Область изобретения
Настоящее изобретение относится к линкерам, используемым в связи с конъюгатами антитело-лекарственное средство (ADC), которые способны осуществлять повышающее и понижающее модулирование внеклеточной и/или внутриклеточной стабильности конъюгатов антитело-лекарственное средство. Изобретение также относится к терапевтическим применениям и режимам лечения с использованием таких клинически предпочтительных конъюгатов антитело-лекарственное средство с модулированной стабильностью. И наконец, изобретение относится к способам получения модулирующих стабильность линкеров и конъюгатов антитело-лекарственное средство с модулированной стабильностью.
Ссылка на перечень последовательностей
Данная заявка подается в электронном виде через EFS-Web и включает в себя представленный в электронном виде перечень последовательностей в формате. txt. Файл в формате. txt под названием "PC72108A_SEQ_LISTING_ST25.txt", содержащий перечень последовательностей, создан 14 августа 2015 года и имеет размер 10 KB. Перечень последовательностей, содержащийся в этом.txt файле, является частью описания изобретения и во всей его полноте включен в данное описание посредством ссылки.
Предшествующий уровень техники
Терапия антителами обеспечивает направленное терапевтическое лечение пациентов с различными расстройствами, такими как различные виды рака и иммунологические заболевания, и поэтому играет важную роль в биологических исследованиях. Были изучены различные подходы направленной терапии антителами, включая конъюгаты антитело-лекарственное средство (ADC) (Chari, R.V., Miller, M.L., and Widdison, W.C. (2014) Antibidy-drug conjugates: an emerging concept in cancer therapy. Angewandte Chemie 53, 3796-827; Senter, P.D., and Sievers, E.L. (2012) The discovery and development of ADCETRIS® (brentuximab vedotin) for use in relapsed Hodgkin lymphoma и systemic anaplastic large cell lymphoma. Nature biotechnology 30, 631-7; Lambert, J.M. (2013) Drug-conjugated antibobies for the tratment of cancer. British journal of clinical pharmacology 76, 248-62).
В случае ADC (называемых также иммуноконъюгатами в некоторых ситуациях) "полезные нагрузки", имеющие небольшую молекулу, которые часто представляют собой небольшие цитотоксические молекулы (группировки лекарственных средств), ковалентно связаны (конъюгированы) с антителами для неправленной локальной доставки группировок лекарственных средств в опухоли. Общепринятые способы конъюгирования для получения ADC включают химическую модификацию либо по аминам боковых цепей лизина, либо по сульфгидрильным группам цистеина, активированным посредством восстановления межцепочечных дисульфидных связей. Брентуксимаб ведотин и KADCYLA® (адо-трастузумаб эмтазин) - это два примера ADC, полученных этими общепринятыми способами.
Ферментативные подходы с использованием трансглутаминазы для получения ADC также были изучены. Трансглутаминазы принадлежат семейству ферментов, которые катализируют присоединение ацила к первичному амину. Конъюгирование с использованием трансглутаминазы обеспечивает преимущества, которые заключаются в высокой селективности, упрощении реакционных методик и мягких реакционных условиях (смотри, например, Strop et al., Chemistry & Biology, 20:161-167 (2013); и Farias et al., Bioconj. Chem. 25(2):240-250 (2014). В US 2013-0230543 и US 2013-0122020 описано опосредованное трансглутаминазой сайт-специфическое конъюгирование антител и небольших молекул.
Общепринятые способы конъюгирования ADC приводят к получению ADC, имеющих свойственную им стабильность в данной биологической системе. Доставка полезной нагрузки в желаемую область, обычно опухоль, зависит от свойств полезной нагрузки, ковалентного линкера, антитела и биологической системы, в которую вводят ADC. Часто значительное количество полезной нагрузки высвобождается из менее чем оптимально стабильного ADC преждевременно, например в плазме крови, и поэтому необходимы более высокие уровни дозировки ADC для достижения желаемого воздействия полезной нагрузки в области опухоли. В других обстоятельствах использование чрезмерно стабильного ADC приводит к менее чем оптимальной активности высвобождения полезной нагрузки в являющихся мишенью клетках опухоли.
Протеолитически расщепляемые пептидные связи, использующие элемент удаления пара-аминобензилоксикарбонила (РАВС), широко применяются в исследованиях конъюгатов антитело-лекарственное средство (ADC) с 2002 года (Dubowchik, G.M. et al. Bioconjugate Chem. 2002, 13, 855-869). Такие связи предположительно подвергаются расщеплению после интернализации в нацеленные на антиген клетки и воздействия на ADC деструктивного протеолитического окружения, находящегося в эндосомальных и лизосомальных органеллах. Связанные с цистеином варианты этих дипептид-РАВС линкеров запатентованы (US 6,214,345 В2), и было продемонстрировано, что аминосодержащие варианты этих и других связей подходят для сайт-специфического конъюгирования с остатками глутамина с использованием ферментативного конъюгирования, промотируемого микробной трансглутаминазой, а также с использованием агликозилированных антител и сконструированных вариантов антител, которые эффективно конъюгируются с использованием ферментативного метода конъюгирования (WO 2012/059882 А2).
Все публикации, патенты и патентные заявки, процитированные в данном описании, включены в него посредством ссылки во всей их полноте для всех целей в той степени, как если бы каждая(ый) отдельная(ый) публикация, патент и патентная заявка были конкретно и отдельно указаны как включенные посредством ссылки. В случае если один или более из включенных литературных и подобных материалов отличаются от данной заявки или противоречат данной заявке, включая, без ограничения, определенные термины, использование терминов, описанные методы или т.п., то преимущество имеет текст данной заявки.
Краткое изложение сущности изобретения
Настоящее изобретение в целом относится к ADC, получаемым, как правило, в результате опосредованного трансглутаминазой конъюгирования, которые обладают свойствами стабильности, изменяющимися вследствие присутствия модулирующей группировки на линкерах ADC. Авторы изобретения неожиданно обнаружили, что такая модулирующая группировка, расположенная в конкретном положении на линкере ADC, и более конкретно некоторые химические отличительные признаки, включенные в эти модулирующие группировки, обладает способностью либо увеличивать, либо уменьшать стабильность ADC, по желанию, чтобы  или меньше относительное количество полезной нагрузки ADC высвобождалась в желаемом месте действия.
или меньше относительное количество полезной нагрузки ADC высвобождалась в желаемом месте действия.
Таким образом, предложены конкретные замещения на аминосодержащем конце конъюгирования, часто представляющие собой ацилированные производные лизина, которые могут иметь сильное влияние на стабильность in vitro в плазме крови и экспозицию результирующих ADC in vivo. Продемонстрировано, что стабильность конъюгата можно модулировать, варьируя природу заместителей, присутствующих на аминосодержащих концах конъюгирования, а также что заместитель может быть выбран для оптимизации стабильности каждого уникального сайта конъюгирования. Таким образом, данное изобретение позволяет осуществлять корректирвоку стабильности ADC in vivo и, следовательно, обеспечивает модулирование параметров, которые влияют как на эффективность, так и безопасность in vivo, и, следовательно, дает возможность оптимизировать терапевтический индекс ADC.
Краткое описание графических материалов
На Фиг. 1A-1I представлены исследования in vitro цитотоксичности химерного анти-Trop2 (опухоль-ассоциированный трансдуктор кальциевого сигнала 2 или антиген трофобластов 2) антитела, конъюгированного по ряду сайтов с амино-капроил (С6) vcAur0101 цитотоксической полезной нагрузкой.
На Фиг. 1J-1O представлены исследования in vitro цитотоксичности химерного анти-Trop2 антитела, конъюгированного по сайтам LCQ04 и L11B с конструкциями линкер-полезная нагрузка Примеров 2, 3 и 5.
На Фиг. 2 представлена стабильность in vivo ADC соединений Примеров 12 и 14 (Фиг. 2А), ADC соединений Примеров 16 и 13 (Фиг. 2В) и ADC соединений Примеров 11, 18 и 17 (Фиг. 2С), связанных через сайт LCQ04 на анти-Trop2 антителе. Пунктирные линии означают концентрацию полезной нагрузки, сплошные линии означают концентрацию антитела.
На Фиг. 3 представлено подтверждение мышиной карбоксилэстеразы 1с как фермента, ответственного за расщепление линкера vc-pabc в плазме крови, путем сравнения линии мышей с нокаутированным геном с56/bl6 ces1c-/-, гетерозиготной линии с56/bl6 ces1c+/- и линии дикого типа с56/bl6 ces1c+/+.
Подробное описание изобретения
Настоящее изобретение в целом относится к конъюгатам антитело-лекарственное средство (ADC), имеющим желаемую степень стабильности. Конкретно, согласно изобретению предложены соединения формулы (I):

где:
М представляет собой модулятор стабильности;
Р представляет собой пептидную последовательность, которая содержит один или более остатков глутамина;
Q представляет собой остаток глутамина, присутствующий в Р;
каждый Е независимо выбран из группы, состоящей из -C(R1)2-, -O-C(R1)2-С(R1)2-, где r равно по меньшей мере 2, и -C(R1)2-C(R1)2-O-, где s равно по меньшей мере 1;
каждый R1 независимо выбран из группы, состоящей из Н, прямого или разветвленного C1-C6алкила, прямого или разветвленного С2-С6алкенила и прямого или разветвленного С2-С6алкинила;
каждый X независимо представляет собой аминокислоту, где каждая аминокислота X является одной и той же или другой;
каждый Y независимо представляет собой аминокислоту, где каждая аминокислота Y является одной и той же или другой;
каждый Z независимо представляет собой спейсерный элемент, где каждый спейсерный элемент является одним и тем же или другим;
m равно 0-5, n равно 1-5, р равно 0-2, q равно 0-10, r равно 0-2, и s равно 0-2, где q+r+s=2 или более; и
D представляет собой цитотоксический агент или другой терапевтический агент, или D представляет собой визуализирующий агент.
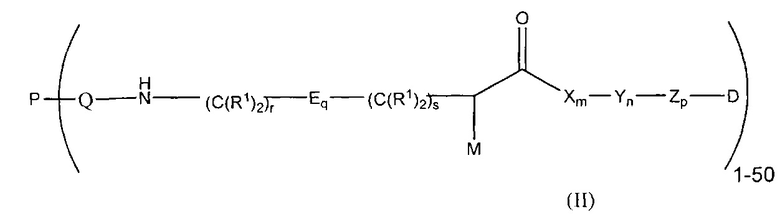
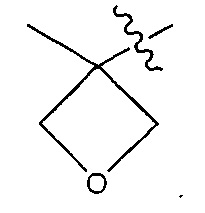
Воплощения изобретения также включают соединения формулы (II):
 ,
,
где:
М представляет собой модулятор стабильности;
Р представляет собой пептидную последовательность, которая содержит один или более остатков глутамина;
Q представляет собой один из указанных остатков глутамина, присутствующих в Р;
каждый Е независимо выбран из группы, состоящей из -C(R1)2-, -O-C(R1)2-C(R1)2- где r равно по меньшей мере 2, и -C(R1)2-C(R1)2-O-, где s равно по меньшей мере 1;
каждый R1 независимо выбран из группы, состоящей из Н, прямого или разветвленного C1-C6алкила, прямого или разветвленного С2-С6алкенила и прямого или разветвленного С2-С6алкинила;
каждый X независимо представляет собой аминокислоту, где каждая аминокислота X является одной и той же или другой;
каждый Y независимо представляет собой аминокислоту, где каждая аминокислота Y является одной и той же или другой;
каждый Z независимо представляет собой спейсерный элемент, где каждый спейсерный элемент является одним и тем же или другим;
m равно 0-5, n равно 1-5, р равно 0-2, q равно 0-10, r равно 0-2, и s равно 0-2, где q+r+s=2 или более; и
D представляет собой цитотоксический агент или другой терапевтический агент, или D представляет собой визуализирующий агент.
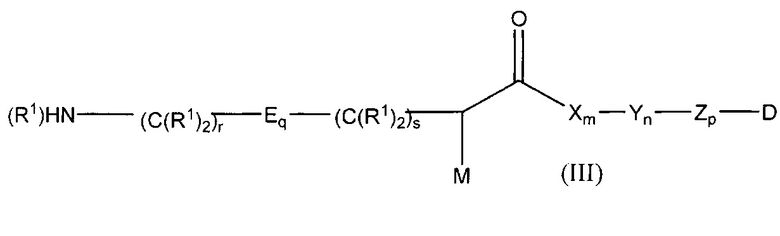
Дополнительные воплощения включают соединения формулы (III):
 ,
,
полезные для изготовления конъюгатов антитело-лекарственное средство, где:
М представляет собой модулятор стабильности;
Р представляет собой пептидную последовательность, которая содержит один или более остатков глутамина;
Q представляет собой один из указанных остатков глутамина, присутствующих в Р;
каждый Е независимо выбран из группы, состоящей из -C(R1)2-, -O-C(R1)2-C(R1)2- где r равно по меньшей мере 2, и -C(R1)2-C(R1)2-O-, где s равно по меньшей мере 1;
каждый R1 независимо выбран из группы, состоящей из Н, прямого или разветвленного C1-С6алкила, прямого или разветвленного С2-С6алкенила и прямого или разветвленного С2-С6алкинила;
каждый X независимо представляет собой аминокислоту, где каждая аминокислота X является одной и той же или другой;
каждый Y независимо представляет собой аминокислоту, где каждая аминокислота Y является одной и той же или другой;
каждый Z независимо представляет собой спейсерный элемент, где каждый спейсерный элемент является одним и тем же или другим;
m равно 0-5, n равно 1-5, р равно 0-2, q равно 0-10, r равно 0-2, и s равно 0-2, где q+r+s=2 или более; и
D представляет собой цитотоксический агент или другой терапевтический агент, или D представляет собой визуализирующий агент.
Воплощения изобретения включают соединения, которые описаны в данном документе, где М представляет собой -М1-М2, где М1 представляет собой -NR1-C(O)-, -NR1-S(O)2- или отсутствует, и М2 выбран из группы, состоящей из замещенного метила, -С2-С20алкила, -С1-С20гетероалкила, -С2-С6алкенила, -С2-Сбалкинила, C1-C6алкокси, карбокси, -N(R1)2, -С6-С14арила, -С6-С14гетероарила, -С1-С10гетероциклила и -С3-С10карбоциклила, и где М2 возможно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из -C1-С6алкила, -C1-С6алкенила, -C1-C6алкинила, галогена, C1-С6алкокси, гидроксила, -N(R1)2, -C(O)N(R1)2, -NO2, -С6-С14арила, -С6-С14гетероарила, -C1-С10гетероциклила, -С3-С10карбоциклила, карбокси, -SH, групп -S(C1-C6алкил), -S(C6-C14арил), -S(С6-С14гетероарил), -S(С1-С10гетероциклил), -S(С3-С10карбоциклил), -С1-С8алкил-С(O)-С1-С8алкил, -С1-С8алкил-С(O)-Н, -С1-С8алкил-С(O)-O-С1-С8алкил, -NR1-C(O)-N(R1)2, -С1-С8алкил-O-С(O)-N(R1)2, -С1-С8 алкил-S(O)2-N(R1)2, -С1-С8алкил-S(O)2-ОН, -С1-С8алкил-S(O)2-С1-С8алкил и -С1-С8алкил-S(O)-С1-С8алкил; при условии, что М2 не является незамещенным метилом, когда М1 представляет собой -NH-C(O)-, и -(C(R1)2)r-Eq-(C(R1)2)s- представляет собой прямоцепочечный С4-алкил.
Воплощения изобретения включают соединения, описанные в данном документе, где М представляет собой -М1-М2, где М1 представляет собой -NR1-C(O)-, -NR1-S(O)2- или отсутствует, и М2 выбран из группы, состоящей из -С2-С10алкила, -С1-С10 гетероалкила, -С6-С14арила, -С6-С14гетероарила, -С1-С10гетероциклила и -С3-С10карбоциклила, и где М2 возможно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из -C1-С6алкила, галогена, -С6-С14арила и -С6-С14 гетероарила.
Дополнительные воплощения включают соединения, описанные в данном документе, где М2 выбран из группы, состоящей из

В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:
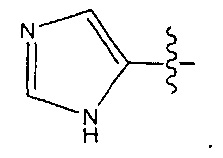
В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:
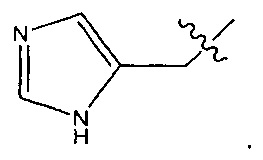
В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:

В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:
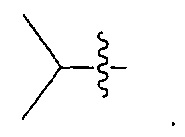
В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:

В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:

В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:
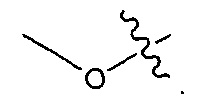
В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:
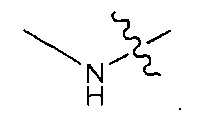
В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:
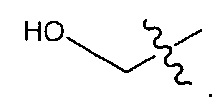
В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:
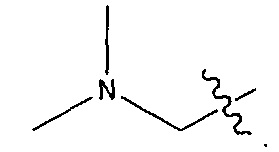
В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:

В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:
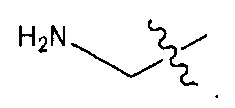
В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:
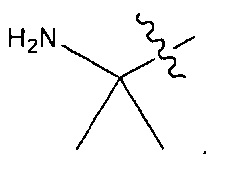
В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:
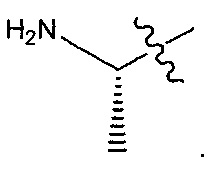
В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:
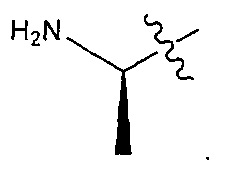
В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:
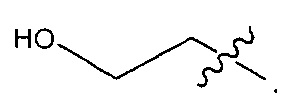
В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:
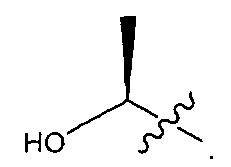
В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-мюдулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:
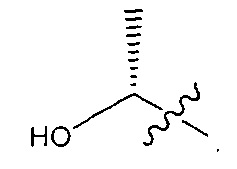
В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:
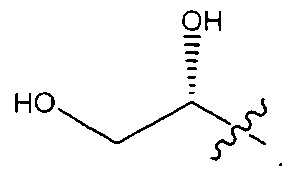
В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:
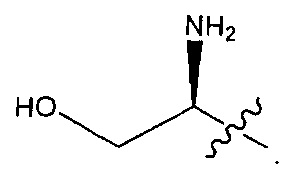
В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:
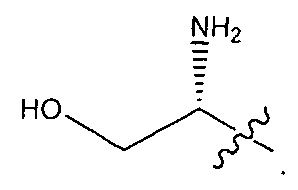
В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:
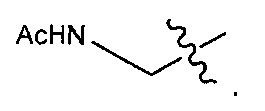
В некоторых воплощениях ADC по настоящему изобретению содержит линкерную систему на основе лизина с модифицированный ацил-модулирующей группировкой, где модифицированный ацил-модулирующая группировка представляет собой:
В некоторых воплощениях предложен способ модулирования стабильности ADC in vivo, включающий стадии выбора модулятора М, способного модулировать внеклеточную стабильность указанного соединения; включения указанного модулятора М в указанный конъюгат; и введения указанного конъюгата пациенту.
В некоторых воплощениях стабильность модулируют так, что относительное количество цитотоксического агента, высвобождаемого вне клеток-мишеней, по сравнению с цитотоксическим агентом, высвобождаемым внутри клеток-мишеней, увеличивают.
В некоторых воплощениях стабильность модулируют так, что относительное количество цитотоксического агента, высвобождаемого вне клеток-мишеней, по сравнению с цитотоксическим агентом, высвобождаемым внутри клеток-мишеней, уменьшают.
В некоторых воплощениях стабильность модулируют так, что относительное количество цитотоксического агента, высвобождаемого внутри клеток-мишеней, по сравнению с цитотоксическим агентом, высвобождаемым вне клеток-мишеней, увеличивают.
В некоторых воплощениях стабильность модулируют так, что относительное количество цитотоксического агента, высвобождаемого внутри клеток-мишеней, по сравнению с цитотоксическим агентом, высвобождаемым вне клеток-мишеней, уменьшают.
В некоторых воплощениях предложенные способы дополнительно включают стадию очистки, где ADC очищают хроматографией. В некоторых воплощениях трансглутаминаза представляет собой микробную, очищенную или сконструированную трансглутаминазу.
В другом аспекте изобретения предложен способ лечения рака у нуждающегося в этом субъекта, включающий введение субъекту эффективного количества фармацевтической композиции, содержащей ADC или ADCы, описанные в данном документе.
В другом аспекте изобретения предложен способ ингибирования роста или прогрессирования опухоли у нуждающегося в этом субъекта, включающий введение субъекту эффективного количества фармацевтической композиции, содержащей ADC, описанный в данном документе.
В другом аспекте изобретения предложен способ диагностирования рака у субъекта с подозрением на рак, включающий а) приведение образца субъекта в контакт с ADC, описанным в данном документе, в условиях, которые приводят к связыванию ADC с белком, ассоциированным с раком, и б) определение связывания ADC с белком, ассоциированным с раком.
В некоторых воплощениях антитело в ADC, как описано в данном документе, представляет собой моноклональное антитело, поликлональное антитело, человеческое антитело, гуманизированное антитело, химерное антитело, биспецифическое антитело, минитело, диатело или фрагмент антитела.
Воплощения изобретения включают соединения, описанные в данном документе, где цитотоксический агент D выбран из группы, состоящей из антрациклина, ауристатина, сплицеостатина, димера CBI (1-(хлорметил)-2,3-дигидро-1Н-бензо[е]индол)/CPI (циклопропа[с]пирроло[3,2-е]индол) (включая "смешанные" димеры, содержащие как CBI, так и CPI компоненты, как описано в предварительной патентной заявке США 61/932,118), калихеамицина, дуокармицина, энедиина, гелданамицина, майтанзина, пуромицина, таксана, алкалоида барвинка, SN-38, тубулизина, гемиастерлина, камптотецина, комбретастатина, доластатина, димера индолинобензодиазепина, димера пирролобензодиазепина и пладиенолида и их стереоизомеров, изостеров, аналогов или производных. Например, воплощения включают соединеия, где цитотоксический агент D представляет собой ауристатин, выбранный из группы, состоящей из долестатина, MMAD, ММАЕ, MMAF, PF-06380101, PF-06463377 и PF-06456780.
Дополнительные воплощения изобретения включают соединения, где D представляет собой нечто иное, чем цитотоксический агент, например где D представляет собой группировку, имеющую терапевтические свойства (т.е. терапевтический агент), и включает пептид(ы), белок(белки), нуклеиновую(ые) кислоту(ы), фактор(ы) роста, противовирусный(ые) агент(ы) или иммунологический(ие) агент(ы), или D представляет собой фторофор(ы) или другой(ие) визуализирующий(ие) агент(ы). Как и в случае, когда D представляет собой цитотоксический агент, изобретение охватывает модулирование стабильности соединений, когда D является иным, чем цитотоксический агент.
Дополнительные воплощения изобретения включают соединения, где Р представляет собой пептид, содержащий аминокислотную последовательность, выбранную из группы, состоящей из Q, LQG, LLQGG (SEQ ID NO: 1), LLQG (SEQ ID NO: 2), LSLSQG (SEQ ID NO: 3), GGGLLQGG (SEQ ID NO: 4), GLLQG (SEQ ID NO: 5), LLQ, GSPLAQSHGG (SEQ ID NO: 6), GLLQGGG (SEQ ID NO: 7), GLLQGG (SEQ ID NO: 8), GLLQ (SEQ ID NO: 9), LLQLLQGA (SEQ ID NO: 10), LLQGA (SEQ ID NO: 11), LLQYQGA (SEQ ID NO: 12), LLQGSG (SEQ ID NO: 13), LLQYQG (SEQ ID NO: 14), LLQLLQG (SEQ ID NO: 15), SLLQG (SEQ ID NO: 16), LLQLQ (SEQ ID NO: 17), LLQLLQ (SEQ ID NO: 18), LLQGR (SEQ ID NO: 19), LLQGPP (SEQ ID NO: 20), LLQGPA (SEQ ID NO: 21), GGLLQGPP (SEQ ID NO: 22), GGLLQGA (SEQ ID NO: 23), LLQGA (SEQ ID NO: 24), LLQGPGK (SEQ ID NO: 25), LLQGPG (SEQ ID NO: 26), LLQGP (SEQ ID NO: 27), LLQP (SEQ ID NO: 28), LLQPGK (SEQ ID NO: 29), LLQAPGK (SEQ ID NO: 30), LLQGAPG (SEQ ID NO: 31), LLQGAP (SEQ ID NO: 32), LLQGPA (SEQ ID NO: 33), LLQGPP (SEQ ID NO: 34), GGLLQGPP (SEQ ID NO: 35) и LLQLQG (SEQ ID NO: 36).
Воплощения изобретения также включают соединения, описанные в данном документе, где Р представляет собой пептид, содержащий аминокислотную последовательность XXQX(SEQ ID NO: 37), где X представляет собой любую аминокислоту.
Воплощения изобретения включают соединения, описанные в данном документе, где стабильность указанного соединения уменьшена по меньшей мере 1-, 5-, 10-, 25-, 50-, 75- или 100-кратно относительно соответствующего соединения, не имеющего указанного модулятора М.
Воплощения изобретения включают соединения, описанные в данном документе, где стабильность указанного соединения увеличена по меньшей мере 1-, 5-, 10-, 25-, 50-, 75- или 100-кратно относительно соответствующего соединения, не имеющего указанного модулятора М.
Воплощения изобретения включают соединения, описанные в данном документе, где X-Y выбран из группы, состоящей из Gly, β-Ala, Val-Cit, Phe-Lys, Val-Lys, Phe-Phe-Lys, Ala-Lys, Phe-Cit, Leu-Cit, Ala-Cit, Trp-Cit, Phe-Ala, Gly-Phe-Leu-Gly, Ala-Leu-Ala-Leu, Phe-N9-тозил-Arg, Phe-N9-нитро-Arg, Val-Ala и Ala-Ala-Asn.
Воплощения изобретения включают соединения, описанные в данном документе, где Р выбран из группы, состоящей из моноклонального антитела, поликлонального антитела, человеческого антитела, гуманизированного антитела, химерного антитела, биспецифического антитела, минитела и фрагмента антитела.
Воплощения изобретения включают соединения, описанные в данном документе, где антитело выбрано из трастузумаба, трастузумаба мутантов (например трастузумаба мутантов, раскрытых в данном документе или в международной патентной заявке PCT/IB2012/056234), ореговомаба, эдреколомаба, цетуксимаба, гуманизированного моноклонального антитела к рецептору витронектина (αvβ3), алемтузумаба, анти-HLA-DR антител, включающих гуманизированное анти-HLA-DR антитело для лечения неходжкинской лимфомы, 131I Lym-1, анти-HLA-Dr10 антител, включающих мышиное анти-HLA-Dr10 антитело для лечения неходжкинской лимфомы, анти-cd33 антител, анти-cd22 антител, включающих гуманизированное анти-CD22 mAb для лечения болезни Ходжкина или неходжкинской лимфомы, лабетузумаба, бевацизумаба, ибритумомаба тиуксетана, офатумумаба, панитумумаба, ритуксимаба, тозитумомаба, ипилимумаба и гемтузумаба.
Дополнительные воплощения изобретения включают соединения, описанные в данном документе, где каждый Q независимо представляет собой остаток глутамина, эндогенный для пептидной последовательности Р, или остаток глутамина, предоставленный в последовательности сконструированной метки на указанной пептидной последовательности Р (где метка может представлять собой множественную аминокислотную последовательность, содержащую глутамин, или просто глутамин).
В еще одном аспекте изобретения антитело служит в качестве макромолекулярного носителя, с функцией направленной доставки или без нее, облегчающего контролируемое высвобождение полезной нагрузки D с помощью расщепляемого линкера с модулированной стабильностью.
Воплощения изобретения включают соединения, описанные в данном документе, где каждый Q представляет собой остаток глутамина, эндогенный для указанной пептидной последовательности Р.
Воплощения изобретения включают соединения, описанные в данном документе, каждый Q представляет собой остаток глутамина, предоставленный в последовательности сконструированной метки на указанной пептидной последовательности Р (и снова, где метка может представлять собой множественную аминокислотную последовательность, содержащую глутамин, или просто глутамин).
Дополнительные воплощения изобретения включают способы модулирования in vivo стабильности конъюгата антитело-лекарственное средство формулы (II):
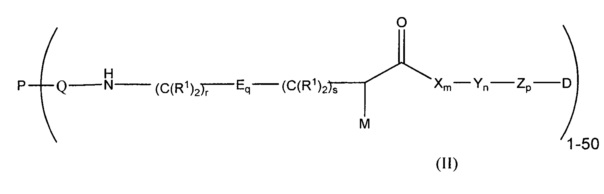 ,
,
где:
М представляет собой модулятор стабильности;
Р представляет собой пептидную последовательность, которая содержит один или более остатков глутамина;
Q представляет собой один из указанных остатков глутамина, присутствующих в Р;
каждый Е независимо выбран из группы, состоящей из -C(R1)2-, -O-C(R1)2-C(R1)2- где r равно по меньшей мере 2, и -C(R1)2-C(R1)2-O- где s равно по меньшей мере 1;
каждый R1 независимо выбран из группы, состоящей из Н, прямого или разветвленного C1-С6алкила, прямого или разветвленного С2-С6алкенила и прямого или разветвленного С2-С6алкинила;
каждый X независимо представляет собой аминокислоту, где каждая аминокислота X является одной и той же или другой;
каждый Y независимо представляет собой аминокислоту, где каждая аминокислота Y является одной и той же или другой;
каждый Z независимо представляет собой спейсерный элемент, где каждый спейсерный элемент является одним и тем же или другим;
m равно 0-5, n равно 1-5, р равно 0-2, q равно 0-10, r равно 0-2, и s равно 0-2, где q+r+s=2 или более; и
D представляет собой цитотоксический агент;
включающий стадии
выбора модулятора М, способного модулировать внеклеточную стабильность указанного соединения;
включения указанного модулятора М в указанный конъюгат; и
введения указанного конъюгата пациенту.
Воплощения изобретения включают способы лечения, как описано в данном документе, при которых относительное количество цитотоксического агента, высвобождаемого вне клеток-мишеней, по сравнению с цитотоксическим агентом, высвобождаемым внутри клеток-мишеней, увеличивают.
Воплощения изобретения включают способы лечения, как описано в данном документе, при которых относительное количество цитотоксического агента, высвобождаемого вне клеток-мишеней, по сравнению с цитотоксическим агентом, высвобождаемым внутри клеток-мишеней, уменьшают.
Воплощения изобретения включают способы лечения, как описано в данном документе, при которых относительное количество цитотоксического агента, высвобождаемого внутри клеток-мишеней, по сравнению с цитотоксическим агентом, высвобождаемым вне клеток-мишеней, увеличивают.
Воплощения изобретения включают способы лечения, как описано в данном документе, при которых относительное количество цитотоксического агента, высвобождаемого внутри клеток-мишеней, по сравнению с цитотоксическим агентом, высвобождаемым вне клеток-мишеней, уменьшают.
Дополнительные воплощения изобретения включают способы синтеза соединения формулы (II):
 ,
,
где:
М представляет собой модулятор стабильности;
Р представляет собой пептидную последовательность, которая содержит один или более остатков глутамина;
Q представляет собой один из указанных остатков глутамина, присутствующих в Р;
каждый Е независимо выбран из группы, состоящей из -C(R1)2-, -O-C(R1)2-C(R1)2- где r равно по меньшей мере 2, и -C(R1)2-C(R1)2-O- где s равно по меньшей мере 1;
каждый R1 независимо выбран из группы, состоящей из Н, прямого или разветвленного C1-С6алкила, прямого или разветвленного С2-С6алкенила и прямого или разветвленного С2-С6алкинила;
каждый X независимо представляет собой аминокислоту, где каждая аминокислота X является одной и той же или другой;
каждый Y независимо представляет собой аминокислоту, где каждая аминокислота Y является одной и той же или другой;
каждый Z независимо представляет собой спейсерный элемент, где каждый спейсерный элемент является одним и тем же или другим;
m равно 0-5, n is 0-5, р равно 0-2, q равно 0-10, r равно 0-2, и s равно 0-2, где q+r+s=2 или более; и
D представляет собой цитотоксический агент;
включающий стадии
получения некоторого количества первого соединения, имеющего структуру
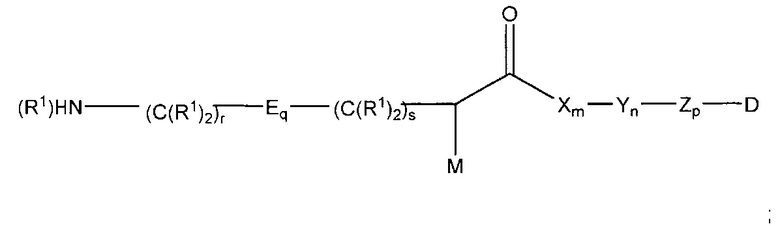
получения некоторого количества второго соединения, которое содержит пептидную последовательность, содержащую глутамин; и
осуществления взаимодействия указанных количеств первого и второго соединений в присутствии трансглутаминазы.
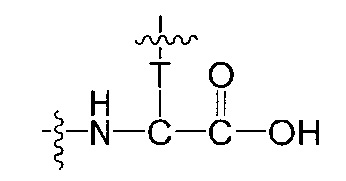
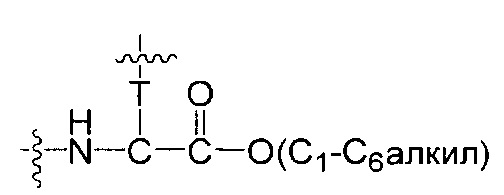
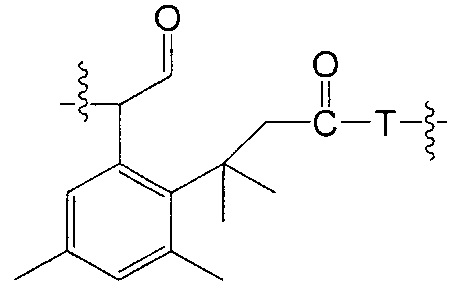
Воплощения изобретения включают соединения, описанные в данном документе, где Z выбран из группы, состоящей из РАВ-OH (пара-аминобензилкарбамоил), РАВ-ОН (пара-аминокарбамоилокси),
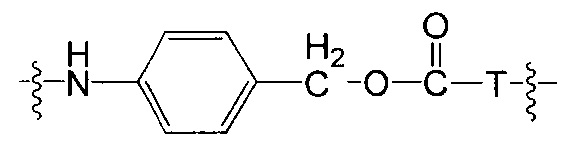 ,
, 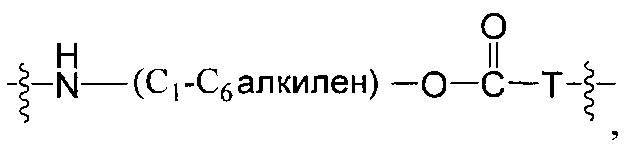
 ,
,  и
и
 ,
,
где Т представляет собой О, NH или S.
Воплощения изобретения включают соединения, описанные в данном документе, где Z представляет собой Z1-Z2, где Z1 выбран из группы, состоящей из пара-аминобензилкарбамоила (РАВС), пара-аминобензилокси, орто-аминобензилкарбамоила, орто-аминобензилокси, -NH-U-C(R1)2OCO- и -NH-U-C(R1)2O-; Z2 отсутствует или выбран из группы, состоящей из групп -N(R1)2-(C1-С6-алкилен)-ОСО-, -N(R1)2-(C1-C6-алкилен)-N(R1)2CO-, -N(R1)2-(C1-C6-алкилен)-SCO-, -N(R1)2-(С3-С9-циклоалкилен)-ОСО-, -N(R1)2-(С3-С9-циклоалкилен)-N(R1)2CO- и -N(R1)2-(С3-С9-алкилен)-SCO-; где U представляет собой ароматическое или гетероароматическое С5-С20 кольцо, возможно замещенное дополнительными заместителями в количестве вплоть до пяти, выбранными из группы, состоящей из -C1-C6алкила, -C1-C6алкенила, -C1-C6алкинила, галогена, С1-С6алкокси, гидроксила, -N(R1)2, -C(O)N(R1)2, -NO2, -С6-С14арила, -С6-С14гетероарила, -C1-С10гетероциклила, -С3-С10карбоциклила, карбокси, -SH, групп -S(С1-С6алкил), -S(C6-C14арил), -S(С6-С14 гетероарил), -S(С1-С10гетероциклил), -S(С3-С10карбоциклил), -С1-С8алкил-С(O)-С1-С8алкил, -С1-С8алкил-С(O)-Н, -С1-С8алкил-С(O)-O-С1-С8алкил, -NR1-C(O)-N(R1)2, -С1-С8алкил-O-С(O)-N(R1)2, -C1-C8алкил-S(O)2-N(R1)2, -С1-С8алкил-S(O)2-ОН, -С1-С8алкил-S(O)2-С1-С8алкил и -С1-С8алкил-S(O)-С1-С8алкил. Заместители -NH-и -C(R1)2OCO- или -C(R1)2O- на U должны быть расположены таким образом, чтобы обеспечивалось эффективное удаление Z2-D при расщеплении связи Y-Z (смотри, например, US 7,754,681 В2 и US 6214345 В2).
Термин "алкил", сам по себе или как часть другого термина, относится к прямоцепочечному или разветвленному насыщенному углеводороду, имеющему указанное количество атомов углерода (например, "С1-С8алкил" относится к алкильной группе, имеющей от 1 до 8 атомов углерода). Когда количество атомов углерода не указано, тогда алкильная группа имеет от 1 до 8 атомов углерода. Репрезентативные прямоцепочечные С1-С8алкилы включают, без ограничения, метил, этил, н-пропил, н-бутил, н-пентил, н-гексил, н-гептил и н-октил, а разветвленные С1-С8алкилы включают, без ограничения, -изопропил, -втор-бутил, -изобутил, -трет-бутил, -изопентил и -2-метилбутил; ненасыщенные С2-С8алкилы включают, без ограничения, винил, аллил, 1-бутенил, 2-бутенил, изобутиленил, 1-пентенил, 2-пентенил, 3-метил-1-бутенил, 2-метил-2-бутенил, 2,3-диметил-2-бутенил, 1-гексил, 2-гексил, 3-гексил, ацетиленил, пропинил, 1-бутинил, 2-бутинил, 1-пентинил, 2-пентинил и 3-метил-1-бутинил.
Термин "гетероалкил", сам по себе или в сочетании с другим термином, означает, если не указано иное, стабильный углеводород с прямой или разветвленной цепью или их комбинациями, полностью насыщенный или содержащий от 1 до 3 степеней ненасыщения, состоящий из указанного количества атомов углерода и от одного до трех гетероатомов, выбранных из группы, состоящей из О, N, Si и S, и где атомы азота и серы возможно могут быть окисленными, и гетероатом азота возможно может быть кватернизированным. Гетероатомы О, N и S могут быть расположены в любом внутреннем положении гетероалкильной группы. Гетероатом Si может быть расположен в любом положении гетероалкильной группы, включая положение, в котором алкильная группа присоединена к остальной части молекулы. Вплоть до двух гетероатомов могут следовать друг за другом.
Термин "арил", сам по себе или как часть другого термина, означает, если не указано иное, замещенный или незамещенный одновалентный карбоциклический ароматический углеводородный радикал из 6-20, предпочтительно 6-14, атомов углерода, образующийся в результате удаления одного атома водорода с единственного атома углерода родительской ароматической кольцевой системы. Типичные арильные группы включают, без ограничения, радикалы, образующиеся из бензола, замещенного бензола, нафталина, антрацена, бифенила и т.п. Арильная группа может быть замещена одной или более предпочтительно 1-5 следующими группами: С1-С8алкил, -O-(С1-С8алкил), -C(O)R', -OC(O)R', -C(O)OR', -C(O)NH2, -C(O)NHR', -C(O)N(R')2, -NHC(O)R', -S(O)2R', -S(O)R', -ОН, галоген, -N3, -NH2, -NH(R'), -N(R')2 и -CN; где каждый R' независимо выбран из -Н, С1-С8алкила и незамещенного арила.
Термин "гетероциклил", сам по себе или как часть другого термина, если не указано иное, относится к одновалентной, замещенной или незамещенной, ароматической или неароматической моноциклической, бициклической или трициклической кольцевой системе, имеющей от 1 до 10, предпочтительно от 3 до 8, атомов углерода (также упоминаемых как члены кольца) и от одного до четырех гетероатомов-членов кольца, независимо выбранных из N, О, Р или S, и образующийся в результате удаления одного атома водорода с кольцевого атома родительской кольцевой системы. Один или более атомов N, С или S в гетероциклиле могут быть окисленными. Кольцо, которое содержит гетероатом, может быть ароматическим или неароматическим. Если не указано иное, гетероциклил соединен с его боковой группой по любому гетероатому или атому углерода, который приводит к стабильной структуре. Репрезентативные примеры С1-С10 гетероциклила включают, без ограничения, тетрагидрофуранил, оксетанил, пиранил, пирродидинил, пиперидинил, пиперазинил, бензофуранил, бензотиофен, бензотиазолил, индолил, бензопиразолил, пирролил, тиофенил (тиофен), фуранил, тиазолил, имидазолил, пиразолил, триазолил, хинолинил, включая такие группировки, как 1,2,3,4-тетрагидрохинолинил, пиримидинил, пиридинил, пиридонил, пиразинил, пиридазинил, изотиазолил, изоксазолил, тетразолил, эпоксид, оксетан и BODIPY (4,4-дифтор-4-бора-3а,4а-диаза-s-индацен) (замещенный или незамещенный). C1-С10гетероциклил может быть замещен группами в количестве вплоть до семи, включающими, без ограничения, С1-С8алкил, С1-С8гетероалкил, -OR', арил, -C(O)R', -OC(O)R', -C(O)OR', -C(O)NH2, -C(O)NHR', -C(O)N(R')2, -NHC(O)R', -S(=O)2R', -S(O)R', галоген, -N3, -NH2, -NH(R'), -N(R')2 и -CN, где каждый R' независимо выбран из -Н, С1-С8алкила, С1-С8гетероалкила и арила. В некоторых воплощениях замещенный гетероциклил может также содержать одну или более групп -NHC(=NH)NH2, -NHCONH2, -S(=O)2R' и -SR'.
Термин "карбоциклил", сам по себе или как часть другого термина, если не указано иное, относится к а 3-, 4-, 5-, 6-, 7- или 8-членному одновалентному, замещенному или незамещенному, насыщенному или ненасыщенному неароматическому моноциклическому или бициклическому карбоциклическому кольцу, образующемуся в результате удаления одного атома водорода с кольцевого атома родительской кольцевой системы. Репрезентативные карбоциклилы включают, без ограничения, циклопропил, циклобутил, циклопентил, циклопентадиенил, циклогексил, циклогексенил, 1,3-циклогексадиенил, 1,4-циклогексадиенил, циклогептил, 1,3-циклогептадиенил, 1,3,5-циклогептадиенил, циклооктил, циклооктадиенил, бицикло(1.1.1.)пентан и бицикло(2.2.2.)октан. Карбоциклическая группа может быть незамещенной или может быть замещена группами в количестве вплоть до семи, включающими, без ограничения, С1-С8алкил, С1-С8 гетероалкил, -OR', арил, -C(O)R', -OC(O)R', -C(O)OR', -C(O)NH2, -C(O)NHR', -C(O)N(R')2, -NHC(O)R', -S(=O)2R', -S(=O)R', -OH, -галоген, -N3, -NH2, -NH(R'), -N(R')2 и -CN, где каждый R' такой, как указано выше.
Термин "алкенил-", если не указано иное, относится к ненасыщенному углеводороду с прямой или разветвленной цепью, содержащему по меньшей мере одну двойную связь и предпочтительно от 2 до 10 атомов углерода. Примеры группы алкенил- включают, без ограничения, этилен, пропилен, 1-бутилен, 2-бутилен, изобутилен, втор-бутилен, 1-пентен, 2-пентен, изопентен, 1-гексен, 2-гексен, 3-гексен, изогексен, 1-гептен, 2-гептен, 3-гептен, 1-октен, 2-октен, 3-октен, 4-октен, 1-нонен, 2-нонен, 3-нонен, 4-нонен, 1-децен, 2-децен, 3-децен, 4-децен и 5-децен. Группа алкенил- может быть незамещенной или замещенной.
Термин "алкокси-" относится к группе алкил-О-, где алкил такой, как определено в данном документе. Иллюстративные группы алкокси- включают, без ограничения, метокси, этокси, н-пропокси, 1-пропокси, н-бутокси и трет-бутокси. Группа алкокси может быть незамещенной или замещенной.
Термин "алкинил-" относится к ненасыщенному углеводороду с прямой или разветвленной цепью, содержащему по меньшей мере одну тройную связь и предпочтительно от 2 до 10 атомов углерода. Примеры группы алкинил-включают, без ограничения, ацетилен, пропин, 1-бутин, 2-бутин, изобутин, втор-бутин, 1-пентин, 2-пентин, изопентин, 1-гексин, 2-гексин, 3-гексин, изогексин, 1-гептин, 2-гептин, 3-гептин, 1-октин, 2-октин, 3-октин,. 4-октин, 1-нонин, 2-нонин, 3-нонин, 4-нонин, 1-децин, 2-децин, 3-децин, 4-децин и 5-децин. Группа алкинил- может быть незамещенной или замещенной.
Термин "гетероарил-" относится к 5-10-членным моно- и бициклическим ароматическим группам, содержащим по меньшей мере один гетероатом, выбранный из атомов кислорода, серы и азота и, как правило, от 1 до 9 атомов углерода. Примеры моноциклических гетероарильных радикалов включают, без ограничения, оксазинил, тиазинил, диазинил, триазинил, тиадиазолил, тетразинил, имидазолил, тетразолил, изоксазолил, фуранил, фуразанил, оксазолил, тиазолил, тиофенил, пиразолил, триазолил, пиримидинил, N-пиридил, 2-пиридил, 3-пиридил и 4-пиридил. Примеры бициклических гетероарильных радикалов включают, без ограничения, бензимидазолил, индолил, изохинолинил, бензофуранил, бензотиофенил, индазолил, хинолинил, хиназолинил, пуринил, бензизоксазолил, бензоксазолил, бензотиазолил, бензодиазолил, бензотриазолил, изоиндолил и индазолил. Гетероарильная группа может быть незамещенной или замещенной.
Термин "карбоксил-" относится к группе, обычно алкильной группе, как она определена в данном документе, которая присоединена к родительской структуре через атом кислорода карбоксильной функциональной группы (С(О)-О-). Примеры карбоксильных групп включают ацетокси, пропионокси, пропилкарбоксил и изопентилкарбоксил.
"Замещенный", например в связи с такой химической группировкой, как "замещенный алкил", если не указано иное, означает группировку, в которой один или более атомов водорода, каждый независимо, заменены заместителем. Типичные заместители включают, без ограничения, -X, -R, -О-, -OR, -SR, -S-, -NR2, -NR3, =NR, -CX3, -CN, -OCN, -SCN, -N=C=O, -NCS, -NO, -NO2, =N2, -N3, -NRC(=O)R, -C(=O)NR2, -SO3-, -SO3H, -S(=O)2R, -OS(=O)2OR, -S(=O)2NR, -S(=O)R, -OP(=O)(OR)2, -P(=O)(OR)2, -PO32-, PO3H2, -AsO2H2, -C(=O)R, -C(=O)X, -C(=S)R, -CO2R, -CO2-, -C(=S)OR, -C(=O)SR, -C(=S)SR, -C(=O)NR2, -C(=S)NR2 или -C(=NR)NR2, где каждый X независимо представляет собой галоген: -F, -Cl, -Br или -I; и каждый R независимо представляет собой -Н, С1-С20алкил, C1-С20гетероалкил, С6-С20арил, C1-С10гетероциклил, защитную группу или пролекарственную группировку.
Предложены также способы лечения рака, ингибирования роста или прогрессирования опухолей, ингибирования метастазирования раковых клеток или опухолей или вызывания регрессии опухоли у нуждающегося в этом субъекта, включающие введение субъекту эффективного количества фармацевтической композиции, содержащей ADC, описанные в данном документе.
Общие методы и определения
Если не дано иного определения, научные и технические термины, использованные в связи с настоящим изобретением, имеют значения, которые являются общеупотребительными и понятными специалистам в данной области. Кроме того, если контекст не требует иного, термины в единственном числе охватывают формы множественного числа, а термины во множественном числе охватывают формы единственного числа. Как правило, номенклатура, использованная в связи с клеточной и тканевой культурой, молекулярной биологией, иммунологией, микробиологией, генетикой и химией белков и нуклеиновых кислот и гибридизацией и их методами, описанными в данном документе, общеизвестны и обычно используются в данной области.
Способы и методы по настоящему изобретению обычно осуществляют в соответствии со способами, общеизвестными в данной области и так, как описано в различных общих и более конкретных источниках информации, которые цитируются и обсуждаются по всему описанию настоящего изобретения, если не указано иное (смотри, например, Sambrook J. & Russell D. Molecular Cloning: A Laboratory Manual, 3rd ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. (2000); Ausubel et al., Short Protocols in Molecular Biology: A Compendium of Methods from Current Protocols in Molecular Biology, Wiley, John & Sons, Inc. (2002); Harlow and Lane Using Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. (1998); и Coligan et al., Short Protocols in Protein Science, Wiley, John & Sons, Inc. (2003)). Ферментативные реакции и методы очистки осуществляют согласно спецификациям производителя, как обычно принято в данной области или как описано в данном документе. Номенклатура, использованная в связи с молекулярной биологией, биохимией, иммунологией, аналитической химией, синтетической органической химией и медицинской и фармацевтической химией, и лабораторные методики и методы молекулярной биологии, биохимии, иммунологии, аналитической химии, синтетической органической химии и медицинской и фармацевтической химии, описанные в данном документе, общеизвестны и широко используются в данной области. Следует иметь в виду, что везде в данном описании изобретения и в формуле изобретения слово "содержать" или его варианты, такие как "содержит" или "содержащий", подразумевают включение определенного целого числа или группы целых чисел, но не исключение какого-либо другого целого числа или группы целых чисел.
Термины "глутаминосодержащая метка", "глутаминная метка," "Q-содержащий метка", "Q-метка" или "трансглутаминазная метка" в данном описании относятся к полипептиду или белку, содержащему один или более остатков Gln, или могут относиться к единственному глутамину или остатку глутамина.
В данном описании термины "сайт-специфичность" "сайт-специфически конъюгированный" или "сайт-специфически перекрестно связанный" относятся к специфическому конъюгированию или перекрестному связыванию аминосодержащей группировки с антителом по конкретному сайту (например по различным положениям, перечисленным в Таблице 1) через глутаминосодержащую метку, эндогенный глутамин и/или эндогенный глутамин, сделанный реакционно-способным посредством конструирования антитела или сконструированной трансглутаминазы. Сайт-специфичность может быть измерена различными методами, включая, без ограничения, масс-спектрометрию (например, масс-спектрометрию с матрично-активированной лазерной десорбционной ионизацией (МАЛДИ-МС), масс-спектрометрию с распылительной ионизацией (ЭРИ-МС), тандемную масс-спектрометрию (МС-МС) и время-пролетную масс-спектрометрию (ВП-МС)), хроматографию гидрофобного взаимодействия, ионообменную хроматографию, сайт-направленный мутагенез, флуоресцентное мечение, эксклюзионную хроматографию и рентгеновскую кристаллографию.
Термины "нагрузка" или "лекарственная нагрузка" или "полезная нагрузка" означают или относятся к среднему количеству полезных нагрузок ("полезная нагрузка" и "полезные нагрузки" здесь используются взаимозаменяемо с "лекарственным средством" и "лекарственными средствами") на антитело в молекуле ADC. Лекарственная нагрузка может быть в переделах от 1 до 20 лекарственных средств на антитело. Иногда это называется DAR, т.е. соотношение лекарственного средства и антитела. Соотношение лекарственное средство-антитело (DAR) у ADC по настоящему изобретению составляет от примерно 1 до примерно 60. В некоторых воплощениях DAR равно по меньшей мере примерно любому из 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45,46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59 и 60. Среднее количество лекарственных средств на антитело, или значение DAR, может быть охарактеризовано различными стандартными методами, такими как спектроскопия в УФ/видимом диапазоне, масс-спектрометрия, ELISA (твердофазный иммуноферментный анализ) и ВЭЖХ (высокоэффективная жидкостная хроматография). Количественное значение DAR также может быть определено. В некоторых случаях разделение, очистка и определение характеристик гомогенных ADC, имеющих конкретное значение DAR, могут быть осуществлены такими методами, как обращенно-фазная ВЭЖХ или электрофорез. DAR может быть ограничено количеством сайтов присоединения на антителе. Например, если сайтом присоединения является глутамин, как в настоящем изобретении, то антитело может иметь только один или несколько подходящих остатков глутамина, через которые линкерная единица может быть присоединена. Как правило, меньше чем теоретический максимум лекарственных веществ конъюгируются с антителом в ходе реакции конъюгирования.
В данном документе термин "эндогенный глутамин (Q), сделанный реакционноспособным" относится к эндогенному глутамину, который сделан доступным, экспонированным или реакционноспособным с аминосодержащей группировкой в присутствии трансглутаминазы в результате конструирования антитела (например, в результате ферментативного дегликозилирования и/или аминокислотной модификации) или сконструированной трансглутаминазой.
В данном документе термин "биосовместимый полимер" относится к полимеру (например, повторяющимся мономерным или структурным звеньям), который пригоден для терапевтического или медицинского лечения реципиента (например, человека) и не вызывает никаких нежелательных местных или системных эффектов у реципиента. Биосовместимый полимер (синтетический, рекомбинантный или нативный) может представлять собой растворимый в воде полимер или нерастворимый в воде полимер. Биосовместимый полимер может также представлять собой линейный или разветвленный полимер.
В данном документе термин "антитело" представляет собой молекулу иммуноглобулина, способную связываться с конкретной мишенью, такой как углевод, полинуклеотид, липид, полипептид и т.д., через по меньшей мере один сайт распознавания антигена, локализованный в вариабельной области молекулы иммуноглобулина. В данном документе, если из контекста не следует иное, этот термин охватывает не только интактные поликлональные или моноклональные антитела, но и их фрагменты (такие как Fab, Fab', F(ab')2, Fv), одноцепочечые (ScFv) и однодоменные антитела, включая акульи и верблюжьи), и слитые белки, содержащие участок антитела, мультивалентные антитела (например, COVX-BODY™), мультиспецифические антитела (например, биспецифические антитела, если только они проявляют желаемую биологическую активность) и фрагменты антител, как описано в данном документе, и любую другую модифицированную конфигурацию молекулы иммуноглобулина, которая содержит антигенраспознающий сайт. Антитело включает антитело любого класса, такое как IgG, IgA или IgM (или их подклассы), и антитело какого-либо конкретного класса не требуется. В зависимости от аминокислотной последовательности константного домена тяжелых цепей антитела иммуноглобулины могут быть отнесены к разным классам. Существует пять основных классов иммуноглобулинов: IgA, IgD, IgE, IgG и IgM, и некоторые из них дополнительно могут быть разделены на подклассы (изотипы), например IgG1, IgG2, IgG3, IgG4, IgA1 и IgA2. Константные домены тяжелых цепей, которые соответствуют разным классам иммуноглобулинов, называются альфа, дельта, эпсилон, гамма и мю соответственно. Субъединичные структуры и трехмерные конфигурации разных классов иммуноглобулинов общеизвестны. В одном аспекте иммуноглобулин представляет собой человеческий, мышиный, обезьяний или кроличий иммуноглобулин.
Термин "Fab-содержащий полипептид" в данном документе относится к полипептиду, содержащему Fab фрагмент, Fab' фрагмент или "(Fab')2 фрагмент". Fab-содержащий полипептид может содержать часть или всю шарнирную последовательность дикого типа (обычно на карбоксильном конце Fab участка полипептида). Fab-содержащий полипептид может быть получен или выделен из любого подходящего иммуноглобулина, например из по меньшей мере одного из различных подтипов IgG1, IgG2, IgG3 или IgG4, или из IgA, IgE, IgD или IgM. Fab-содержащий полипептид может представлять собой Fab-содержащий слитый полипептид, где один или более полипептидов связаны с Fab-содержащим полипептидом. Fab слияние объединяет Fab полипептид иммуноглобулина с партнером слияния, который, как правило, может представлять собой любой белок, полипептид или небольшую молекулу. Фактически, любой белок или любая небольшая молекула может быть связан(а) с Fab полипептидом с образованием Fab-содержащего слитого полипептида. Fab-содержащие партнеры слияния могут включать, без ограничения, область рецептора, связывающуюся с мишенью, молекулу адгезии, лиганд, фермент, цитокин, хемокин или какой-либо другой белок или домен белка.
"Fab фрагмент" состоит из одной легкой цепи и СН1 и вариабельных областей одной тяжелой цепи. Тяжелая цепь Fab молекулы не может образовывать дисульфидную связь с другой тяжелой цепью молекулы.
"Fab' фрагмент" содержит одну легкую цепь и участок одной тяжелой цепи, который содержит VH домен и СН1 домен, а также область между СН1 и СН2 доменами, так что межцепочечная дисульфидная связь может быть образована между двумя тяжелыми цепями двух Fab' фрагментов с образованием F(ab')2 молекулы.
"F(ab')2 фрагмент" содержит две легких цепи и две тяжелых цепи, содержащие участок константной области между СН1 и СН2 доменами, так что межцепочечная дисульфидная связь может быть образована между двумя тяжелыми цепями. Таким образом, F(ab')2 фрагмент состоит из двух Fab' фрагментов, которые удерживаются вместе дисульфидной связью между двумя тяжелыми цепями.
"Фрагменты антитела" в данном документе содержат только участок интактного антитела, где этот участок предпочтительно сохраняет по меньшей мере одну, предпочтительно больше или все функции, в норме ассоциированные с этим участком, когда присутствует в интактном антителе.
"Мультиспецифическое антитело" представляет собой антитело, которое направленно воздействует на более чем один антиген или эпитоп. "Биспецифическое", "с двойной специфичностью" или "бифункциональное" антитело представляет собой гибридное антитело, имеющее два сайта связывания разных антигенов. Биспецифические антитела представляют собой разновидность мультиспецифического антитела и могут быть получены различными методами, включающими, без ограничения, слияние гибридом, связывание Fab' фрагментов или мутации на шарнирном и СН3 доменах антитела (смотри, например, Songsivilai & Lachmann, Clin. Exp. Immunol. 79:315-321 (1990); Kostelny et al., J. Immunol. 148:1547-1553 (1992); и Strop et al., J. Mol. Biol. 420(3):204-219 (2012)). Два связывающих сайта биспецифического антитела будут связываться с двумя разными эпитопами, которые могут находиться на одном и том же белке-мишени или на разных белках-мишенях.
Термин "моноклональное антитело" в данном документе относится к антителу, полученному из популяции по существу гомогенных антител, т.е. индивидуальные антитела, составляющие популяцию, идентичны за исключением возможных природных мутаций, которые могут присутствовать в незначительных количествах. Моноклональные антитела являются высокоспецифическими, будучи направленными против единственного антигена. Кроме того, в отличие от препаратов на основе поликлональных антител, которые обычно включают разные антитела, направленные против разных детерминант (эпитопов), каждое моноклональное антитело направлено против единственной детерминанты на антигене.
Моноклональные антитела в некоторых воплощениях конкретно могут включать "химерные" антитела, в которых участок тяжелой и/или легкой цепи идентичен или гомологичен соответствующим последовательностям в антителах, происходящих из конкретного вида или принадлежащих конкретному классу или подклассу антител, а остальная часть цепи(ей) идентична или гомологична соответствующим последовательностям в антителах, происходящих из другого вида или принадлежащих другому классу или подклассу антитела, а также фрагменты таких антител, если только они проявляют желаемую биологическую активность (патент США №4816567; и Morrisoon et al., Proc. Natl. Acad. Sci. USA 81:6851-6855 (1984)).
"Гуманизированные" формы антител, не являющихся человеческими (например, мышиных), представляют собой химерные антитела, которые содержат минимальную последовательность из иммуноглобулина, не являющимся человеческим. В основном, гуманизированные антитела являются человеческими иммуноглобулинами (антитело-реципиент), в которых остатки из гипервариабельной области реципиента заменены остатками из гипревариабельной области вида, не являющегося человеческим (антитело-донор), такого как мышь, крыса, кролик или примат, не являющийся человеком, имеющими желаемые специфичность, аффинность и мощность. В некоторых случаях остатки каркасной области (FR) человеческого иммуноглобулина заменены соответствующим остатками, не являющимися человеческими. Гуманизированные антитела могут также содержать остатки, которые не обнаруживаются в антителе-реципиенте или в антителе-доноре. Такие модификации производят для дополнительного улучшения характеристик антитела. Как правило, гуманизированное антитело будет содержать по существу все из по меньшей мере одного и обычно двух вариабельных доменов, в которых все или по существу все гипервариабельные петли соответствуют гипервариабельным петлям иммуноглобулина, не являющегося человеческим, и все или по существу все FR являются каркасными областями последовательности человеческого иммуноглобулина. Гуманизированное антитело возможно будет также содержать по меньшей мере участок константной области иммуноглобулина (Fc), обычно человеческого иммуноглобулина. Дополнительные подробности смотри в Jones et al., Nature 321:522-525 (1986); Riechmann et al., Nature 332:323-329 (1988); и Presta, Curr. Op. Struct. Biol. 2:593-596 (1992), а также в процитированных в них следующих обзорных статьях и источниках информации: Vaswani and Hamilton, Ann. Allergy, Asthma & Immunol. 1:105-115 (1998); Harris, Biochem. Soc. Transactions 23:1035-1038 (1995); Hurle and Gross, Curr. Op. Biotech. 5:428-433 (1994).
"Человеческое антитело" представляет собой антитело, имеющее аминокислотную последовательность, которая соответствует аминокислотной последовательности антитела, продуцированного человеком и/или созданного с использованием любого метода получения человеческих антител, как описано в данном документе. Это определение человеческого антитела специально исключает гуманизированное антитело, содержащее антиген-связывающие остатки, не являющиеся человеческими.
"Шарнирная область", "шарнирная последовательность" и их варианты в данном документе имеют смысловое значение, известное в данной области, которое иллюстрируется, например, в Janeway et al., ImmunoBiology: the immune system in health and disease, (Elsevier Science Ltd., NY) (4th ed., 1999); Bloom et al., Protein Science (1997), 6:407-415; Humphreys et al., J. Immunol. Methods (1997), 209: 193-202.
Термин "Fc-содержащий полипептид" в данном документе относится к полипептиду (например, антителу или иммуноадгезину), содержащему карбоксильные концевые полипептидные последовательности тяжелой цепи иммуноглобулина. Fc-содержащий полипептид может содержать нативные или вариантные Fc-области (т.е. последовательности). Fc-область иммуноглобулина обычно содержит два константных домена, СН2 домен и СН3 домен, и возможно содержит СН4 домен. Fc-содержащий полипептид может содержать часть или всю шарнирную последовательность дикого типа (обычно на амино-конце Fc-содержащего полипептида). Fc-содержащий полипептид может также представлять собой димер. Fc-содержащий полипептид может быть получен или может происходить из любого подходящего иммуноглобулина, например из по меньшей мере одного из различных подтипов IgG1, IgG2, IgG3 или IgG4, или из IgA, IgE, IgD или IgM. Хотя границы Fc-области тяжелой цепи иммуноглобулина могут варьировать, например Fc-область тяжелой цепи человеческого IgG обычно определяют как участок от аминокислотного остатка в положении Glu216, или от Ala231, до его карбоксильного конца. Нумерация остатков в Fc-области является нумерацией из EU указателя согласно Kabat (Kabat et al., Subsequencies of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md., 1991).
Fc-содержащий полипептид может представлять собой Fc-содержащий слитый полипептид, где один или более полипептидов связаны с Fc-содержащим полипептидом. Fc слияние объединяет Fc полипептид иммуноглобулина с партнером слияния, который, как правило, может представлять собой любой белок, полипептид или небольшую молекулу. Фактически, любой белок или любая небольшая молекула могут быть связаны с Fc-областью с образованием Fc-содержащего слитого полипептида. Fc-содержащие партнеры слияния могут включать, без ограничения, область рецептора, связывающую мишень, молекулу адгезии, лиганд, фермент, цитокин, хемокин или какой-либо другой белок или домен белка.
В данном документе термин "иммуноадгезин" означает антителоподобные или иммуноглобулиноподобные молекулы, которые соединяют "связывающий домен" гетерологичного белка ("адгезина", например рецептора, лиганда или фермента) с эффекторным компонентом константных доменов иммуноглобулина (т.е. Fc доменом). Структурно иммуноадгезины содержат слияние аминокислотной последовательности адгезина с желаемой специфичностью связывания, которое является иным, чем сайт распознавания и связывания антигена (антигенсвязывающий сайт) антитела (т.е. является "гетерологичным"), и последовательности константного домена иммуноглобулина. Последовательность константного домена иммуноглобулина в иммуноадгезине может быть получена из любого иммуноглобулина, например подтипов IgG1, IgG2, IgG3 или IgG4, IgA, IgE, IgD или IgM.
Термины "полипептид", "олигопептид", "пептид" и "белок" в данном документе использованы взаимозаменяемо и относятся к цепям из аминокислот любой длины, предпочтительно относительно коротким (например, 10-100 аминокислот). Цепь может быть линейной или разветвленной, она может содержать модифицированные аминокислоты и/или может прерываться молекулами, не являющимися аминокислотами. Эти термины также охватывают аминокислотную цепь, которая была модифицирована естественно или в результате вмешательства, например образования дисульфидных связей, гликозилирования, липидирования, ацетилирования, фосфорилирования или любой другой манипуляции или модификации, такой как конъюгирование с компонентом-меткой. Это определение также охватывает, например, полипептиды, содержащие один или более аналогов аминокислот (включая, например, неприродные аминокислоты и т.д.), а также другие модификации, известные в данной области. Понятно, что полипептиды могут существовать как одноцепочечные или как ассоциированные цепи.
В данном документе термины "аминокислота дикого типа", "IgG дикого типа" или "mAb дикого типа" относятся к последовательности аминокислот или нуклеиновых кислот, которая существует в природе в определенной популяции (например, человека, мышей, крыс, клеток и т.д.).
В данном документе термин "эффективность конъюгирования" или "эффективность перекрестного связывания" представляет собой соотношение между экспериментально измеренными количествами ADC, описанным в данном документе, и максимально ожидаемым количеством ADC. Эффективность конъюгирования или эффективность перекрестного связывания может быть измерена различными методами, известными специалистам в данной области, такими как хроматография гидрофобного взаимодействия. Эффективность конъюгирования может быть измерена также при разной температуре, например при комнатной температуре или при 37°С.
Термин "эффекторная функция" относится к биологическим активностям, присущим Fc-области антитела. Примеры эффекторных функций антител включают, без ограничения, антитело-зависимую клеточно-опосредованную цитотоксичность (ADCC), связывание Fc рецептора, комплемент-зависимую цитотоксичность (CDC), фагоцитоз, связывание C1q и понижающую регуляцию клеточных поверхностных рецепторов (например, В-клеточного рецептора; BCR) (смотри, например, патент США №6737056). Такие эффекторные функции обычно требуют, чтобы Fc-область была соединена со связывающим доменом (например, вариабельным доменом антитела), и могут быть оценены с использованием различных анализов, известных в данной области для оценки таких эффекторных функций антител. Примером измерения эффекторной функции является измерение посредством связывания Fcγ3 и/или C1q.
В данном документе "антитело-зависимая клеточно-опосредованная цитотоксичность", или "ADCC", относится к клеточно-опосредованной реакции, при которой неспецифические цитотоксические клетки, которые экспрессируют Fc рецепторы (FcR) (например, естественные клетки-киллеры (NK), нейтрофилы и макрофаги), распознают связанное антитело на клетке-мишени и после этого вызывают лизис клетки-мишени. ADCC активность молекулы, представляющей интерес, может быть оценена с использованием анализа ADCC in vitro, такого как анализ, описанный в патенте США №5500362 или в патенте США №5821337. Полезные эффекторные клетки для таких анализов включают мононуклеарные клетки периферической крови (РВМС) и NK клетки. Альтернативно, или дополнительно, ADCC активность молекулы, представляющей интерес, может быть оценена in vivo, например на животной модели, такой как модель, описанная в Clynes et al., 1998, PNAS (USA), 95:652-656.
"Комплемент-зависимая цитотоксичность", или "CDC", относится к лизированию мишени в присутствии комплемента. Путь активации комплемента инициируется посредством связывания первого компонента системы комплемента (C1q) с молекулой (например, антителом) в комплексе с распознанным антигеном. Оценка активации комплемента, например CDC анализ, описана в Gazzano-Santoro et al., J. Immunol. Methods, 202: 163 (1996), может быть осуществлена.
В данном документе "Fc рецептор" и "FcR" означает рецептор, который связывается с Fc-областью антитела. Предпочтительный FcR имеет нативную последовательность человеческого FcR. Более того, предпочтительным FcR является FcR, который связывает IgG антитело (гамма-рецептор) и включает рецепторы подклассов FcγRI, FcγRII, FcγRIII и FcγRIV, в том числе аллельные варианты и альтернативно сплайсированные формы этих рецепторов. FcyRII рецепторы включают FcyRIIA ("активирующий рецептор") и FcγRIIB ("ингибирующий рецептор"), имеющие сходные аминокислотные последовательности, которые различаются, главным образом, их цитоплазматическими доменами. FcR рассмотрены в Ravetch and Kinet, 1991, Ann. Rev. Immunol., 9:457-92; Capel et al., 1994, Immunomethods, 4:25-34; de Haas et al., 1995, J. Lab. Clin. Med., 126:330-41; Nimmerjahn et al., 2005, Immunity 23:2-4. "FcR" также включает неонатальный рецептор, FcRn, который ответственней за перенос материнских IgG в плод (Guyer et al., 1976, J. Immunol., 117:587; и Kim et al., 1994, J. Immunol., 24:249).
Термин "очищать" и его грамматические варианты означает удаление, полностью или частично, по меньшей мере одной примеси из смеси, содержащей ADC и одну или более примесей, которое в результате повышает уровень чистоты ADC в композиции (т.е. снижает количество (м.д. (миллионные доли)) примеси(ей) в композиции).
Упоминание "примерно" перед значением или параметром включает (и описывает) воплощения, которые относятся к этому значению или параметру как таковому. Например, описание с указанием "примерно X" включает описание "X." Числовые диапазоны включают в себя числа, определяющие диапазон.
"Индивидуум" или "субъект" представляет собой млекопитающее, более предпочтительно человека. Млекопитающие также включают, без ограничения, сельскохозяйственных животных, спортивных животных, домашних питомцев, приматов, лошадей, собак, кошек, мышей и крыс.
Понятно, что везде, где воплощения описаны языком "содержащий", иные аналогичные воплощения, описанные терминами "состоящий из" и/или "по существу состоящий из" также предусмотрены.
Там где аспекты или воплощения изобретения описаны языком группы Маркуша или иной группировкой альтернатив, настоящее изобретение охватывает не только всю группу, перечисленную в целом, но и каждый член группы индивидуально и все возможные подгруппы основной группы, а также основную группу, в которой отсутствуют один или более членов группы. Настоящее изобретение также предусматривает явное исключение одного или более любых членов группы в заявленном изобретении.
Обозначения остатков в этой заявке основаны на схеме нумерации EU константного домена (Edelman et al., Proc. Natl. Acad. Sci. USA, 63(1):78-85 (1969)).
Если не дано иного определения, все технические и научные термины, использованные в данном документе, имеют значения, обычно понятные специалисту в области, к которой данное изобретение относится. Иллюстративные способы и вещества описаны в данном документе, хотя способы и вещества, подобные или эквивалентные описанным, также могут быть использованы на практике или при тестировании настоящего изобретения. Все публикации и другие источники информации, упомянутые в данном описании, включены в него во всей их полноте посредством ссылки. В случае конфликта, текст настоящего описание изобретения, включая определения, будет иметь преимущество. Хотя в данном описании процитирован целый ряд документов, это цитирование не является признанием того, что любой из этих документов является частью обычных общих знаний в данной области. Следует иметь в виду, что использованное в описании изобретения и формуле изобретения слово "содержать" или его варианты, такие как "содержит" или "содержащий", подразумевает включение указанного целого числа или группы целых чисел, но не исключение какого-либо другого целого числа или группы целых чисел. Если контекст не требует иного, термины в единственном числе будут охватывать формы множественного числа, а термины во множественном числе будут охватывать форму единственного числа. Вещества, способы и примеры являются только иллюстративными и не должны рассматриваться как ограничивающие.
Трансглутаминазы являются белок-глутамин гамма-глутамилтрансферазами, которые обычно катализируют рН-зависимое трансамидирование остатков глутамина с остатками лизина. Трансглутаминаза, использованная в изобретении, описанном в данном документе, может быть получена или изготовлена из множества различных источников, или может быть сконструирована для катализирования трансамидирования одного или более остатков эндогенного глутамина с одним или более остатками лизина или другой аминосодержащей группировкой. В некоторых воплощениях трансглутаминаза представляет собой кальций-зависимую трансглутаминазу, которой необходим кальций, чтобы индуцировать конформационные изменения фермента и обеспечивать активность фермента. Например, трансглутаминаза может быть выделена из печени морской свинки и получена через коммерческие источники (например, Sigma-Aldrich (St Louis, МО) и MP Biomedicals (Irvine, CA)). В некоторых воплощениях трансглутаминаза представляет собой кальций-независимую трансглутаминазу, которой не нужен кальций, чтобы индуцировать конформационные изменения фермента и обеспечивать активность фермента. В некоторых воплощениях трансглутаминаза представляет собой микробную трансглутаминазу, получаемую из микробного генома, такую как трансглутаминаза из Streptoverticillium или Streptomices (например, Streptomyces mobarensis или Streptoverticillium mobarensis). Коммерчески доступная кальций-независимая трансглутаминаза, такая как ACTIVA™ (Ajinomoto, Japan), является подходящей для настоящего изобретения. В некоторых воплощениях трансглутаминаза представляет собой белок млекопитающего (например, трансглутаминазу человека), бактериальный белок, растительный белок, белок грибов (например, трансглутаминазы из Oomycetes и Actinomicetes) или прокариотический белок. В некоторых воплощениях трансглутаминаза представляет собой трансглутаминазу из Micrococcus, Clostridium, Turolpsis, Rhizopus, Monascus или Bacillus.
В некоторых воплощениях трансглутаминаза, используемая в изобретении, описанном в данном документе, представляет собой сконструированную трансглутаминазу, которая катализирует трансамидирование одного или более остатков эндогенного глутамина в антителе с одним или более остатками лизина или другими аминосодержащими группировками. Например, один или более аминокислотных остатков дикого типа в природной трансглутаминазе могут быть исключены или заменены или замещены другим(и) аминокислотным(ыми) остатком(ами) для получения сконструированной трансглутаминазы.
В некоторых воплощениях трансглутаминаза, используемая в изобретении, описанном в данном документе, может также представлять собой рекомбинантный белок, полученный с использованием рекомбинантных методов, известных специалистам в данной области. В некоторых воплощениях трансглутаминаза, используемая в изобретении, описанном в данном документе, может представлять собой очищенный белок. Например, очищенная трансглутаминаза имеет чистоту по меньшей мере примерно 50%. В данном документе "чистый" или "очищенный" белок относится к белку (например, трансглутаминазе), не содержащему других белков-примесей. В некоторых воплощениях очищенная трансглутаминаза имеет чистоту по меньшей мере примерно 55%-60%, 60%-65%, 65%-70%, 70%-75%, 75%-80%, 80%-85%, 85%-90%, 90%-95%, 95%-98% или 99%. В некоторых воплощениях очищенная трансглутаминаза имеет чистоту примерно 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99% или 100%.
В некоторых воплощениях ADC по настоящему изобретению содержит по меньшей мере 1 эндогенный глутамин, сделанный реакционно-сопособным в реакции трансамидирования посредством конструирования антитела или сконструированной трансглутаминазой. В некоторых воплощениях конструирование антитела представляет собой дегликозилирование антитела (например, ферментативное дегликозилирование) или аминокислотную модификацию, включающую делецию, вставку, замену, мутацию аминокислот или любую их комбинацию на антителе. Например, аминокислоту дикого типа Asn (N) в положении 297 в антителе замещают или заменяют аминокислотой Ala (А), что приводит к агликозилированию в положении 297 и реакционно-способному эндогенному глутамину (Q) в положении 295. В качестве другого примера, аминокислотная модификация в антителе представляет собой замену аминокислоты с N на Q в положении 297, приводящую к агликозилированию в положении 297, реакционно-способному эндогенному Q в положении 295 и сайт-специфическому конъюгированию между N297Q и Q295 и одной или более аминосодержащими группировками в этих двух сайтах в присутствии трансглутаминазы.
В некоторых воплощениях ADC по настоящему изобретению содержит глутаминосодержащую метку, сконструированную по меньшей мере по одному или более чем одному положениям, включающим, без ограничения, 1) карбоксильный конец легкой цепи, тяжелой цепи или как легкой цепи, так и тяжелой цепи; 2) аминоконец легкой цепи, тяжелой цепи или как легкой цепи, так и тяжелой цепи; и 3) S60-R61, R108, Т135, S160, S168, S190-S192, Р189-S192, G200-S202, K222-Т225, K222-Т223, Т223, L251-S254, M252-I253, Е294-N297, E293-N297, N297 и/или G385, где глутаминосодержащая метка вставлена в антитело или заменяет одну или более эндогенных аминокислот в антителе.
Примеры конкретной глутаминосодержащей метки и ее соответствующего сконструированного положения приведены в Таблице 1 и описаны в WO 2012/059882 и WO 2015/015448, которые включены в данное описание посредством ссылки.
В некоторых воплощениях антитело в ADC содержит аминокислотную модификацию в положении 222, 340 или 370 (нумерация EU) по сравнению с антителом дикого типа в том же положении. В некоторых воплощениях модификация представляет собой делецию, вставку, замещение, мутацию аминокислот или любую их комбинацию. В некоторых воплощениях замещение представляет собой замену аминокислоты дикого типа другой аминокислотой (например, аминокислотой не дикого типа). В некоторых воплощениях другая аминокислота (например, не дикого типа) или вставленная аминокислота представляет собой Arg (например, K222R, K340R или K370R). В некоторых воплощениях вставка представляет собой вставку одной или более аминокислот (например, вставку одной, двух, трех или более аминокислот). В некоторых воплощениях другая (например, не дикого типа) аминокислота представляет собой Ala, Asn, Asp, Cys, Glu, Gln, Gly, His, Ile, Leu, Met, Phe, Pro, Ser, Thr, Trp, Tyr или Val.
Соответственно, в некоторых воплощениях ADC по настоящему изобретению содержит а) замены аминокислот в положениях N297Q и K222R, где аминосодержащая группировка сайт-специфически конъюгирована с эндогенным глутамином в положении 295 и замещенным глутамином в положении 297; и/или б) одну или более глутаминосодержащих меток, где аминосодержащая группировка сайт-специфически конъюгирована с глутаминосодержащей(ими) меткой(ами) по карбоксильному концу легкой цепи антитела. В некоторых воплощениях соотношение лекарственное средство-антитело (DAR) равно примерно 3-9. В некоторых воплощениях глутаминосодержащая метка на карбоксильном конце легкой цепи антитела представляет собой GGLLQGA (SEQ ID NO: 23).
В некоторых воплощениях ADC по настоящему изобретению содержит а) замены аминокислот в положениях N297Q и K222R, где аминосодержащая группировка сайт-специфически конъюгирована с эндогенным глутамином в положении 295 и замещенным глутамином в положении 297; б) одну или более глутаминосодержащих меток, где аминосодержащая группировка сайт-специфически конъюгирована с глутаминосодержащей(ими) меткой(ами) по карбоксильному концу легкой цепи антитела; и/или в) одну или более глутаминосодержащих меток, где аминосодержащая группировка дополнительно сайт-специфически конъюгирована с глутаминосодержащей меткой по одному или более положениям, выбранным из группы, состоящей из S60-R61, R108, Т135, S160, S168, S190-S192, P189-S192, G200-S202, K222-Т225, K222-Т223, Т223, L251-S254, M252-I253, E294-N297, E293-N297, N297 и G385, в антителе, где глутаминосодержащая метка вставлена в антитело или заменяет одну или более эндогенных аминокислот в антителе. В некоторых воплощениях соотношение лекарственное средство-антитело равно по меньшей мере примерно 6. Например, в некоторых воплощениях ADC по настоящему изобретению также содержит а) замены аминокислот в положениях N297Q и K222R, где аминосодержащая группировка сайт-специфически конъюгирована с эндогенным глутамином в положении 295 и замещенным глутамином в положении 297; б) одну или более глутаминосодержащих меток, где аминосодержащая группировка сайт-специфически конъюгирована с глутаминосодержащей(ими) меткой(ами) по карбоксильному концу легкой цепи антитела; в) одну или более глутаминных меток, где аминосодержащая группировка дополнительно сайт-специфически конъюгирована с глутаминосодержащей меткой по карбоксильному концу тяжелой цепи антитела; и где соотношение лекарственное средство-антитело (DAR) равно примерно 6-9. В некоторых воплощениях глутаминосодержащая метка на карбоксильном конце легкой цепи антитела представляет собой GGLLQGA (SEQ ID NO: 23), и глутаминосодержащая метка на карбоксильном конце тяжелой цепи антитела представляет собой LLQGA (SEQ ID NO: 11).
В одном варианте ADC по настоящему изобретению содержит а) замены аминокислот в положениях N297Q и K222R, где аминосодержащая группировка сайт-специфически конъюгирована с эндогенным глутамином по положению 295 и замещенным глутамином по положению 297; б) одну или более глутаминосодержащих меток, где аминосодержащая группировка сайт-специфически конъюгирована с глутаминосодержащей(ими) меткой(ами) по карбоксильному концу легкой цепи антитела; с) одну или более глутаминосодержащих меток, где аминосодержащая группировка дополнительно сайт-специфически конъюгирована с глутаминосодержащей меткой, вставленной после положения аминокислоты Т135 в тяжелой цепи антитела; и где соотношение лекарственное средство-антитело (DAR) равно примерно 6-9. В некоторых воплощениях глутаминосодержащая метка на карбоксильном конце легкой цепи антитела представляет собой GGLLQGA (SEQ ID NO: 23), и глутаминосодержащая метка, вставленная после Т135 в тяжелой цепи антитела, представляет собой LLQG (SEQ ID NO: 2).
В другом варианте ADC по настоящему изобретению содержит а) замены аминокислот в положениях N297Q и K222R, где аминосодержащая группировка сайт-специфически конъюгирована с эндогенным глутамином по положению 295 и замещенным глутамином по положению 297; б) одну или более глутаминосодержащих меток, где аминосодержащая группировка сайт-специфически конъюгирована с глутаминосодержащей меткой(ами) по карбоксильному концу легкой цепи антитела; с) одну или более глутаминосодержащих меток, где аминосодержащая группировка дополнительно сайт-специфически конъюгирована с глутаминосодержащей меткой по положениям аминокислот G200-S202 в тяжелой цепи антитела; и где соотношение лекарственное средство-антитело (DAR) равно примерно 6-9. В некоторых воплощениях глутаминосодержащая метка на карбоксильном конце легкой цепи антитела представляет собой GGLLQGA (SEQ ID NO: 23); и глутаминосодержащая метка, вставленная после Т135 в тяжелой цепи антитела, представляет собой LLQG (SEQ ID NO: 2).
В некоторых воплощениях глутаминосодержащая метка для ADC, описанного в данном документе, содержит аминокислотную последовательность XXQX (SEQ ID NO: 37), где X может представлять собой стандартную или нестандартную аминокислоту, как описано в данном документе. Например, в некоторых воплощениях X представляет собой L (Leu), A (Ala), G (Gly), S (Ser), V (Val), F (Phe), Y (Tyr), H (His), R (Arg), N (Asn), E (Glu), D (Asp), С (Cys), Q (Gln), I (Ile), M (Met), P (Pro), T (Thr), K (Lys) или W (Trp).
В некоторых воплощениях глутаминосодержащая метка содержит аминокислотную последовательность, выбранную из группы, состоящей из Q, LQG, LLQGG (SEQ ID NO: 1), LLQG (SEQ ID NO: 2), LSLSQG (SEQ ID NO: 3), GGGLLQGG (SEQ ID NO: 4), GLLQG (SEQ ID NO: 5), LLQ, GSPLAQSHGG (SEQ ID NO: 6), GLLQGGG (SEQ ID NO: 7), GLLQGG (SEQ ID NO: 8), GLLQ (SEQ ID NO: 9), LLQLLQGA (SEQ ID NO: 10), LLQGA (SEQ ID NO: 11), LLQYQGA (SEQ ID NO: 12), LLQGSG (SEQ ID NO: 13), LLQYQG (SEQ ID NO: 14), LLQLLQG (SEQ ID NO: 15), SLLQG (SEQ ID NO: 16), LLQLQ (SEQ ID NO: 17), LLQLLQ (SEQ ID NO: 18), LLQGR (SEQ ID NO: 19), LLQGPP (SEQ ID NO: 20), LLQGPA (SEQ ID NO: 21), GGLLQGPP (SEQ ID NO: 22), GGLLQGA (SEQ ID NO: 23), LLQGA (SEQ ID NO: 24), LLQGPGK (SEQ ID NO: 25), LLQGPG (SEQ ID NO: 26), LLQGP (SEQ ID NO: 27), LLQP (SEQ ID NO: 28), LLQPGK (SEQ ID NO: 29), LLQAPGK (SEQ ID NO: 30), LLQGAPG (SEQ ID NO: 31), LLQGAP (SEQ ID NO: 32), LLQGPA (SEQ ID NO: 33), LLQGPP (SEQ ID NO: 34), GGLLQGPP (SEQ ID NO: 35) и LLQLQG (SEQ ID NO: 36).
В некоторых воплощениях глутаминосодержащая метка содержит аминокислотную последовательность LLQGA (SEQ ID NO: 24), LQG, GGLLQGA (SEQ ID NO: 23), LLQGPA (SEQ ID NO: 33), LLQGPP (SEQ ID NO: 34), GGLLQGPP (SEQ ID NO: 35), LLQGSG (SEQ ID NO: 13), LLQG (SEQ ID NO: 2), LLQYQG (SEQ ID NO: 14), LLQLLQG (SEQ ID NO: 15), LLQLQG (SEQ ID NO: 36), LLQLLQ (SEQ ID NO: 18), LLQLQ (SEQ ID NO: 17), LLQGR (SEQ ID NO: 19), LLQYQGA (SEQ ID NO: 12), SLLQG (SEQ ID NO: 16) или LLQLLQGA (SEQ ID NO: 10).
В некоторых воплощениях глутаминосодержащая метка не содержит аминокислотную последовательность, выбранную из группы, состоящей из LGGQGGG (SEQ ID NO: 41), GGGQGGL (SEQ ID NO: 42), GXGQGGG (SEQ ID NO: 43),GGXQGGG (SEQ ID NO: 44), GGGQXGG (SEQ ID NO: 45) и GGGQGXG (SEQ ID NO: 46), где X представляет собой G, A, S, L, V, F, Y, R, N или Е). Другие типичные метки также описаны, например, в US 2013/0230543 и US 2013/0122020.
В некоторых воплощениях антитело в ADC, описанных в данном документе, содержит аминокислотную модификацию по последнему положению аминокислот на карбоксильном конце по сравнению с антителом дикого типа в том же положении. В некоторых воплощениях модификация представляет собой делецию, вставку, замещение, мутацию аминокислот или любую их комбинацию. В некоторых воплощениях замещение представляет собой замену аминокислоты дикого типа другой аминокислотой (например, аминокислотой не дикого типа). В некоторых воплощениях вставка представляет собой вставку одной или более аминокислот (например, вставку одной, двух, трех или более аминокислот). В некоторых воплощениях другая аминокислота (например, не дикого типа) или вставленная аминокислота представляет собой Arg. В некоторых воплощениях другая (например, не дикого типа) аминокислота представляет собой Ala, Asn, Asp, Cys, Glu, Gln, Gly, His, Ile, Leu, Met, Phe, Pro, Ser, Thr, Trp, Tyr или Val. Например, в некоторых воплощениях последняя аминокислота на карбоксильном конце антитела (например, тяжелой цепи антитела) может быть удалена, и глутаминосодержащая метка, сконструированная на С-конце полипептида, содержит аминокислотную последовательность LLQGA (SEQ ID NO: 11) или GGLLQGA (SEQ ID NO: 23).
В некоторых воплощениях антитело содержит аминокислотную модификацию по положению первой аминокислоты на амино конце по сравнению с антителом дикого типа в том же положении. В некоторых воплощениях модификация представляет собой делецию, вставку, замещение, мутацию аминокислот или любую их комбинацию. В некоторых воплощениях замещение представляет собой замену аминокислоты дикого типа другой (например, аминокислотой не дикого типа). В некоторых воплощениях вставка представляет собой вставку аминокислоты. В некоторых воплощениях аминокислота не дикого типа или вставленная аминокислота представляет собой Arg. В некоторых воплощениях другая (не дикого типа или вставленная) аминокислота представляет собой Ala, Asn, Asp, Cys, Glu, Gln, Gly, His, Ile, Leu, Met, Phe, Pro, Ser, Thr, Trp, Tyr или Val.
В некоторых воплощениях ADC, описанный в данном документе, содержит полномерную тяжелую цепь антитела и легкую цепь антитела. В некоторых воплощениях антитело, описанное в данном документе, представляет собой моноклональное антитело, поликлональное антитело, человеческое антитело, гуманизированное антитело, химерное антитело, биспецифическое антитело, минитело, диатело или фрагмент антитела. В некоторых воплощениях антитело представляет собой IgG. В некоторых воплощениях IgG выбран из группы, состоящей из IgG1, IgG2, IgG3 и IgG4. В некоторых воплощениях антитело представляет собой IgA, IgE, IgD или IgM. В некоторых воплощениях эффекторная функция (например, измеренная посредством связывания Fcγ3 и/или C1q) ADC, описанных в данном документе, снижена не более чем примерно 1-кратно, 2-кратно, 3-кратно, 4-кратно или 5-кратно по сравнению с антителом дикого типа. В некоторых воплощениях антитело ADC представляет собой IgG, где эффекторная функция IgG снижена не более чем примерно 2-кратно по сравнению с IgG дикого типа. В других воплощениях эффекторная функция IgG снижена примерно 2-кратно по сравнению с IgG дикого типа. В других воплощениях эффекторная функция IgG снижена более чем примерно 2-кратно по сравнению с IgG дикого типа. В некоторых воплощениях антитело ADC представляет собой IgG, где эффекторная функция IgG снижена не более чем примерно 1-кратно по сравнению с IgG дикого типа. В других воплощениях эффекторная функция IgG снижена примерно 1-кратно по сравнению с IgG дикого типа. В некоторых воплощениях эффекторная функция IgG снижена более чем примерно 1-кратно, 3-кратно, 4-кратно или 5-кратно по сравнению с IgG дикого типа.
Количество агентов-аминосодержащих группировок, которое может быть конъюгировано с антителом, зависит от 1) количества глутаминосодержащих меток, которые связаны с антителом/вставлены в антитело, а также количества глутаминов на глутаминосодержащей метке; и/или 2) количества эндогенных глутаминов на антителе (т.е. нативных глутаминов без конструирования, таких как глутамины в вариабельных доменах, CDR (область, определяющая комплементарность) и т.д.); и/или 3) количества эндогенных глутаминов, сделанных реакционно-способными посредством конструирования антитела, как описано в данном докумнете, или сконструированной трансглутаминазой. Например, две аминосодержащие группировки могут быть сайт-специфически конъюгированы с антителом по карбоксильным концам двух легких цепей, и четыре аминосодержащие группировки могут быть сайт-специфически конъюгированы с антителом по положениям Q295 и N297Q. В некоторых воплощениях аминосодержащая группировка может быть одной и той же или другой в каждом положении конъюгирования.
Примеры цитотоксического агента включают, без ограничения, антрациклин, ауристатин (например, долестатин, MMAD, ММАЕ, MMAF, PF-06380101, PF-06463377 и PF-06456780), сплицеостатин, димер CBI/CPI (включая "смешанные" димеры, содержащие как CBI, так и CPI компоненты, как описано в предварительной патентной заявке США 61/932,118), калихеамицин, дуокармицин, энедиин, гелданамицин, майтанзин, пуромицин, таксан, алкалоид барвинка, SN-38, тубулизин, гемиастерлин, камптотецин, комбретастатин, доластатин, димер индолинобензодиазепина, димер пирролобензодиазепина и пладиенолид и их стереоизомеры, изостеры, аналоги или производные.
Антрациклины получают из бактерий Strepomyces и используют для лечения широкого спектра видов рака, таких как лейкозы, лимфомы, рак молочной железы, рак матки, рак яичника и рак легкого. Примеры антрациклинов включают, без ограничения, даунорубицин, доксорубицин (т.е. адриамицин), эпирубицин, идарубицин, валрубицин и митоксантрон.
Доластатины и их пептидные аналоги и производные, ауристатины, являются высокоактивными антимитотическими агентами, которые, как было показано, обладают противораковой и противогрибковой активностью (смотри, например, патент США №5663149 и Pettit et al., Antimicrob. Agents Chemother. 42:2961-2965, 1998). Примеры доластатинов и ауристатинов включают, без ограничения, доластатин 10, ауристатин Е, ауристатин ЕВ (АЕВ), ауристатин EFP (AEFP), MMAD (монометил-ауристатин D или монометил-доластатин 10), MMAF (монометил-ауристатин F или N-метилвалин-валин-долаизолейин-долапроин-фенилаланин), ММАЕ (монометил-ауристатин Е или N-метилвалин-валин-долаизолейин-долапроин-норэфедрин), 5-бензоилвалериановая кислота-АЕ эфир (AEVB) и другие новые ауристатины (такие как ауристатины, описанные в публикации заявки США №2013/0129753). В некоторых воплощениях ауристатин представляет собой 0101 (2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид), имеющий следующую структуру:
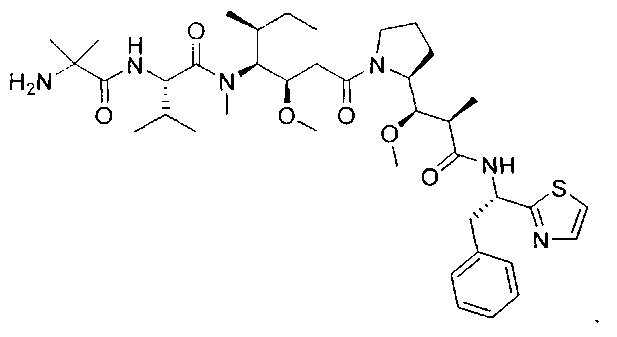
В некоторых воплощениях ауристатин представляет собой 3377 (N,2-диметилаланил-N-{(1S,2R)-4-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбоксил-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирродидин-1-ил}-2-метокси-1-[(1S)-1-метилпропил]-4-оксобутил}-N-метил-L-валинамид), имеющий следующую структуру:
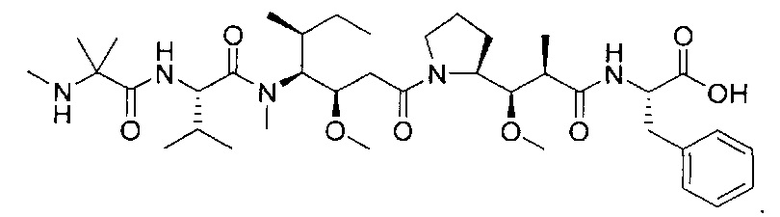
В других воплощениях ауристатин представляет собой 0131 (2-метил-L-пролил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирродидин-1-ил}-3-метокси-5-метил-1-оксогенптан-4-ил]-N-метил-L-валинамид), имеющий следующую структуру:
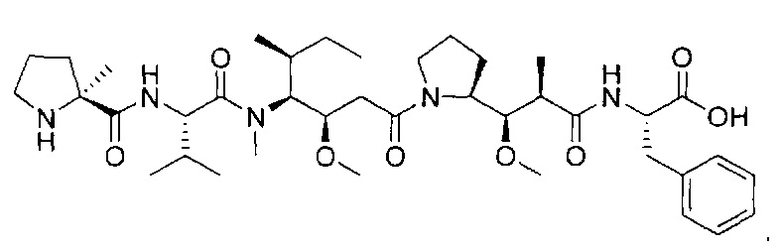
В других воплощениях ауристатин представляет собой 0121(2-метил-L-пропил-N-[(3R,4S,5S)-4(2S)-2-[(1R,2R)-3-{[(2S)-1-метокси-1-оксо-3-сфенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирродидин-1-ил}-3-метокси-5-метил-1-оксогенптан-4-ил]-N-метил-L-валинамид), имеющий следующую структуру:
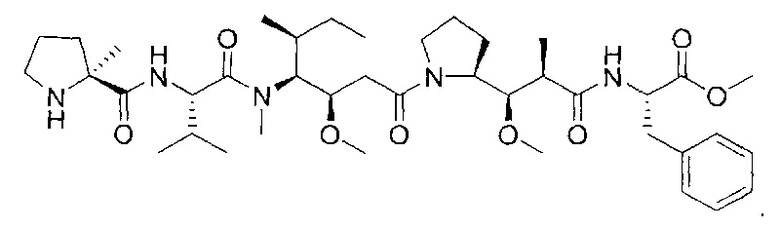
Камптотецин представляет собой цитотоксический хинолиновый алкалоид, который ингибирует фермент топоизомеразу I. Примеры камптотецина и его производных включают, без ограничения, топотекан и иринотекан и их метаболиты, такие как SN-38.
Комбретастатины являются природными фенолами со свойствами разрушения сосудов в опухолях. Иллюстративные комбретастатины и их производные включают, без ограничения, комбретастатин А-4 (СА-4) и омбрабулин.
Дуокармицин и СС-1065 являются ДНК алкилирующими агентами с цитотоксической активностью (смотри Boger and Johnson, PNAS 92:3642-3649 (1995)). Иллюстративные доластатины и ауристатины включают, без ограничения, (+)-докармицин А и (+)-дуокармицин SA, и (+)-СС-1065.
Энедиины представляют собой класс противоопухолевых бактериальных продуктов, характеризующихся либо девяти-, либо десятичленными кольцами или присутствием в циклических системах сопряженных связей типа тройная-двойная-тройная. Иллюстративные энедиины включают, без ограничения, калихеамицин, эсперамицин, унциаламицин, динемицин и их производные.
Гемиастерлин и его аналоги (например, HTI-286) связываются с тубулином, нарушают нормальную динамику микротрубочек и, в стехиометрических количествах, деполимеризуют микротрубочки.
Гелданамицины являются бензохиноновыми ансамициновыми антибиотиками, которые связываются с Hsp90 (белок теплового шока 90), и применяются в качестве противоопухолевых лекарственных средств. Иллюстративные гелданамицины включают, без ограничения, 17-AAG (17-N-аллиламино-17-деметоксигелданамицин) и 17-DMAG (17-диметиламиноэтиламино-17-деметоксигелданамицин).
Майтанзины или их производные майтанзиноиды ингибируют пролиферацию клеток, ингибируя образование микротрубочек во время митоза посредством ингибирования полимеризации тубулина (смотри Remillard et al., Science 189:1002-1005 (1975)). Иллюстративные майтанзины и майтанзиноиды включают, без ограничения, мертанзин (DM1) и его производные, а также ансамитоцин.
Димеры пирролобензодиазепина (PBD) и димеры индолинобензодиазепина (IGN) являются противоопухолевыми агентами, которые содержат одну или более иммунных функциональных групп, или их эквиваленты, которые связываются с дуплексом ДНК. Молекулы PBD и IGN основаны на природном продукте атрамицине и взаимодействуют с ДНК последовательность-селективным образом с предпочтением к пурин-гуанин-пуриновым последовательностям. Иллюстративные PBD и их аналоги включают, без ограничения, SJG-136.
Сплицеостатины и пладиенолиды являются противоопухолевыми соединениями, которые ингибируют сплайсинг и взаимодействуют с сплицеосомой, SF3b. Примеры сплицеостатинов включают, без ограничения, сплицеостатин A, FR901464. Примеры пладиенолидов включают, без ограничения, пладиенолид В, пладиенолид D и Е7107.
Таксаны представляют собой дитерпены, которые действуют в качестве антитубулиновых агентов или митотических ингибиторов. Иллюстративные таксаны включают, без ограничения, паклитаксел (например, TAXOL®) и доцетаксел (TAXOTERE®).
Алкалоиды барвинка также являются антитубулиновыми агентами. Иллюстративные алкалоиды барвинка включают, без ограничения, винкристин, винбластин, виндезин и винорелбин.
В некоторых воплощениях группировка агента представляет собой полипептид токсина (или белок токсина). Примеры полипептида токсина включают, без ограничения, цепь дифтерийного токсина А, несвязывающие активные фрагменты дифтерийного токсина, цепь экзотоксина А, цепь рицина А, цепь абрина А, цепь модеццина А, альфа-сарцин, белки Aleurites fordii, белки диантина, белки Phytolaca americana (PAPI, PAPII и PAP-S), ингибитор Momordica charantia, курцин, кротин, ингибитор Sapaonaria officinalis, гелонин, митогеллин, рестриктоцин, феномицин, эномицин, трикотецены, пептиды ингибиторного цистинового узла (ICK) (например, кератотоксины) и конотоксин (например, KIIIA или SmIIIa).
Способы конъюгирования ADC
Конъюгирование анти-Trop2 антител с конструкциями линкер-полезная нагрузка по настоящему изобретению осуществляют способами, при которых количество антитела доводят до 5 мг/мл в буфере, содержащем 25 мМ Tris-HCl, рН 8,0-8,5. Количество конструкции линкер-полезная нагрузка добавляют в 5-50-кратном или 5-500-кратном молярном избытке относительно антитела, и ферментативную реакцию инициируют добавлением 2% (масс./об.) бактериальной трансглутаминазы (Ajinomoto Active TI, Japan) и инкубируют при слабом встряхивании при 37°С в течение 16-24 часов. ADC очищают с использованием Butyl Sepharose™ High Performance (Butyl HP, GE Healthcare Biosciences), регулируя реакционную смесь для получения буферной композиции 0,75 М сульфата аммония, 25 мМ фосфата калия, рН 7 (Буфер А). Вещество наносят на Butyl HP, промывают 5 CV (колоночный объем) Буфера А и элюируют 20 CV с линейным градиентом в 25 мМ фосфат калия, рН 7. Фракции, содержащие ADC, собирают, подвергают диализу против PBS (забуференный фосфатами физиологический раствор), концентрируют с использованием центрифужной фильтровальной установки 10 kDa Amicon Ultra (Millipore Corporation) и подвергают стерильной фильтрации через фильтр 0,2 мкм. Альтернативно, ADC очищают с использованием смолы MabSelect Protein A (GE Healthcare Biosciences) согласно стандартным протоколам и буферного обмена в PBS. Известны другие способы конъюгирования ADC, в том числе способы, описанные в WO 2012/059882.
Способ использования конъюгатов антитело-лекарственное средство по изобретению
ADC по настоящему изобретению полезны в различных применениях, включающих, без ограничения, способы терапевтического лечения и диагностические способы.
В одном аспекте изобретения предложен способ лечения рака у субъекта. Соответственно, в некоторых воплощениях предложен способ лечения рака у нуждающегося в этом субъекта, включающий введение субъекту эффективного количества композиции (например, фармацевтической композиции), содержащей ADC, описанный в данном документе. В данном документе рак включает, без ограничения, солидный рак (такой как рак мочевого пузыря, молочной железы, шейки матки, хориокарцинома, рак ободочной кишки, пищевода, желудка, глиобластома, рак в области головы и шеи, рак почки, печени, легкого (например, немелкоклеточный рак легкого (NSCLC)), рак ротовой полости, рак яичника, поджелудочной железы, предстательной железы и кожи); и гемобластоз (liquid cancer) (такой как острый лимфобластный лейкоз (ALL), хронический лимфоцитарный лейкоз (CLL), острый миелогенный лейкоз (AML), хронический миелогенный лейкоз (CML), волосковоклеточый лейкоз (HCL), Т-клеточный пролимфоцитарный лейкоз (Т-PLL), лейкоз из больших гранулярных лимфоцитов, Т-клеточный лейкоз взрослых и множественная миелома).
В некоторых воплощениях предложен способ ингибирования роста или прогрессирования опухолей у нуждающегося в этом субъекта, включающий введение субъекту эффективного количества композиции, содержащей ADC, описанные в данном документе. В других воплощениях предложен способ ингибирования метастазирования раковых клеток или опухолей (например, солидных опухолей или гемобластозов) у нуждающегося в этом субъекта, включающий введение субъекту эффективного количества композиции, содержащей ADC, описанные в данном документе. В других воплощениях предложен способ индуцирования регрессии опухоли у нуждающегося в этом субъекта, включающий введение субъекту эффективного количества композиции, содержащей ADC, описанные в данном документе.
Группировка агента в ADC, описанных в данном документе, может представлять собой детектируемую группировку, такую как визуализирующий агент и метка фермент-субстрат. ADC, описанные в данном документе, могут быть также использованы для диагностических анализов in vivo, таких как визуализация in vivo (например, PET (позитронно-эмиссионная томография) или SPECT (однофотонная эмиссионная компьютерная томография)), или окрашивающий реагент.
В некоторых воплощениях способы, описанные в данном документе, дополнительно включают стадию лечения субъекта дополнительной формой терапии. В некоторых воплощениях дополнительная форма терапии представляет собой дополнительную противораковую терапию, включающую, без ограничения, химиотерапию, облучение, хирургическое вмешательство, гормональную терапию и/или дополнительную иммунотерапию.
В некоторых воплощениях дополнительная форма терапии включает введение одного или более терапевтических агентов в дополнение к ADC, описанным в данном документе. Терапевтические агенты включают, без ограничения, второй ADC (например, стандартный ADC, такой как брентуксимаб ведотин (ADCETRIS®) и адо-трастузумаб эмтазин (KADCYLA®)), антитело (например, анти-VEGF антитело, анти-HER2 антитело, анти-CD25 антитело и/или анти-CD20 антитело), ингибитор ангиогенеза, цитотоксический агент (например, доцетаксел, цисплатин, доксорубицин, митомицин, тамоксифен или фторурацил) и противовоспалительный агент (например, преднизон и прогестерон).
Фармацевтические композиции
Согласно настоящему изобретению также предложена фармацевтическая композиция, содержащая ADC, описанные в данном документе, в фармацевтически приемлемом эксципиенте или носителе. ADC можно вводить сами по себе или в комбинации с одним или более другими ADC по изобретению или в комбинации с одним или более другими лекарственными средствами (или в виде любой их комбинации). Например, ADC по настоящему изобретению можно вводить в комбинации со стандартными ADC (т.е. не разработанными со свойствами модулирования стабильности). Таким образом, способы и применения по изобретению также охватывают воплощения комбинаций (совместного введения) с другими активными агентами, как подробно описано ниже.
В данном документе термины "совместное введение", "соместно введенный" или "в комбинации с" означают и относятся к следующему: (1) непрерывное введение комбинации ADC, раскрытого в данном документе, и терапевтического(их) агента(ов) пациенту, нуждающемуся в лечении, когда такие компоненты приготовлены вместе в стандартной лекарственной форме, которая высвобождает указанные компоненты по существу одновременно в организме указанного пациента; (2) по существу одновременное введение такой комбинации ADC, раскрытого в данном документе, и терапевтического(их) агента(ов) пациенту, нуждающемуся в лечении, когда такие компоненты приготовлены отдельно друг от друга в отдельных лекарственных формах, которые указанный пациент принимает по существу одновременно, при этом указанные компоненты высвобождаются у указанного пациента по существу одновременно; (3) последовательное введение такой комбинации ADC, раскрытого в данном документе, и терапевтического(их) агента(ов) пациенту, нуждающемуся в лечении, когда такие компоненты приготовлены отдельно друг от друга в отдельных лекарственных формах, которые указанный пациент принимает последовательно через значительные интервалы времени между каждым введением, при этом указанные компоненты высвобождаются у указанного пациента по существу в разное время; и (4) последовательное введение такой комбинации ADC, раскрытого в данном документе, и терапевтического(их) агента(ов) пациенту, нуждающемуся в лечении, когда такие компоненты приготовлены вместе в стандартной лекарственной форме, которая высвобождает указанные компоненты контролируемым образом, при этом они параллельно, последовательно и/или перекрываясь высвобождаются у указанного пациента одновременно и/или в разное время, причем каждую часть можно вводить либо одинаковым путем, либо разными путями.
Как правило, ADC, раскрытые в данном документе, пригодны для введения в виде композиции совместно с одним или более фармацевтически приемлемым(ыми) эксципиентом(ами). Термин "эксципиент" в данном документе использован для описания любого ингредиента, иного чем соединение(я) по изобретению. Выбор эксципиента(ов) в большой степени зависит от таких факторов, как конкретный способ введения, влияние эксципиента на растворимость и стабильность и характер лекарственной формы. В данном документе "фармацевтически приемлемый эксципиент" включает любые и все растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые агенты, изотонические и задерживающие всасывание агенты и т.п., которые являются физиологически совместимыми. Некоторые примеры фармацевтически приемлемых эксципиентов представляют собой воду, физиологический раствор, забуференный фосфатами физиологический раствор, декстрозу, глицерин, этанол и т.п., а также их комбинации. В некоторых воплощениях изотонические агенты, включая, без ограничения, сахара, полиспирты (например, маннит, сорбит) или хлорид натрия включены в фармацевтическую композицию. Дополнительные примеры фармацевтически приемлемых веществ включают, без ограничения, увлажняющие агенты или незначительные количества вспомогательных веществ, таких как увлажняющие или эмульгируюие агенты, консерванты или буферы, которые увеличивают срок хранения или эффективность антитела.
В некоторых воплощениях ADC, описанные в данном документе, могут быть деиммунизированы для снижения иммуногенности после введения субъекту с использованием известных методов, таких как методы, описанные, например, в публикациях заявок РСТ WO 98/52976 и WO 00/34317.
Фармацевтические композиции по настоящему изобретению и способы их получения очевидны для специалистов в данной области. Такие композиции и способы их получения можно найти, например, в Remington's Pharmaceutical Sciences, 22nd Edition (Mack Publishing Company, 2012). Фармацевтические композиции предпочтительно изготавливают согласно условиям GMP (надлежащая практика организации производства).
Фармацевтическая композиция по изобретению может быть получена, упакована или может продаваться в нерасфасованном виде, в виде стандартной лекарственной дозы или в виде множества стандартных лекарственных доз. В данном документе "стандартная доза" представляет собой дискретное количество фармацевтической композиции, содержащей предопределенное количество активного ингредиента. Количество активного ингредиента обычно равно дозировке активного ингредиента, которую можно будет вводить субъекту, или удобную долю такой дозировки, такую как, например, половина или одна треть такой дозировки. Любой способ введения пептидов, белков или антител, приемлемый в данной области, может быть использован соответствующим образом для сконструированных полипептидных конъюгатов, раскрытых в данном документе.
Фармацевтические композиции по изобретению обычно подходят для парентерального введения. Парентеральное введение фармацевтической композиции включает любой путь введения, характеризующийся физическим разрывом ткани субъекта и введением фармацевтической композиции через разрыв в ткани, что приводит в результате к прямому введению в кровоток, в мышцу или во внутренний орган. Например, парентеральное введение включает, без ограничения, введение фармацевтической композиции инъекцированием композиции, внесением композиции через хирургический разрез, внесением композиции через тканепроникающую нехирургическую рану и т.п. В частности, парентеральное введение включает, без ограничения, подкожные, интраперитонеальные, внутримышечные, интрастернальные, внутривенную, интраартериальные, интратекальные, интравентрикулярные, интрауретральные, интракраниальные, интрасиновиальные инъекции или инфузии и методы инфузии при почечном диализе. В некоторых воплощениях парентеральное введение представляет собой внутривенный или подкожный путь введения.
Препараты фармацевтической композиции, подходящей для парентерального введения, обычно содержат активный ингредиент, объединенный с фармацевтически приемлемым носителем, таким как стерильная вода или стерильный изотонический физиологический раствор. Такие препараты могут быть приготовлены, упакованы или могут продаваться в форме, подходящей для болюсного введения или для непрерывного введения. Инъецируемые препараты могут быть приготовлены, упакованы или могут продаваться стандартной лекарственной форме, такой как ампулы или многодозовые контейнеры, содержащие консервант. Препараты для парентерального введение включают, без ограничения, суспензии, растворы, эмульсии в масляных или водных носителях, пасты и т.п. Такие препараты дополнительно могут содержать один или более дополнительных ингредиентов, включающих, без ограничения, суспендирующие, стабилизирующие или диспергирующие агенты. В одном воплощении препарата для парентерального введения активный ингредиент представлен в сухой (т.е. порошкообразной или гранулированной) форме для разведения подходящим носителем (например стерильной апирогенной водой) перед парентеральным введением разведенной композиции. Парентеральные препараты также включают водные растворы, которые могут содержать эксципиенты, такие как соли, углеводы и буферные агенты (предпочтительно до рН от 3 до 9), но для некоторых применений они могут быть стабильно приготовлены в виде стерильного неводного раствора или в виде высушенной формы, предназначенной для использования совместно с подходящим носителем, таким как стерильная, апирогенная вода.
Иллюстративные формы для перентерального введения включают растворы или суспензии в стерильных водных растворах, например водных растворах пропиленгликоля или декстрозы. Такие лекарственные формы могут быть соответствующим образом забуферены, если это желательно. Другие парентерально вводимые препараты, которые полезны, включают препараты, которые содержат активный ингредиент в микрокристаллической форме или в липосомальном препарате. Препараты для парентерального введения могут быть приготовлены для немедленного и/или модифицированного высвобождения. Препараты модифицированного высвобождения включают препараты контролируемого, отсроченного, длительного, импульсного, направленного и программируемого высвобождения. Например, в одном аспекте стерильные инъкционные растворы могут быть приготовлены путем включения сконструированного Fc-содержащего полипептида, например конъюгата антитело-лекарственное средство или биспецифического антитела, в требуемом количестве в подходящем растворителе с одним ингредиентом или комбинацией ингредиентов, перечисленных выше, которые требуются, с последующей стерилизацией. Как правило, дисперсии получают путем введения активного соединения в стерильный носитель, который содержит основную дисперсионную среду и другие необходимые ингредиенты из перечисленных выше. В случае стерильных порошков для приготовления стерильных инъекционных растворов предпочтительными способами приготовления являются вакуумная сушка и сублимационная сушка, которые обеспечивают получение порошка активного ингредиента плюс любой дополнительный желаемый ингредиент из его предварительно профильтрованного стерильного раствора. Надлежащую текучесть раствора можно поддерживать, например, используя покрытие, такое как лецитин, поддерживая требуемый размер частиц в случае дисперсии и используя поверхностно-активные вещества. Пролонгированное всасывание инъекционных композиций может быть обеспечено путем включения в композицию агента, который замедляет всасывание, например, моностеаратные соли и желатин.
Режимы дозировок можно регулировать для обеспечения оптимального желаемого ответа. Например, можно вводить однократный болюс, можно вводить несколько разделенных доз с течением времени, или дозу можно пропорционально снижать или увеличивать, если этого требует терапевтическая ситуация. Особенно предпочтительным является приготовление парентеральных композиций в стандартной лекарственной форме для облегчения введения и единообразия дозировки. Стандартная лекарственная форма в данном документе относится к физически дискретным единицам, пригодным в качестве единичных (унитарных) дозировок для пациентов/субъектов, которых лечат, причем каждая единица содержит предопределенное количество активного соединения, рассчитанное для продуцирования желаемого терапевтического эффекта, совместно с требуемым фармацевтическим носителем. Детализация для стандартных лекарственных форм по изобретению обычно диктуется и напрямую зависит от (а) уникальных характеристик группировки агента (например, небольшие молекулы, такие как цитотоксический агент) и конкретного терапевтического или профилактического эффекта, которого требуется достичь, и (б) ограничений, характерных в области компаундирования такого активного соединения для лечения чувствительных индивидуумов.
Таким образом, специалисту должно быть понятно из приведенного описания, что доза и режим введения доз регулируются в соответствии с методами, известными в терапевтических областях. То есть максимальная переносимая доза легко может быть установлена, и эффективное количество, приносящее заметную терапевтическую пользу пациенту, также может быть определено, как и возможны временные требования по введению каждого агента для обеспечения обнаружимой терапевтической пользы для пациента. Соответственно, хотя некоторые дозы и режимы введения проиллюстрированы примерами в данном описании, эти примеры никоим образом не ограничивают дозу и режим введения, которые могут быть назначены пациенту при осуществлении настоящего изобретения на практике.
Следует отметить, что значения дозировок могут варьировать в зависимости от типа и тяжести состояния, которое лечат, и могут включать однократные и многократные дозы. Следует также иметь в виду, что для любого конкретного субъекта конкретные режимы дозировки следует корректировать со временем согласно потребностям индивидуума и профессиональному суждению лица, осуществляющего введение или наблюдающего за введением композиций, и что диапазоны дозировки, изложенные в данном описании, являются только иллюстративными и не ограничивают объем или практику заявленной композиции. Кроме того, режим дозировки композиций по данному изобретению может основываться на целом ряде различных факторов, включая тип заболевания, возраст, вес, пол, медицинское состояние пациента, тяжесть состояния, путь введения и конкретное используемое антитело. Таким образом, режим дозировки может варьировать в широких пределах, но может быть определен рутинно с импользованием стандартных методов. Например, дозы могут быть скорректированы на основе фармакокинетических или фармакодинамических параметров, которые могут включать клинические эффекты, такие как токсические эффекты, и/или лабораторные значения. Таким образом, настоящее изобретение охватывает увеличение дозы для пациента, которое определяет специалист. Определение подходящих дозировок и режимов известно в релевантной области и, как будет понятно специалисту, охвачено сведениями, раскрытыми в данном документе.
Для введения субъектам-людям суммарная ежемесячная доза ADC, раскрытого в данном документе, обычно находится в диапазоне от примерно 0,01 мг до примерно 1200 мг на пациента в зависимости, разумеется, от способа введения. Например, может быть необходима внутривенная месячная доза от примерно 1 до примерно 1000 мг/пациент. Суммарную месячную дозу можно вводить однократной дозой или разделенными дозами, и она может, по усмотрению лечащего врача, выходить за пределы обычного диапазона, указанного в данном документе.
Иллюстративный, не являющийся ограничивающим диапазон для терапевтически или профилактически эффективного количества ADC, раскрытого в данном документе, составляет от примерно 0,01 до примерно 1000 мг/пациент/месяц. В некоторых воплощениях ADC можно вводить в дозе от примерно 1 до примерно 200 или от примерно 1 до примерно 150 мг/пациент/месяц. В некоторых воплощениях пациентом является человек.
Наборы
Согласно изобретению предложены также наборы (или продукты производства) для использования в лечении расстройств, описанных выше. Наборы по изобретению включают в себя один или более контейнеров, содержащих очищенный ADC и инструкции по применению конъюгата для лечения заболевания. Например, инструкции содержат описание введения ADC для лечения заболевания, такого как рак (например, солидный рак или гемобластоз). Набор может дополнительно включать в себя описание выбора индивидуума, подходящего для лечения, исходя из идентификации, имеет ли индивидуум данное заболевание и данную стадию заболевания.
Инструкции, относящиеся к применению ADC, обычно содержат информацию о дозировке, схеме введения доз и пути введения для назначаемого лечения. Контейнеры могут представлять собой контейнеры для стандартных доз, нерасфасованные упаковки (например, многодозовые упаковки) или контейнеры для субъединичных доз. Инструкции, поставляемые в наборах по изобретению, обычно представляют собой письменные инструкции на этикетке или в виде вкладыша в упаковку (например, бумажного листка, вложенного в набор), но машиночитаемые инструкции (например, инструкции на магнитном или оптическом запоминающем диске) также приемлемы.
Наборы по изобретению находятся в подходящей упаковке. Подходящие упаковки включают, без ограничения, ампулы, бутыли, баночки, гибкие упаковки (например, герметичные миларовые или пластиковые мешки) и т.п. Также предусмотрены упаковки для использования совместно со специальным устройством, таким как ингалятор, устройство для введения в нос (например, распылитель) или инфузионное устрйоство, такое мини-насос. Набор может иметь стерильный порт доступа (например, контейнер может представлять собой мешок с внутривенным раствором или флакон, имеющий пробку, протыкаемую гиподермической инъекционной иглой). Контейнер также может иметь стерильный порт доступа (например, контейнер может представлять собой мешок с внутривенным раствором или флакон, имеющий пробку, протыкаемую гиподермической инъекционной иглой). По меньшей мере один активный агент в композиции представляет собой ADC с более высокой нагрузкой, как описано в данном документе. Контейнер может дополнительно содержать второй фармацевтически активный агент.
В наборах возможно могут быть предоставлены дополнительные компоненты, такие как буферы и поясняющая информация. Обычно набор содержит контейнер и этикетку или вкладыш(и) в упаковку на или вместе с контейнером.
ПРИМЕРЫ
Следует иметь в виду, что примеры и воплощения, описанные в данном документе, приведены только в иллюстративных целях, и что различные модификации или изменения, которые будут понятны специалистам в данной области в свете данного описания, входят в замысел и объем данной заявки.
Условия ВЭЖХ и ЖХ/МС, использованные для анализов:
Условия аналитической ВЭЖХ: колонка Phenomenex Luna С18 (2), 150×3,0 мм, 5 мкм; подвижная фаза А: 0,02% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 0% до 100% В за 8,5 минут, затем 100% В в течение 1,5 минут; скорость потока: 1,5 мл/минута; температура: не контролировали; детектирование: DAD (детектор на диодной матрице) 210, 254 нм; впрыскиваемый объем: 10 мкл; прибор: Agilent 1100 ВЭЖХ.
Условия аналитической ЖХ/МС: Waters Acquity UPLC HSS ТЗ, C18, 2,1×50 мм, 1,7 мкм; подвижная фаза А: 0,1% муравьиной кислоты в воде (об./об.); подвижная фаза В: 0,1% муравьиной кислоты в ацетонитриле (об./об.); градиент: 5% В за 0,1 минуты, от 5% до 95% В за 2,5 минут, 95% В за 0,35 минуты; скорость потока: 1,25 мл/минута; температура: 60°С; детектирование: 200-450 нм; МС (+) диапазон 100-2000 дальтон; впрыскиваемый объем: 5 мкл; прибор: Waters Acquity.
Условия препаративной ВЭЖХ, использованные для очисток:
Метод А: колонка Phenomenex Luna С18, 100×30 мм, 5 мкм; подвижная фаза А: 0,02% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 5% до 50% В за 20 минут; скорость потока: 20 мл/минута; температура: не контролировали; детектирование: DAD 210, 254 нм; впрыскиваемый объем: вплоть до 5 мл; прибор: Gilson 215.
Метод В: колонка Phenomenex Luna С18, 100×30 мм, 5 мкм; подвижная фаза А: 0,02% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 20% до 80% В за 20 минут; скорость потока: 20 мл/минута; температура: не контролировали; детектирование: DAD 210, 254 нм; впрыскиваемый объем: вплоть до 5 мл; прибор: Gilson 215.
Метод С: колонка Phenomenex Luna С18, 100×30 мм, 5 мкм; подвижная фаза А: 0,02% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 5% до 70% В за 20 минут; скорость потока: 20 мл/минута; температура: не контролировали; детектирование: DAD 210, 254 нм; впрыскиваемый объем: вплоть до 5 мл; прибор: Gilson 215.
Метод D: колонка Phenomenex Luna С18, 100×30 мм, 5 мкм; подвижная фаза А: 0,02% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 10% до 70% В за 20 минут; скорость потока: 20 мл/минута; температура: не контролировали; детектирование: DAD 210, 254 нм; впрыскиваемый объем: вплоть до 5 мл; прибор: Gilson 215.
Метод Е: колонка Phenomenex Luna С18, 100×30 мм, 5 мкм; подвижная фаза А: 0,02% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 30% до 85% В за 20 минут; скорость потока: 20 мл/минута; температура: не контролировали; детектирование: DAD 210, 254 нм; впрыскиваемый объем: вплоть до 5 мл; прибор: Gilson 215.
Метод F: колонка Phenomenex Luna С18, 100×30 мм, 5 мкм; подвижная фаза А: 0,02% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 10% до 90% В за 20 минут; скорость потока: 20 мл/минута; температура: не контролировали; детектирование: DAD 210, 254 нм; впрыскиваемый объем: вплоть до 5 мл; прибор: Gilson 215.
Метод G: колонка Phenomenex Luna С18, 100×30 мм, 5 мкм; подвижная фаза А: 0,02% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 10% до 95% В за 20 минут; скорость потока: 20 мл/минута; температура: не контролировали; детектирование: DAD 210, 254 нм; впрыскиваемый объем: вплоть до 5 мл; прибор: Gilson 215.
Метод Н: колонка Phenomenex Luna С18, 100×30 мм, 5 мкм; подвижная фаза А: 0,02% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 25% до 65% В за 20 минут; скорость потока: 20 мл/минута; температура: не контролировали; детектирование: DAD 210, 254 нм; впрыскиваемый объем: вплоть до 5 мл; прибор: Gilson 215.
Метод I: колонка Phenomenex Synergi Max С18, 250×50 мм, 10 мкм; подвижная фаза А: 0,2% муравьиной кислоты в воде (об./об.); подвижная фаза В: ацетонитрил; градиент: от 33,0% до 58% В за 25 минут, затем увеличение до 100% В за 2 минут, выдержка в течение 5 минут; скорость потока: 80 мл/минута; температура: не контролировали; детектирование: DAD 220 нм.
Метод J: колонка DIKMA Diamonsil(2) С18, 200×20 мм, 5 мкм; подвижная фаза А: 0,225% муравьиной кислоты в воде (об./об.); подвижная фаза В: ацетонитрил; градиент: от 53% до 73% В за 11 минут, затем увеличение до 100% В за 0,5 минут, выдержка в течение 2 минут; скорость потока: 35 мл/минута; температура: не контролировали; детектирование: DAD 220 нм.
Метод K: колонка Phenomenex Luna С18, 100×30 мм, 5 мкм; подвижная фаза А: 0,02% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 20% до 90% В за 20 минут; скорость потока: 20 мл/минута; температура: не контролировали; детектирование: DAD 210, 254 нм; впрыскиваемый объем: вплоть до 5 мл; прибор: Gilson 215.
Метод L: колонка Phenomenex Luna С18, 100×30 мм, 5 мкм; подвижная фаза А: 0,02% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 10% до 80% В за 20 минут; скорость потока: 20 мл/минута; температура: не контролировали; детектирование: DAD 210, 254 нм; впрыскиваемый объем: вплоть до 5 мл; прибор: Gilson 215.
Метод М: колонка Phenomenex Luna С18, 150×21,2 мм, 5 мкм; подвижная фаза А: 0,02% уксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% уксусной кислоты в ацетонитриле (об./об.); градиент: от 1% до 100% В за 8,5 минут, затем выдержка при 100% В в течение 2 минут; скорость потока: 27 мл/минута; температура: 25°С; детектирование: DAD 215 нм, 254 нм; МС (+) диапазон 150-2000 дальтон; впрыскиваемый объем: вплоть до 5 мл; прибор: 305 RP Waters FractionLynx LCMS.
Метод N: колонка Phenomenex Luna C18, 100×30 мм, 5 мкм; подвижная фаза А: 0,02% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 50% до 100% В за 20 минут; скорость потока: 20 мл/минута; температура: не контролировали; детектирование: DAD 210, 254 нм; впрыскиваемый объем: вплоть до 5 мл; прибор: Gilson 215.
Метод О: колонка Phenomenex Luna С18, 150×21,2 мм, 5 мкм; подвижная фаза А: 0,02% уксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% уксусной кислоты в ацетонитриле (об./об.); градиент: от 35% до 50% В за 8,5 минут, затем выдержка при 100% В в течение 2 минут; скорость потока: 27 мл/минута; температура: 25°С; детектирование: DAD 215 нм, 254 нм; МС (+) диапазон 150-2000 дальтон; впрыскиваемый объем: вплоть до 5 мл; прибор: 305 RP Waters FractionLynx LCMS.
Метод Р: колонка Phenomenex Luna С18, 100×30 мм, 5 мкм; подвижная фаза А: 0,02% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 25% до 100% В за 20 минут; скорость потока: 20 мл/минута; температура: не контролировали; детектирование: DAD 210, 254 нм; впрыскиваемый объем: вплоть до 5 мл; прибор: Gilson 215.
Метод Q: колонка Phenomenex Luna С18, 100×30 мм, 5 мкм; подвижная фаза А: 0,02% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 20% до 100% В за 20 минут; скорость потока: 20 мл/минута; температура: не контролировали; детектирование: DAD 210, 254 нм; впрыскиваемый объем: вплоть до 5 мл; прибор: Gilson 215.
Метод R: колонка Phenomenex Luna С18, 150×21,2 мм, 5 мкм; подвижная фаза А: 0,02% уксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% уксусной кислоты в ацетонитриле (об./об.); градиент: от 35% до 50% В за 8,5 минут, затем выдержка при 100% В в течение 2 минут; скорость потока: 27 мл/минута; температура: 25°С; детектирование: DAD 215 нм, 254 нм; МС (+) диапазон 150-2000 дальтон; впрыскиваемый объем: вплоть до 5 мл; прибор: 305 RP Waters FractionLynx LCMS.
Метод S: колонка Phenomenex Luna C18, 100×30 мм, 5 мкм; подвижная фаза А: 0,02% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 20% до 90% В за 20 минут; скорость потока: 20 мл/минута; температура: не контролировали; детектирование: DAD 210, 254 нм; впрыскиваемый объем: вплоть до 5 мл; прибор: Gilson 215.
Метод Т: колонка Phenomenex Synergi Max С18, 250×50 мм, 10 мкм; подвижная фаза А: 0,2% муравьиной кислоты в воде (об./об.); подвижная фаза В: ацетонитрил; градиент: от 35,0% до 65% В за 30 минут, затем увеличение до 100% В за 2 минуты, выдержка в течение 5 минут; скорость потока: 30 мл/минута; температура: не контролировали; детектирование: DAD 220 нм.
Метод U: колонка Isco MPLC, RediSep 43 г С18 Gold; подвижная фаза А: 0,02% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: 20-100% В за 20 минут; скорость потока: 40 мл/мин; температура: не контролировали; DAD 214, 254 нм.
Метод V: колонка Isco MPLC, RediSep 43 г С18 Gold; подвижная фаза А: 0,02% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,02% трифторуксусной кислоты в ацетонитриле (об./об.); градиент 10-80% В за 20 минут; скорость потока: 40 мл/мин; температура: не контролировали; DAD 214, 254 нм.
Метод W: колонка Phenomenex Synergi Max-RP 250×50 мм, 10 мкм; подвижная фаза А: 0,1% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,1% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 24% до 54% В за 30 минут; скорость потока: 30 мл/минута; температура: не контролировали; детектирование: DAD 210, 254 нм; впрыскиваемый объем: вплоть до 5 мл.
Метод X: колонка Luna С18 150×25, 5 мкм; подвижная фаза А: 0,2% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,2% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 30% до 50% В за 11 минут; скорость потока: 35 мл/минута; температура: не контролировали; детектирование: DAD 210, 254 нм; впрыскиваемый объем: вплоть до 5 мл.
Метод Y: колонка Phenomenex Synergi Max-RP 250×50 мм, 100×30 мм, 10 мкм; подвижная фаза А: 0,1% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,1% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 30% до 60% В за 22 минуты; скорость потока: 80 мл/минута; температура: не контролировали; детектирование: DAD 210, 254 нм; впрыскиваемый объем: вплоть до 5 мл.
Метод Z: колонка Phenomenex Synergi Max-RP 250×50 мм, 10 мкм; подвижная фаза А: 0,1% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,1% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 30% до 60% В за 21,5 минуты; скорость потока: 80 мл/минута; температура: не контролировали; детектирование: DAD 210, 254 нм; впрыскиваемый объем: вплоть до 5 мл.
Метод АА: колонка Phenomenex Synergi Max-RP 250×80, 10 мкм; подвижная фаза А: 0,1% трифторуксусной кислоты в воде (об./об.); подвижная фаза В: 0,1% трифторуксусной кислоты в ацетонитриле (об./об.); градиент: от 40% до 70% В за 30 минут; скорость потока: 30 мл/минута; температура: не контролировали; детектирование: DAD 220 нм; впрыскиваемый объем: вплоть до 5 мл.
Пример 1: Получение N~2~-бензоил-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (6)
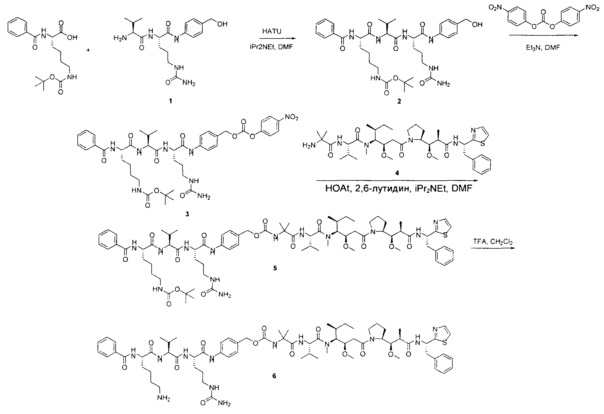
Стадия 1: Синтез N~2~-бензоил-N~6~-(трет-бутоксикарбонил)-L-лизил-L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (2)
В раствор N~2~-бензоил-N~6~-(трет-бутоксикарбонил)-L-лизина (100 мг, 0,285 ммоль) и L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (1, 108 мг, 0,285 ммоль) в 1 мл N,N-диметилформамида добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 111 мг, 0,285 ммоль), затем N,N-диизопропилэтиламин (201 мкл, 149 мг, 1,14 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 16 часов при комнатной температуре. Реакционную смесь затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод А). Фракции, содержащие продукт, лиофилизировали с получением 127 мг (63%) целевого продукта. ЖХ/МС m/z 712,4 [М+Н+]; 710,4 [М-Н+]; время удерживания = 1,30 минуты.
Стадия 2: Синтез N~2~-бензоил-N~6~-(трет-бутоксикарбонил)-L-лизил-L-валил-N~5~-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]-L-орнитинамида (3)
Раствор N~2~-бензоил-N~6~-(трет-бутоксикарбонил)-L-лизил-L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (2, 125 мг, 0,352 ммоль) в 1 мл N,N-диметилформамида обрабатывали пара-нитрофенилкарбонатом (107 мг, 0,352 ммоль) и триэтиламином (101 мкл, 73,4 мг, 0,704 ммоль). Смесь перемешивали в течение двух часов при комнатной температуре и затем концентрировали почти досуха и очищали хроматографией на силикагеле, используя элюирование с градиентом от 5% до 20% метанола в дихлорметане с получением 129 мг (83,6%) целевого продукта. ЖХ/МС m/z 877,4 [М+Н+]; 899,3 [M+Na+]; время удерживания = 1,73 минуты.
Стадия 3: Синтез N~2~-бензоил-N~6~-(трет-бутоксикарбонил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (5)
В раствор 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (4, 20 мг, 0,027 ммоль), N~2~-бензоил-N~6~-(трет-бутоксикарбонил)-L-лизил-L-валил-N~5~-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]-L-орнитинамида (3, 28 мг, 0,032 ммоль) в 0,2 мл N,N-диметилформамида добавляли 2,6-лутидин (5,8 мг, 0,032 ммоль), N,N-диизопропилэтиламин (9 мкл, 7 мг, 0,054 ммоль) и 3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ол (HOAt, 4,4 мг, 0,032 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 16 часов при комнатной температуре. Реакционную смесь затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод В). Фракции, содержащие продукт, лиофилизировали с получением 30 мг (75%) целевого продукта. ЖХ/МС m/z 1481,8 [М+Н+]; 1478,8 [М-Н+]; время удерживания = 1,91 минуты.
Стадия 4: Синтез N~2~-бензоил-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (6)
В раствор N~2~-бензоил-N~6~-(трет-бутоксикарбонил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (5, 30 мг, 0,02 ммоль) в 2,5 мл дихлорметана добавляли 2,5 мл 10%-ного раствора трифторуксусной кислоты в дихлорметане. Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 2,25 часов при комнатной температуре. Реакционную смесь затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод С). Фракции, содержащие продукт, лиофилизировали с получением 22 мг (74%) целевого продукта. ЖХ/МС m/z 1381,7 [М+Н+]; время удерживания = 1,54 минуты. Аналитическая ВЭЖХ: время удерживания = 6,00 минут.
Пример 2: Получение N~2~(2,2-диметилпропаноил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (12)
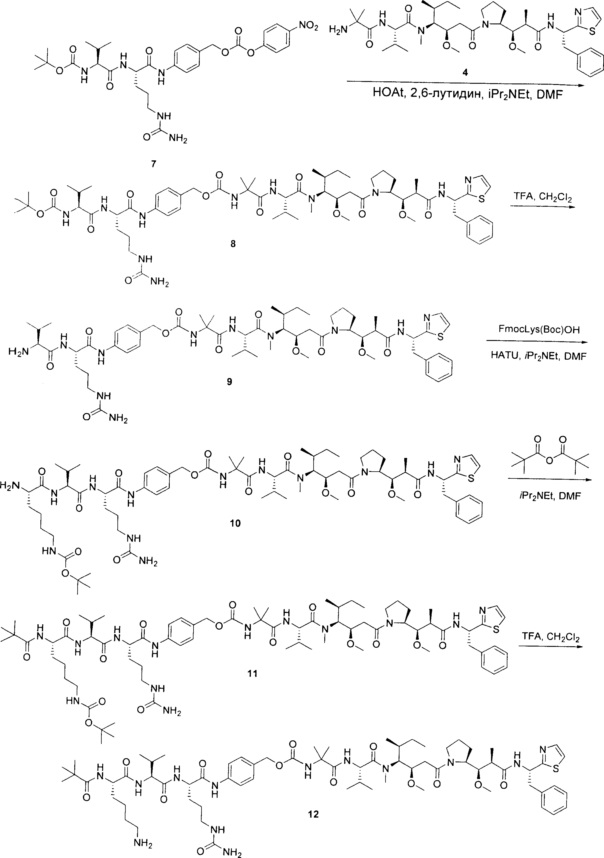
Стадия 1: Синтез N-(трет-бутоксикарбонил)-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (8)
В раствор 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (4, 80 мг, 0,11 ммоль) и N-(трет-бутоксикарбонил)-L-валил-N~5~-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]-L-орнитинамида (7, 76,7 мг, 0,119 ммоль) в 0,5 мл N,N-диметилформамида добавляли 2,6-лутидин (46,3 мг, 0,432 ммоль), N,N-диизопропилзтиламин (67,7 мкл, 56,4 мг, 0,432 ммоль) и 3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ол (HOAt, 17,7 мг, 0,130 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 16 часов при 45°С. Реакционную смесь затем концентрировали и очищали хроматографией на силикагеле, используя элюирование с градиентом от 5% до 20% метанола в дихлорметане с получением 90 мг (67%) целевого продукта. ЖХ/МС m/z 1248,7 [М+Н+]; время удерживания = 1,84 минут.
Стадия 2: Синтез L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (9)
В раствор N-(трет-бутоксикарбонил)-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (8, 91 мг, 0,072 ммоль) в 5 мл дихлорметана добавляли 2 мл 20%-ного раствора трифторуксусной кислоты в дихлорметане. Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 2,75 часов при комнатной температуре. Реакционную смесь затем концентрировали с получением 91 мг (100%) целевого продукта, который напрямую использовали не следующей стадии без очистки. ЖХ/МС m/z 1148,7 [М+Н+]; время удерживания = 1.43 минут.
Стадия 3: Синтез N~6~-(трет-бутоксикарбонил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (10)
В раствор N~6~-(трет-бутоксикарбонил)-N~2~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизина (50,6 мг, 0,108 ммоль) в 0,5 мл N,N-диметилформамида добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 41,6 мг, 0,108 ммоль), затем N,N-диизопропилэтиламин (51 мкл, 37,6 мг, 0,288 ммоль), и эту смесь перемешивали в течение 15 минут при комнатной температуре. Затем добавляли раствор L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (9, 91 мг, 0,072 ммоль) в 1 мл N,N-диметилформамида. Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 2 часов при комнатной температуре, после чего медленно добавляли диэтиламин (2 мл, 1,41 г, 19,3 ммоль). Реакционную смесь перемешивали в течение 16 часов и затем подкисляли добавлением трифторуксусной кислоты, концентрировали и очищали обращенно-фазной ВЭЖХ (Метод D). Фракции, содержащие продукт, лиофилизировали с получением 99 мг (92%) целевого продукта. ЖХ/МС m/z 1376,8 [М+Н+]; 1374,8 [М-Н+]; время удерживания = 1,40 минут.
Стадия 4: Синтез N~6~-(трет-бутоксикарбонил)-N~2~-(2,2-Диметилпропаноил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (11)
В раствор N~6~-(трет-бутоксикарбонил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида трифторацетата (10, 21,5 мг, 0,015 ммоль) и пивалинового ангидрида (5,6 мг, 0,030 ммоль) в 0,2 мл N,N-диметилформамида добавляли N,N-диизопропилэтиламин (26 мкл, 19,4 мг, 0,15 ммоль), и эту смесь перемешивали при комнатной температуре в течение 45 минут. Раствор затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод E). Фракции, содержащие продукт, лиофилизировали с получением 21,9 мг (100%) целевого продукта. ЖХ/МС m/z 1460,7 [М+Н+]; 1458,5 [M-Н+]; время удерживания = 1,93 минут.
Стадия 5: Синтез N~2~-(2,2-Диметилпропаноил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (12)
В раствор N~6~-(трет-бутоксикарбонил)-N~2~-(2,2-диметилпропаноил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (11, 21,9 мг, 0,015 ммоль) в 1 мл дихлорметана добавляли 0,33 мл трифторуксусной кислоты. Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 1 часа при комнатной температуре. Реакционную смесь затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод F). Фракции, содержащие продукт, лиофилизировали с получением 8 мг (36%) целевого продукта. ЖХ/МС m/z 1360,7 [М+Н+]; время удерживания = 1,45 минут. Аналитическая ВЭЖХ: время удерживания = 6,01 минут.
Пример 3: Получение N~2-(фенилацетил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (14)
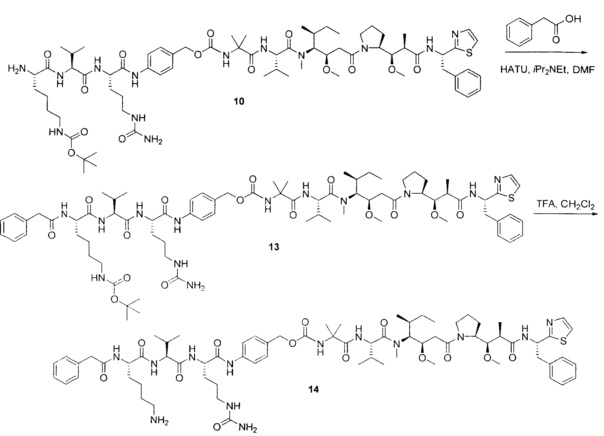
Стадия 1: Синтез N~6~-(трет-бутоксикарбонил)-N~2~-(фенилацетил)-1-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (13)
В раствор фенилуксусной кислоты (15 мг, 0,11 ммоль) в 0,2 мл N,N-диметилформамида добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 43,1 мг, 0,11 ммоль), затем N,N-диизопропилэтиламин (20 мкл, 14,5 мг, 0,11 ммоль), и эту смесь перемешивали при комнатной температуре в течение 45 минут. Добавляли раствор N~6~-(трет-бутоксикарбонил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида трифторацетата (10, 20 мг, 0,013 ммоль) в 0,2 мл N,N-диметилформамида, и эту смесь перемешивали при комнатной температуре в течение 16 часов. Раствор затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод G). Фракции, содержащие продукт, лиофилизировали с получением 10 мг (52%) целевого продукта. ЖХ/МС m/z 1495,3 [М+Н+]; 1493,1 [М-Н+]; время удерживания = 1,93 минут.
Стадия 2: Синтез N~2~-(фенилацетил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (14)
В раствор N~6~-(трет-бутоксикарбонил)-N~2~-(фенилацетил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (13, 10 мг, 0,007 ммоль) в 1 мл дихлорметана добавляли 1 мл 20%-ного раствора трифторуксусной кислоты в дихлорметане. Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 2 часов при комнатной температуре. Реакционную смесь затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод G). Фракции, содержащие продукт, лиофилизировали с получением 10 мг (100%) целевого продукта. ЖХ/МС m/z 1394,7 [М+Н+]; время удерживания = 1,52 минут. Аналитическая ВЭЖХ: время удерживания = 5,39 минут.
Пример 4: Получение N~2~-(1Н-имидазол-5-илкарбонил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (16)
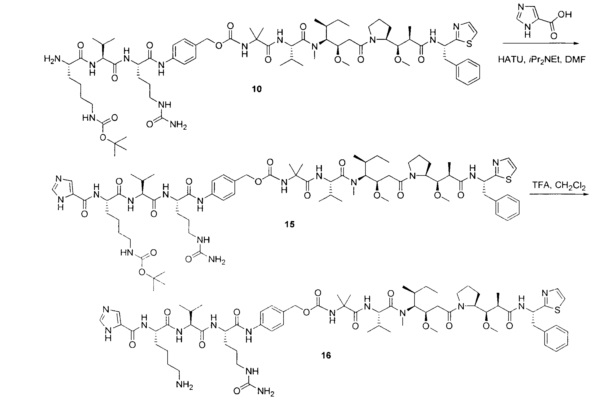
Стадия 1: Синтез N~6~-(трет-бутоксикарбонил)-N~2~-(1Н-имидазол-5-илкарбонил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (15)
В раствор N~6~-(трет-бутоксикарбонил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-1-орнитинамида трифторацетата (10, 20,5 мг, 0,014 ммоль) и 1Н-имидазол-5-карбоновой кислоты (15,7 мг, 0,14 ммоль) в 0,2 мл N,N-диметилформамида добавляли N,N-диизопропилэтиламин (25 мкл, 18,3 мг, 0,14 ммоль), и эту смесь перемешивали при комнатной температуре в течение 5 минут. Добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 54,3 мг, 0,14 ммоль), и смесь перемешивали в течение еще часа при комнатной температуре. Раствор затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод G). Фракции, содержащие продукт, лиофилизировали с получением 13 мг (59%) целевого продукта. ЖХ/МС m/z 1471,6 [М+Н+]; 1469,0 [М-Н+]; время удерживания = 1,70 минут.
Стадия 2: Синтез N~2~-(1Н-имидазол-5-илкарбонил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (16)
В раствор N~6~-(трет-бутоксикарбонил)-N~2~-(1Н-имидазол-5-илкарбонил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (15, 13 мг, 0,008 ммоль) в 5 мл дихлорметана добавляли 2 мл 20%-ного раствора трифторуксусной кислоты в дихлорметане. Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 15 минут при комнатной температуре. Реакционную смесь затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод D). Фракции, содержащие продукт, лиофилизировали с получением 6 мг (50%) целевого продукта. ЖХ/МС m/z 1370,7 [М+Н+]; 1368,6 [М-Н+]; время удерживания = 1,39 минут. Аналитическая ВЭЖХ: время удерживания = 5,30 минут.
Пример 5: Получение N~2~-(1Н-имидазол-5-илацетил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (18)
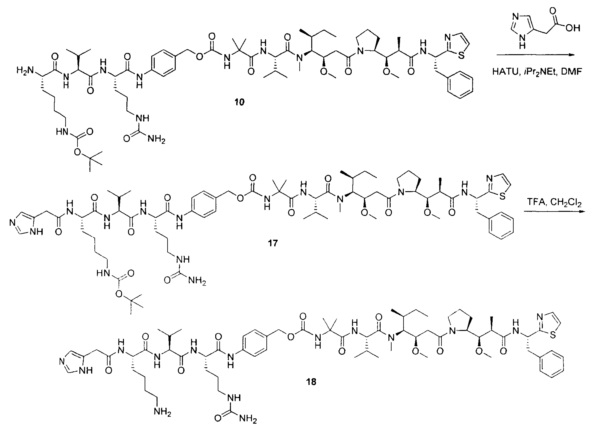
Стадия 1: Синтез N~6~-(трет-бутоксикарбонил)-N~2~-(1Н-имидазол-5-илацетил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (17)
В раствор N~6~-(трет-бутоксикарбонил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5)5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида трифторацетата (10, 20 мг, 0,013 ммоль) и 2-(1Н-имидазол-5-ил)уксусной кислоты (30,0 мг, 0,20 ммоль) в 0,2 мл N,N-диметилформамида добавляли N,N-диизопропилэтиламин (23 мкл, 17 мг, 0,13 ммоль), и эту смесь перемешивали при комнатной температуре в течение 5 минут. Добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 50,4 мг, 0,13 ммоль), и смесь перемешивали в течение еще часа при комнатной температуре. Раствор затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод G). Фракции, содержащие продукт, лиофилизировали с получением 13,6 мг (65%) целевого продукта. ЖХ/МС m/z 1485,6 [М+Н+]; 1483,5 [М-Н+]; время удерживания = 1,44 минут.
Стадия 2: Синтез N~2~-(1Н-имидазол-5-илацетил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (18)
В раствор N~6~-(трет-бутоксикарбонил)-N~2~-(1Н-имидазол-5-илацетил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (17, 13,6 мг, 0,009 ммоль) в 5 мл дихлорметана добавляли 2 мл 20%-ного раствора трифторуксусной кислоты в дихлорметане. Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 15 минут при комнатной температуре. Реакционную смесь затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод D). Фракции, содержащие продукт, лиофилизировали с получением 13 мг (96%) целевого продукта. ЖХ/МС m/z 1384,9 [М+Н+]; 1382,8 [М-Н+]; время удерживания = 1,26 минут. Аналитическая ВЭЖХ: время удерживания = 5,21 минут.
Пример 6: Получение N~2~-(3-метилбутаноил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (19)
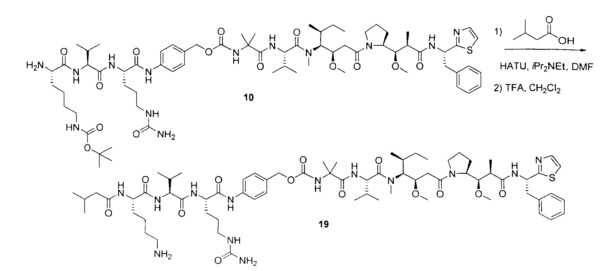
Раствор 3-метилбутановой кислоты (10,2 мг, 0,100 ммоль) в 0,4 мл N,N-диметилформамида обрабатывали N,N-диизопропилэтиламином (35 мкл, 26,1 мг, 0,200 ммоль) и гексафторфосфатом O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 77,6 мг, 0,200 ммоль), и эту смесь перемешивали при комнатной температуре в течение пяти минут. Добавляли раствор N~6~-(трет-бутоксикарбонил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида трифторацетата (10, 30 мг, 0,020 ммоль) в 0,4 мл N,N-диметилформамида, и эту смесь перемешивали при комнатной температуре в течение 1 часа. Раствор затем концентрировали почти досуха, затем растворяли в 2 мл дихлорметана и обрабатывали 0,5 мл трифторуксусной кислоты. После перемешивания при комнатной температуре в течение еще 30 минут реакционную смесь концентрировали и очищали обращенно-фазной ВЭЖХ (Метод Н). Фракции, содержащие продукт, лиофилизировали с получением 13 мг (44%) целевого продукта. ЖХ/МС m/z 1360,8 [М+Н+]; 1358,4 [М-Н+]; время удерживания = 1,49 минут. Аналитическая ВЭЖХ: время удерживания = 5,98 минут.
Пример 7: Получение N~2~-(2-метилпропаноил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (25)
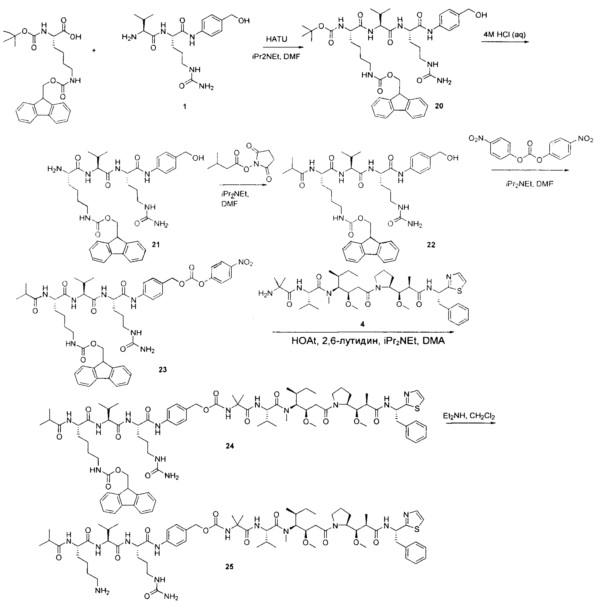
Стадия 1: Синтез N~2~-(трет-бутоксикарбонил)-N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизил-L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (20)
В раствор L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (1, 6,5 г, 17,1 ммоль) и N~2~-(трет-бутоксикарбонил)-N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизина (8,03 г, 17,1 ммоль) в 200 мл N,N-диметилформамида добавляли N,N-диизопропилэтиламин (3,32 г, 25,7 ммоль) и гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 7,8 г, 20,56 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение 12 часов и затем концентрировали и очищали хроматографией на диоксиде кремния, используя элюирование с градиентом от 5% до 17% метанола в дихлорметане, с получением 4,00 г (28,2%) целевого продукта в виде белого твердого вещества.
Стадия 2: Синтез N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизил-L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (21)
Смесь N~2~-(трет-бутоксикарбонил)-N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизил-1-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (20, 4,8 г, 5,78 ммоль) в 96 мл 4М водной соляной кислоты перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь доводили до рН 7,5 медленным добавлением насыщенного водного раствора K2CO3, и затем полученную суспензию фильтровали. Твердое вещество собирали и сушили в вакууме с получением 4,00 г (94,9%) целевого продукта в виде желтого твердого вещества.
Стадия 3: Синтез N~6~-[(9Н-флуорен-9-илметокси)карбонил]-N~2~-(2-метилпропаноил)-L-лизил-L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (22)
В раствор N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизил-L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (21, 500 мг, 0,685 ммоль) и 1-[(2-метилпропаноил)окси]пирродидин-2,5-диона (190 мг, 1,03 ммоль) в 20 мл N,N-диметилформамида добавляли N,N-диизопропилэтиламин (221 мг, 1,7 ммоль). После перемешивания в течение 2 часов при комнатной температуре смесь концентрировали и очищали обращенно-фазной ВЭЖХ (Метод I) с получением 100 мг (18,2%) целевого продукта в виде белого твердого вещества.
Стадия 4: Синтез N~6~-[(9Н-флуорен-9-илметокси)карбонил]-N~2~-(2-метилпропаноил)-1-лизил-1-валил-N~5~-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]-L-орнитинамида (23)
В раствор N~6~-[(9Н-флуорен-9-илметокси)карбонил]-N~2~-(2-метилпропаноил)-L-лизил-L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (22, 100 мг, 0,125 ммоль) и пара-нитрофенилкарбоната (42 мг, 0,138 ммоль) в 5 мл N,N-диметилформамида добавляли N,N-диизопропилэтиламин (32 мг, 0,25 ммоль), и эту смесь перемешивали при комнатной температуре в течение 12 часов. Раствор затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод J) с получением 110 мг (91,7%) целевого продукта в виде белого твердого вещества. МСВР (масс-спектрометрия высокого разрешения) m/z для C50H60N8O12: 987,4036 (M+Na)+. 1Н ЯМР (400 МГц, DMSO): δ 10.12 (s, 1Н), 8.30 (m, 2Н), 8.18 (m, 1Н), 7.88 (m, 3H), 7.64 (m, 6Н), 7.27 (m, 6Н), 6.00 (s, 1Н), 5.44 (s, 2Н), 5.23 (s, 2Н), 4.21 (m, 6Н), 2.95 (m, 5Н), 1.99 (m, 1Н), 1.24 (m, 12Н), 0.99 (d, 6Н), 0.85 (d, 6Н);
Стадия 4: Синтез N~6~-[(9Н-флуорен-9-илметокси)карбонил]-N~2~-(2-метилпропаноил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (24)
В раствор 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (4, 27,5 мг, 0,032 ммоль) и N~6~-[(9Н-флуорен-9-илметокси)карбонил]-N~2~-(2-метилпропаноил)-L-лизил-L-валил-N~5~-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]-L-орнитинамида (23, 36,7 мг, 0,038 ммоль) в 1 мл N,N-диметилацетамида добавляли 2,6-лутидин (13,7 мг, 0,128 ммоль), N,N-диизопропилэтиламин (23 мкл, 16,7 мг, 0,128 ммоль) и 3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ол (HOAt, 5,2 мг, 0,038 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 16 часов при комнатной температуре. Раствор затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод K). Фракции, содержащие продукт, лиофилизировали с получением 30 мг (60%) целевого продукта.
Стадия 5: Синтез N~2~-(2-метилпропаноил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (25)
Раствор N~6~-[(9Н-флуорен-9-илметокси)карбонил]-N~2~-(2-метил пропаноил)-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-гриоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (24, 30 мг, 0,019 ммоль) в 1 мл дихлорметана обрабатывали 1 мл диэтиламина. Раствор перемешивали при комнатной температуре в течение пяти часов и затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод K). Фракции, содержащие продукт, лиофилизировали с получением 6,0 мг (21%) целевого продукта. ЖХ/МС m/z 1346,9 [М+Н+]; время удерживания = 1,47 минут. Аналитическая ВЭЖХ: время удерживания = 5,87 минут.
Методика для Примеров 8-18 и 49-71: Конъюгирование, очистка сайт-специфических анти-Trop2 ADC
Для конъюгирования анти-Trop2 антител с конструкциями линкер-полезная нагрузка (LP) Примеров 1-7 указанное количество антитела доводили до 5 мг/мл в буфере, содержащем 25 мМ Tris-HCl, рН 8,0-8,5. Указанное количество LP добавляли в 5-50-кратном молярном избытке относительно количества антитела, и ферментативную реакцию инициировали добавлением 2% (масс./об.) бактериальной трансглутаминазы (Ajinomoto Activa TI, Japan), и реакционную смесь инкубировали при слабом встряхивании при 37°С в течение 16-24 часов. ADC очищали с использованием Butyl Sepharose™ High Performance (Butyl HP, GE Healthcare Biosciences), регулируя реакционную смесь для получения буферной композиции 0,75 М сульфата аммония, 25 мМ фосфата калия, рН 7 (Буфер А). Вещество наносили на Butyl HP, промывали 5 CV Буфера А и элюировали 20 CV с линейным градиентом в 25 мМ фосфата калия, рН 7. Фракции, содержащие ADC, собирали, подвергали диализу против PBS, концентрировали с использованием центрифужной фильтровальной установки 10 kDa Amicon Ultra (Millipore Corporation) и подвергали стерильной фильтрации через фильтр 0,2 мкм. Альтернативно, ADC очищали с использованем смолы MabSelect Protein A (GE Healthcare Biosciences) согласно стандартным протоколам, и буферным обменом в PBS.
Следующие ADC (присоединенные в различных положениях и на различных метках на анти-Trop2 антителе, как указано в Таблицах 2-7) были получены с использованием описанного выше метода (в скобках указан остаток глутамина, причем волнистые линии показывают точки присоединения к остатку анти-Trop2 антитела):
Сравнительный Пример 8: C6vcMMAD:
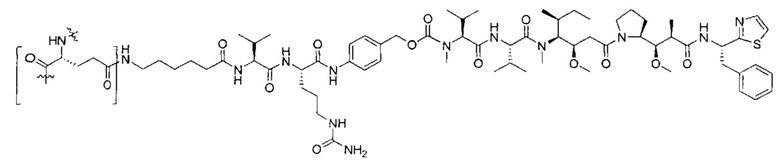 Сравнительный Пример 9: AcLys-vcMMAD:
Сравнительный Пример 9: AcLys-vcMMAD:
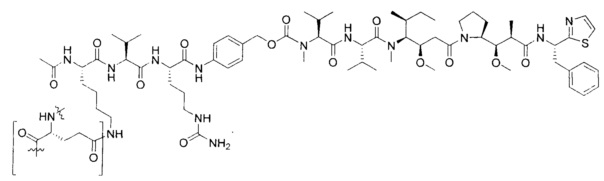
Сравнительный Пример 10: C6vc0101:
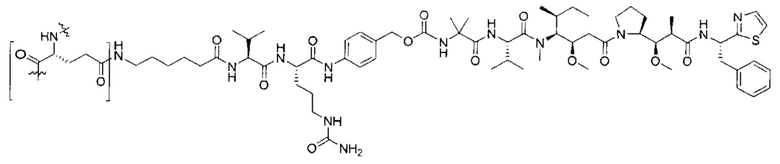 Сравнительный Пример 11: AcLys-vc0101:
Сравнительный Пример 11: AcLys-vc0101:
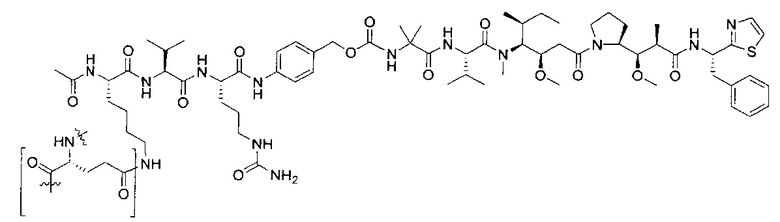
Пример 12:
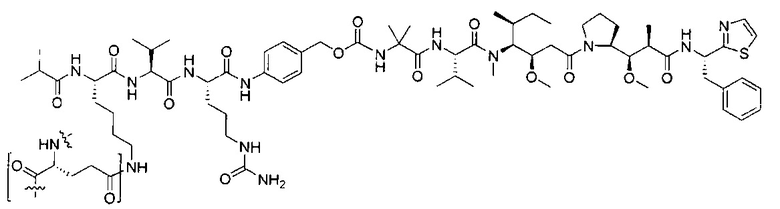
Пример 13:
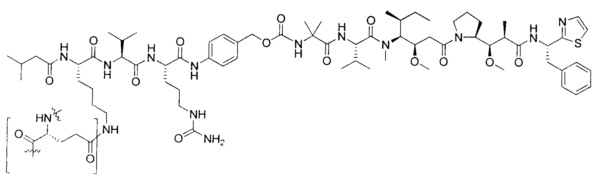
Пример 14:
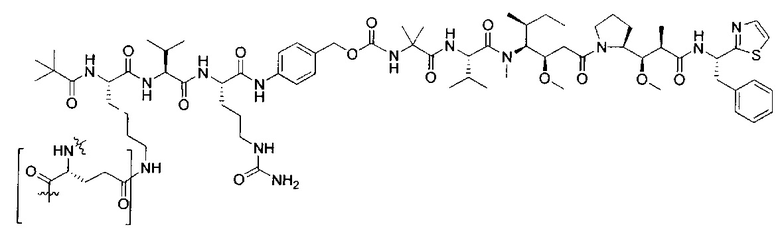
Пример 15:
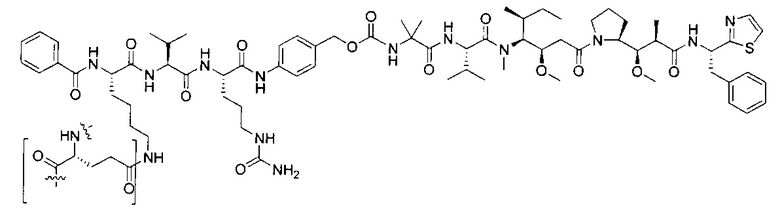
Пример 16:
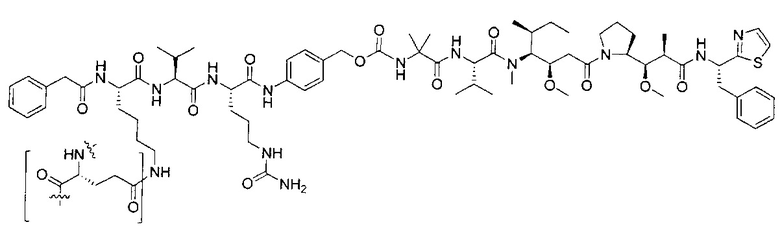
Пример 17:
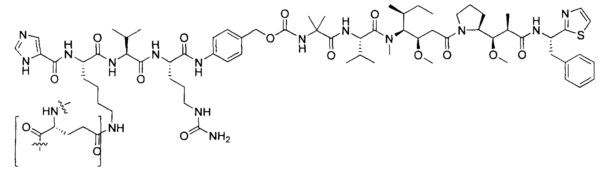
Пример 18:
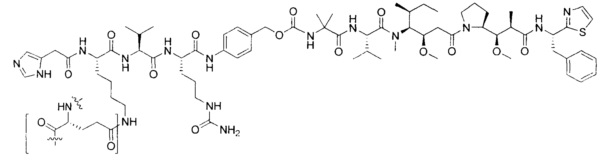
Пример 19: Анализ стабильности in vivo - Мышь
Образцы плазмы крови получали от мышей в точки времени от 1 до 10 суток после введения однократной дозы ADC 9 мг/кг. Образцы разводили равным объемом 1х PBS, и ADC выделяли с использованием гранул MabSelect или Trop2 антигена, свзанного с CNBr-активированной сефарозой Sepharose (GE Healthcare), используя стандартные протоколы. Соотношения лекарственное средство-антитело оценивали до и после экспонирования in vivo с использованием методов HIC (хроматография гидрофобного взаимодействия) и масс-спектрометрии.
Пример 20: Анализ стабильности in vivo - Крыса
Образцы плазмы крови получали от крыс в точки времени от 1 до 10 суток после введения однократной дозы ADC 9 мг/кг. Образцы разводили равным объемом 1x PBS, и ADC выделяли с использованием гранул MabSelect или Trop2 антигена, свзанногой с CNBr-активированной Sepharose (GE Healthcare), используя стандартные протоколы. Соотношения лекарственное средство-антитело оценивали до и после экспонирования in vivo с использованием методов HIC и масс-спектрометрии.
Пример 21 А: Анализ стабильности in vitro - Плазма крови мыши
100-200 мкг ADC инкубировали в плазме крови мыши (предоставленной Bioreclamation), дополненной 1х PBS до конечной концентрации ADC 0,125 мг/мл. После инкубирования в течение 3-7 суток образцы разводили равным объемом 1х PBS, и ADC выделяли с использованием Trop2 антигена, связанного с CNBr-активированной Sepharose (GE Healthcare), используя стандартные протоколы. Соотношения лекарственное средство-антитело оценивали до и после экспонирования in vivo с использованием методов HIC и масс-спектрометрии.
Пример 21В: Идентификация фермента в плазме крови мыши, ответственного за расщепление линкера
Для классификации фермента плазмы крови мыши анализы стабильности in vitro проводили в присутствии различных ингибиторов протеаз с использованием конъюгата анти-Trop2 L11B С6 vc0101 в качестве субстрата. Результаты показали, что фермент плазмы крови мыши по всей вероятности представляет собой серингидролазу, ингибируемую 1 мМ Pefabloc (Roche) и 100 мкМ BNPP (Sigma). Чтобы идентифицировать фермент, авторы изобретения повышали активность гидролиза линкера анти-Trop2 L11B С6 vc0101 путем фракционирования сыворотки мышей Balb/c с использованием ряда стадий. Во-первых, сыворотку Balb/c (BioreclamationIVT) обедняли IgG и альбумином, используя коммерчески доступные смолы MabSelect Protein А и Protein G (GE Healthcare), а затем набор для обеднения мышиной сыворотки альбумином Qproteome Murine Albumin Depletion Kit (Qiagen). Обедненную сыворотку затем подвергали последовательной преципитации с сульфатом аммония. Фракции с наивысшей активностью гидролазы линкера затем наносили на катионообменную колонку HiTrap SP (GE Healthcare), используя постепенные градиентые элюирования хлоридом натрия. После этого SP фракции с наивысшей активностью наносили на анионообменную колонку HiTrap Q (GE Healthcare), используя постепенные градиентные элюирования хлоридом натрия. Фракции с наивысшей активностью гидролазы затем объединяли и фракционировали по размерам, используя колонку Superdex 200 (GE Healthcare). Протеомический анализ собранных Superdex 200 фракций с наивысшей активностью выявил карбоксилэстеразу 1С, которая способна гидролизовать амидные связи и которая может быть ингибирована Pefabloc и BNPP. Для провеки корректной индентификации мышиной карбоксилэстеразы 1С в качестве фермента плазмы крови, ответственного за расщепления линкера, рекомбинантный белок эксперссировали в клетках Expi 293 и очищали с использованием HIS метки, следуя стандартным протоколам. Было подтверждено, что очищенный белок расщепляет множество конъюгатов с линкерами на основе VC с одинаковой относительной активностью, как показано для плазмы крови мыши.
Пример 22: Анализ стабильности in vitro - Плазма крови крысы
100-200 мкг ADC инкубировали в плазме крови крысы (предоставленной Bioreclamation), дополненной 1х PBS до конечной концентрации ADC 0,125 мг/мл. После инкубирования в течение 3-7 суток образцы разводили равным объемом 1х PBS, и ADC выделяли с использованием Trop2 антигена, связанного с CNBr-активированной Sepharose (GE Healthcare), согласно стандартным протоколам. Соотношения лекарственное средство-антитело оценивали до и после экспонирования in vitro с использованием методов HIC и масс-спектрометрии.
Пример 23: Анализ стабильности in vitro - Плазма крови яванского макака
100-200 мкг ADC инкубировали в плазме крови яванского макака (предоставленной Bioreclamation), дополненной 1х PBS до конечной концентрации ADC 0,125 мг/мл. После инкубирования в течение 3-7 суток образцы разводили равным объемом 1х PBS, и ADC выделяли с использованием Trop2 антигена, связанного с CNBr-активированной Sepharose (GE Healthcare), согласно стандартным протоколам. Соотношения лекарственное средство-антитело оценивали до и после экспонирования in vitro с использованием методов HIC и масс-спектрометрии.
Пример 24: Анализ цитотоксичности in vitro
Как показано на Фиг. 1 (А-I), исследования цитотоксичности in vitro химерных анти-Trop2 антител, конъюгированных по целому ряду сайтов с цитотоксической полезной нагрузкой аминокапроил (С6) vc Aur0101, осуществляли с использованием клеток ВхРС3, экспрессирующих мишень. Необработанные соединения (сплошная линия) и их метаболиты, выделенные после 4,5 суток обработки в мышиной плазме крови (прерывистая линия), тестировали одновременно для определения любых изменений цитотоксичности. ВхРс3 представляет собой линию раковых клеток с высокими уровнями экспрессии мишени (Trop-2 +++). Клетки высевали на белые луночные планшеты с прозрачным дном в количестве 2000 клеток на лунку за 24 часа до обработки. Клетки обрабатывали 4-кратными последовательными разведениями конъюгатов антитело-лекарственное средство в трех повторах. Жизнеспособность клеток определяли люминисцентным анализом жизнеспособности клеток CellTiter-Glo® Luminescent Cell Viability Assay 96 (Promega, Madison, WI) через 96 часов после обработки. Относительную жизнеспособность клеток определяли в процентах от необработанного контроля.
Также, как показано на Фиг. 1 (J-О), исследования цитотоксичности in vitro химерных анти-Trop2 антител, конъюгированных по сайтам LCQ04 или L11B с одной из трех конструкций линкер-полезная нагрузка (линкер-полезная нагрузка Примеров 2, 3 и 5), осуществляли с использованием клеток ВхРС3, экспрессирующих мишень. Необработанные соединения (сплошная линия) и их метаболиты, выделенные после 4,5 суток обработки в мышиной плазме крови (прерывистая линия), тестировали одновременно для определения любых изменений цитотоксичности. ВхРс3 представляет собой линию раковых клеток с высокими уровнями экспрессии мишени (Trop-2 +++). Клетки высевали на белые луночные планшеты с прозрачным дном в количестве 2000 клеток на лунку за 24 часа до обработки. Клетки обрабатывали 4-кратными последовательными разведениями конъюгатов антитело-лекарственное средство в трех повторах. Жизнеспособность клеток определяли люминисцентным анализом жизнеспособности клеток CellTiter-Glo® Luminescent Cell Viability Assay 96 (Promega, Madison, WI) через 96 часов после обработки. Относительную жизнеспособность клеток определяли в процентах от необработанного контроля.
Пример 25: Получение N~2~-(гидроксиацетил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (30)
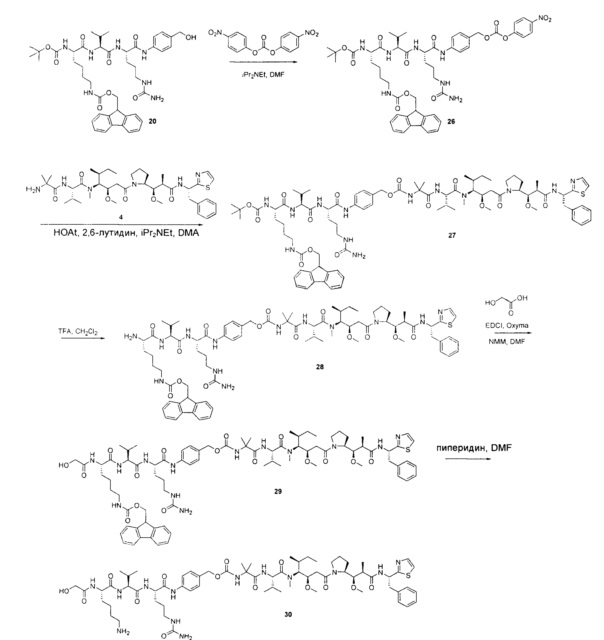
Стадия 1: Синтез N~2~-(трет-бутоксикарбонил)-N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизил-L-валил-N~5~-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]-L-орнитинамида (26)
В раствор N~2~-(трет-бутоксикарбонил)-N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизил-L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (20, 998 мг, 1.20 ммоль) и пара-нитрофенилкарбоната (748 мг, 2,41 ммоль) в 7 мл N,N-диметилформамида и в 7 мл дихлорметана добавляли N,N-диизопропилэтиламин (452 мкл, 0,328 г, 2,41 ммоль), и эту смесь перемешивали при комнатной температуре в течение 3 часов. Остаток разбавляли этилацетатом и диэтиловым эфиром, и полученную суспензию перемешивали в течение 30 минут. Твердое вещество выделяли фильтрованием и промывали диэтиловым эфиром несколько раз с получением после сушки на воздухе целевого продукта в виде желтого твердого вещества (1319 мг), которое использовали без дополнительной очистки. ЖХ/МС m/z 995,5 [М+Н+]; время удерживания = 1,02 минут. Аналитическая ВЭЖХ: время удерживания = 8,181 минут.
Стадия 2: Синтез N~2~-(трет-бутоксикарбонил)-N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (27)
В раствор 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (4, 371 мг, 0,499 ммоль) и N~2~-(трет-бутоксикарбонил)-N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизил-L-валил-N~5~-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]-L-орнитинамида (26, 802 мг, 0,64 ммоль) в 2,5 мл N,N-диметилацетамида добавляли 2,6-лутидин (216 мкл, 200 мг, 1,9 ммоль), N,N-диизопропилэтиламин (326 мкл, 240 мг, 1,9 ммоль) и 3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ол (HOAt, 17,7 мг, 0,130 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 90 минут при 50°С и затем при комнатной температуре в течение 16 часов. Реакционную смесь затем очищали обращенно-фазной хроматографией (Метод U) с получением после лиофилизации 339 мг (42%, 2 стадии) целевого продукта в виде белого твердого вещества. ЖХ/МС m/z 1599,9 [М+Н+]; время удерживания = 1,09 минут. Аналитическая ВЭЖХ: время удерживания = 8,163 минут.
Стадия 3: Синтез N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (28)
В суспензию N~2~-(трет-бутоксикарбонил)-N~6~-[(9Н-флуорен-9-ил метокси)карбонил]-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5-карбамоил-L-орнитинамида (27, 439 мг, 0,275 ммоль) в 2 мл дихлорметана добавляли 1 мл трифторуксусной кислоты. Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 20 минут при комнатной температуре. Реакционную смесь затем концентрировали и очищали обращенно-фазной хроматографией (Метод V) с получением после лиофилизации 410 мг (96%). ЖХ/МС m/z 1499,8 [М+Н+]; время удерживания = 0,86 минут. Аналитическая ВЭЖХ: время удерживания = 6,778 минут.
Стадия 4: Синтез N~6~-[(9Н-флуорен-9-илметокси)карбонил]-N~2~-(гидроксиацетил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (29)
В раствор гидроксиуксусной кислоты (14,7 мг, 0,193 ммоль) в 0,3 мл N,N-диметилформамида добавляли гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида (EDCI, 7,99 мг, 0,0417 ммоль), этил(гидроксиимино)цианоацетат (Oxyma, 20 мг, 0,14 ммоль) и N-метилморфолин (40 мкл, 37 мг, 0,36 ммоль). Полученный желтый раствор перемешивали в течение 20 минут и затем добавляли раствор N~6~-[(9H-флуорен-9-илметокси)карбонил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (28, 50 мг, 0,032 ммоль) в 0,5 мл N,N-диметилформамида. Эту смесь контролировали методом ЖХ/МС и перемешивали в течение часа при комнатной температуре и затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод F). Фракции, содержащие продукт, лиофилизировали с получением после лиофилизации 12,2 мг (24%). ЖХ/МС m/z 1557,8 [М+Н+]; время удерживания = 0,98 минут. Аналитическая ВЭЖХ: время удерживания = 7.371 минут.
Стадия 5: Синтез N~2~-(гидроксиацетил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (30)
В раствор N~6~-[(9Н-флуорен-9-илметокси)карбонил]-N~2~-(гидроксиацетил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (29, 12,2 мг, 0,00784 ммоль) в 0,3 мл N,N-диметилформамида добавляли пиперидин (0,1 мл, 90 мг, 1 ммоль), и эту смесь перемешивали при комнатной температуре в течение часа. Раствор затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод Е). Фракции, содержащие продукт, лиофилизировали с получением 8 мг (70%) целевого продукта. ЖХ/МС m/z 1334,8 [М+Н+]; время удерживания = 0,76 минут. Аналитическая ВЭЖХ: время удерживания = 5,788 минут.
Пример 26: Получение N~2~-(метилкарбамоил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (33)
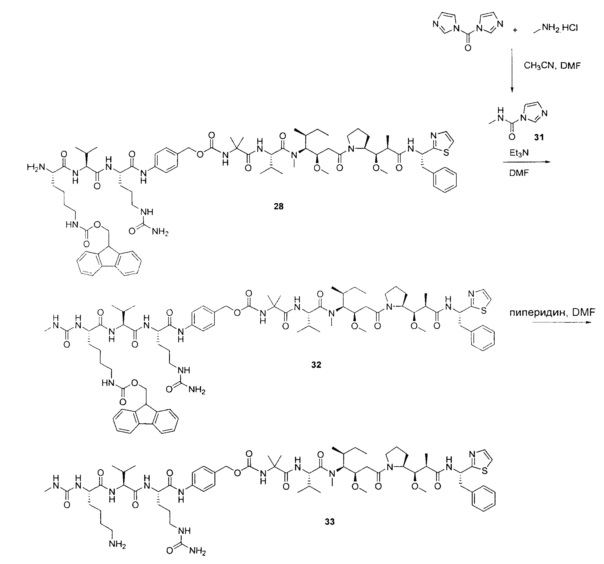
Стадия 1: Синтез N-метил-1Н-имидазол-1-карбоксамида (31)
Смесь 1,1'-карбонилдиимидазола (1,76 г, 10,7 ммоль) и гидрохлорида метиламина (661 мг, 9,7 ммоль) растворяли в 1,8 мл N,N-диметилформамида и 5,5 мл ацетонитрила. Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 3,25 часов при комнатной температуре. Реакционную смесь затем концентрировали и очищали хроматографией на силикагеле, используя элюирование с градиентом от 0% до 10% метанола в дихлорметане, с получением 696 мг (57%) целевого продукта. ЖХ/МС m/z 126,0 [М+Н+]; время удерживания = 0,17 минут. 1Н ЯМР (400 МГц, DMSO-d6) δ=8.22 (s, 1Н), 7.65 (s, 1Н), 7.03 (s, 1Н), 2.83 (d, J=4.3 Гц, 3H).
Стадия 2: Синтез N~6~-[(9Н-флуорен-9-илметокси)карбонил]-N~2~-(метил карбамоил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (32)
В смесь N-метил-1Н-имидазол-1-карбоксамида (40,1 мг, 0,321 ммоль) и N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (28, 50 мг, 0,032 ммоль) в 0,5 мл N,N-диметилформамида добавляли триэтиламин (44,7 мкл, 32,5 мг, 0,321 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 1,5 часов при комнатной температуре, а затем разбавляли диметилсульфоксидом и очищали обращенно-фазной ВЭЖХ (Метод L) с получением после лиофилизации 8,8 мг (18%) целевого продукта. ЖХ/МС m/z 1556,8 [М+Н+]; время удерживания = 0,99 минут. Аналитическая ВЭЖХ: время удерживания = 7,521 минут.
Стадия 3: Синтез N~2~-(метилкарбамоил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (33)
В раствор N~6~-[(9Н-флуорен-9-илметокси)карбонил]-N~2~-(метилкарбамоил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (32, 8,8 мг, 0,0057 ммоль) в 0,3 мл N,N-диметилформамида добавляли пиперидин (0,1 мл, 90 мг, 1 ммоль), и эту смесь перемешивали при комнатной температуре в течение 1,5 часов. Раствор затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод Е, затем М). Фракции, содержащие продукт, лиофилизировали с получением 4,6 мг (58%) целевого продукта. ЖХ/МС m/z 1333,7 [М+Н+]; время удерживания = 0,76 минут. Аналитическая ВЭЖХ: время удерживания = 5,825 минут.
Пример 27: Получение N-метилглицил-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (34)
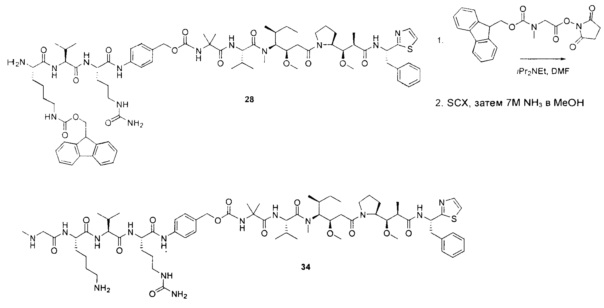
В раствор 2,5-диоксопирродидин-1-ил-N-[(9Н-флуорен-9-илметокси)карбонил]-N-метилглицината (11,9 мг, 0,0416 ммоль) в 0,1 мл N,N-диметилформамида добавляли N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамид (28, 50 мг, 0,032 ммоль) и N,N-диизопропилэтиламин (17 мкл, 13 мг, 0,096 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение часа при комнатной температуре (ЖХ/МС m/z 1670,71 [М+Н+]; время удерживания = 1,03 минут). Смесь концентрировали под сильным потоком азота, и остаток растворяли в метаноле и пропускали через SCX колонку (предварительно промытую 2 колоночными объемами метанола). Картридж промывали колоночным объемом метанола, и продукт оставляли на колонке на ночь. Колонку промывали 7М раствором аммиака в метаноле, после концентрирования остаток очищали обращенно-фазной ВЭЖХ (Метод L). Фракции, содержащие продукт, лиофилизировали с получением 4,2 мг (8%) целевого продукта. ЖХ/МС m/z 1347,51 [М+Н+]; время удерживания = 0,68 минут. Аналитическая ВЭЖХ: время удерживания = 5,486 минут.
Пример 28: Получение N,N-диметилглицил-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (35)

В раствор N,N-диметилглицина (4,5 мг, 0,044 ммоль) в 0,1 мл N,N-диметилформамида добавляли 0,1 мл исходного раствора гидрохлорида N-(3-диметиламинопропил)-N'-этилкарбодиимида (EDCI, 79,9 мг, 0,417 ммоль), этил(гидроксиимино)цианоацетат (Оксиmа, 10 мг, 0,07 ммоль) и N-метилморфолин (10 мкл, 9 мг, 0,09 ммоль) в 1 мл N,N-диметилформамида. Полученный желтый раствор перемешивали в течение 15 минут, затем добавляли раствор N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (28, 50 мг, 0,032 ммоль) в 0,5 мл N,N-диметилформамида. Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 40 минут при комнатной температуре и снова добавляли N-метилморфолин (50 мкл, 45 мг, 0,45 ммоль). Смесь перемешивали в течение 16 часов, концентрировали и очищали обращенно-фазной ВЭЖХ (Метод L). Фракции, содержащие продукт, лиофилизировали с получением 5,4 мг (11%) целевого продукта. ЖХ/МС m/z 1361,8 [М+Н+]; время удерживания = 0,71 минут. Аналитическая ВЭЖХ: время удерживания = 5,481 минут.
Пример 29: Получение глицил-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (37)
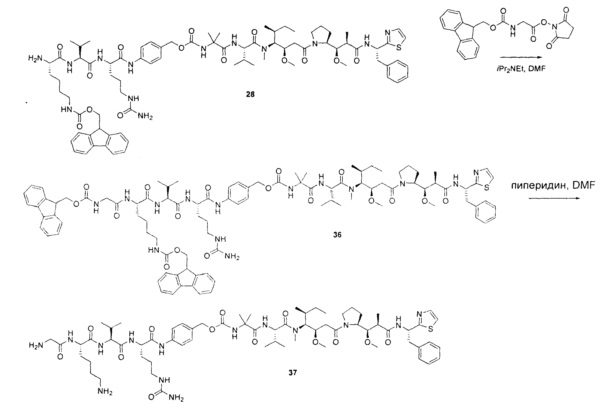
Стадия 1: Синтез N-[(9Н-флуорен-9-илметокси)карбонил]глицил-N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (36)
В раствор N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (28, 65,0 мг, 0,043 ммоль) в 0,650 мл N,N-диметилформамида добавляли 2,5-диоксопирродидин-1-ил-N-[(9Н-флуорен-9-илметокси)карбонил]глицинат (68,4 мг, 0,173 ммоль) и N,N-диизопропилэтиламин (23,1 мг, 0,173 ммоль, 30,8 мкл). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 2 часов при комнатной температуре и напрямую очищали обращенно-фазной ВЭЖХ (Метод N). Фракции, содержащие продукт, лиофилизировали с получением 22,3 мг (29%) целевого продукта. ЖХ/МС m/z 1777,2 [М+Н+]; время удерживания = 1,10 минут. Аналитическая ВЭЖХ: время удерживания = 8,544 минут.
Стадия 2: Синтез глицил-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил~L-орнитинамида (37)
В раствор N-[(9Н-флуорен-9-илметокси)карбонил]глицил-N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (36, 20 мг, 0,011 ммоль) в 0,6 мл N,N-диметилацетамида добавляли пиперидин (0,3 мл, 270 мг, 3 ммоль), и эту смесь перемешивали при комнатной температуре в течение 15 минут. Раствор затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод L, затем М). Фракции, содержащие продукт, лиофилизировали с получением 14,4 мг (90%) целевого продукта. ЖХ/МС m/z 1333,8 [М+Н+]; время удерживания = 0,70 минут. Аналитическая ВЭЖХ: время удерживания = 5,825 минут.
Пример 30: Получение N~2~-[(3-метилоксетан-3-ил)карбонил]-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (39)
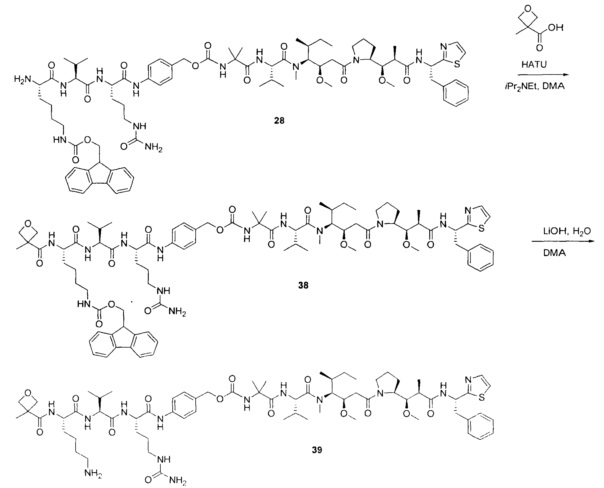
Стадия 1: Синтез N~6~-[(9Н-флуорен-9-илметокси)карбонил]-N~2~-[(3-метилоксетан-3-ил)карбонил]-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (38)
В бане лед-вода в раствор N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (28, 50,0 мг, 0,031 ммоль) в 0,5 мл N,N-диметилацетамида добавляли 3-метилоксетан-3-карбоновую кислоту (22 мг, 0,19 ммоль), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 65 мг, 0,17 ммоль) и N,N-диизопропилэтиламин (35 мкл, 26 мг, 0,20 ммоль). Смесь перемешивали в течение дополнительных 40 минут в бане лед-вода. Раствор затем напрямую очищали обращенно-фазной ВЭЖХ (Метод Р). Фракции, содержащие продукт, лиофилизировали с получением 37,4 мг (55%) целевого продукта. ЖХ/МС m/z 1597,8 [М+Н+]; время удерживания = 1,00 минут. Аналитическая ВЭЖХ: время удерживания = 7,567 минут.
Стадия 2: Синтез N~2~-[(3-метилоксетан-3-ил)карбонил]-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (39)
В раствор N~6~-[(9Н-флуорен-9-илметокси)карбонил]-N~2-[(3-метилоксетан-3-ил)карбонил]-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (38, 27,4 мг, 0,0172 ммоль) в 0,5 мл N,N-диметилацетамида добавляли водный раствор гидроксида лития (411 мкл, 41,1 мг, 0,0343 ммоль), и эту смесь перемешивали при комнатной температуре в течение часа. Раствор затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод L). Фракции, содержащие продукт, лиофилизировали с получением 9,3 мг (36%) целевого продукта. ЖХ/МС m/z 1374,4 [М+Н+]; время удерживания = 0,77 минут. Аналитическая ВЭЖХ: время удерживания = 5,841 минут.
Пример 31: Получение 2-метилаланил-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (41)
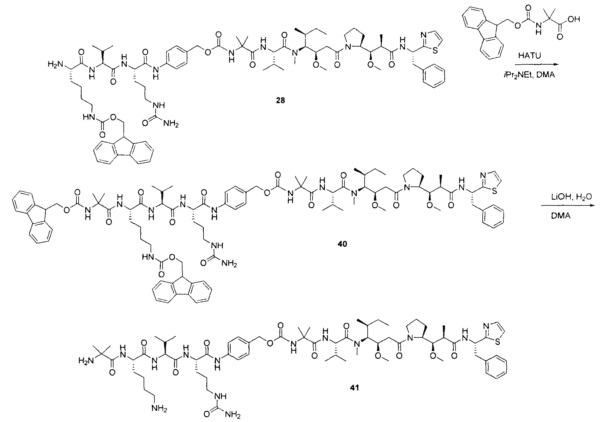
Стадия 1: Синтез N-[(9Н-флуорен-9-илметокси)карбонил]-2-метилаланил-N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамид (40)
В бане лед-вода в раствор N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5-карбамоил-L-орнитинамида (28, 50,0 мг, 0,031 ммоль) в 0,5 мл N,N-диметилацетамида добавляли N-[(9Н-флуорен-9-илметокси)карбонил]-2-метилаланин (17,5 мг, 0,0538 ммоль), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 25,9 мг, 0,0681 ммоль) и N,N-диизопропилэтиламин (25 мкл, 19 мг, 0,14 ммоль). Эту смесь перемешивали в течение дополнительных 40 минут в бане лед-вода. Раствор затем напрямую очищали обращенно-фазной ВЭЖХ (Метод Q). Фракции, содержащие продукт, лиофилизировали с получением 56 мг (100%) целевого продукта. ЖХ/МС m/z 1806,9 [М+Н+]; время удерживания = 1,12 минут. Аналитическая ВЭЖХ: время удерживания = 8,817 минут.
Стадия 2: Синтез 2-метилаланил-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (40)
В раствор N-[(9Н-флуорен-9-илметокси)карбонил]-2-метилаланил-N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (40, 56 мг, 0,031 ммоль) в 0,5 мл N,N-диметилацетамида добавляли водный раствор гидроксида лития (100 мкл, 10 мг, 0,4 ммоль), и эту смесь перемешивали при комнатной температуре в течение часа. Раствор затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод L). Фракции, содержащие продукт, лиофилизировали с получением 7,6 мг (15%) целевого продукта. ЖХ/МС m/z 1384,2 [M+Na+]; время удерживания = 0,71 минут. Аналитическая ВЭЖХ: время удерживания = 5,467 минут.
Пример 32: Получение N~2~-(метоксикарбонил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (43)
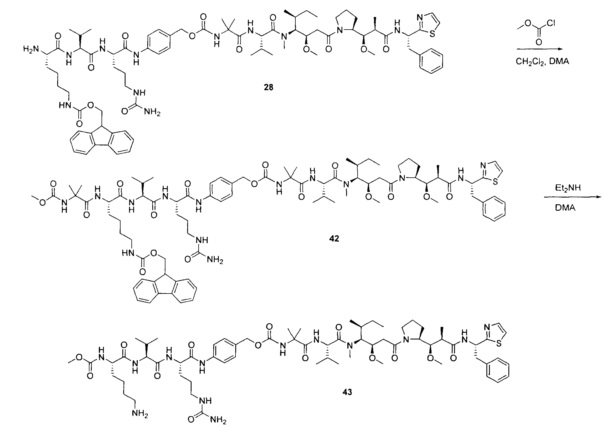
Стадия 1: Синтез N-(метоксикарбонил)-2-метилаланил-N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (42)
В раствор N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (28, 30,0 мг, 0,019 ммоль) в 0,3 мл N,N-диметилацетамида и 0,2 мл дихлорметана добавляли метилхлорформиат (29 мкл, 35,2 мг, 0,372 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 2 часов при комнатной температуре. Через 2 ч добавляли N,N-диизопропилэтиламин (10,0 мкл, 7,5 мг, 0,056 ммоль), и смесь перемешивали в течение еще 16 часов. ЖХ/МС все еще показывала, что реакция не заверешена, поэтому добавляли метилхлорформиат (60 мкл, 73 мг, 0,78 ммоль) и затем N,N-диизопропилэтиламин (10 мкл, 7,5 мг, 0,056 ммоль). После перемешивания при комнатной температуре в течение еще 4 часов смесь напрямую очищали обращенно-фазной ВЭЖХ (Метод Q). Фракции, содержащие продукт, лиофилизировали с получением 6,4 мг (22%) целевого продукта. ЖХ/МС m/z 1557,9 [М+Н+]; время удерживания = 1,05 минут. Аналитическая ВЭЖХ: время удерживания = 7,709 минут.
Стадия 2: Синтез N~2~-(метоксикарбонил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (43)
В раствор N-(метоксикарбонил)-2-метилаланил-N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (42, 6,4 мг, 0,0041 ммоль) в 0,5 мл N,N-диметилацетамида добавляли диэтиламин (0,3 мл, 200 мг, 3 ммоль), и эту смесь перемешивали при комнатной температуре в течение 3 часов. Раствор затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод L). Фракции, содержащие продукт, лиофилизировали с получением 2,6 мг (44%) целевого продукта. ЖХ/МС m/z 1334,8 [М+Н+]; время удерживания = 0,83 минут. Аналитическая ВЭЖХ: время удерживания = 5,908 минут.
Пример 33: Получение L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (44)
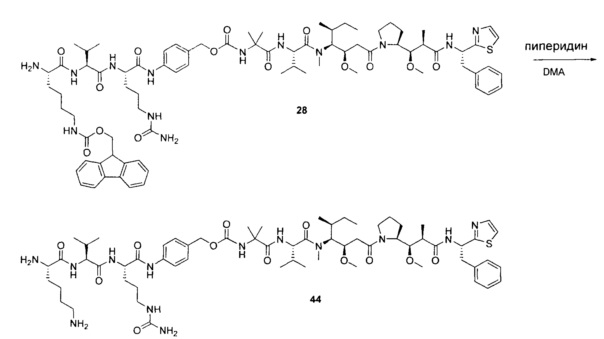
В раствор N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (28, 20,0 мг, 0,012 ммоль) в 0,2 мл N,N-диметилацетамида добавляли пиперидин (0,1 мл, 90 мг, 1 ммоль), и эту смесь перемешивали при комнатной температуре в течение 4 часов. Раствор затем напрямую очищали обращенно-фазной ВЭЖХ (Метод L, затем R). Фракции, содержащие продукт, лиофилизировали с получением 7,6 мг (44%) целевого продукта. ЖХ/МС m/z 1276,7 [М+Н+]; время удерживания = 0,70 минут. Аналитическая ВЭЖХ: время удерживания = 5,516 минут.
Пример 34: Получение L-аланил-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (46)

Стадия 1: Синтез N-[(9Н-флуорен-9-илметокси)карбонил]-L-аланил-N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (45)
В бане лед-вода в раствор N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5-карбамоил-L-орнитинамида (28, 8,7 мг, 0,0054 ммоль) в 0,5 мл N,N-диметилацетамида добавляли 2,5-диоксопирродидин-1-ил-N-[(9Н-флуорен-9-илметокси)карбонил]-L-аланинат ((8,81 мг, 0,0216 ммоль) и N,N-диизопропилэтиламин (10,0 мкл, 7,5 мг, 0,056 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 16 часов при комнатной температуре и напрямую очищали обращенно-фазной ВЭЖХ (Метод S). Фракции, содержащие продукт, лиофилизировали с получением 5,3 мг (55%) целевого продукта. ЖХ/МС m/z 1791,7 [М+Н+]; время удерживания = 1,11 минут. Аналитическая ВЭЖХ: время удерживания = 8,655 минут.
Стадия 2: Синтез L-аланил-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (46)
В раствор N-[(9Н-флуорен-9-илметокси)карбонил]-L-аланил-N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (45, 5,3 мг, 0,0030 ммоль) в 0,5 мл N,N-диметилацетамида добавляли водный раствор гидроксида лития (210 мкл, 20 мг, 0,9 ммоль), и эту смесь перемешивали при комнатной температуре в течение часа. Раствор затем концентрировали и очищали обращенно-фазной ВЭЖХ (Метод L). Фракции, содержащие продукт, лиофилизировали с получением 3,4 мг (73%) целевого продукта. ЖХ/МС m/z 1347,7 [М+Н]+; время удерживания = 0,69 минут. Аналитическая ВЭЖХ: время удерживания = 5,487 минут.
Пример 35: Получение L-аланил-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (48)
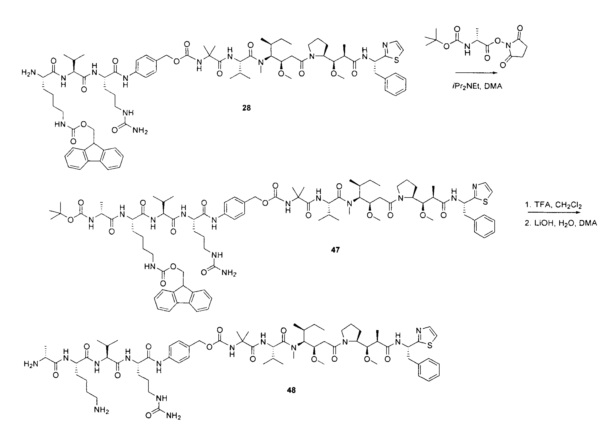
Стадия 1: Синтез N-(трет-бутоксикарбонил)-D-аланил-N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (47)
В бане лед-вода в раствор N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (28, 50 мг, 0,03 ммоль) в 0,5 мл N,N-диметилацетамида добавляли 2,5-диоксопирродидин-1-ил-N-(трет-бутоксикарбонил)-D-аланинат (15,7 мг, 0,0548 ммоль) и N,N-диизопропилэтиламин (22,0 мкл, 16 мг, 0,12 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 16 часов при комнатной температуре и напрямую очищали обращенно-фазной ВЭЖХ (Метод S). Фракции, содержащие продукт, лиофилизировали с получением 20,0 мг (40%) целевого продукта. ЖХ/МС m/z 1791,7 [М+Н+]; время удерживания = 1,08 минут. Аналитическая ВЭЖХ: время удерживания = 8,024 минут.
Стадия 2: Синтез D-аланил-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (48)
В раствор N-(трет-бутоксикарбонил)-D-аланил-N~6~-[(9Н-флуорен-9-ил метокси)карбонил]-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (47, 20,0 мг, 0,012 ммоль) в 1 мл ацетонитрила добавляли 0,5 мл трифторуксусной кислоты. Эту смесь перемешивали при комнатной температуре в течение 15 минут, затем концентрировали. Остаток растворяли в 0,5 мл N,N-диметилацетамида и охлаждали в бане лед-вода. В него добавляли водный раствор гидроксида лития (50,3 мкл, 5,3 мг, 0,12 ммоль), и эту смесь перемешивали при комнатной температуре в течение 30 минут. Раствор затем напрямую очищали обращенно-фазной ВЭЖХ (Метод L). Фракции, содержащие продукт, лиофилизировали с получением 8,9 мг (47%) целевого продукта. ЖХ/МС m/z 1347,9 [М+Н]+; время удерживания = 0,77 минут. Аналитическая ВЭЖХ: время удерживания = 5,429 минут.
Пример 36: Получение L-альфа-аспартил-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (50)
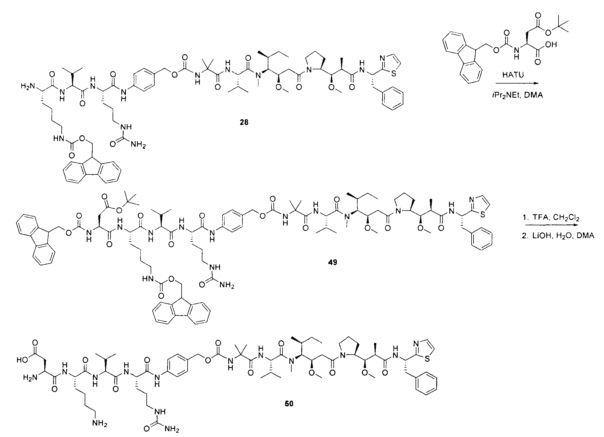
Стадия 1: Синтез трет-бутил-(6S,9S,12S,15S)-1-амино-6-({4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}карбамоил)-15-{[(9Н-флуорен-9-илметокси)карбонил]амино}-12-(4-{[(9Н-флуорен-9-илметокси)карбонил]амино}бутил)-1,8,11,14-тетраоксо-9-(пропан-2-ил)-2,7,10,13-тетраазагептадекан-17-оата (49)
В бане лед-вода в раствор N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (28, 50 мг, 0,03 ммоль) в 0,5 мл N,N-диметилацетамида добавляли (2S)-4-трет-бутокси-2-{[(9Н-флуорен-9-илметокси)карбонил]амино}-4-оксобутановую кислоту (20,9 мг, 0,0722 ммоль), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 28,0 мг, 0,074 ммоль) и N,N-диизопропилэтиламин (22,0 мкл, 16 мг, 0,12 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение часа и напрямую очищали обращенно-фазной ВЭЖХ (Метод Q). Фракции, содержащие продукт, лиофилизировали с получением 41 мг (70%) целевого продукта. ЖХ/МС m/z 1771,2 [М+Н+]; время удерживания = 1,16 минут. Аналитическая ВЭЖХ: время удерживания = 8,591 минут.
Стадия 2: Синтез L-альфа-аспартил-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (50)
В раствор трет-бутил-(6S,9S,12S,15S)-1-амино-6-({4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}карбамоил)-15-{[(9Н-флуорен-9-илметокси)карбонил]амино}-12-(4-{[(9Н-флуорен-9-илметокси)карбонил]амино}бутил)-1,8,11,14-тетраоксо-9-(пропан-2-ил)-2,7,10,13-тетраазагептадекан-17-оата (49, 41,0 мг, 0,023 ммоль) в 1 мл ацетонитрила добавляли 0,5 мл трифторуксусной кислоты. Эту смесь перемешивали при комнатной температуре в течение 25 минут (ЖХ/МС m/z 1671,0 [М+Н]+; время удерживания = 0,93 минут), затем концентрировали. Остаток растворяли в 0,5 мл N,N-диметилацетамида и охлаждали в бане лед-вода. К нему добавляли водный раствор гидроксида лития (50,3 мкл, 5,3 мг, 0,12 ммоль), и эту смесь перемешивали при комнатной температуре в течение 20 минут. Раствор затем напрямую очищали обращенно-фазной ВЭЖХ (Метод Q). Фракции, содержащие продукт, лиофилизировали с получением 1,1 мг (3%) целевого продукта. ЖХ/МС m/z 1392,1 [М+Н]+; время удерживания = 0,80 минут. Аналитическая ВЭЖХ: время удерживания = 5,309 минут.
Пример 37: Получение N-ацетилглицил-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (52)
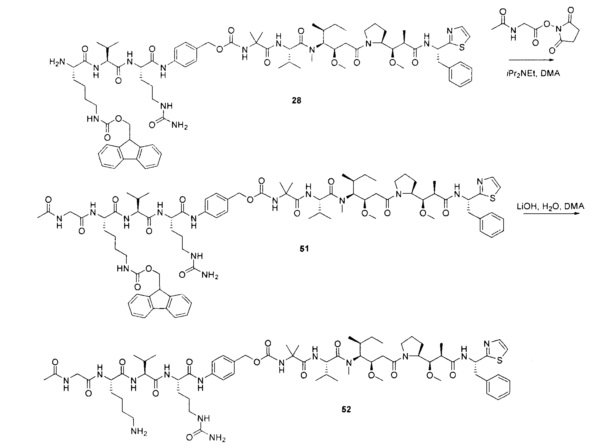
Стадия 1: Синтез N-ацетилглицил-N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (51)
В бане лед-вода в раствор N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (28, 30 мг, 0,02 ммоль) в 0,3 мл N,N-диметилацетамида добавляли 2,5-диоксопирродидин-1-ил-N-ацетилглицинат (13,7 мг, 0,0640 ммоль) и N,N-диизопропилэтиламин (22 мкл, 16 мг, 0,12 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 16 часов при комнатной температуре и напрямую очищали обращенно-фазной ВЭЖХ (Метод Q). Фракции, содержащие продукт, лиофилизировали с получением 17,4 мг (60%) целевого продукта. ЖХ/МС m/z 1598,9 [М+Н+]; время удерживания = 1,04 минут. Аналитическая ВЭЖХ: время удерживания = 7,326 минут.
Стадия 2: Синтез N-ацетилглицил-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (52)
В раствор N-ацетилглицил-N~6~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (51, 17,4 мг, 0,0109 ммоль) в 0,5 мл N,N-диметилацетамида добавляли водный раствор гидроксида лития (100 мкл, 10 мг, 0,2 ммоль), и эту смесь перемешивали при комнатной температуре в течение 20 минут. Раствор затем напрямую очищали обращенно-фазной ВЭЖХ (Метод В). Фракции, содержащие продукт, лиофилизировали с получением 9,4 мг (58%) целевого продукта. ЖХ/МС m/z 1398,1 [M+Na+]; время удерживания = 0,84 минут. Аналитическая ВЭЖХ: время удерживания = 5,715 минут.
Пример 38: Получение L-серил-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (57)
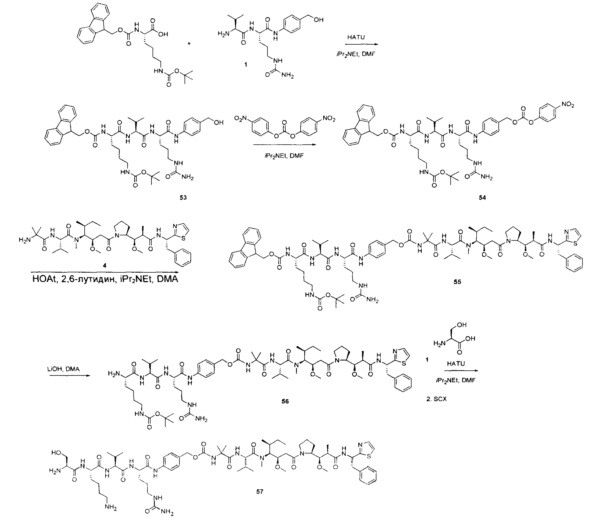
Стадия 1: Синтез N~6~-(трет-бутоксикарбонил)-N~2~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизилвалил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (53)
В бане лед-вода в раствор N~6~-(трет-бутоксикарбонил)-N~2~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизина (1090 мг, 2,32 ммоль) в 15 мл N,N-диметилформамида добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 882 мг, 2,32 ммоль). Через 40 минут добавляли 1-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамид (1, 800 мг, 2,11 ммоль), затем N,N-диизопропилэтиламин (1,114 мл, 817 мг, 6,32 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 2 часов при комнатной температуре. Реакционную смесь затем вливали в метил-трет-бутиловый эфир (150 мл), перемешивали в течение 15 минут, затем фильтровали. Полученное твердое вещество промывали смесью дихлорметан:метил-трет-бутиловый эфир:метанол (10:10:2) с получением неочищенного продукта (1,3 г) в виде серого твердого вещества. Это твердое ведщество очищали обращенно-фазной ВЭЖХ (Метод T). Фракции, содержащие продукт, лиофилизировали с получением 570 мг (33%) целевого продукта. Аналитическая ВЭЖХ: время удерживания = 4,55 минут. 1Н ЯМР (400 МГц, DMSO-d6) δ=9.97 (br. s., 1Н), 8.14 (d, J=6.5 Гц, 1H), 7.90 (d, J=7.5 Гц, 2H), 7.73 (br. s., 3H), 7.54 (d, J=8.5 Гц, 3H), 7.42 (t, J=7.3 Гц, 2H), 7.33 (t, J=7.3 Гц, 2H), 7.23 (d, J=8.0 Гц, 2H), 6.78 (br. s., 1H), 5.98 (br. s., 1H), 5.43 (br. s., 2H), 5.11 (br. s., 1H), 4.45-4.34 (m, 3H), 4.34-4.15 (m, 4H), 4.02 (br. s., 1H), 3.02 (d, J=5.5 Гц, 1H), 2.89 (br. s., 3H), 1.98 (d, J=6.5 Гц, 1H), 1.61 (br. s., 3H), 1.52 (br. s., 1H), 1.42-1.30 (m, 13H), 1.24 (br. s., 2H), 0.83 (d, J=12.0 Гц, 3H), 0.85 (d, J=11.5 Гц, 3H).
Стадия 2: Синтез N~6~-(трет-бутоксикарбонил)-N~2~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизилвалил-N~5~-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]-L-орнитинамида (54)
В бане лед-вода в раствор N~6~-(трет-бутоксикарбонил)-N~2~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизилвалил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (53, 570 мг, 0,687 ммоль) и пара-нитрофенилкарбоната (418 мг, 1,37 ммоль) в 10 мл N,N-диметилформамида добавляли N,N-диизопропилэтиламин (257 мкл, 178 мг, 1,37 ммоль), и эту смесь перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь затем вливали в метил-трет-бутиловый эфир (150 мл), перемешивали в течение 15 минут, затем фильтровали. Полученное твердое вещество диспергировали в дихлорметане (20 мл) в течение 20 минут, затем фильтровали с получением 400 мг (58%) целевого продукта. ЖХ/МС m/z 996,0 [М+Н+]; время удерживания = 4,829 минут. Аналитическая ВЭЖХ: время удерживания = 5,43 минут. 1Н ЯМР (400 МГц, DMSO-d6) δ=10.14 (s, 1Н), 8.36-8.29 (m, 2Н), 8.18 (d, J=7.5 Гц, 1Н), 7.90 (d, J=7.5 Гц, 2Н), 7.74 (d, J=7.0 Гц, 2Н), 7.66 (d, J=8.0 Гц, 2Н), 7.62-7.50 (m, 2Н), 7.42 (d, J=7.0 Гц, 3H), 7.38-7.11 (m, 3H), 6.78 (br. s., 1Н), 6.00 (br. s., 1Н), 5.44 (br. s., 2Н), 5.26 (s, 2Н), 4.39 (br. s., 1 Н), 4.35-4.16 (m, 4Н), 4.04 (br. s., 1Н), 3.05 (d, J=7.0 Гц, 1Н), 2.90 (br. s., 3H), 2.74 (s, 1Н), 2.34 (br. s., 1H), 2.10 (s, 1H), 1.99 (d, J=6.0 Гц, 1H), 1.68 (br. s., 1H), 1.62 (br. s., 2H), 1.53 (br. s., 2H), 1.38 (s, 11H), 1.42-1.20 (m, 1H), 1.42-1.20 (m, 1H), 0.85 (d, J=12.5 Гц, 3H), 0.86 (d, J=12.5 Гц, 3H).
Стадия 3: Синтез N~6~-(трет-бутоксикарбонил)-N~2~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (55)
В раствор 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (4, 440 мг, 0,59 ммоль) и N~6~-(трет-бутоксикарбонил)-N~2~-[(9Н-флуорен-9-илметокси)карбонил]-L-лизилвалил-N~5~-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]-L-орнитинамида (54, 658,0 мг, 0,661 ммоль) в 1,3 мл N,N-диметилацетамида добавляли 2,6-лутидин (137 мкл, 130 мг, 1,2 ммоль), N,N-диизопропилэтиламин (207,0 мкл, 160 мг, 1,2 ммоль) и 3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ол (HOAt, 92,1 мг, 0,677 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 2,5 часов при 50°С и затем при комнатной температуре в течение 16 часов. Реакционную смесь затем очищали обращенно-фазной хроматографией (Метод U) с получением после лиофилизации 630 мг (67%) целевого продукта в виде белого твердого вещества. ЖХ/МС m/z 1600,6 [М+Н+]; время удерживания = 1,07 минут. Аналитическая ВЭЖХ: время удерживания = 8,213 минут.
Стадия 4: Синтез N~6~-(трет-бутоксикарбонил)-1-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (56)
В бане лед-вода в раствор N~6~-(трет-бутоксикарбонил)-N~2~-[(9Н-флуорен-9-ил метокси)карбонил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (55, 630 мг, 0,394 ммоль) в 5 мл N,N-диметилацетамида добавляли раствор гидроксида лития (29,4 мг, 1,23 ммоль) в 0,5 мл воды, и эту смесь перемешивали при комнатной температуре в течение 30 минут, после чего добавляли гидроксид лития (40,4 мг, 1,69 ммоль). В сумме через 3 часа раствор охлаждали в бане лед-вода, затем гасили уксусной кислотой (0,2 мл) и очищали обращенно-фазной ВЭЖХ (Method 2). Фракции, содержащие продукт, лиофилизировали с получением 350 мг (60%) целевого продукта. ЖХ/МС m/z 1377,6 [М+Н+]; время удерживания = 0,77 минут. Аналитическая ВЭЖХ: время удерживания = 6,181 минут.
Стадия 5: Синтез L-серил-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (57)
В бане лед-вода в раствор N~6~-(трет-бутоксикарбонил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (56, 50,0 мг, 0,0335 ммоль) в 0,5 мл N,N-диметилацетамида добавляли N-(трет-бутоксикарбонил)-L-серин (6,88 мг, 0,0335 ммоль) в 0,1 мл N,N-диметилацетамида, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 15,3 мг, 0,0402 ммоль) в 0,1 мл N,N-диметилацетамида и N,N-диизопропилэтиламин (17,9 мкл, 13,4 мг, 0,101 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 45 минут при комнатной температуре, затем концентрировали под сильным потоком азота с получением неочищенного остатка (ЖХ/МС m/z 1565,6 [М+Н+], время удерживания = 0,96 минут). Этот желтый остаток растворяли в 1,2 мл метанола и наносили поровну на два SCX картриджа, предварительно элюированных 3 колоночными объемами метанола. Колонки промывали 3 колоночными объемами метанола (следы продукта отсутствовали в промывочных растворах согласно ЖХ/МС). Продукт оставляли на колонке на ночь. Целевой продукт элюировали 2 колоночными объемами 1 н. триэтиламина в метаноле. После концентрирования при пониженном давлении остаток затем очищали обращенно-фазной хроматографией (Метод V) с получением после лиофилизации 8,3 мг (16%) целевого продукта. ЖХ/МС m/z 1364,5 [М+Н+], время удерживания = 0,68 минут. Аналитическая ВЭЖХ: время удерживания = 5,464 минут.
Пример 39: Получение D-серил-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (58)
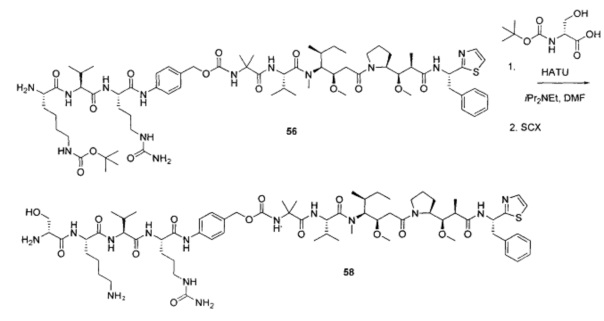
В бане лед-вода в раствор N~6~-(трет-бутоксикарбонил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (56, 50,0 мг, 0,0335 ммоль) в 0,5 мл N,N-диметилацетамида добавляли N-(трет-бутоксикарбонил)-D-серин (6,88 мг, 0,0335 ммоль) в 0,1 мл N,N-диметилацетамида, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 15,3 мг, 0,0402 ммоль ммоль) в 0,1 мл N,N-диметилацетамида и N,N-диизопропилэтиламин (17,9 мкл, 13,4 мг, 0,101 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 45 минут при комнатной температуре, затем концентрировали под сильным потоком азота с получением неочищенного остатка (ЖХ/МС m/z 1565,6 [М+Н+], время удерживания = 0,96 минут). Этот желтый остаток растворяли в 1,2 мл метанола и наносили поровну на два SCX картриджа, предварительно элюированных 3 колоночными объемами метанола. Колонки промывали 3 колоночными объемами метанола (следы продукта отсутствовали в промывочных растворах согласно ЖХ/МС). Продукт оставляли на колонке на ночь. Целевой продукт элюировали 2 колоночными объемами 1 н. триэтиламина в метаноле. После концентрирования при пониженном давлении остаток затем очищали обращенно-фазной хроматографией (Метод V) с получением после лиофилизации 10,4 мг (19%) целевого продукта. ЖХ/МС m/z 1364,5 [М+Н+], время удерживания = 0,67 минут. Аналитическая ВЭЖХ: время удерживания = 5,424 минут.
Пример 40: Получение N~2~-[(2S)-2-гидроксипропаноил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5-карбамоил-L-орнитинамида (59)
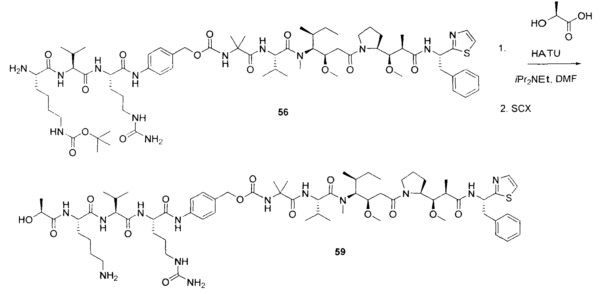
В бане лед-вода в раствор N~6~-(трет-бутоксикарбонил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (56, 50,0 мг, 0,0335 ммоль) в 0,5 мл N,N-диметилацетамида добавляли (2S)-2-гидроксипропановую кислоту (3,02 мг, 0,0335 ммоль) в 0,1 мл N,N-диметилацетамида, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 15,3 мг, 0,0402 ммоль) в 0,1 мл N,N-диметилацетамида и N,N-диизопропилэтиламин (17,9 мкл, 13,4 мг, 0,101 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 45 минут при комнатной температуре, затем концентрировали в сильном потоке азота с получением неочищенного остатка (ЖХ/МС m/z 1449,5 [М+Н+], время удерживания = 0,91 минут). Этот желтый остаток растворяли в 1,2 мл метанола и наносили поровну на два SCX картриджа, предварительно элюированных 3 колоночными объемами метанола. Колонки промывали 3 колоночными объемами метанола (следы продукта отсутствовали в промывочных растворах соглансно ЖХ/МС). Продукт оставляли на колонке на ночь. Целевые продукты элюировали 2 колоночными объемами 1 н. триэтиламина в метаноле. После концентрирования при пониженном давлении остаток затем очищали обращенно-фазной хроматографией (Метод V) с получением после лиофилизации 12,9 мг (26%) целевого продукта. ЖХ/МС m/z 1349,4 [М+Н+], время удерживания = 0,74 минут. Аналитическая ВЭЖХ: время удерживания = 5,813 минут.
Пример 41: Получение N~2~-[(2R)-2-гидроксипропаноил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (60)
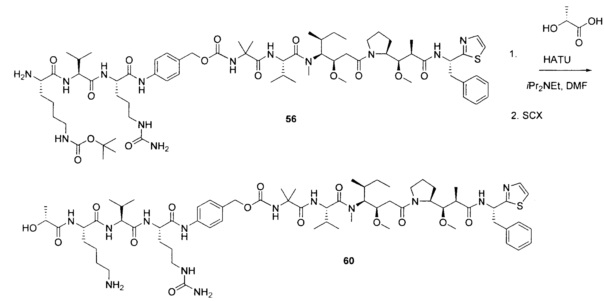
В бане лед-вода в раствор N~6~-(трет-бутоксикарбонил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (56, 50,0 мг, 0,0335 ммоль) в 0,5 мл N,N-диметилацетамида добавляли (2R)-2-гидроксипропановую кислоту (3,01 мг, 0,0334 ммоль) в 0,1 мл N,N-диметилацетамида, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 15,3 мг, 0,0402 ммоль ммоль) в 0,1 мл N,N-диметилацетамида и N,N-диизопропилэтиламин (17,9 мкл, 13,4 мг, 0,101 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 45 минут при комнатной температуре, затем концентрировали в сильном потоке азота с получением неочищенного остатка (ЖХ/МС m/z 1449,4 [М+Н+], время удерживания = 0,90 минут). Этот остаток затем очищали обращенно-фазной хроматографией (Метод V) с получением после лиофилизации 29,6 мг (60%) целевого продукта. ЖХ/МС m/z 1349,4 [М+Н+], время удерживания = 0,75 минут. Аналитическая ВЭЖХ: время удерживания = 5,740 минут.
Пример 42: Получение N~2~-[(2S)-2,3-дигидроксипропаноил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (62)
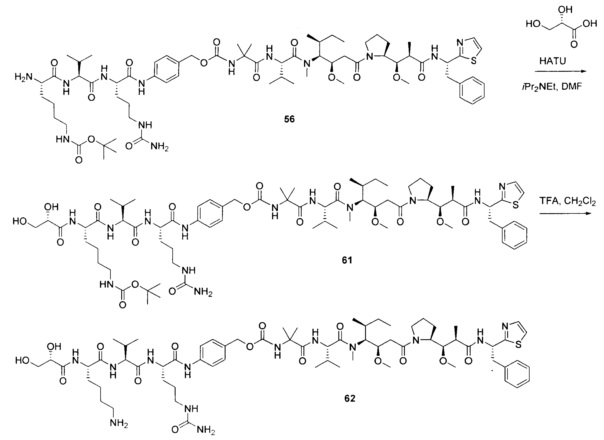
Стадия 1: Синтез N~6~-(трет-бутоксикарбонил)-N~2~-[(2S)-2,3-дигидроксипропаноил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (61)
В бане лед-вода в раствор N~6~-(трет-бутоксикарбонил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (56, 50,0 мг, 0,0335 ммоль) в 0,5 мл N,N-диметилацетамида добавляли (2S)-2,3-дигидроксипропановую кислотуу (4,02 мг, 0,0379 ммоль) в 0,1 мл N,N-диметилацетамида, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 15,3 мг, 0,0402 ммоль) в 0,1 мл N,N-диметилацетамида и N,N-диизопропилэтиламин (17,9 мкл, 13,4 мг, 0,101 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 45 минут при комнатной температуре, затем концентрировали в сильном потоке азота. Желтый остаток затем очищали обращенно-фазной хроматографией (Метод U) с получением после лиофилизации 10,8 мг (22%) целевого продукта. ЖХ/МС m/z 1465,5 [М+Н+], время удерживания = 0,89 минут. Аналитическая ВЭЖХ: время удерживания = 6,607 минут.
Стадия 2: Синтез N~2~-[(2S)-2,3-дигидроксипропаноил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (62)
В суспензию N~6~-(трет-бутоксикарбонил)-N~2~-[(2S)-2,3-дигидроксипропаноил]-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (61, 10,8 мг, 0,0074 ммоль) в 1 мл дихлорметана добавляли 0,5 мл трифторуксусной кислоты. Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 20 минут при комнатной температуре. Реакционную смесь затем концентрировали и очищали обращенно-фазной хроматографией (Метод V) с получением после лиофилизации 3,6 мг (33%). ЖХ/МС m/z 1365,4 [М+Н+]; время удерживания = 0,74 минут. Аналитическая ВЭЖХ: время удерживания = 5,749 минут.
Пример 43: Получение N~2~-(3-гидроксипропаноил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (64)
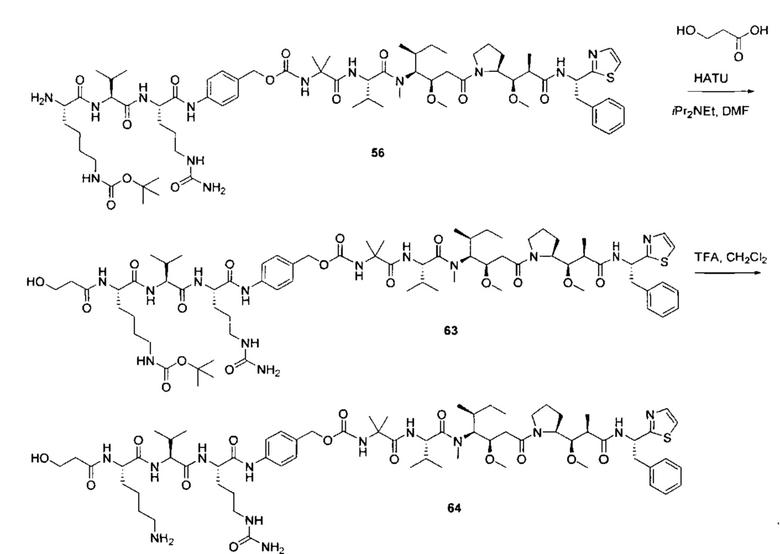
Стадия 1: Синтез N~6~-(трел-бутоксикарбонил)-N~2~-(3-гидроксипропаноил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (63)
В бане лед-вода в раствор N~6~-(трет-бутоксикарбонил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (56, 50,0 мг, 0,0335 ммоль) в 0,5 мл N,N-диметилацетамида добавляли 3-гидроксипропановую кислоту (4,01 мг, 0,0445 ммоль) в 0,1 мл N,N-диметилацетамида, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 15,3 мг, 0,0402 ммоль) в 0,1 мл N,N-диметилацетамида и N,N-диизопропилэтиламин (17,9 мкл, 13,4 мг, 0,101 ммоль). Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 45 минут при комнатной температуре, затем концентрировали в сильном потоке азота. Желтый остаток затем очищали обращенно-фазной хроматографией (Метод U) с получением после лиофилизации 13,9 мг (29%) целевого продукта. ЖХ/МС m/z 1449,6 [М+Н+], время удерживания = 0,91 минут. Аналитическая ВЭЖХ: время удерживания = 6,721 минут.
Стадия 2: Синтез N~2~-(3-гидроксипропаноил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (64)
В суспензию N~6~-(трет-бутоксикарбонил)-N~2~-(3-гидроксипропаноил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида (63, 13,9 мг, 0,0096 ммоль) в 1 мл дихлорметана добавляли 0,5 мл трифторуксусной кислоты. Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 20 минут при комнатной температуре. Реакционную смесь затем концентрировали и очищали обращенно-фазной хроматографией (Метод V) с получением после лиофилизации 4,1 мг (29%). ЖХ/МС m/z 1349,4 [М+Н+]; время удерживания = 0,75 минут. Аналитическая ВЭЖХ: время удерживания = 5,758 минут.
Пример 44: Получение N~2~-(метилсульфонил)лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида, трифторуксусной соли (70)
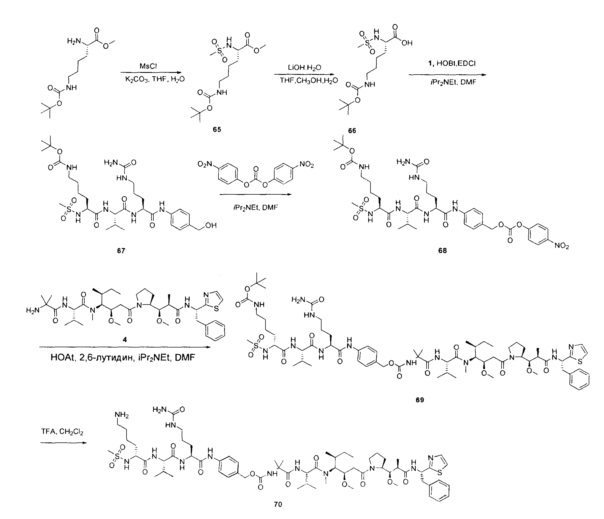
Стадия 1: Синтез метил-N~6~-(трет-бутоксикарбонил)-N~2~-(метилсульфонил)-L-лизината (65)
В охлажденный на люду раствор метил-N~6~-(трет-бутоксикарбонил)-L-лизината (2,0 г, 6,73 ммоль) в смеси тетрагидрофуран:вода (1:1, 20 мл) добавляли карбонат калия (4,6 г, 33,7 ммоль), затем по каплям добавляли мезилхлорид (0,62 мл, 8,08 ммоль). Эту смесь перемешивали при комнатной температуре в течение 3 часов, и смесь затем фильтровали. Фильтрат экстрагировали этилацетатом три раза. Объединенные экстракты промывали рассолом, сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле, используя элюирование с градиентом от 0% до 2% метанола в дихлорметане, с получением 2,0 г (87%) целевого продукта в виде бесцветного масла. 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ=5.23 (d, J=9.0 Гц, 1Н), 4.62 (br. s., 1Н), 4.10 (dt, J=5.0, 8.5 Гц, 1H), 3.78 (s, 3H), 3.16-3.04 (m, 2Н), 2.95 (s, 3H), 1.88-1.78 (m, 2Н), 1.77-1.64 (m, 1Н), 1.59-1.47 (m, 3H), 1.45 (s, 9Н).
Стадия 2: Синтез N~6~-(трет-бутоксикарбонил)-N~2~-(метилсульфонил)-L-лизина (66)
В перемешиваемый раствор метил-N~6~-(трет-бутоксикарбонил)-N~-(метилсульфонил)-L-лизината (65, 2,0 г, 5,90 ммоль) в смеси тетрагидрофуран:вода:метанол (1:1:1, 30 мл) добавляли моногидрат гидроксида лития (496 мг, 11,8 ммоль). Эту смесь перемешивали при комнатной температуре в течение 3 часов. Смесь концентрировали при пониженном давлении. Оставшийся водный слой экстрагировали дважды этилацетатом. Водный слой подкисляли до рН 3 1 н. водным раствором соляной кислоты и затем экстрагировали дважды смесью изопропиловый спирт:этилацетат (1:5). Объединеные органические экстракты промывали рассолом, сушили над сульфатом натрия и концентрировали при пониженном давлении с получением 1,3 г (68%) целевого продукта в виде белого твердого вещества. МС m/z 323,1 [M-Н+]; 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ=6.40 (br. s., 1Н), 5.81-5.66 (m, 1Н), 4.79 (br. s., 1Н), 3.23-3.06 (m, 2Н), 2.99 (s, 3H), 1.96-1.71 (m, 2Н), 1.55-1.39 (m, 13Н).
Стадия 3: Синтез N~6~-(трет-бутоксикарбонил)-N~2~-(метилсульфонил)-L-лизил-L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]орнитинамида (67)
В охлажденный на льду раствор N~6~-(трет-бутоксикарбонил)-N~2~-(метилсульфонил)-L-лизина (66, 1,0 г, 3,09 ммоль) и L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (1, 1,28 г, 3,40 ммоль) в 20 мл N,N-диметилформамида добавляли N,N-диизопропилэтиламин (600 мкл, 438 мг, 3,40 ммоль), гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида (649 мг, 3,40 ммоль) и гидрат 1-гидроксибензотриазола (459 мг, 3,40 ммоль). Смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 16 часов. Смесь затем по каплям добавляли в 150 мл трет-бутил-метилового эфира. Твердое вещество очищали обращенно-фазной ВЭЖХ. Фракции, содержащие продукт, лиофилизировали с получением 580 мг (28%) целевого продукта в виде белого твердого вещества.
Стадия 4: Синтез N~6~-(трет-бутоксикарбонил)-N~2~-(метилсульфонил)-L-лизил-L-валил-N~5~-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]орнитинамида (68)
В перемешиваемый раствор N~6~-(трет-бутоксикарбонил)-N~2~-(метилсульфонил)-L-лизил-L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]орнитинамида (67, 580 мг, 0,85 ммоль) и пара-нитрофенилкарбоната (770 мг, 2,56 ммоль) в 10 мл N,N-диметилформамида добавляли N,N-диизопропилэтиламин (451 мкл, 330 мг, 2,56 ммоль). Эту смесь перемешивали при комнатной температуре в течение 16 часов. Смесь по каплям добавляли в 150 мл трет-бутил-метилового эфира. Твердое вещество очищали обращенно-фазной ВЭЖХ (Метод XXX). Фракции, содержащие продукт, лиофилизировали с получением 200 мг (28%) целевого продукта в виде белого твердого вещества. МС m/z 851,4 [М+Н+]. 1Н ЯМР (400 МГц, DMSO-d6) δ=10.16 (s, 1Н), 8.32 (d, J=9.5 Гц, 2Н), 8.26 (d, J=6.5 Гц, 1Н), 7.95 (t, J=7.4 Гц, 1Н), 7.65 (d, J=8.5 Гц, 2Н), 7.58 (t, J=5.9 Гц, 2Н), 7.45 (d, J=9.0 Гц, 1Н), 7.42 (d, J=8.5 Гц, 2Н), 6.79 (t, J=5.5 Гц, 1Н), 6.00 (t, J=5.8 Гц, 1Н), 5.45 (s, 2Н), 4.40 (d, J=5.0 Гц, 1Н), 4.33-4.06 (m, 1Н), 3.84 (d, J=4.5 Гц, 1Н), 3.30 (br. s., 1Н), 3.09-2.99 (m, 1Н), 2.98-2.93 (m, 1Н), 2.93-2.86 (m, 2Н), 2.83 (s, 3H), 2.01 (d, J=6.5 Гц, 1Н), 1.59 (br. s., 3H), 1.47 (d, J=13.1 Гц, 2Н), 1.38 (s, 13Н), 0.89 (d, J=7.0 Гц, 3H), 0.85 (d, J=7.0 Гц, 3H).
Стадия 5: Синтез N~6~-(трет-бутоксикарбонил)-N~2~-(метилсульфонил)-D-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида (69)
В раствор 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (4, 52,0 мг, 0,070 ммоль) и N~6~-(трет-бутоксикарбонил)-N~2~-(метилсульфонил)-L-лизил-L-валил-N~5~-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]орнитинамида (68, 60,6 мг, 0,0710 ммоль) в 2 мл N,N-диметилацетамида добавляли 2,6-лутидин (31 мкл, 28,3 мг, 0,264 ммоль), N,N-диизопропилэтиламин (46 мкл, 34,1 мг, 0,264 ммоль) и 3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ол (9 мг, 0,066 ммоль). Эту смесь перемешивали в течение 16 часов при 45°С. Реакционную смесь затем очищали обращенно-фазной ВЭЖХ (Метод S) с получением после лиофилизации 70 мг (69%) целевого продукта в виде белого твердого вещества. ЖХ/МС m/z 1455,09 [М+Н+], 1453,77 [М-Н+]; время удерживания = 1,80 минут.
Стадия 6: Синтез N~2~-(метилсульфонил)лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида, трифторуксусной соли (70)
В раствор N~6~-(трет-бутоксикарбонил)-N~2~-(метилсульфонил)-D-лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида (69, 30 мг, 0,021 ммоль) в 0,8 мл дихлорметана добавляли 0,2 мл трифторуксусной кислоты. Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 30 минут при комнатной температуре. Раствор затем напрямую очищали обращенно-фазной ВЭЖХ (Метод В). Фракции, содержащие продукт, лиофилизировали с получением после лиофилизации 15 мг (48%). ЖХ/МС m/z 1355,4 [М+Н+], 1353,3 [М-Н+]; время удерживания = 1,42 минут.
Пример 45: Получение N~2~-(фенилсульфонил)лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида, трифторуксусной соли (75)
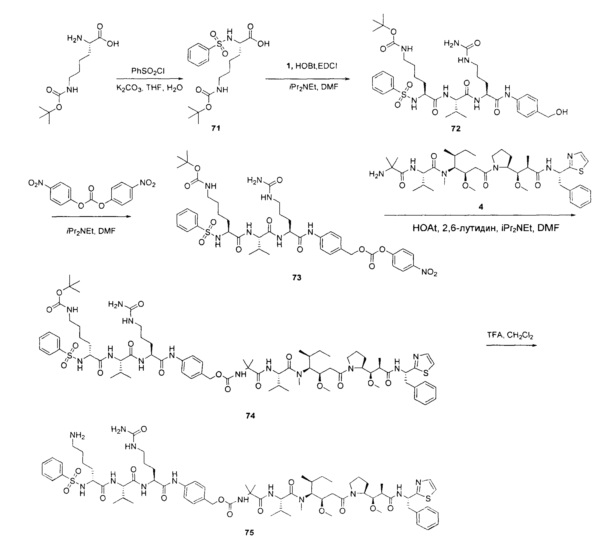
Стадия 1: Синтез N~6~-(трет-бутоксикарбонил)-N~2~-(фенилсульфонил)-L-лизина (71)
В охлажденный на льду раствор N~6~-(трет-бутоксикарбонил)-L-лизина (3,0 г, 12,2 ммоль) в смеси тетрагидрофуран:вода (1:1, 100 мл) добавляли карбонат калия (8,4 г, 61,0 ммоль), затем по каплям добавляли бензолсульфонилхлорид (1,86 мл, 14,6 ммоль). Эту смесь перемешивали при комнатной температуре в течение 3 часов и затем фильтровали. Фильтрат экстрагировали три раза смесью этилацетат:изопропиловый спирт 10:1. Объединенные экстракты промывали рассолом, сушили над сульфатом натрия и концентрировали при пониженном давлении. К остатку добавляли 100 мл трет-бутил-метилового эфира, и смесь перемешивали в течение 30 минут. Твердое вещество собирали фильтрованием и очищали обращенно-фазной ВЭЖХ. Фракции, содержащие продукт, лиофилизировали с получением 538 мг (11%) целевого продукта в виде белого твердого вещества. МС m/z 408,9 [M+Na+]. 1H ЯМР (400 МГц, DMSO-d6) δ=7.76 (d, J=7.5 Гц, 2Н), 7.63-7.50 (m, 3H), 3.61-3.53 (m, 1Н), 2.82-2.73 (m, 2Н), 1.61-1.40 (m, 2Н), 1.35 (s, 9Н), 1.28-1.06 (m, 4Н).
Стадия 2: Синтез N~6~-(трет-бутоксикарбонил)-N~2~-(фенилсульфонил)-L-лизил-L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]орнитинамида (72)
В охлажденный на льду раствор N~6~-(трет-бутоксикарбонил)-N~2~-(фенилсульфонил)-L-лизина (71, 1,2 г, 3,10 ммоль) и L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (1, 1,30 г, 3,41 ммоль) в 20 мл N,N-диметилформамида добавляли N,N-диизопропилэтиламин (600 мкл, 438 мг, 3,40 ммоль), гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодимида (650 мг, 3,41 ммоль) и гидрат 1-гидроксибензотриазола (460 мг, 3,41 ммоль). Эту смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 16 часов. Смесь по каплям добавляли в 150 мл трет-бутил-метилового эфира. Твердое вещество выделяли фильтрованием и затем очищали обращенно-фазной ВЭЖХ. Фракции, содержащие продукт, лиофилизировали с получением 850 мг (37%) целевого продукта в виде белого твердого вещества.
Стадия 3: Синтез N~6~-(трет-бутоксикарбонил)-N~-2~-(фенилсульфонил)-L-лизил-L-валил-N~5~-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]орнитинамида (73)
В перемешиваемый раствор N~6~-(трет-бутоксикарбонил)-N~2~-(фенилсульфонил)-L-лизил-L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]орнитинамида (72, 850 мг, 1,14 ммоль) и пара-нитрофенилкарбоната (853 мг, 2,85 ммоль) в 15 мл N,N-диметилформамида добавляли N,N-диизопропилэтиламин (501 мкл, 365 мг, 2,85 ммоль). Эту смесь перемешивали при комнатной температуре в течение 16 часов. Смесь по каплям добавляли в 150 мл трет-бутил-метилового эфира. Твердое вещество очищали обращенно-фазной ВЭЖХ. Фракции, содержащие продукт, лиофилизировали с получением 450 мг (43%) целевого продукта в виде белого твердого вещества. МС m/z 935,1 [M+Na+]; 1Н ЯМР (400 МГц, DMSO-d6) δ=10.11 (s, 1H), 8.35-8.27 (m, 2H), 8.10 (d, J=7.0 Гц, 1H), 7.94 (d, J=8.5 Гц, 1H), 7.83 (d, J=8.5 Гц, 1H), 7.76 (d, J=7.5 Гц, 2H), 7.63 (d, J=8.5 Гц, 2H), 7.60-7.48 (m, 5H), 7.40 (t, J=5.8 Гц, 2H), 6.70 (s, J=5.6, 5.6 Гц, 1H), 6.01-5.93 (m, 1H), 5.43 (s, 2H), 4.36 (d, J=6.0 Гц, 1H), 4.01 (t, J=7.5 Гц, 1H), 3.78 (d, J=5.0 Гц, 1H), 3.01 (d, J=6.0 Гц, 1H), 2.97-2.88 (m, 1H), 2.76 (q, J=6.5 Гц, 2H), 1.86 (d, J=6.5 Гц, 1H), 1.44 (d, J=5.5 Гц, 4H), 1.41-1.40 (m, 1H), 1.36 (s, 9H), 1.31-1.13 (m, 3H), 1.05 (br. s., 1H), 0.75 (d, J=7.0 Гц, 3H), 0.73 (d, J=7.0 Гц, 3H).
Стадия 4: Синтез N~6~-(трет-бутоксикарбонил)-N~2~-(фенилсульфонил)лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида (74)
В раствор 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамиада (4, 52,0 мг, 0,070 ммоль) и N~6~-(трет-бутоксикарбонил)-N~2~-(фенилсульфонил)-L-лизил-L-валил-N~5~-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]орнитинамида (73, 60,4 мг, 0,066 ммоль) в 2 мл N,N-диметилацетамида добавляли 2,6-лутидин (31 мкл, 28,3 мг, 0,264 ммоль), N,N-диизопропилэтиламин (46 мкл, 34,1 мг, 0,264 ммоль) и 3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ол (9 мг, 0,066 ммоль). Эту смесь перемешивали в течение 16 часов при 45°С. Реакционную смесь затем очищали обращенно-фазной ВЭЖХ (Метод S) с получением после лиофилизации 85 мг (85%) целевого продукта в виде белого твердого вещества. ЖХ/МС m/z 1518,4 [М+Н+], 1516,3 [М-Н+]; время удерживания = 1,93 минут.
Стадия 5: Синтез N~2~-(фенилсульфонил)лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида, трифторуксусной соли (75)
В раствор N~6~-(трет-бутоксикарбонил)-N~2~-(фенилсульфонил)лизил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида (75, 82 мг, 0,054 ммоль) в 0,8 мл дихлорметана добавляли 0,2 мл трифторуксусной кислоты. Эту смесь контролировали методом ЖХ/МС и перемешивали в течение 30 минут при комнатной температуре. Раствор затем напрямую очищали обращенно-фазной ВЭЖХ (Метод В). Фракции, содержащие продукт, лиофилизировали с получением после лиофилизации 35 мг (42%). ЖХ/МС m/z 1416,87 [М+Н+]; время удерживания = 1,35 минут.
Пример 46: Получение N-ацетил-О-(2-аминоэтил)-D-серил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида, соли с трифторуксусной кислотой (83)
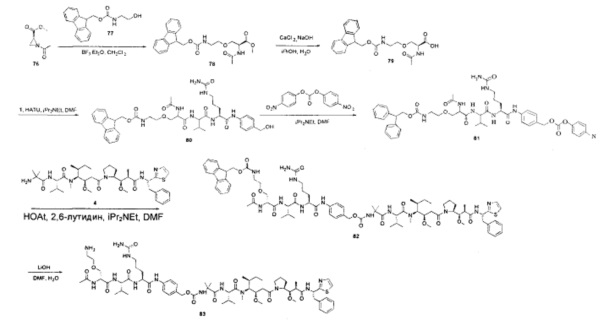
Стадия 1: Синтез метил-N-ацетил-O-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино}этил)-L-серината (78)
В суспензию 9Н-флуорен-9-илметил-(2-гидроксиэтил)карбамата (1000 мг, 3,53 ммоль) и метил-(2S)-1-ацетилазиридин-2-карбоксилата (US 20110021482, 758 мг, 5,29 ммоль) в 20 мл дихлорметана при -30°С по каплям добавляли диэтилэфират трифторида бора (532 мкл, 601 мг, 4,24 ммоль). Смесь перемешивали при этой температуре в течение 2 часов, затем оставляли нагреваться до комнатной температуры. Еще через час в реакционную смесь добавляли насыщенный водный раствор бикарбоната натрия и разбавляли дихлорметаном (80 мл). Органическую фазу отделяли и промывали рассолом, сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали обращенно-фазной ВЭЖХ (Метод W). Соответствующие фракции объединяли, и органические растворители удаляли при пониженном давлении. Оставшуюся водную смесь экстрагировали этилацетатом (2×100 мл). Объединенные экстракты промывали рассолом, сушили над сульфатом натрия и концентрировали. Остаток выпаривали совместно с дихлорметаном (50 мл) один раз с получением указанного в заголовке соединения, 160 мг (11%), в виде светло-желтой смолы. МС m/z 448,9 [M+Na+]; 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ=7.78 (d, J=7.5 Гц, 2Н), 7.60 (d, J=7.5 Гц, 2Н), 7.42 (t, J=7.5 Гц, 2Н), 7.37-7.31 (m, 2Н), 6.66 (br. s., 1Н), 5.07 (br. s., 1Н), 4.89-4.72 (m, 1Н), 4.45 (d, J=6.5 Гц, 2Н), 4.23 (d, J=6.5 Гц, 1Н), 3.88 (d, J=6.5 Гц, 1Н), 3.72 (br. s., 4Н), 3.54 (br. s., 2Н), 3.36 (br. s., 2H), 2.09 (s, 3H).
Стадия 2: Синтез N-ацетил-O-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино}этил)-L-серина (79)
В раствор N-ацетил-O-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино}этил)-L-серината (78, 160 мг, 0,375 ммоль) в смеси изопропанол:вода (7:3, 10 мл) добавляли хлорид кальция (833 г, 7,5 ммоль). Как только смесь стала прозрачной, добавляли гидроксид натрия (18 мг, 0,450 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 3 часов. Смесь затем подкисляли до рН~1-2 3М водным раствором соляной кислоты и разбавляли водой (10 мл). Смесь экстрагировали этилацетатом (20 мл), и органическую фазу выделяли и концентрировали при пониженном давлении для удаления большей части изопропанола. Водную фазу экстрагировали этилацетатом (2×100 мл). Объединенные органические слои промывали рассолом, сушили над сульфатом натрия и концентрировали с получением 107 мг (96%) целевого соединения в виде синего твердого вещества, которое использовали на следующей стадии без дополнительной очистки. МС m/z 434,8 [M+Na+]; 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ=8.48 (d, J=9.0 Гц, 1Н), 7.78 (d, J=7.5 Гц, 2Н), 7.56 (d, J=7.5 Гц, 2Н), 7.47-7.37 (m, 2Н), 7.35-7.29 (m, 2Н), 7.15 (t, J=6.1 Гц, 1Н), 5.03 (d, J=9.0 Гц, 1Н), 4.51 (dd, J=6.8, 10.8 Гц, 1Н), 4.41 (dd, J=7.0, 10.5 Гц, 2Н), 4.30-4.23 (m, 1Н), 4.32-4.21 (m, 1Н), 4.21-4.09 (m, 1Н), 3.57 (t, J=8.3 Гц, 3H), 3.53-3.45 (m, 2Н), 3.40 (br. s., 1Н), 3.31 (d, J=9.0 Гц, 1Н), 2.00 (s, 3H).
Стадия 3: Синтез N-ацетил-O-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино}этил)-L-серилвалил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (80)
В раствор N-ацетил-O-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино}этил)-L-серина (79, 105 мг, 0,255 ммоль) и L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (1, 96,6 мг, 0,255 ммоль) в 3 мл N,N-диметилформамида добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (106 мг, 0,280 ммоль), затем N,N-диизопропилэтиламин (98,7 мг, 0,764 ммоль). Эту желтую прозрачную смесь перемешивали при комнатной температуре в течение 2 часов. Смесь разбавляли трет-бутил-метиловым эфиром (60 мл) и перемешивали в течение 30 минут. Твердое вещество выделяли фильтрованием и затем очищали обращенно-фазной ВЭЖХ (Метод X). Фракции, содержащие продукт, лиофилизировали с получением 84 мг (43%) целевого продукта в виде белого твердого вещества. МС m/z 774,2 [М+Н+].
Стадия 4: Синтез N-ацетил-O-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино}этил)серилвалил-N~5~-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]-L-орнитинамида (81)
В перемешиваемый раствор N-ацетил-O-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино}этил)-L-серилвалил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (80, 200 мг, 0,258 ммоль) и пара-нитрофенилкарбоната (853 мг, 2,85 ммоль) в 5 мл N,N-диметилформамида добавляли N,N-диизопропилэтиламин (100,8 мкл, 73,5 мг, 0,569 ммоль). Эту смесь перемешивали при комнатной температуре в течение 3 часов, затем снова добавляли пара-нитрофенилкарбонат (78,6 мг, 0,258 ммоль). Еще через час смесь по каплям добавляли в 100 мл трет-бутил-метилового эфира. Твердое вещество выделяли фильтрованием, промывали некоторым количеством трет-бутил-метилового эфира с получением после сушки на воздухе 180 мг (74%) целевого продукта в виде желтого твердого вещества. МС m/z 939,2 [М+Н+] 961,1 [M+Na+].
Стадия 5: Синтез N-ацетил-O-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино}этил)-D-серил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида (82)
В раствор 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (4, 60,0 мг, 0,081 ммоль) и N-ацетил-O-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино}этил)серилвалил-N~5~-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]-L-орнитинамида (81, 121 мг, 0,188 ммоль) в 2,5 мл N,N-диметилацетамида добавляли 2,6-лутидин (37,4 мкл, 34,6 мг, 0,323 ммоль) N,N-диизопропилэтиламин (42,2 мкл, 31,3 мг, 0,242 ммоль) и 3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ол (11,0 мг, 0,081 ммоль). Эту смесь перемешивали в течение 6 часов при 45°С. Реакционную смесь затем очищали обращенно-фазной ВЭЖХ (Метод 6) с получением после лиофилизации 62 мг (50%) целевого продукта в виде белого твердого вещества. ЖХ/МС m/z 1543,3 [М+Н+]; время удерживания = 2,25 минут. Аналитическая ВЭЖХ: время удерживания = 7,435 минут.
Стадия 6: Синтез N-ацетил-О-(2-аминоэтил)-D-серил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида, соли с трифторуксусной кислотой (83)
В раствор N-ацетил-O-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино}этил)-D-серил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5-~-карбамоилорнитинамида (82, 42 мг, 0,027 ммоль) в 2,5 мл N,N-диметилформамида добавляли водный раствор гидроксида лития (3,26 мг, 0,136 ммоль), и эту смесь перемешивали при комнатной температуре в течение часа. Раствор затем концентрировали и очищали обращенно-фазной ВЭЖХ {Метод L). Фракции, содержащие продукт, лиофилизировали с получением 29 мг (74%) целевого продукта. ЖХ/МС m/z 1321,2 [М+Н+]; время удерживания = 1,30 минут. Аналитическая ВЭЖХ: время удерживания = 5,804 минут.
Пример 47: N-ацетил-O-[2-(2-аминоэтокси)этил]-L-серил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида, соли с трифторуксусной кислотой (90)
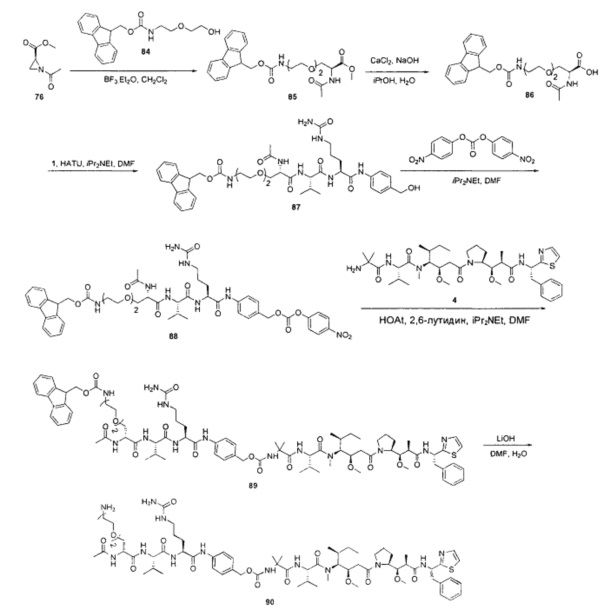
Стадия 1: Синтез метил-N-ацетил-O-[2-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино}этокси)этил]-L-серината (85)
В раствор 9Н-флуорен-9-илметил[2-(2-гидроксиэтокси)этил]карбамата (84, 7,55 г, 23,1 ммоль) и метил-(2S)-1-ацетилазиридин-2-карбоксилата (758 мг, 5,29 ммоль) в 130 мл дихлорметана по каплям добавляли диэтилэфират трифторида бора (3,94 мл, 4,46 г, 31,4 ммоль). Через час в реакционную смесь добавляли насыщенный водный раствор бикарбоната натрия. Органическую фазу отделяли и промывали рассолом, сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали обращенно фазной ВЭЖХ (Метод Y). Соответствующие фракции объединяли, и органические растворители удаляли при пониженном давлении. Оставшуюся водную смесь экстрагировали дихлорметаном (2×100 мл). Объединенные экстракты промывали рассолом, сушили над сульфатом натрия и концентрировали при пониженном давлении с получением указанного в заголовке соединения, 3,8 г (26%), в виде бесцветного масла. МС m/z 493,0 [M+Na+]; 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ=7.77 (d, J=7.5 Гц, 2Н), 7.62 (d, J=7.0 Гц, 2Н), 7.44-7.37 (m, 2Н), 7.35-7.29 (m, 2Н), 6.69 (d, J=6.5 Гц, 1Н), 5.49 (br. s., 1Н), 4.78 (d, J=8.0 Гц, 1Н), 4.42 (d, J=6.5 Гц, 2Н), 4.25 (d, J=7.0 Гц, 1Н), 3.95 (d, J=10.0 Гц, 1Н), 3.75 (s, 3H), 3.66-3.50 (m, 6Н), 3.41 (br. s., 2Н), 2.04 (s, 3H).
Стадия 2: Синтез N-ацетил-O-[2-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино} этокси)этил]-L-серина (86)
В раствор метил-N-ацетил-O-[2-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино}этокси)этил]-L-серината (85, 3200 мг, 6,8 ммоль) в 90 мл изопропанола и 40 мл воды добавляли хлорид кальция (15,1 г, 136 ммоль). Как только смесь стала прозрачной, добавляли гидроксид натрия (326 мг, 8,16 ммоль). Через 2 часа смесь затем подкисляли до рН~1-2 3М водным раствором соляной кислоты и разбавляли водой (300 мл). Смесь экстрагировали этилацетатом (3×250 мл), и органическую фазу выделяли и концентрировали при пониженном давлении с получением 3000 мг (97%) целевого соединения в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. МС m/z 456,9 [М+Н+]; 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ=7.77 (d, J=7.5 Гц, 2Н), 7.63-7.53 (m, 2Н), 7.44-7.38 (m, 2Н), 7.36-7.30 (m, 2Н), 6.65 (d, J=6.0 Гц, 1Н), 4.77 (br. s., 1Н), 4.51 (br. s., 1H), 4.43 (d, J=6.5 Гц, 1H), 4.26 (d, J=6.0 Гц, 1H), 3.95 (br. s., 1H), 3.76 (br. s„ 1H), 3.70-3.57 (m, 3H), 3.54 (d, J=7.5 Гц, 2H), 3.41 (d, J=13.1 Гц, 2H), 3.29 (br. s., 1H), 2.05 (br. s., 3H).
Стадия 3: Синтез N-ацетил-O-[2-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино}этокси)этил]-L-серилвалил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (87)
В раствор N-ацетил-O-[2-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино}этокси)этил]-L-серина (86, 3000 мг, 6,57 ммоль) в 45 мл N,N-диметилформамида добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU, 3000 мг, 7,89 ммоль). Через 20 минут добавляли L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамид (1, 2490 мг, 6,57 ммоль) и затем N,N-диизопропилэтиламин (2550 мг, 19,7 ммоль). Через 3 часа смесь вливали в смесь трет-бутил-метилового эфира (500 мл) и петролейного эфира (200 мл) и перемешивали в течение 20 минут. Твердое вещество выделяли фильтрованием и затем очищали хроматографией на силикагеле, используя элюирование с градиентом от 1% до 20% метанола в дихлорметане, с получением 3100 мг (58%) целевого продукта в виде белого твердого вещества. МС m/z 818,1[М+Н+].
Стадия 4: Синтез N-ацетил-O-[2-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино}этокси)этил]-L-серилвалил-N~5~-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]-L-орнитинамида (88)
В охлажденный на льду перемешиваемый раствор N-ацетил-O-[2-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино}этокси)этил]-L-серилвалил-N~5-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (87, 500 мг, 0,611 ммоль) и пара-нитрофенилкарбоната (279 мг, 0,917 ммоль) в 6 мл N,N-диметилформамида добавляли N,N-диизопропилэтиламин (216,7 мкл, 158 мг, 1,22 ммоль). Эту смесь перемешивали при комнатной температуре в течение 5 часов, затем снова добавляли пара-нитрофенилкарбонат (100 мг, 0,538 ммоль). Еще через 12 часов смесь вливали в смесь тредл-бутил-метилового эфира (60 мл) и петролейного эфира (20 мл) и перемешивали в течение 15 минут. Твердое вещество выделяли фильтрованием и затем очищали хроматографией на силикагеле, используя элюирование с градиентом от 1% до 10% метанола в дихлорметане, с получением 300 мг (50%) целевого продукта в виде белого твердого вещества. МС m/z 983,5 [М+Н+].
Стадия 5: Синтез N-ацетил-O-[2-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино}этокси)этил]-L-серил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида (89)
В раствор 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (4, 60,0 мг, 0,081 ммоль) и N-ацетил-O-[2-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино}этокси)этил]-L-серилвалил-N~5~-карбамоил-N-[4-({[(4-нитрофенокси)карбонил]окси}метил)фенил]-L-орнитинамида (88, 75,4 мг, 0,077 ммоль) в 2,5 мл N,N-диметилацетамида добавляли 2,6-лутидин (37,4 мкл, 34,6 мг, 0,323 ммоль), N,N-диизопропилэтиламин (42,2 мкл, 31,3 мг, 0,242 ммоль) и 3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ол (HOAt, 11,0 мг, 0,081 ммоль). Эту смесь перемешивали в течение 16 часов при 45°С. Реакционную смесь затем очищали обращенно-фазной ВЭЖХ (Метод G) с получением после лиофилизации 76 мг (58%) целевого продукта в виде белого твердого вещества. ЖХ/МС m/z 1587,3 [М+Н+]; время удерживания = 2,26 минут. Аналитическая ВЭЖХ: время удерживания = 7,459 минут.
Стадия 6: Синтез N-ацетил-O-[2-(2-аминоэтокси)этил]-L-серил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида, соли с трифторуксусной кислотой (90)
В раствор N-ацетил-O-[2-(2-{[(9Н-флуорен-9-илметокси)карбонил]амино}этокси)этил]-L-серил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида (89, 40 мг, 0,025 ммоль) в 2,5 мл N,N-диметилформамида добавляли водный раствор гидроксида лития (3,02 мг, 0,126 ммоль), и эту смесь перемешивали при комнатной температуре в течение 30 минут. Раствор затем очищали обращенно-фазной ВЭЖХ (Метод D). Фракции, содержащие продукт, лиофилизировали с получением 27 мг (72%) целевого продукта. ЖХ/МС m/z 1365,2 [М+Н+]; время удерживания = 1,26 минут. Аналитическая ВЭЖХ: время удерживания = 5,768 минут.
Пример 48: Получение N~2~-(3-гидроксипропаноил)-L-лизилвалил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоил-L-орнитинамида, соли с трифторуксусной кислотой (97)
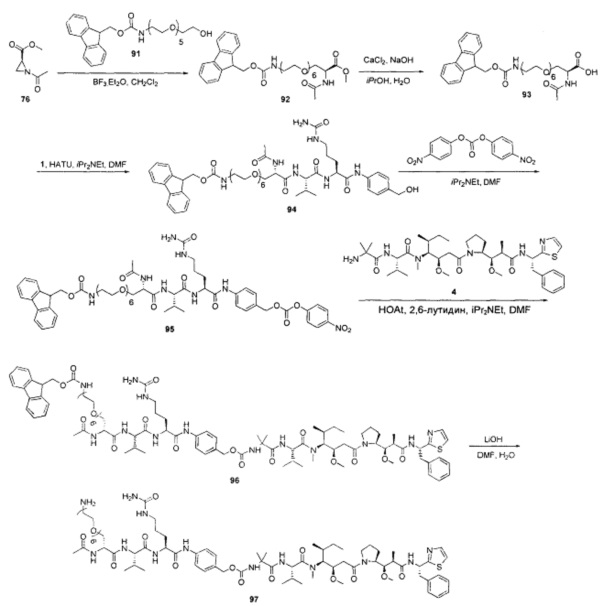
Стадия 1: Синтез метил-N-ацетил-O-[1-(9Н-флуорен-9-ил)-3-оксо-2,7,10,13,16,19-гексаокса-4-азагеникозан-21-ил]-L-серината (92)
В раствор 9Н-флуорен-9-илметил-(17-гидрокси-3,6,9,12,15-пентагексагептадец-1-ил)карбамата (91, 7600 мг, 15,09 ммоль) и метил-(2S)-1-ацетилазиридин-2-карбоксилата (4320 мг, 30,2 ммоль) в 150 мл дихлорметана по каплям добавляли диэтилэфират трифторида бора (2,84 мл, 3210 мг, 22,6 ммоль). Через 2,5 часа реакционную смесь промывали насыщенным водным раствором бикарбоната натрия, затем рассолом, сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали обращенно-фазной ВЭЖХ (Метод Z). Соответствующие фракции объединяли, и органические растворители удаляли при пониженном давлении. Оставшуюся водную смесь экстрагировали этилацетатом (2×100 мл). Объединенные экстракты сушили над сульфатом натрия и концентрировали с получением указанного в заголовке соединения, 5 г (51%), в виде масла. МС m/z 647,1 [М+Н+]; 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ=7.76 (d, J=7.5 Гц, 2Н), 7.61 (d, J=7.5 Гц, 2Н), 7.40 (t, J=7.5 Гц, 2Н), 7.31 (t, J=7.4 Гц, 2Н), 6.72 (d, J=7.5 Гц, 1Н), 5.47 (br. s., 1Н), 4.73 (td, J=3.5, 8.0 Гц, 1Н), 4.41 (d, J=7.0 Гц, 2Н), 4.26-4.19 (m, 1Н), 3.94 (dd, J=3.5, 10.0 Гц, 1Н), 3.75 (s, 3H), 3.70 (dd, J=3.5, 10.0 Гц, 1Н), 3.67-3.56 (m, 22Н), 3.40 (q, J=5.2 Гц, 2Н), 2.07 (s, 3H).
Стадия 2: Синтез N-ацетил-O-[1-(9Н-флуорен-9-ил)-3-оксо-2,7,10,13,16,19-гексаокса-4-азагеникозан-21-ил]-L-серин (93)
В раствор метил-N-ацетил-O-[1-(9Н-флуорен-9-ил)-3-оксо-2,7,10,13,16,19-гексаокса-4-азагеникозан-21-ил]-L-серината (92, 4200 мг, 6,49 ммоль) в смеси изопропанол:вода (7:3, 170 мл) добавляли хлорид кальция (14,4 г, 130 ммоль). Как только смесь стала прозрачной, добавляли гидроксид натрия (312 мг, 7,79 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 3 часов. Смесь затем подкисляли до рН~1-2 3М водным раствором соляной кислоты и разбавляли водой (100 мл). Смесь экстрагировали этилацетатом (100 мл), и органическую фазу выделяли и концентрировали при пониженном давлении для удаления большей части изопропанола. Водную фазу экстрагировали этилацетатом (2×100 мл). Объединенные органические слои промывали рассолом, сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток затем очищали хроматографией на силикагеле, используя элюирование с градиентом от 0% до 15% метанола в дихлорметане, с получением 3,4 г (83%) целевого соединения в виде желтого масла. МС m/z 633,1 [М+Н+].
Стадия 3: Синтез N-ацетил-O-[1-(9Н-флуорен-9-ил)-3-оксо-2,7,10,13,16,19-гексаокса-4-азагеникозан-21-ил]-L-серилвалил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (94)
В охлажденный на льду раствор N-ацетил-O-[1-(9Н-флуорен-9-ил)-3-оксо-2,7,10,13,16,19-гексаокса-4-азагеникозан-21-ил]-L-серина (93, 1,20 г, 1,90 ммоль) и L-валил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (1, 720 мг, 1,90 ммоль) в 15 мл N,N-диметилформамида добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (794 мг, 2,09 ммоль), затем N,N-диизопропилэтиламин (736 мг, 5,69 ммоль). Эту желтую прозрачную смесь перемешивали при комнатной температуре в течение 2 часов. Смесь разбавляли этилацетатом (3×30 мл), промывали водой (60 мл), сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток затем очищали хроматографией на силикагеле, используя элюирование с градиентом от 0% до 15% метанола в дихлорметане, с получением 1,5 г (80%) целевого продукта в виде бесцветного стекла. МС m/z 994,8 [М+Н+].
Стадия 4: Синтез N-ацетил-O-[2-(2-аминоэтокси)этил]-L-серил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида (95)
В ледяной бане в пермешиваемый раствор N-ацетил-O-[1-(9Н-флуорен-9-ил)-3-оксо-2,7,10,13,16,19-гексаокса-4-азагеникозан-21-ил]-L-серилвалил-N~5~-карбамоил-N-[4-(гидроксиметил)фенил]-L-орнитинамида (94, 1,5 г, 1,5 ммоль) и пара-нитрофенилкарбоната (918 мг, 3,02 ммоль) в 15 мл N,N-диметилформамида добавляли N,N-диизопропилэтиламин (402 мкл, 293 мг, 2,26 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, затем снова добавляли пара-нитрофенилкарбонат (918 мг, 3,02 ммоль). Еще через 3 часа смесь разбавляли этилацетатом (30 мл) и промывали водой (40 мл). Водную фазу экстрагировали этилацетатом (30 мл). Объединенные органические слои концентрировали при пониженном давлении, затем очищали хроматографией на силикагеле, используя элюирование с градиентом от 0% до 15% метанола в дихлорметане, затем очищали обращенно-фазной ВЭЖХ (Метод АА) с получением после лиофилизации 944 мг (54%) целевого продукта в виде белого твердого вещества. МС m/z 1159,8 [М+Н+].
Стадия 5: Синтез N-ацетил-O-[1-(9Н-флуорен-9-ил)-3-оксо-2,7,10,13,16,19-гексаокса-4-азагеникозан-21-ил]-L-серил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7110-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида (96)
В раствор 2-метилаланил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-1-валинамида (4, 55,0 мг, 0,074 ммоль) и N-ацетил-O-[2-(2-аминоэтокси)этил]-L-серил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида (95, 85,8 мг, 0,074 ммоль) в 2,5 мл N,N-диметилацетамида добавляли 2,6-лутидин (34,3 мкл, 31,7 мг, 0,296 ммоль), N,N-диизопропилэтиламин (38,7 мкл, 28,7 мг, 0,222 ммоль) и 3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ол (10,1 мг, 0,074 ммоль). Эту смесь перемешивали в течение 6 часов при 45°С. Реакционную смесь затем очищали обращенно-фазной ВЭЖХ (Метод G) с получением после лиофилизации 90 мг (69%) целевого продукта в виде белого твердого вещества. ЖХ/МС m/z 1764,4 [М+Н+]; время удерживания = 1,99 минут. Аналитическая ВЭЖХ: время удерживания = 7,476 минут.
Стадия 6: Синтез N-ацетил-O-(17-амино-3,6,9,12,15-пентаоксагептадец-1-ил)-L-серил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида, соли с трифторуксусной кислотой (97)
В раствор N-ацетил-О-[1-(9Н-флуорен-9-ил)-3-оксо-2,7,10,13,16,19-гексаокса-4-азагеникозан-21-ил]-L-серил-L-валил-N-{4-[(8S,11S,12R)-11-[(2S)-бутан-2-ил]-12-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]амино}пропил]пирродидин-1-ил}-2-оксоэтил)-5,5,10-триметил-3,6,9-триоксо-8-(пропан-2-ил)-2,13-диокса-4,7,10-триазатетрадец-1-ил]фенил}-N~5~-карбамоилорнитинамида (96, 46 мг, 0,026 ммоль) в 2,5 мл N,N-диметилформамида добавляли водный раствор гидроксида лития (3,1 мг, 0,130 ммоль), и эту смесь перемешивали при комнатной температуре в течение 20 минут. Раствор затем концентрировали и очищали обращенно-фазной ВЭЖХ {Метод D, затем Метод Н). Фракции, содержащие продукт, лиофилизировали с получением 15 мг (35%) целевого продукта. ЖХ/МС m/z 1542,2 [М+Н+]; время удерживания = 1,41 минут. Аналитическая ВЭЖХ: время удерживания = 5,836 минут.
Приведенные ниже дополнительные ADC (с присоединением по различным положениям и на различных метках на анти-Trop2 антителе, как указано в Таблицах 2-7) были получены с использованием вышеуказанного метода конъюгирования (заключенная в скобки часть представляет собой остаток глутамина, при этом волнистые линии показывают точки присоединения к остальной части анти-Trop2 антитела):
Пример 49:
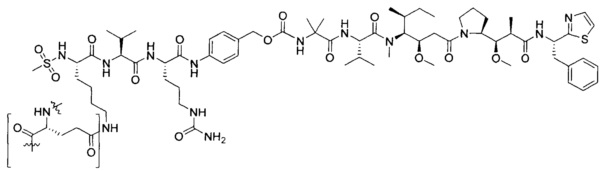
Пример 50:
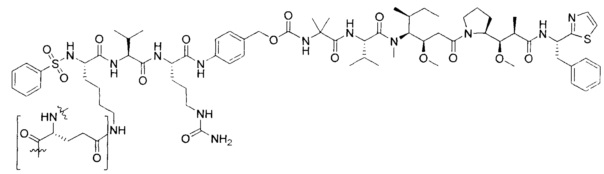
Пример 51:
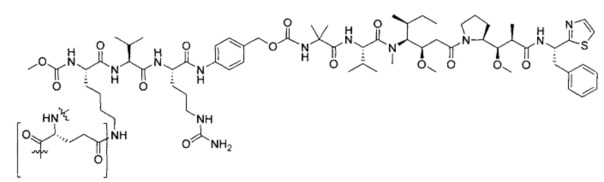
Пример 52:
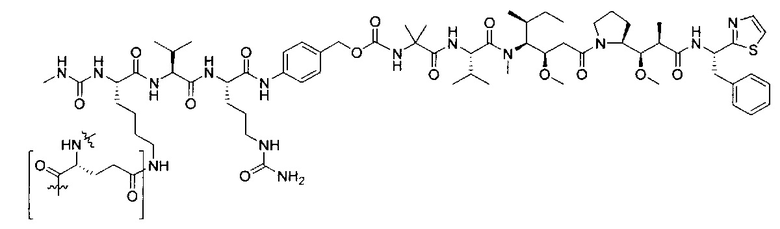
Пример 53:

Пример 54:
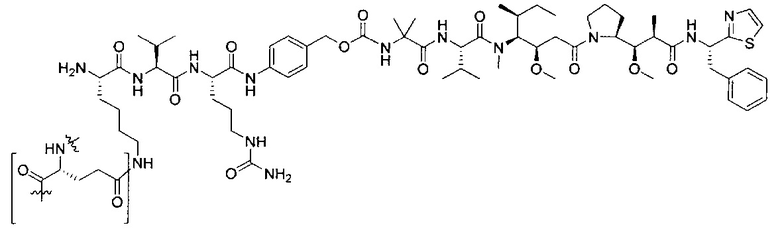
Пример 55:
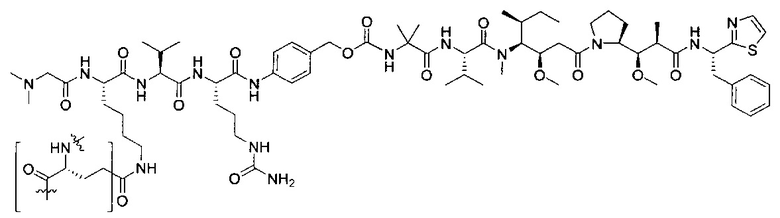
Пример 56:
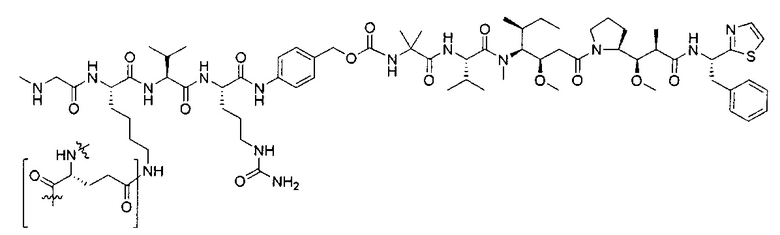
Пример 57:
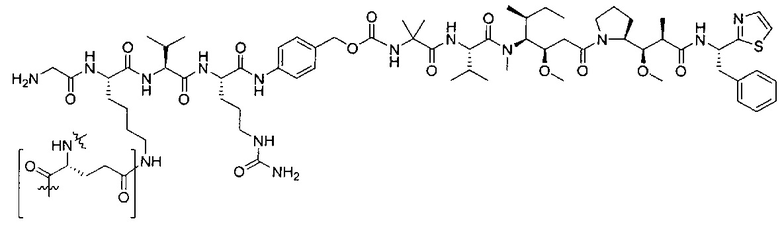
Пример 58:
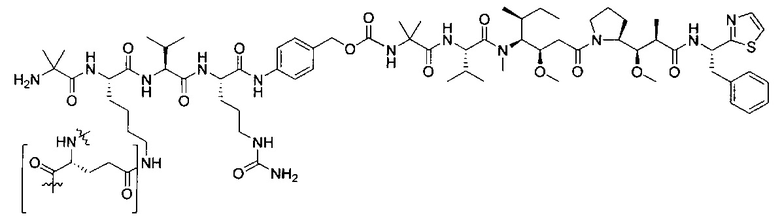
Пример 59:
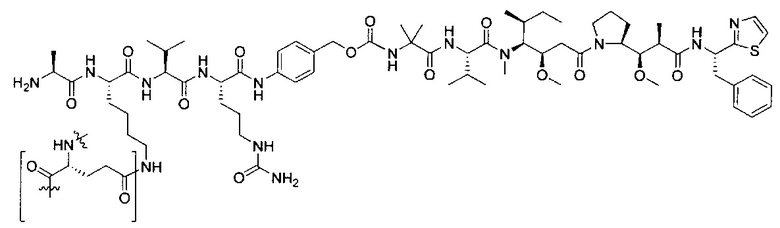
Пример 60:
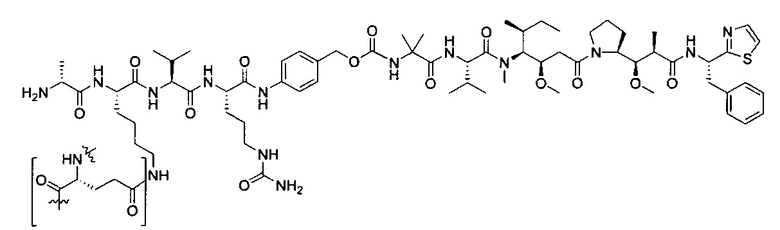
Пример 61:

Пример 62:

Пример 63:
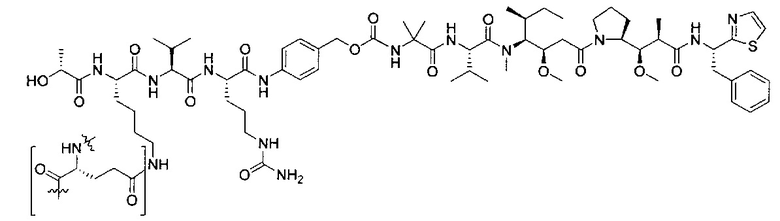
Пример 64:
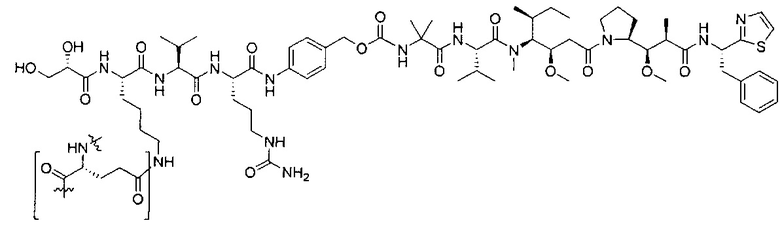
Пример 65:
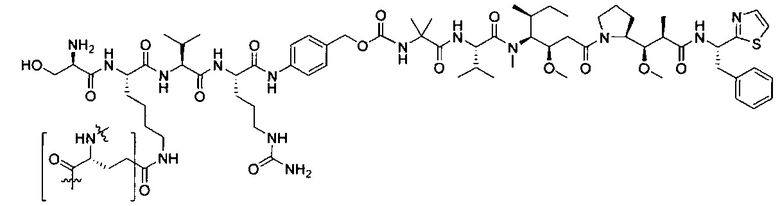
Пример 66:

Пример 67:
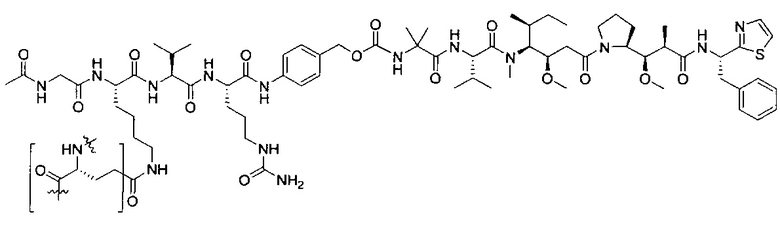
Пример 68:
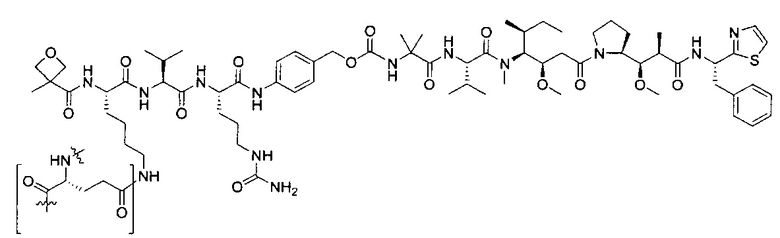
Пример 69:
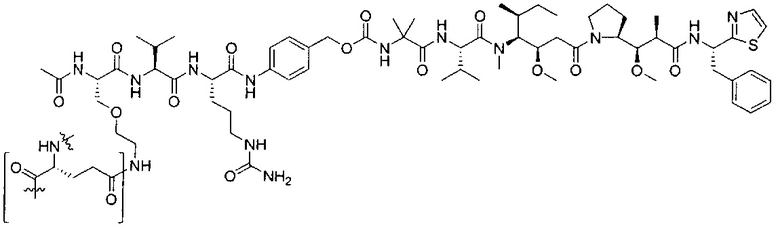
Пример 70:
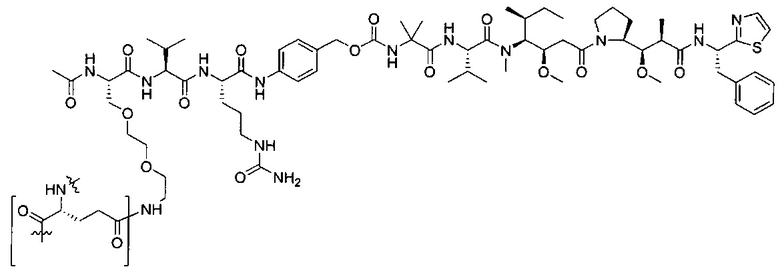
Пример 71:
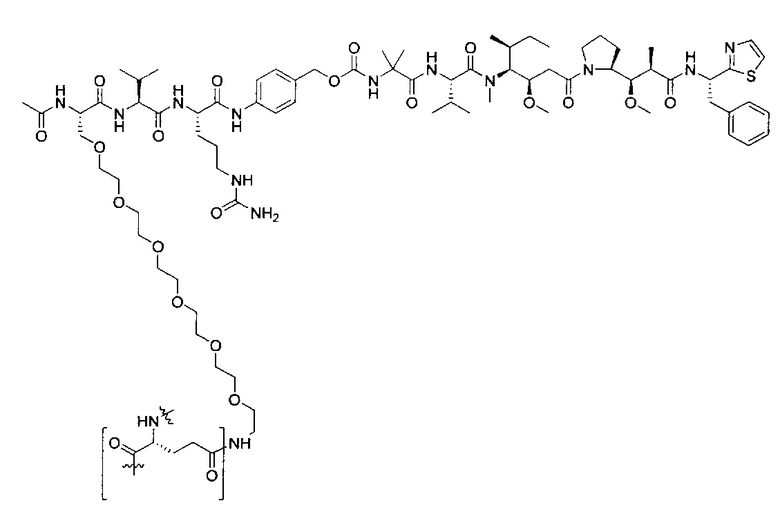
Пример 72. Подтверждение мышиной карбоксилэстеразы 1С в качестве фермента, ответственного за расщепление линкера VC-PABC в плазме крови
Образцы плазмы крови из линии мышей с нокаутированным геном C56/BL6 Ces1C-/-, гетерозиготной линии C56/BL6 Ces1C+/- и линии дикого типа C56/BL6 Ces1C+/+ использовали для тестирования стабильности конъюгата анти-Trop2 L11B-C6-VC-PABC-Aur0101. 56 мкг ADC инкубировали в плазме крови в 35% плазме, дополненной 1х PBS до конечной концентрации ADC 0,125 мг/мл. После инкубирования в течение 18 часов ADC выделяли с использованием M1S1 антигена, связанного с CNBr-активированной Sepharose (GE Healthcare), используя стандартные протоколы. Значения DAR оценивали до и после инкубирования, используя HIC анализ. Стабильность конъюгатов (в %) вычисляли в виде % DAR, оставшегося после инкубирования (смотри Фиг. 3).
БИОЛОГИЧЕСКИЕ ДАННЫЕ
Данные по стабильности, полученные по методикам, изложенным в Примерах 8-18, суммированы в Таблицах 2-7.

Стабильность конъюгатов in vivo у мыши. Значения DAR для конъюгатов, очищенных из животных, которым вводили дозу 9 мг/кг, сравнивали со значениями DAR до экспозиции in vivo. Значения стабильности, указанные выше, выражены в виде % DAR, оставшегося после заданного периода экспозиции (показаны в скобках).
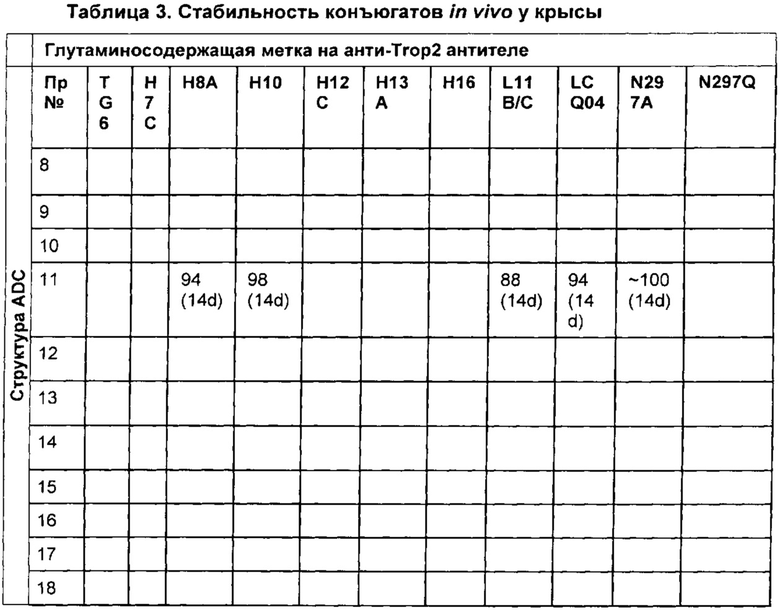
Стабильность конъюгатов in vivo у крысы. Значения DAR для конъюгатов, очищенных из животных, которым вводили дозу 9 мг/кг, сравнивали со значениями DAR до экспозиции in vivo. Значения стабильности, указанные выше, выражены в виде % DAR, оставшегося после заданного периода экспозиции (показаны в скобках).


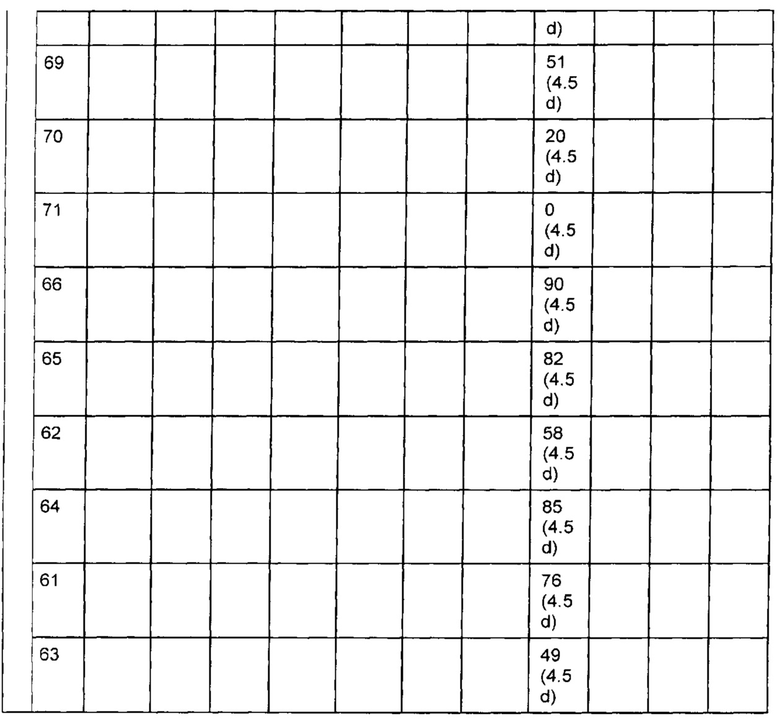
Стабильность конъюгатов in vitro в плазме крови мыши. Значения DAR для конъюгатов, очищенных после инкубирования плазмы сравнивали со значениями DAR до обработки плазмы. Значения стабильности, указанные выше, выражены в виде % DAR, оставшегося после заданного периода инкубирования (показаны в скобках).
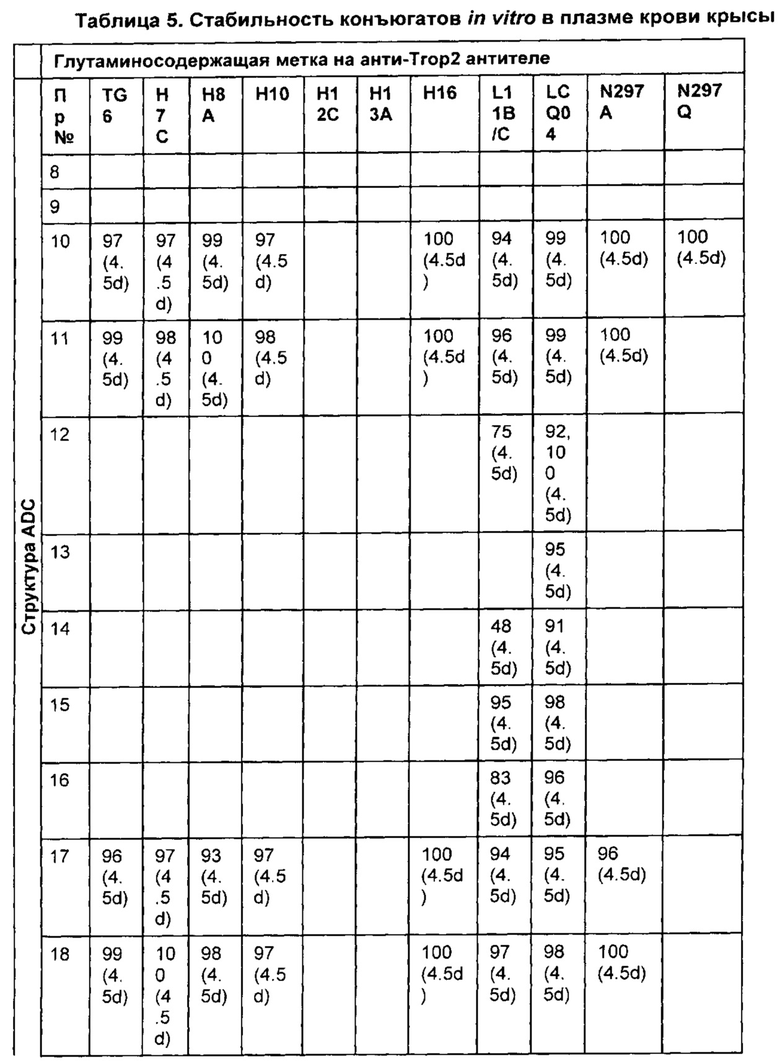
Стабильность конъюгатов in vitro в плазме крови крысы. Значения DAR для конъюгатов, очищенных после инкубирования плазмы сравнивали со значениями DAR до обработки плазмы. Значения стабильности, указанные выше, выражены в виде % DAR, оставшегося после заданного периода инкубирования (показаны в скобках).
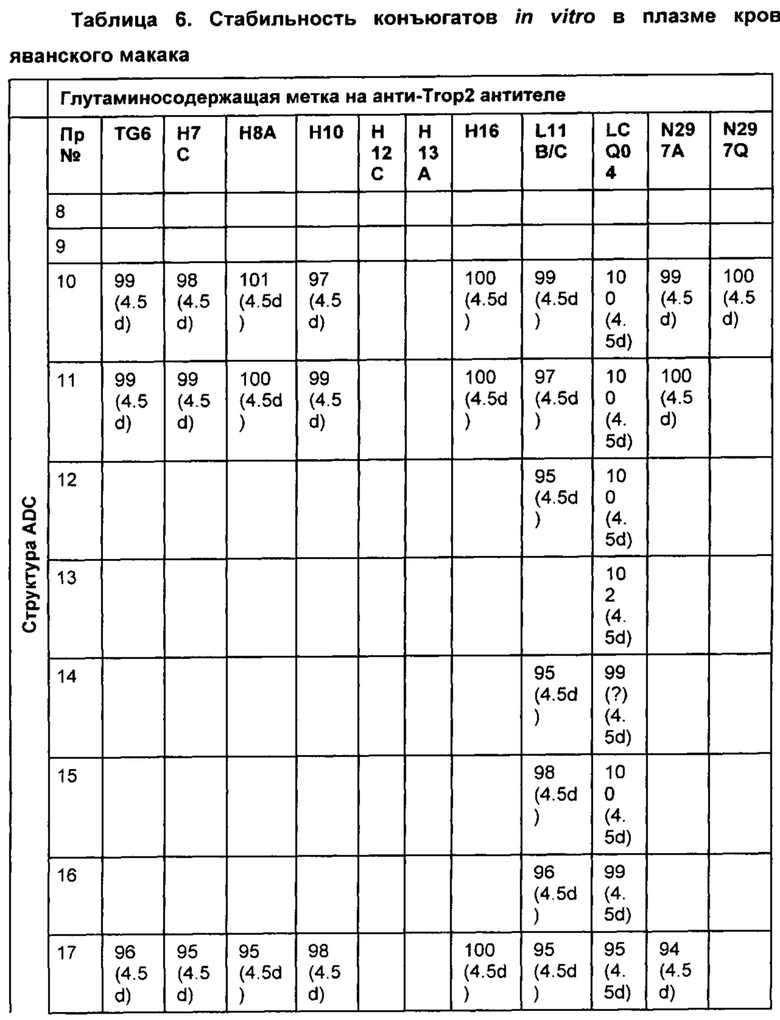

Стабильность конъюгатов in vitro в плазме крови яванского макака. Значения DAR для конъюгатов, очищенных после инкубирования плазмы сравнивали со значениями DAR до обработки плазмы. Значения стабильности, указанные выше, выражены в виде % DAR, оставшегося после заданного периода инкубирования (показаны в скобках).
Таблица 7А. Экспериментальные значения DAR с вычисленным % стабильности
Все значения DAR были определены в результате оценки количества лекарственнызх средств, присоединенных к антителу, используя HIC или масс-спектрометрический анализ. Стабильность конъюгатов вычислена в виде % DAR, оставшегося после инкубирования в плазме крови в течение 3 или 4,5 суток.
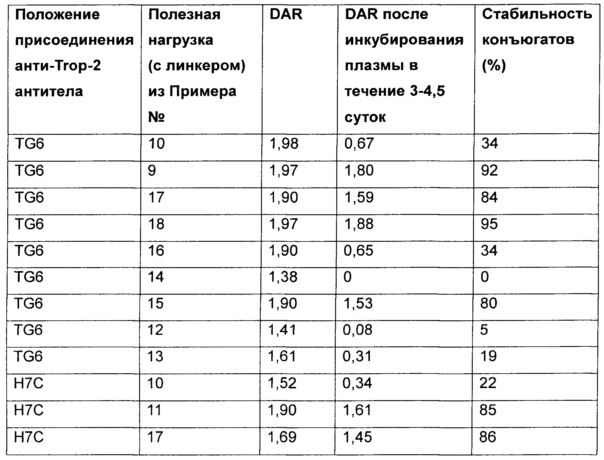
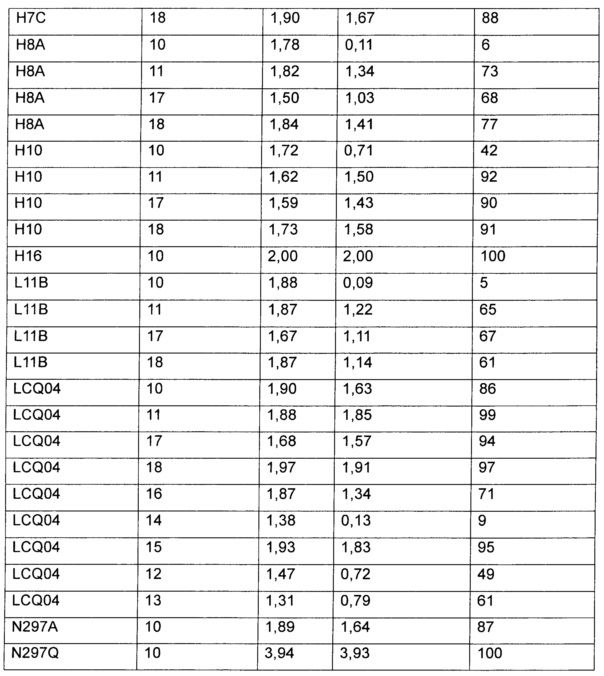
Таблица 7В. Дополнительные экспериментальные значения DAR с вычисленным % стабильности
Все значения DAR были определены в результате оценки количества лекарственнызх средств, присоединенных к антителу, используя HIC или масс-спектрометрический анализ. Стабильность конъюгатов вычислена в виде % DAR, оставшегося после инкубирования в плазме крови в течение 3 или 4,5 суток.
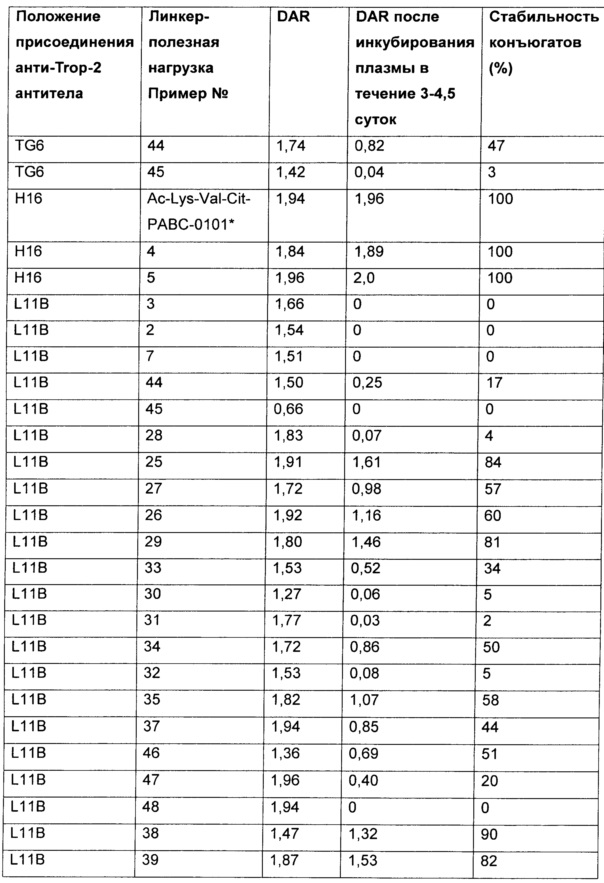
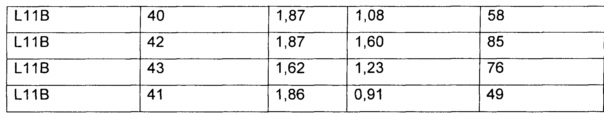
*Получена, как описано WO 2012/059882 и WO 2015/015448.
Таблица 8. Дополнительные экспериментальные значения DAR с вычисленным % стабильности; конструкции линкер-полезная нагрузка, конъюгированные с антителом Combo_Rd4_0.6nM_C29 с глутаминосодержащей меткой LCQ05
Способы конъюгирования были описаны в данном документе для анти-Trop2 антител. Антитело Combo_Rd4_0.6nM_C29 представляет собой анти-ВСМА антитело, как описано в предварительной патентной заявке США №62/146,843. После инкубирования в плазме крови мыши SCID соединения очищали с использованием смолы MabSelect Protein A (GE Healthcare) согласно стандартным способам. Все значения DAR были определены в результате оценки количества лекарственных средств, присоединенных к антителу, используя HIC анализ. Стабильность конъюгатов вычислена в виде % DAR, оставшегося после инкубирования в плазме крови мыши SCID в течение 4,5 суток.
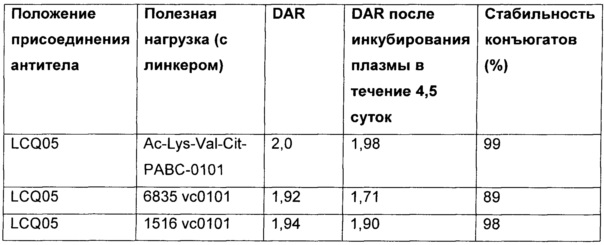
Хотя раскрытые сведения описаны со ссылкой на различные применения, способы и композиции, должно быть понятно, что различные изменения и модификации могут быть сделаны, не отклоняясь от идей, описанных в данном документе, и от заявленного изобретения. Приведенные выше примеры представлены для лучшей иллюстрации раскрытых идей и не ограничивают объем идей, представленных в данном документе. Несмотря не то, что идеи настоящего изобретения описаны на основе этих иллюстративных воплощений, специалисту будет понятно, что многочисленные варианты и модификации этих иллюстративных воплощений возможны без излишнего экспериментирования. Все такие варианты и модификации входят в объем настоящего изобретения.
Все источники информации, процитированные в данном описании, включая патенты, патентные заявки, статьи, книги и т.п., и источники информации, процитированные в них, во всей их полноте включены в данное описание посредством ссылки. В случае если один или более из включенных литературных и подобных материалов отличаются от данной заявки или противоречат данной заявкой, включая, без ограничения, определенные термины, использование терминов, описанные методы или т.п., то преимущестов имеет текст данной заявки.
В приведенном выше описании и в Примерах подробно изложены некоторые конкретные воплощения изобретения и описан наилучший вариант, предусмотренный авторами изобретения. Следует иметь в виду, однако, что независимо от того, насколько подробным может быть изложенное выше в тексте, изобретение может быть осуществлено на практике многими путями, и изобретение следует толковать в соответствии с прилагаемой формулой изобретения и ее эквивалентами.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНЪЮГАТ ЛИГАНДА С ЦИТОТОКСИЧЕСКИМ ЛЕКАРСТВЕННЫМ СРЕДСТВОМ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2708461C2 |
| ПАРА-АМИНОБЕНЗИЛЬНЫЕ ЛИНКЕРЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В КОНЪЮГАТАХ | 2021 |
|
RU2839675C1 |
| ЦИТОТОКСИЧЕСКИЕ ПЕПТИДЫ И ИХ КОНЪЮГАТЫ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2012 |
|
RU2586885C2 |
| ПРОИЗВОДНЫЕ КАЛИХЕАМИЦИНА И ИХ КОНЪЮГАТЫ "АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО" | 2018 |
|
RU2732568C1 |
| КОНЪЮГАТ АНТИТЕЛА И ЛЕКАРСТВЕННОГО СРЕДСТВА И ЕГО ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ РАКА | 2015 |
|
RU2685259C2 |
| Аналоги сплицеостатина | 2013 |
|
RU2618523C2 |
| КОНЪЮГАТ АНТИТЕЛА ПРОТИВ IGF-1R С ЛЕКАРСТВЕННЫМ СРЕДСТВОМ И ЕГО ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ РАКА | 2015 |
|
RU2692563C2 |
| ЛИГАНДЫ, ТРОПНЫЕ К ПРОСТАТИЧЕСКОМУ СПЕЦИФИЧЕСКОМУ МЕМБРАННОМУ АНТИГЕНУ И ИХ ПРИМЕНЕНИЕ ДЛЯ ПОЛУЧЕНИЯ ДВОЙНЫХ КОНЪЮГАТОВ С ТЕРАПЕВТИЧЕСКИМИ АГЕНТАМИ НА ИХ ОСНОВЕ ДЛЯ КОМБИНИРОВАННОЙ ТЕРАПИИ ПСМА ЭКСПРЕССИРУЮЩИХ ОПУХОЛЕЙ | 2023 |
|
RU2841078C1 |
| КОНЪЮГАТЫ АНТИТЕЛА И ЛЕКАРСТВЕННОГО СРЕДСТВА (ADC) И КОНЪЮГАТЫ АНТИТЕЛА И ПРОЛЕКАРСТВА (APDC), СОДЕРЖАЩИЕ ФЕРМЕНТАТИВНО РАСЩЕПЛЯЕМЫЕ ГРУППЫ | 2016 |
|
RU2751512C2 |
| НОВЫЕ КОНЪЮГАТЫ СВЯЗЫВАЮЩЕЕ СОЕДИНЕНИЕ - АКТИВНОЕ СОЕДИНЕНИЕ (ADC) И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2610336C2 |









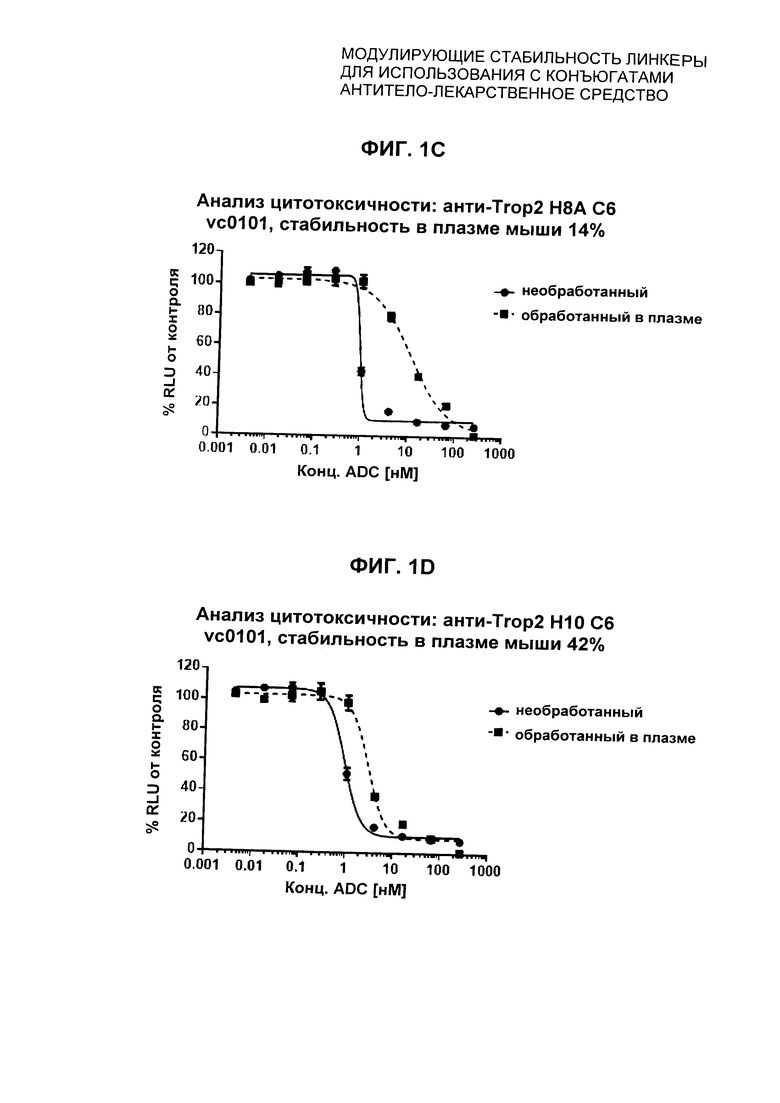
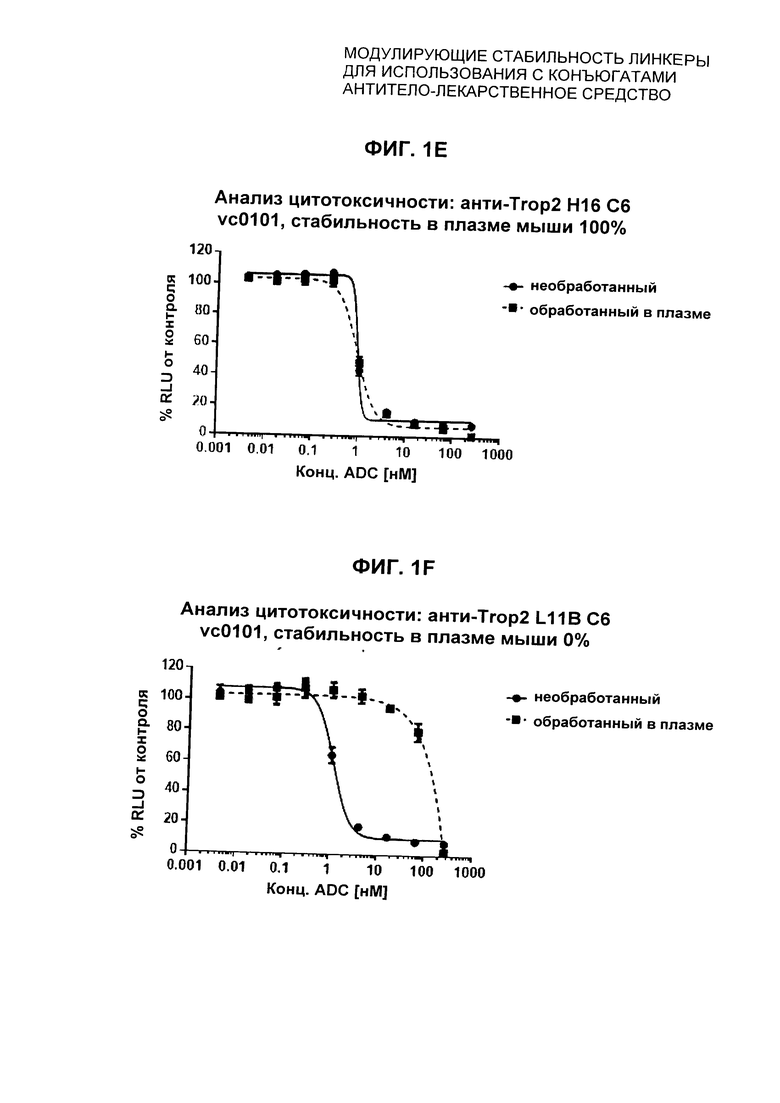
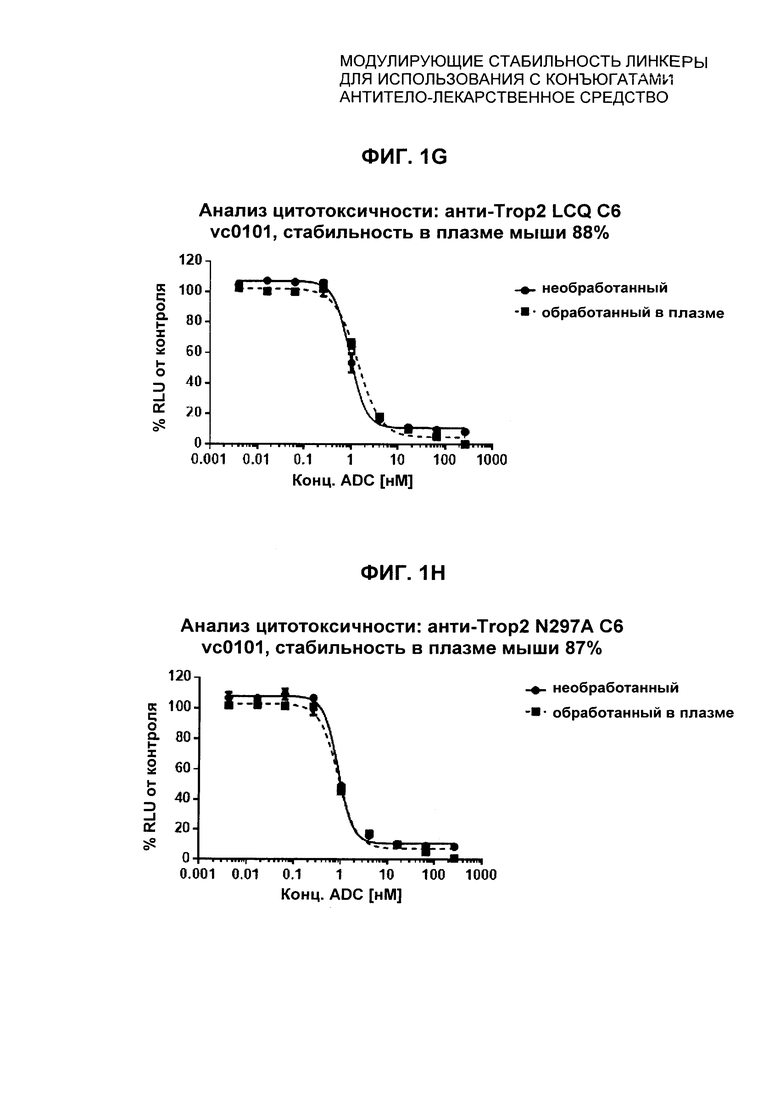
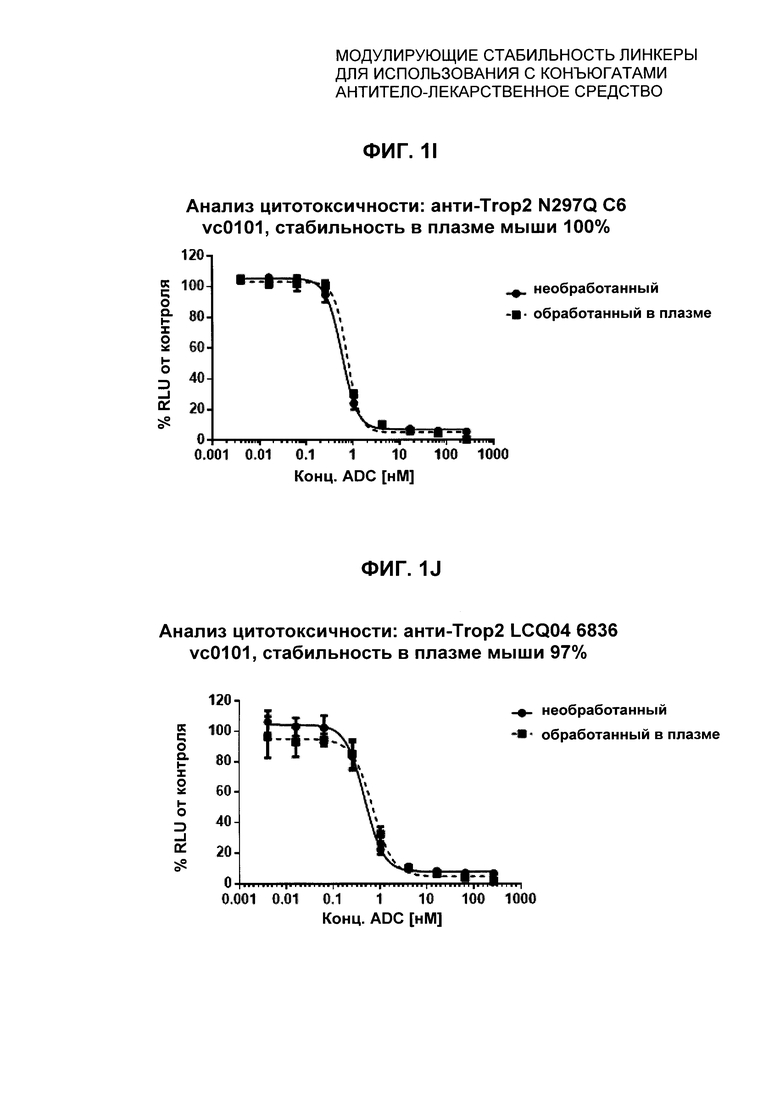
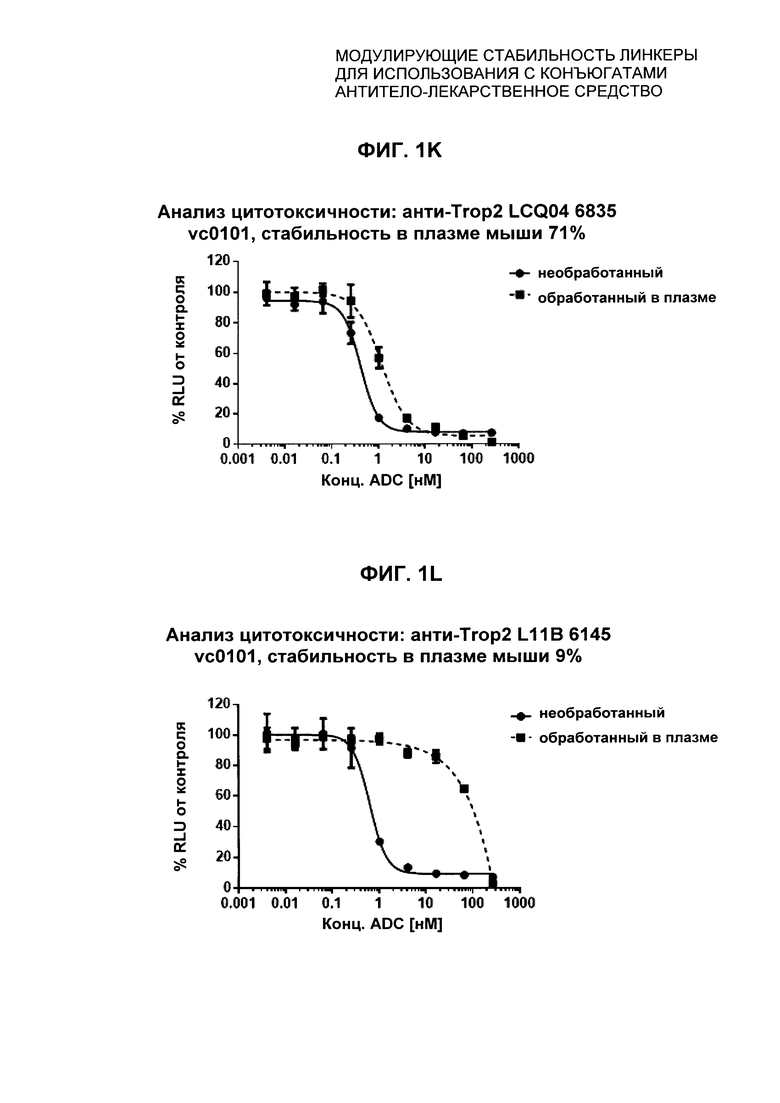
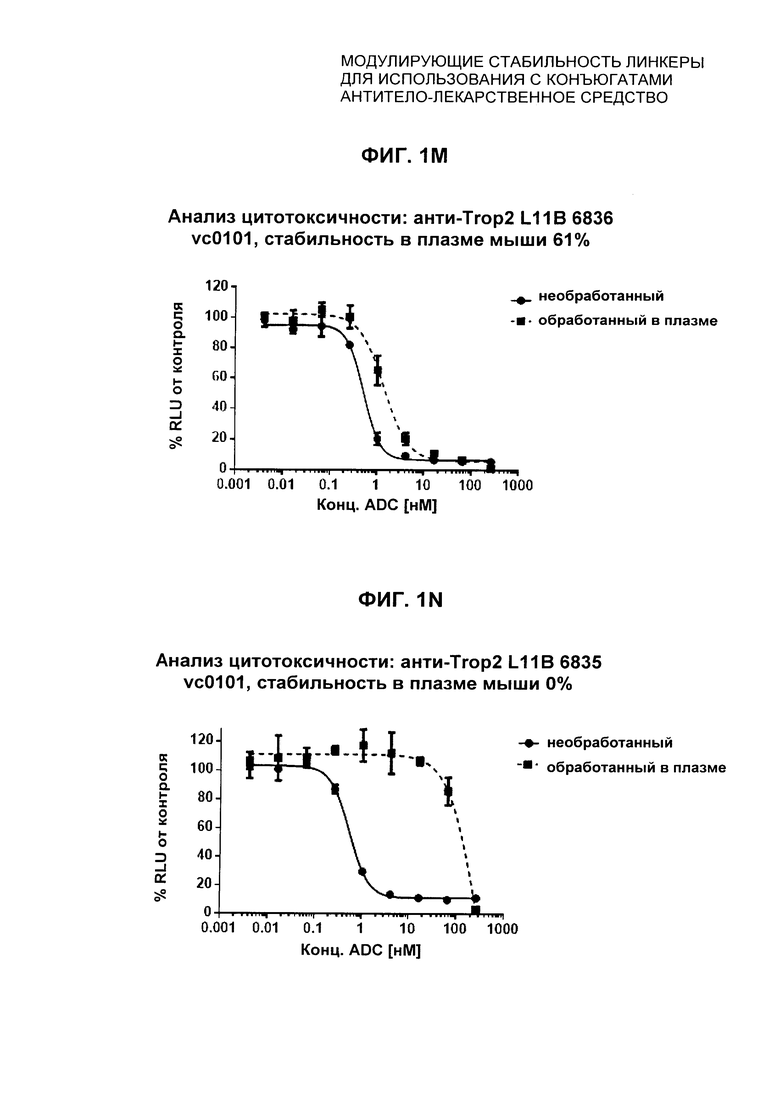
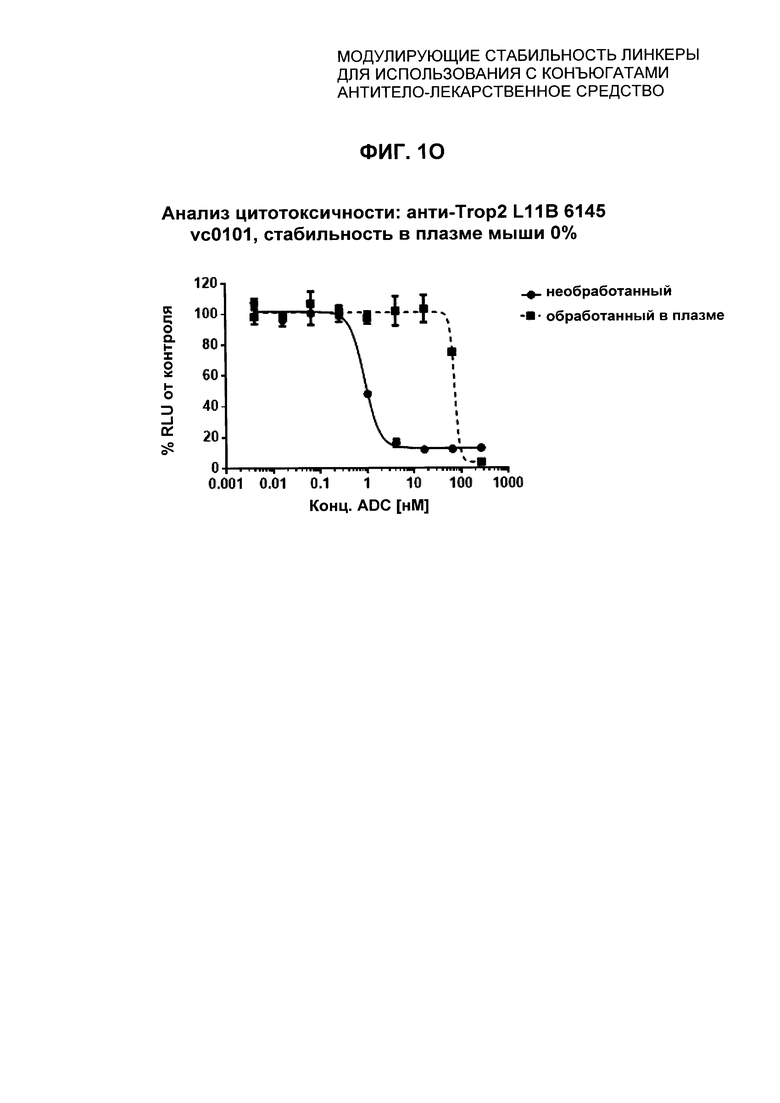
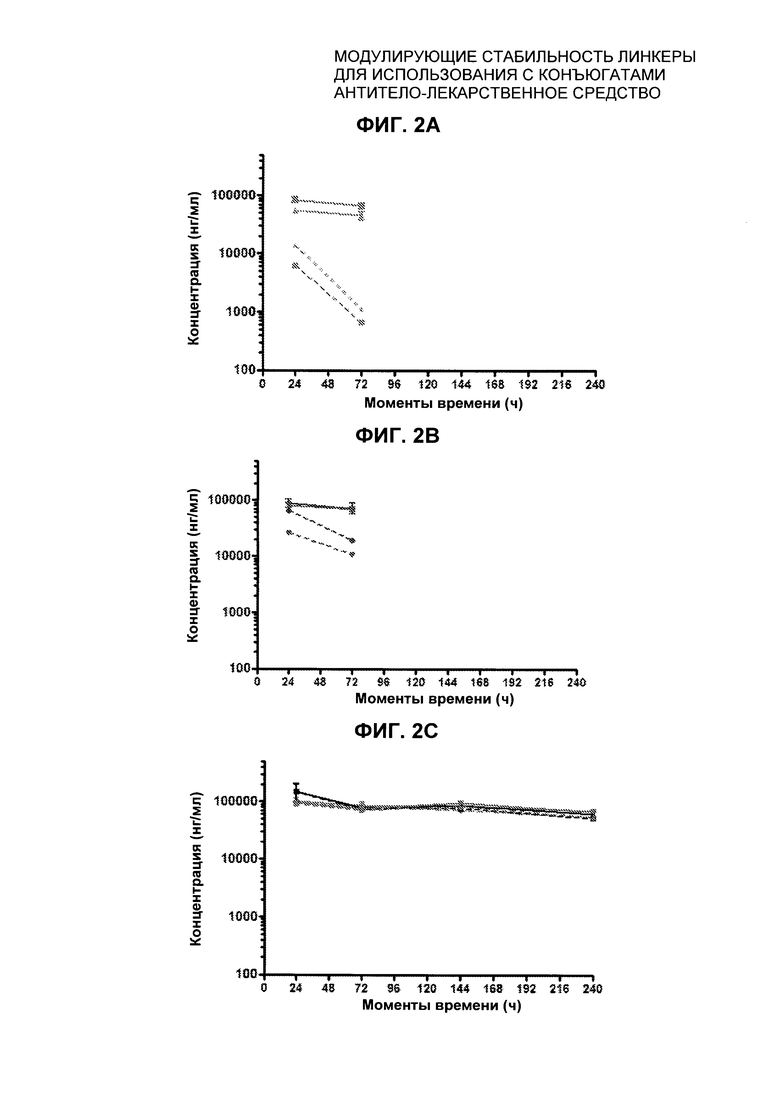
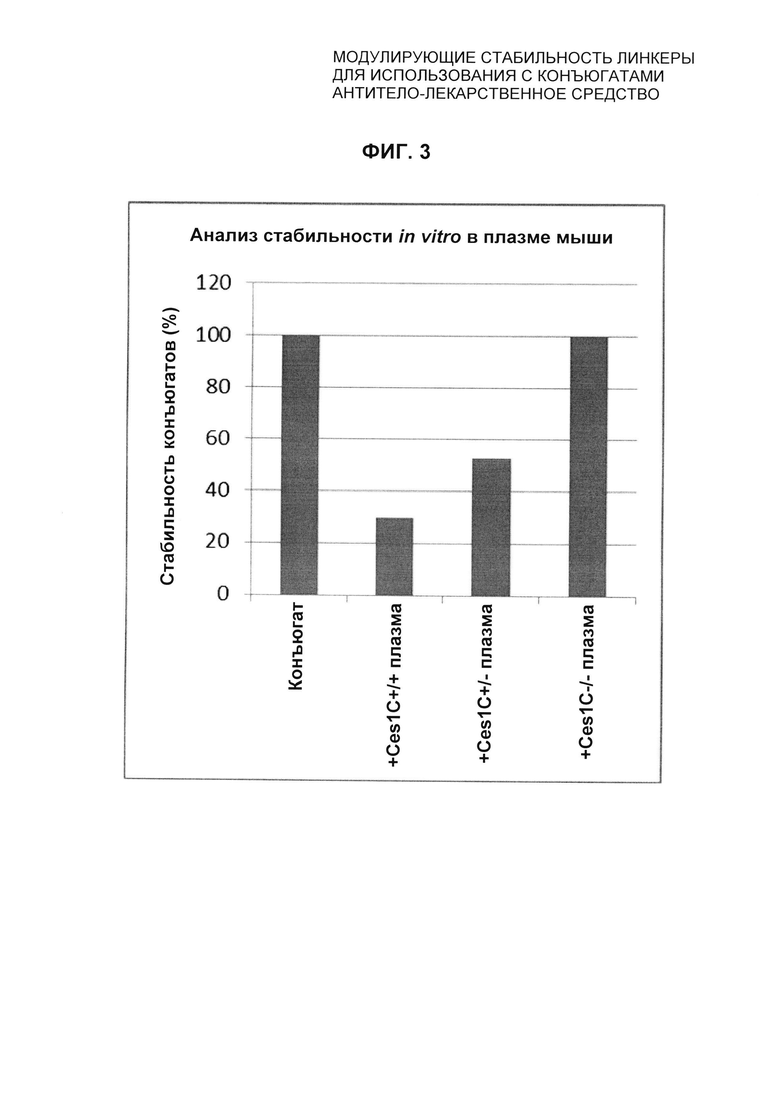
Группа изобретений относится к биотехнологии и медицине, в частности к конъюгатам антитело-лекарственное средство (ADC) с модулированной стабильностью, модулирующие стабильность линкерные компоненты для получения этих ADC, способ лечения рака с использованием указанных конъюгатов и способ их получения. Конъюгат антитело-лекарственное средство имеет структуру (I): 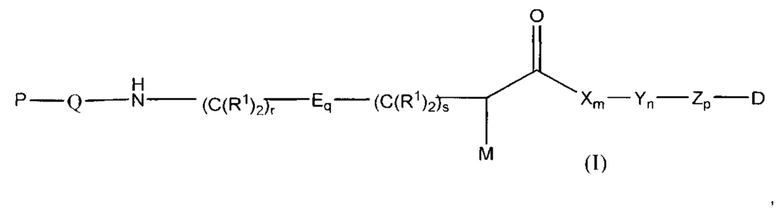
или структуру (II), а линкерный компонент - структуру (III), как определено в формуле изобретения. Конъюгат антитело-лекарственное средство получают в результате опосредованного трансглутаминазой конъюгирования. Способ лечения рака у нуждающегося в этом субъекта включает введение субъекту эффективного количества ADC. Линкеры (III), используемые в конъюгатах ADC, способны осуществлять повышающее и понижающее модулирование внеклеточной и/или внутриклеточной стабильности конъюгатов, чтобы большее или меньшее относительное количество полезной нагрузки ADC высвобождалось в желаемом месте действия. Изобретения позволяют осуществлять корректировку стабильности ADC in vivo и дают возможность оптимизировать терапевтический индекс ADC. 7 н. и 12 з.п. ф-лы, 3 ил., 9 табл., 72 пр.
1. Соединение формулы (I):
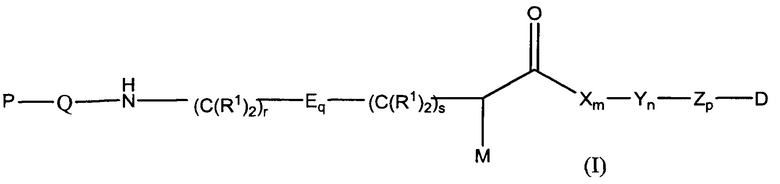
или его фармацевтически приемлемая(ый) соль или сольват;
где:
М представляет собой -М1-М2, где М1 представляет собой -NR1-C(O)-, -NR1-S(O)2-или отсутствует, и М2 выбран из группы, состоящей из -С2-С20алкила, С1-С6алкокси, -N(R1)2, -С6-С14арила, -С3-С10 насыщенного карбоциклического кольца, группы 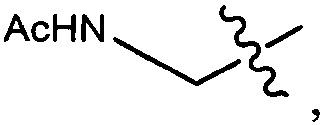 5-10 членного моно- или бициклического гетероарила, и неароматического -С3-С8гетероциклила, где указанный гетероарил содержит по меньшей мере один гетероатом азота и указанный гетероциклил содержит один гетероатом, выбранный из N, О, P и S; и где М2 возможно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из -C1-С6алкила, C1-С6алкокси, гидроксила, -N(R1)2, -С6-С14арила, -С3-С10 насыщенного карбоциклического кольца, группы -NR1-C(O)-N(R1)2, 5-10 членного моно- или бициклического гетероарила, и неароматического -С3-С8гетероциклила, где указанный гетероарил содержит по меньшей мере один гетероатом азота и указанный гетероциклил содержит один гетероатом, выбранный из N, О, P и S;
5-10 членного моно- или бициклического гетероарила, и неароматического -С3-С8гетероциклила, где указанный гетероарил содержит по меньшей мере один гетероатом азота и указанный гетероциклил содержит один гетероатом, выбранный из N, О, P и S; и где М2 возможно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из -C1-С6алкила, C1-С6алкокси, гидроксила, -N(R1)2, -С6-С14арила, -С3-С10 насыщенного карбоциклического кольца, группы -NR1-C(O)-N(R1)2, 5-10 членного моно- или бициклического гетероарила, и неароматического -С3-С8гетероциклила, где указанный гетероарил содержит по меньшей мере один гетероатом азота и указанный гетероциклил содержит один гетероатом, выбранный из N, О, P и S;
или М2 представляет собой метил, замещенный одним заместителем, выбранным из группы, состоящей из гидроксила, N(R1)2, -С6-С14арила, 5-10 членного моно- или бициклического гетероарила, где указанный гетероарил содержит по меньшей мере один гетероатом азота;
P представляет собой пептидную последовательность, которая содержит один или более остатков глутамина, где P выбран из группы, состоящей из моноклонального антитела, поликлонального антитела, человеческого антитела, гуманизированного антитела, химерного антитела, биспецифического антитела, минитела и фрагмента антитела;
Q представляет собой остаток глутамина, присутствующий в P;
каждый E независимо выбран из группы, состоящей из -C(R1)2-, -O-C(R1)2-C(R1)2-, где r равно 2, и -C(R1)2-C(R1)2-O-, где s равно 1-2;
каждый R1 независимо выбран из группы, состоящей из Н, прямого или разветвленного C1-С6алкила, прямого или разветвленного С2-С6алкенила и прямого или разветвленного С2-С6алкинила;
каждый X независимо представляет собой аминокислоту, где каждая аминокислота X является одной и той же или другой;
каждый Y независимо представляет собой аминокислоту, где каждая аминокислота Y является одной и той же или другой;
где X-Y выбран из группы, состоящей из Gly, β-Аlа, Val-Cit, Phe-Lys, Val-Lys, Phe-Phe-Lys, Ala-Lys, Phe-Cit, Leu-Cit, Ala-Cit, Trp-Cit, Phe-Ala, Gly-Phe-Leu-Gly, Ala-Leu-Ala-Leu, Phe-N9-тозил-Arg, Phe-N9-нитро-Arg, Val-Ala и Ala-Ala-Asn;
каждый Z независимо представляет собой спейсерный элемент, представляющий собой 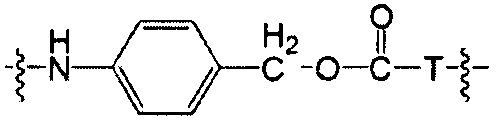 , где T представляет собой O, NH или S, где каждый спейсерный элемент является одним и тем же или другим;
, где T представляет собой O, NH или S, где каждый спейсерный элемент является одним и тем же или другим;
m равно 0-5, n равно 1-5, p равно 0-2, q равно 0-10, r равно 0-2, и s равно 0-2, где q+r+s=2 или более; и
D представляет собой цитотоксический агент.
2. Соединение формулы (II):

или его фармацевтически приемлемая(ый) соль или сольват;
где:
М представляет собой -М1-М2, где М1 представляет собой -NR1-C(O)-, -NR1-S(O)2- или отсутствует, и М2 выбран из группы, состоящей из -С2-С20алкила, С1-С6алкокси, -N(R1)2, -С6-С14арила, -С3-С10 насыщенного карбоциклического кольца, группы 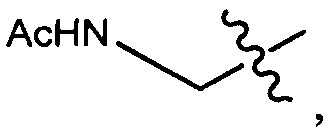 5-10 членного моно- или бициклического гетероарила, и неароматического -С3-С8гетероциклила, где указанный гетероарил содержит по меньшей мере один гетероатом азота и указанный гетероциклил содержит один гетероатом, выбранный из N, О, Р и S; и где М2 возможно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из -C1-С6алкила, C1-С6алкокси, гидроксила, -N(R1)2, -С6-С14арила, -С3-С10 насыщенного карбоциклического кольца, группы -NRl-C(O)-N(R1)2, 5-10 членного моно- или бициклического гетероарила, и неароматического -С3-С8гетероциклила, где указанный гетероарил содержит по меньшей мере один гетероатом азота и указанный гетероциклил содержит один гетероатом, выбранный из N, О, Р и S;
5-10 членного моно- или бициклического гетероарила, и неароматического -С3-С8гетероциклила, где указанный гетероарил содержит по меньшей мере один гетероатом азота и указанный гетероциклил содержит один гетероатом, выбранный из N, О, Р и S; и где М2 возможно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из -C1-С6алкила, C1-С6алкокси, гидроксила, -N(R1)2, -С6-С14арила, -С3-С10 насыщенного карбоциклического кольца, группы -NRl-C(O)-N(R1)2, 5-10 членного моно- или бициклического гетероарила, и неароматического -С3-С8гетероциклила, где указанный гетероарил содержит по меньшей мере один гетероатом азота и указанный гетероциклил содержит один гетероатом, выбранный из N, О, Р и S;
или М2 представляет собой метил, замещенный одним заместителем, выбранным из группы, состоящей из гидроксила, N(R1)2, -С6-С14арила, 5-10 членного моно- или бициклического гетероарила, где указанный гетероарил содержит по меньшей мере один гетероатом азота;
P представляет собой пептидную последовательность, которая содержит один или более остатков глутамина, где Р выбран из группы, состоящей из моноклонального антитела, поликлонального антитела, человеческого антитела, гуманизированного антитела, химерного антитела, биспецифического антитела, минитела и фрагмента антитела;
Q представляет собой один из указанных остатков глутамина, присутствующих в Р;
каждый E независимо выбран из группы, состоящей из -C(R1)2-, -O-C(R1)2-C(R1)2-, где r равно 2, и -C(R1)2-C(R1)2-O-, где s равно 1-2;
каждый R1 независимо выбран из группы, состоящей из Н, прямого или разветвленного С1-С6алкила, прямого или разветвленного С2-С6алкенила и прямого или разветвленного С2-С6алкинила;
каждый X независимо представляет собой аминокислоту, где каждая аминокислота X является одной и той же или другой;
каждый Y независимо представляет собой аминокислоту, где каждая аминокислота Y является одной и той же или другой;
где X-Y выбран из группы, состоящей из Gly, β-Ala, Val-Cit, Phe-Lys, Val-Lys, Phe-Phe-Lys, Ala-Lys, Phe-Cit, Leu-Cit, Ala-Cit, Trp-Cit, Phe-Ala, Gly-Phe-Leu-Gly, Ala-Leu-Ala-Leu, Phe-N9-тозил-Arg, Phe-N9-нитро-Arg, Val-Ala и Ala-Ala-Asn;
каждый Z независимо представляет собой спейсерный элемент, представляющий собой 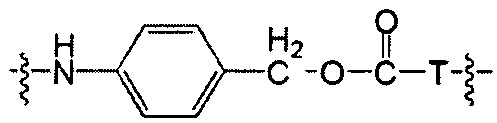 , где T представляет собой O, NH или S, где каждый спейсерный элемент является одним и тем же или другим;
, где T представляет собой O, NH или S, где каждый спейсерный элемент является одним и тем же или другим;
m равно 0-5, n равно 1-5, p равно 0-2, q равно 0-10, r равно 0-2, и s равно 0-2, где q+r+s=2 или более; и
D представляет собой цитотоксический агент.
3. Соединение формулы (III):
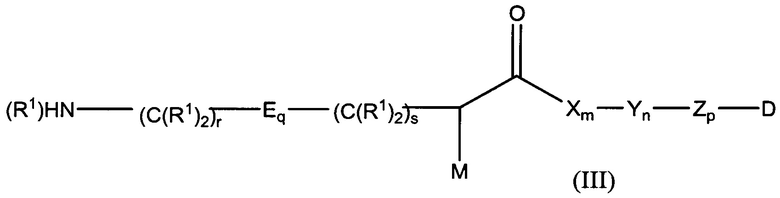
или его фармацевтически приемлемая(ый) соль или сольват;
где:
М представляет собой -М1-М2, где М1 представляет собой -NR1-C(O)-, -NR1-S(O)2- или отсутствует, и М2 выбран из группы, состоящей из -С2-С20алкила, C1-С6алкокси, -N(R1)2, -С6-С14арила, -С3-С10 насыщенного карбоциклического кольца, группы 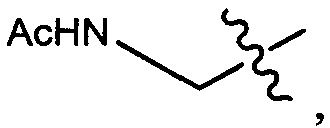 5-10 членного моно- или бициклического гетероарила, и неароматического -С3-С8гетероциклила, где указанный гетероарил содержит по меньшей мере один гетероатом азота и указанный гетероциклил содержит один гетероатом, выбранный из N, О, Р и S; и где М2 возможно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из -C1-С6алкила, C1-С6алкокси, гидроксила, -N(R1)2, -С6-С14арила, -С3-С10 насыщенного карбоциклического кольца, группы -NR1-C(O)-N(R1)2, 5-10 членного моно- или бициклического гетероарила, и неароматического -С3-С8гетероциклила, где указанный гетероарил содержит по меньшей мере один гетероатом азота и указанный гетероциклил содержит один гетероатом, выбранный из N, О, Р и S;
5-10 членного моно- или бициклического гетероарила, и неароматического -С3-С8гетероциклила, где указанный гетероарил содержит по меньшей мере один гетероатом азота и указанный гетероциклил содержит один гетероатом, выбранный из N, О, Р и S; и где М2 возможно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из -C1-С6алкила, C1-С6алкокси, гидроксила, -N(R1)2, -С6-С14арила, -С3-С10 насыщенного карбоциклического кольца, группы -NR1-C(O)-N(R1)2, 5-10 членного моно- или бициклического гетероарила, и неароматического -С3-С8гетероциклила, где указанный гетероарил содержит по меньшей мере один гетероатом азота и указанный гетероциклил содержит один гетероатом, выбранный из N, О, Р и S;
или М2 представляет собой метил, замещенный одним заместителем, выбранным из группы, состоящей из гидроксила, N(R1)2, -С6-С14арила, 5-10 членного моно- или бициклического гетероарила, где указанный гетероарил содержит по меньшей мере один гетероатом азота;
каждый E независимо выбран из группы, состоящей из -C(R1)2-, -O-C(R1)2-C(R1)2-, где r равно 2, и -C(R1)2-C(R1)2-O-, где s равно 1-2;
каждый R1 независимо выбран из группы, состоящей из Н, прямого или разветвленного C1-С6алкила, прямого или разветвленного С2-С6алкенила и прямого или разветвленного С2-С6алкинила;
каждый X независимо представляет собой аминокислоту, где каждая аминокислота X является одной и той же или другой;
каждый Y независимо представляет собой аминокислоту, где каждая аминокислота Y является одной и той же или другой;
где X-Y выбран из группы, состоящей из Gly, β-Alа, Val-Cit, Phe-Lys, Val-Lys, Phe-Phe-Lys, Ala-Lys, Phe-Cit, Leu-Cit, Ala-Cit, Trp-Cit, Phe-Ala, Gly-Phe-Leu-Gly, Ala-Leu-Ala-Leu, Phe-N9-тозил-Arg, Phe-N9-нитро-Arg, Val-Ala и Ala-Ala-Asn;
каждый Z независимо представляет собой спейсерный элемент, представляющий собой 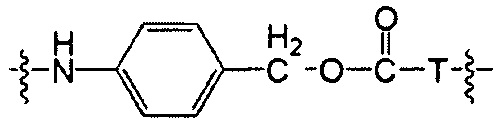 , где T представляет собой O, NH или S, где каждый спейсерный элемент является одним и тем же или другим;
, где T представляет собой O, NH или S, где каждый спейсерный элемент является одним и тем же или другим;
m равно 0-5, n равно 1-5, p равно 0-2, q равно 0-10, r равно 0-2, и s равно 0-2, где q+r+s=2 или более; и
D представляет собой цитотоксический агент.
4. Соединение по любому из пп.1-3, где М представляет собой -М1-М2, где М1 представляет собой -NR1-C(O)-, -NR1-S(O)2- или отсутствует, и М2 выбран из группы, состоящей из -С2-С10алкила -С6-С14арила, 5-10 членного моноциклического или бициклического гетероарила, неароматического -С3-С8гетероциклила и -С3-С10 насыщенного карбоциклического кольца, и где М2 возможно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из -C1-С6алкила, -С6-С14арила и 5-10 членного моно- или бициклического гетероарила.
5. Соединение по п.4, где М2 выбран из группы, состоящей из:
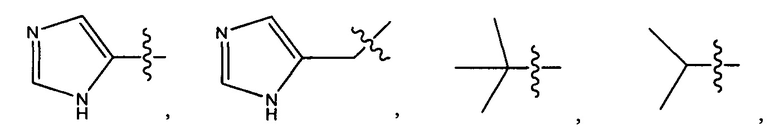
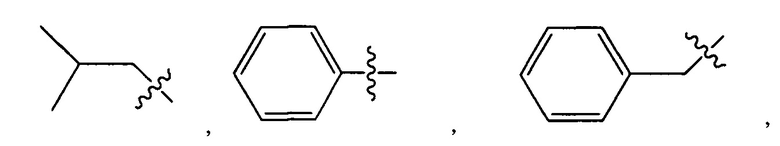

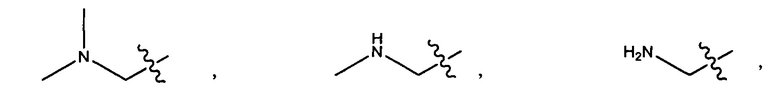
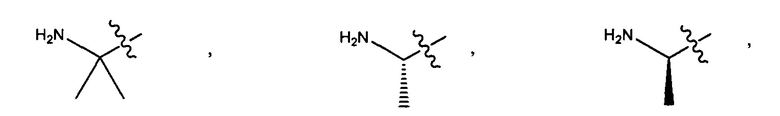


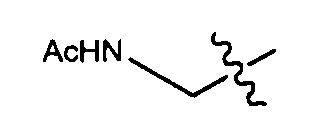 и
и 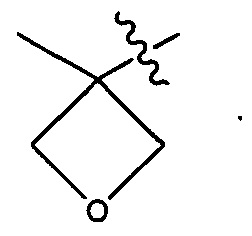 .
.
6. Соединение по любому из пп.1-5, где цитотоксический агент D выбран из группы, состоящей из антрациклина, ауристатина, сплицеостатина, димера CBI (1-(хлорметил)-2,3-дигидро-1Н-бензо[е]индол)/CPI (циклопропа[с]пирроло[3,2-е]индол), калихеамицина, дуокармицина, энедиина, гелданамицина, майтанзина, пуромицина, таксана, алкалоида барвинка, SN-38, тубулизина, гемиастерлина, камптотецина, комбретастатина, доластатина, димера индолинобензодиазепина, димера пирролобензодиазепина и пладиенолида и их стереоизомеров, изостеров, аналогов или производных.
7. Соединение по любому из пп.1-6, где цитотоксический агент D представляет собой ауристатин, выбранный из группы, состоящей из долестатина, MMAD, ММАЕ, MMAF, PF-06380101, PF-06463377 и PF-06456780.
8. Соединение по п.1 или 2, где P представляет собой пептид, содержащий аминокислотную последовательность, выбранную из группы, состоящей из LQG, LLQGG (SEQ ID NO: 1), LLQG (SEQ ID NO: 2), LSLSQG (SEQ ID NO: 3), GGGLLQGG (SEQ ID NO: 4), GLLQG (SEQ ID NO: 5), LLQ, GSPLAQSHGG (SEQ ID NO: 6), GLLQGGG (SEQ ID NO: 7), GLLQGG (SEQ ID NO: 8), GLLQ (SEQ ID NO: 9), LLQLLQGA (SEQ ID NO: 10), LLQGA (SEQ ID NO: 11), LLQYQGA (SEQ ID NO: 12), LLQGSG (SEQ ID NO: 13), LLQYQG (SEQ ID NO: 14), LLQLLQG (SEQ ID NO: 15), SLLQG (SEQ ID NO: 16), LLQLQ (SEQ ID NO: 17), LLQLLQ (SEQ ID NO: 18), LLQGR (SEQ ID NO: 19), LLQGPP (SEQ ID NO: 20), LLQGPA (SEQ ID NO: 21), GGLLQGPP (SEQ ID NO: 22), GGLLQGA (SEQ ID NO: 23), LLQGA (SEQ ID NO: 24), LLQGPGK (SEQ ID NO: 25), LLQGPG (SEQ ID NO: 26), LLQGP (SEQ ID NO: 27), LLQP (SEQ ID NO: 28), LLQPGK (SEQ ID NO: 29), LLQAPGK (SEQ ID NO: 30), LLQGAPG (SEQ ID NO: 31), LLQGAP (SEQ ID NO: 32), LLQGPA (SEQ ID NO: 33), LLQGPP (SEQ ID NO: 34), GGLLQGPP (SEQ ID NO: 35), LLQLQG (SEQ ID NO: 36) и XXQX(SEQ ID NO: 37), где X представляет собой любую аминокислоту.
9. Соединение, выбранное из группы, состоящей из:
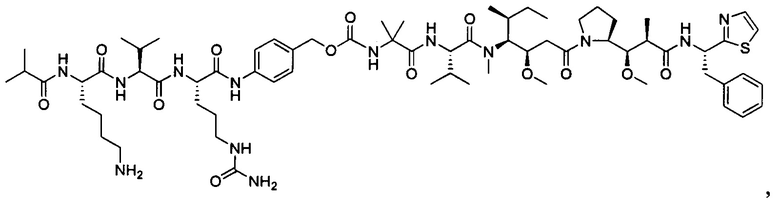
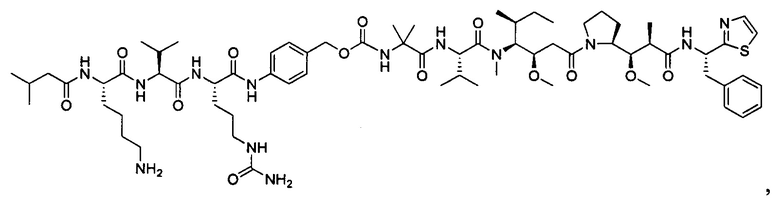
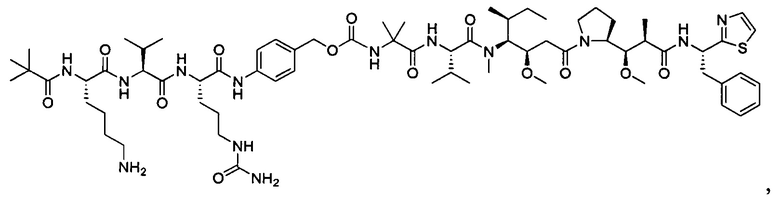
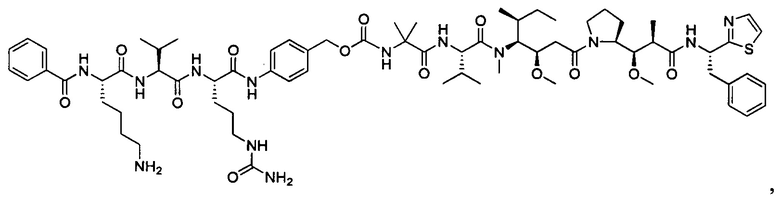
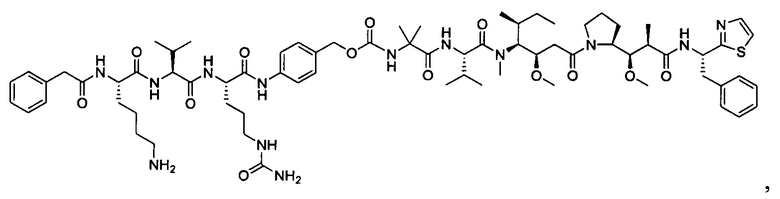
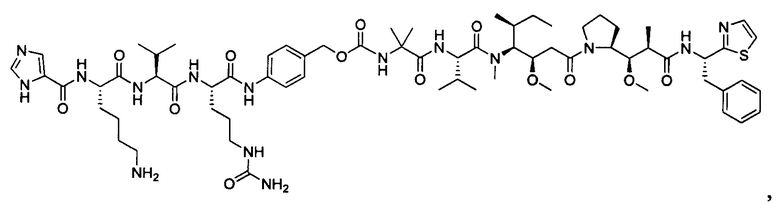
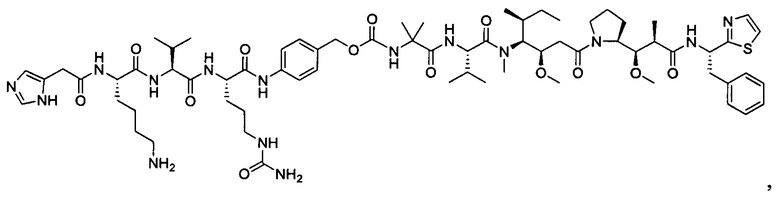

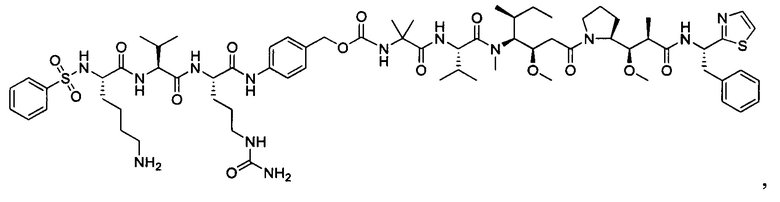
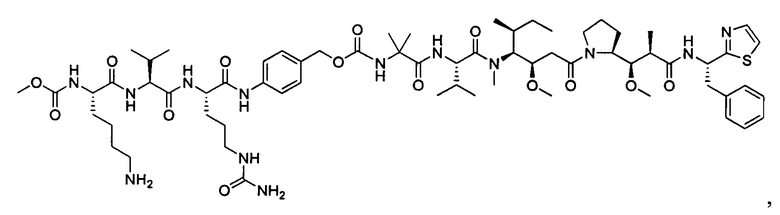
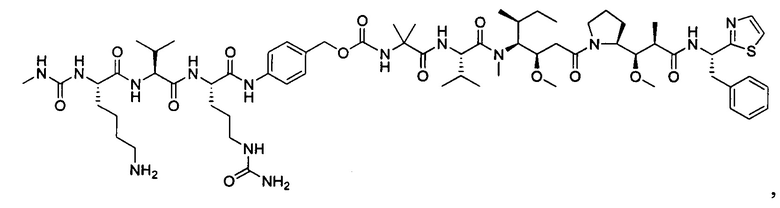
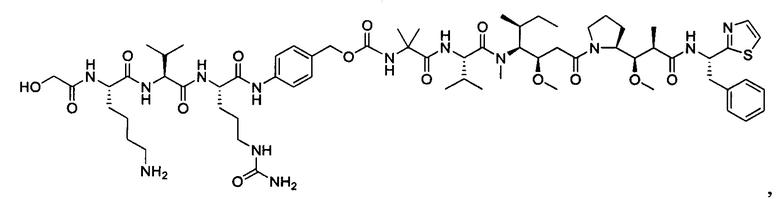
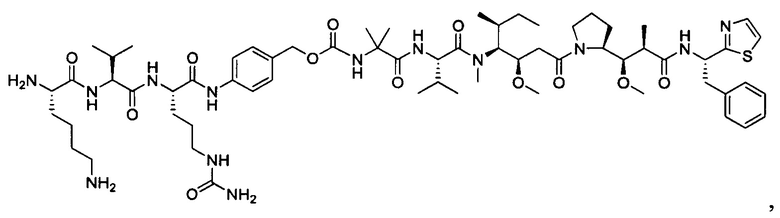
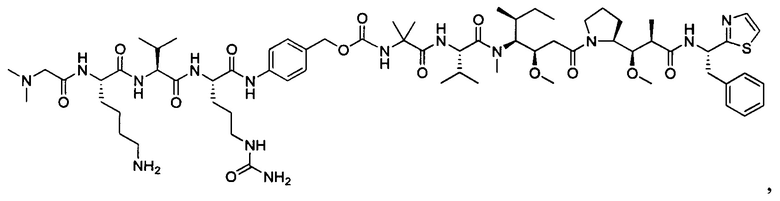
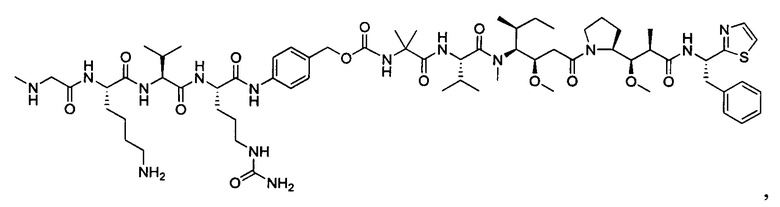
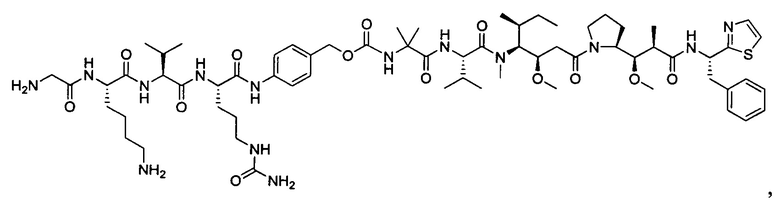

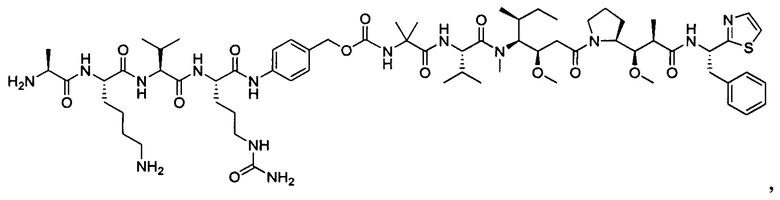
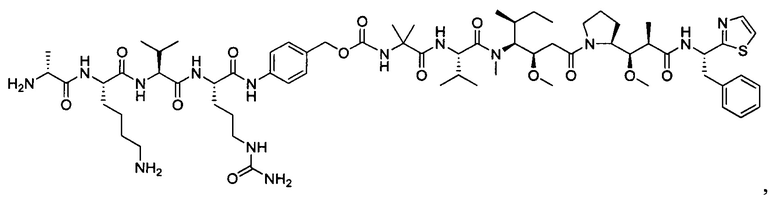
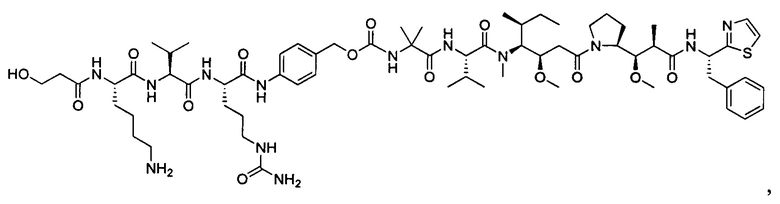
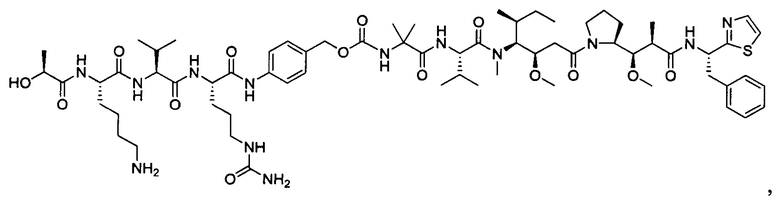

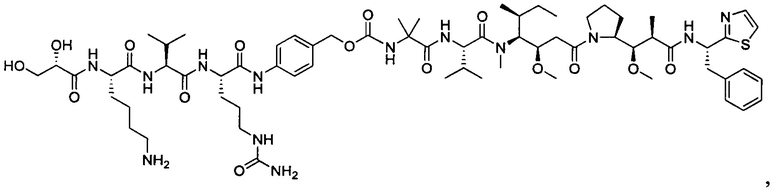
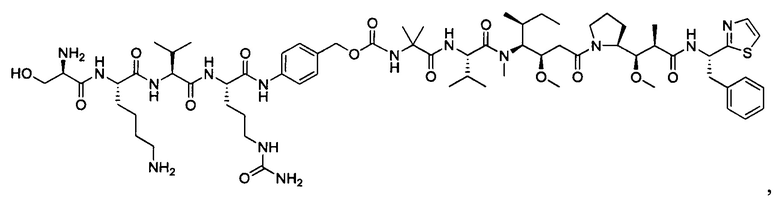
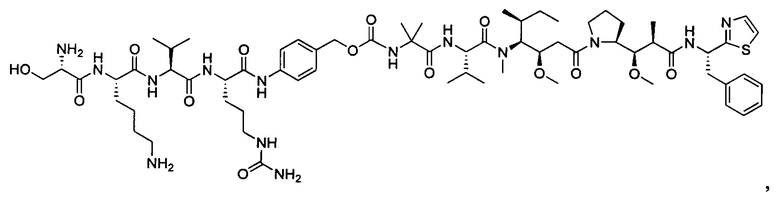
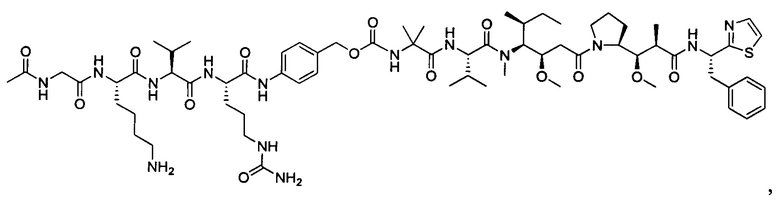

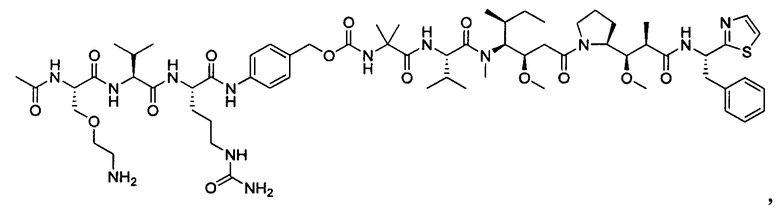
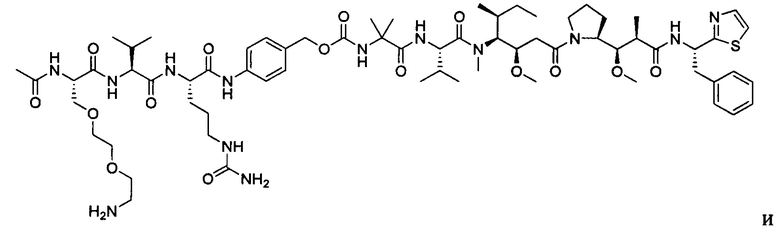

или его фармацевтически приемлемая(ый) соль или сольват.
10. Соединение, выбранное из группы, состоящей из:
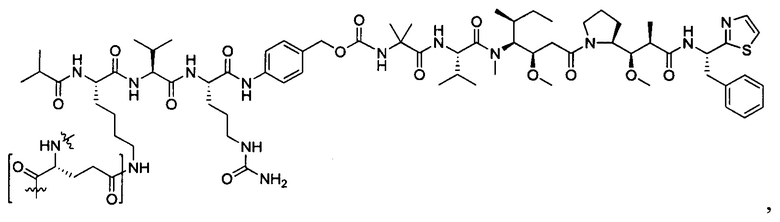
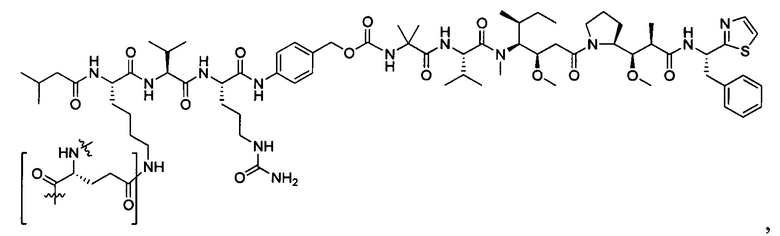
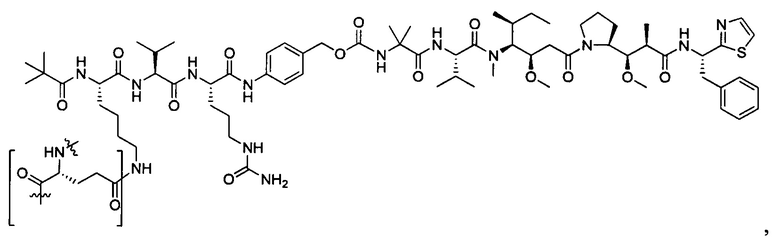
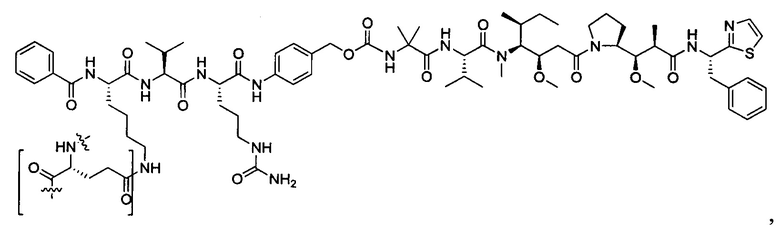
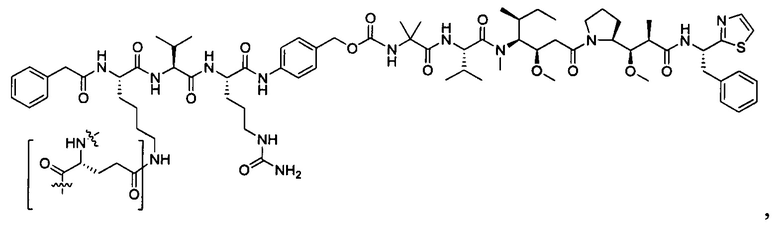
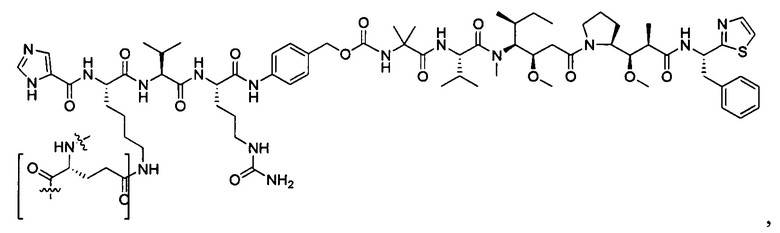
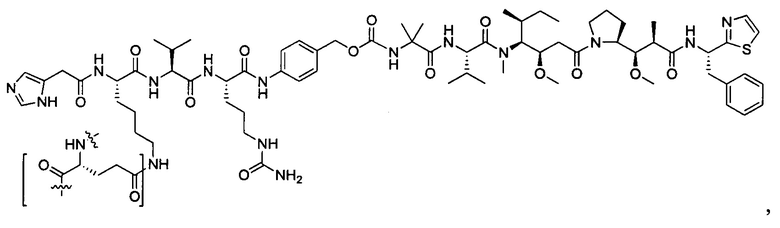
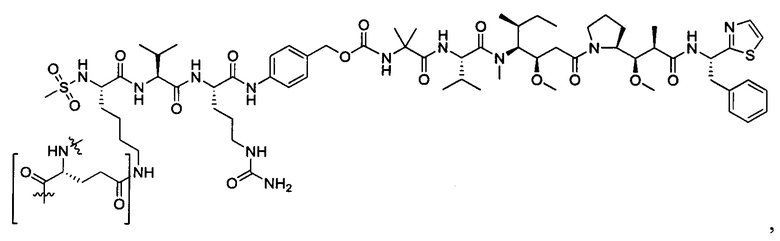
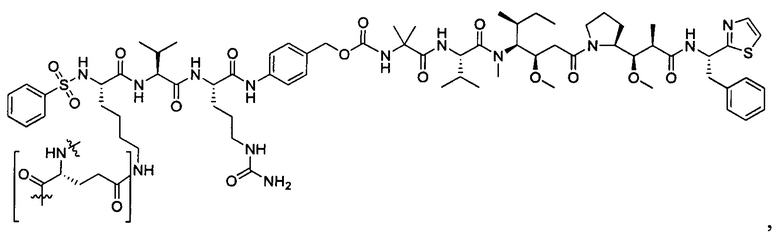
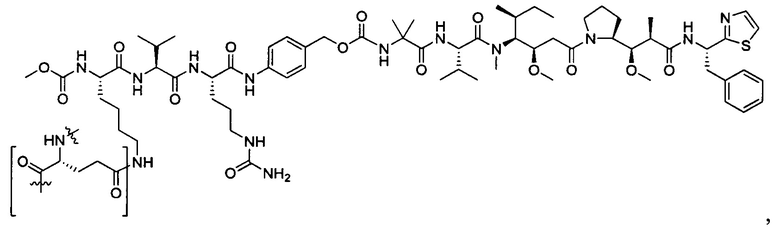
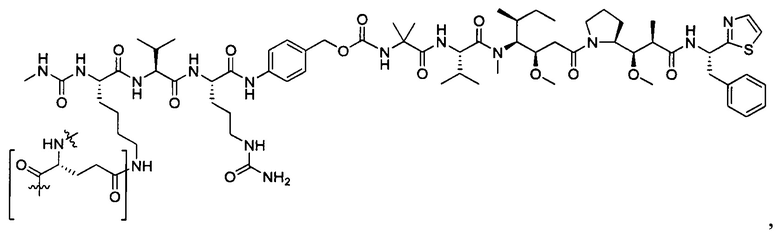
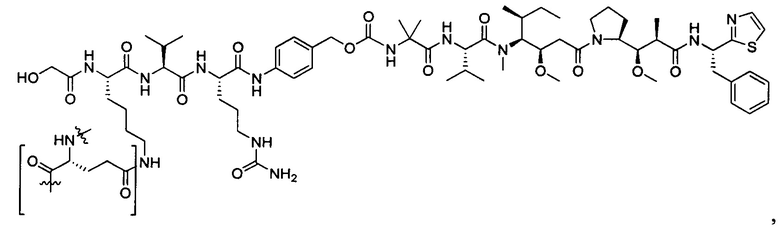
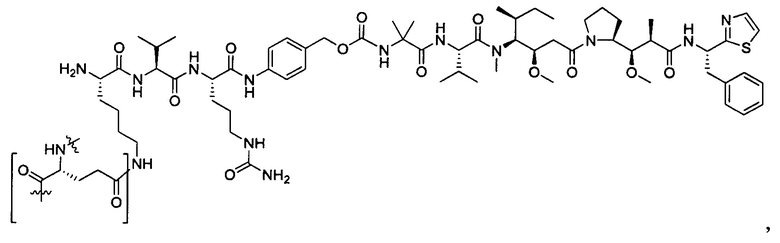


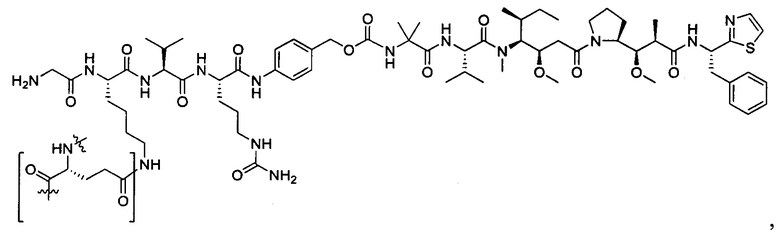
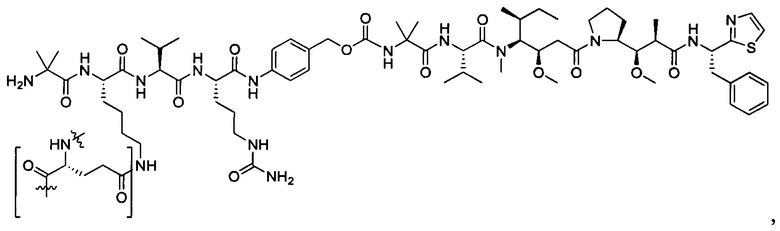
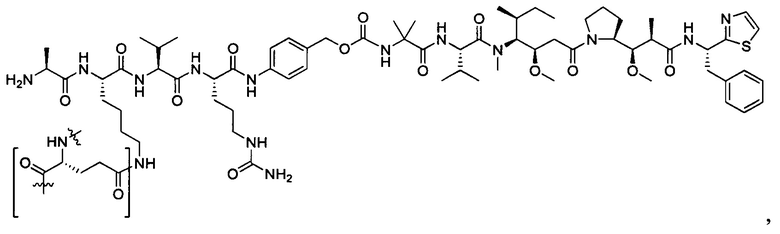

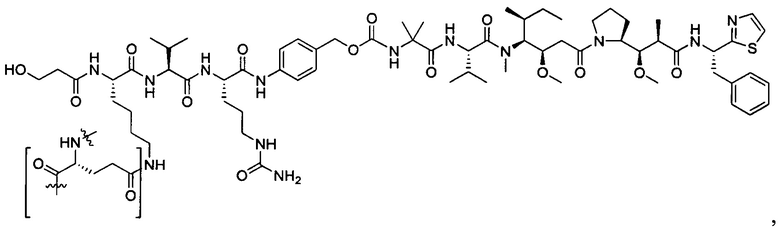
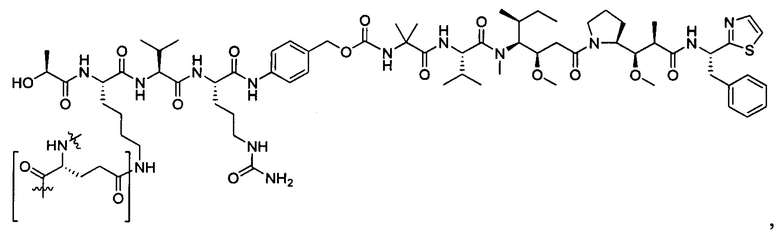

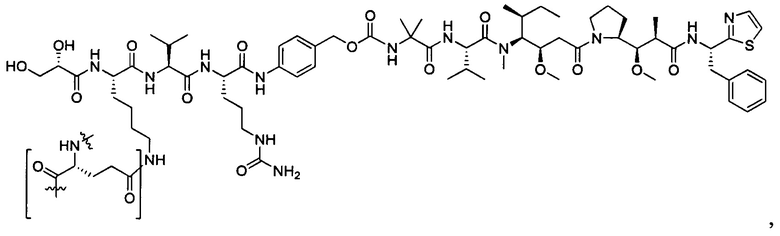
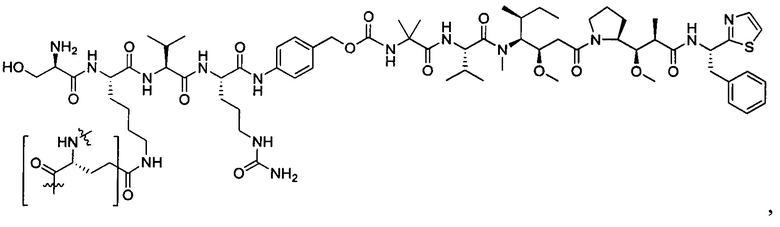
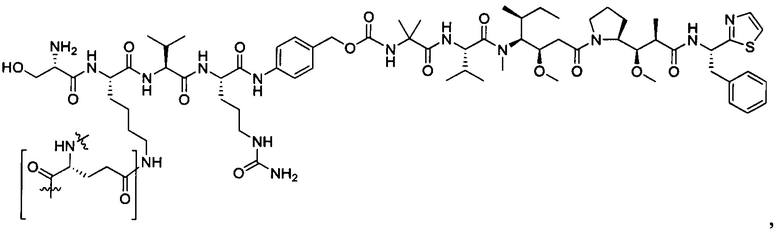
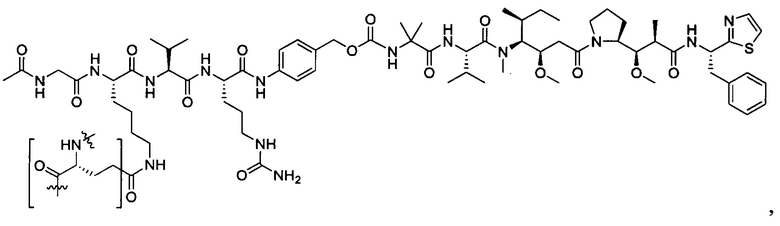
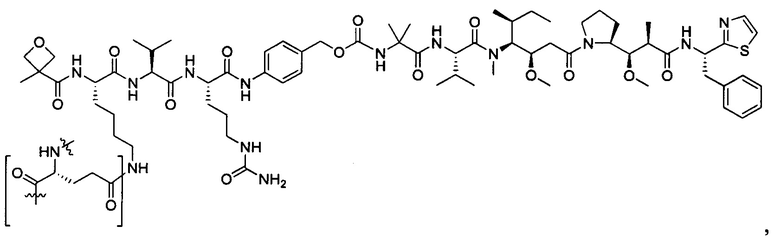
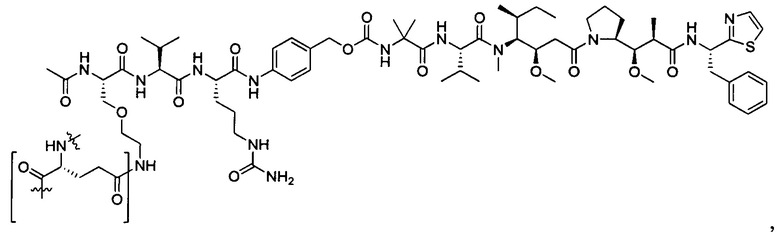
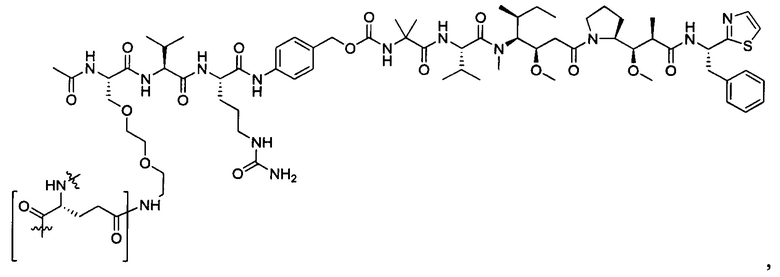
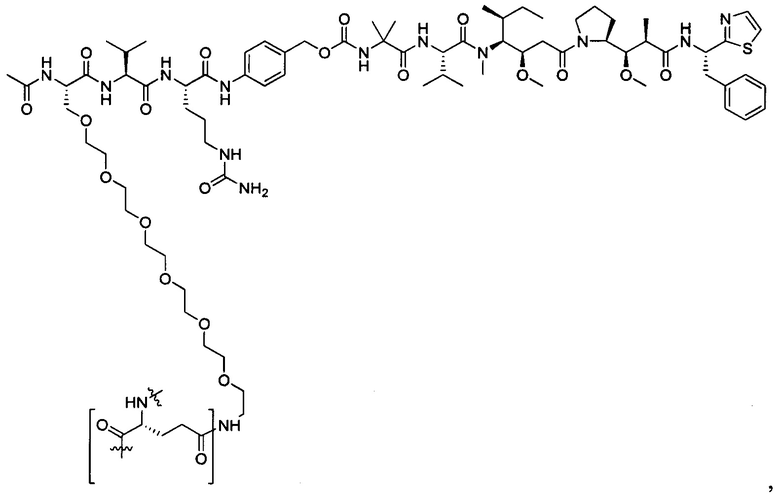
или его фармацевтически приемлемая(ый) соль или сольват, где указанная в скобках часть соединения представляет собой остаток глутамина в пептидной последовательности.
11. Соединение по п.1 или 2, где стабильность указанного соединения уменьшается на 5-50% по сравнению с соответствующим соединением, не имеющим указанного модулятора М.
12. Соединение по п.1 или 2, где стабильность указанного соединения увеличивается на 5-50% по сравнению с соответствующим соединением, не имеющим указанного модулятора М.
13. Соединение по п.1 или 2, где указанное антитело выбрано из группы, состоящей из трастузумаба, трастузумаба мутантов, ореговомаба, эдреколомаба, цетуксимаба, гуманизированного моноклонального антитела к рецептору витронектина (αvβ3), алемтузумаба, анти-HLA-DR антител, 1311 Lym-1, анти-HLA-Dr10 антител, анти-cd33 антител, анти-cd22 антител, лабетузумаба, бевацизумаба, ибритумомаба тиуксетана, офатумумаба, панитумумаба, ритуксимаба, тозитумомаба, ипилимумаба и гемтузумаба.
14. Соединение по п.1 или 2, где каждый Q независимо представляет собой остаток глутамина, эндогенный для указанной пептидной последовательности P, или остаток глутамина, предоставленный в последовательности сконструированной метки на указанной пептидной последовательности Р.
15. Соединение по п.1 или 2, где один или более Q представляет собой остаток глутамина, эндогенный для указанной пептидной последовательности Р.
16. Соединение по п.1 или 2, где один или более Q представляет собой остаток глутамина, предоставленный в последовательности сконструированной метки на указанной пептидной последовательности Р.
17. Соединение по п.16, где одна или более чем одна метка представляет собой единственную аминокислоту глутамин.
18. Способ лечения рака, включающий введение эффективного количества соединения по п. 1 или 2 пациенту.
19. Способ синтеза соединения следующей формулы:
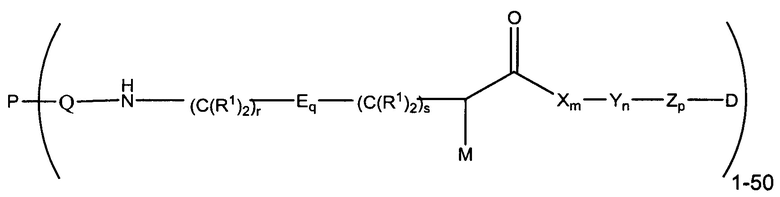
где:
М представляет собой -М1-М2, где М1 представляет собой -NR1-C(O)-, -NR1-S(O)2-или отсутствует, и М2 выбран из группы, состоящей из -С2-С20алкила, C1-С6алкокси, -N(R1)2, -С6-С14арила, -С3-С10 насыщенного карбоциклического кольца, группы 5-10 членного моно- или бициклического гетероарила, и неароматического -С3-С8гетероциклила, где указанный гетероарил содержит по меньшей мере один гетероатом азота и указанный гетероциклил содержит один гетероатом, выбранный из N, О, Р и S; и где М2 возможно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из -C1-С6алкила, C1-С6алкокси, гидроксила, -N(R1)2, -С6-С14арила, -С3-С10 насыщенного карбоциклического кольца, группы -NR1-C(O)-N(R1)2, 5-10 членного моно- или бициклического гетероарила, и неароматического -С3-С8гетероциклила, где указанный гетероарил содержит по меньшей мере один гетероатом азота и указанный гетероциклил содержит один гетероатом, выбранный из N, О, Р и S;
или М2 представляет собой метил, замещенный одним заместителем, выбранным из группы, состоящей из гидроксила, N(R1)2, -С6-С14арила, 5-10 членного моно- или бициклического гетероарила, где указанный гетероарил содержит по меньшей мере один гетероатом азота;
P представляет собой пептидную последовательность, которая содержит один или более остатков глутамина, где Р выбран из группы, состоящей из моноклонального антитела, поликлонального антитела, человеческого антитела, гуманизированного антитела, химерного антитела, биспецифического антитела, минитела и фрагмента антитела;
Q представляет собой один из указанных остатков глутамина, присутствующих в Р;
каждый E независимо выбран из группы, состоящей из -C(R1)2-, -O-C(R1)2-C(R1)2-, где r равно 2, и -C(R1)2-C(R1)2-O-, где s равно 1-2;
каждый R1 независимо выбран из группы, состоящей из Н, прямого или разветвленного С1-С6алкила, прямого или разветвленного С2-С6алкенила и прямого или разветвленного С2-С6алкинила;
каждый X независимо представляет собой аминокислоту, где каждая аминокислота X является одной и той же или другой;
каждый Y независимо представляет собой аминокислоту, где каждая аминокислота Y является одной и той же или другой;
где X-Y выбран из группы, состоящей из Gly, β-Ala, Val-Cit, Phe-Lys, Val-Lys, Phe-Phe-Lys, Ala-Lys, Phe-Cit, Leu-Cit, Ala-Cit, Trp-Cit, Phe-Ala, Gly-Phe-Leu-Gly, Ala-Leu-Ala-Leu, Phe-N9-тозил-Arg, Phe-N9-нитро-Arg, Val-Ala и Ala-Ala-Asn;
каждый Z независимо представляет собой спейсерный элемент, представляющий собой 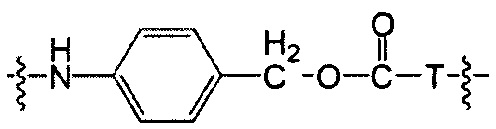 , где T представляет собой O, NH или S, где каждый спейсерный элемент является одним и тем же или другим;
, где T представляет собой O, NH или S, где каждый спейсерный элемент является одним и тем же или другим;
m равно 0-5, n равно 1-5, p равно 0-2, q равно 0-10, r равно 0-2, и s равно 0-2, где q+r+s=2 или более; и
D представляет собой цитотоксический агент;
включающий стадии:
получения некоторого количества первого соединения, имеющего структуру
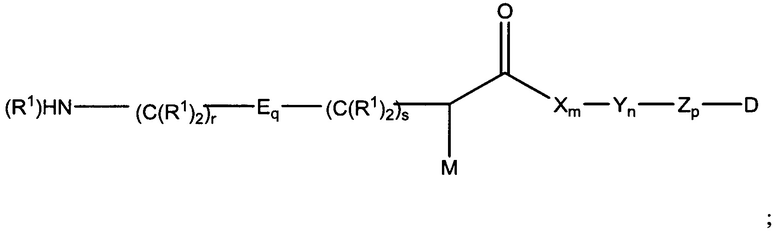
получения некоторого количества второго соединения, которое содержит пептидную последовательность, содержащую один или более остатков глутамина, где пептидная последовательность выбрана из группы, состоящей из моноклонального антитела, поликлонального антитела, человеческого антитела, гуманизированного антитела, химерного антитела, биспецифического антитела, минитела и фрагмента антитела; и
осуществления взаимодействия указанных количеств первого и второго соединений в присутствии трансглутаминазы.
| WO 2013072813 A2, 23.05.2013 | |||
| WO 2012059882 A2, 10.05.2012 | |||
| ХИМИЧЕСКИЕ ЛИНКЕРЫ И ИХ КОНЪЮГАТЫ | 2005 |
|
RU2402548C2 |

