Область, к которой относится изобретение
Представлены новое изоиндолиновое производное, включающая его фармацевтическая композиция и применение.
Предпосылки создания изобретения
Циклический аденозин–3',5'–монофосфат (цАМФ) играет важную роль в качестве вторичного мессенджера в клетках. Внутриклеточный гидролиз цАМФ до аденозин–5'–монофосфата (АМФ) связан со многими воспалительными заболеваниями, включая, но не ограничиваясь этим, псориаз, аллергический ринит, шок, наследственный аллергический дерматит, болезнь Крона, респираторный дистресс–синдром взрослых (ARDS), эозинофильную гранулему, аллергический конъюнктивит, остеоартрит и язвенный колит. Фосфодиэстераза (PDE) циклических нуклеотидов является важным фактором, контролирующим уровень цАМФ. Известно, что семейство PDE включает 11 членов. Хотя PDE1, PDE2, PDE3, PDE4 и PDE7 все используют цАМФ в качестве субстрата, только PDE4 и PDE7 являются высокоселективными для гидролиза цАМФ. Поэтому ингибиторы PDE, особенно ингибиторы PDE4, рассматриваются как энхансеры цАМФ. Иммунные клетки содержат PDE3 и PDE4, из которых PDE4 широко распространен в человеческих моноцитах. Поэтому ингибирование PDE4 является целью терапевтических вмешательств при различных патологических процессах. Исследования показали, что введение ингибиторов PDE4 восстанавливает память в животных моделях, в том числе при болезни Альцгеймера. Было показано, что PDE4 является основным регулятором циклического АМФ в гладких мышцах дыхательных путей и воспалительных клетках. Ингибиторы PDE4 можно использовать для лечения различных заболеваний, включая аллергические и воспалительные заболевания, диабет, заболевания центральной нервной системы, боль и так далее.
Фактор некроза опухоли–α (TNF–α) представляет собой разновидность провоспалительного цитокина, который играет важную роль в иммунном гомеостазе, воспалении и защите хозяина. Как было показано, TNF–α является одним из основных медиаторов воспаления. Неконтролируемая активность TNF–α или сверхродукция TNF–α связаны с патологией различных заболеваний, включая, но не ограничиваясь этим, рак и воспалительные заболевания. Дисрегуляция TNF–α также может привести к аутоиммунным заболеваниям, синдрому токсического шока, кахексии, артриту, псориазу, ВИЧ–инфекции и СПИДу, заболеваниям нервной системы и центральной нервной системы, сепсису, застойной сердечной недостаточности, отторжению трансплантата и вирусным инфекциям. Таким образом, снижение уровня TNF–α или регулирование активности TNF–α является многообещающей стратегией при лечении многих иммунологических, воспалительных и злокачественных заболеваний (например, рака и воспаления).
Таким образом, соединения, способные ингибировать PDE4 и/или TNF–α, могут лечить различные заболевания. Например, Апремиласт представляет собой низкомолекулярный ингибитор PDE4 и иммуномодулятор, который ингибирует PDE4 и TNF–α, и он был одобрен FDA для лечения псориатического артрита и бляшечного псориаза. Однако Апремиласт имеет побочные эффекты на центральную нервную систему и желудочно–кишечные побочные эффекты, такие как головная боль, тошнота и рвота и желудочная секреция. Таким образом, клинически необходимо продолжать поиск ингибиторов PDE4 с оптимизированными функциональными характеристиками.
Сущность изобретения
Настоящее изобретение обеспечивает соединение формулы I или его фармацевтически приемлемую соль, сольват, полиморф, сокристалл, стереоизомер, изотопное соединение, метаболит или пролекарство:

где, атом углерода, помеченный звездочкой *, представляет собой центр асимметрии;
R1 и R2 независимо представляют собой H, D, замещенный или незамещенный (C1–C6)алкил, замещенный или незамещенный (C3–C6)циклоалкил, R6–S(O)2– или R6–C(O)–; или R1 и R2 и атом азота, с которым они вместе связаны, образуют 5–7–членный гетероцикл, содержащий N;
R6 представляет собой замещенный или незамещенный (C3–C6)циклоалкил; или (C1–C6)алкил, который необязательно замещен одной или более группами, выбранными из D, галогена, гидроксила, амино, (C1–C6)алкиламино и (C1–C6)алкокси или бензилокси;
X1, X2, X3, X4, X5 и X6 независимо представляют собой CH, CD, CR7 или N;
R7 представляет собой галоген или циано;
R3 и R4 независимо представляют собой H, замещенный или незамещенный (C1–C6)алкил, замещенный или незамещенный (C3–C6)циклоалкил или замещенный или незамещенный (C1–C6)алкил– (C3–C6)циклоалкил; или R3 и R4 и атом кислорода, с которым они вместе связаны, образуют 5–7–членный гетероцикл, содержащий O;
R5 представляет собой замещенный или незамещенный (C1–C6)алкил;
R10, R11 и R12 независимо представляют собой H или D;
Заместитель в замещенном (C1–C6)алкиле, замещенном (C3–C6)циклоалкиле или замещенном (C1–C6)алкил– (C3–C6)циклоалкиле представляет собой один или более (например, 1–6, предпочтительно 1–5) выбранных из группы, состоящей из: D, галогена, гидроксила, амино, (C1–C6)алкиламино и (C1–C6)алкокси, бензилокси; когда присутствуют несколько заместителей, эти заместители являются одинаковыми или отличными друг от друга;
при условии, что один из X1, X2, X3, X4, X5 и X6 представляет собой N; или по меньшей мере один из X1, X2, X3, X4, X5 и X6 представляет собой CR7.
Предпочтительно, центр асимметрии относится к (S)– конфигурации углерода, (R)– конфигурации углерода или рацемату, более предпочтительно к (S)– конфигурации углерода.
В предпочтительном варианте осуществления (C1–C6)алкил в замещенном или незамещенном (C1–C6)алкиле, замещенном или незамещенном (C1–C6)алкил– (C3–C6)циклоалкиле или (C1–C6)алкиламино предпочтительно представляет собой (C1–C4)алкил. (C1–C4)Алкил предпочтительно представляет собой метил, этил, изопропил, н–пропил, н–бутил, изобутил или трет–бутил. Замещенный (C1–C6)алкил в замещенном или незамещенном (C1–C6)алкиле или замещенном или незамещенном (C1–C6)алкил– (C3–C6)циклоалкиле предпочтительно замещен одним или более галогенами или D. В предпочтительном варианте осуществления замещенный (C1–C6)алкил предпочтительно представляет собой CD3, CH2D, CHD2, C2D5, CH2CD3 или CHF2.
В предпочтительном варианте осуществления (C1–C6)алкокси предпочтительно представляет собой (C1–C4)алкокси. (C1–C4)Алкокси предпочтительно представляет собой метокси, этокси, изопропокси, н–пропокси, н–бутокси, изобутокси или трет–бутокси.
В предпочтительном варианте осуществления (C3–C6)циклоалкил в замещенном или незамещенном (C3–C6)циклоалкиле и замещенном или незамещенном (C1–C6)алкил– (C3–C6)циклоалкиле предпочтительно представляет собой циклопропил, циклобутил, циклопентил или циклогексил.
В предпочтительном варианте осуществления галоген предпочтительно представляет собой фтор, хлор, бром или йод, более предпочтительно фтор, хлор или бром.
В предпочтительном варианте осуществления (C1–C6)алкиламино представляет собой  , где один из Ra и Rb представляет собой H, другой представляет собой (C1–C6)алкил; или Ra и Rb независимо представляют собой (C1–C6)алкил.
, где один из Ra и Rb представляет собой H, другой представляет собой (C1–C6)алкил; или Ra и Rb независимо представляют собой (C1–C6)алкил.
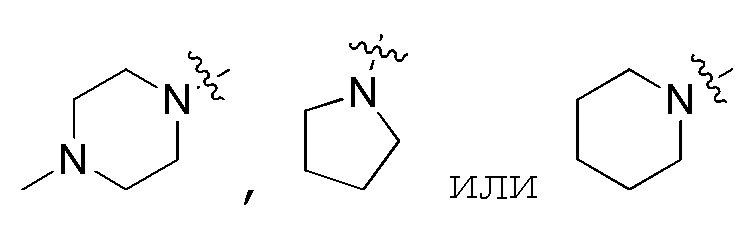
В предпочтительном варианте осуществления, когда R1 и R2 и атом азота, с которым они вместе связаны, образуют 5–7–членный гетероцикл, содержащий N, указанный 5–7–членный гетероцикл, содержащий N, предпочтительно выбран из 
 .
.
В предпочтительном варианте осуществления, когда R3 и R4 и атом кислорода, с которым они вместе связаны, образуют 5–7–членный гетероцикл, содержащий O, указанный 5–7–членный гетероцикл, содержащий O, предпочтительно представляет собой  .
.
В предпочтительном варианте осуществления X1 представляет собой N, X2, X3, X4, X5 и X6 независимо представляют собой CH, CD или CR7;
В предпочтительном варианте осуществления X2 представляет собой N, X1, X3, X4, X5 и X6 независимо представляют собой CH, CD или CR7;
В предпочтительном варианте осуществления X3 представляет собой N, X1, X2, X4, X5 и X6 независимо представляют собой CH, CD или CR7;
В предпочтительном варианте осуществления X4 представляет собой N, X1, X2, X3, X5 и X6 независимо представляют собой CH, CD или CR7;
В предпочтительном варианте осуществления X5 представляет собой N, X1, X2, X3, X4 и X6 независимо представляют собой CH, CD или CR7;
В предпочтительном варианте осуществления X6 представляет собой N, X1, X2, X3, X4 и X5 независимо представляют собой CH, CD или CR7.
В предпочтительном варианте осуществления X6 представляет собой N, X1, X2, X3 независимо представляют собой CH, CD или CR7, X4, X5 независимо представляют собой CH, CD;
В предпочтительном варианте осуществления X6 представляет собой N, X1 представляет собой CR7, X2, X3, X4 и X5 независимо представляют собой CH или CD. В другом варианте осуществления X6 представляет собой N, X1 представляет собой CR7, X2, X3, X4 и X5 представляют собой CH.
В предпочтительном варианте осуществления X6 представляет собой N, X2 представляет собой CR7, X1, X3, X4 и X5 независимо представляют собой CH или CD. Еще в одном варианте осуществления X6 представляет собой N, X2 представляет собой CR7, X1, X3, X4 и X5 представляет собой CH.
В предпочтительном варианте осуществления X6 представляет собой N, X3 представляет собой CR7, X1, X2, X4 и X5 независимо представляют собой CH или CD. Еще в одном варианте осуществления X6 представляет собой N, X3 представляет собой CR7, X1, X2, X4 и X5 представляет собой CН.
В предпочтительном варианте осуществления один из R1 и R2 представляет собой H или D, другой представляет собой R6–S(O)2– или R6–C(O)–. В следующем варианте осуществления один из R1 и R2 представляет собой Н, другой представляет собой R6–C(O)–.
В предпочтительном варианте осуществления R6 представляет собой (C3–C6)циклоалкил; или (C1–C4)алкил, который необязательно замещен одним или более заместителями, выбранными из D, галогена, гидроксила, амино, (C1–C4)алкиламино, (C1–C4)алкокси, бензилокси. Предпочтительно R6 представляет собой (C3–C6)циклоалкил; или (C1–C4)алкил, который необязательно замещен одним или более заместителями, выбранными из (C1–C4)алкокси, бензилокси. Более предпочтительно R6 представляет собой циклопропил, метил, этил, гидроксиметил, бензилоксиметил, метоксиметил, изобутил, диметиламинометил, изопропил, CD3 или C2D5.
В предпочтительном варианте осуществления R7 представляет собой фтор, хлор, бром или циано.
В предпочтительном варианте осуществления R3 и R4 независимо представляют собой водород, замещенный или незамещенный (C1–C6)алкил. Замещенный (C1–C6)алкил может представлять собой (C1–C6)алкил, замещенный одним или более галогенами или D. Предпочтительно R3 и R4 независимо представляют собой H, метил, этил, пропил, изопропил, CD3, CH2D, CHD2, C2D5, CH2CD3 или CHF2.
В предпочтительном варианте осуществления R5 представляет собой замещенный или незамещенный (C1–C6)алкил. В более предпочтительном варианте осуществления R5 представляет собой метил, этил, пропил, изопропил, CD3, CH2D, CHD2, C2D5 или CH2CD3.
В предпочтительном варианте осуществления X3 представляет собой CR7, X1, X2, X4, X5 и X6 независимо представляют собой CH или CD; R7 представляет собой фтор, хлор или циано.
В предпочтительном варианте осуществления X2 представляет собой CR7, X1, X3, X4, X5 и X6 независимо представляют собой CH или CD; R7 представляет собой фтор, хлор или циано.
В предпочтительном варианте осуществления X1 представляет собой CR7, X2, X3, X4, X5 и X6 независимо представляют собой CH или CD; R7 представляет собой фтор, хлор или циано.
В предпочтительном варианте осуществления X3 представляет собой CR7, X1, X2, X4, X5 и X6 независимо представляют собой CH или CD; R7 представляет собой фтор, хлор или циано; один из R3 и R4 представляет собой замещенный (C1–C6)алкил, замещенный (C3–C6)циклоалкил или замещенный (C1–C6)алкил– (C3–C6)циклоалкил. Предпочтительно один из R3 и R4 представляет собой (C1–C6)алкил, замещенный одним или более галогенами или D. Более предпочтительно один из R3 и R4 представляет собой CD3 или CHF2.
В предпочтительном варианте осуществления X2 представляет собой CR7, X1, X3, X4, X5 и X6 независимо представляют собой CH или CD; R7 представляет собой фтор, хлор или циано; один из R3 и R4 представляет собой замещенный (C1–C6)алкил, замещенный (C3–C6)циклоалкил или замещенный (C1–C6)алкил– (C3–C6)циклоалкил. Предпочтительно один из R3 и R4 представляет собой (C1–C6)алкил, замещенный одним или более галогенами или D. Более предпочтительно один из R3 и R4 представляет собой CD3 или CHF2.
В предпочтительном варианте осуществления X1 представляет собой CR7, X2, X3, X4, X5 и X6 независимо представляют собой CH или CD; R7 представляет собой фтор, хлор или циано; один из R3 и R4 представляет собой замещенный (C1–C6)алкил, замещенный (C3–C6)циклоалкил или замещенный (C1–C6)алкил– (C3–C6)циклоалкил. Предпочтительно один из R3 и R4 представляет собой (C1–C6)алкил, замещенный одним или более галогенами или D. Более предпочтительно один из R3 и R4 представляет собой CD3 или CHF2.
В предпочтительном варианте осуществления X3 представляет собой CR7, X1, X2, X4, X5 и X6 независимо представляют собой CH или CD; R7 представляет собой фтор, хлор или циано; один из R3 и R4 представляет собой CН3, CD3, C2H5, C2D5, CH2CD3 или CHF2, другой представляет собой CD3 или CHF2.
В предпочтительном варианте осуществления X3 представляет собой CR7, X1, X2, X4, X5 и X6 независимо представляют собой CH или CD; R7 представляет собой фтор, хлор или циано; R3 представляет собой CD3 или CHF2, R4 представляет собой CН3, CD3, C2H5, C2D5 или CH2CD3.
В предпочтительном варианте осуществления соединение формулы I выбрано из следующих соединений:





Настоящее изобретение также предоставляет способ получения соединения формулы I, который выбран из способа A или способа B: способ A включает следующие стадии: соединение формулы I–A и соединение формулы I–B подвергают взаимодействию следующим образом с получением соединения формулы I;
 ;
;
где, X1, X2, X3, X4, X5, X6, R1, R2, R3, R4, R5, R10, R11 и R12 имеют значения, определенные выше.
Предпочтительно соединение формулы I–A и соединение формулы I–B подвергают взаимодействию в присутствии кислоты. Кислота представляет собой обычную кислоту для такой реакции в области органического синтеза и предпочтительно представляет собой уксусную кислоту.
В способе получения соединения формулы I реакционные условия могут представлять собой обычные условия для такой реакции в области органического синтеза. В этой реакции количество кислоты может конкретно не ограничиваться при условии, что оно не влияет на реакцию. Количество соединения формулы I–A и соединения формулы I–B может быть выбрано в соответствии с обычным количеством такой реакции в области органического синтеза. Температура реакции может быть обычной температурой для такой реакции в данной области, предпочтительно, 10oC–120oC.
способ B включает следующие стадии: соединение формулы I–3 и соединение формулы I–4 подвергают взаимодействию следующим образом с получением соединения формулы I;

где, X1, X2, X3, X4, X5, X6, R1, R2, R3, R4, R5, R10, R11 и R12 имеют значения, определенные выше; Y представляет собой уходящую группу, такую как галоген.
В другом варианте осуществления способа B соединение формулы I–4 можно заменить соединением формулы I–4ʽ (R6–S(O)2–Y) с получением соединения формулы I, где группы имеют значения, определенные выше.
Способы получения соединения формулы I также можно осуществить путем обращения к обычным способам получения такого типа соединений в области органического синтеза. Условия и стадии, связанные с химическими реакциями, можно осуществить путем обращения к обычным условиям и стадиям таких реакций в области органического синтеза, и соединения, полученные вышеуказанными способами, также можно дополнительно модифицировать в периферических положениях для получения других целевых соединений по настоящему изобретению.
Настоящее изобретение также обеспечивает промежуточное соединение для синтеза соединения формулы I, такое как соединения формулы I–3,
 ;
;
где, X1, X2, X3, X4, X5, X6, R1, R2, R3, R4, R5, R10, R11 и R12 имеют значения, определенные выше.
Изобретение также предоставляет фармацевтическую композицию, содержащую одно или более из соединений формулы I, его фармацевтически приемлемую соль, сольват, полиморф, сокристалл, стереоизомер, изотопное соединение, метаболит или пролекарство, и один или более фармацевтически приемлемых эксципиентов. Фармацевтическая композиция также может дополнительно содержать другое терапевтическое средство с фармакологической активностью. Другое терапевтическое средство может включать, но не ограничивается этим, лекарственные средства против ангиогенеза, иммуномодуляторы, иммунотерапевтические лекарственные средства, химиотерапевтические лекарственные средства, гормональные соединения, противоопухолевые лекарственные средства или противовоспалительные лекарственные средства.
Фармацевтически приемлемый эксципиент может представлять собой такое вещество, которое широко используют в области получения лекарственных средств. Эксципиент в основном используют для предоставления безопасной, стабильной и функционализированной фармацевтической композиции, и также может обеспечить способ, когда активные ингредиенты растворяются с желаемой скоростью после приема средства субъектом, или способствует эффективной абсорбции активных ингредиентов после введения композиции субъекту. Эксципиент может быть инертным наполнителем или таким, которое обеспечивает некоторые функции, такие как стабилизация общего значения рН композиции или предотвращение деградации активного ингредиента композиции. Фармацевтически приемлемый эксципиент может включать один или более из следующих эксципиентов: связующее, суспендирующий агент, эмульгатор, разбавитель, наполнитель, гранулирующий агент, адгезив, дезинтегрирующий агент, смазывающий агент, антиадгезивный агент, вещество, обеспечивающее скольжение, смачивающий агент, гелеобразующий агент, замедлитель абсорбции, ингибитор растворения или упрочняющий агент, адсорбент, буфер, хелатирующий агент, консервант, краситель, корригирующий агент и подсластитель.
Фармацевтическая композиция по изобретению может быть получена на основе информации, представленной в настоящем документе, в соответствии с любым способом, известным специалисту в данной области техники. Например, фармацевтическая композиция может быть получена путем смешивания одного или более из соединений формулы I или его фармацевтически приемлемой соли, сольвата, полиморфа, сокристалла, стереоизомера, изотопного соединения, метаболита или пролекарства, с одним или более фармацевтически приемлемыми эксципиентами на основе общей технологии получения лекарственных средств. Эти технологии включают, но ими не ограничиваются, обычное смешивание, растворение, гранулирование, эмульгирование, растирание в порошок, “обертывание“, “встраивание“ или сушку сублимацией.
Фармацевтическая композиция, согласно изобретению, может быть получена для введения любым способом, включая инъекцию (внутривенную), мукозальный способ введения, пероральное введение (твердый и жидкий препарат), ингаляцию, офтальмическое введение, ректальное введение, местное или парентеральное введение (инфузия, инъекция, имплантация, подкожное введение, введение через вену, введение через артерию, внутримышечное введение). Фармацевтическая композиция по изобретению также может представлять собой лекарственную форму с контролируемым высвобождением или замедленным высвобождением. Примеры твердого перорального препарата включают, но ими не ограничиваются, порошок, капсулу, каплету, мягкую капсулу или таблетку. Примеры жидкого препарата для перорального или мукозального введения включают, но ими не ограничиваясь, суспензию, эмульсию, эликсир и раствор. Примеры препарата для местного применения включают, но ими не ограничиваются, эмульсию, гель, мазь, крем, пластырь, пасту, пену, лосьон, капли или сывороточный препарат. Примеры препарата для парентерального введения включают, но ими не ограничиваются, раствор для инъекций, сухой препарат, который может быть растворен или суспендирован в фармацевтически приемлемом носителе, инъекцируемую суспензию и инъекцируемую эмульсию. Примеры других подходящих препаратов фармацевтической композиции включают, но ими не ограничиваются, глазные капли и другие офтальмологические препараты; аэрозоль, такой как назальный спрей или ингаляция; жидкие лекарственные формы, подходящие для парентерального введения; суппозиторий и пастилка.
Терапевтическое или профилактическое количество одного или более соединений формулы I или его фармацевтически приемлемой соли, сольвата, полиморфа, сокристалла, стереоизомера, изотопного соединения, метаболита или пролекарства, любой его фармацевтической композиции или препарата и т.д. можно вводить субъекту в течение периода (медикаментозный цикл доставки), за которым следует период, свободный от соединения (немедикаментозный цикл доставки). Медикаментозный цикл доставки и немедикаментозный цикл доставки может быть повторен в течение необходимого времени. Необходимая продолжительность и время медикаментозного цикла доставки и немедикаментозного цикла доставки зависят от типа и/или тяжести заболевания, расстройства или состояния, подвергаемого лечению или профилактики, а также пола, возраста, массы субъекта и других параметров (например, биологических, физических и физиологических параметров субъекта и т.д.). Специалист в данной области может достаточно точно определить подходящую продолжительность и время медикаментозного цикла доставки и немедикаментозного цикла доставки на основе данных, описанных в настоящем документе.
Настоящее изобретение дополнительно предоставляет способ регулирования генерации или активности PDE4 или TNF–α, который включает введение нуждающемуся субъекту терапевтически эффективного количества одного или более соединений формулы I или фармацевтически приемлемой соли, сольвата, полиморфа, сокристалла, стереоизомера, изотопного соединения, метаболита и пролекарства такого соединения или их фармацевтической композиции.
Настоящее изобретение дополнительно предоставляет применение одного или более соединений формулы I, их фармацевтически приемлемых солей, сольватов, полиморфов, сокристаллов, стереоизомеров, изотопных соединений, метаболитов и пролекарств для получения лекарственного препарата для регулирования генерации или активности PDE4 и/или TNF–α.
Настоящее изобретение дополнительно предоставляет соединение формулы I, его фармацевтически приемлемую соль, сольват, полиморф, сокристалл, стереоизомер, изотопное соединение, метаболит и пролекарство для применения в регулировании генерации или активности PDE4 и/или TNF–α.
В одном из вариантов осуществления, в случае, когда термин “регулировать“ используется для описания активности или генерации определенной молекулы, он относится к ингибированию активности или генерации молекулы. В другом варианте осуществления, когда термин “регулировать“ используется для описания активности или синтеза определенной молекулы, он относится к увеличению или усилению активности или синтеза молекулы. Однако в другом варианте осуществления, в случае, когда термин “регулировать“ используется для описания активности или синтеза определенной молекулы, он относится к уменьшению или увеличению активности или синтеза молекулы.
В другом аспекте предоставлен способ лечения или профилактики заболевания, расстройства или состояния, вызванного аномальной генерацией или регуляцией PDE4 и/или TNF–α, включающий введение субъекту терапевтически или профилактически эффективного количества соединения формулы I, его фармацевтически приемлемой соли, сольвата, стереоизомера, изотопного соединения, метаболита или пролекарства, или их фармацевтической композиции.
Настоящее изобретение дополнительно предоставляет применение одного или более соединений формулы I, его фармацевтически приемлемой соли, сольвата, полиморфа, сокристалла, стереоизомера, изотопного соединения, метаболита и пролекарства для получения лекарственного средства для лечения или профилактики заболевания, расстройства или состояния, связанного с аномальной генерацией или регуляцией PDE4 и/или TNF–α.
Настоящее изобретение дополнительно предоставляет соединение формулы I, его фармацевтически приемлемую соль, сольват, полиморф, сокристалл, стереоизомер, изотопное соединение, метаболит и пролекарство для применения в лечении или профилактике заболевания, расстройства или состояния, связанного с аномальной генерацией или регуляцией PDE4 и/или TNF–α.
В соответствии со способом или применением по настоящему изобретению, примеры заболевания, расстройства или состояния, связанного с аномальной генерацией или регуляцией PDE4 и/или TNF–α, включают, но не ограничиваются этим, раковые заболевания, воспалительные заболевания, заболевания и расстройства, связанные с нежелательным ангиогенезом, боли, синдром макулярной дегенерации (MD), кожные заболевания, кератоз, заболевания дыхательной системы (такие как астма или ХОБЛ), иммунодефицитные заболевания, заболевания центральной нервной системы (ЦНС), аутоиммунные заболевания, атеросклероз, наследственные заболевания, аллергию, вирусы, нарушения сна и связанные с ними синдром. Хорошо известные в данной области примеры заболевания, расстройства или состояния включают, но ими не ограничиваются, заболевания, расстройства или состояния, которые описаны в публикациях PCT патентов WO 02012015986 и WO2006018182 и в публикации патента США 0100204227.
В одном варианте осуществления примерами заболевания, расстройства или состояния, связанного с аномальной генерацией или регуляцией PDE4 и/или TNF–α, являются псориатический артрит и бляшковидный псориаз.
Способ лечения или профилактики заболевания, расстройства или состояния по изобретению включает введение одного или более соединениий формулы I, его фармацевтически приемлемой соли, сольвата, стереоизомера, изотопного соединения, метаболита и пролекарства субъекту любыми подходящими способами, такими как инъекция, мукозальный способ, пероральный способ, ингаляционный способ, введение через глаз, ректальный способ, с помощью имплантата длительного действия, с помощью липосомы, эмульсии или методом замедленного высвобождения.
Специалист в данной области понимает, что терапевтически эффективное или профилактически эффективное количество соединения, используемое в изобретении, может изменяться в зависимости от характеристик субъекта, таких как возраст, диета, здоровье и т.д., тяжесть, осложнение и тип заболевания, нарушение или состояние, подлежащее лечению или предотвращению, и в зависимости от используемого препарата и т.д. На основании описания изобретения специалист в данной области может легко определить терапевтически эффективное или профилактически эффективное количество соединения, которое необходимо вводить субъекту с тем, чтобы вызвать желаемый биологический или медицинский ответ у субъекта.
В настоящей заявке цитируются или описаны различные публикации, статьи и патенты, причем цель цитирования или описания этих ссылок или включения этих ссылок полностью или рассмотрение этих ссылок, заключается в том, чтобы проиллюстрировать уровень техники изобретения, а не признание того, что содержание этих ссылок обуславливает отчасти предшествующий уровень техники изобретения.
Если не указано иное, технические и научные термины, используемые в настоящем документе, имеют те же значения, которые обычно известны специалистам в данной области техники. В иных случаях, некоторые термины, используемые в настоящем документе, имеют значения, указанные в настоящем описании. Все патенты, опубликованные заявки и публикации, цитируемые в настоящем документе, включены в настоящий документ посредством ссылки так же, как подробно определено в настоящем документе. Следует отметить, что, если не указано иное в контексте, форма единственного числа, используемая в настоящем документе и в прилагаемых пунктах формулы, охватывает значение формы множественного числа.
Если специально не указано иное, соотношения (включая проценты) или части, используемые в настоящем документе, приведены по массе.
При использовании совместно с числовой переменной термины "около" и "приблизительно" обычно означают, что значение этой переменной и все значения этой переменной находятся в пределах экспериментальной погрешности (например, в пределах 95% доверительного интервала для среднего значения) или±10% указанного значения или шире.
Предполагается, что выражения "содержащий", "включающий", "имеющий" и подобные являются неограничивающими и не исключают дополнительные не указанные элементы, стадии или компоненты. Выражение "состоящий из" исключает любой элемент, стадию или ингредиент, который не указан. Выражение "состоящий по существу из" означает, что объем ограничен указанными элементами, стадиями или компонентами, и необязательно существующими элементами, стадиями или компонентами, которые существенно не влияют на основные и новые характеристики заявленного объекта изобретения. Следует понимать, что выражение "включающий" охватывает выражение "состоящий по существу из" и "состоящий из".
Термин "замещенный" или "замещает" означает, что любой один или более атомов водорода на конкретном атоме заменены заместителем, если валентное состояние конкретного атома является нормальным и замещенное соединение является стабильным.
Как используется в настоящем документе, в случае, когда конкретная соль, композиция и эксципиент и т.д. обозначаются как “фармацевтически приемлемые“, это означает, что соль, композиция или эксципиент и т.д. обычно нетоксичны, безопасны и пригодны для введения субъекту, предпочтительно млекопитающему, более предпочтительно человеку.
Термин “фармацевтически приемлемая соль“, используемый в настоящем документе, относится к фармацевтически приемлемой органической или неорганической соли. Примеры соли включают, но ими не ограничиваются, сульфат, цитрат, ацетат, оксалат, хлорид, бромид, йодид, нитрат, гидросульфат, фосфат, кислый фосфат, изоникотинат, лактат, салицилат, кислый цитрат, тартрат, олеат, таннат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат, этансульфонат, бензосульфонат, п–толуолсульфонат и эмбонат (то есть 1–1–метилен–бис(2–гидроксил–3–нафтоат)). Соединения по изобретению могут быть использованы для образования фармацевтически приемлемых солей с различными аминокислотами. Подходящая соль щелочного металла включает, но ими не ограничивается, соль алюминия, соль кальция, соль лития, соль магния, соль калия, натриевую соль, соль цинка, соль висмута и соль диэтаноламина.
Используемый в настоящем документе термин “метаболит“ относится к активному веществу, продуцируемому молекулой лекарственного средства, которая подверглась изменениям химической структуры in vivo, причем активное вещество обычно является производным вышеуказанной молекулы лекарственного средства, а также может быть химически модифицировано.
Как используется в настоящем документе, и если не указано иное, термин “полиморф“ относится к одному или более видам кристаллической структуры, образованной различными расположениями молекул в пространственной решетке при кристаллизации.
Используемый в настоящем документе термин “сокристалл“ относится к многокомпонентной системе, содержащей одну или более молекул API (активный фармацевтический ингредиент) и одну или более целевых молекул (или лиганд). В сокристалле молекулы API и целевые молекулы (или лиганд) существуют в виде твердых веществ при комнатной температуре, когда они используются отдельно в своей чистой форме (чтобы отличить сокристалл от сольвата или гидрата). Из этого конкретного определения исключаются соли, в которых происходит значительный или полный протонный обмен между молекулами API и гостевыми молекулами. В сокристалле API и лиганды взаимодействуют через водородные связи и другие возможные нековалентные взаимодействия. Отмечается, что сам сокристалл может образовывать сольваты, включая гидраты.
Используемый в настоящем документе термин “сольват“ относится к кристаллической форме соединения формулы I или его фармацевтически приемлемой соли, полиморфа, сокристалла, стереоизомера, изотопного соединения, метаболита или пролекарства, который также имеет одну или более молекул растворителя, включенных в кристаллическую структуру. Сольват может включать стехиометрическое количество или нестехиометрическое количество растворителя, а молекула растворителя в растворителе может иметь упорядоченное или неупорядоченное расположение. Сольват, содержащий нестехиометрическое количество молекул растворителя, может быть образован путем потери, по меньшей мере, одной молекулы растворителя (но не всех) из сольвата. В конкретном варианте осуществления, сольват относится к гидрату, что означает, что кристалл соединения дополнительно включает молекулу воды и воду используют в качестве растворителя.
Как используется в настоящем документе и если не указано иное, термин “пролекарство“ относится к производному соединения, содержащему биологически реакционноспособную функциональную группу, причем биологически реакционноспособная функциональная группа может быть отщеплена от соединения, или может быть осуществлено взаимодействие другими способами с получением соединения в биологических условиях (in vivo или in vitro). Обычно пролекарство неактивно или, по меньшей мере, имеет более низкую активность, чем соединение, что заставляет соединение проявлять свою активность после его отщепления от биологически активной функциональной группы. Для получения соединения, биологически реакционноспособную функциональную группу можно гидролизовать или окислять в биологических условиях. Например, пролекарство может содержать биологически гидролизуемую группу. Примеры биологически гидролизуемой группы включают, но ими не ограничиваются, биологически гидролизуемый фосфат, биологически гидролизуемый сложный эфир, биологически гидролизуемый амид, биологически гидролизуемый сложный эфир угольной кислоты, биологически гидролизуемый карбамат и биологически гидролизуемый уреид.
Соединение формулы I по изобретению, его фармацевтически приемлемая соль, сольват, полиморф, сокристалл, стереоизомер, изотопное соединение, метаболит или пролекарство могут содержать один или более асимметричных центров (“стереоизомер“). Используемый в настоящем документе термин “стереоизомер“ относится ко всем стереоизомерам, включая энантиомер, диастереоизомер, эпимер, эндо–экзо–изомер, атропоизомер, региоизомер, цис– и транс–изомер. “Стереоизомер“ в настоящем документе также включает “чистый стереоизомер“ и “обогащенный стереоизомер“ или “рацемический изомер“ различных вышеуказанных стереоизомеров. Эти стереоизомеры могут быть получены в соответствии с методом асимметрического синтеза или разделены, очищены и обогащены с помощью хирального способа разделения (включая, но ими не ограничиваясь, тонкослойную хроматографию, центробежную хроматографию, колоночную хроматографию, газовую хроматографию, жидкостную хроматографию высокого давления и т.д.), а также путем хирального разделения посредством связывания (химическое связывание и т.д.) или солеобразования (физическое связывание и т.д.) с другим хиральным(и) соединением(ями). Термин “чистый стереоизомер“ в настоящем документе подразумевает, что массовое содержание стереоизомера соединения составляет не менее 95% относительно других стереоизомеров соединения. Термин “обогащенный стереоизомер“ в настоящем документе подразумевает, что массовое содержание стереоизомера соединения составляет не менее 50% относительно других стереоизомеров соединения. Термин “рацемический изомер“ в настоящем документе подразумевает, что массовое содержание стереоизомера соединения равно массовому содержанию другого стереоизомера соединения.
Термин “изотопное соединение“, используемый в настоящем документе, подразумевает, что присутствует один или более природных или искусственных изотопов атомов, содержащихся в соединении формулы I или его фармацевтически приемлемой соли, сольвате, полиморфе, сокристалле, стереоизомере, метаболите или пролекарстве. Искусственные изотопы атомов включают, но ими не ограничиваются, дейтерий (2H или D), тритий (3H или Т), иод–125 (125I), фосфор–32 (32P), углерод–13 (13C) или углерод–14 (14С). Вышеуказанное изотопное соединение также может быть использовано в качестве терапевтического или диагностического средства (то есть средства для внутреннего обнаружения) или инструмента исследования. Все изотопные варианты соединения по изобретению, независимо от того, являются ли они радиоактивными, включены в объем настоящего изобретения.
Термин “обогащенный изотопом“, используемый в настоящем документе, подразумевает, что имеется один или более искусственных изотопов атомов, содержащихся в соединении формулы I или его фармацевтически приемлемой соли, сольвате, полиморфе, сокристалле, стереоизомере, изотопном соединении, метаболите или пролекарстве. Термин “обогащенный изотопом“ также подразумевает, что соединение формулы I или его фармацевтически приемлемая соль, сольват, полиморф, сокристалл, стереоизомер, изотопное соединение, метаболит или пролекарственное соединение, содержат, по меньшей мере, один искусственный атом изотопа.
Используемый в настоящем документе термин “пациент“ или “субъект“ относится к любому животному, которое должно быть обработано или было обработано соединением или композицией в соответствии с вариантом осуществления изобретения, причем предпочтительным является млекопитающее, и наиболее предпочтительным является человек. Термин “млекопитающее“, используемый в настоящем документе, включает любых млекопитающих. Примеры млекопитающих включают, но ими не ограничиваются, крупный рогатый скот, лошадь, овцу, свинью, кошку, собаку, мышей, крысу, кролика, морскую свинку, обезьяну, человека и т.д. Наиболее предпочтительным является человек. Термины “субъект“ и “пациент“ используются в настоящем документе взаимозаменяемо.
В одном из вариантов осуществления, термины “лечить“ и “лечение“ относятся к облегчению, предотвращению или устранению заболевания или расстройства или, по меньшей мере, одного из его идентифицируемых симптомов, например, лечение злокачественной опухоли путем уменьшения или стабилизации симптомов злокачественной опухоли или заболевания. В другом варианте осуществления, “лечить“ или “лечение“ относится к облегчению, предотвращению или устранению, по меньшей мере, одного измеряемого параметра заболевания или расстройства в организме, который подвергается лечению, причем заболевание или расстройство может быть не выявлено у млекопитающих. Однако в другом варианте осуществления, термин “лечить“ или “лечение“ относится к замедлению развития заболевания или расстройства по физическим параметрам, например стабилизация идентифицируемых симптомов, или физиологическим, например стабилизация физических параметров, или по обоим этим параметрам. В другом варианте осуществления, термин “лечить“ или “лечение“ относится к сдерживанию начала заболевания или расстройства.
В некоторых вариантах осуществления, соединение вводят в профилактических целях. Используемый в настоящем документе термин “предотвращать“ или “профилактика“ относится к снижению риска данного заболевания или симптома. В предпочтительном способе варианта осуществления, назначенное соединение вводят субъекту с профилактической целью, например, субъекту с семейным анамнезом или склонностью к злокачественной опухоли или аутоиммунному заболеванию.
Используемый в настоящем документе термин “терапевтически эффективное количество“ относится к количеству соединения или композиции, которое может вызывать биологический или медицинский ответ (который стремятся получить исследователи, ветеринары, врачи или другие клиницисты) в тканевой системе, у животного или человека, который может включать облегчение симптомов заболевания или симптома, подвергаемого лечению. В предпочтительном варианте осуществления терапевтически эффективное количество является количеством, достаточным для эффективного лечения, улучшения или профилактики заболевания, расстройства или состояния, связанного с аномальной генерацией или регуляцией PDE4 и/или TNF–α.
Термин “профилактически эффективное количество“ относится к количеству активного соединения или агента (которое стремятся получить исследователи, ветеринары, врачи или другие клиницисты), которое может сжерживать начало заболевания у субъекта. Профилактически эффективное количество соединения относится к количеству терапевтического средства, используемого отдельно или в сочетании с другим активным соединением, которое может обеспечить терапевтический эффект для лечения или профилактики заболевания, расстройства или состояния.
Если не указано иное, используемые в настоящем документе термины в форме единственного числа также включают термины в форме множественного числа.
Если не указано иное, термин “или“ или “и“ используется в настоящем документе в виде “и/или“.
Если не указано иное, “ “ или “
“ или “ “ в конкретной группе в настоящем документе относится к положению присоединения.
“ в конкретной группе в настоящем документе относится к положению присоединения.
Термин “необязательный“ или “необязательно“ означает, что событие или обстоятельство, описанное далее, может произойти или может не произойти. Этот термин охватывает случаи, когда событие или обстоятельство может произойти или может не произойти. Например, “необязательное замещение“ или “необязательно замещенный“ охватывает случаи, которые являются незамещенными или замещенными.
Термин “Cm–Cn“ или “Cm–n“, используемый в настоящем документе, означает m–n атомов углерода в этой части. Например, “C1–C6 алкил“ относится к алкилу с 1–6 атомами углерода. Диапазон числовых значений в настоящем документе охватывает целые числа в данном диапазоне и поддиапазоны, образованные этими целыми числами. Например, "C1–6" или "C1–C6" означает, что группа может иметь 1, 2, 3, 4, 5 или 6 атомов углерода. Соответственно, "C1–6 алкил" охватывает "C2–5", "C1–4", "C2–4" и C1, C2, C3, C4, C5, C6 и т.д.
Термин “один или более“ или “по меньшей мере один“, используемый в настоящем документе, относится к 1, 2, 3, 4, 5, 6, 7, 8, 9 или более.
Если не указано иное, термин "гетеро" относится к гетероатому или гетероатомной группе (то есть, атомной группе, содержащей гетероатомы), то есть к атому, отличному от углерода и водорода, или атомной группе, содержащей эти атомы. Предпочтительно, гетероатомы независимо выбраны из кислорода, азота, серы и т.д. В варианте осуществления, в котором присутствуют два или более гетероатомов, эти два или более гетероатомов могут быть одинаковыми, или часть или все из этих двух или более гетероатомов могут быть отличными друг от друга.
Термин "алкил", при использовании отдельно или в комбинации с другими терминами, относится к насыщенным алифатическим углеводородным группам, состоящим из прямых или разветвленных цепей атомов углерода и водорода, которые связаны с остальной частью молекулы простой связью. "Алкил" включает, например, C1–C6 алкил. Неограничивающие примеры включают, но не ограничиваются этим, метил, этил, пропил, изопропил, н–бутил, изобутил, втор–бутил, трет–бутил, пентил, гексил и т.д. Алкильные группы, описанные в настоящем документе, могут быть необязательно замещенными.
Термин "алкокси", при использовании отдельно или в комбинации с другими терминами, относится к описанному выше "алкилу", который связан с остальной частью молекулы посредством "–O–", где алкил такой, как определен выше. "Алкокси" включает, например, C1–C6 алкокси. Алкокси, описанный в настоящем документе, может быть необязательно замещенным.
Термин "C3–C6 циклоалкил", при использовании отдельно или в комбинации с другими терминами, относится к насыщенному одновалентному углеводородному кольцу, содержащему 3, 4, 5 или 6 атомов углерода ("C3–C6 циклоалкил"), примерами которого являются циклопропил, циклобутил, циклопентил или циклогексил и т.д. C3–C6 Циклоалкил, описанный в настоящем документе, может быть необязательно замещенным.
Термин “(C1–C6)алкиламино“ относится к  , где один из Ra и Rb представляет собой Н, другой представляет собой (C1–C6)алкил; или Ra и Rb независимо представляют собой (C1–C6)алкил.
, где один из Ra и Rb представляет собой Н, другой представляет собой (C1–C6)алкил; или Ra и Rb независимо представляют собой (C1–C6)алкил.
Термин “гетероцикл“ или “гетероциклил“ относится к группе насыщенных или ненасыщенных моноциклических или полициклических систем, в которых один или более кольцевых атомов представляют собой гетероатомы, выбранные из N, O, S, а остальные представляют собой С. В настоящем документе, когда присутствует более чем один гетероатом, гетероатомы могут быть одинаковыми или отличными друг от друга. "5–7–Членный гетероцикл" относится к гетероциклам, содержащим 5–7 кольцевых атомов, где один или более, предпочтительно 1 или 2 кольцевых атома независимо выбраны из О, N и S, а остальные кольцевые атомы представляют собой С. Гетероцикл или гетероциклил, описанные в настоящем документе, могут быть необязательно замещенными.
Соответственно, термин "5–7–членный гетероцикл, содержащий N" относится к вышеуказанному "5–7–членному гетероциклу", где по меньшей мере один гетероатом представляет собой N. Примером является  .
.
Аналогично, термин "5–7–членный гетероцикл, содержащий O" относится к вышеуказанному "5–7–членному гетероциклу", где по меньшей мере один гетероатом представляет собой O. Примером является  .
.
Термин “гало“ или “галоген“ относится к фтору, хлору, брому или йоду.
Термин “гидроксил“ относится к –OH.
Термин “циано“ относится к –CN.
Термин “амино“ относится к –NH2. Амино, описанный в настоящем документе, может быть необязательно замещенным, например, замещенным одним или более C1–6 алкилами.
Термин "замещенный" используемый в настоящем документе относится к необязательному замещению одного или более атомов водорода указанного атома данной группой при условии, что нормальная валентность указанного атома в текущей ситуации не превышена и что замещение образует стабильное соединение. Комбинации заместителей и/или переменных допускаются только тогда, когда такие комбинации образуют стабильные соединения. В настоящем документе примеры заместителей включают, но не ограничиваются этим, дейтерий (D), бензилокси, алкиламин, C1–6 алкил, галоген, C1–6 алкокси, галогенированный C1–6 алкил, галогенированный C1–6 алкокси, гетероциклил, нитро, циано, гидроксил, карбоксил, амино, сульфонил, C3–C6 циклоалкил и т.д.
Дейтерий (D или 2H) является стабильным нерадиоактивным изотопом водорода, его атомная масса составляет 2,0144. В естественном состоянии водород существует в форме изотопной смеси H (водород или протий), D (2H или дейтерий) и T (3H или тритий), где содержание дейтерия составляет 0,0156%. Согласно общим техническим знаниям в этой области, во всех соединениях, структуры которых содержат природные атомы водорода, атом водорода фактически представляет собой смесь H, D и Т. Поэтому, если соединение содержит дейтерий, относительное содержание которого превышает его относительное содержание в природе 0,0156% в любом положении, эти соединения следует рассматривать как неприродные или обогащенные дейтерием, и поэтому эти соединения являются новыми по сравнению с его необогащенными аналогами.
В изобретении, “обогащенное дейтерием“ соединение относится к соединению формулы I или его фармацевтически приемлемой соли, сольвату, полиморфу, сокристату, стереоизомеру, изотопному соединению, метаболиту или пролекарству, где относительное содержание дейтерия больше, чем его природное относительное содержание в любом соответствующем положении. Поэтому в “обогащенном дейтерием“ соединении относительное содержание дейтерия в любом из соответствующих положений составляет вероятно от 0,0156% до 100%. Обогащенное дейтерием положение представлено D, тогда как необогащенное дейтерием положение представлено H. Согласно общим техническим знаниям в данной области, символ H может быть элиминирован в необогащенном дейтерием положении. Примером способа получения обогащенного дейтерием соединения является замещение водорода дейтерием или использование исходного вещества, обогащенного дейтерием, для синтеза соединения.
В изобретении, процентное содержание дейтерия в обогащенном дейтерием веществе или относительное содержание дейтерия относится к молярному процентному содержанию.
В изобретении, “необогащенный дейтерием“ относится к водороду в естественном состоянии, который находится в форме смеси изотопов H (водород или протий), D (2H или дейтерий) и T (3H или тритий).
Каждое из указанных выше предпочтительных условий может сочетаться любым способом без отхода от общеизвестных знаний в данной области техники и, таким образом, образуя различные предпочтительные варианты осуществления изобретения.
Реагенты и исходные вещества, используемые в настоящем документе, являются коммерчески доступными.
Положительные эффекты настоящего изобретения состоят в том, что соединение формулы I может регулировать генерацию и/или активность PDE4 и/или TNF–α с тем, чтобы эффективно лечить рак и воспалительные заболевания. Кроме того, соединение по настоящему изобретению обладает меньшей токсичностью и хорошей безопасностью.
Подробное описание вариантов осуществления
Изобретение будет дополнительно проиллюстрировано следующими примерами, но подразумевается, что изобретение не ограничивается объемом этих примеров. Экспериментальными методами, которые подробно не описаны в следующих примерах, являются методы, соответствующие обычным методам и условиям или соответствующие руководствам по использованию продуктов.
В следующих примерах “в течение ночи“ означает 10–16 часов, предпочтительно 12 часов. Температура кипения с обратным холодильником относится к температуре кипения растворителя при атмосфеоном давлении.
Пример 1 Синтез соединения 101

Стадия 1. Синтез соединения 101–C
К раствору соединения 101–A (3–фтор–6–нитрофталевая кислота) (1,9 г, 8,3 ммоль) в MeOH (50 мл) добавляли Pd/C (200 мг, 10%, 50% воды). Смесь перемешивали при 25°C в течение ночи в атмосфере H2 (50 фунтов/дюйм2 (3,515 кг/см2)). После завершения реакции смесь фильтровали через слой целита и фильтрат концентрировали с получением соединения 101–C (3–амино–6–фторфталевой кислоты) (1,6 г, выход: 97%) в виде желтого твердого вещества. 1H ЯМР (300 МГц, DMSO–d6) δ 7,09 (т, J=9,0 Гц, 1H), 6,74 (дд, J=6,0, 5,1 Гц, 1H).
Стадия 2. Синтез соединения 101–E
Раствор 101–C (500 мг, 2,5 ммоль) в Ac2O (8 мл) перемешивали при 25°C в течение ночи. Затем растворитель выпаривали с получением соединения 101–E [N–(7–фтор–1,3–диоксо–1,3–дигидро изобензофуран–4–ил)ацетамид] (320 мг, выход: 57%) в виде желтого твердого вещества. 1H ЯМР (300 МГц, DMSO–d6) δ 9,94 (с, 1H), 8,34 (дд, J=9,3, 3,9 Гц, 1H), 7,79 (т, J=9,0 Гц, 1H), 2,16 (с, 3H).
Стадия 3. Синтез соединения 101
Раствор 101–E (380 мг, 1,7 ммоль), 1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этанамин (CAS № 253168–94–4) (465 мг, 1,7 ммоль) в AcOH (10 мл) перемешивали при 100°C в течение ночи. Реакционную смесь упаривали и очищали препаративной ВЭЖХ (система NH4HCO3/CH3CN) и затем лиофилизировали с получением соединения 101 (N–(2–(1– (3–этокси–4–метоксифенил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид)) (412 мг, выход: 51%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6) δ 9,74 (с, 1H), 8,37–8,42 (м, 1H), 7,64 (т, J=9,0 Гц, 1H), 7,04 (с, 1H), 6,90–6,99 (м, 2H), 5,74 (дд, J=10,2, 4,5 Гц, 1H), 4,11–4,33 (м, 2H), 4,04–3,97 (м, 2H), 3,72 (с, 3H), 2,99 (с, 3H), 2,15 (с, 3H), 1,30 (т, J=6,6 Гц, 3H). МС: 477 ([M–1] +).
Синтез исходного вещества 101–A
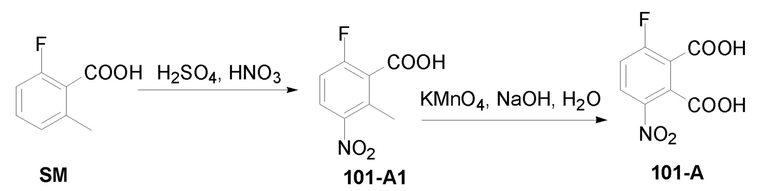
К раствору 2–фтор–6–метилбензойной кислоты (CAS № 90259–27–1) (14 г, 90,9 ммоль) в 110 мл конц.H2SO4 добавляли по каплям дымящую HNO3 (5 мл) в 20 мл конц.H2SO4 при –15°C. Затем смесь перемешивали при 0°C в течение 2 часов. Смесь выливали в дробленый лед при перемешивании. Полученное твердое вещество собирали и растворяли в EtOAc (200 мл), промывали водой (100мл ×2), сушили над Na2SO4, фильтровали и концентрировали досуха с получением соединения 101–A1 (6–фтор–2–метил–3–нитробензойная кислота) (15,3 г, выход: 85%) в виде белого твердого вещества. 1H ЯМР (300 МГц, CDCl3) δ 8,01 (с, 1H), 7,18 (с, 1H), 2,63 (с, 3H).
К раствору соединения 101–A1 (6–фтор–2–метил–3–нитробензойная кислота) (13,6 г, 68 ммоль) в 150 мл H2O добавляли NaOH (8,2 г, 205 ммоль), раствор перемешивали при 80°C в течение 3 часов. Добавляли по порциям KMnO4 (86 г, 547 ммоль) в течение 3 часов. Затем смесь перемешивали при 80°C в течение еще 30 минут. Раствор фильтровали и промывали горячей водой (80 мл ×3). Охлаждали водой со льдом и подкисляли 2N раствором HCl до pH=1. Экстрагировали при помощи EtOAc(200 мл ×5), объединенную EtOAc фазу промывали водой (300 мл ×2), насыщенным солевым раствором (300 мл), сушили над Na2SO4, фильтровали и концентрировали досуха с получением продукта 101–A (3–фтор–6–нитрофталевая кислота) (4,5 г, выход: 29%) в виде белого твердого вещества. 1H ЯМР (300 МГц, DMSO) δ 8,28–8,24 (м, 1H), 7,8 (т, J=9,0 Гц, 1H).
Пример 2 Синтез соединения 102

К раствору соединения 102–A ((S)–1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этанамин (S)–2–ацетамидо–4–метилпентаноат) (CAS № 608141–43–1) (300 мг, 0,67 ммоль) в AcOH (15 мл) добавляли соединение 101–E (N–(7–фтор–1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамид) (157 мг, 0,7 ммоль) и осуществляли взаимодействие при 120°C в течение ночи. Реакционную смесь упаривали досуха на роторном испарителе, очищали препаративной ВЭЖХ и лиофилизировали с получением соединения 102 ((S)–N–(2–(1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид)) (197 мг, выход: 61%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6) δ 9,73 (с, 1H), 8,41–8,44 (м, 1H), 7,65 (т, J=9,2 Гц, 1H), 7,06 (д, J=2,0 Гц, 1H), 6,93–7,01 (м, 2H), 5,76 (дд, J=10,4, 4,4 Гц, 1H), 4,31 (дд, J=14,4, 10,4 Гц, 1H), 4,16 (дд, J=14,4, 4,4 Гц, 1H), 4,02 (кв., J=7,2 Гц, 2H), 3,74 (с, 3H), 3,02 (с, 3H), 2,17 (с, 3H), 1,32 (т, J=7,2 Гц, 3H). ЖХ–МС: 496 ([M+18]+).
Пример 3 Синтез соединения 103
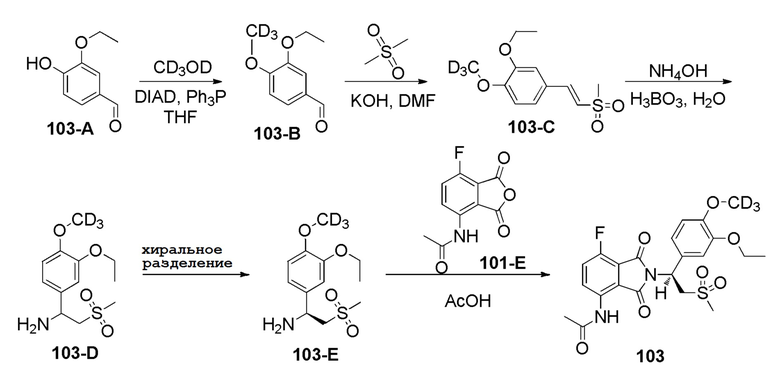
Стадия 1. Синтез соединения 103–B
К раствору соединения 103–A (3–этокси–4–гидроксибензальдегид, CAS № 121–32–4) (10,1 г, 60,77 ммоль), CD3OD (2,4 г, 66,9 ммоль) и Ph3P (19,12 г, 73 ммоль) в THF (250 мл) медленно добавляли DIAD (14,75 г, 73 ммоль) при 0°C. Затем смесь перемешивали при 30°C в течение 2 часов. Растворитель удаляли путем выпаривания и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью PE:EtOAc (4:1), с получением продукта 103–B (3–этокси–4–d3–метоксибензальдегид) в виде бесцветного масла (11 г, 99%).
1H ЯМР (300 МГц, CDCl3) δ 9,85 (с, 1H), 7,41–7,46 (м, 2H), 6,98 (д, J=8,1 Гц, 1H), 4,18 (кв., J=6,9 Гц, 2H), 1,50 (т, J=6,9 Гц, 3H).
Стадия 2. Синтез соединения 103–C
Раствор диметилсульфона (14,1 г, 150,3 ммоль), KOH (5,05 г, 90,1 ммоль) в DMF (150 мл) перемешивали в течение 15 минут при 30°C. Соединение 103–B (11 г, 60,1 ммоль) медленно добавляли к смеси. Смесь перемешивали в течение 3 часов при 60°C. Смесь гасили при помощи NH4Cl (300 мл), экстрагировали при помощи EtOAc (200 мл ×2). Объединенную органическую фазу промывали насыщенным солевым раствором (200 мл ×2), сушили над Na2SO4, фильтровали и концентрировали под вакуумом с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле, элюируя смесью PE:EtOAc (2:1), с получением продукта 103–C (2–этокси–1–d3–метокси–4–(2–(метилсульфонил)винил)бензол) в виде желтого твердого вещества (6,5 г, 42%).
1H ЯМР (300 МГц, CDCl3) δ 7,55 (д, J=15,3 Гц, 1H), 7,12 (д, J=2,8 Гц, 1H), 7,10 (д, J=1,8 Гц, 1H), 6,88–7,02 (м, 1H), 6,76 (д, J=15,3 Гц, 1H), 4,13 (кв., J=6,9 Гц, 2H), 2,99 (с, 3H), 1,50 (т, J=6,9 Гц, 3H).
Стадия 3. Синтез соединения 103–D
Раствор H3BO3 (0,775 г, 12,5 ммоль) в H2O (25 мл) перемешивали при 60°C в течение 30 минут. Затем добавляли 103–C (6,5 г, 25 ммоль) и NH4OH (250 мл). Смесь перемешивали при 80°C в закрытой емкости в течение 3 дней. Смесь экстрагировали при помощи DCM (150 мл ×3), объединенную органическую фазу промывали 2 N раствором HCl (150 мл ×2). Объединенную водную фазу доводили до pH=10 при помощи NaOH, затем экстрагировали при помощи DCM (150 мл ×2). Объединенный органический раствор сушили, фильтровали и концентрировали под вакуумом с получением продукта 103–D (1-(3–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этанамин) (3,5 г, 51%).
1H ЯМР (300 МГц, CDCl3) δ 6,83–6,93 (м, 3H), 4,60 (дд, J=9,3, 3,3 Гц, 1H), 4,11 (кв., J=6,9 Гц, 2H), 3,20–3,37 (м, 2H), 2,91 (с, 3H), 1,83 (с, 2H), 1,47 (т, J=6,9 Гц, 3H).
Стадия 4. Синтез соединения 103–E
Соединение 103–D (3,5 г, 12,68 ммоль) подвергали хиральному разделению с получением соединения 103–E ((S)– 1– (3–этокси–4–d3–метоксифенил)– 2– (метилсульфонил)этанамин) (0,9 г, эи:95,2%).
Метод разделения:
Колонка: chiralpak IA, 5мкм, 4,6×250 мм.
Подвижная фаза: Hex:IPA:DEA=70:30:0,2
Скорость потока (F): 1,0 мл/мин
Длина волны (W): 230 нм
Температура (T): температура окружающей среды
Стадия 5. Синтез соединения 103
Смесь соединения 101–E (161 мг, 0,72 ммоль) и соединения 103–E (200 мг, 0,72 ммоль) в HOAc (5 мл) подвергали взаимодействию при 110°C в течение ночи. Смесь концентрировали досуха при пониженном давлении, затем остаток очищали при помощи препаративной ВЭЖХ с получением соединения 103 ((S)–N–(2–(1–(3–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид)) (160 мг, 46%).
1H ЯМР (400 МГц, DMSO–d6) δ 9,74 (с, 1H), 8,42 (дд, J=9,6, 4,0 Гц, 1H), 7,65 (т, J=9,2 Гц, 1H), 7,06 (д, J=1,6 Гц, 1H), 6,92–7,01 (м, 2H), 5,76 (дд, J=10,4, 4,4 Гц, 1H), 4,31 (дд, J=14,4, 10,8 Гц, 1H), 4,16 (дд, J=14,4, 4,4 Гц, 1H), 4,02 (кв., J=7,2 Гц, 2H), 3,02 (с, 3H), 2,18 (с, 3H), 1,32 (т, J=6,8 Гц, 3H). ЖХМС: [(M+18)]+=499,0.
Пример 4 Синтез соединений 104, 105, 106
Соединения 104, 105 и 106 синтезировали в соответствии со способом синтеза соединения 103 в примере 3 с использованием соответствующих субстратов.

(R)–N–(2–(1–(3–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,76 (с, 1H), 8,42 (дд, J=9,2, 4,0 Гц, 1H), 7,66 (т, J=9,2 Гц, 1H), 7,06 (д, J=2,0 Гц, 1H), 6,93–7,01 (м, 2H), 5,76 (дд, J=10,8, 4,4 Гц, 1H), 4,15–4,34 (м, 2H), 4,02 (кв., J=6,8 Гц, 2H), 3,02 (с, 3H), 2,18 (с, 3H), 1,33 (т, J=6,8 Гц, 3H). ЖХМС: [(M+18)]+=499,0.

(S)–N–(2–(1–(3,4–d6–диметоксифенил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,76 (с, 1H), 8,40–8,43 (м, 1H), 7,66 (т, J=9,2 Гц, 1H), 7,06 (д, J=2,0 Гц, 1H), 6,93–7,01 (м, 2H), 5,77 (дд, J=10,8, 4,4 Гц, 1H), 4,28–4,35 (м, 1H), 4,15–4,19 (м, 1H), 3,03 (с, 3H), 2,18 (с, 3H). ЖХМС: [(M+18)]+=488,0.

(R)–N–(2–(1–(3,4–d6–диметоксифенил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,77 (с, 1H), 8,42 (дд, J=9,6, 4,0 Гц, 1H), 7,66 (т, J=9,2 Гц, 1H), 7,06 (д, J=1,6 Гц, 1H), 6,93–7,02 (м, 2H), 5,75–5,79 (м, 1H), 4,28–4,37 (м, 1H), 4,15–4,20 (м, 1H), 3,03 (с, 3H), 2,17 (с, 3H). ЖХМС: [(M+18)]+=488,0.
Соединения 108 и 109 можно получить в соответствии со способом синтеза соединения 103 в примере 3.

N–(2–(1–(3,4–d6–диметоксифенил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид

N–(2–(1–(3–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
Пример 5. Синтез соединения 107

Соединение 107 ((S)–N–(2–(1–(3–d5–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид) синтезировали в соответствии со способом соединения 103 в примере 3, за исключением того, что использовали промежуточное соединение 107–H вместо соединения 103–B.
1H ЯМР (400 МГц, DMSO) δ 9,76 (с, 1H), 8,42 (дд, J=9,2, 3,8 Гц, 1H), 7,66 (т, J=9,0 Гц, 1H), 7,06 (д, J=2,1 Гц, 1H), 7,00–6,92 (м, 2H), 5,76 (дд, J=10,3, 4,4 Гц, 1H), 4,34–4,14 (м, 2H), 3,02 (с, 3H), 2,18 (с, 3H). ЖХМС: [(M+18)]+=504,0.
Синтез промежуточного соединения 107–H:
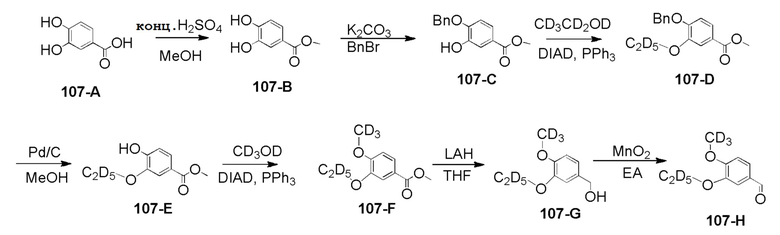
Стадия 1. Синтез соединения 107–B
К раствору соединения 107–A (CAS №99–50–3) (50 г, 0,325 моль) в MeOH (300 мл) медленно добавляли конц. H2SO4 (50 мл). Затем смесь нагревали до температуры кипения с обратным холодильником в течение ночи. Растворитель удаляли. Остаток разбавляли водой (500 мл), экстрагировали при помощи EA (300 мл ×2), промывали насыщенным солевым раствором (300 мл ×2), сушили и концентрировали с получением продукта 107–B в виде белого твердого вещества (54,5 г, 100%).
1H ЯМР (300 МГц, DMSO) δ 9,77 (с, 1H), 9,34 (с, 1H), 7,35–7,29 (м, 2H), 6,80 (д, J=8,2 Гц, 1H), 3,76 (с, 3H).
Стадия 2. Синтез соединения 107–C
К раствору соединения 107–B (54,5 г, 0,32 моль) в MeCN (1,2 л) добавляли K2CO3 (63 г, 0,455 моль). Затем смесь перемешивали при 30°C в течение 0,5 часа. Медленно добавляли BnBr (78 г, 0,455 моль) в MeCN (0,3 л). Смесь перемешивали при 30°C в течение ночи. Твердое вещество удаляли. Растворитель удаляли. Остаток разбавляли при помощи EA (этилацетат, 50 мл) и PE (петролейный эфир, 100 мл), перемешивали при 30°C в течение 15 минут, фильтровали. Лепешку очищали растиранием в порошок с EA (50 мл) и PE (100 мл) в течение ночи с получением продукта 107–C в виде белого твердого вещества (28,8 г, 35%).
1H ЯМР (300 МГц, CDCl3) δ 7,63–7,59 (м, 2H), 7,42–7,38 (м, 5H), 6,95 (д, J=8,3 Гц, 1H), 5,76 (с, 1H), 5,18 (с, 2H), 3,89 (с, 3H).
Стадия 3. Синтез соединения 107–D
К раствору соединения 107–C (18 г, 69,8 ммоль), CD3CD2OD (4,4 г, 83,8 ммоль) и Ph3P (23,8 г, 90,7 ммоль) в THF (300 мл) при 0°C медленно добавляли DIAD (диизопропилазодикарбоксилат, 18,34 г, 90,7 ммоль). Затем смесь перемешивали при 30°C в течение ночи. Растворитель удаляли. Остаток очищали хроматографией на силикагеле, элюируя смесью PE:EA=50:1, с получением продукта 107–D в виде белого твердого вещества (16,7 г, 82%).
1H ЯМР (300 МГц, CDCl3) δ 7,62–7,59 (м, 2H), 7,46–7,30 (м, 5H), 6,92 (д, J=8,3 Гц, 1H), 5,22 (с, 2H), 3,89 (с, 3H).
Стадия 4. Синтез соединения 107–E
К раствору соединения 107–D (16,7 г, 57,3 ммоль) в MeOH (300 мл) добавляли Pd/C (1,67 г, 10%). Затем смесь перемешивали при 30°C в течение ночи в атмосфере H2 (50 фунтов/дюйм2 (3,515 кг/см2)). Смесь фильтровали. Растворитель удаляли с получением продукта 107–E в виде белого твердого вещества (11,52 г, 100%).
1H ЯМР (400 МГц, CDCl3) δ 7,62 (дд, J=8,3, 1,8 Гц, 1H), 7,53 (д, J=1,8 Гц, 1H), 6,94 (д, J=8 Гц, 1H), 6,11 (с, 1H), 3,88 (с, 3H).
Стадия 5. Синтез соединения 107–F
К раствору соединения 107–E (11,52 г, 57,3 ммоль), CD3OD (2,5 г, 69,6 ммоль) и Ph3P (19,8 г, 75,4 ммоль) в THF (300 мл) при 0°C медленно добавляли DIAD (15,3 г, 75,4 ммоль). Затем смесь перемешивали при 30°C в течение ночи. Растворитель удаляли. Остаток очищали хроматографией на силикагеле, элюируя смесью PE:EA=10:1, с получением продукта 107–F в виде белого твердого вещества (12,5 г, 100%).
1H ЯМР (300 МГц, CDCl3) δ 7,67 (дд, J=8,4, 0,9 Гц, 1H), 7,54 (с, 1H), 6,88 (д, J=8,4 Гц, 1H), 3,89 (с, 3H).
Стадия 6. Синтез соединения 107–G
К раствору соединения 107–F (12,5 г, 57,3 ммоль) в THF (200 мл) при 0°C медленно добавляли LAH (3,3 г, 86 ммоль). Затем смесь перемешивали при 30°C в течение 2 часов. Медленно добавляли воду (4 мл) для гашения реакции. Затем медленно добавляли водный раствор NaOH (8 мл, 20%), перемешивали в течение 0,5 часа. Смесь фильтровали, концентрировали с получением остатка, который очищали хроматографией на силикагеле, элюируя смесью PE:EA=2:1, с получением продукта в виде бесцветного масла 107–G (10,6 г, 97%).
1H ЯМР (300 МГц, CDCl3) δ 6,91–6,81 (м, 3H), 4,59 (с, 2H).
Стадия 7. Синтез соединения 107–H
К раствору соединения 107–G (10,6 г, 55,7 ммоль) в EA (200 мл) добавляли MnO2 (48,5 г, 557 ммоль). Затем смесь перемешивали при 25°C в течение ночи. Смесь фильтровали, концентрировали досуха с получением остатка, который очищали растиранием в порошок со смесью PE:EA=5:1 (18 мл) при 0°C в течение 0,25 часа, с получением продукта 107–H (7,08 г, 67%) в виде белого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 9,83 (с, 1H), 7,45–7,39 (м, 2H), 6,96 (д, J=8,2 Гц, 1H).
Соединение 110 можно получить в соответствии со способом синтеза соединения 107.

N–(2–(1–(3–d5–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
Пример 6. Синтез соединения 201

Соединение 201–A (5–фтор–3–нитрофталевая кислота) синтезировали в соответствии со способом синтеза соединения 301–E в примере 10, за исключением того, что использовали соответствующее исходное вещество метил-5–фтор–2–метил–3–нитробензоат вместо соединения 301–A. Метил-5–фтор–2–метил–3–нитробензоат синтезировали в соответствии со способом синтеза исходного вещества 301–A, за исключением того, что 5–фтор–2–метилбензойную кислоту (CAS № 33184–16–6) использовали вместо соединения 301–A1 (4–фтор–2–метилбензойная кислота).
Стадия 1. Синтез соединения 201–B
К раствору соединения 201–A (900 мг) в MeOH (15 мл) добавляли 10% Pd/C (180 мг, 50% масс.) в атмосфере азота. Смесь перемешивали в атмосфере H2 (50 фунтов/дюйм2 (3,515 кг/см2)) в течение ночи. Смесь фильтровали и концентрировали при пониженном давлении с получением соединения 201–B (3–амино–5–фторфталевая кислота, 774 мг) в виде желтого твердого вещества.
1H ЯМР (DMSO–d6, 400 МГц): δ 6,58–6,62 (м, 1H), 6,41–6,44 (м, 1H).
Стадия 2. Синтез соединения 201–C
Раствор 201–B (100 мг, 0,5 ммоль) в Ac2O (4 мл) перемешивали при 25°C в течение ночи. Реакционную смесь упаривали досуха при пониженном давлении с получением соединения 201–C (N–(6–фтор–1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамид, 70 мг, выход: 63%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6) δ 9,87–9,92 (м, 1H), 8,27–8,35 (м, 1H), 7,72–7,74 (м, 1H), 2,24 (с, 3H).
Стадия 3. Синтез соединения 201
Раствор соединения 201–C (70 мг, 0,3 ммоль) и 1– (3–этокси–4–метоксифенил)– 2– (метилсульфонил)этанамина (CAS № 253168–94–4, 86 мг, 0,3 ммоль) в AcOH (6 мл) перемешивали в течение ночи при 70°C. Реакционную смесь упаривали досуха, затем очищали препаративной ВЭЖХ (система NH4HCO3/ацетонитрил), затем лиофилизировали с получением соединения 201 (N–(2–(1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этил)–6–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид, 42 мг, выход: 30%) в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6) δ 9,77 (с, 1H), 8,24 (дд, J=12,0,1,8 Гц, 1H), 7,48 (дд, J=6,9,1,8 Гц, 1H), 7,04 (д, J=0,9 Гц, 1H), 6,90–6,98 (м, 2H), 5,73–5,77 (м, 1H), 4,29–4,34 (м, 1H), 4,10–4,17 (м, 1H), 3,96–4,03 (м, 2H), 3,72 (с, 3H), 3,00 (с, 3H), 2,20 (с, 3H), 1,30 (т, J=7,2 Гц, 3H). МС: 477 ([M–1] +).
Пример 7. Синтез соединения 202

Стадия 1. Синтез соединения 102–B
К раствору соединения 102–A ((S)–1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этанамин (S)–2–ацетамидо–4–метилпентаноат, 800 мг, 1,79 ммоль) в H2O (10 мл) добавляли насыщенный водный раствор Na2CO3 до pH=10, затем смесь экстрагировали при помощи EtOAc (30 мл ×2). Объединенный EtOAc раствор сушили, фильтровали и концентрировали с получением соединения 102–B ((S)–1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этанамин, 460 мг, выход: 94%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6) δ 7,02 (с, 1H), 6,89 (с, 2H), 4,27 (дд, J=9,3, 3,6 Гц, 1H), 3,99–4,06 (м, 2H), 3,73 (с, 3H), 3,20–3,45 (м, 2H), 2,95 (с, 3H), 2,16 (с, 2H), 1,27–1,35 (м, 3H).
Стадия 2. Синтез соединения 202–C
К раствору соединения 102–B в DMF (15 мл) добавляли соединение 201–A (3–нитро–5–фторфталевая кислота, 386 мг, 1,68 ммоль) и HATU (1–[бис(диметиламино)метилен]–1H–1,2,3–триазоло[4,5–b]пиридиний 3–оксидгексафторфосфат, 1,4 г, 3,7 ммоль) и DIEA (N,N–диизопропилэтиламин, 760 мг, 5,88 ммоль), затем смесь перемешивали при 25°C в течение ночи. К реакционной смеси добавляли H2O (10 мл) и затем перемешивали в течение 15 минут, экстрагировали при помощи EtOAc (100 мл). EtOAc раствор промывали насыщенным солевым раствором (20 мл ×2), затем сушили, фильтровали и концентрировали под вакуумом с получением неочищенного продукта. Остаток очищали колоночной хроматографией на силикагеле с использованием PE:EtOAc(3:1-1:1) с получением соединения 202–C((S)–2–(1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этил)–6–фтор–4–нитроизоиндолин–1,3–дион, 330 мг, выход: 42%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6) δ 8,36 (дд, J=8,8, 2,4 Гц, 1H), 8,20 (дд, J=10,8, 2,2 Гц, 1H), 7,09 (д, J=1,6 Гц, 1H), 7,01 (дд, J=8,4, 2,0 Гц, 1H), 6,94 (д, J=8,8 Гц, 1H), 5,79 (дд, J=9,6, 5,6 Гц, 1H), 4,18–4,31 (м, 2H), 3,99–4,06 (м, 2H), 3,74 (с, 3H), 2,98 (с, 3H), 1,32 (т, J=6,8 Гц, 3H).
Стадия 3. Синтез соединения 202–D
К смеси соединения 202–C (330 мг, 0,704 ммоль) в EtOAc (20 мл) добавляли Pd/C (10%, 50% H2O, 40 мг) и затем перемешивали в атмосфере H2 (50 фунтов/дюйм2 (3,515 кг/см2)) в течение 4 часов при 25°C. Смесь фильтровали и концентрировали с получением соединения 202–D ((S)–4–амино–2–(1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этил)–6–фторизоиндолин–1,3–дион, 289 мг, выход: 94%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6) δ 7,06 (с, 1H), 6,93 (д, J=0,4 Гц, 2H), 6,80 (дд, J=7,2, 2,0 Гц, 1H), 6,70–6,73 (м, 3H), 5,71 (дд, J=10,8, 4,4 Гц, 1H), 4,33 (дд, J=14,4, 10,4 Гц, 1H), 3,99–4,10 (м, 3H), 3,73 (с, 3H), 3,00 (с, 3H), 1,32 (т, J=7,2 Гц, 3H).
Стадия 4. Синтез соединения 202
К раствору соединения 202–D (289 мг, 0,66 ммоль) в пиридине (30 мл) добавляли AC2O (5 мл), смесь нагревали до 70°C и перемешивали в течение ночи. Смесь концентрировали. Затем добавляли CH3CN (10 мл ×2) и смесь концентрировали еще два раза с получением остатка, который очищали колоночной хроматографией на силикагеле, элюируя смесью (PE:EtOAc=1:1), с получением неочищенного продукта (200 мг), который дополнительно очищали препаративной ВЭЖХ, с получением продукта 202 ((S)–N–(2–(1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этил)–6–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид, 84 мг, выход: 26%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6) δ 9,77 (с, 1H), 8,26 (дд, J=12,4, 2,4 Гц, 1H), 7,48 (дд, J=6,8, 2,0 Гц, 1H), 7,07 (д, J=2,0 Гц, 1H), 6,93–7,00 (м, 2H), 5,77 (дд, J=10,4, 4,0 Гц, 1H), 4,32 (дд, J=14,4, 10,8 Гц, 1H), 4,15 (дд, J=14,4, 4,4 Гц, 1H), 4,02 (кв., J=7,2 Гц, 2H), 3,74 (с, 3H), 3,01 (с, 3H), 2,22 (с, 3H), 1,32 (т, J=7,2 Гц, 3H). ЖХМС: 496,0 ([M+18]+).
Пример 8. Синтез соединения 203

Стадия 1. Синтез соединения 203–C
К смеси соединения 103–E ((S)–1–(3–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этанамин) (680 мг, 2,46 ммоль) в DMF (30 мл) добавляли соединение 201–A (3–нитро–5–фторфталевая кислота, 564 мг, 2,46 ммоль), HATU (2,06 г, 5,41 ммоль) и DIEA (1,1 г, 9,61 ммоль) при 25°C, затем смесь перемешивали при 25°C в течение ночи. К реакционной смеси добавляли H2O (15 мл) и перемешивали в течение 15 минут, затем экстрагировали при помощи EtOAc (150 мл). Органическую фазу промывали насыщенным солевым раствором (50 мл ×3), сушили, фильтровали и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали колоночной хроматографией на силикагеле с использованием PE:EtOAc(3:1~1:1) с получением соединения 203–C ((S)–2–(1–(3–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этил)–6–фтор–4–нитроизоиндолин–1,3–дион, 605 мг, выход: 52%) в виде желтого твердого вещества.
1H ЯМР(300 МГц, DMSO–d6): δ 8,34–8,38 (м, 1H), 8,19–8,22 (м, 1H), 7,09 (с, 1H), 6,92–7,02 (м, 2H), 5,76–5,79 (м, 1H), 4,21–4,27 (м, 2H), 3,98–4,04 (м, 2H), 2,98 (с, 3H), 1,29–1,34 (м, 3H).
Стадия 2. Синтез соединения 203–D
К смеси соединения 203–C (605 мг, 1,29 ммоль) в EtOAc (20 мл) добавляли Pd/C (10%, 50% H2O, 60 мг), осуществляли взаимодействие в течение 4 часов при 25°C в атмосфере H2 (50 фунтов/дюйм2 (3,515 кг/см2)). Смесь фильтровали и концентрировали с получением соединения 203–D ((S)–4–амино–2–(1–(3–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этил)–6–фторизоиндолин–1,3–дион, 518 мг, выход: 91%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6) δ 7,06 (с, 1H), 6,78–6,93 (м, 2H), 6,70–6,74 (м, 4H), 5,71 (дд, J=10,48, 4,4 Гц, 1H), 4,30–4,33 (m 1H), 3,98–4,10 (м, 3H), 3,00 (с, 3H), 1,32 (т, J=6,8 Гц, 3H).
Стадия 3. Синтез соединения 203
К раствору соединения 203–D (247 мг, 0,56 ммоль) в CH3COOH (6 мл) добавляли Ac2O (3 мл). Смесь нагревали до 85°C и осущесвляли взаимодействие в течение 5 часов, затем концентрировали и очищали препаративной ВЭЖХ с получением продукта. К продукту добавляли гексан (5 мл) и смесь перемешивали в течение 2 часов, затем фильтровали с получением соединения 203 ((S)–N–(2–(1–(3–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этил)–6–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид, 123 мг, выход: 46%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6) δ 9,77 (с, 1H), 8,26 (дд, J=12,0, 2,0 Гц, 1H), 7,49 (дд, J=6,8, 2,0 Гц, 1H), 7,07 (д, J=1,6 Гц, 1H), 6,92–7,00 (м, 2H), 5,77 (дд, J=10,4, 4,4 Гц, 1H), 4,32 (дд, J=14,4, 10,4 Гц, 1H), 4,15 (дд, J=14,4, 4,4 Гц, 1H), 4,02 (кв., J=6,8 Гц, 2H), 3,02 (с, 3H), 2,22 (с, 3H), 1,32 (т, J=6,8 Гц, 3H). ЖХМС: 499,0 ([M+18]+).
Пример 9. Синтез соединений 204, 205, 206 и 207
Соединения 204, 205, 206 и 207 синтезировали в соответствии со способом синтеза соединения 203 в примере 8, с соответствующими субстратами для замены соединения 103–E.
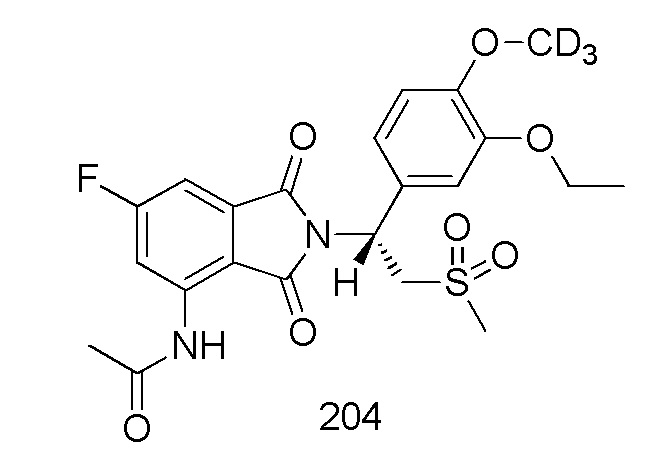
(R)–N–(2–(1–(3–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этил)–6–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,79 (с, 1H), 8,26 (дд, J=12,0, 2,4 Гц, 1H), 7,50 (дд, J=6,8, 2,4 Гц, 1H), 7,07 (д, J=2,0 Гц, 1H), 6,92–7,00 (м, 2H), 5,77 (дд, J=10,4, 4,4 Гц, 1H), 4,14–4,35 (м, 2H), 4,02 (кв., J=6,8 Гц, 2H), 3,02 (с, 3H), 2,22 (с, 3H), 1,32 (т, J=6,8 Гц, 3H). ЖХМС: 499,0 ([M+18]+)

(S)–N(2–(1–(3,4–d6–диметоксифенил)–2–(метилсульфонил)этил)–6–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,80 (с, 1H), 8,26 (дд, J=12,0, 2,0 Гц, 1H), 7,51 (дд, J=7,2, 2,4 Гц, 1H), 7,07 (д, J=2,0 Гц, 1H), 6,92–7,01 (м, 2H), 5,78 (дд, J=10,8, 4,4 Гц, 1H), 4,33 (дд, J=14,8, 10,8 Гц, 1H), 4,16 (дд, J=10,8, 4,4 Гц, 1H), 3,02 (с, 3H), 2,22 (с, 3H). ЖХМС: 488,0 ([M+18]+)

(R)–N–(2–(1–(3,4–d6–диметоксифенил)–2–(метилсульфонил)этил)–6–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,79 (с, 1H), 8,26 (дд, J=12,0, 2,4 Гц, 1H), 7,50 (дд, J=6,8, 2,0 Гц, 1H), 7,07 (д, J=2,0, 1H), 6,92–7,00 (м, 2H), 5,78 (дд, J=10,8, 4,4 Гц, 1H), 4,30–4,36 (м, 1H), 4,14–4,19 (м, 1H), 3,02 (с, 3H), 2,22 (с, 3H). ЖХМС: 488,0 ([M+18]+)

(S)–N–(2–(1–(3–d5–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этил)–6–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,79 (с, 1H), 8,26 (дд, J=12,0, 2,4 Гц, 1H), 7,50 (дд, J=6,8, 2,4 Гц, 1H), 7,06 (д, J=2,0 Гц, 1H), 6,92–6,99 (м, 2H), 5,77 (дд, J=10,4, 4,4 Гц, 1H), 4,14–4,35 (м, 2H), 3,02 (с, 3H), 2,22 (с, 3H). ЖХМС: 504,0 ([M+18]+)
Соединения 208, 209 и 210 можно синтезировать в соответствии со способом синтеза соединения 203 в примере 8 с соответствующими субстратами для замены соединения 103–E.

N–(2–(1–(3–d5–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этил)–6–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид

N–(2–(1–(3–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этил)–6–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид

N–(2–(1–(3,4–d6–диметоксифенил)–2–(метилсульфонил)этил)–6–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
Пример 10. Синтез соединения 301

Стадия 1. Синтез соединения 301–C
Раствор соединения 301–A (метил-4–фтор–2–метил–3–нитробензоат, 3,0 г, 14,1 ммоль), NaOH (1,6 г, 42,3 ммоль) в H2O/MeOH (30 мл/30 мл) перемешивали при 25°C в течение ночи. Затем смесь доводили до pH=5, экстрагировали при помощи EtOAc (100 мл ×3), промывали насыщенным солевым раствором (100 мл ×2), сушили, фильтровали и концентрировали с получением соединения 301–C (4–фтор–2–метил–3–нитробензойная кислота, 2,8 г, выход: 100%) в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6) δ 8,06–8,11 (м, 1H), 8,18 (т, J=9,0 Гц, 1H), 2,47 (с, 3H).
Стадия 2. Синтез соединения 301–E
К раствору соединения 301–C (2,8 г, 14,1 ммоль), NaOH (1,6 г, 42 ммоль) в H2O (30 мл) добавляли по порциям KMnO4 (17,7 г, 112 ммоль) в течение 3 часов при 85°C и затем смесь перемешивали в течение 3 часов при 85°C. Затем смесь фильтровали и лепешку промывали при помощи H2O (50 мл ×3). Фильтрат доводили до pH=1, экстрагировали при помощи EtOAc (100 мл ×3), промывали насыщенным солевым раствором (100 мл ×2), сушили, фильтровали и концентрировали с получением соединения 301–E (4–фтор–3–нитрофталевая кислота, 900 мг, выход: 28%) в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6) δ 8,12–8,17 (м, 1H), 7,75–7,81 (м, 1H).
Стадия 3. Синтез соединения 301–G
К раствору соединения 301–E (900 мг, 3,9 ммоль) в MeOH (30 мл) добавляли Pd/C (180 мг, 10%, 50% воды). Смесь перемешивали при 25°C в течение ночи в атмосфере H2 (50 фунтов/дюйм2 (3,515 кг/см2)). После завершения реакции смесь фильтровали через слой целита и фильтрат концентрировали с получением соединения 301–G (3–амино–4–фторфталевая кислота, 700 мг, неочищенное вещество) в виде желтого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6) δ 7,13–7,20 (м, 1H), 6,76–6,80 (м, 1H).
Стадия 4. Синтез соединения 301–H
Раствор соединения 301–G (300 мг, 1,5 ммоль), 1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этанамина (356 мг, 1,5 ммоль), AcOH (660 мг, 15 ммоль), Et3N (758 мг, 7,5 ммоль) в CH3CN (20 мл) перемешивали при 80°C в течение ночи в атмосфере N2. Затем растворитель удаляли, остаток очищали колоночной хроматографией на силикагеле (PE/EtOAc=2/1) с получением соединения 301–H (4–амино–2–(1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этил)–5–фторизоиндолин–1,3–дион, 150 мг, выход: 23%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6) δ 7,37–7,44 (м, 1H), 7,07 (с, 1H), 6,99–7,03 (м, 1H), 6,94 (с, 2H), 6,55 (д, J=5,7 Гц, 1H), 5,70–5,75 (м, 1H), 4,30–4,38 (м, 1H), 4,12–4,13 (м, 1H), 3,97–4,08 (м, 2H), 3,74 (с, 3H), 3,01 (с, 3H), 1,33 (т, J=7,2 Гц, 3H).
Стадия 5. Синтез соединения 301
Раствор соединения 301–H (100 мг, 0,23 ммоль) в Ac2O (6 мл) перемешивали при 70°C в течение ночи. Реакционную смесь упаривали досуха и очищали препаративной ВЭЖХ (система NH4HCO3/ацетонитрил), затем лиофилизировали с получением соединения 301 (N–(2–(1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этил)–5–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид, 37 мг, выход: 34%) в виде белого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6) δ 10,16 (с, 1H), 7,69–7,82 (м, 1H), 7,07 (с, 1H), 6,91–6,94 (м, 2H), 5,72–5,77 (м, 1H), 4,31–4,36 (м, 1H), 4,10–4,16 (м, 1H), 3,97–4,05 (м, 2H), 3,73 (с, 3H), 2,99 (с, 3H), 2,09 (с, 3H), 1,32 (т, J=7,2 Гц, 3H). МС: 477 ([M–1]+).
Синтез исходного вещества 301–A

Соединение 301–A1 (4–фтор–2–метилбензойная кислота, CAS № 321–21–1, 100 г, 649 ммоль) добавляли к 660 мл дымящей HNO3 по каплям для поддержания температуры ниже 10°C. Смесь перемешивали в течение 1-2 часов. Смесь выливали в воду со льдом (2,4 л) и перемешивали в течение 30 минут. Полученное твердое вещество фильтровали и промывали холодной водой и затем растворяли в 1,5 л EtOAc. EtOAc фазу промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и затем фильтровали. Твердый Na2SO4 промывали при помощи EtOAc (200мл ×3). Объединенную EtOAc фазу концентрировали с получением неочищенного продукта 301–A2, который использовали непосредственно на следующей стадии без очистки.
К раствору соединения 301–A2 (110 г, 502 ммоль) в 1,5 л метанола добавляли 20 мл конц. H2SO4. Смесь нагревали до температуры кипения с обратным холодильником в течение ночи. Смесь охлаждали до комнатной температуры, затем концентрировали до около 100 мл и затем разбавляли при помощи 500 мл холодной воды. Смесь экстрагировали при помощи EtOAc (500 мл ×3). Объединенную органическую фазу промывали насыщ.NaHCO3, водой и насыщенным солевым раствором, сушили над Na2SO4, фильтровали и промывали при помощи EtOAc, EtOAc фазу концентрировали досуха с получением неочищенного продукта. Неочищенный продукт перекристаллизовывали из смеси PE/EtOAc (10:1, 400 мл) для удаления большинства основных побочных продуктов, остаток очищали колоночной хроматографией на силикагеле (PE/EtOAc: 100:1) с получением продукта 301–A (метил-4–фтор–2–метил–3–нитробензоат, 20 г, две стадии, выход: 19%).
1H ЯМР (DMSO–d6, 300 МГц): 8,08 (дд, J=5,7, 9,0 Гц, 1H), 7,58 (т, J=9,0 Гц, 1H),, 3,85 (с, 3H), δ 2,45 (с, 3H)
Пример 11. Синтез соединения 302

Стадия 1. Синтез соединения 302–C
К смеси соединения 102–B ((S)–1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этанамин, 954 мг, 3,49 ммоль) в DMF (60 мл) добавляли соединение 301–E (3–нитро–4–фторфталевая кислота, 800 мг, 3,49 ммоль), HATU (CAS № 148893–10–1, 2,08 г, 5,478 ммоль) и DIEA (CAS № 7087–68–5, 1,58 г, 12,2 ммоль) при 5°C. Реакционную смесь перемешивали при 25°C в течение ночи. К реакционной смеси добавляли H2O (20 мл), перемешивали в течение 15 минут, экстрагировали при помощи EtOAc (100 мл). EtOAc раствор промывали насыщенным солевым раствором (50 мл ×3), сушили, фильтровали и концентрировали с получением неочищенного продукта. Остаток очищали колоночной хроматографией на силикагеле с использованием PE:EtOAc (от 2:1 до 1:1) с получением соединения 302–C ((S)–2–(1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этил)–5–фтор–4–нитроизоиндолин–1,3–дион, 396 мг, выход: 25%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6) δ 8,24 (дд, J=8,4, 4,0 Гц, 1H), 8,09 (дд, J=10,0, 8,4 Гц, 1H), 7,09 (д, J=2,4 Гц, 1H), 7,01 (дд, J=8,4, 2,0 Гц, 1H), 6,94 (д, J=8,0 Гц, 1H), 5,78 (дд, J=9,6, 5,6 Гц, 1H), 4,22–4,26 (м, 2H), 4,00–4,06 (м, 2H), 3,74 (с, 3H), 2,99 (с, 3H), 1,32 (т, J=7,2 Гц, 3H).
Стадия 2. Синтез соединения 302–D
К смеси соединения 302–C (396 мг, 0,85 ммоль) в EtOAc (15 мл) добавляли Pd/C (50 мг, 10%, 50% H2O), реакционную смесь перемешивали в атмосфере H2(50 фунтов/дюйм2 (3,515 кг/см2)) в течение 4 часов при 25°C. Смесь фильтровали и концентрировали с получением соединения 302–D ((S)–4–амино–2–(1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этил)–5–фторизоиндолин–1,3–дион, 327 мг, выход: 88%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6): δ 7,37–7,43 (м, 1H), 6,93–7,06 (м, 4H), 6,52 (с, 2H), 5,70–5,73 (м, 1H), 4,29–4,37 (м, 1H), 3,99–4,04 (м, 3H), 3,73 (с, 3H), 3,00 (с, 3H), 1,29–1,34 (м, 3H).
Стадия 3. Синтез соединения 302
К раствору соединения 302–D (132 мг, 0,302 ммоль) в HOAc (4 мл) добавляли AC2O (2 мл), смесь нагревали до 85°C и осуществляли взаимодействие при 85°C в течение 5 часов. Эту смесь концентрировали и разбавляли при помощи EtOAc (30мл). EtOAc раствор промывали насыщенным водным раствором NaHCO3 (15 мл), сушили над Na2SO4, затем концентрировали с получением неочищенного продукта, который очищали препаративной ВЭЖХ, с получением соединения 302 ((S)–N–(2–(1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этил)–5–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид, 31 мг, выход: 21%) в виде белого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6) δ 10,13 (с, 1H), 7,79 (дд, J=8,0, 4,4 Гц, 1H), 7,72 (дд, J=10,4, 8,4 Гц, 1H), 7,07 (д, J=2,0 Гц, 1H), 6,92–6,95 (м, 2H), 5,75 (дд, J=10,4, 4,8 Гц, 1H), 4,11–4,32 (м, 2H), 4,01 (кв., J=7,2 Гц, 2H), 3,73 (с, 3H), 2,99 (с, 3H), 2,09 (с, 3H), 1,32 (т, J=7,2 Гц, 3H). ЖХМС: 476,9 ([M–1]–).
Пример 12. Синтез соединения 401

Стадия 1. Синтез соединения 401–A
Получение раствора LDA (диизопропиламид лития):
К раствору диизопропиламина (35 мл, 0,25 моль) в 100 мл безводного THF по каплям добавляли n–BuLi (2,5 N, 96 мл, 0,24 моль) при –30°C в атмосфере N2, температуру поддерживали ниже –30°C. Реакционный раствор перемешивали при –30°C в течение 15 минут и затем при 0°C в течение 30 минут.
Раствор 2–фторпиридина (CAS № 372–48–5, 19,42 г, 0,2 моль) в 100 мл безводного THF охлаждали до –70°C в атмосфере N2. Вышеуказанный раствор LDA добавляли по каплям к этому раствору, при этом температуру поддерживали ниже –70°C. Затем раствор перемешивали при –70°C в течение 1 часа. К реакционной смеси по каплям добавляли раствор I2 (61 г, 0,24 моль) в 50 мл безводного THF и затем реакционную смесь перемешивали при –75°C в течение 1 часа. Реакцию гасили насыщенным раствором NH4Cl и перемешивали при 25°C в течение 30 минут. THF удаляли путем выпаривания. Остаток экстрагировали при помощи EtOAc (500 мл ×2). Объединенный EtOAc раствор промывали водой и насыщенным солевым раствором, сушили над Na2SO4. Фильтровали и концентрировали досуха с получением неочищенного продукта 401–A (2–фтор–4–йодпиридин, 30 г, выход: 67%).
1H ЯМР (300МГц, DMSO–d6): δ 8,37–8,43 (м, 1H), 8,21–8,22 (м, 1H), 7,13–7,18 (м, 1H).
Стадия 2. Синтез соединения 401–B
Получение раствора LDA:
К раствору диизопропиламина (17 мл, 96,9 ммоль) в 300 мл безводного THF по каплям добавляли n–BuLi (46,5 мл, 116 ммоль) при –30°C в атмосфере N2, температуру поддерживали ниже –30°C. Раствор перемешивали при –30°C в течение 15 минут и затем при 0°C в течение 30 минут.
Раствор соединения 401–A (21,6 г, 96,9 ммоль) в 100 мл безводного THF охлаждали до –70°C. Вышеуказанный раствор LDA добавляли по каплям к этому раствору, при этом температуру поддерживали ниже –70°C. Затем раствор перемешивали при –70°C в течение 1 часа. К раствору добавляли по каплям этилформиат (10 мл, 121 ммоль) и медленно нагревали до –50°C в течение 1 часа. Реакционную смесь гасили насыщенным раствором NH4Cl и перемешивали при 25°C в течение 30 минут. THF удаляли путем выпаривания и реакционный раствор экстрагировали при помощи EtOAc (300 мл ×2). Объединенную органическую фазу промывали водой и насыщенным солевым раствором, сушили над Na2SO4. Na2SO4 фильтровали и органическую фазу концентрировали досуха и остаток очищали колоночной хроматографией на силикагеле (PE/EtOAc: 50:1–10:1) с получением продукта 401–B (2–фтор–4–йодникотинальдегид, 13,0 г, выход: 53%).
1H ЯМР (300 МГц, DMSO–d6): δ 10,15 (с, 1H), 7,97 (д, J=5,1 Гц, 1H), 7,87 (д, J=5,1 Гц, 1H).
Стадия 3. Синтез соединения 401–C
К раствору соединения 401–B (13,3 г, 53 ммоль) в 466 мл t–BuOH и 133 мл воды, охлажденному льдом, добавляли 2–метил–2–бутен (13,3 г, 53 ммоль), Na2HPO4 (70 г, 583 ммоль), затем добавляли по порциям NaClO2 (24g, 265 ммоль). Реакционную смесь перемешивали при 25°C в течение 1,5 часов, затем разбавляли 800 мл DCM и подкисляли раствором 6 N/HCl до pH=2. Органическую фазу отделяли и водную фазу экстрагировали при помощи DCM/MeOH (20:1, 1000 мл ×2). Объединенную органическую фазу промывали насыщенным солевым раствором, сушили над Na2SO4. Фильтровали и концентрировали досуха. Остаток кристаллизовали из DCM/PE(1:1) с получением продукта 401–C (2–фтор–4–йодникотиновая кислота, 11,5 г, выход: 81%).
1H ЯМР (300 МГц, DMSO–d6): δ 14,26 (ушир. с, 1H), 8,02 (д, J=4,2 Гц, 1H), 7,94 (дд, J=3,9, 0,6 Гц, 1H).
Стадия 4. Синтез 401–D
К раствору соединения 401–C (10,3 г, 38,6 ммоль) в 40 мл MeOH и Et2O (40 мл), охлаждаемому водой со льдом, добавляли по каплям TMSCH2N2 (29 мл, 57,9 ммоль). Смесь перемешивали при 25°C в течение ночи. Затем добавляли ледяную воду для гашения реакции. Растворитель удаляли путем выпаривания и добавляли насыщ. NaHCO3 и смесь перемешивали в течение 30 минут. Смесь экстрагировали при помощи EtOAc (100 мл ×3). Объединенную органическую фазу промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали досуха с получением продукта 401–D (метил-2–фтор–4–йодникотинат, 9,6 г, выход: 88%).
1H ЯМР (300 МГц, DMSO–d6): δ 8,06 (д, J=5,4 Гц, 1H), 7,97 (д, J=5,1 Гц, 1H), 3,93 (с, 3H).
Стадия 5. Синтез соединения 401–E
К раствору соединения 401–D (9,6 г, 34,1 ммоль) и 2,4–диметоксибензиламина (7,41 г, 44,3 ммоль) в 50 мл DMSO добавляли K2CO3 (11,8 г, 68,2 ммоль). Смесь перемешивали при 25°C в течение 5 часов, затем разбавляли 500 мл EtOAc. Смесь промывали водой и насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали досуха и остаток очищали колоночной хроматографией на силикагеле (PE/EtOAc: 50:1–10:1) с получением продукта 401–E (метил-2–((2,4–диметоксибензил)амино)–4–йодникотинат, 10,66 г, выход: 73%).
1H ЯМР (400 МГц, DMSO–d6): δ 7,69 (д, J=5,2 Гц, 1H), 7,07 (д, J=5,2 Гц, 1H), 7,00–7,03 (м, 2H), 6,55 (д, J=2,4 Гц, 1H), 6,44 (дд, J=8,4, 2,4Гц, 1H), 4,44 (д, J=6,0 Гц, 2H), 3,87 (с, 3H), 3,81 (с, 3H), 3,72 (с, 3H).
Стадия 6. Синтез соединения 401–F
К раствору соединения 401–E (10,66 г, 24,9 ммоль) в 80 мл DCM, охлаждаемому водой со льдом, добавляли 30 мл TFA. Смесь перемешивали при 25°C в течение 3 часов, концентрировали досуха. Смесь подщелачивали насыщенным раствором NaHCO3. Твердое вещество фильтровали и промывали ледяной водой и сушили с получением продукта 401–F (метил-2–амино–4–йодникотинат, 5,54 г, выход: 80%).
1H ЯМР (300 МГц, DMSO–d6): δ 7,65 (д, J=5,1 Гц, 1H), 7,07 (д, J=5,4 Гц, 1H), 6,43 (с, 2H), 3,83 (с, 3H).
Стадия 7. Синтез соединения 401–G
Раствор соединения 401–F (5,54 г, 0,02 ммоль) в 100 мл HOAc и 50 мл Ac2O перемешивали при 80°C в течение ночи. Смесь концентрировали досуха и очищали колоночной хроматографией на силикагеле (PE/EtOAc: 1:1–1:2) с получением продукта 401–G (метил-2–ацетамидо–4–йодникотинат, 2,7 г, 42%).
1H ЯМР (300 МГц, DMSO–d6): δ 10,56 (с, 1H), 8,08 (д, J=5,1 Гц, 1H), 7,82 (д, J=5,4 Гц, 1H), 3,74 (с, 3H), 2,01 (с, 3H).
Стадия 8. Синтез соединения 401–H
К раствору соединения 401–G (3,2 г, 10 ммоль) в 50 мл MeOH добавляли DIEA (3,3 мл, 20 ммоль) и PdCl2(PPh3)2 (702 мг, 1 ммоль). Смесь перемешивали в атмосфере 50 МПа CO при 100°C в течение ночи. Охлаждали до 25°C и концентрировали досуха и очищали колоночной хроматографией на силикагеле (PE/EtOAc: 5:1~2:1) с получением продукта 401–H (диметил-2–аминопиридин–3,4–дикарбоксилат, 1,8 г, выход:86%).
1H ЯМР (300 МГц, DMSO–d6): δ 8,24 (д, J=4,8 Гц, 1H), 6,97 (с, 2H), 6,67 (д, J=4,8 Гц, 1H), 3,80 (с, 3H), 3,78 (с, 3H).
Стадия 9. Синтез соединения 401–I
Раствор соединения 401–H (400 мг, 1,9 ммоль) в 20 мл 20% KOH и 20 мл THF перемешивали при 25°C в течение ночи. Экстрагировали при помощи TMBE (50 мл), органическую фазу отделяли и водную фазу подкисляли 2N раствором HCl до pH=2. Полученное твердое вещество фильтровали и промывали ледяной водой и сушили с получением продукта 401–I (2–аминопиридин–3,4–дикарбоновая кислота, 285 мг, выход: 82%)
1H ЯМР (300 МГц, DMSO–d6): δ 8,15 (д, J=4,8 Гц, 1H), 6,59 (д, J=4,8 Гц, 1H),
Стадия 10. Синтез соединения 401
Раствор соединения 401–I (500 мг, 2,75 ммоль) в 20 мл Ac2O нагревали до температуры кипения с обратным холодильником в течение 3 часов. Затем смесь охлаждали до комнатной температуры и концентрировали досуха, остаток растворяли в 20 мл HOAc, затем добавляли 1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этанамин (750 мг, 2,75 ммоль). Реакционную смесь кипятили с обратным холодильником в течение ночи и затем охлаждали до 25°C и добавляли 20 мл Ac2O и перемешивали при 85°C в течение еще 5 часов. Смесь охлаждали до 25°C и концентрировали досуха и очищали при помощи препаративной ВЭЖХ с получением продукта 401 (N–(2–(1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этил)–1,3–диоксо–2,3–дигидро–1H–пирроло[3,4–c]пиридин–4–ил)ацетамид, 735 мг, выход: 58%).
1H ЯМР (300 МГц, DMSO–d6): δ 10,51 (с, 1H), 8,88 (д, J=4,8 Гц, 1H), 7,67 (с, 1H), 7,09 (с, 1H), 6,92–7,00 (м, 2H), 5,73–5,78 (м, 1H), 4,25–4,33 (м, 1H), 4,10–4,17 (м, 1H), 3,98–4,06 (м, 2H), 3,73 (с, 3H), 2,97 (с, 3H), 2,17 (с, 3H), 1,32 (т, J=7,2 Гц, 3H). ЖХМС:[(M+1)]+=461,9.
Пример 13. Синтез соединения 601

Стадия 1. Синтез соединения 601–B
К раствору соединения 601–A (3–гидроксипиколиновая кислота, CAS № 874–24–8, 60 г, 430 ммоль) в MeOH (600 мл) медленно добавляли конц. H2SO4 (60 мл). Затем смесь нагревали до 80°C в течение ночи. Доводили до pH=7 с использованием твердого Na2CO3, смесь фильтровали и лепешку промывали при помощи EtOAc (500 мл). Фильтрат концентрировали с получением соединения 601–B (метил-3–гидроксипиколинат, 40 г, выход: 61%) в виде белого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 10,60 (с, 1H), 8,27 (д, J=3,7 Гц, 1H), 7,35–7,44 (м, 2H), 4,05 (с, 3H).
Стадия 2. Синтез соединения 601–C
К раствору соединения 601–B (30 г, 196 ммоль) в H2O (1500 мл) медленно добавляли Br2 (94 г, 590 ммоль). Смесь перемешивали при 30°C в течение ночи. Затем смесь экстрагировали при помощи DCM (500 мл ×2), промывали насыщенным солевым раствором (500 мл), сушили, фильтровали и концентрировали с получением соединения 601–C (метил-4,6–дибром–3–гидроксипиколинат, 49 г, выход: 81%) в виде белого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 11,35 (с, 1H), 7,87 (с, 1H), 4,07 (с, 3H).
Стадия 3. Синтез соединения 601–D
Раствор соединения 601–C (49 г, 157 ммоль), BnBr (80,5 г, 472 ммоль) и K2CO3 (98 г, 710 ммоль) в CH3CN (1 л) кипятили с обратным холодильником в течение ночи. Смесь охлаждали до 25°C. Твердое вещество удаляли фильтрованием и фильтрат упаривали с получением остатка, который очищали путем растирания в порошок с PE:EtOAc=10:1 (100 мл) при 30°C в течение 1 часа, с получением соединения 601–D (метил-3–(бензилокси)–4,6–дибромпиколинат, 43 г, выход: 68%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CDCl3): δ 7,89 (с, 1H), 7,39–7,54 (м, 5H), 5,15 (с, 2H), 3,94 (с, 3H).
Стадия 4. Синтез 601–E
К раствору соединения 601–D (43 г, 107 ммоль), HCOONa (8,7 г, 128 ммоль) в DMF (1 л) добавляли Pd(PPh3)4 (6,2 г, 5,35 ммоль). Смесь перемешивали при 80°C в течение ночи. Добавляли H2O (5 л) и смесь экстрагировали при помощи EtOAc (1л ×2). Объединенный EtOAc раствор промывали насыщенным солевым раствором (1л ×2), сушили и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле, элюируя смесью PE:EtOAc =10:1, с получением продукта 601–E (метил-3–(бензилокси)–4–бромпиколинат, 14,8 г, 43%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 8,27 (дд, J=4,8, 0,6 Гц, 1H), 7,71 (дд, J=4,8, 0,6 Гц, 1H), 7,55 (д, J=6,9 Гц, 2H), 7,37–7,45 (м, 3H), 5,17 (с, 2H), 3,95 (с, 3H).
Стадия 5. Синтез соединения 601–F
К раствору соединения 601–E (14,7 г, 45,63 ммоль), 2,4–диметоксибензиламина (9,92 г, 59,32 ммоль), xantphos (1,58 г, 2,74 ммоль), Cs2CO3 (22,3 г, 68,5 ммоль) в толуоле (350 мл) добавляли Pd2(dba)3 (0,84 г, 0,913 ммоль) в атмосфере N2. Затем смесь нагревали до 100°C в течение 2 дней. Реакционный раствор концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле, элюируя смесью PE:EtOAc (1:1), с получением продукта 601–F (этил-3–(бензилокси)–4–((2,4–диметоксибензил)амино)пиколинат, 8,4 г, 58%) в виде оранжевого масла.
1H ЯМР (300 МГц, CDCl3) δ 8,12 (д, J=5,7 Гц, 1H), 7,35–7,44 (м, 5H), 7,00 (д, J=8,4 Гц, 1H), 6,64 (д, J=5,1 Гц, 1H), 6,40–6,45 (м, 2H), 5,26–5,31 (м, 1H), 4,99 (с, 2H), 4,40–4,48 (м, 2H), 4,24 (д, J=6,0 Гц, 2H), 3,80 (с, 3H), 3,75 (с, 3H), 1,41 (т, J=7,2 Гц, 3H).
Стадия 6. Синтез соединения 601–G
Раствор соединения 601–F (7,33 г, 17,4 ммоль) в DCM (дихлорметан, 60 мл) и TFA (трифторуксусная кислота, 30 мл) перемешивали при 30°C в течение 4 часов. Добавляли H2O (50 мл), экстрагировали при помощи DCM (50 мл ×2). Объединенный DCM раствор сушили и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле, элюируя смесью PE:EtOAc (1:1), с получением продукта 601–G (этил-4–амино–3–(бензилокси)пиколинат, 4,0 г, 85%) в виде коричневого масла.
1H ЯМР (300 МГц, CDCl3) δ 8,08 (д, J=5,1 Гц, 1H), 7,36–7,48 (м, 5H), 6,71 (д, J=5,1 Гц, 1H), 5,02 (с, 2H), 4,40–4,48 (м, 4H), 1,41 (т, J=7,2 Гц, 3H).
Стадия 7. Синтез соединения 601–H
Раствор соединения 601–G (4,0 г, 15,5 ммоль) в Ac2O (40 мл) перемешивали при 100°C в течение ночи. Смесь концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле, элюируя смесью PE:EtOAc (1:1), с получением продукта 601–H (этил-4–ацетамидо–3–(бензилокси)пиколинат, 3,17 г, 69%) в виде желтого масла.
1H ЯМР (300 МГц, CDCl3) δ,38–8,438 (м, 2H), 7,66 (с, 1H), 7,43 (с, 5H), 5,10 (с, 2H), 4,51 (кв., J=7,2 Гц, 2H), 1,86 (с, 3H), 1,47 (т, J=7,2 Гц, 3H).
Стадия 8. Синтез соединения 601–I
К смеси соединения 601–H (3,17 г, 10,01 ммоль) в MeOH (30 мл) и EtOAc (30 мл) добавляли Pd/C (10%, 50% H2O, 0,32 г). Затем смесь перемешивали при 30°C в атмосфере H2 (50 фунтов/дюйм2 (3,515 кг/см2)) в течение ночи. Смесь фильтровали, фильтрат концентрировали с получением неочищенного продукта 601–I (этил-4–ацетамидо–3–гидроксипиколинат, 1,67 г, 74%) в виде коричневого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6) δ 10,99 (ушир. с, 1H), 9,78 (с, 1H), 8,27 (д, J=5,6 Гц, 1H), 8,07 (с, 1H), 4,40 (кв., J=7,2 Гц, 2H), 2,19 (с, 3H), 1,35 (т, J=7,2 Гц, 3H).
Стадия 9. Синтез соединения 601–J
К смеси соединения 601–I (1,53 г, 6,8 ммоль) в DMF (30 мл) добавляли Et3N (1,44 г, 14,28 ммоль) и N,N–бис(трифторметилсульфонил)анилин (3,83 г, 10,7 ммоль), смесь перемешивали при 25°C в течение 3 часов. Добавляли воду (500 мл) и смесь экстрагировали при помощи EtOAc (200 мл ×2). Объединенный EtOAc раствор промывали насыщенным солевым раствором (200 мл ×2), сушили и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле, элюируя смесью PE:EtOAc (2:1), с получением продукта 601–J (этил-4–ацетамидо–3–(((трифторметил)сульфонил)окси)пиколинат, 2,0 г, 83%) в виде бесцветного масла.
1H ЯМР (300 МГц, CDCl3) δ 8,56–8,61 (м, 2H), 7,87 (с, 1H), 4,49 (кв., J=7,2 Гц, 2H), 2,29 (с, 3H), 1,44 (т, J=7,2 Гц, 3H).
Стадия 10. Синтез соединения 601–K
Смесь соединения 601–J (1,85 г, 5,2 ммоль), Pd(OAc)2 (233 мг, 1,04 ммоль), DPPP (1,3–бис(дифенилфосфино)пропан, 429 мг, 1,04 ммоль) и Et3N (1,16 г, 11,5 ммоль) в DMSO (1,84 мл) и EtOH (50 мл) нагревали до 70°C в течение ночи. Смесь фильтровали, фильтрат концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле, элюируя смесью PE:EtOAc (1:1), с получением продукта 601–K (диэтил-4–ацетамидопиридин–2,3–дикарбоксилат, 1,0 г, 69%) в виде бесцветного масла.
1H ЯМР (300 МГц, CDCl3): δ 10,50 (с, 1H), 8,56–8,66 (м, 2H), 4,35–4,46 (м, 4H), 2,26 (с, 3H), 1,35–1,44 (м, 6H).
Стадия 11. Синтез соединения 601–L
Смесь соединения 601–K (1,0 г, 3,6 ммоль) в KOH (20%, 50 мл, водн.) и THF (50 мл) перемешивали при 25°C в течение 3 часов. Растворитель удаляли и остаток разбавляли MeOH (100 мл), перемешивали при 50°C в течение 1 часа, фильтровали. Фильтрат концентрировали с получением продукта 601–L (4–аминопиридин–2,3–дикарбоновая кислота, 0,9 г, неочищенное вещество) в виде белого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6) δ 8,52–9,26 (м, 2H), 8,11 (д, J=6,8 Гц, 1H), 7,19 (д, J=6,8 Гц, 1H).
Стадия 12. Синтез соединения 601–M
Смесь соединения 601–L (0,9 г) в Ac2O (30 мл) нагревали до 80°C в течение ночи. Затем растворитель удаляли с получением продукта 601–M (N–(5,7–диоксо–5,7–дигидрофуро[3,4–b]пиридин–4–ил)ацетамид, 0,6 г) в виде коричневого масла.
Стадия 13. Синтез соединения 601
Смесь соединения 601–M (0,6 г) и [1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этанамин] (0,8 г, 2,9 ммоль) в AcOH (20 мл) нагревали до 120°C в течение 3 часов. Растворитель удаляли с получением неочищенного продукта, который очищали препаративной ВЭЖХ, с получением целевого продукта 601 (N–(6–(1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этил)–5,7–диоксо–6,7–дигидро–5H–пирроло[3,4–b]пиридин–4–ил)ацетамид, 71 мг, выход для 3 стадий 4%).
1H ЯМР (400 МГц, DMSO–d6) δ 9,79 (с, 1H), 8,80 (д, J=5,6 Гц, 1H), 8,36 (д, J=6,0 Гц, 1H), 7,08 (д, J=2,0 Гц, 1H), 7,02 (дд, J=8,4, 2,0 Гц, 1H), 6,94 (д, J=8,4 Гц, 1H), 5,82 (дд, J=10,4, 4,8 Гц, 1H), 4,17–4,33 (м, 2H), 4,03 (кв., J=7,2 Гц, 2H), 3,74 (с, 3H), 3,02 (с, 3H), 2,26 (с, 3H), 1,32 (т, J=7,2 Гц, 3H). ЖХМС [(M+1)]+=462,0.
Пример 14. Синтез соединения 701

Стадия 1. Синтез соединения 701–B
К раствору DMSO (3,79 г, 40,3 ммоль) в 150 мл безводного THF, охлажденному до 0°C ледяной водой, медленно по каплям добавляли n–BuLi (2,5 M, 16,1 мл, 40,3 ммоль) в атмосфере N2. Затем реакционную смесь перемешивали в ледяной бане в течение 2 часов. К этому раствору по каплям добавляли раствор 701–A (6–этокси–5–метоксипиколинонитрил, 2,87 г, 16,1 ммоль) в 30 мл безводного THF. Затем смесь перемешивали при 0°C на ледяной бане в течение 2 часов. Смесь гасили ледяной водой и THF удаляли путем выпаривания. Смесь экстрагировали при помощи EtOAc (500 мл ×3). Объединенную органическую фазу промывали водой и насыщенным солевым раствором, сушили над Na2SO4, концентрировали досуха и кристаллизовали из PE/EtOAc (2:1) с получением продукта 701–B (1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этенамин, 3,5 г, выход: 80%).
1H ЯМР (300МГц, DMSO–d6): δ 7,52 (д, J=8,1 Гц, 1H), 7,35 (д, J=8,4 Гц, 1H), 6,80 (с, 2H), 5,55 (с, 1H), 4,40 (кв., J=6,9 Гц, 2H), 3,83 (с, 3H), 3,00 (с, 3H), 1,34 (т, J=6,9 Гц, 3H).
Стадия 2: синтез соединения 701–C
К раствору соединения 701–B (4,2 г, 15,4 ммоль) в 100 мл THF, охлаждаемому ледяной водой, добавляли 2N раствор HCl (50 мл). Смесь перемешивали при 25°C в течение ночи. THF выпаривали и смесь подщелачивали насыщенным раствором NaHCO3, затем экстрагировали при помощи EtOAc (200 мл ×3). Объединенную органическую фазу промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали досуха с получением продукта 701–C (1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этенон, 4,0 г, выход: 95%)
1H ЯМР (300 МГц, DMSO–d6): δ 7,73 (д, J=8,1 Гц, 1H), 7,46 (д, J=8,1Гц, 1H), 5,07 (с, 2H), 4,45 (кв., J=7,2 Гц, 2H), 3,89 (с, 3H), 3,15 (с, 3H), 1,38 (т, J=7,2 Гц, 3H).
Стадия 3. Синтез соединения 701–D
К раствору соединения 701–C (4,0 г, 14,6 ммоль) в 100 мл MeOH, охлаждаемому ледяной водой, добавляли по порциям NaBH4 (1,11 г, 29,3 ммоль) и перемешивали при 25°C в течение 2 часов. Затем смесь гасили 2 N раствором HCl (20 мл) и перемешивали в течение 30 минут, концентрировали досуха и подщелачивали насыщенным раствором NaHCO3, затем экстрагировали смесью DCM/MeOH (20:1, 400 мл ×2). Объединенную органическую фазу промывали насыщенным солевым раствором, сушили над Na2SO4, концентрировали досуха с получением неочищенного продукта 701–D (1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанол, 4,0 г, выход:100%), который использовали непосредственно на следующей стадии.
Стадия 4. Синтез соединения 701–E
К раствору соединения 701–D (4,0 г, 14,53 ммоль) в 100 мл DCM, охлаждаемому ледяной водой, добавляли Et3N (4,0 мл, 29,0 ммоль), затем добавляли по каплям MsCl (1,7 мл, 21,8 ммоль). Смесь перемешивали при 25°C в течение ночи и затем гасили ледяной водой и перемешивали в течение 30 минут, затем экстрагировали при помощи DCM. Объединенную органическую фазу промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и концентрировали досуха и кристаллизовали из PE/EtOAc (1:1) с получением продукта 701–E (2–этокси–3–метокси–6–(2–(метилсульфонил)винил)пиридин, 1,8 г, выход: 48%).
1H ЯМР (300 МГц, DMSO–d6): δ 7,37 (д, J=6,3 Гц, 2H), 7,31 (с, 2H), 4,40 (кв., J=6,9 Гц, 2H), 3,83 (с, 3H), 3,11 (с, 3H), 1,35 (т, J=6,9 Гц, 3H).
Стадия 5. Синтез соединения 701–F
Раствор B(OH)3 (70 мг, 0,81 ммоль) в 5 мл воды нагревали до 50°C и перемешивали в течение 15 минут. Добавляли соединение 701–E (140 мг, 0,54 ммоль) и перемешивали в течение 30 минут и затем добавляли 20 мл NH3.H2O. Смесь перемешивали при 80°C в герметично закрытой пробирке в течение 3 дней. Затем смесь охлаждали до 25°C и концентрировали досуха, подщелачивали насыщенным раствором NaHCO3 и перемешивали в течение 30 минут, затем экстрагировали при помощи DCM/MeOH (50 мл ×3), объединенную органическую фазу промывали насыщенным солевым раствором, сушили над Na2SO4, концентрировали досуха с получением неочищенного продукта 701–F (1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин, 110 мг).
1H ЯМР (400 МГц, DMSO–d6): δ 7,24 (д, J=8,0 Гц, 1H), 6,98 (д, J=8,0 Гц, 1H), 4,31–4,39 (м, 2H), 4,21–4,25 (м, 1H), 3,76 (с, 3H), 3,36–3,43 (м, 2H), 3,02 (с, 3H), 2,32 (с, 2H), 1,32 (т, J=7,2 Гц, 3H).
Стадия 6. Синтез соединения 701
К раствору соединения 701–F (110 мг, 0,4 ммоль) в 20 мл HOAc добавляли (N–(1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамид, CAS № 6296–53–3, 82 мг, 0,4 ммоль). Смесь нагревали до температуры кипения с обратным холодильником в течение ночи, затем охлаждали до 25°C и концентрировали досуха и очищали препаративной ВЭЖХ с получением продукта 701 (N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид, 123 мг, выход для 2 стадий: 49%).
1H ЯМР (400 МГц, DMSO–d6): δ 9,71 (с, 1H), 8,47 (д, J=8,4 Гц, 1H), 7,80–7,84 (м, 1H), 7,60 (д, J=7,2 Гц, 1H), 7,27 (д, J=8,0 Гц, 1H), 7,02 (д, J=8,0 Гц, 1H), 5,81 (дд, J=10,8, 4,0 Гц, 1H), 4,30–4,34 (м, 1H), 4,15–4,22 (м,,3H), 3,76 (с, 3H), 3,09 (с, 3H), 2,18 (с, 3H), 1,17 (дд, J=7,2 Гц, 3H). ЖХМС:[(M+1)]+=462,0.
Синтез исходного вещества 701–A:

Синтез соединения 701–A2:
К раствору соединения 701–A1 (2–бром–пиридин–3–ол, CAS № 6602–32–0, 60 г, 0,35 моль) в H2O (600 мл) добавляли K2CO3 (96,7 г, 0,7 моль), I2 (90,7 г, 0,357 моль). Реакционную смесь перемешивали при 15°C в течение ночи. Смесь доводили до pH=5 3N раствором HCl. Полученное твердое вещество собирали фильтрацией, промывали водой (200 мл ×3), сушили с получением соединения 701–A2 (2–бром–6–йодпиридин–3–ол, 101 г, выход: 97%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6) δ 11,11 (с, 1H), 7,62 (д, J=8,4 Гц, 1H), 7,03 (д, J=8,1 Гц, 1H).
Синтез соединения 701–A3:
К раствору соединения 701–A2 (2–бром–6–йодпиридин–3–ол, 101 г, 0,337 моль) в 200 мл DMF добавляли K2CO3 (70 г, 0,506 моль) и смесь перемешивали в течение 30 минут. Добавляли CH3I (57,4 г, 0,404 моль), затем смесь перемешивали при 100°C в течение 2 часов. Реакционную смесь выливали в 2 л H2O, перемешивали в течение 1 часа, фильтровали, промывали водой (500 мл ×2), собирали твердое вещество, которое суспендировали в PE:EtOAc=2:1 (300 мл) при 15°C в течение 1 часа, с получением продукта 701–A3 (2–бром–6–йод–3–метоксипиридин, 74 г, выход: 70%) в виде коричневого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6) δ 7,79 (д, J=8,4 Гц, 1H), 7,28 (д, J=8,1 Гц, 1H), 3,87 (с, 3H).
Синтез соединения 701–A4:
К раствору EtOH (1,5 л) добавляли 27,6 г Na, затем смесь перемешивали при 15°C, до растворения твердого вещества. Добавляли соединение 701–A3 (2–бром–6–йод–3–метоксипиридин, 37,7 г, 0,12 моль) и смесь нагревали до 100°C в течение ночи. Растворитель удаляли. Остаток разбавляли при помощи EtOAc (1 л) и раствор промывали водой (1л ×2), сушили над Na2SO4, концентрировали досуха с получением продукта 701–A4 (2–этокси–6–йод–3–метоксипиридин, 31,6 г, выход: 94%) в виде коричневого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6) δ 7,32 (д, J=8,0 Гц, 1H), 7,05 (д, J=8,0 Гц, 1H), 4,26 (кв., J=7,2 Гц, 2H), 3,76 (с, 3H), 1,31 (т, J=7,2 Гц, 3H).
Синтез соединения 701–A
К раствору соединения 701–A4 [2–этокси–6–йод–3–метоксипиридин] (31,6 г, 0,113 моль) в 300 мл DMF добавляли CuCN (12,2 г, 0,136 моль). Смесь нагревали до 150°C в течение 2 часов и затем разбавляли водой (1 л) и экстрагировали при помощи EtOAc (500 мл ×2). Органическую фазу промывали насыщенным солевым раствором (500 мл ×3), сушили над Na2SO4, концентрировали досуха с получением соединения 701–A (6–этокси–5–метоксипиколинонитрил, 20 г, выход: 99%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6) δ 7,63 (д, J=8,1 Гц, 1H), 7,39 (д, J=7,8 Гц, 1H), 4,31 (кв., J=7,2 Гц, 2H), 3,86 (с, 3H), 1,32 (т, J=7,2 Гц, 3H).
Пример 15. Синтез соединения 801

Стадия 1. Синтез соединения 801–B
К раствору соединения 801–A (пиридин–2,3–диол, CAS № 16867–04–2, 24 г, 216 ммоль) и NaOBut (20,75 г, 216 ммоль) в EtOH (200 мл) добавляли йодэтан (37 г, 237,6 ммоль). Смесь перемешивали при 85°C в течение ночи. Растворитель удаляли выпариванием. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью DCM:MeOH (50:1), с получением продукта 801–B (3–этоксипиридин–2–ол, 12,2 г, 40%) в виде коричневого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6) δ 11,52 (с, 1H), 6,90–6,92 (м, 1H), 6,77 (д, J=7,2 Гц, 1H), 6,06 (дд, J=14,0, 6,8 Гц, 1H), 3,91 (кв., J=6,8 Гц, 2H), 1,30 (т, J=6,8 Гц, 3H).
Стадия 2. Синтез соединения 801–C
Раствор соединения 801–B (12,2 г, 87,7 ммоль) в POCl3 (160 мл) перемешивали при 80°C в течение ночи. Растворитель удаляли выпариванием. Остаток разбавляли водой (200 мл) и смесь доводили до pH=8 при помощи NaHCO3, затем экстрагировали при помощи DCM (200 мл ×2). Объединенный органический раствор сушили над NaSO4, фильтровали и концентрировали с получением неочищенного вещества, которое очищали колоночной хроматографией на силикагеле, элюируя смесью PE:EtOAc (4:1), с получением продукта 801–C (2–хлор–3–этоксипиридин, 12,1 г, 87%) в виде желтого масла.
1H ЯМР (300 МГц, CDCl3) δ 7,93 (т, J=3,0 Гц, 1H), 7,15–7,18 (м, 2H), 4,09 (кв., J=7,2 Гц, 2H), 1,46 (т, J=7,2 Гц, 3H).
Стадия 3. Синтез соединения 801–D
Na (13,1 г, 571 ммоль) осторожно добавляли к MeOH (250 мл) и перемешивали при 30°C до растворения Na. Затем к смеси добавляли соединение 801–C (9,0 г, 57,14 ммоль) и смесь кипятили с обратным холодильником в течение 2 дней. Растворитель удаляли. Остаток разбавляли при помощи DCM (300 мл), промывали водой (200 мл ×2), сушили и концентрировали с получением продукта 801–D (3–этокси–2–метоксипиридин, 7,2 г, выход: 83%) в виде желтого масла.
1H ЯМР (300 МГц, CDCl3) δ 7,71 (дд, J=5,1, 1,5 Гц, 1H), 7,02 (дд, J=7,8, 1,2 Гц, 1H), 6,80 (дд, J=7,8, 5,1 Гц, 1H), 4,03–4,12 (м, 5H), 1,46 (т, J=6,9 Гц, 3H).
Стадия 4. Синтез соединения 801–E
К раствору соединения 801–D (7,2 г, 47 ммоль) и NaOAc (4,6 г, 56,4 ммоль) в AcOH (120 мл) медленно добавляли Br2 (9,0 г, 56,4 ммоль) в AcOH (20 мл) при 10°C. Смесь перемешивали при 30°C в течение ночи. Реакционную смесь выливали в лед (300 г), экстрагировали при помощи MTBE (метил-трет–бутиловый эфир, 100 мл ×2). Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле, элюируя смесью PE:EtOAc (20:1), с получением продукта 801–E (5–бром–3–этокси–2–метоксипиридин, 8,4 г, 77%) в виде желтого масла.
1H ЯМР (300 МГц, CDCl3) δ 7,77 (д, J=2,1 Гц, 1H), 7,13 (д, J=1,8 Гц, 1H), 4,05–4,11 (м, 2H), 3,98 (с, 3H), 1,44–1,51 (м, 3H).
Стадия 5. Синтез соединения 801–F
К раствору соединения 801–E (8,4 г, 36,2 ммоль) в THF (150 мл) медленно добавляли n–BuLi (17,4 мл, 2,5 M) при –70°C. Смесь перемешивали при –70°C в течение 1,5 часов. К реакционной смеси добавляли DMF (7 мл, 90,5 ммоль) и перемешивали при –70°C в течение 0,5 часа. Смесь гасили при помощи NH4Cl (100 мл), экстрагировали при помощи EtOAc (100 мл ×2). Объединенный EtOAc раствор промывали насыщенным солевым раствором (100 мл), сушили над NaSO4, фильтровали и концентрировали с получением неочищенного продукта 801–F (5–этокси–6–метоксиникотинальдегид, 6,5 г) в виде красного твердого вещества.
1H ЯМР (400 МГц, CDCl3) δ 9,85 (с, 1H), 8,11 (д, J=2,0 Гц, 1H), 7,38 (д, J=2,0 Гц, 1H), 4,07–4,13 (м, 2H), 4,04 (с, 3H), 1,41–1,47 (м, 3H).
Стадия 6. Синтез соединения 801–G
К раствору диметилсульфона (30,8 г, 328 ммоль), KOH (1,84 г, 32,8 ммоль) в DMF (400 мл) медленно добавляли 801–F (5,94 г, 32,8 ммоль) в DMF (100 мл). Смесь перемешивали при 30°C в течение 3 часов. Смесь гасили при помощи NH4Cl (500 мл), экстрагировали при помощи EtOAc (500 мл ×2). Объединенную органическую фазу промывали насыщенным солевым раствором (500 мл ×2), сушили над Na2SO4 затем фильтровали и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле, элюируя смесью PE:EtOAc (2:1), с получением продукта 801–G (3–этокси–2–метокси–5–(2–(метилсульфонил)винил)пиридин, 0,71 г, 8,5%) в виде коричневого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 7,87 (д, J=1,8 Гц, 1H), 7,58 (д, J=15,3 Гц, 1H), 7,13 (д, J=1,8 Гц, 1H), 6,80 (д, J=15,3 Гц, 1H), 4,09–4,16 (м, 2H), 4,06 (с, 3H), 3,05 (с, 3H), 1,52 (т, J=6,9 Гц, 3H).
Стадия 7. Синтез соединения 801–H
Раствор H3BO3 (0,368 г, 5,95 ммоль) в H2O (6 мл) перемешивали при 50°C в течение 15 минут. Добавляли соединение 801–G (1,1 г, 4,3 ммоль) и перемешивали при 50°C в течение 15 минут. Затем добавляли NH4OH (60 мл), реакционную смесь перемешивали при 80°C в герметично закрытой пробирке в течение 3 дней. Смесь экстрагировали при помощи DCM (50 мл ×3), объединенную органическую фазу экстрагировали 2 N раствором HCl (50 мл ×2). Водную фазу доводили до pH=10 при помощи NaOH, экстрагировали при помощи DCM (100 мл ×2). Объединенный DCM раствор сушили и концентрировали с получением продукта 801–H (1– (5–этокси–6–метоксипиридин–3–ил)– 2– (метилсульфонил)этанамин, 0,71 г, 66%).
1H ЯМР (300 МГц, CDCl3) δ 7,71 (с, 1H), 7,12 (с, 1H), 4,67 (д, J=9,6 Гц, 1H), 4,11 (кв., J=6,9 Гц, 2H), 4,01 (с, 3H), 3,09–3,40 (м, 2H), 2,99 (с, 3H), 1,88 (с, 2H), 1,49 (т, J=6,9 Гц, 3H).
Стадия 8. Синтез соединения 801
Смесь соединения (N–(1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамид, 112 мг, 0,55 ммоль) и соединения 801–H (150 мг, 0,55 ммоль) в HOAc (5 мл) нагревали до 110°C в течение ночи. Концентрировали досуха при пониженном давлении, остаток очищали препаративной ВЭЖХ с получением соединения 801 (N–(2–(1–(5–этокси–6–метоксипиридин–3–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид, 66 мг, 26%) в виде белого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6) δ 9,70 (с, 1H), 8,44 (д, J=8,4 Гц, 1H), 7,78–7,81 (м, 2H), 7,57 (д, J=6,8 Гц, 1H), 7,39 (с, 1H), 5,83 (дд, J=10,4, 4,4 Гц, 1H), 4,32–4,38 (м, 1H), 4,15–4,20 (м, 1H), 4,05–4,10 (м, 2H), 3,85 (с, 3H), 3,04 (с, 3H), 2,19 (с, 3H), 1,34 (т, J=6,8 Гц, 3H). ЖХМС: [(M+1)]+=462,0.
Пример 16. Синтез соединения 901
Стадия 1. Синтез соединения 901–B
К раствору соединения 901–A (4–этокси–5–метоксипиколинальдегид, 8,4 г, 46,36 ммоль) в 150 мл MeOH добавляли гидроксиламин гидрохлорид (3,87 г, 55,63 ммоль) и NaOAc (4,56 г, 55,63 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 3 часов. Охлаждали до 25°C и концентрировали досуха, затем разбавляли 30 мл холодной воды и экстрагировали при помощи EtOAc. Объединенный EtOAc раствор промывали водой и насыщенным солевым раствором. Сушили над Na2SO4, концентрировали досуха с получением продукта 901–B (4–этокси–5–метоксипиколинальдегидоксим, 8,0 г, выход: 88%).
1H ЯМР (400 МГц, DMSO–d6): δ 11,40 (с, 1H), 8,15 (с, 1H), 7,97 (с, 1H), 7,30 (с, 1H), 4,14 (кв., J=7,2 Гц, 2H), 3,88 (с, 1H), 1,36 (т, J=7,2 Гц, 3H)
Стадия 2. Синтез соединения 901–C
Раствор соединения 901–B (8,0 г, 40,8 ммоль) в 60 мл Ac2O перемешивали в герметично закрытой пробирке в микроволновом реакторе при 170°C в течение 30 минут, затем охлаждали до 25°C и концентрировали досуха и осаждали из PE/EtOAc (1:1) с получением желаемого продукта 901–C (4–этокси–5–метоксипиколинoнитрил, 3,6 г, выход: 40%).
1H ЯМР (300 МГц, DMSO–d6): δ 8,33 (с, 1H), 7,70 (с, 1H), 4,18 (кв., J=6,9 Гц, 2H), 3,93 (с, 1H), 1,35 (т, J=6,9 Гц, 3H).
Стадия 3. Синтез соединения 901–D
Раствор диметилсульфона (4,7 г, 50 ммоль) в 200 мл безводного THF охлаждали до 0°C в атмосфере N2 и к раствору медленно добавляли по каплям n–BuLi (20 мл, 50 ммоль). Затем смесь перемешивали в ледяной бане в течение 2 часов. К этому раствору добавляли по каплям раствор соединения 901–C (3,56 г, 20 ммоль) в 50 мл безводного THF на ледяной бане. Смесь перемешивали на ледяной бане в течение 2 часов, затем гасили ледяной водой. THF выпаривали и затем смесь экстрагировали при помощи EtOAc (500 мл ×3), объединенный EtOAc раствор промывали водой и насыщенным солевым раствором, сушили над Na2SO4, концентрировали досуха с получением неочищенного продукта 901–D (1–(4–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этенамин, 5,5 г), который использовали непосредственно на следующей стадии без очистки.
Стадия 4. Синтез соединения 901–E
К раствору соединения 901–D (5,0 г, 18,4 ммоль) в 140 мл THF, охлаждаемому ледяной водой, добавляли 2N раствор HCl (60 мл). Смесь перемешивали при 25°C в течение ночи. THF выпаривали и остаток подщелачивали насыщенным раствором NaHCO3. Смесь экстрагировали при помощи EtOAc (200 мл ×3). Объединенный EtOAc раствор промывали насыщенным солевым раствором, сушили над Na2SO4. Концентрировали досуха с получением продукта 901–E (1–(4–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этенон, 6,5 г).
1H ЯМР (400 МГц, DMSO–d6): δ 8,38 (с, 1H), 7,60 (с, 1H), 5,15 (с, 2H), 4,22 (кв., J=6,8 Гц, 2H), 3,99 (с, 3H), 3,17 (с, 3H), 1,37 (т, J=6,8 Гц, 3H).
Стадия 5. Синтез соединения 901–F
К раствору соединения 901–E (6,5 г, неочищенное, 23,8 ммоль) в 100 мл MeOH, охлажденного ледяной водой, добавляли по порциям NaBH4 (1,8 г, 47,6 ммоль). Смесь перемешивали при 25°C в течение 2 часов, затем гасили 2 N раствором HCl (30 мл) и перемешивали в течение 30 минут. Смесь концентрировали досуха и подщелачивали насыщенным раствором NaHCO3 и экстрагировали смесью DCM/MeOH (20:1, 400 мл ×2). Объединенную органическую фазу промывали насыщенным солевым раствором. Сушили над Na2SO4, концентрировали досуха с получением неочищенного продукта 901–F (1–(4–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанол, 5,5 г).
Стадия 6. Синтез соединения 901–G
К раствору соединения 901–F (5,5 г, 20 ммоль) в 100 мл DCM, охлажденному ледяной водой, добавляли Et3N (5,6 мл, 40 ммоль), затем по каплям добавляли MsCl (2,3 мл, 30 ммоль). Смесь перемешивали при 25°C в течение ночи, затем гасили ледяной водой и перемешивали в течение 30 минут. Экстрагировали при помощи DCM (100 мл ×2), объединенную органическую фазу промывали водой и насыщенным солевым раствором. Сушили над Na2SO4. Концентрировали досуха и кристаллизовали из PE/EtOAc (1:1) с получением продукта 901–G (4–этокси–5–метокси–2–(2–(метилсульфонил)винил)пиридин, 4,0 г, выход: 34% в течение 4 стадии).
1H ЯМР (300 МГц, DMSO–d6): δ 8,26 (с, 1H), 7,39–7,59 (м, 3H), 4,12–4,18 (м, 2H), 3,90 (с, 3H), 3,12 (с, 3H), 1,36 (т, J=6,9 Гц, 3H).
Стадия 7. Синтез соединения 901–H
Раствор B(OH)3 (93 мг, 1,5 ммоль) в 5 мл воды нагревали до 50°C в течение 15 минут. Добавляли соединение 901–G (515 мг, 2,0 ммоль) и смесь перемешивали в течение 30 минут. Затем добавляли 30 мл NH3.H2O. Смесь перемешивали при 80°C в герметично закрытой пробирке в течение 3 дней. Охлаждали до 25°C и концентрировали досуха, подщелачивали насыщенным раствором NaHCO3 и перемешивали при 25°C в течение 30 минут. Смесь экстрагировали при помощи DCM/MeOH (50 мл ×3), объединенную органическую фазу промывали насыщенным солевым раствором, сушили над Na2SO4, концентрировали досуха с получением неочищенного продукта 901–H (1–(4–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин, 470 мг, выход: 86%).
1H ЯМР (400 МГц, DMSO–d6): δ 8,07 (с, 1H), 7,17 (с, 1H), 4,29–4,32 (м, 1H), 4,13 (кв., J=7,2 Гц, 2H), 3,82 (с, 3H), 3,37–3,46 (м, 2H), 3,05 (с, 3H), 1,36 (т, J=7,2 Гц, 3H).
Стадия 8. Синтез соединения 901
К раствору соединения 901–H (220 мг, 0,8 ммоль) в 20 мл HOAc добавляли N– (1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамид (164 мг, 0,8 ммоль), затем смесь кипятили с обратным холодильником в течение ночи. Затем смесь охлаждали до 25°C и концентрировали досуха и очищали препаративной ВЭЖХ с получением продукта 901 (N–(2–(1–(4–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид, 100 мг, выход: 27%).
1H ЯМР(400 МГц, DMSO–d6): δ 9,71 (с, 1H), 8,48 (д, J=8,4 Гц, 1H), 8,08 (с, 1H), 7,79–7,83 (м, 1H), 7,59 (д, J=6,8 Гц, 1H), 7,10 (с, 1H), 5,86 (дд, J=10,8, 3,6 Гц, 1H), 5,86 (дд, J=3,6, 7,2Гц, 1H), 4,40 (дд, J=14,8, 3,6 Гц, 1H), 4,16–4,19 (м, 1H), 4,10 (кв., J=7,2 Гц, 2H), 3,82 (с, 3H), 3,09 (с, 3H), 2,19 (с, 3H), 1,32 (т, J=7,2 Гц, 3H). ЖХМС: [(M+1)]+=461,9.
Синтез исходного вещества 901–A

Синтез соединения 901–A2
Раствор соединения 901–A1 (5–гидрокси–2– (гидроксиметил)– 4H–пиран–4–он, CAS № 501–30–4, 60 г, 422 ммоль) в 300 мл H2O и KOH (30 г) охлаждали до 10°C. По каплям добавляли диметиловый эфир серной кислоты (53,2 г, 422 ммоль) при 10°C. Смесь перемешивали при 10°C в течение 1 часа, затем фильтровали, собирали твердое вещество, промывали ацетоном (40 мл), сушили с получением продукта 901–A2 (2–(гидроксиметил)–5–метокси–4H–пиран–4–он, 17,5 г, выход: 26,6%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, DMSO) δ 8,08 (с, 1H), 6,30 (с, 1H), 5,79 (ушир. с, 1H), 4,30 (с, 2H), 3,65 (с, 3H).
Синтез соединения 901–A3
В плотно закрываемую пробирку добавляли соединение 901–A2 (2–(гидроксиметил)–5–метокси–4H–пиран–4–он, 32 г, 205 ммоль) и NH4OH (160 мл). Пробирку герметично закрывали и нагревали до 90°C в течение 3 часов. Смесь концентрировали, затем добавляли MeOH (200 мл) и активированный уголь (15 г). Смесь нагревали до температуры кипения с обратным холодильником в течение 0,5 часа. Смесь фильтровали через слой целита и концентрировали досуха с получением продукта 901–A3 (2–(гидроксиметил)–5–метоксипиридин–4(1H)–он, 27,8 г, выход: 87%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, DMSO) δ 7,38 (с, 1H), 6,22 (с, 1H), 4,36 (с, 2H), 3,67 (с, 3H).
Синтез соединения 901–A4
К раствору соединения 901–A3 [2–(гидроксиметил)–5–метоксипиридин–4(1H)–он, 27,8 г, 179 ммоль) в DMF (280 мл) добавляли Cs2CO3 (64,2 г, 197 ммоль). Смесь нагревали до 85°C и затем добавляли йодэтан (29,3 г, 188 ммоль). Смесь перемешивали при 85°C в течение 2 часов. Смесь фильтровали. Фильтрат разбавляли водой (300 мл), экстрагировали смесью DCM:MeOH=10:1 (300 мл ×4), органическую фазу сушили и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле, элюируя при помощи EtOAc, с получением продукта 901–A4 ((4–этокси–5–метоксипиридин–2–ил)метанол, 17,5 г, выход: 53%) в виде желтого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6) δ 8,04 (с, 1H), 7,04 (с, 1H), 5,29 (т, J=6,0 Гц, 1H), 4,44 (д, J=5,6 Гц, 2H), 4,10 (кв., J=7,2 Гц, 2H), 3,81 (с, 3H), 1,36 (т, J=7,2 Гц, 3H).
Синтез соединения 901–A
К раствору соединения 901–A4 ((4–этокси–5–метоксипиридин–2–ил)метанол, 10 г, 54,6 ммоль) в CHCl3 (400 мл) добавляли MnO2 (47 г, 546 ммоль). Смесь нагревали до 65°C и осуществляли взаимодействие в течение 2 часов. Смесь фильтровали, концентрировали с получением продукта 901–A (4–этокси–5–метоксипиколинальдегид, 9 г, выход: 91%) в виде серого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6) δ 9,82 (с, 1H), 8,40 (с, 1H), 7,45 (с, 1H), 4,23–4,16 (м, 2H), 3,97 (с, 3H), 1,36 (т, J=6,9 Гц, 3H).
Пример 17. Синтез соединения 702 и 703
Синтез промежуточных соединений 701–F1 и 701–F2

Соединение 701–F (6,6 г, 24 ммоль) подвергали хиральному разделению с получением продукта 701–F1 ((R)–1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин, 2,5 г, эи: 98,42%) и 701–F2 ((S)–1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин, 1,7 г, эи: 98,29%).
Метод разделения:
Колонка: chiralpak IA 5мкм 4,6 ×250 мм
Подвижная фаза: Hex:EtOH:DEA=70:30:0,2
Скорость потока (F): 1,0 мл/мин
Длина волны (W): 230 нм
Температура (T): 30°C
Синтез соединения 702
Соединение 702 синтезировали в соответствии со способом соединения 701 в примере 14, за исключением того, что использовали промежуточное соединение 701–F2 вместо соединения 701–F.

(S)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,73 (с, 1H), 8,47 (д, J=8,4 Гц, 1H), 7,82 (т, J=8,0 Гц, 1H), 7,60 (д, J=7,2 Гц, 1H), 7,27 (д, J=8,0 Гц, 1H), 7,02 (д, J=8,0 Гц, 1H), 5,81 (дд, J=10,8, 3,2 Гц, 1H), 4,15–4,35 (м, 4H), 3,76 (с, 3H), 3,10 (с, 3H), 2,18 (с, 3H), 1,17 (т, J=7,2 Гц, 3H). ЖХ–МС: 462,2 ([M+1]+).
Синтез соединения 703
Соединение 703 синтезировали в соответствии со способом соединения 701 в примере 14, за исключением того, что использовали промежуточное соединение 701–F1 вместо соединения 701–F.

(R)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,73 (с, 1H), 8,47 (д, J=8,4 Гц, 1H), 7,82 (т, J=8,0 Гц, 1H), 7,60 (д, J=7,6 Гц, 1H), 7,27 (д, J=8,4 Гц, 1H), 7,02 (д, J=7,6 Гц, 1H), 5,81 (дд, J=10,8, 3,6 Гц, 1H), 4,15–4,35 (м, 4H), 3,76 (с, 3H), 3,10 (с, 3H), 2,18 (с, 3H), 1,17 (т, J=7,2 Гц, 3H). ЖХ–МС: 462,1 ([M+1]+).
Пример 18. Синтез соединений 704, 705 и 706
Соединения 705 и 706 синтезировали в соответствии со способом соединения 701 в примере 14, за исключением того, что использовали промежуточное соединение 701–F2 ((S)–1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) или 701–F1 ((R)–1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин), соответственно, вместо соединения 701–F и использовали промежуточное соединение 101–E (N–(7–фтор–1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамид) вместо N– (1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамида.

(S)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,77 (с, 1H), 8,45 (дд, J=9,2, 3,6 Гц, 1H), 7,69 (т, J=9,2 Гц, 1H), 7,28 (д, J=8,4 Гц, 1H), 7,05 (д, J=8,4 Гц, 1H), 5,78–5,81 (м, 1H), 4,12–4,36 (м, 4H), 3,77 (с, 3H), 3,10 (с, 3H), 2,16 (с, 3H), 1,20 (т, J=6,8 Гц, 3H). ЖХМС: [(M+1)]+=480,2
(R)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,78 (с, 1H), 8,44–8,46 (м, 1H), 7,69 (т, J=8,8 Гц, 1H), 7,28 (д, J=8,0 Гц, 1H), 7,06 (д, J=8,0 Гц, 1H), 5,80 (д, J=8,0 Гц, 1H), 4,12–4,35 (м, 4H), 3,77 (с, 3H), 3,10 (с, 3H), 2,17 (с, 3H), 1,20 (т, J=6,8 Гц, 3H). ЖХМС: [(M+1)]+=480,0
Соединение 704 можно синтезировать в соответствии со способом синтеза соединения 701 в примере 14, за исключением того, что используют промежуточное соединение 101–E вместо N– (1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамида.

N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
Пример 19. Синтез соединений 707, 708 и 709
Соединение 708 и 709 синтезировали в соответствии со способом синтеза соединения 701 в примере 14, за исключением того, что использовали промежуточное соединение 701–F2 ((S)–1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) или 701–F1 ((R)–1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин), соответственно, вместо соединения 701–F и использовали промежуточное соединение 201–C (N– (6–фтор–1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамид) вместо N–(1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамида.
(S)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–6–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,81 (с, 1H), 8,29 (дд, J=12,0, 1,6 Гц, 1H), 7,54 (дд, J=6,8, 1,6 Гц, 1H), 7,28 (д, J=8,4 Гц, 1H), 7,04 (д, J=8,0 Гц, 1H), 5,80 (дд, J=10,8, 3,2 Гц, 1H), 4,13–4,36 (м, 4H), 3,76 (с, 3H), 3,10 (с, 3H), 2,21 (с, 3H), 1,18 (т, J=7,2 Гц, 3H). ЖХ–МС: 479,9 ([M+1]+).
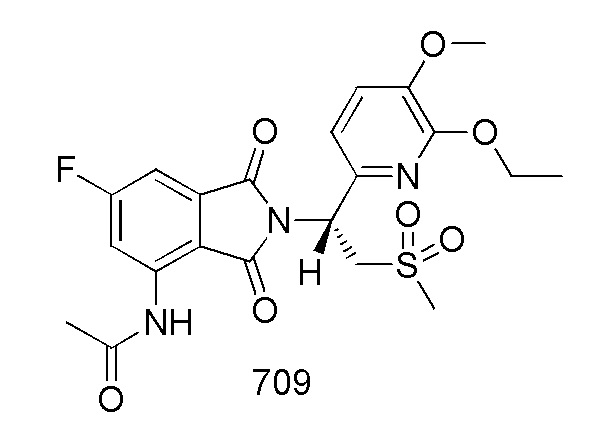
(R)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–6–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,80 (с, 1H), 8,29 (дд, J=12,0, 2,0 Гц, 1H), 7,54 (дд, J=6,4, 2,0 Гц, 1H), 7,28 (д, J=8,0 Гц, 1H), 7,04 (д, J=8,0 Гц, 1H), 5,81 (дд, J=10,4, 3,2 Гц, 1H), 4,16–4,32 (м, 4H), 3,76 (с, 3H), 3,10 (с, 3H), 2,21 (с, 3H), 1,19 (т, J=7,2 Гц, 3H). ЖХ–МС: 480,2 ([M+1]+).
Соединение 707 можно синтезировать в соответствии со способом синтеза соединения 701 в примере 14, за исключением того, что используют промежуточное соединение 201–C (N– (6–фтор–1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамид) вместо N–(1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамида.

N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–6–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
Пример 20. Синтез соединения 712

К раствору N–(1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамида (117 мг, 0,568 ммоль) в AcOH (5мл) добавляли соединение 712–A1 (150 мг, 0,54 ммоль), смесь перемешивали при 80°C в течение ночи. Реакционный раствор концентрировали досуха на роторном испарителе и очищали препаративной ВЭЖХ и лиофилизировали с получением соединения 712 ((R)–N–(2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид, 185 мг).
1H ЯМР (400 МГц, DMSO–d6) δ 9,72 (с, 1H), 8,478 (д, J=8,4 Гц, 1H), 7,82 (т, J=8,0 Гц,1H), 7,60 (д, J=7,2 Гц, 1H), 7,27 (д, J=8,0 Гц, 1H), 7,01 (д, J=8,4 Гц, 1H), 5,81 (дд, J=10,8, 3,2 Гц, 1H), 4,35–4,30 (м, 1H), 4,22–4,15 (м, 3H), 3,09 (с, 3H), 2,18 (с, 3H), 1,17 (т, J=7,2 Гц, 3H). ЖХМС: ([M+H]+). 465,0. эи%=100%.
Синтез промежуточного соединения 712–A

Стадия 1. Синтез соединения B
К раствору соединения A (2–хлор–6–йодпиридин–3–ол, CAS № 185220–68–2, 5 г, 19,6 ммоль) в 14 мл DMF добавляли K2CO3 (4,06 г, 29,4 ммоль). Смесь перемешивали в течение 30 минут. Добавляли CD3I (3,41 г, 23,5 ммоль). Смесь перемешивали при 100°C в течение 2 часов. Смесь охлаждали и выливали в 100 мл воды, перемешивали в течение 0,5 часа, фильтровали, промывали водой (200 мл ×2), собирали твердое вещество, сушили в вакууме с получением соединения B (2–хлор–6–йод–3–d3–метоксипиридин, 4,87 г, выход: 91%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6) δ 7,81 (д, J=8,4 Гц, 1H), 7,36 (д, J=8,4 Гц, 1H).
Стадия 2. Синтез соединения C
NaOEt (8,53 г, 125,3 ммоль) медленно добавляли по порциям к безводному EtOH (100 мл) при 10°C. Внутреннюю температуру повышали до 35°C, когда добавление было завершено. Затем добавляли соединение B (2–хлор–6–йод–3–d3–метоксипиридин, 4,87 г, 17,9 ммоль) и смесь нагревали до 100°C и перемешивали в течение 2 часов. Смесь охлаждали и растворитель удаляли. Остаток разбавляли EtOAc (100 мл) и ледяной водой (100 мл), затем перемешивали и разделяли. Органическую фазу промывали водой (100 мл) и насыщенным солевым раствором (100 мл), затем сушили над Na2SO4, фильтровали и концентрировали досуха с получением соединения C (2–этокси–6–йод–3–d3–метоксипиридин, 4,86 г, выход: 96%) в виде коричневого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6) δ 7,30 (дд, J=8,1, 1,2 Гц, 1H), 7,03 (дд, J=8,1, 1,2 Гц, 1H), 4,28–4,21 (м, 2H), 1,32–1,27 (м, 3H).
Стадия 3. Синтез соединения D
К раствору соединения C (2–этокси–6–йод–3–d3–метоксипиридин, 4,86 г, 17,2 ммоль) в DMF (50 мл, N2 барботировали в течение 0,5 часа) добавляли DIEA (3,33 г, 25,8 ммоль), метансульфонилэтен (2,19 г, 20,6 ммоль), S–Phos (2–дициклогексилфосфин–2',6'–диметоксибифенил, 1,09 г, 2,58 ммоль) и Pd2(dba)3 (1,06 г, 1,5 ммоль). Смесь продували N2 4 раза и перемешивали при 110°C в течение 4 часов под защитой N2. Затем смесь охлаждали, фильтровали и фильтрат концентрировали на роторном испарителе с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (PE:EtOAc=4:1–1:1), с получением соединения D (2–этокси–3–d3–метокси–6– (2– (метилсульфонил)винил)пиридин, 3,6 г, 80%) в виде не совсем белого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6): δ 7,43–7,31 (м, 4H), 4,43–4,36 (м, 2H), 3,11 (с, 3H), 1,34 (т, J=7,2 Гц, 3H).
Стадия 4. Синтез соединения 712–A
B(OH)3 (2,49 г, 40,3 ммоль), диоксан (40 мл), соединение D (2–этокси–3–d3–метокси–6– (2– (метилсульфонил)винил)пиридин, 7,0 г, 26,9 ммоль) и NH3.H2O (200 мл) добавляли в герметично закрываемую пробирку и смесь перемешивали при 100°C в течение 60 часов. Затем герметично закрытую пробирку охлаждали, выкачивали воздух и открывали. Реакционный раствор концентрировали до 50 мл, затем осажденное твердое вещество фильтровали и промывали при помощи H2O. Фильтрат концентрировали досуха на роторном испарителе с получением неочищенного вещества, которое суспендировали в PE/EtOAc (3:1, 40 мл), с получением 712–A (1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин, 5,3 г, выход: 71%) в виде белого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6): δ 7,23 (д, J=7,8 Гц, 1H), 6,97 (д, J=8,1 Гц, 1H), 4,33 (кв., J=6,9 Гц, 2H), 4,23–4,19 (м, 1H), 3,76 (с, 3H), 3,38–3,36 (м, 2H), 3,02 (с, 3H), 2,21 (ушир. с, 2H), 1,31 (т, J=6,9 Гц, 3H).
Стадия 5. Хиральное разделение
712–A (1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) очищали хиральной препаративной ВЭЖХ с получением соединения 712–A1 ((R)–1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) и соединения 712–A2 ((S)–1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин).
Условия хирального разделения: Колонка: CHIRALPAK IA, размер частиц: 5мкм; длина волны: 230 нм; подвижная фаза: гексан/EtOH/(0,2%TEA)=70/30[об/об(0,2%TEA)]; температура: 30oC.
Пример 21. Синтез соединений 710 и 711
Соединение 711 синтезировали в соответствии со способом синтеза соединения 712 в примере 20, за исключением того, что использовали соответствующий субстрат 712–A2 вместо соединения 712–A1.

(S)–N–(2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,72 (с, 1H), 8,47 (д, J=8,4 Гц, 1H), 7,82 (т, J=8,4 Гц,1H), 7,60 (д, J=7,2 Гц, 1H), 7,27 (д, J=8,0 Гц, 1H), 7,02 (д, J=8,4 Гц, 1H), 5,81 (дд, J=10,4, 3,6 Гц, 1H), 4,32 (дд, J=14,4, 3,6 Гц, 1H), 4,22–4,15 (м, 3H), 3,09 (с, 3H), 2,18 (с, 3H), 1,17 (т, J=7,2 Гц, 3H). ЖХМС: ([M+H]+).=465,0. эи%=98,3%
Соединение 710 можно синтезировать в соответствии со способом синтеза соединения 712 в примере 20, за исключением того, что используют соответствующий субстрат 712–A вместо соединения 712–A1.

N–(2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид)
Пример 22. Синтез соединения 716, 718 и 719
Соединение 719 синтезировали в соответствии со способом синтеза соединения 712 в примере 20, за исключением того, что использовали соответствующий субстрат 101–E (N–(7–фтор–1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамид) вместо N–(1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамида.
Соединение 718 синтезировали в соответствии со способом синтеза соединения 712 в примере 20, за исключением того, что использовали соответствующий субстрат 101–E вместо N– (1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамида и использовали соответствующий субстрат 712–A2 ((S)–1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) вместо 712–A1 ((R)–1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин)).
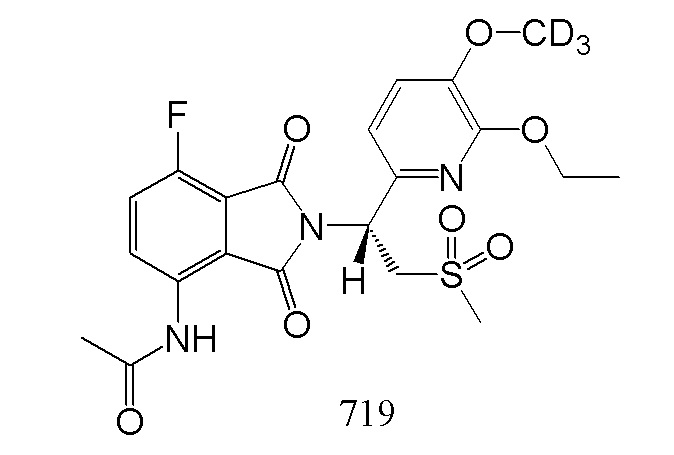
(R)–N–(2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид)
1H ЯМР (400 МГц, DMSO–d6) δ 9,76 (с, 1H), 8,45 (дд, J=9,2, 3,6Гц, 1H), 7,69 (т, J=9,2 Гц, 1H), 7,27 (д, J=7,6 Гц, 1H), 7,05 (д, J=8,0 Гц, 1H), 5,79 (дд, J=10,8, 3,6 Гц, 1H), 4,35–4,31 (м, 1H), 4,23–4,12 (м, 3H), 3,10 (с, 3H), 2,16 (с, 3H), 1,20 (т, J=7,2 Гц, 3H).
ЖХМС: ([M+H]+)=483,0. эи%=99,6%

(S)–N–(2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид)
1H ЯМР (400 МГц, DMSO–d6) δ 9,76 (с, 1H), 8,45 (дд, J=9,2, 3,6 Гц, 1H), 7,69 (т, J=9,2 Гц, 1H), 7,27 (д, J=8,0 Гц, 1H), 7,05 (д, J=8,0 Гц, 1H), 5,79 (дд, J=10,8, 4,0 Гц, 1H), 4,33 (дд, J=14,8, 4,0 Гц, 1H), 4,23–4,13 (м, 3H), 3,10 (с, 3H), 2,16 (с, 3H), 1,20 (т, J=6,8 Гц, 3H). ЖХМС: ([M+H]+)=482,9. эи%=98,5%
Соединение 716 можно синтезировать в соответствии со способом синтеза соединения 712 в примере 20, за исключением того, что используют соответствующий субстрат 101–E (N–(7–фтор–1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамид) вместо N– (1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамида и используют соответствующий субстрат 712–A (1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) вместо соединения 712–A1 ((R)–1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин)).

N–(2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид)
Пример 23. Синтез соединений 722, 724 и 725
Соединение 725 синтезировали в соответствии со способом синтеза соединения 712 в примере 20, за исключением того, что использовали соответствующий субстрат 201–C (N–(6–фтор–1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамид) вместо N–(1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамида.
Соединение 724 синтезировали в соответствии со способом синтеза соединения 712 в примере 20, за исключением того, что использовали соответствующий субстрат 201–C (N– (6–фтор–1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамид) вместо N– (1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамида и использовали соответствующий субстрат 712–A2 ((S)–1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) вместо соединения 712–A1 ((R)–1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин).
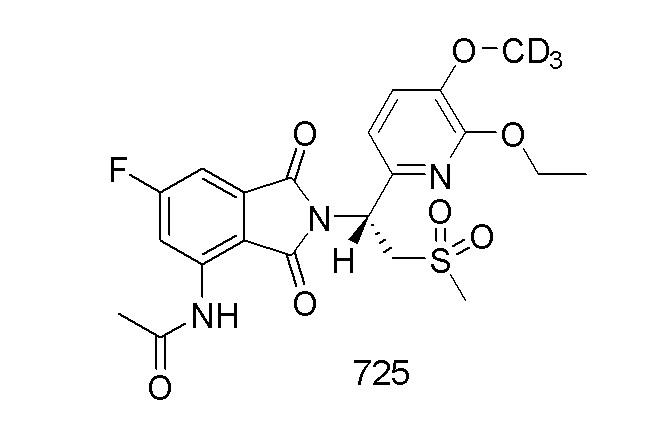
(R)–N–(2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–6–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,79 (с, 1H), 8,29 (дд, J=12,0, 2,0 Гц, 1H), 7,53 (дд, J=6,8, 2,4 Гц, 1H), 7,27 (д, J=8,0 Гц, 1H), 7,03 (д, J=8,4 Гц, 1H), 5,80 (дд, J=10,8, 3,6 Гц, 1H), 4,33 (дд, J=14,8, 4,0 Гц, 1H), 4,21–4,14 (м, 3H), 3,09 (с, 3H), 2,21 (с, 3H), 1,19 (т, J=7,2 Гц, 3H). ЖХМС: ([M+H]+)=483,0. эи%=100%

(S)–N–(2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–6–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,79 (с, 1H), 8,31–8,27 (м, 1H), 7,53 (дд, J=6,8, 2,4 Гц, 1H), 7,27 (д, J=8,4 Гц, 1H), 7,03 (д, J=8,0 Гц, 1H), 5,80 (дд, J=10,8, 3,6 Гц, 1H), 4,33 (дд, J=14,4, 3,6 Гц, 1H), 4,21–4,14 (м, 3H), 3,10 (с, 3H), 2,21 (с, 3H), 1,19 (т, J=7,2 Гц, 3H). ЖХМС: ([M+H]+)=483,2. эи%=98,4%
Соединение 722 можно синтезировать в соответствии со способом синтеза соединения 712 в примере 20, за исключением того, что используют соответствующий субстрат 201–C (N–(6–фтор–1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамид) вместо N–(1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамида и используют соответствующий субстрат 712–A (1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) вместо соединения 712–A1 ((R)–1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин).
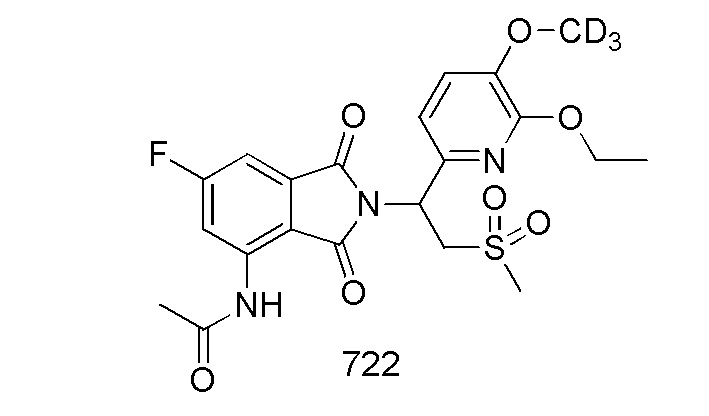
N–(2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–6–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
Пример 24. Синтез соединения 111
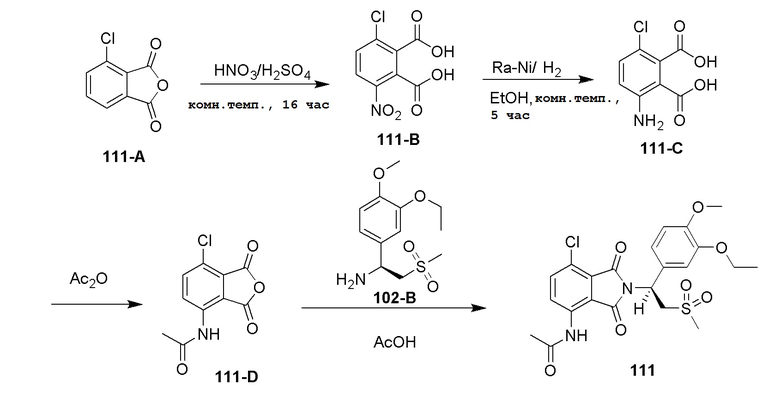
Стадия 1. Синтез соединения 111–B
К смеси HNO3 и H2SO4 (20 мл:50 мл) добавляли соединение 111–A (3–хлорфталевый ангидрид, CAS № 117–21–5, 25,0 г, 137 ммоль) небольшими порциями при 0°C. Смесь перемешивали при 25°C в течение 12 часов, затем охлаждали до 0°C с последующим добавлением колотого льда. Твердое вещество фильтровали и сушили с получением соединения 111–B (3–хлор–6–нитрофталевая кислота, 21,0 г, 63% выход) в виде белого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6):δ 14,34 (ушир. с, 2H), 8,17 (д, J=8,8 Гц, 1H), 7,93 (д, J=8,8 Гц, 1H).
Стадия 2. Синтез соединения 111–C
К раствору соединения 111–B (3–хлор–6–нитрофталевая кислота, 1,0 г, 4,08 ммоль) в EtOH (130 мл) добавляли Ni Ренея (1 г) под защитой N2. Смесь продували при помощи H2 и перемешивали под давлением баллона H2 в течение 5 часов при 25°C. Реакционную смесь фильтровали через слой целита под защитой N2 и лепешку промывали при помощи EtOH и фильтрат концентрировали с получением соединения 111–C (3–амино–6–хлорфталевая кислота, 0,75 г, 85%) в виде светло–зеленого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6): δ7,35–7,14 (м, 1H), 6,86–6,67 (м,1H).
Стадия 3. Синтез соединения 111–D
Смесь соединения 111–C (3–амино–6–хлорфталевая кислота, 0,75 г, 3,49 ммоль) в Ac2O (10 мл) перемешивали при 120°C в течение ночи. Реакционный раствор концентрировали на роторном испарителе, затем осажденное твердое вещество фильтровали и промывали петролейным эфиром с получением продукта 111–D (N–(7–хлор–1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамид) в виде бледно–желтого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6) δ 9,86 (с, 1H), 8,38 (д, J=8,7 Гц, 1H), 7,93 (д, J=9,0 Гц, 1H).
Стадия 4. Синтез соединения 111
Смесь соединения 111–D (N–(7–хлор–1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамид, 100 мг, 0,418 ммоль) и соединения 102–B ((S)–1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этанамин, 115 мг, 0,418 ммоль) в AcOH (2 мл) перемешивали при 80°C в течение 16 часов. Смесь охлаждали, затем твердое вещество осаждали. Твердое вещество фильтровали и промывали EtOAc, н–гексаном, сушили в вакууме с получением соединения 111 ((S)–N–(7–хлор–2–(1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид, 155 мг, 75% выход).
1H ЯМР (400 МГц, DMSO–d6) δ 9,77 (с, 1H), 8,46 (д, J=9,2 Гц, 1H), 7,80 (д, J=9,2 Гц, 1H), 7,07 (д, J=1,2 Гц, 1H), 7,01–6,93 (м, 2H), 5,78 (дд, J=10,0, 4,4 Гц, 1H), 4,35–4,29 (м, 1H), 4,20–4,16 (м, 1H), 4,03 (кв., J=6,8 Гц, 2H), 3,74 (с, 3H), 3,03 (с, 3H), 2,20 (с, 3H), 1,33 (т, J=6,8 Гц, 3H). ЖХМС: [(M–H)]– =493,1. эи%=100%.
Пример 25. Синтез соединений 112, 113 и 114
Соединение 112 можно получить в соответствии со способом синтеза соединения 111, за исключением того, что использовали соответствующий субстрат 1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этанамин вместо соединения 102–B ((S)–1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этанамин).
N–(7–хлор–2–(1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид
Синтез соединения 113
Соединение 113 можно получить в соответствии со способом синтеза соединения 111, за исключением того, что использовали соответствующий субстрат 103–E ((S)–1–(3–этокси–4–d3–метоксифенил)– 2– (метилсульфонил)этанамин) вместо соединения 102–B ((S)–1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этанамин).

(S)–N–(7–хлор–2–(1–(3–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,77 (с, 1H), 8,46 (д, J=9,2 Гц, 1H), 7,80 (д, J=9,2 Гц, 1H), 7,06 (с, 1H), 7,01–6,93 (м, 2H), 5,78 (дд, J=10,0 Гц, 4,4 Гц, 1H), 4,32–4,28 (м, 1H), 4,20–4,15 (м, 1H), 4,02 (кв., J=6,8 Гц, 2H), 3,02 (с, 3H), 2,20 (с, 3H), 1,32 (т, J=6,8 Гц, 3H). ЖХМС: [(M–H)]– =496,1. эи%=99,5%.
Соединение 114 можно получить в соответствии со способом синтеза соединения 111, за исключением того, что использовали соответствующий субстрат 103–D (1–(3–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этанамин) вместо соединения 102–B ((S)–1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этанамин).

(N–(7–хлор–2–(1–(3–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид)
Пример 26. Синтез соединений 728, 729 и 730
Соединение 728 синтезировали в соответствии со способом синтеза соединения 111 в примере 24, за исключением того, что использовали соответствующий субстрат 701–F (1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) вместо соединения 102–B ((S)–1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этанамин).

N–(7–хлор–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид)
1H ЯМР (400 МГц, DMSO–d6) δ 9,79 (с, 1H), 8,49 (д, J=9,2 Гц, 1H), 7,84 (д, J=8,8 Гц, 1H), 7,28 (д, J=8,0 Гц, 1H), 7,06 (д, J=8,0 Гц, 1H), 5,81 (дд, J=10,4, 3,2 Гц, 1H), 4,36–4,32 (м, 1H), 4,23–4,17 (м, 3H), 3,77 (с, 3H), 3,10 (с, 3H), 2,19 (с, 3H), 1,19 (т, J=6,8 Гц, 3H). ЖХМС: [(M–H)]– =496,2.
Соединение 729 и 730 синтезировали в соответствии со способом синтеза соединения 111 в примере 24, за исключением того, что использовали соответствующий субстрат 701–F2 ((S)–1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) или 701–F1 ((R)–1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) вместо соединения 102–B ((S)–1–(3–этокси–4–метоксифенил)–2–метилсульфонил)этанамин), соответственно.

(S)–N–(7–хлор–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)1,3–диоксоизоиндолин–4–ил)ацетамид

(R)–N–(7–хлор–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид
Пример 27. Синтез соединения 731

К смеси соединения 728 (N–(7–хлор–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид, 150 мг, 0,303 ммоль), Zn(CN)2(222 мг, 1,90 ммоль), Zn порошка (100 мг, 1,54 ммоль) и dppf (76 мг, 0,137 ммоль) в DMA (3 мл) добавляли Pd2(dba)3 (105 мг, 0,115 ммоль). Смесь продували при помощи N2 и перемешивали при 140°C в течение 2 часов при микроволновом облучении, затем охлаждали и фильтровали. Фильтрат концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (EtOAc:петролейный эфир=2/1) с получением неочищенного соединения, которое дополнительно очищали препаративной ВЭЖХ, с получением соединения 731 (N– (7–циано–2– (1– (6–этокси–5–метоксипиридин–2–ил)– 2– (метилсульфонил)этил)– 1,3–диоксоизоиндолин–4–ил)ацетамид, 62,0 мг, 42% выход).
1H ЯМР (400 МГц, DMSO–d6) δ 9,93 (с, 1H), 8,64 (д, J=8,8 Гц, 1H), 8,23 (д, J=8,8 Гц, 1H), 7,28 (д, J=8,0 Гц, 1H), 7,09 (д, J=8,0 Гц, 1H), 5,83 (дд, J=10,4, 3,6 Гц, 1H), 4,38–4,34 (м, 1H), 4,25–4,16 (м, 3H), 3,77 (с, 3H), 3,11 (с, 3H), 2,24 (с, 3H), 1,21 (т, J=7,2 Гц, 3H). ЖХМС: [(M+H)]+=487,2
Пример 28. Синтез соединения 732 и 733
Соединение 729 преобразовывали в соединение 732 в соответствии со способом, описанным для соединения 731 в примере 27.

(S)–N–(7–циано–2–(1–(6–этокси–5–метоксипиридин–2–ил)– 2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,93 (с, 1H), 8,65 (д, J=8,8 Гц, 1H), 8,23 (д, J=8,8 Гц, 1H), 7,28 (д, J=8,0 Гц, 1H), 7,09 (д, J=8,0 Гц, 1H), 5,83 (дд, J=10,8 Гц, 4,0 Гц, 1H), 4,35–4,34 (м, 1H), 4,25–4,17 (м, 3H), 3,77 (с, 3H), 3,11 (с, 3H), 2,24 (с, 3H), 1,21 (т, J=6,8 Гц, 3H). ЖХМС: [(M+H)]+=487,2. эи%: 100%.
Соединение 733 преобразовывали из соединения 730 в соответствии со способом, описанным для соединения 731 в примере 27.

(R)–N–(7–циано–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид
Пример 29. Синтез соединений 115 и 116
Соединение 111 преобразовывали в соединение 115 в соответствии со способом, описанным для соединения 731 в примере 27.

(S)–N–(7–циано–2–(1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,91 (с, 1H), 8,62 (дд, J=8,8, 3,2 Гц, 1H), 8,19 (д, J=8,4 Гц, 1H), 7,08 (д, J=1,6 Гц, 1H), 7,03 (дд, J=8,4, 1,6 Гц, 1H), 6,95 (д, J=8,8 Гц, 1H), 5,79 (дд, J=10,0, 4,8 Гц, 1H), 4,33–4,18 (м, 2H), 4,03 (кв., J=6,8 Гц, 2H), 3,74 (с, 3H), 3,02 (с, 3H), 2,25 (с, 3H), 1,33 (т, J=7,2 Гц, 3H). ЖХМС: [M+NH4+]=503,0. эи%: 100%.
Соединение 116 преобразовывали из соединения 112 в соответствии со способом, описанным для соединения 731 в примере 27.

N–(7–циано–2–(1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид
Пример 30. Синтез соединения 734

Стадия 1. Синтез соединения 734–B
К смеси соединения 734–A (5–бром–2–метил–3–нитробензойная кислота, CAS № 107650–20–4, 16,0 г, 0,062 моль) в H2O (240 мл) добавляли NaOH (4,92 г, 0,122 моль). Смесь нагревали до 80°C, затем добавляли по порциям KMnO4 (39 г, 0,123 моль) в течение 3 часов. Затем смесь перемешивали в течение 30 минут. Реакционный раствор фильтровали и лепешку промывали горячей водой (200 мл). Фильтрат доводили до pH=1 2N раствором HCl, экстрагировали при помощи EtOAc (300 мл ×3), объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме с получением соединения 734–B (5–бром–3–нитрофталевая кислота, 9,0 г, выход:51%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, DMSO) δ 14,0 (ушир. с, 1H), 8,52 (с, 1H), 8,33 (с, 1H).
Стадия 2. Синтез соединения 734–C
К раствору соединения 734–B (5–бром–3–нитрофталевая кислота, 5,0 г, 17,24 ммоль) в EtOH (150 мл) добавляли Ni Ренея (5 г) в атмосфере N2. Смесь продували при помощи H2 и перемешивали под давлением баллона H2 в течение 7 часов при 25°C. Реакционную смесь фильтровали через слой целита и лепешку промывали при помощи EtOH. Фильтрат концентрировали досуха с получением соединения 734–C (3–амино–5–бромфталевая кислота, 5,0 г, неочищенное) в виде светло–зеленого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6): δ7,02–7,01 (м, 1H), 6,79–6,77 (м,1H), 1,03 (ушир. с, 2H).
Стадия 3. Синтез соединения 734–D
Смесь соединения 734–C (3–амино–5–бромфталевая кислота, 5,0 г) в Ac2O (50 мл) перемешивали при 120°C в течение ночи. Реакционный раствор концентрировали на роторном испарителе и твердое вещество осаждали, затем фильтровали и промывали твердое вещество при помощи EtOAc с получением продукта 734–D (N–(6–бром–1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамид, 2,1 г, выход: 38%) в виде бледно–желтого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6) δ 9,90 (с, 1H), 8,63 (с, 1H), 8,00 (с, 1H), 2,98 (с, 3H), 2,22 (с, 3H).
Стадия 4. Синтез соединения 734–F
Смесь соединения 734–D (N–(6–фтор–1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамид, 500 мг, 1,767 ммоль) и соединения 701–F2 ((S)–1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин, 484 мг, 1,767 ммоль) в AcOH (5 мл) перемешивали при 80°C в течение 2 часов. Затем добавляли H2O (30 мл) и смесь экстрагировали при помощи EtOAc (30 мл ×2). Органические слои промывали насыщенным солевым раствором, сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (PE:EtOAc=1:1) с получением соединения 734–F ((S)–N–(6–бром–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид, 0,9 г, выход: 95%) в виде белого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6) δ 9,77 (с, 1H), 8,69 (д, J=1,2 Гц, 1H), 7,81 (д, J=1,2 Гц, 1H), 7,27 (д, J=8,4 Гц, 1H), 7,03 (д, J=8,0 Гц, 1H), 5,79 (м, 1H), 4,31–4,30 (м, 1H), 4,20–4,16 (м, 3H), 3,76 (с, 3H), 3,09 (с, 3H), 2,20 (с, 3H), 1,19 (т, J=7,2 Гц, 3H).
Стадия 5. Синтез соединения 734
К дегазированной смеси соединения 734–F ((S)–N–(6–бром–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид, 450 мг, 0,833 ммоль), Zn(CN)2 (292 мг, 2,50 ммоль) и dppf (185 мг, 0,333 ммоль) в DMA (8 мл) добавляли Pd2(dba)3 (305 мг, 0,333 ммоль). Смесь продували при помощи N2 и перемешивали при 110°C в течение 1 часа при микроволновом облучении. Реакционную смесь охлаждали, фильтровали и фильтрат концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле (EtOAc:петролейный эфир=1:1) с получением неочищенного соединения, которое дополнительно очищали препаративной ВЭЖХ с получением соединения 734 ((S)–N–(6–циано–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид, 220,0 мг, 54% выход) в виде желтого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6) δ 9,94 (д, J=0,8 Гц, 1H), 8,14 (д, J=1,2 Гц, 1H), 7,27 (д, J=8,0 Гц, 1H), 7,06 (д, J=8,0 Гц, 1H), 5,83 (дд, J=10,8, 3,6 Гц, 1H), 4,35 (дд, J=14,4, 3,6 Гц, 1H), 4,22–4,16 (м, 3H), 3,77 (с, 3H), 3,10 (с, 3H), 2,22 (с, 3H), 1,20 (т, J=7,2 Гц, 3H). ЖХМС (ESI) [M+H]+=486,9.
Соединения 735 и 736 можно синтезировать в соответствии со способом синтеза соединения 734, за исключением того, что используют соответствующий субстрат 701–F1 ((R)–1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) и 701–F (1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) вместо соединения 701–F2, соответственно.
(R)–N–(6–циано–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид
N–(6–циано–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид
Пример 31. Синтез соединения 737

Стадия 1. Синтез соединения 737–C
К смеси соединения 701–F (5 г, 18,2 ммоль) в DMF (300мл) добавляли соединение 737–B (3–нитрофталевая кислота, CAS № 603–11–2, 3,85 г, 18,2 ммоль), HATU (15,2 г, 40 ммоль) и DIEA (8,23 г, 63,7 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли H2O (150 мл) и перемешивали в течение 15 минут, затем экстрагировали при помощи EtOAc (400 мл) и органическую фазу промывали насыщенным солевым раствором (300 мл ×2), сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией, элюируя смесью PE:EA (2:1-1:1), с получением соединения 737–C (4,5 г, выход: 55%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, DMSO) δ 8,35 (д, J=8,4 Гц, 1H), 8,24 (д, J=7,8 Гц, 1H), 8,11 (т, J=8,1 Гц, 1H), 7,27 (д, J=7,5 Гц, 1H), 7,08 (д, J=7,5 Гц, 1H), 5,89–5,80 (м, 1H), 4,39–4,31 (м, 1H), 4,22–4,12 (м, 3H), 3,76 (с, 3H), 3,09 (с, 3H), 1,18 (т, J=7,2 Гц, 3H).
Стадия 2. Синтез соединения 737–D
К смеси соединения 737–C (4,5 г, 0,01 ммоль) в этилацетате (200 мл) добавляли 20% Pd/C (800 мг) и перемешивали при комнатной температуре в течение 7 часов в атмосфере H2 (50 фунтов/дюйм2 (3,515 кг/см2)). Смесь фильтровали и концентрировали с получением соединения 737–D (4,09 г, выход:97%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, DMSO) δ 7,45 (т, J=7,5 Гц, 1H), 7,26 (д, J=8,1 Гц, 1H), 7,04–6,90 (м, 3H), 6,49 (с, 2H), 5,79–5,71 (м, 1H), 4,25–4,16 (м, 4H), 3,76 (с, 3H), 3,08 (с, 3H), 1,20 (т, J=6,9 Гц, 3H).
Стадия 3. Синтез соединения 737–E
К раствору соединения 737–D (4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион, 500 мг, 1,19 ммоль) в THF (15 мл) добавляли Et3N (606 мг, 6 ммоль), MsCl (187 мг, 1,79 ммоль) и DMAP (290 мг, 2,38 ммоль). Смесь перемешивали при 75°C в течение 16 часов. Смесь концентрировали и экстрагировали при помощи EtOAc (30 мл), промывали 1N раствором HCl, насыщенным солевым раствором, сушили над Na2SO4, концентрировали с получением неочищенного вещества, затем очищали колоночной хроматографией на силикагеле (PE:EtOAc=1:1) и препаративной ВЭЖХ с получением 350 мг смеси 737–E (N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)–N–(метилсульфонил)метансульфонамид) и (N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)метансульфонамид) в виде желтого твердого вещества.
Стадия 4. Синтез соединения 737
К раствору 737–E (300 мг) в CH3CN добавляли раствор NaOH (2N, 0,6 мл), смесь перемешивали при 25°C в течение 1 часа. Затем смесь доводили до pH=8 раствором HCl (1N), экстрагировали при помощи EtOAc, сушили над Na2SO4 и фильтрат концентрировали с получением неочищенного вещества, которое очищали препаративной ВЭЖХ, с получением соединения 737 (N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)метансульфонамид, 103 мг, 20% за 2 стадии).
1H ЯМР (400 МГц, DMSO–d6) δ 9,33 (с, 1H), 7,86–7,78 (м, 2H), 7,63 (д, J=6,8 Гц, 1H), 7,27 (д, J=8,0 Гц, 1H), 7,02 (д, J=8,4 Гц, 1H), 5,80 (дд, J=10,0, 3,6 Гц, 1H), 4,34–4,29 (м, 1H), 4,22–4,16 (м, 3H), 3,76 (с, 3H), 3,28 (с, 3H), 3,09 (с, 3H), 1,18 (т, J=7,2 Гц, 3H), ЖХМС (ESI) [M+H]+=498,1.
Соединение 737–D2 синтезировали в соответствии со способом синтеза соединения 737–D, за исключением того, что использовали соединение 701–F2 ((S)–1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) вместо соединения 701–F.

(S)–4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион
1H ЯМР (300 МГц, DMSO–d6) δ 7,48–7,43 (м, 1H), 7,27–7,24 (м, 1H), 7,02–6,92 (м, 3H), 6,49 (с, 2H), 5,77–5,73 (м, 1H), 4,24–4,19 (м, 4H), 3,76 (с, 3H), 3,07 (с, 3H), 1,23–1,17 (м, 3H).
Соединение 737–D1 можно синтезировать в соответствии со способом синтеза соединения 737–D, за исключением того, что используют соединение 701–F1 ((R)–1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) вместо соединения 701–F.

(R)–4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион
Соединение 738 можно синтезировать в соответствии со способом синтеза соединения 737, за исключением того, что используют соединение 737–D2 вместо соединения 737–D.

(S)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)метансульфонамид)
Пример 32. Синтез соединения 742

Раствор соединения 737–D (4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион, 479 мг, 1,143 ммоль) в DCM (20 мл) охлаждали до 0°C, добавляли соединение 742–A (2– (бензилокси)ацетилхлорид, CAS № 19810–31–2, 1,26 г, 6,86 ммоль) и DIEA (1,28 г, 9,94 ммоль), затем смесь перемешивали при 0°C в течение 1 часа. Затем смесь концентрировали и очищали колоночной хроматографией на силикагеле (PE:EtOAc =3:1–2:1) с получением продукта (625 мг, выход: 96%) в виде желтого твердого вещества. 125 мг продукта очищали препаративной ВЭЖХ с получением соединения 742 (2–(бензилокси)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид, 53 мг).
1H ЯМР (400 МГц, DMSO–d6) δ 10,42 (с, 1H), 8,71 (д, J=8,4 Гц, 1H), 7,86 (т, J=8,0 Гц, 1H), 7,62 (д, J=7,2 Гц, 1H), 7,46 (д, J=6,4 Гц, 2H), 7,35–7,27 (м, 3H), 7,05 (д, J=8,0 Гц, 1H), 5,83 (дд, J=10,8, 3,6 Гц, 1H), 4,69 (с, 2H), 4,36–4,32 (м, 1H), 4,20–4,16 (м, 5H), 3,76 (с, 3H), 3,10 (с, 3H), 1,17 (т, J=7,2 Гц, 3H). ЖХМС: [M+H]+=568,0.
Соединения 743 и 744 можно синтезировать в соответствии со способом синтеза соединения 742, за исключением того, что используют соединения 737–D2 и 737–D1, соответственно, вместо соединения 737–D.

(S)–2–(бензилокси)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид

(R)–2–(бензилокси)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид
Пример 33. Синтез соединения 739

К смеси соединения 742 (2–(бензилокси)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид, 500 мг, 0,88 ммоль) в DMF (50 мл) добавляли Pd/C (20%, 50% масс., 50 мг) и перемешивали при 25°C в течение 12 часов в атмосфере H2 (50 фунтов/дюйм2 (3,515 кг/см2)). Смесь фильтровали и концентрировали и очищали препаративной ВЭЖХ с получением соединения 739 (N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)–2–гидроксиацетамид, 160 мг, выход: 32%).
1H ЯМР (400 МГц, DMSO–d6) δ 10,63 (с, 1H), 8,78 (д, J=8,4 Гц, 1H), 7,86 (т, J=8,4 Гц, 1H), 7,61 (д, J=7,2 Гц, 1H), 7,27 (д, J=8,4 Гц, 1H), 7,04 (д, J=8,0 Гц, 1H), 6,34 (т, J=5,6 Гц, 1H), 5,82 (дд, J=10,8, 3,6 Гц, 1H), 4,35–4,31 (м, 1H), 4,22–4,16 (м, 3H), 4,07 (д, J=5,6 Гц, 2H), 3,76 (с, 3H), 3,10 (с, 3H), 1,17 (т, J=7,2 Гц, 3H). ЖХМС: [M+H]+=478,2.
Соединения 740 и 741 можно получить из соединений 743 и 744, соответственно, в соответствии со способом синтеза соединения 739.

(S)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)–2–гидроксиацетамид

(R)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)–2–гидроксиацетамид
Пример 34. Синтез соединения 748
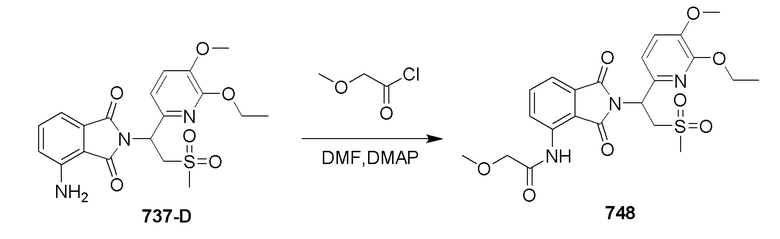
Раствор соединения 737–D (4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион, 100 мг, 0,238 ммоль) в DMF (10 мл) охлаждали до 0°C, добавляли 2–метоксиацетилхлорид (CAS № 38870–89–2, 148 мг, 1,367 ммоль) и DMAP (45,8 мг, 0,357 ммоль) и смесь перемешивали при 0°C в течение 2 часов. Затем реакционную смесь гасили при помощи HCl (1M, 20 мл) и экстрагировали при помощи EtOAc (20 мл). Органическую фазу концентрировали и очищали препаративной ВЭЖХ с получением соединения 748 (N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)–2–метоксиацетамид, 30 мг, выход: 26%).
1H ЯМР (400 МГц, DMSO–d6) δ 10,29 (с, 1H), 8,72 (д, J=8,4 Гц, 1H), 7,86 (т, J=8,4 Гц, 1H), 7,62 (д, J=6,8 Гц, 1H), 7,27 (д, J=8,0 Гц, 1H), 7,04 (д, J=8,0 Гц, 1H), 5,81 (дд, J=10,8, 3,6 Гц, 1H), 4,34–4,30 (м, 1H), 4,23–4,15 (м, 3H), 4,10 (с, 2H), 3,76 (с, 3H), 3,45 (с, 3H), 3,10 (с, 3H), 1,17 (т, J=7,2 Гц, 3H). ЖХМС: [M+H]+=492,2.
Соединения 749 и 750 можно синтезировать в соответствии со способом синтеза соединения 748, за исключением того, что используют соединения 737–D2 и 737–D1 вместо соединения 737–D, соответственно.

(S)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)–2–метоксиацетамид

(R)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)–2–метоксиацетамид
Пример 35. Синтез соединения 745

Соединение 745 (N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)–2–метоксиацетамид) синтезировали в соответствии со способом синтеза 748 в примере 34, за исключением того, что использовали соответствующее соединение 745–D вместо соединения 737–D (4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион).
1H ЯМР (400 МГц, DMSO–d6) δ 10,28 (с, 1H), 8,74 (дд, J=9,6, 4,0 Гц, 1H), 7,73 (т, J=9,2 Гц, 1H), 7,27 (д, J=8,0 Гц, 1H), 7,07 (д, J=8,4 Гц, 1H), 5,80 (дд, J=10,8,=3,6 Гц, 1H), 4,35–4,14 (м, 4H), 4,10 (с, 2H), 3,77 (с, 3H), 3,45 (с, 3H), 3,10 (с, 3H), 1,21 (т, J=7,2 Гц, 3H). ЖХМС: [M+H]+=510,2.
Синтез соединения 745–D
Соединение 745–D (4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фторизоиндолин–1,3–дион) синтезировали в соответствии со способом синтеза соединения 737–D в примере 31, за исключением того, что использовали исходное вещество 101–A (3–фтор–6–нитрофталевая кислота) вместо соединения 737–B (3–нитрофталевая кислота).
1H ЯМР (300 МГц, DMSO–d6) δ 7,35 (т, J =9,0 Гц, 1H), 7,26 (д, J=8,1 Гц, 1H), 7,07–6,95 (м, 2H), 6,46 (ушир. с, 2H), 5,75–5,70 (м, 1H), 4,28–4,16 (м, 4H), 3,75 (с, 3H), 3,08 (с, 3H), 2,68 (с, 3H), 1,35–1,17 (м, 3H).
Соединения 745–D2 и 745–D1 синтезировали в соответствии со способом синтеза соединения 737–D в примере 31, за исключением того, что использовали исходное вещество 101–A (3–фтор–6–нитрофталевая кислота) вместо соединения 737–B (3–нитрофталевая кислота), и использовали соединение 701–F2 ((S)–1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) или 701–F1 ((R)–1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) вместо соединения 701–F (1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин), соответственно.

(S)–4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фторизоиндолин–1,3–дион
1H ЯМР (300 МГц, DMSO–d6) δ 7,35–7,25 (м, 2H), 7,07–7,04 (м, 2H), 6,46–6,44 (м, 2H), 5,77–5,72 (м, 1H), 4,25–4,17 (м, 4H), 4,04–4,02 (м, 1H), 3,76 (с, 3H), 3,08 (с, 3H), 1,23–1,15 (м, 3H).

(R)–4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фторизоиндолин–1,3–дион
Синтез соединений 746 и 747
Соединения 746 и 747 можно синтезировать в соответствии со способом синтеза соединения 745, за исключением того, что используют соединение 745–D2 или 745–D1 вместо соединения 745–D.

(S)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)–2–метоксиацетамид

(R)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)–2–метоксиацетамид)
Пример 36. Синтез соединения 751

Стадия 1. Синтез соединения 751–B
2– (Диметиламино)уксусную кислоту (CAS № 1118–68–9, 1,50 г, 14,56 моль) в SOCl2 (15 мл) перемешивали при 75°C в течение 2 часов. Реакционную смесь концентрировали с получением соединения 751–B (2– (диметиламино)ацетилхлорид, 1,90 г, неочищенное вещество), которое использовали на следующей стадии без дополнительной очистки.
Стадия 2. Синтез соединения 751
К раствору соединения 745–D (4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фторизоиндолин–1,3–дион, 300 мг, 0,68 ммоль) в THF (10 мл) добавляли соединение 751–B (2– (диметиламино)ацетилхлорид, 432 мг, 2,74 ммоль). Смесь перемешивали при 75°C в течение 1 часа, затем охлаждали до 25°C. Добавляли водный раствор NaHCO3 (30 мл) и смесь экстрагировали при помощи EtOAc (50 мл ×2). Органические слои промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и фильтрат концентрировали с получением неочищенного продукта, который очищали препаративной ВЭЖХ с получением соединения 751 (2–(диметиламино)– N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид, 120 мг, выход: 34%).
1H ЯМР (300 МГц, DMSO–d6) δ 10,84 (с, 1H), 8,80–8,77 (м, 1H), 7,71 (т, J=8,7 Гц,1H), 7,28 (д, J=6,9 Гц, 1H), 7,08 (д, J=6,9 Гц, 1H), 5,83–5,79 (м, 1H), 4,31–4,20 (м, 4H), 3,77 (с, 3H), 3,18–3,11 (м, 5H), 2,32 (с, 6H), 1,20 (т, J=6,3 Гц, 3H). ЖХМС: [M+H]+=523,2.
Соединения 752 и 753 можно синтезировать в соответствии со способом синтеза соединения 751, за исключением того, что используют соединение 745–D2 или 745–D1, соответственно, вместо соединения 745–D.

(S)–2–(диметиламино)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид

(R)–2–(диметиламино)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид
Пример 37. Синтез соединения 754
Соединение 754 синтезировали в соответствии со способом синтеза соединения 751 в примере 36, за исключением того, что использовали соответствующий субстрат 737–D (4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион) вместо соединения 745–D.
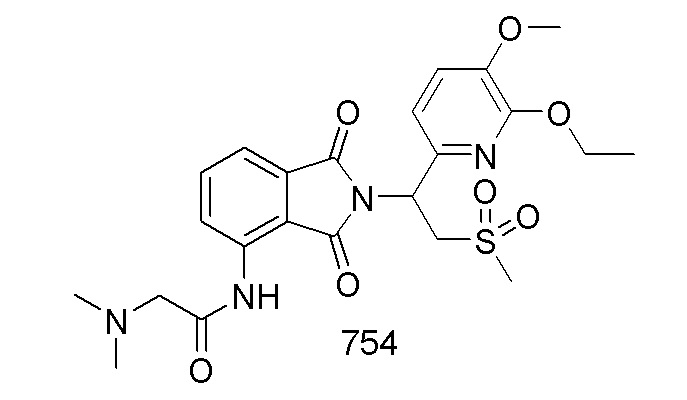
2–(диметиламино)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид)
1H ЯМР (300 МГц, DMSO–d6) δ 10,84 (с, 1H), 8,75 (д, J=8,4 Гц, 1H), 7,86–7,82 (м, 1H), 7,59 (д, J=6,4 Гц, 1H), 7,28 (д, J=8,4 Гц, 1H), 7,04 (д, J=8,4 Гц, 1H), 5,81–5,70 (м, 1H), 4,31–4,16 (м, 4H), 3,76 (с, 3H), 3,16 (с, 2H), 3,10 (с, 3H), 2,32 (с, 6H), 1,16 (т, J=7,2 Гц, 3H). ЖХМС: [M+H]+=505,2.
Соединения 755 и 756 можно синтезировать в соответствии со способом синтеза соединения 751 в примере 36, за исключением того, что используют соответствующий субстрат 737–D2 ((S)–4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион) или 737–D1 ((R)– 4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион), соответственно, вместо соединения 745–D.

(S)–2–(диметиламино)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид)

(R)–2–(диметиламино)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)ацетамид
Пример 38. Синтез соединения 757
Соединение 757 синтезировали в соответствии со способом синтеза соединения 751 в примере 36, за исключением того, что использовали изовалерилхлорид (CAS № 108–12–3) вместо соединения 751–B (2– (диметиламино)ацетилхлорид).

N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)–3–метилбутанамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,71 (с, 1H), 8,47 (дд, J=9,2, 3,2 Гц, 1H), 7,69 (т, J=9,2 Гц, 1H), 7,27 (д, J=8,0 Гц, 1H), 7,05 (д, J=8,0 Гц, 1H), 5,79 (дд, J=10,8, 3,6 Гц, 1H), 4,33 (дд,=14,4, 2,8 Гц,1H), 4,22–4,13 (м, 3H), 3,77 (с, 3H), 3,10 (с, 3H), 2,33 (д, J=6,8 Гц, 2H), 2,09–2,06 (м, 1H), 1,19 (т, J=7,2 Гц, 3H), 0,94 (д, J=6,8 Гц, 6H). ЖХМС: [M+H]+=522,2
Соединения 758 и 759 можно синтезировать в соответствии со способом синтеза соединения 751 в примере 36, за исключением того, что используют изовалерилхлорид вместо соединения 751–B (2– (диметиламино)ацетилхлорид) и используют 745–D2 ((S)–4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фторизоиндолин–1,3–дион)) или 745–D1 ((R)–4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фторизоиндолин–1,3–дион), соответственно, вместо соединения 745–D.

(S)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)–3–метилбутанамид

(R)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)–3–метилбутанамид
Пример 39. Синтез соединения 760
Соединение 760 синтезировали в соответствии со способом синтеза соединения 751 в примере 36, за исключением того, что использовали изовалерилхлорид вместо соединения 751–B (2–(диметиламино)ацетилхлорид) и использовали соединение 737–D (4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион) вместо соединения 745–D (4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фторизоиндолин–1,3–дион).

N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)–3–метилбутанамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,68 (с, 1H), 8,49 (д, J=8,4 Гц, 1H), 7,82 (т, J=8,0 Гц, 1H), 7,60 (д, J=7,2 Гц, 1H), 7,28 (д, J=8,0 Гц, 1H), 7,03 (д, J=8,4 Гц, 1H), 5,81 (дд, J=10,8, 3,6 Гц, 1H), 4,32 (дд, J=14,4, 3,6 Гц, 1H), 4,23–4,15 (м, 3H), 3,76 (с, 3H), 3,10 (с, 3H), 2,34 (д, J=6,8 Гц, 2H), 2,10–2,07 (м, 1H), 1,16 (т, J=7,2 Гц, 3H), 0,95 (д, J=6,8 Гц, 6H). ЖХМС: [M+H]+=504,0.
Соединения 761 и 762 можно синтезировать в соответствии со способом синтеза соединения 751 в примере 36, за исключением того, что используют изовалерилхлорид вместо соединения 751–B (2–(диметиламино)ацетилхлорид) и используют соединение 737–D2 ((S)–4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион) или 737–D1 ((R)–4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион), соответственно, вместо соединения 745–D.

(S)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)–3–метилбутанамид
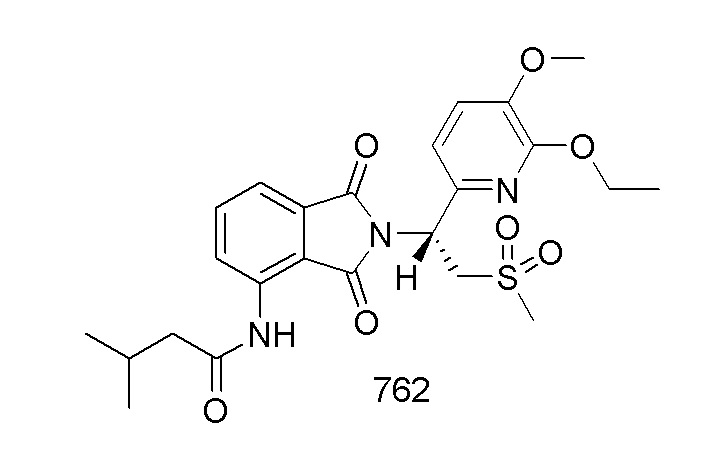
(R)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)–3–метилбутанамид
Пример 40. Синтез соединения 763

К раствору соединения 745–D (4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фторизоиндолин–1,3–дион, 260 мг, 0,59 ммоль) в THF (3 мл) добавляли циклопропанкарбонилхлорид (CAS № 4023–34–1, 124 мг, 1,19 ммоль) и перемешивали при 75°C в течение 1,5 часов. Смесь концентрировали и очищали препаративной ВЭЖХ с получением продукта 763 (119 мг, выход: 40%).
1H ЯМР (400 МГц, DMSO–d6) δ 10,03 (с, 1H), 8,43–8,40 (м, 1H), 7,68 (т, J=9,2 Гц,1H), 7,27 (д, J=8,4 Гц, 1H), 7,06 (д, J=8,4 Гц, 1H), 5,80 (дд, J=10,4, 3,6 Гц, 1H), 4,36–4,31 (м, 1H), 4,24–4,13 (м, 3H), 3,77 (с, 3H), 3,10 (с, 3H), 2,00–1,97 (м, 1H), 1,20 (т, J=7,2 Гц, 3H), 0,87 (д, J=5,2 Гц, 4H). ЖХМС: [M+H]+=506,2.
Соединение 764 синтезировали в соответствии со способом синтеза соединения 763 в примере 40, за исключением того, что использовали соединение 745–D2 ((S)–4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фторизоиндолин–1,3–дион) вместо соединения 745–D.

(S)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)циклопропанкарбоксамид)
1H ЯМР (400 МГц, DMSO–d6) δ 10,02 (с, 1H), 8,42 (дд, J=9,2, 4,0 Гц, 1H), 7,67 (т, J=9,2 Гц, 1H), 7,27 (д, J=8,0 Гц, 1H), 7,07 (д, J=7,6 Гц, 1H), 5,82–5,80 (м, 1H), 4,32–4,31 (м, 1H), 4,24–4,18 (м, 3H), 3,77 (с, 3H), 3,10 (с, 3H), 2,07–1,96 (м, 1H), 1,20 (т, J=7,2 Гц, 3H), 0,88–0,86 (м, 4H). ЖХМС: [M+H]+=506,2.
Соединение 765 синтезировали в соответствии со способом синтеза соединения 763 в примере 40, за исключением того, что использовали соединение 745–D1 ((R)–4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион) вместо соединения 745–D.

(R)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)циклопропанкарбоксамид
Пример 41. Синтез соединения 767
Синтез промежуточного соединения 767–D2

Соединение 767–D2 ((S)–4–амино–2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фторизоиндолин–1,3–дион)) синтезировали в соответствии со способом синтеза соединения 737–D в примере 31, за исключением того, что использовали соединение 712–A2 ((S)–1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) вместо соединения 701–F (1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) и использовали соединение 101–A (3–фтор–6–нитрофталевая кислота) вместо соединения 737–B (3–нитрофталевая кислота).
1H ЯМР (300 МГц, DMSO–d6) δ 7,38–7,33 (м, 1H), 7,26 (д, J=10,5 Гц, 1H), 7,09–7,03 (м, 1H), 6,98–6,95 (м, 1H), 6,46 (с, 2H), 5,77–5,71 (м, 1H), 4,31–4,16 (м, 4H), 3,08 (с, 3H), 1,23–1,15 (м, 3H).
Синтез соединений 767–D и 767–D1
Соединения 767–D и 767–D1 можно синтезировать в соответствии со способом синтеза соединения 737–D в примере 31, за исключением того, что используют соединение 101–A (3–фтор–6–нитрофталевая кислота) вместо соединения 737–B (3–нитрофталевая кислота) и используют соединение 712–A (1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) или 712–A1 ((R)–1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин), соответственно, вместо соединения 701–F (1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин).

4–амино–2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фторизоиндолин–1,3–дион

(R)–4–амино–2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фторизоиндолин–1,3–дион

Соединение 767 ((S)–N–(2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)циклопропанкарбоксамид) синтезировали в соответствии со способом синтеза соединения 763 в примере 40, за исключением того, что соединение использовали соединение 767–D2 вместо соединения 745–D.
1H ЯМР (400 МГц, DMSO–d6) δ 10,02 (с, 1H), 8,43–8,40 (м, 1H), 7,67 (т, J=9,2 Гц, 1H), 7,27 (д, J=8,0 Гц, 1H), 7,06 (д, J=8,0 Гц, 1H), 5,82–5,78 (м, 1H), 4,33 (дд, J=14,4, 3,6 Гц, 1H), 4,22–4,13 (м, 3H), 3,10 (с, 3H), 2,01–1,95 (м, 1H), 1,20 (т, J=7,2 Гц, 3H), 0,90–0,86 (м, 4H). ЖХМС: 509,2 ([M+H]+). эи%=98,9%
Соединение 766 можно синтезировать в соответствии со способом синтеза соединения 763 в примере 40, за исключением того, что используют соединение 767–D вместо соединения 745–D.

N–(2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)циклопропанкарбоксамид
Соединение 768 можно синтезировать в соответствии со способом синтеза соединения 763 в примере 40, за исключением того, что используют соединение 767–D1 вместо соединения 745–D.

(R)–N–(2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)циклопропанкарбоксамид
Пример 42. Синтез соединения 770
Соединение 770 синтезировали в соответствии со способом синтеза соединения 763 в примере 40, за исключением того, что использовали соединение 737–D2 ((S)–4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион) вместо соединения 745–D.

(S)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)циклопропанкарбоксамид
1H ЯМР (400 МГц, DMSO–d6) δ 9,98 (с, 1H), 8,43 (д, J=8,4 Гц, 1H), 7,80 (т, J=8,0 Гц, 1H), 7,59 (д, J=7,6 Гц, 1H), 7,27 (д, J=8,4 Гц, 1H), 7,03 (д, J=8,0 Гц, 1H), 5,82 (дд, J=10,8 Гц, 3,6 Гц, 1H), 4,31–4,30 (м, 1H), 4,23–4,16 (м, 3H), 3,76 (с, 3H), 3,10 (с, 3H), 2,00–1,97 (м, 1H), 1,17 (т, J=7,2 Гц, 3H), 0,88–0,87 (м, 4H). ЖХМС: 487,9 ([M+H]+).
Соединения 769 и 771 можно синтезировать в соответствии со способом синтеза соединения 763 в примере 40, за исключением того, что используют соединение 737–D (4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион) или 737–D1 ((R)–4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион), соответственно, вместо соединения 745–D.

N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)циклопропанкарбоксамид

(R)–N–(2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)циклопропанкарбоксамид
Пример 43. Синтез соединения 773
Синтез промежуточного соединения 773–D2

Соединение 773–D2 ((S)–4–амино–2–(1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион) синтезировали в соответствии со способом синтеза соединения 737–D в примере 31, за исключением того, что использовали соединение 712–A2 ((S)–1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) вместо соединения 701–F (1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин).
1H ЯМР (300 МГц, DMSO–d6) δ 7,48–7,43 (м, 1H), 7,25 (д, J=8,4 Гц, 1H), 7,01–6,91 (м, 3H), 6,49(с, 2H), 5,78–5,72 (м, 1H), 4,30–4,19 (м, 4H), 3,07 (с, 3H), 1,22–1,15 (м, 3H).
Синтез соединений 773–D и 773–D1
Соединения 773–D и 773–D1 можно синтезировать в соответствии со способом синтеза соединения 737–D в примере 31, за исключением того, что используют соединение 712–A (1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) или 712–A1 ((R)–1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин), соответственно, вместо соединения 701–F.

4–амино–2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион
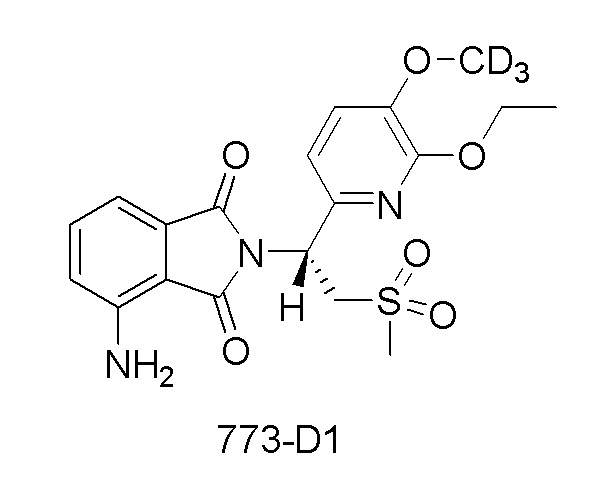
(R)–4–амино–2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион
Соединение 773 синтезировали в соответствии со способом синтеза соединения 763 в примере 40, за исключением того, что использовали соединение 773–D2 вместо соединения 745–D.

(S)–N–(2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)циклопропанкарбоксамид)
1H ЯМР (400 МГц, DMSO–d6) δ 9,99 (с, 1H), 8,43 (д, J=8,4 Гц, 1H), 7,80 (т, J=8,0 Гц,1H), 7,59 (д, J=6,8 Гц, 1H), 7,27 (д, J=7,6 Гц, 1H), 7,04–7,02 (м, 1H), 5,83–5,80 (м, 1H), 4,36–4,29 (м, 1H), 4,23–4,16 (м, 3H), 3,10 (с, 3H), 2,02–1,96 (м, 1H), 1,17 (т, J=7,2 Гц, 3H), 0,88 (д, J=6,0 Гц, 4H). ЖХМС: ([M+H]+)=491,2 эи%=99,0%
Соединения 772 и 774 можно синтезировать в соответствии со способом синтеза соединения 763 в примере 40, за исключением того, что используют соединение 773–D или 773–D1, соответственно, вместо соединения 745–D.

N–(2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)циклопропанкарбоксамид

(R)–N–(2–(1–(6–этокси–5–d3–метоксипиридин–2–ил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)циклопропанкарбоксамид
Пример 44. Синтез соединения 118

Соединение 118 ((S)–N–(2–(1–(3–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)циклопропанкарбоксамид) синтезировали в соответствии со способом синтеза соединения 763 в примере 40, за исключением того, что использовали соединение 118–D2 вместо соединения 745–D.
1H ЯМР (400 МГц, DMSO–d6) δ 10,01 (с, 1H), 8,38 (дд, J=9,2, 3,6 Гц, 1H), 7,64 (т, J=9,2 Гц, 1H), 7,07 (с, 1H), 7,01–6,93 (м, 2H), 5,77 (дд, J1=10,4, 4,4 Гц, 1H), 4,31 (дд, J1=14,4, 10,8 Гц, 1H), 4,18–4,14 (м, 1H), 4,03 (кв., J=6,8 Гц, 2H), 3,02 (с, 3H), 2,00–1,97 (м, 1H), 1,33 (т, J=6,8 Гц, 3H), 0,89–0,87 (м, 4H). ЖХМС: ([M+H]+)=507,9.
Синтез соединения 118–D2
Соединение 118–D2 ((S)–4–амино–2–(1–(3–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этил)–7–фторизоиндолин–1,3–дион) синтезировали в соответствии со способом синтеза соединения 737–D в примере 31, за исключением того, что использовали соединение 103–E ((S)–1–(3–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этанамин) вместо соединения 701–F (1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин) и использовали соединение 101–A (3–фтор–6–нитрофталевая кислота) вместо соединения 737–B (3–нитрофталевая кислота).
1H ЯМР (300 МГц, DMSO–d6) δ 7,33 (т, J=9,0 Гц, 1H), 7,06–7,01 (м, 2H), 6,95–6,93 (м, 2H), 6,46 (с, 2H), 5,73–5,69 (м, 1H), 4,37–4,29 (м, 1H), 4,12–3,98 (м, 3H), 3,01 (с, 3H), 1,36–1,30 (м, 3H).
Пример 45. Синтез соединения 120
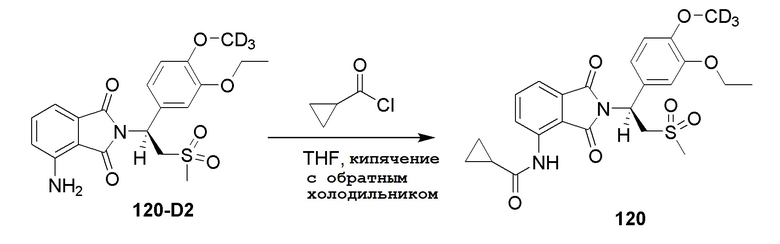
Соединение 120 ((S)–N–(2–(1–(3–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этил)–1,3–диоксоизоиндолин–4–ил)циклопропанкарбоксамид) синтезировали в соответствии со способом синтеза соединения 763 в примере 40, за исключением того, что использовали соединение 120–D2 вместо соединения 745–D.
1H ЯМР (400 МГц, DMSO–d6) δ 9,98 (с, 1H), 8,40 (д, J=8,4 Гц, 1H), 7,78 (т, J=8,0 Гц, 1H), 7,56 (д, J=7,2 Гц, 1H), 7,08 (д, J=2,0 Гц, 1H), 7,01–6,92 (м, 2H), 5,79 (дд, J=10,4, 4,4 Гц, 1H), 4,35 (дд, J=14,4, 10,8 Гц, 1H), 4,15 (дд, J=14,4, 4,4 Гц, 1H), 4,02 (кв., J=7,2 Гц, 2H), 3,02 (с, 3H), 2,00–1,94 (м, 1H), 1,32 (т, J=7,2 Гц, 3H), 0,90–0,88 (м, 4H). ЖХМС: ([M+H]+)=490,0.
Синтез соединения 120–D2
Соединение 120–D2 ((S)–4–амино–2–(1–(3–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этил)изоиндолин–1,3–дион) синтезировали в соответствии со способом синтеза соединения 737–D в примере 31, за исключением того, что использовали соединение 103–E ((S)–1–(3–этокси–4–d3–метоксифенил)–2–(метилсульфонил)этанамин) вместо соединения 701–F (1–(6–этокси–5–метоксипиридин–2–ил)–2–(метилсульфонил)этанамин).
1H ЯМР (300 МГц, DMSO–d6) δ 7,46–7,41 (м, 1H), 7,06 (с, 1H), 6,99–6,93 (м, 4H), 6,52–6,50 (м, 2H), 6,46 (с, 2H), 5,74–5,69 (м, 1H), 4,35–4,31 (м, 1H), 4,11–3,97 (м, 3H), 3,00 (с, 3H), 1,32 (т, J=6,9 Гц, 3H).
Пример 46. Синтез соединения 502

Стадия 1. Синтез соединения 502–B
Смесь соединения 502–A (5–аминопиридин–3,4–дикарбоновая кислота, 0,3 г, 1,32 ммоль) в Ac2O (8 мл) перемешивали при 120°C в течение ночи. Растворитель удаляли с получением неочищенного продукта 502–B (N–(1,3–диоксо–1,3–дигидрофуро[3,4–c]пиридин–7–ил)ацетамид, 0,35 г).
1H ЯМР (300 МГц, DMSO–d6) δ 10,47 (с, 1H), 9,45 (с, 1H), 9,03 (с, 1H), 2,22 (с, 3H).
Стадия 2. Синтез соединения 502
Смесь соединения 502–B (N–(1,3–диоксо–1,3–дигидрофуро[3,4–c]пиридин–7–ил)ацетамид, 0,3 г, неочищенное вещество) и соединения 102–B ((S)–1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этанамин, 0,30 г, 1,10 ммоль) в AcOH (6 мл) перемешивали при 70°C в течение 3 часов. Растворитель удаляли с получением неочищенного продукта, который очищали препаративной ВЭЖХ с получением соединения 502 ((S)–N–(2–(1–(3–этокси–4–метоксифенил)–2–(метилсульфонил)этил)–1,3–диоксо–2,3–дигидро–1H–пирроло[3,4–c]пиридин–7–ил)ацетамид, 144 мг, выход: 28%).
1H ЯМР (400 МГц, DMSO–d6) δ 9,98 (с, 1H), 9,52 (с, 1H), 8,83 (с, 1H), 7,08 (д, J=2,0 Гц, 1H), 7,01 (дд, J=8,4, 2,0 Гц, 1H), 6,94 (д, J=8,4 Гц, 1H), 5,79 (дд, J=10,4, 4,8 Гц, 1H), 4,30–4,17 (м, 2H), 4,02 (кв., J=6,8 Гц, 2H), 3,74 (с, 3H), 3,01 (с, 3H), 2,21 (с, 3H), 1,32 (т, J=6,8 Гц, 3H). ЖХМС: [M+H]+=461,9.
Синтез исходного вещества 502–A

Синтез соединения 502–A2
При –60°C, к раствору 2,2,6,6–тетраметилпиперидина (16,8 г, 118,8 ммоль) в 200 мл THF медленно по каплям добавляли n–BuLi (2,5 M, 44 мл, 108,9 ммоль) и перемешивали в течение 15 минут. Затем добавляли соединение 502–A1 (5–бромникотиновая кислота, 10 г, 49,5 ммоль) и перемешивали при –60°C в течение 0,5 часа. Затем реакционную смесь барботировали сухим CO2 при 25°C в течение 3 часов. Добавляли воду (150 мл) для гашения реакции. THF удаляли. Водную фазу доводили до pH=3 1N раствором HCl, концентрировали и полученное осажденное твердое вещество собирали фильтрованием и сушили с получением соединения 502–A2 (5–бромпиридин–3,4–дикарбоновая кислота, 12,5 г) в виде коричневого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6) δ 9,02–9,03 (м, 2H), 8,73 (ушир. с, 2H).
Синтез соединения 502–A3
К раствору соединения 502–A2 (9,3 г, 37,8 ммоль) в 100 мл DMF добавляли K2CO3 (26,1 г, 189 ммоль) и перемешивали при 25°C в течение 0,5 часа. Затем к смеси добавляли CH3I (5,9 мл, 94,5 ммоль) при 0°C. Смесь перемешивали при 25°C в течение 3 часов. Смесь выливали в воду (600 мл), экстрагировали при помощи EtOAc (200 мл ×2), промывали насыщенным солевым раствором (200 мл ×2), сушили и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле, элюируя смесью PE:EtOAc=6:1, с получением продукта 502–A3 (диметил-5–бромпиридин–3,4–дикарбоксилат, 1,59 г, 15%) в виде желтого твердого вещества.
1H ЯМР (300 МГц, DMSO–d6) δ 9,13 (с, 1H), 9,12 (с, 1H), 3,93 (с, 3H), 3,90 (с, 3H).
Синтез соединения 502–A4
К раствору соединения 502–A3 (1,59 г, 5,8 ммоль) в 70 мл толуола добавляли 2,4–диметоксибензиламин (1,46 г, 8,75 ммоль), Pd2(dba)3 (0,532 г, 0,58 ммоль), Xantphos (1,0 г, 1,74 ммоль), Cs2CO3 (3,8 г, 11,6 ммоль). Затем смесь перемешивали при 105°C в течение ночи. Смесь охлаждали до 25°C, фильтровали и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле, элюируя смесью PE:EtOAc=10:1–3:1, с получением соединения 502–A4 (диметил-5– ((2,4–диметоксибензил)амино)пиридин–3,4–дикарбоксилат, 1,92 г, 92%) в виде коричневого масла.
1H ЯМР (300 МГц, CDCl3) δ 8,33 (с, 1H), 8,13 (с, 1H), 7,16 (д, J=8,1 Гц, 1H), 6,56–6,60 (м, 1H), 6,43–6,49 (м, 2H), 4,42 (д, J=6,0 Гц, 2H), 3,90–3,81 (м, 12H).
Синтез соединения 502–A5
К раствору соединения 502–A4 (1,92 г, 5,33 ммоль) в 30 мл DCM медленно по каплям добавляли TFA (9 мл) при 0°C. Затем смесь перемешивали при 25°C в течение 2 часов. Растворитель удаляли. Остаток разбавляли водой (100 мл), доводили до pH=8 при помощи Na2CO3, экстрагировали при помощи DCM (100 мл ×2), сушили и концентрировали с получением продукта 502–A5 (диметил-5–аминопиридин–3,4–дикарбоксилат, 1,15 г) в виде коричневого масла.
1H ЯМР (300 МГц, DMSO–d6) δ 8,32 (с, 1H), 7,99 (с, 1H), 6,18 (с, 2H), 3,81 (с, 6H).
Синтез соединения 502–A
К смеси соединения 502–A5 (0,8 г, 3,8 ммоль) в 60 мл THF добавляли KOH (20%, 60 мл). Затем смесь перемешивали при 25°C в течение 3 часов. Растворитель удаляли. Остаток разбавляли водой (20 мл), экстрагировали при помощи EtOAc (20 мл ×2). Водную фазу доводили до pH=3 2N раствором HCl. Растворитель удаляли. Остаток разбавляли EtOH (50 мл), перемешивали при 25°C в течение 1 часа, фильтровали, концентрировали с получением соединения 502–A (5–аминопиридин–3,4–дикарбоновая кислота, 1,2 г) в виде желтого твердого вещества.
Пример 47. Синтез соединения 121

Стадия 1. Синтез соединения 121–B
К раствору соединения 103–A (3–этокси–4–гидроксибензальдегид, 10 г, 60,2 ммоль) и Cs2CO3 (29,43 г, 90,3 ммоль) в DMF (70 мл) и H2O (70 мл) добавляли натрий 2–хлор–2,2–дифторацетат (23 г, 150,5 ммоль). Смесь перемешивали при 100°C в течение ночи. Добавляли воду (500 мл) и смесь экстрагировали при помощи EtOAc (100 мл ×2). Органическую фазу промывали насыщенным солевым раствором (100 мл ×2), сушили, концентрировали и очищали колоночной хроматографией на силикагеле (PE:EtOAc=10:1) с получением соединения 121–B (4– (дифторметокси)– 3–этоксибензальдегид, 2,5 г, 19%) в виде желтого масла.
1H ЯМР (300 МГц, CDCl3) δ 9,92 (с, 1H), 7,48–7,43 (м, 2H), 7,30 (д, J=6,8Гц, 1H), 6,70 (тд, J=99,6, 0,9 Гц, 1H), 4,20–4,14 (м, 2H), 1,50–1,45 (м, 3H).
Стадия 2. Синтез соединения 121–C
Раствор DMSO (2,72 г, 29 ммоль), KOH (0,97 г, 17,3 ммоль) в DMF (50 мл) перемешивали при 30°C в течение 30 минут. К смеси медленно добавляли соединение 121–B (4–(дифторметокси)–3–этоксибензальдегид, 2,5 г, 11,65 ммоль) и перемешивали при 30°C в течение 3 часов. Смесь гасили насыщенным раствором NH4Cl (50 мл), экстрагировали при помощи EtOAc (100 мл ×3), промывали насыщенным солевым раствором (100 мл ×3), объединенную органическую фазу сушили, фильтровали и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией на силикагеле, элюируя смесью PE: EtOAc=5:1–1:1, с получением соединения 121–C (1–(дифторметокси)–2–этокси–4–(2–(метилсульфонил)винил)бензол, 0,45 г, 13%) в виде желтого масла.
1H ЯМР (300 МГц, CDCl3) δ 7,57 (д, J=15,3 Гц, 1H), 7,27–6,39 (м, 5H), 44,14(кв., J=6,9 Гц, 2H), 3,05 (с, 3H), 1,49 (т, J=6,9 Гц, 3H).
Стадия 3. Синтез соединения 121–D
Раствор H3BO3 (0,19 г, 3,08 ммоль) и соединения 121–C (1–(дифторметокси)–2–этокси–4–(2–(метилсульфонил)винил)бензол, 0,45 г, 1,54 ммоль) в NH4OH (60 мл) и диоксане (10 мл) перемешивали в герметично закрытой пробирке в течение 3 дней при 100°C. Смесь экстрагировали при помощи EtOAc (50 мл ×3), органический слой промывали 1N раствором HCl (50 мл ×2), доводили до pH=12 при помощи NaOH, экстрагировали при помощи EtOAc (50 мл ×3), сушили и концентрировали с получением продукта 121–D (1–(4–(дифторметокси)–3–этоксифенил)–2–(метилсульфонил)этанамин, 0,29 г, 61%) в виде бесцветного масла.
1H ЯМР (400 МГц, DMSO–d6) δ 7,22–6,96 (м, 3H), 4,34–4,31 (м, 1H), 4,11 (кв., J=7,2 Гц, 2H), 3,48–3,42 (м, 1H), 3,30–3,25 (м, 1H), 3,02 (с, 3H), 2,33 (ушир. с, 2H), 1,35 (т, J=7,2 Гц, 3H).
Стадия 4. Синтез соединения 121
Смесь соединения 121–D (1–(4–(дифторметокси)–3–этоксифенил)–2–(метилсульфонил)этанамин, 280 мг, 0,906 ммоль) и соединения 101–E (N–(7–фтор–1,3–диоксо–1,3–дигидроизобензофуран–4–ил)ацетамид, 202 мг, 0,906 ммоль) в HOAc (10 мл) перемешивали при 80°C в течение ночи. Затем смесь концентрировали досуха при пониженном давлении. Остаток очищали препаративной ВЭЖХ с получением соединения 121 (N–(2–(1–(4–(дифторметокси)–3–этоксифенил)–2–(метилсульфонил)этил)–7–фтор–1,3–диоксоизоиндолин–4–ил)ацетамид, 235 мг, 50%) в виде белого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6) δ 9,76 (с, 1H), 8,44–8,41 (м, 1H), 7,67 (т, J=9,2 Гц, 1H), 7,25–6,88 (м, 4H), 5,83 (дд, J=10,4, 4,4 Гц, 1H), 4,34–4,19 (м, 2H), 4,12 (кв., J=7,2 Гц, 2H), 3,06 (с, 3H), 2,17 (с, 3H), 1,35 (т, J=7,2 Гц, 3H). ЖХМС: [M+H]+=514,9.
Пример эффективности 1. Анализ ингибирования активности PDE4
Оценивали ингибирующий эффект, значение IC50, соединения на PDE4A1A, PDE4B1 и PDE4D3.
Материалы для эксперимента:
Фермент: PDE4A1A (BPS, № по каталогу 60040); PDE4B1 (BPS, № по каталогу 60041); PDE4D3 (BPS, № по каталогу 60046).
Соединение в качестве положительного контроля: Trequinsin (Sigma, Кат. № T2057).
Реакционный планшет: 384–луночный планшет (Perkin Elmer, Кат.№ 6007279).
Оборудование: счетчик Wallac Victor Multi–lable (Perkin Elmer).
Стадии эксперимента:
I. Получение 1× реакционного раствора и терминирующего раствора;
II. Ферментативная реакция PDE;
1) PDE растворяли в 1× реакционном растворе с образованием 2× раствора фермента.
2) FAM–цАМФ растворяли в 1× реакционном растворе с образованием 2× раствора субстрата.
3) Echo 550 использовали для переноса соответствующего объема соединения в растворе DMSO в реакционный планшет.
4) 2× раствор фермента добавляли в соответствующую лунку реакционного планшета и инкубировали с раствором соединения при комнатной температуре в течение 15 минут.
5) 2× раствор субстрата добавляли в соответствующую лунку реакционного планшета для инициирования реакции.
6) Реакционный планшет инкубировали при комнатной температуре в течение 30 минут и останавливали инкубацию путем добавления терминирующего раствора, затем инкубировали при комнатной температуре в течение 60 минут.
III. Считывание на Victor;
IV. Подбор кривой;
Процент ингибирования рассчитывали при помощи Excel; IC50 рассчитывали при помощи GraphPad Prism.
Соответствующие структуры кодов соединений по настоящему изобретению являются такими, как описано выше, и структуры эталонных соединений являются следующими:
Результаты эксперимента:
Пример эффективности 2. Анализ активности TNF–α
I. Восстановление PBMC и Стадии культивирования клеток:
(1) Восстановление клеток:
1) Перемешивание осуществляли непрерывно на водяной бане при 37°С для быстрого оттаивания клеток.
2) Клетки осторожно добавляли в 15 мл центрифужную пробирку, в которую затем добавляли 10 мл свежей предварительно нагретой восстановительной среды и затем осуществляли центрифугирование при 1000 об/мин в течение 10 мин.
3) Надосадочную жидкость сливали и осуществляли ресуспендирование с 10 мл свежей предварительно нагретой полной среды RPMI 1640.
(2) Посев в 96–луночный планшет:
1) Рассчитывали общее количество клеток, необходимое для эксперимента и доводили до соответствующей концентрации клеток на мл. 100 мкл и 105 клеток на лунку.
2) Клеточную суспензию разбавляли соответствующим объемом среды для культивирования клеток.
3) Клеточную суспензию добавляли в одноразовую стерильную лунку для образца.
4) 100 мкл клеточной суспензии добавляли в каждую лунку 96–луночного планшета.
5) Планшет инкубировали в инкубаторе при 37°C, 5% CO2 в течение 2 часов.
(3) Стадии получения соединения:
1) LPS: 1 мг/мл исходного раствора разбавляли водой, разделяли на аликвоты и хранили при –80°C. Перед каждым испытанием рабочий раствор LPS разбавляли из исходного раствора бессывороточной средой RPMI 1640.
2) Испытываемое соединение
20 мМ исходного раствора растворяли в DMSO и соединение проверяли на растворимость, разделяли на аликвоты и хранили при –80°C.
(4) Получение градиента соединения 8X:
Серию градиента концентрации соединения разбавляли DMSO: получали 10 мМ, 2 мМ, 0,4 мМ, 80 мкМ, 16 мкМ, 3,2 мкМ, 0,64 мкМ, 0,128 мкМ и затем соединения 125–кратно разбавляли бессывороточной RPMI 1640 средой до конечной 8X. Конечная концентрация DMSO в клеточной культуре составляла 0,1%.
(5) Экспериментальные процедуры обработки соединения и сбор супернатантов:
1) Посев клеток: свежие клетки высевали в 96–луночные планшеты для культивирования клеток в соответствии с описанной выше процедурой, 100 мкл и 105 клеток на лунку, и затем инкубировали в инкубаторе при 37°C, 5% CO2 в течение 2 часов.
2) Подготовка соединений: Перед испытанием соединения добавляли в планшеты в соответствии с приведенным выше описанием. Дозу соединения в 8Х концентрации получали с использованием бессывороточной RPMI 1640 среды и все градиенты раствора добавляли в планшет с соединениями.
3) Добавление соединения: 16,7 мкл раствора соединения в рабочей концентрации добавляли в каждую лунку планшета для культивирования клеток. Планшет инкубировали в инкубаторе при 37°C, 5% CO2 в течение 1 часа.
4) Добавляли 16,7 мкл 8X LPS на лунку (конечная концентрация LPS представляет собой EC80, количество каждой РВМС, которое необходимо определить). Планшет инкубировали в течение 18 часов в инкубаторе при 37°C, 5% CO2.
5) 80 мкл супернатанта на лунку собирали и затем подвергали ELISA анализу TNF–α. Собранный супернатант можно хранить при –80°С. Надосадочную жидкость необходимо разбавлять в различных соотношениях, чтобы экспериментальная доза не превышала линейный диапазон стандартной кривой TNF–α, в зависимости от количества TNF–α, высвобождаемого у разных доноров. Типично 20–100 мкл супернатанта разбавляли до 200 мкл и затем использовали для экспериментов ELISA.
(6) Стадии ELISA TNF–α:
Процедуру испытания ELISA TNF–α осуществляют в соответствии с экспериментальной процедурой с использованием набора BD human TNF–α ELISA.
План эксперимента:
Четыре соединения на планшет. Осуществляли 5–кратное разбавление, начиная с 10 мкМ, с 8 градиентами и делали параллельные лунки. TNF–α стандарт добавляли в каждый планшет. (1ая лунка, начиная с 500 пг/мл, 2–кратное разбавление, 7 градиентов)
Для ZPE (0% ингибирование) использовали 15 пг/мл LPS+0,1% DMSO, в то время как для HPE (100% ингибирование) использовали только 0,1% DMSO.
Рассчитывали статистику степени ингибирования. Степень ингибирования (%)=[1– (Max–Min)/(Испытываем.соед.– Min)]×100%. IC50 использовали для оценки концентрации испытываемого соединения (нМ) при 50% ингибировании.
Пример эффективности 3. Испытание фармакокинетических (PK) параметров
1. Цель испытания
Испытываемые соединения вводили внутривенно или внутрижелудочно однократно крысам SD. Образцы крови собирали в разные моменты времени. ЖХ–МС/МС использовали для определения концентрации испытываемых соединений в плазме крыс после введения испытываемых соединений и расчета соответствующих PK параметров.
2. План эксперимента
2.1. Подготовка испытываемых соединений
Испытываемые соединения рассчитывали на основании свободных радикалов и преобразовывали только по чистоте.
2.1.1. Группа внутривенной инъекции
Подходящее количество испытываемого соединения добавляли к 5% DMSO+95% HP–бета–CD (20%) с получением раствора 0,6 мг/мл для внутривенного введения.
2.1.2. Группа перорального введения
Подходящее количество испытываемого соединения добавляли к 5% DMSO+95% HP–бета–CD (20%) с получением раствора 1 мг/мл для внутрижелудочного введения.
2.2. Доза и путь введения
Самцов крыс Sprague–Dawley приобретали у Shanghai Xipuer–Bikai Laboratory Animal Co., Ltd. В каждой группе было по 3 крысы. Одну группу использовали в качестве контроля для сбора чистой плазмы. Каждая из других групп получала испытываемое соединение путем внутривенной инъекции (доза составляла 3 мг/кг) или внутрижелудочного введения (доза составляла 10 мг/кг). Гепарин натрия использовали для антикоагуляции. Анализировали концентрацию испытываемого соединения в образце крови.
Голодание в течение 10–14 часов перед пероральным введением испытываемых соединений и возобновление питания через 4 часа после перорального введения испытываемых соединений.
2.3. Детальное клиническое наблюдение
Группа, получавшая внутривенную инъекцию. До и после введения не наблюдалось никаких явных аномальных состояний в каждый момент сбора крови.
Группа перорального введения: мягкий стул наблюдали через 4–8 часов после введения в каждой группе, и все животные восстанавливались на следующий день.
2.4. Сбор и обработка образцов
Временные точки взятия образцов крови были стедующими: внутривенная инъекция: до введения и через 0,083 часа, 0,25 часа, 0,5 часа, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 24 часа после введения; пероральное введение: до введения и через 0,25 часа, 0,5 часа, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 24 часа после введения. Образцы крови собирали путем пункции яремной вены. Для каждого образца собрирали около 0,25 мл крови. Гепарин–натрий использовали для антикоагуляции, и образцы помещали на лед после сбора. Образцы крови помещали на лед после сбора и плазму отделяли центрифугированием (условия центрифугирования: 8000 об/мин, 6 мин, 2–8°С). Собранную плазму хранили при –80°C до анализа.
3. Аналитические методы
3.1. Лекарственные средства и реагенты
Испытываемые соединения: предоставлены компанией Kangpu Biopharmaceuticals, Ltd.
Внутренний стандарт толуолсульфонилмочевина: предоставлен учреждением, осуществляющим испытания.
Метанол (Burdick & Jackson, ВЭЖХ), ацетонитрил (Burdick & Jackson, ВЭЖХ), муравьиная кислота (J & K), вода представляет собой сверхчистую воду.
3.2. Оборудование
Сверхвысокоэффективная жидкостная хроматография (Waters, ACQUITY UPLC), включая бинарное устройство для подачи растворителя (ACQUITY UPLC Binary Solvent Manager), пробоотборник (ACQUITY UPLC Autosampler Mod.), органайзер образцов с высокой пропускной способностью (ACQUTIY UPLC Sample Organizer), высокотемпературное отделение колонки (ACQUITY UPLC Column Heater HT). Масс–спектрометр (API 4000, Bio System Inc, USA), источник электрораспылительной ионизации (ESI), серийный квадрупольный масс–анализатор. Система обработки данных представляет собой программу Analyst (American Applied Biosystems Inc., version 1.5.1).
3.3. Аналитические методы
ЖХ–МС/МС определение.
Предварительная обработка образца
50 мкл образца плазмы добавляли в 1,5–мл центрифужную пробирку и к образцу добавляли 250 мкл раствора внутреннего стандарта (такой же объем метанола добавляли к чистому образцу вместо внутреннего стандарта). Образец смешивали вихревым способом и центрифугировали в течение 5 минут при 14000 об/мин. Супернатант 200 мкл добавляли в 96–луночный планшет для ЖХ–МС/МС анализа.
4. Фармакокинетические результаты
Фармакокинетические параметры испытываемых соединений рассчитывали с использованием некомпартментной модели WinNonlin v5.2, программы для расчета фармакокинетики, на основе данных концентрации в плазме. Результаты эксперимента приведены в таблице ниже.
(п/о)
(п/о)
(в/в)
(п/о)
соединение 2
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ БРОМДОМЕНА | 2012 |
|
RU2671571C1 |
| ИНГИБИТОРЫ БРОМДОМЕНА | 2012 |
|
RU2647592C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2734390C2 |
| 3-КАРБОНИЛАМИНО-5-ЦИКЛОПЕНТИЛ-1H-ПИРАЗОЛЬНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ ИНГИБИТОРОВ CDK2 | 2020 |
|
RU2797889C2 |
| ПРОИЗВОДНЫЕ АРИЛАМИДОВ В КАЧЕСТВЕ БЛОКАТОРОВ TTX-S | 2011 |
|
RU2535671C1 |
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ ДЛЯ ИНГИБИРОВАНИЯ SUV39H2 | 2016 |
|
RU2729187C1 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| ЗАМЕЩЕННЫЕ ПИРИДИНОВЫЕ И ПИРАЗИНОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE4 | 2014 |
|
RU2802185C2 |
| СОЕДИНЕНИЯ, ЯВЛЯЮЩИЕСЯ АНТАГОНИСТАМИ А3-АДЕНОЗИНОВОГО РЕЦЕПТОРА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2737157C2 |
| ДЕСТРУКТОРЫ БЕЛКА MDM2 | 2017 |
|
RU2743432C2 |
Изобретение относится к соединению формулы I

или его фармацевтической приемлемой соли, стереоизомеру или изотопному соединению, где атом углерода, помеченный звездочкой *, представляет собой центр асимметрии; R1 и R2 независимо представляют собой H, R6-S(O)2- или R6-C(O)-; R6 представляет собой незамещенный (C3)циклоалкил; или (C1-C6)алкил, который необязательно замещен одной или более группами, выбранными из D, галогена, гидроксила, амино, (C1-C6)алкиламино, (C1-C6)алкокси и бензилокси; X1, X2, X3, X4, X5 и X6 независимо представляют собой CH, CR7 или N; R7 представляет собой галоген или циано; R3 и R4 независимо представляют собой H, замещенный или незамещенный (C1-C6)алкил; R5 представляет собой незамещенный (C1-C6)алкил; R10, R11 и R12 независимо представляют собой H или D; заместитель в замещенном (C1-C6)алкиле представляет собой один или более, выбранных из группы, состоящей из: D, галогена, гидроксила, амино, (C1-C6)алкиламино, (C1-C6)алкокси и бензилокси; когда присутствуют несколько заместителей, эти заместители являются одинаковыми или отличными друг от друга; при условии, что: один из X1, X2, X3, X4, X5 и X6 представляет собой N, которе может регулировать генерацию и/или активность PDE4 и/или TNF–α. 6 н. и 14 з.п. ф-лы, 47 пр., 2 табл.
1. Соединение формулы I или его фармацевтически приемлемая соль, стереоизомер или изотопное соединение

где атом углерода, помеченный звездочкой *, представляет собой центр асимметрии;
R1 и R2 независимо представляют собой H, R6-S(O)2- или R6-C(O)-;
R6 представляет собой незамещенный (C3)циклоалкил; или (C1-C6)алкил, который необязательно замещен одной или более группами, выбранными из D, галогена, гидроксила, амино, (C1-C6)алкиламино, (C1-C6)алкокси и бензилокси;
X1, X2, X3, X4, X5 и X6 независимо представляют собой CH, CR7 или N;
R7 представляет собой галоген или циано;
R3 и R4 независимо представляют собой H, замещенный или незамещенный (C1-C6)алкил;
R5 представляет собой незамещенный (C1-C6)алкил;
R10, R11 и R12 независимо представляют собой H или D;
заместитель в замещенном (C1-C6)алкиле представляет собой один или более, выбранных из группы, состоящей из: D, галогена, гидроксила, амино, (C1-C6)алкиламино, (C1-C6)алкокси и бензилокси; когда присутствуют несколько заместителей, эти заместители являются одинаковыми или отличными друг от друга;
при условии, что: один из X1, X2, X3, X4, X5 и X6 представляет собой N.
2. Соединение формулы I по п. 1 или его фармацевтически приемлемая соль, стереоизомер или изотопное соединение, где
X1 представляет собой N, X2, X3, X4, X5 и X6 независимо представляют собой CH или CR7; или
X2 представляет собой N, X1, X3, X4, X5 и X6 независимо представляют собой CH или CR7; или
X3 представляет собой N, X1, X2, X4, X5 и X6 независимо представляют собой CH или CR7; или
X4 представляет собой N, X1, X2, X3, X5 и X6 независимо представляют собой CH или CR7; или
X5 представляет собой N, X1, X2, X3, X4 и X6 независимо представляют собой CH или CR7; или
X6 представляет собой N, X1, X2, X3, X4 и X5 независимо представляют собой CH или CR7.
3. Соединение формулы I по п. 1 или 2 или его фармацевтически приемлемая соль, стереоизомер или изотопное соединение, где
X6 представляет собой N, X1, X2, X3 независимо представляют собой CH или CR7, X4, X5 независимо представляют собой CH; или
X6 представляет собой N, X1 представляет собой CR7, X2, X3, X4 и X5 независимо представляют собой CH; или
X6 представляет собой N, X2 представляет собой CR7, X1, X3, X4 и X5 независимо представляют собой CH; или
X6 представляет собой N, X3 представляет собой CR7, X1, X2, X4 и X5 независимо представляют собой CH.
4. Соединение формулы I по любому одному из пп. 1-3 или его фармацевтически приемлемая соль, стереоизомер или изотопное соединение, где
один из R1 и R2 представляет собой H, другой представляет собой R6-S(O)2- или R6-C(O)-.
5. Соединение формулы I по любому одному из пп. 1-4 или его фармацевтически приемлемая соль, стереоизомер или изотопное соединение, где
R6 представляет собой (C3)циклоалкил; или (C1-C4)алкил, который необязательно замещен одним или более заместителями, выбранными из группы, состоящей из D, галогена, гидроксила, амино, (C1-C4)алкиламино, (C1-C4)алкокси и бензилокси,
предпочтительно R6 представляет собой циклопропил, метил, этил, гидроксиметил, бензилоксиметил, метоксиметил, изобутил, диметиламинометил, изопропил, CD3 или C2D5.
6. Соединение формулы I по любому одному из пп. 1-5 или его фармацевтически приемлемая соль, стереоизомер или изотопное соединение, где
R3 и R4 независимо представляют собой H, замещенный или незамещенный (C1-C6)алкил;
предпочтительно R3 и R4 независимо представляют собой H, метил, этил, пропил, изопропил, CD3, CH2D, CHD2, C2D5, CH2CD3 или CHF2.
7. Соединение формулы I по любому одному из пп. 1-6 или его фармацевтически приемлемая соль, стереоизомер или изотопное соединение, где
R5 представляет собой метил, этил, пропил или изопропил.
8. Соединение формулы I по любому одному из пп. 1-7 или его фармацевтически приемлемая соль, стереоизомер или изотопное соединение, где
R7 представляет собой фтор, хлор, бром или циано.
9. Соединение формулы I по любому одному из пп. 1-8 или его фармацевтически приемлемая соль, стереоизомер или изотопное соединение, где
X6 представляет собой N, X1, X2, X3, X4 и X5 независимо представляют собой CH или CR7;
R7 представляет собой галоген или циано.
10. Соединение формулы I по любому одному из пп. 1-9 или его фармацевтически приемлемая соль, стереоизомер или изотопное соединение, где
R10 и R11 представляют собой H.
11. Соединение формулы I по любому одному из пп. 1-10 или его фармацевтически приемлемая соль, стереоизомер или изотопное соединение, где
R12 представляет собой H.
12. Соединение формулы I по любому одному из пп. 1-11 или его фармацевтически приемлемая соль, стереоизомер или изотопное соединение, где
центр асимметрии относится к углероду, имеющему (S)-конфигурацию.
13. Соединение формулы I по любому одному из пп. 1-12 или его фармацевтически приемлемая соль, стереоизомер или изотопное соединение, где соединение формулы I представляет собой любое из следующих соединений:





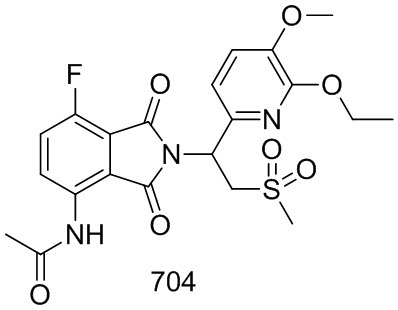


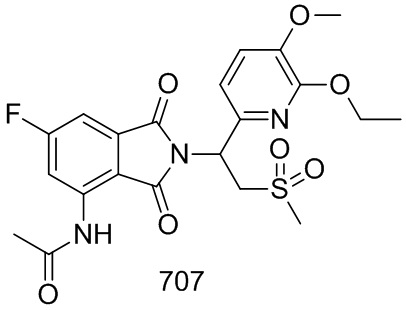

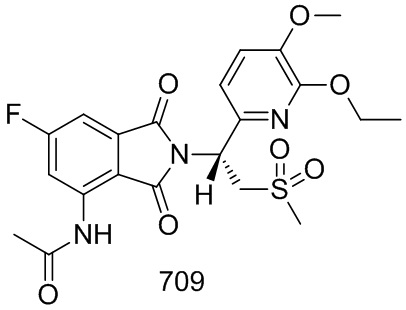

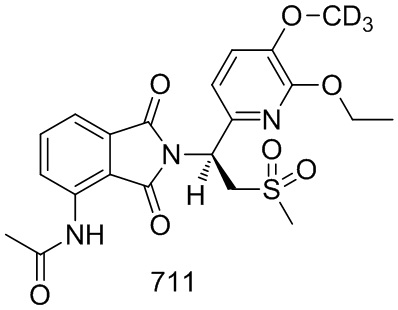







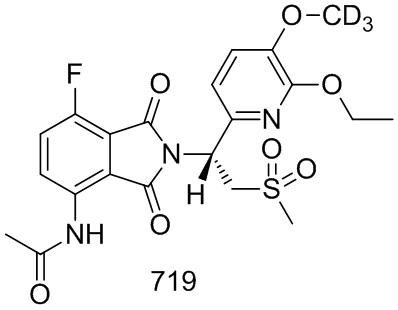

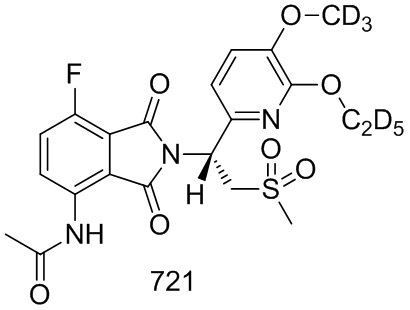

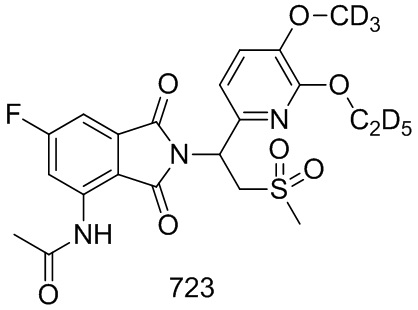


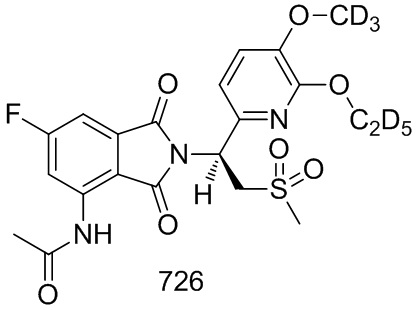







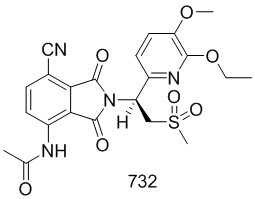

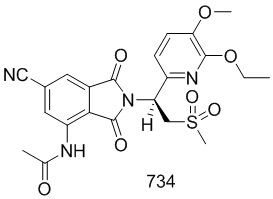


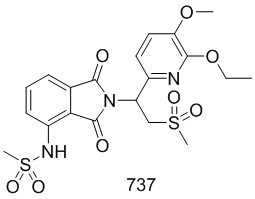
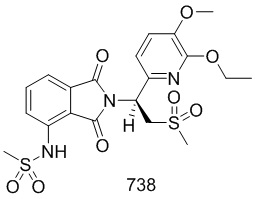



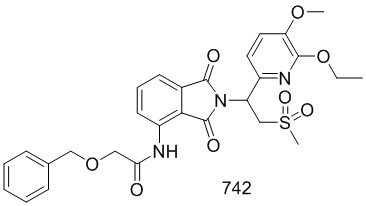





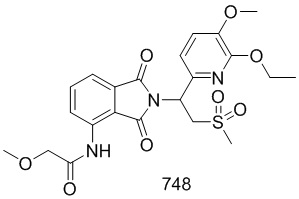
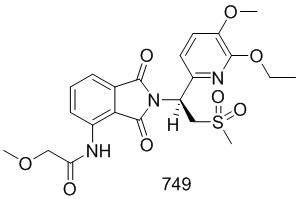














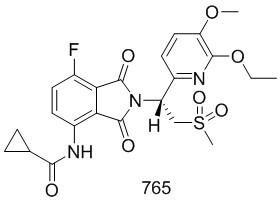







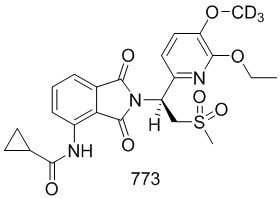
 .
.
14. Способ получения соединения формулы I по любому одному из пп. 1-13, включающий следующие стадии:
соединение формулы I-A и соединение формулы I-B подвергают взаимодействию следующим образом с получением соединения формулы I;

где соединение формулы I-A и соединение формулы I-B подвергают взаимодействию в присутствии кислоты;
температура реакции составляет 10–120 °C; и
где X1, X2, X3, X4, X5, X6, R1, R2, R3, R4, R5, R10, R11 и R12 имеют значения, определенные в любом из пп. 1-13.
15. Способ по п. 14,
где соединение формулы I-A и соединение формулы I-B подвергают взаимодействию в присутствии уксусной кислоты.
16. Способ получения соединения формулы I по любому одному из пп. 1-13, включающий следующие стадии:
соединение формулы I-3 и соединение формулы I-4 подвергают взаимодействию следующим образом с получением соединения формулы I;

где температура реакции составляет 0–75 °C; и
где X1, X2, X3, X4, X5, X6, R1, R2, R3, R4, R5, R10, R11 и R12 имеют значения, определенные в любом из пп. 1-13; Y представляет собой галоген.
17. Соединение формулы I-3:

где X1, X2, X3, X4, X5, X6, R1, R2, R3, R4, R5, R10, R11 и R12 имеют значения, определенные в любом из пп. 1-13.
18. Фармацевтическая композиция, ингибирующая генерацию или активность PDE4 и/или TNF–α, содержащая терапевтически эффективное количество одного или более соединений формулы I по любому из пп. 1-13, его фармацевтически приемлемой соли, стереоизомера или изотопного соединения и один или более фармацевтических эксципиентов.
19. Применение одного или более соединений формулы I по любому из пп. 1-13, его фармацевтически приемлемой соли, стереоизомера или изотопного соединения для получения лекарственного средства для ингибирования генерации или активности PDE4 и/или TNF-α или для получения лекарственного средства для лечения или профилактики заболевания, расстройства или состояния, связанного с аномальной генерацией или регуляцией PDE4 и/или TNF-α.
20. Применение по п. 19, где
заболевание, расстройство или состояние представляет собой псориатический артрит или бляшковидный псориаз.
| WO 2000025777 A1, 11.05.2000 | |||
| WO 2004060313 A2, 22.07.2004 | |||
| WO 2015175773 A1, 19.11.2015 | |||
| WO 2015175956 A1, 19.11.2015 | |||
| WO 2014072259 A1, 15.05.2014 | |||
| Man, H.-W., Schafer, P., Wong, L | |||
| M., Patterson, R | |||
| T., Corral, L | |||
| G., Raymon, H | |||
| Muller, G | |||
| W | |||
| Колосоуборка | 1923 |
|
SU2009A1 |
| Discovery of (S)-N-{ | |||

