Настоящее изобретение относится к пара-аминобензил-линкерным соединениям, пригодным для связывания частей лекарственного средства с антителами, к соединениям линкер-лекарственное средство, в которых указанные пара-аминобензил-линкерные соединения ковалентно связаны с частями лекарственного средства, и к конъюгатам антитело-лекарственное средство, содержащим указанные пара-аминобензил-линкерные соединения, ковалентно связанные с лекарственным средством, при этом указанное лекарственное средство ферментативно отщепляется от конъюгата в конкретном типе клеток или тканей, являющихся мишенями для указанного антитела.
Предпосылки создания изобретения
Таргетная терапия предназначена для снижения неспецифической токсичности и повышения эффективности по сравнению с традиционной терапией. Этот подход воплощается в мощной нацеливающей способности моноклональных антител или антигенсвязывающих фрагментов специфически доставлять сильнодействующие конъюгированные низкомолекулярные терапевтические средства к антиген-положительным клеткам. Чтобы решить проблему токсичности, химиотерапевтические средства (лекарственные средства)были соединены с молекулами-мишенями, такими как антитела или лиганды белковых рецепторов, которые с высокой степенью специфичности связываются с опухолевыми клетками с образованием соединений, называемых конъюгатами антитело-лекарственное средство (ADC) или иммуноконъюгатами (Chau и соавт. Lancet 2019, 394, 793-804). Теоретически ADC или иммуноконъюгаты должны быть менее токсичными, потому что они направляют цитотоксическое лекарственное средство на больные клетки, которые сверхэкспрессируют конкретный антиген или рецептор клеточной поверхности по сравнению с нормальными клетками. Многообещающие достижения с ADC или иммуноконъюгатами показали, что цитотоксические лекарственные средства связаны с антителами через линкер, который расщепляется в месте опухоли или внутри опухолевых клеток. Такие достижения могут быть применены к любым заболеваниям, требующим адресной доставки (Tumey, Innovations for Next-Generation Antibody-Drug Conjugates. Humana Press 2018, 187-214). Недавние примеры также демонстрируют интерес к иммуноопосредованным воспалительным заболеваниям (Wang и соавт. J Am Chem Soc. 2015, 137, 3229-32), заболеваниям, связанным с мышцами (Sugo и соавт. J. Control. Release 2016, 237, 1-13) и заболевания, вызванные бактериальной инфекцией (Mariathasan и соавт.Trends Mol. Med. 2017, 23, 135-149). Более того, ADC не ограничиваются цитотоксическими лекарственными средствами и могут быть использованы для доставки специфических визуализирующих средств (Dammes и соавт. Theranostics 2020, 10, 938-955) или олигонуклеотидов (Dovgan и соавт. Bioconjug. Chem. 2019, 30, 2483-2501) к антиген-положительным клеткам.
Линкеры могут влиять на физико-химические свойства ADC. Поскольку многие цитотоксические средства гидрофобны по своей природе, их связывание с антителом с помощью дополнительного гидрофобного фрагмента может привести к агрегации. Агрегаты ADC нерастворимы и часто ограничивают достижимую нагрузку лекарственного средства на антитело, что может негативно повлиять на активность ADC. Белковые агрегаты биологических препаратов в целом также связаны с повышенной иммуногенностью.
Химический раствор для адресной доставки лекарственных средств, например, цитотоксических или цитостатических лекарственных средств, конъюгированных с клеточно-специфическими лигандами, представляет собой «саморасщепляющийся линкер», РАВС или РАВ (пара-аминобензилоксикарбонил или пара-аминобензил), присоединяющий фрагмент лекарственного средства к лиганду в конъюгате (Alouane и соавт. Angew. Chem. Int. Ed. 2015, 54, 7492-7509; Bargh и соавт. Chem. Soc. Rev. 2019, 48, 4361-4374). Линкерная единица РАВ также упоминается как электронный каскадный спейсер. Амидная связь, соединяющая карбокси-конец пептидной единицы и пара-аминобензил РАВС или РАВ, может быть субстратом и расщепляться некоторыми протеазами. Ароматический амин становится донором электронов и инициирует электронный каскад, который приводит к вытеснению уходящей группы, которая высвобождает свободное лекарственного средство после удаления диоксида углерода (de Groot и соавт. J. Org. Chem. 2001, 66, 8815-8830). При расщеплении пептидной связи, примыкающей к РАВС или РАВ, то есть внутриклеточным ферментом, лекарственное средство высвобождается из лиганда, при этом никакая оставшаяся часть линкера не связывается (de Groot и соавт. Molecular Cancer Therapeutics 2002, 1, 901-911; de Groot и соавт. J. Med. Chem. 1999, 42, 5277-5283).
Линкеры, содержащие пара-аминобензильную или пара-аминобензилоксикарбонильную (РАВ или РАВС) единицу в сочетании с пептидной единицей, были разработаны с «саморасщепляемым» или «саморасщепляющимся» механизмом 1,6-элиминирования и фрагментации ферментативных, гидролитических или других метаболических условиях для высвобождения части лекарственного средства из нацеливающего лиганда, такого как антитело (ЕР 1718667; WO 2016/040684). Для использования единицы РАВ в пролекарственных средствах и конъюгатах см. также: Alouane и соавт. Angew. Chem. Int. Ed. 2015, 54, 7492-7509; Bargh и соавт. Chem. Soc. Rev. 2019, 48, 4361-4374; Dal Corso и соавт.Chem. Eur. J. 2019, 25,14740-14757.
Ограничения саморасщепляющихся линкеров типа РАВ заключаются в склонности вызывать плохую растворимость и агрегацию конъюгатов. Например, в заявке WO 2017/214282, саморасщепляющиеся линкеры типа РАВ, содержащие несколько гидрофильных групп (таких как глюкорониды, полигидроксилированные боковые цепи …), приводят к высокой агрегации конъюгатов (>15%). Кроме того, некоторые содержащие РАВ конъюгаты могут быть неподходящими субстратами для определенных расщепляющих ферментов или расщепляются слишком медленно для достижения эффективности. Следовательно, было бы желательно улучшить свойства конъюгатов антитело-лекарственное средство путем оптимизации структуры саморасщепляющегося линкера для уменьшения агрегации при сохранении фармакологической активности и стабильности. Неожиданно оказалось, что эти новые пара-аминобензильные линкеры, содержащие пара-аминобензильную единицу и пептидный элемент, как раскрыто в настоящей заявке, устраняют вышеупомянутые недостатки, и их использование приводит к ADC с низкими уровнями агрегации, хорошей стабильностью, желаемыми уровнями содержания лекарственного средства и ожидаемой фармакологической активностью, как показано в следующих примерах.
Краткое описание фигур
На фигуре 1 показана сайт-специфическая конъюгация антител с использованием бактериальной трансглютаминазы (LAR2).
На фигуре 2 показана конъюгация с использованием 4 межцепочечных дисульфидных мостиков антитела.
На фигуре 3 показана конъюгация с использованием технологии повторного образования мостиков, порождающего фрагментацию.
Определения
Термин «антитело» или «Ab» используют в самом широком смысле для обозначения молекулы иммуноглобулина, которая распознает и специфически связывается с мишенью, такой как белок, полипептид, углевод, полинуклеотид, липид или их комбинации через по меньшей мере один участок распознавания антигена в пределах вариабельной области молекулы иммуноглобулина. Антитело может быть поликлональным или моноклональным, многоцепочечным или одноцепочечным, или интактным иммуноглобулином, и может быть получено из природных источников или из рекомбинантных источников. «Интактное» антитело представляет собой гликопротеин, который обычно содержит по меньшей мере две тяжелые (Н) цепи и две легкие (L) цепи, соединенные между собой дисульфидными связями. Каждая тяжелая цепь состоит из вариабельной области тяжелой цепи (обозначаемой в настоящей заявке как VH) и константной области тяжелой цепи. Константная область тяжелой цепи включает в себя три домена, CH1, СН2 и CH3. Каждая легкая цепь состоит из вариабельной области легкой цепи (обозначаемой в настоящей заявке как VL) и константной области легкой цепи. Константная область легкой цепи состоит из одного домена, CL. Области VH и VL могут быть дополнительно подразделены на области гипервариабельности, называемые определяющими комплементарность областями (CDR), перемежающиеся с более консервативными областями, называемыми каркасными областями (FR). Каждая VH и VL состоит из трех CDR и четырех FR, расположенных от амино-конца к карбоксильному концу в следующем порядке: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. Вариабельные области тяжелой и легкой цепей содержат связывающий домен, взаимодействующий с антигеном. Константные области антител могут опосредовать связывание иммуноглобулина с тканями или факторами хозяина, включая различные клетки иммунной системы (например, эффекторные клетки) и первый компонент (C1q) классической системы комплемента. Антитело может быть моноклональным антителом, человеческим антителом, гуманизированным антителом, верблюжьим антителом или химерным антителом. Антитела могут относиться к любому изотипу (например, IgG, IgE, IgM, IgD, IgA и IgY), классу (например, IgG1, IgG2, IgG3, IgG4, IgA1 и IgA2) или подклассу. Антитело может быть интактным антителом или его антигенсвязывающим фрагментом.
Термин «фрагмент антитела» или «антигенсвязывающий фрагмент» или «фрагмент», используемый в настоящей заявке, относится по меньшей мере к одной части антитела, которая сохраняет способность специфически взаимодействовать с (например, путем связывания, стерических затруднений, стабилизации/дестабилизации, пространственного распределения)эпитопом антигена. Антигенсвязывающие фрагменты также могут сохранять способность интернализоваться в антигенэкспрессирующую клетку. В некоторых вариантах осуществления антигенсвязывающие фрагменты также сохраняют иммунную эффекторную активность. Термины «антитело», «фрагмент антитела», «антигенсвязывающий фрагмент» и т.п.предназначены для охвата использования связывающих доменов из антител в контексте более крупных макромолекул, таких как ADC. Было показано, что фрагменты полноразмерного антитела могут выполнять антигенсвязывающую функцию полноразмерного антитела. Примеры фрагментов антител включают в себя, но не ограничиваются ими, фрагменты Fab, Fab', F(ab')2, Fv, фрагменты антител scFv, связанные дисульфидной связью Fvs (sdFv), фрагмент Fd, состоящий из доменов VH и СН1, линейные антитела, однодоменные антитела, такие как sdAb (или VL, или VH), домены VHH верблюдовых, мультиспецифические антитела, образованные из фрагментов антител, таких как бивалентный фрагмент, содержащий два фрагмента Fab, связанных дисульфидным мостиком в шарнирной области, и изолированный CDR или другие связывающие эпитоп фрагменты антитела. Антигенсвязывающий фрагмент также может быть включен в однодоменные антитела, макситела, минитела, нанотела, интратела, диатела, триантела, тетратела, конструкции биспецифических или мультиспецифических антител, ADC, v-NAR и бис-scFv (см., например, Holliger and Hudson Nat. Biotechnol. 2005, 23, 1126-1136). Антигенсвязывающие фрагменты также можно прививать в каркасы на основе полипептидов, таких как фибронектин типа III (Fn3) (см. патент США №6,703,199, в котором описаны минитела полипептида фибронектина).
Термин «scFv» относится к слитому белку, содержащему по меньшей мере один антигенсвязывающий фрагмент, содержащий вариабельную область легкой цепи, и по меньшей мере один антигенсвязывающий фрагмент, содержащий вариабельную область тяжелой цепи, при этом вариабельные области легкой и тяжелой цепей соединены непрерывно, например, посредством синтетического линкера, например, короткого гибкого полипептидного линкера, и способны экспрессироваться в виде одноцепочечного полипептида, и где scFv сохраняет специфичность интактного антитела из которого оно получено. Если не указано иное, scFv может иметь вариабельные области VL и VH в любом порядке, например, относительно N-концевого и С-концевого концов полипептида, scFv может содержать VL-линкер-VH или может содержать VH-линкер-VL. Антигенсвязывающие фрагменты получают с использованием обычных способов, известных специалистам в данной области, и связывающие фрагменты проверяют на применимость (например, аффинность связывания, интернализацию) таким же образом, как и интактные антитела. Антигенсвязывающие фрагменты, например, могут быть получены расщеплением интактного белка, например, протеазным или химическим расщеплением.
Термин «лекарственное средство» или «средство», или «терапевтическое лекарственное средство» или «терапевтическое средство», используемый в настоящей заявке, относится к химическому соединению, смеси химических соединений, биологической макромолекуле, экстракту, полученному из биологических материалов, или комбинации из двух или более из них, которые способны модулировать биологический процесс и/или обладают биологической активностью. В частности, термин «лекарственное средство» может представлять собой «химиотерапевтическое средство» или «противораковое средство».
Термин «химиотерапевтическое средство» или «противораковое средство» используют в настоящей заявке для обозначения всех средств, которые эффективны при лечении рака (независимо от механизма действия). Ингибирование метастазирования или ангиогенеза часто является свойством химиотерапевтического средства. К химиотерапевтическим средствам относят антитела, биологические молекулы и малые молекулы. Химиотерапевтическое средство может быть цитотоксическим или цитостатическим средством. Примеры химиотерапевтического средства включают в себя, без каких-либо ограничений, монометилауристатин Е, ауристатин Е, SN-38, доксорубицин и т.д.
Термин «цитостатическое средство» относится к средству, которое ингибирует или подавляет клеточный рост и/или размножение клеток.
Термин «цитотоксическое средство» относится к веществу, которое вызывает гибель клеток, главным образом, путем вмешательства в экспрессионную активность и/или функционирование клетки.
Используемый в настоящей заявке термин «галоген(С1-С4)алкил» означает (С1-С4)алкильный радикал, который замещен одним или несколькими атомами галогена, такими как фтор, хлор, бром или йод, включая, но не ограничиваясь ими, фторметил, фторметил, дифторметил, трифторметил, 2-фторэтил, 2,2-дифторэтил, 2,2,2-трифторэтил, 2,2,2-трифтор-1,1-диметилэтил, 2,2,2-трихлорэтил, 3-фторпропил, 4-фторбутил, хлорметил, трихлорметил, йодометил и бромметил.
Термин «защитная группа» относится к заместителю, который обычно используют для блокирования или защиты конкретной функциональной группы при взаимодействии с другими функциональными группами соединения. Например, «аминозащитная группа» представляет собой заместитель, присоединенный к аминогруппе, который блокирует или защищает функциональную аминогруппу в соединении. Подходящие аминозащитные группы включают в себя ацетил, трифторацетил, m-бутоксикарбонил (Boc), бензилоксикарбонил (CBz) и 9-флуоренилметиленоксикарбонил (Fmoc). Аналогично, «гидроксизащитная группа» относится к заместителю гидроксильной группы, который блокирует или защищает гидроксильную функциональность. Подходящие гидроксизащитные группы включают ацетил, бензил, бензоил, тетрагидропиранил и триалкилсилил. Кроме того, «карбонилзащитная группа» относится к заместителю карбонильной группы, который блокирует или защищает карбонильную функциональность. Подходящие карбонилзащитные группы включают в себя ацеталь, ацилаль и дитианы. Подобным образом, «карбоксильная защитная группа» относится к заместителю карбоксильной группы, который блокирует или защищает карбоксильную функциональность. Подходящие карбоксильные защитные группы включают в себя метиловый эфир, бензиловый эфир, трет-бутиловый эфир, силиловый эфир и ортоэфир. Общее описание защитных групп и их использование см. в Т. W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
Используемый в настоящей заявке термин «фрагмент» означает определенный сегмент, фрагмент или функциональную группу молекулы или соединения. Химические фрагменты иногда обозначаются как химические структурные элементы, которые встроены или присоединены (например, заместитель или вариабельная группа) к молекуле, соединению или химической формуле.
Термин «мостиковый спейсер», используемый в настоящей заявке, относится к одному или нескольким линкерным компонентам, которые ковалентно присоединены друг к другу с образованием двухвалентного фрагмента, который связывает двухвалентный пептид А1-А2 с реакционноспособной группой. В некоторых вариантах осуществления «мостиковый спейсер» содержит карбонильную группу, присоединенную к N-концу группы А2 через амидную связь. В некоторых вариантах осуществления «мостиковый спейсер» содержит аминогруппу, присоединенную к карбонильной группе группы А2 через амидную связь. В некоторых вариантах осуществления мостиковый спейсер содержит группу полиоксиэтилена (ПЭГ). В других вариантах осуществления мостиковый спейсер содержит бутаноильную, пентаноильную, гексаноильную, гептаноильную или октаноильную группу. В некоторых вариантах осуществления мостиковый спейсер содержит гексаноильную группу (т.е. группу -СО-(СН2)5-). В предпочтительных вариантах осуществления мостиковый спейсер может содержать: -CO-СН2-СН2-PEG1-, -CO-СН2-PEG3- или -NH-СН2-СН2-PEG1-.
Термин «полиоксиэтилен», «полиэтиленгликоль» или «ПЭГ», используемый в настоящей заявке, относится к конфигурации с линейной цепью, разветвленной цепью или звездообразной конфигурации, состоящей из групп (ОСН2СН2). В некоторых вариантах осуществления группа полиоксиэтилена или ПЭГ представляет собой *-(OCH2CH2)t-**, где t равно 1-20, и где «*» указывает конец, направленный к группе А2-А1, а «**» указывает точку присоединения к реакционноспособной группе. В некоторых вариантах осуществления, мостиковый спейсер содержит группу ПЭГ, которая представляет собой *-(ОСН2СН2)t-**, где t равно 1-20, и где «*» указывает точку присоединения к группе -(СН2)n-С(O)-, в которой карбонильная группа присоединена к N-концу группы А2, а n равно 1-5, и «**» указывает точку присоединения к реакционноспособной группе. В некоторых вариантах осуществления мостиковый спейсер содержит группу ПЭГ, которая представляет собой *-(OCH2CH2)t-**, где t равно 1-20, и где «*» указывает точку присоединения к группе -(СН2)n-NH-, в которой аминогруппа присоединена к карбонильной группе группы А2 и n равно 1-5, а «**» указывает точку присоединения к реакционноспособной группе. Например, термин «ПЭГ3», используемый в настоящей заявке, означает, что t равно 3.
Термин «реакционноспособная группа», используемый в настоящей заявке, представляет собой функциональную группу, способную образовывать ковалентную связь с функциональной группой антитела или фрагмента антитела. В некоторых вариантах осуществления реакционноспособную группу выбирают, но не ограничиваются ими, из тиола, малеимида, диброммалеимида, галогенацетамида, азида, алкина, циклооктена, триарилфосфина, оксаноборнадиена, циклооктина, диарилтетразина, моноарилтетразина, норборнена, альдегида, гидроксиламина, гидразина, NH2-NH-C(=O)-, кетона, винилсульфона, азиридина, амино, аминокислотного остатка. В предпочтительном варианте осуществления реакционноспособную группу выбирают из малеимидной группы, диброммалеимидной группы, тиольной группы, циклооктиновой группы или азидогруппы, более предпочтительно из малеимидной группы, диброммалеимидной группы или азидогруппы. Например, малеимидные или диброммалеимидные группы могут иметь структуру:
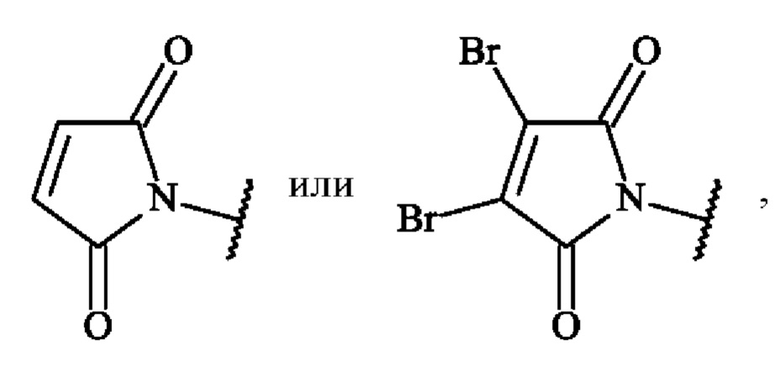 соответственно
соответственно
Например, азидогруппа может иметь структуру: -N=N+=N-.
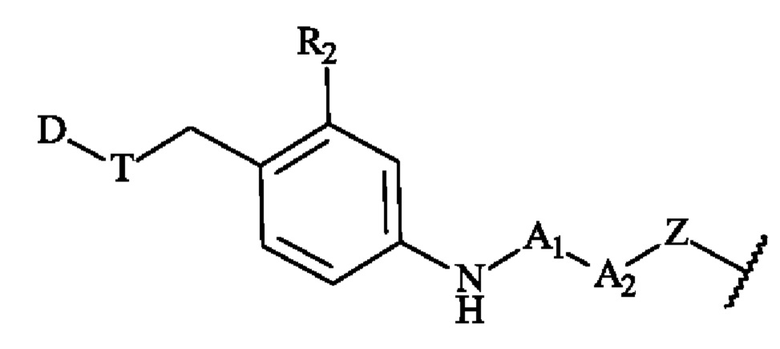
Термин «группа присоединения», используемый в настоящей заявке, относится к двухвалентной или трехвалентной части, которая связывает мостиковый спейсер с антителом или его фрагментом. Группа присоединения представляет собой двухвалентную или трехвалентную часть, образованную реакцией между реакционноспособной группой и функциональной группой на антителе или его фрагменте. В некоторых вариантах осуществления реакционноспособную группу для образования группы присоединения выбирают, но не ограничивается ими, из тиола, малеимида, диброммалеимида, галогенацетамида, азида, алкина, циклооктена, триарилфосфина, оксаноборнадиена, циклооктина, диарилтетразина, моноарилтетразина, норборнена, альдегида, гидроксиламина, гидразина, NH2-NH-C(=O)-, кетона, винилсульфона, азиридина, амино, аминокислотного остатка. В предпочтительном варианте осуществления реакционноспособную группу для образования группы присоединения выбирают из малеимидной группы, диброммалеимидной группы, тиольной группы, циклооктиновой группы или азидогруппы, более предпочтительно из малеимидной группы, диброммалеимидной группы или азидогруппы.
Термин «предшественник спейсерного звена» или «предшественник спейсерного звена Z'» или «Z'», используемый в настоящей заявке, относится к компоненту, содержащему мостиковый спейсер и реакционноспособную группу.
Термин «спейсерное звено» или «спейсерное звено Z» или «Z», используемый в настоящей заявке, относится к компоненту, содержащему мостиковый спейсер и группу присоединения.
Термин «конъюгат» или «конъюгат антитело-лекарственное средство», или «ADC» или «иммуноконъюгат», используемый в настоящей заявке, относится к антителу, которое ковалентно присоединено к части лекарственного средства через пара-аминобензильный линкер формулы (I). В одном варианте осуществления конъюгат антитело-лекарственное средство формулы (III), где спейсерное звено Z связано с антителом Ab через амидную группу, может быть получен путем взаимодействия свободной функциональной аминогруппы на антителе Ab с активным сложным эфиром, содержащим предшественник спейсерного звена Z.
Например, карбоксильная группа на предшественнике спейсерного звена Z' может быть активирована путем взаимодействия с N-гидроксисукцинимидом, а затем взаимодействия с Ab-NH2 с образованием конъюгата, в котором Ab и Z связаны посредством амидной группы. Полезные функциональные группы на антителе для связывания со спейсерным звеном Z, или естественным путем или посредством химической обработки включают в себя, но не ограничиваются ими, сульфгидрильную (-SH), амино, гидроксильную, аномерную гидроксильную группу углевода и карбоксильную группу. В одном варианте осуществления реакционноспособные функциональные группы на нантителе представляют собой сульфгидрильную и аминогруппу. Сульфгидрильные группы могут образовываться путем восстановления внутримолекулярной цистеиндисульфидной связи антитела. Альтернативно, сульфгидрильные группы могут быть получены реакцией аминогруппы лизиновой части антитела с использованием 2-иминотиолана (реагент Траута) или другого реагента, образующего сульфгидрильные группы. В другом варианте осуществления спейсерное звено Z связано с антителом Ab дисульфидной связью между атомом серы антитела и атомом серы спейсерного звена Z. В еще одном варианте осуществления реакционноспособная группа спейсерного звена Z содержит реакционноспособный участок, который может образовывать связь с первичной или вторичной аминогруппой антитела. Примеры этих реакционноспособных участков включают в себя, но не ограничиваются ими, активированные сложные эфиры, такие как сложные эфиры сукцинимида, сложные эфиры 4-нитрофенила, сложные эфиры пентафторфенила, сложные эфиры тетрафторфенила, ангидриды, хлорангидриды, сульфонилхлориды, изоцианаты и изотиоцианаты. В еще одном варианте осуществления реакционноспособная группа спейсерного звена Z взаимодействует с альдегидной, ацетальной или кетальной группой на сахаре (углеводе) гликозилированного антитела. Например, углевод можно слегка окислить с использованием реагента, такого как периодат натрия, и полученную единицу (-СНО) окисленного углевода можно конденсировать с помощью спейсерного звена, которое содержит функциональные группы, такие как гидразид, оксим, первичный или вторичный амин, гидразин, тиосемикарбазон, карбоксилат гидразина и арилгидразид, такие как описанные у Kaneko и соавт. Bioconjugate Chem. 1991, 2, 133-141.
Генерация ADC может быть выполнена способами, известными специалистам в данной области техники. В типичном варианте осуществления изобретение относится к ADC формулы (III), полученному контактированием соединения линкер-лекарственное средство формулы (II) с антителом (или его фрагментом), имеющим реакционноспособную сульфгидрильную, амино- или карбоксильную часть, в подходящих условиях для осуществления конденсации реакционноспособной части с частью предшественника спейсерного звена Z' соединения линкер-лекарственное средство формулы (II), где Z' превращается в Z в результате указанного контакта.
В некоторых вариантах осуществления ADC формулы (III) включает спейсерное звено Z, которое представляет собой двухвалентную часть, соединяющую N-конец группы А2-А1 с антителом. В некоторых вариантах осуществления ADC формулы (III) включает спейсерное звено Z, которое представляет собой двухвалентную часть, соединяющую карбонильную группу группы А2-А1 с антителом.
Спейсерное звено Z ковалентно связано с функциональной группой, отходящей от антитела, такой как амин (например, -NH2 из остатка Lys), карбоксильная (-СООН из остатка Asp или Glu) или сульфгидрильной (например, -SH из остатка Cys), которая образует амид или тиоэфирную или дисульфидную группу. Конъюгат антитело-лекарственное средство в соответствии с изобретением, в котором предшественник спейсерного звена Z' вступает в реакцию с сульфгидрильной функциональной группой антитела (например, содержащим Cys пептидом или восстановленным антителом)с образованием тиоэфирной связи, включает в себя конъюгат, представленный следующей формулой:
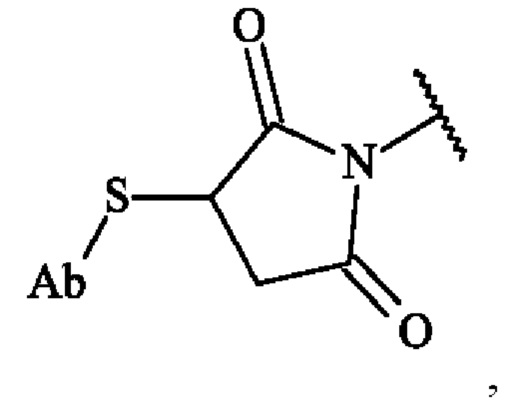
в которой Ab представляет собой антитело или его антигенсвязывающий фрагмент. В другом варианте осуществления конъюгат антитело-лекарственное средство в соответствии с изобретением, в котором предшественник спейсерного звена Z' вступает в реакцию с двумя сульфгидрильными функциональными группами антитела с образованием тиоэфирных связей, включает в себя конъюгат, представленный следующей формулой:
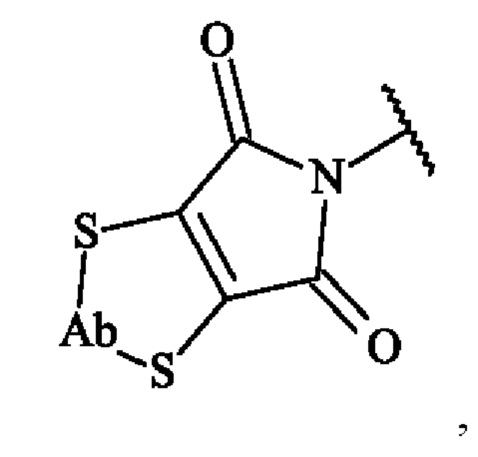
в которой Ab представляет собой антитело или его антигенсвязывающий фрагмент.
В некоторых вариантах осуществления антитело или его антигенсвязывающий фрагмент функционализированы для получения функциональной группы, которая вступает в реакцию с промежуточным соединением линкер-лекарственное средство. В некоторых вариантах осуществления антитело или его антигенсвязывающий фрагмент получают с бактериальной трансглютаминазой (BTG), которая представляет собой реакционноспособные глютамины, специфически функционализированные амином, содержащим циклооктиновую BCN (N-[(1R,8S,9s)-бицикло[6.1.0]нон-4-ин-9-илметилоксикарбонил]-1,8-диамино-3,6- диоксаоктан)часть. В некоторых вариантах осуществления ADC в соответствии с изобретением, в котором предшественник спейсерного звена Z' вступает в реакцию с глютаминовой функциональной группой антитела с образованием амидной связи, включает в себя конъюгат, представленный следующей формулой:
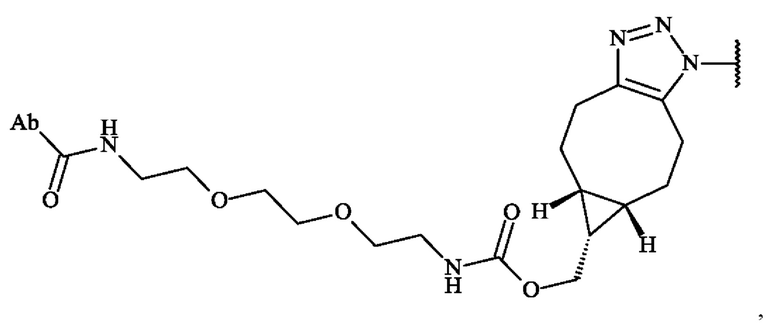
в которой Ab представляет собой антитело или его антигенсвязывающий фрагмент.
Термин «содержание лекарственного средства» представлен буквой р, и также упоминается в настоящей заявке как соотношение лекарственного средства к антителу (DAR или соотношение лекарственного средства: антитела). Содержание лекарственного средства может варьироваться от 1 до 8 частей лекарственного средства на антитело или антигенсвязывающий фрагмент. В некоторых вариантах осуществления более высокое содержание лекарственного средства (например, р>8) может вызывать агрегацию, нерастворимость, токсичность или потерю клеточной проницаемости определенных конъюгатов антитело-лекарственное средство. Более высокое содержание лекарственного средства может также негативно влиять на фармакокинетику (например, клиренс) некоторых ADC. В некоторых вариантах осуществления более низкое содержание лекарственного средства (например, р<2) может снижать действенность некоторых ADC против клеток, экспрессирующих мишени. В некоторых вариантах осуществления р представляет собой целое число от 2 до 8. В некоторых вариантах осуществления р равно 2, или р равно 4, или р равно 8.
Содержание лекарственного средства ADC можно контролировать различными способами, например, но не ограничиваясь этим, путем ограничения молярного избытка промежуточного соединения лекарственное средство-линкер или линкерного реагента по отношению к антителу или путем ограничения времени или температуры реакции конъюгации.
В некоторых вариантах осуществления содержание лекарственного средства в смеси ADC, полученной в результате реакции конъюгации, составляет от 1 до 8 частей лекарственного средства, присоединенных к одному антителу или антигенсвязывающему фрагменту. Среднее количество частей лекарственного средства на антитело или антигенсвязывающий фрагмент (т.е. среднее содержание лекарственного средства или среднее р) можно рассчитать любым общепринятым методом, известным в данной области, например, с помощью масс-спектрометрии (например, жидкостной хроматографии-масс-спектрометрии (ЖХ-МС)) и/или высокоэффективной жидкостной хроматографии (например, HIC-ВЭЖХ). В некоторых вариантах осуществления среднее количество частей лекарственного средства на антитело или антигенсвязывающий фрагмент составляет приблизительно от 1,5 до приблизительно 2,5, или от приблизительно 7,0 до приблизительно 8. В некоторых вариантах осуществления среднее количество частей лекарственного средства на антитело или антигенсвязывающий фрагмент составляет приблизительно 2. В некоторых вариантах осуществления среднее количество частей лекарственного средства на антитело или антигенсвязывающий фрагмент составляет 2. В некоторых вариантах осуществления среднее количество частей лекарственного средства на антитело или антигенсвязывающий фрагмент составляет приблизительно 4. В некоторых вариантах осуществления среднее количество частей лекарственного средства на антитело или антигенсвязывающий фрагмент составляет 4. В некоторых вариантах осуществления, среднее количество частей лекарственного средства на антитело или антигенсвязывающий фрагмент составляет приблизительно 8. В некоторых вариантах осуществления среднее количество частей лекарственного средства на антитело или антигенсвязывающий фрагмент составляет 8.
Подробное описание изобретения
Согласно первому аспекту изобретения предложено пара-аминобензил-линкерное соединение формулы (I):
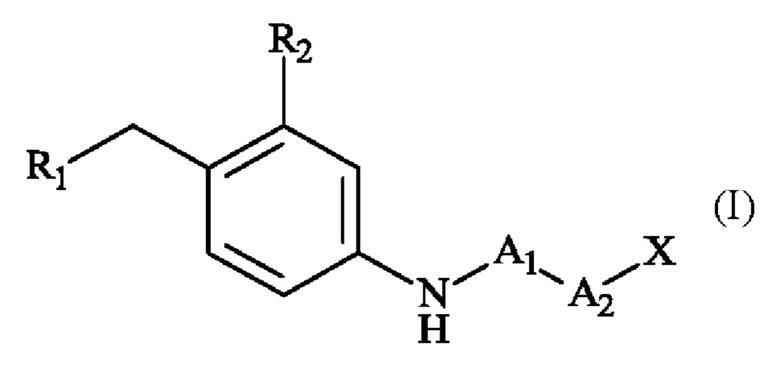
в которой:
• R1 представляет собой гидр оксигруппу или атом галогена,
• R2 представляет собой группу -S(O)2(OH), группу -S(O)2(O-М+), линейную или разветвленную группу - (С1-С4)алкил-S(O)2(ОН), линейную или разветвленную группу -(С1-С4)алкил-S(O)2(O-М+), линейную или разветвленную группу -галоген(С1-С4)алкил-S(O)2(ОН), или линейную или разветвленную группу -галоген(С1-С4)алкил-S(O)2(O-М+);
• A1 представляет собой группу -С(O)-СН(R3)-NH-;
• А2 представляет собой группу -C(O)-CH(R4)-NH-, группу 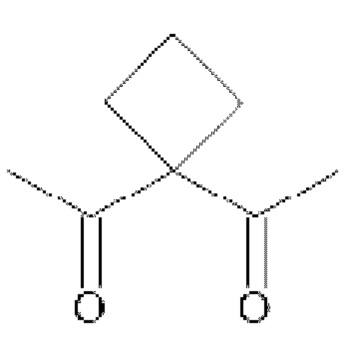 группу
группу 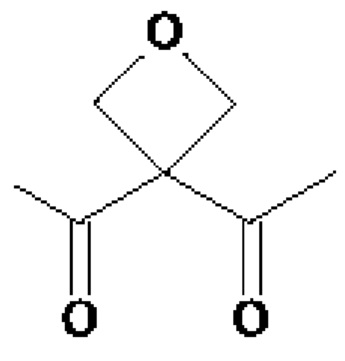 или группу
или группу 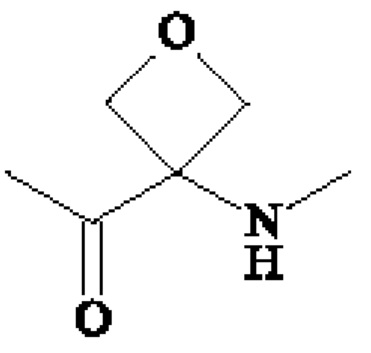
• R3 и R4, независимо друг от друга, представляют собой боковую цепь аминокислоты,
• X представляет собой атом водорода, гидроксигруппу или защитную группу,
• М+ представляет собой фармацевтически приемлемый одновалентный катион.
В предпочтительном варианте осуществления R1 представляет собой гидр окси группу, атом брома, атом хлора или атом йода. Более предпочтительно, R1 представляет собой гидр окси групп у, атом хлора или атом йода. Еще более предпочтительно R1 представляет собой гидроксигруппу.
Предпочтительно R2 представляет собой группу-S(O)2(ОН), группу -S(O)2(O-M+), группу -СН2-S(O)2(OH), группу -СН2-S(O)2(O-M+), группу -СН2-СН2-S(O)2(OH), группу -СН2-СН2-S(O)2(O-M+), группу -СН2-СН2-СН2-S(O)2(OH) или группу -СН2-СН2-СН2-S(O)2(O-M+). Примеры М+ включают в себя Li+, Na+ или К+. В предпочтительном варианте осуществления М+ представляет собой Na+.
В конкретном варианте осуществления A1 представляет собой группу -C(O)-CH(R3)-NH- и А2 представляет собой группу -C(O)-CH(R4)-NH-, у которой R3 и R4, независимо друг от друга представляют собой боковую цепь аминокислоты. Углерод, несущий боковую цепь, может иметь конфигурацию D или L (R или S). Предпочтительно углерод, несущий боковую цепь, имеет конфигурацию L. В конкретном варианте осуществления R3 и R4, независимо друг от друга, представляют собой, но не ограничиваются ими, водород, метил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, фенил, бензил, п-гидроксибензил,
-СН2ОН, -CH2SH, -CH2SeH -СН2СН2ОН, -CH2CH2SH, -СН(ОН)СН3, -CH2SCH3, -CH2CH2SCH3, -CH2CONH2, -СН2СООН, -CH2CH2CONH2, -СН2СН2СООН, -(СН2)3NHC(=NH)NH2, -(СН2)3NH2, -(СН2)3NHCOCH3, -(СН2)3NHCHO, -(СН2)4NHC(=NH)NH2, -(СН2)4NH2, -(СН2)4NHCO-(3-метил-3,4-дигидро-2Н-пиррол-2-ил), -(СН2)4NHCO-(3,4-дигидро-2H-пиррол-2-ил), -(СН2)4NHCOCH3, -(СН2)4NHCHO, -(СН2)3NHCONH2, -(СН2)4NHCONH2, -CH2CH2CH(OH)CH2NH2, 4-имидазолилметил, 3-индолилметил.
В предпочтительном варианте осуществления группа А1-А2 образует а дипептид, содержащий 2 аминокислотных звена, где указанные аминокислотные звенья выбраны из природных и неприродных аминокислот, предпочтительно природных аминокислот. Указанная аминокислотное звено может представлять собой аланин (Ala), цистеин (Cys), аспарагиновую кислоту (Asp), глютаминовую кислоту (Glu), фенилаланин (Phe), глицин (Gly), гистидин (His), изолейцин (Ile), лизин (Lys), лейцин (Leu), метионин (Met), аспарагин (Asn), пролин (Pro), глютамин (Gln), аргинин (Arg), серии (Ser), треонин (Thr), валин (Val), триптофан (Trp), тирозин (Туг), цитруллин (Cit), норвалин (Nva), норлейцин (Nle), селеноцистеин (Втор), пирролизин (Pyl), гомосерин, гомоцистеин и десметилпирролизин.
В одном варианте осуществления, когда A1 представляет собой аминокислотную единицу пролина (Pro), A1 определяется формулой:
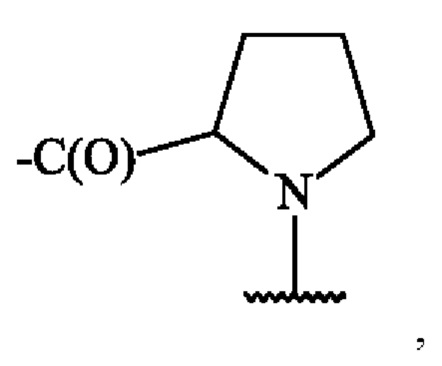
в которой карбонильная группа присоединена к аминогруппе пара-аминобензильной части, а волнистая линия указывает место ковалентного присоединения к карбонильной группе группы А2.
В одном варианте осуществления, когда А2 представляет собой аминокислотную единицу пролина (Pro), А2 определяется формулой:
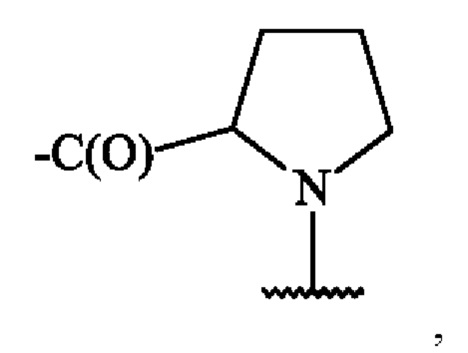
в которой карбонильная группа присоединена к аминогруппе группы A1, а волнистая линия указывает место ковалентного присоединения к группе X, группе Z' или группе Z, как определено в изобретении.
В конкретном варианте осуществления группа A1-A2 может представлять собой Val-Cit; Cit-Val; Ala-Ala; Ala-Cit; Cit-Ala; Asn-Cit; Cit-Asn; Cit-Cit; Val-Glu; Glu-Val; Ser-Cit; Cit-Ser; Lys-Cit; Cit-Lys; Asp-Cit; Cit-Asp; Ala-Val; Val-Ala; Phe-Ala; Ala-Phe; Phe-Lys; Lys-Phe; Val-Lys; Lys-Val; Ala-Lys; Lys-Ala; Phe-Cit; Cit-Phe; Leu-Cit; Cit-Leu; Ile-Cit; Cit-Ile; Phe-Arg; Arg-Phe; Cit-Trp; Trp-Cit. В предпочтительном варианте осуществления группа A1-A2 представляет собой Cit-Val или Ala-Val.
Предпочтительно A1 представляет собой группу -C(O)-CH(R3)-NH-, в которой R3 представляет собой группу -(СН2)3-NH-CO-NH2 или метальную группу.
Преимущественно А2 представляет собой группу -C(O)-CH(R4)-NH-, в которой R4 представляет собой изопропильную группу;
группу 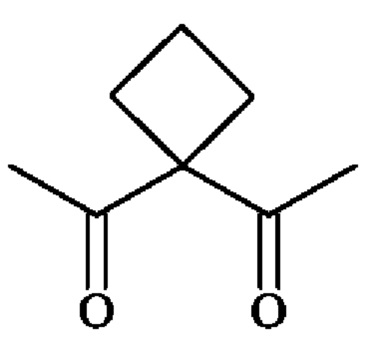 ; группу
; группу 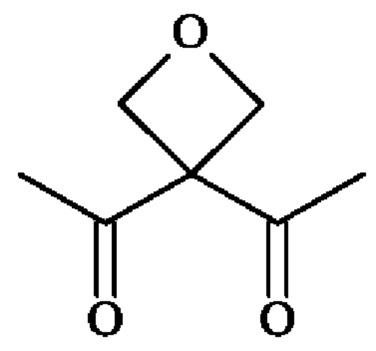 ; или группу
; или группу
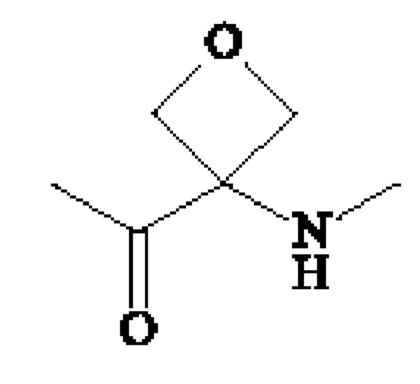
В предпочтительном варианте осуществления A1 представляет собой группу -С(O)-СН(R3)-NH-, в которой R3 представляет собой группу -(СН2)3-NH-CO-NH2 или метильную группу, и А2 представляет собой группу -С(O)-СН(R4)-NH-, в которой R4 представляет собой изопропильную группу.
В другом предпочтительном варианте осуществления A1 представляет собой группу -С(O)-СН(R3)-NH-, в которой R3 представляет собой группу -(СН2)3-NH-CO-NH2, и А2 представляет собой группу
 или группу
или группу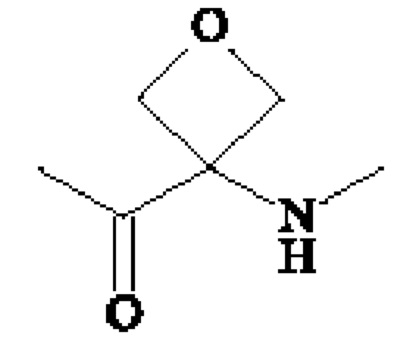
В одном варианте осуществления X представляет собой атом водорода или защитную группу. Предпочтительно X представляет собой атом водорода или защитную группу для аминофункциональности, карбонильной функциональности или карбоксильной функциональности. Более предпочтительно X представляет собой атом водорода или защитную группу, выбранную из Fmoc, Boc или CBz. Еще более предпочтительно, X представляет собой защитную группу Fmoc.
Предпочтительные пара-аминобензил-линкерные соединения формулы (I) представляют собой:
- 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-(гидроксиметил)бензолсульфонат натрия;
- 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]пропаноил]амино]-2-(гидрокеиметил)бензолсульфонат натрия;
- 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]пропаноил]амино]-2-(гидроксиметил)бензолсульфо новую кислоту;
- [5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-(гидроксиметил)фенил]метансульфонат натрия;
-2-(хлорметил)-5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]бензолсульфоновую кислоту;
-2-(хлорметил)-5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]пропаноил]амино]бензолсульфоновую кислоту;
- 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-(йодметил)бензолсульфоновую кислоту;
- N-{[(9H-флуорен-9-ил)метокси]карбонил}-1-валил-N5-карбамоил-N-[4-(гидроксиметил)-3-(2-сульфонатоэтил)фенил]-1-орнитинамид натрия;
- N-{[(9H-флуорен-9-ил)метокси]карбонил}-1-валил-N5-карбамоил-N-[4-(гидроксиметил)-3-(3-сульфопропил)фенил]-1-орнитинамид.
Изобретение также относится к способу получения пара-аминобензил-линкерных соединений формулы (I), отличающемуся тем, что в качестве исходного вещества используют соединение формулы (IV):
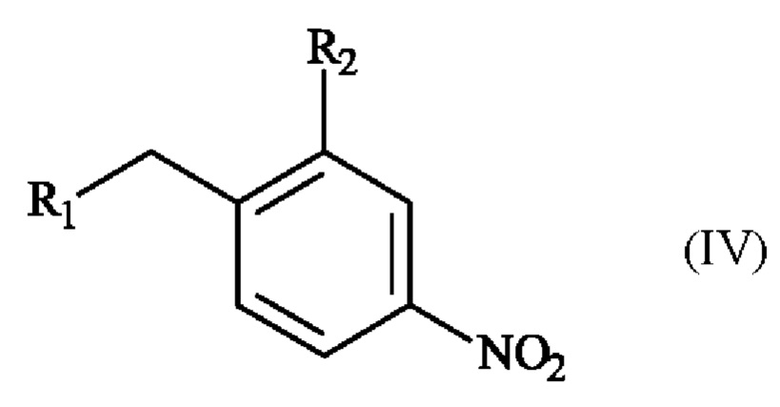
в которой R1 и R2 определены для формулы (I),
которое подвергают реакции восстановления с получением соединения формулы (V):
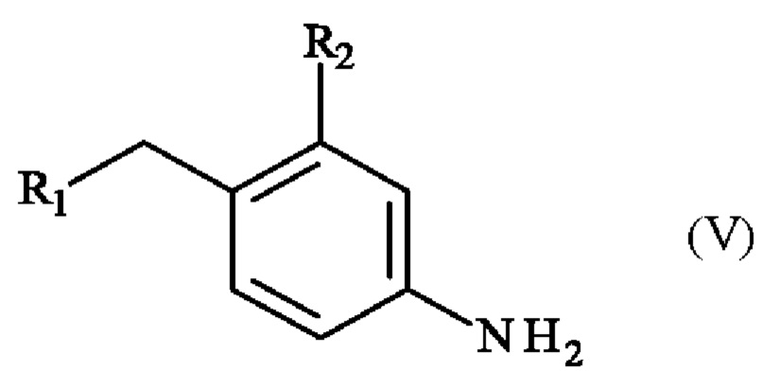
в которой R1 и R2 имеют определения, приведенные в настоящей заявке выше,
соединение формулы (V), которое далее подвергают сочетанию с соединением формулы (VI):
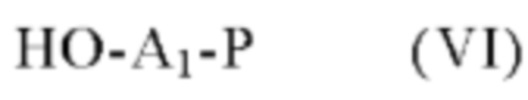
в которой Р представляет собой аминозащитную группу и A1 имеет определения, приведенные в формуле (I),
с получением соединения формулы (VII)
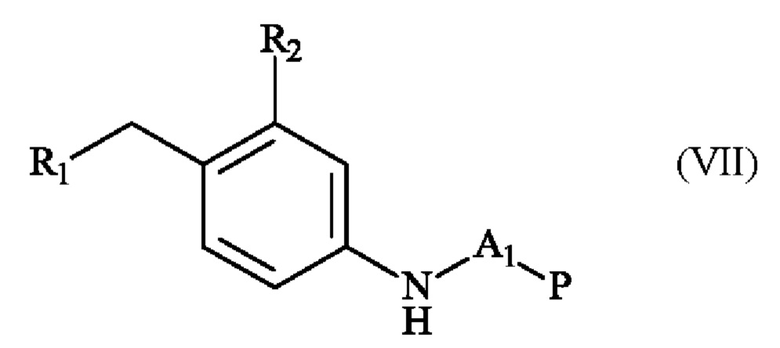
в которой A1, Р, R1 и R2 имеют определения, приведенные в настоящей заявке выше,
соединение формулы (VII), которое далее подвергают сочетанию с соединением формулы (VIII):
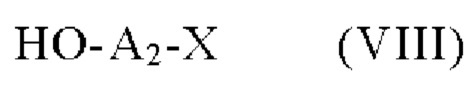
в которой X и А2 определены в формуле (I),
с получением пара-аминобензил-линкерного соединения формулы (I).
Альтернативный способ получения пара-аминобензил-линкерных соединений формулы (I)отличается тем, что в качестве исходного вещества используют соединение формулы (V), полученное из соединения формулы (IV), которое подвергают сочетанию с соединением формулы (IX):
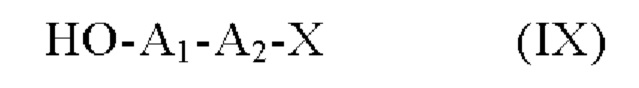
в которой A1, А2 и X определены в формуле (I),
с получением пара-аминобензил-линкерного соединения формулы (I).
В одном варианте осуществления пара-аминобензил-линкерное соединение формулы (I), полученное в конце вышеупомянутых способов, может быть дополнительно очищено в соответствии с общепринятой методикой разделения, которое при желании может быть превращено, в его аддитивные соли с фармацевтически приемлемой кислотой или основанием, которые могут быть разделены на их изомеры в соответствии с общепринятой методикой разделения, при этом следует понимать, что в любой момент, который считается подходящим в ходе описанного выше способа, некоторые группы (гидрокси, амино …) исходных реагентов или промежуточных продуктов синтеза могут быть защищены, впоследствии сняты защитные группы и функционализированы, как того требует синтез. Соединения формул (IV), (VI), (VIII) и (IX) или имеются в продаже, или могут быть получены специалистом в данной области с использованием обычных химических реакций, описанных в литературных источниках.
Изобретение также относится к применению пара-аминобензил-линкерных соединений формулы (I) в получении конъюгата антитело-лекарственное средство. Более конкретно изобретение относится к применению пара-аминобензил-линкерного соединения формулы (I), выбранного из:
- 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-(гидроксиметил)бензолсульфоната натрия,
- 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]пропаноил]амино]-2-(гидроксиметил)бензолсульфоната натрия,
- 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]пропаноил]амино]-2-(гидроксиметил)бензолсульфоновой кислоты,
- [5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-(гидроксиметил)фенил]метансульфоната натрия,
-2-(хлорметил)-5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]бензолсульфоновой кислоты или
-2-(хлорметил)-5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]пропаноил]амино]бензолсульфоновой кислоты,
- 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-(йодметил)бензолсульфоновой кислоты,
- N-{[(9H-флуорен-9-ил)метокси]карбонил}-1-валил-N5-карбамоил-N-[4-(гидроксиметил)-3-(2-сульфонатоэтил)фенил]-1-орнитинамид натрия или
- N-{[(9H-флуорен-9-ил)метокси]карбонил}-1-валил-N5-карбамоил-N-[4-(гидроксиметил)-3-(3-сульфопропил)фенил]-1-орнитинамида,
в получении конъюгата антитело-лекарственное средство. Во втором аспекте изобретение относится к соединению линкер-лекарственное средство формулы (II):
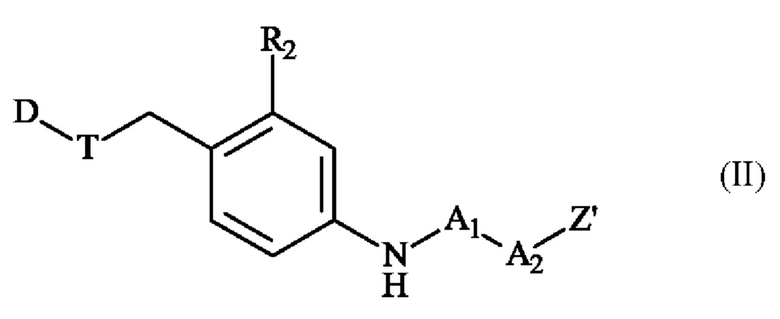
в которой:
• D представляет собой часть лекарственного средства;
• Т представляет собой связь, -O-C(O)-N(CH3)-СН2-СН2-N(CH3)-C(O)-*, -О-*, -NR5-*, -NR5-C(O)-* или -О-С(О)-*, в которой * указывает место присоединения к D;
• R2 представляет собой группу -S(O)2(OH), группу -S(O)2(O-M+), линейную или разветвленную группу -(С1-С4)алкил-S(O)2(ОН), линейную или разветвленную группу -(С1-С4)алкил-S(O)2(O-М), линейную или разветвленную группу -галоген(С1-С4)алкил-S(O)2(ОН) или линейную или разветвленную группу -галоген(С1-С4)алкил-S(O)2(O-М+);
• A1 представляет собой группу -C(O)-CH(R3)-NH-;
• A2 представляет собой группу -C(O)-CH(R4)-NH-, группу 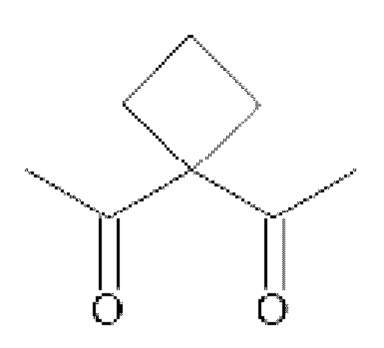 группу
группу 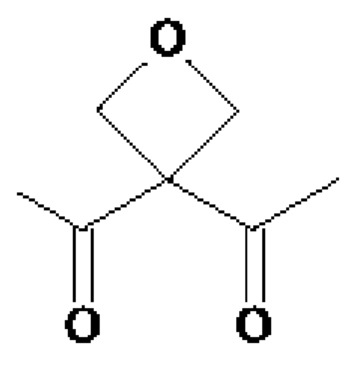 или группу
или группу 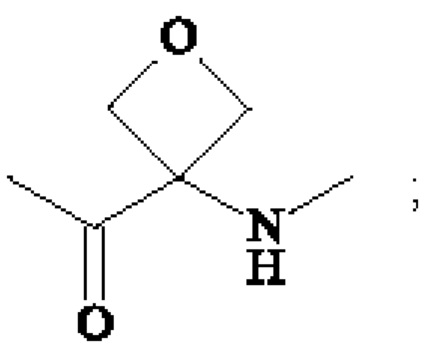
• R3 и R4 независимо друг от друга представляют собой боковую цепь аминокислоты,
• R5 представляет собой атом водорода или группу (С1-С4)алкила,
• Z' представляет собой предшественник спейсерного звена,
• М+ представляет собой фармацевтически приемлемый одновалентный катион.
В одном варианте осуществления во втором аспекте изобретения D представляет собой часть лекарственного средства, содержащую атом азота или атом кислорода, при этом D непосредственно связан с Т через указанный атом азота или атом кислорода части лекарственного средства. В некоторых вариантах осуществления D представляет собой кватернизованный третичный амин, содержащий часть лекарственного средства. В конкретном варианте осуществления, D представляет собой монометил ауристатин Е (название ИЮПАК: (S)-N-((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-3-метокси-5-метил-1-оксогептан-4-ил)-N,3-диметил- 2-((S)-3-метил-2-(метиламино)бутанамидо)бутанамид или ММАЕ), ауристатин Е (название ИЮПАК: (2S)-2-[[(2S)-2-(диметиламино)-3-метил-бутаноил]амино]-N-[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1-метилпропил]-4-оксо-бутил]-N,3-диметил-бутанамид), SN-38 (название ИЮПАК: (4S)-4,11-диэтил-4,9-дигидрокси-1H-пирано[3',4':6,7]индолизино[1,2-b]хинолин-3,14(4H,12Н)-дион), или доксорубицин (название ИЮПАК: (1S,3S)-3,5,12-тригидрокси-3-(гидроксиацетил)-10-метокси-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил 3-амино-2,3,6-тридеокси-α-1-ликсо-гексопиранозид).
Предпочтительно, Т представляет собой связь или -О-С(О)-*, в которой * указывает место присоединения к D. В некоторых вариантах осуществления, когда Т представляет собой связь, D представляет собой кватернизованный третичный амин, содержащий часть лекарственного средства.
Предпочтительно во втором аспекте изобретения R2 представляет собой группу -S(O)2(OH), группу -S(O)2(O-M+), группу -СН2-S(O)2(OH), группу -СН2-S(O)2(O-M+), группу -СН2-СН2-S(O)2(OH), группу -СН2-СН2-S(O)2(O-M+), группу -СН2-СН2-СН2-S(O)2(OH) или группу -СН2-СН2-СН2-S(O)2(O-M+). В предпочтительном варианте осуществления второго аспекта изобретения М+ представляет собой Na+.
Преимущественно во втором аспекте изобретения A1 представляет собой группу -C(O)-CH(R3)-NH-, в которой R3 представляет собой группу -(СН2)3-NH-CO-NH2 или метальную группу.
В предпочтительном варианте осуществления второго аспекта изобретения, А2 представляет собой группу-C(O)-CH(R4)-NH-, в которой R4 представляет собой изопропильную группу;
группу 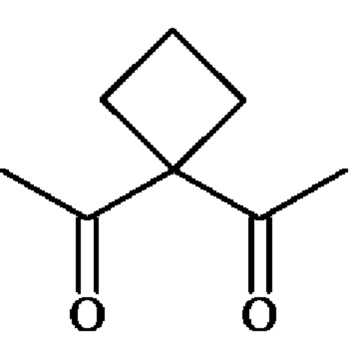 ; группу
; группу 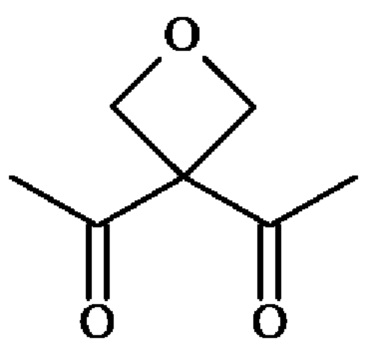
или группу 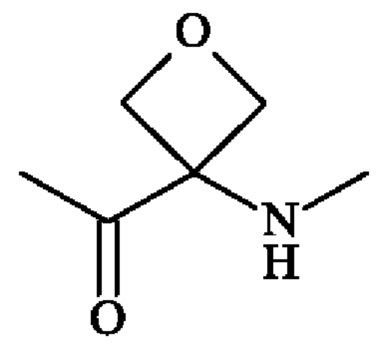
В другом предпочтительном варианте осуществления второго аспекта изобретения A1 представляет собой группу -C(O)-CH(R3)-NH-, в которой R3 представляет собой группу -(СН2)3-NH-CO-NH2, и А2 представляет собой
группу 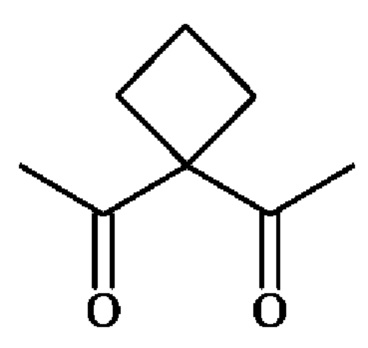 или группу
или группу 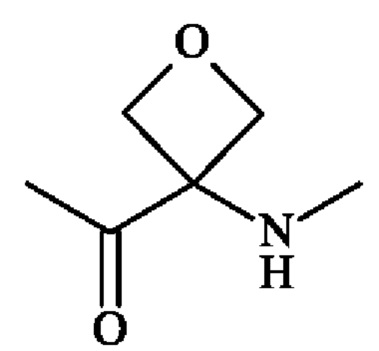 .
.
В другом предпочтительном варианте осуществления второго аспекта изобретения A1 представляет собой группу -C(O)-CH(R3)-NH- и А2 представляет собой группу -C(O)-CH(R4)-NH-, в которой R3 и R4, независимо друг от друга, представляют собой боковую цепь аминокислоты. В частности, когда R3 и R4 представляют собой боковую цепь аминокислоты, углерод, несущий боковую цепь, имеет конфигурацию L. В предпочтительном варианте осуществления A1 представляет собой группу -C(O)-CH(R3)-NH-, в которой R3 представляет собой группу -(СН2)3-NH-CO-NH2 или метальную группу и А2 представляет собой группу -C(O)-CH(R4)-NH-, в которой R4 представляет собой изопропильную группу. Предпочтительно во втором аспекте изобретения группа A1-A2 представляет собой Cit-Val или Ala-Val.
Предпочтительно предшественник спейсерного звена Z' выбирают из:
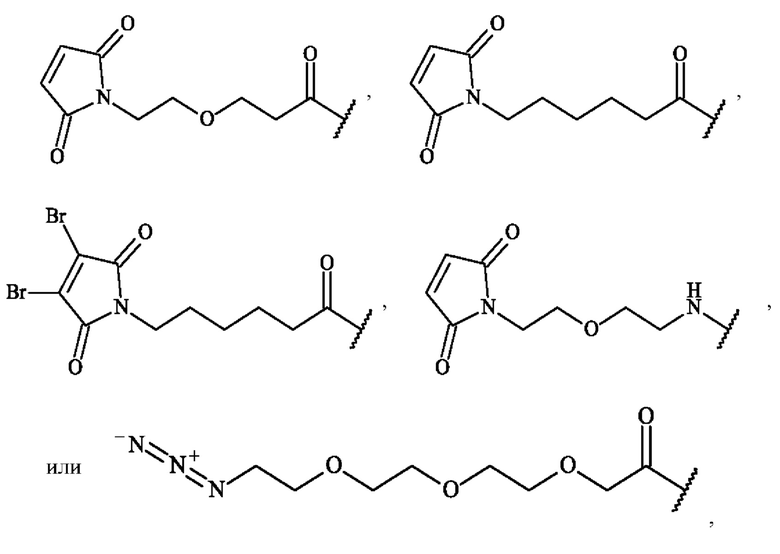
в которой волнистая линия указывает место ковалентного присоединения к N-концу или карбонильной группе группы А2.
Предпочтительное соединение линкер-лекарственное средство формулы (II) выбирают из:
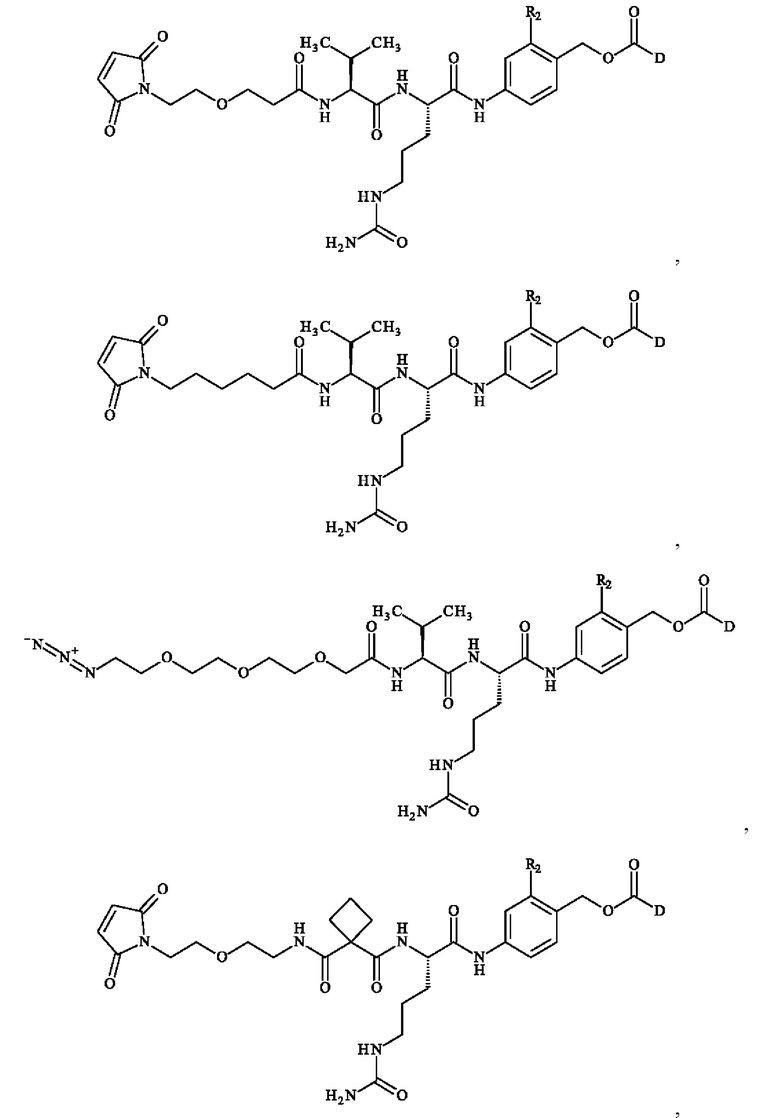
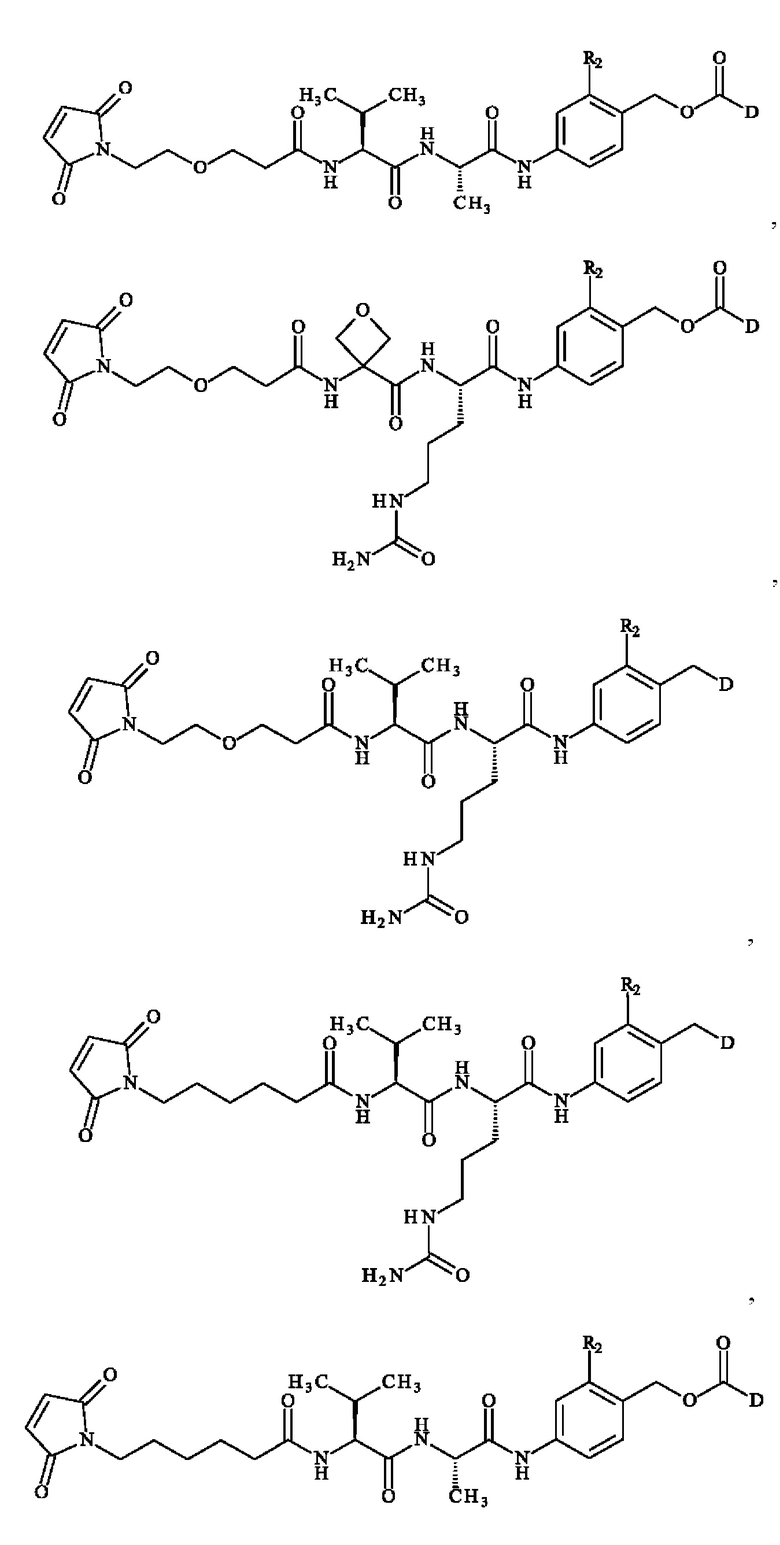
где R2 и D имеют определения, приведенные в настоящей заявке выше.
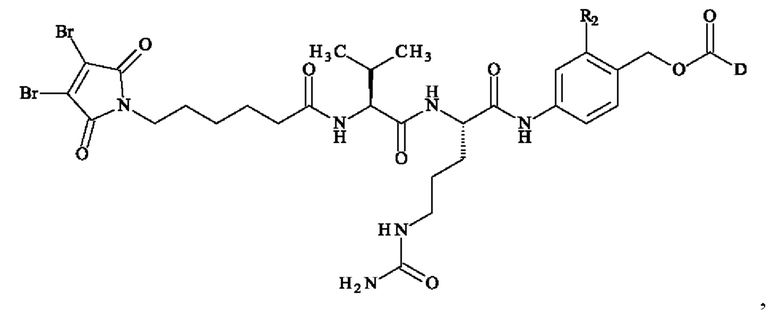
Более предпочтительное соединение линкер-лекарственное средство формулы (II) выбирают из:
причем R2 и D имеют определения, приведенные в настоящей заявке выше.
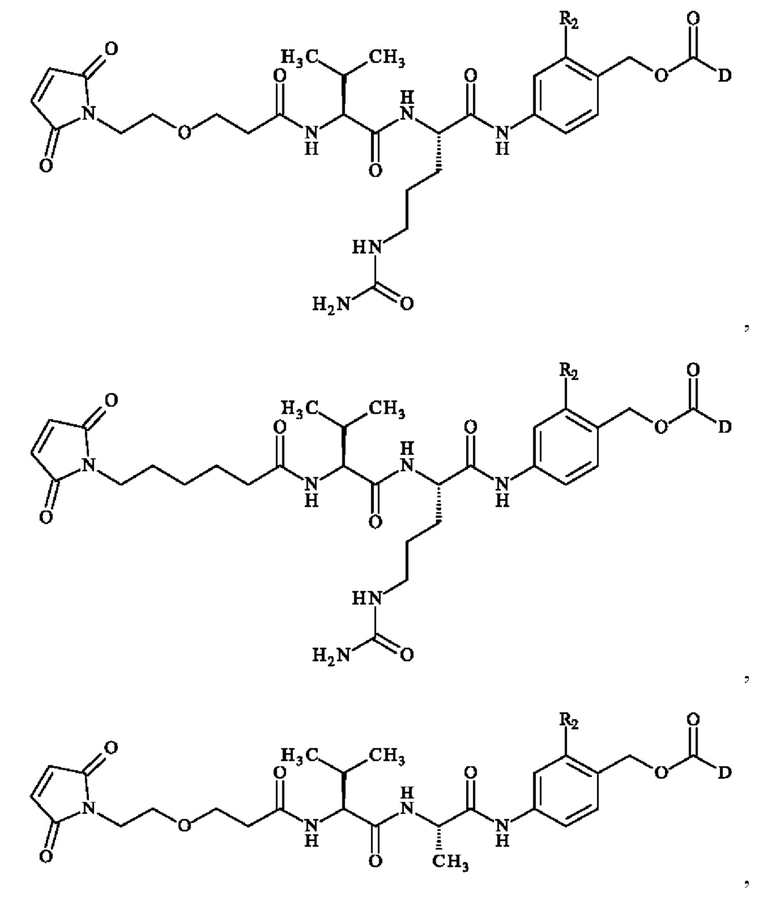
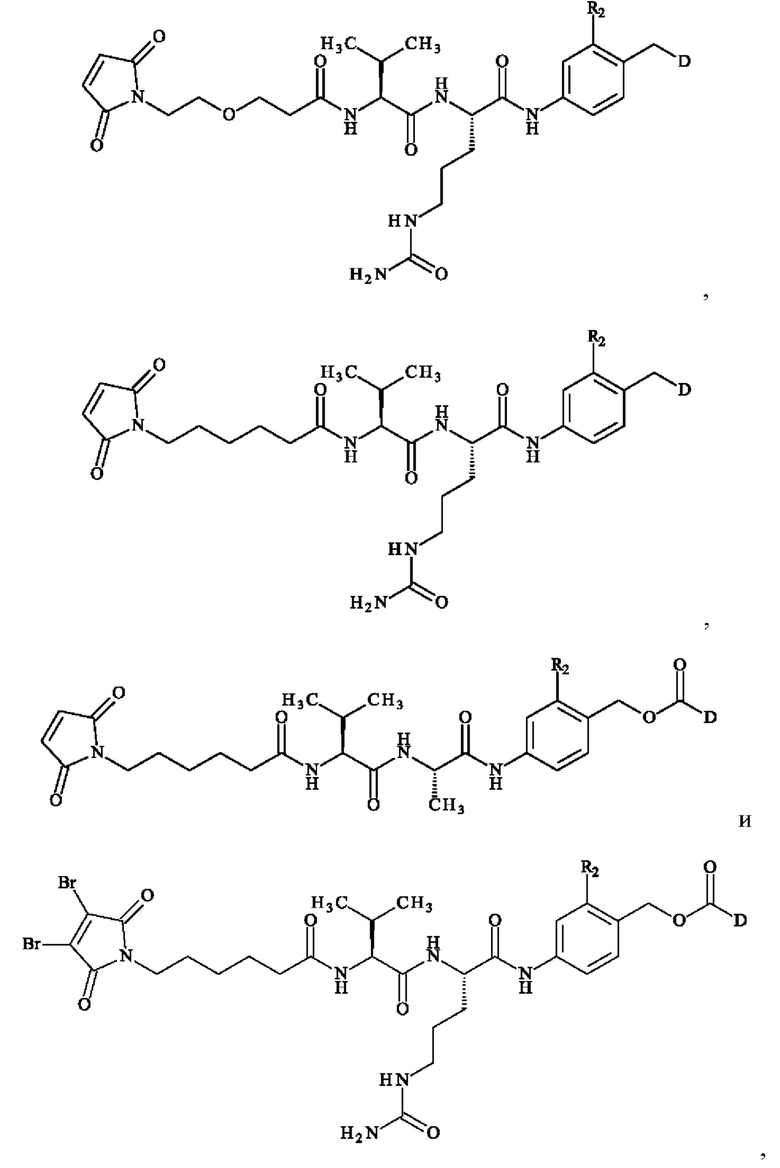
В предпочтительном варианте осуществления соединение линкер -лекарственное средство формулы (II) выбирают из:
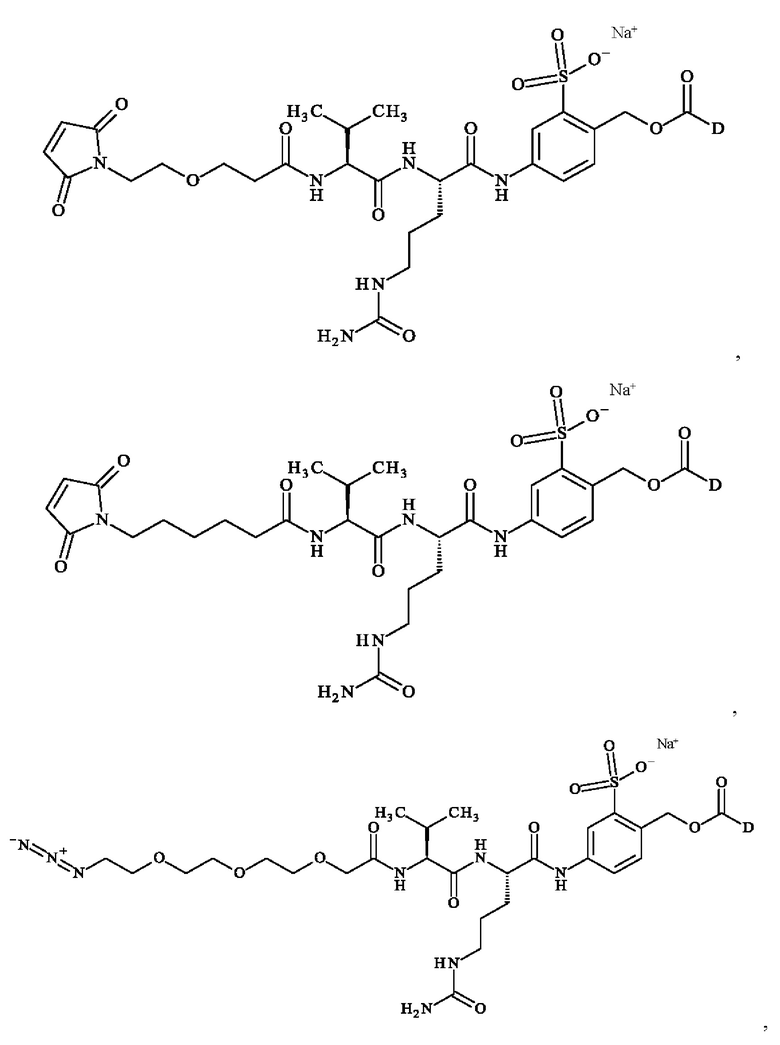
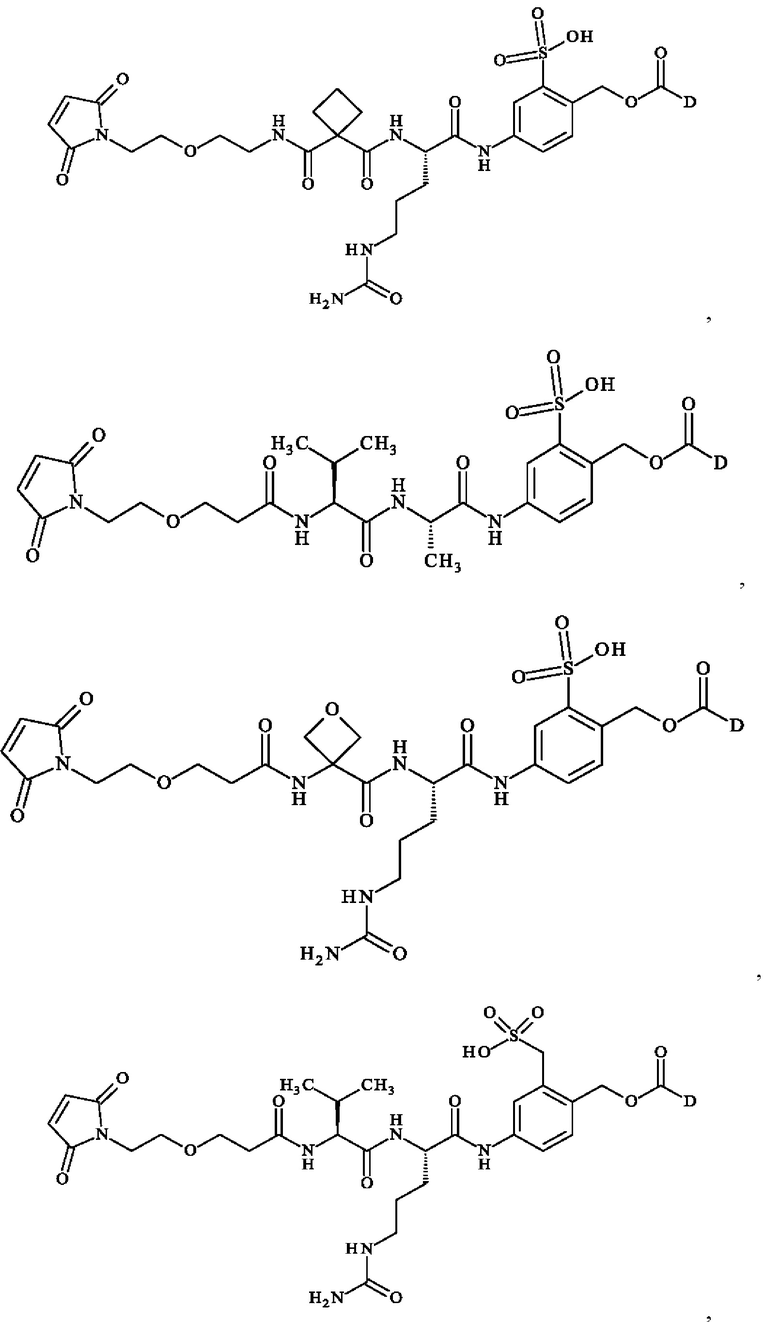
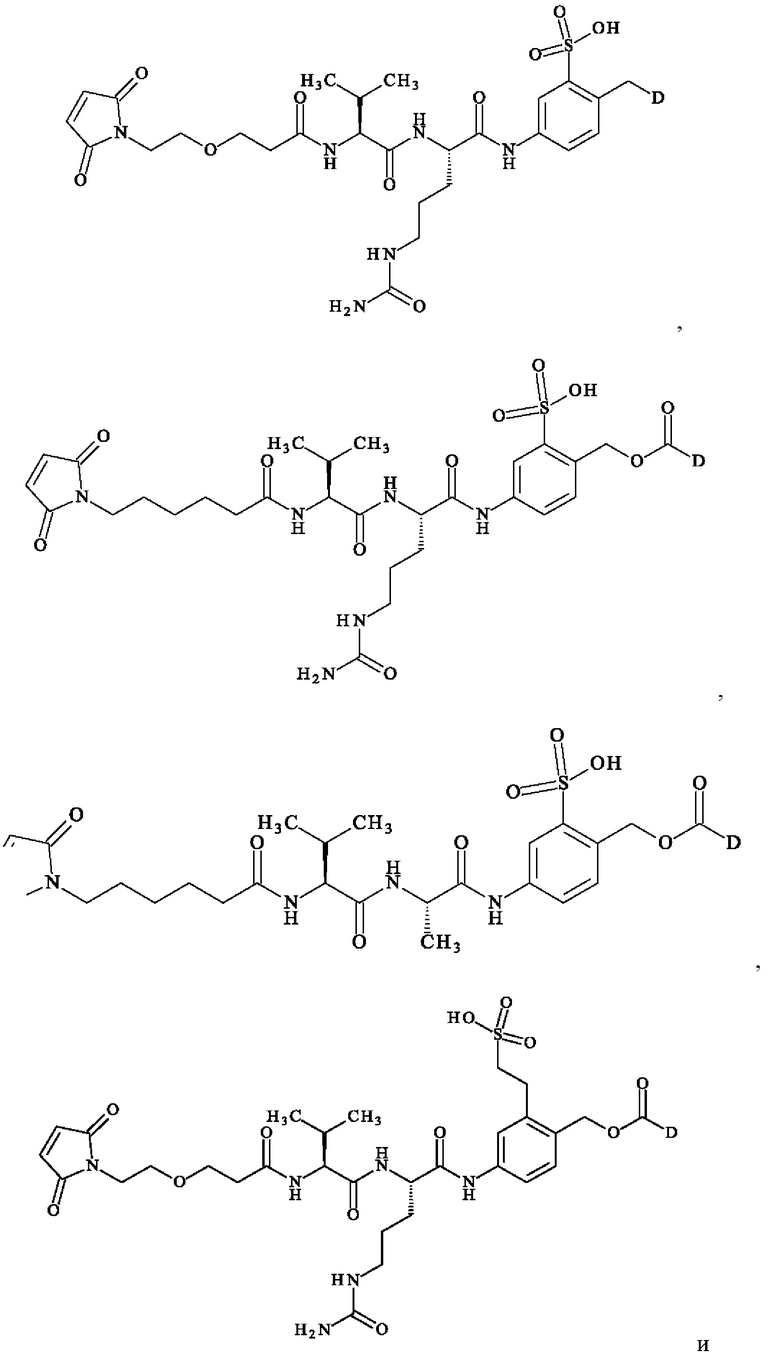
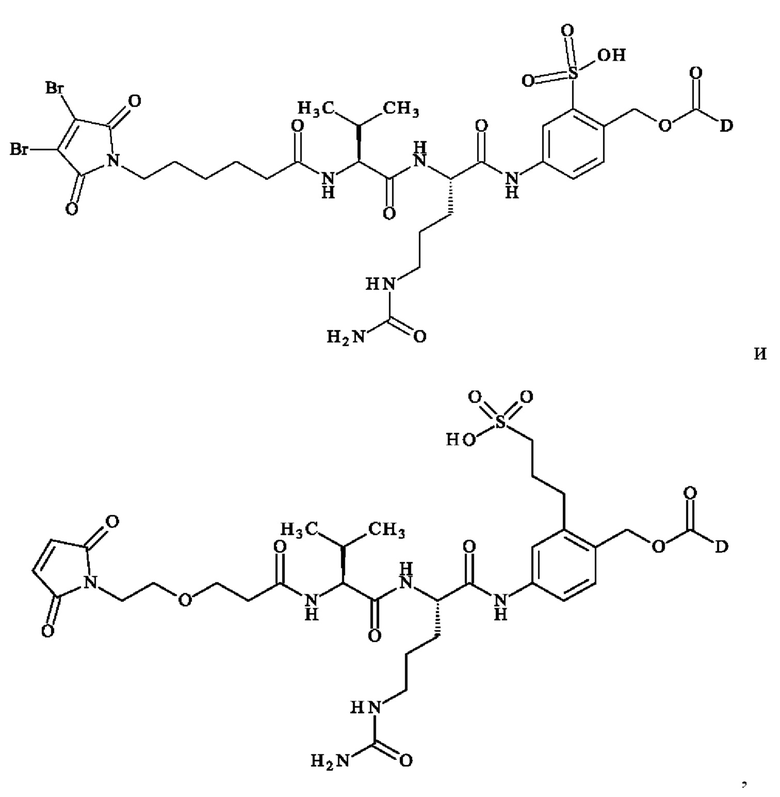
при этом D имеет определение, приведенное в настоящей заявке ранее. В конкретном варианте осуществления предпочтительные соединения линкер-лекарственное средство формулы (II) представляют собой:
- 5-[[(2S)-2-[[(2S)-2-[3-[2-(2,5-диоксопиррол-1-ил)этокси]пропаноиламино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфонат натрия;
- 5-[[(2S)-2-[[(2S)-2-[6-(2,5-диоксопиррол-1-ил)гексаноиламино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфонат натрия;
- 5-[[(2S)-2-[[(2S)-2-[[2-[2-[2-(2-азидоэтокси)этокси]этокси]ацетил]амино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфонат натрия;
- 5-[[(2S)-2-[[1-[2-[2-(2,5-диоксопиррол-1-ил)этокси]этилкарбамоил]циклобутанкарбонил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)- 1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1 S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновую кислоту;
- 5-[[(2S)-2-[[(2S)-2-[3-[2-(2,5-диоксопиррол- 1-ил)этокси]пропаноиламино]-3-метил-бутаноил]амино]пропаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфонат натрия;
- 5-[[(2S)-2-[[(2S)-2-[3-[2-(2,5-диоксопиррол- 1-ил)этокси]пропаноиламино]-3-метил-бутаноил]амино]пропаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновую кислоту;
- 5-[[(2S)-2-[[3-[2-[2-(2,5-диоксопиррол-1-ил)этокси]этилкарбамоил]оксетан-3-карбонил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновую кислоту;
- [5-[[(2S)-2-[[(2S)-2-[3-[2-(2,5-диоксопиррол-1-ил)этокси]пропаноиламино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)- 1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]фенил]метансульфоновую кислоту;
- [4-[[(2S)-2-[[(2S)-2-[3-[2-(2,5-диоксопиррол-1-ил)этокси]пропаноиламино]-3-метил-бутаноил]амино]- 5-уреидо-пентаноил]амино]-2-сульфо-фенил]метил-[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1Е,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-диметил-аммония; 2,2,2-трифторацетат;
- N-({[4-({N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-1-валил-1-аланил}амино)-2-сульфофенил]метокси}карбонил)-N-метил-1-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил- 1-оксогептан-4-ил]-N-метил-1-валинамид;
- N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-1-валил-N5-карбамоил-N-(4-{[({[(4S)-4,11-диэтил-9-гидрокси-3,14-диоксо-3,4,12,14-тетрагидро-1H-пирано[3',4':6,7]индолизино[1,2-b]хинолин-4-ил]окси} карбонил)окси]метил}-3-сульфофенил)-1-орнитинамид;
-(1S',3S')-3,5,12-тригидрокси-3-(гидроксиацетил)-10-метокси-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил 2,3,6-тридеокси-3-[({[4-({N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-1-валил-N5-карбамоил-1-орнитил}амино)-2-сульфофенил]метокси}карбонил)амино]-α-1-ликсо-гексопиранозид;
-N-{3-[2-(2,3-диоксо-2,5-дигидро-1H-пиррол-1-ил)этокси)пропаноил}-1-валил-N-{4-[(5S,SS,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1S,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-оксоэтил)-4,10-диметил-3,6,9-триоксо-5,8-ди(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадекан-1-ил]-3-(2-сульфоэтил)фенил}-N5-карбамоил-1-орнитинамид;
- 5-[[(2S)-2-[[(2S)-2-[6-(3,4-дибром-2,5-диоксо-пиррол-1-ил)гексаноиламино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин- 1-ил]-2-метокси-1-[(1S)- 1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновую кислоту.
Также изобретение относится к способу получения соединений линкер-лекарственное средство формулы (II), при этом отличается тем, что в качестве исходного вещества используют соединение формулы (I):
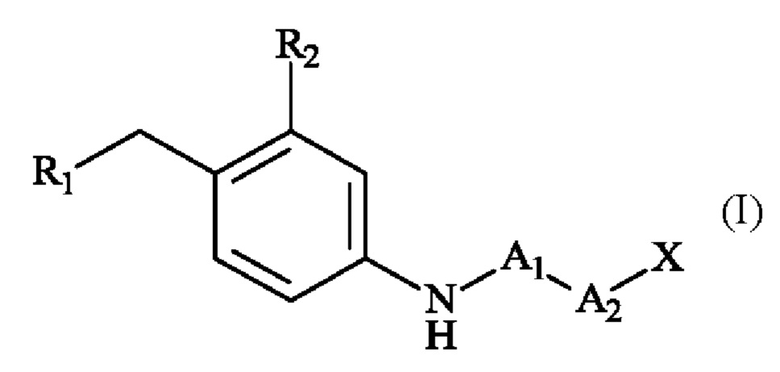
в которой R1, R2, A1, А2 и X имеют приведенные выше определения,
которое затем модифицируется с получением соединения формулы (X):
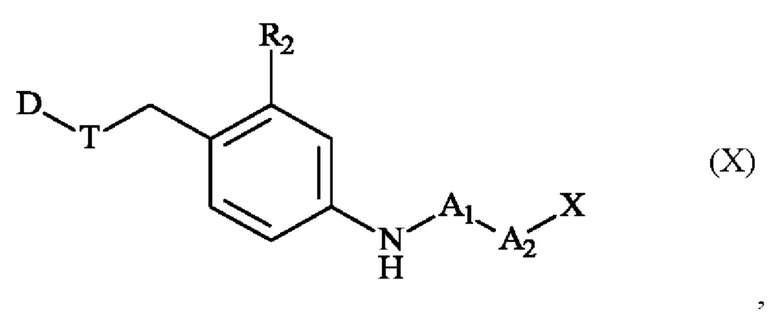
в которой A1, А2, D, R2, Т и X определены в формуле (II),
соединение формулы (X), которое затем после удаления защитной группы X подвергают сочетанию с соединением формулы (XI):

в которой Z' имеет определения, приведенные в формуле (II),
с получением соединения линкер-лекарственное средство формулы (II).
Альтернативный способ получения соединений линкер-лекарственное средство формулы (II) отличается тем, что в качестве исходного вещества используют соединение формулы (VII), полученное в две стадии из соединения формулы (IV), которое подвергают сочетанию с группой D-T, чтобы получить соединение формулы (XII):
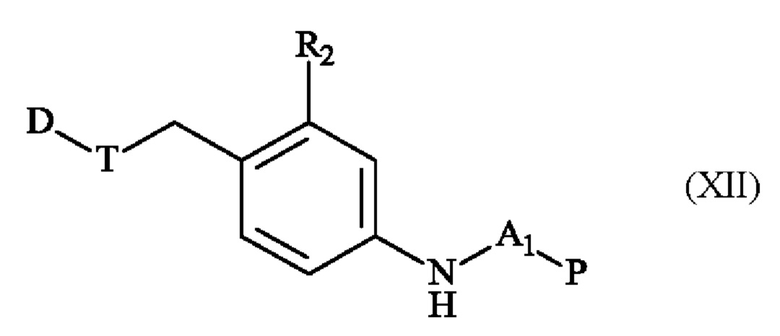
в которой A1, D, Р, R2 и Т имеют определения, приведенные в настоящей заявке выше,
соединение формулы (XII), которое далее подвергают сочетанию с соединением формулы (XIII):
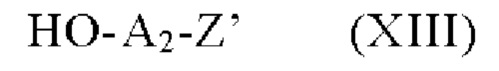
в которой Z и А2 определены в формуле (I),
чтобы получить соединение линкер-лекарственное средство формулы (II).
В одном варианте осуществления соединение линкер-лекарственное средство формулы (II), полученное в конце вышеупомянутых способов, может быть дополнительно очищено в соответствии с общепринятой методикой разделения, которое при желании может быть превращено, в его аддитивные соли с фармацевтически приемлемой кислотой или основанием, которые могут быть разделены на их изомеры в соответствии с общепринятой методикой разделения, при этом следует понимать, что в любой момент, который считается подходящим в ходе описанного выше способа, некоторые группы (гидрокси, амино …) исходных реагентов или промежуточных продуктов синтеза могут быть защищены, впоследствии сняты защитные группы и функционализированы, как того требует синтез. Соединения формул (IV), (XI) и (XIII) или имеются в продаже, или могут быть получены специалистом в данной области с использованием обычных химических реакций, описанных в литературных источниках.
Изобретение также относится к применению соединения линкер-лекарственное средство формулы (II) в получении конъюгата антитело-лекарственное средство.
В третьем аспекте изобретения предложен конъюгат антитело-лекарственное средство (ADC) формулы (III):
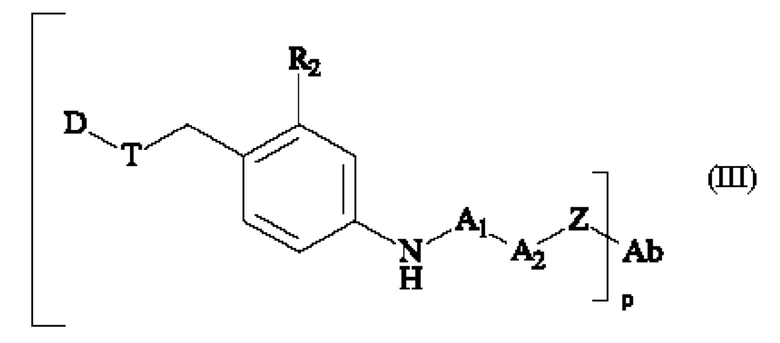
в которой
• Ab представляет собой антитело или его антигенсвязывающий фрагмент,
• D представляет собой часть лекарственного средства,
• Т представляет собой связь, -O-C(O)-N(CH3)-СН2-СН2-N(CH3)-C(O)-*, -О-*, -NR5-*, -NR5-C(O)-* или -О-С(О)-*, где * указывает место присоединения к D,
• Z представляет собой спейсерное звено,
• A1 представляет собой группу -C(O)-CH(R3)-NH-;
• А2 представляет собой группу -C(O)-CH(R4)-NH-, группу 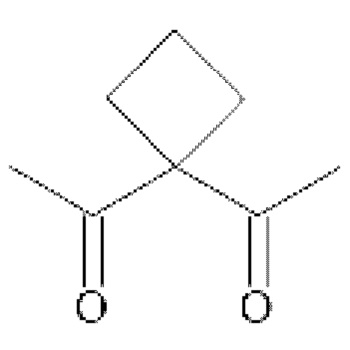
Группу 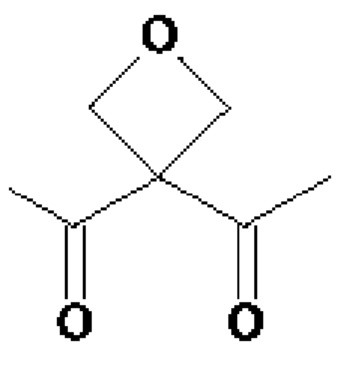 или группу
или группу 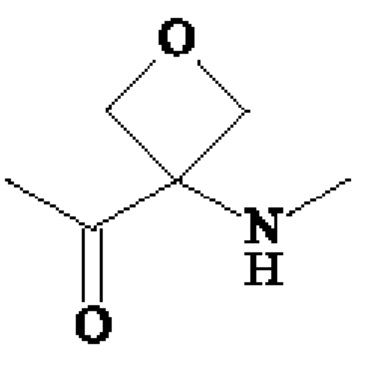 ;
;
• R2 представляет собой группу -S(O)2(OH), группу -S(O)2(O-M+), линейную или разветвленную группу -(С1-С4)алкил-S(O)2(ОН), линейную или разветвленную группу -(С1-С4)алкил-S(O)2(O-М+), линейную или разветвленную группу -галоген(С1-С4)алкил-S(O)2(ОН) или линейную или разветвленную группу -галоген(С1-С4)алкил-S(O)2(O-М+),
• R3 и R4, независимо друг от друга, представляют собой боковую цепь аминокислоты;
• R5 представляет собой группу (С1-С4)алкила;
• М+ представляет собой фармацевтически приемлемый одновалентный катион; и
• р является целым числом от 1 до 8.
В одном варианте осуществления в третьем аспекте изобретения D представляет собой часть лекарственного средства, содержащую атом азота или атом кислорода, при этом D непосредственно связан с Т через указанный атом азота или атом кислорода части лекарственного средства. В некоторых вариантах осуществления в третьем аспекте изобретения D представляет собой кватернизованный третичный амин, содержащий часть лекарственного средства. В конкретном варианте осуществления в третьем аспекте изобретения D представляет собой монометил ауристатин Е (название ИЮПАК: (S)-N-((3R,4S,5S)-1-((5)-2-((1S,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-3-метокси-5-метил-1-оксогептан-4-ил)-N,3-диметил-2-((S)-3-метил-2-(метиламино)бутанамидо)бутанамид или ММАЕ), ауристатин Е (название ИЮПАК: (2S)-2-[[(2S)-2-(диметиламино)-3-метил-бутаноил]амино]-N-[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-N,3-диметил-бутанамид), SN-38 (название ИЮПАК: (4S)-4,11-диэтил-4,9-дигидрокси-1H-пирано[3',4':6,7]индолизино[1,2-b]хинолин-3,14(4H,12H)-дион), или доксорубицин (название ИЮПАК: (1S,3S)-3,5,12-тригидрокси-3-(гидроксиацетил)-10-метокси-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил 3-амино-2,3,6-тридеокси-α-L-ликсо-гексопиранозид).
Предпочтительно в третьем аспекте изобретения Т представляет собой связь или -О-С(О)-*, в которой * указывает место присоединения к D. В некоторых вариант осуществления в третьем аспекте изобретения, когда Т представляет собой связь, D представляет собой кватернизованный третичный амин, содержащий часть лекарственного средства.
Предпочтительно в третьем аспекте изобретения R2 представляет собой группу -S(O)2(OH), группу -S(O)2(O-M+), группу -СН2-S(O)2(OH), группу -СН2-S(O)2(O-M+), группу -СН2-СН2-S(O)2(OH), группу -СН2-СН2-S(O)2(O-M+), группу -СН2-СН2-СН2-S(O)2(OH) или группу -СН2-СН2-СН2-S(O)2(O-M+). В предпочтительном варианте осуществления третьего аспекта изобретения М+ представляет собой Na+.
Преимущественно в третьем аспекте изобретения A1 представляет собой группу -C(O)-CH(R3)-NH-, в которой R3 представляет собой группу -(СН2)3-NH-CO-NH2 или метальную группу.
В предпочтительном варианте осуществления третьего аспекта изобретения, А2 представляет собой группу -C(O)-CH(R4)-NH-, в которой R4 представляет собой изопропильную группу;
группу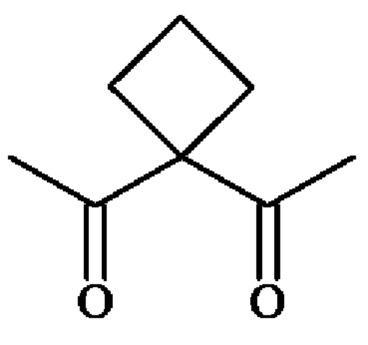 ; группу
; группу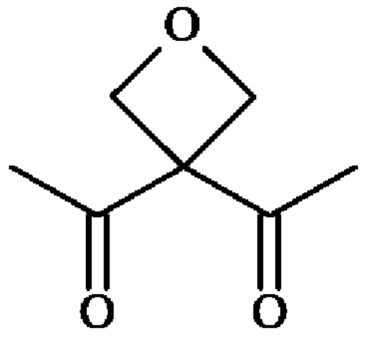
или группу 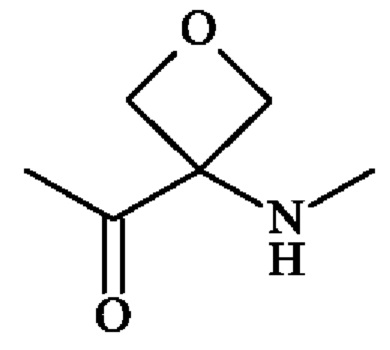
В другом предпочтительном варианте осуществления третьего аспекта изобретения, A1 представляет собой группу -C(O)-CH(R3)-NH-, в которой R3 представляет собой группу -(СН2)3-NH-CO-NH2 и А2 представляет собой группу
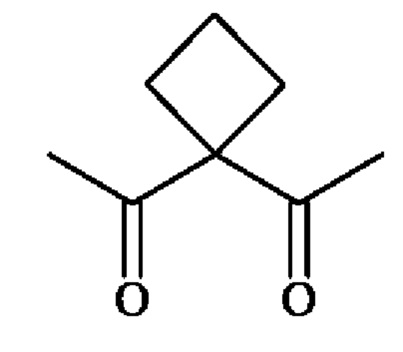 или группу
или группу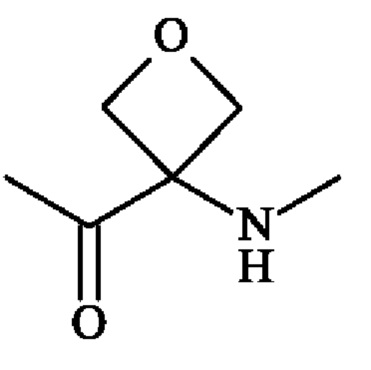
В другом предпочтительном варианте осуществления третьего аспекта изобретения, A1 представляет собой группу -C(O)-CH(R3)-NH- и А2 представляет собой группу -C(O)-CH(R4)-NH-, в которой R3 и R4, независимо друг от друга, представляют собой боковую цепь аминокислоты. В частности, когда R3 и R4 представляют собой боковую цепь аминокислоты, углерод, несущий боковую цепь, имеет конфигурацию L. В предпочтительном варианте осуществления A1 представляет собой группу -C(O)-CH(R3)-NH-, в которой R3 представляет собой группу -(СН2)3-NH-CO-NH2 или метальную группу и А2 представляет собой группу -C(O)-CH(R4)-NH-, в которой R4 представляет собой изопропильную группу. Предпочтительно в третьем аспекте изобретения группа A1-A2 представляет собой Cit-Val или Ala-Val.
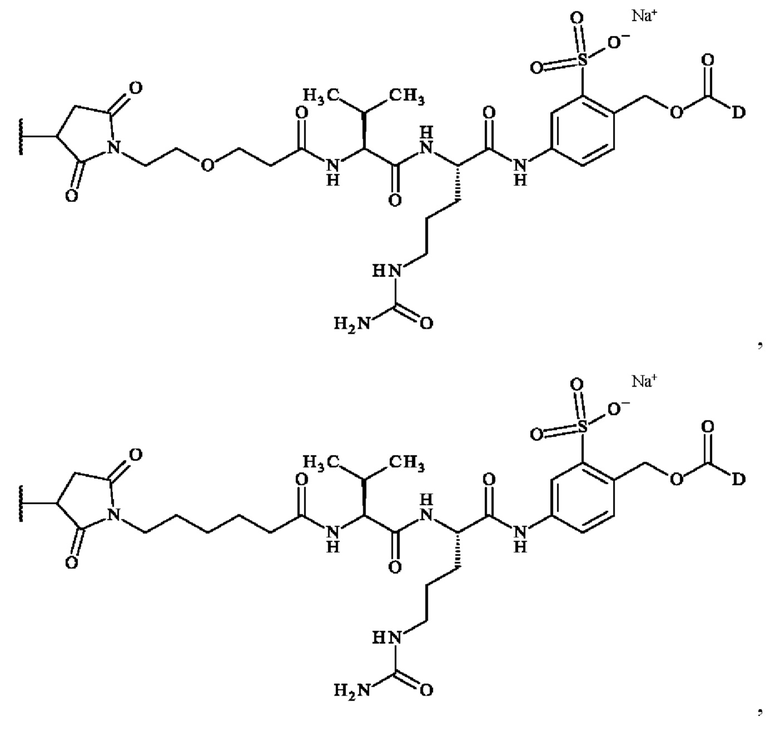
В предпочтительном варианте осуществления ADC формулы (III) образован из соединения линкер-лекарственное средство формулы (II), выбранного из:
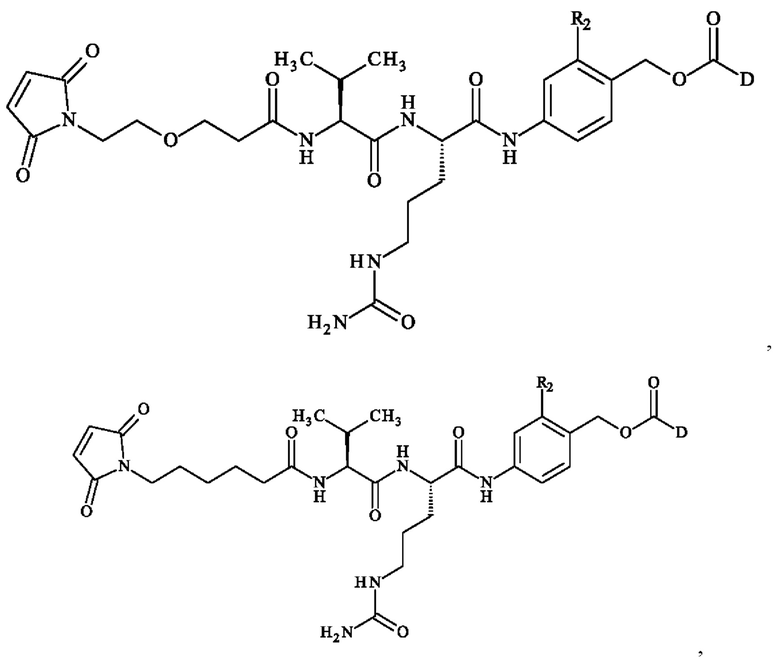
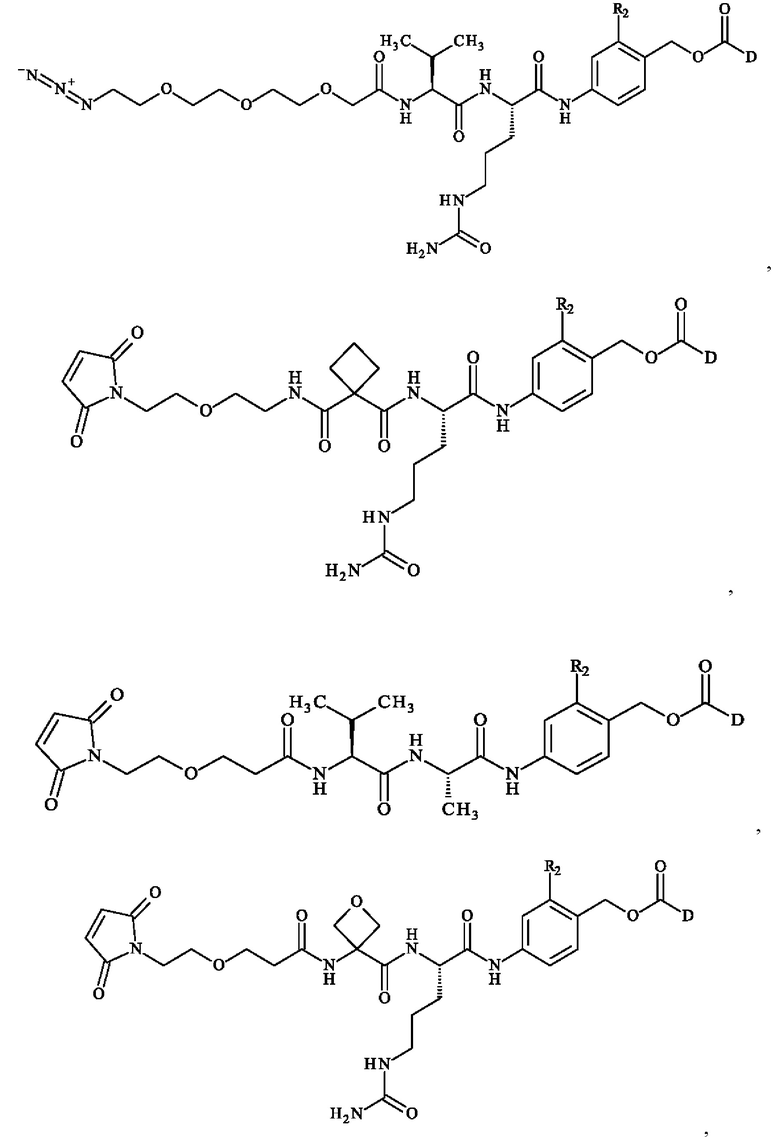
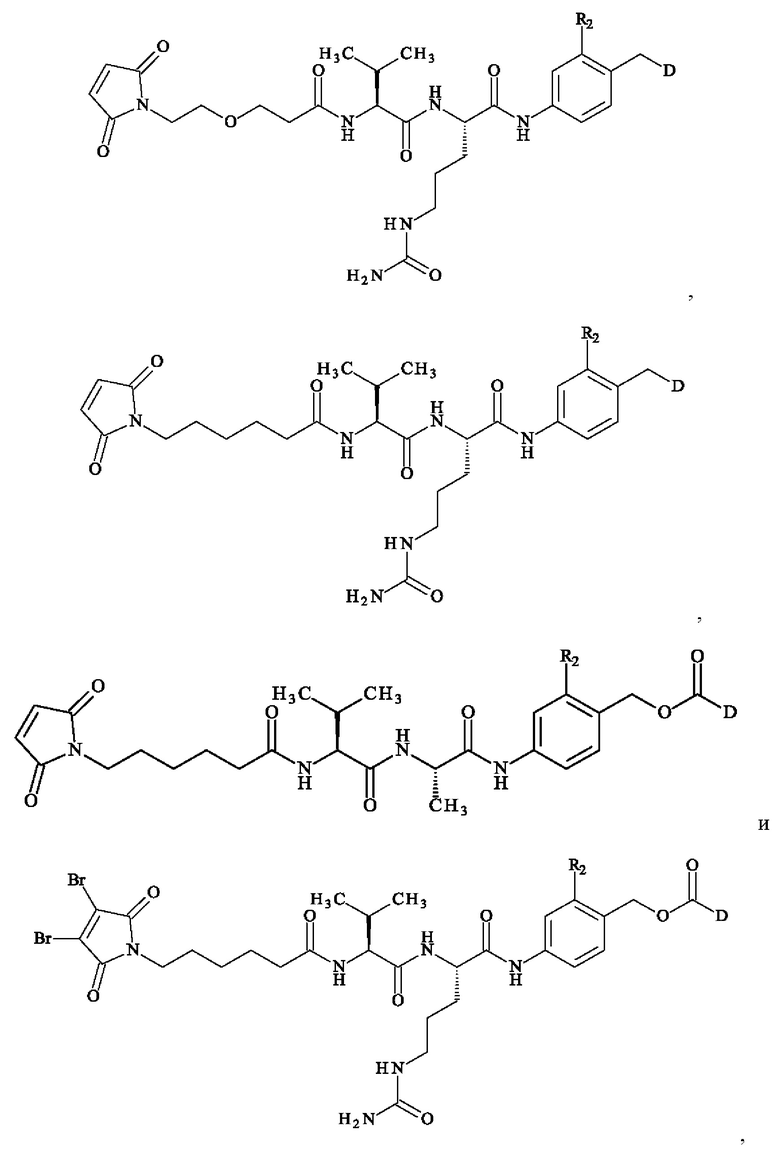
в которой R2 и D имеют определения, приведенные в настоящей заявке выше.
Более предпочтительно ADC формулы (III) образован из соединения линкер-лекарственное средство формулы (II), выбранного из:
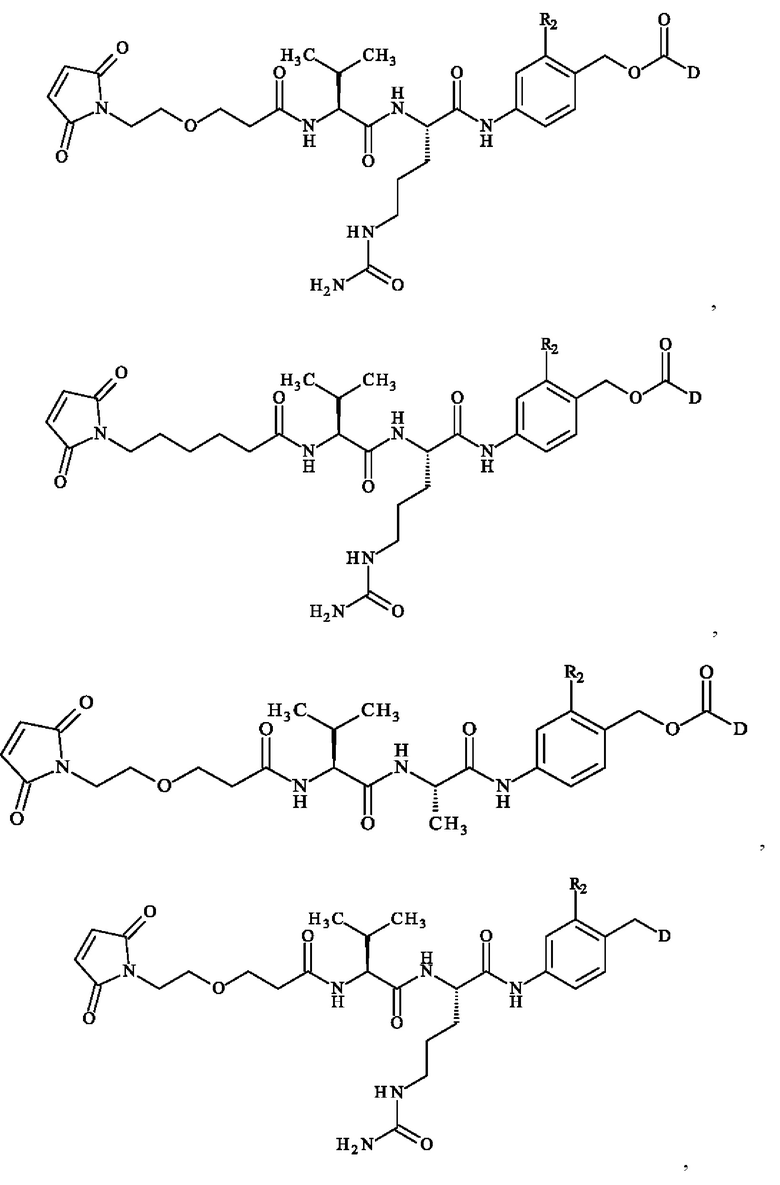
в которой R2 и D имеют определения, приведенные в настоящей заявке выше.
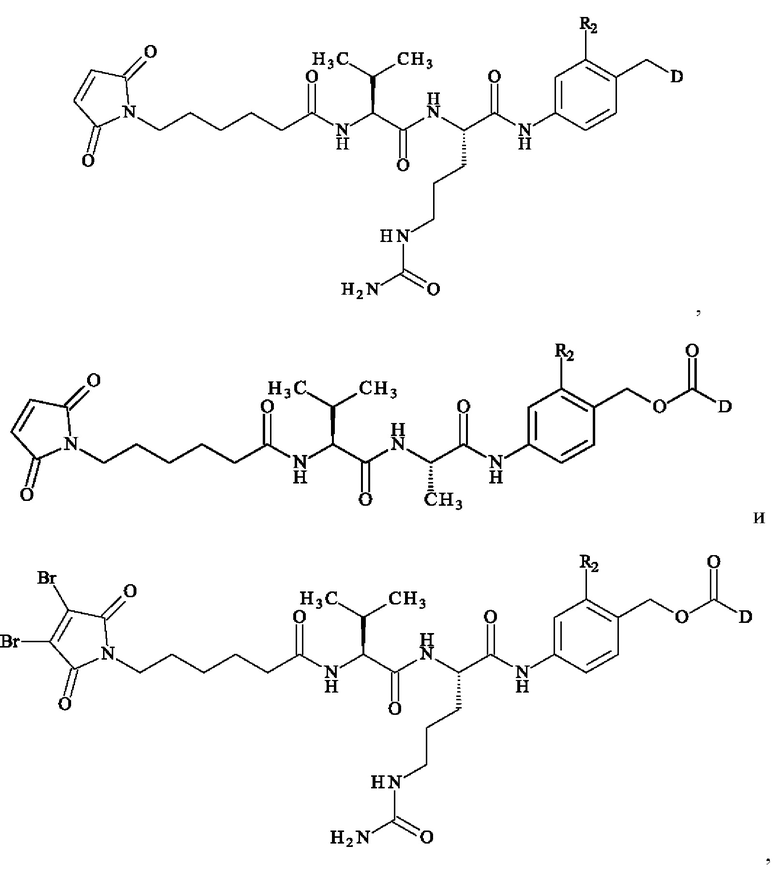
В предпочтительном варианте осуществления ADC формулы (III) образован из соединения линкер-лекарственное средство формулы (II), выбранного из:
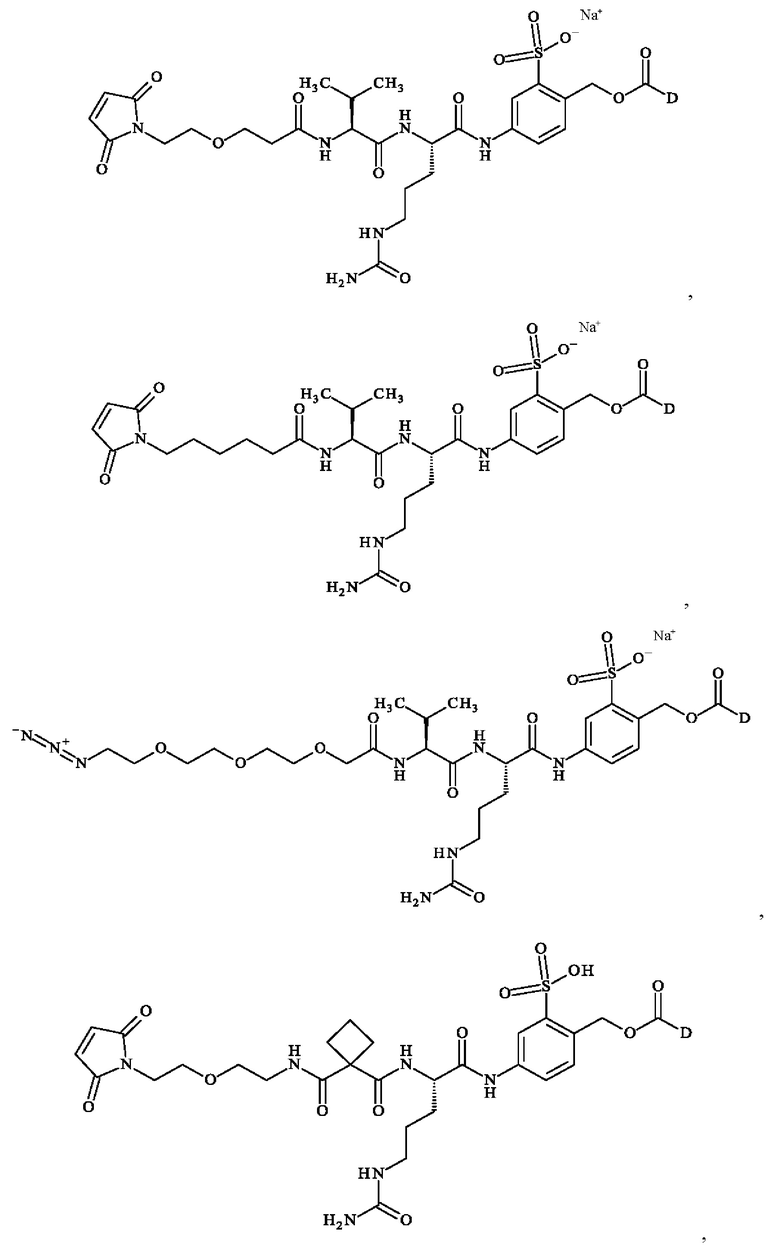
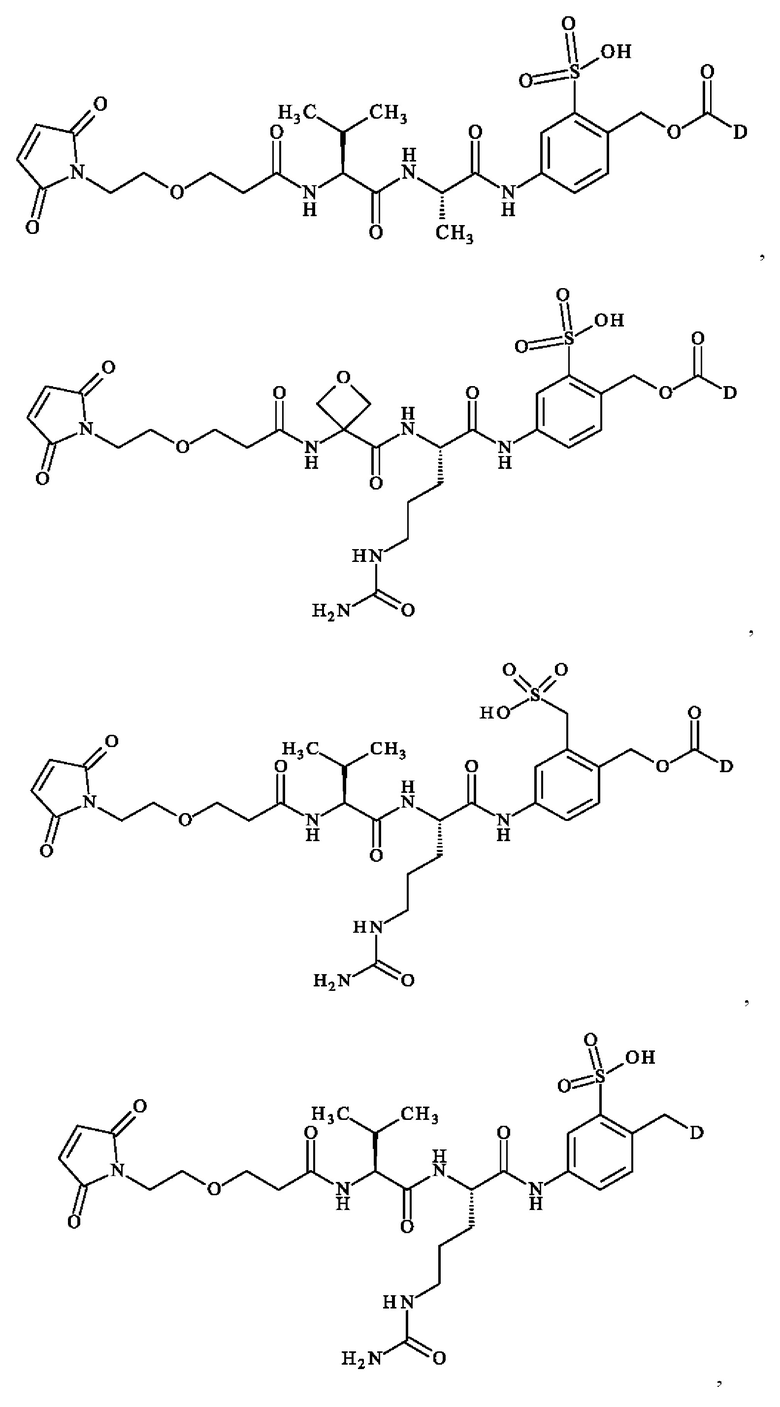
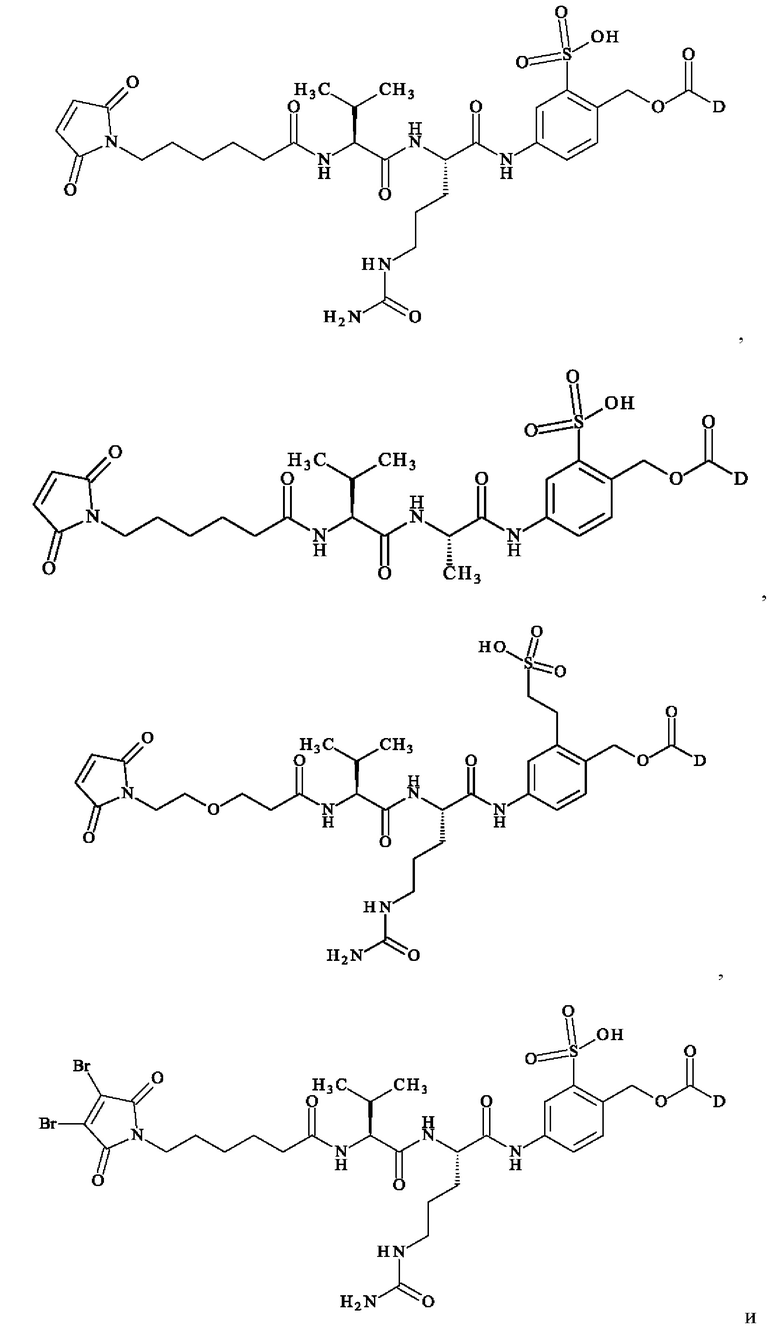
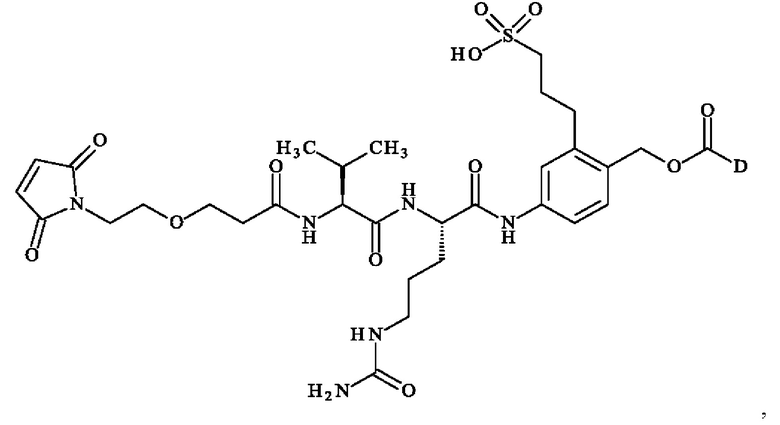
в которой D имеет определение, приведенное в настоящей заявке ранее.
В конкретном варианте осуществления изобретение обеспечивает конъюгат антитело-лекарственное средство формулы (III), в которой
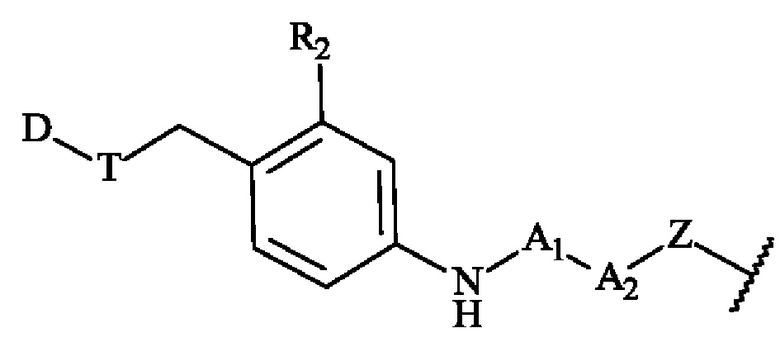
содержит формулу, выбранную из:
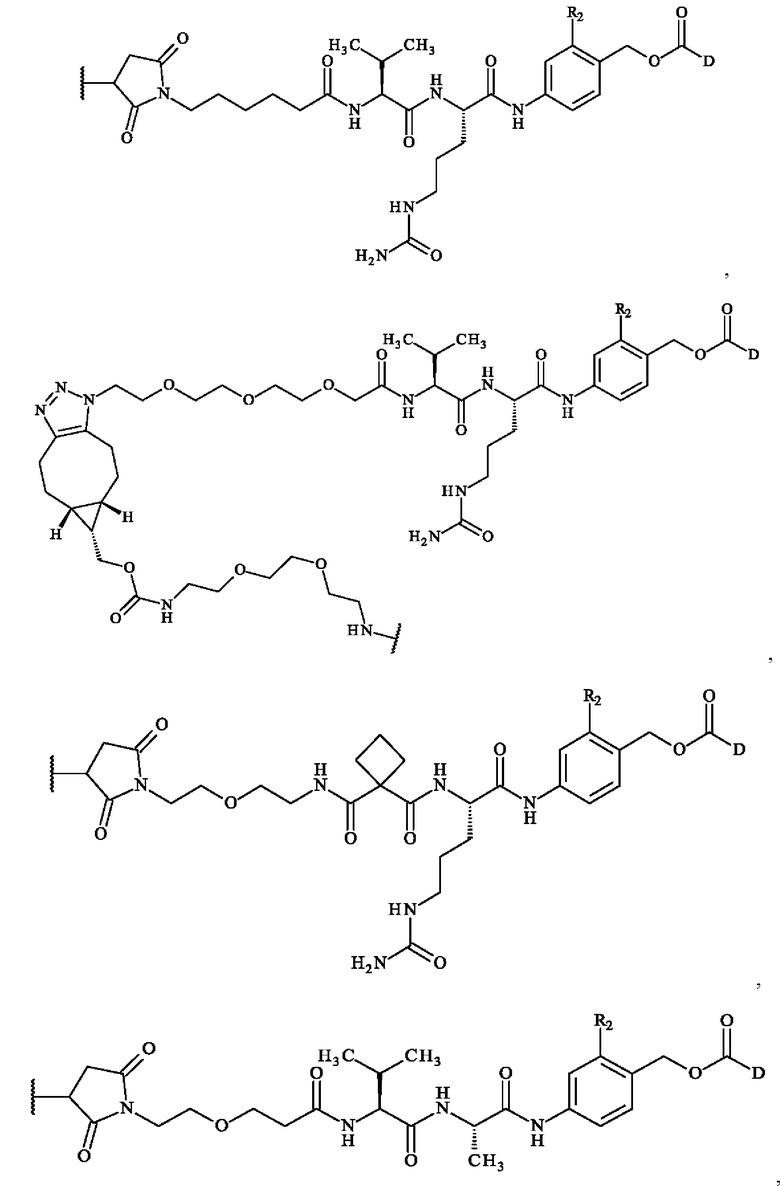
где волнистая линия указывает место ковалентного связывания с антителом и R2 и D имеют определения, приведенные в настоящей заявке выше.
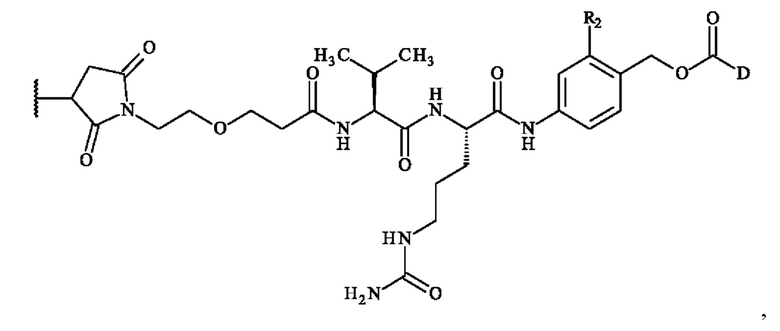
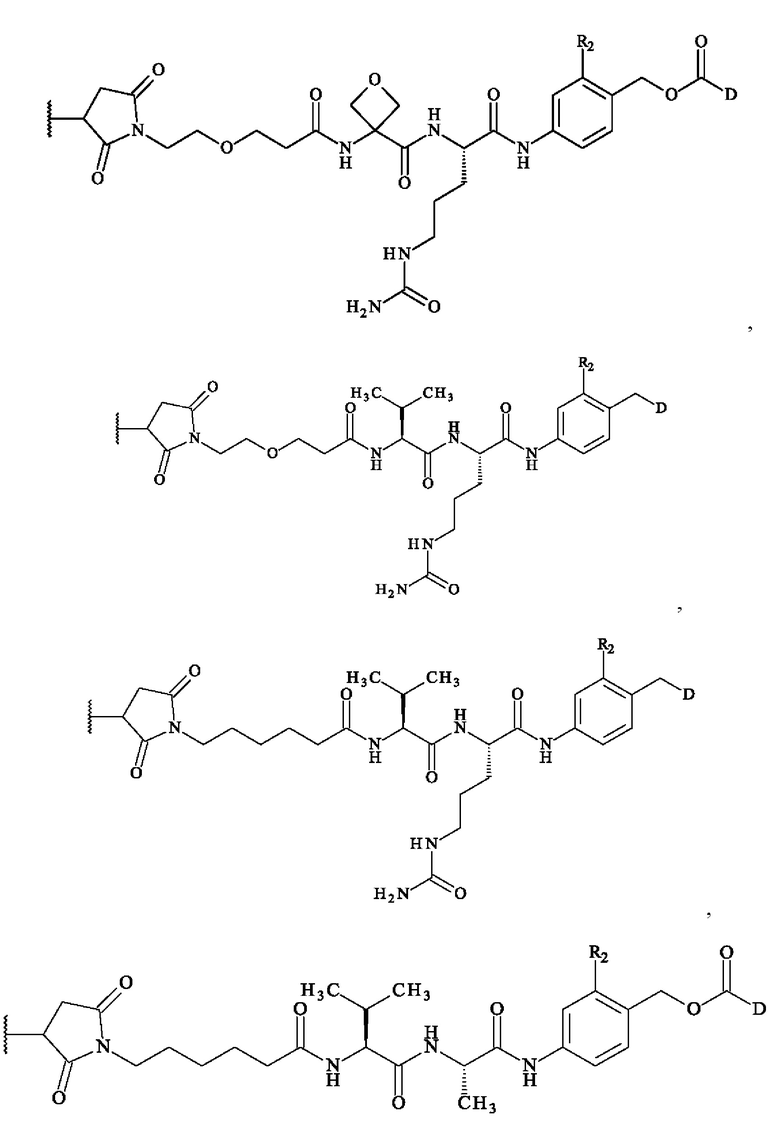
В предпочтительном варианте осуществления изобретение обеспечивает конъюгат антитело-лекарственное средство формулы (III), в которой
содержит формулу, выбранную из:
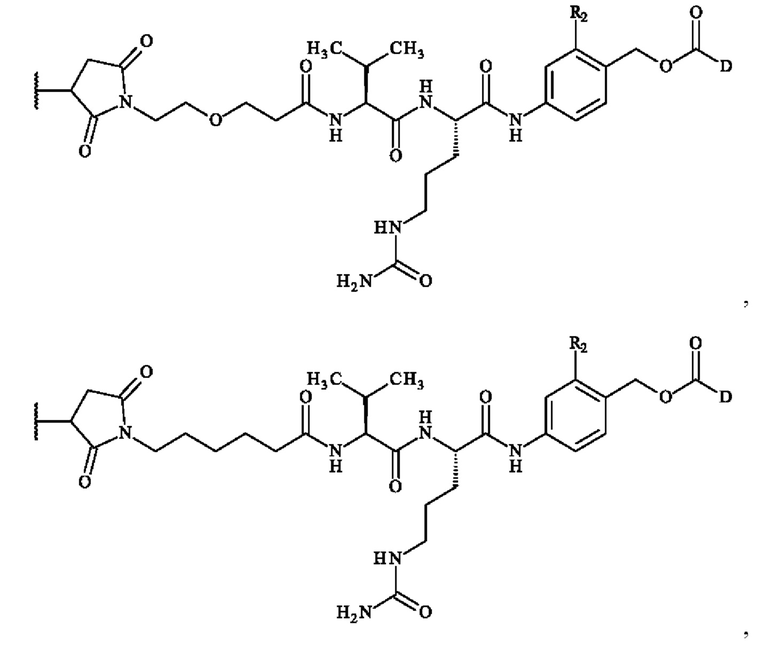
при этом волнистая линия указывает место ковалентного связывания с антителом и R2 и D имеют определения, приведенные в настоящей заявке выше.
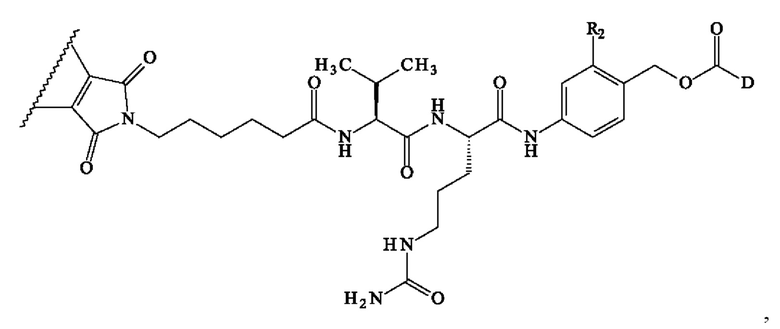
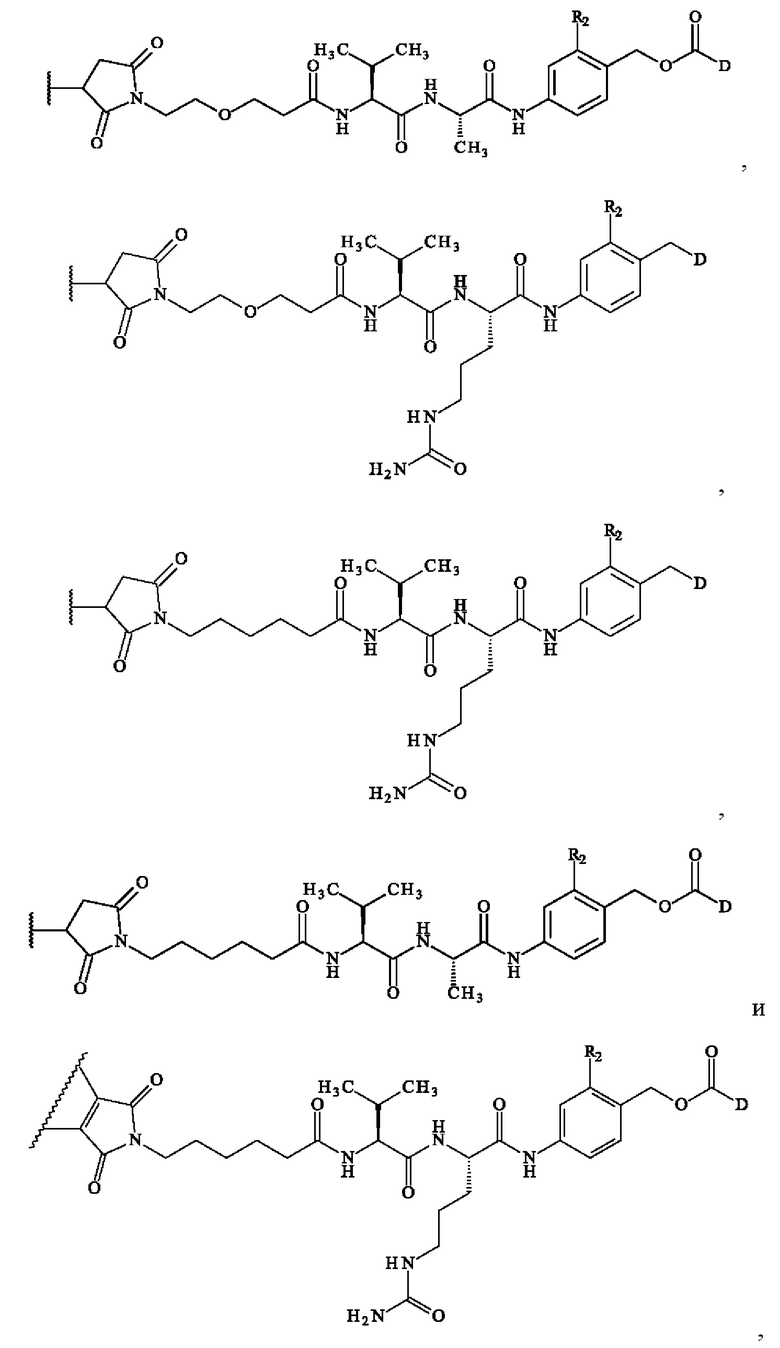
В предпочтительном варианте осуществления изобретение обеспечивает конъюгат антитело-лекарственное средство формулы (III), в которой
содержит формулу, выбранную из:
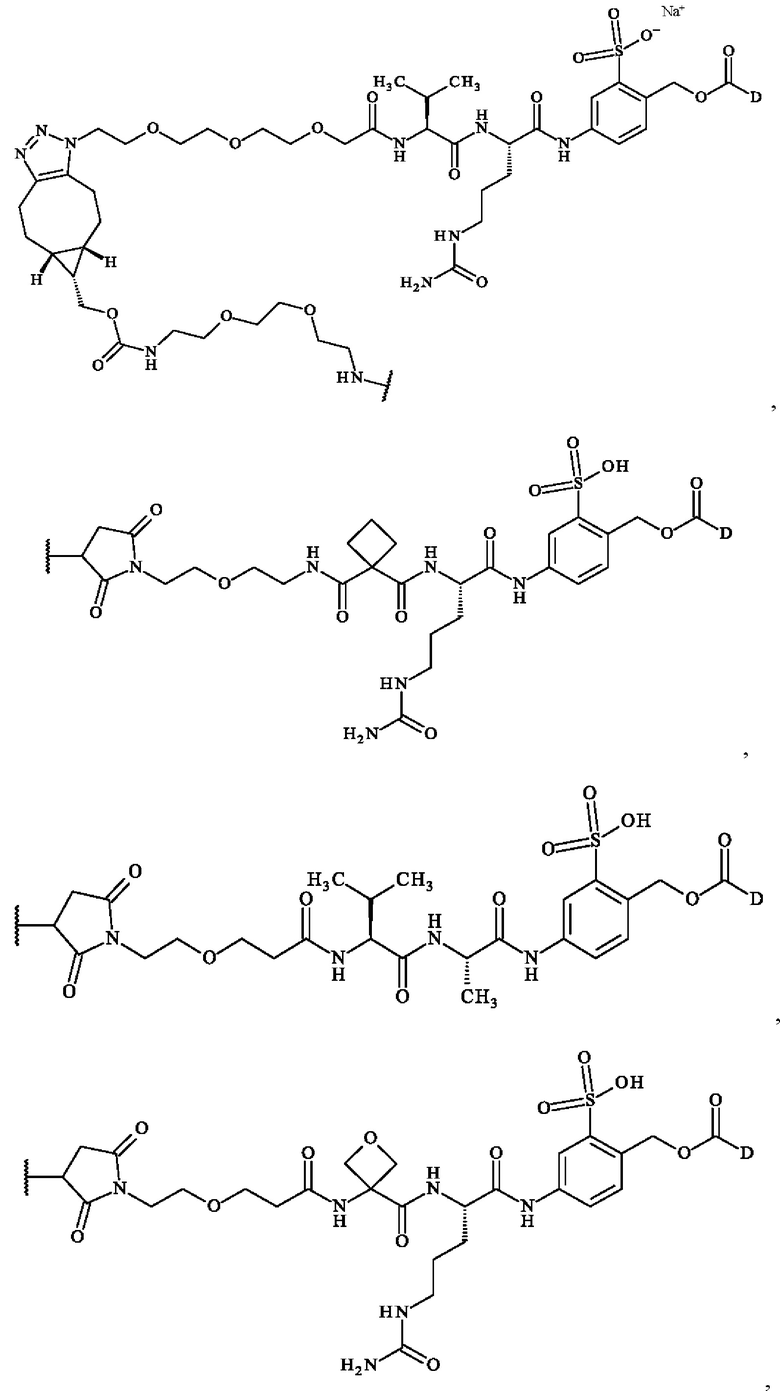
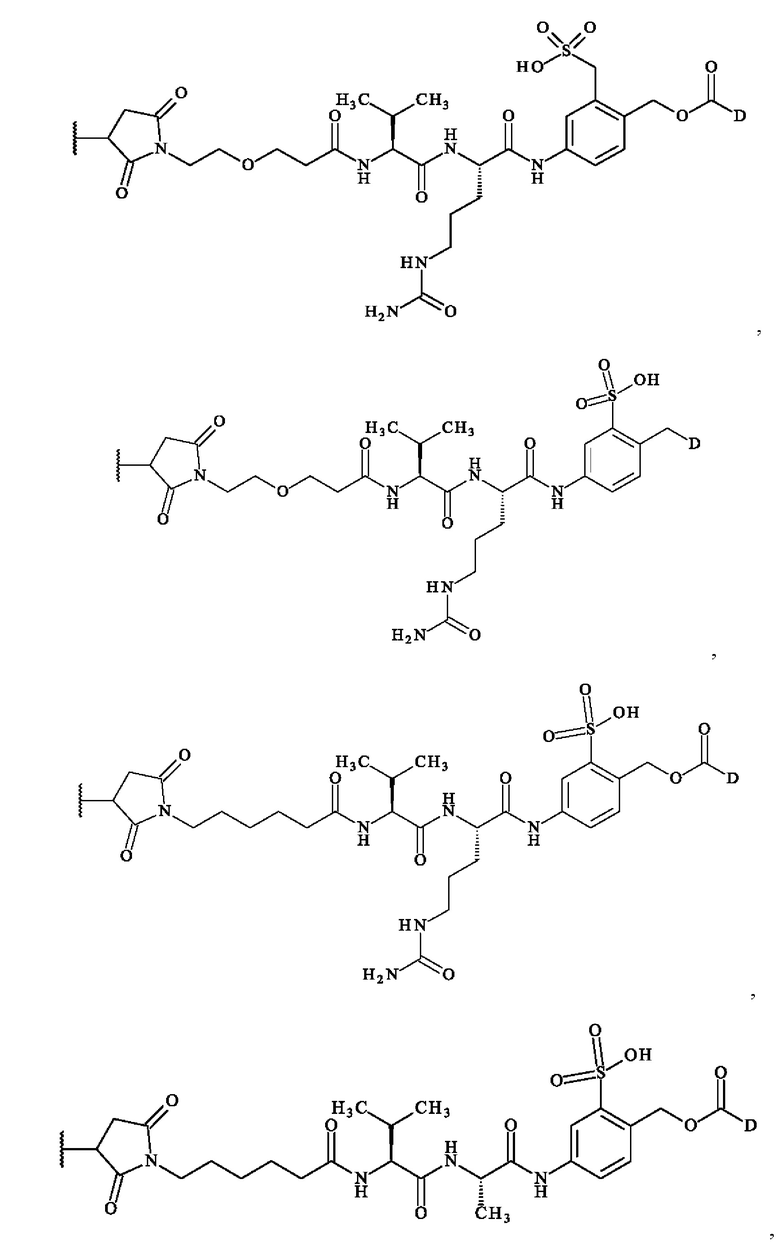
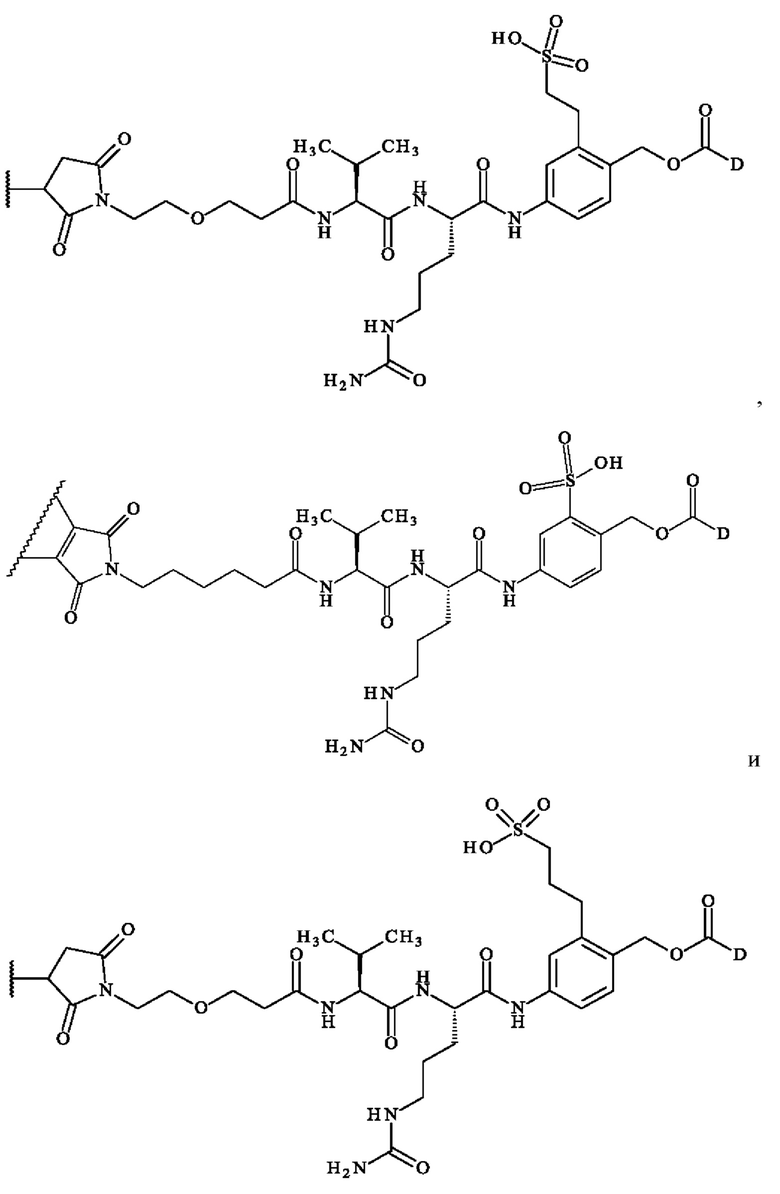
где волнистая линия указывает место ковалентного связывания с антителом и D имеет определение, приведенное в настоящей заявке ранее.
ADC в соответствии с изобретением применимы для лечения заболеваний у млекопитающих, например, у нуждающегося в этом пациента. Соответственно, ADC можно использовать в различных условиях для лечения расстройства, связанного с экспрессией антигена, с которым связывается антитело ADC, например, рака. ADC можно использовать для доставки лекарственного средства в клетку-мишень. Не ограничиваясь какой-либо теорией, в одном варианте осуществления антитело ADC связывается с или ассоциируется с антигеном или рецептором клеточной поверхности, и после связывания ADC может быть поглощен (интернализирован) внутри указанной клетки через антиген- или рецептор-опосредованное эндоцитоз или другой механизм интернализации. Антиген может быть присоединен к указанной клетке или может быть белком внеклеточного матрикса, ассоциированным с указанной клеткой. Оказавшись внутри клетки, посредством ферментативного или неферментативного механизма расщепления, в зависимости от компонентов линкерной системы, лекарственное средство высвобождается внутри клетки. В альтернативном варианте осуществления лекарственное средство отщепляется от ADC вблизи указанной клетки, и лекарственное средство впоследствии проникает в клетку.
Настоящее изобретение относится к фармацевтическим композициям, содержащим описанную в настоящей заявке композицию ADC и фармацевтически приемлемый носитель. Фармацевтические композиции могут находиться в любой форме, которая позволяет вводить ADC пациенту для лечения расстройства, связанного с экспрессией антигена, с которым связывается антитело ADC. В частности, настоящее изобретение относится к фармацевтическим композициям, содержащим по меньшей мере один конъюгат антитело-лекарственное средство формулы (III) в комбинации с одним или несколькими фармацевтически приемлемыми вспомогательными веществами. Например, фармацевтические композиции могут быть в виде жидкости или лиофилизированного твердого вещества. Среди фармацевтических композиций в соответствии с изобретением можно особо упомянуть те, которые подходят для перорального, парентерального, назального, чрескожного или транскожного, ректального, перлингвального, глазного или респираторного введения. Предпочтительным путем введения является парентеральный. Парентеральное введение включает в себя подкожные инъекции, методики внутривенной, внутримышечной и интрастернальной инъекции или инфузии. В предпочтительных вариантах осуществления фармацевтическую композицию, содержащую ADC, вводят внутривенно в виде жидкого раствора. Жидкость может быть пригодна для доставки путем инъекции. В композицию для введения путем инъекции также могут быть включены одно или несколько поверхностно-активных веществ, консервантов, смачивающих средств, диспергирующих средств, суспендирующих средств, буферов, стабилизаторов, разбавителей, смазывающих веществ, связующих веществ, дезинтегрирующих средств, абсорбентов и изотонических средств.
В другом аспекте настоящее изобретение относится к применению любого из конъюгатов антитело-лекарственное средств, определенных выше, для производства фармацевтического препарата для лечения нуждающегося в этом млекопитающего. В другом аспекте настоящее изобретение относится к любому из конъюгатов антитело-лекарственное средство, определенных выше, для применения при лечении нуждающегося в этом млекопитающего. Изобретение также относится к способам лечения нуждающегося в этом млекопитающего, при этом способ включает в себя введение млекопитающему фармацевтической композиции в терапевтически эффективной дозе.
Изобретение далее пояснено с помощью следующих примеров. Эти примеры предназначены для иллюстративных целей и не предназначены для ограничения объема изобретения.
Сокращения


Вещества и методы/общие процедуры
Все реагенты, полученные из коммерческих источников, использовали без дополнительной очистки. Безводные растворители получали из коммерческих источников и использовали без дополнительной сушки. Флэш-хроматографию выполняли на предварительно заполненных силикагелевых картриджах CombiFlash Rf (Teledyne ISCO)(Macherey-Nagel Chromabond Flash). Тонкослойную хроматографию проводили на пластинах 5 х 10 см, покрытых силикагелем Merck Type 60 F254. Микроволновый нагрев проводили на приборе СЕМ Discover®.
Измерения 1H-ЯМР проводили на спектрометре Bruker Avance 400 МГц или 500 МГц Avance Neo, с использованием ДМСО-d6 (также обозначаемого как дмсо-d6 или ДМСО) или CDCl3 в качестве растворителя. Данные 1H ЯМР представлены в виде дельта-значений, выраженных в частях на миллион (част. на млн.), с использованием остаточного пика растворителя (2,50 част. на млн. для ДМСО-d6 и 7,26 част. на млн. для CDCl3) в качестве внутреннего стандарта. Паттерны расщепления обозначаются как: s (синглет), d (дуплет), t (триплет), q (квартет), m (мультиплет), brs (широкий синглет), dd (дуплет дуплетов), brm (широкий мультиплет), td (триплет дуплетов), dt (дуплет триплетов), br dd (широкий дуплет дуплетов).
ИК измерения проводили на Bruker Tensor 27, оснащенном устройством ATR Golden Gate (SPECAC). Измерения ЯМР осуществляли на масс-спектрометре LTQ OrbiTrap Velos Pro (ThermoFisher Scientific). Образцы растворяли в CH3CN/H2O (2/1:об./об.) в диапазоне концентраций приблизительно от 0,01 до 0,05 мг/мл и вводили в источник путем впрыскивания 2 мкл в поток со скоростью 0,1 мл/мин.
Параметры ионизации ЭРИ были следующими: 3,5 кВ и 350°С переносной ионный капилляр. Все спектры были получены в режиме положительных ионов с разрешающей способностью 30 000 или 60 000 с использованием фиксирующей массы.
УЭЖХ-МС:
Waters Aquity класса А с диодным матричным УФ-детектором «PDA» и массовым устройством «ZQ detector 2» и программным обеспечением MassLinks software.
Детектор ZQ 2: сканирование МС от 0,15 до 6 мин и от 100 до 2372 Да
Детектор PDA: от 190 до 400 нм
Колонка: Acquity СВЭЖХ® ВЕН колонка C18, 1,7 мкм, 130 Å, 2,1x50 мм
Колонку использовали при 40°С со скоростью потока 0,6 мл/мин
Растворитель А: вода + 0,02% ТФУ, Растворитель В: ацетонитрил + 0,02% ТФУ
Градиент от 2% В до 100% В за 5 минут, затем 0,3 минуты промывания посредством 100% В и 0,5 минуты уравновешивания при 2% В для следующего впрыскивания (общий градиент 6 минут).
Преп. ВЭЖХ:
Interchim Puriflash 4100® с максимальным давлением 100 бар и максимальной скоростью потока 250 мл/мин или Interchim Puriflash 4250® с максимальным давлением 250 бар и максимальной скоростью потока 250 мл/мин.
Насос для четвертичного растворителя с возможностью одновременного использования 4 растворителей в градиенте
УФ: 2 длины волны для сбора от 200 до 400 нм
Сбор: пробирки 8 мл или 32 мл
Колонки: Waters Xbridge® 10 мкм
Были использованы 3 метода преп.ВЭЖХ:
1)Метод ТФУ: растворитель: А = вода+0,05% ТФУ, В = ацетонитрил + 0,05% ТФУ, градиент от 5 100% В при 15 - 30 ОК (объем колонки)
2)Метод NH4HCO3: растворитель: А = вода+0,02 М NH4HCO3, В = ацетонитрил/вода 80/20+0.02М NH4HCO3, градиент от 5 100% В при 15 - 30 ОК
3)Нейтральный метод: растворитель: А = вода, В = ацетонитрил, градиент от 5 100% В при 15 - 30 ОК
Все фракции, содержащие чистое соединение, объединяли и подвергали прямой лиофилизации с получением соединения в виде аморфного порошка.
Очистка препаративной СФХ:
Препаративную хиральную СФХ осуществляли на системе PIC solution Prep200. Образец растворяли в этаноле в концентрации 150 мг/мл. Подвижную фазу выдерживали изократически при 40% этанол/CO2. Прибор был снабжен колонкой Chiralpak IA и петлей на 3 мл. ABPR (автоматический регулятор обратного давления) был установлен на 100 бар.
Химические названия ИЮПАК были созданы с использованием программного обеспечения Biovia Draw версии 18.1.NET или программного обеспечения ACD/Name 2018.2.2 (версия файла N50E41, Build 103230, 21 июля 2018 г.)
Получение 1: (2,5-диоксопирролидин-1-ил)1-[2-[2-(2,5-диоксопиррол-1-ил)этокси]этилкарбамоил]циклобутанкарбоксилат
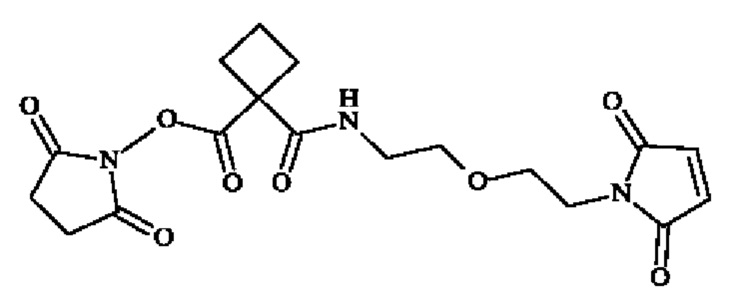
Стадия 1: трет-Бутил 1-[2-[2-(2,5-диоксопиррол-1-ил)этокси]этилкарбамоил]циклобутанкарбоксилат
В раствор 1-трет-бутоксикарбонилциклобутанкарбоновой кислоты (58.6 мг; 0,293 ммоль) в DCM (5,85 мл) последовательно добавляли 1-[2-(2-аминоэтокси)этил]пиррол-2,5-дион (53,9 мг; 0,293 ммоль), EDC (84,2 мг; 0,439 ммоль), НОВт (59,3 мг; 0.439 ммоль) и DIPEA (204 мкл; 1,17 ммоль). Реакционную смесь перемешивали при КТ в течение 18 часов и затем концентрировали досуха и солюбилизировали в DMF (1 мл). После солюбилизации остатка в DMF (1 мл), сырой продукт очищали с помощью обращенно-фазовой С18 преп. ВЭЖХ путем прямого осаждения реакционной смеси на колонку Xbridge® и используя метод ТФУ, чтобы получить указанное в заголовке соединение (57,3 мг; 0.156 ммоль). ИК (см-1): 3390, 1697/1666. 1H ЯМР (400 МГц, дмсо-d6) δ част. на млн. 7.5 (t, 1Н), 7.02 (s, 2Н), 3.55/3.5 (2t, 4Н), 3.38 (m, 2Н), 3.17 (q, 2Н), 2.33 (m, 4Н), 1.77 (m, 2Н), 1.38 (s, 9Н). СВЭЖХ-МС: МС(ЭРИ): m/z [M+Na]+=389.26, [M+H-tBu]+=311.22
Стадия 2: 1-[2-[2-(2,5-диоксопиррол-1-ил)этокси]этилкарбамоил]циклобутанкарбоноеая кислота
В раствор трет-бутил 1-[2-[2-(2,5-диоксопиррол-1-ил)этокси]этилкарбамоил]циклобутанкарбоксилата (7 мг; 0.0191 ммоль) в DCM (0,175 мл), добавляли ТФУ (51,2 мкл; 0,668 ммоль). Реакционную смесь перемешивали при КТ в течение 3,5 часов, затем концентрировали досуха с получением указанного в заголовке соединения (5,8 мг; 0.0187 ммоль) в виде бесцветного масла. Неочищенный продукт использовали в следующей стадии. СВЭЖХ-МС: МС(ЭРИ): m/z [М+Н]+=311.35, [M+Na]+=333.37
Стадия 3: Получение 1
В раствор 1-[2-[2-(2,5-диоксопиррол-1-ил)этокси]этилкарбамоил]циклобутанкарбоновой кислоты (8,47 мг; 0,0273 ммоль) в DMF (0,560 мл), последовательно добавляли TSTU (9,04 мг; 0.030 ммоль) и DIPEA (9,5 мкл; 0,0540 ммоль). Реакционную смесь перемешивали при КТ в течение 2 часов, чтобы получить раствор из Получения 1 в DMF. Неочищенный продукт будет использован непосредственно для следующих стадий. СВЭЖХ-МС: МС(ЭРИ): m/z [М+Н]+=408.43, [M+Na]+=430.38
Получение 2: (2,5-Диоксопирролидин-1-ил)3-[3-[2-(2,5-диоксопиррол-1-ил)этокси]пропаноиламино]оксетан-3-карбоксилат
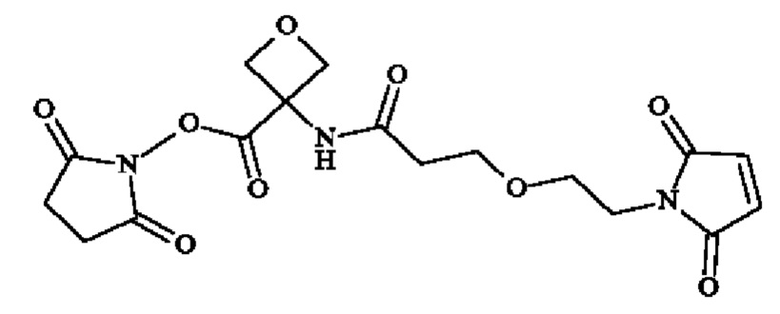
Стадия 1: синтез 3-[3-[2-(2,5-диоксопиррол-1-ил)этокси]пропаноиламино]оксетан-3-карбоновой кислоты
В раствор 3-аминооксетан-3-карбоновой кислоты (115 мг; 0,982 ммоль) в DMF (3,45 мл), последовательно добавляли (2,5-диоксопирролидин-1-ил)3-[2-(2,5-диоксопиррол-1-ил)этокси]пропаноат (274,3 мг; 0,883 ммоль) и DIPEA (855 мкл; 4,91 ммоль). Реакционную смесь перемешивали при КТ в течение 18 часов, затем концентрировали досуха и солюбилизировали в DMF (1 мл). Раствор сырого продукта очищали посредством обращенно-фазовой C18 преп. ВЭЖХ путем прямого осаждения реакционной смеси на колонку Xbridge® и используя метод ТФУ, чтобы получить указанное в заголовке соединение (16 мг; 0.0512 ммоль). СВЭЖХ-МС: МС(ЭРИ): m/z [М+Н]+=313.09, [M+Na]+=335.06. 1Н ЯМР (400 МГц, дмсо-d6) δ част. на млн. 8.92 (s, 1Н), 7.02 (s, 2Н), 4.8/4.45 (2d, 4Н), 3.57/3.48 (2m, 6Н), 2.32 (t, 2Н). IR (см-1): 3700-2300, 1769/1740/1697, 692
Стадия 2: Получение 2
В раствор 3-[2-[2-(2,5-диоксопиррол-1-ил)этокси]этилкарбамоил]оксетан-3-карбоновой кислоты (5.8 мг; 0.0187 ммоль) в DMF (0.380 мл), последовательно добавляли TSTU (6,32 мг; 0,0210 ммоль) и DIPEA (6,7 мкл; 0,0382 ммоль). Реакционную смесь перемешивали при КТ в течение 2 часов, чтобы получить раствор Получения 2 в DMF. Неочищенный продукт будет использован непосредственно для следующих стадий. СВЭЖХ-МС: МС(ЭРИ): m/z [М+Н]+=310.29, [M+Na]+=332.27
ПРИМЕР 1: 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-(гидроксиметил)бензолсульфонат натрия
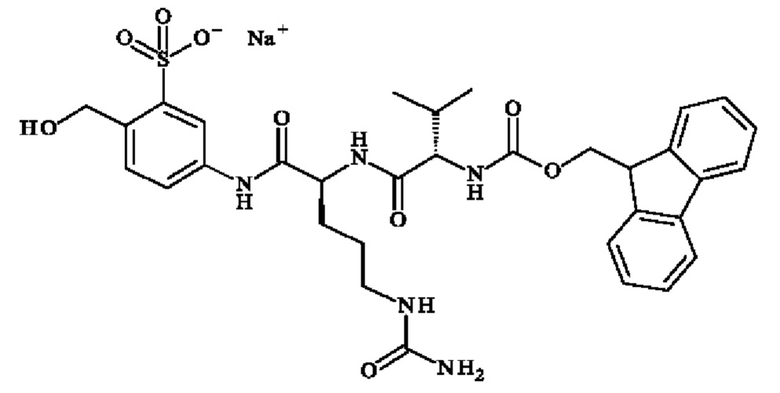
Стадия 1: натрия 2-(гидроксиметил)-5-нитро-бензолсульфонат
В раствор 5-нитро-2-[(Е)-2-(4-нитро-2-сульфо-фенил)винил]
бензолсульфоната натрия (25,0 г; 52,7 ммоль; 1 экв.) в воде (336 мл), вводили поток озона в течение 1,5 часов. После завершения реакции смесь продували аргоном в течение 30 минут для удаления избытка озона. Затем добавляли карбонат натрия (39,1 г; 368 ммоль) и боргидрид натрия (3,99 г; 105 ммоль) и раствор оранжевого цвета перемешивали при КТ в течение 16 часов. Реакционную смесь концентрировали досуха для получения указанного в заголовке соединения (39,9 г; 156 ммоль) в виде коричневого твердого вещества. 1H ЯМР (ДМСО): δ 4.99 (d, 2Н, J=3.6 Гц), 5.36 (m, 1H, J=5.6 Гц), 7.83 (d, 1Н, J=8.4 Гц), 8.21 (d, 1Н, J=8.4 Гц), 8.45 (s, 1H).
Стадия 2: 5-амино-2-(гидроксиметил)бензолсульфонат натрия
Натрия 2-(гидроксиметил)-5-нитро-бензолсульфонат (26,9 г; 105 ммоль) растворяли в воде (403 мл). Затем реакционную смесь продували аргоном. Добавляли Pd/C 10% (2,65 г), затем снова суспензию продували аргоном, после этого водородом. Реакционную смесь перемешивали при КТ в течение 3,5 дней в атмосфере водорода. После фильтрации через Celite® и промывали водой и метанолом, фильтрат концентрировали досуха и трижды выпаривали совместно с толуолом, чтобы удалить оставшиеся следы воды. Очистка колоночной хроматографией на силикагеле с использованием этилацетата/метанола (от 90/10 до 70/30) в качестве элюента давала указанное в заголовке соединение (14,29 г; 63,46 ммоль) в виде бледно-желтого твердого вещества. 1Н ЯМР (ДМСО): δ 4.52 (d, 2Н, J=5.2 Гц), 4.95 (t, 1H, J=5.2 Гц), 5.04 (s, 2Н), 6.42 (d, 1H, J=7.6 Гц), 6.93 (d, 1Н, J=7.6 Гц), 7.03 (s, 1H).
Стадия 3: 5-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-5-уреидо-пентаноил]амино]-2-(гидроксиметил)бензолсульфонат натрия
В раствор Fmoc-1-Cit-OH (№CAS 133174-15-9; 882 мг; 2.22 ммоль) в DMF (32.5 мл), добавляли 5-амино-2-(гидроксиметил)бензолсульфонат натрия (500 мг; 2.22 ммоль), HBTU (1,01 г; 2.66 ммоль) и DIPEA (917 мл; 5,55 ммоль). Реакционную смесь перемешивали при КТ в течение 16 часов, затем концентрировали досуха и выпаривали совместно с водой (2 х 100 мл). Неочищенный продукт очищали колоночной хроматографией на C18 с использованием нейтрального метода, с получением указанного в заголовке соединения (1.0 г; 1.40 ммоль) в виде бледно-красного масла. 1H ЯМР (ДМСО): δ 4.30-4.12 (m, 4Н), 4.74 (d, 2Н, J=4.4 Гц), 5.05 (t, 1Н, J=5.6 Гц), 5.37 (s, 2Н), 5.97 (t, 1Н, J=4.8 Гц), 7.34-7.42 (m, 4Н), 7.62-7.90 (m, 7Н), 8.15 (s, 1H), 10.05 (s, 1H).
Стадия 4: 5-[[(2S)-2-амино-5-уреидо-пентаноил]амино]-2-(гидроксиметил)бензолсульфонат натрия
В раствор 5-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-5-уреидо-пентаноил]амино]-2-(гидроксиметил)бензолсульфоната натрия (11,2 г; 15.73 ммоль) в DMF (224 мл), добавляли пиперидин (3,1 мл; 31.47 ммоль). Реакционную смесь перемешивали при КТ в течение 3 часов, затем добавляли воду (400 мл). Водный слой экстрагировали этилацетатом (2 х 300 мл) и посредством DCM (300 мл). В водный слой добавляли карбонат натрия (5,01 г; 47,1 ммоль; 3 экв.) и смесь перемешивали в течение 3 часов при КТ. Смесь лиофилизировали с получением указанного в заголовке соединения (6,01 г; 15,73 ммоль) в виде белого твердого вещества. 1Н ЯМР (ДМСО): δ 1.55-1.64 (m, 4Н), 2.99-3.01 (m, 2Н), 3.58 (m, 1H), 4.75 (s, 2Н), 5.06 (s, 1H), 5.38 (s, 2Н), 5.98 (t, 1H, J=5.6 Гц), 7.38 (d, 1H, J=8.4 Гц), 7.72 (dd, 1Н, J=8.4 и 2.4 Гц), 7.86 (d, 1Н, J=2.4 Гц), 10.17 (s, 1Н).
Стадия 5: Пример 1
В раствор 5-[[(2S)-2-амино-5-уреидо-пентаноил]амино]-2-(гидроксиметил)бензолсульфоната натрия (6,01 г; 15.73 ммоль) в DMF (150 мл), добавляли Fmoc-1-Val-OSu (№CAS 130878-68-1; 6,85 г; 15,69 ммоль). Бежевый раствор перемешивали при КТ в течение 3 часов, затем реакционную смесь разбавляли насыщенным гидрокарбонатом натрия (100 мл) и водой (100 мл), и концентрировали досуха. Остаток очищали на силикагеле с использованием этилацетата/метанола от 90/10 до 50/50 в качестве элюента с получением указанного в заголовке соединения (4,44 г; 6.31 ммоль) в виде белого твердого вещества. 1H ЯМР (ДМСО): 0.85-0.90 (m, 6Н), 1.31-1.76 (m, 4Н), 1.95-2.06 (m, 1Н), 2.91-3.05 (m, 2Н), 3.95 (t, 1H, J=8.4 Гц), 4.24-4.35 (m, 3Н), 4.37-4.45 (m, 1Н), 4.76 (d, 2Н, J=6 Гц), 5.07 (t, 1H, J=6.4 Гц,), 5.40 (s, 2Н), 6.03 (t, 1Н, J=5.6 Гц), 7.32-7.46 (m, 6Н), 7.67 (d, 1Н, J=8 Гц), 7.76 (t, 2Н, J=7.2 Гц), 7.88-7.91 (m, 3H), 8.12 (d, 1Н, J=7.6 Гц), 10.08 (s, 1H). 13С ЯМР (ДМСО): 18.25, 19.24, 26.70, 29.56, 30.45, 39.50, 46.67, 53.17, 60.01, 60.96, 65.66, 117.85, 119.15, 120.05, 125.36, 127.06, 127.62, 128.09, 134.39, 136.79, 140.67, 143.89, 145.34, 156.08, 158.82, 170.37, 171.16. LCMS (2-100 ацетонитрил/H2O+0,1% муравьиная кислота): 93.85% Tr=8,4 мин. Положительный режим: обнаружено 682.15 (NH+). Отрицательный режим: обнаружено 680.17 (MH-)
ПРИМЕР 2: 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]пропаноил]амино]-2-(гидроксиметил)бензолсульфоновая кислота
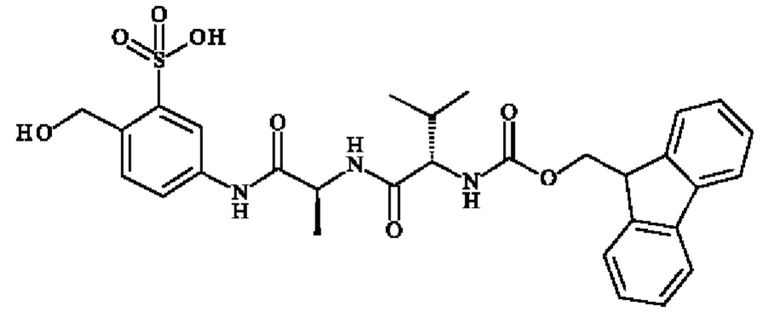
Стадия 1: 5-[[(2S)-2-(трет-бутоксикарбониламино)пропаноил]амино]-2-(гидроксиметил)бензолсульфонат натрия
В раствор Boc-1-Ala-OH (№CAS 15761-38-3; 588 мг; 3,11 ммоль) в DMF (38,6 мл) последовательно добавляли HATU (1,77 г; 4.67 ммоль), 5-амино-2-(гидроксиметил)бензолсульфонат натрия (771 мг; 3,42 ммоль) и DIPEA (1,29 мл; 7,78 ммоль). Реакционную смесь перемешивали в течение 16 часов при КТ, затем концентрировали досуха и выпаривали совместно с водой, чтобы получить сырую реакционную смесь. Полученный остаток очищали колоночной хроматографией на силикагеле, используя этилацетат/метанол от 95:5 до 80:20 в качестве элюента с получением указанного в заголовке соединения (1,17 г; 2,95 ммоль) в виде белого твердого вещества. 1Н ЯМР (ДМСО): δ 1.24 (s, 9Н), 1.38 (m, 3H), 4.05-1.44 (m, 1H), 4.73 (d, 2Н, J=4.8 Гц), 5.04 (t, 1Н, J=5.6 Гц), 6.97-7.02 (m, 1H), 7.33 (d, 1Н, J=8 Гц), 7.65-7.70 (m, 1H), 7.83 (s, 1Н), 9.91 (s, 1Н).
Стадия 2: 5-[[(2S)-2-аминопропаноил]амино]-2-(гидроксиметил)бензолсульфоновая кислота, гидрохлорид
5-[[(2S)-2-(трет-Бутоксикарбониламино)пропаноил]амино]-2-(гидроксиметил)бензолсульфонат натрия (1,17 г; 2,95 ммоль; 1 экв.) суспендировали в растворе 4 н. HCl в диоксане (10 мл). Смесь перемешивали при КТ в течение 2 часов, затем концентрировали досуха с получением неочищенной смеси (982 мг; 2.95 ммоль) в виде белого твердого вещества. 1Н ЯМР (ДМСО): δ 1.45 (d, 3H, J=5.6 Гц), 3.91-4.0 (m, 1Н), 4.76 (s, 2Н), 7.41 (d, 1H, J=7.6 Гц), 7.66 (d, 1H, J=7.6 Гц), 7.85 (s, 1H), 8.17 (s, 2Н), 10.44 (s, 1Н)
Стадия 3: Пример 2
В раствор 5-[[(2S)-2-аминопропаноил]амино]-2-(гидроксиметил)бензолсульфоновой кислоты, гидрохлорид (981 мг; 2,95 ммоль) в DMF (34,5 мл), добавляли Fmoc-1-Val-OSu (№CAS 130878-68-1; 1,29 г; 2,95 ммоль; 1 экв.) и DIPEA (975 мкл; 5.9 ммоль). Смесь перемешивали в течение ночи при КТ, затем концентрировали досуха и выпаривали совместно с водой с получением неочищенной смеси. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле, используя этилацетат / метанол от 95:5 до 80:20 в качестве элюента, чтобы получить Пример 2 (1,28 г; 2.072 ммоль) в виде бесцветного масла. 1Н ЯМР (ДМСО): δ 0.80-0.92 (m, 6Н), 1.30 (d, 3H, J=6.4 Гц), 2.02-2.10 (m, 1H), 4.17-4.31 (m, 3H), 4.37-4.44 (m, 1Н), 4.73 (d, 2Н, J=5.6 Гц), 5.04 (t, 1H, J=6.4 Гц), 7.28-7.36 (m, 3H), 7.37-7.47 (m, 3H), 7.66 (d, 1Н, J=8.4 Гц), 7.71-7.77 (m, 2Н), 7.83-7.85 (m, 1H), 7.88 (d, 2Н, J=7.6 Гц), 8.14 (d, 1H, J=6.4 Гц), 9.99 (s, 1Н).
ПРИМЕР 3: [5-[[(2S)-2-[[(2S)-2-(9Н-Флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-(гидроксиметил)фенил]метансульфонат натрия
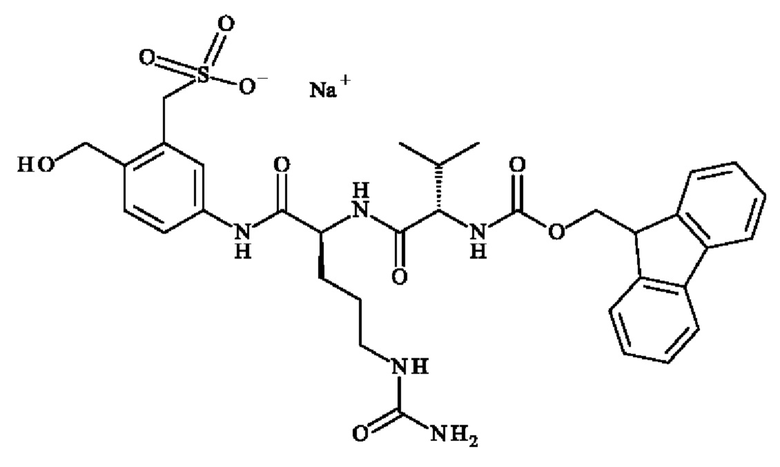
Стадия 1: (2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентановая кислота
Fmoc-1-Val-OSu (№CAS 130878-68-1; 1,59 г; 3,64 ммоль; 1,0 экв.) добавляли при КТ в раствор L(+)-цитруллина (0,67 г; 3,82 ммоль; 1,05 экв.) и бикарбонат натрия (0,32 г; 3,82 ммоль; 1,05 экв.) в смесь из 1,2-диметоксиэтана (9,5 мл), воды (9,5 мл) и ТГФ (4.7 мл). Реакционную смесь перемешивали при КТ в течение 16 часов, затем растворители удаляли в вакууме и остаток подкисляли 1 н. раствором соляной кислоты до рН 1. Белую суспензию фильтровали, промывали водой (3 х 40 мл), диэтиловым эфиром (3 х 40 мл) и выпаривали совместно с ацетонитрилом (2 х 150 мл). Полученное белое твердое вещество очищали с помощью флэш-хроматографии на силикагеле с использованием (DCM/уксусная кислота (99/1))/ метанол (от 10/0 до 7/3) в качестве элюента, затем растирали с диэтиловым эфиром (2 х 30 мл). Полученное твердое вещество растворяли в смеси из метанола (50 мл) и воды (50 мл), концентрировали досуха, выпаривали совместно с ацетонитрилом (100 мл) и сушили с получением указанного в заголовке соединения (0,75 г; 1,5 ммоль) в виде белого твердого вещества. 1Н ЯМР (ДМСО): δ 0.84-0.88 (m, 6Н), 1.34-1.41 (m, 2Н), 1.51-1.73 (m, 2Н), 1.95-2.04 (m, 1H), 2.91-2.96 (m, 2Н), 3.88-3.92 (m, 1H), 4.06-4.11 (m, 1Н), 4.19-4.31 (m, 3H), 5.36 (s, 2Н), 5.91-5.94 (m, 1Н), 7.30-7.34 (m, 2Н), 7.39-7.45 (m, 3H), 7.75 (t, 2Н, J=7.3 Гц), 7.89 (d, 2Н, J=7.4 Гц), 8.02 (brs, 1Н), 12.56 (brs, 1H)
Стадия 2: (2-Метоксикарбонил-5-нитро-фенил)метансульфонат натрия
В раствор сульфита натрия (14,39 г; 114,17 ммоль) в воде (228 мл) добавляли суспензию из метил-2-(бромметил)-4-нитробензоата (10,43 г; 38,06 ммоль) в метаноле (37 мл). Реакционную смесь перемешивали при КТ в течение 20 часов и затем концентрировали досуха. Полученный сырой продукт очищали с помощью флэш-хроматографии на силикагеле с использованием этилацетат/метанол (от 10/0 до 6/4) в качестве элюента с получением указанного в заголовке соединения (13,9 г; 31.07 ммоль) в виде твердого вещества белого цвета. 1H ЯМР (ДМСО): δ 3.82 (s, 3H), 4.29 (s, 2Н), 7.87 (d, 1Н, J=8.6 Гц), 8.16 (dd, 1H, J=2.5 и 8.6 Гц), 8.25 (d, 1Н, J=2.5 Гц)
Стадия 3: [2-(Гидроксиметил)-5-нитро-фенил]метансульфонат натрия
К суспензии (2-метоксикарбонил-5-нитро-фенил)метансульфоната натрия (10.1 г; 22,09 ммоль) в ТГФ (368 мл), добавляли боргидрид лития (0,59 г; 24.3 ммоль). Реакционную смесь перемешивали при КТ в течение 16 часов, затем снова добавляли боргидрид лития (0,27 г; 11,04 ммоль) и реакционную смесь перемешивали при КТ в течение 24 часов. Реакционную смесь разбавляли с метанолом (50 мл) и концентрировали досуха. Сырой продукт очищали с помощью флэш-хроматографии на силикагеле с использованием этилацетат/метанола (от 10/0 до 5/5) в качестве элюента с получением указанного в заголовке соединения (5,09 г; 6.52 ммоль) в виде твердого вещества желтого цвета. 1Н ЯМР (ДМСО): δ 3.90 (s, 2Н), 4.76 (d, 2Н, J=5.7 Гц), 5.38 (t, 1Н, J=5.6 Гц), 7.66 (d, 1Н, J=8.8 Гц), 8.07-8.09 (m, 2Н)
Стадия 4: [5-Амино-2-(гидроксиметил)фенил]метансульфонат натрия В раствор [2-(гидроксиметил)-5-нитро-фенил]метансульфоната натрия (2 г; 3.57 ммоль) в метаноле (102 мл)добавляли Pd/C 10% (0,56 г; 0,53 ммоль) под аргоном. Реакционную смесь продули водородом и перемешивали при КТ при атмосферном давлении водорода в течение 2 часов. Реакционную смесь затем фильтровали через Celite®, промывали метанолом (3 х 50 мл) и концентрировали досуха. Полученный сырой продукт очищали с помощью флэш-хроматографии на C18 с использованием ацетонитрил/воды (от 2/98 до 50/50) с получением указанного в заголовке соединения (1,98 г; 3,46 ммоль) в виде твердого вещества серого цвета. 1Н ЯМР (ДМСО): δ 3.72 (s, 2Н), 4.3 (d, 2Н, J=5.9 Гц), 4.89 (s, 2Н), 4.96 (t, 1H, J=5.9 Гц), 6.38 (dd, 1H, J=2.4 и 8.1 Гц), 6.49 (d, 1H, J=2.4 Гц), 6.9 (d, 1H, J=8.1 Гц)
Стадия 5: Пример 3
В раствор (2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентановой кислоты (543 мг; 1,09 ммоль) в сухом DMF (6 мл), добавляли HATU (600 мг; 1.58 ммоль), гидрокарбонат натрия (102 мг; 1,22 ммоль) и суспензию [5-амино-2-(гидроксиметил)фенил]метансульфоната натрия (650 мг; 1,41 ммоль) в сухом DMF (14 мл). Реакционную смесь перемешивали при КТ в течение 3 часов, а затем выпаривали совместно с диоксаном (200 мл). Сырой продукт очищали с помощью флэш-хроматографии на силикагеле с использованием DCM/метанола (от 10/0 до 5/5) в качестве элюента, а затем с помощью флэш-хроматографии на C18 с использованием ацетонитрил/вода+0,1% муравьиной кислоты (от 2/98 до 50/50) в качестве элюента, чтобы получить после сублимационной сушки Пример 3 (233 мг; 0,32 ммоль) в виде белого твердого вещества. 1Н ЯМР (ДМСО): δ 0.84-0.89 (m, 6Н), 1.32-1.49 (m, 2Н), 1.54-1.73 (m, 2Н), 1.94-2.04 (m, 1H), 2.89-3.09 (m, 2Н), 3.81 (s, 2Н), 3.91-3.95 (m, 1H), 4.20-4.33 (m, 3H), 4.41-4.46 (m, 1Н), 4.47 (s, 2Н), 5.07 (brs, 1H), 5.40 (brs, 2Н), 5.98 (t, 1Н, J=5.7 Гц), 7.21 (d, 1H, J=8.2 Гц), 7.31-7.36 (m, 3H), 7.39-7.45 (m, 3H), 7.63 (dd, 1Н, J=2.2 и 8.3Hz), 7.75 (t, 2Н, J=7.5Hz), 7.89 (d, 2H, J=7.4 Гц), 8.08 (d, 1H, J=7.6 Гц), 10.01-10.8 (m, 1H). ЖХ-МС (2-100 ACN/H2O+0,1% ТФУ): 93.75% Tr=8,1 мин. Отрицательный режим: обнаружено 694.14 (М-Н)
ПРИМЕР 4: 2-(хлорметил)-5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]бензолсульфоновая кислота
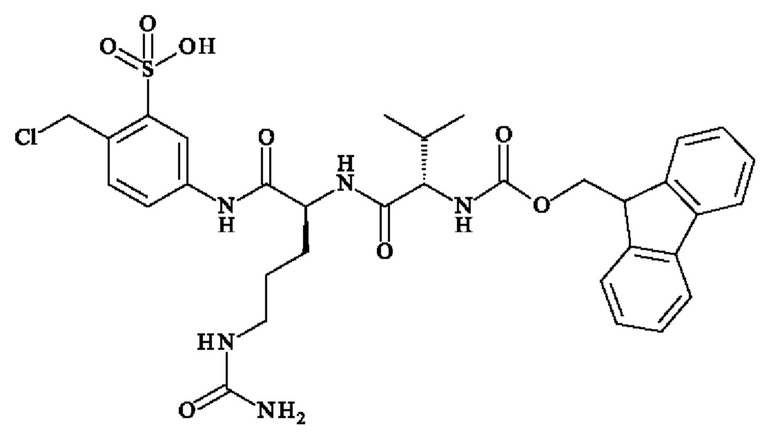
5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-(гидроксиметил)бензолсульфоновую кислоту (свободная кислота из Примера 1; 300 мг, 0,4263 ммоль) растворяли в безводном NMP (6 мл) при КТ. Параллельно готовили раствор SOCl2 (206 мкл) в NMP (6 мл). К реакционной смеси 6 раз добавляли в течение периода времени 75 минут раствор 900 мкл указанного раствора SOCl2. После последнего добавления реакционную смесь перемешивали при КТ в течение 15 минут. Сырой продукт очищали путем прямого осаждения реакционной смеси на колонке Oasis с использованием метода ТФУ с получением указанного в заголовке соединения (138 мг; 0,1971 ммоль) в виде белого порошка. 1Н ЯМР (400 МГц, дмсо-d6) δ част. на млн. 10.15+8.1+7.42+6.0 (s+2d+m, 4Н), 7.9 (m, 3H), 7.75 (m, 3H), 7.42+7.31 (2 m, 5Н), 5.23 (s, 2Н), 4.4 (m, 1Н), 4.3-4.2 (m, 3H), 3.95 (dd, 1Н), 3.0 (m, 2Н), 2.0 (m, 1Н), 1.7+1.6 (2 m, 2Н), 1.48+1.37 (2m, 2Н), 0.88 (2d, 6Н). HR-ЭРИ+: m/z [М+Н]+=700.2199 / 700.2202 [измерено/теоретически]
ПРИМЕР 5: 2-(Хлорметил)-5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]пропаноил]амино]бензолсульфоновая кислота
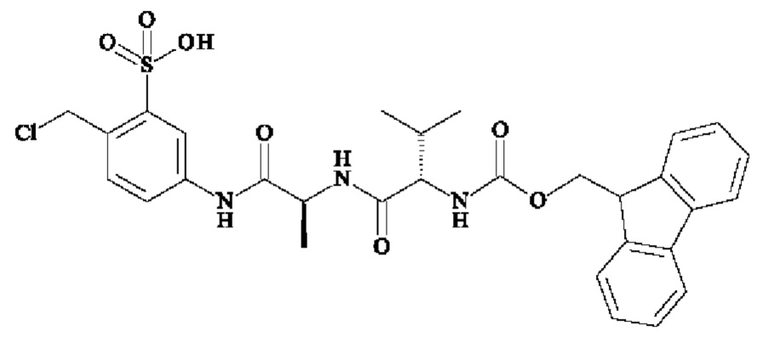
В раствор 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]пропаноил]амино]-2-(гидроксиметил)бензолсульфоната (сульфонат, полученный из Примера 2; 504,1 мг, 0.816 ммоль) в NMP (5 мл), 6 раз в течение 75-минутного периода добавляли раствор SOCl2 (60 мкл 0,816 ммоль) в NMP (500 мкл). Реакционную смесь перемешивали при КТ в течение 15 минут. Сырой продукт очищали путем прямого осаждения реакционной смеси на колонке Oasis с использованием метода ТФУ, чтобы получить (337 мг) в виде белого порошка. ИК длина волны (см-1): от 3600 до 2400, 1688+1648, 1599, 1518, 1022. СВЭЖХ-МС: МС (ЭРИ)m/z [М+Н]+=614.17+616.18 (Cl)
ПРИМЕР 6: 5-[[(2S)-2-[[(2S)-2-[3-[2-(2,5-Диоксопиррол-1-ил)этокси]пропаноиламино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфонат натрия
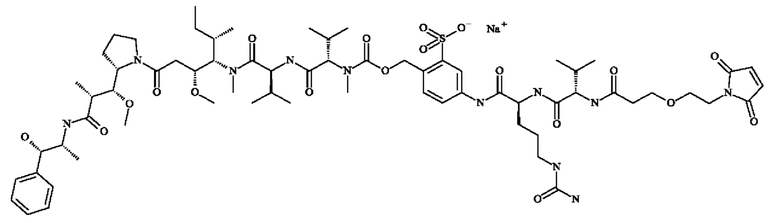
Стадия 1: 5-[[(2S)-2-[[(2S)-2-(9H-Флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[(4-нитрофенокси)карбонилоксиметил]бензолсульфонат натрия
В раствор Примера 1 (450 мг; 0.64 ммоль) в DMF (6 мл)добавляли DIEA (1,34 мл; 7.67 ммоль) и бис(4-нитрофенил)карбонат (778 мг; 2.56 ммоль). Раствор перемешивали при КТ в течение 2 часов и добавляли бис(4-нитрофенил)карбонат (390 мг; 1.28 ммоль). Через 1 час раствор концентрировали под сниженным давлением и остаток очищали с помощью хроматографии на силикагеле (градиент метанола и уксусной кислоты в DCM), чтобы получить указанное в заголовке соединение (523 мг; 0.45 ммоль). 1Н ЯМР (400 МГц, дмсо-d6) δ част. на млн. 10.2/8.1/5.95 (m, 3H), 8.3 (d, 2Н), 7.95 (s, 1Н), 7.9 (d, 2Н), 7.75 (dd, 1Н), 7.75 (m, 2Н), 7.65 (d, 2Н), 7.4 (d, 4Н), 7.35 (d, 1Н), 5.7 (s, 2Н), 5.35 (brs, 2Н), 4.4 (m, 1H), 4.3 (t, 1H), 4.2 (d, 2Н), 3.95 (m, 1H), 3 (m, 2Н), 2 (m, 1Н), 1.8-1.3 (m, 4Н), 0.85 (2d, 6Н)
Стадия 2: 5-[[(2S)-2-[[(2S)-2-амино-3-метил-бутаноил]амино]-5-урейдо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1- [(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновая кислота, соль трифторуксусной кислоты
В раствор ММАЕ (200 мг; 0,28 ммоль) в DMF (5.6 мл), добавляли DIEA (0,19 мл; 1,39 ммоль), затем 5-[[(2S)-2-[[(2S)-2-(9H-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[(4-нитрофенокси)карбонилоксиметил]бензолсульфонат натрия (472 мг; 0,55 ммоль) и HOBt (75 мг; 0,56 ммоль). Раствор перемешивали при КТ в течение 3,5 часов, затем DEA (0,19 мл; 1.39 ммоль) добавляли. Через 2 часа раствор концентрировали под сниженным давлением и остаток очищали с помощью C18 обращенно-фазовой препаративной ВЭЖХ на Xbridge® способом ТФУ, чтобы получить указанное в заголовке соединение (99 мг; 0.08 ммоль). 1Н ЯМР (400 МГц, дмсо-d6) δ част. на млн. 10.2/10 (2s, 1H), 8.56/8.35 (2m, 1H), 8.1 (d, 1H), 7.9 (brs, 1H), 7.88/7.6 (2d, 1Н), 7.68 (brs, 1Н), 7.35-7.13 (m, 5Н), 7.3 (brs, 1Н), 6/5.96 (2t, 1H), 5.5 (m, 2Н), 5.4 (brs, 2Н), 4.8-4.15 (m, 5Н), 4.05-3.85 (m, 2Н), 3.8-3.6 (5s, 9Н), 3.62-3.5 (m, 2Н), 3.1-2.8 (m, 5Н), 3 (brs, 3H), 2.4/2.3 (d+dd, 2Н), 2.2-1.9 (m, 3H), 1.9-1.75 (m, 11Н), 1.05/1 (2d, 9Н), 1-0.75 (m, 21Н)
Стадия 3: Пример 6
В раствор 5-[[(2S)-2-[[(2S)-2-амино-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-мето кси-2-метил-3-оксо-пропил]пирролидин- 1-ил]-2-метокси-1-[(1S)- 1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновой кислоты, соль ТФУ (22 мг; 0,018 ммоль) в DMF (0.18 мл), добавляли DIEA (6,3 мкл мл; 36 мкммоль) и 1-[2-[3-(2,5-диоксопирролидин-1-ил)-3-оксо-пропокси]этил]пиррол-2,5-дион (6,2 мг; 20 мкмоль). Раствор перемешивали при КТ в течение 4,5 часов. Реакционную смесь очищали с помощью C18 обращенно-фазовой препаративной ВЭЖХ на колонке Xbridge® с использованием метода NH4HCO3 с получением Примера 6 (9.6 мг; 6.8 мкмоль). HRMS (ЭРИ)[М+Н]+ обнаружено=1420.7041 (δ=-0,3 част. на млн.)
ПРИМЕР 7: 5-[[(2S)-2-[[(2S)-2-[6-(2,5-Диоксопиррол-1-ил)гексаноиламино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфонат натрия
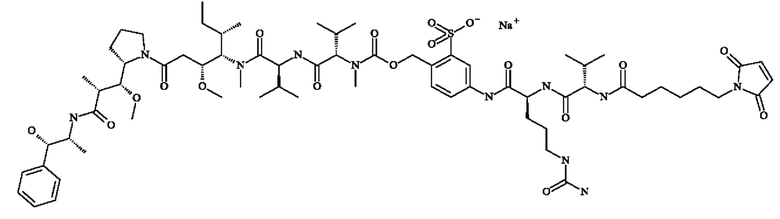
В раствор 5-[[(2S)-2-[[(2S)-2-амино-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-мето кси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновой кислоты, соль ТФУ (полученную из Стадии 2 Примера 6; 30 мг; 0.025 ммоль) в DMF (0.25 мл)добавляли DIEA (8,7 мкл мл; 36 мкммоль) и (2,5-диоксопирролидин-1-ил)6-(2,5-диоксопиррол-1-ил)гексаноат (8,46 мг; 0,027 ммоль). Раствор перемешивали при КТ в течение 3,5 часов. Реакционную смесь очищали с помощью C18 обращенно-фазовой препаративной ВЭЖХ на колонке Xbridge® с использованием метода NH4HCO3 с получением Пример 7 (20 мг; 14.09 мкмоль). HRMS (ЭРИ)[М+Н]+ обнаружено=1418.7253 (δ=0,1 част. на млн.)
ПРИМЕР 8: 5-[[(2S)-2-[[(2S)-2-[[2-[2-[2-(2азидоэтокси)этокси]этокси]ацетил]амино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфонат натрия
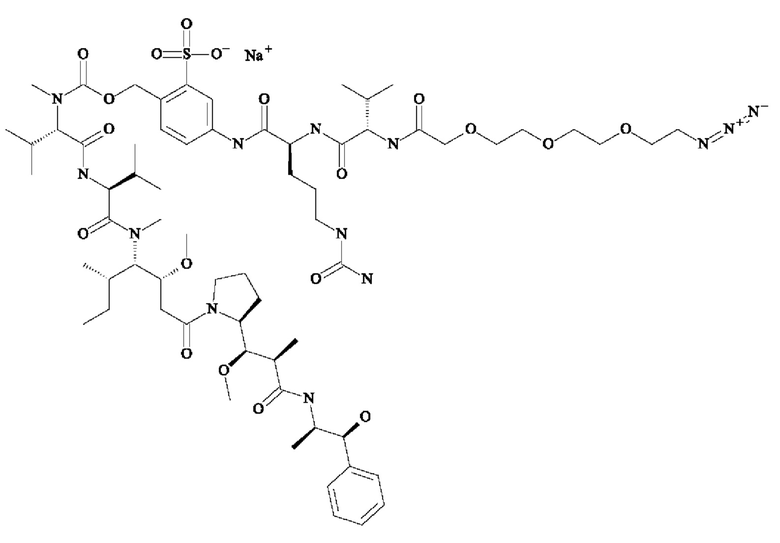
В раствор 2-[2-[2-(2-азидоэтокси)этокси]этокси]уксусной кислоты (6,4 мг; 0.027 ммоль) в DMF (125 мкл), добавляли DIEA (21,7 мкл; 124 мкммоль) и TSTU (7,88 мг; 0,026 ммоль). После завершения активации добавляли раствор 5-[[(2S)-2-[[(2S)-2-амино-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновой кислоты, соль ТФУ (30 мг; 0,025 ммоль) в DMF (0,15 мл). Раствор перемешивали при КТ в течение 2 часов. Реакционную смесь очищали с помощью C18 обращенно-фазовой препаративной ВЭЖХ на колонке Xbridge® с использованием метода NH4HCO3 с получением соединения Примера 8 (16 мг; 11,10 мкмоль). HRMS (ЭРИ)[М+Н]+ обнаружено=1440.7382 (δ=-2,6 част. на млн.)
ПРИМЕР 9: 5-[[(2S)-2-[[1-[2-[2-(2,5-диоксопиррол-1-ил)этокси]этилкарбамоил]циклобутанкарбонил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновая кислота
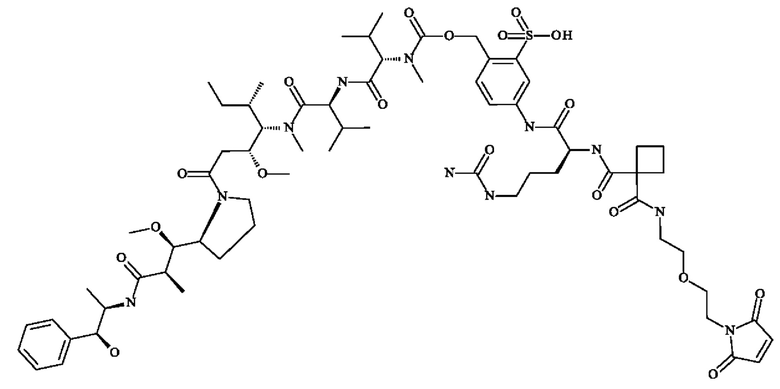
Стадия 1: 5-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-5-уреидо-пентаноил]амино]-2-[(4-нитрофенокси)карбонилоксиметил]бензолсульфонат натрия
В раствор 5-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-5-уреидо-пентаноил]амино]-2-(гидроксиметил)бензолсульфоната натрия (261 мг; 0,448 ммоль) в DMF (5,75 мл), последовательно добавляли бис(4-нитрофенил) карбонат (1,09 г; 3.58 ммоль) и DIPEA (1,87 мл; 10.75 ммоль). Реакционную смесь перемешивали при КТ в течение 16 часов, затем концентрировали досуха с получением неочищенной смеси. Сырой продукт очищали с помощью хроматографии на силикагеле (градиент метанола, содержащий 2% АсОН в DCM)с получением указанного в заголовке соединения (137 мг; 0,183 ммоль) в виде белого твердого вещества. СВЭЖХ-МС: МС(ЭРИ): m/z [М+Н]+=748.48
Стадия 2: 5-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1- [(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфонат натрия
В раствор натриевой соли 5-[[(2S)-2-(9Н-флуорен-9-илметоксикарбонил амино)-5-уреидо-пентаноил]амино]-2-[(4-нитрофенокси)карбонилоксиметил]бензолсульфоновой кислоты (64,5 мг; 0,0863 ммоль) в DMF (1,42 мл), последовательно добавляли ММАЕ (61,9 мг; 0,0863 ммоль), DIPEA (75,1 мкл; 0.431 ммоль) и HOBt (23,3 мг; 0,173 ммоль). Реакционную смесь перемешивали при КТ в течение 16 часов. Сырой продукт очищали с помощью C18 обращенно-фазовой преп. ВЭЖХ путем прямого осаждения реакционной смеси на колонку Xbridge® и используя метод ТФУ, чтобы получить указанное в заголовке соединение (33.5 мг; 0.0253 ммоль). СВЭЖХ-МС: МС(ЭРИ): m/z [М+Н]+=1326.88, [M+Na]+=1348.09
Стадия 3: 5-[[(2S)-2-амино-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фения-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоноеая кислота, соль ТФУ
В раствор 5-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-мето кси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1- [(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоната натрия (36,8 мг; 0.0292 ммоль) в DMF (750 мкл), добавляли пиперидин (23,1 мкл; 0,234 ммоль). Реакционную смесь перемешивали при КТ в течение 17 часов. Раствор сырого продукта очищали посредством C18 обращенно-фазовой преп. ВЭЖХ путем прямого осаждения реакционной смеси на колонку Xbridge® и используя метод ТФУ, чтобы получить указанное в заголовке соединение (17 мг; 0,0139 ммоль). СВЭЖХ-МС: МС(ЭРИ): m/z [М+Н]+=1038.10, [M+Na]+=1059.85
Стадия 4: Пример 9
В раствор 5-[[(2S)-2-амино-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновой кислоты, соль ТФУ (24,5 мг; 0,0215 ммоль) в DMF (735 мкл), последовательно добавляли соединение из Получения 1 (10.5 мг; 0,0258 ммоль) и DIPEA (19 мкл; 0.108 ммоль). Реакционную смесь перемешивали при КТ в течение 16 часов. Раствор сырого продукта очищали посредством C18 обращенно-фазовой преп. ВЭЖХ путем прямого осаждения реакционной смеси на колонку Xbridge® и с использованием метода ТФУ, чтобы получить Пример 9 (19,3 мг; 0,0147 ммоль) в виде белого порошка. НЯ-ЭРИ+: m/z [М+Н]+=1396.7040 / 1396.7074 [измерено/теоретически]
ПРИМЕР 10: 5-[[(2S)-2-[[(2S)-2-[3-[2-(2,5-диоксопиррол-1-ил)этокси]пропаноиламино]-3-метил-бутаноил]амино]пропаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновая кислота
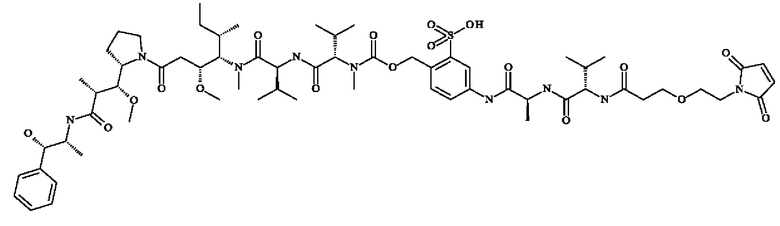
Стадия 1: 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикар6ониламино)-3-метил-бутаноил]амино]пропаноил]амино]-2-[(4-нитрофенокси)карбонилоксиметил]бензолсульфоноеая кислота
К суспензии Примера 2 (1,28 г; 2,07 ммоль) в ТГФ (65 мл), добавляли пиридин (875 мкл; 10,8 ммоль), а затем 4-нитрофенил хлорформиат (1,09 г; 5,41 ммоль). Смесь перемешивали в течение ночи при КТ. Затем добавляли еще 4-нитрофенил хлорформиат (1,09 г; 5,41 ммоль; 2,5 экв.). После 5 часов перемешивания при КТ, смесь концентрировали досуха затем очищали с помощью колоночной хроматографии на C18 с использованием вода/ацетонитрил от 90/10 до 0/100 в качестве элюента за 30 минут. Ацетонитрил из объединенных пробирок удаляли, а остаток лиофилизировали с получением указанного в заголовке соединения (650 мг; 0,83 ммоль) в виде белого твердого вещества. 1Н ЯМР (ДМСО): δ 0.88 (m, 6Н), 1.31 (d, 3H, J=4.8 Гц), 1.97-2.03 (m, 1Н), 3.92 (t, 1Н, J=6.8 Гц), 4.23 (s, 2Н), 4.24-4.34 (m, 1Н), 4.42 (t, 1Н, J=5.6 Гц), 5.69 (s, 2Н), 7.30-7.48 (m, 6Н), 7.62 (d, 2Н, J=8 Гц), 7.72-7.76 (m, 3H), 7.89 (d, 2Н, J=6.4 Гц), 7.94 (s, 1H), 8.18 (d, 1H, J=5.6 Гц), 8.33 (d, 2H, J=7.6 Гц), 10.11 (s, 1H). 13C ЯМР (ДМСО): δ 18.01, 18.26, 19.21, 30.4, 46.66, 49.05, 59.91, 65.67, 67.82, 117.7, 119.1, 120.06, 122.66, 125.37, 126.33, 127.05, 127.62, 128.0, 138.06, 140.67, 143.77, 143.86, 145.1, 146.23, 151.96, 155.47, 156.12, 171.0, 171.15. LCMS (2-100 ACN/H2O+0.05% ТФУ): 90.41% Tr=12,7 мин. Положительный режим 578.41 обнаружено. Отрицательный режим 759,17 обнаружено
Стадия 2: 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]пропаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновая кислота
В раствор 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]пропаноил]амино]-2-[(4-нитрофенокси)карбонилоксиметил]бензолсульфоновая кислота (29,3 мг; 0,0374 ммоль) в DMF (1.20 мл), последовательно добавляли ММАЕ (26.9 мг; 0.0374 ммоль), DIPEA (32.6 мкл; 0.187 ммоль) и HOBt (10.1 мг; 0,075 ммоль). Реакционную смесь перемешивали при КТ в течение 16 часов. Сырой продукт очищали с помощью C18 обращенно-фазовой преп. ВЭЖХ путем прямого осаждения реакционной смеси на колонку Xbridge® и используя метод ТФУ, чтобы получить указанное в заголовке соединение (35.4 мг; 0,0264 ммоль). СВЭЖХ-МС: МС(ЭРИ): m/z [М+Н]+=1340.9
Стадия 3: 5-[[(2S)-2-[[(2S)-2-амино-3-метил-бутаноил]амино]пропаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновая кислота, соль ТФУ
В раствор 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]пропаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфонов ой кислоты (50,1 мг; 0,0374 ммоль) в DMF (1,5 мл), добавляли пиперидин (29,6 мкл; 0,299 ммоль). Реакционную смесь перемешивали при КТ в течение 1 часа. Раствор сырого продукта очищали посредством C18 обращенно-фазовой преп. ВЭЖХ путем прямого осаждения реакционной смеси на колонку Xbridge® и используя метод ТФУ, чтобы получить указанное в заголовке соединение (23 мг; 0,0187 ммоль) в виде белого порошка. НЯ-ЭРИ+: m/z [М+Н]+=1117.6235/1117.6218, [М+2Н]/2+=559.3146/559.3148 [измерено/теоретически]
Стадия 4: Пример 10
В раствор 5-[[(2S)-2-[[(2S)-2-амино-3-метил-бутаноил]амино]пропаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновой кислоты, соль ТФУ (10,4 мг; 0,00845 ммоль) в DMF (0.34 мл), последовательно добавляли (2,5-диоксопирролидин-1-ил)3-[2-(2,5-диоксопиррол-1-ил)этокси]пропаноат (2,9 мг; 0,00929 ммоль) и DIPEA (7,36 мкл; 0,0422 ммоль). Реакционную смесь перемешивали при КТ в течение 16 часов. Сырой продукт очищали с помощью C18 обращенно-фазовой преп. ВЭЖХ путем прямого осаждения реакционной смеси на колонку Xbridge® и с использованием метода ТФУ, чтобы получить Пример 10 (7,7 мг; 0,0059 ммоль) в виде белого порошка. HR-ЭРИ+: m/z [М+Н]+=1312.6701/1312.6750, [измерено/теоретически]
ПРИМЕР 11: 5-[[(2S)-2-[[3-[2-[2-(2,5-диоксопиррол-1-ил)этокси]этилкарбамоил]оксетан-3-карбонил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновая кислота
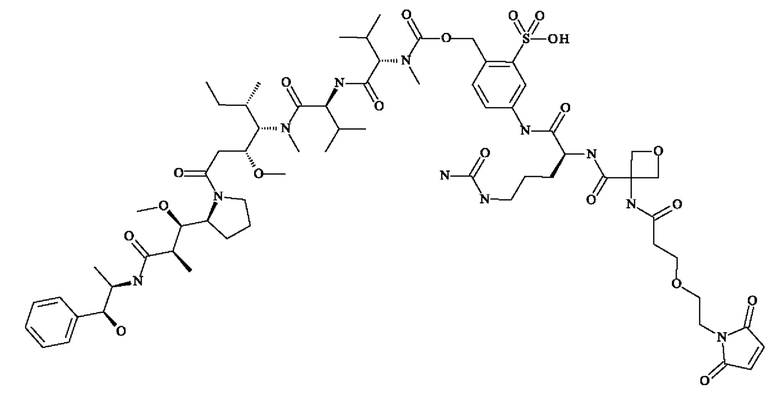
В раствор 5-[[(2S)-2-амино-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновой кислоты; 2,2,2-трифторуксусной кислоты (16 мг; 0,0131 ммоль) в DMF (480 мкл), последовательно добавляли соединение из Получения 2 (10,24 мг; 0.0250 ммоль) и DIPEA (11,4 мкл; 0,0657 ммоль). Реакционную смесь перемешивали при КТ в течение 16 часов. Раствор сырого продукта очищали посредством C18 обращенно-фазовой преп. ВЭЖХ путем прямого осаждения реакционной смеси на колонку Xbridge® и с использованием метода ТФУ, чтобы получить Пример 11 (4,4 мг; 0,0031 ммоль) в виде белого порошка. HR-ЭРИ+: m/z [М+2Н]2+=699.8477 / 699.8472 [измерено/теоретически]
ПРИМЕР 12: [5-[[(2S)-2-[[(2S)-2-[3-[2-(2,5-диоксопиррол-1-ил)этокси]пропаноиламино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1E,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]фенил]метансульфоновая кислота
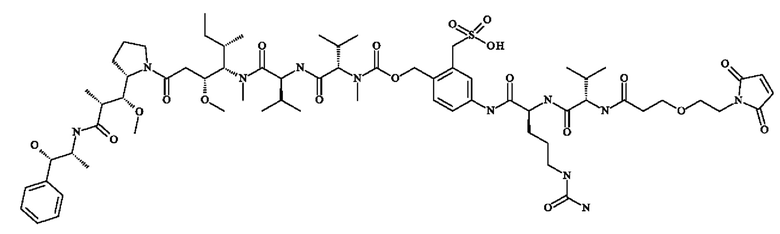
Стадия 1: [5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[(4-нитрофенокси)карбонилоксиметил]фенил]метансульфонат натрия
В раствор Примера 3 (30 мг; 0.0418 ммоль) в DMF (0.4 мл)добавляли DIEA (87 мкл; 70.501 ммоль) и бис(4-нитрофенил)карбонат (51 мг; 0Д67 ммоль). Раствор перемешивали при КТ в течение 2 часов и снова добавляли бис(4-нитрофенил)карбонат (25 мг; 0,08 ммоль). Через 1 час при КТ раствор концентрировали под сниженным давлением, и остаток очищали с помощью хроматографии на силикагеле (градиент метанола в DCM), чтобы получить указанное в заголовке соединение (25 мг; 0,030 ммоль).
Стадия 2: [5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]фенил]метансульфоноеая кислота
В раствор ММАЕ (10 мг; 0.0139 ммоль) в DMF (0.6 мл), добавляли DIEA (12 мкл; 0,0696 ммоль), а затем [5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[(4-нитрофенокси)карбонилоксиметил]фенил]метансульфонат натрия (24 мг; 0,028 ммоль) и HOBt (3,77 мг; 0,0279 ммоль). Раствор перемешивали при КТ в течение 3,5 часов, и раствор непосредственно очищали с помощью C18 обращенно-фазовой препаративной ВЭЖХ на Xbridge® способом ТФУ, чтобы получить указанное в заголовке соединение (5,4 мг; 0,003 ммоль). HRMS (ЭРИ)[М+Н]+ обнаружено=1439.7558 (δ=1.9 част. на млн.)
Стадия 3: [5-[[(2S)-2-[[(2S)-2-амино-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1- [(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]фенил]метан сульфоновая кислота, соль ТФУ
[5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-l-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]фенил]метансульфоновую кислоту (5 мг; 3,4 мкмоль)растворяли в DMF (0.2 мл), затем добавляли DEA (0,7 мкл; 6,8 ммоль). Реакционную смесь перемешивали при КТ в течение 1 часа, и раствор очищали с помощью C18 обращенно-фазовой препаративной ВЭЖХ на Xbridge® способом ТФУ, чтобы получить указанное в заголовке соединение (3,5 мг; 2,6 мкмоль).
Стадия 4: Пример 12
В раствор [5-[[(2S)-2-[[(2S)-2-амино-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]фенил]метансульфоновой кислоты, соль ТФУ (3,5 мг; 2,6 мкмоль) в DMF (15 мкл), добавляли DIEA (2,3 мкл; 13 мкммоль) и 1-[2-[3-(2,5-диоксопирролидин-1-ил)-3-оксо-пропокси]этил]пиррол-2,5-дион (0,9 мг; 2,9 мкмоль). Раствор перемешивали при КТ в течение 2,5 часов. Реакционную смесь очищали с помощью C18 обращенно-фазовой препаративной ВЭЖХ на колонке Xbridge® с использованием метода NH4HCO3 с получением Примера 12 (2,8 мг; 1,8 мкмоль). HRMS (ЭРИ)[М+Н]+ обнаружено=1412.7397 (δ=1.1 част. на млн.)
ПРИМЕР 13: 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-(йодметил)бензолсульфоновая кислота
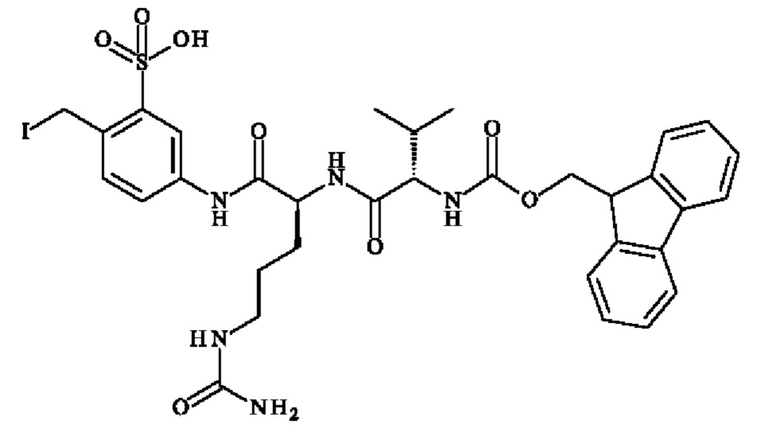
В раствор Примера 4 (100 мг; 123 мкмоль) в растворе в ацетоне (6 мл)добавляли йодид натрия (51 мг; 340 мкмоль). Реакционную смесь перемешивали при КТ в течение 20 часов. Растворитель выпаривали, и соединение использовали в следующей стадии без дополнительной обработки. СВЭЖХ-МС: [М+Н]+ 792.65; [M+Na]+ 814.49
ПРИМЕР 14: [4-[[(2S)-2-[[(2S)-2-[3-[2-(2,5-диоксопиррол-1-ил)этокси]пропаноиламино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-сульфо-фенил]метил-[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-диметил-аммоний; 2,2,2-трифторацетат
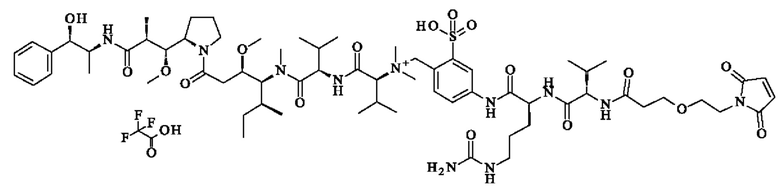
Стадия 1: [4-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-сульфо-фенил]метил-[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-диметил-аммоний; йодид
В раствор (2S)-2-[[(2S)-2-(диметиламино)-3-метил-бутаноил]амино]-N-[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-N,3-диметил-бутанамида (ауристатин Е)(42,1 мг; 57,5 мкмоль) в DMF (10 мл), последовательно добавляли Пример 13 (152 мг; 95,8 мкмоль) и DIPEA (83,4 мкл; 365 мкмоль). Реакционную смесь перемешивали при КТ в течение 17 часов. Желаемый продукт наблюдали с помощью СВЭЖХ-МС и раствор использовали без доработки на следующей стадии. СВЭЖХ-МС: [М+Н]+ 1396.31; [M+Na]+ 1418.37
Стадия 2: [4-[[(2S)-2-[[(2S)-2-амино-3-метил-бутаноил]амино]-5-урейдо -пентаноил]амино]-2-сульфо-фенил]метил-[(1 S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R, 2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-диметил-аммоний;2,2,2-трифторацетат;2,2,2-трифторуксусная кислота
В раствор [4-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-сульфо-фенил]метил-[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-диметил-аммоний; йодид в DMF, добавляли пиперидин (45,4 мкл; 460 мкмоль) и реакционную смесь перемешивали при КТ в течение 1 часа. Сырой продукт очищали с помощью C18 обращенно-фазовой препаративной ВЭЖХ путем прямого осаждения реакционной смеси на колонку Xbridge® и с использованием метода ТФУ, чтобы получить целевой продукт (15 мг; 10,7 мкмоль) в виде белого твердого вещества. СВЭЖХ-МС: [М+Н]+ 1174.51; [M+Na]+ 1197.03
Стадия 3: Пример 14
В раствор [4-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-сульфо-фенил]метил-[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]- 1-мето кси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-диметил-аммония; йодид (15 мг; 10,7 мкмоль) в растворе в DMF (450 мкл), последовательно добавляли (2,5-диоксопирролидин-1-ил)-3-[2-(2,5-диоксопиррол-1-ил)этокси]пропаноат (3,7 мг; 11,8 мкмоль) и DIPEA (9,3 мкл; 53,5 мкмоль). Раствор перемешивали при КТ в течение 1 часа. Сырой продукт очищали с помощью C18 обращенно-фазовой преп. ВЭЖХ путем прямого осаждения реакционной смеси на колонку Xbridge® и с использованием метода ТФУ, чтобы получить Пример 14 (15,7 мг; 12,1 мкмоль) в виде белого твердого вещества. HRMS (ЭРИ)[M-CF3CO2]+1368.7460 (δ=-1.7 част. на млн.)
ПРИМЕР 23: N-({[4-({N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-L-аланил}амино)-2-сульфофенил]метокси}карбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-1-валинамид
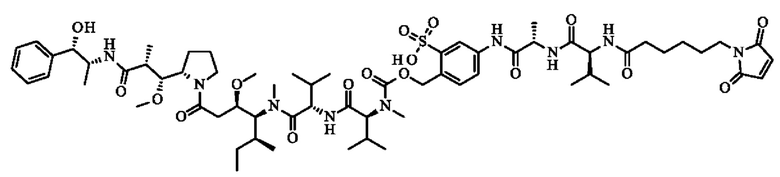
В раствор 5-[[(2S)-2-[[(2S)-2-амино-3-метил-бутаноил]амино]пропаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновой кислоты, соль ТФУ (12.7 мг; 0,0103 ммоль; получено из Стадии 3 Примера 10) в DMF (0.4 мл), добавляли DIEA (9 мкл; 51 мкмоль) и (2,5-диоксопирролидин-1-ил)6-(2,5-диоксопиррол-1-ил)гексаноат (3,5 мг; 11,3 мкмоль). Раствор перемешивали при КТ в течение 6 часов. Реакционную смесь очищали с помощью C18 обращенно-фазовой препаративной ВЭЖХ на колонке Xbridge® с использованием метода ТФУ, чтобы получить Пример 23. IR (см-1): 3278, 1768/1703, 1631, 1159, 829/696. RMN 1H (500 МГц, ДМСО-d6) δ част. на млн. 9.96 (s, 1Н), 8.1 (s, 1Н), 7.87 (m, 2Н), 7.68 (m, 1Н), 7.6 (d, 1H), 7.31 (m, 2H), 7.26 (m, 3H), 7.17 (m, 1H), 6.99 (s, 2H), 5.48 (m, 2H), 4.74 (m, 1H), 4.49 (m, 1H), 4.46 (m, 1H), 4.41 (m, 1H), 4.38 (m, 1H), 4.33 (m, 1H), 4.19 (m, 1H), 3.98 (m, 1H), 3.98 (m, 1H), 3.77 (m, 1H), 3.6 (m, 2H), 3.56 (m, 1H), 3.46 (m, 1H), 3.36 (m, 2H), 3.32 (m, 1H), 3.26 (m, 2H), 3.24 (m, 3H), 3.23 (m, 2H), 3.19 (m, 3H), 3.13 (m, 2H), 3.04 (m, 1H), 2.98 (m, 3H), 2.94 (m, 1H), 2.9 (m, 1H), 2.86 (m, 1H), 2.37 (br dd, 2H), 2.12 (m, 2H), 2.12 (m, 2H), 2.06 (m, 2H), 1.97 (m, 2H), 1.49 (m, 9H), 1.29 (d, 3H), 0.89 (m, 27H). HRMS (ЭРИ)[M+H]+ обнаружено=1310.6893 (δ=-4.5 част. на млн.)
ПРИМЕР 24: N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-(4-{[({[(4S)-4,11-диэтил-9-гидрокси-3,14-диоксо-3,4,12,14-тетрагидро-1H-пирано[3',4':6,7]индолизино[1,2-b]хинолин-4-ил]окси}карбонил)окси]метил}-3-сульфофенил)-L-орнитинамид
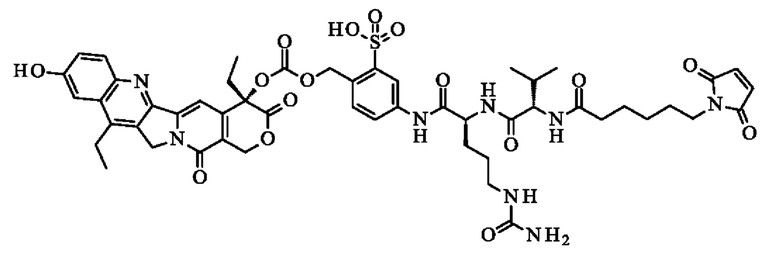
Стадия 1: 5-[[(2S)-2-[[(2S)-2-амино-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-(гидроксиметил)бензолсульфоновая кислота
В раствор Примера 1 (200 мг; 0,284 ммоль) в DMF (8 мл), добавляли DEA (212 мкл; 2.053 ммоль) и реакционную смесь перемешивали при КТ в течение 1 часа. Избыток DEA выпаривали в вакууме и раствор сырого целевого соединения в DMF использовали непосредственно в следующей стадии.
Стадия 2: 5-[[(2S)-2-[[(2S)-2-[6-(2,5-диоксопиррол-1-ил)гексаноиламино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-(гидроксиметил)бензолсульфоновая кислота
В раствор (8 мл)соединения из предыдущей стадии добавляли DIEA (102 мкл; 0,5866 ммоль) и (2,5-диоксопирролидин-1-ил)6-(2,5-диоксопиррол-1-ил)гексаноат (99,4 мг; 0.3226 ммоль). Реакционную смесь перемешивали при КТ в течение 18 часов. Реакционную смесь очищали с помощью C18 обращенно-фазовой препаративной ВЭЖХ на колонке Xbridge® с использованием метода ТФУ, чтобы получить после лиофилизации целевое соединение. ИК (см-1): между 3674 и 2999, 1768 (слабый) + 1699+1641, 1238 / 1141, 825 и 694. RMN 1H (400 МГц, ДМСО-d6) δ част. на млн. 9.97 (s, 1H), 8.04 (d, 1Н), 7.85 (d, 1H), 7.77 (d, 1Н), 7.65 (dd, 1Н), 7.32 (d, 1Н), 6.99 (s, 2Н), 6.02 (m, 1H), 5.18 (m, 1Н), 4.72 (s, 2Н), 4.36 (m, 1H), 4.21 (dd, 1Н), 3.37 (t, 2Н), 2.98 (m, 2Н), 2.16 (m, 2Н), 1.97 (m, 1Н), 1.65 (m, 2Н), 1.39 (m, 8Н), 0.83 (dd, 6Н). HRMS (ЭРИ)[М+Н]+ обнаружено=653.2572 (δ=-4.2 част. на млн.)
Стадия 3: трет-бутил [(19S)-10,19-диэтил-19-гидрокси-14,18-диоксо-1 7-окса-3,13-диазапентацикло[11.8.0.02,11.04,9.015,20]геникоза-1(21), 2,4(9), 5, 7,10,15(20)-гептаен-7-ил]карбонат
К суспензии (19S)-10,19-диэтил-7,19-дигидрокси-17-окса-3,13-диазапентацикло[11.8.0.02,11.04,9.015,20]геникоза-1(21),2,4(9),5,7,10,15(20)-гептаен-14,18-диона (220 мг; 0,5607 ммоль) в DCM (22 мл), добавляли трет-бутоксикарбонил трет-бутил карбонат (128.5 мг; 0,5887 ммоль) в виде порошка и пиридин (91 мкл; 1,121 ммоль). Желтую суспензию перемешивали при КТ в течение 18 часов. Растворитель и избыток пиридина упаривали в вакууме и сырой продукт очищали с помощью хроматографии на силикагеле (DCM/MeOH) с получением ожидаемого соединения в виде желтого порошка. ИК (см-1): 3700-3000, 1753, 1659, 1254/1142. RMN 1H (400 МГц, ДМСО-d6) δ част. на млн. 8.21 (d, 1Н), 8.09 (d, 1H), 7.74 (dd, 1H), 7.33 (s, 1Н), 6.5 (s, 1H), 5.38 (2s, 4Н), 3.2 (q, 2Н), 1.86 (m, 2Н), 1.54 (s, 9Н), 1.3 (t, 3H), 0.88 (t, 3H)
Стадия 4: трет-бутил-[(19S)-19-хлоркарбонилокси-10,19-диэтил-14,18-диоксо-17-окса-3,13-диазапентацикло[11.8.0.02,11.04,9.015,20]геникоза-1(21), 2,4(9), 5, 7,10,15(20)-гептаен- 7-ил]карбонат
В раствор трет-бутил-[(19S)-10,19-диэтил-19-гидрокси-14,18-диоксо-17-окса-3,13-диазапентацикло[11.8.0.02,11.04,9.015,20]геникоза-1(21),2,4(9),5,7,10,15(20)-гептаен-7-ил]карбоната (30 мг; 0.0609 ммоль) в DCM (2 мл)добавляли DMAP (22.3 мг; 0.1827 ммоль), а затем трифосген (7,2 мг; 0,0244 ммоль). Реакционную смесь перемешивали при КТ в течение 30 минут и этот раствор использовали как таковой в следующей стадии.
Стадия 5: 2-[[(19S)-7-трет-бутоксикарбонилокси-10,19-диэтил-14,18-диоксо-17-окса-3,13-диазапентацикло[11.8.0.02,11.04,9.015,20]геникоза-1(21),2,4(9), 5, 7,10,15(20)-гептаен-19-ил]оксикарбонилоксиметил]-5-[[(2S)-2-[[(2S)-2-[6-(2,5-диоксопиррол-1-ил)гексаноиламино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]бензолсульфоновая кислота
В раствор трет-бутил-[(19S)-19-хлоркарбонилокси-10,19-диэтил-14,18-диоксо-17-окса-3,13-диазапентацикло[11.8.0.02,11.04,9.015,20]геникоза-1(21),2,4(9),5,7,10,15(20)-гептаен-7-ил]карбоната в растворе в DCM (2 мл), добавляли DMAP (22,3 мг; 0,1827 ммоль), а затем 5-[[(2S)-2-[[(2S)-2-[6-(2,5-диоксопиррол-1-ил)гексаноиламино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-(гидроксиметил)бензолсульфоновую кислоту (31,80 мг; 0,04873 ммоль; получено согласно Стадии 2 выше). Реакционную смесь перемешивали при КТ в течение 14 часов. Реакционную смесь очищали с помощью C18 обращенно-фазовой препаративной ВЭЖХ на колонке CSH® с использованием метода ТФУ, чтобы получить после лиофилизации целевое соединение.
Стадия 6: Пример 24
В раствор 2-[[(19S)-7-трет-бутоксикарбонилокси-10,19-диэтил-14,18-диоксо-17-окса-3,13-диазапентацикло[11.8.0.02,11.04,9.015,20]геникоза-1(21),2,4(9),5,7,10,15(20)-гептаен-19-ил]оксикарбонилоксиметил]-5-[[(2S)-2-[[(2S)-2-[6-(2,5-диоксопиррол- 1-ил)гексаноиламино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]бензолсульфоновой кислоты (12,5 мг; 1,66 мкмоль) в DCM (2,5 мл), добавляли ТФУ (0.3 мл) и раствор перемешивали при КТ в течение 1,5 часов. Реакционную смесь очищали с помощью C18 обращенно-фазовой препаративной ВЭЖХ на колонке CSH® с использованием метода ТФУ, чтобы получить после лиофилизации Пример 24. HRMS (ЭРИ)[М+Н]+ обнаружено=1071.3768 (δ=0,4 част. на млн.)
ПРИМЕР 25: (1S,3S)-3,5,12-тригидрокси-3-(гидроксиацетил)-10-метокси-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил 2,3,6-тридеокси-3-[({[4-({N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-L-орнитил}амино)-2-сульфофенил]метокси}карбонил)амино]-α-L-ликсо-гексопиранозид
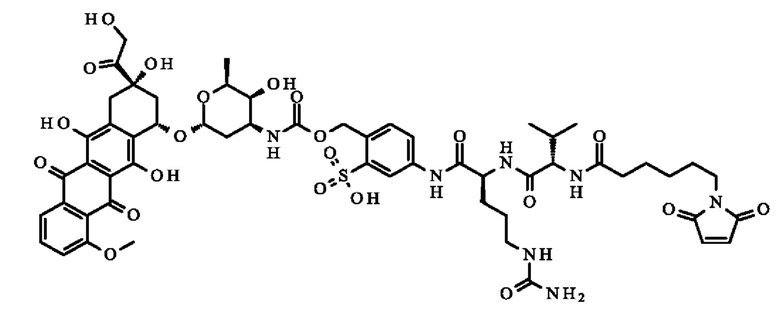
Стадия 1: 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[(2S,3S,4S,6R)-3-гидрокси-2-метил-6-[[(1S,3S)-3,5,12-тригидрокси-3-(2-гидроксиацетил)-10-метокси-6,11-диоксо-2,4-дигидро-1Н-тетрацен-1-ил]окси]тетрагидропиран-4-ил]карбамоилоксиметил]бензолсульфоновая кислота
В раствор 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[(4-нитрофенокси)карбонилоксиметил]бензолсульфоната натрия (157 мг; 0,1854 ммоль; полученный из Стадии 1 Примера 6) в DMF (11 мл), добавляли (7S,9S)-7-[(2R,4S,5S,6S)-4-амино-5-гидрокси-6-метил-тетрагидропиран-2-ил]окси-6,9,11-тригидрокси-9-(2-гидроксиацетил)-4-метокси-8,10-дигидро-7Н-тетрацен-5,12-дион (107,5 мг; 0,1853 ммоль), а затем DIEA (323 мкл; 1,854 ммоль). Красная смесь становилась темно-фиолетовой и через 2 часа превращение завершалось. Этот раствор использовали как таковой в следующей стадии.
Стадия 2: 5-[[(2S)-2-[[(2S)-2-амино-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[(2S,3S,4S, 6R)-3-гидрокси-2-метил-6-[[(1S,3S)-3,5,12-тригидрокси-3-(2-гидроксиацетил)-10-метокси-6,11-диоксо-2,4-дигидро-1H-тетрацен-1-ил]окси]тетрагидропиран-4-ил]карбамоилоксиметил]бензолсульфоновая кислота
В раствор 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[(2S,3S,4S,6R)-3-гидрокси-2-метил-6-[[(1S,3S)-3,5,12-тригидрокси-3-(2-гидроксиацетил)-10-метокси-6,11-диоксо-2,4-дигидро-1Н-тетрацен-1-ил]окси]тетрагидропиран-4-ил]карбамоилоксиметил]бензолсульфоновой кислоты, полученный в предыдущей стадии, добавляли DEA (96 мкл; 0,9268 ммоль). Реакционную смесь перемешивали при КТ в течение 1 часа. Растворитель частично выпаривали и сырой продукт очищали с помощью C18 обращенно-фазовой препаративной ВЭЖХ на колонке X-Bridge® с использованием метода ТФУ, чтобы получить после лиофилизации целевое соединение. ИК (см-1): 3357, 2661, 1666/1608, 1585/1531, 1201, 1082, 1020, 763/709. RMN 1H (500 МГц, ДМСО-d6) δ част. на млн. 14.06 (s, 1Н), 13.29 (s, 1H), 10.15 (s, 1Н), 8.62 (d, 1Н), 8.04 (гл, 3H), 7.93 (d, 1Н), 7.93 (t, 1Н), 7.89 (d, 1H), 7.66 (dd, 1Н), 7.61 (dd, 1H), 7.26 (d, 1Н), 6.88 (d, 1Н), 5.99 (t, 1H), 5.49 (brs, 1H), 5.37 (m, 2Н), 5.25 (brs, 1Н), 4.97 (t, 1H), 4.79 (brm, 2Н), 4.58 (s, 2Н), 4.46 (q, 1Н), 4.18 (q, 1H), 3.99 (s, 3H), 3.75 (m, 1H), 3.63 (m, 1H), 3.51 (brs, 1Н), 2.99 (m, 2Н), 2.99 (m, 2Н), 2.16 (m, 2Н), 2.07 (m, 1Н), 1.87 (td, 1H), 1.72 (m, 1Н), 1.61 (m, 1H), 1.5 (dt, 1Н), 1.45 (m, 1H), 1.37 (m, 1H), 1.14 (d, 3H), 0.93 (2d, 6Н)
Стадия 3: Пример 25
В раствор 5-[[(2S)-2-[[(2S)-2-амино-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[(2S,3S,4S,6R)-3-гидрокси-2-метил-6-[[(1S,3S)-3,5,12-тригидрокси-3-(2-гидроксиацетил)-10-метокси-6,11-диоксо-2,4-дигидро-1Н-тетрацен-1-ил]окси]тетрагидропиран-4-ил]карбамоилоксиметил]бензолсульфоновой кислоты (5 мг; 4,7 мкмоль) в DMF (1 мл), добавляли DIEA (1,6 мкл; 5,2 мкммоль) и (2,5-диоксопирролидин-1-ил)6-(2,5-диоксопиррол-1-ил)гексаноат (1.6 мг; 5.2 мкмоль). Раствор перемешивали при КТ в течение 12 часов. Реакционную смесь очищали с помощью C18 обращенно-фазовой препаративной ВЭЖХ на колонке Xbridge® с использованием метода ТФУ, чтобы получить Пример 25. HRMS (ЭРИ)[М+Н]+ обнаружено=1222.4153 (δ=1,7 част. на млн.)
ПРИМЕР 26: натрия N-{[(9H-флуорен-9-ил)метокси]карбонил}-L-валил-N5-карбамоил-N-[4-(гидроксиметил)-3-(2-сульфонатоэтил)фенил]-L-орнитинамид
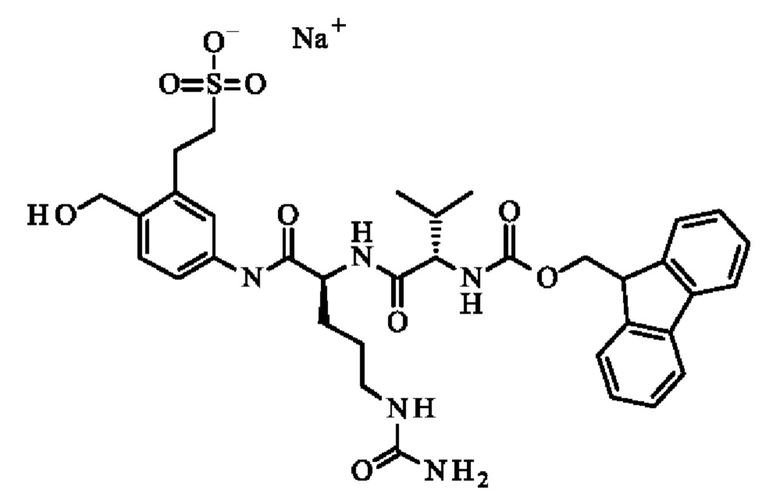
Стадия 1: (2-аллил-4-нитро-фенил)метокси-трет-бутил-диметил-силан
Раствор трет-бутил-[(2-йод-4-нитро-фенил)метокси]-диметил-силана (6,56 г; 16.68 ммоль) и аллилтрибутилолова (7,76 мл; 25.02 ммоль) в 1,4-диоксане (165 мл) продували аргоном (3 раза). Добавляли тетракис(трифенилфосфин)палладий(0)(1,93 г; 1,67 ммоль) и смесь снова продули аргоном (3 раза) и перемешивали при 100°С в течение 18 часов. Реакционную смесь фильтровали через Celite® и концентрировали досуха. Остаток растворяли в DCM (150 мл) и промывали 1М водным раствором гидроксида натрия. Водный слой экстрагировали с DCM и объединенные органические слои промывали соляным раствором, сушили над сульфатом натрия, фильтровали и концентрировали досуха. Полученный сырой продукт очищали с помощью флэш-хроматографии на силикагеле с использованием циклогексан/EtOAc (от 10/0 до 95/5) в качестве элюента, чтобы получить целевое соединение (4.91 г; 15.97 ммоль) в виде масла оранжевого цвета. 1H ЯМР (ДМСО): δ 0.11 (s, 6Н), 0.92 (s, 9Н), 3.49 (d, 2Н, J=6.5 Гц), 4.84 (s, 2Н), 5.04-5.15 (m, 2Н), 5.92-6.02 (m, 1H), 7.69 (d, 1Н, J=8.5 Гц), 8.02 (d, 1Н, J=2.5 Гц), 8.13 (dd, 1Н, J=2.5 и 8.5 Гц)
Стадия 2: 3-[2-[[трет-бутил(диметил)силил]оксиметил]-5-нитро-фенил]пропан-1,2-диол
В раствор (2-аллил-4-нитро-фенил)метокси-трет-бутил-диметил-силана (4.91 г; 15.97 ммоль) в смеси ацетона (147 мл) и воды (20 мл), добавляли 4-метилморфолин N-оксид (3.74 г; 31.94 ммоль) и осмат калия (VI)дигидрат (0.29 г; 0.8 ммоль), и смесь перемешивали при КТ в течение 20 часов. Реакционную смесь гасили насыщенным водным раствором тиосульфата натрия (40 мл) и воды (40 мл), и водный слой экстрагировали посредством EtOAc. Объединенные органические слои промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали досуха. Полученный сырой продукт очищали с помощью флэш-хроматографии на силикагеле с использованием циклогексан / EtOAc (от 10/0 до 5/5) в качестве элюента, чтобы получить целевое соединение (4.65 г; 13.62 ммоль) в виде желтого масла. 1H ЯМР (ДМСО): δ 0.11 (s, 6Н), 0.93 (s, 9Н), 2.58 (dd, 1H, J=8.8 и 14.2 Гц), 2.9 (dd, 1Н, J=3.5 и 14.2 Гц), 3.26-3.30 (m, 1Н), 3.36-3.41 (m, 1Н), 3.61-3.68 (m, 1Н), 4.7 (t, 1H, J=5.7 Гц), 4.75 (d, 1H, J=5.4 Гц), 4.84-4.93 (m, 2Н), 7.65-7.67 (m, 1Н), 8.08-8.11 (m, 2Н)
Стадия 3: 2-[2-[[трет-бутил(диметил)силил]оксиметил]-5-нитро-фенил]ацеталъдегид
В раствор 3-[2-[[трет-бутил(диметил)силил]оксиметил]-5-нитро-фенил]пропан-1,2-диола (3,05 г; 8,22 ммоль) в смеси воды (18 мл) и ТГФ (51 мл) при 0°С, добавляли порционно периодат натрия (5,27 г; 24.65 ммоль). Реакционную смесь перемешивали в течение 2 часов от 0°С до 10°С, затем гасили насыщенным водным раствором тиосульфата натрия (50 мл) и воды (50 мл). Водный слой экстрагировали с EtOAc. Объединенные органические слои промывали соляным раствором, сушили над сульфатом натрия, фильтровали и концентрировали досуха при 30°С, чтобы получить целевое соединение (2,61 г; 8,22 ммоль) в виде желтого масла, используемое как таковое для следующей стадии. 1H ЯМР (ДМСО): δ 0.08 (s, 6Н), 0.90 (s, 9Н), 4.04 (s, 2Н), 4.72 (s, 2Н), 7.7 (d, 1H, J=8.8 Гц), 8.13 (d, 1Н, J=2.5 Гц), 8.18 (dd, 1Н, J=2.4 и 8.5 Гц), 9.69 (s, 1Н)
Стадия 4: 2-[2-[[трет-бутил(диметил)силил]оксиметил]-5-нитро-фенил]этанол
В раствор 2-[2-[[трет-бутил(диметил)силил]оксиметил]-5-нитро-фенил]ацетальдегида (2.61 г;) в МеОН (40 мл), добавляли при 0°С боргидрид натрия (0,47 г; 12.33 ммоль). Реакционную смесь нагревали до КТ и перемешивали в течение 1 часа, затем гасили добавлением рассола (25 мл) и воды (25 мл). Водный слой экстрагировали посредством EtOAc, затем объединенные органические слои промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали досуха. Полученный сырой продукт очищали с помощью флэш-хроматографии на силикагеле с использованием циклогексан / EtOAc (от 10/0 до 6/4), чтобы получить целевое соединение (1,36 г; 4,36 ммоль) в виде желтого твердого вещества. 1H ЯМР (ДМСО): 5 0.11 (s, 6Н), 0.92 (s, 9Н), 2.82 (t, 2H, J=6.4 Гц), 3.67 (q, 2H, J=5.9 Гц), 4.74 (t, 1H, J=5.1 Гц), 4.87 (d, 2H), 7.66 (d, 1H, J=8.6 Гц), 8.08-8.11 (m, 2H)
Стадия 5: [2-(2-бромэтил)-4-нитро-фенил]метокси-трет-бутил-диметил-силан
В раствор 2-[2-[[трет-бутил(диметил)силил]оксиметил]-5-нитро-фенил]этанола (1,26 г; 4,05 ммоль) в ТГФ (25 мл) при 0°С, добавляли TEA (1,12 мл; 8,09 ммоль) и мезилхлорид (0,47 мл; 6,07 ммоль). Реакционной смеси давали медленно нагреваться до КТ, перемешивали в течение 5 часов, затем фильтровали и промывали посредством ТГФ. Фильтрат добавляли в раствор бромида лития (1,76 г; 20,23 ммоль) в ТГФ (25 мл) при 0°С. Реакционной смеси давали медленно нагреваться до КТ, перемешивали в течение 4 дней, затем гасили насыщенным водным раствором хлорида аммония (50 мл). Водный слой экстрагировали с EtOAc. Объединенные органические слои промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали досуха, чтобы получить целевое соединение (1,38 г; 4,03 ммоль) в виде желтого масла использовали как таковой для следующей стадии. 1Н ЯМР (ДМСО): δ 0.12 (s, 6Н), 0.92 (s, 9Н), 3.27 (t, 2Н, J=7.2 Гц), 3.81 (t, 2Н, J=7.2 Гц), 4.87 (s, 2Н), 7.68 (d, 1Н, J=8.4 Гц), 8.13-8.16 (m, 1Н), 8.18-8.19 (m, 1Н)
Стадия 6: [2-(2-бромэтил)-4-нитро-фенил]метанол
В раствор [2-(2-бромэтил)-4-нитро-фенил]метокси-трет-бутил-диметил-силана (1,38 г) в ТГФ (28 мл) и воде (14 мл) при 0°С, добавляли уксусную кислоту (42 мл; 737,28 ммоль). Реакционной смеси давали медленно нагреться до КТ, перемешивали в течение 4 дней, затем разбавляли с водой (100 мл). Водный слой экстрагировали с EtOAc. Объединенные органические слои промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали досуха. Полученный сырой продукт очищали с помощью флэш-хроматографии на силикагеле с использованием циклогексан/EtOAc (от 10/0 до 6/4) в качестве элюента, чтобы получить целевое соединение (0,73 г; 2,81 ммоль) в виде бежевого твердого вещества. 1Н ЯМР (ДМСО): δ 3.27 (t, 2Н, J=7.2 Гц), 3.8 (t, 2Н, J=7.2 Гц), 4.67 (s, 2Н), 5.51 (brs, 1Н), 7.7 (d, 1H, J=8.4 Гц), 8.11-8.14 (m, 1H), 8.15-8.16 (m, 1H)
Стадия 7: 2-[2-(гидроксиметил)-5-нитро-фенил]этилсульфонилоксинатрий
В раствор [2-(2-бромэтил)-4-нитро-фенил]метанола (188 мг; 0,72 ммоль) в EtOH (750 мкл), последовательно добавляли раствор сульфита натрия (273 мг; 2.17 ммоль) в воде (1.1 мл) и йодид тетрабутиламмония (13 мг; 0.036 ммоль). Реакционную смесь нагревали при 70°С под микроволновым облучением в течение 35 минут. Реакционную смесь разбавляли водой (20 мл) и промывали посредством EtOAc. Водный слой концентрировали досуха, и полученный сырой продукт очищали с помощью флэш-хроматографии на силикагеле с использованием DCM / МеОН (от 10/0 до 6/4) в качестве элюента, а затем с помощью флэш-хроматографии на C18 с использованием ACN / вода (от 2/98 до 34/66) в качестве элюента, чтобы получить целевое соединение (80 мг; 0.28 ммоль) в виде белого твердого вещества. 1Н ЯМР (ДМСО): δ 2.66-2.70 (m, 2Н), 2.94-2.98 (m, 2Н), 4.64 (d, 2Н, J=5.3 Гц), 5.47 (t, 1Н, J=5.3 Гц), 7.68 (d, 1Н, J=8.5 Гц), 7.99 (d, 1Н, J=2.3 Гц), 8.06 (dd, 1H, J=2.4 и 8.4 Гц)
Стадия 8: 2-[5-амино-2-(гидроксиметил)фенил]этилсульфонилоксинатрия
В раствор [2-(2-бромэтил)-4-нитро-фенил]метанола (293 мг; 1,03 ммоль) в МеОН (11 мл) и воды (2 мл), добавляли Pd/C 10% (55 мг; 0.052 ммоль) под аргоном. Реакционную смесь продули водородом и перемешивали при КТ при атмосферном давлении водорода в течение 16 часов. Реакционную смесь фильтровали через 40 мкм PTFE фильтр, промывали смесью МеОН/вода (1/1) и концентрировали досуха, чтобы получить целевое соединение (274 мг; 1.03 ммоль) в виде желтого твердого вещества, используемого как таковое в следующей стадии. 1H ЯМР (ДМСО): δ 2.55-2.59 (m, 2Н), 2.75-2.79 (m, 2Н), 4.32 (d, 2Н, J=5.3 Гц), 5.66 (m, 1Н, J=5.2 Гц), 4.86 (brs, 2Н), 6.33-6.35 (m, 2Н), 6.93 (d, 1H, J=7.8 Гц)
Стадия 9: Пример 26
В раствор 2-[5-амино-2-(гидроксиметил)фенил]этилсульфонилоксинатрия (304 мг; 1.15 ммоль) в DMF (6 мл), добавляли (2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентановую кислоту (474 мг; 0.96 ммоль), HATU (472 мг; 1.24 ммоль) и гидрокарбонат натрия (160 мг; 1,91 ммоль). Реакционную смесь перемешивали при КТ в течение 20 часов, разбавляли с водой (50 мл) и промывали посредством EtOAc. Водный слой концентрировали досуха и очищали с помощью флэш-хроматографии на C18 с использованием ACN / вода + ТФУ (0,1%)(от 2/98 до 50/50) в качестве элюента, чтобы после сублимационной сушки получить Пример 26 (83 мг; 0.11 ммоль) в виде бледно-желтого твердого вещества. 1Н ЯМР (ДМСО): δ 0.84-0.88 (m, 6Н), 1.32-1.49 (m, 2Н), 1.54-1.74 (m, 2Н), 1.94-2.04 (m, 1H), 2.58-2.62 (m, 2Н), 2.83-2.87 (m, 2Н), 2.90-3.05 (m, 2Н), 3.93 (t, 1H, J=8 Гц), 4.23-4.34 (m, 3H), 4.37-4.43 (m, 1Н), 4.45 (s, 2Н), 5.34 (brs, 2Н), 5.98 (brs, 1Н), 7.25 (d, 1H, J=8.1 Гц), 7.30-7.34 (m, 2Н), 7.39-7.44 (m, 4Н), 7.74 (t, 2Н, J=7.2 Гц), 7.89 (d, 2Н, J=7.5 Гц), 8.08 (d, 1Н, J=7.5 Гц), 9.90 (s, 1H). LCMS (2-100 ACN / H2O+0,1% ТФУ): 76,60% Tr=8.1 мин. Отрицательный режим 708.45 обнаружено (М-Н)
ПРИМЕР 27: N-{3-[2-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)этокси]пропаноил}-L-валил-N-{4-[(5S,8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-оксоэтил)-4,10-диметил-3,6,9-триоксо-5,8-ди(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадекан-1-ил]-3-(2-сульфоэтил)фенил}-N5-карбамоил-L-орнитинамид
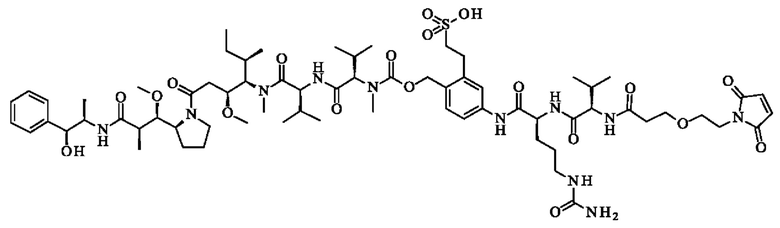
Стадия 1: 2-[5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[(4-нитрофенокси)карбонилоксиметил]фенил]этансульфоновая кислота
В раствор Примера 26 (32 мг, 0,0436 ммоль) в DMF (0,5 мл)добавляли бис(4-нитрофенил)карбонат (53.1 мг; 0,1747 ммоль) и DIEA (91 мкл; 0,5240 ммоль). Раствор перемешивали при КТ в течение 4 часов. После выпаривания досуха под вакуумом сырой продукт очищали с помощью хроматографии на силикагеле, чтобы получить целевое соединение.
Стадия 2: 2-[5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]фенил]этансульфоновой кислоты
В раствор ММАЕ (17 мг, 0,02368 ммоль) в DMF (6 мл), добавляли 2-[5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[(4-нитрофенокси)карбонилоксиметил]фенил]этансульфоновой кислоты (41,4 мг; 0.0473 ммоль), DIEA (20 мкл; 0,1184 ммоль) и HOBt (6,4 мг; 0,047 ммоль). Реакционную смесь перемешивали при КТ в течение ночи и этот раствор было задействован как таковой на следующей стадии.
Стадия 3: 2-[5-[[(2S)-2-[[(2S)-2-амино-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1- [(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]фенил]этансульфоноеая кислота, ТФУ
В раствор 2-[5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]фенил]этансульфоновой кислоты добавляли DEA (24 мкл; 0,2338 ммоль) и раствор перемешивали при КТ в течение 1,5 часов. Реакционную смесь очищали с помощью C18 обращенно-фазовой препаративной ВЭЖХ на колонке CSH® с использованием метода ТФУ, чтобы после лиофилизации получить целевое соединение.
Стадия 4: Пример 27
В раствор 2-[5-[[(2S)-2-[[(2S)-2-амино-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-мето кси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]фенил]этансульфоновой кислоты, ТФУ (9,5 мг; 7,1 ммоль) в DMF (0.2 мл), добавляли DIEA (2.5 мкл; 0.014 ммоль), а затем (2,5-диоксопирролидин-1-ил)3-[2-(2,5-диоксопиррол-1-ил)этокси]пропаноат (2.4 мг; 7,8 мкмоль). Реакционную смесь перемешивали при КТ в течение ночи. Реакционную смесь очищали с помощью C18 обращенно-фазовой препаративной ВЭЖХ на колонке CSH® с использованием метода ТФУ, чтобы после лиофилизации получить Пример 27. HRMS (ЭРИ)[М+Н]+ обнаружено=1426.7443 (δ=-6.7 част. на млн.)
ПРИМЕР 28: 5-[[(2S)-2-[[(2S)-2-[6-(3,4-дибром-2,5-диоксо-пиррол-1-ил)гексаноиламино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновая кислота
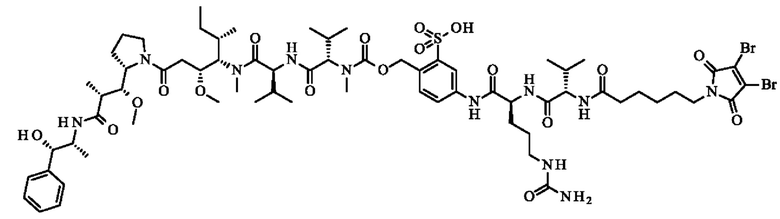
В раствор 6-(3,4-дибром-2,5-диоксо-пиррол-1-ил гексановой кислоты в растворе в DMF (0.2 мл)добавляли HATU (11.3 мг; 29,8 мкмоль) в виде порошка. Реакционную смесь перемешивали при КТ в течение 10 минут и добавляли лутидин (6,3 мкл; 54,2 ммоль). Реакционную смесь перемешивали при КТ в течение 2 часов. Затем добавляли 5-[[(2S)-2-[[(2S)-2-амино-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновую кислоту (10 мг; 8,3 мкмоль; полученную из Стадии 2 Примера 6) и реакционную смесь перемешивали при КТ в течение 15 часов. Реакционную смесь очищали с помощью C18 обращенно-фазовой препаративной ВЭЖХ на колонке X-Bridge® с использованием метода ТФУ, чтобы после лиофилизации получить целевое соединение. HRMS (ЭРИ)[М+Н]+ обнаружено=1552.5595 (δ=-2.5 част. на млн.)
ПРИМЕР 34: N-{[(9H-флуорен-9-ил)метокси]карбонил}-L-валил-N5-карбамоил-N-[4-(гидроксиметил)-3-(3-сульфопропил)фенил]-L-орнитинамид
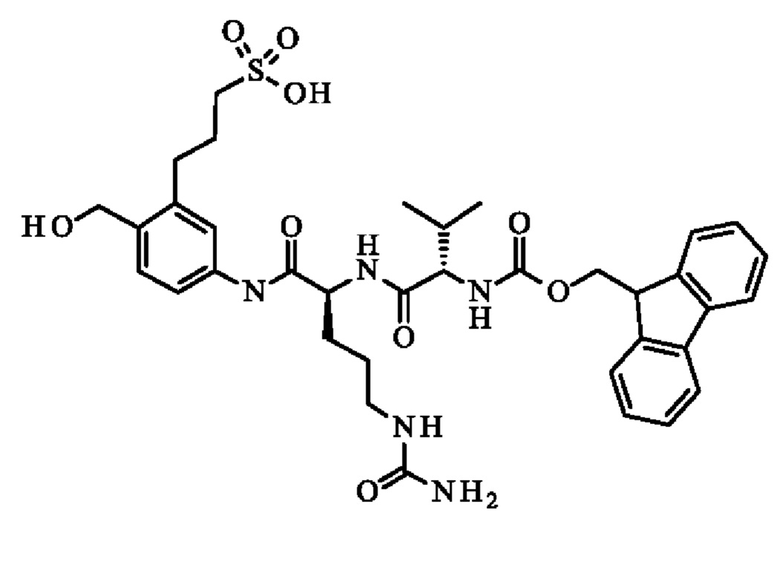
Стадия 1: 2-йод-4-нитробензойная кислота
К раствору 2-амино-4-нитробензойной кислоты (10,0 г; 54.9 ммоль) в ACN (280 мл), добавляли моногидрат n-толуолсульфоновой кислоты (32,0 г; 168 ммоль). Смесь перемешивали при КТ в течение 15 минут, затем в течение 15 минут по каплям добавляли раствор нитрита натрия (8.00 г; 115.9 ммоль) и йодида калия (24.0 г; 144.6 ммоль) в воде (140 мл). Реакционную смесь перемешивали в течение 19 часов. После завершения реакции смесь гасили тиосульфатом натрия (13,02 г; 82,36 ммоль) и подкисляли 3 н. водным раствором хлористого водорода (25 мл). Водный слой экстрагировали посредством DCM и объединенные органические слои промывали 1 н. водным раствором хлороводорода, сушили над сульфатом натрия, фильтровали и концентрировали досуха, чтобы получить указанное в заголовке соединение (15,0 г; 51,2 ммоль) в виде оранжевого порошка. 1Н ЯМР (ДМСО): 7.86 (d, 1Н, J=8.4 Гц), 8.27 (d, 1H, J=8.4 Гц), 8.64 (s, 1H), 13.8 (brs, 1Н)
Стадия 2: (2-йод-4-нитрофенил)метанол
В раствор 2-йод-4-нитробензойной кислоты (5.00 г; 17.1 ммоль) в ТГФ (70 мл), добавляли раствор борана 1 н. в ТГФ (85 мл; 85.0 ммоль). Реакционную смесь перемешивали при 65°С в течение 4 часов. После завершения реакции реакционную смесь охлаждали до КТ и гасили посредством МеОН (200 мл). Смесь перемешивали при КТ в течение 30 минут, затем концентрировали досуха. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с использованием циклогексан / EtOAc (от 80/20 до 50/50) в качестве элюента, чтобы получить указанное в заголовке соединение (3.38 г; 12,1 ммоль) в виде желтого твердого вещества. 1Н ЯМР (ДМСО): 5 4.47 (d, 2Н, J=5.2 Гц), 5.82 (t, 1H, J=5.2 Гц), 7.70 (d, 1Н, J=8.8 Гц), 8.29 (dd, 1H, J=8.8 и 2.0 Гц), 8.54 (d, 1H, J=2.0 Гц)
Стадия 3: (4-амино-2-йодфенил)метанол
К раствору (2-йод-4-нитрофенил)метанола (3,70 г; 13.3 ммоль) в ЕЮН (100 мл) и воде (25 мл) последовательно добавляли железо (3,70 г; 66.3 ммоль) и хлорид аммония (800 мг; 15,0 ммоль). Реакционную смесь перемешивали в течение 3 часов при 80°С. После завершения реакции реакционную смесь фильтровали через Celite®, промывали посредством EtOH и концентрировали досуха. Полученный остаток ресуспендировали в EtOAc и промывали насыщенным раствором гидрокарбоната натрия. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали досуха с получением указанного в заголовке соединения (2.48 г; 9.95 ммоль) в виде желтого масла. 1Н ЯМР (ДМСО): δ 4.28 (d, 2Н, J=5.2 Гц), 4.97 (t, 1Н, J=5.2 Гц), 5.16 (s, 2Н), 6.57 (d, 1H, J=8.4 Гц), 7.02-7.10 (m, 2Н)
Стадия 4: 4-({[трет-бутил(диметил)силил]окси}метил)-3-йоданилин
В раствор (4-амино-2-йодфенил)метанола (3,51 г; 13,4 ммоль) в DCM (150 мл), добавляли имидазол (0,95 г; 14.0 ммоль). Смесь охлаждали до 0°С, затем добавляли по каплям в течение 15 минут раствор трет-бутилхлордиметилсилана (2,40 мл; 13.85 ммоль) в DCM (150 мл). Ледяную баню удаляли и реакционную смесь перемешивали при КТ в течение 16 часов. После завершения реакции реакционную смесь гасили посредством МеОН (20 мл) и концентрировали досуха. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с использованием циклогексан / EtOAc (от 100/0 до 90/10) в качестве элюента, чтобы получить указанное в заголовке соединение (3,64 г; 10.0 ммоль)в виде желтого масла. 1Н ЯМР (ДМСО): δ 0.06 (s, 6Н), 0.88 (s, 9Н), 4.46 (s, 2Н), 5.24 (s, 2Н), 6.55 (d, 1H, J=8.4 Гц), 7.03 (d, 1Н, J=8.4 Гц), 7.05 (s, 1H)
Стадия 5: N-[4-({[трет-бутил(диметил)силил]окси}метил)-3-йодфенил]-N5-карбамоил-N2-{[(9Н-флуорен-9-ил)метокси]карбонил}-L-орнитинамид
В раствор 4-({[трет-бутил(диметил)силил]окси}метил)-3-йоданилина (10,0 г; 27.5 ммоль) в МеОН (70 мл) и DCM (140 мл) последовательно добавляли Fmoc-Cit-OH (12,0 г; 30.28 ммоль) и EEDQ (8,17 г; 33.0 ммоль). Реакционную смесь перемешивали в течение 14 часов при КТ. После завершения реакции, полученный остаток очищали с помощью колоночной хроматографии на силикагеле с использованием DCM / МеОН (от 100/0 до 88/12) в качестве элюента, чтобы получить указанное в заголовке соединение (17,09 г; 22,0 ммоль) в виде белого твердого вещества. 1Н ЯМР (ДМСО): δ 0.09 (s, 6Н), 0.91 (s, 9Н), 1.38-1.48 (m, 2Н), 1.59-1.68 (m, 2Н), 2.93-3.05 (m, 2Н), 4.06-4.15 (m, 1Н), 4.20-4.29 (m, 3H), 4.56 (s, 2Н), 5.41 (s, 2Н), 5.98 (t, 1Н, J=5.5 Гц), 7.30-7.43 (m, 5Н), 7.55 (dd, 1Н, J=8.8 и 2.1 Гц), 7.69 (d, 1H, J=7.8 Гц), 7.74 (dd, 2Н, J=7.2 и 3.4 Гц), 7.89 (d, 2Н, J=7.5 Гц), 8.25 (d, 1H, J=1.5 Гц), 10.12 (s, 1H)
Стадия б: N-[4-({[трет-бутил(диметил)силил]окси}метил)-3-йодфенил]-N5-карбамоил-L-орнитинамид
В раствор N-[4-({[трет-бутил(диметил)силил]окси}метил)-3-йодфенил]-N5-карбамоил-N2-{[(9H-флуорен-9-ил)метокси]карбонил}-L-орнитинамида (17,1 г; 23,0 ммоль) в ТГФ (120 мл)добавляли диметиламин 2М в ТГФ (44,5 мл; 89,0 ммоль). Реакционную смесь перемешивали в течение 15 часов при КТ. После концентрации досуха полученный остаток очищали с помощью колоночной хроматографии на С18 с использованием вода/ACN (от 98/02 до 0/100) в качестве элюента, чтобы получить указанное в заголовке соединение (5.47 г; 10.5 ммоль) в виде белого твердого вещества. 1Н ЯМР (ДМСО): δ 0.0 (s, 6Н), 0.81 (s, 9Н), 1.27-1.38 (гл, 3H), 1.47-1.53 (m, 1Н), 2.83-2.89 (m, 2Н), 3.16-3.19 (m, 1H), 4.46 (s, 2Н), 5.26 (s, 2Н), 5.82 (t, 1H, J=5.6 Гц), 7.24 (d, 1H, J=8.5 Гц), 7.50 (dd, 1Н, J=8,3 и 2.0 Гц), 8.17 (d, 1H, J=2.0 Гц)
Стадия 7: N-{[(9Н-флуорен-9-ил)метокси]карбонил}-L-валил-N-[4-({[трет-бутил(диметил)силил]окси}метил)-3-йодфенил]-N5-карбамоил-L-орнитинамид
В раствор N-[4-({[трет-бутил(диметил)силил]окси}метил)-3-йод фенил]-N5-карбамоил-L-орнитинамида (3,00 г; 5,76 ммоль) в 2-метилтетрагидрофуране (240 мл), последовательно добавляли Fmoc-Val-OSu (8.65 г; 8,65 ммоль) и DIPEA (1.90 мл; 11,5 ммоль). Реакционную смесь перемешивали в течение 15 часов при КТ. Реакционную смесь фильтровали через пористую воронку и извлеченное твердое вещество промывали 2-метилтетрагидрофураном, затем сушили в высоком вакууме с получением указанного в заголовке соединения (3,57 г; 4,24 ммоль) в виде белого твердого вещества. 1Н ЯМР (ДМСО): δ 0.10 (s, 6Н), 0.83-0.95 (m, 15Н), 1.27-1.52 (m, 2Н), 1.52-1.75 (m, 2Н), 1.93-2.07 (m, 1Н), 2.88-3.09 (m, 2Н), 3.93 (t, 1H, J=8.0 Гц), 4.17-4.49 (m, 4Н), 4.56 (s, 2Н), 5.40 (s, 2Н), 5.96 (t, 1H, J=5.6 Гц), 7.27-7.37 (m, 3H), 7.37-7.48 (m, 3H), 7.54 (d, 1Н, J=8.0 Гц), 7.74 (t, 2Н, J=7.2 Гц), 7.88 (d, 2Н, J=7.6 Гц), 8.13 (d, 1Н, J=7.6 Гц), 8.22 (s, 1Н), 10.11 (s, 1H)
Стадия 8: N-{[(9Н-флуорен-9-ил)метокси]карбонил}-L-валил-N-[4-({[трет-бутил(диметил)силил]окси}метил)-3-(3-гидроксипроп-1-ин-1-ил)фенил]-N5-карбамоил-L-орнитинамид
В раствор N-{[(9H-флуорен-9-ил)метокси]карбонил}-L-валил-N-[4-({[трет-бутил(диметил)силил]окси}метил)-3-йодфенил]-N5-карбамоил-L-орнитинамида (4,0 г; 4,55 ммоль) и 2-пропин-1-ола (0.54 мл; 9.10 ммоль) в сухом DMF (36 мл), добавляли DIPEA (2,26 мл; 13,66 ммоль; 3,0 экв.). Реакционную смесь продували аргоном, затем добавляли дихлорид бис(трифенилфосфин)палладия(II)(0,64 г; 0,91 ммоль) и йодид меди(I)(0,17 г; 0,91 ммоль). Реакционную смесь снова продували аргоном и перемешивали при КТ в течение 16 часов. Добавляли насыщенный раствор хлорида аммония (80 мл) и водный слой экстрагировали посредством EtOAc. Объединенные органические слои промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали досуха. Сырой продукт очищали с помощью флэш-хроматографии на силикагеле с использованием DCM / МеОН (от 10/0 до 85/15) в качестве элюента, чтобы получить указанный в заголовке продукт (2,7 г; 3,52 ммоль) в виде коричневого твердого вещества. 1Н ЯМР (ДМСО-d6): 0.08 (s, 6Н), 0.85-0.89 (m, 6Н), 0.90 (s, 9Н), 1.32-1.48 (m, 2Н), 1.54-1.74 (m, 2Н), 1.94-2.03 (m, 1H), 2.89-3.06 (m, 2Н), 3.93 (dd, 1Н, J=7.0 и 9.1 Гц), 4.22-4.33 (m, 5Н), 4.36-4.41 (m, 1H), 4.74 (s, 2Н), 5.35 (t, 1H, J=5.9 Гц), 5.40 (s, 2Н), 5.96 (t, 1H, J=6.0 Гц), 7.32 (t, 2Н, J=7.4 Гц), 7.36-7.43 (m, 4Н), 7.48 (dd, 1Н, J=2.3 и 8.8 Гц), 7.74 (t, 2Н, J=7.7 Гц), 7.78 (s, 1Н), 7.89 (d, 2Н, J=7.8 Гц), 8.14 (d, 1H, J=7.1 Гц), 10.09 (s, 1Н)
Стадия 9: N-{[(9Н-флуорен-9-ил)метокси]карбонил}-L-валил-N-[4-({[трет-бутил(диметил)силил]окси}метил)-3-{3-[(метансульфонил)окси]проп-1-ин-1-ил}фенил]-N5-карбамоил-L-орпитинамид
В раствор N-{[(9H-флуорен-9-ил)метокси]карбонил}-L-валил-N-[4-({[трет-бутил(диметил)силил]окси}метил)-3-(3-гидроксиргор-1-ин-1-ил)фенил]-N5-карбамоил-L-орнитинамида (1,0 г; 1,3 ммоль) в сухом DMF (9 мл), последовательно добавляли при 0°С DIPEA (0,64 мл; 3,9 ммоль) и метансульфонилхлорид (0,15 мл; 1,95 ммоль). Реакционной смеси давали медленно нагреться до КТ и перемешивали в течение 1 часа. Добавляли насыщенный раствор хлорида аммония (25 мл) и водный слой экстрагировали посредством EtOAc. Объединенные органические слои промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали досуха, чтобы получить указанный в заголовке продукт (1,1 г; 1,3 ммоль) в виде оранжевой смолы, используемой как таковой в следующей стадии. 1Н ЯМР (ДМСО-d6): 0.08 (s, 6Н), 0.84-0.89 (m, 6Н), 0.90 (s, 9Н), 1.32-1.49 (m, 2Н), 1.54-1.74 (m, 2Н), 1.94-2.03 (m, 1Н), 2.91-3.05 (m, 2Н), 3.29 (s, 3H), 3.93 (dd, 1Н, J=7.1 и 9.0 Гц), 4.20-4.33 (m, 3H), 4.37-4.42 (m, 1H), 4.75 (s, 2Н), 5.23 (s, 2Н), 5.4 (s, 2Н), 5.96 (t, 1Н, J=5.8 Гц), 7.32 (t, 2Н, J=7.2 Гц), 7.39-7.43 (m, 4Н), 7.56 (dd, 1H, J=2.3 и 8.8 Гц), 7.74 (t, 2Н, J=7.4 Гц), 7.83 (d, 1H, J=2.3 Гц), 7.89 (d, 2Н, J=7.3 Гц), 8.14 (d, 1Н, J=7.3 Гц), 10.15 (s, 1Н)
Стадия 10: N-{[(9Н- флуорен-9-ил)метокси]карбонил}-L-валил-N-{3-[3-(ацетилсульфанил)проп-1-ин-1-ил]-4-({[трет-бутил(диметил)силил]окси}метил)фенил}-N5-карбамоил-L-орнитинамид
В раствор N-{[(9H-флуорен-9-ил)метокси]карбонил}-L-валил-N-[4-({[трет-бутил(диметил)силил]окси}метил)-3-{3-[(метансульфонил)окси]проп-1-ин-1-ил}фенил]-N5-карбамоил-L-орнитинамида (1,1 г; 1.3 ммоль) в сухом DMF (10 мл), добавляли тиоацетат калия (0,3 г; 2.6 ммоль). Реакционную смесь перемешивали при КТ в течение 16 часов. Добавляли насыщенный раствор бикарбоната натрия (35 мл) и водный слой экстрагировали посредством EtOAc. Объединенные органические слои промывали соляным раствором, сушили над сульфатом натрия, фильтровали и концентрировали досуха. Сырой продукт очищали с помощью флэш-хроматографии на силикагеле с использованием DCM / МеОН (от 10/0 до 9/1) в качестве элюента, чтобы получить указанный в заголовке продукт (0,52 г; 0.62 ммоль) в виде желтоватого твердого вещества. 1Н ЯМР (ДМСО-d6): 0.08 (s, 6Н), 0.84-0.89 (m, 6Н), 0.90 (s, 9Н), 1.31-1.49 (m, 2Н), 1.54-1.73 (m, 2Н), 1.95-2.02 (m, 1H), 2.4 (s, 3H), 2.89-3.06 (m, 2Н), 3.93 (dd, 1H, J=6.9 и 9.1 Гц), 3.97 (s, 2Н), 4.20-4.33 (m, 3H), 4.36-4.41 (m, 1Н), 4.69 (s, 2Н), 5.39 (s, 2Н), 5.96 (t, 1Н, J=5.8 Гц), 7.32 (t, 2Н, J=7.2 Гц), 7.35-7.43 (m, 4Н), 7.49 (dd, 1Н, J=2.2 и 8.5 Гц), 7.72-7.76 (m, 3H), 7.89 (d, 2Н, J=7.5 Гц), 8.12 (d, 1H, J=7.4 Гц), 10.08 (s, 1H)
Стадия 11: N-{[(9Н-флуорен-9-ил)метокси]карбонил}-L-валил-N5-карбамоил-N-[4-(гидроксиметил)-3-(3-сульфопроп-1-ил-1-ил)фенил]-L-орнитинамид
К 35% раствору перекиси водорода (1,08 мл; 12,57 ммоль), добавляли муравьиную кислоту (9,68 мл; 256,5 ммоль) при 0°С. Этот раствор перемешивали в течение 1 часа, затем добавляли к N-{[(9H-флуорен-9-ил)метокси]карбонил}-L-валил-N-{3-[3-(ацетилсульфанил)проп-1-ин-1-ил]-4-({[трет-бутил(диметил)силил]окси}метил)фенил}-N5-карбамоил-L-орнитинамид (367 мг; 0,44 ммоль) при КТ. Реакционную смесь перемешивали в течение 2 часов, разбавляли с водой (20 мл) и концентрировали досуха при 35°С. Остаток суспендировали в 1 н. соляной кислоте (25 мл) и промывали посредством DCM / МеОН (7/3). Водный слой концентрировали досуха при 35°С. Сырой продукт очищали с помощью флэш-хроматографии на C18 с использованием ACN / вода + ТФУ (0,1%)(от 2/98 до 50/50) в качестве элюента, чтобы после сублимационной сушки получить продукт, указанный в заголовке (117 мг; 0.13 ммоль) в виде белого твердого вещества. 1Н ЯМР (ДМСО-d6): δ 0.84-0.88 (m, 6Н), 1.33-1.50 (m, 2Н), 1.55-1.75 (m, 2Н), 1.95-2.02 (m, 1Н), 2.89-3.05 (m, 2Н), 3.58 (s, 2Н), 3.91-3.95 (m, 1H), 4.20-4.33 (m, 3H), 4.37-4.42 (m, 1H), 4.55 (s, 2Н), 5.22 (brs, 1Н), 5.38 (s, 2Н), 5.96 (t, 1H, J=5.6 Гц), 7.32 (t, 2Н, J=7.2 Гц), 7.36-7.43 (m, 4Н), 7.48 (dd, 1Н, J=2.4 и 9.2 Гц), 7.65 (d, 1H, J=1.6 Гц), 7.74 (t, 2Н, J=6.8 Гц), 7.89 (d, 2Н, J=7.4 Гц), 8.12 (d, 1H, J=7.6 Гц), 10.03 (s, 1Н). LCMS (2-100 ACN/H2O+0,05% ТФУ): 77,27%, Tr=8,3 мин. Положительный режим 720.26 обнаружено (М+Н+)
Стадия 12: Пример 34
К суспензии N-{[(9H-флуорен-9-ил)метокси]карбонил}-L-валил-N5-карбамоил-N-[4-(гидроксиметил)-3-(3-сульфопроп-1-ин-1-ил)фенил]-L-орнитинамида (10 мг; 0,014 ммоль) в ТГФ (600 мкл) и уксусной кислоты (60 мкл), добавляли под аргоном Pt/C сухой 5% (7 мг; 0,0018 ммоль). Реакционную смесь продували водородом три раза, затем перемешивали при КТ в течение 64 часов. Реакционную смесь фильтровали через 45 мкм фильтр PTFE, промывали посредством МеОН и МеОН / вода (1/1), затем концентрировали досуха, чтобы получить Пример 34 (7 мг) в виде масла оранжевого цвета. LCMS (2-100 ACN/H2O+0.05% FA): 6.93%, Tr=8,8 мин. Отрицательный режим 722.19 обнаружено (М-Н)
Как показано в примерах с 6 по 12, примерах с 14 по 25, примере 27 или примере 28, Примере 34, пара-аминобензил-линкерное соединение формулы (I), может быть использовано для получения соединения линкер-лекарственное средство формулы (II) и для получения конъюгата антитело-лекарственное средство формулы (III) в соответствии с изобретением.
ПРИМЕР А: Конъюгация и аналитическая характеристика ADC
Как используют в данной заявке, конъюгаты антитело-лекарственное средство могут быть идентифицированы с использованием соглашения об именах в общем формате «целевой антиген/антитело-линкер-лекарственное средство». Только для примера, если конъюгат антитело-лекарственное средство упоминается как «мишень Х-пример Y», такой конъюгат будет содержать антитело, которое связывает мишень X, и линкер-лекарственное средство, представленное в Примере Y.
1. Конъюгация
Примерные ADC были синтезированы с использованием одного из способов, описанных ниже. Антитела, использованные в примерном синтезе ADC, были обозначены аббревиатурами Ab Т TG и Ab Т (Таблица 1).
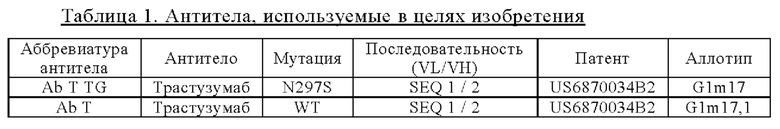
Антитело Т TG было снабжено бактериальной трансглютаминазой (BTG)-реакционноспособными глютаминами, которые были специфически функционализированы аминсодержащим циклооктином BCN (или N-[(1R,8S,9s)-бицикло[6.1.0]нон-4-ин-9-илметилоксикарбонил]-1,8-диамино-3,6-диоксаоктан). Конъюгацию сайт-специфического антитела с частью BCN с использованием бактериальной трансглютаминазы проводили, как описано в Innate Pharma 2013 (презентация на саммите ADC, Сан-Франциско, Калифорния, 15 октября 2013 г.), WO 2017/059160А1 и WO 2016/144608А1. Эти модификации позволили конъюгировать описанные азидсодержащие предшественники с использованием следующего метода А (Фигура 1).
Конъюгаты Ab Т были получены путем полного восстановления четырех межцепочечных дисульфидных связей с последующей конъюгацией с избыточным количеством лекарственного средства (Фигура 2). В целях изобретения водорастворимый третичный фосфин, такой как трис-(2-карбоксиэтил)фосфин (ТСЕР), использовали в качестве восстанавливающего средства. Полное восстановление и конъюгация обеспечивают в первую очередь полностью нагруженные виды с 8 лекарственными средствами на антитело и, в конечном счете, небольшую часть mAb с 6 лекарственными средствами, что объясняет среднее значение DAR 7,1 и 7,7 (Таблица 2).
Некоторые конъюгаты Ab Т были получены с помощью технологии повторного образования мостиков, описанной в способе D ниже. Четыре межцепочечные дисульфидные связи Ab Т были полностью восстановлены с помощью ТСЕР и преобразованы с использованием линкера-полезной нагрузки, функционализированного диброммалеимидом (Фигура 3). Конъюгация приводит к двум конформациям из-за близости цистеина, участвующего в межцепочечных дисульфидных мостиках, на каждой тяжелой цепи антитела.
Способ A (DAR2)
К раствору Ab (1,8 мг/мл; 2.5 мл, 4,5 мг)добавляли диметилсульфоксид (ДМСО, 619 мкл, 20% от объема связывания). Смесь перемешивали в течение 30 секунд на вортексе с последующим добавлением 4-кратного молярного избытка полезной нагрузки линкер-боеголовка (20 мМ, 6 мкл в ДМСО). Реакционную смесь перемешивали при КТ в течение ночи при 64 об/мин. Для удаления неконъюгированного линкер-полезная нагрузка, добавляли 10-кратный молярный избыток смолы Tentagel, содержащей DBCO (0,1-0,2 ммоль/г, Iris Biotech, CS-0477.0500) и смесь перемешивали в течение 6 часов при КТ. Буфер заменяли на PBS IX (Sigma Life Science, Р3813, 10РАК) посредством 3 циклов фильтрации с использованием Vivaspin 20, 50KD, PES (Sartorius Stedim, VS2031), затем стерильно фильтровали через 0,2 мкм стерильный фильтр PES, 25 мм (Whatmann, G896-2502) и хранили при 4°С.
Способ В (DAR8)
Раствор Ab (10,3 мг/мл; 0,5 мл)разбавляли в EDTA, содержащем буфер PBS рН 7,4 (10 мМ; 0,5 мл), чтобы зафиксировать конечную концентрацию EDTA в реакции сочетания 5 мМ. Затем к антителу добавляли ТСЕР (1 мМ в буфере PBS рН 7,4, 849 мкл) с последующей инкубацией при 37°С в течение 2 часов. После восстановления раствор антител охлаждали до 2-8°С и добавляли 20-кратный молярный избыток линкера-полезной нагрузки (5 мМ, 141 мкл в ДМСО). Реакционную смесь инкубировали при 4°С в течение 1,5 часов. Раствор центрифугировали (14000 G при 4°С) в течение 20 минут и его загружали на хроматографическую колонку HiLoad 26/600 Superdex 200 пг (GE Healthcare, 28989336)ЭХ. ADC элюировали посредством 20% DMA в PBS (Sigma Life Science, Р3813, 10РАК)с последующим диализом в 2 цикла (16 и 4 часа) в PBS 1X рН 7,4 (Sigma Life Science, Р3813, 10РАК). Конъюгат концентрировали с использованием Vivaspin 20, 50KD, PES (Sartorius Stedim, VS2031), стерильно фильтровали через 0,2 мкм стерильный фильтр PES, 25 мм (Whatmann, G896-2502) и хранили при 4°С.
Способ С (DAR8)
Раствор Ab (10,3 мг/мл; 0,5 мл)разбавляли в EDTA, содержащем буфер PBS рН 7,4 (10 мМ; 0,5 мл), чтобы зафиксировать конечную концентрацию EDTA в реакции сочетания 5 мМ. Затем к антителу добавляли ТСЕР (1 мМ в буфере PBS рН 7,4; 849 мкл) с последующей инкубацией при 37°С в течение 2 часов. После восстановления раствор антитела охлаждали до 2-8°С и добавляли 20-кратный молярный избыток линкера-полезной нагрузки (5 мМ; 141 мкл в ДМСО). Реакционную смесь инкубировали при 4°С в течение 1,5 часов. Затем конъюгат очищали на смоле rmp с белком A (GE Healthcare, 17-5138-01) с последующим диализом в 2 цикла (16 и 4 часа) в PBS 1X рН 7,4 (Sigma Life Science, Р3813, 10РАК). ADC концентрировали с использованием Vivaspin 20, 50KD, PES (Sartorius Stedim, VS2031), стерильно фильтровали через 0,2 мкм стерильный фильтр PES, 25 мм (Whatmann, G896-2502) и хранили при 4°С.
Способ D (DAR4)
К раствору Ab (5 мг/мл, 0,5 мл) в буфере BBS рН 8 (приготовленном, как описано ниже)добавляли 8-кратный молярный избыток ТСЕР (1 мМ в BBS рН 8, 137 мкл) и реакционную смесь инкубировали при 37°С в течение 2 часов. Затем добавляли 7,5-кратный молярный избыток полезной нагрузки линкера (1 мМ, 129 мкл в DMF) и полученный раствор перемешивали при КТ в течение 1 часа при 600 об./мин. Полученный ADC подвергали диализу в PBS 1X рН 7,4 (Sigma Life Science, Р3813, 10РАК) в течение 16 часов при 4°С. Затем конъюгат очищали на смоле rmp с белком A (GE Healthcare, 17-5138-01)с последующим 2-часовым диализом при КТ в PBS IX рН 7,4 (Sigma Life Science, Р3813, 10РАК). ADC концентрировали с использованием Vivaspin 20, 50KD, PES (Sartorius Stedim, VS2031), стерильно фильтровали через 0,2 мкм стерильный фильтр PES, 25 мм (Whatmann, G896-2502) и хранили при 4°С.
Приготовление буфера BBS рН 8: Na2B4O7. 10 H2O (528 мг)растворяли в деионизированной воде (27,7 мл). Хлорид натрия (63 мг, 25 мМ) и EDTA (16 мг, 1 мМ)солюбилизировали в 22,3 мл 0,1 М соляной кислоты. Затем два раствора смешивали вместе, и полученный раствор непосредственно использовали для стадии конъюгации.
2. Общая методология ЖХ-МС
Отношение лекарственного средства к антителу (DAR) примерных определяли с помощью жидкостной хроматографии - масс-спектрометрии (ЖХ-МС) с использованием следующего способа ЖХ-I или ЖХ-II. Для способа ЖХ-I подвижную фазу А очищали с помощью воды класса МС (Biosolve, Dieuze, Франция, 00232141B1BS), подвижную фазу В - ацетонитрилом класса МС (Biosolve, Dieuze, Франция, 000120410IBS), а подвижную фазу D очищали с помощью воды класса МС с добавлением 1% муравьиной кислоты (FA)(Honeywell/Fluka, Bucharest, Румыния, 56302). Подвижную фазу D фиксировали на уровне 10%, чтобы поддерживать состав подвижной фазы с содержанием 0,1% FA, а температуру колонки устанавливали равной 80°С. Общий способ МС был оптимизирован для всех синтезированных ADC (Таблица 2). Для способа ЖХ-II подвижную фазу А очищали водой класса МС (Biosolve, Dieuze, Франция, 00232141B1BS), а подвижную фазу В ацетонитрилом класса МС (Biosolve, Dieuze, Франция, 0001204101BS), обе содержащие 0,1% муравьиной кислоты (FA)(Honeywell/Fluka, Bucharest, Румыния, 56302). Температуру колонки устанавливали равной 80°С. Общий способ МС был оптимизирован для всех синтезированных ADC (таблица 2).
Способ ЖХ-I: ADC загружали на Bioresolve RP mAb Polyphenyl, 450А, 2,7 мкм, 2,1x150 мм (Waters, Saint-Quentin-en-Yvelines, Франция, 186008946). Для анализа как в интактном, так и в восстановленном состоянии проводили стадию обессоливания в течение 1,5 минут при 20% В со скоростью потока 0,6 мл/мин. Стадию элюирования проводили с градиентом от 1,5 мин при 20% В до 16,5 минут при 70% В со скоростью потока 0,3 мл/мин. Стадия промывки была установлена от 16,8 минут до 18,8 минут при 90% В со скоростью потока 0,6 мл/мин. Наконец, использовали стадию кондиционирования в течение 19,1 минут в течение 1,9 минут при 20% В со скоростью потока 0,6 мл/мин (общее время работы=21 минута).
Способ ЖХ-II: ADC загружали на Bioresolve RP mAb Polyphenyl, 450A, 2,7 мкм, 2,1x150 мм (Waters, Saint- Quentin-en-Yvelines, Франция, 186008946). Для анализа как в интактном, так и в восстановленном состоянии проводили стадию обессоливания в течение 1,5 минут при 20% В со скоростью потока 0,6 мл/мин. Затем стадию элюирования при той же скорости потока с градиентом от 1,5 минут при 20% В до 16,5 минут при 50% В. Следующую стадию промывки устанавливали от 16,8 до 18,8 минут при 100% В с потоком 0,6 мл/мин. Наконец, использовали стадию кондиционирования от 18,8 минут до 19,2 минут от 100% до 20% В, стабилизируя колонку дополнительными 1,9 минутами при 20% В с указанной выше скоростью потока (общее время работы = 21 мин).
Анализ ЖХ-МС проводили с использованием хроматографической системы Waters UPLC H-Class Bio с масс-спектрометром Xevo G2 XS Q-TOF ESI (Waters, Манчестер, Великобритания). ADC анализировали либо в интактном состоянии (без предварительной обработки), либо после восстановления с 5 мМ (конечная концентрация)дитиотреитола DTT (Thermo Scientific, Rockford, IL, 20291). Затем обработанный ADC анализировали с использованием вышеупомянутых ЖХ-I и ЖХ-II (таблица 2). Времяпролетные масс-спектры аналитов с ионизацией электрораспылением получали с использованием программного обеспечения для сбора данных MassLynx™ (Waters, Манчестер, Великобритания). Затем извлеченный спектр интенсивности по отношению к m/z подвергали деконволюции с использованием метода максимальной энтропии (MaxEnt) программного обеспечения MassLynx™ для определения массы каждого интактного вида антитела или каждого восстановленного фрагмента антитела в зависимости от используемой обработки. Наконец, DAR определяли из деконволютивных спектров или УФ-хроматограммы путем суммирования интегрированной площади пика МС (полный ионный ток) или УФ (280 нм) неконъюгированных и конъюгированных данных видов (mAb или ассоциированного фрагмента). Для определения DAR с помощью УФ-хроматограммы относительный процент площади каждого вида умножали на количество присоединенных лекарственных средств. Суммарные взвешенные площади каждого вида были разделены на сумму процентов общей относительной площади, и результаты дали оценку окончательного среднего значения DAR для полного ADC. Для определения DAR по деконволюционным спектрам процент каждого идентифицированного вида рассчитывали по значению пика интенсивности из деконволютивных спектров. Полученный процент умножали на количество присоединенных лекарственных средств. Суммированные результаты дали оценку окончательного среднего значения DAR для полного ADC.
3. Эксклюзионная хроматография
Эксклюзионную хроматографию (ЭХ) проводили для определения качества ADC, процента его агрегации после очистки. Анализ проводили на аналитической колонке Superdex 200 Increase 5/150 GL (GE Healthcare, 28990945) в изократических условиях 100% PBS рН 7,4 (Sigma Life Science, P3813, 10PAK), поток 0,45 мл/мин в течение 12 минут. Процент фракции агрегатов в образце конъюгата определяли количественно на основании площади пика поглощения при 280 нм. Его расчет был основан на соотношении между высокомолекулярным элюентом при 280 нм, деленном на сумму площадей пиков поглощения при той же длине волны высокомолекулярных и мономерных элюентов, умноженных на 100.
4. Хроматография гидрофобного взаимодействия
Хроматографию гидрофобных взаимодействий (HIC) проводили для определения влияния гидрофобности линкера-полезной нагрузки и метода биоконъюгации на антитело. Анализ проводили с использованием колонки TSKgel Butyl-NPR (Tosoh Bioscience, 0014947) с подвижной фазой A (1,5 M сульфата аммония (NH4)2SO4, 25 мМ двухосновного фосфата калия (K2НРО4), доведенного до рН 7) и В (25 мМ двухосновного фосфата калия (К2НРО4), 20% изопропанол, доведенный до рН 7). Элюирование начинали с потока 0,6 мл/мин при 5% В. Градиент повышался от 5% до 100% В за 17 минут, после чего следовала стадия промывки при 100% В в течение 5 минут. Наконец, была проведена стадия кондиционирования при 100%-5% В в течение 2 минут, после чего в течение 2 минут при 5% В.
Для целей изобретения относительное время удерживания (RRT) каждого ADC рассчитывали, используя время удерживания ADC (RT), деленное на RT антитела (Таблица 3).
5. Результаты
Характеристики примерных ADC были обобщены в таблице 2 (обозначены как Примеры 15-22 и Примеры 29-33; сочетание и способ ЖХ-МС, статус агрегации и DAR). Средние значения DAR определяли с использованием описанного выше способа ЖХ-МС ЖХ-I или ЖХ-II (точка 2), а процентное содержание агрегатов измеряли с помощью эксклюзионной хроматографии (ЭХ), описанной выше (точка 3).
Гидрофобность приведенного в качестве примера ADC сравнивали путем расчета относительного времени удерживания с использованием хроматографии HIC, описанной выше (пункт 4). В таблице 3 суммированы значения RRT конъюгатов.
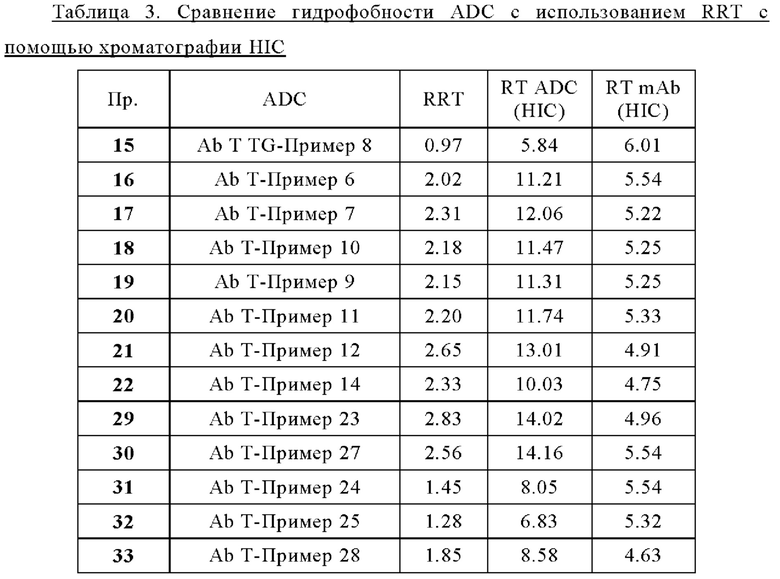
Исследование стабильности проводили для всех приведенных в качестве примера ADC, где конъюгаты инкубировали в условиях ускоренной деградации в буфере PBS при +37°С в течение 1 недели. Для измерения состояния агрегации конъюгатов использовали хроматографию ЭХ. Полученные результаты приведены в таблице 4.
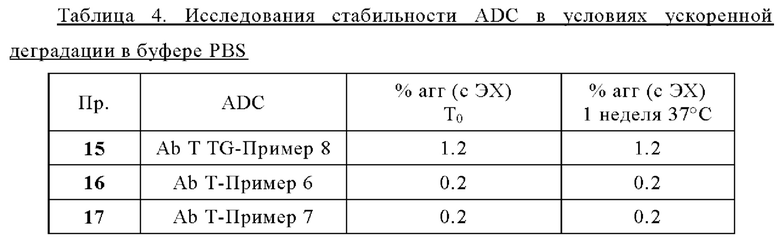
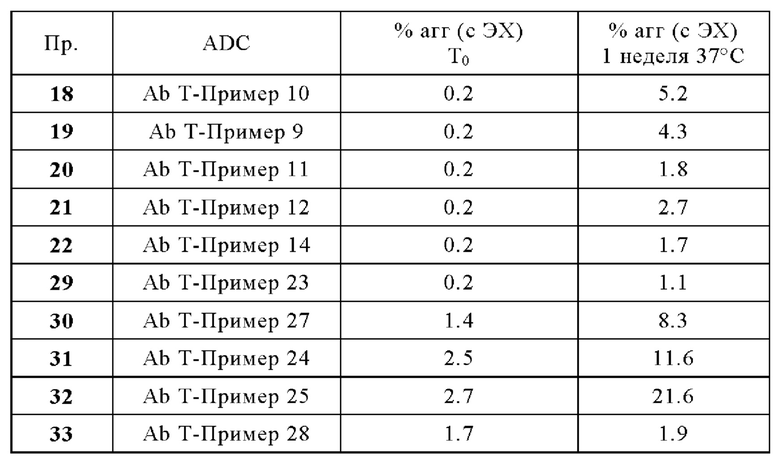
ПРИМЕР В: Влияние примерных ADC на жизнеспособность клеток с использованием анализа CTG
Линии клеток НСС1954 (HER+) и MOLT4 (HER-) культивировали в RPMI с добавлением 10% инактивированной нагреванием фетальной бычьей сыворотки, пенициллина (100 МЕ/мл), стрептомицина (100 мкг/мл) и L-глютамина (2 мМ). Линии клеток культивировали при 37°С во влажной атмосфере, содержащей 5% СО2. Клетки высевали в 96-луночные микропланшеты и подвергали воздействию ADC в течение 120 часов (5-кратные серийные разведения; 9 концентраций в каждой, три повторности). Влияние ADC на жизнеспособность клеток оценивали через 5 дней инкубации при 37°С/5% СО2 путем количественного определения клеточных уровней АТФ с использованием CellTiterGlo при 75 мкл реагента на лунку. Все условия тестировали в трех повторностях. Люминесценцию определяли количественно на многофункциональном считывающем планшете. IC50s рассчитывали с использованием стандартной четырехпараметрической аппроксимации кривой. IC50 определяется как концентрация соединения, при которой сигнал CTG уменьшается до 50% от измеренного для контроля. Каждый эксперимент проводили не менее двух раз с воспроизводимостью результатов.
Как показано в таблице 5, все примерные ADC являются активными в антиген-положительных клетках в анализе CTG.
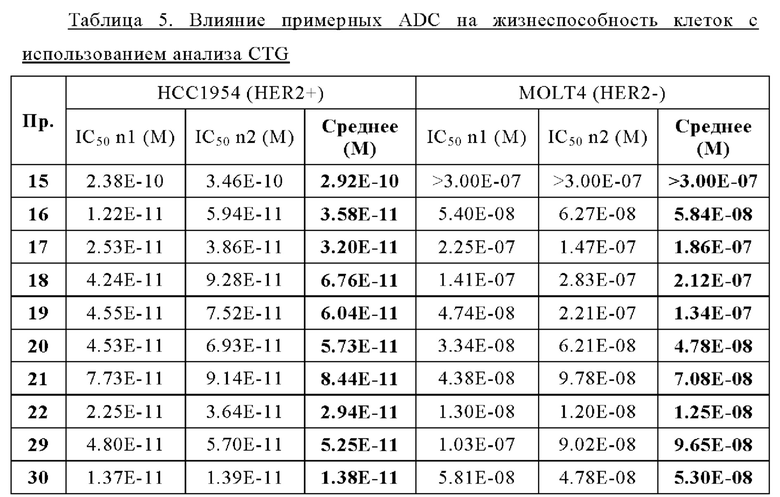
| название | год | авторы | номер документа |
|---|---|---|---|
| Модулирующие стабильность линкеры для использования с конъюгатами антитело-лекарственное средство | 2015 |
|
RU2680238C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| ЗАМЕЩЕННЫЕ АЗОЛЫ, ПРОТИВОВИРУСНЫЙ АКТИВНЫЙ КОМПОНЕНТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2010 |
|
RU2452735C1 |
| ЦИТОТОКСИЧЕСКИЕ ПЕПТИДЫ И ИХ КОНЪЮГАТЫ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2012 |
|
RU2586885C2 |
| ПРОИЗВОДНЫЕ ФЕНИЛКАРБАМАТА В КАЧЕСТВЕ МОДУЛЯТОРОВ ФОРМИЛПЕПТИДНОГО РЕЦЕПТОРА | 2014 |
|
RU2703725C1 |
| НОВЫЕ СОЕДИНЕНИЯ, КОТОРЫЕ ЯВЛЯЮТСЯ ИНГИБИТОРАМИ ERK | 2013 |
|
RU2660429C2 |
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ТЕРАПИИ ПРОТИВ ВИЧ | 2019 |
|
RU2806030C2 |
| НОВЫЕ КОНЪЮГАТЫ СВЯЗЫВАЮЩЕЕ СОЕДИНЕНИЕ - АКТИВНОЕ СОЕДИНЕНИЕ (ADC) И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2610336C2 |
| СОЕДИНЕНИЯ ПИРИДАЗИНАМИДА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ СЕЛЕЗЕНКИ (SYK) | 2013 |
|
RU2627661C2 |
| КОМБИНИРОВАННОЕ ЛЕЧЕНИЕ АГОНИСТОМ ТОЛЛ-ПОДОБНОГО РЕЦЕПТОРА (TLR7) И ИНГИБИТОРОМ СБОРКИ КАПСИДА ВИРУСА ГЕПАТИТА В | 2016 |
|
RU2718917C2 |
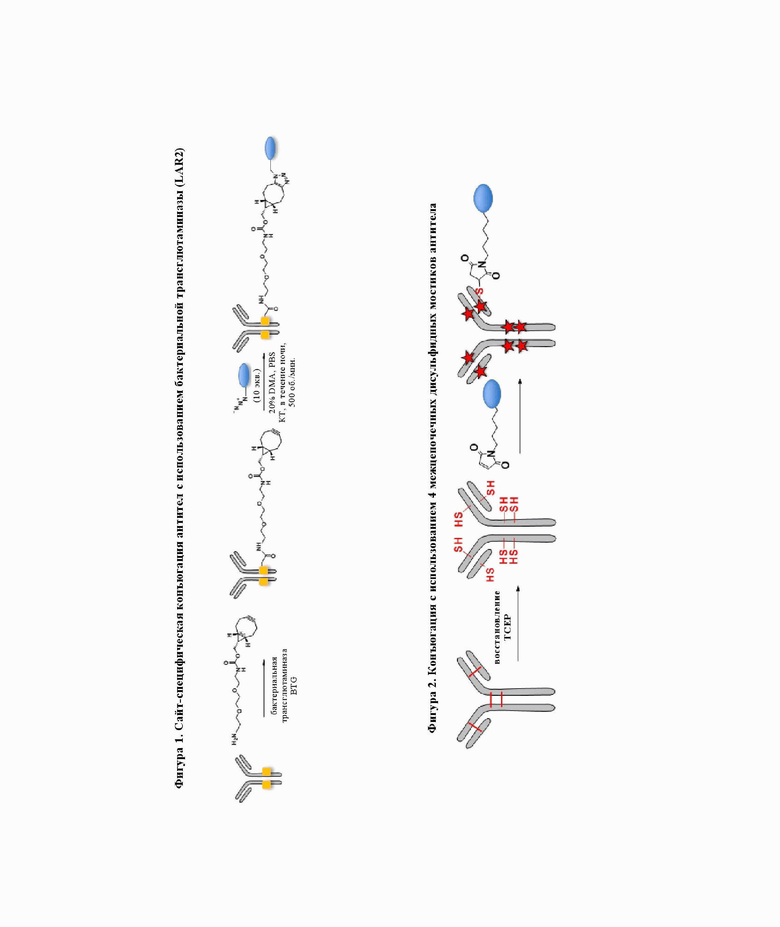
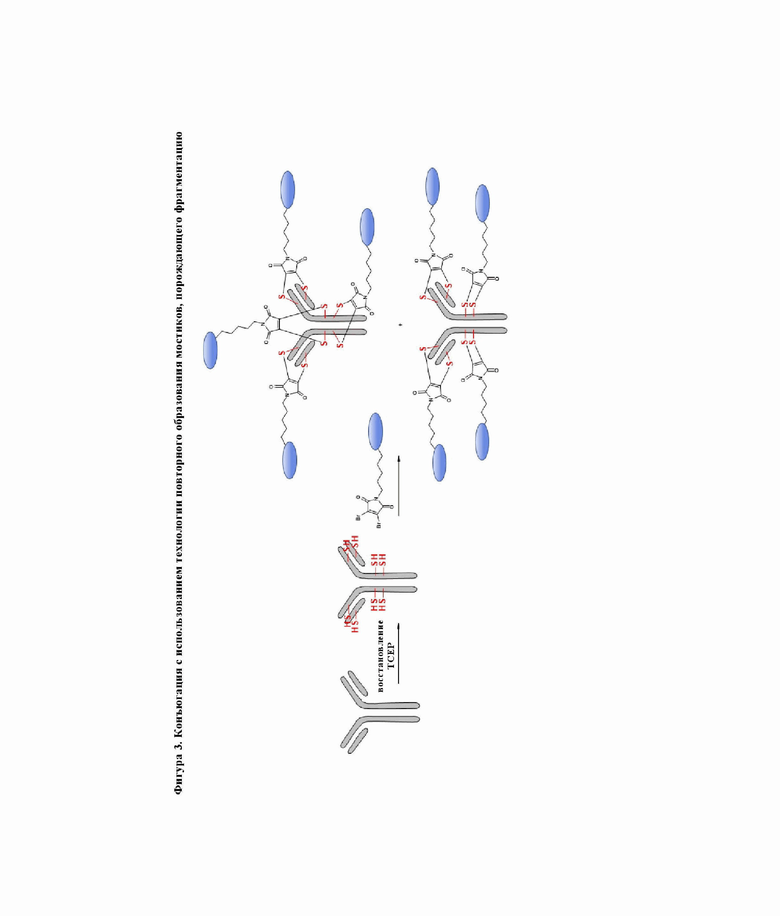
Настоящее изобретение относится к пара-аминобензил-линкерным соединениям формулы (I), где R1, R2, A1, A2, X определены в формуле изобретения, предназначенным для связывания частей лекарственного средства с антителами, к соединениям линкер-лекарственное средство, в которых указанные пара-аминобензил-линкерные соединения ковалентно связаны с частями лекарственного средства, и к конъюгатам антитело-лекарственное средство, содержащим указанные пара-аминобензил-линкерные соединения, ковалентно связанные с лекарственным средством, при этом указанное лекарственное средство ферментативно отщепляется от конъюгата в конкретном типе клеток или тканей, являющихся мишенями для указанного антитела. 4 н. и 13 з.п. ф-лы, 3 ил., 5 табл., 34 пр.
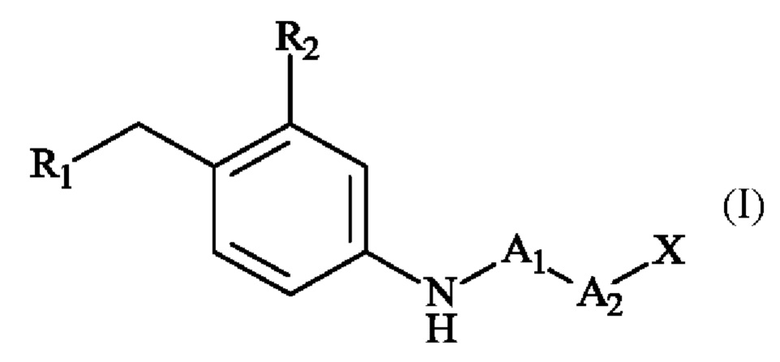
1. Пара-аминобензил-линкерное соединение формулы (I):
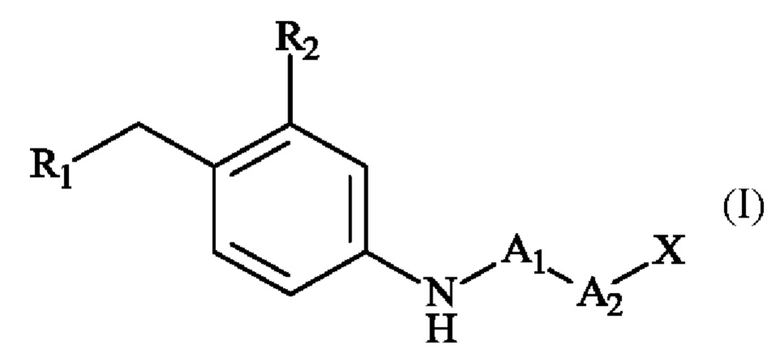
в которой:
• R1 представляет собой гидроксигруппу или атом галогена;
• R2 представляет собой группу -S(O)2(OH), группу -S(O)2(O-M+), линейную или разветвленную группу -(С1-С4)алкил-S(O)2(ОН), линейную или разветвленную группу -(С1-С4)алкил-S(O)2(O-М+), линейную или разветвленную группу -галоген(С1-С4)алкил-S(O)2(ОН) или линейную или разветвленную группу
-галоген(С1-С4)алкил-S(O)2(O-М+);
• A1 представляет собой группу -C(O)-CH(R3)-NH-, в которой R3 представляет собой группу -(CH2)3-NH-CO-NH2 или метальную группу;
• А2 представляет собой группу -C(O)-CH(R4)-NH-, в которой R4 представляет собой изопропильную группу, группу
 , группу
, группу  или группу
или группу  ;
;
• X представляет собой атом водорода или защитную группу, выбранную из Fmoc, Boc или CBz;
• М+ представляет собой фармацевтически приемлемый одновалентный катион.
2. Пара-аминобензил-линкерное соединение по п. 1, где R1 представляет собой гидроксигруппу, атом брома, атом хлора или атом йода.
3. Пара-аминобензил-линкерное соединение по п. 1, где R2 представляет собой группу -S(O)2(OH), группу -S(O)2(O-M+), группу -СН2-S(O)2(OH), группу -CH2-S(O)2(O-M+), группу -CH2-CH2-S(O)2(OH), группу -СН2-CH2-S(O)2(O-M+), группу-CH2-CH2-CH2-S(O)2(OH) или группу -СН2-СН2-СН2-S(O)2(O-M+).
4. Пара-аминобензил-линкерное соединение по п. 1, где A1 представляет собой группу -C(O)-CH(R3)-NH-, в которой R3 представляет собой группу -(CH2)3-NH-CO-NH2 или метальную группу, и А2 представляет собой группу -C(O)-CH(R4)-NH-, в которой R4 представляет собой изопропильную группу.
5. Пара-аминобензил-линкерное соединение по п. 1, которое представляет собой:
- 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-(гидроксиметил)бензолсульфонат натрия;
- 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]пропаноил]амино]-2-(гидроксиметил)бензолсульфонат натрия;
- 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]пропаноил]амино]-2-(гидроксиметил)бензолсульфоновую кислоту;
- [5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-(гидроксиметил)фенил]метансульфонат натрия;
-2-(хлорметил)-5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]бензолсульфоновую кислоту;
-2-(хлорметил)-5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]пропаноил]амино] бензолсульфоновую кислоту;
- 5-[[(2S)-2-[[(2S)-2-(9Н-флуорен-9-илметоксикарбониламино)-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-(йодметил)бензолсульфоновую кислоту;
- N-{[(9H-флуорен-9-ил-метокси]карбонил}L-валил-N5-карбамоил-N-[4-(гидроксиметил)-3-(2-сульфонатоэтил)фенил]L-орнитинамид натрия;
- N-{[(9H-флуорен-9-ил)метокси]карбонил}-L-валил-N5-карбамоил-N-[4-(гидроксиметил)-3-(3-сульфопропил)фенил]-L-орнитинамид.
6. Пара-аминобензил-линкерное соединение формулы (I) по любому из пп. 1-5 для применения в получении конъюгата антитело-лекарственное средство.
7. Соединение линкер-лекарственное средство формулы (II):
в которой:
• D представляет собой часть лекарственного средства;
• Т представляет собой связь или -О-С(О)-*, в которой * указывает место присоединения к D;
• R2 представляет собой группу -S(O)2(OH), группу -S(O)2(O-M+), линейную или разветвленную группу -(С1-С4)алкил-S(O)2(ОН), линейную или разветвленную группу -(С1-С4)алкил-S(O)2(O-М+), линейную или разветвленную группу -галоген(С1-С4)алкил-S(O)2(ОН) или линейную или разветвленную группу -галоген(С1-С4)алкил-S(O)2(O-М+);
• A1 представляет собой группу -C(O)-CH(R3)-NH-, в которой R3 представляет собой группу -(CH2)3-NH-CO-NH2 или метальную группу;
• А2 представляет собой группу -C(O)-CH(R4)-NH-, в которой R4
представляет собой изопропильную группу, группу 
группу  или группу
или группу 
• R5 представляет собой атом водорода или группу (С1-С4)алкила;
• Z’ представляет собой предшественник спейсерного звена;
• М+ представляет собой фармацевтически приемлемый одновалентный катион.
8. Соединение линкер-лекарственное средство по п. 7, где R2 представляет собой группу -S(O)2(OH), группу -S(O)2(O-M+), группу -СН2-S(O)2(ОН), группу -CH2-S(O)2(O-M+), группу -CH2-CH2-S(O)2(OH), группу -СН2-CH2-S(O)2(O-M+), группу -CH2-CH2-CH2-S(O)2(OH) или группу -СН2-СН2-СН2-S(O)2(O-M+).
9. Соединение линкер-лекарственное средство по п. 7, где Z’ представляет собой группу, выбранную из:
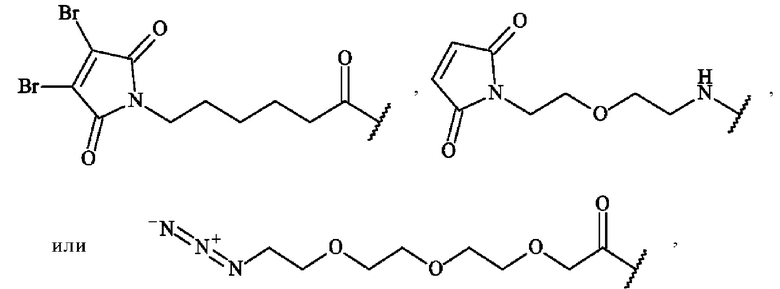
при этом волнистая линия указывает место ковалентного присоединения к N-концу или карбонильной группе группы А2.
10. Соединение линкер-лекарственное средство по п. 7, которое представляет собой:
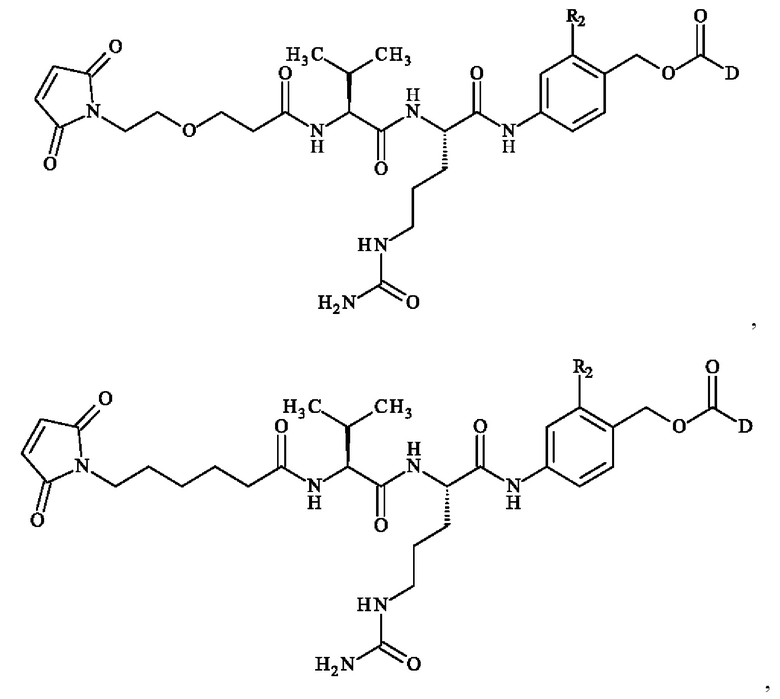
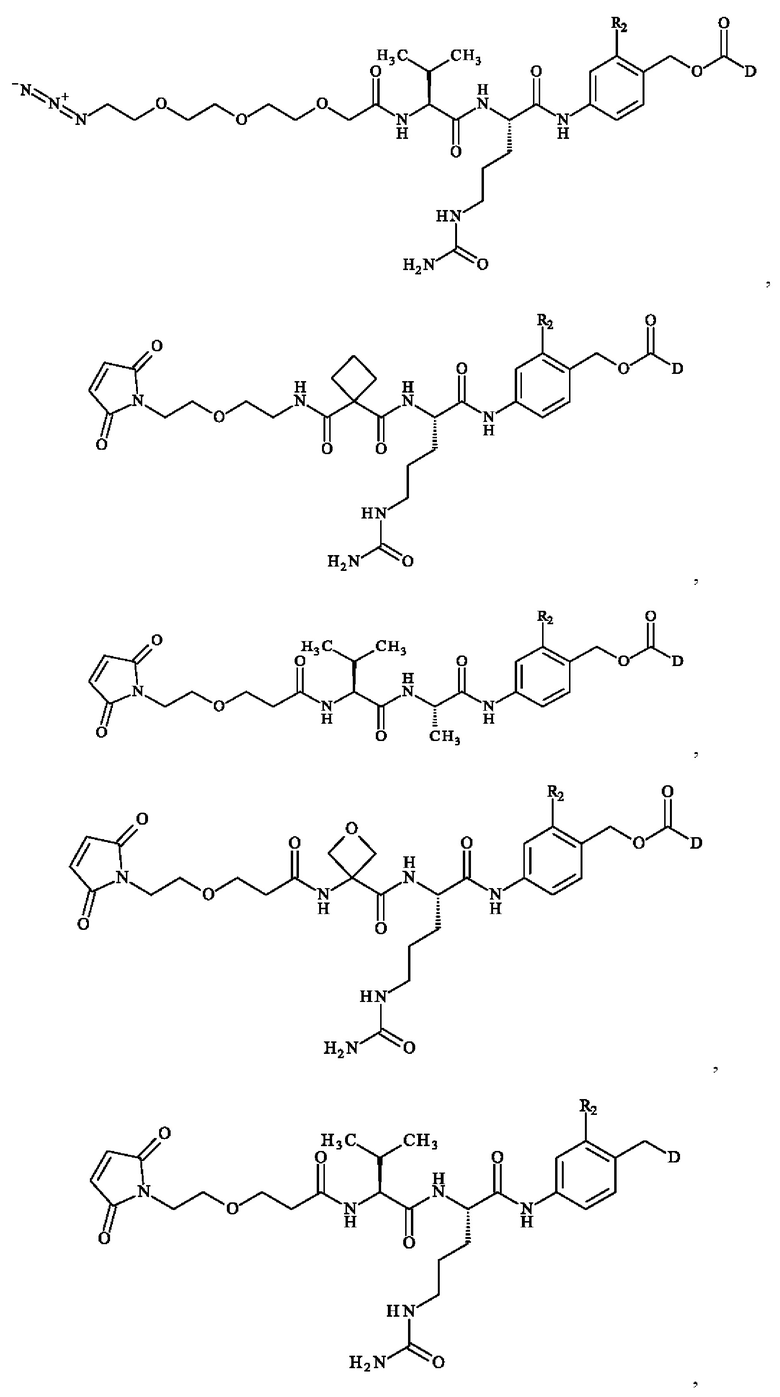
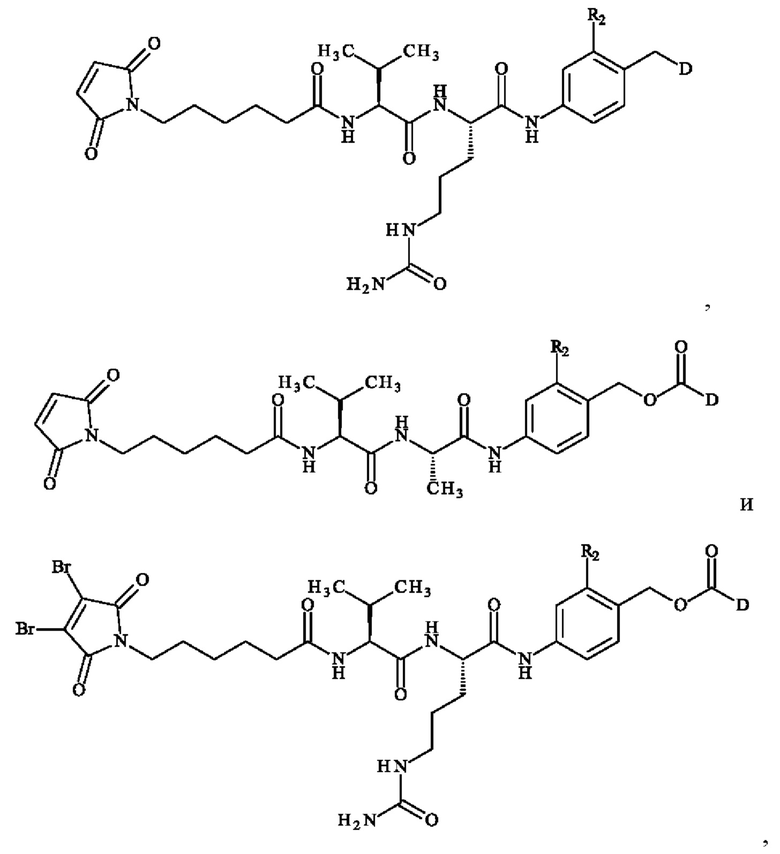
в которой R2 и D имеют определения, указанные в п. 7.
11. Соединение линкер-лекарственное средство по п. 7, которое представляет собой:
- 5-[[(2S)-2-[[(2S)-2-[3-[2-(2,5-диоксопиррол-1-ил)этокси]пропаноиламино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфонат натрия;
- 5-[[(2S)-2-[[(2S)-2-[6-(2,5-диоксопиррол-1-ил)гексаноиламино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфонат натрия;
- 5-[[(2S)-2-[[(2S)-2-[[2-[2-[2-(2азидоэтокси)этокси]этокси]ацетил]амино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфонат натрия;
- 5-[[(2S)-2-[[1-[2-[2-(2,5-диоксопиррол-1-ил)этокси]этилкарбамоил]циклобутанкарбонил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновую кислоту;
- 5-[[(2S)-2-[[(2S)-2-[3-[2-(2,5-диоксопиррол-1-ил)этокси]пропаноиламино]-3-метил-бутаноил]амино]пропаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфонат натрия;
- 5-[[(2S)-2-[[(2S)-2-[3-[2-(2,5-диоксопиррол-1-ил)этокси]пропаноиламино]-3-метил-бутаноил]амино]пропаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновую кислоту;
- 5-[[(2S)-2-[[3-[2-[2-(2,5-диоксопиррол-1-ил)этокси]этилкарбамоил]оксетан-3-карбонил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновую кислоту;
- [5-[[(2S)-2-[[(2S)-2-[3-[2-(2,5-диоксопиррол-1-ил)этокси]пропаноиламино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]фенил]метансульфоновую кислоту;
- [4-[[(2S)-2-[[(2S)-2-[3-[2-(2,5-диоксопиррол-1-ил)этокси]пропаноиламино]-3-метил-бутаноил] амино]-5-уреидо-пентаноил]амино]-2-сульфо-фенил]метил-[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-диметил-аммоний; 2,2,2-трифторацетат;
- N-({[4-({N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-L-аланил}амино)-2-сульфофенил]метокси}карбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид;
- N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-N-(4-{[({[(4S)-4,11-диэтил-9-гидрокси-3,14-диоксо-3,4,12,14-тетрагидро-1H-пирано[3',4':6,7]индолизино[1,2-6]хинолин-4-ил]окси}карбонил)окси]метил}-3-сульфофенил)-L-орнитинамид;
- (1S,3S)-3,5,12-тригидрокси-3-(гидроксиацетил)-10-метокси-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил 2,3,6-тридеокси-3-[({[4-({N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-L-валил-N5-карбамоил-L-орнитил}амино)-2-сульфофенил]метокси}карбонил)амино]-α-L-ликсо-гексопиранозид;
- N-{3-[2-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)этокси]пропаноил}-L-валил-N-{4-[(5S,8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-оксоэтил)-4,10-диметил-3,6,9-триоксо-5,8-ди(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадекан-1-ил]-3-(2-сульфоэтил)фенил}-N5-карбамоил-L-орнитинамид;
- 5-[[(2S)-2-[[(23)-2-[6-(3,4-дибром-2,5-диоксо-пиррол-1-ил)гексаноиламино]-3-метил-бутаноил]амино]-5-уреидо-пентаноил]амино]-2-[[[(1S)-1-[[(1S)-1-[[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-гидрокси-1-метил-2-фенил-этил]амино]-1-метокси-2-метил-3-оксо-пропил]пирролидин-1-ил]-2-метокси-1-[(1S)-1-метилпропил]-4-оксо-бутил]-метил-карбамоил]-2-метил-пропил]карбамоил]-2-метил-пропил]-метил-карбамоил]оксиметил]бензолсульфоновую кислоту.
12. Конъюгат антитело-лекарственное средство формулы (III):
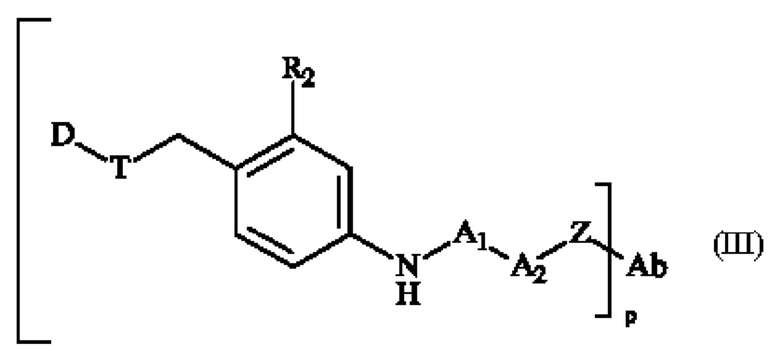
в которой
• Ab представляет собой антитело или его антиген связывающий фрагмент,
• D представляет собой часть лекарственного средства,
• Т представляет собой связь или -О-С(О)-*, в которой * указывает место присоединения к D,
• Z представляет собой спейсерное звено,
• A1 представляет собой группу -C(O)-CH(R3)-NH-, в которой R3 представляет собой группу -(CH2)3-NH-CO-NH2 или метильную группу,
• А2 представляет собой группу -C(O)-CH(R4)-NH-, в которой R4
представляет собой изопропильную группу, группу 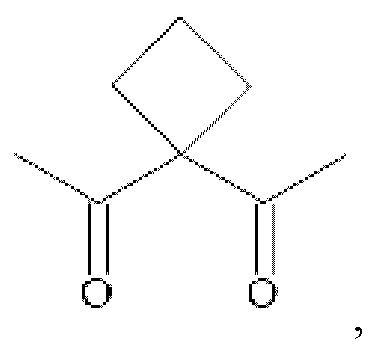 группу
группу 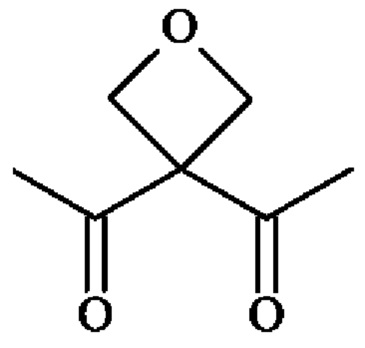 или группу
или группу 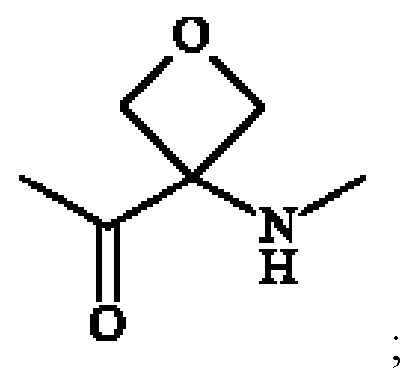
• R2 представляет собой группу -S(O)2(OH), группу -S(O)2(O-M+), линейную или разветвленную группу -(С1-С4)алкил-S(O)2(ОН), линейную или разветвленную группу -(С1-С4)алкил-S(O)2(O-М+), линейную или разветвленную группу -галоген(С1-С4)алкил-S(O)2(ОН) или линейную или разветвленную группу -галоген(С1-С4)алкил-S(O)2(O-М+);
• R5 представляет собой группу (С1-С4)алкила;
• М+ представляет собой фармацевтически приемлемый одновалентный катион; и
• р является целым числом от 1 до 8.
13. Конъюгат антитело-лекарственное средство по п. 12, где R2 представляет собой группу -S(O)2(OH), группу -S(O)2(O-M+), группу -СН2-S(O)2(OH), группу -CH2-S(O)2(O-M+), группу -CH2-CH2-S(O)2(OH), группу -СН2-CH2-S(O)2(O-M+), группу -CH2-CH2-CH2-S(O)2(OH) или группу -СН2-СН2-СН2-S(O)2(O-M+).
14. Конъюгат антитело-лекарственное средство по п. 12, где указанный конъюгат антитело-лекарственное средство образован из соединения линкер-лекарственное средство формулы (II), выбранного из:
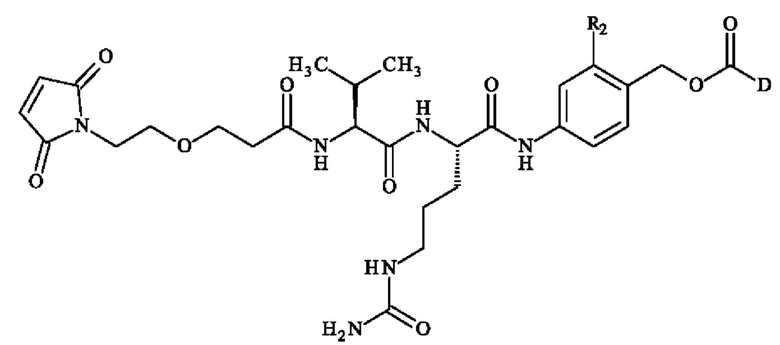 ,
,
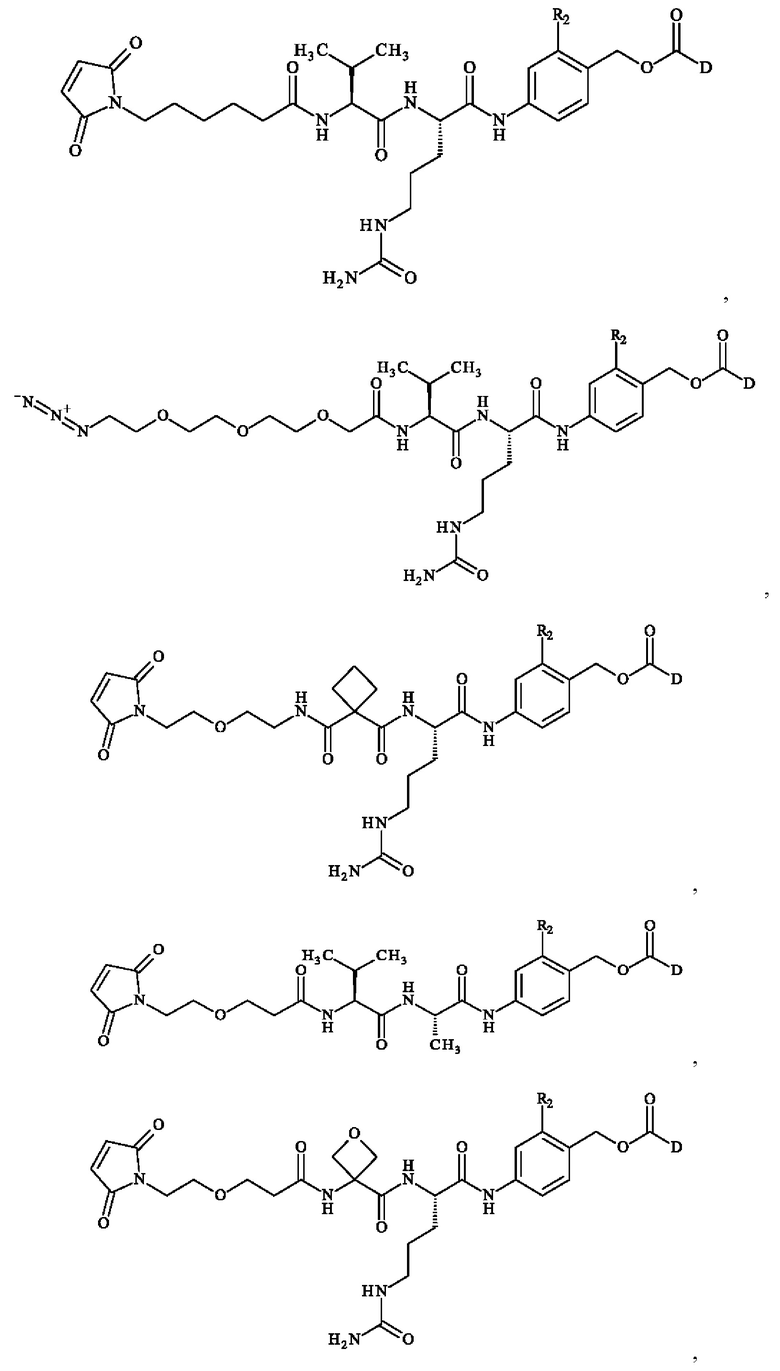
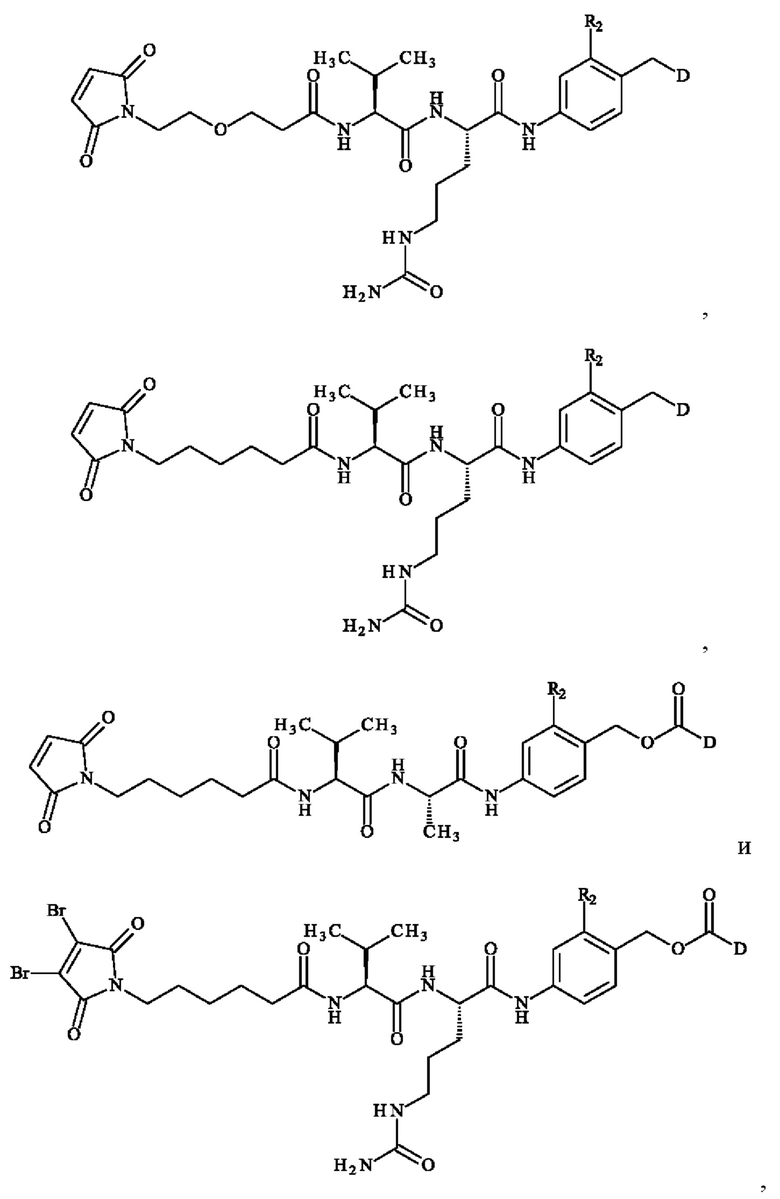
в которой R2 и D имеют определения, указанные в п. 12.
15. Конъюгат антитело-лекарственное средство по п. 12, где указанный конъюгат антитело-лекарственное средство содержит формулу, выбранную из:
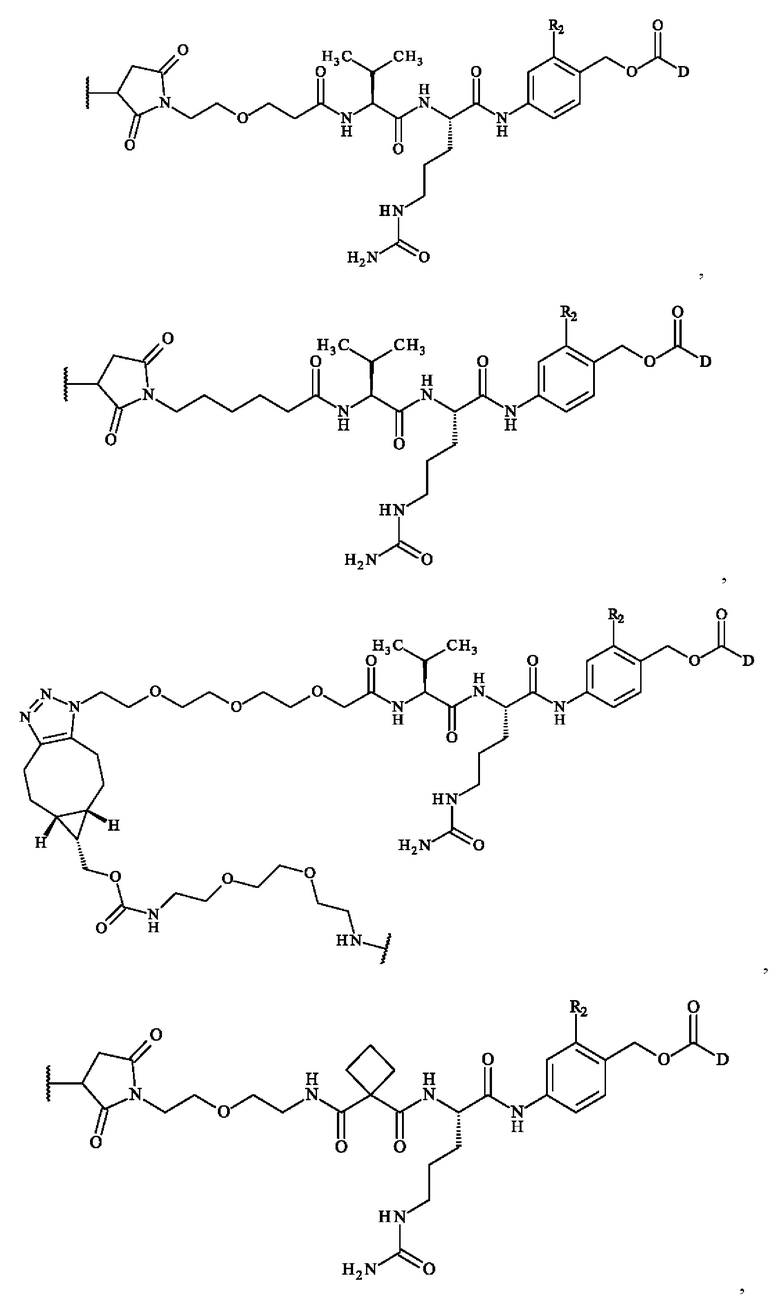
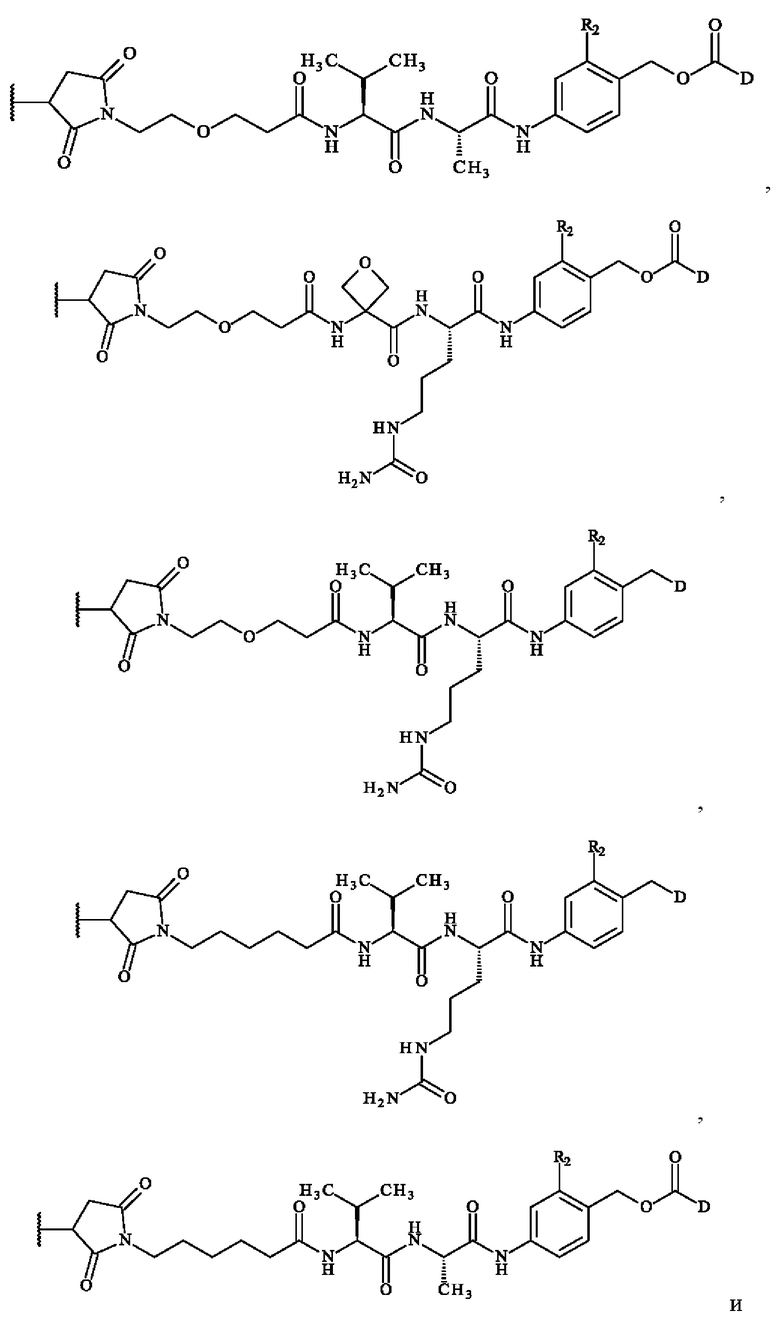
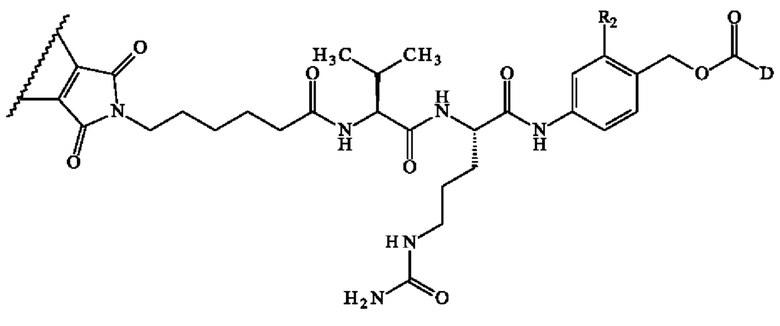 ,
,
в которой волнистая линия указывает место ковалентного связывания с антителом или его антигенсвязывающим фрагментом и D и R2 имеют определения, указанные в п. 12.
16. Фармацевтическая композиция для лечения расстройства, связанного с экспрессией антигена, с которым связывается антитело или его антигенсвязывающий фрагмент ADC, содержащая терапевтически эффективную дозу конъюгата антитело-лекарственного средства по любому из пп. 12-15, и фармацевтически приемлемый носитель.
17. Конъюгат антитело-лекарственное средство по любому из пп. 12-15 для применения в лечении нуждающегося в этом млекопитающего.
| Устройство для сборки калош или других каких-либо изделий | 1930 |
|
SU24730A1 |
| WO 2019108974 A1, 06.06.2019 | |||
| WO 2017214282 A1, 14.12.2017. | |||

