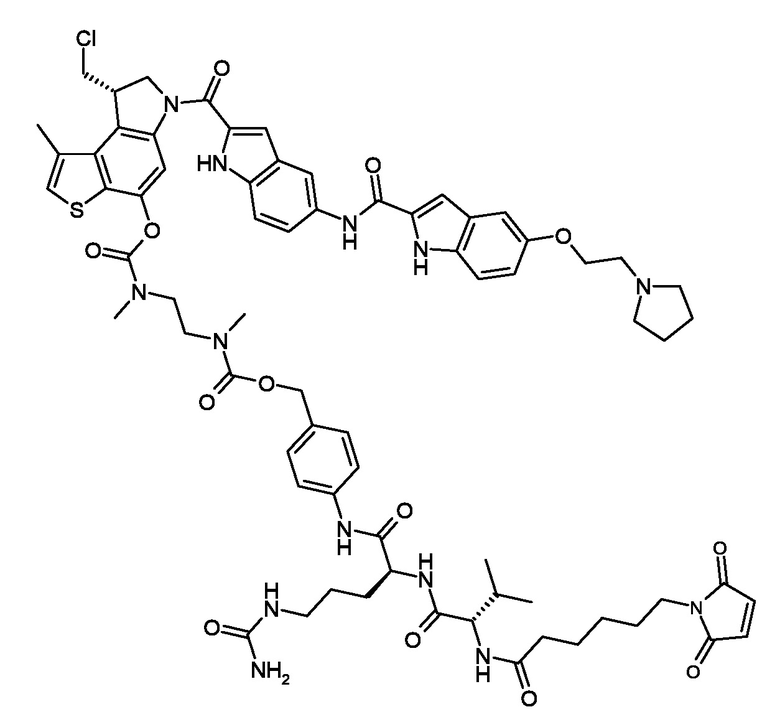
N-[19-(2,5-диокси-2,5-дигидро-1H-пиррол-1-ил)-17-оксо-4,7,10,13-тетраокса-16-азанонадекан-1-оил]-L-валил-N5-карбамоил-N-[4-({[{2-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)амино]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 4a).
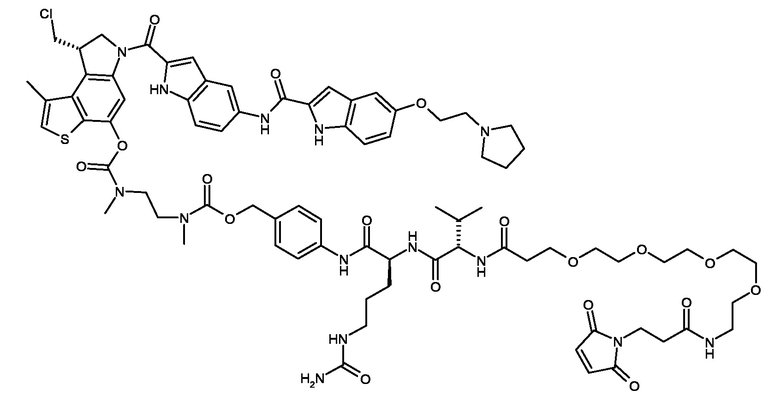
N-[19-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-17-оксо-4,7,10,13-тетраоксо-16-азанонадекан-1-оил]-L-валил-N5-карбамоил-N-[4-({[{2-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-yl}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 5a).
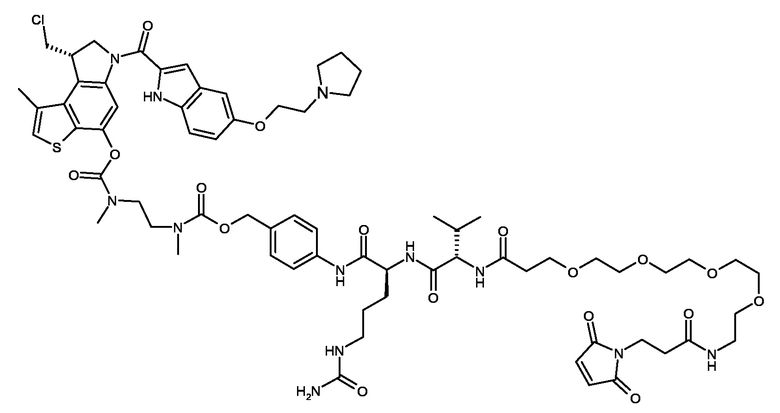
N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[{3-[({[(8R)-8-(хлорметил)-1-метил-6-({5-[({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)амино]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]-2,2-диметилпропил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 1b).
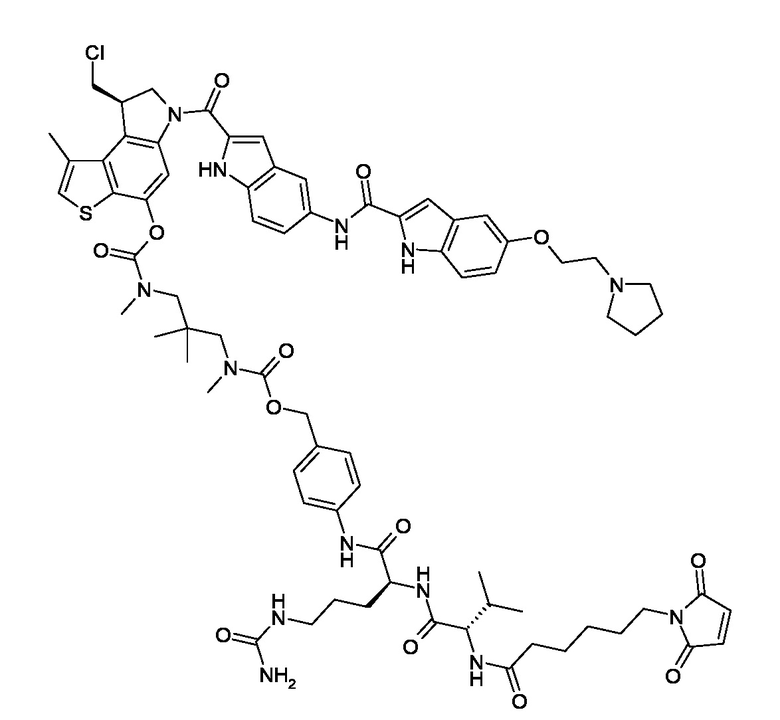
N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[{3-[({[(8R)-8-(хлорметил)-1-метил-6-({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]-2,2-диметилпропил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 2b).
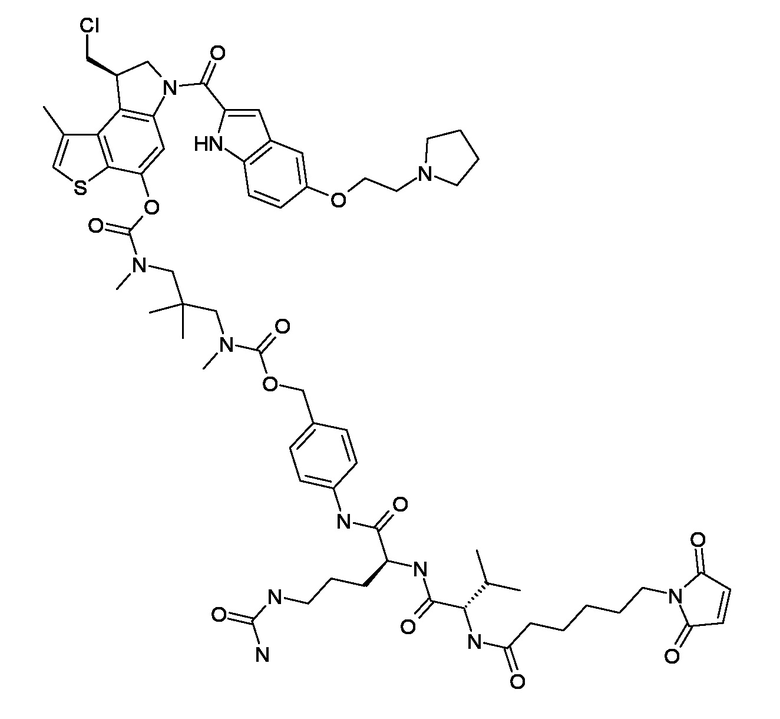
N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[{2-[({[(8R)-8-(хлорметил)-1-метил-6-({5-[({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)амино]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 3b).
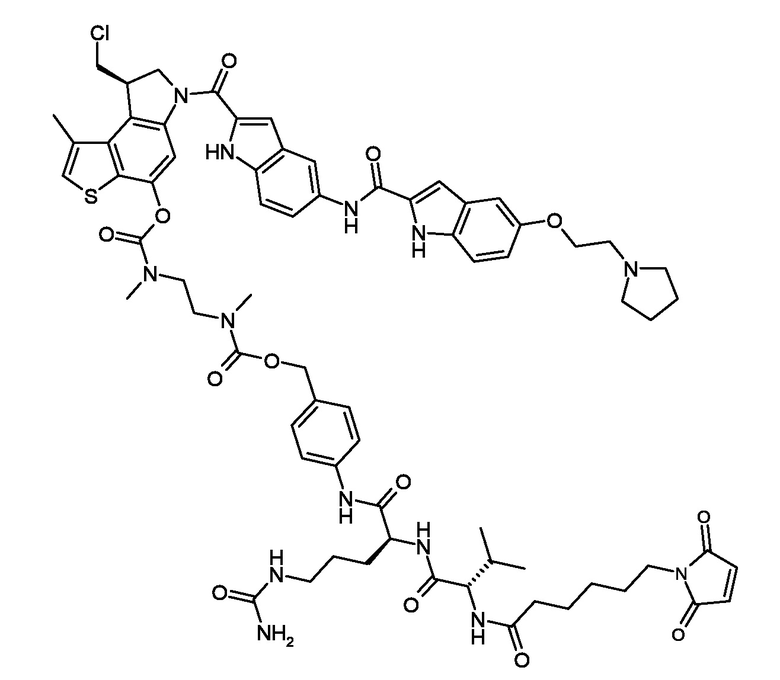
N-[19-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-17-оксо-4,7,10,13-тетраоксо-16-азанонадекан-1-оил]-L-валил-N5-карбамоил-N-[4-({[{2-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)амино]-1H-индол-2-yl}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 4b).
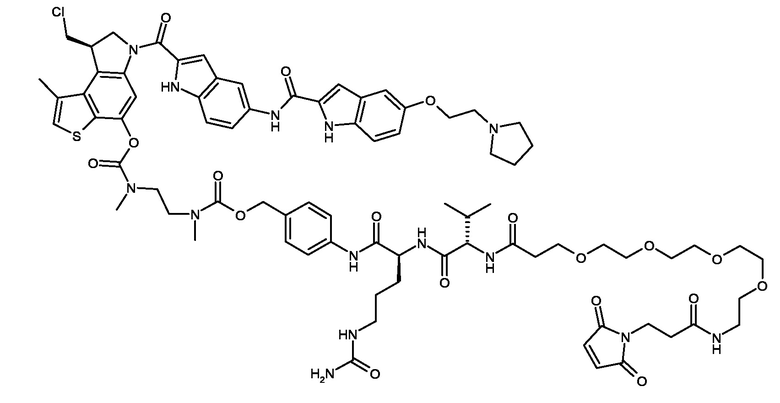
N-[19-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-17-оксо-4,7,10,13-тетраокса-16-азанонадекан-1-оил]-L-валил-N5-карбамоил-N-[4-({[{2-[({[(8R)-8-(хлорметил)-1-метил-6-({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 5b).
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ АЛКИЛИРУЮЩИЕ СРЕДСТВА | 2013 |
|
RU2632206C2 |
| ФУНКЦИОНАЛИЗИРОВАННЫЕ ПРОИЗВОДНЫЕ ТИЕНОИНДОЛА ДЛЯ ЛЕЧЕНИЯ РАКОВОГО ЗАБОЛЕВАНИЯ | 2013 |
|
RU2632199C2 |
| Бифункциональные цитотоксические агенты, содержащие CTI фармакофор | 2016 |
|
RU2682645C1 |
| ФУНКЦИОНАЛИЗИРОВАННЫЕ ПРОИЗВОДНЫЕ МОРФОЛИНИЛАНТРАЦИКЛИНА | 2015 |
|
RU2715902C2 |
| ПАРА-АМИНОБЕНЗИЛЬНЫЕ ЛИНКЕРЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В КОНЪЮГАТАХ | 2021 |
|
RU2839675C1 |
| АНТИТЕЛА К В7-Н3 И КОНЪЮГАТЫ АНТИТЕЛА И ЛЕКАРСТВЕННОГО СРЕДСТВА | 2017 |
|
RU2764651C2 |
| Бифункциональные цитотоксические агенты | 2015 |
|
RU2669807C2 |
| Модулирующие стабильность линкеры для использования с конъюгатами антитело-лекарственное средство | 2015 |
|
RU2680238C2 |
| НОВОЕ ПРОИЗВОДНОЕ ГИДРОКСАМОВОЙ КИСЛОТЫ | 2011 |
|
RU2575129C2 |
| Аналоги сплицеостатина | 2013 |
|
RU2618523C2 |
Изобретение относится к новым способам получения тиено-индольных производных, соответствующих формуле (Ia) или (Ib), с применением асимметрического синтеза, для приготовления ключевых промежуточных соединений
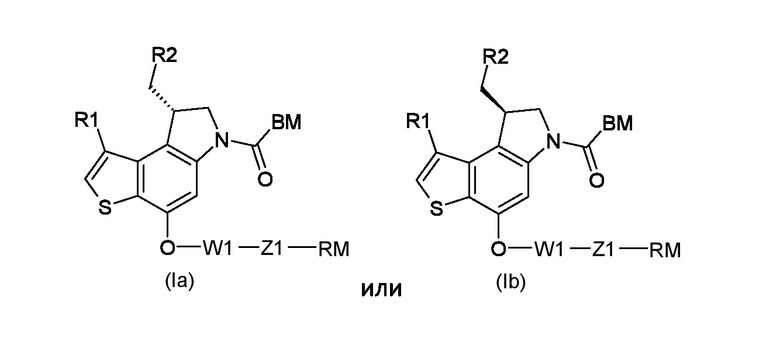
где: R1 представляет собой водород или линейный или разветвленный С1-С6 алкил; R2 представляет собой уходящую группу, выбранную из галогеновых или сульфонатных групп; BM представляет собой ДНК-связывающий фрагмент, соответствующий формуле (II-1) или (II-2)
W1 представляет собой саморасщепляющую систему, соответствующую формуле (III)
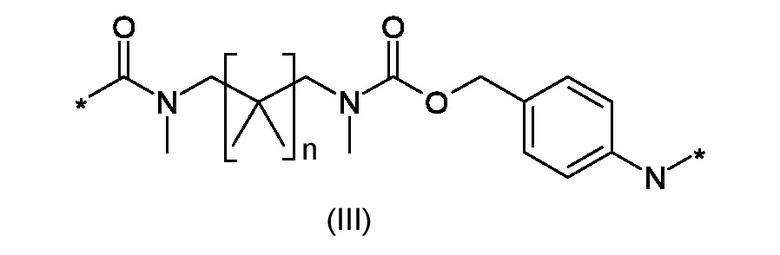
где n это 0 или 1;
Z1 представляет собой линкер, соответствующий формуле (IV-1) или (IV-2)
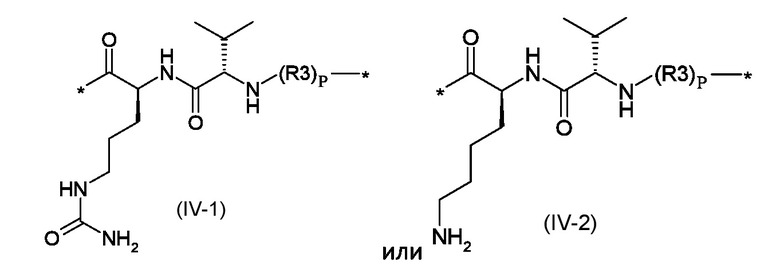
где p это число от 0 до 1, а R3 представляет собой полиоксиэтиленовую цепь, соответствующую формуле (V)
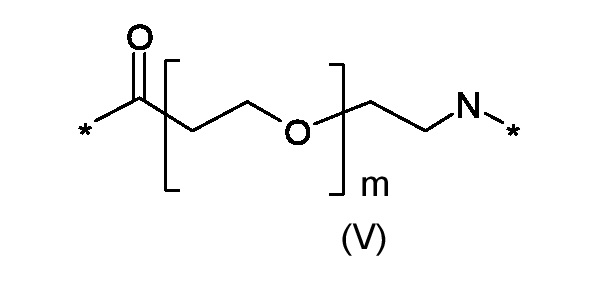
где m это число от 0 до 5; и
RM представляет собой фрагмент, соответствующий формуле (VI)
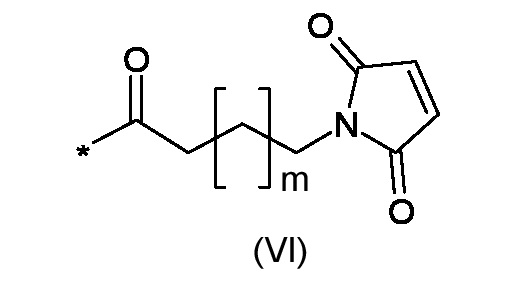
где m является таким, как указано выше. Синтез начинается с N-алкилирования 5-амино-4-гало-3-алкил-1-бензотиофен-7-ол производных энантиочистым глицидил 3-нозилатом, с последующей внутримолекулярной 6-эндо-тет циклизацией с применением алкильных реагентов Гриньяра; Мицунобу активация вторичного спирта способствует внутримолекулярной спироциклизации, давая 4,4a,5,6-тетрагидро-8H-циклопропа[c]тиено[3,2-e]индол-8-он производные; стереоэлектронный контроль региоселективного циклопропанового раскрытия позволил получить ключевые энантиомерно чистые 8-(галометил)-1-алкил-7,8-дигидро-6H-тиено[3,2-e]индол-4-ол промежуточные соединения, из которых могут быть затем получены конечные тиено-индольные производные, соответствующие формуле (Ia) или (Ib). 3 н. и 11 з.п. ф-лы, 3 пр.
1. Способ получения тиено-индольных производных, соответствующих формуле (Ia) или (Ib)
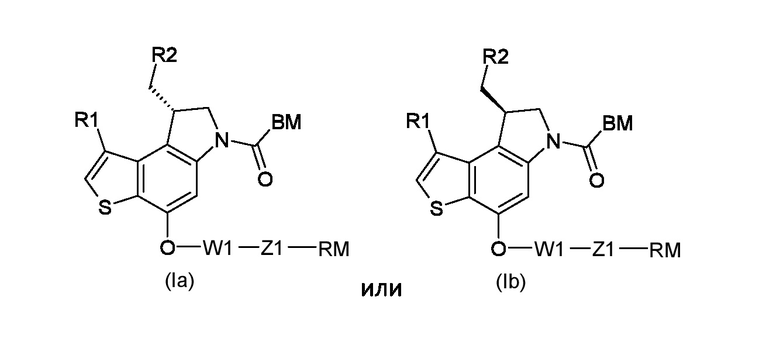
где:
R1 представляет собой водород или линейный или разветвленный С1-С6 алкил;
R2 представляет собой уходящую группу, выбранную из галогеновых или сульфонатных групп;
BM представляет собой ДНК-связывающий фрагмент, соответствующий формуле (II-1) или (II-2)
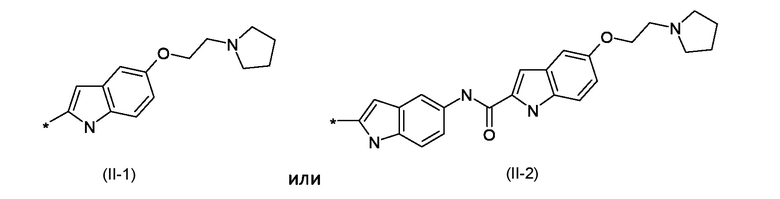
W1 представляет собой саморасщепляющую систему, соответствующую формуле (III)
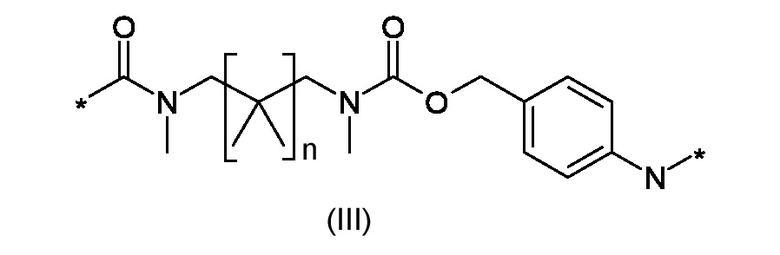
где n это 0 или 1;
Z1 представляет собой линкер, соответствующий формуле (IV-1) или (IV-2)
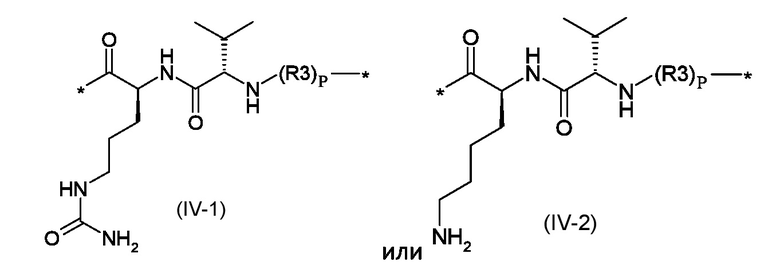
где p это число от 0 до 1, а R3 представляет собой полиоксиэтиленовую цепь, соответствующую формуле (V)
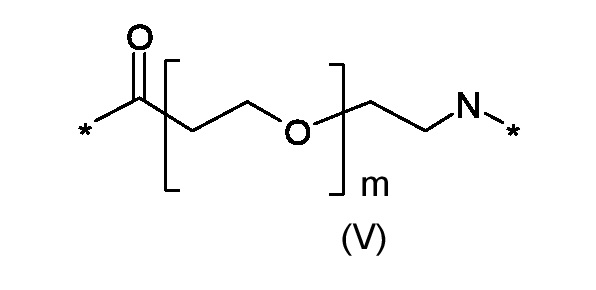
где m это число от 0 до 5; и
RM представляет собой фрагмент, соответствующий формуле (VI)
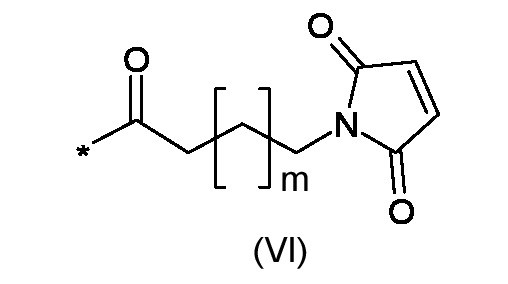
где m является таким, как указано выше;
который включает следующие стадии:
а) алкилирование соединения, представленного формулой (VII)
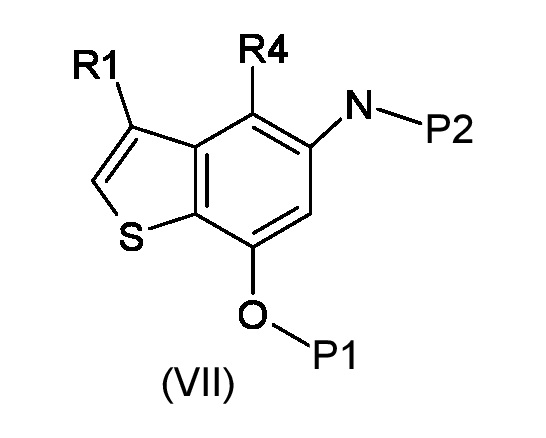
где R4 представляет собой галоген, P1 представляет собой гидроксизащитную группу, P2 представляет собой азотзащитную группу, а R1 является таким, как указано выше, (S)-глицидил 3-нозилатом (VIIIa) или (R)-глицидил 3-нозилатом (VIIIb)
b) реакция полученного соединения, соответствующего формуле (IXa) или (IXb)
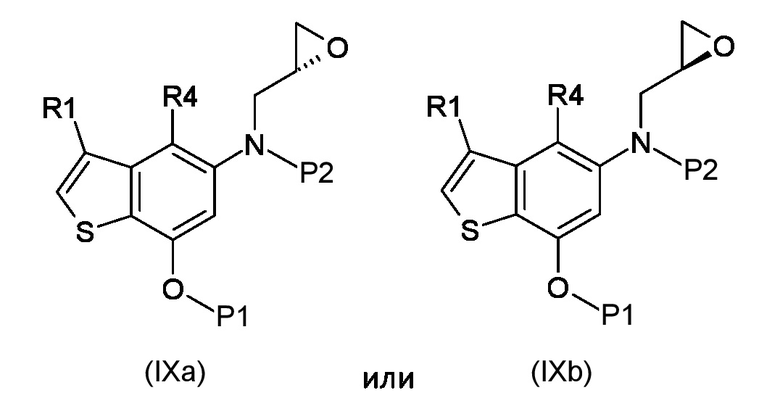
где R1, R4, P1 и P2 являются такими, как указано выше, с металлоорганическим реагентом, выбранным из группы, состоящей из: изо-PrMgCl·LiCl, сек-Bu2MgLi·Cl, MeMgBr, изо-PrMgBr и EtMgBr;
с) удаление гидроксизащитной группы P1 из полученного соединения, соответствующего формуле (Xa) или (Xb)
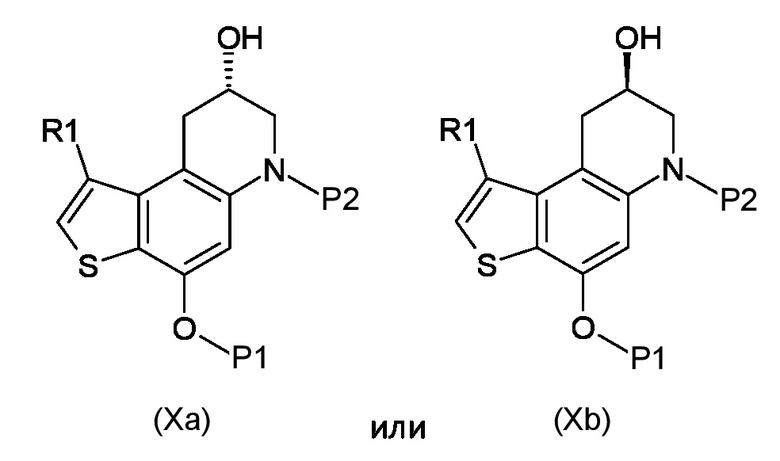
где R1, P1 и P2 являются такими, как указано выше;
d) осуществление внутримолекулярной спироциклизации полученного соединения, соответствующего формуле (XIa) или (XIb)
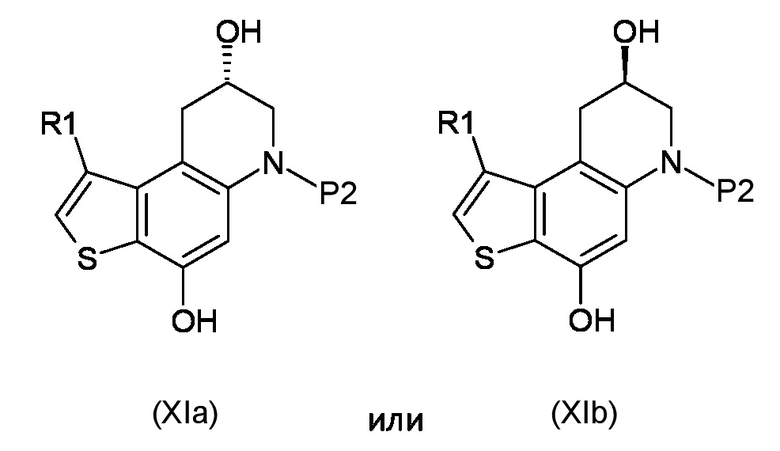
где R1 и P2 являются такими, как указано выше;
e) достижение стереоконтролируемого региоселективного циклопропанового раскрытия полученного соединения, соответствующего формуле (XIIa) или (XIIb)
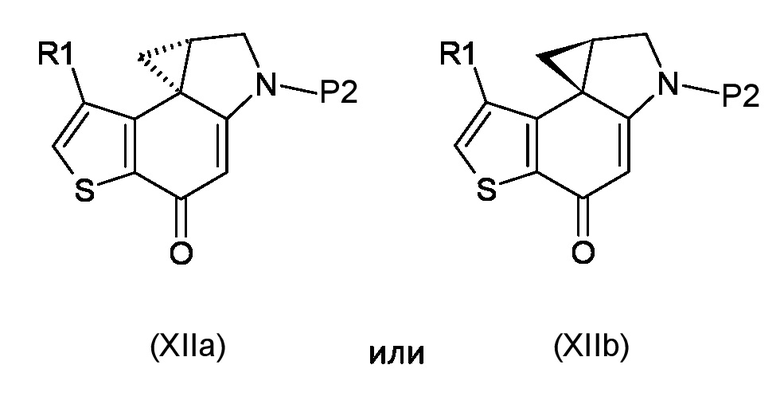
где R1 и P2 являются такими, как указано выше, реакцией с кислотой, которая имеет формулу HR2, где R2 является таким, как указано выше;
f) удаление азотзащитной группы P2 из полученного соединения, соответствующего формуле (XIIIa) или (XIIIb)
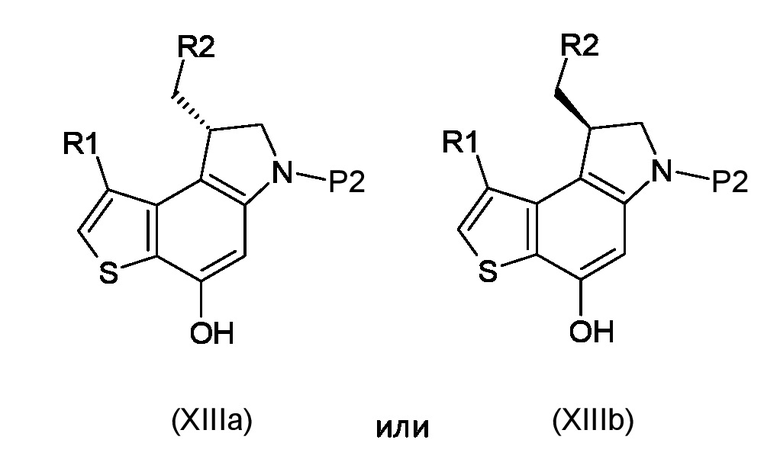
где R1, R2 и P2 являются такими, как указано выше, таким образом, чтобы получить ключевой энантиомерно чистый интермедиат 8-(галометил)-1-алкил-7,8-дигидро-6H-тиено[3,2-e]индол-4-ол интермедиат, соответствующий формуле (XIVa) или (XIVb), соответственно
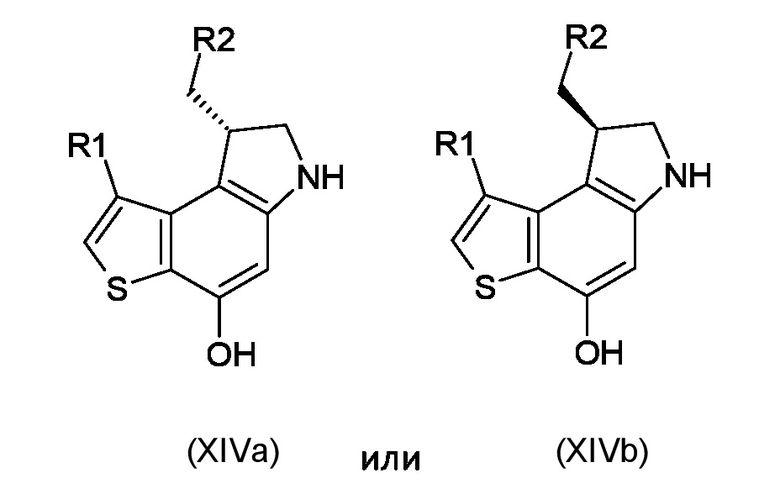
где R1 и R2 являются такими, как указано выше;
стадия g) сочетание промежуточного соединения, соответствующего формуле (XIVa) или (XIVb), с остатком кислоты BM-COOH, таким образом, чтобы получить промежуточное соединение, соответствующее формуле (XVa) или (XVb), соответственно
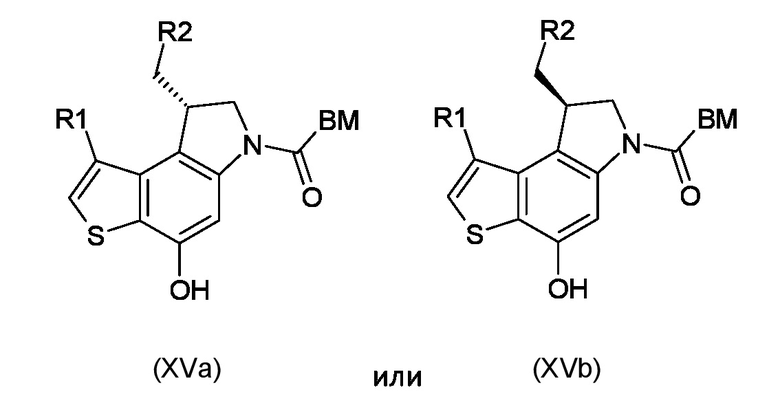
где BM, R1 и R2 являются такими, как указано выше;
стадия h) преобразование промежуточного соединения, соответствующего формуле (XVa) или (XVb), в карбонатные производные, соответствующие формуле (XVIa) или (XVIb), соответственно
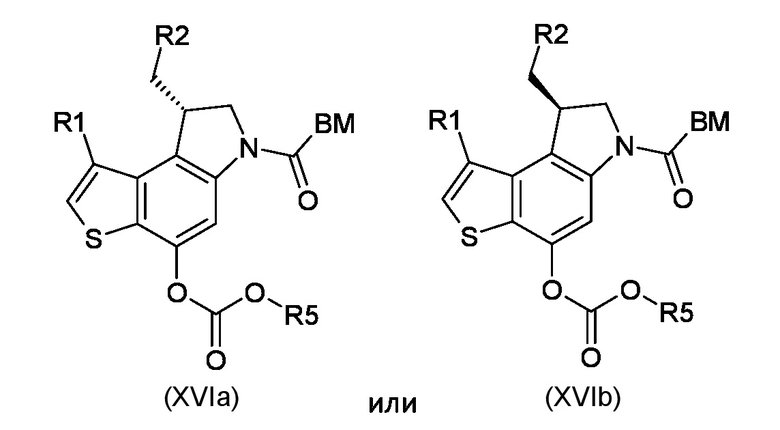
где BM, R1 и R2 являются такими, как указано в пункте 1, а R5 представляет собой сукцинимидильный или 4-нитро-фенильный остаток;
стадия i) взаимодействие полученного промежуточного соединения, соответствующего формуле (XVIa) или (XVIb), с амином, соответствующим формуле (XVII)
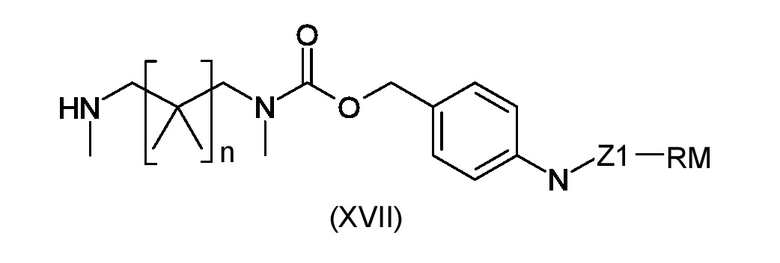
где n, Z1 и RM являются такими, как указано в пункте 1, таким образом, чтобы получить конечные соединения, соответствующие формуле (Ia) или (Ib).
2. Способ по п. 1, который дополнительно включает преобразование тиено-индольных производных, соответствующих формуле (Ia) или (Ib), как определено в п. 1, в фармацевтически приемлимые соли.
3. Способ по пп. 1, 2, для получения тиено-индольных производных, соответствующих формуле (Ia), как обозначено в п. 1.
4. Способ по пп. 1-3, для получения тиено-индольных производных, соответствующих формуле (Ia), где R1 представляет собой метил, а R2 хлор.
5. Способ по пп. 1-4, для получения тиено-индольных производных, соответствующих формуле (Ia), где BM представляет собой ДНК-связывающий фрагмент, соответствующий формуле (II-1), как определено в п. 1.
6. Способ по пп. 1-5, для получения следующих тиено-индолных производных, соответствующих формуле (Ia):
N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[{3-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)амино]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]-2,2-диметилпропил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 1a);
N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[{3-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]-2,2-диметилпропил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 2a);
N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[{2-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)амино]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 3a);
N-[19-(2,5-диокси-2,5-дигидро-1H-пиррол-1-ил)-17-оксо-4,7,10,13-тетраокса-16-азанонадекан-1-оил]-L-валил-N5-карбамоил-N-[4-({[{2-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)амино]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 4a);
N-[19-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-17-оксо-4,7,10,13-тетраоксо-16-азанонадекан-1-оил]-L-валил-N5-карбамоил-N-[4-({[{2-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-yl}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 5a) и
N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-[4-({[{2-[({[(8S)-8-(хлорметил)-1-метил-6-({5-[2-(пирролидин-1-ил)этокси]-1H-индол-2-ил}карбонил)-7,8-дигидро-6H-тиено[3,2-e]индол-4-ил]окси}карбонил)(метил)амино]этил}(метил)карбамоил]окси}метил)фенил]-L-орнитинамид (соединение 6a).
7. Промежуточное соединение, соответствующее формуле (XIa) или (XIb)
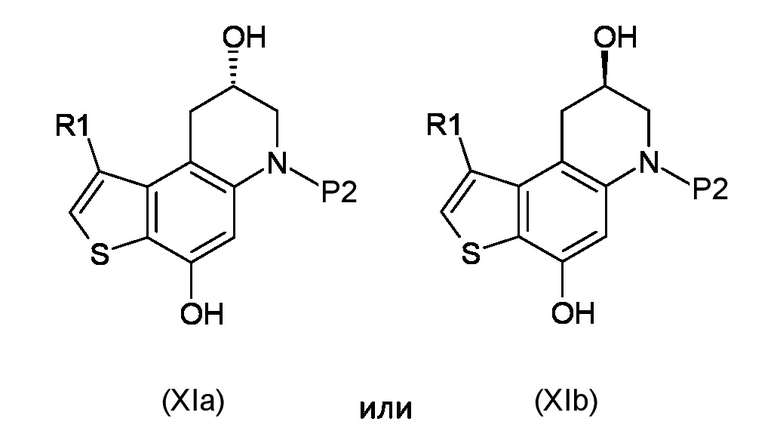
где R1 и P2 являются такими, как указано в п. 1.
8. Промежуточное соединение по п. 7, где R1 является метилом, а P2 трет-бутокси карбонилом.
9. Способ получения соединения, соответствующего формуле (XIa) или (XIb), как определено в п. 7, который содержит следующие стадии:
a) N-алкилирование соединения, соответствующего формуле (VII), как определено в п. 1, энантиочистым соединением, соответствующим формуле (VIIIa) или (VIIIb), для получения соответствующих соединений, соответствующих формуле (IXa) или (IXb), с применением сильно основных условий;
b) внутримолекулярная 6-эндо-тет циклизация полученного соединения, соответствующего формуле (IXa) или (IXb)
где R1, R4, P1 и P2 являются такими, как определено в п. 1, с металлоорганическими реагентами, выбранными из группы, состоящей из: изо-PrMgCl·LiCl, сек-Bu2MgLi·Cl, MeMgBr, изо-PrMgBr и EtMgBr, и
с) селективное удаление гидроксизащитной группы P1, как определено в п. 1.
10. Способ по п. 1 или 9, где реакция соединения, соответствующего формуле (IXa) или (IXb), с металлоорганическим реагентом выполняется при температуре в диапазоне от -5°С до 50°С в органическом растворителе, выбранном из: THF, Et2O, DCM и их смеси.
11. Способ по п. 1, где стадия а) выполняется в сильно основных условиях при температуре в диапазоне от 10°С до 50°С, в органическом растворителе, выбранном из: THF, Et2O, DMA, DMF и их смеси.
12. Способ по п. 1, где стадия d) выполняется с фосфинами и азопроизводными соединениями при температуре в диапазоне от -10°C до 50°C, в органическом растворителе, выбранном из: THF, Et2O, DCM и их смеси.
13. Способ по п. 12, где фосфин представляет собой выбранный из Ph3P и Bu3P, а азопроизводные соединения представляют собой выбранные из: диизопропила, азодикарбоксилата и 1,1'-(азодикарбонил)-дипиперидина.
14. Способ по п. 1, где стадия е) выполняется в контролируемых кислых условиях посредством взаимодействия с кислотой формулы HR2, где R2 представляет собой галоген, при температуре в диапазоне от -80°C до 25°C, в органическом растворителе, выбранном из: EtOAc, Et2O DCM и их смеси.
| WO 2013149948 A1, 10.10.2013 | |||
| ХИМИЧЕСКИЕ ЛИНКЕРЫ И ИХ КОНЪЮГАТЫ | 2005 |
|
RU2402548C2 |
| Uematsu, M., & Boger, D | |||
| L | |||
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Asymmetric Synthesis of a CBI-Based Cyclic N-Acyl O-Amino Phenol Duocarmycin Prodrug | |||
| The Journal of Organic Chemistry, 79(20), 9699-9703 | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Tichenor, M | |||
| S., MacMillan, K | |||
| S., Stover, J | |||
| S., Wolkenberg, S | |||
| E., Pavani, M | |||
| G., Zanella, | |||

